User login
Biologic Therapy in Psoriasis: Navigating the Options
Psoriasis is a T cell–mediated inflammatory disease that manifests as erythematous scaling plaques of the skin. In recent decades, our understanding of psoriasis has transformed from a disease isolated to the skin to a systemic disease impacting the overall health of those affected.
With recent elucidation of the pathways driving psoriasis, development of targeted therapies has resulted in an influx of options to the market. Navigating the options can seem overwhelming even to the seasoned clinician. Becoming familiar with a sound treatment approach during residency will create a foundation for biologic use in psoriasis patients throughout your career. Here we offer an approach to choosing biologic treatments based on individual patient characteristics, including disease severity, comorbidities, and ultimate treatment goals.
Immune Pathogenesis
Although the pathogenesis of psoriasis is complex and outside the scope of this article, we do recommend clinicians keep in mind the current understanding of pathways involved and ways our therapies alter them. Briefly, psoriasis is a T cell–mediated disease in which IL-12 and IL-23 released by activated dendritic cells activate T helper cells including TH1, TH17, and TH22. These cells produce additional cytokines, including IFN-γ, tumor necrosis factor (TNF) α, IL-17, and IL-22, which propagate the immune response and lead to keratinocyte hyperproliferation. In general, psoriasis medications work by altering T-cell activation, effector cytokines, or cytokine receptors.
Comorbidities
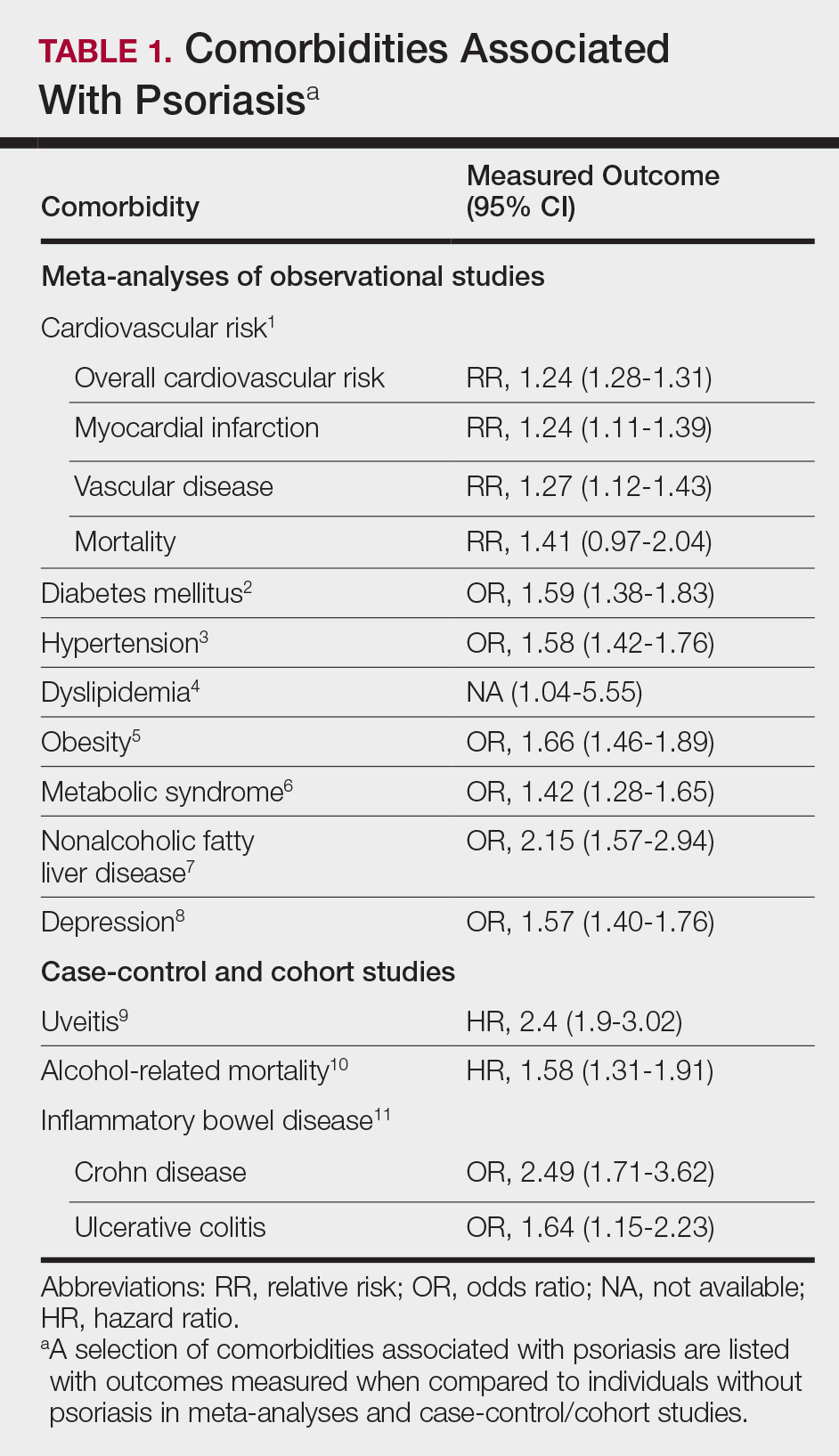
A targeted approach should take into consideration the immune dysregulation shared by psoriasis and associated comorbidities (Table 1). One goal of biologic treatments is to improve comorbidities when possible. At minimum, selected treatments should not exacerbate these conditions.
Treatment Goals
Establishing treatment goals can help shape patient expectations and provide a plan for clinicians. In 2017, the National Psoriasis Foundation published a treat-to-target approach using body surface area (BSA) measurements at baseline, 3 months, and then every 6 months after starting a new treatment.12 The target response is a decrease in psoriasis to 1% or less BSA at 3 months and to maintain this response when evaluated at 6-month intervals. Alternatively, a target of 3% BSA after 3 months is satisfactory if the patient improves by 75% BSA overall. If these targets are not met after 6 months, therapeutic alternatives can be considered.12
Biologic Treatment of Psoriasis
Treatment options for patients with psoriasis depend first on disease severity. Topicals and phototherapy are first line for mild to moderate disease. For moderate to severe disease, addition of systemic agents such as methotrexate, cyclosporine, or acitretin; small-molecular-weight immunomodulators such as apremilast; or biologic medications should be considered. Current biologics available for moderate to severe plaque psoriasis target TNF-α, IL-12/IL-23, IL-23, IL-17A, or IL-17A receptor.
TNF-α Inhibitors
Tumor necrosis factor α inhibitors have been available for treatment of autoimmune disease for nearly 20 years. These medications block either soluble cytokine or membrane-bound cytokine. All are given as subcutaneous injections, except for infliximab, which is a weight-based infusion.
Efficacy
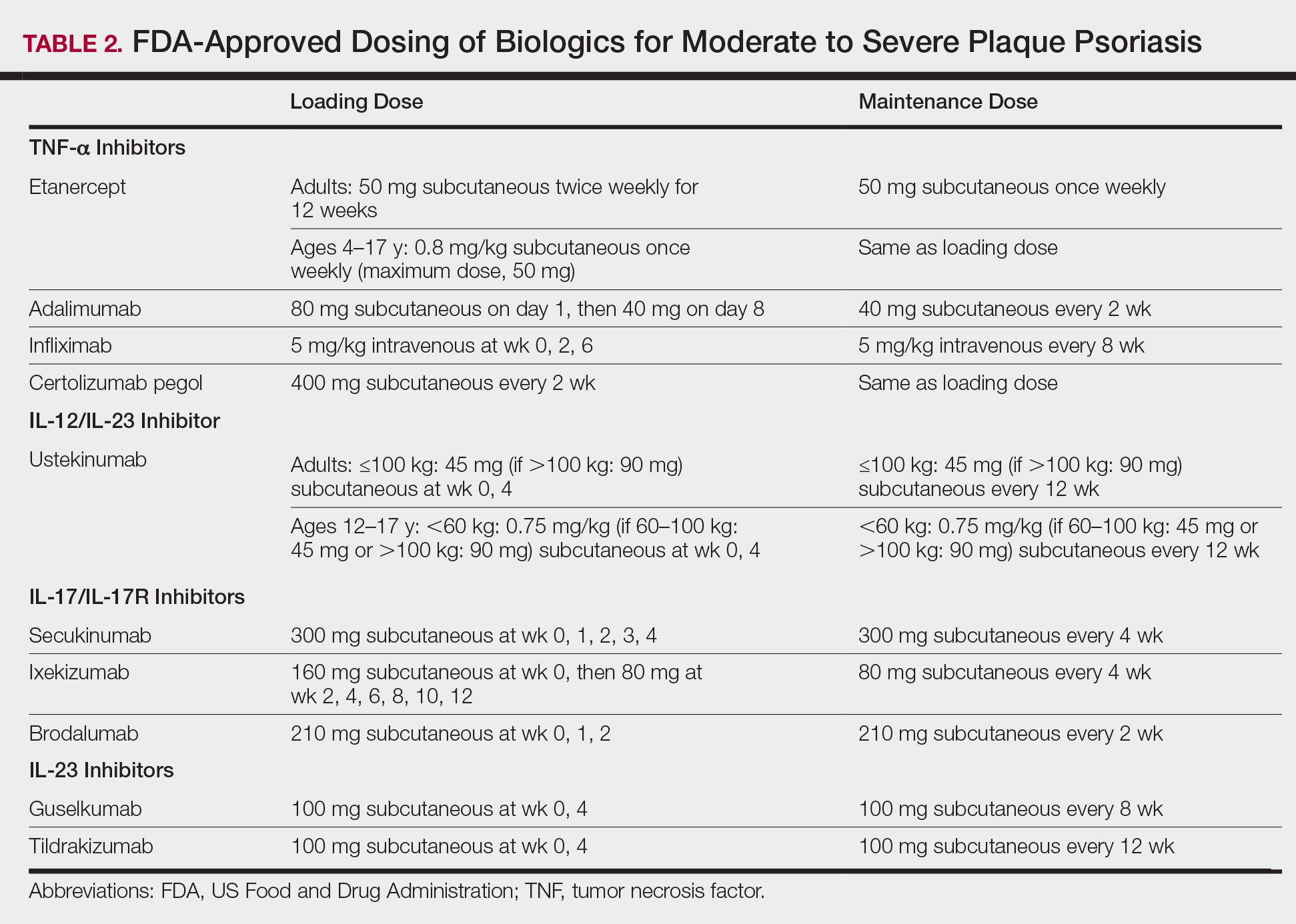
Tumor necrosis factor α inhibitors are the first class to demonstrate long-term efficacy and safety in both psoriasis and psoriatic arthritis (PsA). Etanercept was approved for adults with PsA in 2002 and psoriasis in 2004, and later for pediatric psoriasis (≥4 years of age) in 2016 (Table 2). Although etanercept has a sustained safety profile, the response rates are not as high as other anti–TNF-α inhibitors. Adalimumab is one of the most prescribed biologics, with a total of 10 indications at present, including PsA. Infliximab is an intravenous infusion that demonstrates a rapid and sustained response in most patients. The dose and dosing interval can be adjusted according to response. Certolizumab pegol was approved for PsA in 2013 and for psoriasis in 2018.
Tumor necrosis factor α inhibitors maintain efficacy well and work best when dosed continuously. Both neutralizing and nonneutralizing antibodies form with these agents. Neutralizing antibodies may contribute to decreased efficacy, particularly for the chimeric antibody infliximab. One approach to mitigate loss of efficacy is the short-term addition of low-dose methotrexate (eg, 7.5–15 mg weekly) for 3 to 6 months until response is recaptured.
Safety
To evaluate long-term safety, a multicenter prospective registry study (Psoriasis Longitudinal Assessment and Registry [PSOLAR]) was initiated in 2007 to follow clinical outcomes. Data through 2013 showed no significant increase in rates of infection, malignancy, or major adverse cardiovascular events in more than 12,000 patients.13
Conflicting information exists in the literature regarding risk for malignancy with TNF-α inhibitors. One recent retrospective cohort study suggested a slightly increased risk for malignancies other than nonmelanoma skin cancers in patients on TNF-α inhibitors for more than 12 months (relative risk, 1.54).14 Reports of increased risk for cutaneous squamous cell carcinomas necessitate regular skin checks.15 A potential risk for lymphoma has been noted, though having psoriasis itself imparts an increased risk for Hodgkin and cutaneous T-cell lymphoma.16
Reactivation of tuberculosis and hepatitis have been reported with TNF-α inhibition. Data suggest that infliximab may be associated with more serious infections.13
Demyelinating conditions such as multiple sclerosis have occurred de novo or worsened in patients on TNF-α inhibitors.17 Tumor necrosis factor α blockers should be avoided in patients with decompensated heart failure. Rare cases of liver enzyme elevation and cytopenia have been noted. Additionally, lupuslike syndromes, which are generally reversible upon discontinuation, have occurred in some patients.
Patient Selection
Tumor necrosis factor α inhibitors are the treatment of choice for patients with comorbid PsA. This class halts progression of joint destruction over time.18Select TNF-α inhibitors are indicated for inflammatory bowel disease (IBD) and are a preferred treatment in this patient population. Specifically, adalimumab and infliximab are approved for both Crohn disease (CD) and ulcerative colitis. Certolizumab pegol is approved for CD.
Tumor necrosis factor α is upregulated in obesity, cardiovascular disease, and atherosclerotic plaques. Evidence suggests that TNF-α blockers may lower cardiovascular risk over time.19 For patients with obesity, infliximab is a good option, as it is the only TNF-α inhibitor with weight-based dosing.
In patients with frequent infections or history of hepatitis C, etanercept has been the biologic most commonly used when no alternatives exist, in part due to its shorter half-life.
IL-12/IL-23 Inhibitor
Ustekinumab is a monoclonal antibody that binds the p40 subunit shared by IL-12 and IL-23, blocking their ability to bind receptors. IL-12 and IL-23 play a role in activating naïve T cells to become TH1 or TH17 cells, respectively.
Efficacy and Safety
Clinical trials demonstrate long-term efficacy of ustekinumab, which was approved for psoriasis in 2009, PsA in 2013, and later pediatric psoriasis (≥12 years of age) in 2017. Dosing is listed in Table 2.
Laboratory abnormalities did not arise in trials. Periodic tuberculosis screening is required. Prospective data over 5 years showed very low rates of adverse events (AEs), serious infections, malignancies, and major adverse cardiovascular events.20 Ustekinumab did not worsen or improve demyelinating disease and appears safe in this population.
Patient Selection
Ustekinumab is approved for PsA and is a good option for those who are not candidates for TNF-α and IL-17 inhibitors. Ustekinumab also is approved for CD. The dosing interval of 12 weeks makes ustekinumab convenient for patients. Two dosages exist based on the patient’s weight, offering an advantage to obese patients.
IL-17/IL-17R Inhibitors
Activated TH17 cells produce the IL-17 cytokine family, which stimulates keratinocyte proliferation and dermal inflammation. Secukinumab is a fully human monoclonal antibody, and ixekizumab is a humanized monoclonal antibody; both target IL-17A. Brodalumab targets the IL-17A receptor.
Efficacy and Safety
IL-17 inhibitors showed impressive and rapid responses in trials.21-23 The subsets of patients who responded well and continued treatment in extension trials demonstrated that these treatments maintain efficacy over time.24-26
In addition to tuberculosis reactivation, there is a small increased risk for cutaneous candidiasis with IL-17 inhibitors, which can be managed without stopping treatment. Laboratory abnormalities were limited to mild neutropenia, which was not associated with increased risk for infection.21-23 With ixekizumab, neutropenia was seen more commonly in the first 12 weeks.22
IL-17 is highly expressed in the gut mucosa, and its inhibition is thought to weaken the barrier function of the gut mucosa, promoting inflammation. As a consequence, this class is contraindicated in patients with IBD due to exacerbations of existing IBD and cases of new-onset IBD.21-23 Symptoms of diarrhea, abdominal pain, blood in stool, or nighttime stooling on review of gastrointestinal tract symptoms should prompt further evaluation.
Brodalumab has a unique warning for risk for suicidal ideation and behavior.23 Depression is more common in the psoriasis population in general; therefore, physicians should be aware of this potential comorbidity regardless of the treatment plan. Because the response rates are so impressive with brodalumab, the Risk Evaluation and Mitigation Strategy (REMS) program was established to ensure understanding of this risk so that patients can be appropriately counseled and managed.
Patient Selection
The improvement in psoriasis is rapid and may occur as early as week 2 to 3 of treatment after initiation of IL-17 inhibitors. Ixekizumab and secukinumab also are approved for PsA. Although improvement in joint disease is not as fast as with the anti-TNF inhibitors, notable improvement occurs by week 20 to 24.27
IL-23 Inhibitors
Guselkumab and tildrakizumab are the newest biologics for psoriasis, approved in 2017 and 2018, respectively. Both are monoclonal antibodies against the p19 subunit of IL-23, which blocks activation of TH17 cells.
Efficacy and Safety
Guselkumab and tildrakizumab demonstrated efficacy with minimal AEs or precautions noted thus far.28,29 Infections are again a risk, making tuberculosis testing the only recommended monitoring.
Patient Selection
Both medications offer another effective and safe option for patients with psoriasis. Similar to ustekinumab, the dosing interval of 12 weeks for tildrakizumab is ideal for patients who have needle phobia or are unable to administer their own injections.
Special Populations
Pregnancy
Antibodies cross the placenta as pregnancy progresses, with the highest rate in the third trimester. Certolizumab pegol has shown the lowest concentrations in infant serum, possibly due to its unique structure lacking the fragment crystallizable region required for passage through the placenta.30 For this reason, certolizumab pegol is a treatment of choice if biologic therapy is warranted during pregnancy.
Much of the pregnancy data for the remaining TNF-α inhibitors come from patients with rheumatoid arthritis or CD. In these populations, rates of major birth defects and miscarriages do not differ greatly from untreated women with these conditions.31 One retrospective study of unintentional pregnancies in women receiving ustekinumab showed rates of AEs similar to the general population.32
Pregnancy data for IL-17 or IL-23 inhibitors are largely limited to animal studies. One retrospective study of women exposed to secukinumab early in gestation showed no increased risk for pregnancy-related AEs.33 Discontinuation is still recommended for patients who become pregnant.
Pediatric Patients
Etanercept is approved for pediatric psoriatic patients 4 years and older. Children with juvenile idiopathic arthritis who are 2 years and older can receive etanercept. Ustekinumab is safe and effective for pediatric psoriatic patients 12 years and older, offering a second biologic option in children.
Although not approved for pediatric psoriasis, adalimumab is approved in pediatric CD (≥6 years of age) and for juvenile idiopathic arthritis (≥2 years of age). Infliximab is approved for children 6 years and older with CD or ulcerative colitis.
Monitoring
Periodic tuberculosis screening is recommended for all biologics. For patients with latent tuberculosis, biologics may be restarted after 1 month of treatment of tuberculosis.
Prior to initiation of biologics, patients should be screened for hepatitis with hepatitis B surface antigen and antibody, hepatitis B core antibody, and hepatitis C antibody. Patients at risk for human immunodeficiency virus also should be screened.
Generally, complete blood cell count and comprehensive metabolic profile are advisable prior to starting a biologic. Opinions differ on frequency of repeating laboratory work. Complete blood cell count and comprehensive metabolic profile should be monitored at least every 3 to 6 months in patients on TNF-α inhibitors, and neutrophil count should be monitored during the induction phase of IL-17 inhibitors.
All patients with psoriasis should maintain age-appropriate cancer screenings, especially those on biologics. If malignancy is discovered, biologic medication should be discontinued. Debate exists as to when therapy can be safely restarted following treatment of malignancy. Patients who are considered at low risk for recurrence may opt to restart a biologic after 5 years, or sooner if symptoms warrant.34 This decision should involve the patient’s cancer specialist.
Conclusion
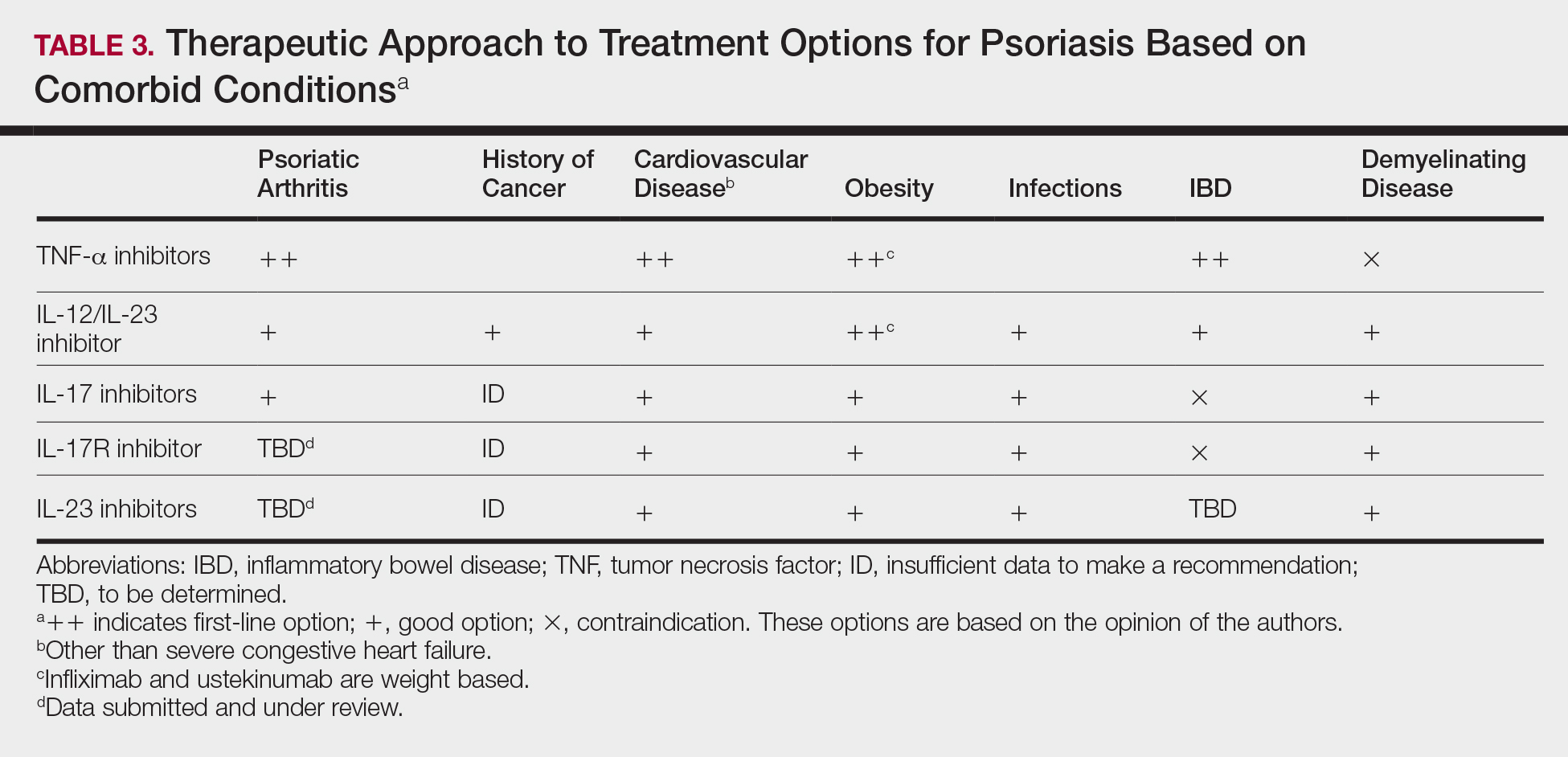
Treatment choices are based on psoriasis type and severity, comorbidities, patient preferences, and drug accessibility. One approach is detailed in Table 3. As research advances the understanding of psoriasis, this field will continue to rapidly change. Knowledge of the immunopathogenesis of psoriasis and its relation to comorbidities can direct your decision-making for individual patients.
- Armstrong AW, Harskamp CT, Armstrong EJ. The association between psoriasis and obesity: a systematic review and meta-analysis of observational studies. Nutr Diabetes. 2012;2:e54.
- Armstrong AW, Harskamp CT, Armstrong EJ. Psoriasis and the risk of diabetes mellitus: a systematic review and meta-analysis. JAMA Dermatol. 2013;149:84-91.
- Armstrong AW, Harskamp CT, Armstrong EJ. The association between psoriasis and hypertension: a systematic review and meta-analysis of observational studies. J Hypertens. 2013;31:433-442; discussion 442-433.
- Candia R, Ruiz A, Torres-Robles R, et al. Risk of non-alcoholic fatty liver disease in patients with psoriasis: a systematic review and meta-analysis. J Eur Acad Dermatol Venereol. 2015;29:656-662.
- Chi CC, Tung TH, Wang J, et al. Risk of uveitis among people with psoriasis: a nationwide cohort study. JAMA Ophthalmol. 2017;135:415-422.
- Cohen AD, Dreiher J, Birkenfeld S. Psoriasis associated with ulcerative colitis and Crohn’s disease. J Eur Acad Dermatol Venereol. 2009;23:561-565.
- Dowlatshahi EA, Wakkee M, Arends LR, et al. The prevalence and odds of depressive symptoms and clinical depression in psoriasis patients: a systematic review and meta-analysis. J Invest Dermatol. 2014;134:1542-1551.
- Gaeta M, Castelvecchio S, Ricci C, et al. Role of psoriasis as independent predictor of cardiovascular disease: a meta-regression analysis. Int J Cardiol. 2013;168:2282-2288.
- Ma C, Harskamp CT, Armstrong EJ, et al. The association between psoriasis and dyslipidaemia: a systematic review. Br J Dermatol. 2013;168:486-495.
- Parisi R, Webb RT, Carr MJ, et al. Alcohol-related mortality in patients with psoriasis: a population-based cohort study. JAMA Dermatol. 2017;153:1256-1262.
- Rodríguez-Zúñiga MJM, García-Perdomo HA. Systematic review and meta-analysis of the association between psoriasis and metabolic syndrome. J Am Acad Dermatol. 2017;77:657-666.e8.
- Armstrong AW, Siegel MP, Bagel J, et al. From the Medical Board of the National Psoriasis Foundation: treatment targets for plaque psoriasis. J Am Acad Dermatol. 2017;76:290-298.
- Gottlieb AB, Kalb RE, Langley RG, et al. Safety observations in 12095 patients with psoriasis enrolled in an international registry (PSOLAR): experience with infliximab and other systemic and biologic therapies. J Drugs Dermatol. 2014;13:1441-1448.
- Fiorentino D, Ho V, Lebwohl MG, et al. Risk of malignancy with systemic psoriasis treatment in the Psoriasis Longitudinal Assessment Registry. J Am Acad Dermatol. 2017;77:845-854.e5.
- van Lümig PP, Menting SP, van den Reek JM, et al. An increased risk of non-melanoma skin cancer during TNF-inhibitor treatment in psoriasis patients compared to rheumatoid arthritis patients probably relates to disease-related factors. J Eur Acad Dermatol Venereol. 2015;29:752-760.
- Gelfand JM, Berlin J, Van Voorhees A, et al. Lymphoma rates are low but increased in patients with psoriasis: results from a population-based cohort study in the United Kingdom. Arch Dermatol. 2003;139:1425-1429.
- Sicotte NL, Voskuhl RR. Onset of multiple sclerosis associated with anti-TNF therapy. Neurology. 2001;57:1885-1888.
- Finckh A, Simard JF, Duryea J, et al. The effectiveness of anti-tumor necrosis factor therapy in preventing progressive radiographic joint damage in rheumatoid arthritis: a population-based study. Arthritis Rheum. 2006;54:54-59.
- Wu JJ, Sundaram M, Cloutier M, et al. The risk of cardiovascular events in psoriasis patients treated with tumor necrosis factor-α inhibitors versus phototherapy: an observational cohort study. J Am Acad Dermatol. 2018;79:60-68.
- Kimball AB, Papp KA, Wasfi Y, et al. Long-term efficacy of ustekinumab in patients with moderate-to-severe psoriasis treated for up to 5 years in the PHOENIX 1 study. J Eur Acad Dermatol Venereol. 2013;27:1535-1545.
- Langley RG, Elewski BE, Lebwohl M, et al. Secukinumab in plaque psoriasis—results of two phase 3 trials. N Engl J Med. 2014;371:326-338.
- Griffiths CE, Reich K, Lebwohl M, et al. Comparison of ixekizumab with etanercept or placebo in moderate-to-severe psoriasis (UNCOVER-2 and UNCOVER-3): results from two phase 3 randomised trials. Lancet. 2015;386:541-551.
- Lebwohl M, Strober B, Menter A, et al. Phase 3 studies comparing brodalumab with ustekinumab in psoriasis. N Engl J Med. 2015;373:1318-1328.
- Bissonnette R, Luger T, Thaçi D, et al. Secukinumab demonstrates high sustained efficacy and a favourable safety profile in patients with moderate-to-severe psoriasis through 5 years of treatment (SCULPTURE Extension Study). J Eur Acad Dermatol Venereol. 2018;32:1507-1514.
- Leonardi C, Maari C, Philipp S, et al. Maintenance of skin clearance with ixekizumab treatment of psoriasis: three-year results from the UNCOVER-3 study. J Am Acad Dermatol. 2018;79:824-830.
- Papp K, Leonardi C, Menter A, et al. Safety and efficacy of brodalumab for psoriasis after 120 weeks of treatment. J Am Acad Dermatol. 2014;71:1183-1190.e1183.
- Gottlieb AB, Strand V, Kishimoto M, et al. Ixekizumab improves patient-reported outcomes up to 52 weeks in bDMARD-naïve patients with active psoriatic arthritis (SPIRIT-P1). Rheumatology (Oxford). 2018;57:1777-1788.
- Blauvelt A, Papp KA, Griffiths CE, et al. Efficacy and safety of guselkumab, an anti-interleukin-23 monoclonal antibody, compared with adalimumab for the continuous treatment of patients with moderate to severe psoriasis: results from the phase III, double-blinded, placebo- and active comparator-controlled VOYAGE 1 trial. J Am Acad Dermatol. 2017;76:405-417.
- Reich K, Papp KA, Blauvelt A, et al. Tildrakizumab versus placebo or etanercept for chronic plaque psoriasis (reSURFACE 1 and reSURFACE 2): results from two randomised controlled, phase 3 trials. Lancet. 2017;390:276-288.
- Mariette X, Förger F, Abraham B, et al. Lack of placental transfer of certolizumab pegol during pregnancy: results from CRIB, a prospective, postmarketing, pharmacokinetic study. Ann Rheum Dis. 2018;77:228-233.
- Komaki F, Komaki Y, Micic D, et al. Outcome of pregnancy and neonatal complications with anti-tumor necrosis factor-α use in females with immune mediated diseases; a systematic review and meta-analysis. J Autoimmun. 2017;76:38-52.
- Götestam Skorpen C, Hoeltzenbein M, Tincani A, et al. The EULAR points to consider for use of antirheumatic drugs before pregnancy, and during pregnancy and lactation. Ann Rheum Dis. 2016;75:795-810.
- Warren RB, Reich K, Langley RG, et al. Secukinumab in pregnancy: outcomes in psoriasis, psoriatic arthritis and ankylosing spondylitis from the global safety database [published online ahead of print June 21, 2018]. Br J Dermatol. doi:10.1111/bjd.16901.
- Elandt K, Aletaha D. Treating rheumatic patients with a malignancy. Arthritis Res Ther. 2011;13:223.
Psoriasis is a T cell–mediated inflammatory disease that manifests as erythematous scaling plaques of the skin. In recent decades, our understanding of psoriasis has transformed from a disease isolated to the skin to a systemic disease impacting the overall health of those affected.
With recent elucidation of the pathways driving psoriasis, development of targeted therapies has resulted in an influx of options to the market. Navigating the options can seem overwhelming even to the seasoned clinician. Becoming familiar with a sound treatment approach during residency will create a foundation for biologic use in psoriasis patients throughout your career. Here we offer an approach to choosing biologic treatments based on individual patient characteristics, including disease severity, comorbidities, and ultimate treatment goals.
Immune Pathogenesis
Although the pathogenesis of psoriasis is complex and outside the scope of this article, we do recommend clinicians keep in mind the current understanding of pathways involved and ways our therapies alter them. Briefly, psoriasis is a T cell–mediated disease in which IL-12 and IL-23 released by activated dendritic cells activate T helper cells including TH1, TH17, and TH22. These cells produce additional cytokines, including IFN-γ, tumor necrosis factor (TNF) α, IL-17, and IL-22, which propagate the immune response and lead to keratinocyte hyperproliferation. In general, psoriasis medications work by altering T-cell activation, effector cytokines, or cytokine receptors.
Comorbidities
A targeted approach should take into consideration the immune dysregulation shared by psoriasis and associated comorbidities (Table 1). One goal of biologic treatments is to improve comorbidities when possible. At minimum, selected treatments should not exacerbate these conditions.
Treatment Goals
Establishing treatment goals can help shape patient expectations and provide a plan for clinicians. In 2017, the National Psoriasis Foundation published a treat-to-target approach using body surface area (BSA) measurements at baseline, 3 months, and then every 6 months after starting a new treatment.12 The target response is a decrease in psoriasis to 1% or less BSA at 3 months and to maintain this response when evaluated at 6-month intervals. Alternatively, a target of 3% BSA after 3 months is satisfactory if the patient improves by 75% BSA overall. If these targets are not met after 6 months, therapeutic alternatives can be considered.12
Biologic Treatment of Psoriasis
Treatment options for patients with psoriasis depend first on disease severity. Topicals and phototherapy are first line for mild to moderate disease. For moderate to severe disease, addition of systemic agents such as methotrexate, cyclosporine, or acitretin; small-molecular-weight immunomodulators such as apremilast; or biologic medications should be considered. Current biologics available for moderate to severe plaque psoriasis target TNF-α, IL-12/IL-23, IL-23, IL-17A, or IL-17A receptor.
