User login
Beta Thalassemia: Pricey Gene Therapy Hits The Mark
With luck, maybe Ms. Ahmed’s son could follow in his aunt’s footsteps and get a stem cell transplant from a compatible family donor. But while little Yusuf Saeed has a twin sister of his own, she wasn’t a match. Without another treatment option, he’d face the prospect of a lifetime not only cut short but burdened by multiple monthly transfusions and severe limitations.
Then came glimpses of hope. One of Yusuf’s physicians at Cohen Children’s Medical Center in Long Island, New York, told Yusuf’s mother about a new kind of gene therapy on the horizon. But it took time to get FDA approval. Yusuf grew older, heading toward his teenage years, when regular transfusions would be a huge burden. “He’s turning 5 and 6, and there’s nothing,” Ms. Ahmed recalled, and the family worried.
Finally, the FDA approved the one-time treatment — betibeglogene autotemcel (beti-cel, Zynteglo) in 2022. By January 2024, the hospital was ready to treat Yusuf. At age 8, he became the first patient in the state of New York to undergo gene therapy for beta thalassemia.
A medical team infused Yusuf with his own stem cells, which had been genetically engineered to boost production of hemoglobin and prevent thalassemia’s devastating effects.
There are caveats about the treatment. It’s an extraordinarily expensive therapy that can be performed at only a few institutions. And it’s so brand new that caveats may not even have appeared yet. Yet, for kids like Yusuf, the gene therapy could transform a life.
“We feel like a weight has been lifted,” Ms. Ahmed said in an interview. “It’s something we’ve been waiting for.”
Anemia Becomes a Lifetime Threat
Among all genetic diseases, thalassemia stands alone. It’s the most common condition caused by a single gene, according to Hanny Al-Samkari, MD, a hematologist/clinical investigator at Massachusetts General Hospital and associate professor of medicine at Harvard Medical School, in Boston, Massachusetts.
Millions of people have the thalassemia trait, especially in southern Europe, the Middle East, southeast Asia, and Africa, Dr. Al-Samkari said. (Yusuf’s parents are from Pakistan.)
The trait, which appears to provide protection against malaria, may cause mild anemia in some cases but is otherwise harmless. However, a child born to parents with the same kind of trait has a high risk of developing alpha thalassemia or beta thalassemia. Like his aunt, Yusuf developed beta thalassemia, which is generally more severe. Yusuf’s bleeding disorder requires him to be transfusion-dependent.
In these patients, the disease disrupts the production of red blood cells in the bone marrow, Dr. Al-Samkari said. Hemoglobin levels can fall to 7 or 8 g/dL, compared with the normal levels of 12-16 g/dL in adults. “They’re chronically anemic, and that low hemoglobin that leads to things you associate with anemia: fatigue, reduced exercise tolerance, mind fog, challenges with work or school, and hypersomnolence.”
In addition, the bones become thinner and more brittle, he said, leading to fractures.
Transfusions are one treatment option, but they’re needed for a lifetime and cause their own problems, such as iron overload. Care of thalassemia patients “becomes quite complex and quite challenging for both families and medical institutions,” Alexis A. Thompson MD, MPH, chief of hematology at Children’s Hospital of Philadelphia, Pennsylvania, said in an interview.
Yusara Ahmed remembers her sister’s endless visits to the hospital after she was diagnosed at age 4. “We were all very traumatized by the hospital environment,” she said. But good news came in 2008, a few years later, when her sister was able to get a stem cell transplant from their brother.
But while stem cell transplants can be curative, most children don’t have a relative who can be a suitable match as a donor, Dr. Thompson said. Now, gene therapy offers another option, by turning a patient into his or her own matched donor.
Stem Cells Out, Stem Cells In
Last year, Yusuf went to Cohen Children’s Medical Center to donate stem cells, which were sent to a laboratory where they were genetically engineered to add copies of the beta-globin gene. Then, in January 2024, the modified stem cells were infused back into Yusuf after he underwent chemotherapy to make room for them in his bone marrow.
In April, a bald-headed Yusuf played with toy dinosaurs while his mother and clinicians met the media at a hospital press conference about his so-far-successful treatment. Early reports about the efficacy of the treatment suggest it may be the proverbial “game changer” for many of the estimated 100,000-plus people in the world who are diagnosed with transfusion-dependent beta thalassemia each year.
Over a median follow-up of 29.5 months, 20 of 22 patients treated with beti-cel no longer needed transfusions, according to a 2022 open-label phase 3 study published in the New England Journal of Medicine. Only one adverse event — thrombocytopenia in one patient — was considered both serious and related to the treatment, the industry-funded trial reported.
Costly Treatment Seems to Be Cost-Effective
As of 2022, gene therapy for transfusion-dependent beta thalassemia was listed as $2.8 million per treatment making it the most expensive single-treatment therapy ever approved in the United States. The price is “extraordinary,” said Dr. Thompson. “For some families, it gives them pause when they first hear about it.”
The hospital makes the case to insurers that covering the treatment is cost-effective in the long run, considering the high cost of traditional treatment, she said. “We’ve been very successful in getting coverage.”
In addition, the independent Institute for Clinical and Economic Review reported in 2022 that the treatment will be cost-effective at the “anticipated price of $2.1 million with an 80% payback option for patients who do not achieve and maintain transfusion independence over a 5-year period.”
Moving Forward, Clinicians Want to Reduce Complications
What’s next for transfusion-dependent beta thalassemia treatment? Earlier this year, the FDA approved a second gene therapy treatment called exagamglogene autotemcel (exa-cel, Casgevy). “We’re just beginning to evaluate individuals for the product, and we intend to make it available for families as well,” Dr. Thompson said.
In the bigger picture, she said gene therapy still has room for improvement. The need for chemotherapy is one target. According to her, it causes most of the complications related to gene therapy.
“Chemotherapy is a part of all gene therapies today because one has to make space in the bone marrow in order to have modified stem cells to come back to settle in and grow,” she said.
One strategy is to reduce the number of stem cells that are required for the therapy to work. “That would essentially eliminate the need for chemotherapy,” she said. “We’re not there yet.”
Another goal is to reduce the small risk of complications from gene therapy itself, she said. “Overall, though, this doesn’t detract us at all from being very excited about how well children are doing with the current approach. We’re very enthusiastic and very confident in recommending it to families.”
Back on Long Island, a Sense of Relief
Several months after his treatment, Yusuf is doing well. His hemoglobin levels are increasing, and his bone marrow has grown back, his mother said. He’s being home-schooled for the time being because he still faces a risk of infection. (Ms. Ahmed, a stay-at-home mom, has worked a teacher and mosque volunteer. Her husband runs a consumer electronics business.)
As Yusuf gets better, his parents hope they’ll soon be able to take a long trip back home to Pakistan to see relatives. They’ll be able to share their son with family along with something else: a sense of relief.
Dr. Al-Samkari discloses consulting for Agios. Dr. Thompson discloses research for Beam, Bluebird Bio, Editas, Novartis, and Novo Nordisk and consulting for Beam, Bluebird Bio, Editas, Roche, and Vertex.
With luck, maybe Ms. Ahmed’s son could follow in his aunt’s footsteps and get a stem cell transplant from a compatible family donor. But while little Yusuf Saeed has a twin sister of his own, she wasn’t a match. Without another treatment option, he’d face the prospect of a lifetime not only cut short but burdened by multiple monthly transfusions and severe limitations.
Then came glimpses of hope. One of Yusuf’s physicians at Cohen Children’s Medical Center in Long Island, New York, told Yusuf’s mother about a new kind of gene therapy on the horizon. But it took time to get FDA approval. Yusuf grew older, heading toward his teenage years, when regular transfusions would be a huge burden. “He’s turning 5 and 6, and there’s nothing,” Ms. Ahmed recalled, and the family worried.
Finally, the FDA approved the one-time treatment — betibeglogene autotemcel (beti-cel, Zynteglo) in 2022. By January 2024, the hospital was ready to treat Yusuf. At age 8, he became the first patient in the state of New York to undergo gene therapy for beta thalassemia.
A medical team infused Yusuf with his own stem cells, which had been genetically engineered to boost production of hemoglobin and prevent thalassemia’s devastating effects.
There are caveats about the treatment. It’s an extraordinarily expensive therapy that can be performed at only a few institutions. And it’s so brand new that caveats may not even have appeared yet. Yet, for kids like Yusuf, the gene therapy could transform a life.
“We feel like a weight has been lifted,” Ms. Ahmed said in an interview. “It’s something we’ve been waiting for.”
Anemia Becomes a Lifetime Threat
Among all genetic diseases, thalassemia stands alone. It’s the most common condition caused by a single gene, according to Hanny Al-Samkari, MD, a hematologist/clinical investigator at Massachusetts General Hospital and associate professor of medicine at Harvard Medical School, in Boston, Massachusetts.
Millions of people have the thalassemia trait, especially in southern Europe, the Middle East, southeast Asia, and Africa, Dr. Al-Samkari said. (Yusuf’s parents are from Pakistan.)
The trait, which appears to provide protection against malaria, may cause mild anemia in some cases but is otherwise harmless. However, a child born to parents with the same kind of trait has a high risk of developing alpha thalassemia or beta thalassemia. Like his aunt, Yusuf developed beta thalassemia, which is generally more severe. Yusuf’s bleeding disorder requires him to be transfusion-dependent.
In these patients, the disease disrupts the production of red blood cells in the bone marrow, Dr. Al-Samkari said. Hemoglobin levels can fall to 7 or 8 g/dL, compared with the normal levels of 12-16 g/dL in adults. “They’re chronically anemic, and that low hemoglobin that leads to things you associate with anemia: fatigue, reduced exercise tolerance, mind fog, challenges with work or school, and hypersomnolence.”
In addition, the bones become thinner and more brittle, he said, leading to fractures.
Transfusions are one treatment option, but they’re needed for a lifetime and cause their own problems, such as iron overload. Care of thalassemia patients “becomes quite complex and quite challenging for both families and medical institutions,” Alexis A. Thompson MD, MPH, chief of hematology at Children’s Hospital of Philadelphia, Pennsylvania, said in an interview.
Yusara Ahmed remembers her sister’s endless visits to the hospital after she was diagnosed at age 4. “We were all very traumatized by the hospital environment,” she said. But good news came in 2008, a few years later, when her sister was able to get a stem cell transplant from their brother.
But while stem cell transplants can be curative, most children don’t have a relative who can be a suitable match as a donor, Dr. Thompson said. Now, gene therapy offers another option, by turning a patient into his or her own matched donor.
Stem Cells Out, Stem Cells In
Last year, Yusuf went to Cohen Children’s Medical Center to donate stem cells, which were sent to a laboratory where they were genetically engineered to add copies of the beta-globin gene. Then, in January 2024, the modified stem cells were infused back into Yusuf after he underwent chemotherapy to make room for them in his bone marrow.
In April, a bald-headed Yusuf played with toy dinosaurs while his mother and clinicians met the media at a hospital press conference about his so-far-successful treatment. Early reports about the efficacy of the treatment suggest it may be the proverbial “game changer” for many of the estimated 100,000-plus people in the world who are diagnosed with transfusion-dependent beta thalassemia each year.
Over a median follow-up of 29.5 months, 20 of 22 patients treated with beti-cel no longer needed transfusions, according to a 2022 open-label phase 3 study published in the New England Journal of Medicine. Only one adverse event — thrombocytopenia in one patient — was considered both serious and related to the treatment, the industry-funded trial reported.
Costly Treatment Seems to Be Cost-Effective
As of 2022, gene therapy for transfusion-dependent beta thalassemia was listed as $2.8 million per treatment making it the most expensive single-treatment therapy ever approved in the United States. The price is “extraordinary,” said Dr. Thompson. “For some families, it gives them pause when they first hear about it.”
The hospital makes the case to insurers that covering the treatment is cost-effective in the long run, considering the high cost of traditional treatment, she said. “We’ve been very successful in getting coverage.”
In addition, the independent Institute for Clinical and Economic Review reported in 2022 that the treatment will be cost-effective at the “anticipated price of $2.1 million with an 80% payback option for patients who do not achieve and maintain transfusion independence over a 5-year period.”
Moving Forward, Clinicians Want to Reduce Complications
What’s next for transfusion-dependent beta thalassemia treatment? Earlier this year, the FDA approved a second gene therapy treatment called exagamglogene autotemcel (exa-cel, Casgevy). “We’re just beginning to evaluate individuals for the product, and we intend to make it available for families as well,” Dr. Thompson said.
In the bigger picture, she said gene therapy still has room for improvement. The need for chemotherapy is one target. According to her, it causes most of the complications related to gene therapy.
“Chemotherapy is a part of all gene therapies today because one has to make space in the bone marrow in order to have modified stem cells to come back to settle in and grow,” she said.
One strategy is to reduce the number of stem cells that are required for the therapy to work. “That would essentially eliminate the need for chemotherapy,” she said. “We’re not there yet.”
Another goal is to reduce the small risk of complications from gene therapy itself, she said. “Overall, though, this doesn’t detract us at all from being very excited about how well children are doing with the current approach. We’re very enthusiastic and very confident in recommending it to families.”
Back on Long Island, a Sense of Relief
Several months after his treatment, Yusuf is doing well. His hemoglobin levels are increasing, and his bone marrow has grown back, his mother said. He’s being home-schooled for the time being because he still faces a risk of infection. (Ms. Ahmed, a stay-at-home mom, has worked a teacher and mosque volunteer. Her husband runs a consumer electronics business.)
As Yusuf gets better, his parents hope they’ll soon be able to take a long trip back home to Pakistan to see relatives. They’ll be able to share their son with family along with something else: a sense of relief.
Dr. Al-Samkari discloses consulting for Agios. Dr. Thompson discloses research for Beam, Bluebird Bio, Editas, Novartis, and Novo Nordisk and consulting for Beam, Bluebird Bio, Editas, Roche, and Vertex.
With luck, maybe Ms. Ahmed’s son could follow in his aunt’s footsteps and get a stem cell transplant from a compatible family donor. But while little Yusuf Saeed has a twin sister of his own, she wasn’t a match. Without another treatment option, he’d face the prospect of a lifetime not only cut short but burdened by multiple monthly transfusions and severe limitations.
Then came glimpses of hope. One of Yusuf’s physicians at Cohen Children’s Medical Center in Long Island, New York, told Yusuf’s mother about a new kind of gene therapy on the horizon. But it took time to get FDA approval. Yusuf grew older, heading toward his teenage years, when regular transfusions would be a huge burden. “He’s turning 5 and 6, and there’s nothing,” Ms. Ahmed recalled, and the family worried.
Finally, the FDA approved the one-time treatment — betibeglogene autotemcel (beti-cel, Zynteglo) in 2022. By January 2024, the hospital was ready to treat Yusuf. At age 8, he became the first patient in the state of New York to undergo gene therapy for beta thalassemia.
A medical team infused Yusuf with his own stem cells, which had been genetically engineered to boost production of hemoglobin and prevent thalassemia’s devastating effects.
There are caveats about the treatment. It’s an extraordinarily expensive therapy that can be performed at only a few institutions. And it’s so brand new that caveats may not even have appeared yet. Yet, for kids like Yusuf, the gene therapy could transform a life.
“We feel like a weight has been lifted,” Ms. Ahmed said in an interview. “It’s something we’ve been waiting for.”
Anemia Becomes a Lifetime Threat
Among all genetic diseases, thalassemia stands alone. It’s the most common condition caused by a single gene, according to Hanny Al-Samkari, MD, a hematologist/clinical investigator at Massachusetts General Hospital and associate professor of medicine at Harvard Medical School, in Boston, Massachusetts.
Millions of people have the thalassemia trait, especially in southern Europe, the Middle East, southeast Asia, and Africa, Dr. Al-Samkari said. (Yusuf’s parents are from Pakistan.)
The trait, which appears to provide protection against malaria, may cause mild anemia in some cases but is otherwise harmless. However, a child born to parents with the same kind of trait has a high risk of developing alpha thalassemia or beta thalassemia. Like his aunt, Yusuf developed beta thalassemia, which is generally more severe. Yusuf’s bleeding disorder requires him to be transfusion-dependent.
In these patients, the disease disrupts the production of red blood cells in the bone marrow, Dr. Al-Samkari said. Hemoglobin levels can fall to 7 or 8 g/dL, compared with the normal levels of 12-16 g/dL in adults. “They’re chronically anemic, and that low hemoglobin that leads to things you associate with anemia: fatigue, reduced exercise tolerance, mind fog, challenges with work or school, and hypersomnolence.”
In addition, the bones become thinner and more brittle, he said, leading to fractures.
Transfusions are one treatment option, but they’re needed for a lifetime and cause their own problems, such as iron overload. Care of thalassemia patients “becomes quite complex and quite challenging for both families and medical institutions,” Alexis A. Thompson MD, MPH, chief of hematology at Children’s Hospital of Philadelphia, Pennsylvania, said in an interview.
Yusara Ahmed remembers her sister’s endless visits to the hospital after she was diagnosed at age 4. “We were all very traumatized by the hospital environment,” she said. But good news came in 2008, a few years later, when her sister was able to get a stem cell transplant from their brother.
But while stem cell transplants can be curative, most children don’t have a relative who can be a suitable match as a donor, Dr. Thompson said. Now, gene therapy offers another option, by turning a patient into his or her own matched donor.
Stem Cells Out, Stem Cells In
Last year, Yusuf went to Cohen Children’s Medical Center to donate stem cells, which were sent to a laboratory where they were genetically engineered to add copies of the beta-globin gene. Then, in January 2024, the modified stem cells were infused back into Yusuf after he underwent chemotherapy to make room for them in his bone marrow.
In April, a bald-headed Yusuf played with toy dinosaurs while his mother and clinicians met the media at a hospital press conference about his so-far-successful treatment. Early reports about the efficacy of the treatment suggest it may be the proverbial “game changer” for many of the estimated 100,000-plus people in the world who are diagnosed with transfusion-dependent beta thalassemia each year.
Over a median follow-up of 29.5 months, 20 of 22 patients treated with beti-cel no longer needed transfusions, according to a 2022 open-label phase 3 study published in the New England Journal of Medicine. Only one adverse event — thrombocytopenia in one patient — was considered both serious and related to the treatment, the industry-funded trial reported.
Costly Treatment Seems to Be Cost-Effective
As of 2022, gene therapy for transfusion-dependent beta thalassemia was listed as $2.8 million per treatment making it the most expensive single-treatment therapy ever approved in the United States. The price is “extraordinary,” said Dr. Thompson. “For some families, it gives them pause when they first hear about it.”
The hospital makes the case to insurers that covering the treatment is cost-effective in the long run, considering the high cost of traditional treatment, she said. “We’ve been very successful in getting coverage.”
In addition, the independent Institute for Clinical and Economic Review reported in 2022 that the treatment will be cost-effective at the “anticipated price of $2.1 million with an 80% payback option for patients who do not achieve and maintain transfusion independence over a 5-year period.”
Moving Forward, Clinicians Want to Reduce Complications
What’s next for transfusion-dependent beta thalassemia treatment? Earlier this year, the FDA approved a second gene therapy treatment called exagamglogene autotemcel (exa-cel, Casgevy). “We’re just beginning to evaluate individuals for the product, and we intend to make it available for families as well,” Dr. Thompson said.
In the bigger picture, she said gene therapy still has room for improvement. The need for chemotherapy is one target. According to her, it causes most of the complications related to gene therapy.
“Chemotherapy is a part of all gene therapies today because one has to make space in the bone marrow in order to have modified stem cells to come back to settle in and grow,” she said.
One strategy is to reduce the number of stem cells that are required for the therapy to work. “That would essentially eliminate the need for chemotherapy,” she said. “We’re not there yet.”
Another goal is to reduce the small risk of complications from gene therapy itself, she said. “Overall, though, this doesn’t detract us at all from being very excited about how well children are doing with the current approach. We’re very enthusiastic and very confident in recommending it to families.”
