User login
Patient Has No Complaints, But Family Is Concerned
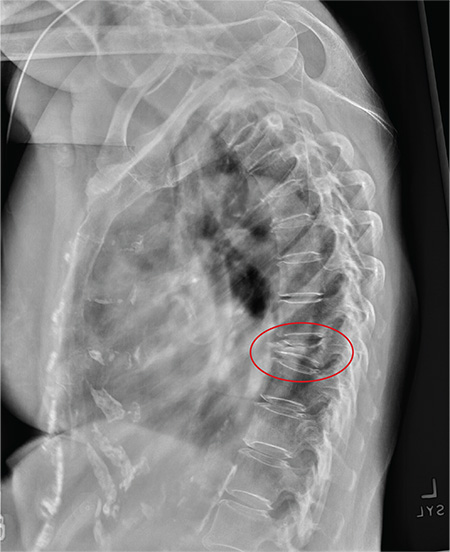
ANSWER
The radiograph shows diffuse osteopenia and spondylosis. Of note is a moderate to severe compression deformity of the T8 vertebral body. However, by plain radiograph, it is age indeterminate as to its acuity. For definitive diagnosis, MRI without contrast is required to assess for marrow edema, which then suggests an acute fracture.
This patient was admitted for further workup. MRI was ultimately obtained and revealed marrow edema within that vertebral body. She was treated with a rigid custom-made brace.
ANSWER
The radiograph shows diffuse osteopenia and spondylosis. Of note is a moderate to severe compression deformity of the T8 vertebral body. However, by plain radiograph, it is age indeterminate as to its acuity. For definitive diagnosis, MRI without contrast is required to assess for marrow edema, which then suggests an acute fracture.
This patient was admitted for further workup. MRI was ultimately obtained and revealed marrow edema within that vertebral body. She was treated with a rigid custom-made brace.
ANSWER
The radiograph shows diffuse osteopenia and spondylosis. Of note is a moderate to severe compression deformity of the T8 vertebral body. However, by plain radiograph, it is age indeterminate as to its acuity. For definitive diagnosis, MRI without contrast is required to assess for marrow edema, which then suggests an acute fracture.
This patient was admitted for further workup. MRI was ultimately obtained and revealed marrow edema within that vertebral body. She was treated with a rigid custom-made brace.
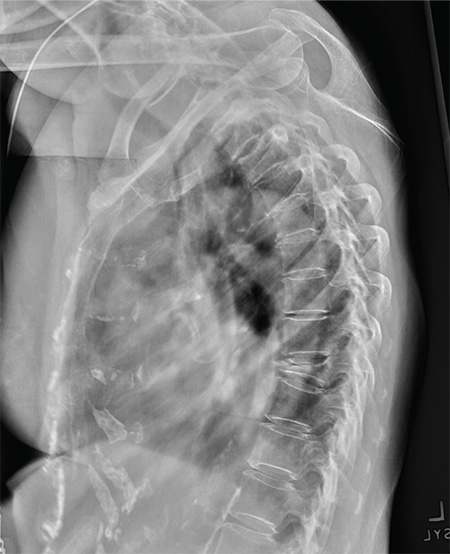
An 80-year-old woman presents to the emergency department for evaluation. Her family reports that she has baseline dementia and resides in an assisted living facility. Staff there report that recently the patient has fallen multiple times. The patient herself does not voice any specific complaints, but her family has noticed she is not as active or walking as much as usual. Medical history is significant for mild hypertension. On physical exam, you note that the patient is awake and alert but oriented only to person. Her vital signs are stable. Primary and secondary survey do not demonstrate any obvious injury or trauma. You order some basic blood work, as well as some imaging studies—including thoracic and lumbar radiographs. The lateral thoracic radiograph is shown. What is your impression?
Alcohol withdrawal syndrome in medical patients
Deprived of alcohol while in the hospital, up to 80% of patients who are alcohol-dependent risk developing alcohol withdrawal syndrome,1 a potentially life-threatening condition. Clinicians should anticipate the syndrome and be ready to treat and prevent its complications.
Because alcoholism is common, nearly every provider will encounter its complications and withdrawal symptoms. Each year, an estimated 1.2 million hospital admissions are related to alcohol abuse, and about 500,000 episodes of withdrawal symptoms are severe enough to require clinical attention.1–3 Nearly 50% of patients with alcohol withdrawal syndrome are middle-class, highly functional individuals, making withdrawal difficult to recognize.1
While acute trauma patients or those with alcohol withdrawal delirium are often admitted directly to an intensive care unit (ICU), many others are at risk for or develop alcohol withdrawal syndrome and are managed initially or wholly on the acute medical unit. While specific statistics have not been published on non-ICU patients with alcohol withdrawal syndrome, they are an important group of patients who need to be well managed to prevent the progression of alcohol withdrawal syndrome to alcohol withdrawal delirium, alcohol withdrawal-induced seizure, and other complications.
This article reviews how to identify and manage alcohol withdrawal symptoms in noncritical, acutely ill medical patients, with practical recommendations for diagnosis and management.
CAN LEAD TO DELIRIUM TREMENS
In people who are physiologically dependent on alcohol, symptoms of withdrawal usually occur after abrupt cessation.4 If not addressed early in the hospitalization, alcohol withdrawal syndrome can progress to alcohol withdrawal delirium (also known as delirium tremens or DTs), in which the mortality rate is 5% to 10%.5,6 Potential mechanisms of DTs include increased dopamine release and dopamine receptor activity, hypersensitivity to N-methyl-d-aspartate, and reduced levels of gamma-aminobutyric acid (GABA).7
Long-term changes are thought to occur in neurons after repeated detoxification from alcohol, a phenomenon called “kindling.” After each detoxification, alcohol craving and obsessive thoughts increase,8 and subsequent episodes of alcohol withdrawal tend to be progressively worse.
Withdrawal symptoms
Alcohol withdrawal syndrome encompasses a spectrum of symptoms and conditions, from minor (eg, insomnia, tremulousness) to severe (seizures, DTs).2 The symptoms typically depend on the amount of alcohol consumed, the time since the last drink, and the number of previous detoxifications.9
The Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition,1 states that to establish a diagnosis of alcohol withdrawal syndrome, a patient must meet four criteria:
- The patient must have ceased or reduced alcohol intake after heavy or prolonged use.
- Two or more of the following must develop within a few hours to a few days: autonomic hyperactivity (sweating or pulse greater than 100 beats per minute); increased hand tremor; insomnia; nausea or vomiting; transient visual, tactile, or auditory hallucinations or illusions; psychomotor agitation; anxiety; grand mal seizure.
- The above symptoms must cause significant distress or functional impairment.
- The symptoms must not be related to another medical condition.
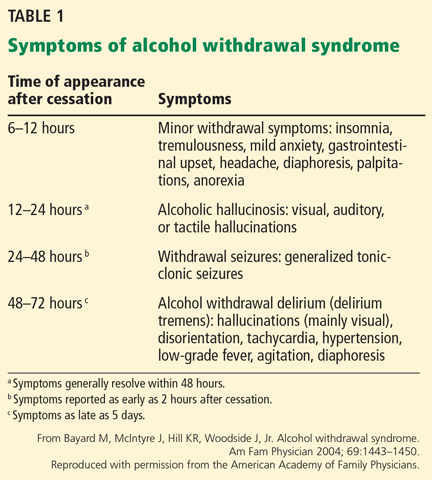
Some of the symptoms described in the second criterion above can occur while the patient still has a measurable blood alcohol level, usually within 6 hours of cessation of drinking.10 Table 1 describes the timetable of onset of symptoms and their severity.2
The elderly may be affected more severely
While the progression of the symptoms described above is commonly used for medical inpatients, the timeline may be different in an elderly patient. Compared with younger patients, elderly patients may have higher blood alcohol concentrations owing to lower total body water, so small amounts of alcohol can produce significant effects.11,12 Brower et al12 found that elderly patients experienced more withdrawal symptoms, especially cognitive impairment, weakness, and high blood pressure, and for 3 days longer.
In the elderly, alcohol may have a greater impact on the central nervous system because of increased permeability of the blood-brain barrier. And importantly, elderly patients tend to have more concomitant diseases and take more medications, all of which can affect alcohol metabolism.
ASSESSMENT SCALES FOR ALCOHOL WITHDRAWAL SYNDROME
A number of clinical scales for evaluating alcohol withdrawal have been developed. Early ones such as the 30-item Total Severity Assessment (TSA) scale and the 11-item Selected Severity Assessment (SSA) scale were limited because they were extremely detailed, burdensome to nursing staff to administer, and contained items such as daily “sleep disturbances” that were not acute enough to meet specific monitoring needs or to guide drug therapy.13,14
The Clinical Institute Withdrawal Assessment for alcohol (CIWA-A) scale, with 15 items, was derived from the SSA scale and includes acute items for assessment as often as every half-hour.15

The CIWA-Ar scale (Table 2) was developed from the CIWA-A scale by Sullivan et al.15 Using both observation and interview, it focuses on 10 areas: nausea and vomiting, tremor, paroxysmal sweats, anxiety, agitation, headache, disorientation, tactile disturbances, auditory disturbances, and visual disturbances. Scores can range from 0 to 67; a higher score indicates worse withdrawal symptoms and outcomes and therefore necessitates escalation of treatment.
The CIWA-Ar scale is now the one most commonly used in clinical trials16–20 and, we believe, in practice. Other scales, including the CIWA-AD and the Alcohol Withdrawal Scale have been validated but are not widely used in practice.14,21
BASELINE ASSESSMENT AND EARLY SUPPORTIVE CARE
A thorough history and physical examination should be performed on admission in patients known to be or suspected of being alcohol-dependent to assess the patient’s affected body systems. The time elapsed since the patient’s last alcohol drink helps predict the onset of withdrawal complications.
Baseline laboratory tests for most patients with suspected alcohol withdrawal syndrome should include a basic blood chemistry panel, complete blood cell count, and possibly an alcohol and toxicology screen, depending on the patient’s history and presentation.
Hydration and nutritional support are important in patients presenting with alcohol withdrawal syndrome. Severe disturbances in electrolytes can lead to serious complications, including cardiac arrhythmia. Close monitoring and electrolyte replacement as needed are recommended for hospitalized alcoholic patients and should follow hospital protocols.22
Thiamine and folic acid status deserve special attention, since long-standing malnutrition is common in alcoholic patients. Thiamine deficiency can result in Wernicke encephalopathy and Korsakoff syndrome, characterized by delirium, ataxia, vision changes, and amnesia. Alcohol withdrawal guidelines recommend giving thiamine intravenously for the first 2 to 5 days after admission.23 In addition, thiamine must be given before any intravenous glucose product, as thiamine is a cofactor in carbohydrate metabolism.23 Folic acid should also be supplemented, as chronic deficiencies may lead to megaloblastic or macrocytic anemia.
CIWA-Ar scale. To provide consistent monitoring and ongoing treatment, clinicians and institutions are encouraged to use a simple assessment scale that detects and quantifies alcohol withdrawal syndrome and that can be used for reassessment after an intervention.21 The CIWA-Ar scale should be used to facilitate “symptom-triggered therapy” in which, depending on the score, the patient receives pharmacologic treatment followed by a scheduled reevaluation.23,24 Most patients with a CIWA-Ar score of 8 or higher benefit from benzodiazepine therapy.16,18,19
PRIMARY DRUG THERAPIES FOR MEDICAL INPATIENTS
Benzodiazepines are the first-line agents
Benzodiazepines are the first-line agents recommended for preventing and treating alcohol withdrawal syndrome.23 Their various pharmacokinetic profiles, wide therapeutic indices, and safety compared with older sedative hypnotics make them the preferred class.23,25 No single benzodiazepine is preferred over the others for treating alcohol withdrawal syndrome: studies have shown benefits using short-acting, intermediate-acting, and long-acting agents. The choice of drug is variable and patient-specific.16,18,26
Benzodiazepines promote and enhance binding of the inhibitory neurotransmitter GABA to GABAA receptors in the central nervous system.27 As a class, benzodiazepines are all structurally related and produce the same effects—namely, sedation, hypnosis, decreased anxiety, muscle relaxation, anterograde amnesia, and anticonvulsant activity.27
The most studied benzodiazepines for treating and preventing alcohol withdrawal syndrome are chlordiazepoxide, oxazepam, and lorazepam,16–20 whereas diazepam was used in older studies.23
Diazepam and chlordiazepoxide are metabolized by oxidation, each sharing the long-acting active metabolite desmethyldiazepam (half-life 72 hours), and short-acting metabolite oxazepam (half-life 8 hours).27 In addition, the parent drugs also have varying pharmacokinetic profiles: diazepam has a half-life of more than 30 hours and chlordiazepoxide a half-life of about 8 hours. Chlordiazepoxide and diazepam’s combination of both long- and short-acting benzodiazepine activity provides long-term efficacy in attenuating withdrawal symptoms, but chlordiazepoxide’s shorter parent half-life allows more frequent dosing.
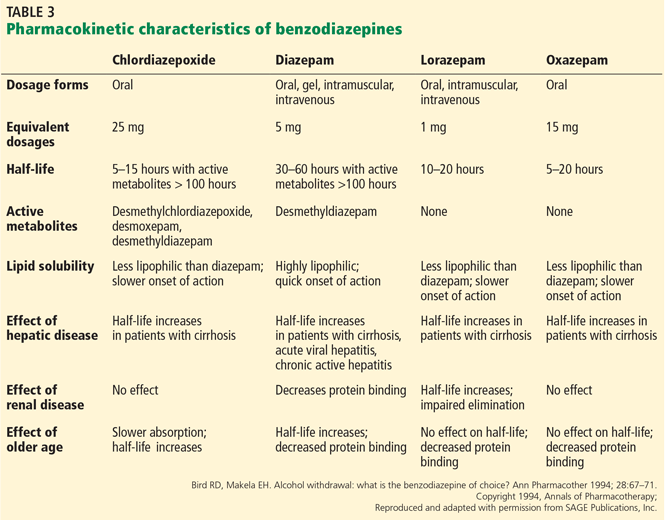
Lorazepam (half-life 10–20 hours) and oxazepam (half-life 5–20 hours) undergo glucuronide conjugation and do not have metabolites.27,28 Table 3 provides a pharmacokinetic summary.27,28
Various dosage regimens are used in giving benzodiazepines, the most common being symptom-triggered therapy, governed by assessment scales, and scheduled around-the-clock therapy.29 Current evidence supports symptom-triggered therapy in most inpatients who are not critically ill, as it can reduce both benzodiazepine use and adverse drug events and can reduce the length of stay.16,19
Trials of symptom-triggered benzodiazepine therapy
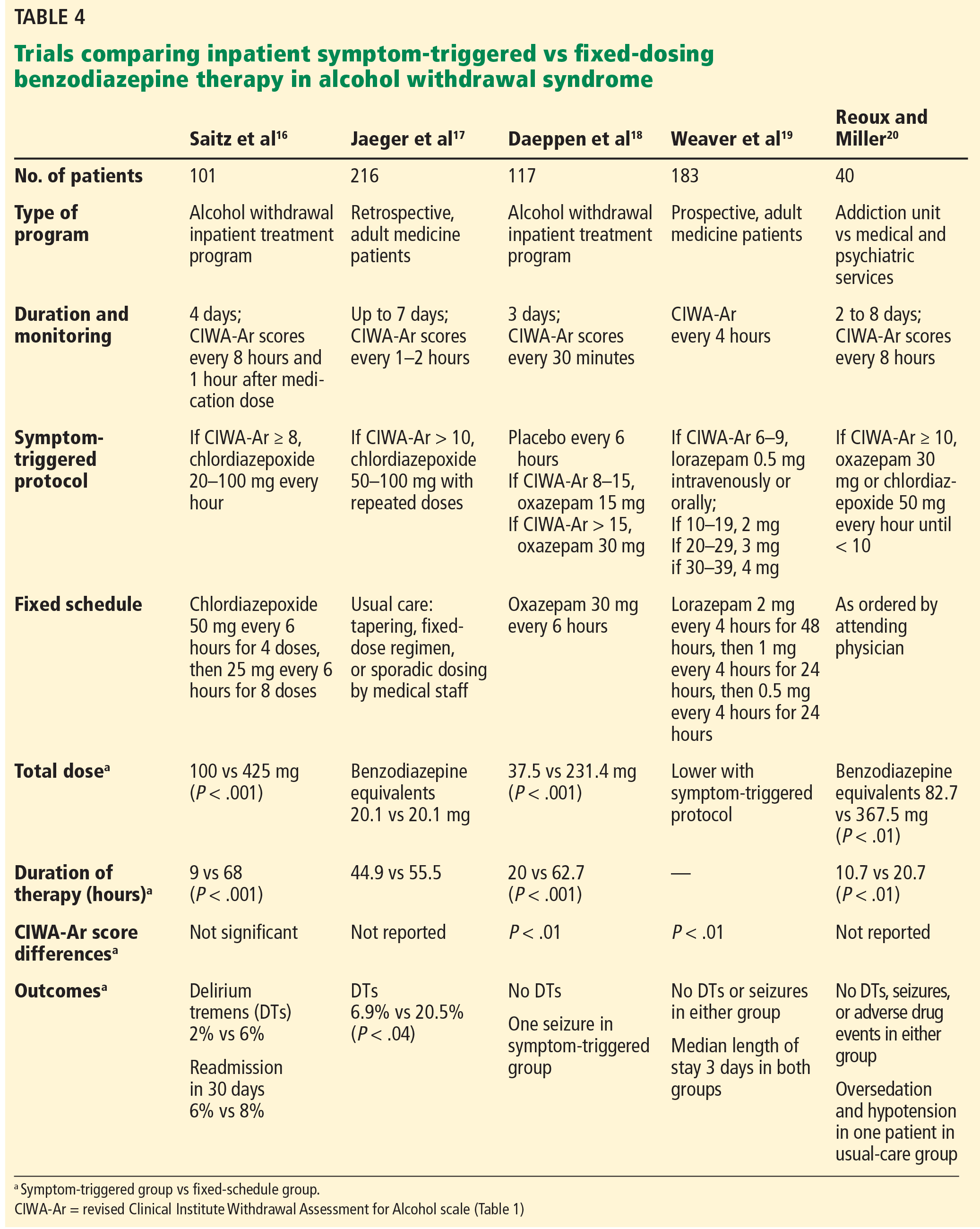
Most inpatient trials of symptom-triggered therapy (Table 4)3,16–20 used the CIWA-Ar scale for monitoring. In some of the studies, benzodiazepines were given if the score was 8 or higher, but others used cut points as high as 15 or higher. Doses:
- Chlordiazepoxide (first dose 25–100 mg)
- Lorazepam (first dose 0.5–2 mg)
- Oxazepam (30 mg).
After each dose, patients were reevaluated at intervals of 30 minutes to 8 hours.
Most of these trials showed no difference in rates of adverse drug events such as seizures, hallucinations, and lethargy with symptom-triggered therapy compared with scheduled therapy.16,18,20 They also found either no difference in the incidence of delirium tremens, or a lower incidence of delirium tremens with symptom-triggered therapy than with scheduled therapy.16–18,20
Weaver et al19 found no difference in length of stay between scheduled therapy and symptom-triggered therapy, but Saitz et al16 reported a median benzodiazepine treatment duration of 9 hours with symptom-triggered therapy vs 68 hours with fixed dosing. Thus, the study by Saitz et al suggests that hospitalization might be shorter with symptom-triggered therapy.
Many of the trials had notable limitations related to the diversity of patients enrolled and the protocols for both symptom-triggered therapy and fixed dosing. Some trials enrolled only inpatients in detoxification programs; others focused on inpatients with acute medical illness. The inpatient alcohol treatment trials16,18 excluded medically ill patients and those with concurrent psychiatric illness,16,18 and one excluded patients with seizures.16 One of the inpatient alcohol treatment program trials16 excluded patients on beta-blockers or clonidine because of concern that these drugs could mask withdrawal symptoms, whereas trials in medically ill patients allowed these same drugs.17,19,20
Most of the patients were men (approximately 75%, but ranging from 74% to 100%), and therefore the study results may not be as applicable to women.16–20 Most participants were middle-aged, with average ages in all studies between 46 and 55. Finally, the studies used a wide range of medications and dosing, with patient monitoring intervals ranging from every 30 minutes to every 8 hours.16–20
In a 2010 Cochrane analysis, Amato et al29 concluded that the limited evidence available favors symptom-triggered regimens over fixed-dosing regimens, but that differences in isolated trials should be interpreted very cautiously.
Therapeutic ethanol
Aside from the lack of evidence to support its use in alcohol withdrawal syndrome, prescribing oral ethanol to alcoholic patients clearly poses an ethical dilemma. However, giving ethanol intravenously has been studied, mostly in surgical trauma patients.30
Early reports described giving intravenous ethanol on a gram-to-gram basis to match the patient’s consumption before admission to prevent alcohol withdrawal syndrome. But later studies reported prevention of alcohol withdrawal syndrome with very small amounts of intravenous ethanol.30,31 While clinical trials have been limited to ICU patients, ethanol infusion at an initial rate of 2.5 to 5 g per hour and titrated up to 10 g per hour has appeared to be safe and effective for preventing alcohol withdrawal syndrome.30,31 The initial infusion rate of 2.5 to 5 g per hour is equivalent to 4 to 10 alcoholic beverages per 24 hours.
Nevertheless, ethanol infusion carries the potential for toxicities (eg, gastric irritation, precipitation of acute hepatic failure, hypoglycemia, pancreatitis, bone marrow suppression, prolonged wound healing) and drug interactions (eg, with anticoagulants and anticonvulsants). Thus, ethanol is neither widely used nor recommended.25,31
ADJUNCTIVE THERAPIES
Many medications are used adjunctively in the acute setting, both for symptoms of alcohol withdrawal syndrome and for agitation.
Haloperidol
No clinical trial has yet examined haloperidol monotherapy in patients with alcohol withdrawal syndrome in either general medical units or intensive care units. Yet haloperidol remains important and is recommended as an adjunct therapy for agitation.23,32 Dosing of haloperidol in protocols for surgical patients ranged from 2 to 5 mg intravenously every 0.5 to 2 hours, with a maximum dosage of 0.5 mg per kg per 24 hours.7,33
Alpha-2 agonists
Alpha-2 agonists are thought to reduce sympathetic overdrive and the autonomic symptoms associated with alcohol withdrawal syndrome, and these agents (primarily clonidine) have been studied in the treatment of alcohol withdrawal syndrome.34,35
Clonidine. In a Swedish study,34 26 men ages 20 to 55 who presented with the tremor, sweating, dysphoria, tension, anxiety, and tachycardia associated with alcohol withdrawal syndrome received clonidine 4 µg per kg twice daily or carbamazepine 200 mg three to four times daily in addition to an antiepileptic. Adjunctive use of a benzodiazepine was allowed at night in both groups. No statistically significant difference in symptom reduction was noted between the two groups, and there was no difference in total benzodiazepine use.
Dexmedetomidine, given intravenously, has been tested as an adjunct to benzodiazepine treatment in severe alcohol withdrawal syndrome. It has been shown to decrease the amount of total benzodiazepine needed compared with benzodiazepine therapy alone, but no differences have been seen in length of hospital stay.36–39 However, research on this drug so far is limited to ICU patients.
Beta-blockers
Beta-blockers have been used in inpatients with alcohol withdrawal syndrome to reduce heart rate and potentially reduce alcohol craving. However, the data are limited and conflicting.
Atenolol 50 to 100 mg daily, in a study in 120 patients, reduced length of stay (4 vs 5 days), reduced benzodiazepine use, and improved vital signs and behavior compared with placebo.40
Propranolol 40 mg every 6 hours reduced arrhythmias but increased hallucinations when used alone in a study in 47 patients.41 When used in combination with chlordiazepoxide, no benefit was seen in arrhythmia reduction.41
Barbiturates and other antiepileptics
Data continue to emerge on antiepileptics as both monotherapy and adjunctive therapy for alcohol withdrawal syndrome. Barbiturates as monotherapy were largely replaced by benzodiazepines in view of the narrow therapeutic index of barbiturates and their full agonist effect on the GABA receptor complex. However, phenobarbital has been evaluated in patients presenting with severe alcohol withdrawal syndrome or resistant alcohol withdrawal (ie, symptoms despite large or repeated doses of benzodiazepines) as an adjunct to benzodiazepines.42,43
In addition, a newer trial44 involved giving a single dose of phenobarbital in the emergency department in combination with a CIWA-Ar–based benzodiazepine protocol, compared with the benzodiazepine protocol alone. The group that received phenobarbital had fewer ICU admissions; its evaluation is ongoing.
The three other medications with the most data are carbamazepine, valproic acid, and gabapentin.45,46 However, the studies were small and the benefit was modest. Although these agents are possible alternatives in protracted alcohol withdrawal syndrome, no definite conclusion can be made regarding their place in therapy.46
RECOMMENDATIONS FOR DRUG THERAPY AND SUPPORTIVE CARE
Which benzodiazepine to use?
No specific benzodiazepine is recommended over the others for managing alcohol withdrawal syndrome, but studies best support the long-acting agent chlordiazepoxide.16,17,20 Other benzodiazepines such as lorazepam and oxazepam have proved to be beneficial, but drugs should be selected on the basis of patient characteristics and drug metabolism.18,19,27
Patients with severe liver dysfunction and the elderly may have slower oxidative metabolism, so the effects of medications that are primarily oxidized, such as chlordiazepoxide and diazepam, may be prolonged. Therefore, lorazepam and oxazepam would be preferred in these groups.47 While most patients with alcohol withdrawal syndrome and liver dysfunction do not have advanced cirrhosis, we recommend liver function testing (serum aspartate aminotransferase, alanine aminotransferase, and alkaline phosphatase levels) and screening for liver disease, given the drug metabolism and package insert caution for use in those with impaired hepatic function.48
Patients with end-stage renal disease (stage 5 chronic kidney disease) or acute kidney injury should not receive parenteral diazepam or lorazepam. The rationale is the potential accumulation of propylene glycol, the solvent used in these formulations.
In the elderly, the Beers list of drugs to avoid in older adults includes benzodiazepines, not differentiating individual benzodiazepines in terms of risk.49 However, chlordiazepoxide may be preferable to diazepam due to its shorter parent half-life and lower lipophilicity.27 Few studies have been done using benzodiazepines in elderly patients with alcohol withdrawal syndrome, but those published have shown either equivalent dosing required compared with younger patients or more severe withdrawal for which they received greater amounts of chlordiazepoxide.9,12 Lorazepam and oxazepam have less potential to accumulate in the elderly compared with the nonelderly due to the drugs’ metabolic profiles; lorazepam is the preferred agent because of its faster onset of action.47 Ultimately, the choice of benzodiazepine in elderly patients with alcohol withdrawal syndrome should be based on patient-specific characteristics.
How should benzodiazepines be dosed?
While the CIWA-Ar thresholds and subsequent dosing of benzodiazepines varied in different studies, we recommend starting benzodiazepine therapy at a CIWA-Ar score of 8 or higher, with subsequent dosing based on score reassessment. Starting doses of benzodiazepines should be chlordiazepoxide 25 to 50 mg, lorazepam 1 to 2 mg, or oxazepam 15 mg.16–20
Subsequent doses should be titrated upward, increasing by 1.5 to 2 times the previous dose and monitored at least every 1 to 2 hours after dose adjustments. Once a patient is stable and the CIWA-Ar score is less than 8, monitoring intervals can be extended to every 4 to 8 hours. If the CIWA-Ar score is more than 20, studies suggest the need for patient reevaluation for transfer to the ICU; however, some health systems have a lower threshold for this intervention.7,14,50

Dosing algorithms and CIWA-Ar goals may vary slightly from institution to institution, but it has been shown that symptom-triggered therapy works best when hospitals have a protocol for it and staff are adequately trained to assess patients with alcohol withdrawal syndrome.7,50,51 Suggestions for dose ranges and symptom-triggered therapy are shown in Table 5.
In case of benzodiazepine overdose or potential benzodiazepine-induced delirium, flumazenil could be considered.52
Patients who should not receive symptom-triggered therapy include immediate postoperative patients in whom clinicians cannot properly assess withdrawal symptoms and patients with a history of DTs.51 While controversy exists regarding the use of symptom-triggered therapy in patients with complicated medical comorbidities, there are data to support symptom-triggered therapy in some ICU patients, as it has resulted in less benzodiazepine use and reduced mechanical ventilation.53,54
There are limited data to support phenobarbital in treating resistant alcohol withdrawal syndrome, either alone or concurrently with benzodiazepines, in escalating doses ranging from 65 to 260 mg, with a maximum daily dose of 520 mg.42,55,56
Haloperidol
For patients exhibiting agitation despite benzodiazepine therapy, giving haloperidol adjunctively can be beneficial.
Haloperidol can be used in medical patients as an adjunctive therapy for agitation, but caution is advised because of the potential for a lowering of the seizure threshold, extrapyramidal effects, and risk of QTc prolongation leading to arrhythmias. Patients considered at highest risk for torsades de pointes may have a QTc of 500 msec or greater.57
Patients should also be screened for factors that have been shown to be independent predictors of QTc prolongation (female sex, diagnosis of myocardial infarction, septic shock or left ventricular dysfunction, other QT-prolonging drugs, age > 68, baseline QTc ≥ 450 msec, and hypokalemia).58 If combined predictors have been identified, it is recommended that haloperidol be avoided.
If haloperidol is to be given, a baseline electrocardiogram and electrolyte panel should be obtained, with daily electrocardiograms thereafter, as well as ongoing review of the patient’s medications to minimize drug interactions that could further increase the risk for QTc prolongation.
Suggested haloperidol dosing is 2 to 5 mg intravenously every 0.5 to 2 hours with a maximum dose of 0.5 mg/kg/24 hours.8,33 A maximum of 35 mg of intravenous haloperidol should be used in a 24-hour period to avoid QTc prolongation.57
Antihypertensive therapy
Many patients receive symptomatic relief of autonomic hyperreactivity with benzodiazepines. However, some may require additional antihypertensive therapy for cardiac adrenergic symptoms (hypertension, tachycardia) if symptoms do not resolve by treating other medical problems commonly seen in patients with alcohol withdrawal syndrome, such as dehydration and electrolyte imbalances.7
Published protocols suggest giving clonidine 0.1 mg orally every hour up to three times as needed until systolic blood pressure is less than 140 mm Hg (less than 160 mm Hg if the patient is over age 60) and diastolic pressure is less than 90 mm Hg.51 Once the patient is stabilized, the dosing can be scheduled to a maximum of 2.4 mg daily.59 However, we believe that the use of clonidine should be restricted to patients who have a substantial increase in blood pressure over baseline or are nearing a hypertensive urgency or emergency (pressures > 180/120 mm Hg) and should not be used to treat other general symptoms associated with alcohol withdrawal syndrome.42
In addition, based on limited evidence, we recommend using beta-blockers only in patients with symptomatic tachycardia or as an adjunct in hypertension management.40,41
Therapies to avoid in acutely ill medical patients
Ethanol is not recommended. Instead, intravenous benzodiazepines should be given in patients presenting with severe alcohol withdrawal syndrome.
Antiepileptics, including valproic acid, carbamazepine, and pregabalin, lack benefit in these patients either as monotherapy or as adjunctive therapy and so are not recommended.45,60–62
Magnesium supplementation (in patients with normal serum magnesium levels) should not be given, as no clinical benefit has been shown.63
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Arlington, VA: American Psychiatric Association, 2013:501.
- Bayard M, McIntyre J, Hill KR, Woodside J Jr. Alcohol withdrawal syndrome. Am Fam Physician 2004; 69:1443–1450.
- Kosten TR, O'Connor PG. Management of drug and alcohol withdrawal. N Engl J Med 2003; 348:1786–1795.
- Isbell H, Fraser HF, Wilker A, Bellevile RE, Eisenman AJ. An experimental study of the etiology of rum fits and delirium tremens. Q J Stud Alcohol 1955; 16:1–33.
- Khan A, Levy P, DeHorn S, Miller W, Compton S. Predictors of mortality in patients with delirium tremens. Acad Emerg Med 2008; 15:788–790.
- Monte R, Rabuñal R, Casariego E, López-Agreda H, Mateos A, Pértega S. Analysis of the factors determining survival of alcoholic withdrawal syndrome patients in a general hospital. Alcohol Alcohol 2010; 45:151–158.
- Stanley KM, Amabile CM, Simpson KN, Couillard D, Norcross ED, Worrall CL. Impact of an alcohol withdrawal syndrome practice guideline on surgical patient outcomes. Pharmacotherapy 2003; 23:843–854.
- Brousse G, Arnaud B, Vorspan F, et al. Alteration of glutamate/GABA balance during acute alcohol withdrawal in emergency department: a prospective analysis. Alcohol Alcohol 2012; 47:501–508.
- Liskow BI, Rinck C, Campbell J, DeSouza C. Alcohol withdrawal in the elderly. J Stud Alcohol 1989; 50:414–421.
- Etherington JM. Emergency management of acute alcohol problems. Part 1: uncomplicated withdrawal. Can Fam Physician 1996; 42:2186–2190.
- Letizia M, Reinbolz M. Identifying and managing acute alcohol withdrawal in the elderly. Geriatr Nurs 2005; 26:176–183.
- Brower KJ, Mudd S, Blow FC, Young JP, Hill EM. Severity and treatment of alcohol withdrawal in elderly versus younger patients. Alcohol Clin Exp Res 1994; 18:196–201.
- Williams D, Lewis J, McBride A. A comparison of rating scales for the alcohol-withdrawal syndrome. Alcohol Alcohol 2001; 36:104–108.
- Reoux JP, Oreskovich MR. A comparison of two versions of the Clinical Institute Withdrawal Assessment for Alcohol: the CIWA-Ar and CIWA-AD. Am J Addict 2006; 15:85–93.
- Sullivan JT, Sykora K, Schneiderman J, Naranjo CA, Sellers EM. Assessment of alcohol withdrawal: the revised Clinical Institute Withdrawal Assessment for alcohol scale (CIWA-Ar). Br J Addict 1989; 84:1353–1357.
- Saitz R, Mayo-Smith MF, Roberts MS, Redmond HA, Bernard DR, Calkins DR. Individualized treatment for alcohol withdrawal. A randomized double-blind controlled trial. JAMA 1994; 272:519–523.
- Jaeger TM, Lohr RH, Pankratz VS. Symptom-triggered therapy for alcohol withdrawal syndrome in medical inpatients. Mayo Clin Proc 2001; 76:695–701.
- Daeppen JB, Gache P, Landry U, et al. Symptom-triggered vs fixed-schedule doses of benzodiazepine for alcohol withdrawal: a randomized treatment trial. Arch Intern Med 2002; 162:1117–1121.
- Weaver MF, Hoffman HJ, Johnson RE, Mauck K. Alcohol withdrawal pharmacotherapy for inpatients with medical comorbidity. J Addict Dis 2006; 25:17–24.
- Reoux JP, Miller K. Routine hospital alcohol detoxification practice compared to symptom triggered management with an Objective Withdrawal Scale (CIWA-Ar). Am J Addict 2000; 9:135–144.
- Wetterling T, Kanitz RD, Besters B, et al. A new rating scale for the assessment of the alcohol-withdrawal syndrome (AWS scale). Alcohol Alcohol 1997; 32:753–760.
- Myrick H, Anton RF. Treatment of alcohol withdrawal. Alcohol Health Res World 1998; 22:38–43.
- Mayo-Smith MF, Beecher LH, Fischer TL, et al; Working Group on the Management of Alcohol Withdrawal Delirium, Practice Guidelines Committee, American Society of Addiction Medicine. Management of alcohol withdrawal delirium. An evidence-based practice guideline. Arch Intern Med 2004; 164:1405–1412.
- Sellers EM, Sullivan JT, Somer G, Sykora K. Characterization of DSM-III-R criteria for uncomplicated alcohol withdrawal provides an empirical basis for DSM-IV. Arch Gen Psychiatry 1991; 48:442–447.
- Sarff M, Gold JA. Alcohol withdrawal syndromes in the intensive care unit. Crit Care Med 2010; 38(suppl):S494–S501.
- Kumar CN, Andrade C, Murthy P. A randomized, double-blind comparison of lorazepam and chlordiazepoxide in patients with uncomplicated alcohol withdrawal. J Stud Alcohol Drugs 2009; 70:467–474.
- Bird RD, Makela EH. Alcohol withdrawal: what is the benzodiazepine of choice? Ann Pharmacother 1994; 28:67–71.
- Perry EC. Inpatient management of acute alcohol withdrawal syndrome. CNS Drugs 2014; 28:401–410.
- Amato L, Minozzi S, Vecchi S, Davoli M. Benzodiazepines for alcohol withdrawal. Cochrane Database Syst Rev 2010; 3:CD005063.
- Weinberg JA, Magnotti LJ, Fischer PE, et al. Comparison of intravenous ethanol versus diazepam for alcohol withdrawal prophylaxis in the trauma ICU: results of a randomized trial. J Trauma 2008; 64:99–104.
- Craft PP, Foil MB, Cunningham PR, Patselas PC, Long-Snyder BM, Collier MS. Intravenous ethanol for alcohol detoxification in trauma patients. South Med J 1994; 87:47–54.
- Ungur LA, Neuner B, John S, Wernecke K, Spies C. Prevention and therapy of alcohol withdrawal on intensive care units: systematic review of controlled trials. Alcohol Clin Exp Res 2013; 37:675–686.
- Lansford CD, Guerriero CH, Kocan MJ, et al. Improved outcomes in patients with head and neck cancer using a standardized care protocol for postoperative alcohol withdrawal. Arch Otolaryngol Head Neck Surg 2008; 134:865–872.
- Walinder J, Balldin J, Bokstrom K, Karlsson I, Lundstrom B, Svensson TH. Clonidine suppression of the alcohol withdrawal syndrome. Drug Alcohol Depend 1981; 8:345–348.
- Muzyk AJ, Fowler JA, Norwood DK, Chilipko A. Role of alpha2-agonists in the treatment of acute alcohol withdrawal. Ann Pharmacother 2011; 45:649–657.
- Crispo AL, Daley MJ, Pepin JL, Harford PH, Brown CV. Comparison of clinical outcomes in nonintubated patients with severe alcohol withdrawal syndrome treated with continuous-infusion sedatives: dexmedetomidine versus benzodiazepines. Pharmacotherapy 2014; 34:910–917.
- VanderWeide LA, Foster CJ, MacLaren R, Kiser TH, Fish DN, Mueller SW. Evaluation of early dexmedetomidine addition to the standard of care for severe alcohol withdrawal in the ICU: a retrospective controlled cohort study. J Intensive Care Med 2014. [Epub ahead of print October 16, 2014]
- Rayner SG, Weinert CR, Peng H, Jepsen S, Broccard AF. Dexmedetomidine as adjunct treatment for severe alcohol withdrawal in the ICU. Ann Intensive Care 2012; 2:12.
- Muzyk AJ, Kerns S, Brudney S, Gagliardi JP. Dexmedetomidine for the treatment of alcohol withdrawal syndrome: rationale and current status of research. CNS Drugs 2013; 27:913–920.
- Kraus ML, Gottlieb LD, Horwitz RI, Anscher M. Randomized clinical trial of atenolol in patients with alcohol withdrawal. N Engl J Med 1985; 313:905–909.
- Zilm DH, Jacob MS, MacLeod SM, Sellers EM, Ti TY. Propranolol and chlordiazepoxide effects on cardiac arrhythmias during alcohol withdrawal. Alcohol Clin Exp Res 1980; 4:400–405.
- Hack JB, Hoffmann RS, Nelson LS. Resistant alcohol withdrawal: does an unexpectedly large sedative requirement identify these patients early? J Med Toxicol 2006; 2:55–60.
- Hayner CE, Wuestefeld NL, Bolton PJ. Phenobarbital treatment in a patient with resistant alcohol withdrawal syndrome. Pharmacotherapy 2009; 29:875–878.
- Rosenson J, Clements C, Simon B, et al. Phenobarbital for acute alcohol withdrawal: a prospective randomized double-blind placebo-controlled study. J Emerg Med 2013; 44:592–598.e2.
- Prince V, Turpin KR. Treatment of alcohol withdrawal syndrome with carbamazepine, gabapentin, and nitrous oxide. Am J Health Syst Pharm 2008; 65:1039–1047.
- Leggio L, Kenna GA, Swift RM. New developments for the pharmacological treatment of alcohol withdrawal syndrome. A focus on non-benzodiazepine GABAergic medications. Prog Neuropsychopharmacol Biol Psychiatry 2008; 32:1106–1117.
- Peppers MP. Benzodiazepines for alcohol withdrawal in the elderly and in patients with liver disease. Pharmacotherapy 1996; 16:49–57.
- Valeant Pharmaceuticals North America LLC. Librium—chlordiazepoxide hydrochloride capsule, gelatin coated. http://dailymed.nlm.nih.gov/dailymed/archives/fdaDrugInfo.cfm?archiveid=125207. Accessed November 20, 2015.
- American Geriatrics Society 2012 Beers Criteria Update Expert Panel. American Geriatrics Society updated Beers Criteria for potentially inappropriate medication use in older adults. J Am Geriatr Soc 2012; 60:616–631.
- Hecksel KA, Bostwick JM, Jaeger TM, Cha SS. Inappropriate use of symptom-triggered therapy for alcohol withdrawal in the general hospital. Mayo Clin Proc 2008; 83:274–279.
- Manasco A, Chang S, Larriviere J, Hamm LL, Glass M. Alcohol withdrawal. South Med J 2012; 105:607–612.
- Moore PW, Donovan JW, Burkhart KK, et al. Safety and efficacy of flumazenil for reversal of iatrogenic benzodiazepine-associated delirium toxicity during treatment of alcohol withdrawal, a retrospective review at one center. J Med Toxicol 2014; 10:126–132.
- Bostwick JM, Lapid MI. False positives on the clinical institute withdrawal assessment for alcohol-revised: is this scale appropriate for use in the medically ill? Psychosomatics 2004; 45:256–261.
- de Wit M, Jones DG, Sessler CN, Zilberberg MD, Weaver MF. Alcohol-use disorders in the critically ill patient. Chest 2010; 138:994–1003.
- Young GP, Rores C, Murphy C, Dailey RH. Intravenous phenobarbital for alcohol withdrawal and convulsions. Ann Emerg Med 1987; 16:847–850.
- Hendey GW, Dery RA, Barnes RL, Snowden B, Mentler P. A prospective, randomized trial of phenobarbital versus benzodiazepines for acute alcohol withdrawal. Am J Emerg Med 2011; 29:382–385.
- Sharma ND, Rosman HS, Padhi ID, Tisdale JE. Torsades de pointes associated with intravenous haloperidol in critically ill patients. Am J Cardiol 1998; 81:238–240.
- Tisdale JE, Jaynes HA, Kingery JR, et al. Development and validation of a risk score to predict QT interval prolongation in hospitalized patients. Circ Cardiovasc Qual Outcomes 2013; 6:479–487.
- Boehringer Ingelheim Pharmaceuticals, Inc. Product Information: Catapres oral tablets, clonidine HCl oral tablets, 2012.
- Reoux JP, Saxon AJ, Malte CA, Baer JS, Sloan KL. Divalproex sodium in alcohol withdrawal: a randomized double-blind placebo-controlled clinical trial. Alcohol Clin Exp Res 2001; 25:1324–1329.
- Malcolm R, Ballenger JC, Sturgis ET, Anton R. Double-blind controlled trial comparing carbamazepine to oxazepam treatment of alcohol withdrawal. Am J Psychiatry 1989; 146:617–621.
- Förg A, Hein J, Volkmar K, et al. Efficacy and safety of pregabalin in the treatment of alcohol withdrawal syndrome: a randomized placebo-controlled trial. Alcohol Alcohol 2012; 47:149–155.
- Wilson A, Vulcano B. A double-blind, placebo-controlled trial of magnesium sulfate in the ethanol withdrawal syndrome. Alcohol Clin Exp Res 1984; 8:542–545.
Deprived of alcohol while in the hospital, up to 80% of patients who are alcohol-dependent risk developing alcohol withdrawal syndrome,1 a potentially life-threatening condition. Clinicians should anticipate the syndrome and be ready to treat and prevent its complications.
Because alcoholism is common, nearly every provider will encounter its complications and withdrawal symptoms. Each year, an estimated 1.2 million hospital admissions are related to alcohol abuse, and about 500,000 episodes of withdrawal symptoms are severe enough to require clinical attention.1–3 Nearly 50% of patients with alcohol withdrawal syndrome are middle-class, highly functional individuals, making withdrawal difficult to recognize.1
While acute trauma patients or those with alcohol withdrawal delirium are often admitted directly to an intensive care unit (ICU), many others are at risk for or develop alcohol withdrawal syndrome and are managed initially or wholly on the acute medical unit. While specific statistics have not been published on non-ICU patients with alcohol withdrawal syndrome, they are an important group of patients who need to be well managed to prevent the progression of alcohol withdrawal syndrome to alcohol withdrawal delirium, alcohol withdrawal-induced seizure, and other complications.
This article reviews how to identify and manage alcohol withdrawal symptoms in noncritical, acutely ill medical patients, with practical recommendations for diagnosis and management.
CAN LEAD TO DELIRIUM TREMENS
In people who are physiologically dependent on alcohol, symptoms of withdrawal usually occur after abrupt cessation.4 If not addressed early in the hospitalization, alcohol withdrawal syndrome can progress to alcohol withdrawal delirium (also known as delirium tremens or DTs), in which the mortality rate is 5% to 10%.5,6 Potential mechanisms of DTs include increased dopamine release and dopamine receptor activity, hypersensitivity to N-methyl-d-aspartate, and reduced levels of gamma-aminobutyric acid (GABA).7
Long-term changes are thought to occur in neurons after repeated detoxification from alcohol, a phenomenon called “kindling.” After each detoxification, alcohol craving and obsessive thoughts increase,8 and subsequent episodes of alcohol withdrawal tend to be progressively worse.
Withdrawal symptoms
Alcohol withdrawal syndrome encompasses a spectrum of symptoms and conditions, from minor (eg, insomnia, tremulousness) to severe (seizures, DTs).2 The symptoms typically depend on the amount of alcohol consumed, the time since the last drink, and the number of previous detoxifications.9
The Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition,1 states that to establish a diagnosis of alcohol withdrawal syndrome, a patient must meet four criteria:
- The patient must have ceased or reduced alcohol intake after heavy or prolonged use.
- Two or more of the following must develop within a few hours to a few days: autonomic hyperactivity (sweating or pulse greater than 100 beats per minute); increased hand tremor; insomnia; nausea or vomiting; transient visual, tactile, or auditory hallucinations or illusions; psychomotor agitation; anxiety; grand mal seizure.
- The above symptoms must cause significant distress or functional impairment.
- The symptoms must not be related to another medical condition.
Some of the symptoms described in the second criterion above can occur while the patient still has a measurable blood alcohol level, usually within 6 hours of cessation of drinking.10 Table 1 describes the timetable of onset of symptoms and their severity.2
The elderly may be affected more severely
While the progression of the symptoms described above is commonly used for medical inpatients, the timeline may be different in an elderly patient. Compared with younger patients, elderly patients may have higher blood alcohol concentrations owing to lower total body water, so small amounts of alcohol can produce significant effects.11,12 Brower et al12 found that elderly patients experienced more withdrawal symptoms, especially cognitive impairment, weakness, and high blood pressure, and for 3 days longer.
In the elderly, alcohol may have a greater impact on the central nervous system because of increased permeability of the blood-brain barrier. And importantly, elderly patients tend to have more concomitant diseases and take more medications, all of which can affect alcohol metabolism.
ASSESSMENT SCALES FOR ALCOHOL WITHDRAWAL SYNDROME
A number of clinical scales for evaluating alcohol withdrawal have been developed. Early ones such as the 30-item Total Severity Assessment (TSA) scale and the 11-item Selected Severity Assessment (SSA) scale were limited because they were extremely detailed, burdensome to nursing staff to administer, and contained items such as daily “sleep disturbances” that were not acute enough to meet specific monitoring needs or to guide drug therapy.13,14
The Clinical Institute Withdrawal Assessment for alcohol (CIWA-A) scale, with 15 items, was derived from the SSA scale and includes acute items for assessment as often as every half-hour.15
The CIWA-Ar scale (Table 2) was developed from the CIWA-A scale by Sullivan et al.15 Using both observation and interview, it focuses on 10 areas: nausea and vomiting, tremor, paroxysmal sweats, anxiety, agitation, headache, disorientation, tactile disturbances, auditory disturbances, and visual disturbances. Scores can range from 0 to 67; a higher score indicates worse withdrawal symptoms and outcomes and therefore necessitates escalation of treatment.