TNF-α Inhibitors
Tumor necrosis factor α inhibitors have been available for treatment of autoimmune disease for nearly 20 years. These medications block either soluble cytokine or membrane-bound cytokine. All are given as subcutaneous injections, except for infliximab, which is a weight-based infusion.
Efficacy
Tumor necrosis factor α inhibitors are the first class to demonstrate long-term efficacy and safety in both psoriasis and psoriatic arthritis (PsA). Etanercept was approved for adults with PsA in 2002 and psoriasis in 2004, and later for pediatric psoriasis (≥4 years of age) in 2016 (Table 2). Although etanercept has a sustained safety profile, the response rates are not as high as other anti–TNF-α inhibitors. Adalimumab is one of the most prescribed biologics, with a total of 10 indications at present, including PsA. Infliximab is an intravenous infusion that demonstrates a rapid and sustained response in most patients. The dose and dosing interval can be adjusted according to response. Certolizumab pegol was approved for PsA in 2013 and for psoriasis in 2018.
Tumor necrosis factor α inhibitors maintain efficacy well and work best when dosed continuously. Both neutralizing and nonneutralizing antibodies form with these agents. Neutralizing antibodies may contribute to decreased efficacy, particularly for the chimeric antibody infliximab. One approach to mitigate loss of efficacy is the short-term addition of low-dose methotrexate (eg, 7.5–15 mg weekly) for 3 to 6 months until response is recaptured.
Safety
To evaluate long-term safety, a multicenter prospective registry study (Psoriasis Longitudinal Assessment and Registry [PSOLAR]) was initiated in 2007 to follow clinical outcomes. Data through 2013 showed no significant increase in rates of infection, malignancy, or major adverse cardiovascular events in more than 12,000 patients.13
Conflicting information exists in the literature regarding risk for malignancy with TNF-α inhibitors. One recent retrospective cohort study suggested a slightly increased risk for malignancies other than nonmelanoma skin cancers in patients on TNF-α inhibitors for more than 12 months (relative risk, 1.54).14 Reports of increased risk for cutaneous squamous cell carcinomas necessitate regular skin checks.15 A potential risk for lymphoma has been noted, though having psoriasis itself imparts an increased risk for Hodgkin and cutaneous T-cell lymphoma.16
Reactivation of tuberculosis and hepatitis have been reported with TNF-α inhibition. Data suggest that infliximab may be associated with more serious infections.13
Demyelinating conditions such as multiple sclerosis have occurred de novo or worsened in patients on TNF-α inhibitors.17 Tumor necrosis factor α blockers should be avoided in patients with decompensated heart failure. Rare cases of liver enzyme elevation and cytopenia have been noted. Additionally, lupuslike syndromes, which are generally reversible upon discontinuation, have occurred in some patients.
Patient Selection
Tumor necrosis factor α inhibitors are the treatment of choice for patients with comorbid PsA. This class halts progression of joint destruction over time.18Select TNF-α inhibitors are indicated for inflammatory bowel disease (IBD) and are a preferred treatment in this patient population. Specifically, adalimumab and infliximab are approved for both Crohn disease (CD) and ulcerative colitis. Certolizumab pegol is approved for CD.
Tumor necrosis factor α is upregulated in obesity, cardiovascular disease, and atherosclerotic plaques. Evidence suggests that TNF-α blockers may lower cardiovascular risk over time.19 For patients with obesity, infliximab is a good option, as it is the only TNF-α inhibitor with weight-based dosing.
In patients with frequent infections or history of hepatitis C, etanercept has been the biologic most commonly used when no alternatives exist, in part due to its shorter half-life.
IL-12/IL-23 Inhibitor
Ustekinumab is a monoclonal antibody that binds the p40 subunit shared by IL-12 and IL-23, blocking their ability to bind receptors. IL-12 and IL-23 play a role in activating naïve T cells to become TH1 or TH17 cells, respectively.
Efficacy and Safety
Clinical trials demonstrate long-term efficacy of ustekinumab, which was approved for psoriasis in 2009, PsA in 2013, and later pediatric psoriasis (≥12 years of age) in 2017. Dosing is listed in Table 2.
Laboratory abnormalities did not arise in trials. Periodic tuberculosis screening is required. Prospective data over 5 years showed very low rates of adverse events (AEs), serious infections, malignancies, and major adverse cardiovascular events.20 Ustekinumab did not worsen or improve demyelinating disease and appears safe in this population.
Patient Selection
Ustekinumab is approved for PsA and is a good option for those who are not candidates for TNF-α and IL-17 inhibitors. Ustekinumab also is approved for CD. The dosing interval of 12 weeks makes ustekinumab convenient for patients. Two dosages exist based on the patient’s weight, offering an advantage to obese patients.
IL-17/IL-17R Inhibitors
Activated TH17 cells produce the IL-17 cytokine family, which stimulates keratinocyte proliferation and dermal inflammation. Secukinumab is a fully human monoclonal antibody, and ixekizumab is a humanized monoclonal antibody; both target IL-17A. Brodalumab targets the IL-17A receptor.
Efficacy and Safety
IL-17 inhibitors showed impressive and rapid responses in trials.21-23 The subsets of patients who responded well and continued treatment in extension trials demonstrated that these treatments maintain efficacy over time.24-26
In addition to tuberculosis reactivation, there is a small increased risk for cutaneous candidiasis with IL-17 inhibitors, which can be managed without stopping treatment. Laboratory abnormalities were limited to mild neutropenia, which was not associated with increased risk for infection.21-23 With ixekizumab, neutropenia was seen more commonly in the first 12 weeks.22
IL-17 is highly expressed in the gut mucosa, and its inhibition is thought to weaken the barrier function of the gut mucosa, promoting inflammation. As a consequence, this class is contraindicated in patients with IBD due to exacerbations of existing IBD and cases of new-onset IBD.21-23 Symptoms of diarrhea, abdominal pain, blood in stool, or nighttime stooling on review of gastrointestinal tract symptoms should prompt further evaluation.
Brodalumab has a unique warning for risk for suicidal ideation and behavior.23 Depression is more common in the psoriasis population in general; therefore, physicians should be aware of this potential comorbidity regardless of the treatment plan. Because the response rates are so impressive with brodalumab, the Risk Evaluation and Mitigation Strategy (REMS) program was established to ensure understanding of this risk so that patients can be appropriately counseled and managed.
Patient Selection
The improvement in psoriasis is rapid and may occur as early as week 2 to 3 of treatment after initiation of IL-17 inhibitors. Ixekizumab and secukinumab also are approved for PsA. Although improvement in joint disease is not as fast as with the anti-TNF inhibitors, notable improvement occurs by week 20 to 24.27
IL-23 Inhibitors
Guselkumab and tildrakizumab are the newest biologics for psoriasis, approved in 2017 and 2018, respectively. Both are monoclonal antibodies against the p19 subunit of IL-23, which blocks activation of TH17 cells.
Efficacy and Safety
Guselkumab and tildrakizumab demonstrated efficacy with minimal AEs or precautions noted thus far.28,29 Infections are again a risk, making tuberculosis testing the only recommended monitoring.
Patient Selection
Both medications offer another effective and safe option for patients with psoriasis. Similar to ustekinumab, the dosing interval of 12 weeks for tildrakizumab is ideal for patients who have needle phobia or are unable to administer their own injections.
Special Populations
Pregnancy
Antibodies cross the placenta as pregnancy progresses, with the highest rate in the third trimester. Certolizumab pegol has shown the lowest concentrations in infant serum, possibly due to its unique structure lacking the fragment crystallizable region required for passage through the placenta.30 For this reason, certolizumab pegol is a treatment of choice if biologic therapy is warranted during pregnancy.
Much of the pregnancy data for the remaining TNF-α inhibitors come from patients with rheumatoid arthritis or CD. In these populations, rates of major birth defects and miscarriages do not differ greatly from untreated women with these conditions.31 One retrospective study of unintentional pregnancies in women receiving ustekinumab showed rates of AEs similar to the general population.32
Pregnancy data for IL-17 or IL-23 inhibitors are largely limited to animal studies. One retrospective study of women exposed to secukinumab early in gestation showed no increased risk for pregnancy-related AEs.33 Discontinuation is still recommended for patients who become pregnant.
Pediatric Patients
Etanercept is approved for pediatric psoriatic patients 4 years and older. Children with juvenile idiopathic arthritis who are 2 years and older can receive etanercept. Ustekinumab is safe and effective for pediatric psoriatic patients 12 years and older, offering a second biologic option in children.
Although not approved for pediatric psoriasis, adalimumab is approved in pediatric CD (≥6 years of age) and for juvenile idiopathic arthritis (≥2 years of age). Infliximab is approved for children 6 years and older with CD or ulcerative colitis.
Monitoring
Periodic tuberculosis screening is recommended for all biologics. For patients with latent tuberculosis, biologics may be restarted after 1 month of treatment of tuberculosis.
Prior to initiation of biologics, patients should be screened for hepatitis with hepatitis B surface antigen and antibody, hepatitis B core antibody, and hepatitis C antibody. Patients at risk for human immunodeficiency virus also should be screened.
Generally, complete blood cell count and comprehensive metabolic profile are advisable prior to starting a biologic. Opinions differ on frequency of repeating laboratory work. Complete blood cell count and comprehensive metabolic profile should be monitored at least every 3 to 6 months in patients on TNF-α inhibitors, and neutrophil count should be monitored during the induction phase of IL-17 inhibitors.
All patients with psoriasis should maintain age-appropriate cancer screenings, especially those on biologics. If malignancy is discovered, biologic medication should be discontinued. Debate exists as to when therapy can be safely restarted following treatment of malignancy. Patients who are considered at low risk for recurrence may opt to restart a biologic after 5 years, or sooner if symptoms warrant.34 This decision should involve the patient’s cancer specialist.
Conclusion
Treatment choices are based on psoriasis type and severity, comorbidities, patient preferences, and drug accessibility. One approach is detailed in Table 3. As research advances the understanding of psoriasis, this field will continue to rapidly change. Knowledge of the immunopathogenesis of psoriasis and its relation to comorbidities can direct your decision-making for individual patients.
Psoriasis is a T cell–mediated inflammatory disease that manifests as erythematous scaling plaques of the skin. In recent decades, our understanding of psoriasis has transformed from a disease isolated to the skin to a systemic disease impacting the overall health of those affected.
With recent elucidation of the pathways driving psoriasis, development of targeted therapies has resulted in an influx of options to the market. Navigating the options can seem overwhelming even to the seasoned clinician. Becoming familiar with a sound treatment approach during residency will create a foundation for biologic use in psoriasis patients throughout your career. Here we offer an approach to choosing biologic treatments based on individual patient characteristics, including disease severity, comorbidities, and ultimate treatment goals.
Immune Pathogenesis
Although the pathogenesis of psoriasis is complex and outside the scope of this article, we do recommend clinicians keep in mind the current understanding of pathways involved and ways our therapies alter them. Briefly, psoriasis is a T cell–mediated disease in which IL-12 and IL-23 released by activated dendritic cells activate T helper cells including TH1, TH17, and TH22. These cells produce additional cytokines, including IFN-γ, tumor necrosis factor (TNF) α, IL-17, and IL-22, which propagate the immune response and lead to keratinocyte hyperproliferation. In general, psoriasis medications work by altering T-cell activation, effector cytokines, or cytokine receptors.
Comorbidities
A targeted approach should take into consideration the immune dysregulation shared by psoriasis and associated comorbidities (Table 1). One goal of biologic treatments is to improve comorbidities when possible. At minimum, selected treatments should not exacerbate these conditions.
Treatment Goals
Establishing treatment goals can help shape patient expectations and provide a plan for clinicians. In 2017, the National Psoriasis Foundation published a treat-to-target approach using body surface area (BSA) measurements at baseline, 3 months, and then every 6 months after starting a new treatment.12 The target response is a decrease in psoriasis to 1% or less BSA at 3 months and to maintain this response when evaluated at 6-month intervals. Alternatively, a target of 3% BSA after 3 months is satisfactory if the patient improves by 75% BSA overall. If these targets are not met after 6 months, therapeutic alternatives can be considered.12
Biologic Treatment of Psoriasis
Treatment options for patients with psoriasis depend first on disease severity. Topicals and phototherapy are first line for mild to moderate disease. For moderate to severe disease, addition of systemic agents such as methotrexate, cyclosporine, or acitretin; small-molecular-weight immunomodulators such as apremilast; or biologic medications should be considered. Current biologics available for moderate to severe plaque psoriasis target TNF-α, IL-12/IL-23, IL-23, IL-17A, or IL-17A receptor.
TNF-α Inhibitors
Tumor necrosis factor α inhibitors have been available for treatment of autoimmune disease for nearly 20 years. These medications block either soluble cytokine or membrane-bound cytokine. All are given as subcutaneous injections, except for infliximab, which is a weight-based infusion.
Efficacy
Tumor necrosis factor α inhibitors are the first class to demonstrate long-term efficacy and safety in both psoriasis and psoriatic arthritis (PsA). Etanercept was approved for adults with PsA in 2002 and psoriasis in 2004, and later for pediatric psoriasis (≥4 years of age) in 2016 (Table 2). Although etanercept has a sustained safety profile, the response rates are not as high as other anti–TNF-α inhibitors. Adalimumab is one of the most prescribed biologics, with a total of 10 indications at present, including PsA. Infliximab is an intravenous infusion that demonstrates a rapid and sustained response in most patients. The dose and dosing interval can be adjusted according to response. Certolizumab pegol was approved for PsA in 2013 and for psoriasis in 2018.
Tumor necrosis factor α inhibitors maintain efficacy well and work best when dosed continuously. Both neutralizing and nonneutralizing antibodies form with these agents. Neutralizing antibodies may contribute to decreased efficacy, particularly for the chimeric antibody infliximab. One approach to mitigate loss of efficacy is the short-term addition of low-dose methotrexate (eg, 7.5–15 mg weekly) for 3 to 6 months until response is recaptured.
Safety
To evaluate long-term safety, a multicenter prospective registry study (Psoriasis Longitudinal Assessment and Registry [PSOLAR]) was initiated in 2007 to follow clinical outcomes. Data through 2013 showed no significant increase in rates of infection, malignancy, or major adverse cardiovascular events in more than 12,000 patients.13
Conflicting information exists in the literature regarding risk for malignancy with TNF-α inhibitors. One recent retrospective cohort study suggested a slightly increased risk for malignancies other than nonmelanoma skin cancers in patients on TNF-α inhibitors for more than 12 months (relative risk, 1.54).14 Reports of increased risk for cutaneous squamous cell carcinomas necessitate regular skin checks.15 A potential risk for lymphoma has been noted, though having psoriasis itself imparts an increased risk for Hodgkin and cutaneous T-cell lymphoma.16
Reactivation of tuberculosis and hepatitis have been reported with TNF-α inhibition. Data suggest that infliximab may be associated with more serious infections.13
Demyelinating conditions such as multiple sclerosis have occurred de novo or worsened in patients on TNF-α inhibitors.17 Tumor necrosis factor α blockers should be avoided in patients with decompensated heart failure. Rare cases of liver enzyme elevation and cytopenia have been noted. Additionally, lupuslike syndromes, which are generally reversible upon discontinuation, have occurred in some patients.
Patient Selection
Tumor necrosis factor α inhibitors are the treatment of choice for patients with comorbid PsA. This class halts progression of joint destruction over time.18Select TNF-α inhibitors are indicated for inflammatory bowel disease (IBD) and are a preferred treatment in this patient population. Specifically, adalimumab and infliximab are approved for both Crohn disease (CD) and ulcerative colitis. Certolizumab pegol is approved for CD.
Tumor necrosis factor α is upregulated in obesity, cardiovascular disease, and atherosclerotic plaques. Evidence suggests that TNF-α blockers may lower cardiovascular risk over time.19 For patients with obesity, infliximab is a good option, as it is the only TNF-α inhibitor with weight-based dosing.
In patients with frequent infections or history of hepatitis C, etanercept has been the biologic most commonly used when no alternatives exist, in part due to its shorter half-life.
IL-12/IL-23 Inhibitor
Ustekinumab is a monoclonal antibody that binds the p40 subunit shared by IL-12 and IL-23, blocking their ability to bind receptors. IL-12 and IL-23 play a role in activating naïve T cells to become TH1 or TH17 cells, respectively.
Efficacy and Safety
Clinical trials demonstrate long-term efficacy of ustekinumab, which was approved for psoriasis in 2009, PsA in 2013, and later pediatric psoriasis (≥12 years of age) in 2017. Dosing is listed in Table 2.
Laboratory abnormalities did not arise in trials. Periodic tuberculosis screening is required. Prospective data over 5 years showed very low rates of adverse events (AEs), serious infections, malignancies, and major adverse cardiovascular events.20 Ustekinumab did not worsen or improve demyelinating disease and appears safe in this population.
Patient Selection
Ustekinumab is approved for PsA and is a good option for those who are not candidates for TNF-α and IL-17 inhibitors. Ustekinumab also is approved for CD. The dosing interval of 12 weeks makes ustekinumab convenient for patients. Two dosages exist based on the patient’s weight, offering an advantage to obese patients.
IL-17/IL-17R Inhibitors
Activated TH17 cells produce the IL-17 cytokine family, which stimulates keratinocyte proliferation and dermal inflammation. Secukinumab is a fully human monoclonal antibody, and ixekizumab is a humanized monoclonal antibody; both target IL-17A. Brodalumab targets the IL-17A receptor.
Efficacy and Safety
IL-17 inhibitors showed impressive and rapid responses in trials.21-23 The subsets of patients who responded well and continued treatment in extension trials demonstrated that these treatments maintain efficacy over time.24-26
In addition to tuberculosis reactivation, there is a small increased risk for cutaneous candidiasis with IL-17 inhibitors, which can be managed without stopping treatment. Laboratory abnormalities were limited to mild neutropenia, which was not associated with increased risk for infection.21-23 With ixekizumab, neutropenia was seen more commonly in the first 12 weeks.22
IL-17 is highly expressed in the gut mucosa, and its inhibition is thought to weaken the barrier function of the gut mucosa, promoting inflammation. As a consequence, this class is contraindicated in patients with IBD due to exacerbations of existing IBD and cases of new-onset IBD.21-23 Symptoms of diarrhea, abdominal pain, blood in stool, or nighttime stooling on review of gastrointestinal tract symptoms should prompt further evaluation.
Brodalumab has a unique warning for risk for suicidal ideation and behavior.23 Depression is more common in the psoriasis population in general; therefore, physicians should be aware of this potential comorbidity regardless of the treatment plan. Because the response rates are so impressive with brodalumab, the Risk Evaluation and Mitigation Strategy (REMS) program was established to ensure understanding of this risk so that patients can be appropriately counseled and managed.
Patient Selection
The improvement in psoriasis is rapid and may occur as early as week 2 to 3 of treatment after initiation of IL-17 inhibitors. Ixekizumab and secukinumab also are approved for PsA. Although improvement in joint disease is not as fast as with the anti-TNF inhibitors, notable improvement occurs by week 20 to 24.27
IL-23 Inhibitors
Guselkumab and tildrakizumab are the newest biologics for psoriasis, approved in 2017 and 2018, respectively. Both are monoclonal antibodies against the p19 subunit of IL-23, which blocks activation of TH17 cells.
Efficacy and Safety
Guselkumab and tildrakizumab demonstrated efficacy with minimal AEs or precautions noted thus far.28,29 Infections are again a risk, making tuberculosis testing the only recommended monitoring.
Patient Selection
Both medications offer another effective and safe option for patients with psoriasis. Similar to ustekinumab, the dosing interval of 12 weeks for tildrakizumab is ideal for patients who have needle phobia or are unable to administer their own injections.
Special Populations
Pregnancy
Antibodies cross the placenta as pregnancy progresses, with the highest rate in the third trimester. Certolizumab pegol has shown the lowest concentrations in infant serum, possibly due to its unique structure lacking the fragment crystallizable region required for passage through the placenta.30 For this reason, certolizumab pegol is a treatment of choice if biologic therapy is warranted during pregnancy.
Much of the pregnancy data for the remaining TNF-α inhibitors come from patients with rheumatoid arthritis or CD. In these populations, rates of major birth defects and miscarriages do not differ greatly from untreated women with these conditions.31 One retrospective study of unintentional pregnancies in women receiving ustekinumab showed rates of AEs similar to the general population.32
Pregnancy data for IL-17 or IL-23 inhibitors are largely limited to animal studies. One retrospective study of women exposed to secukinumab early in gestation showed no increased risk for pregnancy-related AEs.33 Discontinuation is still recommended for patients who become pregnant.
Pediatric Patients
Etanercept is approved for pediatric psoriatic patients 4 years and older. Children with juvenile idiopathic arthritis who are 2 years and older can receive etanercept. Ustekinumab is safe and effective for pediatric psoriatic patients 12 years and older, offering a second biologic option in children.
Although not approved for pediatric psoriasis, adalimumab is approved in pediatric CD (≥6 years of age) and for juvenile idiopathic arthritis (≥2 years of age). Infliximab is approved for children 6 years and older with CD or ulcerative colitis.
Monitoring
Periodic tuberculosis screening is recommended for all biologics. For patients with latent tuberculosis, biologics may be restarted after 1 month of treatment of tuberculosis.
Prior to initiation of biologics, patients should be screened for hepatitis with hepatitis B surface antigen and antibody, hepatitis B core antibody, and hepatitis C antibody. Patients at risk for human immunodeficiency virus also should be screened.
Generally, complete blood cell count and comprehensive metabolic profile are advisable prior to starting a biologic. Opinions differ on frequency of repeating laboratory work. Complete blood cell count and comprehensive metabolic profile should be monitored at least every 3 to 6 months in patients on TNF-α inhibitors, and neutrophil count should be monitored during the induction phase of IL-17 inhibitors.
All patients with psoriasis should maintain age-appropriate cancer screenings, especially those on biologics. If malignancy is discovered, biologic medication should be discontinued. Debate exists as to when therapy can be safely restarted following treatment of malignancy. Patients who are considered at low risk for recurrence may opt to restart a biologic after 5 years, or sooner if symptoms warrant.34 This decision should involve the patient’s cancer specialist.
Conclusion
Treatment choices are based on psoriasis type and severity, comorbidities, patient preferences, and drug accessibility. One approach is detailed in Table 3. As research advances the understanding of psoriasis, this field will continue to rapidly change. Knowledge of the immunopathogenesis of psoriasis and its relation to comorbidities can direct your decision-making for individual patients.
- Armstrong AW, Harskamp CT, Armstrong EJ. The association between psoriasis and obesity: a systematic review and meta-analysis of observational studies. Nutr Diabetes. 2012;2:e54.
- Armstrong AW, Harskamp CT, Armstrong EJ. Psoriasis and the risk of diabetes mellitus: a systematic review and meta-analysis. JAMA Dermatol. 2013;149:84-91.
- Armstrong AW, Harskamp CT, Armstrong EJ. The association between psoriasis and hypertension: a systematic review and meta-analysis of observational studies. J Hypertens. 2013;31:433-442; discussion 442-433.
- Candia R, Ruiz A, Torres-Robles R, et al. Risk of non-alcoholic fatty liver disease in patients with psoriasis: a systematic review and meta-analysis. J Eur Acad Dermatol Venereol. 2015;29:656-662.
- Chi CC, Tung TH, Wang J, et al. Risk of uveitis among people with psoriasis: a nationwide cohort study. JAMA Ophthalmol. 2017;135:415-422.
- Cohen AD, Dreiher J, Birkenfeld S. Psoriasis associated with ulcerative colitis and Crohn’s disease. J Eur Acad Dermatol Venereol. 2009;23:561-565.
- Dowlatshahi EA, Wakkee M, Arends LR, et al. The prevalence and odds of depressive symptoms and clinical depression in psoriasis patients: a systematic review and meta-analysis. J Invest Dermatol. 2014;134:1542-1551.
- Gaeta M, Castelvecchio S, Ricci C, et al. Role of psoriasis as independent predictor of cardiovascular disease: a meta-regression analysis. Int J Cardiol. 2013;168:2282-2288.
- Ma C, Harskamp CT, Armstrong EJ, et al. The association between psoriasis and dyslipidaemia: a systematic review. Br J Dermatol. 2013;168:486-495.
- Parisi R, Webb RT, Carr MJ, et al. Alcohol-related mortality in patients with psoriasis: a population-based cohort study. JAMA Dermatol. 2017;153:1256-1262.
- Rodríguez-Zúñiga MJM, García-Perdomo HA. Systematic review and meta-analysis of the association between psoriasis and metabolic syndrome. J Am Acad Dermatol. 2017;77:657-666.e8.
- Armstrong AW, Siegel MP, Bagel J, et al. From the Medical Board of the National Psoriasis Foundation: treatment targets for plaque psoriasis. J Am Acad Dermatol. 2017;76:290-298.
- Gottlieb AB, Kalb RE, Langley RG, et al. Safety observations in 12095 patients with psoriasis enrolled in an international registry (PSOLAR): experience with infliximab and other systemic and biologic therapies. J Drugs Dermatol. 2014;13:1441-1448.
- Fiorentino D, Ho V, Lebwohl MG, et al. Risk of malignancy with systemic psoriasis treatment in the Psoriasis Longitudinal Assessment Registry. J Am Acad Dermatol. 2017;77:845-854.e5.
- van Lümig PP, Menting SP, van den Reek JM, et al. An increased risk of non-melanoma skin cancer during TNF-inhibitor treatment in psoriasis patients compared to rheumatoid arthritis patients probably relates to disease-related factors. J Eur Acad Dermatol Venereol. 2015;29:752-760.
- Gelfand JM, Berlin J, Van Voorhees A, et al. Lymphoma rates are low but increased in patients with psoriasis: results from a population-based cohort study in the United Kingdom. Arch Dermatol. 2003;139:1425-1429.
- Sicotte NL, Voskuhl RR. Onset of multiple sclerosis associated with anti-TNF therapy. Neurology. 2001;57:1885-1888.
- Finckh A, Simard JF, Duryea J, et al. The effectiveness of anti-tumor necrosis factor therapy in preventing progressive radiographic joint damage in rheumatoid arthritis: a population-based study. Arthritis Rheum. 2006;54:54-59.
- Wu JJ, Sundaram M, Cloutier M, et al. The risk of cardiovascular events in psoriasis patients treated with tumor necrosis factor-α inhibitors versus phototherapy: an observational cohort study. J Am Acad Dermatol. 2018;79:60-68.
- Kimball AB, Papp KA, Wasfi Y, et al. Long-term efficacy of ustekinumab in patients with moderate-to-severe psoriasis treated for up to 5 years in the PHOENIX 1 study. J Eur Acad Dermatol Venereol. 2013;27:1535-1545.
- Langley RG, Elewski BE, Lebwohl M, et al. Secukinumab in plaque psoriasis—results of two phase 3 trials. N Engl J Med. 2014;371:326-338.
- Griffiths CE, Reich K, Lebwohl M, et al. Comparison of ixekizumab with etanercept or placebo in moderate-to-severe psoriasis (UNCOVER-2 and UNCOVER-3): results from two phase 3 randomised trials. Lancet. 2015;386:541-551.
- Lebwohl M, Strober B, Menter A, et al. Phase 3 studies comparing brodalumab with ustekinumab in psoriasis. N Engl J Med. 2015;373:1318-1328.
- Bissonnette R, Luger T, Thaçi D, et al. Secukinumab demonstrates high sustained efficacy and a favourable safety profile in patients with moderate-to-severe psoriasis through 5 years of treatment (SCULPTURE Extension Study). J Eur Acad Dermatol Venereol. 2018;32:1507-1514.
- Leonardi C, Maari C, Philipp S, et al. Maintenance of skin clearance with ixekizumab treatment of psoriasis: three-year results from the UNCOVER-3 study. J Am Acad Dermatol. 2018;79:824-830.
- Papp K, Leonardi C, Menter A, et al. Safety and efficacy of brodalumab for psoriasis after 120 weeks of treatment. J Am Acad Dermatol. 2014;71:1183-1190.e1183.
- Gottlieb AB, Strand V, Kishimoto M, et al. Ixekizumab improves patient-reported outcomes up to 52 weeks in bDMARD-naïve patients with active psoriatic arthritis (SPIRIT-P1). Rheumatology (Oxford). 2018;57:1777-1788.
- Blauvelt A, Papp KA, Griffiths CE, et al. Efficacy and safety of guselkumab, an anti-interleukin-23 monoclonal antibody, compared with adalimumab for the continuous treatment of patients with moderate to severe psoriasis: results from the phase III, double-blinded, placebo- and active comparator-controlled VOYAGE 1 trial. J Am Acad Dermatol. 2017;76:405-417.
- Reich K, Papp KA, Blauvelt A, et al. Tildrakizumab versus placebo or etanercept for chronic plaque psoriasis (reSURFACE 1 and reSURFACE 2): results from two randomised controlled, phase 3 trials. Lancet. 2017;390:276-288.
- Mariette X, Förger F, Abraham B, et al. Lack of placental transfer of certolizumab pegol during pregnancy: results from CRIB, a prospective, postmarketing, pharmacokinetic study. Ann Rheum Dis. 2018;77:228-233.
- Komaki F, Komaki Y, Micic D, et al. Outcome of pregnancy and neonatal complications with anti-tumor necrosis factor-α use in females with immune mediated diseases; a systematic review and meta-analysis. J Autoimmun. 2017;76:38-52.
- Götestam Skorpen C, Hoeltzenbein M, Tincani A, et al. The EULAR points to consider for use of antirheumatic drugs before pregnancy, and during pregnancy and lactation. Ann Rheum Dis. 2016;75:795-810.
- Warren RB, Reich K, Langley RG, et al. Secukinumab in pregnancy: outcomes in psoriasis, psoriatic arthritis and ankylosing spondylitis from the global safety database [published online ahead of print June 21, 2018]. Br J Dermatol. doi:10.1111/bjd.16901.
- Elandt K, Aletaha D. Treating rheumatic patients with a malignancy. Arthritis Res Ther. 2011;13:223.
- Armstrong AW, Harskamp CT, Armstrong EJ. The association between psoriasis and obesity: a systematic review and meta-analysis of observational studies. Nutr Diabetes. 2012;2:e54.
- Armstrong AW, Harskamp CT, Armstrong EJ. Psoriasis and the risk of diabetes mellitus: a systematic review and meta-analysis. JAMA Dermatol. 2013;149:84-91.
- Armstrong AW, Harskamp CT, Armstrong EJ. The association between psoriasis and hypertension: a systematic review and meta-analysis of observational studies. J Hypertens. 2013;31:433-442; discussion 442-433.
- Candia R, Ruiz A, Torres-Robles R, et al. Risk of non-alcoholic fatty liver disease in patients with psoriasis: a systematic review and meta-analysis. J Eur Acad Dermatol Venereol. 2015;29:656-662.
- Chi CC, Tung TH, Wang J, et al. Risk of uveitis among people with psoriasis: a nationwide cohort study. JAMA Ophthalmol. 2017;135:415-422.
- Cohen AD, Dreiher J, Birkenfeld S. Psoriasis associated with ulcerative colitis and Crohn’s disease. J Eur Acad Dermatol Venereol. 2009;23:561-565.
- Dowlatshahi EA, Wakkee M, Arends LR, et al. The prevalence and odds of depressive symptoms and clinical depression in psoriasis patients: a systematic review and meta-analysis. J Invest Dermatol. 2014;134:1542-1551.
- Gaeta M, Castelvecchio S, Ricci C, et al. Role of psoriasis as independent predictor of cardiovascular disease: a meta-regression analysis. Int J Cardiol. 2013;168:2282-2288.
- Ma C, Harskamp CT, Armstrong EJ, et al. The association between psoriasis and dyslipidaemia: a systematic review. Br J Dermatol. 2013;168:486-495.
- Parisi R, Webb RT, Carr MJ, et al. Alcohol-related mortality in patients with psoriasis: a population-based cohort study. JAMA Dermatol. 2017;153:1256-1262.
- Rodríguez-Zúñiga MJM, García-Perdomo HA. Systematic review and meta-analysis of the association between psoriasis and metabolic syndrome. J Am Acad Dermatol. 2017;77:657-666.e8.
- Armstrong AW, Siegel MP, Bagel J, et al. From the Medical Board of the National Psoriasis Foundation: treatment targets for plaque psoriasis. J Am Acad Dermatol. 2017;76:290-298.
- Gottlieb AB, Kalb RE, Langley RG, et al. Safety observations in 12095 patients with psoriasis enrolled in an international registry (PSOLAR): experience with infliximab and other systemic and biologic therapies. J Drugs Dermatol. 2014;13:1441-1448.
- Fiorentino D, Ho V, Lebwohl MG, et al. Risk of malignancy with systemic psoriasis treatment in the Psoriasis Longitudinal Assessment Registry. J Am Acad Dermatol. 2017;77:845-854.e5.
- van Lümig PP, Menting SP, van den Reek JM, et al. An increased risk of non-melanoma skin cancer during TNF-inhibitor treatment in psoriasis patients compared to rheumatoid arthritis patients probably relates to disease-related factors. J Eur Acad Dermatol Venereol. 2015;29:752-760.
- Gelfand JM, Berlin J, Van Voorhees A, et al. Lymphoma rates are low but increased in patients with psoriasis: results from a population-based cohort study in the United Kingdom. Arch Dermatol. 2003;139:1425-1429.
