User login
First recommendations for cancer screening in myositis issued
AT ACR 2022
PHILADELPHIA – The first consensus screening guidelines for patients with idiopathic inflammatory myopathy (IIM) provide recommendations on risk stratification for individuals, basic and enhanced screening protocols, and screening frequency.
The recommendations, issued by the International Myositis Assessment and Clinical Studies Group (IMACS), stratify cancer risk for individual patients into low, intermediate, or high categories based on the IIM disease subtype, autoantibody status, and clinical features, reported Alexander Oldroyd, PhD, MSc, MBChB of the University of Manchester, England.

“There’s a big unmet need for cancer screening. One in four adults with myositis has cancer, either 3 years before or after a diagnosis of myositis. It’s one of the leading causes of death in these patients, and they’re overwhelmingly diagnosed at a late stage, so we need standardized approaches to get early diagnosis,” he said in an interview at the annual meeting of the American College of Rheumatology.
Sharon Kolasinski, MD, of the University of Pennsylvania in Philadelphia, said in an interview that the guideline is a welcome development for rheumatologists. Dr. Kolasinski moderated the session where Dr. Oldroyd described the guideline, but she was not involved in its formulation.
“I think that we all have wondered for a very long time: What is the optimal cancer screening for myositis patients? We all worry that the onset of their diseases is associated with a coincident cancer, or that they will develop it soon,” she said.
Dr. Oldroyd emphasized that all patients with myositis have elevated risk for cancer compared with the general population and that the guideline categories of low, intermediate, and high are relative only to patients with IIM.
International consensus
The data on which the recommendations are based come from a systematic review and meta-analysis by Dr. Oldroyd and colleagues of 69 studies on cancer risk factors and 9 on IIM-specific cancer screening.
The authors of that paper found that the dermatomyositis subtype, older age, male sex, dysphagia, cutaneous ulceration and antitranscriptional intermediary factor-1 gamma (anti-TIF1-gamma) positivity were associated with significantly increased risk of cancer.
In contrast, polymyositis and clinically amyopathic dermatomyositis subtypes, Raynaud’s phenomenon, interstitial lung disease, very high serum creatine kinase or lactate dehydrogenase levels, and positivity for anti-Jo1 or anti-EJ antibodies were associated with significantly reduced risk of cancer.
The consensus recommendations were developed with anonymous contributions from 75 expert participants in 22 countries, with additional input from 3 patient partners.
Do this
The guideline lists 18 recommendations, of which 13 are strong and 5 are conditional.
An example of a strong recommendation is number 3, based on a moderate level of evidences:
“All adult IIM patients, irrespective of cancer risk, should continue to participate in country/region-specific age and sex appropriate cancer screening programs,” the guideline recommends.
Patients with verified inclusion body myositis or juvenile-onset IIM do not, however, require routine screening for myositis-associated cancer, the guideline says (recommendations 1 and 2).
There are also recommendations that all adults with new-onset IIM be tested for myositis-specific and myositis-associated autoantibodies to assist in stratifying patients by risk category.
The guideline divides screening recommendations into basic and enhanced. The basic screening should include a comprehensive history and physical exam, complete blood count, liver functions tests, erythrocyte sedimentation rates/plasma viscosity, serum protein electrophoresis, urinalysis, and chest x-ray.
Adults with IIM who are determined to be at low risk for IIM-related cancer should have basic cancer screening at the time of IIM diagnosis. Adults with intermediate risk should undergo both basic and enhanced screening at the time of IIM diagnosis, and those with high risk should undergo enhanced screening at the time of myositis diagnosis, with basic screening annually for 3 years, the recommendations say.
Consider doing this
Conditional recommendations (“clinicians should consider ...”) include the use of PET/CT for adults at high risk for cancer when an underlying cancer has not been detected at the time of IIM diagnosis. They also include a single screening test for anti-TIF1-gamma positive dermatomyositis patients whose disease onset was after age 40 and who have at least one additional risk factor.
Also conditionally recommended are upper and lower gastrointestinal endoscopy for patients at high risk when an underlying cancer is not found at the time of IIM diagnosis, nasoendoscopy in geographical regions with elevated risk for nasopharyngeal cancers, and screening for all IIM patients with red-flag symptoms or clinical features of cancer, including unexplained weight loss, family history of cancer, smoking, unexplained fever, or night sweats.
Guided steps
“I think clinicians have a lot of questions such as, ‘well, what should I do, when should I do it?’ These are important clinical questions, and we need guidance about this. We need to balance comprehensiveness with cost-effectiveness, and we need expert opinion about what steps we should take now and which should we take later,” Dr. Kolasinski said.
The guideline development process was supported by the University of Manchester, IMACS, National Institute for Health Research (United Kingdom), National Institutes of Health, National Health Service Northern Care Alliance, The Myositis Association, Myositis UK, University of Pittsburgh, Versus Arthritis, and the Center for Musculoskeletal Research. Dr. Oldroyd and Dr. Kolasinski reported having no relevant conflicts of interest.
AT ACR 2022
PHILADELPHIA – The first consensus screening guidelines for patients with idiopathic inflammatory myopathy (IIM) provide recommendations on risk stratification for individuals, basic and enhanced screening protocols, and screening frequency.
The recommendations, issued by the International Myositis Assessment and Clinical Studies Group (IMACS), stratify cancer risk for individual patients into low, intermediate, or high categories based on the IIM disease subtype, autoantibody status, and clinical features, reported Alexander Oldroyd, PhD, MSc, MBChB of the University of Manchester, England.
“There’s a big unmet need for cancer screening. One in four adults with myositis has cancer, either 3 years before or after a diagnosis of myositis. It’s one of the leading causes of death in these patients, and they’re overwhelmingly diagnosed at a late stage, so we need standardized approaches to get early diagnosis,” he said in an interview at the annual meeting of the American College of Rheumatology.
Sharon Kolasinski, MD, of the University of Pennsylvania in Philadelphia, said in an interview that the guideline is a welcome development for rheumatologists. Dr. Kolasinski moderated the session where Dr. Oldroyd described the guideline, but she was not involved in its formulation.
“I think that we all have wondered for a very long time: What is the optimal cancer screening for myositis patients? We all worry that the onset of their diseases is associated with a coincident cancer, or that they will develop it soon,” she said.
Dr. Oldroyd emphasized that all patients with myositis have elevated risk for cancer compared with the general population and that the guideline categories of low, intermediate, and high are relative only to patients with IIM.
International consensus
The data on which the recommendations are based come from a systematic review and meta-analysis by Dr. Oldroyd and colleagues of 69 studies on cancer risk factors and 9 on IIM-specific cancer screening.
The authors of that paper found that the dermatomyositis subtype, older age, male sex, dysphagia, cutaneous ulceration and antitranscriptional intermediary factor-1 gamma (anti-TIF1-gamma) positivity were associated with significantly increased risk of cancer.
In contrast, polymyositis and clinically amyopathic dermatomyositis subtypes, Raynaud’s phenomenon, interstitial lung disease, very high serum creatine kinase or lactate dehydrogenase levels, and positivity for anti-Jo1 or anti-EJ antibodies were associated with significantly reduced risk of cancer.
The consensus recommendations were developed with anonymous contributions from 75 expert participants in 22 countries, with additional input from 3 patient partners.
Do this
The guideline lists 18 recommendations, of which 13 are strong and 5 are conditional.
An example of a strong recommendation is number 3, based on a moderate level of evidences:
“All adult IIM patients, irrespective of cancer risk, should continue to participate in country/region-specific age and sex appropriate cancer screening programs,” the guideline recommends.
Patients with verified inclusion body myositis or juvenile-onset IIM do not, however, require routine screening for myositis-associated cancer, the guideline says (recommendations 1 and 2).
There are also recommendations that all adults with new-onset IIM be tested for myositis-specific and myositis-associated autoantibodies to assist in stratifying patients by risk category.
The guideline divides screening recommendations into basic and enhanced. The basic screening should include a comprehensive history and physical exam, complete blood count, liver functions tests, erythrocyte sedimentation rates/plasma viscosity, serum protein electrophoresis, urinalysis, and chest x-ray.
Adults with IIM who are determined to be at low risk for IIM-related cancer should have basic cancer screening at the time of IIM diagnosis. Adults with intermediate risk should undergo both basic and enhanced screening at the time of IIM diagnosis, and those with high risk should undergo enhanced screening at the time of myositis diagnosis, with basic screening annually for 3 years, the recommendations say.
Consider doing this
Conditional recommendations (“clinicians should consider ...”) include the use of PET/CT for adults at high risk for cancer when an underlying cancer has not been detected at the time of IIM diagnosis. They also include a single screening test for anti-TIF1-gamma positive dermatomyositis patients whose disease onset was after age 40 and who have at least one additional risk factor.
Also conditionally recommended are upper and lower gastrointestinal endoscopy for patients at high risk when an underlying cancer is not found at the time of IIM diagnosis, nasoendoscopy in geographical regions with elevated risk for nasopharyngeal cancers, and screening for all IIM patients with red-flag symptoms or clinical features of cancer, including unexplained weight loss, family history of cancer, smoking, unexplained fever, or night sweats.
Guided steps
“I think clinicians have a lot of questions such as, ‘well, what should I do, when should I do it?’ These are important clinical questions, and we need guidance about this. We need to balance comprehensiveness with cost-effectiveness, and we need expert opinion about what steps we should take now and which should we take later,” Dr. Kolasinski said.
The guideline development process was supported by the University of Manchester, IMACS, National Institute for Health Research (United Kingdom), National Institutes of Health, National Health Service Northern Care Alliance, The Myositis Association, Myositis UK, University of Pittsburgh, Versus Arthritis, and the Center for Musculoskeletal Research. Dr. Oldroyd and Dr. Kolasinski reported having no relevant conflicts of interest.
AT ACR 2022
PHILADELPHIA – The first consensus screening guidelines for patients with idiopathic inflammatory myopathy (IIM) provide recommendations on risk stratification for individuals, basic and enhanced screening protocols, and screening frequency.
The recommendations, issued by the International Myositis Assessment and Clinical Studies Group (IMACS), stratify cancer risk for individual patients into low, intermediate, or high categories based on the IIM disease subtype, autoantibody status, and clinical features, reported Alexander Oldroyd, PhD, MSc, MBChB of the University of Manchester, England.
“There’s a big unmet need for cancer screening. One in four adults with myositis has cancer, either 3 years before or after a diagnosis of myositis. It’s one of the leading causes of death in these patients, and they’re overwhelmingly diagnosed at a late stage, so we need standardized approaches to get early diagnosis,” he said in an interview at the annual meeting of the American College of Rheumatology.
Sharon Kolasinski, MD, of the University of Pennsylvania in Philadelphia, said in an interview that the guideline is a welcome development for rheumatologists. Dr. Kolasinski moderated the session where Dr. Oldroyd described the guideline, but she was not involved in its formulation.
“I think that we all have wondered for a very long time: What is the optimal cancer screening for myositis patients? We all worry that the onset of their diseases is associated with a coincident cancer, or that they will develop it soon,” she said.
Dr. Oldroyd emphasized that all patients with myositis have elevated risk for cancer compared with the general population and that the guideline categories of low, intermediate, and high are relative only to patients with IIM.
International consensus
The data on which the recommendations are based come from a systematic review and meta-analysis by Dr. Oldroyd and colleagues of 69 studies on cancer risk factors and 9 on IIM-specific cancer screening.
The authors of that paper found that the dermatomyositis subtype, older age, male sex, dysphagia, cutaneous ulceration and antitranscriptional intermediary factor-1 gamma (anti-TIF1-gamma) positivity were associated with significantly increased risk of cancer.
In contrast, polymyositis and clinically amyopathic dermatomyositis subtypes, Raynaud’s phenomenon, interstitial lung disease, very high serum creatine kinase or lactate dehydrogenase levels, and positivity for anti-Jo1 or anti-EJ antibodies were associated with significantly reduced risk of cancer.
The consensus recommendations were developed with anonymous contributions from 75 expert participants in 22 countries, with additional input from 3 patient partners.
Do this
The guideline lists 18 recommendations, of which 13 are strong and 5 are conditional.
An example of a strong recommendation is number 3, based on a moderate level of evidences:
“All adult IIM patients, irrespective of cancer risk, should continue to participate in country/region-specific age and sex appropriate cancer screening programs,” the guideline recommends.
Patients with verified inclusion body myositis or juvenile-onset IIM do not, however, require routine screening for myositis-associated cancer, the guideline says (recommendations 1 and 2).
There are also recommendations that all adults with new-onset IIM be tested for myositis-specific and myositis-associated autoantibodies to assist in stratifying patients by risk category.
The guideline divides screening recommendations into basic and enhanced. The basic screening should include a comprehensive history and physical exam, complete blood count, liver functions tests, erythrocyte sedimentation rates/plasma viscosity, serum protein electrophoresis, urinalysis, and chest x-ray.
Adults with IIM who are determined to be at low risk for IIM-related cancer should have basic cancer screening at the time of IIM diagnosis. Adults with intermediate risk should undergo both basic and enhanced screening at the time of IIM diagnosis, and those with high risk should undergo enhanced screening at the time of myositis diagnosis, with basic screening annually for 3 years, the recommendations say.
Consider doing this
Conditional recommendations (“clinicians should consider ...”) include the use of PET/CT for adults at high risk for cancer when an underlying cancer has not been detected at the time of IIM diagnosis. They also include a single screening test for anti-TIF1-gamma positive dermatomyositis patients whose disease onset was after age 40 and who have at least one additional risk factor.
Also conditionally recommended are upper and lower gastrointestinal endoscopy for patients at high risk when an underlying cancer is not found at the time of IIM diagnosis, nasoendoscopy in geographical regions with elevated risk for nasopharyngeal cancers, and screening for all IIM patients with red-flag symptoms or clinical features of cancer, including unexplained weight loss, family history of cancer, smoking, unexplained fever, or night sweats.
Guided steps
“I think clinicians have a lot of questions such as, ‘well, what should I do, when should I do it?’ These are important clinical questions, and we need guidance about this. We need to balance comprehensiveness with cost-effectiveness, and we need expert opinion about what steps we should take now and which should we take later,” Dr. Kolasinski said.
The guideline development process was supported by the University of Manchester, IMACS, National Institute for Health Research (United Kingdom), National Institutes of Health, National Health Service Northern Care Alliance, The Myositis Association, Myositis UK, University of Pittsburgh, Versus Arthritis, and the Center for Musculoskeletal Research. Dr. Oldroyd and Dr. Kolasinski reported having no relevant conflicts of interest.
Novel gene-based therapies for neuromuscular diseases
Neuromuscular diseases (NMDs) are a broad classification of heterogeneous groups of disorders characterized by progressive muscle weakness resulting from muscle or nerve dysfunction.1 Diagnosis is based on symptoms and a full medical history, as well as on muscle and imaging tests (including electromyography, nerve-conduction studies, magnetic resonance imaging, muscle biopsy, and blood tests) to confirm or rule out specific NMDs.2 Early diagnosis of NMDs can be difficult because symptoms overlap with those of many other diseases.
Although individually, NMDs are rare, collectively, they affect approximately 250,000 people in the United States. Disease types vary in regard to cause, symptoms, prevalence, age of onset, progression, and severity. Functional impairment from any NMD can lead to lifelong morbidities and shortened life expectancy.1,3
Treatment options for NMDs are limited; most target symptoms, not disease progression. Although there is a need for safe and effective gene-based therapies for NMDs, there are challenges to developing and delivering such treatments that have impeded clinical success. These include a lack of understanding about disease pathology and drug targets, limited animal model systems, and few reliable biomarkers that are predictive of therapeutic success.4,5

Notwithstanding that challenges remain, our understanding of gene expression in NMDs has greatly advanced in the past few decades. This progress has translated into promising results in the gene-therapy field – thereby setting the stage for therapeutic approaches that use novel gene-delivery and gene-manipulation tools.6 These novel approaches include nonviral strategies, such as antisense oligonucleotides (ASOs), and viral-based strategies, such as adeno-associated virus (AAV)-mediated gene silencing and AAV-mediated gene delivery.
In this article, we highlight advancements in the clinical development of gene-based therapies for NMDs. We focus on amyotrophic lateral sclerosis (ALS), spinal muscular atrophy (SMA), and Duchenne muscular dystrophy (DMD) because of recent clinical successes in developing such therapies.1,6,7 We also catalog completed and ongoing clinical trials for ALS, SMA, and DMD (Tables 1-3).
Amyotrophic lateral sclerosis
ALS is caused by progressive degeneration of upper- and lower-motor neurons, which eventually leads to respiratory failure and death 3 to 5 years after disease onset.7-9 There are two subtypes: Familial ALS (10% of cases) and sporadic ALS (90% of cases). Commonly mutated ALS-associated genes6,8 are:
- Superoxide dismutase type 1 (SOD1).
- Chromosome 9 open reading frame 72 (C9orf72).
- Transactive response DNA-binding protein 43 (TARDBP).
- Fused in sarcoma (FUS).
SOD1-targeted therapy is being studied, with early evidence of clinical success. Mutations in SOD1 account for 10% to 20% of familial ALS cases and 1% to 2% of sporadic ALS cases.6,10 10 Mutations in C9orf72 account for 25 to 40% of familial ALS cases and 7% of sporadic ALS cases.8,9,11 Mutations in TARDBP account for 3% of familial ALS cases and 2% of sporadic cases.12 Mutations in FUS account for 4% of familial ALS cases and 1% of sporadic cases. Overall, these mutant proteins can trigger neurotoxicity, thus inducing motor-neuron death.6,10
Treatment of ALS
Two treatments for ALS are Food and Drug Administration approved: riluzole (Rilutek), approved in 1995, and edaravone (Radicava), approved in 2017.

Riluzole is an oral anti-excitotoxic glutamate antagonist.11 Approval of riluzole was based on the results of two studies that demonstrated a 2- to 3-month survival benefit.10,14 For patients who have difficulty swallowing, an oral suspension (Tiglutik, approved in 2018) and an oral film (Exservan, approved in 2019) are available.
Edaravone is a free-radical scavenger that decreases oxidative stress and is administered intravenously (IV).9,13,14 Findings from clinical trials suggest functional improvement or slower decline in function for some patients.
Although these two agents demonstrate modest therapeutic benefit, neither reverses progression of disease.10,14
Gene-based therapy for ALS
Many non-viral strategies, including antisense oligonucleotide (ASO), monoclonal antibodies, reverse transcriptase inhibitors, and HGF gene replacement therapy are used as therapeutic approaches to SOD1, C9orf72, and FUS gene mutations in ALS patients, and are being evaluated in clinical studies14,15 (Table 113-17).
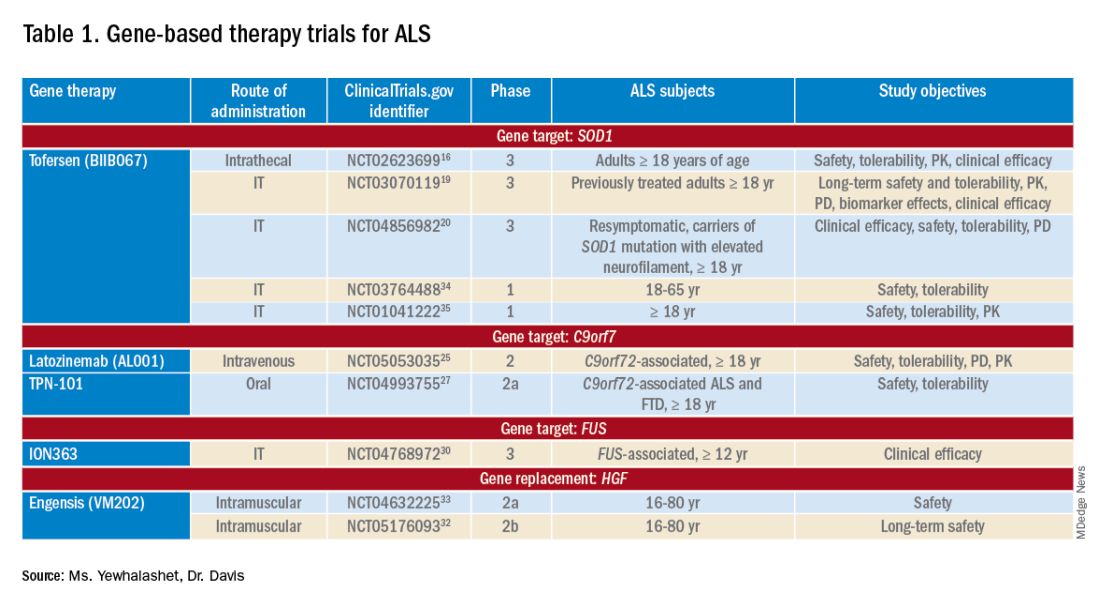
Tofersen, also known as BIIB067, is an investigational ASO, administered by intrathecal (IT) injection, that binds to SOD1 mRNA, thus reducing its protein levels.16 Tofersen was evaluated in the VALOR phase 3 study (ClinicalTrials.gov Identifier: NCT02623699), a three-part randomized, double-blind, placebo-controlled trial: single ascending dose (Part A), multiple ascending dose (B), and fixed dose (C).10 In Parts A and B, 48 participants received five IT injections of tofersen or placebo over 12 weeks and were followed for an additional 12 weeks. Reduction in SOD1 protein production and neurofilament level in cerebrospinal fluid (CSF) (a potential biomarker of motor-neuron degeneration) was observed, which determined the fixed-dose for Part C.16,17
Part C examined the efficacy, safety and tolerability, pharmacokinetics (PK), and pharmacodynamics (PD) of tofersen, compared with placebo, in adults with ALS who had a confirmed SOD1 mutation.17 A total of 108 participants were enrolled; 60 were identified as “faster-progressing”; 48, as “slower-progressing.”18 The primary endpoint of Part C was change from baseline to Week 28 on the Revised ALS Functional Rating Scale (ALSFRS-R) total score. (ALSFRS-R measures overall clinical effect; the score ranges from 0 [no function] to 4 [full function].17)
Tofersen failed to meet the primary efficacy outcome because statistically significant findings were lacking in the faster-progressing population, as measured by joint-rank analysis (difference of 1.2 on the ALSFRS-R score; P = .97). However, trends favoring tofersen were observed across key secondary clinical outcome measures18:
- Change from baseline in CSF SOD1 protein concentration.17 Percent reduction in the total SOD1 protein level was much higher in the tofersen-treated group than in the control group (38% more than controls in the faster-progressing population; 26% more than controls in the slower-progressing population).18
- Change from baseline in neurofilament light-chain concentration in plasma.17,18 Percent reduction in the level of neurofilament light chain was also observed to be higher in the tofersen-treated group than in the control group (67% more than controls in the faster-progressing population and 48% more than controls in the slower-progressing population).18
Because of these encouraging results, VALOR participants were moved to the ongoing open-label extension trial of tofersen (ClinicalTri-als.gov Identifier: NCT03070119), in which both groups were treated with the active agent.
These data suggest that early tofersen treatment might slow decline in faster-progressing patients and stabilize clinical function in slower-progressing patients.18,19 Overall, most adverse events (AEs) in the trial among patients receiving active treatment were of mild or moderate severity, and were largely consistent with either disease progression or lumbar puncture–related complications.18
Because data from VALOR suggested potential benefit from tofersen, the ATLAS trial (ClinicalTrials.gov Identifier: NCT04856982) is investigating the clinical value of presymptomatic treatment and the optimal timing of initiation of therapy.20,21 ATLAS is a phase 3, randomized, placebo-controlled trial that examines the clinical efficacy, safety, and tolerability of tofersen in presymptomatic adult carriers of SOD1 mutation who have an elevated neurofilament light-chain concentration.21 ATLAS will also evaluate the efficacy of tofersen when initiated before, rather than after, ALS manifests clinically. Enrollment is still open for this trial.20,21
Latozinemab, also known as AL001, is a first-in-class monoclonal antibody, administered by IV infusion, that elevates levels of progranulin, a key regulator of the immune activity and lysosomal function in the brain.22,23 Latozinemab limits progranulin endocytosis and degradation by sortilin inhibition.22 Progranulin gene mutations can reduce progranulin expression (by 50 to 70 percent reduction), which may cause neuro-degeneration due to abnormal accumulation of TAR-DNA-binding protein 43 (TDP-43) in the brain cells.22,24 TDP-43 pathology has also been shown to be associated with C9orf72 mutations.23 Although the mechanism is not fully understood, the role of progranulin deficiency in TDP-43 pathology is believed to be associated with neurodegenerative diseases like ALS.11,23,24,43 Previous animal models of chronic neurodegenera-tion have demonstrated how increased progranulin levels can be protective against TDP-43 pathology, increasing neuronal development and survival, thus potentially slowing disease progression.23,24,43 Currently, latozinemab is being investigated in a randomized, double-blind, placebo-controlled, multicenter phase 2 trial (ClinicalTrials.gov Identifier: NCT05053035). Approximately, 45 C90rf72-associated ALS participants (≥ 18 years of age) will receive latozinemab or placebo infusions every 4 weeks (for 24 weeks). Study endpoints include safety, tolerability, PK, PD, as well as plasma, and CSF progranulin levels.25 In previous studies, latozinemab demonstrated encouraging results in frontotemporal dementia (FTD) patients who carry a progranulin mutation. Because FTD was revealed to have significant genetic overlap with ALS, there is disease-modifying potential for latozinemab in ALS patients.23,24
TPN-101 is a nucleoside analog reverse transcriptase inhibitor, administered orally, that was originally developed for human immunodeficiency virus (HIV) treatment. However, due to recent findings suggesting retrotransposon activity contributing to neurodegeneration in TDP-43 mediated diseases, including ALS and FTD, TNP-101 is being repurposed.26 The safety and tolerability of TNP-101 are currently being evaluated in C9orf72-associated ALS and FTD patients (≥ 18 years of age). The study is a randomized, double-blind, placebo-controlled paral-lel-group phase 2a trial (ClinicalTrials.gov Identifier: NCT04993755) The study includes a screening period of 6 weeks, double-blind treatment period of 24 weeks, an open-label treatment period of 24 weeks, and 4 weeks of the post-treatment follow-up visit. Study endpoints include the incidence and severity of spontaneously reported treatment-emergent adverse events (TEAEs) associated with TNP-101 and placebo for a to-tal of 48 weeks.27
ION363 is an investigational ASO, administered by IT injection, that selectively targets one of the FUS mutations (p.P525L), which is responsible for earlier disease onset and rapid ALS progression.28,29 The clinical efficacy of ION363, specifically in clinical function and survival is being assessed in FUS-associated ALS patients (≥ 12 years of age). This randomized phase 3 study (ClinicalTrials.gov Identifier: NCT04768972) includes two parts; part 1 will consist of participants receiving a multi-dose regimen (1 dose every 4-12 weeks) of ION363 or placebo for 61 weeks followed by an open-label extension treatment period in part 2, which will consist of participants receiving ION363 (every 12 weeks) for 85 weeks. The primary endpoint of the study is the change from baseline to day 505 in functional impairment, using ALS Functional Rating Scale-Revised (ALSFRS-R). This measures functional disease severity, specifically in bulbar function, gross motor skills, fine motor skills, and respiratory. The score for all 12 questions can range from 0 (no function) to 4 (full function) with a total possible score of 48.30
Engensis, also known as VM202, is a non-viral gene therapy, administered by intramuscular (IM) injection, that uses a plasmid to deliver the hepatocyte growth factor (HGF) gene to promote HGF protein production. The HGF protein plays a role in angiogenesis, the previous of muscle atrophy, and the promotion of neuronal survival and growth. Based on preclinical studies, increasing HGF protein production has been shown to reduce neurodegeneration, thus potentially halting or slowing ALS progression.31 Currently, the safety of engensis is being evaluated in ALS patients (18-80 years of age) in the REViVALS phase 2a (ClinicalTrials.gov Identifier: NCT04632225)/2b (ClinicalTrial.gov Identifier: NCT05176093).32,33 The ReViVALS trial is a double-blind, randomized, placebo-controlled, multi-center study. The phase 2a study endpoints include the incidence of TEAEs, treatment-emergent serious adverse events (TESAEs), injection site reactions, and clinically significant labor-atory values post-treatment (engensis vs placebo group) for 180 days.33 A phase 2b study will evaluate the long-term safety of engensis for an additional 6 months. Study endpoints include the incidence of AEs, changes from baseline in ALSFRS-R scores to evaluate improvement in muscle function, changes from baseline in quality of life using the ALS patient assessment questionnaire, time to all-cause mortality compared to placebo, etc.32
Spinal muscular atrophy
SMA is a hereditary lower motor-neuron disease caused (in 95% of cases) by deletions or, less commonly, by mutations of the survival motor neuron 1 (SMN1) gene on chromosome 5q13 that encodes the SMN protein.6 Reduction in expression of the SMN protein causes motor neurons to degenerate.36-38 Because of a large inverted duplication in chromosome 5q, two variants of SMN (SMN1 and SMN2) exist on each allele. The paralog gene, SMN2, also produces the SMN protein – although at a lower level (10% to 20% of total SMN protein production) than SMN1 does.
A single nucleotide substitution in SMN2 alters splicing and suppresses transcription of exon 7, resulting in a shortened mRNA strand that yields a truncated SMN protein product.6,37,39 SMA is classified based on age of onset and maximum motor abilities achieved, ranging from the most severe (Type 0) to mildest (Type 4) disease.36,40 Because SMA patients lack functional SMN1 (due to polymorphisms), disease severity is determined by copy numbers of SMN2.6,39
Gene-based therapy for SMA
Three FDA-approved SMN treatments demonstrate clinically meaningful benefit in SMA: SMN2-targeting nusinersen [Spinraza] and risdiplam [Evrysdi], and SMN1-targeting onasemnogene abeparvovec-xioi [Zolgensma]38 Additional approaches to SMA treatment are through SMN-independent therapies, which target muscle and nerve function. Research has strongly suggested that combined SMA therapies, specifically approved SMN-targeted and investigational SMN-independent treatments, such as GYM329 (also known as RO7204239) may be the best strategy to treat all ages, stages, and types of SMA.41 (Table 226-41).
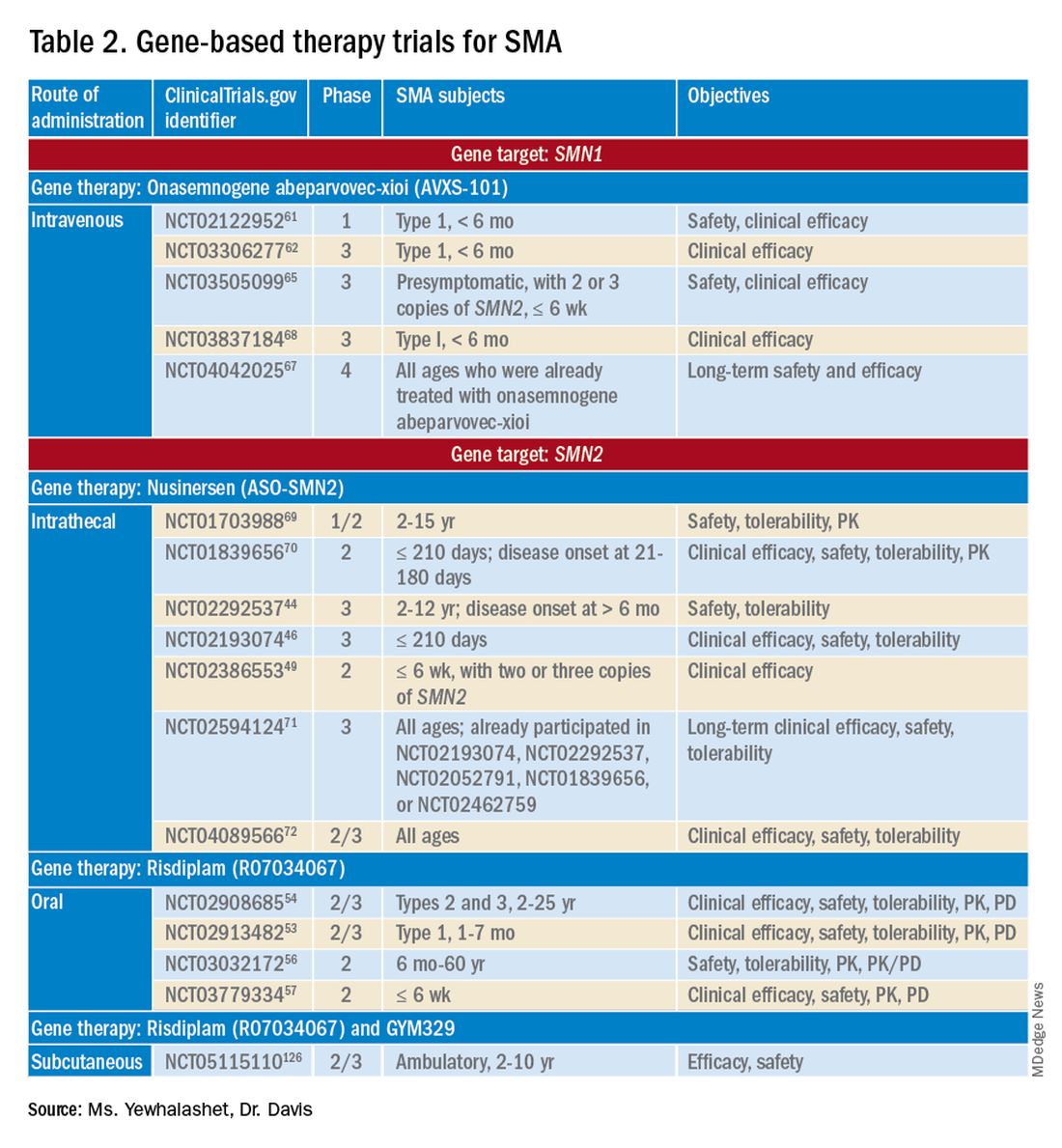
Agents that modulate SMN2. Nusinersen, approved by the FDA in 2016, was the first treatment indicated for all SMA types in pediatric and adult patients.42 The agent is an ASO that targets exon 7 of SMN2, thus stabilizing transcription. Inclusion of exon 7 increases SMN protein production, improving motor function.6,38 Nusinersen is a lifelong treatment that requires IT administration every 4 months because it cannot cross the blood-brain barrier.38,43
Pivotal clinical studies that led to approval of nusinersen include CHERISH (ClinicalTrial.gov Identifier: NCT02292537) and ENDEAR (ClinicalTrial.gov Identifier: NCT02193074) studies.
CHERISH was a phase 3, randomized, double-blind, sham procedure–controlled trial that examined the clinical efficacy and safety of nusinersen in 126 participants with later-onset SMA (2-12 years of age). The primary endpoint was the change from baseline using the Hammersmith Functional Motor Scale Expanded (HFMSE) at 15 months. HFMSE looks at 33 activities to assess improvement in motor function. The study met the primary efficacy outcome, demonstrating statistically significant (P = .0000001) improvement in overall motor function. The nusinersen group showed a 3.9-point increase in the HFMSE score from baseline, which indicates improvement, compared with a 1.0-point decline from baseline in the control group.46,47
ENDEAR was also a randomized, double-blind, sham procedure–controlled phase 3 trial, which investigated the efficacy and safety of nusinersen in 121 participants with early-onset SMA Type 1 (≤ 210 days of age). Coprimary endpoints were:
- Percentage of motor milestones responders, as determined using Section 2 of the Hammersmith Infant Neurological Examination–Part 2.
- Event-free survival (that is, avoidance of combined endpoint of death or permanent ventilation).
ENDEAR met the first primary efficacy outcome, demonstrating statistically significant (P < .0001) improvement in motor milestones (head control, rolling, independent sitting, and standing). By 13 months of age, approximately 51% of nusinersen-treated participants showed improvement, compared with none in the control group.46,47
The second primary endpoint was also met, with a statistically significant (P = .005) 47% decrease in mortality or permanent ventilation use.46-48
The NURTURE (ClinicalTrial.gov Identifier: NCT02386553) study is also investigating the efficacy and safety of nusinersen. An ongoing, open-label, supportive phase 2 trial, NURTURE is evaluating the efficacy and safety of multiple doses of nusinersen in 25 presymptomatic SMA patients (≤ 6 weeks of age). The primary endpoint of this study is time to death or respiratory intervention.49 Interim results demonstrate that 100% of presymptomatic infants are functioning without respiratory intervention after median follow-up of 2.9 years.46-48
Although nusinersen has been shown to be generally safe in clinical studies, development of lumbar puncture–related complications, as well as the need for sedation during IT administration, might affect treatment tolerability in some patients.39
Risdiplam was approved by the FDA in 2020 as the first orally administered small-molecule treatment of SMA (for patients ≤ 2 months of age).52 Risdiplam is a SMN2 splicing modifier, binding to the 5’ splice site of intron 7 and exonic splicing enhancer 2 in exon 7 of SMN2 pre-mRNA. This alternative splicing increases efficiency in SMN2 gene transcription, thus increasing SMN protein production in motor-neuron cells.36 An important advantage of risdiplam is the convenience of oral administration: A large percentage of SMA patients (that is, those with Type 2 disease) have severe scoliosis, which can further complicate therapy or deter patients from using a treatment that is administered through the IT route.40
FDA approval of risdiplam was based on clinical data from two pivotal studies, FIREFISH (ClinicalTrial.gov Identifier: NCT02913482) and SUNFISH (ClinicalTrial.gov Identifier: NCT02908685).53-54
FIREFISH is an open-label, phase 2/3 ongoing trial in infants (1-7 months of age) with SMA Type 1. The study comprises two parts; Part 1 determined the dose of risdiplam used in Part 2, which assessed the efficacy and safety of risdiplam for 24 months. The primary endpoint was the percentage of infants sitting without support for 5 seconds after 12 months of treatment using the gross motor scale of the Bayley Scales of Infant and Toddler Development–Third Edition. A statistically significant (P < .0001) therapeutic benefit was observed in motor milestones. Approximately 29% of infants achieved the motor milestone of independent sitting for 5 seconds, which had not been observed in the natural history of SMA.53-55
SUNFISH is an ongoing randomized, double-blind, placebo-controlled trial of risdiplam in adult and pediatric patients with SMA Types 2 and 3 (2-25 years old). This phase 2/3 study comprises two parts: Part 1 determined the dose (for 12 weeks) to be used for confirmatory Part 2 (for 12 to 24 months). The primary endpoint was the change from baseline on the 32-item Motor Function Measure at 12 months. The study met its primary endpoint, demonstrating statistically significant (P = .0156) improvement in motor function scores, with a 1.36-point increase in the risdiplam group, compared with a 0.19-point decrease in the control group.54,55
Ongoing risdiplam clinical trials also include JEWELFISH (ClinicalTrial.gov Identifier: NCT03032172) and RAINBOW (ClinicalTrial.gov Identifier: NCT03779334).56-57 JEWELFISH is an open-label, phase 2 trial assessing the safety of risdiplam in patients (6 months to 60 years old) who received prior treatment. The study has completed recruitment; results are pending.56 RAINBOW is an ongoing, open-label, single-arm, phase 2 trial, evaluating the clinical efficacy and safety of risdiplam in SMA-presymptomatic newborns (≤ 6 weeks old). The study is open for enrollment.57 Overall, interim results for JEWELFISH and RAINBOW appear promising.
In addition, combined SMA therapies, specifically risdiplam and GYM329 are currently being investigated to address the underlying cause and symptoms of SMA concurrently.58 GYM329, is an investigational anti-myostatin antibody, selectively binding preforms of myostatin - pro-myostatin and latent myostatin, thus improving muscle mass and strength for SMA patients.59 The safety and efficacy of GYM329 in combination with risdiplam is currently being investigated in 180 ambulant participants with SMA (2-10 years of age) in the MANATEE (ClinicalTrial.gov Identifier: NCT05115110) phase 2/3 trial. The MANATEE study is a two-part, seamless, randomized, placebo-controlled, double-blind trial. Part 1 will assess the safety of the combination treatment in approximately 36 participants; participants will receive both GYM329 (every 4 weeks) by subcutaneous (SC) injection into the abdomen and risdiplam (once per day) for 24 weeks followed by a 72-week open-label treatment period. 54,58 The outcome measures include the incidence of AEs, percentage change from baseline in the contractile area of skeletal muscle (in dominant thigh and calf), change from baseline in RHS total score, and incidence of change from baseline in serum concentration (total myostatin, free latent myostatin, and mature myostatin) etc.54 Part 2 will be conducted on 144 participants, specifically assessing the efficacy and safety of the optimal dose of GYM329 selected from Part 1 (combined with risdiplam) for 72 weeks. Once the treatment period is completed in either part, participants can partake in a 2-year open-label extension period.54,58 Other outcome measures include change from baseline in lean muscle mass (assessed by full body dual-energy X- ray absorptiometry (DXA) scan), in time taken to walk/run 10 meters (measured by RHS), in time taken to rise from the floor (measured by RHS), etc.54 Overall, this combination treatment has the potential to further improve SMA patient outcomes and will be further investigated in other patient populations (including non-ambulant patients and a broader age range) in the future.58
An agent that alters SMN1 expression. Onasemnogene abeparvovec-xioi, FDA approved in 2019, was the first gene-replacement therapy indicated for treating SMA in children ≤ 2 years old.60 Treatment utilizes an AAV vector type 9 (AAV9) to deliver a functional copy of SMN1 into target motor-neuron cells, thus increasing SMN protein production and improving motor function. This AAV serotype is ideal because it crosses the blood-brain barrier. Treatment is administered as a one-time IV fusion.38,39,43
FDA approval was based on the STR1VE (ClinicalTrial.gov Identifier: NCT03306277) phase 3 study and START (ClinicalTrial.gov Identifier: NCT02122952) phase 1 study.61,62 START was the first trial to investigate the safety and efficacy of onasemnogene abeparvovec-xioi in SMA Type 1 infants (< 6 months old). Results demonstrated remarkable clinical benefit, including 100% permanent ventilation-free survival and a 92% (11 of 12 patients) rate of improvement in motor function. Improvement in development milestones was also observed: 92% (11 of 12 patients) could sit without support for 5 seconds and 75% (9 of 12) could sit without support for 30 seconds.14,61,63
The efficacy of onasemnogene abeparvovec-xioi seen in STR1VE was consistent with what was observed in START. STRIVE, a phase 3 open-label, single-dose trial, examined treatment efficacy and safety in 22 symptomatic infants (< 6 months old) with SMA Type 1 (one or two SMN2 copies). The primary endpoint was 30 seconds of independent sitting and event-free survival. Patients were followed for as long as 18 months. Treatment showed statistically significant (P < .0001) improvement in motor milestone development and event-free survival, which had not been observed in SMA Type 1 historically. Approximately 59% (13 of 22 patients) could sit independently for 30 seconds at 18 months of age. At 14 months of age, 91% (20 of 22 patients) were alive and achieved independence from ventilatory support.34,35,53
Although many clinical studies suggest that onasemnogene abeparvovec-xioi can slow disease progression, the benefits and risks of long-term effects are still unknown. A 15-year observational study is investigating the long-term therapeutic effects and potential complications of onasemnogene abeparvovec-xioi. Participants in START were invited to enroll in this long-term follow-up study (ClinicalTrial.gov Identifier: NCT04042025).66-67
Duchenne muscular dystrophy
DMD is the most common muscular dystrophy of childhood. With an X-linked pattern of inheritance, DMD is seen mostly in young males (1 in every 3,500 male births).38,39,73 DMD is caused by mutation of the dystrophin encoding gene, or DMD, on the X chromosome. Deletion of one or more exons of DMD prevents production of the dystrophin protein, which leads to muscle degeneration.38,39,43 Common DMD deletion hotspots are exon 51 (20% of cases), exon 53 (13% of cases), exon 44 (11% of cases), and exon 45 (12% of cases).74 Nonsense mutations, which account for another 10% of DMD cases, occur when premature termination codons are found in the DMD gene. Those mutations yield truncated dystrophin protein products.39,66
Therapy for DMD
There are many therapeutic options for DMD, including deflazacort (Emflaza), FDA approved in 2017, which has been shown to reduce inflammation and immune system activity in DMD patients (≥ 5 years old). Deflazacort is a corticosteroid prodrug; its active metabolite acts on the glucocorticoid receptor to exert anti-inflammatory and immunosuppressive effects. Studies have shown that muscle strength scores over 6-12 months and average time to loss of ambulation numerically favored deflazacort over placebo.74,75
Gene-based therapy for DMD
Mutation-specific therapeutic approaches, such as exon skipping and nonsense suppression, have shown promise for the treatment of DMD (Table 358-79):
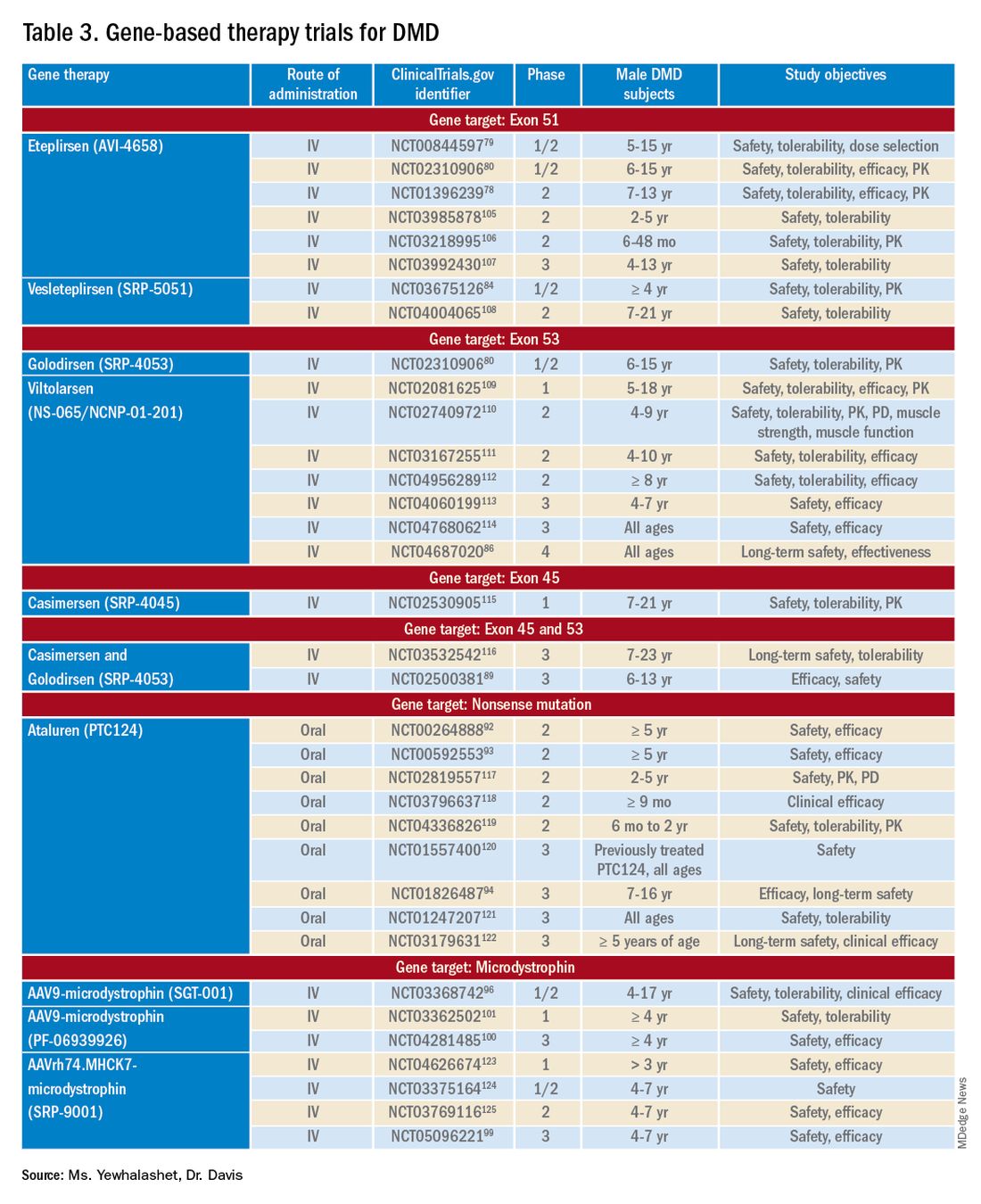
- ASO-mediated exon skipping allows one or more exons to be omitted from the mutated DMD mRNA.74,75 Effective FDA-approved ASOs include golodirsen [Vyondys 53], viltolarsen [Viltepso], and casimersen [Amondys 45].74
- An example of therapeutic suppression of nonsense mutations is ataluren [Translarna], an investigational agent that can promote premature termination codon read-through in DMD patients.66
Another potential treatment approach is through the use of AAV gene transfer to treat DMD. However, because DMD is too large for the AAV vector (packaging size, 5.0 kb), microdystrophin genes (3.5-4 kb, are used as an alternative to fit into a single AAV vector.39,76
Exon skipping targeting exon 51. Eteplirsen, approved in 2016, is indicated for the treatment of DMD patients with the confirmed DMD gene mutation that is amenable to exon 51 skipping. Eteplirsen binds to exon 51 of dystrophin pre-mRNA, causing it to be skipped, thus, restoring the reading frame in patients with DMD gene mutation amenable to exon 51 skipping. This exclusion promotes dystrophin production. Though the dystrophin protein is still functional, it is shortened.38,77 Treatment is administered IV, once a week (over 35-60 minutes). Eteplirsen’s accelerated approval was based on 3 clinical studies (ClinicalTrial.gov Identifier: NCT01396239, NCT01540409, and NCT00844597.) 78-81 The data demonstrated an increased expression of dystrophin in skeletal muscles in some DMD patients treated with eteplirsen. Though the clinical benefit of eteplirsen (including improved motor function) was not established, it was concluded by the FDA that the data were reasonably likely to predict clinical benefit. Continued approval for this indication may depend on the verification of a clinical benefit in confirmatory trials. Ongoing clinical trials include (ClinicalTrial.gov Identifier: NCT03992430 (MIS51ON), NCT03218995, and NCT03218995).77,81,82
Vesleteplirsen, is an investigational agent that is designed for DMD patients who are amendable to exon 51 skip-ping. The mechanism of action of vesleteplirsen appears to be similar to that of eteplirsen.83 The ongoing MOMENTUM (ClinicalTrial.gov Identifier: NCT04004065) phase 2 trial is assessing the safety and tolerability of vesleteplirsen at multiple-ascending dose levels (administered via IV infusion) in 60 participants (7-21 years of age). The study consists of two parts; participants receive escalating dose levels of vesleteplirsen (every 4 weeks) for 72 weeks during part A and participants receive the selected doses from part A (every 4 weeks) for 2 years during part B. Study endpoints include the number of AEs (up to 75 weeks) and the change from baseline to week 28 in dystrophin protein level. 84 Serious AEs of reversible hypomagnesemia were observed in part B, and as a result, the study protocol was amended to include magnesium supplementation and monitoring of magnesium levels.83
Exon skipping targeting exon 53. Golodirsen, FDA approved in 2019, is indicated for the treatment of DMD in patients who have a confirmed DMD mutation that is amenable to exon 53 skipping. The mechanism of action is similar to eteplirsen, however, golodirsen is designed to bind to exon 53.38,39 Treatment is administered by IV infusion over 35-60 minutes.
