User login
Wilson disease
To the Editor: We read the IM Board Review article by Hanouneh et al in the February issue of the Journal with great interest.1 The authors described an interesting case of a young woman presenting with what initially seemed to be jaundice of acute onset, with rapid progression to acute encephalopathy and worsening liver failure. The patient was eventually diagnosed with fulminant Wilson disease and, thankfully, underwent successful liver transplant. We thank the authors for their in-depth review of the common causes of acute liver failure, the general approach to management, and the tailored treatment of Wilson disease in such settings.
However, we believe that several aspects merit further attention. First, on initial presentation and investigation, it would have been important to consider cholestatic hepatobiliary pathologic processes (eg, choledocholithiasis, cholangitis, primary biliary cirrhosis, primary sclerosing cholangitis), given the characteristic liver panel results.
Second, the authors rightly pointed out that hemolytic anemia is common in patients with acute liver failure secondary to Wilson disease. However, it is important to keep in mind that additional testing should include Coombs testing (typically negative in Wilson disease) and examination of the peripheral smear to exclude other etiologies, since such conditions as thrombotic thrombocytopenic purpura may present with multiorgan failure as well.2
Third, the authors report that Kayser-Fleischer rings are pathognomonic for Wilson disease. However, many reports in peer-reviewed medical journals suggest that this may not be the case and the overall clinical picture should be
considered.3
Fourth, while the authors focus their attention on liver transplant, several other treatments deserve mentioning. We agree that liver transplant is considered the only lifesaving treatment. But in certain situations, molecular absorbent recirculation systems and hemodialysis may provide temporary support while awaiting transportation to a liver transplant center or actual liver transplant.4
- Hanouneh MA, Garber A, Tavill AS, Zein NN, Hanouneh IA. A tale of two sisters with liver disease. Cleve Clin J Med 2016; 83:109–115.
- Nguyen TC, Cruz MA, Carcillo JA. Thrombocytopenia-associated multiple organ failure and acute kidney injury. Crit Care Clin 2015; 31:661–674.
- Frommer D, Morris J, Sherlock S, Abrams J, Newman S. Kayser-Fleischer-like rings in patients without Wilson’s disease. Gastroenterology 1977; 72:1331–1335.
- Hamlyn AN, Gollan JL, Douglas AP, Sherlock S. Fulminant Wilson’s disease with haemolysis and renal failure: copper studies and assessment of dialysis regimens. Br Med J 1977; 2:660–663.
To the Editor: We read the IM Board Review article by Hanouneh et al in the February issue of the Journal with great interest.1 The authors described an interesting case of a young woman presenting with what initially seemed to be jaundice of acute onset, with rapid progression to acute encephalopathy and worsening liver failure. The patient was eventually diagnosed with fulminant Wilson disease and, thankfully, underwent successful liver transplant. We thank the authors for their in-depth review of the common causes of acute liver failure, the general approach to management, and the tailored treatment of Wilson disease in such settings.
However, we believe that several aspects merit further attention. First, on initial presentation and investigation, it would have been important to consider cholestatic hepatobiliary pathologic processes (eg, choledocholithiasis, cholangitis, primary biliary cirrhosis, primary sclerosing cholangitis), given the characteristic liver panel results.
Second, the authors rightly pointed out that hemolytic anemia is common in patients with acute liver failure secondary to Wilson disease. However, it is important to keep in mind that additional testing should include Coombs testing (typically negative in Wilson disease) and examination of the peripheral smear to exclude other etiologies, since such conditions as thrombotic thrombocytopenic purpura may present with multiorgan failure as well.2
Third, the authors report that Kayser-Fleischer rings are pathognomonic for Wilson disease. However, many reports in peer-reviewed medical journals suggest that this may not be the case and the overall clinical picture should be
considered.3
Fourth, while the authors focus their attention on liver transplant, several other treatments deserve mentioning. We agree that liver transplant is considered the only lifesaving treatment. But in certain situations, molecular absorbent recirculation systems and hemodialysis may provide temporary support while awaiting transportation to a liver transplant center or actual liver transplant.4
To the Editor: We read the IM Board Review article by Hanouneh et al in the February issue of the Journal with great interest.1 The authors described an interesting case of a young woman presenting with what initially seemed to be jaundice of acute onset, with rapid progression to acute encephalopathy and worsening liver failure. The patient was eventually diagnosed with fulminant Wilson disease and, thankfully, underwent successful liver transplant. We thank the authors for their in-depth review of the common causes of acute liver failure, the general approach to management, and the tailored treatment of Wilson disease in such settings.
However, we believe that several aspects merit further attention. First, on initial presentation and investigation, it would have been important to consider cholestatic hepatobiliary pathologic processes (eg, choledocholithiasis, cholangitis, primary biliary cirrhosis, primary sclerosing cholangitis), given the characteristic liver panel results.
Second, the authors rightly pointed out that hemolytic anemia is common in patients with acute liver failure secondary to Wilson disease. However, it is important to keep in mind that additional testing should include Coombs testing (typically negative in Wilson disease) and examination of the peripheral smear to exclude other etiologies, since such conditions as thrombotic thrombocytopenic purpura may present with multiorgan failure as well.2
Third, the authors report that Kayser-Fleischer rings are pathognomonic for Wilson disease. However, many reports in peer-reviewed medical journals suggest that this may not be the case and the overall clinical picture should be
considered.3
Fourth, while the authors focus their attention on liver transplant, several other treatments deserve mentioning. We agree that liver transplant is considered the only lifesaving treatment. But in certain situations, molecular absorbent recirculation systems and hemodialysis may provide temporary support while awaiting transportation to a liver transplant center or actual liver transplant.4
- Hanouneh MA, Garber A, Tavill AS, Zein NN, Hanouneh IA. A tale of two sisters with liver disease. Cleve Clin J Med 2016; 83:109–115.
- Nguyen TC, Cruz MA, Carcillo JA. Thrombocytopenia-associated multiple organ failure and acute kidney injury. Crit Care Clin 2015; 31:661–674.
- Frommer D, Morris J, Sherlock S, Abrams J, Newman S. Kayser-Fleischer-like rings in patients without Wilson’s disease. Gastroenterology 1977; 72:1331–1335.
- Hamlyn AN, Gollan JL, Douglas AP, Sherlock S. Fulminant Wilson’s disease with haemolysis and renal failure: copper studies and assessment of dialysis regimens. Br Med J 1977; 2:660–663.
- Hanouneh MA, Garber A, Tavill AS, Zein NN, Hanouneh IA. A tale of two sisters with liver disease. Cleve Clin J Med 2016; 83:109–115.
- Nguyen TC, Cruz MA, Carcillo JA. Thrombocytopenia-associated multiple organ failure and acute kidney injury. Crit Care Clin 2015; 31:661–674.
- Frommer D, Morris J, Sherlock S, Abrams J, Newman S. Kayser-Fleischer-like rings in patients without Wilson’s disease. Gastroenterology 1977; 72:1331–1335.
- Hamlyn AN, Gollan JL, Douglas AP, Sherlock S. Fulminant Wilson’s disease with haemolysis and renal failure: copper studies and assessment of dialysis regimens. Br Med J 1977; 2:660–663.
In reply: Wilson disease
In Reply: We thank Dr. Mirrakhimov and colleagues for bringing important questions to our attention.
In terms of the differential diagnosis of cholestatic liver injury, we agree that pathologic processes such choledocholithiasis, cholangitis, primary biliary cirrhosis, and primary sclerosing cholangitis should be generally considered. However, in the case we described, the patient had no abdominal pain or fever, which makes choledocholithiasis or cholangitis very unlikely. Primary biliary cirrhosis and primary sclerosing cholangitis can cause chronic liver disease but should not be considered in the differential diagnosis of acute liver injury (acute hepatitis), such as in the case we described.
We agree that the hemolytic anemia typically seen in patients with Wilson disease is Coombs-negative, and that Coombs testing and a peripheral smear should be performed. Both were negative in our patient.
We also agree with Dr. Mirrakhimov and colleagues that Kayser-Fleischer rings are not necessarily specific for Wilson disease and can be seen in patients with other forms of cholestatic liver disease such as primary biliary cirrhosis. However, Kayser-Fleischer rings are pathognomonic for acute liver failure from Wilson disease. In other words, when Kayser-Fleischer rings are seen in a patient with acute liver failure, the diagnosis is Wilson disease until proven otherwise.
We discussed on page 112 of our article other treatments such as plasmapheresis as adjunctive therapy to bridge patients with acute liver failure secondary to Wilson disease to transplant. However, liver transplant is still the only definitive and potentially curative treatment.
In Reply: We thank Dr. Mirrakhimov and colleagues for bringing important questions to our attention.
In terms of the differential diagnosis of cholestatic liver injury, we agree that pathologic processes such choledocholithiasis, cholangitis, primary biliary cirrhosis, and primary sclerosing cholangitis should be generally considered. However, in the case we described, the patient had no abdominal pain or fever, which makes choledocholithiasis or cholangitis very unlikely. Primary biliary cirrhosis and primary sclerosing cholangitis can cause chronic liver disease but should not be considered in the differential diagnosis of acute liver injury (acute hepatitis), such as in the case we described.
We agree that the hemolytic anemia typically seen in patients with Wilson disease is Coombs-negative, and that Coombs testing and a peripheral smear should be performed. Both were negative in our patient.
We also agree with Dr. Mirrakhimov and colleagues that Kayser-Fleischer rings are not necessarily specific for Wilson disease and can be seen in patients with other forms of cholestatic liver disease such as primary biliary cirrhosis. However, Kayser-Fleischer rings are pathognomonic for acute liver failure from Wilson disease. In other words, when Kayser-Fleischer rings are seen in a patient with acute liver failure, the diagnosis is Wilson disease until proven otherwise.
We discussed on page 112 of our article other treatments such as plasmapheresis as adjunctive therapy to bridge patients with acute liver failure secondary to Wilson disease to transplant. However, liver transplant is still the only definitive and potentially curative treatment.
In Reply: We thank Dr. Mirrakhimov and colleagues for bringing important questions to our attention.
In terms of the differential diagnosis of cholestatic liver injury, we agree that pathologic processes such choledocholithiasis, cholangitis, primary biliary cirrhosis, and primary sclerosing cholangitis should be generally considered. However, in the case we described, the patient had no abdominal pain or fever, which makes choledocholithiasis or cholangitis very unlikely. Primary biliary cirrhosis and primary sclerosing cholangitis can cause chronic liver disease but should not be considered in the differential diagnosis of acute liver injury (acute hepatitis), such as in the case we described.
We agree that the hemolytic anemia typically seen in patients with Wilson disease is Coombs-negative, and that Coombs testing and a peripheral smear should be performed. Both were negative in our patient.
We also agree with Dr. Mirrakhimov and colleagues that Kayser-Fleischer rings are not necessarily specific for Wilson disease and can be seen in patients with other forms of cholestatic liver disease such as primary biliary cirrhosis. However, Kayser-Fleischer rings are pathognomonic for acute liver failure from Wilson disease. In other words, when Kayser-Fleischer rings are seen in a patient with acute liver failure, the diagnosis is Wilson disease until proven otherwise.
We discussed on page 112 of our article other treatments such as plasmapheresis as adjunctive therapy to bridge patients with acute liver failure secondary to Wilson disease to transplant. However, liver transplant is still the only definitive and potentially curative treatment.
In Middle of Trip, Woman Falls
Answer
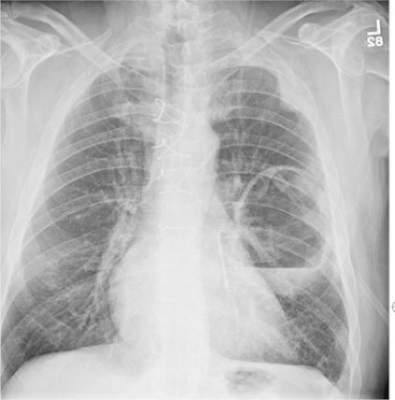
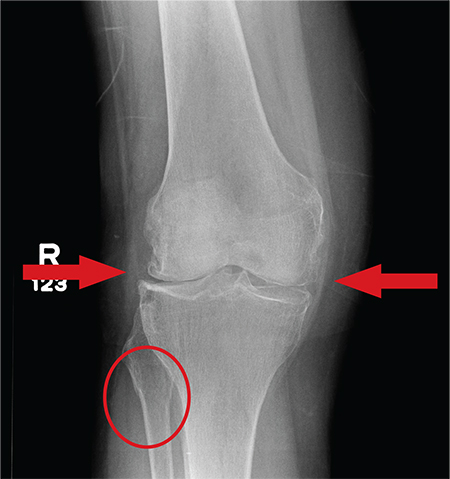
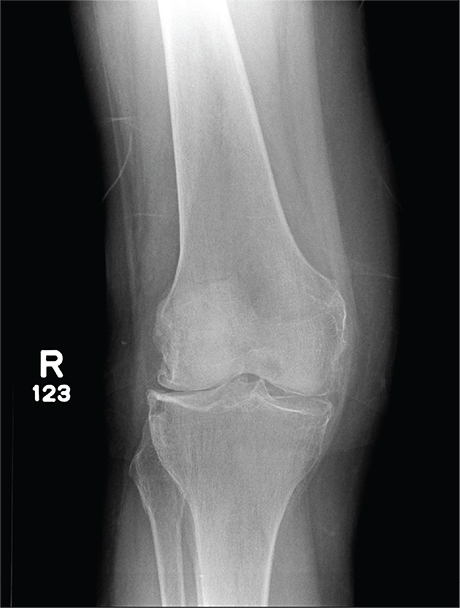
The radiograph has several findings, one of which is a nondisplaced proximal fibula fracture. In addition, there is a moderate suprapatellar joint effusion. The patient also has fairly advanced tricompartment degenerative arthrosis. (To review, the tricompartment comprises all three anatomic areas of the knee: the patellofemoral, lateral tibiofemoral, and medial tibiofemoral joints.)
Answer
The radiograph has several findings, one of which is a nondisplaced proximal fibula fracture. In addition, there is a moderate suprapatellar joint effusion. The patient also has fairly advanced tricompartment degenerative arthrosis. (To review, the tricompartment comprises all three anatomic areas of the knee: the patellofemoral, lateral tibiofemoral, and medial tibiofemoral joints.)
Answer
The radiograph has several findings, one of which is a nondisplaced proximal fibula fracture. In addition, there is a moderate suprapatellar joint effusion. The patient also has fairly advanced tricompartment degenerative arthrosis. (To review, the tricompartment comprises all three anatomic areas of the knee: the patellofemoral, lateral tibiofemoral, and medial tibiofemoral joints.)
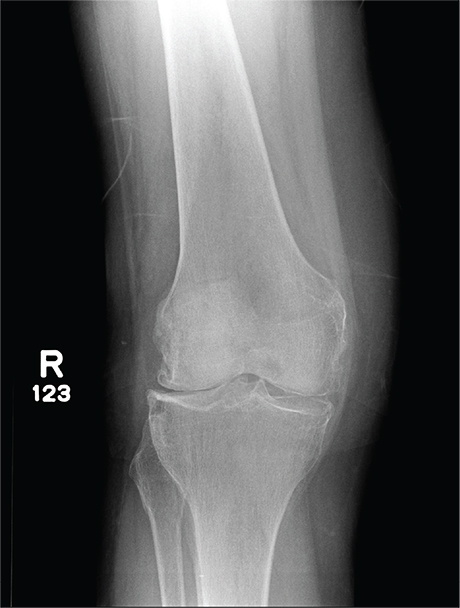
A 70-year-old woman presents to your emergency department for evaluation of right knee pain secondary to a fall. She and her husband, in the process of driving from Florida to their home in California, stopped for the night in your town. The patient states that shortly after getting up this morning, she tripped, lost her balance, and fell. All her weight landed on her right knee; she says it is now “extremely painful” to bear weight on that leg. She also twisted her right ankle, causing additional discomfort. Her medical history is significant for hypertension, which is controlled by medication. On physical exam, you note an elderly female who is uncomfortable but in no obvious distress. Inspection of her right knee shows no obvious deformity but a moderate amount of swelling. The patient has limited range of motion secondary to the swelling. She also has moderate tenderness circumferentially around the knee. There is additional swelling and mild bruising on both the medial and lateral aspects of the right ankle. You obtain a radiograph of the right knee. What is your impression?
Cognitive bias and diagnostic error
To the Editor: I appreciated the article on cognitive biases and diagnostic error by Mull et al in the November 2015 issue.1 They presented an excellent description of the pitfalls of diagnosis as reflected in a case of a patient misdiagnosed with heart failure who ultimately died of pulmonary tuberculosis complicated by pulmonary embolism (the latter possibly from using the wrong form of heparin). To the points they raised, I would like to add a few of my own about diagnosis in general and heart failure in particular.
First, any initial diagnosis not confirmed objectively within the first 24 hours should be questioned, and other possibilities should be investigated. I have found this to be essential for every day’s stay in the hospital and for every outpatient visit. The authors mention checklists as part of the solution to the problem of misdiagnosis, and I would suggest that confirmation of initial diagnoses be built into these checklists.
In the case of a presumptive diagnosis of an acute exacerbation of heart failure treated empirically with diuretics, the diagnosis should be confirmed by the next day’s response to the diuretics, ie, increased urine output, a lower respiratory rate, and a fall in the pro-B-type natriuretic peptide level. Moreover, a change in the radiographic appearance should be seen, and respiratory and pulmonary function should improve after the first 24 hours on oxygen supplementation plus diuretics. Daily patient weights are also critical in determining response to a diuretic, and are rarely done accurately. I order weights and review them daily for patients like this.
Second, it is good to look at things yourself, including the patient, medication lists, laboratory values, and radiographic films. The attending physician should look at the radiographs together with a senior radiologist. Seeing no improvement or change on the second hospital day, or seeing signs incompatible with heart failure, one could order computed tomography of the chest and begin to entertain pulmonary diagnoses.
Even vital signs can be questionable. For example, in the case presented here, with a temperature of 99°F, a heart rate of 105, and a pulse oxygenation saturation of 89%, a respiratory rate of 24 seems unbelievably low. In my experience, the respiratory rate is recorded erroneously most of the time unless it is recorded electronically or checked at the bedside by the physician using a timepiece with a sweep second-hand.
Additionally, I have found that ordering several days’ laboratory tests (eg, complete blood cell counts, chemistry panels) in advance, in many cases, risks missing important findings and wastes time, energy, and the patient’s blood. I have learned to evaluate each patient daily and to order the most pertinent laboratory tests. With electronic medical records, I can check laboratory results as soon as they are available.
- Mull N, Reilly JB, Myers JS. An elderly woman with ‘heart failure’: cognitive biases and diagnostic error. Cleve Clin J Med 2015; 82:745–753.
To the Editor: I appreciated the article on cognitive biases and diagnostic error by Mull et al in the November 2015 issue.1 They presented an excellent description of the pitfalls of diagnosis as reflected in a case of a patient misdiagnosed with heart failure who ultimately died of pulmonary tuberculosis complicated by pulmonary embolism (the latter possibly from using the wrong form of heparin). To the points they raised, I would like to add a few of my own about diagnosis in general and heart failure in particular.
First, any initial diagnosis not confirmed objectively within the first 24 hours should be questioned, and other possibilities should be investigated. I have found this to be essential for every day’s stay in the hospital and for every outpatient visit. The authors mention checklists as part of the solution to the problem of misdiagnosis, and I would suggest that confirmation of initial diagnoses be built into these checklists.
In the case of a presumptive diagnosis of an acute exacerbation of heart failure treated empirically with diuretics, the diagnosis should be confirmed by the next day’s response to the diuretics, ie, increased urine output, a lower respiratory rate, and a fall in the pro-B-type natriuretic peptide level. Moreover, a change in the radiographic appearance should be seen, and respiratory and pulmonary function should improve after the first 24 hours on oxygen supplementation plus diuretics. Daily patient weights are also critical in determining response to a diuretic, and are rarely done accurately. I order weights and review them daily for patients like this.
Second, it is good to look at things yourself, including the patient, medication lists, laboratory values, and radiographic films. The attending physician should look at the radiographs together with a senior radiologist. Seeing no improvement or change on the second hospital day, or seeing signs incompatible with heart failure, one could order computed tomography of the chest and begin to entertain pulmonary diagnoses.
Even vital signs can be questionable. For example, in the case presented here, with a temperature of 99°F, a heart rate of 105, and a pulse oxygenation saturation of 89%, a respiratory rate of 24 seems unbelievably low. In my experience, the respiratory rate is recorded erroneously most of the time unless it is recorded electronically or checked at the bedside by the physician using a timepiece with a sweep second-hand.
Additionally, I have found that ordering several days’ laboratory tests (eg, complete blood cell counts, chemistry panels) in advance, in many cases, risks missing important findings and wastes time, energy, and the patient’s blood. I have learned to evaluate each patient daily and to order the most pertinent laboratory tests. With electronic medical records, I can check laboratory results as soon as they are available.
To the Editor: I appreciated the article on cognitive biases and diagnostic error by Mull et al in the November 2015 issue.1 They presented an excellent description of the pitfalls of diagnosis as reflected in a case of a patient misdiagnosed with heart failure who ultimately died of pulmonary tuberculosis complicated by pulmonary embolism (the latter possibly from using the wrong form of heparin). To the points they raised, I would like to add a few of my own about diagnosis in general and heart failure in particular.
First, any initial diagnosis not confirmed objectively within the first 24 hours should be questioned, and other possibilities should be investigated. I have found this to be essential for every day’s stay in the hospital and for every outpatient visit. The authors mention checklists as part of the solution to the problem of misdiagnosis, and I would suggest that confirmation of initial diagnoses be built into these checklists.
In the case of a presumptive diagnosis of an acute exacerbation of heart failure treated empirically with diuretics, the diagnosis should be confirmed by the next day’s response to the diuretics, ie, increased urine output, a lower respiratory rate, and a fall in the pro-B-type natriuretic peptide level. Moreover, a change in the radiographic appearance should be seen, and respiratory and pulmonary function should improve after the first 24 hours on oxygen supplementation plus diuretics. Daily patient weights are also critical in determining response to a diuretic, and are rarely done accurately. I order weights and review them daily for patients like this.
Second, it is good to look at things yourself, including the patient, medication lists, laboratory values, and radiographic films. The attending physician should look at the radiographs together with a senior radiologist. Seeing no improvement or change on the second hospital day, or seeing signs incompatible with heart failure, one could order computed tomography of the chest and begin to entertain pulmonary diagnoses.
Even vital signs can be questionable. For example, in the case presented here, with a temperature of 99°F, a heart rate of 105, and a pulse oxygenation saturation of 89%, a respiratory rate of 24 seems unbelievably low. In my experience, the respiratory rate is recorded erroneously most of the time unless it is recorded electronically or checked at the bedside by the physician using a timepiece with a sweep second-hand.
Additionally, I have found that ordering several days’ laboratory tests (eg, complete blood cell counts, chemistry panels) in advance, in many cases, risks missing important findings and wastes time, energy, and the patient’s blood. I have learned to evaluate each patient daily and to order the most pertinent laboratory tests. With electronic medical records, I can check laboratory results as soon as they are available.
- Mull N, Reilly JB, Myers JS. An elderly woman with ‘heart failure’: cognitive biases and diagnostic error. Cleve Clin J Med 2015; 82:745–753.
- Mull N, Reilly JB, Myers JS. An elderly woman with ‘heart failure’: cognitive biases and diagnostic error. Cleve Clin J Med 2015; 82:745–753.
In reply: Cognitive bias and diagnostic error
In Reply: We thank Dr. Field for his insights and personal observations related to diagnosis and biases that contribute to diagnostic errors.
Dr. Field’s comment about the importance of revisiting one’s initial working diagnosis is consistent with our proposed diagnostic time out. A diagnostic time out can incorporate a short checklist and aid in debiasing clinicians when findings do not fit the case presentation, such as lack of response to diuretic therapy. Being mindful of slowing down and not necessarily rushing to judgment is another important component.1 Of note, the residents in our case did revisit their initial working diagnosis, as suggested by Dr. Field. Questions from learners have great potential to serve as debiasing instruments and should always be encouraged. Those who do not work with students can do the same by speaking with nurses or other members of the healthcare team, who offer observations that busy physicians might miss.
Our case highlights the problem that we lack objective criteria to diagnose symptomatic heart failure. While B-type natriuretic factor (BNP) has a strong negative predictive value, serial BNP measurements have not been established to be helpful in the management of heart failure.2 Although certain findings on chest radiography have strong positive and negative likelihood associations, the role of serial chest radiographs is less clear.3 Thus, heart failure remains a clinical diagnosis in current practice.
As Dr. Field points out, the accuracy and performance characteristics of diagnostic testing, such as the respiratory rate, need to be considered in conjunction with debiasing strategies to achieve higher diagnostic accuracy. Multiple factors can contribute to low-performing or misinterpreted diagnostic tests, and inaccurate vital signs have been shown to be similarly prone to potential error.4
Finally, we wholeheartedly agree with Dr. Field’s comment on unnecessary testing. High-value care is appropriate care. Using Bayesian reasoning to guide testing, monitoring the treatment course appropriately, and eliminating waste is highly likely to improve both value and diagnostic accuracy. Automated, ritual ordering of daily tests can indicate that thinking has been shut off, leaving clinicians susceptible to premature closure of the diagnostic process as well as the potential for “incidentalomas” to distract them from the right diagnosis, all the while leading to low-value care such as wasteful spending, patient dissatisfaction, and hospital-acquired anemia.5 We believe that deciding on a daily basis what the next day’s tests will be can be another powerful debiasing habit, one with benefits beyond diagnosis.
- Schiff GD. Minimizing diagnostic error: the importance of follow-up and feedback. Am J Med 2008; 121(suppl):S38–S42.
- Yancy CW, Jessup M, Bozkurt B, et al. 2013 ACCF/AHA guideline for the management of heart failure. Circulation 2013; 128:e240–e327.
- Wang CS, FitzGerald JM, Schulzer M, Mak E, Ayas NT. Does this dyspneic patient in the emergency department have congestive heart failure? JAMA 2005; 294:1944–1956.
- Philip KE, Pack E, Cambiano V, Rollmann H, Weil S, O’Beirne J. The accuracy of respiratory rate assessment by doctors in a London teaching hospital: a cross-sectional study. J Clin Monit Comput 2015; 29:455–460.
- Koch CG, Li L, Sun Z, et al. Hospital-acquired anemia: prevalence, outcomes, and healthcare implications. J Hosp Med 2013; 8:506–512.
In Reply: We thank Dr. Field for his insights and personal observations related to diagnosis and biases that contribute to diagnostic errors.
Dr. Field’s comment about the importance of revisiting one’s initial working diagnosis is consistent with our proposed diagnostic time out. A diagnostic time out can incorporate a short checklist and aid in debiasing clinicians when findings do not fit the case presentation, such as lack of response to diuretic therapy. Being mindful of slowing down and not necessarily rushing to judgment is another important component.1 Of note, the residents in our case did revisit their initial working diagnosis, as suggested by Dr. Field. Questions from learners have great potential to serve as debiasing instruments and should always be encouraged. Those who do not work with students can do the same by speaking with nurses or other members of the healthcare team, who offer observations that busy physicians might miss.
Our case highlights the problem that we lack objective criteria to diagnose symptomatic heart failure. While B-type natriuretic factor (BNP) has a strong negative predictive value, serial BNP measurements have not been established to be helpful in the management of heart failure.2 Although certain findings on chest radiography have strong positive and negative likelihood associations, the role of serial chest radiographs is less clear.3 Thus, heart failure remains a clinical diagnosis in current practice.
As Dr. Field points out, the accuracy and performance characteristics of diagnostic testing, such as the respiratory rate, need to be considered in conjunction with debiasing strategies to achieve higher diagnostic accuracy. Multiple factors can contribute to low-performing or misinterpreted diagnostic tests, and inaccurate vital signs have been shown to be similarly prone to potential error.4
Finally, we wholeheartedly agree with Dr. Field’s comment on unnecessary testing. High-value care is appropriate care. Using Bayesian reasoning to guide testing, monitoring the treatment course appropriately, and eliminating waste is highly likely to improve both value and diagnostic accuracy. Automated, ritual ordering of daily tests can indicate that thinking has been shut off, leaving clinicians susceptible to premature closure of the diagnostic process as well as the potential for “incidentalomas” to distract them from the right diagnosis, all the while leading to low-value care such as wasteful spending, patient dissatisfaction, and hospital-acquired anemia.5 We believe that deciding on a daily basis what the next day’s tests will be can be another powerful debiasing habit, one with benefits beyond diagnosis.
In Reply: We thank Dr. Field for his insights and personal observations related to diagnosis and biases that contribute to diagnostic errors.
Dr. Field’s comment about the importance of revisiting one’s initial working diagnosis is consistent with our proposed diagnostic time out. A diagnostic time out can incorporate a short checklist and aid in debiasing clinicians when findings do not fit the case presentation, such as lack of response to diuretic therapy. Being mindful of slowing down and not necessarily rushing to judgment is another important component.1 Of note, the residents in our case did revisit their initial working diagnosis, as suggested by Dr. Field. Questions from learners have great potential to serve as debiasing instruments and should always be encouraged. Those who do not work with students can do the same by speaking with nurses or other members of the healthcare team, who offer observations that busy physicians might miss.
Our case highlights the problem that we lack objective criteria to diagnose symptomatic heart failure. While B-type natriuretic factor (BNP) has a strong negative predictive value, serial BNP measurements have not been established to be helpful in the management of heart failure.2 Although certain findings on chest radiography have strong positive and negative likelihood associations, the role of serial chest radiographs is less clear.3 Thus, heart failure remains a clinical diagnosis in current practice.
As Dr. Field points out, the accuracy and performance characteristics of diagnostic testing, such as the respiratory rate, need to be considered in conjunction with debiasing strategies to achieve higher diagnostic accuracy. Multiple factors can contribute to low-performing or misinterpreted diagnostic tests, and inaccurate vital signs have been shown to be similarly prone to potential error.4
Finally, we wholeheartedly agree with Dr. Field’s comment on unnecessary testing. High-value care is appropriate care. Using Bayesian reasoning to guide testing, monitoring the treatment course appropriately, and eliminating waste is highly likely to improve both value and diagnostic accuracy. Automated, ritual ordering of daily tests can indicate that thinking has been shut off, leaving clinicians susceptible to premature closure of the diagnostic process as well as the potential for “incidentalomas” to distract them from the right diagnosis, all the while leading to low-value care such as wasteful spending, patient dissatisfaction, and hospital-acquired anemia.5 We believe that deciding on a daily basis what the next day’s tests will be can be another powerful debiasing habit, one with benefits beyond diagnosis.
- Schiff GD. Minimizing diagnostic error: the importance of follow-up and feedback. Am J Med 2008; 121(suppl):S38–S42.
- Yancy CW, Jessup M, Bozkurt B, et al. 2013 ACCF/AHA guideline for the management of heart failure. Circulation 2013; 128:e240–e327.
- Wang CS, FitzGerald JM, Schulzer M, Mak E, Ayas NT. Does this dyspneic patient in the emergency department have congestive heart failure? JAMA 2005; 294:1944–1956.
