User login
EADV: Secukinumab shows sustained high efficacy in SCULPTURE through 3 years
COPENHAGEN – Secukinumab maintained consistently high and previously unheard of long-term Psoriasis Area and Severity Index 90 and PASI 100 response rates in psoriasis patients through 3 years of prospective follow-up in an extension of the phase III SCULPTURE trial.
The key to maintaining these high response rates was to give secukinumab (Cosentyx) on a fixed schedule every 4 weeks. SCULPTURE tested the hypothesis that retreatment of initial responders only as needed based upon early evidence of relapse off treatment might be a reasonable alternative strategy. The answer turns out to be emphatically no.

“We can see from the 3-year data that retreatment as needed is probably not a good strategy over the long term. We see four times fewer patients with PASI 90s and 10 times fewer patients with PASI 100s if they are treated as needed. From the patient point of view, quality of life and satisfaction are not optimal with retreatment as needed,” Dr. Diamant Thaci said at the annual congress of the European Academy of Dermatology and Venereology.
The 3-year extension of SCULPTURE also provided reassuring safety data, he noted.
“It’s interesting that with long-term treatment out to 3 years, we don’t see any new safety signals. In fact, the signals we saw in the preapproval studies are slightly diluted here, and now that secukinumab is approved, we’re not seeing them as problems in real life clinical practice,” said Dr. Thaci of University Hospital in Lübeck, Germany.
The 52-week SCULPTURE results were published earlier this year (J Am Acad Dermatol. 2015 Jul;73[1]:27-36). Dr. Thaci’s update extended the findings from 52 weeks through 3 years.
The double-blind phase of the core SCULPTURE study began with 966 patients with moderate to severe plaque psoriasis and a baseline PASI score of about 23. After 12 weeks on subcutaneous secukinumab, the 842 subjects who had achieved 75% or more improvement from their baseline PASI score were randomized to one of four study arms: secukinumab at either 300 mg or 150 mg, administered either every 4 weeks or by retreatment as needed when an individual’s response dropped below PASI 75.
Picking up where the published 52-week results left off, Dr. Thaci focused special attention on the PASI 90 outcomes out to 3 years because he said he believes PASI 90 will become generally accepted as the new treatment target, now that secukinumab is on the market and other supereffective biologic agents are advancing through the developmental pipeline.
At 52 weeks, 68.5% of patients on fixed-dose secukinumab at 300 mg every 4 weeks had a PASI 90 response. At 3 years, after an additional 2 years on this schedule, the PASI 90 rate was still hovering around the two-thirds mark at 63.8%. In contrast, patients on retreatment with 300 mg as needed had a PASI 90 rate of 14.8% at 52 weeks and 13% at 3 years.
Although the Food and Drug Administration recommended the 300-mg dose when it approved secukinumab as a first-in-class biologic in January, the agency added that 150 mg may be acceptable in some patients. But the 150-mg dose was clearly inferior long term in the SCULPTURE extension. At 52 weeks, 50.3% of patients on secukinumab at 150 mg every 4 weeks had a PASI 90 response; at 3 years the rate was 36.8%. With retreatment as needed using 150 mg, the 52-week and 3-year PASI 90 rates were 12% and 11.8%, Dr. Thaci reported.
The PASI 100 (clear skin) response rate at 52 weeks with fixed-dose secukinumab at 300 mg was 43.8%. At 3 years it was 42.6%.
“These excellent responders kept their excellent response by continuing treatment for 3 years,” the dermatologist noted.
The PASI 100 rates with retreatment as needed were 5.3% and 3.8%.
In terms of safety through 3 years of treatment, rates of the adverse events of greatest interest with the use of immunomodulatory therapies – Candida infections, malignancies, serious infections, and major adverse cardiovascular events – were similarly low across all four treatment arms, ranging from 0 to 1.6 events per 100 subject-years. Thus, there was no safety advantage in using the retreatment as needed strategy.
“In the past we were all talking about the risk of Candida infection caused by inhibiting [interleukin]-17. But if you treat patients in real life clinical practice, you may see a signal of Candida infection in the beginning, yet in the long term you don’t see Candida as a significant side effect in our daily practice – and in this trial the incidence decreases,” Dr. Thaci said.
One audience member rose to criticize the way the analysis of the long-term extension of SCULPTURE was structured – namely, that only patients still on secukinumab at the 3-year mark were included in the results. This “as-observed” method inflates the efficacy results, he said. How many patients dropped out over time and weren’t counted? he asked.
Dr. Thaci replied that roughly 90% of patients in the trial at 52 weeks continued on secukinumab out to 3 years.
“It’s not a massive dropout. I wish it were a nonresponder imputation analysis, but in long-term studies, it’s very difficult to show that analysis because the vast majority of the trials that have been presented in the past have been ‘as observed.’ But I don’t think you’ll see a big difference between this as-observed analysis and a nonresponder imputation analysis. This is a very fair and convincing analysis,” he said.
Secukinumab is marketed by Novartis, which sponsored the study. Dr. Thaci serves as an investigator and consultant to Novartis and other pharmaceutical companies.
COPENHAGEN – Secukinumab maintained consistently high and previously unheard of long-term Psoriasis Area and Severity Index 90 and PASI 100 response rates in psoriasis patients through 3 years of prospective follow-up in an extension of the phase III SCULPTURE trial.
The key to maintaining these high response rates was to give secukinumab (Cosentyx) on a fixed schedule every 4 weeks. SCULPTURE tested the hypothesis that retreatment of initial responders only as needed based upon early evidence of relapse off treatment might be a reasonable alternative strategy. The answer turns out to be emphatically no.
“We can see from the 3-year data that retreatment as needed is probably not a good strategy over the long term. We see four times fewer patients with PASI 90s and 10 times fewer patients with PASI 100s if they are treated as needed. From the patient point of view, quality of life and satisfaction are not optimal with retreatment as needed,” Dr. Diamant Thaci said at the annual congress of the European Academy of Dermatology and Venereology.
The 3-year extension of SCULPTURE also provided reassuring safety data, he noted.
“It’s interesting that with long-term treatment out to 3 years, we don’t see any new safety signals. In fact, the signals we saw in the preapproval studies are slightly diluted here, and now that secukinumab is approved, we’re not seeing them as problems in real life clinical practice,” said Dr. Thaci of University Hospital in Lübeck, Germany.
The 52-week SCULPTURE results were published earlier this year (J Am Acad Dermatol. 2015 Jul;73[1]:27-36). Dr. Thaci’s update extended the findings from 52 weeks through 3 years.
The double-blind phase of the core SCULPTURE study began with 966 patients with moderate to severe plaque psoriasis and a baseline PASI score of about 23. After 12 weeks on subcutaneous secukinumab, the 842 subjects who had achieved 75% or more improvement from their baseline PASI score were randomized to one of four study arms: secukinumab at either 300 mg or 150 mg, administered either every 4 weeks or by retreatment as needed when an individual’s response dropped below PASI 75.
Picking up where the published 52-week results left off, Dr. Thaci focused special attention on the PASI 90 outcomes out to 3 years because he said he believes PASI 90 will become generally accepted as the new treatment target, now that secukinumab is on the market and other supereffective biologic agents are advancing through the developmental pipeline.
At 52 weeks, 68.5% of patients on fixed-dose secukinumab at 300 mg every 4 weeks had a PASI 90 response. At 3 years, after an additional 2 years on this schedule, the PASI 90 rate was still hovering around the two-thirds mark at 63.8%. In contrast, patients on retreatment with 300 mg as needed had a PASI 90 rate of 14.8% at 52 weeks and 13% at 3 years.
Although the Food and Drug Administration recommended the 300-mg dose when it approved secukinumab as a first-in-class biologic in January, the agency added that 150 mg may be acceptable in some patients. But the 150-mg dose was clearly inferior long term in the SCULPTURE extension. At 52 weeks, 50.3% of patients on secukinumab at 150 mg every 4 weeks had a PASI 90 response; at 3 years the rate was 36.8%. With retreatment as needed using 150 mg, the 52-week and 3-year PASI 90 rates were 12% and 11.8%, Dr. Thaci reported.
The PASI 100 (clear skin) response rate at 52 weeks with fixed-dose secukinumab at 300 mg was 43.8%. At 3 years it was 42.6%.
“These excellent responders kept their excellent response by continuing treatment for 3 years,” the dermatologist noted.
The PASI 100 rates with retreatment as needed were 5.3% and 3.8%.
In terms of safety through 3 years of treatment, rates of the adverse events of greatest interest with the use of immunomodulatory therapies – Candida infections, malignancies, serious infections, and major adverse cardiovascular events – were similarly low across all four treatment arms, ranging from 0 to 1.6 events per 100 subject-years. Thus, there was no safety advantage in using the retreatment as needed strategy.
“In the past we were all talking about the risk of Candida infection caused by inhibiting [interleukin]-17. But if you treat patients in real life clinical practice, you may see a signal of Candida infection in the beginning, yet in the long term you don’t see Candida as a significant side effect in our daily practice – and in this trial the incidence decreases,” Dr. Thaci said.
One audience member rose to criticize the way the analysis of the long-term extension of SCULPTURE was structured – namely, that only patients still on secukinumab at the 3-year mark were included in the results. This “as-observed” method inflates the efficacy results, he said. How many patients dropped out over time and weren’t counted? he asked.
Dr. Thaci replied that roughly 90% of patients in the trial at 52 weeks continued on secukinumab out to 3 years.
“It’s not a massive dropout. I wish it were a nonresponder imputation analysis, but in long-term studies, it’s very difficult to show that analysis because the vast majority of the trials that have been presented in the past have been ‘as observed.’ But I don’t think you’ll see a big difference between this as-observed analysis and a nonresponder imputation analysis. This is a very fair and convincing analysis,” he said.
Secukinumab is marketed by Novartis, which sponsored the study. Dr. Thaci serves as an investigator and consultant to Novartis and other pharmaceutical companies.
COPENHAGEN – Secukinumab maintained consistently high and previously unheard of long-term Psoriasis Area and Severity Index 90 and PASI 100 response rates in psoriasis patients through 3 years of prospective follow-up in an extension of the phase III SCULPTURE trial.
The key to maintaining these high response rates was to give secukinumab (Cosentyx) on a fixed schedule every 4 weeks. SCULPTURE tested the hypothesis that retreatment of initial responders only as needed based upon early evidence of relapse off treatment might be a reasonable alternative strategy. The answer turns out to be emphatically no.
“We can see from the 3-year data that retreatment as needed is probably not a good strategy over the long term. We see four times fewer patients with PASI 90s and 10 times fewer patients with PASI 100s if they are treated as needed. From the patient point of view, quality of life and satisfaction are not optimal with retreatment as needed,” Dr. Diamant Thaci said at the annual congress of the European Academy of Dermatology and Venereology.
The 3-year extension of SCULPTURE also provided reassuring safety data, he noted.
“It’s interesting that with long-term treatment out to 3 years, we don’t see any new safety signals. In fact, the signals we saw in the preapproval studies are slightly diluted here, and now that secukinumab is approved, we’re not seeing them as problems in real life clinical practice,” said Dr. Thaci of University Hospital in Lübeck, Germany.
The 52-week SCULPTURE results were published earlier this year (J Am Acad Dermatol. 2015 Jul;73[1]:27-36). Dr. Thaci’s update extended the findings from 52 weeks through 3 years.
The double-blind phase of the core SCULPTURE study began with 966 patients with moderate to severe plaque psoriasis and a baseline PASI score of about 23. After 12 weeks on subcutaneous secukinumab, the 842 subjects who had achieved 75% or more improvement from their baseline PASI score were randomized to one of four study arms: secukinumab at either 300 mg or 150 mg, administered either every 4 weeks or by retreatment as needed when an individual’s response dropped below PASI 75.
Picking up where the published 52-week results left off, Dr. Thaci focused special attention on the PASI 90 outcomes out to 3 years because he said he believes PASI 90 will become generally accepted as the new treatment target, now that secukinumab is on the market and other supereffective biologic agents are advancing through the developmental pipeline.
At 52 weeks, 68.5% of patients on fixed-dose secukinumab at 300 mg every 4 weeks had a PASI 90 response. At 3 years, after an additional 2 years on this schedule, the PASI 90 rate was still hovering around the two-thirds mark at 63.8%. In contrast, patients on retreatment with 300 mg as needed had a PASI 90 rate of 14.8% at 52 weeks and 13% at 3 years.
Although the Food and Drug Administration recommended the 300-mg dose when it approved secukinumab as a first-in-class biologic in January, the agency added that 150 mg may be acceptable in some patients. But the 150-mg dose was clearly inferior long term in the SCULPTURE extension. At 52 weeks, 50.3% of patients on secukinumab at 150 mg every 4 weeks had a PASI 90 response; at 3 years the rate was 36.8%. With retreatment as needed using 150 mg, the 52-week and 3-year PASI 90 rates were 12% and 11.8%, Dr. Thaci reported.
The PASI 100 (clear skin) response rate at 52 weeks with fixed-dose secukinumab at 300 mg was 43.8%. At 3 years it was 42.6%.
“These excellent responders kept their excellent response by continuing treatment for 3 years,” the dermatologist noted.
The PASI 100 rates with retreatment as needed were 5.3% and 3.8%.
In terms of safety through 3 years of treatment, rates of the adverse events of greatest interest with the use of immunomodulatory therapies – Candida infections, malignancies, serious infections, and major adverse cardiovascular events – were similarly low across all four treatment arms, ranging from 0 to 1.6 events per 100 subject-years. Thus, there was no safety advantage in using the retreatment as needed strategy.
“In the past we were all talking about the risk of Candida infection caused by inhibiting [interleukin]-17. But if you treat patients in real life clinical practice, you may see a signal of Candida infection in the beginning, yet in the long term you don’t see Candida as a significant side effect in our daily practice – and in this trial the incidence decreases,” Dr. Thaci said.
One audience member rose to criticize the way the analysis of the long-term extension of SCULPTURE was structured – namely, that only patients still on secukinumab at the 3-year mark were included in the results. This “as-observed” method inflates the efficacy results, he said. How many patients dropped out over time and weren’t counted? he asked.
Dr. Thaci replied that roughly 90% of patients in the trial at 52 weeks continued on secukinumab out to 3 years.
“It’s not a massive dropout. I wish it were a nonresponder imputation analysis, but in long-term studies, it’s very difficult to show that analysis because the vast majority of the trials that have been presented in the past have been ‘as observed.’ But I don’t think you’ll see a big difference between this as-observed analysis and a nonresponder imputation analysis. This is a very fair and convincing analysis,” he said.
Secukinumab is marketed by Novartis, which sponsored the study. Dr. Thaci serves as an investigator and consultant to Novartis and other pharmaceutical companies.
AT THE EADV CONGRESS
Key clinical point: Three-year data from a phase III trial provide reassurance regarding the long-term safety and efficacy of secukinumab for psoriasis.
Major finding: The high level of improvement seen in the 52-week core study of secukinumab, at the dose of 300 mg every 4 weeks, in patients with psoriasis was largely maintained with 2 more years of therapy, with a 63.8% PASI-90 rate at 3 years.
Data source: A prospective 2-year extension of a 1-year, randomized double-blind trial of 842 psoriasis patients, who initially achieved at least a PASI-75 response to secukinumab.
Disclosures: The study was sponsored by Novartis. The presenter serves as a paid investigator and consultant for Novartis and other pharmaceutical companies.

