User login
Adalimumab effective in nonpsoriatic peripheral spondyloarthritis
Adalimumab alleviated symptoms and improved physical functions in certain patients with active nonpsoriatic peripheral spondyloarthritis in the first 12 weeks of treatment in the ongoing ABILITY-2 trial.
The findings of the trial, which investigators called the “first global, multicenter, randomized, controlled trial” in patients with nonpsoriatic peripheral spondyloarthritis (SpA), add to the growing list of evidence that tumor necrosis factor inhibitors are effective in treating spondyloarthritis (Arthritis Rheumatol. 2014 Dec. 29 [doi:10.1002/art.39008]).
The randomized, double-blind study examined 165 patients aged at least 18 years with active, nonpsoriatic peripheral SpA. The patients met Assessment of Spondyloarthritis International Society (ASAS) peripheral SpA criteria rather than the “older” European Spondyloarthropathy Study Group (ESSG) criteria and had inadequate response to at least two nonsteroidal anti-inflammatory drugs (NSAIDs) or were intolerant to or had a contraindication for NSAIDs. These subjects were placed into cohorts receiving either 40 mg of adalimumab or a placebo for 12 weeks followed by a 144-week-long open-label period. After randomization, 84 subjects were placed on the adalimumab regimen and the remaining 81 were given a placebo. All patients will enter an open-label, 144-week treatment period on adalimumab, according to lead author Dr. Philip Mease of the University of Washington, Seattle, and his coauthors.

The investigators reported that the groups had similar baseline demographics, including 52%-57% being female and mean ages of 38.5-42.5 years, and disease characteristics, including mean symptom duration of 6.6-7.7 years and time since diagnosis of 3.0-4.2 years.
The investigators used a novel primary outcome measurement that required at least 40% improvement in Patient Global Assessments of Disease Activity (PGA) and Pain (PGA-pain) scores from baseline (with at least 20 mm absolute improvement on a visual analog scale) and at least 40% improvement in one or more of the following areas: swollen joint (SJC) and tender joint counts (TJC); enthesitis count; or dactylitis count. Subjects who achieved the primary endpoint of peripheral SpA response criteria were noted as PSpARC 40; individuals who reached other levels of improvement – 20%, 50%, and 70%, with at least 10, 20, or 30 mm absolute improvement, respectively – were noted as PSpARC 20, PSpARC 50, and PSpARC 70, respectively.
After 12 weeks of treatment, nearly twice as many adalimumab-treated patients achieved PSpARC 40 as did placebo-treated patients (39% [33 of 84] vs. 20% [16 of 81]; P = .006). Also, the proportions of patients who achieved PSpARC 20, PSpARC 50, and PSpARC 70 were higher in subjects on adalimumab than in those receiving the placebo: 56% vs. 37% (P < .05), 34% vs. 11% (P < .001), and 22% vs. 3% (P < .001), respectively.
Significant differences between the two treatments were noted by investigators as early as week 2 of the trial. In subjects who achieved PSpARC 40, investigators recorded a minimum 40% improvement and at least 20-mm improvement in the PGA (54% adalimumab vs. 29% placebo; P < .001) and PGA-pain (54% adalimumab vs. 31% placebo; P = .004), and at least 40% improvement in TJC/SJC (57% adalimumab vs. 30% placebo; P < .001). Adverse effects between both cohorts were not significantly different.
“The safety profile of adalimumab in this patient population was consistent with the well-established safety of adalimumab in multiple immune-mediated diseases, [but] longer-term data are needed to confirm and support the favorable benefit-risk profile of adalimumab in nonpsoriatic peripheral SpA observed in this study,” Dr. Mease and his associates concluded.
In an interview, Dr. Grant Louie of Johns Hopkins Arthritis Center in Baltimore, who was not involved in the study, noted that the results are significant because the investigators used such a “rigorously controlled environment” and a more relevant classification system for diagnosing patients.

“When using the ASAS criteria for classifying peripheral spondyloarthritis, it introduces a little more heterogeneity into the patient population,” said Dr. Louie. “This new system incorporates what used to be referred to as undifferentiated spondyloarthritis as well as patients who have reactive arthritis [and] some with inflammatory bowel disease-associated arthritis, while excluding patients who have psoriatic arthritis and ankylosing spondylitis. So a big advantage is that you can incorporate some of these patients earlier in the process so they can undergo the appropriate treatments.”
However, Dr. Louie explained that he had reservations with how the study measured its primary outcome and how clinicians and researchers will be able to use these data without having an externally validated endpoint.
“The biggest caveat – which the authors themselves do mention in the paper, to their credit – is that they’re introducing a new primary study endpoint,” Dr. Louie said. “This composite score that they’re using, which takes into account factors like tender and swollen joints, has not been validated in any external study. Without knowing if the primary endpoint has validity and reliability, I am a little skeptical.”
Dr. Dafna A. Gladman of the University of Toronto shared some of Dr. Louie’s skepticism of the primary endpoint, but credits the investigators with creating a measure that appears to have “construct validity, as well as sensitivity to change.”
In Dr. Gladman’s editorial (Arthritis Rheumatol. 2014 Dec. 29 [doi:10.1002/art.39014]), she noted that “the development of the measure is not described. Was it developed through a Delphi process? Has it shown face and construct validity in peripheral SpA? Has it shown test retest reliability? What we do know is that this measure distinguished between drug and placebo treated patients, [that] the majority of the response was related to the patient reported outcome which was a prerequisite and to the joint count item, [and] that the individual measures tested in this study showed the same results. One could then conclude that the new measure has construct validity, as well as sensitivity to change and thus would be acceptable as the primary outcome measure in this study.”
The ABILITY-2 study was funded by AbbVie, which markets adalimumab. Two authors are employees of the company. All other authors reported receiving grant/research support, consulting fees, and/or speaking fees from AbbVie and other companies that market biologics.
Adalimumab alleviated symptoms and improved physical functions in certain patients with active nonpsoriatic peripheral spondyloarthritis in the first 12 weeks of treatment in the ongoing ABILITY-2 trial.
The findings of the trial, which investigators called the “first global, multicenter, randomized, controlled trial” in patients with nonpsoriatic peripheral spondyloarthritis (SpA), add to the growing list of evidence that tumor necrosis factor inhibitors are effective in treating spondyloarthritis (Arthritis Rheumatol. 2014 Dec. 29 [doi:10.1002/art.39008]).
The randomized, double-blind study examined 165 patients aged at least 18 years with active, nonpsoriatic peripheral SpA. The patients met Assessment of Spondyloarthritis International Society (ASAS) peripheral SpA criteria rather than the “older” European Spondyloarthropathy Study Group (ESSG) criteria and had inadequate response to at least two nonsteroidal anti-inflammatory drugs (NSAIDs) or were intolerant to or had a contraindication for NSAIDs. These subjects were placed into cohorts receiving either 40 mg of adalimumab or a placebo for 12 weeks followed by a 144-week-long open-label period. After randomization, 84 subjects were placed on the adalimumab regimen and the remaining 81 were given a placebo. All patients will enter an open-label, 144-week treatment period on adalimumab, according to lead author Dr. Philip Mease of the University of Washington, Seattle, and his coauthors.
The investigators reported that the groups had similar baseline demographics, including 52%-57% being female and mean ages of 38.5-42.5 years, and disease characteristics, including mean symptom duration of 6.6-7.7 years and time since diagnosis of 3.0-4.2 years.
The investigators used a novel primary outcome measurement that required at least 40% improvement in Patient Global Assessments of Disease Activity (PGA) and Pain (PGA-pain) scores from baseline (with at least 20 mm absolute improvement on a visual analog scale) and at least 40% improvement in one or more of the following areas: swollen joint (SJC) and tender joint counts (TJC); enthesitis count; or dactylitis count. Subjects who achieved the primary endpoint of peripheral SpA response criteria were noted as PSpARC 40; individuals who reached other levels of improvement – 20%, 50%, and 70%, with at least 10, 20, or 30 mm absolute improvement, respectively – were noted as PSpARC 20, PSpARC 50, and PSpARC 70, respectively.
After 12 weeks of treatment, nearly twice as many adalimumab-treated patients achieved PSpARC 40 as did placebo-treated patients (39% [33 of 84] vs. 20% [16 of 81]; P = .006). Also, the proportions of patients who achieved PSpARC 20, PSpARC 50, and PSpARC 70 were higher in subjects on adalimumab than in those receiving the placebo: 56% vs. 37% (P < .05), 34% vs. 11% (P < .001), and 22% vs. 3% (P < .001), respectively.
Significant differences between the two treatments were noted by investigators as early as week 2 of the trial. In subjects who achieved PSpARC 40, investigators recorded a minimum 40% improvement and at least 20-mm improvement in the PGA (54% adalimumab vs. 29% placebo; P < .001) and PGA-pain (54% adalimumab vs. 31% placebo; P = .004), and at least 40% improvement in TJC/SJC (57% adalimumab vs. 30% placebo; P < .001). Adverse effects between both cohorts were not significantly different.
“The safety profile of adalimumab in this patient population was consistent with the well-established safety of adalimumab in multiple immune-mediated diseases, [but] longer-term data are needed to confirm and support the favorable benefit-risk profile of adalimumab in nonpsoriatic peripheral SpA observed in this study,” Dr. Mease and his associates concluded.
In an interview, Dr. Grant Louie of Johns Hopkins Arthritis Center in Baltimore, who was not involved in the study, noted that the results are significant because the investigators used such a “rigorously controlled environment” and a more relevant classification system for diagnosing patients.
“When using the ASAS criteria for classifying peripheral spondyloarthritis, it introduces a little more heterogeneity into the patient population,” said Dr. Louie. “This new system incorporates what used to be referred to as undifferentiated spondyloarthritis as well as patients who have reactive arthritis [and] some with inflammatory bowel disease-associated arthritis, while excluding patients who have psoriatic arthritis and ankylosing spondylitis. So a big advantage is that you can incorporate some of these patients earlier in the process so they can undergo the appropriate treatments.”
However, Dr. Louie explained that he had reservations with how the study measured its primary outcome and how clinicians and researchers will be able to use these data without having an externally validated endpoint.
“The biggest caveat – which the authors themselves do mention in the paper, to their credit – is that they’re introducing a new primary study endpoint,” Dr. Louie said. “This composite score that they’re using, which takes into account factors like tender and swollen joints, has not been validated in any external study. Without knowing if the primary endpoint has validity and reliability, I am a little skeptical.”
Dr. Dafna A. Gladman of the University of Toronto shared some of Dr. Louie’s skepticism of the primary endpoint, but credits the investigators with creating a measure that appears to have “construct validity, as well as sensitivity to change.”
In Dr. Gladman’s editorial (Arthritis Rheumatol. 2014 Dec. 29 [doi:10.1002/art.39014]), she noted that “the development of the measure is not described. Was it developed through a Delphi process? Has it shown face and construct validity in peripheral SpA? Has it shown test retest reliability? What we do know is that this measure distinguished between drug and placebo treated patients, [that] the majority of the response was related to the patient reported outcome which was a prerequisite and to the joint count item, [and] that the individual measures tested in this study showed the same results. One could then conclude that the new measure has construct validity, as well as sensitivity to change and thus would be acceptable as the primary outcome measure in this study.”
The ABILITY-2 study was funded by AbbVie, which markets adalimumab. Two authors are employees of the company. All other authors reported receiving grant/research support, consulting fees, and/or speaking fees from AbbVie and other companies that market biologics.
Adalimumab alleviated symptoms and improved physical functions in certain patients with active nonpsoriatic peripheral spondyloarthritis in the first 12 weeks of treatment in the ongoing ABILITY-2 trial.
The findings of the trial, which investigators called the “first global, multicenter, randomized, controlled trial” in patients with nonpsoriatic peripheral spondyloarthritis (SpA), add to the growing list of evidence that tumor necrosis factor inhibitors are effective in treating spondyloarthritis (Arthritis Rheumatol. 2014 Dec. 29 [doi:10.1002/art.39008]).
The randomized, double-blind study examined 165 patients aged at least 18 years with active, nonpsoriatic peripheral SpA. The patients met Assessment of Spondyloarthritis International Society (ASAS) peripheral SpA criteria rather than the “older” European Spondyloarthropathy Study Group (ESSG) criteria and had inadequate response to at least two nonsteroidal anti-inflammatory drugs (NSAIDs) or were intolerant to or had a contraindication for NSAIDs. These subjects were placed into cohorts receiving either 40 mg of adalimumab or a placebo for 12 weeks followed by a 144-week-long open-label period. After randomization, 84 subjects were placed on the adalimumab regimen and the remaining 81 were given a placebo. All patients will enter an open-label, 144-week treatment period on adalimumab, according to lead author Dr. Philip Mease of the University of Washington, Seattle, and his coauthors.
