User login
Cutis is a peer-reviewed clinical journal for the dermatologist, allergist, and general practitioner published monthly since 1965. Concise clinical articles present the practical side of dermatology, helping physicians to improve patient care. Cutis is referenced in Index Medicus/MEDLINE and is written and edited by industry leaders.
ass lick
assault rifle
balls
ballsac
black jack
bleach
Boko Haram
bondage
causas
cheap
child abuse
cocaine
compulsive behaviors
cost of miracles
cunt
Daech
display network stats
drug paraphernalia
explosion
fart
fda and death
fda AND warn
fda AND warning
fda AND warns
feom
fuck
gambling
gfc
gun
human trafficking
humira AND expensive
illegal
ISIL
ISIS
Islamic caliphate
Islamic state
madvocate
masturbation
mixed martial arts
MMA
molestation
national rifle association
NRA
nsfw
nuccitelli
pedophile
pedophilia
poker
porn
porn
pornography
psychedelic drug
recreational drug
sex slave rings
shit
slot machine
snort
substance abuse
terrorism
terrorist
texarkana
Texas hold 'em
UFC
section[contains(@class, 'nav-hidden')]
section[contains(@class, 'nav-hidden active')
A peer-reviewed, indexed journal for dermatologists with original research, image quizzes, cases and reviews, and columns.
Enfuvirtide-Induced Cutaneous Amyloidosis
To the Editor:
Cutaneous amyloidosis can be secondary to many causes. We describe a case of amyloidosis that was secondary to the deposition of an antiretroviral drug enfuvirtide and clinically presented as bullae over the anterior abdominal wall.
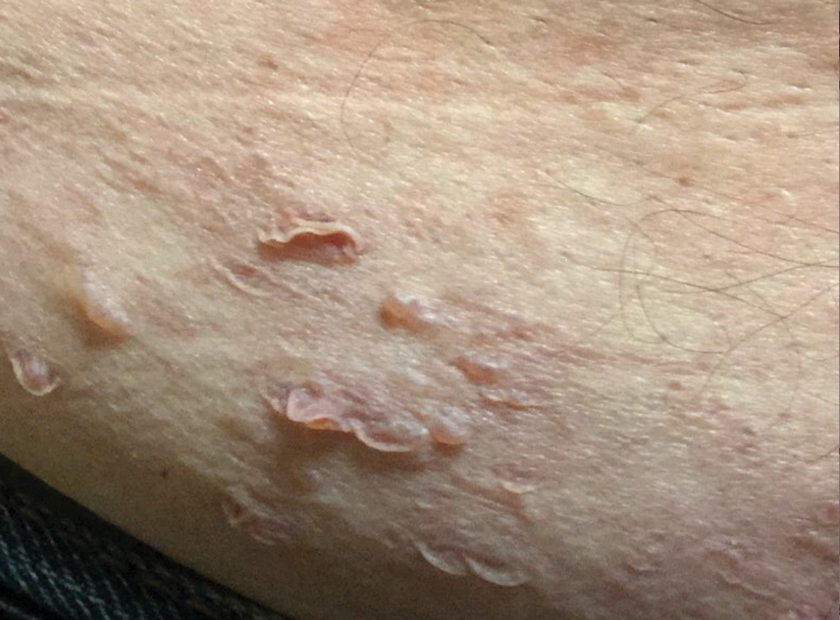
A 65-year-old man with HIV presented with pink vesicles and flaccid bullae on the anterolateral aspect of the lower abdomen (Figure 1) in areas of self-administered subcutaneous injections of enfuvirtide. He reported tissue swelling with a yellow discoloration immediately after injections that would spontaneously subside after a few minutes.
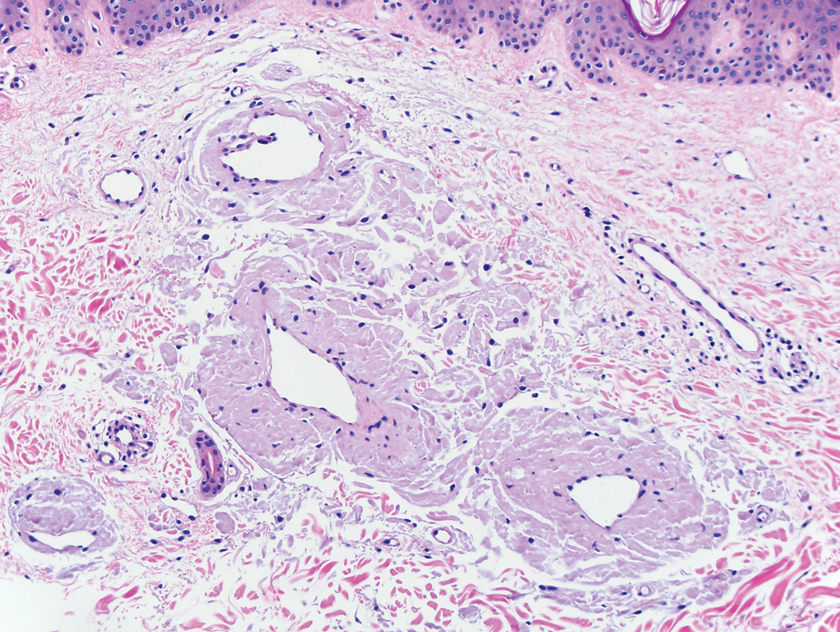
A biopsy from the left lateral abdomen revealed dilated vessels concentrically encompassed by pink globular material and nodular collections of the pink amorphous substance in the upper dermis (Figure 2), which was accompanied by a sparse, perivascular, lymphohistiocytic inflammatory infiltrate; scattered plasma cells; and rare eosinophils in a background of dermal edema. Although Congo red stain was negative, crystal violet revealed metachromatic staining of the globular material that was highlighted as dark violet against a blue background. Given these clinical and histopathologic findings, a diagnosis of drug-induced amyloidosis was made.
Amyloidosis refers to a group of disorders that result from misfolding of proteins in the characteristic beta-pleated sheet structure that can accumulate in various tissues. There are different subtypes of amyloidosis based on the type of protein deposited: immunoglobulin light chain protein (AL); serum amyloid A (AA), an acute-phase reactant accumulating in those with long-standing inflammatory conditions; beta-2 microglobulin (Ab2M) in patients with renal failure; keratin in macular and lichen amyloidosis; pharmaceutical-derived amyloid (eg, enfuvirtide, injectable insulin); and mutated proteins in hereditary amyloidosis such as transthyretin.1 Other familial forms include genetic variants of apolipolipoprotein AII (AApoAI, AApoAII), fibrinogen A alpha chain (AFib), lysozyme (ALys), cystatin C (ACys), and gelsolin (AGel).2
Cutaneous amyloidosis can stem from a systemic disease or arise as a localized phenomenon. Primary cutaneous amyloidosis can present as either macular, lichen, or nodular forms. The pathogenesis of cutaneous nodular amyloidosis differs from that of lichen and macular types and results from deposition of light chain–derived amyloid protein. In contrast, lichen and macular subtypes have keratin-derived amyloid deposits in the papillary dermis and stain positive for keratin antibodies, especially cytokeratins 5 and 6. Primary nodular amyloidosis has a 7% to 50% risk for developing systemic amyloidosis and a 9% risk for local recurrence, hence the necessity to assess for monoclonal gammopathy with urine light chains and serum immunoelectrophoresis.3
Drug-induced amyloidosis is a distinct type of cutaneous amyloidosis that histopathologically resembles nodular amyloidosis. Multiple drugs have been reported in this setting: insulin,4,5 enfuvirtide injections, and liraglutide.6 Enfuvirtide belongs to a class of antiretroviral agents and is a synthetic peptide composed of 36 amino acids. It inhibits the fusion of HIV with the host helper T cell by binding to glycoprotein 41.7 Enfuvirtide-related amyloidosis was described in 3 case reports, 2 that confirmed enfuvirtide as the amyloid constituent by protein analysis.8-10 One study analyzed the amyloid proteome in 50 cases of insulin-derived amyloidosis and 2 cases of enfuvirtide-derived amyloidosis. Laser microdissection–tandem microscopy revealed that the amyloid in such cases was composed of the drug enfuvirtide itself along with deposits of apolipoproteins (E, A-I, A-IV) and serum amyloid P component.4 Additional complications can occur at the site of enfuvirtide injections. A retrospective review of 7 patients with injection-site reactions to enfuvirtide described erythema, induration, and nodules, with histopathologic findings including hypersensitivity reactions and palisaded granulomas resembling granuloma annulare. Amorphous material was noted within histiocytes and in the surrounding connective tissue that was confirmed as enfuvirtide by immunoperoxidase staining.11
In summary, several types of cutaneous amyloidosis occur, including secondary cutaneous involvement by systemic amyloidosis and drug-induced amyloidosis, and notable histopathologic overlap exists between these types. Given the differing treatment requirements depending on the type of cutaneous amyloidosis, obtaining an appropriate clinical history, including the patient’s medication list, is important to ensure the correct diagnosis is reached. Protein analysis with mass spectrometry can be used if the nature of the amyloid remains indeterminate.
- Merlini G, Bellotti V. Molecular mechanisms of amyloidosis. N Engl J Med. 2003;349:583-596.
- Ferri FF. Amyloidosis. In: Ferri F. Ferri’s Clinical Advisor 2016: 5 Books in 1. Elsevier; 2016.
- Kaltoft B, Schmidt G, Lauritzen AF, et al. Primary localised cutaneous amyloidosis—a systematic review. Dan Med J. 2013;60:A4727.
- D’Souza A, Theis JD, Vrana JA, et al. Pharmaceutical amyloidosis associated with subcutaneous insulin and enfuvirtide administration. Amyloid. 2014;21:71-75.
- Sie MP, van der Wiel HE, Smedts FM, et al. Human recombinant insulin and amyloidosis: an unexpected association. Neth J Med. 2010;68:138-140.
- Martins CO, Lezcano C, Yi SS, et al. Novel iatrogenic amyloidosis caused by peptide drug liraglutide: a clinical mimic of AL amyloidosis. Haematologica. 2018;103:E610-E612.