- Sicotte NL, Voskuhl RR. Onset of multiple sclerosis associated with anti-TNF therapy. Neurology. 2001;57:1885-1888.
- Finckh A, Simard JF, Duryea J, et al. The effectiveness of anti-tumor necrosis factor therapy in preventing progressive radiographic joint damage in rheumatoid arthritis: a population-based study. Arthritis Rheum. 2006;54:54-59.
- Wu JJ, Sundaram M, Cloutier M, et al. The risk of cardiovascular events in psoriasis patients treated with tumor necrosis factor-α inhibitors versus phototherapy: an observational cohort study. J Am Acad Dermatol. 2018;79:60-68.
- Kimball AB, Papp KA, Wasfi Y, et al. Long-term efficacy of ustekinumab in patients with moderate-to-severe psoriasis treated for up to 5 years in the PHOENIX 1 study. J Eur Acad Dermatol Venereol. 2013;27:1535-1545.
- Langley RG, Elewski BE, Lebwohl M, et al. Secukinumab in plaque psoriasis—results of two phase 3 trials. N Engl J Med. 2014;371:326-338.
- Griffiths CE, Reich K, Lebwohl M, et al. Comparison of ixekizumab with etanercept or placebo in moderate-to-severe psoriasis (UNCOVER-2 and UNCOVER-3): results from two phase 3 randomised trials. Lancet. 2015;386:541-551.
- Lebwohl M, Strober B, Menter A, et al. Phase 3 studies comparing brodalumab with ustekinumab in psoriasis. N Engl J Med. 2015;373:1318-1328.
- Bissonnette R, Luger T, Thaçi D, et al. Secukinumab demonstrates high sustained efficacy and a favourable safety profile in patients with moderate-to-severe psoriasis through 5 years of treatment (SCULPTURE Extension Study). J Eur Acad Dermatol Venereol. 2018;32:1507-1514.
- Leonardi C, Maari C, Philipp S, et al. Maintenance of skin clearance with ixekizumab treatment of psoriasis: three-year results from the UNCOVER-3 study. J Am Acad Dermatol. 2018;79:824-830.
- Papp K, Leonardi C, Menter A, et al. Safety and efficacy of brodalumab for psoriasis after 120 weeks of treatment. J Am Acad Dermatol. 2014;71:1183-1190.e1183.
- Gottlieb AB, Strand V, Kishimoto M, et al. Ixekizumab improves patient-reported outcomes up to 52 weeks in bDMARD-naïve patients with active psoriatic arthritis (SPIRIT-P1). Rheumatology (Oxford). 2018;57:1777-1788.
- Blauvelt A, Papp KA, Griffiths CE, et al. Efficacy and safety of guselkumab, an anti-interleukin-23 monoclonal antibody, compared with adalimumab for the continuous treatment of patients with moderate to severe psoriasis: results from the phase III, double-blinded, placebo- and active comparator-controlled VOYAGE 1 trial. J Am Acad Dermatol. 2017;76:405-417.
- Reich K, Papp KA, Blauvelt A, et al. Tildrakizumab versus placebo or etanercept for chronic plaque psoriasis (reSURFACE 1 and reSURFACE 2): results from two randomised controlled, phase 3 trials. Lancet. 2017;390:276-288.
- Mariette X, Förger F, Abraham B, et al. Lack of placental transfer of certolizumab pegol during pregnancy: results from CRIB, a prospective, postmarketing, pharmacokinetic study. Ann Rheum Dis. 2018;77:228-233.
- Komaki F, Komaki Y, Micic D, et al. Outcome of pregnancy and neonatal complications with anti-tumor necrosis factor-α use in females with immune mediated diseases; a systematic review and meta-analysis. J Autoimmun. 2017;76:38-52.
- Götestam Skorpen C, Hoeltzenbein M, Tincani A, et al. The EULAR points to consider for use of antirheumatic drugs before pregnancy, and during pregnancy and lactation. Ann Rheum Dis. 2016;75:795-810.
- Warren RB, Reich K, Langley RG, et al. Secukinumab in pregnancy: outcomes in psoriasis, psoriatic arthritis and ankylosing spondylitis from the global safety database [published online ahead of print June 21, 2018]. Br J Dermatol. doi:10.1111/bjd.16901.
- Elandt K, Aletaha D. Treating rheumatic patients with a malignancy. Arthritis Res Ther. 2011;13:223.
Practice Points
- Psoriasis affects millions of Americans and is associated with a growing list of comorbidities.
- With the increasing number of biologic treatment options available, the clinician must keep in mind the immune pathways involved in psoriasis and the ways our therapies alter them.
- Consider disease severity, comorbidities, patient preferences, and drug accessibility when choosing psoriasis treatments.
Update on the Pathophysiology of Psoriasis
Increased understanding of the pathophysiology of psoriasis has been one of the driving forces in the development of new therapies. An understanding of the processes involved is important in the optimal management of the disease. The last 30 years of research and clinical practice have revolutionized our understanding of the pathogenesis of psoriasis as the dysregulation of immunity triggered by environmental and genetic stimuli. Psoriasis was originally regarded as a primary disorder of epidermal hyperproliferation. However, experimental models and clinical results from immunomodulating therapies have refined this perspective in conceptualizing psoriasis as a genetically programmed pathologic interaction among resident skin cells; infiltrating immunocytes; and a host of proinflammatory cytokines, chemokines, and growth factors produced by these immunocytes. Two populations of immunocytes and their respective signaling molecules collaborate in the pathogenesis: (1) innate immunocytes, mediated by antigen-presenting cells (APCs)(including natural killer [NK] T lymphocytes, Langerhans cells, and neutrophils), and (2) acquired or adaptive immunocytes, mediated by mature CD4+ and CD8+ T lymphocytes in the skin. Such dysregulation of immunity and subsequent inflammation is responsible for the development and perpetuation of the clinical plaques and histological inflammatory infiltrate characteristic of psoriasis.
Although psoriasis is considered to be an immune-mediated disease in which intralesional T lymphocytes and their proinflammatory signals trigger primed basal layer keratinocytes to rapidly proliferate, debate and research focus on the stimulus that incites this inflammatory process. Our current understanding considers psoriasis to be triggered by exogenous or endogenous environmental stimuli in genetically susceptible individuals. Such stimuli include group A streptococcal pharyngitis, viremia, allergic drug reactions, antimalarial drugs, lithium, beta-blockers, IFN-α, withdrawal of systemic corticosteroids, local trauma (Köbner phenomenon), and emotional stress. These stimuli correlate with the onset or flares of psoriatic lesions. Psoriasis genetics centers on susceptibility loci and corresponding candidate genes, particularly the psoriasis susceptibility (PSORS) 1 locus on the major histocompatibility complex (MHC) class I region. Current research on the pathogenesis of psoriasis examines the complex interactions among immunologic mechanisms, environmental stimuli, and genetic susceptibility. After discussing the clinical presentation and histopathologic features of psoriasis, we will review the pathophysiology of psoriasis through noteworthy developments, including serendipitous observations, reactions to therapies, clinical trials, and animal model systems that have shaped our view of the disease process. In addition to the classic skin lesions, approximately 23% of psoriasis patients develop psoriatic arthritis, with a 10-year latency after diagnosis of psoriasis.1
Principles of Immunity
The immune system, intended to protect its host from foreign invaders and unregulated cell growth, employs 2 main effector pathways—the innate and the acquired (or adaptive) immune responses—both of which contribute to the pathophysiology of psoriasis.2 Innate immunity responses occur within minutes to hours of antigen exposure but fail to develop memory for when the antigen is encountered again. However, adaptive immunity responses take days to weeks to respond after challenged with an antigen. The adaptive immune cells have the capacity to respond to a greater range of antigens and develop immunologic memory via rearrangement of antigen receptors on B and T cells. These specialized B and T cells can then be promptly mobilized and differentiated into mature effector cells that protect the host from a foreign pathogen.
Innate and adaptive immune responses are highly intertwined; they can initiate, perpetuate, and terminate the immune mechanisms responsible for inflammation. They can modify the nature of the immune response by altering the relative proportions of type 1 (TH1), type 2 (TH2), and the more recently discovered type 17 (TH17) subset of helper T cells and their respective signaling molecules. A TH1 response is essential for a cellular immunologic reaction to intracellular bacteria and viruses or cellular immunity. A TH2 response promotes IgE synthesis, eosinophilia, and mast cell maturation for extracellular parasites and helminthes as well as humoral immunity, while a TH17 response is important for cell-mediated immunity to extracellular bacteria and plays a role in autoimmunity.3 The innate and adaptive immune responses employ common effector molecules such as chemokines and cytokines, which are essential in mediating an immune response.
Implicating Dysregulation of Immunity
Our present appreciation of the pathogenesis of psoriasis is based on the history of trial-and-error therapies; serendipitous discoveries; and the current immune targeting drugs used in a variety of chronic inflammatory conditions, including rheumatoid arthritis, ankylosing spondylitis, and inflammatory bowel disease. Before the mid-1980s, research focused on the hyperproliferative epidermal cells as the primary pathology because a markedly thickened epidermis was indeed demonstrated on histologic specimens. Altered cell-cycle kinetics were thought to be the culprit behind the hyperkeratotic plaques. Thus, initial treatments centered on oncologic and antimitotic therapies used to arrest keratinocyte proliferation with agents such as arsenic, ammoniated mercury, and methotrexate.4
However, a paradigm shift from targeting epidermal keratinocytes to immunocyte populations was recognized when a patient receiving cyclosporine to prevent transplant rejection noted clearing of psoriatic lesions in the 1980s.5 Cyclosporine was observed to inhibit messenger RNA transcription of T-cell cytokines, thereby implicating immunologic dysregulation, specifically T-cell hyperactivity, in the pathogenesis of psoriasis.6 However, the concentrations of oral cyclosporine reached in the epidermis exerted direct effects on keratinocyte proliferation and lymphocyte function in these patients.7 Thus, the question was raised as to whether the keratinocytes or the lymphocytes drove the psoriatic plaques. The use of an IL-2 diphtheria toxin-fusion protein, denileukin diftitox, specific for activated T cells with high-affinity IL-2 receptors and nonreactive with keratinocytes, distinguished which cell type was responsible. This targeted T-cell toxin provided clinical and histological clearing of psoriatic plaques. Thus, T lymphocytes rather than keratinocytes were recognized as the definitive driver behind the psoriatic plaques.8
Additional studies have demonstrated that treatments that induce prolonged clearing of psoriatic lesions without continuous therapy, such as psoralen plus UVA irradiation, decreased the numbers of T cells in plaques by at least 90%.9 However, treatments that require continual therapy for satisfactory clinical results, such as cyclosporine and etretinate, simply suppress T-cell activity and proliferation.10,11 Further evidence has linked cellular immunity with the pathogenesis of psoriasis, defining it as a TH1-type disease. Natural killer T cells were shown to be involved through the use of a severe combined immunodeficient mouse model. They were injected into prepsoriatic skin grafted on immunodeficient mice, creating a psoriatic plaque with an immune response showing cytokines from TH1 cells rather than TH2 cells.12 When psoriatic plaques were treated topically with the toll-like receptor 7 agonist imiquimod, aggravation and spreading of the plaques were noted. The exacerbation of psoriasis was accompanied by an induction of lesional TH1-type interferon produced by plasmacytoid dendritic cell (DC) precursors. Plasmacytoid DCs were observed to compose up to 16% of the total dermal infiltrate in psoriatic skin lesions based on their coexpression of BDCA2 and CD123.13 Additionally, cancer patients being treated with interferon alfa experienced induction of psoriasis.14 Moreover, patients being treated for warts with intralesional interferon alfa developed psoriatic plaques in neighboring prior asymptomatic skin.15 Patients with psoriasis who were treated with interferon gamma, a TH1 cytokine type, also developed new plaques correlating with the sites of injection.16
Intralesional T Lymphocytes
Psoriatic lesions contain a host of innate immunocytes, such as APCs, NK cells, and neutrophils, as well as adaptive T cells and an inflammatory infiltrate. These cells include CD4 and CD8 subtypes in which the CD8+ cells predominate in the epidermis, while CD4+ cells show preference for the dermis.17 There are 2 groups of CD8+ cells: one group migrates to the epidermis, expressing the integrin CD103, while the other group is found in the dermis but may be headed to or from the epidermis. The CD8+ cells residing in the epidermis that express the integrin CD103 are capable of interacting with E-cadherin, which enables these cells to travel to the epidermis and bind resident cells. Immunophenotyping reveals that these mature T cells represent chiefly activated memory cells, including CD2+, CD3+, CD5+, CLA, CD28, and CD45RO+.18 Many of these cells express activation markers such as HLA-DR, CD25, and CD27, in addition to the T-cell receptor (TCR).
T-Lymphocyte Stimulation
Both mature CD4+ and CD8+ T cells can respond to the peptides presented by APCs. Although the specific antigen that these T cells are reacting to has not yet been elucidated, several antigenic stimuli have been proposed, including self-proteins, microbial pathogens, and microbial superantigens. The premise that self-reactive T lymphocytes may contribute to the disease process is derived from the molecular mimicry theory in which an exuberant immune response to a pathogen produces cross-reactivity with self-antigens.19 Considering that infections have been associated with the onset of psoriasis, this theory merits consideration. However, it also has been observed that T cells can be activated without antigens or superantigens but rather with direct contact with accessory cells.20 No single theory has clearly emerged. Researchers continue to search for the inciting stimulus that triggers the T lymphocyte and attempt to determine whether T cells are reacting to a self-derived or non–self-derived antigen.
T-Lymphocyte Signaling
T-cell signaling is a highly coordinated process in which T lymphocytes recognize antigens via presentation by mature APCs in the skin rather than the lymphoid tissues. Such APCs expose antigenic peptides via class I or II MHC molecules for which receptors are present on the T-cell surface. The antigen recognition complex at the T-cell and APC interface, in concert with a host of antigen-independent co-stimulatory signals, regulates T-cell signaling and is referred to as the immunologic synapse. The antigen presentation and network of co-stimulatory and adhesion molecules optimize T-cell activation, and dermal DCs release IL-12 and IL-23 to promote a TH1 and TH17 response, respectively. The growth factors released by these helper T cells sustain neoangiogenesis, stimulate epidermal hyperproliferation, alter epidermal differentiation, and decrease susceptibility to apoptosis that characterizes the erythematous hypertrophic scaling lesions of psoriasis.21 Furthermore, the cytokines produced from the immunologic response, such as tumor necrosis factor (TNF) α, IFN-γ, and IL-2, correspond to cytokines that are upregulated in psoriatic plaques.22
Integral components of the immunologic synapse complex include co-stimulatory signals such as CD28, CD40, CD80, and CD86, as well as adhesion molecules such as cytotoxic T-lymphocyte antigen 4 and lymphocyte function-associated antigen (LFA) 1, which possess corresponding receptors on the T cell. These molecules play a key role in T-cell signaling, as their disruption has been shown to decrease T-cell responsiveness and associated inflammation. The B7 family of molecules routinely interacts with CD28 T cells to co-stimulate T-cell activation. Cytotoxic T-lymphocyte antigen 4 immunoglobulin, an antibody on the T-cell surface, targets B7 and interferes with signaling between B7 and CD28. In psoriatic patients, this blockade was demonstrated to attenuate the T-cell response and correlated with a clinical and histological decrease in psoriasiform hyperplasia.23 Biologic therapies that disrupt the LFA-1 component of the immunologic synapse also have demonstrated efficacy in the treatment of psoriasis. Alefacept is a human LFA-3 fusion protein that binds CD2 on T cells and blocks the interaction between LFA-3 on APCs and CD2 on memory CD45RO+ T cells and induces apoptosis of such T cells. Efalizumab is a human monoclonal antibody to the CD11 chain of LFA-1 that blocks the interaction between LFA-1 on the T cell and intercellular adhesion molecule 1 on an APC or endothelial cell. Both alefacept and efalizumab, 2 formerly marketed biologic therapies, demonstrated remarkable clinical reduction of psoriatic lesions, and alefacept has been shown to produce disease remission for up to 18 months after discontinuation of therapy.24-26
NK T Cells
Natural killer T cells represent a subset of CD3+ T cells present in psoriatic plaques. Although NK T cells possess a TCR, they differ from T cells by displaying NK receptors comprised of lectin and immunoglobulin families. These cells exhibit remarkable specificity and are activated upon recognition of glycolipids presented by CD1d molecules. This process occurs in contrast to CD4+ and CD8+ T cells, which, due to their TCR diversity, respond to peptides processed by APCs and displayed on MHC molecules. Natural killer T cells can be classified into 2 subsets: (1) one group that expresses CD4 and preferentially produces TH1- versus TH2-type cytokines, and (2) another group that lacks CD4 and CD8 that only produces TH1-type cytokines. The innate immune system employs NK T cells early in the immune response because of their direct cytotoxicity and rapid production of cytokines such as IFN-γ, which promotes a TH1 inflammatory response, and IL-4, which promotes the development of TH2 cells. Excessive or dysfunctional NK T cells have been associated with autoimmune diseases such as multiple sclerosis and inflammatory bowel disease as well as allergic contact dermatitis.27-29
In psoriasis, NK T cells are located in the epidermis, closely situated to epidermal keratinocytes, which suggests a role for direct antigen presentation. Furthermore, CD1d is overexpressed throughout the epidermis of psoriatic plaques, whereas normally CD1d expression is confined to terminally differentiated keratinocytes. An in vitro study examining cytokine-based inflammation demonstrative of psoriasis treated cultured CD1d-positive keratinocytes with interferon gamma in the presence of alpha-galactosylceramide of the lectin family.30 Interferon gamma was observed to enhance keratinocyte CD1d expression, and subsequently, CD1d-positive keratinocytes were found to activate NK T cells to produce high levels of IFN-γ, while levels of IL-4 remained undetectable. The preferential production of IFN-γ supports a TH1-mediated mechanism regulated by NK T cells in the immunopathogenesis of psoriasis.
Dendritic Cells
Dendritic cells are APCs that process antigens in the tissues in which they reside, after which they migrate to local lymph nodes where they present their native antigens to T cells. This process allows the T-cell response to be tailored to the appropriate antigens in the corresponding tissues. Immature DCs that capture antigens mature by migrating to the T-cell center of the lymph node where they present their antigens to either MHC molecules or the CD1 family. This presentation results in T-cell proliferation and differentiation that correlates with the required type of T-cell response. Multiple subsets of APCs, including myeloid and plasmacytoid DCs, are highly represented in the epidermis and dermis of psoriatic plaques as compared with normal skin.31 Dermal DCs are thought to be responsible for activating both the TH1 and TH17 infiltrate by secreting IL-12 and IL-23, respectively. This mixed cellular response secretes cytokines and leads to a cascade of events involving keratinocytes, fibroblasts, endothelial cells, and neutrophils that create the cutaneous lesions seen in psoriasis.3
Although DCs play a pivotal role in eliciting an immune response against a foreign invader, they also contribute to the establishment of tolerance. Throughout their maturation, DCs are continuously sensing their environment, which shapes their production of TH1- versus TH2-type cytokines and subsequently the nature of the T-cell response. When challenged with a virus, bacteria, or unchecked cell growth, DCs mature into APCs. However, in the absence of a strong stimulus, DCs fail to mature into APCs and present self-peptides with MHC molecules, thereby creating regulatory T cells involved in peripheral tolerance.32 If this balance between immunogenic APCs and housekeeping T cells is upset, inflammatory conditions such as psoriasis can result.
Cytokines
Cytokines are low-molecular-weight glycoproteins that function as signals to produce inflammation, defense, tissue repair and remodeling, fibrosis, angiogenesis, and restriction of neoplastic growth.33 Cytokines are produced by immunocytes such as lymphocytes and macrophages as well as nonimmunocytes such as endothelial cells and keratinocytes. Proinflammatory cytokines include IL-1, IL-2, the IL-17 family, IFN-γ, and TNF-α, while anti-inflammatory cytokines include IL-4 and IL-10. A relative preponderance of TH1 proinflammatory cytokines or an insufficiency of TH2 anti-inflammatory cytokines induces local inflammation and recruitment of additional immunocyte populations, which produce added cytokines.34 A vicious cycle of inflammation occurs that results in cutaneous manifestations such as a plaque. Psoriatic lesions are characterized by a relative increase of TH1-type (eg, IL-2, IFN-γ, TNF-α, TNF-β) to TH2-type (eg, IL-4, IL-5, IL-6, IL-9, IL-10, IL-13) cytokines and an increase in TH17-type cytokines. Natural killer T cells stimulated by CD1d-overexpressing keratinocytes increase production of proinflammatory IFN-γ without effect on the anti-inflammatory IL-4. In addition to the cytokines produced by T cells, APCs produce IL-18, IL-23, and TNF-α found in the inflammatory infiltrate of psoriatic plaques. Both IL-18 and IL-23 stimulate TH1 cells to produce IFN-γ, and IL-23 stimulates TH17 cells. Clearly, a TH1- and TH17-type pattern governs the immune effector cells and their respective cytokines present in psoriatic skin.
Tumor Necrosis Factor α
Although a network of cytokines is responsible for the inflammation of psoriasis, TNF-α has been implicated as a master proinflammatory cytokine of the innate immune response due to its widespread targets and sources. Tumor necrosis factor α is produced by activated T cells, keratinocytes, NK cells, macrophages, monocytes, Langerhans APCs, and endothelial cells. Psoriatic lesions demonstrate high concentrations of TNF-α, while the synovial fluid of psoriatic arthritis patients demonstrates elevated concentrations of TNF-α, IL-1, IL-6, and IL-8.34 In psoriasis, TNF-α supports the expression of adhesion molecules (intercellular adhesion molecule 1 and P- and E-selectin), angiogenesis via vascular endothelial growth factor, the synthesis of proinflammatory molecules (IL-1, IL-6, IL-8, and nuclear factor κβ), and keratinocyte hyperproliferation via vasoactive intestinal peptide.35
A role for TNF-α in psoriasis treatment was serendipitously discovered in a trial for Crohn disease in which infliximab, a mouse-human IgG1 anti–TNF-α monoclonal antibody, was observed to clear psoriatic plaques in a patient with both Crohn disease and psoriasis.36 Immunotherapies that target TNF-α, including infliximab, etanercept, and adalimumab, demonstrate notable efficacy in the treatment of psoriasis.37-39 Tumor necrosis factor α is regarded as the driver of the inflammatory cycle of psoriasis due to its numerous modes of production, capability to amplify other proinflammatory signals, and the efficacy and rapidity with which it produces clinical improvements in psoriasis.
IL-23/TH17 Axis
A new distinct population of helper T cells has been shown to play an important role in psoriasis. These cells develop with the help of IL-23 (secreted by dermal DCs) and subsequently secrete cytokines such as IL-17; they are, therefore, named TH17 cells. CD161 is considered a surface marker for these cells.40 Strong evidence for this IL-23/TH17 axis has been shown in mouse and human models as well as in genetic studies.
IL-23 is a cytokine that shares the p40 subunit with IL-12 and has been linked to autoimmune diseases in both mice and humans.3 It is required for optimal development of TH17 cells41 from a committed CD4+ T-cell population after exposure to transforming growth factor β1 in combination with other proinflammatory cytokines.42,43 IL-23 messenger RNA is produced at higher levels in inflammatory psoriatic skin lesions versus uninvolved skin,44 and intradermal IL-23 injections in mice produced lesions resembling psoriasis macroscopically and microscopically.45 Furthermore, several systemic therapies have been shown to modulate IL-23 levels and correlate with clinical benefit.3 Alterations in the gene for the IL-23 receptor have been shown to be protective for psoriasis,46-48 and the gene coding for the p40 subunit is associated with psoriasis.46,47
Type 17 helper T cells produce a number of cytokines, such as IL-22, IL-17A, IL-17F, and IL-26; the latter 3 are considered to be specific to this lineage.42 IL-22 acts on outer body barrier tissues, such as the skin, and has antimicrobial activity. Blocking the activity of IL-22 in mice prevented the development of skin lesions,49 and psoriasis patients have elevated levels of IL-22 in the skin and blood.50,51 The IL-17 cytokines induce the expression of proinflammatory cytokines, colony-stimulating factors, and chemokines, and they recruit, mobilize, and activate neutrophils.52 IL-17 messenger RNA was found in lesional psoriatic skin but not unaffected skin,53 and cells isolated from the dermis of psoriatic skin have been shown to produce IL-17.54 IL-17A is not elevated in the serum of psoriatic patients (unlike other autoimmune diseases),55 and it is, therefore, thought that TH17 cells and IL-17A production are localized to the affected psoriatic skin. Consistent with this concept is the finding that treatments such as cyclosporin A and anti-TNF agents decrease proinflammatory cytokines in lesional skin but not in the periphery.56-58 These cytokines released by TH17 cells in addition to those released by TH1 cells act on keratinocytes and produce epidermal hyperproliferation, acanthosis, and hyperparakeratosis characteristic of psoriasis.3
New therapies have been developed to target the IL-23/TH17 axis. Ustekinumab is approved for moderate to severe plaque psoriasis. This treatment’s effect may be sustained for up to 3 years, it is generally well tolerated, and it may be useful for patients refractory to anti-TNF therapy such as etanercept.59 Briakinumab, another blocker of IL-12 and IL-23, was studied in phase 3 clinical trials, but its development was discontinued due to safety concerns.60 Newer drugs targeting the IL-23/TH17 axis include secukinumab, ixekizumab, brodalumab, guselkumab, and tildrakizumab.
Genetic Basis of Psoriasis
Psoriasis is a disease of overactive immunity in genetically susceptible individuals. Because patients exhibit varying skin phenotypes, extracutaneous manifestations, and disease courses, multiple genes resulting from linkage disequilibrium are believed to be involved in the pathogenesis of psoriasis. A decade of genome-wide linkage scans have established that PSORS1 is the strongest susceptibility locus demonstrable through family linkage studies; PSORS1 is responsible for up to 50% of the genetic component of psoriasis.61 More recently, HLA-Cw6 has received the most attention as a candidate gene of the PSORS1 susceptibility locus on the MHC class I region on chromosome 6p21.3.62 This gene may function in antigen presentation via MHC class I, which aids in the activation of the overactive T cells characteristic of psoriatic inflammation.
Studies involving the IL-23/TH17 axis have shown genetics to play a role. Individuals may be protected from psoriasis with a nonsynonymous nucleotide substitution in the IL23R gene,47-49 and certain haplotypes of the IL23R gene are associated with the disease47,49 in addition to other autoimmune conditions.
Genomic scans have shown additional susceptibility loci for psoriasis on chromosomes 1q21, 3q21, 4q32-35, 16q12, and 17q25. Two regions on chromosome 17q were recently localized via mapping, which demonstrated a 6 megabase pairs separation, thereby indicating independent linkage factors. Genes SLC9A3R1 and NAT9 are present in the first region, while RAPTOR is demonstrated in the second region.63SLC9A3R1 and NAT9 are players that regulate signal transduction, the immunologic synapse, and T-cell growth. RAPTOR is involved in T-cell function and growth pathways. Using these genes as an example, we can predict that the alterations of regulatory genes, even those yet undetermined, can enhance T-cell proliferation and inflammation manifested in psoriasis.
Conclusion
Psoriasis is a complex disease whereby multiple exogenous and endogenous stimuli incite already heightened innate immune responses in genetically predetermined individuals. The disease process is a result of a network of cell types, including T cells, DCs, and keratinocytes that, with the production of cytokines, generate a chronic inflammatory state. Our understanding of these cellular interactions and cytokines originates from developments, some meticulously planned, others serendipitous, in the fields of immunology, cell and molecular biology, and genetics. Such progress has fostered the creation of targeted immune therapy that has demonstrated remarkable efficacy in psoriasis treatment. Further study of the underlying pathophysiology of psoriasis may provide additional targets for therapy.
- Gottlieb A. Psoriasis. Dis Manag Clin Outcome. 1998;1:195-202.
- Gaspari AA. Innate and adaptive immunity and the pathophysiology of psoriasis. J Am Acad Dermatol. 2006;54(3 suppl 2):S67-S80.
- Di Cesare A, Di Meglio P, Nestle F. The IL-23/Th17 axis in the immunopathogenesis of psoriasis. J Invest Dermatol. 2009;129:1339-1350.
- Barker J. The pathophysiology of psoriasis. Lancet. 1991;338:227-230.
- Nickoloff BJ, Nestle FO. Recent insights into the immunopathogenesis of psoriasis provide new therapeutic opportunities. J Clin Invest. 2004;113:1664-1675.
- Bos J, Meinardi M, van Joost T, et al. Use of cyclosporine in psoriasis. Lancet. 1989;23:1500-1505.
- Khandke L, Krane J, Ashinoff R, et al. Cyclosporine in psoriasis treatment: inhibition of keratinocyte cell-cycle progression in G1 independent effects on transforming growth factor-alpha/epidermal growth factor receptor pathways. Arch Dermatol. 1991;127:1172-1179.
- Gottlieb S, Gilleaudeau P, Johnson R, et al. Response of psoriasis to a lymphocyte-selective toxin (DAB389IL-2) suggests a primary immune, but not keratinocyte, pathogenic basis. Nat Med. 1995;1:442-447.
- Vallat V, Gilleaudeau P, Battat L, et al. PUVA bath therapy strongly suppresses immunological and epidermal activation in psoriasis: a possible cellular basis for remittive therapy. J Exp Med. 1994;180:283-296.
- Gottlieb A, Grossman R, Khandke L, et al. Studies of the effect of cyclosporine in psoriasis in vivo: combined effects on activated T lymphocytes and epidermal regenerative maturation. J Invest Dermatol. 1992;98:302-309.
- Gottlieb S, Hayes E, Gilleaudeau P, et al. Cellular actions of etretinate in psoriasis: enhanced epidermal differentiation and reduced cell-mediated inflammation are unexpected outcomes. J Cutan Pathol. 1996;23:404-418.
- Nickoloff B, Bonish B, Huang B, et al. Characterization of a T cell line bearing natural killer receptors and capable of creating psoriasis in a SCID mouse model system. J Dermatol Sci. 2000;24:212-225.
- Gillet M, Conrad C, Geiges M, et al. Psoriasis triggered by toll-like receptor 7 agonist imiquimod in the presence of dermal plasmacytoid dendritic cell precursors. Arch Dermatol. 2004;140:1490-1495.
- Funk J, Langeland T, Schrumpf E, et al. Psoriasis induced by interferon-alpha. Br J Dermatol. 1991;125:463-465.
- Shiohara T, Kobayahsi M, Abe K, et al. Psoriasis occurring predominantly on warts: possible involvement of interferon alpha. Arch Dermatol. 1988;124:1816-1821.
- Fierlbeck G, Rassner G, Muller C. Psoriasis induced at the injection site of recombinant interferon gamma: results of immunohistologic investigations. Arch Dermatol. 1990;126:351-355.
- Prinz J. The role of T cells in psoriasis. J Eur Acad Dermatol Venereol. 2003;17(suppl):1-5.
- Bos J, de Rie M. The pathogenesis of psoriasis: immunological facts and speculations. Immunol Today. 1999;20:40-46.
- Wucherpfennig KW, Strominger JL. Molecular mimicry in T cell–mediated autoimmunity: viral peptides activate human T cell clones specific for myelin basic protein. Cell. 1995;80:695-705.
- Geginat J, Campagnaro S, Sallusto F, et al. TCR-independent proliferation and differentiation of human CD4+ T cell subsets induced by cytokines. Adv Exp Med Biol. 2002;512:107-112.
- Kastelan M, Massari L, Brajac I. Apoptosis mediated by cytolytic molecules might be responsible for maintenance of psoriatic plaques. Med Hypotheses. 2006;67:336-337.
- Austin L, Ozawa M, Kikuchi T, et al. The majority of epidermal T cells in psoriasis vulgaris lesions can produce type 1 cytokines, interferon-gamma, interleukin-2, and tumor necrosis factor-alpha, defining TC1 (cytotoxic T lymphocyte) and TH1 effector populations: a type 1 differentiation bias is also measured in circulating blood T cells in psoriatic patients. J Invest Dermatol. 1999;113:752-759.
- Abrams J, Kelley S, Hayes E, et al. Blockade of T lymphocyte costimulation with cytotoxic T lymphocyte-associated antigen 4-immunoglobulin (CTLA4Ig) reverses the cellular pathology of psoriatic plagues, including the activation of keratinocytes, dendritic cells and endothelial cells. J Exp Med. 2000;192:681-694.
- Lebwohl M, Christophers E, Langley R, et al. An international, randomized, double-blind, placebo-controlled phase 3 trial of intramuscular alefacept in patients with chronic plaque psoriasis. Arch Dermatol. 2003;139:719-727.
- Krueger G, Ellis C. Alefacept therapy produces remission for patients with chronic plaque psoriasis. Br J Dermatol. 2003;148:784-788.
- Gordon K, Leonardi C, Tyring S, et al. Efalizumab (anti-CD11a) is safe and effective in the treatment of psoriasis: pooled results of the 12-week first treatment period from 2 phase III trials. J Invest Dermatol. 2002;119:242.
- Singh A, Wilson M, Hong S, et al. Natural killer T cell activation protects mice against experimental autoimmune encephalomyelitis. J Exp Med. 2001;194:1801-1811.
- Saubermann L, Beck P, De Jong Y, et al. Activation of natural killer T cells by alpha-glactosylceramide in the presence of CD1d provides protection against colitis in mice. Gastroenterology. 2000;119:119-128.
- Campos R, Szczepanik M, Itakura A, et al. Cutaneous immunization rapidly activates liver invariant Valpha 14 NKT cells stimulating B-1 B cells to initiate T cell recruitment for elicitation of contact sensitivity. J Exp Med. 2003;198:1785-1796.
