User login
Ingenol mebutate for AKs gets thumbs-up from patients
VANCOUVER – Field therapy for actinic keratoses using topical ingenol mebutate resulted in improved patient-reported outcomes in an observational study, Dr. Thomas L. Diepgen reported at the World Congress of Dermatology.
Topical ingenol mebutate won regulatory approval for the field treatment of actinic keratoses on the strength of four randomized, double-blind placebo-controlled clinical trials, but patients and their physicians need to know how the drug performs in clinical practice. The answer is, quite well, said Dr. Diepgen of the University of Heidelberg (Germany), who presented the results of the observational study emphasizing patient-reported outcomes in 826 patients whose actinic keratoses (AKs) were treated with ingenol mebutate (Picato) in 292 German dermatologists’ offices.

Unlike in randomized clinical trials, where strict eligibility criteria often result in a skewed population of participants, this observational study provided a representative snapshot of German patients seeking AK therapy. Their mean age was 73 years, with a mean 6.2-year duration of AKs and a median baseline of 5 lesions. Eighty percent of patients had previously undergone other types of therapy for the AKs, and 34% of them had a history of nonmelanoma skin cancer.
Participants completed the Skindex-16 quality of life questionnaire at their baseline office visit, and again 8 weeks later. The Skindex-16 doesn’t ask disease-specific questions, but this 16-item questionnaire was employed in the earlier pivotal randomized trials (N. Engl. J. Med. 2012;366:1010-19), and investigators felt they should utilize the same instrument, said Dr. Diepgen.
Scores on the Skindex-16 improved significantly from a mean baseline of 24.3 out of a possible 96 points to 12.1 after 8 weeks.
Similarly, when patients were asked to rate their skin roughness, wrinkling, and/or blotchiness on a 0-3 scale, their mean scores fell from 1.46 at baseline to 0.69 at follow-up. Ninety-eight percent of patients reported no new skin anomalies such as hypopigmentation in the treatment area.
Session cochair Dr. Marc Bourcier of the University of Sherbrooke (Que.) observed that this study underscores that the timing of quality of life assessment makes an enormous difference. Had the assessment taken place at day 4, for example, when ingenol mebutate–induced skin irritation would have been prominent, the results would have been very different. Dr. Diepgen agreed, noting that he and his coinvestigators wanted to evaluate patients’ response to the long-lasting results of the treatment, rather than to the transient experience of the therapy.
The study was sponsored by Leo Pharma. Dr. Diepgen reported having received research grants and speakers fees, and/or serving on advisory boards for Leo and more than a dozen other pharmaceutical companies.
VANCOUVER – Field therapy for actinic keratoses using topical ingenol mebutate resulted in improved patient-reported outcomes in an observational study, Dr. Thomas L. Diepgen reported at the World Congress of Dermatology.
Topical ingenol mebutate won regulatory approval for the field treatment of actinic keratoses on the strength of four randomized, double-blind placebo-controlled clinical trials, but patients and their physicians need to know how the drug performs in clinical practice. The answer is, quite well, said Dr. Diepgen of the University of Heidelberg (Germany), who presented the results of the observational study emphasizing patient-reported outcomes in 826 patients whose actinic keratoses (AKs) were treated with ingenol mebutate (Picato) in 292 German dermatologists’ offices.
Unlike in randomized clinical trials, where strict eligibility criteria often result in a skewed population of participants, this observational study provided a representative snapshot of German patients seeking AK therapy. Their mean age was 73 years, with a mean 6.2-year duration of AKs and a median baseline of 5 lesions. Eighty percent of patients had previously undergone other types of therapy for the AKs, and 34% of them had a history of nonmelanoma skin cancer.
Participants completed the Skindex-16 quality of life questionnaire at their baseline office visit, and again 8 weeks later. The Skindex-16 doesn’t ask disease-specific questions, but this 16-item questionnaire was employed in the earlier pivotal randomized trials (N. Engl. J. Med. 2012;366:1010-19), and investigators felt they should utilize the same instrument, said Dr. Diepgen.
Scores on the Skindex-16 improved significantly from a mean baseline of 24.3 out of a possible 96 points to 12.1 after 8 weeks.
Similarly, when patients were asked to rate their skin roughness, wrinkling, and/or blotchiness on a 0-3 scale, their mean scores fell from 1.46 at baseline to 0.69 at follow-up. Ninety-eight percent of patients reported no new skin anomalies such as hypopigmentation in the treatment area.
Session cochair Dr. Marc Bourcier of the University of Sherbrooke (Que.) observed that this study underscores that the timing of quality of life assessment makes an enormous difference. Had the assessment taken place at day 4, for example, when ingenol mebutate–induced skin irritation would have been prominent, the results would have been very different. Dr. Diepgen agreed, noting that he and his coinvestigators wanted to evaluate patients’ response to the long-lasting results of the treatment, rather than to the transient experience of the therapy.
The study was sponsored by Leo Pharma. Dr. Diepgen reported having received research grants and speakers fees, and/or serving on advisory boards for Leo and more than a dozen other pharmaceutical companies.
VANCOUVER – Field therapy for actinic keratoses using topical ingenol mebutate resulted in improved patient-reported outcomes in an observational study, Dr. Thomas L. Diepgen reported at the World Congress of Dermatology.
Topical ingenol mebutate won regulatory approval for the field treatment of actinic keratoses on the strength of four randomized, double-blind placebo-controlled clinical trials, but patients and their physicians need to know how the drug performs in clinical practice. The answer is, quite well, said Dr. Diepgen of the University of Heidelberg (Germany), who presented the results of the observational study emphasizing patient-reported outcomes in 826 patients whose actinic keratoses (AKs) were treated with ingenol mebutate (Picato) in 292 German dermatologists’ offices.
Unlike in randomized clinical trials, where strict eligibility criteria often result in a skewed population of participants, this observational study provided a representative snapshot of German patients seeking AK therapy. Their mean age was 73 years, with a mean 6.2-year duration of AKs and a median baseline of 5 lesions. Eighty percent of patients had previously undergone other types of therapy for the AKs, and 34% of them had a history of nonmelanoma skin cancer.
Participants completed the Skindex-16 quality of life questionnaire at their baseline office visit, and again 8 weeks later. The Skindex-16 doesn’t ask disease-specific questions, but this 16-item questionnaire was employed in the earlier pivotal randomized trials (N. Engl. J. Med. 2012;366:1010-19), and investigators felt they should utilize the same instrument, said Dr. Diepgen.
Scores on the Skindex-16 improved significantly from a mean baseline of 24.3 out of a possible 96 points to 12.1 after 8 weeks.
Similarly, when patients were asked to rate their skin roughness, wrinkling, and/or blotchiness on a 0-3 scale, their mean scores fell from 1.46 at baseline to 0.69 at follow-up. Ninety-eight percent of patients reported no new skin anomalies such as hypopigmentation in the treatment area.
Session cochair Dr. Marc Bourcier of the University of Sherbrooke (Que.) observed that this study underscores that the timing of quality of life assessment makes an enormous difference. Had the assessment taken place at day 4, for example, when ingenol mebutate–induced skin irritation would have been prominent, the results would have been very different. Dr. Diepgen agreed, noting that he and his coinvestigators wanted to evaluate patients’ response to the long-lasting results of the treatment, rather than to the transient experience of the therapy.
The study was sponsored by Leo Pharma. Dr. Diepgen reported having received research grants and speakers fees, and/or serving on advisory boards for Leo and more than a dozen other pharmaceutical companies.
AT WCD 2015
Key clinical point: Field therapy for actinic keratoses using topical ingenol mebutate resulted in improved patient-reported outcomes.
Major finding: Mean scores on the Skindex-16, which reflects the quality of life impact of a patient’s skin disease, improved significantly from 24.3 pretreatment to 12.1 after 8 weeks.
Data source: A prospective observational study of patient-reported outcomes of ingenol mebutate therapy for actinic keratoses in 826 patients treated in 292 German dermatologists’ offices.
Disclosures: The study was sponsored by Leo Pharma, which markets ingenol mebutate. The presenter reported having received research grants and speakers’ fees, and/or serving on advisory boards for Leo and more than a dozen other pharmaceutical companies.

Simulated daylight PDT advantageous for AKs
VANCOUVER – Indoor simulated daylight photodynamic therapy for actinic keratoses sidesteps the major shortcoming of natural daylight PDT by providing a standardized, dermatologist-controlled light dose that’s not dependent upon the vagaries of weather, season, or outdoor temperature, Dr. Uwe Reinhold reported at the World Congress of Dermatology.
Daylight PDT, in which the photosensitizing agent is activated by natural light, is an increasingly popular concept that originated in Scandinavia but is starting to catch on in the United States. Daylight PDT is less expensive and far less painful than traditional PDT, in which the photosensitizer is activated by a pulsed dye laser or an intense pulsed light device. But on a rainy day or a cold, short, winter day, it can be a problem getting sufficient daylight outdoors to reliably activate the PDT, noted Dr. Reinhold of the Dermatology Center Bonn (Germany) Friedensplatz.
Dr. Reinhold and his colleagues solved that problem by installing a special lamp system on the ceiling of a treatment room in the office. The system enables a dermatologist to simultaneously treat several patients, who receive their 2-hour light dose while seated comfortably in the treatment room reading a book or resting.
Dr. Reinhold presented a retrospective study of 32 patients who underwent simulated daylight PDT (SDL-PDT) in his office. At baseline, the patients had a mean of 5.3 AKs on the scalp and/or face. At follow-up 12 weeks after their second and final SDL-PDT session, they averaged 0.4 AKs. Ninety-three percent of all AKs were cleared, and three-quarters of the patients were completely AK-free.
Traditional PDT is so painful that compliance becomes an issue, Dr. Reinhold noted. In contrast, SDL-PDT, like daylight PDT, is almost pain free. Pain assessment on a 0-10 visual analog scale conducted during the first SDL-PDT session showed mean scores of 0.1, 0.3, and 0.6 at 30, 60, and 90 minutes after illumination began. None of the patients required an analgesic, according to the dermatologist.
The procedure begins with curettage of hyperkeratotic lesions, followed by application of aminolevulinic acid (ALA) gel under occlusion for 30 minutes. Dr. Reinhold uses BF-200 (Ameluz), an ALA manufactured by Biofrontera, a German company, which is popular in Europe but not marketed in the United States. The gel contains 78 mg of ALA per gram. After the 30-minute incubation, the photosensitizer is removed and the special lights are switched on for 2 hours. Protective eye goggles aren’t needed. All patients receive a second treatment session 1 week later.
The lights Dr. Reinhold uses are Indoorlux, marketed by Swiss Red AG. One pair of lights is needed per patient. At a distance of 110-150 cm from the light source, the system produces 15,000-25,000 Lux. The lamps mimic the green and red components of daylight. The combined effective light dose at the wavelengths important in activating protoporphyrin IX so that it can destroy precancerous cells – green/yellow at 570-590 nm and orange/red at 620-640 nm – is 14.3-24.2 J/cm2, depending upon the distance from the light source. That’s comfortably above the 9.4-10.8 J/cm2 other investigators have determined is required for effective natural daylight PDT.
In the United States, however, as in Europe, SDL-PDT is currently an off-label therapy for AK treatment, he noted.
Dr. Reinhold reported serving as a consultant to Biofrontera and receiving speaking fees from the company.
VANCOUVER – Indoor simulated daylight photodynamic therapy for actinic keratoses sidesteps the major shortcoming of natural daylight PDT by providing a standardized, dermatologist-controlled light dose that’s not dependent upon the vagaries of weather, season, or outdoor temperature, Dr. Uwe Reinhold reported at the World Congress of Dermatology.
Daylight PDT, in which the photosensitizing agent is activated by natural light, is an increasingly popular concept that originated in Scandinavia but is starting to catch on in the United States. Daylight PDT is less expensive and far less painful than traditional PDT, in which the photosensitizer is activated by a pulsed dye laser or an intense pulsed light device. But on a rainy day or a cold, short, winter day, it can be a problem getting sufficient daylight outdoors to reliably activate the PDT, noted Dr. Reinhold of the Dermatology Center Bonn (Germany) Friedensplatz.
Dr. Reinhold and his colleagues solved that problem by installing a special lamp system on the ceiling of a treatment room in the office. The system enables a dermatologist to simultaneously treat several patients, who receive their 2-hour light dose while seated comfortably in the treatment room reading a book or resting.
Dr. Reinhold presented a retrospective study of 32 patients who underwent simulated daylight PDT (SDL-PDT) in his office. At baseline, the patients had a mean of 5.3 AKs on the scalp and/or face. At follow-up 12 weeks after their second and final SDL-PDT session, they averaged 0.4 AKs. Ninety-three percent of all AKs were cleared, and three-quarters of the patients were completely AK-free.
Traditional PDT is so painful that compliance becomes an issue, Dr. Reinhold noted. In contrast, SDL-PDT, like daylight PDT, is almost pain free. Pain assessment on a 0-10 visual analog scale conducted during the first SDL-PDT session showed mean scores of 0.1, 0.3, and 0.6 at 30, 60, and 90 minutes after illumination began. None of the patients required an analgesic, according to the dermatologist.
The procedure begins with curettage of hyperkeratotic lesions, followed by application of aminolevulinic acid (ALA) gel under occlusion for 30 minutes. Dr. Reinhold uses BF-200 (Ameluz), an ALA manufactured by Biofrontera, a German company, which is popular in Europe but not marketed in the United States. The gel contains 78 mg of ALA per gram. After the 30-minute incubation, the photosensitizer is removed and the special lights are switched on for 2 hours. Protective eye goggles aren’t needed. All patients receive a second treatment session 1 week later.