- Lazzarin A, Clotet B, Cooper D, et al. Efficacy of enfuvirtide in patients infected with drug-resistant HIV-1 in Europe and Australia. N Engl J Med. 2003;348:2186-2195.
- Naujokas A, Vidal CI, Mercer SE, et al. A novel form of amyloid deposited at the site of enfuvirtide injection. J Cutan Pathol. 2012;39:220-221; quiz 219.
- Mercer S, Whang T, Vidal C, et al. Massive amyloidosis at the site of enfuvirtide (Fuzeon) injection. J Cutan Pathol. 2011;38:93.
- Morilla ME, Kocher J, Harmaty M. Localized amyloidosis at the site of enfuvirtide injection. Ann Intern Med. 2009;151:515-516.
- Ball RA, Kinchelow T; ISR Substudy Group. Injection site reactions with the HIV-1 fusion inhibitor enfuvirtide. J Am Acad Dermatol. 2003;49:826-831.
To the Editor:
Cutaneous amyloidosis can be secondary to many causes. We describe a case of amyloidosis that was secondary to the deposition of an antiretroviral drug enfuvirtide and clinically presented as bullae over the anterior abdominal wall.
A 65-year-old man with HIV presented with pink vesicles and flaccid bullae on the anterolateral aspect of the lower abdomen (Figure 1) in areas of self-administered subcutaneous injections of enfuvirtide. He reported tissue swelling with a yellow discoloration immediately after injections that would spontaneously subside after a few minutes.
A biopsy from the left lateral abdomen revealed dilated vessels concentrically encompassed by pink globular material and nodular collections of the pink amorphous substance in the upper dermis (Figure 2), which was accompanied by a sparse, perivascular, lymphohistiocytic inflammatory infiltrate; scattered plasma cells; and rare eosinophils in a background of dermal edema. Although Congo red stain was negative, crystal violet revealed metachromatic staining of the globular material that was highlighted as dark violet against a blue background. Given these clinical and histopathologic findings, a diagnosis of drug-induced amyloidosis was made.
Amyloidosis refers to a group of disorders that result from misfolding of proteins in the characteristic beta-pleated sheet structure that can accumulate in various tissues. There are different subtypes of amyloidosis based on the type of protein deposited: immunoglobulin light chain protein (AL); serum amyloid A (AA), an acute-phase reactant accumulating in those with long-standing inflammatory conditions; beta-2 microglobulin (Ab2M) in patients with renal failure; keratin in macular and lichen amyloidosis; pharmaceutical-derived amyloid (eg, enfuvirtide, injectable insulin); and mutated proteins in hereditary amyloidosis such as transthyretin.1 Other familial forms include genetic variants of apolipolipoprotein AII (AApoAI, AApoAII), fibrinogen A alpha chain (AFib), lysozyme (ALys), cystatin C (ACys), and gelsolin (AGel).2
Cutaneous amyloidosis can stem from a systemic disease or arise as a localized phenomenon. Primary cutaneous amyloidosis can present as either macular, lichen, or nodular forms. The pathogenesis of cutaneous nodular amyloidosis differs from that of lichen and macular types and results from deposition of light chain–derived amyloid protein. In contrast, lichen and macular subtypes have keratin-derived amyloid deposits in the papillary dermis and stain positive for keratin antibodies, especially cytokeratins 5 and 6. Primary nodular amyloidosis has a 7% to 50% risk for developing systemic amyloidosis and a 9% risk for local recurrence, hence the necessity to assess for monoclonal gammopathy with urine light chains and serum immunoelectrophoresis.3
Drug-induced amyloidosis is a distinct type of cutaneous amyloidosis that histopathologically resembles nodular amyloidosis. Multiple drugs have been reported in this setting: insulin,4,5 enfuvirtide injections, and liraglutide.6 Enfuvirtide belongs to a class of antiretroviral agents and is a synthetic peptide composed of 36 amino acids. It inhibits the fusion of HIV with the host helper T cell by binding to glycoprotein 41.7 Enfuvirtide-related amyloidosis was described in 3 case reports, 2 that confirmed enfuvirtide as the amyloid constituent by protein analysis.8-10 One study analyzed the amyloid proteome in 50 cases of insulin-derived amyloidosis and 2 cases of enfuvirtide-derived amyloidosis. Laser microdissection–tandem microscopy revealed that the amyloid in such cases was composed of the drug enfuvirtide itself along with deposits of apolipoproteins (E, A-I, A-IV) and serum amyloid P component.4 Additional complications can occur at the site of enfuvirtide injections. A retrospective review of 7 patients with injection-site reactions to enfuvirtide described erythema, induration, and nodules, with histopathologic findings including hypersensitivity reactions and palisaded granulomas resembling granuloma annulare. Amorphous material was noted within histiocytes and in the surrounding connective tissue that was confirmed as enfuvirtide by immunoperoxidase staining.11
In summary, several types of cutaneous amyloidosis occur, including secondary cutaneous involvement by systemic amyloidosis and drug-induced amyloidosis, and notable histopathologic overlap exists between these types. Given the differing treatment requirements depending on the type of cutaneous amyloidosis, obtaining an appropriate clinical history, including the patient’s medication list, is important to ensure the correct diagnosis is reached. Protein analysis with mass spectrometry can be used if the nature of the amyloid remains indeterminate.
To the Editor:
Cutaneous amyloidosis can be secondary to many causes. We describe a case of amyloidosis that was secondary to the deposition of an antiretroviral drug enfuvirtide and clinically presented as bullae over the anterior abdominal wall.
A 65-year-old man with HIV presented with pink vesicles and flaccid bullae on the anterolateral aspect of the lower abdomen (Figure 1) in areas of self-administered subcutaneous injections of enfuvirtide. He reported tissue swelling with a yellow discoloration immediately after injections that would spontaneously subside after a few minutes.
A biopsy from the left lateral abdomen revealed dilated vessels concentrically encompassed by pink globular material and nodular collections of the pink amorphous substance in the upper dermis (Figure 2), which was accompanied by a sparse, perivascular, lymphohistiocytic inflammatory infiltrate; scattered plasma cells; and rare eosinophils in a background of dermal edema. Although Congo red stain was negative, crystal violet revealed metachromatic staining of the globular material that was highlighted as dark violet against a blue background. Given these clinical and histopathologic findings, a diagnosis of drug-induced amyloidosis was made.
Amyloidosis refers to a group of disorders that result from misfolding of proteins in the characteristic beta-pleated sheet structure that can accumulate in various tissues. There are different subtypes of amyloidosis based on the type of protein deposited: immunoglobulin light chain protein (AL); serum amyloid A (AA), an acute-phase reactant accumulating in those with long-standing inflammatory conditions; beta-2 microglobulin (Ab2M) in patients with renal failure; keratin in macular and lichen amyloidosis; pharmaceutical-derived amyloid (eg, enfuvirtide, injectable insulin); and mutated proteins in hereditary amyloidosis such as transthyretin.1 Other familial forms include genetic variants of apolipolipoprotein AII (AApoAI, AApoAII), fibrinogen A alpha chain (AFib), lysozyme (ALys), cystatin C (ACys), and gelsolin (AGel).2
Cutaneous amyloidosis can stem from a systemic disease or arise as a localized phenomenon. Primary cutaneous amyloidosis can present as either macular, lichen, or nodular forms. The pathogenesis of cutaneous nodular amyloidosis differs from that of lichen and macular types and results from deposition of light chain–derived amyloid protein. In contrast, lichen and macular subtypes have keratin-derived amyloid deposits in the papillary dermis and stain positive for keratin antibodies, especially cytokeratins 5 and 6. Primary nodular amyloidosis has a 7% to 50% risk for developing systemic amyloidosis and a 9% risk for local recurrence, hence the necessity to assess for monoclonal gammopathy with urine light chains and serum immunoelectrophoresis.3
Drug-induced amyloidosis is a distinct type of cutaneous amyloidosis that histopathologically resembles nodular amyloidosis. Multiple drugs have been reported in this setting: insulin,4,5 enfuvirtide injections, and liraglutide.6 Enfuvirtide belongs to a class of antiretroviral agents and is a synthetic peptide composed of 36 amino acids. It inhibits the fusion of HIV with the host helper T cell by binding to glycoprotein 41.7 Enfuvirtide-related amyloidosis was described in 3 case reports, 2 that confirmed enfuvirtide as the amyloid constituent by protein analysis.8-10 One study analyzed the amyloid proteome in 50 cases of insulin-derived amyloidosis and 2 cases of enfuvirtide-derived amyloidosis. Laser microdissection–tandem microscopy revealed that the amyloid in such cases was composed of the drug enfuvirtide itself along with deposits of apolipoproteins (E, A-I, A-IV) and serum amyloid P component.4 Additional complications can occur at the site of enfuvirtide injections. A retrospective review of 7 patients with injection-site reactions to enfuvirtide described erythema, induration, and nodules, with histopathologic findings including hypersensitivity reactions and palisaded granulomas resembling granuloma annulare. Amorphous material was noted within histiocytes and in the surrounding connective tissue that was confirmed as enfuvirtide by immunoperoxidase staining.11
In summary, several types of cutaneous amyloidosis occur, including secondary cutaneous involvement by systemic amyloidosis and drug-induced amyloidosis, and notable histopathologic overlap exists between these types. Given the differing treatment requirements depending on the type of cutaneous amyloidosis, obtaining an appropriate clinical history, including the patient’s medication list, is important to ensure the correct diagnosis is reached. Protein analysis with mass spectrometry can be used if the nature of the amyloid remains indeterminate.
- Merlini G, Bellotti V. Molecular mechanisms of amyloidosis. N Engl J Med. 2003;349:583-596.
- Ferri FF. Amyloidosis. In: Ferri F. Ferri’s Clinical Advisor 2016: 5 Books in 1. Elsevier; 2016.
- Kaltoft B, Schmidt G, Lauritzen AF, et al. Primary localised cutaneous amyloidosis—a systematic review. Dan Med J. 2013;60:A4727.