- Bonish B, Jullien D, Dutronc Y, et al. Overexpression of CD1d by keratinocytes in psoriasis and CD1d-dependent IFN-gamma production by NK-T cells. J Immunol. 2000;165:4076-4085.
- Deguchi M, Aiba S, Ohtani H, et al. Comparison of the distribution and numbers of antigen-presenting cells among T-lymphocyte-mediated dermatoses: CD1a+, factor XIIIa+, and CD68+ cells in eczematous dermatitis, psoriasis, lichen planus and graft-versus-host disease. Arch Dermatol Res. 2002;294:297-302.
- Bos J, de Rie M, Teunissen M, et al. Psoriasis: dysregulation of innate immunity. Br J Dermatol. 2005;152:1098-1107.
- Trefzer U, Hofmann M, Sterry W, et al. Cytokine and anticytokine therapy in dermatology. Expert Opin Biol Ther. 2003;3:733-743.
- Nickoloff B. The cytokine network in psoriasis. Arch Dermatol. 1991;127:871-884.
- Victor F, Gottlieb A. TNF-alpha and apoptosis: implications for the pathogenesis and treatment of psoriasis. J Drugs Dermatol. 2002;3:264-275.
- Oh C, Das K, Gottlieb A. Treatment with anti-tumour necrosis factor alpha (TNF-alpha) monoclonal antibody dramatically decreases the clinical activity of psoriasis lesions. J Am Acad Dermatol. 2000;42:829-830.
- Reich K, Nestle FO, Papp K, et al; EXPRESS study investigators. Infliximab induction and maintenance therapy for moderate-to-severe psoriasis: a phase III, multicentre, double-blind trial. Lancet. 2005;366:1367-1374.
- Leonardi C, Powers J, Matheson R, et al. Etanercept as monotherapy in patients with psoriasis. N Engl J Med. 2003;349:2014-2022.
- Saini R, Tutrone W, Weinberg J. Advances in therapy for psoriasis: an overview of infliximab, etanercept, efalizumab, alefacept, adalimumab, tazarotene, and pimecrolimus. Curr Pharm Des. 2005;11:273-280.
- Cosmi L, De Palma R, Santarlasci V, et al. Human interleukin 17-producing cells originate from a CD161+CD4+ T cell precursor. J Exp Med. 2008;205:1903-1916.
- de Beaucoudrey L, Puel A, Filipe-Santos O, et al. Mutations in STAT3 and IL12RB1 impair the development of human IL-17-producing T cells. J Exp Med. 2008;205:1543-1550.
- Manel N, Unutmaz D, Littman DR. The differentiation of humanT(H)-17 cells requires transforming growth factor-beta and induction of the nuclear receptor RORgammat. Nat Immunol. 2008;9:641-649.
- Yang L, Anderson DE, Baecher-Allan C, et al. IL-21 and TGF-beta are required for differentiation of human T(H)17 cells. Nature. 2008;454:350-352.
- Lee E, Trepicchio WL, Oestreicher JL, et al. Increased expression of interleukin 23 p19 and p40 in lesional skin of patients with psoriasis vulgaris. J Exp Med. 2004;199:125-130.
- Chan JR, Blumenschein W, Murphy E, et al. IL-23 stimulates epidermal hyperplasia via TNF and IL-20R2-dependent mechanisms with implications for psoriasis pathogenesis. J Exp Med. 2006;203:2557-2587.
- Capon F, Di Meglio P, Szaub J, et al. Sequence variants in the genes for the interleukin-23 receptor (IL23R) and its ligand (IL12B) confer protection against psoriasis. Hum Genet. 2007;122:201-206.
- Cargill M, Schrodi SJ, Chang M, et al. A large-scale genetic association study confirms IL12B and leads to the identification of IL23R as psoriasis-risk genes. Am J Hum Genet. 2007;80:273-290.
- Nair RP, Ruether A, Stuart PE, et al. Polymorphisms of the IL12B and IL23R genes are associated with psoriasis. J Invest Dermatol. 2008;128:1653-1661.
- Ma HL, Liang S, Li J, et al. IL-22 is required for Th17 cell-mediated pathology in a mouse model of psoriasis-like skin inflammation. J Clin Invest. 2008;118:597-607.
- Wolk K, Witte E, Wallace E, et al. IL-22 regulates the expression of genes responsible for antimicrobial defense, cellular differentiation, and mobility in keratinocytes: a potential role in psoriasis. Eur J Immunol. 2006;36:1309-1323.
- Boniface K, Guignouard E, Pedretti N, et al. A role for T cell-derived interleukin 22 in psoriatic skin inflammation. Clin Exp Immunol. 2007;150:407-415.
- Weaver CT, Hatton RD, Mangan PR, et al. IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annu Rev Immunol. 2007;25:821-852.
- Teunissen MB, Koomen CW, de Waal Malefyt R, et al. Interleukin-17 and interferon-gamma synergize in the enhancement of proinflammatory cytokine production by human keratinocytes. J Invest Dermatol. 1998;111:645-649.
- Lowes MA, Kikuchi T, Fuentes-Duculan J, et al. Psoriasis vulgaris lesions contain discrete populations of Th1 and Th17 T cells. J Invest Dermatol. 2008;128:1207-1211.
- Arican O, Aral M, Sasmaz S, et al. Serum levels of TNF-alpha, IFN-gamma, IL-6, IL-8, IL-12, IL-17, and IL-18 in patients with active psoriasis and correlation with disease severity. Mediators Inflamm. 2005;2005:273-279.
- Zaba LC, Cardinale I, Gilleaudeau P, et al. Amelioration of epidermal hyperplasia by TNF inhibition is associated with reduced Th17 responses. J Exp Med. 2007;204:3183-3194.
- Haider AS, Cohen J, Fei J, et al. Insights into gene modulation by therapeutic TNF and IFNgamma antibodies: TNF regulates IFNgamma production by T cells and TNF-regulated genes linked to psoriasis transcriptome. J Invest Dermatol. 2008;128:655-666.
- Haider AS, Lowes MA, Suarez-Farinas M, et al. Identification of cellular pathways of “type 1,” Th17 T cells, and TNF- and inducible nitric oxide synthase-producing dendritic cells in autoimmune inflammation through pharmacogenomic study of cyclosporine A in psoriasis. J Immunol. 2008;180:1913-1920.
- Croxtall JD. Ustekinumab: a review of its use in the management of moderate to severe plaque psoriasis. Drugs. 2011;71:1733-1753.
- Gordon KB, Langely RG, Gottlieb AB, et al. A phase III, randomized, controlled trial of the fully human IL-12/23 mAb briakinumab in moderate-to-severe psoriasis. J Invest Dermatol. 2012;132:304-314.
- Rahman P, Elder JT. Genetic epidemiology of psoriasis and psoriatic arthritis. Ann Rheum Dis. 2005;64(suppl 2):ii37-ii39.
- Elder JT. PSORS1: linking genetics and immunology. J Invest Dermatol. 2006;126:1205-1206.
- Krueger JG, Bowcock A. Psoriasis pathophysiology: current concepts of pathogenesis. Ann Rheum Dis. 2005;64(suppl 2):ii30-ii36.
Increased understanding of the pathophysiology of psoriasis has been one of the driving forces in the development of new therapies. An understanding of the processes involved is important in the optimal management of the disease. The last 30 years of research and clinical practice have revolutionized our understanding of the pathogenesis of psoriasis as the dysregulation of immunity triggered by environmental and genetic stimuli. Psoriasis was originally regarded as a primary disorder of epidermal hyperproliferation. However, experimental models and clinical results from immunomodulating therapies have refined this perspective in conceptualizing psoriasis as a genetically programmed pathologic interaction among resident skin cells; infiltrating immunocytes; and a host of proinflammatory cytokines, chemokines, and growth factors produced by these immunocytes. Two populations of immunocytes and their respective signaling molecules collaborate in the pathogenesis: (1) innate immunocytes, mediated by antigen-presenting cells (APCs)(including natural killer [NK] T lymphocytes, Langerhans cells, and neutrophils), and (2) acquired or adaptive immunocytes, mediated by mature CD4+ and CD8+ T lymphocytes in the skin. Such dysregulation of immunity and subsequent inflammation is responsible for the development and perpetuation of the clinical plaques and histological inflammatory infiltrate characteristic of psoriasis.
Although psoriasis is considered to be an immune-mediated disease in which intralesional T lymphocytes and their proinflammatory signals trigger primed basal layer keratinocytes to rapidly proliferate, debate and research focus on the stimulus that incites this inflammatory process. Our current understanding considers psoriasis to be triggered by exogenous or endogenous environmental stimuli in genetically susceptible individuals. Such stimuli include group A streptococcal pharyngitis, viremia, allergic drug reactions, antimalarial drugs, lithium, beta-blockers, IFN-α, withdrawal of systemic corticosteroids, local trauma (Köbner phenomenon), and emotional stress. These stimuli correlate with the onset or flares of psoriatic lesions. Psoriasis genetics centers on susceptibility loci and corresponding candidate genes, particularly the psoriasis susceptibility (PSORS) 1 locus on the major histocompatibility complex (MHC) class I region. Current research on the pathogenesis of psoriasis examines the complex interactions among immunologic mechanisms, environmental stimuli, and genetic susceptibility. After discussing the clinical presentation and histopathologic features of psoriasis, we will review the pathophysiology of psoriasis through noteworthy developments, including serendipitous observations, reactions to therapies, clinical trials, and animal model systems that have shaped our view of the disease process. In addition to the classic skin lesions, approximately 23% of psoriasis patients develop psoriatic arthritis, with a 10-year latency after diagnosis of psoriasis.1
Principles of Immunity
The immune system, intended to protect its host from foreign invaders and unregulated cell growth, employs 2 main effector pathways—the innate and the acquired (or adaptive) immune responses—both of which contribute to the pathophysiology of psoriasis.2 Innate immunity responses occur within minutes to hours of antigen exposure but fail to develop memory for when the antigen is encountered again. However, adaptive immunity responses take days to weeks to respond after challenged with an antigen. The adaptive immune cells have the capacity to respond to a greater range of antigens and develop immunologic memory via rearrangement of antigen receptors on B and T cells. These specialized B and T cells can then be promptly mobilized and differentiated into mature effector cells that protect the host from a foreign pathogen.
Innate and adaptive immune responses are highly intertwined; they can initiate, perpetuate, and terminate the immune mechanisms responsible for inflammation. They can modify the nature of the immune response by altering the relative proportions of type 1 (TH1), type 2 (TH2), and the more recently discovered type 17 (TH17) subset of helper T cells and their respective signaling molecules. A TH1 response is essential for a cellular immunologic reaction to intracellular bacteria and viruses or cellular immunity. A TH2 response promotes IgE synthesis, eosinophilia, and mast cell maturation for extracellular parasites and helminthes as well as humoral immunity, while a TH17 response is important for cell-mediated immunity to extracellular bacteria and plays a role in autoimmunity.3 The innate and adaptive immune responses employ common effector molecules such as chemokines and cytokines, which are essential in mediating an immune response.
Implicating Dysregulation of Immunity
Our present appreciation of the pathogenesis of psoriasis is based on the history of trial-and-error therapies; serendipitous discoveries; and the current immune targeting drugs used in a variety of chronic inflammatory conditions, including rheumatoid arthritis, ankylosing spondylitis, and inflammatory bowel disease. Before the mid-1980s, research focused on the hyperproliferative epidermal cells as the primary pathology because a markedly thickened epidermis was indeed demonstrated on histologic specimens. Altered cell-cycle kinetics were thought to be the culprit behind the hyperkeratotic plaques. Thus, initial treatments centered on oncologic and antimitotic therapies used to arrest keratinocyte proliferation with agents such as arsenic, ammoniated mercury, and methotrexate.4
However, a paradigm shift from targeting epidermal keratinocytes to immunocyte populations was recognized when a patient receiving cyclosporine to prevent transplant rejection noted clearing of psoriatic lesions in the 1980s.5 Cyclosporine was observed to inhibit messenger RNA transcription of T-cell cytokines, thereby implicating immunologic dysregulation, specifically T-cell hyperactivity, in the pathogenesis of psoriasis.6 However, the concentrations of oral cyclosporine reached in the epidermis exerted direct effects on keratinocyte proliferation and lymphocyte function in these patients.7 Thus, the question was raised as to whether the keratinocytes or the lymphocytes drove the psoriatic plaques. The use of an IL-2 diphtheria toxin-fusion protein, denileukin diftitox, specific for activated T cells with high-affinity IL-2 receptors and nonreactive with keratinocytes, distinguished which cell type was responsible. This targeted T-cell toxin provided clinical and histological clearing of psoriatic plaques. Thus, T lymphocytes rather than keratinocytes were recognized as the definitive driver behind the psoriatic plaques.8
Additional studies have demonstrated that treatments that induce prolonged clearing of psoriatic lesions without continuous therapy, such as psoralen plus UVA irradiation, decreased the numbers of T cells in plaques by at least 90%.9 However, treatments that require continual therapy for satisfactory clinical results, such as cyclosporine and etretinate, simply suppress T-cell activity and proliferation.10,11 Further evidence has linked cellular immunity with the pathogenesis of psoriasis, defining it as a TH1-type disease. Natural killer T cells were shown to be involved through the use of a severe combined immunodeficient mouse model. They were injected into prepsoriatic skin grafted on immunodeficient mice, creating a psoriatic plaque with an immune response showing cytokines from TH1 cells rather than TH2 cells.12 When psoriatic plaques were treated topically with the toll-like receptor 7 agonist imiquimod, aggravation and spreading of the plaques were noted. The exacerbation of psoriasis was accompanied by an induction of lesional TH1-type interferon produced by plasmacytoid dendritic cell (DC) precursors. Plasmacytoid DCs were observed to compose up to 16% of the total dermal infiltrate in psoriatic skin lesions based on their coexpression of BDCA2 and CD123.13 Additionally, cancer patients being treated with interferon alfa experienced induction of psoriasis.14 Moreover, patients being treated for warts with intralesional interferon alfa developed psoriatic plaques in neighboring prior asymptomatic skin.15 Patients with psoriasis who were treated with interferon gamma, a TH1 cytokine type, also developed new plaques correlating with the sites of injection.16
Intralesional T Lymphocytes
Psoriatic lesions contain a host of innate immunocytes, such as APCs, NK cells, and neutrophils, as well as adaptive T cells and an inflammatory infiltrate. These cells include CD4 and CD8 subtypes in which the CD8+ cells predominate in the epidermis, while CD4+ cells show preference for the dermis.17 There are 2 groups of CD8+ cells: one group migrates to the epidermis, expressing the integrin CD103, while the other group is found in the dermis but may be headed to or from the epidermis. The CD8+ cells residing in the epidermis that express the integrin CD103 are capable of interacting with E-cadherin, which enables these cells to travel to the epidermis and bind resident cells. Immunophenotyping reveals that these mature T cells represent chiefly activated memory cells, including CD2+, CD3+, CD5+, CLA, CD28, and CD45RO+.18 Many of these cells express activation markers such as HLA-DR, CD25, and CD27, in addition to the T-cell receptor (TCR).
T-Lymphocyte Stimulation
Both mature CD4+ and CD8+ T cells can respond to the peptides presented by APCs. Although the specific antigen that these T cells are reacting to has not yet been elucidated, several antigenic stimuli have been proposed, including self-proteins, microbial pathogens, and microbial superantigens. The premise that self-reactive T lymphocytes may contribute to the disease process is derived from the molecular mimicry theory in which an exuberant immune response to a pathogen produces cross-reactivity with self-antigens.19 Considering that infections have been associated with the onset of psoriasis, this theory merits consideration. However, it also has been observed that T cells can be activated without antigens or superantigens but rather with direct contact with accessory cells.20 No single theory has clearly emerged. Researchers continue to search for the inciting stimulus that triggers the T lymphocyte and attempt to determine whether T cells are reacting to a self-derived or non–self-derived antigen.
T-Lymphocyte Signaling
T-cell signaling is a highly coordinated process in which T lymphocytes recognize antigens via presentation by mature APCs in the skin rather than the lymphoid tissues. Such APCs expose antigenic peptides via class I or II MHC molecules for which receptors are present on the T-cell surface. The antigen recognition complex at the T-cell and APC interface, in concert with a host of antigen-independent co-stimulatory signals, regulates T-cell signaling and is referred to as the immunologic synapse. The antigen presentation and network of co-stimulatory and adhesion molecules optimize T-cell activation, and dermal DCs release IL-12 and IL-23 to promote a TH1 and TH17 response, respectively. The growth factors released by these helper T cells sustain neoangiogenesis, stimulate epidermal hyperproliferation, alter epidermal differentiation, and decrease susceptibility to apoptosis that characterizes the erythematous hypertrophic scaling lesions of psoriasis.21 Furthermore, the cytokines produced from the immunologic response, such as tumor necrosis factor (TNF) α, IFN-γ, and IL-2, correspond to cytokines that are upregulated in psoriatic plaques.22
Integral components of the immunologic synapse complex include co-stimulatory signals such as CD28, CD40, CD80, and CD86, as well as adhesion molecules such as cytotoxic T-lymphocyte antigen 4 and lymphocyte function-associated antigen (LFA) 1, which possess corresponding receptors on the T cell. These molecules play a key role in T-cell signaling, as their disruption has been shown to decrease T-cell responsiveness and associated inflammation. The B7 family of molecules routinely interacts with CD28 T cells to co-stimulate T-cell activation. Cytotoxic T-lymphocyte antigen 4 immunoglobulin, an antibody on the T-cell surface, targets B7 and interferes with signaling between B7 and CD28. In psoriatic patients, this blockade was demonstrated to attenuate the T-cell response and correlated with a clinical and histological decrease in psoriasiform hyperplasia.23 Biologic therapies that disrupt the LFA-1 component of the immunologic synapse also have demonstrated efficacy in the treatment of psoriasis. Alefacept is a human LFA-3 fusion protein that binds CD2 on T cells and blocks the interaction between LFA-3 on APCs and CD2 on memory CD45RO+ T cells and induces apoptosis of such T cells. Efalizumab is a human monoclonal antibody to the CD11 chain of LFA-1 that blocks the interaction between LFA-1 on the T cell and intercellular adhesion molecule 1 on an APC or endothelial cell. Both alefacept and efalizumab, 2 formerly marketed biologic therapies, demonstrated remarkable clinical reduction of psoriatic lesions, and alefacept has been shown to produce disease remission for up to 18 months after discontinuation of therapy.24-26
NK T Cells
Natural killer T cells represent a subset of CD3+ T cells present in psoriatic plaques. Although NK T cells possess a TCR, they differ from T cells by displaying NK receptors comprised of lectin and immunoglobulin families. These cells exhibit remarkable specificity and are activated upon recognition of glycolipids presented by CD1d molecules. This process occurs in contrast to CD4+ and CD8+ T cells, which, due to their TCR diversity, respond to peptides processed by APCs and displayed on MHC molecules. Natural killer T cells can be classified into 2 subsets: (1) one group that expresses CD4 and preferentially produces TH1- versus TH2-type cytokines, and (2) another group that lacks CD4 and CD8 that only produces TH1-type cytokines. The innate immune system employs NK T cells early in the immune response because of their direct cytotoxicity and rapid production of cytokines such as IFN-γ, which promotes a TH1 inflammatory response, and IL-4, which promotes the development of TH2 cells. Excessive or dysfunctional NK T cells have been associated with autoimmune diseases such as multiple sclerosis and inflammatory bowel disease as well as allergic contact dermatitis.27-29
In psoriasis, NK T cells are located in the epidermis, closely situated to epidermal keratinocytes, which suggests a role for direct antigen presentation. Furthermore, CD1d is overexpressed throughout the epidermis of psoriatic plaques, whereas normally CD1d expression is confined to terminally differentiated keratinocytes. An in vitro study examining cytokine-based inflammation demonstrative of psoriasis treated cultured CD1d-positive keratinocytes with interferon gamma in the presence of alpha-galactosylceramide of the lectin family.30 Interferon gamma was observed to enhance keratinocyte CD1d expression, and subsequently, CD1d-positive keratinocytes were found to activate NK T cells to produce high levels of IFN-γ, while levels of IL-4 remained undetectable. The preferential production of IFN-γ supports a TH1-mediated mechanism regulated by NK T cells in the immunopathogenesis of psoriasis.
Dendritic Cells
Dendritic cells are APCs that process antigens in the tissues in which they reside, after which they migrate to local lymph nodes where they present their native antigens to T cells. This process allows the T-cell response to be tailored to the appropriate antigens in the corresponding tissues. Immature DCs that capture antigens mature by migrating to the T-cell center of the lymph node where they present their antigens to either MHC molecules or the CD1 family. This presentation results in T-cell proliferation and differentiation that correlates with the required type of T-cell response. Multiple subsets of APCs, including myeloid and plasmacytoid DCs, are highly represented in the epidermis and dermis of psoriatic plaques as compared with normal skin.31 Dermal DCs are thought to be responsible for activating both the TH1 and TH17 infiltrate by secreting IL-12 and IL-23, respectively. This mixed cellular response secretes cytokines and leads to a cascade of events involving keratinocytes, fibroblasts, endothelial cells, and neutrophils that create the cutaneous lesions seen in psoriasis.3
Although DCs play a pivotal role in eliciting an immune response against a foreign invader, they also contribute to the establishment of tolerance. Throughout their maturation, DCs are continuously sensing their environment, which shapes their production of TH1- versus TH2-type cytokines and subsequently the nature of the T-cell response. When challenged with a virus, bacteria, or unchecked cell growth, DCs mature into APCs. However, in the absence of a strong stimulus, DCs fail to mature into APCs and present self-peptides with MHC molecules, thereby creating regulatory T cells involved in peripheral tolerance.32 If this balance between immunogenic APCs and housekeeping T cells is upset, inflammatory conditions such as psoriasis can result.
Cytokines
Cytokines are low-molecular-weight glycoproteins that function as signals to produce inflammation, defense, tissue repair and remodeling, fibrosis, angiogenesis, and restriction of neoplastic growth.33 Cytokines are produced by immunocytes such as lymphocytes and macrophages as well as nonimmunocytes such as endothelial cells and keratinocytes. Proinflammatory cytokines include IL-1, IL-2, the IL-17 family, IFN-γ, and TNF-α, while anti-inflammatory cytokines include IL-4 and IL-10. A relative preponderance of TH1 proinflammatory cytokines or an insufficiency of TH2 anti-inflammatory cytokines induces local inflammation and recruitment of additional immunocyte populations, which produce added cytokines.34 A vicious cycle of inflammation occurs that results in cutaneous manifestations such as a plaque. Psoriatic lesions are characterized by a relative increase of TH1-type (eg, IL-2, IFN-γ, TNF-α, TNF-β) to TH2-type (eg, IL-4, IL-5, IL-6, IL-9, IL-10, IL-13) cytokines and an increase in TH17-type cytokines. Natural killer T cells stimulated by CD1d-overexpressing keratinocytes increase production of proinflammatory IFN-γ without effect on the anti-inflammatory IL-4. In addition to the cytokines produced by T cells, APCs produce IL-18, IL-23, and TNF-α found in the inflammatory infiltrate of psoriatic plaques. Both IL-18 and IL-23 stimulate TH1 cells to produce IFN-γ, and IL-23 stimulates TH17 cells. Clearly, a TH1- and TH17-type pattern governs the immune effector cells and their respective cytokines present in psoriatic skin.
Tumor Necrosis Factor α
Although a network of cytokines is responsible for the inflammation of psoriasis, TNF-α has been implicated as a master proinflammatory cytokine of the innate immune response due to its widespread targets and sources. Tumor necrosis factor α is produced by activated T cells, keratinocytes, NK cells, macrophages, monocytes, Langerhans APCs, and endothelial cells. Psoriatic lesions demonstrate high concentrations of TNF-α, while the synovial fluid of psoriatic arthritis patients demonstrates elevated concentrations of TNF-α, IL-1, IL-6, and IL-8.34 In psoriasis, TNF-α supports the expression of adhesion molecules (intercellular adhesion molecule 1 and P- and E-selectin), angiogenesis via vascular endothelial growth factor, the synthesis of proinflammatory molecules (IL-1, IL-6, IL-8, and nuclear factor κβ), and keratinocyte hyperproliferation via vasoactive intestinal peptide.35
A role for TNF-α in psoriasis treatment was serendipitously discovered in a trial for Crohn disease in which infliximab, a mouse-human IgG1 anti–TNF-α monoclonal antibody, was observed to clear psoriatic plaques in a patient with both Crohn disease and psoriasis.36 Immunotherapies that target TNF-α, including infliximab, etanercept, and adalimumab, demonstrate notable efficacy in the treatment of psoriasis.37-39 Tumor necrosis factor α is regarded as the driver of the inflammatory cycle of psoriasis due to its numerous modes of production, capability to amplify other proinflammatory signals, and the efficacy and rapidity with which it produces clinical improvements in psoriasis.
IL-23/TH17 Axis
A new distinct population of helper T cells has been shown to play an important role in psoriasis. These cells develop with the help of IL-23 (secreted by dermal DCs) and subsequently secrete cytokines such as IL-17; they are, therefore, named TH17 cells. CD161 is considered a surface marker for these cells.40 Strong evidence for this IL-23/TH17 axis has been shown in mouse and human models as well as in genetic studies.
IL-23 is a cytokine that shares the p40 subunit with IL-12 and has been linked to autoimmune diseases in both mice and humans.3 It is required for optimal development of TH17 cells41 from a committed CD4+ T-cell population after exposure to transforming growth factor β1 in combination with other proinflammatory cytokines.42,43 IL-23 messenger RNA is produced at higher levels in inflammatory psoriatic skin lesions versus uninvolved skin,44 and intradermal IL-23 injections in mice produced lesions resembling psoriasis macroscopically and microscopically.45 Furthermore, several systemic therapies have been shown to modulate IL-23 levels and correlate with clinical benefit.3 Alterations in the gene for the IL-23 receptor have been shown to be protective for psoriasis,46-48 and the gene coding for the p40 subunit is associated with psoriasis.46,47
Type 17 helper T cells produce a number of cytokines, such as IL-22, IL-17A, IL-17F, and IL-26; the latter 3 are considered to be specific to this lineage.42 IL-22 acts on outer body barrier tissues, such as the skin, and has antimicrobial activity. Blocking the activity of IL-22 in mice prevented the development of skin lesions,49 and psoriasis patients have elevated levels of IL-22 in the skin and blood.50,51 The IL-17 cytokines induce the expression of proinflammatory cytokines, colony-stimulating factors, and chemokines, and they recruit, mobilize, and activate neutrophils.52 IL-17 messenger RNA was found in lesional psoriatic skin but not unaffected skin,53 and cells isolated from the dermis of psoriatic skin have been shown to produce IL-17.54 IL-17A is not elevated in the serum of psoriatic patients (unlike other autoimmune diseases),55 and it is, therefore, thought that TH17 cells and IL-17A production are localized to the affected psoriatic skin. Consistent with this concept is the finding that treatments such as cyclosporin A and anti-TNF agents decrease proinflammatory cytokines in lesional skin but not in the periphery.56-58 These cytokines released by TH17 cells in addition to those released by TH1 cells act on keratinocytes and produce epidermal hyperproliferation, acanthosis, and hyperparakeratosis characteristic of psoriasis.3
New therapies have been developed to target the IL-23/TH17 axis. Ustekinumab is approved for moderate to severe plaque psoriasis. This treatment’s effect may be sustained for up to 3 years, it is generally well tolerated, and it may be useful for patients refractory to anti-TNF therapy such as etanercept.59 Briakinumab, another blocker of IL-12 and IL-23, was studied in phase 3 clinical trials, but its development was discontinued due to safety concerns.60 Newer drugs targeting the IL-23/TH17 axis include secukinumab, ixekizumab, brodalumab, guselkumab, and tildrakizumab.
Genetic Basis of Psoriasis
Psoriasis is a disease of overactive immunity in genetically susceptible individuals. Because patients exhibit varying skin phenotypes, extracutaneous manifestations, and disease courses, multiple genes resulting from linkage disequilibrium are believed to be involved in the pathogenesis of psoriasis. A decade of genome-wide linkage scans have established that PSORS1 is the strongest susceptibility locus demonstrable through family linkage studies; PSORS1 is responsible for up to 50% of the genetic component of psoriasis.61 More recently, HLA-Cw6 has received the most attention as a candidate gene of the PSORS1 susceptibility locus on the MHC class I region on chromosome 6p21.3.62 This gene may function in antigen presentation via MHC class I, which aids in the activation of the overactive T cells characteristic of psoriatic inflammation.
Studies involving the IL-23/TH17 axis have shown genetics to play a role. Individuals may be protected from psoriasis with a nonsynonymous nucleotide substitution in the IL23R gene,47-49 and certain haplotypes of the IL23R gene are associated with the disease47,49 in addition to other autoimmune conditions.
Genomic scans have shown additional susceptibility loci for psoriasis on chromosomes 1q21, 3q21, 4q32-35, 16q12, and 17q25. Two regions on chromosome 17q were recently localized via mapping, which demonstrated a 6 megabase pairs separation, thereby indicating independent linkage factors. Genes SLC9A3R1 and NAT9 are present in the first region, while RAPTOR is demonstrated in the second region.63SLC9A3R1 and NAT9 are players that regulate signal transduction, the immunologic synapse, and T-cell growth. RAPTOR is involved in T-cell function and growth pathways. Using these genes as an example, we can predict that the alterations of regulatory genes, even those yet undetermined, can enhance T-cell proliferation and inflammation manifested in psoriasis.
Conclusion
Psoriasis is a complex disease whereby multiple exogenous and endogenous stimuli incite already heightened innate immune responses in genetically predetermined individuals. The disease process is a result of a network of cell types, including T cells, DCs, and keratinocytes that, with the production of cytokines, generate a chronic inflammatory state. Our understanding of these cellular interactions and cytokines originates from developments, some meticulously planned, others serendipitous, in the fields of immunology, cell and molecular biology, and genetics. Such progress has fostered the creation of targeted immune therapy that has demonstrated remarkable efficacy in psoriasis treatment. Further study of the underlying pathophysiology of psoriasis may provide additional targets for therapy.
Increased understanding of the pathophysiology of psoriasis has been one of the driving forces in the development of new therapies. An understanding of the processes involved is important in the optimal management of the disease. The last 30 years of research and clinical practice have revolutionized our understanding of the pathogenesis of psoriasis as the dysregulation of immunity triggered by environmental and genetic stimuli. Psoriasis was originally regarded as a primary disorder of epidermal hyperproliferation. However, experimental models and clinical results from immunomodulating therapies have refined this perspective in conceptualizing psoriasis as a genetically programmed pathologic interaction among resident skin cells; infiltrating immunocytes; and a host of proinflammatory cytokines, chemokines, and growth factors produced by these immunocytes. Two populations of immunocytes and their respective signaling molecules collaborate in the pathogenesis: (1) innate immunocytes, mediated by antigen-presenting cells (APCs)(including natural killer [NK] T lymphocytes, Langerhans cells, and neutrophils), and (2) acquired or adaptive immunocytes, mediated by mature CD4+ and CD8+ T lymphocytes in the skin. Such dysregulation of immunity and subsequent inflammation is responsible for the development and perpetuation of the clinical plaques and histological inflammatory infiltrate characteristic of psoriasis.
Although psoriasis is considered to be an immune-mediated disease in which intralesional T lymphocytes and their proinflammatory signals trigger primed basal layer keratinocytes to rapidly proliferate, debate and research focus on the stimulus that incites this inflammatory process. Our current understanding considers psoriasis to be triggered by exogenous or endogenous environmental stimuli in genetically susceptible individuals. Such stimuli include group A streptococcal pharyngitis, viremia, allergic drug reactions, antimalarial drugs, lithium, beta-blockers, IFN-α, withdrawal of systemic corticosteroids, local trauma (Köbner phenomenon), and emotional stress. These stimuli correlate with the onset or flares of psoriatic lesions. Psoriasis genetics centers on susceptibility loci and corresponding candidate genes, particularly the psoriasis susceptibility (PSORS) 1 locus on the major histocompatibility complex (MHC) class I region. Current research on the pathogenesis of psoriasis examines the complex interactions among immunologic mechanisms, environmental stimuli, and genetic susceptibility. After discussing the clinical presentation and histopathologic features of psoriasis, we will review the pathophysiology of psoriasis through noteworthy developments, including serendipitous observations, reactions to therapies, clinical trials, and animal model systems that have shaped our view of the disease process. In addition to the classic skin lesions, approximately 23% of psoriasis patients develop psoriatic arthritis, with a 10-year latency after diagnosis of psoriasis.1
Principles of Immunity
The immune system, intended to protect its host from foreign invaders and unregulated cell growth, employs 2 main effector pathways—the innate and the acquired (or adaptive) immune responses—both of which contribute to the pathophysiology of psoriasis.2 Innate immunity responses occur within minutes to hours of antigen exposure but fail to develop memory for when the antigen is encountered again. However, adaptive immunity responses take days to weeks to respond after challenged with an antigen. The adaptive immune cells have the capacity to respond to a greater range of antigens and develop immunologic memory via rearrangement of antigen receptors on B and T cells. These specialized B and T cells can then be promptly mobilized and differentiated into mature effector cells that protect the host from a foreign pathogen.
Innate and adaptive immune responses are highly intertwined; they can initiate, perpetuate, and terminate the immune mechanisms responsible for inflammation. They can modify the nature of the immune response by altering the relative proportions of type 1 (TH1), type 2 (TH2), and the more recently discovered type 17 (TH17) subset of helper T cells and their respective signaling molecules. A TH1 response is essential for a cellular immunologic reaction to intracellular bacteria and viruses or cellular immunity. A TH2 response promotes IgE synthesis, eosinophilia, and mast cell maturation for extracellular parasites and helminthes as well as humoral immunity, while a TH17 response is important for cell-mediated immunity to extracellular bacteria and plays a role in autoimmunity.3 The innate and adaptive immune responses employ common effector molecules such as chemokines and cytokines, which are essential in mediating an immune response.