- Philip KE, Pack E, Cambiano V, Rollmann H, Weil S, O’Beirne J. The accuracy of respiratory rate assessment by doctors in a London teaching hospital: a cross-sectional study. J Clin Monit Comput 2015; 29:455–460.
- Koch CG, Li L, Sun Z, et al. Hospital-acquired anemia: prevalence, outcomes, and healthcare implications. J Hosp Med 2013; 8:506–512.
- Schiff GD. Minimizing diagnostic error: the importance of follow-up and feedback. Am J Med 2008; 121(suppl):S38–S42.
- Yancy CW, Jessup M, Bozkurt B, et al. 2013 ACCF/AHA guideline for the management of heart failure. Circulation 2013; 128:e240–e327.
- Wang CS, FitzGerald JM, Schulzer M, Mak E, Ayas NT. Does this dyspneic patient in the emergency department have congestive heart failure? JAMA 2005; 294:1944–1956.
- Philip KE, Pack E, Cambiano V, Rollmann H, Weil S, O’Beirne J. The accuracy of respiratory rate assessment by doctors in a London teaching hospital: a cross-sectional study. J Clin Monit Comput 2015; 29:455–460.
- Koch CG, Li L, Sun Z, et al. Hospital-acquired anemia: prevalence, outcomes, and healthcare implications. J Hosp Med 2013; 8:506–512.
A guide to managing acute liver failure
When the liver fails, it usually fails gradually. The sudden (acute) onset of liver failure, while less common, demands prompt management, with transfer to an intensive care unit, specific treatment depending on the cause, and consideration of liver transplant, without which the mortality rate is high.
This article reviews the definition, epidemiology, etiology, and management of acute liver failure.
DEFINITIONS
Acute liver failure is defined as a syndrome of acute hepatitis with evidence of abnormal coagulation (eg, an international normalized ratio > 1.5) complicated by the development of mental alteration (encephalopathy) within 26 weeks of the onset of illness in a patient without a history of liver disease.1 In general, patients have no evidence of underlying chronic liver disease, but there are exceptions; patients with Wilson disease, vertically acquired hepatitis B virus infection, or autoimmune hepatitis can present with acute liver failure superimposed on chronic liver disease or even cirrhosis.
The term acute liver failure has replaced older terms such as fulminant hepatic failure, hyperacute liver failure, and subacute liver failure, which were used for prognostic purposes. Patients with hyperacute liver failure (defined as development of encephalopathy within 7 days of onset of illness) generally have a good prognosis with medical management, whereas those with subacute liver failure (defined as development of encephalopathy within 5 to 26 weeks of onset of illness) have a poor prognosis without liver transplant.2,3
NEARLY 2,000 CASES A YEAR
There are nearly 2,000 cases of acute liver failure each year in the United States, and it accounts for 6% of all deaths due to liver disease.4 It is more common in women than in men, and more common in white people than in other races. The peak incidence is at a fairly young age, ie, 35 to 45 years.
CAUSES
The most common cause of acute liver failure in the United States and other Western countries is acetaminophen toxicity, followed by viral hepatitis. In contrast, viral hepatitis is the most common cause in developing countries.5
Acetaminophen toxicity
Patients with acetaminophen-induced liver failure tend to be younger than other patients with acute liver failure.1 Nearly half of them present after intentionally taking a single large dose, while the rest present with unintentional toxicity while taking acetaminophen for pain relief on a long-term basis and ingesting more than the recommended dose.6
After ingestion, 52% to 57% of acetaminophen is converted to glucuronide conjugates, and 30% to 44% is converted to sulfate conjugates. These compounds are nontoxic, water-soluble, and rapidly excreted in the urine.
However, about 5% to 10% of ingested acetaminophen is shunted to the cytochrome P450 system. P450 2E1 is the main isoenzyme involved in acetaminophen metabolism, but 1A2, 3A4, and 2A6 also contribute.7,8 P450 2E1 is the same isoenzyme responsible for ethanol metabolism and is inducible. Thus, regular alcohol consumption can increase P450 2E1 activity, setting the stage under certain circumstances for increased acetaminophen metabolism through this pathway.

Metabolism of acetaminophen through the cytochrome P450 pathway results in production of N-acetyl-p-benzoquinone imine (NAPQI), the compound that damages the liver. NAPQI is rendered nontoxic by binding to glutathione, forming NAPQI-glutathione adducts. Glutathione capacity is limited, however. With too much acetaminophen, glutathione becomes depleted and NAPQI accumulates, binds with proteins to form adducts, and leads to necrosis of hepatocytes (Figure 1).9,10
Acetylcysteine, used in treating acetaminophen toxicity, is a substrate for glutathione synthesis and ultimately increases the amount of glutathione available to bind NAPQI and prevent damage to hepatocytes.11
Acetaminophen is a dose-related toxin. Most ingestions leading to acute liver failure exceed 10 g/day (> 150 mg/kg/day). Moderate chronic ingestion, eg, 4 g/day, usually leads to transient mild elevation of liver enzymes in healthy individuals12 but can in rare cases cause acute liver failure.13

Whitcomb and Block14 retrospectively identified 49 patients who presented with acetaminophen-induced hepatotoxicity in 1987 through 1993; 21 (43%) had been taking acetaminophen for therapeutic purposes. All 49 patients took more than the recommended limit of 4 g/day, many of them while fasting and some while using alcohol. Acute liver failure was seen with ingestion of more than 12 g/day—or more than 10 g/day in alcohol users. The authors attributed the increased risk to activation of cytochrome P450 2E1 by alcohol and depletion of glutathione stores by starvation or alcohol abuse.
Advice to patients taking acetaminophen is given in Table 1.
Other drugs and supplements

A number of other drugs and herbal supplements can also cause acute liver failure (Table 2), the most common being antimicrobial and antiepileptic drugs.15 Of the antimicrobials, antitubercular drugs (especially isoniazid) are believed to be the most common causes, followed by trimethoprim-sulfamethoxazole. Phenytoin is the antiepileptic drug most often implicated in acute liver failure.
Statins can also cause acute liver failure, especially when combined with other hepatotoxic agents.16
The herbal supplements and weight-loss agents Hydroxycut and Herbalife have both been reported to cause acute liver failure, with patients presenting with either the hepatocellular or the cholestatic pattern of liver injury.17 The exact chemical in these supplements that causes liver injury has not yet been determined.
The National Institutes of Health maintains a database of cases of liver failure due to medications and supplements at livertox.nih.gov. The database includes the pattern of hepatic injury, mechanism of injury, management, and outcomes.
Viral hepatitis
Hepatitis B virus is the most common viral cause of acute liver failure and is responsible for about 8% of cases.18
Patients with chronic hepatitis B virus infection—as evidenced by positive hepatitis B surface antigen—can develop acute liver failure if the infection is reactivated by the use of immunosuppressive drugs for solid-organ or bone-marrow transplant or medications such as anti-tumor necrosis agents, rituximab, or chemotherapy. These patients should be treated prophylactically with a nucleoside analogue, which should be continued for 6 months after immunosuppressive therapy is completed.
Hepatitis A virus is responsible for about 4% of cases.18
Hepatitis C virus rarely causes acute liver failure, especially in the absence of hepatitis A and hepatitis B.3,19
Hepatitis E virus, which is endemic in areas of Asia and Africa, can cause liver disease in pregnant women and in young adults who have concomitant liver disease from another cause. It tends to cause acute liver failure more frequently in pregnant women than in the rest of the population and carries a mortality rate of more than 20% in this subgroup.
TT (transfusion-transmitted) virus was reported in the 1990s to cause acute liver failure in about 27% of patients in whom no other cause could be found.20
Other rare viral causes of acute liver failure include Epstein-Barr virus, cytomegalovirus, and herpes simplex virus types 1, 2, and 6.
Other causes
Other causes of acute liver failure include ischemic hepatitis, autoimmune hepatitis, Wilson disease, Budd-Chiari syndrome, and HELLP (hemolysis, elevated liver enzymes and low platelets) syndrome.
MANY PATIENTS NEED LIVER TRANSPLANT

Many patients with acute liver failure ultimately require orthotopic liver transplant,21 especially if they present with severe encephalopathy. Other aspects of treatment vary according to the cause of liver failure (Table 3).
SPECIFIC MANAGEMENT
Management of acetaminophen toxicity
If the time of ingestion is known, checking the acetaminophen level can help determine the cause of acute liver failure and also predict the risk of hepatotoxicity, based on the work of Rumack and Matthew.22 Calculators are available, eg, http://reference.medscape.com/calculator/acetaminophen-toxicity.
If a patient presents with acute liver failure several days after ingesting acetaminophen, the level can be in the nontoxic range, however. In this scenario, measuring acetaminophen-protein adducts can help establish acetaminophen toxicity as the cause, as the adducts last longer in the serum and provide 100% sensitivity and specificity.23 While most laboratories can rapidly measure acetaminophen levels, only a few can measure acetaminophen-protein adducts, and thus this test is not used clinically.
Acetylcysteine is the main drug used for acetaminophen toxicity. Ideally, it should be given within 8 hours of acetaminophen ingestion, but giving it later is also useful.1
Acetylcysteine is available in oral and intravenous forms, the latter for patients who have encephalopathy or cannot tolerate oral intake due to repeated episodes of vomiting.24,25 The oral form is much less costly and is thus preferred over intravenous acetylcysteine in patients who can tolerate oral intake. Intravenous acetylcysteine should be given in a loading dose of 150 mg/kg in 5% dextrose over 15 minutes, followed by a maintenance dose of 50 mg/kg over 4 hours and then 100 mg/kg given over 16 hours.1 No dose adjustment is needed in patients who have renal toxicity (acetaminophen can also be toxic to the kidneys).
Most patients with acetaminophen-induced liver failure survive with medical management alone and do not need a liver transplant.3,26 Cirrhosis does not occur in these patients.
Management of viral acute liver failure
When patients present with acute liver failure, it is necessary to look for a viral cause by serologic testing, including hepatitis A virus IgM antibody, hepatitis B surface antigen, and hepatitis B core IgM antibody.
Hepatitis B can become reactivated in immunocompromised patients, and therefore the hepatitis B virus DNA level should be checked. Detection of hepatitis B virus DNA in a patient previously known to have undetectable hepatitis B virus DNA confirms hepatitis B reactivation.
Patients with hepatitis B-induced acute liver failure should be treated with entecavir or tenofovir. Although this treatment may not change the course of acute liver failure or accelerate the recovery, it can prevent reinfection in the transplanted liver if liver transplant becomes indicated.27–29
Herpes simplex virus should be suspected in patients presenting with anicteric hepatitis with fever. Polymerase chain reaction testing for herpes simplex virus should be done,30 and if positive, patients should be given intravenous acyclovir.31 Despite treatment, herpes simplex virus disease is associated with a very poor prognosis without liver transplant.
Autoimmune hepatitis
The autoantibodies usually seen in autoimmune hepatitis are antinuclear antibody, antismooth muscle antibody, and anti-liver-kidney microsomal antibody, and patients need to be tested for them.
The diagnosis of autoimmune hepatitis can be challenging, as these autoimmune markers can be negative in 5% of patients. Liver biopsy becomes essential to establish the diagnosis in that setting.32
Guidelines advise starting prednisone 40 to 60 mg/day and placing the patient on the liver transplant list.1
Wilson disease
Although it is an uncommon cause of liver failure, Wilson disease needs special attention because it has a poor prognosis. The mortality rate in acute liver failure from Wilson disease reaches 100% without liver transplant.
Wilson disease is caused by a genetic defect that allows copper to accumulate in the liver and other organs. However, diagnosing Wilson disease as the cause of acute liver failure can be challenging because elevated serum and urine copper levels are not specific to Wilson disease and can be seen in patients with acute liver failure from any cause. In addition, the ceruloplasmin level is usually normal or high because it is an acute-phase reactant. Accumulation of copper in the liver parenchyma is usually patchy; therefore, qualitative copper staining on random liver biopsy samples provides low diagnostic yield. Quantitative copper on liver biopsy is the gold standard test to establish the diagnosis, but the test is time-consuming. Kayser-Fleischer rings around the iris are considered pathognomic for Wilson disease when seen with acute liver failure, but they are seen in only about 50% of patients.33
A unique feature of acute Wilson disease is that most patients have very high bilirubin levels and low alkaline phosphatase levels. An alkaline phosphatase-to-bilirubin ratio less than 2 in patients with acute liver failure is highly suggestive of Wilson disease.34
Another clue to the diagnosis is that patients with Wilson disease tend to develop Coombs-negative hemolytic anemia, which leads to a disproportionate elevation in aminotransferase levels, with aspartate aminotransferase being higher than alanine aminotransferase.
Once Wilson disease is suspected, the patient should be listed for liver transplant because death is almost certain without it. For patients awaiting liver transplant, the American Association for the Study of Liver Diseases guidelines recommend certain measures to lower the serum copper level such as albumin dialysis, continuous hemofiltration, plasmapheresis, and plasma exchange,1 but the evidence supporting their use is limited.
NONSPECIFIC MANAGEMENT
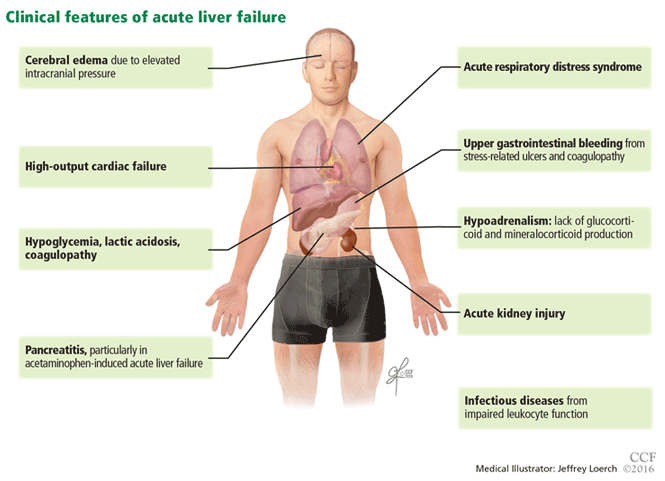
Acute liver failure can affect a number of organs and systems in addition to the liver (Figure 2).
General considerations
Because their condition can rapidly deteriorate, patients with acute liver failure are best managed in intensive care.
Patients who present to a center that does not have the facilities for liver transplant should be transferred to a transplant center as soon as possible, preferably by air. If the patient may not be able to protect the airway, endotracheal intubation should be performed before transfer.
The major causes of death in patients with acute liver failure are cerebral edema and infection. Gastrointestinal bleeding was a major cause of death in the past, but with prophylactic use of histamine H2 receptor blockers and proton pump inhibitors, the incidence of gastrointestinal bleeding has been significantly reduced.
Although initially used only in patients with acetaminophen-induced liver failure, acetylcysteine has also shown benefit in patients with acute liver failure from other causes. In patients with grade 1 or 2 encephalopathy on a scale of 0 (minimal) to 4 (comatose), the transplant-free survival rate is higher when acetylcysteine is given compared with placebo, but this benefit does not extend to patients with a higher grade of encephalopathy.35
Cerebral edema and intracranial hypertension
Cerebral edema is the leading cause of death in patients with acute liver failure, and it develops in nearly 40% of patients.36
The mechanism by which cerebral edema develops is not well understood. Some have proposed that ammonia is converted to glutamine, which causes cerebral edema either directly by its osmotic effect37,38 or indirectly by decreasing other osmolytes, thereby promoting water retention.39
Cerebral edema leads to intracranial hypertension, which can ultimately cause cerebral herniation and death. Because of the high mortality rate associated with cerebral edema, invasive devices were extensively used in the past to monitor intracranial pressure. However, in light of known complications of these devices, including bleeding,40 and lack of evidence of long-term benefit in terms of mortality rates, their use has come under debate.
Treatments. Many treatments are available for cerebral edema and intracranial hypertension. The first step is to elevate the head of the bed about 30 degrees. In addition, hyponatremia should be corrected, as it can worsen cerebral edema.41 If patients are intubated, maintaining a hypercapneic state is advisable to decrease the intracranial pressure.
Of the two pharmacologic options, mannitol is more often used.42 It is given as a bolus dose of 0.5 to 1 g/kg intravenously if the serum osmolality is less than 320 mOsm/L.1 Given the risk of fluid overload with mannitol, caution must be exercised in patients with renal dysfunction. The other pharmacologic option is 3% hypertonic saline.
Therapeutic hypothermia is a newer treatment for cerebral edema. Lowering the body temperature to 32 to 33°C (89.6 to 91.4°F) using cooling blankets decreases intracranial pressure and cerebral blood flow and improves the cerebral perfusion pressure.43 With this treatment, patients should be closely monitored for side effects of infection, coagulopathy, and cardiac arrythmias.1
l-ornithine l-aspartate was successfully used to prevent brain edema in rats, but in humans, no benefit was seen compared with placebo.44,45 The underlying basis for this experimental treatment is that supplemental ornithine and aspartate should increase glutamate synthesis, which should increase the activity of enzyme glutamine synthetase in skeletal muscles. With the increase in enzyme activity, conversion of ammonia to glutamine should increase, thereby decreasing ammonia circulation and thus decreasing cerebral edema.
Patients with cerebral edema have a high incidence of seizures, but prophylactic antiseizure medications such as phenytoin have not been proven to be beneficial.46
Infection
Nearly 80% of patients with acute liver failure develop an infectious complication, which can be attributed to a state of immunodeficiency.47
The respiratory and urinary tracts are the most common sources of infection.48 In patients with bacteremia, Enterococcus species and coagulase-negative Staphylococcus species49 are the commonly isolated organisms. Also, in patients with acute liver failure, fungal infections account for 30% of all infections.50
Infected patients often develop worsening of their encephalopathy51 without fever or elevated white blood cell count.49,52 Thus, in any patient in whom encephalopathy is worsening, an evaluation must be done to rule out infection. In these patients, systemic inflammatory response syndrome is an independent risk factor for death.53
Despite the high mortality rate with infection, whether using antibiotics prophylactically in acute liver failure is beneficial is controversial.54,55
Gastrointestinal bleeding
The current prevalence of upper gastrointestinal bleeding in acute liver failure patients is about 1.5%.56 Coagulopathy and endotracheal intubation are the main risk factors for upper gastrointestinal bleeding in these patients.57 The most common source of bleeding is stress ulcers in the stomach. The ulcers develop from a combination of factors, including decreased blood flow to the mucosa causing ischemia and hypoperfusion-reperfusion injury.
Pharmacologic inhibition of gastric acid secretion has been shown to reduce upper gastrointestinal bleeding in acute liver failure. A histamine H2 receptor blocker or proton pump inhibitor should be given to prevent gastrointestinal bleeding in patients with acute liver failure.1,58
EXPERIMENTAL TREATMENTS
Artificial liver support systems
Membranes and dialysate solutions have been developed to remove toxic substances that are normally metabolized by the liver. Two of these—the molecular adsorbent recycling system (MARS) and the extracorporeal liver assist device (ELAD)—were developed in the late 1990s. MARS consisted of a highly permeable hollow fiber membrane mixed with albumin, and ELAD consisted of porcine hepatocytes attached to microcarriers in the extracapillary space of the hollow fiber membrane. Both systems allowed for transfer of water-soluble and protein-bound toxins in the blood across the membrane and into the dialysate.59 The clinical benefit offered by these devices is controversial,60–62 thus limiting their use to experimental purposes only.
Hepatocyte transplant
Use of hepatocyte transplant as a bridge to liver transplant was tested in 1970s, first in rats and later in humans.63 By reducing the blood ammonia level and improving cerebral perfusion pressure and cardiac function, replacement of 1% to 2% of the total liver cell mass by transplanted hepatocytes acts as a bridge to orthotopic liver transplant.64,65
PROGNOSIS
Different criteria have been used to identify patients with poor prognosis who may eventually need to undergo liver transplant.
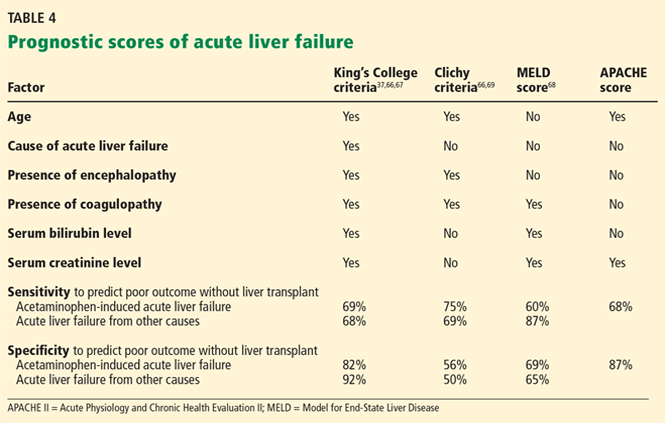
The King’s College criteria system is the most commonly used for prognosis (Table 4).37,66–69 Its main drawback is that it is applicable only in patients with encephalopathy, and when patients reach this stage, their condition often deteriorates rapidly, and they die while awaiting liver transplant.37,66,67
The Model for End-Stage Liver Disease (MELD) score is an alternative to the King’s College criteria. A high MELD score on admission signifies advanced disease, and patients with a high MELD score tend to have a worse prognosis than those with a low score.68
The Acute Physiology and Chronic Health Evaluation (APACHE) II score can also be used, as it is more sensitive than the King’s College criteria.6
The Clichy criteria66,69 can also be used.
Liver biopsy. In addition to helping establish the cause of acute liver failure, liver biopsy can also be used as a prognostic tool. Hepatocellular necrosis greater than 70% on the biopsy predicts death with a specificity of 90% and a sensitivity of 56%.70
Hypophosphatemia has been reported to indicate recovering liver function in patients with acute liver failure.71 As the liver regenerates, its energy requirement increases. To supply the energy, adenosine triphosphate production increases, and phosphorus shifts from the extracellular to the intracellular compartment to meet the need for extra phosphorus during this process. A serum phosphorus level of 2.9 mg/dL or higher appears to indicate a poor prognosis in patients with acute liver failure, as it signifies that adequate hepatocyte regeneration is not occurring.
- Polson J, Lee WM; American Association for the Study of Liver Disease. AASLD position paper: the management of acute liver failure. Hepatology 2005; 41:1179–1197.
- O’Grady JG, Schalm SW, Williams R. Acute liver failure: redefining the syndromes. Lancet 1993; 342:273–275.
- Ostapowicz G, Fontana RJ, Schiodt FV, et al; US Acute Liver Failure Study Group. Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States. Ann Intern Med 2002; 137:947–954.
- Lee WM, Squires RH Jr, Nyberg SL, Doo E, Hoofnagle JH. Acute liver failure: summary of a workshop. Hepatology 2008; 47:1401–1415.
- Acharya SK, Panda SK, Saxena A, Gupta SD. Acute hepatic failure in India: a perspective from the East. J Gastroenterol Hepatol 2000; 15:473–479.
- Larson AM, Polson J, Fontana RJ, et al; Acute Liver Failure Study Group. Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology 2005; 42:1364–1372.
- Patten CJ, Thomas PE, Guy RL, et al. Cytochrome P450 enzymes involved in acetaminophen activation by rat and human liver microsomes and their kinetics. Chem Res Toxicol 1993; 6:511–518.
- Chen W, Koenigs LL, Thompson SJ, et al. Oxidation of acetaminophen to its toxic quinone imine and nontoxic catechol metabolites by baculovirus-expressed and purified human cytochromes P450 2E1 and 2A6. Chem Res Toxicol 1998; 11:295-301.
- Mitchell JR, Jollow DJ, Potter WZ, Gillette JR, Brodie BB. Acetaminophen-induced hepatic necrosis. IV. Protective role of glutathione. J Pharmacol Exp Ther 1973; 187:211–217.
- Schilling A, Corey R, Leonard M, Eghtesad B. Acetaminophen: old drug, new warnings. Cleve Clin J Med 2010; 77:19–27.
- Lauterburg BH, Corcoran GB, Mitchell JR. Mechanism of action of N-acetylcysteine in the protection against the hepatotoxicity of acetaminophen in rats in vivo. J Clin Invest 1983; 71:980–991.
- Watkins PB, Kaplowitz N, Slattery JT, et al. Aminotransferase elevations in healthy adults receiving 4 grams of acetaminophen daily: a randomized controlled trial. JAMA 2006; 296:87–93.
- Schiødt FV, Rochling FA, Casey DL, Lee WM. Acetaminophen toxicity in an urban county hospital. N Engl J Med 1997; 337:1112–1117.
- Whitcomb DC, Block GD. Association of acetaminophen hepatotoxicity with fasting and ethanol use. JAMA 1994; 272:1845–1850.
- Chalasani N, Fontana RJ, Bonkovsky HL, et al; Drug Induced Liver Injury Network (DILIN). Causes, clinical features, and outcomes from a prospective study of drug-induced liver injury in the United States. Gastroenterology 2008; 135:1924–1934 e1–4
- Reuben A, Koch DG, Lee WM; Acute Liver Failure Study Group. Drug-induced acute liver failure: results of a US multicenter, prospective study. Hepatology 2010; 52:2065–2076.
- Stevens T, Qadri A, Zein NN. Two patients with acute liver injury associated with use of the herbal weight-loss supplement hydroxycut. Ann Intern Med 2005; 142:477–478.
- Bernal W, Lee WM, Wendon J, Larsen FS, Williams R. Acute liver failure: a curable disease by 2024? J Hepatol 2015; 62(suppl 1):S112–S120.
- Schiodt FV, Davern TJ, Shakil AO, McGuire B, Samuel G, Lee WM. Viral hepatitis-related acute liver failure. Am J Gastroenterol 2003; 98:448–453.
- Charlton M, Adjei P, Poterucha J, et al. TT-virus infection in North American blood donors, patients with fulminant hepatic failure, and cryptogenic cirrhosis. Hepatology 1998; 28:839–842.
- Bismuth H, Samuel D, Gugenheim J, et al. Emergency liver transplantation for fulminant hepatitis. Ann Intern Med 1987; 107:337–341.
- Rumack BH, Matthew H. Acetaminophen poisoning and toxicity. Pediatrics 1975; 55:871–876.
- Davern TJ 2nd, James LP, Hinson JA, et al; Acute Liver Failure Study Group. Measurement of serum acetaminophen-protein adducts in patients with acute liver failure. Gastroenterology 2006; 130:687–694.
- Perry HE, Shannon MW. Efficacy of oral versus intravenous N-acetylcysteine in acetaminophen overdose: results of an open-label, clinical trial. J Pediatr 1998; 132:149–152.
- Smilkstein MJ, Knapp GL, Kulig KW, Rumack BH. Efficacy of oral N-acetylcysteine in the treatment of acetaminophen overdose. Analysis of the national multicenter study (1976 to 1985). N Engl J Med 1988; 319:1557–1562.
- Makin AJ, Wendon J, Williams R. A 7-year experience of severe acetaminophen-induced hepatotoxicity (1987-1993). Gastroenterology 1995; 109:1907–1916.
- Tsang SW, Chan HL, Leung NW, et al. Lamivudine treatment for fulminant hepatic failure due to acute exacerbation of chronic hepatitis B infection. Aliment Pharmacol Ther 2001; 15:1737–1744.
- Yu JW, Sun LJ, Yan BZ, Kang P, Zhao YH. Lamivudine treatment is associated with improved survival in fulminant hepatitis B. Liver Int 2011; 31:499–506.
- Garg H, Sarin SK, Kumar M, Garg V, Sharma BC, Kumar A. Tenofovir improves the outcome in patients with spontaneous reactivation of hepatitis B presenting as acute-on-chronic liver failure. Hepatology 2011; 53:774–780.
- Pinna AD, Rakela J, Demetris AJ, Fung JJ. Five cases of fulminant hepatitis due to herpes simplex virus in adults. Dig Dis Sci 2002; 47:750–754.
- Farr RW, Short S, Weissman D. Fulminant hepatitis during herpes simplex virus infection in apparently immunocompetent adults: report of two cases and review of the literature. Clin Infect Dis 1997; 24:1191–1194.
- Czaja AJ, Freese DK; American Association for the Study of Liver Disease. Diagnosis and treatment of autoimmune hepatitis. Hepatology 2002; 36:479–497.
- Roberts EA, Schilsky ML. A practice guideline on Wilson disease. Hepatology 2003; 37:1475–1492.
- Berman DH, Leventhal RI, Gavaler JS, Cadoff EM, Van Thiel DH. Clinical differentiation of fulminant Wilsonian hepatitis from other causes of hepatic failure. Gastroenterology 1991; 100:1129–1134.
- Lee WM, Hynan LS, Rossaro L, et al. Intravenous N-acetylcysteine improves transplant-free survival in early stage non-acetaminophen acute liver failure. Gastroenterology 2009; 137:856–864.
- O’Grady JG, Alexander GJ, Hayllar KM, Williams R. Early indicators of prognosis in fulminant hepatic failure. Gastroenterology 1989; 97:439–445.
- Clemmesen JO, Larsen FS, Kondrup J, Hansen BA, Ott P. Cerebral herniation in patients with acute liver failure is correlated with arterial ammonia concentration. Hepatology 1999; 29:648–653.
- Swain M, Butterworth RF, Blei AT. Ammonia and related amino acids in the pathogenesis of brain edema in acute ischemic liver failure in rats. Hepatology 1992; 15:449–453.
- Haussinger D, Laubenberger J, vom Dahl S, et al. Proton magnetic resonance spectroscopy studies on human brain myo-inositol in hypo-osmolarity and hepatic encephalopathy. Gastroenterology 1994; 107:1475–1480.
- Blei AT, Olafsson S, Webster S, Levy R. Complications of intracranial pressure monitoring in fulminant hepatic failure. Lancet 1993; 341:157–158.
- Cordoba J, Gottstein J, Blei AT. Chronic hyponatremia exacerbates ammonia-induced brain edema in rats after portacaval anastomosis. J Hepatol 1998; 29:589–594.
- Canalese J, Gimson AE, Davis C, Mellon PJ, Davis M, Williams R. Controlled trial of dexamethasone and mannitol for the cerebral oedema of fulminant hepatic failure. Gut 1982; 23:625–629.
- Jalan R, SW OD, Deutz NE, Lee A, Hayes PC. Moderate hypothermia for uncontrolled intracranial hypertension in acute liver failure. Lancet 1999; 354:1164–1168.
- Rose C, Michalak A, Rao KV, Quack G, Kircheis G, Butterworth RF. L-ornithine-L-aspartate lowers plasma and cerebrospinal fluid ammonia and prevents brain edema in rats with acute liver failure. Hepatology 1999; 30:636–640.
- Acharya SK, Bhatia V, Sreenivas V, Khanal S, Panda SK. Efficacy of L-ornithine L-aspartate in acute liver failure: a double-blind, randomized, placebo-controlled study. Gastroenterology 2009; 136:2159–2168.
- Bhatia V, Batra Y, Acharya SK. Prophylactic phenytoin does not improve cerebral edema or survival in acute liver failure—a controlled clinical trial. J Hepatol 2004; 41:89–96.
- Canalese J, Gove CD, Gimson AE, Wilkinson SP, Wardle EN, Williams R. Reticuloendothelial system and hepatocytic function in fulminant hepatic failure. Gut 1982; 23:265–269.
- Rolando N, Harvey F, Brahm J, et al. Prospective study of bacterial infection in acute liver failure: an analysis of fifty patients. Hepatology 1990; 11:49–53.