Back on Long Island, a Sense of Relief
Several months after his treatment, Yusuf is doing well. His hemoglobin levels are increasing, and his bone marrow has grown back, his mother said. He’s being home-schooled for the time being because he still faces a risk of infection. (Ms. Ahmed, a stay-at-home mom, has worked a teacher and mosque volunteer. Her husband runs a consumer electronics business.)
As Yusuf gets better, his parents hope they’ll soon be able to take a long trip back home to Pakistan to see relatives. They’ll be able to share their son with family along with something else: a sense of relief.
Dr. Al-Samkari discloses consulting for Agios. Dr. Thompson discloses research for Beam, Bluebird Bio, Editas, Novartis, and Novo Nordisk and consulting for Beam, Bluebird Bio, Editas, Roche, and Vertex.
Myeloma: VRd Plus Isatuximab Improves Outcomes
Patients who took isatuximab (Sarclisa) plus bortezomib, lenalidomide, and dexamethasone (VRd) reached higher estimated progression-free survival at a median 59.7 months vs. those who took VRd alone (63.2% vs. 45.2%, respectively, 98.5% CI, hazard ratio [HR] = 0.60, P < .001), reported Thierry Facon, MD, professor of hematology at Lille University Hospital, France, and colleagues at the annual meeting of the American Society of Clinical Oncology in Chicago. The study was simultaneously published in The New England Journal of Medicine.
“The significant progression-free benefit observed with Sarclisa with combination therapy compared to VRd is important and encouraging for patients with newly diagnosed multiple myeloma,” Dr. Facon said in an interview. The findings demonstrated the VRd-isatuximab’s potential as “a first-in-class combination to address gaps in care for newly diagnosed multiple myeloma transplant-ineligible patients,” he said.
According to Dr. Facon, more than 180,000 people worldwide are diagnosed with MM each year, he said, making it the second-most common hematologic malignancy.
“There is a need for new frontline therapeutic options for all MM patients,” he said. “Effective frontline therapy has the potential to modify the course of the disease, which is a key outcome for transplant-ineligible patients who often face high rates of attrition in later lines of therapy.”
For the industry-funded IMROZ study, researchers recruited patients aged 18-80 at 93 sites in 21 nations from 2017-2019. All were ineligible for transplant due to comorbidities or being aged 65 or older. Exclusions included Eastern Cooperative Oncology Group (ECOG) performance status scores of more than 2.
The subjects were randomly assigned in a 3-to-2 ratio to isatuximab-VRd (n = 265) or VRd alone (n = 181) and received four induction cycles (6 weeks per cycle) followed by 4-week cycles of continuous treatment until disease progression, unacceptable adverse event, or other criteria for discontinuation. If progression occurred, patients could be switched from the VRd-only group to the isatuximab-VRd group.
The median age in both the isatuximab-VRd and VRd groups was 72. The percentages of women were 46.0% and 48.1%, respectively, and 72.5% and 72.4%, respectively, were White. The next largest race/ethnic group was Asian (11.7% and 9.4%, respectively). Almost all had ECOG status of 0 or 1 (88.7% and 89.5%, respectively).
At study cut-off in September 2023, the percentages of subjects in the isatuximab-VRd and VRd groups who were still receiving treatment were 47.2% and 24.3%, respectively.
An intention-to-treat analysis found that the two groups had similar rates of overall response (91.3% for isatuximab-VRd vs. 92.3% for VRd), but the isatuximab-VRd group had higher complete or better response (74.7% vs. 64.1%, P = .01).
The percentage of patients who were minimal residual disease (MRD)-negative and had a complete response was also higher in the VRd-isatuximab group vs. the VRd group (55.5% vs. 40.9%, respectively, P = .003). A total of 26.0% of patients in the VRd-isatuximab group died vs. 32.6% in the VRd group; the estimated overall survival rates at 60 months were 72.3% and 66.3%, respectively, HR = 0.78, 99.97% CI).
As for adverse events, grade 5 events were more common in the VRd-isatuximab group (11.0% vs. 5.5%), as were deaths within the first 60 days of treatment (1.5% vs. 0.6%). “The difference was driven in part by different treatment exposures,” the researchers reported. Treatment-emergent events led to treatment discontinuation in 22.8% and 26.0% of patients, respectively.
“The safety and tolerability of Sarclisa observed was consistent with the established safety profile of Sarclisa and VRd with no new safety signals observed,” Dr. Facon said.
In an interview, Zandra Klippel, MD, global product head for multiple myeloma at Sanofi — the maker of isatuximab and funder of the study — said the Food and Drug Administration has accepted a priority review application for the investigational use of isatuximab in combination with VRd for the treatment of patients with transplant-ineligible, newly diagnosed MM.
“Our FDA approval date is expected on September 27, 2024,” Dr. Klippel said. “If all goes well, we anticipate launching as early as 2024 in the US and rolling out in other key countries starting in 2025 and continuing through 2026.”
Dr. Klippel added that isatuximab “continues to be evaluated in multiple ongoing phase 3 clinical trials in combination with current standard treatments across the MM treatment continuum.”
In an interview, Sagar Lonial, MD, chair and professor of hematology and medical oncology and chief medical officer at Winship Cancer Institute at Emory University in Atlanta, said the study is “important.”
However, Dr. Lonial, who is familiar with the findings but didn’t take part in the study, said it’s difficult to understand the impact of the treatment on frail patients. It appears that the combination treatment may be good for frail patients, he said, “but I need to better understand the magnitude of the benefit in that subset a little more.”
As for adverse events, he said “they are what would be expected for a trial like this.”
Pneumonia and COVID-19 infections were higher in the VRd-isatuximab group, he said, and “we know in general that vaccine responses are blocked by CD38 antibodies.” This can be managed, he said, via intravenous immunoglobulin support.
Manni Mohyuddin, MD, assistant professor at Huntsman Cancer Institute in Utah, said in an interview that the findings suggest that in older, fit patients, “you can get fairly good outcomes without use of transplant.”
In the United States, many more patients in the cohort would have been considered transplant-eligible, he said, and not eliminated from consideration for transplant due to age over 65. However, as patients age, “you get more diminishing returns for transplants,” said Dr. Mohyuddin, who is familiar with the study findings but didn’t take part in the research.
All the drugs in the new combination are FDA approved, he said, although the combination isn’t. “I suspect this will make it to our guidelines very soon and then be reimbursed by insurance companies and Medicare.”
The study was funded by Sanofi and an M.D. Anderson Cancer Center support grant. Dr. Facon has no disclosures. Other study authors report multiple ties relationships with various drug makers. Dr. Lonial disclosed ties with Takeda, Amgen, Novartis, BMS, GSK, AbbVie, Genentech, Pfizer, Regeneron, Janssen, AstraZeneca, and TG Therapeutics). Dr. Mohyuddin disclosed a relationship with Janssen.
Patients who took isatuximab (Sarclisa) plus bortezomib, lenalidomide, and dexamethasone (VRd) reached higher estimated progression-free survival at a median 59.7 months vs. those who took VRd alone (63.2% vs. 45.2%, respectively, 98.5% CI, hazard ratio [HR] = 0.60, P < .001), reported Thierry Facon, MD, professor of hematology at Lille University Hospital, France, and colleagues at the annual meeting of the American Society of Clinical Oncology in Chicago. The study was simultaneously published in The New England Journal of Medicine.
“The significant progression-free benefit observed with Sarclisa with combination therapy compared to VRd is important and encouraging for patients with newly diagnosed multiple myeloma,” Dr. Facon said in an interview. The findings demonstrated the VRd-isatuximab’s potential as “a first-in-class combination to address gaps in care for newly diagnosed multiple myeloma transplant-ineligible patients,” he said.
According to Dr. Facon, more than 180,000 people worldwide are diagnosed with MM each year, he said, making it the second-most common hematologic malignancy.
“There is a need for new frontline therapeutic options for all MM patients,” he said. “Effective frontline therapy has the potential to modify the course of the disease, which is a key outcome for transplant-ineligible patients who often face high rates of attrition in later lines of therapy.”
For the industry-funded IMROZ study, researchers recruited patients aged 18-80 at 93 sites in 21 nations from 2017-2019. All were ineligible for transplant due to comorbidities or being aged 65 or older. Exclusions included Eastern Cooperative Oncology Group (ECOG) performance status scores of more than 2.
The subjects were randomly assigned in a 3-to-2 ratio to isatuximab-VRd (n = 265) or VRd alone (n = 181) and received four induction cycles (6 weeks per cycle) followed by 4-week cycles of continuous treatment until disease progression, unacceptable adverse event, or other criteria for discontinuation. If progression occurred, patients could be switched from the VRd-only group to the isatuximab-VRd group.
The median age in both the isatuximab-VRd and VRd groups was 72. The percentages of women were 46.0% and 48.1%, respectively, and 72.5% and 72.4%, respectively, were White. The next largest race/ethnic group was Asian (11.7% and 9.4%, respectively). Almost all had ECOG status of 0 or 1 (88.7% and 89.5%, respectively).
At study cut-off in September 2023, the percentages of subjects in the isatuximab-VRd and VRd groups who were still receiving treatment were 47.2% and 24.3%, respectively.
An intention-to-treat analysis found that the two groups had similar rates of overall response (91.3% for isatuximab-VRd vs. 92.3% for VRd), but the isatuximab-VRd group had higher complete or better response (74.7% vs. 64.1%, P = .01).
The percentage of patients who were minimal residual disease (MRD)-negative and had a complete response was also higher in the VRd-isatuximab group vs. the VRd group (55.5% vs. 40.9%, respectively, P = .003). A total of 26.0% of patients in the VRd-isatuximab group died vs. 32.6% in the VRd group; the estimated overall survival rates at 60 months were 72.3% and 66.3%, respectively, HR = 0.78, 99.97% CI).
As for adverse events, grade 5 events were more common in the VRd-isatuximab group (11.0% vs. 5.5%), as were deaths within the first 60 days of treatment (1.5% vs. 0.6%). “The difference was driven in part by different treatment exposures,” the researchers reported. Treatment-emergent events led to treatment discontinuation in 22.8% and 26.0% of patients, respectively.
“The safety and tolerability of Sarclisa observed was consistent with the established safety profile of Sarclisa and VRd with no new safety signals observed,” Dr. Facon said.
In an interview, Zandra Klippel, MD, global product head for multiple myeloma at Sanofi — the maker of isatuximab and funder of the study — said the Food and Drug Administration has accepted a priority review application for the investigational use of isatuximab in combination with VRd for the treatment of patients with transplant-ineligible, newly diagnosed MM.
“Our FDA approval date is expected on September 27, 2024,” Dr. Klippel said. “If all goes well, we anticipate launching as early as 2024 in the US and rolling out in other key countries starting in 2025 and continuing through 2026.”
Dr. Klippel added that isatuximab “continues to be evaluated in multiple ongoing phase 3 clinical trials in combination with current standard treatments across the MM treatment continuum.”
In an interview, Sagar Lonial, MD, chair and professor of hematology and medical oncology and chief medical officer at Winship Cancer Institute at Emory University in Atlanta, said the study is “important.”
However, Dr. Lonial, who is familiar with the findings but didn’t take part in the study, said it’s difficult to understand the impact of the treatment on frail patients. It appears that the combination treatment may be good for frail patients, he said, “but I need to better understand the magnitude of the benefit in that subset a little more.”
As for adverse events, he said “they are what would be expected for a trial like this.”
Pneumonia and COVID-19 infections were higher in the VRd-isatuximab group, he said, and “we know in general that vaccine responses are blocked by CD38 antibodies.” This can be managed, he said, via intravenous immunoglobulin support.
Manni Mohyuddin, MD, assistant professor at Huntsman Cancer Institute in Utah, said in an interview that the findings suggest that in older, fit patients, “you can get fairly good outcomes without use of transplant.”
In the United States, many more patients in the cohort would have been considered transplant-eligible, he said, and not eliminated from consideration for transplant due to age over 65. However, as patients age, “you get more diminishing returns for transplants,” said Dr. Mohyuddin, who is familiar with the study findings but didn’t take part in the research.
All the drugs in the new combination are FDA approved, he said, although the combination isn’t. “I suspect this will make it to our guidelines very soon and then be reimbursed by insurance companies and Medicare.”
The study was funded by Sanofi and an M.D. Anderson Cancer Center support grant. Dr. Facon has no disclosures. Other study authors report multiple ties relationships with various drug makers. Dr. Lonial disclosed ties with Takeda, Amgen, Novartis, BMS, GSK, AbbVie, Genentech, Pfizer, Regeneron, Janssen, AstraZeneca, and TG Therapeutics). Dr. Mohyuddin disclosed a relationship with Janssen.
Patients who took isatuximab (Sarclisa) plus bortezomib, lenalidomide, and dexamethasone (VRd) reached higher estimated progression-free survival at a median 59.7 months vs. those who took VRd alone (63.2% vs. 45.2%, respectively, 98.5% CI, hazard ratio [HR] = 0.60, P < .001), reported Thierry Facon, MD, professor of hematology at Lille University Hospital, France, and colleagues at the annual meeting of the American Society of Clinical Oncology in Chicago. The study was simultaneously published in The New England Journal of Medicine.
“The significant progression-free benefit observed with Sarclisa with combination therapy compared to VRd is important and encouraging for patients with newly diagnosed multiple myeloma,” Dr. Facon said in an interview. The findings demonstrated the VRd-isatuximab’s potential as “a first-in-class combination to address gaps in care for newly diagnosed multiple myeloma transplant-ineligible patients,” he said.
According to Dr. Facon, more than 180,000 people worldwide are diagnosed with MM each year, he said, making it the second-most common hematologic malignancy.
“There is a need for new frontline therapeutic options for all MM patients,” he said. “Effective frontline therapy has the potential to modify the course of the disease, which is a key outcome for transplant-ineligible patients who often face high rates of attrition in later lines of therapy.”
For the industry-funded IMROZ study, researchers recruited patients aged 18-80 at 93 sites in 21 nations from 2017-2019. All were ineligible for transplant due to comorbidities or being aged 65 or older. Exclusions included Eastern Cooperative Oncology Group (ECOG) performance status scores of more than 2.
The subjects were randomly assigned in a 3-to-2 ratio to isatuximab-VRd (n = 265) or VRd alone (n = 181) and received four induction cycles (6 weeks per cycle) followed by 4-week cycles of continuous treatment until disease progression, unacceptable adverse event, or other criteria for discontinuation. If progression occurred, patients could be switched from the VRd-only group to the isatuximab-VRd group.
The median age in both the isatuximab-VRd and VRd groups was 72. The percentages of women were 46.0% and 48.1%, respectively, and 72.5% and 72.4%, respectively, were White. The next largest race/ethnic group was Asian (11.7% and 9.4%, respectively). Almost all had ECOG status of 0 or 1 (88.7% and 89.5%, respectively).
At study cut-off in September 2023, the percentages of subjects in the isatuximab-VRd and VRd groups who were still receiving treatment were 47.2% and 24.3%, respectively.
An intention-to-treat analysis found that the two groups had similar rates of overall response (91.3% for isatuximab-VRd vs. 92.3% for VRd), but the isatuximab-VRd group had higher complete or better response (74.7% vs. 64.1%, P = .01).
The percentage of patients who were minimal residual disease (MRD)-negative and had a complete response was also higher in the VRd-isatuximab group vs. the VRd group (55.5% vs. 40.9%, respectively, P = .003). A total of 26.0% of patients in the VRd-isatuximab group died vs. 32.6% in the VRd group; the estimated overall survival rates at 60 months were 72.3% and 66.3%, respectively, HR = 0.78, 99.97% CI).
As for adverse events, grade 5 events were more common in the VRd-isatuximab group (11.0% vs. 5.5%), as were deaths within the first 60 days of treatment (1.5% vs. 0.6%). “The difference was driven in part by different treatment exposures,” the researchers reported. Treatment-emergent events led to treatment discontinuation in 22.8% and 26.0% of patients, respectively.
“The safety and tolerability of Sarclisa observed was consistent with the established safety profile of Sarclisa and VRd with no new safety signals observed,” Dr. Facon said.
In an interview, Zandra Klippel, MD, global product head for multiple myeloma at Sanofi — the maker of isatuximab and funder of the study — said the Food and Drug Administration has accepted a priority review application for the investigational use of isatuximab in combination with VRd for the treatment of patients with transplant-ineligible, newly diagnosed MM.
“Our FDA approval date is expected on September 27, 2024,” Dr. Klippel said. “If all goes well, we anticipate launching as early as 2024 in the US and rolling out in other key countries starting in 2025 and continuing through 2026.”
Dr. Klippel added that isatuximab “continues to be evaluated in multiple ongoing phase 3 clinical trials in combination with current standard treatments across the MM treatment continuum.”
In an interview, Sagar Lonial, MD, chair and professor of hematology and medical oncology and chief medical officer at Winship Cancer Institute at Emory University in Atlanta, said the study is “important.”
However, Dr. Lonial, who is familiar with the findings but didn’t take part in the study, said it’s difficult to understand the impact of the treatment on frail patients. It appears that the combination treatment may be good for frail patients, he said, “but I need to better understand the magnitude of the benefit in that subset a little more.”
As for adverse events, he said “they are what would be expected for a trial like this.”
Pneumonia and COVID-19 infections were higher in the VRd-isatuximab group, he said, and “we know in general that vaccine responses are blocked by CD38 antibodies.” This can be managed, he said, via intravenous immunoglobulin support.
Manni Mohyuddin, MD, assistant professor at Huntsman Cancer Institute in Utah, said in an interview that the findings suggest that in older, fit patients, “you can get fairly good outcomes without use of transplant.”
In the United States, many more patients in the cohort would have been considered transplant-eligible, he said, and not eliminated from consideration for transplant due to age over 65. However, as patients age, “you get more diminishing returns for transplants,” said Dr. Mohyuddin, who is familiar with the study findings but didn’t take part in the research.
All the drugs in the new combination are FDA approved, he said, although the combination isn’t. “I suspect this will make it to our guidelines very soon and then be reimbursed by insurance companies and Medicare.”
The study was funded by Sanofi and an M.D. Anderson Cancer Center support grant. Dr. Facon has no disclosures. Other study authors report multiple ties relationships with various drug makers. Dr. Lonial disclosed ties with Takeda, Amgen, Novartis, BMS, GSK, AbbVie, Genentech, Pfizer, Regeneron, Janssen, AstraZeneca, and TG Therapeutics). Dr. Mohyuddin disclosed a relationship with Janssen.
FROM ASCO 2024
MCL: Dual Therapy ‘Promising’ in Patients With TP53 mutations
In first-line patients (n = 29) and relapsed/refractory patients (n = 45) with TP53 mutations, complete response rates were 55% and 58%, respectively, reported hematologist-oncologist Michael Wang, MD, of the University of Texas MD Anderson Cancer Center, and colleagues, at the annual meeting of the American Society of Clinical Oncology (ASCO) in Chicago.
“These results are encouraging in light of the poor responses and shorter survival outcomes with standard chemotherapy,” Dr. Wang said in an ASCO presentation.
The current standard of care for relapsed/refractory MCL includes Bruton tyrosine kinase inhibitors for first relapse and CAR T-cell therapy (CAR-T) for second relapse in eligible patients or pirtobrutinib (Jaypirca) in patients ineligible for CAR T-cell therapy, Ohio State University Comprehensive Cancer Center hematology specialist Narendranath Epperla, MD, MS, said in an interview. Dr. Epperla is familiar with the new study findings but didn’t take part in the research.
Options for third relapse and beyond include clinical trial, rituximab [Rituxan] and lenalidomide [Revlimid], and bortezomib [Velcade],” Dr. Epperla said. “Venetoclax is not currently FDA-approved but can also be considered at third relapse.”