EADV: Spotlight on alexithymia in psoriasis
COPENHAGEN – Alexithymia – difficulty in recognizing and describing one’s emotions – is exceptionally common among psoriasis patients and may represent a novel therapeutic target, according to Dr. Carle Paul.
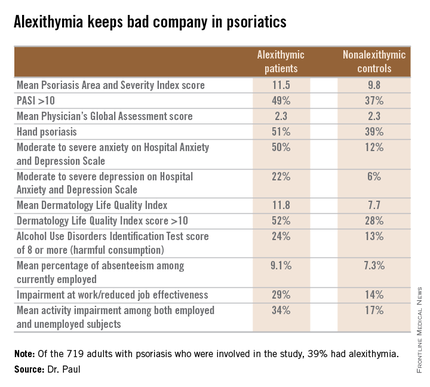
“We found a significant association between alexithymia and more severe psoriasis, anxiety, depression, decreased quality of life, harmful alcohol consumption, and work impairment,” reported Dr. Paul, professor and chairman of the department of dermatology at the University of Toulouse (France).

Alexithymia, a personality construct sometimes referred to as “emotional blindness,” was first described by psychologists in the 1970s. The previous glaring lack of data on the prevalence and consequences of alexithymia in psoriasis patients served as the impetus for the ongoing EPIDEPSO study (Epidemiological Study in Patients With Recently Diagnosed Psoriasis), a prospective 1-year international, epidemiologic, noninterventional observational study involving 719 adults with moderate to severe plaque psoriasis of less than 10 years’ duration. Dr. Paul presented the baseline findings at the at the annual congress of the European Academy of Dermatology and Venereology.
The first noteworthy finding was the strikingly high prevalence of alexithymia in this group of psoriasis patients: 39% of the 719 patients had alexithymia, as defined by a score of 61 or more on the validated, 20-item Toronto Alexithymia Scale.
Patients with alexithymia had slightly but significantly more severe psoriasis as evidenced by their mean Psoriasis Area and Severity Index score of 11.5, compared with 9.8 in unaffected patients. Hand psoriasis was more common in alexithymic patients by a margin of 51%-39%, although the prevalence of psoriasis of the face and neck was similar in the two groups.
Alexithymia was associated with significantly higher rates of several forms of psychiatric comorbidity and problems in living as assessed by validated tests. One comorbid condition stood out above the rest.
“The most striking feature is the very close relationship between alexithymia and anxiety,” according to Dr. Paul. “Among alexithymic patients, 50% had moderate to severe anxiety, as measured by the Hospital Anxiety and Depression Scale–A, whereas in psoriasis patients without alexithymia, this proportion was 12%.”
“Alexithymia identifies a patient population with a high burden of psoriasis, but at the same time, they have difficulty in expressing their emotions and feelings to their doctor. And I think this may explain the fact that some psoriasis patients have difficulties in interacting with doctors, because even though they have a high psoriasis burden, they cannot express how much they suffer from the disease,” the dermatologist continued.
Audience members at this standing-room-only EADV session on new research findings in psoriasis were clearly intrigued by the novel findings about a psychological condition unfamiliar to most. They wanted to know if the Toronto Alexithymia Scale is suitable for use in everyday clinical practice. The answer is yes, Dr. Paul replied, but the practical importance of identifying the large subgroup of patients with alexithymia has yet to be determined.
“We want to find out if we can modify alexithymia with interventions, but we don’t have the prospective data yet,” he added.
Even if alexithymia turns out to be a fixed characteristic not amenable to intervention, however, EPIDEPSO has shown that it could be useful as a red flag because it keeps company with several psychiatric conditions, which are anxiety, depression, and alcohol abuse.
What about causality? others asked. Does alexithymia cause anxiety and depression, or does the emotional toll of psoriasis promote anxiety and depression, which then causes alexithymia? Dr. Paul responded that it’s impossible to say at this point because these initial EPIDEPSO data are cross-sectional; however, the 1-year follow-up may yield insight.
EPIDEPSO is funded by Janssen. Dr. Paul reported receiving a research grant from the company to conduct the study.
COPENHAGEN – Alexithymia – difficulty in recognizing and describing one’s emotions – is exceptionally common among psoriasis patients and may represent a novel therapeutic target, according to Dr. Carle Paul.
“We found a significant association between alexithymia and more severe psoriasis, anxiety, depression, decreased quality of life, harmful alcohol consumption, and work impairment,” reported Dr. Paul, professor and chairman of the department of dermatology at the University of Toulouse (France).
Alexithymia, a personality construct sometimes referred to as “emotional blindness,” was first described by psychologists in the 1970s. The previous glaring lack of data on the prevalence and consequences of alexithymia in psoriasis patients served as the impetus for the ongoing EPIDEPSO study (Epidemiological Study in Patients With Recently Diagnosed Psoriasis), a prospective 1-year international, epidemiologic, noninterventional observational study involving 719 adults with moderate to severe plaque psoriasis of less than 10 years’ duration. Dr. Paul presented the baseline findings at the at the annual congress of the European Academy of Dermatology and Venereology.
The first noteworthy finding was the strikingly high prevalence of alexithymia in this group of psoriasis patients: 39% of the 719 patients had alexithymia, as defined by a score of 61 or more on the validated, 20-item Toronto Alexithymia Scale.
Patients with alexithymia had slightly but significantly more severe psoriasis as evidenced by their mean Psoriasis Area and Severity Index score of 11.5, compared with 9.8 in unaffected patients. Hand psoriasis was more common in alexithymic patients by a margin of 51%-39%, although the prevalence of psoriasis of the face and neck was similar in the two groups.
Alexithymia was associated with significantly higher rates of several forms of psychiatric comorbidity and problems in living as assessed by validated tests. One comorbid condition stood out above the rest.
“The most striking feature is the very close relationship between alexithymia and anxiety,” according to Dr. Paul. “Among alexithymic patients, 50% had moderate to severe anxiety, as measured by the Hospital Anxiety and Depression Scale–A, whereas in psoriasis patients without alexithymia, this proportion was 12%.”
“Alexithymia identifies a patient population with a high burden of psoriasis, but at the same time, they have difficulty in expressing their emotions and feelings to their doctor. And I think this may explain the fact that some psoriasis patients have difficulties in interacting with doctors, because even though they have a high psoriasis burden, they cannot express how much they suffer from the disease,” the dermatologist continued.
Audience members at this standing-room-only EADV session on new research findings in psoriasis were clearly intrigued by the novel findings about a psychological condition unfamiliar to most. They wanted to know if the Toronto Alexithymia Scale is suitable for use in everyday clinical practice. The answer is yes, Dr. Paul replied, but the practical importance of identifying the large subgroup of patients with alexithymia has yet to be determined.
“We want to find out if we can modify alexithymia with interventions, but we don’t have the prospective data yet,” he added.
Even if alexithymia turns out to be a fixed characteristic not amenable to intervention, however, EPIDEPSO has shown that it could be useful as a red flag because it keeps company with several psychiatric conditions, which are anxiety, depression, and alcohol abuse.
What about causality? others asked. Does alexithymia cause anxiety and depression, or does the emotional toll of psoriasis promote anxiety and depression, which then causes alexithymia? Dr. Paul responded that it’s impossible to say at this point because these initial EPIDEPSO data are cross-sectional; however, the 1-year follow-up may yield insight.
EPIDEPSO is funded by Janssen. Dr. Paul reported receiving a research grant from the company to conduct the study.
COPENHAGEN – Alexithymia – difficulty in recognizing and describing one’s emotions – is exceptionally common among psoriasis patients and may represent a novel therapeutic target, according to Dr. Carle Paul.
“We found a significant association between alexithymia and more severe psoriasis, anxiety, depression, decreased quality of life, harmful alcohol consumption, and work impairment,” reported Dr. Paul, professor and chairman of the department of dermatology at the University of Toulouse (France).
Alexithymia, a personality construct sometimes referred to as “emotional blindness,” was first described by psychologists in the 1970s. The previous glaring lack of data on the prevalence and consequences of alexithymia in psoriasis patients served as the impetus for the ongoing EPIDEPSO study (Epidemiological Study in Patients With Recently Diagnosed Psoriasis), a prospective 1-year international, epidemiologic, noninterventional observational study involving 719 adults with moderate to severe plaque psoriasis of less than 10 years’ duration. Dr. Paul presented the baseline findings at the at the annual congress of the European Academy of Dermatology and Venereology.
The first noteworthy finding was the strikingly high prevalence of alexithymia in this group of psoriasis patients: 39% of the 719 patients had alexithymia, as defined by a score of 61 or more on the validated, 20-item Toronto Alexithymia Scale.
Patients with alexithymia had slightly but significantly more severe psoriasis as evidenced by their mean Psoriasis Area and Severity Index score of 11.5, compared with 9.8 in unaffected patients. Hand psoriasis was more common in alexithymic patients by a margin of 51%-39%, although the prevalence of psoriasis of the face and neck was similar in the two groups.
Alexithymia was associated with significantly higher rates of several forms of psychiatric comorbidity and problems in living as assessed by validated tests. One comorbid condition stood out above the rest.
“The most striking feature is the very close relationship between alexithymia and anxiety,” according to Dr. Paul. “Among alexithymic patients, 50% had moderate to severe anxiety, as measured by the Hospital Anxiety and Depression Scale–A, whereas in psoriasis patients without alexithymia, this proportion was 12%.”
“Alexithymia identifies a patient population with a high burden of psoriasis, but at the same time, they have difficulty in expressing their emotions and feelings to their doctor. And I think this may explain the fact that some psoriasis patients have difficulties in interacting with doctors, because even though they have a high psoriasis burden, they cannot express how much they suffer from the disease,” the dermatologist continued.
Audience members at this standing-room-only EADV session on new research findings in psoriasis were clearly intrigued by the novel findings about a psychological condition unfamiliar to most. They wanted to know if the Toronto Alexithymia Scale is suitable for use in everyday clinical practice. The answer is yes, Dr. Paul replied, but the practical importance of identifying the large subgroup of patients with alexithymia has yet to be determined.
“We want to find out if we can modify alexithymia with interventions, but we don’t have the prospective data yet,” he added.
Even if alexithymia turns out to be a fixed characteristic not amenable to intervention, however, EPIDEPSO has shown that it could be useful as a red flag because it keeps company with several psychiatric conditions, which are anxiety, depression, and alcohol abuse.
What about causality? others asked. Does alexithymia cause anxiety and depression, or does the emotional toll of psoriasis promote anxiety and depression, which then causes alexithymia? Dr. Paul responded that it’s impossible to say at this point because these initial EPIDEPSO data are cross-sectional; however, the 1-year follow-up may yield insight.
EPIDEPSO is funded by Janssen. Dr. Paul reported receiving a research grant from the company to conduct the study.
AT THE EADV CONGRESS
Key clinical point: Alexithymia is strikingly common among psoriasis patients and is associated with multiple psychiatric comorbidities and problems in living.
Major finding: Thirty-nine percent of a large cohort of psoriasis patients met the criteria for alexithymia, an inability to identify and describe one’s emotions. Affected patients had markedly higher rates of anxiety, depression, problem drinking, and impairments of quality of life and work productivity.
Data source: The EPIDEPSO study, an ongoing 1-year prospective, observational international study involving 719 adults with moderate to severe psoriasis of less than 10 years’ duration.
Disclosures: The EPIDEPSO study is funded by Janssen, which provided the presenter with a research grant.

EADV: Best treatments for great saphenous vein reflux
COPENHAGEN – Superior 5-year outcomes for great saphenous vein reflux were achieved with conventional surgery and endovenous laser ablation as compared with ultrasound-guided foam sclerotherapy in a randomized trial, Dr. Simone van der Velden reported at the annual congress of the European Academy of Dermatology and Venereology.
The multicenter study included 224 randomized legs belonging to symptomatic patients with a target great saphenous vein diameter of at least 5 mm. If deemed necessary, patients could undergo one re-treatment at 3 or 12 months after their initial procedure. At 5 years of follow-up, 86% of the treated legs were available for long-term evaluation, noted Dr. van der Velden of Erasmus University Medical Center in Rotterdam, the Netherlands.