The investigators reported that the groups had similar baseline demographics, including 52%-57% being female and mean ages of 38.5-42.5 years, and disease characteristics, including mean symptom duration of 6.6-7.7 years and time since diagnosis of 3.0-4.2 years.
The investigators used a novel primary outcome measurement that required at least 40% improvement in Patient Global Assessments of Disease Activity (PGA) and Pain (PGA-pain) scores from baseline (with at least 20 mm absolute improvement on a visual analog scale) and at least 40% improvement in one or more of the following areas: swollen joint (SJC) and tender joint counts (TJC); enthesitis count; or dactylitis count. Subjects who achieved the primary endpoint of peripheral SpA response criteria were noted as PSpARC 40; individuals who reached other levels of improvement – 20%, 50%, and 70%, with at least 10, 20, or 30 mm absolute improvement, respectively – were noted as PSpARC 20, PSpARC 50, and PSpARC 70, respectively.
After 12 weeks of treatment, nearly twice as many adalimumab-treated patients achieved PSpARC 40 as did placebo-treated patients (39% [33 of 84] vs. 20% [16 of 81]; P = .006). Also, the proportions of patients who achieved PSpARC 20, PSpARC 50, and PSpARC 70 were higher in subjects on adalimumab than in those receiving the placebo: 56% vs. 37% (P < .05), 34% vs. 11% (P < .001), and 22% vs. 3% (P < .001), respectively.
Significant differences between the two treatments were noted by investigators as early as week 2 of the trial. In subjects who achieved PSpARC 40, investigators recorded a minimum 40% improvement and at least 20-mm improvement in the PGA (54% adalimumab vs. 29% placebo; P < .001) and PGA-pain (54% adalimumab vs. 31% placebo; P = .004), and at least 40% improvement in TJC/SJC (57% adalimumab vs. 30% placebo; P < .001). Adverse effects between both cohorts were not significantly different.
“The safety profile of adalimumab in this patient population was consistent with the well-established safety of adalimumab in multiple immune-mediated diseases, [but] longer-term data are needed to confirm and support the favorable benefit-risk profile of adalimumab in nonpsoriatic peripheral SpA observed in this study,” Dr. Mease and his associates concluded.
In an interview, Dr. Grant Louie of Johns Hopkins Arthritis Center in Baltimore, who was not involved in the study, noted that the results are significant because the investigators used such a “rigorously controlled environment” and a more relevant classification system for diagnosing patients.
“When using the ASAS criteria for classifying peripheral spondyloarthritis, it introduces a little more heterogeneity into the patient population,” said Dr. Louie. “This new system incorporates what used to be referred to as undifferentiated spondyloarthritis as well as patients who have reactive arthritis [and] some with inflammatory bowel disease-associated arthritis, while excluding patients who have psoriatic arthritis and ankylosing spondylitis. So a big advantage is that you can incorporate some of these patients earlier in the process so they can undergo the appropriate treatments.”
However, Dr. Louie explained that he had reservations with how the study measured its primary outcome and how clinicians and researchers will be able to use these data without having an externally validated endpoint.
“The biggest caveat – which the authors themselves do mention in the paper, to their credit – is that they’re introducing a new primary study endpoint,” Dr. Louie said. “This composite score that they’re using, which takes into account factors like tender and swollen joints, has not been validated in any external study. Without knowing if the primary endpoint has validity and reliability, I am a little skeptical.”
Dr. Dafna A. Gladman of the University of Toronto shared some of Dr. Louie’s skepticism of the primary endpoint, but credits the investigators with creating a measure that appears to have “construct validity, as well as sensitivity to change.”
In Dr. Gladman’s editorial (Arthritis Rheumatol. 2014 Dec. 29 [doi:10.1002/art.39014]), she noted that “the development of the measure is not described. Was it developed through a Delphi process? Has it shown face and construct validity in peripheral SpA? Has it shown test retest reliability? What we do know is that this measure distinguished between drug and placebo treated patients, [that] the majority of the response was related to the patient reported outcome which was a prerequisite and to the joint count item, [and] that the individual measures tested in this study showed the same results. One could then conclude that the new measure has construct validity, as well as sensitivity to change and thus would be acceptable as the primary outcome measure in this study.”
The ABILITY-2 study was funded by AbbVie, which markets adalimumab. Two authors are employees of the company. All other authors reported receiving grant/research support, consulting fees, and/or speaking fees from AbbVie and other companies that market biologics.
FROM ARTHRITIS & RHEUMATOLOGY
Key clinical point: Patients with active nonpsoriatic peripheral spondyloarthritis who have not responded to treatment with NSAIDs might consider treatment with adalimumab.
Major finding: After 12 weeks, significantly more subjects in the adalimumab cohort achieved the primary outcome of at least 40% improvement in peripheral SpA response criteria than did those receiving placebo: 39% vs. 20%, respectively (P = .006).
Data source: The randomized, double-blind ABILITY-2 trial of 165 patients with active nonpsoriatic peripheral spondyloarthritis.
Disclosures: The trial was funded by AbbVie, which markets adalimumab. Two authors are employees of the company. All other authors reported ties with AbbVie and other companies that market biologics.

Annual costs of psoriasis costs top $112 billion
The total economic burden of psoriasis in the United States is at least $112 billion per year, and possibly as high as $135 billion, investigators estimated in a study published Jan. 7 in JAMA Dermatology.
Dr. Elizabeth A. Brezinski of the University of California, Davis, in Sacramento, and her associates, reviewed 22 studies conducted between Jan. 1, 2008, and Sept. 20, 2013, adjusting the results to 2013 dollars.
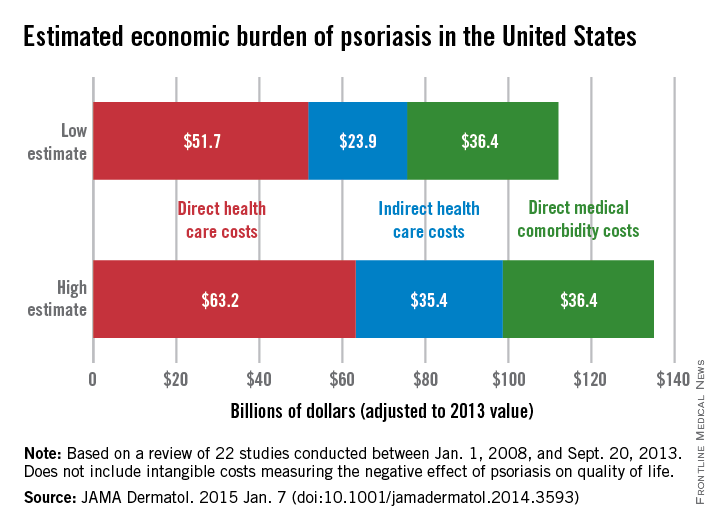
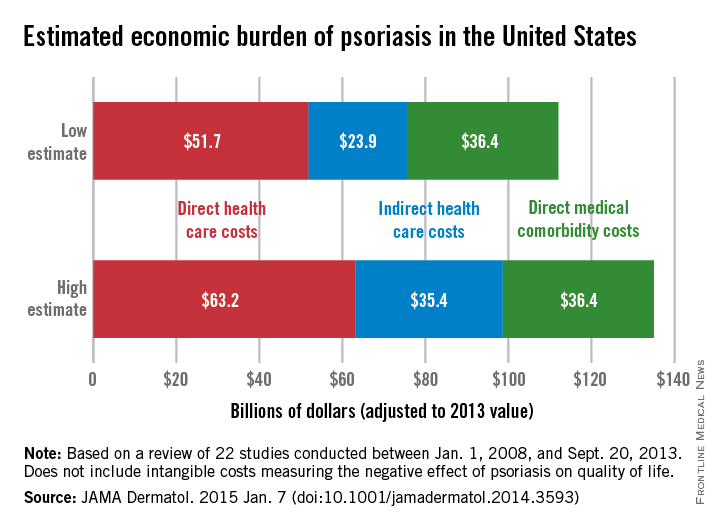
Estimates for the direct medical cost of psoriasis care ranged from $51.7 billion to $63.2 billion per year. Indirect costs from absenteeism or working while sick contributed another $23.9-$35.4 billion, with comorbidity costs estimated at $36.4 billion annually (JAMA Dermatol. 2014 Jan. 7 [doi:10.1001/jamadermatol.2014.3593]).
Intangible costs on quality of life, which were not included in the annual figures, were estimated to be $85.1 billion over the lifetimes of the psoriasis patient population (7.4 million as of 2013), they said.
“Defining the economic burden of psoriasis from a societal perspective is the foundation for innovating and providing access to cost-effective therapies that will result in improved patient outcomes,” Dr. Brezinski and her coauthors wrote.
One of the researchers reported serving as an investigator for, or consultant to, AbbVie, Amgen, Celgene, Janssen, Lilly, Merck, Pfizer, and UCB. No other disclosures were reported.
The total economic burden of psoriasis in the United States is at least $112 billion per year, and possibly as high as $135 billion, investigators estimated in a study published Jan. 7 in JAMA Dermatology.
Dr. Elizabeth A. Brezinski of the University of California, Davis, in Sacramento, and her associates, reviewed 22 studies conducted between Jan. 1, 2008, and Sept. 20, 2013, adjusting the results to 2013 dollars.
Estimates for the direct medical cost of psoriasis care ranged from $51.7 billion to $63.2 billion per year. Indirect costs from absenteeism or working while sick contributed another $23.9-$35.4 billion, with comorbidity costs estimated at $36.4 billion annually (JAMA Dermatol. 2014 Jan. 7 [doi:10.1001/jamadermatol.2014.3593]).
Intangible costs on quality of life, which were not included in the annual figures, were estimated to be $85.1 billion over the lifetimes of the psoriasis patient population (7.4 million as of 2013), they said.
“Defining the economic burden of psoriasis from a societal perspective is the foundation for innovating and providing access to cost-effective therapies that will result in improved patient outcomes,” Dr. Brezinski and her coauthors wrote.
One of the researchers reported serving as an investigator for, or consultant to, AbbVie, Amgen, Celgene, Janssen, Lilly, Merck, Pfizer, and UCB. No other disclosures were reported.
The total economic burden of psoriasis in the United States is at least $112 billion per year, and possibly as high as $135 billion, investigators estimated in a study published Jan. 7 in JAMA Dermatology.
Dr. Elizabeth A. Brezinski of the University of California, Davis, in Sacramento, and her associates, reviewed 22 studies conducted between Jan. 1, 2008, and Sept. 20, 2013, adjusting the results to 2013 dollars.
Estimates for the direct medical cost of psoriasis care ranged from $51.7 billion to $63.2 billion per year. Indirect costs from absenteeism or working while sick contributed another $23.9-$35.4 billion, with comorbidity costs estimated at $36.4 billion annually (JAMA Dermatol. 2014 Jan. 7 [doi:10.1001/jamadermatol.2014.3593]).
Intangible costs on quality of life, which were not included in the annual figures, were estimated to be $85.1 billion over the lifetimes of the psoriasis patient population (7.4 million as of 2013), they said.
“Defining the economic burden of psoriasis from a societal perspective is the foundation for innovating and providing access to cost-effective therapies that will result in improved patient outcomes,” Dr. Brezinski and her coauthors wrote.
One of the researchers reported serving as an investigator for, or consultant to, AbbVie, Amgen, Celgene, Janssen, Lilly, Merck, Pfizer, and UCB. No other disclosures were reported.
FROM JAMA DERMATOLOGY
Rx for specialists: Know how ACA affects patients’ ability to pay for meds
ORLANDO – Despite recent, significant shifts in health care coverage thanks to the Affordable Care Act, many specialists are unaware of how patients pay for pricey prescriptions such as biologics.
One reason is that clinicians just haven’t been paying enough attention, according to Dr. Kim L. Isaacs, codirector of the multidiscipline treatment and research center for inflammatory bowel disease at the University of North Carolina.
“It gets complicated because we’re taking care of patients, so it’s hard to think about the financial end of things as well, and it lands on the patient’s lap,” Dr. Isaacs said in an interview after her presentation, “Navigating the Affordable Care Act,” at a conference on inflammatory bowel diseases sponsored by the Crohn’s and Colitis Foundation of America.
But not knowing how and if chronically ill patients can afford to pay for their medications can impact their compliance, and even their disease states.