The lights Dr. Reinhold uses are Indoorlux, marketed by Swiss Red AG. One pair of lights is needed per patient. At a distance of 110-150 cm from the light source, the system produces 15,000-25,000 Lux. The lamps mimic the green and red components of daylight. The combined effective light dose at the wavelengths important in activating protoporphyrin IX so that it can destroy precancerous cells – green/yellow at 570-590 nm and orange/red at 620-640 nm – is 14.3-24.2 J/cm2, depending upon the distance from the light source. That’s comfortably above the 9.4-10.8 J/cm2 other investigators have determined is required for effective natural daylight PDT.
In the United States, however, as in Europe, SDL-PDT is currently an off-label therapy for AK treatment, he noted.
Dr. Reinhold reported serving as a consultant to Biofrontera and receiving speaking fees from the company.
VANCOUVER – Indoor simulated daylight photodynamic therapy for actinic keratoses sidesteps the major shortcoming of natural daylight PDT by providing a standardized, dermatologist-controlled light dose that’s not dependent upon the vagaries of weather, season, or outdoor temperature, Dr. Uwe Reinhold reported at the World Congress of Dermatology.
Daylight PDT, in which the photosensitizing agent is activated by natural light, is an increasingly popular concept that originated in Scandinavia but is starting to catch on in the United States. Daylight PDT is less expensive and far less painful than traditional PDT, in which the photosensitizer is activated by a pulsed dye laser or an intense pulsed light device. But on a rainy day or a cold, short, winter day, it can be a problem getting sufficient daylight outdoors to reliably activate the PDT, noted Dr. Reinhold of the Dermatology Center Bonn (Germany) Friedensplatz.
Dr. Reinhold and his colleagues solved that problem by installing a special lamp system on the ceiling of a treatment room in the office. The system enables a dermatologist to simultaneously treat several patients, who receive their 2-hour light dose while seated comfortably in the treatment room reading a book or resting.
Dr. Reinhold presented a retrospective study of 32 patients who underwent simulated daylight PDT (SDL-PDT) in his office. At baseline, the patients had a mean of 5.3 AKs on the scalp and/or face. At follow-up 12 weeks after their second and final SDL-PDT session, they averaged 0.4 AKs. Ninety-three percent of all AKs were cleared, and three-quarters of the patients were completely AK-free.
Traditional PDT is so painful that compliance becomes an issue, Dr. Reinhold noted. In contrast, SDL-PDT, like daylight PDT, is almost pain free. Pain assessment on a 0-10 visual analog scale conducted during the first SDL-PDT session showed mean scores of 0.1, 0.3, and 0.6 at 30, 60, and 90 minutes after illumination began. None of the patients required an analgesic, according to the dermatologist.
The procedure begins with curettage of hyperkeratotic lesions, followed by application of aminolevulinic acid (ALA) gel under occlusion for 30 minutes. Dr. Reinhold uses BF-200 (Ameluz), an ALA manufactured by Biofrontera, a German company, which is popular in Europe but not marketed in the United States. The gel contains 78 mg of ALA per gram. After the 30-minute incubation, the photosensitizer is removed and the special lights are switched on for 2 hours. Protective eye goggles aren’t needed. All patients receive a second treatment session 1 week later.
The lights Dr. Reinhold uses are Indoorlux, marketed by Swiss Red AG. One pair of lights is needed per patient. At a distance of 110-150 cm from the light source, the system produces 15,000-25,000 Lux. The lamps mimic the green and red components of daylight. The combined effective light dose at the wavelengths important in activating protoporphyrin IX so that it can destroy precancerous cells – green/yellow at 570-590 nm and orange/red at 620-640 nm – is 14.3-24.2 J/cm2, depending upon the distance from the light source. That’s comfortably above the 9.4-10.8 J/cm2 other investigators have determined is required for effective natural daylight PDT.
In the United States, however, as in Europe, SDL-PDT is currently an off-label therapy for AK treatment, he noted.
Dr. Reinhold reported serving as a consultant to Biofrontera and receiving speaking fees from the company.
AT WCD 2015
Key clinical point: A field containing multiple actinic keratoses can be treated virtually painlessly using lamps that simulate daylight to activate photodynamic therapy.
Major finding: 3 months after simulated daylight PDT, the mean number of AKs in treated patients was reduced from 5.3 at baseline to 0.4.
Data source: This was a retrospective study including 32 patients whose actinic keratoses was treated using simulated daylight PDT.
Disclosures: The study was supported by Biofrontera. The presenter reported serving as a consultant to and receiving speaking fees from the company.
WCD: Tofacitinib’s benefits for psoriasis persist for 2 years
VANCOUVER – Oral tofacitinib for adults with moderate to severe plaque psoriasis demonstrated sustained efficacy and a “generally manageable” safety profile throughout 2 years of follow-up in a large, multicenter ongoing long-term extension study, Dr. Matthias Augustin reported at the World Congress of Dermatology.
One month after enrolling in the phase III, open-label trial (known as OPT Extend), and going on the investigational oral Janus kinase inhibitor at 10 mg b.i.d., 56.2% of the 2,847 participants showed a PASI 75 response – that is, at least a 75% reduction from their baseline Psoriasis Area and Severity Index score. After 2 years in the OPT Extend study, 64.5% of the 1,912 patients remaining in the study were PASI 75 responders.

Similarly, 56.3% of patients went from moderate or severe disease at baseline to “clear” or “almost clear” by Physician Global Assessment (PGA) at 1 month, and 53.9% of participants had a PGA score of 0 or 1 at 24 months, added Dr. Augustin, professor of dermatology at the University of Hamburg-Eppendorf and director of the German Center for Health Services Research in Dermatology and Nursing.
Psoriasis patients who had participated in any of four earlier phase III or two phase II randomized, controlled trials of tofacitinib were eligible to participate in OPT Extend. Participants were placed on tofacitinib at 10 mg b.i.d. for the first 2 months of the open-label study; then investigators adjusted their dose individually – either 5 or 10 mg b.i.d. – at each follow-up visit based on efficacy and side effects. For purposes of OPT Extend, patients’ baseline PASI and PGA scores were defined as the values recorded on the day they were randomized in the earlier round of trials.
Through 2 years of follow-up in this interim analysis of OPT Extend, less than 10% of subjects discontinued tofacitinib because of adverse events. The adverse events were the same as the ones that arose in the earlier, briefer randomized trials, the longest of which lasted 1 year. No signs of cumulative organ toxicity were evident during the additional follow-up.
Slightly more than 4% of all adverse events were categorized as severe, while 62% were mild. The most frequent adverse events were nasopharyngitis in 16% of patients, increased creatinine phosphokinase in 10%, and upper respiratory tract infections in 7%. Serious infections requiring systemic antibiotics or hospitalization occurred in 1.8% of patients on tofacitinib for 2 years, the most common of which were pneumonia, herpes zoster, and urinary tract infection. Herpes zoster occurred in 3.5% of participants, with most cases being mild or moderate. Malignancies other than nonmelanoma skin cancers occurred in 1.2% of subjects.
In terms of laboratory findings of interest, patients’ LDL/HDL ratio remained stable throughout 24 months on tofacitinib. Nor were there any clinically meaningful changes in average levels of other laboratory parameters, including hemoglobin, creatinine phosphokinase, lymphocytes, and neutrophils. A lymphocyte count below 500/mm3 occurred in 0.4% of patients at some point; however, it was unrelated to duration of exposure to tofacitinib and wasn’t linked to an increased infection rate.
Dr. Augustin said an unmet need exists for effective and nontoxic oral medications for moderate to severe psoriasis, because many patients would prefer not to take an injectable biologic. Tofacitinib is aimed at filling that need, and serving as an easier, less toxic initial therapy. However, it is essential that an optimal oral agent have a favorable safety profile because psoriasis is a lifelong disease with an average lifetime duration in excess of 40 years in adults and more than 60 years in affected children, he said.
Pfizer, which is developing the drug, has filed for marketing approval for tofacitinib for treatment of adults with moderate to severe chronic plaque psoriasis who are candidates for systemic therapy or phototherapy. The Food and Drug Administration has announced its intent to issue a ruling on the application in October 2015.
The OPT Expect study is funded by Pfizer. Dr. Augustin is an adviser to and/or a recipient of research grants from Pfizer and more than a dozen other medical companies.
VANCOUVER – Oral tofacitinib for adults with moderate to severe plaque psoriasis demonstrated sustained efficacy and a “generally manageable” safety profile throughout 2 years of follow-up in a large, multicenter ongoing long-term extension study, Dr. Matthias Augustin reported at the World Congress of Dermatology.
One month after enrolling in the phase III, open-label trial (known as OPT Extend), and going on the investigational oral Janus kinase inhibitor at 10 mg b.i.d., 56.2% of the 2,847 participants showed a PASI 75 response – that is, at least a 75% reduction from their baseline Psoriasis Area and Severity Index score. After 2 years in the OPT Extend study, 64.5% of the 1,912 patients remaining in the study were PASI 75 responders.
Similarly, 56.3% of patients went from moderate or severe disease at baseline to “clear” or “almost clear” by Physician Global Assessment (PGA) at 1 month, and 53.9% of participants had a PGA score of 0 or 1 at 24 months, added Dr. Augustin, professor of dermatology at the University of Hamburg-Eppendorf and director of the German Center for Health Services Research in Dermatology and Nursing.
Psoriasis patients who had participated in any of four earlier phase III or two phase II randomized, controlled trials of tofacitinib were eligible to participate in OPT Extend. Participants were placed on tofacitinib at 10 mg b.i.d. for the first 2 months of the open-label study; then investigators adjusted their dose individually – either 5 or 10 mg b.i.d. – at each follow-up visit based on efficacy and side effects. For purposes of OPT Extend, patients’ baseline PASI and PGA scores were defined as the values recorded on the day they were randomized in the earlier round of trials.
Through 2 years of follow-up in this interim analysis of OPT Extend, less than 10% of subjects discontinued tofacitinib because of adverse events. The adverse events were the same as the ones that arose in the earlier, briefer randomized trials, the longest of which lasted 1 year. No signs of cumulative organ toxicity were evident during the additional follow-up.
Slightly more than 4% of all adverse events were categorized as severe, while 62% were mild. The most frequent adverse events were nasopharyngitis in 16% of patients, increased creatinine phosphokinase in 10%, and upper respiratory tract infections in 7%. Serious infections requiring systemic antibiotics or hospitalization occurred in 1.8% of patients on tofacitinib for 2 years, the most common of which were pneumonia, herpes zoster, and urinary tract infection. Herpes zoster occurred in 3.5% of participants, with most cases being mild or moderate. Malignancies other than nonmelanoma skin cancers occurred in 1.2% of subjects.
In terms of laboratory findings of interest, patients’ LDL/HDL ratio remained stable throughout 24 months on tofacitinib. Nor were there any clinically meaningful changes in average levels of other laboratory parameters, including hemoglobin, creatinine phosphokinase, lymphocytes, and neutrophils. A lymphocyte count below 500/mm3 occurred in 0.4% of patients at some point; however, it was unrelated to duration of exposure to tofacitinib and wasn’t linked to an increased infection rate.
Dr. Augustin said an unmet need exists for effective and nontoxic oral medications for moderate to severe psoriasis, because many patients would prefer not to take an injectable biologic. Tofacitinib is aimed at filling that need, and serving as an easier, less toxic initial therapy. However, it is essential that an optimal oral agent have a favorable safety profile because psoriasis is a lifelong disease with an average lifetime duration in excess of 40 years in adults and more than 60 years in affected children, he said.
Pfizer, which is developing the drug, has filed for marketing approval for tofacitinib for treatment of adults with moderate to severe chronic plaque psoriasis who are candidates for systemic therapy or phototherapy. The Food and Drug Administration has announced its intent to issue a ruling on the application in October 2015.
The OPT Expect study is funded by Pfizer. Dr. Augustin is an adviser to and/or a recipient of research grants from Pfizer and more than a dozen other medical companies.
VANCOUVER – Oral tofacitinib for adults with moderate to severe plaque psoriasis demonstrated sustained efficacy and a “generally manageable” safety profile throughout 2 years of follow-up in a large, multicenter ongoing long-term extension study, Dr. Matthias Augustin reported at the World Congress of Dermatology.
One month after enrolling in the phase III, open-label trial (known as OPT Extend), and going on the investigational oral Janus kinase inhibitor at 10 mg b.i.d., 56.2% of the 2,847 participants showed a PASI 75 response – that is, at least a 75% reduction from their baseline Psoriasis Area and Severity Index score. After 2 years in the OPT Extend study, 64.5% of the 1,912 patients remaining in the study were PASI 75 responders.
Similarly, 56.3% of patients went from moderate or severe disease at baseline to “clear” or “almost clear” by Physician Global Assessment (PGA) at 1 month, and 53.9% of participants had a PGA score of 0 or 1 at 24 months, added Dr. Augustin, professor of dermatology at the University of Hamburg-Eppendorf and director of the German Center for Health Services Research in Dermatology and Nursing.
Psoriasis patients who had participated in any of four earlier phase III or two phase II randomized, controlled trials of tofacitinib were eligible to participate in OPT Extend. Participants were placed on tofacitinib at 10 mg b.i.d. for the first 2 months of the open-label study; then investigators adjusted their dose individually – either 5 or 10 mg b.i.d. – at each follow-up visit based on efficacy and side effects. For purposes of OPT Extend, patients’ baseline PASI and PGA scores were defined as the values recorded on the day they were randomized in the earlier round of trials.
Through 2 years of follow-up in this interim analysis of OPT Extend, less than 10% of subjects discontinued tofacitinib because of adverse events. The adverse events were the same as the ones that arose in the earlier, briefer randomized trials, the longest of which lasted 1 year. No signs of cumulative organ toxicity were evident during the additional follow-up.