- D’Souza A, Theis JD, Vrana JA, et al. Pharmaceutical amyloidosis associated with subcutaneous insulin and enfuvirtide administration. Amyloid. 2014;21:71-75.
- Sie MP, van der Wiel HE, Smedts FM, et al. Human recombinant insulin and amyloidosis: an unexpected association. Neth J Med. 2010;68:138-140.
- Martins CO, Lezcano C, Yi SS, et al. Novel iatrogenic amyloidosis caused by peptide drug liraglutide: a clinical mimic of AL amyloidosis. Haematologica. 2018;103:E610-E612.
- Lazzarin A, Clotet B, Cooper D, et al. Efficacy of enfuvirtide in patients infected with drug-resistant HIV-1 in Europe and Australia. N Engl J Med. 2003;348:2186-2195.
- Naujokas A, Vidal CI, Mercer SE, et al. A novel form of amyloid deposited at the site of enfuvirtide injection. J Cutan Pathol. 2012;39:220-221; quiz 219.
- Mercer S, Whang T, Vidal C, et al. Massive amyloidosis at the site of enfuvirtide (Fuzeon) injection. J Cutan Pathol. 2011;38:93.
- Morilla ME, Kocher J, Harmaty M. Localized amyloidosis at the site of enfuvirtide injection. Ann Intern Med. 2009;151:515-516.
- Ball RA, Kinchelow T; ISR Substudy Group. Injection site reactions with the HIV-1 fusion inhibitor enfuvirtide. J Am Acad Dermatol. 2003;49:826-831.
- Merlini G, Bellotti V. Molecular mechanisms of amyloidosis. N Engl J Med. 2003;349:583-596.
- Ferri FF. Amyloidosis. In: Ferri F. Ferri’s Clinical Advisor 2016: 5 Books in 1. Elsevier; 2016.
- Kaltoft B, Schmidt G, Lauritzen AF, et al. Primary localised cutaneous amyloidosis—a systematic review. Dan Med J. 2013;60:A4727.
- D’Souza A, Theis JD, Vrana JA, et al. Pharmaceutical amyloidosis associated with subcutaneous insulin and enfuvirtide administration. Amyloid. 2014;21:71-75.
- Sie MP, van der Wiel HE, Smedts FM, et al. Human recombinant insulin and amyloidosis: an unexpected association. Neth J Med. 2010;68:138-140.
- Martins CO, Lezcano C, Yi SS, et al. Novel iatrogenic amyloidosis caused by peptide drug liraglutide: a clinical mimic of AL amyloidosis. Haematologica. 2018;103:E610-E612.
- Lazzarin A, Clotet B, Cooper D, et al. Efficacy of enfuvirtide in patients infected with drug-resistant HIV-1 in Europe and Australia. N Engl J Med. 2003;348:2186-2195.
- Naujokas A, Vidal CI, Mercer SE, et al. A novel form of amyloid deposited at the site of enfuvirtide injection. J Cutan Pathol. 2012;39:220-221; quiz 219.
- Mercer S, Whang T, Vidal C, et al. Massive amyloidosis at the site of enfuvirtide (Fuzeon) injection. J Cutan Pathol. 2011;38:93.
- Morilla ME, Kocher J, Harmaty M. Localized amyloidosis at the site of enfuvirtide injection. Ann Intern Med. 2009;151:515-516.
- Ball RA, Kinchelow T; ISR Substudy Group. Injection site reactions with the HIV-1 fusion inhibitor enfuvirtide. J Am Acad Dermatol. 2003;49:826-831.
Practice Points
- There are multiple types of cutaneous amyloidosis, and proper diagnosis is essential to direct treatment and follow-up care.
- Medication-associated amyloidosis is a rare type of amyloidosis that is not associated with systemic amyloidosis and is treated by switching to alternative medicines.
Asymptomatic Discolored Lesions on the Groin
The Diagnosis: Lichen Planus Pigmentosus-Inversus
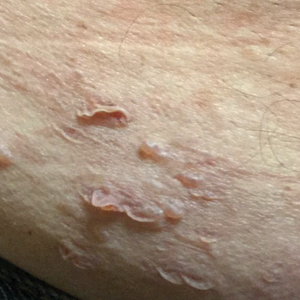
Histopathologic examination revealed hyperkeratosis with dense, bandlike, lymphocytic inflammation at the dermoepidermal junction with associated melanin-containing macrophages in the papillary dermis (Figure 1). The physical examination and histopathology were consistent with a diagnosis of lichen planus pigmentosus-inversus (LPPI). Treatment was discussed with the patient, with options including phototherapy, tacrolimus, or a high-dose steroid. Given that the lesions were asymptomatic and not bothersome, the patient denied treatment and agreed to routine follow-up.
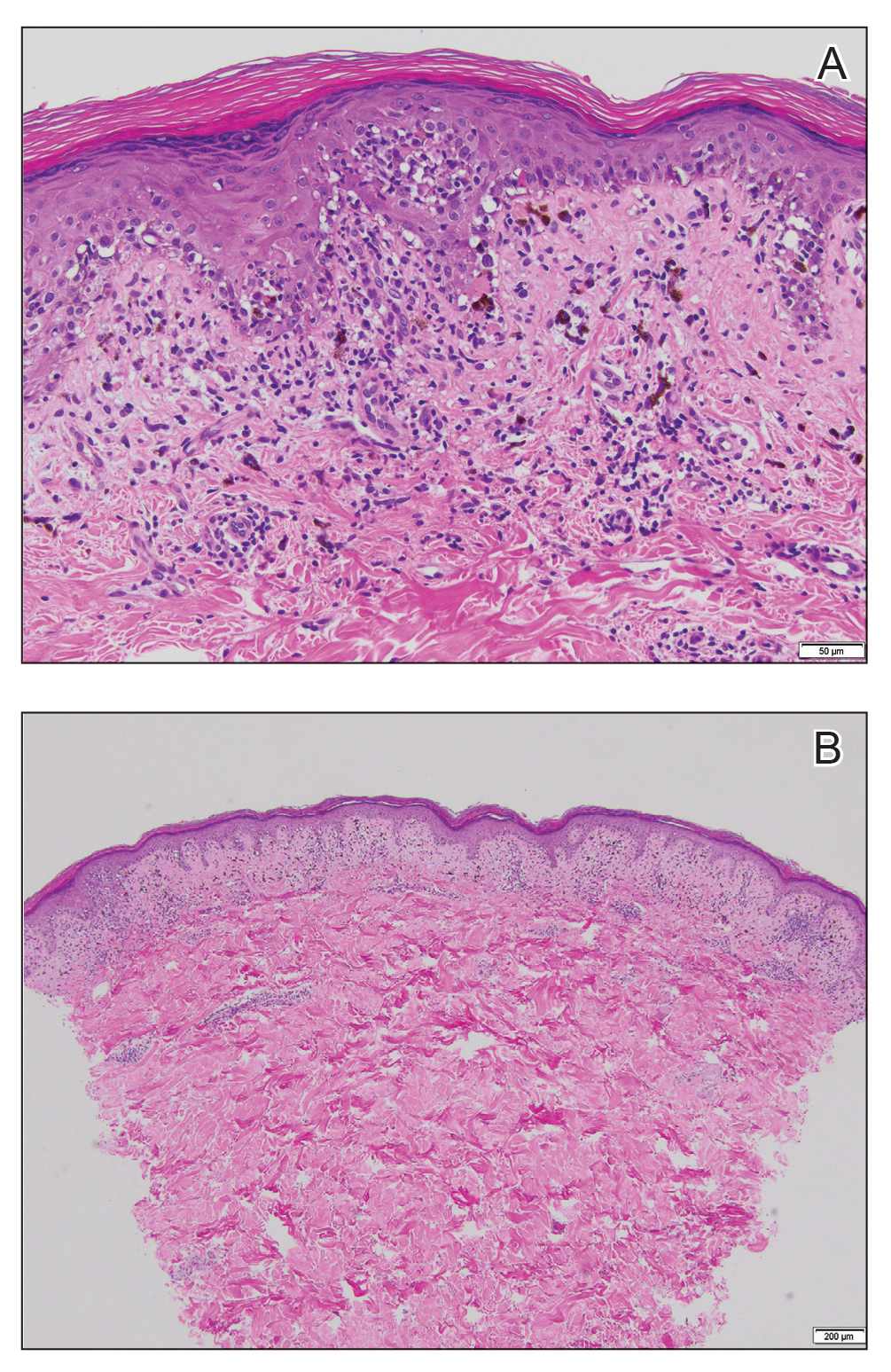
The first case of LPPI was reported in 20011; since then, approximately 100 cases have been reported in the literature.2 A rare variant of lichen planus, LPPI predominantly occurs in middle-aged women.2,3 Lichen planus pigmentosus-inversus is characterized by well-circumscribed, brown macules confined to non-sun-exposed intertriginous areas such as the axillae and groin.2 Although the rash remains localized, multiple lesions could arise in the same area, such as the groin as seen in our patient (Figure 2). Unlike in lichen planus, the oral mucosa, nails, and scalp are not affected. Furthermore, pruritus typically is absent in most cases of LPPI.2,4 Histopathologic findings include an atrophic epidermis with lichenoid infiltrates of lymphocytes and histocytes as well as substantial pigmentary incontinence with melanin-containing macrophages in the papillary dermis.4,5
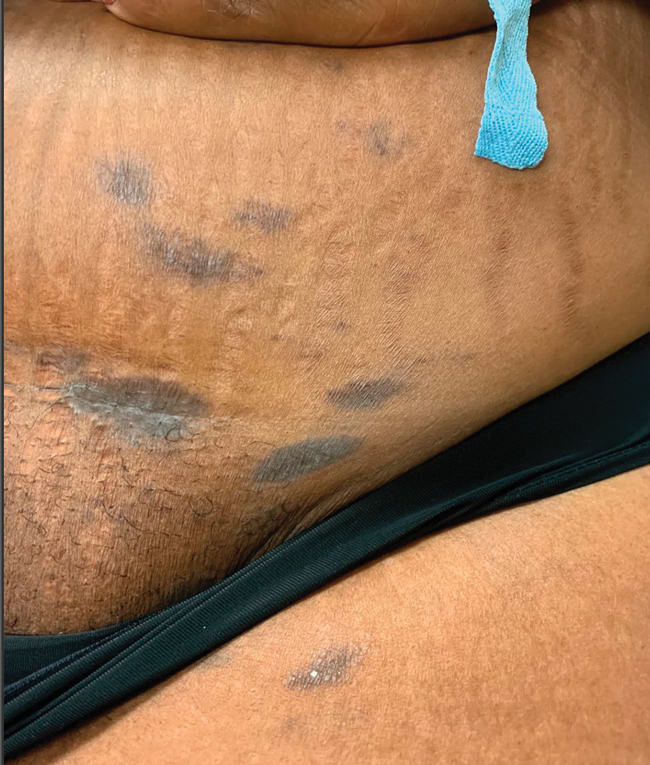
Given the gender, age, and clinical features of our patient, this case represents a classic scenario of LPPI. It currently is unknown if ethnicity plays a role in the disorder. Lichen planus pigmentosus-inversus initially was thought to be more prevalent in White patients; however, studies have been reported in individuals with darker skin.1,2
The main differential diagnosis includes erythema dyschromicum perstans, postinflammatory hyperpigmentation, and lichen planus. Although erythema dyschromicum perstans develops in individuals with darker skin, lesions are restricted to the upper torso and limbs.2-4 In both lichen planus and lichen actinicus, skin findings primarily develop in sun-exposed areas, such as the face, neck, and hands.4,6 Given the negative history of trauma, postinflammatory hyperpigmentation was unlikely in our patient. Furthermore, a distinguishing characteristic of LPPI is the deposition of melanin deep within the dermal layer.3
Lesions developing in nonexposed intertriginous skin makes LPPI unique and distinguishes it from other more common conditions. The lesions commonly are hyperpigmented and are not as pruritic as other lichen-associated conditions. Lichen planus pigmentosus-inversus often persists for months, and the rash generally is resistant to treatment.2,5 Topical tacrolimus and high-dose steroids may improve symptoms, though results have varied substantially. In addition, some cases have resolved spontaneously.1,4,6,7 Because LPPI is asymptomatic and benign, spontaneous resolution and routine care is a reasonable treatment strategy. Some cases have supported this strategy as safe and high-value care.2
- Mohamed M, Korbi M, Hammedi F, et al. Lichen planus pigmentosus inversus: a series of 10 Tunisian patients. Int J Dermatol. 2016;55:1088-1091.