Dr. Isaacs compared the histories of two patients. The first one purchased through the ACA’s health insurance marketplace provision, and regardless of her preexisting condition, she was able to receive and afford treatment for her Crohn’s disease for the first time in a decade. The second patient purchased ACA-sanctioned insurance but it still wasn’t enough to cover the costs of her care.
“She told me she had thought the Silver Plan would work for her, but that she still couldn’t afford her medications, and I was thinking, ‘What’s a Silver Plan?’ ” Dr. Isaacs said. “I didn’t have a clue.”
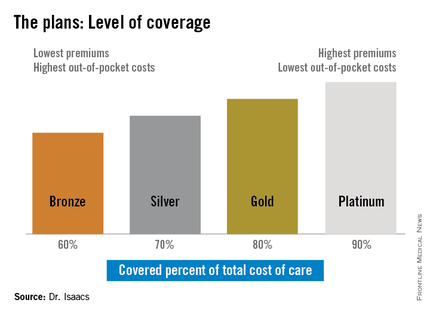
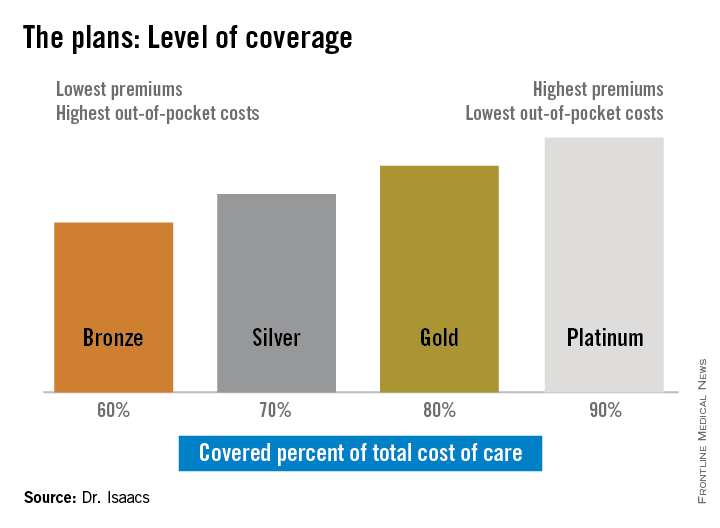
The discrepancy between the patients’ plans prompted Dr. Isaacs to investigate whether what she was prescribing was practical under her patients’ various levels of coverage. She discovered that under the ACA, the individual mandate requiring all Americans to purchase some form of health insurance means many have turned to a variety of state- and federally-sponsored health insurance marketplaces that offer coverage plans ranging from Bronze to Platinum.
“If you ask [most specialists] what the Gold Plan is, most of them won’t have heard of it, they don’t know,” she said.

But she said that physicians need to be aware of whether the drugs they are prescribing are on the formulary used by their patients’ respective plans since the different insurance providers that back the various plans often don’t cover the same medications.
In addition, the so-called “donut hole,” the annually adjusted gap in prescription drug coverage for Medicare patients, is not scheduled to close until 2020. While the gap exists, Medicare patients have a set amount of annual drug coverage after which the patient shares a substantial portion of the cost until the following year when the process begins again.
“For our IBD patients, this is very important because for some of our more expensive drugs, there may be a period of time [annually] when the drugs are not covered,” Dr. Isaacs said.
The same could be true for other specialties such as oncology, rheumatology, and neurology, where disease states require high-cost specialty drugs; however, some patient advocacy groups will assist patients in paying for their treatment, she said.
Patients who purchase their health plans through the government-sponsored insurance marketplaces are usually eligible for subsidies, depending on their income and where they fall in relation to the federally set poverty level, said Dr. Isaacs.
Another important, basic point, she added, is that providers need to ensure that they are on their patients’ chosen plans. “If you’re not, then you need to be sure there is someone else who is on the plan who can take care of your patient.”
Dr. Isaacs reported she has affiliations with AbbVie, Janssen, Millennium, Takeda, UCB, and others.
On Twitter @whitneymcknight
ORLANDO – Despite recent, significant shifts in health care coverage thanks to the Affordable Care Act, many specialists are unaware of how patients pay for pricey prescriptions such as biologics.
One reason is that clinicians just haven’t been paying enough attention, according to Dr. Kim L. Isaacs, codirector of the multidiscipline treatment and research center for inflammatory bowel disease at the University of North Carolina.
“It gets complicated because we’re taking care of patients, so it’s hard to think about the financial end of things as well, and it lands on the patient’s lap,” Dr. Isaacs said in an interview after her presentation, “Navigating the Affordable Care Act,” at a conference on inflammatory bowel diseases sponsored by the Crohn’s and Colitis Foundation of America.
But not knowing how and if chronically ill patients can afford to pay for their medications can impact their compliance, and even their disease states.
Dr. Isaacs compared the histories of two patients. The first one purchased through the ACA’s health insurance marketplace provision, and regardless of her preexisting condition, she was able to receive and afford treatment for her Crohn’s disease for the first time in a decade. The second patient purchased ACA-sanctioned insurance but it still wasn’t enough to cover the costs of her care.
“She told me she had thought the Silver Plan would work for her, but that she still couldn’t afford her medications, and I was thinking, ‘What’s a Silver Plan?’ ” Dr. Isaacs said. “I didn’t have a clue.”
The discrepancy between the patients’ plans prompted Dr. Isaacs to investigate whether what she was prescribing was practical under her patients’ various levels of coverage. She discovered that under the ACA, the individual mandate requiring all Americans to purchase some form of health insurance means many have turned to a variety of state- and federally-sponsored health insurance marketplaces that offer coverage plans ranging from Bronze to Platinum.
“If you ask [most specialists] what the Gold Plan is, most of them won’t have heard of it, they don’t know,” she said.
But she said that physicians need to be aware of whether the drugs they are prescribing are on the formulary used by their patients’ respective plans since the different insurance providers that back the various plans often don’t cover the same medications.
In addition, the so-called “donut hole,” the annually adjusted gap in prescription drug coverage for Medicare patients, is not scheduled to close until 2020. While the gap exists, Medicare patients have a set amount of annual drug coverage after which the patient shares a substantial portion of the cost until the following year when the process begins again.
“For our IBD patients, this is very important because for some of our more expensive drugs, there may be a period of time [annually] when the drugs are not covered,” Dr. Isaacs said.
The same could be true for other specialties such as oncology, rheumatology, and neurology, where disease states require high-cost specialty drugs; however, some patient advocacy groups will assist patients in paying for their treatment, she said.
Patients who purchase their health plans through the government-sponsored insurance marketplaces are usually eligible for subsidies, depending on their income and where they fall in relation to the federally set poverty level, said Dr. Isaacs.
Another important, basic point, she added, is that providers need to ensure that they are on their patients’ chosen plans. “If you’re not, then you need to be sure there is someone else who is on the plan who can take care of your patient.”
Dr. Isaacs reported she has affiliations with AbbVie, Janssen, Millennium, Takeda, UCB, and others.
On Twitter @whitneymcknight
ORLANDO – Despite recent, significant shifts in health care coverage thanks to the Affordable Care Act, many specialists are unaware of how patients pay for pricey prescriptions such as biologics.
One reason is that clinicians just haven’t been paying enough attention, according to Dr. Kim L. Isaacs, codirector of the multidiscipline treatment and research center for inflammatory bowel disease at the University of North Carolina.
“It gets complicated because we’re taking care of patients, so it’s hard to think about the financial end of things as well, and it lands on the patient’s lap,” Dr. Isaacs said in an interview after her presentation, “Navigating the Affordable Care Act,” at a conference on inflammatory bowel diseases sponsored by the Crohn’s and Colitis Foundation of America.
But not knowing how and if chronically ill patients can afford to pay for their medications can impact their compliance, and even their disease states.
Dr. Isaacs compared the histories of two patients. The first one purchased through the ACA’s health insurance marketplace provision, and regardless of her preexisting condition, she was able to receive and afford treatment for her Crohn’s disease for the first time in a decade. The second patient purchased ACA-sanctioned insurance but it still wasn’t enough to cover the costs of her care.
“She told me she had thought the Silver Plan would work for her, but that she still couldn’t afford her medications, and I was thinking, ‘What’s a Silver Plan?’ ” Dr. Isaacs said. “I didn’t have a clue.”
The discrepancy between the patients’ plans prompted Dr. Isaacs to investigate whether what she was prescribing was practical under her patients’ various levels of coverage. She discovered that under the ACA, the individual mandate requiring all Americans to purchase some form of health insurance means many have turned to a variety of state- and federally-sponsored health insurance marketplaces that offer coverage plans ranging from Bronze to Platinum.
“If you ask [most specialists] what the Gold Plan is, most of them won’t have heard of it, they don’t know,” she said.
But she said that physicians need to be aware of whether the drugs they are prescribing are on the formulary used by their patients’ respective plans since the different insurance providers that back the various plans often don’t cover the same medications.
In addition, the so-called “donut hole,” the annually adjusted gap in prescription drug coverage for Medicare patients, is not scheduled to close until 2020. While the gap exists, Medicare patients have a set amount of annual drug coverage after which the patient shares a substantial portion of the cost until the following year when the process begins again.
“For our IBD patients, this is very important because for some of our more expensive drugs, there may be a period of time [annually] when the drugs are not covered,” Dr. Isaacs said.
The same could be true for other specialties such as oncology, rheumatology, and neurology, where disease states require high-cost specialty drugs; however, some patient advocacy groups will assist patients in paying for their treatment, she said.
Patients who purchase their health plans through the government-sponsored insurance marketplaces are usually eligible for subsidies, depending on their income and where they fall in relation to the federally set poverty level, said Dr. Isaacs.
Another important, basic point, she added, is that providers need to ensure that they are on their patients’ chosen plans. “If you’re not, then you need to be sure there is someone else who is on the plan who can take care of your patient.”
Dr. Isaacs reported she has affiliations with AbbVie, Janssen, Millennium, Takeda, UCB, and others.
On Twitter @whitneymcknight
EXPERT ANALYSIS FROM 2014 ADVANCES IN IBD
Immunogenicity to TNF-alpha blockers varies in psoriatic arthritis
Immunogenicity to three TNF-alpha blocking agents seems to vary substantially among patients with psoriatic arthritis, and the use of methotrexate appears to attenuate the presence of antidrug antibodies, according to findings from a cross-sectional study.
Although researchers have looked at the immunogenicity of TNF-alpha blockers in patients with diseases such as rheumatoid arthritis (RA), ankylosing spondylitis, psoriasis, and inflammatory bowel disease, their immunogenicity in psoriatic arthritis (PsA) patients has not been fully investigated, noted the current study’s investigators, led by Dr. Michael Zisapel of the department of rheumatology at Tel Aviv University (J. Rheumatol. 2014 Nov. 15 [doi:10.3899/jrheum.140685]).
The prevalence of antidrug antibodies (ADAb) to TNF-alpha blockers has been reported to be 20%-40% in RA, 25%-64% in ankylosing spondylitis, and about 33% in psoriasis, and evidence has shown that this is significantly reduced by the use of methotrexate, they said.
The study involved 93 patients with PsA who were taking adalimumab (n = 48), infliximab (n = 24), and etanercept (n = 21). A quarter of the patients were taking methotrexate at an average dose of 13.3 mg/week.
Overall, 77% of the patients had therapeutic drug levels. The prevalence of immunogenicity in the entire group was 33.3%, and one-fifth of patients had high concentrations of antibodies.
High levels of ADAb were found in 29% of patients taking adalimumab and 21% of the patients taking infliximab. No ADAb levels were found in patients taking etanercept.
Interestingly, the patients taking adalimumab demonstrated an immunogenicity of 54%, but only half of them (29%) showed high concentrations of antibodies.
“This finding suggests that a variety of levels might be found that might affect the significance of immunogenicity differently among patients producing ADAb,” the investigators wrote.
Fewer methotrexate-treated patients had high ADAb concentrations, compared with patients not taking methotrexate (16.7% vs. 21.7%).
A clear correlation was found between the presence of immunogenicity, lower drug levels, and decreased clinical response in PsA patients, just as other studies have found in RA and ankylosing spondylitis patients, the authors noted.
“Our results suggest that the use of methotrexate should be strongly considered in addition to monoclonal antibodies,” they said.
Limitations of their study included the small number of patients and the use of a bridging ELISA test which is reliable but considered to be less accurate than radioimmunoassay.
No disclosure information was available.
Immunogenicity to three TNF-alpha blocking agents seems to vary substantially among patients with psoriatic arthritis, and the use of methotrexate appears to attenuate the presence of antidrug antibodies, according to findings from a cross-sectional study.
