User login
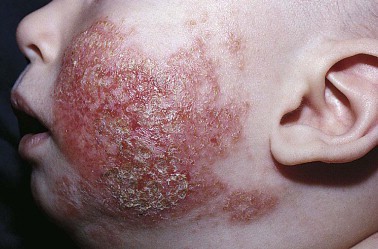
Pimecrolimus Shines in Large Infant Atopy Study
PRAGUE – Long-term therapy utilizing a pimecrolimus-based strategy for mild to moderate atopic dermatitis beginning in infancy proved as effective and safe as conventional therapy with topical corticosteroids in a 5-year randomized study.
"Given that pimecrolimus cream 1% is not associated with topical corticosteroid-specific side effects, such as hypothalamic-pituitary-adrenal axis suppression or skin atrophy, which can limit their long-term use, pimecrolimus cream may represent the drug of choice for long-term atopic dermatitis management," said Dr. Thomas A. Luger, professor and chairman of the department of dermatology at the University of Müenster (Germany).
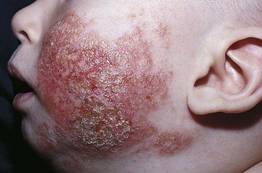
Currently, pimecrolimus is not approved for use in patients under 2 years of age, he noted.
Dr. Luger, who is an investigator on the study’s steering committee, reported on 2,418 infants with mild to moderate atopic dermatitis randomized to daily use of an emollient for their dry skin plus either pimecrolimus cream 1% (Elidel) or low-potency to midpotency topical steroids on an as-needed basis. The participants’ mean age at enrollment was 7 months. Fifty-three percent of infants had moderate atopic dermatitis based on the Investigator’s Global Assessment; the rest had mild disease.
This was a 5-year, international, open-label randomized study designed to reflect real world, as-needed use of topical medications. Patients in the pimecrolimus group applied the topical calcineurin inhibitor at the first sign of an atopic dermatitis flare. If pimecrolimus didn’t control the itching, redness, and/or lesion elevation to the parents’ satisfaction, the child was switched to a low-potency topical steroid. If that didn’t quell the episode, a midpotency topical steroid was used. And if the child’s skin disease worsened despite the midpotency topical steroid, the patient went to the clinic. If the physician determined systemic therapy was needed, the child was removed from the study.
Treatment success was defined as a score of 0 or 1, meaning clear or almost clear, on the Investigator’s Global Assessment. By week 3 of the study, more than 50% of patients in both treatment arms met that standard. The success rate gradually increased over a 5-year period to about 90% overall in both groups and greater than 95% for facial atopic dermatitis.
At baseline, the participants’ mean total body surface area affected by atopic dermatitis was 17%, and 22% of infants had a total body surface area of 30% or greater. By week 3, however, the mean total body surface area had dropped below 5% in both groups. The total body surface area progressively improved, reaching a mean of 0% at week 78 and staying that way for the remainder of the 5-year study, Dr. Luger reported at the annual congress of the European Academy of Dermatology and Venereology.
Of children in the pimecrolimus cohort, 70% completed the 5-year study, as did 72% in the topical steroid arm. During the course of the study, 38% of completers in the pimecrolimus arm used a low-potency topical steroid, and 41% used a midpotency topical steroid.
The two treatment strategies proved similarly well tolerated. The rate of discontinuation because of adverse events was 0.6% in the pimecrolimus cohort and 1.1% in the topical steroids group. The incidence and time to onset of all adverse events were similar in the two study arms. Serious adverse events, the majority of which were infections, occurred in 20% of the pimecrolimus group and 17% of the topical steroids group.
The study was funded by Novartis, which markets Elidel. Dr. Luger is a consultant to and a paid speaker for the pharmaceutical company.
PRAGUE – Long-term therapy utilizing a pimecrolimus-based strategy for mild to moderate atopic dermatitis beginning in infancy proved as effective and safe as conventional therapy with topical corticosteroids in a 5-year randomized study.
"Given that pimecrolimus cream 1% is not associated with topical corticosteroid-specific side effects, such as hypothalamic-pituitary-adrenal axis suppression or skin atrophy, which can limit their long-term use, pimecrolimus cream may represent the drug of choice for long-term atopic dermatitis management," said Dr. Thomas A. Luger, professor and chairman of the department of dermatology at the University of Müenster (Germany).
Currently, pimecrolimus is not approved for use in patients under 2 years of age, he noted.
Dr. Luger, who is an investigator on the study’s steering committee, reported on 2,418 infants with mild to moderate atopic dermatitis randomized to daily use of an emollient for their dry skin plus either pimecrolimus cream 1% (Elidel) or low-potency to midpotency topical steroids on an as-needed basis. The participants’ mean age at enrollment was 7 months. Fifty-three percent of infants had moderate atopic dermatitis based on the Investigator’s Global Assessment; the rest had mild disease.
This was a 5-year, international, open-label randomized study designed to reflect real world, as-needed use of topical medications. Patients in the pimecrolimus group applied the topical calcineurin inhibitor at the first sign of an atopic dermatitis flare. If pimecrolimus didn’t control the itching, redness, and/or lesion elevation to the parents’ satisfaction, the child was switched to a low-potency topical steroid. If that didn’t quell the episode, a midpotency topical steroid was used. And if the child’s skin disease worsened despite the midpotency topical steroid, the patient went to the clinic. If the physician determined systemic therapy was needed, the child was removed from the study.
Treatment success was defined as a score of 0 or 1, meaning clear or almost clear, on the Investigator’s Global Assessment. By week 3 of the study, more than 50% of patients in both treatment arms met that standard. The success rate gradually increased over a 5-year period to about 90% overall in both groups and greater than 95% for facial atopic dermatitis.
At baseline, the participants’ mean total body surface area affected by atopic dermatitis was 17%, and 22% of infants had a total body surface area of 30% or greater. By week 3, however, the mean total body surface area had dropped below 5% in both groups. The total body surface area progressively improved, reaching a mean of 0% at week 78 and staying that way for the remainder of the 5-year study, Dr. Luger reported at the annual congress of the European Academy of Dermatology and Venereology.
Of children in the pimecrolimus cohort, 70% completed the 5-year study, as did 72% in the topical steroid arm. During the course of the study, 38% of completers in the pimecrolimus arm used a low-potency topical steroid, and 41% used a midpotency topical steroid.
The two treatment strategies proved similarly well tolerated. The rate of discontinuation because of adverse events was 0.6% in the pimecrolimus cohort and 1.1% in the topical steroids group. The incidence and time to onset of all adverse events were similar in the two study arms. Serious adverse events, the majority of which were infections, occurred in 20% of the pimecrolimus group and 17% of the topical steroids group.
The study was funded by Novartis, which markets Elidel. Dr. Luger is a consultant to and a paid speaker for the pharmaceutical company.
PRAGUE – Long-term therapy utilizing a pimecrolimus-based strategy for mild to moderate atopic dermatitis beginning in infancy proved as effective and safe as conventional therapy with topical corticosteroids in a 5-year randomized study.
"Given that pimecrolimus cream 1% is not associated with topical corticosteroid-specific side effects, such as hypothalamic-pituitary-adrenal axis suppression or skin atrophy, which can limit their long-term use, pimecrolimus cream may represent the drug of choice for long-term atopic dermatitis management," said Dr. Thomas A. Luger, professor and chairman of the department of dermatology at the University of Müenster (Germany).
Currently, pimecrolimus is not approved for use in patients under 2 years of age, he noted.
Dr. Luger, who is an investigator on the study’s steering committee, reported on 2,418 infants with mild to moderate atopic dermatitis randomized to daily use of an emollient for their dry skin plus either pimecrolimus cream 1% (Elidel) or low-potency to midpotency topical steroids on an as-needed basis. The participants’ mean age at enrollment was 7 months. Fifty-three percent of infants had moderate atopic dermatitis based on the Investigator’s Global Assessment; the rest had mild disease.
This was a 5-year, international, open-label randomized study designed to reflect real world, as-needed use of topical medications. Patients in the pimecrolimus group applied the topical calcineurin inhibitor at the first sign of an atopic dermatitis flare. If pimecrolimus didn’t control the itching, redness, and/or lesion elevation to the parents’ satisfaction, the child was switched to a low-potency topical steroid. If that didn’t quell the episode, a midpotency topical steroid was used. And if the child’s skin disease worsened despite the midpotency topical steroid, the patient went to the clinic. If the physician determined systemic therapy was needed, the child was removed from the study.
Treatment success was defined as a score of 0 or 1, meaning clear or almost clear, on the Investigator’s Global Assessment. By week 3 of the study, more than 50% of patients in both treatment arms met that standard. The success rate gradually increased over a 5-year period to about 90% overall in both groups and greater than 95% for facial atopic dermatitis.
At baseline, the participants’ mean total body surface area affected by atopic dermatitis was 17%, and 22% of infants had a total body surface area of 30% or greater. By week 3, however, the mean total body surface area had dropped below 5% in both groups. The total body surface area progressively improved, reaching a mean of 0% at week 78 and staying that way for the remainder of the 5-year study, Dr. Luger reported at the annual congress of the European Academy of Dermatology and Venereology.
Of children in the pimecrolimus cohort, 70% completed the 5-year study, as did 72% in the topical steroid arm. During the course of the study, 38% of completers in the pimecrolimus arm used a low-potency topical steroid, and 41% used a midpotency topical steroid.
The two treatment strategies proved similarly well tolerated. The rate of discontinuation because of adverse events was 0.6% in the pimecrolimus cohort and 1.1% in the topical steroids group. The incidence and time to onset of all adverse events were similar in the two study arms. Serious adverse events, the majority of which were infections, occurred in 20% of the pimecrolimus group and 17% of the topical steroids group.
The study was funded by Novartis, which markets Elidel. Dr. Luger is a consultant to and a paid speaker for the pharmaceutical company.
AT THE ANNUAL CONGRESS OF THE EUROPEAN ACADEMY OF DERMATOLOGY AND VENEREOLOGY
Major Finding: After 5 years of participation in a study of pimecrolimus- versus topical corticosteroid-based treatment strategies for infants with atopic dermatitis, about 90% of patients in both study arms were rated clear or almost clear.
Data Source: This was a randomized, international, prospective open-label study involving 2,418 infants with mild to moderate atopic dermatitis.
Disclosures: The study was funded by Novartis, which markets Elidel. Dr. Luger is a consultant to and a paid speaker for the pharmaceutical company.
Psoriasis Plus Diabetes Equals Heightened Vascular Risk
PRAGUE – Patients with diabetes and psoriasis are significantly more likely to develop new-onset, diabetes-related microvascular and macrovascular complications than are psoriasis-free patients with diabetes, according to the results of a large study.
"In view of this greater likelihood of developing microvascular and macrovascular complications, clinicians may wish to consider closer disease management of diabetic patients with psoriasis," said Dr. April W. Armstrong, director of clinical research and teledermatology for the department of dermatology at the University of California Davis Health System. The study is the first to examine the impact of comorbid psoriasis and diabetes – two diseases characterized by systemic inflammation – on the risk of diabetic vascular complications.
She and her coinvestigators identified 6,164 adult patients with psoriasis and diabetes in the Thomson Reuters MarketScan medical records database covering 2000-2006. A limitation of the database is its lack of information regarding how well the participants’ diabetes was controlled, Dr. Armstrong noted.
The patients were matched to an equal number of control patients (psoriasis-free patients with diabetes) based on sex, diabetes type, prior diabetic complications, diabetic complications, and a vascular complications propensity score. Then, the researchers analyzed the risk of incident diabetes-related microvascular and macrovascular complications over the subsequent 1, 3, and 5 years.
At 12 months’ follow-up, 18.3% of the patients with diabetes and psoriasis had developed new-onset diabetic microvascular complications – that is, retinopathy, neuropathy, or nephropathy – compared with 16.5% of controls. Similarly, 18.6% of patients with diabetes and psoriasis developed microvascular complications within this time frame, compared with 15.9% of controls.
Over the full 5 years of follow-up, 29.2% of the patients with diabetes and psoriasis experienced incident microvascular and 28.6% developed macrovascular complications, compared with 26.0% and 25.7% of control patients. This translated to a highly significant adjusted 14% increased relative risk of incident microvascular complications in the cohort with comorbid diabetes and psoriasis, as well as a 13% increased incidence of macrovascular complications such as MI, heart failure, or stroke, Dr. Armstrong reported at the annual congress of the European Academy of Dermatology and Venereology.
At baseline, investigators classified 73% of the 6,164 psoriasis patients who also had diabetes as having mild psoriasis based on their use of topical therapies only, while the remaining 27% had moderate to severe psoriasis defined by their use of systemic medications or phototherapy.
Next, the researchers sought to find out whether the risk of diabetic vascular complications in patients with comorbid psoriasis climbed with increasing severity of their dermatologic disease. The answer turned out to be "sort of," noted Dr. Armstrong.
That is, the adjusted 5-year risk of incident diabetic microvascular complications was 13% greater in patients with mild psoriasis than in psoriasis-free patients with diabetes, and 16% greater in those with moderate to severe psoriasis. The results were consistent with the notion that more severe psoriasis, with its greater attendant systemic inflammation, spells greater vascular risk.
However, the risk for macrovascular complications wasn’t as clear cut. While the risk of diabetic macrovascular complications was increased 16% in patients with diabetes and mild psoriasis, the relative risk in those with moderate to severe psoriasis was only 10% more than in psoriasis-free patients with diabetes – and that degree of elevation didn’t achieve statistical significance.
One plausible explanation for this discordant finding, according to Dr. Armstrong, is that the use of systemic therapies in the cohort with moderate to severe psoriasis quelled their systemic inflammation, which dampened their risk for macrovascular disease to a level similar to that seen in patients with diabetes without psoriasis, which, it must be emphasized, is still significantly greater than in the general population without diabetes.
Dr. Armstrong reported being the recipient of psoriasis research grants from Abbott Laboratories, which sponsored this study, as well as from Amgen and Janssen Pharmaceuticals.
PRAGUE – Patients with diabetes and psoriasis are significantly more likely to develop new-onset, diabetes-related microvascular and macrovascular complications than are psoriasis-free patients with diabetes, according to the results of a large study.
"In view of this greater likelihood of developing microvascular and macrovascular complications, clinicians may wish to consider closer disease management of diabetic patients with psoriasis," said Dr. April W. Armstrong, director of clinical research and teledermatology for the department of dermatology at the University of California Davis Health System. The study is the first to examine the impact of comorbid psoriasis and diabetes – two diseases characterized by systemic inflammation – on the risk of diabetic vascular complications.