Implicating Dysregulation of Immunity
Our present appreciation of the pathogenesis of psoriasis is based on the history of trial-and-error therapies; serendipitous discoveries; and the current immune targeting drugs used in a variety of chronic inflammatory conditions, including rheumatoid arthritis, ankylosing spondylitis, and inflammatory bowel disease. Before the mid-1980s, research focused on the hyperproliferative epidermal cells as the primary pathology because a markedly thickened epidermis was indeed demonstrated on histologic specimens. Altered cell-cycle kinetics were thought to be the culprit behind the hyperkeratotic plaques. Thus, initial treatments centered on oncologic and antimitotic therapies used to arrest keratinocyte proliferation with agents such as arsenic, ammoniated mercury, and methotrexate.4
However, a paradigm shift from targeting epidermal keratinocytes to immunocyte populations was recognized when a patient receiving cyclosporine to prevent transplant rejection noted clearing of psoriatic lesions in the 1980s.5 Cyclosporine was observed to inhibit messenger RNA transcription of T-cell cytokines, thereby implicating immunologic dysregulation, specifically T-cell hyperactivity, in the pathogenesis of psoriasis.6 However, the concentrations of oral cyclosporine reached in the epidermis exerted direct effects on keratinocyte proliferation and lymphocyte function in these patients.7 Thus, the question was raised as to whether the keratinocytes or the lymphocytes drove the psoriatic plaques. The use of an IL-2 diphtheria toxin-fusion protein, denileukin diftitox, specific for activated T cells with high-affinity IL-2 receptors and nonreactive with keratinocytes, distinguished which cell type was responsible. This targeted T-cell toxin provided clinical and histological clearing of psoriatic plaques. Thus, T lymphocytes rather than keratinocytes were recognized as the definitive driver behind the psoriatic plaques.8
Additional studies have demonstrated that treatments that induce prolonged clearing of psoriatic lesions without continuous therapy, such as psoralen plus UVA irradiation, decreased the numbers of T cells in plaques by at least 90%.9 However, treatments that require continual therapy for satisfactory clinical results, such as cyclosporine and etretinate, simply suppress T-cell activity and proliferation.10,11 Further evidence has linked cellular immunity with the pathogenesis of psoriasis, defining it as a TH1-type disease. Natural killer T cells were shown to be involved through the use of a severe combined immunodeficient mouse model. They were injected into prepsoriatic skin grafted on immunodeficient mice, creating a psoriatic plaque with an immune response showing cytokines from TH1 cells rather than TH2 cells.12 When psoriatic plaques were treated topically with the toll-like receptor 7 agonist imiquimod, aggravation and spreading of the plaques were noted. The exacerbation of psoriasis was accompanied by an induction of lesional TH1-type interferon produced by plasmacytoid dendritic cell (DC) precursors. Plasmacytoid DCs were observed to compose up to 16% of the total dermal infiltrate in psoriatic skin lesions based on their coexpression of BDCA2 and CD123.13 Additionally, cancer patients being treated with interferon alfa experienced induction of psoriasis.14 Moreover, patients being treated for warts with intralesional interferon alfa developed psoriatic plaques in neighboring prior asymptomatic skin.15 Patients with psoriasis who were treated with interferon gamma, a TH1 cytokine type, also developed new plaques correlating with the sites of injection.16
Intralesional T Lymphocytes
Psoriatic lesions contain a host of innate immunocytes, such as APCs, NK cells, and neutrophils, as well as adaptive T cells and an inflammatory infiltrate. These cells include CD4 and CD8 subtypes in which the CD8+ cells predominate in the epidermis, while CD4+ cells show preference for the dermis.17 There are 2 groups of CD8+ cells: one group migrates to the epidermis, expressing the integrin CD103, while the other group is found in the dermis but may be headed to or from the epidermis. The CD8+ cells residing in the epidermis that express the integrin CD103 are capable of interacting with E-cadherin, which enables these cells to travel to the epidermis and bind resident cells. Immunophenotyping reveals that these mature T cells represent chiefly activated memory cells, including CD2+, CD3+, CD5+, CLA, CD28, and CD45RO+.18 Many of these cells express activation markers such as HLA-DR, CD25, and CD27, in addition to the T-cell receptor (TCR).
T-Lymphocyte Stimulation
Both mature CD4+ and CD8+ T cells can respond to the peptides presented by APCs. Although the specific antigen that these T cells are reacting to has not yet been elucidated, several antigenic stimuli have been proposed, including self-proteins, microbial pathogens, and microbial superantigens. The premise that self-reactive T lymphocytes may contribute to the disease process is derived from the molecular mimicry theory in which an exuberant immune response to a pathogen produces cross-reactivity with self-antigens.19 Considering that infections have been associated with the onset of psoriasis, this theory merits consideration. However, it also has been observed that T cells can be activated without antigens or superantigens but rather with direct contact with accessory cells.20 No single theory has clearly emerged. Researchers continue to search for the inciting stimulus that triggers the T lymphocyte and attempt to determine whether T cells are reacting to a self-derived or non–self-derived antigen.
T-Lymphocyte Signaling
T-cell signaling is a highly coordinated process in which T lymphocytes recognize antigens via presentation by mature APCs in the skin rather than the lymphoid tissues. Such APCs expose antigenic peptides via class I or II MHC molecules for which receptors are present on the T-cell surface. The antigen recognition complex at the T-cell and APC interface, in concert with a host of antigen-independent co-stimulatory signals, regulates T-cell signaling and is referred to as the immunologic synapse. The antigen presentation and network of co-stimulatory and adhesion molecules optimize T-cell activation, and dermal DCs release IL-12 and IL-23 to promote a TH1 and TH17 response, respectively. The growth factors released by these helper T cells sustain neoangiogenesis, stimulate epidermal hyperproliferation, alter epidermal differentiation, and decrease susceptibility to apoptosis that characterizes the erythematous hypertrophic scaling lesions of psoriasis.21 Furthermore, the cytokines produced from the immunologic response, such as tumor necrosis factor (TNF) α, IFN-γ, and IL-2, correspond to cytokines that are upregulated in psoriatic plaques.22
Integral components of the immunologic synapse complex include co-stimulatory signals such as CD28, CD40, CD80, and CD86, as well as adhesion molecules such as cytotoxic T-lymphocyte antigen 4 and lymphocyte function-associated antigen (LFA) 1, which possess corresponding receptors on the T cell. These molecules play a key role in T-cell signaling, as their disruption has been shown to decrease T-cell responsiveness and associated inflammation. The B7 family of molecules routinely interacts with CD28 T cells to co-stimulate T-cell activation. Cytotoxic T-lymphocyte antigen 4 immunoglobulin, an antibody on the T-cell surface, targets B7 and interferes with signaling between B7 and CD28. In psoriatic patients, this blockade was demonstrated to attenuate the T-cell response and correlated with a clinical and histological decrease in psoriasiform hyperplasia.23 Biologic therapies that disrupt the LFA-1 component of the immunologic synapse also have demonstrated efficacy in the treatment of psoriasis. Alefacept is a human LFA-3 fusion protein that binds CD2 on T cells and blocks the interaction between LFA-3 on APCs and CD2 on memory CD45RO+ T cells and induces apoptosis of such T cells. Efalizumab is a human monoclonal antibody to the CD11 chain of LFA-1 that blocks the interaction between LFA-1 on the T cell and intercellular adhesion molecule 1 on an APC or endothelial cell. Both alefacept and efalizumab, 2 formerly marketed biologic therapies, demonstrated remarkable clinical reduction of psoriatic lesions, and alefacept has been shown to produce disease remission for up to 18 months after discontinuation of therapy.24-26
NK T Cells
Natural killer T cells represent a subset of CD3+ T cells present in psoriatic plaques. Although NK T cells possess a TCR, they differ from T cells by displaying NK receptors comprised of lectin and immunoglobulin families. These cells exhibit remarkable specificity and are activated upon recognition of glycolipids presented by CD1d molecules. This process occurs in contrast to CD4+ and CD8+ T cells, which, due to their TCR diversity, respond to peptides processed by APCs and displayed on MHC molecules. Natural killer T cells can be classified into 2 subsets: (1) one group that expresses CD4 and preferentially produces TH1- versus TH2-type cytokines, and (2) another group that lacks CD4 and CD8 that only produces TH1-type cytokines. The innate immune system employs NK T cells early in the immune response because of their direct cytotoxicity and rapid production of cytokines such as IFN-γ, which promotes a TH1 inflammatory response, and IL-4, which promotes the development of TH2 cells. Excessive or dysfunctional NK T cells have been associated with autoimmune diseases such as multiple sclerosis and inflammatory bowel disease as well as allergic contact dermatitis.27-29
In psoriasis, NK T cells are located in the epidermis, closely situated to epidermal keratinocytes, which suggests a role for direct antigen presentation. Furthermore, CD1d is overexpressed throughout the epidermis of psoriatic plaques, whereas normally CD1d expression is confined to terminally differentiated keratinocytes. An in vitro study examining cytokine-based inflammation demonstrative of psoriasis treated cultured CD1d-positive keratinocytes with interferon gamma in the presence of alpha-galactosylceramide of the lectin family.30 Interferon gamma was observed to enhance keratinocyte CD1d expression, and subsequently, CD1d-positive keratinocytes were found to activate NK T cells to produce high levels of IFN-γ, while levels of IL-4 remained undetectable. The preferential production of IFN-γ supports a TH1-mediated mechanism regulated by NK T cells in the immunopathogenesis of psoriasis.
Dendritic Cells
Dendritic cells are APCs that process antigens in the tissues in which they reside, after which they migrate to local lymph nodes where they present their native antigens to T cells. This process allows the T-cell response to be tailored to the appropriate antigens in the corresponding tissues. Immature DCs that capture antigens mature by migrating to the T-cell center of the lymph node where they present their antigens to either MHC molecules or the CD1 family. This presentation results in T-cell proliferation and differentiation that correlates with the required type of T-cell response. Multiple subsets of APCs, including myeloid and plasmacytoid DCs, are highly represented in the epidermis and dermis of psoriatic plaques as compared with normal skin.31 Dermal DCs are thought to be responsible for activating both the TH1 and TH17 infiltrate by secreting IL-12 and IL-23, respectively. This mixed cellular response secretes cytokines and leads to a cascade of events involving keratinocytes, fibroblasts, endothelial cells, and neutrophils that create the cutaneous lesions seen in psoriasis.3
Although DCs play a pivotal role in eliciting an immune response against a foreign invader, they also contribute to the establishment of tolerance. Throughout their maturation, DCs are continuously sensing their environment, which shapes their production of TH1- versus TH2-type cytokines and subsequently the nature of the T-cell response. When challenged with a virus, bacteria, or unchecked cell growth, DCs mature into APCs. However, in the absence of a strong stimulus, DCs fail to mature into APCs and present self-peptides with MHC molecules, thereby creating regulatory T cells involved in peripheral tolerance.32 If this balance between immunogenic APCs and housekeeping T cells is upset, inflammatory conditions such as psoriasis can result.
Cytokines
Cytokines are low-molecular-weight glycoproteins that function as signals to produce inflammation, defense, tissue repair and remodeling, fibrosis, angiogenesis, and restriction of neoplastic growth.33 Cytokines are produced by immunocytes such as lymphocytes and macrophages as well as nonimmunocytes such as endothelial cells and keratinocytes. Proinflammatory cytokines include IL-1, IL-2, the IL-17 family, IFN-γ, and TNF-α, while anti-inflammatory cytokines include IL-4 and IL-10. A relative preponderance of TH1 proinflammatory cytokines or an insufficiency of TH2 anti-inflammatory cytokines induces local inflammation and recruitment of additional immunocyte populations, which produce added cytokines.34 A vicious cycle of inflammation occurs that results in cutaneous manifestations such as a plaque. Psoriatic lesions are characterized by a relative increase of TH1-type (eg, IL-2, IFN-γ, TNF-α, TNF-β) to TH2-type (eg, IL-4, IL-5, IL-6, IL-9, IL-10, IL-13) cytokines and an increase in TH17-type cytokines. Natural killer T cells stimulated by CD1d-overexpressing keratinocytes increase production of proinflammatory IFN-γ without effect on the anti-inflammatory IL-4. In addition to the cytokines produced by T cells, APCs produce IL-18, IL-23, and TNF-α found in the inflammatory infiltrate of psoriatic plaques. Both IL-18 and IL-23 stimulate TH1 cells to produce IFN-γ, and IL-23 stimulates TH17 cells. Clearly, a TH1- and TH17-type pattern governs the immune effector cells and their respective cytokines present in psoriatic skin.
Tumor Necrosis Factor α
Although a network of cytokines is responsible for the inflammation of psoriasis, TNF-α has been implicated as a master proinflammatory cytokine of the innate immune response due to its widespread targets and sources. Tumor necrosis factor α is produced by activated T cells, keratinocytes, NK cells, macrophages, monocytes, Langerhans APCs, and endothelial cells. Psoriatic lesions demonstrate high concentrations of TNF-α, while the synovial fluid of psoriatic arthritis patients demonstrates elevated concentrations of TNF-α, IL-1, IL-6, and IL-8.34 In psoriasis, TNF-α supports the expression of adhesion molecules (intercellular adhesion molecule 1 and P- and E-selectin), angiogenesis via vascular endothelial growth factor, the synthesis of proinflammatory molecules (IL-1, IL-6, IL-8, and nuclear factor κβ), and keratinocyte hyperproliferation via vasoactive intestinal peptide.35
A role for TNF-α in psoriasis treatment was serendipitously discovered in a trial for Crohn disease in which infliximab, a mouse-human IgG1 anti–TNF-α monoclonal antibody, was observed to clear psoriatic plaques in a patient with both Crohn disease and psoriasis.36 Immunotherapies that target TNF-α, including infliximab, etanercept, and adalimumab, demonstrate notable efficacy in the treatment of psoriasis.37-39 Tumor necrosis factor α is regarded as the driver of the inflammatory cycle of psoriasis due to its numerous modes of production, capability to amplify other proinflammatory signals, and the efficacy and rapidity with which it produces clinical improvements in psoriasis.
IL-23/TH17 Axis
A new distinct population of helper T cells has been shown to play an important role in psoriasis. These cells develop with the help of IL-23 (secreted by dermal DCs) and subsequently secrete cytokines such as IL-17; they are, therefore, named TH17 cells. CD161 is considered a surface marker for these cells.40 Strong evidence for this IL-23/TH17 axis has been shown in mouse and human models as well as in genetic studies.
IL-23 is a cytokine that shares the p40 subunit with IL-12 and has been linked to autoimmune diseases in both mice and humans.3 It is required for optimal development of TH17 cells41 from a committed CD4+ T-cell population after exposure to transforming growth factor β1 in combination with other proinflammatory cytokines.42,43 IL-23 messenger RNA is produced at higher levels in inflammatory psoriatic skin lesions versus uninvolved skin,44 and intradermal IL-23 injections in mice produced lesions resembling psoriasis macroscopically and microscopically.45 Furthermore, several systemic therapies have been shown to modulate IL-23 levels and correlate with clinical benefit.3 Alterations in the gene for the IL-23 receptor have been shown to be protective for psoriasis,46-48 and the gene coding for the p40 subunit is associated with psoriasis.46,47
Type 17 helper T cells produce a number of cytokines, such as IL-22, IL-17A, IL-17F, and IL-26; the latter 3 are considered to be specific to this lineage.42 IL-22 acts on outer body barrier tissues, such as the skin, and has antimicrobial activity. Blocking the activity of IL-22 in mice prevented the development of skin lesions,49 and psoriasis patients have elevated levels of IL-22 in the skin and blood.50,51 The IL-17 cytokines induce the expression of proinflammatory cytokines, colony-stimulating factors, and chemokines, and they recruit, mobilize, and activate neutrophils.52 IL-17 messenger RNA was found in lesional psoriatic skin but not unaffected skin,53 and cells isolated from the dermis of psoriatic skin have been shown to produce IL-17.54 IL-17A is not elevated in the serum of psoriatic patients (unlike other autoimmune diseases),55 and it is, therefore, thought that TH17 cells and IL-17A production are localized to the affected psoriatic skin. Consistent with this concept is the finding that treatments such as cyclosporin A and anti-TNF agents decrease proinflammatory cytokines in lesional skin but not in the periphery.56-58 These cytokines released by TH17 cells in addition to those released by TH1 cells act on keratinocytes and produce epidermal hyperproliferation, acanthosis, and hyperparakeratosis characteristic of psoriasis.3
New therapies have been developed to target the IL-23/TH17 axis. Ustekinumab is approved for moderate to severe plaque psoriasis. This treatment’s effect may be sustained for up to 3 years, it is generally well tolerated, and it may be useful for patients refractory to anti-TNF therapy such as etanercept.59 Briakinumab, another blocker of IL-12 and IL-23, was studied in phase 3 clinical trials, but its development was discontinued due to safety concerns.60 Newer drugs targeting the IL-23/TH17 axis include secukinumab, ixekizumab, brodalumab, guselkumab, and tildrakizumab.
Genetic Basis of Psoriasis
Psoriasis is a disease of overactive immunity in genetically susceptible individuals. Because patients exhibit varying skin phenotypes, extracutaneous manifestations, and disease courses, multiple genes resulting from linkage disequilibrium are believed to be involved in the pathogenesis of psoriasis. A decade of genome-wide linkage scans have established that PSORS1 is the strongest susceptibility locus demonstrable through family linkage studies; PSORS1 is responsible for up to 50% of the genetic component of psoriasis.61 More recently, HLA-Cw6 has received the most attention as a candidate gene of the PSORS1 susceptibility locus on the MHC class I region on chromosome 6p21.3.62 This gene may function in antigen presentation via MHC class I, which aids in the activation of the overactive T cells characteristic of psoriatic inflammation.
Studies involving the IL-23/TH17 axis have shown genetics to play a role. Individuals may be protected from psoriasis with a nonsynonymous nucleotide substitution in the IL23R gene,47-49 and certain haplotypes of the IL23R gene are associated with the disease47,49 in addition to other autoimmune conditions.
Genomic scans have shown additional susceptibility loci for psoriasis on chromosomes 1q21, 3q21, 4q32-35, 16q12, and 17q25. Two regions on chromosome 17q were recently localized via mapping, which demonstrated a 6 megabase pairs separation, thereby indicating independent linkage factors. Genes SLC9A3R1 and NAT9 are present in the first region, while RAPTOR is demonstrated in the second region.63SLC9A3R1 and NAT9 are players that regulate signal transduction, the immunologic synapse, and T-cell growth. RAPTOR is involved in T-cell function and growth pathways. Using these genes as an example, we can predict that the alterations of regulatory genes, even those yet undetermined, can enhance T-cell proliferation and inflammation manifested in psoriasis.
Conclusion
Psoriasis is a complex disease whereby multiple exogenous and endogenous stimuli incite already heightened innate immune responses in genetically predetermined individuals. The disease process is a result of a network of cell types, including T cells, DCs, and keratinocytes that, with the production of cytokines, generate a chronic inflammatory state. Our understanding of these cellular interactions and cytokines originates from developments, some meticulously planned, others serendipitous, in the fields of immunology, cell and molecular biology, and genetics. Such progress has fostered the creation of targeted immune therapy that has demonstrated remarkable efficacy in psoriasis treatment. Further study of the underlying pathophysiology of psoriasis may provide additional targets for therapy.
- Gottlieb A. Psoriasis. Dis Manag Clin Outcome. 1998;1:195-202.
- Gaspari AA. Innate and adaptive immunity and the pathophysiology of psoriasis. J Am Acad Dermatol. 2006;54(3 suppl 2):S67-S80.
- Di Cesare A, Di Meglio P, Nestle F. The IL-23/Th17 axis in the immunopathogenesis of psoriasis. J Invest Dermatol. 2009;129:1339-1350.
- Barker J. The pathophysiology of psoriasis. Lancet. 1991;338:227-230.
- Nickoloff BJ, Nestle FO. Recent insights into the immunopathogenesis of psoriasis provide new therapeutic opportunities. J Clin Invest. 2004;113:1664-1675.
- Bos J, Meinardi M, van Joost T, et al. Use of cyclosporine in psoriasis. Lancet. 1989;23:1500-1505.
- Khandke L, Krane J, Ashinoff R, et al. Cyclosporine in psoriasis treatment: inhibition of keratinocyte cell-cycle progression in G1 independent effects on transforming growth factor-alpha/epidermal growth factor receptor pathways. Arch Dermatol. 1991;127:1172-1179.
- Gottlieb S, Gilleaudeau P, Johnson R, et al. Response of psoriasis to a lymphocyte-selective toxin (DAB389IL-2) suggests a primary immune, but not keratinocyte, pathogenic basis. Nat Med. 1995;1:442-447.
- Vallat V, Gilleaudeau P, Battat L, et al. PUVA bath therapy strongly suppresses immunological and epidermal activation in psoriasis: a possible cellular basis for remittive therapy. J Exp Med. 1994;180:283-296.
- Gottlieb A, Grossman R, Khandke L, et al. Studies of the effect of cyclosporine in psoriasis in vivo: combined effects on activated T lymphocytes and epidermal regenerative maturation. J Invest Dermatol. 1992;98:302-309.
- Gottlieb S, Hayes E, Gilleaudeau P, et al. Cellular actions of etretinate in psoriasis: enhanced epidermal differentiation and reduced cell-mediated inflammation are unexpected outcomes. J Cutan Pathol. 1996;23:404-418.
- Nickoloff B, Bonish B, Huang B, et al. Characterization of a T cell line bearing natural killer receptors and capable of creating psoriasis in a SCID mouse model system. J Dermatol Sci. 2000;24:212-225.
- Gillet M, Conrad C, Geiges M, et al. Psoriasis triggered by toll-like receptor 7 agonist imiquimod in the presence of dermal plasmacytoid dendritic cell precursors. Arch Dermatol. 2004;140:1490-1495.
- Funk J, Langeland T, Schrumpf E, et al. Psoriasis induced by interferon-alpha. Br J Dermatol. 1991;125:463-465.
- Shiohara T, Kobayahsi M, Abe K, et al. Psoriasis occurring predominantly on warts: possible involvement of interferon alpha. Arch Dermatol. 1988;124:1816-1821.
- Fierlbeck G, Rassner G, Muller C. Psoriasis induced at the injection site of recombinant interferon gamma: results of immunohistologic investigations. Arch Dermatol. 1990;126:351-355.
- Prinz J. The role of T cells in psoriasis. J Eur Acad Dermatol Venereol. 2003;17(suppl):1-5.
- Bos J, de Rie M. The pathogenesis of psoriasis: immunological facts and speculations. Immunol Today. 1999;20:40-46.
- Wucherpfennig KW, Strominger JL. Molecular mimicry in T cell–mediated autoimmunity: viral peptides activate human T cell clones specific for myelin basic protein. Cell. 1995;80:695-705.
- Geginat J, Campagnaro S, Sallusto F, et al. TCR-independent proliferation and differentiation of human CD4+ T cell subsets induced by cytokines. Adv Exp Med Biol. 2002;512:107-112.
- Kastelan M, Massari L, Brajac I. Apoptosis mediated by cytolytic molecules might be responsible for maintenance of psoriatic plaques. Med Hypotheses. 2006;67:336-337.
- Austin L, Ozawa M, Kikuchi T, et al. The majority of epidermal T cells in psoriasis vulgaris lesions can produce type 1 cytokines, interferon-gamma, interleukin-2, and tumor necrosis factor-alpha, defining TC1 (cytotoxic T lymphocyte) and TH1 effector populations: a type 1 differentiation bias is also measured in circulating blood T cells in psoriatic patients. J Invest Dermatol. 1999;113:752-759.
- Abrams J, Kelley S, Hayes E, et al. Blockade of T lymphocyte costimulation with cytotoxic T lymphocyte-associated antigen 4-immunoglobulin (CTLA4Ig) reverses the cellular pathology of psoriatic plagues, including the activation of keratinocytes, dendritic cells and endothelial cells. J Exp Med. 2000;192:681-694.
- Lebwohl M, Christophers E, Langley R, et al. An international, randomized, double-blind, placebo-controlled phase 3 trial of intramuscular alefacept in patients with chronic plaque psoriasis. Arch Dermatol. 2003;139:719-727.
- Krueger G, Ellis C. Alefacept therapy produces remission for patients with chronic plaque psoriasis. Br J Dermatol. 2003;148:784-788.
- Gordon K, Leonardi C, Tyring S, et al. Efalizumab (anti-CD11a) is safe and effective in the treatment of psoriasis: pooled results of the 12-week first treatment period from 2 phase III trials. J Invest Dermatol. 2002;119:242.
- Singh A, Wilson M, Hong S, et al. Natural killer T cell activation protects mice against experimental autoimmune encephalomyelitis. J Exp Med. 2001;194:1801-1811.
- Saubermann L, Beck P, De Jong Y, et al. Activation of natural killer T cells by alpha-glactosylceramide in the presence of CD1d provides protection against colitis in mice. Gastroenterology. 2000;119:119-128.
- Campos R, Szczepanik M, Itakura A, et al. Cutaneous immunization rapidly activates liver invariant Valpha 14 NKT cells stimulating B-1 B cells to initiate T cell recruitment for elicitation of contact sensitivity. J Exp Med. 2003;198:1785-1796.
- Bonish B, Jullien D, Dutronc Y, et al. Overexpression of CD1d by keratinocytes in psoriasis and CD1d-dependent IFN-gamma production by NK-T cells. J Immunol. 2000;165:4076-4085.
- Deguchi M, Aiba S, Ohtani H, et al. Comparison of the distribution and numbers of antigen-presenting cells among T-lymphocyte-mediated dermatoses: CD1a+, factor XIIIa+, and CD68+ cells in eczematous dermatitis, psoriasis, lichen planus and graft-versus-host disease. Arch Dermatol Res. 2002;294:297-302.
- Bos J, de Rie M, Teunissen M, et al. Psoriasis: dysregulation of innate immunity. Br J Dermatol. 2005;152:1098-1107.
- Trefzer U, Hofmann M, Sterry W, et al. Cytokine and anticytokine therapy in dermatology. Expert Opin Biol Ther. 2003;3:733-743.
- Nickoloff B. The cytokine network in psoriasis. Arch Dermatol. 1991;127:871-884.
- Victor F, Gottlieb A. TNF-alpha and apoptosis: implications for the pathogenesis and treatment of psoriasis. J Drugs Dermatol. 2002;3:264-275.
- Oh C, Das K, Gottlieb A. Treatment with anti-tumour necrosis factor alpha (TNF-alpha) monoclonal antibody dramatically decreases the clinical activity of psoriasis lesions. J Am Acad Dermatol. 2000;42:829-830.
- Reich K, Nestle FO, Papp K, et al; EXPRESS study investigators. Infliximab induction and maintenance therapy for moderate-to-severe psoriasis: a phase III, multicentre, double-blind trial. Lancet. 2005;366:1367-1374.
- Leonardi C, Powers J, Matheson R, et al. Etanercept as monotherapy in patients with psoriasis. N Engl J Med. 2003;349:2014-2022.
- Saini R, Tutrone W, Weinberg J. Advances in therapy for psoriasis: an overview of infliximab, etanercept, efalizumab, alefacept, adalimumab, tazarotene, and pimecrolimus. Curr Pharm Des. 2005;11:273-280.
- Cosmi L, De Palma R, Santarlasci V, et al. Human interleukin 17-producing cells originate from a CD161+CD4+ T cell precursor. J Exp Med. 2008;205:1903-1916.
- de Beaucoudrey L, Puel A, Filipe-Santos O, et al. Mutations in STAT3 and IL12RB1 impair the development of human IL-17-producing T cells. J Exp Med. 2008;205:1543-1550.
- Manel N, Unutmaz D, Littman DR. The differentiation of humanT(H)-17 cells requires transforming growth factor-beta and induction of the nuclear receptor RORgammat. Nat Immunol. 2008;9:641-649.
- Yang L, Anderson DE, Baecher-Allan C, et al. IL-21 and TGF-beta are required for differentiation of human T(H)17 cells. Nature. 2008;454:350-352.
- Lee E, Trepicchio WL, Oestreicher JL, et al. Increased expression of interleukin 23 p19 and p40 in lesional skin of patients with psoriasis vulgaris. J Exp Med. 2004;199:125-130.
- Chan JR, Blumenschein W, Murphy E, et al. IL-23 stimulates epidermal hyperplasia via TNF and IL-20R2-dependent mechanisms with implications for psoriasis pathogenesis. J Exp Med. 2006;203:2557-2587.
- Capon F, Di Meglio P, Szaub J, et al. Sequence variants in the genes for the interleukin-23 receptor (IL23R) and its ligand (IL12B) confer protection against psoriasis. Hum Genet. 2007;122:201-206.
- Cargill M, Schrodi SJ, Chang M, et al. A large-scale genetic association study confirms IL12B and leads to the identification of IL23R as psoriasis-risk genes. Am J Hum Genet. 2007;80:273-290.
- Nair RP, Ruether A, Stuart PE, et al. Polymorphisms of the IL12B and IL23R genes are associated with psoriasis. J Invest Dermatol. 2008;128:1653-1661.
- Ma HL, Liang S, Li J, et al. IL-22 is required for Th17 cell-mediated pathology in a mouse model of psoriasis-like skin inflammation. J Clin Invest. 2008;118:597-607.
- Wolk K, Witte E, Wallace E, et al. IL-22 regulates the expression of genes responsible for antimicrobial defense, cellular differentiation, and mobility in keratinocytes: a potential role in psoriasis. Eur J Immunol. 2006;36:1309-1323.
- Boniface K, Guignouard E, Pedretti N, et al. A role for T cell-derived interleukin 22 in psoriatic skin inflammation. Clin Exp Immunol. 2007;150:407-415.
- Weaver CT, Hatton RD, Mangan PR, et al. IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annu Rev Immunol. 2007;25:821-852.
- Teunissen MB, Koomen CW, de Waal Malefyt R, et al. Interleukin-17 and interferon-gamma synergize in the enhancement of proinflammatory cytokine production by human keratinocytes. J Invest Dermatol. 1998;111:645-649.
- Lowes MA, Kikuchi T, Fuentes-Duculan J, et al. Psoriasis vulgaris lesions contain discrete populations of Th1 and Th17 T cells. J Invest Dermatol. 2008;128:1207-1211.
- Arican O, Aral M, Sasmaz S, et al. Serum levels of TNF-alpha, IFN-gamma, IL-6, IL-8, IL-12, IL-17, and IL-18 in patients with active psoriasis and correlation with disease severity. Mediators Inflamm. 2005;2005:273-279.
- Zaba LC, Cardinale I, Gilleaudeau P, et al. Amelioration of epidermal hyperplasia by TNF inhibition is associated with reduced Th17 responses. J Exp Med. 2007;204:3183-3194.
- Haider AS, Cohen J, Fei J, et al. Insights into gene modulation by therapeutic TNF and IFNgamma antibodies: TNF regulates IFNgamma production by T cells and TNF-regulated genes linked to psoriasis transcriptome. J Invest Dermatol. 2008;128:655-666.
- Haider AS, Lowes MA, Suarez-Farinas M, et al. Identification of cellular pathways of “type 1,” Th17 T cells, and TNF- and inducible nitric oxide synthase-producing dendritic cells in autoimmune inflammation through pharmacogenomic study of cyclosporine A in psoriasis. J Immunol. 2008;180:1913-1920.
- Croxtall JD. Ustekinumab: a review of its use in the management of moderate to severe plaque psoriasis. Drugs. 2011;71:1733-1753.
- Gordon KB, Langely RG, Gottlieb AB, et al. A phase III, randomized, controlled trial of the fully human IL-12/23 mAb briakinumab in moderate-to-severe psoriasis. J Invest Dermatol. 2012;132:304-314.
- Rahman P, Elder JT. Genetic epidemiology of psoriasis and psoriatic arthritis. Ann Rheum Dis. 2005;64(suppl 2):ii37-ii39.
- Elder JT. PSORS1: linking genetics and immunology. J Invest Dermatol. 2006;126:1205-1206.
- Krueger JG, Bowcock A. Psoriasis pathophysiology: current concepts of pathogenesis. Ann Rheum Dis. 2005;64(suppl 2):ii30-ii36.
- Gottlieb A. Psoriasis. Dis Manag Clin Outcome. 1998;1:195-202.
- Gaspari AA. Innate and adaptive immunity and the pathophysiology of psoriasis. J Am Acad Dermatol. 2006;54(3 suppl 2):S67-S80.
- Di Cesare A, Di Meglio P, Nestle F. The IL-23/Th17 axis in the immunopathogenesis of psoriasis. J Invest Dermatol. 2009;129:1339-1350.
- Barker J. The pathophysiology of psoriasis. Lancet. 1991;338:227-230.
- Nickoloff BJ, Nestle FO. Recent insights into the immunopathogenesis of psoriasis provide new therapeutic opportunities. J Clin Invest. 2004;113:1664-1675.
- Bos J, Meinardi M, van Joost T, et al. Use of cyclosporine in psoriasis. Lancet. 1989;23:1500-1505.
- Khandke L, Krane J, Ashinoff R, et al. Cyclosporine in psoriasis treatment: inhibition of keratinocyte cell-cycle progression in G1 independent effects on transforming growth factor-alpha/epidermal growth factor receptor pathways. Arch Dermatol. 1991;127:1172-1179.