The primary endpoint was obliteration or absence of the treated great saphenous vein segment. This was achieved with conventional surgery in 85% of treated cases, in 77% of legs treated with endovenous laser ablation (EVLA), and in 23% with ultrasound-guided foam sclerotherapy (UGFS).
Absence of above-the-knee greater saphenous vein reflux – a secondary endpoint – was achieved in 85% of the conventional surgery group and in 82% of the EVLA group, both of which were significantly better results than the 41% response with UGFS.
Another secondary endpoint was grade II neovascularization. Here again, both conventional surgery and EVLA outperformed UGFS, with rates of 17%, 13%, and 4%, respectively. In contrast, there was no significant difference between the three treatment groups in terms of the presence of refluxing tributaries above or below knee level, she continued.
Scores on the disease-specific Chronic Venous Insufficiency quality of life Questionnaire (CIVIQ) deteriorated over time in the UGFS group, improved in the EVLA-treated patients, and remained stable in the conventional surgery group.
Conventional surgery was performed under general anesthesia and entailed high ligation of the saphenofemoral junction and phlebectomy of tributaries. In contrast, EVLA was done under local tumescent anesthesia using a 940-nm laser. The laser fiber was introduced at knee level, positioned 1-2 cm below the saphenofemoral junction, and delivered an energy of roughly 60 Joules/cm2.
For UGFS, operators utilized a foam comprising 1 mL of sodium tetradecyl sulfate per 3 mL of air. A maximum of 10 mL of foam could be injected per treatment session, depending upon the diameter of the great saphenous vein and length of the refluxing trunk. Phlebectomies in this group were performed only in the event of patient complaints.
Of note, patients in the minimally invasive UGFS group required re-treatment three times more often than did those in the other two study arms.
Dr. van der Velden said she has heard from some UGFS partisans that she and her coinvestigators may have undertreated patients in that study arm because they didn’t routinely perform phlebectomies of the tributaries, and the average amount of foam they injected, about 4.5 mL, was on the low side.
The study was sponsored by Erasmus University. Dr. van der Velden reported having no financial conflicts of interest.
COPENHAGEN – Superior 5-year outcomes for great saphenous vein reflux were achieved with conventional surgery and endovenous laser ablation as compared with ultrasound-guided foam sclerotherapy in a randomized trial, Dr. Simone van der Velden reported at the annual congress of the European Academy of Dermatology and Venereology.
The multicenter study included 224 randomized legs belonging to symptomatic patients with a target great saphenous vein diameter of at least 5 mm. If deemed necessary, patients could undergo one re-treatment at 3 or 12 months after their initial procedure. At 5 years of follow-up, 86% of the treated legs were available for long-term evaluation, noted Dr. van der Velden of Erasmus University Medical Center in Rotterdam, the Netherlands.
The primary endpoint was obliteration or absence of the treated great saphenous vein segment. This was achieved with conventional surgery in 85% of treated cases, in 77% of legs treated with endovenous laser ablation (EVLA), and in 23% with ultrasound-guided foam sclerotherapy (UGFS).
Absence of above-the-knee greater saphenous vein reflux – a secondary endpoint – was achieved in 85% of the conventional surgery group and in 82% of the EVLA group, both of which were significantly better results than the 41% response with UGFS.
Another secondary endpoint was grade II neovascularization. Here again, both conventional surgery and EVLA outperformed UGFS, with rates of 17%, 13%, and 4%, respectively. In contrast, there was no significant difference between the three treatment groups in terms of the presence of refluxing tributaries above or below knee level, she continued.
Scores on the disease-specific Chronic Venous Insufficiency quality of life Questionnaire (CIVIQ) deteriorated over time in the UGFS group, improved in the EVLA-treated patients, and remained stable in the conventional surgery group.
Conventional surgery was performed under general anesthesia and entailed high ligation of the saphenofemoral junction and phlebectomy of tributaries. In contrast, EVLA was done under local tumescent anesthesia using a 940-nm laser. The laser fiber was introduced at knee level, positioned 1-2 cm below the saphenofemoral junction, and delivered an energy of roughly 60 Joules/cm2.
For UGFS, operators utilized a foam comprising 1 mL of sodium tetradecyl sulfate per 3 mL of air. A maximum of 10 mL of foam could be injected per treatment session, depending upon the diameter of the great saphenous vein and length of the refluxing trunk. Phlebectomies in this group were performed only in the event of patient complaints.
Of note, patients in the minimally invasive UGFS group required re-treatment three times more often than did those in the other two study arms.
Dr. van der Velden said she has heard from some UGFS partisans that she and her coinvestigators may have undertreated patients in that study arm because they didn’t routinely perform phlebectomies of the tributaries, and the average amount of foam they injected, about 4.5 mL, was on the low side.
The study was sponsored by Erasmus University. Dr. van der Velden reported having no financial conflicts of interest.
COPENHAGEN – Superior 5-year outcomes for great saphenous vein reflux were achieved with conventional surgery and endovenous laser ablation as compared with ultrasound-guided foam sclerotherapy in a randomized trial, Dr. Simone van der Velden reported at the annual congress of the European Academy of Dermatology and Venereology.
The multicenter study included 224 randomized legs belonging to symptomatic patients with a target great saphenous vein diameter of at least 5 mm. If deemed necessary, patients could undergo one re-treatment at 3 or 12 months after their initial procedure. At 5 years of follow-up, 86% of the treated legs were available for long-term evaluation, noted Dr. van der Velden of Erasmus University Medical Center in Rotterdam, the Netherlands.
The primary endpoint was obliteration or absence of the treated great saphenous vein segment. This was achieved with conventional surgery in 85% of treated cases, in 77% of legs treated with endovenous laser ablation (EVLA), and in 23% with ultrasound-guided foam sclerotherapy (UGFS).
Absence of above-the-knee greater saphenous vein reflux – a secondary endpoint – was achieved in 85% of the conventional surgery group and in 82% of the EVLA group, both of which were significantly better results than the 41% response with UGFS.
Another secondary endpoint was grade II neovascularization. Here again, both conventional surgery and EVLA outperformed UGFS, with rates of 17%, 13%, and 4%, respectively. In contrast, there was no significant difference between the three treatment groups in terms of the presence of refluxing tributaries above or below knee level, she continued.
Scores on the disease-specific Chronic Venous Insufficiency quality of life Questionnaire (CIVIQ) deteriorated over time in the UGFS group, improved in the EVLA-treated patients, and remained stable in the conventional surgery group.
Conventional surgery was performed under general anesthesia and entailed high ligation of the saphenofemoral junction and phlebectomy of tributaries. In contrast, EVLA was done under local tumescent anesthesia using a 940-nm laser. The laser fiber was introduced at knee level, positioned 1-2 cm below the saphenofemoral junction, and delivered an energy of roughly 60 Joules/cm2.
For UGFS, operators utilized a foam comprising 1 mL of sodium tetradecyl sulfate per 3 mL of air. A maximum of 10 mL of foam could be injected per treatment session, depending upon the diameter of the great saphenous vein and length of the refluxing trunk. Phlebectomies in this group were performed only in the event of patient complaints.
Of note, patients in the minimally invasive UGFS group required re-treatment three times more often than did those in the other two study arms.
Dr. van der Velden said she has heard from some UGFS partisans that she and her coinvestigators may have undertreated patients in that study arm because they didn’t routinely perform phlebectomies of the tributaries, and the average amount of foam they injected, about 4.5 mL, was on the low side.
The study was sponsored by Erasmus University. Dr. van der Velden reported having no financial conflicts of interest.
AT THE EADV CONGRESS
Key clinical point: Long-term outcomes for treatment of great saphenous vein reflux were significantly better with conventional surgery or endovenous laser ablation than with ultrasound-guided foam sclerotherapy.
Major finding: Obliteration or absence of the treated great saphenous vein segment was achieved with conventional surgery in 85% of treated cases, with endovenous laser ablation (EVLA) in 77% of legs treated, and with ultrasound-guided foam sclerotherapy (UGFS) in 23%.
Data source: This multicenter clinical trial with 5-year follow-up included 224 legs randomized to one of three popular treatments for great saphenous varicose veins.
Disclosures: The study was sponsored by Erasmus University. The presenter reported having no financial conflicts of interest.

EADV: Another promising topical for atopic dermatitis
COPENHAGEN – A novel topical nonsteroidal inhibitor of phosphodiesterase-4 showed a favorable efficacy to side effect ratio in a phase II study of adolescents and adults with mild to moderate atopic dermatitis, Dr. Jon M. Hanifin reported at the annual congress of the European Academy of Dermatology and Venereology.
“For topical agents, I think a 30% improvement with few side effects is a desirable balance,” observed Dr. Hanifin of Oregon Health and Sciences University, Portland.
The investigational agent, known for now as OPA-15406, is formulated as a twice-daily ointment. It is highly selective for the phosphodiesterase-4 (PDE-4) B subtype.

Dr. Hanifin noted that these are “exciting times” in the development of new topical therapies for atopic dermatitis (AD). In addition to the successful phase II study of OPA-15406 he presented, a highlight of the EADV congress was the strongly positive pivotal phase III data presented for crisaborole, another nonsteroidal topical PDE-4 inhibitor, albeit with a different mechanism of action.
For Dr. Hanifin these developments are particularly satisfying personally because 33 years ago as a young investigator – before the term ‘translational science’ had come into vogue – he and his research team made the seminal observation that increased phosphodiesterase activity is “a basic biochemical characteristic relevant to skin immunocellular regulation in atopic disease” (J Allergy Clin Immunol. 1982 Dec;70[6]:452-7).
The 8-week, multicenter, randomized, double-blind phase II OPA-15406 dose-ranging study included 121 patients, 70% with moderate AD, the rest with mild disease. About 20% were adolescents, the rest adults. Their mean baseline Eczema Area Severity Index (EASI) score was 9.5, with a self-reported pruritus score of 61 on a 0-100 scale. Participants were randomized to twice-daily application of OPA-15406 at 0.3% or 1% or the vehicle ointment as a control.
A significant treatment effect was seen within the first week. At 1 week, the mean EASI score was reduced by 31% from baseline in the 1% OPA-15406 group, 15% with the 0.3% formulation, and 6% with vehicle. The active treatment remained significantly more effective than vehicle throughout the 8-week period.
Another measure of efficacy – an Investigator’s Global Assessment (IGA) score of 0 (clear) or 1 along with at least a 2-point improvement on the 0-5 scale at week 4 – was met by 21% of patients on 1% OPA-15406, 15% on the 0.3% formulation, and 2.7% on vehicle. By the less stringent standard of an IGA of 0 or 1 plus at least a 1-point reduction at week 4, the success rates were 30%, 24%, and 10%.
Dr. Hanifin said the 1% formulation is the one likely to advance to phase III studies, given its superior efficacy. This was particularly evident with regard to itch. At the first evaluation, after just 1 week of treatment, pruritus scores showed a mean 30% reduction with 1% OPA-15406 versus no significant change in controls. He added that his clinical impression is that many patients experience a significant improvement in itching within the first 24 hours on the 1% formulation, an observation that deserves formal study.
Scores on the Dermatology Life Quality Index and Children’s Dermatology Life Quality Index were significantly better in the active therapy arms than with vehicle as early as week 1.
The adverse events findings were “not very exciting,” according to the dermatologist. No treatment-related serious adverse events occurred. The two most common adverse events leading to treatment discontinuation – worsening AD and pruritus – occurred more often in the vehicle-treated controls.
Session cochair Dr. Jacek Szepietowski called the pruritus results particularly impressive.
“It’s very difficult to do successful clinical trials of topicals for atopic dermatitis because the vehicle alone, especially if it’s an ointment, can have favorable effects which increase over time both on EASI and pruritus,” commented Dr. Szepietowski, professor and head of the department of dermatology, venereology, and allergology at the Medical University of Wroclaw (Poland).
The study was sponsored by Otsuka Pharmaceuticals. Dr. Hanifin reported serving as a consultant to and paid investigator for the company.
COPENHAGEN – A novel topical nonsteroidal inhibitor of phosphodiesterase-4 showed a favorable efficacy to side effect ratio in a phase II study of adolescents and adults with mild to moderate atopic dermatitis, Dr. Jon M. Hanifin reported at the annual congress of the European Academy of Dermatology and Venereology.
“For topical agents, I think a 30% improvement with few side effects is a desirable balance,” observed Dr. Hanifin of Oregon Health and Sciences University, Portland.
The investigational agent, known for now as OPA-15406, is formulated as a twice-daily ointment. It is highly selective for the phosphodiesterase-4 (PDE-4) B subtype.
Dr. Hanifin noted that these are “exciting times” in the development of new topical therapies for atopic dermatitis (AD). In addition to the successful phase II study of OPA-15406 he presented, a highlight of the EADV congress was the strongly positive pivotal phase III data presented for crisaborole, another nonsteroidal topical PDE-4 inhibitor, albeit with a different mechanism of action.
For Dr. Hanifin these developments are particularly satisfying personally because 33 years ago as a young investigator – before the term ‘translational science’ had come into vogue – he and his research team made the seminal observation that increased phosphodiesterase activity is “a basic biochemical characteristic relevant to skin immunocellular regulation in atopic disease” (J Allergy Clin Immunol. 1982 Dec;70[6]:452-7).
The 8-week, multicenter, randomized, double-blind phase II OPA-15406 dose-ranging study included 121 patients, 70% with moderate AD, the rest with mild disease. About 20% were adolescents, the rest adults. Their mean baseline Eczema Area Severity Index (EASI) score was 9.5, with a self-reported pruritus score of 61 on a 0-100 scale. Participants were randomized to twice-daily application of OPA-15406 at 0.3% or 1% or the vehicle ointment as a control.
A significant treatment effect was seen within the first week. At 1 week, the mean EASI score was reduced by 31% from baseline in the 1% OPA-15406 group, 15% with the 0.3% formulation, and 6% with vehicle. The active treatment remained significantly more effective than vehicle throughout the 8-week period.
Another measure of efficacy – an Investigator’s Global Assessment (IGA) score of 0 (clear) or 1 along with at least a 2-point improvement on the 0-5 scale at week 4 – was met by 21% of patients on 1% OPA-15406, 15% on the 0.3% formulation, and 2.7% on vehicle. By the less stringent standard of an IGA of 0 or 1 plus at least a 1-point reduction at week 4, the success rates were 30%, 24%, and 10%.
Dr. Hanifin said the 1% formulation is the one likely to advance to phase III studies, given its superior efficacy. This was particularly evident with regard to itch. At the first evaluation, after just 1 week of treatment, pruritus scores showed a mean 30% reduction with 1% OPA-15406 versus no significant change in controls. He added that his clinical impression is that many patients experience a significant improvement in itching within the first 24 hours on the 1% formulation, an observation that deserves formal study.
Scores on the Dermatology Life Quality Index and Children’s Dermatology Life Quality Index were significantly better in the active therapy arms than with vehicle as early as week 1.
The adverse events findings were “not very exciting,” according to the dermatologist. No treatment-related serious adverse events occurred. The two most common adverse events leading to treatment discontinuation – worsening AD and pruritus – occurred more often in the vehicle-treated controls.
Session cochair Dr. Jacek Szepietowski called the pruritus results particularly impressive.
“It’s very difficult to do successful clinical trials of topicals for atopic dermatitis because the vehicle alone, especially if it’s an ointment, can have favorable effects which increase over time both on EASI and pruritus,” commented Dr. Szepietowski, professor and head of the department of dermatology, venereology, and allergology at the Medical University of Wroclaw (Poland).
The study was sponsored by Otsuka Pharmaceuticals. Dr. Hanifin reported serving as a consultant to and paid investigator for the company.
COPENHAGEN – A novel topical nonsteroidal inhibitor of phosphodiesterase-4 showed a favorable efficacy to side effect ratio in a phase II study of adolescents and adults with mild to moderate atopic dermatitis, Dr. Jon M. Hanifin reported at the annual congress of the European Academy of Dermatology and Venereology.
“For topical agents, I think a 30% improvement with few side effects is a desirable balance,” observed Dr. Hanifin of Oregon Health and Sciences University, Portland.
The investigational agent, known for now as OPA-15406, is formulated as a twice-daily ointment. It is highly selective for the phosphodiesterase-4 (PDE-4) B subtype.
Dr. Hanifin noted that these are “exciting times” in the development of new topical therapies for atopic dermatitis (AD). In addition to the successful phase II study of OPA-15406 he presented, a highlight of the EADV congress was the strongly positive pivotal phase III data presented for crisaborole, another nonsteroidal topical PDE-4 inhibitor, albeit with a different mechanism of action.
For Dr. Hanifin these developments are particularly satisfying personally because 33 years ago as a young investigator – before the term ‘translational science’ had come into vogue – he and his research team made the seminal observation that increased phosphodiesterase activity is “a basic biochemical characteristic relevant to skin immunocellular regulation in atopic disease” (J Allergy Clin Immunol. 1982 Dec;70[6]:452-7).
The 8-week, multicenter, randomized, double-blind phase II OPA-15406 dose-ranging study included 121 patients, 70% with moderate AD, the rest with mild disease. About 20% were adolescents, the rest adults. Their mean baseline Eczema Area Severity Index (EASI) score was 9.5, with a self-reported pruritus score of 61 on a 0-100 scale. Participants were randomized to twice-daily application of OPA-15406 at 0.3% or 1% or the vehicle ointment as a control.
A significant treatment effect was seen within the first week. At 1 week, the mean EASI score was reduced by 31% from baseline in the 1% OPA-15406 group, 15% with the 0.3% formulation, and 6% with vehicle. The active treatment remained significantly more effective than vehicle throughout the 8-week period.
Another measure of efficacy – an Investigator’s Global Assessment (IGA) score of 0 (clear) or 1 along with at least a 2-point improvement on the 0-5 scale at week 4 – was met by 21% of patients on 1% OPA-15406, 15% on the 0.3% formulation, and 2.7% on vehicle. By the less stringent standard of an IGA of 0 or 1 plus at least a 1-point reduction at week 4, the success rates were 30%, 24%, and 10%.
Dr. Hanifin said the 1% formulation is the one likely to advance to phase III studies, given its superior efficacy. This was particularly evident with regard to itch. At the first evaluation, after just 1 week of treatment, pruritus scores showed a mean 30% reduction with 1% OPA-15406 versus no significant change in controls. He added that his clinical impression is that many patients experience a significant improvement in itching within the first 24 hours on the 1% formulation, an observation that deserves formal study.
Scores on the Dermatology Life Quality Index and Children’s Dermatology Life Quality Index were significantly better in the active therapy arms than with vehicle as early as week 1.
The adverse events findings were “not very exciting,” according to the dermatologist. No treatment-related serious adverse events occurred. The two most common adverse events leading to treatment discontinuation – worsening AD and pruritus – occurred more often in the vehicle-treated controls.
Session cochair Dr. Jacek Szepietowski called the pruritus results particularly impressive.
“It’s very difficult to do successful clinical trials of topicals for atopic dermatitis because the vehicle alone, especially if it’s an ointment, can have favorable effects which increase over time both on EASI and pruritus,” commented Dr. Szepietowski, professor and head of the department of dermatology, venereology, and allergology at the Medical University of Wroclaw (Poland).
The study was sponsored by Otsuka Pharmaceuticals. Dr. Hanifin reported serving as a consultant to and paid investigator for the company.
AT THE EADV CONGRESS
Key clinical point: The pipeline for nonsteroidal topical therapies for pediatric and adult atopic dermatitis shows great promise.
Major finding: Atopic dermatitis patients showed a mean 31% reduction in Eczema Area Severity Index scores after 1 week on topical 1% OPA-15406 ointment, compared with a 6% decrease on vehicle alone.
Data source: This phase II multicenter, double-blind, 8-week randomized trial included 121 adolescents and adults with mild to moderate atopic dermatitis.
Disclosures: The study was sponsored by Otsuka Pharmaceuticals. The presenter reported serving as a consultant and paid investigator for the company.