She and her coinvestigators identified 6,164 adult patients with psoriasis and diabetes in the Thomson Reuters MarketScan medical records database covering 2000-2006. A limitation of the database is its lack of information regarding how well the participants’ diabetes was controlled, Dr. Armstrong noted.
The patients were matched to an equal number of control patients (psoriasis-free patients with diabetes) based on sex, diabetes type, prior diabetic complications, diabetic complications, and a vascular complications propensity score. Then, the researchers analyzed the risk of incident diabetes-related microvascular and macrovascular complications over the subsequent 1, 3, and 5 years.
At 12 months’ follow-up, 18.3% of the patients with diabetes and psoriasis had developed new-onset diabetic microvascular complications – that is, retinopathy, neuropathy, or nephropathy – compared with 16.5% of controls. Similarly, 18.6% of patients with diabetes and psoriasis developed microvascular complications within this time frame, compared with 15.9% of controls.
Over the full 5 years of follow-up, 29.2% of the patients with diabetes and psoriasis experienced incident microvascular and 28.6% developed macrovascular complications, compared with 26.0% and 25.7% of control patients. This translated to a highly significant adjusted 14% increased relative risk of incident microvascular complications in the cohort with comorbid diabetes and psoriasis, as well as a 13% increased incidence of macrovascular complications such as MI, heart failure, or stroke, Dr. Armstrong reported at the annual congress of the European Academy of Dermatology and Venereology.
At baseline, investigators classified 73% of the 6,164 psoriasis patients who also had diabetes as having mild psoriasis based on their use of topical therapies only, while the remaining 27% had moderate to severe psoriasis defined by their use of systemic medications or phototherapy.
Next, the researchers sought to find out whether the risk of diabetic vascular complications in patients with comorbid psoriasis climbed with increasing severity of their dermatologic disease. The answer turned out to be "sort of," noted Dr. Armstrong.
That is, the adjusted 5-year risk of incident diabetic microvascular complications was 13% greater in patients with mild psoriasis than in psoriasis-free patients with diabetes, and 16% greater in those with moderate to severe psoriasis. The results were consistent with the notion that more severe psoriasis, with its greater attendant systemic inflammation, spells greater vascular risk.
However, the risk for macrovascular complications wasn’t as clear cut. While the risk of diabetic macrovascular complications was increased 16% in patients with diabetes and mild psoriasis, the relative risk in those with moderate to severe psoriasis was only 10% more than in psoriasis-free patients with diabetes – and that degree of elevation didn’t achieve statistical significance.
One plausible explanation for this discordant finding, according to Dr. Armstrong, is that the use of systemic therapies in the cohort with moderate to severe psoriasis quelled their systemic inflammation, which dampened their risk for macrovascular disease to a level similar to that seen in patients with diabetes without psoriasis, which, it must be emphasized, is still significantly greater than in the general population without diabetes.
Dr. Armstrong reported being the recipient of psoriasis research grants from Abbott Laboratories, which sponsored this study, as well as from Amgen and Janssen Pharmaceuticals.
PRAGUE – Patients with diabetes and psoriasis are significantly more likely to develop new-onset, diabetes-related microvascular and macrovascular complications than are psoriasis-free patients with diabetes, according to the results of a large study.
"In view of this greater likelihood of developing microvascular and macrovascular complications, clinicians may wish to consider closer disease management of diabetic patients with psoriasis," said Dr. April W. Armstrong, director of clinical research and teledermatology for the department of dermatology at the University of California Davis Health System. The study is the first to examine the impact of comorbid psoriasis and diabetes – two diseases characterized by systemic inflammation – on the risk of diabetic vascular complications.
She and her coinvestigators identified 6,164 adult patients with psoriasis and diabetes in the Thomson Reuters MarketScan medical records database covering 2000-2006. A limitation of the database is its lack of information regarding how well the participants’ diabetes was controlled, Dr. Armstrong noted.
The patients were matched to an equal number of control patients (psoriasis-free patients with diabetes) based on sex, diabetes type, prior diabetic complications, diabetic complications, and a vascular complications propensity score. Then, the researchers analyzed the risk of incident diabetes-related microvascular and macrovascular complications over the subsequent 1, 3, and 5 years.
At 12 months’ follow-up, 18.3% of the patients with diabetes and psoriasis had developed new-onset diabetic microvascular complications – that is, retinopathy, neuropathy, or nephropathy – compared with 16.5% of controls. Similarly, 18.6% of patients with diabetes and psoriasis developed microvascular complications within this time frame, compared with 15.9% of controls.
Over the full 5 years of follow-up, 29.2% of the patients with diabetes and psoriasis experienced incident microvascular and 28.6% developed macrovascular complications, compared with 26.0% and 25.7% of control patients. This translated to a highly significant adjusted 14% increased relative risk of incident microvascular complications in the cohort with comorbid diabetes and psoriasis, as well as a 13% increased incidence of macrovascular complications such as MI, heart failure, or stroke, Dr. Armstrong reported at the annual congress of the European Academy of Dermatology and Venereology.
At baseline, investigators classified 73% of the 6,164 psoriasis patients who also had diabetes as having mild psoriasis based on their use of topical therapies only, while the remaining 27% had moderate to severe psoriasis defined by their use of systemic medications or phototherapy.
Next, the researchers sought to find out whether the risk of diabetic vascular complications in patients with comorbid psoriasis climbed with increasing severity of their dermatologic disease. The answer turned out to be "sort of," noted Dr. Armstrong.
That is, the adjusted 5-year risk of incident diabetic microvascular complications was 13% greater in patients with mild psoriasis than in psoriasis-free patients with diabetes, and 16% greater in those with moderate to severe psoriasis. The results were consistent with the notion that more severe psoriasis, with its greater attendant systemic inflammation, spells greater vascular risk.
However, the risk for macrovascular complications wasn’t as clear cut. While the risk of diabetic macrovascular complications was increased 16% in patients with diabetes and mild psoriasis, the relative risk in those with moderate to severe psoriasis was only 10% more than in psoriasis-free patients with diabetes – and that degree of elevation didn’t achieve statistical significance.
One plausible explanation for this discordant finding, according to Dr. Armstrong, is that the use of systemic therapies in the cohort with moderate to severe psoriasis quelled their systemic inflammation, which dampened their risk for macrovascular disease to a level similar to that seen in patients with diabetes without psoriasis, which, it must be emphasized, is still significantly greater than in the general population without diabetes.
Dr. Armstrong reported being the recipient of psoriasis research grants from Abbott Laboratories, which sponsored this study, as well as from Amgen and Janssen Pharmaceuticals.
AT THE ANNUAL CONGRESS OF THE EUROPEAN ACADEMY OF DERMATOLOGY AND VENEREOLOGY
Major Finding: Patients with diabetes and psoriasis had 5-year risks of new-onset microvascular and macrovascular diabetic complications that were 14% and 13% greater, respectively, than in matched psoriasis-free patients with diabetes.
Data Source: This was a retrospective study of 6,164 psoriasis patients with diabetes and an equal number of matched psoriasis-free patients with diabetes.
Disclosures: Dr. Armstrong reported being the recipient of psoriasis research grants from Abbott Laboratories, which sponsored this study, as well as from Amgen and Janssen Pharmaceuticals.
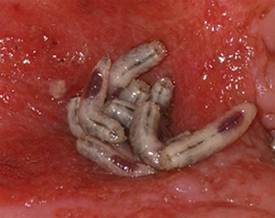
Maggots Prove Wound-Cleaning Worth
PRAGUE – Maggot debridement of wounds proved significantly faster, less painful, and less labor intensive than surgical debridement and conventional dressings in a randomized, multicenter clinical trial.
"There was quite an amazing debridement after 7 days of maggot therapy," Dr. Kristina Opletalova said of the phase III study findings presented at the annual congress of the European Academy of Dermatology and Venereology.
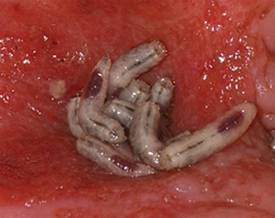
She reported on 119 patients hospitalized for 2 weeks for treatment of nonhealing sloughy wounds, most of which were venous ulcers on the lower limbs. Participants were randomized to maggot therapy or to thrice-weekly surgical debridement with topical anesthesia and conventional dressings.
The maggot therapy was administered via a novel delivery system: 80 maggots of Lucilia sericata were bagged in a special two-layer dressing, known as a Vitapad (BioMonde Laboratories), which allowed the critters to move and feed on the wound surface and kept them from escaping. The dressing was changed twice weekly.
Patients were blindfolded for all dressing changes so they didn’t know which study arm they were in. Wound sloughing was analyzed using computerized planimetry software, and other end points were assessed by an investigator blinded to treatment arm.
After 7 days of therapy, the wound status was significantly better in the maggot debridement group. The mean percentage of slough in wounds was 54.5%, compared with 66.5% in controls, but the significantly faster debridement didn’t boost the final healing rate. The day 15 percentage of slough in the wounds didn’t differ significantly between the two groups: 55.4% with maggot therapy and 53.8% in controls.
"So we think maggot debridement therapy should be stopped after 1 week and other types of dressings should then be used," said Dr. Opletalova, a dermatologist at the University of Caen (France).

Pain scores, which were assessed on a weekly basis, were similarly low in the two groups, but it must be noted that the control group received topical anesthetic and the maggot therapy group did not. Nursing time was four-fold greater in the control group. The study didn’t include a formal cost analysis, but the markedly reduced nursing time in the maggot therapy group is likely to spell cost savings, she said.
Maggot therapy was well tolerated. Patients expressed no reticence about it. A similar number of patients in both study arms reported a crawling sensation on their wounds.
Dr. Opletalova said that maggot therapy is likely to be particularly useful in patients with wounds that need rapid debridement, such as those with diabetes, or to prepare a wound for skin grafting or when there is an increased risk for infection.
Further details of the recently published randomized trial can be found in the Archives of Dermatology (2012;148:432-8).
The study was supported by university hospital research funding and by a grant from the French Society of Dermatology. Dr. Opletalova reported having no financial conflicts.
PRAGUE – Maggot debridement of wounds proved significantly faster, less painful, and less labor intensive than surgical debridement and conventional dressings in a randomized, multicenter clinical trial.
"There was quite an amazing debridement after 7 days of maggot therapy," Dr. Kristina Opletalova said of the phase III study findings presented at the annual congress of the European Academy of Dermatology and Venereology.
She reported on 119 patients hospitalized for 2 weeks for treatment of nonhealing sloughy wounds, most of which were venous ulcers on the lower limbs. Participants were randomized to maggot therapy or to thrice-weekly surgical debridement with topical anesthesia and conventional dressings.
The maggot therapy was administered via a novel delivery system: 80 maggots of Lucilia sericata were bagged in a special two-layer dressing, known as a Vitapad (BioMonde Laboratories), which allowed the critters to move and feed on the wound surface and kept them from escaping. The dressing was changed twice weekly.
Patients were blindfolded for all dressing changes so they didn’t know which study arm they were in. Wound sloughing was analyzed using computerized planimetry software, and other end points were assessed by an investigator blinded to treatment arm.
After 7 days of therapy, the wound status was significantly better in the maggot debridement group. The mean percentage of slough in wounds was 54.5%, compared with 66.5% in controls, but the significantly faster debridement didn’t boost the final healing rate. The day 15 percentage of slough in the wounds didn’t differ significantly between the two groups: 55.4% with maggot therapy and 53.8% in controls.
"So we think maggot debridement therapy should be stopped after 1 week and other types of dressings should then be used," said Dr. Opletalova, a dermatologist at the University of Caen (France).
Pain scores, which were assessed on a weekly basis, were similarly low in the two groups, but it must be noted that the control group received topical anesthetic and the maggot therapy group did not. Nursing time was four-fold greater in the control group. The study didn’t include a formal cost analysis, but the markedly reduced nursing time in the maggot therapy group is likely to spell cost savings, she said.
Maggot therapy was well tolerated. Patients expressed no reticence about it. A similar number of patients in both study arms reported a crawling sensation on their wounds.
Dr. Opletalova said that maggot therapy is likely to be particularly useful in patients with wounds that need rapid debridement, such as those with diabetes, or to prepare a wound for skin grafting or when there is an increased risk for infection.
Further details of the recently published randomized trial can be found in the Archives of Dermatology (2012;148:432-8).
The study was supported by university hospital research funding and by a grant from the French Society of Dermatology. Dr. Opletalova reported having no financial conflicts.
PRAGUE – Maggot debridement of wounds proved significantly faster, less painful, and less labor intensive than surgical debridement and conventional dressings in a randomized, multicenter clinical trial.
"There was quite an amazing debridement after 7 days of maggot therapy," Dr. Kristina Opletalova said of the phase III study findings presented at the annual congress of the European Academy of Dermatology and Venereology.
She reported on 119 patients hospitalized for 2 weeks for treatment of nonhealing sloughy wounds, most of which were venous ulcers on the lower limbs. Participants were randomized to maggot therapy or to thrice-weekly surgical debridement with topical anesthesia and conventional dressings.
The maggot therapy was administered via a novel delivery system: 80 maggots of Lucilia sericata were bagged in a special two-layer dressing, known as a Vitapad (BioMonde Laboratories), which allowed the critters to move and feed on the wound surface and kept them from escaping. The dressing was changed twice weekly.
Patients were blindfolded for all dressing changes so they didn’t know which study arm they were in. Wound sloughing was analyzed using computerized planimetry software, and other end points were assessed by an investigator blinded to treatment arm.
After 7 days of therapy, the wound status was significantly better in the maggot debridement group. The mean percentage of slough in wounds was 54.5%, compared with 66.5% in controls, but the significantly faster debridement didn’t boost the final healing rate. The day 15 percentage of slough in the wounds didn’t differ significantly between the two groups: 55.4% with maggot therapy and 53.8% in controls.
"So we think maggot debridement therapy should be stopped after 1 week and other types of dressings should then be used," said Dr. Opletalova, a dermatologist at the University of Caen (France).
Pain scores, which were assessed on a weekly basis, were similarly low in the two groups, but it must be noted that the control group received topical anesthetic and the maggot therapy group did not. Nursing time was four-fold greater in the control group. The study didn’t include a formal cost analysis, but the markedly reduced nursing time in the maggot therapy group is likely to spell cost savings, she said.
Maggot therapy was well tolerated. Patients expressed no reticence about it. A similar number of patients in both study arms reported a crawling sensation on their wounds.
Dr. Opletalova said that maggot therapy is likely to be particularly useful in patients with wounds that need rapid debridement, such as those with diabetes, or to prepare a wound for skin grafting or when there is an increased risk for infection.