The CIWA-Ar scale is now the one most commonly used in clinical trials16–20 and, we believe, in practice. Other scales, including the CIWA-AD and the Alcohol Withdrawal Scale have been validated but are not widely used in practice.14,21
BASELINE ASSESSMENT AND EARLY SUPPORTIVE CARE
A thorough history and physical examination should be performed on admission in patients known to be or suspected of being alcohol-dependent to assess the patient’s affected body systems. The time elapsed since the patient’s last alcohol drink helps predict the onset of withdrawal complications.
Baseline laboratory tests for most patients with suspected alcohol withdrawal syndrome should include a basic blood chemistry panel, complete blood cell count, and possibly an alcohol and toxicology screen, depending on the patient’s history and presentation.
Hydration and nutritional support are important in patients presenting with alcohol withdrawal syndrome. Severe disturbances in electrolytes can lead to serious complications, including cardiac arrhythmia. Close monitoring and electrolyte replacement as needed are recommended for hospitalized alcoholic patients and should follow hospital protocols.22
Thiamine and folic acid status deserve special attention, since long-standing malnutrition is common in alcoholic patients. Thiamine deficiency can result in Wernicke encephalopathy and Korsakoff syndrome, characterized by delirium, ataxia, vision changes, and amnesia. Alcohol withdrawal guidelines recommend giving thiamine intravenously for the first 2 to 5 days after admission.23 In addition, thiamine must be given before any intravenous glucose product, as thiamine is a cofactor in carbohydrate metabolism.23 Folic acid should also be supplemented, as chronic deficiencies may lead to megaloblastic or macrocytic anemia.
CIWA-Ar scale. To provide consistent monitoring and ongoing treatment, clinicians and institutions are encouraged to use a simple assessment scale that detects and quantifies alcohol withdrawal syndrome and that can be used for reassessment after an intervention.21 The CIWA-Ar scale should be used to facilitate “symptom-triggered therapy” in which, depending on the score, the patient receives pharmacologic treatment followed by a scheduled reevaluation.23,24 Most patients with a CIWA-Ar score of 8 or higher benefit from benzodiazepine therapy.16,18,19
PRIMARY DRUG THERAPIES FOR MEDICAL INPATIENTS
Benzodiazepines are the first-line agents
Benzodiazepines are the first-line agents recommended for preventing and treating alcohol withdrawal syndrome.23 Their various pharmacokinetic profiles, wide therapeutic indices, and safety compared with older sedative hypnotics make them the preferred class.23,25 No single benzodiazepine is preferred over the others for treating alcohol withdrawal syndrome: studies have shown benefits using short-acting, intermediate-acting, and long-acting agents. The choice of drug is variable and patient-specific.16,18,26
Benzodiazepines promote and enhance binding of the inhibitory neurotransmitter GABA to GABAA receptors in the central nervous system.27 As a class, benzodiazepines are all structurally related and produce the same effects—namely, sedation, hypnosis, decreased anxiety, muscle relaxation, anterograde amnesia, and anticonvulsant activity.27
The most studied benzodiazepines for treating and preventing alcohol withdrawal syndrome are chlordiazepoxide, oxazepam, and lorazepam,16–20 whereas diazepam was used in older studies.23
Diazepam and chlordiazepoxide are metabolized by oxidation, each sharing the long-acting active metabolite desmethyldiazepam (half-life 72 hours), and short-acting metabolite oxazepam (half-life 8 hours).27 In addition, the parent drugs also have varying pharmacokinetic profiles: diazepam has a half-life of more than 30 hours and chlordiazepoxide a half-life of about 8 hours. Chlordiazepoxide and diazepam’s combination of both long- and short-acting benzodiazepine activity provides long-term efficacy in attenuating withdrawal symptoms, but chlordiazepoxide’s shorter parent half-life allows more frequent dosing.
Lorazepam (half-life 10–20 hours) and oxazepam (half-life 5–20 hours) undergo glucuronide conjugation and do not have metabolites.27,28 Table 3 provides a pharmacokinetic summary.27,28
Various dosage regimens are used in giving benzodiazepines, the most common being symptom-triggered therapy, governed by assessment scales, and scheduled around-the-clock therapy.29 Current evidence supports symptom-triggered therapy in most inpatients who are not critically ill, as it can reduce both benzodiazepine use and adverse drug events and can reduce the length of stay.16,19
Trials of symptom-triggered benzodiazepine therapy
Most inpatient trials of symptom-triggered therapy (Table 4)3,16–20 used the CIWA-Ar scale for monitoring. In some of the studies, benzodiazepines were given if the score was 8 or higher, but others used cut points as high as 15 or higher. Doses:
- Chlordiazepoxide (first dose 25–100 mg)
- Lorazepam (first dose 0.5–2 mg)
- Oxazepam (30 mg).
After each dose, patients were reevaluated at intervals of 30 minutes to 8 hours.
Most of these trials showed no difference in rates of adverse drug events such as seizures, hallucinations, and lethargy with symptom-triggered therapy compared with scheduled therapy.16,18,20 They also found either no difference in the incidence of delirium tremens, or a lower incidence of delirium tremens with symptom-triggered therapy than with scheduled therapy.16–18,20
Weaver et al19 found no difference in length of stay between scheduled therapy and symptom-triggered therapy, but Saitz et al16 reported a median benzodiazepine treatment duration of 9 hours with symptom-triggered therapy vs 68 hours with fixed dosing. Thus, the study by Saitz et al suggests that hospitalization might be shorter with symptom-triggered therapy.
Many of the trials had notable limitations related to the diversity of patients enrolled and the protocols for both symptom-triggered therapy and fixed dosing. Some trials enrolled only inpatients in detoxification programs; others focused on inpatients with acute medical illness. The inpatient alcohol treatment trials16,18 excluded medically ill patients and those with concurrent psychiatric illness,16,18 and one excluded patients with seizures.16 One of the inpatient alcohol treatment program trials16 excluded patients on beta-blockers or clonidine because of concern that these drugs could mask withdrawal symptoms, whereas trials in medically ill patients allowed these same drugs.17,19,20
Most of the patients were men (approximately 75%, but ranging from 74% to 100%), and therefore the study results may not be as applicable to women.16–20 Most participants were middle-aged, with average ages in all studies between 46 and 55. Finally, the studies used a wide range of medications and dosing, with patient monitoring intervals ranging from every 30 minutes to every 8 hours.16–20
In a 2010 Cochrane analysis, Amato et al29 concluded that the limited evidence available favors symptom-triggered regimens over fixed-dosing regimens, but that differences in isolated trials should be interpreted very cautiously.
Therapeutic ethanol
Aside from the lack of evidence to support its use in alcohol withdrawal syndrome, prescribing oral ethanol to alcoholic patients clearly poses an ethical dilemma. However, giving ethanol intravenously has been studied, mostly in surgical trauma patients.30
Early reports described giving intravenous ethanol on a gram-to-gram basis to match the patient’s consumption before admission to prevent alcohol withdrawal syndrome. But later studies reported prevention of alcohol withdrawal syndrome with very small amounts of intravenous ethanol.30,31 While clinical trials have been limited to ICU patients, ethanol infusion at an initial rate of 2.5 to 5 g per hour and titrated up to 10 g per hour has appeared to be safe and effective for preventing alcohol withdrawal syndrome.30,31 The initial infusion rate of 2.5 to 5 g per hour is equivalent to 4 to 10 alcoholic beverages per 24 hours.
Nevertheless, ethanol infusion carries the potential for toxicities (eg, gastric irritation, precipitation of acute hepatic failure, hypoglycemia, pancreatitis, bone marrow suppression, prolonged wound healing) and drug interactions (eg, with anticoagulants and anticonvulsants). Thus, ethanol is neither widely used nor recommended.25,31
ADJUNCTIVE THERAPIES
Many medications are used adjunctively in the acute setting, both for symptoms of alcohol withdrawal syndrome and for agitation.
Haloperidol
No clinical trial has yet examined haloperidol monotherapy in patients with alcohol withdrawal syndrome in either general medical units or intensive care units. Yet haloperidol remains important and is recommended as an adjunct therapy for agitation.23,32 Dosing of haloperidol in protocols for surgical patients ranged from 2 to 5 mg intravenously every 0.5 to 2 hours, with a maximum dosage of 0.5 mg per kg per 24 hours.7,33
Alpha-2 agonists
Alpha-2 agonists are thought to reduce sympathetic overdrive and the autonomic symptoms associated with alcohol withdrawal syndrome, and these agents (primarily clonidine) have been studied in the treatment of alcohol withdrawal syndrome.34,35
Clonidine. In a Swedish study,34 26 men ages 20 to 55 who presented with the tremor, sweating, dysphoria, tension, anxiety, and tachycardia associated with alcohol withdrawal syndrome received clonidine 4 µg per kg twice daily or carbamazepine 200 mg three to four times daily in addition to an antiepileptic. Adjunctive use of a benzodiazepine was allowed at night in both groups. No statistically significant difference in symptom reduction was noted between the two groups, and there was no difference in total benzodiazepine use.
Dexmedetomidine, given intravenously, has been tested as an adjunct to benzodiazepine treatment in severe alcohol withdrawal syndrome. It has been shown to decrease the amount of total benzodiazepine needed compared with benzodiazepine therapy alone, but no differences have been seen in length of hospital stay.36–39 However, research on this drug so far is limited to ICU patients.
Beta-blockers
Beta-blockers have been used in inpatients with alcohol withdrawal syndrome to reduce heart rate and potentially reduce alcohol craving. However, the data are limited and conflicting.
Atenolol 50 to 100 mg daily, in a study in 120 patients, reduced length of stay (4 vs 5 days), reduced benzodiazepine use, and improved vital signs and behavior compared with placebo.40
Propranolol 40 mg every 6 hours reduced arrhythmias but increased hallucinations when used alone in a study in 47 patients.41 When used in combination with chlordiazepoxide, no benefit was seen in arrhythmia reduction.41
Barbiturates and other antiepileptics
Data continue to emerge on antiepileptics as both monotherapy and adjunctive therapy for alcohol withdrawal syndrome. Barbiturates as monotherapy were largely replaced by benzodiazepines in view of the narrow therapeutic index of barbiturates and their full agonist effect on the GABA receptor complex. However, phenobarbital has been evaluated in patients presenting with severe alcohol withdrawal syndrome or resistant alcohol withdrawal (ie, symptoms despite large or repeated doses of benzodiazepines) as an adjunct to benzodiazepines.42,43
In addition, a newer trial44 involved giving a single dose of phenobarbital in the emergency department in combination with a CIWA-Ar–based benzodiazepine protocol, compared with the benzodiazepine protocol alone. The group that received phenobarbital had fewer ICU admissions; its evaluation is ongoing.
The three other medications with the most data are carbamazepine, valproic acid, and gabapentin.45,46 However, the studies were small and the benefit was modest. Although these agents are possible alternatives in protracted alcohol withdrawal syndrome, no definite conclusion can be made regarding their place in therapy.46
RECOMMENDATIONS FOR DRUG THERAPY AND SUPPORTIVE CARE
Which benzodiazepine to use?
No specific benzodiazepine is recommended over the others for managing alcohol withdrawal syndrome, but studies best support the long-acting agent chlordiazepoxide.16,17,20 Other benzodiazepines such as lorazepam and oxazepam have proved to be beneficial, but drugs should be selected on the basis of patient characteristics and drug metabolism.18,19,27
Patients with severe liver dysfunction and the elderly may have slower oxidative metabolism, so the effects of medications that are primarily oxidized, such as chlordiazepoxide and diazepam, may be prolonged. Therefore, lorazepam and oxazepam would be preferred in these groups.47 While most patients with alcohol withdrawal syndrome and liver dysfunction do not have advanced cirrhosis, we recommend liver function testing (serum aspartate aminotransferase, alanine aminotransferase, and alkaline phosphatase levels) and screening for liver disease, given the drug metabolism and package insert caution for use in those with impaired hepatic function.48
Patients with end-stage renal disease (stage 5 chronic kidney disease) or acute kidney injury should not receive parenteral diazepam or lorazepam. The rationale is the potential accumulation of propylene glycol, the solvent used in these formulations.
In the elderly, the Beers list of drugs to avoid in older adults includes benzodiazepines, not differentiating individual benzodiazepines in terms of risk.49 However, chlordiazepoxide may be preferable to diazepam due to its shorter parent half-life and lower lipophilicity.27 Few studies have been done using benzodiazepines in elderly patients with alcohol withdrawal syndrome, but those published have shown either equivalent dosing required compared with younger patients or more severe withdrawal for which they received greater amounts of chlordiazepoxide.9,12 Lorazepam and oxazepam have less potential to accumulate in the elderly compared with the nonelderly due to the drugs’ metabolic profiles; lorazepam is the preferred agent because of its faster onset of action.47 Ultimately, the choice of benzodiazepine in elderly patients with alcohol withdrawal syndrome should be based on patient-specific characteristics.
How should benzodiazepines be dosed?
While the CIWA-Ar thresholds and subsequent dosing of benzodiazepines varied in different studies, we recommend starting benzodiazepine therapy at a CIWA-Ar score of 8 or higher, with subsequent dosing based on score reassessment. Starting doses of benzodiazepines should be chlordiazepoxide 25 to 50 mg, lorazepam 1 to 2 mg, or oxazepam 15 mg.16–20
Subsequent doses should be titrated upward, increasing by 1.5 to 2 times the previous dose and monitored at least every 1 to 2 hours after dose adjustments. Once a patient is stable and the CIWA-Ar score is less than 8, monitoring intervals can be extended to every 4 to 8 hours. If the CIWA-Ar score is more than 20, studies suggest the need for patient reevaluation for transfer to the ICU; however, some health systems have a lower threshold for this intervention.7,14,50
Dosing algorithms and CIWA-Ar goals may vary slightly from institution to institution, but it has been shown that symptom-triggered therapy works best when hospitals have a protocol for it and staff are adequately trained to assess patients with alcohol withdrawal syndrome.7,50,51 Suggestions for dose ranges and symptom-triggered therapy are shown in Table 5.
In case of benzodiazepine overdose or potential benzodiazepine-induced delirium, flumazenil could be considered.52
Patients who should not receive symptom-triggered therapy include immediate postoperative patients in whom clinicians cannot properly assess withdrawal symptoms and patients with a history of DTs.51 While controversy exists regarding the use of symptom-triggered therapy in patients with complicated medical comorbidities, there are data to support symptom-triggered therapy in some ICU patients, as it has resulted in less benzodiazepine use and reduced mechanical ventilation.53,54
There are limited data to support phenobarbital in treating resistant alcohol withdrawal syndrome, either alone or concurrently with benzodiazepines, in escalating doses ranging from 65 to 260 mg, with a maximum daily dose of 520 mg.42,55,56
Haloperidol
For patients exhibiting agitation despite benzodiazepine therapy, giving haloperidol adjunctively can be beneficial.
Haloperidol can be used in medical patients as an adjunctive therapy for agitation, but caution is advised because of the potential for a lowering of the seizure threshold, extrapyramidal effects, and risk of QTc prolongation leading to arrhythmias. Patients considered at highest risk for torsades de pointes may have a QTc of 500 msec or greater.57
Patients should also be screened for factors that have been shown to be independent predictors of QTc prolongation (female sex, diagnosis of myocardial infarction, septic shock or left ventricular dysfunction, other QT-prolonging drugs, age > 68, baseline QTc ≥ 450 msec, and hypokalemia).58 If combined predictors have been identified, it is recommended that haloperidol be avoided.
If haloperidol is to be given, a baseline electrocardiogram and electrolyte panel should be obtained, with daily electrocardiograms thereafter, as well as ongoing review of the patient’s medications to minimize drug interactions that could further increase the risk for QTc prolongation.
Suggested haloperidol dosing is 2 to 5 mg intravenously every 0.5 to 2 hours with a maximum dose of 0.5 mg/kg/24 hours.8,33 A maximum of 35 mg of intravenous haloperidol should be used in a 24-hour period to avoid QTc prolongation.57
Antihypertensive therapy
Many patients receive symptomatic relief of autonomic hyperreactivity with benzodiazepines. However, some may require additional antihypertensive therapy for cardiac adrenergic symptoms (hypertension, tachycardia) if symptoms do not resolve by treating other medical problems commonly seen in patients with alcohol withdrawal syndrome, such as dehydration and electrolyte imbalances.7
Published protocols suggest giving clonidine 0.1 mg orally every hour up to three times as needed until systolic blood pressure is less than 140 mm Hg (less than 160 mm Hg if the patient is over age 60) and diastolic pressure is less than 90 mm Hg.51 Once the patient is stabilized, the dosing can be scheduled to a maximum of 2.4 mg daily.59 However, we believe that the use of clonidine should be restricted to patients who have a substantial increase in blood pressure over baseline or are nearing a hypertensive urgency or emergency (pressures > 180/120 mm Hg) and should not be used to treat other general symptoms associated with alcohol withdrawal syndrome.42
In addition, based on limited evidence, we recommend using beta-blockers only in patients with symptomatic tachycardia or as an adjunct in hypertension management.40,41
Therapies to avoid in acutely ill medical patients
Ethanol is not recommended. Instead, intravenous benzodiazepines should be given in patients presenting with severe alcohol withdrawal syndrome.
Antiepileptics, including valproic acid, carbamazepine, and pregabalin, lack benefit in these patients either as monotherapy or as adjunctive therapy and so are not recommended.45,60–62
Magnesium supplementation (in patients with normal serum magnesium levels) should not be given, as no clinical benefit has been shown.63
Deprived of alcohol while in the hospital, up to 80% of patients who are alcohol-dependent risk developing alcohol withdrawal syndrome,1 a potentially life-threatening condition. Clinicians should anticipate the syndrome and be ready to treat and prevent its complications.
Because alcoholism is common, nearly every provider will encounter its complications and withdrawal symptoms. Each year, an estimated 1.2 million hospital admissions are related to alcohol abuse, and about 500,000 episodes of withdrawal symptoms are severe enough to require clinical attention.1–3 Nearly 50% of patients with alcohol withdrawal syndrome are middle-class, highly functional individuals, making withdrawal difficult to recognize.1
While acute trauma patients or those with alcohol withdrawal delirium are often admitted directly to an intensive care unit (ICU), many others are at risk for or develop alcohol withdrawal syndrome and are managed initially or wholly on the acute medical unit. While specific statistics have not been published on non-ICU patients with alcohol withdrawal syndrome, they are an important group of patients who need to be well managed to prevent the progression of alcohol withdrawal syndrome to alcohol withdrawal delirium, alcohol withdrawal-induced seizure, and other complications.
This article reviews how to identify and manage alcohol withdrawal symptoms in noncritical, acutely ill medical patients, with practical recommendations for diagnosis and management.
CAN LEAD TO DELIRIUM TREMENS
In people who are physiologically dependent on alcohol, symptoms of withdrawal usually occur after abrupt cessation.4 If not addressed early in the hospitalization, alcohol withdrawal syndrome can progress to alcohol withdrawal delirium (also known as delirium tremens or DTs), in which the mortality rate is 5% to 10%.5,6 Potential mechanisms of DTs include increased dopamine release and dopamine receptor activity, hypersensitivity to N-methyl-d-aspartate, and reduced levels of gamma-aminobutyric acid (GABA).7
Long-term changes are thought to occur in neurons after repeated detoxification from alcohol, a phenomenon called “kindling.” After each detoxification, alcohol craving and obsessive thoughts increase,8 and subsequent episodes of alcohol withdrawal tend to be progressively worse.
Withdrawal symptoms
Alcohol withdrawal syndrome encompasses a spectrum of symptoms and conditions, from minor (eg, insomnia, tremulousness) to severe (seizures, DTs).2 The symptoms typically depend on the amount of alcohol consumed, the time since the last drink, and the number of previous detoxifications.9
The Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition,1 states that to establish a diagnosis of alcohol withdrawal syndrome, a patient must meet four criteria:
- The patient must have ceased or reduced alcohol intake after heavy or prolonged use.
- Two or more of the following must develop within a few hours to a few days: autonomic hyperactivity (sweating or pulse greater than 100 beats per minute); increased hand tremor; insomnia; nausea or vomiting; transient visual, tactile, or auditory hallucinations or illusions; psychomotor agitation; anxiety; grand mal seizure.
- The above symptoms must cause significant distress or functional impairment.
- The symptoms must not be related to another medical condition.
Some of the symptoms described in the second criterion above can occur while the patient still has a measurable blood alcohol level, usually within 6 hours of cessation of drinking.10 Table 1 describes the timetable of onset of symptoms and their severity.2
The elderly may be affected more severely
While the progression of the symptoms described above is commonly used for medical inpatients, the timeline may be different in an elderly patient. Compared with younger patients, elderly patients may have higher blood alcohol concentrations owing to lower total body water, so small amounts of alcohol can produce significant effects.11,12 Brower et al12 found that elderly patients experienced more withdrawal symptoms, especially cognitive impairment, weakness, and high blood pressure, and for 3 days longer.
In the elderly, alcohol may have a greater impact on the central nervous system because of increased permeability of the blood-brain barrier. And importantly, elderly patients tend to have more concomitant diseases and take more medications, all of which can affect alcohol metabolism.
ASSESSMENT SCALES FOR ALCOHOL WITHDRAWAL SYNDROME
A number of clinical scales for evaluating alcohol withdrawal have been developed. Early ones such as the 30-item Total Severity Assessment (TSA) scale and the 11-item Selected Severity Assessment (SSA) scale were limited because they were extremely detailed, burdensome to nursing staff to administer, and contained items such as daily “sleep disturbances” that were not acute enough to meet specific monitoring needs or to guide drug therapy.13,14
The Clinical Institute Withdrawal Assessment for alcohol (CIWA-A) scale, with 15 items, was derived from the SSA scale and includes acute items for assessment as often as every half-hour.15
The CIWA-Ar scale (Table 2) was developed from the CIWA-A scale by Sullivan et al.15 Using both observation and interview, it focuses on 10 areas: nausea and vomiting, tremor, paroxysmal sweats, anxiety, agitation, headache, disorientation, tactile disturbances, auditory disturbances, and visual disturbances. Scores can range from 0 to 67; a higher score indicates worse withdrawal symptoms and outcomes and therefore necessitates escalation of treatment.
The CIWA-Ar scale is now the one most commonly used in clinical trials16–20 and, we believe, in practice. Other scales, including the CIWA-AD and the Alcohol Withdrawal Scale have been validated but are not widely used in practice.14,21
BASELINE ASSESSMENT AND EARLY SUPPORTIVE CARE
A thorough history and physical examination should be performed on admission in patients known to be or suspected of being alcohol-dependent to assess the patient’s affected body systems. The time elapsed since the patient’s last alcohol drink helps predict the onset of withdrawal complications.
Baseline laboratory tests for most patients with suspected alcohol withdrawal syndrome should include a basic blood chemistry panel, complete blood cell count, and possibly an alcohol and toxicology screen, depending on the patient’s history and presentation.
Hydration and nutritional support are important in patients presenting with alcohol withdrawal syndrome. Severe disturbances in electrolytes can lead to serious complications, including cardiac arrhythmia. Close monitoring and electrolyte replacement as needed are recommended for hospitalized alcoholic patients and should follow hospital protocols.22
Thiamine and folic acid status deserve special attention, since long-standing malnutrition is common in alcoholic patients. Thiamine deficiency can result in Wernicke encephalopathy and Korsakoff syndrome, characterized by delirium, ataxia, vision changes, and amnesia. Alcohol withdrawal guidelines recommend giving thiamine intravenously for the first 2 to 5 days after admission.23 In addition, thiamine must be given before any intravenous glucose product, as thiamine is a cofactor in carbohydrate metabolism.23 Folic acid should also be supplemented, as chronic deficiencies may lead to megaloblastic or macrocytic anemia.
CIWA-Ar scale. To provide consistent monitoring and ongoing treatment, clinicians and institutions are encouraged to use a simple assessment scale that detects and quantifies alcohol withdrawal syndrome and that can be used for reassessment after an intervention.21 The CIWA-Ar scale should be used to facilitate “symptom-triggered therapy” in which, depending on the score, the patient receives pharmacologic treatment followed by a scheduled reevaluation.23,24 Most patients with a CIWA-Ar score of 8 or higher benefit from benzodiazepine therapy.16,18,19
PRIMARY DRUG THERAPIES FOR MEDICAL INPATIENTS
Benzodiazepines are the first-line agents
Benzodiazepines are the first-line agents recommended for preventing and treating alcohol withdrawal syndrome.23 Their various pharmacokinetic profiles, wide therapeutic indices, and safety compared with older sedative hypnotics make them the preferred class.23,25 No single benzodiazepine is preferred over the others for treating alcohol withdrawal syndrome: studies have shown benefits using short-acting, intermediate-acting, and long-acting agents. The choice of drug is variable and patient-specific.16,18,26
Benzodiazepines promote and enhance binding of the inhibitory neurotransmitter GABA to GABAA receptors in the central nervous system.27 As a class, benzodiazepines are all structurally related and produce the same effects—namely, sedation, hypnosis, decreased anxiety, muscle relaxation, anterograde amnesia, and anticonvulsant activity.27
The most studied benzodiazepines for treating and preventing alcohol withdrawal syndrome are chlordiazepoxide, oxazepam, and lorazepam,16–20 whereas diazepam was used in older studies.23
Diazepam and chlordiazepoxide are metabolized by oxidation, each sharing the long-acting active metabolite desmethyldiazepam (half-life 72 hours), and short-acting metabolite oxazepam (half-life 8 hours).27 In addition, the parent drugs also have varying pharmacokinetic profiles: diazepam has a half-life of more than 30 hours and chlordiazepoxide a half-life of about 8 hours. Chlordiazepoxide and diazepam’s combination of both long- and short-acting benzodiazepine activity provides long-term efficacy in attenuating withdrawal symptoms, but chlordiazepoxide’s shorter parent half-life allows more frequent dosing.
Lorazepam (half-life 10–20 hours) and oxazepam (half-life 5–20 hours) undergo glucuronide conjugation and do not have metabolites.27,28 Table 3 provides a pharmacokinetic summary.27,28
Various dosage regimens are used in giving benzodiazepines, the most common being symptom-triggered therapy, governed by assessment scales, and scheduled around-the-clock therapy.29 Current evidence supports symptom-triggered therapy in most inpatients who are not critically ill, as it can reduce both benzodiazepine use and adverse drug events and can reduce the length of stay.16,19
Trials of symptom-triggered benzodiazepine therapy
Most inpatient trials of symptom-triggered therapy (Table 4)3,16–20 used the CIWA-Ar scale for monitoring. In some of the studies, benzodiazepines were given if the score was 8 or higher, but others used cut points as high as 15 or higher. Doses:
- Chlordiazepoxide (first dose 25–100 mg)
- Lorazepam (first dose 0.5–2 mg)
- Oxazepam (30 mg).
After each dose, patients were reevaluated at intervals of 30 minutes to 8 hours.
Most of these trials showed no difference in rates of adverse drug events such as seizures, hallucinations, and lethargy with symptom-triggered therapy compared with scheduled therapy.16,18,20 They also found either no difference in the incidence of delirium tremens, or a lower incidence of delirium tremens with symptom-triggered therapy than with scheduled therapy.16–18,20
Weaver et al19 found no difference in length of stay between scheduled therapy and symptom-triggered therapy, but Saitz et al16 reported a median benzodiazepine treatment duration of 9 hours with symptom-triggered therapy vs 68 hours with fixed dosing. Thus, the study by Saitz et al suggests that hospitalization might be shorter with symptom-triggered therapy.
Many of the trials had notable limitations related to the diversity of patients enrolled and the protocols for both symptom-triggered therapy and fixed dosing. Some trials enrolled only inpatients in detoxification programs; others focused on inpatients with acute medical illness. The inpatient alcohol treatment trials16,18 excluded medically ill patients and those with concurrent psychiatric illness,16,18 and one excluded patients with seizures.16 One of the inpatient alcohol treatment program trials16 excluded patients on beta-blockers or clonidine because of concern that these drugs could mask withdrawal symptoms, whereas trials in medically ill patients allowed these same drugs.17,19,20
Most of the patients were men (approximately 75%, but ranging from 74% to 100%), and therefore the study results may not be as applicable to women.16–20 Most participants were middle-aged, with average ages in all studies between 46 and 55. Finally, the studies used a wide range of medications and dosing, with patient monitoring intervals ranging from every 30 minutes to every 8 hours.16–20
In a 2010 Cochrane analysis, Amato et al29 concluded that the limited evidence available favors symptom-triggered regimens over fixed-dosing regimens, but that differences in isolated trials should be interpreted very cautiously.
Therapeutic ethanol
Aside from the lack of evidence to support its use in alcohol withdrawal syndrome, prescribing oral ethanol to alcoholic patients clearly poses an ethical dilemma. However, giving ethanol intravenously has been studied, mostly in surgical trauma patients.30
Early reports described giving intravenous ethanol on a gram-to-gram basis to match the patient’s consumption before admission to prevent alcohol withdrawal syndrome. But later studies reported prevention of alcohol withdrawal syndrome with very small amounts of intravenous ethanol.30,31 While clinical trials have been limited to ICU patients, ethanol infusion at an initial rate of 2.5 to 5 g per hour and titrated up to 10 g per hour has appeared to be safe and effective for preventing alcohol withdrawal syndrome.30,31 The initial infusion rate of 2.5 to 5 g per hour is equivalent to 4 to 10 alcoholic beverages per 24 hours.
Nevertheless, ethanol infusion carries the potential for toxicities (eg, gastric irritation, precipitation of acute hepatic failure, hypoglycemia, pancreatitis, bone marrow suppression, prolonged wound healing) and drug interactions (eg, with anticoagulants and anticonvulsants). Thus, ethanol is neither widely used nor recommended.25,31
ADJUNCTIVE THERAPIES
Many medications are used adjunctively in the acute setting, both for symptoms of alcohol withdrawal syndrome and for agitation.
Haloperidol
No clinical trial has yet examined haloperidol monotherapy in patients with alcohol withdrawal syndrome in either general medical units or intensive care units. Yet haloperidol remains important and is recommended as an adjunct therapy for agitation.23,32 Dosing of haloperidol in protocols for surgical patients ranged from 2 to 5 mg intravenously every 0.5 to 2 hours, with a maximum dosage of 0.5 mg per kg per 24 hours.7,33
Alpha-2 agonists
Alpha-2 agonists are thought to reduce sympathetic overdrive and the autonomic symptoms associated with alcohol withdrawal syndrome, and these agents (primarily clonidine) have been studied in the treatment of alcohol withdrawal syndrome.34,35
Clonidine. In a Swedish study,34 26 men ages 20 to 55 who presented with the tremor, sweating, dysphoria, tension, anxiety, and tachycardia associated with alcohol withdrawal syndrome received clonidine 4 µg per kg twice daily or carbamazepine 200 mg three to four times daily in addition to an antiepileptic. Adjunctive use of a benzodiazepine was allowed at night in both groups. No statistically significant difference in symptom reduction was noted between the two groups, and there was no difference in total benzodiazepine use.
Dexmedetomidine, given intravenously, has been tested as an adjunct to benzodiazepine treatment in severe alcohol withdrawal syndrome. It has been shown to decrease the amount of total benzodiazepine needed compared with benzodiazepine therapy alone, but no differences have been seen in length of hospital stay.36–39 However, research on this drug so far is limited to ICU patients.
Beta-blockers
Beta-blockers have been used in inpatients with alcohol withdrawal syndrome to reduce heart rate and potentially reduce alcohol craving. However, the data are limited and conflicting.
Atenolol 50 to 100 mg daily, in a study in 120 patients, reduced length of stay (4 vs 5 days), reduced benzodiazepine use, and improved vital signs and behavior compared with placebo.40
Propranolol 40 mg every 6 hours reduced arrhythmias but increased hallucinations when used alone in a study in 47 patients.41 When used in combination with chlordiazepoxide, no benefit was seen in arrhythmia reduction.41
Barbiturates and other antiepileptics
Data continue to emerge on antiepileptics as both monotherapy and adjunctive therapy for alcohol withdrawal syndrome. Barbiturates as monotherapy were largely replaced by benzodiazepines in view of the narrow therapeutic index of barbiturates and their full agonist effect on the GABA receptor complex. However, phenobarbital has been evaluated in patients presenting with severe alcohol withdrawal syndrome or resistant alcohol withdrawal (ie, symptoms despite large or repeated doses of benzodiazepines) as an adjunct to benzodiazepines.42,43
In addition, a newer trial44 involved giving a single dose of phenobarbital in the emergency department in combination with a CIWA-Ar–based benzodiazepine protocol, compared with the benzodiazepine protocol alone. The group that received phenobarbital had fewer ICU admissions; its evaluation is ongoing.
The three other medications with the most data are carbamazepine, valproic acid, and gabapentin.45,46 However, the studies were small and the benefit was modest. Although these agents are possible alternatives in protracted alcohol withdrawal syndrome, no definite conclusion can be made regarding their place in therapy.46
RECOMMENDATIONS FOR DRUG THERAPY AND SUPPORTIVE CARE
Which benzodiazepine to use?
No specific benzodiazepine is recommended over the others for managing alcohol withdrawal syndrome, but studies best support the long-acting agent chlordiazepoxide.16,17,20 Other benzodiazepines such as lorazepam and oxazepam have proved to be beneficial, but drugs should be selected on the basis of patient characteristics and drug metabolism.18,19,27
Patients with severe liver dysfunction and the elderly may have slower oxidative metabolism, so the effects of medications that are primarily oxidized, such as chlordiazepoxide and diazepam, may be prolonged. Therefore, lorazepam and oxazepam would be preferred in these groups.47 While most patients with alcohol withdrawal syndrome and liver dysfunction do not have advanced cirrhosis, we recommend liver function testing (serum aspartate aminotransferase, alanine aminotransferase, and alkaline phosphatase levels) and screening for liver disease, given the drug metabolism and package insert caution for use in those with impaired hepatic function.48
Patients with end-stage renal disease (stage 5 chronic kidney disease) or acute kidney injury should not receive parenteral diazepam or lorazepam. The rationale is the potential accumulation of propylene glycol, the solvent used in these formulations.
In the elderly, the Beers list of drugs to avoid in older adults includes benzodiazepines, not differentiating individual benzodiazepines in terms of risk.49 However, chlordiazepoxide may be preferable to diazepam due to its shorter parent half-life and lower lipophilicity.27 Few studies have been done using benzodiazepines in elderly patients with alcohol withdrawal syndrome, but those published have shown either equivalent dosing required compared with younger patients or more severe withdrawal for which they received greater amounts of chlordiazepoxide.9,12 Lorazepam and oxazepam have less potential to accumulate in the elderly compared with the nonelderly due to the drugs’ metabolic profiles; lorazepam is the preferred agent because of its faster onset of action.47 Ultimately, the choice of benzodiazepine in elderly patients with alcohol withdrawal syndrome should be based on patient-specific characteristics.
How should benzodiazepines be dosed?
While the CIWA-Ar thresholds and subsequent dosing of benzodiazepines varied in different studies, we recommend starting benzodiazepine therapy at a CIWA-Ar score of 8 or higher, with subsequent dosing based on score reassessment. Starting doses of benzodiazepines should be chlordiazepoxide 25 to 50 mg, lorazepam 1 to 2 mg, or oxazepam 15 mg.16–20
Subsequent doses should be titrated upward, increasing by 1.5 to 2 times the previous dose and monitored at least every 1 to 2 hours after dose adjustments. Once a patient is stable and the CIWA-Ar score is less than 8, monitoring intervals can be extended to every 4 to 8 hours. If the CIWA-Ar score is more than 20, studies suggest the need for patient reevaluation for transfer to the ICU; however, some health systems have a lower threshold for this intervention.7,14,50
Dosing algorithms and CIWA-Ar goals may vary slightly from institution to institution, but it has been shown that symptom-triggered therapy works best when hospitals have a protocol for it and staff are adequately trained to assess patients with alcohol withdrawal syndrome.7,50,51 Suggestions for dose ranges and symptom-triggered therapy are shown in Table 5.
In case of benzodiazepine overdose or potential benzodiazepine-induced delirium, flumazenil could be considered.52
Patients who should not receive symptom-triggered therapy include immediate postoperative patients in whom clinicians cannot properly assess withdrawal symptoms and patients with a history of DTs.51 While controversy exists regarding the use of symptom-triggered therapy in patients with complicated medical comorbidities, there are data to support symptom-triggered therapy in some ICU patients, as it has resulted in less benzodiazepine use and reduced mechanical ventilation.53,54
There are limited data to support phenobarbital in treating resistant alcohol withdrawal syndrome, either alone or concurrently with benzodiazepines, in escalating doses ranging from 65 to 260 mg, with a maximum daily dose of 520 mg.42,55,56
Haloperidol
For patients exhibiting agitation despite benzodiazepine therapy, giving haloperidol adjunctively can be beneficial.
Haloperidol can be used in medical patients as an adjunctive therapy for agitation, but caution is advised because of the potential for a lowering of the seizure threshold, extrapyramidal effects, and risk of QTc prolongation leading to arrhythmias. Patients considered at highest risk for torsades de pointes may have a QTc of 500 msec or greater.57
Patients should also be screened for factors that have been shown to be independent predictors of QTc prolongation (female sex, diagnosis of myocardial infarction, septic shock or left ventricular dysfunction, other QT-prolonging drugs, age > 68, baseline QTc ≥ 450 msec, and hypokalemia).58 If combined predictors have been identified, it is recommended that haloperidol be avoided.
If haloperidol is to be given, a baseline electrocardiogram and electrolyte panel should be obtained, with daily electrocardiograms thereafter, as well as ongoing review of the patient’s medications to minimize drug interactions that could further increase the risk for QTc prolongation.
Suggested haloperidol dosing is 2 to 5 mg intravenously every 0.5 to 2 hours with a maximum dose of 0.5 mg/kg/24 hours.8,33 A maximum of 35 mg of intravenous haloperidol should be used in a 24-hour period to avoid QTc prolongation.57
Antihypertensive therapy
Many patients receive symptomatic relief of autonomic hyperreactivity with benzodiazepines. However, some may require additional antihypertensive therapy for cardiac adrenergic symptoms (hypertension, tachycardia) if symptoms do not resolve by treating other medical problems commonly seen in patients with alcohol withdrawal syndrome, such as dehydration and electrolyte imbalances.7
Published protocols suggest giving clonidine 0.1 mg orally every hour up to three times as needed until systolic blood pressure is less than 140 mm Hg (less than 160 mm Hg if the patient is over age 60) and diastolic pressure is less than 90 mm Hg.51 Once the patient is stabilized, the dosing can be scheduled to a maximum of 2.4 mg daily.59 However, we believe that the use of clonidine should be restricted to patients who have a substantial increase in blood pressure over baseline or are nearing a hypertensive urgency or emergency (pressures > 180/120 mm Hg) and should not be used to treat other general symptoms associated with alcohol withdrawal syndrome.42
In addition, based on limited evidence, we recommend using beta-blockers only in patients with symptomatic tachycardia or as an adjunct in hypertension management.40,41
Therapies to avoid in acutely ill medical patients
Ethanol is not recommended. Instead, intravenous benzodiazepines should be given in patients presenting with severe alcohol withdrawal syndrome.
Antiepileptics, including valproic acid, carbamazepine, and pregabalin, lack benefit in these patients either as monotherapy or as adjunctive therapy and so are not recommended.45,60–62
Magnesium supplementation (in patients with normal serum magnesium levels) should not be given, as no clinical benefit has been shown.63
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Arlington, VA: American Psychiatric Association, 2013:501.
- Bayard M, McIntyre J, Hill KR, Woodside J Jr. Alcohol withdrawal syndrome. Am Fam Physician 2004; 69:1443–1450.
- Kosten TR, O'Connor PG. Management of drug and alcohol withdrawal. N Engl J Med 2003; 348:1786–1795.
- Isbell H, Fraser HF, Wilker A, Bellevile RE, Eisenman AJ. An experimental study of the etiology of rum fits and delirium tremens. Q J Stud Alcohol 1955; 16:1–33.
- Khan A, Levy P, DeHorn S, Miller W, Compton S. Predictors of mortality in patients with delirium tremens. Acad Emerg Med 2008; 15:788–790.
- Monte R, Rabuñal R, Casariego E, López-Agreda H, Mateos A, Pértega S. Analysis of the factors determining survival of alcoholic withdrawal syndrome patients in a general hospital. Alcohol Alcohol 2010; 45:151–158.
- Stanley KM, Amabile CM, Simpson KN, Couillard D, Norcross ED, Worrall CL. Impact of an alcohol withdrawal syndrome practice guideline on surgical patient outcomes. Pharmacotherapy 2003; 23:843–854.
- Brousse G, Arnaud B, Vorspan F, et al. Alteration of glutamate/GABA balance during acute alcohol withdrawal in emergency department: a prospective analysis. Alcohol Alcohol 2012; 47:501–508.
- Liskow BI, Rinck C, Campbell J, DeSouza C. Alcohol withdrawal in the elderly. J Stud Alcohol 1989; 50:414–421.
- Etherington JM. Emergency management of acute alcohol problems. Part 1: uncomplicated withdrawal. Can Fam Physician 1996; 42:2186–2190.
- Letizia M, Reinbolz M. Identifying and managing acute alcohol withdrawal in the elderly. Geriatr Nurs 2005; 26:176–183.
- Brower KJ, Mudd S, Blow FC, Young JP, Hill EM. Severity and treatment of alcohol withdrawal in elderly versus younger patients. Alcohol Clin Exp Res 1994; 18:196–201.
- Williams D, Lewis J, McBride A. A comparison of rating scales for the alcohol-withdrawal syndrome. Alcohol Alcohol 2001; 36:104–108.
- Reoux JP, Oreskovich MR. A comparison of two versions of the Clinical Institute Withdrawal Assessment for Alcohol: the CIWA-Ar and CIWA-AD. Am J Addict 2006; 15:85–93.
- Sullivan JT, Sykora K, Schneiderman J, Naranjo CA, Sellers EM. Assessment of alcohol withdrawal: the revised Clinical Institute Withdrawal Assessment for alcohol scale (CIWA-Ar). Br J Addict 1989; 84:1353–1357.
- Saitz R, Mayo-Smith MF, Roberts MS, Redmond HA, Bernard DR, Calkins DR. Individualized treatment for alcohol withdrawal. A randomized double-blind controlled trial. JAMA 1994; 272:519–523.
- Jaeger TM, Lohr RH, Pankratz VS. Symptom-triggered therapy for alcohol withdrawal syndrome in medical inpatients. Mayo Clin Proc 2001; 76:695–701.
- Daeppen JB, Gache P, Landry U, et al. Symptom-triggered vs fixed-schedule doses of benzodiazepine for alcohol withdrawal: a randomized treatment trial. Arch Intern Med 2002; 162:1117–1121.
- Weaver MF, Hoffman HJ, Johnson RE, Mauck K. Alcohol withdrawal pharmacotherapy for inpatients with medical comorbidity. J Addict Dis 2006; 25:17–24.
- Reoux JP, Miller K. Routine hospital alcohol detoxification practice compared to symptom triggered management with an Objective Withdrawal Scale (CIWA-Ar). Am J Addict 2000; 9:135–144.
- Wetterling T, Kanitz RD, Besters B, et al. A new rating scale for the assessment of the alcohol-withdrawal syndrome (AWS scale). Alcohol Alcohol 1997; 32:753–760.
- Myrick H, Anton RF. Treatment of alcohol withdrawal. Alcohol Health Res World 1998; 22:38–43.
- Mayo-Smith MF, Beecher LH, Fischer TL, et al; Working Group on the Management of Alcohol Withdrawal Delirium, Practice Guidelines Committee, American Society of Addiction Medicine. Management of alcohol withdrawal delirium. An evidence-based practice guideline. Arch Intern Med 2004; 164:1405–1412.
- Sellers EM, Sullivan JT, Somer G, Sykora K. Characterization of DSM-III-R criteria for uncomplicated alcohol withdrawal provides an empirical basis for DSM-IV. Arch Gen Psychiatry 1991; 48:442–447.
- Sarff M, Gold JA. Alcohol withdrawal syndromes in the intensive care unit. Crit Care Med 2010; 38(suppl):S494–S501.
- Kumar CN, Andrade C, Murthy P. A randomized, double-blind comparison of lorazepam and chlordiazepoxide in patients with uncomplicated alcohol withdrawal. J Stud Alcohol Drugs 2009; 70:467–474.
- Bird RD, Makela EH. Alcohol withdrawal: what is the benzodiazepine of choice? Ann Pharmacother 1994; 28:67–71.
- Perry EC. Inpatient management of acute alcohol withdrawal syndrome. CNS Drugs 2014; 28:401–410.
- Amato L, Minozzi S, Vecchi S, Davoli M. Benzodiazepines for alcohol withdrawal. Cochrane Database Syst Rev 2010; 3:CD005063.
- Weinberg JA, Magnotti LJ, Fischer PE, et al. Comparison of intravenous ethanol versus diazepam for alcohol withdrawal prophylaxis in the trauma ICU: results of a randomized trial. J Trauma 2008; 64:99–104.
- Craft PP, Foil MB, Cunningham PR, Patselas PC, Long-Snyder BM, Collier MS. Intravenous ethanol for alcohol detoxification in trauma patients. South Med J 1994; 87:47–54.
- Ungur LA, Neuner B, John S, Wernecke K, Spies C. Prevention and therapy of alcohol withdrawal on intensive care units: systematic review of controlled trials. Alcohol Clin Exp Res 2013; 37:675–686.
- Lansford CD, Guerriero CH, Kocan MJ, et al. Improved outcomes in patients with head and neck cancer using a standardized care protocol for postoperative alcohol withdrawal. Arch Otolaryngol Head Neck Surg 2008; 134:865–872.
- Walinder J, Balldin J, Bokstrom K, Karlsson I, Lundstrom B, Svensson TH. Clonidine suppression of the alcohol withdrawal syndrome. Drug Alcohol Depend 1981; 8:345–348.
- Muzyk AJ, Fowler JA, Norwood DK, Chilipko A. Role of alpha2-agonists in the treatment of acute alcohol withdrawal. Ann Pharmacother 2011; 45:649–657.
- Crispo AL, Daley MJ, Pepin JL, Harford PH, Brown CV. Comparison of clinical outcomes in nonintubated patients with severe alcohol withdrawal syndrome treated with continuous-infusion sedatives: dexmedetomidine versus benzodiazepines. Pharmacotherapy 2014; 34:910–917.
- VanderWeide LA, Foster CJ, MacLaren R, Kiser TH, Fish DN, Mueller SW. Evaluation of early dexmedetomidine addition to the standard of care for severe alcohol withdrawal in the ICU: a retrospective controlled cohort study. J Intensive Care Med 2014. [Epub ahead of print October 16, 2014]
- Rayner SG, Weinert CR, Peng H, Jepsen S, Broccard AF. Dexmedetomidine as adjunct treatment for severe alcohol withdrawal in the ICU. Ann Intensive Care 2012; 2:12.
- Muzyk AJ, Kerns S, Brudney S, Gagliardi JP. Dexmedetomidine for the treatment of alcohol withdrawal syndrome: rationale and current status of research. CNS Drugs 2013; 27:913–920.
- Kraus ML, Gottlieb LD, Horwitz RI, Anscher M. Randomized clinical trial of atenolol in patients with alcohol withdrawal. N Engl J Med 1985; 313:905–909.
- Zilm DH, Jacob MS, MacLeod SM, Sellers EM, Ti TY. Propranolol and chlordiazepoxide effects on cardiac arrhythmias during alcohol withdrawal. Alcohol Clin Exp Res 1980; 4:400–405.
- Hack JB, Hoffmann RS, Nelson LS. Resistant alcohol withdrawal: does an unexpectedly large sedative requirement identify these patients early? J Med Toxicol 2006; 2:55–60.
- Hayner CE, Wuestefeld NL, Bolton PJ. Phenobarbital treatment in a patient with resistant alcohol withdrawal syndrome. Pharmacotherapy 2009; 29:875–878.
- Rosenson J, Clements C, Simon B, et al. Phenobarbital for acute alcohol withdrawal: a prospective randomized double-blind placebo-controlled study. J Emerg Med 2013; 44:592–598.e2.