Better therapies are needed for a number of reasons, including poor outcomes in high-risk patients, such as those with TP53 mutations and those who progress following CAR T, Dr. Epperla said. Also, “as the novel agents are being moved into earlier lines of therapy, there remains an unmet need in those who progress on these agents with fewer options in the relapsed setting.”
At last December’s American Society of Hematology annual meeting, Dr. Wang and colleagues reported on the primary analysis results from the Sympatico study. Patients with relapsed/refractory MCL after 1-5 prior therapies were randomly assigned to receive 560 mg of ibrutinib once daily with either placebo (n = 133) or 400 mg daily of venetoclax after ramp-up (n = 134) for 2 years. Then subjects continued taking ibrutinib alone until their disease progressed or they reached unacceptable toxicity.
At a median follow-up of 51.2 months, median progression-free survival was longer in the ibrutinib-venetoclax group vs. ibrutinib alone (hazard ratio [HR] = 0.65, 95% CI, P = .0052).
The new analysis pools several cohorts of patients with TP53 mutations who all took the combination therapy: 5 from a safety run-in phase, 40 from the randomized phase, and 29 from a first-line cohort (median age at baseline = 67).
Median overall survival was not reached in the first-line group and 35.0 months in the relapsed/refractory group (total = 47.1 months). Median progress-free survival in the groups was 22.0 months and 20.9 months, respectively, and median duration of response was 20.5 months and 26.5 months, respectively.
With regard to the new findings, “it is good to see the responses with ibrutinib and venetoclax were deep and durable,” Dr. Epperla said. The combination treatment “provides a good alternative option for TP53-mutated MCL patients who are ineligible for CAR-T.”
Dr. Epperla added that the findings about the addition of ibrutinib could apply to newer-generation Bruton tyrosine kinase inhibitors that have relatively better safety profiles.
However, Dr. Epperla cautioned that the treatment needs to be weighed against the toxicity and cost of the regimen of ibrutinib and venetoclax for 2 years then single-agent ibrutinib until progression or unacceptable toxicity.
This news organization reported in 2023 that estimated net spending on ibrutinib per Medicare data increased by nearly half from 2014-2020, reaching $11,980 in 2020 vs. $7,787 for venetoclax.
Dr. Epperla also noted that “there are newer therapies that are emerging, such as T-cell-engaging bispecific antibodies, and they have shown promising results.”
In an interview, Brad S. Kahl, MD, a hematologist-oncologist at Washington University, St. Louis, said the improvement in outcomes are “modestly significant.”
Dr. Kahl, who is familiar with the study findings but didn’t take part in the research, said it is “worth adding the venetoclax, particularly in these biologically high risk patients with p53 mutations. Venetoclax is not FDA-approved, so insurance approval will need to be determined on a case-by-case basis. The combination is very expensive.”
Dr. Kahl agreed with Dr. Epperla that the findings could be extrapolated to other Bruton tyrosine kinase inhibitors.
The study was funded by Pharmacyclics, an AbbVie Company. Dr. Epperla disclosed relationships with BeiGene and Eli Lilly. Dr. Kahl reported ties with AstraZeneca, BeiGene, Abbvie, and Genentech.
In first-line patients (n = 29) and relapsed/refractory patients (n = 45) with TP53 mutations, complete response rates were 55% and 58%, respectively, reported hematologist-oncologist Michael Wang, MD, of the University of Texas MD Anderson Cancer Center, and colleagues, at the annual meeting of the American Society of Clinical Oncology (ASCO) in Chicago.
“These results are encouraging in light of the poor responses and shorter survival outcomes with standard chemotherapy,” Dr. Wang said in an ASCO presentation.
The current standard of care for relapsed/refractory MCL includes Bruton tyrosine kinase inhibitors for first relapse and CAR T-cell therapy (CAR-T) for second relapse in eligible patients or pirtobrutinib (Jaypirca) in patients ineligible for CAR T-cell therapy, Ohio State University Comprehensive Cancer Center hematology specialist Narendranath Epperla, MD, MS, said in an interview. Dr. Epperla is familiar with the new study findings but didn’t take part in the research.
Options for third relapse and beyond include clinical trial, rituximab [Rituxan] and lenalidomide [Revlimid], and bortezomib [Velcade],” Dr. Epperla said. “Venetoclax is not currently FDA-approved but can also be considered at third relapse.”
Better therapies are needed for a number of reasons, including poor outcomes in high-risk patients, such as those with TP53 mutations and those who progress following CAR T, Dr. Epperla said. Also, “as the novel agents are being moved into earlier lines of therapy, there remains an unmet need in those who progress on these agents with fewer options in the relapsed setting.”
At last December’s American Society of Hematology annual meeting, Dr. Wang and colleagues reported on the primary analysis results from the Sympatico study. Patients with relapsed/refractory MCL after 1-5 prior therapies were randomly assigned to receive 560 mg of ibrutinib once daily with either placebo (n = 133) or 400 mg daily of venetoclax after ramp-up (n = 134) for 2 years. Then subjects continued taking ibrutinib alone until their disease progressed or they reached unacceptable toxicity.
At a median follow-up of 51.2 months, median progression-free survival was longer in the ibrutinib-venetoclax group vs. ibrutinib alone (hazard ratio [HR] = 0.65, 95% CI, P = .0052).
The new analysis pools several cohorts of patients with TP53 mutations who all took the combination therapy: 5 from a safety run-in phase, 40 from the randomized phase, and 29 from a first-line cohort (median age at baseline = 67).
Median overall survival was not reached in the first-line group and 35.0 months in the relapsed/refractory group (total = 47.1 months). Median progress-free survival in the groups was 22.0 months and 20.9 months, respectively, and median duration of response was 20.5 months and 26.5 months, respectively.
With regard to the new findings, “it is good to see the responses with ibrutinib and venetoclax were deep and durable,” Dr. Epperla said. The combination treatment “provides a good alternative option for TP53-mutated MCL patients who are ineligible for CAR-T.”
Dr. Epperla added that the findings about the addition of ibrutinib could apply to newer-generation Bruton tyrosine kinase inhibitors that have relatively better safety profiles.
However, Dr. Epperla cautioned that the treatment needs to be weighed against the toxicity and cost of the regimen of ibrutinib and venetoclax for 2 years then single-agent ibrutinib until progression or unacceptable toxicity.
This news organization reported in 2023 that estimated net spending on ibrutinib per Medicare data increased by nearly half from 2014-2020, reaching $11,980 in 2020 vs. $7,787 for venetoclax.
Dr. Epperla also noted that “there are newer therapies that are emerging, such as T-cell-engaging bispecific antibodies, and they have shown promising results.”
In an interview, Brad S. Kahl, MD, a hematologist-oncologist at Washington University, St. Louis, said the improvement in outcomes are “modestly significant.”
Dr. Kahl, who is familiar with the study findings but didn’t take part in the research, said it is “worth adding the venetoclax, particularly in these biologically high risk patients with p53 mutations. Venetoclax is not FDA-approved, so insurance approval will need to be determined on a case-by-case basis. The combination is very expensive.”
Dr. Kahl agreed with Dr. Epperla that the findings could be extrapolated to other Bruton tyrosine kinase inhibitors.
The study was funded by Pharmacyclics, an AbbVie Company. Dr. Epperla disclosed relationships with BeiGene and Eli Lilly. Dr. Kahl reported ties with AstraZeneca, BeiGene, Abbvie, and Genentech.
In first-line patients (n = 29) and relapsed/refractory patients (n = 45) with TP53 mutations, complete response rates were 55% and 58%, respectively, reported hematologist-oncologist Michael Wang, MD, of the University of Texas MD Anderson Cancer Center, and colleagues, at the annual meeting of the American Society of Clinical Oncology (ASCO) in Chicago.
“These results are encouraging in light of the poor responses and shorter survival outcomes with standard chemotherapy,” Dr. Wang said in an ASCO presentation.
The current standard of care for relapsed/refractory MCL includes Bruton tyrosine kinase inhibitors for first relapse and CAR T-cell therapy (CAR-T) for second relapse in eligible patients or pirtobrutinib (Jaypirca) in patients ineligible for CAR T-cell therapy, Ohio State University Comprehensive Cancer Center hematology specialist Narendranath Epperla, MD, MS, said in an interview. Dr. Epperla is familiar with the new study findings but didn’t take part in the research.
Options for third relapse and beyond include clinical trial, rituximab [Rituxan] and lenalidomide [Revlimid], and bortezomib [Velcade],” Dr. Epperla said. “Venetoclax is not currently FDA-approved but can also be considered at third relapse.”
Better therapies are needed for a number of reasons, including poor outcomes in high-risk patients, such as those with TP53 mutations and those who progress following CAR T, Dr. Epperla said. Also, “as the novel agents are being moved into earlier lines of therapy, there remains an unmet need in those who progress on these agents with fewer options in the relapsed setting.”
At last December’s American Society of Hematology annual meeting, Dr. Wang and colleagues reported on the primary analysis results from the Sympatico study. Patients with relapsed/refractory MCL after 1-5 prior therapies were randomly assigned to receive 560 mg of ibrutinib once daily with either placebo (n = 133) or 400 mg daily of venetoclax after ramp-up (n = 134) for 2 years. Then subjects continued taking ibrutinib alone until their disease progressed or they reached unacceptable toxicity.
At a median follow-up of 51.2 months, median progression-free survival was longer in the ibrutinib-venetoclax group vs. ibrutinib alone (hazard ratio [HR] = 0.65, 95% CI, P = .0052).
The new analysis pools several cohorts of patients with TP53 mutations who all took the combination therapy: 5 from a safety run-in phase, 40 from the randomized phase, and 29 from a first-line cohort (median age at baseline = 67).
Median overall survival was not reached in the first-line group and 35.0 months in the relapsed/refractory group (total = 47.1 months). Median progress-free survival in the groups was 22.0 months and 20.9 months, respectively, and median duration of response was 20.5 months and 26.5 months, respectively.
With regard to the new findings, “it is good to see the responses with ibrutinib and venetoclax were deep and durable,” Dr. Epperla said. The combination treatment “provides a good alternative option for TP53-mutated MCL patients who are ineligible for CAR-T.”
Dr. Epperla added that the findings about the addition of ibrutinib could apply to newer-generation Bruton tyrosine kinase inhibitors that have relatively better safety profiles.
However, Dr. Epperla cautioned that the treatment needs to be weighed against the toxicity and cost of the regimen of ibrutinib and venetoclax for 2 years then single-agent ibrutinib until progression or unacceptable toxicity.
This news organization reported in 2023 that estimated net spending on ibrutinib per Medicare data increased by nearly half from 2014-2020, reaching $11,980 in 2020 vs. $7,787 for venetoclax.
Dr. Epperla also noted that “there are newer therapies that are emerging, such as T-cell-engaging bispecific antibodies, and they have shown promising results.”
In an interview, Brad S. Kahl, MD, a hematologist-oncologist at Washington University, St. Louis, said the improvement in outcomes are “modestly significant.”
Dr. Kahl, who is familiar with the study findings but didn’t take part in the research, said it is “worth adding the venetoclax, particularly in these biologically high risk patients with p53 mutations. Venetoclax is not FDA-approved, so insurance approval will need to be determined on a case-by-case basis. The combination is very expensive.”
Dr. Kahl agreed with Dr. Epperla that the findings could be extrapolated to other Bruton tyrosine kinase inhibitors.
The study was funded by Pharmacyclics, an AbbVie Company. Dr. Epperla disclosed relationships with BeiGene and Eli Lilly. Dr. Kahl reported ties with AstraZeneca, BeiGene, Abbvie, and Genentech.
FROM ASCO 2024
CAR T for B-ALL: Game Changer For Young Patients?
It’s becoming more common for patients with less severe disease to undergo the treatment, often bypassing hematopoietic stem cell transplantation (HSCT), and survival is on the rise.
From 2018 to 2022, the percentage of patients in an international cohort who had disease burden of ≥50% fell from 18% to 4%, researchers reported at the annual meeting of the American Society of Clinical Oncology (ASCO) in Chicago. Median relapse-free survival in patients who didn’t undergo post-infusion HSCT grew from 18 months in 2018 to 27 months in 2020. It was not estimable in 2021.
“This introduction of the therapy is changing the treatment landscape of how we look at refractory B-ALL, where the standard of care previously would be to proceed to transplant. This therapy is actually reducing the use of transplant, which has lots of morbidity and mortality associated with it,” Texas Children’s Cancer Center hematologist-oncologist Rayne H. Rouce, MD, who led the study, said in an interview.
Tisagenlecleucel received Food and Drug Administration approval in 2017, said Nirali N. Shah, MD, MHSc, head of the Pediatric Oncology Branch’s Hematologic Malignancies Section at the National Cancer Institute, in an interview. Dr. Shah is familiar with the study findings but didn’t take part in the research.
Remission rates have been around 60%-70%, Dr. Shah said, although that rate is “likely higher” now because of gains in experience and improvement in disease burden prior to therapy.
The new findings fill a knowledge gap about real-world outcomes since a lot of the prior data was based on investigational CAR T-cell products, she said.
The noninterventional, prospective, longitudinal study, funded by tisagenlecleucel manufacturer Novartis, tracked 974 patients up to age 25 who received tisagenlecleucel in the United States, Canada, Korea, and Taiwan.
The study found that between 2018 and 2022:
- The percentage of patients who received treatment while in morphological complete remission grew from 34% to 51%.
- The percentages who were in third or greater relapse fell from 14% to 2%.
- The percentages undergoing ≥1 HSCT before tisagenlecleucel infusion fell from 37% to 15%.
- Overall, 34.5% of 911 patients received post-infusion HSCT.
In the big picture, the findings suggest that the therapy can be considered more than “a last resort for patients in a second or greater relapse or who are refractory,” Dr. Rouce said. By offering CAR T-cell therapy to earlier-stage patients, she said, “when they’re less sick, when they have less comorbidities, and when their organs are functioning better, we could potentially save them from having to go on to a transplant.”
Dr. Shah said the findings indicate that “a substantial number of patients are surviving. It’s remarkable actually. Prior to tisagenlecleucel, patients had dismal outcomes from standard chemotherapy.”
She added that the study suggests “providers are getting much more comfortable with getting their patients in the best shape prior to getting CAR T-cell therapy. Outcomes are improving as providers expand the use of CAR T-cell therapy to patients who are less heavily pretreated and have lower disease burden.”
Moving forward, “at some point there will likely be a plateau in terms of how good the outcomes can be.” And there will be discussion of the role of HSCT.
“We’ll figure out some of the nuances about which patients need transplants and which can avoid them. But curative potential is growing. With or without transplant, this is ultimately going to lead to a much higher fraction of patients being cured who previously would not have been cured,” she said. “That’s the bottom line.”
As for adverse effects, Dr. Shah said “disease burden has a pretty direct relationship with side effects and toxicities. If you have more disease, you have more severe side effects.”
Reducing disease burden will reduce side effects, she said. Also, “we’re getting a lot better at managing these toxicities. Eliminating some of the more toxic chemotherapy through earlier use of CAR T-cells in chemotherapy-refractory patients may well help reduce therapy burden and improve long-term survival outcomes, she added.
As for cost, drugs.com reports that the therapy runs to more than $612,000 per infusion. But Dr. Shah said insurers are covering the treatment. She added that there are efforts to expand the indication so CAR T-cell therapy can be used earlier in patients who are chemotherapy-refractory.
Novartis funded the study. Dr. Shah discloses ties with Lentigen, VOR, and CARGO, ImmunoACT, and Sobi. Dr. Rouce reports relationships with Pfizer and Novartis.
It’s becoming more common for patients with less severe disease to undergo the treatment, often bypassing hematopoietic stem cell transplantation (HSCT), and survival is on the rise.
From 2018 to 2022, the percentage of patients in an international cohort who had disease burden of ≥50% fell from 18% to 4%, researchers reported at the annual meeting of the American Society of Clinical Oncology (ASCO) in Chicago. Median relapse-free survival in patients who didn’t undergo post-infusion HSCT grew from 18 months in 2018 to 27 months in 2020. It was not estimable in 2021.
“This introduction of the therapy is changing the treatment landscape of how we look at refractory B-ALL, where the standard of care previously would be to proceed to transplant. This therapy is actually reducing the use of transplant, which has lots of morbidity and mortality associated with it,” Texas Children’s Cancer Center hematologist-oncologist Rayne H. Rouce, MD, who led the study, said in an interview.
Tisagenlecleucel received Food and Drug Administration approval in 2017, said Nirali N. Shah, MD, MHSc, head of the Pediatric Oncology Branch’s Hematologic Malignancies Section at the National Cancer Institute, in an interview. Dr. Shah is familiar with the study findings but didn’t take part in the research.
Remission rates have been around 60%-70%, Dr. Shah said, although that rate is “likely higher” now because of gains in experience and improvement in disease burden prior to therapy.
The new findings fill a knowledge gap about real-world outcomes since a lot of the prior data was based on investigational CAR T-cell products, she said.
The noninterventional, prospective, longitudinal study, funded by tisagenlecleucel manufacturer Novartis, tracked 974 patients up to age 25 who received tisagenlecleucel in the United States, Canada, Korea, and Taiwan.
The study found that between 2018 and 2022:
- The percentage of patients who received treatment while in morphological complete remission grew from 34% to 51%.
- The percentages who were in third or greater relapse fell from 14% to 2%.
- The percentages undergoing ≥1 HSCT before tisagenlecleucel infusion fell from 37% to 15%.
- Overall, 34.5% of 911 patients received post-infusion HSCT.
In the big picture, the findings suggest that the therapy can be considered more than “a last resort for patients in a second or greater relapse or who are refractory,” Dr. Rouce said. By offering CAR T-cell therapy to earlier-stage patients, she said, “when they’re less sick, when they have less comorbidities, and when their organs are functioning better, we could potentially save them from having to go on to a transplant.”
Dr. Shah said the findings indicate that “a substantial number of patients are surviving. It’s remarkable actually. Prior to tisagenlecleucel, patients had dismal outcomes from standard chemotherapy.”
She added that the study suggests “providers are getting much more comfortable with getting their patients in the best shape prior to getting CAR T-cell therapy. Outcomes are improving as providers expand the use of CAR T-cell therapy to patients who are less heavily pretreated and have lower disease burden.”
Moving forward, “at some point there will likely be a plateau in terms of how good the outcomes can be.” And there will be discussion of the role of HSCT.
“We’ll figure out some of the nuances about which patients need transplants and which can avoid them. But curative potential is growing. With or without transplant, this is ultimately going to lead to a much higher fraction of patients being cured who previously would not have been cured,” she said. “That’s the bottom line.”
As for adverse effects, Dr. Shah said “disease burden has a pretty direct relationship with side effects and toxicities. If you have more disease, you have more severe side effects.”
Reducing disease burden will reduce side effects, she said. Also, “we’re getting a lot better at managing these toxicities. Eliminating some of the more toxic chemotherapy through earlier use of CAR T-cells in chemotherapy-refractory patients may well help reduce therapy burden and improve long-term survival outcomes, she added.
As for cost, drugs.com reports that the therapy runs to more than $612,000 per infusion. But Dr. Shah said insurers are covering the treatment. She added that there are efforts to expand the indication so CAR T-cell therapy can be used earlier in patients who are chemotherapy-refractory.
Novartis funded the study. Dr. Shah discloses ties with Lentigen, VOR, and CARGO, ImmunoACT, and Sobi. Dr. Rouce reports relationships with Pfizer and Novartis.
It’s becoming more common for patients with less severe disease to undergo the treatment, often bypassing hematopoietic stem cell transplantation (HSCT), and survival is on the rise.
From 2018 to 2022, the percentage of patients in an international cohort who had disease burden of ≥50% fell from 18% to 4%, researchers reported at the annual meeting of the American Society of Clinical Oncology (ASCO) in Chicago. Median relapse-free survival in patients who didn’t undergo post-infusion HSCT grew from 18 months in 2018 to 27 months in 2020. It was not estimable in 2021.