Further details of the recently published randomized trial can be found in the Archives of Dermatology (2012;148:432-8).
The study was supported by university hospital research funding and by a grant from the French Society of Dermatology. Dr. Opletalova reported having no financial conflicts.
AT THE ANNUAL CONGRESS OF THE EUROPEAN ACADEMY OF DERMATOLOGY AND VENEREOLOGY
Major Finding: After 7 days of therapy, the mean percentage of slough in wounds was 54.5% in the maggot debridement group, compared with 66.5% in the control group.
Data Source: The phase-III findings come from a randomized, multicenter, blinded, prospective clinical trial involving 119 hospitalized patients.
Disclosures: The study was supported by university research funding and by a grant from the French Society of Dermatology. Dr. Opletalova reported having no financial conflicts.
Low-Dose Isotretinoin Tames Adult Acne
PRAGUE – Oral isotretinoin dosed at 5 mg per day proved to be highly effective, fast acting, and well tolerated for persistent, low-grade, adult acne in a randomized, double-blind clinical trial.
Results of this study provide physicians with evidence supporting the use of low-dose isotretinoin in the management of adult acne, Dr. Marius Rademaker said at the annual congress of the European Academy of Dermatology and Venereology.

"There’s a high degree of dissatisfaction with treatment among adult acne sufferers because of their usual slow response to the traditional acne therapies, the poor clearance, and the very high relapse rate when you stop treatment. The standards of a woman of 35 with adult acne are quite different from those of a 15-year-old. I think people no longer want 70% improvement, they want 100% clearance," said Dr. Rademaker, a dermatologist at Waikato Hospital in Hamilton, New Zealand.
There have been few randomized, controlled studies focusing on treatment of adult acne, he reported, adding that he could find no studies involving systemic antibiotics for acne in adults. He found a few studies on topical retinoid trials, but they only included a minority of adults. And, he found no studies assessing the effectiveness of low dose isotretinoin for adults.
Therefore, he conducted a randomized trial of isotretinoin at 5 mg/day to determine if a lower dose would be as effective and would have fewer adverse events than the standard dose of 0.5-1.0 mg/kg per day. Avoiding relapse upon discontinuation of isotretinoin appears to be more a function of the duration of sebaceous gland suppression – longer is better – than of cumulative dose, he added.
He reported on 58 adults aged 25-55 with low-grade, indolent acne that had persisted since adolescence. Nearly 90% were women. Participants were randomized double-blind to 16 weeks of isotretinoin 5 mg/day or placebo, followed by an additional 16 weeks of open-label isotretinoin in both study arms. The primary end point was the change in the number of facial acne lesions between baseline and week 16.
The acne lesion count in the isotretinoin group was reduced by half within the first 4 weeks of the study, from a mean baseline of 10.6 lesions. By week 16, the group’s mean acne lesion count had dropped to 3.2. After a further 16 weeks of open-label therapy, it had fallen to 1.3.
In contrast, the mean acne lesion count in the control group didn’t change significantly over the first 16 weeks from a baseline of 9.7 lesions. After a subsequent 16 weeks of open-label isotretinoin, acne count dropped to 3.9.
A secondary outcome measure was change in Dermatology Life Quality Index (DLQI) scores. From a mean baseline score of 4.8, indicative of moderate skin disease–related disability, the score fell to 1.3 after 16 weeks of double-blind isotretinoin and to 1.2 after another 16 weeks of open-label therapy. The mean DLQI score of 4.9 was unchanged in the control group after 16 weeks of placebo, but dropped to 2.3 after 16 weeks of open-label isotretinoin.
In an interview, Dr. Neil S. Goldberg, a dermatologist in Bronxville, N.Y, questioned the blindness of any study involving isotretinoin. "No isotretinoin study can be blinded. The side effects of isotretinoin, even low dose, are just too obvious."

The most common side effect in the study was dry lips, which nearly two-thirds of patients reported while on isotretinoin. Dry skin, musculoskeletal aches and pains, dry eyes, and fatigue were less frequently reported. One patient withdrew from the study because of anxiety and mood changes that may or may not have been treatment related, Dr. Rademaker said.
He added that in his experience, after a year of treatment at 5 mg/day, virtually all patients with persistent low-grade adult acne no longer have any acne. He is pursing the possibilities of conducting a long-term, follow-up study.
"Isotretinoin seems to me to be just as valuable a treatment in adults as in teenagers," said Dr. Steven R. Feldman, professor of dermatology at Wake Forest Baptist Medical Center in Winston-Salem, N.C. "My standard approach is to use it in the same way, though using it in lower doses for longer periods of time is reasonable and often effective with fewer side effects." Caution is warranted, however, when prescribing isotretinoin to women of childbearing potential because of the drug’s known teratogenicity.

In response to audience questions about restrictions placed upon isotretinoin prescribing in New Zealand, Dr. Rademaker replied that his country’s pregnancy prevention plan requires patient education but not mandatory pregnancy tests. "And our pregnancy rates [for females on isotretinoin] are lower than in Europe or the U.S.," he said.
"iPledge makes everything harder. It inhibits flexibility. In Australia – where my two nieces and nephew live – when they started isotretinoin, the doctor just gave them 6 months of isotretinoin and sent them on their way," said Dr. Goldberg.
Clinical acne is present in 45% of women aged 21-30 years, 26% of women aged 31-40, and 12% of women aged 41-50, according to the results of a recent cross-sectional study of 2,895 U.S. females aged 10-70 years (J. Womens Health [Larchmt] 2012;21:223-30).
Dr. Rademaker’s investigator-initiated study was sponsored by Douglas Pharmaceuticals (an isotretinoin manufacturer in New Zealand). He had no other conflicts of interest. Dr. Feldman reported significant financial relationships with several pharmaceutical companies. Dr. Goldberg had no financial conflicts to report.
In the United States, 5 mg of isotretinoin is not available, so dermatologists usually prescribe 10 mg per day as a low dose, according to Dr. Hilary E. Baldwin. She noted that she prescribes low-dose isotretinoin for adults, but not for getting patients from 70% to 100% clear.
 |
|
She added that she often prescribes isotretinoin for women who tend to get one to two large acne lesions per week. "Topical products are ineffective on this type of lesion and it is hard to justify long-term use of antibiotics for 1-2 lesions per week. Antibiotic resistance is of crucial importance in these patients. Alternatives to long term/low dose isotretinoin include oral contraceptives and spironolactone. However, some adult patients exceed the safe-age contraindications for OCPs," she said.
Dr. Baldwin is vice chair of dermatology at the State University of New York, Brooklyn. She has received research funds from and/or serves as a consultant to Allergan, Coria, Galderma, GlaxoSmithKline, Graceway Pharmaceuticals, L’Oreal, Ortho Dermatologics, Medicis, and Sanofi-Aventis.
In the United States, 5 mg of isotretinoin is not available, so dermatologists usually prescribe 10 mg per day as a low dose, according to Dr. Hilary E. Baldwin. She noted that she prescribes low-dose isotretinoin for adults, but not for getting patients from 70% to 100% clear.
|
|
She added that she often prescribes isotretinoin for women who tend to get one to two large acne lesions per week. "Topical products are ineffective on this type of lesion and it is hard to justify long-term use of antibiotics for 1-2 lesions per week. Antibiotic resistance is of crucial importance in these patients. Alternatives to long term/low dose isotretinoin include oral contraceptives and spironolactone. However, some adult patients exceed the safe-age contraindications for OCPs," she said.
Dr. Baldwin is vice chair of dermatology at the State University of New York, Brooklyn. She has received research funds from and/or serves as a consultant to Allergan, Coria, Galderma, GlaxoSmithKline, Graceway Pharmaceuticals, L’Oreal, Ortho Dermatologics, Medicis, and Sanofi-Aventis.
In the United States, 5 mg of isotretinoin is not available, so dermatologists usually prescribe 10 mg per day as a low dose, according to Dr. Hilary E. Baldwin. She noted that she prescribes low-dose isotretinoin for adults, but not for getting patients from 70% to 100% clear.
|
|
She added that she often prescribes isotretinoin for women who tend to get one to two large acne lesions per week. "Topical products are ineffective on this type of lesion and it is hard to justify long-term use of antibiotics for 1-2 lesions per week. Antibiotic resistance is of crucial importance in these patients. Alternatives to long term/low dose isotretinoin include oral contraceptives and spironolactone. However, some adult patients exceed the safe-age contraindications for OCPs," she said.
Dr. Baldwin is vice chair of dermatology at the State University of New York, Brooklyn. She has received research funds from and/or serves as a consultant to Allergan, Coria, Galderma, GlaxoSmithKline, Graceway Pharmaceuticals, L’Oreal, Ortho Dermatologics, Medicis, and Sanofi-Aventis.
PRAGUE – Oral isotretinoin dosed at 5 mg per day proved to be highly effective, fast acting, and well tolerated for persistent, low-grade, adult acne in a randomized, double-blind clinical trial.
Results of this study provide physicians with evidence supporting the use of low-dose isotretinoin in the management of adult acne, Dr. Marius Rademaker said at the annual congress of the European Academy of Dermatology and Venereology.
"There’s a high degree of dissatisfaction with treatment among adult acne sufferers because of their usual slow response to the traditional acne therapies, the poor clearance, and the very high relapse rate when you stop treatment. The standards of a woman of 35 with adult acne are quite different from those of a 15-year-old. I think people no longer want 70% improvement, they want 100% clearance," said Dr. Rademaker, a dermatologist at Waikato Hospital in Hamilton, New Zealand.
There have been few randomized, controlled studies focusing on treatment of adult acne, he reported, adding that he could find no studies involving systemic antibiotics for acne in adults. He found a few studies on topical retinoid trials, but they only included a minority of adults. And, he found no studies assessing the effectiveness of low dose isotretinoin for adults.
Therefore, he conducted a randomized trial of isotretinoin at 5 mg/day to determine if a lower dose would be as effective and would have fewer adverse events than the standard dose of 0.5-1.0 mg/kg per day. Avoiding relapse upon discontinuation of isotretinoin appears to be more a function of the duration of sebaceous gland suppression – longer is better – than of cumulative dose, he added.
He reported on 58 adults aged 25-55 with low-grade, indolent acne that had persisted since adolescence. Nearly 90% were women. Participants were randomized double-blind to 16 weeks of isotretinoin 5 mg/day or placebo, followed by an additional 16 weeks of open-label isotretinoin in both study arms. The primary end point was the change in the number of facial acne lesions between baseline and week 16.
The acne lesion count in the isotretinoin group was reduced by half within the first 4 weeks of the study, from a mean baseline of 10.6 lesions. By week 16, the group’s mean acne lesion count had dropped to 3.2. After a further 16 weeks of open-label therapy, it had fallen to 1.3.
In contrast, the mean acne lesion count in the control group didn’t change significantly over the first 16 weeks from a baseline of 9.7 lesions. After a subsequent 16 weeks of open-label isotretinoin, acne count dropped to 3.9.
A secondary outcome measure was change in Dermatology Life Quality Index (DLQI) scores. From a mean baseline score of 4.8, indicative of moderate skin disease–related disability, the score fell to 1.3 after 16 weeks of double-blind isotretinoin and to 1.2 after another 16 weeks of open-label therapy. The mean DLQI score of 4.9 was unchanged in the control group after 16 weeks of placebo, but dropped to 2.3 after 16 weeks of open-label isotretinoin.
In an interview, Dr. Neil S. Goldberg, a dermatologist in Bronxville, N.Y, questioned the blindness of any study involving isotretinoin. "No isotretinoin study can be blinded. The side effects of isotretinoin, even low dose, are just too obvious."
The most common side effect in the study was dry lips, which nearly two-thirds of patients reported while on isotretinoin. Dry skin, musculoskeletal aches and pains, dry eyes, and fatigue were less frequently reported. One patient withdrew from the study because of anxiety and mood changes that may or may not have been treatment related, Dr. Rademaker said.
He added that in his experience, after a year of treatment at 5 mg/day, virtually all patients with persistent low-grade adult acne no longer have any acne. He is pursing the possibilities of conducting a long-term, follow-up study.
"Isotretinoin seems to me to be just as valuable a treatment in adults as in teenagers," said Dr. Steven R. Feldman, professor of dermatology at Wake Forest Baptist Medical Center in Winston-Salem, N.C. "My standard approach is to use it in the same way, though using it in lower doses for longer periods of time is reasonable and often effective with fewer side effects." Caution is warranted, however, when prescribing isotretinoin to women of childbearing potential because of the drug’s known teratogenicity.
In response to audience questions about restrictions placed upon isotretinoin prescribing in New Zealand, Dr. Rademaker replied that his country’s pregnancy prevention plan requires patient education but not mandatory pregnancy tests. "And our pregnancy rates [for females on isotretinoin] are lower than in Europe or the U.S.," he said.
"iPledge makes everything harder. It inhibits flexibility. In Australia – where my two nieces and nephew live – when they started isotretinoin, the doctor just gave them 6 months of isotretinoin and sent them on their way," said Dr. Goldberg.
Clinical acne is present in 45% of women aged 21-30 years, 26% of women aged 31-40, and 12% of women aged 41-50, according to the results of a recent cross-sectional study of 2,895 U.S. females aged 10-70 years (J. Womens Health [Larchmt] 2012;21:223-30).
Dr. Rademaker’s investigator-initiated study was sponsored by Douglas Pharmaceuticals (an isotretinoin manufacturer in New Zealand). He had no other conflicts of interest. Dr. Feldman reported significant financial relationships with several pharmaceutical companies. Dr. Goldberg had no financial conflicts to report.
PRAGUE – Oral isotretinoin dosed at 5 mg per day proved to be highly effective, fast acting, and well tolerated for persistent, low-grade, adult acne in a randomized, double-blind clinical trial.
Results of this study provide physicians with evidence supporting the use of low-dose isotretinoin in the management of adult acne, Dr. Marius Rademaker said at the annual congress of the European Academy of Dermatology and Venereology.
"There’s a high degree of dissatisfaction with treatment among adult acne sufferers because of their usual slow response to the traditional acne therapies, the poor clearance, and the very high relapse rate when you stop treatment. The standards of a woman of 35 with adult acne are quite different from those of a 15-year-old. I think people no longer want 70% improvement, they want 100% clearance," said Dr. Rademaker, a dermatologist at Waikato Hospital in Hamilton, New Zealand.
There have been few randomized, controlled studies focusing on treatment of adult acne, he reported, adding that he could find no studies involving systemic antibiotics for acne in adults. He found a few studies on topical retinoid trials, but they only included a minority of adults. And, he found no studies assessing the effectiveness of low dose isotretinoin for adults.