Slightly more than 4% of all adverse events were categorized as severe, while 62% were mild. The most frequent adverse events were nasopharyngitis in 16% of patients, increased creatinine phosphokinase in 10%, and upper respiratory tract infections in 7%. Serious infections requiring systemic antibiotics or hospitalization occurred in 1.8% of patients on tofacitinib for 2 years, the most common of which were pneumonia, herpes zoster, and urinary tract infection. Herpes zoster occurred in 3.5% of participants, with most cases being mild or moderate. Malignancies other than nonmelanoma skin cancers occurred in 1.2% of subjects.
In terms of laboratory findings of interest, patients’ LDL/HDL ratio remained stable throughout 24 months on tofacitinib. Nor were there any clinically meaningful changes in average levels of other laboratory parameters, including hemoglobin, creatinine phosphokinase, lymphocytes, and neutrophils. A lymphocyte count below 500/mm3 occurred in 0.4% of patients at some point; however, it was unrelated to duration of exposure to tofacitinib and wasn’t linked to an increased infection rate.
Dr. Augustin said an unmet need exists for effective and nontoxic oral medications for moderate to severe psoriasis, because many patients would prefer not to take an injectable biologic. Tofacitinib is aimed at filling that need, and serving as an easier, less toxic initial therapy. However, it is essential that an optimal oral agent have a favorable safety profile because psoriasis is a lifelong disease with an average lifetime duration in excess of 40 years in adults and more than 60 years in affected children, he said.
Pfizer, which is developing the drug, has filed for marketing approval for tofacitinib for treatment of adults with moderate to severe chronic plaque psoriasis who are candidates for systemic therapy or phototherapy. The Food and Drug Administration has announced its intent to issue a ruling on the application in October 2015.
The OPT Expect study is funded by Pfizer. Dr. Augustin is an adviser to and/or a recipient of research grants from Pfizer and more than a dozen other medical companies.
AT WCD 2015
Key clinical point: Oral tofacitinib showed sustained efficacy for moderate to severe plaque psoriasis at 24 months in a large ongoing phase III study.
Major finding: At month 1 of the OPT Extend study, 56.2% of patients on tofacitinib had a PASI 75 response; at month 24, the PASI 75 rate was 64.5%.
Data source: The OPT Extend study is an ongoing, phase III multicenter open-label study of long-term therapy with oral tofacitinib at 5 or 10 mg b.i.d. in 2,847 patients with moderate to severe psoriasis.
Disclosures: The study is funded by Pfizer. The presenter is an adviser to and recipient of research grants from that company as well as others.

IHCC: Childhood migraine hurts school performance
VALENCIA, SPAIN – Pediatric migraine often has a markedly adverse effect upon school performance and psychosocial adjustment, according to a landmark series of Brazilian studies.
“This is a series of articles that has certainly changed my own practice,” Dr. Kenneth J. Mack said at the International Headache Congress.
The Brazilian investigators are following a nationally representative sample of 5,671 Brazilian 5- to 12-year-olds. They found the prevalence of episodic migraine was 9%, while 17.6% of the youths were categorized as having probable migraine, 12.8% had periodic tensiontype headaches, and 0.6% had chronic migraine as defined by migraine headaches on at least 15 days per month, a rate Dr. Mack said was disturbingly high in such a young population.

In their most recent analysis, the Brazilian investigators reported that the children with episodic or chronic migraine had significantly higher rates of abnormal scores on various measures of psychosocial adjustment. They also had increased rates of attention-deficit/hyperactivity disorder, dyslexia, and other learning disabilities (Headache 2015;55 [suppl. 1]:39-50).
Previously, the investigators reported that the children with migraine were at significantly elevated risk of impaired school performance, as assessed by their teachers as well as on standardized tests of academic competencies. The more frequent, severe, and/or longer-lasting the migraines, the poorer the school performance (Neurology 2012;79:1881-8).
“I’ve always asked about school problems, but since these studies I’ve spent much more time delving into school problems in younger kids with chronic headaches. And I’m surprised by how often they really do have school problems – how often they need extra support and help at school,” said Dr. Mack, professor of neurology and pediatrics at the Mayo Clinic in Rochester, Minn.
Another recent research focus in the field of pediatric headache has been its seasonality, which appears to be strongly tied to the school year, he added at the meeting sponsored by the International Headache Society and the American Headache Society.
One good way to look at how severe headaches are and when they occur is by examining pediatric emergency department visits. In a single-center study of 2,731 visits to a Pittsburgh pediatric emergency department by children aged 4 years and older with a chief complaint of headache, there was a clear peak incidence in September and October – the start of the school year – and a nadir in May and June, when school lets out for the summer. This applied to headaches of all types and to patients of all ages and races and to both sexes (Pediatr. Emerg. Care 2014;30:174-6).
This report was soon followed by a nationwide study of ED visits recorded in the U.S. National Hospital Ambulatory Medical Care Survey. The investigators concluded that 5- to 18-year-olds in the United States collectively make an estimated 250,000 ED visits annually related to various types of primary headache. Indeed, headache accounted for 2.1% of all ED visits in this age group during the study years. Looking at rates on a month-by-month basis, the investigators found that ED visits for headache were highest in September (odds ratio, 1.64) and January (OR, 1.92), when children and adolescents start the academic year and return from winter break, respectively, and lowest in April (OR, 0.42) (Cephalalgia 2014;34:473-8).
In an effort to nail down the apparent link between seeking care for a headache and the school year, Dr. Mack and a coinvestigator, in a soon-to-be-published study, looked retrospectively at 103 pediatric patients diagnosed with new daily persistent headache. They focused on this particular type of headache because its onset can be traced to a particular day. Twenty-two percent of patients presented in September and another 18% in January, the school-start months. And just 1 of 103 patients presented in May or June, Dr. Mack reported.
He said he had no relevant financial conflicts.
VALENCIA, SPAIN – Pediatric migraine often has a markedly adverse effect upon school performance and psychosocial adjustment, according to a landmark series of Brazilian studies.
“This is a series of articles that has certainly changed my own practice,” Dr. Kenneth J. Mack said at the International Headache Congress.
The Brazilian investigators are following a nationally representative sample of 5,671 Brazilian 5- to 12-year-olds. They found the prevalence of episodic migraine was 9%, while 17.6% of the youths were categorized as having probable migraine, 12.8% had periodic tensiontype headaches, and 0.6% had chronic migraine as defined by migraine headaches on at least 15 days per month, a rate Dr. Mack said was disturbingly high in such a young population.
In their most recent analysis, the Brazilian investigators reported that the children with episodic or chronic migraine had significantly higher rates of abnormal scores on various measures of psychosocial adjustment. They also had increased rates of attention-deficit/hyperactivity disorder, dyslexia, and other learning disabilities (Headache 2015;55 [suppl. 1]:39-50).
Previously, the investigators reported that the children with migraine were at significantly elevated risk of impaired school performance, as assessed by their teachers as well as on standardized tests of academic competencies. The more frequent, severe, and/or longer-lasting the migraines, the poorer the school performance (Neurology 2012;79:1881-8).
“I’ve always asked about school problems, but since these studies I’ve spent much more time delving into school problems in younger kids with chronic headaches. And I’m surprised by how often they really do have school problems – how often they need extra support and help at school,” said Dr. Mack, professor of neurology and pediatrics at the Mayo Clinic in Rochester, Minn.
Another recent research focus in the field of pediatric headache has been its seasonality, which appears to be strongly tied to the school year, he added at the meeting sponsored by the International Headache Society and the American Headache Society.
One good way to look at how severe headaches are and when they occur is by examining pediatric emergency department visits. In a single-center study of 2,731 visits to a Pittsburgh pediatric emergency department by children aged 4 years and older with a chief complaint of headache, there was a clear peak incidence in September and October – the start of the school year – and a nadir in May and June, when school lets out for the summer. This applied to headaches of all types and to patients of all ages and races and to both sexes (Pediatr. Emerg. Care 2014;30:174-6).
This report was soon followed by a nationwide study of ED visits recorded in the U.S. National Hospital Ambulatory Medical Care Survey. The investigators concluded that 5- to 18-year-olds in the United States collectively make an estimated 250,000 ED visits annually related to various types of primary headache. Indeed, headache accounted for 2.1% of all ED visits in this age group during the study years. Looking at rates on a month-by-month basis, the investigators found that ED visits for headache were highest in September (odds ratio, 1.64) and January (OR, 1.92), when children and adolescents start the academic year and return from winter break, respectively, and lowest in April (OR, 0.42) (Cephalalgia 2014;34:473-8).
In an effort to nail down the apparent link between seeking care for a headache and the school year, Dr. Mack and a coinvestigator, in a soon-to-be-published study, looked retrospectively at 103 pediatric patients diagnosed with new daily persistent headache. They focused on this particular type of headache because its onset can be traced to a particular day. Twenty-two percent of patients presented in September and another 18% in January, the school-start months. And just 1 of 103 patients presented in May or June, Dr. Mack reported.
He said he had no relevant financial conflicts.
VALENCIA, SPAIN – Pediatric migraine often has a markedly adverse effect upon school performance and psychosocial adjustment, according to a landmark series of Brazilian studies.
“This is a series of articles that has certainly changed my own practice,” Dr. Kenneth J. Mack said at the International Headache Congress.
The Brazilian investigators are following a nationally representative sample of 5,671 Brazilian 5- to 12-year-olds. They found the prevalence of episodic migraine was 9%, while 17.6% of the youths were categorized as having probable migraine, 12.8% had periodic tensiontype headaches, and 0.6% had chronic migraine as defined by migraine headaches on at least 15 days per month, a rate Dr. Mack said was disturbingly high in such a young population.
In their most recent analysis, the Brazilian investigators reported that the children with episodic or chronic migraine had significantly higher rates of abnormal scores on various measures of psychosocial adjustment. They also had increased rates of attention-deficit/hyperactivity disorder, dyslexia, and other learning disabilities (Headache 2015;55 [suppl. 1]:39-50).
Previously, the investigators reported that the children with migraine were at significantly elevated risk of impaired school performance, as assessed by their teachers as well as on standardized tests of academic competencies. The more frequent, severe, and/or longer-lasting the migraines, the poorer the school performance (Neurology 2012;79:1881-8).
“I’ve always asked about school problems, but since these studies I’ve spent much more time delving into school problems in younger kids with chronic headaches. And I’m surprised by how often they really do have school problems – how often they need extra support and help at school,” said Dr. Mack, professor of neurology and pediatrics at the Mayo Clinic in Rochester, Minn.
Another recent research focus in the field of pediatric headache has been its seasonality, which appears to be strongly tied to the school year, he added at the meeting sponsored by the International Headache Society and the American Headache Society.
One good way to look at how severe headaches are and when they occur is by examining pediatric emergency department visits. In a single-center study of 2,731 visits to a Pittsburgh pediatric emergency department by children aged 4 years and older with a chief complaint of headache, there was a clear peak incidence in September and October – the start of the school year – and a nadir in May and June, when school lets out for the summer. This applied to headaches of all types and to patients of all ages and races and to both sexes (Pediatr. Emerg. Care 2014;30:174-6).
This report was soon followed by a nationwide study of ED visits recorded in the U.S. National Hospital Ambulatory Medical Care Survey. The investigators concluded that 5- to 18-year-olds in the United States collectively make an estimated 250,000 ED visits annually related to various types of primary headache. Indeed, headache accounted for 2.1% of all ED visits in this age group during the study years. Looking at rates on a month-by-month basis, the investigators found that ED visits for headache were highest in September (odds ratio, 1.64) and January (OR, 1.92), when children and adolescents start the academic year and return from winter break, respectively, and lowest in April (OR, 0.42) (Cephalalgia 2014;34:473-8).
In an effort to nail down the apparent link between seeking care for a headache and the school year, Dr. Mack and a coinvestigator, in a soon-to-be-published study, looked retrospectively at 103 pediatric patients diagnosed with new daily persistent headache. They focused on this particular type of headache because its onset can be traced to a particular day. Twenty-two percent of patients presented in September and another 18% in January, the school-start months. And just 1 of 103 patients presented in May or June, Dr. Mack reported.
He said he had no relevant financial conflicts.
EXPERT ANALYSIS FROM IHC 2015

AAS: New approach underway to ID teens at high suicide risk
ATLANTA – The potential utility of the medical emergency department as a key venue in which to conduct youth suicide screening is now well established, and a federally funded effort to develop and validate an optimal screening tool designed specifically for this purpose is underway.
Suicide is the second-leading cause of death among 12- to 17-year-olds in the United States. Routine screening for suicide risk in medical EDs would solve a major challenge in preventing these deaths: namely, that adolescents – especially older male teens, who are at highest suicide risk – seldom seek mental health care. Yet, roughly one-third of adolescents visit the ED per year, Cheryl A. King, Ph.D., explained at the annual conference of the American Association of Suicidology.

“They come in for exacerbation of common medical illnesses and sports injuries, but they’re also coming in for things like alcohol poisoning, car accidents, and fistfights,” noted Dr. King, professor of psychology and psychiatry at the University of Michigan, Ann Arbor.
These ED visits offer a particularly good opportunity to screen for suicide risk in adolescent males, who account for 80% of all youth suicides in the United States. Yet, because they are so much less likely than teen girls to share thoughts of suicide, males comprise only 25%-35% of teens hospitalized for suicide risk, she continued.
Dr. King has been a leader in the exploration of the medical ED as a venue for routine screening for high risk for suicidal behavior in adolescents. She and others have shown that such screening is productive. But there’s a problem: ED personnel already feel plenty busy and time crushed without taking on a huge new responsibility. And screening instruments designed to miss few teens at risk will have a high false-positive rate. “I don’t want to set the screen-positive threshold so low that 40% of all youth who come in with a broken thumb that was jammed in a car door screen positive, because then what will the ED physicians say to me? They may not even want to talk to me,” she said.