- Lichen planus pigmentosus-inversus: a rare variant of lichen planus. J Am Acad Dermatol. 2015;72(suppl 1):AB239. https://doi.org /10.1016/j.jaad.2015.02.959
- Chen S, Sun W, Zhou G, et al. Lichen planus pigmentosus-inversus: report of three Chinese cases and review of the published work. J Dermatol. 2015;42:77-80.
- Tabanlıoǧlu-Onan D, Íncel-Uysal P, Öktem A, et al. Lichen planus pigmentosus-inversus: a peculiar variant of lichen planus. Dermatologica Sinica. 2017;35:210-212.
- Barros HR, Almeida JR, Mattos e Dinato SL, et al. Lichen planus pigmentosus inversus. An Bras Dermatol. 2013;88(6 suppl 1):146-149.
- Bennàssar A, Mas A, Julià M, et al. Annular plaques in the skin folds: 4 cases of lichen planus pigmentosus-inversus [in Spanish]. Actas Dermosifiliogr. 2009;100:602-605.
- Ghorbel HH, Badri T, Ben Brahim E, et al. Lichen planus pigmentosus inversus. Indian J Dermatol Venereol Leprol. 2014;80:580.
The Diagnosis: Lichen Planus Pigmentosus-Inversus
Histopathologic examination revealed hyperkeratosis with dense, bandlike, lymphocytic inflammation at the dermoepidermal junction with associated melanin-containing macrophages in the papillary dermis (Figure 1). The physical examination and histopathology were consistent with a diagnosis of lichen planus pigmentosus-inversus (LPPI). Treatment was discussed with the patient, with options including phototherapy, tacrolimus, or a high-dose steroid. Given that the lesions were asymptomatic and not bothersome, the patient denied treatment and agreed to routine follow-up.
The first case of LPPI was reported in 20011; since then, approximately 100 cases have been reported in the literature.2 A rare variant of lichen planus, LPPI predominantly occurs in middle-aged women.2,3 Lichen planus pigmentosus-inversus is characterized by well-circumscribed, brown macules confined to non-sun-exposed intertriginous areas such as the axillae and groin.2 Although the rash remains localized, multiple lesions could arise in the same area, such as the groin as seen in our patient (Figure 2). Unlike in lichen planus, the oral mucosa, nails, and scalp are not affected. Furthermore, pruritus typically is absent in most cases of LPPI.2,4 Histopathologic findings include an atrophic epidermis with lichenoid infiltrates of lymphocytes and histocytes as well as substantial pigmentary incontinence with melanin-containing macrophages in the papillary dermis.4,5
Given the gender, age, and clinical features of our patient, this case represents a classic scenario of LPPI. It currently is unknown if ethnicity plays a role in the disorder. Lichen planus pigmentosus-inversus initially was thought to be more prevalent in White patients; however, studies have been reported in individuals with darker skin.1,2
The main differential diagnosis includes erythema dyschromicum perstans, postinflammatory hyperpigmentation, and lichen planus. Although erythema dyschromicum perstans develops in individuals with darker skin, lesions are restricted to the upper torso and limbs.2-4 In both lichen planus and lichen actinicus, skin findings primarily develop in sun-exposed areas, such as the face, neck, and hands.4,6 Given the negative history of trauma, postinflammatory hyperpigmentation was unlikely in our patient. Furthermore, a distinguishing characteristic of LPPI is the deposition of melanin deep within the dermal layer.3
Lesions developing in nonexposed intertriginous skin makes LPPI unique and distinguishes it from other more common conditions. The lesions commonly are hyperpigmented and are not as pruritic as other lichen-associated conditions. Lichen planus pigmentosus-inversus often persists for months, and the rash generally is resistant to treatment.2,5 Topical tacrolimus and high-dose steroids may improve symptoms, though results have varied substantially. In addition, some cases have resolved spontaneously.1,4,6,7 Because LPPI is asymptomatic and benign, spontaneous resolution and routine care is a reasonable treatment strategy. Some cases have supported this strategy as safe and high-value care.2
The Diagnosis: Lichen Planus Pigmentosus-Inversus
Histopathologic examination revealed hyperkeratosis with dense, bandlike, lymphocytic inflammation at the dermoepidermal junction with associated melanin-containing macrophages in the papillary dermis (Figure 1). The physical examination and histopathology were consistent with a diagnosis of lichen planus pigmentosus-inversus (LPPI). Treatment was discussed with the patient, with options including phototherapy, tacrolimus, or a high-dose steroid. Given that the lesions were asymptomatic and not bothersome, the patient denied treatment and agreed to routine follow-up.
The first case of LPPI was reported in 20011; since then, approximately 100 cases have been reported in the literature.2 A rare variant of lichen planus, LPPI predominantly occurs in middle-aged women.2,3 Lichen planus pigmentosus-inversus is characterized by well-circumscribed, brown macules confined to non-sun-exposed intertriginous areas such as the axillae and groin.2 Although the rash remains localized, multiple lesions could arise in the same area, such as the groin as seen in our patient (Figure 2). Unlike in lichen planus, the oral mucosa, nails, and scalp are not affected. Furthermore, pruritus typically is absent in most cases of LPPI.2,4 Histopathologic findings include an atrophic epidermis with lichenoid infiltrates of lymphocytes and histocytes as well as substantial pigmentary incontinence with melanin-containing macrophages in the papillary dermis.4,5
Given the gender, age, and clinical features of our patient, this case represents a classic scenario of LPPI. It currently is unknown if ethnicity plays a role in the disorder. Lichen planus pigmentosus-inversus initially was thought to be more prevalent in White patients; however, studies have been reported in individuals with darker skin.1,2
The main differential diagnosis includes erythema dyschromicum perstans, postinflammatory hyperpigmentation, and lichen planus. Although erythema dyschromicum perstans develops in individuals with darker skin, lesions are restricted to the upper torso and limbs.2-4 In both lichen planus and lichen actinicus, skin findings primarily develop in sun-exposed areas, such as the face, neck, and hands.4,6 Given the negative history of trauma, postinflammatory hyperpigmentation was unlikely in our patient. Furthermore, a distinguishing characteristic of LPPI is the deposition of melanin deep within the dermal layer.3
Lesions developing in nonexposed intertriginous skin makes LPPI unique and distinguishes it from other more common conditions. The lesions commonly are hyperpigmented and are not as pruritic as other lichen-associated conditions. Lichen planus pigmentosus-inversus often persists for months, and the rash generally is resistant to treatment.2,5 Topical tacrolimus and high-dose steroids may improve symptoms, though results have varied substantially. In addition, some cases have resolved spontaneously.1,4,6,7 Because LPPI is asymptomatic and benign, spontaneous resolution and routine care is a reasonable treatment strategy. Some cases have supported this strategy as safe and high-value care.2
- Mohamed M, Korbi M, Hammedi F, et al. Lichen planus pigmentosus inversus: a series of 10 Tunisian patients. Int J Dermatol. 2016;55:1088-1091.
- Lichen planus pigmentosus-inversus: a rare variant of lichen planus. J Am Acad Dermatol. 2015;72(suppl 1):AB239. https://doi.org /10.1016/j.jaad.2015.02.959
- Chen S, Sun W, Zhou G, et al. Lichen planus pigmentosus-inversus: report of three Chinese cases and review of the published work. J Dermatol. 2015;42:77-80.