Therefore, he conducted a randomized trial of isotretinoin at 5 mg/day to determine if a lower dose would be as effective and would have fewer adverse events than the standard dose of 0.5-1.0 mg/kg per day. Avoiding relapse upon discontinuation of isotretinoin appears to be more a function of the duration of sebaceous gland suppression – longer is better – than of cumulative dose, he added.
He reported on 58 adults aged 25-55 with low-grade, indolent acne that had persisted since adolescence. Nearly 90% were women. Participants were randomized double-blind to 16 weeks of isotretinoin 5 mg/day or placebo, followed by an additional 16 weeks of open-label isotretinoin in both study arms. The primary end point was the change in the number of facial acne lesions between baseline and week 16.
The acne lesion count in the isotretinoin group was reduced by half within the first 4 weeks of the study, from a mean baseline of 10.6 lesions. By week 16, the group’s mean acne lesion count had dropped to 3.2. After a further 16 weeks of open-label therapy, it had fallen to 1.3.
In contrast, the mean acne lesion count in the control group didn’t change significantly over the first 16 weeks from a baseline of 9.7 lesions. After a subsequent 16 weeks of open-label isotretinoin, acne count dropped to 3.9.
A secondary outcome measure was change in Dermatology Life Quality Index (DLQI) scores. From a mean baseline score of 4.8, indicative of moderate skin disease–related disability, the score fell to 1.3 after 16 weeks of double-blind isotretinoin and to 1.2 after another 16 weeks of open-label therapy. The mean DLQI score of 4.9 was unchanged in the control group after 16 weeks of placebo, but dropped to 2.3 after 16 weeks of open-label isotretinoin.
In an interview, Dr. Neil S. Goldberg, a dermatologist in Bronxville, N.Y, questioned the blindness of any study involving isotretinoin. "No isotretinoin study can be blinded. The side effects of isotretinoin, even low dose, are just too obvious."
The most common side effect in the study was dry lips, which nearly two-thirds of patients reported while on isotretinoin. Dry skin, musculoskeletal aches and pains, dry eyes, and fatigue were less frequently reported. One patient withdrew from the study because of anxiety and mood changes that may or may not have been treatment related, Dr. Rademaker said.
He added that in his experience, after a year of treatment at 5 mg/day, virtually all patients with persistent low-grade adult acne no longer have any acne. He is pursing the possibilities of conducting a long-term, follow-up study.
"Isotretinoin seems to me to be just as valuable a treatment in adults as in teenagers," said Dr. Steven R. Feldman, professor of dermatology at Wake Forest Baptist Medical Center in Winston-Salem, N.C. "My standard approach is to use it in the same way, though using it in lower doses for longer periods of time is reasonable and often effective with fewer side effects." Caution is warranted, however, when prescribing isotretinoin to women of childbearing potential because of the drug’s known teratogenicity.
In response to audience questions about restrictions placed upon isotretinoin prescribing in New Zealand, Dr. Rademaker replied that his country’s pregnancy prevention plan requires patient education but not mandatory pregnancy tests. "And our pregnancy rates [for females on isotretinoin] are lower than in Europe or the U.S.," he said.
"iPledge makes everything harder. It inhibits flexibility. In Australia – where my two nieces and nephew live – when they started isotretinoin, the doctor just gave them 6 months of isotretinoin and sent them on their way," said Dr. Goldberg.
Clinical acne is present in 45% of women aged 21-30 years, 26% of women aged 31-40, and 12% of women aged 41-50, according to the results of a recent cross-sectional study of 2,895 U.S. females aged 10-70 years (J. Womens Health [Larchmt] 2012;21:223-30).
Dr. Rademaker’s investigator-initiated study was sponsored by Douglas Pharmaceuticals (an isotretinoin manufacturer in New Zealand). He had no other conflicts of interest. Dr. Feldman reported significant financial relationships with several pharmaceutical companies. Dr. Goldberg had no financial conflicts to report.
AT THE ANNUAL CONGRESS OF THE EUROPEAN ACADEMY OF DERMATOLOGY AND VENEREOLOGY
Major Finding: Sixteen weeks of low-dose isotretinoin at 5 mg/day in patients with persistent, low-grade adult acne resulted in a reduction in mean facial lesion count from 10.6 at baseline to 3.2, with a further 16 weeks of open-label isotretinoin dropping the lesion count to 1.3.
Data Source: This was a randomized, double-blind, placebo-controlled trial involving 58 adults aged 25-55 with persistent, low-grade acne since adolescence.
Disclosures: Dr. Rademaker’s investigator-initiated study was sponsored by Douglas Pharmaceuticals (an isotretinoin manufacturer in New Zealand). He had no other conflicts of interest.

Brodalumab Knocks Psoriasis Out of Park
PRAGUE – In a 48-week, phase-II study of brodalumab – a novel selective interleukin-17 inhibitor – 60% of psoriasis patients achieved a Psoriasis Area and Severity Index score of 100, reported Dr. Kim A. Papp.
There were 181 patients with moderate to severe plaque psoriasis in the multicenter, open-label study, and nearly 100% achieved at least a PASI 75 response, 80% achieved at least a PASI 90 response, and 60% achieved a PASI 100, Dr. Papp said at the annual congress of the European Academy of Dermatology and Venereology. Equally important, responses were maintained through week 48, essentially undiminished, he said.

Dr. Papp, director of research at Probity Medical Research, Waterloo, Ont., presented the results of the open-label extension of a previously published phase II, 12-week study (N. Engl. J. Med. 2012;366:1181-9). At the end of the 12-week study, participants were observed off therapy until their psoriasis relapsed, defined as a 50% loss of therapeutic benefit. At that point, they were treated with 210 mg of brodalumab administered subcutaneously every 2 weeks for 48 weeks. The peak response was achieved between weeks 8 and 12 or 16; few study dropouts occurred.
"The side effect profile was varied and included a mix of serious infections and development of atrial fibrillation and other cardiac disorders. Certainly, there were no obvious signals to suggest that we should have undue concerns going into the planned phase III program," said Dr. Papp.
Session cochair Dr. Peter van de Kerkhof was enthusiastic about the brodalumab data. "These are absolutely fabulous results. It’s really very impressive. This study gives us a lead as to where we’re going: to a new phase in biologic therapy where we get an efficacy which we couldn’t have dreamed of 10 years ago. A new bar is set," said Dr. van de Kerkhof, professor and chairman of the department of dermatology at Radboud University in Nijmegen, the Netherlands.
Brodalumab (Amgen) is a human monoclonal antibody directed against the interleukin-17A receptor as a means of quelling inflammatory cytokines. Another humanized monoclonal antibody that neutralizes interleukin-17, ixekizumab (Eli Lilly), was also the subject of a recently published, encouraging phase II study (N. Engl. J. Med. 2012;366:1190-9). Secukinumab (Novartis) is another fully-human monoclonal antibody directed against IL-17A, which has completed several successful phase II studies. These anti-IL-17 biologics are being considered as possible new therapies for other inflammatory diseases, including psoriatic arthritis and rheumatoid arthritis.
Dr. van de Kerkhof cautioned that from a safety standpoint, it will be important to scrutinize the upcoming large, definitive phase III clinical trials of these agents for evidence of an increase in infections, particularly Staphylococcus aureus and Candida albicans.
Interleukin-17A and IL-17F play roles in the normal host-immune defenses against microorganisms. Patients with genetic defects in IL-17 tend to have problems with recurrent chronic mucocutaneous candidiasis. Moreover, individuals with chronic mucocutaneous candidiasis have been shown to have antibodies to IL-17A, IL-17F, and IL-22, he noted.
The study was sponsored by Amgen. Dr. Papp reported receiving research funds from and serving as a consultant to Amgen and numerous other pharmaceutical companies. Dr. van de Kerkhof has received research funding and consultancy fees from more than a dozen pharmaceutical companies.
PRAGUE – In a 48-week, phase-II study of brodalumab – a novel selective interleukin-17 inhibitor – 60% of psoriasis patients achieved a Psoriasis Area and Severity Index score of 100, reported Dr. Kim A. Papp.
There were 181 patients with moderate to severe plaque psoriasis in the multicenter, open-label study, and nearly 100% achieved at least a PASI 75 response, 80% achieved at least a PASI 90 response, and 60% achieved a PASI 100, Dr. Papp said at the annual congress of the European Academy of Dermatology and Venereology. Equally important, responses were maintained through week 48, essentially undiminished, he said.
Dr. Papp, director of research at Probity Medical Research, Waterloo, Ont., presented the results of the open-label extension of a previously published phase II, 12-week study (N. Engl. J. Med. 2012;366:1181-9). At the end of the 12-week study, participants were observed off therapy until their psoriasis relapsed, defined as a 50% loss of therapeutic benefit. At that point, they were treated with 210 mg of brodalumab administered subcutaneously every 2 weeks for 48 weeks. The peak response was achieved between weeks 8 and 12 or 16; few study dropouts occurred.
"The side effect profile was varied and included a mix of serious infections and development of atrial fibrillation and other cardiac disorders. Certainly, there were no obvious signals to suggest that we should have undue concerns going into the planned phase III program," said Dr. Papp.
Session cochair Dr. Peter van de Kerkhof was enthusiastic about the brodalumab data. "These are absolutely fabulous results. It’s really very impressive. This study gives us a lead as to where we’re going: to a new phase in biologic therapy where we get an efficacy which we couldn’t have dreamed of 10 years ago. A new bar is set," said Dr. van de Kerkhof, professor and chairman of the department of dermatology at Radboud University in Nijmegen, the Netherlands.
Brodalumab (Amgen) is a human monoclonal antibody directed against the interleukin-17A receptor as a means of quelling inflammatory cytokines. Another humanized monoclonal antibody that neutralizes interleukin-17, ixekizumab (Eli Lilly), was also the subject of a recently published, encouraging phase II study (N. Engl. J. Med. 2012;366:1190-9). Secukinumab (Novartis) is another fully-human monoclonal antibody directed against IL-17A, which has completed several successful phase II studies. These anti-IL-17 biologics are being considered as possible new therapies for other inflammatory diseases, including psoriatic arthritis and rheumatoid arthritis.
Dr. van de Kerkhof cautioned that from a safety standpoint, it will be important to scrutinize the upcoming large, definitive phase III clinical trials of these agents for evidence of an increase in infections, particularly Staphylococcus aureus and Candida albicans.
Interleukin-17A and IL-17F play roles in the normal host-immune defenses against microorganisms. Patients with genetic defects in IL-17 tend to have problems with recurrent chronic mucocutaneous candidiasis. Moreover, individuals with chronic mucocutaneous candidiasis have been shown to have antibodies to IL-17A, IL-17F, and IL-22, he noted.
The study was sponsored by Amgen. Dr. Papp reported receiving research funds from and serving as a consultant to Amgen and numerous other pharmaceutical companies. Dr. van de Kerkhof has received research funding and consultancy fees from more than a dozen pharmaceutical companies.
PRAGUE – In a 48-week, phase-II study of brodalumab – a novel selective interleukin-17 inhibitor – 60% of psoriasis patients achieved a Psoriasis Area and Severity Index score of 100, reported Dr. Kim A. Papp.
There were 181 patients with moderate to severe plaque psoriasis in the multicenter, open-label study, and nearly 100% achieved at least a PASI 75 response, 80% achieved at least a PASI 90 response, and 60% achieved a PASI 100, Dr. Papp said at the annual congress of the European Academy of Dermatology and Venereology. Equally important, responses were maintained through week 48, essentially undiminished, he said.
Dr. Papp, director of research at Probity Medical Research, Waterloo, Ont., presented the results of the open-label extension of a previously published phase II, 12-week study (N. Engl. J. Med. 2012;366:1181-9). At the end of the 12-week study, participants were observed off therapy until their psoriasis relapsed, defined as a 50% loss of therapeutic benefit. At that point, they were treated with 210 mg of brodalumab administered subcutaneously every 2 weeks for 48 weeks. The peak response was achieved between weeks 8 and 12 or 16; few study dropouts occurred.
"The side effect profile was varied and included a mix of serious infections and development of atrial fibrillation and other cardiac disorders. Certainly, there were no obvious signals to suggest that we should have undue concerns going into the planned phase III program," said Dr. Papp.
Session cochair Dr. Peter van de Kerkhof was enthusiastic about the brodalumab data. "These are absolutely fabulous results. It’s really very impressive. This study gives us a lead as to where we’re going: to a new phase in biologic therapy where we get an efficacy which we couldn’t have dreamed of 10 years ago. A new bar is set," said Dr. van de Kerkhof, professor and chairman of the department of dermatology at Radboud University in Nijmegen, the Netherlands.
Brodalumab (Amgen) is a human monoclonal antibody directed against the interleukin-17A receptor as a means of quelling inflammatory cytokines. Another humanized monoclonal antibody that neutralizes interleukin-17, ixekizumab (Eli Lilly), was also the subject of a recently published, encouraging phase II study (N. Engl. J. Med. 2012;366:1190-9). Secukinumab (Novartis) is another fully-human monoclonal antibody directed against IL-17A, which has completed several successful phase II studies. These anti-IL-17 biologics are being considered as possible new therapies for other inflammatory diseases, including psoriatic arthritis and rheumatoid arthritis.
Dr. van de Kerkhof cautioned that from a safety standpoint, it will be important to scrutinize the upcoming large, definitive phase III clinical trials of these agents for evidence of an increase in infections, particularly Staphylococcus aureus and Candida albicans.
Interleukin-17A and IL-17F play roles in the normal host-immune defenses against microorganisms. Patients with genetic defects in IL-17 tend to have problems with recurrent chronic mucocutaneous candidiasis. Moreover, individuals with chronic mucocutaneous candidiasis have been shown to have antibodies to IL-17A, IL-17F, and IL-22, he noted.
The study was sponsored by Amgen. Dr. Papp reported receiving research funds from and serving as a consultant to Amgen and numerous other pharmaceutical companies. Dr. van de Kerkhof has received research funding and consultancy fees from more than a dozen pharmaceutical companies.
AT THE ANNUAL CONGRESS OF THE EUROPEAN ACADEMY OF DERMATOLOGY AND VENEREOLOGY
Major Finding: After 48 weeks of treatment with brodalumab, 60% of patients with moderate to severe psoriasis displayed a PASI 100 response.
Data Source: This was a multicenter, open-label study involving 181 psoriasis patients who received 210 mg of brodalumab administered subcutaneously every 2 weeks.