- Rolando N, Wade JJ, Stangou A, et al. Prospective study comparing the efficacy of prophylactic parenteral antimicrobials, with or without enteral decontamination, in patients with acute liver failure. Liver Transpl Surg 1996; 2:8–13.
- Rolando N, Harvey F, Brahm J, et al. Fungal infection: a common, unrecognised complication of acute liver failure. J Hepatol 1991; 12:1–9.
- Vaquero J, Polson J, Chung C, et al. Infection and the progression of hepatic encephalopathy in acute liver failure. Gastroenterology 2003; 125:755–764.
- Rolando N, Philpott-Howard J, Williams R. Bacterial and fungal infection in acute liver failure. Semin Liver Dis 1996; 16:389–402.
- Rolando N, Wade J, Davalos M, Wendon J, Philpott-Howard J, Williams R. The systemic inflammatory response syndrome in acute liver failure. Hepatology 2000; 32:734–739.
- Rolando N, Gimson A, Wade J, Philpott- Howard J, Casewell M, Williams R. Prospective controlled trial of selective parenteral and enteral antimicrobial regimen in fulminant liver failure. Hepatology 1993; 17:196–201.
- Karvellas CJ, Cavazos J, Battenhouse H, et al; US Acute Liver Failure Study Group. Effects of antimicrobial prophylaxis and blood stream infections in patients with acute liver failure: a retrospective cohort study. Clin Gastroenterol Hepatol 2014; 12:1942–1949.
- Acharya SK, Dasarathy S, Kumer TL, et al. Fulminant hepatitis in a tropical population: clinical course, cause, and early predictors of outcome. Hepatology 1996; 23:1148–1155.
- Cook DJ, Fuller HD, Guyatt GH, et al. Risk factors for gastrointestinal bleeding in critically ill patients. Canadian Critical Care Trials Group. N Engl J Med 1994; 330:377–381.
- MacDougall BR, Williams R. H2-receptor antagonist in the prevention of acute gastrointestinal hemorrhage in fulminant hepatic failure: a controlled trial. Gastroenterology 1978; 74:464–465.
- Stange J, Mitzner SR, Risler T, et al. Molecular adsorbent recycling system (MARS): clinical results of a new membrane-based blood purification system for bioartificial liver support. Artif Organs 1999; 23:319–330.
- Vaid A, Chewich H, Balk EM, Jaber BL. Molecular adsorbent recirculating system as artificial support therapy for liver failure: a meta-analysis. ASAIO J 2012; 58:51–59.
- Khuroo MS, Khuroo MS, Farahat KL. Molecular adsorbent recirculating system for acute and acute-on-chronic liver failure: a meta-analysis. Liver Transpl 2004; 10:1099–1106.
- Kjaergard LL, Liu J, Als-Nielsen B, Gluud C. Artificial and bioartificial support systems for acute and acute-on-chronic liver failure: a systematic review. JAMA 2003; 289:217–222.
- Sommer BG, Sutherland DE, Matas AJ, Simmons RL, Najarian JS. Hepatocellular transplantation for treatment of D-galactosamine-induced acute liver failure in rats. Transplant Proc 1979; 11:578–584.
- Demetriou AA, Reisner A, Sanchez J, Levenson SM, Moscioni AD, Chowdhury JR. Transplantation of microcarrier-attached hepatocytes into 90% partially hepatectomized rats. Hepatology 1988; 8:1006–1009.
- Strom SC, Fisher RA, Thompson MT, et al. Hepatocyte transplantation as a bridge to orthotopic liver transplantation in terminal liver failure. Transplantation 1997; 63:559–569.
- Pauwels A, Mostefa-Kara N, Florent C, Levy VG. Emergency liver transplantation for acute liver failure. Evaluation of London and Clichy criteria. J Hepatol 1993; 17:124–127.
- Anand AC, Nightingale P, Neuberger JM. Early indicators of prognosis in fulminant hepatic failure: an assessment of the King's criteria. J Hepatol 1997; 26:62–68.
- Schmidt LE, Larsen FS. MELD score as a predictor of liver failure and death in patients with acetaminophen-induced liver injury. Hepatology 2007; 45:789–796.
- Bernuau J, Goudeau A, Poynard T, et al. Multivariate analysis of prognostic factors in fulminant hepatitis B. Hepatology 1986; 6:648–651.
- Donaldson BW, Gopinath R, Wanless IR, et al. The role of transjugular liver biopsy in fulminant liver failure: relation to other prognostic indicators. Hepatology 1993; 18:1370–1376.
- Schmidt LE, Dalhoff K. Serum phosphate is an early predictor of outcome in severe acetaminophen-induced hepatotoxicity. Hepatology 2002; 36:659–665.
When the liver fails, it usually fails gradually. The sudden (acute) onset of liver failure, while less common, demands prompt management, with transfer to an intensive care unit, specific treatment depending on the cause, and consideration of liver transplant, without which the mortality rate is high.
This article reviews the definition, epidemiology, etiology, and management of acute liver failure.
DEFINITIONS
Acute liver failure is defined as a syndrome of acute hepatitis with evidence of abnormal coagulation (eg, an international normalized ratio > 1.5) complicated by the development of mental alteration (encephalopathy) within 26 weeks of the onset of illness in a patient without a history of liver disease.1 In general, patients have no evidence of underlying chronic liver disease, but there are exceptions; patients with Wilson disease, vertically acquired hepatitis B virus infection, or autoimmune hepatitis can present with acute liver failure superimposed on chronic liver disease or even cirrhosis.
The term acute liver failure has replaced older terms such as fulminant hepatic failure, hyperacute liver failure, and subacute liver failure, which were used for prognostic purposes. Patients with hyperacute liver failure (defined as development of encephalopathy within 7 days of onset of illness) generally have a good prognosis with medical management, whereas those with subacute liver failure (defined as development of encephalopathy within 5 to 26 weeks of onset of illness) have a poor prognosis without liver transplant.2,3
NEARLY 2,000 CASES A YEAR
There are nearly 2,000 cases of acute liver failure each year in the United States, and it accounts for 6% of all deaths due to liver disease.4 It is more common in women than in men, and more common in white people than in other races. The peak incidence is at a fairly young age, ie, 35 to 45 years.
CAUSES
The most common cause of acute liver failure in the United States and other Western countries is acetaminophen toxicity, followed by viral hepatitis. In contrast, viral hepatitis is the most common cause in developing countries.5
Acetaminophen toxicity
Patients with acetaminophen-induced liver failure tend to be younger than other patients with acute liver failure.1 Nearly half of them present after intentionally taking a single large dose, while the rest present with unintentional toxicity while taking acetaminophen for pain relief on a long-term basis and ingesting more than the recommended dose.6
After ingestion, 52% to 57% of acetaminophen is converted to glucuronide conjugates, and 30% to 44% is converted to sulfate conjugates. These compounds are nontoxic, water-soluble, and rapidly excreted in the urine.
However, about 5% to 10% of ingested acetaminophen is shunted to the cytochrome P450 system. P450 2E1 is the main isoenzyme involved in acetaminophen metabolism, but 1A2, 3A4, and 2A6 also contribute.7,8 P450 2E1 is the same isoenzyme responsible for ethanol metabolism and is inducible. Thus, regular alcohol consumption can increase P450 2E1 activity, setting the stage under certain circumstances for increased acetaminophen metabolism through this pathway.
Metabolism of acetaminophen through the cytochrome P450 pathway results in production of N-acetyl-p-benzoquinone imine (NAPQI), the compound that damages the liver. NAPQI is rendered nontoxic by binding to glutathione, forming NAPQI-glutathione adducts. Glutathione capacity is limited, however. With too much acetaminophen, glutathione becomes depleted and NAPQI accumulates, binds with proteins to form adducts, and leads to necrosis of hepatocytes (Figure 1).9,10
Acetylcysteine, used in treating acetaminophen toxicity, is a substrate for glutathione synthesis and ultimately increases the amount of glutathione available to bind NAPQI and prevent damage to hepatocytes.11
Acetaminophen is a dose-related toxin. Most ingestions leading to acute liver failure exceed 10 g/day (> 150 mg/kg/day). Moderate chronic ingestion, eg, 4 g/day, usually leads to transient mild elevation of liver enzymes in healthy individuals12 but can in rare cases cause acute liver failure.13
Whitcomb and Block14 retrospectively identified 49 patients who presented with acetaminophen-induced hepatotoxicity in 1987 through 1993; 21 (43%) had been taking acetaminophen for therapeutic purposes. All 49 patients took more than the recommended limit of 4 g/day, many of them while fasting and some while using alcohol. Acute liver failure was seen with ingestion of more than 12 g/day—or more than 10 g/day in alcohol users. The authors attributed the increased risk to activation of cytochrome P450 2E1 by alcohol and depletion of glutathione stores by starvation or alcohol abuse.
Advice to patients taking acetaminophen is given in Table 1.
Other drugs and supplements
A number of other drugs and herbal supplements can also cause acute liver failure (Table 2), the most common being antimicrobial and antiepileptic drugs.15 Of the antimicrobials, antitubercular drugs (especially isoniazid) are believed to be the most common causes, followed by trimethoprim-sulfamethoxazole. Phenytoin is the antiepileptic drug most often implicated in acute liver failure.
Statins can also cause acute liver failure, especially when combined with other hepatotoxic agents.16
The herbal supplements and weight-loss agents Hydroxycut and Herbalife have both been reported to cause acute liver failure, with patients presenting with either the hepatocellular or the cholestatic pattern of liver injury.17 The exact chemical in these supplements that causes liver injury has not yet been determined.
The National Institutes of Health maintains a database of cases of liver failure due to medications and supplements at livertox.nih.gov. The database includes the pattern of hepatic injury, mechanism of injury, management, and outcomes.
Viral hepatitis
Hepatitis B virus is the most common viral cause of acute liver failure and is responsible for about 8% of cases.18
Patients with chronic hepatitis B virus infection—as evidenced by positive hepatitis B surface antigen—can develop acute liver failure if the infection is reactivated by the use of immunosuppressive drugs for solid-organ or bone-marrow transplant or medications such as anti-tumor necrosis agents, rituximab, or chemotherapy. These patients should be treated prophylactically with a nucleoside analogue, which should be continued for 6 months after immunosuppressive therapy is completed.
Hepatitis A virus is responsible for about 4% of cases.18
Hepatitis C virus rarely causes acute liver failure, especially in the absence of hepatitis A and hepatitis B.3,19
Hepatitis E virus, which is endemic in areas of Asia and Africa, can cause liver disease in pregnant women and in young adults who have concomitant liver disease from another cause. It tends to cause acute liver failure more frequently in pregnant women than in the rest of the population and carries a mortality rate of more than 20% in this subgroup.
TT (transfusion-transmitted) virus was reported in the 1990s to cause acute liver failure in about 27% of patients in whom no other cause could be found.20
Other rare viral causes of acute liver failure include Epstein-Barr virus, cytomegalovirus, and herpes simplex virus types 1, 2, and 6.
Other causes
Other causes of acute liver failure include ischemic hepatitis, autoimmune hepatitis, Wilson disease, Budd-Chiari syndrome, and HELLP (hemolysis, elevated liver enzymes and low platelets) syndrome.
MANY PATIENTS NEED LIVER TRANSPLANT
Many patients with acute liver failure ultimately require orthotopic liver transplant,21 especially if they present with severe encephalopathy. Other aspects of treatment vary according to the cause of liver failure (Table 3).
SPECIFIC MANAGEMENT
Management of acetaminophen toxicity
If the time of ingestion is known, checking the acetaminophen level can help determine the cause of acute liver failure and also predict the risk of hepatotoxicity, based on the work of Rumack and Matthew.22 Calculators are available, eg, http://reference.medscape.com/calculator/acetaminophen-toxicity.
If a patient presents with acute liver failure several days after ingesting acetaminophen, the level can be in the nontoxic range, however. In this scenario, measuring acetaminophen-protein adducts can help establish acetaminophen toxicity as the cause, as the adducts last longer in the serum and provide 100% sensitivity and specificity.23 While most laboratories can rapidly measure acetaminophen levels, only a few can measure acetaminophen-protein adducts, and thus this test is not used clinically.
Acetylcysteine is the main drug used for acetaminophen toxicity. Ideally, it should be given within 8 hours of acetaminophen ingestion, but giving it later is also useful.1
Acetylcysteine is available in oral and intravenous forms, the latter for patients who have encephalopathy or cannot tolerate oral intake due to repeated episodes of vomiting.24,25 The oral form is much less costly and is thus preferred over intravenous acetylcysteine in patients who can tolerate oral intake. Intravenous acetylcysteine should be given in a loading dose of 150 mg/kg in 5% dextrose over 15 minutes, followed by a maintenance dose of 50 mg/kg over 4 hours and then 100 mg/kg given over 16 hours.1 No dose adjustment is needed in patients who have renal toxicity (acetaminophen can also be toxic to the kidneys).
Most patients with acetaminophen-induced liver failure survive with medical management alone and do not need a liver transplant.3,26 Cirrhosis does not occur in these patients.
Management of viral acute liver failure
When patients present with acute liver failure, it is necessary to look for a viral cause by serologic testing, including hepatitis A virus IgM antibody, hepatitis B surface antigen, and hepatitis B core IgM antibody.
Hepatitis B can become reactivated in immunocompromised patients, and therefore the hepatitis B virus DNA level should be checked. Detection of hepatitis B virus DNA in a patient previously known to have undetectable hepatitis B virus DNA confirms hepatitis B reactivation.
Patients with hepatitis B-induced acute liver failure should be treated with entecavir or tenofovir. Although this treatment may not change the course of acute liver failure or accelerate the recovery, it can prevent reinfection in the transplanted liver if liver transplant becomes indicated.27–29
Herpes simplex virus should be suspected in patients presenting with anicteric hepatitis with fever. Polymerase chain reaction testing for herpes simplex virus should be done,30 and if positive, patients should be given intravenous acyclovir.31 Despite treatment, herpes simplex virus disease is associated with a very poor prognosis without liver transplant.
Autoimmune hepatitis
The autoantibodies usually seen in autoimmune hepatitis are antinuclear antibody, antismooth muscle antibody, and anti-liver-kidney microsomal antibody, and patients need to be tested for them.
The diagnosis of autoimmune hepatitis can be challenging, as these autoimmune markers can be negative in 5% of patients. Liver biopsy becomes essential to establish the diagnosis in that setting.32
Guidelines advise starting prednisone 40 to 60 mg/day and placing the patient on the liver transplant list.1
Wilson disease
Although it is an uncommon cause of liver failure, Wilson disease needs special attention because it has a poor prognosis. The mortality rate in acute liver failure from Wilson disease reaches 100% without liver transplant.
Wilson disease is caused by a genetic defect that allows copper to accumulate in the liver and other organs. However, diagnosing Wilson disease as the cause of acute liver failure can be challenging because elevated serum and urine copper levels are not specific to Wilson disease and can be seen in patients with acute liver failure from any cause. In addition, the ceruloplasmin level is usually normal or high because it is an acute-phase reactant. Accumulation of copper in the liver parenchyma is usually patchy; therefore, qualitative copper staining on random liver biopsy samples provides low diagnostic yield. Quantitative copper on liver biopsy is the gold standard test to establish the diagnosis, but the test is time-consuming. Kayser-Fleischer rings around the iris are considered pathognomic for Wilson disease when seen with acute liver failure, but they are seen in only about 50% of patients.33
A unique feature of acute Wilson disease is that most patients have very high bilirubin levels and low alkaline phosphatase levels. An alkaline phosphatase-to-bilirubin ratio less than 2 in patients with acute liver failure is highly suggestive of Wilson disease.34
Another clue to the diagnosis is that patients with Wilson disease tend to develop Coombs-negative hemolytic anemia, which leads to a disproportionate elevation in aminotransferase levels, with aspartate aminotransferase being higher than alanine aminotransferase.
Once Wilson disease is suspected, the patient should be listed for liver transplant because death is almost certain without it. For patients awaiting liver transplant, the American Association for the Study of Liver Diseases guidelines recommend certain measures to lower the serum copper level such as albumin dialysis, continuous hemofiltration, plasmapheresis, and plasma exchange,1 but the evidence supporting their use is limited.
NONSPECIFIC MANAGEMENT
Acute liver failure can affect a number of organs and systems in addition to the liver (Figure 2).
General considerations
Because their condition can rapidly deteriorate, patients with acute liver failure are best managed in intensive care.
Patients who present to a center that does not have the facilities for liver transplant should be transferred to a transplant center as soon as possible, preferably by air. If the patient may not be able to protect the airway, endotracheal intubation should be performed before transfer.
The major causes of death in patients with acute liver failure are cerebral edema and infection. Gastrointestinal bleeding was a major cause of death in the past, but with prophylactic use of histamine H2 receptor blockers and proton pump inhibitors, the incidence of gastrointestinal bleeding has been significantly reduced.
Although initially used only in patients with acetaminophen-induced liver failure, acetylcysteine has also shown benefit in patients with acute liver failure from other causes. In patients with grade 1 or 2 encephalopathy on a scale of 0 (minimal) to 4 (comatose), the transplant-free survival rate is higher when acetylcysteine is given compared with placebo, but this benefit does not extend to patients with a higher grade of encephalopathy.35
Cerebral edema and intracranial hypertension
Cerebral edema is the leading cause of death in patients with acute liver failure, and it develops in nearly 40% of patients.36
The mechanism by which cerebral edema develops is not well understood. Some have proposed that ammonia is converted to glutamine, which causes cerebral edema either directly by its osmotic effect37,38 or indirectly by decreasing other osmolytes, thereby promoting water retention.39
Cerebral edema leads to intracranial hypertension, which can ultimately cause cerebral herniation and death. Because of the high mortality rate associated with cerebral edema, invasive devices were extensively used in the past to monitor intracranial pressure. However, in light of known complications of these devices, including bleeding,40 and lack of evidence of long-term benefit in terms of mortality rates, their use has come under debate.
Treatments. Many treatments are available for cerebral edema and intracranial hypertension. The first step is to elevate the head of the bed about 30 degrees. In addition, hyponatremia should be corrected, as it can worsen cerebral edema.41 If patients are intubated, maintaining a hypercapneic state is advisable to decrease the intracranial pressure.
Of the two pharmacologic options, mannitol is more often used.42 It is given as a bolus dose of 0.5 to 1 g/kg intravenously if the serum osmolality is less than 320 mOsm/L.1 Given the risk of fluid overload with mannitol, caution must be exercised in patients with renal dysfunction. The other pharmacologic option is 3% hypertonic saline.
Therapeutic hypothermia is a newer treatment for cerebral edema. Lowering the body temperature to 32 to 33°C (89.6 to 91.4°F) using cooling blankets decreases intracranial pressure and cerebral blood flow and improves the cerebral perfusion pressure.43 With this treatment, patients should be closely monitored for side effects of infection, coagulopathy, and cardiac arrythmias.1
l-ornithine l-aspartate was successfully used to prevent brain edema in rats, but in humans, no benefit was seen compared with placebo.44,45 The underlying basis for this experimental treatment is that supplemental ornithine and aspartate should increase glutamate synthesis, which should increase the activity of enzyme glutamine synthetase in skeletal muscles. With the increase in enzyme activity, conversion of ammonia to glutamine should increase, thereby decreasing ammonia circulation and thus decreasing cerebral edema.
Patients with cerebral edema have a high incidence of seizures, but prophylactic antiseizure medications such as phenytoin have not been proven to be beneficial.46
Infection
Nearly 80% of patients with acute liver failure develop an infectious complication, which can be attributed to a state of immunodeficiency.47
The respiratory and urinary tracts are the most common sources of infection.48 In patients with bacteremia, Enterococcus species and coagulase-negative Staphylococcus species49 are the commonly isolated organisms. Also, in patients with acute liver failure, fungal infections account for 30% of all infections.50
Infected patients often develop worsening of their encephalopathy51 without fever or elevated white blood cell count.49,52 Thus, in any patient in whom encephalopathy is worsening, an evaluation must be done to rule out infection. In these patients, systemic inflammatory response syndrome is an independent risk factor for death.53
Despite the high mortality rate with infection, whether using antibiotics prophylactically in acute liver failure is beneficial is controversial.54,55
Gastrointestinal bleeding
The current prevalence of upper gastrointestinal bleeding in acute liver failure patients is about 1.5%.56 Coagulopathy and endotracheal intubation are the main risk factors for upper gastrointestinal bleeding in these patients.57 The most common source of bleeding is stress ulcers in the stomach. The ulcers develop from a combination of factors, including decreased blood flow to the mucosa causing ischemia and hypoperfusion-reperfusion injury.
Pharmacologic inhibition of gastric acid secretion has been shown to reduce upper gastrointestinal bleeding in acute liver failure. A histamine H2 receptor blocker or proton pump inhibitor should be given to prevent gastrointestinal bleeding in patients with acute liver failure.1,58
EXPERIMENTAL TREATMENTS
Artificial liver support systems
Membranes and dialysate solutions have been developed to remove toxic substances that are normally metabolized by the liver. Two of these—the molecular adsorbent recycling system (MARS) and the extracorporeal liver assist device (ELAD)—were developed in the late 1990s. MARS consisted of a highly permeable hollow fiber membrane mixed with albumin, and ELAD consisted of porcine hepatocytes attached to microcarriers in the extracapillary space of the hollow fiber membrane. Both systems allowed for transfer of water-soluble and protein-bound toxins in the blood across the membrane and into the dialysate.59 The clinical benefit offered by these devices is controversial,60–62 thus limiting their use to experimental purposes only.
Hepatocyte transplant
Use of hepatocyte transplant as a bridge to liver transplant was tested in 1970s, first in rats and later in humans.63 By reducing the blood ammonia level and improving cerebral perfusion pressure and cardiac function, replacement of 1% to 2% of the total liver cell mass by transplanted hepatocytes acts as a bridge to orthotopic liver transplant.64,65
PROGNOSIS
Different criteria have been used to identify patients with poor prognosis who may eventually need to undergo liver transplant.
The King’s College criteria system is the most commonly used for prognosis (Table 4).37,66–69 Its main drawback is that it is applicable only in patients with encephalopathy, and when patients reach this stage, their condition often deteriorates rapidly, and they die while awaiting liver transplant.37,66,67
The Model for End-Stage Liver Disease (MELD) score is an alternative to the King’s College criteria. A high MELD score on admission signifies advanced disease, and patients with a high MELD score tend to have a worse prognosis than those with a low score.68
The Acute Physiology and Chronic Health Evaluation (APACHE) II score can also be used, as it is more sensitive than the King’s College criteria.6
The Clichy criteria66,69 can also be used.
Liver biopsy. In addition to helping establish the cause of acute liver failure, liver biopsy can also be used as a prognostic tool. Hepatocellular necrosis greater than 70% on the biopsy predicts death with a specificity of 90% and a sensitivity of 56%.70
Hypophosphatemia has been reported to indicate recovering liver function in patients with acute liver failure.71 As the liver regenerates, its energy requirement increases. To supply the energy, adenosine triphosphate production increases, and phosphorus shifts from the extracellular to the intracellular compartment to meet the need for extra phosphorus during this process. A serum phosphorus level of 2.9 mg/dL or higher appears to indicate a poor prognosis in patients with acute liver failure, as it signifies that adequate hepatocyte regeneration is not occurring.
When the liver fails, it usually fails gradually. The sudden (acute) onset of liver failure, while less common, demands prompt management, with transfer to an intensive care unit, specific treatment depending on the cause, and consideration of liver transplant, without which the mortality rate is high.
This article reviews the definition, epidemiology, etiology, and management of acute liver failure.
DEFINITIONS
Acute liver failure is defined as a syndrome of acute hepatitis with evidence of abnormal coagulation (eg, an international normalized ratio > 1.5) complicated by the development of mental alteration (encephalopathy) within 26 weeks of the onset of illness in a patient without a history of liver disease.1 In general, patients have no evidence of underlying chronic liver disease, but there are exceptions; patients with Wilson disease, vertically acquired hepatitis B virus infection, or autoimmune hepatitis can present with acute liver failure superimposed on chronic liver disease or even cirrhosis.
The term acute liver failure has replaced older terms such as fulminant hepatic failure, hyperacute liver failure, and subacute liver failure, which were used for prognostic purposes. Patients with hyperacute liver failure (defined as development of encephalopathy within 7 days of onset of illness) generally have a good prognosis with medical management, whereas those with subacute liver failure (defined as development of encephalopathy within 5 to 26 weeks of onset of illness) have a poor prognosis without liver transplant.2,3
NEARLY 2,000 CASES A YEAR
There are nearly 2,000 cases of acute liver failure each year in the United States, and it accounts for 6% of all deaths due to liver disease.4 It is more common in women than in men, and more common in white people than in other races. The peak incidence is at a fairly young age, ie, 35 to 45 years.
CAUSES
The most common cause of acute liver failure in the United States and other Western countries is acetaminophen toxicity, followed by viral hepatitis. In contrast, viral hepatitis is the most common cause in developing countries.5
Acetaminophen toxicity
Patients with acetaminophen-induced liver failure tend to be younger than other patients with acute liver failure.1 Nearly half of them present after intentionally taking a single large dose, while the rest present with unintentional toxicity while taking acetaminophen for pain relief on a long-term basis and ingesting more than the recommended dose.6
After ingestion, 52% to 57% of acetaminophen is converted to glucuronide conjugates, and 30% to 44% is converted to sulfate conjugates. These compounds are nontoxic, water-soluble, and rapidly excreted in the urine.
However, about 5% to 10% of ingested acetaminophen is shunted to the cytochrome P450 system. P450 2E1 is the main isoenzyme involved in acetaminophen metabolism, but 1A2, 3A4, and 2A6 also contribute.7,8 P450 2E1 is the same isoenzyme responsible for ethanol metabolism and is inducible. Thus, regular alcohol consumption can increase P450 2E1 activity, setting the stage under certain circumstances for increased acetaminophen metabolism through this pathway.
Metabolism of acetaminophen through the cytochrome P450 pathway results in production of N-acetyl-p-benzoquinone imine (NAPQI), the compound that damages the liver. NAPQI is rendered nontoxic by binding to glutathione, forming NAPQI-glutathione adducts. Glutathione capacity is limited, however. With too much acetaminophen, glutathione becomes depleted and NAPQI accumulates, binds with proteins to form adducts, and leads to necrosis of hepatocytes (Figure 1).9,10
Acetylcysteine, used in treating acetaminophen toxicity, is a substrate for glutathione synthesis and ultimately increases the amount of glutathione available to bind NAPQI and prevent damage to hepatocytes.11
Acetaminophen is a dose-related toxin. Most ingestions leading to acute liver failure exceed 10 g/day (> 150 mg/kg/day). Moderate chronic ingestion, eg, 4 g/day, usually leads to transient mild elevation of liver enzymes in healthy individuals12 but can in rare cases cause acute liver failure.13
Whitcomb and Block14 retrospectively identified 49 patients who presented with acetaminophen-induced hepatotoxicity in 1987 through 1993; 21 (43%) had been taking acetaminophen for therapeutic purposes. All 49 patients took more than the recommended limit of 4 g/day, many of them while fasting and some while using alcohol. Acute liver failure was seen with ingestion of more than 12 g/day—or more than 10 g/day in alcohol users. The authors attributed the increased risk to activation of cytochrome P450 2E1 by alcohol and depletion of glutathione stores by starvation or alcohol abuse.
Advice to patients taking acetaminophen is given in Table 1.
Other drugs and supplements
A number of other drugs and herbal supplements can also cause acute liver failure (Table 2), the most common being antimicrobial and antiepileptic drugs.15 Of the antimicrobials, antitubercular drugs (especially isoniazid) are believed to be the most common causes, followed by trimethoprim-sulfamethoxazole. Phenytoin is the antiepileptic drug most often implicated in acute liver failure.
Statins can also cause acute liver failure, especially when combined with other hepatotoxic agents.16
The herbal supplements and weight-loss agents Hydroxycut and Herbalife have both been reported to cause acute liver failure, with patients presenting with either the hepatocellular or the cholestatic pattern of liver injury.17 The exact chemical in these supplements that causes liver injury has not yet been determined.
The National Institutes of Health maintains a database of cases of liver failure due to medications and supplements at livertox.nih.gov. The database includes the pattern of hepatic injury, mechanism of injury, management, and outcomes.
Viral hepatitis
Hepatitis B virus is the most common viral cause of acute liver failure and is responsible for about 8% of cases.18
Patients with chronic hepatitis B virus infection—as evidenced by positive hepatitis B surface antigen—can develop acute liver failure if the infection is reactivated by the use of immunosuppressive drugs for solid-organ or bone-marrow transplant or medications such as anti-tumor necrosis agents, rituximab, or chemotherapy. These patients should be treated prophylactically with a nucleoside analogue, which should be continued for 6 months after immunosuppressive therapy is completed.
Hepatitis A virus is responsible for about 4% of cases.18
Hepatitis C virus rarely causes acute liver failure, especially in the absence of hepatitis A and hepatitis B.3,19
Hepatitis E virus, which is endemic in areas of Asia and Africa, can cause liver disease in pregnant women and in young adults who have concomitant liver disease from another cause. It tends to cause acute liver failure more frequently in pregnant women than in the rest of the population and carries a mortality rate of more than 20% in this subgroup.
TT (transfusion-transmitted) virus was reported in the 1990s to cause acute liver failure in about 27% of patients in whom no other cause could be found.20
Other rare viral causes of acute liver failure include Epstein-Barr virus, cytomegalovirus, and herpes simplex virus types 1, 2, and 6.
Other causes
Other causes of acute liver failure include ischemic hepatitis, autoimmune hepatitis, Wilson disease, Budd-Chiari syndrome, and HELLP (hemolysis, elevated liver enzymes and low platelets) syndrome.
MANY PATIENTS NEED LIVER TRANSPLANT
Many patients with acute liver failure ultimately require orthotopic liver transplant,21 especially if they present with severe encephalopathy. Other aspects of treatment vary according to the cause of liver failure (Table 3).
SPECIFIC MANAGEMENT
Management of acetaminophen toxicity
If the time of ingestion is known, checking the acetaminophen level can help determine the cause of acute liver failure and also predict the risk of hepatotoxicity, based on the work of Rumack and Matthew.22 Calculators are available, eg, http://reference.medscape.com/calculator/acetaminophen-toxicity.
If a patient presents with acute liver failure several days after ingesting acetaminophen, the level can be in the nontoxic range, however. In this scenario, measuring acetaminophen-protein adducts can help establish acetaminophen toxicity as the cause, as the adducts last longer in the serum and provide 100% sensitivity and specificity.23 While most laboratories can rapidly measure acetaminophen levels, only a few can measure acetaminophen-protein adducts, and thus this test is not used clinically.
Acetylcysteine is the main drug used for acetaminophen toxicity. Ideally, it should be given within 8 hours of acetaminophen ingestion, but giving it later is also useful.1
Acetylcysteine is available in oral and intravenous forms, the latter for patients who have encephalopathy or cannot tolerate oral intake due to repeated episodes of vomiting.24,25 The oral form is much less costly and is thus preferred over intravenous acetylcysteine in patients who can tolerate oral intake. Intravenous acetylcysteine should be given in a loading dose of 150 mg/kg in 5% dextrose over 15 minutes, followed by a maintenance dose of 50 mg/kg over 4 hours and then 100 mg/kg given over 16 hours.1 No dose adjustment is needed in patients who have renal toxicity (acetaminophen can also be toxic to the kidneys).