Approval of golodirsen was based primarily on a two-part, phase 1/2 clinical trial (ClinicalTrial.gov Identifier: NCT02310906). Part 1 was a randomized, placebo-controlled, dose-titration study that assessed multiple-dose efficacy in 12 DMD male patients, 6 to 15 years old, with deletions that were amenable to exon 53 skipping.
Part 2 was an open-label trial in 12 DMD patients from Part 1 of the trial plus 13 newly enrolled male DMD patients who were also amenable to exon 53 skipping and who had not already received treatment. Primary endpoints were change from baseline in total distance walked during the 6-minute walk test at Week 144 and dystrophin protein levels (measured by western blot testing) at Week 48. A statistically significant increase in the mean dystrophin level was observed, from a baseline 0.10% mean dystrophin level to a 1.02% mean dystrophin level after 48 weeks of treatment (P < .001). Common reported adverse events associated with golodirsen were headache, fever, abdominal pain, rash, and dermatitis. Renal toxicity was observed in preclinical studies of golodirsen but not in clinical studies.80,85
Viltolarsen, approved in 2020, is also indicated for the treatment of DMD in patients with deletions amenable to exon 53 skipping. The mechanism of action and administration (IV infusion over 60 minutes) are similar to that of golodirsen.
Approval of viltolarsen was based on two phase 2 clinical trials (ClinicalTrial.gov Identifier: NCT02740972 and NCT03167255) in a total of 32 patients. NCT02740972 was a randomized, double-blind, placebo-controlled, dose-finding study that evaluated the clinical efficacy of viltolarsen in 16 male DMD patients (4-9 years old) for 24 weeks.
NCT03167255 was an open-label study that evaluated the safety and tolerability of viltolarsen in DMD male patients (5-18 years old) for 192 weeks. The efficacy endpoint was the change in dystrophin production from baseline after 24 weeks of treatment. A statistically significant increase in the mean dystrophin level was observed, from a 0.6% mean dystrophin level at baseline to a 5.9% mean dystrophin level at Week 25 (P = .01). The most common adverse events observed were upper respiratory tract infection, cough, fever, and injection-site reaction.86-87
Exon skipping targeting exon 45. Casimersen was approved in 2021 for the treatment of DMD in patients with deletions amenable to exon 45 skipping.88 Treatment is administered by IV infusion over 30-60 minutes. Approval was based on an increase in dystrophin production in skeletal muscle in treated patients. Clinical benefit was reported in interim results from the ESSENCE (ClinicalTrial.gov Identifier: NCT02500381) study, an ongoing double-blind, placebo-controlled phase 3 trial that is evaluating the efficacy of casimersen, compared with placebo, in male participants (6-13 years old) for 48 weeks. Efficacy is based on the change from baseline dystrophin intensity level, determined by immunohistochemistry, at Week 48.
Interim results from ESSENCE show a statistically significant increase in dystrophin production in the casimersen group, from a 0.9% mean dystrophin level at baseline to a 1.7% mean dystrophin level at Week 48 (P = .004); in the control group, a 0.54% mean dystrophin level at baseline increased to a 0.76% mean dystrophin level at Week 48 (P = .09). Common adverse events have included respiratory tract infection, headache, arthralgia, fever, and oropharyngeal pain. Renal toxicity was observed in preclinical data but not in clinical studies.60,84
Targeting nonsense mutations. Ataluren is an investigational, orally administered nonsense mutation suppression therapy (through the read-through of stop codons).37 Early clinical evidence supporting the use of ataluren in DMD was seen in an open-label, dose-ranging, phase 2a study (ClinicalTrial.gov Identifier: NCT00264888) in male DMD patients (≥ 5 years old) caused by nonsense mutation. The study demonstrated a modest (61% ) increase in dystrophin expression in 23 of 38 patients after 28 days of treatment.37,91,92
However, a phase 2b randomized, double-blind, placebo-controlled trial (ClinicalTrial.gov Identifier: NCT00592553) and a subsequent confirmatory ACT DMD phase 3 study (ClinicalTrial.gov Identifier: NCT01826487) did not meet their primary endpoint of improvement in ambulation after 48 weeks as measured by the 6-minute walk test.37,93,94 In ACT DMD, approximately 74% of the ataluren group did not experience disease progression, compared with 56% of the control group (P = 0386), measured by a change in the 6-minute walk test, which assessed ambulatory decline.37,95
Based on limited data showing that ataluren is effective and well tolerated, the European Medicines Agency has given conditional approval for clinical use of the drug in Europe. However, ataluren was rejected by the FDA as a candidate therapy for DMD in the United States.22 Late-stage clinical studies of ataluren are ongoing in the United States.
AAV gene transfer with microdystrophin. Limitations on traditional gene-replacement therapy prompted exploration of gene-editing strategies for treating DMD, including using AAV-based vectors to transfer microdystrophin, an engineered version of DMD, into target muscles.43 The microdystrophin gene is designed to produce a functional, truncated form of dystrophin, thus improving muscular function.
There are 3 ongoing investigational microdystrophin gene therapies that are in clinical development (ClinicalTrial.gov Identifier: NCT03368742 (IGNITE DMD), NCT04281485 (CIFFREO), and NCT05096221 (EMBARK)).38,82
IGNITE DMD is a phase 1/2 randomized, controlled, single-ascending dose trial evaluating the safety and efficacy of a SGT-001, single IV infusion of AAV9 vector containing a microdystrophin construct in DMD patients (4-17 years old) for 12 months. At the conclusion of the trial, treatment and control groups will be followed for 5 years. The primary efficacy endpoint is the change from baseline in microdystrophin protein production in muscle-biopsy material, using western blot testing.96 Long-term interim data on biopsy findings from three patients demonstrated clinical evidence of durable microdystrophin protein expression after 2 years of treatment.96,97
The CIFFREO trial will assess the safety and efficacy of the PF-06939926 microdystrophin gene therapy, an investigational AAV9 containing microdystrophin, in approximately 99 ambulatory DMD patients (4-7 years of age). The study is a randomized, double-blind, placebo-controlled, multicenter phase 3 trial. The primary efficacy end-point is the change from baseline in the North Star Ambulatory Assessment (NSAA), which measures gross motor function. This will be assessed at 52 weeks; all study participants will be followed for a total of 5 years post-treatment.98,99,100 Due to unexpected patient death (in a non-ambulatory cohort) in the phase 1b (in a non-ambulatory cohort) in the phase 1b (ClinicalTrial.gov Identifier: (NCT03362502) trial, microdystrophin gene therapy was immediately placed on clinical hold.101,102 The amended study protocol required that all participants undergo one week of in-hospital observation after receiving treatment.102
The EMBARK study is a global, randomized, double-blind, placebo-controlled, phase 3 trial that is evaluating the safety and efficacy of SRP-9001, which is a rAAVrh74.MHCK7.microdystrophin gene therapy. The AAV vector (rAAVrh74) contains the microdystrophin construct, driven by the skeletal and cardiac muscle–specific promoter, MHCK7.98,99 In the EMBARK study, approximately 120 participants with DMD (4-7 years of age) will be enrolled. The primary efficacy endpoint includes the change from baseline to week 52 in the NSAA total score.99 Based on SRP-9001, data demonstrating consistent statistically significant functional improvements in NSAA total scores and timed function tests (after one-year post- treatment) in DMD patients from previous studies and an integrated analysis from multiple studies (ClinicalTrial.gov Identifier: NCT03375164, NCT03769116, and NCT04626674), the ongoing EMBARK has great promise.103,104
Challenges ahead, but advancements realized
Novel gene-based therapies show significant potential for transforming the treatment of NMDs. The complex pathologies of NMDs have been a huge challenge to disease management in an area once considered unremediable by gene-based therapy. However, advancements in precision medicine – specifically, gene-delivery systems (for example, AAV9 and AAVrh74 vectors) combined with gene modification strategies (ASOs and AAV-mediated silencing) – have the potential to, first, revolutionize standards of care for sporadic and inherited NMDs and, second, significantly reduce disease burden.6
What will be determined to be the “best” therapeutic approach will, likely, vary from NMD to NMD; further investigation is required to determine which agents offer optimal clinical efficacy and safety profiles.43 Furthermore, the key to therapeutic success will continue to be early detection and diagnosis – first, by better understanding disease pathology and drug targets and, second, by validation of reliable biomarkers that are predictive of therapeutic benefit.4,5
To sum up, development challenges remain, but therapeutic approaches to ALS, SMA, and DMD that utilize novel gene-delivery and gene-manipulation tools show great promise.
Ms. Yewhalashet is a student in the masters of business and science program, with a concentration in healthcare economics, at Keck Graduate Institute Henry E. Riggs School of Applied Life Sciences, Claremont, Calif. Dr. Davis is professor of practice in clinical and regulatory affairs, Keck Graduate Institute Henry E. Riggs School of Applied Life Sciences.
References
1. Aitken M et al. Understanding neuromuscular disease care. IQVIA [Internet]. Oct 30, 2018. Accessed Mar 1, 2022. https://www.iqvia.com/insights/the-iqvia-institute/reports/understanding-neuromuscular-disease-care.
2. National Institute of Neurological Disorders and Stroke. Neurological diagnostic tests and procedures fact sheet. Updated Nov 15, 2021. Ac-cessed Mar 1, 2022. http://www.ninds.nih.gov/Disorders/Patient-Caregiver-Education/Fact-Sheets/Neurological-Diagnostic-Tests-and-Procedures-Fact.
3. Deenen JCW et al. The epidemiology of neuromuscular disorders: A comprehensive overview of the literature. J Neuromuscul Dis. 2015;2(1):73-85.
4. Cavazzoni P. The path forward: Advancing treatments and cures for neurodegenerative diseases. U.S. Food and Drug Administration. Jul 29, 2021. Accessed Mar 1, 2022. http://www.fda.gov/news-events/congressional-testimony/path-forward-advancing-treatments-and-cures-neurodegenerative-diseases-07292021.
5. Martier R, Konstantinova P. Gene therapy for neurodegenerative diseases: Slowing down the ticking clock. Front Neurosci. 2020 Sep 18;14:580179. doi: 10.3389/fnins.2020.580179.
6. Sun J, Roy S. Gene-based therapies for neurodegenerative diseases. Nat Neurosci. 2021 Mar;24(3):297-311. doi:10.1038/s41593-020-00778-1.
7. Amado DA, Davidson BL. Gene therapy for ALS: A review. Mol Ther. 2021 Dec 1;29(12):3345-58. doi:10.1016/j.ymthe.2021.04.008.
8. Yun Y, Ha Y. CRISPR/Cas9-mediated gene correction to understand ALS. Int J Mol Sci. 2020;21(11):3801. doi:10.3390/ijms21113801.
9. National Institute of Neurological Disorders and Stroke. Amyotrophic lateral sclerosis (ALS) fact sheet. Updated Nov 15, 2021. Accessed Mar 1, 2022. http://www.ninds.nih.gov/Disorders/Patient-Caregiver-Education/Fact-Sheets/Amyotrophic-Lateral-Sclerosis-ALS-Fact-Sheet.
10. Cappella M et al. Gene therapy for ALS – A perspective. Int J Mol Sci. 2019;20(18):4388. doi:10.3390/ijms20184388.
11. Abramzon YA, Fratta P, Traynor BJ, Chia R. The Overlapping Genetics of Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Front Neurosci. 2020;14. Accessed August 18, 2022. https://www.frontiersin.org/articles/10.3389/fnins.2020.00042
12. Giannini M, Bayona-Feliu A, Sproviero D, Barroso SI, Cereda C, Aguilera A. TDP-43 mutations link Amyotrophic Lateral Sclerosis with R-loop homeostasis and R loop-mediated DNA damage. PLOS Genet. 2020;16(12):e1009260. doi:10.1371/journal.pgen.1009260
13. FDA-approved drugs for treating ALS. The ALS Association [Internet]. Accessed Mar 1, 2022. http://www.als.org/navigating-als/living-with-als/fda-approved-drugs.
14. Jensen TL et al. Current and future prospects for gene therapy for rare genetic diseases affecting the brain and spinal cord. Front Mol Neurosci. 2021 Oct 6;14:695937. doi:10.3389/fnmol.2021.695937.
15. ALS Gene Targeted Therapies. The ALS Association. Accessed August 22, 2022. https://www.als.org/understanding-als/who-gets-als/genetic-testing/als-gene-targeted-therapies
16. Tofersen for ALS clears phase 1/2 trial, now in phase 3. Advances in Motion. Massachusetts General Hospital [Internet]. Sep 30, 2020. Accessed Mar 1, 2022. https://advances.massgeneral.org/neuro/journal.aspx?id=1699.17. Biogen. A study to evaluate the efficacy, safety, tol-erability, pharmacokinetics, and pharmacodynamics of BIIB067 administered to adult subjects with amyotrophic lateral sclerosis and confirmed superoxide dismutase 1 mutation. ClinicalTrials.gov Identifier: NCT02623699. Updated Jul 25, 2021. Accessed Feb 17, 2022. https://clinicaltrials.gov/ct2/show/NCT02623699.
18. Biogen. Biogen announces topline results from the tofersen phase 3 study and its open-label Extension in SOD1-ALS. Press release. Oct 17, 2021. Accessed Mar 1, 2022. https://investors.biogen.com/news-releases/news-release-details/biogen-announces-topline-results-tofersen-phase-3-study-and-its.
19. Biogen. An extension study to assess the long-term safety, tolerability, pharmacokinetics, and effect on disease progression of BIIB067 ad-ministered to previously treated adults with amyotrophic lateral sclerosis caused by superoxide dismutase 1 mutation. ClinicalTrials.gov Identi-fier: NCT03070119. Updated Sep 10, 2021. Accessed Feb 17, 2022. https://clinicaltrials.gov/ct2/show/NCT03070119.
20. MS MW. #AANAM – ATLAS Trial to Assess Tofersen in Presymptomatic SOD1 ALS. Accessed February 19, 2022. https://alsnewstoday.com/news-posts/2021/04/23/aanam-atlas-clinical-trial- tofersen-presymptomatic-sod1-als-patients/
21.Biogen. A phase 3 randomized, placebo-controlled trial with a longitudinal natural history run-in and open-label extension to evaluate BIIB067 initiated in clinically presymptomatic adults with a confirmed superoxide dismutase 1 mutation. ClinicalTrials.gov Identifier: NCT04856982. Updated Feb 18, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT04856982.
22. Latozinemab | ALZFORUM. Accessed August 19, 2022. https://www.alzforum.org/therapeutics/latozinemab
23. Alector Presents AL001 (latozinemab) Data from the FTD-C9orf72 Cohort of the INFRONT-2 Phase 2 Clinical Trial | Alector. Accessed August 18, 2022. https://investors.alector.com/news- releas-es/news-release-details/alector-presents-al001-latozinemab-data-ftd-c9orf72-cohort/
24. Alector Announces First Participant Dosed in Phase 2 Study Evaluating AL001 in Amyotrophic Lateral Sclerosis (ALS) | Alector. Accessed August 18, 2022. https://investors.alector.com/news- releases/news-release-details/lector-announces-first-participant-dosed-phase-2-study-0/ 25. A Phase 2 Study to Evaluate AL001 in C9orf72-Associated ALS - Full Text View - ClinicalTrials.gov. Accessed August 19, 2022. https://clinicaltrials.gov/ct2/show/NCT05053035
26.TPN-101 | ALZFORUM. Accessed August 19, 2022. https://www.alzforum.org/therapeutics/tpn- 101
27. Transposon Therapeutics, Inc. A Phase 2a Study of TPN-101 in Patients With Amyotrophic Lateral Sclerosis (ALS) and/or Frontotemporal Dementia (FTD) Associated With Hexanucleotide Repeat Expansion in the C9orf72 Gene (C9ORF72 ALS/FTD). clinicaltrials.gov; 2022. Ac-cessed August 17, 2022. https://clinicaltrials.gov/ct2/show/NCT04993755
28. Kerk SY, Bai Y, Smith J, et al. Homozygous ALS-linked FUS P525L mutations cell- autonomously perturb transcriptome profile and chem-oreceptor signaling in human iPSC microglia. Stem Cell Rep. 2022;17(3):678-692. doi:10.1016/j.stemcr.2022.01.004
29. ION363 | ALZFORUM. Accessed August 19, 2022. https://www.alzforum.org/therapeutics/ion363 30. Ionis Pharmaceuticals, Inc. A Phase 1-3 Study to Evaluate the Efficacy, Safety, Pharmacokinetics and Pharmacodynamics of Intrathecally Administered ION363 in Amyo-trophic Lateral Sclerosis Patients With Fused in Sarcoma Mutations (FUS-ALS). clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT04768972
31. PhD LF. Engensis (VM202) - ALS News Today. Accessed August 19, 2022. https://alsnewstoday.com/vm202/
32. Helixmith Co., Ltd. A 6-Month Extension Study Following Protocol VMALS-002-2 (A Phase 2a, Double-Blind, Randomized, Place-bo-Controlled, Multicenter Study to Assess the Safety of Engensis in Participants With Amyotrophic Lateral Sclerosis). clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT05176093 33. Safety of Engensis in Participants With Amyotrophic Lateral Sclerosis - Full Text View - ClinicalTrials.gov. Accessed August 19, 2022. https://clinicaltrials.gov/ct2/show/NCT04632225
34. Biogen. A phase 1, safety, tolerability, and distribution study of a microdose of radiolabeled BIIB067 co-administered with BIIB067 to healthy adults. ClinicalTrials.gov Identifier: NCT03764488. Updated Jul 19, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03764488.
35. Ionis Pharmaceuticals Inc. A phase 1, double-blind, placebo-controlled, dose-escalation study of the safety, tolerability, and pharmacokinet-ics of ISIS 333611 administered intrathecally to patients with familial amyotrophic lateral sclerosis due to superoxide dismutase 1 gene muta-tions. ClinicalTrials.gov Identifier: NCT01041222. Updated Apr 13, 2012. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT01041222.
36. Messina S, Sframeli M. New treatments in spinal muscular atrophy: Positive results and new challenges. J Clin Med. 2020;9(7):2222. doi:10.3390/jcm9072222.
37. Scoto M et al. Genetic therapies for inherited neuromuscular disorders. Lancet Child Adolesc Health. 2018 Aug;2(8):600-9. doi:10.1016/S2352-4642(18)30140-8.
38. Abreu NJ, Waldrop MA. Overview of gene therapy in spinal muscular atrophy and Duchenne muscular dystrophy. Pediatr Pulmonol. 2021 Apr;56(4):710-20. doi:10.1002/ppul.25055.
39. Brandsema J, Cappa R. Genetically targeted therapies for inherited neuromuscular disorders. Practical Neurology [Internet]. Jul/Aug 2021:69-73. Accessed Mar 1, 2022. https://practicalneurology.com/articles/2021-july-aug/genetically-targeted-therapies-for-inherited-neuromuscular-disorders/pdf.
40. Ojala KS et al. In search of a cure: The development of therapeutics to alter the progression of spinal muscular atrophy. Brain Sci. 2021;11(2):194. doi:10.3390/brainsci11020194.
41. McCall S. Cure SMA Releases Updated Drug Pipeline. Cure SMA. Published December 13, 2021. Accessed August 21, 2022. https://www.curesma.org/cure-sma-releases-updated-drug-pipeline- 2021/ 42. FDA approves first drug for spinal muscular atrophy. U.S. Food and Drug Administration. News release. Dec 23, 2016. Accessed Mar 1, 2022. http://www.fda.gov/news-events/press-announcements/fda-approves-first-drug-spinal-muscular-atrophy.43. Kirschner J. Postnatal gene therapy for neuromuscular diseases – Opportunities and limitations. J Perinat Med. 2021 Sep;49(8):1011-5. doi:10.1515/jpm-2021-0435.
43. Terryn J, Verfaillie CM, Van Damme P. Tweaking Progranulin Expression: Therapeutic Avenues and Opportunities. Front Mol Neurosci. 2021;14. Accessed September 4, 2022. https://www.frontiersin.org/articles/10.3389/fnmol.2021.71303144.
44. Biogen. A phase 3, randomized, double-blind, sham-procedure controlled study to assess the clinical efficacy and safety of ISIS 396443 administered intrathecally in patients with later-onset spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT02292537. Updated Feb 17, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/study/NCT02292537.
45. Why Spinraza/later-onset studies. SPINRAZA® (nusinersen) [Internet]. Accessed Mar 1, 2022. www.spinraza.com/en_us/home/why-spinraza/later-onset-studies.html#scroll-tabs.
46. Biogen. A Phase 3, Randomized, Double-Blind, Sham-Procedure Controlled Study to Assess the Clinical Efficacy and Safety of ISIS 396443 Administered Intrathecally in Patients With Infantile- Onset Spinal Muscular Atrophy. clinicaltrials.gov; 2021. Accessed February 10, 2022. https://clinicaltrials.gov/ct2/show/results/NCT02193074
47. Early-onset SMA (Type 1) | SPINRAZA® (nusinersen). Accessed Mar 1, 2022. https://www.spinraza-hcp.com/en_us/home/why-spinraza/about-spinraza.html.
48. Finkel RS et al; ENDEAR Study Group. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N Engl J Med. 2017;377(18):1723-32. doi: 10.1056/NEJMoa1702752.
49. Biogen. An open-label study to assess the efficacy, safety, tolerability, and pharmacokinetics of multiple doses of ISIS 396443 delivered intrathecally to subjects with genetically diagnosed and presymptomatic spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT02386553. Updated Nov 18, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02386553.
50. De Vivo DC et al; NURTURE Study Group. Nusinersen initiated in infants during the presymptomatic stage of spinal muscular atrophy: In-terim efficacy and safety results from the phase 2 NURTURE study. Neuromuscul Disord. 2019 Nov;29(11):842-56. doi:10.1016/j.nmd.2019.09.007.
51. Why Spinraza/presymptomatic study. SPINRAZA® (nusinersen) [Internet]. Accessed Feb 22, 2022. www.spinraza.com/en_us/home/why-spinraza/presymptomatic-study.html#scroll-tabs.
52. FDA approves oral treatment for spinal muscular atrophy. U.S. Food and Drug Administration. News release. Aug 7, 2020. Accessed Mar 1, 2022. http://www.fda.gov/news-events/press-announcements/fda-approves-oral-treatment-spinal-muscular-atrophy.
53. Hoffmann-La Roche. A two-part seamless, open-label, multicenter study to investigate the safety, tolerability, pharmacokinetics, pharmaco-dynamics and efficacy of risdiplam (RO7034067) in infants with type 1 spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT02913482. Updated Jan 21, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02913482.
54. Hoffmann-La Roche. A two-part seamless, multi-center randomized, placebo-controlled, double-blind study to investigate the safety, tolera-bility, pharmacokinetics, pharmacodynamics and efficacy of risdiplam (RO7034067) in type 2 and 3 spinal muscular atrophy patients. Clinical-Trials.gov Identifier: NCT02908685. Updated Dec 28, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02908685.
55. Genentech. Genentech’s risdiplam shows significant improvement in survival and motor milestones in infants with type 1 spinal muscular atrophy (SMA). Press release. Apr 27, 2020. Accessed Mar 1, 2022. http://www.gene.com/media/press-releases/14847/2020-04-27/genentechs-risdiplam-shows-significant-i
56. Hoffmann-La Roche. An open-label study to investigate the safety, tolerability, and pharmacokinetics/pharmacodynamics of risdiplam (RO7034067) in adult and pediatric patients with spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT03032172. Updated Jan 27, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03032172.
57. Hoffmann-La Roche. An open-label study of risdiplam in infants with genetically diagnosed and presymptomatic spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT03779334. Updated Jan 27, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03779334.
58. McCall S. Update on Genentech/Roche Initiation of MANATEE Clinical Study. Cure SMA. Published October 20, 2021. Accessed August 20, 2022. https://www.curesma.org/update-on- genentech-roche-initiation-of-manatee-clinical-study/
59. Abati E, Manini A, Comi GP, Corti S. Inhibition of myostatin and related signaling pathways for the treatment of muscle atrophy in motor neuron diseases. Cell Mol Life Sci. 2022;79(7):374. doi:10.1007/s00018-022-04408-w
60. FDA approves innovative gene therapy to treat pediatric patients with spinal muscular atrophy, a rare disease and leading genetic cause of infant mortality. U.S. Food and Drug Administration. News release. May 24, 2019. Accessed Mar 1, 2022. http://www.fda.gov/news-events/press-announcements/fda-approves-innovative-gene-therapy-treat-pediatric-patients-spinal-muscular-atrophy-rare-disease.
61. Novartis Gene Therapies. Phase I gene transfer clinical trial for spinal muscular atrophy type 1 delivering AVXS-101. ClinicalTrials.gov Identifier: NCT02122952. Updated Jun 14, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02122952.
62. Novartis Gene Therapies. Phase 3, open-label, single-arm, single-dose gene replacement therapy clinical trial for patients with spinal mus-cular atrophy type 1 with one or two SMN2 copies delivering AVXS-101 by intravenous infusion. ClinicalTrials.gov Identifier: NCT03306277. Updated Jun 14, 2021. Accessed Feb 21, 2022. https://clinicaltrials.gov/ct2/show/NCT03306277.
63. Mendell JR et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N Engl J Med. 2017;377(18):1713-22. doi:10.1056/NEJMoa1706198.
64. Symptomatic study results. ZOLGENSMA [Internet]. Updated Nov 2021. Accessed Mar 1, 2022. Error! Hyperlink reference not valid..
65. Novartis Gene Therapies. A global study of a single, one-time dose of AVXS-101 delivered to infants with genetically diagnosed and pre-symptomatic spinal muscular atrophy with multiple copies of SMN2. ClinicalTrials.gov Identifier: NCT03505099. Updated Jan 1, 2022. Ac-cessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03505099.
66. Chiu W et al. Current genetics and potential gene-targeting therapeutics for neuromuscular diseases. Int J Mol Sci. 2020 Dec;21(24):9589. doi:10.3390/ijms21249589.
67. Novartis Gene Therapies. A long-term follow-up study of patients in the clinical trials for spinal muscular atrophy receiving AVXS-101. Clini-calTrials.gov Identifier: NCT04042025. Updated Jun 9, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT04042025.
68. Novartis Gene Therapies. Phase 3, open-label, single-arm, single-dose gene replacement therapy clinical trial for patients with spinal mus-cular atrophy type 1 with one or two SMN2 copies delivering AVXS-101 by intravenous infusion. ClinicalTrials.gov Identifier: NCT0383718. Up-dated Jan 11, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03837184.
69. Biogen. An open-label, dose escalation study to assess the safety, tolerability and dose-range finding of multiple doses of ISIS 396443 de-livered intrathecally to patients with spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT01703988. Updated Apr 13, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT01703988.
70. Biogen. A study to assess the efficacy, safety, tolerability, and pharmacokinetics of multiple doses of ISIS 396443 delivered intrathecally to patients with infantile-onset spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT01839656. Updated Feb 17, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT01839656.
71. Biogen. An open-label extension study for patients with spinal muscular atrophy who previously participated in investigational studies of ISIS 396443. ClinicalTrials.gov Identifier: NCT02594124. Updated Nov 15, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02594124.
72. Biogen. Escalating dose and randomized, controlled study of nusinersen (BIIB058) in participants with spinal muscular atrophy. ClinicalTri-als.gov Identifier: NCT04089566. Updated Feb 24, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT04089566.
73. National Center for Advancing Translational Sciences. Duchenne muscular dystrophy. Genetic and Rare Diseases Information Center. Up-dated Nov 2, 2020. Accessed Mar 1, 2022. https://rarediseases.info.nih.gov/diseases/6291/duchenne-muscular-dystrophy.
74. Matsuo M. Antisense oligonucleotide-mediated exon-skipping therapies: Precision medicine spreading from Duchenne muscular dystrophy. JMA J. 2021 Jul 15;4(3):232-40. doi:10.31662/jmaj.2021-0019.
75. FDA approves drug to treat Duchenne muscular dystrophy. U.S. Food and Drug Administration. News release. Feb 9, 2017. Accessed Mar 1, 2022. http://www.fda.gov/news-events/press-announcements/fda-approves-drug-treat-duchenne-muscular-dystrophy.74.
76. Duan D. Dystrophin gene replacement and gene repair therapy for Duchenne muscular dystrophy in 2016: An interview. Hum Gene Ther Clin Dev. 2016 Mar;27(1):9-18. doi:10.1089/humc.2016.001.
77. EXONDYS 51®. Parent Project Muscular Dystrophy. Accessed August 21, 2022. https://www.parentprojectmd.org/drug-development-pipeline/exondys-51/
78. Sarepta Therapeutics, Inc. A Randomized, Double-Blind, Placebo-Controlled, Multiple Dose Efficacy, Safety, Tolerability and Pharmacoki-netics Study of AVI-4658(Eteplirsen),in the Treatment of Ambulant Subjects With Duchenne Muscular Dystrophy. clinicaltrials.gov; 2020. Ac-cessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT01396239
79. Sarepta Therapeutics, Inc. Clinical Study to Assess the Safety Fo AVI-4658 in Subjects With Duchenne Muscular Dystrophy Due to a Frame-Shift Mutation Amenable to Correction by Skipping Exon 51. clinicaltrials.gov; 2015. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/study/NCT00844597
80. Sarepta Therapeutics, Inc. A 2-part, randomized, double-blind, placebo-controlled, dose-titration, safety, tolerability, and pharmacokinetics study (Part 1) followed by an open-label efficacy and safety evaluation (Part 2) of SRP-4053 in patients with Duchenne muscular dystrophy amenable to exon 53 skipping. ClinicalTrials.gov Identifier: NCT02310906. Updated Oct 19, 2020. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/results/NCT02310906.
81. Commissioner O of the. FDA grants accelerated approval to first drug for Duchenne muscular dystrophy. FDA. Published March 24, 2020. Accessed August 21, 2022. hDuchenne Muscular Dystrophy Amenable to Exon 51-Skipping Treatment. clinicaltrials.gov; 2022. Accessed Au-gust 18, 2022. https://clinicaltrials.gov/ct2/show/NCT04004065
109. National Center of Neurology and Psychiatry, Japan. Exploratory study of NS-065/NCNP-01 in Duchenne muscular dystrophy. ClinicalTri-als.gov Identifier: NCT02081625; Updated Feb 26, 2020. Accessed Mar 2, 2022. https://clinicaltrialsttps://www.fda.gov/news-events/press-announcements/fda-grants-accelerated-approval-first-drug-duchenne-muscular- dys-trophy
82. Duchenne Drug Development Pipeline. Parent Project Muscular Dystrophy. Accessed August 21, 2022. https://www.parentprojectmd.org/duchenne-drug-development-pipeline/
83. Sarepta Therapeutics Provides Update on SRP-5051 for the Treatment of Duchenne Muscular Dystrophy | Sarepta Therapeutics, Inc. Ac-cessed August 22, 2022. https://investorrelations.sarepta.com/news-releases/news-release-details/sarepta-therapeutics- pro-vides-update-srp-5051-treatment-duchenne
84. Sarepta Therapeutics, Inc. An Open-Label Extension Study for Patients With Duchenne Muscular Dystrophy Who Participated in Studies of SRP-5051. clinicaltrials.gov; 2021. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT03675126
85. VYONDYS 53. Prescribing information. Sarepta Therapeutics Inc.; 2019. Accessed Mar 2, 2022. http://www.accessdata.fda.gov/drugsatfda_docs/label/2019/211970s000lbl.pdf.
86. NS Pharma Inc. Long-term use of viltolarsen in boys with Duchenne muscular dystrophy in clinical practice (VILT-502). ClinicalTrials.gov Identifier: NCT04687020. Updated Nov 22, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT04687020.
87. VILTEPSO. Prescribing information. NS Pharma; 2020. Accessed Mar 2, 2022. http://www.accessdata.fda.gov/drugsatfda_docs/label/2020/212154s000lbl.pdf.
88. FDA approves targeted treatment for rare Duchenne muscular dystrophy mutation. U.S. Food and Drug Administration. News release. Feb 25, 2021. Accessed Mar 1, 2022. http://www.fda.gov/news-events/press-announcements/fda-approves-targeted-treatment-rare-duchenne-muscular-dystrophy-mutation-0.
89. Sarepta Therapeutics Inc. A double-blind, placebo-controlled, multi-center study with an open-label extension to evaluate the efficacy and safety of SRP-4045 and SRP-4053 in patients with Duchenne muscular dystrophy. Clinicaltrials.gov Identifier: NCT02500381. Updated Aug 19, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02500381.
90. AMONDYS 45. Prescribing information. Sarepta Therapeutics Inc.; 2021. Accessed Feb 22, 2022. http://www.accessdata.fda.gov/drugsatfda_docs/label/2021/213026lbl.pdf.
91. Finkel RS et al. Phase 2a study of ataluren-mediated dystrophin production in patients with nonsense mutation Duchenne muscular dys-trophy. PLoS ONE. 2013;8(12):e81302. doi:10.1371/journal.pone.0081302.
92. PTC Therapeutics. A phase 2 study of PTC124 as an oral treatment for nonsense-mutation-mediated Duchenne muscular dystrophy. Clini-calTrials.gov Identifier: NCT00264888. Updated Jan 14, 2009. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT00264888.
93. PTC Therapeutics. A phase 2B efficacy and safety study of PTC124 in subjects with nonsense-mutation-mediated Duchenne and Becker muscular dystrophy. ClinicalTrials.gov Identifier: NCT00592553. Updated Apr 7, 2020. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT00592553.
94. PTC Therapeutics. A phase 3 efficacy and safety study of ataluren in patients with nonsense mutation dystrophinopathy. ClinicalTrials.gov Identifier: NCT01826487. Updated Aug 4, 2020. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT01826487.
95. Bushby K et al; PTC124-GD-007-DMD Study Group. Ataluren treatment of patients with nonsense mutation dystrophinopathy. Muscle Nerve. 2014 Oct;50(4):477-87. doi:10.1002/mus.24332.
96. Solid Biosciences LLC. A randomized, controlled, open-label, single-ascending dose, phase I/II study to investigate the safety and tolerabil-ity, and efficacy of intravenous SGT-001 in male adolescents and children with Duchenne muscular dystrophy. ClinicalTrials.gov Identifier: NCT03368742. Updated Aug 24, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03368742.
97. Solid Biosciences reports 1.5-year data from patients in the ongoing IGNITE DMD phase I/II clinical trial of SGT-001. Press release. Solid Biosciences. Sep 27, 2021. Accessed Mar 2, 2022. http://www.solidbio.com/about/media/press-releases/solid-biosciences-reports-1-5-year-data-from-patients-in-the-ongoing-ignite-dmd-phase-i-ii-clinical-trial-of-sgt-001.
98. Potter RA et al. Dose-escalation study of systemically delivered rAAVrh74.MHCK7.microdystrophin in the mdx mouse model of Duchenne muscular dystrophy. Hum Gene Ther. 2021 Apr;32(7-8):375-89. doi:10.1089/hum.2019.255.
99. Sarepta Therapeutics, Inc. A Phase 3 Multinational, Randomized, Double-Blind, Placebo- Controlled Systemic Gene Delivery Study to Evaluate the Safety and Efficacy of SRP-9001 in Patients With Duchenne Muscular Dystrophy (EMBARK). clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT05096221
100. Pfizer. A PHASE 3, MULTICENTER, RANDOMIZED, DOUBLE-BLIND, PLACEBO CONTROLLED STUDY TO EVALUATE THE SAFETY AND EFFICACY OF PF 06939926 FOR THE TREATMENT OF DUCHENNE MUSCULAR DYSTROPHY. clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT04281485
101. Pfizer. A phase 1B multicenter open-label, single ascending dose study to evaluate the safety and tolerability of PF-06939926 in ambula-tory and non-ambulatory subjects with Duchenne muscular dystrophy. ClinicalTrials.gov Identifier: NCT03362502. Updated Mar 2, 2022. Ac-cessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03362502.
102. MS MW. Phase 3 CIFFREO DMD Gene Therapy Trial Slated to Begin in June in US. Accessed August 21, 2022. https://musculardystrophynews.com/news/phase-3-trial-of-pfizers-gene-therapy- expected-to-open-in-us-in-june/
103. SRP-9001. Parent Project Muscular Dystrophy. Accessed August 22, 2022. https://www.parentprojectmd.org/drug-development-pipeline/srp-9001-micro-dystrophin-gene- transfer/
104. Sarepta Therapeutics’ Investigational Gene Therapy SRP-9001 for Duchenne Muscular Dystrophy Demonstrates Significant Functional Improvements Across Multiple Studies | Sarepta Therapeutics, Inc. Accessed August 22, 2022. https://investorrelations.sarepta.com/news-releases/news-release- details/sarepta-therapeutics-investigational-gene-therapy-srp-9001
105. Sarepta Therapeutics, Inc. An Open-Label Safety, Tolerability, and Efficacy Study of Eteplirsen in Patients With Duchenne Muscular Dys-trophy Who Have Completed Study 4658-102.clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT03985878
106. Sarepta Therapeutics, Inc. An Open-Label Safety, Tolerability, and Pharmacokinetics Study of Eteplirsen in Young Patients With Duchenne Mus-cular Dystrophy Amenable to Exon 51 Skipping. clinicaltrials.gov; 2021. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT03218995
107.Sarepta Therapeutics, Inc. A Randomized, Double-Blind, Dose Finding and Comparison Study of the Safety and Efficacy of a High Dose of Eteplirsen, Preceded by an Open-Label Dose Escalation, in Patients With Duchenne Muscular Dystrophy With Deletion Mutations Amenable to Exon 51 Skipping. clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT03992430
108. Sarepta Therapeutics, Inc. A Phase 2, Two-Part, Multiple-Ascending-Dose Study of SRP-5051 for Dose Determination, Then Dose Ex-pansion, in Patients With .gov/ct2/show/NCT02081625.
110. NS Pharma Inc. A phase II, dose finding study to assess the safety, tolerability, pharmacokinetics, and pharmacodynamics of NS-065/NCNP-01 in boys with Duchenne muscular dystrophy (DMD). ClinicalTrials.gov Identifier: NCT02740972. Updated Dec 7, 2021. Ac-cessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT02740972.
111. NS Pharma Inc. A phase II, open-label, extension study to assess the safety and efficacy of NS-065/NCNP-01 in boys with Duchenne muscular dystrophy (DMD). ClinicalTrials.gov Identifier: NCT03167255. Updated Nov 24, 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03167255.
112. NS Pharma Inc. A phase 2 open label study to assess the safety, tolerability, and efficacy of viltolarsen in ambulant and non-ambulant boys with Duchenne muscular dystrophy (DMD) compared with natural history controls. ClinicalTrials.gov Identifier: NCT04956289. Updated Feb 1, 2022. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT04956289.
113. NS Pharma Inc. A phase 3 randomized, double-blind, placebo-controlled, multi-center study to assess the efficacy and safety of viltolarsen in ambulant boys with Duchenne muscular dystrophy (DMD). ClinicalTrials.gov Identifier: NCT04060199. Updated Nov 16, 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT04060199.
114. NS Pharma Inc. A phase 3, multi-center, open-label extension study to assess the safety and efficacy of viltolarsen in ambulant boys with Duchenne muscular dystrophy (DMD). ClinicalTrials.gov Identifier: NCT04768062. Updated Nov 16, 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT04768062.
115. Sarepta Therapeutics Inc. A randomized, double-blind, placebo-controlled, dose-titration, safety, tolerability, and pharmacokinetics study followed by an open-label safety and efficacy evaluation of SRP-4045 in advanced-stage patients with Duchenne muscular dystrophy amena-ble to exon 45 skipping. ClinicalTrials.gov Identifier: NCT02530905. Updated May 17, 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT02530905.
116. Sarepta Therapeutics Inc. Long-term, open-label extension study for patients with Duchenne muscular dystrophy enrolled in clinical trials evaluating casimersen or golodirsen. ClinicalTrials.gov Identifier: NCT03532542. Updated Dec 20, 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03532542.
117. PTC Therapeutics. A phase 2 study of the safety, pharmacokinetics, and pharmacodynamics of ataluren (PTC124®) in patients aged ≥2 to <5 years old with nonsense mutation dystrophinopathy. ClinicalTrials.gov Identifier: NCT02819557. Updated Aug 28, 2020. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT02819557.
118. PTC Therapeutics. Phase 2, non-interventional, clinical study to assess dystrophin levels in subjects with nonsense mutation Duchenne muscular dystrophy who have been treated with ataluren for ≥ 9 months. ClinicalTrials.gov Identifier: NCT03796637. Updated Apr 10, 2020. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03796637.
119. PTC Therapeutics. An Open-Label Study Evaluating the Safety and Pharmacokinetics of Ataluren in Children From ≥6 Months to <2 Years of Age With Nonsense Mutation Duchenne Muscular Dystrophy. clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT04336826 120. PTC Therapeutics. An open-label study for previously treated ataluren (PTC124®) pa-tients with nonsense mutation dystrophinopathy. ClinicalTrials.gov Identifier: NCT01557400. Updated Nov 25, 2020. Accessed Feb 21, 2022. https://clinicaltrials.gov/ct2/show/NCT01557400.
121. PTC Therapeutics. An open-label, safety study for ataluren (PTC124) patients with nonsense mutation dystrophinopathy. ClinicalTrials.gov Identifier: NCT01247207. Updated Feb 16, 2022. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT01247207.
122. PTC Therapeutics. A phase 3, randomized, double-blind, placebo-controlled efficacy and safety study of ataluren in patients with non-sense mutation Duchenne muscular dystrophy and open-label extension. ClinicalTrials.gov Identifier: NCT03179631. Updated Feb 8, 2022. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03179631.
123. Sarepta Therapeutics, Inc. An Open-Label, Systemic Gene Delivery Study Using Commercial Process Material to Evaluate the Safety of and Expression From SRP-9001 in Subjects With Duchenne Muscular Dystrophy (ENDEAVOR). clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT04626674
124. Sarepta Therapeutics, Inc. Systemic Gene Delivery Phase I/IIa Clinical Trial for Duchenne Muscular Dystrophy Using RAA-Vrh74.MHCK7.Micro-Dystrophin (MicroDys-IV-001). clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT03375164
125. Sarepta Therapeutics Inc. A multicenter, randomized, double-blind, placebo-controlled trial for Duchenne muscular dystrophy using SRP-9001. ClinicalTrials.gov Identifier: NCT03769116. Updated Dec 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03769116.
126. Hoffmann-La Roche. A Two-Part, Seamless, Multi-Center, Randomized, Placebo-Controlled, Double-Blind Study to Investigate the Safety, Tolerability, Pharmacokinetics, Pharmacodynamics and Efficacy of RO7204239 in Combination With Risdiplam (RO7034067) in Ambulant Pa-tients With Spinal Muscular Atrophy. clinicaltrials.gov; 2022. Accessed September 1, 2022. https://clinicaltrials.gov/ct2/show/NCT05115110
Neuromuscular diseases (NMDs) are a broad classification of heterogeneous groups of disorders characterized by progressive muscle weakness resulting from muscle or nerve dysfunction.1 Diagnosis is based on symptoms and a full medical history, as well as on muscle and imaging tests (including electromyography, nerve-conduction studies, magnetic resonance imaging, muscle biopsy, and blood tests) to confirm or rule out specific NMDs.2 Early diagnosis of NMDs can be difficult because symptoms overlap with those of many other diseases.