“This introduction of the therapy is changing the treatment landscape of how we look at refractory B-ALL, where the standard of care previously would be to proceed to transplant. This therapy is actually reducing the use of transplant, which has lots of morbidity and mortality associated with it,” Texas Children’s Cancer Center hematologist-oncologist Rayne H. Rouce, MD, who led the study, said in an interview.
Tisagenlecleucel received Food and Drug Administration approval in 2017, said Nirali N. Shah, MD, MHSc, head of the Pediatric Oncology Branch’s Hematologic Malignancies Section at the National Cancer Institute, in an interview. Dr. Shah is familiar with the study findings but didn’t take part in the research.
Remission rates have been around 60%-70%, Dr. Shah said, although that rate is “likely higher” now because of gains in experience and improvement in disease burden prior to therapy.
The new findings fill a knowledge gap about real-world outcomes since a lot of the prior data was based on investigational CAR T-cell products, she said.
The noninterventional, prospective, longitudinal study, funded by tisagenlecleucel manufacturer Novartis, tracked 974 patients up to age 25 who received tisagenlecleucel in the United States, Canada, Korea, and Taiwan.
The study found that between 2018 and 2022:
- The percentage of patients who received treatment while in morphological complete remission grew from 34% to 51%.
- The percentages who were in third or greater relapse fell from 14% to 2%.
- The percentages undergoing ≥1 HSCT before tisagenlecleucel infusion fell from 37% to 15%.
- Overall, 34.5% of 911 patients received post-infusion HSCT.
In the big picture, the findings suggest that the therapy can be considered more than “a last resort for patients in a second or greater relapse or who are refractory,” Dr. Rouce said. By offering CAR T-cell therapy to earlier-stage patients, she said, “when they’re less sick, when they have less comorbidities, and when their organs are functioning better, we could potentially save them from having to go on to a transplant.”
Dr. Shah said the findings indicate that “a substantial number of patients are surviving. It’s remarkable actually. Prior to tisagenlecleucel, patients had dismal outcomes from standard chemotherapy.”
She added that the study suggests “providers are getting much more comfortable with getting their patients in the best shape prior to getting CAR T-cell therapy. Outcomes are improving as providers expand the use of CAR T-cell therapy to patients who are less heavily pretreated and have lower disease burden.”
Moving forward, “at some point there will likely be a plateau in terms of how good the outcomes can be.” And there will be discussion of the role of HSCT.
“We’ll figure out some of the nuances about which patients need transplants and which can avoid them. But curative potential is growing. With or without transplant, this is ultimately going to lead to a much higher fraction of patients being cured who previously would not have been cured,” she said. “That’s the bottom line.”
As for adverse effects, Dr. Shah said “disease burden has a pretty direct relationship with side effects and toxicities. If you have more disease, you have more severe side effects.”
Reducing disease burden will reduce side effects, she said. Also, “we’re getting a lot better at managing these toxicities. Eliminating some of the more toxic chemotherapy through earlier use of CAR T-cells in chemotherapy-refractory patients may well help reduce therapy burden and improve long-term survival outcomes, she added.
As for cost, drugs.com reports that the therapy runs to more than $612,000 per infusion. But Dr. Shah said insurers are covering the treatment. She added that there are efforts to expand the indication so CAR T-cell therapy can be used earlier in patients who are chemotherapy-refractory.
Novartis funded the study. Dr. Shah discloses ties with Lentigen, VOR, and CARGO, ImmunoACT, and Sobi. Dr. Rouce reports relationships with Pfizer and Novartis.
FROM ASCO 2024
New Drug Combo Boosts PFS
At a median follow-up of 4 years, progression-free survival for the new treatment, known as BrECADD, was 94.3% vs. 90.9% for BEACOPP (hazard ratio, 0.66, 95% CI, P = .035), researchers led by Peter Borchmann, MD, assistant medical director of hematology and oncology at the University Hospital of Cologne, Germany, reported at the annual meeting of the American Society of Clinical Oncology (ASCO).
“These results are really striking,” said hematologist-oncologist Oreofe O. Odejide, MD, MPH, of the Dana-Farber Cancer Institute and Harvard Medical School, Boston, who was not involved in the study and commented on it during an ASCO news briefing. “This is really poised to impact the standard-of-care treatment for patients with advanced-stage classical Hodgkin lymphoma.”
As Dr. Borchmann explained at the briefing, Hodgkin lymphoma is the most common cancer among young adults. “The median age at onset is around 30 years, and it can be primarily cured with chemotherapy. Intensified chemotherapy probably is better primary lymphoma control than less intensive treatment, but this comes at the cost of treatment-related adverse events.”
Dr. Borchmann and colleagues developed the existing treatment known as BEACOPP, a combination of bleomycin, etoposide, doxorubicin, cyclophosphamide, vincristine, procarbazine, and prednisone. “It’s our standard of care due to its high primary cure rate, which is reflected by compelling progression-free survival,” he said.
However, he said, “it’s a high burden of treatment.” The investigational treatment, BrECADD, includes six drugs instead of seven: brentuximab vedotin, etoposide, cyclophosphamide, doxorubicin, dacarbazine, and dexamethasone. Two of the additions — brentuximab vedotin and dacarbazine — are cancer drugs, and dexamethasone is a steroid. There is one fewer cancer drug in the new formulation.
In the international HD21 trial (9 countries, 233 sites), researchers recruited patients aged 18-60 who received four or six cycles of either BEACOPP or BrECADD. The doses were guided by PET2 findings.
In the intention-to-treat cohort of 1,482 subjects (median age 31.1, 44% female), 742 were assigned to BrECADD and 740 to BEACOPP.
There were few early treatment failures in the BrECADD group vs. BEACOPP. The numbers who had primary progression within the first 3 months were 5 vs. 15, respectively, and the numbers reaching early relapse between months 3 and 12 were 11 vs. 23, respectively.
Four-year overall survival rates in the groups were nearly identical at 98.5% for BrECADD and 98.2% for BEACOPP. In regard to fertility, follicle-stimulating hormone recovery rates after 1 year were higher in the BrECADD group in both men (67% vs. 24%, respectively) and women (89% vs. 68%, respectively). Birth rates were also higher in the BrECADD group (n = 60 vs. n = 43 in the BEACOPP group).
Nearly two-thirds of those in BrECADD group (64%) required 12 weeks of therapy — four cycles. As for treatment-related morbidity toxicities, they were less common in the BrECADD group vs. the BEACOPP group (42% vs. 59%, respectively, P < .0001), and 1% of BrECADD-treated had them at 1 year.
Oncologist Julie R. Gralow, MD, chief medical officer and executive vice president of ASCO, welcomed the findings at the ACO news briefing. “By replacing some pretty toxic chemo with an antibody-drug conjugate [brentuximab vedotin], and changing the regimen a bit, and using PET scan to determine the number of cycles received, the long-term outcomes were maintained, if not even improved upon,” said Dr. Dr. Gralow, who was not involved in the study.
In addition, she said, the findings about fertility are good news because “these are young people who probably haven’t started a family yet, and we’re increasing the odds that they will be able to do so after survival.”
Moving forward, she said, “we will need to have some discussion on how this relates to ABVD, which is a more commonly used regimen in the United States right now.” ABVD refers to a combination of doxorubicin hydrochloride, bleomycin sulfate, vinblastine sulfate, and dacarbazine.
Takeda funded the study. Dr. Borchmann reported ties with BMS, GmbH & Co, Incyte, MSD/Merck, Roche, Takeda/Millennium, Miltenyi, Amgen, and Novartis. Some of the other study authors reported various disclosures. Dr. Odejide and Dr. Gralow have no disclosures.
At a median follow-up of 4 years, progression-free survival for the new treatment, known as BrECADD, was 94.3% vs. 90.9% for BEACOPP (hazard ratio, 0.66, 95% CI, P = .035), researchers led by Peter Borchmann, MD, assistant medical director of hematology and oncology at the University Hospital of Cologne, Germany, reported at the annual meeting of the American Society of Clinical Oncology (ASCO).
“These results are really striking,” said hematologist-oncologist Oreofe O. Odejide, MD, MPH, of the Dana-Farber Cancer Institute and Harvard Medical School, Boston, who was not involved in the study and commented on it during an ASCO news briefing. “This is really poised to impact the standard-of-care treatment for patients with advanced-stage classical Hodgkin lymphoma.”
As Dr. Borchmann explained at the briefing, Hodgkin lymphoma is the most common cancer among young adults. “The median age at onset is around 30 years, and it can be primarily cured with chemotherapy. Intensified chemotherapy probably is better primary lymphoma control than less intensive treatment, but this comes at the cost of treatment-related adverse events.”
Dr. Borchmann and colleagues developed the existing treatment known as BEACOPP, a combination of bleomycin, etoposide, doxorubicin, cyclophosphamide, vincristine, procarbazine, and prednisone. “It’s our standard of care due to its high primary cure rate, which is reflected by compelling progression-free survival,” he said.
However, he said, “it’s a high burden of treatment.” The investigational treatment, BrECADD, includes six drugs instead of seven: brentuximab vedotin, etoposide, cyclophosphamide, doxorubicin, dacarbazine, and dexamethasone. Two of the additions — brentuximab vedotin and dacarbazine — are cancer drugs, and dexamethasone is a steroid. There is one fewer cancer drug in the new formulation.
In the international HD21 trial (9 countries, 233 sites), researchers recruited patients aged 18-60 who received four or six cycles of either BEACOPP or BrECADD. The doses were guided by PET2 findings.
In the intention-to-treat cohort of 1,482 subjects (median age 31.1, 44% female), 742 were assigned to BrECADD and 740 to BEACOPP.
There were few early treatment failures in the BrECADD group vs. BEACOPP. The numbers who had primary progression within the first 3 months were 5 vs. 15, respectively, and the numbers reaching early relapse between months 3 and 12 were 11 vs. 23, respectively.
Four-year overall survival rates in the groups were nearly identical at 98.5% for BrECADD and 98.2% for BEACOPP. In regard to fertility, follicle-stimulating hormone recovery rates after 1 year were higher in the BrECADD group in both men (67% vs. 24%, respectively) and women (89% vs. 68%, respectively). Birth rates were also higher in the BrECADD group (n = 60 vs. n = 43 in the BEACOPP group).
Nearly two-thirds of those in BrECADD group (64%) required 12 weeks of therapy — four cycles. As for treatment-related morbidity toxicities, they were less common in the BrECADD group vs. the BEACOPP group (42% vs. 59%, respectively, P < .0001), and 1% of BrECADD-treated had them at 1 year.
Oncologist Julie R. Gralow, MD, chief medical officer and executive vice president of ASCO, welcomed the findings at the ACO news briefing. “By replacing some pretty toxic chemo with an antibody-drug conjugate [brentuximab vedotin], and changing the regimen a bit, and using PET scan to determine the number of cycles received, the long-term outcomes were maintained, if not even improved upon,” said Dr. Dr. Gralow, who was not involved in the study.
In addition, she said, the findings about fertility are good news because “these are young people who probably haven’t started a family yet, and we’re increasing the odds that they will be able to do so after survival.”
Moving forward, she said, “we will need to have some discussion on how this relates to ABVD, which is a more commonly used regimen in the United States right now.” ABVD refers to a combination of doxorubicin hydrochloride, bleomycin sulfate, vinblastine sulfate, and dacarbazine.
Takeda funded the study. Dr. Borchmann reported ties with BMS, GmbH & Co, Incyte, MSD/Merck, Roche, Takeda/Millennium, Miltenyi, Amgen, and Novartis. Some of the other study authors reported various disclosures. Dr. Odejide and Dr. Gralow have no disclosures.
At a median follow-up of 4 years, progression-free survival for the new treatment, known as BrECADD, was 94.3% vs. 90.9% for BEACOPP (hazard ratio, 0.66, 95% CI, P = .035), researchers led by Peter Borchmann, MD, assistant medical director of hematology and oncology at the University Hospital of Cologne, Germany, reported at the annual meeting of the American Society of Clinical Oncology (ASCO).
“These results are really striking,” said hematologist-oncologist Oreofe O. Odejide, MD, MPH, of the Dana-Farber Cancer Institute and Harvard Medical School, Boston, who was not involved in the study and commented on it during an ASCO news briefing. “This is really poised to impact the standard-of-care treatment for patients with advanced-stage classical Hodgkin lymphoma.”
As Dr. Borchmann explained at the briefing, Hodgkin lymphoma is the most common cancer among young adults. “The median age at onset is around 30 years, and it can be primarily cured with chemotherapy. Intensified chemotherapy probably is better primary lymphoma control than less intensive treatment, but this comes at the cost of treatment-related adverse events.”
Dr. Borchmann and colleagues developed the existing treatment known as BEACOPP, a combination of bleomycin, etoposide, doxorubicin, cyclophosphamide, vincristine, procarbazine, and prednisone. “It’s our standard of care due to its high primary cure rate, which is reflected by compelling progression-free survival,” he said.
However, he said, “it’s a high burden of treatment.” The investigational treatment, BrECADD, includes six drugs instead of seven: brentuximab vedotin, etoposide, cyclophosphamide, doxorubicin, dacarbazine, and dexamethasone. Two of the additions — brentuximab vedotin and dacarbazine — are cancer drugs, and dexamethasone is a steroid. There is one fewer cancer drug in the new formulation.
In the international HD21 trial (9 countries, 233 sites), researchers recruited patients aged 18-60 who received four or six cycles of either BEACOPP or BrECADD. The doses were guided by PET2 findings.
In the intention-to-treat cohort of 1,482 subjects (median age 31.1, 44% female), 742 were assigned to BrECADD and 740 to BEACOPP.
There were few early treatment failures in the BrECADD group vs. BEACOPP. The numbers who had primary progression within the first 3 months were 5 vs. 15, respectively, and the numbers reaching early relapse between months 3 and 12 were 11 vs. 23, respectively.
Four-year overall survival rates in the groups were nearly identical at 98.5% for BrECADD and 98.2% for BEACOPP. In regard to fertility, follicle-stimulating hormone recovery rates after 1 year were higher in the BrECADD group in both men (67% vs. 24%, respectively) and women (89% vs. 68%, respectively). Birth rates were also higher in the BrECADD group (n = 60 vs. n = 43 in the BEACOPP group).
Nearly two-thirds of those in BrECADD group (64%) required 12 weeks of therapy — four cycles. As for treatment-related morbidity toxicities, they were less common in the BrECADD group vs. the BEACOPP group (42% vs. 59%, respectively, P < .0001), and 1% of BrECADD-treated had them at 1 year.
Oncologist Julie R. Gralow, MD, chief medical officer and executive vice president of ASCO, welcomed the findings at the ACO news briefing. “By replacing some pretty toxic chemo with an antibody-drug conjugate [brentuximab vedotin], and changing the regimen a bit, and using PET scan to determine the number of cycles received, the long-term outcomes were maintained, if not even improved upon,” said Dr. Dr. Gralow, who was not involved in the study.
In addition, she said, the findings about fertility are good news because “these are young people who probably haven’t started a family yet, and we’re increasing the odds that they will be able to do so after survival.”
Moving forward, she said, “we will need to have some discussion on how this relates to ABVD, which is a more commonly used regimen in the United States right now.” ABVD refers to a combination of doxorubicin hydrochloride, bleomycin sulfate, vinblastine sulfate, and dacarbazine.
Takeda funded the study. Dr. Borchmann reported ties with BMS, GmbH & Co, Incyte, MSD/Merck, Roche, Takeda/Millennium, Miltenyi, Amgen, and Novartis. Some of the other study authors reported various disclosures. Dr. Odejide and Dr. Gralow have no disclosures.
FROM ASCO 2024
In HPV-Positive Head and Neck Cancer, Treatment Is a Quandary
The topic of head and neck cancer is especially timely since the disease is evolving. A hematologist/oncologist with the Association of VA Hematology/Oncology (AVAHO) told colleagues that specialists are grappling with how to de-escalate treatment.
Molly Tokaz, MD, of Veterans Affairs Puget Sound Health Care and the University of Washington said tobacco is fading as a cause as fewer people smoke, and that human papillomavirus (HPV) is triggering more cases. HPV-positive patients have better prognoses, raising the prospect that their treatment could be adjusted.
“Instead of increasing the amount of therapy we're giving, we’re trying to peel it back,” she said. “If they’re going to respond no matter what we do, why are we going in with these huge weapons of mass destruction if we can get the same results with something more like a light infantry?”
Tokaz spoke about deescalating therapy at a May 2024 regional AVAHO meeting in Seattle that was focused on head and neck cancer. She elaborated on her presentation in an interview with Federal Practitioner. according to Tokaz, 90% of head and neck cancers are mucosal squamous cell carcinomas (SCC). HPV is associated specifically with nasopharyngeal cancer, which is distinct from SCC, and oropharyngeal cancer, which has been linked to better prognoses.
HPV-positive head and neck cancer is a unique entity with its own epidemiology, clinical prognosis, and treatment. “Patients tend to be younger without the same number of comorbid conditions,” Tokaz said. “Some of them are never smokers or light smokers. So, it's a different demographic than we’ve seen traditionally.”
The bad news is that HPV-associated head and neck cancer numbers are on the rise. Fortunately, outcomes tend to be better for the HPV-positive forms.
As for therapy for head and neck cancer, immunotherapy and targeted therapy play smaller roles than in some other cancers because the form tends to be diagnosed in early stages before metastases appear. Surgery, chemotherapy, and radiation remain the major treatments. According to Tokaz’s presentation, surgery, or radiation—often with minimal adjuvant chemotherapy—can be appropriate for the earliest stage I and II cases of head and neck SCC. (She noted that HPV-positive oropharyngeal squamous cell carcinoma has its own staging system.)
Stage I and II cases make up 15% of new diagnoses and have a 5-year survival rate of > 70%. “In the earliest days, our main role was to make radiation work better and reduce it while adding a minimum amount of toxicity mutations,” she said. “Chemotherapy can help, but it’s only demonstrated improvement in overall survival in patients with positive surgical margins and extracapsular extension.”
In Stage III, IVA, and IVB cases, which make up 70% of new diagnoses, chemotherapy plus radiation is recommended. Five-year survival drops to 30% to 50%. Finally, 10% of new diagnoses are Stage IVC, which is incurable and median survival is < 1 year.
Since HPV-positive patients generally have better prognoses, oncologists are considering how to adjust their treatment. However, Tokaz notes that clinical trials have not shown a benefit from less intensive treatment in these patients. “At this point, we still treat them the same way as HPV-negative patients. But it's an ongoing area of research.”
Researchers are also exploring how to optimize regimens in patients ineligible for treatment with the chemotherapy agent cisplatin. “These folks have been traditionally excluded from clinical trials because they’re sicker,” Tokaz explained. “Researchers normally want the fittest and the best patients [in trials]. If you give a drug to someone with a lot of other comorbid conditions, they might not do as well with it, and it makes your drug look bad.”
Figuring out how to treat these patients is an especially urgent task in head and neck cancer because so many patients are frail and have comorbidities. More globally, Tokaz said the rise of HPV-related head and neck cancer highlights the importance of HPV vaccination, which is crucial for preventing cervical and anal cancer in addition to head and neck cancer. “HPV vaccination for children and young adults is crucial.”
Molly Tokaz, MD, reported no relevant financial relationships.
The topic of head and neck cancer is especially timely since the disease is evolving. A hematologist/oncologist with the Association of VA Hematology/Oncology (AVAHO) told colleagues that specialists are grappling with how to de-escalate treatment.
Molly Tokaz, MD, of Veterans Affairs Puget Sound Health Care and the University of Washington said tobacco is fading as a cause as fewer people smoke, and that human papillomavirus (HPV) is triggering more cases. HPV-positive patients have better prognoses, raising the prospect that their treatment could be adjusted.