Most patients with acetaminophen-induced liver failure survive with medical management alone and do not need a liver transplant.3,26 Cirrhosis does not occur in these patients.
Management of viral acute liver failure
When patients present with acute liver failure, it is necessary to look for a viral cause by serologic testing, including hepatitis A virus IgM antibody, hepatitis B surface antigen, and hepatitis B core IgM antibody.
Hepatitis B can become reactivated in immunocompromised patients, and therefore the hepatitis B virus DNA level should be checked. Detection of hepatitis B virus DNA in a patient previously known to have undetectable hepatitis B virus DNA confirms hepatitis B reactivation.
Patients with hepatitis B-induced acute liver failure should be treated with entecavir or tenofovir. Although this treatment may not change the course of acute liver failure or accelerate the recovery, it can prevent reinfection in the transplanted liver if liver transplant becomes indicated.27–29
Herpes simplex virus should be suspected in patients presenting with anicteric hepatitis with fever. Polymerase chain reaction testing for herpes simplex virus should be done,30 and if positive, patients should be given intravenous acyclovir.31 Despite treatment, herpes simplex virus disease is associated with a very poor prognosis without liver transplant.
Autoimmune hepatitis
The autoantibodies usually seen in autoimmune hepatitis are antinuclear antibody, antismooth muscle antibody, and anti-liver-kidney microsomal antibody, and patients need to be tested for them.
The diagnosis of autoimmune hepatitis can be challenging, as these autoimmune markers can be negative in 5% of patients. Liver biopsy becomes essential to establish the diagnosis in that setting.32
Guidelines advise starting prednisone 40 to 60 mg/day and placing the patient on the liver transplant list.1
Wilson disease
Although it is an uncommon cause of liver failure, Wilson disease needs special attention because it has a poor prognosis. The mortality rate in acute liver failure from Wilson disease reaches 100% without liver transplant.
Wilson disease is caused by a genetic defect that allows copper to accumulate in the liver and other organs. However, diagnosing Wilson disease as the cause of acute liver failure can be challenging because elevated serum and urine copper levels are not specific to Wilson disease and can be seen in patients with acute liver failure from any cause. In addition, the ceruloplasmin level is usually normal or high because it is an acute-phase reactant. Accumulation of copper in the liver parenchyma is usually patchy; therefore, qualitative copper staining on random liver biopsy samples provides low diagnostic yield. Quantitative copper on liver biopsy is the gold standard test to establish the diagnosis, but the test is time-consuming. Kayser-Fleischer rings around the iris are considered pathognomic for Wilson disease when seen with acute liver failure, but they are seen in only about 50% of patients.33
A unique feature of acute Wilson disease is that most patients have very high bilirubin levels and low alkaline phosphatase levels. An alkaline phosphatase-to-bilirubin ratio less than 2 in patients with acute liver failure is highly suggestive of Wilson disease.34
Another clue to the diagnosis is that patients with Wilson disease tend to develop Coombs-negative hemolytic anemia, which leads to a disproportionate elevation in aminotransferase levels, with aspartate aminotransferase being higher than alanine aminotransferase.
Once Wilson disease is suspected, the patient should be listed for liver transplant because death is almost certain without it. For patients awaiting liver transplant, the American Association for the Study of Liver Diseases guidelines recommend certain measures to lower the serum copper level such as albumin dialysis, continuous hemofiltration, plasmapheresis, and plasma exchange,1 but the evidence supporting their use is limited.
NONSPECIFIC MANAGEMENT
Acute liver failure can affect a number of organs and systems in addition to the liver (Figure 2).
General considerations
Because their condition can rapidly deteriorate, patients with acute liver failure are best managed in intensive care.
Patients who present to a center that does not have the facilities for liver transplant should be transferred to a transplant center as soon as possible, preferably by air. If the patient may not be able to protect the airway, endotracheal intubation should be performed before transfer.
The major causes of death in patients with acute liver failure are cerebral edema and infection. Gastrointestinal bleeding was a major cause of death in the past, but with prophylactic use of histamine H2 receptor blockers and proton pump inhibitors, the incidence of gastrointestinal bleeding has been significantly reduced.
Although initially used only in patients with acetaminophen-induced liver failure, acetylcysteine has also shown benefit in patients with acute liver failure from other causes. In patients with grade 1 or 2 encephalopathy on a scale of 0 (minimal) to 4 (comatose), the transplant-free survival rate is higher when acetylcysteine is given compared with placebo, but this benefit does not extend to patients with a higher grade of encephalopathy.35
Cerebral edema and intracranial hypertension
Cerebral edema is the leading cause of death in patients with acute liver failure, and it develops in nearly 40% of patients.36
The mechanism by which cerebral edema develops is not well understood. Some have proposed that ammonia is converted to glutamine, which causes cerebral edema either directly by its osmotic effect37,38 or indirectly by decreasing other osmolytes, thereby promoting water retention.39
Cerebral edema leads to intracranial hypertension, which can ultimately cause cerebral herniation and death. Because of the high mortality rate associated with cerebral edema, invasive devices were extensively used in the past to monitor intracranial pressure. However, in light of known complications of these devices, including bleeding,40 and lack of evidence of long-term benefit in terms of mortality rates, their use has come under debate.
Treatments. Many treatments are available for cerebral edema and intracranial hypertension. The first step is to elevate the head of the bed about 30 degrees. In addition, hyponatremia should be corrected, as it can worsen cerebral edema.41 If patients are intubated, maintaining a hypercapneic state is advisable to decrease the intracranial pressure.
Of the two pharmacologic options, mannitol is more often used.42 It is given as a bolus dose of 0.5 to 1 g/kg intravenously if the serum osmolality is less than 320 mOsm/L.1 Given the risk of fluid overload with mannitol, caution must be exercised in patients with renal dysfunction. The other pharmacologic option is 3% hypertonic saline.
Therapeutic hypothermia is a newer treatment for cerebral edema. Lowering the body temperature to 32 to 33°C (89.6 to 91.4°F) using cooling blankets decreases intracranial pressure and cerebral blood flow and improves the cerebral perfusion pressure.43 With this treatment, patients should be closely monitored for side effects of infection, coagulopathy, and cardiac arrythmias.1
l-ornithine l-aspartate was successfully used to prevent brain edema in rats, but in humans, no benefit was seen compared with placebo.44,45 The underlying basis for this experimental treatment is that supplemental ornithine and aspartate should increase glutamate synthesis, which should increase the activity of enzyme glutamine synthetase in skeletal muscles. With the increase in enzyme activity, conversion of ammonia to glutamine should increase, thereby decreasing ammonia circulation and thus decreasing cerebral edema.
Patients with cerebral edema have a high incidence of seizures, but prophylactic antiseizure medications such as phenytoin have not been proven to be beneficial.46
Infection
Nearly 80% of patients with acute liver failure develop an infectious complication, which can be attributed to a state of immunodeficiency.47
The respiratory and urinary tracts are the most common sources of infection.48 In patients with bacteremia, Enterococcus species and coagulase-negative Staphylococcus species49 are the commonly isolated organisms. Also, in patients with acute liver failure, fungal infections account for 30% of all infections.50
Infected patients often develop worsening of their encephalopathy51 without fever or elevated white blood cell count.49,52 Thus, in any patient in whom encephalopathy is worsening, an evaluation must be done to rule out infection. In these patients, systemic inflammatory response syndrome is an independent risk factor for death.53
Despite the high mortality rate with infection, whether using antibiotics prophylactically in acute liver failure is beneficial is controversial.54,55
Gastrointestinal bleeding
The current prevalence of upper gastrointestinal bleeding in acute liver failure patients is about 1.5%.56 Coagulopathy and endotracheal intubation are the main risk factors for upper gastrointestinal bleeding in these patients.57 The most common source of bleeding is stress ulcers in the stomach. The ulcers develop from a combination of factors, including decreased blood flow to the mucosa causing ischemia and hypoperfusion-reperfusion injury.
Pharmacologic inhibition of gastric acid secretion has been shown to reduce upper gastrointestinal bleeding in acute liver failure. A histamine H2 receptor blocker or proton pump inhibitor should be given to prevent gastrointestinal bleeding in patients with acute liver failure.1,58
EXPERIMENTAL TREATMENTS
Artificial liver support systems
Membranes and dialysate solutions have been developed to remove toxic substances that are normally metabolized by the liver. Two of these—the molecular adsorbent recycling system (MARS) and the extracorporeal liver assist device (ELAD)—were developed in the late 1990s. MARS consisted of a highly permeable hollow fiber membrane mixed with albumin, and ELAD consisted of porcine hepatocytes attached to microcarriers in the extracapillary space of the hollow fiber membrane. Both systems allowed for transfer of water-soluble and protein-bound toxins in the blood across the membrane and into the dialysate.59 The clinical benefit offered by these devices is controversial,60–62 thus limiting their use to experimental purposes only.
Hepatocyte transplant
Use of hepatocyte transplant as a bridge to liver transplant was tested in 1970s, first in rats and later in humans.63 By reducing the blood ammonia level and improving cerebral perfusion pressure and cardiac function, replacement of 1% to 2% of the total liver cell mass by transplanted hepatocytes acts as a bridge to orthotopic liver transplant.64,65
PROGNOSIS
Different criteria have been used to identify patients with poor prognosis who may eventually need to undergo liver transplant.
The King’s College criteria system is the most commonly used for prognosis (Table 4).37,66–69 Its main drawback is that it is applicable only in patients with encephalopathy, and when patients reach this stage, their condition often deteriorates rapidly, and they die while awaiting liver transplant.37,66,67
The Model for End-Stage Liver Disease (MELD) score is an alternative to the King’s College criteria. A high MELD score on admission signifies advanced disease, and patients with a high MELD score tend to have a worse prognosis than those with a low score.68
The Acute Physiology and Chronic Health Evaluation (APACHE) II score can also be used, as it is more sensitive than the King’s College criteria.6
The Clichy criteria66,69 can also be used.
Liver biopsy. In addition to helping establish the cause of acute liver failure, liver biopsy can also be used as a prognostic tool. Hepatocellular necrosis greater than 70% on the biopsy predicts death with a specificity of 90% and a sensitivity of 56%.70
Hypophosphatemia has been reported to indicate recovering liver function in patients with acute liver failure.71 As the liver regenerates, its energy requirement increases. To supply the energy, adenosine triphosphate production increases, and phosphorus shifts from the extracellular to the intracellular compartment to meet the need for extra phosphorus during this process. A serum phosphorus level of 2.9 mg/dL or higher appears to indicate a poor prognosis in patients with acute liver failure, as it signifies that adequate hepatocyte regeneration is not occurring.
- Polson J, Lee WM; American Association for the Study of Liver Disease. AASLD position paper: the management of acute liver failure. Hepatology 2005; 41:1179–1197.
- O’Grady JG, Schalm SW, Williams R. Acute liver failure: redefining the syndromes. Lancet 1993; 342:273–275.
- Ostapowicz G, Fontana RJ, Schiodt FV, et al; US Acute Liver Failure Study Group. Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States. Ann Intern Med 2002; 137:947–954.
- Lee WM, Squires RH Jr, Nyberg SL, Doo E, Hoofnagle JH. Acute liver failure: summary of a workshop. Hepatology 2008; 47:1401–1415.
- Acharya SK, Panda SK, Saxena A, Gupta SD. Acute hepatic failure in India: a perspective from the East. J Gastroenterol Hepatol 2000; 15:473–479.
- Larson AM, Polson J, Fontana RJ, et al; Acute Liver Failure Study Group. Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology 2005; 42:1364–1372.
- Patten CJ, Thomas PE, Guy RL, et al. Cytochrome P450 enzymes involved in acetaminophen activation by rat and human liver microsomes and their kinetics. Chem Res Toxicol 1993; 6:511–518.
- Chen W, Koenigs LL, Thompson SJ, et al. Oxidation of acetaminophen to its toxic quinone imine and nontoxic catechol metabolites by baculovirus-expressed and purified human cytochromes P450 2E1 and 2A6. Chem Res Toxicol 1998; 11:295-301.
- Mitchell JR, Jollow DJ, Potter WZ, Gillette JR, Brodie BB. Acetaminophen-induced hepatic necrosis. IV. Protective role of glutathione. J Pharmacol Exp Ther 1973; 187:211–217.
- Schilling A, Corey R, Leonard M, Eghtesad B. Acetaminophen: old drug, new warnings. Cleve Clin J Med 2010; 77:19–27.
- Lauterburg BH, Corcoran GB, Mitchell JR. Mechanism of action of N-acetylcysteine in the protection against the hepatotoxicity of acetaminophen in rats in vivo. J Clin Invest 1983; 71:980–991.
- Watkins PB, Kaplowitz N, Slattery JT, et al. Aminotransferase elevations in healthy adults receiving 4 grams of acetaminophen daily: a randomized controlled trial. JAMA 2006; 296:87–93.
- Schiødt FV, Rochling FA, Casey DL, Lee WM. Acetaminophen toxicity in an urban county hospital. N Engl J Med 1997; 337:1112–1117.
- Whitcomb DC, Block GD. Association of acetaminophen hepatotoxicity with fasting and ethanol use. JAMA 1994; 272:1845–1850.
- Chalasani N, Fontana RJ, Bonkovsky HL, et al; Drug Induced Liver Injury Network (DILIN). Causes, clinical features, and outcomes from a prospective study of drug-induced liver injury in the United States. Gastroenterology 2008; 135:1924–1934 e1–4
- Reuben A, Koch DG, Lee WM; Acute Liver Failure Study Group. Drug-induced acute liver failure: results of a US multicenter, prospective study. Hepatology 2010; 52:2065–2076.
- Stevens T, Qadri A, Zein NN. Two patients with acute liver injury associated with use of the herbal weight-loss supplement hydroxycut. Ann Intern Med 2005; 142:477–478.
- Bernal W, Lee WM, Wendon J, Larsen FS, Williams R. Acute liver failure: a curable disease by 2024? J Hepatol 2015; 62(suppl 1):S112–S120.
- Schiodt FV, Davern TJ, Shakil AO, McGuire B, Samuel G, Lee WM. Viral hepatitis-related acute liver failure. Am J Gastroenterol 2003; 98:448–453.
- Charlton M, Adjei P, Poterucha J, et al. TT-virus infection in North American blood donors, patients with fulminant hepatic failure, and cryptogenic cirrhosis. Hepatology 1998; 28:839–842.
- Bismuth H, Samuel D, Gugenheim J, et al. Emergency liver transplantation for fulminant hepatitis. Ann Intern Med 1987; 107:337–341.
- Rumack BH, Matthew H. Acetaminophen poisoning and toxicity. Pediatrics 1975; 55:871–876.
- Davern TJ 2nd, James LP, Hinson JA, et al; Acute Liver Failure Study Group. Measurement of serum acetaminophen-protein adducts in patients with acute liver failure. Gastroenterology 2006; 130:687–694.
- Perry HE, Shannon MW. Efficacy of oral versus intravenous N-acetylcysteine in acetaminophen overdose: results of an open-label, clinical trial. J Pediatr 1998; 132:149–152.
- Smilkstein MJ, Knapp GL, Kulig KW, Rumack BH. Efficacy of oral N-acetylcysteine in the treatment of acetaminophen overdose. Analysis of the national multicenter study (1976 to 1985). N Engl J Med 1988; 319:1557–1562.
- Makin AJ, Wendon J, Williams R. A 7-year experience of severe acetaminophen-induced hepatotoxicity (1987-1993). Gastroenterology 1995; 109:1907–1916.
- Tsang SW, Chan HL, Leung NW, et al. Lamivudine treatment for fulminant hepatic failure due to acute exacerbation of chronic hepatitis B infection. Aliment Pharmacol Ther 2001; 15:1737–1744.
- Yu JW, Sun LJ, Yan BZ, Kang P, Zhao YH. Lamivudine treatment is associated with improved survival in fulminant hepatitis B. Liver Int 2011; 31:499–506.
- Garg H, Sarin SK, Kumar M, Garg V, Sharma BC, Kumar A. Tenofovir improves the outcome in patients with spontaneous reactivation of hepatitis B presenting as acute-on-chronic liver failure. Hepatology 2011; 53:774–780.
- Pinna AD, Rakela J, Demetris AJ, Fung JJ. Five cases of fulminant hepatitis due to herpes simplex virus in adults. Dig Dis Sci 2002; 47:750–754.
- Farr RW, Short S, Weissman D. Fulminant hepatitis during herpes simplex virus infection in apparently immunocompetent adults: report of two cases and review of the literature. Clin Infect Dis 1997; 24:1191–1194.
- Czaja AJ, Freese DK; American Association for the Study of Liver Disease. Diagnosis and treatment of autoimmune hepatitis. Hepatology 2002; 36:479–497.
- Roberts EA, Schilsky ML. A practice guideline on Wilson disease. Hepatology 2003; 37:1475–1492.
- Berman DH, Leventhal RI, Gavaler JS, Cadoff EM, Van Thiel DH. Clinical differentiation of fulminant Wilsonian hepatitis from other causes of hepatic failure. Gastroenterology 1991; 100:1129–1134.
- Lee WM, Hynan LS, Rossaro L, et al. Intravenous N-acetylcysteine improves transplant-free survival in early stage non-acetaminophen acute liver failure. Gastroenterology 2009; 137:856–864.
- O’Grady JG, Alexander GJ, Hayllar KM, Williams R. Early indicators of prognosis in fulminant hepatic failure. Gastroenterology 1989; 97:439–445.
- Clemmesen JO, Larsen FS, Kondrup J, Hansen BA, Ott P. Cerebral herniation in patients with acute liver failure is correlated with arterial ammonia concentration. Hepatology 1999; 29:648–653.
- Swain M, Butterworth RF, Blei AT. Ammonia and related amino acids in the pathogenesis of brain edema in acute ischemic liver failure in rats. Hepatology 1992; 15:449–453.
- Haussinger D, Laubenberger J, vom Dahl S, et al. Proton magnetic resonance spectroscopy studies on human brain myo-inositol in hypo-osmolarity and hepatic encephalopathy. Gastroenterology 1994; 107:1475–1480.
- Blei AT, Olafsson S, Webster S, Levy R. Complications of intracranial pressure monitoring in fulminant hepatic failure. Lancet 1993; 341:157–158.
- Cordoba J, Gottstein J, Blei AT. Chronic hyponatremia exacerbates ammonia-induced brain edema in rats after portacaval anastomosis. J Hepatol 1998; 29:589–594.
- Canalese J, Gimson AE, Davis C, Mellon PJ, Davis M, Williams R. Controlled trial of dexamethasone and mannitol for the cerebral oedema of fulminant hepatic failure. Gut 1982; 23:625–629.
- Jalan R, SW OD, Deutz NE, Lee A, Hayes PC. Moderate hypothermia for uncontrolled intracranial hypertension in acute liver failure. Lancet 1999; 354:1164–1168.
- Rose C, Michalak A, Rao KV, Quack G, Kircheis G, Butterworth RF. L-ornithine-L-aspartate lowers plasma and cerebrospinal fluid ammonia and prevents brain edema in rats with acute liver failure. Hepatology 1999; 30:636–640.
- Acharya SK, Bhatia V, Sreenivas V, Khanal S, Panda SK. Efficacy of L-ornithine L-aspartate in acute liver failure: a double-blind, randomized, placebo-controlled study. Gastroenterology 2009; 136:2159–2168.
- Bhatia V, Batra Y, Acharya SK. Prophylactic phenytoin does not improve cerebral edema or survival in acute liver failure—a controlled clinical trial. J Hepatol 2004; 41:89–96.
- Canalese J, Gove CD, Gimson AE, Wilkinson SP, Wardle EN, Williams R. Reticuloendothelial system and hepatocytic function in fulminant hepatic failure. Gut 1982; 23:265–269.
- Rolando N, Harvey F, Brahm J, et al. Prospective study of bacterial infection in acute liver failure: an analysis of fifty patients. Hepatology 1990; 11:49–53.
- Rolando N, Wade JJ, Stangou A, et al. Prospective study comparing the efficacy of prophylactic parenteral antimicrobials, with or without enteral decontamination, in patients with acute liver failure. Liver Transpl Surg 1996; 2:8–13.
- Rolando N, Harvey F, Brahm J, et al. Fungal infection: a common, unrecognised complication of acute liver failure. J Hepatol 1991; 12:1–9.
- Vaquero J, Polson J, Chung C, et al. Infection and the progression of hepatic encephalopathy in acute liver failure. Gastroenterology 2003; 125:755–764.
- Rolando N, Philpott-Howard J, Williams R. Bacterial and fungal infection in acute liver failure. Semin Liver Dis 1996; 16:389–402.
- Rolando N, Wade J, Davalos M, Wendon J, Philpott-Howard J, Williams R. The systemic inflammatory response syndrome in acute liver failure. Hepatology 2000; 32:734–739.
- Rolando N, Gimson A, Wade J, Philpott- Howard J, Casewell M, Williams R. Prospective controlled trial of selective parenteral and enteral antimicrobial regimen in fulminant liver failure. Hepatology 1993; 17:196–201.
- Karvellas CJ, Cavazos J, Battenhouse H, et al; US Acute Liver Failure Study Group. Effects of antimicrobial prophylaxis and blood stream infections in patients with acute liver failure: a retrospective cohort study. Clin Gastroenterol Hepatol 2014; 12:1942–1949.
- Acharya SK, Dasarathy S, Kumer TL, et al. Fulminant hepatitis in a tropical population: clinical course, cause, and early predictors of outcome. Hepatology 1996; 23:1148–1155.
- Cook DJ, Fuller HD, Guyatt GH, et al. Risk factors for gastrointestinal bleeding in critically ill patients. Canadian Critical Care Trials Group. N Engl J Med 1994; 330:377–381.
- MacDougall BR, Williams R. H2-receptor antagonist in the prevention of acute gastrointestinal hemorrhage in fulminant hepatic failure: a controlled trial. Gastroenterology 1978; 74:464–465.
- Stange J, Mitzner SR, Risler T, et al. Molecular adsorbent recycling system (MARS): clinical results of a new membrane-based blood purification system for bioartificial liver support. Artif Organs 1999; 23:319–330.
- Vaid A, Chewich H, Balk EM, Jaber BL. Molecular adsorbent recirculating system as artificial support therapy for liver failure: a meta-analysis. ASAIO J 2012; 58:51–59.
- Khuroo MS, Khuroo MS, Farahat KL. Molecular adsorbent recirculating system for acute and acute-on-chronic liver failure: a meta-analysis. Liver Transpl 2004; 10:1099–1106.
- Kjaergard LL, Liu J, Als-Nielsen B, Gluud C. Artificial and bioartificial support systems for acute and acute-on-chronic liver failure: a systematic review. JAMA 2003; 289:217–222.
- Sommer BG, Sutherland DE, Matas AJ, Simmons RL, Najarian JS. Hepatocellular transplantation for treatment of D-galactosamine-induced acute liver failure in rats. Transplant Proc 1979; 11:578–584.
- Demetriou AA, Reisner A, Sanchez J, Levenson SM, Moscioni AD, Chowdhury JR. Transplantation of microcarrier-attached hepatocytes into 90% partially hepatectomized rats. Hepatology 1988; 8:1006–1009.
- Strom SC, Fisher RA, Thompson MT, et al. Hepatocyte transplantation as a bridge to orthotopic liver transplantation in terminal liver failure. Transplantation 1997; 63:559–569.
- Pauwels A, Mostefa-Kara N, Florent C, Levy VG. Emergency liver transplantation for acute liver failure. Evaluation of London and Clichy criteria. J Hepatol 1993; 17:124–127.
- Anand AC, Nightingale P, Neuberger JM. Early indicators of prognosis in fulminant hepatic failure: an assessment of the King's criteria. J Hepatol 1997; 26:62–68.
- Schmidt LE, Larsen FS. MELD score as a predictor of liver failure and death in patients with acetaminophen-induced liver injury. Hepatology 2007; 45:789–796.
- Bernuau J, Goudeau A, Poynard T, et al. Multivariate analysis of prognostic factors in fulminant hepatitis B. Hepatology 1986; 6:648–651.
- Donaldson BW, Gopinath R, Wanless IR, et al. The role of transjugular liver biopsy in fulminant liver failure: relation to other prognostic indicators. Hepatology 1993; 18:1370–1376.
- Schmidt LE, Dalhoff K. Serum phosphate is an early predictor of outcome in severe acetaminophen-induced hepatotoxicity. Hepatology 2002; 36:659–665.
- Polson J, Lee WM; American Association for the Study of Liver Disease. AASLD position paper: the management of acute liver failure. Hepatology 2005; 41:1179–1197.
- O’Grady JG, Schalm SW, Williams R. Acute liver failure: redefining the syndromes. Lancet 1993; 342:273–275.
- Ostapowicz G, Fontana RJ, Schiodt FV, et al; US Acute Liver Failure Study Group. Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States. Ann Intern Med 2002; 137:947–954.
- Lee WM, Squires RH Jr, Nyberg SL, Doo E, Hoofnagle JH. Acute liver failure: summary of a workshop. Hepatology 2008; 47:1401–1415.
- Acharya SK, Panda SK, Saxena A, Gupta SD. Acute hepatic failure in India: a perspective from the East. J Gastroenterol Hepatol 2000; 15:473–479.
- Larson AM, Polson J, Fontana RJ, et al; Acute Liver Failure Study Group. Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology 2005; 42:1364–1372.
- Patten CJ, Thomas PE, Guy RL, et al. Cytochrome P450 enzymes involved in acetaminophen activation by rat and human liver microsomes and their kinetics. Chem Res Toxicol 1993; 6:511–518.
- Chen W, Koenigs LL, Thompson SJ, et al. Oxidation of acetaminophen to its toxic quinone imine and nontoxic catechol metabolites by baculovirus-expressed and purified human cytochromes P450 2E1 and 2A6. Chem Res Toxicol 1998; 11:295-301.
- Mitchell JR, Jollow DJ, Potter WZ, Gillette JR, Brodie BB. Acetaminophen-induced hepatic necrosis. IV. Protective role of glutathione. J Pharmacol Exp Ther 1973; 187:211–217.
- Schilling A, Corey R, Leonard M, Eghtesad B. Acetaminophen: old drug, new warnings. Cleve Clin J Med 2010; 77:19–27.
- Lauterburg BH, Corcoran GB, Mitchell JR. Mechanism of action of N-acetylcysteine in the protection against the hepatotoxicity of acetaminophen in rats in vivo. J Clin Invest 1983; 71:980–991.
- Watkins PB, Kaplowitz N, Slattery JT, et al. Aminotransferase elevations in healthy adults receiving 4 grams of acetaminophen daily: a randomized controlled trial. JAMA 2006; 296:87–93.
- Schiødt FV, Rochling FA, Casey DL, Lee WM. Acetaminophen toxicity in an urban county hospital. N Engl J Med 1997; 337:1112–1117.
- Whitcomb DC, Block GD. Association of acetaminophen hepatotoxicity with fasting and ethanol use. JAMA 1994; 272:1845–1850.
- Chalasani N, Fontana RJ, Bonkovsky HL, et al; Drug Induced Liver Injury Network (DILIN). Causes, clinical features, and outcomes from a prospective study of drug-induced liver injury in the United States. Gastroenterology 2008; 135:1924–1934 e1–4
- Reuben A, Koch DG, Lee WM; Acute Liver Failure Study Group. Drug-induced acute liver failure: results of a US multicenter, prospective study. Hepatology 2010; 52:2065–2076.
- Stevens T, Qadri A, Zein NN. Two patients with acute liver injury associated with use of the herbal weight-loss supplement hydroxycut. Ann Intern Med 2005; 142:477–478.
- Bernal W, Lee WM, Wendon J, Larsen FS, Williams R. Acute liver failure: a curable disease by 2024? J Hepatol 2015; 62(suppl 1):S112–S120.
- Schiodt FV, Davern TJ, Shakil AO, McGuire B, Samuel G, Lee WM. Viral hepatitis-related acute liver failure. Am J Gastroenterol 2003; 98:448–453.
- Charlton M, Adjei P, Poterucha J, et al. TT-virus infection in North American blood donors, patients with fulminant hepatic failure, and cryptogenic cirrhosis. Hepatology 1998; 28:839–842.
- Bismuth H, Samuel D, Gugenheim J, et al. Emergency liver transplantation for fulminant hepatitis. Ann Intern Med 1987; 107:337–341.
- Rumack BH, Matthew H. Acetaminophen poisoning and toxicity. Pediatrics 1975; 55:871–876.
- Davern TJ 2nd, James LP, Hinson JA, et al; Acute Liver Failure Study Group. Measurement of serum acetaminophen-protein adducts in patients with acute liver failure. Gastroenterology 2006; 130:687–694.
- Perry HE, Shannon MW. Efficacy of oral versus intravenous N-acetylcysteine in acetaminophen overdose: results of an open-label, clinical trial. J Pediatr 1998; 132:149–152.
- Smilkstein MJ, Knapp GL, Kulig KW, Rumack BH. Efficacy of oral N-acetylcysteine in the treatment of acetaminophen overdose. Analysis of the national multicenter study (1976 to 1985). N Engl J Med 1988; 319:1557–1562.
- Makin AJ, Wendon J, Williams R. A 7-year experience of severe acetaminophen-induced hepatotoxicity (1987-1993). Gastroenterology 1995; 109:1907–1916.
- Tsang SW, Chan HL, Leung NW, et al. Lamivudine treatment for fulminant hepatic failure due to acute exacerbation of chronic hepatitis B infection. Aliment Pharmacol Ther 2001; 15:1737–1744.
- Yu JW, Sun LJ, Yan BZ, Kang P, Zhao YH. Lamivudine treatment is associated with improved survival in fulminant hepatitis B. Liver Int 2011; 31:499–506.
- Garg H, Sarin SK, Kumar M, Garg V, Sharma BC, Kumar A. Tenofovir improves the outcome in patients with spontaneous reactivation of hepatitis B presenting as acute-on-chronic liver failure. Hepatology 2011; 53:774–780.
- Pinna AD, Rakela J, Demetris AJ, Fung JJ. Five cases of fulminant hepatitis due to herpes simplex virus in adults. Dig Dis Sci 2002; 47:750–754.
- Farr RW, Short S, Weissman D. Fulminant hepatitis during herpes simplex virus infection in apparently immunocompetent adults: report of two cases and review of the literature. Clin Infect Dis 1997; 24:1191–1194.
- Czaja AJ, Freese DK; American Association for the Study of Liver Disease. Diagnosis and treatment of autoimmune hepatitis. Hepatology 2002; 36:479–497.
- Roberts EA, Schilsky ML. A practice guideline on Wilson disease. Hepatology 2003; 37:1475–1492.
- Berman DH, Leventhal RI, Gavaler JS, Cadoff EM, Van Thiel DH. Clinical differentiation of fulminant Wilsonian hepatitis from other causes of hepatic failure. Gastroenterology 1991; 100:1129–1134.
- Lee WM, Hynan LS, Rossaro L, et al. Intravenous N-acetylcysteine improves transplant-free survival in early stage non-acetaminophen acute liver failure. Gastroenterology 2009; 137:856–864.