- Prince V, Turpin KR. Treatment of alcohol withdrawal syndrome with carbamazepine, gabapentin, and nitrous oxide. Am J Health Syst Pharm 2008; 65:1039–1047.
- Leggio L, Kenna GA, Swift RM. New developments for the pharmacological treatment of alcohol withdrawal syndrome. A focus on non-benzodiazepine GABAergic medications. Prog Neuropsychopharmacol Biol Psychiatry 2008; 32:1106–1117.
- Peppers MP. Benzodiazepines for alcohol withdrawal in the elderly and in patients with liver disease. Pharmacotherapy 1996; 16:49–57.
- Valeant Pharmaceuticals North America LLC. Librium—chlordiazepoxide hydrochloride capsule, gelatin coated. http://dailymed.nlm.nih.gov/dailymed/archives/fdaDrugInfo.cfm?archiveid=125207. Accessed November 20, 2015.
- American Geriatrics Society 2012 Beers Criteria Update Expert Panel. American Geriatrics Society updated Beers Criteria for potentially inappropriate medication use in older adults. J Am Geriatr Soc 2012; 60:616–631.
- Hecksel KA, Bostwick JM, Jaeger TM, Cha SS. Inappropriate use of symptom-triggered therapy for alcohol withdrawal in the general hospital. Mayo Clin Proc 2008; 83:274–279.
- Manasco A, Chang S, Larriviere J, Hamm LL, Glass M. Alcohol withdrawal. South Med J 2012; 105:607–612.
- Moore PW, Donovan JW, Burkhart KK, et al. Safety and efficacy of flumazenil for reversal of iatrogenic benzodiazepine-associated delirium toxicity during treatment of alcohol withdrawal, a retrospective review at one center. J Med Toxicol 2014; 10:126–132.
- Bostwick JM, Lapid MI. False positives on the clinical institute withdrawal assessment for alcohol-revised: is this scale appropriate for use in the medically ill? Psychosomatics 2004; 45:256–261.
- de Wit M, Jones DG, Sessler CN, Zilberberg MD, Weaver MF. Alcohol-use disorders in the critically ill patient. Chest 2010; 138:994–1003.
- Young GP, Rores C, Murphy C, Dailey RH. Intravenous phenobarbital for alcohol withdrawal and convulsions. Ann Emerg Med 1987; 16:847–850.
- Hendey GW, Dery RA, Barnes RL, Snowden B, Mentler P. A prospective, randomized trial of phenobarbital versus benzodiazepines for acute alcohol withdrawal. Am J Emerg Med 2011; 29:382–385.
- Sharma ND, Rosman HS, Padhi ID, Tisdale JE. Torsades de pointes associated with intravenous haloperidol in critically ill patients. Am J Cardiol 1998; 81:238–240.
- Tisdale JE, Jaynes HA, Kingery JR, et al. Development and validation of a risk score to predict QT interval prolongation in hospitalized patients. Circ Cardiovasc Qual Outcomes 2013; 6:479–487.
- Boehringer Ingelheim Pharmaceuticals, Inc. Product Information: Catapres oral tablets, clonidine HCl oral tablets, 2012.
- Reoux JP, Saxon AJ, Malte CA, Baer JS, Sloan KL. Divalproex sodium in alcohol withdrawal: a randomized double-blind placebo-controlled clinical trial. Alcohol Clin Exp Res 2001; 25:1324–1329.
- Malcolm R, Ballenger JC, Sturgis ET, Anton R. Double-blind controlled trial comparing carbamazepine to oxazepam treatment of alcohol withdrawal. Am J Psychiatry 1989; 146:617–621.
- Förg A, Hein J, Volkmar K, et al. Efficacy and safety of pregabalin in the treatment of alcohol withdrawal syndrome: a randomized placebo-controlled trial. Alcohol Alcohol 2012; 47:149–155.
- Wilson A, Vulcano B. A double-blind, placebo-controlled trial of magnesium sulfate in the ethanol withdrawal syndrome. Alcohol Clin Exp Res 1984; 8:542–545.
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Arlington, VA: American Psychiatric Association, 2013:501.
- Bayard M, McIntyre J, Hill KR, Woodside J Jr. Alcohol withdrawal syndrome. Am Fam Physician 2004; 69:1443–1450.
- Kosten TR, O'Connor PG. Management of drug and alcohol withdrawal. N Engl J Med 2003; 348:1786–1795.
- Isbell H, Fraser HF, Wilker A, Bellevile RE, Eisenman AJ. An experimental study of the etiology of rum fits and delirium tremens. Q J Stud Alcohol 1955; 16:1–33.
- Khan A, Levy P, DeHorn S, Miller W, Compton S. Predictors of mortality in patients with delirium tremens. Acad Emerg Med 2008; 15:788–790.
- Monte R, Rabuñal R, Casariego E, López-Agreda H, Mateos A, Pértega S. Analysis of the factors determining survival of alcoholic withdrawal syndrome patients in a general hospital. Alcohol Alcohol 2010; 45:151–158.
- Stanley KM, Amabile CM, Simpson KN, Couillard D, Norcross ED, Worrall CL. Impact of an alcohol withdrawal syndrome practice guideline on surgical patient outcomes. Pharmacotherapy 2003; 23:843–854.
- Brousse G, Arnaud B, Vorspan F, et al. Alteration of glutamate/GABA balance during acute alcohol withdrawal in emergency department: a prospective analysis. Alcohol Alcohol 2012; 47:501–508.
- Liskow BI, Rinck C, Campbell J, DeSouza C. Alcohol withdrawal in the elderly. J Stud Alcohol 1989; 50:414–421.
- Etherington JM. Emergency management of acute alcohol problems. Part 1: uncomplicated withdrawal. Can Fam Physician 1996; 42:2186–2190.
- Letizia M, Reinbolz M. Identifying and managing acute alcohol withdrawal in the elderly. Geriatr Nurs 2005; 26:176–183.
- Brower KJ, Mudd S, Blow FC, Young JP, Hill EM. Severity and treatment of alcohol withdrawal in elderly versus younger patients. Alcohol Clin Exp Res 1994; 18:196–201.
- Williams D, Lewis J, McBride A. A comparison of rating scales for the alcohol-withdrawal syndrome. Alcohol Alcohol 2001; 36:104–108.
- Reoux JP, Oreskovich MR. A comparison of two versions of the Clinical Institute Withdrawal Assessment for Alcohol: the CIWA-Ar and CIWA-AD. Am J Addict 2006; 15:85–93.
- Sullivan JT, Sykora K, Schneiderman J, Naranjo CA, Sellers EM. Assessment of alcohol withdrawal: the revised Clinical Institute Withdrawal Assessment for alcohol scale (CIWA-Ar). Br J Addict 1989; 84:1353–1357.
- Saitz R, Mayo-Smith MF, Roberts MS, Redmond HA, Bernard DR, Calkins DR. Individualized treatment for alcohol withdrawal. A randomized double-blind controlled trial. JAMA 1994; 272:519–523.
- Jaeger TM, Lohr RH, Pankratz VS. Symptom-triggered therapy for alcohol withdrawal syndrome in medical inpatients. Mayo Clin Proc 2001; 76:695–701.
- Daeppen JB, Gache P, Landry U, et al. Symptom-triggered vs fixed-schedule doses of benzodiazepine for alcohol withdrawal: a randomized treatment trial. Arch Intern Med 2002; 162:1117–1121.
- Weaver MF, Hoffman HJ, Johnson RE, Mauck K. Alcohol withdrawal pharmacotherapy for inpatients with medical comorbidity. J Addict Dis 2006; 25:17–24.
- Reoux JP, Miller K. Routine hospital alcohol detoxification practice compared to symptom triggered management with an Objective Withdrawal Scale (CIWA-Ar). Am J Addict 2000; 9:135–144.
- Wetterling T, Kanitz RD, Besters B, et al. A new rating scale for the assessment of the alcohol-withdrawal syndrome (AWS scale). Alcohol Alcohol 1997; 32:753–760.
- Myrick H, Anton RF. Treatment of alcohol withdrawal. Alcohol Health Res World 1998; 22:38–43.
- Mayo-Smith MF, Beecher LH, Fischer TL, et al; Working Group on the Management of Alcohol Withdrawal Delirium, Practice Guidelines Committee, American Society of Addiction Medicine. Management of alcohol withdrawal delirium. An evidence-based practice guideline. Arch Intern Med 2004; 164:1405–1412.
- Sellers EM, Sullivan JT, Somer G, Sykora K. Characterization of DSM-III-R criteria for uncomplicated alcohol withdrawal provides an empirical basis for DSM-IV. Arch Gen Psychiatry 1991; 48:442–447.
- Sarff M, Gold JA. Alcohol withdrawal syndromes in the intensive care unit. Crit Care Med 2010; 38(suppl):S494–S501.
- Kumar CN, Andrade C, Murthy P. A randomized, double-blind comparison of lorazepam and chlordiazepoxide in patients with uncomplicated alcohol withdrawal. J Stud Alcohol Drugs 2009; 70:467–474.
- Bird RD, Makela EH. Alcohol withdrawal: what is the benzodiazepine of choice? Ann Pharmacother 1994; 28:67–71.
- Perry EC. Inpatient management of acute alcohol withdrawal syndrome. CNS Drugs 2014; 28:401–410.
- Amato L, Minozzi S, Vecchi S, Davoli M. Benzodiazepines for alcohol withdrawal. Cochrane Database Syst Rev 2010; 3:CD005063.
- Weinberg JA, Magnotti LJ, Fischer PE, et al. Comparison of intravenous ethanol versus diazepam for alcohol withdrawal prophylaxis in the trauma ICU: results of a randomized trial. J Trauma 2008; 64:99–104.
- Craft PP, Foil MB, Cunningham PR, Patselas PC, Long-Snyder BM, Collier MS. Intravenous ethanol for alcohol detoxification in trauma patients. South Med J 1994; 87:47–54.
- Ungur LA, Neuner B, John S, Wernecke K, Spies C. Prevention and therapy of alcohol withdrawal on intensive care units: systematic review of controlled trials. Alcohol Clin Exp Res 2013; 37:675–686.
- Lansford CD, Guerriero CH, Kocan MJ, et al. Improved outcomes in patients with head and neck cancer using a standardized care protocol for postoperative alcohol withdrawal. Arch Otolaryngol Head Neck Surg 2008; 134:865–872.
- Walinder J, Balldin J, Bokstrom K, Karlsson I, Lundstrom B, Svensson TH. Clonidine suppression of the alcohol withdrawal syndrome. Drug Alcohol Depend 1981; 8:345–348.
- Muzyk AJ, Fowler JA, Norwood DK, Chilipko A. Role of alpha2-agonists in the treatment of acute alcohol withdrawal. Ann Pharmacother 2011; 45:649–657.
- Crispo AL, Daley MJ, Pepin JL, Harford PH, Brown CV. Comparison of clinical outcomes in nonintubated patients with severe alcohol withdrawal syndrome treated with continuous-infusion sedatives: dexmedetomidine versus benzodiazepines. Pharmacotherapy 2014; 34:910–917.
- VanderWeide LA, Foster CJ, MacLaren R, Kiser TH, Fish DN, Mueller SW. Evaluation of early dexmedetomidine addition to the standard of care for severe alcohol withdrawal in the ICU: a retrospective controlled cohort study. J Intensive Care Med 2014. [Epub ahead of print October 16, 2014]
- Rayner SG, Weinert CR, Peng H, Jepsen S, Broccard AF. Dexmedetomidine as adjunct treatment for severe alcohol withdrawal in the ICU. Ann Intensive Care 2012; 2:12.
- Muzyk AJ, Kerns S, Brudney S, Gagliardi JP. Dexmedetomidine for the treatment of alcohol withdrawal syndrome: rationale and current status of research. CNS Drugs 2013; 27:913–920.
- Kraus ML, Gottlieb LD, Horwitz RI, Anscher M. Randomized clinical trial of atenolol in patients with alcohol withdrawal. N Engl J Med 1985; 313:905–909.
- Zilm DH, Jacob MS, MacLeod SM, Sellers EM, Ti TY. Propranolol and chlordiazepoxide effects on cardiac arrhythmias during alcohol withdrawal. Alcohol Clin Exp Res 1980; 4:400–405.
- Hack JB, Hoffmann RS, Nelson LS. Resistant alcohol withdrawal: does an unexpectedly large sedative requirement identify these patients early? J Med Toxicol 2006; 2:55–60.
- Hayner CE, Wuestefeld NL, Bolton PJ. Phenobarbital treatment in a patient with resistant alcohol withdrawal syndrome. Pharmacotherapy 2009; 29:875–878.
- Rosenson J, Clements C, Simon B, et al. Phenobarbital for acute alcohol withdrawal: a prospective randomized double-blind placebo-controlled study. J Emerg Med 2013; 44:592–598.e2.
- Prince V, Turpin KR. Treatment of alcohol withdrawal syndrome with carbamazepine, gabapentin, and nitrous oxide. Am J Health Syst Pharm 2008; 65:1039–1047.
- Leggio L, Kenna GA, Swift RM. New developments for the pharmacological treatment of alcohol withdrawal syndrome. A focus on non-benzodiazepine GABAergic medications. Prog Neuropsychopharmacol Biol Psychiatry 2008; 32:1106–1117.
- Peppers MP. Benzodiazepines for alcohol withdrawal in the elderly and in patients with liver disease. Pharmacotherapy 1996; 16:49–57.
- Valeant Pharmaceuticals North America LLC. Librium—chlordiazepoxide hydrochloride capsule, gelatin coated. http://dailymed.nlm.nih.gov/dailymed/archives/fdaDrugInfo.cfm?archiveid=125207. Accessed November 20, 2015.
- American Geriatrics Society 2012 Beers Criteria Update Expert Panel. American Geriatrics Society updated Beers Criteria for potentially inappropriate medication use in older adults. J Am Geriatr Soc 2012; 60:616–631.
- Hecksel KA, Bostwick JM, Jaeger TM, Cha SS. Inappropriate use of symptom-triggered therapy for alcohol withdrawal in the general hospital. Mayo Clin Proc 2008; 83:274–279.
- Manasco A, Chang S, Larriviere J, Hamm LL, Glass M. Alcohol withdrawal. South Med J 2012; 105:607–612.
- Moore PW, Donovan JW, Burkhart KK, et al. Safety and efficacy of flumazenil for reversal of iatrogenic benzodiazepine-associated delirium toxicity during treatment of alcohol withdrawal, a retrospective review at one center. J Med Toxicol 2014; 10:126–132.
- Bostwick JM, Lapid MI. False positives on the clinical institute withdrawal assessment for alcohol-revised: is this scale appropriate for use in the medically ill? Psychosomatics 2004; 45:256–261.
- de Wit M, Jones DG, Sessler CN, Zilberberg MD, Weaver MF. Alcohol-use disorders in the critically ill patient. Chest 2010; 138:994–1003.
- Young GP, Rores C, Murphy C, Dailey RH. Intravenous phenobarbital for alcohol withdrawal and convulsions. Ann Emerg Med 1987; 16:847–850.
- Hendey GW, Dery RA, Barnes RL, Snowden B, Mentler P. A prospective, randomized trial of phenobarbital versus benzodiazepines for acute alcohol withdrawal. Am J Emerg Med 2011; 29:382–385.
- Sharma ND, Rosman HS, Padhi ID, Tisdale JE. Torsades de pointes associated with intravenous haloperidol in critically ill patients. Am J Cardiol 1998; 81:238–240.
- Tisdale JE, Jaynes HA, Kingery JR, et al. Development and validation of a risk score to predict QT interval prolongation in hospitalized patients. Circ Cardiovasc Qual Outcomes 2013; 6:479–487.
- Boehringer Ingelheim Pharmaceuticals, Inc. Product Information: Catapres oral tablets, clonidine HCl oral tablets, 2012.
- Reoux JP, Saxon AJ, Malte CA, Baer JS, Sloan KL. Divalproex sodium in alcohol withdrawal: a randomized double-blind placebo-controlled clinical trial. Alcohol Clin Exp Res 2001; 25:1324–1329.
- Malcolm R, Ballenger JC, Sturgis ET, Anton R. Double-blind controlled trial comparing carbamazepine to oxazepam treatment of alcohol withdrawal. Am J Psychiatry 1989; 146:617–621.
- Förg A, Hein J, Volkmar K, et al. Efficacy and safety of pregabalin in the treatment of alcohol withdrawal syndrome: a randomized placebo-controlled trial. Alcohol Alcohol 2012; 47:149–155.
- Wilson A, Vulcano B. A double-blind, placebo-controlled trial of magnesium sulfate in the ethanol withdrawal syndrome. Alcohol Clin Exp Res 1984; 8:542–545.
KEY POINTS
- Patients diagnosed with or suspected of having alcohol withdrawal syndrome need a thorough history and physical examination, appropriate laboratory tests, and monitoring using the revised Clinical Institute Withdrawal Assessment for Alcohol scale (CIWA-Ar) or a similar scale.
- For most patients, benzodiazepines should be given in a symptom-triggered fashion, using the CIWA-Ar score as a monitoring tool. Alternatively, scheduled benzodiazepine dosing should be considered for patients with a history of alcohol withdrawal delirium or for patients in whom withdrawal symptoms cannot be easily assessed.
- The choice of benzodiazepine should be individualized, based on the half-life of the drug, comorbid diseases, and monitoring plans.
- Many patients with alcohol withdrawal syndrome require fluid and electrolyte replacement, as well as adjunctive therapies such as haloperidol for delirium and antihypertensives for cardiac or adrenergic symptoms. No standard currently exists for drug dosing, administration, and assessment protocols in these patients. Therefore, clinicians are adapting study designs and assessment scales to meet patients’ individual needs.

Autoantibody-mediated encephalitis: Not just paraneoplastic, not just limbic, and not untreatable
A 79-year-old woman with a history of breast cancer in remission and hypertension presented to a local emergency department because of subacute memory loss and compulsive shopping. Her serum sodium concentration was 127 mmol/L (reference range 132–148). Computed tomography (CT) and magnetic resonance imaging (MRI) of the brain were normal, and she was sent home.
Three days later, she experienced a generalized tonic-clonic seizure that evolved into status epilepticus. She was intubated and admitted to the intensive care unit. Cerebrospinal fluid analysis was normal, and infectious causes of encephalitis were ruled out. MRI showed increased signal in both hippocampi (Figure 1). Her seizures were refractory to treatment, and she was given pentobarbital to induce a coma.
Serum evaluation of neuronal antibodies revealed elevated titers of the voltage-gated potassium channel (VGKC) complex antibody, with subsequent subtyping confirming the leucine-rich glioma-inactivated protein 1 (LGI1) protein as the antigenic target.

She received a 5-day course of intravenous immunoglobulin and methylprednisolone, pentobarbital was withdrawn, and the seizures did not recur, but weeks later she remained comatose. Positron emission tomography (PET) of the brain revealed hypermetabolism in the medial and anterior aspects of both temporal lobes. She underwent five sessions of plasma exchange, after which she began to improve and follow commands. She was ultimately discharged to an acute rehabilitation facility after a 4-week hospital stay.
She received infusions of intravenous immunoglobulin twice a month for 6 months. At her last follow-up visit, she was seizure-free and neurologically intact except for mild inattention.
NEWLY RECOGNIZED DISEASES
Although autoantibody-mediated encephalitic syndromes were first described more than 50 years ago,1,2 their autoimmune basis was not recognized until the early 1980s.3 In the past 10 years, a flood of novel clinical syndromes associated with neuronal autoantibodies has been described that may be markedly improved or even completely resolved with immunotherapy. In cases of unexplained seizure, encephalitis, or acute-onset psychiatric syndromes, suspecting these syndromes can lead to diagnosis, treatment, and a good outcome.
This review describes the key clinical autoantibody-mediated encephalitic syndromes, explains the better-characterized antibody associations, and discusses their diagnosis and treatment.
CLASSIFIED ANATOMICALLY, IMMUNOLOGICALLY, OR EPONYMOUSLY
Autoantibody-mediated encephalitis is also known as autoimmune-mediated encephalitis, autoimmune-mediated limbic encephalitis, and autoimmune synaptic encephalitis.
How to categorize these syndromes is still in flux: they can be listed by the area of the brain affected, the antibody involved, or the name of the discoverer (eg, Morvan syndrome).
Autoantibodies identified in autoimmune encephalitis fall under two broad categories:
- Those targeting intracellular (intranuclear or intracytoplasmic) antigens; the syndromes they cause are more likely to be paraneoplastic and less responsive to immunotherapy
- Those targeting antigens on the neuronal surface: the syndromes they cause are less likely to be paraneoplastic and are more responsive to immunotherapy.4
SYNDROMES DEFINED BY BRAIN AREA AFFECTED
Below, we provide examples of neurologic syndromes of autoantibody-mediated encephalitis according to the region of the brain most affected, ie, the limbic system, the brainstem, or the cerebellum (Figure 2).
LIMBIC ENCEPHALITIS
Memory loss, behavioral changes, seizures
Patients with limbic encephalitis (such as the patient described in the vignette above) present with symptoms attributed to dysfunction of mesial temporal lobe structures, most notably the hippocampus. Prominent symptoms include short-term memory loss, behavioral disturbances such as agitation and confusion, and psychiatric problems such as depression and psychosis. Recurrent seizures are a salient feature and, not uncommonly, progress to status epilepticus.
Antibodies are not all cancer-associated
Cerebrospinal fluid analysis can be normal or show abnormalities suggesting immune activation, eg, slight pleocytosis, elevated protein, increased immunoglobulin G synthesis, and oligoclonal banding.5
In many cases, an autoantibody is found in the blood or in the cerebrospinal fluid. Some patients may express more than one autoantibody, so the traditional view of “one antibody, one syndrome” is incorrect.
Although initially identified as a rare paraneoplastic disorder, limbic encephalitis sometimes occurs in the absence of malignancy.
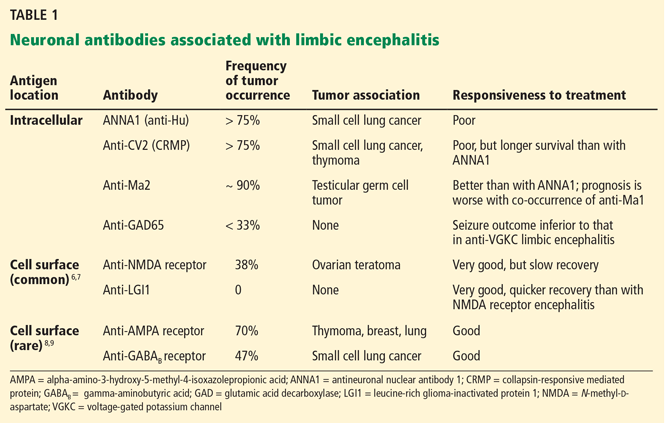
Multiple antibodies have been linked to the syndrome (Table 1).6–9 The “classic” antibodies initially found in paraneoplastic forms are now generally viewed as nonpathogenic, in part because they are directed against intracellular antigens. Neuronal injury in paraneoplastic limbic encephalitis is believed to be mediated by cytotoxic T lymphocytes, with neuronal autoantibodies being produced after the injury.4 Recently defined antibodies, such as those targeting the N-methyl-d-aspartate (NMDA) receptor6 and the LGI1 protein,7 are now understood to be common causes of limbic encephalitis.
Imaging usually shows limbic focal changes
Structural MRI or functional fluorodeoxyglucose (FDG)-PET imaging may show focal changes in limbic system structures, such as the mesial temporal lobes. It is now recognized that other cortical areas may be involved, and the term “limbic encephalitis” may give way to “cortical” or “focal encephalitis.”
In about 60% of patients, MRI shows hyperintense fluid-attenuated inversion recovery (FLAIR) or T2 signal changes in the mesial temporal lobes, likely reflecting inflammatory changes.4,10,11 On FDG-PET, hypermetabolism may be observed in the mesial temporal lobes early in the disease despite normal findings on MRI.12 Hypometabolism, either diffuse or localized to the mesial temporal lobes, eventually sets in, likely reflecting cytotoxic injury in the aftermath of prolonged inflammation or seizures.
Consider other causes
Before diagnosing limbic encephalitis, it is essential to evaluate for infectious meningoencephalitis, especially herpes simplex viral encephalitis. Thiamine deficiency (Wernicke encephalopathy), drug intoxication, prion disease, Hashimoto encephalopathy, tumor, and subclinical status epilepticus should also be considered. Some of these conditions are associated with the same neuronal autoantibodies detected in limbic encephalitis. Further complicating the picture, case reports have shown the presence of serum neuronal autoantibodies—VGKC complex13–15 and NMDA-receptor antibodies16,17—in confirmed cases of prion disease. In addition, adequately treated herpes simplex viral encephalitis can precipitate the production of NMDA-receptor antibodies and their characteristic syndrome.18–20
BRAINSTEM ENCEPHALITIS
The brainstem—the midbrain, pons, and medulla—can be affected, either in isolation or more commonly as part of a more widespread autoantibody-mediated encephalitis. Symptoms and signs include eye movement abnormalities, ptosis, dysphagia, dysarthria, ataxia, facial palsy, vertigo, hearing impairment, reduced consciousness, and hypoventilation.21
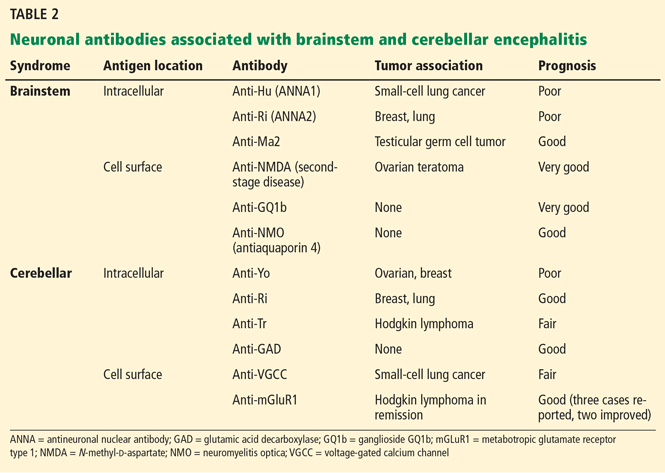
Anti-Hu, anti-Ri, and anti-Ma2 antibodies are most commonly associated with brainstem encephalitis (Table 2). Anti-Ma2-associated encephalitis may improve after a combination of immunotherapy and tumor removal21; the others have a poor prognosis.
Neuromyelitis optica spectrum disorders
Neuromyelitis optica spectrum disorders most commonly involve demyelination affecting the optic nerves and spinal cord, leading to unilateral or bilateral optic neuritis and transverse myelitis spanning three or more vertebral segments.22 The initial clinical manifestation may be an encephalitic pattern, affecting predominantly the brainstem in a restricted fashion,22 or the central nervous system in a more diffuse pattern, mimicking either acute disseminated encephalomyelitis or, in less severe cases, posterior reversible encephalopathy syndrome.23
Testing for antiaquaporin-4 antibody, also known as neuromyelitis optica immunoglobulin G, is the single most decisive laboratory test for diagnosing neuromyelitis optica spectrum disorders, so serum and cerebrospinal fluid evaluation for this autoantibody should be considered when caring for a patient whose clinical picture suggests brainstem encephalitis.22
Bickerstaff brainstem encephalitis
Bickerstaff brainstem encephalitis was first described more than half a century ago in patients with postinfectious ataxia, ophthalmoparesis, and altered consciousness. This rare disease was later found to be associated with antiganglioside GQ1b (anti-GQ1b) autoantibody. MRI is normal in about 90% of cases, so recognizing the clinical presentation and analyzing anti-GQ1b serum titers are critical to diagnosis.
Recovery is usually spontaneous and complete and can be hastened by immunotherapy, especially intravenous immunoglobulin.24
Other causes of brainstem encephalitis
The differential diagnosis of a presentation of brainstem encephalitis includes:
- Infectious causes, the most common being Listeria species followed by enterovirus 71 and herpes simplex virus.25 Tuberculosis, brucellosis, and Whipple disease should also be considered.
- Primary central nervous system inflammatory and demyelinating conditions, eg, multiple sclerosis and acute disseminated encephalomyelitis.
- Systemic inflammatory conditions, eg, Behçet disease, systemic lupus erythematosus, and sarcoidosis.
- Direct brainstem neoplastic involvement, as might occur in primary central nervous system lymphoma or leptomeningeal carcinomatosis.
CEREBELLAR SYNDROME
Patients with autoantibody-mediated encephalitis localized predominantly to the cerebellum typically present with dizziness, vertigo, and unsteady gait, progressing eventually to limb and gait ataxia.4 Symptoms are often subacute, progressing over weeks.
Multiple neuronal autoantibodies have been found to occur with cerebellar encephalitis (Table 2). In most cases, they are paraneoplastic and considered not to be pathogenic, given the intracellular location of their target antigen.4 In such cases, the syndrome is more accurately described as autoantibody-associated rather than autoantibody-mediated. Only in a minority of cases have neuronal autoantibodies been demonstrated to be directly pathogenic, ie, antimetabotropic glutamate receptor type 1 (anti-mGluR1) antibody-associated cerebellitis26 and antiglutamic acid decarboxylase (anti-GAD)-associated cerebellar ataxia.27
Differential diagnosis of cerebellar syndromes
The differential diagnosis of autoantibody-associated cerebellar syndromes is broad and includes:
- Alcohol-induced atrophy
- Drug-induced cerebellar atrophy (eg, from lithium, phenytoin, gabapentin, metronidazole, amiodarone, carbamazepine)
- Vitamin B1 and E deficiency
- Hypothyroidism, hypoparathyroidism
- Neurodegenerative disease (eg, prion disease, multiple system atrophy)
- Parainfectious causes (eg, after infection with Epstein-Barr virus)
- Immune-mediated diseases (Miller-Fisher syndrome, associated with anti-GQ1b antibodies, and antigliadin-associated ataxia, which can occur in isolation or as part of celiac disease).4
SYNDROMES ASSOCIATED WITH SPECIFIC ANTIBODIES
A few of the autoantibody-mediated encephalitic syndromes have specific antibody associations and characteristic clinical presentations. The most prominent of these syndromes are VGKC complex antibody encephalitis (as in the patient described at the beginning of this article) and anti-NMDA receptor encephalitis.
VGKC COMPLEX ANTIBODY-MEDIATED LIMBIC ENCEPHALITIS
VGKC complex antibodies, initially reported to be associated with the peripheral nerve hyperexcitability disorder neuromyotonia, were subsequently found in Morvan syndrome.28,29 Patients with this syndrome often present with autonomic dysfunction and peripheral nerve hyperexcitability but also develop insomnia, confusion, hallucinations, and memory loss. Drawing on the clinical overlap between Morvan syndrome and limbic encephalitis, Buckley et al30 were the first to report VGKC complex antibodies in two cases of limbic encephalitis.
VGKC complex antibodies are now understood to be associated with a wide variety of neurologic conditions, including chronic idiopathic pain, epilepsy,31 movement disorders, cranial nerve abnormalities, autonomic dysfunction,32 and gut dysmotility.33 In contrast, these antibodies are rare in healthy people.34 Limbic encephalitis associated with VGKC complex antibody usually lacks cerebellar and brainstem dysfunction, which may help distinguish it from other types of autoantibody-mediated limbic encephalitis.12
VGKC complex antibody does not bind to the potassium channel itself. Instead it recognizes other constituents of the channel complex, most notably LGI1 and contactin-associated protein 2 (CASPR2). LGI1 antibody is more commonly associated with limbic encephalitis—as illustrated in our case study—in addition to a distinctive type of seizure affecting the arm and face (faciobrachial dystonic seizure).34 The CASPR2 antibody, on the other hand, more often correlates with peripheral nerve manifestations and Morvan syndrome.29 Hyponatremia is commonly seen on serum chemical analysis and provides a clue that these syndromes are present.12
Good response to immunotherapy
A critical change in therapy came as clinicians realized that seizures were often refractory to standard antiepileptic drugs but responded well to immunotherapies. On the basis of these observations, sera of patients with long-standing epilepsy have been reanalyzed to look for neuronal autoantibodies.31 These antibodies should be checked in cases of new-onset refractory status epilepticus of unknown origin that does not respond to antiepileptic medications.
About half of patients with VGKC complex antibody-mediated limbic encephalitis have normal findings on brain MRI.5 Seven of 10 patients who were prospectively followed for VGKC complex antibody-mediated faciobrachial dystonic seizures had normal brain MRIs.35
VGKC complex antibody-mediated limbic encephalitis does not usually recur.36 Most cases are nonparaneoplastic, as evidenced by failure to detect a single active tumor in 64 patients after a median follow-up of 3 years. The prognosis is generally favorable except in cases with coexisting tumors.12
ANTI-NMDA RECEPTOR ENCEPHALITIS
Often associated with ovarian teratoma
Anti-NMDA receptor encephalitis typically affects women in their 20s and 30s, and about half of patients have an ovarian teratoma. It can also occur in younger patients and in men, in whom it is less likely to be associated with a neoplasm.37
Typical initial symptoms include striking and often stereotyped neuropsychiatric disturbances manifesting as psychosis, confusion, seizures, and amnesia. After 1 to 2 weeks, new symptoms set in, including reduced consciousness, movement disorders (ranging from orolingualfacial dyskinesia to rigidity and choreoathetosis), autonomic dysfunction, and hypoventilation, often prompting admission to the intensive care unit.38
Although the outcome is favorable in most cases, recovery, in contrast to VGKC complex antibody-mediated limbic encephalitis, is slow and may take longer than 1 year. Up to a quarter of patients have a relapse, underscoring the importance of maintenance immunotherapy.
It is important to undertake an intensive search for possible ovarian and extraovarian teratomas in young women with this syndrome—including CT of the pelvis, vaginal ultrasonography, and PET imaging—as removal of the teratoma may be curative.37
DIAGNOSIS OF AUTOANTIBODY-MEDIATED ENCEPHALITIS
Critical to diagnosing autoantibody-mediated encephalitis is awareness of these disorders. Since antibody testing may be very specific and is not usually part of the standard batteries of tests, a high level of suspicion is needed. Patients may present to different specialists in different settings; therefore, clinicians in pediatrics, rheumatology, psychiatry, and intensive care medicine need to be aware of these syndromes to avoid delay and misdiagnosis.
Clinical features suggesting autoantibody-mediated encephalitis include:
- Acute or subacute onset of a neurologic syndrome
- New-onset refractory status epilepticus of unknown etiology
- Acute or subacute psychiatric illness with unexpected progression to neurologic symptoms or delirium
- Unusual movement disorders not conforming to standard syndromes
- Cognitive impairment, psychosis, or behavioral or language disorders with atypical findings on imaging or cerebrospinal fluid analysis.
Imaging. Diagnosis of autoantibody-mediated encephalitis focuses on evidence suggesting an inflammatory central nervous system syndrome. MRI may show hyperintense signals on T2, FLAIR, or diffusion-weighted imaging changes in various brain regions. In many cases, however, MRI is negative despite severe clinical symptoms. In a study of 72 patients suspected of having autoimmune dementia of various etiologies, including but not restricted to antineuronal surface antibody-mediated causes, Flanagan et al39 identified atypical neuroimaging findings in only 29%. PET imaging may show hypermetabolism in certain brain areas correlating to clinical syndromes but is often difficult to obtain in a timely fashion.
Cerebrospinal fluid is often abnormal, showing elevated protein, increased immunoglobulin G synthesis, or oligoclonal banding. As with imaging studies, the cerebrospinal fluid may be normal despite severe clinical manifestations.
Electroencephalography may show focal slowing or seizure activity. Neuropsychologic testing may show different patterns of abnormalities.
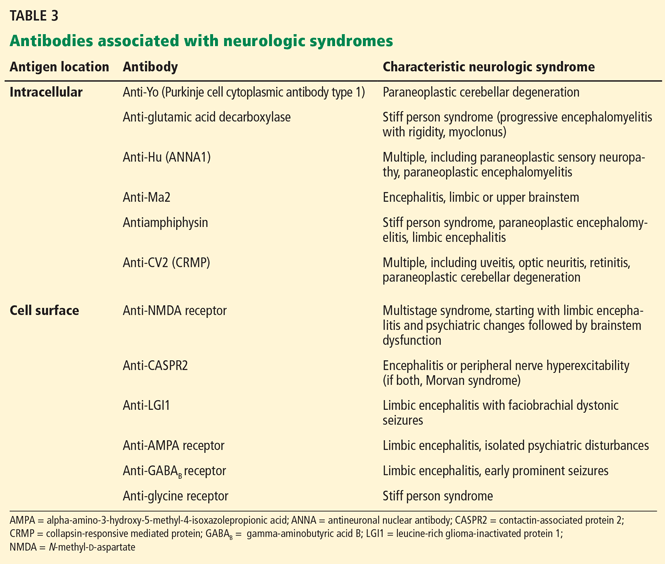
Antibody testing. None of these tests can be used in isolation, and the diagnosis of autoantibody-mediated encephalitis hinges on recognizing a clinical syndrome and ordering supportive testing. Specific antibodies are more likely in different clinical syndromes and should be sought (Table 3).
Patients who have autoantibody-mediated encephalitis may test negative for autoantibodies for many possible reasons:
- Blood testing for antibodies may be less sensitive than cerebrospinal fluid testing
- Antibody titers may vary in the course of the disease
- The patient may be expressing an antibody that is less often tested for (eg, anti-AMPA receptor or antigamma-aminobutyric acid B) or one that has not yet been isolated.
Evaluating for malignancy is recommended in all cases of autoantibody-mediated encephalitis. The initial workup may involve CT of the chest, abdomen, and pelvis, as well as mammography in women and serum prostate-specific antigen testing and testicular ultrasonography in men. Ordering FDG-PET in cases in which CT is negative or inconclusive increases cancer detection.40 If no cancer is found, close tumor surveillance—every 3 to 6 months—is recommended for at least 2 years.41
TREATMENT
Owing in large part to the rarity of autoantibody-mediated encephalitides, no randomized trials of therapy have been performed. Treatment at present is guided mostly by case series and expert consensus, which suggest first-line therapy with intravenous immunoglobulin, high-dose corticosteroids, plasmapheresis, or a combination.
Different syndromes and antibody-related disorders respond differently to therapy. Syndromes associated with antibodies against intracellular antigens tend to be more resistant to immune therapy than cell surface antigen-related syndromes.4
Tiered approach
Combined treatment with intravenous immunoglobulin and high-dose corticosteroids may be superior to treatment with steroids alone for LGI1-antibody mediated limbic encephalitis.42
In cases refractory to first-line (“tier 1”) therapy, second-line immunotherapy with drugs affecting B-cell populations (eg, rituximab, cyclophosphamide, and mycophenolate mofetil) has been used.
A tiered approach has been most extensively studied for anti-NMDA-receptor encephalitis, with better outcomes found using second-line therapy.43
Treatment strategies for these disorders will likely evolve over time with additional experience.
Outpatient management
Once the patient is discharged from the hospital, a multidisciplinary approach to care is recommended, including physical rehabilitation, speech therapy, neuropsychiatric and neuroimmunologic follow-up, and annual surveillance for malignancies.
- Brierley JB, Corsellis JAN, Hierons R, Nevin S. Subacute encephalitis of later adult life mainly affecting the limbic areas. Brain 1960; 83:357–368.
- Corsellis JA, Goldberg GJ, Norton AR. “Limbic encephalitis” and its association with carcinoma. Brain 1968; 91:481–496.
- Greenlee JE, Brashear HR. Antibodies to cerebellar Purkinje cells in patients with paraneoplastic cerebellar degeneration and ovarian carcinoma. Ann Neurol 1983; 14:609–613.
- Rosenfeld MR, Dalmau JO. Paraneoplastic disorders of the CNS and autoimmune synaptic encephalitis. Continuum (Minneap Minn) 2012; 18:366–383.
- Irani SR, Gelfand JM, Al-Diwani A, Vincent A. Cell-surface central nervous system autoantibodies: clinical relevance and emerging paradigms. Ann Neurol 2014; 76:168–184.
- Dalmau J, Tüzün E, Wu HY, et al. Paraneoplastic anti-N-methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol 2007; 61:25–36.
- Irani SR, Alexander S, Waters P, et al. Antibodies to Kv1 potassium channel-complex proteins leucine-rich, glioma inactivated 1 protein and contactin-associated protein-2 in limbic encephalitis, Morvan’s syndrome and acquired neuromyotonia. Brain 2010; 133:2734–2748.
- Lancaster E, Lai M, Peng X, et al. Antibodies to the GABA(B) receptor in limbic encephalitis with seizures: case series and characterisation of the antigen. Lancet Neurol 2010; 9:67–76.
- Lai M, Hughes EG, Peng X, et al. AMPA receptor antibodies in limbic encephalitis alter synaptic receptor location. Ann Neurol 2009; 65:424–434.
- Zuliani L, Graus F, Giometto B, Bien C, Vincent A. Central nervous system neuronal surface antibody associated syndromes: review and guidelines for recognition. J Neurol Neurosurg Psychiatry 2012; 83:638–645.
- Ances BM, Vitaliani R, Taylor RA, et al. Treatment-responsive limbic encephalitis identified by neuropil antibodies: MRI and PET correlates. Brain 2005; 128:1764–1777.
- Vincent A, Buckley C, Schott JM, et al. Potassium channel antibody-associated encephalopathy: a potentially immunotherapy-responsive form of limbic encephalitis. Brain 2004; 127:701–712.
- Jammoul A, Lederman RJ, Tavee J, Li Y. Presence of voltage-gated potassium channel complex antibody in a case of genetic prion disease. BMJ Case Rep 2014; pii:bcr2013201622.
- Angus-Leppan H, Rudge P, Mead S, Collinge J, Vincent A. Autoantibodies in sporadic Creutzfeldt-Jakob disease. JAMA Neurol 2013; 70:919–922.
- Fujita K, Yuasa T, Watanabe O, et al. Voltage-gated potassium channel complex antibodies in Creutzfeldt-Jakob disease. J Neurol 2012; 259:2249–2250.
- Fujita K, Yuasa T, Takahashi Y, et al. Antibodies to N-methyl-D-aspartate glutamate receptors in Creutzfeldt–Jakob disease patients. J Neuroimmunol 2012; 251:90–93.
- Mackay G, Ahmad K, Stone J, et al. NMDA receptor autoantibodies in sporadic Creutzfeldt-Jakob disease. J Neurol 2012; 259:1979–1981.
- Leypoldt F, Titulaer MJ, Aguilar E, et al. Herpes simplex virus–1 encephalitis can trigger anti-NMDA receptor encephalitis: case report. Neurology 2013; 81:1637–1639.
- Desena A, Graves D, Warnack W, Greenberg BM. Herpes simplex encephalitis as a potential cause of anti-N-methyl-D-aspartate receptor antibody encephalitis: report of 2 cases. JAMA Neurol 2014; 71:344–346.
- Armangue T, Leypoldt F, Málaga I, et al. Herpes simplex virus encephalitis is a trigger of brain autoimmunity. Ann Neurol 2014; 75:317–323.
- Blaes F. Paraneoplastic brain stem encephalitis. Curr Treat Options Neurol 2013; 15:201–209.
- Wildemann B, Jarius S. The expanding range of autoimmune disorders of the nervous system. Lancet Neurol 2013; 12:22–24.
- Kim W, Kim SH, Lee SH, Li XF, Kim HJ. Brain abnormalities as an initial manifestation of neuromyelitis optica spectrum disorder. Mult Scler 2011; 17:1107–1112.
- Shahrizaila N, Yuki N. Bickerstaff brainstem encephalitis and Fisher syndrome: anti-GQ1b antibody syndrome. J Neurol Neurosurg Psychiatry 2013; 84:576–583.
- Jubelt B, Mihai C, Li MT, Veerapaneni P. Rhombencephalitis/brainstem encephalitis. Curr Neurol Neurosci Rep 2011; 11:543–552.
- Sillevis Smitt P, Kinoshita A, De Leeuw B, et al. Paraneoplastic cerebellar ataxia due to autoantibodies against a glutamate receptor. N Engl J Med 2000; 342:21–27.
- Ishida K, Mitoma H, Son SY, et al. Selective suppression of cerebellar GABAergic transmission by an autoantibody to glutamic acid decarboxylase. Ann Neurol 1999; 46:263–267.
- Hart IK, Waters C, Vincent A, et al. Autoantibodies detected to expressed K+ channels are implicated in neuromyotonia. Ann Neurol 1997; 41:238–246.
- Barber P, Anderson NE, Vincent A. Morvan’s syndrome associated with voltage-gated K+ channel antibodies. Neurology 2000; 54:771–772.
- Buckley C, Oger J, Clover L, et al. Potassium channel antibodies in two patients with reversible limbic encephalitis. Ann Neurol 2001; 50:73–78.
- Majoie HJ, de Baets M, Renier W, Lang B, Vincent A. Antibodies to voltage-gated potassium and calcium channels in epilepsy. Epilepsy Res 2006; 71:135–141.
- Tan KM, Lennon VA, Klein CJ, Boeve BF, Pittock SJ. Clinical spectrum of voltage-gated potassium channel autoimmunity. Neurology 2008; 70:1883–1890.
- Knowles CH, Lang B, Clover L, et al. A role for autoantibodies in some cases of acquired non-paraneoplastic gut dysmotility. Scand J Gastroenterol 2002; 37:166–170.
- Irani SR, Michell AW, Lang B, et al. Faciobrachial dystonic seizures precede Lgi1 antibody limbic encephalitis. Ann Neurol 2011; 69:892–900.
- Irani SR, Stagg CJ, Schott JM, et al. Faciobrachial dystonic seizures: the influence of immunotherapy on seizure control and prevention of cognitive impairment in a broadening phenotype. Brain 2013: 136:3151–3162.
- Vincent A, Bien CG, Irani SR, Waters P. Autoantibodies associated with diseases of the CNS: new developments and future challenges. Lancet Neurol 2011; 10:759–772.
- Dalmau J, Lancaster E, Martinez-Hernandez E, Rosenfeld MR, Balice-Gordon R. Clinical experience and laboratory investigations in patients with anti-NMDAR encephalitis. Lancet Neurol 2011; 10:63–74.
- Irani SR, Bera K, Waters P, et al. N-methyl-D-aspartate antibody encephalitis: temporal progression of clinical and paraclinical observations in a predominantly non-paraneoplastic disorder of both sexes. Brain 2010; 133:1655–1667.
- Flanagan EP, McKeon A, Lennon VA, et al. Autoimmune dementia: clinical course and predictors of immunotherapy response. Mayo Clin Proc 2010; 85:881–897.
- Younes-Mhenni S, Janier MF, Cinotti L, et al. FDG-PET improves tumour detection in patients with paraneoplastic neurological syndromes. Brain 2004; 127:2331–2338.
- Lancaster E, Martinez-Hernandez E, Dalmau J. Encephalitis and antibodies to synaptic and neuronal cell surface proteins. Neurology 2011; 77:179–189.
- Shin YW, Lee ST, Shin JW, et al. VGKC-complex/LGI1-antibody encephalitis: clinical manifestations and response to immunotherapy. J Neuroimmunol 2013; 265:75–81.
- Titulaer MJ, McCracken L, Gabilondo I, et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet Neurol 2013; 12:157–165.
A 79-year-old woman with a history of breast cancer in remission and hypertension presented to a local emergency department because of subacute memory loss and compulsive shopping. Her serum sodium concentration was 127 mmol/L (reference range 132–148). Computed tomography (CT) and magnetic resonance imaging (MRI) of the brain were normal, and she was sent home.
Three days later, she experienced a generalized tonic-clonic seizure that evolved into status epilepticus. She was intubated and admitted to the intensive care unit. Cerebrospinal fluid analysis was normal, and infectious causes of encephalitis were ruled out. MRI showed increased signal in both hippocampi (Figure 1). Her seizures were refractory to treatment, and she was given pentobarbital to induce a coma.
Serum evaluation of neuronal antibodies revealed elevated titers of the voltage-gated potassium channel (VGKC) complex antibody, with subsequent subtyping confirming the leucine-rich glioma-inactivated protein 1 (LGI1) protein as the antigenic target.
She received a 5-day course of intravenous immunoglobulin and methylprednisolone, pentobarbital was withdrawn, and the seizures did not recur, but weeks later she remained comatose. Positron emission tomography (PET) of the brain revealed hypermetabolism in the medial and anterior aspects of both temporal lobes. She underwent five sessions of plasma exchange, after which she began to improve and follow commands. She was ultimately discharged to an acute rehabilitation facility after a 4-week hospital stay.