“Instead of increasing the amount of therapy we're giving, we’re trying to peel it back,” she said. “If they’re going to respond no matter what we do, why are we going in with these huge weapons of mass destruction if we can get the same results with something more like a light infantry?”
Tokaz spoke about deescalating therapy at a May 2024 regional AVAHO meeting in Seattle that was focused on head and neck cancer. She elaborated on her presentation in an interview with Federal Practitioner. according to Tokaz, 90% of head and neck cancers are mucosal squamous cell carcinomas (SCC). HPV is associated specifically with nasopharyngeal cancer, which is distinct from SCC, and oropharyngeal cancer, which has been linked to better prognoses.
HPV-positive head and neck cancer is a unique entity with its own epidemiology, clinical prognosis, and treatment. “Patients tend to be younger without the same number of comorbid conditions,” Tokaz said. “Some of them are never smokers or light smokers. So, it's a different demographic than we’ve seen traditionally.”
The bad news is that HPV-associated head and neck cancer numbers are on the rise. Fortunately, outcomes tend to be better for the HPV-positive forms.
As for therapy for head and neck cancer, immunotherapy and targeted therapy play smaller roles than in some other cancers because the form tends to be diagnosed in early stages before metastases appear. Surgery, chemotherapy, and radiation remain the major treatments. According to Tokaz’s presentation, surgery, or radiation—often with minimal adjuvant chemotherapy—can be appropriate for the earliest stage I and II cases of head and neck SCC. (She noted that HPV-positive oropharyngeal squamous cell carcinoma has its own staging system.)
Stage I and II cases make up 15% of new diagnoses and have a 5-year survival rate of > 70%. “In the earliest days, our main role was to make radiation work better and reduce it while adding a minimum amount of toxicity mutations,” she said. “Chemotherapy can help, but it’s only demonstrated improvement in overall survival in patients with positive surgical margins and extracapsular extension.”
In Stage III, IVA, and IVB cases, which make up 70% of new diagnoses, chemotherapy plus radiation is recommended. Five-year survival drops to 30% to 50%. Finally, 10% of new diagnoses are Stage IVC, which is incurable and median survival is < 1 year.
Since HPV-positive patients generally have better prognoses, oncologists are considering how to adjust their treatment. However, Tokaz notes that clinical trials have not shown a benefit from less intensive treatment in these patients. “At this point, we still treat them the same way as HPV-negative patients. But it's an ongoing area of research.”
Researchers are also exploring how to optimize regimens in patients ineligible for treatment with the chemotherapy agent cisplatin. “These folks have been traditionally excluded from clinical trials because they’re sicker,” Tokaz explained. “Researchers normally want the fittest and the best patients [in trials]. If you give a drug to someone with a lot of other comorbid conditions, they might not do as well with it, and it makes your drug look bad.”
Figuring out how to treat these patients is an especially urgent task in head and neck cancer because so many patients are frail and have comorbidities. More globally, Tokaz said the rise of HPV-related head and neck cancer highlights the importance of HPV vaccination, which is crucial for preventing cervical and anal cancer in addition to head and neck cancer. “HPV vaccination for children and young adults is crucial.”
Molly Tokaz, MD, reported no relevant financial relationships.
The topic of head and neck cancer is especially timely since the disease is evolving. A hematologist/oncologist with the Association of VA Hematology/Oncology (AVAHO) told colleagues that specialists are grappling with how to de-escalate treatment.
Molly Tokaz, MD, of Veterans Affairs Puget Sound Health Care and the University of Washington said tobacco is fading as a cause as fewer people smoke, and that human papillomavirus (HPV) is triggering more cases. HPV-positive patients have better prognoses, raising the prospect that their treatment could be adjusted.
“Instead of increasing the amount of therapy we're giving, we’re trying to peel it back,” she said. “If they’re going to respond no matter what we do, why are we going in with these huge weapons of mass destruction if we can get the same results with something more like a light infantry?”
Tokaz spoke about deescalating therapy at a May 2024 regional AVAHO meeting in Seattle that was focused on head and neck cancer. She elaborated on her presentation in an interview with Federal Practitioner. according to Tokaz, 90% of head and neck cancers are mucosal squamous cell carcinomas (SCC). HPV is associated specifically with nasopharyngeal cancer, which is distinct from SCC, and oropharyngeal cancer, which has been linked to better prognoses.
HPV-positive head and neck cancer is a unique entity with its own epidemiology, clinical prognosis, and treatment. “Patients tend to be younger without the same number of comorbid conditions,” Tokaz said. “Some of them are never smokers or light smokers. So, it's a different demographic than we’ve seen traditionally.”
The bad news is that HPV-associated head and neck cancer numbers are on the rise. Fortunately, outcomes tend to be better for the HPV-positive forms.
As for therapy for head and neck cancer, immunotherapy and targeted therapy play smaller roles than in some other cancers because the form tends to be diagnosed in early stages before metastases appear. Surgery, chemotherapy, and radiation remain the major treatments. According to Tokaz’s presentation, surgery, or radiation—often with minimal adjuvant chemotherapy—can be appropriate for the earliest stage I and II cases of head and neck SCC. (She noted that HPV-positive oropharyngeal squamous cell carcinoma has its own staging system.)
Stage I and II cases make up 15% of new diagnoses and have a 5-year survival rate of > 70%. “In the earliest days, our main role was to make radiation work better and reduce it while adding a minimum amount of toxicity mutations,” she said. “Chemotherapy can help, but it’s only demonstrated improvement in overall survival in patients with positive surgical margins and extracapsular extension.”
In Stage III, IVA, and IVB cases, which make up 70% of new diagnoses, chemotherapy plus radiation is recommended. Five-year survival drops to 30% to 50%. Finally, 10% of new diagnoses are Stage IVC, which is incurable and median survival is < 1 year.
Since HPV-positive patients generally have better prognoses, oncologists are considering how to adjust their treatment. However, Tokaz notes that clinical trials have not shown a benefit from less intensive treatment in these patients. “At this point, we still treat them the same way as HPV-negative patients. But it's an ongoing area of research.”
Researchers are also exploring how to optimize regimens in patients ineligible for treatment with the chemotherapy agent cisplatin. “These folks have been traditionally excluded from clinical trials because they’re sicker,” Tokaz explained. “Researchers normally want the fittest and the best patients [in trials]. If you give a drug to someone with a lot of other comorbid conditions, they might not do as well with it, and it makes your drug look bad.”
Figuring out how to treat these patients is an especially urgent task in head and neck cancer because so many patients are frail and have comorbidities. More globally, Tokaz said the rise of HPV-related head and neck cancer highlights the importance of HPV vaccination, which is crucial for preventing cervical and anal cancer in addition to head and neck cancer. “HPV vaccination for children and young adults is crucial.”
Molly Tokaz, MD, reported no relevant financial relationships.
Head and Neck Cancer in Spotlight at AVAHO Regional Meeting
In the US Department of Veterans Affairs (VA) health care system, head and neck cancer is one of the most complex oncologic conditions to treat because so many medical professionals are involved in its care. Specialists in speech therapy, nutrition, lymphedema, and dentistry are all part of the picture.
“It takes a complete team to treat cancer in a comprehensive manner, and specialists work hand-in-hand,” said Cindy Bowman, MSN, RN, OCN, president of the Association of VA Hematology/Oncology (AVAHO).
AVAHO held a regional meeting in Seattle on May 4, 2024, that was entirely devoted to head and neck cancer. “The goal was to help the VA oncology professionals gain a global view of how various team members can seamlessly work together,” said Bowman, an oncology nurse navigator and coordinator of the Cancer Care Navigation Program at Bay Pines VA Healthcare System in the Tampa-St. Petersburg, FL area.
According to a 2017 report, 2031 cases of head and neck cancer were diagnosed in 2010 among VA patients, accounting for 4.4% of all cancers. “Veterans are especially vulnerable to this type of cancer for several reasons, such as high rates of smoking and alcohol use,” Bowman said. In addition, she said veterans who served in parts of Southeast Asia, North Africa, and the Middle East are at higher risk of nasopharyngeal carcinoma, which has been linked to Epstein-Barr virus infections in those regions.
Radiation treatment were a significant topic at the regional meeting, and 1 session was focused on the importance of prompt care. “Head and neck cancers are very aggressive,” Bowman said. “The sooner we identify them, the sooner we get treatment started.”
Attendees also heard from a speech therapist and a dietician, who discussed a collaborative approach to improving treatment outcomes. “These are two very important pieces of the puzzle.” Bowman said.
On the nutrition front, a lot of newly diagnosed patients already have malnutrition because they have been having difficulty swallowing. So right up front, a registered dietician works with them and individualizes their nutrition treatment plans all the way into recovery. Some of these folks will end up with their relationship with their dietitian for many years.
“Speech therapists work with patients to design swallowing and tongue exercises that target their individual cancer.” Bowman said. The goal is to prevent the need for a feeding tube.
Another session at the regional conference focused on lymphedema—swelling that can develop due to radiation treatment. “All patients with head and neck cancer should be sent to a lymphedema specialist prior to starting treatment since the specialists can prevent this from happening by giving the patients tools, such as compression garments,” Bowman said. “This way, we don’t end up with somebody 15 or 20 years from now coming back and saying they’re not able to move their neck or unable to swallow the right way.”
Another session highlighted the important role of dental care for patients with head and neck cancer. “We send patients to the dentist prior to ever starting anything. We know that radiation therapy can cause osteoradionecrosis, in which people’s teeth begin to crumble. Fortunately, the VA is now covering dentures for these patients, and they automatically get dental care coverage.” Bowman said.
“In the big picture,” she said, “Attendees should come out of the regional meeting with new insight into the importance of teamwork in head and neck cancer care. We need to make sure that all the pieces to the puzzle are there, and everybody is working together to expedite care for the veterans so that they have the best outcomes possible.”
In the US Department of Veterans Affairs (VA) health care system, head and neck cancer is one of the most complex oncologic conditions to treat because so many medical professionals are involved in its care. Specialists in speech therapy, nutrition, lymphedema, and dentistry are all part of the picture.
“It takes a complete team to treat cancer in a comprehensive manner, and specialists work hand-in-hand,” said Cindy Bowman, MSN, RN, OCN, president of the Association of VA Hematology/Oncology (AVAHO).
AVAHO held a regional meeting in Seattle on May 4, 2024, that was entirely devoted to head and neck cancer. “The goal was to help the VA oncology professionals gain a global view of how various team members can seamlessly work together,” said Bowman, an oncology nurse navigator and coordinator of the Cancer Care Navigation Program at Bay Pines VA Healthcare System in the Tampa-St. Petersburg, FL area.
According to a 2017 report, 2031 cases of head and neck cancer were diagnosed in 2010 among VA patients, accounting for 4.4% of all cancers. “Veterans are especially vulnerable to this type of cancer for several reasons, such as high rates of smoking and alcohol use,” Bowman said. In addition, she said veterans who served in parts of Southeast Asia, North Africa, and the Middle East are at higher risk of nasopharyngeal carcinoma, which has been linked to Epstein-Barr virus infections in those regions.
Radiation treatment were a significant topic at the regional meeting, and 1 session was focused on the importance of prompt care. “Head and neck cancers are very aggressive,” Bowman said. “The sooner we identify them, the sooner we get treatment started.”
Attendees also heard from a speech therapist and a dietician, who discussed a collaborative approach to improving treatment outcomes. “These are two very important pieces of the puzzle.” Bowman said.
On the nutrition front, a lot of newly diagnosed patients already have malnutrition because they have been having difficulty swallowing. So right up front, a registered dietician works with them and individualizes their nutrition treatment plans all the way into recovery. Some of these folks will end up with their relationship with their dietitian for many years.
“Speech therapists work with patients to design swallowing and tongue exercises that target their individual cancer.” Bowman said. The goal is to prevent the need for a feeding tube.
Another session at the regional conference focused on lymphedema—swelling that can develop due to radiation treatment. “All patients with head and neck cancer should be sent to a lymphedema specialist prior to starting treatment since the specialists can prevent this from happening by giving the patients tools, such as compression garments,” Bowman said. “This way, we don’t end up with somebody 15 or 20 years from now coming back and saying they’re not able to move their neck or unable to swallow the right way.”
Another session highlighted the important role of dental care for patients with head and neck cancer. “We send patients to the dentist prior to ever starting anything. We know that radiation therapy can cause osteoradionecrosis, in which people’s teeth begin to crumble. Fortunately, the VA is now covering dentures for these patients, and they automatically get dental care coverage.” Bowman said.
“In the big picture,” she said, “Attendees should come out of the regional meeting with new insight into the importance of teamwork in head and neck cancer care. We need to make sure that all the pieces to the puzzle are there, and everybody is working together to expedite care for the veterans so that they have the best outcomes possible.”
In the US Department of Veterans Affairs (VA) health care system, head and neck cancer is one of the most complex oncologic conditions to treat because so many medical professionals are involved in its care. Specialists in speech therapy, nutrition, lymphedema, and dentistry are all part of the picture.
“It takes a complete team to treat cancer in a comprehensive manner, and specialists work hand-in-hand,” said Cindy Bowman, MSN, RN, OCN, president of the Association of VA Hematology/Oncology (AVAHO).
AVAHO held a regional meeting in Seattle on May 4, 2024, that was entirely devoted to head and neck cancer. “The goal was to help the VA oncology professionals gain a global view of how various team members can seamlessly work together,” said Bowman, an oncology nurse navigator and coordinator of the Cancer Care Navigation Program at Bay Pines VA Healthcare System in the Tampa-St. Petersburg, FL area.
According to a 2017 report, 2031 cases of head and neck cancer were diagnosed in 2010 among VA patients, accounting for 4.4% of all cancers. “Veterans are especially vulnerable to this type of cancer for several reasons, such as high rates of smoking and alcohol use,” Bowman said. In addition, she said veterans who served in parts of Southeast Asia, North Africa, and the Middle East are at higher risk of nasopharyngeal carcinoma, which has been linked to Epstein-Barr virus infections in those regions.
Radiation treatment were a significant topic at the regional meeting, and 1 session was focused on the importance of prompt care. “Head and neck cancers are very aggressive,” Bowman said. “The sooner we identify them, the sooner we get treatment started.”
Attendees also heard from a speech therapist and a dietician, who discussed a collaborative approach to improving treatment outcomes. “These are two very important pieces of the puzzle.” Bowman said.
On the nutrition front, a lot of newly diagnosed patients already have malnutrition because they have been having difficulty swallowing. So right up front, a registered dietician works with them and individualizes their nutrition treatment plans all the way into recovery. Some of these folks will end up with their relationship with their dietitian for many years.
“Speech therapists work with patients to design swallowing and tongue exercises that target their individual cancer.” Bowman said. The goal is to prevent the need for a feeding tube.
Another session at the regional conference focused on lymphedema—swelling that can develop due to radiation treatment. “All patients with head and neck cancer should be sent to a lymphedema specialist prior to starting treatment since the specialists can prevent this from happening by giving the patients tools, such as compression garments,” Bowman said. “This way, we don’t end up with somebody 15 or 20 years from now coming back and saying they’re not able to move their neck or unable to swallow the right way.”
Another session highlighted the important role of dental care for patients with head and neck cancer. “We send patients to the dentist prior to ever starting anything. We know that radiation therapy can cause osteoradionecrosis, in which people’s teeth begin to crumble. Fortunately, the VA is now covering dentures for these patients, and they automatically get dental care coverage.” Bowman said.
“In the big picture,” she said, “Attendees should come out of the regional meeting with new insight into the importance of teamwork in head and neck cancer care. We need to make sure that all the pieces to the puzzle are there, and everybody is working together to expedite care for the veterans so that they have the best outcomes possible.”
Global Quest to Cut CAR T Costs
In the United States, a stand-alone device could greatly reduce the expense of producing modified immune cells. In India, researchers hope homegrown technology is the key to getting costs under control. In Latin America, a partnership between the Brazilian government and a US nonprofit may be just the ticket.
At stake is expanded access to CAR T-cell therapy, a form of immunotherapy that in just the past few years has revolutionized the care of hematologic cancers.
“Among patients with lymphoma, leukemia, and myeloma, anywhere between 30% to 50% reach long-term remission after one CAR T-cell infusion,” Mayo Clinic–Rochester hematologist/oncologist Saad J. Kenderian, MB, ChB, said in an interview. “It’s such an important therapy.”
However, only a small percentage of eligible patients in the United States — perhaps 20% or fewer — are receiving the treatment, he added.
A 2024 report suggested that many patients in the United States who may benefit aren’t being treated because of a range of possible reasons, including high prices, manufacturing logistics, and far distance from the limited number of institutions offering the therapy.
“Taken together, the real-world cost of CAR T-cell therapy can range from $700,000 to $1 million, which may make the treatment unaffordable to those patients without robust financial and/or social support,” the report authors noted.
Outside Western countries, access to the therapy is even more limited, because of its exorbitant price. The 2024 report noted that “there is a wide use of CAR T-cell therapy in Europe and China, but access is limited in developing countries in Southeast Asia, Africa, and Latin America.”
Harnessing the Power of T-Cells
Several types of CAR T-cell therapy have been approved by the US Food and Drug Administration (FDA) for patients with relapsed/refractory blood cancers such as follicular lymphoma, large B-cell lymphoma, multiple myeloma, and B-cell precursor acute lymphoblastic leukemia. A 2023 review analyzed clinical trials and reported that complete response rates were 40%-54% in aggressive B-cell lymphoma, 67% in mantle cell lymphoma, and 69%-74% in indolent B-cell lymphoma.
Pediatric hematologist/oncologist Kirsten Williams, MD, who specializes in pediatric blood and marrow transplant and cellular therapy at the Aflac Cancer and Blood Disorders Center of Children’s Healthcare of Atlanta, described CAR T-cell therapy as “a very unique form of immunotherapy” that harnesses the power of the immune system’s T-cells.
These cells are effective tumor killers, but they typically aren’t assigned to control cancer, she said in an interview. “We have very few of them, and most of our T cells are focused on killing various viruses,” she said. The therapy “allows us to take the T cell that would have killed the flu or mono and instead target leukemia, B-cell leukemia, or lymphoma.”
As she explained, “T cells are collected by a machine that reserves white blood cells and gives back the rest of the blood to the patient. We insert a gene into the T cells that encodes for a B-cell receptor. This receptor acts as a GPS signal, pulling T cells to the cancer so that they can kill it.”
In addition, “with this genetic change, we also add some things that allow the T cell to be stronger, to have a higher signal to kill the cancer cell once it locks on.”
The therapy is unique for each patient, Dr. Williams said. “We have collected and modified your specific T cells, and they can now only be infused into you. If we try to give your product to someone else, those cells would either cause harm by attacking the patient or would be immediately killed by that patient’s own immune system. This is very different than all the other kinds of therapies. When you take other medicines, it doesn’t matter who receives that pill.”
Treatment: Individual, Complex, and Costly
Why is CAR T-cell therapy so expensive? While only a single treatment is needed, the T cells have to go through an “individualized, bespoke manufacturing” process that’s “highly technical,” pediatric oncologist Stephan A. Grupp, MD, PhD, section chief of the Cellular Therapy and Transplant Section at Children’s Hospital of Philadelphia, said in an interview. As he explained, the cells for a single patient have to go through the same testing as with a drug that might be given to 1,000 people.
“The first thing we need to do is collect the cells from a patient,” said Dr. Williams. “For adults, that process involves putting in two big IVs — one in each arm — and then pulling the blood through a machine. This typically involves an 8-hour collection in the hospital and very highly specialized people to oversee the collection process.”
Secondly, at some institutions, “the cells get sent to a company where they undergo the process where the gene is inserted,” she said. “This process needs to be done in a very sterile environment so there’s no infections, and it needs to have a lot of oversight.”