- O’Grady JG, Alexander GJ, Hayllar KM, Williams R. Early indicators of prognosis in fulminant hepatic failure. Gastroenterology 1989; 97:439–445.
- Clemmesen JO, Larsen FS, Kondrup J, Hansen BA, Ott P. Cerebral herniation in patients with acute liver failure is correlated with arterial ammonia concentration. Hepatology 1999; 29:648–653.
- Swain M, Butterworth RF, Blei AT. Ammonia and related amino acids in the pathogenesis of brain edema in acute ischemic liver failure in rats. Hepatology 1992; 15:449–453.
- Haussinger D, Laubenberger J, vom Dahl S, et al. Proton magnetic resonance spectroscopy studies on human brain myo-inositol in hypo-osmolarity and hepatic encephalopathy. Gastroenterology 1994; 107:1475–1480.
- Blei AT, Olafsson S, Webster S, Levy R. Complications of intracranial pressure monitoring in fulminant hepatic failure. Lancet 1993; 341:157–158.
- Cordoba J, Gottstein J, Blei AT. Chronic hyponatremia exacerbates ammonia-induced brain edema in rats after portacaval anastomosis. J Hepatol 1998; 29:589–594.
- Canalese J, Gimson AE, Davis C, Mellon PJ, Davis M, Williams R. Controlled trial of dexamethasone and mannitol for the cerebral oedema of fulminant hepatic failure. Gut 1982; 23:625–629.
- Jalan R, SW OD, Deutz NE, Lee A, Hayes PC. Moderate hypothermia for uncontrolled intracranial hypertension in acute liver failure. Lancet 1999; 354:1164–1168.
- Rose C, Michalak A, Rao KV, Quack G, Kircheis G, Butterworth RF. L-ornithine-L-aspartate lowers plasma and cerebrospinal fluid ammonia and prevents brain edema in rats with acute liver failure. Hepatology 1999; 30:636–640.
- Acharya SK, Bhatia V, Sreenivas V, Khanal S, Panda SK. Efficacy of L-ornithine L-aspartate in acute liver failure: a double-blind, randomized, placebo-controlled study. Gastroenterology 2009; 136:2159–2168.
- Bhatia V, Batra Y, Acharya SK. Prophylactic phenytoin does not improve cerebral edema or survival in acute liver failure—a controlled clinical trial. J Hepatol 2004; 41:89–96.
- Canalese J, Gove CD, Gimson AE, Wilkinson SP, Wardle EN, Williams R. Reticuloendothelial system and hepatocytic function in fulminant hepatic failure. Gut 1982; 23:265–269.
- Rolando N, Harvey F, Brahm J, et al. Prospective study of bacterial infection in acute liver failure: an analysis of fifty patients. Hepatology 1990; 11:49–53.
- Rolando N, Wade JJ, Stangou A, et al. Prospective study comparing the efficacy of prophylactic parenteral antimicrobials, with or without enteral decontamination, in patients with acute liver failure. Liver Transpl Surg 1996; 2:8–13.
- Rolando N, Harvey F, Brahm J, et al. Fungal infection: a common, unrecognised complication of acute liver failure. J Hepatol 1991; 12:1–9.
- Vaquero J, Polson J, Chung C, et al. Infection and the progression of hepatic encephalopathy in acute liver failure. Gastroenterology 2003; 125:755–764.
- Rolando N, Philpott-Howard J, Williams R. Bacterial and fungal infection in acute liver failure. Semin Liver Dis 1996; 16:389–402.
- Rolando N, Wade J, Davalos M, Wendon J, Philpott-Howard J, Williams R. The systemic inflammatory response syndrome in acute liver failure. Hepatology 2000; 32:734–739.
- Rolando N, Gimson A, Wade J, Philpott- Howard J, Casewell M, Williams R. Prospective controlled trial of selective parenteral and enteral antimicrobial regimen in fulminant liver failure. Hepatology 1993; 17:196–201.
- Karvellas CJ, Cavazos J, Battenhouse H, et al; US Acute Liver Failure Study Group. Effects of antimicrobial prophylaxis and blood stream infections in patients with acute liver failure: a retrospective cohort study. Clin Gastroenterol Hepatol 2014; 12:1942–1949.
- Acharya SK, Dasarathy S, Kumer TL, et al. Fulminant hepatitis in a tropical population: clinical course, cause, and early predictors of outcome. Hepatology 1996; 23:1148–1155.
- Cook DJ, Fuller HD, Guyatt GH, et al. Risk factors for gastrointestinal bleeding in critically ill patients. Canadian Critical Care Trials Group. N Engl J Med 1994; 330:377–381.
- MacDougall BR, Williams R. H2-receptor antagonist in the prevention of acute gastrointestinal hemorrhage in fulminant hepatic failure: a controlled trial. Gastroenterology 1978; 74:464–465.
- Stange J, Mitzner SR, Risler T, et al. Molecular adsorbent recycling system (MARS): clinical results of a new membrane-based blood purification system for bioartificial liver support. Artif Organs 1999; 23:319–330.
- Vaid A, Chewich H, Balk EM, Jaber BL. Molecular adsorbent recirculating system as artificial support therapy for liver failure: a meta-analysis. ASAIO J 2012; 58:51–59.
- Khuroo MS, Khuroo MS, Farahat KL. Molecular adsorbent recirculating system for acute and acute-on-chronic liver failure: a meta-analysis. Liver Transpl 2004; 10:1099–1106.
- Kjaergard LL, Liu J, Als-Nielsen B, Gluud C. Artificial and bioartificial support systems for acute and acute-on-chronic liver failure: a systematic review. JAMA 2003; 289:217–222.
- Sommer BG, Sutherland DE, Matas AJ, Simmons RL, Najarian JS. Hepatocellular transplantation for treatment of D-galactosamine-induced acute liver failure in rats. Transplant Proc 1979; 11:578–584.
- Demetriou AA, Reisner A, Sanchez J, Levenson SM, Moscioni AD, Chowdhury JR. Transplantation of microcarrier-attached hepatocytes into 90% partially hepatectomized rats. Hepatology 1988; 8:1006–1009.
- Strom SC, Fisher RA, Thompson MT, et al. Hepatocyte transplantation as a bridge to orthotopic liver transplantation in terminal liver failure. Transplantation 1997; 63:559–569.
- Pauwels A, Mostefa-Kara N, Florent C, Levy VG. Emergency liver transplantation for acute liver failure. Evaluation of London and Clichy criteria. J Hepatol 1993; 17:124–127.
- Anand AC, Nightingale P, Neuberger JM. Early indicators of prognosis in fulminant hepatic failure: an assessment of the King's criteria. J Hepatol 1997; 26:62–68.
- Schmidt LE, Larsen FS. MELD score as a predictor of liver failure and death in patients with acetaminophen-induced liver injury. Hepatology 2007; 45:789–796.
- Bernuau J, Goudeau A, Poynard T, et al. Multivariate analysis of prognostic factors in fulminant hepatitis B. Hepatology 1986; 6:648–651.
- Donaldson BW, Gopinath R, Wanless IR, et al. The role of transjugular liver biopsy in fulminant liver failure: relation to other prognostic indicators. Hepatology 1993; 18:1370–1376.
- Schmidt LE, Dalhoff K. Serum phosphate is an early predictor of outcome in severe acetaminophen-induced hepatotoxicity. Hepatology 2002; 36:659–665.
KEY POINTS
- In the United States, the most common cause of acute liver failure is acetaminophen toxicity, followed by viral hepatitis.
- Testing for the cause of acute liver failure needs to start as soon as possible so that specific treatment can be initiated and the patient can be placed on the transplant list if needed.
- Acetylcysteine and either a proton pump inhibitor or a histamine H2 receptor blocker should be given to all patients with acute liver failure. Liver transplant is the cornerstone of therapy in patients not responding to other treatments.
- There are a number of prognostic scores for acute liver failure, but each has limitations.
Woman, 35, With Jaundice and Altered Mental Status
IN THIS ARTICLE
- Results of case patient's initial laboratory work-up
- Top 10 prescription medications associated with idiosyncratic disease
- Outcome for the case patient
A 35-year-old African-American woman presented to the emergency department (ED) after being found disoriented and lethargic in her apartment by her friends. Given her altered mental status, the history of present illness was limited and informed mainly by her mother and friends. She had been unreachable by telephone for three days, and friends grew concerned when she was absent from work on two consecutive days. After obtaining access to her apartment, they found her in the bathroom jaundiced, incoherent, and surrounded by nonbloody, nonbilious vomit. She had no prior significant medical history, no documented daily medication, and no recent travel. Of note, previous medical contact was limited, and she did not have an established primary care provider. Additionally, there was no contributory family history, including autoimmune illness or liver disease.
ED presentation was marked by indications of grade 4 encephalopathy, including unresponsiveness to noxious stimuli. Initial laboratory work-up was notable for significantly elevated liver function test results (see Table 1). Based on her international normalized ratio (INR), total bilirubin, and creatinine, her initial Model for End-Stage Liver Disease score was 39, correlating to an 83% three-month mortality rate.1 Autoimmune marker testing revealed a positive antinuclear antibody (ANA), elevated immunoglobulin G (IgG), elevated smooth muscle antibody (IgG), normal antimitochondrial antibody, and normal anti-liver/kidney microsome antibody (IgG). Viral hepatitis serologies, including A, B, C, and E, were unremarkable. Ceruloplasmin and iron saturation were within normal limits. Acetaminophen, salicylate, and ethanol levels were negligible. Pregnancy testing and urine toxin testing were negative. Thyroid function tests were normal. Infectious work-up, including pan-culture, remained negative. Syphilis, herpes simplex virus (HSV), HIV, and varicella zoster testing were unremarkable.
CT of the head was not consistent with cerebral edema. CT of the abdomen and pelvis showed evidence of chronic pancreatitis and trace perihepatic ascites. She was intubated for airway protection and transferred to the medical ICU.
On liver biopsy, the patient was found to have acute hepatitis with centrilobular necrosis, approximately 30% to 40%, and prominent cholestasis. Histologically, these findings were reported as most consistent with drug-induced liver injury. Given her comatose state, coagulopathy, and extremely limited life expectancy without liver transplantation, the patient was listed for transplant as a status 1A candidate with fulminant hepatic failure.
She was placed on propofol and N-acetylcysteine infusions in addition to supportive IV resuscitation. The patient’s synthetic and neurocognitive function improved gradually over several weeks, and she was able to provide collateral history. She denied taking any prescription medications or having any ongoing medical issues. She did report that for two months prior to admission she had been taking an oral beauty supplement designed to enhance hair, skin, and nails. She obtained the supplement online. She could not recall the week leading up to admission, but she did note increasing malaise and fatigue beginning two weeks prior to admission. She denied any recreational drug or alcohol use.
Continue for discussion >>
DISCUSSION
Drug-induced liver injury (DILI) is a relatively uncommon occurrence in the United States.2 It is estimated to occur in approximately 20 individuals per 100,000 persons per year.2 However, DILI incidence secondary to herbal and dietary supplement use appears to be on the rise in the US. In a prospective study conducted by the Drug-Induced Liver Injury Network (DILIN) that included patients with liver injury referred to eight DILIN centers between 2004 and 2013, the proportion of DILI cases caused by herbal and dietary supplements increased from 7% to 20% over the study period.3
DILI can be subclassified into intrinsic and idiosyncratic. Intrinsic DILI results from substances causing a predictable time course and natural history. Substances causing a varied, unpredictable occurrence of DILI in susceptible individuals are idiosyncratic.4 Overall, acetaminophen overdose is the most common cause of DILI.2 However, the most common idiosyncratic offending agents, taken at FDA-approved dosages, are antimicrobials (see Table 2).5 The second most common offending agents are herbal and dietary supplements.5

In a retrospective cohort study evaluating all cases of acute liver failure (ALF) over a six-year period in an integrated health care system, the leading cause of ALF was DILI.6 Of the 32 patients with confirmed drug-induced ALF in this study, the majority of cases (18) were associated with acetaminophen. Herbal and dietary supplements were implicated in six cases, with miscellaneous medications accounting for the remaining eight cases.6 In terms of outcomes, 18.8% of patients with ALF due to DILI underwent liver transplantation, 68.8% were discharged, and 12.5% died during hospitalization.6
DILI disproportionately affects women and minorities7;although the etiology is unclear, it is hypothesized that increased use of antibiotics may play a role among women.2 Providers should be aware of the increased risk for DILI in these populations and consider this diagnosis in the appropriate setting.
Teasing out the diagnosis
DILI is a diagnosis of exclusion, aided in large part by the history and physical exam.4 An extensive history may alert the health care provider to a potential offending substance as well as provide information on timing of exposure.4 DILI should be suspected in patients with persistently elevated liver enzymes, unremarkable work-up for all other underlying liver disease (including autoimmune and viral serologies), and negative abdominal imaging.4 In particular, acute hepatitis C virus (HCV) and hepatitis E virus (HEV) infection mimic the clinical presentation of DILI and should be excluded with HCV RNA and IgM anti-HEV testing, with reflex HEV RNA testing to confirm positive IgM anti-HEV results.8,9 Liver biopsy is rarely indicated for the diagnosis of DILI.2
The presentation of DILI ranges from asymptomatic, with mildly abnormal results on liver function testing, to fulminant hepatic failure. Acetaminophen is the most frequently reported cause of intrinsic DILI in the US, playing a role in approximately half of all ALF cases.10 DILI can be further subdivided according to the pattern of liver test abnormalities as hepatocellular, mixed, or cholestatic based on the ratio of ALT to alkaline phosphatase (R value).2 Utilizing the formula serum ALT/upper limit of normal (ULN) divided by the serum alkaline phosphatase/ULN to determine R value, liver test abnormalities are defined as hepatocellular (R > 5), mixed (R = 2-5), and cholestatic (R < 2).4 These liver test patterns can be used to predict prognosis (see “Prognosis: Hy’s law”). In a prospective, longitudinal study, DILIN found that chronic DILI was present in 18% of the study population at 6 months following onset.5 Patients with the cholestatic presentation were more likely to develop chronic DILI than were those with the hepatocellular or mixed pattern. Furthermore, the hepatocellular pattern on presentation was associated with greater mortality.5 Patients with the mixed pattern had the most favorable outcomes. Another prospective cohort study found that persistently elevated liver enzymes in DILI patients at 12 months is associated with older age and the cholestatic pattern of liver test abnormalities at presentation, in particular, alkaline phosphatase elevation.11 However, neither length of therapy nor type of offending medication was associated with long-term liver test abnormalities.11
Managing DILI and ALF
In all DILI cases, immediate discontinuation of the offending agent is the initial treatment recommendation.2 Patients presenting with DILI who have an accompanying bilirubin level > 2 mg/dL should be referred to a hepatology specialist due to an increased risk for ALF.2 ALF is defined as coagulopathy to INR ≥ 1.5 and hepatic encephalopathy within 26 weeks of initial symptom onset in individuals without known underlying liver disease, with the exception of autoimmune hepatitis, Wilson disease, and reactivation of hepatitis B.12-15 Fulminant hepatic failure is further specified as encephalopathy occurring within 8 weeks of jaundice onset.12
Patients presenting with ALF should be transferred to an intensive care setting, preferably within a liver transplant center, for supportive care and potential liver transplant evaluation.12 CT of the head should be used to rule out other etiologies for altered mental status.16N-Acetylcysteine is the treatment of choice for acetaminophen-induced ALF, and it has also been shown to improve transplant-free survival outcomes in patients with non-acetaminophen–related early ALF.17 Infectious work-up and continuous monitoring are essential in ALF care, since up to 80% of patients with ALF will develop a bacterial infection.18 A comprehensive infectious work-up should include pan-culture of blood, urine, and sputum in addition to assessment for Epstein-Barr virus, cytomegalovirus, and HSV.4,18 For irreversible ALF, liver transplantation remains the only validated treatment option.12,19
Prognosis: Hy’s law
Hy’s law refers to a method used in clinical trials to assess a drug’s likelihood of causing severe hepatotoxicity; it is also used to predict which patients with DILI will develop ALF.12,20 According to Hy’s law, patients with AST or ALT elevations three times ULN and total bilirubin elevations two times ULN are at increased risk for ALF.In a retrospective cohort study of more than 15,000 patients with DILI, the Hy’s law criteria were found to have high specificity but low sensitivity for detecting individuals at risk for ALF.15 An alternative model, the Drug-Induced Liver Toxicity ALF Score, uses platelet count and bilirubin level to identify patients at risk for ALF with high sensitivity.15
Patient education
Effective patient education is essential to decreasing DILI incidence at a time when herbal and dietary supplement consumption is increasing. Patients will often bring herbal and dietary supplements to their providers to obtain a safety profile prior to initiation. In these cases, it is essential to reinforce with patients the absence of federal regulation of these products. It should be stressed to patients that, due to the lack of government oversight, it is impossible to confidently identify the entirety and quantity of ingredients in these supplements. Furthermore, there is no existing protocol for surveillance or adverse event reporting for these products.21 Because these products are not routinely or systematically studied, even health care providers have no evidence on which to base monitoring or usage recommendations. Providers may direct patients to the National Institutes of Health’s LiverTox website (livertox.NIH.gov) to review prior case reports of hepatotoxicity for specific dietary and herbal supplements.
Level of education is associated with knowledge of the potential for overdose when taking OTC medications that contain acetaminophen.22 As a result, health care providers should strongly reinforce with patients the importance of reading all medication labels and abiding by the listed administration directions. In particular, providers should emphasize that the maximum daily dosage of acetaminophen is 4 g.23 For patients with chronic liver disease, a more conservative recommendation is warranted. Generally, patients with cirrhosis may be advised to consume up to 2 g/d of acetaminophen as a firstline treatment for pain. However, providers should ensure acetaminophen ingestion is limited to a brief period.24
Additionally, it is important to educate patients that many combination OTC medications contain acetaminophen. Of note, chronic opioid users are more likely to accurately identify OTC medications containing acetaminophen, compared with acute opioid users.22 These findings should compel health care providers to deliver in-depth education for all patients, particularly those with less education or experience with medications. Education on avoidance of offending medications, including medications within the same class, when appropriate, is essential for quality patient care.2
Continue to outcome for the case patient >>
OUTCOME FOR THE CASE PATIENT
Following discharge, the patient was monitored closely with regular clinic visits and blood work. Her liver test results improved gradually, with consideration of a repeat biopsy to evaluate for overlap or missed autoimmune disease. Her repeat ANA was negative and IgG was within normal limits. Within three months of admission, her liver tests normalized and repeat biopsy was deferred.
Upon review of the herbal beauty supplement the patient reported taking, shark cartilage was noted as a primary ingredient. In a case report, shark cartilage was identified as a hepatotoxin.25 The patient was advised never to ingest the offending supplement, or any other substances not regulated by the FDA, again. Furthermore, the offending medication was listed as a medication allergy in her electronic health record.
CONCLUSION
It is crucial to emphasize to patients the potential hepatotoxicity of medications and herbal and dietary supplements, especially OTC medications that pose an overdose risk. Patients should review all new supplements with their providers prior to therapy initiation. With known hepatotoxins, providers should closely monitor patients for liver injury while treatment is ongoing. In suspected cases of DILI, a thorough history and physical exam will greatly inform the diagnosis. In the majority of cases, the suspect medication should be discontinued immediately, with subsequent assessment of liver response. Identification of DILI early in the course increases the likelihood of full hepatic recovery and improves patient outcomes.
References
1. Kamath PS, Wiesner RH, Malinchoc M, et al. A model to predict survival in patients with end-stage liver disease. Hepatology. 2001;33(2):464-470.
2. Leise MD, Poterucha JJ, Talwalkar JA. Drug-induced liver injury. Mayo Clin Proc. 2014;89(1):95-106.
3. Navarro VJ, Barnhart H, Bonkovsky HL, et al. Liver injury from herbals and dietary supplements in the US Drug-Induced Liver Injury Network. Hepatology. 2014;60(4):1399-1408.
4. Chalasani NP, Hayashi PH, Bonkovsky HL, et al. ACG Clinical Guideline: the diagnosis and management of idiosyncratic drug-induced liver injury. Am J Gastroenterol. 2014;109(7):950-966.
5. Chalasani N, Bonkovsky HL, Fontana R, et al; United States Drug Induced Liver Injury Network. Features and outcomes of 899 patients with drug-induced liver injury: the DILIN prospective study. Gastroenterology. 2015;148(7):1340-1352.
6. Goldberg DS, Forde KA, Carbonari DM, et al. Population-representative incidence of drug-induced acute liver failure based on an analysis of an integrated health care system. Gastroenterology. 2015;148(7):1353-1361.
7. Reuben A, Koch DG, Lee WM. Drug-induced acute liver failure: results of a US multicenter, prospective study. Hepatology. 2010;52(6):2065-2076.
8. Davern TJ, Chalasani N, Fontana RJ, et al; Drug-Induced Liver Injury Network (DILIN). Acute hepatitis E infection accounts for some cases of suspected drug-induced liver injury. Gastroenterology. 2011;141(5):1665-1672.e1-9.
9. Chalasani N, Fontana RJ, Bonkovsky HL, et al. Causes, clinical features, and outcomes from a prospective study of drug-induced liver injury in the United States. Gastroenterology. 2008;135(6):1924-1934.
10. Fisher K, Vuppalanchi R, Saxena R. Drug-induced liver injury. Arch Pathol Lab Med. 2015;139(7):876-887.
11. Fontana RJ, Hayashi PH, Barnhart H, et al. Persistent liver biochemistry abnormalities are more common in older patients and those with cholestatic drug induced liver injury. Am J Gastroenterol. 2015;110(10):1450-1459.
12. Punzalan CS, Barry CT. Acute liver failure: diagnosis and management. J Intensive Care Med. 2015 Oct 6. [Epub ahead of print]
13. Bower WA, Johns M, Margolis HS, et al. Population-based surveillance for acute liver failure. Am J Gastroenterol. 2007;102(11):2459-2463.
14. O’Grady JG, Schalm SW, Williams R. Acute liver failure: redefining the syndromes. Lancet. 1993;342(8866):273-275.
15. Lo Re V III, Haynes K, Forde KA, et al. Risk of acute liver failure in patients with drug-induced liver injury: evaluation of Hy’s law and a new prognostic model. Clin Gastroenterol Hepatol. 2015;13(13):2360-2368.
16. Polson J, Lee WM; American Association for the Study of Liver Diseases. AASLD position paper: the management of acute liver failure. Hepatology. 2005;41:1179-1197.
17. Lee WM, Hynan LS, Rossaro L, et al. Intravenous N-acetylcysteine improves transplant-free survival in early stage non-acetaminophen acute liver failure. Gastroenterology. 2009;137(3):856-864.
18. Rolando N, Harvey F, Brahm J. Prospective study of bacterial infection in acute liver failure: an analysis of fifty patients. Hepatology. 1990;11(1):49-53.
19. Panackel C, Thomas R, Sebastian B, Mathai SK. Recent advances in management of acute liver failure. Indian J Crit Care Med. 2015;19(1):27-33.
20. Temple R. Hy’s law: predicting serious hepatotoxicity. Pharmacoepidemiol Drug Saf. 2006;15(4):241-243.
21. Bunchorntavakul C, Reddy K. Review article: herbal and dietary supplement hepatotoxicity. Aliment Pharmacol Ther. 2012;37(1):3-17.
22. Boudreau DM, Wirtz H, Von Korff M, et al. A survey of adult awareness and use of medicine containing acetaminophen. Pharmacoepidemiol Drug Saf. 2013;22(3):229-240.
23. Burns MJ, Friedman SL, Larson AM. Acetaminophen (paracetamol) poisoning in adults: pathophysiology, presentation, and diagnosis. UpToDate. www.uptodate.com/contents/acetaminophen-paracetamol-poisoning-in-adults-pathophysiology-presentation-and-diagnosis. Accessed May 20, 2016.
24. Lewis JH, Stine JG. Review article: prescribing medications in patients with cirrhosis—a practical guide. Aliment Pharmacol Ther. 2013;37(12):1132-1156.
25. Ashar B, Vargo E. Shark cartilage-induced hepatitis. Ann Intern Med. 1996;125(9):780-781.
IN THIS ARTICLE
- Results of case patient's initial laboratory work-up
- Top 10 prescription medications associated with idiosyncratic disease
- Outcome for the case patient
A 35-year-old African-American woman presented to the emergency department (ED) after being found disoriented and lethargic in her apartment by her friends. Given her altered mental status, the history of present illness was limited and informed mainly by her mother and friends. She had been unreachable by telephone for three days, and friends grew concerned when she was absent from work on two consecutive days. After obtaining access to her apartment, they found her in the bathroom jaundiced, incoherent, and surrounded by nonbloody, nonbilious vomit. She had no prior significant medical history, no documented daily medication, and no recent travel. Of note, previous medical contact was limited, and she did not have an established primary care provider. Additionally, there was no contributory family history, including autoimmune illness or liver disease.
ED presentation was marked by indications of grade 4 encephalopathy, including unresponsiveness to noxious stimuli. Initial laboratory work-up was notable for significantly elevated liver function test results (see Table 1). Based on her international normalized ratio (INR), total bilirubin, and creatinine, her initial Model for End-Stage Liver Disease score was 39, correlating to an 83% three-month mortality rate.1 Autoimmune marker testing revealed a positive antinuclear antibody (ANA), elevated immunoglobulin G (IgG), elevated smooth muscle antibody (IgG), normal antimitochondrial antibody, and normal anti-liver/kidney microsome antibody (IgG). Viral hepatitis serologies, including A, B, C, and E, were unremarkable. Ceruloplasmin and iron saturation were within normal limits. Acetaminophen, salicylate, and ethanol levels were negligible. Pregnancy testing and urine toxin testing were negative. Thyroid function tests were normal. Infectious work-up, including pan-culture, remained negative. Syphilis, herpes simplex virus (HSV), HIV, and varicella zoster testing were unremarkable.
CT of the head was not consistent with cerebral edema. CT of the abdomen and pelvis showed evidence of chronic pancreatitis and trace perihepatic ascites. She was intubated for airway protection and transferred to the medical ICU.
On liver biopsy, the patient was found to have acute hepatitis with centrilobular necrosis, approximately 30% to 40%, and prominent cholestasis. Histologically, these findings were reported as most consistent with drug-induced liver injury. Given her comatose state, coagulopathy, and extremely limited life expectancy without liver transplantation, the patient was listed for transplant as a status 1A candidate with fulminant hepatic failure.
She was placed on propofol and N-acetylcysteine infusions in addition to supportive IV resuscitation. The patient’s synthetic and neurocognitive function improved gradually over several weeks, and she was able to provide collateral history. She denied taking any prescription medications or having any ongoing medical issues. She did report that for two months prior to admission she had been taking an oral beauty supplement designed to enhance hair, skin, and nails. She obtained the supplement online. She could not recall the week leading up to admission, but she did note increasing malaise and fatigue beginning two weeks prior to admission. She denied any recreational drug or alcohol use.
Continue for discussion >>
DISCUSSION
Drug-induced liver injury (DILI) is a relatively uncommon occurrence in the United States.2 It is estimated to occur in approximately 20 individuals per 100,000 persons per year.2 However, DILI incidence secondary to herbal and dietary supplement use appears to be on the rise in the US. In a prospective study conducted by the Drug-Induced Liver Injury Network (DILIN) that included patients with liver injury referred to eight DILIN centers between 2004 and 2013, the proportion of DILI cases caused by herbal and dietary supplements increased from 7% to 20% over the study period.3
DILI can be subclassified into intrinsic and idiosyncratic. Intrinsic DILI results from substances causing a predictable time course and natural history. Substances causing a varied, unpredictable occurrence of DILI in susceptible individuals are idiosyncratic.4 Overall, acetaminophen overdose is the most common cause of DILI.2 However, the most common idiosyncratic offending agents, taken at FDA-approved dosages, are antimicrobials (see Table 2).5 The second most common offending agents are herbal and dietary supplements.5
In a retrospective cohort study evaluating all cases of acute liver failure (ALF) over a six-year period in an integrated health care system, the leading cause of ALF was DILI.6 Of the 32 patients with confirmed drug-induced ALF in this study, the majority of cases (18) were associated with acetaminophen. Herbal and dietary supplements were implicated in six cases, with miscellaneous medications accounting for the remaining eight cases.6 In terms of outcomes, 18.8% of patients with ALF due to DILI underwent liver transplantation, 68.8% were discharged, and 12.5% died during hospitalization.6
DILI disproportionately affects women and minorities7;although the etiology is unclear, it is hypothesized that increased use of antibiotics may play a role among women.2 Providers should be aware of the increased risk for DILI in these populations and consider this diagnosis in the appropriate setting.
Teasing out the diagnosis
DILI is a diagnosis of exclusion, aided in large part by the history and physical exam.4 An extensive history may alert the health care provider to a potential offending substance as well as provide information on timing of exposure.4 DILI should be suspected in patients with persistently elevated liver enzymes, unremarkable work-up for all other underlying liver disease (including autoimmune and viral serologies), and negative abdominal imaging.4 In particular, acute hepatitis C virus (HCV) and hepatitis E virus (HEV) infection mimic the clinical presentation of DILI and should be excluded with HCV RNA and IgM anti-HEV testing, with reflex HEV RNA testing to confirm positive IgM anti-HEV results.8,9 Liver biopsy is rarely indicated for the diagnosis of DILI.2
The presentation of DILI ranges from asymptomatic, with mildly abnormal results on liver function testing, to fulminant hepatic failure. Acetaminophen is the most frequently reported cause of intrinsic DILI in the US, playing a role in approximately half of all ALF cases.10 DILI can be further subdivided according to the pattern of liver test abnormalities as hepatocellular, mixed, or cholestatic based on the ratio of ALT to alkaline phosphatase (R value).2 Utilizing the formula serum ALT/upper limit of normal (ULN) divided by the serum alkaline phosphatase/ULN to determine R value, liver test abnormalities are defined as hepatocellular (R > 5), mixed (R = 2-5), and cholestatic (R < 2).4 These liver test patterns can be used to predict prognosis (see “Prognosis: Hy’s law”). In a prospective, longitudinal study, DILIN found that chronic DILI was present in 18% of the study population at 6 months following onset.5 Patients with the cholestatic presentation were more likely to develop chronic DILI than were those with the hepatocellular or mixed pattern. Furthermore, the hepatocellular pattern on presentation was associated with greater mortality.5 Patients with the mixed pattern had the most favorable outcomes. Another prospective cohort study found that persistently elevated liver enzymes in DILI patients at 12 months is associated with older age and the cholestatic pattern of liver test abnormalities at presentation, in particular, alkaline phosphatase elevation.11 However, neither length of therapy nor type of offending medication was associated with long-term liver test abnormalities.11
Managing DILI and ALF
In all DILI cases, immediate discontinuation of the offending agent is the initial treatment recommendation.2 Patients presenting with DILI who have an accompanying bilirubin level > 2 mg/dL should be referred to a hepatology specialist due to an increased risk for ALF.2 ALF is defined as coagulopathy to INR ≥ 1.5 and hepatic encephalopathy within 26 weeks of initial symptom onset in individuals without known underlying liver disease, with the exception of autoimmune hepatitis, Wilson disease, and reactivation of hepatitis B.12-15 Fulminant hepatic failure is further specified as encephalopathy occurring within 8 weeks of jaundice onset.12
Patients presenting with ALF should be transferred to an intensive care setting, preferably within a liver transplant center, for supportive care and potential liver transplant evaluation.12 CT of the head should be used to rule out other etiologies for altered mental status.16N-Acetylcysteine is the treatment of choice for acetaminophen-induced ALF, and it has also been shown to improve transplant-free survival outcomes in patients with non-acetaminophen–related early ALF.17 Infectious work-up and continuous monitoring are essential in ALF care, since up to 80% of patients with ALF will develop a bacterial infection.18 A comprehensive infectious work-up should include pan-culture of blood, urine, and sputum in addition to assessment for Epstein-Barr virus, cytomegalovirus, and HSV.4,18 For irreversible ALF, liver transplantation remains the only validated treatment option.12,19
Prognosis: Hy’s law
Hy’s law refers to a method used in clinical trials to assess a drug’s likelihood of causing severe hepatotoxicity; it is also used to predict which patients with DILI will develop ALF.12,20 According to Hy’s law, patients with AST or ALT elevations three times ULN and total bilirubin elevations two times ULN are at increased risk for ALF.In a retrospective cohort study of more than 15,000 patients with DILI, the Hy’s law criteria were found to have high specificity but low sensitivity for detecting individuals at risk for ALF.15 An alternative model, the Drug-Induced Liver Toxicity ALF Score, uses platelet count and bilirubin level to identify patients at risk for ALF with high sensitivity.15
Patient education
Effective patient education is essential to decreasing DILI incidence at a time when herbal and dietary supplement consumption is increasing. Patients will often bring herbal and dietary supplements to their providers to obtain a safety profile prior to initiation. In these cases, it is essential to reinforce with patients the absence of federal regulation of these products. It should be stressed to patients that, due to the lack of government oversight, it is impossible to confidently identify the entirety and quantity of ingredients in these supplements. Furthermore, there is no existing protocol for surveillance or adverse event reporting for these products.21 Because these products are not routinely or systematically studied, even health care providers have no evidence on which to base monitoring or usage recommendations. Providers may direct patients to the National Institutes of Health’s LiverTox website (livertox.NIH.gov) to review prior case reports of hepatotoxicity for specific dietary and herbal supplements.