She received infusions of intravenous immunoglobulin twice a month for 6 months. At her last follow-up visit, she was seizure-free and neurologically intact except for mild inattention.
NEWLY RECOGNIZED DISEASES
Although autoantibody-mediated encephalitic syndromes were first described more than 50 years ago,1,2 their autoimmune basis was not recognized until the early 1980s.3 In the past 10 years, a flood of novel clinical syndromes associated with neuronal autoantibodies has been described that may be markedly improved or even completely resolved with immunotherapy. In cases of unexplained seizure, encephalitis, or acute-onset psychiatric syndromes, suspecting these syndromes can lead to diagnosis, treatment, and a good outcome.
This review describes the key clinical autoantibody-mediated encephalitic syndromes, explains the better-characterized antibody associations, and discusses their diagnosis and treatment.
CLASSIFIED ANATOMICALLY, IMMUNOLOGICALLY, OR EPONYMOUSLY
Autoantibody-mediated encephalitis is also known as autoimmune-mediated encephalitis, autoimmune-mediated limbic encephalitis, and autoimmune synaptic encephalitis.
How to categorize these syndromes is still in flux: they can be listed by the area of the brain affected, the antibody involved, or the name of the discoverer (eg, Morvan syndrome).
Autoantibodies identified in autoimmune encephalitis fall under two broad categories:
- Those targeting intracellular (intranuclear or intracytoplasmic) antigens; the syndromes they cause are more likely to be paraneoplastic and less responsive to immunotherapy
- Those targeting antigens on the neuronal surface: the syndromes they cause are less likely to be paraneoplastic and are more responsive to immunotherapy.4
SYNDROMES DEFINED BY BRAIN AREA AFFECTED
Below, we provide examples of neurologic syndromes of autoantibody-mediated encephalitis according to the region of the brain most affected, ie, the limbic system, the brainstem, or the cerebellum (Figure 2).
LIMBIC ENCEPHALITIS
Memory loss, behavioral changes, seizures
Patients with limbic encephalitis (such as the patient described in the vignette above) present with symptoms attributed to dysfunction of mesial temporal lobe structures, most notably the hippocampus. Prominent symptoms include short-term memory loss, behavioral disturbances such as agitation and confusion, and psychiatric problems such as depression and psychosis. Recurrent seizures are a salient feature and, not uncommonly, progress to status epilepticus.
Antibodies are not all cancer-associated
Cerebrospinal fluid analysis can be normal or show abnormalities suggesting immune activation, eg, slight pleocytosis, elevated protein, increased immunoglobulin G synthesis, and oligoclonal banding.5
In many cases, an autoantibody is found in the blood or in the cerebrospinal fluid. Some patients may express more than one autoantibody, so the traditional view of “one antibody, one syndrome” is incorrect.
Although initially identified as a rare paraneoplastic disorder, limbic encephalitis sometimes occurs in the absence of malignancy.
Multiple antibodies have been linked to the syndrome (Table 1).6–9 The “classic” antibodies initially found in paraneoplastic forms are now generally viewed as nonpathogenic, in part because they are directed against intracellular antigens. Neuronal injury in paraneoplastic limbic encephalitis is believed to be mediated by cytotoxic T lymphocytes, with neuronal autoantibodies being produced after the injury.4 Recently defined antibodies, such as those targeting the N-methyl-d-aspartate (NMDA) receptor6 and the LGI1 protein,7 are now understood to be common causes of limbic encephalitis.
Imaging usually shows limbic focal changes
Structural MRI or functional fluorodeoxyglucose (FDG)-PET imaging may show focal changes in limbic system structures, such as the mesial temporal lobes. It is now recognized that other cortical areas may be involved, and the term “limbic encephalitis” may give way to “cortical” or “focal encephalitis.”
In about 60% of patients, MRI shows hyperintense fluid-attenuated inversion recovery (FLAIR) or T2 signal changes in the mesial temporal lobes, likely reflecting inflammatory changes.4,10,11 On FDG-PET, hypermetabolism may be observed in the mesial temporal lobes early in the disease despite normal findings on MRI.12 Hypometabolism, either diffuse or localized to the mesial temporal lobes, eventually sets in, likely reflecting cytotoxic injury in the aftermath of prolonged inflammation or seizures.
Consider other causes
Before diagnosing limbic encephalitis, it is essential to evaluate for infectious meningoencephalitis, especially herpes simplex viral encephalitis. Thiamine deficiency (Wernicke encephalopathy), drug intoxication, prion disease, Hashimoto encephalopathy, tumor, and subclinical status epilepticus should also be considered. Some of these conditions are associated with the same neuronal autoantibodies detected in limbic encephalitis. Further complicating the picture, case reports have shown the presence of serum neuronal autoantibodies—VGKC complex13–15 and NMDA-receptor antibodies16,17—in confirmed cases of prion disease. In addition, adequately treated herpes simplex viral encephalitis can precipitate the production of NMDA-receptor antibodies and their characteristic syndrome.18–20
BRAINSTEM ENCEPHALITIS
The brainstem—the midbrain, pons, and medulla—can be affected, either in isolation or more commonly as part of a more widespread autoantibody-mediated encephalitis. Symptoms and signs include eye movement abnormalities, ptosis, dysphagia, dysarthria, ataxia, facial palsy, vertigo, hearing impairment, reduced consciousness, and hypoventilation.21
Anti-Hu, anti-Ri, and anti-Ma2 antibodies are most commonly associated with brainstem encephalitis (Table 2). Anti-Ma2-associated encephalitis may improve after a combination of immunotherapy and tumor removal21; the others have a poor prognosis.
Neuromyelitis optica spectrum disorders
Neuromyelitis optica spectrum disorders most commonly involve demyelination affecting the optic nerves and spinal cord, leading to unilateral or bilateral optic neuritis and transverse myelitis spanning three or more vertebral segments.22 The initial clinical manifestation may be an encephalitic pattern, affecting predominantly the brainstem in a restricted fashion,22 or the central nervous system in a more diffuse pattern, mimicking either acute disseminated encephalomyelitis or, in less severe cases, posterior reversible encephalopathy syndrome.23
Testing for antiaquaporin-4 antibody, also known as neuromyelitis optica immunoglobulin G, is the single most decisive laboratory test for diagnosing neuromyelitis optica spectrum disorders, so serum and cerebrospinal fluid evaluation for this autoantibody should be considered when caring for a patient whose clinical picture suggests brainstem encephalitis.22
Bickerstaff brainstem encephalitis
Bickerstaff brainstem encephalitis was first described more than half a century ago in patients with postinfectious ataxia, ophthalmoparesis, and altered consciousness. This rare disease was later found to be associated with antiganglioside GQ1b (anti-GQ1b) autoantibody. MRI is normal in about 90% of cases, so recognizing the clinical presentation and analyzing anti-GQ1b serum titers are critical to diagnosis.
Recovery is usually spontaneous and complete and can be hastened by immunotherapy, especially intravenous immunoglobulin.24
Other causes of brainstem encephalitis
The differential diagnosis of a presentation of brainstem encephalitis includes:
- Infectious causes, the most common being Listeria species followed by enterovirus 71 and herpes simplex virus.25 Tuberculosis, brucellosis, and Whipple disease should also be considered.
- Primary central nervous system inflammatory and demyelinating conditions, eg, multiple sclerosis and acute disseminated encephalomyelitis.
- Systemic inflammatory conditions, eg, Behçet disease, systemic lupus erythematosus, and sarcoidosis.
- Direct brainstem neoplastic involvement, as might occur in primary central nervous system lymphoma or leptomeningeal carcinomatosis.
CEREBELLAR SYNDROME
Patients with autoantibody-mediated encephalitis localized predominantly to the cerebellum typically present with dizziness, vertigo, and unsteady gait, progressing eventually to limb and gait ataxia.4 Symptoms are often subacute, progressing over weeks.
Multiple neuronal autoantibodies have been found to occur with cerebellar encephalitis (Table 2). In most cases, they are paraneoplastic and considered not to be pathogenic, given the intracellular location of their target antigen.4 In such cases, the syndrome is more accurately described as autoantibody-associated rather than autoantibody-mediated. Only in a minority of cases have neuronal autoantibodies been demonstrated to be directly pathogenic, ie, antimetabotropic glutamate receptor type 1 (anti-mGluR1) antibody-associated cerebellitis26 and antiglutamic acid decarboxylase (anti-GAD)-associated cerebellar ataxia.27
Differential diagnosis of cerebellar syndromes
The differential diagnosis of autoantibody-associated cerebellar syndromes is broad and includes:
- Alcohol-induced atrophy
- Drug-induced cerebellar atrophy (eg, from lithium, phenytoin, gabapentin, metronidazole, amiodarone, carbamazepine)
- Vitamin B1 and E deficiency
- Hypothyroidism, hypoparathyroidism
- Neurodegenerative disease (eg, prion disease, multiple system atrophy)
- Parainfectious causes (eg, after infection with Epstein-Barr virus)
- Immune-mediated diseases (Miller-Fisher syndrome, associated with anti-GQ1b antibodies, and antigliadin-associated ataxia, which can occur in isolation or as part of celiac disease).4
SYNDROMES ASSOCIATED WITH SPECIFIC ANTIBODIES
A few of the autoantibody-mediated encephalitic syndromes have specific antibody associations and characteristic clinical presentations. The most prominent of these syndromes are VGKC complex antibody encephalitis (as in the patient described at the beginning of this article) and anti-NMDA receptor encephalitis.
VGKC COMPLEX ANTIBODY-MEDIATED LIMBIC ENCEPHALITIS
VGKC complex antibodies, initially reported to be associated with the peripheral nerve hyperexcitability disorder neuromyotonia, were subsequently found in Morvan syndrome.28,29 Patients with this syndrome often present with autonomic dysfunction and peripheral nerve hyperexcitability but also develop insomnia, confusion, hallucinations, and memory loss. Drawing on the clinical overlap between Morvan syndrome and limbic encephalitis, Buckley et al30 were the first to report VGKC complex antibodies in two cases of limbic encephalitis.
VGKC complex antibodies are now understood to be associated with a wide variety of neurologic conditions, including chronic idiopathic pain, epilepsy,31 movement disorders, cranial nerve abnormalities, autonomic dysfunction,32 and gut dysmotility.33 In contrast, these antibodies are rare in healthy people.34 Limbic encephalitis associated with VGKC complex antibody usually lacks cerebellar and brainstem dysfunction, which may help distinguish it from other types of autoantibody-mediated limbic encephalitis.12
VGKC complex antibody does not bind to the potassium channel itself. Instead it recognizes other constituents of the channel complex, most notably LGI1 and contactin-associated protein 2 (CASPR2). LGI1 antibody is more commonly associated with limbic encephalitis—as illustrated in our case study—in addition to a distinctive type of seizure affecting the arm and face (faciobrachial dystonic seizure).34 The CASPR2 antibody, on the other hand, more often correlates with peripheral nerve manifestations and Morvan syndrome.29 Hyponatremia is commonly seen on serum chemical analysis and provides a clue that these syndromes are present.12
Good response to immunotherapy
A critical change in therapy came as clinicians realized that seizures were often refractory to standard antiepileptic drugs but responded well to immunotherapies. On the basis of these observations, sera of patients with long-standing epilepsy have been reanalyzed to look for neuronal autoantibodies.31 These antibodies should be checked in cases of new-onset refractory status epilepticus of unknown origin that does not respond to antiepileptic medications.
About half of patients with VGKC complex antibody-mediated limbic encephalitis have normal findings on brain MRI.5 Seven of 10 patients who were prospectively followed for VGKC complex antibody-mediated faciobrachial dystonic seizures had normal brain MRIs.35
VGKC complex antibody-mediated limbic encephalitis does not usually recur.36 Most cases are nonparaneoplastic, as evidenced by failure to detect a single active tumor in 64 patients after a median follow-up of 3 years. The prognosis is generally favorable except in cases with coexisting tumors.12
ANTI-NMDA RECEPTOR ENCEPHALITIS
Often associated with ovarian teratoma
Anti-NMDA receptor encephalitis typically affects women in their 20s and 30s, and about half of patients have an ovarian teratoma. It can also occur in younger patients and in men, in whom it is less likely to be associated with a neoplasm.37
Typical initial symptoms include striking and often stereotyped neuropsychiatric disturbances manifesting as psychosis, confusion, seizures, and amnesia. After 1 to 2 weeks, new symptoms set in, including reduced consciousness, movement disorders (ranging from orolingualfacial dyskinesia to rigidity and choreoathetosis), autonomic dysfunction, and hypoventilation, often prompting admission to the intensive care unit.38
Although the outcome is favorable in most cases, recovery, in contrast to VGKC complex antibody-mediated limbic encephalitis, is slow and may take longer than 1 year. Up to a quarter of patients have a relapse, underscoring the importance of maintenance immunotherapy.
It is important to undertake an intensive search for possible ovarian and extraovarian teratomas in young women with this syndrome—including CT of the pelvis, vaginal ultrasonography, and PET imaging—as removal of the teratoma may be curative.37
DIAGNOSIS OF AUTOANTIBODY-MEDIATED ENCEPHALITIS
Critical to diagnosing autoantibody-mediated encephalitis is awareness of these disorders. Since antibody testing may be very specific and is not usually part of the standard batteries of tests, a high level of suspicion is needed. Patients may present to different specialists in different settings; therefore, clinicians in pediatrics, rheumatology, psychiatry, and intensive care medicine need to be aware of these syndromes to avoid delay and misdiagnosis.
Clinical features suggesting autoantibody-mediated encephalitis include:
- Acute or subacute onset of a neurologic syndrome
- New-onset refractory status epilepticus of unknown etiology
- Acute or subacute psychiatric illness with unexpected progression to neurologic symptoms or delirium
- Unusual movement disorders not conforming to standard syndromes
- Cognitive impairment, psychosis, or behavioral or language disorders with atypical findings on imaging or cerebrospinal fluid analysis.
Imaging. Diagnosis of autoantibody-mediated encephalitis focuses on evidence suggesting an inflammatory central nervous system syndrome. MRI may show hyperintense signals on T2, FLAIR, or diffusion-weighted imaging changes in various brain regions. In many cases, however, MRI is negative despite severe clinical symptoms. In a study of 72 patients suspected of having autoimmune dementia of various etiologies, including but not restricted to antineuronal surface antibody-mediated causes, Flanagan et al39 identified atypical neuroimaging findings in only 29%. PET imaging may show hypermetabolism in certain brain areas correlating to clinical syndromes but is often difficult to obtain in a timely fashion.
Cerebrospinal fluid is often abnormal, showing elevated protein, increased immunoglobulin G synthesis, or oligoclonal banding. As with imaging studies, the cerebrospinal fluid may be normal despite severe clinical manifestations.
Electroencephalography may show focal slowing or seizure activity. Neuropsychologic testing may show different patterns of abnormalities.
Antibody testing. None of these tests can be used in isolation, and the diagnosis of autoantibody-mediated encephalitis hinges on recognizing a clinical syndrome and ordering supportive testing. Specific antibodies are more likely in different clinical syndromes and should be sought (Table 3).
Patients who have autoantibody-mediated encephalitis may test negative for autoantibodies for many possible reasons:
- Blood testing for antibodies may be less sensitive than cerebrospinal fluid testing
- Antibody titers may vary in the course of the disease
- The patient may be expressing an antibody that is less often tested for (eg, anti-AMPA receptor or antigamma-aminobutyric acid B) or one that has not yet been isolated.
Evaluating for malignancy is recommended in all cases of autoantibody-mediated encephalitis. The initial workup may involve CT of the chest, abdomen, and pelvis, as well as mammography in women and serum prostate-specific antigen testing and testicular ultrasonography in men. Ordering FDG-PET in cases in which CT is negative or inconclusive increases cancer detection.40 If no cancer is found, close tumor surveillance—every 3 to 6 months—is recommended for at least 2 years.41
TREATMENT
Owing in large part to the rarity of autoantibody-mediated encephalitides, no randomized trials of therapy have been performed. Treatment at present is guided mostly by case series and expert consensus, which suggest first-line therapy with intravenous immunoglobulin, high-dose corticosteroids, plasmapheresis, or a combination.
Different syndromes and antibody-related disorders respond differently to therapy. Syndromes associated with antibodies against intracellular antigens tend to be more resistant to immune therapy than cell surface antigen-related syndromes.4
Tiered approach
Combined treatment with intravenous immunoglobulin and high-dose corticosteroids may be superior to treatment with steroids alone for LGI1-antibody mediated limbic encephalitis.42
In cases refractory to first-line (“tier 1”) therapy, second-line immunotherapy with drugs affecting B-cell populations (eg, rituximab, cyclophosphamide, and mycophenolate mofetil) has been used.
A tiered approach has been most extensively studied for anti-NMDA-receptor encephalitis, with better outcomes found using second-line therapy.43
Treatment strategies for these disorders will likely evolve over time with additional experience.
Outpatient management
Once the patient is discharged from the hospital, a multidisciplinary approach to care is recommended, including physical rehabilitation, speech therapy, neuropsychiatric and neuroimmunologic follow-up, and annual surveillance for malignancies.
A 79-year-old woman with a history of breast cancer in remission and hypertension presented to a local emergency department because of subacute memory loss and compulsive shopping. Her serum sodium concentration was 127 mmol/L (reference range 132–148). Computed tomography (CT) and magnetic resonance imaging (MRI) of the brain were normal, and she was sent home.
Three days later, she experienced a generalized tonic-clonic seizure that evolved into status epilepticus. She was intubated and admitted to the intensive care unit. Cerebrospinal fluid analysis was normal, and infectious causes of encephalitis were ruled out. MRI showed increased signal in both hippocampi (Figure 1). Her seizures were refractory to treatment, and she was given pentobarbital to induce a coma.
Serum evaluation of neuronal antibodies revealed elevated titers of the voltage-gated potassium channel (VGKC) complex antibody, with subsequent subtyping confirming the leucine-rich glioma-inactivated protein 1 (LGI1) protein as the antigenic target.
She received a 5-day course of intravenous immunoglobulin and methylprednisolone, pentobarbital was withdrawn, and the seizures did not recur, but weeks later she remained comatose. Positron emission tomography (PET) of the brain revealed hypermetabolism in the medial and anterior aspects of both temporal lobes. She underwent five sessions of plasma exchange, after which she began to improve and follow commands. She was ultimately discharged to an acute rehabilitation facility after a 4-week hospital stay.
She received infusions of intravenous immunoglobulin twice a month for 6 months. At her last follow-up visit, she was seizure-free and neurologically intact except for mild inattention.
NEWLY RECOGNIZED DISEASES
Although autoantibody-mediated encephalitic syndromes were first described more than 50 years ago,1,2 their autoimmune basis was not recognized until the early 1980s.3 In the past 10 years, a flood of novel clinical syndromes associated with neuronal autoantibodies has been described that may be markedly improved or even completely resolved with immunotherapy. In cases of unexplained seizure, encephalitis, or acute-onset psychiatric syndromes, suspecting these syndromes can lead to diagnosis, treatment, and a good outcome.
This review describes the key clinical autoantibody-mediated encephalitic syndromes, explains the better-characterized antibody associations, and discusses their diagnosis and treatment.
CLASSIFIED ANATOMICALLY, IMMUNOLOGICALLY, OR EPONYMOUSLY
Autoantibody-mediated encephalitis is also known as autoimmune-mediated encephalitis, autoimmune-mediated limbic encephalitis, and autoimmune synaptic encephalitis.
How to categorize these syndromes is still in flux: they can be listed by the area of the brain affected, the antibody involved, or the name of the discoverer (eg, Morvan syndrome).
Autoantibodies identified in autoimmune encephalitis fall under two broad categories:
- Those targeting intracellular (intranuclear or intracytoplasmic) antigens; the syndromes they cause are more likely to be paraneoplastic and less responsive to immunotherapy
- Those targeting antigens on the neuronal surface: the syndromes they cause are less likely to be paraneoplastic and are more responsive to immunotherapy.4
SYNDROMES DEFINED BY BRAIN AREA AFFECTED
Below, we provide examples of neurologic syndromes of autoantibody-mediated encephalitis according to the region of the brain most affected, ie, the limbic system, the brainstem, or the cerebellum (Figure 2).
LIMBIC ENCEPHALITIS
Memory loss, behavioral changes, seizures
Patients with limbic encephalitis (such as the patient described in the vignette above) present with symptoms attributed to dysfunction of mesial temporal lobe structures, most notably the hippocampus. Prominent symptoms include short-term memory loss, behavioral disturbances such as agitation and confusion, and psychiatric problems such as depression and psychosis. Recurrent seizures are a salient feature and, not uncommonly, progress to status epilepticus.
Antibodies are not all cancer-associated
Cerebrospinal fluid analysis can be normal or show abnormalities suggesting immune activation, eg, slight pleocytosis, elevated protein, increased immunoglobulin G synthesis, and oligoclonal banding.5
In many cases, an autoantibody is found in the blood or in the cerebrospinal fluid. Some patients may express more than one autoantibody, so the traditional view of “one antibody, one syndrome” is incorrect.
Although initially identified as a rare paraneoplastic disorder, limbic encephalitis sometimes occurs in the absence of malignancy.
Multiple antibodies have been linked to the syndrome (Table 1).6–9 The “classic” antibodies initially found in paraneoplastic forms are now generally viewed as nonpathogenic, in part because they are directed against intracellular antigens. Neuronal injury in paraneoplastic limbic encephalitis is believed to be mediated by cytotoxic T lymphocytes, with neuronal autoantibodies being produced after the injury.4 Recently defined antibodies, such as those targeting the N-methyl-d-aspartate (NMDA) receptor6 and the LGI1 protein,7 are now understood to be common causes of limbic encephalitis.
Imaging usually shows limbic focal changes
Structural MRI or functional fluorodeoxyglucose (FDG)-PET imaging may show focal changes in limbic system structures, such as the mesial temporal lobes. It is now recognized that other cortical areas may be involved, and the term “limbic encephalitis” may give way to “cortical” or “focal encephalitis.”
In about 60% of patients, MRI shows hyperintense fluid-attenuated inversion recovery (FLAIR) or T2 signal changes in the mesial temporal lobes, likely reflecting inflammatory changes.4,10,11 On FDG-PET, hypermetabolism may be observed in the mesial temporal lobes early in the disease despite normal findings on MRI.12 Hypometabolism, either diffuse or localized to the mesial temporal lobes, eventually sets in, likely reflecting cytotoxic injury in the aftermath of prolonged inflammation or seizures.
Consider other causes
Before diagnosing limbic encephalitis, it is essential to evaluate for infectious meningoencephalitis, especially herpes simplex viral encephalitis. Thiamine deficiency (Wernicke encephalopathy), drug intoxication, prion disease, Hashimoto encephalopathy, tumor, and subclinical status epilepticus should also be considered. Some of these conditions are associated with the same neuronal autoantibodies detected in limbic encephalitis. Further complicating the picture, case reports have shown the presence of serum neuronal autoantibodies—VGKC complex13–15 and NMDA-receptor antibodies16,17—in confirmed cases of prion disease. In addition, adequately treated herpes simplex viral encephalitis can precipitate the production of NMDA-receptor antibodies and their characteristic syndrome.18–20
BRAINSTEM ENCEPHALITIS
The brainstem—the midbrain, pons, and medulla—can be affected, either in isolation or more commonly as part of a more widespread autoantibody-mediated encephalitis. Symptoms and signs include eye movement abnormalities, ptosis, dysphagia, dysarthria, ataxia, facial palsy, vertigo, hearing impairment, reduced consciousness, and hypoventilation.21
Anti-Hu, anti-Ri, and anti-Ma2 antibodies are most commonly associated with brainstem encephalitis (Table 2). Anti-Ma2-associated encephalitis may improve after a combination of immunotherapy and tumor removal21; the others have a poor prognosis.
Neuromyelitis optica spectrum disorders
Neuromyelitis optica spectrum disorders most commonly involve demyelination affecting the optic nerves and spinal cord, leading to unilateral or bilateral optic neuritis and transverse myelitis spanning three or more vertebral segments.22 The initial clinical manifestation may be an encephalitic pattern, affecting predominantly the brainstem in a restricted fashion,22 or the central nervous system in a more diffuse pattern, mimicking either acute disseminated encephalomyelitis or, in less severe cases, posterior reversible encephalopathy syndrome.23
Testing for antiaquaporin-4 antibody, also known as neuromyelitis optica immunoglobulin G, is the single most decisive laboratory test for diagnosing neuromyelitis optica spectrum disorders, so serum and cerebrospinal fluid evaluation for this autoantibody should be considered when caring for a patient whose clinical picture suggests brainstem encephalitis.22
Bickerstaff brainstem encephalitis
Bickerstaff brainstem encephalitis was first described more than half a century ago in patients with postinfectious ataxia, ophthalmoparesis, and altered consciousness. This rare disease was later found to be associated with antiganglioside GQ1b (anti-GQ1b) autoantibody. MRI is normal in about 90% of cases, so recognizing the clinical presentation and analyzing anti-GQ1b serum titers are critical to diagnosis.
Recovery is usually spontaneous and complete and can be hastened by immunotherapy, especially intravenous immunoglobulin.24
Other causes of brainstem encephalitis
The differential diagnosis of a presentation of brainstem encephalitis includes:
- Infectious causes, the most common being Listeria species followed by enterovirus 71 and herpes simplex virus.25 Tuberculosis, brucellosis, and Whipple disease should also be considered.
- Primary central nervous system inflammatory and demyelinating conditions, eg, multiple sclerosis and acute disseminated encephalomyelitis.
- Systemic inflammatory conditions, eg, Behçet disease, systemic lupus erythematosus, and sarcoidosis.
- Direct brainstem neoplastic involvement, as might occur in primary central nervous system lymphoma or leptomeningeal carcinomatosis.
CEREBELLAR SYNDROME
Patients with autoantibody-mediated encephalitis localized predominantly to the cerebellum typically present with dizziness, vertigo, and unsteady gait, progressing eventually to limb and gait ataxia.4 Symptoms are often subacute, progressing over weeks.
Multiple neuronal autoantibodies have been found to occur with cerebellar encephalitis (Table 2). In most cases, they are paraneoplastic and considered not to be pathogenic, given the intracellular location of their target antigen.4 In such cases, the syndrome is more accurately described as autoantibody-associated rather than autoantibody-mediated. Only in a minority of cases have neuronal autoantibodies been demonstrated to be directly pathogenic, ie, antimetabotropic glutamate receptor type 1 (anti-mGluR1) antibody-associated cerebellitis26 and antiglutamic acid decarboxylase (anti-GAD)-associated cerebellar ataxia.27
Differential diagnosis of cerebellar syndromes
The differential diagnosis of autoantibody-associated cerebellar syndromes is broad and includes:
- Alcohol-induced atrophy
- Drug-induced cerebellar atrophy (eg, from lithium, phenytoin, gabapentin, metronidazole, amiodarone, carbamazepine)
- Vitamin B1 and E deficiency
- Hypothyroidism, hypoparathyroidism
- Neurodegenerative disease (eg, prion disease, multiple system atrophy)
- Parainfectious causes (eg, after infection with Epstein-Barr virus)
- Immune-mediated diseases (Miller-Fisher syndrome, associated with anti-GQ1b antibodies, and antigliadin-associated ataxia, which can occur in isolation or as part of celiac disease).4
SYNDROMES ASSOCIATED WITH SPECIFIC ANTIBODIES
A few of the autoantibody-mediated encephalitic syndromes have specific antibody associations and characteristic clinical presentations. The most prominent of these syndromes are VGKC complex antibody encephalitis (as in the patient described at the beginning of this article) and anti-NMDA receptor encephalitis.
VGKC COMPLEX ANTIBODY-MEDIATED LIMBIC ENCEPHALITIS
VGKC complex antibodies, initially reported to be associated with the peripheral nerve hyperexcitability disorder neuromyotonia, were subsequently found in Morvan syndrome.28,29 Patients with this syndrome often present with autonomic dysfunction and peripheral nerve hyperexcitability but also develop insomnia, confusion, hallucinations, and memory loss. Drawing on the clinical overlap between Morvan syndrome and limbic encephalitis, Buckley et al30 were the first to report VGKC complex antibodies in two cases of limbic encephalitis.
VGKC complex antibodies are now understood to be associated with a wide variety of neurologic conditions, including chronic idiopathic pain, epilepsy,31 movement disorders, cranial nerve abnormalities, autonomic dysfunction,32 and gut dysmotility.33 In contrast, these antibodies are rare in healthy people.34 Limbic encephalitis associated with VGKC complex antibody usually lacks cerebellar and brainstem dysfunction, which may help distinguish it from other types of autoantibody-mediated limbic encephalitis.12
VGKC complex antibody does not bind to the potassium channel itself. Instead it recognizes other constituents of the channel complex, most notably LGI1 and contactin-associated protein 2 (CASPR2). LGI1 antibody is more commonly associated with limbic encephalitis—as illustrated in our case study—in addition to a distinctive type of seizure affecting the arm and face (faciobrachial dystonic seizure).34 The CASPR2 antibody, on the other hand, more often correlates with peripheral nerve manifestations and Morvan syndrome.29 Hyponatremia is commonly seen on serum chemical analysis and provides a clue that these syndromes are present.12
Good response to immunotherapy
A critical change in therapy came as clinicians realized that seizures were often refractory to standard antiepileptic drugs but responded well to immunotherapies. On the basis of these observations, sera of patients with long-standing epilepsy have been reanalyzed to look for neuronal autoantibodies.31 These antibodies should be checked in cases of new-onset refractory status epilepticus of unknown origin that does not respond to antiepileptic medications.
About half of patients with VGKC complex antibody-mediated limbic encephalitis have normal findings on brain MRI.5 Seven of 10 patients who were prospectively followed for VGKC complex antibody-mediated faciobrachial dystonic seizures had normal brain MRIs.35
VGKC complex antibody-mediated limbic encephalitis does not usually recur.36 Most cases are nonparaneoplastic, as evidenced by failure to detect a single active tumor in 64 patients after a median follow-up of 3 years. The prognosis is generally favorable except in cases with coexisting tumors.12
ANTI-NMDA RECEPTOR ENCEPHALITIS
Often associated with ovarian teratoma
Anti-NMDA receptor encephalitis typically affects women in their 20s and 30s, and about half of patients have an ovarian teratoma. It can also occur in younger patients and in men, in whom it is less likely to be associated with a neoplasm.37
Typical initial symptoms include striking and often stereotyped neuropsychiatric disturbances manifesting as psychosis, confusion, seizures, and amnesia. After 1 to 2 weeks, new symptoms set in, including reduced consciousness, movement disorders (ranging from orolingualfacial dyskinesia to rigidity and choreoathetosis), autonomic dysfunction, and hypoventilation, often prompting admission to the intensive care unit.38
Although the outcome is favorable in most cases, recovery, in contrast to VGKC complex antibody-mediated limbic encephalitis, is slow and may take longer than 1 year. Up to a quarter of patients have a relapse, underscoring the importance of maintenance immunotherapy.
It is important to undertake an intensive search for possible ovarian and extraovarian teratomas in young women with this syndrome—including CT of the pelvis, vaginal ultrasonography, and PET imaging—as removal of the teratoma may be curative.37
DIAGNOSIS OF AUTOANTIBODY-MEDIATED ENCEPHALITIS
Critical to diagnosing autoantibody-mediated encephalitis is awareness of these disorders. Since antibody testing may be very specific and is not usually part of the standard batteries of tests, a high level of suspicion is needed. Patients may present to different specialists in different settings; therefore, clinicians in pediatrics, rheumatology, psychiatry, and intensive care medicine need to be aware of these syndromes to avoid delay and misdiagnosis.
Clinical features suggesting autoantibody-mediated encephalitis include:
- Acute or subacute onset of a neurologic syndrome
- New-onset refractory status epilepticus of unknown etiology
- Acute or subacute psychiatric illness with unexpected progression to neurologic symptoms or delirium
- Unusual movement disorders not conforming to standard syndromes
- Cognitive impairment, psychosis, or behavioral or language disorders with atypical findings on imaging or cerebrospinal fluid analysis.
Imaging. Diagnosis of autoantibody-mediated encephalitis focuses on evidence suggesting an inflammatory central nervous system syndrome. MRI may show hyperintense signals on T2, FLAIR, or diffusion-weighted imaging changes in various brain regions. In many cases, however, MRI is negative despite severe clinical symptoms. In a study of 72 patients suspected of having autoimmune dementia of various etiologies, including but not restricted to antineuronal surface antibody-mediated causes, Flanagan et al39 identified atypical neuroimaging findings in only 29%. PET imaging may show hypermetabolism in certain brain areas correlating to clinical syndromes but is often difficult to obtain in a timely fashion.
Cerebrospinal fluid is often abnormal, showing elevated protein, increased immunoglobulin G synthesis, or oligoclonal banding. As with imaging studies, the cerebrospinal fluid may be normal despite severe clinical manifestations.
Electroencephalography may show focal slowing or seizure activity. Neuropsychologic testing may show different patterns of abnormalities.
Antibody testing. None of these tests can be used in isolation, and the diagnosis of autoantibody-mediated encephalitis hinges on recognizing a clinical syndrome and ordering supportive testing. Specific antibodies are more likely in different clinical syndromes and should be sought (Table 3).
Patients who have autoantibody-mediated encephalitis may test negative for autoantibodies for many possible reasons:
- Blood testing for antibodies may be less sensitive than cerebrospinal fluid testing
- Antibody titers may vary in the course of the disease
- The patient may be expressing an antibody that is less often tested for (eg, anti-AMPA receptor or antigamma-aminobutyric acid B) or one that has not yet been isolated.
Evaluating for malignancy is recommended in all cases of autoantibody-mediated encephalitis. The initial workup may involve CT of the chest, abdomen, and pelvis, as well as mammography in women and serum prostate-specific antigen testing and testicular ultrasonography in men. Ordering FDG-PET in cases in which CT is negative or inconclusive increases cancer detection.40 If no cancer is found, close tumor surveillance—every 3 to 6 months—is recommended for at least 2 years.41
TREATMENT
Owing in large part to the rarity of autoantibody-mediated encephalitides, no randomized trials of therapy have been performed. Treatment at present is guided mostly by case series and expert consensus, which suggest first-line therapy with intravenous immunoglobulin, high-dose corticosteroids, plasmapheresis, or a combination.
Different syndromes and antibody-related disorders respond differently to therapy. Syndromes associated with antibodies against intracellular antigens tend to be more resistant to immune therapy than cell surface antigen-related syndromes.4
Tiered approach
Combined treatment with intravenous immunoglobulin and high-dose corticosteroids may be superior to treatment with steroids alone for LGI1-antibody mediated limbic encephalitis.42
In cases refractory to first-line (“tier 1”) therapy, second-line immunotherapy with drugs affecting B-cell populations (eg, rituximab, cyclophosphamide, and mycophenolate mofetil) has been used.
A tiered approach has been most extensively studied for anti-NMDA-receptor encephalitis, with better outcomes found using second-line therapy.43
Treatment strategies for these disorders will likely evolve over time with additional experience.
Outpatient management
Once the patient is discharged from the hospital, a multidisciplinary approach to care is recommended, including physical rehabilitation, speech therapy, neuropsychiatric and neuroimmunologic follow-up, and annual surveillance for malignancies.
- Brierley JB, Corsellis JAN, Hierons R, Nevin S. Subacute encephalitis of later adult life mainly affecting the limbic areas. Brain 1960; 83:357–368.
- Corsellis JA, Goldberg GJ, Norton AR. “Limbic encephalitis” and its association with carcinoma. Brain 1968; 91:481–496.
- Greenlee JE, Brashear HR. Antibodies to cerebellar Purkinje cells in patients with paraneoplastic cerebellar degeneration and ovarian carcinoma. Ann Neurol 1983; 14:609–613.
- Rosenfeld MR, Dalmau JO. Paraneoplastic disorders of the CNS and autoimmune synaptic encephalitis. Continuum (Minneap Minn) 2012; 18:366–383.
- Irani SR, Gelfand JM, Al-Diwani A, Vincent A. Cell-surface central nervous system autoantibodies: clinical relevance and emerging paradigms. Ann Neurol 2014; 76:168–184.
- Dalmau J, Tüzün E, Wu HY, et al. Paraneoplastic anti-N-methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol 2007; 61:25–36.
- Irani SR, Alexander S, Waters P, et al. Antibodies to Kv1 potassium channel-complex proteins leucine-rich, glioma inactivated 1 protein and contactin-associated protein-2 in limbic encephalitis, Morvan’s syndrome and acquired neuromyotonia. Brain 2010; 133:2734–2748.
- Lancaster E, Lai M, Peng X, et al. Antibodies to the GABA(B) receptor in limbic encephalitis with seizures: case series and characterisation of the antigen. Lancet Neurol 2010; 9:67–76.
- Lai M, Hughes EG, Peng X, et al. AMPA receptor antibodies in limbic encephalitis alter synaptic receptor location. Ann Neurol 2009; 65:424–434.
- Zuliani L, Graus F, Giometto B, Bien C, Vincent A. Central nervous system neuronal surface antibody associated syndromes: review and guidelines for recognition. J Neurol Neurosurg Psychiatry 2012; 83:638–645.
- Ances BM, Vitaliani R, Taylor RA, et al. Treatment-responsive limbic encephalitis identified by neuropil antibodies: MRI and PET correlates. Brain 2005; 128:1764–1777.
- Vincent A, Buckley C, Schott JM, et al. Potassium channel antibody-associated encephalopathy: a potentially immunotherapy-responsive form of limbic encephalitis. Brain 2004; 127:701–712.
- Jammoul A, Lederman RJ, Tavee J, Li Y. Presence of voltage-gated potassium channel complex antibody in a case of genetic prion disease. BMJ Case Rep 2014; pii:bcr2013201622.
- Angus-Leppan H, Rudge P, Mead S, Collinge J, Vincent A. Autoantibodies in sporadic Creutzfeldt-Jakob disease. JAMA Neurol 2013; 70:919–922.
- Fujita K, Yuasa T, Watanabe O, et al. Voltage-gated potassium channel complex antibodies in Creutzfeldt-Jakob disease. J Neurol 2012; 259:2249–2250.
- Fujita K, Yuasa T, Takahashi Y, et al. Antibodies to N-methyl-D-aspartate glutamate receptors in Creutzfeldt–Jakob disease patients. J Neuroimmunol 2012; 251:90–93.
- Mackay G, Ahmad K, Stone J, et al. NMDA receptor autoantibodies in sporadic Creutzfeldt-Jakob disease. J Neurol 2012; 259:1979–1981.
- Leypoldt F, Titulaer MJ, Aguilar E, et al. Herpes simplex virus–1 encephalitis can trigger anti-NMDA receptor encephalitis: case report. Neurology 2013; 81:1637–1639.
- Desena A, Graves D, Warnack W, Greenberg BM. Herpes simplex encephalitis as a potential cause of anti-N-methyl-D-aspartate receptor antibody encephalitis: report of 2 cases. JAMA Neurol 2014; 71:344–346.
- Armangue T, Leypoldt F, Málaga I, et al. Herpes simplex virus encephalitis is a trigger of brain autoimmunity. Ann Neurol 2014; 75:317–323.
- Blaes F. Paraneoplastic brain stem encephalitis. Curr Treat Options Neurol 2013; 15:201–209.
- Wildemann B, Jarius S. The expanding range of autoimmune disorders of the nervous system. Lancet Neurol 2013; 12:22–24.
- Kim W, Kim SH, Lee SH, Li XF, Kim HJ. Brain abnormalities as an initial manifestation of neuromyelitis optica spectrum disorder. Mult Scler 2011; 17:1107–1112.
- Shahrizaila N, Yuki N. Bickerstaff brainstem encephalitis and Fisher syndrome: anti-GQ1b antibody syndrome. J Neurol Neurosurg Psychiatry 2013; 84:576–583.
- Jubelt B, Mihai C, Li MT, Veerapaneni P. Rhombencephalitis/brainstem encephalitis. Curr Neurol Neurosci Rep 2011; 11:543–552.
- Sillevis Smitt P, Kinoshita A, De Leeuw B, et al. Paraneoplastic cerebellar ataxia due to autoantibodies against a glutamate receptor. N Engl J Med 2000; 342:21–27.
- Ishida K, Mitoma H, Son SY, et al. Selective suppression of cerebellar GABAergic transmission by an autoantibody to glutamic acid decarboxylase. Ann Neurol 1999; 46:263–267.
- Hart IK, Waters C, Vincent A, et al. Autoantibodies detected to expressed K+ channels are implicated in neuromyotonia. Ann Neurol 1997; 41:238–246.
- Barber P, Anderson NE, Vincent A. Morvan’s syndrome associated with voltage-gated K+ channel antibodies. Neurology 2000; 54:771–772.
- Buckley C, Oger J, Clover L, et al. Potassium channel antibodies in two patients with reversible limbic encephalitis. Ann Neurol 2001; 50:73–78.
- Majoie HJ, de Baets M, Renier W, Lang B, Vincent A. Antibodies to voltage-gated potassium and calcium channels in epilepsy. Epilepsy Res 2006; 71:135–141.
- Tan KM, Lennon VA, Klein CJ, Boeve BF, Pittock SJ. Clinical spectrum of voltage-gated potassium channel autoimmunity. Neurology 2008; 70:1883–1890.
- Knowles CH, Lang B, Clover L, et al. A role for autoantibodies in some cases of acquired non-paraneoplastic gut dysmotility. Scand J Gastroenterol 2002; 37:166–170.
- Irani SR, Michell AW, Lang B, et al. Faciobrachial dystonic seizures precede Lgi1 antibody limbic encephalitis. Ann Neurol 2011; 69:892–900.
- Irani SR, Stagg CJ, Schott JM, et al. Faciobrachial dystonic seizures: the influence of immunotherapy on seizure control and prevention of cognitive impairment in a broadening phenotype. Brain 2013: 136:3151–3162.
- Vincent A, Bien CG, Irani SR, Waters P. Autoantibodies associated with diseases of the CNS: new developments and future challenges. Lancet Neurol 2011; 10:759–772.
- Dalmau J, Lancaster E, Martinez-Hernandez E, Rosenfeld MR, Balice-Gordon R. Clinical experience and laboratory investigations in patients with anti-NMDAR encephalitis. Lancet Neurol 2011; 10:63–74.
- Irani SR, Bera K, Waters P, et al. N-methyl-D-aspartate antibody encephalitis: temporal progression of clinical and paraclinical observations in a predominantly non-paraneoplastic disorder of both sexes. Brain 2010; 133:1655–1667.
- Flanagan EP, McKeon A, Lennon VA, et al. Autoimmune dementia: clinical course and predictors of immunotherapy response. Mayo Clin Proc 2010; 85:881–897.
- Younes-Mhenni S, Janier MF, Cinotti L, et al. FDG-PET improves tumour detection in patients with paraneoplastic neurological syndromes. Brain 2004; 127:2331–2338.
- Lancaster E, Martinez-Hernandez E, Dalmau J. Encephalitis and antibodies to synaptic and neuronal cell surface proteins. Neurology 2011; 77:179–189.
- Shin YW, Lee ST, Shin JW, et al. VGKC-complex/LGI1-antibody encephalitis: clinical manifestations and response to immunotherapy. J Neuroimmunol 2013; 265:75–81.
- Titulaer MJ, McCracken L, Gabilondo I, et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet Neurol 2013; 12:157–165.
- Brierley JB, Corsellis JAN, Hierons R, Nevin S. Subacute encephalitis of later adult life mainly affecting the limbic areas. Brain 1960; 83:357–368.
- Corsellis JA, Goldberg GJ, Norton AR. “Limbic encephalitis” and its association with carcinoma. Brain 1968; 91:481–496.
- Greenlee JE, Brashear HR. Antibodies to cerebellar Purkinje cells in patients with paraneoplastic cerebellar degeneration and ovarian carcinoma. Ann Neurol 1983; 14:609–613.
- Rosenfeld MR, Dalmau JO. Paraneoplastic disorders of the CNS and autoimmune synaptic encephalitis. Continuum (Minneap Minn) 2012; 18:366–383.
- Irani SR, Gelfand JM, Al-Diwani A, Vincent A. Cell-surface central nervous system autoantibodies: clinical relevance and emerging paradigms. Ann Neurol 2014; 76:168–184.
- Dalmau J, Tüzün E, Wu HY, et al. Paraneoplastic anti-N-methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol 2007; 61:25–36.
- Irani SR, Alexander S, Waters P, et al. Antibodies to Kv1 potassium channel-complex proteins leucine-rich, glioma inactivated 1 protein and contactin-associated protein-2 in limbic encephalitis, Morvan’s syndrome and acquired neuromyotonia. Brain 2010; 133:2734–2748.
- Lancaster E, Lai M, Peng X, et al. Antibodies to the GABA(B) receptor in limbic encephalitis with seizures: case series and characterisation of the antigen. Lancet Neurol 2010; 9:67–76.
- Lai M, Hughes EG, Peng X, et al. AMPA receptor antibodies in limbic encephalitis alter synaptic receptor location. Ann Neurol 2009; 65:424–434.
- Zuliani L, Graus F, Giometto B, Bien C, Vincent A. Central nervous system neuronal surface antibody associated syndromes: review and guidelines for recognition. J Neurol Neurosurg Psychiatry 2012; 83:638–645.
- Ances BM, Vitaliani R, Taylor RA, et al. Treatment-responsive limbic encephalitis identified by neuropil antibodies: MRI and PET correlates. Brain 2005; 128:1764–1777.
- Vincent A, Buckley C, Schott JM, et al. Potassium channel antibody-associated encephalopathy: a potentially immunotherapy-responsive form of limbic encephalitis. Brain 2004; 127:701–712.
- Jammoul A, Lederman RJ, Tavee J, Li Y. Presence of voltage-gated potassium channel complex antibody in a case of genetic prion disease. BMJ Case Rep 2014; pii:bcr2013201622.
- Angus-Leppan H, Rudge P, Mead S, Collinge J, Vincent A. Autoantibodies in sporadic Creutzfeldt-Jakob disease. JAMA Neurol 2013; 70:919–922.
- Fujita K, Yuasa T, Watanabe O, et al. Voltage-gated potassium channel complex antibodies in Creutzfeldt-Jakob disease. J Neurol 2012; 259:2249–2250.
- Fujita K, Yuasa T, Takahashi Y, et al. Antibodies to N-methyl-D-aspartate glutamate receptors in Creutzfeldt–Jakob disease patients. J Neuroimmunol 2012; 251:90–93.
- Mackay G, Ahmad K, Stone J, et al. NMDA receptor autoantibodies in sporadic Creutzfeldt-Jakob disease. J Neurol 2012; 259:1979–1981.
- Leypoldt F, Titulaer MJ, Aguilar E, et al. Herpes simplex virus–1 encephalitis can trigger anti-NMDA receptor encephalitis: case report. Neurology 2013; 81:1637–1639.
- Desena A, Graves D, Warnack W, Greenberg BM. Herpes simplex encephalitis as a potential cause of anti-N-methyl-D-aspartate receptor antibody encephalitis: report of 2 cases. JAMA Neurol 2014; 71:344–346.
- Armangue T, Leypoldt F, Málaga I, et al. Herpes simplex virus encephalitis is a trigger of brain autoimmunity. Ann Neurol 2014; 75:317–323.
- Blaes F. Paraneoplastic brain stem encephalitis. Curr Treat Options Neurol 2013; 15:201–209.
- Wildemann B, Jarius S. The expanding range of autoimmune disorders of the nervous system. Lancet Neurol 2013; 12:22–24.
- Kim W, Kim SH, Lee SH, Li XF, Kim HJ. Brain abnormalities as an initial manifestation of neuromyelitis optica spectrum disorder. Mult Scler 2011; 17:1107–1112.
- Shahrizaila N, Yuki N. Bickerstaff brainstem encephalitis and Fisher syndrome: anti-GQ1b antibody syndrome. J Neurol Neurosurg Psychiatry 2013; 84:576–583.
- Jubelt B, Mihai C, Li MT, Veerapaneni P. Rhombencephalitis/brainstem encephalitis. Curr Neurol Neurosci Rep 2011; 11:543–552.