Although researchers have looked at the immunogenicity of TNF-alpha blockers in patients with diseases such as rheumatoid arthritis (RA), ankylosing spondylitis, psoriasis, and inflammatory bowel disease, their immunogenicity in psoriatic arthritis (PsA) patients has not been fully investigated, noted the current study’s investigators, led by Dr. Michael Zisapel of the department of rheumatology at Tel Aviv University (J. Rheumatol. 2014 Nov. 15 [doi:10.3899/jrheum.140685]).
The prevalence of antidrug antibodies (ADAb) to TNF-alpha blockers has been reported to be 20%-40% in RA, 25%-64% in ankylosing spondylitis, and about 33% in psoriasis, and evidence has shown that this is significantly reduced by the use of methotrexate, they said.
The study involved 93 patients with PsA who were taking adalimumab (n = 48), infliximab (n = 24), and etanercept (n = 21). A quarter of the patients were taking methotrexate at an average dose of 13.3 mg/week.
Overall, 77% of the patients had therapeutic drug levels. The prevalence of immunogenicity in the entire group was 33.3%, and one-fifth of patients had high concentrations of antibodies.
High levels of ADAb were found in 29% of patients taking adalimumab and 21% of the patients taking infliximab. No ADAb levels were found in patients taking etanercept.
Interestingly, the patients taking adalimumab demonstrated an immunogenicity of 54%, but only half of them (29%) showed high concentrations of antibodies.
“This finding suggests that a variety of levels might be found that might affect the significance of immunogenicity differently among patients producing ADAb,” the investigators wrote.
Fewer methotrexate-treated patients had high ADAb concentrations, compared with patients not taking methotrexate (16.7% vs. 21.7%).
A clear correlation was found between the presence of immunogenicity, lower drug levels, and decreased clinical response in PsA patients, just as other studies have found in RA and ankylosing spondylitis patients, the authors noted.
“Our results suggest that the use of methotrexate should be strongly considered in addition to monoclonal antibodies,” they said.
Limitations of their study included the small number of patients and the use of a bridging ELISA test which is reliable but considered to be less accurate than radioimmunoassay.
No disclosure information was available.
Immunogenicity to three TNF-alpha blocking agents seems to vary substantially among patients with psoriatic arthritis, and the use of methotrexate appears to attenuate the presence of antidrug antibodies, according to findings from a cross-sectional study.
Although researchers have looked at the immunogenicity of TNF-alpha blockers in patients with diseases such as rheumatoid arthritis (RA), ankylosing spondylitis, psoriasis, and inflammatory bowel disease, their immunogenicity in psoriatic arthritis (PsA) patients has not been fully investigated, noted the current study’s investigators, led by Dr. Michael Zisapel of the department of rheumatology at Tel Aviv University (J. Rheumatol. 2014 Nov. 15 [doi:10.3899/jrheum.140685]).
The prevalence of antidrug antibodies (ADAb) to TNF-alpha blockers has been reported to be 20%-40% in RA, 25%-64% in ankylosing spondylitis, and about 33% in psoriasis, and evidence has shown that this is significantly reduced by the use of methotrexate, they said.
The study involved 93 patients with PsA who were taking adalimumab (n = 48), infliximab (n = 24), and etanercept (n = 21). A quarter of the patients were taking methotrexate at an average dose of 13.3 mg/week.
Overall, 77% of the patients had therapeutic drug levels. The prevalence of immunogenicity in the entire group was 33.3%, and one-fifth of patients had high concentrations of antibodies.
High levels of ADAb were found in 29% of patients taking adalimumab and 21% of the patients taking infliximab. No ADAb levels were found in patients taking etanercept.
Interestingly, the patients taking adalimumab demonstrated an immunogenicity of 54%, but only half of them (29%) showed high concentrations of antibodies.
“This finding suggests that a variety of levels might be found that might affect the significance of immunogenicity differently among patients producing ADAb,” the investigators wrote.
Fewer methotrexate-treated patients had high ADAb concentrations, compared with patients not taking methotrexate (16.7% vs. 21.7%).
A clear correlation was found between the presence of immunogenicity, lower drug levels, and decreased clinical response in PsA patients, just as other studies have found in RA and ankylosing spondylitis patients, the authors noted.
“Our results suggest that the use of methotrexate should be strongly considered in addition to monoclonal antibodies,” they said.
Limitations of their study included the small number of patients and the use of a bridging ELISA test which is reliable but considered to be less accurate than radioimmunoassay.
No disclosure information was available.
FROM JOURNAL OF RHEUMATOLOGY
Key clinical point: The use of methotrexate should be strongly considered in addition to TNF-alpha blockers in patients with psoriatic arthritis to reduce the presence and influence of antidrug antibodies.
Major finding: High levels of antidrug antibodies were found in 29% of patients taking adalimumab, 21% of the patients taking infliximab, and no patients taking etanercept.
Data source: A cross-sectional study of 93 consecutive psoriatic arthritis patients.
Disclosures: No disclosure information was available.
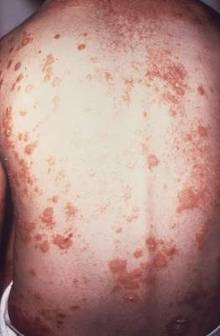
Psoriasis patients have higher rate of low back pain
The prevalence of low back pain appears to be higher in people with psoriasis than in the general population, according to an analysis of national survey data.
The findings may change the way psoriasis patients are managed when they present to primary care or specialty clinics with sudden-onset back and buttock pain, said first author of the study, Dr. Nicole Thom of the division of rheumatology at Cedars-Sinai Medical Center, Los Angeles, and her colleagues (Arthritis Care Res. 2014 Dec. 2 [doi:10.1002/acr.22528]).
Using data from the 2009-2010 U.S. National Health and Nutrition Examination Survey of 6,684 adults, the researchers identified 5,103 people who had answered questions on back pain. A total of 148 had psoriasis and 5 had psoriatic arthritis (PsA).
People with psoriasis/PsA had a significantly higher prevalence of axial pain as measured using the 3-month duration criteria, compared with people without the disease (31.1% vs. 18.9%; P = .04). They were also more likely to have alternating buttock pain (7.2% vs. 2.4%; P = .03) and meet Berlin 7b and 8a criteria for inflammatory back pain (P = .04 and P = .02, respectively). The prevalence of spondyloarthritis was significantly higher in the psoriasis/PsA group when using Amor or European Spondyloarthritis Study Group criteria (14.3% vs. 1.5%; P = .001). Sudden onset of axial pain was also higher in the psoriasis/PsA group (23.3% vs. 13.0%; P = .01), the researchers reported.
“The internist or family medicine physician should include inflammatory back pain in their differential diagnosis,” the study authors suggest.
With more and more research continuing to support multiple comorbidities in psoriasis, it also raises the question as to whether rheumatologists, dermatologists, and other health care professionals should be screening for them, they said.
The work was supported in part by grants from the National Institute of Arthritis and Musculoskeletal and Skin Diseases and a National Center for Research Resources grant to the Clinical Translational Science Institute at the University of California, Los Angeles.
The prevalence of low back pain appears to be higher in people with psoriasis than in the general population, according to an analysis of national survey data.
The findings may change the way psoriasis patients are managed when they present to primary care or specialty clinics with sudden-onset back and buttock pain, said first author of the study, Dr. Nicole Thom of the division of rheumatology at Cedars-Sinai Medical Center, Los Angeles, and her colleagues (Arthritis Care Res. 2014 Dec. 2 [doi:10.1002/acr.22528]).
Using data from the 2009-2010 U.S. National Health and Nutrition Examination Survey of 6,684 adults, the researchers identified 5,103 people who had answered questions on back pain. A total of 148 had psoriasis and 5 had psoriatic arthritis (PsA).
People with psoriasis/PsA had a significantly higher prevalence of axial pain as measured using the 3-month duration criteria, compared with people without the disease (31.1% vs. 18.9%; P = .04). They were also more likely to have alternating buttock pain (7.2% vs. 2.4%; P = .03) and meet Berlin 7b and 8a criteria for inflammatory back pain (P = .04 and P = .02, respectively). The prevalence of spondyloarthritis was significantly higher in the psoriasis/PsA group when using Amor or European Spondyloarthritis Study Group criteria (14.3% vs. 1.5%; P = .001). Sudden onset of axial pain was also higher in the psoriasis/PsA group (23.3% vs. 13.0%; P = .01), the researchers reported.
“The internist or family medicine physician should include inflammatory back pain in their differential diagnosis,” the study authors suggest.
With more and more research continuing to support multiple comorbidities in psoriasis, it also raises the question as to whether rheumatologists, dermatologists, and other health care professionals should be screening for them, they said.
The work was supported in part by grants from the National Institute of Arthritis and Musculoskeletal and Skin Diseases and a National Center for Research Resources grant to the Clinical Translational Science Institute at the University of California, Los Angeles.
The prevalence of low back pain appears to be higher in people with psoriasis than in the general population, according to an analysis of national survey data.
The findings may change the way psoriasis patients are managed when they present to primary care or specialty clinics with sudden-onset back and buttock pain, said first author of the study, Dr. Nicole Thom of the division of rheumatology at Cedars-Sinai Medical Center, Los Angeles, and her colleagues (Arthritis Care Res. 2014 Dec. 2 [doi:10.1002/acr.22528]).
Using data from the 2009-2010 U.S. National Health and Nutrition Examination Survey of 6,684 adults, the researchers identified 5,103 people who had answered questions on back pain. A total of 148 had psoriasis and 5 had psoriatic arthritis (PsA).
People with psoriasis/PsA had a significantly higher prevalence of axial pain as measured using the 3-month duration criteria, compared with people without the disease (31.1% vs. 18.9%; P = .04). They were also more likely to have alternating buttock pain (7.2% vs. 2.4%; P = .03) and meet Berlin 7b and 8a criteria for inflammatory back pain (P = .04 and P = .02, respectively). The prevalence of spondyloarthritis was significantly higher in the psoriasis/PsA group when using Amor or European Spondyloarthritis Study Group criteria (14.3% vs. 1.5%; P = .001). Sudden onset of axial pain was also higher in the psoriasis/PsA group (23.3% vs. 13.0%; P = .01), the researchers reported.
“The internist or family medicine physician should include inflammatory back pain in their differential diagnosis,” the study authors suggest.
With more and more research continuing to support multiple comorbidities in psoriasis, it also raises the question as to whether rheumatologists, dermatologists, and other health care professionals should be screening for them, they said.
The work was supported in part by grants from the National Institute of Arthritis and Musculoskeletal and Skin Diseases and a National Center for Research Resources grant to the Clinical Translational Science Institute at the University of California, Los Angeles.
FROM ARTHRITIS CARE & RESEARCH
Ixekizumab linked to decrease in psoriasis-related sexual difficulties
AMSTERDAM – Patient complaints of psoriasis-related sexual difficulties decreased significantly in response to treatment with the investigational biologic agent ixekizumab in a phase 2 dose-ranging study.
Sexual problems attributed by psoriasis patients to their skin condition are a common, underappreciated, and understudied problem. Most physicians simply don’t ask. But when they do, as was done formally in this study, it turned out that at baseline roughly one-third of the 142 participating subjects with moderate to severe chronic plaque psoriasis reported some degree of sexual problems they believed due specifically to their skin disease, Dr. Lyn Guenther reported at the annual congress of the European Academy of Dermatology and Venereology.
That rate dropped precipitously and in dose-dependent fashion in response to subcutaneous therapy with ixekizumab, a super-potent psoriasis medication directed against interleukin 17A, according to Dr. Guenther, professor of dermatology at the University of Western Ontario, London.
The study entailed double-blind randomization of the 142 participants to subcutaneous injections of ixekizumab at 10, 25, 75, or 150 mg or placebo at 0, 2, 4, 8, 12, and 16 weeks. The standard primary outcome -- the proportion of patients with a PASI 75 improvement at 12 weeks -- has previously been reported as highly positive (N. Engl. J. Med. 2012;366:1190-9).
For her secondary analysis of psoriasis-related sexual difficulties and their response to treatment, Dr. Guenther used as her metric the patients’ response to item 9 on the Dermatology Life Quality Index, which all subjects completed at weeks 0, 8, and 16. Item 9 asks the extent to which the responder’s skin “caused any sexual difficulties” during the past week. The options range from 0 (none at all) to 3 (very much). She categorized a response of 1 or more as evidence of sexual difficulties. And because of the relatively small sample size in this study, she lumped together as the low-dose therapy group those patients assigned to ixekizumab at 10 or 25 mg, with the high-dose group being comprised of patients on 75 or
After 8 weeks on low-dose ixekizumab, the proportion of patients reporting any skin disease-related sexual difficulties within the past week fell from a baseline of 30% to 16%. After 8 weeks of high-dose ixekizumab, the figure was just 7%. Those rates remained unchanged at 16 weeks. In contrast, the placebo group remained unchanged over time, with 32% of patients still reporting sexual difficulties caused by their skin disease at week 8.