Although individually, NMDs are rare, collectively, they affect approximately 250,000 people in the United States. Disease types vary in regard to cause, symptoms, prevalence, age of onset, progression, and severity. Functional impairment from any NMD can lead to lifelong morbidities and shortened life expectancy.1,3
Treatment options for NMDs are limited; most target symptoms, not disease progression. Although there is a need for safe and effective gene-based therapies for NMDs, there are challenges to developing and delivering such treatments that have impeded clinical success. These include a lack of understanding about disease pathology and drug targets, limited animal model systems, and few reliable biomarkers that are predictive of therapeutic success.4,5
Notwithstanding that challenges remain, our understanding of gene expression in NMDs has greatly advanced in the past few decades. This progress has translated into promising results in the gene-therapy field – thereby setting the stage for therapeutic approaches that use novel gene-delivery and gene-manipulation tools.6 These novel approaches include nonviral strategies, such as antisense oligonucleotides (ASOs), and viral-based strategies, such as adeno-associated virus (AAV)-mediated gene silencing and AAV-mediated gene delivery.
In this article, we highlight advancements in the clinical development of gene-based therapies for NMDs. We focus on amyotrophic lateral sclerosis (ALS), spinal muscular atrophy (SMA), and Duchenne muscular dystrophy (DMD) because of recent clinical successes in developing such therapies.1,6,7 We also catalog completed and ongoing clinical trials for ALS, SMA, and DMD (Tables 1-3).
Amyotrophic lateral sclerosis
ALS is caused by progressive degeneration of upper- and lower-motor neurons, which eventually leads to respiratory failure and death 3 to 5 years after disease onset.7-9 There are two subtypes: Familial ALS (10% of cases) and sporadic ALS (90% of cases). Commonly mutated ALS-associated genes6,8 are:
- Superoxide dismutase type 1 (SOD1).
- Chromosome 9 open reading frame 72 (C9orf72).
- Transactive response DNA-binding protein 43 (TARDBP).
- Fused in sarcoma (FUS).
SOD1-targeted therapy is being studied, with early evidence of clinical success. Mutations in SOD1 account for 10% to 20% of familial ALS cases and 1% to 2% of sporadic ALS cases.6,10 10 Mutations in C9orf72 account for 25 to 40% of familial ALS cases and 7% of sporadic ALS cases.8,9,11 Mutations in TARDBP account for 3% of familial ALS cases and 2% of sporadic cases.12 Mutations in FUS account for 4% of familial ALS cases and 1% of sporadic cases. Overall, these mutant proteins can trigger neurotoxicity, thus inducing motor-neuron death.6,10
Treatment of ALS
Two treatments for ALS are Food and Drug Administration approved: riluzole (Rilutek), approved in 1995, and edaravone (Radicava), approved in 2017.
Riluzole is an oral anti-excitotoxic glutamate antagonist.11 Approval of riluzole was based on the results of two studies that demonstrated a 2- to 3-month survival benefit.10,14 For patients who have difficulty swallowing, an oral suspension (Tiglutik, approved in 2018) and an oral film (Exservan, approved in 2019) are available.
Edaravone is a free-radical scavenger that decreases oxidative stress and is administered intravenously (IV).9,13,14 Findings from clinical trials suggest functional improvement or slower decline in function for some patients.
Although these two agents demonstrate modest therapeutic benefit, neither reverses progression of disease.10,14
Gene-based therapy for ALS
Many non-viral strategies, including antisense oligonucleotide (ASO), monoclonal antibodies, reverse transcriptase inhibitors, and HGF gene replacement therapy are used as therapeutic approaches to SOD1, C9orf72, and FUS gene mutations in ALS patients, and are being evaluated in clinical studies14,15 (Table 113-17).
Tofersen, also known as BIIB067, is an investigational ASO, administered by intrathecal (IT) injection, that binds to SOD1 mRNA, thus reducing its protein levels.16 Tofersen was evaluated in the VALOR phase 3 study (ClinicalTrials.gov Identifier: NCT02623699), a three-part randomized, double-blind, placebo-controlled trial: single ascending dose (Part A), multiple ascending dose (B), and fixed dose (C).10 In Parts A and B, 48 participants received five IT injections of tofersen or placebo over 12 weeks and were followed for an additional 12 weeks. Reduction in SOD1 protein production and neurofilament level in cerebrospinal fluid (CSF) (a potential biomarker of motor-neuron degeneration) was observed, which determined the fixed-dose for Part C.16,17
Part C examined the efficacy, safety and tolerability, pharmacokinetics (PK), and pharmacodynamics (PD) of tofersen, compared with placebo, in adults with ALS who had a confirmed SOD1 mutation.17 A total of 108 participants were enrolled; 60 were identified as “faster-progressing”; 48, as “slower-progressing.”18 The primary endpoint of Part C was change from baseline to Week 28 on the Revised ALS Functional Rating Scale (ALSFRS-R) total score. (ALSFRS-R measures overall clinical effect; the score ranges from 0 [no function] to 4 [full function].17)
Tofersen failed to meet the primary efficacy outcome because statistically significant findings were lacking in the faster-progressing population, as measured by joint-rank analysis (difference of 1.2 on the ALSFRS-R score; P = .97). However, trends favoring tofersen were observed across key secondary clinical outcome measures18:
- Change from baseline in CSF SOD1 protein concentration.17 Percent reduction in the total SOD1 protein level was much higher in the tofersen-treated group than in the control group (38% more than controls in the faster-progressing population; 26% more than controls in the slower-progressing population).18
- Change from baseline in neurofilament light-chain concentration in plasma.17,18 Percent reduction in the level of neurofilament light chain was also observed to be higher in the tofersen-treated group than in the control group (67% more than controls in the faster-progressing population and 48% more than controls in the slower-progressing population).18
Because of these encouraging results, VALOR participants were moved to the ongoing open-label extension trial of tofersen (ClinicalTri-als.gov Identifier: NCT03070119), in which both groups were treated with the active agent.
These data suggest that early tofersen treatment might slow decline in faster-progressing patients and stabilize clinical function in slower-progressing patients.18,19 Overall, most adverse events (AEs) in the trial among patients receiving active treatment were of mild or moderate severity, and were largely consistent with either disease progression or lumbar puncture–related complications.18
Because data from VALOR suggested potential benefit from tofersen, the ATLAS trial (ClinicalTrials.gov Identifier: NCT04856982) is investigating the clinical value of presymptomatic treatment and the optimal timing of initiation of therapy.20,21 ATLAS is a phase 3, randomized, placebo-controlled trial that examines the clinical efficacy, safety, and tolerability of tofersen in presymptomatic adult carriers of SOD1 mutation who have an elevated neurofilament light-chain concentration.21 ATLAS will also evaluate the efficacy of tofersen when initiated before, rather than after, ALS manifests clinically. Enrollment is still open for this trial.20,21
Latozinemab, also known as AL001, is a first-in-class monoclonal antibody, administered by IV infusion, that elevates levels of progranulin, a key regulator of the immune activity and lysosomal function in the brain.22,23 Latozinemab limits progranulin endocytosis and degradation by sortilin inhibition.22 Progranulin gene mutations can reduce progranulin expression (by 50 to 70 percent reduction), which may cause neuro-degeneration due to abnormal accumulation of TAR-DNA-binding protein 43 (TDP-43) in the brain cells.22,24 TDP-43 pathology has also been shown to be associated with C9orf72 mutations.23 Although the mechanism is not fully understood, the role of progranulin deficiency in TDP-43 pathology is believed to be associated with neurodegenerative diseases like ALS.11,23,24,43 Previous animal models of chronic neurodegenera-tion have demonstrated how increased progranulin levels can be protective against TDP-43 pathology, increasing neuronal development and survival, thus potentially slowing disease progression.23,24,43 Currently, latozinemab is being investigated in a randomized, double-blind, placebo-controlled, multicenter phase 2 trial (ClinicalTrials.gov Identifier: NCT05053035). Approximately, 45 C90rf72-associated ALS participants (≥ 18 years of age) will receive latozinemab or placebo infusions every 4 weeks (for 24 weeks). Study endpoints include safety, tolerability, PK, PD, as well as plasma, and CSF progranulin levels.25 In previous studies, latozinemab demonstrated encouraging results in frontotemporal dementia (FTD) patients who carry a progranulin mutation. Because FTD was revealed to have significant genetic overlap with ALS, there is disease-modifying potential for latozinemab in ALS patients.23,24
TPN-101 is a nucleoside analog reverse transcriptase inhibitor, administered orally, that was originally developed for human immunodeficiency virus (HIV) treatment. However, due to recent findings suggesting retrotransposon activity contributing to neurodegeneration in TDP-43 mediated diseases, including ALS and FTD, TNP-101 is being repurposed.26 The safety and tolerability of TNP-101 are currently being evaluated in C9orf72-associated ALS and FTD patients (≥ 18 years of age). The study is a randomized, double-blind, placebo-controlled paral-lel-group phase 2a trial (ClinicalTrials.gov Identifier: NCT04993755) The study includes a screening period of 6 weeks, double-blind treatment period of 24 weeks, an open-label treatment period of 24 weeks, and 4 weeks of the post-treatment follow-up visit. Study endpoints include the incidence and severity of spontaneously reported treatment-emergent adverse events (TEAEs) associated with TNP-101 and placebo for a to-tal of 48 weeks.27
ION363 is an investigational ASO, administered by IT injection, that selectively targets one of the FUS mutations (p.P525L), which is responsible for earlier disease onset and rapid ALS progression.28,29 The clinical efficacy of ION363, specifically in clinical function and survival is being assessed in FUS-associated ALS patients (≥ 12 years of age). This randomized phase 3 study (ClinicalTrials.gov Identifier: NCT04768972) includes two parts; part 1 will consist of participants receiving a multi-dose regimen (1 dose every 4-12 weeks) of ION363 or placebo for 61 weeks followed by an open-label extension treatment period in part 2, which will consist of participants receiving ION363 (every 12 weeks) for 85 weeks. The primary endpoint of the study is the change from baseline to day 505 in functional impairment, using ALS Functional Rating Scale-Revised (ALSFRS-R). This measures functional disease severity, specifically in bulbar function, gross motor skills, fine motor skills, and respiratory. The score for all 12 questions can range from 0 (no function) to 4 (full function) with a total possible score of 48.30
Engensis, also known as VM202, is a non-viral gene therapy, administered by intramuscular (IM) injection, that uses a plasmid to deliver the hepatocyte growth factor (HGF) gene to promote HGF protein production. The HGF protein plays a role in angiogenesis, the previous of muscle atrophy, and the promotion of neuronal survival and growth. Based on preclinical studies, increasing HGF protein production has been shown to reduce neurodegeneration, thus potentially halting or slowing ALS progression.31 Currently, the safety of engensis is being evaluated in ALS patients (18-80 years of age) in the REViVALS phase 2a (ClinicalTrials.gov Identifier: NCT04632225)/2b (ClinicalTrial.gov Identifier: NCT05176093).32,33 The ReViVALS trial is a double-blind, randomized, placebo-controlled, multi-center study. The phase 2a study endpoints include the incidence of TEAEs, treatment-emergent serious adverse events (TESAEs), injection site reactions, and clinically significant labor-atory values post-treatment (engensis vs placebo group) for 180 days.33 A phase 2b study will evaluate the long-term safety of engensis for an additional 6 months. Study endpoints include the incidence of AEs, changes from baseline in ALSFRS-R scores to evaluate improvement in muscle function, changes from baseline in quality of life using the ALS patient assessment questionnaire, time to all-cause mortality compared to placebo, etc.32
Spinal muscular atrophy
SMA is a hereditary lower motor-neuron disease caused (in 95% of cases) by deletions or, less commonly, by mutations of the survival motor neuron 1 (SMN1) gene on chromosome 5q13 that encodes the SMN protein.6 Reduction in expression of the SMN protein causes motor neurons to degenerate.36-38 Because of a large inverted duplication in chromosome 5q, two variants of SMN (SMN1 and SMN2) exist on each allele. The paralog gene, SMN2, also produces the SMN protein – although at a lower level (10% to 20% of total SMN protein production) than SMN1 does.
A single nucleotide substitution in SMN2 alters splicing and suppresses transcription of exon 7, resulting in a shortened mRNA strand that yields a truncated SMN protein product.6,37,39 SMA is classified based on age of onset and maximum motor abilities achieved, ranging from the most severe (Type 0) to mildest (Type 4) disease.36,40 Because SMA patients lack functional SMN1 (due to polymorphisms), disease severity is determined by copy numbers of SMN2.6,39
Gene-based therapy for SMA
Three FDA-approved SMN treatments demonstrate clinically meaningful benefit in SMA: SMN2-targeting nusinersen [Spinraza] and risdiplam [Evrysdi], and SMN1-targeting onasemnogene abeparvovec-xioi [Zolgensma]38 Additional approaches to SMA treatment are through SMN-independent therapies, which target muscle and nerve function. Research has strongly suggested that combined SMA therapies, specifically approved SMN-targeted and investigational SMN-independent treatments, such as GYM329 (also known as RO7204239) may be the best strategy to treat all ages, stages, and types of SMA.41 (Table 226-41).
Agents that modulate SMN2. Nusinersen, approved by the FDA in 2016, was the first treatment indicated for all SMA types in pediatric and adult patients.42 The agent is an ASO that targets exon 7 of SMN2, thus stabilizing transcription. Inclusion of exon 7 increases SMN protein production, improving motor function.6,38 Nusinersen is a lifelong treatment that requires IT administration every 4 months because it cannot cross the blood-brain barrier.38,43
Pivotal clinical studies that led to approval of nusinersen include CHERISH (ClinicalTrial.gov Identifier: NCT02292537) and ENDEAR (ClinicalTrial.gov Identifier: NCT02193074) studies.
CHERISH was a phase 3, randomized, double-blind, sham procedure–controlled trial that examined the clinical efficacy and safety of nusinersen in 126 participants with later-onset SMA (2-12 years of age). The primary endpoint was the change from baseline using the Hammersmith Functional Motor Scale Expanded (HFMSE) at 15 months. HFMSE looks at 33 activities to assess improvement in motor function. The study met the primary efficacy outcome, demonstrating statistically significant (P = .0000001) improvement in overall motor function. The nusinersen group showed a 3.9-point increase in the HFMSE score from baseline, which indicates improvement, compared with a 1.0-point decline from baseline in the control group.46,47
ENDEAR was also a randomized, double-blind, sham procedure–controlled phase 3 trial, which investigated the efficacy and safety of nusinersen in 121 participants with early-onset SMA Type 1 (≤ 210 days of age). Coprimary endpoints were:
- Percentage of motor milestones responders, as determined using Section 2 of the Hammersmith Infant Neurological Examination–Part 2.
- Event-free survival (that is, avoidance of combined endpoint of death or permanent ventilation).
ENDEAR met the first primary efficacy outcome, demonstrating statistically significant (P < .0001) improvement in motor milestones (head control, rolling, independent sitting, and standing). By 13 months of age, approximately 51% of nusinersen-treated participants showed improvement, compared with none in the control group.46,47
The second primary endpoint was also met, with a statistically significant (P = .005) 47% decrease in mortality or permanent ventilation use.46-48
The NURTURE (ClinicalTrial.gov Identifier: NCT02386553) study is also investigating the efficacy and safety of nusinersen. An ongoing, open-label, supportive phase 2 trial, NURTURE is evaluating the efficacy and safety of multiple doses of nusinersen in 25 presymptomatic SMA patients (≤ 6 weeks of age). The primary endpoint of this study is time to death or respiratory intervention.49 Interim results demonstrate that 100% of presymptomatic infants are functioning without respiratory intervention after median follow-up of 2.9 years.46-48
Although nusinersen has been shown to be generally safe in clinical studies, development of lumbar puncture–related complications, as well as the need for sedation during IT administration, might affect treatment tolerability in some patients.39
Risdiplam was approved by the FDA in 2020 as the first orally administered small-molecule treatment of SMA (for patients ≤ 2 months of age).52 Risdiplam is a SMN2 splicing modifier, binding to the 5’ splice site of intron 7 and exonic splicing enhancer 2 in exon 7 of SMN2 pre-mRNA. This alternative splicing increases efficiency in SMN2 gene transcription, thus increasing SMN protein production in motor-neuron cells.36 An important advantage of risdiplam is the convenience of oral administration: A large percentage of SMA patients (that is, those with Type 2 disease) have severe scoliosis, which can further complicate therapy or deter patients from using a treatment that is administered through the IT route.40
FDA approval of risdiplam was based on clinical data from two pivotal studies, FIREFISH (ClinicalTrial.gov Identifier: NCT02913482) and SUNFISH (ClinicalTrial.gov Identifier: NCT02908685).53-54
FIREFISH is an open-label, phase 2/3 ongoing trial in infants (1-7 months of age) with SMA Type 1. The study comprises two parts; Part 1 determined the dose of risdiplam used in Part 2, which assessed the efficacy and safety of risdiplam for 24 months. The primary endpoint was the percentage of infants sitting without support for 5 seconds after 12 months of treatment using the gross motor scale of the Bayley Scales of Infant and Toddler Development–Third Edition. A statistically significant (P < .0001) therapeutic benefit was observed in motor milestones. Approximately 29% of infants achieved the motor milestone of independent sitting for 5 seconds, which had not been observed in the natural history of SMA.53-55
SUNFISH is an ongoing randomized, double-blind, placebo-controlled trial of risdiplam in adult and pediatric patients with SMA Types 2 and 3 (2-25 years old). This phase 2/3 study comprises two parts: Part 1 determined the dose (for 12 weeks) to be used for confirmatory Part 2 (for 12 to 24 months). The primary endpoint was the change from baseline on the 32-item Motor Function Measure at 12 months. The study met its primary endpoint, demonstrating statistically significant (P = .0156) improvement in motor function scores, with a 1.36-point increase in the risdiplam group, compared with a 0.19-point decrease in the control group.54,55
Ongoing risdiplam clinical trials also include JEWELFISH (ClinicalTrial.gov Identifier: NCT03032172) and RAINBOW (ClinicalTrial.gov Identifier: NCT03779334).56-57 JEWELFISH is an open-label, phase 2 trial assessing the safety of risdiplam in patients (6 months to 60 years old) who received prior treatment. The study has completed recruitment; results are pending.56 RAINBOW is an ongoing, open-label, single-arm, phase 2 trial, evaluating the clinical efficacy and safety of risdiplam in SMA-presymptomatic newborns (≤ 6 weeks old). The study is open for enrollment.57 Overall, interim results for JEWELFISH and RAINBOW appear promising.
In addition, combined SMA therapies, specifically risdiplam and GYM329 are currently being investigated to address the underlying cause and symptoms of SMA concurrently.58 GYM329, is an investigational anti-myostatin antibody, selectively binding preforms of myostatin - pro-myostatin and latent myostatin, thus improving muscle mass and strength for SMA patients.59 The safety and efficacy of GYM329 in combination with risdiplam is currently being investigated in 180 ambulant participants with SMA (2-10 years of age) in the MANATEE (ClinicalTrial.gov Identifier: NCT05115110) phase 2/3 trial. The MANATEE study is a two-part, seamless, randomized, placebo-controlled, double-blind trial. Part 1 will assess the safety of the combination treatment in approximately 36 participants; participants will receive both GYM329 (every 4 weeks) by subcutaneous (SC) injection into the abdomen and risdiplam (once per day) for 24 weeks followed by a 72-week open-label treatment period. 54,58 The outcome measures include the incidence of AEs, percentage change from baseline in the contractile area of skeletal muscle (in dominant thigh and calf), change from baseline in RHS total score, and incidence of change from baseline in serum concentration (total myostatin, free latent myostatin, and mature myostatin) etc.54 Part 2 will be conducted on 144 participants, specifically assessing the efficacy and safety of the optimal dose of GYM329 selected from Part 1 (combined with risdiplam) for 72 weeks. Once the treatment period is completed in either part, participants can partake in a 2-year open-label extension period.54,58 Other outcome measures include change from baseline in lean muscle mass (assessed by full body dual-energy X- ray absorptiometry (DXA) scan), in time taken to walk/run 10 meters (measured by RHS), in time taken to rise from the floor (measured by RHS), etc.54 Overall, this combination treatment has the potential to further improve SMA patient outcomes and will be further investigated in other patient populations (including non-ambulant patients and a broader age range) in the future.58
An agent that alters SMN1 expression. Onasemnogene abeparvovec-xioi, FDA approved in 2019, was the first gene-replacement therapy indicated for treating SMA in children ≤ 2 years old.60 Treatment utilizes an AAV vector type 9 (AAV9) to deliver a functional copy of SMN1 into target motor-neuron cells, thus increasing SMN protein production and improving motor function. This AAV serotype is ideal because it crosses the blood-brain barrier. Treatment is administered as a one-time IV fusion.38,39,43
FDA approval was based on the STR1VE (ClinicalTrial.gov Identifier: NCT03306277) phase 3 study and START (ClinicalTrial.gov Identifier: NCT02122952) phase 1 study.61,62 START was the first trial to investigate the safety and efficacy of onasemnogene abeparvovec-xioi in SMA Type 1 infants (< 6 months old). Results demonstrated remarkable clinical benefit, including 100% permanent ventilation-free survival and a 92% (11 of 12 patients) rate of improvement in motor function. Improvement in development milestones was also observed: 92% (11 of 12 patients) could sit without support for 5 seconds and 75% (9 of 12) could sit without support for 30 seconds.14,61,63
The efficacy of onasemnogene abeparvovec-xioi seen in STR1VE was consistent with what was observed in START. STRIVE, a phase 3 open-label, single-dose trial, examined treatment efficacy and safety in 22 symptomatic infants (< 6 months old) with SMA Type 1 (one or two SMN2 copies). The primary endpoint was 30 seconds of independent sitting and event-free survival. Patients were followed for as long as 18 months. Treatment showed statistically significant (P < .0001) improvement in motor milestone development and event-free survival, which had not been observed in SMA Type 1 historically. Approximately 59% (13 of 22 patients) could sit independently for 30 seconds at 18 months of age. At 14 months of age, 91% (20 of 22 patients) were alive and achieved independence from ventilatory support.34,35,53
Although many clinical studies suggest that onasemnogene abeparvovec-xioi can slow disease progression, the benefits and risks of long-term effects are still unknown. A 15-year observational study is investigating the long-term therapeutic effects and potential complications of onasemnogene abeparvovec-xioi. Participants in START were invited to enroll in this long-term follow-up study (ClinicalTrial.gov Identifier: NCT04042025).66-67
Duchenne muscular dystrophy
DMD is the most common muscular dystrophy of childhood. With an X-linked pattern of inheritance, DMD is seen mostly in young males (1 in every 3,500 male births).38,39,73 DMD is caused by mutation of the dystrophin encoding gene, or DMD, on the X chromosome. Deletion of one or more exons of DMD prevents production of the dystrophin protein, which leads to muscle degeneration.38,39,43 Common DMD deletion hotspots are exon 51 (20% of cases), exon 53 (13% of cases), exon 44 (11% of cases), and exon 45 (12% of cases).74 Nonsense mutations, which account for another 10% of DMD cases, occur when premature termination codons are found in the DMD gene. Those mutations yield truncated dystrophin protein products.39,66
Therapy for DMD
There are many therapeutic options for DMD, including deflazacort (Emflaza), FDA approved in 2017, which has been shown to reduce inflammation and immune system activity in DMD patients (≥ 5 years old). Deflazacort is a corticosteroid prodrug; its active metabolite acts on the glucocorticoid receptor to exert anti-inflammatory and immunosuppressive effects. Studies have shown that muscle strength scores over 6-12 months and average time to loss of ambulation numerically favored deflazacort over placebo.74,75
Gene-based therapy for DMD
Mutation-specific therapeutic approaches, such as exon skipping and nonsense suppression, have shown promise for the treatment of DMD (Table 358-79):
- ASO-mediated exon skipping allows one or more exons to be omitted from the mutated DMD mRNA.74,75 Effective FDA-approved ASOs include golodirsen [Vyondys 53], viltolarsen [Viltepso], and casimersen [Amondys 45].74
- An example of therapeutic suppression of nonsense mutations is ataluren [Translarna], an investigational agent that can promote premature termination codon read-through in DMD patients.66
Another potential treatment approach is through the use of AAV gene transfer to treat DMD. However, because DMD is too large for the AAV vector (packaging size, 5.0 kb), microdystrophin genes (3.5-4 kb, are used as an alternative to fit into a single AAV vector.39,76
Exon skipping targeting exon 51. Eteplirsen, approved in 2016, is indicated for the treatment of DMD patients with the confirmed DMD gene mutation that is amenable to exon 51 skipping. Eteplirsen binds to exon 51 of dystrophin pre-mRNA, causing it to be skipped, thus, restoring the reading frame in patients with DMD gene mutation amenable to exon 51 skipping. This exclusion promotes dystrophin production. Though the dystrophin protein is still functional, it is shortened.38,77 Treatment is administered IV, once a week (over 35-60 minutes). Eteplirsen’s accelerated approval was based on 3 clinical studies (ClinicalTrial.gov Identifier: NCT01396239, NCT01540409, and NCT00844597.) 78-81 The data demonstrated an increased expression of dystrophin in skeletal muscles in some DMD patients treated with eteplirsen. Though the clinical benefit of eteplirsen (including improved motor function) was not established, it was concluded by the FDA that the data were reasonably likely to predict clinical benefit. Continued approval for this indication may depend on the verification of a clinical benefit in confirmatory trials. Ongoing clinical trials include (ClinicalTrial.gov Identifier: NCT03992430 (MIS51ON), NCT03218995, and NCT03218995).77,81,82
Vesleteplirsen, is an investigational agent that is designed for DMD patients who are amendable to exon 51 skip-ping. The mechanism of action of vesleteplirsen appears to be similar to that of eteplirsen.83 The ongoing MOMENTUM (ClinicalTrial.gov Identifier: NCT04004065) phase 2 trial is assessing the safety and tolerability of vesleteplirsen at multiple-ascending dose levels (administered via IV infusion) in 60 participants (7-21 years of age). The study consists of two parts; participants receive escalating dose levels of vesleteplirsen (every 4 weeks) for 72 weeks during part A and participants receive the selected doses from part A (every 4 weeks) for 2 years during part B. Study endpoints include the number of AEs (up to 75 weeks) and the change from baseline to week 28 in dystrophin protein level. 84 Serious AEs of reversible hypomagnesemia were observed in part B, and as a result, the study protocol was amended to include magnesium supplementation and monitoring of magnesium levels.83
Exon skipping targeting exon 53. Golodirsen, FDA approved in 2019, is indicated for the treatment of DMD in patients who have a confirmed DMD mutation that is amenable to exon 53 skipping. The mechanism of action is similar to eteplirsen, however, golodirsen is designed to bind to exon 53.38,39 Treatment is administered by IV infusion over 35-60 minutes.
Approval of golodirsen was based primarily on a two-part, phase 1/2 clinical trial (ClinicalTrial.gov Identifier: NCT02310906). Part 1 was a randomized, placebo-controlled, dose-titration study that assessed multiple-dose efficacy in 12 DMD male patients, 6 to 15 years old, with deletions that were amenable to exon 53 skipping.
Part 2 was an open-label trial in 12 DMD patients from Part 1 of the trial plus 13 newly enrolled male DMD patients who were also amenable to exon 53 skipping and who had not already received treatment. Primary endpoints were change from baseline in total distance walked during the 6-minute walk test at Week 144 and dystrophin protein levels (measured by western blot testing) at Week 48. A statistically significant increase in the mean dystrophin level was observed, from a baseline 0.10% mean dystrophin level to a 1.02% mean dystrophin level after 48 weeks of treatment (P < .001). Common reported adverse events associated with golodirsen were headache, fever, abdominal pain, rash, and dermatitis. Renal toxicity was observed in preclinical studies of golodirsen but not in clinical studies.80,85
Viltolarsen, approved in 2020, is also indicated for the treatment of DMD in patients with deletions amenable to exon 53 skipping. The mechanism of action and administration (IV infusion over 60 minutes) are similar to that of golodirsen.
Approval of viltolarsen was based on two phase 2 clinical trials (ClinicalTrial.gov Identifier: NCT02740972 and NCT03167255) in a total of 32 patients. NCT02740972 was a randomized, double-blind, placebo-controlled, dose-finding study that evaluated the clinical efficacy of viltolarsen in 16 male DMD patients (4-9 years old) for 24 weeks.
NCT03167255 was an open-label study that evaluated the safety and tolerability of viltolarsen in DMD male patients (5-18 years old) for 192 weeks. The efficacy endpoint was the change in dystrophin production from baseline after 24 weeks of treatment. A statistically significant increase in the mean dystrophin level was observed, from a 0.6% mean dystrophin level at baseline to a 5.9% mean dystrophin level at Week 25 (P = .01). The most common adverse events observed were upper respiratory tract infection, cough, fever, and injection-site reaction.86-87
Exon skipping targeting exon 45. Casimersen was approved in 2021 for the treatment of DMD in patients with deletions amenable to exon 45 skipping.88 Treatment is administered by IV infusion over 30-60 minutes. Approval was based on an increase in dystrophin production in skeletal muscle in treated patients. Clinical benefit was reported in interim results from the ESSENCE (ClinicalTrial.gov Identifier: NCT02500381) study, an ongoing double-blind, placebo-controlled phase 3 trial that is evaluating the efficacy of casimersen, compared with placebo, in male participants (6-13 years old) for 48 weeks. Efficacy is based on the change from baseline dystrophin intensity level, determined by immunohistochemistry, at Week 48.
Interim results from ESSENCE show a statistically significant increase in dystrophin production in the casimersen group, from a 0.9% mean dystrophin level at baseline to a 1.7% mean dystrophin level at Week 48 (P = .004); in the control group, a 0.54% mean dystrophin level at baseline increased to a 0.76% mean dystrophin level at Week 48 (P = .09). Common adverse events have included respiratory tract infection, headache, arthralgia, fever, and oropharyngeal pain. Renal toxicity was observed in preclinical data but not in clinical studies.60,84
Targeting nonsense mutations. Ataluren is an investigational, orally administered nonsense mutation suppression therapy (through the read-through of stop codons).37 Early clinical evidence supporting the use of ataluren in DMD was seen in an open-label, dose-ranging, phase 2a study (ClinicalTrial.gov Identifier: NCT00264888) in male DMD patients (≥ 5 years old) caused by nonsense mutation. The study demonstrated a modest (61% ) increase in dystrophin expression in 23 of 38 patients after 28 days of treatment.37,91,92
However, a phase 2b randomized, double-blind, placebo-controlled trial (ClinicalTrial.gov Identifier: NCT00592553) and a subsequent confirmatory ACT DMD phase 3 study (ClinicalTrial.gov Identifier: NCT01826487) did not meet their primary endpoint of improvement in ambulation after 48 weeks as measured by the 6-minute walk test.37,93,94 In ACT DMD, approximately 74% of the ataluren group did not experience disease progression, compared with 56% of the control group (P = 0386), measured by a change in the 6-minute walk test, which assessed ambulatory decline.37,95
Based on limited data showing that ataluren is effective and well tolerated, the European Medicines Agency has given conditional approval for clinical use of the drug in Europe. However, ataluren was rejected by the FDA as a candidate therapy for DMD in the United States.22 Late-stage clinical studies of ataluren are ongoing in the United States.
AAV gene transfer with microdystrophin. Limitations on traditional gene-replacement therapy prompted exploration of gene-editing strategies for treating DMD, including using AAV-based vectors to transfer microdystrophin, an engineered version of DMD, into target muscles.43 The microdystrophin gene is designed to produce a functional, truncated form of dystrophin, thus improving muscular function.
There are 3 ongoing investigational microdystrophin gene therapies that are in clinical development (ClinicalTrial.gov Identifier: NCT03368742 (IGNITE DMD), NCT04281485 (CIFFREO), and NCT05096221 (EMBARK)).38,82
IGNITE DMD is a phase 1/2 randomized, controlled, single-ascending dose trial evaluating the safety and efficacy of a SGT-001, single IV infusion of AAV9 vector containing a microdystrophin construct in DMD patients (4-17 years old) for 12 months. At the conclusion of the trial, treatment and control groups will be followed for 5 years. The primary efficacy endpoint is the change from baseline in microdystrophin protein production in muscle-biopsy material, using western blot testing.96 Long-term interim data on biopsy findings from three patients demonstrated clinical evidence of durable microdystrophin protein expression after 2 years of treatment.96,97
The CIFFREO trial will assess the safety and efficacy of the PF-06939926 microdystrophin gene therapy, an investigational AAV9 containing microdystrophin, in approximately 99 ambulatory DMD patients (4-7 years of age). The study is a randomized, double-blind, placebo-controlled, multicenter phase 3 trial. The primary efficacy end-point is the change from baseline in the North Star Ambulatory Assessment (NSAA), which measures gross motor function. This will be assessed at 52 weeks; all study participants will be followed for a total of 5 years post-treatment.98,99,100 Due to unexpected patient death (in a non-ambulatory cohort) in the phase 1b (in a non-ambulatory cohort) in the phase 1b (ClinicalTrial.gov Identifier: (NCT03362502) trial, microdystrophin gene therapy was immediately placed on clinical hold.101,102 The amended study protocol required that all participants undergo one week of in-hospital observation after receiving treatment.102
The EMBARK study is a global, randomized, double-blind, placebo-controlled, phase 3 trial that is evaluating the safety and efficacy of SRP-9001, which is a rAAVrh74.MHCK7.microdystrophin gene therapy. The AAV vector (rAAVrh74) contains the microdystrophin construct, driven by the skeletal and cardiac muscle–specific promoter, MHCK7.98,99 In the EMBARK study, approximately 120 participants with DMD (4-7 years of age) will be enrolled. The primary efficacy endpoint includes the change from baseline to week 52 in the NSAA total score.99 Based on SRP-9001, data demonstrating consistent statistically significant functional improvements in NSAA total scores and timed function tests (after one-year post- treatment) in DMD patients from previous studies and an integrated analysis from multiple studies (ClinicalTrial.gov Identifier: NCT03375164, NCT03769116, and NCT04626674), the ongoing EMBARK has great promise.103,104
Challenges ahead, but advancements realized
Novel gene-based therapies show significant potential for transforming the treatment of NMDs. The complex pathologies of NMDs have been a huge challenge to disease management in an area once considered unremediable by gene-based therapy. However, advancements in precision medicine – specifically, gene-delivery systems (for example, AAV9 and AAVrh74 vectors) combined with gene modification strategies (ASOs and AAV-mediated silencing) – have the potential to, first, revolutionize standards of care for sporadic and inherited NMDs and, second, significantly reduce disease burden.6
What will be determined to be the “best” therapeutic approach will, likely, vary from NMD to NMD; further investigation is required to determine which agents offer optimal clinical efficacy and safety profiles.43 Furthermore, the key to therapeutic success will continue to be early detection and diagnosis – first, by better understanding disease pathology and drug targets and, second, by validation of reliable biomarkers that are predictive of therapeutic benefit.4,5
To sum up, development challenges remain, but therapeutic approaches to ALS, SMA, and DMD that utilize novel gene-delivery and gene-manipulation tools show great promise.
Ms. Yewhalashet is a student in the masters of business and science program, with a concentration in healthcare economics, at Keck Graduate Institute Henry E. Riggs School of Applied Life Sciences, Claremont, Calif. Dr. Davis is professor of practice in clinical and regulatory affairs, Keck Graduate Institute Henry E. Riggs School of Applied Life Sciences.
References
1. Aitken M et al. Understanding neuromuscular disease care. IQVIA [Internet]. Oct 30, 2018. Accessed Mar 1, 2022. https://www.iqvia.com/insights/the-iqvia-institute/reports/understanding-neuromuscular-disease-care.
2. National Institute of Neurological Disorders and Stroke. Neurological diagnostic tests and procedures fact sheet. Updated Nov 15, 2021. Ac-cessed Mar 1, 2022. http://www.ninds.nih.gov/Disorders/Patient-Caregiver-Education/Fact-Sheets/Neurological-Diagnostic-Tests-and-Procedures-Fact.
3. Deenen JCW et al. The epidemiology of neuromuscular disorders: A comprehensive overview of the literature. J Neuromuscul Dis. 2015;2(1):73-85.
4. Cavazzoni P. The path forward: Advancing treatments and cures for neurodegenerative diseases. U.S. Food and Drug Administration. Jul 29, 2021. Accessed Mar 1, 2022. http://www.fda.gov/news-events/congressional-testimony/path-forward-advancing-treatments-and-cures-neurodegenerative-diseases-07292021.
5. Martier R, Konstantinova P. Gene therapy for neurodegenerative diseases: Slowing down the ticking clock. Front Neurosci. 2020 Sep 18;14:580179. doi: 10.3389/fnins.2020.580179.
6. Sun J, Roy S. Gene-based therapies for neurodegenerative diseases. Nat Neurosci. 2021 Mar;24(3):297-311. doi:10.1038/s41593-020-00778-1.
7. Amado DA, Davidson BL. Gene therapy for ALS: A review. Mol Ther. 2021 Dec 1;29(12):3345-58. doi:10.1016/j.ymthe.2021.04.008.
8. Yun Y, Ha Y. CRISPR/Cas9-mediated gene correction to understand ALS. Int J Mol Sci. 2020;21(11):3801. doi:10.3390/ijms21113801.
9. National Institute of Neurological Disorders and Stroke. Amyotrophic lateral sclerosis (ALS) fact sheet. Updated Nov 15, 2021. Accessed Mar 1, 2022. http://www.ninds.nih.gov/Disorders/Patient-Caregiver-Education/Fact-Sheets/Amyotrophic-Lateral-Sclerosis-ALS-Fact-Sheet.
10. Cappella M et al. Gene therapy for ALS – A perspective. Int J Mol Sci. 2019;20(18):4388. doi:10.3390/ijms20184388.
11. Abramzon YA, Fratta P, Traynor BJ, Chia R. The Overlapping Genetics of Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Front Neurosci. 2020;14. Accessed August 18, 2022. https://www.frontiersin.org/articles/10.3389/fnins.2020.00042
12. Giannini M, Bayona-Feliu A, Sproviero D, Barroso SI, Cereda C, Aguilera A. TDP-43 mutations link Amyotrophic Lateral Sclerosis with R-loop homeostasis and R loop-mediated DNA damage. PLOS Genet. 2020;16(12):e1009260. doi:10.1371/journal.pgen.1009260
13. FDA-approved drugs for treating ALS. The ALS Association [Internet]. Accessed Mar 1, 2022. http://www.als.org/navigating-als/living-with-als/fda-approved-drugs.
14. Jensen TL et al. Current and future prospects for gene therapy for rare genetic diseases affecting the brain and spinal cord. Front Mol Neurosci. 2021 Oct 6;14:695937. doi:10.3389/fnmol.2021.695937.
15. ALS Gene Targeted Therapies. The ALS Association. Accessed August 22, 2022. https://www.als.org/understanding-als/who-gets-als/genetic-testing/als-gene-targeted-therapies
16. Tofersen for ALS clears phase 1/2 trial, now in phase 3. Advances in Motion. Massachusetts General Hospital [Internet]. Sep 30, 2020. Accessed Mar 1, 2022. https://advances.massgeneral.org/neuro/journal.aspx?id=1699.17. Biogen. A study to evaluate the efficacy, safety, tol-erability, pharmacokinetics, and pharmacodynamics of BIIB067 administered to adult subjects with amyotrophic lateral sclerosis and confirmed superoxide dismutase 1 mutation. ClinicalTrials.gov Identifier: NCT02623699. Updated Jul 25, 2021. Accessed Feb 17, 2022. https://clinicaltrials.gov/ct2/show/NCT02623699.
18. Biogen. Biogen announces topline results from the tofersen phase 3 study and its open-label Extension in SOD1-ALS. Press release. Oct 17, 2021. Accessed Mar 1, 2022. https://investors.biogen.com/news-releases/news-release-details/biogen-announces-topline-results-tofersen-phase-3-study-and-its.
19. Biogen. An extension study to assess the long-term safety, tolerability, pharmacokinetics, and effect on disease progression of BIIB067 ad-ministered to previously treated adults with amyotrophic lateral sclerosis caused by superoxide dismutase 1 mutation. ClinicalTrials.gov Identi-fier: NCT03070119. Updated Sep 10, 2021. Accessed Feb 17, 2022. https://clinicaltrials.gov/ct2/show/NCT03070119.
20. MS MW. #AANAM – ATLAS Trial to Assess Tofersen in Presymptomatic SOD1 ALS. Accessed February 19, 2022. https://alsnewstoday.com/news-posts/2021/04/23/aanam-atlas-clinical-trial- tofersen-presymptomatic-sod1-als-patients/
21.Biogen. A phase 3 randomized, placebo-controlled trial with a longitudinal natural history run-in and open-label extension to evaluate BIIB067 initiated in clinically presymptomatic adults with a confirmed superoxide dismutase 1 mutation. ClinicalTrials.gov Identifier: NCT04856982. Updated Feb 18, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT04856982.
22. Latozinemab | ALZFORUM. Accessed August 19, 2022. https://www.alzforum.org/therapeutics/latozinemab
23. Alector Presents AL001 (latozinemab) Data from the FTD-C9orf72 Cohort of the INFRONT-2 Phase 2 Clinical Trial | Alector. Accessed August 18, 2022. https://investors.alector.com/news- releas-es/news-release-details/alector-presents-al001-latozinemab-data-ftd-c9orf72-cohort/
24. Alector Announces First Participant Dosed in Phase 2 Study Evaluating AL001 in Amyotrophic Lateral Sclerosis (ALS) | Alector. Accessed August 18, 2022. https://investors.alector.com/news- releases/news-release-details/lector-announces-first-participant-dosed-phase-2-study-0/ 25. A Phase 2 Study to Evaluate AL001 in C9orf72-Associated ALS - Full Text View - ClinicalTrials.gov. Accessed August 19, 2022. https://clinicaltrials.gov/ct2/show/NCT05053035
26.TPN-101 | ALZFORUM. Accessed August 19, 2022. https://www.alzforum.org/therapeutics/tpn- 101
27. Transposon Therapeutics, Inc. A Phase 2a Study of TPN-101 in Patients With Amyotrophic Lateral Sclerosis (ALS) and/or Frontotemporal Dementia (FTD) Associated With Hexanucleotide Repeat Expansion in the C9orf72 Gene (C9ORF72 ALS/FTD). clinicaltrials.gov; 2022. Ac-cessed August 17, 2022. https://clinicaltrials.gov/ct2/show/NCT04993755
28. Kerk SY, Bai Y, Smith J, et al. Homozygous ALS-linked FUS P525L mutations cell- autonomously perturb transcriptome profile and chem-oreceptor signaling in human iPSC microglia. Stem Cell Rep. 2022;17(3):678-692. doi:10.1016/j.stemcr.2022.01.004
29. ION363 | ALZFORUM. Accessed August 19, 2022. https://www.alzforum.org/therapeutics/ion363 30. Ionis Pharmaceuticals, Inc. A Phase 1-3 Study to Evaluate the Efficacy, Safety, Pharmacokinetics and Pharmacodynamics of Intrathecally Administered ION363 in Amyo-trophic Lateral Sclerosis Patients With Fused in Sarcoma Mutations (FUS-ALS). clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT04768972
31. PhD LF. Engensis (VM202) - ALS News Today. Accessed August 19, 2022. https://alsnewstoday.com/vm202/
32. Helixmith Co., Ltd. A 6-Month Extension Study Following Protocol VMALS-002-2 (A Phase 2a, Double-Blind, Randomized, Place-bo-Controlled, Multicenter Study to Assess the Safety of Engensis in Participants With Amyotrophic Lateral Sclerosis). clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT05176093 33. Safety of Engensis in Participants With Amyotrophic Lateral Sclerosis - Full Text View - ClinicalTrials.gov. Accessed August 19, 2022. https://clinicaltrials.gov/ct2/show/NCT04632225
34. Biogen. A phase 1, safety, tolerability, and distribution study of a microdose of radiolabeled BIIB067 co-administered with BIIB067 to healthy adults. ClinicalTrials.gov Identifier: NCT03764488. Updated Jul 19, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03764488.
35. Ionis Pharmaceuticals Inc. A phase 1, double-blind, placebo-controlled, dose-escalation study of the safety, tolerability, and pharmacokinet-ics of ISIS 333611 administered intrathecally to patients with familial amyotrophic lateral sclerosis due to superoxide dismutase 1 gene muta-tions. ClinicalTrials.gov Identifier: NCT01041222. Updated Apr 13, 2012. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT01041222.
36. Messina S, Sframeli M. New treatments in spinal muscular atrophy: Positive results and new challenges. J Clin Med. 2020;9(7):2222. doi:10.3390/jcm9072222.
37. Scoto M et al. Genetic therapies for inherited neuromuscular disorders. Lancet Child Adolesc Health. 2018 Aug;2(8):600-9. doi:10.1016/S2352-4642(18)30140-8.
38. Abreu NJ, Waldrop MA. Overview of gene therapy in spinal muscular atrophy and Duchenne muscular dystrophy. Pediatr Pulmonol. 2021 Apr;56(4):710-20. doi:10.1002/ppul.25055.
39. Brandsema J, Cappa R. Genetically targeted therapies for inherited neuromuscular disorders. Practical Neurology [Internet]. Jul/Aug 2021:69-73. Accessed Mar 1, 2022. https://practicalneurology.com/articles/2021-july-aug/genetically-targeted-therapies-for-inherited-neuromuscular-disorders/pdf.
40. Ojala KS et al. In search of a cure: The development of therapeutics to alter the progression of spinal muscular atrophy. Brain Sci. 2021;11(2):194. doi:10.3390/brainsci11020194.
41. McCall S. Cure SMA Releases Updated Drug Pipeline. Cure SMA. Published December 13, 2021. Accessed August 21, 2022. https://www.curesma.org/cure-sma-releases-updated-drug-pipeline- 2021/ 42. FDA approves first drug for spinal muscular atrophy. U.S. Food and Drug Administration. News release. Dec 23, 2016. Accessed Mar 1, 2022. http://www.fda.gov/news-events/press-announcements/fda-approves-first-drug-spinal-muscular-atrophy.43. Kirschner J. Postnatal gene therapy for neuromuscular diseases – Opportunities and limitations. J Perinat Med. 2021 Sep;49(8):1011-5. doi:10.1515/jpm-2021-0435.
43. Terryn J, Verfaillie CM, Van Damme P. Tweaking Progranulin Expression: Therapeutic Avenues and Opportunities. Front Mol Neurosci. 2021;14. Accessed September 4, 2022. https://www.frontiersin.org/articles/10.3389/fnmol.2021.71303144.
44. Biogen. A phase 3, randomized, double-blind, sham-procedure controlled study to assess the clinical efficacy and safety of ISIS 396443 administered intrathecally in patients with later-onset spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT02292537. Updated Feb 17, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/study/NCT02292537.