In an early proof-of-concept study, she and her colleagues developed and tested a high-bar screen, one in which a positive result required self-reported serious suicidal ideation within the past 2 weeks, a suicide attempt within the past month, or co-occurring depression plus alcohol/substance misuse. Applying the screen to 298 adolescents who presented to an ED, the investigators found a 16% screen-positive rate (Acad. Emerg. Med. 2009;16:1234-41).
The investigators went on to survey pools of adolescents and parents as to the acceptability of routine ED screening for suicide risk, and got a thumbs-up from both groups (Ped. Emerg. Care 2012;28:626-32).
Next, Dr. King and her colleagues established that their screening tool actually predicted a high near-term risk for suicidal behavior. In a separate study of 81 adolescents aged 14-19 who were screen positive, 15 (18.5%) engaged in some type of suicidal behavior in the 2 months following their ED visit (J. Child Adolesc. Psychopharmacol. 2015;25:100-8).
Another promising screening tool for use in youths presenting to EDs is the ASQ (Ask Suicide-Screening Questions), a four-question test developed at the National Institute of Mental Health (NIMH). It assesses current thoughts of wishing to die, being better off dead, suicidal ideation, and past attempts (Arch. Pediatr. Adolesc. Med. 2012;166:1170-16); however, the predictive validity of this scale in terms of future suicide attempts has yet to be validated, according to Dr. King.
The clinician-administered Columbia–Suicide Severity Rating Scale has demonstrated predictive validity, but it is likely to prove too comprehensive and time consuming for routine use as a screening tool in the ED. However, several specific items incorporated in the scale – namely, the duration element of the intensity scale score and a lifetime history of nonsuicidal self-injury – showed predictive validity as a streamlined screening tool in a study by Dr. King and her colleagues (Pediatr. Emerg. Care 2015;31:88-94).
The most recent development in ED screening is the National Institute of Mental Health–funded ED-STARS (Emergency Department Screening for Teens at Risk for Suicide) project. Dr. King is a codirector of this multicenter effort to develop the optimal suicide risk screening tool for adolescents in the ED, along with a triage algorithm ED physicians can follow.
The tool being developed is a computerized adaptive screen. Rather than answering a fixed list of screening questions, patients will take a very brief, personalized test administered via computer. The adaptive aspect involves an algorithm in which the next question posed depends upon the individual’s response to the question before. This screening instrument is being paired with the Implicit Association Test developed by investigators at Harvard University, Cambridge, Mass.
ED-STARS is being carried out by the Pediatric Emergency Care Applied Research Network. If the project proves successful, the plan is to extend the screening tool into school settings.
Dr. King’s research efforts are funded by the NIMH. She reported having no relevant financial conflicts.
ATLANTA – The potential utility of the medical emergency department as a key venue in which to conduct youth suicide screening is now well established, and a federally funded effort to develop and validate an optimal screening tool designed specifically for this purpose is underway.
Suicide is the second-leading cause of death among 12- to 17-year-olds in the United States. Routine screening for suicide risk in medical EDs would solve a major challenge in preventing these deaths: namely, that adolescents – especially older male teens, who are at highest suicide risk – seldom seek mental health care. Yet, roughly one-third of adolescents visit the ED per year, Cheryl A. King, Ph.D., explained at the annual conference of the American Association of Suicidology.
“They come in for exacerbation of common medical illnesses and sports injuries, but they’re also coming in for things like alcohol poisoning, car accidents, and fistfights,” noted Dr. King, professor of psychology and psychiatry at the University of Michigan, Ann Arbor.
These ED visits offer a particularly good opportunity to screen for suicide risk in adolescent males, who account for 80% of all youth suicides in the United States. Yet, because they are so much less likely than teen girls to share thoughts of suicide, males comprise only 25%-35% of teens hospitalized for suicide risk, she continued.
Dr. King has been a leader in the exploration of the medical ED as a venue for routine screening for high risk for suicidal behavior in adolescents. She and others have shown that such screening is productive. But there’s a problem: ED personnel already feel plenty busy and time crushed without taking on a huge new responsibility. And screening instruments designed to miss few teens at risk will have a high false-positive rate. “I don’t want to set the screen-positive threshold so low that 40% of all youth who come in with a broken thumb that was jammed in a car door screen positive, because then what will the ED physicians say to me? They may not even want to talk to me,” she said.
In an early proof-of-concept study, she and her colleagues developed and tested a high-bar screen, one in which a positive result required self-reported serious suicidal ideation within the past 2 weeks, a suicide attempt within the past month, or co-occurring depression plus alcohol/substance misuse. Applying the screen to 298 adolescents who presented to an ED, the investigators found a 16% screen-positive rate (Acad. Emerg. Med. 2009;16:1234-41).
The investigators went on to survey pools of adolescents and parents as to the acceptability of routine ED screening for suicide risk, and got a thumbs-up from both groups (Ped. Emerg. Care 2012;28:626-32).
Next, Dr. King and her colleagues established that their screening tool actually predicted a high near-term risk for suicidal behavior. In a separate study of 81 adolescents aged 14-19 who were screen positive, 15 (18.5%) engaged in some type of suicidal behavior in the 2 months following their ED visit (J. Child Adolesc. Psychopharmacol. 2015;25:100-8).
Another promising screening tool for use in youths presenting to EDs is the ASQ (Ask Suicide-Screening Questions), a four-question test developed at the National Institute of Mental Health (NIMH). It assesses current thoughts of wishing to die, being better off dead, suicidal ideation, and past attempts (Arch. Pediatr. Adolesc. Med. 2012;166:1170-16); however, the predictive validity of this scale in terms of future suicide attempts has yet to be validated, according to Dr. King.
The clinician-administered Columbia–Suicide Severity Rating Scale has demonstrated predictive validity, but it is likely to prove too comprehensive and time consuming for routine use as a screening tool in the ED. However, several specific items incorporated in the scale – namely, the duration element of the intensity scale score and a lifetime history of nonsuicidal self-injury – showed predictive validity as a streamlined screening tool in a study by Dr. King and her colleagues (Pediatr. Emerg. Care 2015;31:88-94).
The most recent development in ED screening is the National Institute of Mental Health–funded ED-STARS (Emergency Department Screening for Teens at Risk for Suicide) project. Dr. King is a codirector of this multicenter effort to develop the optimal suicide risk screening tool for adolescents in the ED, along with a triage algorithm ED physicians can follow.
The tool being developed is a computerized adaptive screen. Rather than answering a fixed list of screening questions, patients will take a very brief, personalized test administered via computer. The adaptive aspect involves an algorithm in which the next question posed depends upon the individual’s response to the question before. This screening instrument is being paired with the Implicit Association Test developed by investigators at Harvard University, Cambridge, Mass.
ED-STARS is being carried out by the Pediatric Emergency Care Applied Research Network. If the project proves successful, the plan is to extend the screening tool into school settings.
Dr. King’s research efforts are funded by the NIMH. She reported having no relevant financial conflicts.
ATLANTA – The potential utility of the medical emergency department as a key venue in which to conduct youth suicide screening is now well established, and a federally funded effort to develop and validate an optimal screening tool designed specifically for this purpose is underway.
Suicide is the second-leading cause of death among 12- to 17-year-olds in the United States. Routine screening for suicide risk in medical EDs would solve a major challenge in preventing these deaths: namely, that adolescents – especially older male teens, who are at highest suicide risk – seldom seek mental health care. Yet, roughly one-third of adolescents visit the ED per year, Cheryl A. King, Ph.D., explained at the annual conference of the American Association of Suicidology.
“They come in for exacerbation of common medical illnesses and sports injuries, but they’re also coming in for things like alcohol poisoning, car accidents, and fistfights,” noted Dr. King, professor of psychology and psychiatry at the University of Michigan, Ann Arbor.
These ED visits offer a particularly good opportunity to screen for suicide risk in adolescent males, who account for 80% of all youth suicides in the United States. Yet, because they are so much less likely than teen girls to share thoughts of suicide, males comprise only 25%-35% of teens hospitalized for suicide risk, she continued.
Dr. King has been a leader in the exploration of the medical ED as a venue for routine screening for high risk for suicidal behavior in adolescents. She and others have shown that such screening is productive. But there’s a problem: ED personnel already feel plenty busy and time crushed without taking on a huge new responsibility. And screening instruments designed to miss few teens at risk will have a high false-positive rate. “I don’t want to set the screen-positive threshold so low that 40% of all youth who come in with a broken thumb that was jammed in a car door screen positive, because then what will the ED physicians say to me? They may not even want to talk to me,” she said.
In an early proof-of-concept study, she and her colleagues developed and tested a high-bar screen, one in which a positive result required self-reported serious suicidal ideation within the past 2 weeks, a suicide attempt within the past month, or co-occurring depression plus alcohol/substance misuse. Applying the screen to 298 adolescents who presented to an ED, the investigators found a 16% screen-positive rate (Acad. Emerg. Med. 2009;16:1234-41).
The investigators went on to survey pools of adolescents and parents as to the acceptability of routine ED screening for suicide risk, and got a thumbs-up from both groups (Ped. Emerg. Care 2012;28:626-32).
Next, Dr. King and her colleagues established that their screening tool actually predicted a high near-term risk for suicidal behavior. In a separate study of 81 adolescents aged 14-19 who were screen positive, 15 (18.5%) engaged in some type of suicidal behavior in the 2 months following their ED visit (J. Child Adolesc. Psychopharmacol. 2015;25:100-8).
Another promising screening tool for use in youths presenting to EDs is the ASQ (Ask Suicide-Screening Questions), a four-question test developed at the National Institute of Mental Health (NIMH). It assesses current thoughts of wishing to die, being better off dead, suicidal ideation, and past attempts (Arch. Pediatr. Adolesc. Med. 2012;166:1170-16); however, the predictive validity of this scale in terms of future suicide attempts has yet to be validated, according to Dr. King.
The clinician-administered Columbia–Suicide Severity Rating Scale has demonstrated predictive validity, but it is likely to prove too comprehensive and time consuming for routine use as a screening tool in the ED. However, several specific items incorporated in the scale – namely, the duration element of the intensity scale score and a lifetime history of nonsuicidal self-injury – showed predictive validity as a streamlined screening tool in a study by Dr. King and her colleagues (Pediatr. Emerg. Care 2015;31:88-94).
The most recent development in ED screening is the National Institute of Mental Health–funded ED-STARS (Emergency Department Screening for Teens at Risk for Suicide) project. Dr. King is a codirector of this multicenter effort to develop the optimal suicide risk screening tool for adolescents in the ED, along with a triage algorithm ED physicians can follow.
The tool being developed is a computerized adaptive screen. Rather than answering a fixed list of screening questions, patients will take a very brief, personalized test administered via computer. The adaptive aspect involves an algorithm in which the next question posed depends upon the individual’s response to the question before. This screening instrument is being paired with the Implicit Association Test developed by investigators at Harvard University, Cambridge, Mass.
ED-STARS is being carried out by the Pediatric Emergency Care Applied Research Network. If the project proves successful, the plan is to extend the screening tool into school settings.
Dr. King’s research efforts are funded by the NIMH. She reported having no relevant financial conflicts.
EXPERT ANALYSIS FROM THE ANNUAL AAS CONFERENCE

EuroPCR: OCT stenting guidance may decrease MIs
PARIS – Optical coherence tomography guidance of percutaneous coronary intervention resulted in a change in PCI strategy in two-thirds of patients in the multicenter ILUMIEN I study.
“We were surprised by the high rate at which the OCT [optical coherence tomography] findings influenced practice. Physician decision making was influenced by OCT findings pre-PCI and/or post PCI in 65% of patients, mostly those with more complex disease,” Dr. William Wijns reported at the annual congress of the European Association of Percutaneous Cardiovascular Interventions.
Another unexpected finding: The acute MI rate through 1 year of follow-up was zero among patients whose cardiologists altered their initial stenting strategy in response to the pre-PCI OCT findings and then performed post-PCI stent optimization because they deemed the initial deployment unacceptable based upon the post-PCI OCT findings.

In contrast, the MI rates were 10.3%-13.2% when cardiologists didn’t alter their strategy in response to either of the OCT results or when they altered it only once, based upon either the pre- or post-PCI OCT images. The great majority of these MIs occurred periprocedurally.
“These were true MIs with symptoms, not just enzyme bumps. The reduced MI rate in the subgroup of patients in whom operators changed the procedure based on OCT data, both pre- and post-PCI, was a surprise. The more you work on the artery, the more you’d expect to have troponin increases, at least,” observed Dr. Wijns of the cardiovascular center at Aalst, Belgium, and principal investigator in ILUMIEN I.
He was quick to add that the observed disparity in MI rates based upon the extent to which interventional cardiologists acted upon OCT findings was the result of a post hoc analysis and therefore must be considered merely hypothesis generating. It is, however, an exciting hypothesis, and one which will be tested prospectively in future randomized trials.
ILUMIEN I was a 40-center, 418-patient, prospective, randomized, observational study conducted in the United States, Europe, and Asia. The purpose of the study was to learn what impact OCT imaging had on procedural technique and to identify OCT findings that predict clinical outcomes.
All participants underwent paired fractional flow reserve and OCT studies at the time of angiography prior to their planned PCI and once again immediately post PCI. If the post-PCI imaging showed a suboptimal initial result – stent underexpansion with greater than 20% in-stent residual diameter stenosis, malapposition, flow-limiting edge dissection, or thrombus and/or tissue protrusion causing flow reduction – cardiologists had the option of optimizing the results. If they elected to do so, then OCT imaging was performed once again post optimization to see if in fact the technical outcomes had been improved as assessed in a core laboratory.