- Sillevis Smitt P, Kinoshita A, De Leeuw B, et al. Paraneoplastic cerebellar ataxia due to autoantibodies against a glutamate receptor. N Engl J Med 2000; 342:21–27.
- Ishida K, Mitoma H, Son SY, et al. Selective suppression of cerebellar GABAergic transmission by an autoantibody to glutamic acid decarboxylase. Ann Neurol 1999; 46:263–267.
- Hart IK, Waters C, Vincent A, et al. Autoantibodies detected to expressed K+ channels are implicated in neuromyotonia. Ann Neurol 1997; 41:238–246.
- Barber P, Anderson NE, Vincent A. Morvan’s syndrome associated with voltage-gated K+ channel antibodies. Neurology 2000; 54:771–772.
- Buckley C, Oger J, Clover L, et al. Potassium channel antibodies in two patients with reversible limbic encephalitis. Ann Neurol 2001; 50:73–78.
- Majoie HJ, de Baets M, Renier W, Lang B, Vincent A. Antibodies to voltage-gated potassium and calcium channels in epilepsy. Epilepsy Res 2006; 71:135–141.
- Tan KM, Lennon VA, Klein CJ, Boeve BF, Pittock SJ. Clinical spectrum of voltage-gated potassium channel autoimmunity. Neurology 2008; 70:1883–1890.
- Knowles CH, Lang B, Clover L, et al. A role for autoantibodies in some cases of acquired non-paraneoplastic gut dysmotility. Scand J Gastroenterol 2002; 37:166–170.
- Irani SR, Michell AW, Lang B, et al. Faciobrachial dystonic seizures precede Lgi1 antibody limbic encephalitis. Ann Neurol 2011; 69:892–900.
- Irani SR, Stagg CJ, Schott JM, et al. Faciobrachial dystonic seizures: the influence of immunotherapy on seizure control and prevention of cognitive impairment in a broadening phenotype. Brain 2013: 136:3151–3162.
- Vincent A, Bien CG, Irani SR, Waters P. Autoantibodies associated with diseases of the CNS: new developments and future challenges. Lancet Neurol 2011; 10:759–772.
- Dalmau J, Lancaster E, Martinez-Hernandez E, Rosenfeld MR, Balice-Gordon R. Clinical experience and laboratory investigations in patients with anti-NMDAR encephalitis. Lancet Neurol 2011; 10:63–74.
- Irani SR, Bera K, Waters P, et al. N-methyl-D-aspartate antibody encephalitis: temporal progression of clinical and paraclinical observations in a predominantly non-paraneoplastic disorder of both sexes. Brain 2010; 133:1655–1667.
- Flanagan EP, McKeon A, Lennon VA, et al. Autoimmune dementia: clinical course and predictors of immunotherapy response. Mayo Clin Proc 2010; 85:881–897.
- Younes-Mhenni S, Janier MF, Cinotti L, et al. FDG-PET improves tumour detection in patients with paraneoplastic neurological syndromes. Brain 2004; 127:2331–2338.
- Lancaster E, Martinez-Hernandez E, Dalmau J. Encephalitis and antibodies to synaptic and neuronal cell surface proteins. Neurology 2011; 77:179–189.
- Shin YW, Lee ST, Shin JW, et al. VGKC-complex/LGI1-antibody encephalitis: clinical manifestations and response to immunotherapy. J Neuroimmunol 2013; 265:75–81.
- Titulaer MJ, McCracken L, Gabilondo I, et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet Neurol 2013; 12:157–165.
KEY POINTS
- Autoantibody-mediated encephalitis accounts for a portion of cases of unexplained status epilepticus, encephalitis, and acute-onset psychiatric symptoms.
- Magnetic resonance imaging and cerebrospinal fluid analysis may be normal early in the disease course.
- Patients can express more than one autoantibody and present with more than one neuronal syndrome.
- Syndromes in which antibodies attack antigens on the surface of neurons are more likely to respond to immunotherapy than those involving intracellular antigens.
- Anti-N-methyl-d-aspartate receptor encephalitis typically presents with psychosis, seizures, and movement disorders in young women and is often associated with an ovarian teratoma.
- Limbic encephalitis, mediated by antibody to the voltage-gated potassium channel complex, is typically nonneoplastic and responds well to immunotherapy.
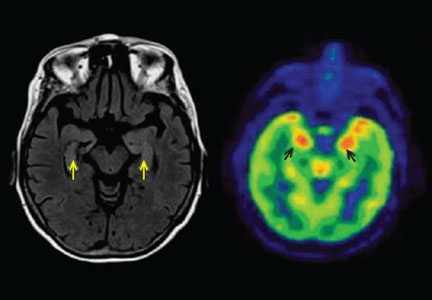
Eventration of the diaphragm presenting as spontaneous pneumothorax
A 25-year-old man with a 2-day history of upper respiratory tract infection presents to the emergency department with the sudden onset of right-sided back and chest pain and shortness of breath after a severe coughing fit.
He is morbidly obese, is a long-time smoker, and has had recurrent exacerbations of asthma with frequent upper respiratory tract infections. He has no history of recent trauma.
A review of systems reveals no significant impairment in exercise tolerance. He has been able to continue doing manual labor at his job as a railroad worker.
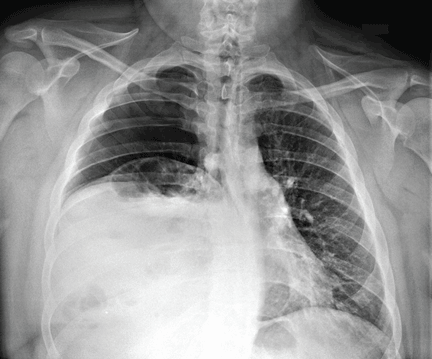
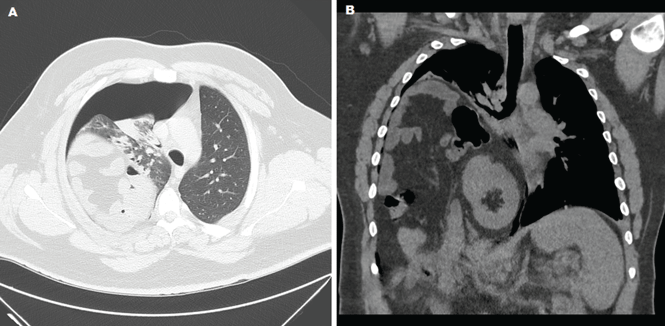
Radiography shows a large right pneumothorax and an elevated right diaphragm (Figure 1). Computed tomography (CT) (Figure 2) reveals a right anterior apical pneumothorax with hypoplastic lung and significant elevation of the right diaphragm with fat, bowel, and kidney within the right thorax. He is hemodynamically stable and shows no signs of bowel obstruction.
The physical examination is normal except for diminished breath sounds on the right side. He is diagnosed with congenital diaphragmatic hernia and spontaneous pneumothorax. A 10-F locking pigtail catheter is inserted under CT guidance, leading to complete resolution of the pneumothorax. He is discharged home the next day with a plan for elective repair of the hernia.
Two months later, he returns for scheduled right thoracotomy to repair the hernia. However, while preparing the chest cavity, the surgeon finds no diaphragmatic hernia and no intra-abdominal content—but rather, a severely elevated and thinned-out diaphragm with uninterrupted continuity. The diagnosis is changed to congenital diaphragmatic eventration, and plication of the diaphragm is performed with a series of interrupted, pledgeted polypropylene sutures.
CONGENITAL EVENTRATION OF THE DIAPHRAGM
Congenital diaphragmatic eventration is a rare developmental defect of the central, muscular portion of the diaphragm. The true prevalence is not known, but early reports identified this condition in less than 0.1% of adult.1
Symptomatic patients usually experience dyspnea secondary to ventilation-perfusion mismatch resulting from chronic atelectasis and lung hypoplasia, as well as impaired ventilation resulting from the limited caudal migration of the diaphragm.2,3 Increased susceptibility to recurrent upper respiratory tract infections and pneumonia is also a common feature.
Although rare, spontaneous pneumothorax can develop in patients such as ours, whose lengthy history of smoking and asthma predisposed him to the development of emphysema-like blebs and bullae and to subsequent rupture of blebs brought on by vigorous coughing that caused an involuntary Valsalva maneuver.4
As in our patient, distinguishing congenital diaphragmatic eventration from hernia preoperatively can be difficult with plain chest radiography. Spiral CT with multiplanar reconstruction or with magnetic resonance imaging can help establish the diagnosis.3 However, a severely attenuated diaphragm can be difficult to visualize on CT, as in our patient, leading to a presumptive diagnosis of diaphragmatic hernia. In such situations, the diagnosis of eventration can only be made intraoperatively.
Surgical repair is indicated only for patients with symptoms. Other potential causes of the symptoms should first be ruled out, however, including primary pulmonary disease, cardiac dysfunction, and morbid obesity.
- Chin EF, Lynn RB. Surgery of eventration of the diaphragm. J Thorac Surg 1956; 32:6–14.
- Ridyard JB, Stewart RM. Regional lung function in unilateral diaphragmatic paralysis. Thorax 1976; 31:438–442.
- Shen C, Che G. Congenital eventration of hemidiaphragm in an adult. Ann Thorac Surg 2012; 94:e137–e139.
- Porpodis K, Zarogoulidis P, Spyratos D, et al. Pneumothorax and asthma. J Thorac Dis 2014; 6(suppl 1):S152–S161.
A 25-year-old man with a 2-day history of upper respiratory tract infection presents to the emergency department with the sudden onset of right-sided back and chest pain and shortness of breath after a severe coughing fit.
He is morbidly obese, is a long-time smoker, and has had recurrent exacerbations of asthma with frequent upper respiratory tract infections. He has no history of recent trauma.
A review of systems reveals no significant impairment in exercise tolerance. He has been able to continue doing manual labor at his job as a railroad worker.
Radiography shows a large right pneumothorax and an elevated right diaphragm (Figure 1). Computed tomography (CT) (Figure 2) reveals a right anterior apical pneumothorax with hypoplastic lung and significant elevation of the right diaphragm with fat, bowel, and kidney within the right thorax. He is hemodynamically stable and shows no signs of bowel obstruction.
The physical examination is normal except for diminished breath sounds on the right side. He is diagnosed with congenital diaphragmatic hernia and spontaneous pneumothorax. A 10-F locking pigtail catheter is inserted under CT guidance, leading to complete resolution of the pneumothorax. He is discharged home the next day with a plan for elective repair of the hernia.
Two months later, he returns for scheduled right thoracotomy to repair the hernia. However, while preparing the chest cavity, the surgeon finds no diaphragmatic hernia and no intra-abdominal content—but rather, a severely elevated and thinned-out diaphragm with uninterrupted continuity. The diagnosis is changed to congenital diaphragmatic eventration, and plication of the diaphragm is performed with a series of interrupted, pledgeted polypropylene sutures.
CONGENITAL EVENTRATION OF THE DIAPHRAGM
Congenital diaphragmatic eventration is a rare developmental defect of the central, muscular portion of the diaphragm. The true prevalence is not known, but early reports identified this condition in less than 0.1% of adult.1
Symptomatic patients usually experience dyspnea secondary to ventilation-perfusion mismatch resulting from chronic atelectasis and lung hypoplasia, as well as impaired ventilation resulting from the limited caudal migration of the diaphragm.2,3 Increased susceptibility to recurrent upper respiratory tract infections and pneumonia is also a common feature.
Although rare, spontaneous pneumothorax can develop in patients such as ours, whose lengthy history of smoking and asthma predisposed him to the development of emphysema-like blebs and bullae and to subsequent rupture of blebs brought on by vigorous coughing that caused an involuntary Valsalva maneuver.4
As in our patient, distinguishing congenital diaphragmatic eventration from hernia preoperatively can be difficult with plain chest radiography. Spiral CT with multiplanar reconstruction or with magnetic resonance imaging can help establish the diagnosis.3 However, a severely attenuated diaphragm can be difficult to visualize on CT, as in our patient, leading to a presumptive diagnosis of diaphragmatic hernia. In such situations, the diagnosis of eventration can only be made intraoperatively.
Surgical repair is indicated only for patients with symptoms. Other potential causes of the symptoms should first be ruled out, however, including primary pulmonary disease, cardiac dysfunction, and morbid obesity.
A 25-year-old man with a 2-day history of upper respiratory tract infection presents to the emergency department with the sudden onset of right-sided back and chest pain and shortness of breath after a severe coughing fit.
He is morbidly obese, is a long-time smoker, and has had recurrent exacerbations of asthma with frequent upper respiratory tract infections. He has no history of recent trauma.
A review of systems reveals no significant impairment in exercise tolerance. He has been able to continue doing manual labor at his job as a railroad worker.
Radiography shows a large right pneumothorax and an elevated right diaphragm (Figure 1). Computed tomography (CT) (Figure 2) reveals a right anterior apical pneumothorax with hypoplastic lung and significant elevation of the right diaphragm with fat, bowel, and kidney within the right thorax. He is hemodynamically stable and shows no signs of bowel obstruction.
The physical examination is normal except for diminished breath sounds on the right side. He is diagnosed with congenital diaphragmatic hernia and spontaneous pneumothorax. A 10-F locking pigtail catheter is inserted under CT guidance, leading to complete resolution of the pneumothorax. He is discharged home the next day with a plan for elective repair of the hernia.
Two months later, he returns for scheduled right thoracotomy to repair the hernia. However, while preparing the chest cavity, the surgeon finds no diaphragmatic hernia and no intra-abdominal content—but rather, a severely elevated and thinned-out diaphragm with uninterrupted continuity. The diagnosis is changed to congenital diaphragmatic eventration, and plication of the diaphragm is performed with a series of interrupted, pledgeted polypropylene sutures.
CONGENITAL EVENTRATION OF THE DIAPHRAGM
Congenital diaphragmatic eventration is a rare developmental defect of the central, muscular portion of the diaphragm. The true prevalence is not known, but early reports identified this condition in less than 0.1% of adult.1
Symptomatic patients usually experience dyspnea secondary to ventilation-perfusion mismatch resulting from chronic atelectasis and lung hypoplasia, as well as impaired ventilation resulting from the limited caudal migration of the diaphragm.2,3 Increased susceptibility to recurrent upper respiratory tract infections and pneumonia is also a common feature.
Although rare, spontaneous pneumothorax can develop in patients such as ours, whose lengthy history of smoking and asthma predisposed him to the development of emphysema-like blebs and bullae and to subsequent rupture of blebs brought on by vigorous coughing that caused an involuntary Valsalva maneuver.4
As in our patient, distinguishing congenital diaphragmatic eventration from hernia preoperatively can be difficult with plain chest radiography. Spiral CT with multiplanar reconstruction or with magnetic resonance imaging can help establish the diagnosis.3 However, a severely attenuated diaphragm can be difficult to visualize on CT, as in our patient, leading to a presumptive diagnosis of diaphragmatic hernia. In such situations, the diagnosis of eventration can only be made intraoperatively.
Surgical repair is indicated only for patients with symptoms. Other potential causes of the symptoms should first be ruled out, however, including primary pulmonary disease, cardiac dysfunction, and morbid obesity.
- Chin EF, Lynn RB. Surgery of eventration of the diaphragm. J Thorac Surg 1956; 32:6–14.
- Ridyard JB, Stewart RM. Regional lung function in unilateral diaphragmatic paralysis. Thorax 1976; 31:438–442.
- Shen C, Che G. Congenital eventration of hemidiaphragm in an adult. Ann Thorac Surg 2012; 94:e137–e139.
- Porpodis K, Zarogoulidis P, Spyratos D, et al. Pneumothorax and asthma. J Thorac Dis 2014; 6(suppl 1):S152–S161.
- Chin EF, Lynn RB. Surgery of eventration of the diaphragm. J Thorac Surg 1956; 32:6–14.
- Ridyard JB, Stewart RM. Regional lung function in unilateral diaphragmatic paralysis. Thorax 1976; 31:438–442.
- Shen C, Che G. Congenital eventration of hemidiaphragm in an adult. Ann Thorac Surg 2012; 94:e137–e139.
- Porpodis K, Zarogoulidis P, Spyratos D, et al. Pneumothorax and asthma. J Thorac Dis 2014; 6(suppl 1):S152–S161.
Case Studies in Toxicology: The Perilous Pursuit of Perfection
Case
A 37-year-old woman had undergone outpatient tumescent liposuction of her abdominal adipose tissue. Upon initiation of the procedure, she developed acute confusion followed by agitation and hallucinations and was transported by emergency medical services to the nearest hospital.
Upon arrival to the ED, the patient was mildly agitated. Her initial vital signs were: blood pressure, 122/79 mm Hg; heart rate, 57 beats/minute; respiratory rate, 18 breaths/minute; and temperature, 96.8°F. Oxygen saturation was 100% on room air.
On examination, the patient was awake and oriented, but was restless and perseverating. She described the sensation of “dying and coming back to life.” The patient’s pupils were equal, round, and normally reactive; her skin was neither diaphoretic nor dry; her heart rate and rhythm were normal; and her lungs were clear to auscultation bilaterally. The skin of the patient’s abdomen was pale, cool, and clammy, but otherwise soft with normoactive bowel sounds. An electrocardiogram revealed sinus rhythm with normal QRS and QTc intervals.
What is tumescent liposuction?

The tumescent technique (the word tumescent derived from the Latin word meaning “to swell”) involves the infiltration of a large volume of a dilute solution of lidocaine and epinephrine into the subcutaneous fat until it becomes distended and tense. The administration of this solution allows for the removal of significant adipose tissue with minimal blood loss and without general anesthesia. Advocates of tumescent liposuction argue that this dilution and subcutaneous infiltration alter the pharmacokinetics of lidocaine, providing a safe delivery of large doses of the drug. In addition, a substantial quantity of the infiltrated lidocaine is suctioned out with the fat removal. Klein3 specifically recommends administering a dilution of 1 g lidocaine in 1,000 mL of saline (0.1%) to the patient in aliquots of up to 2 g lidocaine. Such a high dose may approach or surpass 35 mg/kg, far exceeding the recommended lidocaine infiltration dose of 7 mg/kg when mixed with epinephrine.
Several studies have investigated the safety of the tumescent technique and the risk of lidocaine toxicity. In a review of pharmacokinetic studies, patients received between 10.5 and 67.7 mg/kg of lidocaine with a reported maximum serum lidocaine concentration in all patients of only 2.93 mcg/mL (therapeutic, 1-5 mcg/mL).4 However, peak serum lidocaine concentrations may not occur for up to 28 hours following tumescent infiltration.4 Despite the purported safety findings of this and other studies, there are also reports of tumescent anesthesia-associated toxicity and fatalities.5,6
Case Continuation
The patient’s mental status slowly improved throughout her hospital stay and she was reportedly at baseline by the following morning. Her serum lidocaine concentration drawn at presentation to the ED was 9.7 mcg/mL.
What are the clinical effects of lidocaine toxicity?
The clinical effects of lidocaine overdose are dependent on both the magnitude of the exposure and the rate at which it occurs. The central nervous system (CNS) and heart are the organ systems primarily affected by lidocaine. As a local anesthetic, lidocaine inhibits the action potential formation in electrically excitable cells. This reversible inhibition of the voltage-gated sodium channels prevents the influx of positively charged sodium ions and the resultant depolarization of the cell. Lidocaine, initially or at low concentrations, has a quiescent effect on neurons, which explains its therapeutic use as a local anesthetic.
Similarly, lidocaine initially reduces impulse propagation through the cardiac conduction system and can be used to treat rhythm disturbances. When used for the suppression of cardiac dysrhythmias, the therapeutic serum concentration of lidocaine is 1 to 5 mcg/mL. Subjective symptoms of lidocaine toxicity arising from therapeutic use (primarily when it is used to manage dysrhythmia) include light-headedness, disorientation, confusion, and psychosis, and are associated with serum concentrations of 3 to 6 mcg/mL. As the concentration increases, the clinical effects of lidocaine appear to shift from inhibitory to excitatory. At serum concentrations of 5 to 9 mcg/mL, objective symptoms, such as excitation, tremor, and seizure predominate. As the concentration continues to rise, coma, respiratory depression, cardiovascular (CV) collapse, and death may occur.7
The exact mechanism of this toxicity transition from inhibitory effect of sodium channel blockade to excitatory effect is not well understood. Some suggest a preferential inhibition of inhibitory interneurons in the CNS is responsible.8 Another potential mechanism is a concentration-dependent inhibition of the potassium rectifier channel.9 Inhibition of the efflux of positively charged potassium ions would result in slowing cellular repolarization, leaving the cells in a relatively excitable state. In the CNS, this produces seizures; in the heart, it may result in dysrhythmia. Cardiovascular collapse may occur with very high serum concentrations of lidocaine or following a rapid serum increase—eg, after a large intravenous (IV) bolus dose,7 which can potentially result from an unintentional intravascular injection during tumescent liposuction.
What is the treatment for lidocaine toxicity?
The first step in the treatment of lidocaine-associated CNS toxicity is the discontinuation of the drug. Failure to appropriately recognize the symptoms of early lidocaine toxicity may result in the progression to severe CNS effects and eventual CV collapse. Benzodiazepines should be used as needed for mild symptoms. Seizures should be treated aggressively with benzodiazepines or barbiturates, while ensuring maintenance of oxygenation, ventilation, and perfusion.7
In cases of lidocaine-associated CV toxicity, treatment begins with airway management, oxygen administration, and life support. Potential antidotal treatment of severe local anesthetic-associated CV toxicity involves the rapid administration of IV fat emulsion, or “lipid rescue.” Although best studied for bupivacaine toxicity, the exact mechanism of IV fat emulsion as an antidote is not completely understood. However, in the treatment of local anesthetic toxicity, lipid rescue is believed to offer a “sink” to remove the lipid-soluble anesthetics from their site of action and trap them within the vascular space. Suggested dosing of 20% lipid solution is a bolus of 1.5 mL/kg over a 1-minute period, followed by 0.25mL/kg per minute or 15 mL/kg per hour to run over 30 to 60 minutes.10
Case Conclusion
The patient made a full recovery and was discharged home in normal condition. Her healthcare provider was informed about the complication of the procedure.
Dr Hines is a senior toxicology fellow, department of emergency medicine, New York University School of Medicine. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
- Lozinski A, Huq NS. Tumescent liposuction. Clin Plastic Surg. 2013;40(4):593-613.
- Klein JA. Tumescent technique chronicles: local anesthesia, liposuction, and beyond. Dermatol Surg. 1995;21(5):449-457.
- Klein JA. Tumescent technique for regional anesthesia permits lidocaine doses of 35 mg/kg for liposuction. J Dermatol Surg Oncol. 1990;16(3):248-263.
- Conroy PH, O’Rourke J. Tumescent anesthesia. The Surgeon. 2012;210-201.
- Rao RR, Fly SF, Hoffman RS. Deaths related to liposuction. N Engl J Med. 1999;340(19):1471-1475.
- Martinez MA, Ballesteros S, Segura LJ, Garcia M. Reporting a fatality during tumescent liposuction. Forensic Sci Int. 2008;178(1):e11-e-16.
- Schwartz DR, Kaufman B. Local anesthestics. In: Hoffman RS, Howland MA, Lewin NA, Nelson LS, Goldfrank LR. Goldfrank’s Toxicologic Emergencies. 10th ed. New York, NY: McGraw Hill; 2015:921-930.
- Tanaka K, Yamasaki M. Blocking of cortical inhibitory synapses by intravenous lidoaine. Nature. 1966;209(5019):207-208.
- Friederich P, Benzenberg D, Urban BW. Bupivacaine inhibits human neuronal Kv3 channels in SH-SY5Y human neuroblastoma cells. Br J Anaesth. 2002;88(6):864-866.
- Bania TC. Antidotes in depth, intravenous fat emulsions. In: Hoffman RS, Howland MA, Lewin NA, Nelson LS, Goldfrank LR. Goldfrank’s Toxicologic Emergencies. 10th ed. New York, NY: McGraw Hill; 2015:931-937.
Case
A 37-year-old woman had undergone outpatient tumescent liposuction of her abdominal adipose tissue. Upon initiation of the procedure, she developed acute confusion followed by agitation and hallucinations and was transported by emergency medical services to the nearest hospital.
Upon arrival to the ED, the patient was mildly agitated. Her initial vital signs were: blood pressure, 122/79 mm Hg; heart rate, 57 beats/minute; respiratory rate, 18 breaths/minute; and temperature, 96.8°F. Oxygen saturation was 100% on room air.
On examination, the patient was awake and oriented, but was restless and perseverating. She described the sensation of “dying and coming back to life.” The patient’s pupils were equal, round, and normally reactive; her skin was neither diaphoretic nor dry; her heart rate and rhythm were normal; and her lungs were clear to auscultation bilaterally. The skin of the patient’s abdomen was pale, cool, and clammy, but otherwise soft with normoactive bowel sounds. An electrocardiogram revealed sinus rhythm with normal QRS and QTc intervals.
What is tumescent liposuction?
The tumescent technique (the word tumescent derived from the Latin word meaning “to swell”) involves the infiltration of a large volume of a dilute solution of lidocaine and epinephrine into the subcutaneous fat until it becomes distended and tense. The administration of this solution allows for the removal of significant adipose tissue with minimal blood loss and without general anesthesia. Advocates of tumescent liposuction argue that this dilution and subcutaneous infiltration alter the pharmacokinetics of lidocaine, providing a safe delivery of large doses of the drug. In addition, a substantial quantity of the infiltrated lidocaine is suctioned out with the fat removal. Klein3 specifically recommends administering a dilution of 1 g lidocaine in 1,000 mL of saline (0.1%) to the patient in aliquots of up to 2 g lidocaine. Such a high dose may approach or surpass 35 mg/kg, far exceeding the recommended lidocaine infiltration dose of 7 mg/kg when mixed with epinephrine.
Several studies have investigated the safety of the tumescent technique and the risk of lidocaine toxicity. In a review of pharmacokinetic studies, patients received between 10.5 and 67.7 mg/kg of lidocaine with a reported maximum serum lidocaine concentration in all patients of only 2.93 mcg/mL (therapeutic, 1-5 mcg/mL).4 However, peak serum lidocaine concentrations may not occur for up to 28 hours following tumescent infiltration.4 Despite the purported safety findings of this and other studies, there are also reports of tumescent anesthesia-associated toxicity and fatalities.5,6
Case Continuation
The patient’s mental status slowly improved throughout her hospital stay and she was reportedly at baseline by the following morning. Her serum lidocaine concentration drawn at presentation to the ED was 9.7 mcg/mL.
What are the clinical effects of lidocaine toxicity?
The clinical effects of lidocaine overdose are dependent on both the magnitude of the exposure and the rate at which it occurs. The central nervous system (CNS) and heart are the organ systems primarily affected by lidocaine. As a local anesthetic, lidocaine inhibits the action potential formation in electrically excitable cells. This reversible inhibition of the voltage-gated sodium channels prevents the influx of positively charged sodium ions and the resultant depolarization of the cell. Lidocaine, initially or at low concentrations, has a quiescent effect on neurons, which explains its therapeutic use as a local anesthetic.
Similarly, lidocaine initially reduces impulse propagation through the cardiac conduction system and can be used to treat rhythm disturbances. When used for the suppression of cardiac dysrhythmias, the therapeutic serum concentration of lidocaine is 1 to 5 mcg/mL. Subjective symptoms of lidocaine toxicity arising from therapeutic use (primarily when it is used to manage dysrhythmia) include light-headedness, disorientation, confusion, and psychosis, and are associated with serum concentrations of 3 to 6 mcg/mL. As the concentration increases, the clinical effects of lidocaine appear to shift from inhibitory to excitatory. At serum concentrations of 5 to 9 mcg/mL, objective symptoms, such as excitation, tremor, and seizure predominate. As the concentration continues to rise, coma, respiratory depression, cardiovascular (CV) collapse, and death may occur.7
The exact mechanism of this toxicity transition from inhibitory effect of sodium channel blockade to excitatory effect is not well understood. Some suggest a preferential inhibition of inhibitory interneurons in the CNS is responsible.8 Another potential mechanism is a concentration-dependent inhibition of the potassium rectifier channel.9 Inhibition of the efflux of positively charged potassium ions would result in slowing cellular repolarization, leaving the cells in a relatively excitable state. In the CNS, this produces seizures; in the heart, it may result in dysrhythmia. Cardiovascular collapse may occur with very high serum concentrations of lidocaine or following a rapid serum increase—eg, after a large intravenous (IV) bolus dose,7 which can potentially result from an unintentional intravascular injection during tumescent liposuction.
What is the treatment for lidocaine toxicity?
The first step in the treatment of lidocaine-associated CNS toxicity is the discontinuation of the drug. Failure to appropriately recognize the symptoms of early lidocaine toxicity may result in the progression to severe CNS effects and eventual CV collapse. Benzodiazepines should be used as needed for mild symptoms. Seizures should be treated aggressively with benzodiazepines or barbiturates, while ensuring maintenance of oxygenation, ventilation, and perfusion.7
In cases of lidocaine-associated CV toxicity, treatment begins with airway management, oxygen administration, and life support. Potential antidotal treatment of severe local anesthetic-associated CV toxicity involves the rapid administration of IV fat emulsion, or “lipid rescue.” Although best studied for bupivacaine toxicity, the exact mechanism of IV fat emulsion as an antidote is not completely understood. However, in the treatment of local anesthetic toxicity, lipid rescue is believed to offer a “sink” to remove the lipid-soluble anesthetics from their site of action and trap them within the vascular space. Suggested dosing of 20% lipid solution is a bolus of 1.5 mL/kg over a 1-minute period, followed by 0.25mL/kg per minute or 15 mL/kg per hour to run over 30 to 60 minutes.10
Case Conclusion
The patient made a full recovery and was discharged home in normal condition. Her healthcare provider was informed about the complication of the procedure.
Dr Hines is a senior toxicology fellow, department of emergency medicine, New York University School of Medicine. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
Case
A 37-year-old woman had undergone outpatient tumescent liposuction of her abdominal adipose tissue. Upon initiation of the procedure, she developed acute confusion followed by agitation and hallucinations and was transported by emergency medical services to the nearest hospital.
Upon arrival to the ED, the patient was mildly agitated. Her initial vital signs were: blood pressure, 122/79 mm Hg; heart rate, 57 beats/minute; respiratory rate, 18 breaths/minute; and temperature, 96.8°F. Oxygen saturation was 100% on room air.
On examination, the patient was awake and oriented, but was restless and perseverating. She described the sensation of “dying and coming back to life.” The patient’s pupils were equal, round, and normally reactive; her skin was neither diaphoretic nor dry; her heart rate and rhythm were normal; and her lungs were clear to auscultation bilaterally. The skin of the patient’s abdomen was pale, cool, and clammy, but otherwise soft with normoactive bowel sounds. An electrocardiogram revealed sinus rhythm with normal QRS and QTc intervals.
What is tumescent liposuction?
The tumescent technique (the word tumescent derived from the Latin word meaning “to swell”) involves the infiltration of a large volume of a dilute solution of lidocaine and epinephrine into the subcutaneous fat until it becomes distended and tense. The administration of this solution allows for the removal of significant adipose tissue with minimal blood loss and without general anesthesia. Advocates of tumescent liposuction argue that this dilution and subcutaneous infiltration alter the pharmacokinetics of lidocaine, providing a safe delivery of large doses of the drug. In addition, a substantial quantity of the infiltrated lidocaine is suctioned out with the fat removal. Klein3 specifically recommends administering a dilution of 1 g lidocaine in 1,000 mL of saline (0.1%) to the patient in aliquots of up to 2 g lidocaine. Such a high dose may approach or surpass 35 mg/kg, far exceeding the recommended lidocaine infiltration dose of 7 mg/kg when mixed with epinephrine.
Several studies have investigated the safety of the tumescent technique and the risk of lidocaine toxicity. In a review of pharmacokinetic studies, patients received between 10.5 and 67.7 mg/kg of lidocaine with a reported maximum serum lidocaine concentration in all patients of only 2.93 mcg/mL (therapeutic, 1-5 mcg/mL).4 However, peak serum lidocaine concentrations may not occur for up to 28 hours following tumescent infiltration.4 Despite the purported safety findings of this and other studies, there are also reports of tumescent anesthesia-associated toxicity and fatalities.5,6
Case Continuation
The patient’s mental status slowly improved throughout her hospital stay and she was reportedly at baseline by the following morning. Her serum lidocaine concentration drawn at presentation to the ED was 9.7 mcg/mL.
What are the clinical effects of lidocaine toxicity?
The clinical effects of lidocaine overdose are dependent on both the magnitude of the exposure and the rate at which it occurs. The central nervous system (CNS) and heart are the organ systems primarily affected by lidocaine. As a local anesthetic, lidocaine inhibits the action potential formation in electrically excitable cells. This reversible inhibition of the voltage-gated sodium channels prevents the influx of positively charged sodium ions and the resultant depolarization of the cell. Lidocaine, initially or at low concentrations, has a quiescent effect on neurons, which explains its therapeutic use as a local anesthetic.
Similarly, lidocaine initially reduces impulse propagation through the cardiac conduction system and can be used to treat rhythm disturbances. When used for the suppression of cardiac dysrhythmias, the therapeutic serum concentration of lidocaine is 1 to 5 mcg/mL. Subjective symptoms of lidocaine toxicity arising from therapeutic use (primarily when it is used to manage dysrhythmia) include light-headedness, disorientation, confusion, and psychosis, and are associated with serum concentrations of 3 to 6 mcg/mL. As the concentration increases, the clinical effects of lidocaine appear to shift from inhibitory to excitatory. At serum concentrations of 5 to 9 mcg/mL, objective symptoms, such as excitation, tremor, and seizure predominate. As the concentration continues to rise, coma, respiratory depression, cardiovascular (CV) collapse, and death may occur.7
The exact mechanism of this toxicity transition from inhibitory effect of sodium channel blockade to excitatory effect is not well understood. Some suggest a preferential inhibition of inhibitory interneurons in the CNS is responsible.8 Another potential mechanism is a concentration-dependent inhibition of the potassium rectifier channel.9 Inhibition of the efflux of positively charged potassium ions would result in slowing cellular repolarization, leaving the cells in a relatively excitable state. In the CNS, this produces seizures; in the heart, it may result in dysrhythmia. Cardiovascular collapse may occur with very high serum concentrations of lidocaine or following a rapid serum increase—eg, after a large intravenous (IV) bolus dose,7 which can potentially result from an unintentional intravascular injection during tumescent liposuction.
What is the treatment for lidocaine toxicity?
The first step in the treatment of lidocaine-associated CNS toxicity is the discontinuation of the drug. Failure to appropriately recognize the symptoms of early lidocaine toxicity may result in the progression to severe CNS effects and eventual CV collapse. Benzodiazepines should be used as needed for mild symptoms. Seizures should be treated aggressively with benzodiazepines or barbiturates, while ensuring maintenance of oxygenation, ventilation, and perfusion.7
In cases of lidocaine-associated CV toxicity, treatment begins with airway management, oxygen administration, and life support. Potential antidotal treatment of severe local anesthetic-associated CV toxicity involves the rapid administration of IV fat emulsion, or “lipid rescue.” Although best studied for bupivacaine toxicity, the exact mechanism of IV fat emulsion as an antidote is not completely understood. However, in the treatment of local anesthetic toxicity, lipid rescue is believed to offer a “sink” to remove the lipid-soluble anesthetics from their site of action and trap them within the vascular space. Suggested dosing of 20% lipid solution is a bolus of 1.5 mL/kg over a 1-minute period, followed by 0.25mL/kg per minute or 15 mL/kg per hour to run over 30 to 60 minutes.10
Case Conclusion
The patient made a full recovery and was discharged home in normal condition. Her healthcare provider was informed about the complication of the procedure.
Dr Hines is a senior toxicology fellow, department of emergency medicine, New York University School of Medicine. Dr Nelson, editor of “Case Studies in Toxicology,” is a professor in the department of emergency medicine and director of the medical toxicology fellowship program at the New York University School of Medicine and the New York City Poison Control Center. He is also associate editor, toxicology, of the EMERGENCY MEDICINE editorial board.
- Lozinski A, Huq NS. Tumescent liposuction. Clin Plastic Surg. 2013;40(4):593-613.
- Klein JA. Tumescent technique chronicles: local anesthesia, liposuction, and beyond. Dermatol Surg. 1995;21(5):449-457.
- Klein JA. Tumescent technique for regional anesthesia permits lidocaine doses of 35 mg/kg for liposuction. J Dermatol Surg Oncol. 1990;16(3):248-263.
- Conroy PH, O’Rourke J. Tumescent anesthesia. The Surgeon. 2012;210-201.
- Rao RR, Fly SF, Hoffman RS. Deaths related to liposuction. N Engl J Med. 1999;340(19):1471-1475.
- Martinez MA, Ballesteros S, Segura LJ, Garcia M. Reporting a fatality during tumescent liposuction. Forensic Sci Int. 2008;178(1):e11-e-16.
- Schwartz DR, Kaufman B. Local anesthestics. In: Hoffman RS, Howland MA, Lewin NA, Nelson LS, Goldfrank LR. Goldfrank’s Toxicologic Emergencies. 10th ed. New York, NY: McGraw Hill; 2015:921-930.
- Tanaka K, Yamasaki M. Blocking of cortical inhibitory synapses by intravenous lidoaine. Nature. 1966;209(5019):207-208.
- Friederich P, Benzenberg D, Urban BW. Bupivacaine inhibits human neuronal Kv3 channels in SH-SY5Y human neuroblastoma cells. Br J Anaesth. 2002;88(6):864-866.
- Bania TC. Antidotes in depth, intravenous fat emulsions. In: Hoffman RS, Howland MA, Lewin NA, Nelson LS, Goldfrank LR. Goldfrank’s Toxicologic Emergencies. 10th ed. New York, NY: McGraw Hill; 2015:931-937.
- Lozinski A, Huq NS. Tumescent liposuction. Clin Plastic Surg. 2013;40(4):593-613.
- Klein JA. Tumescent technique chronicles: local anesthesia, liposuction, and beyond. Dermatol Surg. 1995;21(5):449-457.
- Klein JA. Tumescent technique for regional anesthesia permits lidocaine doses of 35 mg/kg for liposuction. J Dermatol Surg Oncol. 1990;16(3):248-263.
- Conroy PH, O’Rourke J. Tumescent anesthesia. The Surgeon. 2012;210-201.
- Rao RR, Fly SF, Hoffman RS. Deaths related to liposuction. N Engl J Med. 1999;340(19):1471-1475.
- Martinez MA, Ballesteros S, Segura LJ, Garcia M. Reporting a fatality during tumescent liposuction. Forensic Sci Int. 2008;178(1):e11-e-16.
- Schwartz DR, Kaufman B. Local anesthestics. In: Hoffman RS, Howland MA, Lewin NA, Nelson LS, Goldfrank LR. Goldfrank’s Toxicologic Emergencies. 10th ed. New York, NY: McGraw Hill; 2015:921-930.
- Tanaka K, Yamasaki M. Blocking of cortical inhibitory synapses by intravenous lidoaine. Nature. 1966;209(5019):207-208.
- Friederich P, Benzenberg D, Urban BW. Bupivacaine inhibits human neuronal Kv3 channels in SH-SY5Y human neuroblastoma cells. Br J Anaesth. 2002;88(6):864-866.
- Bania TC. Antidotes in depth, intravenous fat emulsions. In: Hoffman RS, Howland MA, Lewin NA, Nelson LS, Goldfrank LR. Goldfrank’s Toxicologic Emergencies. 10th ed. New York, NY: McGraw Hill; 2015:931-937.

A young man with an unusual cause of palpitations
A 23-year-old man presents to the emergency department with the sudden onset of palpitations, lightheadedness, and dyspnea, accompanied by weakness and nausea, which started earlier in the evening. He estimates that he has experienced 15 similar episodes, lasting minutes to hours, since the age of 16, with the last one 3 years ago. These episodes typically end by themselves or with self-induced vomiting and lying supine. The current episode did not resolve with these maneuvers.
He has never received medical attention for these symptoms. He has no chest pain, orthopnea, paroxysmal nocturnal dyspnea, lower extremity edema, or syncope. He has had no recent illness, contacts with sick people, or travel.
The patient’s history includes a “childhood heart murmur,” which resolved, and also mild asthma. He is otherwise healthy but has not received regular medical care. He used to play competitive soccer but quit because playing made his symptoms of dyspnea on exertion and palpitations much worse.
He uses marijuana frequently and alcohol occasionally. He does not smoke tobacco or use other recreational drugs. Other than infrequent use of albuterol, he does not take any prescription or over-the-counter medications. He has no allergies. He knows of no family history of arrhythmia or sudden cardiac death.
Physical examination. On initial examination, his temperature is 36.4°C (97.5°F), heart rate 230 bpm, systolic blood pressure 60 mm Hg, respiratory rate 30 breaths per minute, oxygen saturation 100% while breathing room air, and body mass index 25 kg/m2.

He is awake, anxious, and appears ill. He speaks only in short sentences. A focused cardiac examination reveals a regular tachycardia with no appreciable murmur or extra heart sounds; the apical impulse is not displaced. His lungs are clear. His abdomen is soft and nontender. He has 2+ pulses on a scale of 0 to 4+, with no peripheral edema.
His initial electrocardiogram (ECG) (Figure 1) shows a heart rate of 260 bpm and a regular wide complex tachycardia, defined as a rate greater than 100 bpm and a QRS complex wider than 0.12 seconds.
FOCUS ON REGULAR WIDE COMPLEX TACHYCARDIA
1. Which of the following is not in the differential diagnosis of regular wide complex tachycardia?
- Monomorphic ventricular tachycardia
- Orthodromic atrioventricular reentrant tachycardia
- Antidromic atrioventricular reentrant tachycardia
- Sinus tachycardia with bundle branch block
Orthodromic atrioventricular reentrant tachycardia is not in the differential diagnosis.
Wide complex tachycardia can occur when the impulse originates outside the normal conduction system or when there is abnormal ventricular activation through the atrioventricular (AV) node and His-Purkinje system.
The main distinction to make when diagnosing the cause of a wide complex tachycardia is between the following:
Monomorphic ventricular tachycardia, which originates from a single ventricular focus that depolarizes the adjacent myocardium in a stepwise fashion, causing a wide QRS complex that does not begin in the native conduction system, and
Sinus tachycardia with bundle branch block, ie, supraventricular tachycardia with aberrant conduction within the normal conduction system.

Three different conduction patterns are seen with atrioventricular reentrant tachycardia (Figure 2):
Sinus depolarization (Figure 2), in which the atrial impulse travels down the AV node, and the accessory pathway can be hidden and not contribute to the surface ECG.
Orthodromic atrioventricular reentrant tachycardia (Figure2), in which the depolarizing impulse travels antegrade down the AV node, then propagates from the ventricle back to the atria via the accessory pathway, resulting in a narrow QRS.
Antidromic atrioventricular reentrant tachycardia (Figure 2), in which the depolarization travels antegrade down the accessory pathway then propagates from the ventricle back to the atria via the AV node, resulting in a wide complex QRS with a delta wave.
Important features of the patient’s electrocardiogram (Figure 1) are consistent with antidromic atrioventricular reentrant tachycardia:
- “Buried” retrograde P waves, which are best seen in the continuous strip of lead II as a positive deflection notching in the negative nadir of the wave
- The PR segment is short, suggesting retrograde atrial depolarization
- The P wave is followed by a slow slurred upstroke (delta wave), best seen in lead I.
Treatment depends on diagnosis
Distinguishing supraventricular tachycardia from ventricular tachycardia is important, as the treatments differ. Supraventricular tachycardia is treated with adenosine, calcium channel blockers, and beta-blockers, which are not only ineffective for ventricular tachycardia, but rarely may precipitate hemodynamic deterioration.
Also important is distinguishing pre-excitation atrial fibrillation from other types of supraventricular tachycardia with aberrancy, because the nodal blockade used to treat other causes of the condition may worsen the tachycardia via the accessory pathway. If pre-excitation atrial fibrillation is suspected on the basis of an irregular wide complex tachycardia with delta waves on ECG, then procainamide—a sodium channel blocker that affects the cardiac action potential and prolongs the refractory period of the accessory pathway—can be used to help control the arrhythmia.1
Brugada criteria aid diagnosis
In 1991, Brugada et al2 devised an algorithm to differentiate ventricular tachycardia from supraventricular tachycardia with aberrancy in the setting of regular wide complex tachycardia (Figure 3). It has a sensitivity of 98.7% and a specificity of 96.5% for diagnosing ventricular tachycardia and 96.5% sensitivity and 98.7% specificity for diagnosing supraventricular tachycardia with aberrant conduction. Using the algorithm, only 11 (ie, 2%) of the 544 tachycardias in their study were misclassified.2–4
The Brugada algorithm consists of four criteria, with the presence of any leading to a diagnosis of ventricular tachycardia:
- Absence of an RS complex in all precordial leads (the QRS complexes in precordial leads have all negative or all positive deflections).
- An RS interval in at least one precordial lead of at least 100 ms (the interval is measured from the onset of R to the nadir of the S wave).
- AV dissociation, as determined by the existence of P waves marching out independent of the QRS complexes, capture beats (narrow QRS complexes resulting from the rare occasion when an intrinsic P wave conducts down the native pathway), or fusion beats (combined capture beat and ventricular beat, resulting in a different morphology than most of the wide QRS complexes present).
- Leads V1, V2, and V6 satisfying the classic morphologic criteria for ventricular tachycardia.
If none of these criteria are met, supraventricular tachycardia is diagnosed.
In our patient, we can further confirm the diagnosis of antidromic atrioventricular reentrant tachycardia by using Brugada criteria to exclude ventricular tachycardia (Figure 3): the ECG (Figure 1) shows an RS complex in multiple precordial leads, the maximum RS interval is less than 100 ms in the precordial leads, there is no evidence of AV dissociation (lead II in the continuous strip shows buried P waves associated with QRS), and morphologic criteria are not met for ventricular tachycardia in leads V1, V2, and V6.
IS CARDIOVERSION NEEDED?
According to the American Heart Association guidelines for advanced cardiopulmonary life support, patients with tachyarrhythmias who are hemodynamically unstable should undergo cardioversion immediately.5
Our patient, who has a heart rate faster than 200 bpm and a systolic blood pressure of only 60 mm Hg, undergoes synchronized cardioversion in the emergency department. Immediately afterward, his ECG (Figure 4) demonstrates sinus rhythm with pre-excitation consistent with type B Wolff-Parkinson-White syndrome.

Once he is hemodynamically stable, a more thorough physical examination is performed. Examination of the head, ears, eyes, nose, and throat is unremarkable. He has no jugular venous distention or carotid bruits. His lungs are clear to auscultation bilaterally, without wheezes. His cardiac examination shows a regular rate and rhythm, normal first and second heart sounds, and no murmurs, rubs, or gallops.
WHICH DIAGNOSTIC STUDIES ARE NEEDED?
Laboratory tests
In an otherwise healthy young patient presenting with an arrhythmia, the initial laboratory workup should focus on a precipitating illness or a disease state that may incite an arrhythmia.
Our patient is evaluated for infection or septic shock (white blood cell count with differential), anemia (hemoglobin), thyrotoxicosis (thyroid-stimulating hormone and free thyroxine levels), drug abuse (urine toxicology screen), and cardiac syndromes including structural heart disease and myocardial injury (cardiac enzymes and B-type natriuretic peptide).6
His initial laboratory tests show normal electrolyte levels and renal function, leukocytosis with a white blood cell count of 15.6 × 109/L (normal 4.0–10.0), mildly elevated thyroid-stimulating hormone, and a negative urine toxicology screen.
Transthoracic echocardiography
For a young patient presenting with pre-excitation on ECG and hemodynamic instability, transthoracic echocardiography to evaluate chamber size and look for structural abnormalities is a reasonable option.

Our patient undergoes transthoracic echocardiography, which demonstrates normal left ventricular size and function with a left ventricular ejection fraction of 69%, moderate right atrial enlargement, and mild right ventricular enlargement (Figure 5). The septal leaflet of the tricuspid valve is apically displaced, and there is mild regurgitation.
DIAGNOSIS: EBSTEIN ANOMALY
These findings are consistent with Ebstein anomaly. It can be recognized on transthoracic echocardiography as adherence of the septal and posterior tricuspid valve leaflets to the myocardium due to failure of the tissue to detach during embryogenesis, apical displacement of the annulus, right atrial enlargement, and right ventricular enlargement.7–10 Apical displacement of the tricuspid valve is a hallmark finding and must be more than 20 mm or 8 mm/m2 of body surface area to make the diagnosis.11–13 ECG often demonstrates right atrial enlargement, first-degree atrial ventricular block, and right bundle branch block.