Disclosures: This study was sponsored by Amgen. Dr. Papp reported receiving research funds from and serving as a consultant to Amgen and numerous other pharmaceutical companies. Dr. van de Kerkhof has received research funding and consultancy fees from more than a dozen pharmaceutical companies.

Botox for Chronic Migraine Brings Cost Savings
LONDON – Roughly 40% of the cost of 6 months of onabotulinumtoxinA for the treatment of chronic migraine is offset by resultant decreased use of emergency departments, urgent care facilities, and migraine-related hospitalizations, according to a prospective, real-world, cost-benefit study conducted in clinical practice.
"The associated savings offset a reasonable proportion of the cost of treatment for the entire group – nonresponders as well as responders – and there were a fair number of nonresponders because this was a severely affected group," Dr. John F. Rothrock said at the European Headache and Migraine Trust International Congress.

Moreover, the 40% figure undoubtedly underestimates the total savings by a considerable margin because it includes only migraine-related direct medical costs for emergency department and urgent care visits and hospitalizations. Additional savings would be expected as a result of reduced need for abortive medications and prophylactic therapies, as well as decreased work absenteeism, which is a "gigantic" indirect cost associated with chronic migraine, noted Dr. Rothrock, professor and chief of neurosciences at the University of Nevada, Reno.
Chronic migraine, defined as migraine headaches an average of at least 15 days per month, affects 1%-2% of U.S. adults. The costs, both economic and in terms of diminished quality of life, are enormous. Botox is the only Food and Drug Administration (FDA)-approved therapy for this common disorder.
But onabotulinumtoxinA (Botox) is also an expensive therapy, and that’s what prompted Dr. Rothrock to systematically study the treatment’s real-world economic impact.
He reported on 230 consecutive chronic migraine patients treated with Botox using the same protocol as in the two pivotal PREEMPT (Phase III Research Evaluating Migraine Prophylaxis Therapy) studies (Cephalalgia 2010;30:793-803 and 804-14) that led to FDA approval of an indication for Botox in treating chronic migraine: namely, injections at baseline, 3 months, and 6 months.
Nearly three-quarters of participants in Dr. Rothrock’s study were rated as having "very severe disability" using the MIDAS (Migraine Disability Assessment Score) system. The majority of patients had a median 36-month history of daily or near-daily headache. Subjects had tried a median of three appropriately dosed prophylactic agents before they received Botox. Thirty-seven percent of participants had symptomatic headache medication overuse.
Forty-eight percent of patients had a positive treatment response to Botox as defined by at least a 50% reduction in the number of headache days per month during month 6, compared with the month prior to starting treatment. In other words, they were successfully converted from chronic to episodic migraine.
The cost of a Botox treatment session, including the drug and reimbursement for the procedure, was estimated at $1,300. Costs for emergency department visits and other end points were based upon national averages.
The reduction in direct medical costs in the overall group during the 6 months after starting Botox was a mean $1,025, compared with the 6 months prior to beginning therapy. This was driven mainly by a mean 0.92 fewer emergency department visits and 0.39 fewer urgent care visits.
Asked when he typically resorts to Botox in patients with migraine, Dr. Rothrock replied that while many physicians reserve it for end-of-the-line therapy, as an advocate of evidence-based medicine, he disagrees with that approach.
"There’s clear evidence that Botox works. It’s the only FDA-approved treatment in the United States for chronic migraine, period," said Dr. Rothrock, who is the editor-in-chief of the journal Headache.
"But I think if you look at the literature concerning topiramate you’ll find that topiramate owns a pretty good evidence base," the neurologist continued. "My approach to the chronic migraine patient who’s topiramate naïve, which is a rare thing, is to try topiramate first. And if they tolerate it well, which is often a problem, and they respond well, then that’s great. I do that for cost reasons. It’s a lot cheaper than beginning Botox. But if they don’t respond to topiramate or they can’t tolerate it and they can’t then try zonisamide, a molecularly similar drug that has a middling-weak evidence base, then I’ll go straight to Botox. It’s my second-line therapy."
Aside from topiramate, the myriad other drugs used off-label for prophylaxis of chronic migraine have virtually no supporting evidence for that application, Dr. Rothrock observed.
"And I would maintain that the longer the patient remains in chronic migraine, the tougher will be your job to get them out of chronic migraine. So we can fiddle around with verapamil, et cetera, for chronic migraine, but I really would urge you to consider putting Botox very close to the top of your treatment armamentarium," he concluded.
The study was supported by a grant from Allergan. Dr. Rothrock reported serving as a consultant to the company
LONDON – Roughly 40% of the cost of 6 months of onabotulinumtoxinA for the treatment of chronic migraine is offset by resultant decreased use of emergency departments, urgent care facilities, and migraine-related hospitalizations, according to a prospective, real-world, cost-benefit study conducted in clinical practice.
"The associated savings offset a reasonable proportion of the cost of treatment for the entire group – nonresponders as well as responders – and there were a fair number of nonresponders because this was a severely affected group," Dr. John F. Rothrock said at the European Headache and Migraine Trust International Congress.
Moreover, the 40% figure undoubtedly underestimates the total savings by a considerable margin because it includes only migraine-related direct medical costs for emergency department and urgent care visits and hospitalizations. Additional savings would be expected as a result of reduced need for abortive medications and prophylactic therapies, as well as decreased work absenteeism, which is a "gigantic" indirect cost associated with chronic migraine, noted Dr. Rothrock, professor and chief of neurosciences at the University of Nevada, Reno.
Chronic migraine, defined as migraine headaches an average of at least 15 days per month, affects 1%-2% of U.S. adults. The costs, both economic and in terms of diminished quality of life, are enormous. Botox is the only Food and Drug Administration (FDA)-approved therapy for this common disorder.
But onabotulinumtoxinA (Botox) is also an expensive therapy, and that’s what prompted Dr. Rothrock to systematically study the treatment’s real-world economic impact.
He reported on 230 consecutive chronic migraine patients treated with Botox using the same protocol as in the two pivotal PREEMPT (Phase III Research Evaluating Migraine Prophylaxis Therapy) studies (Cephalalgia 2010;30:793-803 and 804-14) that led to FDA approval of an indication for Botox in treating chronic migraine: namely, injections at baseline, 3 months, and 6 months.
Nearly three-quarters of participants in Dr. Rothrock’s study were rated as having "very severe disability" using the MIDAS (Migraine Disability Assessment Score) system. The majority of patients had a median 36-month history of daily or near-daily headache. Subjects had tried a median of three appropriately dosed prophylactic agents before they received Botox. Thirty-seven percent of participants had symptomatic headache medication overuse.
Forty-eight percent of patients had a positive treatment response to Botox as defined by at least a 50% reduction in the number of headache days per month during month 6, compared with the month prior to starting treatment. In other words, they were successfully converted from chronic to episodic migraine.
The cost of a Botox treatment session, including the drug and reimbursement for the procedure, was estimated at $1,300. Costs for emergency department visits and other end points were based upon national averages.
The reduction in direct medical costs in the overall group during the 6 months after starting Botox was a mean $1,025, compared with the 6 months prior to beginning therapy. This was driven mainly by a mean 0.92 fewer emergency department visits and 0.39 fewer urgent care visits.
Asked when he typically resorts to Botox in patients with migraine, Dr. Rothrock replied that while many physicians reserve it for end-of-the-line therapy, as an advocate of evidence-based medicine, he disagrees with that approach.
"There’s clear evidence that Botox works. It’s the only FDA-approved treatment in the United States for chronic migraine, period," said Dr. Rothrock, who is the editor-in-chief of the journal Headache.
"But I think if you look at the literature concerning topiramate you’ll find that topiramate owns a pretty good evidence base," the neurologist continued. "My approach to the chronic migraine patient who’s topiramate naïve, which is a rare thing, is to try topiramate first. And if they tolerate it well, which is often a problem, and they respond well, then that’s great. I do that for cost reasons. It’s a lot cheaper than beginning Botox. But if they don’t respond to topiramate or they can’t tolerate it and they can’t then try zonisamide, a molecularly similar drug that has a middling-weak evidence base, then I’ll go straight to Botox. It’s my second-line therapy."
Aside from topiramate, the myriad other drugs used off-label for prophylaxis of chronic migraine have virtually no supporting evidence for that application, Dr. Rothrock observed.
"And I would maintain that the longer the patient remains in chronic migraine, the tougher will be your job to get them out of chronic migraine. So we can fiddle around with verapamil, et cetera, for chronic migraine, but I really would urge you to consider putting Botox very close to the top of your treatment armamentarium," he concluded.
The study was supported by a grant from Allergan. Dr. Rothrock reported serving as a consultant to the company
LONDON – Roughly 40% of the cost of 6 months of onabotulinumtoxinA for the treatment of chronic migraine is offset by resultant decreased use of emergency departments, urgent care facilities, and migraine-related hospitalizations, according to a prospective, real-world, cost-benefit study conducted in clinical practice.
"The associated savings offset a reasonable proportion of the cost of treatment for the entire group – nonresponders as well as responders – and there were a fair number of nonresponders because this was a severely affected group," Dr. John F. Rothrock said at the European Headache and Migraine Trust International Congress.
Moreover, the 40% figure undoubtedly underestimates the total savings by a considerable margin because it includes only migraine-related direct medical costs for emergency department and urgent care visits and hospitalizations. Additional savings would be expected as a result of reduced need for abortive medications and prophylactic therapies, as well as decreased work absenteeism, which is a "gigantic" indirect cost associated with chronic migraine, noted Dr. Rothrock, professor and chief of neurosciences at the University of Nevada, Reno.
Chronic migraine, defined as migraine headaches an average of at least 15 days per month, affects 1%-2% of U.S. adults. The costs, both economic and in terms of diminished quality of life, are enormous. Botox is the only Food and Drug Administration (FDA)-approved therapy for this common disorder.
But onabotulinumtoxinA (Botox) is also an expensive therapy, and that’s what prompted Dr. Rothrock to systematically study the treatment’s real-world economic impact.
He reported on 230 consecutive chronic migraine patients treated with Botox using the same protocol as in the two pivotal PREEMPT (Phase III Research Evaluating Migraine Prophylaxis Therapy) studies (Cephalalgia 2010;30:793-803 and 804-14) that led to FDA approval of an indication for Botox in treating chronic migraine: namely, injections at baseline, 3 months, and 6 months.
Nearly three-quarters of participants in Dr. Rothrock’s study were rated as having "very severe disability" using the MIDAS (Migraine Disability Assessment Score) system. The majority of patients had a median 36-month history of daily or near-daily headache. Subjects had tried a median of three appropriately dosed prophylactic agents before they received Botox. Thirty-seven percent of participants had symptomatic headache medication overuse.
Forty-eight percent of patients had a positive treatment response to Botox as defined by at least a 50% reduction in the number of headache days per month during month 6, compared with the month prior to starting treatment. In other words, they were successfully converted from chronic to episodic migraine.
The cost of a Botox treatment session, including the drug and reimbursement for the procedure, was estimated at $1,300. Costs for emergency department visits and other end points were based upon national averages.
The reduction in direct medical costs in the overall group during the 6 months after starting Botox was a mean $1,025, compared with the 6 months prior to beginning therapy. This was driven mainly by a mean 0.92 fewer emergency department visits and 0.39 fewer urgent care visits.
Asked when he typically resorts to Botox in patients with migraine, Dr. Rothrock replied that while many physicians reserve it for end-of-the-line therapy, as an advocate of evidence-based medicine, he disagrees with that approach.
"There’s clear evidence that Botox works. It’s the only FDA-approved treatment in the United States for chronic migraine, period," said Dr. Rothrock, who is the editor-in-chief of the journal Headache.
"But I think if you look at the literature concerning topiramate you’ll find that topiramate owns a pretty good evidence base," the neurologist continued. "My approach to the chronic migraine patient who’s topiramate naïve, which is a rare thing, is to try topiramate first. And if they tolerate it well, which is often a problem, and they respond well, then that’s great. I do that for cost reasons. It’s a lot cheaper than beginning Botox. But if they don’t respond to topiramate or they can’t tolerate it and they can’t then try zonisamide, a molecularly similar drug that has a middling-weak evidence base, then I’ll go straight to Botox. It’s my second-line therapy."
Aside from topiramate, the myriad other drugs used off-label for prophylaxis of chronic migraine have virtually no supporting evidence for that application, Dr. Rothrock observed.
"And I would maintain that the longer the patient remains in chronic migraine, the tougher will be your job to get them out of chronic migraine. So we can fiddle around with verapamil, et cetera, for chronic migraine, but I really would urge you to consider putting Botox very close to the top of your treatment armamentarium," he concluded.
The study was supported by a grant from Allergan. Dr. Rothrock reported serving as a consultant to the company
AT THE EUROPEAN HEADACHE AND MIGRAINE TRUST INTERNATIONAL CONGRESS
Major Finding: OnabotulinumtoxinA (Botox) therapy for chronic migraine resulted in significantly fewer emergency department and urgent care visits over the course of 6 months, compared with the 6 months prior to initiating therapy. This decreased use offset 40% of the treatment cost.
Data Source: This was a prospective, open-label, single-center study conducted in 230 consecutive patients with chronic migraine.
Disclosures: The study was supported by a grant from Allergan. Dr. Rothrock reported serving as a consultant to the company.

Botox May Greatly Reduce Migraine Chronicity
LONDON – Twenty-five percent of chronic migraine patients experience at least a 75% reduction in headache days per month after 6 months of treatment with onabotulinumtoxinA, according to a new analysis of data from the landmark PREEMPT trials.
"That’s what I tell my patients to expect: If they go on Botox [onabotulinumtoxinA], they have a 1 in 4 chance of a 75% or greater reduction in headache symptoms over 6 months of therapy," Dr. David W. Dodick said at the European Headache and Migraine Trust International Congress.

PREEMPT (Phase III Research Evaluating Migraine Prophylaxis Therapy) consisted of two pivotal trials that resulted in Food and Drug Administration licensure of Botox as the only approved treatment for chronic migraine, an often-disabling disorder that affects 1%-2% of the U.S. adult population.
PREEMPT involved 1,384 adults with chronic migraine as defined by the International Classification of Headache Disorders, 2nd edition, criteria – basically, an average of at least 15 days of headache per month – at 122 U.S. and European sites. Participants were randomized double-blind to Botox or placebo injections at baseline, 12, and 24 weeks. Then the placebo group was crossed over to open-label Botox, which all subjects received at weeks 36 and 48.
The previously reported primary end point was the change in frequency of headache days per month between baseline and 6 months. Botox significantly outperformed placebo, achieving a mean 8.4-day decrease in headache days per month from the pretreatment baseline of 19.9 days, compared with a 6.6-day drop with placebo (Cephalalgia 2010;30:793-803 and 804-14).