The degree to which a patient’s psoriasis improved in response to therapy with the humanized monoclonal antibody was closely related to the reduction in skin-related sexual problems. Among the 76 ixekizumab-treated patients who achieved a PASI 75 response at week 16, the rate of self-reported sexual difficulties within the previous week was 7%. For those with less than a PASI 75 response, the rate was 24%.
An impressively high 38% percent of patients on high-dose ixekizumab achieved a PASI 100 response. Only 5% of them reported any skin-related sexual difficulties at week 8, as did 9% at week 16.
Dr. Guenther also looked at the data restricting the analysis to patients with more severe baseline sexual impairment as defined by a response of 2 or 3 on item 9 of the DLQI. Among patients on high-dose izekizumab, the rate dropped from 10.5% at baseline to 1.8% at week 8 to zero at week 16. For patients on low-dose izekizumab, the progression was 13.8% to 8.8% to 3.5% at week 16. Rates remained unchanged over time in the control group.
Although this phase 2 study was limited in size, Dr. Guenther found much the same thing earlier in the much larger phase 3 PHOENIX 1 and 2 trials of ustekinumab (Stelara), which together featured 1,334 psoriasis patients randomized to the human anti-interkelukin-12/23 monoclonal antibody. In that analysis, the proportion of ustekinumab-treated patients with impaired sexual function as assessed by DLQI item 9 plunged from 22.4% at baseline to 2.7% at week 12, compared with no change in placebo-treated controls. The bigger the PASI improvement, the greater the reduction in psoriasis-related sexual dysfunction (J. Eur. Acad. Dermatol. Venereol. 2011;25:851-7).
The phase 2 ixekizumab study was funded by Eli Lilly. Dr. Guenther is a consultant to the company. Positive primary outcomes in three pivotal phase 3 clinical trials of ixekizumab totalling nearly 3,900 randomized psoriasis patients have since been reported. The company plans to apply for marketing approval of the biologic in the first half of 2015.
AMSTERDAM – Patient complaints of psoriasis-related sexual difficulties decreased significantly in response to treatment with the investigational biologic agent ixekizumab in a phase 2 dose-ranging study.
Sexual problems attributed by psoriasis patients to their skin condition are a common, underappreciated, and understudied problem. Most physicians simply don’t ask. But when they do, as was done formally in this study, it turned out that at baseline roughly one-third of the 142 participating subjects with moderate to severe chronic plaque psoriasis reported some degree of sexual problems they believed due specifically to their skin disease, Dr. Lyn Guenther reported at the annual congress of the European Academy of Dermatology and Venereology.
That rate dropped precipitously and in dose-dependent fashion in response to subcutaneous therapy with ixekizumab, a super-potent psoriasis medication directed against interleukin 17A, according to Dr. Guenther, professor of dermatology at the University of Western Ontario, London.
The study entailed double-blind randomization of the 142 participants to subcutaneous injections of ixekizumab at 10, 25, 75, or 150 mg or placebo at 0, 2, 4, 8, 12, and 16 weeks. The standard primary outcome -- the proportion of patients with a PASI 75 improvement at 12 weeks -- has previously been reported as highly positive (N. Engl. J. Med. 2012;366:1190-9).
For her secondary analysis of psoriasis-related sexual difficulties and their response to treatment, Dr. Guenther used as her metric the patients’ response to item 9 on the Dermatology Life Quality Index, which all subjects completed at weeks 0, 8, and 16. Item 9 asks the extent to which the responder’s skin “caused any sexual difficulties” during the past week. The options range from 0 (none at all) to 3 (very much). She categorized a response of 1 or more as evidence of sexual difficulties. And because of the relatively small sample size in this study, she lumped together as the low-dose therapy group those patients assigned to ixekizumab at 10 or 25 mg, with the high-dose group being comprised of patients on 75 or
After 8 weeks on low-dose ixekizumab, the proportion of patients reporting any skin disease-related sexual difficulties within the past week fell from a baseline of 30% to 16%. After 8 weeks of high-dose ixekizumab, the figure was just 7%. Those rates remained unchanged at 16 weeks. In contrast, the placebo group remained unchanged over time, with 32% of patients still reporting sexual difficulties caused by their skin disease at week 8.
The degree to which a patient’s psoriasis improved in response to therapy with the humanized monoclonal antibody was closely related to the reduction in skin-related sexual problems. Among the 76 ixekizumab-treated patients who achieved a PASI 75 response at week 16, the rate of self-reported sexual difficulties within the previous week was 7%. For those with less than a PASI 75 response, the rate was 24%.
An impressively high 38% percent of patients on high-dose ixekizumab achieved a PASI 100 response. Only 5% of them reported any skin-related sexual difficulties at week 8, as did 9% at week 16.
Dr. Guenther also looked at the data restricting the analysis to patients with more severe baseline sexual impairment as defined by a response of 2 or 3 on item 9 of the DLQI. Among patients on high-dose izekizumab, the rate dropped from 10.5% at baseline to 1.8% at week 8 to zero at week 16. For patients on low-dose izekizumab, the progression was 13.8% to 8.8% to 3.5% at week 16. Rates remained unchanged over time in the control group.
Although this phase 2 study was limited in size, Dr. Guenther found much the same thing earlier in the much larger phase 3 PHOENIX 1 and 2 trials of ustekinumab (Stelara), which together featured 1,334 psoriasis patients randomized to the human anti-interkelukin-12/23 monoclonal antibody. In that analysis, the proportion of ustekinumab-treated patients with impaired sexual function as assessed by DLQI item 9 plunged from 22.4% at baseline to 2.7% at week 12, compared with no change in placebo-treated controls. The bigger the PASI improvement, the greater the reduction in psoriasis-related sexual dysfunction (J. Eur. Acad. Dermatol. Venereol. 2011;25:851-7).
The phase 2 ixekizumab study was funded by Eli Lilly. Dr. Guenther is a consultant to the company. Positive primary outcomes in three pivotal phase 3 clinical trials of ixekizumab totalling nearly 3,900 randomized psoriasis patients have since been reported. The company plans to apply for marketing approval of the biologic in the first half of 2015.
AMSTERDAM – Patient complaints of psoriasis-related sexual difficulties decreased significantly in response to treatment with the investigational biologic agent ixekizumab in a phase 2 dose-ranging study.
Sexual problems attributed by psoriasis patients to their skin condition are a common, underappreciated, and understudied problem. Most physicians simply don’t ask. But when they do, as was done formally in this study, it turned out that at baseline roughly one-third of the 142 participating subjects with moderate to severe chronic plaque psoriasis reported some degree of sexual problems they believed due specifically to their skin disease, Dr. Lyn Guenther reported at the annual congress of the European Academy of Dermatology and Venereology.
That rate dropped precipitously and in dose-dependent fashion in response to subcutaneous therapy with ixekizumab, a super-potent psoriasis medication directed against interleukin 17A, according to Dr. Guenther, professor of dermatology at the University of Western Ontario, London.
The study entailed double-blind randomization of the 142 participants to subcutaneous injections of ixekizumab at 10, 25, 75, or 150 mg or placebo at 0, 2, 4, 8, 12, and 16 weeks. The standard primary outcome -- the proportion of patients with a PASI 75 improvement at 12 weeks -- has previously been reported as highly positive (N. Engl. J. Med. 2012;366:1190-9).
For her secondary analysis of psoriasis-related sexual difficulties and their response to treatment, Dr. Guenther used as her metric the patients’ response to item 9 on the Dermatology Life Quality Index, which all subjects completed at weeks 0, 8, and 16. Item 9 asks the extent to which the responder’s skin “caused any sexual difficulties” during the past week. The options range from 0 (none at all) to 3 (very much). She categorized a response of 1 or more as evidence of sexual difficulties. And because of the relatively small sample size in this study, she lumped together as the low-dose therapy group those patients assigned to ixekizumab at 10 or 25 mg, with the high-dose group being comprised of patients on 75 or
After 8 weeks on low-dose ixekizumab, the proportion of patients reporting any skin disease-related sexual difficulties within the past week fell from a baseline of 30% to 16%. After 8 weeks of high-dose ixekizumab, the figure was just 7%. Those rates remained unchanged at 16 weeks. In contrast, the placebo group remained unchanged over time, with 32% of patients still reporting sexual difficulties caused by their skin disease at week 8.
The degree to which a patient’s psoriasis improved in response to therapy with the humanized monoclonal antibody was closely related to the reduction in skin-related sexual problems. Among the 76 ixekizumab-treated patients who achieved a PASI 75 response at week 16, the rate of self-reported sexual difficulties within the previous week was 7%. For those with less than a PASI 75 response, the rate was 24%.
An impressively high 38% percent of patients on high-dose ixekizumab achieved a PASI 100 response. Only 5% of them reported any skin-related sexual difficulties at week 8, as did 9% at week 16.
Dr. Guenther also looked at the data restricting the analysis to patients with more severe baseline sexual impairment as defined by a response of 2 or 3 on item 9 of the DLQI. Among patients on high-dose izekizumab, the rate dropped from 10.5% at baseline to 1.8% at week 8 to zero at week 16. For patients on low-dose izekizumab, the progression was 13.8% to 8.8% to 3.5% at week 16. Rates remained unchanged over time in the control group.
Although this phase 2 study was limited in size, Dr. Guenther found much the same thing earlier in the much larger phase 3 PHOENIX 1 and 2 trials of ustekinumab (Stelara), which together featured 1,334 psoriasis patients randomized to the human anti-interkelukin-12/23 monoclonal antibody. In that analysis, the proportion of ustekinumab-treated patients with impaired sexual function as assessed by DLQI item 9 plunged from 22.4% at baseline to 2.7% at week 12, compared with no change in placebo-treated controls. The bigger the PASI improvement, the greater the reduction in psoriasis-related sexual dysfunction (J. Eur. Acad. Dermatol. Venereol. 2011;25:851-7).
The phase 2 ixekizumab study was funded by Eli Lilly. Dr. Guenther is a consultant to the company. Positive primary outcomes in three pivotal phase 3 clinical trials of ixekizumab totalling nearly 3,900 randomized psoriasis patients have since been reported. The company plans to apply for marketing approval of the biologic in the first half of 2015.
AT THE EADV CONGRESS
Key clinical point: Psoriasis patients have a high rate of sexual problems they attribute to their skin disease, and which decrease with effective psoriasis therapy.
Major finding: The prevalence of self-reported psoriasis-related sexual difficulties within the previous week fell from 32% at baseline to 7% among patients with a PASI 75 response to ixekizumab at week 16.
Data source: This was a double-blind, phase 2, dose-ranging study involving 142 patients with moderate to severe chronic plaque psoriasis randomized to 16 weeks of ixekizumab or placebo.
Disclosures: The study was funded by Eli Lilly. The presenter is a consultant to the company.
Consider phototherapy for certain psoriasis patients
LAS VEGAS – Psoriasis patients who are unresponsive to topical therapy, do not have psoriatic arthritis, and can make regular office visits may be candidates for successful treatment with phototherapy.
A patient with type II skin and extensive plaque psoriasis could be treated with a targeted NB-UVB laser, Dr. Kenneth B. Gordon said at Skin Disease Education Foundation’s annual Las Vegas Dermatology Seminar.
However, it’s important to evaluate each patient individually on phototherapy decisions, rather than relying on published protocols, noted Dr. Gordon of Northwestern University Feinberg School of Medicine in Chicago.
The advantages of the NB-UVB laser for psoriasis include a high rate of response (data show more than 75% of patients achieve PASI 75), the ability to avoid treating uninvolved skin, and potential long-term clearing of lesions, he said.
For someone with extensive plaque psoriasis, Dr. Gordon said he would start with 3 treatments per week, at 200 mJ, and increase the fluence by 25 mJ if necessary. “A good response can be expected in 4-6 weeks,” he said.
As for long-term treatment, “I leave it up to the patient but offer long-term maintenance,” said Dr. Gordon. Long-term efficacy of NB-UVB for plaque psoriasis has not been well studied, and safety data are unknown. “It is unclear whether there is a skin cancer risk,” he said. Reducing the number of exposures for a patient on maintenance therapy may be an option, he added.
Short-term treatment with NB-UVB can be effective in some psoriasis patients, such as in cases of an acute flare of guttate psoriasis, Dr. Gordon noted. Psoralen and UV light therapy (PUVA) is another option, he said.