45. Why Spinraza/later-onset studies. SPINRAZA® (nusinersen) [Internet]. Accessed Mar 1, 2022. www.spinraza.com/en_us/home/why-spinraza/later-onset-studies.html#scroll-tabs.
46. Biogen. A Phase 3, Randomized, Double-Blind, Sham-Procedure Controlled Study to Assess the Clinical Efficacy and Safety of ISIS 396443 Administered Intrathecally in Patients With Infantile- Onset Spinal Muscular Atrophy. clinicaltrials.gov; 2021. Accessed February 10, 2022. https://clinicaltrials.gov/ct2/show/results/NCT02193074
47. Early-onset SMA (Type 1) | SPINRAZA® (nusinersen). Accessed Mar 1, 2022. https://www.spinraza-hcp.com/en_us/home/why-spinraza/about-spinraza.html.
48. Finkel RS et al; ENDEAR Study Group. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N Engl J Med. 2017;377(18):1723-32. doi: 10.1056/NEJMoa1702752.
49. Biogen. An open-label study to assess the efficacy, safety, tolerability, and pharmacokinetics of multiple doses of ISIS 396443 delivered intrathecally to subjects with genetically diagnosed and presymptomatic spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT02386553. Updated Nov 18, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02386553.
50. De Vivo DC et al; NURTURE Study Group. Nusinersen initiated in infants during the presymptomatic stage of spinal muscular atrophy: In-terim efficacy and safety results from the phase 2 NURTURE study. Neuromuscul Disord. 2019 Nov;29(11):842-56. doi:10.1016/j.nmd.2019.09.007.
51. Why Spinraza/presymptomatic study. SPINRAZA® (nusinersen) [Internet]. Accessed Feb 22, 2022. www.spinraza.com/en_us/home/why-spinraza/presymptomatic-study.html#scroll-tabs.
52. FDA approves oral treatment for spinal muscular atrophy. U.S. Food and Drug Administration. News release. Aug 7, 2020. Accessed Mar 1, 2022. http://www.fda.gov/news-events/press-announcements/fda-approves-oral-treatment-spinal-muscular-atrophy.
53. Hoffmann-La Roche. A two-part seamless, open-label, multicenter study to investigate the safety, tolerability, pharmacokinetics, pharmaco-dynamics and efficacy of risdiplam (RO7034067) in infants with type 1 spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT02913482. Updated Jan 21, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02913482.
54. Hoffmann-La Roche. A two-part seamless, multi-center randomized, placebo-controlled, double-blind study to investigate the safety, tolera-bility, pharmacokinetics, pharmacodynamics and efficacy of risdiplam (RO7034067) in type 2 and 3 spinal muscular atrophy patients. Clinical-Trials.gov Identifier: NCT02908685. Updated Dec 28, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02908685.
55. Genentech. Genentech’s risdiplam shows significant improvement in survival and motor milestones in infants with type 1 spinal muscular atrophy (SMA). Press release. Apr 27, 2020. Accessed Mar 1, 2022. http://www.gene.com/media/press-releases/14847/2020-04-27/genentechs-risdiplam-shows-significant-i
56. Hoffmann-La Roche. An open-label study to investigate the safety, tolerability, and pharmacokinetics/pharmacodynamics of risdiplam (RO7034067) in adult and pediatric patients with spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT03032172. Updated Jan 27, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03032172.
57. Hoffmann-La Roche. An open-label study of risdiplam in infants with genetically diagnosed and presymptomatic spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT03779334. Updated Jan 27, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03779334.
58. McCall S. Update on Genentech/Roche Initiation of MANATEE Clinical Study. Cure SMA. Published October 20, 2021. Accessed August 20, 2022. https://www.curesma.org/update-on- genentech-roche-initiation-of-manatee-clinical-study/
59. Abati E, Manini A, Comi GP, Corti S. Inhibition of myostatin and related signaling pathways for the treatment of muscle atrophy in motor neuron diseases. Cell Mol Life Sci. 2022;79(7):374. doi:10.1007/s00018-022-04408-w
60. FDA approves innovative gene therapy to treat pediatric patients with spinal muscular atrophy, a rare disease and leading genetic cause of infant mortality. U.S. Food and Drug Administration. News release. May 24, 2019. Accessed Mar 1, 2022. http://www.fda.gov/news-events/press-announcements/fda-approves-innovative-gene-therapy-treat-pediatric-patients-spinal-muscular-atrophy-rare-disease.
61. Novartis Gene Therapies. Phase I gene transfer clinical trial for spinal muscular atrophy type 1 delivering AVXS-101. ClinicalTrials.gov Identifier: NCT02122952. Updated Jun 14, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02122952.
62. Novartis Gene Therapies. Phase 3, open-label, single-arm, single-dose gene replacement therapy clinical trial for patients with spinal mus-cular atrophy type 1 with one or two SMN2 copies delivering AVXS-101 by intravenous infusion. ClinicalTrials.gov Identifier: NCT03306277. Updated Jun 14, 2021. Accessed Feb 21, 2022. https://clinicaltrials.gov/ct2/show/NCT03306277.
63. Mendell JR et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N Engl J Med. 2017;377(18):1713-22. doi:10.1056/NEJMoa1706198.
64. Symptomatic study results. ZOLGENSMA [Internet]. Updated Nov 2021. Accessed Mar 1, 2022. Error! Hyperlink reference not valid..
65. Novartis Gene Therapies. A global study of a single, one-time dose of AVXS-101 delivered to infants with genetically diagnosed and pre-symptomatic spinal muscular atrophy with multiple copies of SMN2. ClinicalTrials.gov Identifier: NCT03505099. Updated Jan 1, 2022. Ac-cessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03505099.
66. Chiu W et al. Current genetics and potential gene-targeting therapeutics for neuromuscular diseases. Int J Mol Sci. 2020 Dec;21(24):9589. doi:10.3390/ijms21249589.
67. Novartis Gene Therapies. A long-term follow-up study of patients in the clinical trials for spinal muscular atrophy receiving AVXS-101. Clini-calTrials.gov Identifier: NCT04042025. Updated Jun 9, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT04042025.
68. Novartis Gene Therapies. Phase 3, open-label, single-arm, single-dose gene replacement therapy clinical trial for patients with spinal mus-cular atrophy type 1 with one or two SMN2 copies delivering AVXS-101 by intravenous infusion. ClinicalTrials.gov Identifier: NCT0383718. Up-dated Jan 11, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03837184.
69. Biogen. An open-label, dose escalation study to assess the safety, tolerability and dose-range finding of multiple doses of ISIS 396443 de-livered intrathecally to patients with spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT01703988. Updated Apr 13, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT01703988.
70. Biogen. A study to assess the efficacy, safety, tolerability, and pharmacokinetics of multiple doses of ISIS 396443 delivered intrathecally to patients with infantile-onset spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT01839656. Updated Feb 17, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT01839656.
71. Biogen. An open-label extension study for patients with spinal muscular atrophy who previously participated in investigational studies of ISIS 396443. ClinicalTrials.gov Identifier: NCT02594124. Updated Nov 15, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02594124.
72. Biogen. Escalating dose and randomized, controlled study of nusinersen (BIIB058) in participants with spinal muscular atrophy. ClinicalTri-als.gov Identifier: NCT04089566. Updated Feb 24, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT04089566.
73. National Center for Advancing Translational Sciences. Duchenne muscular dystrophy. Genetic and Rare Diseases Information Center. Up-dated Nov 2, 2020. Accessed Mar 1, 2022. https://rarediseases.info.nih.gov/diseases/6291/duchenne-muscular-dystrophy.
74. Matsuo M. Antisense oligonucleotide-mediated exon-skipping therapies: Precision medicine spreading from Duchenne muscular dystrophy. JMA J. 2021 Jul 15;4(3):232-40. doi:10.31662/jmaj.2021-0019.
75. FDA approves drug to treat Duchenne muscular dystrophy. U.S. Food and Drug Administration. News release. Feb 9, 2017. Accessed Mar 1, 2022. http://www.fda.gov/news-events/press-announcements/fda-approves-drug-treat-duchenne-muscular-dystrophy.74.
76. Duan D. Dystrophin gene replacement and gene repair therapy for Duchenne muscular dystrophy in 2016: An interview. Hum Gene Ther Clin Dev. 2016 Mar;27(1):9-18. doi:10.1089/humc.2016.001.
77. EXONDYS 51®. Parent Project Muscular Dystrophy. Accessed August 21, 2022. https://www.parentprojectmd.org/drug-development-pipeline/exondys-51/
78. Sarepta Therapeutics, Inc. A Randomized, Double-Blind, Placebo-Controlled, Multiple Dose Efficacy, Safety, Tolerability and Pharmacoki-netics Study of AVI-4658(Eteplirsen),in the Treatment of Ambulant Subjects With Duchenne Muscular Dystrophy. clinicaltrials.gov; 2020. Ac-cessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT01396239
79. Sarepta Therapeutics, Inc. Clinical Study to Assess the Safety Fo AVI-4658 in Subjects With Duchenne Muscular Dystrophy Due to a Frame-Shift Mutation Amenable to Correction by Skipping Exon 51. clinicaltrials.gov; 2015. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/study/NCT00844597
80. Sarepta Therapeutics, Inc. A 2-part, randomized, double-blind, placebo-controlled, dose-titration, safety, tolerability, and pharmacokinetics study (Part 1) followed by an open-label efficacy and safety evaluation (Part 2) of SRP-4053 in patients with Duchenne muscular dystrophy amenable to exon 53 skipping. ClinicalTrials.gov Identifier: NCT02310906. Updated Oct 19, 2020. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/results/NCT02310906.
81. Commissioner O of the. FDA grants accelerated approval to first drug for Duchenne muscular dystrophy. FDA. Published March 24, 2020. Accessed August 21, 2022. hDuchenne Muscular Dystrophy Amenable to Exon 51-Skipping Treatment. clinicaltrials.gov; 2022. Accessed Au-gust 18, 2022. https://clinicaltrials.gov/ct2/show/NCT04004065
109. National Center of Neurology and Psychiatry, Japan. Exploratory study of NS-065/NCNP-01 in Duchenne muscular dystrophy. ClinicalTri-als.gov Identifier: NCT02081625; Updated Feb 26, 2020. Accessed Mar 2, 2022. https://clinicaltrialsttps://www.fda.gov/news-events/press-announcements/fda-grants-accelerated-approval-first-drug-duchenne-muscular- dys-trophy
82. Duchenne Drug Development Pipeline. Parent Project Muscular Dystrophy. Accessed August 21, 2022. https://www.parentprojectmd.org/duchenne-drug-development-pipeline/
83. Sarepta Therapeutics Provides Update on SRP-5051 for the Treatment of Duchenne Muscular Dystrophy | Sarepta Therapeutics, Inc. Ac-cessed August 22, 2022. https://investorrelations.sarepta.com/news-releases/news-release-details/sarepta-therapeutics- pro-vides-update-srp-5051-treatment-duchenne
84. Sarepta Therapeutics, Inc. An Open-Label Extension Study for Patients With Duchenne Muscular Dystrophy Who Participated in Studies of SRP-5051. clinicaltrials.gov; 2021. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT03675126
85. VYONDYS 53. Prescribing information. Sarepta Therapeutics Inc.; 2019. Accessed Mar 2, 2022. http://www.accessdata.fda.gov/drugsatfda_docs/label/2019/211970s000lbl.pdf.
86. NS Pharma Inc. Long-term use of viltolarsen in boys with Duchenne muscular dystrophy in clinical practice (VILT-502). ClinicalTrials.gov Identifier: NCT04687020. Updated Nov 22, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT04687020.
87. VILTEPSO. Prescribing information. NS Pharma; 2020. Accessed Mar 2, 2022. http://www.accessdata.fda.gov/drugsatfda_docs/label/2020/212154s000lbl.pdf.
88. FDA approves targeted treatment for rare Duchenne muscular dystrophy mutation. U.S. Food and Drug Administration. News release. Feb 25, 2021. Accessed Mar 1, 2022. http://www.fda.gov/news-events/press-announcements/fda-approves-targeted-treatment-rare-duchenne-muscular-dystrophy-mutation-0.
89. Sarepta Therapeutics Inc. A double-blind, placebo-controlled, multi-center study with an open-label extension to evaluate the efficacy and safety of SRP-4045 and SRP-4053 in patients with Duchenne muscular dystrophy. Clinicaltrials.gov Identifier: NCT02500381. Updated Aug 19, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02500381.
90. AMONDYS 45. Prescribing information. Sarepta Therapeutics Inc.; 2021. Accessed Feb 22, 2022. http://www.accessdata.fda.gov/drugsatfda_docs/label/2021/213026lbl.pdf.
91. Finkel RS et al. Phase 2a study of ataluren-mediated dystrophin production in patients with nonsense mutation Duchenne muscular dys-trophy. PLoS ONE. 2013;8(12):e81302. doi:10.1371/journal.pone.0081302.
92. PTC Therapeutics. A phase 2 study of PTC124 as an oral treatment for nonsense-mutation-mediated Duchenne muscular dystrophy. Clini-calTrials.gov Identifier: NCT00264888. Updated Jan 14, 2009. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT00264888.
93. PTC Therapeutics. A phase 2B efficacy and safety study of PTC124 in subjects with nonsense-mutation-mediated Duchenne and Becker muscular dystrophy. ClinicalTrials.gov Identifier: NCT00592553. Updated Apr 7, 2020. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT00592553.
94. PTC Therapeutics. A phase 3 efficacy and safety study of ataluren in patients with nonsense mutation dystrophinopathy. ClinicalTrials.gov Identifier: NCT01826487. Updated Aug 4, 2020. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT01826487.
95. Bushby K et al; PTC124-GD-007-DMD Study Group. Ataluren treatment of patients with nonsense mutation dystrophinopathy. Muscle Nerve. 2014 Oct;50(4):477-87. doi:10.1002/mus.24332.
96. Solid Biosciences LLC. A randomized, controlled, open-label, single-ascending dose, phase I/II study to investigate the safety and tolerabil-ity, and efficacy of intravenous SGT-001 in male adolescents and children with Duchenne muscular dystrophy. ClinicalTrials.gov Identifier: NCT03368742. Updated Aug 24, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03368742.
97. Solid Biosciences reports 1.5-year data from patients in the ongoing IGNITE DMD phase I/II clinical trial of SGT-001. Press release. Solid Biosciences. Sep 27, 2021. Accessed Mar 2, 2022. http://www.solidbio.com/about/media/press-releases/solid-biosciences-reports-1-5-year-data-from-patients-in-the-ongoing-ignite-dmd-phase-i-ii-clinical-trial-of-sgt-001.
98. Potter RA et al. Dose-escalation study of systemically delivered rAAVrh74.MHCK7.microdystrophin in the mdx mouse model of Duchenne muscular dystrophy. Hum Gene Ther. 2021 Apr;32(7-8):375-89. doi:10.1089/hum.2019.255.
99. Sarepta Therapeutics, Inc. A Phase 3 Multinational, Randomized, Double-Blind, Placebo- Controlled Systemic Gene Delivery Study to Evaluate the Safety and Efficacy of SRP-9001 in Patients With Duchenne Muscular Dystrophy (EMBARK). clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT05096221
100. Pfizer. A PHASE 3, MULTICENTER, RANDOMIZED, DOUBLE-BLIND, PLACEBO CONTROLLED STUDY TO EVALUATE THE SAFETY AND EFFICACY OF PF 06939926 FOR THE TREATMENT OF DUCHENNE MUSCULAR DYSTROPHY. clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT04281485
101. Pfizer. A phase 1B multicenter open-label, single ascending dose study to evaluate the safety and tolerability of PF-06939926 in ambula-tory and non-ambulatory subjects with Duchenne muscular dystrophy. ClinicalTrials.gov Identifier: NCT03362502. Updated Mar 2, 2022. Ac-cessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03362502.
102. MS MW. Phase 3 CIFFREO DMD Gene Therapy Trial Slated to Begin in June in US. Accessed August 21, 2022. https://musculardystrophynews.com/news/phase-3-trial-of-pfizers-gene-therapy- expected-to-open-in-us-in-june/
103. SRP-9001. Parent Project Muscular Dystrophy. Accessed August 22, 2022. https://www.parentprojectmd.org/drug-development-pipeline/srp-9001-micro-dystrophin-gene- transfer/
104. Sarepta Therapeutics’ Investigational Gene Therapy SRP-9001 for Duchenne Muscular Dystrophy Demonstrates Significant Functional Improvements Across Multiple Studies | Sarepta Therapeutics, Inc. Accessed August 22, 2022. https://investorrelations.sarepta.com/news-releases/news-release- details/sarepta-therapeutics-investigational-gene-therapy-srp-9001
105. Sarepta Therapeutics, Inc. An Open-Label Safety, Tolerability, and Efficacy Study of Eteplirsen in Patients With Duchenne Muscular Dys-trophy Who Have Completed Study 4658-102.clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT03985878
106. Sarepta Therapeutics, Inc. An Open-Label Safety, Tolerability, and Pharmacokinetics Study of Eteplirsen in Young Patients With Duchenne Mus-cular Dystrophy Amenable to Exon 51 Skipping. clinicaltrials.gov; 2021. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT03218995
107.Sarepta Therapeutics, Inc. A Randomized, Double-Blind, Dose Finding and Comparison Study of the Safety and Efficacy of a High Dose of Eteplirsen, Preceded by an Open-Label Dose Escalation, in Patients With Duchenne Muscular Dystrophy With Deletion Mutations Amenable to Exon 51 Skipping. clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT03992430
108. Sarepta Therapeutics, Inc. A Phase 2, Two-Part, Multiple-Ascending-Dose Study of SRP-5051 for Dose Determination, Then Dose Ex-pansion, in Patients With .gov/ct2/show/NCT02081625.
110. NS Pharma Inc. A phase II, dose finding study to assess the safety, tolerability, pharmacokinetics, and pharmacodynamics of NS-065/NCNP-01 in boys with Duchenne muscular dystrophy (DMD). ClinicalTrials.gov Identifier: NCT02740972. Updated Dec 7, 2021. Ac-cessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT02740972.
111. NS Pharma Inc. A phase II, open-label, extension study to assess the safety and efficacy of NS-065/NCNP-01 in boys with Duchenne muscular dystrophy (DMD). ClinicalTrials.gov Identifier: NCT03167255. Updated Nov 24, 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03167255.
112. NS Pharma Inc. A phase 2 open label study to assess the safety, tolerability, and efficacy of viltolarsen in ambulant and non-ambulant boys with Duchenne muscular dystrophy (DMD) compared with natural history controls. ClinicalTrials.gov Identifier: NCT04956289. Updated Feb 1, 2022. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT04956289.
113. NS Pharma Inc. A phase 3 randomized, double-blind, placebo-controlled, multi-center study to assess the efficacy and safety of viltolarsen in ambulant boys with Duchenne muscular dystrophy (DMD). ClinicalTrials.gov Identifier: NCT04060199. Updated Nov 16, 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT04060199.
114. NS Pharma Inc. A phase 3, multi-center, open-label extension study to assess the safety and efficacy of viltolarsen in ambulant boys with Duchenne muscular dystrophy (DMD). ClinicalTrials.gov Identifier: NCT04768062. Updated Nov 16, 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT04768062.
115. Sarepta Therapeutics Inc. A randomized, double-blind, placebo-controlled, dose-titration, safety, tolerability, and pharmacokinetics study followed by an open-label safety and efficacy evaluation of SRP-4045 in advanced-stage patients with Duchenne muscular dystrophy amena-ble to exon 45 skipping. ClinicalTrials.gov Identifier: NCT02530905. Updated May 17, 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT02530905.
116. Sarepta Therapeutics Inc. Long-term, open-label extension study for patients with Duchenne muscular dystrophy enrolled in clinical trials evaluating casimersen or golodirsen. ClinicalTrials.gov Identifier: NCT03532542. Updated Dec 20, 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03532542.
117. PTC Therapeutics. A phase 2 study of the safety, pharmacokinetics, and pharmacodynamics of ataluren (PTC124®) in patients aged ≥2 to <5 years old with nonsense mutation dystrophinopathy. ClinicalTrials.gov Identifier: NCT02819557. Updated Aug 28, 2020. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT02819557.
118. PTC Therapeutics. Phase 2, non-interventional, clinical study to assess dystrophin levels in subjects with nonsense mutation Duchenne muscular dystrophy who have been treated with ataluren for ≥ 9 months. ClinicalTrials.gov Identifier: NCT03796637. Updated Apr 10, 2020. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03796637.
119. PTC Therapeutics. An Open-Label Study Evaluating the Safety and Pharmacokinetics of Ataluren in Children From ≥6 Months to <2 Years of Age With Nonsense Mutation Duchenne Muscular Dystrophy. clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT04336826 120. PTC Therapeutics. An open-label study for previously treated ataluren (PTC124®) pa-tients with nonsense mutation dystrophinopathy. ClinicalTrials.gov Identifier: NCT01557400. Updated Nov 25, 2020. Accessed Feb 21, 2022. https://clinicaltrials.gov/ct2/show/NCT01557400.
121. PTC Therapeutics. An open-label, safety study for ataluren (PTC124) patients with nonsense mutation dystrophinopathy. ClinicalTrials.gov Identifier: NCT01247207. Updated Feb 16, 2022. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT01247207.
122. PTC Therapeutics. A phase 3, randomized, double-blind, placebo-controlled efficacy and safety study of ataluren in patients with non-sense mutation Duchenne muscular dystrophy and open-label extension. ClinicalTrials.gov Identifier: NCT03179631. Updated Feb 8, 2022. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03179631.
123. Sarepta Therapeutics, Inc. An Open-Label, Systemic Gene Delivery Study Using Commercial Process Material to Evaluate the Safety of and Expression From SRP-9001 in Subjects With Duchenne Muscular Dystrophy (ENDEAVOR). clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT04626674
124. Sarepta Therapeutics, Inc. Systemic Gene Delivery Phase I/IIa Clinical Trial for Duchenne Muscular Dystrophy Using RAA-Vrh74.MHCK7.Micro-Dystrophin (MicroDys-IV-001). clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT03375164
125. Sarepta Therapeutics Inc. A multicenter, randomized, double-blind, placebo-controlled trial for Duchenne muscular dystrophy using SRP-9001. ClinicalTrials.gov Identifier: NCT03769116. Updated Dec 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03769116.
126. Hoffmann-La Roche. A Two-Part, Seamless, Multi-Center, Randomized, Placebo-Controlled, Double-Blind Study to Investigate the Safety, Tolerability, Pharmacokinetics, Pharmacodynamics and Efficacy of RO7204239 in Combination With Risdiplam (RO7034067) in Ambulant Pa-tients With Spinal Muscular Atrophy. clinicaltrials.gov; 2022. Accessed September 1, 2022. https://clinicaltrials.gov/ct2/show/NCT05115110
Neuromuscular diseases (NMDs) are a broad classification of heterogeneous groups of disorders characterized by progressive muscle weakness resulting from muscle or nerve dysfunction.1 Diagnosis is based on symptoms and a full medical history, as well as on muscle and imaging tests (including electromyography, nerve-conduction studies, magnetic resonance imaging, muscle biopsy, and blood tests) to confirm or rule out specific NMDs.2 Early diagnosis of NMDs can be difficult because symptoms overlap with those of many other diseases.
Although individually, NMDs are rare, collectively, they affect approximately 250,000 people in the United States. Disease types vary in regard to cause, symptoms, prevalence, age of onset, progression, and severity. Functional impairment from any NMD can lead to lifelong morbidities and shortened life expectancy.1,3
Treatment options for NMDs are limited; most target symptoms, not disease progression. Although there is a need for safe and effective gene-based therapies for NMDs, there are challenges to developing and delivering such treatments that have impeded clinical success. These include a lack of understanding about disease pathology and drug targets, limited animal model systems, and few reliable biomarkers that are predictive of therapeutic success.4,5
Notwithstanding that challenges remain, our understanding of gene expression in NMDs has greatly advanced in the past few decades. This progress has translated into promising results in the gene-therapy field – thereby setting the stage for therapeutic approaches that use novel gene-delivery and gene-manipulation tools.6 These novel approaches include nonviral strategies, such as antisense oligonucleotides (ASOs), and viral-based strategies, such as adeno-associated virus (AAV)-mediated gene silencing and AAV-mediated gene delivery.
In this article, we highlight advancements in the clinical development of gene-based therapies for NMDs. We focus on amyotrophic lateral sclerosis (ALS), spinal muscular atrophy (SMA), and Duchenne muscular dystrophy (DMD) because of recent clinical successes in developing such therapies.1,6,7 We also catalog completed and ongoing clinical trials for ALS, SMA, and DMD (Tables 1-3).
Amyotrophic lateral sclerosis
ALS is caused by progressive degeneration of upper- and lower-motor neurons, which eventually leads to respiratory failure and death 3 to 5 years after disease onset.7-9 There are two subtypes: Familial ALS (10% of cases) and sporadic ALS (90% of cases). Commonly mutated ALS-associated genes6,8 are:
- Superoxide dismutase type 1 (SOD1).
- Chromosome 9 open reading frame 72 (C9orf72).
- Transactive response DNA-binding protein 43 (TARDBP).
- Fused in sarcoma (FUS).
SOD1-targeted therapy is being studied, with early evidence of clinical success. Mutations in SOD1 account for 10% to 20% of familial ALS cases and 1% to 2% of sporadic ALS cases.6,10 10 Mutations in C9orf72 account for 25 to 40% of familial ALS cases and 7% of sporadic ALS cases.8,9,11 Mutations in TARDBP account for 3% of familial ALS cases and 2% of sporadic cases.12 Mutations in FUS account for 4% of familial ALS cases and 1% of sporadic cases. Overall, these mutant proteins can trigger neurotoxicity, thus inducing motor-neuron death.6,10
Treatment of ALS
Two treatments for ALS are Food and Drug Administration approved: riluzole (Rilutek), approved in 1995, and edaravone (Radicava), approved in 2017.
Riluzole is an oral anti-excitotoxic glutamate antagonist.11 Approval of riluzole was based on the results of two studies that demonstrated a 2- to 3-month survival benefit.10,14 For patients who have difficulty swallowing, an oral suspension (Tiglutik, approved in 2018) and an oral film (Exservan, approved in 2019) are available.
Edaravone is a free-radical scavenger that decreases oxidative stress and is administered intravenously (IV).9,13,14 Findings from clinical trials suggest functional improvement or slower decline in function for some patients.
Although these two agents demonstrate modest therapeutic benefit, neither reverses progression of disease.10,14
Gene-based therapy for ALS
Many non-viral strategies, including antisense oligonucleotide (ASO), monoclonal antibodies, reverse transcriptase inhibitors, and HGF gene replacement therapy are used as therapeutic approaches to SOD1, C9orf72, and FUS gene mutations in ALS patients, and are being evaluated in clinical studies14,15 (Table 113-17).
Tofersen, also known as BIIB067, is an investigational ASO, administered by intrathecal (IT) injection, that binds to SOD1 mRNA, thus reducing its protein levels.16 Tofersen was evaluated in the VALOR phase 3 study (ClinicalTrials.gov Identifier: NCT02623699), a three-part randomized, double-blind, placebo-controlled trial: single ascending dose (Part A), multiple ascending dose (B), and fixed dose (C).10 In Parts A and B, 48 participants received five IT injections of tofersen or placebo over 12 weeks and were followed for an additional 12 weeks. Reduction in SOD1 protein production and neurofilament level in cerebrospinal fluid (CSF) (a potential biomarker of motor-neuron degeneration) was observed, which determined the fixed-dose for Part C.16,17
Part C examined the efficacy, safety and tolerability, pharmacokinetics (PK), and pharmacodynamics (PD) of tofersen, compared with placebo, in adults with ALS who had a confirmed SOD1 mutation.17 A total of 108 participants were enrolled; 60 were identified as “faster-progressing”; 48, as “slower-progressing.”18 The primary endpoint of Part C was change from baseline to Week 28 on the Revised ALS Functional Rating Scale (ALSFRS-R) total score. (ALSFRS-R measures overall clinical effect; the score ranges from 0 [no function] to 4 [full function].17)
Tofersen failed to meet the primary efficacy outcome because statistically significant findings were lacking in the faster-progressing population, as measured by joint-rank analysis (difference of 1.2 on the ALSFRS-R score; P = .97). However, trends favoring tofersen were observed across key secondary clinical outcome measures18:
- Change from baseline in CSF SOD1 protein concentration.17 Percent reduction in the total SOD1 protein level was much higher in the tofersen-treated group than in the control group (38% more than controls in the faster-progressing population; 26% more than controls in the slower-progressing population).18
- Change from baseline in neurofilament light-chain concentration in plasma.17,18 Percent reduction in the level of neurofilament light chain was also observed to be higher in the tofersen-treated group than in the control group (67% more than controls in the faster-progressing population and 48% more than controls in the slower-progressing population).18
Because of these encouraging results, VALOR participants were moved to the ongoing open-label extension trial of tofersen (ClinicalTri-als.gov Identifier: NCT03070119), in which both groups were treated with the active agent.
These data suggest that early tofersen treatment might slow decline in faster-progressing patients and stabilize clinical function in slower-progressing patients.18,19 Overall, most adverse events (AEs) in the trial among patients receiving active treatment were of mild or moderate severity, and were largely consistent with either disease progression or lumbar puncture–related complications.18
Because data from VALOR suggested potential benefit from tofersen, the ATLAS trial (ClinicalTrials.gov Identifier: NCT04856982) is investigating the clinical value of presymptomatic treatment and the optimal timing of initiation of therapy.20,21 ATLAS is a phase 3, randomized, placebo-controlled trial that examines the clinical efficacy, safety, and tolerability of tofersen in presymptomatic adult carriers of SOD1 mutation who have an elevated neurofilament light-chain concentration.21 ATLAS will also evaluate the efficacy of tofersen when initiated before, rather than after, ALS manifests clinically. Enrollment is still open for this trial.20,21
Latozinemab, also known as AL001, is a first-in-class monoclonal antibody, administered by IV infusion, that elevates levels of progranulin, a key regulator of the immune activity and lysosomal function in the brain.22,23 Latozinemab limits progranulin endocytosis and degradation by sortilin inhibition.22 Progranulin gene mutations can reduce progranulin expression (by 50 to 70 percent reduction), which may cause neuro-degeneration due to abnormal accumulation of TAR-DNA-binding protein 43 (TDP-43) in the brain cells.22,24 TDP-43 pathology has also been shown to be associated with C9orf72 mutations.23 Although the mechanism is not fully understood, the role of progranulin deficiency in TDP-43 pathology is believed to be associated with neurodegenerative diseases like ALS.11,23,24,43 Previous animal models of chronic neurodegenera-tion have demonstrated how increased progranulin levels can be protective against TDP-43 pathology, increasing neuronal development and survival, thus potentially slowing disease progression.23,24,43 Currently, latozinemab is being investigated in a randomized, double-blind, placebo-controlled, multicenter phase 2 trial (ClinicalTrials.gov Identifier: NCT05053035). Approximately, 45 C90rf72-associated ALS participants (≥ 18 years of age) will receive latozinemab or placebo infusions every 4 weeks (for 24 weeks). Study endpoints include safety, tolerability, PK, PD, as well as plasma, and CSF progranulin levels.25 In previous studies, latozinemab demonstrated encouraging results in frontotemporal dementia (FTD) patients who carry a progranulin mutation. Because FTD was revealed to have significant genetic overlap with ALS, there is disease-modifying potential for latozinemab in ALS patients.23,24
TPN-101 is a nucleoside analog reverse transcriptase inhibitor, administered orally, that was originally developed for human immunodeficiency virus (HIV) treatment. However, due to recent findings suggesting retrotransposon activity contributing to neurodegeneration in TDP-43 mediated diseases, including ALS and FTD, TNP-101 is being repurposed.26 The safety and tolerability of TNP-101 are currently being evaluated in C9orf72-associated ALS and FTD patients (≥ 18 years of age). The study is a randomized, double-blind, placebo-controlled paral-lel-group phase 2a trial (ClinicalTrials.gov Identifier: NCT04993755) The study includes a screening period of 6 weeks, double-blind treatment period of 24 weeks, an open-label treatment period of 24 weeks, and 4 weeks of the post-treatment follow-up visit. Study endpoints include the incidence and severity of spontaneously reported treatment-emergent adverse events (TEAEs) associated with TNP-101 and placebo for a to-tal of 48 weeks.27
ION363 is an investigational ASO, administered by IT injection, that selectively targets one of the FUS mutations (p.P525L), which is responsible for earlier disease onset and rapid ALS progression.28,29 The clinical efficacy of ION363, specifically in clinical function and survival is being assessed in FUS-associated ALS patients (≥ 12 years of age). This randomized phase 3 study (ClinicalTrials.gov Identifier: NCT04768972) includes two parts; part 1 will consist of participants receiving a multi-dose regimen (1 dose every 4-12 weeks) of ION363 or placebo for 61 weeks followed by an open-label extension treatment period in part 2, which will consist of participants receiving ION363 (every 12 weeks) for 85 weeks. The primary endpoint of the study is the change from baseline to day 505 in functional impairment, using ALS Functional Rating Scale-Revised (ALSFRS-R). This measures functional disease severity, specifically in bulbar function, gross motor skills, fine motor skills, and respiratory. The score for all 12 questions can range from 0 (no function) to 4 (full function) with a total possible score of 48.30
Engensis, also known as VM202, is a non-viral gene therapy, administered by intramuscular (IM) injection, that uses a plasmid to deliver the hepatocyte growth factor (HGF) gene to promote HGF protein production. The HGF protein plays a role in angiogenesis, the previous of muscle atrophy, and the promotion of neuronal survival and growth. Based on preclinical studies, increasing HGF protein production has been shown to reduce neurodegeneration, thus potentially halting or slowing ALS progression.31 Currently, the safety of engensis is being evaluated in ALS patients (18-80 years of age) in the REViVALS phase 2a (ClinicalTrials.gov Identifier: NCT04632225)/2b (ClinicalTrial.gov Identifier: NCT05176093).32,33 The ReViVALS trial is a double-blind, randomized, placebo-controlled, multi-center study. The phase 2a study endpoints include the incidence of TEAEs, treatment-emergent serious adverse events (TESAEs), injection site reactions, and clinically significant labor-atory values post-treatment (engensis vs placebo group) for 180 days.33 A phase 2b study will evaluate the long-term safety of engensis for an additional 6 months. Study endpoints include the incidence of AEs, changes from baseline in ALSFRS-R scores to evaluate improvement in muscle function, changes from baseline in quality of life using the ALS patient assessment questionnaire, time to all-cause mortality compared to placebo, etc.32
Spinal muscular atrophy
SMA is a hereditary lower motor-neuron disease caused (in 95% of cases) by deletions or, less commonly, by mutations of the survival motor neuron 1 (SMN1) gene on chromosome 5q13 that encodes the SMN protein.6 Reduction in expression of the SMN protein causes motor neurons to degenerate.36-38 Because of a large inverted duplication in chromosome 5q, two variants of SMN (SMN1 and SMN2) exist on each allele. The paralog gene, SMN2, also produces the SMN protein – although at a lower level (10% to 20% of total SMN protein production) than SMN1 does.
A single nucleotide substitution in SMN2 alters splicing and suppresses transcription of exon 7, resulting in a shortened mRNA strand that yields a truncated SMN protein product.6,37,39 SMA is classified based on age of onset and maximum motor abilities achieved, ranging from the most severe (Type 0) to mildest (Type 4) disease.36,40 Because SMA patients lack functional SMN1 (due to polymorphisms), disease severity is determined by copy numbers of SMN2.6,39
Gene-based therapy for SMA
Three FDA-approved SMN treatments demonstrate clinically meaningful benefit in SMA: SMN2-targeting nusinersen [Spinraza] and risdiplam [Evrysdi], and SMN1-targeting onasemnogene abeparvovec-xioi [Zolgensma]38 Additional approaches to SMA treatment are through SMN-independent therapies, which target muscle and nerve function. Research has strongly suggested that combined SMA therapies, specifically approved SMN-targeted and investigational SMN-independent treatments, such as GYM329 (also known as RO7204239) may be the best strategy to treat all ages, stages, and types of SMA.41 (Table 226-41).
Agents that modulate SMN2. Nusinersen, approved by the FDA in 2016, was the first treatment indicated for all SMA types in pediatric and adult patients.42 The agent is an ASO that targets exon 7 of SMN2, thus stabilizing transcription. Inclusion of exon 7 increases SMN protein production, improving motor function.6,38 Nusinersen is a lifelong treatment that requires IT administration every 4 months because it cannot cross the blood-brain barrier.38,43
Pivotal clinical studies that led to approval of nusinersen include CHERISH (ClinicalTrial.gov Identifier: NCT02292537) and ENDEAR (ClinicalTrial.gov Identifier: NCT02193074) studies.
CHERISH was a phase 3, randomized, double-blind, sham procedure–controlled trial that examined the clinical efficacy and safety of nusinersen in 126 participants with later-onset SMA (2-12 years of age). The primary endpoint was the change from baseline using the Hammersmith Functional Motor Scale Expanded (HFMSE) at 15 months. HFMSE looks at 33 activities to assess improvement in motor function. The study met the primary efficacy outcome, demonstrating statistically significant (P = .0000001) improvement in overall motor function. The nusinersen group showed a 3.9-point increase in the HFMSE score from baseline, which indicates improvement, compared with a 1.0-point decline from baseline in the control group.46,47
ENDEAR was also a randomized, double-blind, sham procedure–controlled phase 3 trial, which investigated the efficacy and safety of nusinersen in 121 participants with early-onset SMA Type 1 (≤ 210 days of age). Coprimary endpoints were:
- Percentage of motor milestones responders, as determined using Section 2 of the Hammersmith Infant Neurological Examination–Part 2.
- Event-free survival (that is, avoidance of combined endpoint of death or permanent ventilation).
ENDEAR met the first primary efficacy outcome, demonstrating statistically significant (P < .0001) improvement in motor milestones (head control, rolling, independent sitting, and standing). By 13 months of age, approximately 51% of nusinersen-treated participants showed improvement, compared with none in the control group.46,47
The second primary endpoint was also met, with a statistically significant (P = .005) 47% decrease in mortality or permanent ventilation use.46-48
The NURTURE (ClinicalTrial.gov Identifier: NCT02386553) study is also investigating the efficacy and safety of nusinersen. An ongoing, open-label, supportive phase 2 trial, NURTURE is evaluating the efficacy and safety of multiple doses of nusinersen in 25 presymptomatic SMA patients (≤ 6 weeks of age). The primary endpoint of this study is time to death or respiratory intervention.49 Interim results demonstrate that 100% of presymptomatic infants are functioning without respiratory intervention after median follow-up of 2.9 years.46-48
Although nusinersen has been shown to be generally safe in clinical studies, development of lumbar puncture–related complications, as well as the need for sedation during IT administration, might affect treatment tolerability in some patients.39
Risdiplam was approved by the FDA in 2020 as the first orally administered small-molecule treatment of SMA (for patients ≤ 2 months of age).52 Risdiplam is a SMN2 splicing modifier, binding to the 5’ splice site of intron 7 and exonic splicing enhancer 2 in exon 7 of SMN2 pre-mRNA. This alternative splicing increases efficiency in SMN2 gene transcription, thus increasing SMN protein production in motor-neuron cells.36 An important advantage of risdiplam is the convenience of oral administration: A large percentage of SMA patients (that is, those with Type 2 disease) have severe scoliosis, which can further complicate therapy or deter patients from using a treatment that is administered through the IT route.40
FDA approval of risdiplam was based on clinical data from two pivotal studies, FIREFISH (ClinicalTrial.gov Identifier: NCT02913482) and SUNFISH (ClinicalTrial.gov Identifier: NCT02908685).53-54
FIREFISH is an open-label, phase 2/3 ongoing trial in infants (1-7 months of age) with SMA Type 1. The study comprises two parts; Part 1 determined the dose of risdiplam used in Part 2, which assessed the efficacy and safety of risdiplam for 24 months. The primary endpoint was the percentage of infants sitting without support for 5 seconds after 12 months of treatment using the gross motor scale of the Bayley Scales of Infant and Toddler Development–Third Edition. A statistically significant (P < .0001) therapeutic benefit was observed in motor milestones. Approximately 29% of infants achieved the motor milestone of independent sitting for 5 seconds, which had not been observed in the natural history of SMA.53-55
SUNFISH is an ongoing randomized, double-blind, placebo-controlled trial of risdiplam in adult and pediatric patients with SMA Types 2 and 3 (2-25 years old). This phase 2/3 study comprises two parts: Part 1 determined the dose (for 12 weeks) to be used for confirmatory Part 2 (for 12 to 24 months). The primary endpoint was the change from baseline on the 32-item Motor Function Measure at 12 months. The study met its primary endpoint, demonstrating statistically significant (P = .0156) improvement in motor function scores, with a 1.36-point increase in the risdiplam group, compared with a 0.19-point decrease in the control group.54,55
Ongoing risdiplam clinical trials also include JEWELFISH (ClinicalTrial.gov Identifier: NCT03032172) and RAINBOW (ClinicalTrial.gov Identifier: NCT03779334).56-57 JEWELFISH is an open-label, phase 2 trial assessing the safety of risdiplam in patients (6 months to 60 years old) who received prior treatment. The study has completed recruitment; results are pending.56 RAINBOW is an ongoing, open-label, single-arm, phase 2 trial, evaluating the clinical efficacy and safety of risdiplam in SMA-presymptomatic newborns (≤ 6 weeks old). The study is open for enrollment.57 Overall, interim results for JEWELFISH and RAINBOW appear promising.
In addition, combined SMA therapies, specifically risdiplam and GYM329 are currently being investigated to address the underlying cause and symptoms of SMA concurrently.58 GYM329, is an investigational anti-myostatin antibody, selectively binding preforms of myostatin - pro-myostatin and latent myostatin, thus improving muscle mass and strength for SMA patients.59 The safety and efficacy of GYM329 in combination with risdiplam is currently being investigated in 180 ambulant participants with SMA (2-10 years of age) in the MANATEE (ClinicalTrial.gov Identifier: NCT05115110) phase 2/3 trial. The MANATEE study is a two-part, seamless, randomized, placebo-controlled, double-blind trial. Part 1 will assess the safety of the combination treatment in approximately 36 participants; participants will receive both GYM329 (every 4 weeks) by subcutaneous (SC) injection into the abdomen and risdiplam (once per day) for 24 weeks followed by a 72-week open-label treatment period. 54,58 The outcome measures include the incidence of AEs, percentage change from baseline in the contractile area of skeletal muscle (in dominant thigh and calf), change from baseline in RHS total score, and incidence of change from baseline in serum concentration (total myostatin, free latent myostatin, and mature myostatin) etc.54 Part 2 will be conducted on 144 participants, specifically assessing the efficacy and safety of the optimal dose of GYM329 selected from Part 1 (combined with risdiplam) for 72 weeks. Once the treatment period is completed in either part, participants can partake in a 2-year open-label extension period.54,58 Other outcome measures include change from baseline in lean muscle mass (assessed by full body dual-energy X- ray absorptiometry (DXA) scan), in time taken to walk/run 10 meters (measured by RHS), in time taken to rise from the floor (measured by RHS), etc.54 Overall, this combination treatment has the potential to further improve SMA patient outcomes and will be further investigated in other patient populations (including non-ambulant patients and a broader age range) in the future.58
An agent that alters SMN1 expression. Onasemnogene abeparvovec-xioi, FDA approved in 2019, was the first gene-replacement therapy indicated for treating SMA in children ≤ 2 years old.60 Treatment utilizes an AAV vector type 9 (AAV9) to deliver a functional copy of SMN1 into target motor-neuron cells, thus increasing SMN protein production and improving motor function. This AAV serotype is ideal because it crosses the blood-brain barrier. Treatment is administered as a one-time IV fusion.38,39,43
FDA approval was based on the STR1VE (ClinicalTrial.gov Identifier: NCT03306277) phase 3 study and START (ClinicalTrial.gov Identifier: NCT02122952) phase 1 study.61,62 START was the first trial to investigate the safety and efficacy of onasemnogene abeparvovec-xioi in SMA Type 1 infants (< 6 months old). Results demonstrated remarkable clinical benefit, including 100% permanent ventilation-free survival and a 92% (11 of 12 patients) rate of improvement in motor function. Improvement in development milestones was also observed: 92% (11 of 12 patients) could sit without support for 5 seconds and 75% (9 of 12) could sit without support for 30 seconds.14,61,63
The efficacy of onasemnogene abeparvovec-xioi seen in STR1VE was consistent with what was observed in START. STRIVE, a phase 3 open-label, single-dose trial, examined treatment efficacy and safety in 22 symptomatic infants (< 6 months old) with SMA Type 1 (one or two SMN2 copies). The primary endpoint was 30 seconds of independent sitting and event-free survival. Patients were followed for as long as 18 months. Treatment showed statistically significant (P < .0001) improvement in motor milestone development and event-free survival, which had not been observed in SMA Type 1 historically. Approximately 59% (13 of 22 patients) could sit independently for 30 seconds at 18 months of age. At 14 months of age, 91% (20 of 22 patients) were alive and achieved independence from ventilatory support.34,35,53
Although many clinical studies suggest that onasemnogene abeparvovec-xioi can slow disease progression, the benefits and risks of long-term effects are still unknown. A 15-year observational study is investigating the long-term therapeutic effects and potential complications of onasemnogene abeparvovec-xioi. Participants in START were invited to enroll in this long-term follow-up study (ClinicalTrial.gov Identifier: NCT04042025).66-67
Duchenne muscular dystrophy
DMD is the most common muscular dystrophy of childhood. With an X-linked pattern of inheritance, DMD is seen mostly in young males (1 in every 3,500 male births).38,39,73 DMD is caused by mutation of the dystrophin encoding gene, or DMD, on the X chromosome. Deletion of one or more exons of DMD prevents production of the dystrophin protein, which leads to muscle degeneration.38,39,43 Common DMD deletion hotspots are exon 51 (20% of cases), exon 53 (13% of cases), exon 44 (11% of cases), and exon 45 (12% of cases).74 Nonsense mutations, which account for another 10% of DMD cases, occur when premature termination codons are found in the DMD gene. Those mutations yield truncated dystrophin protein products.39,66
Therapy for DMD
There are many therapeutic options for DMD, including deflazacort (Emflaza), FDA approved in 2017, which has been shown to reduce inflammation and immune system activity in DMD patients (≥ 5 years old). Deflazacort is a corticosteroid prodrug; its active metabolite acts on the glucocorticoid receptor to exert anti-inflammatory and immunosuppressive effects. Studies have shown that muscle strength scores over 6-12 months and average time to loss of ambulation numerically favored deflazacort over placebo.74,75
Gene-based therapy for DMD
Mutation-specific therapeutic approaches, such as exon skipping and nonsense suppression, have shown promise for the treatment of DMD (Table 358-79):
- ASO-mediated exon skipping allows one or more exons to be omitted from the mutated DMD mRNA.74,75 Effective FDA-approved ASOs include golodirsen [Vyondys 53], viltolarsen [Viltepso], and casimersen [Amondys 45].74
- An example of therapeutic suppression of nonsense mutations is ataluren [Translarna], an investigational agent that can promote premature termination codon read-through in DMD patients.66
Another potential treatment approach is through the use of AAV gene transfer to treat DMD. However, because DMD is too large for the AAV vector (packaging size, 5.0 kb), microdystrophin genes (3.5-4 kb, are used as an alternative to fit into a single AAV vector.39,76
Exon skipping targeting exon 51. Eteplirsen, approved in 2016, is indicated for the treatment of DMD patients with the confirmed DMD gene mutation that is amenable to exon 51 skipping. Eteplirsen binds to exon 51 of dystrophin pre-mRNA, causing it to be skipped, thus, restoring the reading frame in patients with DMD gene mutation amenable to exon 51 skipping. This exclusion promotes dystrophin production. Though the dystrophin protein is still functional, it is shortened.38,77 Treatment is administered IV, once a week (over 35-60 minutes). Eteplirsen’s accelerated approval was based on 3 clinical studies (ClinicalTrial.gov Identifier: NCT01396239, NCT01540409, and NCT00844597.) 78-81 The data demonstrated an increased expression of dystrophin in skeletal muscles in some DMD patients treated with eteplirsen. Though the clinical benefit of eteplirsen (including improved motor function) was not established, it was concluded by the FDA that the data were reasonably likely to predict clinical benefit. Continued approval for this indication may depend on the verification of a clinical benefit in confirmatory trials. Ongoing clinical trials include (ClinicalTrial.gov Identifier: NCT03992430 (MIS51ON), NCT03218995, and NCT03218995).77,81,82
Vesleteplirsen, is an investigational agent that is designed for DMD patients who are amendable to exon 51 skip-ping. The mechanism of action of vesleteplirsen appears to be similar to that of eteplirsen.83 The ongoing MOMENTUM (ClinicalTrial.gov Identifier: NCT04004065) phase 2 trial is assessing the safety and tolerability of vesleteplirsen at multiple-ascending dose levels (administered via IV infusion) in 60 participants (7-21 years of age). The study consists of two parts; participants receive escalating dose levels of vesleteplirsen (every 4 weeks) for 72 weeks during part A and participants receive the selected doses from part A (every 4 weeks) for 2 years during part B. Study endpoints include the number of AEs (up to 75 weeks) and the change from baseline to week 28 in dystrophin protein level. 84 Serious AEs of reversible hypomagnesemia were observed in part B, and as a result, the study protocol was amended to include magnesium supplementation and monitoring of magnesium levels.83
Exon skipping targeting exon 53. Golodirsen, FDA approved in 2019, is indicated for the treatment of DMD in patients who have a confirmed DMD mutation that is amenable to exon 53 skipping. The mechanism of action is similar to eteplirsen, however, golodirsen is designed to bind to exon 53.38,39 Treatment is administered by IV infusion over 35-60 minutes.