Dr. Dodick’s report on the proportion of patients achieving at least a 75% reduction in headache days per month was just one of several secondary end points in PREEMPT presented at the congress for the first time. Some PREEMPT investigators indicated they found these secondary outcome measures more clinically relevant than the primary study end point.

"The primary end point was perhaps one I wouldn’t have chosen myself," said Dr. John F. Rothrock, professor and chief of neurosciences at the University of Nevada, Reno, and editor-in-chief of the journal Headache. "The fact that you lose 1.8 headache days per month if you treat your patient with Botox is not really very exciting to me. It doesn’t lead me to be very enthusiastic about implementing a therapy that costs as much as Botox."
"A far more interesting end point to me – and one that was a secondary assessment in PREEMPT – was in how many patients do you achieve remission from chronic migraine back to episodic migraine with implementation of Botox therapy? Because that’s really what we’re after as clinicians: not just lopping off a couple of headache days per month, but how often can I wipe the slate clean and take that patient who has headache on more days than not – and in some cases, daily – and give them a headache-free or nearly headache-free existence off prophylactic medications and only occasionally using abortive therapy?" he continued.
The good news is that at the study’s end at week 56, roughly 70% of patients had achieved a 50% or greater reduction in headache days per month, compared with baseline. The divergence between active treatment and placebo in this secondary outcome measure was already significant at the first assessment, just 4 weeks after the first treatment session. The divergence grew through the first 6 months, then diminished once the former placebo group went on Botox, Dr. Rothrock noted.
The one potentially treatment-related serious adverse event seen in the study was status migrainosus requiring hospitalization. The incidence was 0.1% in the Botox arm and zero in the placebo group.
"I’ve now treated just short of 1,000 patients with Botox using the PREEMPT protocol, and it’s really unusual to run into a clinically significant side effect. The most common side effect in the PREEMPT trials was neck pain and stiffness. Patients talk about a wobbly neck or bobble head, a weakness in the neck. Other than that, it’s a very clean therapy, especially compared to other things that we use to try to suppress chronic migraine and get it to remit to episodic migraine," the neurologist observed.
Dr. Dodick agreed that the primary end point in PREEMPT doesn’t come close to capturing the whole story regarding Botox therapy.
"My own clinical experience using it for the last 12 years is that some patients have a significant reduction in headache days, while others who’ve had one continuous headache continue to do so with zero days reduction, but the severity is dramatically reduced. Their consumption of acute medications decreases and their response to medications is enhanced. So if you look at just one clinical end point – headache days per month – you may miss a dramatic improvement that occurs with no reduction in days," said Dr. Dodick, professor of neurology at the Mayo Clinic, Phoenix, and a past president of the American Headache Society.
Another newly analyzed secondary end point was the response rate per treatment cycle in PREEMPT. Dr. Dodick reported that 49% of participants responded to the first Botox injection with at 50% or greater reduction in their frequency of headache days per month. An additional 11% achieved this benchmark only after the second treatment cycle, and another 10% did so after the third.
"Just because a patient doesn’t respond to one injection doesn’t mean we shouldn’t try a second. I generally don’t go beyond two nonresponding injections. But don’t stop after one," he urged.
That advice is consistent with the results of an appraisal by the U.K. National Institute for Health and Clinical Excellence (NICE), which deemed Botox for chronic migraine "an appropriate use of NHS [National Health Service] resources," with the proviso that the treatment be stopped if it hasn’t achieved a 30% reduction in the number of headache days per month after two cycles.
THE PREEMPT trials and the new secondary analyses were sponsored by Allergan. Both Dr. Dodick and Dr. Rothrock reported receiving research funds from and consulting for the company.
LONDON – Twenty-five percent of chronic migraine patients experience at least a 75% reduction in headache days per month after 6 months of treatment with onabotulinumtoxinA, according to a new analysis of data from the landmark PREEMPT trials.
"That’s what I tell my patients to expect: If they go on Botox [onabotulinumtoxinA], they have a 1 in 4 chance of a 75% or greater reduction in headache symptoms over 6 months of therapy," Dr. David W. Dodick said at the European Headache and Migraine Trust International Congress.
PREEMPT (Phase III Research Evaluating Migraine Prophylaxis Therapy) consisted of two pivotal trials that resulted in Food and Drug Administration licensure of Botox as the only approved treatment for chronic migraine, an often-disabling disorder that affects 1%-2% of the U.S. adult population.
PREEMPT involved 1,384 adults with chronic migraine as defined by the International Classification of Headache Disorders, 2nd edition, criteria – basically, an average of at least 15 days of headache per month – at 122 U.S. and European sites. Participants were randomized double-blind to Botox or placebo injections at baseline, 12, and 24 weeks. Then the placebo group was crossed over to open-label Botox, which all subjects received at weeks 36 and 48.
The previously reported primary end point was the change in frequency of headache days per month between baseline and 6 months. Botox significantly outperformed placebo, achieving a mean 8.4-day decrease in headache days per month from the pretreatment baseline of 19.9 days, compared with a 6.6-day drop with placebo (Cephalalgia 2010;30:793-803 and 804-14).
Dr. Dodick’s report on the proportion of patients achieving at least a 75% reduction in headache days per month was just one of several secondary end points in PREEMPT presented at the congress for the first time. Some PREEMPT investigators indicated they found these secondary outcome measures more clinically relevant than the primary study end point.
"The primary end point was perhaps one I wouldn’t have chosen myself," said Dr. John F. Rothrock, professor and chief of neurosciences at the University of Nevada, Reno, and editor-in-chief of the journal Headache. "The fact that you lose 1.8 headache days per month if you treat your patient with Botox is not really very exciting to me. It doesn’t lead me to be very enthusiastic about implementing a therapy that costs as much as Botox."
"A far more interesting end point to me – and one that was a secondary assessment in PREEMPT – was in how many patients do you achieve remission from chronic migraine back to episodic migraine with implementation of Botox therapy? Because that’s really what we’re after as clinicians: not just lopping off a couple of headache days per month, but how often can I wipe the slate clean and take that patient who has headache on more days than not – and in some cases, daily – and give them a headache-free or nearly headache-free existence off prophylactic medications and only occasionally using abortive therapy?" he continued.
The good news is that at the study’s end at week 56, roughly 70% of patients had achieved a 50% or greater reduction in headache days per month, compared with baseline. The divergence between active treatment and placebo in this secondary outcome measure was already significant at the first assessment, just 4 weeks after the first treatment session. The divergence grew through the first 6 months, then diminished once the former placebo group went on Botox, Dr. Rothrock noted.
The one potentially treatment-related serious adverse event seen in the study was status migrainosus requiring hospitalization. The incidence was 0.1% in the Botox arm and zero in the placebo group.
"I’ve now treated just short of 1,000 patients with Botox using the PREEMPT protocol, and it’s really unusual to run into a clinically significant side effect. The most common side effect in the PREEMPT trials was neck pain and stiffness. Patients talk about a wobbly neck or bobble head, a weakness in the neck. Other than that, it’s a very clean therapy, especially compared to other things that we use to try to suppress chronic migraine and get it to remit to episodic migraine," the neurologist observed.
Dr. Dodick agreed that the primary end point in PREEMPT doesn’t come close to capturing the whole story regarding Botox therapy.
"My own clinical experience using it for the last 12 years is that some patients have a significant reduction in headache days, while others who’ve had one continuous headache continue to do so with zero days reduction, but the severity is dramatically reduced. Their consumption of acute medications decreases and their response to medications is enhanced. So if you look at just one clinical end point – headache days per month – you may miss a dramatic improvement that occurs with no reduction in days," said Dr. Dodick, professor of neurology at the Mayo Clinic, Phoenix, and a past president of the American Headache Society.
Another newly analyzed secondary end point was the response rate per treatment cycle in PREEMPT. Dr. Dodick reported that 49% of participants responded to the first Botox injection with at 50% or greater reduction in their frequency of headache days per month. An additional 11% achieved this benchmark only after the second treatment cycle, and another 10% did so after the third.
"Just because a patient doesn’t respond to one injection doesn’t mean we shouldn’t try a second. I generally don’t go beyond two nonresponding injections. But don’t stop after one," he urged.
That advice is consistent with the results of an appraisal by the U.K. National Institute for Health and Clinical Excellence (NICE), which deemed Botox for chronic migraine "an appropriate use of NHS [National Health Service] resources," with the proviso that the treatment be stopped if it hasn’t achieved a 30% reduction in the number of headache days per month after two cycles.
THE PREEMPT trials and the new secondary analyses were sponsored by Allergan. Both Dr. Dodick and Dr. Rothrock reported receiving research funds from and consulting for the company.
LONDON – Twenty-five percent of chronic migraine patients experience at least a 75% reduction in headache days per month after 6 months of treatment with onabotulinumtoxinA, according to a new analysis of data from the landmark PREEMPT trials.
"That’s what I tell my patients to expect: If they go on Botox [onabotulinumtoxinA], they have a 1 in 4 chance of a 75% or greater reduction in headache symptoms over 6 months of therapy," Dr. David W. Dodick said at the European Headache and Migraine Trust International Congress.
PREEMPT (Phase III Research Evaluating Migraine Prophylaxis Therapy) consisted of two pivotal trials that resulted in Food and Drug Administration licensure of Botox as the only approved treatment for chronic migraine, an often-disabling disorder that affects 1%-2% of the U.S. adult population.
PREEMPT involved 1,384 adults with chronic migraine as defined by the International Classification of Headache Disorders, 2nd edition, criteria – basically, an average of at least 15 days of headache per month – at 122 U.S. and European sites. Participants were randomized double-blind to Botox or placebo injections at baseline, 12, and 24 weeks. Then the placebo group was crossed over to open-label Botox, which all subjects received at weeks 36 and 48.
The previously reported primary end point was the change in frequency of headache days per month between baseline and 6 months. Botox significantly outperformed placebo, achieving a mean 8.4-day decrease in headache days per month from the pretreatment baseline of 19.9 days, compared with a 6.6-day drop with placebo (Cephalalgia 2010;30:793-803 and 804-14).
Dr. Dodick’s report on the proportion of patients achieving at least a 75% reduction in headache days per month was just one of several secondary end points in PREEMPT presented at the congress for the first time. Some PREEMPT investigators indicated they found these secondary outcome measures more clinically relevant than the primary study end point.
"The primary end point was perhaps one I wouldn’t have chosen myself," said Dr. John F. Rothrock, professor and chief of neurosciences at the University of Nevada, Reno, and editor-in-chief of the journal Headache. "The fact that you lose 1.8 headache days per month if you treat your patient with Botox is not really very exciting to me. It doesn’t lead me to be very enthusiastic about implementing a therapy that costs as much as Botox."
"A far more interesting end point to me – and one that was a secondary assessment in PREEMPT – was in how many patients do you achieve remission from chronic migraine back to episodic migraine with implementation of Botox therapy? Because that’s really what we’re after as clinicians: not just lopping off a couple of headache days per month, but how often can I wipe the slate clean and take that patient who has headache on more days than not – and in some cases, daily – and give them a headache-free or nearly headache-free existence off prophylactic medications and only occasionally using abortive therapy?" he continued.
The good news is that at the study’s end at week 56, roughly 70% of patients had achieved a 50% or greater reduction in headache days per month, compared with baseline. The divergence between active treatment and placebo in this secondary outcome measure was already significant at the first assessment, just 4 weeks after the first treatment session. The divergence grew through the first 6 months, then diminished once the former placebo group went on Botox, Dr. Rothrock noted.
The one potentially treatment-related serious adverse event seen in the study was status migrainosus requiring hospitalization. The incidence was 0.1% in the Botox arm and zero in the placebo group.
"I’ve now treated just short of 1,000 patients with Botox using the PREEMPT protocol, and it’s really unusual to run into a clinically significant side effect. The most common side effect in the PREEMPT trials was neck pain and stiffness. Patients talk about a wobbly neck or bobble head, a weakness in the neck. Other than that, it’s a very clean therapy, especially compared to other things that we use to try to suppress chronic migraine and get it to remit to episodic migraine," the neurologist observed.
Dr. Dodick agreed that the primary end point in PREEMPT doesn’t come close to capturing the whole story regarding Botox therapy.
"My own clinical experience using it for the last 12 years is that some patients have a significant reduction in headache days, while others who’ve had one continuous headache continue to do so with zero days reduction, but the severity is dramatically reduced. Their consumption of acute medications decreases and their response to medications is enhanced. So if you look at just one clinical end point – headache days per month – you may miss a dramatic improvement that occurs with no reduction in days," said Dr. Dodick, professor of neurology at the Mayo Clinic, Phoenix, and a past president of the American Headache Society.
Another newly analyzed secondary end point was the response rate per treatment cycle in PREEMPT. Dr. Dodick reported that 49% of participants responded to the first Botox injection with at 50% or greater reduction in their frequency of headache days per month. An additional 11% achieved this benchmark only after the second treatment cycle, and another 10% did so after the third.
"Just because a patient doesn’t respond to one injection doesn’t mean we shouldn’t try a second. I generally don’t go beyond two nonresponding injections. But don’t stop after one," he urged.
That advice is consistent with the results of an appraisal by the U.K. National Institute for Health and Clinical Excellence (NICE), which deemed Botox for chronic migraine "an appropriate use of NHS [National Health Service] resources," with the proviso that the treatment be stopped if it hasn’t achieved a 30% reduction in the number of headache days per month after two cycles.
THE PREEMPT trials and the new secondary analyses were sponsored by Allergan. Both Dr. Dodick and Dr. Rothrock reported receiving research funds from and consulting for the company.
AT THE EUROPEAN HEADACHE AND MIGRAINE TRUST INTERNATIONAL CONGRESS
Major Finding: One-quarter of patients with chronic migraine treated with three sets of onabotulinumtoxinA (Botox) injections over the course of 6 months experienced a 75% or greater reduction in headache days per month.
Data Source: This was a newly analyzed secondary end point in the randomized, double-blind, placebo-controlled PREEMPT trials, which included 1,384 patients at 122 sites.
Disclosures: The PREEMPT studies were sponsored by Allergan. Both Dr. Dodick and Dr. Rothrock reported receiving research funds from and consulting for the company.

Pediatricians Called Upon for More STD Screening
VAIL, COLO. – National data indicate general pediatricians now provide care for more adolescents than ever before – and that means they need to keep current with new trends in STD management, according to Dr. Ann-Christine Nyquist.