Ebstein anomaly is a rare embryonic developmental abnormality of the tricuspid valve. It occurs in 1 to 5 of 200,000 live births, accounting for approximately 0.5% of all congenital heart disease.14,15 Most cases are sporadic and result from failure of the ventricle to delaminate during embryogenesis of the tricuspid valve, resulting in apical displacement of either the septal, posterior, or, very rarely, anterior leaflet of the tricuspid valve.7,8 The prevalence is higher in infants whose mothers took lithium during early pregnancy.16
2. Which of the following is not a common finding associated with Ebstein anomaly?
- Apical displacement of the septal leaflet of the tricuspid valve
- Wolff-Parkinson-White syndrome
- Accessory bypass tract
- Tachyarrhythmias
- Increased risk of sudden death
- Left-sided heart failure
The answer is left-sided heart failure. Ebstein anomaly is associated with increased risk of tachyarrhythmias, right-sided heart failure, and sudden death.7,8,17,18 In Ebstein anomaly, the tricuspid valve forms closer to the apex, so the part of the right ventricle that is superior to the displaced tricuspid valve functions as the right atrium, thus the term “atrialized” right ventricle. These abnormalities create an environment for accessory pathways, most commonly type B Wolff-Parkinson-White syndrome.19 Biventricular dysfunction can occur in rare severe cases.7,8,18
Our patient is found to have an accessory tract-mediated antidromic atrioventricular reentrant tachycardia in the setting of Wolff-Parkinson-White syndrome and Ebstein anomaly. This is further confirmed with an electrophysiology study demonstrating a right posterior accessory pathway.
TREATMENT FOR EBSTEIN ANOMALY
3. Which treatment is advised for Ebstein anomaly?
- Observation alone
- Standard heart failure medications
- Radiofrequency catheter ablation
- Tricuspid valve repair or replacement
- Biventricular reconstruction
- Heart transplant
The answer is all of the above. Observation alone is advised for patients with mild symptoms, no evidence of right-to-left shunting, and only mild cardiomegaly. Medical management includes an angiotensin-converting enzyme inhibitor or angiotensin receptor blocker, a beta-blocker, and diuretics. Radiofrequency catheter ablation is the first-line therapy for patients with symptomatic Wolff-Parkinson-White syndrome.20 A patient who develops worsening right-sided heart failure, cyanosis, paradoxical emboli, or frequent tachyarrhythmias should be considered for corrective surgery, which may include tricuspid valve repair or replacement, or biventricular reconstruction.7,8,21 Cardiac transplant is reserved for severe cases.8
On hospital day 4, our patient undergoes successful radiofrequency catheter ablation without complications. At follow-up 3 months later, he continues to do well, with resolution of his symptoms and no further evidence of pre-excitation. His postprocedure ECG no longer shows delta waves.
TAKE-HOME POINTS
- For a patient with regular wide complex tachycardia, the first step is to assess hemodynamic stability. If the patient is hemodynamically unstable, emergent cardioversion is indicated.
- The differential diagnosis for regular wide complex tachycardia includes supraventricular tachycardia with aberrancy (orthodromic atrioventricular reentrant tachycardia, antidromic atrioventricular reentrant tachycardia, atrial tachycardia), and ventricular tachycardia.
- When pre-excited atrial fibrillation is suspected, AV nodal blocking agents should be avoided, as they may worsen tachyarrhythmia. Sodium channel blockers such as procainamide can help slow down the conduction of the accessory pathway.
- Ebstein anomaly is diagnosed on transthoracic echocardiography as apical displacement of the tricuspid valve resulting in atrialization of the right ventricle.
- Patients with Ebstein anomaly have a higher risk of death from right-sided heart failure and tachyarrhythmias, most commonly type B Wolff-Parkinson-White syndrome.
- Ebstein anomaly is medically managed with standard heart failure medications, including neurohormonal blockade therapies.
- Patients with Ebstein anomaly and cyanosis require surgical intervention with either valve repair or replacement.
Acknowledgment: We thank Dr. William Collins for his contribution in reviewing the manuscript and his technical expertise in developing some of the figures.
- January CT, Wann L, Alpert JS, et al; ACC/AHA Task Force Members. 2014 AHA/ACC/HRS Guideline for the Management of Patients With Atrial Fibrillation: Executive Summary: A Report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the Heart Rhythm Society. Circulation 2014; 130:2071–2104.
- Brugada P, Brugada J, Mont L, Smeets J, Andries EW. A new approach to the differential diagnosis of a regular tachycardia with a wide QRS complex. Circulation 1991; 83:1649–1659.
- Alzand BS, Crijns HJ. Diagnostic criteria of broad QRS complex tachycardia: decades of evolution. Europace 2011; 13:465–472.
- Wellens HJ, Bar FW, Lie KI. The value of the electrocardiogram in the differential diagnosis of a tachycardia with a widened QRS complex. Am J Med 1978; 64:27–33.
- Field JM, Hazinski MF, Sayre MR, et al. Part 1: executive summary: 2010 American Heart Association Guidelines for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care. Circulation 2010; 122(suppl 3):S640–S656.
- Walkey AJ, Wiener RS, Ghobrial JM, Curtis LH, Benjamin EJ. Incident stroke and mortality associated with new-onset atrial fibrillation in patients hospitalized with severe sepsis. JAMA 2011; 306:2248–2254.
- Attenhofer Jost CH, Connolly HM, Edwards WD, Hayes D, Warnes CA, Danielson GK. Ebstein’s anomaly - review of a multifaceted congenital cardiac condition. Swiss Med Wkly 2005; 135:269–281.
- Attenhofer Jost CH, Connolly HM, Dearani JA, Edwards WD, Danielson GK. Ebstein’s anomaly. Circulation 2007; 115:277–285.
- Oechslin E, Buchholz S, Jenni R. Ebstein’s anomaly in adults: Doppler-echocardiographic evaluation. Thorac Cardiovasc Surg 2000; 48:209–213.
- Ali SK, Nimeri NA. Clinical and echocardiographic features of Ebstein’s malformation in Sudanese patients. Cardiol Young 2006; 16:147–151.
- Edwards WD. Embryology and pathologic features of Ebstein’s anomaly. Prog Pediatr Cardiol 1993; 2:5–15.
- Shiina A, Seward JB, Edwards WD, Hagler DJ, Tajik AJ. Two dimensional echocardiographic spectrum of Ebstein’s anomaly: detailed anatomic assessment. J Am Coll Cardiol 1984; 3:356–370.
- Gussenhoven EJ, Stewart PA, Becker AE, Essed CE, Ligtvoet KM, De Villeneuve VH. “Offsetting” of the septal tricuspid leaflet in normal hearts and in hearts with Ebstein’s anomaly. Anatomic and echographic correlation. Am J Cardiol 1984; 54:172–176.
- Brickner ME, Hillis LD, Lange RA. Congenital heart disease in adults. Second of two parts. N Engl J Med 2000; 342:334–342.
- Report of the New England Regional Infant Cardiac Program. Pediatrics 1980; 65:375–461.
- Cohen LS, Friedman JM, Jefferson JW, Johnson EM, Weiner ML. A reevaluation of risk of in utero exposure to lithium. JAMA 1994; 271:146–150.
- Paranon S, Acar P. Ebstein’s anomaly of the tricuspid valve: from fetus to adult: congenital heart disease. Heart 2008; 94:237–243.
- Watson H. Natural history of Ebstein’s anomaly of tricuspid valve in childhood and adolescence. An international co-operative study of 505 cases. Br Heart J 1974; 36:417–427.
- Delhaas T, Sarvaas GJ, Rijlaarsdam ME, et al. A multicenter, long-term study on arrhythmias in children with Ebstein anomaly. Pediatr Cardiol 2010; 31:229–223.
- Tischenko A, Fox DJ, Yee R, et al. When should we recommend catheter ablation for patients with the Wolff-Parkinson-White syndrome? Curr Opin Cardiol 2008; 23:32–37.
- Misaki T, Watanabe G, Iwa T, et al. Surgical treatment of patients with Wolff-Parkinson-White syndrome and associated Ebstein’s anomaly. J Thorac Cardiovasc Surg 1995; 110:1702–1707.
A 23-year-old man presents to the emergency department with the sudden onset of palpitations, lightheadedness, and dyspnea, accompanied by weakness and nausea, which started earlier in the evening. He estimates that he has experienced 15 similar episodes, lasting minutes to hours, since the age of 16, with the last one 3 years ago. These episodes typically end by themselves or with self-induced vomiting and lying supine. The current episode did not resolve with these maneuvers.
He has never received medical attention for these symptoms. He has no chest pain, orthopnea, paroxysmal nocturnal dyspnea, lower extremity edema, or syncope. He has had no recent illness, contacts with sick people, or travel.
The patient’s history includes a “childhood heart murmur,” which resolved, and also mild asthma. He is otherwise healthy but has not received regular medical care. He used to play competitive soccer but quit because playing made his symptoms of dyspnea on exertion and palpitations much worse.
He uses marijuana frequently and alcohol occasionally. He does not smoke tobacco or use other recreational drugs. Other than infrequent use of albuterol, he does not take any prescription or over-the-counter medications. He has no allergies. He knows of no family history of arrhythmia or sudden cardiac death.
Physical examination. On initial examination, his temperature is 36.4°C (97.5°F), heart rate 230 bpm, systolic blood pressure 60 mm Hg, respiratory rate 30 breaths per minute, oxygen saturation 100% while breathing room air, and body mass index 25 kg/m2.
He is awake, anxious, and appears ill. He speaks only in short sentences. A focused cardiac examination reveals a regular tachycardia with no appreciable murmur or extra heart sounds; the apical impulse is not displaced. His lungs are clear. His abdomen is soft and nontender. He has 2+ pulses on a scale of 0 to 4+, with no peripheral edema.
His initial electrocardiogram (ECG) (Figure 1) shows a heart rate of 260 bpm and a regular wide complex tachycardia, defined as a rate greater than 100 bpm and a QRS complex wider than 0.12 seconds.
FOCUS ON REGULAR WIDE COMPLEX TACHYCARDIA
1. Which of the following is not in the differential diagnosis of regular wide complex tachycardia?
- Monomorphic ventricular tachycardia
- Orthodromic atrioventricular reentrant tachycardia
- Antidromic atrioventricular reentrant tachycardia
- Sinus tachycardia with bundle branch block
Orthodromic atrioventricular reentrant tachycardia is not in the differential diagnosis.
Wide complex tachycardia can occur when the impulse originates outside the normal conduction system or when there is abnormal ventricular activation through the atrioventricular (AV) node and His-Purkinje system.
The main distinction to make when diagnosing the cause of a wide complex tachycardia is between the following:
Monomorphic ventricular tachycardia, which originates from a single ventricular focus that depolarizes the adjacent myocardium in a stepwise fashion, causing a wide QRS complex that does not begin in the native conduction system, and
Sinus tachycardia with bundle branch block, ie, supraventricular tachycardia with aberrant conduction within the normal conduction system.
Three different conduction patterns are seen with atrioventricular reentrant tachycardia (Figure 2):
Sinus depolarization (Figure 2), in which the atrial impulse travels down the AV node, and the accessory pathway can be hidden and not contribute to the surface ECG.
Orthodromic atrioventricular reentrant tachycardia (Figure2), in which the depolarizing impulse travels antegrade down the AV node, then propagates from the ventricle back to the atria via the accessory pathway, resulting in a narrow QRS.
Antidromic atrioventricular reentrant tachycardia (Figure 2), in which the depolarization travels antegrade down the accessory pathway then propagates from the ventricle back to the atria via the AV node, resulting in a wide complex QRS with a delta wave.
Important features of the patient’s electrocardiogram (Figure 1) are consistent with antidromic atrioventricular reentrant tachycardia:
- “Buried” retrograde P waves, which are best seen in the continuous strip of lead II as a positive deflection notching in the negative nadir of the wave
- The PR segment is short, suggesting retrograde atrial depolarization
- The P wave is followed by a slow slurred upstroke (delta wave), best seen in lead I.
Treatment depends on diagnosis
Distinguishing supraventricular tachycardia from ventricular tachycardia is important, as the treatments differ. Supraventricular tachycardia is treated with adenosine, calcium channel blockers, and beta-blockers, which are not only ineffective for ventricular tachycardia, but rarely may precipitate hemodynamic deterioration.
Also important is distinguishing pre-excitation atrial fibrillation from other types of supraventricular tachycardia with aberrancy, because the nodal blockade used to treat other causes of the condition may worsen the tachycardia via the accessory pathway. If pre-excitation atrial fibrillation is suspected on the basis of an irregular wide complex tachycardia with delta waves on ECG, then procainamide—a sodium channel blocker that affects the cardiac action potential and prolongs the refractory period of the accessory pathway—can be used to help control the arrhythmia.1
Brugada criteria aid diagnosis
In 1991, Brugada et al2 devised an algorithm to differentiate ventricular tachycardia from supraventricular tachycardia with aberrancy in the setting of regular wide complex tachycardia (Figure 3). It has a sensitivity of 98.7% and a specificity of 96.5% for diagnosing ventricular tachycardia and 96.5% sensitivity and 98.7% specificity for diagnosing supraventricular tachycardia with aberrant conduction. Using the algorithm, only 11 (ie, 2%) of the 544 tachycardias in their study were misclassified.2–4
The Brugada algorithm consists of four criteria, with the presence of any leading to a diagnosis of ventricular tachycardia:
- Absence of an RS complex in all precordial leads (the QRS complexes in precordial leads have all negative or all positive deflections).
- An RS interval in at least one precordial lead of at least 100 ms (the interval is measured from the onset of R to the nadir of the S wave).
- AV dissociation, as determined by the existence of P waves marching out independent of the QRS complexes, capture beats (narrow QRS complexes resulting from the rare occasion when an intrinsic P wave conducts down the native pathway), or fusion beats (combined capture beat and ventricular beat, resulting in a different morphology than most of the wide QRS complexes present).
- Leads V1, V2, and V6 satisfying the classic morphologic criteria for ventricular tachycardia.
If none of these criteria are met, supraventricular tachycardia is diagnosed.
In our patient, we can further confirm the diagnosis of antidromic atrioventricular reentrant tachycardia by using Brugada criteria to exclude ventricular tachycardia (Figure 3): the ECG (Figure 1) shows an RS complex in multiple precordial leads, the maximum RS interval is less than 100 ms in the precordial leads, there is no evidence of AV dissociation (lead II in the continuous strip shows buried P waves associated with QRS), and morphologic criteria are not met for ventricular tachycardia in leads V1, V2, and V6.
IS CARDIOVERSION NEEDED?
According to the American Heart Association guidelines for advanced cardiopulmonary life support, patients with tachyarrhythmias who are hemodynamically unstable should undergo cardioversion immediately.5
Our patient, who has a heart rate faster than 200 bpm and a systolic blood pressure of only 60 mm Hg, undergoes synchronized cardioversion in the emergency department. Immediately afterward, his ECG (Figure 4) demonstrates sinus rhythm with pre-excitation consistent with type B Wolff-Parkinson-White syndrome.
Once he is hemodynamically stable, a more thorough physical examination is performed. Examination of the head, ears, eyes, nose, and throat is unremarkable. He has no jugular venous distention or carotid bruits. His lungs are clear to auscultation bilaterally, without wheezes. His cardiac examination shows a regular rate and rhythm, normal first and second heart sounds, and no murmurs, rubs, or gallops.
WHICH DIAGNOSTIC STUDIES ARE NEEDED?
Laboratory tests
In an otherwise healthy young patient presenting with an arrhythmia, the initial laboratory workup should focus on a precipitating illness or a disease state that may incite an arrhythmia.
Our patient is evaluated for infection or septic shock (white blood cell count with differential), anemia (hemoglobin), thyrotoxicosis (thyroid-stimulating hormone and free thyroxine levels), drug abuse (urine toxicology screen), and cardiac syndromes including structural heart disease and myocardial injury (cardiac enzymes and B-type natriuretic peptide).6
His initial laboratory tests show normal electrolyte levels and renal function, leukocytosis with a white blood cell count of 15.6 × 109/L (normal 4.0–10.0), mildly elevated thyroid-stimulating hormone, and a negative urine toxicology screen.
Transthoracic echocardiography
For a young patient presenting with pre-excitation on ECG and hemodynamic instability, transthoracic echocardiography to evaluate chamber size and look for structural abnormalities is a reasonable option.
Our patient undergoes transthoracic echocardiography, which demonstrates normal left ventricular size and function with a left ventricular ejection fraction of 69%, moderate right atrial enlargement, and mild right ventricular enlargement (Figure 5). The septal leaflet of the tricuspid valve is apically displaced, and there is mild regurgitation.
DIAGNOSIS: EBSTEIN ANOMALY
These findings are consistent with Ebstein anomaly. It can be recognized on transthoracic echocardiography as adherence of the septal and posterior tricuspid valve leaflets to the myocardium due to failure of the tissue to detach during embryogenesis, apical displacement of the annulus, right atrial enlargement, and right ventricular enlargement.7–10 Apical displacement of the tricuspid valve is a hallmark finding and must be more than 20 mm or 8 mm/m2 of body surface area to make the diagnosis.11–13 ECG often demonstrates right atrial enlargement, first-degree atrial ventricular block, and right bundle branch block.
Ebstein anomaly is a rare embryonic developmental abnormality of the tricuspid valve. It occurs in 1 to 5 of 200,000 live births, accounting for approximately 0.5% of all congenital heart disease.14,15 Most cases are sporadic and result from failure of the ventricle to delaminate during embryogenesis of the tricuspid valve, resulting in apical displacement of either the septal, posterior, or, very rarely, anterior leaflet of the tricuspid valve.7,8 The prevalence is higher in infants whose mothers took lithium during early pregnancy.16
2. Which of the following is not a common finding associated with Ebstein anomaly?
- Apical displacement of the septal leaflet of the tricuspid valve
- Wolff-Parkinson-White syndrome
- Accessory bypass tract
- Tachyarrhythmias
- Increased risk of sudden death
- Left-sided heart failure
The answer is left-sided heart failure. Ebstein anomaly is associated with increased risk of tachyarrhythmias, right-sided heart failure, and sudden death.7,8,17,18 In Ebstein anomaly, the tricuspid valve forms closer to the apex, so the part of the right ventricle that is superior to the displaced tricuspid valve functions as the right atrium, thus the term “atrialized” right ventricle. These abnormalities create an environment for accessory pathways, most commonly type B Wolff-Parkinson-White syndrome.19 Biventricular dysfunction can occur in rare severe cases.7,8,18
Our patient is found to have an accessory tract-mediated antidromic atrioventricular reentrant tachycardia in the setting of Wolff-Parkinson-White syndrome and Ebstein anomaly. This is further confirmed with an electrophysiology study demonstrating a right posterior accessory pathway.
TREATMENT FOR EBSTEIN ANOMALY
3. Which treatment is advised for Ebstein anomaly?
- Observation alone
- Standard heart failure medications
- Radiofrequency catheter ablation
- Tricuspid valve repair or replacement
- Biventricular reconstruction
- Heart transplant
The answer is all of the above. Observation alone is advised for patients with mild symptoms, no evidence of right-to-left shunting, and only mild cardiomegaly. Medical management includes an angiotensin-converting enzyme inhibitor or angiotensin receptor blocker, a beta-blocker, and diuretics. Radiofrequency catheter ablation is the first-line therapy for patients with symptomatic Wolff-Parkinson-White syndrome.20 A patient who develops worsening right-sided heart failure, cyanosis, paradoxical emboli, or frequent tachyarrhythmias should be considered for corrective surgery, which may include tricuspid valve repair or replacement, or biventricular reconstruction.7,8,21 Cardiac transplant is reserved for severe cases.8
On hospital day 4, our patient undergoes successful radiofrequency catheter ablation without complications. At follow-up 3 months later, he continues to do well, with resolution of his symptoms and no further evidence of pre-excitation. His postprocedure ECG no longer shows delta waves.
TAKE-HOME POINTS
- For a patient with regular wide complex tachycardia, the first step is to assess hemodynamic stability. If the patient is hemodynamically unstable, emergent cardioversion is indicated.
- The differential diagnosis for regular wide complex tachycardia includes supraventricular tachycardia with aberrancy (orthodromic atrioventricular reentrant tachycardia, antidromic atrioventricular reentrant tachycardia, atrial tachycardia), and ventricular tachycardia.
- When pre-excited atrial fibrillation is suspected, AV nodal blocking agents should be avoided, as they may worsen tachyarrhythmia. Sodium channel blockers such as procainamide can help slow down the conduction of the accessory pathway.
- Ebstein anomaly is diagnosed on transthoracic echocardiography as apical displacement of the tricuspid valve resulting in atrialization of the right ventricle.
- Patients with Ebstein anomaly have a higher risk of death from right-sided heart failure and tachyarrhythmias, most commonly type B Wolff-Parkinson-White syndrome.
- Ebstein anomaly is medically managed with standard heart failure medications, including neurohormonal blockade therapies.
- Patients with Ebstein anomaly and cyanosis require surgical intervention with either valve repair or replacement.
Acknowledgment: We thank Dr. William Collins for his contribution in reviewing the manuscript and his technical expertise in developing some of the figures.
A 23-year-old man presents to the emergency department with the sudden onset of palpitations, lightheadedness, and dyspnea, accompanied by weakness and nausea, which started earlier in the evening. He estimates that he has experienced 15 similar episodes, lasting minutes to hours, since the age of 16, with the last one 3 years ago. These episodes typically end by themselves or with self-induced vomiting and lying supine. The current episode did not resolve with these maneuvers.
He has never received medical attention for these symptoms. He has no chest pain, orthopnea, paroxysmal nocturnal dyspnea, lower extremity edema, or syncope. He has had no recent illness, contacts with sick people, or travel.
The patient’s history includes a “childhood heart murmur,” which resolved, and also mild asthma. He is otherwise healthy but has not received regular medical care. He used to play competitive soccer but quit because playing made his symptoms of dyspnea on exertion and palpitations much worse.
He uses marijuana frequently and alcohol occasionally. He does not smoke tobacco or use other recreational drugs. Other than infrequent use of albuterol, he does not take any prescription or over-the-counter medications. He has no allergies. He knows of no family history of arrhythmia or sudden cardiac death.
Physical examination. On initial examination, his temperature is 36.4°C (97.5°F), heart rate 230 bpm, systolic blood pressure 60 mm Hg, respiratory rate 30 breaths per minute, oxygen saturation 100% while breathing room air, and body mass index 25 kg/m2.
He is awake, anxious, and appears ill. He speaks only in short sentences. A focused cardiac examination reveals a regular tachycardia with no appreciable murmur or extra heart sounds; the apical impulse is not displaced. His lungs are clear. His abdomen is soft and nontender. He has 2+ pulses on a scale of 0 to 4+, with no peripheral edema.
His initial electrocardiogram (ECG) (Figure 1) shows a heart rate of 260 bpm and a regular wide complex tachycardia, defined as a rate greater than 100 bpm and a QRS complex wider than 0.12 seconds.
FOCUS ON REGULAR WIDE COMPLEX TACHYCARDIA
1. Which of the following is not in the differential diagnosis of regular wide complex tachycardia?
- Monomorphic ventricular tachycardia
- Orthodromic atrioventricular reentrant tachycardia
- Antidromic atrioventricular reentrant tachycardia
- Sinus tachycardia with bundle branch block
Orthodromic atrioventricular reentrant tachycardia is not in the differential diagnosis.
Wide complex tachycardia can occur when the impulse originates outside the normal conduction system or when there is abnormal ventricular activation through the atrioventricular (AV) node and His-Purkinje system.
The main distinction to make when diagnosing the cause of a wide complex tachycardia is between the following:
Monomorphic ventricular tachycardia, which originates from a single ventricular focus that depolarizes the adjacent myocardium in a stepwise fashion, causing a wide QRS complex that does not begin in the native conduction system, and
Sinus tachycardia with bundle branch block, ie, supraventricular tachycardia with aberrant conduction within the normal conduction system.
Three different conduction patterns are seen with atrioventricular reentrant tachycardia (Figure 2):
Sinus depolarization (Figure 2), in which the atrial impulse travels down the AV node, and the accessory pathway can be hidden and not contribute to the surface ECG.
Orthodromic atrioventricular reentrant tachycardia (Figure2), in which the depolarizing impulse travels antegrade down the AV node, then propagates from the ventricle back to the atria via the accessory pathway, resulting in a narrow QRS.
Antidromic atrioventricular reentrant tachycardia (Figure 2), in which the depolarization travels antegrade down the accessory pathway then propagates from the ventricle back to the atria via the AV node, resulting in a wide complex QRS with a delta wave.
Important features of the patient’s electrocardiogram (Figure 1) are consistent with antidromic atrioventricular reentrant tachycardia:
- “Buried” retrograde P waves, which are best seen in the continuous strip of lead II as a positive deflection notching in the negative nadir of the wave
- The PR segment is short, suggesting retrograde atrial depolarization
- The P wave is followed by a slow slurred upstroke (delta wave), best seen in lead I.
Treatment depends on diagnosis
Distinguishing supraventricular tachycardia from ventricular tachycardia is important, as the treatments differ. Supraventricular tachycardia is treated with adenosine, calcium channel blockers, and beta-blockers, which are not only ineffective for ventricular tachycardia, but rarely may precipitate hemodynamic deterioration.
Also important is distinguishing pre-excitation atrial fibrillation from other types of supraventricular tachycardia with aberrancy, because the nodal blockade used to treat other causes of the condition may worsen the tachycardia via the accessory pathway. If pre-excitation atrial fibrillation is suspected on the basis of an irregular wide complex tachycardia with delta waves on ECG, then procainamide—a sodium channel blocker that affects the cardiac action potential and prolongs the refractory period of the accessory pathway—can be used to help control the arrhythmia.1
Brugada criteria aid diagnosis
In 1991, Brugada et al2 devised an algorithm to differentiate ventricular tachycardia from supraventricular tachycardia with aberrancy in the setting of regular wide complex tachycardia (Figure 3). It has a sensitivity of 98.7% and a specificity of 96.5% for diagnosing ventricular tachycardia and 96.5% sensitivity and 98.7% specificity for diagnosing supraventricular tachycardia with aberrant conduction. Using the algorithm, only 11 (ie, 2%) of the 544 tachycardias in their study were misclassified.2–4
The Brugada algorithm consists of four criteria, with the presence of any leading to a diagnosis of ventricular tachycardia:
- Absence of an RS complex in all precordial leads (the QRS complexes in precordial leads have all negative or all positive deflections).
- An RS interval in at least one precordial lead of at least 100 ms (the interval is measured from the onset of R to the nadir of the S wave).
- AV dissociation, as determined by the existence of P waves marching out independent of the QRS complexes, capture beats (narrow QRS complexes resulting from the rare occasion when an intrinsic P wave conducts down the native pathway), or fusion beats (combined capture beat and ventricular beat, resulting in a different morphology than most of the wide QRS complexes present).
- Leads V1, V2, and V6 satisfying the classic morphologic criteria for ventricular tachycardia.
If none of these criteria are met, supraventricular tachycardia is diagnosed.
In our patient, we can further confirm the diagnosis of antidromic atrioventricular reentrant tachycardia by using Brugada criteria to exclude ventricular tachycardia (Figure 3): the ECG (Figure 1) shows an RS complex in multiple precordial leads, the maximum RS interval is less than 100 ms in the precordial leads, there is no evidence of AV dissociation (lead II in the continuous strip shows buried P waves associated with QRS), and morphologic criteria are not met for ventricular tachycardia in leads V1, V2, and V6.
IS CARDIOVERSION NEEDED?
According to the American Heart Association guidelines for advanced cardiopulmonary life support, patients with tachyarrhythmias who are hemodynamically unstable should undergo cardioversion immediately.5
Our patient, who has a heart rate faster than 200 bpm and a systolic blood pressure of only 60 mm Hg, undergoes synchronized cardioversion in the emergency department. Immediately afterward, his ECG (Figure 4) demonstrates sinus rhythm with pre-excitation consistent with type B Wolff-Parkinson-White syndrome.
Once he is hemodynamically stable, a more thorough physical examination is performed. Examination of the head, ears, eyes, nose, and throat is unremarkable. He has no jugular venous distention or carotid bruits. His lungs are clear to auscultation bilaterally, without wheezes. His cardiac examination shows a regular rate and rhythm, normal first and second heart sounds, and no murmurs, rubs, or gallops.
WHICH DIAGNOSTIC STUDIES ARE NEEDED?
Laboratory tests
In an otherwise healthy young patient presenting with an arrhythmia, the initial laboratory workup should focus on a precipitating illness or a disease state that may incite an arrhythmia.
Our patient is evaluated for infection or septic shock (white blood cell count with differential), anemia (hemoglobin), thyrotoxicosis (thyroid-stimulating hormone and free thyroxine levels), drug abuse (urine toxicology screen), and cardiac syndromes including structural heart disease and myocardial injury (cardiac enzymes and B-type natriuretic peptide).6
His initial laboratory tests show normal electrolyte levels and renal function, leukocytosis with a white blood cell count of 15.6 × 109/L (normal 4.0–10.0), mildly elevated thyroid-stimulating hormone, and a negative urine toxicology screen.
Transthoracic echocardiography
For a young patient presenting with pre-excitation on ECG and hemodynamic instability, transthoracic echocardiography to evaluate chamber size and look for structural abnormalities is a reasonable option.
Our patient undergoes transthoracic echocardiography, which demonstrates normal left ventricular size and function with a left ventricular ejection fraction of 69%, moderate right atrial enlargement, and mild right ventricular enlargement (Figure 5). The septal leaflet of the tricuspid valve is apically displaced, and there is mild regurgitation.
DIAGNOSIS: EBSTEIN ANOMALY
These findings are consistent with Ebstein anomaly. It can be recognized on transthoracic echocardiography as adherence of the septal and posterior tricuspid valve leaflets to the myocardium due to failure of the tissue to detach during embryogenesis, apical displacement of the annulus, right atrial enlargement, and right ventricular enlargement.7–10 Apical displacement of the tricuspid valve is a hallmark finding and must be more than 20 mm or 8 mm/m2 of body surface area to make the diagnosis.11–13 ECG often demonstrates right atrial enlargement, first-degree atrial ventricular block, and right bundle branch block.
Ebstein anomaly is a rare embryonic developmental abnormality of the tricuspid valve. It occurs in 1 to 5 of 200,000 live births, accounting for approximately 0.5% of all congenital heart disease.14,15 Most cases are sporadic and result from failure of the ventricle to delaminate during embryogenesis of the tricuspid valve, resulting in apical displacement of either the septal, posterior, or, very rarely, anterior leaflet of the tricuspid valve.7,8 The prevalence is higher in infants whose mothers took lithium during early pregnancy.16
2. Which of the following is not a common finding associated with Ebstein anomaly?
- Apical displacement of the septal leaflet of the tricuspid valve
- Wolff-Parkinson-White syndrome
- Accessory bypass tract
- Tachyarrhythmias
- Increased risk of sudden death
- Left-sided heart failure
The answer is left-sided heart failure. Ebstein anomaly is associated with increased risk of tachyarrhythmias, right-sided heart failure, and sudden death.7,8,17,18 In Ebstein anomaly, the tricuspid valve forms closer to the apex, so the part of the right ventricle that is superior to the displaced tricuspid valve functions as the right atrium, thus the term “atrialized” right ventricle. These abnormalities create an environment for accessory pathways, most commonly type B Wolff-Parkinson-White syndrome.19 Biventricular dysfunction can occur in rare severe cases.7,8,18
Our patient is found to have an accessory tract-mediated antidromic atrioventricular reentrant tachycardia in the setting of Wolff-Parkinson-White syndrome and Ebstein anomaly. This is further confirmed with an electrophysiology study demonstrating a right posterior accessory pathway.
TREATMENT FOR EBSTEIN ANOMALY
3. Which treatment is advised for Ebstein anomaly?
- Observation alone
- Standard heart failure medications
- Radiofrequency catheter ablation
- Tricuspid valve repair or replacement
- Biventricular reconstruction
- Heart transplant
The answer is all of the above. Observation alone is advised for patients with mild symptoms, no evidence of right-to-left shunting, and only mild cardiomegaly. Medical management includes an angiotensin-converting enzyme inhibitor or angiotensin receptor blocker, a beta-blocker, and diuretics. Radiofrequency catheter ablation is the first-line therapy for patients with symptomatic Wolff-Parkinson-White syndrome.20 A patient who develops worsening right-sided heart failure, cyanosis, paradoxical emboli, or frequent tachyarrhythmias should be considered for corrective surgery, which may include tricuspid valve repair or replacement, or biventricular reconstruction.7,8,21 Cardiac transplant is reserved for severe cases.8
On hospital day 4, our patient undergoes successful radiofrequency catheter ablation without complications. At follow-up 3 months later, he continues to do well, with resolution of his symptoms and no further evidence of pre-excitation. His postprocedure ECG no longer shows delta waves.
TAKE-HOME POINTS
- For a patient with regular wide complex tachycardia, the first step is to assess hemodynamic stability. If the patient is hemodynamically unstable, emergent cardioversion is indicated.
- The differential diagnosis for regular wide complex tachycardia includes supraventricular tachycardia with aberrancy (orthodromic atrioventricular reentrant tachycardia, antidromic atrioventricular reentrant tachycardia, atrial tachycardia), and ventricular tachycardia.
- When pre-excited atrial fibrillation is suspected, AV nodal blocking agents should be avoided, as they may worsen tachyarrhythmia. Sodium channel blockers such as procainamide can help slow down the conduction of the accessory pathway.
- Ebstein anomaly is diagnosed on transthoracic echocardiography as apical displacement of the tricuspid valve resulting in atrialization of the right ventricle.
- Patients with Ebstein anomaly have a higher risk of death from right-sided heart failure and tachyarrhythmias, most commonly type B Wolff-Parkinson-White syndrome.
- Ebstein anomaly is medically managed with standard heart failure medications, including neurohormonal blockade therapies.
- Patients with Ebstein anomaly and cyanosis require surgical intervention with either valve repair or replacement.
Acknowledgment: We thank Dr. William Collins for his contribution in reviewing the manuscript and his technical expertise in developing some of the figures.
- January CT, Wann L, Alpert JS, et al; ACC/AHA Task Force Members. 2014 AHA/ACC/HRS Guideline for the Management of Patients With Atrial Fibrillation: Executive Summary: A Report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the Heart Rhythm Society. Circulation 2014; 130:2071–2104.
- Brugada P, Brugada J, Mont L, Smeets J, Andries EW. A new approach to the differential diagnosis of a regular tachycardia with a wide QRS complex. Circulation 1991; 83:1649–1659.
- Alzand BS, Crijns HJ. Diagnostic criteria of broad QRS complex tachycardia: decades of evolution. Europace 2011; 13:465–472.
- Wellens HJ, Bar FW, Lie KI. The value of the electrocardiogram in the differential diagnosis of a tachycardia with a widened QRS complex. Am J Med 1978; 64:27–33.
- Field JM, Hazinski MF, Sayre MR, et al. Part 1: executive summary: 2010 American Heart Association Guidelines for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care. Circulation 2010; 122(suppl 3):S640–S656.
- Walkey AJ, Wiener RS, Ghobrial JM, Curtis LH, Benjamin EJ. Incident stroke and mortality associated with new-onset atrial fibrillation in patients hospitalized with severe sepsis. JAMA 2011; 306:2248–2254.
- Attenhofer Jost CH, Connolly HM, Edwards WD, Hayes D, Warnes CA, Danielson GK. Ebstein’s anomaly - review of a multifaceted congenital cardiac condition. Swiss Med Wkly 2005; 135:269–281.
- Attenhofer Jost CH, Connolly HM, Dearani JA, Edwards WD, Danielson GK. Ebstein’s anomaly. Circulation 2007; 115:277–285.
- Oechslin E, Buchholz S, Jenni R. Ebstein’s anomaly in adults: Doppler-echocardiographic evaluation. Thorac Cardiovasc Surg 2000; 48:209–213.
- Ali SK, Nimeri NA. Clinical and echocardiographic features of Ebstein’s malformation in Sudanese patients. Cardiol Young 2006; 16:147–151.
- Edwards WD. Embryology and pathologic features of Ebstein’s anomaly. Prog Pediatr Cardiol 1993; 2:5–15.
- Shiina A, Seward JB, Edwards WD, Hagler DJ, Tajik AJ. Two dimensional echocardiographic spectrum of Ebstein’s anomaly: detailed anatomic assessment. J Am Coll Cardiol 1984; 3:356–370.
- Gussenhoven EJ, Stewart PA, Becker AE, Essed CE, Ligtvoet KM, De Villeneuve VH. “Offsetting” of the septal tricuspid leaflet in normal hearts and in hearts with Ebstein’s anomaly. Anatomic and echographic correlation. Am J Cardiol 1984; 54:172–176.
- Brickner ME, Hillis LD, Lange RA. Congenital heart disease in adults. Second of two parts. N Engl J Med 2000; 342:334–342.
- Report of the New England Regional Infant Cardiac Program. Pediatrics 1980; 65:375–461.
- Cohen LS, Friedman JM, Jefferson JW, Johnson EM, Weiner ML. A reevaluation of risk of in utero exposure to lithium. JAMA 1994; 271:146–150.
- Paranon S, Acar P. Ebstein’s anomaly of the tricuspid valve: from fetus to adult: congenital heart disease. Heart 2008; 94:237–243.
- Watson H. Natural history of Ebstein’s anomaly of tricuspid valve in childhood and adolescence. An international co-operative study of 505 cases. Br Heart J 1974; 36:417–427.
- Delhaas T, Sarvaas GJ, Rijlaarsdam ME, et al. A multicenter, long-term study on arrhythmias in children with Ebstein anomaly. Pediatr Cardiol 2010; 31:229–223.
- Tischenko A, Fox DJ, Yee R, et al. When should we recommend catheter ablation for patients with the Wolff-Parkinson-White syndrome? Curr Opin Cardiol 2008; 23:32–37.
- Misaki T, Watanabe G, Iwa T, et al. Surgical treatment of patients with Wolff-Parkinson-White syndrome and associated Ebstein’s anomaly. J Thorac Cardiovasc Surg 1995; 110:1702–1707.
- January CT, Wann L, Alpert JS, et al; ACC/AHA Task Force Members. 2014 AHA/ACC/HRS Guideline for the Management of Patients With Atrial Fibrillation: Executive Summary: A Report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the Heart Rhythm Society. Circulation 2014; 130:2071–2104.
- Brugada P, Brugada J, Mont L, Smeets J, Andries EW. A new approach to the differential diagnosis of a regular tachycardia with a wide QRS complex. Circulation 1991; 83:1649–1659.
- Alzand BS, Crijns HJ. Diagnostic criteria of broad QRS complex tachycardia: decades of evolution. Europace 2011; 13:465–472.
- Wellens HJ, Bar FW, Lie KI. The value of the electrocardiogram in the differential diagnosis of a tachycardia with a widened QRS complex. Am J Med 1978; 64:27–33.
- Field JM, Hazinski MF, Sayre MR, et al. Part 1: executive summary: 2010 American Heart Association Guidelines for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care. Circulation 2010; 122(suppl 3):S640–S656.
- Walkey AJ, Wiener RS, Ghobrial JM, Curtis LH, Benjamin EJ. Incident stroke and mortality associated with new-onset atrial fibrillation in patients hospitalized with severe sepsis. JAMA 2011; 306:2248–2254.
- Attenhofer Jost CH, Connolly HM, Edwards WD, Hayes D, Warnes CA, Danielson GK. Ebstein’s anomaly - review of a multifaceted congenital cardiac condition. Swiss Med Wkly 2005; 135:269–281.
- Attenhofer Jost CH, Connolly HM, Dearani JA, Edwards WD, Danielson GK. Ebstein’s anomaly. Circulation 2007; 115:277–285.
- Oechslin E, Buchholz S, Jenni R. Ebstein’s anomaly in adults: Doppler-echocardiographic evaluation. Thorac Cardiovasc Surg 2000; 48:209–213.
- Ali SK, Nimeri NA. Clinical and echocardiographic features of Ebstein’s malformation in Sudanese patients. Cardiol Young 2006; 16:147–151.
- Edwards WD. Embryology and pathologic features of Ebstein’s anomaly. Prog Pediatr Cardiol 1993; 2:5–15.
- Shiina A, Seward JB, Edwards WD, Hagler DJ, Tajik AJ. Two dimensional echocardiographic spectrum of Ebstein’s anomaly: detailed anatomic assessment. J Am Coll Cardiol 1984; 3:356–370.
- Gussenhoven EJ, Stewart PA, Becker AE, Essed CE, Ligtvoet KM, De Villeneuve VH. “Offsetting” of the septal tricuspid leaflet in normal hearts and in hearts with Ebstein’s anomaly. Anatomic and echographic correlation. Am J Cardiol 1984; 54:172–176.
- Brickner ME, Hillis LD, Lange RA. Congenital heart disease in adults. Second of two parts. N Engl J Med 2000; 342:334–342.
- Report of the New England Regional Infant Cardiac Program. Pediatrics 1980; 65:375–461.
- Cohen LS, Friedman JM, Jefferson JW, Johnson EM, Weiner ML. A reevaluation of risk of in utero exposure to lithium. JAMA 1994; 271:146–150.
- Paranon S, Acar P. Ebstein’s anomaly of the tricuspid valve: from fetus to adult: congenital heart disease. Heart 2008; 94:237–243.
- Watson H. Natural history of Ebstein’s anomaly of tricuspid valve in childhood and adolescence. An international co-operative study of 505 cases. Br Heart J 1974; 36:417–427.
- Delhaas T, Sarvaas GJ, Rijlaarsdam ME, et al. A multicenter, long-term study on arrhythmias in children with Ebstein anomaly. Pediatr Cardiol 2010; 31:229–223.
- Tischenko A, Fox DJ, Yee R, et al. When should we recommend catheter ablation for patients with the Wolff-Parkinson-White syndrome? Curr Opin Cardiol 2008; 23:32–37.
- Misaki T, Watanabe G, Iwa T, et al. Surgical treatment of patients with Wolff-Parkinson-White syndrome and associated Ebstein’s anomaly. J Thorac Cardiovasc Surg 1995; 110:1702–1707.
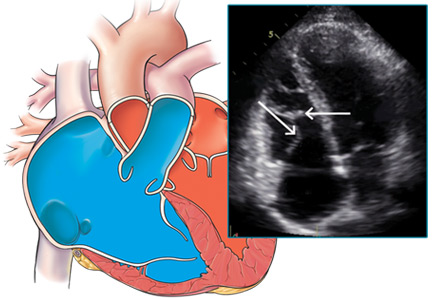
An alerting sign: Enlarged cardiac silhouette
A 75-year-old woman with a history of hypertension and left-lung lobectomy for a carcinoid tumor 10 years ago presented with a 2-week history of progressive cough, dyspnea, and fatigue. Her heart rate was 159 beats per minute with an irregularly irregular rhythm, and her respiratory rate was 36 breaths per minute. Her blood pressure was 140/90 mm Hg. Examination revealed decreased breath sounds and dullness on percussion at the left lung base, jugular venous distention with a positive hepatojugular reflux sign, and an enlarged liver. Electrocardiography showed atrial fibrillation. Chest radiography (Figure 1) revealed enlargement of the cardiac silhouette, with a disproportionately increased transverse diameter, and an obscured left costophrenic angle. A radiograph taken 13 months earlier (Figure 1) had shown a normal cardiothoracic ratio.
EVALUATION OF PERICARDIAL EFFUSION
Pericardial effusion should be suspected in patients presenting with symptoms of impaired cardiac function such as fatigue, dyspnea, nausea, palpitations, lightheadedness, cough, and hoarseness. Patients may also present with chest pain, often decreased by sitting up and leaning forward and exacerbated by lying supine.
Physical examination may reveal distant heart sounds, an absent or displaced apical impulse, dullness and increased fremitus beneath the angle of the left scapula (the Ewart sign), pulsus paradoxus, and nonspecific findings such as tachycardia and hypotension. Jugular venous distention, hepatojugular reflux, and peripheral edema suggest impaired cardiac function.
A chest radiograph showing unexplained new symmetric cardiomegaly (which is often globe-shaped) without signs of pulmonary congestion1 or with a left dominant pleural effusion is an indicator of pericardial effusion, as in our patient. Pericardial fluid may be seen outlining the heart between the epicardial and mediastinal fat, posterior to the sternum in a lateral view.
Other common causes of cardiomegaly include hypertension, congestive heart failure, valvular disease, cardiomyopathy, ischemic heart disease, and pulmonary disease.
Once pericardial effusion is suspected, the next step is to confirm its presence and determine its hemodynamic significance. Transthoracic echocardiography is the imaging test of choice to confirm effusion, as it can be done rapidly and in unstable patients.2
If transthoracic echocardiography is nondiagnostic but suspicion is high, further evaluation may include transesophageal echocardiography,3 computed tomography, or magnetic resonance imaging.
MAKING THE DIAGNOSIS
Pericardial effusion can occur as part of various diseases involving the pericardium, eg, acute pericarditis, myocarditis, autoimmune disease, postmyocardial infarction, malignancy, aortic dissection, and chest trauma. It can also be associated with certain drugs.
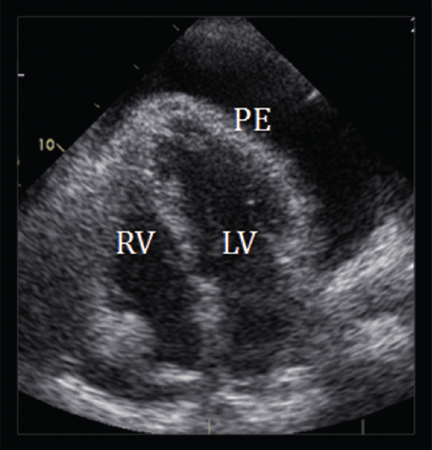
In our patient, echocardiography (Figure 2, Figure 3) demonstrated a large amount of pericardial fluid, and 820 mL of red fluid was aspirated by pericardiocentesis, resulting in relief of her respiratory symptoms. Subcostal two-dimensional echocardiography demonstrated rocking of the heart and intermittent right-ventricular collapse (watch video at www.ccjm.org). Flow cytometry demonstrated 10% kappa+ monoclonal cells. Bone marrow biopsy with immunohistochemical staining revealed infiltration by CD20+, CD5+, CD23+, and BCL1– cells, compatible with small lymphocytic lymphoma.
MALIGNANT PERICARDIAL EFFUSION
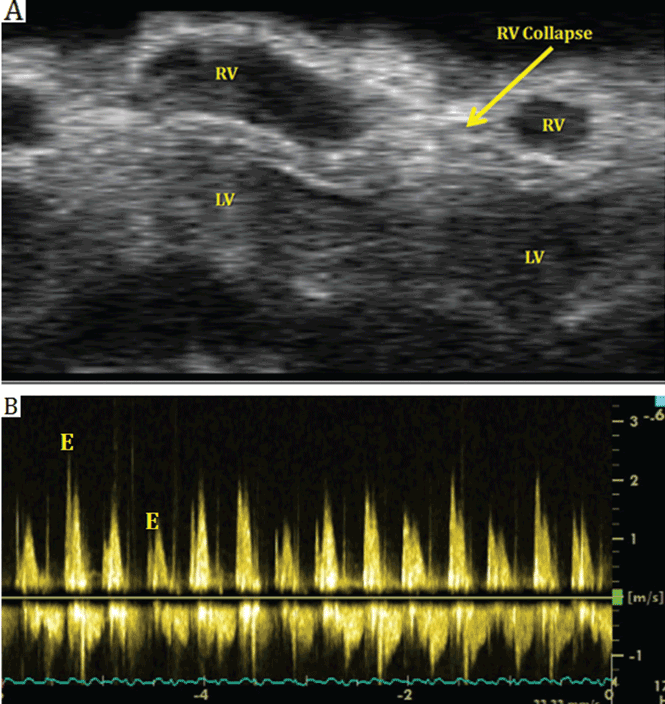
Pericardial disease can be the first manifestation of malignancy,4 more often when the patient presents with a large pericardial effusion or tamponade. Malignant tumors of the lung, breast, and esophagus—as well as lymphoma, leukemia, and melanoma—often spread to the pericardium directly or through the lymphatic vessels or bloodstream.4 In our patient, corticosteroid treatment was initiated, and echocardiography at a follow-up visit 2 months later showed no pericardial fluid.