Pre-PCI measurements of fractional flow reserve and OCT were successfully accomplished in 91% and 98% of patients, respectively. Armed with the fractional flow reserve data, the interventional cardiologists developed their initial PCI strategy. Then they received the OCT results. Based upon these preprocedural OCT findings, cardiologists changed their PCI strategy in 57% of cases.
Post-PCI fractional flow reserve and OCT results were acquired in 83% and 98% of patients, respectively. Based upon what interventionalists saw as an unacceptable initial PCI result apparent upon the second OCT findings, they performed PCI optimization in 27% of patients.
OCT is known to have superior resolution, compared with angiography or, for that matter, intravascular ultrasound, so it’s not surprising that analysis of the post-PCI OCT findings at the central core laboratory identified a high rate of abnormal findings following what interventionalists deemed a successful result based upon angiographic appearance. Malapposition was present in 32% of cases, stent underexpansion in 27%, edge dissection in 32%, malapposition plus edge dissection in 9%, and tissue or thrombus protrusion in 4%.
Cardiologists performed PCI optimization based upon the second OCT findings in 106 patients. The third and final round of OCT in those patients showed that OCT-guided optimization achieved a sharp decrease in the rates of malapposition and malapposition plus edge dissection.
The 1-year major adverse cardiovascular event rate ranged from a low of 11.5% in the 65 patients who had a change in PCI strategy based upon the preprocedural OCT findings and who also underwent OCT-guided post-PCI optimization to 15.9% in the 137 patients who had neither. Stent thrombosis rates were very low in all four groups, as was in-hospital mortality.
Dr. Wijns noted that analysis of OCT guidance parameters predictive of 1-year clinical outcomes is ongoing.
The ILUMIEN I study was sponsored by St. Jude Medical. Dr. Wijns is a consultant to the company.
PARIS – Optical coherence tomography guidance of percutaneous coronary intervention resulted in a change in PCI strategy in two-thirds of patients in the multicenter ILUMIEN I study.
“We were surprised by the high rate at which the OCT [optical coherence tomography] findings influenced practice. Physician decision making was influenced by OCT findings pre-PCI and/or post PCI in 65% of patients, mostly those with more complex disease,” Dr. William Wijns reported at the annual congress of the European Association of Percutaneous Cardiovascular Interventions.
Another unexpected finding: The acute MI rate through 1 year of follow-up was zero among patients whose cardiologists altered their initial stenting strategy in response to the pre-PCI OCT findings and then performed post-PCI stent optimization because they deemed the initial deployment unacceptable based upon the post-PCI OCT findings.
In contrast, the MI rates were 10.3%-13.2% when cardiologists didn’t alter their strategy in response to either of the OCT results or when they altered it only once, based upon either the pre- or post-PCI OCT images. The great majority of these MIs occurred periprocedurally.
“These were true MIs with symptoms, not just enzyme bumps. The reduced MI rate in the subgroup of patients in whom operators changed the procedure based on OCT data, both pre- and post-PCI, was a surprise. The more you work on the artery, the more you’d expect to have troponin increases, at least,” observed Dr. Wijns of the cardiovascular center at Aalst, Belgium, and principal investigator in ILUMIEN I.
He was quick to add that the observed disparity in MI rates based upon the extent to which interventional cardiologists acted upon OCT findings was the result of a post hoc analysis and therefore must be considered merely hypothesis generating. It is, however, an exciting hypothesis, and one which will be tested prospectively in future randomized trials.
ILUMIEN I was a 40-center, 418-patient, prospective, randomized, observational study conducted in the United States, Europe, and Asia. The purpose of the study was to learn what impact OCT imaging had on procedural technique and to identify OCT findings that predict clinical outcomes.
All participants underwent paired fractional flow reserve and OCT studies at the time of angiography prior to their planned PCI and once again immediately post PCI. If the post-PCI imaging showed a suboptimal initial result – stent underexpansion with greater than 20% in-stent residual diameter stenosis, malapposition, flow-limiting edge dissection, or thrombus and/or tissue protrusion causing flow reduction – cardiologists had the option of optimizing the results. If they elected to do so, then OCT imaging was performed once again post optimization to see if in fact the technical outcomes had been improved as assessed in a core laboratory.
Pre-PCI measurements of fractional flow reserve and OCT were successfully accomplished in 91% and 98% of patients, respectively. Armed with the fractional flow reserve data, the interventional cardiologists developed their initial PCI strategy. Then they received the OCT results. Based upon these preprocedural OCT findings, cardiologists changed their PCI strategy in 57% of cases.
Post-PCI fractional flow reserve and OCT results were acquired in 83% and 98% of patients, respectively. Based upon what interventionalists saw as an unacceptable initial PCI result apparent upon the second OCT findings, they performed PCI optimization in 27% of patients.
OCT is known to have superior resolution, compared with angiography or, for that matter, intravascular ultrasound, so it’s not surprising that analysis of the post-PCI OCT findings at the central core laboratory identified a high rate of abnormal findings following what interventionalists deemed a successful result based upon angiographic appearance. Malapposition was present in 32% of cases, stent underexpansion in 27%, edge dissection in 32%, malapposition plus edge dissection in 9%, and tissue or thrombus protrusion in 4%.
Cardiologists performed PCI optimization based upon the second OCT findings in 106 patients. The third and final round of OCT in those patients showed that OCT-guided optimization achieved a sharp decrease in the rates of malapposition and malapposition plus edge dissection.
The 1-year major adverse cardiovascular event rate ranged from a low of 11.5% in the 65 patients who had a change in PCI strategy based upon the preprocedural OCT findings and who also underwent OCT-guided post-PCI optimization to 15.9% in the 137 patients who had neither. Stent thrombosis rates were very low in all four groups, as was in-hospital mortality.
Dr. Wijns noted that analysis of OCT guidance parameters predictive of 1-year clinical outcomes is ongoing.
The ILUMIEN I study was sponsored by St. Jude Medical. Dr. Wijns is a consultant to the company.
PARIS – Optical coherence tomography guidance of percutaneous coronary intervention resulted in a change in PCI strategy in two-thirds of patients in the multicenter ILUMIEN I study.
“We were surprised by the high rate at which the OCT [optical coherence tomography] findings influenced practice. Physician decision making was influenced by OCT findings pre-PCI and/or post PCI in 65% of patients, mostly those with more complex disease,” Dr. William Wijns reported at the annual congress of the European Association of Percutaneous Cardiovascular Interventions.
Another unexpected finding: The acute MI rate through 1 year of follow-up was zero among patients whose cardiologists altered their initial stenting strategy in response to the pre-PCI OCT findings and then performed post-PCI stent optimization because they deemed the initial deployment unacceptable based upon the post-PCI OCT findings.
In contrast, the MI rates were 10.3%-13.2% when cardiologists didn’t alter their strategy in response to either of the OCT results or when they altered it only once, based upon either the pre- or post-PCI OCT images. The great majority of these MIs occurred periprocedurally.
“These were true MIs with symptoms, not just enzyme bumps. The reduced MI rate in the subgroup of patients in whom operators changed the procedure based on OCT data, both pre- and post-PCI, was a surprise. The more you work on the artery, the more you’d expect to have troponin increases, at least,” observed Dr. Wijns of the cardiovascular center at Aalst, Belgium, and principal investigator in ILUMIEN I.
He was quick to add that the observed disparity in MI rates based upon the extent to which interventional cardiologists acted upon OCT findings was the result of a post hoc analysis and therefore must be considered merely hypothesis generating. It is, however, an exciting hypothesis, and one which will be tested prospectively in future randomized trials.
ILUMIEN I was a 40-center, 418-patient, prospective, randomized, observational study conducted in the United States, Europe, and Asia. The purpose of the study was to learn what impact OCT imaging had on procedural technique and to identify OCT findings that predict clinical outcomes.
All participants underwent paired fractional flow reserve and OCT studies at the time of angiography prior to their planned PCI and once again immediately post PCI. If the post-PCI imaging showed a suboptimal initial result – stent underexpansion with greater than 20% in-stent residual diameter stenosis, malapposition, flow-limiting edge dissection, or thrombus and/or tissue protrusion causing flow reduction – cardiologists had the option of optimizing the results. If they elected to do so, then OCT imaging was performed once again post optimization to see if in fact the technical outcomes had been improved as assessed in a core laboratory.
Pre-PCI measurements of fractional flow reserve and OCT were successfully accomplished in 91% and 98% of patients, respectively. Armed with the fractional flow reserve data, the interventional cardiologists developed their initial PCI strategy. Then they received the OCT results. Based upon these preprocedural OCT findings, cardiologists changed their PCI strategy in 57% of cases.
Post-PCI fractional flow reserve and OCT results were acquired in 83% and 98% of patients, respectively. Based upon what interventionalists saw as an unacceptable initial PCI result apparent upon the second OCT findings, they performed PCI optimization in 27% of patients.
OCT is known to have superior resolution, compared with angiography or, for that matter, intravascular ultrasound, so it’s not surprising that analysis of the post-PCI OCT findings at the central core laboratory identified a high rate of abnormal findings following what interventionalists deemed a successful result based upon angiographic appearance. Malapposition was present in 32% of cases, stent underexpansion in 27%, edge dissection in 32%, malapposition plus edge dissection in 9%, and tissue or thrombus protrusion in 4%.
Cardiologists performed PCI optimization based upon the second OCT findings in 106 patients. The third and final round of OCT in those patients showed that OCT-guided optimization achieved a sharp decrease in the rates of malapposition and malapposition plus edge dissection.
The 1-year major adverse cardiovascular event rate ranged from a low of 11.5% in the 65 patients who had a change in PCI strategy based upon the preprocedural OCT findings and who also underwent OCT-guided post-PCI optimization to 15.9% in the 137 patients who had neither. Stent thrombosis rates were very low in all four groups, as was in-hospital mortality.
Dr. Wijns noted that analysis of OCT guidance parameters predictive of 1-year clinical outcomes is ongoing.
The ILUMIEN I study was sponsored by St. Jude Medical. Dr. Wijns is a consultant to the company.
AT EUROPCR 2015
Key clinical point: OCT findings result in a change in PCI strategy in the majority of patients undergoing coronary stenting.
Major finding: OCT imaging results obtained pre- and post PCI altered interventional cardiologists’ PCI strategy in 65% of treated patients.
Data source: This was a three-continent, 40-center, 418-patient, prospective, nonrandomized, observational study.
Disclosures: The ILUMIEN I study was sponsored by St. Jude Medical. The presenter is a consultant to the company.

EuroPCR: New Sapien 3 TAVI valve findings show ‘wow’ factor
PARIS – Thirty-day outcomes of the first European, all-transfemoral-approach study of the latest-generation Sapien 3 heart valve for transcatheter aortic valve implantation in intermediate-risk elderly patients with severe aortic stenosis included a 1.0% mortality rate and a mere 2.3% rate of moderate paravalvular aortic regurgitation, with no severe aortic regurgitation and a mild aortic regurgitation rate of 26%.
These initial results from the Sapien 3 CE IR study are highly concordant with the impressive results of two U.S. studies using the Sapien 3 valve for transcatheter aortic valve implantation (TAVI) reported earlier this year at the American College of Cardiology meeting in San Diego; one study was of 1,076 intermediate–surgical risk patients and the other involved 583 high-risk patients.
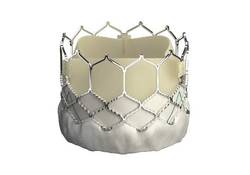
“These results represent at least parity with the best reported surgical outcomes. If we step forward, these favorable results suggest that Sapien 3 TAVI may be expected to challenge surgical aortic valve replacement as the gold standard therapy in elderly patients with aortic stenosis,” Dr. Alec Vahanian declared in presenting the 30-day Sapien 3 CE IR study results at the annual congress of the European Association of Percutaneous Cardiovascular Interventions.
At present, the Sapien 3 valve is approved in Europe for treatment of high-risk or inoperable patients with severe aortic stenosis, but not for intermediate-risk patients such as those in the Sapien 3 CE IR study. The valve remains investigational in the United States, although its manufacturer Edwards Lifesciences, has filed for Food and Drug Administration approval of the device in high-risk patients.
The European Sapien 3 CE IR study includes 101 patients, mean age 84.4 years, with a Society of Thoracic Surgeons (STS) risk score of 5.2%. All underwent TAVI via a transfemoral approach. Fifty-five percent did so under conscious sedation, in contrast to the U.S. trial in intermediate-risk patients, where fewer than 20% had conscious sedation.
To put the observed 30-day all-cause mortality rate of 1.0% in perspective, it’s the lowest seen in the 11 clinical trials performed over the years with the three generations of the Sapien valve. The mortality rate is in line with that found in the much larger U.S. trial in intermediate-risk patients, where the subgroup treated via a transfemoral approach had a 1.1% mortality rate, observed Dr. Vahanian, head of cardiology at Bichat University Hospital in Paris.
The technical procedural success rate in the European trial was 98%. There was no coronary obstruction, valve embolization, or annular rupture.
The 30-day overall stroke incidence was 4%, including a 2% incidence of disabling stroke.
The incidence of vascular complications was low: a major vascular complication rate of 2%, with life-threatening bleeding in 2% of patients. No acute MIs occurred within 30 days, the acute kidney injury rate was 2%, and new-onset atrial fibrillation occurred in 6.9% of patients. Four percent of patients required a new permanent pacemaker, a rate lower than in the U.S. study. There have been no cases of worsening heart failure.
Plus, patients feel a lot better: While 64% were New York Heart Association class III or IV at baseline, 90% were class I or II after 1 month, the cardiologist continued.
In terms of key hemodynamic outcomes, the valve area doubled after TAVI, while the mean gradient plunged from close to 50 mm Hg at baseline to 12 mm Hg 1 month post procedure.