EADV: Long-term apremilast results show what to expect for psoriasis
COPENHAGEN – What can physicians and their psoriasis patients realistically expect from long-term apremilast therapy?
“I think the take-home number is a 50%-plus PASI-75 response after 1 year among patients with moderate to severe plaque psoriasis,” Dr. Kristian Reich said at the annual congress of the European Academy of Dermatology and Venereology.
He presented what he considers to be the first solid data addressing this key question. The data come from a new analysis of the 52-week results of the LIBERATE trial.
“Due to the randomized withdrawal design and long-term treatment only of responders in the original pivotal phase III ESTEEM program, I think we’ve had no real understanding from clinical trials of what the long-term efficacy of apremilast [Otezla] is. Of course, with psoriasis being a chronic disease, this is really what we want to know. And we can get this data very nicely from the LIBERATE study. I should point out that although LIBERATE is a relatively small study, every single patient is included here. So I think this is a very robust analysis of 1-year efficacy data,” said Dr. Reich, managing partner of Dermatologikum Hamburg (Germany).

In contrast to the inclusive analysis performed in LIBERATE, in the pivotal ESTEEM I trial a mere 77 of 562 psoriasis patients randomized to apremilast were on the oral phosphodiesterase type 4 inhibitor for the full 52-week study period, with 61% of that superselect subgroup showing a PASI-75 response (J Am Acad Dermatol. 2015 Jul;73[1]:37-49).
LIBERATE included 250 psoriasis patients. Their baseline Psoriasis Area and Severity Index (PASI) score was about 20, with 27% body surface area involved, a body mass index of 29 kg/m2, 89 kg of body weight, and a mean Dermatology Life Quality Index (DLQI) score of 13. Seventy percent to 80% of subjects had previously used conventional systemic therapies, but no one was allowed to have prior use of biologic therapies.
Patients were randomized to one of three study arms: 16 weeks of oral apremilast at the approved dose of 30 mg twice daily plus a weekly placebo injection, subcutaneous etanercept (Enbrel) at 50 mg once weekly plus a placebo tablet, or dual placebos. At 16 weeks, all patients were switched to apremilast at 30 mg twice daily with no placebos through week 52.
The PASI-75 response rate at week 16 was 11.9% in the placebo group, 39.8% with apremilast, and 48.2% with etanercept.
“I am surprised to see in the more recent studies the high response to etanercept. This is fantastic data. Etanercept seems to ripen like old wine. It’s getting better the longer we have it,” the dermatologist commented.
The PASI-75 rates at 1 year were 46.4% with placebo/apremilast, 50.6% with apremilast throughout, and 55.4% with etanercept/apremilast.
The mean improvement in DLQI from baseline to 52 weeks – a secondary endpoint – was 6.6 points with placebo/apremilast, 8.0 with apremilast/apremilast, and 8.9 points with etanercept/apremilast. A noteworthy finding was that during the first 16 weeks apremilast brought a clinically meaningful improvement in quality of life significantly faster than etanercept did, with a significant difference seen between the two study arms even in the first 1-2 weeks.
The likely explanation for this benefit is that the mean reduction in pruritus visual analog scale scores was significantly greater with apremilast than etanercept through the first 8 weeks. Although pruritus is traditionally thought of more in the context of atopic dermatitis, it’s actually also the No. 1 complaint among psoriasis patients, according to Dr. Reich.
“This is a special thing with apremilast, that the pruritus really goes down very significantly early on. It raises an interesting question about what the role of phosphodiesterase-4 inhibition might be in pruritus. I couldn’t give you a molecular explanation, but I think because pruritus is so annoying and it definitely affects quality of life, this could be a possible explanation for why there is more rapid improvement in quality of life independent of the PASI improvement,” he said.
Switching from etanercept to apremilast didn’t result in any clinically significant safety findings through week 52. No meaningful laboratory changes occurred during 52 weeks of monitoring. There were no cases of suicidal ideation, no increase in serious infections, and only a single cardiac event. The most common apremilast-associated side effects seen in LIBERATE were loose stools in 8%-15% of patients, nausea in 10%, and headache in 13%. These adverse events were mild to moderate in nature and decreased in prevalence over time. The maximum weight loss noted over the course of 52 weeks occurred in the placebo-to-apremilast group, with a mean 1.3-kg reduction.
“I would call the LIBERATE results a validation of the earlier data showing a very clean safety profile with this drug,” Dr. Reich said.
This is reflected in the product labeling, which unlike other systemic therapies for psoriasis includes no requirements for laboratory monitoring or tuberculosis testing.
Both the LIBERATE and the ESTEEM trials were sponsored by Celgene. Dr. Reich received research grants as an investigator in both programs.
COPENHAGEN – What can physicians and their psoriasis patients realistically expect from long-term apremilast therapy?
“I think the take-home number is a 50%-plus PASI-75 response after 1 year among patients with moderate to severe plaque psoriasis,” Dr. Kristian Reich said at the annual congress of the European Academy of Dermatology and Venereology.
He presented what he considers to be the first solid data addressing this key question. The data come from a new analysis of the 52-week results of the LIBERATE trial.
“Due to the randomized withdrawal design and long-term treatment only of responders in the original pivotal phase III ESTEEM program, I think we’ve had no real understanding from clinical trials of what the long-term efficacy of apremilast [Otezla] is. Of course, with psoriasis being a chronic disease, this is really what we want to know. And we can get this data very nicely from the LIBERATE study. I should point out that although LIBERATE is a relatively small study, every single patient is included here. So I think this is a very robust analysis of 1-year efficacy data,” said Dr. Reich, managing partner of Dermatologikum Hamburg (Germany).
In contrast to the inclusive analysis performed in LIBERATE, in the pivotal ESTEEM I trial a mere 77 of 562 psoriasis patients randomized to apremilast were on the oral phosphodiesterase type 4 inhibitor for the full 52-week study period, with 61% of that superselect subgroup showing a PASI-75 response (J Am Acad Dermatol. 2015 Jul;73[1]:37-49).
LIBERATE included 250 psoriasis patients. Their baseline Psoriasis Area and Severity Index (PASI) score was about 20, with 27% body surface area involved, a body mass index of 29 kg/m2, 89 kg of body weight, and a mean Dermatology Life Quality Index (DLQI) score of 13. Seventy percent to 80% of subjects had previously used conventional systemic therapies, but no one was allowed to have prior use of biologic therapies.
Patients were randomized to one of three study arms: 16 weeks of oral apremilast at the approved dose of 30 mg twice daily plus a weekly placebo injection, subcutaneous etanercept (Enbrel) at 50 mg once weekly plus a placebo tablet, or dual placebos. At 16 weeks, all patients were switched to apremilast at 30 mg twice daily with no placebos through week 52.
The PASI-75 response rate at week 16 was 11.9% in the placebo group, 39.8% with apremilast, and 48.2% with etanercept.
“I am surprised to see in the more recent studies the high response to etanercept. This is fantastic data. Etanercept seems to ripen like old wine. It’s getting better the longer we have it,” the dermatologist commented.
The PASI-75 rates at 1 year were 46.4% with placebo/apremilast, 50.6% with apremilast throughout, and 55.4% with etanercept/apremilast.
The mean improvement in DLQI from baseline to 52 weeks – a secondary endpoint – was 6.6 points with placebo/apremilast, 8.0 with apremilast/apremilast, and 8.9 points with etanercept/apremilast. A noteworthy finding was that during the first 16 weeks apremilast brought a clinically meaningful improvement in quality of life significantly faster than etanercept did, with a significant difference seen between the two study arms even in the first 1-2 weeks.
The likely explanation for this benefit is that the mean reduction in pruritus visual analog scale scores was significantly greater with apremilast than etanercept through the first 8 weeks. Although pruritus is traditionally thought of more in the context of atopic dermatitis, it’s actually also the No. 1 complaint among psoriasis patients, according to Dr. Reich.
“This is a special thing with apremilast, that the pruritus really goes down very significantly early on. It raises an interesting question about what the role of phosphodiesterase-4 inhibition might be in pruritus. I couldn’t give you a molecular explanation, but I think because pruritus is so annoying and it definitely affects quality of life, this could be a possible explanation for why there is more rapid improvement in quality of life independent of the PASI improvement,” he said.
Switching from etanercept to apremilast didn’t result in any clinically significant safety findings through week 52. No meaningful laboratory changes occurred during 52 weeks of monitoring. There were no cases of suicidal ideation, no increase in serious infections, and only a single cardiac event. The most common apremilast-associated side effects seen in LIBERATE were loose stools in 8%-15% of patients, nausea in 10%, and headache in 13%. These adverse events were mild to moderate in nature and decreased in prevalence over time. The maximum weight loss noted over the course of 52 weeks occurred in the placebo-to-apremilast group, with a mean 1.3-kg reduction.
“I would call the LIBERATE results a validation of the earlier data showing a very clean safety profile with this drug,” Dr. Reich said.
This is reflected in the product labeling, which unlike other systemic therapies for psoriasis includes no requirements for laboratory monitoring or tuberculosis testing.
Both the LIBERATE and the ESTEEM trials were sponsored by Celgene. Dr. Reich received research grants as an investigator in both programs.
COPENHAGEN – What can physicians and their psoriasis patients realistically expect from long-term apremilast therapy?
“I think the take-home number is a 50%-plus PASI-75 response after 1 year among patients with moderate to severe plaque psoriasis,” Dr. Kristian Reich said at the annual congress of the European Academy of Dermatology and Venereology.
He presented what he considers to be the first solid data addressing this key question. The data come from a new analysis of the 52-week results of the LIBERATE trial.
“Due to the randomized withdrawal design and long-term treatment only of responders in the original pivotal phase III ESTEEM program, I think we’ve had no real understanding from clinical trials of what the long-term efficacy of apremilast [Otezla] is. Of course, with psoriasis being a chronic disease, this is really what we want to know. And we can get this data very nicely from the LIBERATE study. I should point out that although LIBERATE is a relatively small study, every single patient is included here. So I think this is a very robust analysis of 1-year efficacy data,” said Dr. Reich, managing partner of Dermatologikum Hamburg (Germany).
In contrast to the inclusive analysis performed in LIBERATE, in the pivotal ESTEEM I trial a mere 77 of 562 psoriasis patients randomized to apremilast were on the oral phosphodiesterase type 4 inhibitor for the full 52-week study period, with 61% of that superselect subgroup showing a PASI-75 response (J Am Acad Dermatol. 2015 Jul;73[1]:37-49).
LIBERATE included 250 psoriasis patients. Their baseline Psoriasis Area and Severity Index (PASI) score was about 20, with 27% body surface area involved, a body mass index of 29 kg/m2, 89 kg of body weight, and a mean Dermatology Life Quality Index (DLQI) score of 13. Seventy percent to 80% of subjects had previously used conventional systemic therapies, but no one was allowed to have prior use of biologic therapies.
Patients were randomized to one of three study arms: 16 weeks of oral apremilast at the approved dose of 30 mg twice daily plus a weekly placebo injection, subcutaneous etanercept (Enbrel) at 50 mg once weekly plus a placebo tablet, or dual placebos. At 16 weeks, all patients were switched to apremilast at 30 mg twice daily with no placebos through week 52.
The PASI-75 response rate at week 16 was 11.9% in the placebo group, 39.8% with apremilast, and 48.2% with etanercept.
“I am surprised to see in the more recent studies the high response to etanercept. This is fantastic data. Etanercept seems to ripen like old wine. It’s getting better the longer we have it,” the dermatologist commented.
The PASI-75 rates at 1 year were 46.4% with placebo/apremilast, 50.6% with apremilast throughout, and 55.4% with etanercept/apremilast.
The mean improvement in DLQI from baseline to 52 weeks – a secondary endpoint – was 6.6 points with placebo/apremilast, 8.0 with apremilast/apremilast, and 8.9 points with etanercept/apremilast. A noteworthy finding was that during the first 16 weeks apremilast brought a clinically meaningful improvement in quality of life significantly faster than etanercept did, with a significant difference seen between the two study arms even in the first 1-2 weeks.
The likely explanation for this benefit is that the mean reduction in pruritus visual analog scale scores was significantly greater with apremilast than etanercept through the first 8 weeks. Although pruritus is traditionally thought of more in the context of atopic dermatitis, it’s actually also the No. 1 complaint among psoriasis patients, according to Dr. Reich.
“This is a special thing with apremilast, that the pruritus really goes down very significantly early on. It raises an interesting question about what the role of phosphodiesterase-4 inhibition might be in pruritus. I couldn’t give you a molecular explanation, but I think because pruritus is so annoying and it definitely affects quality of life, this could be a possible explanation for why there is more rapid improvement in quality of life independent of the PASI improvement,” he said.
Switching from etanercept to apremilast didn’t result in any clinically significant safety findings through week 52. No meaningful laboratory changes occurred during 52 weeks of monitoring. There were no cases of suicidal ideation, no increase in serious infections, and only a single cardiac event. The most common apremilast-associated side effects seen in LIBERATE were loose stools in 8%-15% of patients, nausea in 10%, and headache in 13%. These adverse events were mild to moderate in nature and decreased in prevalence over time. The maximum weight loss noted over the course of 52 weeks occurred in the placebo-to-apremilast group, with a mean 1.3-kg reduction.
“I would call the LIBERATE results a validation of the earlier data showing a very clean safety profile with this drug,” Dr. Reich said.
This is reflected in the product labeling, which unlike other systemic therapies for psoriasis includes no requirements for laboratory monitoring or tuberculosis testing.
Both the LIBERATE and the ESTEEM trials were sponsored by Celgene. Dr. Reich received research grants as an investigator in both programs.
AT THE EADV CONGRESS
Key clinical point: Patients on apremilast for moderate to severe plaque psoriasis can reasonably anticipate a 50%-plus likelihood of a PASI-75 response after 52 weeks of treatment.
Major finding: A 75% or greater improvement in Psoriasis Area and Severity Index scores was documented in 50.6% of patients after 52 weeks on apremilast and in 55.4% who underwent a protocol-mandated switch from etanercept to apremilast after 16 weeks on the injectable biologic agent.
Data source: A prospective 52-week study randomizing 250 patients with moderate to severe psoriasis to 16 weeks of oral apremilast, etanercept, or placebo followed by 36 weeks of apremilast for all.
Disclosures: The LIBERATE trial was sponsored by Celgene. The presenter received a research grant from the company.