Approval of golodirsen was based primarily on a two-part, phase 1/2 clinical trial (ClinicalTrial.gov Identifier: NCT02310906). Part 1 was a randomized, placebo-controlled, dose-titration study that assessed multiple-dose efficacy in 12 DMD male patients, 6 to 15 years old, with deletions that were amenable to exon 53 skipping.
Part 2 was an open-label trial in 12 DMD patients from Part 1 of the trial plus 13 newly enrolled male DMD patients who were also amenable to exon 53 skipping and who had not already received treatment. Primary endpoints were change from baseline in total distance walked during the 6-minute walk test at Week 144 and dystrophin protein levels (measured by western blot testing) at Week 48. A statistically significant increase in the mean dystrophin level was observed, from a baseline 0.10% mean dystrophin level to a 1.02% mean dystrophin level after 48 weeks of treatment (P < .001). Common reported adverse events associated with golodirsen were headache, fever, abdominal pain, rash, and dermatitis. Renal toxicity was observed in preclinical studies of golodirsen but not in clinical studies.80,85
Viltolarsen, approved in 2020, is also indicated for the treatment of DMD in patients with deletions amenable to exon 53 skipping. The mechanism of action and administration (IV infusion over 60 minutes) are similar to that of golodirsen.
Approval of viltolarsen was based on two phase 2 clinical trials (ClinicalTrial.gov Identifier: NCT02740972 and NCT03167255) in a total of 32 patients. NCT02740972 was a randomized, double-blind, placebo-controlled, dose-finding study that evaluated the clinical efficacy of viltolarsen in 16 male DMD patients (4-9 years old) for 24 weeks.
NCT03167255 was an open-label study that evaluated the safety and tolerability of viltolarsen in DMD male patients (5-18 years old) for 192 weeks. The efficacy endpoint was the change in dystrophin production from baseline after 24 weeks of treatment. A statistically significant increase in the mean dystrophin level was observed, from a 0.6% mean dystrophin level at baseline to a 5.9% mean dystrophin level at Week 25 (P = .01). The most common adverse events observed were upper respiratory tract infection, cough, fever, and injection-site reaction.86-87
Exon skipping targeting exon 45. Casimersen was approved in 2021 for the treatment of DMD in patients with deletions amenable to exon 45 skipping.88 Treatment is administered by IV infusion over 30-60 minutes. Approval was based on an increase in dystrophin production in skeletal muscle in treated patients. Clinical benefit was reported in interim results from the ESSENCE (ClinicalTrial.gov Identifier: NCT02500381) study, an ongoing double-blind, placebo-controlled phase 3 trial that is evaluating the efficacy of casimersen, compared with placebo, in male participants (6-13 years old) for 48 weeks. Efficacy is based on the change from baseline dystrophin intensity level, determined by immunohistochemistry, at Week 48.
Interim results from ESSENCE show a statistically significant increase in dystrophin production in the casimersen group, from a 0.9% mean dystrophin level at baseline to a 1.7% mean dystrophin level at Week 48 (P = .004); in the control group, a 0.54% mean dystrophin level at baseline increased to a 0.76% mean dystrophin level at Week 48 (P = .09). Common adverse events have included respiratory tract infection, headache, arthralgia, fever, and oropharyngeal pain. Renal toxicity was observed in preclinical data but not in clinical studies.60,84
Targeting nonsense mutations. Ataluren is an investigational, orally administered nonsense mutation suppression therapy (through the read-through of stop codons).37 Early clinical evidence supporting the use of ataluren in DMD was seen in an open-label, dose-ranging, phase 2a study (ClinicalTrial.gov Identifier: NCT00264888) in male DMD patients (≥ 5 years old) caused by nonsense mutation. The study demonstrated a modest (61% ) increase in dystrophin expression in 23 of 38 patients after 28 days of treatment.37,91,92
However, a phase 2b randomized, double-blind, placebo-controlled trial (ClinicalTrial.gov Identifier: NCT00592553) and a subsequent confirmatory ACT DMD phase 3 study (ClinicalTrial.gov Identifier: NCT01826487) did not meet their primary endpoint of improvement in ambulation after 48 weeks as measured by the 6-minute walk test.37,93,94 In ACT DMD, approximately 74% of the ataluren group did not experience disease progression, compared with 56% of the control group (P = 0386), measured by a change in the 6-minute walk test, which assessed ambulatory decline.37,95
Based on limited data showing that ataluren is effective and well tolerated, the European Medicines Agency has given conditional approval for clinical use of the drug in Europe. However, ataluren was rejected by the FDA as a candidate therapy for DMD in the United States.22 Late-stage clinical studies of ataluren are ongoing in the United States.
AAV gene transfer with microdystrophin. Limitations on traditional gene-replacement therapy prompted exploration of gene-editing strategies for treating DMD, including using AAV-based vectors to transfer microdystrophin, an engineered version of DMD, into target muscles.43 The microdystrophin gene is designed to produce a functional, truncated form of dystrophin, thus improving muscular function.
There are 3 ongoing investigational microdystrophin gene therapies that are in clinical development (ClinicalTrial.gov Identifier: NCT03368742 (IGNITE DMD), NCT04281485 (CIFFREO), and NCT05096221 (EMBARK)).38,82
IGNITE DMD is a phase 1/2 randomized, controlled, single-ascending dose trial evaluating the safety and efficacy of a SGT-001, single IV infusion of AAV9 vector containing a microdystrophin construct in DMD patients (4-17 years old) for 12 months. At the conclusion of the trial, treatment and control groups will be followed for 5 years. The primary efficacy endpoint is the change from baseline in microdystrophin protein production in muscle-biopsy material, using western blot testing.96 Long-term interim data on biopsy findings from three patients demonstrated clinical evidence of durable microdystrophin protein expression after 2 years of treatment.96,97
The CIFFREO trial will assess the safety and efficacy of the PF-06939926 microdystrophin gene therapy, an investigational AAV9 containing microdystrophin, in approximately 99 ambulatory DMD patients (4-7 years of age). The study is a randomized, double-blind, placebo-controlled, multicenter phase 3 trial. The primary efficacy end-point is the change from baseline in the North Star Ambulatory Assessment (NSAA), which measures gross motor function. This will be assessed at 52 weeks; all study participants will be followed for a total of 5 years post-treatment.98,99,100 Due to unexpected patient death (in a non-ambulatory cohort) in the phase 1b (in a non-ambulatory cohort) in the phase 1b (ClinicalTrial.gov Identifier: (NCT03362502) trial, microdystrophin gene therapy was immediately placed on clinical hold.101,102 The amended study protocol required that all participants undergo one week of in-hospital observation after receiving treatment.102
The EMBARK study is a global, randomized, double-blind, placebo-controlled, phase 3 trial that is evaluating the safety and efficacy of SRP-9001, which is a rAAVrh74.MHCK7.microdystrophin gene therapy. The AAV vector (rAAVrh74) contains the microdystrophin construct, driven by the skeletal and cardiac muscle–specific promoter, MHCK7.98,99 In the EMBARK study, approximately 120 participants with DMD (4-7 years of age) will be enrolled. The primary efficacy endpoint includes the change from baseline to week 52 in the NSAA total score.99 Based on SRP-9001, data demonstrating consistent statistically significant functional improvements in NSAA total scores and timed function tests (after one-year post- treatment) in DMD patients from previous studies and an integrated analysis from multiple studies (ClinicalTrial.gov Identifier: NCT03375164, NCT03769116, and NCT04626674), the ongoing EMBARK has great promise.103,104
Challenges ahead, but advancements realized
Novel gene-based therapies show significant potential for transforming the treatment of NMDs. The complex pathologies of NMDs have been a huge challenge to disease management in an area once considered unremediable by gene-based therapy. However, advancements in precision medicine – specifically, gene-delivery systems (for example, AAV9 and AAVrh74 vectors) combined with gene modification strategies (ASOs and AAV-mediated silencing) – have the potential to, first, revolutionize standards of care for sporadic and inherited NMDs and, second, significantly reduce disease burden.6
What will be determined to be the “best” therapeutic approach will, likely, vary from NMD to NMD; further investigation is required to determine which agents offer optimal clinical efficacy and safety profiles.43 Furthermore, the key to therapeutic success will continue to be early detection and diagnosis – first, by better understanding disease pathology and drug targets and, second, by validation of reliable biomarkers that are predictive of therapeutic benefit.4,5
To sum up, development challenges remain, but therapeutic approaches to ALS, SMA, and DMD that utilize novel gene-delivery and gene-manipulation tools show great promise.
Ms. Yewhalashet is a student in the masters of business and science program, with a concentration in healthcare economics, at Keck Graduate Institute Henry E. Riggs School of Applied Life Sciences, Claremont, Calif. Dr. Davis is professor of practice in clinical and regulatory affairs, Keck Graduate Institute Henry E. Riggs School of Applied Life Sciences.
References
1. Aitken M et al. Understanding neuromuscular disease care. IQVIA [Internet]. Oct 30, 2018. Accessed Mar 1, 2022. https://www.iqvia.com/insights/the-iqvia-institute/reports/understanding-neuromuscular-disease-care.
2. National Institute of Neurological Disorders and Stroke. Neurological diagnostic tests and procedures fact sheet. Updated Nov 15, 2021. Ac-cessed Mar 1, 2022. http://www.ninds.nih.gov/Disorders/Patient-Caregiver-Education/Fact-Sheets/Neurological-Diagnostic-Tests-and-Procedures-Fact.
3. Deenen JCW et al. The epidemiology of neuromuscular disorders: A comprehensive overview of the literature. J Neuromuscul Dis. 2015;2(1):73-85.
4. Cavazzoni P. The path forward: Advancing treatments and cures for neurodegenerative diseases. U.S. Food and Drug Administration. Jul 29, 2021. Accessed Mar 1, 2022. http://www.fda.gov/news-events/congressional-testimony/path-forward-advancing-treatments-and-cures-neurodegenerative-diseases-07292021.
5. Martier R, Konstantinova P. Gene therapy for neurodegenerative diseases: Slowing down the ticking clock. Front Neurosci. 2020 Sep 18;14:580179. doi: 10.3389/fnins.2020.580179.
6. Sun J, Roy S. Gene-based therapies for neurodegenerative diseases. Nat Neurosci. 2021 Mar;24(3):297-311. doi:10.1038/s41593-020-00778-1.
7. Amado DA, Davidson BL. Gene therapy for ALS: A review. Mol Ther. 2021 Dec 1;29(12):3345-58. doi:10.1016/j.ymthe.2021.04.008.
8. Yun Y, Ha Y. CRISPR/Cas9-mediated gene correction to understand ALS. Int J Mol Sci. 2020;21(11):3801. doi:10.3390/ijms21113801.
9. National Institute of Neurological Disorders and Stroke. Amyotrophic lateral sclerosis (ALS) fact sheet. Updated Nov 15, 2021. Accessed Mar 1, 2022. http://www.ninds.nih.gov/Disorders/Patient-Caregiver-Education/Fact-Sheets/Amyotrophic-Lateral-Sclerosis-ALS-Fact-Sheet.
10. Cappella M et al. Gene therapy for ALS – A perspective. Int J Mol Sci. 2019;20(18):4388. doi:10.3390/ijms20184388.
11. Abramzon YA, Fratta P, Traynor BJ, Chia R. The Overlapping Genetics of Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Front Neurosci. 2020;14. Accessed August 18, 2022. https://www.frontiersin.org/articles/10.3389/fnins.2020.00042
12. Giannini M, Bayona-Feliu A, Sproviero D, Barroso SI, Cereda C, Aguilera A. TDP-43 mutations link Amyotrophic Lateral Sclerosis with R-loop homeostasis and R loop-mediated DNA damage. PLOS Genet. 2020;16(12):e1009260. doi:10.1371/journal.pgen.1009260
13. FDA-approved drugs for treating ALS. The ALS Association [Internet]. Accessed Mar 1, 2022. http://www.als.org/navigating-als/living-with-als/fda-approved-drugs.
14. Jensen TL et al. Current and future prospects for gene therapy for rare genetic diseases affecting the brain and spinal cord. Front Mol Neurosci. 2021 Oct 6;14:695937. doi:10.3389/fnmol.2021.695937.
15. ALS Gene Targeted Therapies. The ALS Association. Accessed August 22, 2022. https://www.als.org/understanding-als/who-gets-als/genetic-testing/als-gene-targeted-therapies
16. Tofersen for ALS clears phase 1/2 trial, now in phase 3. Advances in Motion. Massachusetts General Hospital [Internet]. Sep 30, 2020. Accessed Mar 1, 2022. https://advances.massgeneral.org/neuro/journal.aspx?id=1699.17. Biogen. A study to evaluate the efficacy, safety, tol-erability, pharmacokinetics, and pharmacodynamics of BIIB067 administered to adult subjects with amyotrophic lateral sclerosis and confirmed superoxide dismutase 1 mutation. ClinicalTrials.gov Identifier: NCT02623699. Updated Jul 25, 2021. Accessed Feb 17, 2022. https://clinicaltrials.gov/ct2/show/NCT02623699.
18. Biogen. Biogen announces topline results from the tofersen phase 3 study and its open-label Extension in SOD1-ALS. Press release. Oct 17, 2021. Accessed Mar 1, 2022. https://investors.biogen.com/news-releases/news-release-details/biogen-announces-topline-results-tofersen-phase-3-study-and-its.
19. Biogen. An extension study to assess the long-term safety, tolerability, pharmacokinetics, and effect on disease progression of BIIB067 ad-ministered to previously treated adults with amyotrophic lateral sclerosis caused by superoxide dismutase 1 mutation. ClinicalTrials.gov Identi-fier: NCT03070119. Updated Sep 10, 2021. Accessed Feb 17, 2022. https://clinicaltrials.gov/ct2/show/NCT03070119.
20. MS MW. #AANAM – ATLAS Trial to Assess Tofersen in Presymptomatic SOD1 ALS. Accessed February 19, 2022. https://alsnewstoday.com/news-posts/2021/04/23/aanam-atlas-clinical-trial- tofersen-presymptomatic-sod1-als-patients/
21.Biogen. A phase 3 randomized, placebo-controlled trial with a longitudinal natural history run-in and open-label extension to evaluate BIIB067 initiated in clinically presymptomatic adults with a confirmed superoxide dismutase 1 mutation. ClinicalTrials.gov Identifier: NCT04856982. Updated Feb 18, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT04856982.
22. Latozinemab | ALZFORUM. Accessed August 19, 2022. https://www.alzforum.org/therapeutics/latozinemab
23. Alector Presents AL001 (latozinemab) Data from the FTD-C9orf72 Cohort of the INFRONT-2 Phase 2 Clinical Trial | Alector. Accessed August 18, 2022. https://investors.alector.com/news- releas-es/news-release-details/alector-presents-al001-latozinemab-data-ftd-c9orf72-cohort/
24. Alector Announces First Participant Dosed in Phase 2 Study Evaluating AL001 in Amyotrophic Lateral Sclerosis (ALS) | Alector. Accessed August 18, 2022. https://investors.alector.com/news- releases/news-release-details/lector-announces-first-participant-dosed-phase-2-study-0/ 25. A Phase 2 Study to Evaluate AL001 in C9orf72-Associated ALS - Full Text View - ClinicalTrials.gov. Accessed August 19, 2022. https://clinicaltrials.gov/ct2/show/NCT05053035
26.TPN-101 | ALZFORUM. Accessed August 19, 2022. https://www.alzforum.org/therapeutics/tpn- 101
27. Transposon Therapeutics, Inc. A Phase 2a Study of TPN-101 in Patients With Amyotrophic Lateral Sclerosis (ALS) and/or Frontotemporal Dementia (FTD) Associated With Hexanucleotide Repeat Expansion in the C9orf72 Gene (C9ORF72 ALS/FTD). clinicaltrials.gov; 2022. Ac-cessed August 17, 2022. https://clinicaltrials.gov/ct2/show/NCT04993755
28. Kerk SY, Bai Y, Smith J, et al. Homozygous ALS-linked FUS P525L mutations cell- autonomously perturb transcriptome profile and chem-oreceptor signaling in human iPSC microglia. Stem Cell Rep. 2022;17(3):678-692. doi:10.1016/j.stemcr.2022.01.004
29. ION363 | ALZFORUM. Accessed August 19, 2022. https://www.alzforum.org/therapeutics/ion363 30. Ionis Pharmaceuticals, Inc. A Phase 1-3 Study to Evaluate the Efficacy, Safety, Pharmacokinetics and Pharmacodynamics of Intrathecally Administered ION363 in Amyo-trophic Lateral Sclerosis Patients With Fused in Sarcoma Mutations (FUS-ALS). clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT04768972
31. PhD LF. Engensis (VM202) - ALS News Today. Accessed August 19, 2022. https://alsnewstoday.com/vm202/
32. Helixmith Co., Ltd. A 6-Month Extension Study Following Protocol VMALS-002-2 (A Phase 2a, Double-Blind, Randomized, Place-bo-Controlled, Multicenter Study to Assess the Safety of Engensis in Participants With Amyotrophic Lateral Sclerosis). clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT05176093 33. Safety of Engensis in Participants With Amyotrophic Lateral Sclerosis - Full Text View - ClinicalTrials.gov. Accessed August 19, 2022. https://clinicaltrials.gov/ct2/show/NCT04632225
34. Biogen. A phase 1, safety, tolerability, and distribution study of a microdose of radiolabeled BIIB067 co-administered with BIIB067 to healthy adults. ClinicalTrials.gov Identifier: NCT03764488. Updated Jul 19, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03764488.
35. Ionis Pharmaceuticals Inc. A phase 1, double-blind, placebo-controlled, dose-escalation study of the safety, tolerability, and pharmacokinet-ics of ISIS 333611 administered intrathecally to patients with familial amyotrophic lateral sclerosis due to superoxide dismutase 1 gene muta-tions. ClinicalTrials.gov Identifier: NCT01041222. Updated Apr 13, 2012. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT01041222.
36. Messina S, Sframeli M. New treatments in spinal muscular atrophy: Positive results and new challenges. J Clin Med. 2020;9(7):2222. doi:10.3390/jcm9072222.
37. Scoto M et al. Genetic therapies for inherited neuromuscular disorders. Lancet Child Adolesc Health. 2018 Aug;2(8):600-9. doi:10.1016/S2352-4642(18)30140-8.
38. Abreu NJ, Waldrop MA. Overview of gene therapy in spinal muscular atrophy and Duchenne muscular dystrophy. Pediatr Pulmonol. 2021 Apr;56(4):710-20. doi:10.1002/ppul.25055.
39. Brandsema J, Cappa R. Genetically targeted therapies for inherited neuromuscular disorders. Practical Neurology [Internet]. Jul/Aug 2021:69-73. Accessed Mar 1, 2022. https://practicalneurology.com/articles/2021-july-aug/genetically-targeted-therapies-for-inherited-neuromuscular-disorders/pdf.
40. Ojala KS et al. In search of a cure: The development of therapeutics to alter the progression of spinal muscular atrophy. Brain Sci. 2021;11(2):194. doi:10.3390/brainsci11020194.
41. McCall S. Cure SMA Releases Updated Drug Pipeline. Cure SMA. Published December 13, 2021. Accessed August 21, 2022. https://www.curesma.org/cure-sma-releases-updated-drug-pipeline- 2021/ 42. FDA approves first drug for spinal muscular atrophy. U.S. Food and Drug Administration. News release. Dec 23, 2016. Accessed Mar 1, 2022. http://www.fda.gov/news-events/press-announcements/fda-approves-first-drug-spinal-muscular-atrophy.43. Kirschner J. Postnatal gene therapy for neuromuscular diseases – Opportunities and limitations. J Perinat Med. 2021 Sep;49(8):1011-5. doi:10.1515/jpm-2021-0435.
43. Terryn J, Verfaillie CM, Van Damme P. Tweaking Progranulin Expression: Therapeutic Avenues and Opportunities. Front Mol Neurosci. 2021;14. Accessed September 4, 2022. https://www.frontiersin.org/articles/10.3389/fnmol.2021.71303144.
44. Biogen. A phase 3, randomized, double-blind, sham-procedure controlled study to assess the clinical efficacy and safety of ISIS 396443 administered intrathecally in patients with later-onset spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT02292537. Updated Feb 17, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/study/NCT02292537.
45. Why Spinraza/later-onset studies. SPINRAZA® (nusinersen) [Internet]. Accessed Mar 1, 2022. www.spinraza.com/en_us/home/why-spinraza/later-onset-studies.html#scroll-tabs.
46. Biogen. A Phase 3, Randomized, Double-Blind, Sham-Procedure Controlled Study to Assess the Clinical Efficacy and Safety of ISIS 396443 Administered Intrathecally in Patients With Infantile- Onset Spinal Muscular Atrophy. clinicaltrials.gov; 2021. Accessed February 10, 2022. https://clinicaltrials.gov/ct2/show/results/NCT02193074
47. Early-onset SMA (Type 1) | SPINRAZA® (nusinersen). Accessed Mar 1, 2022. https://www.spinraza-hcp.com/en_us/home/why-spinraza/about-spinraza.html.
48. Finkel RS et al; ENDEAR Study Group. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N Engl J Med. 2017;377(18):1723-32. doi: 10.1056/NEJMoa1702752.
49. Biogen. An open-label study to assess the efficacy, safety, tolerability, and pharmacokinetics of multiple doses of ISIS 396443 delivered intrathecally to subjects with genetically diagnosed and presymptomatic spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT02386553. Updated Nov 18, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02386553.
50. De Vivo DC et al; NURTURE Study Group. Nusinersen initiated in infants during the presymptomatic stage of spinal muscular atrophy: In-terim efficacy and safety results from the phase 2 NURTURE study. Neuromuscul Disord. 2019 Nov;29(11):842-56. doi:10.1016/j.nmd.2019.09.007.
51. Why Spinraza/presymptomatic study. SPINRAZA® (nusinersen) [Internet]. Accessed Feb 22, 2022. www.spinraza.com/en_us/home/why-spinraza/presymptomatic-study.html#scroll-tabs.
52. FDA approves oral treatment for spinal muscular atrophy. U.S. Food and Drug Administration. News release. Aug 7, 2020. Accessed Mar 1, 2022. http://www.fda.gov/news-events/press-announcements/fda-approves-oral-treatment-spinal-muscular-atrophy.
53. Hoffmann-La Roche. A two-part seamless, open-label, multicenter study to investigate the safety, tolerability, pharmacokinetics, pharmaco-dynamics and efficacy of risdiplam (RO7034067) in infants with type 1 spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT02913482. Updated Jan 21, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02913482.
54. Hoffmann-La Roche. A two-part seamless, multi-center randomized, placebo-controlled, double-blind study to investigate the safety, tolera-bility, pharmacokinetics, pharmacodynamics and efficacy of risdiplam (RO7034067) in type 2 and 3 spinal muscular atrophy patients. Clinical-Trials.gov Identifier: NCT02908685. Updated Dec 28, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02908685.
55. Genentech. Genentech’s risdiplam shows significant improvement in survival and motor milestones in infants with type 1 spinal muscular atrophy (SMA). Press release. Apr 27, 2020. Accessed Mar 1, 2022. http://www.gene.com/media/press-releases/14847/2020-04-27/genentechs-risdiplam-shows-significant-i
56. Hoffmann-La Roche. An open-label study to investigate the safety, tolerability, and pharmacokinetics/pharmacodynamics of risdiplam (RO7034067) in adult and pediatric patients with spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT03032172. Updated Jan 27, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03032172.
57. Hoffmann-La Roche. An open-label study of risdiplam in infants with genetically diagnosed and presymptomatic spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT03779334. Updated Jan 27, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03779334.
58. McCall S. Update on Genentech/Roche Initiation of MANATEE Clinical Study. Cure SMA. Published October 20, 2021. Accessed August 20, 2022. https://www.curesma.org/update-on- genentech-roche-initiation-of-manatee-clinical-study/
59. Abati E, Manini A, Comi GP, Corti S. Inhibition of myostatin and related signaling pathways for the treatment of muscle atrophy in motor neuron diseases. Cell Mol Life Sci. 2022;79(7):374. doi:10.1007/s00018-022-04408-w
60. FDA approves innovative gene therapy to treat pediatric patients with spinal muscular atrophy, a rare disease and leading genetic cause of infant mortality. U.S. Food and Drug Administration. News release. May 24, 2019. Accessed Mar 1, 2022. http://www.fda.gov/news-events/press-announcements/fda-approves-innovative-gene-therapy-treat-pediatric-patients-spinal-muscular-atrophy-rare-disease.
61. Novartis Gene Therapies. Phase I gene transfer clinical trial for spinal muscular atrophy type 1 delivering AVXS-101. ClinicalTrials.gov Identifier: NCT02122952. Updated Jun 14, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02122952.
62. Novartis Gene Therapies. Phase 3, open-label, single-arm, single-dose gene replacement therapy clinical trial for patients with spinal mus-cular atrophy type 1 with one or two SMN2 copies delivering AVXS-101 by intravenous infusion. ClinicalTrials.gov Identifier: NCT03306277. Updated Jun 14, 2021. Accessed Feb 21, 2022. https://clinicaltrials.gov/ct2/show/NCT03306277.
63. Mendell JR et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N Engl J Med. 2017;377(18):1713-22. doi:10.1056/NEJMoa1706198.
64. Symptomatic study results. ZOLGENSMA [Internet]. Updated Nov 2021. Accessed Mar 1, 2022. Error! Hyperlink reference not valid..
65. Novartis Gene Therapies. A global study of a single, one-time dose of AVXS-101 delivered to infants with genetically diagnosed and pre-symptomatic spinal muscular atrophy with multiple copies of SMN2. ClinicalTrials.gov Identifier: NCT03505099. Updated Jan 1, 2022. Ac-cessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03505099.
66. Chiu W et al. Current genetics and potential gene-targeting therapeutics for neuromuscular diseases. Int J Mol Sci. 2020 Dec;21(24):9589. doi:10.3390/ijms21249589.
67. Novartis Gene Therapies. A long-term follow-up study of patients in the clinical trials for spinal muscular atrophy receiving AVXS-101. Clini-calTrials.gov Identifier: NCT04042025. Updated Jun 9, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT04042025.
68. Novartis Gene Therapies. Phase 3, open-label, single-arm, single-dose gene replacement therapy clinical trial for patients with spinal mus-cular atrophy type 1 with one or two SMN2 copies delivering AVXS-101 by intravenous infusion. ClinicalTrials.gov Identifier: NCT0383718. Up-dated Jan 11, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03837184.
69. Biogen. An open-label, dose escalation study to assess the safety, tolerability and dose-range finding of multiple doses of ISIS 396443 de-livered intrathecally to patients with spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT01703988. Updated Apr 13, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT01703988.
70. Biogen. A study to assess the efficacy, safety, tolerability, and pharmacokinetics of multiple doses of ISIS 396443 delivered intrathecally to patients with infantile-onset spinal muscular atrophy. ClinicalTrials.gov Identifier: NCT01839656. Updated Feb 17, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT01839656.
71. Biogen. An open-label extension study for patients with spinal muscular atrophy who previously participated in investigational studies of ISIS 396443. ClinicalTrials.gov Identifier: NCT02594124. Updated Nov 15, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02594124.
72. Biogen. Escalating dose and randomized, controlled study of nusinersen (BIIB058) in participants with spinal muscular atrophy. ClinicalTri-als.gov Identifier: NCT04089566. Updated Feb 24, 2022. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT04089566.
73. National Center for Advancing Translational Sciences. Duchenne muscular dystrophy. Genetic and Rare Diseases Information Center. Up-dated Nov 2, 2020. Accessed Mar 1, 2022. https://rarediseases.info.nih.gov/diseases/6291/duchenne-muscular-dystrophy.
74. Matsuo M. Antisense oligonucleotide-mediated exon-skipping therapies: Precision medicine spreading from Duchenne muscular dystrophy. JMA J. 2021 Jul 15;4(3):232-40. doi:10.31662/jmaj.2021-0019.
75. FDA approves drug to treat Duchenne muscular dystrophy. U.S. Food and Drug Administration. News release. Feb 9, 2017. Accessed Mar 1, 2022. http://www.fda.gov/news-events/press-announcements/fda-approves-drug-treat-duchenne-muscular-dystrophy.74.
76. Duan D. Dystrophin gene replacement and gene repair therapy for Duchenne muscular dystrophy in 2016: An interview. Hum Gene Ther Clin Dev. 2016 Mar;27(1):9-18. doi:10.1089/humc.2016.001.
77. EXONDYS 51®. Parent Project Muscular Dystrophy. Accessed August 21, 2022. https://www.parentprojectmd.org/drug-development-pipeline/exondys-51/
78. Sarepta Therapeutics, Inc. A Randomized, Double-Blind, Placebo-Controlled, Multiple Dose Efficacy, Safety, Tolerability and Pharmacoki-netics Study of AVI-4658(Eteplirsen),in the Treatment of Ambulant Subjects With Duchenne Muscular Dystrophy. clinicaltrials.gov; 2020. Ac-cessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT01396239
79. Sarepta Therapeutics, Inc. Clinical Study to Assess the Safety Fo AVI-4658 in Subjects With Duchenne Muscular Dystrophy Due to a Frame-Shift Mutation Amenable to Correction by Skipping Exon 51. clinicaltrials.gov; 2015. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/study/NCT00844597
80. Sarepta Therapeutics, Inc. A 2-part, randomized, double-blind, placebo-controlled, dose-titration, safety, tolerability, and pharmacokinetics study (Part 1) followed by an open-label efficacy and safety evaluation (Part 2) of SRP-4053 in patients with Duchenne muscular dystrophy amenable to exon 53 skipping. ClinicalTrials.gov Identifier: NCT02310906. Updated Oct 19, 2020. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/results/NCT02310906.
81. Commissioner O of the. FDA grants accelerated approval to first drug for Duchenne muscular dystrophy. FDA. Published March 24, 2020. Accessed August 21, 2022. hDuchenne Muscular Dystrophy Amenable to Exon 51-Skipping Treatment. clinicaltrials.gov; 2022. Accessed Au-gust 18, 2022. https://clinicaltrials.gov/ct2/show/NCT04004065
109. National Center of Neurology and Psychiatry, Japan. Exploratory study of NS-065/NCNP-01 in Duchenne muscular dystrophy. ClinicalTri-als.gov Identifier: NCT02081625; Updated Feb 26, 2020. Accessed Mar 2, 2022. https://clinicaltrialsttps://www.fda.gov/news-events/press-announcements/fda-grants-accelerated-approval-first-drug-duchenne-muscular- dys-trophy
82. Duchenne Drug Development Pipeline. Parent Project Muscular Dystrophy. Accessed August 21, 2022. https://www.parentprojectmd.org/duchenne-drug-development-pipeline/
83. Sarepta Therapeutics Provides Update on SRP-5051 for the Treatment of Duchenne Muscular Dystrophy | Sarepta Therapeutics, Inc. Ac-cessed August 22, 2022. https://investorrelations.sarepta.com/news-releases/news-release-details/sarepta-therapeutics- pro-vides-update-srp-5051-treatment-duchenne
84. Sarepta Therapeutics, Inc. An Open-Label Extension Study for Patients With Duchenne Muscular Dystrophy Who Participated in Studies of SRP-5051. clinicaltrials.gov; 2021. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT03675126
85. VYONDYS 53. Prescribing information. Sarepta Therapeutics Inc.; 2019. Accessed Mar 2, 2022. http://www.accessdata.fda.gov/drugsatfda_docs/label/2019/211970s000lbl.pdf.
86. NS Pharma Inc. Long-term use of viltolarsen in boys with Duchenne muscular dystrophy in clinical practice (VILT-502). ClinicalTrials.gov Identifier: NCT04687020. Updated Nov 22, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT04687020.
87. VILTEPSO. Prescribing information. NS Pharma; 2020. Accessed Mar 2, 2022. http://www.accessdata.fda.gov/drugsatfda_docs/label/2020/212154s000lbl.pdf.
88. FDA approves targeted treatment for rare Duchenne muscular dystrophy mutation. U.S. Food and Drug Administration. News release. Feb 25, 2021. Accessed Mar 1, 2022. http://www.fda.gov/news-events/press-announcements/fda-approves-targeted-treatment-rare-duchenne-muscular-dystrophy-mutation-0.
89. Sarepta Therapeutics Inc. A double-blind, placebo-controlled, multi-center study with an open-label extension to evaluate the efficacy and safety of SRP-4045 and SRP-4053 in patients with Duchenne muscular dystrophy. Clinicaltrials.gov Identifier: NCT02500381. Updated Aug 19, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT02500381.
90. AMONDYS 45. Prescribing information. Sarepta Therapeutics Inc.; 2021. Accessed Feb 22, 2022. http://www.accessdata.fda.gov/drugsatfda_docs/label/2021/213026lbl.pdf.
91. Finkel RS et al. Phase 2a study of ataluren-mediated dystrophin production in patients with nonsense mutation Duchenne muscular dys-trophy. PLoS ONE. 2013;8(12):e81302. doi:10.1371/journal.pone.0081302.
92. PTC Therapeutics. A phase 2 study of PTC124 as an oral treatment for nonsense-mutation-mediated Duchenne muscular dystrophy. Clini-calTrials.gov Identifier: NCT00264888. Updated Jan 14, 2009. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT00264888.
93. PTC Therapeutics. A phase 2B efficacy and safety study of PTC124 in subjects with nonsense-mutation-mediated Duchenne and Becker muscular dystrophy. ClinicalTrials.gov Identifier: NCT00592553. Updated Apr 7, 2020. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT00592553.
94. PTC Therapeutics. A phase 3 efficacy and safety study of ataluren in patients with nonsense mutation dystrophinopathy. ClinicalTrials.gov Identifier: NCT01826487. Updated Aug 4, 2020. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT01826487.
95. Bushby K et al; PTC124-GD-007-DMD Study Group. Ataluren treatment of patients with nonsense mutation dystrophinopathy. Muscle Nerve. 2014 Oct;50(4):477-87. doi:10.1002/mus.24332.
96. Solid Biosciences LLC. A randomized, controlled, open-label, single-ascending dose, phase I/II study to investigate the safety and tolerabil-ity, and efficacy of intravenous SGT-001 in male adolescents and children with Duchenne muscular dystrophy. ClinicalTrials.gov Identifier: NCT03368742. Updated Aug 24, 2021. Accessed Mar 1, 2022. https://clinicaltrials.gov/ct2/show/NCT03368742.
97. Solid Biosciences reports 1.5-year data from patients in the ongoing IGNITE DMD phase I/II clinical trial of SGT-001. Press release. Solid Biosciences. Sep 27, 2021. Accessed Mar 2, 2022. http://www.solidbio.com/about/media/press-releases/solid-biosciences-reports-1-5-year-data-from-patients-in-the-ongoing-ignite-dmd-phase-i-ii-clinical-trial-of-sgt-001.
98. Potter RA et al. Dose-escalation study of systemically delivered rAAVrh74.MHCK7.microdystrophin in the mdx mouse model of Duchenne muscular dystrophy. Hum Gene Ther. 2021 Apr;32(7-8):375-89. doi:10.1089/hum.2019.255.
99. Sarepta Therapeutics, Inc. A Phase 3 Multinational, Randomized, Double-Blind, Placebo- Controlled Systemic Gene Delivery Study to Evaluate the Safety and Efficacy of SRP-9001 in Patients With Duchenne Muscular Dystrophy (EMBARK). clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT05096221
100. Pfizer. A PHASE 3, MULTICENTER, RANDOMIZED, DOUBLE-BLIND, PLACEBO CONTROLLED STUDY TO EVALUATE THE SAFETY AND EFFICACY OF PF 06939926 FOR THE TREATMENT OF DUCHENNE MUSCULAR DYSTROPHY. clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT04281485
101. Pfizer. A phase 1B multicenter open-label, single ascending dose study to evaluate the safety and tolerability of PF-06939926 in ambula-tory and non-ambulatory subjects with Duchenne muscular dystrophy. ClinicalTrials.gov Identifier: NCT03362502. Updated Mar 2, 2022. Ac-cessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03362502.
102. MS MW. Phase 3 CIFFREO DMD Gene Therapy Trial Slated to Begin in June in US. Accessed August 21, 2022. https://musculardystrophynews.com/news/phase-3-trial-of-pfizers-gene-therapy- expected-to-open-in-us-in-june/
103. SRP-9001. Parent Project Muscular Dystrophy. Accessed August 22, 2022. https://www.parentprojectmd.org/drug-development-pipeline/srp-9001-micro-dystrophin-gene- transfer/
104. Sarepta Therapeutics’ Investigational Gene Therapy SRP-9001 for Duchenne Muscular Dystrophy Demonstrates Significant Functional Improvements Across Multiple Studies | Sarepta Therapeutics, Inc. Accessed August 22, 2022. https://investorrelations.sarepta.com/news-releases/news-release- details/sarepta-therapeutics-investigational-gene-therapy-srp-9001
105. Sarepta Therapeutics, Inc. An Open-Label Safety, Tolerability, and Efficacy Study of Eteplirsen in Patients With Duchenne Muscular Dys-trophy Who Have Completed Study 4658-102.clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT03985878
106. Sarepta Therapeutics, Inc. An Open-Label Safety, Tolerability, and Pharmacokinetics Study of Eteplirsen in Young Patients With Duchenne Mus-cular Dystrophy Amenable to Exon 51 Skipping. clinicaltrials.gov; 2021. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT03218995
107.Sarepta Therapeutics, Inc. A Randomized, Double-Blind, Dose Finding and Comparison Study of the Safety and Efficacy of a High Dose of Eteplirsen, Preceded by an Open-Label Dose Escalation, in Patients With Duchenne Muscular Dystrophy With Deletion Mutations Amenable to Exon 51 Skipping. clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT03992430
108. Sarepta Therapeutics, Inc. A Phase 2, Two-Part, Multiple-Ascending-Dose Study of SRP-5051 for Dose Determination, Then Dose Ex-pansion, in Patients With .gov/ct2/show/NCT02081625.
110. NS Pharma Inc. A phase II, dose finding study to assess the safety, tolerability, pharmacokinetics, and pharmacodynamics of NS-065/NCNP-01 in boys with Duchenne muscular dystrophy (DMD). ClinicalTrials.gov Identifier: NCT02740972. Updated Dec 7, 2021. Ac-cessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT02740972.
111. NS Pharma Inc. A phase II, open-label, extension study to assess the safety and efficacy of NS-065/NCNP-01 in boys with Duchenne muscular dystrophy (DMD). ClinicalTrials.gov Identifier: NCT03167255. Updated Nov 24, 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03167255.
112. NS Pharma Inc. A phase 2 open label study to assess the safety, tolerability, and efficacy of viltolarsen in ambulant and non-ambulant boys with Duchenne muscular dystrophy (DMD) compared with natural history controls. ClinicalTrials.gov Identifier: NCT04956289. Updated Feb 1, 2022. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT04956289.
113. NS Pharma Inc. A phase 3 randomized, double-blind, placebo-controlled, multi-center study to assess the efficacy and safety of viltolarsen in ambulant boys with Duchenne muscular dystrophy (DMD). ClinicalTrials.gov Identifier: NCT04060199. Updated Nov 16, 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT04060199.
114. NS Pharma Inc. A phase 3, multi-center, open-label extension study to assess the safety and efficacy of viltolarsen in ambulant boys with Duchenne muscular dystrophy (DMD). ClinicalTrials.gov Identifier: NCT04768062. Updated Nov 16, 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT04768062.
115. Sarepta Therapeutics Inc. A randomized, double-blind, placebo-controlled, dose-titration, safety, tolerability, and pharmacokinetics study followed by an open-label safety and efficacy evaluation of SRP-4045 in advanced-stage patients with Duchenne muscular dystrophy amena-ble to exon 45 skipping. ClinicalTrials.gov Identifier: NCT02530905. Updated May 17, 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT02530905.
116. Sarepta Therapeutics Inc. Long-term, open-label extension study for patients with Duchenne muscular dystrophy enrolled in clinical trials evaluating casimersen or golodirsen. ClinicalTrials.gov Identifier: NCT03532542. Updated Dec 20, 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03532542.
117. PTC Therapeutics. A phase 2 study of the safety, pharmacokinetics, and pharmacodynamics of ataluren (PTC124®) in patients aged ≥2 to <5 years old with nonsense mutation dystrophinopathy. ClinicalTrials.gov Identifier: NCT02819557. Updated Aug 28, 2020. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT02819557.
118. PTC Therapeutics. Phase 2, non-interventional, clinical study to assess dystrophin levels in subjects with nonsense mutation Duchenne muscular dystrophy who have been treated with ataluren for ≥ 9 months. ClinicalTrials.gov Identifier: NCT03796637. Updated Apr 10, 2020. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03796637.
119. PTC Therapeutics. An Open-Label Study Evaluating the Safety and Pharmacokinetics of Ataluren in Children From ≥6 Months to <2 Years of Age With Nonsense Mutation Duchenne Muscular Dystrophy. clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT04336826 120. PTC Therapeutics. An open-label study for previously treated ataluren (PTC124®) pa-tients with nonsense mutation dystrophinopathy. ClinicalTrials.gov Identifier: NCT01557400. Updated Nov 25, 2020. Accessed Feb 21, 2022. https://clinicaltrials.gov/ct2/show/NCT01557400.
121. PTC Therapeutics. An open-label, safety study for ataluren (PTC124) patients with nonsense mutation dystrophinopathy. ClinicalTrials.gov Identifier: NCT01247207. Updated Feb 16, 2022. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT01247207.
122. PTC Therapeutics. A phase 3, randomized, double-blind, placebo-controlled efficacy and safety study of ataluren in patients with non-sense mutation Duchenne muscular dystrophy and open-label extension. ClinicalTrials.gov Identifier: NCT03179631. Updated Feb 8, 2022. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03179631.
123. Sarepta Therapeutics, Inc. An Open-Label, Systemic Gene Delivery Study Using Commercial Process Material to Evaluate the Safety of and Expression From SRP-9001 in Subjects With Duchenne Muscular Dystrophy (ENDEAVOR). clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT04626674
124. Sarepta Therapeutics, Inc. Systemic Gene Delivery Phase I/IIa Clinical Trial for Duchenne Muscular Dystrophy Using RAA-Vrh74.MHCK7.Micro-Dystrophin (MicroDys-IV-001). clinicaltrials.gov; 2022. Accessed August 18, 2022. https://clinicaltrials.gov/ct2/show/NCT03375164
125. Sarepta Therapeutics Inc. A multicenter, randomized, double-blind, placebo-controlled trial for Duchenne muscular dystrophy using SRP-9001. ClinicalTrials.gov Identifier: NCT03769116. Updated Dec 2021. Accessed Mar 2, 2022. https://clinicaltrials.gov/ct2/show/NCT03769116.
126. Hoffmann-La Roche. A Two-Part, Seamless, Multi-Center, Randomized, Placebo-Controlled, Double-Blind Study to Investigate the Safety, Tolerability, Pharmacokinetics, Pharmacodynamics and Efficacy of RO7204239 in Combination With Risdiplam (RO7034067) in Ambulant Pa-tients With Spinal Muscular Atrophy. clinicaltrials.gov; 2022. Accessed September 1, 2022. https://clinicaltrials.gov/ct2/show/NCT05115110
Spinal muscular atrophy: Patient care in the age of genetically targeted therapy
In 2016, the U.S. Food and Drug Administration approved nusinersen, the first treatment for spinal muscular atrophy (SMA). Until then, SMA had a mortality rate nearly double that of the general population.1 Two-thirds of patients were symptomatic within 6 months of birth and, in the absence of mechanical ventilation and other support, had a nearly 100% mortality rate by age 2.2
Five years later, there are three approved treatments for SMA, all of which have been shown to slow or even halt disease progression in many patients. Neurologists, whose SMA patient population once consisted almost entirely of children, are now treating more adults with the disease. Indeed, more than half of all people alive with SMA in the United States today are adults, according to Cure SMA.
“Managing SMA used to be clinic follow-ups where we were doing our best supportive care and watching people fall apart before our eyes,” said John Brandsema, MD, a physician and neuromuscular section head at the Children’s Hospital of Philadelphia. “Today, what we see in the vast majority of people is that they are either the same as they were before – which is completely against the natural history of this disease and something to be celebrated – or that people are really better with their function. It totally changes everything in the clinic.”