Level of education is associated with knowledge of the potential for overdose when taking OTC medications that contain acetaminophen.22 As a result, health care providers should strongly reinforce with patients the importance of reading all medication labels and abiding by the listed administration directions. In particular, providers should emphasize that the maximum daily dosage of acetaminophen is 4 g.23 For patients with chronic liver disease, a more conservative recommendation is warranted. Generally, patients with cirrhosis may be advised to consume up to 2 g/d of acetaminophen as a firstline treatment for pain. However, providers should ensure acetaminophen ingestion is limited to a brief period.24
Additionally, it is important to educate patients that many combination OTC medications contain acetaminophen. Of note, chronic opioid users are more likely to accurately identify OTC medications containing acetaminophen, compared with acute opioid users.22 These findings should compel health care providers to deliver in-depth education for all patients, particularly those with less education or experience with medications. Education on avoidance of offending medications, including medications within the same class, when appropriate, is essential for quality patient care.2
Continue to outcome for the case patient >>
OUTCOME FOR THE CASE PATIENT
Following discharge, the patient was monitored closely with regular clinic visits and blood work. Her liver test results improved gradually, with consideration of a repeat biopsy to evaluate for overlap or missed autoimmune disease. Her repeat ANA was negative and IgG was within normal limits. Within three months of admission, her liver tests normalized and repeat biopsy was deferred.
Upon review of the herbal beauty supplement the patient reported taking, shark cartilage was noted as a primary ingredient. In a case report, shark cartilage was identified as a hepatotoxin.25 The patient was advised never to ingest the offending supplement, or any other substances not regulated by the FDA, again. Furthermore, the offending medication was listed as a medication allergy in her electronic health record.
CONCLUSION
It is crucial to emphasize to patients the potential hepatotoxicity of medications and herbal and dietary supplements, especially OTC medications that pose an overdose risk. Patients should review all new supplements with their providers prior to therapy initiation. With known hepatotoxins, providers should closely monitor patients for liver injury while treatment is ongoing. In suspected cases of DILI, a thorough history and physical exam will greatly inform the diagnosis. In the majority of cases, the suspect medication should be discontinued immediately, with subsequent assessment of liver response. Identification of DILI early in the course increases the likelihood of full hepatic recovery and improves patient outcomes.
References
1. Kamath PS, Wiesner RH, Malinchoc M, et al. A model to predict survival in patients with end-stage liver disease. Hepatology. 2001;33(2):464-470.
2. Leise MD, Poterucha JJ, Talwalkar JA. Drug-induced liver injury. Mayo Clin Proc. 2014;89(1):95-106.
3. Navarro VJ, Barnhart H, Bonkovsky HL, et al. Liver injury from herbals and dietary supplements in the US Drug-Induced Liver Injury Network. Hepatology. 2014;60(4):1399-1408.
4. Chalasani NP, Hayashi PH, Bonkovsky HL, et al. ACG Clinical Guideline: the diagnosis and management of idiosyncratic drug-induced liver injury. Am J Gastroenterol. 2014;109(7):950-966.
5. Chalasani N, Bonkovsky HL, Fontana R, et al; United States Drug Induced Liver Injury Network. Features and outcomes of 899 patients with drug-induced liver injury: the DILIN prospective study. Gastroenterology. 2015;148(7):1340-1352.
6. Goldberg DS, Forde KA, Carbonari DM, et al. Population-representative incidence of drug-induced acute liver failure based on an analysis of an integrated health care system. Gastroenterology. 2015;148(7):1353-1361.
7. Reuben A, Koch DG, Lee WM. Drug-induced acute liver failure: results of a US multicenter, prospective study. Hepatology. 2010;52(6):2065-2076.
8. Davern TJ, Chalasani N, Fontana RJ, et al; Drug-Induced Liver Injury Network (DILIN). Acute hepatitis E infection accounts for some cases of suspected drug-induced liver injury. Gastroenterology. 2011;141(5):1665-1672.e1-9.
9. Chalasani N, Fontana RJ, Bonkovsky HL, et al. Causes, clinical features, and outcomes from a prospective study of drug-induced liver injury in the United States. Gastroenterology. 2008;135(6):1924-1934.
10. Fisher K, Vuppalanchi R, Saxena R. Drug-induced liver injury. Arch Pathol Lab Med. 2015;139(7):876-887.
11. Fontana RJ, Hayashi PH, Barnhart H, et al. Persistent liver biochemistry abnormalities are more common in older patients and those with cholestatic drug induced liver injury. Am J Gastroenterol. 2015;110(10):1450-1459.
12. Punzalan CS, Barry CT. Acute liver failure: diagnosis and management. J Intensive Care Med. 2015 Oct 6. [Epub ahead of print]
13. Bower WA, Johns M, Margolis HS, et al. Population-based surveillance for acute liver failure. Am J Gastroenterol. 2007;102(11):2459-2463.
14. O’Grady JG, Schalm SW, Williams R. Acute liver failure: redefining the syndromes. Lancet. 1993;342(8866):273-275.
15. Lo Re V III, Haynes K, Forde KA, et al. Risk of acute liver failure in patients with drug-induced liver injury: evaluation of Hy’s law and a new prognostic model. Clin Gastroenterol Hepatol. 2015;13(13):2360-2368.
16. Polson J, Lee WM; American Association for the Study of Liver Diseases. AASLD position paper: the management of acute liver failure. Hepatology. 2005;41:1179-1197.
17. Lee WM, Hynan LS, Rossaro L, et al. Intravenous N-acetylcysteine improves transplant-free survival in early stage non-acetaminophen acute liver failure. Gastroenterology. 2009;137(3):856-864.
18. Rolando N, Harvey F, Brahm J. Prospective study of bacterial infection in acute liver failure: an analysis of fifty patients. Hepatology. 1990;11(1):49-53.
19. Panackel C, Thomas R, Sebastian B, Mathai SK. Recent advances in management of acute liver failure. Indian J Crit Care Med. 2015;19(1):27-33.
20. Temple R. Hy’s law: predicting serious hepatotoxicity. Pharmacoepidemiol Drug Saf. 2006;15(4):241-243.
21. Bunchorntavakul C, Reddy K. Review article: herbal and dietary supplement hepatotoxicity. Aliment Pharmacol Ther. 2012;37(1):3-17.
22. Boudreau DM, Wirtz H, Von Korff M, et al. A survey of adult awareness and use of medicine containing acetaminophen. Pharmacoepidemiol Drug Saf. 2013;22(3):229-240.
23. Burns MJ, Friedman SL, Larson AM. Acetaminophen (paracetamol) poisoning in adults: pathophysiology, presentation, and diagnosis. UpToDate. www.uptodate.com/contents/acetaminophen-paracetamol-poisoning-in-adults-pathophysiology-presentation-and-diagnosis. Accessed May 20, 2016.
24. Lewis JH, Stine JG. Review article: prescribing medications in patients with cirrhosis—a practical guide. Aliment Pharmacol Ther. 2013;37(12):1132-1156.
25. Ashar B, Vargo E. Shark cartilage-induced hepatitis. Ann Intern Med. 1996;125(9):780-781.
IN THIS ARTICLE
- Results of case patient's initial laboratory work-up
- Top 10 prescription medications associated with idiosyncratic disease
- Outcome for the case patient
A 35-year-old African-American woman presented to the emergency department (ED) after being found disoriented and lethargic in her apartment by her friends. Given her altered mental status, the history of present illness was limited and informed mainly by her mother and friends. She had been unreachable by telephone for three days, and friends grew concerned when she was absent from work on two consecutive days. After obtaining access to her apartment, they found her in the bathroom jaundiced, incoherent, and surrounded by nonbloody, nonbilious vomit. She had no prior significant medical history, no documented daily medication, and no recent travel. Of note, previous medical contact was limited, and she did not have an established primary care provider. Additionally, there was no contributory family history, including autoimmune illness or liver disease.
ED presentation was marked by indications of grade 4 encephalopathy, including unresponsiveness to noxious stimuli. Initial laboratory work-up was notable for significantly elevated liver function test results (see Table 1). Based on her international normalized ratio (INR), total bilirubin, and creatinine, her initial Model for End-Stage Liver Disease score was 39, correlating to an 83% three-month mortality rate.1 Autoimmune marker testing revealed a positive antinuclear antibody (ANA), elevated immunoglobulin G (IgG), elevated smooth muscle antibody (IgG), normal antimitochondrial antibody, and normal anti-liver/kidney microsome antibody (IgG). Viral hepatitis serologies, including A, B, C, and E, were unremarkable. Ceruloplasmin and iron saturation were within normal limits. Acetaminophen, salicylate, and ethanol levels were negligible. Pregnancy testing and urine toxin testing were negative. Thyroid function tests were normal. Infectious work-up, including pan-culture, remained negative. Syphilis, herpes simplex virus (HSV), HIV, and varicella zoster testing were unremarkable.
CT of the head was not consistent with cerebral edema. CT of the abdomen and pelvis showed evidence of chronic pancreatitis and trace perihepatic ascites. She was intubated for airway protection and transferred to the medical ICU.
On liver biopsy, the patient was found to have acute hepatitis with centrilobular necrosis, approximately 30% to 40%, and prominent cholestasis. Histologically, these findings were reported as most consistent with drug-induced liver injury. Given her comatose state, coagulopathy, and extremely limited life expectancy without liver transplantation, the patient was listed for transplant as a status 1A candidate with fulminant hepatic failure.
She was placed on propofol and N-acetylcysteine infusions in addition to supportive IV resuscitation. The patient’s synthetic and neurocognitive function improved gradually over several weeks, and she was able to provide collateral history. She denied taking any prescription medications or having any ongoing medical issues. She did report that for two months prior to admission she had been taking an oral beauty supplement designed to enhance hair, skin, and nails. She obtained the supplement online. She could not recall the week leading up to admission, but she did note increasing malaise and fatigue beginning two weeks prior to admission. She denied any recreational drug or alcohol use.
Continue for discussion >>
DISCUSSION
Drug-induced liver injury (DILI) is a relatively uncommon occurrence in the United States.2 It is estimated to occur in approximately 20 individuals per 100,000 persons per year.2 However, DILI incidence secondary to herbal and dietary supplement use appears to be on the rise in the US. In a prospective study conducted by the Drug-Induced Liver Injury Network (DILIN) that included patients with liver injury referred to eight DILIN centers between 2004 and 2013, the proportion of DILI cases caused by herbal and dietary supplements increased from 7% to 20% over the study period.3
DILI can be subclassified into intrinsic and idiosyncratic. Intrinsic DILI results from substances causing a predictable time course and natural history. Substances causing a varied, unpredictable occurrence of DILI in susceptible individuals are idiosyncratic.4 Overall, acetaminophen overdose is the most common cause of DILI.2 However, the most common idiosyncratic offending agents, taken at FDA-approved dosages, are antimicrobials (see Table 2).5 The second most common offending agents are herbal and dietary supplements.5
In a retrospective cohort study evaluating all cases of acute liver failure (ALF) over a six-year period in an integrated health care system, the leading cause of ALF was DILI.6 Of the 32 patients with confirmed drug-induced ALF in this study, the majority of cases (18) were associated with acetaminophen. Herbal and dietary supplements were implicated in six cases, with miscellaneous medications accounting for the remaining eight cases.6 In terms of outcomes, 18.8% of patients with ALF due to DILI underwent liver transplantation, 68.8% were discharged, and 12.5% died during hospitalization.6
DILI disproportionately affects women and minorities7;although the etiology is unclear, it is hypothesized that increased use of antibiotics may play a role among women.2 Providers should be aware of the increased risk for DILI in these populations and consider this diagnosis in the appropriate setting.
Teasing out the diagnosis
DILI is a diagnosis of exclusion, aided in large part by the history and physical exam.4 An extensive history may alert the health care provider to a potential offending substance as well as provide information on timing of exposure.4 DILI should be suspected in patients with persistently elevated liver enzymes, unremarkable work-up for all other underlying liver disease (including autoimmune and viral serologies), and negative abdominal imaging.4 In particular, acute hepatitis C virus (HCV) and hepatitis E virus (HEV) infection mimic the clinical presentation of DILI and should be excluded with HCV RNA and IgM anti-HEV testing, with reflex HEV RNA testing to confirm positive IgM anti-HEV results.8,9 Liver biopsy is rarely indicated for the diagnosis of DILI.2
The presentation of DILI ranges from asymptomatic, with mildly abnormal results on liver function testing, to fulminant hepatic failure. Acetaminophen is the most frequently reported cause of intrinsic DILI in the US, playing a role in approximately half of all ALF cases.10 DILI can be further subdivided according to the pattern of liver test abnormalities as hepatocellular, mixed, or cholestatic based on the ratio of ALT to alkaline phosphatase (R value).2 Utilizing the formula serum ALT/upper limit of normal (ULN) divided by the serum alkaline phosphatase/ULN to determine R value, liver test abnormalities are defined as hepatocellular (R > 5), mixed (R = 2-5), and cholestatic (R < 2).4 These liver test patterns can be used to predict prognosis (see “Prognosis: Hy’s law”). In a prospective, longitudinal study, DILIN found that chronic DILI was present in 18% of the study population at 6 months following onset.5 Patients with the cholestatic presentation were more likely to develop chronic DILI than were those with the hepatocellular or mixed pattern. Furthermore, the hepatocellular pattern on presentation was associated with greater mortality.5 Patients with the mixed pattern had the most favorable outcomes. Another prospective cohort study found that persistently elevated liver enzymes in DILI patients at 12 months is associated with older age and the cholestatic pattern of liver test abnormalities at presentation, in particular, alkaline phosphatase elevation.11 However, neither length of therapy nor type of offending medication was associated with long-term liver test abnormalities.11
Managing DILI and ALF
In all DILI cases, immediate discontinuation of the offending agent is the initial treatment recommendation.2 Patients presenting with DILI who have an accompanying bilirubin level > 2 mg/dL should be referred to a hepatology specialist due to an increased risk for ALF.2 ALF is defined as coagulopathy to INR ≥ 1.5 and hepatic encephalopathy within 26 weeks of initial symptom onset in individuals without known underlying liver disease, with the exception of autoimmune hepatitis, Wilson disease, and reactivation of hepatitis B.12-15 Fulminant hepatic failure is further specified as encephalopathy occurring within 8 weeks of jaundice onset.12
Patients presenting with ALF should be transferred to an intensive care setting, preferably within a liver transplant center, for supportive care and potential liver transplant evaluation.12 CT of the head should be used to rule out other etiologies for altered mental status.16N-Acetylcysteine is the treatment of choice for acetaminophen-induced ALF, and it has also been shown to improve transplant-free survival outcomes in patients with non-acetaminophen–related early ALF.17 Infectious work-up and continuous monitoring are essential in ALF care, since up to 80% of patients with ALF will develop a bacterial infection.18 A comprehensive infectious work-up should include pan-culture of blood, urine, and sputum in addition to assessment for Epstein-Barr virus, cytomegalovirus, and HSV.4,18 For irreversible ALF, liver transplantation remains the only validated treatment option.12,19
Prognosis: Hy’s law
Hy’s law refers to a method used in clinical trials to assess a drug’s likelihood of causing severe hepatotoxicity; it is also used to predict which patients with DILI will develop ALF.12,20 According to Hy’s law, patients with AST or ALT elevations three times ULN and total bilirubin elevations two times ULN are at increased risk for ALF.In a retrospective cohort study of more than 15,000 patients with DILI, the Hy’s law criteria were found to have high specificity but low sensitivity for detecting individuals at risk for ALF.15 An alternative model, the Drug-Induced Liver Toxicity ALF Score, uses platelet count and bilirubin level to identify patients at risk for ALF with high sensitivity.15
Patient education
Effective patient education is essential to decreasing DILI incidence at a time when herbal and dietary supplement consumption is increasing. Patients will often bring herbal and dietary supplements to their providers to obtain a safety profile prior to initiation. In these cases, it is essential to reinforce with patients the absence of federal regulation of these products. It should be stressed to patients that, due to the lack of government oversight, it is impossible to confidently identify the entirety and quantity of ingredients in these supplements. Furthermore, there is no existing protocol for surveillance or adverse event reporting for these products.21 Because these products are not routinely or systematically studied, even health care providers have no evidence on which to base monitoring or usage recommendations. Providers may direct patients to the National Institutes of Health’s LiverTox website (livertox.NIH.gov) to review prior case reports of hepatotoxicity for specific dietary and herbal supplements.
Level of education is associated with knowledge of the potential for overdose when taking OTC medications that contain acetaminophen.22 As a result, health care providers should strongly reinforce with patients the importance of reading all medication labels and abiding by the listed administration directions. In particular, providers should emphasize that the maximum daily dosage of acetaminophen is 4 g.23 For patients with chronic liver disease, a more conservative recommendation is warranted. Generally, patients with cirrhosis may be advised to consume up to 2 g/d of acetaminophen as a firstline treatment for pain. However, providers should ensure acetaminophen ingestion is limited to a brief period.24
Additionally, it is important to educate patients that many combination OTC medications contain acetaminophen. Of note, chronic opioid users are more likely to accurately identify OTC medications containing acetaminophen, compared with acute opioid users.22 These findings should compel health care providers to deliver in-depth education for all patients, particularly those with less education or experience with medications. Education on avoidance of offending medications, including medications within the same class, when appropriate, is essential for quality patient care.2
Continue to outcome for the case patient >>
OUTCOME FOR THE CASE PATIENT
Following discharge, the patient was monitored closely with regular clinic visits and blood work. Her liver test results improved gradually, with consideration of a repeat biopsy to evaluate for overlap or missed autoimmune disease. Her repeat ANA was negative and IgG was within normal limits. Within three months of admission, her liver tests normalized and repeat biopsy was deferred.
Upon review of the herbal beauty supplement the patient reported taking, shark cartilage was noted as a primary ingredient. In a case report, shark cartilage was identified as a hepatotoxin.25 The patient was advised never to ingest the offending supplement, or any other substances not regulated by the FDA, again. Furthermore, the offending medication was listed as a medication allergy in her electronic health record.
CONCLUSION
It is crucial to emphasize to patients the potential hepatotoxicity of medications and herbal and dietary supplements, especially OTC medications that pose an overdose risk. Patients should review all new supplements with their providers prior to therapy initiation. With known hepatotoxins, providers should closely monitor patients for liver injury while treatment is ongoing. In suspected cases of DILI, a thorough history and physical exam will greatly inform the diagnosis. In the majority of cases, the suspect medication should be discontinued immediately, with subsequent assessment of liver response. Identification of DILI early in the course increases the likelihood of full hepatic recovery and improves patient outcomes.
References
1. Kamath PS, Wiesner RH, Malinchoc M, et al. A model to predict survival in patients with end-stage liver disease. Hepatology. 2001;33(2):464-470.
2. Leise MD, Poterucha JJ, Talwalkar JA. Drug-induced liver injury. Mayo Clin Proc. 2014;89(1):95-106.
3. Navarro VJ, Barnhart H, Bonkovsky HL, et al. Liver injury from herbals and dietary supplements in the US Drug-Induced Liver Injury Network. Hepatology. 2014;60(4):1399-1408.
4. Chalasani NP, Hayashi PH, Bonkovsky HL, et al. ACG Clinical Guideline: the diagnosis and management of idiosyncratic drug-induced liver injury. Am J Gastroenterol. 2014;109(7):950-966.
5. Chalasani N, Bonkovsky HL, Fontana R, et al; United States Drug Induced Liver Injury Network. Features and outcomes of 899 patients with drug-induced liver injury: the DILIN prospective study. Gastroenterology. 2015;148(7):1340-1352.
6. Goldberg DS, Forde KA, Carbonari DM, et al. Population-representative incidence of drug-induced acute liver failure based on an analysis of an integrated health care system. Gastroenterology. 2015;148(7):1353-1361.
7. Reuben A, Koch DG, Lee WM. Drug-induced acute liver failure: results of a US multicenter, prospective study. Hepatology. 2010;52(6):2065-2076.
8. Davern TJ, Chalasani N, Fontana RJ, et al; Drug-Induced Liver Injury Network (DILIN). Acute hepatitis E infection accounts for some cases of suspected drug-induced liver injury. Gastroenterology. 2011;141(5):1665-1672.e1-9.
9. Chalasani N, Fontana RJ, Bonkovsky HL, et al. Causes, clinical features, and outcomes from a prospective study of drug-induced liver injury in the United States. Gastroenterology. 2008;135(6):1924-1934.
10. Fisher K, Vuppalanchi R, Saxena R. Drug-induced liver injury. Arch Pathol Lab Med. 2015;139(7):876-887.
11. Fontana RJ, Hayashi PH, Barnhart H, et al. Persistent liver biochemistry abnormalities are more common in older patients and those with cholestatic drug induced liver injury. Am J Gastroenterol. 2015;110(10):1450-1459.
12. Punzalan CS, Barry CT. Acute liver failure: diagnosis and management. J Intensive Care Med. 2015 Oct 6. [Epub ahead of print]
13. Bower WA, Johns M, Margolis HS, et al. Population-based surveillance for acute liver failure. Am J Gastroenterol. 2007;102(11):2459-2463.
14. O’Grady JG, Schalm SW, Williams R. Acute liver failure: redefining the syndromes. Lancet. 1993;342(8866):273-275.
15. Lo Re V III, Haynes K, Forde KA, et al. Risk of acute liver failure in patients with drug-induced liver injury: evaluation of Hy’s law and a new prognostic model. Clin Gastroenterol Hepatol. 2015;13(13):2360-2368.
16. Polson J, Lee WM; American Association for the Study of Liver Diseases. AASLD position paper: the management of acute liver failure. Hepatology. 2005;41:1179-1197.
17. Lee WM, Hynan LS, Rossaro L, et al. Intravenous N-acetylcysteine improves transplant-free survival in early stage non-acetaminophen acute liver failure. Gastroenterology. 2009;137(3):856-864.
18. Rolando N, Harvey F, Brahm J. Prospective study of bacterial infection in acute liver failure: an analysis of fifty patients. Hepatology. 1990;11(1):49-53.
19. Panackel C, Thomas R, Sebastian B, Mathai SK. Recent advances in management of acute liver failure. Indian J Crit Care Med. 2015;19(1):27-33.
20. Temple R. Hy’s law: predicting serious hepatotoxicity. Pharmacoepidemiol Drug Saf. 2006;15(4):241-243.
21. Bunchorntavakul C, Reddy K. Review article: herbal and dietary supplement hepatotoxicity. Aliment Pharmacol Ther. 2012;37(1):3-17.
22. Boudreau DM, Wirtz H, Von Korff M, et al. A survey of adult awareness and use of medicine containing acetaminophen. Pharmacoepidemiol Drug Saf. 2013;22(3):229-240.
23. Burns MJ, Friedman SL, Larson AM. Acetaminophen (paracetamol) poisoning in adults: pathophysiology, presentation, and diagnosis. UpToDate. www.uptodate.com/contents/acetaminophen-paracetamol-poisoning-in-adults-pathophysiology-presentation-and-diagnosis. Accessed May 20, 2016.
24. Lewis JH, Stine JG. Review article: prescribing medications in patients with cirrhosis—a practical guide. Aliment Pharmacol Ther. 2013;37(12):1132-1156.
25. Ashar B, Vargo E. Shark cartilage-induced hepatitis. Ann Intern Med. 1996;125(9):780-781.
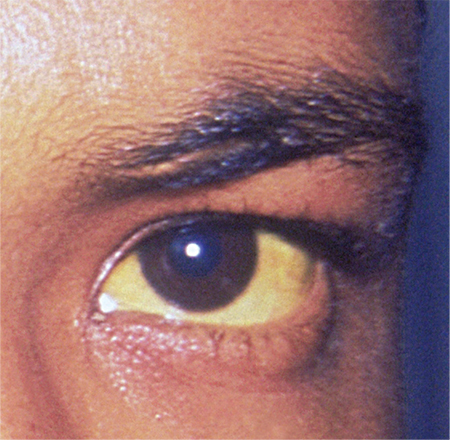
This is not an acute coronary syndrome
A 72-year-old woman presented to the emergency room with persistent substernal chest discomfort. The initial electrocardiogram (ECG) showed 2.0-mm ST-segment elevation in leads II, III, and aVF, and 1.0-mm ST-segment elevation in lead V6 (Figure 1). The index troponin T level was 1.5 ng/mL (reference range < 0.01). ST-elevation myocardial infarction protocols were activated, and she was taken for urgent catheterization.
Coronary angiography showed normal coronary arteries. However, intraprocedural left ventriculography identified circumferential midventricular and apical akinesis with compensatory basal hyperkinesis (Figure 2).
Further inquiry into the patient’s medical history revealed that she had been experiencing psychological distress brought on by the failure of businesses she owned.
Transthoracic echocardiography subsequently verified a depressed ejection fraction (30%) with prominent apical and midventricular wall akinesis, thus confirming the diagnosis of stress cardiomyopathy. She was discharged home on a low-dose beta-blocker and an angiotensin-converting enzyme inhibitor, and within 6 weeks her systolic function had completely returned to normal.
STRESS CARDIOMYOPATHY: CAUSES, DIAGNOSIS, PROGNOSIS
Stress cardiomyopathy—also called broken heart syndrome, stress-induced cardiomyopathy, takotsubo cardiomyopathy, and apical ballooning syndrome—is an increasingly recognized acquired cardiomyopathy that typically affects older postmenopausal women exposed to a triggering stressor such as severe medical illness, major surgery, or a psychologically stressful life event.1,2
Our patient’s acute presentation is a classic example of how stress cardiomyopathy can be indistinguishable from acute coronary syndrome. It has been estimated that 1% to 2% of patients presenting with an initial diagnosis of acute coronary syndrome actually have stress cardiomyopathy.2
Diagnostic criteria
The diagnosis of stress cardiomyopathy can be established when coronary angiography reveals nonobstructive coronary arteries in patients with abnormal ventricular wall motion identified on echocardiography or ventriculography, or both. These findings are part of the proposed diagnostic criteria2:
- Transient hypokinesis, akinesis, or dyskinesis of the left ventricle midsegments, with or without apical involvement; regional wall motion abnormalities extending beyond a single epicardial vascular distribution; usually, a psychological or physiologic stressor is present
- No obstructive coronary disease or no angiographic evidence of acute plaque rupture
- New abnormalities on ECG, or modest elevation in cardiac enzymes
- No evidence of pheochromocytoma or myocarditis.2
Other characteristics that help to differentiate stress cardiomyopathy from acute coronary syndrome include a prolonged QTc interval, attenuation of the QRS amplitude, and a decreased troponin-ejection fraction product.3–5
Prognosis
The prognosis is generally excellent, with most patients achieving full recovery of myocardial function within several weeks.2 However, in the acute setting, there are relatively high rates of acute heart failure (44% to 46%), left ventricular outflow tract obstruction (19%), and unstable ventricular arrhythmias (3.4%), including torsades de pointes.1,2,6,7
Stress cardiomyopathy recurs in approximately 11% of patients within 4 years.8 Death is considered a rare complication but has occurred in as many as 8% of reported cases.1
- Bybee KA, Kara T, Prasad A, et al. Systematic review: transient left ventricular apical ballooning: a syndrome that mimics ST-segment elevation myocardial infarction. Ann Intern Med 2004; 141:858–865.
- Prasad A, Lerman A, Rihal CS. Apical ballooning syndrome (takotsubo or stress cardiomyopathy): a mimic of acute myocardial infarction. Am Heart J 2008; 155:408–417.
- Bennett J, Ferdinande B, Kayaert P, et al. Time course of electrocardiographic changes in transient left ventricular ballooning syndrome. Int J Cardiol 2013; 169:276–280.
- Madias JE. Transient attenuation of the amplitude of the QRS complexes in the diagnosis of takotsubo syndrome. Eur Heart J Acute Cardiovasc Care 2014; 3:28–36.
- Nascimento FO, Yang S, Larrauri-Reyes M, et al. Usefulness of the troponin-ejection fraction product to differentiate stress cardiomyopathy from ST-segment elevation myocardial infarction. Am J Cardiol 2014; 113:429–433.
- De Backer O, Debonnaire P, Gevaert S, Missault L, Gheeraert P, Muyldermans L. Prevalence, associated factors and management implications of left ventricular outflow tract obstruction in takotsubo cardiomyopathy: a two-year, two-center experience. BMC Cardiovasc Disord 2014; 14:147.
- Syed FF, Asirvatham SJ, Francis J. Arrhythmia occurrence with takotsubo cardiomyopathy: a literature review. Europace 2011; 13:780–788.
- Elesber AA, Prasad A, Lennon RJ, Wright RS, Lerman A, Rihal CS. Four-year recurrence rate and prognosis of the apical ballooning syndrome. J Am Coll Cardiol 2007; 50:448–452.
A 72-year-old woman presented to the emergency room with persistent substernal chest discomfort. The initial electrocardiogram (ECG) showed 2.0-mm ST-segment elevation in leads II, III, and aVF, and 1.0-mm ST-segment elevation in lead V6 (Figure 1). The index troponin T level was 1.5 ng/mL (reference range < 0.01). ST-elevation myocardial infarction protocols were activated, and she was taken for urgent catheterization.