- Tabanlıoǧlu-Onan D, Íncel-Uysal P, Öktem A, et al. Lichen planus pigmentosus-inversus: a peculiar variant of lichen planus. Dermatologica Sinica. 2017;35:210-212.
- Barros HR, Almeida JR, Mattos e Dinato SL, et al. Lichen planus pigmentosus inversus. An Bras Dermatol. 2013;88(6 suppl 1):146-149.
- Bennàssar A, Mas A, Julià M, et al. Annular plaques in the skin folds: 4 cases of lichen planus pigmentosus-inversus [in Spanish]. Actas Dermosifiliogr. 2009;100:602-605.
- Ghorbel HH, Badri T, Ben Brahim E, et al. Lichen planus pigmentosus inversus. Indian J Dermatol Venereol Leprol. 2014;80:580.
- Mohamed M, Korbi M, Hammedi F, et al. Lichen planus pigmentosus inversus: a series of 10 Tunisian patients. Int J Dermatol. 2016;55:1088-1091.
- Lichen planus pigmentosus-inversus: a rare variant of lichen planus. J Am Acad Dermatol. 2015;72(suppl 1):AB239. https://doi.org /10.1016/j.jaad.2015.02.959
- Chen S, Sun W, Zhou G, et al. Lichen planus pigmentosus-inversus: report of three Chinese cases and review of the published work. J Dermatol. 2015;42:77-80.
- Tabanlıoǧlu-Onan D, Íncel-Uysal P, Öktem A, et al. Lichen planus pigmentosus-inversus: a peculiar variant of lichen planus. Dermatologica Sinica. 2017;35:210-212.
- Barros HR, Almeida JR, Mattos e Dinato SL, et al. Lichen planus pigmentosus inversus. An Bras Dermatol. 2013;88(6 suppl 1):146-149.
- Bennàssar A, Mas A, Julià M, et al. Annular plaques in the skin folds: 4 cases of lichen planus pigmentosus-inversus [in Spanish]. Actas Dermosifiliogr. 2009;100:602-605.
- Ghorbel HH, Badri T, Ben Brahim E, et al. Lichen planus pigmentosus inversus. Indian J Dermatol Venereol Leprol. 2014;80:580.
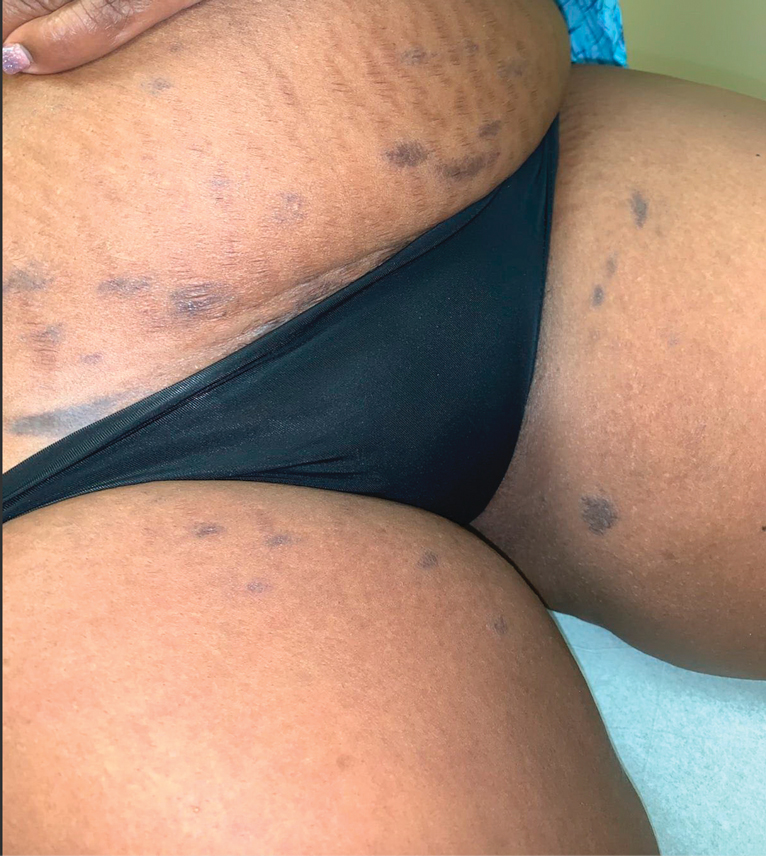
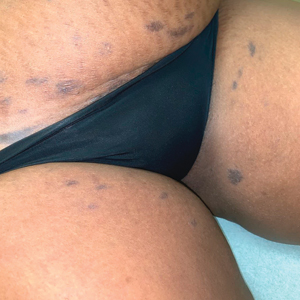
A 45-year-old African American woman presented with an asymptomatic rash that had worsened over the month prior to presentation. It initially began on the upper thighs and then spread to the abdomen, groin, and buttocks. The rash was mildly pruritic and had grown both in size and number of lesions. She had not tried any new over-the-counter medications. Her medical history was notable for late-stage breast cancer diagnosed 4 years prior that was treated with radiation and neoadjuvant NeoPACT—carboplatin, docetaxel, and pembrolizumab. One year prior to presentation, she underwent a lumpectomy that was complicated by gas gangrene of the finger. She has been in remission since the surgery. Physical examination at the current presentation was remarkable for multiple well-circumscribed, hyperpigmented macules on the medial thighs, lower abdomen, and buttocks. Syphilis antibody screening was negative.
Permanent Alopecia in Breast Cancer Patients: Role of Taxanes and Endocrine Therapies
Anagen effluvium during chemotherapy is common, typically beginning within 1 month of treatment onset and resolving by 6 months after the final course.1 Permanent chemotherapy-induced alopecia (PCIA), in which hair loss persists beyond 6 months after chemotherapy without recovery to original density, was first reported in patients following high-dose chemotherapy regimens for allogeneic bone marrow transplantation.2 There are now increasing reports of PCIA in patients with breast cancer; at least 400 such cases have been documented.3-16 In addition to chemotherapy, patients often receive adjuvant endocrine therapy with selective estrogen receptor modulators, aromatase inhibitors, or gonadotropin-releasing hormone agonists.5-16 Endocrine therapies also can lead to alopecia, but their role in PCIA has not been well defined.15,16 We describe 3 patients with breast cancer who experienced PCIA following chemotherapy with taxanes with or without endocrine therapies. We also review the literature on non–bone marrow transplantation PCIA to better characterize this entity and explore the role of endocrine therapies in PCIA.
Case Reports
Patient 1
A 62-year-old woman with a history of stage II invasive ductal carcinoma presented with persistent hair loss 5 years after completing chemotherapy. She underwent 6 cycles of docetaxel and carboplatin along with radiation therapy as well as 1 year of trastuzumab and did not receive endocrine therapy. At the current presentation, she reported patchy hair regrowth that gradually filled in but failed to return to full density. Physical examination revealed the hair was diffusely thin, especially bitemporally (Figures 1A and 1B), and she did not experience any loss of body hair. She had no family history of hair loss. Her medical history was notable for hypertension, chronic obstructive bronchitis, osteopenia, and depression. Her thyroid stimulating hormone (TSH) level was within reference range. Medications included lisinopril, metoprolol, escitalopram, and trazodone. A biopsy from the occipital scalp showed nonscarring alopecia with variation of hair follicle size, a decreased number of hair follicles, and a decreased anagen to telogen ratio (Figure 1C). She was treated with clobetasol solution and minoxidil solution 5% for 1 year with mild improvement. She experienced no further hair loss but did not regain original hair density.