- Gottlieb S, Gilleaudeau P, Johnson R, et al. Response of psoriasis to a lymphocyte-selective toxin (DAB389IL-2) suggests a primary immune, but not keratinocyte, pathogenic basis. Nat Med. 1995;1:442-447.
- Vallat V, Gilleaudeau P, Battat L, et al. PUVA bath therapy strongly suppresses immunological and epidermal activation in psoriasis: a possible cellular basis for remittive therapy. J Exp Med. 1994;180:283-296.
- Gottlieb A, Grossman R, Khandke L, et al. Studies of the effect of cyclosporine in psoriasis in vivo: combined effects on activated T lymphocytes and epidermal regenerative maturation. J Invest Dermatol. 1992;98:302-309.
- Gottlieb S, Hayes E, Gilleaudeau P, et al. Cellular actions of etretinate in psoriasis: enhanced epidermal differentiation and reduced cell-mediated inflammation are unexpected outcomes. J Cutan Pathol. 1996;23:404-418.
- Nickoloff B, Bonish B, Huang B, et al. Characterization of a T cell line bearing natural killer receptors and capable of creating psoriasis in a SCID mouse model system. J Dermatol Sci. 2000;24:212-225.
- Gillet M, Conrad C, Geiges M, et al. Psoriasis triggered by toll-like receptor 7 agonist imiquimod in the presence of dermal plasmacytoid dendritic cell precursors. Arch Dermatol. 2004;140:1490-1495.
- Funk J, Langeland T, Schrumpf E, et al. Psoriasis induced by interferon-alpha. Br J Dermatol. 1991;125:463-465.
- Shiohara T, Kobayahsi M, Abe K, et al. Psoriasis occurring predominantly on warts: possible involvement of interferon alpha. Arch Dermatol. 1988;124:1816-1821.
- Fierlbeck G, Rassner G, Muller C. Psoriasis induced at the injection site of recombinant interferon gamma: results of immunohistologic investigations. Arch Dermatol. 1990;126:351-355.
- Prinz J. The role of T cells in psoriasis. J Eur Acad Dermatol Venereol. 2003;17(suppl):1-5.
- Bos J, de Rie M. The pathogenesis of psoriasis: immunological facts and speculations. Immunol Today. 1999;20:40-46.
- Wucherpfennig KW, Strominger JL. Molecular mimicry in T cell–mediated autoimmunity: viral peptides activate human T cell clones specific for myelin basic protein. Cell. 1995;80:695-705.
- Geginat J, Campagnaro S, Sallusto F, et al. TCR-independent proliferation and differentiation of human CD4+ T cell subsets induced by cytokines. Adv Exp Med Biol. 2002;512:107-112.
- Kastelan M, Massari L, Brajac I. Apoptosis mediated by cytolytic molecules might be responsible for maintenance of psoriatic plaques. Med Hypotheses. 2006;67:336-337.
- Austin L, Ozawa M, Kikuchi T, et al. The majority of epidermal T cells in psoriasis vulgaris lesions can produce type 1 cytokines, interferon-gamma, interleukin-2, and tumor necrosis factor-alpha, defining TC1 (cytotoxic T lymphocyte) and TH1 effector populations: a type 1 differentiation bias is also measured in circulating blood T cells in psoriatic patients. J Invest Dermatol. 1999;113:752-759.
- Abrams J, Kelley S, Hayes E, et al. Blockade of T lymphocyte costimulation with cytotoxic T lymphocyte-associated antigen 4-immunoglobulin (CTLA4Ig) reverses the cellular pathology of psoriatic plagues, including the activation of keratinocytes, dendritic cells and endothelial cells. J Exp Med. 2000;192:681-694.
- Lebwohl M, Christophers E, Langley R, et al. An international, randomized, double-blind, placebo-controlled phase 3 trial of intramuscular alefacept in patients with chronic plaque psoriasis. Arch Dermatol. 2003;139:719-727.
- Krueger G, Ellis C. Alefacept therapy produces remission for patients with chronic plaque psoriasis. Br J Dermatol. 2003;148:784-788.
- Gordon K, Leonardi C, Tyring S, et al. Efalizumab (anti-CD11a) is safe and effective in the treatment of psoriasis: pooled results of the 12-week first treatment period from 2 phase III trials. J Invest Dermatol. 2002;119:242.
- Singh A, Wilson M, Hong S, et al. Natural killer T cell activation protects mice against experimental autoimmune encephalomyelitis. J Exp Med. 2001;194:1801-1811.
- Saubermann L, Beck P, De Jong Y, et al. Activation of natural killer T cells by alpha-glactosylceramide in the presence of CD1d provides protection against colitis in mice. Gastroenterology. 2000;119:119-128.
- Campos R, Szczepanik M, Itakura A, et al. Cutaneous immunization rapidly activates liver invariant Valpha 14 NKT cells stimulating B-1 B cells to initiate T cell recruitment for elicitation of contact sensitivity. J Exp Med. 2003;198:1785-1796.
- Bonish B, Jullien D, Dutronc Y, et al. Overexpression of CD1d by keratinocytes in psoriasis and CD1d-dependent IFN-gamma production by NK-T cells. J Immunol. 2000;165:4076-4085.
- Deguchi M, Aiba S, Ohtani H, et al. Comparison of the distribution and numbers of antigen-presenting cells among T-lymphocyte-mediated dermatoses: CD1a+, factor XIIIa+, and CD68+ cells in eczematous dermatitis, psoriasis, lichen planus and graft-versus-host disease. Arch Dermatol Res. 2002;294:297-302.
- Bos J, de Rie M, Teunissen M, et al. Psoriasis: dysregulation of innate immunity. Br J Dermatol. 2005;152:1098-1107.
- Trefzer U, Hofmann M, Sterry W, et al. Cytokine and anticytokine therapy in dermatology. Expert Opin Biol Ther. 2003;3:733-743.
- Nickoloff B. The cytokine network in psoriasis. Arch Dermatol. 1991;127:871-884.
- Victor F, Gottlieb A. TNF-alpha and apoptosis: implications for the pathogenesis and treatment of psoriasis. J Drugs Dermatol. 2002;3:264-275.
- Oh C, Das K, Gottlieb A. Treatment with anti-tumour necrosis factor alpha (TNF-alpha) monoclonal antibody dramatically decreases the clinical activity of psoriasis lesions. J Am Acad Dermatol. 2000;42:829-830.
- Reich K, Nestle FO, Papp K, et al; EXPRESS study investigators. Infliximab induction and maintenance therapy for moderate-to-severe psoriasis: a phase III, multicentre, double-blind trial. Lancet. 2005;366:1367-1374.
- Leonardi C, Powers J, Matheson R, et al. Etanercept as monotherapy in patients with psoriasis. N Engl J Med. 2003;349:2014-2022.
- Saini R, Tutrone W, Weinberg J. Advances in therapy for psoriasis: an overview of infliximab, etanercept, efalizumab, alefacept, adalimumab, tazarotene, and pimecrolimus. Curr Pharm Des. 2005;11:273-280.
- Cosmi L, De Palma R, Santarlasci V, et al. Human interleukin 17-producing cells originate from a CD161+CD4+ T cell precursor. J Exp Med. 2008;205:1903-1916.
- de Beaucoudrey L, Puel A, Filipe-Santos O, et al. Mutations in STAT3 and IL12RB1 impair the development of human IL-17-producing T cells. J Exp Med. 2008;205:1543-1550.
- Manel N, Unutmaz D, Littman DR. The differentiation of humanT(H)-17 cells requires transforming growth factor-beta and induction of the nuclear receptor RORgammat. Nat Immunol. 2008;9:641-649.
- Yang L, Anderson DE, Baecher-Allan C, et al. IL-21 and TGF-beta are required for differentiation of human T(H)17 cells. Nature. 2008;454:350-352.
- Lee E, Trepicchio WL, Oestreicher JL, et al. Increased expression of interleukin 23 p19 and p40 in lesional skin of patients with psoriasis vulgaris. J Exp Med. 2004;199:125-130.
- Chan JR, Blumenschein W, Murphy E, et al. IL-23 stimulates epidermal hyperplasia via TNF and IL-20R2-dependent mechanisms with implications for psoriasis pathogenesis. J Exp Med. 2006;203:2557-2587.
- Capon F, Di Meglio P, Szaub J, et al. Sequence variants in the genes for the interleukin-23 receptor (IL23R) and its ligand (IL12B) confer protection against psoriasis. Hum Genet. 2007;122:201-206.
- Cargill M, Schrodi SJ, Chang M, et al. A large-scale genetic association study confirms IL12B and leads to the identification of IL23R as psoriasis-risk genes. Am J Hum Genet. 2007;80:273-290.
- Nair RP, Ruether A, Stuart PE, et al. Polymorphisms of the IL12B and IL23R genes are associated with psoriasis. J Invest Dermatol. 2008;128:1653-1661.
- Ma HL, Liang S, Li J, et al. IL-22 is required for Th17 cell-mediated pathology in a mouse model of psoriasis-like skin inflammation. J Clin Invest. 2008;118:597-607.
- Wolk K, Witte E, Wallace E, et al. IL-22 regulates the expression of genes responsible for antimicrobial defense, cellular differentiation, and mobility in keratinocytes: a potential role in psoriasis. Eur J Immunol. 2006;36:1309-1323.
- Boniface K, Guignouard E, Pedretti N, et al. A role for T cell-derived interleukin 22 in psoriatic skin inflammation. Clin Exp Immunol. 2007;150:407-415.
- Weaver CT, Hatton RD, Mangan PR, et al. IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annu Rev Immunol. 2007;25:821-852.
- Teunissen MB, Koomen CW, de Waal Malefyt R, et al. Interleukin-17 and interferon-gamma synergize in the enhancement of proinflammatory cytokine production by human keratinocytes. J Invest Dermatol. 1998;111:645-649.
- Lowes MA, Kikuchi T, Fuentes-Duculan J, et al. Psoriasis vulgaris lesions contain discrete populations of Th1 and Th17 T cells. J Invest Dermatol. 2008;128:1207-1211.
- Arican O, Aral M, Sasmaz S, et al. Serum levels of TNF-alpha, IFN-gamma, IL-6, IL-8, IL-12, IL-17, and IL-18 in patients with active psoriasis and correlation with disease severity. Mediators Inflamm. 2005;2005:273-279.
- Zaba LC, Cardinale I, Gilleaudeau P, et al. Amelioration of epidermal hyperplasia by TNF inhibition is associated with reduced Th17 responses. J Exp Med. 2007;204:3183-3194.
- Haider AS, Cohen J, Fei J, et al. Insights into gene modulation by therapeutic TNF and IFNgamma antibodies: TNF regulates IFNgamma production by T cells and TNF-regulated genes linked to psoriasis transcriptome. J Invest Dermatol. 2008;128:655-666.
- Haider AS, Lowes MA, Suarez-Farinas M, et al. Identification of cellular pathways of “type 1,” Th17 T cells, and TNF- and inducible nitric oxide synthase-producing dendritic cells in autoimmune inflammation through pharmacogenomic study of cyclosporine A in psoriasis. J Immunol. 2008;180:1913-1920.
- Croxtall JD. Ustekinumab: a review of its use in the management of moderate to severe plaque psoriasis. Drugs. 2011;71:1733-1753.
- Gordon KB, Langely RG, Gottlieb AB, et al. A phase III, randomized, controlled trial of the fully human IL-12/23 mAb briakinumab in moderate-to-severe psoriasis. J Invest Dermatol. 2012;132:304-314.
- Rahman P, Elder JT. Genetic epidemiology of psoriasis and psoriatic arthritis. Ann Rheum Dis. 2005;64(suppl 2):ii37-ii39.
- Elder JT. PSORS1: linking genetics and immunology. J Invest Dermatol. 2006;126:1205-1206.
- Krueger JG, Bowcock A. Psoriasis pathophysiology: current concepts of pathogenesis. Ann Rheum Dis. 2005;64(suppl 2):ii30-ii36.
Practice Points
- Psoriasis is a systemic inflammatory disease.
- We now have an increased understanding of the specific cytokines involved in the disease.
- Therapies have been developed to target these cytokines.
Food for Thought
This special issue is dedicated to resident education on psoriasis. With that in mind, we hope to address many topics of interest to those in training. Over the years, diet has been a hot topic among psoriasis patients. They want to know how diet affects psoriasis and what changes can be made to their diet to improve their condition. Although they have expected specific answers, my response has usually been that they should, of course, eat an overall healthy and balanced diet, and lose weight if necessary. I have continued, however, that no specific diet has been recommended. However, now we have some information that may start to give us some answers.
The Mediterranean diet has been regarded as a healthy regimen.1 This diet emphasizes eating primarily plant-based foods, such as fruits and vegetables; whole grains; legumes; and nuts. Other recommendations include replacing butter with healthy fats such as olive oil and canola oil, using herbs and spices instead of salt to flavor foods, and limiting red meat to no more than a few times a month.1
As we know, psoriasis is a chronic inflammatory disease. The Mediterranean diet has been shown to reduce chronic inflammation and has a positive effect on the risk for metabolic syndrome and cardiovascular events.1 Phan et al1 hypothesized a positive effect of the Mediterranean diet on psoriasis. They performed a study to assess the association between a score that reflects the adhesion to a Mediterranean diet (MEDI-LITE) and the onset and/or severity of psoriasis.1
The NutriNet-Santé program is an ongoing, observational, web-based questionnaire cohort study launched in France in May 2009.1 Data were collected and analyzed between April 2017 and June 2017. Individuals with psoriasis were identified utilizing a validated online questionnaire and then categorized by disease severity into 1 of 3 groups: severe psoriasis, nonsevere psoriasis, and psoriasis free.1
During the initial 2 years of participation in the cohort, data on dietary intake (including alcohol) were gathered to calculate the MEDI-LITE score, ranging from 0 (no adherence) to 18 (maximum adherence).1 Of the 158,361 total web-based participants, 35,735 (23%) replied to the psoriasis questionnaire.1 Of the respondents, 3557 (10%) individuals reported having psoriasis. The condition was severe in 878 cases (24.7%), and 299 (8.4%) incident cases were recorded (cases occurring >2 years after participant inclusion in the cohort). After adjustment for confounding factors, the investigators found a significant inverse relationship between the MEDI-LITE score and having severe psoriasis (odds ratio [OR], 0.71; 95% CI, 0.55-0.92 for the MEDI-LITE score’s second tertile [score of 8 to 9]; and OR, 0.78; 95% CI, 0.59-1.01 for the third tertile [score of 10 to 18]).1
The authors noted that patients with severe psoriasis displayed low levels of adherence to the Mediterranean diet.1 They commented that this finding supports the hypothesis that the Mediterranean diet may slow the progression of psoriasis. If these findings are confirmed, adherence to a Mediterranean diet should be integrated into the routine management of moderate to severe psoriasis.1 These findings are by no means definitive, but it is a first step in helping us define more specific dietary recommendations for psoriasis.
This issue includes several articles looking at various facets of psoriasis important to residents, including the pathophysiology of psoriasis,2 treatment approach using biologic therapies,3 risk factors and triggers for psoriasis,4 and the psychosocial impact of psoriasis.5 We hope that you find this issue enjoyable and informative.
- Phan C, Touvier M, Kesse-Guyot E, et al. Association between Mediterranean anti-inflammatory dietary profile and severity of psoriasis: results from the NutriNet-Santé cohort [published online July 25, 2018]. JAMA Dermatol. doi:10.1001/jamadermatol.2018.2127.
- Hugh JM, Weinberg JM. Update on the pathophysiology of psoriasis. Cutis. 2018;102(suppl 5):6-12.
- McKay C, Kondratuk KE, Miller JP, et al. Biologic therapy in psoriasis: navigating the options. Cutis. 2018;102(suppl 5):13-17.
- Lee EB, Wu KK, Lee MP, et al. Psoriasis risk factors and triggers. Cutis. 2018;102(suppl 5):18-20.
- Kolli SS, Amin SD, Pona A, et al. Psychosocial impact of psoriasis: a review for dermatology residents. Cutis. 2018;102(suppl 5):21-25.
This special issue is dedicated to resident education on psoriasis. With that in mind, we hope to address many topics of interest to those in training. Over the years, diet has been a hot topic among psoriasis patients. They want to know how diet affects psoriasis and what changes can be made to their diet to improve their condition. Although they have expected specific answers, my response has usually been that they should, of course, eat an overall healthy and balanced diet, and lose weight if necessary. I have continued, however, that no specific diet has been recommended. However, now we have some information that may start to give us some answers.
The Mediterranean diet has been regarded as a healthy regimen.1 This diet emphasizes eating primarily plant-based foods, such as fruits and vegetables; whole grains; legumes; and nuts. Other recommendations include replacing butter with healthy fats such as olive oil and canola oil, using herbs and spices instead of salt to flavor foods, and limiting red meat to no more than a few times a month.1
As we know, psoriasis is a chronic inflammatory disease. The Mediterranean diet has been shown to reduce chronic inflammation and has a positive effect on the risk for metabolic syndrome and cardiovascular events.1 Phan et al1 hypothesized a positive effect of the Mediterranean diet on psoriasis. They performed a study to assess the association between a score that reflects the adhesion to a Mediterranean diet (MEDI-LITE) and the onset and/or severity of psoriasis.1
The NutriNet-Santé program is an ongoing, observational, web-based questionnaire cohort study launched in France in May 2009.1 Data were collected and analyzed between April 2017 and June 2017. Individuals with psoriasis were identified utilizing a validated online questionnaire and then categorized by disease severity into 1 of 3 groups: severe psoriasis, nonsevere psoriasis, and psoriasis free.1
During the initial 2 years of participation in the cohort, data on dietary intake (including alcohol) were gathered to calculate the MEDI-LITE score, ranging from 0 (no adherence) to 18 (maximum adherence).1 Of the 158,361 total web-based participants, 35,735 (23%) replied to the psoriasis questionnaire.1 Of the respondents, 3557 (10%) individuals reported having psoriasis. The condition was severe in 878 cases (24.7%), and 299 (8.4%) incident cases were recorded (cases occurring >2 years after participant inclusion in the cohort). After adjustment for confounding factors, the investigators found a significant inverse relationship between the MEDI-LITE score and having severe psoriasis (odds ratio [OR], 0.71; 95% CI, 0.55-0.92 for the MEDI-LITE score’s second tertile [score of 8 to 9]; and OR, 0.78; 95% CI, 0.59-1.01 for the third tertile [score of 10 to 18]).1
The authors noted that patients with severe psoriasis displayed low levels of adherence to the Mediterranean diet.1 They commented that this finding supports the hypothesis that the Mediterranean diet may slow the progression of psoriasis. If these findings are confirmed, adherence to a Mediterranean diet should be integrated into the routine management of moderate to severe psoriasis.1 These findings are by no means definitive, but it is a first step in helping us define more specific dietary recommendations for psoriasis.
This issue includes several articles looking at various facets of psoriasis important to residents, including the pathophysiology of psoriasis,2 treatment approach using biologic therapies,3 risk factors and triggers for psoriasis,4 and the psychosocial impact of psoriasis.5 We hope that you find this issue enjoyable and informative.
This special issue is dedicated to resident education on psoriasis. With that in mind, we hope to address many topics of interest to those in training. Over the years, diet has been a hot topic among psoriasis patients. They want to know how diet affects psoriasis and what changes can be made to their diet to improve their condition. Although they have expected specific answers, my response has usually been that they should, of course, eat an overall healthy and balanced diet, and lose weight if necessary. I have continued, however, that no specific diet has been recommended. However, now we have some information that may start to give us some answers.
The Mediterranean diet has been regarded as a healthy regimen.1 This diet emphasizes eating primarily plant-based foods, such as fruits and vegetables; whole grains; legumes; and nuts. Other recommendations include replacing butter with healthy fats such as olive oil and canola oil, using herbs and spices instead of salt to flavor foods, and limiting red meat to no more than a few times a month.1
As we know, psoriasis is a chronic inflammatory disease. The Mediterranean diet has been shown to reduce chronic inflammation and has a positive effect on the risk for metabolic syndrome and cardiovascular events.1 Phan et al1 hypothesized a positive effect of the Mediterranean diet on psoriasis. They performed a study to assess the association between a score that reflects the adhesion to a Mediterranean diet (MEDI-LITE) and the onset and/or severity of psoriasis.1
The NutriNet-Santé program is an ongoing, observational, web-based questionnaire cohort study launched in France in May 2009.1 Data were collected and analyzed between April 2017 and June 2017. Individuals with psoriasis were identified utilizing a validated online questionnaire and then categorized by disease severity into 1 of 3 groups: severe psoriasis, nonsevere psoriasis, and psoriasis free.1
During the initial 2 years of participation in the cohort, data on dietary intake (including alcohol) were gathered to calculate the MEDI-LITE score, ranging from 0 (no adherence) to 18 (maximum adherence).1 Of the 158,361 total web-based participants, 35,735 (23%) replied to the psoriasis questionnaire.1 Of the respondents, 3557 (10%) individuals reported having psoriasis. The condition was severe in 878 cases (24.7%), and 299 (8.4%) incident cases were recorded (cases occurring >2 years after participant inclusion in the cohort). After adjustment for confounding factors, the investigators found a significant inverse relationship between the MEDI-LITE score and having severe psoriasis (odds ratio [OR], 0.71; 95% CI, 0.55-0.92 for the MEDI-LITE score’s second tertile [score of 8 to 9]; and OR, 0.78; 95% CI, 0.59-1.01 for the third tertile [score of 10 to 18]).1
The authors noted that patients with severe psoriasis displayed low levels of adherence to the Mediterranean diet.1 They commented that this finding supports the hypothesis that the Mediterranean diet may slow the progression of psoriasis. If these findings are confirmed, adherence to a Mediterranean diet should be integrated into the routine management of moderate to severe psoriasis.1 These findings are by no means definitive, but it is a first step in helping us define more specific dietary recommendations for psoriasis.
This issue includes several articles looking at various facets of psoriasis important to residents, including the pathophysiology of psoriasis,2 treatment approach using biologic therapies,3 risk factors and triggers for psoriasis,4 and the psychosocial impact of psoriasis.5 We hope that you find this issue enjoyable and informative.
- Phan C, Touvier M, Kesse-Guyot E, et al. Association between Mediterranean anti-inflammatory dietary profile and severity of psoriasis: results from the NutriNet-Santé cohort [published online July 25, 2018]. JAMA Dermatol. doi:10.1001/jamadermatol.2018.2127.
- Hugh JM, Weinberg JM. Update on the pathophysiology of psoriasis. Cutis. 2018;102(suppl 5):6-12.
- McKay C, Kondratuk KE, Miller JP, et al. Biologic therapy in psoriasis: navigating the options. Cutis. 2018;102(suppl 5):13-17.
- Lee EB, Wu KK, Lee MP, et al. Psoriasis risk factors and triggers. Cutis. 2018;102(suppl 5):18-20.
- Kolli SS, Amin SD, Pona A, et al. Psychosocial impact of psoriasis: a review for dermatology residents. Cutis. 2018;102(suppl 5):21-25.
- Phan C, Touvier M, Kesse-Guyot E, et al. Association between Mediterranean anti-inflammatory dietary profile and severity of psoriasis: results from the NutriNet-Santé cohort [published online July 25, 2018]. JAMA Dermatol. doi:10.1001/jamadermatol.2018.2127.
- Hugh JM, Weinberg JM. Update on the pathophysiology of psoriasis. Cutis. 2018;102(suppl 5):6-12.
- McKay C, Kondratuk KE, Miller JP, et al. Biologic therapy in psoriasis: navigating the options. Cutis. 2018;102(suppl 5):13-17.
- Lee EB, Wu KK, Lee MP, et al. Psoriasis risk factors and triggers. Cutis. 2018;102(suppl 5):18-20.
- Kolli SS, Amin SD, Pona A, et al. Psychosocial impact of psoriasis: a review for dermatology residents. Cutis. 2018;102(suppl 5):21-25.
Higher BMI associated with greater loss of gray matter volume in MS
ATLANTA – Among patients with relapsing-remitting multiple sclerosis, higher body mass index, but not vitamin D status, appears to be related to greater loss of gray matter brain volume over time, results from a 5-year analysis showed.

“We had previously known that obesity is a risk factor for developing MS, and among those who already have the disease, obesity-related comorbidities are associated with increased morbidity and mortality,” lead study author Ellen M. Mowry, MD, said in an interview at the annual meeting of the American Neurological Association. “Loss of brain tissue, especially as measured by reduced volume of gray matter noted on brain MRI, is predictive of long-term disability in MS. While we await the results of confirmatory studies and randomized trials, this study adds to the growing body of evidence suggesting there may be a role for modification of lifestyle factors in mitigating longer-term MS-related disability risk.”
In an effort to determine if body mass index (BMI) or vitamin D status is associated with longer-term MRI measures of neurodegeneration, Dr. Mowry and her colleagues drew from 469 patients participating in a longitudinal MS cohort study at the University of California, San Francisco, known as EPIC. Participants had clinical evaluations, brain MRI, and blood draws annually and were followed for 5 years. The main outcomes of interest were BMI and serum 25-hydroxyvitamin D levels measured over the time period, and their relationship to brain volume.
At baseline, the mean age of patients was 42 years, 70% were female, their mean BMI was 25 kg/m2, and their mean serum vitamin D level was 27.8 ng/mL. Dr. Mowry, a neurologist at Johns Hopkins University, Baltimore, and her colleagues found that over time, each 1-kg/m2 higher BMI was independently associated with reduced gray matter in multivariate models (–1.1 mL; P = .001). In addition, each 1-kg/m2 higher BMI over time was independently associated with greater declines in normalized brain parenchymal brain volume (–1.1 mL; P = .039). Elevated vitamin D levels, however, did not appear to be meaningfully associated with brain volumes.
Dr. Mowry acknowledged certain limitations of the study, including its nonrandomized design. “Such a trial may be warranted but I believe will be challenging to conduct,” she said. “Also, this cohort was designed to assess the association of genes with brain MRI outcomes, and so the people included were racially homogeneous – only Caucasians were included. Since MS risk is especially high among African Americans in recent years, and African Americans appear overall to have a higher risk of long-term disability, it is important to evaluate these and other prognostic factors amongst a more representative group of people with MS.”
The study received funding support from the National Institutes of Health, GlaxoSmithKline, and Biogen. Dr. Mowry disclosed that she has received medication from Teva for use in a clinical trial. In addition, she has been the primary investigator for studies sponsored by Biogen and Sun Pharma, and has conducted investigator-initiated studies sponsored by Genzyme and Biogen.
SOURCE: Ann Neurol. 2018;84[S22]:S206-7. Abstract M250.
ATLANTA – Among patients with relapsing-remitting multiple sclerosis, higher body mass index, but not vitamin D status, appears to be related to greater loss of gray matter brain volume over time, results from a 5-year analysis showed.
“We had previously known that obesity is a risk factor for developing MS, and among those who already have the disease, obesity-related comorbidities are associated with increased morbidity and mortality,” lead study author Ellen M. Mowry, MD, said in an interview at the annual meeting of the American Neurological Association. “Loss of brain tissue, especially as measured by reduced volume of gray matter noted on brain MRI, is predictive of long-term disability in MS. While we await the results of confirmatory studies and randomized trials, this study adds to the growing body of evidence suggesting there may be a role for modification of lifestyle factors in mitigating longer-term MS-related disability risk.”
In an effort to determine if body mass index (BMI) or vitamin D status is associated with longer-term MRI measures of neurodegeneration, Dr. Mowry and her colleagues drew from 469 patients participating in a longitudinal MS cohort study at the University of California, San Francisco, known as EPIC. Participants had clinical evaluations, brain MRI, and blood draws annually and were followed for 5 years. The main outcomes of interest were BMI and serum 25-hydroxyvitamin D levels measured over the time period, and their relationship to brain volume.
At baseline, the mean age of patients was 42 years, 70% were female, their mean BMI was 25 kg/m2, and their mean serum vitamin D level was 27.8 ng/mL. Dr. Mowry, a neurologist at Johns Hopkins University, Baltimore, and her colleagues found that over time, each 1-kg/m2 higher BMI was independently associated with reduced gray matter in multivariate models (–1.1 mL; P = .001). In addition, each 1-kg/m2 higher BMI over time was independently associated with greater declines in normalized brain parenchymal brain volume (–1.1 mL; P = .039). Elevated vitamin D levels, however, did not appear to be meaningfully associated with brain volumes.
Dr. Mowry acknowledged certain limitations of the study, including its nonrandomized design. “Such a trial may be warranted but I believe will be challenging to conduct,” she said. “Also, this cohort was designed to assess the association of genes with brain MRI outcomes, and so the people included were racially homogeneous – only Caucasians were included. Since MS risk is especially high among African Americans in recent years, and African Americans appear overall to have a higher risk of long-term disability, it is important to evaluate these and other prognostic factors amongst a more representative group of people with MS.”
The study received funding support from the National Institutes of Health, GlaxoSmithKline, and Biogen. Dr. Mowry disclosed that she has received medication from Teva for use in a clinical trial. In addition, she has been the primary investigator for studies sponsored by Biogen and Sun Pharma, and has conducted investigator-initiated studies sponsored by Genzyme and Biogen.
SOURCE: Ann Neurol. 2018;84[S22]:S206-7. Abstract M250.
ATLANTA – Among patients with relapsing-remitting multiple sclerosis, higher body mass index, but not vitamin D status, appears to be related to greater loss of gray matter brain volume over time, results from a 5-year analysis showed.
“We had previously known that obesity is a risk factor for developing MS, and among those who already have the disease, obesity-related comorbidities are associated with increased morbidity and mortality,” lead study author Ellen M. Mowry, MD, said in an interview at the annual meeting of the American Neurological Association. “Loss of brain tissue, especially as measured by reduced volume of gray matter noted on brain MRI, is predictive of long-term disability in MS. While we await the results of confirmatory studies and randomized trials, this study adds to the growing body of evidence suggesting there may be a role for modification of lifestyle factors in mitigating longer-term MS-related disability risk.”
In an effort to determine if body mass index (BMI) or vitamin D status is associated with longer-term MRI measures of neurodegeneration, Dr. Mowry and her colleagues drew from 469 patients participating in a longitudinal MS cohort study at the University of California, San Francisco, known as EPIC. Participants had clinical evaluations, brain MRI, and blood draws annually and were followed for 5 years. The main outcomes of interest were BMI and serum 25-hydroxyvitamin D levels measured over the time period, and their relationship to brain volume.
At baseline, the mean age of patients was 42 years, 70% were female, their mean BMI was 25 kg/m2, and their mean serum vitamin D level was 27.8 ng/mL. Dr. Mowry, a neurologist at Johns Hopkins University, Baltimore, and her colleagues found that over time, each 1-kg/m2 higher BMI was independently associated with reduced gray matter in multivariate models (–1.1 mL; P = .001). In addition, each 1-kg/m2 higher BMI over time was independently associated with greater declines in normalized brain parenchymal brain volume (–1.1 mL; P = .039). Elevated vitamin D levels, however, did not appear to be meaningfully associated with brain volumes.
Dr. Mowry acknowledged certain limitations of the study, including its nonrandomized design. “Such a trial may be warranted but I believe will be challenging to conduct,” she said. “Also, this cohort was designed to assess the association of genes with brain MRI outcomes, and so the people included were racially homogeneous – only Caucasians were included. Since MS risk is especially high among African Americans in recent years, and African Americans appear overall to have a higher risk of long-term disability, it is important to evaluate these and other prognostic factors amongst a more representative group of people with MS.”
The study received funding support from the National Institutes of Health, GlaxoSmithKline, and Biogen. Dr. Mowry disclosed that she has received medication from Teva for use in a clinical trial. In addition, she has been the primary investigator for studies sponsored by Biogen and Sun Pharma, and has conducted investigator-initiated studies sponsored by Genzyme and Biogen.
SOURCE: Ann Neurol. 2018;84[S22]:S206-7. Abstract M250.
AT ANA 2018
Key clinical point: Higher body mass in MS patients appears to be related to greater brain atrophy over time.
Major finding: Over time, each 1-kg/m2 higher BMI was independently associated with reduced gray matter in multivariate models (–1.1 mL; P = .001).
Study details: An analysis of 469 patients participating in a longitudinal MS cohort study.
Disclosures: The study received funding support from the National Institutes of Health, GlaxoSmithKline, and Biogen. Dr. Mowry disclosed that she has received medication from Teva for use in a clinical trial. In addition, she has been the primary investigator for studies sponsored by Biogen and Sun Pharma, and has conducted investigator-initiated studies sponsored by Genzyme and Biogen.
Source: Ann Neurol. 2018;84[S22]:S206-7. Abstract M250.
“Unique” Challenges for Screening Native American Women
American Indian/Alaska Native (AI/AN) women face the same barriers as all low-income minority women face in accessing preventive care, but according to researchers from Rutgers University in New Jersey and University of Arizona, they also face “unique challenges and circumstances.” The researchers reviewed 18 studies to find out more about facilitators of, and barriers to, breast cancer screening.
Low-income women are more likely to be diagnosed at a later stage and to die of breast cancer, one study found. The factors are well known: cost, lack of a usual source of care, lack of insurance, distance from a facility, and lack of transportation.
However, the researchers of the meta-analysis say, “compounding these barriers,” AI/AN women expressed the belief that preventive care is not a priority, especially when it is their own preventive care. Moreover, some barriers that might be unique to the AI/AN women included concern with “manifest destiny”: the assumption that thinking or talking about breast cancer can cause it, for instance. One study examined “traditionality” and found women who could be seen as more traditional, defining themselves as living an “Indian way of life,” were less likely to be current with screening. Other women expressed mistrust in the technology of screening or spoke about perception of discrimination in the health care system.