Coronary angiography showed normal coronary arteries. However, intraprocedural left ventriculography identified circumferential midventricular and apical akinesis with compensatory basal hyperkinesis (Figure 2).
Further inquiry into the patient’s medical history revealed that she had been experiencing psychological distress brought on by the failure of businesses she owned.
Transthoracic echocardiography subsequently verified a depressed ejection fraction (30%) with prominent apical and midventricular wall akinesis, thus confirming the diagnosis of stress cardiomyopathy. She was discharged home on a low-dose beta-blocker and an angiotensin-converting enzyme inhibitor, and within 6 weeks her systolic function had completely returned to normal.
STRESS CARDIOMYOPATHY: CAUSES, DIAGNOSIS, PROGNOSIS
Stress cardiomyopathy—also called broken heart syndrome, stress-induced cardiomyopathy, takotsubo cardiomyopathy, and apical ballooning syndrome—is an increasingly recognized acquired cardiomyopathy that typically affects older postmenopausal women exposed to a triggering stressor such as severe medical illness, major surgery, or a psychologically stressful life event.1,2
Our patient’s acute presentation is a classic example of how stress cardiomyopathy can be indistinguishable from acute coronary syndrome. It has been estimated that 1% to 2% of patients presenting with an initial diagnosis of acute coronary syndrome actually have stress cardiomyopathy.2
Diagnostic criteria
The diagnosis of stress cardiomyopathy can be established when coronary angiography reveals nonobstructive coronary arteries in patients with abnormal ventricular wall motion identified on echocardiography or ventriculography, or both. These findings are part of the proposed diagnostic criteria2:
- Transient hypokinesis, akinesis, or dyskinesis of the left ventricle midsegments, with or without apical involvement; regional wall motion abnormalities extending beyond a single epicardial vascular distribution; usually, a psychological or physiologic stressor is present
- No obstructive coronary disease or no angiographic evidence of acute plaque rupture
- New abnormalities on ECG, or modest elevation in cardiac enzymes
- No evidence of pheochromocytoma or myocarditis.2
Other characteristics that help to differentiate stress cardiomyopathy from acute coronary syndrome include a prolonged QTc interval, attenuation of the QRS amplitude, and a decreased troponin-ejection fraction product.3–5
Prognosis
The prognosis is generally excellent, with most patients achieving full recovery of myocardial function within several weeks.2 However, in the acute setting, there are relatively high rates of acute heart failure (44% to 46%), left ventricular outflow tract obstruction (19%), and unstable ventricular arrhythmias (3.4%), including torsades de pointes.1,2,6,7
Stress cardiomyopathy recurs in approximately 11% of patients within 4 years.8 Death is considered a rare complication but has occurred in as many as 8% of reported cases.1
A 72-year-old woman presented to the emergency room with persistent substernal chest discomfort. The initial electrocardiogram (ECG) showed 2.0-mm ST-segment elevation in leads II, III, and aVF, and 1.0-mm ST-segment elevation in lead V6 (Figure 1). The index troponin T level was 1.5 ng/mL (reference range < 0.01). ST-elevation myocardial infarction protocols were activated, and she was taken for urgent catheterization.
Coronary angiography showed normal coronary arteries. However, intraprocedural left ventriculography identified circumferential midventricular and apical akinesis with compensatory basal hyperkinesis (Figure 2).
Further inquiry into the patient’s medical history revealed that she had been experiencing psychological distress brought on by the failure of businesses she owned.
Transthoracic echocardiography subsequently verified a depressed ejection fraction (30%) with prominent apical and midventricular wall akinesis, thus confirming the diagnosis of stress cardiomyopathy. She was discharged home on a low-dose beta-blocker and an angiotensin-converting enzyme inhibitor, and within 6 weeks her systolic function had completely returned to normal.
STRESS CARDIOMYOPATHY: CAUSES, DIAGNOSIS, PROGNOSIS
Stress cardiomyopathy—also called broken heart syndrome, stress-induced cardiomyopathy, takotsubo cardiomyopathy, and apical ballooning syndrome—is an increasingly recognized acquired cardiomyopathy that typically affects older postmenopausal women exposed to a triggering stressor such as severe medical illness, major surgery, or a psychologically stressful life event.1,2
Our patient’s acute presentation is a classic example of how stress cardiomyopathy can be indistinguishable from acute coronary syndrome. It has been estimated that 1% to 2% of patients presenting with an initial diagnosis of acute coronary syndrome actually have stress cardiomyopathy.2
Diagnostic criteria
The diagnosis of stress cardiomyopathy can be established when coronary angiography reveals nonobstructive coronary arteries in patients with abnormal ventricular wall motion identified on echocardiography or ventriculography, or both. These findings are part of the proposed diagnostic criteria2:
- Transient hypokinesis, akinesis, or dyskinesis of the left ventricle midsegments, with or without apical involvement; regional wall motion abnormalities extending beyond a single epicardial vascular distribution; usually, a psychological or physiologic stressor is present
- No obstructive coronary disease or no angiographic evidence of acute plaque rupture
- New abnormalities on ECG, or modest elevation in cardiac enzymes
- No evidence of pheochromocytoma or myocarditis.2
Other characteristics that help to differentiate stress cardiomyopathy from acute coronary syndrome include a prolonged QTc interval, attenuation of the QRS amplitude, and a decreased troponin-ejection fraction product.3–5
Prognosis
The prognosis is generally excellent, with most patients achieving full recovery of myocardial function within several weeks.2 However, in the acute setting, there are relatively high rates of acute heart failure (44% to 46%), left ventricular outflow tract obstruction (19%), and unstable ventricular arrhythmias (3.4%), including torsades de pointes.1,2,6,7
Stress cardiomyopathy recurs in approximately 11% of patients within 4 years.8 Death is considered a rare complication but has occurred in as many as 8% of reported cases.1
- Bybee KA, Kara T, Prasad A, et al. Systematic review: transient left ventricular apical ballooning: a syndrome that mimics ST-segment elevation myocardial infarction. Ann Intern Med 2004; 141:858–865.
- Prasad A, Lerman A, Rihal CS. Apical ballooning syndrome (takotsubo or stress cardiomyopathy): a mimic of acute myocardial infarction. Am Heart J 2008; 155:408–417.
- Bennett J, Ferdinande B, Kayaert P, et al. Time course of electrocardiographic changes in transient left ventricular ballooning syndrome. Int J Cardiol 2013; 169:276–280.
- Madias JE. Transient attenuation of the amplitude of the QRS complexes in the diagnosis of takotsubo syndrome. Eur Heart J Acute Cardiovasc Care 2014; 3:28–36.
- Nascimento FO, Yang S, Larrauri-Reyes M, et al. Usefulness of the troponin-ejection fraction product to differentiate stress cardiomyopathy from ST-segment elevation myocardial infarction. Am J Cardiol 2014; 113:429–433.
- De Backer O, Debonnaire P, Gevaert S, Missault L, Gheeraert P, Muyldermans L. Prevalence, associated factors and management implications of left ventricular outflow tract obstruction in takotsubo cardiomyopathy: a two-year, two-center experience. BMC Cardiovasc Disord 2014; 14:147.
- Syed FF, Asirvatham SJ, Francis J. Arrhythmia occurrence with takotsubo cardiomyopathy: a literature review. Europace 2011; 13:780–788.
- Elesber AA, Prasad A, Lennon RJ, Wright RS, Lerman A, Rihal CS. Four-year recurrence rate and prognosis of the apical ballooning syndrome. J Am Coll Cardiol 2007; 50:448–452.
- Bybee KA, Kara T, Prasad A, et al. Systematic review: transient left ventricular apical ballooning: a syndrome that mimics ST-segment elevation myocardial infarction. Ann Intern Med 2004; 141:858–865.
- Prasad A, Lerman A, Rihal CS. Apical ballooning syndrome (takotsubo or stress cardiomyopathy): a mimic of acute myocardial infarction. Am Heart J 2008; 155:408–417.
- Bennett J, Ferdinande B, Kayaert P, et al. Time course of electrocardiographic changes in transient left ventricular ballooning syndrome. Int J Cardiol 2013; 169:276–280.
- Madias JE. Transient attenuation of the amplitude of the QRS complexes in the diagnosis of takotsubo syndrome. Eur Heart J Acute Cardiovasc Care 2014; 3:28–36.
- Nascimento FO, Yang S, Larrauri-Reyes M, et al. Usefulness of the troponin-ejection fraction product to differentiate stress cardiomyopathy from ST-segment elevation myocardial infarction. Am J Cardiol 2014; 113:429–433.
- De Backer O, Debonnaire P, Gevaert S, Missault L, Gheeraert P, Muyldermans L. Prevalence, associated factors and management implications of left ventricular outflow tract obstruction in takotsubo cardiomyopathy: a two-year, two-center experience. BMC Cardiovasc Disord 2014; 14:147.
- Syed FF, Asirvatham SJ, Francis J. Arrhythmia occurrence with takotsubo cardiomyopathy: a literature review. Europace 2011; 13:780–788.
- Elesber AA, Prasad A, Lennon RJ, Wright RS, Lerman A, Rihal CS. Four-year recurrence rate and prognosis of the apical ballooning syndrome. J Am Coll Cardiol 2007; 50:448–452.
An 85-year-old woman with respiratory failure and positional hypoxemia
An 85-year-old woman was brought to our intensive care unit because of worsening hypoxemia over the past day. About 3 weeks earlier she had been diagnosed with acute bilateral pulmonary emboli in the distal branches of the left and right lower lobes and right middle lobe, for which she was receiving anticoagulation therapy.
At presentation she had generalized fatigue and dyspnea at rest that was worse with exertion, but she denied having fever, chest pain, or cough. Her medical history included hypertension, hyperlipidemia, hypothyroidism, stage 1 breast cancer in remission, thromboembolic stroke, and myasthenia gravis. Before her hospital admission, she had been taking rosuvastatin, metoprolol tartrate, pyridostigmine, prednisone, furosemide, levothyroxine, and rivaroxaban. She did not smoke, she was retired, and she had not traveled recently.
Her blood pressure was 135/66 mm Hg, pulse 73 beats per minute, respiratory rate 16, temperature 35.4ºC (95.7ºF), and oxygen saturation 88% while receiving oxygen at 6 L/min via nasal cannula. Physical examination revealed mild edema in the lower extremities and basilar decreased breath sounds. She had no finger clubbing or cyanosis and was not using accessory muscles to breathe. Of note, her oxygen saturation remained more than 93% when she was recumbent but sharply dropped to less than 85% when she was upright.
Laboratory values
Results of initial laboratory testing were as follows:
- Sodium 138 mmol/L (reference range 132–148)
- Potassium 4.2 mmol/L (3.5–5.0)
- Chloride 99 mmol/L (98–111)
- Bicarbonate 29 mmol/L (23–32)
- Creatinine 0.52 mg/dL (0.7–1.4).
- White blood cell count 11.06 × 109/L (3.7–11.0)
- Hemoglobin 12.6 g/dL (12–16)
- Platelet count 211 × 109/L (150–400).
- International normalized ratio 1.4.
Electrocardiography and imaging studies
Standard 12-lead electrocardiography showed normal sinus rhythm with left axis deviation and left ventricular hypertrophy.
Chest radiography showed bilateral interstitial opacities and small pleural effusions.
Computed tomography (CT) of the chest with contrast, compared with a CT scan done 20 days earlier, showed that the pulmonary emboli had resolved.
Arterial blood gases
In view of her positional hypoxemia, blood for arterial blood gas measurements was drawn in the supine and upright positions.
Supine, with a fraction of inspired oxygen (Fio2) via high-flow nasal cannula of 45%, her values were:
- pH 7.45 (reference range 7.35–7.45)
- Pco2 34 mm Hg (36–46)
- Po2 81 mm Hg (85–95)
- Bicarbonate 23 mmol/L (22–26).
Upright, her hypoxemia was significantly worse:
- pH 7.46
- Pco2 33 mm Hg
- Po2 57 mm Hg
- Bicarbonate 23 mmol/L.
The methemoglobin level was normal on both measurements.
During her stay in the intensive care unit, she required up to 100% Fio2 because of persistent hypoxemia.
CAUSES OF HYPOXEMIA
1. So far, the patient’s laboratory tests and imaging studies point to which of the following as the most likely cause of her severe hypoxemia?
- Ventilation-perfusion (V/Q) mismatch
- Diffusion abnormality
- Hypoventilation
- Shunting
- None of the above
The arterial blood gas measurements suggested the possibility of shunting as the cause, although further imaging would be needed to confirm that.
V/Q mismatch can occur in respiratory failure due to pulmonary embolism, pulmonary edema, or shunting. If ventilation is preserved but perfusion is impaired, the V/Q ratio approaches infinity (dead-space ventilation), a situation that can be seen in pulmonary embolism. If perfusion is preserved and ventilation is impaired, the V/Q ratio approaches zero, which is consistent with a physiologic shunt.
Hypoxemia may improve in less severe forms of V/Q mismatch. In our patient, the repeat CT with contrast showed that her pulmonary embolism had resolved, so this is probably not the cause of her severe hypoxemia.
Diffusion abnormalities are due to defects in the lung parenchyma, such as in chronic obstructive pulmonary disease, interstitial lung disease, and lung fibrosis.
Hypoxemia from diffusion defects is usually aggravated by a precipitating factor that increases oxygen demand, and it usually improves with oxygen supplementation. This is unlikely in our patient, as she did not have a history of chronic interstitial lung disease and CT showed no evidence of severe lung parenchymal disease.
Hypoventilation is usually due to drugs that cause respiratory depression, to stroke, or to neuromuscular diseases such as myasthenia gravis that can cause respiratory muscle weakness. It results in elevation of Pco2 and, if not corrected, respiratory acidosis.
Our patient had a diagnosis of myasthenia gravis, though hypoventilation is unlikely in her case because she had a normal respiratory rate and low Pco2 values.
Shunting can be physiologic or anatomic and can occur in the heart or the lungs. In physiologic shunting, severe V/Q mismatch can occur when ventilation is affected, as in severe pulmonary edema and pneumonia. In anatomic shunting, a defect such as an atrial septal defect or a pulmonary arteriovenous malformation allows blood to bypass areas of ventilation from the venous to the arterial circulation, preventing it from being oxygenated. In true anatomic shunting, supplemental oxygen with 100% Fio2 has little effect, whereas in V/Q mismatch it can raise the arterial oxygen saturation.
Our patient’s radiograph did not suggest severe pneumonia or pulmonary edema, which makes these unlikely causes of her hypoxemia. At this point, because of her positional hypoxemia, further evaluation with contrast-enhanced echocardiography was needed to evaluate for anatomic shunting in the heart or lungs.
FURTHER TESTING
Transthoracic echocardiography (TTE) with agitated saline with a Valsalva maneuver was performed. Normally, no bubbles are seen in the left-sided chambers after intravenous injection of agitated saline contrast, whereas bubbles on the left side suggest an intracardiac or intrapulmonary shunt. In our patient, this test was negative, and her right ventricular systolic pressure was normal.
2. What further testing should be considered to evaluate our patient’s hypoxemia?
- High-resolution chest CT
- Transesophageal echocardiography (TEE)
- Pulmonary function testing
- Repeated arterial blood gas measurement
- Edrophonium testing
Repeat imaging with high-resolution CT would likely not provide additional information and would expose the patient to additional radiation without adding much clinical benefit.
TEE could help further evaluate the intracardiac anatomy and look for shunting, which may be missed on TTE because of suboptimal positioning or image quality.
Pulmonary function testing is useful in establishing the baseline function and impairment in respiratory volumes. If an acute myasthenic crisis is suspected, measuring the negative inspiratory force and the forced vital capacity can be useful in monitoring worsening respiratory muscle weakness and assessing the need for mechanical ventilation.
In our patient, it is unlikely that pulmonary function testing would help, since her acute respiratory failure was probably not caused by neuromuscular weakness.
Repeated arterial blood gas measurement would likely only confirm that the patient still has positional hypoxemia but would not help sort through the differential diagnosis.
Edrophonium testing is useful in diagnosing myasthenia gravis and differentiating it from other neuromuscular diseases, such as Lambert-Eaton syndrome. Edrophonium, a reversible acetylcholinesterase inhibitor, prevents degradation of acetylcholine and prolongs its effect at the synaptic cleft, thus improving muscle weakness.
Our patient has already been diagnosed with myasthenia gravis, so this test is not likely to uncover the cause of her hypoxemia.
Because we still strongly suspected a shunt, TEE was performed with intravenous injection of agitated saline. TEE with the patient upright revealed intracardiac right-to-left shunting through a patent foramen ovale. The midesophageal view after saline injection showed a large interatrial septal aneurysm with total excursion of 2 cm, and right-to-left shunting within the first beat, consistent with an intracardiac shunt (Figure 1). Color Doppler imaging (Figure 2) demonstrated turbulent flow through the patent foramen ovale, consistent with right-to-left shunting, and also showed the patent foramen ovale in a closed position (Figure 3).

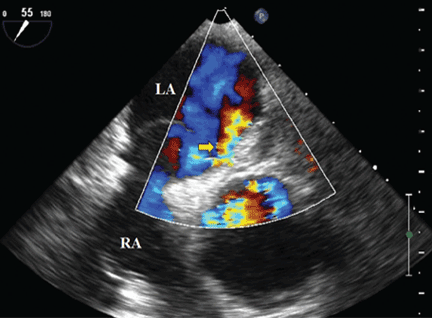
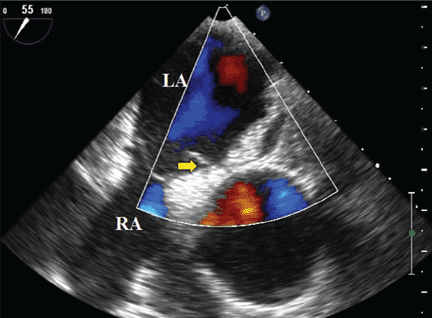
3. Which is now most likely the cause of our patient’s hypoxemia?
- Chronic thromboembolic pulmonary hypertension
- Myasthenic crisis
- Platypnea-orthodeoxia syndrome due to the patent foramen ovale
- Methemoglobinemia
Chronic thromboembolic pulmonary hypertension is usually a long-term result of untreated or inadequately treated thromboembolic disease (eg, pulmonary emboli), which causes vascular remodeling and pulmonary arteriopathy, which in turn leads to increased pulmonary vascular resistance and pulmonary hypertension.
This is unlikely the cause of our patient’s acute hypoxemia, as her symptoms did not suggest it. Moreover, an elevated right ventricular systolic pressure on TTE would suggest pulmonary hypertension, but TTE did not show this, and repeat chest CT indicated that her pulmonary embolism had been adequately treated and had resolved. A V/Q scan and right heart catheterization would help rule out chronic thromboembolic pulmonary hypertension, although these were not done in our patient.
Myasthenic crisis is the progressive fatiguing and paralysis of respiratory muscles ultimately requiring mechanical ventilation to sustain life. It is often brought on by infection or drug therapy.
Our patient did not require intubation and she had no signs or symptoms of myasthenic crisis such as ptosis, dysphagia, or dysarthria. She had a negative inspiratory force of −21 cm H2O, and pulmonary function testing 4 days before her hospital admission had shown a forced vital capacity of 1.84 L, making myasthenic crisis an unlikely cause of her respiratory failure.
Platypnea-orthodeoxia syndrome is a syndrome of dyspnea (platypnea) and hypoxemia (orthodeoxia) that is induced by sitting upright or standing and resolves when lying down. It is a result of right-to-left intracardiac or intrapulmonary shunting in the presence of an anatomic defect and a functional element causing redirection of shunt flow through the anatomic defect in an upright position.1 It is associated with specific cardiac, pulmonary, and hepatic diseases, such as atrial septal defect, pulmonary arteriovenous malformation, and hepatopulmonary syndrome.2 It can occur even if right-sided chamber pressures are normal, and several mechanisms of the underlying pathophysiology have been described.3
Platypnea-orthodeoxia syndrome can be triggered by an event that causes a spontaneous transient elevation of right atrial pressure and pulmonary hypertension, such as our patient’s acute pulmonary embolism. Increased right-to-left shunting occurs in an upright position, causing preferential redirection of flow from the inferior vena cava through the interatrial septum and the patent foramen ovale.4
Our patient was elderly and, like one in every four people in the world, she had had a patent foramen ovale since the day she was born. Never causing a problem, it had remained undiagnosed until complicated by platypnea-orthodeoxia syndrome after her recent pulmonary embolism.
Methemoglobinemia. Methemoglobin has a lower affinity for oxygen than normal hemoglobin. Elevations usually occur with medications such as anesthetics and nitrates and can be diagnosed through an elevated level on arterial blood gas testing.
Our patient did not have elevated methemoglobin on her blood gas measurements on admission; therefore, this is unlikely to be the diagnosis.
CASE CONCLUDED
Percutaneous closure of the patent foramen ovale with a 30-mm Amplatzer Cribriform Occluder brought significant improvement in our patient’s functional status and arterial oxygenation saturation, and 2 weeks later at follow-up she no longer needed supplemental oxygen. TEE 6 months later showed an intact closure device and no interatrial shunting.
WHEN HYPOXEMIA DOES NOT RESPOND TO OXYGEN
In the intensive care unit, time is critical, and when hypoxia is refractory to high Fio2, shunting should be considered.
In the acute-care setting, platypnea-orthodeoxia syndrome can be identified quickly by pulse oximetry and serial blood gas measurements in the upright and supine positions. A drop in arterial oxygenation in the upright position vs the supine position helps confirm the diagnosis.
Other conditions in the differential diagnosis of this syndrome include recurrent pulmonary embolism, acute respiratory distress syndrome, interstitial pulmonary fibrosis, intrapulmonary shunting due to arteriovenous malformation, and diaphragm paralysis due to neuromuscular disease.
In our patient, positional blood gas measurements demonstrated a significant drop in arterial oxygen saturation from the supine to the upright position, raising our suspicion of shunting. It helped narrow the differential diagnosis and guided our selection of additional diagnostic tests.
The initial chest radiograph in our patient was normal. TTE did not reveal shunting and showed a normal right ventricular systolic pressure. TTE with agitated saline also failed to reveal shunting. Because of suboptimal positioning and image quality, TTE may miss the shunting physiology, and that is why we proceeded to positional TEE, which can better evaluate the hemodynamic effects of positional changes on patent foramen ovale and shunting.
MORE ABOUT PATENT FORAMEN OVALE
The prevalence of patent foramen ovale is estimated at 27% in the general population, but it is usually not symptomatic. It can be associated with atrial septal aneurysm and Chiari network malformations. When associated with atrial septal aneurysm, it carries a higher risk of stroke.5
Our patient had a large atrial septal aneurysm with a septal excursion of 2 cm as well as a history of thromboembolic stroke, which was likely associated with the patent foramen ovale and the atrial septal aneurysm.
Atrial septal aneurysm is rare, with a prevalence of 1% at autopsy and 1.9% by TTE. It is defined by a septal excursion of at least 10 mm and a base diameter of at least 15 mm and is more frequently detected on TEE than on TTE.6
Studies have shown that contrast and color Doppler TEE are superior to TTE for detecting patent foramen ovale.7 Tilt-table TEE with contrast enhancement has also been used to better demonstrate the morphology of the interatrial septum and the degree of shunting due to the separation between the septum primum and septum secundum causing the patent foramen ovale.8 Contrast-enhanced transcranial Doppler has also been shown comparable to contrast TEE to detect interatrial shunting. However, TEE provides additional anatomic information.9
In our patient, atrial septal aneurysm and patent foramen ovale were exaggerated by upright positioning, which opened the aneurysm and increased the shunting through the patent foramen ovale.
The treatment of choice in symptomatic patients with platypnea-orthodeoxia syndrome is directed at the underlying cause, in this case closure of the foramen ovale. This treatment has been shown to be safe and effective in these patients,10 but caution should be used when considering foramen ovale closure in patients with pulmonary hypertension.11
In patients with irreversible or severe pulmonary hypertension, closure of the patent foramen ovale can exacerbate right heart dysfunction and lead to right heart failure. There are situations when closure of a patent foramen ovale can be considered in pulmonary hypertension; however, each decision is individualized, and caution must be used. A detailed discussion is beyond the scope of this paper.
A thorough history and physical examination are important in differentiating the various causes of hypoxemia. Appropriate diagnostic testing is needed along with prompt treatment of the underlying cause of platypnea-orthodeoxia syndrome.
- Cheng TO. Mechanisms of platypnea-orthodeoxia: what causes water to flow uphill? Circulation 2002; 105:e47.
- Natalie AA, Nichols L, Bump GM. Platypnea-orthodeoxia, an uncommon presentation of patent foramen ovale. Am J Med Sci 2010; 339:78–80.
- Acharya SS, Kartan R. A case of orthodeoxia caused by an atrial septal aneurysm. Chest 2000; 118:871–874.
- Irwin B, Ray S. Patent foramen ovale—assessment and treatment. Cardiovasc Ther 2012; 30:e128–e135.
- Mas JL, Zuber M. Recurrent cerebrovascular events in patients with patent foramen ovale, atrial septal aneurysm, or both and cryptogenic stroke or transient ischemic attack. French Study Group on Patent Foramen Ovale and Atrial Septal Aneurysm. Am Heart J 1995; 130:1083–1088.
- Kerut EK, Norfleet WT, Plotnick GD, Giles TD. Patent foramen ovale: a review of associated conditions and the impact of physiological size. J Am Coll Cardiol 2001; 38:613–623.
- Hausmann D, Mügge A, Becht I, Daniel WG. Diagnosis of patent foramen ovale by transesophageal echocardiography and association with cerebral and peripheral embolic events. Am J Cardiol 1992; 70:668–672.
- Roxas-Timonera M, Larracas C, Gersony D, Di Tullio M, Keller A, Homma S. Patent foramen ovale presenting as platypnea-orthodeoxia: diagnosis by transesophageal echocardiography. J Am Soc Echocardiogr 2001; 14:1039–1041.
- Sloan MA, Alexandrov AV, Tegeler CH, et al; Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Assessment: transcranial Doppler ultrasonography: report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology 2004; 62:1468–1481.
- Blanche C, Noble S, Roffi M, et al. Platypnea-orthodeoxia syndrome in the elderly treated by percutaneous patent foramen ovale closure: a case series and literature review. Eur J Intern Med 2013; 24:813–817.
- Tobis J, Shenoda M. Percutaneous treatment of patent foramen ovale and atrial septal defects. J Am Coll Cardiol 2012; 60:1722–1732.
An 85-year-old woman was brought to our intensive care unit because of worsening hypoxemia over the past day. About 3 weeks earlier she had been diagnosed with acute bilateral pulmonary emboli in the distal branches of the left and right lower lobes and right middle lobe, for which she was receiving anticoagulation therapy.
At presentation she had generalized fatigue and dyspnea at rest that was worse with exertion, but she denied having fever, chest pain, or cough. Her medical history included hypertension, hyperlipidemia, hypothyroidism, stage 1 breast cancer in remission, thromboembolic stroke, and myasthenia gravis. Before her hospital admission, she had been taking rosuvastatin, metoprolol tartrate, pyridostigmine, prednisone, furosemide, levothyroxine, and rivaroxaban. She did not smoke, she was retired, and she had not traveled recently.
Her blood pressure was 135/66 mm Hg, pulse 73 beats per minute, respiratory rate 16, temperature 35.4ºC (95.7ºF), and oxygen saturation 88% while receiving oxygen at 6 L/min via nasal cannula. Physical examination revealed mild edema in the lower extremities and basilar decreased breath sounds. She had no finger clubbing or cyanosis and was not using accessory muscles to breathe. Of note, her oxygen saturation remained more than 93% when she was recumbent but sharply dropped to less than 85% when she was upright.
Laboratory values
Results of initial laboratory testing were as follows:
- Sodium 138 mmol/L (reference range 132–148)
- Potassium 4.2 mmol/L (3.5–5.0)
- Chloride 99 mmol/L (98–111)
- Bicarbonate 29 mmol/L (23–32)
- Creatinine 0.52 mg/dL (0.7–1.4).
- White blood cell count 11.06 × 109/L (3.7–11.0)
- Hemoglobin 12.6 g/dL (12–16)
- Platelet count 211 × 109/L (150–400).
- International normalized ratio 1.4.
Electrocardiography and imaging studies
Standard 12-lead electrocardiography showed normal sinus rhythm with left axis deviation and left ventricular hypertrophy.
Chest radiography showed bilateral interstitial opacities and small pleural effusions.
Computed tomography (CT) of the chest with contrast, compared with a CT scan done 20 days earlier, showed that the pulmonary emboli had resolved.
Arterial blood gases
In view of her positional hypoxemia, blood for arterial blood gas measurements was drawn in the supine and upright positions.
Supine, with a fraction of inspired oxygen (Fio2) via high-flow nasal cannula of 45%, her values were:
- pH 7.45 (reference range 7.35–7.45)
- Pco2 34 mm Hg (36–46)
- Po2 81 mm Hg (85–95)
- Bicarbonate 23 mmol/L (22–26).
Upright, her hypoxemia was significantly worse:
- pH 7.46
- Pco2 33 mm Hg
- Po2 57 mm Hg
- Bicarbonate 23 mmol/L.
The methemoglobin level was normal on both measurements.
During her stay in the intensive care unit, she required up to 100% Fio2 because of persistent hypoxemia.
CAUSES OF HYPOXEMIA
1. So far, the patient’s laboratory tests and imaging studies point to which of the following as the most likely cause of her severe hypoxemia?
- Ventilation-perfusion (V/Q) mismatch
- Diffusion abnormality
- Hypoventilation
- Shunting
- None of the above
The arterial blood gas measurements suggested the possibility of shunting as the cause, although further imaging would be needed to confirm that.
V/Q mismatch can occur in respiratory failure due to pulmonary embolism, pulmonary edema, or shunting. If ventilation is preserved but perfusion is impaired, the V/Q ratio approaches infinity (dead-space ventilation), a situation that can be seen in pulmonary embolism. If perfusion is preserved and ventilation is impaired, the V/Q ratio approaches zero, which is consistent with a physiologic shunt.
Hypoxemia may improve in less severe forms of V/Q mismatch. In our patient, the repeat CT with contrast showed that her pulmonary embolism had resolved, so this is probably not the cause of her severe hypoxemia.
Diffusion abnormalities are due to defects in the lung parenchyma, such as in chronic obstructive pulmonary disease, interstitial lung disease, and lung fibrosis.
Hypoxemia from diffusion defects is usually aggravated by a precipitating factor that increases oxygen demand, and it usually improves with oxygen supplementation. This is unlikely in our patient, as she did not have a history of chronic interstitial lung disease and CT showed no evidence of severe lung parenchymal disease.
Hypoventilation is usually due to drugs that cause respiratory depression, to stroke, or to neuromuscular diseases such as myasthenia gravis that can cause respiratory muscle weakness. It results in elevation of Pco2 and, if not corrected, respiratory acidosis.
Our patient had a diagnosis of myasthenia gravis, though hypoventilation is unlikely in her case because she had a normal respiratory rate and low Pco2 values.