Finally, “after the cells are generated, they are typically frozen and shipped back to the site where the patient is at the hospital,” she said. “Then we give chemotherapy to the patient, which prepares the patient’s blood system. It removes some of the T-cells that are there, allowing for the T cells that we’re about to infuse to quickly be activated, find the cancer, and kill it.”
Side effects can boost costs even more. “Unfortunately, some significant toxicities can occur after we infuse these cells,” Dr. Williams noted. “Patients can have trouble breathing and sometimes need ventilatory support. They can have trouble maintaining their blood pressure and become swollen as fluid seeps into tissues. Or they can have high fevers and organ dysfunction. Many of those patients go to the intensive care unit, which is obviously expensive as well.”
Taking Gene Therapy In-House
As Dr. Williams explained, one way to reduce costs is to “perform the genetic manipulation and expansion of the cells outside of a company.” Several academic institutions in the United States are embracing this approach, including Children’s Hospital of Philadelphia, which is experimenting with an automated device developed by the German company Miltenyi Biotec and known as the CliniMACS Prodigy machine.
“The current manufacturing process is very manual and requires a lot of interaction with the product and highly trained personnel,” Dr. Grupp said. “If you have an automated device, you have those cells in the device over the 7 to 12 days that you actually need to grow the cells. There’s much less interaction, so you need fewer trained personnel.”
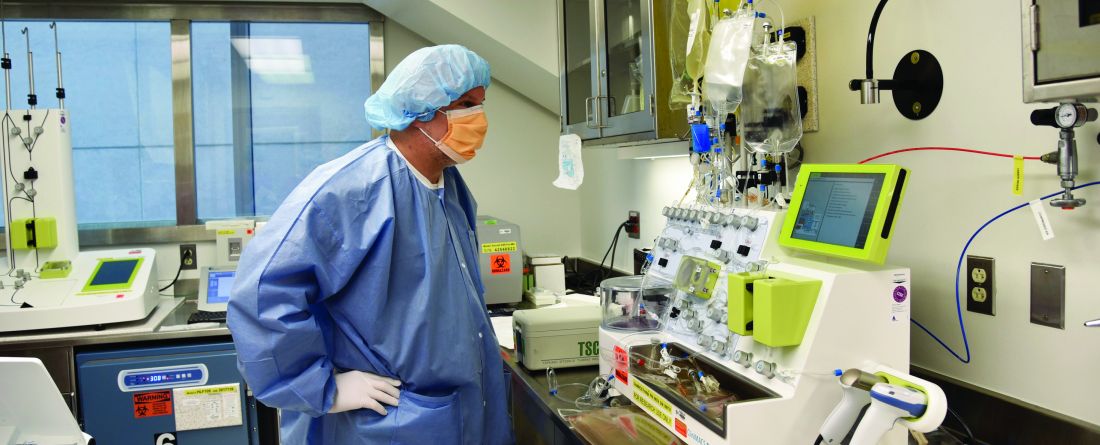
The device is experimental and not yet FDA approved, Dr. Grupp noted, so that patients are all in clinical trials. Children’s Hospital of Philadelphia has treated more than a dozen patients with the device, he said.
Another member of Children’s Hospital of Philadelphia’s CAR T-cell team told WHYY-FM that a single patient’s treatment would run about $30,000 for labor and testing, but not other expenses such as facility costs.
Dr. Grupp estimated that about half a dozen of these devices are in use in the United States, and many more worldwide. “They’re all just like we are — at the absolute beginning. We’ve only been doing this for about a year.”
In the big picture, Dr. Grupp said, “this is where cell therapy is going. Whether it’s point of care or not, automated cell manufacturing is the obvious next step.”
India: Big Hopes for Homegrown Technology
In India, researchers are hoping that their homegrown approach to CAR T-cell therapy will expand access by greatly lowering treatment prices.
Last fall, India’s equivalent of the FDA-granted approval for actalycabtagene autoleucel (NexCAR19), which was developed by Indian scientists who worked closely with the US National Institutes of Health (NIH). The therapy’s developer is a company called ImmunoACT.
In an interview, ImmunoACT founder Rahul Purwar, PhD, MSc, associate professor at Indian Institute of Technology Bombay, said the treatment costs about $40,000. The price is much lower than in the United States because staffing, facility construction, and maintenance are less expensive in India, he said.
Results of small early clinical trials have been promising, with complete responses in 68% of 38 lymphoma patients and 72% of 15 leukemia patients. Updated data will be presented at the annual American Society of Hematology meeting in December 2024, Dr. Purwar said.
According to the NIH, at first ImmunoACT hopes to treat about 1,200 patients a year. The immediate goal is to “focus and stabilize our operation in India,” Dr. Purwar said. “Then, if opportunities come, we will try to bring CAR T to all who might benefit from these technologies. A majority of countries don’t have access to these technologies.”
A US-Brazil Partnership Holds Promise
Meanwhile, a US nonprofit known as Caring Cross announced this year that it has partnered with Fundação Oswaldo Cruz (Fiocruz), a Brazilian government foundation, to manufacture CAR T cells at point-of-care in South America.
“Our model is different than traditional biotech/pharma,” Boro Dropulic, PhD, MBA, cofounder and executive director of Caring Cross, said in an interview. “Our goal is to develop technologies and transfer them to organizations like Fiocruz to enable them to manufacture these transformative therapies for patients in their regions. We believe this model is an important solution for therapies that are priced so high that they are not accessible to many patients that need them, particularly underserved populations and those in low- and middle-income countries.”
According to Dr. Dropulic: “We have developed a production process where the material cost is about $20,000 per dose.” When labor and infrastructure costs are added, the total expense won’t be more than $37,000-$47,500, he said.
The research process for the CAR T-cell technology is at an earlier stage than in India. Scientists plan to start clinical trials of the technology in the United States by the end of 2024 and then begin them in Brazil in 2025, after safety and efficacy have been demonstrated. The first trial, a phase I/II study, will enroll about 20 patients, Dr. Dropulic said.
Dr. Kenderian reported ties with Novartis, Capstan Bio, Kite/Gilead, Juno/BMS, Humanigen, Tolero, Leah Labs, Lentigen, Luminary, Sunesis/Viracta, Morphosys, Troque, Carisma, Sendero, and LifEngine. Dr. Williams disclosed grants from National Institutes of Health and philanthropic organizations. Dr. Grupp reported relationships with Novartis, Kite, Vertex and Servier, Roche, GSK, Humanigen, CBMG, Eureka, Janssen/JNJ, Jazz, Adaptimmune, TCR2, Cellectis, Juno, Allogene, and Cabaletta. Dr. Purwar is the founder of ImmunoACT. Dr. Dropulic serves as executive director of Caring Cross and CEO of Vector BioMed, which provides vectors for gene therapy.
In the United States, a stand-alone device could greatly reduce the expense of producing modified immune cells. In India, researchers hope homegrown technology is the key to getting costs under control. In Latin America, a partnership between the Brazilian government and a US nonprofit may be just the ticket.
At stake is expanded access to CAR T-cell therapy, a form of immunotherapy that in just the past few years has revolutionized the care of hematologic cancers.
“Among patients with lymphoma, leukemia, and myeloma, anywhere between 30% to 50% reach long-term remission after one CAR T-cell infusion,” Mayo Clinic–Rochester hematologist/oncologist Saad J. Kenderian, MB, ChB, said in an interview. “It’s such an important therapy.”
However, only a small percentage of eligible patients in the United States — perhaps 20% or fewer — are receiving the treatment, he added.
A 2024 report suggested that many patients in the United States who may benefit aren’t being treated because of a range of possible reasons, including high prices, manufacturing logistics, and far distance from the limited number of institutions offering the therapy.
“Taken together, the real-world cost of CAR T-cell therapy can range from $700,000 to $1 million, which may make the treatment unaffordable to those patients without robust financial and/or social support,” the report authors noted.
Outside Western countries, access to the therapy is even more limited, because of its exorbitant price. The 2024 report noted that “there is a wide use of CAR T-cell therapy in Europe and China, but access is limited in developing countries in Southeast Asia, Africa, and Latin America.”
Harnessing the Power of T-Cells
Several types of CAR T-cell therapy have been approved by the US Food and Drug Administration (FDA) for patients with relapsed/refractory blood cancers such as follicular lymphoma, large B-cell lymphoma, multiple myeloma, and B-cell precursor acute lymphoblastic leukemia. A 2023 review analyzed clinical trials and reported that complete response rates were 40%-54% in aggressive B-cell lymphoma, 67% in mantle cell lymphoma, and 69%-74% in indolent B-cell lymphoma.
Pediatric hematologist/oncologist Kirsten Williams, MD, who specializes in pediatric blood and marrow transplant and cellular therapy at the Aflac Cancer and Blood Disorders Center of Children’s Healthcare of Atlanta, described CAR T-cell therapy as “a very unique form of immunotherapy” that harnesses the power of the immune system’s T-cells.
These cells are effective tumor killers, but they typically aren’t assigned to control cancer, she said in an interview. “We have very few of them, and most of our T cells are focused on killing various viruses,” she said. The therapy “allows us to take the T cell that would have killed the flu or mono and instead target leukemia, B-cell leukemia, or lymphoma.”
As she explained, “T cells are collected by a machine that reserves white blood cells and gives back the rest of the blood to the patient. We insert a gene into the T cells that encodes for a B-cell receptor. This receptor acts as a GPS signal, pulling T cells to the cancer so that they can kill it.”
In addition, “with this genetic change, we also add some things that allow the T cell to be stronger, to have a higher signal to kill the cancer cell once it locks on.”
The therapy is unique for each patient, Dr. Williams said. “We have collected and modified your specific T cells, and they can now only be infused into you. If we try to give your product to someone else, those cells would either cause harm by attacking the patient or would be immediately killed by that patient’s own immune system. This is very different than all the other kinds of therapies. When you take other medicines, it doesn’t matter who receives that pill.”
Treatment: Individual, Complex, and Costly
Why is CAR T-cell therapy so expensive? While only a single treatment is needed, the T cells have to go through an “individualized, bespoke manufacturing” process that’s “highly technical,” pediatric oncologist Stephan A. Grupp, MD, PhD, section chief of the Cellular Therapy and Transplant Section at Children’s Hospital of Philadelphia, said in an interview. As he explained, the cells for a single patient have to go through the same testing as with a drug that might be given to 1,000 people.
“The first thing we need to do is collect the cells from a patient,” said Dr. Williams. “For adults, that process involves putting in two big IVs — one in each arm — and then pulling the blood through a machine. This typically involves an 8-hour collection in the hospital and very highly specialized people to oversee the collection process.”
Secondly, at some institutions, “the cells get sent to a company where they undergo the process where the gene is inserted,” she said. “This process needs to be done in a very sterile environment so there’s no infections, and it needs to have a lot of oversight.”
Finally, “after the cells are generated, they are typically frozen and shipped back to the site where the patient is at the hospital,” she said. “Then we give chemotherapy to the patient, which prepares the patient’s blood system. It removes some of the T-cells that are there, allowing for the T cells that we’re about to infuse to quickly be activated, find the cancer, and kill it.”
Side effects can boost costs even more. “Unfortunately, some significant toxicities can occur after we infuse these cells,” Dr. Williams noted. “Patients can have trouble breathing and sometimes need ventilatory support. They can have trouble maintaining their blood pressure and become swollen as fluid seeps into tissues. Or they can have high fevers and organ dysfunction. Many of those patients go to the intensive care unit, which is obviously expensive as well.”
Taking Gene Therapy In-House
As Dr. Williams explained, one way to reduce costs is to “perform the genetic manipulation and expansion of the cells outside of a company.” Several academic institutions in the United States are embracing this approach, including Children’s Hospital of Philadelphia, which is experimenting with an automated device developed by the German company Miltenyi Biotec and known as the CliniMACS Prodigy machine.
“The current manufacturing process is very manual and requires a lot of interaction with the product and highly trained personnel,” Dr. Grupp said. “If you have an automated device, you have those cells in the device over the 7 to 12 days that you actually need to grow the cells. There’s much less interaction, so you need fewer trained personnel.”
The device is experimental and not yet FDA approved, Dr. Grupp noted, so that patients are all in clinical trials. Children’s Hospital of Philadelphia has treated more than a dozen patients with the device, he said.
Another member of Children’s Hospital of Philadelphia’s CAR T-cell team told WHYY-FM that a single patient’s treatment would run about $30,000 for labor and testing, but not other expenses such as facility costs.
Dr. Grupp estimated that about half a dozen of these devices are in use in the United States, and many more worldwide. “They’re all just like we are — at the absolute beginning. We’ve only been doing this for about a year.”
In the big picture, Dr. Grupp said, “this is where cell therapy is going. Whether it’s point of care or not, automated cell manufacturing is the obvious next step.”
India: Big Hopes for Homegrown Technology
In India, researchers are hoping that their homegrown approach to CAR T-cell therapy will expand access by greatly lowering treatment prices.
Last fall, India’s equivalent of the FDA-granted approval for actalycabtagene autoleucel (NexCAR19), which was developed by Indian scientists who worked closely with the US National Institutes of Health (NIH). The therapy’s developer is a company called ImmunoACT.
In an interview, ImmunoACT founder Rahul Purwar, PhD, MSc, associate professor at Indian Institute of Technology Bombay, said the treatment costs about $40,000. The price is much lower than in the United States because staffing, facility construction, and maintenance are less expensive in India, he said.
Results of small early clinical trials have been promising, with complete responses in 68% of 38 lymphoma patients and 72% of 15 leukemia patients. Updated data will be presented at the annual American Society of Hematology meeting in December 2024, Dr. Purwar said.
According to the NIH, at first ImmunoACT hopes to treat about 1,200 patients a year. The immediate goal is to “focus and stabilize our operation in India,” Dr. Purwar said. “Then, if opportunities come, we will try to bring CAR T to all who might benefit from these technologies. A majority of countries don’t have access to these technologies.”
A US-Brazil Partnership Holds Promise
Meanwhile, a US nonprofit known as Caring Cross announced this year that it has partnered with Fundação Oswaldo Cruz (Fiocruz), a Brazilian government foundation, to manufacture CAR T cells at point-of-care in South America.
“Our model is different than traditional biotech/pharma,” Boro Dropulic, PhD, MBA, cofounder and executive director of Caring Cross, said in an interview. “Our goal is to develop technologies and transfer them to organizations like Fiocruz to enable them to manufacture these transformative therapies for patients in their regions. We believe this model is an important solution for therapies that are priced so high that they are not accessible to many patients that need them, particularly underserved populations and those in low- and middle-income countries.”
According to Dr. Dropulic: “We have developed a production process where the material cost is about $20,000 per dose.” When labor and infrastructure costs are added, the total expense won’t be more than $37,000-$47,500, he said.
The research process for the CAR T-cell technology is at an earlier stage than in India. Scientists plan to start clinical trials of the technology in the United States by the end of 2024 and then begin them in Brazil in 2025, after safety and efficacy have been demonstrated. The first trial, a phase I/II study, will enroll about 20 patients, Dr. Dropulic said.
Dr. Kenderian reported ties with Novartis, Capstan Bio, Kite/Gilead, Juno/BMS, Humanigen, Tolero, Leah Labs, Lentigen, Luminary, Sunesis/Viracta, Morphosys, Troque, Carisma, Sendero, and LifEngine. Dr. Williams disclosed grants from National Institutes of Health and philanthropic organizations. Dr. Grupp reported relationships with Novartis, Kite, Vertex and Servier, Roche, GSK, Humanigen, CBMG, Eureka, Janssen/JNJ, Jazz, Adaptimmune, TCR2, Cellectis, Juno, Allogene, and Cabaletta. Dr. Purwar is the founder of ImmunoACT. Dr. Dropulic serves as executive director of Caring Cross and CEO of Vector BioMed, which provides vectors for gene therapy.
In the United States, a stand-alone device could greatly reduce the expense of producing modified immune cells. In India, researchers hope homegrown technology is the key to getting costs under control. In Latin America, a partnership between the Brazilian government and a US nonprofit may be just the ticket.
At stake is expanded access to CAR T-cell therapy, a form of immunotherapy that in just the past few years has revolutionized the care of hematologic cancers.
“Among patients with lymphoma, leukemia, and myeloma, anywhere between 30% to 50% reach long-term remission after one CAR T-cell infusion,” Mayo Clinic–Rochester hematologist/oncologist Saad J. Kenderian, MB, ChB, said in an interview. “It’s such an important therapy.”
However, only a small percentage of eligible patients in the United States — perhaps 20% or fewer — are receiving the treatment, he added.
A 2024 report suggested that many patients in the United States who may benefit aren’t being treated because of a range of possible reasons, including high prices, manufacturing logistics, and far distance from the limited number of institutions offering the therapy.
“Taken together, the real-world cost of CAR T-cell therapy can range from $700,000 to $1 million, which may make the treatment unaffordable to those patients without robust financial and/or social support,” the report authors noted.
Outside Western countries, access to the therapy is even more limited, because of its exorbitant price. The 2024 report noted that “there is a wide use of CAR T-cell therapy in Europe and China, but access is limited in developing countries in Southeast Asia, Africa, and Latin America.”
Harnessing the Power of T-Cells
Several types of CAR T-cell therapy have been approved by the US Food and Drug Administration (FDA) for patients with relapsed/refractory blood cancers such as follicular lymphoma, large B-cell lymphoma, multiple myeloma, and B-cell precursor acute lymphoblastic leukemia. A 2023 review analyzed clinical trials and reported that complete response rates were 40%-54% in aggressive B-cell lymphoma, 67% in mantle cell lymphoma, and 69%-74% in indolent B-cell lymphoma.
Pediatric hematologist/oncologist Kirsten Williams, MD, who specializes in pediatric blood and marrow transplant and cellular therapy at the Aflac Cancer and Blood Disorders Center of Children’s Healthcare of Atlanta, described CAR T-cell therapy as “a very unique form of immunotherapy” that harnesses the power of the immune system’s T-cells.
These cells are effective tumor killers, but they typically aren’t assigned to control cancer, she said in an interview. “We have very few of them, and most of our T cells are focused on killing various viruses,” she said. The therapy “allows us to take the T cell that would have killed the flu or mono and instead target leukemia, B-cell leukemia, or lymphoma.”
As she explained, “T cells are collected by a machine that reserves white blood cells and gives back the rest of the blood to the patient. We insert a gene into the T cells that encodes for a B-cell receptor. This receptor acts as a GPS signal, pulling T cells to the cancer so that they can kill it.”
In addition, “with this genetic change, we also add some things that allow the T cell to be stronger, to have a higher signal to kill the cancer cell once it locks on.”
The therapy is unique for each patient, Dr. Williams said. “We have collected and modified your specific T cells, and they can now only be infused into you. If we try to give your product to someone else, those cells would either cause harm by attacking the patient or would be immediately killed by that patient’s own immune system. This is very different than all the other kinds of therapies. When you take other medicines, it doesn’t matter who receives that pill.”
Treatment: Individual, Complex, and Costly
Why is CAR T-cell therapy so expensive? While only a single treatment is needed, the T cells have to go through an “individualized, bespoke manufacturing” process that’s “highly technical,” pediatric oncologist Stephan A. Grupp, MD, PhD, section chief of the Cellular Therapy and Transplant Section at Children’s Hospital of Philadelphia, said in an interview. As he explained, the cells for a single patient have to go through the same testing as with a drug that might be given to 1,000 people.
“The first thing we need to do is collect the cells from a patient,” said Dr. Williams. “For adults, that process involves putting in two big IVs — one in each arm — and then pulling the blood through a machine. This typically involves an 8-hour collection in the hospital and very highly specialized people to oversee the collection process.”
Secondly, at some institutions, “the cells get sent to a company where they undergo the process where the gene is inserted,” she said. “This process needs to be done in a very sterile environment so there’s no infections, and it needs to have a lot of oversight.”