Among those changes are a more proactive approach to rehabilitation and an even greater emphasis on personalized medicine and multidisciplinary care. But there is also a need for updated treatment guidelines, a new classification system to measure disease severity, specific biomarkers to guide therapy choices, more data on long-term efficacy of existing therapeutics, new medications to complement those therapies, and a deeper understanding of a disease that may have treatment options but still has no cure.
Advances in early diagnosis
Patients with SMA lack a working copy of the survival motor neuron 1 (SMN1) gene, which provides instructions for producing a protein called SMN that is critical for the maintenance and function of motor neurons. Without this protein, motor neurons eventually die, causing debilitating and progressive muscle weakness that affects the ability to walk, eat, and breathe. SMA is rare, affecting about 1 in 10,000 newborns.
In approximately 96% of patients, SMA is caused by homozygous loss of the SMN1 gene. People with SMA have at least one copy of the SMN2 gene, sometimes called a “backup” gene, that also produces SMN protein. However, a single nucleotide difference between SMN2 and SMN1 causes about 90% of the protein produced by SMN2 to be truncated and less stable. Even with multiple copies of SMN2 present, as is the case with many infants with SMA, the amount of functional protein produced isn’t enough to compensate for the loss of SMN1.3
All three approved medications are SMN up-regulators and work to increase the amount of functional SMN protein. Starting these medications early, even before symptoms present, is critical to preserve motor function. Early treatment depends on early diagnosis, which became more widespread after 2018 when SMA was added to the federally Recommended Uniform Screening Panel for newborns. As of July 1, 2022, 47 states have incorporated SMA newborn screening into their state panel, ensuring that 97% of all infants born in the United States undergo SMA screening shortly after birth. Screening in the remaining states – Hawaii, Nevada, and South Carolina – and Washington, D.C. is expected by mid-2023.
SMA newborn screening is a PCR-based assay that detects homozygous SMN1 gene deletion found in about 95% of all people with SMA. The remaining 5% of cases are caused by various genetic mutations that can only be detected with gene sequencing. In these cases, and in children who don’t undergo SMA newborn screening, the disease is usually identified when symptoms are noticed by a parent, pediatrician, or primary care provider. But a study found that in 2018 only 52.7% of pediatricians correctly identified genetic testing as a requirement for a definitive diagnosis of SMA; in 2019, with a larger sample size, that number decreased to 45%.4 The lack of awareness of diagnostic requirements for SMA could contribute to delays in diagnosis, said Mary Schroth, MD, chief medical officer for Cure SMA and a coauthor of the study.
“In our world, suspicion of SMA in an infant is an emergency situation,” Dr. Schroth said. “These babies need to be referred immediately and have genetic testing so that treatment can begin as soon as possible.”
Based on the study findings, Dr. Schroth and others with Cure SMA launched a new tool in 2021 designed to help pediatricians, primary care physicians, and parents identify early signs of SMA, so that a referral to a pediatric neurologist happens quickly. Called SMArt Moves, the educational resource features videos and a checklist to help increase early detection in infants who had a negative SMA newborn screening result or did not receive SMA screening at birth.5
Who to treat, when, and with which treatment
For many patients, having multiple effective treatment options means that SMA is no longer a fatal disease in early childhood, but one that can be managed into adolescence and adulthood. The question for clinicians is, who do they treat, when, and with which treatment?
Studies have long shown that the number of copies of the backup gene that a patient has is inversely associated with disease severity.6 In 2018, a group of SMA experts published a treatment algorithm to help guide decision-making following a positive SMA newborn screening.7 The treatment guidelines were updated in 2020 based on clinical trial data for presymptomatic infants, and current recommendations include immediate treatment for infants with two to four copies of the SMN2 gene.8 For patients with only one copy of SMN2, most of whom will likely be symptomatic at birth, the guidelines recommend that treatment decisions be made jointly between the clinician and the family.7,8
Some suggest that the number of SMN2 copies a patient has should also be a factor in determining phenotype, which has started a conversation on the development of a new classification system.9 The original classification system for disease severity – Types 0-4 – was based on age of onset and degree of motor function achieved, with Type 0 developing prenatally and being the most severe and Type 4 developing in adulthood. Type 1 is the most common, affecting more than half of all people with SMA, followed by Types 2-4. In 2018, updated consensus care guidelines offered a revised classification system that better reflected disease progression in the age of therapy. The functional motor outcomes include nonsitters (historically Type I), sitters (historically Type 2/3), and walkers (historically Type 3/4).10,11 These guidelines are a start, but clinicians say more revision is needed.
“Types 1, 2, 3, 4 were based on function – getting to a certain point and then losing it, but now that we can treat this disease, people will shift categories based on therapeutic response or based on normal development that is possible now that the neurologic piece has been stabilized,” Dr. Brandsema said. “We need to completely change our thinking around all these different aspects of SMA management.”
While discussions of a new classification system for SMA are underway, another effort to update treatment recommendations is closer to completion. Led by Cure SMA, a group of about 50 physician experts in the United States and Europe who specialize in SMA are revising guidelines for diagnosis and treatment, the first time the recommendations have been updated since 2018. The updated recommendations, which should be published later this year, will focus on diagnosis and treatment considerations.
“We have three treatments that are available, and there are specific FDA indications for each of those, but it’s not totally clear just how those medications should be used or applied to different clinical situations,” said Dr. Schroth. “We’re in a rapid phase of learning right now in the SMA community, trying to understand how these treatments alter physiology and disease outcomes and how to best use the tools that we now have available to us. In parallel with clinical treatments, we have to be doing the best care we can to optimize the outcomes for those treatments.”
Research advances in 2021
Although all three drugs approved to treat SMA – nusinersen (Spinraza; Biogen), onasemnogene abeparvovec-xioi gene replacement therapy (Zolgensma; Novartis Gene Therapies), and risdiplam (Evrysdi, Genentech/Roche) – are highly effective, there are still unanswered questions and unmet needs. New research findings from 2021 focused on higher dosing, different drug-delivery methods, combination therapy, and complementary therapeutics to address SMA comorbidities.
Higher-dose nusinersen. The first drug approved to treat SMA, nusinersen is an antisense oligonucleotide approved for all ages and all SMA types. It works by altering splicing of the SMN2 gene pre-mRNA to make more complete SMN protein. Given as an intrathecal (IT) injection, four “loading doses” are administered within the first 2 months of treatment, followed by a maintenance dose every 4 months for the duration of the individual’s life.
Reports from patients of waning effects of nusinersen just prior to follow-up treatment have led some clinicians to ask if a higher dose may be needed. A study underway seeks to address that issue.
DEVOTE is a phase 2/3 trial to study the safety and efficacy of high-dose nusinersen in patients with SMA. Preliminary findings reported in 2021 found no adverse events among patients treated with 28 mg of nusinersen for 161-257 days.12 Another analysis from this trial found that higher doses are associated with greater decrease of plasma phosphorylated neurofilament heavy chain (pNF-H) levels in patients with SMA and may lead to clinically meaningful improvement in motor function beyond that observed with the approved 12 mg dose.13 The trial is ongoing.
Another trial, ASCEND, is a phase 3B study assessing higher dose nusinersen in patients previously treated with risdiplam. Recruitment for that trial began in October 2021.
Long-term efficacy and IT administration of SMA therapy. Several studies are looking at the long-term efficacy and alternate routes of administration of onasemnogene abeparvovec and other SMA therapies.
A one-time gene replacement therapy delivered via an IV infusion replaces the function of the missing or nonworking SMN1 gene with a new, working copy of the SMN1 gene. FDA approved in 2019, it is authorized for use in patients with SMA up to 2 years of age.
The latest data from an ongoing, long-term follow-up safety study of onasemnogene abeparvovec, published in May 2021, suggest that the treatment’s effects persist more than 5 years after treatment. Researchers followed 13 infants with symptomatic SMA type 1 since the beginning of the phase 1 clinical trial of the gene transfer therapy. All patients who received the therapeutic dose maintained their baseline motor function, and two of the patients actually improved without other SMN-targeted treatment. At a median 6.2 years after they received treatment, all were alive and none needed permanent ventilation.14
After a 2-year hold by the FDA, a study of IT administration of onasemnogene abeparvovec is now enrolling patients. Citing concerns from animal studies that IT administration might result in dorsal root ganglia injury, the FDA issued a partial hold on the STRONG trial in 2019. Following positive study results in nonhuman primates, the FDA announced the trial can continue. Novartis is launching a new phase 3 STEER trial to test the drug delivered intrathecally in patients aged 2-18 years with Type 2 SMA. IT administration could allow the gene therapy to be used safely and effectively in more patients with SMA.
Efficacy of risdiplam in more patients. The first oral treatment for SMA was approved by the FDA in 2020. It’s given once per day in patients with SMA of all ages and disease types. The drug increases functional SMN protein production by the SMN2 gene.
A July 2021 publication of the results of the FIREFISH study found that infants with Type I SMA treated with risdiplam for 12 months were significantly more likely to achieve motor milestones, such as sitting without support, compared with untreated infants with Type 1 SMA.15 Risdiplam is also effective in older patients with Type 2 or 3 SMA, according to results published in December from the SUNFISH clinical trial.16 Another study, RAINBOWFISH, is studying safety and efficacy at 24 months in presymptomatic infants started on treatment at up to 6 weeks of age.
The efficacy of risdiplam in previously treated patients is the subject of JEWELFISH, an ongoing study in patients 6 months to 60 years with SMA. Preliminary data presented at the 2020 Virtual SMA Research and Clinical Care Meeting suggest treatment with risdiplam led to a median two-fold increase in the amount of blood SMN protein levels after 4 weeks, which was sustained for at least 24 months.17
Combination therapy. Among the more eagerly awaited results are those from studies of combination therapies, including those that combine approved SMN up-regulators with new non–SMN-targeted therapeutics.
“We’re seeing that while these three approved therapies have dramatic results, especially for infants who are treated presymptomatically, there are still unmet medical needs in those patients, particularly for older teens and adults whose disease may have progressed before they were able to start therapy,” said Jackie Glascock, PhD, vice president of research for Cure SMA.
Of particular interest are studies of myostatin inhibitors, therapeutics that block the production of the protein myostatin. Myostatin acts on muscle cells to reduce muscle growth. Animal studies suggest that inhibiting myostatin increases muscle mass, which could be important in patients with muscle loss due to SMA.
Three experimental myostatin inhibitors are currently in clinical trials. MANATEE is a global phase 2-3 trial that aims to evaluate the safety and efficacy of the antimyostatin antibody GYM329 (RO7204239) in combination with risdiplam. SAPPHIRE is a phase 3 trial of apitegromab (SRK-015) in combination with nusinersen or risdiplam. RESILIANT is a phase 3 trial of tadefgrobep alfa in combination with other treatments.
A trial is underway to study the efficacy and safety of nusinersen in patients with persistent symptoms of SMA after treatment with the gene therapy. The phase 4 study, RESPOND, is enrolling children aged 2-36 months.
What’s needed next
Despite the advances in treatment and patient care, Dr. Brandsema, Dr. Schroth, and Dr. Glascock note that there remain unmet needs in the SMA community in a variety of areas.
Increased focus on adults with SMA. Before nusinersen, treatment of SMA mainly involved treating its symptoms. Many patients stopped seeing their neurologist, relying more heavily on pulmonary care specialists and/or primary care providers to address breathing, nutrition, and mobility problems. “Now with the approval of these treatments, they’re coming back to see their neurologists and are becoming more visible in the SMA community,” Dr. Schroth said.
Despite this re-emergence, a 2020 meta-analysis of studies on adults with SMA found a paucity of data on physical and occupational therapy, respiratory management, mental health care, and palliative care.18
“There is just so much work we need to do in the area of adult clinical care of SMA.”
Treatment algorithms. While the development of the newborn screening algorithm and revised patient care guidelines are helpful resources, clinicians still face uncertainty when choosing which therapy will work best for their patients. Treatment algorithms that help clinicians figure out what therapy or combination of therapies will offer the best outcomes for individual patients are desperately needed, Dr. Brandsema said.
“Each person’s experience of this disease is so unique to the individual based partly on their genetics and partly on the factors about what got them into care and how compliant they are with everything we’re trying to do to help them,” he said. “Biomarkers would help clinicians create personalized treatment plans for each patient.”
More basic science. While scientists have a good understanding of the SMN gene, there are many unanswered questions about the function of the SMN protein and its relationship to motor neuron loss. SMN is a ubiquitously expressed protein, and its function in other cell types is largely unknown. Despite all of the research advances, there is much basic science left to be done.
“We are strongly advocating to regulatory authorities that these aren’t cures and we need to continue to invest in the basic research,” Dr. Glascock said. “These biological questions that pertain to SMN and its function and expression really drive drug development. I really think that understanding those pathways better will lead us to more druggable targets.”
Two deaths from liver failure linked to spinal muscular atrophy drug
Two children taking the gene therapy drug onasemnogene abeparvovec (Zolgensma, Novartis) for spinal muscular atrophy (SMA) have died from acute liver failure, according to a statement issued by the drug’s manufacturer.
The patients were 4 months and 28 months of age and lived in Russia and Kazakhstan. They died 5-6 weeks after infusion with Zolgensma and approximately 1-10 days after the initiation of a corticosteroid taper.
These are the first known fatal cases of acute liver failure associated with the drug, which the company notes was a known side effect included in the product label and in a boxed warning in the United States.
“Following two recent patient fatalities, and in alignment with health authorities, we will be updating the labeling to specify that fatal acute liver failure has been reported,” the statement reads.
“While this is important safety information, it is not a new safety signal,” it adds.
Rare genetic disorder
SMA is a rare genetic disorder that affects about 1 in 10,000 newborns. Patients with SMA lack a working copy of the survival motor neuron 1 (SMN1) gene, which encodes a protein called SMN that is critical for the maintenance and function of motor neurons.
Without this protein, motor neurons eventually die, causing debilitating and progressive muscle weakness that affects the ability to walk, eat, and breathe.
Zolgensma, a one-time gene replacement therapy delivered via intravenous infusion, replaces the function of the missing or nonworking SMN1 gene with a new, working copy of the SMN1 gene.
The first gene therapy treatment for SMA, it was approved by the U.S. Food and Drug Administration in 2019 for patients with SMA up to 2 years of age. It is also the most expensive drug in the world, costing about $2.1 million for a one-time treatment.
“We have notified health authorities in all markets where Zolgensma is used, including the FDA, and are communicating to relevant healthcare professionals as an additional step in markets where this action is supported by health authorities,” the manufacturer’s statement says.
Studies have suggested that the treatment’s effects persist more than 5 years after infusion.
Clinical trials currently underway by Novartis are studying the drug’s long-term efficacy and safety and its potential use in older patients.
The company is also leading the phase 3 clinical trial STEER to test intrathecal (IT) administration of the drug in patients ages 2-18 years who have type 2 SMA.
That trial began late last year after the FDA lifted a 2-year partial hold on an earlier study. The FDA halted the STRONG trial in 2019, citing concerns from animal studies that IT administration may result in dorsal root ganglia injury. The partial hold was released last fall following positive study results in nonhuman primates.
None of the current trials will be affected by the two deaths reported, according to a Novartis spokesperson.
Kelli Whitlock Burton is a staff writer/reporter for Medscape Neurology and MDedge Neurology.
References
1. Viscidi E et al. Comparative all-cause mortality among a large population of patients with spinal muscular atrophy versus matched controls. Neurol Ther. 2022 Mar;11(1):449-457. doi: 10.1007/s40120-021-00307-7.
2. Finkel RS et al. Observational study of spinal muscular atrophy type I and implications for clinical trials. Neurology. 2014 Aug 26;83(9):810-817. doi: 10.1212/WNL.0000000000000741.
3. Klotz J et al. Advances in the therapy of spinal muscular atrophy. J Pediatr. 2021 Sep;236:13-20.e1. doi: 10.1016/j.jpeds.2021.06.033.
4. Curry M et al. Awareness screening and referral patterns among pediatricians in the United States related to early clinical features of spinal muscular atrophy (SMA). BMC Pediatr. 2021 May;21(1):236. doi: 10.1186/s12887-021-02692-2.
5. SMArt Moves. https://smartmoves.curesma.org/
6. Swoboda KJ et al. Natural history of denervation in SMA: Relation to age, SMN2 copy number, and function. Ann Neurol. 2005 May;57(5):704-12. doi: 10.1002/ana.20473.
7. Glascock J et al. Treatment algorithm for infants diagnosed with spinal muscular atrophy through newborn screening. J Neuromuscul Dis. 2018;5(2):145-158. doi: 10.3233/JND-180304.
8. Glascock J et al. Revised recommendations for the treatment of infants diagnosed with spinal muscular atrophy via newborn screening who have 4 copies of SMN2. J Neuromuscul Dis. 2020;7(2):97-100. doi: 10.3233/JND-190468.
9. Talbot K, Tizzano EF. The clinical landscape for SMA in a new therapeutic era. Gene Ther. 2017 Sep;24(9):529-533. doi: 10.1038/gt.2017.52.
10. Mercuri E et al. Diagnosis and management of spinal muscular atrophy: Part 1: Recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscul Disord. 2018 Feb;28(2):103-115. doi: 10.1016/j.nmd.2017.11.005.
11. Finkel RS et al. Diagnosis and management of spinal muscular atrophy: Part 2: Pulmonary and acute care; medications, supplements and immunizations; other organ systems; and ethics. Neuromuscul Disord. 2018 Mar;28(3):197-207. doi: 10.1016/j.nmd.2017.11.004.
12. Pascual SI et al. Ongoing phase 2/3 DEVOTE (232SM203) randomized, controlled study to explore high-dose nusinersen in SMA: Part A interim results and Part B enrollment update. Presented at MDA Clinical and Scientific Conference 2021, Mar 15-18.
13. Finkel RS et al. Scientific rationale for a higher dose of nusinersen. Presented at 2021 Cure SMA Annual Meeting, Jun 9-11. Abstract P46.
14. Mendell JR et al. Five-year extension results of the phase 1 START trial of onasemnogene abeparvovec in spinal muscular atrophy. JAMA Neurol. 2021 Jul;78(7):834-841. doi: 10.1001/jamaneurol.2021.1272.
15. Darras BT et al. Risdiplam-treated infants with type 1 spinal muscular atrophy versus historical controls. N Engl J Med. 2021 Jul 29;385(5):427-435. doi: 10.1056/NEJMoa2102047.
16. Mercuri E et al. Safety and efficacy of once-daily risdiplam in type 2 and non-ambulant type 3 spinal muscular atrophy (SUNFISH part 2): A phase 3, double-blind, randomised, placebo-controlled trial. Lancet Neurol. 2022 Jan;21(1):42-52. doi: 10.1016/S1474-4422(21)00367-7. Erratum in: Lancet Neurol. 2022 Feb;21(2):e2. doi: 10.1016/S1474-4422(22)00006-0. Correction in: Lancet Neurol. 2022 Mar;21(3):e3. doi: 10.1016/S1474-4422(22)00038-2.
17. Genentech announces 2-year risdiplam data from SUNFISH and new data from JEWELFISH in infants, children and adults with SMA. https://www.curesma.org/genentech-risdiplam-data-conference-2020/
18. Wan HWY et al. Health, wellbeing and lived experiences of adults with SMA: a scoping systematic review. Orphanet J Rare Dis. 2020;15(1):70. doi: 10.1186/s13023-020-1339-3.
In 2016, the U.S. Food and Drug Administration approved nusinersen, the first treatment for spinal muscular atrophy (SMA). Until then, SMA had a mortality rate nearly double that of the general population.1 Two-thirds of patients were symptomatic within 6 months of birth and, in the absence of mechanical ventilation and other support, had a nearly 100% mortality rate by age 2.2
Five years later, there are three approved treatments for SMA, all of which have been shown to slow or even halt disease progression in many patients. Neurologists, whose SMA patient population once consisted almost entirely of children, are now treating more adults with the disease. Indeed, more than half of all people alive with SMA in the United States today are adults, according to Cure SMA.
“Managing SMA used to be clinic follow-ups where we were doing our best supportive care and watching people fall apart before our eyes,” said John Brandsema, MD, a physician and neuromuscular section head at the Children’s Hospital of Philadelphia. “Today, what we see in the vast majority of people is that they are either the same as they were before – which is completely against the natural history of this disease and something to be celebrated – or that people are really better with their function. It totally changes everything in the clinic.”
Among those changes are a more proactive approach to rehabilitation and an even greater emphasis on personalized medicine and multidisciplinary care. But there is also a need for updated treatment guidelines, a new classification system to measure disease severity, specific biomarkers to guide therapy choices, more data on long-term efficacy of existing therapeutics, new medications to complement those therapies, and a deeper understanding of a disease that may have treatment options but still has no cure.
Advances in early diagnosis
Patients with SMA lack a working copy of the survival motor neuron 1 (SMN1) gene, which provides instructions for producing a protein called SMN that is critical for the maintenance and function of motor neurons. Without this protein, motor neurons eventually die, causing debilitating and progressive muscle weakness that affects the ability to walk, eat, and breathe. SMA is rare, affecting about 1 in 10,000 newborns.
In approximately 96% of patients, SMA is caused by homozygous loss of the SMN1 gene. People with SMA have at least one copy of the SMN2 gene, sometimes called a “backup” gene, that also produces SMN protein. However, a single nucleotide difference between SMN2 and SMN1 causes about 90% of the protein produced by SMN2 to be truncated and less stable. Even with multiple copies of SMN2 present, as is the case with many infants with SMA, the amount of functional protein produced isn’t enough to compensate for the loss of SMN1.3
All three approved medications are SMN up-regulators and work to increase the amount of functional SMN protein. Starting these medications early, even before symptoms present, is critical to preserve motor function. Early treatment depends on early diagnosis, which became more widespread after 2018 when SMA was added to the federally Recommended Uniform Screening Panel for newborns. As of July 1, 2022, 47 states have incorporated SMA newborn screening into their state panel, ensuring that 97% of all infants born in the United States undergo SMA screening shortly after birth. Screening in the remaining states – Hawaii, Nevada, and South Carolina – and Washington, D.C. is expected by mid-2023.
SMA newborn screening is a PCR-based assay that detects homozygous SMN1 gene deletion found in about 95% of all people with SMA. The remaining 5% of cases are caused by various genetic mutations that can only be detected with gene sequencing. In these cases, and in children who don’t undergo SMA newborn screening, the disease is usually identified when symptoms are noticed by a parent, pediatrician, or primary care provider. But a study found that in 2018 only 52.7% of pediatricians correctly identified genetic testing as a requirement for a definitive diagnosis of SMA; in 2019, with a larger sample size, that number decreased to 45%.4 The lack of awareness of diagnostic requirements for SMA could contribute to delays in diagnosis, said Mary Schroth, MD, chief medical officer for Cure SMA and a coauthor of the study.
“In our world, suspicion of SMA in an infant is an emergency situation,” Dr. Schroth said. “These babies need to be referred immediately and have genetic testing so that treatment can begin as soon as possible.”
Based on the study findings, Dr. Schroth and others with Cure SMA launched a new tool in 2021 designed to help pediatricians, primary care physicians, and parents identify early signs of SMA, so that a referral to a pediatric neurologist happens quickly. Called SMArt Moves, the educational resource features videos and a checklist to help increase early detection in infants who had a negative SMA newborn screening result or did not receive SMA screening at birth.5
Who to treat, when, and with which treatment
For many patients, having multiple effective treatment options means that SMA is no longer a fatal disease in early childhood, but one that can be managed into adolescence and adulthood. The question for clinicians is, who do they treat, when, and with which treatment?
Studies have long shown that the number of copies of the backup gene that a patient has is inversely associated with disease severity.6 In 2018, a group of SMA experts published a treatment algorithm to help guide decision-making following a positive SMA newborn screening.7 The treatment guidelines were updated in 2020 based on clinical trial data for presymptomatic infants, and current recommendations include immediate treatment for infants with two to four copies of the SMN2 gene.8 For patients with only one copy of SMN2, most of whom will likely be symptomatic at birth, the guidelines recommend that treatment decisions be made jointly between the clinician and the family.7,8
Some suggest that the number of SMN2 copies a patient has should also be a factor in determining phenotype, which has started a conversation on the development of a new classification system.9 The original classification system for disease severity – Types 0-4 – was based on age of onset and degree of motor function achieved, with Type 0 developing prenatally and being the most severe and Type 4 developing in adulthood. Type 1 is the most common, affecting more than half of all people with SMA, followed by Types 2-4. In 2018, updated consensus care guidelines offered a revised classification system that better reflected disease progression in the age of therapy. The functional motor outcomes include nonsitters (historically Type I), sitters (historically Type 2/3), and walkers (historically Type 3/4).10,11 These guidelines are a start, but clinicians say more revision is needed.
“Types 1, 2, 3, 4 were based on function – getting to a certain point and then losing it, but now that we can treat this disease, people will shift categories based on therapeutic response or based on normal development that is possible now that the neurologic piece has been stabilized,” Dr. Brandsema said. “We need to completely change our thinking around all these different aspects of SMA management.”
While discussions of a new classification system for SMA are underway, another effort to update treatment recommendations is closer to completion. Led by Cure SMA, a group of about 50 physician experts in the United States and Europe who specialize in SMA are revising guidelines for diagnosis and treatment, the first time the recommendations have been updated since 2018. The updated recommendations, which should be published later this year, will focus on diagnosis and treatment considerations.
“We have three treatments that are available, and there are specific FDA indications for each of those, but it’s not totally clear just how those medications should be used or applied to different clinical situations,” said Dr. Schroth. “We’re in a rapid phase of learning right now in the SMA community, trying to understand how these treatments alter physiology and disease outcomes and how to best use the tools that we now have available to us. In parallel with clinical treatments, we have to be doing the best care we can to optimize the outcomes for those treatments.”
Research advances in 2021
Although all three drugs approved to treat SMA – nusinersen (Spinraza; Biogen), onasemnogene abeparvovec-xioi gene replacement therapy (Zolgensma; Novartis Gene Therapies), and risdiplam (Evrysdi, Genentech/Roche) – are highly effective, there are still unanswered questions and unmet needs. New research findings from 2021 focused on higher dosing, different drug-delivery methods, combination therapy, and complementary therapeutics to address SMA comorbidities.
Higher-dose nusinersen. The first drug approved to treat SMA, nusinersen is an antisense oligonucleotide approved for all ages and all SMA types. It works by altering splicing of the SMN2 gene pre-mRNA to make more complete SMN protein. Given as an intrathecal (IT) injection, four “loading doses” are administered within the first 2 months of treatment, followed by a maintenance dose every 4 months for the duration of the individual’s life.
Reports from patients of waning effects of nusinersen just prior to follow-up treatment have led some clinicians to ask if a higher dose may be needed. A study underway seeks to address that issue.
DEVOTE is a phase 2/3 trial to study the safety and efficacy of high-dose nusinersen in patients with SMA. Preliminary findings reported in 2021 found no adverse events among patients treated with 28 mg of nusinersen for 161-257 days.12 Another analysis from this trial found that higher doses are associated with greater decrease of plasma phosphorylated neurofilament heavy chain (pNF-H) levels in patients with SMA and may lead to clinically meaningful improvement in motor function beyond that observed with the approved 12 mg dose.13 The trial is ongoing.
Another trial, ASCEND, is a phase 3B study assessing higher dose nusinersen in patients previously treated with risdiplam. Recruitment for that trial began in October 2021.
Long-term efficacy and IT administration of SMA therapy. Several studies are looking at the long-term efficacy and alternate routes of administration of onasemnogene abeparvovec and other SMA therapies.
A one-time gene replacement therapy delivered via an IV infusion replaces the function of the missing or nonworking SMN1 gene with a new, working copy of the SMN1 gene. FDA approved in 2019, it is authorized for use in patients with SMA up to 2 years of age.
The latest data from an ongoing, long-term follow-up safety study of onasemnogene abeparvovec, published in May 2021, suggest that the treatment’s effects persist more than 5 years after treatment. Researchers followed 13 infants with symptomatic SMA type 1 since the beginning of the phase 1 clinical trial of the gene transfer therapy. All patients who received the therapeutic dose maintained their baseline motor function, and two of the patients actually improved without other SMN-targeted treatment. At a median 6.2 years after they received treatment, all were alive and none needed permanent ventilation.14
After a 2-year hold by the FDA, a study of IT administration of onasemnogene abeparvovec is now enrolling patients. Citing concerns from animal studies that IT administration might result in dorsal root ganglia injury, the FDA issued a partial hold on the STRONG trial in 2019. Following positive study results in nonhuman primates, the FDA announced the trial can continue. Novartis is launching a new phase 3 STEER trial to test the drug delivered intrathecally in patients aged 2-18 years with Type 2 SMA. IT administration could allow the gene therapy to be used safely and effectively in more patients with SMA.
Efficacy of risdiplam in more patients. The first oral treatment for SMA was approved by the FDA in 2020. It’s given once per day in patients with SMA of all ages and disease types. The drug increases functional SMN protein production by the SMN2 gene.
A July 2021 publication of the results of the FIREFISH study found that infants with Type I SMA treated with risdiplam for 12 months were significantly more likely to achieve motor milestones, such as sitting without support, compared with untreated infants with Type 1 SMA.15 Risdiplam is also effective in older patients with Type 2 or 3 SMA, according to results published in December from the SUNFISH clinical trial.16 Another study, RAINBOWFISH, is studying safety and efficacy at 24 months in presymptomatic infants started on treatment at up to 6 weeks of age.
The efficacy of risdiplam in previously treated patients is the subject of JEWELFISH, an ongoing study in patients 6 months to 60 years with SMA. Preliminary data presented at the 2020 Virtual SMA Research and Clinical Care Meeting suggest treatment with risdiplam led to a median two-fold increase in the amount of blood SMN protein levels after 4 weeks, which was sustained for at least 24 months.17
Combination therapy. Among the more eagerly awaited results are those from studies of combination therapies, including those that combine approved SMN up-regulators with new non–SMN-targeted therapeutics.
“We’re seeing that while these three approved therapies have dramatic results, especially for infants who are treated presymptomatically, there are still unmet medical needs in those patients, particularly for older teens and adults whose disease may have progressed before they were able to start therapy,” said Jackie Glascock, PhD, vice president of research for Cure SMA.
Of particular interest are studies of myostatin inhibitors, therapeutics that block the production of the protein myostatin. Myostatin acts on muscle cells to reduce muscle growth. Animal studies suggest that inhibiting myostatin increases muscle mass, which could be important in patients with muscle loss due to SMA.
Three experimental myostatin inhibitors are currently in clinical trials. MANATEE is a global phase 2-3 trial that aims to evaluate the safety and efficacy of the antimyostatin antibody GYM329 (RO7204239) in combination with risdiplam. SAPPHIRE is a phase 3 trial of apitegromab (SRK-015) in combination with nusinersen or risdiplam. RESILIANT is a phase 3 trial of tadefgrobep alfa in combination with other treatments.
A trial is underway to study the efficacy and safety of nusinersen in patients with persistent symptoms of SMA after treatment with the gene therapy. The phase 4 study, RESPOND, is enrolling children aged 2-36 months.
What’s needed next
Despite the advances in treatment and patient care, Dr. Brandsema, Dr. Schroth, and Dr. Glascock note that there remain unmet needs in the SMA community in a variety of areas.
Increased focus on adults with SMA. Before nusinersen, treatment of SMA mainly involved treating its symptoms. Many patients stopped seeing their neurologist, relying more heavily on pulmonary care specialists and/or primary care providers to address breathing, nutrition, and mobility problems. “Now with the approval of these treatments, they’re coming back to see their neurologists and are becoming more visible in the SMA community,” Dr. Schroth said.
Despite this re-emergence, a 2020 meta-analysis of studies on adults with SMA found a paucity of data on physical and occupational therapy, respiratory management, mental health care, and palliative care.18
“There is just so much work we need to do in the area of adult clinical care of SMA.”
Treatment algorithms. While the development of the newborn screening algorithm and revised patient care guidelines are helpful resources, clinicians still face uncertainty when choosing which therapy will work best for their patients. Treatment algorithms that help clinicians figure out what therapy or combination of therapies will offer the best outcomes for individual patients are desperately needed, Dr. Brandsema said.
“Each person’s experience of this disease is so unique to the individual based partly on their genetics and partly on the factors about what got them into care and how compliant they are with everything we’re trying to do to help them,” he said. “Biomarkers would help clinicians create personalized treatment plans for each patient.”
More basic science. While scientists have a good understanding of the SMN gene, there are many unanswered questions about the function of the SMN protein and its relationship to motor neuron loss. SMN is a ubiquitously expressed protein, and its function in other cell types is largely unknown. Despite all of the research advances, there is much basic science left to be done.
“We are strongly advocating to regulatory authorities that these aren’t cures and we need to continue to invest in the basic research,” Dr. Glascock said. “These biological questions that pertain to SMN and its function and expression really drive drug development. I really think that understanding those pathways better will lead us to more druggable targets.”
Two deaths from liver failure linked to spinal muscular atrophy drug
Two children taking the gene therapy drug onasemnogene abeparvovec (Zolgensma, Novartis) for spinal muscular atrophy (SMA) have died from acute liver failure, according to a statement issued by the drug’s manufacturer.
The patients were 4 months and 28 months of age and lived in Russia and Kazakhstan. They died 5-6 weeks after infusion with Zolgensma and approximately 1-10 days after the initiation of a corticosteroid taper.
These are the first known fatal cases of acute liver failure associated with the drug, which the company notes was a known side effect included in the product label and in a boxed warning in the United States.
“Following two recent patient fatalities, and in alignment with health authorities, we will be updating the labeling to specify that fatal acute liver failure has been reported,” the statement reads.
“While this is important safety information, it is not a new safety signal,” it adds.
Rare genetic disorder
SMA is a rare genetic disorder that affects about 1 in 10,000 newborns. Patients with SMA lack a working copy of the survival motor neuron 1 (SMN1) gene, which encodes a protein called SMN that is critical for the maintenance and function of motor neurons.
Without this protein, motor neurons eventually die, causing debilitating and progressive muscle weakness that affects the ability to walk, eat, and breathe.
Zolgensma, a one-time gene replacement therapy delivered via intravenous infusion, replaces the function of the missing or nonworking SMN1 gene with a new, working copy of the SMN1 gene.
The first gene therapy treatment for SMA, it was approved by the U.S. Food and Drug Administration in 2019 for patients with SMA up to 2 years of age. It is also the most expensive drug in the world, costing about $2.1 million for a one-time treatment.
“We have notified health authorities in all markets where Zolgensma is used, including the FDA, and are communicating to relevant healthcare professionals as an additional step in markets where this action is supported by health authorities,” the manufacturer’s statement says.
Studies have suggested that the treatment’s effects persist more than 5 years after infusion.
Clinical trials currently underway by Novartis are studying the drug’s long-term efficacy and safety and its potential use in older patients.
The company is also leading the phase 3 clinical trial STEER to test intrathecal (IT) administration of the drug in patients ages 2-18 years who have type 2 SMA.
That trial began late last year after the FDA lifted a 2-year partial hold on an earlier study. The FDA halted the STRONG trial in 2019, citing concerns from animal studies that IT administration may result in dorsal root ganglia injury. The partial hold was released last fall following positive study results in nonhuman primates.
None of the current trials will be affected by the two deaths reported, according to a Novartis spokesperson.
Kelli Whitlock Burton is a staff writer/reporter for Medscape Neurology and MDedge Neurology.
References
1. Viscidi E et al. Comparative all-cause mortality among a large population of patients with spinal muscular atrophy versus matched controls. Neurol Ther. 2022 Mar;11(1):449-457. doi: 10.1007/s40120-021-00307-7.
2. Finkel RS et al. Observational study of spinal muscular atrophy type I and implications for clinical trials. Neurology. 2014 Aug 26;83(9):810-817. doi: 10.1212/WNL.0000000000000741.
3. Klotz J et al. Advances in the therapy of spinal muscular atrophy. J Pediatr. 2021 Sep;236:13-20.e1. doi: 10.1016/j.jpeds.2021.06.033.
4. Curry M et al. Awareness screening and referral patterns among pediatricians in the United States related to early clinical features of spinal muscular atrophy (SMA). BMC Pediatr. 2021 May;21(1):236. doi: 10.1186/s12887-021-02692-2.
5. SMArt Moves. https://smartmoves.curesma.org/
6. Swoboda KJ et al. Natural history of denervation in SMA: Relation to age, SMN2 copy number, and function. Ann Neurol. 2005 May;57(5):704-12. doi: 10.1002/ana.20473.
7. Glascock J et al. Treatment algorithm for infants diagnosed with spinal muscular atrophy through newborn screening. J Neuromuscul Dis. 2018;5(2):145-158. doi: 10.3233/JND-180304.
8. Glascock J et al. Revised recommendations for the treatment of infants diagnosed with spinal muscular atrophy via newborn screening who have 4 copies of SMN2. J Neuromuscul Dis. 2020;7(2):97-100. doi: 10.3233/JND-190468.
9. Talbot K, Tizzano EF. The clinical landscape for SMA in a new therapeutic era. Gene Ther. 2017 Sep;24(9):529-533. doi: 10.1038/gt.2017.52.
10. Mercuri E et al. Diagnosis and management of spinal muscular atrophy: Part 1: Recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscul Disord. 2018 Feb;28(2):103-115. doi: 10.1016/j.nmd.2017.11.005.
11. Finkel RS et al. Diagnosis and management of spinal muscular atrophy: Part 2: Pulmonary and acute care; medications, supplements and immunizations; other organ systems; and ethics. Neuromuscul Disord. 2018 Mar;28(3):197-207. doi: 10.1016/j.nmd.2017.11.004.
12. Pascual SI et al. Ongoing phase 2/3 DEVOTE (232SM203) randomized, controlled study to explore high-dose nusinersen in SMA: Part A interim results and Part B enrollment update. Presented at MDA Clinical and Scientific Conference 2021, Mar 15-18.
13. Finkel RS et al. Scientific rationale for a higher dose of nusinersen. Presented at 2021 Cure SMA Annual Meeting, Jun 9-11. Abstract P46.
14. Mendell JR et al. Five-year extension results of the phase 1 START trial of onasemnogene abeparvovec in spinal muscular atrophy. JAMA Neurol. 2021 Jul;78(7):834-841. doi: 10.1001/jamaneurol.2021.1272.
15. Darras BT et al. Risdiplam-treated infants with type 1 spinal muscular atrophy versus historical controls. N Engl J Med. 2021 Jul 29;385(5):427-435. doi: 10.1056/NEJMoa2102047.
16. Mercuri E et al. Safety and efficacy of once-daily risdiplam in type 2 and non-ambulant type 3 spinal muscular atrophy (SUNFISH part 2): A phase 3, double-blind, randomised, placebo-controlled trial. Lancet Neurol. 2022 Jan;21(1):42-52. doi: 10.1016/S1474-4422(21)00367-7. Erratum in: Lancet Neurol. 2022 Feb;21(2):e2. doi: 10.1016/S1474-4422(22)00006-0. Correction in: Lancet Neurol. 2022 Mar;21(3):e3. doi: 10.1016/S1474-4422(22)00038-2.
17. Genentech announces 2-year risdiplam data from SUNFISH and new data from JEWELFISH in infants, children and adults with SMA. https://www.curesma.org/genentech-risdiplam-data-conference-2020/
18. Wan HWY et al. Health, wellbeing and lived experiences of adults with SMA: a scoping systematic review. Orphanet J Rare Dis. 2020;15(1):70. doi: 10.1186/s13023-020-1339-3.
In 2016, the U.S. Food and Drug Administration approved nusinersen, the first treatment for spinal muscular atrophy (SMA). Until then, SMA had a mortality rate nearly double that of the general population.1 Two-thirds of patients were symptomatic within 6 months of birth and, in the absence of mechanical ventilation and other support, had a nearly 100% mortality rate by age 2.2
Five years later, there are three approved treatments for SMA, all of which have been shown to slow or even halt disease progression in many patients. Neurologists, whose SMA patient population once consisted almost entirely of children, are now treating more adults with the disease. Indeed, more than half of all people alive with SMA in the United States today are adults, according to Cure SMA.
“Managing SMA used to be clinic follow-ups where we were doing our best supportive care and watching people fall apart before our eyes,” said John Brandsema, MD, a physician and neuromuscular section head at the Children’s Hospital of Philadelphia. “Today, what we see in the vast majority of people is that they are either the same as they were before – which is completely against the natural history of this disease and something to be celebrated – or that people are really better with their function. It totally changes everything in the clinic.”
Among those changes are a more proactive approach to rehabilitation and an even greater emphasis on personalized medicine and multidisciplinary care. But there is also a need for updated treatment guidelines, a new classification system to measure disease severity, specific biomarkers to guide therapy choices, more data on long-term efficacy of existing therapeutics, new medications to complement those therapies, and a deeper understanding of a disease that may have treatment options but still has no cure.
Advances in early diagnosis
Patients with SMA lack a working copy of the survival motor neuron 1 (SMN1) gene, which provides instructions for producing a protein called SMN that is critical for the maintenance and function of motor neurons. Without this protein, motor neurons eventually die, causing debilitating and progressive muscle weakness that affects the ability to walk, eat, and breathe. SMA is rare, affecting about 1 in 10,000 newborns.
In approximately 96% of patients, SMA is caused by homozygous loss of the SMN1 gene. People with SMA have at least one copy of the SMN2 gene, sometimes called a “backup” gene, that also produces SMN protein. However, a single nucleotide difference between SMN2 and SMN1 causes about 90% of the protein produced by SMN2 to be truncated and less stable. Even with multiple copies of SMN2 present, as is the case with many infants with SMA, the amount of functional protein produced isn’t enough to compensate for the loss of SMN1.3
All three approved medications are SMN up-regulators and work to increase the amount of functional SMN protein. Starting these medications early, even before symptoms present, is critical to preserve motor function. Early treatment depends on early diagnosis, which became more widespread after 2018 when SMA was added to the federally Recommended Uniform Screening Panel for newborns. As of July 1, 2022, 47 states have incorporated SMA newborn screening into their state panel, ensuring that 97% of all infants born in the United States undergo SMA screening shortly after birth. Screening in the remaining states – Hawaii, Nevada, and South Carolina – and Washington, D.C. is expected by mid-2023.
SMA newborn screening is a PCR-based assay that detects homozygous SMN1 gene deletion found in about 95% of all people with SMA. The remaining 5% of cases are caused by various genetic mutations that can only be detected with gene sequencing. In these cases, and in children who don’t undergo SMA newborn screening, the disease is usually identified when symptoms are noticed by a parent, pediatrician, or primary care provider. But a study found that in 2018 only 52.7% of pediatricians correctly identified genetic testing as a requirement for a definitive diagnosis of SMA; in 2019, with a larger sample size, that number decreased to 45%.4 The lack of awareness of diagnostic requirements for SMA could contribute to delays in diagnosis, said Mary Schroth, MD, chief medical officer for Cure SMA and a coauthor of the study.
“In our world, suspicion of SMA in an infant is an emergency situation,” Dr. Schroth said. “These babies need to be referred immediately and have genetic testing so that treatment can begin as soon as possible.”
Based on the study findings, Dr. Schroth and others with Cure SMA launched a new tool in 2021 designed to help pediatricians, primary care physicians, and parents identify early signs of SMA, so that a referral to a pediatric neurologist happens quickly. Called SMArt Moves, the educational resource features videos and a checklist to help increase early detection in infants who had a negative SMA newborn screening result or did not receive SMA screening at birth.5
Who to treat, when, and with which treatment
For many patients, having multiple effective treatment options means that SMA is no longer a fatal disease in early childhood, but one that can be managed into adolescence and adulthood. The question for clinicians is, who do they treat, when, and with which treatment?
Studies have long shown that the number of copies of the backup gene that a patient has is inversely associated with disease severity.6 In 2018, a group of SMA experts published a treatment algorithm to help guide decision-making following a positive SMA newborn screening.7 The treatment guidelines were updated in 2020 based on clinical trial data for presymptomatic infants, and current recommendations include immediate treatment for infants with two to four copies of the SMN2 gene.8 For patients with only one copy of SMN2, most of whom will likely be symptomatic at birth, the guidelines recommend that treatment decisions be made jointly between the clinician and the family.7,8
Some suggest that the number of SMN2 copies a patient has should also be a factor in determining phenotype, which has started a conversation on the development of a new classification system.9 The original classification system for disease severity – Types 0-4 – was based on age of onset and degree of motor function achieved, with Type 0 developing prenatally and being the most severe and Type 4 developing in adulthood. Type 1 is the most common, affecting more than half of all people with SMA, followed by Types 2-4. In 2018, updated consensus care guidelines offered a revised classification system that better reflected disease progression in the age of therapy. The functional motor outcomes include nonsitters (historically Type I), sitters (historically Type 2/3), and walkers (historically Type 3/4).10,11 These guidelines are a start, but clinicians say more revision is needed.
“Types 1, 2, 3, 4 were based on function – getting to a certain point and then losing it, but now that we can treat this disease, people will shift categories based on therapeutic response or based on normal development that is possible now that the neurologic piece has been stabilized,” Dr. Brandsema said. “We need to completely change our thinking around all these different aspects of SMA management.”
While discussions of a new classification system for SMA are underway, another effort to update treatment recommendations is closer to completion. Led by Cure SMA, a group of about 50 physician experts in the United States and Europe who specialize in SMA are revising guidelines for diagnosis and treatment, the first time the recommendations have been updated since 2018. The updated recommendations, which should be published later this year, will focus on diagnosis and treatment considerations.
“We have three treatments that are available, and there are specific FDA indications for each of those, but it’s not totally clear just how those medications should be used or applied to different clinical situations,” said Dr. Schroth. “We’re in a rapid phase of learning right now in the SMA community, trying to understand how these treatments alter physiology and disease outcomes and how to best use the tools that we now have available to us. In parallel with clinical treatments, we have to be doing the best care we can to optimize the outcomes for those treatments.”
Research advances in 2021
Although all three drugs approved to treat SMA – nusinersen (Spinraza; Biogen), onasemnogene abeparvovec-xioi gene replacement therapy (Zolgensma; Novartis Gene Therapies), and risdiplam (Evrysdi, Genentech/Roche) – are highly effective, there are still unanswered questions and unmet needs. New research findings from 2021 focused on higher dosing, different drug-delivery methods, combination therapy, and complementary therapeutics to address SMA comorbidities.
Higher-dose nusinersen. The first drug approved to treat SMA, nusinersen is an antisense oligonucleotide approved for all ages and all SMA types. It works by altering splicing of the SMN2 gene pre-mRNA to make more complete SMN protein. Given as an intrathecal (IT) injection, four “loading doses” are administered within the first 2 months of treatment, followed by a maintenance dose every 4 months for the duration of the individual’s life.
Reports from patients of waning effects of nusinersen just prior to follow-up treatment have led some clinicians to ask if a higher dose may be needed. A study underway seeks to address that issue.
DEVOTE is a phase 2/3 trial to study the safety and efficacy of high-dose nusinersen in patients with SMA. Preliminary findings reported in 2021 found no adverse events among patients treated with 28 mg of nusinersen for 161-257 days.12 Another analysis from this trial found that higher doses are associated with greater decrease of plasma phosphorylated neurofilament heavy chain (pNF-H) levels in patients with SMA and may lead to clinically meaningful improvement in motor function beyond that observed with the approved 12 mg dose.13 The trial is ongoing.