EADV: Hidradenitis suppurativa carries high cardiovascular risk
COPENHAGEN – Hidradenitis suppurativa, a common, chronic, inflammatory scarring skin disease of the hair follicles, is a red flag signaling elevated levels of multiple cardiovascular risk factors, according to a systematic review and meta-analysis.
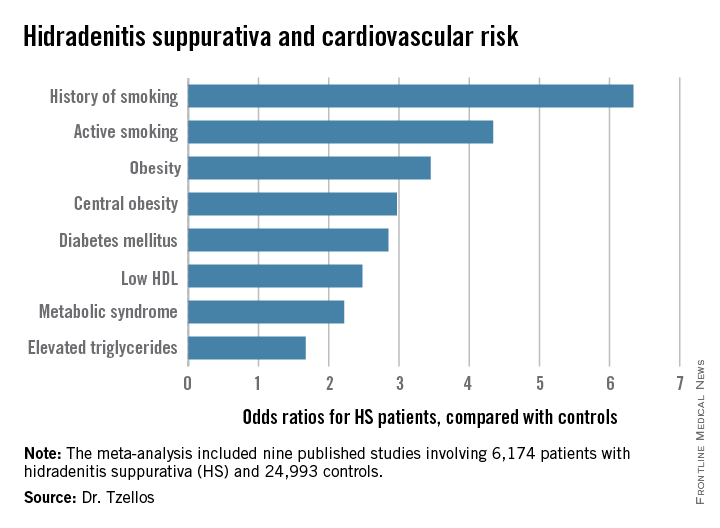
“The need for screening of hidradenitis suppurativa patients for modifiable cardiovascular risk is emphasized,” Dr. Thrasyvoulos Tzellos said in presenting the findings at the annual congress of the European Academy of Dermatology and Venereology.
For such a common and dramatically destructive disease, hidradenitis suppurativa (HS) was underresearched until recently. Investigative interest grew as the tumor necrosis factor inhibitor adalimumab (Humira) underwent development as a novel therapy for what has been traditionally a notoriously difficult to treat disease. The biologic agent received Food and Drug Administration marketing approval in October as the first and only approved treatment for HS.
Dr. Tzellos’s meta-analysis included nine published studies totaling 6,174 HS patients and 24,993 controls. Five studies were case control, and the other four were cross sectional. An indicator of the recent explosive research interest in HS can be seen in the fact that 80% of all the HS patients included in the meta-analysis come from two studies published within just the last year, one from Massachusetts General Hospital (J Am Acad Dermatol. 2014 Dec;71[6]:1144-50) and the other from Israel (Br J Dermatol. 2015 Aug;173[2]:464-70).
Not all the studies examined the same cardiovascular risk factors. For example, only six of nine studies looked at diabetes mellitus as an endpoint. Of those studies that did, diabetes occurred in 856 of 5,685 HS patients, a rate 2.85-fold higher than in controls, according to Dr. Tzellos of University Hospital of North Norway in Troms.
The only cardiovascular risk factor examined that was not significantly more common among patients with HS than controls was hypertension. The 1.57-fold increased likelihood of hypertension among HS patients didn’t achieve statistical significance.
Although patients whose HS was treated exclusively in outpatient settings had significantly higher levels of cardiovascular risk factors than did controls, risk levels were consistently higher still in patients who had been hospitalized for HS.
A meta-analysis such as this cannot address causality, leaving open the question of whether increased cardiovascular risk factors are intrinsic to HS, or the debilitating recurrent skin disease causes affected patients to take a defeatest attitude toward maintenance of a healthy lifestyle.
Dr. Tzellos reported having no financial conflicts regarding this meta-analysis, carried out with academic funding.
COPENHAGEN – Hidradenitis suppurativa, a common, chronic, inflammatory scarring skin disease of the hair follicles, is a red flag signaling elevated levels of multiple cardiovascular risk factors, according to a systematic review and meta-analysis.
“The need for screening of hidradenitis suppurativa patients for modifiable cardiovascular risk is emphasized,” Dr. Thrasyvoulos Tzellos said in presenting the findings at the annual congress of the European Academy of Dermatology and Venereology.
For such a common and dramatically destructive disease, hidradenitis suppurativa (HS) was underresearched until recently. Investigative interest grew as the tumor necrosis factor inhibitor adalimumab (Humira) underwent development as a novel therapy for what has been traditionally a notoriously difficult to treat disease. The biologic agent received Food and Drug Administration marketing approval in October as the first and only approved treatment for HS.
Dr. Tzellos’s meta-analysis included nine published studies totaling 6,174 HS patients and 24,993 controls. Five studies were case control, and the other four were cross sectional. An indicator of the recent explosive research interest in HS can be seen in the fact that 80% of all the HS patients included in the meta-analysis come from two studies published within just the last year, one from Massachusetts General Hospital (J Am Acad Dermatol. 2014 Dec;71[6]:1144-50) and the other from Israel (Br J Dermatol. 2015 Aug;173[2]:464-70).
Not all the studies examined the same cardiovascular risk factors. For example, only six of nine studies looked at diabetes mellitus as an endpoint. Of those studies that did, diabetes occurred in 856 of 5,685 HS patients, a rate 2.85-fold higher than in controls, according to Dr. Tzellos of University Hospital of North Norway in Troms.
The only cardiovascular risk factor examined that was not significantly more common among patients with HS than controls was hypertension. The 1.57-fold increased likelihood of hypertension among HS patients didn’t achieve statistical significance.
Although patients whose HS was treated exclusively in outpatient settings had significantly higher levels of cardiovascular risk factors than did controls, risk levels were consistently higher still in patients who had been hospitalized for HS.
A meta-analysis such as this cannot address causality, leaving open the question of whether increased cardiovascular risk factors are intrinsic to HS, or the debilitating recurrent skin disease causes affected patients to take a defeatest attitude toward maintenance of a healthy lifestyle.
Dr. Tzellos reported having no financial conflicts regarding this meta-analysis, carried out with academic funding.
COPENHAGEN – Hidradenitis suppurativa, a common, chronic, inflammatory scarring skin disease of the hair follicles, is a red flag signaling elevated levels of multiple cardiovascular risk factors, according to a systematic review and meta-analysis.
“The need for screening of hidradenitis suppurativa patients for modifiable cardiovascular risk is emphasized,” Dr. Thrasyvoulos Tzellos said in presenting the findings at the annual congress of the European Academy of Dermatology and Venereology.
For such a common and dramatically destructive disease, hidradenitis suppurativa (HS) was underresearched until recently. Investigative interest grew as the tumor necrosis factor inhibitor adalimumab (Humira) underwent development as a novel therapy for what has been traditionally a notoriously difficult to treat disease. The biologic agent received Food and Drug Administration marketing approval in October as the first and only approved treatment for HS.
Dr. Tzellos’s meta-analysis included nine published studies totaling 6,174 HS patients and 24,993 controls. Five studies were case control, and the other four were cross sectional. An indicator of the recent explosive research interest in HS can be seen in the fact that 80% of all the HS patients included in the meta-analysis come from two studies published within just the last year, one from Massachusetts General Hospital (J Am Acad Dermatol. 2014 Dec;71[6]:1144-50) and the other from Israel (Br J Dermatol. 2015 Aug;173[2]:464-70).
Not all the studies examined the same cardiovascular risk factors. For example, only six of nine studies looked at diabetes mellitus as an endpoint. Of those studies that did, diabetes occurred in 856 of 5,685 HS patients, a rate 2.85-fold higher than in controls, according to Dr. Tzellos of University Hospital of North Norway in Troms.
The only cardiovascular risk factor examined that was not significantly more common among patients with HS than controls was hypertension. The 1.57-fold increased likelihood of hypertension among HS patients didn’t achieve statistical significance.
Although patients whose HS was treated exclusively in outpatient settings had significantly higher levels of cardiovascular risk factors than did controls, risk levels were consistently higher still in patients who had been hospitalized for HS.
A meta-analysis such as this cannot address causality, leaving open the question of whether increased cardiovascular risk factors are intrinsic to HS, or the debilitating recurrent skin disease causes affected patients to take a defeatest attitude toward maintenance of a healthy lifestyle.
Dr. Tzellos reported having no financial conflicts regarding this meta-analysis, carried out with academic funding.
AT THE EADV CONGRESS
Key clinical point: Be vigilant in screening for modifiable cardiovascular risk factors in patients with hidradenitis suppurativa.
Major finding: Hidradenitis suppurativa patients were 2.85-fold more likely than controls to have diabetes, 2.22-fold more likely to have metabolic syndrome, and 4.34-fold more likely to be active smokers.
Data source: A meta-analysis of nine published studies totaling 6,174 hidradenitis suppurativa patients and 24,993 controls.
Disclosures: The presenter reported having no financial conflicts regarding this meta-analysis, carried out with academic funding.
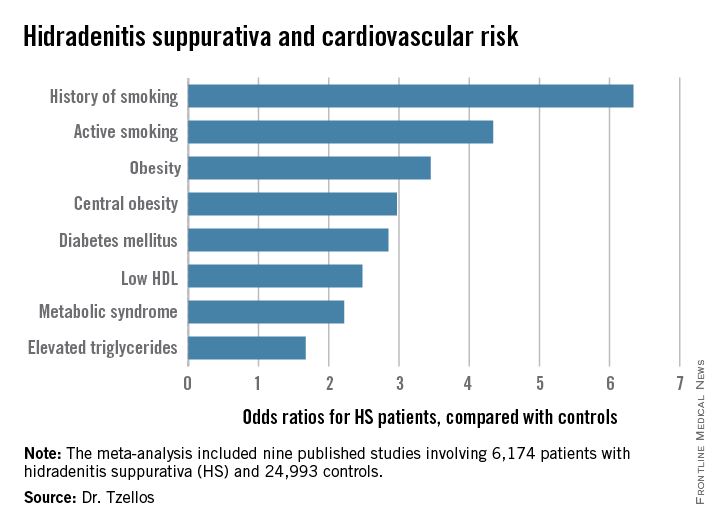
EADV: Ixekizumab promising for psoriatic arthritis
COPENHAGEN – Ixekizumab not only shows considerable promise for the treatment of moderate to severe psoriasis, it looks like it may be a winner for comorbid psoriatic arthritis, too.
The investigational IgG4 humanized monoclonal antibody directed against interleukin-17A brought marked improvements in joint pain, systemic inflammatory burden, and quality of life as well as skin disease in patients with both psoriasis and self-reported psoriatic arthritis in a combined analysis of three phase III clinical trials, Dr. Alice B. Gottlieb reported at the annual congress of the European Academy of Dermatology and Venereology.
Of the 3,126 patients with moderate to severe psoriasis who participated in the 12-week, phase III UNCOVER-1, -2, and -3 trials, 751 (24%) also had self-reported psoriatic arthritis; her analysis focused on them.