The Sapien 3 valve has the lowest profile of any heart valve. It is typically delivered through a 14-French expandable sheath. The device features a skirt of fabric at the bottom of the frame that’s designed to minimize paravalvular leak.
“I’m very impressed with the data you present. Fantastic!” declared session chair Dr. Carlos E. Ruiz of Lenox Hill Hospital in New York. “Obviously, this valve has raised the bar to a level that will be very hard for other valve technologies to emulate.”

Session cochair Dr. A. Pieter Kappetein, professor of cardiothoracic surgery at Erasmus University in Rotterdam, the Netherlands, posed a question: If you have patients with a mean age of 84, an STS score that would project a 30-day mortality of 5.2% with surgical aortic valve replacement, and yet you only have 1% mortality with Sapien 3 TAVI, why bother with a randomized study before widespread adoption of the less invasive procedure as the treatment of choice in intermediate-risk patients?
Dr. Vahanian replied that this is a time to accumulate evidence. The plan is to follow the study participants for 5 years, a long-term follow-up he views as essential when extending TAVI beyond a high-risk population with limited life expectancy to a less sick group of patients with a longer remaining lifetime. He added that a randomized trial of surgery vs. TAVI is coming in the near future, and the results will provide a solid basis for definitive new practice guidelines. In the meantime, individual patient management decisions are made by heart teams – and the heart teams are keeping up to date regarding the emerging impressive evidence favoring TAVI.
The Sapien 3 studies are sponsored by Edwards Lifesciences. Dr. Vahanian is a consultant to the company.
PARIS – Thirty-day outcomes of the first European, all-transfemoral-approach study of the latest-generation Sapien 3 heart valve for transcatheter aortic valve implantation in intermediate-risk elderly patients with severe aortic stenosis included a 1.0% mortality rate and a mere 2.3% rate of moderate paravalvular aortic regurgitation, with no severe aortic regurgitation and a mild aortic regurgitation rate of 26%.
These initial results from the Sapien 3 CE IR study are highly concordant with the impressive results of two U.S. studies using the Sapien 3 valve for transcatheter aortic valve implantation (TAVI) reported earlier this year at the American College of Cardiology meeting in San Diego; one study was of 1,076 intermediate–surgical risk patients and the other involved 583 high-risk patients.
“These results represent at least parity with the best reported surgical outcomes. If we step forward, these favorable results suggest that Sapien 3 TAVI may be expected to challenge surgical aortic valve replacement as the gold standard therapy in elderly patients with aortic stenosis,” Dr. Alec Vahanian declared in presenting the 30-day Sapien 3 CE IR study results at the annual congress of the European Association of Percutaneous Cardiovascular Interventions.
At present, the Sapien 3 valve is approved in Europe for treatment of high-risk or inoperable patients with severe aortic stenosis, but not for intermediate-risk patients such as those in the Sapien 3 CE IR study. The valve remains investigational in the United States, although its manufacturer Edwards Lifesciences, has filed for Food and Drug Administration approval of the device in high-risk patients.
The European Sapien 3 CE IR study includes 101 patients, mean age 84.4 years, with a Society of Thoracic Surgeons (STS) risk score of 5.2%. All underwent TAVI via a transfemoral approach. Fifty-five percent did so under conscious sedation, in contrast to the U.S. trial in intermediate-risk patients, where fewer than 20% had conscious sedation.
To put the observed 30-day all-cause mortality rate of 1.0% in perspective, it’s the lowest seen in the 11 clinical trials performed over the years with the three generations of the Sapien valve. The mortality rate is in line with that found in the much larger U.S. trial in intermediate-risk patients, where the subgroup treated via a transfemoral approach had a 1.1% mortality rate, observed Dr. Vahanian, head of cardiology at Bichat University Hospital in Paris.
The technical procedural success rate in the European trial was 98%. There was no coronary obstruction, valve embolization, or annular rupture.
The 30-day overall stroke incidence was 4%, including a 2% incidence of disabling stroke.
The incidence of vascular complications was low: a major vascular complication rate of 2%, with life-threatening bleeding in 2% of patients. No acute MIs occurred within 30 days, the acute kidney injury rate was 2%, and new-onset atrial fibrillation occurred in 6.9% of patients. Four percent of patients required a new permanent pacemaker, a rate lower than in the U.S. study. There have been no cases of worsening heart failure.
Plus, patients feel a lot better: While 64% were New York Heart Association class III or IV at baseline, 90% were class I or II after 1 month, the cardiologist continued.
In terms of key hemodynamic outcomes, the valve area doubled after TAVI, while the mean gradient plunged from close to 50 mm Hg at baseline to 12 mm Hg 1 month post procedure.
The Sapien 3 valve has the lowest profile of any heart valve. It is typically delivered through a 14-French expandable sheath. The device features a skirt of fabric at the bottom of the frame that’s designed to minimize paravalvular leak.
“I’m very impressed with the data you present. Fantastic!” declared session chair Dr. Carlos E. Ruiz of Lenox Hill Hospital in New York. “Obviously, this valve has raised the bar to a level that will be very hard for other valve technologies to emulate.”
Session cochair Dr. A. Pieter Kappetein, professor of cardiothoracic surgery at Erasmus University in Rotterdam, the Netherlands, posed a question: If you have patients with a mean age of 84, an STS score that would project a 30-day mortality of 5.2% with surgical aortic valve replacement, and yet you only have 1% mortality with Sapien 3 TAVI, why bother with a randomized study before widespread adoption of the less invasive procedure as the treatment of choice in intermediate-risk patients?
Dr. Vahanian replied that this is a time to accumulate evidence. The plan is to follow the study participants for 5 years, a long-term follow-up he views as essential when extending TAVI beyond a high-risk population with limited life expectancy to a less sick group of patients with a longer remaining lifetime. He added that a randomized trial of surgery vs. TAVI is coming in the near future, and the results will provide a solid basis for definitive new practice guidelines. In the meantime, individual patient management decisions are made by heart teams – and the heart teams are keeping up to date regarding the emerging impressive evidence favoring TAVI.
The Sapien 3 studies are sponsored by Edwards Lifesciences. Dr. Vahanian is a consultant to the company.
PARIS – Thirty-day outcomes of the first European, all-transfemoral-approach study of the latest-generation Sapien 3 heart valve for transcatheter aortic valve implantation in intermediate-risk elderly patients with severe aortic stenosis included a 1.0% mortality rate and a mere 2.3% rate of moderate paravalvular aortic regurgitation, with no severe aortic regurgitation and a mild aortic regurgitation rate of 26%.
These initial results from the Sapien 3 CE IR study are highly concordant with the impressive results of two U.S. studies using the Sapien 3 valve for transcatheter aortic valve implantation (TAVI) reported earlier this year at the American College of Cardiology meeting in San Diego; one study was of 1,076 intermediate–surgical risk patients and the other involved 583 high-risk patients.
“These results represent at least parity with the best reported surgical outcomes. If we step forward, these favorable results suggest that Sapien 3 TAVI may be expected to challenge surgical aortic valve replacement as the gold standard therapy in elderly patients with aortic stenosis,” Dr. Alec Vahanian declared in presenting the 30-day Sapien 3 CE IR study results at the annual congress of the European Association of Percutaneous Cardiovascular Interventions.
At present, the Sapien 3 valve is approved in Europe for treatment of high-risk or inoperable patients with severe aortic stenosis, but not for intermediate-risk patients such as those in the Sapien 3 CE IR study. The valve remains investigational in the United States, although its manufacturer Edwards Lifesciences, has filed for Food and Drug Administration approval of the device in high-risk patients.
The European Sapien 3 CE IR study includes 101 patients, mean age 84.4 years, with a Society of Thoracic Surgeons (STS) risk score of 5.2%. All underwent TAVI via a transfemoral approach. Fifty-five percent did so under conscious sedation, in contrast to the U.S. trial in intermediate-risk patients, where fewer than 20% had conscious sedation.
To put the observed 30-day all-cause mortality rate of 1.0% in perspective, it’s the lowest seen in the 11 clinical trials performed over the years with the three generations of the Sapien valve. The mortality rate is in line with that found in the much larger U.S. trial in intermediate-risk patients, where the subgroup treated via a transfemoral approach had a 1.1% mortality rate, observed Dr. Vahanian, head of cardiology at Bichat University Hospital in Paris.
The technical procedural success rate in the European trial was 98%. There was no coronary obstruction, valve embolization, or annular rupture.
The 30-day overall stroke incidence was 4%, including a 2% incidence of disabling stroke.
The incidence of vascular complications was low: a major vascular complication rate of 2%, with life-threatening bleeding in 2% of patients. No acute MIs occurred within 30 days, the acute kidney injury rate was 2%, and new-onset atrial fibrillation occurred in 6.9% of patients. Four percent of patients required a new permanent pacemaker, a rate lower than in the U.S. study. There have been no cases of worsening heart failure.
Plus, patients feel a lot better: While 64% were New York Heart Association class III or IV at baseline, 90% were class I or II after 1 month, the cardiologist continued.
In terms of key hemodynamic outcomes, the valve area doubled after TAVI, while the mean gradient plunged from close to 50 mm Hg at baseline to 12 mm Hg 1 month post procedure.
The Sapien 3 valve has the lowest profile of any heart valve. It is typically delivered through a 14-French expandable sheath. The device features a skirt of fabric at the bottom of the frame that’s designed to minimize paravalvular leak.
“I’m very impressed with the data you present. Fantastic!” declared session chair Dr. Carlos E. Ruiz of Lenox Hill Hospital in New York. “Obviously, this valve has raised the bar to a level that will be very hard for other valve technologies to emulate.”
Session cochair Dr. A. Pieter Kappetein, professor of cardiothoracic surgery at Erasmus University in Rotterdam, the Netherlands, posed a question: If you have patients with a mean age of 84, an STS score that would project a 30-day mortality of 5.2% with surgical aortic valve replacement, and yet you only have 1% mortality with Sapien 3 TAVI, why bother with a randomized study before widespread adoption of the less invasive procedure as the treatment of choice in intermediate-risk patients?
Dr. Vahanian replied that this is a time to accumulate evidence. The plan is to follow the study participants for 5 years, a long-term follow-up he views as essential when extending TAVI beyond a high-risk population with limited life expectancy to a less sick group of patients with a longer remaining lifetime. He added that a randomized trial of surgery vs. TAVI is coming in the near future, and the results will provide a solid basis for definitive new practice guidelines. In the meantime, individual patient management decisions are made by heart teams – and the heart teams are keeping up to date regarding the emerging impressive evidence favoring TAVI.
The Sapien 3 studies are sponsored by Edwards Lifesciences. Dr. Vahanian is a consultant to the company.
AT EUROPCR 2015
Key clinical point: Short-term outcomes following transcatheter aortic valve implantation using the Sapien 3 valve in patients with severe aortic stenosis at intermediate surgical risk are the best ever reported with TAVI or surgical valve replacement.
Major finding: Key 30-day outcomes after TAVI using the Sapien 3 valve via a transfemoral approach in intermediate–surgical risk patients with severe aortic stenosis included 1% overall mortality, a 2% rate of disabling stroke, and no severe paravalvular aortic regurgitation.
Data source: The European Sapien 3 CE IR study is a nonrandomized study of 101 intermediate–surgical risk octogenarians with severe aortic stenosis who underwent TAVI with the Sapien 3 valve via a transfemoral approach.
Disclosures: The European Sapien 3 CE IR study is sponsored by Edwards Lifesciences. The presenter is a consultant to the company.
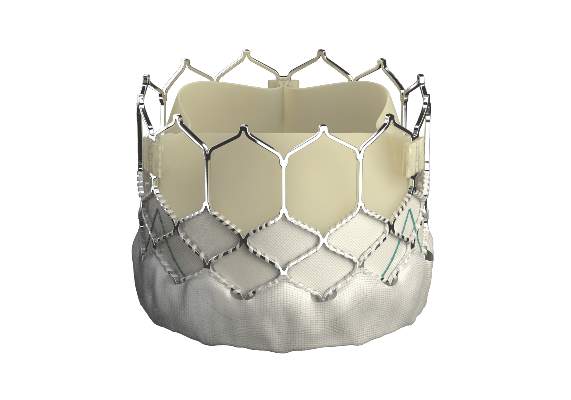
EuroPCR: Which TAVI vascular closure device is safest?
PARIS – The Perclose ProGlide vascular closure device resulted in significantly fewer major vascular complications than its chief competitor, the Prostar XL, in patients undergoing transcatheter aortic valve implantation by a percutaneous transfemoral approach in the multicenter CONTROL trial.
“ProGlide-based vascular closure is associated with significantly lower rates of arterial rupture and hematomas, lower rates of major vascular complications, bleeding, acute kidney injury, and shorter hospital stay,” Dr. Israel M. Barbash reported in presenting the CONTROL results at the annual congress of the European Association of Percutaneous Cardiovascular Interventions.
Serious vascular complications remain a major concern when transcatheter aortic valve implantation (TAVI) is performed via a percutaneous transfemoral approach. The CONTROL study was conducted to determine whether the two closure devices most widely used for this purpose worldwide – the Prostar XL and Perclose ProGlide, both marketed by Abbott Vascular – differ in their adverse event rates.
CONTROL was a nine-center, international, retrospective, nonrandomized, matched-pairs comparison study. Starting with a pool of 3,138 percutaneous transfemoral TAVI patients, investigators used propensity score matching on nine variables to narrow the study population to 1,270 patients in 635 closely matched pairs.
The variables used in the matching process fell into three categories: comorbid conditions, including diabetes, coronary artery disease, and peripheral vascular disease; arterial factors such as tortuosity and calcification; and sheath type and size. Most patients underwent TAVI with a Cook Check-flo Performer or Edwards expandable eSheath and received a CoreValve or Sapien XT heart valve.