Phototherapy also has a role in managing psoriasis in conjunction with retinoids, Dr. Gordon said. He cited an example of a 59-year-old woman with palmar psoriasis. For this patient, he said he would recommend starting with acitretin for approximately 1 month, if the patient tolerates it. “If the acitretin works by itself, no need to add phototherapy,” he said. However, if UVB is added, account for the photosensitizing agent, he emphasized. “Decrease the starting dose by 25-50 mJ or decrease the skin type by one,” he said.
Some psoriasis patients will not benefit from phototherapy, particularly those with erythroderma, said Dr. Gordon. “If skin is highly inflamed, phototherapy could induce easy burning, Koebnerization,” or other complications, he noted.
Dr. Gordon disclosed that his department at Northwestern derives income from phototherapy, but he personally does not. He also disclosed receiving research grants from and/or serving as a consultant to multiple companies including AbbVie, Amgen, Celgene, Eli Lilly, and Janssen.
LAS VEGAS – Psoriasis patients who are unresponsive to topical therapy, do not have psoriatic arthritis, and can make regular office visits may be candidates for successful treatment with phototherapy.
A patient with type II skin and extensive plaque psoriasis could be treated with a targeted NB-UVB laser, Dr. Kenneth B. Gordon said at Skin Disease Education Foundation’s annual Las Vegas Dermatology Seminar.
However, it’s important to evaluate each patient individually on phototherapy decisions, rather than relying on published protocols, noted Dr. Gordon of Northwestern University Feinberg School of Medicine in Chicago.
The advantages of the NB-UVB laser for psoriasis include a high rate of response (data show more than 75% of patients achieve PASI 75), the ability to avoid treating uninvolved skin, and potential long-term clearing of lesions, he said.
For someone with extensive plaque psoriasis, Dr. Gordon said he would start with 3 treatments per week, at 200 mJ, and increase the fluence by 25 mJ if necessary. “A good response can be expected in 4-6 weeks,” he said.
As for long-term treatment, “I leave it up to the patient but offer long-term maintenance,” said Dr. Gordon. Long-term efficacy of NB-UVB for plaque psoriasis has not been well studied, and safety data are unknown. “It is unclear whether there is a skin cancer risk,” he said. Reducing the number of exposures for a patient on maintenance therapy may be an option, he added.
Short-term treatment with NB-UVB can be effective in some psoriasis patients, such as in cases of an acute flare of guttate psoriasis, Dr. Gordon noted. Psoralen and UV light therapy (PUVA) is another option, he said.
Phototherapy also has a role in managing psoriasis in conjunction with retinoids, Dr. Gordon said. He cited an example of a 59-year-old woman with palmar psoriasis. For this patient, he said he would recommend starting with acitretin for approximately 1 month, if the patient tolerates it. “If the acitretin works by itself, no need to add phototherapy,” he said. However, if UVB is added, account for the photosensitizing agent, he emphasized. “Decrease the starting dose by 25-50 mJ or decrease the skin type by one,” he said.
Some psoriasis patients will not benefit from phototherapy, particularly those with erythroderma, said Dr. Gordon. “If skin is highly inflamed, phototherapy could induce easy burning, Koebnerization,” or other complications, he noted.
Dr. Gordon disclosed that his department at Northwestern derives income from phototherapy, but he personally does not. He also disclosed receiving research grants from and/or serving as a consultant to multiple companies including AbbVie, Amgen, Celgene, Eli Lilly, and Janssen.
LAS VEGAS – Psoriasis patients who are unresponsive to topical therapy, do not have psoriatic arthritis, and can make regular office visits may be candidates for successful treatment with phototherapy.
A patient with type II skin and extensive plaque psoriasis could be treated with a targeted NB-UVB laser, Dr. Kenneth B. Gordon said at Skin Disease Education Foundation’s annual Las Vegas Dermatology Seminar.
However, it’s important to evaluate each patient individually on phototherapy decisions, rather than relying on published protocols, noted Dr. Gordon of Northwestern University Feinberg School of Medicine in Chicago.
The advantages of the NB-UVB laser for psoriasis include a high rate of response (data show more than 75% of patients achieve PASI 75), the ability to avoid treating uninvolved skin, and potential long-term clearing of lesions, he said.
For someone with extensive plaque psoriasis, Dr. Gordon said he would start with 3 treatments per week, at 200 mJ, and increase the fluence by 25 mJ if necessary. “A good response can be expected in 4-6 weeks,” he said.
As for long-term treatment, “I leave it up to the patient but offer long-term maintenance,” said Dr. Gordon. Long-term efficacy of NB-UVB for plaque psoriasis has not been well studied, and safety data are unknown. “It is unclear whether there is a skin cancer risk,” he said. Reducing the number of exposures for a patient on maintenance therapy may be an option, he added.
Short-term treatment with NB-UVB can be effective in some psoriasis patients, such as in cases of an acute flare of guttate psoriasis, Dr. Gordon noted. Psoralen and UV light therapy (PUVA) is another option, he said.
Phototherapy also has a role in managing psoriasis in conjunction with retinoids, Dr. Gordon said. He cited an example of a 59-year-old woman with palmar psoriasis. For this patient, he said he would recommend starting with acitretin for approximately 1 month, if the patient tolerates it. “If the acitretin works by itself, no need to add phototherapy,” he said. However, if UVB is added, account for the photosensitizing agent, he emphasized. “Decrease the starting dose by 25-50 mJ or decrease the skin type by one,” he said.
Some psoriasis patients will not benefit from phototherapy, particularly those with erythroderma, said Dr. Gordon. “If skin is highly inflamed, phototherapy could induce easy burning, Koebnerization,” or other complications, he noted.
Dr. Gordon disclosed that his department at Northwestern derives income from phototherapy, but he personally does not. He also disclosed receiving research grants from and/or serving as a consultant to multiple companies including AbbVie, Amgen, Celgene, Eli Lilly, and Janssen.
EXPERT ANALYSIS FROM SDEF LAS VEGAS DERMATOLOGY SEMINAR
Oral tofacitinib scores against psoriasis in phase III trial
AMSTERDAM – Oral tofacitinib for psoriasis proved as effective as – and in certain domains better than – subcutaneous etanercept in improving patient-reported quality of life endpoints in a phase III clinical trial.
The double-blind study included 1,101 patients with moderate to severe chronic plaque psoriasis who were randomized to 12 weeks of the oral Janus kinase inhibitor tofacitinib at 5 mg twice daily, 10 mg twice daily, subcutaneous etanercept (Enbrel) at 50 mg twice weekly, or placebo. The findings were presented by Dr. Fernando Valenzuela at the annual congress of the European Academy of Dermatology and Venereology.
Earlier in 2014, at the annual meeting of the American Academy of Dermatology in Denver, Dr. Valenzuela presented the phase III study’s primary results, in which the oral small molecule proved noninferior to the tumor necrosis factor inhibitor in improving PASI scores. At the EADV Congress in Amsterdam, he focused on the secondary outcomes, which arguably matter more to patients than do changes in PASI scores, namely, measures of quality of life and itchiness.
From a baseline Itch Severity Item score of 5, indicative of moderate to severe itching, week 12 scores fell by a mean of 3.2, 4.0, 3.5, and 0.4 points, respectively, in patients on low- or high-dose tofacitinib, etanercept, and placebo. The improvement in itch in patients randomized to tofacitinib at 10 mg twice daily was not only greater than with etanercept, it occurred faster as well, with significant reduction in itch scores documented on day 2 of therapy, reported Dr. Valenzuela, a dermatologist at the University of Chile in Santiago.
The median baseline Dermatology Life Quality Index (DLQI) score was 12. Significant improvement was seen from week 4 in all three active treatment study arms. By week 12, DLQI scores had dropped by an average of 7.3 points in patients on tofacitinib at 5 mg twice daily, 9.7 points in those on 10 mg twice daily, 9.0 points in etanercept-treated patients, and 1.9 points with placebo. Seventy-eight percent of patients on tofacitinib at 10 mg twice daily experienced a 5-point or larger drop in the DLQI by week 12, as did 75% of those on etanercept, 66% of those on low-dose tofacitinib, and 32% of those on placebo.
At baseline, 30% of patients rated their psoriasis as moderate and 70% rated their psoriasis as severe based on Patient Global Assessment. By week 12, more than 50% of patients on tofacitinib at 10 mg twice daily or etanercept rated their skin as clear or almost clear.
More than 70% of patients in all three active treatment arms indicated at week 12 that they were satisfied with their medication. Satisfaction scores were highest, and equally so, in those on high-dose tofacitinib and etanercept.
“Patients using tofacitinib at the low dose had not that good an improvement. It was better improvement than with placebo, but it doesn’t look like etanercept,” Dr. Valenzuela concluded.
Tofacitinib is approved as Xeljanz for the treatment of rheumatoid arthritis, but remains investigational for psoriasis. According to a company statement, Pfizer intends to submit a supplemental New Drug Application (sNDA) to the Food and Drug Administration by early 2015.
The study was funded by Pfizer. Dr. Valenzuela is an adviser to Pfizer, AbbVie, Eli Lilly, Janssen, Merck, and Novartis.
AMSTERDAM – Oral tofacitinib for psoriasis proved as effective as – and in certain domains better than – subcutaneous etanercept in improving patient-reported quality of life endpoints in a phase III clinical trial.
The double-blind study included 1,101 patients with moderate to severe chronic plaque psoriasis who were randomized to 12 weeks of the oral Janus kinase inhibitor tofacitinib at 5 mg twice daily, 10 mg twice daily, subcutaneous etanercept (Enbrel) at 50 mg twice weekly, or placebo. The findings were presented by Dr. Fernando Valenzuela at the annual congress of the European Academy of Dermatology and Venereology.
Earlier in 2014, at the annual meeting of the American Academy of Dermatology in Denver, Dr. Valenzuela presented the phase III study’s primary results, in which the oral small molecule proved noninferior to the tumor necrosis factor inhibitor in improving PASI scores. At the EADV Congress in Amsterdam, he focused on the secondary outcomes, which arguably matter more to patients than do changes in PASI scores, namely, measures of quality of life and itchiness.
From a baseline Itch Severity Item score of 5, indicative of moderate to severe itching, week 12 scores fell by a mean of 3.2, 4.0, 3.5, and 0.4 points, respectively, in patients on low- or high-dose tofacitinib, etanercept, and placebo. The improvement in itch in patients randomized to tofacitinib at 10 mg twice daily was not only greater than with etanercept, it occurred faster as well, with significant reduction in itch scores documented on day 2 of therapy, reported Dr. Valenzuela, a dermatologist at the University of Chile in Santiago.
The median baseline Dermatology Life Quality Index (DLQI) score was 12. Significant improvement was seen from week 4 in all three active treatment study arms. By week 12, DLQI scores had dropped by an average of 7.3 points in patients on tofacitinib at 5 mg twice daily, 9.7 points in those on 10 mg twice daily, 9.0 points in etanercept-treated patients, and 1.9 points with placebo. Seventy-eight percent of patients on tofacitinib at 10 mg twice daily experienced a 5-point or larger drop in the DLQI by week 12, as did 75% of those on etanercept, 66% of those on low-dose tofacitinib, and 32% of those on placebo.
At baseline, 30% of patients rated their psoriasis as moderate and 70% rated their psoriasis as severe based on Patient Global Assessment. By week 12, more than 50% of patients on tofacitinib at 10 mg twice daily or etanercept rated their skin as clear or almost clear.
More than 70% of patients in all three active treatment arms indicated at week 12 that they were satisfied with their medication. Satisfaction scores were highest, and equally so, in those on high-dose tofacitinib and etanercept.
“Patients using tofacitinib at the low dose had not that good an improvement. It was better improvement than with placebo, but it doesn’t look like etanercept,” Dr. Valenzuela concluded.
Tofacitinib is approved as Xeljanz for the treatment of rheumatoid arthritis, but remains investigational for psoriasis. According to a company statement, Pfizer intends to submit a supplemental New Drug Application (sNDA) to the Food and Drug Administration by early 2015.
The study was funded by Pfizer. Dr. Valenzuela is an adviser to Pfizer, AbbVie, Eli Lilly, Janssen, Merck, and Novartis.
AMSTERDAM – Oral tofacitinib for psoriasis proved as effective as – and in certain domains better than – subcutaneous etanercept in improving patient-reported quality of life endpoints in a phase III clinical trial.