Finally, “after the cells are generated, they are typically frozen and shipped back to the site where the patient is at the hospital,” she said. “Then we give chemotherapy to the patient, which prepares the patient’s blood system. It removes some of the T-cells that are there, allowing for the T cells that we’re about to infuse to quickly be activated, find the cancer, and kill it.”
Side effects can boost costs even more. “Unfortunately, some significant toxicities can occur after we infuse these cells,” Dr. Williams noted. “Patients can have trouble breathing and sometimes need ventilatory support. They can have trouble maintaining their blood pressure and become swollen as fluid seeps into tissues. Or they can have high fevers and organ dysfunction. Many of those patients go to the intensive care unit, which is obviously expensive as well.”
Taking Gene Therapy In-House
As Dr. Williams explained, one way to reduce costs is to “perform the genetic manipulation and expansion of the cells outside of a company.” Several academic institutions in the United States are embracing this approach, including Children’s Hospital of Philadelphia, which is experimenting with an automated device developed by the German company Miltenyi Biotec and known as the CliniMACS Prodigy machine.
“The current manufacturing process is very manual and requires a lot of interaction with the product and highly trained personnel,” Dr. Grupp said. “If you have an automated device, you have those cells in the device over the 7 to 12 days that you actually need to grow the cells. There’s much less interaction, so you need fewer trained personnel.”
The device is experimental and not yet FDA approved, Dr. Grupp noted, so that patients are all in clinical trials. Children’s Hospital of Philadelphia has treated more than a dozen patients with the device, he said.
Another member of Children’s Hospital of Philadelphia’s CAR T-cell team told WHYY-FM that a single patient’s treatment would run about $30,000 for labor and testing, but not other expenses such as facility costs.
Dr. Grupp estimated that about half a dozen of these devices are in use in the United States, and many more worldwide. “They’re all just like we are — at the absolute beginning. We’ve only been doing this for about a year.”
In the big picture, Dr. Grupp said, “this is where cell therapy is going. Whether it’s point of care or not, automated cell manufacturing is the obvious next step.”
India: Big Hopes for Homegrown Technology
In India, researchers are hoping that their homegrown approach to CAR T-cell therapy will expand access by greatly lowering treatment prices.
Last fall, India’s equivalent of the FDA-granted approval for actalycabtagene autoleucel (NexCAR19), which was developed by Indian scientists who worked closely with the US National Institutes of Health (NIH). The therapy’s developer is a company called ImmunoACT.
In an interview, ImmunoACT founder Rahul Purwar, PhD, MSc, associate professor at Indian Institute of Technology Bombay, said the treatment costs about $40,000. The price is much lower than in the United States because staffing, facility construction, and maintenance are less expensive in India, he said.
Results of small early clinical trials have been promising, with complete responses in 68% of 38 lymphoma patients and 72% of 15 leukemia patients. Updated data will be presented at the annual American Society of Hematology meeting in December 2024, Dr. Purwar said.
According to the NIH, at first ImmunoACT hopes to treat about 1,200 patients a year. The immediate goal is to “focus and stabilize our operation in India,” Dr. Purwar said. “Then, if opportunities come, we will try to bring CAR T to all who might benefit from these technologies. A majority of countries don’t have access to these technologies.”
A US-Brazil Partnership Holds Promise
Meanwhile, a US nonprofit known as Caring Cross announced this year that it has partnered with Fundação Oswaldo Cruz (Fiocruz), a Brazilian government foundation, to manufacture CAR T cells at point-of-care in South America.
“Our model is different than traditional biotech/pharma,” Boro Dropulic, PhD, MBA, cofounder and executive director of Caring Cross, said in an interview. “Our goal is to develop technologies and transfer them to organizations like Fiocruz to enable them to manufacture these transformative therapies for patients in their regions. We believe this model is an important solution for therapies that are priced so high that they are not accessible to many patients that need them, particularly underserved populations and those in low- and middle-income countries.”
According to Dr. Dropulic: “We have developed a production process where the material cost is about $20,000 per dose.” When labor and infrastructure costs are added, the total expense won’t be more than $37,000-$47,500, he said.
The research process for the CAR T-cell technology is at an earlier stage than in India. Scientists plan to start clinical trials of the technology in the United States by the end of 2024 and then begin them in Brazil in 2025, after safety and efficacy have been demonstrated. The first trial, a phase I/II study, will enroll about 20 patients, Dr. Dropulic said.
Dr. Kenderian reported ties with Novartis, Capstan Bio, Kite/Gilead, Juno/BMS, Humanigen, Tolero, Leah Labs, Lentigen, Luminary, Sunesis/Viracta, Morphosys, Troque, Carisma, Sendero, and LifEngine. Dr. Williams disclosed grants from National Institutes of Health and philanthropic organizations. Dr. Grupp reported relationships with Novartis, Kite, Vertex and Servier, Roche, GSK, Humanigen, CBMG, Eureka, Janssen/JNJ, Jazz, Adaptimmune, TCR2, Cellectis, Juno, Allogene, and Cabaletta. Dr. Purwar is the founder of ImmunoACT. Dr. Dropulic serves as executive director of Caring Cross and CEO of Vector BioMed, which provides vectors for gene therapy.
New Trial Deepens Debate Over Late-Preterm Steroids
The early cancellation of a trial in southern India suggests that the use of antenatal steroids to prevent respiratory complications after late-preterm birth — a recommended practice in the United States — may not be effective in the developing world.
As reported in Obstetrics & Gynecology, researchers led by Hilda Yenuberi, MD, of Christian Medical College, Vellore, Tamil Nadu, India, stopped the randomized, triple-blinded, placebo-controlled CLAP (Corticosteroids in Late Pregnancy) study at 70% enrollment. An interim analysis found no benefit from prescribing betamethasone vs placebo to women at risk of late-preterm delivery between 34 and 36 and 6/7 weeks of gestation (primary outcome of respiratory distress: 4.9% vs 4.8%, respectively, relative risk [RR], 1.03; 95% CI, 0.57-1.84; number needed to treat = 786).
“These findings may suggest differing efficacy of antenatal corticosteroids in developing countries compared with developed countries ... that should be considered when late-preterm antenatal corticosteroids are administered,” the researchers wrote.
The use of steroids in patients at risk of delivery before 34 weeks is widely accepted as a way to prevent neonatal respiratory distress, a common and potentially deadly condition in premature infants whose lungs are not fully developed. However, there’s debate over steroid treatment in women who are expected to deliver later than 34 weeks but still preterm.
As the study notes, “the American College of Obstetricians and Gynecologists recommends a single course of betamethasone for pregnant individuals at risk of delivering between 34 and 36 6/7 weeks of gestation on the basis of the ALPS (Antenatal Late Preterm Steroid) trial.”
But other randomized trials have reached different conclusions, and steroids are not without risks. Studies have linked prenatal steroids to neurosensory disorders in babies, meaning they’re more likely to need hearing aids and eyeglasses, said Kellie Murphy, MD, MSc, professor of obstetrics and gynecology, University of Toronto, Toronto, Ontario, Canada, in an interview. Dr. Murphy, who was not involved in the new trial, added that there are links between steroids and greater likelihood of poorer performance in school,
For the new study, conducted from 2020 to 2022 at Christian Medical College and Hospital in Vellore, India, researchers randomly assigned 423 patients to betamethasone (410 in the interim analysis; average age, 26.8 years) and 424 to placebo (415 in the interim analysis; average age, 26.2 years).
The average age of participants was 26.8 years. All were between 34 and 36 6/7 weeks of gestation and expected to give birth within the next week. A quarter of participants delivered at term, which the authors wrote “may have influenced the primary outcome.” The total number of neonates was 883, including 58 twin pregnancies.
There was no significant difference in respiratory distress between groups, “defined as need for oxygen or continuous positive airway pressure or mechanical ventilation for at least 2 hours in the first 72 hours of life.” There also were no significant differences in maternal outcomes such as chorioamnionitis or length of hospitalization or neonatal secondary outcomes such as transient tachypnea of the newborn, respiratory distress syndrome, necrotizing enterocolitis, sepsis, hyperbilirubinemia, stillbirth, and early neonatal death.
Serious adverse events occurred in four neonates but none were linked to the intervention.
The study doesn’t discuss cost, but a 2019 report suggests that use of betamethasone to prevent neonatal respiratory distress is cost-effective.
“Our findings are contradictory to those of a systematic review, the major contributor of which was the ALPS trial,” the authors of the new study reported. “The primary outcome of the ALPS trial, the composite of neonatal treatment in the first 72 hours, was significantly less in the group who received betamethasone (11.6%), compared with the placebo group (14.4%; relative risk [RR], 0.80; 95% CI, 0.66-0.97).”
The study authors, who didn’t respond to requests for comment, noted that their trial included twin pregnancies and patients with gestational diabetes; the ALPS trial did not.
Perinatologist Cynthia Gyamfi-Bannerman, MD, MS, chair and professor of Obstetrics, Gynecology, and Reproductive Sciences at the University of California,San Diego, and principal investigator of the ALPS study, said in an interview that the inclusion of twins in the new trial is “a fundamental flaw.”
“Because antenatal corticosteroids have not been shown to be useful in twins at any gestational age, it is not surprising that including twins likely moved the findings to the null in this study,” she said. “Twins were purposefully excluded from the ALPS trial for this reason.”
According to the new study, “the primary outcome among singleton neonates occurred in 4.8% (18/374) who received betamethasone and 5.1% (20/393) who received placebo (RR, 0.94; 95% CI, 0.51-1.75)
What should clinicians take from the study findings? In an accompanying commentary, Blair J. Wylie, MD, MPH, of Columbia University Medical Center, New York, NY, and Syed Asad Ali, MBBS, MPH, of Aga Khan University, Karachi, Pakistan, wrote that, “in settings similar to the US-based ALPS trial, the practice of administering a course of late-preterm antenatal corticosteroids should be continued, as espoused by our professional organizations.”
However, the new study suggests that “research in high-resource environments may not be generalizable to low-resource settings,” they write.
Neonatologist Elizabeth Asztalos, MD, MSc, an associate scientist with Sunnybrook Health Sciences Center in Toronto, Canada, said in an interview that she doesn’t worry about pregnant mothers not getting steroids later than 34 weeks. “We have tools in our armamentarium in the NICU setting to help babies if they need it,” said Dr. Asztalos, who didn’t take part in the new trial. “We can put them on CPAP if they have wet lung. If they have an element of respiratory distress, we can give them surfactants. These bigger babies have more ability to recover from all this compared to a baby who was born at 24, 25, 26 weeks.”
For her part, the University of Toronto’s Dr. Murphy said decision-making about late-preterm steroids is complicated. “You don’t want to miss the opportunity to give to provide benefits for the patients” via steroids, she said. “But on the flip side, it’s a double-edged sword. It’s not easy. It’s not straightforward.”
In the big picture, she said, “people need to be really clear why they’re giving an intervention and what they hope to achieve.”
Christian Medical College supported the study. The authors, Dr. Murphy, Dr. Asztalos, and commentary co-author Dr. Ali have no disclosures. Dr. Gyamfi-Bannerman discloses being principal investigator of the ALPS trial. Commentary co-author Dr. Wylie serves on the ultrasound quality assurance committee of a trial discussed in the commentary.
The early cancellation of a trial in southern India suggests that the use of antenatal steroids to prevent respiratory complications after late-preterm birth — a recommended practice in the United States — may not be effective in the developing world.
As reported in Obstetrics & Gynecology, researchers led by Hilda Yenuberi, MD, of Christian Medical College, Vellore, Tamil Nadu, India, stopped the randomized, triple-blinded, placebo-controlled CLAP (Corticosteroids in Late Pregnancy) study at 70% enrollment. An interim analysis found no benefit from prescribing betamethasone vs placebo to women at risk of late-preterm delivery between 34 and 36 and 6/7 weeks of gestation (primary outcome of respiratory distress: 4.9% vs 4.8%, respectively, relative risk [RR], 1.03; 95% CI, 0.57-1.84; number needed to treat = 786).
“These findings may suggest differing efficacy of antenatal corticosteroids in developing countries compared with developed countries ... that should be considered when late-preterm antenatal corticosteroids are administered,” the researchers wrote.
The use of steroids in patients at risk of delivery before 34 weeks is widely accepted as a way to prevent neonatal respiratory distress, a common and potentially deadly condition in premature infants whose lungs are not fully developed. However, there’s debate over steroid treatment in women who are expected to deliver later than 34 weeks but still preterm.
As the study notes, “the American College of Obstetricians and Gynecologists recommends a single course of betamethasone for pregnant individuals at risk of delivering between 34 and 36 6/7 weeks of gestation on the basis of the ALPS (Antenatal Late Preterm Steroid) trial.”
But other randomized trials have reached different conclusions, and steroids are not without risks. Studies have linked prenatal steroids to neurosensory disorders in babies, meaning they’re more likely to need hearing aids and eyeglasses, said Kellie Murphy, MD, MSc, professor of obstetrics and gynecology, University of Toronto, Toronto, Ontario, Canada, in an interview. Dr. Murphy, who was not involved in the new trial, added that there are links between steroids and greater likelihood of poorer performance in school,
For the new study, conducted from 2020 to 2022 at Christian Medical College and Hospital in Vellore, India, researchers randomly assigned 423 patients to betamethasone (410 in the interim analysis; average age, 26.8 years) and 424 to placebo (415 in the interim analysis; average age, 26.2 years).
The average age of participants was 26.8 years. All were between 34 and 36 6/7 weeks of gestation and expected to give birth within the next week. A quarter of participants delivered at term, which the authors wrote “may have influenced the primary outcome.” The total number of neonates was 883, including 58 twin pregnancies.
There was no significant difference in respiratory distress between groups, “defined as need for oxygen or continuous positive airway pressure or mechanical ventilation for at least 2 hours in the first 72 hours of life.” There also were no significant differences in maternal outcomes such as chorioamnionitis or length of hospitalization or neonatal secondary outcomes such as transient tachypnea of the newborn, respiratory distress syndrome, necrotizing enterocolitis, sepsis, hyperbilirubinemia, stillbirth, and early neonatal death.
Serious adverse events occurred in four neonates but none were linked to the intervention.
The study doesn’t discuss cost, but a 2019 report suggests that use of betamethasone to prevent neonatal respiratory distress is cost-effective.
“Our findings are contradictory to those of a systematic review, the major contributor of which was the ALPS trial,” the authors of the new study reported. “The primary outcome of the ALPS trial, the composite of neonatal treatment in the first 72 hours, was significantly less in the group who received betamethasone (11.6%), compared with the placebo group (14.4%; relative risk [RR], 0.80; 95% CI, 0.66-0.97).”
The study authors, who didn’t respond to requests for comment, noted that their trial included twin pregnancies and patients with gestational diabetes; the ALPS trial did not.
Perinatologist Cynthia Gyamfi-Bannerman, MD, MS, chair and professor of Obstetrics, Gynecology, and Reproductive Sciences at the University of California,San Diego, and principal investigator of the ALPS study, said in an interview that the inclusion of twins in the new trial is “a fundamental flaw.”
“Because antenatal corticosteroids have not been shown to be useful in twins at any gestational age, it is not surprising that including twins likely moved the findings to the null in this study,” she said. “Twins were purposefully excluded from the ALPS trial for this reason.”
According to the new study, “the primary outcome among singleton neonates occurred in 4.8% (18/374) who received betamethasone and 5.1% (20/393) who received placebo (RR, 0.94; 95% CI, 0.51-1.75)
What should clinicians take from the study findings? In an accompanying commentary, Blair J. Wylie, MD, MPH, of Columbia University Medical Center, New York, NY, and Syed Asad Ali, MBBS, MPH, of Aga Khan University, Karachi, Pakistan, wrote that, “in settings similar to the US-based ALPS trial, the practice of administering a course of late-preterm antenatal corticosteroids should be continued, as espoused by our professional organizations.”
However, the new study suggests that “research in high-resource environments may not be generalizable to low-resource settings,” they write.
Neonatologist Elizabeth Asztalos, MD, MSc, an associate scientist with Sunnybrook Health Sciences Center in Toronto, Canada, said in an interview that she doesn’t worry about pregnant mothers not getting steroids later than 34 weeks. “We have tools in our armamentarium in the NICU setting to help babies if they need it,” said Dr. Asztalos, who didn’t take part in the new trial. “We can put them on CPAP if they have wet lung. If they have an element of respiratory distress, we can give them surfactants. These bigger babies have more ability to recover from all this compared to a baby who was born at 24, 25, 26 weeks.”
For her part, the University of Toronto’s Dr. Murphy said decision-making about late-preterm steroids is complicated. “You don’t want to miss the opportunity to give to provide benefits for the patients” via steroids, she said. “But on the flip side, it’s a double-edged sword. It’s not easy. It’s not straightforward.”
In the big picture, she said, “people need to be really clear why they’re giving an intervention and what they hope to achieve.”
Christian Medical College supported the study. The authors, Dr. Murphy, Dr. Asztalos, and commentary co-author Dr. Ali have no disclosures. Dr. Gyamfi-Bannerman discloses being principal investigator of the ALPS trial. Commentary co-author Dr. Wylie serves on the ultrasound quality assurance committee of a trial discussed in the commentary.
The early cancellation of a trial in southern India suggests that the use of antenatal steroids to prevent respiratory complications after late-preterm birth — a recommended practice in the United States — may not be effective in the developing world.
As reported in Obstetrics & Gynecology, researchers led by Hilda Yenuberi, MD, of Christian Medical College, Vellore, Tamil Nadu, India, stopped the randomized, triple-blinded, placebo-controlled CLAP (Corticosteroids in Late Pregnancy) study at 70% enrollment. An interim analysis found no benefit from prescribing betamethasone vs placebo to women at risk of late-preterm delivery between 34 and 36 and 6/7 weeks of gestation (primary outcome of respiratory distress: 4.9% vs 4.8%, respectively, relative risk [RR], 1.03; 95% CI, 0.57-1.84; number needed to treat = 786).
“These findings may suggest differing efficacy of antenatal corticosteroids in developing countries compared with developed countries ... that should be considered when late-preterm antenatal corticosteroids are administered,” the researchers wrote.
The use of steroids in patients at risk of delivery before 34 weeks is widely accepted as a way to prevent neonatal respiratory distress, a common and potentially deadly condition in premature infants whose lungs are not fully developed. However, there’s debate over steroid treatment in women who are expected to deliver later than 34 weeks but still preterm.
As the study notes, “the American College of Obstetricians and Gynecologists recommends a single course of betamethasone for pregnant individuals at risk of delivering between 34 and 36 6/7 weeks of gestation on the basis of the ALPS (Antenatal Late Preterm Steroid) trial.”
But other randomized trials have reached different conclusions, and steroids are not without risks. Studies have linked prenatal steroids to neurosensory disorders in babies, meaning they’re more likely to need hearing aids and eyeglasses, said Kellie Murphy, MD, MSc, professor of obstetrics and gynecology, University of Toronto, Toronto, Ontario, Canada, in an interview. Dr. Murphy, who was not involved in the new trial, added that there are links between steroids and greater likelihood of poorer performance in school,
For the new study, conducted from 2020 to 2022 at Christian Medical College and Hospital in Vellore, India, researchers randomly assigned 423 patients to betamethasone (410 in the interim analysis; average age, 26.8 years) and 424 to placebo (415 in the interim analysis; average age, 26.2 years).
The average age of participants was 26.8 years. All were between 34 and 36 6/7 weeks of gestation and expected to give birth within the next week. A quarter of participants delivered at term, which the authors wrote “may have influenced the primary outcome.” The total number of neonates was 883, including 58 twin pregnancies.
There was no significant difference in respiratory distress between groups, “defined as need for oxygen or continuous positive airway pressure or mechanical ventilation for at least 2 hours in the first 72 hours of life.” There also were no significant differences in maternal outcomes such as chorioamnionitis or length of hospitalization or neonatal secondary outcomes such as transient tachypnea of the newborn, respiratory distress syndrome, necrotizing enterocolitis, sepsis, hyperbilirubinemia, stillbirth, and early neonatal death.