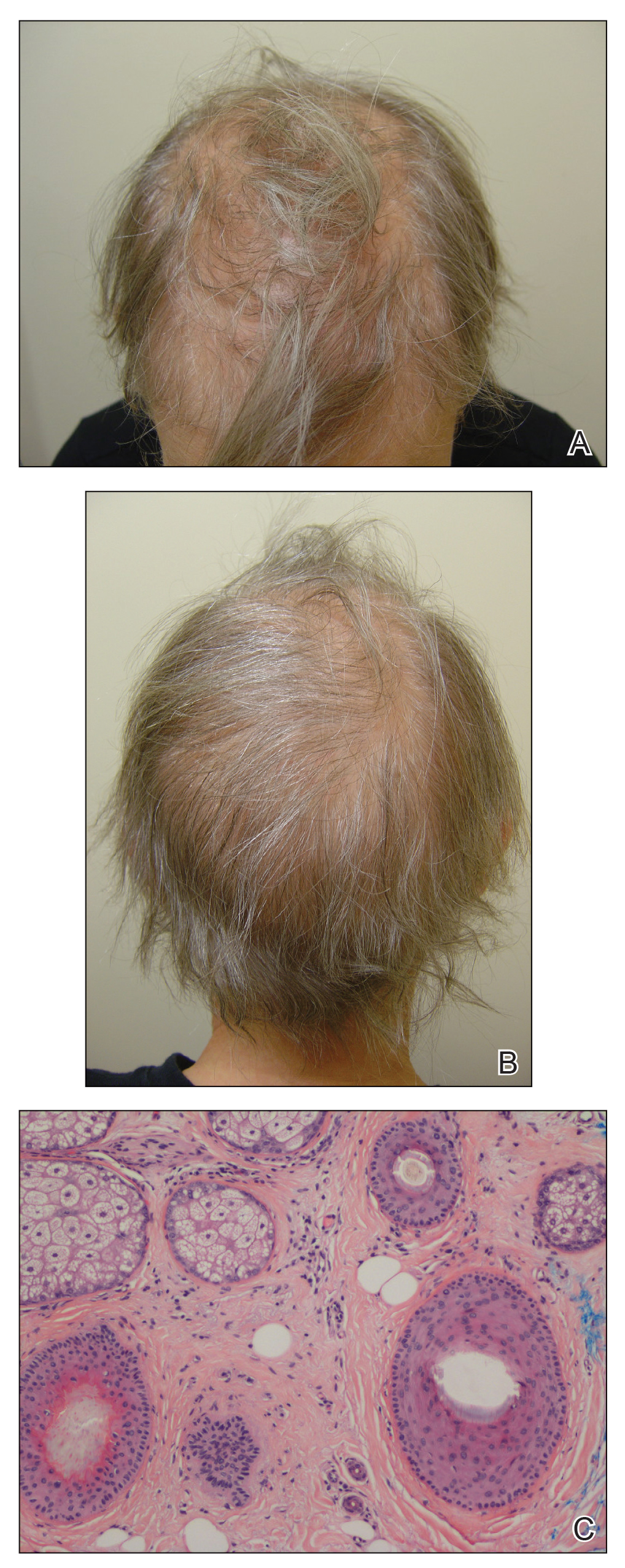
Patient 2
A 35-year-old woman with a history of stage II invasive ductal carcinoma presented with persistent hair loss 10 months after chemotherapy. She underwent 4 cycles of doxorubicin and cyclophosphamide followed by 4 cycles of paclitaxel and was started on trastuzumab. Tamoxifen was initiated 1 month after completing chemotherapy. She received radiation therapy the following month and continued trastuzumab for 1 year. At the current presentation, the patient noted that hair regrowth had started 1 month after the last course of chemotherapy but had progressed slowly. She denied body hair loss. Physical examination revealed diffuse thinning, especially over the crown, with scattered broken hairs throughout the scalp and several miniaturized hairs over the crown. She was evaluated as grade 3 on the Sinclair clinical grading scale used to evaluate female pattern hair loss (FPHL).17 Her family history was remarkable for FPHL in her maternal grandmother. She had no notable medical history, her TSH was normal, and she was taking tamoxifen and trastuzumab. Biopsy was not performed. The patient was started on minoxidil solution 2% and had mild improvement with no further broken-off hairs after 10 months. At that point, she was evaluated as grade 2 to 3 on the Sinclair scale.17
Patient 3
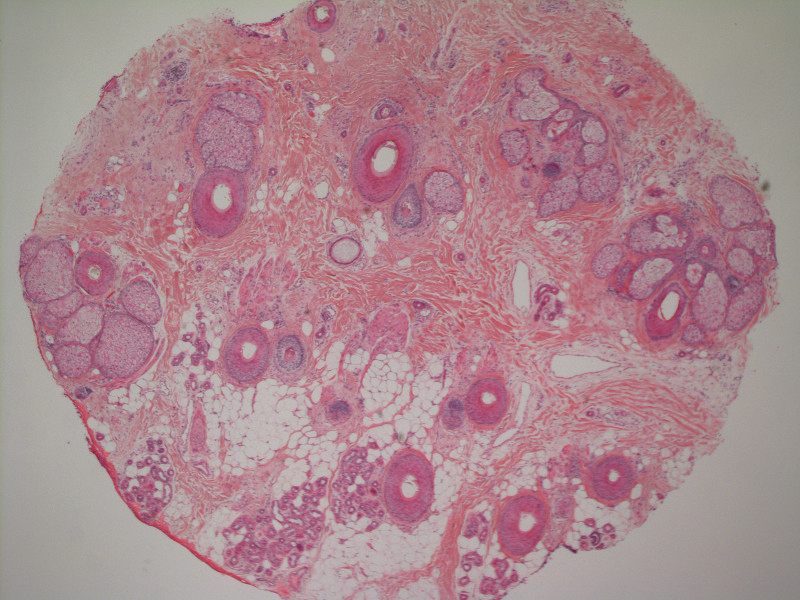
A 51-year-old woman with a history of papillary carcinoma and extensive ductal carcinoma in situ presented with persistent hair loss for 3.5 years following chemotherapy for recurrent breast cancer. After her initial diagnosis in the left breast, she received cyclophosphamide, methotrexate, and 5-fluorouracil but did not receive endocrine therapy. Her hair thinned during chemotherapy but returned to normal density within 1 year. She had a recurrence of the cancer in the right breast 14 years later and received 6 cycles of chemotherapy with cyclophosphamide and docetaxel followed by radiation therapy. After this course, her hair loss incompletely recovered. One year after chemotherapy, she underwent bilateral salpingo-oophorectomy and started anastrozole. Three months later, she noticed increased shedding and progressive thinning of the hair. Physical examination revealed diffuse thinning that was most pronounced over the crown. She also experienced lateral thinning of the eyebrows, decreased eyelashes, and dystrophic fingernails. Fluocinonide solution was discontinued by the patient due to scalp burning. She had a brother with bitemporal recession. Her medical history was notable for Hashimoto thyroiditis, vitamin D deficiency, and peripheral neuropathy. Her TSH occasionally was elevated, and she was intermittently on levothyroxine; however, her free T4 was maintained within reference range on all records. Her medications at the time of evaluation were anastrozole and gabapentin. Biopsies taken from the right and left temporal scalp revealed decreased follicle density with a majority of follicles in anagen, scattered miniaturized follicles, and a mild perivascular and perifollicular lymphoid infiltrate. Mild dermal fibrosis was present without evidence of frank scarring (Figure 2). She declined treatment, and there was no change in her condition over 3 years of follow-up.
Comment
Classification of Chemotherapy-Induced Hair Loss
Chemotherapy-induced alopecia is typically an anagen effluvium that is reversed within 6 months following the final course of chemotherapy. When incomplete regrowth persists, the patient is considered to have PCIA.1 The pathophysiology of PCIA is unclear.
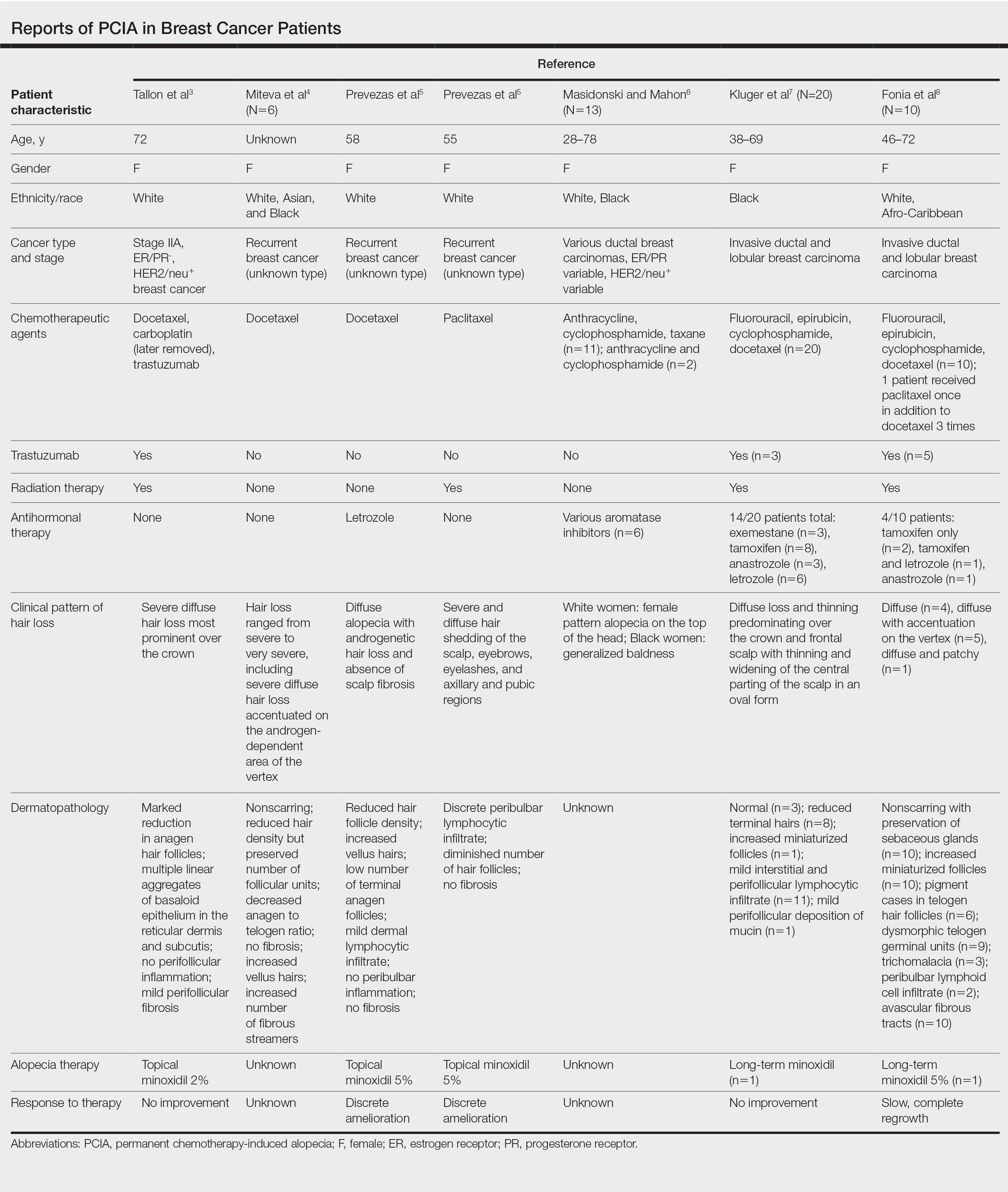
Traditional grading for chemotherapy-induced alopecia does not account for the patterns of loss seen in PCIA, of which the most common appears to be a female pattern with accentuated hair loss in androgen-dependent regions of the scalp.18 Other patterns include a diffuse type with body hair loss, patchy alopecia, and complete alopecia with or without body hair loss (Table).3-8 Whether these patterns all can be attributed to chemotherapy remains to be explored.
Breast Cancer Therapies Causing PCIA
The main agents thought to be responsible for PCIA in breast cancer patients are taxanes. The role of endocrine therapies has not been well explored. Trastuzumab lacks several of the common side effects of chemotherapy due to its specificity for the HER2/neu receptor and has not been found to increase the rate of hair loss when combined with standard chemotherapy.19,20 Although radiation therapy has the potential to damage hair follicles, and a dose-dependent relationship has been described for temporary and permanent alopecia at irradiated sites, permanent alopecia predominantly has been reported with cranial radiation used in the treatment of intracranial malignancies.21 The role of radiation therapy of the breasts in PCIA is unclear, as its inclusion in therapy has not been consistently reported in the literature.