The authoritative source on STD diagnosis, treatment, and prevention is the Centers for Disease Control and Prevention’s "Sexually Transmitted Diseases Treatment Guidelines, 2010." But the "2010" is misleading; the guidelines are actually a living document that is constantly being updated. Thus, it’s best to consult the online version (www.cdc.gov/std/treatment) rather than a hard copy, noted Dr. Nyquist, professor of pediatrics at the University of Colorado at Denver.
As evidence that pediatricians need to be adept at STD management, she cited a major study funded by the American Board of Pediatrics showing that during the years 2000-2006, the proportion of office visits to general pediatricians made by the nation’s 11- to 17-year-olds climbed from 38% to 53%.
Meanwhile, the proportion of office visits to family physicians and other nonpediatric generalists declined from 38% to 30%, while the proportion of visits to nonpediatric specialists dropped from 28% to 22% and visits to pediatric specialists inched up from 2% to 5% (J. Pediatr. 2010;157:148-52e1), DrNyquist noted at a conference on pediatric infectious diseases, sponsored by Children’s Hospital Colorado.
The data came from the CDC’s National Ambulatory Medical Care Survey, with the analysis conducted by pediatricians at the University of Michigan, Ann Arbor. The study was a follow-up to an earlier report on national trends showing who is providing medical care for America’s children, conducted by the same research group (Arch. Pediatr. Adolesc. Med. 2004;158:22-6). The latest study confirms an accelerating trend seen during 1980-2000 for a greater proportion of health care visits for children and adolescents in the United States being provided by general pediatricians.
Nearly 50% of all STDs occur in 15- to 24-year-olds. The CDC guidelines recommend annual screening for chlamydia and gonorrhea in all sexually active females under age 25; the available data aren’t strong enough at present to support routine annual screening of all similarly aged males.
The preferred screening assay is a DNA-based nucleic acid amplification test because of its superior sensitivity and specificity. The assay can be done using a urine sample or a vaginal, rectal, or pharyngeal swab specimen. That’s a boon for pediatricians who are uncomfortable doing a pelvic exam to collect a cervical specimen.
It’s vital to talk with the sexually active adolescent about his or her specific sexual behaviors in order to know what sites to test. The considerable diversity in patterns of adolescent sexual initiation was underscored in a recent study of nearly 14,000 18-year-old participants in the National Longitudinal Study of Adolescent Health. Four in five had engaged in vaginal intercourse, oral-genital sexual activity, and/or anal intercourse by age 18. One in 10 reported anal intercourse. One-third of the youths initiated two or more of the sexual behaviors within a 1-year period (Am. J. Public Health 2012;102:1221-8).
Dr. Nyquist reported having no relevant financial conflicts.
VAIL, COLO. – National data indicate general pediatricians now provide care for more adolescents than ever before – and that means they need to keep current with new trends in STD management, according to Dr. Ann-Christine Nyquist.
The authoritative source on STD diagnosis, treatment, and prevention is the Centers for Disease Control and Prevention’s "Sexually Transmitted Diseases Treatment Guidelines, 2010." But the "2010" is misleading; the guidelines are actually a living document that is constantly being updated. Thus, it’s best to consult the online version (www.cdc.gov/std/treatment) rather than a hard copy, noted Dr. Nyquist, professor of pediatrics at the University of Colorado at Denver.
As evidence that pediatricians need to be adept at STD management, she cited a major study funded by the American Board of Pediatrics showing that during the years 2000-2006, the proportion of office visits to general pediatricians made by the nation’s 11- to 17-year-olds climbed from 38% to 53%.
Meanwhile, the proportion of office visits to family physicians and other nonpediatric generalists declined from 38% to 30%, while the proportion of visits to nonpediatric specialists dropped from 28% to 22% and visits to pediatric specialists inched up from 2% to 5% (J. Pediatr. 2010;157:148-52e1), DrNyquist noted at a conference on pediatric infectious diseases, sponsored by Children’s Hospital Colorado.
The data came from the CDC’s National Ambulatory Medical Care Survey, with the analysis conducted by pediatricians at the University of Michigan, Ann Arbor. The study was a follow-up to an earlier report on national trends showing who is providing medical care for America’s children, conducted by the same research group (Arch. Pediatr. Adolesc. Med. 2004;158:22-6). The latest study confirms an accelerating trend seen during 1980-2000 for a greater proportion of health care visits for children and adolescents in the United States being provided by general pediatricians.
Nearly 50% of all STDs occur in 15- to 24-year-olds. The CDC guidelines recommend annual screening for chlamydia and gonorrhea in all sexually active females under age 25; the available data aren’t strong enough at present to support routine annual screening of all similarly aged males.
The preferred screening assay is a DNA-based nucleic acid amplification test because of its superior sensitivity and specificity. The assay can be done using a urine sample or a vaginal, rectal, or pharyngeal swab specimen. That’s a boon for pediatricians who are uncomfortable doing a pelvic exam to collect a cervical specimen.
It’s vital to talk with the sexually active adolescent about his or her specific sexual behaviors in order to know what sites to test. The considerable diversity in patterns of adolescent sexual initiation was underscored in a recent study of nearly 14,000 18-year-old participants in the National Longitudinal Study of Adolescent Health. Four in five had engaged in vaginal intercourse, oral-genital sexual activity, and/or anal intercourse by age 18. One in 10 reported anal intercourse. One-third of the youths initiated two or more of the sexual behaviors within a 1-year period (Am. J. Public Health 2012;102:1221-8).
Dr. Nyquist reported having no relevant financial conflicts.
VAIL, COLO. – National data indicate general pediatricians now provide care for more adolescents than ever before – and that means they need to keep current with new trends in STD management, according to Dr. Ann-Christine Nyquist.
The authoritative source on STD diagnosis, treatment, and prevention is the Centers for Disease Control and Prevention’s "Sexually Transmitted Diseases Treatment Guidelines, 2010." But the "2010" is misleading; the guidelines are actually a living document that is constantly being updated. Thus, it’s best to consult the online version (www.cdc.gov/std/treatment) rather than a hard copy, noted Dr. Nyquist, professor of pediatrics at the University of Colorado at Denver.
As evidence that pediatricians need to be adept at STD management, she cited a major study funded by the American Board of Pediatrics showing that during the years 2000-2006, the proportion of office visits to general pediatricians made by the nation’s 11- to 17-year-olds climbed from 38% to 53%.
Meanwhile, the proportion of office visits to family physicians and other nonpediatric generalists declined from 38% to 30%, while the proportion of visits to nonpediatric specialists dropped from 28% to 22% and visits to pediatric specialists inched up from 2% to 5% (J. Pediatr. 2010;157:148-52e1), DrNyquist noted at a conference on pediatric infectious diseases, sponsored by Children’s Hospital Colorado.
The data came from the CDC’s National Ambulatory Medical Care Survey, with the analysis conducted by pediatricians at the University of Michigan, Ann Arbor. The study was a follow-up to an earlier report on national trends showing who is providing medical care for America’s children, conducted by the same research group (Arch. Pediatr. Adolesc. Med. 2004;158:22-6). The latest study confirms an accelerating trend seen during 1980-2000 for a greater proportion of health care visits for children and adolescents in the United States being provided by general pediatricians.
Nearly 50% of all STDs occur in 15- to 24-year-olds. The CDC guidelines recommend annual screening for chlamydia and gonorrhea in all sexually active females under age 25; the available data aren’t strong enough at present to support routine annual screening of all similarly aged males.
The preferred screening assay is a DNA-based nucleic acid amplification test because of its superior sensitivity and specificity. The assay can be done using a urine sample or a vaginal, rectal, or pharyngeal swab specimen. That’s a boon for pediatricians who are uncomfortable doing a pelvic exam to collect a cervical specimen.
It’s vital to talk with the sexually active adolescent about his or her specific sexual behaviors in order to know what sites to test. The considerable diversity in patterns of adolescent sexual initiation was underscored in a recent study of nearly 14,000 18-year-old participants in the National Longitudinal Study of Adolescent Health. Four in five had engaged in vaginal intercourse, oral-genital sexual activity, and/or anal intercourse by age 18. One in 10 reported anal intercourse. One-third of the youths initiated two or more of the sexual behaviors within a 1-year period (Am. J. Public Health 2012;102:1221-8).
Dr. Nyquist reported having no relevant financial conflicts.
EXPERT ANALYSIS FROM A CONFERENCE ON PEDIATRIC INFECTIOUS DISEASES
Epic Progress Seen in Reducing Pneumococcal Infections
VAIL, COLO. - American medicine emphatically surpassed the Healthy People 2010 goal for reduction of invasive Streptococcus pneumoniae infections well ahead of schedule in both of the highest-risk target groups: children under age 5 years and seniors. And the tougher Healthy People 2020 objectives are already well within striking distance.
"We get an A+ on this," Dr. Mary P. Glodé commented at the annual pediatric infectious diseases conference sponsored by Children’s Hospital Colorado.

The Healthy People 2010 objective was to reduce the incidence of invasive S. pneumoniae infections to 46 cases per 100,000 among children under age 5 years and to 42 per 100,000 persons age 65 or older. The actual 2010 rates were 19 and 36 per 100,000, respectively.
Between 1999 and 2010, the annual rate of invasive pneumococcal disease in children younger age 5 plummeted by 86% as a consequence of the licensure in 2000 of the pneumococcal conjugate vaccine 7 (PCV 7) vaccine.
Most impressively, the rate also fell by 50% during that period among seniors, even though they didn’t receive the vaccine. This is ascribed to herd immunity. The presumed mechanism is that once immunized, young children were far less likely to become colonized by virulent pneumococcal serotypes, with resultant diminished opportunity for transmission of the pathogens to older children and adults, explained Dr. Glodé, professor of pediatrics and head of the section of pediatric infectious disease at the University of Colorado, Denver, and Children’s Hospital Colorado.
The Healthy People 2020 goal is to further reduce the incidence of invasive pneumococcal infections in children under age 5 from the 2010 rate of 19 down to 12 per 100,000, and in seniors from the 2010 figure of 36 per 100,000 to 31 per 100,000.
The expectation is that the target in children will be handily reached, and early on, as a consequence of the spring 2010 recommendation by the American Academy of Pediatrics and the Centers for Disease Control and Prevention Advisory Committee on Immunization Practices (ACIP) that all children aged 2-59 months be routinely vaccinated with the PCV 13 vaccine, which contains the PCV 7 serotypes plus six others causing invasive disease. Early unpublished CDC data suggest there has already been some decline in disease in children as a result of the added serotypes included in PCV 13, especially in cases involving serotypes 19A and 7F, according to Dr. Glodé.
The difficult unanswered question concerns the best way to get the nation’s seniors to the Healthy People 2020 target. The rate of invasive pneumococcal disease is higher in persons aged 65 and older than in any other age group, as is the associated mortality. Indeed, of an estimated 44,000 cases of invasive pneumococcal disease in the United States during 2009, 37,000 were in adults, including 15,000 cases in individuals age 65 or more.
Late last year the Food and Drug Administration licensed the PCV 13 vaccine for use in people aged 50 and up. But that doesn’t necessarily mean it will see widespread use. There has been no official recommendation from the ACIP that the vaccine be routinely used in this population. The expert panel has noted that to date there are no data showing that the PCV 13 vaccine is effective in preventing pneumococcal pneumonia in adults, although a Dutch trial in 85,000 people over age 65 is underway.
The committee also noted that one-quarter of all cases of invasive pneumococcal disease in seniors are caused by 11 serotypes in the PPSV 23 vaccine that are not included in the PCV 13 vaccine, which further complicates the situation. The PPSV 23 vaccine has been approved since 1983 and is recommended for use in all adults over age 65 and in younger adults with certain underlying medical conditions, including diabetes and chronic lung disease.
Also, if the serotypes contained in the PCV 13 vaccine are going to largely go away in the senior population as a consequence of universal pediatric immunization, which could happen based upon the earlier herd immunity experience noted with the PCV 7 vaccine, then it may not be reasonable to give PCV 13 to all older adults, Dr. Glodé noted.
She reported serving on the data safety monitoring board for trials of an unrelated Pfizer vaccine.
VAIL, COLO. - American medicine emphatically surpassed the Healthy People 2010 goal for reduction of invasive Streptococcus pneumoniae infections well ahead of schedule in both of the highest-risk target groups: children under age 5 years and seniors. And the tougher Healthy People 2020 objectives are already well within striking distance.
"We get an A+ on this," Dr. Mary P. Glodé commented at the annual pediatric infectious diseases conference sponsored by Children’s Hospital Colorado.
The Healthy People 2010 objective was to reduce the incidence of invasive S. pneumoniae infections to 46 cases per 100,000 among children under age 5 years and to 42 per 100,000 persons age 65 or older. The actual 2010 rates were 19 and 36 per 100,000, respectively.
Between 1999 and 2010, the annual rate of invasive pneumococcal disease in children younger age 5 plummeted by 86% as a consequence of the licensure in 2000 of the pneumococcal conjugate vaccine 7 (PCV 7) vaccine.
Most impressively, the rate also fell by 50% during that period among seniors, even though they didn’t receive the vaccine. This is ascribed to herd immunity. The presumed mechanism is that once immunized, young children were far less likely to become colonized by virulent pneumococcal serotypes, with resultant diminished opportunity for transmission of the pathogens to older children and adults, explained Dr. Glodé, professor of pediatrics and head of the section of pediatric infectious disease at the University of Colorado, Denver, and Children’s Hospital Colorado.
The Healthy People 2020 goal is to further reduce the incidence of invasive pneumococcal infections in children under age 5 from the 2010 rate of 19 down to 12 per 100,000, and in seniors from the 2010 figure of 36 per 100,000 to 31 per 100,000.
The expectation is that the target in children will be handily reached, and early on, as a consequence of the spring 2010 recommendation by the American Academy of Pediatrics and the Centers for Disease Control and Prevention Advisory Committee on Immunization Practices (ACIP) that all children aged 2-59 months be routinely vaccinated with the PCV 13 vaccine, which contains the PCV 7 serotypes plus six others causing invasive disease. Early unpublished CDC data suggest there has already been some decline in disease in children as a result of the added serotypes included in PCV 13, especially in cases involving serotypes 19A and 7F, according to Dr. Glodé.
The difficult unanswered question concerns the best way to get the nation’s seniors to the Healthy People 2020 target. The rate of invasive pneumococcal disease is higher in persons aged 65 and older than in any other age group, as is the associated mortality. Indeed, of an estimated 44,000 cases of invasive pneumococcal disease in the United States during 2009, 37,000 were in adults, including 15,000 cases in individuals age 65 or more.