She was quick to note that the UNCOVER trials were primarily psoriasis studies that relied upon patient self-report of psoriatic arthritis. Nevertheless, it seems likely that the great majority of self-reported psoriatic arthritis patients really did have the rheumatologic disease, since the mean baseline C-reactive protein (CRP) level in that group was 8.43 mg/L, a level far higher than expected in patients with psoriasis only.
In any case, more-rigorous phase III studies of ixekizumab conducted specifically in patients with formally rheumatologist-diagnosed psoriatic arthritis and treated in rheumatology practices are due to be presented at the annual meeting of the American College of Rheumatology in November. And while Dr. Gottlieb wasn’t at liberty to discuss those results, she did hint that the data will be strongly positive.
“If you’re happy about these UNCOVER findings, you’ll be ecstatic about those,” predicted Dr. Gottlieb, professor of dermatology and dermatologist in chief at Tufts Medical Center, Boston.
Also coming up at the American College of Rheumatology meeting will be the results of the first-ever head-to-head comparison of an IL-17 inhibitor versus a tumor necrosis factor–alpha blocker in psoriatic arthritis patients. While at present most physicians consider a TNF inhibitor to be the treatment of choice in patients with psoriatic arthritis, that view may change as a result of the forthcoming comparative study, according to the dermatologist.
In each of the three phase III UNCOVER studies, patients were randomized to 12 weeks of subcutaneous ixekizumab at 80 mg every 2 or 4 weeks following a 160-mg loading dose, or to placebo. At baseline, the subgroup with self-reported psoriatic arthritis had a mean Psoriasis Area and Severity Index ( PASI) 0f about 21, a self-rated joint pain severity of 50 on a 0-100 scale, a CRP of 8.43 mg/L, and a Dermatology Life Quality Index (DLQI) score of 14.
Joint pain decreased dramatically in the two ixekizumab groups as early at 2 weeks into the trial, at which point, patients on treatment every 2 weeks averaged a 13.1-point reduction from baseline, with a similar 14.1-point drop noted in those on an every 4 weeks schedule. At week 12, the mean reductions from baseline were 25.2 and 26.8 points, compared with a 1.1-point increase in joint pain among placebo-treated controls.
Inflammatory burden plunged quickly, as evidenced by mean reductions in CRP of 4.63 mg/L and 4.33 mg/L at week 1 with biweekly and monthly dosing, respectively. These reductions were then maintained through week 12.
In terms of improvement in skin symptoms, with ixekizumab dosed every 2 weeks, the PASI 75 response was 89.8% at 12 weeks, the PASI 90 response was 69.3%, and the PASI 100 response (clear skin) was 37.1%. In patients treated every 4 weeks, the rates were 81.1%, 60.8%, and 34.7%.
“There’s good news in both groups, but I think the news is even better in the every-2-weeks group,” Dr. Gottlieb commented.
The mean 12-week decrease from baseline in DLQI was 11.8 points with biweekly dosing and 10.5 with 4-week dosing, compared with 0.8 points in controls. That’s impressive given that a 5-point reduction in DLQI is deemed clinically meaningful, the dermatologist observed. At 12 weeks, 56.5% of patients on ixekizumab every 2 weeks had a DLQI of 0 or 1, as did 54% on monthly dosing and 1.5% of controls.
The ixekizumab-treated groups also showed what Dr. Gottlieb described as “dramatic” improvements – in the 4+ to 5+ point range – in both the mental and physical component scores on the SF-36, another widely used quality of life measure.
Improvements in skin and self-reported joint symptoms appeared to correlate. Patients with a PASI 50 to less-than PASI 75 response had a mean 17-point reduction in joint pain scores, while those with a PASI 75 to less-than PASI 90 response averaged a 25.1-point improvement in joint pain, patients with a PASI 90 to less-than PASI 100 skin response averaged a 27.6-point reduction, and PASI 100 responders had a mean 30.3-point reduction in joint pain scores.
“Obviously, one needs to look at this more carefully in a phase III psoriatic arthritis study. That’ll provide a more robust answer. But this gives a hint,” the dermatologist said.
The UNCOVER program was sponsored by Eli Lilly. Dr. Gottlieb serves as an adviser to Lilly and numerous other pharmaceutical companies.
COPENHAGEN – Ixekizumab not only shows considerable promise for the treatment of moderate to severe psoriasis, it looks like it may be a winner for comorbid psoriatic arthritis, too.
The investigational IgG4 humanized monoclonal antibody directed against interleukin-17A brought marked improvements in joint pain, systemic inflammatory burden, and quality of life as well as skin disease in patients with both psoriasis and self-reported psoriatic arthritis in a combined analysis of three phase III clinical trials, Dr. Alice B. Gottlieb reported at the annual congress of the European Academy of Dermatology and Venereology.
Of the 3,126 patients with moderate to severe psoriasis who participated in the 12-week, phase III UNCOVER-1, -2, and -3 trials, 751 (24%) also had self-reported psoriatic arthritis; her analysis focused on them.
She was quick to note that the UNCOVER trials were primarily psoriasis studies that relied upon patient self-report of psoriatic arthritis. Nevertheless, it seems likely that the great majority of self-reported psoriatic arthritis patients really did have the rheumatologic disease, since the mean baseline C-reactive protein (CRP) level in that group was 8.43 mg/L, a level far higher than expected in patients with psoriasis only.
In any case, more-rigorous phase III studies of ixekizumab conducted specifically in patients with formally rheumatologist-diagnosed psoriatic arthritis and treated in rheumatology practices are due to be presented at the annual meeting of the American College of Rheumatology in November. And while Dr. Gottlieb wasn’t at liberty to discuss those results, she did hint that the data will be strongly positive.
“If you’re happy about these UNCOVER findings, you’ll be ecstatic about those,” predicted Dr. Gottlieb, professor of dermatology and dermatologist in chief at Tufts Medical Center, Boston.
Also coming up at the American College of Rheumatology meeting will be the results of the first-ever head-to-head comparison of an IL-17 inhibitor versus a tumor necrosis factor–alpha blocker in psoriatic arthritis patients. While at present most physicians consider a TNF inhibitor to be the treatment of choice in patients with psoriatic arthritis, that view may change as a result of the forthcoming comparative study, according to the dermatologist.
In each of the three phase III UNCOVER studies, patients were randomized to 12 weeks of subcutaneous ixekizumab at 80 mg every 2 or 4 weeks following a 160-mg loading dose, or to placebo. At baseline, the subgroup with self-reported psoriatic arthritis had a mean Psoriasis Area and Severity Index ( PASI) 0f about 21, a self-rated joint pain severity of 50 on a 0-100 scale, a CRP of 8.43 mg/L, and a Dermatology Life Quality Index (DLQI) score of 14.
Joint pain decreased dramatically in the two ixekizumab groups as early at 2 weeks into the trial, at which point, patients on treatment every 2 weeks averaged a 13.1-point reduction from baseline, with a similar 14.1-point drop noted in those on an every 4 weeks schedule. At week 12, the mean reductions from baseline were 25.2 and 26.8 points, compared with a 1.1-point increase in joint pain among placebo-treated controls.
Inflammatory burden plunged quickly, as evidenced by mean reductions in CRP of 4.63 mg/L and 4.33 mg/L at week 1 with biweekly and monthly dosing, respectively. These reductions were then maintained through week 12.
In terms of improvement in skin symptoms, with ixekizumab dosed every 2 weeks, the PASI 75 response was 89.8% at 12 weeks, the PASI 90 response was 69.3%, and the PASI 100 response (clear skin) was 37.1%. In patients treated every 4 weeks, the rates were 81.1%, 60.8%, and 34.7%.
“There’s good news in both groups, but I think the news is even better in the every-2-weeks group,” Dr. Gottlieb commented.
The mean 12-week decrease from baseline in DLQI was 11.8 points with biweekly dosing and 10.5 with 4-week dosing, compared with 0.8 points in controls. That’s impressive given that a 5-point reduction in DLQI is deemed clinically meaningful, the dermatologist observed. At 12 weeks, 56.5% of patients on ixekizumab every 2 weeks had a DLQI of 0 or 1, as did 54% on monthly dosing and 1.5% of controls.
The ixekizumab-treated groups also showed what Dr. Gottlieb described as “dramatic” improvements – in the 4+ to 5+ point range – in both the mental and physical component scores on the SF-36, another widely used quality of life measure.
Improvements in skin and self-reported joint symptoms appeared to correlate. Patients with a PASI 50 to less-than PASI 75 response had a mean 17-point reduction in joint pain scores, while those with a PASI 75 to less-than PASI 90 response averaged a 25.1-point improvement in joint pain, patients with a PASI 90 to less-than PASI 100 skin response averaged a 27.6-point reduction, and PASI 100 responders had a mean 30.3-point reduction in joint pain scores.
“Obviously, one needs to look at this more carefully in a phase III psoriatic arthritis study. That’ll provide a more robust answer. But this gives a hint,” the dermatologist said.
The UNCOVER program was sponsored by Eli Lilly. Dr. Gottlieb serves as an adviser to Lilly and numerous other pharmaceutical companies.
COPENHAGEN – Ixekizumab not only shows considerable promise for the treatment of moderate to severe psoriasis, it looks like it may be a winner for comorbid psoriatic arthritis, too.
The investigational IgG4 humanized monoclonal antibody directed against interleukin-17A brought marked improvements in joint pain, systemic inflammatory burden, and quality of life as well as skin disease in patients with both psoriasis and self-reported psoriatic arthritis in a combined analysis of three phase III clinical trials, Dr. Alice B. Gottlieb reported at the annual congress of the European Academy of Dermatology and Venereology.
Of the 3,126 patients with moderate to severe psoriasis who participated in the 12-week, phase III UNCOVER-1, -2, and -3 trials, 751 (24%) also had self-reported psoriatic arthritis; her analysis focused on them.
She was quick to note that the UNCOVER trials were primarily psoriasis studies that relied upon patient self-report of psoriatic arthritis. Nevertheless, it seems likely that the great majority of self-reported psoriatic arthritis patients really did have the rheumatologic disease, since the mean baseline C-reactive protein (CRP) level in that group was 8.43 mg/L, a level far higher than expected in patients with psoriasis only.
In any case, more-rigorous phase III studies of ixekizumab conducted specifically in patients with formally rheumatologist-diagnosed psoriatic arthritis and treated in rheumatology practices are due to be presented at the annual meeting of the American College of Rheumatology in November. And while Dr. Gottlieb wasn’t at liberty to discuss those results, she did hint that the data will be strongly positive.
“If you’re happy about these UNCOVER findings, you’ll be ecstatic about those,” predicted Dr. Gottlieb, professor of dermatology and dermatologist in chief at Tufts Medical Center, Boston.
Also coming up at the American College of Rheumatology meeting will be the results of the first-ever head-to-head comparison of an IL-17 inhibitor versus a tumor necrosis factor–alpha blocker in psoriatic arthritis patients. While at present most physicians consider a TNF inhibitor to be the treatment of choice in patients with psoriatic arthritis, that view may change as a result of the forthcoming comparative study, according to the dermatologist.
In each of the three phase III UNCOVER studies, patients were randomized to 12 weeks of subcutaneous ixekizumab at 80 mg every 2 or 4 weeks following a 160-mg loading dose, or to placebo. At baseline, the subgroup with self-reported psoriatic arthritis had a mean Psoriasis Area and Severity Index ( PASI) 0f about 21, a self-rated joint pain severity of 50 on a 0-100 scale, a CRP of 8.43 mg/L, and a Dermatology Life Quality Index (DLQI) score of 14.
Joint pain decreased dramatically in the two ixekizumab groups as early at 2 weeks into the trial, at which point, patients on treatment every 2 weeks averaged a 13.1-point reduction from baseline, with a similar 14.1-point drop noted in those on an every 4 weeks schedule. At week 12, the mean reductions from baseline were 25.2 and 26.8 points, compared with a 1.1-point increase in joint pain among placebo-treated controls.
Inflammatory burden plunged quickly, as evidenced by mean reductions in CRP of 4.63 mg/L and 4.33 mg/L at week 1 with biweekly and monthly dosing, respectively. These reductions were then maintained through week 12.
In terms of improvement in skin symptoms, with ixekizumab dosed every 2 weeks, the PASI 75 response was 89.8% at 12 weeks, the PASI 90 response was 69.3%, and the PASI 100 response (clear skin) was 37.1%. In patients treated every 4 weeks, the rates were 81.1%, 60.8%, and 34.7%.
“There’s good news in both groups, but I think the news is even better in the every-2-weeks group,” Dr. Gottlieb commented.
The mean 12-week decrease from baseline in DLQI was 11.8 points with biweekly dosing and 10.5 with 4-week dosing, compared with 0.8 points in controls. That’s impressive given that a 5-point reduction in DLQI is deemed clinically meaningful, the dermatologist observed. At 12 weeks, 56.5% of patients on ixekizumab every 2 weeks had a DLQI of 0 or 1, as did 54% on monthly dosing and 1.5% of controls.
The ixekizumab-treated groups also showed what Dr. Gottlieb described as “dramatic” improvements – in the 4+ to 5+ point range – in both the mental and physical component scores on the SF-36, another widely used quality of life measure.
Improvements in skin and self-reported joint symptoms appeared to correlate. Patients with a PASI 50 to less-than PASI 75 response had a mean 17-point reduction in joint pain scores, while those with a PASI 75 to less-than PASI 90 response averaged a 25.1-point improvement in joint pain, patients with a PASI 90 to less-than PASI 100 skin response averaged a 27.6-point reduction, and PASI 100 responders had a mean 30.3-point reduction in joint pain scores.
“Obviously, one needs to look at this more carefully in a phase III psoriatic arthritis study. That’ll provide a more robust answer. But this gives a hint,” the dermatologist said.
The UNCOVER program was sponsored by Eli Lilly. Dr. Gottlieb serves as an adviser to Lilly and numerous other pharmaceutical companies.
AT THE EADV CONGRESS
Key clinical point: The IL-17A inhibitor ixekizumab appears to be dramatically effective in psoriatic arthritis as well as in psoriasis.
Major finding: Self-reported joint pain scores in psoriatic arthritis patients were cut in half after 12 weeks of ixekizumab while remaining unmoved in placebo-treated controls.
Data source: This was an analysis of treatment outcomes in 751 self-reported psoriatic arthritis patients who participated in three 12-week phase III randomized clinical trials of ixekizumab vs. placebo.
Disclosures: The UNCOVER clinical trials program was sponsored by Eli Lilly. The presenter serves as an adviser to Lilly and numerous other pharmaceutical companies.

Investigational Biologic Rocks Psoriasis World
COPENHAGEN – An investigational biologic agent that selectively inhibits the p19 subunit of interleukin-23 was the talk of the 2015 EADV congress based upon its striking outperformance of ustekinumab in a head-to-head phase II study in psoriasis patients.
Not only did the investigational agent, known for now as BI 655066, achieve substantially higher rates of PASI 90 – clear or almost clear skin – and PASI 100, but it did so much faster and maintained those stellar results far longer off treatment than with ustekinumab (Stelara), Dr. Kim A. Papp reported at the annual congress of the European Academy of Dermatology and Venereology.