Docetaxel is known to cause chemotherapy-induced alopecia, with an 83.4% incidence in phase 2 trials; however, it also appears to be related to PCIA.20 A PubMed search of articles indexed for MEDLINE was performed using the terms permanent chemotherapy induced alopecia, chemotherapy, docetaxel, endocrine therapies, hair loss, alopecia, and breast cancer. More than 400 cases of PCIA related to chemotherapy in breast cancer patients have been reported in the literature from a combination of case reports/series, retrospective surveys, and at least one prospective study. Data from some of the more detailed reports (n=52) are summarized in the Table. In the single-center, 3-year prospective study of women given adjuvant taxane-based or non–taxane-based chemotherapy, those who received taxane therapy were more likely to develop PCIA (odds ratio, 8.01).9
All 3 of our patients received taxanes. Interestingly, patient 3 underwent 2 rounds of chemotherapy 14 years apart and experienced full regrowth of the hair after the first course of taxane-free chemotherapy but experienced persistent hair loss following docetaxel treatment. Adjuvant endocrine therapies also may contribute to PCIA. A review of the side effects of endocrine therapies revealed an incidence of alopecia that was higher than expected; tamoxifen was the greatest offender. Additionally, using endocrine treatments in combination was found to have a synergistic effect on alopecia.18 Adjuvant endocrine therapy was used in patients 2 and 3. Although endocrine therapies appear to have a milder effect on hair loss compared to chemotherapy, these medications are continued for a longer duration, potentially contributing to the severity of hair loss and prolonging the time to regrowth.
Furthermore, endocrine therapies used in breast cancer treatment decrease estrogen levels or antagonize estrogen receptors, creating an environment of relative hyperandrogenism that may contribute to FPHL in genetically susceptible women.18 Although taxanes may cause irreversible hair loss in these patients, the action of endocrine therapies on the remaining hair follicles may affect the typical female pattern seen clinically. Patients 2 and 3 who presented with FPHL received adjuvant endocrine therapies and had positive family history, while patient 1 did not. Of note, patient 3 experienced worsening hair loss following the addition of anastrozole, which suggests a contribution of endocrine therapy to her PCIA. Our limited cases do not allow for evaluation of a worsened outcome with the combination of taxanes and endocrine therapies; however, we suggest further evaluation for a synergistic effect that may be contributing to PCIA.
Conclusion
Permanent alopecia in breast cancer patients appears to be a true potential adverse effect of taxanes and endocrine therapies, and it is important to characterize it appropriately so that its mechanism can be understood and appropriate treatment and counseling can take place. Although it may not influence clinical decision-making, patients should be informed that hair loss with chemotherapy can be permanent. Treatment with scalp cooling can reduce the risk for severe chemotherapy-induced alopecia, but it is unclear if it reduces risk for PCIA.12,15 Topical or oral minoxidil may be helpful in the treatment of PCIA once it has developed.7,8,15,22 Better characterization of these cases may elucidate risk factors for developing permanent alopecia, allowing for more appropriate risk stratification, counseling, and treatment.
- Dorr VJ. A practitioner’s guide to cancer-related alopecia. Semin Oncol. 1998;25:562-570.
- Machado M, Moreb JS, Khan SA. Six cases of permanent alopecia after various conditioning regimens commonly used in hematopoietic stem cell transplantation. Bone Marrow Transplant. 2007;40:979-982.
- Tallon B, Blanchard E, Goldberg LJ. Permanent chemotherapy-induced alopecia: case report and review of the literature. J Am Acad Dermatol. 2010;63:333-336.
- Miteva M, Misciali C, Fanti PA, et al. Permanent alopecia after systemic chemotherapy: a clinicopathological study of 10 cases. Am J Dermatopathol. 2011;33:345-350.
- Prevezas C, Matard B, Pinquier L, et al. Irreversible and severe alopecia following docetaxel or paclitaxel cytotoxic therapy for breast cancer. Br J Dermatol. 2009;160:883-885.
- Masidonski P, Mahon SM. Permanent alopecia in women being treated for breast cancer. Clin J Oncol Nurs. 2009;13:13-14.
- Kluger N, Jacot W, Frouin E, et al. Permanent scalp alopecia related to breast cancer chemotherapy by sequential fluorouracil/epirubicin/cyclophosphamide (FEC) and docetaxel: a prospective study of 20 patients. Ann Oncol. 2012;23:2879-2884.
- Fonia A, Cota C, Setterfield JF, et al. Permanent alopecia in patients with breast cancer after taxane chemotherapy and adjuvant hormonal therapy: clinicopathologic findings in a cohort of 10 patients. J Am Acad Dermatol. 2017;76:948-957.
- Kang D, Kim IR, Choi EK, et al. Permanent chemotherapy-induced alopecia in patients with breast cancer: a 3-year prospective cohort study [published online August 17, 2018]. Oncologist. 2019;24:414-420.
- Chan J, Adderley H, Alameddine M, et al. Permanent hair loss associated with taxane chemotherapy use in breast cancer: a retrospective survey at two tertiary UK cancer centres [published online December 22, 2020]. Eur J Cancer Care (Engl). doi:10.1111/ecc.13395
- Bourgeois H, Denis F, Kerbrat P, et al. Long term persistent alopecia and suboptimal hair regrowth after adjuvant chemotherapy for breast cancer: alert for an emerging side effect: ALOPERS Observatory. Cancer Res. 2009;69(24 suppl). doi:10.1158/0008-5472.SABCS-09-3174
- Bertrand M, Mailliez A, Vercambre S, et al. Permanent chemotherapy induced alopecia in early breast cancer patients after (neo)adjuvant chemotherapy: long term follow up. Cancer Res. 2013;73(24 suppl). doi:10.1158/0008-5472.SABCS13-P3-09-15
- Kim S, Park HS, Kim JY, et al. Irreversible chemotherapy-induced alopecia in breast cancer patient. Cancer Res. 2016;76(4 suppl). doi:10.1158/1538-7445.SABCS15-P1-15-04
- Thorp NJ, Swift F, Arundell D, et al. Long term hair loss in patients with early breast cancer receiving docetaxel chemotherapy. Cancer Res. 2015;75(9 suppl). doi:10.1158/1538-7445.SABCS14-P5-17-04
- Freites-Martinez A, Shapiro J, van den Hurk C, et al. Hair disorders in cancer survivors. J Am Acad Dermatol. 2019;80:1199-1213.
- Freites-Martinez A, Chan D, Sibaud V, et al. Assessment of quality of life and treatment outcomes of patients with persistent postchemotherapy alopecia. JAMA Dermatol. 2019;155:724-728.
- Sinclair R, Jolley D, Mallari R, et al. The reliability of horizontally sectioned scalp biopsies in the diagnosis of chronic diffuse telogen hair loss in women. J Am Acad Dermatol. 2004;51:189-199.
- Saggar V, Wu S, Dickler MN, et al. Alopecia with endocrine therapies in patients with cancer. Oncologist. 2013;18:1126-1134.
- Yeager CE, Olsen EA. Treatment of chemotherapy-induced alopecia. Dermatol Ther. 2011;24:432-442.
- Baselga J. Clinical trials of single-agent trastuzumab (Herceptin). Semin Oncol. 2000;27(5 suppl 9):20-26.
- Lawenda BD, Gagne HM, Gierga DP, et al. Permanent alopecia after cranial irradiation: dose-response relationship. Int J Radiat Oncol Biol Phys. 2004;60:879-887.
- Yang X, Thai KE. Treatment of permanent chemotherapy-induced alopecia with low dose oral minoxidil [published online May 13, 2015]. Australas J Dermatol. 2016;57:E130-E132.
Anagen effluvium during chemotherapy is common, typically beginning within 1 month of treatment onset and resolving by 6 months after the final course.1 Permanent chemotherapy-induced alopecia (PCIA), in which hair loss persists beyond 6 months after chemotherapy without recovery to original density, was first reported in patients following high-dose chemotherapy regimens for allogeneic bone marrow transplantation.2 There are now increasing reports of PCIA in patients with breast cancer; at least 400 such cases have been documented.3-16 In addition to chemotherapy, patients often receive adjuvant endocrine therapy with selective estrogen receptor modulators, aromatase inhibitors, or gonadotropin-releasing hormone agonists.5-16 Endocrine therapies also can lead to alopecia, but their role in PCIA has not been well defined.15,16 We describe 3 patients with breast cancer who experienced PCIA following chemotherapy with taxanes with or without endocrine therapies. We also review the literature on non–bone marrow transplantation PCIA to better characterize this entity and explore the role of endocrine therapies in PCIA.
Case Reports
Patient 1
A 62-year-old woman with a history of stage II invasive ductal carcinoma presented with persistent hair loss 5 years after completing chemotherapy. She underwent 6 cycles of docetaxel and carboplatin along with radiation therapy as well as 1 year of trastuzumab and did not receive endocrine therapy. At the current presentation, she reported patchy hair regrowth that gradually filled in but failed to return to full density. Physical examination revealed the hair was diffusely thin, especially bitemporally (Figures 1A and 1B), and she did not experience any loss of body hair. She had no family history of hair loss. Her medical history was notable for hypertension, chronic obstructive bronchitis, osteopenia, and depression. Her thyroid stimulating hormone (TSH) level was within reference range. Medications included lisinopril, metoprolol, escitalopram, and trazodone. A biopsy from the occipital scalp showed nonscarring alopecia with variation of hair follicle size, a decreased number of hair follicles, and a decreased anagen to telogen ratio (Figure 1C). She was treated with clobetasol solution and minoxidil solution 5% for 1 year with mild improvement. She experienced no further hair loss but did not regain original hair density.