The major vascular complication rate was 2% in the ProGlide group, and more than threefold higher at roughly 7.5% in the Prostar group. Rates of life-threatening bleeding and major bleeding by the Valve Academic Research Consortium-2 (VARC-2) definitions also were significantly higher with the use of the Prostar device. In addition, rates of hematoma and femoral artery rupture were both threefold higher in the Prostar group.
Acute kidney injury occurred in 6.6% of the Prostar group, compared with 2.7% with ProGlide. The median hospital length of stay was a full day longer in the Prostar group: 6 days versus 5 days, according to Dr. Barbash of Sheba Medical Center in Tel Hashomer, Israel.
Despite the consistently higher adverse event rates documented in the Prostar group, in-hospital mortality rates didn’t differ between the two study arms. There was, however, a trend for lower in-hospital mortality in the ProGlide group by a margin of 3.5% versus 4.9% with Prostar; this difference might well have achieved statistical significance with larger patient numbers, the cardiologist continued.
A learning curve was evident for the ProGlide device: after an operator’s first 20 cases, the vascular complication rate dropped sharply. In contrast, the high adverse event rates associated with the Prostar device didn’t decrease significantly no matter how much experience with the device an operator gained.
“It looks like ‘Goodby Prostar, hello ProGlide.’ That’s what your data say to me,” said session cochair Dr. A. Pieter Kappetein, professor of cardiothoracic surgery at Erasmus University in Rotterdam, the Netherlands.
Asked if he thinks a randomized trial is warranted in light of the clear and consistent messages provided by the CONTROL study, Dr. Barbash replied, yes, but not yet.
“Sheath sizes are decreasing and new players will come into the vascular closure device market soon. When they do, that will be the time for a randomized trial comparing the new ones to the older ones,” he added.
CONTROL was an investigator-initiated study. Dr. Barbash reported having no financial conflicts.
PARIS – The Perclose ProGlide vascular closure device resulted in significantly fewer major vascular complications than its chief competitor, the Prostar XL, in patients undergoing transcatheter aortic valve implantation by a percutaneous transfemoral approach in the multicenter CONTROL trial.
“ProGlide-based vascular closure is associated with significantly lower rates of arterial rupture and hematomas, lower rates of major vascular complications, bleeding, acute kidney injury, and shorter hospital stay,” Dr. Israel M. Barbash reported in presenting the CONTROL results at the annual congress of the European Association of Percutaneous Cardiovascular Interventions.
Serious vascular complications remain a major concern when transcatheter aortic valve implantation (TAVI) is performed via a percutaneous transfemoral approach. The CONTROL study was conducted to determine whether the two closure devices most widely used for this purpose worldwide – the Prostar XL and Perclose ProGlide, both marketed by Abbott Vascular – differ in their adverse event rates.
CONTROL was a nine-center, international, retrospective, nonrandomized, matched-pairs comparison study. Starting with a pool of 3,138 percutaneous transfemoral TAVI patients, investigators used propensity score matching on nine variables to narrow the study population to 1,270 patients in 635 closely matched pairs.
The variables used in the matching process fell into three categories: comorbid conditions, including diabetes, coronary artery disease, and peripheral vascular disease; arterial factors such as tortuosity and calcification; and sheath type and size. Most patients underwent TAVI with a Cook Check-flo Performer or Edwards expandable eSheath and received a CoreValve or Sapien XT heart valve.
The major vascular complication rate was 2% in the ProGlide group, and more than threefold higher at roughly 7.5% in the Prostar group. Rates of life-threatening bleeding and major bleeding by the Valve Academic Research Consortium-2 (VARC-2) definitions also were significantly higher with the use of the Prostar device. In addition, rates of hematoma and femoral artery rupture were both threefold higher in the Prostar group.
Acute kidney injury occurred in 6.6% of the Prostar group, compared with 2.7% with ProGlide. The median hospital length of stay was a full day longer in the Prostar group: 6 days versus 5 days, according to Dr. Barbash of Sheba Medical Center in Tel Hashomer, Israel.
Despite the consistently higher adverse event rates documented in the Prostar group, in-hospital mortality rates didn’t differ between the two study arms. There was, however, a trend for lower in-hospital mortality in the ProGlide group by a margin of 3.5% versus 4.9% with Prostar; this difference might well have achieved statistical significance with larger patient numbers, the cardiologist continued.
A learning curve was evident for the ProGlide device: after an operator’s first 20 cases, the vascular complication rate dropped sharply. In contrast, the high adverse event rates associated with the Prostar device didn’t decrease significantly no matter how much experience with the device an operator gained.
“It looks like ‘Goodby Prostar, hello ProGlide.’ That’s what your data say to me,” said session cochair Dr. A. Pieter Kappetein, professor of cardiothoracic surgery at Erasmus University in Rotterdam, the Netherlands.
Asked if he thinks a randomized trial is warranted in light of the clear and consistent messages provided by the CONTROL study, Dr. Barbash replied, yes, but not yet.
“Sheath sizes are decreasing and new players will come into the vascular closure device market soon. When they do, that will be the time for a randomized trial comparing the new ones to the older ones,” he added.
CONTROL was an investigator-initiated study. Dr. Barbash reported having no financial conflicts.
PARIS – The Perclose ProGlide vascular closure device resulted in significantly fewer major vascular complications than its chief competitor, the Prostar XL, in patients undergoing transcatheter aortic valve implantation by a percutaneous transfemoral approach in the multicenter CONTROL trial.
“ProGlide-based vascular closure is associated with significantly lower rates of arterial rupture and hematomas, lower rates of major vascular complications, bleeding, acute kidney injury, and shorter hospital stay,” Dr. Israel M. Barbash reported in presenting the CONTROL results at the annual congress of the European Association of Percutaneous Cardiovascular Interventions.
Serious vascular complications remain a major concern when transcatheter aortic valve implantation (TAVI) is performed via a percutaneous transfemoral approach. The CONTROL study was conducted to determine whether the two closure devices most widely used for this purpose worldwide – the Prostar XL and Perclose ProGlide, both marketed by Abbott Vascular – differ in their adverse event rates.
CONTROL was a nine-center, international, retrospective, nonrandomized, matched-pairs comparison study. Starting with a pool of 3,138 percutaneous transfemoral TAVI patients, investigators used propensity score matching on nine variables to narrow the study population to 1,270 patients in 635 closely matched pairs.
The variables used in the matching process fell into three categories: comorbid conditions, including diabetes, coronary artery disease, and peripheral vascular disease; arterial factors such as tortuosity and calcification; and sheath type and size. Most patients underwent TAVI with a Cook Check-flo Performer or Edwards expandable eSheath and received a CoreValve or Sapien XT heart valve.
The major vascular complication rate was 2% in the ProGlide group, and more than threefold higher at roughly 7.5% in the Prostar group. Rates of life-threatening bleeding and major bleeding by the Valve Academic Research Consortium-2 (VARC-2) definitions also were significantly higher with the use of the Prostar device. In addition, rates of hematoma and femoral artery rupture were both threefold higher in the Prostar group.
Acute kidney injury occurred in 6.6% of the Prostar group, compared with 2.7% with ProGlide. The median hospital length of stay was a full day longer in the Prostar group: 6 days versus 5 days, according to Dr. Barbash of Sheba Medical Center in Tel Hashomer, Israel.
Despite the consistently higher adverse event rates documented in the Prostar group, in-hospital mortality rates didn’t differ between the two study arms. There was, however, a trend for lower in-hospital mortality in the ProGlide group by a margin of 3.5% versus 4.9% with Prostar; this difference might well have achieved statistical significance with larger patient numbers, the cardiologist continued.
A learning curve was evident for the ProGlide device: after an operator’s first 20 cases, the vascular complication rate dropped sharply. In contrast, the high adverse event rates associated with the Prostar device didn’t decrease significantly no matter how much experience with the device an operator gained.
“It looks like ‘Goodby Prostar, hello ProGlide.’ That’s what your data say to me,” said session cochair Dr. A. Pieter Kappetein, professor of cardiothoracic surgery at Erasmus University in Rotterdam, the Netherlands.
Asked if he thinks a randomized trial is warranted in light of the clear and consistent messages provided by the CONTROL study, Dr. Barbash replied, yes, but not yet.
“Sheath sizes are decreasing and new players will come into the vascular closure device market soon. When they do, that will be the time for a randomized trial comparing the new ones to the older ones,” he added.
CONTROL was an investigator-initiated study. Dr. Barbash reported having no financial conflicts.
AT EuroPCR 2015
Key clinical point: Key outcomes are significantly better with the Perclose ProGlide vascular closure device than the Prostar XL in patients undergoing transcatheter aortic valve implantation via a percutaneous transfemoral approach.
Major finding: Patients who underwent transcatheter aortic valve implantation via a percutaneous transfemoral approach with vascular closure using the Perclose ProGlide device had fewer major vascular complications and a 1-day shorter hospital length of stay than with the Prostar XL closure device.
Data source: CONTROL was a retrospective, propensity score-matched comparison of outcomes in 635 pairs of patients. One patient in each pair was treated with the Perclose ProGlide vascular closure device, the other with the Prostar XL.
Disclosures: The CONTROL study was investigator initiated. The presenter reported having no financial conflicts.
IHC: Promising new drug cuts pain in trigeminal neuralgia
VALENCIA, SPAIN – The prospect of a new and effective oral medication for trigeminal neuralgia with much better patient tolerability than what’s currently available caused a stir at the International Headache Congress.
In a double-blind, randomized, 7-week, phase II, proof-of-concept study of 31 patients with frequent daily paroxysmic episodes of trigeminal neuralgia, the investigational agent known for now as CNV1014802 had a treatment failure rate that was half the rate seen in placebo-treated controls, Dr. Joanna Zakrzewska reported at the meeting, which was sponsored by the International Headache Society and the American Headache Society.

“We saw remarkably few adverse events. Drug-related adverse events were confined to a couple of episodes of mild dizziness/headache. There was no cognitive impairment. With the good tolerability, we think these results are encouraging enough to plan further randomized controlled trials,” said Dr. Zakrzewska of University College London.
Trigeminal neuralgia is an uncommon but severe form of unilateral episodic facial pain typically occurring in sudden, stabbing bursts lasting less than 2 minutes. The current first-line treatments – carbamazepine and other anticonvulsants – are quite effective, but often poorly tolerated, have significant drug-drug interactions, and require lengthy titration. All of this adds up to a high treatment failure rate.
CNV1014802 is a state-dependent sodium channel blocker that can be started at its target dose and has selectivity for the Nav 1.7 sodium channel.
The proof-of-concept study had an unusual design. All 31 participants were known to be responders to the novel agent because they were among the 70% of patients who responded to the drug during an open-label screening period. They were then randomized double-blind to CNV1014802 at 150 mg three times a day or placebo.
The primary endpoint in the study was treatment failure, defined as at least a 50% increase from baseline in frequency of paroxysms, a 50% worsening of the associated pain severity, and a physician global impression that a patient was “much worse” or “very much worse.” This occurred in one-third of patients on the novel drug and 64% of those on placebo. The time to treatment failure, when it occurred, was significantly longer in the active treatment group as well, according to Dr. Zakrzewska.
The treatment failure rate was 33% in patients with trigeminal neuralgia randomized to oral CNV1014802 and 64% in placebo-treated controls.
Patients in the active treatment arm experienced a mean 60% decrease in their number of pain paroxysms per day through week 7 as recorded in patient diaries, compared with a 13% reduction in placebo-treated controls. The CNV1014802 group also had a mean 55% reduction in pain severity versus an 18% improvement in controls.
The study results met with an enthusiastic audience response. “This is fantastic news for patients with trigeminal neuralgia,” said Dr. Peter J. Goadsby, chair of the British Association for the Study of Headache, who was charged with summing up the IHC Congress highlights.
Dr. Zakrzewska received a research grant from Convergence Pharmaceuticals, which sponsored the study.
VALENCIA, SPAIN – The prospect of a new and effective oral medication for trigeminal neuralgia with much better patient tolerability than what’s currently available caused a stir at the International Headache Congress.
In a double-blind, randomized, 7-week, phase II, proof-of-concept study of 31 patients with frequent daily paroxysmic episodes of trigeminal neuralgia, the investigational agent known for now as CNV1014802 had a treatment failure rate that was half the rate seen in placebo-treated controls, Dr. Joanna Zakrzewska reported at the meeting, which was sponsored by the International Headache Society and the American Headache Society.
“We saw remarkably few adverse events. Drug-related adverse events were confined to a couple of episodes of mild dizziness/headache. There was no cognitive impairment. With the good tolerability, we think these results are encouraging enough to plan further randomized controlled trials,” said Dr. Zakrzewska of University College London.
Trigeminal neuralgia is an uncommon but severe form of unilateral episodic facial pain typically occurring in sudden, stabbing bursts lasting less than 2 minutes. The current first-line treatments – carbamazepine and other anticonvulsants – are quite effective, but often poorly tolerated, have significant drug-drug interactions, and require lengthy titration. All of this adds up to a high treatment failure rate.
CNV1014802 is a state-dependent sodium channel blocker that can be started at its target dose and has selectivity for the Nav 1.7 sodium channel.
The proof-of-concept study had an unusual design. All 31 participants were known to be responders to the novel agent because they were among the 70% of patients who responded to the drug during an open-label screening period. They were then randomized double-blind to CNV1014802 at 150 mg three times a day or placebo.
The primary endpoint in the study was treatment failure, defined as at least a 50% increase from baseline in frequency of paroxysms, a 50% worsening of the associated pain severity, and a physician global impression that a patient was “much worse” or “very much worse.” This occurred in one-third of patients on the novel drug and 64% of those on placebo. The time to treatment failure, when it occurred, was significantly longer in the active treatment group as well, according to Dr. Zakrzewska.