Another trial, ASCEND, is a phase 3B study assessing higher dose nusinersen in patients previously treated with risdiplam. Recruitment for that trial began in October 2021.
Long-term efficacy and IT administration of SMA therapy. Several studies are looking at the long-term efficacy and alternate routes of administration of onasemnogene abeparvovec and other SMA therapies.
A one-time gene replacement therapy delivered via an IV infusion replaces the function of the missing or nonworking SMN1 gene with a new, working copy of the SMN1 gene. FDA approved in 2019, it is authorized for use in patients with SMA up to 2 years of age.
The latest data from an ongoing, long-term follow-up safety study of onasemnogene abeparvovec, published in May 2021, suggest that the treatment’s effects persist more than 5 years after treatment. Researchers followed 13 infants with symptomatic SMA type 1 since the beginning of the phase 1 clinical trial of the gene transfer therapy. All patients who received the therapeutic dose maintained their baseline motor function, and two of the patients actually improved without other SMN-targeted treatment. At a median 6.2 years after they received treatment, all were alive and none needed permanent ventilation.14
After a 2-year hold by the FDA, a study of IT administration of onasemnogene abeparvovec is now enrolling patients. Citing concerns from animal studies that IT administration might result in dorsal root ganglia injury, the FDA issued a partial hold on the STRONG trial in 2019. Following positive study results in nonhuman primates, the FDA announced the trial can continue. Novartis is launching a new phase 3 STEER trial to test the drug delivered intrathecally in patients aged 2-18 years with Type 2 SMA. IT administration could allow the gene therapy to be used safely and effectively in more patients with SMA.
Efficacy of risdiplam in more patients. The first oral treatment for SMA was approved by the FDA in 2020. It’s given once per day in patients with SMA of all ages and disease types. The drug increases functional SMN protein production by the SMN2 gene.
A July 2021 publication of the results of the FIREFISH study found that infants with Type I SMA treated with risdiplam for 12 months were significantly more likely to achieve motor milestones, such as sitting without support, compared with untreated infants with Type 1 SMA.15 Risdiplam is also effective in older patients with Type 2 or 3 SMA, according to results published in December from the SUNFISH clinical trial.16 Another study, RAINBOWFISH, is studying safety and efficacy at 24 months in presymptomatic infants started on treatment at up to 6 weeks of age.
The efficacy of risdiplam in previously treated patients is the subject of JEWELFISH, an ongoing study in patients 6 months to 60 years with SMA. Preliminary data presented at the 2020 Virtual SMA Research and Clinical Care Meeting suggest treatment with risdiplam led to a median two-fold increase in the amount of blood SMN protein levels after 4 weeks, which was sustained for at least 24 months.17
Combination therapy. Among the more eagerly awaited results are those from studies of combination therapies, including those that combine approved SMN up-regulators with new non–SMN-targeted therapeutics.
“We’re seeing that while these three approved therapies have dramatic results, especially for infants who are treated presymptomatically, there are still unmet medical needs in those patients, particularly for older teens and adults whose disease may have progressed before they were able to start therapy,” said Jackie Glascock, PhD, vice president of research for Cure SMA.
Of particular interest are studies of myostatin inhibitors, therapeutics that block the production of the protein myostatin. Myostatin acts on muscle cells to reduce muscle growth. Animal studies suggest that inhibiting myostatin increases muscle mass, which could be important in patients with muscle loss due to SMA.
Three experimental myostatin inhibitors are currently in clinical trials. MANATEE is a global phase 2-3 trial that aims to evaluate the safety and efficacy of the antimyostatin antibody GYM329 (RO7204239) in combination with risdiplam. SAPPHIRE is a phase 3 trial of apitegromab (SRK-015) in combination with nusinersen or risdiplam. RESILIANT is a phase 3 trial of tadefgrobep alfa in combination with other treatments.
A trial is underway to study the efficacy and safety of nusinersen in patients with persistent symptoms of SMA after treatment with the gene therapy. The phase 4 study, RESPOND, is enrolling children aged 2-36 months.
What’s needed next
Despite the advances in treatment and patient care, Dr. Brandsema, Dr. Schroth, and Dr. Glascock note that there remain unmet needs in the SMA community in a variety of areas.
Increased focus on adults with SMA. Before nusinersen, treatment of SMA mainly involved treating its symptoms. Many patients stopped seeing their neurologist, relying more heavily on pulmonary care specialists and/or primary care providers to address breathing, nutrition, and mobility problems. “Now with the approval of these treatments, they’re coming back to see their neurologists and are becoming more visible in the SMA community,” Dr. Schroth said.
Despite this re-emergence, a 2020 meta-analysis of studies on adults with SMA found a paucity of data on physical and occupational therapy, respiratory management, mental health care, and palliative care.18
“There is just so much work we need to do in the area of adult clinical care of SMA.”
Treatment algorithms. While the development of the newborn screening algorithm and revised patient care guidelines are helpful resources, clinicians still face uncertainty when choosing which therapy will work best for their patients. Treatment algorithms that help clinicians figure out what therapy or combination of therapies will offer the best outcomes for individual patients are desperately needed, Dr. Brandsema said.
“Each person’s experience of this disease is so unique to the individual based partly on their genetics and partly on the factors about what got them into care and how compliant they are with everything we’re trying to do to help them,” he said. “Biomarkers would help clinicians create personalized treatment plans for each patient.”
More basic science. While scientists have a good understanding of the SMN gene, there are many unanswered questions about the function of the SMN protein and its relationship to motor neuron loss. SMN is a ubiquitously expressed protein, and its function in other cell types is largely unknown. Despite all of the research advances, there is much basic science left to be done.
“We are strongly advocating to regulatory authorities that these aren’t cures and we need to continue to invest in the basic research,” Dr. Glascock said. “These biological questions that pertain to SMN and its function and expression really drive drug development. I really think that understanding those pathways better will lead us to more druggable targets.”
Two deaths from liver failure linked to spinal muscular atrophy drug
Two children taking the gene therapy drug onasemnogene abeparvovec (Zolgensma, Novartis) for spinal muscular atrophy (SMA) have died from acute liver failure, according to a statement issued by the drug’s manufacturer.
The patients were 4 months and 28 months of age and lived in Russia and Kazakhstan. They died 5-6 weeks after infusion with Zolgensma and approximately 1-10 days after the initiation of a corticosteroid taper.
These are the first known fatal cases of acute liver failure associated with the drug, which the company notes was a known side effect included in the product label and in a boxed warning in the United States.
“Following two recent patient fatalities, and in alignment with health authorities, we will be updating the labeling to specify that fatal acute liver failure has been reported,” the statement reads.
“While this is important safety information, it is not a new safety signal,” it adds.
Rare genetic disorder
SMA is a rare genetic disorder that affects about 1 in 10,000 newborns. Patients with SMA lack a working copy of the survival motor neuron 1 (SMN1) gene, which encodes a protein called SMN that is critical for the maintenance and function of motor neurons.
Without this protein, motor neurons eventually die, causing debilitating and progressive muscle weakness that affects the ability to walk, eat, and breathe.
Zolgensma, a one-time gene replacement therapy delivered via intravenous infusion, replaces the function of the missing or nonworking SMN1 gene with a new, working copy of the SMN1 gene.
The first gene therapy treatment for SMA, it was approved by the U.S. Food and Drug Administration in 2019 for patients with SMA up to 2 years of age. It is also the most expensive drug in the world, costing about $2.1 million for a one-time treatment.
“We have notified health authorities in all markets where Zolgensma is used, including the FDA, and are communicating to relevant healthcare professionals as an additional step in markets where this action is supported by health authorities,” the manufacturer’s statement says.
Studies have suggested that the treatment’s effects persist more than 5 years after infusion.
Clinical trials currently underway by Novartis are studying the drug’s long-term efficacy and safety and its potential use in older patients.
The company is also leading the phase 3 clinical trial STEER to test intrathecal (IT) administration of the drug in patients ages 2-18 years who have type 2 SMA.
That trial began late last year after the FDA lifted a 2-year partial hold on an earlier study. The FDA halted the STRONG trial in 2019, citing concerns from animal studies that IT administration may result in dorsal root ganglia injury. The partial hold was released last fall following positive study results in nonhuman primates.
None of the current trials will be affected by the two deaths reported, according to a Novartis spokesperson.
Kelli Whitlock Burton is a staff writer/reporter for Medscape Neurology and MDedge Neurology.
References
1. Viscidi E et al. Comparative all-cause mortality among a large population of patients with spinal muscular atrophy versus matched controls. Neurol Ther. 2022 Mar;11(1):449-457. doi: 10.1007/s40120-021-00307-7.
2. Finkel RS et al. Observational study of spinal muscular atrophy type I and implications for clinical trials. Neurology. 2014 Aug 26;83(9):810-817. doi: 10.1212/WNL.0000000000000741.
3. Klotz J et al. Advances in the therapy of spinal muscular atrophy. J Pediatr. 2021 Sep;236:13-20.e1. doi: 10.1016/j.jpeds.2021.06.033.
4. Curry M et al. Awareness screening and referral patterns among pediatricians in the United States related to early clinical features of spinal muscular atrophy (SMA). BMC Pediatr. 2021 May;21(1):236. doi: 10.1186/s12887-021-02692-2.
5. SMArt Moves. https://smartmoves.curesma.org/
6. Swoboda KJ et al. Natural history of denervation in SMA: Relation to age, SMN2 copy number, and function. Ann Neurol. 2005 May;57(5):704-12. doi: 10.1002/ana.20473.
7. Glascock J et al. Treatment algorithm for infants diagnosed with spinal muscular atrophy through newborn screening. J Neuromuscul Dis. 2018;5(2):145-158. doi: 10.3233/JND-180304.
8. Glascock J et al. Revised recommendations for the treatment of infants diagnosed with spinal muscular atrophy via newborn screening who have 4 copies of SMN2. J Neuromuscul Dis. 2020;7(2):97-100. doi: 10.3233/JND-190468.
9. Talbot K, Tizzano EF. The clinical landscape for SMA in a new therapeutic era. Gene Ther. 2017 Sep;24(9):529-533. doi: 10.1038/gt.2017.52.
10. Mercuri E et al. Diagnosis and management of spinal muscular atrophy: Part 1: Recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscul Disord. 2018 Feb;28(2):103-115. doi: 10.1016/j.nmd.2017.11.005.
11. Finkel RS et al. Diagnosis and management of spinal muscular atrophy: Part 2: Pulmonary and acute care; medications, supplements and immunizations; other organ systems; and ethics. Neuromuscul Disord. 2018 Mar;28(3):197-207. doi: 10.1016/j.nmd.2017.11.004.
12. Pascual SI et al. Ongoing phase 2/3 DEVOTE (232SM203) randomized, controlled study to explore high-dose nusinersen in SMA: Part A interim results and Part B enrollment update. Presented at MDA Clinical and Scientific Conference 2021, Mar 15-18.
13. Finkel RS et al. Scientific rationale for a higher dose of nusinersen. Presented at 2021 Cure SMA Annual Meeting, Jun 9-11. Abstract P46.
14. Mendell JR et al. Five-year extension results of the phase 1 START trial of onasemnogene abeparvovec in spinal muscular atrophy. JAMA Neurol. 2021 Jul;78(7):834-841. doi: 10.1001/jamaneurol.2021.1272.
15. Darras BT et al. Risdiplam-treated infants with type 1 spinal muscular atrophy versus historical controls. N Engl J Med. 2021 Jul 29;385(5):427-435. doi: 10.1056/NEJMoa2102047.
16. Mercuri E et al. Safety and efficacy of once-daily risdiplam in type 2 and non-ambulant type 3 spinal muscular atrophy (SUNFISH part 2): A phase 3, double-blind, randomised, placebo-controlled trial. Lancet Neurol. 2022 Jan;21(1):42-52. doi: 10.1016/S1474-4422(21)00367-7. Erratum in: Lancet Neurol. 2022 Feb;21(2):e2. doi: 10.1016/S1474-4422(22)00006-0. Correction in: Lancet Neurol. 2022 Mar;21(3):e3. doi: 10.1016/S1474-4422(22)00038-2.
17. Genentech announces 2-year risdiplam data from SUNFISH and new data from JEWELFISH in infants, children and adults with SMA. https://www.curesma.org/genentech-risdiplam-data-conference-2020/
18. Wan HWY et al. Health, wellbeing and lived experiences of adults with SMA: a scoping systematic review. Orphanet J Rare Dis. 2020;15(1):70. doi: 10.1186/s13023-020-1339-3.
Racial disparities in preventive services use seen among patients with spina bifida or cerebral palsy
Black adults also had lower odds of having a bone density screening, compared with White adults. Plus, comorbidities were highest among the Black patients, according to the paper, which was published in Annals of Family Medicine.
Elham Mahmoudi, PhD, and her coauthors examined private insurance claims from 11,635 patients with cerebral palsy (CP) or spina bifida over ten years from 2007 to 2017. The researchers analyzed comorbidities and compared the rates of different psychological, cardiometabolic, and musculoskeletal conditions among these patients.
Only 23% of Hispanic participants and 18% of Black participants attended an annual wellness visit, compared with 32% of the White participants.
Only 1% of Black and 2% of White participants received any bone density screening (odds ratio = 0.54, 95% confidence interval [CI], 0.31-0.95), a service that is essential for catching a patient’s potential risk for osteoporosis and fractures.
According to the researchers, patients accessed services such as bone density scans, cholesterol assessments, diabetes screenings, and annual wellness visits less than recommended for people with those chronic conditions.
“People with spina bifida and cerebral palsy have complex care needs. We know through our work that chronic conditions are much higher among them compared with adults without disabilities,” Dr. Mahmoudi, associate professor in the department of family medicine at University of Michigan, Ann Arbor, said in an interview. “I was surprised to see even with private insurance, the rate of using preventative services is so low among White people and minority populations.”
Comorbidities highest in Black participants
Black adults had the highest comorbidity score of 2.5, and Hispanic adults had the lowest comorbidity score of 1.8. For White adults in the study, the comorbidity score was 2.0.
Osteoporosis, a common concern for people with spina bifida or cerebral palsy, was detected in around 4% of all participants. Osteoarthritis was detected in 13.38% of Black participants, versus 8.53% of Hispanic participants and 11.09% of White participants.
Diabetes and hypertension were more common among Black participants than among Hispanic and White participants. The percentages of Black patients with hypertension and diabetes were 16.5% and 39.89%, respectively. Among the Hispanic and White adults, the percentages with hypertension were 22.3% and 28.2%, respectively, according to the paper.
Disparities in access
Jamil Paden, racial and health equity manager at the Christopher and Dana Reeve Foundation, said getting access to literature, transportation, tables, chairs, weigh scales, and imaging equipment that accommodate the needs of people with disabilities are some of the biggest challenges for people with disabilities who are trying to receive care.

“It’s not a one size fits all, we have to recognize that if someone doesn’t see themselves in a particular place, then it makes it more challenging for them to feel comfortable speaking up and saying things about their health, which would prevent a person from saying something early on,” Mr. Paden said in an interview. “That particular issue will continue to grow and become more of a health risk, or health challenge down the line.”
Mr. Paden emphasized intersections between class, race, and circumstances which can, together, make health care less equitable for people with disabilities, especially in underserved communities and communities of color. He urged health care providers to distance their practices from a “one size fits all” approach to treatment and engage in their patients’ individual lives and communities.
“It’s not enough to just say, Hey, you have a disability. So let me treat your disability ... You have to recognize that although a patient may have a dire diagnosis, they also are a person of color, and they have to navigate different aspects of life from their counterparts,” he said.
Dr. Mahmoudi said patient and provider understanding of the disability is often lacking. She recommended advocating for patients, noting that giving both patients and providers the tools to further educate themselves and apply that to their regular visits is a good first step.
“Just having access to a facility doesn’t mean they will get the services they need. Preventative services that are recommended for people with disabilities differ from the general population. Providers should be educated about that and the patient needs to be educated about that,” she added.
“Patients who do not approach clinicians get lost in the system. Maybe many facilities are not disability friendly, or they need health literacy. If they don’t know they are at risk for osteoporosis, for example, then they won’t ask,” Dr. Mahmoudi said.
The study was funded by The National Institute on Disability, Independent Living, and Rehabilitation Research. Dr. Mahmoudi and Mr. Paden report no relevant financial relationships.
Black adults also had lower odds of having a bone density screening, compared with White adults. Plus, comorbidities were highest among the Black patients, according to the paper, which was published in Annals of Family Medicine.
Elham Mahmoudi, PhD, and her coauthors examined private insurance claims from 11,635 patients with cerebral palsy (CP) or spina bifida over ten years from 2007 to 2017. The researchers analyzed comorbidities and compared the rates of different psychological, cardiometabolic, and musculoskeletal conditions among these patients.
Only 23% of Hispanic participants and 18% of Black participants attended an annual wellness visit, compared with 32% of the White participants.
Only 1% of Black and 2% of White participants received any bone density screening (odds ratio = 0.54, 95% confidence interval [CI], 0.31-0.95), a service that is essential for catching a patient’s potential risk for osteoporosis and fractures.
According to the researchers, patients accessed services such as bone density scans, cholesterol assessments, diabetes screenings, and annual wellness visits less than recommended for people with those chronic conditions.
“People with spina bifida and cerebral palsy have complex care needs. We know through our work that chronic conditions are much higher among them compared with adults without disabilities,” Dr. Mahmoudi, associate professor in the department of family medicine at University of Michigan, Ann Arbor, said in an interview. “I was surprised to see even with private insurance, the rate of using preventative services is so low among White people and minority populations.”
Comorbidities highest in Black participants
Black adults had the highest comorbidity score of 2.5, and Hispanic adults had the lowest comorbidity score of 1.8. For White adults in the study, the comorbidity score was 2.0.
Osteoporosis, a common concern for people with spina bifida or cerebral palsy, was detected in around 4% of all participants. Osteoarthritis was detected in 13.38% of Black participants, versus 8.53% of Hispanic participants and 11.09% of White participants.
Diabetes and hypertension were more common among Black participants than among Hispanic and White participants. The percentages of Black patients with hypertension and diabetes were 16.5% and 39.89%, respectively. Among the Hispanic and White adults, the percentages with hypertension were 22.3% and 28.2%, respectively, according to the paper.
Disparities in access
Jamil Paden, racial and health equity manager at the Christopher and Dana Reeve Foundation, said getting access to literature, transportation, tables, chairs, weigh scales, and imaging equipment that accommodate the needs of people with disabilities are some of the biggest challenges for people with disabilities who are trying to receive care.
“It’s not a one size fits all, we have to recognize that if someone doesn’t see themselves in a particular place, then it makes it more challenging for them to feel comfortable speaking up and saying things about their health, which would prevent a person from saying something early on,” Mr. Paden said in an interview. “That particular issue will continue to grow and become more of a health risk, or health challenge down the line.”
Mr. Paden emphasized intersections between class, race, and circumstances which can, together, make health care less equitable for people with disabilities, especially in underserved communities and communities of color. He urged health care providers to distance their practices from a “one size fits all” approach to treatment and engage in their patients’ individual lives and communities.
“It’s not enough to just say, Hey, you have a disability. So let me treat your disability ... You have to recognize that although a patient may have a dire diagnosis, they also are a person of color, and they have to navigate different aspects of life from their counterparts,” he said.
Dr. Mahmoudi said patient and provider understanding of the disability is often lacking. She recommended advocating for patients, noting that giving both patients and providers the tools to further educate themselves and apply that to their regular visits is a good first step.
“Just having access to a facility doesn’t mean they will get the services they need. Preventative services that are recommended for people with disabilities differ from the general population. Providers should be educated about that and the patient needs to be educated about that,” she added.
“Patients who do not approach clinicians get lost in the system. Maybe many facilities are not disability friendly, or they need health literacy. If they don’t know they are at risk for osteoporosis, for example, then they won’t ask,” Dr. Mahmoudi said.
The study was funded by The National Institute on Disability, Independent Living, and Rehabilitation Research. Dr. Mahmoudi and Mr. Paden report no relevant financial relationships.
Black adults also had lower odds of having a bone density screening, compared with White adults. Plus, comorbidities were highest among the Black patients, according to the paper, which was published in Annals of Family Medicine.
Elham Mahmoudi, PhD, and her coauthors examined private insurance claims from 11,635 patients with cerebral palsy (CP) or spina bifida over ten years from 2007 to 2017. The researchers analyzed comorbidities and compared the rates of different psychological, cardiometabolic, and musculoskeletal conditions among these patients.
Only 23% of Hispanic participants and 18% of Black participants attended an annual wellness visit, compared with 32% of the White participants.
Only 1% of Black and 2% of White participants received any bone density screening (odds ratio = 0.54, 95% confidence interval [CI], 0.31-0.95), a service that is essential for catching a patient’s potential risk for osteoporosis and fractures.
According to the researchers, patients accessed services such as bone density scans, cholesterol assessments, diabetes screenings, and annual wellness visits less than recommended for people with those chronic conditions.
“People with spina bifida and cerebral palsy have complex care needs. We know through our work that chronic conditions are much higher among them compared with adults without disabilities,” Dr. Mahmoudi, associate professor in the department of family medicine at University of Michigan, Ann Arbor, said in an interview. “I was surprised to see even with private insurance, the rate of using preventative services is so low among White people and minority populations.”
Comorbidities highest in Black participants
Black adults had the highest comorbidity score of 2.5, and Hispanic adults had the lowest comorbidity score of 1.8. For White adults in the study, the comorbidity score was 2.0.
Osteoporosis, a common concern for people with spina bifida or cerebral palsy, was detected in around 4% of all participants. Osteoarthritis was detected in 13.38% of Black participants, versus 8.53% of Hispanic participants and 11.09% of White participants.
Diabetes and hypertension were more common among Black participants than among Hispanic and White participants. The percentages of Black patients with hypertension and diabetes were 16.5% and 39.89%, respectively. Among the Hispanic and White adults, the percentages with hypertension were 22.3% and 28.2%, respectively, according to the paper.
Disparities in access
Jamil Paden, racial and health equity manager at the Christopher and Dana Reeve Foundation, said getting access to literature, transportation, tables, chairs, weigh scales, and imaging equipment that accommodate the needs of people with disabilities are some of the biggest challenges for people with disabilities who are trying to receive care.
“It’s not a one size fits all, we have to recognize that if someone doesn’t see themselves in a particular place, then it makes it more challenging for them to feel comfortable speaking up and saying things about their health, which would prevent a person from saying something early on,” Mr. Paden said in an interview. “That particular issue will continue to grow and become more of a health risk, or health challenge down the line.”
Mr. Paden emphasized intersections between class, race, and circumstances which can, together, make health care less equitable for people with disabilities, especially in underserved communities and communities of color. He urged health care providers to distance their practices from a “one size fits all” approach to treatment and engage in their patients’ individual lives and communities.
“It’s not enough to just say, Hey, you have a disability. So let me treat your disability ... You have to recognize that although a patient may have a dire diagnosis, they also are a person of color, and they have to navigate different aspects of life from their counterparts,” he said.
Dr. Mahmoudi said patient and provider understanding of the disability is often lacking. She recommended advocating for patients, noting that giving both patients and providers the tools to further educate themselves and apply that to their regular visits is a good first step.
“Just having access to a facility doesn’t mean they will get the services they need. Preventative services that are recommended for people with disabilities differ from the general population. Providers should be educated about that and the patient needs to be educated about that,” she added.
“Patients who do not approach clinicians get lost in the system. Maybe many facilities are not disability friendly, or they need health literacy. If they don’t know they are at risk for osteoporosis, for example, then they won’t ask,” Dr. Mahmoudi said.
The study was funded by The National Institute on Disability, Independent Living, and Rehabilitation Research. Dr. Mahmoudi and Mr. Paden report no relevant financial relationships.
FROM ANNALS OF FAMILY MEDICINE
ALS drug gets FDA panel thumbs-up after rare second look
In a rare second review of a new drug application,
By a vote of 7-2, the FDA Peripheral and Central Nervous System Drugs Advisory Committee reversed course on AMX0035 (Amylyx Pharmaceuticals), a combination of sodium phenylbutyrate and taurursodiol.
The panel previously voted 6-4 to reject the drug, ruling that data provided by Amylyx had failed to demonstrate that the survival benefit reported in the only clinical trial of AMX0035 so far was a direct result of the drug.
This time, two panelists who previously voted no were swayed by the drug maker’s new analysis of previously presented research, more than 1,300 public comments in support of the drug, supportive testimony from ALS patients and clinicians, and assurances from company executives that Amylyx would pull the drug from the market if results of an ongoing phase 3 clinical trial show the drug doesn’t work.
“As in March, today we have to have an internal dialogue between our scientific scrutiny and clinical compassion,” said Liana G. Apostolova, MD, from Indiana University, Indianapolis, who originally voted against the application.
“Today I also saw additional confirmatory evidence that was not unequivocally persuasive but was nonetheless reassuring,” Dr. Apostolova said. “Because of that I am voting in support of AMX0035.”
A rare second chance
ALS (Lou Gehrig’s disease) is a progressive, fatal neurodegenerative disease affecting nerve cells in the brain and spinal cord that causes loss of motor control. It is rare, affecting about 30,000 people in the United States with another 5,000 new cases diagnosed each year. Most people with the disease die within 2 years of diagnosis.
The FDA has approved two therapies for ALS, but both have limited efficacy.
Typically, FDA approval requires two large studies or one study with a “very persuasive” effect on survival.
Amylyx’s application is based on a single study, the multicenter, two-phase CENTAUR trial. In that trial, 137 people with ALS received AMX0035 or placebo for 24 weeks.
Researchers found that patients receiving AMX0035 had a 25% slower decline in function, compared with the those taking placebo. A change of 20% or more is considered clinically meaningful.
The investigators also found a statistically significant median difference of 4.8 months in time to death, first hospitalization, or tracheostomy/permanent assisted ventilation in the group originally assigned to receive AMX0035 compared with the group originally assigned to receive placebo (hazard ratio, 0.62; P = .023).
In the panel’s previous vote against the drug application, members cited several issues with the study, concluding that it did not offer persuasive or robust evidence of efficacy. They also cited missing data assumptions in the primary analysis, issues of randomization and imbalances in concomitant use of riluzole and edaravone, the two FDA-approved drugs for ALS.
The FDA later requested additional information from Amylyx, delayed its final ruling on the new drug application to Sept. 29, and called for a second review meeting – a virtually unheard-of move.
An FDA review posted in advance of the meeting Sept. 29 had hinted at a different outcome. In that report, regulators said new data from Amylyx were not “sufficiently independent or persuasive” to establish effectiveness.
However, FDA officials in the meeting stressed the importance of considering unmet medical need in ALS in the panel’s decision-making process.
“Recognizing the substantial unmet medical need in ALS, we feel that it is important that the committee is afforded the opportunity to consider this new information, along with the information presented at the prior meeting, in that context,” Billy Dunn, MD, director of the FDA Office of Neuroscience, said during the meeting.
Panelists heard additional data that Amylyx claims confirms the results of the CENTAUR study, including new analyses of the previously submitted survival data and new data from that study and an open-label extension.
They also provided new information on a biomarker data from a phase 2 study of AMX0035 to treat Alzheimer’s disease.
“I think we note the limitations of the analyses, but we still haven’t taken it off the table that they could be considered as confirmatory evidence and that’s why we’re here today,” said Teresa Buracchio, MD, director of the division of neurology for the FDA.
Two members of the panel who voted no in March stuck with that position at the Sept. 29 meeting.
“Unfortunately, I don’t believe the new evidence we’ve reviewed, while promising, combined with that prior evidence, constitutes substantial evidence of effectiveness,” said panelist Caleb Alexander, MD, a professor of epidemiology and medicine at the Johns Hopkins University Center for Drug Safety and Effectiveness, Baltimore.
Dr. Alexander, who also voted no in March, said that post hoc data presented at the meeting were not enough to assuage concerns that led him and others to reject the drug in March.
A challenging situation
Amylyx is currently leading the 48-week international, phase 3, placebo-controlled PHOENIX clinical trial of AMX0035. The study has enrolled about half of its 600-patient target.
“Undoubtedly, the results of the phase 3 study would be highly informative for a regulatory decision on the current ... review for AMX0035,” said Emily Freilich, MD, of the FDA.
However, results aren’t expected until late 2023 or early 2024, which “places the agency in a challenging situation of potentially making a regulatory decision that may not be subsequently confirmed by the results of the ongoing study.”
In June, Amylyx received conditional approval in Canada for the drug, but final approval depends on the outcome of the PHOENIX trial. The FDA does not offer a conditional approval track.
“If AMX0035 is not approved now, the FDA anticipated decision will likely happen in 2025, underscoring the critical importance of today’s outcome,” said Tammy Sarnelli, MPAHC, global head of Regulatory Affairs for Amylyx Pharmaceuticals.
If the FDA were to approve AMX0035 and results from the PHOENIX trial ultimately fail to prove efficacy, Justin Klee, co-CEO and cofounder of Amylyx Pharmaceuticals, said the company would withdraw the drug.
“To be clear, if PHOENIX is not successful, we will do what is right for patients, which includes voluntarily removing the product from the market,” Mr. Klee said.
Regardless of the company’s decision, FDA officials noted that the agency does have the ability to recall a drug from the market if studies show that it no longer meets requirements for approval.
“The FDA, with all due respect, significantly understates the complexity and likelihood of their pulling a product from the market,” Dr. Alexander said. “Whether or not they can ultimately pull a product from the market is no substitute for the evidentiary thresholds that are required for market access.”
A version of this article first appeared on Medscape.com.
In a rare second review of a new drug application,
By a vote of 7-2, the FDA Peripheral and Central Nervous System Drugs Advisory Committee reversed course on AMX0035 (Amylyx Pharmaceuticals), a combination of sodium phenylbutyrate and taurursodiol.
The panel previously voted 6-4 to reject the drug, ruling that data provided by Amylyx had failed to demonstrate that the survival benefit reported in the only clinical trial of AMX0035 so far was a direct result of the drug.
This time, two panelists who previously voted no were swayed by the drug maker’s new analysis of previously presented research, more than 1,300 public comments in support of the drug, supportive testimony from ALS patients and clinicians, and assurances from company executives that Amylyx would pull the drug from the market if results of an ongoing phase 3 clinical trial show the drug doesn’t work.
“As in March, today we have to have an internal dialogue between our scientific scrutiny and clinical compassion,” said Liana G. Apostolova, MD, from Indiana University, Indianapolis, who originally voted against the application.
“Today I also saw additional confirmatory evidence that was not unequivocally persuasive but was nonetheless reassuring,” Dr. Apostolova said. “Because of that I am voting in support of AMX0035.”
A rare second chance
ALS (Lou Gehrig’s disease) is a progressive, fatal neurodegenerative disease affecting nerve cells in the brain and spinal cord that causes loss of motor control. It is rare, affecting about 30,000 people in the United States with another 5,000 new cases diagnosed each year. Most people with the disease die within 2 years of diagnosis.
The FDA has approved two therapies for ALS, but both have limited efficacy.
Typically, FDA approval requires two large studies or one study with a “very persuasive” effect on survival.
Amylyx’s application is based on a single study, the multicenter, two-phase CENTAUR trial. In that trial, 137 people with ALS received AMX0035 or placebo for 24 weeks.
Researchers found that patients receiving AMX0035 had a 25% slower decline in function, compared with the those taking placebo. A change of 20% or more is considered clinically meaningful.
The investigators also found a statistically significant median difference of 4.8 months in time to death, first hospitalization, or tracheostomy/permanent assisted ventilation in the group originally assigned to receive AMX0035 compared with the group originally assigned to receive placebo (hazard ratio, 0.62; P = .023).
In the panel’s previous vote against the drug application, members cited several issues with the study, concluding that it did not offer persuasive or robust evidence of efficacy. They also cited missing data assumptions in the primary analysis, issues of randomization and imbalances in concomitant use of riluzole and edaravone, the two FDA-approved drugs for ALS.
The FDA later requested additional information from Amylyx, delayed its final ruling on the new drug application to Sept. 29, and called for a second review meeting – a virtually unheard-of move.
An FDA review posted in advance of the meeting Sept. 29 had hinted at a different outcome. In that report, regulators said new data from Amylyx were not “sufficiently independent or persuasive” to establish effectiveness.
However, FDA officials in the meeting stressed the importance of considering unmet medical need in ALS in the panel’s decision-making process.
“Recognizing the substantial unmet medical need in ALS, we feel that it is important that the committee is afforded the opportunity to consider this new information, along with the information presented at the prior meeting, in that context,” Billy Dunn, MD, director of the FDA Office of Neuroscience, said during the meeting.
Panelists heard additional data that Amylyx claims confirms the results of the CENTAUR study, including new analyses of the previously submitted survival data and new data from that study and an open-label extension.
They also provided new information on a biomarker data from a phase 2 study of AMX0035 to treat Alzheimer’s disease.
“I think we note the limitations of the analyses, but we still haven’t taken it off the table that they could be considered as confirmatory evidence and that’s why we’re here today,” said Teresa Buracchio, MD, director of the division of neurology for the FDA.
Two members of the panel who voted no in March stuck with that position at the Sept. 29 meeting.
“Unfortunately, I don’t believe the new evidence we’ve reviewed, while promising, combined with that prior evidence, constitutes substantial evidence of effectiveness,” said panelist Caleb Alexander, MD, a professor of epidemiology and medicine at the Johns Hopkins University Center for Drug Safety and Effectiveness, Baltimore.
Dr. Alexander, who also voted no in March, said that post hoc data presented at the meeting were not enough to assuage concerns that led him and others to reject the drug in March.
A challenging situation
Amylyx is currently leading the 48-week international, phase 3, placebo-controlled PHOENIX clinical trial of AMX0035. The study has enrolled about half of its 600-patient target.
“Undoubtedly, the results of the phase 3 study would be highly informative for a regulatory decision on the current ... review for AMX0035,” said Emily Freilich, MD, of the FDA.
However, results aren’t expected until late 2023 or early 2024, which “places the agency in a challenging situation of potentially making a regulatory decision that may not be subsequently confirmed by the results of the ongoing study.”
In June, Amylyx received conditional approval in Canada for the drug, but final approval depends on the outcome of the PHOENIX trial. The FDA does not offer a conditional approval track.
“If AMX0035 is not approved now, the FDA anticipated decision will likely happen in 2025, underscoring the critical importance of today’s outcome,” said Tammy Sarnelli, MPAHC, global head of Regulatory Affairs for Amylyx Pharmaceuticals.
If the FDA were to approve AMX0035 and results from the PHOENIX trial ultimately fail to prove efficacy, Justin Klee, co-CEO and cofounder of Amylyx Pharmaceuticals, said the company would withdraw the drug.
“To be clear, if PHOENIX is not successful, we will do what is right for patients, which includes voluntarily removing the product from the market,” Mr. Klee said.
Regardless of the company’s decision, FDA officials noted that the agency does have the ability to recall a drug from the market if studies show that it no longer meets requirements for approval.
“The FDA, with all due respect, significantly understates the complexity and likelihood of their pulling a product from the market,” Dr. Alexander said. “Whether or not they can ultimately pull a product from the market is no substitute for the evidentiary thresholds that are required for market access.”
A version of this article first appeared on Medscape.com.
In a rare second review of a new drug application,
By a vote of 7-2, the FDA Peripheral and Central Nervous System Drugs Advisory Committee reversed course on AMX0035 (Amylyx Pharmaceuticals), a combination of sodium phenylbutyrate and taurursodiol.
The panel previously voted 6-4 to reject the drug, ruling that data provided by Amylyx had failed to demonstrate that the survival benefit reported in the only clinical trial of AMX0035 so far was a direct result of the drug.
This time, two panelists who previously voted no were swayed by the drug maker’s new analysis of previously presented research, more than 1,300 public comments in support of the drug, supportive testimony from ALS patients and clinicians, and assurances from company executives that Amylyx would pull the drug from the market if results of an ongoing phase 3 clinical trial show the drug doesn’t work.
“As in March, today we have to have an internal dialogue between our scientific scrutiny and clinical compassion,” said Liana G. Apostolova, MD, from Indiana University, Indianapolis, who originally voted against the application.
“Today I also saw additional confirmatory evidence that was not unequivocally persuasive but was nonetheless reassuring,” Dr. Apostolova said. “Because of that I am voting in support of AMX0035.”
A rare second chance
ALS (Lou Gehrig’s disease) is a progressive, fatal neurodegenerative disease affecting nerve cells in the brain and spinal cord that causes loss of motor control. It is rare, affecting about 30,000 people in the United States with another 5,000 new cases diagnosed each year. Most people with the disease die within 2 years of diagnosis.
The FDA has approved two therapies for ALS, but both have limited efficacy.
Typically, FDA approval requires two large studies or one study with a “very persuasive” effect on survival.
Amylyx’s application is based on a single study, the multicenter, two-phase CENTAUR trial. In that trial, 137 people with ALS received AMX0035 or placebo for 24 weeks.
Researchers found that patients receiving AMX0035 had a 25% slower decline in function, compared with the those taking placebo. A change of 20% or more is considered clinically meaningful.
The investigators also found a statistically significant median difference of 4.8 months in time to death, first hospitalization, or tracheostomy/permanent assisted ventilation in the group originally assigned to receive AMX0035 compared with the group originally assigned to receive placebo (hazard ratio, 0.62; P = .023).
In the panel’s previous vote against the drug application, members cited several issues with the study, concluding that it did not offer persuasive or robust evidence of efficacy. They also cited missing data assumptions in the primary analysis, issues of randomization and imbalances in concomitant use of riluzole and edaravone, the two FDA-approved drugs for ALS.
The FDA later requested additional information from Amylyx, delayed its final ruling on the new drug application to Sept. 29, and called for a second review meeting – a virtually unheard-of move.
An FDA review posted in advance of the meeting Sept. 29 had hinted at a different outcome. In that report, regulators said new data from Amylyx were not “sufficiently independent or persuasive” to establish effectiveness.
However, FDA officials in the meeting stressed the importance of considering unmet medical need in ALS in the panel’s decision-making process.
“Recognizing the substantial unmet medical need in ALS, we feel that it is important that the committee is afforded the opportunity to consider this new information, along with the information presented at the prior meeting, in that context,” Billy Dunn, MD, director of the FDA Office of Neuroscience, said during the meeting.
Panelists heard additional data that Amylyx claims confirms the results of the CENTAUR study, including new analyses of the previously submitted survival data and new data from that study and an open-label extension.
They also provided new information on a biomarker data from a phase 2 study of AMX0035 to treat Alzheimer’s disease.
“I think we note the limitations of the analyses, but we still haven’t taken it off the table that they could be considered as confirmatory evidence and that’s why we’re here today,” said Teresa Buracchio, MD, director of the division of neurology for the FDA.
Two members of the panel who voted no in March stuck with that position at the Sept. 29 meeting.
“Unfortunately, I don’t believe the new evidence we’ve reviewed, while promising, combined with that prior evidence, constitutes substantial evidence of effectiveness,” said panelist Caleb Alexander, MD, a professor of epidemiology and medicine at the Johns Hopkins University Center for Drug Safety and Effectiveness, Baltimore.
Dr. Alexander, who also voted no in March, said that post hoc data presented at the meeting were not enough to assuage concerns that led him and others to reject the drug in March.
A challenging situation
Amylyx is currently leading the 48-week international, phase 3, placebo-controlled PHOENIX clinical trial of AMX0035. The study has enrolled about half of its 600-patient target.
“Undoubtedly, the results of the phase 3 study would be highly informative for a regulatory decision on the current ... review for AMX0035,” said Emily Freilich, MD, of the FDA.
However, results aren’t expected until late 2023 or early 2024, which “places the agency in a challenging situation of potentially making a regulatory decision that may not be subsequently confirmed by the results of the ongoing study.”
In June, Amylyx received conditional approval in Canada for the drug, but final approval depends on the outcome of the PHOENIX trial. The FDA does not offer a conditional approval track.
“If AMX0035 is not approved now, the FDA anticipated decision will likely happen in 2025, underscoring the critical importance of today’s outcome,” said Tammy Sarnelli, MPAHC, global head of Regulatory Affairs for Amylyx Pharmaceuticals.
If the FDA were to approve AMX0035 and results from the PHOENIX trial ultimately fail to prove efficacy, Justin Klee, co-CEO and cofounder of Amylyx Pharmaceuticals, said the company would withdraw the drug.
“To be clear, if PHOENIX is not successful, we will do what is right for patients, which includes voluntarily removing the product from the market,” Mr. Klee said.
Regardless of the company’s decision, FDA officials noted that the agency does have the ability to recall a drug from the market if studies show that it no longer meets requirements for approval.
“The FDA, with all due respect, significantly understates the complexity and likelihood of their pulling a product from the market,” Dr. Alexander said. “Whether or not they can ultimately pull a product from the market is no substitute for the evidentiary thresholds that are required for market access.”
A version of this article first appeared on Medscape.com.
Polio: The unwanted sequel
Summer, since 1975, is traditionally a time for the BIG blockbusters to hit theaters. Some are new, others are sequels in successful franchises. Some anticipated, some not as much.
And, in summer 2022, we have the least-wanted sequel in modern history – Polio II: The Return.
Of course, this sequel isn’t in the theaters (unless the concessions staff isn’t washing their hands), definitely isn’t funny, and could potentially cost a lot more money than the latest Marvel Cinematic Universe flick.

Personally and professionally, I’m in the middle generation on the disease. I’m young enough that I never had to worry about catching it or having afflicted classmates. But, as a doctor, I’m old enough to still see the consequences. Like most neurologists, I have a handful of patients who had childhood polio, and still deal with the chronic weakness (and consequent pain and orthopedic issues it brings). Signing off on braces and other mobility aids for them is still commonplace.
One of my attendings in residency was the renowned Parkinson’s disease expert Abraham Lieberman. On rounds it was impossible not to notice his marked limp, a consequence of childhood polio, and he’d tell us what it was like, being a 6-year-old boy and dealing with the disease. You learn as much from hearing firsthand experiences as you do from textbooks.
And now the virus is showing up again. A few victims, a lot of virions circulating in waste water, but it shouldn’t be there at all.
We aren’t in the era when schoolchildren died or were crippled by it. Elementary school kids today don’t see classmates catch polio and never return to school, or see their grieving parents.
To take 1 year: More than 3,000 American children died of polio in 1952, and more than 21,000 were left with lifelong paralysis – many of them still among us.
When you think of an iron lung, you think of polio.
Those were the casualties in a war to save future generations from this, along with smallpox and other horrors.
But today, that war is mostly forgotten. And now scientific evidence is drowned out by whatever’s on Facebook and the hard-earned miracle of vaccination is ignored in favor of a nonmedical “social influencer” on YouTube.
So The majority of the population likely has nothing to worry about. But there may be segments that are hit hard, and when they are they will never accept the obvious reasons why. It will be part of a cover-up, or a conspiracy, or whatever the guy on Parler told them it was.
As doctors, we’re in the middle. We have to give patients the best recommendations we can, based on learning, evidence, and experience, but at the same time have to recognize their autonomy. I’m not following someone around to make sure they get vaccinated, or take the medication I prescribed.
But we’re also the ones who can be held legally responsible for bad outcomes, regardless of the actual facts of the matter. On the flip side, you don’t hear about someone suing a Facebook “influencer” for doling out inaccurate, potentially fatal, medical advice.
So cracks appear in herd immunity, and leaks will happen.
A few generations of neurologists, including mine, have completed training without considering polio in a differential diagnosis. It would, of course, get bandied about in grand rounds or at the conference table, but none of us really took it seriously. To us residents it was more of historical note. “Gone with the Wind” and the “Wizard of Oz” both came out in 1939, and while we all knew of them, none of us were going to be watching them at the theaters.
Unlike them, though, polio is trying make it back to prime time. It’s a sequel nobody wanted.
But here it is.
Dr. Block has a solo neurology practice in Scottsdale, Ariz.
Summer, since 1975, is traditionally a time for the BIG blockbusters to hit theaters. Some are new, others are sequels in successful franchises. Some anticipated, some not as much.
And, in summer 2022, we have the least-wanted sequel in modern history – Polio II: The Return.
Of course, this sequel isn’t in the theaters (unless the concessions staff isn’t washing their hands), definitely isn’t funny, and could potentially cost a lot more money than the latest Marvel Cinematic Universe flick.
Personally and professionally, I’m in the middle generation on the disease. I’m young enough that I never had to worry about catching it or having afflicted classmates. But, as a doctor, I’m old enough to still see the consequences. Like most neurologists, I have a handful of patients who had childhood polio, and still deal with the chronic weakness (and consequent pain and orthopedic issues it brings). Signing off on braces and other mobility aids for them is still commonplace.
One of my attendings in residency was the renowned Parkinson’s disease expert Abraham Lieberman. On rounds it was impossible not to notice his marked limp, a consequence of childhood polio, and he’d tell us what it was like, being a 6-year-old boy and dealing with the disease. You learn as much from hearing firsthand experiences as you do from textbooks.
And now the virus is showing up again. A few victims, a lot of virions circulating in waste water, but it shouldn’t be there at all.
We aren’t in the era when schoolchildren died or were crippled by it. Elementary school kids today don’t see classmates catch polio and never return to school, or see their grieving parents.
To take 1 year: More than 3,000 American children died of polio in 1952, and more than 21,000 were left with lifelong paralysis – many of them still among us.
When you think of an iron lung, you think of polio.
Those were the casualties in a war to save future generations from this, along with smallpox and other horrors.
But today, that war is mostly forgotten. And now scientific evidence is drowned out by whatever’s on Facebook and the hard-earned miracle of vaccination is ignored in favor of a nonmedical “social influencer” on YouTube.
So The majority of the population likely has nothing to worry about. But there may be segments that are hit hard, and when they are they will never accept the obvious reasons why. It will be part of a cover-up, or a conspiracy, or whatever the guy on Parler told them it was.
As doctors, we’re in the middle. We have to give patients the best recommendations we can, based on learning, evidence, and experience, but at the same time have to recognize their autonomy. I’m not following someone around to make sure they get vaccinated, or take the medication I prescribed.
But we’re also the ones who can be held legally responsible for bad outcomes, regardless of the actual facts of the matter. On the flip side, you don’t hear about someone suing a Facebook “influencer” for doling out inaccurate, potentially fatal, medical advice.
So cracks appear in herd immunity, and leaks will happen.
A few generations of neurologists, including mine, have completed training without considering polio in a differential diagnosis. It would, of course, get bandied about in grand rounds or at the conference table, but none of us really took it seriously. To us residents it was more of historical note. “Gone with the Wind” and the “Wizard of Oz” both came out in 1939, and while we all knew of them, none of us were going to be watching them at the theaters.
Unlike them, though, polio is trying make it back to prime time. It’s a sequel nobody wanted.
But here it is.
Dr. Block has a solo neurology practice in Scottsdale, Ariz.
Summer, since 1975, is traditionally a time for the BIG blockbusters to hit theaters. Some are new, others are sequels in successful franchises. Some anticipated, some not as much.
And, in summer 2022, we have the least-wanted sequel in modern history – Polio II: The Return.