The double-blind study included 1,101 patients with moderate to severe chronic plaque psoriasis who were randomized to 12 weeks of the oral Janus kinase inhibitor tofacitinib at 5 mg twice daily, 10 mg twice daily, subcutaneous etanercept (Enbrel) at 50 mg twice weekly, or placebo. The findings were presented by Dr. Fernando Valenzuela at the annual congress of the European Academy of Dermatology and Venereology.
Earlier in 2014, at the annual meeting of the American Academy of Dermatology in Denver, Dr. Valenzuela presented the phase III study’s primary results, in which the oral small molecule proved noninferior to the tumor necrosis factor inhibitor in improving PASI scores. At the EADV Congress in Amsterdam, he focused on the secondary outcomes, which arguably matter more to patients than do changes in PASI scores, namely, measures of quality of life and itchiness.
From a baseline Itch Severity Item score of 5, indicative of moderate to severe itching, week 12 scores fell by a mean of 3.2, 4.0, 3.5, and 0.4 points, respectively, in patients on low- or high-dose tofacitinib, etanercept, and placebo. The improvement in itch in patients randomized to tofacitinib at 10 mg twice daily was not only greater than with etanercept, it occurred faster as well, with significant reduction in itch scores documented on day 2 of therapy, reported Dr. Valenzuela, a dermatologist at the University of Chile in Santiago.
The median baseline Dermatology Life Quality Index (DLQI) score was 12. Significant improvement was seen from week 4 in all three active treatment study arms. By week 12, DLQI scores had dropped by an average of 7.3 points in patients on tofacitinib at 5 mg twice daily, 9.7 points in those on 10 mg twice daily, 9.0 points in etanercept-treated patients, and 1.9 points with placebo. Seventy-eight percent of patients on tofacitinib at 10 mg twice daily experienced a 5-point or larger drop in the DLQI by week 12, as did 75% of those on etanercept, 66% of those on low-dose tofacitinib, and 32% of those on placebo.
At baseline, 30% of patients rated their psoriasis as moderate and 70% rated their psoriasis as severe based on Patient Global Assessment. By week 12, more than 50% of patients on tofacitinib at 10 mg twice daily or etanercept rated their skin as clear or almost clear.
More than 70% of patients in all three active treatment arms indicated at week 12 that they were satisfied with their medication. Satisfaction scores were highest, and equally so, in those on high-dose tofacitinib and etanercept.
“Patients using tofacitinib at the low dose had not that good an improvement. It was better improvement than with placebo, but it doesn’t look like etanercept,” Dr. Valenzuela concluded.
Tofacitinib is approved as Xeljanz for the treatment of rheumatoid arthritis, but remains investigational for psoriasis. According to a company statement, Pfizer intends to submit a supplemental New Drug Application (sNDA) to the Food and Drug Administration by early 2015.
The study was funded by Pfizer. Dr. Valenzuela is an adviser to Pfizer, AbbVie, Eli Lilly, Janssen, Merck, and Novartis.
AT THE EADV CONGRESS
Key clinical point: Multiple measures of quality of life and disease burden improved similarly in psoriasis patients regardless of whether they were on subcutaneous etanercept or the oral Janus kinase inhibitor tofacitinib.
Major finding: From a median baseline Dermatology Life Quality Index score of 12, scores improved by an average of 7.3 points after 12 weeks of tofacitinib at 5 mg twice daily, 9.7 points with tofacitinib at 10 mg twice daily, 9.0 points with etanercept at 50 mg twice weekly, and 1.9 points with placebo.
Data source: A phase III randomized, double-blind prospective study involving 1,101 patients with moderate to severe chronic plaque psoriasis.
Disclosures: The presenter is an adviser to Pfizer, which sponsored the study, and other pharmaceutical companies.
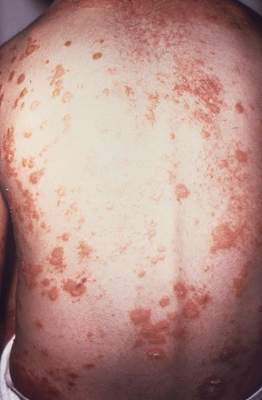
Secukinumab tames psoriatic arthritis in FUTURE 2 trial
BOSTON – Secukinumab improved quality of life and symptoms in patients with psoriatic arthritis, regardless of whether they had received prior anti-tumor-necrosis factor therapy or were concurrently receiving methotrexate, Dr. Ian B. McInnes reported at the annual meeting of the American College of Rheumatology.
A 300-mg dose of the investigational drug, which is given subcutaneously, proved to be the most effective dose, Dr. McInnes, of the University of Glasgow, Scotland, said in his presentation of the 24-week results of the FUTURE 2 study. The primary endpoint, a response of at least ACR20, was achieved by 54% of 100 patients given the 300-mg dose and by 51% of 100 patients given the 150-mg dose. The 75-mg dose was far less effective, with a 29% response in 99 patients; the 98 patients given placebo had a 15% response.
At the 150-mg and 300-mg doses, the rates of ACR20 responses were comparable whether or not patients were also taking concomitant methotrexate. Further, the drug’s safety profile was comparable to placebo, Dr. McInnes said.

An ACR50 was achieved by 35% of patients given secukinumab at either 300 mg or 150 mg, and by 18% of those given 75 mg and 7% of those given placebo. About 20% of patients given the higher doses had an ACR70 response, as did 6% of those given 75 mg and 1% of those given placebo.
At the 300-mg dose, secukinumab also resolved dactylitis and enthesitis in approximately half of the affected patients.
Mean improvements in quality of life based on patients’ SF 36 PCS score (Short Form-36 Physical Component Summary) at 24 weeks from baseline were 7.25 in those on the 300-mg dose and 6.39 in those on the 150-mg dose. Those on the 75-mg dose had a 4.38-point mean improvement, whereas patients on placebo had a 1.34-point mean improvement in SF 36 PCS.
No safety signals were noted; adverse events were few and comparable to placebo. Five subjects on the active drug had mild to moderate candidal infections that responded to oral therapy. Neutropenia occurred in one patient in the 300-mg dose group and in one patient in the placebo group, but was transient and patients continued on therapy.
Secukinumab is a fully-human IgG1k monoclonal antibody that selectively targets IL-17A. The drug, manufactured by Novartis, was unanimously recommended for approval by an advisory committee to the Food and Drug Administration. It was administered weekly as a subcutaneous injection for the first 4 weeks of the study, then given again at week 8 and once every 4 weeks thereafter in patients assigned to one of the secukinumab arms of the double-blind, randomized study. Patient assigned to the placebo group were either responders and assigned to receive secukinumab at week 24 and every 4 weeks thereafter or were nonresponders assigned to receive secukinumab at week 16 and every 4 weeks thereafter. Only patients with at least 20% reductions in the number of tender joints or swollen joints continue to receive the drug beyond 1 year.
To be eligible for the study, patients needed to have a diagnosis of active psoriatic arthritis classified by CASPAR criteria and tenderness in at least 3 of 78 joints and swelling of at least 3 of 76 joints. They additionally needed to have an inadequate response to nonsteroidal anti-inflammatory drugs, methotrexate, or anti-TNF therapy
The primary endpoint of the study was ACR20 response at 24 weeks. Secondary endpoints included PASI 75 and PASI 90 responses, change in DAS28-CRP (28-joint Disease Activity Score using C-reactive protein) from baseline, change in SF-36 PCS and HAQ-DI (Health Assessment Questionnaire-Disability Index) from baseline, ACR50 response, the proportion of subjects with dactylitis and enthesitis, and overall safety and tolerability.
As of press time, the FDA was expected to take additional action in January 2015, according to Novartis.
The study was sponsored by Novartis, the maker of secukinumab. Dr. McInnes receives consulting fees from Novartis as well as multiple other pharmaceutical companies.
BOSTON – Secukinumab improved quality of life and symptoms in patients with psoriatic arthritis, regardless of whether they had received prior anti-tumor-necrosis factor therapy or were concurrently receiving methotrexate, Dr. Ian B. McInnes reported at the annual meeting of the American College of Rheumatology.
A 300-mg dose of the investigational drug, which is given subcutaneously, proved to be the most effective dose, Dr. McInnes, of the University of Glasgow, Scotland, said in his presentation of the 24-week results of the FUTURE 2 study. The primary endpoint, a response of at least ACR20, was achieved by 54% of 100 patients given the 300-mg dose and by 51% of 100 patients given the 150-mg dose. The 75-mg dose was far less effective, with a 29% response in 99 patients; the 98 patients given placebo had a 15% response.
At the 150-mg and 300-mg doses, the rates of ACR20 responses were comparable whether or not patients were also taking concomitant methotrexate. Further, the drug’s safety profile was comparable to placebo, Dr. McInnes said.
An ACR50 was achieved by 35% of patients given secukinumab at either 300 mg or 150 mg, and by 18% of those given 75 mg and 7% of those given placebo. About 20% of patients given the higher doses had an ACR70 response, as did 6% of those given 75 mg and 1% of those given placebo.
At the 300-mg dose, secukinumab also resolved dactylitis and enthesitis in approximately half of the affected patients.
Mean improvements in quality of life based on patients’ SF 36 PCS score (Short Form-36 Physical Component Summary) at 24 weeks from baseline were 7.25 in those on the 300-mg dose and 6.39 in those on the 150-mg dose. Those on the 75-mg dose had a 4.38-point mean improvement, whereas patients on placebo had a 1.34-point mean improvement in SF 36 PCS.
No safety signals were noted; adverse events were few and comparable to placebo. Five subjects on the active drug had mild to moderate candidal infections that responded to oral therapy. Neutropenia occurred in one patient in the 300-mg dose group and in one patient in the placebo group, but was transient and patients continued on therapy.
Secukinumab is a fully-human IgG1k monoclonal antibody that selectively targets IL-17A. The drug, manufactured by Novartis, was unanimously recommended for approval by an advisory committee to the Food and Drug Administration. It was administered weekly as a subcutaneous injection for the first 4 weeks of the study, then given again at week 8 and once every 4 weeks thereafter in patients assigned to one of the secukinumab arms of the double-blind, randomized study. Patient assigned to the placebo group were either responders and assigned to receive secukinumab at week 24 and every 4 weeks thereafter or were nonresponders assigned to receive secukinumab at week 16 and every 4 weeks thereafter. Only patients with at least 20% reductions in the number of tender joints or swollen joints continue to receive the drug beyond 1 year.
To be eligible for the study, patients needed to have a diagnosis of active psoriatic arthritis classified by CASPAR criteria and tenderness in at least 3 of 78 joints and swelling of at least 3 of 76 joints. They additionally needed to have an inadequate response to nonsteroidal anti-inflammatory drugs, methotrexate, or anti-TNF therapy
The primary endpoint of the study was ACR20 response at 24 weeks. Secondary endpoints included PASI 75 and PASI 90 responses, change in DAS28-CRP (28-joint Disease Activity Score using C-reactive protein) from baseline, change in SF-36 PCS and HAQ-DI (Health Assessment Questionnaire-Disability Index) from baseline, ACR50 response, the proportion of subjects with dactylitis and enthesitis, and overall safety and tolerability.
As of press time, the FDA was expected to take additional action in January 2015, according to Novartis.
The study was sponsored by Novartis, the maker of secukinumab. Dr. McInnes receives consulting fees from Novartis as well as multiple other pharmaceutical companies.
BOSTON – Secukinumab improved quality of life and symptoms in patients with psoriatic arthritis, regardless of whether they had received prior anti-tumor-necrosis factor therapy or were concurrently receiving methotrexate, Dr. Ian B. McInnes reported at the annual meeting of the American College of Rheumatology.
A 300-mg dose of the investigational drug, which is given subcutaneously, proved to be the most effective dose, Dr. McInnes, of the University of Glasgow, Scotland, said in his presentation of the 24-week results of the FUTURE 2 study. The primary endpoint, a response of at least ACR20, was achieved by 54% of 100 patients given the 300-mg dose and by 51% of 100 patients given the 150-mg dose. The 75-mg dose was far less effective, with a 29% response in 99 patients; the 98 patients given placebo had a 15% response.
At the 150-mg and 300-mg doses, the rates of ACR20 responses were comparable whether or not patients were also taking concomitant methotrexate. Further, the drug’s safety profile was comparable to placebo, Dr. McInnes said.
An ACR50 was achieved by 35% of patients given secukinumab at either 300 mg or 150 mg, and by 18% of those given 75 mg and 7% of those given placebo. About 20% of patients given the higher doses had an ACR70 response, as did 6% of those given 75 mg and 1% of those given placebo.
At the 300-mg dose, secukinumab also resolved dactylitis and enthesitis in approximately half of the affected patients.