Serious adverse events occurred in four neonates but none were linked to the intervention.
The study doesn’t discuss cost, but a 2019 report suggests that use of betamethasone to prevent neonatal respiratory distress is cost-effective.
“Our findings are contradictory to those of a systematic review, the major contributor of which was the ALPS trial,” the authors of the new study reported. “The primary outcome of the ALPS trial, the composite of neonatal treatment in the first 72 hours, was significantly less in the group who received betamethasone (11.6%), compared with the placebo group (14.4%; relative risk [RR], 0.80; 95% CI, 0.66-0.97).”
The study authors, who didn’t respond to requests for comment, noted that their trial included twin pregnancies and patients with gestational diabetes; the ALPS trial did not.
Perinatologist Cynthia Gyamfi-Bannerman, MD, MS, chair and professor of Obstetrics, Gynecology, and Reproductive Sciences at the University of California,San Diego, and principal investigator of the ALPS study, said in an interview that the inclusion of twins in the new trial is “a fundamental flaw.”
“Because antenatal corticosteroids have not been shown to be useful in twins at any gestational age, it is not surprising that including twins likely moved the findings to the null in this study,” she said. “Twins were purposefully excluded from the ALPS trial for this reason.”
According to the new study, “the primary outcome among singleton neonates occurred in 4.8% (18/374) who received betamethasone and 5.1% (20/393) who received placebo (RR, 0.94; 95% CI, 0.51-1.75)
What should clinicians take from the study findings? In an accompanying commentary, Blair J. Wylie, MD, MPH, of Columbia University Medical Center, New York, NY, and Syed Asad Ali, MBBS, MPH, of Aga Khan University, Karachi, Pakistan, wrote that, “in settings similar to the US-based ALPS trial, the practice of administering a course of late-preterm antenatal corticosteroids should be continued, as espoused by our professional organizations.”
However, the new study suggests that “research in high-resource environments may not be generalizable to low-resource settings,” they write.
Neonatologist Elizabeth Asztalos, MD, MSc, an associate scientist with Sunnybrook Health Sciences Center in Toronto, Canada, said in an interview that she doesn’t worry about pregnant mothers not getting steroids later than 34 weeks. “We have tools in our armamentarium in the NICU setting to help babies if they need it,” said Dr. Asztalos, who didn’t take part in the new trial. “We can put them on CPAP if they have wet lung. If they have an element of respiratory distress, we can give them surfactants. These bigger babies have more ability to recover from all this compared to a baby who was born at 24, 25, 26 weeks.”
For her part, the University of Toronto’s Dr. Murphy said decision-making about late-preterm steroids is complicated. “You don’t want to miss the opportunity to give to provide benefits for the patients” via steroids, she said. “But on the flip side, it’s a double-edged sword. It’s not easy. It’s not straightforward.”
In the big picture, she said, “people need to be really clear why they’re giving an intervention and what they hope to achieve.”
Christian Medical College supported the study. The authors, Dr. Murphy, Dr. Asztalos, and commentary co-author Dr. Ali have no disclosures. Dr. Gyamfi-Bannerman discloses being principal investigator of the ALPS trial. Commentary co-author Dr. Wylie serves on the ultrasound quality assurance committee of a trial discussed in the commentary.
FROM OBSTETRICS & GYNECOLOGY
Consider Skin Cancer, Infection Risks in Solid Organ Transplant Recipients
SAN DIEGO — because of their suppressed immune systems.
“There are over 450,000 people with a solid organ transplant living in the United States. If you do the math, that works out to about 40 organ transplant recipients for every dermatologist, so there’s a lot of them out there for us to take care of,” Sean Christensen, MD, PhD, associate professor of dermatology, Yale University, New Haven, Connecticut, said at the annual meeting of the American Academy of Dermatology (AAD). “If we expand that umbrella to include all types of immunosuppression, that’s over 4 million adults in the US.”
Dr. Christensen encouraged dermatologists to be aware of the varying risks for immunosuppressive drugs and best screening practices for these patients, and to take advantage of a validated skin cancer risk assessment tool for transplant patients.
During his presentation, he highlighted five classes of immunosuppressive drugs and their associated skin cancer risks:
- Calcineurin inhibitors (tacrolimus or cyclosporine), which cause severe immune suppression and pose a severe skin cancer risk. They may also cause gingival hyperplasia and sebaceous hyperplasia.
- Antimetabolites (mycophenolate mofetil or azathioprine), which cause moderate to severe immune suppression and pose a severe skin cancer risk.
- Mammalian target of rapamycin inhibitors (sirolimus or everolimus), which cause severe immune suppression and pose a moderate skin cancer risk. They also impair wound healing.
- Corticosteroids (prednisone), which cause mild to severe immune suppression and pose a minimal skin cancer risk.
- A decoy receptor protein (belatacept), which causes severe immune suppression and poses a mild skin cancer risk.
“Most of our solid-organ transplant recipients will be on both a calcineurin inhibitor and an antimetabolite,” Dr. Christensen said. “In addition to the skin cancer risk associated with immunosuppression, there is an additive risk” that is a direct effect of these medications on the skin. “That means our transplant recipients have a severely and disproportionate increase in skin cancer,” he noted.
Up to half of solid-organ transplant recipients will develop skin cancer, Dr. Christensen said. These patients have a sixfold to 10-fold increased risk for basal cell carcinoma (BCC), a 35- to 65-fold increased risk for squamous cell carcinoma (SCC), a twofold to sevenfold increased risk for melanoma, and a 16- to 100-fold increased risk for Merkel cell carcinoma.
Transplant recipients with SCC, he said, have a twofold to threefold higher risk for metastasis (4%-8% nodal metastasis) and twofold to fivefold higher risk for death (2%-7% mortality) from SCC.
As for other kinds of immunosuppression, HIV positivity, treatment with 6-mercaptopurine or azathioprine (for inflammatory bowel disease and rheumatoid arthritis), and antitumor necrosis factor agents (for psoriasis, inflammatory bowel disease, and rheumatoid arthritis) have been linked in studies to a higher risk for nonmelanoma skin cancer.
Dr. Christensen also highlighted graft-versus-host disease (GVHD). “It does look like there is a disproportionate and increased risk of SCC of the oropharynx and of the skin in patients who have chronic GVHD. This is probably due to a combination of both the immunosuppressive medications that are required but also from chronic and ongoing inflammation in the skin.”
Chronic GVHD has been linked to a 5.3-fold increase in the risk for SCC and a twofold increase in the risk for BCC, he added.
Moreover, new medications for treating GVHD have been linked to an increased risk for SCC, including a 3.2-fold increased risk for SCC associated with ruxolitinib, a Janus kinase (JAK) 1 and JAK2 inhibitor, in a study of patients with polycythemia vera and myelofibrosis; and a case report of SCC in a patient treated with belumosudil, a rho-associated coiled-coil-containing protein kinase-2 kinase inhibitor, for chronic GVHD. Risk for SCC appears to increase based on duration of use with voriconazole, an antifungal, which, he said, is a potent photosynthesizer.
Dr. Christensen also noted the higher risk for infections in immunocompromised patients and added that these patients can develop inflammatory disease despite immunosuppression:
Staphylococcus, Streptococcus, and Dermatophytes are the most common skin pathogens in these patients. There’s a significantly increased risk for reactivation of herpes simplex, varicella-zoster viruses, and cytomegalovirus. Opportunistic and disseminated fungal infections, such as mycobacteria, Candida, histoplasma, cryptococcus, aspergillus, and mucormycosis, can also appear.
More than 80% of transplant recipients develop molluscum and verruca vulgaris/human papillomavirus infection. They may also develop noninfectious inflammatory dermatoses.
Risk Calculator
What can dermatologists do to help transplant patients? Dr. Christensen highlighted the Skin and UV Neoplasia Transplant Risk Assessment Calculator, which predicts skin cancer risk based on points given for race, gender, skin cancer history, age at transplant, and site of transplant.
The tool, validated in a 2023 study of transplant recipients in Europe, is available online and as an app. It makes recommendations to users about when patients should have initial skin screening exams. Those with the most risk — 45% at 5 years — should be screened within 6 months. “We can use [the tool] to triage these cases when we first meet them and get them plugged into the appropriate care,” Dr. Christensen said.
He recommended seeing high-risk patients at least annually. Patients with a prior SCC and a heavy burden of actinic keratosis should be followed more frequently, he said.
In regard to SCC, he highlighted a 2024 study of solid organ transplant recipients that found the risk for a second SCC after a first SCC was 74%, the risk for a third SCC after a second SCC was 83%, and the risk for another SCC after five SCCs was 92%.
Dr. Christensen disclosed relationships with Canfield Scientific Inc. (consulting), Inhibitor Therapeutics (advisory board), and Sol-Gel Technologies Ltd. (grants/research funding).
A version of this article first appeared on Medscape.com.
SAN DIEGO — because of their suppressed immune systems.
“There are over 450,000 people with a solid organ transplant living in the United States. If you do the math, that works out to about 40 organ transplant recipients for every dermatologist, so there’s a lot of them out there for us to take care of,” Sean Christensen, MD, PhD, associate professor of dermatology, Yale University, New Haven, Connecticut, said at the annual meeting of the American Academy of Dermatology (AAD). “If we expand that umbrella to include all types of immunosuppression, that’s over 4 million adults in the US.”
Dr. Christensen encouraged dermatologists to be aware of the varying risks for immunosuppressive drugs and best screening practices for these patients, and to take advantage of a validated skin cancer risk assessment tool for transplant patients.
During his presentation, he highlighted five classes of immunosuppressive drugs and their associated skin cancer risks:
- Calcineurin inhibitors (tacrolimus or cyclosporine), which cause severe immune suppression and pose a severe skin cancer risk. They may also cause gingival hyperplasia and sebaceous hyperplasia.
- Antimetabolites (mycophenolate mofetil or azathioprine), which cause moderate to severe immune suppression and pose a severe skin cancer risk.
- Mammalian target of rapamycin inhibitors (sirolimus or everolimus), which cause severe immune suppression and pose a moderate skin cancer risk. They also impair wound healing.
- Corticosteroids (prednisone), which cause mild to severe immune suppression and pose a minimal skin cancer risk.
- A decoy receptor protein (belatacept), which causes severe immune suppression and poses a mild skin cancer risk.
“Most of our solid-organ transplant recipients will be on both a calcineurin inhibitor and an antimetabolite,” Dr. Christensen said. “In addition to the skin cancer risk associated with immunosuppression, there is an additive risk” that is a direct effect of these medications on the skin. “That means our transplant recipients have a severely and disproportionate increase in skin cancer,” he noted.
Up to half of solid-organ transplant recipients will develop skin cancer, Dr. Christensen said. These patients have a sixfold to 10-fold increased risk for basal cell carcinoma (BCC), a 35- to 65-fold increased risk for squamous cell carcinoma (SCC), a twofold to sevenfold increased risk for melanoma, and a 16- to 100-fold increased risk for Merkel cell carcinoma.
Transplant recipients with SCC, he said, have a twofold to threefold higher risk for metastasis (4%-8% nodal metastasis) and twofold to fivefold higher risk for death (2%-7% mortality) from SCC.
As for other kinds of immunosuppression, HIV positivity, treatment with 6-mercaptopurine or azathioprine (for inflammatory bowel disease and rheumatoid arthritis), and antitumor necrosis factor agents (for psoriasis, inflammatory bowel disease, and rheumatoid arthritis) have been linked in studies to a higher risk for nonmelanoma skin cancer.
Dr. Christensen also highlighted graft-versus-host disease (GVHD). “It does look like there is a disproportionate and increased risk of SCC of the oropharynx and of the skin in patients who have chronic GVHD. This is probably due to a combination of both the immunosuppressive medications that are required but also from chronic and ongoing inflammation in the skin.”
Chronic GVHD has been linked to a 5.3-fold increase in the risk for SCC and a twofold increase in the risk for BCC, he added.
Moreover, new medications for treating GVHD have been linked to an increased risk for SCC, including a 3.2-fold increased risk for SCC associated with ruxolitinib, a Janus kinase (JAK) 1 and JAK2 inhibitor, in a study of patients with polycythemia vera and myelofibrosis; and a case report of SCC in a patient treated with belumosudil, a rho-associated coiled-coil-containing protein kinase-2 kinase inhibitor, for chronic GVHD. Risk for SCC appears to increase based on duration of use with voriconazole, an antifungal, which, he said, is a potent photosynthesizer.
Dr. Christensen also noted the higher risk for infections in immunocompromised patients and added that these patients can develop inflammatory disease despite immunosuppression:
Staphylococcus, Streptococcus, and Dermatophytes are the most common skin pathogens in these patients. There’s a significantly increased risk for reactivation of herpes simplex, varicella-zoster viruses, and cytomegalovirus. Opportunistic and disseminated fungal infections, such as mycobacteria, Candida, histoplasma, cryptococcus, aspergillus, and mucormycosis, can also appear.
More than 80% of transplant recipients develop molluscum and verruca vulgaris/human papillomavirus infection. They may also develop noninfectious inflammatory dermatoses.
Risk Calculator
What can dermatologists do to help transplant patients? Dr. Christensen highlighted the Skin and UV Neoplasia Transplant Risk Assessment Calculator, which predicts skin cancer risk based on points given for race, gender, skin cancer history, age at transplant, and site of transplant.
The tool, validated in a 2023 study of transplant recipients in Europe, is available online and as an app. It makes recommendations to users about when patients should have initial skin screening exams. Those with the most risk — 45% at 5 years — should be screened within 6 months. “We can use [the tool] to triage these cases when we first meet them and get them plugged into the appropriate care,” Dr. Christensen said.
He recommended seeing high-risk patients at least annually. Patients with a prior SCC and a heavy burden of actinic keratosis should be followed more frequently, he said.
In regard to SCC, he highlighted a 2024 study of solid organ transplant recipients that found the risk for a second SCC after a first SCC was 74%, the risk for a third SCC after a second SCC was 83%, and the risk for another SCC after five SCCs was 92%.
Dr. Christensen disclosed relationships with Canfield Scientific Inc. (consulting), Inhibitor Therapeutics (advisory board), and Sol-Gel Technologies Ltd. (grants/research funding).
A version of this article first appeared on Medscape.com.
SAN DIEGO — because of their suppressed immune systems.
“There are over 450,000 people with a solid organ transplant living in the United States. If you do the math, that works out to about 40 organ transplant recipients for every dermatologist, so there’s a lot of them out there for us to take care of,” Sean Christensen, MD, PhD, associate professor of dermatology, Yale University, New Haven, Connecticut, said at the annual meeting of the American Academy of Dermatology (AAD). “If we expand that umbrella to include all types of immunosuppression, that’s over 4 million adults in the US.”
Dr. Christensen encouraged dermatologists to be aware of the varying risks for immunosuppressive drugs and best screening practices for these patients, and to take advantage of a validated skin cancer risk assessment tool for transplant patients.
During his presentation, he highlighted five classes of immunosuppressive drugs and their associated skin cancer risks:
- Calcineurin inhibitors (tacrolimus or cyclosporine), which cause severe immune suppression and pose a severe skin cancer risk. They may also cause gingival hyperplasia and sebaceous hyperplasia.
- Antimetabolites (mycophenolate mofetil or azathioprine), which cause moderate to severe immune suppression and pose a severe skin cancer risk.
- Mammalian target of rapamycin inhibitors (sirolimus or everolimus), which cause severe immune suppression and pose a moderate skin cancer risk. They also impair wound healing.
- Corticosteroids (prednisone), which cause mild to severe immune suppression and pose a minimal skin cancer risk.
- A decoy receptor protein (belatacept), which causes severe immune suppression and poses a mild skin cancer risk.
“Most of our solid-organ transplant recipients will be on both a calcineurin inhibitor and an antimetabolite,” Dr. Christensen said. “In addition to the skin cancer risk associated with immunosuppression, there is an additive risk” that is a direct effect of these medications on the skin. “That means our transplant recipients have a severely and disproportionate increase in skin cancer,” he noted.
Up to half of solid-organ transplant recipients will develop skin cancer, Dr. Christensen said. These patients have a sixfold to 10-fold increased risk for basal cell carcinoma (BCC), a 35- to 65-fold increased risk for squamous cell carcinoma (SCC), a twofold to sevenfold increased risk for melanoma, and a 16- to 100-fold increased risk for Merkel cell carcinoma.
Transplant recipients with SCC, he said, have a twofold to threefold higher risk for metastasis (4%-8% nodal metastasis) and twofold to fivefold higher risk for death (2%-7% mortality) from SCC.
As for other kinds of immunosuppression, HIV positivity, treatment with 6-mercaptopurine or azathioprine (for inflammatory bowel disease and rheumatoid arthritis), and antitumor necrosis factor agents (for psoriasis, inflammatory bowel disease, and rheumatoid arthritis) have been linked in studies to a higher risk for nonmelanoma skin cancer.
Dr. Christensen also highlighted graft-versus-host disease (GVHD). “It does look like there is a disproportionate and increased risk of SCC of the oropharynx and of the skin in patients who have chronic GVHD. This is probably due to a combination of both the immunosuppressive medications that are required but also from chronic and ongoing inflammation in the skin.”
Chronic GVHD has been linked to a 5.3-fold increase in the risk for SCC and a twofold increase in the risk for BCC, he added.
Moreover, new medications for treating GVHD have been linked to an increased risk for SCC, including a 3.2-fold increased risk for SCC associated with ruxolitinib, a Janus kinase (JAK) 1 and JAK2 inhibitor, in a study of patients with polycythemia vera and myelofibrosis; and a case report of SCC in a patient treated with belumosudil, a rho-associated coiled-coil-containing protein kinase-2 kinase inhibitor, for chronic GVHD. Risk for SCC appears to increase based on duration of use with voriconazole, an antifungal, which, he said, is a potent photosynthesizer.
Dr. Christensen also noted the higher risk for infections in immunocompromised patients and added that these patients can develop inflammatory disease despite immunosuppression:
Staphylococcus, Streptococcus, and Dermatophytes are the most common skin pathogens in these patients. There’s a significantly increased risk for reactivation of herpes simplex, varicella-zoster viruses, and cytomegalovirus. Opportunistic and disseminated fungal infections, such as mycobacteria, Candida, histoplasma, cryptococcus, aspergillus, and mucormycosis, can also appear.
More than 80% of transplant recipients develop molluscum and verruca vulgaris/human papillomavirus infection. They may also develop noninfectious inflammatory dermatoses.
Risk Calculator
What can dermatologists do to help transplant patients? Dr. Christensen highlighted the Skin and UV Neoplasia Transplant Risk Assessment Calculator, which predicts skin cancer risk based on points given for race, gender, skin cancer history, age at transplant, and site of transplant.
The tool, validated in a 2023 study of transplant recipients in Europe, is available online and as an app. It makes recommendations to users about when patients should have initial skin screening exams. Those with the most risk — 45% at 5 years — should be screened within 6 months. “We can use [the tool] to triage these cases when we first meet them and get them plugged into the appropriate care,” Dr. Christensen said.
He recommended seeing high-risk patients at least annually. Patients with a prior SCC and a heavy burden of actinic keratosis should be followed more frequently, he said.
In regard to SCC, he highlighted a 2024 study of solid organ transplant recipients that found the risk for a second SCC after a first SCC was 74%, the risk for a third SCC after a second SCC was 83%, and the risk for another SCC after five SCCs was 92%.
Dr. Christensen disclosed relationships with Canfield Scientific Inc. (consulting), Inhibitor Therapeutics (advisory board), and Sol-Gel Technologies Ltd. (grants/research funding).
A version of this article first appeared on Medscape.com.
FROM AAD 2024