- Khandaker MH, Espinosa RE, Nishimura RA, et al. Pericardial disease: diagnosis and management. Mayo Clin Proc 2010; 85:572–593.
- Cheitlin MD, Armstrong WF, Aurigemma GP, et al; American College of Cardiology; American Heart Association; American Society of Echocardiography. ACC/AHA/ASE 2003 guideline update for the clinical application of echocardiography: summary article: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (ACC/AHA/ASE Committee to Update the 1997 Guidelines for the Clinical Application of Echocardiography). Circulation 2003; 108:1146–1162.
- Verhaert D, Gabriel RS, Johnston D, Lytle BW, Desai MY, Klein AL. The role of multimodality imaging in the management of pericardial disease. Circ Cardiovascular Imaging 2010; 3:333–343.
- Burazor I, Imazio M, Markel G, Adler Y. Malignant pericardial effusion. Cardiology 2013; 124:224–232.
A 75-year-old woman with a history of hypertension and left-lung lobectomy for a carcinoid tumor 10 years ago presented with a 2-week history of progressive cough, dyspnea, and fatigue. Her heart rate was 159 beats per minute with an irregularly irregular rhythm, and her respiratory rate was 36 breaths per minute. Her blood pressure was 140/90 mm Hg. Examination revealed decreased breath sounds and dullness on percussion at the left lung base, jugular venous distention with a positive hepatojugular reflux sign, and an enlarged liver. Electrocardiography showed atrial fibrillation. Chest radiography (Figure 1) revealed enlargement of the cardiac silhouette, with a disproportionately increased transverse diameter, and an obscured left costophrenic angle. A radiograph taken 13 months earlier (Figure 1) had shown a normal cardiothoracic ratio.
EVALUATION OF PERICARDIAL EFFUSION
Pericardial effusion should be suspected in patients presenting with symptoms of impaired cardiac function such as fatigue, dyspnea, nausea, palpitations, lightheadedness, cough, and hoarseness. Patients may also present with chest pain, often decreased by sitting up and leaning forward and exacerbated by lying supine.
Physical examination may reveal distant heart sounds, an absent or displaced apical impulse, dullness and increased fremitus beneath the angle of the left scapula (the Ewart sign), pulsus paradoxus, and nonspecific findings such as tachycardia and hypotension. Jugular venous distention, hepatojugular reflux, and peripheral edema suggest impaired cardiac function.
A chest radiograph showing unexplained new symmetric cardiomegaly (which is often globe-shaped) without signs of pulmonary congestion1 or with a left dominant pleural effusion is an indicator of pericardial effusion, as in our patient. Pericardial fluid may be seen outlining the heart between the epicardial and mediastinal fat, posterior to the sternum in a lateral view.
Other common causes of cardiomegaly include hypertension, congestive heart failure, valvular disease, cardiomyopathy, ischemic heart disease, and pulmonary disease.
Once pericardial effusion is suspected, the next step is to confirm its presence and determine its hemodynamic significance. Transthoracic echocardiography is the imaging test of choice to confirm effusion, as it can be done rapidly and in unstable patients.2
If transthoracic echocardiography is nondiagnostic but suspicion is high, further evaluation may include transesophageal echocardiography,3 computed tomography, or magnetic resonance imaging.
MAKING THE DIAGNOSIS
Pericardial effusion can occur as part of various diseases involving the pericardium, eg, acute pericarditis, myocarditis, autoimmune disease, postmyocardial infarction, malignancy, aortic dissection, and chest trauma. It can also be associated with certain drugs.
In our patient, echocardiography (Figure 2, Figure 3) demonstrated a large amount of pericardial fluid, and 820 mL of red fluid was aspirated by pericardiocentesis, resulting in relief of her respiratory symptoms. Subcostal two-dimensional echocardiography demonstrated rocking of the heart and intermittent right-ventricular collapse (watch video at www.ccjm.org). Flow cytometry demonstrated 10% kappa+ monoclonal cells. Bone marrow biopsy with immunohistochemical staining revealed infiltration by CD20+, CD5+, CD23+, and BCL1– cells, compatible with small lymphocytic lymphoma.
MALIGNANT PERICARDIAL EFFUSION
Pericardial disease can be the first manifestation of malignancy,4 more often when the patient presents with a large pericardial effusion or tamponade. Malignant tumors of the lung, breast, and esophagus—as well as lymphoma, leukemia, and melanoma—often spread to the pericardium directly or through the lymphatic vessels or bloodstream.4 In our patient, corticosteroid treatment was initiated, and echocardiography at a follow-up visit 2 months later showed no pericardial fluid.
A 75-year-old woman with a history of hypertension and left-lung lobectomy for a carcinoid tumor 10 years ago presented with a 2-week history of progressive cough, dyspnea, and fatigue. Her heart rate was 159 beats per minute with an irregularly irregular rhythm, and her respiratory rate was 36 breaths per minute. Her blood pressure was 140/90 mm Hg. Examination revealed decreased breath sounds and dullness on percussion at the left lung base, jugular venous distention with a positive hepatojugular reflux sign, and an enlarged liver. Electrocardiography showed atrial fibrillation. Chest radiography (Figure 1) revealed enlargement of the cardiac silhouette, with a disproportionately increased transverse diameter, and an obscured left costophrenic angle. A radiograph taken 13 months earlier (Figure 1) had shown a normal cardiothoracic ratio.
EVALUATION OF PERICARDIAL EFFUSION
Pericardial effusion should be suspected in patients presenting with symptoms of impaired cardiac function such as fatigue, dyspnea, nausea, palpitations, lightheadedness, cough, and hoarseness. Patients may also present with chest pain, often decreased by sitting up and leaning forward and exacerbated by lying supine.
Physical examination may reveal distant heart sounds, an absent or displaced apical impulse, dullness and increased fremitus beneath the angle of the left scapula (the Ewart sign), pulsus paradoxus, and nonspecific findings such as tachycardia and hypotension. Jugular venous distention, hepatojugular reflux, and peripheral edema suggest impaired cardiac function.
A chest radiograph showing unexplained new symmetric cardiomegaly (which is often globe-shaped) without signs of pulmonary congestion1 or with a left dominant pleural effusion is an indicator of pericardial effusion, as in our patient. Pericardial fluid may be seen outlining the heart between the epicardial and mediastinal fat, posterior to the sternum in a lateral view.
Other common causes of cardiomegaly include hypertension, congestive heart failure, valvular disease, cardiomyopathy, ischemic heart disease, and pulmonary disease.
Once pericardial effusion is suspected, the next step is to confirm its presence and determine its hemodynamic significance. Transthoracic echocardiography is the imaging test of choice to confirm effusion, as it can be done rapidly and in unstable patients.2
If transthoracic echocardiography is nondiagnostic but suspicion is high, further evaluation may include transesophageal echocardiography,3 computed tomography, or magnetic resonance imaging.
MAKING THE DIAGNOSIS
Pericardial effusion can occur as part of various diseases involving the pericardium, eg, acute pericarditis, myocarditis, autoimmune disease, postmyocardial infarction, malignancy, aortic dissection, and chest trauma. It can also be associated with certain drugs.
In our patient, echocardiography (Figure 2, Figure 3) demonstrated a large amount of pericardial fluid, and 820 mL of red fluid was aspirated by pericardiocentesis, resulting in relief of her respiratory symptoms. Subcostal two-dimensional echocardiography demonstrated rocking of the heart and intermittent right-ventricular collapse (watch video at www.ccjm.org). Flow cytometry demonstrated 10% kappa+ monoclonal cells. Bone marrow biopsy with immunohistochemical staining revealed infiltration by CD20+, CD5+, CD23+, and BCL1– cells, compatible with small lymphocytic lymphoma.
MALIGNANT PERICARDIAL EFFUSION
Pericardial disease can be the first manifestation of malignancy,4 more often when the patient presents with a large pericardial effusion or tamponade. Malignant tumors of the lung, breast, and esophagus—as well as lymphoma, leukemia, and melanoma—often spread to the pericardium directly or through the lymphatic vessels or bloodstream.4 In our patient, corticosteroid treatment was initiated, and echocardiography at a follow-up visit 2 months later showed no pericardial fluid.
- Khandaker MH, Espinosa RE, Nishimura RA, et al. Pericardial disease: diagnosis and management. Mayo Clin Proc 2010; 85:572–593.
- Cheitlin MD, Armstrong WF, Aurigemma GP, et al; American College of Cardiology; American Heart Association; American Society of Echocardiography. ACC/AHA/ASE 2003 guideline update for the clinical application of echocardiography: summary article: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (ACC/AHA/ASE Committee to Update the 1997 Guidelines for the Clinical Application of Echocardiography). Circulation 2003; 108:1146–1162.
- Verhaert D, Gabriel RS, Johnston D, Lytle BW, Desai MY, Klein AL. The role of multimodality imaging in the management of pericardial disease. Circ Cardiovascular Imaging 2010; 3:333–343.
- Burazor I, Imazio M, Markel G, Adler Y. Malignant pericardial effusion. Cardiology 2013; 124:224–232.
- Khandaker MH, Espinosa RE, Nishimura RA, et al. Pericardial disease: diagnosis and management. Mayo Clin Proc 2010; 85:572–593.
- Cheitlin MD, Armstrong WF, Aurigemma GP, et al; American College of Cardiology; American Heart Association; American Society of Echocardiography. ACC/AHA/ASE 2003 guideline update for the clinical application of echocardiography: summary article: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (ACC/AHA/ASE Committee to Update the 1997 Guidelines for the Clinical Application of Echocardiography). Circulation 2003; 108:1146–1162.
- Verhaert D, Gabriel RS, Johnston D, Lytle BW, Desai MY, Klein AL. The role of multimodality imaging in the management of pericardial disease. Circ Cardiovascular Imaging 2010; 3:333–343.
- Burazor I, Imazio M, Markel G, Adler Y. Malignant pericardial effusion. Cardiology 2013; 124:224–232.
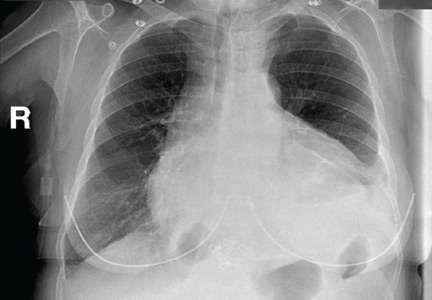
Secondary Survey of Trauma Patient
ANSWER
The radiograph shows a slightly displaced fracture of the distal fourth metacarpal head. No other injuries are present.
The patient’s hand was left in the splint, and orthopedic evaluation was obtained.
ANSWER
The radiograph shows a slightly displaced fracture of the distal fourth metacarpal head. No other injuries are present.
The patient’s hand was left in the splint, and orthopedic evaluation was obtained.
ANSWER
The radiograph shows a slightly displaced fracture of the distal fourth metacarpal head. No other injuries are present.
The patient’s hand was left in the splint, and orthopedic evaluation was obtained.
You are assisting in the evaluation and management of a trauma patient who was brought to your facility earlier today, following a motor vehicle collision. He is estimated to be in his 30s and is presumed to have been ejected, as he was found outside the vehicle. He was intubated in the field, and primary survey and resuscitation have been completed. History is otherwise unknown. The patient’s vital signs are currently stable. As you perform your secondary survey, you note that his right hand and wrist appear to be moderately swollen. He has been placed in a splint. You order a radiograph of the hand. What is your impression?
Revenge of the Deer Stand
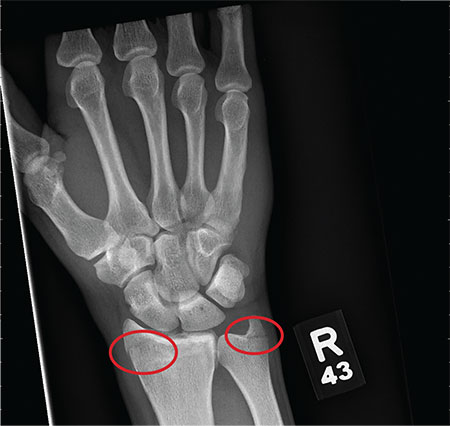
ANSWER
The radiograph shows a fracture of both the distal radius and distal ulna. The patient was placed in a splint and referred to orthopedics for outpatient follow-up.
ANSWER
The radiograph shows a fracture of both the distal radius and distal ulna. The patient was placed in a splint and referred to orthopedics for outpatient follow-up.
ANSWER
The radiograph shows a fracture of both the distal radius and distal ulna. The patient was placed in a splint and referred to orthopedics for outpatient follow-up.
As you begin your shift, knowing deer season has started in your area, you wonder how long it will be before you see your first deer stand casualty of the day. With your first patient, you wonder no more: A 30-year-old man presents for evaluation of right wrist pain after falling from his deer stand. He says one of the straps holding the stand broke, causing him to fall forward and land on his outstretched hands. His medical history is unremarkable. Inspection of the right wrist shows no obvious deformity. No significant swelling is present. There is decreased range of motion and localized tenderness over the radius and ulna. Good pulses and capillary refill are noted. You obtain a radiograph of the wrist. What is your impression?
Recreational cannabis use: Pleasures and pitfalls
Clinicians may be encountering more cannabis users than before, and may be encountering users with complications hitherto unseen. Several trends may explain this phenomenon: the legal status of cannabis is changing, cannabis today is more potent than in the past, and enthusiasts are conjuring new ways to enjoy this substance.
This article discusses the history, pharmacology, and potential complications of cannabis use.
A LONG AND TANGLED HISTORY
Cannabis is a broad term that refers to the cannabis plant and its preparations, such as marijuana and hashish, as well as to a family of more than 60 bioactive substances called cannabinoids. It is the most commonly used illegal drug in the world, with an estimated 160 million users. Each year, about 2.4 million people in the United States use it for the first time.1,2
Cannabis has been used throughout the world for recreational and spiritual purposes for nearly 5,000 years, beginning with the fabled Celestial Emperors of China. The tangled history of cannabis in America began in the 17th century, when farmers were required by law to grow it as a fiber crop. It later found its way into the US Pharmacopeia for a wide range of indications. During the long prelude to Prohibition in the latter half of the 19th century, the US government became increasingly suspicious of mind-altering substances and began restricting its prescription in 1934, culminating in its designation by the US Food and Drug Administration as a schedule I controlled substance in 1970.
Investigation into the potential medical uses for the different chemicals within cannabis is ongoing, as is debate over its changing legality and usefulness to society. The apparent cognitive dissonance surrounding the use and advocacy of medical marijuana is beyond the scope of this review,3 which will instead restrict itself to what is known of the cannabinoids and to the recreational use of cannabis.
THC IS THE PRINCIPAL PSYCHOACTIVE MOLECULE
Delta-9 tetrahydrocannabinol (THC), first isolated in 1964, was identified as the principal psychoactive constituent of cannabis in 2002.4
Two G-protein–linked cannabinoid receptors cloned in the 1990s—CB1 and CB2—were found to be a part of a system of endocannabinoid receptors present throughout the body, from the brain to the immune system to the vas deferens.5 Both receptors inhibit cellular excitation by activating inwardly rectifying potassium channels. These receptors are mostly absent in the brainstem, which may explain why cannabis use rarely causes life-threatening autonomic dysfunction. Although the intoxicating effects of marijuana are mediated by CB1 receptors, the specific mechanisms underlying the cannabis “high” are unclear.6
CANNABINOIDS ARE LIPID-SOLUBLE
The rate of absorption of cannabinoids depends on the route of administration and the type of cannabis product used. When cannabis products are smoked, up to 35% of THC is available, and the average time to peak serum concentration is 8 minutes.7 The peak concentration depends on the dose.
On the other hand, when cannabis products (eg, nabilone, dronabinol) are ingested, absorption is unpredictable because THC is unstable in gastric acid and undergoes first-pass metabolism in the liver, which reduces the drug’s bioavailability. Up to 20% of an ingested dose of THC is absorbed, and the time to peak serum concentration averages between 2 and 4 hours. Consequently, many users prefer to smoke cannabis as a means to control the desired effects.
Cannabinoids are lipid-soluble. They accumulate in fatty tissue in a biphasic pattern, initially moving into highly vascularized tissue such as the liver before accumulating in less well-vascularized tissue such as fat. They are then slowly released from fatty tissue as the fat turns over. THC itself has a volume of distribution of about 2.5 to 3.5 L/kg. It crosses the placenta and enters breast milk.8
THC is metabolized by the cytochrome P450 system, primarily by the enzymes CYP2C9 and CYP3A4. Its primary metabolite, 11-hydroxy-delta-9 THC, is also active, but subsequent metabolism produces many other inactive metabolites. THC is eliminated in feces and urine, and its half-life ranges from 2 to nearly 60 hours.8
A LITTLE ABOUT PLANTS AND STREET NAMES
The plant from which THC and nearly a hundred other chemicals, including cannabinoids, are derived has been called many things over the years:
Hemp is a tall fibrous plant grown for rope and fabric that was used as legal tender in early America. In the mid-19th century, there were over 16 million acres of hemp plantations. Hemp contains very low THC concentrations.
Cannabis is an annual flowering herb that is predominantly diecious (ie, there are male and female plants). After a centuries-long debate among taxonomists, the two principal species are considered to be C sativa and C indica, although today many cannabis cultivars are grown by a great number of breeding enthusiasts.
Concentrations of THC vary widely among cannabis cultivars, ranging historically from around 5% to today’s highly selectively bred species containing more than 30%. Concentrations in seized cannabis have been measured as high as 37%, although the average is around 11%.9 This concentration is defined by the percent of THC per dried mass of plant material tested, usually via gas chromatography.
Hashish is a solid or resinous preparation of the trichomes, or glandular hairs, that grow on the cannabis plant, chiefly on its flowers. Various methods to separate the trichomes from the rest of the plant result in a powder called kief that is then compressed into blocks or bricks. THC concentrations as high as 66% have been measured in nondomestic sources of hashish.9
Hash oil is a further purification, produced by using solvents to dissolve the resin and by filtering out remaining plant material. Evaporating the solvent produces hash oil, sometimes called butane hash oil or honey oil. This process has recently led to an increasing number of home explosions, as people attempt to make the product themselves but do not take suitable precautions when using flammable solvents such as butane. THC concentrations as high as 81% have been measured in nondomestic sources of hash oil.9
Other names for hash oil are dab, wax, and budder. Cannabis enthusiasts refer to the use of hash oil as dabbing, which involves heating a small amount (dab) of the product using a variety of paraphernalia and inhaling the vapor.
IT’S ALL ABOUT GETTING HIGH
For recreational users, the experience has always been about being intoxicated—getting high. The psychological effects range broadly from positive to negative and vary both within and between users, depending on the dose and route of administration. Additional factors that influence the psychological effects include the social and physical settings of drug use and even the user’s expectations. One user’s high is another user’s acute toxic effect.
Although subjective reports of the cannabis experience vary greatly, it typically begins with a feeling of dizziness or lightheadedness followed by a relaxed calm and a feeling of being somewhat “disconnected.” There is a quickening of the sense of humor, described by some as a fatuous euphoria; often there is silly giggling. Awareness of the senses and of music may be increased. Appetite increases, and time seems to pass quickly. Eventually, the user becomes drowsy and experiences decreased attention and difficulty maintaining a coherent conversation. Slowed reaction time and decreased psychomotor activity may also occur. The user may drift into daydreams and eventually fall asleep.
Common negative acute effects of getting high can include mild to severe anxiety and feeling tense or agitated. Clumsiness, headache, and confusion are also possible. Lingering effects the following day may include dry mouth, dry eyes, fatigue, slowed thinking, and slowed recall.6
ACUTE PHYSICAL EFFECTS
Acute physical effects of cannabis use include a rapid onset of increased airway conductance, decreased intraocular pressure, and conjunctival injection. A single cannabis cigarette can also induce cardiovascular effects including a dose-dependent increase in heart rate and blood pressure. Chronic users, however, can experience a decreased heart rate, lower blood pressure, and postural hypotension.
In a personal communication, colleagues in Colorado—where recreational use of cannabis was legalized in 2012—described a sharp increase (from virtually none) in the number of adults presenting to the emergency department with cannabis intoxication since 2012. Their patients experienced palpitations, light-headedness, and severe ataxia lasting as long as 12 hours, possibly reflecting the greater potency of current cannabis products. Most of these patients required only supportive care.
Other acute adverse cardiovascular reactions that have been reported include atrial fibrillation, ventricular tachycardia, and a fivefold increased risk of myocardial infarction in the 60 minutes following cannabis use, which subsequently drops sharply to baseline levels.10 Investigations into the cardiovascular effects of cannabis are often complicated by concurrent use of other drugs such as tobacco or cocaine. Possible mechanisms of injury include alterations in coronary microcirculation or slowed coronary flow. In fact, one author found that cannabis users with a history of myocardial infarction had a risk of death 4.2 times higher than users with no history of myocardial infarction.11,12
In children, acute toxicity has been reported from a variety of exposures to cannabis and hashish, including a report of an increase in pediatric cannabis exposures following the changes in Colorado state laws.13 Most of these patients had altered mental status ranging from drowsiness to coma; one report describes a child who experienced a first-time seizure. These patients unfortunately often underwent extensive evaluations such as brain imaging and lumbar puncture, and mechanical ventilation to protect the airway. Earlier consideration of cannabis exposure in these patients might have limited unnecessary testing. Supportive care is usually all that is needed, and most of these patients fully recover.13–17
CHRONIC EFFECTS
Cannabinoids cause a variety of adverse effects, but the ultimate risk these changes pose to human health has been difficult to calculate. Long-term studies are confounded by possible inaccuracies of patient self-reporting of cannabis use, poor control of covariates, and disparate methodologies.
For more than a century, cannabis use has been reported to cause both acute psychotic symptoms and persistent psychotic disorders.18 But the strength of this relationship is modest. Cannabis is more likely a component cause that, in addition to other factors (eg, specific genetic polymorphisms), contributes to the risk of schizophrenia. Individuals with prodromal symptoms and those who have experienced discrete episodes of psychosis related to cannabis use should be discouraged from using cannabis and cannabinoids.19–21
Mounting evidence implicates chronic cannabis use as a cause of long-term medical problems including chronic bronchitis,22 elevated rates of myocardial infarction and dysrhythmias,11 bone loss,23 and cancers at eight different sites including the lung, head, and neck.24 In view of these chronic effects, healthcare providers should caution their patients about cannabis use, as we do about other drugs such as tobacco.
WITHDRAWAL SYNDROME RECOGNIZED
Until recently, neither clinicians nor users recognized a withdrawal syndrome associated with chronic use of cannabis, probably because this syndrome is not as severe as withdrawal from other controlled substances such as opioids or sedative-hypnotics. A number of studies, however, have reported subtle cannabis withdrawal symptoms that are similar to those associated with tobacco withdrawal.
As such, the fifth and latest edition of the Diagnostic and Statistical Manual of Mental Disorders (DSM-5)25 characterized withdrawal from cannabis use in 2013. The DSM-5 criteria require cessation of heavy or prolonged use of cannabis (ie, daily or almost daily over a period of at least a few months) and three or more of the following withdrawal symptoms:
- Irritability and anger
- Nervousness
- Sleep difficulty or insomnia
- Decreased appetite or weight loss
- Restlessness
- Depressed mood
- Physical symptoms causing discomfort.
Medical treatment of cannabis withdrawal has included a range of antidepressants, mood stabilizers, and alpha-2-adrenergic agonists, all of which have limited success.26 Symptoms of cannabis withdrawal tend to be most intense soon after cessation and decline over the next few weeks.27
CANNABINOID HYPEREMESIS SYNDROME
First reported in 2004,28 cannabinoid hyperemesis syndrome is a recurrent disorder, the pathophysiology of which is poorly understood. It has three phases.
The first phase is a prodrome that may last months or years and is characterized by morning nausea, fear of vomiting, and abdominal discomfort. During this phase, the patient maintains normal eating patterns and may well increase his or her cannabis use due to its well-known antiemetic effects.
The second phase is the hyperemetic phase, characterized by intense, incapacitating emesis with episodes of vomiting throughout the day. These symptoms can be relieved only with frequent hot baths, a feature that distinguishes cannabinoid hyperemesis syndrome from other vomiting syndromes. Hot-water bathing is reported to be a compulsive but learned behavior in which the patient learns that only hot water will provide relief. The extent of relief depends on the temperature of the water—the hotter, the better. Symptoms recur as the water cools.28 Patients often present to the emergency department repeatedly with recurrent symptoms and may remain misdiagnosed or subjected to repeated extensive evaluation including laboratory testing and imaging, which are usually not revealing. If the patient has not been accurately diagnosed, there may be reported weight loss of at least 5 kg.
The third phase, recovery, may take several months to complete, possibly because of the prolonged terminal elimination time of cannabinoids. Complete cessation of cannabis use, including synthetic cannabinoids, is usually necessary.29
Diagnostic criteria for cannabinoid hyperemesis syndrome have been suggested, based on a retrospective case series that included 98 patients.30 The most common features of these affected patients were:
- Severe cyclical vomiting, predominantly in the morning
- Resolution of symptoms with cessation of cannabis use
- Symptomatic relief with hot showers or baths
- Abdominal pain
- At least weekly use of cannabis.
Interestingly, long-term cannabis use has been cited as a critical identifying feature of these patients, with the duration of cannabis use ranging from 10 to 16 years.31,32 Other reports show greater variability in duration of cannabis use before the onset of cannabinoid hyperemesis syndrome. In the large study noted above,30 32% of users reported their duration of cannabis use to be less than 1 year, rendering this criterion less useful.
How can cannabis both cause and prevent vomiting?
The body controls nausea and vomiting via complex circuitry in the brain and gut that involves many neurotransmitters (eg, dopamine, serotonin, substance P) that interact with receptors such as CB1, 5-HT1–4, alpha adrenergic receptors, and mu receptors. Interestingly, cannabis use has antiemetic properties mediated by CB1 with a still unclear additional role of CB2 receptors. Data point to the existence of an underlying antiemetic tone mediated by the endocannabinoid system.
Unfortunately, the mechanism by which cannabinoid hyperemesis syndrome occurs is unknown and represents a paradoxical effect against the otherwise antiemetic effects of cannabis. Several theories have been proposed, including delayed gastric emptying, although only a third of patients demonstrated this on scintigraphy in one study.30 Other theories include disturbance of the hypothalamic-pituitary axis, a buildup of highly lipophilic THC in the brain, and a down-regulation of cannabinoid receptors that results from chronic exposure.30 Given that this syndrome has been recognized only relatively recently, one author has suggested the cause may be recent horticultural developments.5
Treating cannabinoid hyperemesis syndrome is difficult
Treatment of cannabinoid hyperemesis syndrome is notoriously difficult, with many authors reporting resistance to the usual first-line antiemetic drugs. Generally, treatment should include hydration and acid-suppression therapy because endoscopic evaluation of several patients has revealed varying degrees of esophagitis and gastritis.29
Antiemetic therapy should target receptors known to mediate nausea and vomiting. In some cases, antiemetic drugs are more effective when used in combination. Agents include the serotonergic receptor antagonists ondansetron and granisetron, the dopamine antagonists prochlorperazine and metoclopramide, and even haloperidol.33,34 Benzodiazepines may be effective by causing sedation, anxiolysis, and depression of the vomiting center.34,35 Two antihistamines—dimenhydrinate and diphenhydramine—have antiemetic effects, perhaps by inhibiting acetylcholine.34
Aprepitant is a neurokinin-1 antagonist that inhibits the action of substance P. When combined with a corticosteroid and a serotonin antagonist, it relieves nausea and vomiting in chemotherapy patients.34,36
Corticosteroids such as dexamethasone are potent antiemetics thought to inhibit prostaglandin synthesis.34
Capsaicin cream applied to the abdomen has also been reported to relieve symptoms, possibly through an interaction between the TRPv1 receptor and the endocannabinoid system.37,38
DIAGNOSTIC TESTING
Cannabinoids are detectable in plasma and urine, with urine testing being more common.
Common laboratory methods include the enzyme-multiplied immunoassay technique (EMIT) and radioimmunoassay. Gas chromatography coupled with mass spectrometry is the most specific assay; it is used for confirmation and is the reference method.
EMIT is a qualitative urine test that detects 9-carboxy-THC as well as other THC metabolites. These urine tests detect all metabolites, and the result is reported as positive if the total concentration is greater than or equal to a prespecified threshold level, such as 20 ng/mL or 50 ng/mL. A positive test does not denote intoxication, nor does the test identify the source of THC (eg, cannabis, dronabinol, butane hash oil). EMIT does not detect nabilone. The National Institute on Drug Abuse guidelines for urine testing specify a test threshold concentration of 50 ng/mL for screening and 15 ng/mL for confirmation.

Many factors affect the detection of THC metabolites and their presence and duration in urine: dose, duration of use, route of exposure, hydration status, urine volume and concentration, and urine pH. THC metabolites have been detected in urine using gas chromatography-mass spectrometry for up to 7 days after smoking one marijuana cigarette.7 Chronic users have also been reported to have positive urine EMIT tests for up to 46 days after cannabis cessation.39 Detection may be further complicated in chronic users: in one study, users produced both negative and positive specimens over 24 days, suggesting that diet and exercise may influence clearance.40 Also, many factors are known to produce false-positive and false-negative results for these immunoassays (Table 1).39,41
In the United States, penalties for driving under the influence of cannabis vary from state to state, and laws specify plasma testing for quantitative analysis. Some states use a threshold of 5 ng/mL in plasma to imply driving under the influence, whereas others use any detectable amount. Currently, there are no generally accepted guidelines for storage and testing of blood samples, despite the known instability of analytes.42
Saliva, hair, and sweat can also be used for cannabinoid testing. Saliva is easy to collect, can be tested for metabolites to rule out passive cannabis exposure, and can be positive for up to 1 day after exposure. Calculating a blood or plasma concentration from a saliva sample is not possible, however.
Hair testing can also rule out passive exposure, but THC binds very little to melanin, resulting in very low concentrations requiring sensitive tests, such as gas chromatography with tandem mass spectrometry.
Only one device is commercially available for sweat testing; further work is needed to elucidate sweat excretion pharmacokinetics and the limitations of the collection devices.43
CLINICAL MANAGEMENT IS GENERALLY SUPPORTIVE
Historically, clinical toxicity from recreational cannabis use is rarely serious or severe and generally responds to supportive care. Reports of cannabis exposure to poison centers are one-tenth of those reported for ethanol exposures annually.44 Gastrointestinal decontamination with activated charcoal is not recommended, even for orally administered cannabis, since the risks outweigh the expected benefits. Agitation or anxiety may be treated with benzodiazepines as needed. There is no antidote for cannabis toxicity. The ever-increasing availability of high-concentration THC preparations may prompt more aggressive supportive measures in the future.
SYNTHETIC MARIJUANA ALTERNATIVES
Available since the early 2000s, herbal marijuana alternatives are legally sold as incense or potpourri and are often labeled “not for human consumption.” They are known by such brand names as K2 and Spice and contain blends of herbs adulterated with synthetic cannabinoid chemicals developed by researchers exploring the receptor-ligand binding of the endocannabinoid system.
Clinical effects, generally psychiatric, include paranoia, anxiety, agitation, delusions, and psychosis. There are also reports of patients who arrive with sympathomimetic toxicity, some of whom develop bradycardia and hypotension, and some who progress to acute renal failure, seizures, and death. Detection of these products is difficult as they do not react on EMIT testing for THC metabolites and require either gas chromatography-mass spectrometry or liquid chromatography with tandem mass spectrometry.45–48
- Substance Abuse and Mental Health Services Administration. Results from the 2012 National Survey on Drug Use and Health: Summary of National Findings, NSDUH Series H-46, HHS Publication No. (SMA) 13-4795. www.samhsa.gov/data/sites/default/files/NSDUHresultsPDFWHTML2013/Web/NSDUHresults2013.pdf. Accessed October 2, 2015.
- United Nations Office on Drugs and Crime. 2008 World Drug Report. www.unodc.org/documents/wdr/WDR_2008/WDR_2008_eng_web.pdf. Accessed October 2, 2015.
- American Society of Addiction Medicine (ASAM). Public policy statement on medical marijuana. www.asam.org/docs/publicy-policy-statements/1medical-marijuana-4-10.pdf?sfvrsn=0. Accessed October 2, 2015.
- Howlett AC, Barth F, Bonner TI, et al. International Union of Pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol Rev 2002; 54:161–202.
- Sharkey KA, Darmani NA, Parker LA. Regulation of nausea and vomiting by cannabinoids and the endocannabinoid system. Eur J Pharmacol 2014; 722:134–146.
- Iversen L. Cannabis and the brain. Brain 2003; 126:1252–1270.
- Huestis MA, Henningfield JE, Cone EJ. Blood cannabinoids. I. Absorption of THC and formation of 11-OH-THC and THCCOOH during and after smoking marijuana. J Anal Toxicol 1992; 16:276–282.
- Grotenhermen F. Pharmacokinetics and pharmacodynamics of cannabinoids. Clin Pharmacokinet 2003; 42:327–360.
- Mehmedic Z, Chandra S, Slade D, et al. Potency trends of Δ9-THC and other cannabinoids in confiscated cannabis preparations from 1993 to 2008. J Forensic Sci 2010; 55:1209–1217.
- Mittleman MA, Lewis RA, Maclure M, Sherwood JB, Muller JE. Triggering myocardial infarction by marijuana. Circulation 2001; 103:2805–2809.
- Mukamal KJ, Maclure M, Muller JE, Mittleman MA. An exploratory prospective study of marijuana use and mortality following acute myocardial infarction. Am Heart J 2008; 155:465–470.
- Thomas G, Kloner RA, Rezkalla S. Adverse cardiovascular, cerebrovascular, and peripheral vascular effects of marijuana inhalation: what cardiologists need to know. Am J Cardiol 2014; 113:187–190.
- Wang GS, Roosevelt G, Heard K. Pediatric marijuana exposures in a medical marijuana state. JAMA Pediatr 2013; 167:630–633.
- Carstairs SD, Fujinaka MK, Keeney GE, Ly BT. Prolonged coma in a child due to hashish ingestion with quantitation of THC metabolites in urine. J Emerg Med 2011; 41:e69–e71.
- Le Garrec S, Dauger S, Sachs P. Cannabis poisoning in children. Intensive Care Med 2014; 40:1394–1395.
- Ragab AR, Al-Mazroua MK. Passive cannabis smoking resulting in coma in a 16-month old infant. J Clin Case Rep 2012;2:237.
- Robinson K. Beyond resinable doubt? J Clin Forensic Med 2005;12:164–166.
- Burns JK. Pathways from cannabis to psychosis: a review of the evidence. Front Psychiatry 2013;4:128.
- Di Forti M, Sallis H, Allegri F, et al. Daily use, especially of high-potency cannabis, drives the earlier onset of psychosis in cannabis users. Schizophr Bull 2014; 40:1509–1517.
- Moore TH, Zammit S, Lingford-Hughes A, et al. Cannabis use and risk of psychotic or affective mental health outcomes: a systematic review. Lancet 2007; 370:319–328.
- Wilkinson ST, Radhakrishnan R, D'Souza DC. Impact of cannabis use on the development of psychotic disorders. Curr Addict Rep 2014;1:115–128.
- Aldington S, Williams M, Nowitz M, et al. Effects of cannabis on pulmonary structure, function and symptoms. Thorax 2007; 62:1058–1063.
- George KL, Saltman LH, Stein GS, Lian JB, Zurier RB. Ajulemic acid, a nonpsychoactive cannabinoid acid, suppresses osteoclastogenesis in mononuclear precursor cells and induces apoptosis in mature osteoclast-like cells. J Cell Physiol 2008; 214:714–720.
- Reece AS. Chronic toxicology of cannabis. Clin Toxicol (Phila) 2009; 47:517–524.
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Arlington, VA: American Psychiatric Publishing; 2013.
- Allsop DJ, Copeland J, Lintzeris N, et al. Nabiximols as an agonist replacement therapy during cannabis withdrawal: a randomized clinical trial. JAMA Psychiatry 2014; 71:281–291.
- Hesse M, Thylstrup B. Time-course of the DSM-5 cannabis withdrawal symptoms in poly-substance abusers. BMC Psychiatry 2013; 13:258.
- Allen JH, de Moore GM, Heddle R, Twartz JC. Cannabinoid hyperemesis: cyclical hyperemesis in association with chronic cannabis abuse. Gut 2004; 53:1566–1570.
- Galli JA, Sawaya RA, Friedenberg FK. Cannabinoid hyperemesis syndrome. Curr Drug Abuse Rev 2011; 4:241–249.
- Simonetto DA, Oxentenko AS, Herman ML, Szostek JH. Cannabinoid hyperemesis: a case series of 98 patients. Mayo Clin Proc 2012; 87:114–119.
- Soriano-Co M, Batke M, Cappell MS. The cannabis hyperemesis syndrome characterized by persistent nausea and vomiting, abdominal pain, and compulsive bathing associated with chronic marijuana use: a report of eight cases in the United States. Dig Dis Sci 2010; 55:3113–3119.
- Wallace EA, Andrews SE, Garmany CL, Jelley MJ. Cannabinoid hyperemesis syndrome: literature review and proposed diagnosis and treatment algorithm. South Med J 2011; 104:659–664.
- Hickey JL, Witsil JC, Mycyk MB. Haloperidol for treatment of cannabinoid hyperemesis syndrome. Am J Emerg Med 2013; 31:1003.e5–1003.e6.
- Perwitasari DA, Gelderblom H, Atthobari J, et al. Anti-emetic drugs in oncology: pharmacology and individualization by pharmacogenetics. Int J Clin Pharm 2011; 33:33–43.
- Cox B, Chhabra A, Adler M, Simmons J, Randlett D. Cannabinoid hyperemesis syndrome: case report of a paradoxical reaction with heavy marijuana use. Case Rep Med 2012; 2012:757696.
- Sakurai M, Mori T, Kato J, et al. Efficacy of aprepitant in preventing nausea and vomiting due to high-dose melphalan-based conditioning for allogeneic hematopoietic stem cell transplantation. Int J Hematol 2014; 99:457–462.
- Lapoint J. Case series of patients treated for cannabinoid hyperemesis syndrome with capsaicin cream. Clin Tox 2014; 52:707. Abstract #53.
- Biary R, Oh A, Lapoint J, Nelson LS, Hoffman RS, Howland MA. Topical capsaicin cream used as a therapy for cannabinoid hyperemesis syndrome. Clin Tox 2014; 52:787. Abstract #232.
- Moeller KE, Lee KC, Kissack JC. Urine drug screening: practical guide for clinicians. Mayo Clin Proc 2008; 83:66–76.
- Lowe RH, Abraham TT, Darwin WD, Herning R, Cadet JL, Huestis MA. Extended urinary delta9-tetrahydrocannabinol excretion in chronic cannabis users precludes use as a biomarker of new drug exposure. Drug Alcohol Depend 2009; 105:24–32.
- Paul BD, Jacobs A. Effects of oxidizing adulterants on detection of 11-nor-delta9-THC-9-carboxylic acid in urine. J Anal Toxicol 2002; 26:460–463.
- Schwope DM, Karschner EL, Gorelick DA, Huestis MA. Identification of recent cannabis use: whole-blood and plasma free and glucuronidated cannabinoid pharmacokinetics following controlled smoked cannabis administration. Clin Chem 2011; 57:1406-1414.
- Huestis MA, Smith ML. Cannabinoid pharmacokinetics and disposition in alternative matrices. In: Pertwee R, ed. Handbook of Cannabis. Oxford, United Kingdom: Oxford University Press; 2014:296–316.
- Mowry JB, Spyker DA, Cantilena LR Jr, Bailey JE, Ford M. 2012 Annual Report of the American Association of Poison Control Centers’ National Poison Data System (NPDS): 30th Annual Report. Clin Toxicol (Phila) 2013; 51:949–1229.
- Rosenbaum CD, Carreiro SP, Babu KM. Here today, gone tomorrow…and back again? A review of herbal marijuana alternatives (K2, Spice), synthetic cathinones (bath salts), kratom, Salvia divinorum, methoxetamine, and piperazines. J Med Toxicol 2012; 8:15–32.
- Gurney SMR, Scott KS, Kacinko SL, Presley BC, Logan BK. Pharmacology, toxicology, and adverse effects of synthetic cannabinoid drugs. Forensic Sci Rev 2014; 26:53–78.
- McKeever RG, Vearrier D, Jacobs D, LaSala G, Okaneku J, Greenberg MI. K2-not the spice of life; synthetic cannabinoids and ST elevation myocardial infarction: a case report. J Med Toxicol 2015; 11:129–131.
- Schneir AB, Baumbacher T. Convulsions associated with the use of a synthetic cannabinoid product. J Med Toxicol 2012; 8:62–64.
Clinicians may be encountering more cannabis users than before, and may be encountering users with complications hitherto unseen. Several trends may explain this phenomenon: the legal status of cannabis is changing, cannabis today is more potent than in the past, and enthusiasts are conjuring new ways to enjoy this substance.
This article discusses the history, pharmacology, and potential complications of cannabis use.
A LONG AND TANGLED HISTORY
Cannabis is a broad term that refers to the cannabis plant and its preparations, such as marijuana and hashish, as well as to a family of more than 60 bioactive substances called cannabinoids. It is the most commonly used illegal drug in the world, with an estimated 160 million users. Each year, about 2.4 million people in the United States use it for the first time.1,2
Cannabis has been used throughout the world for recreational and spiritual purposes for nearly 5,000 years, beginning with the fabled Celestial Emperors of China. The tangled history of cannabis in America began in the 17th century, when farmers were required by law to grow it as a fiber crop. It later found its way into the US Pharmacopeia for a wide range of indications. During the long prelude to Prohibition in the latter half of the 19th century, the US government became increasingly suspicious of mind-altering substances and began restricting its prescription in 1934, culminating in its designation by the US Food and Drug Administration as a schedule I controlled substance in 1970.
Investigation into the potential medical uses for the different chemicals within cannabis is ongoing, as is debate over its changing legality and usefulness to society. The apparent cognitive dissonance surrounding the use and advocacy of medical marijuana is beyond the scope of this review,3 which will instead restrict itself to what is known of the cannabinoids and to the recreational use of cannabis.
THC IS THE PRINCIPAL PSYCHOACTIVE MOLECULE
Delta-9 tetrahydrocannabinol (THC), first isolated in 1964, was identified as the principal psychoactive constituent of cannabis in 2002.4
Two G-protein–linked cannabinoid receptors cloned in the 1990s—CB1 and CB2—were found to be a part of a system of endocannabinoid receptors present throughout the body, from the brain to the immune system to the vas deferens.5 Both receptors inhibit cellular excitation by activating inwardly rectifying potassium channels. These receptors are mostly absent in the brainstem, which may explain why cannabis use rarely causes life-threatening autonomic dysfunction. Although the intoxicating effects of marijuana are mediated by CB1 receptors, the specific mechanisms underlying the cannabis “high” are unclear.6
CANNABINOIDS ARE LIPID-SOLUBLE
The rate of absorption of cannabinoids depends on the route of administration and the type of cannabis product used. When cannabis products are smoked, up to 35% of THC is available, and the average time to peak serum concentration is 8 minutes.7 The peak concentration depends on the dose.
On the other hand, when cannabis products (eg, nabilone, dronabinol) are ingested, absorption is unpredictable because THC is unstable in gastric acid and undergoes first-pass metabolism in the liver, which reduces the drug’s bioavailability. Up to 20% of an ingested dose of THC is absorbed, and the time to peak serum concentration averages between 2 and 4 hours. Consequently, many users prefer to smoke cannabis as a means to control the desired effects.
Cannabinoids are lipid-soluble. They accumulate in fatty tissue in a biphasic pattern, initially moving into highly vascularized tissue such as the liver before accumulating in less well-vascularized tissue such as fat. They are then slowly released from fatty tissue as the fat turns over. THC itself has a volume of distribution of about 2.5 to 3.5 L/kg. It crosses the placenta and enters breast milk.8
THC is metabolized by the cytochrome P450 system, primarily by the enzymes CYP2C9 and CYP3A4. Its primary metabolite, 11-hydroxy-delta-9 THC, is also active, but subsequent metabolism produces many other inactive metabolites. THC is eliminated in feces and urine, and its half-life ranges from 2 to nearly 60 hours.8
A LITTLE ABOUT PLANTS AND STREET NAMES
The plant from which THC and nearly a hundred other chemicals, including cannabinoids, are derived has been called many things over the years:
Hemp is a tall fibrous plant grown for rope and fabric that was used as legal tender in early America. In the mid-19th century, there were over 16 million acres of hemp plantations. Hemp contains very low THC concentrations.
Cannabis is an annual flowering herb that is predominantly diecious (ie, there are male and female plants). After a centuries-long debate among taxonomists, the two principal species are considered to be C sativa and C indica, although today many cannabis cultivars are grown by a great number of breeding enthusiasts.
Concentrations of THC vary widely among cannabis cultivars, ranging historically from around 5% to today’s highly selectively bred species containing more than 30%. Concentrations in seized cannabis have been measured as high as 37%, although the average is around 11%.9 This concentration is defined by the percent of THC per dried mass of plant material tested, usually via gas chromatography.
Hashish is a solid or resinous preparation of the trichomes, or glandular hairs, that grow on the cannabis plant, chiefly on its flowers. Various methods to separate the trichomes from the rest of the plant result in a powder called kief that is then compressed into blocks or bricks. THC concentrations as high as 66% have been measured in nondomestic sources of hashish.9
Hash oil is a further purification, produced by using solvents to dissolve the resin and by filtering out remaining plant material. Evaporating the solvent produces hash oil, sometimes called butane hash oil or honey oil. This process has recently led to an increasing number of home explosions, as people attempt to make the product themselves but do not take suitable precautions when using flammable solvents such as butane. THC concentrations as high as 81% have been measured in nondomestic sources of hash oil.9
Other names for hash oil are dab, wax, and budder. Cannabis enthusiasts refer to the use of hash oil as dabbing, which involves heating a small amount (dab) of the product using a variety of paraphernalia and inhaling the vapor.
IT’S ALL ABOUT GETTING HIGH
For recreational users, the experience has always been about being intoxicated—getting high. The psychological effects range broadly from positive to negative and vary both within and between users, depending on the dose and route of administration. Additional factors that influence the psychological effects include the social and physical settings of drug use and even the user’s expectations. One user’s high is another user’s acute toxic effect.
Although subjective reports of the cannabis experience vary greatly, it typically begins with a feeling of dizziness or lightheadedness followed by a relaxed calm and a feeling of being somewhat “disconnected.” There is a quickening of the sense of humor, described by some as a fatuous euphoria; often there is silly giggling. Awareness of the senses and of music may be increased. Appetite increases, and time seems to pass quickly. Eventually, the user becomes drowsy and experiences decreased attention and difficulty maintaining a coherent conversation. Slowed reaction time and decreased psychomotor activity may also occur. The user may drift into daydreams and eventually fall asleep.
Common negative acute effects of getting high can include mild to severe anxiety and feeling tense or agitated. Clumsiness, headache, and confusion are also possible. Lingering effects the following day may include dry mouth, dry eyes, fatigue, slowed thinking, and slowed recall.6
ACUTE PHYSICAL EFFECTS
Acute physical effects of cannabis use include a rapid onset of increased airway conductance, decreased intraocular pressure, and conjunctival injection. A single cannabis cigarette can also induce cardiovascular effects including a dose-dependent increase in heart rate and blood pressure. Chronic users, however, can experience a decreased heart rate, lower blood pressure, and postural hypotension.
In a personal communication, colleagues in Colorado—where recreational use of cannabis was legalized in 2012—described a sharp increase (from virtually none) in the number of adults presenting to the emergency department with cannabis intoxication since 2012. Their patients experienced palpitations, light-headedness, and severe ataxia lasting as long as 12 hours, possibly reflecting the greater potency of current cannabis products. Most of these patients required only supportive care.