Mean improvements in quality of life based on patients’ SF 36 PCS score (Short Form-36 Physical Component Summary) at 24 weeks from baseline were 7.25 in those on the 300-mg dose and 6.39 in those on the 150-mg dose. Those on the 75-mg dose had a 4.38-point mean improvement, whereas patients on placebo had a 1.34-point mean improvement in SF 36 PCS.
No safety signals were noted; adverse events were few and comparable to placebo. Five subjects on the active drug had mild to moderate candidal infections that responded to oral therapy. Neutropenia occurred in one patient in the 300-mg dose group and in one patient in the placebo group, but was transient and patients continued on therapy.
Secukinumab is a fully-human IgG1k monoclonal antibody that selectively targets IL-17A. The drug, manufactured by Novartis, was unanimously recommended for approval by an advisory committee to the Food and Drug Administration. It was administered weekly as a subcutaneous injection for the first 4 weeks of the study, then given again at week 8 and once every 4 weeks thereafter in patients assigned to one of the secukinumab arms of the double-blind, randomized study. Patient assigned to the placebo group were either responders and assigned to receive secukinumab at week 24 and every 4 weeks thereafter or were nonresponders assigned to receive secukinumab at week 16 and every 4 weeks thereafter. Only patients with at least 20% reductions in the number of tender joints or swollen joints continue to receive the drug beyond 1 year.
To be eligible for the study, patients needed to have a diagnosis of active psoriatic arthritis classified by CASPAR criteria and tenderness in at least 3 of 78 joints and swelling of at least 3 of 76 joints. They additionally needed to have an inadequate response to nonsteroidal anti-inflammatory drugs, methotrexate, or anti-TNF therapy
The primary endpoint of the study was ACR20 response at 24 weeks. Secondary endpoints included PASI 75 and PASI 90 responses, change in DAS28-CRP (28-joint Disease Activity Score using C-reactive protein) from baseline, change in SF-36 PCS and HAQ-DI (Health Assessment Questionnaire-Disability Index) from baseline, ACR50 response, the proportion of subjects with dactylitis and enthesitis, and overall safety and tolerability.
As of press time, the FDA was expected to take additional action in January 2015, according to Novartis.
The study was sponsored by Novartis, the maker of secukinumab. Dr. McInnes receives consulting fees from Novartis as well as multiple other pharmaceutical companies.
AT THE ACR ANNUAL MEETING
Key clinical point: Secukinumab could prove to be a major new therapy for psoriatic arthritis.
Major finding: After 24 weeks, a response of at least ACR20 was achieved by 54% of 100 patients given the 300-mg dose of secukinumab.
Data source: The FUTURE 2 study group of nearly 400 patients with psoriatic arthritis who were randomized either to one of three doses of secukinumab or placebo.
Disclosures: The study was sponsored by Novartis, the maker of secukinumab. Dr. McInnes receives consulting fees from Novartis as well as multiple other pharmaceutical companies.

Few psoriatic arthritis patients achieve minimal disease activity on methotrexate
BOSTON – It’s time to test whether methotrexate is really up to snuff as a first-line therapy for psoriatic arthritis, Canadian investigators say.
They base that recommendation on findings from a retrospective study showing that fewer than 18% of patients with psoriatic arthritis treated with methotrexate achieved minimal disease activity (MDA) after 6 months.

They also found evidence to suggest that physicians may overestimate the benefits of methotrexate for psoriatic arthritis.
“Physician-dependent measures reveal good response to methotrexate at 6 months, but patient-reported measures are less responsive, possibly due to side effects, back pain, disease, [and/or] disability,” said Dr. Barry J. Sheane of the division of rheumatology in the department of medicine at the University of Toronto.
The data on the effects of methotrexate in psoriatic arthritis are considerably less impressive than are those seen with tumor necrosis factor–alpha inhibitors (TNFi), Dr. Sheane said. For example, at 24 weeks, 39% of patients treated with adalimumab (Humira) and 52% treated with infliximab (Remicade) had achieved MDA, he noted.
“We suggest that a randomized controlled trial of methotrexate compared to a TNF inhibitor is warranted,” Dr. Sheane said at the annual meeting of the American College of Rheumatology.
He and his colleagues reviewed records on 204 consecutive patients treated for psoriatic arthritis with methotrexate from January 2004 through April 2014. The patients, who had a mean duration of psoriatic arthritis of 6.2 years, were all naive to biological antirheumatic drugs and were initiating methotrexate therapy.
Of the 204 patients, 167 had data sufficient for a 6-month analysis.
The investigators defined MDA after 6 months on methotrexate, the primary endpoint, as the presence of at least five out of the following seven domains: 0-1 tender joints, 0-1 swollen joints, Psoriasis Area Severity Index (PASI) 1 or less or body surface area involved 3% or less, 0-1 tender entheseal points, Health Assessment Questionnaire (HAQ) score of 0.5 or less, patient global disease activity Visual Analog Scale score of 20 or lower, and patient pain Visual Analog Scale score of 15 or lower.
They found that 17.4% of patients (29 of 167) met the physician-rated criteria for MDA at 6 months, even though 82.6% of the patients (138) had a PASI score of 1 or lower, and 58.1% (97) had a swollen joint count of 1 or fewer.
When the patients were asked for their assessment of global disease activity, 13.2% (22) rated disease activity with a prespecified score of 20 or lower. Only 12% of patients (20) reported an HAQ score of 0.5 or less.
The mean methotrexate dose was similar between patients who achieved MDA (17.8 mg/week) and those who did not (17.3 mg/week). The median dose was 17.5 mg/week in each group.
After controlling for sex, baseline sacroiliitis, and duration of psoriasis and psoriatic arthritis at the start of methotrexate in a multivariate model, the authors found that only dactylitis, inflammatory back pain, and mechanical back pain were significantly associated with a lower probability of patients reaching MDA. The variables that did not prove to be associated with achieving MDA after 6 months on methotrexate included erythrocyte sedimentation rate, presence of nail disease, number of clinically damaged joints, and body mass index.
The study was internally funded. Dr. Sheane reported having no relevant disclosures.
BOSTON – It’s time to test whether methotrexate is really up to snuff as a first-line therapy for psoriatic arthritis, Canadian investigators say.
They base that recommendation on findings from a retrospective study showing that fewer than 18% of patients with psoriatic arthritis treated with methotrexate achieved minimal disease activity (MDA) after 6 months.
They also found evidence to suggest that physicians may overestimate the benefits of methotrexate for psoriatic arthritis.
“Physician-dependent measures reveal good response to methotrexate at 6 months, but patient-reported measures are less responsive, possibly due to side effects, back pain, disease, [and/or] disability,” said Dr. Barry J. Sheane of the division of rheumatology in the department of medicine at the University of Toronto.
The data on the effects of methotrexate in psoriatic arthritis are considerably less impressive than are those seen with tumor necrosis factor–alpha inhibitors (TNFi), Dr. Sheane said. For example, at 24 weeks, 39% of patients treated with adalimumab (Humira) and 52% treated with infliximab (Remicade) had achieved MDA, he noted.
“We suggest that a randomized controlled trial of methotrexate compared to a TNF inhibitor is warranted,” Dr. Sheane said at the annual meeting of the American College of Rheumatology.
He and his colleagues reviewed records on 204 consecutive patients treated for psoriatic arthritis with methotrexate from January 2004 through April 2014. The patients, who had a mean duration of psoriatic arthritis of 6.2 years, were all naive to biological antirheumatic drugs and were initiating methotrexate therapy.
Of the 204 patients, 167 had data sufficient for a 6-month analysis.
The investigators defined MDA after 6 months on methotrexate, the primary endpoint, as the presence of at least five out of the following seven domains: 0-1 tender joints, 0-1 swollen joints, Psoriasis Area Severity Index (PASI) 1 or less or body surface area involved 3% or less, 0-1 tender entheseal points, Health Assessment Questionnaire (HAQ) score of 0.5 or less, patient global disease activity Visual Analog Scale score of 20 or lower, and patient pain Visual Analog Scale score of 15 or lower.
They found that 17.4% of patients (29 of 167) met the physician-rated criteria for MDA at 6 months, even though 82.6% of the patients (138) had a PASI score of 1 or lower, and 58.1% (97) had a swollen joint count of 1 or fewer.
When the patients were asked for their assessment of global disease activity, 13.2% (22) rated disease activity with a prespecified score of 20 or lower. Only 12% of patients (20) reported an HAQ score of 0.5 or less.
The mean methotrexate dose was similar between patients who achieved MDA (17.8 mg/week) and those who did not (17.3 mg/week). The median dose was 17.5 mg/week in each group.
After controlling for sex, baseline sacroiliitis, and duration of psoriasis and psoriatic arthritis at the start of methotrexate in a multivariate model, the authors found that only dactylitis, inflammatory back pain, and mechanical back pain were significantly associated with a lower probability of patients reaching MDA. The variables that did not prove to be associated with achieving MDA after 6 months on methotrexate included erythrocyte sedimentation rate, presence of nail disease, number of clinically damaged joints, and body mass index.
The study was internally funded. Dr. Sheane reported having no relevant disclosures.
BOSTON – It’s time to test whether methotrexate is really up to snuff as a first-line therapy for psoriatic arthritis, Canadian investigators say.
They base that recommendation on findings from a retrospective study showing that fewer than 18% of patients with psoriatic arthritis treated with methotrexate achieved minimal disease activity (MDA) after 6 months.
They also found evidence to suggest that physicians may overestimate the benefits of methotrexate for psoriatic arthritis.
“Physician-dependent measures reveal good response to methotrexate at 6 months, but patient-reported measures are less responsive, possibly due to side effects, back pain, disease, [and/or] disability,” said Dr. Barry J. Sheane of the division of rheumatology in the department of medicine at the University of Toronto.
The data on the effects of methotrexate in psoriatic arthritis are considerably less impressive than are those seen with tumor necrosis factor–alpha inhibitors (TNFi), Dr. Sheane said. For example, at 24 weeks, 39% of patients treated with adalimumab (Humira) and 52% treated with infliximab (Remicade) had achieved MDA, he noted.
“We suggest that a randomized controlled trial of methotrexate compared to a TNF inhibitor is warranted,” Dr. Sheane said at the annual meeting of the American College of Rheumatology.
He and his colleagues reviewed records on 204 consecutive patients treated for psoriatic arthritis with methotrexate from January 2004 through April 2014. The patients, who had a mean duration of psoriatic arthritis of 6.2 years, were all naive to biological antirheumatic drugs and were initiating methotrexate therapy.
Of the 204 patients, 167 had data sufficient for a 6-month analysis.
The investigators defined MDA after 6 months on methotrexate, the primary endpoint, as the presence of at least five out of the following seven domains: 0-1 tender joints, 0-1 swollen joints, Psoriasis Area Severity Index (PASI) 1 or less or body surface area involved 3% or less, 0-1 tender entheseal points, Health Assessment Questionnaire (HAQ) score of 0.5 or less, patient global disease activity Visual Analog Scale score of 20 or lower, and patient pain Visual Analog Scale score of 15 or lower.
They found that 17.4% of patients (29 of 167) met the physician-rated criteria for MDA at 6 months, even though 82.6% of the patients (138) had a PASI score of 1 or lower, and 58.1% (97) had a swollen joint count of 1 or fewer.
When the patients were asked for their assessment of global disease activity, 13.2% (22) rated disease activity with a prespecified score of 20 or lower. Only 12% of patients (20) reported an HAQ score of 0.5 or less.
The mean methotrexate dose was similar between patients who achieved MDA (17.8 mg/week) and those who did not (17.3 mg/week). The median dose was 17.5 mg/week in each group.
After controlling for sex, baseline sacroiliitis, and duration of psoriasis and psoriatic arthritis at the start of methotrexate in a multivariate model, the authors found that only dactylitis, inflammatory back pain, and mechanical back pain were significantly associated with a lower probability of patients reaching MDA. The variables that did not prove to be associated with achieving MDA after 6 months on methotrexate included erythrocyte sedimentation rate, presence of nail disease, number of clinically damaged joints, and body mass index.
The study was internally funded. Dr. Sheane reported having no relevant disclosures.
AT THE ACR ANNUAL MEETING
Key clinical point: It may be time to rethink methotrexate’s role as first-line therapy for psoriatic arthritis.
Major finding: Only 17.4% of patients with psoriatic arthritis treated with methotrexate achieved minimal disease activity at 6 months.
Data source: Retrospective study of 204 patients, 167 of whom had data sufficient for an efficacy and dosing analysis at 6 months.
Disclosures: The study was internally funded. Dr. Sheane reported having no relevant disclosures.