Although this population has access to screening through IHS facilities, women who also have insurance (typically Medicaid) are more likely to get screened. Women in rural areas who lived near an IHS facility were more likely than were urban women to get mammograms. The researchers suggest this could be because rural women are more likely to be isolated from other mammogram facilities. Too, the IHS is “chronically underfunded,” the researchers note, likely a cause of the health disparities and limiting scope of services.
Their review made clear that efforts to intervene with AI/AN women to increase breast cancer screening have been limited, the researchers say. The intervention studies they reviewed “were not successful in improving screening rates or adherence.” The qualitative studies, on the other hand, suggest that women may be more responsive to locally supportive, targeted, and culturally appropriate interventions that respect traditionality yet encourage trust in the medical system.
American Indian/Alaska Native (AI/AN) women face the same barriers as all low-income minority women face in accessing preventive care, but according to researchers from Rutgers University in New Jersey and University of Arizona, they also face “unique challenges and circumstances.” The researchers reviewed 18 studies to find out more about facilitators of, and barriers to, breast cancer screening.
Low-income women are more likely to be diagnosed at a later stage and to die of breast cancer, one study found. The factors are well known: cost, lack of a usual source of care, lack of insurance, distance from a facility, and lack of transportation.
However, the researchers of the meta-analysis say, “compounding these barriers,” AI/AN women expressed the belief that preventive care is not a priority, especially when it is their own preventive care. Moreover, some barriers that might be unique to the AI/AN women included concern with “manifest destiny”: the assumption that thinking or talking about breast cancer can cause it, for instance. One study examined “traditionality” and found women who could be seen as more traditional, defining themselves as living an “Indian way of life,” were less likely to be current with screening. Other women expressed mistrust in the technology of screening or spoke about perception of discrimination in the health care system.
Although this population has access to screening through IHS facilities, women who also have insurance (typically Medicaid) are more likely to get screened. Women in rural areas who lived near an IHS facility were more likely than were urban women to get mammograms. The researchers suggest this could be because rural women are more likely to be isolated from other mammogram facilities. Too, the IHS is “chronically underfunded,” the researchers note, likely a cause of the health disparities and limiting scope of services.
Their review made clear that efforts to intervene with AI/AN women to increase breast cancer screening have been limited, the researchers say. The intervention studies they reviewed “were not successful in improving screening rates or adherence.” The qualitative studies, on the other hand, suggest that women may be more responsive to locally supportive, targeted, and culturally appropriate interventions that respect traditionality yet encourage trust in the medical system.
American Indian/Alaska Native (AI/AN) women face the same barriers as all low-income minority women face in accessing preventive care, but according to researchers from Rutgers University in New Jersey and University of Arizona, they also face “unique challenges and circumstances.” The researchers reviewed 18 studies to find out more about facilitators of, and barriers to, breast cancer screening.
Low-income women are more likely to be diagnosed at a later stage and to die of breast cancer, one study found. The factors are well known: cost, lack of a usual source of care, lack of insurance, distance from a facility, and lack of transportation.
However, the researchers of the meta-analysis say, “compounding these barriers,” AI/AN women expressed the belief that preventive care is not a priority, especially when it is their own preventive care. Moreover, some barriers that might be unique to the AI/AN women included concern with “manifest destiny”: the assumption that thinking or talking about breast cancer can cause it, for instance. One study examined “traditionality” and found women who could be seen as more traditional, defining themselves as living an “Indian way of life,” were less likely to be current with screening. Other women expressed mistrust in the technology of screening or spoke about perception of discrimination in the health care system.
Although this population has access to screening through IHS facilities, women who also have insurance (typically Medicaid) are more likely to get screened. Women in rural areas who lived near an IHS facility were more likely than were urban women to get mammograms. The researchers suggest this could be because rural women are more likely to be isolated from other mammogram facilities. Too, the IHS is “chronically underfunded,” the researchers note, likely a cause of the health disparities and limiting scope of services.
Their review made clear that efforts to intervene with AI/AN women to increase breast cancer screening have been limited, the researchers say. The intervention studies they reviewed “were not successful in improving screening rates or adherence.” The qualitative studies, on the other hand, suggest that women may be more responsive to locally supportive, targeted, and culturally appropriate interventions that respect traditionality yet encourage trust in the medical system.
Use of topical agents before RT may be safe
Moderate application of topical therapies directly prior to radiotherapy (RT) treatment sessions may be safe and might exhibit minimal effects on delivered treatment dose, investigators report.
The researchers found that although avoiding topical agents prior to radiation treatments was widespread, preclinical data indicate that there is no difference in the radiation dose on the skin with or without a 1- to 2-mm-thick layer of topical agent.
In an online survey, which queried 133 patients and 108 clinicians in relation to existing practices surrounding topical agent use, 83.4% of patients were informed to discontinue application of topical therapies directly before RT treatment sessions. In addition, 54.1% were told to clean and remove any enduring topical agents before treatment. Among clinicians, 91.4% reported to have received or given advice to stop application of topicals before obtaining RT treatment, Brian C. Baumann, MD, and his associates reported in JAMA Oncology.
However, in a preclinical study using a mouse- and tissue-equivalent phantom model to determine the dosimetric effects of concomitant topical agent use with daily RT treatments, Dr. Baumann, of Washington University in St. Louis, and his colleagues determined that when a topical agent was given prior to RT at a thickness below 2 mm, no changes were seen in radiation treatment dose, irrespective of depth, photon and electron energy level, or beam angle.
However, a proportionally thicker covering (3 mm or more) caused a bolus effect at the surface, which resulted in electron dose increases of 2%-5%, and photon increases of 15%-35%, when compared with controls.
Investigators measured radiation dose and photon beam intensity at various surface depths after applying two commonly used topical therapies, a healing ointment of 41% petrolatum or silver sulfadiazine cream 1%. The agents were administered using a thick (1-2 mm) application and proportionally thicker (3 mm or more) covering.
“Thin or moderately applied topical agents, even if applied just before RT, may have minimal influence on skin dose regardless of beam energy or beam incidence,” the investigators wrote. However, the findings do suggest that “applying very thick amounts of a topical agent before RT may increase the surface dose and should be avoided,” they said.
The authors reported that the study was funded by development funds from the University of Pennsylvania, Philadelphia. One of the coauthors, James M. Metz, MD, disclosed service on advisory boards for Ion Beam Applications and Varian Medical Systems for proton therapy; however, these roles were not relevant to this study.
SOURCE: Baumann BC et al. JAMA Oncol. 2018 Oct 18. doi: 10.1001/jamaoncol.2018.4292.
The common practice of discontinuing topical therapies immediately prior to radiotherapy (RT) is likely an accepted myth, and through allowance of these agents, quality of life for patients may improve, according to Simon A. Brown, MD, and Chelsea C. Pinnix, MD, PhD.
The dogma stems from a period known as the “orthovoltage era,” which started in the 1920s, lasting into the 1950s. During this time, radiation oncologists recommended against the use of topical therapies or related agents directly preceding RT sessions, Dr. Brown and Dr. Pinnix wrote in invited commentary. The assumption was that using these agents could lead to increased dermatologic toxicities, because of the alleged bolus effects, or interactions with metal salts present in the topical. Bolus effects are sometimes beneficial, by reducing the delivered treatment dose in deeper tissues; but they also may be harmful, if unanticipated.
A similar study that took place in 1997, in which a group of researchers from the Medical College of Georgia, Augusta, investigated links between various topical agents and irradiation surface dose using a 6-MV photon beam. The results, similar to those reported by Dr. Baumann and his colleagues, showed that surface doses were affected only if agents were applied in a very thick manner, beyond what is considered normal. In addition, metal salts contained within the topical agents did not alter administered surface dose.
Taken together, the commentators stated that the common proposition that topical therapies must be avoided prior to RT is likely not relevant in many clinical situations.
Dr. Brown is affiliated with the department of radiation medicine at Oregon Health & Science University in Portland and Dr. Pinnix is with the department of radiation oncology at the University of Texas at the MD Anderson Cancer Center in Houston. These comments are adapted from their invited commentary.
The common practice of discontinuing topical therapies immediately prior to radiotherapy (RT) is likely an accepted myth, and through allowance of these agents, quality of life for patients may improve, according to Simon A. Brown, MD, and Chelsea C. Pinnix, MD, PhD.
The dogma stems from a period known as the “orthovoltage era,” which started in the 1920s, lasting into the 1950s. During this time, radiation oncologists recommended against the use of topical therapies or related agents directly preceding RT sessions, Dr. Brown and Dr. Pinnix wrote in invited commentary. The assumption was that using these agents could lead to increased dermatologic toxicities, because of the alleged bolus effects, or interactions with metal salts present in the topical. Bolus effects are sometimes beneficial, by reducing the delivered treatment dose in deeper tissues; but they also may be harmful, if unanticipated.
A similar study that took place in 1997, in which a group of researchers from the Medical College of Georgia, Augusta, investigated links between various topical agents and irradiation surface dose using a 6-MV photon beam. The results, similar to those reported by Dr. Baumann and his colleagues, showed that surface doses were affected only if agents were applied in a very thick manner, beyond what is considered normal. In addition, metal salts contained within the topical agents did not alter administered surface dose.
Taken together, the commentators stated that the common proposition that topical therapies must be avoided prior to RT is likely not relevant in many clinical situations.
Dr. Brown is affiliated with the department of radiation medicine at Oregon Health & Science University in Portland and Dr. Pinnix is with the department of radiation oncology at the University of Texas at the MD Anderson Cancer Center in Houston. These comments are adapted from their invited commentary.
The common practice of discontinuing topical therapies immediately prior to radiotherapy (RT) is likely an accepted myth, and through allowance of these agents, quality of life for patients may improve, according to Simon A. Brown, MD, and Chelsea C. Pinnix, MD, PhD.
The dogma stems from a period known as the “orthovoltage era,” which started in the 1920s, lasting into the 1950s. During this time, radiation oncologists recommended against the use of topical therapies or related agents directly preceding RT sessions, Dr. Brown and Dr. Pinnix wrote in invited commentary. The assumption was that using these agents could lead to increased dermatologic toxicities, because of the alleged bolus effects, or interactions with metal salts present in the topical. Bolus effects are sometimes beneficial, by reducing the delivered treatment dose in deeper tissues; but they also may be harmful, if unanticipated.
A similar study that took place in 1997, in which a group of researchers from the Medical College of Georgia, Augusta, investigated links between various topical agents and irradiation surface dose using a 6-MV photon beam. The results, similar to those reported by Dr. Baumann and his colleagues, showed that surface doses were affected only if agents were applied in a very thick manner, beyond what is considered normal. In addition, metal salts contained within the topical agents did not alter administered surface dose.
Taken together, the commentators stated that the common proposition that topical therapies must be avoided prior to RT is likely not relevant in many clinical situations.
Dr. Brown is affiliated with the department of radiation medicine at Oregon Health & Science University in Portland and Dr. Pinnix is with the department of radiation oncology at the University of Texas at the MD Anderson Cancer Center in Houston. These comments are adapted from their invited commentary.
Moderate application of topical therapies directly prior to radiotherapy (RT) treatment sessions may be safe and might exhibit minimal effects on delivered treatment dose, investigators report.
The researchers found that although avoiding topical agents prior to radiation treatments was widespread, preclinical data indicate that there is no difference in the radiation dose on the skin with or without a 1- to 2-mm-thick layer of topical agent.
In an online survey, which queried 133 patients and 108 clinicians in relation to existing practices surrounding topical agent use, 83.4% of patients were informed to discontinue application of topical therapies directly before RT treatment sessions. In addition, 54.1% were told to clean and remove any enduring topical agents before treatment. Among clinicians, 91.4% reported to have received or given advice to stop application of topicals before obtaining RT treatment, Brian C. Baumann, MD, and his associates reported in JAMA Oncology.
However, in a preclinical study using a mouse- and tissue-equivalent phantom model to determine the dosimetric effects of concomitant topical agent use with daily RT treatments, Dr. Baumann, of Washington University in St. Louis, and his colleagues determined that when a topical agent was given prior to RT at a thickness below 2 mm, no changes were seen in radiation treatment dose, irrespective of depth, photon and electron energy level, or beam angle.
However, a proportionally thicker covering (3 mm or more) caused a bolus effect at the surface, which resulted in electron dose increases of 2%-5%, and photon increases of 15%-35%, when compared with controls.
Investigators measured radiation dose and photon beam intensity at various surface depths after applying two commonly used topical therapies, a healing ointment of 41% petrolatum or silver sulfadiazine cream 1%. The agents were administered using a thick (1-2 mm) application and proportionally thicker (3 mm or more) covering.
“Thin or moderately applied topical agents, even if applied just before RT, may have minimal influence on skin dose regardless of beam energy or beam incidence,” the investigators wrote. However, the findings do suggest that “applying very thick amounts of a topical agent before RT may increase the surface dose and should be avoided,” they said.
The authors reported that the study was funded by development funds from the University of Pennsylvania, Philadelphia. One of the coauthors, James M. Metz, MD, disclosed service on advisory boards for Ion Beam Applications and Varian Medical Systems for proton therapy; however, these roles were not relevant to this study.
SOURCE: Baumann BC et al. JAMA Oncol. 2018 Oct 18. doi: 10.1001/jamaoncol.2018.4292.
Moderate application of topical therapies directly prior to radiotherapy (RT) treatment sessions may be safe and might exhibit minimal effects on delivered treatment dose, investigators report.
The researchers found that although avoiding topical agents prior to radiation treatments was widespread, preclinical data indicate that there is no difference in the radiation dose on the skin with or without a 1- to 2-mm-thick layer of topical agent.
In an online survey, which queried 133 patients and 108 clinicians in relation to existing practices surrounding topical agent use, 83.4% of patients were informed to discontinue application of topical therapies directly before RT treatment sessions. In addition, 54.1% were told to clean and remove any enduring topical agents before treatment. Among clinicians, 91.4% reported to have received or given advice to stop application of topicals before obtaining RT treatment, Brian C. Baumann, MD, and his associates reported in JAMA Oncology.
However, in a preclinical study using a mouse- and tissue-equivalent phantom model to determine the dosimetric effects of concomitant topical agent use with daily RT treatments, Dr. Baumann, of Washington University in St. Louis, and his colleagues determined that when a topical agent was given prior to RT at a thickness below 2 mm, no changes were seen in radiation treatment dose, irrespective of depth, photon and electron energy level, or beam angle.
However, a proportionally thicker covering (3 mm or more) caused a bolus effect at the surface, which resulted in electron dose increases of 2%-5%, and photon increases of 15%-35%, when compared with controls.
Investigators measured radiation dose and photon beam intensity at various surface depths after applying two commonly used topical therapies, a healing ointment of 41% petrolatum or silver sulfadiazine cream 1%. The agents were administered using a thick (1-2 mm) application and proportionally thicker (3 mm or more) covering.
“Thin or moderately applied topical agents, even if applied just before RT, may have minimal influence on skin dose regardless of beam energy or beam incidence,” the investigators wrote. However, the findings do suggest that “applying very thick amounts of a topical agent before RT may increase the surface dose and should be avoided,” they said.
The authors reported that the study was funded by development funds from the University of Pennsylvania, Philadelphia. One of the coauthors, James M. Metz, MD, disclosed service on advisory boards for Ion Beam Applications and Varian Medical Systems for proton therapy; however, these roles were not relevant to this study.
SOURCE: Baumann BC et al. JAMA Oncol. 2018 Oct 18. doi: 10.1001/jamaoncol.2018.4292.
REPORTING FROM JAMA ONCOLOGY
Key clinical point: Modest application of topical agents preceding radiotherapy (RT) treatment may be safe for patients.
Major finding: When common topicals were applied at a thickness of less than 2 mm, negligible effects on radiation dose were seen.
Study details: An online survey consisting of 133 patients and 108 clinicians, in addition to a tissue-equivalent phantom and mouse model preclinical study.
Disclosures: The study was funded by development funds from the University of Pennsylvania, Philadelphia. The authors had no disclosures relevant to this study.
Source: Baumann BC et al. JAMA Oncol. 2018 Oct 18. doi: 10.1001/jamaoncol.2018.4292.
Novel Nordic Study Reveals Diclofenac’s Cardiovascular Risks
Risk of major adverse cardiovascular events was increased by 50%, compared with no therapy.
In the largest analysis ever of cardiovascular risk associated with the initiation of nonsteroidal anti-inflammatory drugs (NSAIDs), diclofenac was associated with higher risk for adverse cardiovascular outcomes. The study findings were published online September 4 in BMJ.
Those beginning diclofenac had a 50% increased 30-day risk for a composite outcome of major adverse cardiovascular events (MACE), compared with individuals who did not initiate an NSAID or acetaminophen (95% confidence interval for incidence rate ratio, 1.4–1.7).
The risk was still significantly elevated when the study’s first author, Morten Schmidt, MD, PhD, of the Department of Clinical Epidemiology at Aarhus University in Denmark, and his colleagues compared diclofenac initiation with beginning other NSAIDs or acetaminophen. Compared with that associated with ibuprofen or acetaminophen, the MACE risk was elevated 20% in diclofenac initiators (95% CI, 1.1–1.3 for both). Initiating diclofenac was associated with 30% greater risk for MACE, compared with initiating naproxen (95% CI, 1.1–1.5).
“Diclofenac is the most frequently used NSAID in low-, middle-, and high-income countries and is available over the counter in most countries; therefore, its cardiovascular risk profile is of major clinical and public health importance,” said Dr. Schmidt and his coauthors.
In all, the study included 1,370,832 individuals who initiated diclofenac, 3,878,454 ibuprofen initiators, 291,490 naproxen initiators, and 764,781 acetaminophen initiators. Those starting diclofenac were compared with those starting other medications and with 1,303,209 individuals who sought health care but did not start one of the medications.
Novel Methodology
The researchers used the longstanding and comprehensive Danish health registry system to their advantage in designing a cohort trial that was modeled to resemble a clinical trial. For each month, beginning in 1996 and continuing through 2016, Dr. Schmidt and his collaborators assembled propensity-matched cohorts of individuals to compare each study group. The study design achieved many of the aims of a clinical trial while working within the ethical constraints of studying medications known to elevate cardiovascular risk.
For each 30-day period, the investigators tracked and compared cardiovascular outcomes for each group. Each month, data for a new cohort were collected, beginning a new “clinical trial.” Individuals could be included in more than one month’s worth of “trial” data as long as they continued to meet inclusion criteria.
The completeness of Danish health data meant that the researchers were confident in data about comorbidities, other prescription medications, and outcomes.
Dr. Schmidt and his colleagues performed subgroup and sensitivity analyses to look at the extent to which preexisting risks for cardiovascular disease mediated MACE risk on diclofenac initiation. They found that diclofenac initiators in the highest risk group had as many as 40 excess cardiovascular events per year—about half of them fatal—that were attributable to starting the medication. Although that group had the highest absolute risk, “the relative risks were highest in those with the lowest baseline risk,” said the investigators.
Secondary outcomes for the study included the association between medication use or non-use and each component of the composite primary outcome. These components included first-time occurrences of the nonfatal end points of atrial fibrillation or flutter, ischemic stroke, heart failure, and myocardial infarction. Cardiac death included death from any cardiac cause.
“Supporting use of a combined end point, event rates consistently increased for all individual outcomes” for diclofenac initiators, compared with those who did not start an NSAID, said Dr. Schmidt and his colleagues.
Individuals were excluded if they had known cardiovascular, kidney, liver, or ulcer disease and if they had malignancy or serious mental health diagnoses such as dementia or schizophrenia. Participants, mean age 48 to 56, had to be at least 18 and could not have filled a prescription for an NSAID within the previous 12 months. Men made up 36.6% to 46.3% of the cohorts.
Dr. Schmidt and his collaborators said that in comparison with other NSAIDs, the short half-life of diclofenac means that a supratherapeutic plasma concentration of diclofenac soon after initiation achieves not just cyclooxygenase-2 (COX-2), but also COX-1 inhibition. However, after those high levels fall, patients taking diclofenac spend a substantial period of time with unopposed COX-2 inhibition, a prothrombotic state that also is associated with blood pressure elevation, atherogenesis, and worsening of heart failure.
Diclofenac and ibuprofen entailed similar gastrointestinal bleeding risks, and both medications were associated with a higher risk of bleeding than were ibuprofen, acetaminophen, and no medication.
Public Health Implications
“Comparing diclofenac initiation with no NSAID initiation, the consistency between our results and those of previous meta-analyses of both trial and observational data provides strong evidence to guide clinical decision making,” said Dr. Schmidt and his coauthors.
“Considering its cardiovascular and gastrointestinal risks, however, there is little justification to initiate diclofenac treatment before other traditional NSAIDs,” noted the investigators. “It is time to acknowledge the potential health risk of diclofenac and to reduce its use.”
The study was funded by the Department of Clinical Epidemiology Research Foundation, University of Aarhus, and by the Program for Clinical Research Infrastructure, funded by the Lundbeck Foundation, Novo Nordisk Foundation, and the Danish Research Council. The authors reported that they had no relevant conflicts of interest.
—Kari Oakes
Suggested Reading
Schmidt M, Sørensen HT, Pedersen L. Diclofenac use and cardiovascular risks: series of nationwide cohort studies. BMJ. 2018;362:k3426.
For expert commentary on this article, please visit our Migraine Resource Center online at https://www.mdedge.com/neurologyreviews/migraineresourcecenter/article/177937/headache-migraine/physician-commentary
Risk of major adverse cardiovascular events was increased by 50%, compared with no therapy.
Risk of major adverse cardiovascular events was increased by 50%, compared with no therapy.
In the largest analysis ever of cardiovascular risk associated with the initiation of nonsteroidal anti-inflammatory drugs (NSAIDs), diclofenac was associated with higher risk for adverse cardiovascular outcomes. The study findings were published online September 4 in BMJ.
Those beginning diclofenac had a 50% increased 30-day risk for a composite outcome of major adverse cardiovascular events (MACE), compared with individuals who did not initiate an NSAID or acetaminophen (95% confidence interval for incidence rate ratio, 1.4–1.7).
The risk was still significantly elevated when the study’s first author, Morten Schmidt, MD, PhD, of the Department of Clinical Epidemiology at Aarhus University in Denmark, and his colleagues compared diclofenac initiation with beginning other NSAIDs or acetaminophen. Compared with that associated with ibuprofen or acetaminophen, the MACE risk was elevated 20% in diclofenac initiators (95% CI, 1.1–1.3 for both). Initiating diclofenac was associated with 30% greater risk for MACE, compared with initiating naproxen (95% CI, 1.1–1.5).
“Diclofenac is the most frequently used NSAID in low-, middle-, and high-income countries and is available over the counter in most countries; therefore, its cardiovascular risk profile is of major clinical and public health importance,” said Dr. Schmidt and his coauthors.
In all, the study included 1,370,832 individuals who initiated diclofenac, 3,878,454 ibuprofen initiators, 291,490 naproxen initiators, and 764,781 acetaminophen initiators. Those starting diclofenac were compared with those starting other medications and with 1,303,209 individuals who sought health care but did not start one of the medications.
Novel Methodology
The researchers used the longstanding and comprehensive Danish health registry system to their advantage in designing a cohort trial that was modeled to resemble a clinical trial. For each month, beginning in 1996 and continuing through 2016, Dr. Schmidt and his collaborators assembled propensity-matched cohorts of individuals to compare each study group. The study design achieved many of the aims of a clinical trial while working within the ethical constraints of studying medications known to elevate cardiovascular risk.
For each 30-day period, the investigators tracked and compared cardiovascular outcomes for each group. Each month, data for a new cohort were collected, beginning a new “clinical trial.” Individuals could be included in more than one month’s worth of “trial” data as long as they continued to meet inclusion criteria.
The completeness of Danish health data meant that the researchers were confident in data about comorbidities, other prescription medications, and outcomes.
Dr. Schmidt and his colleagues performed subgroup and sensitivity analyses to look at the extent to which preexisting risks for cardiovascular disease mediated MACE risk on diclofenac initiation. They found that diclofenac initiators in the highest risk group had as many as 40 excess cardiovascular events per year—about half of them fatal—that were attributable to starting the medication. Although that group had the highest absolute risk, “the relative risks were highest in those with the lowest baseline risk,” said the investigators.
Secondary outcomes for the study included the association between medication use or non-use and each component of the composite primary outcome. These components included first-time occurrences of the nonfatal end points of atrial fibrillation or flutter, ischemic stroke, heart failure, and myocardial infarction. Cardiac death included death from any cardiac cause.
“Supporting use of a combined end point, event rates consistently increased for all individual outcomes” for diclofenac initiators, compared with those who did not start an NSAID, said Dr. Schmidt and his colleagues.
Individuals were excluded if they had known cardiovascular, kidney, liver, or ulcer disease and if they had malignancy or serious mental health diagnoses such as dementia or schizophrenia. Participants, mean age 48 to 56, had to be at least 18 and could not have filled a prescription for an NSAID within the previous 12 months. Men made up 36.6% to 46.3% of the cohorts.
Dr. Schmidt and his collaborators said that in comparison with other NSAIDs, the short half-life of diclofenac means that a supratherapeutic plasma concentration of diclofenac soon after initiation achieves not just cyclooxygenase-2 (COX-2), but also COX-1 inhibition. However, after those high levels fall, patients taking diclofenac spend a substantial period of time with unopposed COX-2 inhibition, a prothrombotic state that also is associated with blood pressure elevation, atherogenesis, and worsening of heart failure.
Diclofenac and ibuprofen entailed similar gastrointestinal bleeding risks, and both medications were associated with a higher risk of bleeding than were ibuprofen, acetaminophen, and no medication.
Public Health Implications
“Comparing diclofenac initiation with no NSAID initiation, the consistency between our results and those of previous meta-analyses of both trial and observational data provides strong evidence to guide clinical decision making,” said Dr. Schmidt and his coauthors.
“Considering its cardiovascular and gastrointestinal risks, however, there is little justification to initiate diclofenac treatment before other traditional NSAIDs,” noted the investigators. “It is time to acknowledge the potential health risk of diclofenac and to reduce its use.”
The study was funded by the Department of Clinical Epidemiology Research Foundation, University of Aarhus, and by the Program for Clinical Research Infrastructure, funded by the Lundbeck Foundation, Novo Nordisk Foundation, and the Danish Research Council. The authors reported that they had no relevant conflicts of interest.
—Kari Oakes
Suggested Reading
Schmidt M, Sørensen HT, Pedersen L. Diclofenac use and cardiovascular risks: series of nationwide cohort studies. BMJ. 2018;362:k3426.
For expert commentary on this article, please visit our Migraine Resource Center online at https://www.mdedge.com/neurologyreviews/migraineresourcecenter/article/177937/headache-migraine/physician-commentary
In the largest analysis ever of cardiovascular risk associated with the initiation of nonsteroidal anti-inflammatory drugs (NSAIDs), diclofenac was associated with higher risk for adverse cardiovascular outcomes. The study findings were published online September 4 in BMJ.
Those beginning diclofenac had a 50% increased 30-day risk for a composite outcome of major adverse cardiovascular events (MACE), compared with individuals who did not initiate an NSAID or acetaminophen (95% confidence interval for incidence rate ratio, 1.4–1.7).
The risk was still significantly elevated when the study’s first author, Morten Schmidt, MD, PhD, of the Department of Clinical Epidemiology at Aarhus University in Denmark, and his colleagues compared diclofenac initiation with beginning other NSAIDs or acetaminophen. Compared with that associated with ibuprofen or acetaminophen, the MACE risk was elevated 20% in diclofenac initiators (95% CI, 1.1–1.3 for both). Initiating diclofenac was associated with 30% greater risk for MACE, compared with initiating naproxen (95% CI, 1.1–1.5).
“Diclofenac is the most frequently used NSAID in low-, middle-, and high-income countries and is available over the counter in most countries; therefore, its cardiovascular risk profile is of major clinical and public health importance,” said Dr. Schmidt and his coauthors.
In all, the study included 1,370,832 individuals who initiated diclofenac, 3,878,454 ibuprofen initiators, 291,490 naproxen initiators, and 764,781 acetaminophen initiators. Those starting diclofenac were compared with those starting other medications and with 1,303,209 individuals who sought health care but did not start one of the medications.
Novel Methodology
The researchers used the longstanding and comprehensive Danish health registry system to their advantage in designing a cohort trial that was modeled to resemble a clinical trial. For each month, beginning in 1996 and continuing through 2016, Dr. Schmidt and his collaborators assembled propensity-matched cohorts of individuals to compare each study group. The study design achieved many of the aims of a clinical trial while working within the ethical constraints of studying medications known to elevate cardiovascular risk.
For each 30-day period, the investigators tracked and compared cardiovascular outcomes for each group. Each month, data for a new cohort were collected, beginning a new “clinical trial.” Individuals could be included in more than one month’s worth of “trial” data as long as they continued to meet inclusion criteria.
The completeness of Danish health data meant that the researchers were confident in data about comorbidities, other prescription medications, and outcomes.
Dr. Schmidt and his colleagues performed subgroup and sensitivity analyses to look at the extent to which preexisting risks for cardiovascular disease mediated MACE risk on diclofenac initiation. They found that diclofenac initiators in the highest risk group had as many as 40 excess cardiovascular events per year—about half of them fatal—that were attributable to starting the medication. Although that group had the highest absolute risk, “the relative risks were highest in those with the lowest baseline risk,” said the investigators.
Secondary outcomes for the study included the association between medication use or non-use and each component of the composite primary outcome. These components included first-time occurrences of the nonfatal end points of atrial fibrillation or flutter, ischemic stroke, heart failure, and myocardial infarction. Cardiac death included death from any cardiac cause.
“Supporting use of a combined end point, event rates consistently increased for all individual outcomes” for diclofenac initiators, compared with those who did not start an NSAID, said Dr. Schmidt and his colleagues.
Individuals were excluded if they had known cardiovascular, kidney, liver, or ulcer disease and if they had malignancy or serious mental health diagnoses such as dementia or schizophrenia. Participants, mean age 48 to 56, had to be at least 18 and could not have filled a prescription for an NSAID within the previous 12 months. Men made up 36.6% to 46.3% of the cohorts.
Dr. Schmidt and his collaborators said that in comparison with other NSAIDs, the short half-life of diclofenac means that a supratherapeutic plasma concentration of diclofenac soon after initiation achieves not just cyclooxygenase-2 (COX-2), but also COX-1 inhibition. However, after those high levels fall, patients taking diclofenac spend a substantial period of time with unopposed COX-2 inhibition, a prothrombotic state that also is associated with blood pressure elevation, atherogenesis, and worsening of heart failure.
Diclofenac and ibuprofen entailed similar gastrointestinal bleeding risks, and both medications were associated with a higher risk of bleeding than were ibuprofen, acetaminophen, and no medication.
Public Health Implications
“Comparing diclofenac initiation with no NSAID initiation, the consistency between our results and those of previous meta-analyses of both trial and observational data provides strong evidence to guide clinical decision making,” said Dr. Schmidt and his coauthors.
“Considering its cardiovascular and gastrointestinal risks, however, there is little justification to initiate diclofenac treatment before other traditional NSAIDs,” noted the investigators. “It is time to acknowledge the potential health risk of diclofenac and to reduce its use.”
The study was funded by the Department of Clinical Epidemiology Research Foundation, University of Aarhus, and by the Program for Clinical Research Infrastructure, funded by the Lundbeck Foundation, Novo Nordisk Foundation, and the Danish Research Council. The authors reported that they had no relevant conflicts of interest.
—Kari Oakes
Suggested Reading
Schmidt M, Sørensen HT, Pedersen L. Diclofenac use and cardiovascular risks: series of nationwide cohort studies. BMJ. 2018;362:k3426.
For expert commentary on this article, please visit our Migraine Resource Center online at https://www.mdedge.com/neurologyreviews/migraineresourcecenter/article/177937/headache-migraine/physician-commentary
Study: Problems persist with APMs
Physicians continue to support advanced alternative payment models despite the fact that operational issues have not improved over the last 4 years and new ones have cropped up, according to a follow-up survey conducted by the RAND Corporation for the American Medical Association.
“All the things we heard in 2014 were still present in 2018. Both the challenges that practices had experienced back in 2014 having to do with data timeliness, data completeness and accuracy, payment model execution, all those challenges persisted,” Mark W. Friedberg, MD, senior physician policy researcher at RAND, said in an interview.
RAND surveyed 31 practices of varying practice size and specialty across six geographic regions, some of which participated in the 2014 survey. Supplemental information was provided by interviews with 32 market observers, 8 health plan leaders, 10 hospital and hospital system leaders, 10 state and local medical society leaders, and 4 chapter leaders with MGMA (formerly the Medical Group Management Association).
“We had thought we would hear that the problem had gotten a little bit better since there has been some investment in trying to tamp down the wide range of measures that are involved in these alternative payment models,” Dr. Friedberg said. “We did not see any evidence of that having any effect on the practices that participated in this study this time around.”
Indeed, concerns reported in 2014 were again reported in 2018, along with a new set of concerns, including the perceived pace of change in alternative payment models (APMs), the complexity of APMs, and physician concerns over two-sided risk models.
“Practices, especially those that participated both times, said in 2014 we had these challenges [of rapid changes in APM models] and since then, things have just gotten a lot faster,” he said, noting that doctors are complaining of models that are going through changes, sometimes without much warning. “They are changing quite rapidly from year to year. If you look at the MACRA QPP [Quality Payment Program] for example, that model changes every year to some extent and those things are hard for them to keep up with.”
Running hand in hand with the change is the complexity of the changes, a result of expanding performance measures and uncertainty with thresholds for penalties and rewards and in some ways has had little impact on improving care.