Other acute adverse cardiovascular reactions that have been reported include atrial fibrillation, ventricular tachycardia, and a fivefold increased risk of myocardial infarction in the 60 minutes following cannabis use, which subsequently drops sharply to baseline levels.10 Investigations into the cardiovascular effects of cannabis are often complicated by concurrent use of other drugs such as tobacco or cocaine. Possible mechanisms of injury include alterations in coronary microcirculation or slowed coronary flow. In fact, one author found that cannabis users with a history of myocardial infarction had a risk of death 4.2 times higher than users with no history of myocardial infarction.11,12
In children, acute toxicity has been reported from a variety of exposures to cannabis and hashish, including a report of an increase in pediatric cannabis exposures following the changes in Colorado state laws.13 Most of these patients had altered mental status ranging from drowsiness to coma; one report describes a child who experienced a first-time seizure. These patients unfortunately often underwent extensive evaluations such as brain imaging and lumbar puncture, and mechanical ventilation to protect the airway. Earlier consideration of cannabis exposure in these patients might have limited unnecessary testing. Supportive care is usually all that is needed, and most of these patients fully recover.13–17
CHRONIC EFFECTS
Cannabinoids cause a variety of adverse effects, but the ultimate risk these changes pose to human health has been difficult to calculate. Long-term studies are confounded by possible inaccuracies of patient self-reporting of cannabis use, poor control of covariates, and disparate methodologies.
For more than a century, cannabis use has been reported to cause both acute psychotic symptoms and persistent psychotic disorders.18 But the strength of this relationship is modest. Cannabis is more likely a component cause that, in addition to other factors (eg, specific genetic polymorphisms), contributes to the risk of schizophrenia. Individuals with prodromal symptoms and those who have experienced discrete episodes of psychosis related to cannabis use should be discouraged from using cannabis and cannabinoids.19–21
Mounting evidence implicates chronic cannabis use as a cause of long-term medical problems including chronic bronchitis,22 elevated rates of myocardial infarction and dysrhythmias,11 bone loss,23 and cancers at eight different sites including the lung, head, and neck.24 In view of these chronic effects, healthcare providers should caution their patients about cannabis use, as we do about other drugs such as tobacco.
WITHDRAWAL SYNDROME RECOGNIZED
Until recently, neither clinicians nor users recognized a withdrawal syndrome associated with chronic use of cannabis, probably because this syndrome is not as severe as withdrawal from other controlled substances such as opioids or sedative-hypnotics. A number of studies, however, have reported subtle cannabis withdrawal symptoms that are similar to those associated with tobacco withdrawal.
As such, the fifth and latest edition of the Diagnostic and Statistical Manual of Mental Disorders (DSM-5)25 characterized withdrawal from cannabis use in 2013. The DSM-5 criteria require cessation of heavy or prolonged use of cannabis (ie, daily or almost daily over a period of at least a few months) and three or more of the following withdrawal symptoms:
- Irritability and anger
- Nervousness
- Sleep difficulty or insomnia
- Decreased appetite or weight loss
- Restlessness
- Depressed mood
- Physical symptoms causing discomfort.
Medical treatment of cannabis withdrawal has included a range of antidepressants, mood stabilizers, and alpha-2-adrenergic agonists, all of which have limited success.26 Symptoms of cannabis withdrawal tend to be most intense soon after cessation and decline over the next few weeks.27
CANNABINOID HYPEREMESIS SYNDROME
First reported in 2004,28 cannabinoid hyperemesis syndrome is a recurrent disorder, the pathophysiology of which is poorly understood. It has three phases.
The first phase is a prodrome that may last months or years and is characterized by morning nausea, fear of vomiting, and abdominal discomfort. During this phase, the patient maintains normal eating patterns and may well increase his or her cannabis use due to its well-known antiemetic effects.
The second phase is the hyperemetic phase, characterized by intense, incapacitating emesis with episodes of vomiting throughout the day. These symptoms can be relieved only with frequent hot baths, a feature that distinguishes cannabinoid hyperemesis syndrome from other vomiting syndromes. Hot-water bathing is reported to be a compulsive but learned behavior in which the patient learns that only hot water will provide relief. The extent of relief depends on the temperature of the water—the hotter, the better. Symptoms recur as the water cools.28 Patients often present to the emergency department repeatedly with recurrent symptoms and may remain misdiagnosed or subjected to repeated extensive evaluation including laboratory testing and imaging, which are usually not revealing. If the patient has not been accurately diagnosed, there may be reported weight loss of at least 5 kg.
The third phase, recovery, may take several months to complete, possibly because of the prolonged terminal elimination time of cannabinoids. Complete cessation of cannabis use, including synthetic cannabinoids, is usually necessary.29
Diagnostic criteria for cannabinoid hyperemesis syndrome have been suggested, based on a retrospective case series that included 98 patients.30 The most common features of these affected patients were:
- Severe cyclical vomiting, predominantly in the morning
- Resolution of symptoms with cessation of cannabis use
- Symptomatic relief with hot showers or baths
- Abdominal pain
- At least weekly use of cannabis.
Interestingly, long-term cannabis use has been cited as a critical identifying feature of these patients, with the duration of cannabis use ranging from 10 to 16 years.31,32 Other reports show greater variability in duration of cannabis use before the onset of cannabinoid hyperemesis syndrome. In the large study noted above,30 32% of users reported their duration of cannabis use to be less than 1 year, rendering this criterion less useful.
How can cannabis both cause and prevent vomiting?
The body controls nausea and vomiting via complex circuitry in the brain and gut that involves many neurotransmitters (eg, dopamine, serotonin, substance P) that interact with receptors such as CB1, 5-HT1–4, alpha adrenergic receptors, and mu receptors. Interestingly, cannabis use has antiemetic properties mediated by CB1 with a still unclear additional role of CB2 receptors. Data point to the existence of an underlying antiemetic tone mediated by the endocannabinoid system.
Unfortunately, the mechanism by which cannabinoid hyperemesis syndrome occurs is unknown and represents a paradoxical effect against the otherwise antiemetic effects of cannabis. Several theories have been proposed, including delayed gastric emptying, although only a third of patients demonstrated this on scintigraphy in one study.30 Other theories include disturbance of the hypothalamic-pituitary axis, a buildup of highly lipophilic THC in the brain, and a down-regulation of cannabinoid receptors that results from chronic exposure.30 Given that this syndrome has been recognized only relatively recently, one author has suggested the cause may be recent horticultural developments.5
Treating cannabinoid hyperemesis syndrome is difficult
Treatment of cannabinoid hyperemesis syndrome is notoriously difficult, with many authors reporting resistance to the usual first-line antiemetic drugs. Generally, treatment should include hydration and acid-suppression therapy because endoscopic evaluation of several patients has revealed varying degrees of esophagitis and gastritis.29
Antiemetic therapy should target receptors known to mediate nausea and vomiting. In some cases, antiemetic drugs are more effective when used in combination. Agents include the serotonergic receptor antagonists ondansetron and granisetron, the dopamine antagonists prochlorperazine and metoclopramide, and even haloperidol.33,34 Benzodiazepines may be effective by causing sedation, anxiolysis, and depression of the vomiting center.34,35 Two antihistamines—dimenhydrinate and diphenhydramine—have antiemetic effects, perhaps by inhibiting acetylcholine.34
Aprepitant is a neurokinin-1 antagonist that inhibits the action of substance P. When combined with a corticosteroid and a serotonin antagonist, it relieves nausea and vomiting in chemotherapy patients.34,36
Corticosteroids such as dexamethasone are potent antiemetics thought to inhibit prostaglandin synthesis.34
Capsaicin cream applied to the abdomen has also been reported to relieve symptoms, possibly through an interaction between the TRPv1 receptor and the endocannabinoid system.37,38
DIAGNOSTIC TESTING
Cannabinoids are detectable in plasma and urine, with urine testing being more common.
Common laboratory methods include the enzyme-multiplied immunoassay technique (EMIT) and radioimmunoassay. Gas chromatography coupled with mass spectrometry is the most specific assay; it is used for confirmation and is the reference method.
EMIT is a qualitative urine test that detects 9-carboxy-THC as well as other THC metabolites. These urine tests detect all metabolites, and the result is reported as positive if the total concentration is greater than or equal to a prespecified threshold level, such as 20 ng/mL or 50 ng/mL. A positive test does not denote intoxication, nor does the test identify the source of THC (eg, cannabis, dronabinol, butane hash oil). EMIT does not detect nabilone. The National Institute on Drug Abuse guidelines for urine testing specify a test threshold concentration of 50 ng/mL for screening and 15 ng/mL for confirmation.
Many factors affect the detection of THC metabolites and their presence and duration in urine: dose, duration of use, route of exposure, hydration status, urine volume and concentration, and urine pH. THC metabolites have been detected in urine using gas chromatography-mass spectrometry for up to 7 days after smoking one marijuana cigarette.7 Chronic users have also been reported to have positive urine EMIT tests for up to 46 days after cannabis cessation.39 Detection may be further complicated in chronic users: in one study, users produced both negative and positive specimens over 24 days, suggesting that diet and exercise may influence clearance.40 Also, many factors are known to produce false-positive and false-negative results for these immunoassays (Table 1).39,41
In the United States, penalties for driving under the influence of cannabis vary from state to state, and laws specify plasma testing for quantitative analysis. Some states use a threshold of 5 ng/mL in plasma to imply driving under the influence, whereas others use any detectable amount. Currently, there are no generally accepted guidelines for storage and testing of blood samples, despite the known instability of analytes.42
Saliva, hair, and sweat can also be used for cannabinoid testing. Saliva is easy to collect, can be tested for metabolites to rule out passive cannabis exposure, and can be positive for up to 1 day after exposure. Calculating a blood or plasma concentration from a saliva sample is not possible, however.
Hair testing can also rule out passive exposure, but THC binds very little to melanin, resulting in very low concentrations requiring sensitive tests, such as gas chromatography with tandem mass spectrometry.
Only one device is commercially available for sweat testing; further work is needed to elucidate sweat excretion pharmacokinetics and the limitations of the collection devices.43
CLINICAL MANAGEMENT IS GENERALLY SUPPORTIVE
Historically, clinical toxicity from recreational cannabis use is rarely serious or severe and generally responds to supportive care. Reports of cannabis exposure to poison centers are one-tenth of those reported for ethanol exposures annually.44 Gastrointestinal decontamination with activated charcoal is not recommended, even for orally administered cannabis, since the risks outweigh the expected benefits. Agitation or anxiety may be treated with benzodiazepines as needed. There is no antidote for cannabis toxicity. The ever-increasing availability of high-concentration THC preparations may prompt more aggressive supportive measures in the future.
SYNTHETIC MARIJUANA ALTERNATIVES
Available since the early 2000s, herbal marijuana alternatives are legally sold as incense or potpourri and are often labeled “not for human consumption.” They are known by such brand names as K2 and Spice and contain blends of herbs adulterated with synthetic cannabinoid chemicals developed by researchers exploring the receptor-ligand binding of the endocannabinoid system.
Clinical effects, generally psychiatric, include paranoia, anxiety, agitation, delusions, and psychosis. There are also reports of patients who arrive with sympathomimetic toxicity, some of whom develop bradycardia and hypotension, and some who progress to acute renal failure, seizures, and death. Detection of these products is difficult as they do not react on EMIT testing for THC metabolites and require either gas chromatography-mass spectrometry or liquid chromatography with tandem mass spectrometry.45–48
Clinicians may be encountering more cannabis users than before, and may be encountering users with complications hitherto unseen. Several trends may explain this phenomenon: the legal status of cannabis is changing, cannabis today is more potent than in the past, and enthusiasts are conjuring new ways to enjoy this substance.
This article discusses the history, pharmacology, and potential complications of cannabis use.
A LONG AND TANGLED HISTORY
Cannabis is a broad term that refers to the cannabis plant and its preparations, such as marijuana and hashish, as well as to a family of more than 60 bioactive substances called cannabinoids. It is the most commonly used illegal drug in the world, with an estimated 160 million users. Each year, about 2.4 million people in the United States use it for the first time.1,2
Cannabis has been used throughout the world for recreational and spiritual purposes for nearly 5,000 years, beginning with the fabled Celestial Emperors of China. The tangled history of cannabis in America began in the 17th century, when farmers were required by law to grow it as a fiber crop. It later found its way into the US Pharmacopeia for a wide range of indications. During the long prelude to Prohibition in the latter half of the 19th century, the US government became increasingly suspicious of mind-altering substances and began restricting its prescription in 1934, culminating in its designation by the US Food and Drug Administration as a schedule I controlled substance in 1970.
Investigation into the potential medical uses for the different chemicals within cannabis is ongoing, as is debate over its changing legality and usefulness to society. The apparent cognitive dissonance surrounding the use and advocacy of medical marijuana is beyond the scope of this review,3 which will instead restrict itself to what is known of the cannabinoids and to the recreational use of cannabis.
THC IS THE PRINCIPAL PSYCHOACTIVE MOLECULE
Delta-9 tetrahydrocannabinol (THC), first isolated in 1964, was identified as the principal psychoactive constituent of cannabis in 2002.4
Two G-protein–linked cannabinoid receptors cloned in the 1990s—CB1 and CB2—were found to be a part of a system of endocannabinoid receptors present throughout the body, from the brain to the immune system to the vas deferens.5 Both receptors inhibit cellular excitation by activating inwardly rectifying potassium channels. These receptors are mostly absent in the brainstem, which may explain why cannabis use rarely causes life-threatening autonomic dysfunction. Although the intoxicating effects of marijuana are mediated by CB1 receptors, the specific mechanisms underlying the cannabis “high” are unclear.6
CANNABINOIDS ARE LIPID-SOLUBLE
The rate of absorption of cannabinoids depends on the route of administration and the type of cannabis product used. When cannabis products are smoked, up to 35% of THC is available, and the average time to peak serum concentration is 8 minutes.7 The peak concentration depends on the dose.
On the other hand, when cannabis products (eg, nabilone, dronabinol) are ingested, absorption is unpredictable because THC is unstable in gastric acid and undergoes first-pass metabolism in the liver, which reduces the drug’s bioavailability. Up to 20% of an ingested dose of THC is absorbed, and the time to peak serum concentration averages between 2 and 4 hours. Consequently, many users prefer to smoke cannabis as a means to control the desired effects.
Cannabinoids are lipid-soluble. They accumulate in fatty tissue in a biphasic pattern, initially moving into highly vascularized tissue such as the liver before accumulating in less well-vascularized tissue such as fat. They are then slowly released from fatty tissue as the fat turns over. THC itself has a volume of distribution of about 2.5 to 3.5 L/kg. It crosses the placenta and enters breast milk.8
THC is metabolized by the cytochrome P450 system, primarily by the enzymes CYP2C9 and CYP3A4. Its primary metabolite, 11-hydroxy-delta-9 THC, is also active, but subsequent metabolism produces many other inactive metabolites. THC is eliminated in feces and urine, and its half-life ranges from 2 to nearly 60 hours.8
A LITTLE ABOUT PLANTS AND STREET NAMES
The plant from which THC and nearly a hundred other chemicals, including cannabinoids, are derived has been called many things over the years:
Hemp is a tall fibrous plant grown for rope and fabric that was used as legal tender in early America. In the mid-19th century, there were over 16 million acres of hemp plantations. Hemp contains very low THC concentrations.
Cannabis is an annual flowering herb that is predominantly diecious (ie, there are male and female plants). After a centuries-long debate among taxonomists, the two principal species are considered to be C sativa and C indica, although today many cannabis cultivars are grown by a great number of breeding enthusiasts.
Concentrations of THC vary widely among cannabis cultivars, ranging historically from around 5% to today’s highly selectively bred species containing more than 30%. Concentrations in seized cannabis have been measured as high as 37%, although the average is around 11%.9 This concentration is defined by the percent of THC per dried mass of plant material tested, usually via gas chromatography.
Hashish is a solid or resinous preparation of the trichomes, or glandular hairs, that grow on the cannabis plant, chiefly on its flowers. Various methods to separate the trichomes from the rest of the plant result in a powder called kief that is then compressed into blocks or bricks. THC concentrations as high as 66% have been measured in nondomestic sources of hashish.9
Hash oil is a further purification, produced by using solvents to dissolve the resin and by filtering out remaining plant material. Evaporating the solvent produces hash oil, sometimes called butane hash oil or honey oil. This process has recently led to an increasing number of home explosions, as people attempt to make the product themselves but do not take suitable precautions when using flammable solvents such as butane. THC concentrations as high as 81% have been measured in nondomestic sources of hash oil.9
Other names for hash oil are dab, wax, and budder. Cannabis enthusiasts refer to the use of hash oil as dabbing, which involves heating a small amount (dab) of the product using a variety of paraphernalia and inhaling the vapor.
IT’S ALL ABOUT GETTING HIGH
For recreational users, the experience has always been about being intoxicated—getting high. The psychological effects range broadly from positive to negative and vary both within and between users, depending on the dose and route of administration. Additional factors that influence the psychological effects include the social and physical settings of drug use and even the user’s expectations. One user’s high is another user’s acute toxic effect.
Although subjective reports of the cannabis experience vary greatly, it typically begins with a feeling of dizziness or lightheadedness followed by a relaxed calm and a feeling of being somewhat “disconnected.” There is a quickening of the sense of humor, described by some as a fatuous euphoria; often there is silly giggling. Awareness of the senses and of music may be increased. Appetite increases, and time seems to pass quickly. Eventually, the user becomes drowsy and experiences decreased attention and difficulty maintaining a coherent conversation. Slowed reaction time and decreased psychomotor activity may also occur. The user may drift into daydreams and eventually fall asleep.
Common negative acute effects of getting high can include mild to severe anxiety and feeling tense or agitated. Clumsiness, headache, and confusion are also possible. Lingering effects the following day may include dry mouth, dry eyes, fatigue, slowed thinking, and slowed recall.6
ACUTE PHYSICAL EFFECTS
Acute physical effects of cannabis use include a rapid onset of increased airway conductance, decreased intraocular pressure, and conjunctival injection. A single cannabis cigarette can also induce cardiovascular effects including a dose-dependent increase in heart rate and blood pressure. Chronic users, however, can experience a decreased heart rate, lower blood pressure, and postural hypotension.
In a personal communication, colleagues in Colorado—where recreational use of cannabis was legalized in 2012—described a sharp increase (from virtually none) in the number of adults presenting to the emergency department with cannabis intoxication since 2012. Their patients experienced palpitations, light-headedness, and severe ataxia lasting as long as 12 hours, possibly reflecting the greater potency of current cannabis products. Most of these patients required only supportive care.
Other acute adverse cardiovascular reactions that have been reported include atrial fibrillation, ventricular tachycardia, and a fivefold increased risk of myocardial infarction in the 60 minutes following cannabis use, which subsequently drops sharply to baseline levels.10 Investigations into the cardiovascular effects of cannabis are often complicated by concurrent use of other drugs such as tobacco or cocaine. Possible mechanisms of injury include alterations in coronary microcirculation or slowed coronary flow. In fact, one author found that cannabis users with a history of myocardial infarction had a risk of death 4.2 times higher than users with no history of myocardial infarction.11,12
In children, acute toxicity has been reported from a variety of exposures to cannabis and hashish, including a report of an increase in pediatric cannabis exposures following the changes in Colorado state laws.13 Most of these patients had altered mental status ranging from drowsiness to coma; one report describes a child who experienced a first-time seizure. These patients unfortunately often underwent extensive evaluations such as brain imaging and lumbar puncture, and mechanical ventilation to protect the airway. Earlier consideration of cannabis exposure in these patients might have limited unnecessary testing. Supportive care is usually all that is needed, and most of these patients fully recover.13–17
CHRONIC EFFECTS
Cannabinoids cause a variety of adverse effects, but the ultimate risk these changes pose to human health has been difficult to calculate. Long-term studies are confounded by possible inaccuracies of patient self-reporting of cannabis use, poor control of covariates, and disparate methodologies.
For more than a century, cannabis use has been reported to cause both acute psychotic symptoms and persistent psychotic disorders.18 But the strength of this relationship is modest. Cannabis is more likely a component cause that, in addition to other factors (eg, specific genetic polymorphisms), contributes to the risk of schizophrenia. Individuals with prodromal symptoms and those who have experienced discrete episodes of psychosis related to cannabis use should be discouraged from using cannabis and cannabinoids.19–21
Mounting evidence implicates chronic cannabis use as a cause of long-term medical problems including chronic bronchitis,22 elevated rates of myocardial infarction and dysrhythmias,11 bone loss,23 and cancers at eight different sites including the lung, head, and neck.24 In view of these chronic effects, healthcare providers should caution their patients about cannabis use, as we do about other drugs such as tobacco.
WITHDRAWAL SYNDROME RECOGNIZED
Until recently, neither clinicians nor users recognized a withdrawal syndrome associated with chronic use of cannabis, probably because this syndrome is not as severe as withdrawal from other controlled substances such as opioids or sedative-hypnotics. A number of studies, however, have reported subtle cannabis withdrawal symptoms that are similar to those associated with tobacco withdrawal.
As such, the fifth and latest edition of the Diagnostic and Statistical Manual of Mental Disorders (DSM-5)25 characterized withdrawal from cannabis use in 2013. The DSM-5 criteria require cessation of heavy or prolonged use of cannabis (ie, daily or almost daily over a period of at least a few months) and three or more of the following withdrawal symptoms:
- Irritability and anger
- Nervousness
- Sleep difficulty or insomnia
- Decreased appetite or weight loss
- Restlessness
- Depressed mood
- Physical symptoms causing discomfort.
Medical treatment of cannabis withdrawal has included a range of antidepressants, mood stabilizers, and alpha-2-adrenergic agonists, all of which have limited success.26 Symptoms of cannabis withdrawal tend to be most intense soon after cessation and decline over the next few weeks.27
CANNABINOID HYPEREMESIS SYNDROME
First reported in 2004,28 cannabinoid hyperemesis syndrome is a recurrent disorder, the pathophysiology of which is poorly understood. It has three phases.
The first phase is a prodrome that may last months or years and is characterized by morning nausea, fear of vomiting, and abdominal discomfort. During this phase, the patient maintains normal eating patterns and may well increase his or her cannabis use due to its well-known antiemetic effects.
The second phase is the hyperemetic phase, characterized by intense, incapacitating emesis with episodes of vomiting throughout the day. These symptoms can be relieved only with frequent hot baths, a feature that distinguishes cannabinoid hyperemesis syndrome from other vomiting syndromes. Hot-water bathing is reported to be a compulsive but learned behavior in which the patient learns that only hot water will provide relief. The extent of relief depends on the temperature of the water—the hotter, the better. Symptoms recur as the water cools.28 Patients often present to the emergency department repeatedly with recurrent symptoms and may remain misdiagnosed or subjected to repeated extensive evaluation including laboratory testing and imaging, which are usually not revealing. If the patient has not been accurately diagnosed, there may be reported weight loss of at least 5 kg.
The third phase, recovery, may take several months to complete, possibly because of the prolonged terminal elimination time of cannabinoids. Complete cessation of cannabis use, including synthetic cannabinoids, is usually necessary.29
Diagnostic criteria for cannabinoid hyperemesis syndrome have been suggested, based on a retrospective case series that included 98 patients.30 The most common features of these affected patients were:
- Severe cyclical vomiting, predominantly in the morning
- Resolution of symptoms with cessation of cannabis use
- Symptomatic relief with hot showers or baths
- Abdominal pain
- At least weekly use of cannabis.
Interestingly, long-term cannabis use has been cited as a critical identifying feature of these patients, with the duration of cannabis use ranging from 10 to 16 years.31,32 Other reports show greater variability in duration of cannabis use before the onset of cannabinoid hyperemesis syndrome. In the large study noted above,30 32% of users reported their duration of cannabis use to be less than 1 year, rendering this criterion less useful.
How can cannabis both cause and prevent vomiting?
The body controls nausea and vomiting via complex circuitry in the brain and gut that involves many neurotransmitters (eg, dopamine, serotonin, substance P) that interact with receptors such as CB1, 5-HT1–4, alpha adrenergic receptors, and mu receptors. Interestingly, cannabis use has antiemetic properties mediated by CB1 with a still unclear additional role of CB2 receptors. Data point to the existence of an underlying antiemetic tone mediated by the endocannabinoid system.
Unfortunately, the mechanism by which cannabinoid hyperemesis syndrome occurs is unknown and represents a paradoxical effect against the otherwise antiemetic effects of cannabis. Several theories have been proposed, including delayed gastric emptying, although only a third of patients demonstrated this on scintigraphy in one study.30 Other theories include disturbance of the hypothalamic-pituitary axis, a buildup of highly lipophilic THC in the brain, and a down-regulation of cannabinoid receptors that results from chronic exposure.30 Given that this syndrome has been recognized only relatively recently, one author has suggested the cause may be recent horticultural developments.5
Treating cannabinoid hyperemesis syndrome is difficult
Treatment of cannabinoid hyperemesis syndrome is notoriously difficult, with many authors reporting resistance to the usual first-line antiemetic drugs. Generally, treatment should include hydration and acid-suppression therapy because endoscopic evaluation of several patients has revealed varying degrees of esophagitis and gastritis.29
Antiemetic therapy should target receptors known to mediate nausea and vomiting. In some cases, antiemetic drugs are more effective when used in combination. Agents include the serotonergic receptor antagonists ondansetron and granisetron, the dopamine antagonists prochlorperazine and metoclopramide, and even haloperidol.33,34 Benzodiazepines may be effective by causing sedation, anxiolysis, and depression of the vomiting center.34,35 Two antihistamines—dimenhydrinate and diphenhydramine—have antiemetic effects, perhaps by inhibiting acetylcholine.34
Aprepitant is a neurokinin-1 antagonist that inhibits the action of substance P. When combined with a corticosteroid and a serotonin antagonist, it relieves nausea and vomiting in chemotherapy patients.34,36
Corticosteroids such as dexamethasone are potent antiemetics thought to inhibit prostaglandin synthesis.34
Capsaicin cream applied to the abdomen has also been reported to relieve symptoms, possibly through an interaction between the TRPv1 receptor and the endocannabinoid system.37,38
DIAGNOSTIC TESTING
Cannabinoids are detectable in plasma and urine, with urine testing being more common.
Common laboratory methods include the enzyme-multiplied immunoassay technique (EMIT) and radioimmunoassay. Gas chromatography coupled with mass spectrometry is the most specific assay; it is used for confirmation and is the reference method.
EMIT is a qualitative urine test that detects 9-carboxy-THC as well as other THC metabolites. These urine tests detect all metabolites, and the result is reported as positive if the total concentration is greater than or equal to a prespecified threshold level, such as 20 ng/mL or 50 ng/mL. A positive test does not denote intoxication, nor does the test identify the source of THC (eg, cannabis, dronabinol, butane hash oil). EMIT does not detect nabilone. The National Institute on Drug Abuse guidelines for urine testing specify a test threshold concentration of 50 ng/mL for screening and 15 ng/mL for confirmation.
Many factors affect the detection of THC metabolites and their presence and duration in urine: dose, duration of use, route of exposure, hydration status, urine volume and concentration, and urine pH. THC metabolites have been detected in urine using gas chromatography-mass spectrometry for up to 7 days after smoking one marijuana cigarette.7 Chronic users have also been reported to have positive urine EMIT tests for up to 46 days after cannabis cessation.39 Detection may be further complicated in chronic users: in one study, users produced both negative and positive specimens over 24 days, suggesting that diet and exercise may influence clearance.40 Also, many factors are known to produce false-positive and false-negative results for these immunoassays (Table 1).39,41
In the United States, penalties for driving under the influence of cannabis vary from state to state, and laws specify plasma testing for quantitative analysis. Some states use a threshold of 5 ng/mL in plasma to imply driving under the influence, whereas others use any detectable amount. Currently, there are no generally accepted guidelines for storage and testing of blood samples, despite the known instability of analytes.42
Saliva, hair, and sweat can also be used for cannabinoid testing. Saliva is easy to collect, can be tested for metabolites to rule out passive cannabis exposure, and can be positive for up to 1 day after exposure. Calculating a blood or plasma concentration from a saliva sample is not possible, however.
Hair testing can also rule out passive exposure, but THC binds very little to melanin, resulting in very low concentrations requiring sensitive tests, such as gas chromatography with tandem mass spectrometry.
Only one device is commercially available for sweat testing; further work is needed to elucidate sweat excretion pharmacokinetics and the limitations of the collection devices.43
CLINICAL MANAGEMENT IS GENERALLY SUPPORTIVE
Historically, clinical toxicity from recreational cannabis use is rarely serious or severe and generally responds to supportive care. Reports of cannabis exposure to poison centers are one-tenth of those reported for ethanol exposures annually.44 Gastrointestinal decontamination with activated charcoal is not recommended, even for orally administered cannabis, since the risks outweigh the expected benefits. Agitation or anxiety may be treated with benzodiazepines as needed. There is no antidote for cannabis toxicity. The ever-increasing availability of high-concentration THC preparations may prompt more aggressive supportive measures in the future.
SYNTHETIC MARIJUANA ALTERNATIVES
Available since the early 2000s, herbal marijuana alternatives are legally sold as incense or potpourri and are often labeled “not for human consumption.” They are known by such brand names as K2 and Spice and contain blends of herbs adulterated with synthetic cannabinoid chemicals developed by researchers exploring the receptor-ligand binding of the endocannabinoid system.
Clinical effects, generally psychiatric, include paranoia, anxiety, agitation, delusions, and psychosis. There are also reports of patients who arrive with sympathomimetic toxicity, some of whom develop bradycardia and hypotension, and some who progress to acute renal failure, seizures, and death. Detection of these products is difficult as they do not react on EMIT testing for THC metabolites and require either gas chromatography-mass spectrometry or liquid chromatography with tandem mass spectrometry.45–48
- Substance Abuse and Mental Health Services Administration. Results from the 2012 National Survey on Drug Use and Health: Summary of National Findings, NSDUH Series H-46, HHS Publication No. (SMA) 13-4795. www.samhsa.gov/data/sites/default/files/NSDUHresultsPDFWHTML2013/Web/NSDUHresults2013.pdf. Accessed October 2, 2015.
- United Nations Office on Drugs and Crime. 2008 World Drug Report. www.unodc.org/documents/wdr/WDR_2008/WDR_2008_eng_web.pdf. Accessed October 2, 2015.
- American Society of Addiction Medicine (ASAM). Public policy statement on medical marijuana. www.asam.org/docs/publicy-policy-statements/1medical-marijuana-4-10.pdf?sfvrsn=0. Accessed October 2, 2015.
- Howlett AC, Barth F, Bonner TI, et al. International Union of Pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol Rev 2002; 54:161–202.
- Sharkey KA, Darmani NA, Parker LA. Regulation of nausea and vomiting by cannabinoids and the endocannabinoid system. Eur J Pharmacol 2014; 722:134–146.
- Iversen L. Cannabis and the brain. Brain 2003; 126:1252–1270.
- Huestis MA, Henningfield JE, Cone EJ. Blood cannabinoids. I. Absorption of THC and formation of 11-OH-THC and THCCOOH during and after smoking marijuana. J Anal Toxicol 1992; 16:276–282.
- Grotenhermen F. Pharmacokinetics and pharmacodynamics of cannabinoids. Clin Pharmacokinet 2003; 42:327–360.
- Mehmedic Z, Chandra S, Slade D, et al. Potency trends of Δ9-THC and other cannabinoids in confiscated cannabis preparations from 1993 to 2008. J Forensic Sci 2010; 55:1209–1217.
- Mittleman MA, Lewis RA, Maclure M, Sherwood JB, Muller JE. Triggering myocardial infarction by marijuana. Circulation 2001; 103:2805–2809.
- Mukamal KJ, Maclure M, Muller JE, Mittleman MA. An exploratory prospective study of marijuana use and mortality following acute myocardial infarction. Am Heart J 2008; 155:465–470.
- Thomas G, Kloner RA, Rezkalla S. Adverse cardiovascular, cerebrovascular, and peripheral vascular effects of marijuana inhalation: what cardiologists need to know. Am J Cardiol 2014; 113:187–190.
- Wang GS, Roosevelt G, Heard K. Pediatric marijuana exposures in a medical marijuana state. JAMA Pediatr 2013; 167:630–633.
- Carstairs SD, Fujinaka MK, Keeney GE, Ly BT. Prolonged coma in a child due to hashish ingestion with quantitation of THC metabolites in urine. J Emerg Med 2011; 41:e69–e71.
- Le Garrec S, Dauger S, Sachs P. Cannabis poisoning in children. Intensive Care Med 2014; 40:1394–1395.
- Ragab AR, Al-Mazroua MK. Passive cannabis smoking resulting in coma in a 16-month old infant. J Clin Case Rep 2012;2:237.
- Robinson K. Beyond resinable doubt? J Clin Forensic Med 2005;12:164–166.
- Burns JK. Pathways from cannabis to psychosis: a review of the evidence. Front Psychiatry 2013;4:128.
- Di Forti M, Sallis H, Allegri F, et al. Daily use, especially of high-potency cannabis, drives the earlier onset of psychosis in cannabis users. Schizophr Bull 2014; 40:1509–1517.
- Moore TH, Zammit S, Lingford-Hughes A, et al. Cannabis use and risk of psychotic or affective mental health outcomes: a systematic review. Lancet 2007; 370:319–328.
- Wilkinson ST, Radhakrishnan R, D'Souza DC. Impact of cannabis use on the development of psychotic disorders. Curr Addict Rep 2014;1:115–128.
- Aldington S, Williams M, Nowitz M, et al. Effects of cannabis on pulmonary structure, function and symptoms. Thorax 2007; 62:1058–1063.
- George KL, Saltman LH, Stein GS, Lian JB, Zurier RB. Ajulemic acid, a nonpsychoactive cannabinoid acid, suppresses osteoclastogenesis in mononuclear precursor cells and induces apoptosis in mature osteoclast-like cells. J Cell Physiol 2008; 214:714–720.
- Reece AS. Chronic toxicology of cannabis. Clin Toxicol (Phila) 2009; 47:517–524.
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Arlington, VA: American Psychiatric Publishing; 2013.
- Allsop DJ, Copeland J, Lintzeris N, et al. Nabiximols as an agonist replacement therapy during cannabis withdrawal: a randomized clinical trial. JAMA Psychiatry 2014; 71:281–291.
- Hesse M, Thylstrup B. Time-course of the DSM-5 cannabis withdrawal symptoms in poly-substance abusers. BMC Psychiatry 2013; 13:258.
- Allen JH, de Moore GM, Heddle R, Twartz JC. Cannabinoid hyperemesis: cyclical hyperemesis in association with chronic cannabis abuse. Gut 2004; 53:1566–1570.
- Galli JA, Sawaya RA, Friedenberg FK. Cannabinoid hyperemesis syndrome. Curr Drug Abuse Rev 2011; 4:241–249.
- Simonetto DA, Oxentenko AS, Herman ML, Szostek JH. Cannabinoid hyperemesis: a case series of 98 patients. Mayo Clin Proc 2012; 87:114–119.
- Soriano-Co M, Batke M, Cappell MS. The cannabis hyperemesis syndrome characterized by persistent nausea and vomiting, abdominal pain, and compulsive bathing associated with chronic marijuana use: a report of eight cases in the United States. Dig Dis Sci 2010; 55:3113–3119.
- Wallace EA, Andrews SE, Garmany CL, Jelley MJ. Cannabinoid hyperemesis syndrome: literature review and proposed diagnosis and treatment algorithm. South Med J 2011; 104:659–664.
- Hickey JL, Witsil JC, Mycyk MB. Haloperidol for treatment of cannabinoid hyperemesis syndrome. Am J Emerg Med 2013; 31:1003.e5–1003.e6.
- Perwitasari DA, Gelderblom H, Atthobari J, et al. Anti-emetic drugs in oncology: pharmacology and individualization by pharmacogenetics. Int J Clin Pharm 2011; 33:33–43.
- Cox B, Chhabra A, Adler M, Simmons J, Randlett D. Cannabinoid hyperemesis syndrome: case report of a paradoxical reaction with heavy marijuana use. Case Rep Med 2012; 2012:757696.
- Sakurai M, Mori T, Kato J, et al. Efficacy of aprepitant in preventing nausea and vomiting due to high-dose melphalan-based conditioning for allogeneic hematopoietic stem cell transplantation. Int J Hematol 2014; 99:457–462.
- Lapoint J. Case series of patients treated for cannabinoid hyperemesis syndrome with capsaicin cream. Clin Tox 2014; 52:707. Abstract #53.
- Biary R, Oh A, Lapoint J, Nelson LS, Hoffman RS, Howland MA. Topical capsaicin cream used as a therapy for cannabinoid hyperemesis syndrome. Clin Tox 2014; 52:787. Abstract #232.
- Moeller KE, Lee KC, Kissack JC. Urine drug screening: practical guide for clinicians. Mayo Clin Proc 2008; 83:66–76.
- Lowe RH, Abraham TT, Darwin WD, Herning R, Cadet JL, Huestis MA. Extended urinary delta9-tetrahydrocannabinol excretion in chronic cannabis users precludes use as a biomarker of new drug exposure. Drug Alcohol Depend 2009; 105:24–32.
- Paul BD, Jacobs A. Effects of oxidizing adulterants on detection of 11-nor-delta9-THC-9-carboxylic acid in urine. J Anal Toxicol 2002; 26:460–463.
- Schwope DM, Karschner EL, Gorelick DA, Huestis MA. Identification of recent cannabis use: whole-blood and plasma free and glucuronidated cannabinoid pharmacokinetics following controlled smoked cannabis administration. Clin Chem 2011; 57:1406-1414.
- Huestis MA, Smith ML. Cannabinoid pharmacokinetics and disposition in alternative matrices. In: Pertwee R, ed. Handbook of Cannabis. Oxford, United Kingdom: Oxford University Press; 2014:296–316.
- Mowry JB, Spyker DA, Cantilena LR Jr, Bailey JE, Ford M. 2012 Annual Report of the American Association of Poison Control Centers’ National Poison Data System (NPDS): 30th Annual Report. Clin Toxicol (Phila) 2013; 51:949–1229.
- Rosenbaum CD, Carreiro SP, Babu KM. Here today, gone tomorrow…and back again? A review of herbal marijuana alternatives (K2, Spice), synthetic cathinones (bath salts), kratom, Salvia divinorum, methoxetamine, and piperazines. J Med Toxicol 2012; 8:15–32.
- Gurney SMR, Scott KS, Kacinko SL, Presley BC, Logan BK. Pharmacology, toxicology, and adverse effects of synthetic cannabinoid drugs. Forensic Sci Rev 2014; 26:53–78.
- McKeever RG, Vearrier D, Jacobs D, LaSala G, Okaneku J, Greenberg MI. K2-not the spice of life; synthetic cannabinoids and ST elevation myocardial infarction: a case report. J Med Toxicol 2015; 11:129–131.
- Schneir AB, Baumbacher T. Convulsions associated with the use of a synthetic cannabinoid product. J Med Toxicol 2012; 8:62–64.
- Substance Abuse and Mental Health Services Administration. Results from the 2012 National Survey on Drug Use and Health: Summary of National Findings, NSDUH Series H-46, HHS Publication No. (SMA) 13-4795. www.samhsa.gov/data/sites/default/files/NSDUHresultsPDFWHTML2013/Web/NSDUHresults2013.pdf. Accessed October 2, 2015.
- United Nations Office on Drugs and Crime. 2008 World Drug Report. www.unodc.org/documents/wdr/WDR_2008/WDR_2008_eng_web.pdf. Accessed October 2, 2015.
- American Society of Addiction Medicine (ASAM). Public policy statement on medical marijuana. www.asam.org/docs/publicy-policy-statements/1medical-marijuana-4-10.pdf?sfvrsn=0. Accessed October 2, 2015.
- Howlett AC, Barth F, Bonner TI, et al. International Union of Pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol Rev 2002; 54:161–202.
- Sharkey KA, Darmani NA, Parker LA. Regulation of nausea and vomiting by cannabinoids and the endocannabinoid system. Eur J Pharmacol 2014; 722:134–146.
- Iversen L. Cannabis and the brain. Brain 2003; 126:1252–1270.
- Huestis MA, Henningfield JE, Cone EJ. Blood cannabinoids. I. Absorption of THC and formation of 11-OH-THC and THCCOOH during and after smoking marijuana. J Anal Toxicol 1992; 16:276–282.
- Grotenhermen F. Pharmacokinetics and pharmacodynamics of cannabinoids. Clin Pharmacokinet 2003; 42:327–360.
- Mehmedic Z, Chandra S, Slade D, et al. Potency trends of Δ9-THC and other cannabinoids in confiscated cannabis preparations from 1993 to 2008. J Forensic Sci 2010; 55:1209–1217.
- Mittleman MA, Lewis RA, Maclure M, Sherwood JB, Muller JE. Triggering myocardial infarction by marijuana. Circulation 2001; 103:2805–2809.
- Mukamal KJ, Maclure M, Muller JE, Mittleman MA. An exploratory prospective study of marijuana use and mortality following acute myocardial infarction. Am Heart J 2008; 155:465–470.
- Thomas G, Kloner RA, Rezkalla S. Adverse cardiovascular, cerebrovascular, and peripheral vascular effects of marijuana inhalation: what cardiologists need to know. Am J Cardiol 2014; 113:187–190.
- Wang GS, Roosevelt G, Heard K. Pediatric marijuana exposures in a medical marijuana state. JAMA Pediatr 2013; 167:630–633.
- Carstairs SD, Fujinaka MK, Keeney GE, Ly BT. Prolonged coma in a child due to hashish ingestion with quantitation of THC metabolites in urine. J Emerg Med 2011; 41:e69–e71.
- Le Garrec S, Dauger S, Sachs P. Cannabis poisoning in children. Intensive Care Med 2014; 40:1394–1395.
- Ragab AR, Al-Mazroua MK. Passive cannabis smoking resulting in coma in a 16-month old infant. J Clin Case Rep 2012;2:237.
- Robinson K. Beyond resinable doubt? J Clin Forensic Med 2005;12:164–166.
- Burns JK. Pathways from cannabis to psychosis: a review of the evidence. Front Psychiatry 2013;4:128.
- Di Forti M, Sallis H, Allegri F, et al. Daily use, especially of high-potency cannabis, drives the earlier onset of psychosis in cannabis users. Schizophr Bull 2014; 40:1509–1517.
- Moore TH, Zammit S, Lingford-Hughes A, et al. Cannabis use and risk of psychotic or affective mental health outcomes: a systematic review. Lancet 2007; 370:319–328.
- Wilkinson ST, Radhakrishnan R, D'Souza DC. Impact of cannabis use on the development of psychotic disorders. Curr Addict Rep 2014;1:115–128.
- Aldington S, Williams M, Nowitz M, et al. Effects of cannabis on pulmonary structure, function and symptoms. Thorax 2007; 62:1058–1063.
- George KL, Saltman LH, Stein GS, Lian JB, Zurier RB. Ajulemic acid, a nonpsychoactive cannabinoid acid, suppresses osteoclastogenesis in mononuclear precursor cells and induces apoptosis in mature osteoclast-like cells. J Cell Physiol 2008; 214:714–720.
- Reece AS. Chronic toxicology of cannabis. Clin Toxicol (Phila) 2009; 47:517–524.
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th ed. Arlington, VA: American Psychiatric Publishing; 2013.
- Allsop DJ, Copeland J, Lintzeris N, et al. Nabiximols as an agonist replacement therapy during cannabis withdrawal: a randomized clinical trial. JAMA Psychiatry 2014; 71:281–291.
- Hesse M, Thylstrup B. Time-course of the DSM-5 cannabis withdrawal symptoms in poly-substance abusers. BMC Psychiatry 2013; 13:258.
- Allen JH, de Moore GM, Heddle R, Twartz JC. Cannabinoid hyperemesis: cyclical hyperemesis in association with chronic cannabis abuse. Gut 2004; 53:1566–1570.
- Galli JA, Sawaya RA, Friedenberg FK. Cannabinoid hyperemesis syndrome. Curr Drug Abuse Rev 2011; 4:241–249.
- Simonetto DA, Oxentenko AS, Herman ML, Szostek JH. Cannabinoid hyperemesis: a case series of 98 patients. Mayo Clin Proc 2012; 87:114–119.
- Soriano-Co M, Batke M, Cappell MS. The cannabis hyperemesis syndrome characterized by persistent nausea and vomiting, abdominal pain, and compulsive bathing associated with chronic marijuana use: a report of eight cases in the United States. Dig Dis Sci 2010; 55:3113–3119.
- Wallace EA, Andrews SE, Garmany CL, Jelley MJ. Cannabinoid hyperemesis syndrome: literature review and proposed diagnosis and treatment algorithm. South Med J 2011; 104:659–664.
- Hickey JL, Witsil JC, Mycyk MB. Haloperidol for treatment of cannabinoid hyperemesis syndrome. Am J Emerg Med 2013; 31:1003.e5–1003.e6.
- Perwitasari DA, Gelderblom H, Atthobari J, et al. Anti-emetic drugs in oncology: pharmacology and individualization by pharmacogenetics. Int J Clin Pharm 2011; 33:33–43.
- Cox B, Chhabra A, Adler M, Simmons J, Randlett D. Cannabinoid hyperemesis syndrome: case report of a paradoxical reaction with heavy marijuana use. Case Rep Med 2012; 2012:757696.
- Sakurai M, Mori T, Kato J, et al. Efficacy of aprepitant in preventing nausea and vomiting due to high-dose melphalan-based conditioning for allogeneic hematopoietic stem cell transplantation. Int J Hematol 2014; 99:457–462.
- Lapoint J. Case series of patients treated for cannabinoid hyperemesis syndrome with capsaicin cream. Clin Tox 2014; 52:707. Abstract #53.
- Biary R, Oh A, Lapoint J, Nelson LS, Hoffman RS, Howland MA. Topical capsaicin cream used as a therapy for cannabinoid hyperemesis syndrome. Clin Tox 2014; 52:787. Abstract #232.
- Moeller KE, Lee KC, Kissack JC. Urine drug screening: practical guide for clinicians. Mayo Clin Proc 2008; 83:66–76.
- Lowe RH, Abraham TT, Darwin WD, Herning R, Cadet JL, Huestis MA. Extended urinary delta9-tetrahydrocannabinol excretion in chronic cannabis users precludes use as a biomarker of new drug exposure. Drug Alcohol Depend 2009; 105:24–32.
- Paul BD, Jacobs A. Effects of oxidizing adulterants on detection of 11-nor-delta9-THC-9-carboxylic acid in urine. J Anal Toxicol 2002; 26:460–463.
- Schwope DM, Karschner EL, Gorelick DA, Huestis MA. Identification of recent cannabis use: whole-blood and plasma free and glucuronidated cannabinoid pharmacokinetics following controlled smoked cannabis administration. Clin Chem 2011; 57:1406-1414.
- Huestis MA, Smith ML. Cannabinoid pharmacokinetics and disposition in alternative matrices. In: Pertwee R, ed. Handbook of Cannabis. Oxford, United Kingdom: Oxford University Press; 2014:296–316.
- Mowry JB, Spyker DA, Cantilena LR Jr, Bailey JE, Ford M. 2012 Annual Report of the American Association of Poison Control Centers’ National Poison Data System (NPDS): 30th Annual Report. Clin Toxicol (Phila) 2013; 51:949–1229.
- Rosenbaum CD, Carreiro SP, Babu KM. Here today, gone tomorrow…and back again? A review of herbal marijuana alternatives (K2, Spice), synthetic cathinones (bath salts), kratom, Salvia divinorum, methoxetamine, and piperazines. J Med Toxicol 2012; 8:15–32.
- Gurney SMR, Scott KS, Kacinko SL, Presley BC, Logan BK. Pharmacology, toxicology, and adverse effects of synthetic cannabinoid drugs. Forensic Sci Rev 2014; 26:53–78.
- McKeever RG, Vearrier D, Jacobs D, LaSala G, Okaneku J, Greenberg MI. K2-not the spice of life; synthetic cannabinoids and ST elevation myocardial infarction: a case report. J Med Toxicol 2015; 11:129–131.
- Schneir AB, Baumbacher T. Convulsions associated with the use of a synthetic cannabinoid product. J Med Toxicol 2012; 8:62–64.
KEY POINTS
- Cannabis has been used throughout history and has become increasingly available for recreational purposes, despite its current classification as a schedule I controlled substance.
- Although severe acute toxicity has been reported, it is relatively rare, and most users’ casual experiences are benign.
- Internists are most likely to see complications such as cannabinoid hyperemesis syndrome and cardiovascular problems that cannot be resolved sufficiently in the emergency department.
- Screening urine testing is usually done by enzyme multiplied immunoassay, whereas confirmatory testing is done with gas chromatography-mass spectrometry, which is more specific.