“These data make an irrefutable case for IL-23 as instrumental in the expression of psoriasis,” declared Dr. Papp, president of Probity Medical Research in Waterloo, Ont.
“What these data show is that selective blockade of IL-23 p19 clearly provides a remarkable response, whether we measure it as PASI 75, PASI 90, or PASI 100, compared with ustekinumab, which provides a very robust response itself. And when we look at the safety profile – admittedly in a small number of patients and for a short period of time – we see there are no real differences in safety signals. Obviously, we’re very encouraged to move forward with long-term phase III investigations,” Dr. Papp added.
The multicenter, double-blind phase II study included 166 patients with moderate to severe plaque psoriasis who were randomized to one of four treatment arms: BI 655066 at 90 or 180 mg given subcutaneously at weeks 0, 4, and 16; weight-based ustekinumab at 45 or 90 mg given on the same schedule; or a single 18-mg dose of BI 655066. Dr. Papp presented the results at 36 weeks, fully 20 weeks after the final dose was given.
Significantly more patients taking the investigational agent achieved PASI targets than did those taking ustekinumab, a highly effective medication. Here are the eye-popping 36-week results:
• PASI 90: 81% with BI 655066 at 180 mg, 69% with the lower dose, 30% with ustekinumab, and 7% with a single 18-mg dose of BI 655066.
• PASI 100: 54% and 43% with high- and low-dose BI 655066, respectively, 15% with ustekinumab, and 0% with single-dose BI 655066.
• PASI 75: 93% and 88% with high- and low-dose BI 655066; 55% with ustekinumab; and 19% after single-dose BI 655066.
One of the most intriguing study findings, according to Dr. Papp, is that the slope of the PASI 100 response curve in BI 655066–treated patients was still rising at week 20, a month after the final dose.
“It suggests that if you were to continue treating these patients, you might anticipate an even higher response,” the dermatologist said.
Another notable finding was the accelerated speed of response to the investigational agent: 57 days after the first dose of BI 655066, 50% of patients had a PASI 90 response.
“That means from the time you initiate therapy, you can expect half of your patients to get to PASI 90 in less than 2 months time,” Dr. Papp observed.
In contrast, it took 117 days – basically twice as long – for half of the ustekinumab group to achieve PASI 90.
Turning to the durability of response, Dr. Papp noted that it took 169 days for half of patients in the ustekinumab group to lose their PASI 90 response after treatment stopped, compared with 225 days in the BI 655066 90-mg group. That endpoint was never reached in the BI 655066 180-mg group before the study’s end.
Audience reaction to the study results was “wow.”
“Obviously, the data are terrific. Your mother couldn’t have invented better data,” declared Dr. Alice B. Gottlieb, professor of dermatology and dermatologist in chief at Tufts Medical Center, Boston.
She posed a question: Why are the BI 655066 response rates so much higher and longer lasting than with other biologics, including ustekinumab, which inhibits both IL-23 and IL-12?
“I think one key factor in the durability of the response is the pathway,” Dr. Papp replied. “Exactly why blockade of IL-23 p19 leads to this durable clinical response is unclear, but we can posit that we’re depopulating the Th-17 cells. IL-23 is necessary for Th-17 cell survival. So if you block the water, you ain’t going to grow the grass. I’m speculating here, but I think we’re removing that clone of cells, and it’s only once we start to allow them to repopulate that we then see recurrence of the disease.”
BI 655066 is being developed by Boehringer Ingelheim. Dr. Papp serves as an investigator for and consultant to Boehringer Ingelheim and numerous other pharmaceutical companies.
COPENHAGEN – An investigational biologic agent that selectively inhibits the p19 subunit of interleukin-23 was the talk of the 2015 EADV congress based upon its striking outperformance of ustekinumab in a head-to-head phase II study in psoriasis patients.
Not only did the investigational agent, known for now as BI 655066, achieve substantially higher rates of PASI 90 – clear or almost clear skin – and PASI 100, but it did so much faster and maintained those stellar results far longer off treatment than with ustekinumab (Stelara), Dr. Kim A. Papp reported at the annual congress of the European Academy of Dermatology and Venereology.
“These data make an irrefutable case for IL-23 as instrumental in the expression of psoriasis,” declared Dr. Papp, president of Probity Medical Research in Waterloo, Ont.
“What these data show is that selective blockade of IL-23 p19 clearly provides a remarkable response, whether we measure it as PASI 75, PASI 90, or PASI 100, compared with ustekinumab, which provides a very robust response itself. And when we look at the safety profile – admittedly in a small number of patients and for a short period of time – we see there are no real differences in safety signals. Obviously, we’re very encouraged to move forward with long-term phase III investigations,” Dr. Papp added.
The multicenter, double-blind phase II study included 166 patients with moderate to severe plaque psoriasis who were randomized to one of four treatment arms: BI 655066 at 90 or 180 mg given subcutaneously at weeks 0, 4, and 16; weight-based ustekinumab at 45 or 90 mg given on the same schedule; or a single 18-mg dose of BI 655066. Dr. Papp presented the results at 36 weeks, fully 20 weeks after the final dose was given.
Significantly more patients taking the investigational agent achieved PASI targets than did those taking ustekinumab, a highly effective medication. Here are the eye-popping 36-week results:
• PASI 90: 81% with BI 655066 at 180 mg, 69% with the lower dose, 30% with ustekinumab, and 7% with a single 18-mg dose of BI 655066.
• PASI 100: 54% and 43% with high- and low-dose BI 655066, respectively, 15% with ustekinumab, and 0% with single-dose BI 655066.
• PASI 75: 93% and 88% with high- and low-dose BI 655066; 55% with ustekinumab; and 19% after single-dose BI 655066.
One of the most intriguing study findings, according to Dr. Papp, is that the slope of the PASI 100 response curve in BI 655066–treated patients was still rising at week 20, a month after the final dose.
“It suggests that if you were to continue treating these patients, you might anticipate an even higher response,” the dermatologist said.
Another notable finding was the accelerated speed of response to the investigational agent: 57 days after the first dose of BI 655066, 50% of patients had a PASI 90 response.
“That means from the time you initiate therapy, you can expect half of your patients to get to PASI 90 in less than 2 months time,” Dr. Papp observed.
In contrast, it took 117 days – basically twice as long – for half of the ustekinumab group to achieve PASI 90.
Turning to the durability of response, Dr. Papp noted that it took 169 days for half of patients in the ustekinumab group to lose their PASI 90 response after treatment stopped, compared with 225 days in the BI 655066 90-mg group. That endpoint was never reached in the BI 655066 180-mg group before the study’s end.
Audience reaction to the study results was “wow.”
“Obviously, the data are terrific. Your mother couldn’t have invented better data,” declared Dr. Alice B. Gottlieb, professor of dermatology and dermatologist in chief at Tufts Medical Center, Boston.
She posed a question: Why are the BI 655066 response rates so much higher and longer lasting than with other biologics, including ustekinumab, which inhibits both IL-23 and IL-12?
“I think one key factor in the durability of the response is the pathway,” Dr. Papp replied. “Exactly why blockade of IL-23 p19 leads to this durable clinical response is unclear, but we can posit that we’re depopulating the Th-17 cells. IL-23 is necessary for Th-17 cell survival. So if you block the water, you ain’t going to grow the grass. I’m speculating here, but I think we’re removing that clone of cells, and it’s only once we start to allow them to repopulate that we then see recurrence of the disease.”
BI 655066 is being developed by Boehringer Ingelheim. Dr. Papp serves as an investigator for and consultant to Boehringer Ingelheim and numerous other pharmaceutical companies.
COPENHAGEN – An investigational biologic agent that selectively inhibits the p19 subunit of interleukin-23 was the talk of the 2015 EADV congress based upon its striking outperformance of ustekinumab in a head-to-head phase II study in psoriasis patients.
Not only did the investigational agent, known for now as BI 655066, achieve substantially higher rates of PASI 90 – clear or almost clear skin – and PASI 100, but it did so much faster and maintained those stellar results far longer off treatment than with ustekinumab (Stelara), Dr. Kim A. Papp reported at the annual congress of the European Academy of Dermatology and Venereology.
“These data make an irrefutable case for IL-23 as instrumental in the expression of psoriasis,” declared Dr. Papp, president of Probity Medical Research in Waterloo, Ont.
“What these data show is that selective blockade of IL-23 p19 clearly provides a remarkable response, whether we measure it as PASI 75, PASI 90, or PASI 100, compared with ustekinumab, which provides a very robust response itself. And when we look at the safety profile – admittedly in a small number of patients and for a short period of time – we see there are no real differences in safety signals. Obviously, we’re very encouraged to move forward with long-term phase III investigations,” Dr. Papp added.
The multicenter, double-blind phase II study included 166 patients with moderate to severe plaque psoriasis who were randomized to one of four treatment arms: BI 655066 at 90 or 180 mg given subcutaneously at weeks 0, 4, and 16; weight-based ustekinumab at 45 or 90 mg given on the same schedule; or a single 18-mg dose of BI 655066. Dr. Papp presented the results at 36 weeks, fully 20 weeks after the final dose was given.
Significantly more patients taking the investigational agent achieved PASI targets than did those taking ustekinumab, a highly effective medication. Here are the eye-popping 36-week results:
• PASI 90: 81% with BI 655066 at 180 mg, 69% with the lower dose, 30% with ustekinumab, and 7% with a single 18-mg dose of BI 655066.
• PASI 100: 54% and 43% with high- and low-dose BI 655066, respectively, 15% with ustekinumab, and 0% with single-dose BI 655066.
• PASI 75: 93% and 88% with high- and low-dose BI 655066; 55% with ustekinumab; and 19% after single-dose BI 655066.
One of the most intriguing study findings, according to Dr. Papp, is that the slope of the PASI 100 response curve in BI 655066–treated patients was still rising at week 20, a month after the final dose.
“It suggests that if you were to continue treating these patients, you might anticipate an even higher response,” the dermatologist said.
Another notable finding was the accelerated speed of response to the investigational agent: 57 days after the first dose of BI 655066, 50% of patients had a PASI 90 response.
“That means from the time you initiate therapy, you can expect half of your patients to get to PASI 90 in less than 2 months time,” Dr. Papp observed.
In contrast, it took 117 days – basically twice as long – for half of the ustekinumab group to achieve PASI 90.
Turning to the durability of response, Dr. Papp noted that it took 169 days for half of patients in the ustekinumab group to lose their PASI 90 response after treatment stopped, compared with 225 days in the BI 655066 90-mg group. That endpoint was never reached in the BI 655066 180-mg group before the study’s end.
Audience reaction to the study results was “wow.”
“Obviously, the data are terrific. Your mother couldn’t have invented better data,” declared Dr. Alice B. Gottlieb, professor of dermatology and dermatologist in chief at Tufts Medical Center, Boston.
She posed a question: Why are the BI 655066 response rates so much higher and longer lasting than with other biologics, including ustekinumab, which inhibits both IL-23 and IL-12?
“I think one key factor in the durability of the response is the pathway,” Dr. Papp replied. “Exactly why blockade of IL-23 p19 leads to this durable clinical response is unclear, but we can posit that we’re depopulating the Th-17 cells. IL-23 is necessary for Th-17 cell survival. So if you block the water, you ain’t going to grow the grass. I’m speculating here, but I think we’re removing that clone of cells, and it’s only once we start to allow them to repopulate that we then see recurrence of the disease.”
BI 655066 is being developed by Boehringer Ingelheim. Dr. Papp serves as an investigator for and consultant to Boehringer Ingelheim and numerous other pharmaceutical companies.
AT THE EADV CONGRESS

EADV: Investigational biologic rocks psoriasis world
COPENHAGEN – An investigational biologic agent that selectively inhibits the p19 subunit of interleukin-23 was the talk of the 2015 EADV congress based upon its striking outperformance of ustekinumab in a head-to-head phase II study in psoriasis patients.
Not only did the investigational agent, known for now as BI 655066, achieve substantially higher rates of PASI 90 – clear or almost clear skin – and PASI 100, but it did so much faster and maintained those stellar results far longer off treatment than with ustekinumab (Stelara), Dr. Kim A. Papp reported at the annual congress of the European Academy of Dermatology and Venereology.