Dr. Friedberg noted that some practices are hiring people to examine APMs to devise strategic ways to choose and report data for maximum return.
“In a practice, for example, if their quality of care was already very good, what these folks ended up doing was help them choose measures and work the attribution algorithms in a strategic way to either guarantee a bonus or minimize the risk of incurring a penalty,” he said.
He also noted that practices appear to becoming more risk averse.
“We heard a lot more of the following thing, which is that if [practices] were in a two-sided risk model, several of them reported trying and succeeding in some cases offloading the downside risk to partners,” Dr. Friedberg reported. “And what this resulted in was that the practice, even though from the payer’s perspective they are in a two-sided model, the practice was actually in a one-sided model with a partner who is taking all of the downside risk and a portion of the upside risk, leaving a small upside risk proposition that remained for the practice.”
He said the range of partners that were absorbing the downside risk included hospitals, device manufacturers, consulting companies, or private equity firms.
Despite the concerns surrounding APMs, Dr. Friedberg said that “we did not hear practices broadly saying that they just weren’t interested in alternative payment models. In general, practices still remained pretty enthusiastic about these alternative payment models in theory. If they could be made simpler, if the pace of change weren’t quite so fast, that they would have a chance to really do some important care improvements in alternative payment models.”
He noted some of the surveyed practices were able to make investments in care as a direct result of participating in APMs, such as in behavioral health capabilities in primary care, for example, leading to quality of care improvements.
However, these issues could reveal a future unwillingness to participate in APMs, especially two-sided risk models, something at least the Centers for Medicare & Medicaid Services are pushing for as a stated goal of the QPP is to get practices to participate in APMs and take on more risk.
The growing aversion to taking on downside risk could lead practices to simply stay in fee for service and simply take the payment penalty because it is a fixed amount that can be planned for, as opposed to the fluctuations of bonuses and penalties that comes with a rapidly changing APM environment, Dr. Friedberg said.
Going forward, the report makes a number of recommendations to help create an environment that would potentially make APMs more successful, including simplifying the models; creating stable, predictable, and moderately paced pathways to APM participation; making data available in a more timely fashion; minimizing downside risk or helping practices better manage it; and designing APMs that will encourage clinical changes to help improve the effectiveness of care delivered.
Physicians continue to support advanced alternative payment models despite the fact that operational issues have not improved over the last 4 years and new ones have cropped up, according to a follow-up survey conducted by the RAND Corporation for the American Medical Association.
“All the things we heard in 2014 were still present in 2018. Both the challenges that practices had experienced back in 2014 having to do with data timeliness, data completeness and accuracy, payment model execution, all those challenges persisted,” Mark W. Friedberg, MD, senior physician policy researcher at RAND, said in an interview.
RAND surveyed 31 practices of varying practice size and specialty across six geographic regions, some of which participated in the 2014 survey. Supplemental information was provided by interviews with 32 market observers, 8 health plan leaders, 10 hospital and hospital system leaders, 10 state and local medical society leaders, and 4 chapter leaders with MGMA (formerly the Medical Group Management Association).
“We had thought we would hear that the problem had gotten a little bit better since there has been some investment in trying to tamp down the wide range of measures that are involved in these alternative payment models,” Dr. Friedberg said. “We did not see any evidence of that having any effect on the practices that participated in this study this time around.”
Indeed, concerns reported in 2014 were again reported in 2018, along with a new set of concerns, including the perceived pace of change in alternative payment models (APMs), the complexity of APMs, and physician concerns over two-sided risk models.
“Practices, especially those that participated both times, said in 2014 we had these challenges [of rapid changes in APM models] and since then, things have just gotten a lot faster,” he said, noting that doctors are complaining of models that are going through changes, sometimes without much warning. “They are changing quite rapidly from year to year. If you look at the MACRA QPP [Quality Payment Program] for example, that model changes every year to some extent and those things are hard for them to keep up with.”
Running hand in hand with the change is the complexity of the changes, a result of expanding performance measures and uncertainty with thresholds for penalties and rewards and in some ways has had little impact on improving care.
Dr. Friedberg noted that some practices are hiring people to examine APMs to devise strategic ways to choose and report data for maximum return.
“In a practice, for example, if their quality of care was already very good, what these folks ended up doing was help them choose measures and work the attribution algorithms in a strategic way to either guarantee a bonus or minimize the risk of incurring a penalty,” he said.
He also noted that practices appear to becoming more risk averse.
“We heard a lot more of the following thing, which is that if [practices] were in a two-sided risk model, several of them reported trying and succeeding in some cases offloading the downside risk to partners,” Dr. Friedberg reported. “And what this resulted in was that the practice, even though from the payer’s perspective they are in a two-sided model, the practice was actually in a one-sided model with a partner who is taking all of the downside risk and a portion of the upside risk, leaving a small upside risk proposition that remained for the practice.”
He said the range of partners that were absorbing the downside risk included hospitals, device manufacturers, consulting companies, or private equity firms.
Despite the concerns surrounding APMs, Dr. Friedberg said that “we did not hear practices broadly saying that they just weren’t interested in alternative payment models. In general, practices still remained pretty enthusiastic about these alternative payment models in theory. If they could be made simpler, if the pace of change weren’t quite so fast, that they would have a chance to really do some important care improvements in alternative payment models.”
He noted some of the surveyed practices were able to make investments in care as a direct result of participating in APMs, such as in behavioral health capabilities in primary care, for example, leading to quality of care improvements.
However, these issues could reveal a future unwillingness to participate in APMs, especially two-sided risk models, something at least the Centers for Medicare & Medicaid Services are pushing for as a stated goal of the QPP is to get practices to participate in APMs and take on more risk.
The growing aversion to taking on downside risk could lead practices to simply stay in fee for service and simply take the payment penalty because it is a fixed amount that can be planned for, as opposed to the fluctuations of bonuses and penalties that comes with a rapidly changing APM environment, Dr. Friedberg said.
Going forward, the report makes a number of recommendations to help create an environment that would potentially make APMs more successful, including simplifying the models; creating stable, predictable, and moderately paced pathways to APM participation; making data available in a more timely fashion; minimizing downside risk or helping practices better manage it; and designing APMs that will encourage clinical changes to help improve the effectiveness of care delivered.
Physicians continue to support advanced alternative payment models despite the fact that operational issues have not improved over the last 4 years and new ones have cropped up, according to a follow-up survey conducted by the RAND Corporation for the American Medical Association.
“All the things we heard in 2014 were still present in 2018. Both the challenges that practices had experienced back in 2014 having to do with data timeliness, data completeness and accuracy, payment model execution, all those challenges persisted,” Mark W. Friedberg, MD, senior physician policy researcher at RAND, said in an interview.
RAND surveyed 31 practices of varying practice size and specialty across six geographic regions, some of which participated in the 2014 survey. Supplemental information was provided by interviews with 32 market observers, 8 health plan leaders, 10 hospital and hospital system leaders, 10 state and local medical society leaders, and 4 chapter leaders with MGMA (formerly the Medical Group Management Association).
“We had thought we would hear that the problem had gotten a little bit better since there has been some investment in trying to tamp down the wide range of measures that are involved in these alternative payment models,” Dr. Friedberg said. “We did not see any evidence of that having any effect on the practices that participated in this study this time around.”
Indeed, concerns reported in 2014 were again reported in 2018, along with a new set of concerns, including the perceived pace of change in alternative payment models (APMs), the complexity of APMs, and physician concerns over two-sided risk models.
“Practices, especially those that participated both times, said in 2014 we had these challenges [of rapid changes in APM models] and since then, things have just gotten a lot faster,” he said, noting that doctors are complaining of models that are going through changes, sometimes without much warning. “They are changing quite rapidly from year to year. If you look at the MACRA QPP [Quality Payment Program] for example, that model changes every year to some extent and those things are hard for them to keep up with.”
Running hand in hand with the change is the complexity of the changes, a result of expanding performance measures and uncertainty with thresholds for penalties and rewards and in some ways has had little impact on improving care.
Dr. Friedberg noted that some practices are hiring people to examine APMs to devise strategic ways to choose and report data for maximum return.
“In a practice, for example, if their quality of care was already very good, what these folks ended up doing was help them choose measures and work the attribution algorithms in a strategic way to either guarantee a bonus or minimize the risk of incurring a penalty,” he said.
He also noted that practices appear to becoming more risk averse.
“We heard a lot more of the following thing, which is that if [practices] were in a two-sided risk model, several of them reported trying and succeeding in some cases offloading the downside risk to partners,” Dr. Friedberg reported. “And what this resulted in was that the practice, even though from the payer’s perspective they are in a two-sided model, the practice was actually in a one-sided model with a partner who is taking all of the downside risk and a portion of the upside risk, leaving a small upside risk proposition that remained for the practice.”
He said the range of partners that were absorbing the downside risk included hospitals, device manufacturers, consulting companies, or private equity firms.
Despite the concerns surrounding APMs, Dr. Friedberg said that “we did not hear practices broadly saying that they just weren’t interested in alternative payment models. In general, practices still remained pretty enthusiastic about these alternative payment models in theory. If they could be made simpler, if the pace of change weren’t quite so fast, that they would have a chance to really do some important care improvements in alternative payment models.”
He noted some of the surveyed practices were able to make investments in care as a direct result of participating in APMs, such as in behavioral health capabilities in primary care, for example, leading to quality of care improvements.
However, these issues could reveal a future unwillingness to participate in APMs, especially two-sided risk models, something at least the Centers for Medicare & Medicaid Services are pushing for as a stated goal of the QPP is to get practices to participate in APMs and take on more risk.
The growing aversion to taking on downside risk could lead practices to simply stay in fee for service and simply take the payment penalty because it is a fixed amount that can be planned for, as opposed to the fluctuations of bonuses and penalties that comes with a rapidly changing APM environment, Dr. Friedberg said.
Going forward, the report makes a number of recommendations to help create an environment that would potentially make APMs more successful, including simplifying the models; creating stable, predictable, and moderately paced pathways to APM participation; making data available in a more timely fashion; minimizing downside risk or helping practices better manage it; and designing APMs that will encourage clinical changes to help improve the effectiveness of care delivered.
Intervention may improve genetic testing for HBOC
Researchers from general obstetrics and gynecology (ob.gyn.) practices in New York and Connecticut have shown that (HBOC).

Genetic screening and testing can reduce the morbidity and mortality from breast, ovarian, and endometrial cancers through prevention and early detection. Mark S. DeFrancesco, MD, of Westwood Women’s Health, Waterbury, Conn., and his colleagues reported that, in spite of the American College of Obstetricians and Gynecologists’ recommendation for ob.gyns. to regularly screen, counsel, and refer accordingly for HBOC (Obstet Gynecol. 2015;125:153843), the “incorporation of hereditary cancer risk assessment and testing remains underutilized in the [ob.gyn.] setting.” The authors have addressed this issue in their own practice with promising results and important caveats (Obstet Gynecol 2018;132:1121-9).
The intervention included a process evaluation, improvements to patient work flow, and training of providers by genetic counselors and engineering personnel from the testing laboratory (Myriad Genetics), which provided support for the study. Patients in the study completed a family history questionnaire and, those meeting National Comprehensive Cancer Center Network criteria for genetic testing, were given pretest counseling and offered testing on the same day or referral for testing within 2 weeks.
Of the 3,811 women who completed the questionnaire, 24% (906) met NCCN criteria, 90% of whom were offered testing. However, only 52% (165) of patients who agreed to testing underwent genetic evaluation. This included 70% of patients who were offered same-day testing and 35% of patients who were offered a referral appointment for testing.
Conversations about HBOC and genetic testing can be complicated and may not be a patient’s initial priority. The authors should be commended for identifying the vast majority of high-risk patients. However, only half of patients meeting criteria completed testing and 10% who should have been offered testing were not – numbers still well below target.
Incorporation of family history questionnaires should become commonplace in the generalist’s office and optimizing EHRs may be an opportunity for rapid risk interpretation. As the success of same-day genetic testing was striking, opportunities for partnerships with insurance companies and private laboratories are likely needed to make this a more feasible option. Lastly, assessing women’s knowledge and attitudes around genetic testing could help to more specifically address barriers to testing in future interventions.
Improving genetic screening and testing completion rates requires a coordinated effort. Using tools and applications to optimize convenience (same-day testing, telemedicine) and simplification (electronic screening platforms), we can strive to appropriately detect all women at high risk for hereditary breast and ovarian cancers.
Michelle Lightfoot is a gynecologic oncology fellow at the Ohio State University in Columbus. She has no relevant financial disclosures. Email her at obnews@mdedge.com.
Researchers from general obstetrics and gynecology (ob.gyn.) practices in New York and Connecticut have shown that (HBOC).
Genetic screening and testing can reduce the morbidity and mortality from breast, ovarian, and endometrial cancers through prevention and early detection. Mark S. DeFrancesco, MD, of Westwood Women’s Health, Waterbury, Conn., and his colleagues reported that, in spite of the American College of Obstetricians and Gynecologists’ recommendation for ob.gyns. to regularly screen, counsel, and refer accordingly for HBOC (Obstet Gynecol. 2015;125:153843), the “incorporation of hereditary cancer risk assessment and testing remains underutilized in the [ob.gyn.] setting.” The authors have addressed this issue in their own practice with promising results and important caveats (Obstet Gynecol 2018;132:1121-9).
The intervention included a process evaluation, improvements to patient work flow, and training of providers by genetic counselors and engineering personnel from the testing laboratory (Myriad Genetics), which provided support for the study. Patients in the study completed a family history questionnaire and, those meeting National Comprehensive Cancer Center Network criteria for genetic testing, were given pretest counseling and offered testing on the same day or referral for testing within 2 weeks.
Of the 3,811 women who completed the questionnaire, 24% (906) met NCCN criteria, 90% of whom were offered testing. However, only 52% (165) of patients who agreed to testing underwent genetic evaluation. This included 70% of patients who were offered same-day testing and 35% of patients who were offered a referral appointment for testing.
Conversations about HBOC and genetic testing can be complicated and may not be a patient’s initial priority. The authors should be commended for identifying the vast majority of high-risk patients. However, only half of patients meeting criteria completed testing and 10% who should have been offered testing were not – numbers still well below target.
Incorporation of family history questionnaires should become commonplace in the generalist’s office and optimizing EHRs may be an opportunity for rapid risk interpretation. As the success of same-day genetic testing was striking, opportunities for partnerships with insurance companies and private laboratories are likely needed to make this a more feasible option. Lastly, assessing women’s knowledge and attitudes around genetic testing could help to more specifically address barriers to testing in future interventions.
Improving genetic screening and testing completion rates requires a coordinated effort. Using tools and applications to optimize convenience (same-day testing, telemedicine) and simplification (electronic screening platforms), we can strive to appropriately detect all women at high risk for hereditary breast and ovarian cancers.
Michelle Lightfoot is a gynecologic oncology fellow at the Ohio State University in Columbus. She has no relevant financial disclosures. Email her at obnews@mdedge.com.
Researchers from general obstetrics and gynecology (ob.gyn.) practices in New York and Connecticut have shown that (HBOC).
Genetic screening and testing can reduce the morbidity and mortality from breast, ovarian, and endometrial cancers through prevention and early detection. Mark S. DeFrancesco, MD, of Westwood Women’s Health, Waterbury, Conn., and his colleagues reported that, in spite of the American College of Obstetricians and Gynecologists’ recommendation for ob.gyns. to regularly screen, counsel, and refer accordingly for HBOC (Obstet Gynecol. 2015;125:153843), the “incorporation of hereditary cancer risk assessment and testing remains underutilized in the [ob.gyn.] setting.” The authors have addressed this issue in their own practice with promising results and important caveats (Obstet Gynecol 2018;132:1121-9).
The intervention included a process evaluation, improvements to patient work flow, and training of providers by genetic counselors and engineering personnel from the testing laboratory (Myriad Genetics), which provided support for the study. Patients in the study completed a family history questionnaire and, those meeting National Comprehensive Cancer Center Network criteria for genetic testing, were given pretest counseling and offered testing on the same day or referral for testing within 2 weeks.
Of the 3,811 women who completed the questionnaire, 24% (906) met NCCN criteria, 90% of whom were offered testing. However, only 52% (165) of patients who agreed to testing underwent genetic evaluation. This included 70% of patients who were offered same-day testing and 35% of patients who were offered a referral appointment for testing.
Conversations about HBOC and genetic testing can be complicated and may not be a patient’s initial priority. The authors should be commended for identifying the vast majority of high-risk patients. However, only half of patients meeting criteria completed testing and 10% who should have been offered testing were not – numbers still well below target.
Incorporation of family history questionnaires should become commonplace in the generalist’s office and optimizing EHRs may be an opportunity for rapid risk interpretation. As the success of same-day genetic testing was striking, opportunities for partnerships with insurance companies and private laboratories are likely needed to make this a more feasible option. Lastly, assessing women’s knowledge and attitudes around genetic testing could help to more specifically address barriers to testing in future interventions.
Improving genetic screening and testing completion rates requires a coordinated effort. Using tools and applications to optimize convenience (same-day testing, telemedicine) and simplification (electronic screening platforms), we can strive to appropriately detect all women at high risk for hereditary breast and ovarian cancers.
Michelle Lightfoot is a gynecologic oncology fellow at the Ohio State University in Columbus. She has no relevant financial disclosures. Email her at obnews@mdedge.com.
Physician Commentary: Neurology Community Responds to Diclofenac Cardiovascular Risks
In July 2018, The BMJ published a study examining the cardiovascular risks of diclofenac initiation compared with initiation of other traditional non-steroidal anti-inflammatory drugs, initiation of paracetamol, and no initiation. The results showed a 50% increase in adverse events among diclofenac initiators compared with non-initiators (as well as a 20% increase over paracetamol/ibuprofen initiators, and 30% increase over naproxen initiators). (Read the full study here). Here, I asked several of my colleagues to weigh in on the results of this study and its implications for our practices, and then I share my own thoughts on these findings:

Stewart J. Tepper, MD, FAHS
Professor of Neurology
Geisel School of Medicine at Dartmouth
There have been previous studies and meta-analyses demonstrating the cardiovascular risks of diclofenac. This very large cohort study highlights the magnitude of effects for both those patients at high risk and at low risk for cardiovascular disease. Diclofenac has many advantages for migraine treatment, such as a rapid onset of action in its liquid form, but it has higher risks for major cardiac events than most currently available nonsteroidal anti-inflammatory drugs (NSAIDs). As providers, we must be judicious in diclofenac use and informative with our patients.

Marcelo Bigal, MD, PhD
Chief Medical Officer, Purdue Pharma
It is well established that NSAIDs are associated with increased risk of poor cardiovascular outcomes. This study offers powerful evidence that the risk after frequent diclofenac use is disproportionally increased relative to other commonly used NSAIDs, such as ibuprofen or naproxen. It is relevant to discuss the implications of the findings for the treatment of migraine.
The acute treatment of migraine associated with attack-related disability should favor triptans as first line therapy, not NSAIDs. Because triptans are vasoconstrictive medications, unmet needs exist in patients at cardiovascular risk. Anti-CGRP acute migraine therapies, as well as “ditans” (5HT-1f antagonist) are under regulatory review and may address the needs of these patients. In the context of acute migraine therapy, diclofenac and NSAIDs are typically used instead of triptans, or with triptans when additional efficacy is needed. We certainly find that the use of diclofenac in these situations should be judicious, and reserved to those who clearly need it, have infrequent migraine attacks, and are otherwise healthy.
Diclofenac is also often used in the emergency department in many countries as a rescue therapy. In a series of clinical trials where we tested most commonly used drugs in this setting in Brazil, we found that efficacies were 83.6% for intravenous dipyrone, 66.7% for intramuscular diclofenac and 81.8% for intravenous chlorpromazine. We continue to believe that diclofenac is an important, non-sedative and non-opioid option for the management of headaches in the emergency department, assuming that at discharge, patients would receive proper guidance on the management of migraine without relying on frequent use of NSAIDs.

Jack Schim, MD
Co-Director, The Headache Center of Southern California
This article supports findings of prior epidemiologic studies correlating exposure to NSAIDs with increased cerebrovascular and cardiovascular risk. Prior studies have shown a dose-related response in risk associated with NSAID therapy, supporting a causal association. However, while relative risk is significantly higher in individuals with NSAID exposure, the absolute risk remains very low. The greater risk from NSAIDs continues to be to the kidneys, and to the stomach.
As with all therapies, we need to weigh the advantages and disadvantages of NSAID therapy with our headache patients. All medications carry their own risks. For acute treatment of migraines, our primary tool, triptans, are contraindicated in a significant subset of individuals, including patients with ischemic coronary artery as well as those with history of stroke or transient ischemic attack (TIA). The alternatives, NSAIDs, dopamine blocking agents, have utility and risks.
Diclofenac powder to be dissolved in water is an effective abortive for migraine for many individuals. In general, our patients have intermittent exposure, preferably not more than 2 days per week. For the appropriate individual, NSAIDS, including diclofenac, remain an important tool in the acute care armamentarium.

Rob Cowan, MD, FAAN, FAHS
Higgins Professor of Neurology and Neurosciences
Chief, Division of Headache Medicine, Dept. of Neurology and Neurosciences
Director, Stanford University School of Medicine
These kinds of large, population-based studies must be interpreted with caution. While they may emulate the protocol of prospective studies, they lack proper inclusion/exclusion criteria, particularly with respect to indication. It may be reasonable to assume that the population of diclofenac users is "sicker" than the general population and the population that is using cheaper, more accessible NSAIDs or paracetamol. Without knowing the access and economic issues in Denmark, it is difficult to weigh these variables in the study. Thus, while it is certainly an important issue to explore (the relative risks and benefits of a given medication within a class), the absence of a well-designed, prospective study precludes any definitive conclusion regarding relative safety and risk profile for Diclofenac.
+++
These are great comments by my colleagues. My impression after seeing the data and reading my colleague’s comments, is that diclofenac may be riskier than other NSAIDs in this study; but when used properly in generally healthy migraineurs, it is probably more effective than dangerous when evaluating the risk/benefit ratio. When diclofenac is used as an oral solution (Cambia), 2 days per week or less, in a patient without serious gastrointestinal, renal, cardiac or hypertensive issues, it appears to pose little risk to the patient. When given to the wrong patient, or when taken too frequently, is could be dangerous. What I really like about this preparation is that it causes fewer adverse events compared to triptans and works very quickly. It can be used when triptans have been used enough that week or if they tend to cause significant adverse events when taken. We can use diclofenac for our headache patients, but we should remain vigilant to give it cautiously and only to patients who have no contraindication to its use.
Please write to us at Neurology Reviews Migraine Resource Center (mrc@mdedge.com) with your opinions.
Alan M. Rapoport, M.D.
Editor-in-Chief
Migraine Resource Center
Clinical Professor of Neurology
The David Geffen School of Medicine at UCLA
Los Angeles, California
In July 2018, The BMJ published a study examining the cardiovascular risks of diclofenac initiation compared with initiation of other traditional non-steroidal anti-inflammatory drugs, initiation of paracetamol, and no initiation. The results showed a 50% increase in adverse events among diclofenac initiators compared with non-initiators (as well as a 20% increase over paracetamol/ibuprofen initiators, and 30% increase over naproxen initiators). (Read the full study here). Here, I asked several of my colleagues to weigh in on the results of this study and its implications for our practices, and then I share my own thoughts on these findings:
Stewart J. Tepper, MD, FAHS
Professor of Neurology
Geisel School of Medicine at Dartmouth
There have been previous studies and meta-analyses demonstrating the cardiovascular risks of diclofenac. This very large cohort study highlights the magnitude of effects for both those patients at high risk and at low risk for cardiovascular disease. Diclofenac has many advantages for migraine treatment, such as a rapid onset of action in its liquid form, but it has higher risks for major cardiac events than most currently available nonsteroidal anti-inflammatory drugs (NSAIDs). As providers, we must be judicious in diclofenac use and informative with our patients.
Marcelo Bigal, MD, PhD
Chief Medical Officer, Purdue Pharma
It is well established that NSAIDs are associated with increased risk of poor cardiovascular outcomes. This study offers powerful evidence that the risk after frequent diclofenac use is disproportionally increased relative to other commonly used NSAIDs, such as ibuprofen or naproxen. It is relevant to discuss the implications of the findings for the treatment of migraine.
The acute treatment of migraine associated with attack-related disability should favor triptans as first line therapy, not NSAIDs. Because triptans are vasoconstrictive medications, unmet needs exist in patients at cardiovascular risk. Anti-CGRP acute migraine therapies, as well as “ditans” (5HT-1f antagonist) are under regulatory review and may address the needs of these patients. In the context of acute migraine therapy, diclofenac and NSAIDs are typically used instead of triptans, or with triptans when additional efficacy is needed. We certainly find that the use of diclofenac in these situations should be judicious, and reserved to those who clearly need it, have infrequent migraine attacks, and are otherwise healthy.
Diclofenac is also often used in the emergency department in many countries as a rescue therapy. In a series of clinical trials where we tested most commonly used drugs in this setting in Brazil, we found that efficacies were 83.6% for intravenous dipyrone, 66.7% for intramuscular diclofenac and 81.8% for intravenous chlorpromazine. We continue to believe that diclofenac is an important, non-sedative and non-opioid option for the management of headaches in the emergency department, assuming that at discharge, patients would receive proper guidance on the management of migraine without relying on frequent use of NSAIDs.
Jack Schim, MD
Co-Director, The Headache Center of Southern California
This article supports findings of prior epidemiologic studies correlating exposure to NSAIDs with increased cerebrovascular and cardiovascular risk. Prior studies have shown a dose-related response in risk associated with NSAID therapy, supporting a causal association. However, while relative risk is significantly higher in individuals with NSAID exposure, the absolute risk remains very low. The greater risk from NSAIDs continues to be to the kidneys, and to the stomach.
As with all therapies, we need to weigh the advantages and disadvantages of NSAID therapy with our headache patients. All medications carry their own risks. For acute treatment of migraines, our primary tool, triptans, are contraindicated in a significant subset of individuals, including patients with ischemic coronary artery as well as those with history of stroke or transient ischemic attack (TIA). The alternatives, NSAIDs, dopamine blocking agents, have utility and risks.
Diclofenac powder to be dissolved in water is an effective abortive for migraine for many individuals. In general, our patients have intermittent exposure, preferably not more than 2 days per week. For the appropriate individual, NSAIDS, including diclofenac, remain an important tool in the acute care armamentarium.
Rob Cowan, MD, FAAN, FAHS
Higgins Professor of Neurology and Neurosciences
Chief, Division of Headache Medicine, Dept. of Neurology and Neurosciences
Director, Stanford University School of Medicine
These kinds of large, population-based studies must be interpreted with caution. While they may emulate the protocol of prospective studies, they lack proper inclusion/exclusion criteria, particularly with respect to indication. It may be reasonable to assume that the population of diclofenac users is "sicker" than the general population and the population that is using cheaper, more accessible NSAIDs or paracetamol. Without knowing the access and economic issues in Denmark, it is difficult to weigh these variables in the study. Thus, while it is certainly an important issue to explore (the relative risks and benefits of a given medication within a class), the absence of a well-designed, prospective study precludes any definitive conclusion regarding relative safety and risk profile for Diclofenac.
+++
These are great comments by my colleagues. My impression after seeing the data and reading my colleague’s comments, is that diclofenac may be riskier than other NSAIDs in this study; but when used properly in generally healthy migraineurs, it is probably more effective than dangerous when evaluating the risk/benefit ratio. When diclofenac is used as an oral solution (Cambia), 2 days per week or less, in a patient without serious gastrointestinal, renal, cardiac or hypertensive issues, it appears to pose little risk to the patient. When given to the wrong patient, or when taken too frequently, is could be dangerous. What I really like about this preparation is that it causes fewer adverse events compared to triptans and works very quickly. It can be used when triptans have been used enough that week or if they tend to cause significant adverse events when taken. We can use diclofenac for our headache patients, but we should remain vigilant to give it cautiously and only to patients who have no contraindication to its use.
Please write to us at Neurology Reviews Migraine Resource Center (mrc@mdedge.com) with your opinions.
Alan M. Rapoport, M.D.
Editor-in-Chief
Migraine Resource Center
Clinical Professor of Neurology
The David Geffen School of Medicine at UCLA
Los Angeles, California
In July 2018, The BMJ published a study examining the cardiovascular risks of diclofenac initiation compared with initiation of other traditional non-steroidal anti-inflammatory drugs, initiation of paracetamol, and no initiation. The results showed a 50% increase in adverse events among diclofenac initiators compared with non-initiators (as well as a 20% increase over paracetamol/ibuprofen initiators, and 30% increase over naproxen initiators). (Read the full study here). Here, I asked several of my colleagues to weigh in on the results of this study and its implications for our practices, and then I share my own thoughts on these findings:
Stewart J. Tepper, MD, FAHS
Professor of Neurology
Geisel School of Medicine at Dartmouth
There have been previous studies and meta-analyses demonstrating the cardiovascular risks of diclofenac. This very large cohort study highlights the magnitude of effects for both those patients at high risk and at low risk for cardiovascular disease. Diclofenac has many advantages for migraine treatment, such as a rapid onset of action in its liquid form, but it has higher risks for major cardiac events than most currently available nonsteroidal anti-inflammatory drugs (NSAIDs). As providers, we must be judicious in diclofenac use and informative with our patients.
Marcelo Bigal, MD, PhD
Chief Medical Officer, Purdue Pharma
It is well established that NSAIDs are associated with increased risk of poor cardiovascular outcomes. This study offers powerful evidence that the risk after frequent diclofenac use is disproportionally increased relative to other commonly used NSAIDs, such as ibuprofen or naproxen. It is relevant to discuss the implications of the findings for the treatment of migraine.
The acute treatment of migraine associated with attack-related disability should favor triptans as first line therapy, not NSAIDs. Because triptans are vasoconstrictive medications, unmet needs exist in patients at cardiovascular risk. Anti-CGRP acute migraine therapies, as well as “ditans” (5HT-1f antagonist) are under regulatory review and may address the needs of these patients. In the context of acute migraine therapy, diclofenac and NSAIDs are typically used instead of triptans, or with triptans when additional efficacy is needed. We certainly find that the use of diclofenac in these situations should be judicious, and reserved to those who clearly need it, have infrequent migraine attacks, and are otherwise healthy.
Diclofenac is also often used in the emergency department in many countries as a rescue therapy. In a series of clinical trials where we tested most commonly used drugs in this setting in Brazil, we found that efficacies were 83.6% for intravenous dipyrone, 66.7% for intramuscular diclofenac and 81.8% for intravenous chlorpromazine. We continue to believe that diclofenac is an important, non-sedative and non-opioid option for the management of headaches in the emergency department, assuming that at discharge, patients would receive proper guidance on the management of migraine without relying on frequent use of NSAIDs.
Jack Schim, MD
Co-Director, The Headache Center of Southern California
This article supports findings of prior epidemiologic studies correlating exposure to NSAIDs with increased cerebrovascular and cardiovascular risk. Prior studies have shown a dose-related response in risk associated with NSAID therapy, supporting a causal association. However, while relative risk is significantly higher in individuals with NSAID exposure, the absolute risk remains very low. The greater risk from NSAIDs continues to be to the kidneys, and to the stomach.
As with all therapies, we need to weigh the advantages and disadvantages of NSAID therapy with our headache patients. All medications carry their own risks. For acute treatment of migraines, our primary tool, triptans, are contraindicated in a significant subset of individuals, including patients with ischemic coronary artery as well as those with history of stroke or transient ischemic attack (TIA). The alternatives, NSAIDs, dopamine blocking agents, have utility and risks.
Diclofenac powder to be dissolved in water is an effective abortive for migraine for many individuals. In general, our patients have intermittent exposure, preferably not more than 2 days per week. For the appropriate individual, NSAIDS, including diclofenac, remain an important tool in the acute care armamentarium.
Rob Cowan, MD, FAAN, FAHS
Higgins Professor of Neurology and Neurosciences
Chief, Division of Headache Medicine, Dept. of Neurology and Neurosciences
Director, Stanford University School of Medicine
These kinds of large, population-based studies must be interpreted with caution. While they may emulate the protocol of prospective studies, they lack proper inclusion/exclusion criteria, particularly with respect to indication. It may be reasonable to assume that the population of diclofenac users is "sicker" than the general population and the population that is using cheaper, more accessible NSAIDs or paracetamol. Without knowing the access and economic issues in Denmark, it is difficult to weigh these variables in the study. Thus, while it is certainly an important issue to explore (the relative risks and benefits of a given medication within a class), the absence of a well-designed, prospective study precludes any definitive conclusion regarding relative safety and risk profile for Diclofenac.
+++
These are great comments by my colleagues. My impression after seeing the data and reading my colleague’s comments, is that diclofenac may be riskier than other NSAIDs in this study; but when used properly in generally healthy migraineurs, it is probably more effective than dangerous when evaluating the risk/benefit ratio. When diclofenac is used as an oral solution (Cambia), 2 days per week or less, in a patient without serious gastrointestinal, renal, cardiac or hypertensive issues, it appears to pose little risk to the patient. When given to the wrong patient, or when taken too frequently, is could be dangerous. What I really like about this preparation is that it causes fewer adverse events compared to triptans and works very quickly. It can be used when triptans have been used enough that week or if they tend to cause significant adverse events when taken. We can use diclofenac for our headache patients, but we should remain vigilant to give it cautiously and only to patients who have no contraindication to its use.
Please write to us at Neurology Reviews Migraine Resource Center (mrc@mdedge.com) with your opinions.
Alan M. Rapoport, M.D.
Editor-in-Chief
Migraine Resource Center
Clinical Professor of Neurology
The David Geffen School of Medicine at UCLA
Los Angeles, California