The treatment failure rate was 33% in patients with trigeminal neuralgia randomized to oral CNV1014802 and 64% in placebo-treated controls.
Patients in the active treatment arm experienced a mean 60% decrease in their number of pain paroxysms per day through week 7 as recorded in patient diaries, compared with a 13% reduction in placebo-treated controls. The CNV1014802 group also had a mean 55% reduction in pain severity versus an 18% improvement in controls.
The study results met with an enthusiastic audience response. “This is fantastic news for patients with trigeminal neuralgia,” said Dr. Peter J. Goadsby, chair of the British Association for the Study of Headache, who was charged with summing up the IHC Congress highlights.
Dr. Zakrzewska received a research grant from Convergence Pharmaceuticals, which sponsored the study.
VALENCIA, SPAIN – The prospect of a new and effective oral medication for trigeminal neuralgia with much better patient tolerability than what’s currently available caused a stir at the International Headache Congress.
In a double-blind, randomized, 7-week, phase II, proof-of-concept study of 31 patients with frequent daily paroxysmic episodes of trigeminal neuralgia, the investigational agent known for now as CNV1014802 had a treatment failure rate that was half the rate seen in placebo-treated controls, Dr. Joanna Zakrzewska reported at the meeting, which was sponsored by the International Headache Society and the American Headache Society.
“We saw remarkably few adverse events. Drug-related adverse events were confined to a couple of episodes of mild dizziness/headache. There was no cognitive impairment. With the good tolerability, we think these results are encouraging enough to plan further randomized controlled trials,” said Dr. Zakrzewska of University College London.
Trigeminal neuralgia is an uncommon but severe form of unilateral episodic facial pain typically occurring in sudden, stabbing bursts lasting less than 2 minutes. The current first-line treatments – carbamazepine and other anticonvulsants – are quite effective, but often poorly tolerated, have significant drug-drug interactions, and require lengthy titration. All of this adds up to a high treatment failure rate.
CNV1014802 is a state-dependent sodium channel blocker that can be started at its target dose and has selectivity for the Nav 1.7 sodium channel.
The proof-of-concept study had an unusual design. All 31 participants were known to be responders to the novel agent because they were among the 70% of patients who responded to the drug during an open-label screening period. They were then randomized double-blind to CNV1014802 at 150 mg three times a day or placebo.
The primary endpoint in the study was treatment failure, defined as at least a 50% increase from baseline in frequency of paroxysms, a 50% worsening of the associated pain severity, and a physician global impression that a patient was “much worse” or “very much worse.” This occurred in one-third of patients on the novel drug and 64% of those on placebo. The time to treatment failure, when it occurred, was significantly longer in the active treatment group as well, according to Dr. Zakrzewska.
The treatment failure rate was 33% in patients with trigeminal neuralgia randomized to oral CNV1014802 and 64% in placebo-treated controls.
Patients in the active treatment arm experienced a mean 60% decrease in their number of pain paroxysms per day through week 7 as recorded in patient diaries, compared with a 13% reduction in placebo-treated controls. The CNV1014802 group also had a mean 55% reduction in pain severity versus an 18% improvement in controls.
The study results met with an enthusiastic audience response. “This is fantastic news for patients with trigeminal neuralgia,” said Dr. Peter J. Goadsby, chair of the British Association for the Study of Headache, who was charged with summing up the IHC Congress highlights.
Dr. Zakrzewska received a research grant from Convergence Pharmaceuticals, which sponsored the study.
AT IHC 2015
Key clinical point: Novel sodium channel blocker could fill treatment gap in trigeminal neuralgia.
Major finding: The treatment failure rate was 33% in patients with trigeminal neuralgia randomized to oral CNV1014802 and 64% in placebo-treated controls.
Data source: A double-blind, randomized, 7-week, phase II, proof-of-concept study of 31 patients.
Disclosures: Dr. Zakrzewska received a research grant from Convergence Pharmaceuticals, which sponsored the study.

IHC: IV dihydroergotamine linked to increased thrombosis risk
VALENCIA, SPAIN – Inpatient high-dose intravenous dihydroergotamine delivered through a peripherally inserted central catheter or midline catheter for treatment of refractory headache appears to be associated with an increased risk of venous thrombosis, according to a retrospective case-control study.
“In patients with PICC or midline catheter placement, a low threshold for consideration of ultrasound investigation and consideration of prophylactic anticoagulation are appropriate. But patients with peripheral IVs had a very low thrombosis rate, so it seems reasonable to continue not to anticoagulate these patients,” Dr. Amy Tso said at the International Headache Congress.

Most headache centers rely upon a 5-day inpatient course of IV dihydroergotamine (DHE) as their workhorse therapy for the most refractory patients with chronic migraine or chronic cluster headache. But at the Headache Center of the University of California, San Francisco, where this internationally popular regimen was pioneered (Neurology 2011;77:1827-32), investigators have concluded that the risk of venous thrombosis when the drug is delivered by PICC or midline catheter is suspiciously high in what is a generally healthy ambulatory patient population aside from their severe headaches.
Dr. Tso, a neurologist at the university, presented a new retrospective analysis of 604 consecutive admissions for this therapy in 450 adult and pediatric patients. ‘Venous thrombosis occurred in 6.8% of 264 severe headache patients whose dihydroergotamine was administered through a PICC line or midline catheter. The venous thrombosis rate was 6.4% in 157 patients treated through a PICC line and 7.5% in 107 whose DHE was given through a midline catheter. Eleven of the 18 affected patients had deep vein thrombosis warranting 3 months of oral anticoagulation, and 2 others developed pulmonary embolism.
In contrast, venous thrombosis occurred in 1 of 272 patients treated via a peripheral IV line.
In this study, venous thrombosis risk was unrelated to total DHE dose, age, gender, or the presence or absence of aura.
Since most studies of VTE risk in patients with a PICC or midline catheter have been conducted in cancer patients getting chemotherapy, patients on antibiotics for serious infections, or individuals on total parenteral nutrition – populations very different from headache patients – Dr. Tso opted to use as a control group for her study of 56 headache patients who received a 10-day inpatient course of IV lidocaine at the UCSF Headache Center; not one of them developed venous thrombosis, regardless of whether the lidocaine was given through a PICC, peripheral IV line, or midline catheter, she reported at the meeting, which was sponsored by the International Headache Society and the American Headache Society.
Physicians routinely use a peripheral IV line at the start of the 5-day course of inpatient DHE, during which patients receive 1 mg of the drug every 8 hours. However, DHE is a potent vasoconstrictor, and when the veins clamp down, as occurs often, it’s common practice to switch to a PICC or midline catheter to maintain vascular access and continue treatment.
“I certainly don’t think this new information about venous thrombosis risk would lead us to not give the therapy. The data supporting the use of this protocol in treating chronic migraine and chronic cluster headache have established that it’s so useful,” she said in an interview. “But I’ll have more reticence to put in a central line, and when I do I’ll have a lower threshold for getting an ultrasound when a patient complains of discomfort.”
Dr. Tso reported having no financial conflicts regarding this study.
VALENCIA, SPAIN – Inpatient high-dose intravenous dihydroergotamine delivered through a peripherally inserted central catheter or midline catheter for treatment of refractory headache appears to be associated with an increased risk of venous thrombosis, according to a retrospective case-control study.
“In patients with PICC or midline catheter placement, a low threshold for consideration of ultrasound investigation and consideration of prophylactic anticoagulation are appropriate. But patients with peripheral IVs had a very low thrombosis rate, so it seems reasonable to continue not to anticoagulate these patients,” Dr. Amy Tso said at the International Headache Congress.
Most headache centers rely upon a 5-day inpatient course of IV dihydroergotamine (DHE) as their workhorse therapy for the most refractory patients with chronic migraine or chronic cluster headache. But at the Headache Center of the University of California, San Francisco, where this internationally popular regimen was pioneered (Neurology 2011;77:1827-32), investigators have concluded that the risk of venous thrombosis when the drug is delivered by PICC or midline catheter is suspiciously high in what is a generally healthy ambulatory patient population aside from their severe headaches.
Dr. Tso, a neurologist at the university, presented a new retrospective analysis of 604 consecutive admissions for this therapy in 450 adult and pediatric patients. ‘Venous thrombosis occurred in 6.8% of 264 severe headache patients whose dihydroergotamine was administered through a PICC line or midline catheter. The venous thrombosis rate was 6.4% in 157 patients treated through a PICC line and 7.5% in 107 whose DHE was given through a midline catheter. Eleven of the 18 affected patients had deep vein thrombosis warranting 3 months of oral anticoagulation, and 2 others developed pulmonary embolism.
In contrast, venous thrombosis occurred in 1 of 272 patients treated via a peripheral IV line.
In this study, venous thrombosis risk was unrelated to total DHE dose, age, gender, or the presence or absence of aura.
Since most studies of VTE risk in patients with a PICC or midline catheter have been conducted in cancer patients getting chemotherapy, patients on antibiotics for serious infections, or individuals on total parenteral nutrition – populations very different from headache patients – Dr. Tso opted to use as a control group for her study of 56 headache patients who received a 10-day inpatient course of IV lidocaine at the UCSF Headache Center; not one of them developed venous thrombosis, regardless of whether the lidocaine was given through a PICC, peripheral IV line, or midline catheter, she reported at the meeting, which was sponsored by the International Headache Society and the American Headache Society.
Physicians routinely use a peripheral IV line at the start of the 5-day course of inpatient DHE, during which patients receive 1 mg of the drug every 8 hours. However, DHE is a potent vasoconstrictor, and when the veins clamp down, as occurs often, it’s common practice to switch to a PICC or midline catheter to maintain vascular access and continue treatment.
“I certainly don’t think this new information about venous thrombosis risk would lead us to not give the therapy. The data supporting the use of this protocol in treating chronic migraine and chronic cluster headache have established that it’s so useful,” she said in an interview. “But I’ll have more reticence to put in a central line, and when I do I’ll have a lower threshold for getting an ultrasound when a patient complains of discomfort.”
Dr. Tso reported having no financial conflicts regarding this study.
VALENCIA, SPAIN – Inpatient high-dose intravenous dihydroergotamine delivered through a peripherally inserted central catheter or midline catheter for treatment of refractory headache appears to be associated with an increased risk of venous thrombosis, according to a retrospective case-control study.
“In patients with PICC or midline catheter placement, a low threshold for consideration of ultrasound investigation and consideration of prophylactic anticoagulation are appropriate. But patients with peripheral IVs had a very low thrombosis rate, so it seems reasonable to continue not to anticoagulate these patients,” Dr. Amy Tso said at the International Headache Congress.
Most headache centers rely upon a 5-day inpatient course of IV dihydroergotamine (DHE) as their workhorse therapy for the most refractory patients with chronic migraine or chronic cluster headache. But at the Headache Center of the University of California, San Francisco, where this internationally popular regimen was pioneered (Neurology 2011;77:1827-32), investigators have concluded that the risk of venous thrombosis when the drug is delivered by PICC or midline catheter is suspiciously high in what is a generally healthy ambulatory patient population aside from their severe headaches.
Dr. Tso, a neurologist at the university, presented a new retrospective analysis of 604 consecutive admissions for this therapy in 450 adult and pediatric patients. ‘Venous thrombosis occurred in 6.8% of 264 severe headache patients whose dihydroergotamine was administered through a PICC line or midline catheter. The venous thrombosis rate was 6.4% in 157 patients treated through a PICC line and 7.5% in 107 whose DHE was given through a midline catheter. Eleven of the 18 affected patients had deep vein thrombosis warranting 3 months of oral anticoagulation, and 2 others developed pulmonary embolism.
In contrast, venous thrombosis occurred in 1 of 272 patients treated via a peripheral IV line.
In this study, venous thrombosis risk was unrelated to total DHE dose, age, gender, or the presence or absence of aura.
Since most studies of VTE risk in patients with a PICC or midline catheter have been conducted in cancer patients getting chemotherapy, patients on antibiotics for serious infections, or individuals on total parenteral nutrition – populations very different from headache patients – Dr. Tso opted to use as a control group for her study of 56 headache patients who received a 10-day inpatient course of IV lidocaine at the UCSF Headache Center; not one of them developed venous thrombosis, regardless of whether the lidocaine was given through a PICC, peripheral IV line, or midline catheter, she reported at the meeting, which was sponsored by the International Headache Society and the American Headache Society.
Physicians routinely use a peripheral IV line at the start of the 5-day course of inpatient DHE, during which patients receive 1 mg of the drug every 8 hours. However, DHE is a potent vasoconstrictor, and when the veins clamp down, as occurs often, it’s common practice to switch to a PICC or midline catheter to maintain vascular access and continue treatment.
“I certainly don’t think this new information about venous thrombosis risk would lead us to not give the therapy. The data supporting the use of this protocol in treating chronic migraine and chronic cluster headache have established that it’s so useful,” she said in an interview. “But I’ll have more reticence to put in a central line, and when I do I’ll have a lower threshold for getting an ultrasound when a patient complains of discomfort.”
Dr. Tso reported having no financial conflicts regarding this study.
AT IHC 2015
Key clinical point: Multiday inpatient IV dihydroergotamine delivered by PICC or midline catheter carries an increased risk of venous thrombosis.
Major finding: Venous thrombosis occurred in 6.8% of 264 severe headache patients whose dihydroergotamine was administered through a PICC line or midline catheter.
Data source: A retrospective, single-center, case-control study of 604 admissions involving 450 patients with chronic refractory headache who underwent a 5-day inpatient course of IV dihydroergotamine.
Disclosures: Dr. Tso reported no financial conflicts regarding this study.