“These data make an irrefutable case for IL-23 as instrumental in the expression of psoriasis,” declared Dr. Papp, president of Probity Medical Research in Waterloo, Ont.
“What these data show is that selective blockade of IL-23 p19 clearly provides a remarkable response, whether we measure it as PASI 75, PASI 90, or PASI 100, compared with ustekinumab, which provides a very robust response itself. And when we look at the safety profile – admittedly in a small number of patients and for a short period of time – we see there are no real differences in safety signals. Obviously, we’re very encouraged to move forward with long-term phase III investigations,” Dr. Papp added.
The multicenter, double-blind phase II study included 166 patients with moderate to severe plaque psoriasis who were randomized to one of four treatment arms: BI 655066 at 90 or 180 mg given subcutaneously at weeks 0, 4, and 16; weight-based ustekinumab at 45 or 90 mg given on the same schedule; or a single 18-mg dose of BI 655066. Dr. Papp presented the results at 36 weeks, fully 20 weeks after the final dose was given.
Significantly more patients taking the investigational agent achieved PASI targets than did those taking ustekinumab, a highly effective medication. Here are the eye-popping 36-week results:
• PASI 90: 81% with BI 655066 at 180 mg, 69% with the lower dose, 30% with ustekinumab, and 7% with a single 18-mg dose of BI 655066.
• PASI 100: 54% and 43% with high- and low-dose BI 655066, respectively, 15% with ustekinumab, and 0% with single-dose BI 655066.
• PASI 75: 93% and 88% with high- and low-dose BI 655066; 55% with ustekinumab; and 19% after single-dose BI 655066.
One of the most intriguing study findings, according to Dr. Papp, is that the slope of the PASI 100 response curve in BI 655066–treated patients was still rising at week 20, a month after the final dose.
“It suggests that if you were to continue treating these patients, you might anticipate an even higher response,” the dermatologist said.
Another notable finding was the accelerated speed of response to the investigational agent: 57 days after the first dose of BI 655066, 50% of patients had a PASI 90 response.
“That means from the time you initiate therapy, you can expect half of your patients to get to PASI 90 in less than 2 months time,” Dr. Papp observed.
In contrast, it took 117 days – basically twice as long – for half of the ustekinumab group to achieve PASI 90.
Turning to the durability of response, Dr. Papp noted that it took 169 days for half of patients in the ustekinumab group to lose their PASI 90 response after treatment stopped, compared with 225 days in the BI 655066 90-mg group. That endpoint was never reached in the BI 655066 180-mg group before the study’s end.
Audience reaction to the study results was “wow.”
“Obviously, the data are terrific. Your mother couldn’t have invented better data,” declared Dr. Alice B. Gottlieb, professor of dermatology and dermatologist in chief at Tufts Medical Center, Boston.
She posed a question: Why are the BI 655066 response rates so much higher and longer lasting than with other biologics, including ustekinumab, which inhibits both IL-23 and IL-12?
“I think one key factor in the durability of the response is the pathway,” Dr. Papp replied. “Exactly why blockade of IL-23 p19 leads to this durable clinical response is unclear, but we can posit that we’re depopulating the Th-17 cells. IL-23 is necessary for Th-17 cell survival. So if you block the water, you ain’t going to grow the grass. I’m speculating here, but I think we’re removing that clone of cells, and it’s only once we start to allow them to repopulate that we then see recurrence of the disease.”
BI 655066 is being developed by Boehringer Ingelheim. Dr. Papp serves as an investigator for and consultant to Boehringer Ingelheim and numerous other pharmaceutical companies.
COPENHAGEN – An investigational biologic agent that selectively inhibits the p19 subunit of interleukin-23 was the talk of the 2015 EADV congress based upon its striking outperformance of ustekinumab in a head-to-head phase II study in psoriasis patients.
Not only did the investigational agent, known for now as BI 655066, achieve substantially higher rates of PASI 90 – clear or almost clear skin – and PASI 100, but it did so much faster and maintained those stellar results far longer off treatment than with ustekinumab (Stelara), Dr. Kim A. Papp reported at the annual congress of the European Academy of Dermatology and Venereology.
“These data make an irrefutable case for IL-23 as instrumental in the expression of psoriasis,” declared Dr. Papp, president of Probity Medical Research in Waterloo, Ont.
“What these data show is that selective blockade of IL-23 p19 clearly provides a remarkable response, whether we measure it as PASI 75, PASI 90, or PASI 100, compared with ustekinumab, which provides a very robust response itself. And when we look at the safety profile – admittedly in a small number of patients and for a short period of time – we see there are no real differences in safety signals. Obviously, we’re very encouraged to move forward with long-term phase III investigations,” Dr. Papp added.
The multicenter, double-blind phase II study included 166 patients with moderate to severe plaque psoriasis who were randomized to one of four treatment arms: BI 655066 at 90 or 180 mg given subcutaneously at weeks 0, 4, and 16; weight-based ustekinumab at 45 or 90 mg given on the same schedule; or a single 18-mg dose of BI 655066. Dr. Papp presented the results at 36 weeks, fully 20 weeks after the final dose was given.
Significantly more patients taking the investigational agent achieved PASI targets than did those taking ustekinumab, a highly effective medication. Here are the eye-popping 36-week results:
• PASI 90: 81% with BI 655066 at 180 mg, 69% with the lower dose, 30% with ustekinumab, and 7% with a single 18-mg dose of BI 655066.
• PASI 100: 54% and 43% with high- and low-dose BI 655066, respectively, 15% with ustekinumab, and 0% with single-dose BI 655066.
• PASI 75: 93% and 88% with high- and low-dose BI 655066; 55% with ustekinumab; and 19% after single-dose BI 655066.
One of the most intriguing study findings, according to Dr. Papp, is that the slope of the PASI 100 response curve in BI 655066–treated patients was still rising at week 20, a month after the final dose.
“It suggests that if you were to continue treating these patients, you might anticipate an even higher response,” the dermatologist said.
Another notable finding was the accelerated speed of response to the investigational agent: 57 days after the first dose of BI 655066, 50% of patients had a PASI 90 response.
“That means from the time you initiate therapy, you can expect half of your patients to get to PASI 90 in less than 2 months time,” Dr. Papp observed.
In contrast, it took 117 days – basically twice as long – for half of the ustekinumab group to achieve PASI 90.
Turning to the durability of response, Dr. Papp noted that it took 169 days for half of patients in the ustekinumab group to lose their PASI 90 response after treatment stopped, compared with 225 days in the BI 655066 90-mg group. That endpoint was never reached in the BI 655066 180-mg group before the study’s end.
Audience reaction to the study results was “wow.”
“Obviously, the data are terrific. Your mother couldn’t have invented better data,” declared Dr. Alice B. Gottlieb, professor of dermatology and dermatologist in chief at Tufts Medical Center, Boston.
She posed a question: Why are the BI 655066 response rates so much higher and longer lasting than with other biologics, including ustekinumab, which inhibits both IL-23 and IL-12?
“I think one key factor in the durability of the response is the pathway,” Dr. Papp replied. “Exactly why blockade of IL-23 p19 leads to this durable clinical response is unclear, but we can posit that we’re depopulating the Th-17 cells. IL-23 is necessary for Th-17 cell survival. So if you block the water, you ain’t going to grow the grass. I’m speculating here, but I think we’re removing that clone of cells, and it’s only once we start to allow them to repopulate that we then see recurrence of the disease.”
BI 655066 is being developed by Boehringer Ingelheim. Dr. Papp serves as an investigator for and consultant to Boehringer Ingelheim and numerous other pharmaceutical companies.
COPENHAGEN – An investigational biologic agent that selectively inhibits the p19 subunit of interleukin-23 was the talk of the 2015 EADV congress based upon its striking outperformance of ustekinumab in a head-to-head phase II study in psoriasis patients.
Not only did the investigational agent, known for now as BI 655066, achieve substantially higher rates of PASI 90 – clear or almost clear skin – and PASI 100, but it did so much faster and maintained those stellar results far longer off treatment than with ustekinumab (Stelara), Dr. Kim A. Papp reported at the annual congress of the European Academy of Dermatology and Venereology.
“These data make an irrefutable case for IL-23 as instrumental in the expression of psoriasis,” declared Dr. Papp, president of Probity Medical Research in Waterloo, Ont.
“What these data show is that selective blockade of IL-23 p19 clearly provides a remarkable response, whether we measure it as PASI 75, PASI 90, or PASI 100, compared with ustekinumab, which provides a very robust response itself. And when we look at the safety profile – admittedly in a small number of patients and for a short period of time – we see there are no real differences in safety signals. Obviously, we’re very encouraged to move forward with long-term phase III investigations,” Dr. Papp added.
The multicenter, double-blind phase II study included 166 patients with moderate to severe plaque psoriasis who were randomized to one of four treatment arms: BI 655066 at 90 or 180 mg given subcutaneously at weeks 0, 4, and 16; weight-based ustekinumab at 45 or 90 mg given on the same schedule; or a single 18-mg dose of BI 655066. Dr. Papp presented the results at 36 weeks, fully 20 weeks after the final dose was given.
Significantly more patients taking the investigational agent achieved PASI targets than did those taking ustekinumab, a highly effective medication. Here are the eye-popping 36-week results:
• PASI 90: 81% with BI 655066 at 180 mg, 69% with the lower dose, 30% with ustekinumab, and 7% with a single 18-mg dose of BI 655066.
• PASI 100: 54% and 43% with high- and low-dose BI 655066, respectively, 15% with ustekinumab, and 0% with single-dose BI 655066.
• PASI 75: 93% and 88% with high- and low-dose BI 655066; 55% with ustekinumab; and 19% after single-dose BI 655066.
One of the most intriguing study findings, according to Dr. Papp, is that the slope of the PASI 100 response curve in BI 655066–treated patients was still rising at week 20, a month after the final dose.
“It suggests that if you were to continue treating these patients, you might anticipate an even higher response,” the dermatologist said.
Another notable finding was the accelerated speed of response to the investigational agent: 57 days after the first dose of BI 655066, 50% of patients had a PASI 90 response.
“That means from the time you initiate therapy, you can expect half of your patients to get to PASI 90 in less than 2 months time,” Dr. Papp observed.
In contrast, it took 117 days – basically twice as long – for half of the ustekinumab group to achieve PASI 90.
Turning to the durability of response, Dr. Papp noted that it took 169 days for half of patients in the ustekinumab group to lose their PASI 90 response after treatment stopped, compared with 225 days in the BI 655066 90-mg group. That endpoint was never reached in the BI 655066 180-mg group before the study’s end.
Audience reaction to the study results was “wow.”
“Obviously, the data are terrific. Your mother couldn’t have invented better data,” declared Dr. Alice B. Gottlieb, professor of dermatology and dermatologist in chief at Tufts Medical Center, Boston.
She posed a question: Why are the BI 655066 response rates so much higher and longer lasting than with other biologics, including ustekinumab, which inhibits both IL-23 and IL-12?
“I think one key factor in the durability of the response is the pathway,” Dr. Papp replied. “Exactly why blockade of IL-23 p19 leads to this durable clinical response is unclear, but we can posit that we’re depopulating the Th-17 cells. IL-23 is necessary for Th-17 cell survival. So if you block the water, you ain’t going to grow the grass. I’m speculating here, but I think we’re removing that clone of cells, and it’s only once we start to allow them to repopulate that we then see recurrence of the disease.”
BI 655066 is being developed by Boehringer Ingelheim. Dr. Papp serves as an investigator for and consultant to Boehringer Ingelheim and numerous other pharmaceutical companies.
AT THE EADV CONGRESS
Key clinical point: An investigational inhibitor of the p19 subunit of interleukin-23 is generating efficacy results never before seen in psoriasis.
Major finding: The PASI 90 response rate was 81% fully 20 weeks after the third and final dose of BI 655066 at 180 mg, compared with 30% for ustekinumab.
Data source: This prospective, multicenter, double-blind study included 166 patients with moderate to severe plaque psoriasis who were randomized to various doses of ustekinumab or BI 655066 and followed for 36 weeks.
Disclosures: The study was sponsored by Boehringer Ingelheim. The presenter serves as an investigator for and consultant to Boehringer Ingelheim and numerous other pharmaceutical companies.

EADV: Comorbid spondyloarthropathy common in hidradenitis suppurativa
COPENHAGEN – Back pain is surprisingly common in patients with hidradenitis suppurativa, and more than half of affected patients showed MRI evidence of axial spondyloarthropathy, Dr. Sylke Schneider-Burrus reported at the Annual Congress of the European Academy of Dermatology and Venereology.
“Our study demonstrates that back pain and spondyloarthropathy are very common among hidradenitis suppurativa patients and that neither history nor clinical parameters provide any hints for the presence of spondyloarthropathy. Therefore, we strongly suggest that hidradenitis suppurativa patients should be evaluated for spondyloarthropathy and affected patients should be treated systemically with TNF-alpha blockers in order to avoid chronic joint alterations,” said Dr. Schneider-Burrus, a dermatologist at Charite University Hospital in Berlin.

Hidradenitis suppurativa (HS) is a chronic, recurrent, scarring, inflammatory skin disease of the hair follicles. It causes painful, purulent, foul-smelling fistulating sinuses in the axillae, groin, and perianal region.
Because several other chronic inflammatory diseases affecting epithelial tissue have been associated with increased rates of axial spondyloarthropathy – notably, Crohn’s disease, ulcerative colitis, and psoriasis – Dr. Schneider-Burrus and coinvestigators wondered whether that might true of HS as well.
She presented a survey of 100 HS patients. To her surprise, fully 71% indicated they suffer from back pain, with lower back complaints predominating.
Forty-eight HS patients with back pain consented to undergo a pelvic MRI exam. Fifteen of the 48 (32%) showed clear MRI evidence of spondyloarthropathy, including sacroiliac erosions and subchondral sclerosis, while another 12 showed active sacroiliac synovitis and other acute inflammatory changes.
No significant differences were found between HS patients with and without axial spondyloarthropathy in terms of age at onset of HS, disease duration, HS severity as reflected in Sartorius score, age at MRI, body mass index, or smoking status.
Dr. Schneider-Burrus reported serving as a paid investigator for and consultant to Novartis and AbbVie.
COPENHAGEN – Back pain is surprisingly common in patients with hidradenitis suppurativa, and more than half of affected patients showed MRI evidence of axial spondyloarthropathy, Dr. Sylke Schneider-Burrus reported at the Annual Congress of the European Academy of Dermatology and Venereology.
“Our study demonstrates that back pain and spondyloarthropathy are very common among hidradenitis suppurativa patients and that neither history nor clinical parameters provide any hints for the presence of spondyloarthropathy. Therefore, we strongly suggest that hidradenitis suppurativa patients should be evaluated for spondyloarthropathy and affected patients should be treated systemically with TNF-alpha blockers in order to avoid chronic joint alterations,” said Dr. Schneider-Burrus, a dermatologist at Charite University Hospital in Berlin.
Hidradenitis suppurativa (HS) is a chronic, recurrent, scarring, inflammatory skin disease of the hair follicles. It causes painful, purulent, foul-smelling fistulating sinuses in the axillae, groin, and perianal region.
Because several other chronic inflammatory diseases affecting epithelial tissue have been associated with increased rates of axial spondyloarthropathy – notably, Crohn’s disease, ulcerative colitis, and psoriasis – Dr. Schneider-Burrus and coinvestigators wondered whether that might true of HS as well.
She presented a survey of 100 HS patients. To her surprise, fully 71% indicated they suffer from back pain, with lower back complaints predominating.
Forty-eight HS patients with back pain consented to undergo a pelvic MRI exam. Fifteen of the 48 (32%) showed clear MRI evidence of spondyloarthropathy, including sacroiliac erosions and subchondral sclerosis, while another 12 showed active sacroiliac synovitis and other acute inflammatory changes.
No significant differences were found between HS patients with and without axial spondyloarthropathy in terms of age at onset of HS, disease duration, HS severity as reflected in Sartorius score, age at MRI, body mass index, or smoking status.
Dr. Schneider-Burrus reported serving as a paid investigator for and consultant to Novartis and AbbVie.
COPENHAGEN – Back pain is surprisingly common in patients with hidradenitis suppurativa, and more than half of affected patients showed MRI evidence of axial spondyloarthropathy, Dr. Sylke Schneider-Burrus reported at the Annual Congress of the European Academy of Dermatology and Venereology.
“Our study demonstrates that back pain and spondyloarthropathy are very common among hidradenitis suppurativa patients and that neither history nor clinical parameters provide any hints for the presence of spondyloarthropathy. Therefore, we strongly suggest that hidradenitis suppurativa patients should be evaluated for spondyloarthropathy and affected patients should be treated systemically with TNF-alpha blockers in order to avoid chronic joint alterations,” said Dr. Schneider-Burrus, a dermatologist at Charite University Hospital in Berlin.
Hidradenitis suppurativa (HS) is a chronic, recurrent, scarring, inflammatory skin disease of the hair follicles. It causes painful, purulent, foul-smelling fistulating sinuses in the axillae, groin, and perianal region.
Because several other chronic inflammatory diseases affecting epithelial tissue have been associated with increased rates of axial spondyloarthropathy – notably, Crohn’s disease, ulcerative colitis, and psoriasis – Dr. Schneider-Burrus and coinvestigators wondered whether that might true of HS as well.
She presented a survey of 100 HS patients. To her surprise, fully 71% indicated they suffer from back pain, with lower back complaints predominating.
Forty-eight HS patients with back pain consented to undergo a pelvic MRI exam. Fifteen of the 48 (32%) showed clear MRI evidence of spondyloarthropathy, including sacroiliac erosions and subchondral sclerosis, while another 12 showed active sacroiliac synovitis and other acute inflammatory changes.
No significant differences were found between HS patients with and without axial spondyloarthropathy in terms of age at onset of HS, disease duration, HS severity as reflected in Sartorius score, age at MRI, body mass index, or smoking status.
Dr. Schneider-Burrus reported serving as a paid investigator for and consultant to Novartis and AbbVie.
AT THE EADV CONGRESS
Key clinical point: Axial spondyloarthropathy is extremely common in patients with hidradenitis suppurativa.
Major finding: Seventy-one percent of surveyed hidradenitis suppurativa patients reported suffering from back pain, and 56% of affected patients showed MRI evidence of axial spondyloarthropathy.
Data source: A back pain survey of 100 patients with hidradenitis suppurativa along with pelvic MRI exams in the 48 who reported back pain.
Disclosures: The presenter reported serving as a paid investigator for and consultant to Novartis and AbbVie.