Of course, this sequel isn’t in the theaters (unless the concessions staff isn’t washing their hands), definitely isn’t funny, and could potentially cost a lot more money than the latest Marvel Cinematic Universe flick.
Personally and professionally, I’m in the middle generation on the disease. I’m young enough that I never had to worry about catching it or having afflicted classmates. But, as a doctor, I’m old enough to still see the consequences. Like most neurologists, I have a handful of patients who had childhood polio, and still deal with the chronic weakness (and consequent pain and orthopedic issues it brings). Signing off on braces and other mobility aids for them is still commonplace.
One of my attendings in residency was the renowned Parkinson’s disease expert Abraham Lieberman. On rounds it was impossible not to notice his marked limp, a consequence of childhood polio, and he’d tell us what it was like, being a 6-year-old boy and dealing with the disease. You learn as much from hearing firsthand experiences as you do from textbooks.
And now the virus is showing up again. A few victims, a lot of virions circulating in waste water, but it shouldn’t be there at all.
We aren’t in the era when schoolchildren died or were crippled by it. Elementary school kids today don’t see classmates catch polio and never return to school, or see their grieving parents.
To take 1 year: More than 3,000 American children died of polio in 1952, and more than 21,000 were left with lifelong paralysis – many of them still among us.
When you think of an iron lung, you think of polio.
Those were the casualties in a war to save future generations from this, along with smallpox and other horrors.
But today, that war is mostly forgotten. And now scientific evidence is drowned out by whatever’s on Facebook and the hard-earned miracle of vaccination is ignored in favor of a nonmedical “social influencer” on YouTube.
So The majority of the population likely has nothing to worry about. But there may be segments that are hit hard, and when they are they will never accept the obvious reasons why. It will be part of a cover-up, or a conspiracy, or whatever the guy on Parler told them it was.
As doctors, we’re in the middle. We have to give patients the best recommendations we can, based on learning, evidence, and experience, but at the same time have to recognize their autonomy. I’m not following someone around to make sure they get vaccinated, or take the medication I prescribed.
But we’re also the ones who can be held legally responsible for bad outcomes, regardless of the actual facts of the matter. On the flip side, you don’t hear about someone suing a Facebook “influencer” for doling out inaccurate, potentially fatal, medical advice.
So cracks appear in herd immunity, and leaks will happen.
A few generations of neurologists, including mine, have completed training without considering polio in a differential diagnosis. It would, of course, get bandied about in grand rounds or at the conference table, but none of us really took it seriously. To us residents it was more of historical note. “Gone with the Wind” and the “Wizard of Oz” both came out in 1939, and while we all knew of them, none of us were going to be watching them at the theaters.
Unlike them, though, polio is trying make it back to prime time. It’s a sequel nobody wanted.
But here it is.
Dr. Block has a solo neurology practice in Scottsdale, Ariz.
First-ever Huntington staging system may jump-start drug development for early-stage disease
Researchers liken the Huntington’s disease Integrated Staging System (HD-ISS) to the system currently used to stage cancer. It groups patients according to their underlying biological, clinical, and functional characteristics.
It also includes criteria to biologically define Huntington’s disease stages across the entire disease spectrum, from birth to death, which is something that has not been done before. For now, the HD-ISS is only intended for research, but it could one day be modified for use in the clinic, investigators wrote.
“This systematization is of critical importance to select the most appropriate target population for clinical trials and studies,” said co-investigator Cristina Sampaio, MD, chief medical officer at the CHDI Foundation, Princeton, N.J.
“By providing a methodology to precisely define cases early in the neurodegenerative process, the HD-ISS will be instrumental in conducting trials in the very early disease stages,” Dr. Sampaio added.
The position paper was published in the July issue of the Lancet Neurology.
New approach needed
There is no approved therapy to slow Huntington’s disease progression. Clinical trials currently enroll patients with demonstrable symptoms, which limits the ability to test therapeutics that could delay or prevent neurodegeneration.
Huntington’s disease is rare, occurring in about 2.7 per 100,000 individuals worldwide. It is caused by a mutation in the HTT gene involving a DNA segment known as a CAG trinucleotide repeat.
Currently, Huntington’s disease is diagnosed on the basis of clinical signs that emerge late in the disease course, an approach developed before the discovery of the HTT gene and the development of the genetic test for the CAG mutation.
The disease phase prior to diagnosis has been described as presymptomatic, premanifest, or prodromal. However, the three terms have varying definitions that make it difficult to compare study results across trials.
Because drug development had focused on the overt motor sign phase of the disease, there was no real need for an evidence-based staging system that classified disease phases from birth, the investigators noted.
“Now, the research community and regulators recognize that it is critical to conduct trials early in the disease when no signs or overt symptoms are measurable,” Dr. Sampaio said.
Defining disease stages
Work on the staging system was done through the Huntington’s Disease Regulatory Science Consortium, an international project begun in 2018 among biotech and pharma companies, academic institutions, and nonprofit research and advocacy organizations.
Overall, more than 50 clinicians and researchers were involved in developing the HD-ISS.
Using modeling data from four large observational studies that included patients with Huntington’s disease and control groups, researchers identified four different stages of Huntington’s disease:
- Stage 0: Begins at birth with identification of HTT gene mutations but no detectable pathologic changes.
- Stage 1: Begins when biomarker changes are detected via MRI by a volume decrease in six brain areas.
- Stage 2: Begins when clinical signs of Huntington’s disease are present, as determined through motor and cognitive assessments.
- Stage 3: Begins when functional decline is evident, with worsening on the Independence Scale and the Total Functional Capacity of the Unified Huntington’s Disease Rating Scale.
Applying the HD-ISS to clinical trials requires the collection of information routinely recorded in Huntington’s disease research, as well as some additional data, but researchers say its application is straightforward.
The HD-ISS uses a numerical staging system similar to that used in the U.S. Food and Drug Administration’s guidance for Alzheimer’s disease (AD) and integrates the prodromal, presymptomatic, or premanifest phase of the disease. This distinguishes it from earlier classification systems.
The HD-ISS can be adapted if new Huntington’s disease biomarkers are identified.
“As research results are generated, this will further validate the HD-ISS and potentially lead to the development of a derivative, and possibly simplified, system for clinical practice,” Dr. Sampaio said.
The new system goes further than a more recent proposal from the Movement Disorder Society task force, which addresses earlier stages in Huntington’s disease but doesn’t consider objective biomarker data.
Question of timing
Commenting on the findings, Erin Furr-Stimming, MD, neurologist and director of the Huntington’s Disease Society of America Center of Excellence with McGovern Medical School, UTHealth, Houston, said targeting early-stage disease will be key.
“Similar to more common neurodegenerative diseases such as Alzheimer’s disease and Parkinson’s disease, there is a period of at least a decade when changes are occurring in the nervous system, prior to the manifestation of clinical symptoms and signs significant enough to warrant a clinical diagnosis,” Dr. Furr-Stimming said.
She noted that multiple trials of disease-modifying agents for Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease have failed for a multitude of reasons, “but one consistent question that is relevant to all these diseases is that of timing: Should we intervene and test these therapies earlier?
“The premanifest or prodromal period may be the ideal time to intervene with a disease-modifying therapy, prior to onset of any neurodegeneration,” Dr. Furr-Stimming said.
The CHDI Foundation provided financial support to the Critical Path Institute for the Huntington’s Disease Regulatory Science Consortium, including all working group efforts. Dr. Sampio is an employee of and receives salary from CHDI Management. She has also received consultancy honorariums (unrelated to HD) from Pfizer, Kyowa Kirin, vTv Therapeutics, GW Pharmaceuticals, Neuraly, Neuroderm, Green Valley Pharmaceuticals, and Pinteon Pharmaceuticals. A full list of disclosures for the other researchers is in the original article. Dr. Furr-Stimming reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Researchers liken the Huntington’s disease Integrated Staging System (HD-ISS) to the system currently used to stage cancer. It groups patients according to their underlying biological, clinical, and functional characteristics.
It also includes criteria to biologically define Huntington’s disease stages across the entire disease spectrum, from birth to death, which is something that has not been done before. For now, the HD-ISS is only intended for research, but it could one day be modified for use in the clinic, investigators wrote.
“This systematization is of critical importance to select the most appropriate target population for clinical trials and studies,” said co-investigator Cristina Sampaio, MD, chief medical officer at the CHDI Foundation, Princeton, N.J.
“By providing a methodology to precisely define cases early in the neurodegenerative process, the HD-ISS will be instrumental in conducting trials in the very early disease stages,” Dr. Sampaio added.
The position paper was published in the July issue of the Lancet Neurology.
New approach needed
There is no approved therapy to slow Huntington’s disease progression. Clinical trials currently enroll patients with demonstrable symptoms, which limits the ability to test therapeutics that could delay or prevent neurodegeneration.
Huntington’s disease is rare, occurring in about 2.7 per 100,000 individuals worldwide. It is caused by a mutation in the HTT gene involving a DNA segment known as a CAG trinucleotide repeat.
Currently, Huntington’s disease is diagnosed on the basis of clinical signs that emerge late in the disease course, an approach developed before the discovery of the HTT gene and the development of the genetic test for the CAG mutation.
The disease phase prior to diagnosis has been described as presymptomatic, premanifest, or prodromal. However, the three terms have varying definitions that make it difficult to compare study results across trials.
Because drug development had focused on the overt motor sign phase of the disease, there was no real need for an evidence-based staging system that classified disease phases from birth, the investigators noted.
“Now, the research community and regulators recognize that it is critical to conduct trials early in the disease when no signs or overt symptoms are measurable,” Dr. Sampaio said.
Defining disease stages
Work on the staging system was done through the Huntington’s Disease Regulatory Science Consortium, an international project begun in 2018 among biotech and pharma companies, academic institutions, and nonprofit research and advocacy organizations.
Overall, more than 50 clinicians and researchers were involved in developing the HD-ISS.
Using modeling data from four large observational studies that included patients with Huntington’s disease and control groups, researchers identified four different stages of Huntington’s disease:
- Stage 0: Begins at birth with identification of HTT gene mutations but no detectable pathologic changes.
- Stage 1: Begins when biomarker changes are detected via MRI by a volume decrease in six brain areas.
- Stage 2: Begins when clinical signs of Huntington’s disease are present, as determined through motor and cognitive assessments.
- Stage 3: Begins when functional decline is evident, with worsening on the Independence Scale and the Total Functional Capacity of the Unified Huntington’s Disease Rating Scale.
Applying the HD-ISS to clinical trials requires the collection of information routinely recorded in Huntington’s disease research, as well as some additional data, but researchers say its application is straightforward.
The HD-ISS uses a numerical staging system similar to that used in the U.S. Food and Drug Administration’s guidance for Alzheimer’s disease (AD) and integrates the prodromal, presymptomatic, or premanifest phase of the disease. This distinguishes it from earlier classification systems.
The HD-ISS can be adapted if new Huntington’s disease biomarkers are identified.
“As research results are generated, this will further validate the HD-ISS and potentially lead to the development of a derivative, and possibly simplified, system for clinical practice,” Dr. Sampaio said.
The new system goes further than a more recent proposal from the Movement Disorder Society task force, which addresses earlier stages in Huntington’s disease but doesn’t consider objective biomarker data.
Question of timing
Commenting on the findings, Erin Furr-Stimming, MD, neurologist and director of the Huntington’s Disease Society of America Center of Excellence with McGovern Medical School, UTHealth, Houston, said targeting early-stage disease will be key.
“Similar to more common neurodegenerative diseases such as Alzheimer’s disease and Parkinson’s disease, there is a period of at least a decade when changes are occurring in the nervous system, prior to the manifestation of clinical symptoms and signs significant enough to warrant a clinical diagnosis,” Dr. Furr-Stimming said.
She noted that multiple trials of disease-modifying agents for Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease have failed for a multitude of reasons, “but one consistent question that is relevant to all these diseases is that of timing: Should we intervene and test these therapies earlier?
“The premanifest or prodromal period may be the ideal time to intervene with a disease-modifying therapy, prior to onset of any neurodegeneration,” Dr. Furr-Stimming said.
The CHDI Foundation provided financial support to the Critical Path Institute for the Huntington’s Disease Regulatory Science Consortium, including all working group efforts. Dr. Sampio is an employee of and receives salary from CHDI Management. She has also received consultancy honorariums (unrelated to HD) from Pfizer, Kyowa Kirin, vTv Therapeutics, GW Pharmaceuticals, Neuraly, Neuroderm, Green Valley Pharmaceuticals, and Pinteon Pharmaceuticals. A full list of disclosures for the other researchers is in the original article. Dr. Furr-Stimming reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Researchers liken the Huntington’s disease Integrated Staging System (HD-ISS) to the system currently used to stage cancer. It groups patients according to their underlying biological, clinical, and functional characteristics.
It also includes criteria to biologically define Huntington’s disease stages across the entire disease spectrum, from birth to death, which is something that has not been done before. For now, the HD-ISS is only intended for research, but it could one day be modified for use in the clinic, investigators wrote.
“This systematization is of critical importance to select the most appropriate target population for clinical trials and studies,” said co-investigator Cristina Sampaio, MD, chief medical officer at the CHDI Foundation, Princeton, N.J.
“By providing a methodology to precisely define cases early in the neurodegenerative process, the HD-ISS will be instrumental in conducting trials in the very early disease stages,” Dr. Sampaio added.
The position paper was published in the July issue of the Lancet Neurology.
New approach needed
There is no approved therapy to slow Huntington’s disease progression. Clinical trials currently enroll patients with demonstrable symptoms, which limits the ability to test therapeutics that could delay or prevent neurodegeneration.
Huntington’s disease is rare, occurring in about 2.7 per 100,000 individuals worldwide. It is caused by a mutation in the HTT gene involving a DNA segment known as a CAG trinucleotide repeat.
Currently, Huntington’s disease is diagnosed on the basis of clinical signs that emerge late in the disease course, an approach developed before the discovery of the HTT gene and the development of the genetic test for the CAG mutation.
The disease phase prior to diagnosis has been described as presymptomatic, premanifest, or prodromal. However, the three terms have varying definitions that make it difficult to compare study results across trials.
Because drug development had focused on the overt motor sign phase of the disease, there was no real need for an evidence-based staging system that classified disease phases from birth, the investigators noted.
“Now, the research community and regulators recognize that it is critical to conduct trials early in the disease when no signs or overt symptoms are measurable,” Dr. Sampaio said.
Defining disease stages
Work on the staging system was done through the Huntington’s Disease Regulatory Science Consortium, an international project begun in 2018 among biotech and pharma companies, academic institutions, and nonprofit research and advocacy organizations.
Overall, more than 50 clinicians and researchers were involved in developing the HD-ISS.
Using modeling data from four large observational studies that included patients with Huntington’s disease and control groups, researchers identified four different stages of Huntington’s disease:
- Stage 0: Begins at birth with identification of HTT gene mutations but no detectable pathologic changes.
- Stage 1: Begins when biomarker changes are detected via MRI by a volume decrease in six brain areas.
- Stage 2: Begins when clinical signs of Huntington’s disease are present, as determined through motor and cognitive assessments.
- Stage 3: Begins when functional decline is evident, with worsening on the Independence Scale and the Total Functional Capacity of the Unified Huntington’s Disease Rating Scale.
Applying the HD-ISS to clinical trials requires the collection of information routinely recorded in Huntington’s disease research, as well as some additional data, but researchers say its application is straightforward.
The HD-ISS uses a numerical staging system similar to that used in the U.S. Food and Drug Administration’s guidance for Alzheimer’s disease (AD) and integrates the prodromal, presymptomatic, or premanifest phase of the disease. This distinguishes it from earlier classification systems.
The HD-ISS can be adapted if new Huntington’s disease biomarkers are identified.
“As research results are generated, this will further validate the HD-ISS and potentially lead to the development of a derivative, and possibly simplified, system for clinical practice,” Dr. Sampaio said.
The new system goes further than a more recent proposal from the Movement Disorder Society task force, which addresses earlier stages in Huntington’s disease but doesn’t consider objective biomarker data.
Question of timing
Commenting on the findings, Erin Furr-Stimming, MD, neurologist and director of the Huntington’s Disease Society of America Center of Excellence with McGovern Medical School, UTHealth, Houston, said targeting early-stage disease will be key.
“Similar to more common neurodegenerative diseases such as Alzheimer’s disease and Parkinson’s disease, there is a period of at least a decade when changes are occurring in the nervous system, prior to the manifestation of clinical symptoms and signs significant enough to warrant a clinical diagnosis,” Dr. Furr-Stimming said.
She noted that multiple trials of disease-modifying agents for Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease have failed for a multitude of reasons, “but one consistent question that is relevant to all these diseases is that of timing: Should we intervene and test these therapies earlier?
“The premanifest or prodromal period may be the ideal time to intervene with a disease-modifying therapy, prior to onset of any neurodegeneration,” Dr. Furr-Stimming said.
The CHDI Foundation provided financial support to the Critical Path Institute for the Huntington’s Disease Regulatory Science Consortium, including all working group efforts. Dr. Sampio is an employee of and receives salary from CHDI Management. She has also received consultancy honorariums (unrelated to HD) from Pfizer, Kyowa Kirin, vTv Therapeutics, GW Pharmaceuticals, Neuraly, Neuroderm, Green Valley Pharmaceuticals, and Pinteon Pharmaceuticals. A full list of disclosures for the other researchers is in the original article. Dr. Furr-Stimming reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM THE LANCET NEUROLOGY
FDA unveils 5-year plan for ALS and other neurodegenerative diseases
The agency’s Action Plan for Rare Neurodegenerative Diseases including Amyotrophic Lateral Sclerosis (ALS) aims to advance the development of safe and effective medical products and facilitate patient access to novel treatments.
“The effects of rare neurodegenerative diseases are devastating, with very few effective therapeutic options available to patients. We recognize the urgent need for new treatments that can both improve and extend the lives of people diagnosed with these diseases,” FDA Commissioner Robert M. Califf, MD, said in a news release.
“To face that challenge and to accelerate drug development, we need innovative approaches to better understand these diseases while also building on current scientific and research capabilities,” Dr. Califf acknowledged.
“This action plan, especially including the use of public-private partnerships and direct involvement of patients, will ensure the FDA is working toward meeting the task set forth by Congress to enhance the quality of life for those suffering by facilitating access to new therapies,” Dr. Califf added.
Blueprint to ‘aggressively’ move forward
The action plan represents a “blueprint” for how the agency will “aggressively” move forward to address challenges in drug development for rare neurodegenerative diseases to improve patient health, the FDA said.
The plan was created in accordance with provisions in the Accelerating Access to Critical Therapies for ALS Act (ACT for ALS) that President Biden signed into law in late 2021.
Targeted activities include establishing the FDA Rare Neurodegenerative Diseases Task Force and the public-private partnership for rare neurodegenerative diseases, developing disease-specific science strategies over the next 5 years, and leveraging ongoing FDA regulatory science efforts.
The ALS Science Strategy is part of the plan focused specifically on ALS. It provides a “forward-leaning” framework for FDA activities, which include efforts to improve characterization of disease pathogenesis and natural history, boost clinical trial infrastructure and agility to enable early selection of promising therapeutic candidates for further development, optimize clinical trial design, improve access to the trials, streamline clinical trial operations, and reduce the time and cost of drug development.
The FDA says patient engagement, public workshops, research projects, coordination across FDA centers and offices, and collaboration with the National Institutes of Health will be key to the success of implementation of the ALS Science Strategy.
A version of this article first appeared on Medscape.com.
The agency’s Action Plan for Rare Neurodegenerative Diseases including Amyotrophic Lateral Sclerosis (ALS) aims to advance the development of safe and effective medical products and facilitate patient access to novel treatments.
“The effects of rare neurodegenerative diseases are devastating, with very few effective therapeutic options available to patients. We recognize the urgent need for new treatments that can both improve and extend the lives of people diagnosed with these diseases,” FDA Commissioner Robert M. Califf, MD, said in a news release.
“To face that challenge and to accelerate drug development, we need innovative approaches to better understand these diseases while also building on current scientific and research capabilities,” Dr. Califf acknowledged.
“This action plan, especially including the use of public-private partnerships and direct involvement of patients, will ensure the FDA is working toward meeting the task set forth by Congress to enhance the quality of life for those suffering by facilitating access to new therapies,” Dr. Califf added.
Blueprint to ‘aggressively’ move forward
The action plan represents a “blueprint” for how the agency will “aggressively” move forward to address challenges in drug development for rare neurodegenerative diseases to improve patient health, the FDA said.
The plan was created in accordance with provisions in the Accelerating Access to Critical Therapies for ALS Act (ACT for ALS) that President Biden signed into law in late 2021.
Targeted activities include establishing the FDA Rare Neurodegenerative Diseases Task Force and the public-private partnership for rare neurodegenerative diseases, developing disease-specific science strategies over the next 5 years, and leveraging ongoing FDA regulatory science efforts.
The ALS Science Strategy is part of the plan focused specifically on ALS. It provides a “forward-leaning” framework for FDA activities, which include efforts to improve characterization of disease pathogenesis and natural history, boost clinical trial infrastructure and agility to enable early selection of promising therapeutic candidates for further development, optimize clinical trial design, improve access to the trials, streamline clinical trial operations, and reduce the time and cost of drug development.
The FDA says patient engagement, public workshops, research projects, coordination across FDA centers and offices, and collaboration with the National Institutes of Health will be key to the success of implementation of the ALS Science Strategy.
A version of this article first appeared on Medscape.com.
The agency’s Action Plan for Rare Neurodegenerative Diseases including Amyotrophic Lateral Sclerosis (ALS) aims to advance the development of safe and effective medical products and facilitate patient access to novel treatments.
“The effects of rare neurodegenerative diseases are devastating, with very few effective therapeutic options available to patients. We recognize the urgent need for new treatments that can both improve and extend the lives of people diagnosed with these diseases,” FDA Commissioner Robert M. Califf, MD, said in a news release.
“To face that challenge and to accelerate drug development, we need innovative approaches to better understand these diseases while also building on current scientific and research capabilities,” Dr. Califf acknowledged.
“This action plan, especially including the use of public-private partnerships and direct involvement of patients, will ensure the FDA is working toward meeting the task set forth by Congress to enhance the quality of life for those suffering by facilitating access to new therapies,” Dr. Califf added.
Blueprint to ‘aggressively’ move forward
The action plan represents a “blueprint” for how the agency will “aggressively” move forward to address challenges in drug development for rare neurodegenerative diseases to improve patient health, the FDA said.
The plan was created in accordance with provisions in the Accelerating Access to Critical Therapies for ALS Act (ACT for ALS) that President Biden signed into law in late 2021.
Targeted activities include establishing the FDA Rare Neurodegenerative Diseases Task Force and the public-private partnership for rare neurodegenerative diseases, developing disease-specific science strategies over the next 5 years, and leveraging ongoing FDA regulatory science efforts.
The ALS Science Strategy is part of the plan focused specifically on ALS. It provides a “forward-leaning” framework for FDA activities, which include efforts to improve characterization of disease pathogenesis and natural history, boost clinical trial infrastructure and agility to enable early selection of promising therapeutic candidates for further development, optimize clinical trial design, improve access to the trials, streamline clinical trial operations, and reduce the time and cost of drug development.
The FDA says patient engagement, public workshops, research projects, coordination across FDA centers and offices, and collaboration with the National Institutes of Health will be key to the success of implementation of the ALS Science Strategy.
A version of this article first appeared on Medscape.com.
Tofersen linked to slow, positive effects in ALS
caused by superoxide dismutase 1 (SOD1) gene mutations.
The 1-year results, presented at the European Network for the Cure of Amyotrophic Lateral Sclerosis (ENCALS) 2022 meeting, show a deceleration in functional decline that is similar, but “more pronounced” than the previously reported 6-month results, which were not statistically significant, said lead investigator Timothy Miller, MD, PhD, professor of neurology and director of the ALS Center, Washington University, St. Louis.
“What I thought we saw in the first data cut is confirmed by what we saw in the longer data,” he said in an interview. “There were trends [showing] those treated with tofersen did a bit better, but it was hard to be sure. It was hard to be confident in what we were seeing at that early time point.”
Now, with 6 more months of data, Dr. Miller says he is confident that tofersen is slowing down the neurodegenerative disease process. “I see results that I’m encouraged by,” he said. “As a clinician who treats people with ALS with this mutation I would want this drug to be available to people that I see in my clinic.”
One-year VALOR study results
The primary efficacy objective of the VALOR study was to show the 28-week impact of 100 mg tofersen (three doses given about 2 weeks apart, then five doses given every 4 weeks), versus placebo, on function, measured on the Amyotrophic Lateral Sclerosis Functional Rating Scale–Revised (ALSFRS-R). The open-label extension switched placebo-treated patients to tofersen (delayed-start group) and continued to compare them with the early-start group up to 1 year. This open-label extension phase included 49 patients who had been on early-start tofersen and 18 patients in the delayed-start group.
For the primary endpoint, change from baseline in 48-point ALSFRS-R score, there was a statistically significant benefit for the early-start patients with these patients scoring 3.5 points higher than the delayed-start group (P = .0272, 95% confidence interval [CI], 0.4-6.7). This means that both groups declined in function, which is expected in ALS, but the early-start group declined more slowly.
There was also a benefit associated with early-start tofersen for a number of secondary endpoints, including change from baseline in total SOD1 cerebrospinal fluid concentration (CSF SOD1), plasma neurofilament light chain (NfL) levels, and respiratory function.
“This drug targets the MRNA of SOD1, so it lowers the MRNA and then the SOD1 protein falls,” explained Dr. Miller, adding that these levels dropped 21% in the delayed start group, and 33% in the early-start group. “I think the data pretty clearly show that [tofersen] does what it is supposed to do, and that is the first step.”
Neurofilament light chain, a marker of neurodegeneration, also dropped by 41% in the delayed-start group, and 51% in the early-start group.
Respiratory function, as measured by percent predicted slow vital capacity (SVC), also declined 9.2% more slowly in the early- versus delayed-start group (P = .0159).
Finally, muscle strength, as measured by handheld dynamometry (HHD) score, declined more slowly in the early-start group compared with the late-start group, with an adjusted mean difference in score of 2.8 (P = 0.0186).
Dr. Miller said that the data show that it takes time for tofersen to impact clinical function, but there are signs of benefit before that. “I think what you see is that just starting on the drug, the first thing that happens is SOD1 goes down, the next thing is that neurofilament decreases, but clinical function is not yet changing. It takes time. What I see in these data is that it takes time for us to see that effect on clinical function.”
The bigger picture
While acknowledging that tofersen acts on a genetic mutation found in only about 2% of ALS, Dr. Miller said the study findings carry significance for the wider ALS patient population.
“Assuming we agree there is a clinical effect here, assuming we agree that there is real stabilization of clinical function, I think if we agree on that point then we know that ALS is now a treatable disorder. And that’s a really important point. I’m not sure that we knew that before,” he said. “Yes, there are FDA-approved medications that slow down ALS a bit, but they don’t stabilize it, and if we get the target correct – and we have the correct genetic target here – there can be a substantial influence on slowing down the disease, so that’s one thing to learn for the whole ALS community.”
What lies ahead?
Asked to comment on the study, Richard Bedlack, MD, PhD, who was not involved in the research, said the findings are important and show “clinically meaningful” results. “Based on the new benefit-to-burden ratio, I believe most of my patients with SOD1 mutations will want to try this drug. I would like to be able to offer it to them. But I am curious to see what the FDA will do with these data,” said Dr. Bedlack, professor of neurology at Duke University in Durham, N.C., and director of the Duke ALS Clinic.
“Sometimes that open-label extension gives us time to see differences between patients who initially got drug and those who initially got placebo. That seems to be the case with tofersen here, and it was also the case with AMX0035 [Amylyx Pharmaceuticals Inc.], which did not show a survival benefit in the first 6 months but did in the open-label extension.” A recent FDA advisory board panel concluded there was insufficient evidence of benefit for AMX0035, he noted. “I wonder if the same concern will be raised here, necessitating confirmation in another trial. I hope not, but only time will tell.”
Dr. Miller added that these results “highlight how difficult ALS drug development still is. Among the many uncertainties in setting up a trial (targets, doses, inclusion criteria, outcomes), we still do not know how long we need to treat patients in order to see statistically significant changes in the clinical measures we use (ALSFRS-R, respiratory function, strength, survival, etc.). Most American studies are 6 months long and most European studies are 12 months long. Longer studies may be more likely to show benefits on certain measures (e.g., survival), but they cost more, they are challenged by dropouts as the disease progresses, and the idea of randomizing someone to a placebo for a whole year is psychologically difficult for patients, families, and many clinicians (myself included). So, we are seeing more studies like this one where the first 6 months are randomized, blinded, and placebo controlled, and then there is an open-label extension that lasts many months more.”
The study was sponsored by Biogen. Writing and editorial support was provided by Excel Scientific Solutions. Tofersen was discovered by Ionis Pharmaceuticals Inc. Dr. Miller disclosed ties with Biogen, Ionis Pharmaceuticals Inc., Cytokinetics, C2N, Disarm Therapeutics, and UCB Pharma. Dr. Bedlack disclosed ties with Biogen.
caused by superoxide dismutase 1 (SOD1) gene mutations.
The 1-year results, presented at the European Network for the Cure of Amyotrophic Lateral Sclerosis (ENCALS) 2022 meeting, show a deceleration in functional decline that is similar, but “more pronounced” than the previously reported 6-month results, which were not statistically significant, said lead investigator Timothy Miller, MD, PhD, professor of neurology and director of the ALS Center, Washington University, St. Louis.
“What I thought we saw in the first data cut is confirmed by what we saw in the longer data,” he said in an interview. “There were trends [showing] those treated with tofersen did a bit better, but it was hard to be sure. It was hard to be confident in what we were seeing at that early time point.”
Now, with 6 more months of data, Dr. Miller says he is confident that tofersen is slowing down the neurodegenerative disease process. “I see results that I’m encouraged by,” he said. “As a clinician who treats people with ALS with this mutation I would want this drug to be available to people that I see in my clinic.”
One-year VALOR study results
The primary efficacy objective of the VALOR study was to show the 28-week impact of 100 mg tofersen (three doses given about 2 weeks apart, then five doses given every 4 weeks), versus placebo, on function, measured on the Amyotrophic Lateral Sclerosis Functional Rating Scale–Revised (ALSFRS-R). The open-label extension switched placebo-treated patients to tofersen (delayed-start group) and continued to compare them with the early-start group up to 1 year. This open-label extension phase included 49 patients who had been on early-start tofersen and 18 patients in the delayed-start group.
For the primary endpoint, change from baseline in 48-point ALSFRS-R score, there was a statistically significant benefit for the early-start patients with these patients scoring 3.5 points higher than the delayed-start group (P = .0272, 95% confidence interval [CI], 0.4-6.7). This means that both groups declined in function, which is expected in ALS, but the early-start group declined more slowly.
There was also a benefit associated with early-start tofersen for a number of secondary endpoints, including change from baseline in total SOD1 cerebrospinal fluid concentration (CSF SOD1), plasma neurofilament light chain (NfL) levels, and respiratory function.
“This drug targets the MRNA of SOD1, so it lowers the MRNA and then the SOD1 protein falls,” explained Dr. Miller, adding that these levels dropped 21% in the delayed start group, and 33% in the early-start group. “I think the data pretty clearly show that [tofersen] does what it is supposed to do, and that is the first step.”
Neurofilament light chain, a marker of neurodegeneration, also dropped by 41% in the delayed-start group, and 51% in the early-start group.
Respiratory function, as measured by percent predicted slow vital capacity (SVC), also declined 9.2% more slowly in the early- versus delayed-start group (P = .0159).
Finally, muscle strength, as measured by handheld dynamometry (HHD) score, declined more slowly in the early-start group compared with the late-start group, with an adjusted mean difference in score of 2.8 (P = 0.0186).
Dr. Miller said that the data show that it takes time for tofersen to impact clinical function, but there are signs of benefit before that. “I think what you see is that just starting on the drug, the first thing that happens is SOD1 goes down, the next thing is that neurofilament decreases, but clinical function is not yet changing. It takes time. What I see in these data is that it takes time for us to see that effect on clinical function.”
The bigger picture
While acknowledging that tofersen acts on a genetic mutation found in only about 2% of ALS, Dr. Miller said the study findings carry significance for the wider ALS patient population.
“Assuming we agree there is a clinical effect here, assuming we agree that there is real stabilization of clinical function, I think if we agree on that point then we know that ALS is now a treatable disorder. And that’s a really important point. I’m not sure that we knew that before,” he said. “Yes, there are FDA-approved medications that slow down ALS a bit, but they don’t stabilize it, and if we get the target correct – and we have the correct genetic target here – there can be a substantial influence on slowing down the disease, so that’s one thing to learn for the whole ALS community.”
What lies ahead?
Asked to comment on the study, Richard Bedlack, MD, PhD, who was not involved in the research, said the findings are important and show “clinically meaningful” results. “Based on the new benefit-to-burden ratio, I believe most of my patients with SOD1 mutations will want to try this drug. I would like to be able to offer it to them. But I am curious to see what the FDA will do with these data,” said Dr. Bedlack, professor of neurology at Duke University in Durham, N.C., and director of the Duke ALS Clinic.
“Sometimes that open-label extension gives us time to see differences between patients who initially got drug and those who initially got placebo. That seems to be the case with tofersen here, and it was also the case with AMX0035 [Amylyx Pharmaceuticals Inc.], which did not show a survival benefit in the first 6 months but did in the open-label extension.” A recent FDA advisory board panel concluded there was insufficient evidence of benefit for AMX0035, he noted. “I wonder if the same concern will be raised here, necessitating confirmation in another trial. I hope not, but only time will tell.”
Dr. Miller added that these results “highlight how difficult ALS drug development still is. Among the many uncertainties in setting up a trial (targets, doses, inclusion criteria, outcomes), we still do not know how long we need to treat patients in order to see statistically significant changes in the clinical measures we use (ALSFRS-R, respiratory function, strength, survival, etc.). Most American studies are 6 months long and most European studies are 12 months long. Longer studies may be more likely to show benefits on certain measures (e.g., survival), but they cost more, they are challenged by dropouts as the disease progresses, and the idea of randomizing someone to a placebo for a whole year is psychologically difficult for patients, families, and many clinicians (myself included). So, we are seeing more studies like this one where the first 6 months are randomized, blinded, and placebo controlled, and then there is an open-label extension that lasts many months more.”
The study was sponsored by Biogen. Writing and editorial support was provided by Excel Scientific Solutions. Tofersen was discovered by Ionis Pharmaceuticals Inc. Dr. Miller disclosed ties with Biogen, Ionis Pharmaceuticals Inc., Cytokinetics, C2N, Disarm Therapeutics, and UCB Pharma. Dr. Bedlack disclosed ties with Biogen.
caused by superoxide dismutase 1 (SOD1) gene mutations.
The 1-year results, presented at the European Network for the Cure of Amyotrophic Lateral Sclerosis (ENCALS) 2022 meeting, show a deceleration in functional decline that is similar, but “more pronounced” than the previously reported 6-month results, which were not statistically significant, said lead investigator Timothy Miller, MD, PhD, professor of neurology and director of the ALS Center, Washington University, St. Louis.
“What I thought we saw in the first data cut is confirmed by what we saw in the longer data,” he said in an interview. “There were trends [showing] those treated with tofersen did a bit better, but it was hard to be sure. It was hard to be confident in what we were seeing at that early time point.”
Now, with 6 more months of data, Dr. Miller says he is confident that tofersen is slowing down the neurodegenerative disease process. “I see results that I’m encouraged by,” he said. “As a clinician who treats people with ALS with this mutation I would want this drug to be available to people that I see in my clinic.”
One-year VALOR study results
The primary efficacy objective of the VALOR study was to show the 28-week impact of 100 mg tofersen (three doses given about 2 weeks apart, then five doses given every 4 weeks), versus placebo, on function, measured on the Amyotrophic Lateral Sclerosis Functional Rating Scale–Revised (ALSFRS-R). The open-label extension switched placebo-treated patients to tofersen (delayed-start group) and continued to compare them with the early-start group up to 1 year. This open-label extension phase included 49 patients who had been on early-start tofersen and 18 patients in the delayed-start group.
For the primary endpoint, change from baseline in 48-point ALSFRS-R score, there was a statistically significant benefit for the early-start patients with these patients scoring 3.5 points higher than the delayed-start group (P = .0272, 95% confidence interval [CI], 0.4-6.7). This means that both groups declined in function, which is expected in ALS, but the early-start group declined more slowly.
There was also a benefit associated with early-start tofersen for a number of secondary endpoints, including change from baseline in total SOD1 cerebrospinal fluid concentration (CSF SOD1), plasma neurofilament light chain (NfL) levels, and respiratory function.
“This drug targets the MRNA of SOD1, so it lowers the MRNA and then the SOD1 protein falls,” explained Dr. Miller, adding that these levels dropped 21% in the delayed start group, and 33% in the early-start group. “I think the data pretty clearly show that [tofersen] does what it is supposed to do, and that is the first step.”
Neurofilament light chain, a marker of neurodegeneration, also dropped by 41% in the delayed-start group, and 51% in the early-start group.
Respiratory function, as measured by percent predicted slow vital capacity (SVC), also declined 9.2% more slowly in the early- versus delayed-start group (P = .0159).
Finally, muscle strength, as measured by handheld dynamometry (HHD) score, declined more slowly in the early-start group compared with the late-start group, with an adjusted mean difference in score of 2.8 (P = 0.0186).
Dr. Miller said that the data show that it takes time for tofersen to impact clinical function, but there are signs of benefit before that. “I think what you see is that just starting on the drug, the first thing that happens is SOD1 goes down, the next thing is that neurofilament decreases, but clinical function is not yet changing. It takes time. What I see in these data is that it takes time for us to see that effect on clinical function.”
The bigger picture
While acknowledging that tofersen acts on a genetic mutation found in only about 2% of ALS, Dr. Miller said the study findings carry significance for the wider ALS patient population.
“Assuming we agree there is a clinical effect here, assuming we agree that there is real stabilization of clinical function, I think if we agree on that point then we know that ALS is now a treatable disorder. And that’s a really important point. I’m not sure that we knew that before,” he said. “Yes, there are FDA-approved medications that slow down ALS a bit, but they don’t stabilize it, and if we get the target correct – and we have the correct genetic target here – there can be a substantial influence on slowing down the disease, so that’s one thing to learn for the whole ALS community.”
What lies ahead?
Asked to comment on the study, Richard Bedlack, MD, PhD, who was not involved in the research, said the findings are important and show “clinically meaningful” results. “Based on the new benefit-to-burden ratio, I believe most of my patients with SOD1 mutations will want to try this drug. I would like to be able to offer it to them. But I am curious to see what the FDA will do with these data,” said Dr. Bedlack, professor of neurology at Duke University in Durham, N.C., and director of the Duke ALS Clinic.
“Sometimes that open-label extension gives us time to see differences between patients who initially got drug and those who initially got placebo. That seems to be the case with tofersen here, and it was also the case with AMX0035 [Amylyx Pharmaceuticals Inc.], which did not show a survival benefit in the first 6 months but did in the open-label extension.” A recent FDA advisory board panel concluded there was insufficient evidence of benefit for AMX0035, he noted. “I wonder if the same concern will be raised here, necessitating confirmation in another trial. I hope not, but only time will tell.”
Dr. Miller added that these results “highlight how difficult ALS drug development still is. Among the many uncertainties in setting up a trial (targets, doses, inclusion criteria, outcomes), we still do not know how long we need to treat patients in order to see statistically significant changes in the clinical measures we use (ALSFRS-R, respiratory function, strength, survival, etc.). Most American studies are 6 months long and most European studies are 12 months long. Longer studies may be more likely to show benefits on certain measures (e.g., survival), but they cost more, they are challenged by dropouts as the disease progresses, and the idea of randomizing someone to a placebo for a whole year is psychologically difficult for patients, families, and many clinicians (myself included). So, we are seeing more studies like this one where the first 6 months are randomized, blinded, and placebo controlled, and then there is an open-label extension that lasts many months more.”
The study was sponsored by Biogen. Writing and editorial support was provided by Excel Scientific Solutions. Tofersen was discovered by Ionis Pharmaceuticals Inc. Dr. Miller disclosed ties with Biogen, Ionis Pharmaceuticals Inc., Cytokinetics, C2N, Disarm Therapeutics, and UCB Pharma. Dr. Bedlack disclosed ties with Biogen.
FROM THE ENCALS MEETING 2022
FDA expands indication for spinal muscular atrophy drug
As previously reported, the FDA first approved oral risdiplam for SMA in children older than age 2 years in 2020.
The FDA expanded the indication for risdiplam to include babies younger than 2 months old because of interim safety and efficacy data from the ongoing RAINBOWFISH study. It includes 25 babies from birth to 6 weeks of age at first dose, all of whom have genetically diagnosed SMA but are not yet presenting with symptoms.
After 12 months of risdiplam treatment, the majority of presymptomatic infants with SMA reached key motor milestones, Genentech said in a news release.
Of the six babies with two or three copies of the SMN2 gene, all were able to sit after 1 year of active treatment, roughly two-thirds could stand, and half could walk independently.
All babies were alive at 12 months without permanent ventilation.
“The approval of Evrysdi for presymptomatic babies is particularly important, as early treatment of SMA, before symptoms start to arise, can help babies to achieve motor milestones,” Richard Finkel, MD, principal investigator of the trial, said in the release.
“With the inclusion of SMA in newborn screening programs, this approval provides the opportunity to start treating at home with Evrysdi soon after the diagnosis is confirmed,” added Dr. Finkel, who is director of the experimental neuroscience program, St. Jude Children’s Research Hospital, Memphis.
From newborns to older adults?
SMA is a rare and often fatal genetic disease that causes muscle weakness and progressive loss of movement.
SMA, which affects about 1 in 10,000 babies, is caused by a mutation in the survival motor neuron 1 (SMN1) gene. The gene encodes the SMN protein, which is critical for the maintenance and function of motor neurons.
Risdiplam is an orally administered, centrally and peripherally distributed small molecule that modulates survival motor neuron 2 (SMN2) premessenger RNA splicing to increase SMN protein levels.
As part of the label extension, the prescribing information for risdiplam has also been updated to include 2-year pooled data from parts 1 and 2 of the FIREFISH study, which demonstrated long-term efficacy and safety in symptomatic infants with Type 1 SMA, the company noted.
“Because of its efficacy in multiple settings, Evrysdi is now available for people with SMA, from presymptomatic newborns to older adults,” Levi Garraway, MD, PhD, chief medical officer and head of global product development at Genentech, said in the release.
“We are proud of this achievement, which has the potential to make a real difference to those living with SMA and their caregivers,” Dr. Garraway added.
A version of this article first appeared on Medscape.com.
As previously reported, the FDA first approved oral risdiplam for SMA in children older than age 2 years in 2020.
The FDA expanded the indication for risdiplam to include babies younger than 2 months old because of interim safety and efficacy data from the ongoing RAINBOWFISH study. It includes 25 babies from birth to 6 weeks of age at first dose, all of whom have genetically diagnosed SMA but are not yet presenting with symptoms.
After 12 months of risdiplam treatment, the majority of presymptomatic infants with SMA reached key motor milestones, Genentech said in a news release.
Of the six babies with two or three copies of the SMN2 gene, all were able to sit after 1 year of active treatment, roughly two-thirds could stand, and half could walk independently.
All babies were alive at 12 months without permanent ventilation.
“The approval of Evrysdi for presymptomatic babies is particularly important, as early treatment of SMA, before symptoms start to arise, can help babies to achieve motor milestones,” Richard Finkel, MD, principal investigator of the trial, said in the release.
“With the inclusion of SMA in newborn screening programs, this approval provides the opportunity to start treating at home with Evrysdi soon after the diagnosis is confirmed,” added Dr. Finkel, who is director of the experimental neuroscience program, St. Jude Children’s Research Hospital, Memphis.
From newborns to older adults?
SMA is a rare and often fatal genetic disease that causes muscle weakness and progressive loss of movement.
SMA, which affects about 1 in 10,000 babies, is caused by a mutation in the survival motor neuron 1 (SMN1) gene. The gene encodes the SMN protein, which is critical for the maintenance and function of motor neurons.
Risdiplam is an orally administered, centrally and peripherally distributed small molecule that modulates survival motor neuron 2 (SMN2) premessenger RNA splicing to increase SMN protein levels.
As part of the label extension, the prescribing information for risdiplam has also been updated to include 2-year pooled data from parts 1 and 2 of the FIREFISH study, which demonstrated long-term efficacy and safety in symptomatic infants with Type 1 SMA, the company noted.
“Because of its efficacy in multiple settings, Evrysdi is now available for people with SMA, from presymptomatic newborns to older adults,” Levi Garraway, MD, PhD, chief medical officer and head of global product development at Genentech, said in the release.
“We are proud of this achievement, which has the potential to make a real difference to those living with SMA and their caregivers,” Dr. Garraway added.
A version of this article first appeared on Medscape.com.
As previously reported, the FDA first approved oral risdiplam for SMA in children older than age 2 years in 2020.
The FDA expanded the indication for risdiplam to include babies younger than 2 months old because of interim safety and efficacy data from the ongoing RAINBOWFISH study. It includes 25 babies from birth to 6 weeks of age at first dose, all of whom have genetically diagnosed SMA but are not yet presenting with symptoms.
After 12 months of risdiplam treatment, the majority of presymptomatic infants with SMA reached key motor milestones, Genentech said in a news release.
Of the six babies with two or three copies of the SMN2 gene, all were able to sit after 1 year of active treatment, roughly two-thirds could stand, and half could walk independently.
All babies were alive at 12 months without permanent ventilation.
“The approval of Evrysdi for presymptomatic babies is particularly important, as early treatment of SMA, before symptoms start to arise, can help babies to achieve motor milestones,” Richard Finkel, MD, principal investigator of the trial, said in the release.
“With the inclusion of SMA in newborn screening programs, this approval provides the opportunity to start treating at home with Evrysdi soon after the diagnosis is confirmed,” added Dr. Finkel, who is director of the experimental neuroscience program, St. Jude Children’s Research Hospital, Memphis.
From newborns to older adults?
SMA is a rare and often fatal genetic disease that causes muscle weakness and progressive loss of movement.
SMA, which affects about 1 in 10,000 babies, is caused by a mutation in the survival motor neuron 1 (SMN1) gene. The gene encodes the SMN protein, which is critical for the maintenance and function of motor neurons.
Risdiplam is an orally administered, centrally and peripherally distributed small molecule that modulates survival motor neuron 2 (SMN2) premessenger RNA splicing to increase SMN protein levels.
As part of the label extension, the prescribing information for risdiplam has also been updated to include 2-year pooled data from parts 1 and 2 of the FIREFISH study, which demonstrated long-term efficacy and safety in symptomatic infants with Type 1 SMA, the company noted.
“Because of its efficacy in multiple settings, Evrysdi is now available for people with SMA, from presymptomatic newborns to older adults,” Levi Garraway, MD, PhD, chief medical officer and head of global product development at Genentech, said in the release.
“We are proud of this achievement, which has the potential to make a real difference to those living with SMA and their caregivers,” Dr. Garraway added.
A version of this article first appeared on Medscape.com.