Late last year the Food and Drug Administration licensed the PCV 13 vaccine for use in people aged 50 and up. But that doesn’t necessarily mean it will see widespread use. There has been no official recommendation from the ACIP that the vaccine be routinely used in this population. The expert panel has noted that to date there are no data showing that the PCV 13 vaccine is effective in preventing pneumococcal pneumonia in adults, although a Dutch trial in 85,000 people over age 65 is underway.
The committee also noted that one-quarter of all cases of invasive pneumococcal disease in seniors are caused by 11 serotypes in the PPSV 23 vaccine that are not included in the PCV 13 vaccine, which further complicates the situation. The PPSV 23 vaccine has been approved since 1983 and is recommended for use in all adults over age 65 and in younger adults with certain underlying medical conditions, including diabetes and chronic lung disease.
Also, if the serotypes contained in the PCV 13 vaccine are going to largely go away in the senior population as a consequence of universal pediatric immunization, which could happen based upon the earlier herd immunity experience noted with the PCV 7 vaccine, then it may not be reasonable to give PCV 13 to all older adults, Dr. Glodé noted.
She reported serving on the data safety monitoring board for trials of an unrelated Pfizer vaccine.
VAIL, COLO. - American medicine emphatically surpassed the Healthy People 2010 goal for reduction of invasive Streptococcus pneumoniae infections well ahead of schedule in both of the highest-risk target groups: children under age 5 years and seniors. And the tougher Healthy People 2020 objectives are already well within striking distance.
"We get an A+ on this," Dr. Mary P. Glodé commented at the annual pediatric infectious diseases conference sponsored by Children’s Hospital Colorado.
The Healthy People 2010 objective was to reduce the incidence of invasive S. pneumoniae infections to 46 cases per 100,000 among children under age 5 years and to 42 per 100,000 persons age 65 or older. The actual 2010 rates were 19 and 36 per 100,000, respectively.
Between 1999 and 2010, the annual rate of invasive pneumococcal disease in children younger age 5 plummeted by 86% as a consequence of the licensure in 2000 of the pneumococcal conjugate vaccine 7 (PCV 7) vaccine.
Most impressively, the rate also fell by 50% during that period among seniors, even though they didn’t receive the vaccine. This is ascribed to herd immunity. The presumed mechanism is that once immunized, young children were far less likely to become colonized by virulent pneumococcal serotypes, with resultant diminished opportunity for transmission of the pathogens to older children and adults, explained Dr. Glodé, professor of pediatrics and head of the section of pediatric infectious disease at the University of Colorado, Denver, and Children’s Hospital Colorado.
The Healthy People 2020 goal is to further reduce the incidence of invasive pneumococcal infections in children under age 5 from the 2010 rate of 19 down to 12 per 100,000, and in seniors from the 2010 figure of 36 per 100,000 to 31 per 100,000.
The expectation is that the target in children will be handily reached, and early on, as a consequence of the spring 2010 recommendation by the American Academy of Pediatrics and the Centers for Disease Control and Prevention Advisory Committee on Immunization Practices (ACIP) that all children aged 2-59 months be routinely vaccinated with the PCV 13 vaccine, which contains the PCV 7 serotypes plus six others causing invasive disease. Early unpublished CDC data suggest there has already been some decline in disease in children as a result of the added serotypes included in PCV 13, especially in cases involving serotypes 19A and 7F, according to Dr. Glodé.
The difficult unanswered question concerns the best way to get the nation’s seniors to the Healthy People 2020 target. The rate of invasive pneumococcal disease is higher in persons aged 65 and older than in any other age group, as is the associated mortality. Indeed, of an estimated 44,000 cases of invasive pneumococcal disease in the United States during 2009, 37,000 were in adults, including 15,000 cases in individuals age 65 or more.
Late last year the Food and Drug Administration licensed the PCV 13 vaccine for use in people aged 50 and up. But that doesn’t necessarily mean it will see widespread use. There has been no official recommendation from the ACIP that the vaccine be routinely used in this population. The expert panel has noted that to date there are no data showing that the PCV 13 vaccine is effective in preventing pneumococcal pneumonia in adults, although a Dutch trial in 85,000 people over age 65 is underway.
The committee also noted that one-quarter of all cases of invasive pneumococcal disease in seniors are caused by 11 serotypes in the PPSV 23 vaccine that are not included in the PCV 13 vaccine, which further complicates the situation. The PPSV 23 vaccine has been approved since 1983 and is recommended for use in all adults over age 65 and in younger adults with certain underlying medical conditions, including diabetes and chronic lung disease.
Also, if the serotypes contained in the PCV 13 vaccine are going to largely go away in the senior population as a consequence of universal pediatric immunization, which could happen based upon the earlier herd immunity experience noted with the PCV 7 vaccine, then it may not be reasonable to give PCV 13 to all older adults, Dr. Glodé noted.
She reported serving on the data safety monitoring board for trials of an unrelated Pfizer vaccine.
EXPERT ANALYSIS FROM THE ANNUAL PEDIATRIC INFECTIOUS DISEASES CONFERENCE SPONSORED BY CHILDREN'S HOSPITAL COLORADO
Guideline Reflects New Thinking on Renal Scarring
VAIL, COLO. – The most recent American Academy of Pediatrics guidelines on management of urinary tract infections do an abrupt about-face by recommending that a voiding cystourethrogram no longer be routinely performed after a first febrile UTI in children aged 2 months to 2 years.
"This is confusing. On a dime, we have changed our recommendations regarding radiographic imaging," noted Dr. John W. Ogle, vice chair and director of pediatrics at Denver Health.

The previous 1999 AAP guidelines on UTIs stated unequivocally that "infants and children 2 months through 2 years of age who have the expected response to antimicrobials should have a sonogram and either voiding cystourethrogram or radionuclide scan at the earliest convenient time" (Pediatrics 1999;10[4 Pt 1]:843-52).
This sharp shift away from this stance in the current guidelines reflects new thinking regarding the pathogenesis of renal scarring, Dr. Ogle said at a conference on pediatric infectious diseases sponsored by Children’s Hospital Colorado.
"The assumption that has been made in the past is that UTI in the presence of reflux is the primary thing that leads to renal scarring. That’s not so clear anymore. Some of what we call scarring may be congenital cortical defects in the kidney that we discover because we’ve studied the child. There are good studies demonstrating that you can have scarring from pyelonephritis with complete absence of reflux," said Dr. Ogle, who is also a professor of pediatrics at the University of Colorado, Denver.
The operative hypothesis up until the current guidelines were released in 2011 (Pediatrics 2011;128:595-610) was that identifying reflux via a voiding cystourethrogram (VCUG) allowed physicians to intervene surgically or with prophylactic antimicrobials to prevent further reflux nephropathy with further scarring.
"The evidence seems to be quite clear now that the interventions don’t change those important outcomes of renal scarring. So why look for the reflux? Why do the study, with its cost and discomfort, if you don’t have data that an intervention is going to positively affect the child?" Dr. Ogle said.
A key piece of evidence that triggered the change in AAP recommendations was a Cochrane review of 11 studies totaling 1,148 children. The analysis concluded that correction of vesicoureteral reflux by surgery or medical interventions did not reduce the risk of renal scarring (Cochrane Database Syst. Rev. 2007 July 18;(3):CD001532).
The shift in the AAP guidelines has generated controversy among urologists, some of whom are in agreement with the change while others are opposed.
"I think for pediatricians, adopting these guidelines is an easy step," according to Dr. Ogle. "Many pediatricians in office practice had already adopted this years ago. They have not routinely done a VCUG on every child that presented with a febrile UTI. So for them, the impact of the 2011 Academy guidelines has been to say, ‘What you’ve been doing for the last 10 years is probably appropriate.’ "
He predicted that the current guidelines won’t be the final word on the topic of imaging in patients with febrile UTIs.
"What the guidelines don’t tell us is who exactly you should worry about. They don’t tell you which first-time UTIs you should consider VCUG in, and what’s the strategy for second-time UTIs. So stay tuned, there’s going to be further debate with regard to this, and hopefully further evidence," Dr. Ogle said.
"Remember," he continued, "the great history of American medicine is that first we adopt strategies for interventions in medical conditions, then we study them to see if our interventions are right."
Dr. Ogle reported having no relevant financial conflicts.
VAIL, COLO. – The most recent American Academy of Pediatrics guidelines on management of urinary tract infections do an abrupt about-face by recommending that a voiding cystourethrogram no longer be routinely performed after a first febrile UTI in children aged 2 months to 2 years.
"This is confusing. On a dime, we have changed our recommendations regarding radiographic imaging," noted Dr. John W. Ogle, vice chair and director of pediatrics at Denver Health.
The previous 1999 AAP guidelines on UTIs stated unequivocally that "infants and children 2 months through 2 years of age who have the expected response to antimicrobials should have a sonogram and either voiding cystourethrogram or radionuclide scan at the earliest convenient time" (Pediatrics 1999;10[4 Pt 1]:843-52).
This sharp shift away from this stance in the current guidelines reflects new thinking regarding the pathogenesis of renal scarring, Dr. Ogle said at a conference on pediatric infectious diseases sponsored by Children’s Hospital Colorado.
"The assumption that has been made in the past is that UTI in the presence of reflux is the primary thing that leads to renal scarring. That’s not so clear anymore. Some of what we call scarring may be congenital cortical defects in the kidney that we discover because we’ve studied the child. There are good studies demonstrating that you can have scarring from pyelonephritis with complete absence of reflux," said Dr. Ogle, who is also a professor of pediatrics at the University of Colorado, Denver.
The operative hypothesis up until the current guidelines were released in 2011 (Pediatrics 2011;128:595-610) was that identifying reflux via a voiding cystourethrogram (VCUG) allowed physicians to intervene surgically or with prophylactic antimicrobials to prevent further reflux nephropathy with further scarring.
"The evidence seems to be quite clear now that the interventions don’t change those important outcomes of renal scarring. So why look for the reflux? Why do the study, with its cost and discomfort, if you don’t have data that an intervention is going to positively affect the child?" Dr. Ogle said.
A key piece of evidence that triggered the change in AAP recommendations was a Cochrane review of 11 studies totaling 1,148 children. The analysis concluded that correction of vesicoureteral reflux by surgery or medical interventions did not reduce the risk of renal scarring (Cochrane Database Syst. Rev. 2007 July 18;(3):CD001532).
The shift in the AAP guidelines has generated controversy among urologists, some of whom are in agreement with the change while others are opposed.
"I think for pediatricians, adopting these guidelines is an easy step," according to Dr. Ogle. "Many pediatricians in office practice had already adopted this years ago. They have not routinely done a VCUG on every child that presented with a febrile UTI. So for them, the impact of the 2011 Academy guidelines has been to say, ‘What you’ve been doing for the last 10 years is probably appropriate.’ "
He predicted that the current guidelines won’t be the final word on the topic of imaging in patients with febrile UTIs.
"What the guidelines don’t tell us is who exactly you should worry about. They don’t tell you which first-time UTIs you should consider VCUG in, and what’s the strategy for second-time UTIs. So stay tuned, there’s going to be further debate with regard to this, and hopefully further evidence," Dr. Ogle said.
"Remember," he continued, "the great history of American medicine is that first we adopt strategies for interventions in medical conditions, then we study them to see if our interventions are right."
Dr. Ogle reported having no relevant financial conflicts.
VAIL, COLO. – The most recent American Academy of Pediatrics guidelines on management of urinary tract infections do an abrupt about-face by recommending that a voiding cystourethrogram no longer be routinely performed after a first febrile UTI in children aged 2 months to 2 years.
"This is confusing. On a dime, we have changed our recommendations regarding radiographic imaging," noted Dr. John W. Ogle, vice chair and director of pediatrics at Denver Health.
The previous 1999 AAP guidelines on UTIs stated unequivocally that "infants and children 2 months through 2 years of age who have the expected response to antimicrobials should have a sonogram and either voiding cystourethrogram or radionuclide scan at the earliest convenient time" (Pediatrics 1999;10[4 Pt 1]:843-52).
This sharp shift away from this stance in the current guidelines reflects new thinking regarding the pathogenesis of renal scarring, Dr. Ogle said at a conference on pediatric infectious diseases sponsored by Children’s Hospital Colorado.
"The assumption that has been made in the past is that UTI in the presence of reflux is the primary thing that leads to renal scarring. That’s not so clear anymore. Some of what we call scarring may be congenital cortical defects in the kidney that we discover because we’ve studied the child. There are good studies demonstrating that you can have scarring from pyelonephritis with complete absence of reflux," said Dr. Ogle, who is also a professor of pediatrics at the University of Colorado, Denver.
The operative hypothesis up until the current guidelines were released in 2011 (Pediatrics 2011;128:595-610) was that identifying reflux via a voiding cystourethrogram (VCUG) allowed physicians to intervene surgically or with prophylactic antimicrobials to prevent further reflux nephropathy with further scarring.
"The evidence seems to be quite clear now that the interventions don’t change those important outcomes of renal scarring. So why look for the reflux? Why do the study, with its cost and discomfort, if you don’t have data that an intervention is going to positively affect the child?" Dr. Ogle said.
A key piece of evidence that triggered the change in AAP recommendations was a Cochrane review of 11 studies totaling 1,148 children. The analysis concluded that correction of vesicoureteral reflux by surgery or medical interventions did not reduce the risk of renal scarring (Cochrane Database Syst. Rev. 2007 July 18;(3):CD001532).
The shift in the AAP guidelines has generated controversy among urologists, some of whom are in agreement with the change while others are opposed.
"I think for pediatricians, adopting these guidelines is an easy step," according to Dr. Ogle. "Many pediatricians in office practice had already adopted this years ago. They have not routinely done a VCUG on every child that presented with a febrile UTI. So for them, the impact of the 2011 Academy guidelines has been to say, ‘What you’ve been doing for the last 10 years is probably appropriate.’ "
He predicted that the current guidelines won’t be the final word on the topic of imaging in patients with febrile UTIs.
"What the guidelines don’t tell us is who exactly you should worry about. They don’t tell you which first-time UTIs you should consider VCUG in, and what’s the strategy for second-time UTIs. So stay tuned, there’s going to be further debate with regard to this, and hopefully further evidence," Dr. Ogle said.
"Remember," he continued, "the great history of American medicine is that first we adopt strategies for interventions in medical conditions, then we study them to see if our interventions are right."
Dr. Ogle reported having no relevant financial conflicts.
EXPERT ANALYSIS FROM A CONFERENCE ON PEDIATRIC INFECTIOUS DISEASES SPONSORED BY CHILDREN'S HOSPITAL COLORADO