Shunting can be physiologic or anatomic and can occur in the heart or the lungs. In physiologic shunting, severe V/Q mismatch can occur when ventilation is affected, as in severe pulmonary edema and pneumonia. In anatomic shunting, a defect such as an atrial septal defect or a pulmonary arteriovenous malformation allows blood to bypass areas of ventilation from the venous to the arterial circulation, preventing it from being oxygenated. In true anatomic shunting, supplemental oxygen with 100% Fio2 has little effect, whereas in V/Q mismatch it can raise the arterial oxygen saturation.
Our patient’s radiograph did not suggest severe pneumonia or pulmonary edema, which makes these unlikely causes of her hypoxemia. At this point, because of her positional hypoxemia, further evaluation with contrast-enhanced echocardiography was needed to evaluate for anatomic shunting in the heart or lungs.
FURTHER TESTING
Transthoracic echocardiography (TTE) with agitated saline with a Valsalva maneuver was performed. Normally, no bubbles are seen in the left-sided chambers after intravenous injection of agitated saline contrast, whereas bubbles on the left side suggest an intracardiac or intrapulmonary shunt. In our patient, this test was negative, and her right ventricular systolic pressure was normal.
2. What further testing should be considered to evaluate our patient’s hypoxemia?
- High-resolution chest CT
- Transesophageal echocardiography (TEE)
- Pulmonary function testing
- Repeated arterial blood gas measurement
- Edrophonium testing
Repeat imaging with high-resolution CT would likely not provide additional information and would expose the patient to additional radiation without adding much clinical benefit.
TEE could help further evaluate the intracardiac anatomy and look for shunting, which may be missed on TTE because of suboptimal positioning or image quality.
Pulmonary function testing is useful in establishing the baseline function and impairment in respiratory volumes. If an acute myasthenic crisis is suspected, measuring the negative inspiratory force and the forced vital capacity can be useful in monitoring worsening respiratory muscle weakness and assessing the need for mechanical ventilation.
In our patient, it is unlikely that pulmonary function testing would help, since her acute respiratory failure was probably not caused by neuromuscular weakness.
Repeated arterial blood gas measurement would likely only confirm that the patient still has positional hypoxemia but would not help sort through the differential diagnosis.
Edrophonium testing is useful in diagnosing myasthenia gravis and differentiating it from other neuromuscular diseases, such as Lambert-Eaton syndrome. Edrophonium, a reversible acetylcholinesterase inhibitor, prevents degradation of acetylcholine and prolongs its effect at the synaptic cleft, thus improving muscle weakness.
Our patient has already been diagnosed with myasthenia gravis, so this test is not likely to uncover the cause of her hypoxemia.
Because we still strongly suspected a shunt, TEE was performed with intravenous injection of agitated saline. TEE with the patient upright revealed intracardiac right-to-left shunting through a patent foramen ovale. The midesophageal view after saline injection showed a large interatrial septal aneurysm with total excursion of 2 cm, and right-to-left shunting within the first beat, consistent with an intracardiac shunt (Figure 1). Color Doppler imaging (Figure 2) demonstrated turbulent flow through the patent foramen ovale, consistent with right-to-left shunting, and also showed the patent foramen ovale in a closed position (Figure 3).
3. Which is now most likely the cause of our patient’s hypoxemia?
- Chronic thromboembolic pulmonary hypertension
- Myasthenic crisis
- Platypnea-orthodeoxia syndrome due to the patent foramen ovale
- Methemoglobinemia
Chronic thromboembolic pulmonary hypertension is usually a long-term result of untreated or inadequately treated thromboembolic disease (eg, pulmonary emboli), which causes vascular remodeling and pulmonary arteriopathy, which in turn leads to increased pulmonary vascular resistance and pulmonary hypertension.
This is unlikely the cause of our patient’s acute hypoxemia, as her symptoms did not suggest it. Moreover, an elevated right ventricular systolic pressure on TTE would suggest pulmonary hypertension, but TTE did not show this, and repeat chest CT indicated that her pulmonary embolism had been adequately treated and had resolved. A V/Q scan and right heart catheterization would help rule out chronic thromboembolic pulmonary hypertension, although these were not done in our patient.
Myasthenic crisis is the progressive fatiguing and paralysis of respiratory muscles ultimately requiring mechanical ventilation to sustain life. It is often brought on by infection or drug therapy.
Our patient did not require intubation and she had no signs or symptoms of myasthenic crisis such as ptosis, dysphagia, or dysarthria. She had a negative inspiratory force of −21 cm H2O, and pulmonary function testing 4 days before her hospital admission had shown a forced vital capacity of 1.84 L, making myasthenic crisis an unlikely cause of her respiratory failure.
Platypnea-orthodeoxia syndrome is a syndrome of dyspnea (platypnea) and hypoxemia (orthodeoxia) that is induced by sitting upright or standing and resolves when lying down. It is a result of right-to-left intracardiac or intrapulmonary shunting in the presence of an anatomic defect and a functional element causing redirection of shunt flow through the anatomic defect in an upright position.1 It is associated with specific cardiac, pulmonary, and hepatic diseases, such as atrial septal defect, pulmonary arteriovenous malformation, and hepatopulmonary syndrome.2 It can occur even if right-sided chamber pressures are normal, and several mechanisms of the underlying pathophysiology have been described.3
Platypnea-orthodeoxia syndrome can be triggered by an event that causes a spontaneous transient elevation of right atrial pressure and pulmonary hypertension, such as our patient’s acute pulmonary embolism. Increased right-to-left shunting occurs in an upright position, causing preferential redirection of flow from the inferior vena cava through the interatrial septum and the patent foramen ovale.4
Our patient was elderly and, like one in every four people in the world, she had had a patent foramen ovale since the day she was born. Never causing a problem, it had remained undiagnosed until complicated by platypnea-orthodeoxia syndrome after her recent pulmonary embolism.
Methemoglobinemia. Methemoglobin has a lower affinity for oxygen than normal hemoglobin. Elevations usually occur with medications such as anesthetics and nitrates and can be diagnosed through an elevated level on arterial blood gas testing.
Our patient did not have elevated methemoglobin on her blood gas measurements on admission; therefore, this is unlikely to be the diagnosis.
CASE CONCLUDED
Percutaneous closure of the patent foramen ovale with a 30-mm Amplatzer Cribriform Occluder brought significant improvement in our patient’s functional status and arterial oxygenation saturation, and 2 weeks later at follow-up she no longer needed supplemental oxygen. TEE 6 months later showed an intact closure device and no interatrial shunting.
WHEN HYPOXEMIA DOES NOT RESPOND TO OXYGEN
In the intensive care unit, time is critical, and when hypoxia is refractory to high Fio2, shunting should be considered.
In the acute-care setting, platypnea-orthodeoxia syndrome can be identified quickly by pulse oximetry and serial blood gas measurements in the upright and supine positions. A drop in arterial oxygenation in the upright position vs the supine position helps confirm the diagnosis.
Other conditions in the differential diagnosis of this syndrome include recurrent pulmonary embolism, acute respiratory distress syndrome, interstitial pulmonary fibrosis, intrapulmonary shunting due to arteriovenous malformation, and diaphragm paralysis due to neuromuscular disease.
In our patient, positional blood gas measurements demonstrated a significant drop in arterial oxygen saturation from the supine to the upright position, raising our suspicion of shunting. It helped narrow the differential diagnosis and guided our selection of additional diagnostic tests.
The initial chest radiograph in our patient was normal. TTE did not reveal shunting and showed a normal right ventricular systolic pressure. TTE with agitated saline also failed to reveal shunting. Because of suboptimal positioning and image quality, TTE may miss the shunting physiology, and that is why we proceeded to positional TEE, which can better evaluate the hemodynamic effects of positional changes on patent foramen ovale and shunting.
MORE ABOUT PATENT FORAMEN OVALE
The prevalence of patent foramen ovale is estimated at 27% in the general population, but it is usually not symptomatic. It can be associated with atrial septal aneurysm and Chiari network malformations. When associated with atrial septal aneurysm, it carries a higher risk of stroke.5
Our patient had a large atrial septal aneurysm with a septal excursion of 2 cm as well as a history of thromboembolic stroke, which was likely associated with the patent foramen ovale and the atrial septal aneurysm.
Atrial septal aneurysm is rare, with a prevalence of 1% at autopsy and 1.9% by TTE. It is defined by a septal excursion of at least 10 mm and a base diameter of at least 15 mm and is more frequently detected on TEE than on TTE.6
Studies have shown that contrast and color Doppler TEE are superior to TTE for detecting patent foramen ovale.7 Tilt-table TEE with contrast enhancement has also been used to better demonstrate the morphology of the interatrial septum and the degree of shunting due to the separation between the septum primum and septum secundum causing the patent foramen ovale.8 Contrast-enhanced transcranial Doppler has also been shown comparable to contrast TEE to detect interatrial shunting. However, TEE provides additional anatomic information.9
In our patient, atrial septal aneurysm and patent foramen ovale were exaggerated by upright positioning, which opened the aneurysm and increased the shunting through the patent foramen ovale.
The treatment of choice in symptomatic patients with platypnea-orthodeoxia syndrome is directed at the underlying cause, in this case closure of the foramen ovale. This treatment has been shown to be safe and effective in these patients,10 but caution should be used when considering foramen ovale closure in patients with pulmonary hypertension.11
In patients with irreversible or severe pulmonary hypertension, closure of the patent foramen ovale can exacerbate right heart dysfunction and lead to right heart failure. There are situations when closure of a patent foramen ovale can be considered in pulmonary hypertension; however, each decision is individualized, and caution must be used. A detailed discussion is beyond the scope of this paper.
A thorough history and physical examination are important in differentiating the various causes of hypoxemia. Appropriate diagnostic testing is needed along with prompt treatment of the underlying cause of platypnea-orthodeoxia syndrome.
An 85-year-old woman was brought to our intensive care unit because of worsening hypoxemia over the past day. About 3 weeks earlier she had been diagnosed with acute bilateral pulmonary emboli in the distal branches of the left and right lower lobes and right middle lobe, for which she was receiving anticoagulation therapy.
At presentation she had generalized fatigue and dyspnea at rest that was worse with exertion, but she denied having fever, chest pain, or cough. Her medical history included hypertension, hyperlipidemia, hypothyroidism, stage 1 breast cancer in remission, thromboembolic stroke, and myasthenia gravis. Before her hospital admission, she had been taking rosuvastatin, metoprolol tartrate, pyridostigmine, prednisone, furosemide, levothyroxine, and rivaroxaban. She did not smoke, she was retired, and she had not traveled recently.
Her blood pressure was 135/66 mm Hg, pulse 73 beats per minute, respiratory rate 16, temperature 35.4ºC (95.7ºF), and oxygen saturation 88% while receiving oxygen at 6 L/min via nasal cannula. Physical examination revealed mild edema in the lower extremities and basilar decreased breath sounds. She had no finger clubbing or cyanosis and was not using accessory muscles to breathe. Of note, her oxygen saturation remained more than 93% when she was recumbent but sharply dropped to less than 85% when she was upright.
Laboratory values
Results of initial laboratory testing were as follows:
- Sodium 138 mmol/L (reference range 132–148)
- Potassium 4.2 mmol/L (3.5–5.0)
- Chloride 99 mmol/L (98–111)
- Bicarbonate 29 mmol/L (23–32)
- Creatinine 0.52 mg/dL (0.7–1.4).
- White blood cell count 11.06 × 109/L (3.7–11.0)
- Hemoglobin 12.6 g/dL (12–16)
- Platelet count 211 × 109/L (150–400).
- International normalized ratio 1.4.
Electrocardiography and imaging studies
Standard 12-lead electrocardiography showed normal sinus rhythm with left axis deviation and left ventricular hypertrophy.
Chest radiography showed bilateral interstitial opacities and small pleural effusions.
Computed tomography (CT) of the chest with contrast, compared with a CT scan done 20 days earlier, showed that the pulmonary emboli had resolved.
Arterial blood gases
In view of her positional hypoxemia, blood for arterial blood gas measurements was drawn in the supine and upright positions.
Supine, with a fraction of inspired oxygen (Fio2) via high-flow nasal cannula of 45%, her values were:
- pH 7.45 (reference range 7.35–7.45)
- Pco2 34 mm Hg (36–46)
- Po2 81 mm Hg (85–95)
- Bicarbonate 23 mmol/L (22–26).
Upright, her hypoxemia was significantly worse:
- pH 7.46
- Pco2 33 mm Hg
- Po2 57 mm Hg
- Bicarbonate 23 mmol/L.
The methemoglobin level was normal on both measurements.
During her stay in the intensive care unit, she required up to 100% Fio2 because of persistent hypoxemia.
CAUSES OF HYPOXEMIA
1. So far, the patient’s laboratory tests and imaging studies point to which of the following as the most likely cause of her severe hypoxemia?
- Ventilation-perfusion (V/Q) mismatch
- Diffusion abnormality
- Hypoventilation
- Shunting
- None of the above
The arterial blood gas measurements suggested the possibility of shunting as the cause, although further imaging would be needed to confirm that.
V/Q mismatch can occur in respiratory failure due to pulmonary embolism, pulmonary edema, or shunting. If ventilation is preserved but perfusion is impaired, the V/Q ratio approaches infinity (dead-space ventilation), a situation that can be seen in pulmonary embolism. If perfusion is preserved and ventilation is impaired, the V/Q ratio approaches zero, which is consistent with a physiologic shunt.
Hypoxemia may improve in less severe forms of V/Q mismatch. In our patient, the repeat CT with contrast showed that her pulmonary embolism had resolved, so this is probably not the cause of her severe hypoxemia.
Diffusion abnormalities are due to defects in the lung parenchyma, such as in chronic obstructive pulmonary disease, interstitial lung disease, and lung fibrosis.
Hypoxemia from diffusion defects is usually aggravated by a precipitating factor that increases oxygen demand, and it usually improves with oxygen supplementation. This is unlikely in our patient, as she did not have a history of chronic interstitial lung disease and CT showed no evidence of severe lung parenchymal disease.
Hypoventilation is usually due to drugs that cause respiratory depression, to stroke, or to neuromuscular diseases such as myasthenia gravis that can cause respiratory muscle weakness. It results in elevation of Pco2 and, if not corrected, respiratory acidosis.
Our patient had a diagnosis of myasthenia gravis, though hypoventilation is unlikely in her case because she had a normal respiratory rate and low Pco2 values.
Shunting can be physiologic or anatomic and can occur in the heart or the lungs. In physiologic shunting, severe V/Q mismatch can occur when ventilation is affected, as in severe pulmonary edema and pneumonia. In anatomic shunting, a defect such as an atrial septal defect or a pulmonary arteriovenous malformation allows blood to bypass areas of ventilation from the venous to the arterial circulation, preventing it from being oxygenated. In true anatomic shunting, supplemental oxygen with 100% Fio2 has little effect, whereas in V/Q mismatch it can raise the arterial oxygen saturation.
Our patient’s radiograph did not suggest severe pneumonia or pulmonary edema, which makes these unlikely causes of her hypoxemia. At this point, because of her positional hypoxemia, further evaluation with contrast-enhanced echocardiography was needed to evaluate for anatomic shunting in the heart or lungs.
FURTHER TESTING
Transthoracic echocardiography (TTE) with agitated saline with a Valsalva maneuver was performed. Normally, no bubbles are seen in the left-sided chambers after intravenous injection of agitated saline contrast, whereas bubbles on the left side suggest an intracardiac or intrapulmonary shunt. In our patient, this test was negative, and her right ventricular systolic pressure was normal.
2. What further testing should be considered to evaluate our patient’s hypoxemia?
- High-resolution chest CT
- Transesophageal echocardiography (TEE)
- Pulmonary function testing
- Repeated arterial blood gas measurement
- Edrophonium testing
Repeat imaging with high-resolution CT would likely not provide additional information and would expose the patient to additional radiation without adding much clinical benefit.
TEE could help further evaluate the intracardiac anatomy and look for shunting, which may be missed on TTE because of suboptimal positioning or image quality.
Pulmonary function testing is useful in establishing the baseline function and impairment in respiratory volumes. If an acute myasthenic crisis is suspected, measuring the negative inspiratory force and the forced vital capacity can be useful in monitoring worsening respiratory muscle weakness and assessing the need for mechanical ventilation.
In our patient, it is unlikely that pulmonary function testing would help, since her acute respiratory failure was probably not caused by neuromuscular weakness.
Repeated arterial blood gas measurement would likely only confirm that the patient still has positional hypoxemia but would not help sort through the differential diagnosis.
Edrophonium testing is useful in diagnosing myasthenia gravis and differentiating it from other neuromuscular diseases, such as Lambert-Eaton syndrome. Edrophonium, a reversible acetylcholinesterase inhibitor, prevents degradation of acetylcholine and prolongs its effect at the synaptic cleft, thus improving muscle weakness.
Our patient has already been diagnosed with myasthenia gravis, so this test is not likely to uncover the cause of her hypoxemia.
Because we still strongly suspected a shunt, TEE was performed with intravenous injection of agitated saline. TEE with the patient upright revealed intracardiac right-to-left shunting through a patent foramen ovale. The midesophageal view after saline injection showed a large interatrial septal aneurysm with total excursion of 2 cm, and right-to-left shunting within the first beat, consistent with an intracardiac shunt (Figure 1). Color Doppler imaging (Figure 2) demonstrated turbulent flow through the patent foramen ovale, consistent with right-to-left shunting, and also showed the patent foramen ovale in a closed position (Figure 3).
3. Which is now most likely the cause of our patient’s hypoxemia?
- Chronic thromboembolic pulmonary hypertension
- Myasthenic crisis
- Platypnea-orthodeoxia syndrome due to the patent foramen ovale
- Methemoglobinemia
Chronic thromboembolic pulmonary hypertension is usually a long-term result of untreated or inadequately treated thromboembolic disease (eg, pulmonary emboli), which causes vascular remodeling and pulmonary arteriopathy, which in turn leads to increased pulmonary vascular resistance and pulmonary hypertension.
This is unlikely the cause of our patient’s acute hypoxemia, as her symptoms did not suggest it. Moreover, an elevated right ventricular systolic pressure on TTE would suggest pulmonary hypertension, but TTE did not show this, and repeat chest CT indicated that her pulmonary embolism had been adequately treated and had resolved. A V/Q scan and right heart catheterization would help rule out chronic thromboembolic pulmonary hypertension, although these were not done in our patient.
Myasthenic crisis is the progressive fatiguing and paralysis of respiratory muscles ultimately requiring mechanical ventilation to sustain life. It is often brought on by infection or drug therapy.
Our patient did not require intubation and she had no signs or symptoms of myasthenic crisis such as ptosis, dysphagia, or dysarthria. She had a negative inspiratory force of −21 cm H2O, and pulmonary function testing 4 days before her hospital admission had shown a forced vital capacity of 1.84 L, making myasthenic crisis an unlikely cause of her respiratory failure.
Platypnea-orthodeoxia syndrome is a syndrome of dyspnea (platypnea) and hypoxemia (orthodeoxia) that is induced by sitting upright or standing and resolves when lying down. It is a result of right-to-left intracardiac or intrapulmonary shunting in the presence of an anatomic defect and a functional element causing redirection of shunt flow through the anatomic defect in an upright position.1 It is associated with specific cardiac, pulmonary, and hepatic diseases, such as atrial septal defect, pulmonary arteriovenous malformation, and hepatopulmonary syndrome.2 It can occur even if right-sided chamber pressures are normal, and several mechanisms of the underlying pathophysiology have been described.3
Platypnea-orthodeoxia syndrome can be triggered by an event that causes a spontaneous transient elevation of right atrial pressure and pulmonary hypertension, such as our patient’s acute pulmonary embolism. Increased right-to-left shunting occurs in an upright position, causing preferential redirection of flow from the inferior vena cava through the interatrial septum and the patent foramen ovale.4
Our patient was elderly and, like one in every four people in the world, she had had a patent foramen ovale since the day she was born. Never causing a problem, it had remained undiagnosed until complicated by platypnea-orthodeoxia syndrome after her recent pulmonary embolism.
Methemoglobinemia. Methemoglobin has a lower affinity for oxygen than normal hemoglobin. Elevations usually occur with medications such as anesthetics and nitrates and can be diagnosed through an elevated level on arterial blood gas testing.
Our patient did not have elevated methemoglobin on her blood gas measurements on admission; therefore, this is unlikely to be the diagnosis.
CASE CONCLUDED
Percutaneous closure of the patent foramen ovale with a 30-mm Amplatzer Cribriform Occluder brought significant improvement in our patient’s functional status and arterial oxygenation saturation, and 2 weeks later at follow-up she no longer needed supplemental oxygen. TEE 6 months later showed an intact closure device and no interatrial shunting.
WHEN HYPOXEMIA DOES NOT RESPOND TO OXYGEN
In the intensive care unit, time is critical, and when hypoxia is refractory to high Fio2, shunting should be considered.
In the acute-care setting, platypnea-orthodeoxia syndrome can be identified quickly by pulse oximetry and serial blood gas measurements in the upright and supine positions. A drop in arterial oxygenation in the upright position vs the supine position helps confirm the diagnosis.
Other conditions in the differential diagnosis of this syndrome include recurrent pulmonary embolism, acute respiratory distress syndrome, interstitial pulmonary fibrosis, intrapulmonary shunting due to arteriovenous malformation, and diaphragm paralysis due to neuromuscular disease.
In our patient, positional blood gas measurements demonstrated a significant drop in arterial oxygen saturation from the supine to the upright position, raising our suspicion of shunting. It helped narrow the differential diagnosis and guided our selection of additional diagnostic tests.
The initial chest radiograph in our patient was normal. TTE did not reveal shunting and showed a normal right ventricular systolic pressure. TTE with agitated saline also failed to reveal shunting. Because of suboptimal positioning and image quality, TTE may miss the shunting physiology, and that is why we proceeded to positional TEE, which can better evaluate the hemodynamic effects of positional changes on patent foramen ovale and shunting.
MORE ABOUT PATENT FORAMEN OVALE
The prevalence of patent foramen ovale is estimated at 27% in the general population, but it is usually not symptomatic. It can be associated with atrial septal aneurysm and Chiari network malformations. When associated with atrial septal aneurysm, it carries a higher risk of stroke.5
Our patient had a large atrial septal aneurysm with a septal excursion of 2 cm as well as a history of thromboembolic stroke, which was likely associated with the patent foramen ovale and the atrial septal aneurysm.
Atrial septal aneurysm is rare, with a prevalence of 1% at autopsy and 1.9% by TTE. It is defined by a septal excursion of at least 10 mm and a base diameter of at least 15 mm and is more frequently detected on TEE than on TTE.6
Studies have shown that contrast and color Doppler TEE are superior to TTE for detecting patent foramen ovale.7 Tilt-table TEE with contrast enhancement has also been used to better demonstrate the morphology of the interatrial septum and the degree of shunting due to the separation between the septum primum and septum secundum causing the patent foramen ovale.8 Contrast-enhanced transcranial Doppler has also been shown comparable to contrast TEE to detect interatrial shunting. However, TEE provides additional anatomic information.9
In our patient, atrial septal aneurysm and patent foramen ovale were exaggerated by upright positioning, which opened the aneurysm and increased the shunting through the patent foramen ovale.
The treatment of choice in symptomatic patients with platypnea-orthodeoxia syndrome is directed at the underlying cause, in this case closure of the foramen ovale. This treatment has been shown to be safe and effective in these patients,10 but caution should be used when considering foramen ovale closure in patients with pulmonary hypertension.11
In patients with irreversible or severe pulmonary hypertension, closure of the patent foramen ovale can exacerbate right heart dysfunction and lead to right heart failure. There are situations when closure of a patent foramen ovale can be considered in pulmonary hypertension; however, each decision is individualized, and caution must be used. A detailed discussion is beyond the scope of this paper.
A thorough history and physical examination are important in differentiating the various causes of hypoxemia. Appropriate diagnostic testing is needed along with prompt treatment of the underlying cause of platypnea-orthodeoxia syndrome.
- Cheng TO. Mechanisms of platypnea-orthodeoxia: what causes water to flow uphill? Circulation 2002; 105:e47.
- Natalie AA, Nichols L, Bump GM. Platypnea-orthodeoxia, an uncommon presentation of patent foramen ovale. Am J Med Sci 2010; 339:78–80.
- Acharya SS, Kartan R. A case of orthodeoxia caused by an atrial septal aneurysm. Chest 2000; 118:871–874.
- Irwin B, Ray S. Patent foramen ovale—assessment and treatment. Cardiovasc Ther 2012; 30:e128–e135.
- Mas JL, Zuber M. Recurrent cerebrovascular events in patients with patent foramen ovale, atrial septal aneurysm, or both and cryptogenic stroke or transient ischemic attack. French Study Group on Patent Foramen Ovale and Atrial Septal Aneurysm. Am Heart J 1995; 130:1083–1088.
- Kerut EK, Norfleet WT, Plotnick GD, Giles TD. Patent foramen ovale: a review of associated conditions and the impact of physiological size. J Am Coll Cardiol 2001; 38:613–623.
- Hausmann D, Mügge A, Becht I, Daniel WG. Diagnosis of patent foramen ovale by transesophageal echocardiography and association with cerebral and peripheral embolic events. Am J Cardiol 1992; 70:668–672.
- Roxas-Timonera M, Larracas C, Gersony D, Di Tullio M, Keller A, Homma S. Patent foramen ovale presenting as platypnea-orthodeoxia: diagnosis by transesophageal echocardiography. J Am Soc Echocardiogr 2001; 14:1039–1041.
- Sloan MA, Alexandrov AV, Tegeler CH, et al; Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Assessment: transcranial Doppler ultrasonography: report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology 2004; 62:1468–1481.
- Blanche C, Noble S, Roffi M, et al. Platypnea-orthodeoxia syndrome in the elderly treated by percutaneous patent foramen ovale closure: a case series and literature review. Eur J Intern Med 2013; 24:813–817.
- Tobis J, Shenoda M. Percutaneous treatment of patent foramen ovale and atrial septal defects. J Am Coll Cardiol 2012; 60:1722–1732.
- Cheng TO. Mechanisms of platypnea-orthodeoxia: what causes water to flow uphill? Circulation 2002; 105:e47.
- Natalie AA, Nichols L, Bump GM. Platypnea-orthodeoxia, an uncommon presentation of patent foramen ovale. Am J Med Sci 2010; 339:78–80.
- Acharya SS, Kartan R. A case of orthodeoxia caused by an atrial septal aneurysm. Chest 2000; 118:871–874.
- Irwin B, Ray S. Patent foramen ovale—assessment and treatment. Cardiovasc Ther 2012; 30:e128–e135.
- Mas JL, Zuber M. Recurrent cerebrovascular events in patients with patent foramen ovale, atrial septal aneurysm, or both and cryptogenic stroke or transient ischemic attack. French Study Group on Patent Foramen Ovale and Atrial Septal Aneurysm. Am Heart J 1995; 130:1083–1088.
- Kerut EK, Norfleet WT, Plotnick GD, Giles TD. Patent foramen ovale: a review of associated conditions and the impact of physiological size. J Am Coll Cardiol 2001; 38:613–623.
- Hausmann D, Mügge A, Becht I, Daniel WG. Diagnosis of patent foramen ovale by transesophageal echocardiography and association with cerebral and peripheral embolic events. Am J Cardiol 1992; 70:668–672.
- Roxas-Timonera M, Larracas C, Gersony D, Di Tullio M, Keller A, Homma S. Patent foramen ovale presenting as platypnea-orthodeoxia: diagnosis by transesophageal echocardiography. J Am Soc Echocardiogr 2001; 14:1039–1041.
- Sloan MA, Alexandrov AV, Tegeler CH, et al; Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Assessment: transcranial Doppler ultrasonography: report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology 2004; 62:1468–1481.
- Blanche C, Noble S, Roffi M, et al. Platypnea-orthodeoxia syndrome in the elderly treated by percutaneous patent foramen ovale closure: a case series and literature review. Eur J Intern Med 2013; 24:813–817.
- Tobis J, Shenoda M. Percutaneous treatment of patent foramen ovale and atrial septal defects. J Am Coll Cardiol 2012; 60:1722–1732.
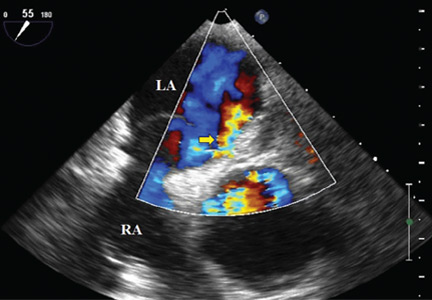
Is Postop Lethargy Cause for Concern?
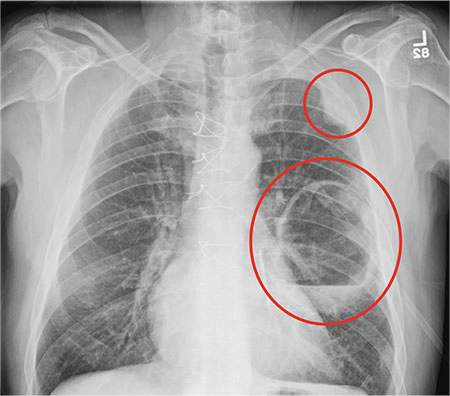
Answer
The radiograph shows a large cavitary lesion within the left mid-lung with evidence of an air fluid level. This finding is strongly suggestive of a postoperative abscess or empyema. Secondarily, there is some pleural thickening within the left lateral apex region. This can be suggestive of scarring or possibly a neoplasm.
The patient was admitted to the ICU for a sepsis workup, and Interventional Radiology was consulted to evaluate for CT-guided drain placement.
Answer
The radiograph shows a large cavitary lesion within the left mid-lung with evidence of an air fluid level. This finding is strongly suggestive of a postoperative abscess or empyema. Secondarily, there is some pleural thickening within the left lateral apex region. This can be suggestive of scarring or possibly a neoplasm.
The patient was admitted to the ICU for a sepsis workup, and Interventional Radiology was consulted to evaluate for CT-guided drain placement.
Answer
The radiograph shows a large cavitary lesion within the left mid-lung with evidence of an air fluid level. This finding is strongly suggestive of a postoperative abscess or empyema. Secondarily, there is some pleural thickening within the left lateral apex region. This can be suggestive of scarring or possibly a neoplasm.
The patient was admitted to the ICU for a sepsis workup, and Interventional Radiology was consulted to evaluate for CT-guided drain placement.
A 65-year-old man is transported to your emergency department from a local rehabilitation hospital. He is three weeks status post cardiac bypass surgery as well as “some other valve procedure.” In the past two to three days, staff members report, the patient has been less active and has not participated in therapy. This morning, he was found to be lethargic, and that is what prompted the call to 911. Examination reveals a lethargic male who has little verbal communication beyond moaning and groaning. His vital signs include a temperature of 36°C; blood pressure, 90/40 mm Hg; and heart rate, 135 beats/min. His O2 saturation is 90% on room air. Inspection of the patient’s chest reveals a recent, healing midline sternotomy incision. There is no overt redness or swelling. On auscultation, you note decreased breath sounds on the left side, with some coarse crackles. As you initiate your facility’s sepsis protocol order set, a stat portable chest radiograph is obtained. What is your impression?