Patient 2
A 35-year-old woman with a history of stage II invasive ductal carcinoma presented with persistent hair loss 10 months after chemotherapy. She underwent 4 cycles of doxorubicin and cyclophosphamide followed by 4 cycles of paclitaxel and was started on trastuzumab. Tamoxifen was initiated 1 month after completing chemotherapy. She received radiation therapy the following month and continued trastuzumab for 1 year. At the current presentation, the patient noted that hair regrowth had started 1 month after the last course of chemotherapy but had progressed slowly. She denied body hair loss. Physical examination revealed diffuse thinning, especially over the crown, with scattered broken hairs throughout the scalp and several miniaturized hairs over the crown. She was evaluated as grade 3 on the Sinclair clinical grading scale used to evaluate female pattern hair loss (FPHL).17 Her family history was remarkable for FPHL in her maternal grandmother. She had no notable medical history, her TSH was normal, and she was taking tamoxifen and trastuzumab. Biopsy was not performed. The patient was started on minoxidil solution 2% and had mild improvement with no further broken-off hairs after 10 months. At that point, she was evaluated as grade 2 to 3 on the Sinclair scale.17
Patient 3
A 51-year-old woman with a history of papillary carcinoma and extensive ductal carcinoma in situ presented with persistent hair loss for 3.5 years following chemotherapy for recurrent breast cancer. After her initial diagnosis in the left breast, she received cyclophosphamide, methotrexate, and 5-fluorouracil but did not receive endocrine therapy. Her hair thinned during chemotherapy but returned to normal density within 1 year. She had a recurrence of the cancer in the right breast 14 years later and received 6 cycles of chemotherapy with cyclophosphamide and docetaxel followed by radiation therapy. After this course, her hair loss incompletely recovered. One year after chemotherapy, she underwent bilateral salpingo-oophorectomy and started anastrozole. Three months later, she noticed increased shedding and progressive thinning of the hair. Physical examination revealed diffuse thinning that was most pronounced over the crown. She also experienced lateral thinning of the eyebrows, decreased eyelashes, and dystrophic fingernails. Fluocinonide solution was discontinued by the patient due to scalp burning. She had a brother with bitemporal recession. Her medical history was notable for Hashimoto thyroiditis, vitamin D deficiency, and peripheral neuropathy. Her TSH occasionally was elevated, and she was intermittently on levothyroxine; however, her free T4 was maintained within reference range on all records. Her medications at the time of evaluation were anastrozole and gabapentin. Biopsies taken from the right and left temporal scalp revealed decreased follicle density with a majority of follicles in anagen, scattered miniaturized follicles, and a mild perivascular and perifollicular lymphoid infiltrate. Mild dermal fibrosis was present without evidence of frank scarring (Figure 2). She declined treatment, and there was no change in her condition over 3 years of follow-up.
Comment
Classification of Chemotherapy-Induced Hair Loss
Chemotherapy-induced alopecia is typically an anagen effluvium that is reversed within 6 months following the final course of chemotherapy. When incomplete regrowth persists, the patient is considered to have PCIA.1 The pathophysiology of PCIA is unclear.
Traditional grading for chemotherapy-induced alopecia does not account for the patterns of loss seen in PCIA, of which the most common appears to be a female pattern with accentuated hair loss in androgen-dependent regions of the scalp.18 Other patterns include a diffuse type with body hair loss, patchy alopecia, and complete alopecia with or without body hair loss (Table).3-8 Whether these patterns all can be attributed to chemotherapy remains to be explored.
Breast Cancer Therapies Causing PCIA
The main agents thought to be responsible for PCIA in breast cancer patients are taxanes. The role of endocrine therapies has not been well explored. Trastuzumab lacks several of the common side effects of chemotherapy due to its specificity for the HER2/neu receptor and has not been found to increase the rate of hair loss when combined with standard chemotherapy.19,20 Although radiation therapy has the potential to damage hair follicles, and a dose-dependent relationship has been described for temporary and permanent alopecia at irradiated sites, permanent alopecia predominantly has been reported with cranial radiation used in the treatment of intracranial malignancies.21 The role of radiation therapy of the breasts in PCIA is unclear, as its inclusion in therapy has not been consistently reported in the literature.
Docetaxel is known to cause chemotherapy-induced alopecia, with an 83.4% incidence in phase 2 trials; however, it also appears to be related to PCIA.20 A PubMed search of articles indexed for MEDLINE was performed using the terms permanent chemotherapy induced alopecia, chemotherapy, docetaxel, endocrine therapies, hair loss, alopecia, and breast cancer. More than 400 cases of PCIA related to chemotherapy in breast cancer patients have been reported in the literature from a combination of case reports/series, retrospective surveys, and at least one prospective study. Data from some of the more detailed reports (n=52) are summarized in the Table. In the single-center, 3-year prospective study of women given adjuvant taxane-based or non–taxane-based chemotherapy, those who received taxane therapy were more likely to develop PCIA (odds ratio, 8.01).9
All 3 of our patients received taxanes. Interestingly, patient 3 underwent 2 rounds of chemotherapy 14 years apart and experienced full regrowth of the hair after the first course of taxane-free chemotherapy but experienced persistent hair loss following docetaxel treatment. Adjuvant endocrine therapies also may contribute to PCIA. A review of the side effects of endocrine therapies revealed an incidence of alopecia that was higher than expected; tamoxifen was the greatest offender. Additionally, using endocrine treatments in combination was found to have a synergistic effect on alopecia.18 Adjuvant endocrine therapy was used in patients 2 and 3. Although endocrine therapies appear to have a milder effect on hair loss compared to chemotherapy, these medications are continued for a longer duration, potentially contributing to the severity of hair loss and prolonging the time to regrowth.
Furthermore, endocrine therapies used in breast cancer treatment decrease estrogen levels or antagonize estrogen receptors, creating an environment of relative hyperandrogenism that may contribute to FPHL in genetically susceptible women.18 Although taxanes may cause irreversible hair loss in these patients, the action of endocrine therapies on the remaining hair follicles may affect the typical female pattern seen clinically. Patients 2 and 3 who presented with FPHL received adjuvant endocrine therapies and had positive family history, while patient 1 did not. Of note, patient 3 experienced worsening hair loss following the addition of anastrozole, which suggests a contribution of endocrine therapy to her PCIA. Our limited cases do not allow for evaluation of a worsened outcome with the combination of taxanes and endocrine therapies; however, we suggest further evaluation for a synergistic effect that may be contributing to PCIA.
Conclusion
Permanent alopecia in breast cancer patients appears to be a true potential adverse effect of taxanes and endocrine therapies, and it is important to characterize it appropriately so that its mechanism can be understood and appropriate treatment and counseling can take place. Although it may not influence clinical decision-making, patients should be informed that hair loss with chemotherapy can be permanent. Treatment with scalp cooling can reduce the risk for severe chemotherapy-induced alopecia, but it is unclear if it reduces risk for PCIA.12,15 Topical or oral minoxidil may be helpful in the treatment of PCIA once it has developed.7,8,15,22 Better characterization of these cases may elucidate risk factors for developing permanent alopecia, allowing for more appropriate risk stratification, counseling, and treatment.
Anagen effluvium during chemotherapy is common, typically beginning within 1 month of treatment onset and resolving by 6 months after the final course.1 Permanent chemotherapy-induced alopecia (PCIA), in which hair loss persists beyond 6 months after chemotherapy without recovery to original density, was first reported in patients following high-dose chemotherapy regimens for allogeneic bone marrow transplantation.2 There are now increasing reports of PCIA in patients with breast cancer; at least 400 such cases have been documented.3-16 In addition to chemotherapy, patients often receive adjuvant endocrine therapy with selective estrogen receptor modulators, aromatase inhibitors, or gonadotropin-releasing hormone agonists.5-16 Endocrine therapies also can lead to alopecia, but their role in PCIA has not been well defined.15,16 We describe 3 patients with breast cancer who experienced PCIA following chemotherapy with taxanes with or without endocrine therapies. We also review the literature on non–bone marrow transplantation PCIA to better characterize this entity and explore the role of endocrine therapies in PCIA.
Case Reports
Patient 1
A 62-year-old woman with a history of stage II invasive ductal carcinoma presented with persistent hair loss 5 years after completing chemotherapy. She underwent 6 cycles of docetaxel and carboplatin along with radiation therapy as well as 1 year of trastuzumab and did not receive endocrine therapy. At the current presentation, she reported patchy hair regrowth that gradually filled in but failed to return to full density. Physical examination revealed the hair was diffusely thin, especially bitemporally (Figures 1A and 1B), and she did not experience any loss of body hair. She had no family history of hair loss. Her medical history was notable for hypertension, chronic obstructive bronchitis, osteopenia, and depression. Her thyroid stimulating hormone (TSH) level was within reference range. Medications included lisinopril, metoprolol, escitalopram, and trazodone. A biopsy from the occipital scalp showed nonscarring alopecia with variation of hair follicle size, a decreased number of hair follicles, and a decreased anagen to telogen ratio (Figure 1C). She was treated with clobetasol solution and minoxidil solution 5% for 1 year with mild improvement. She experienced no further hair loss but did not regain original hair density.
Patient 2
A 35-year-old woman with a history of stage II invasive ductal carcinoma presented with persistent hair loss 10 months after chemotherapy. She underwent 4 cycles of doxorubicin and cyclophosphamide followed by 4 cycles of paclitaxel and was started on trastuzumab. Tamoxifen was initiated 1 month after completing chemotherapy. She received radiation therapy the following month and continued trastuzumab for 1 year. At the current presentation, the patient noted that hair regrowth had started 1 month after the last course of chemotherapy but had progressed slowly. She denied body hair loss. Physical examination revealed diffuse thinning, especially over the crown, with scattered broken hairs throughout the scalp and several miniaturized hairs over the crown. She was evaluated as grade 3 on the Sinclair clinical grading scale used to evaluate female pattern hair loss (FPHL).17 Her family history was remarkable for FPHL in her maternal grandmother. She had no notable medical history, her TSH was normal, and she was taking tamoxifen and trastuzumab. Biopsy was not performed. The patient was started on minoxidil solution 2% and had mild improvement with no further broken-off hairs after 10 months. At that point, she was evaluated as grade 2 to 3 on the Sinclair scale.17
Patient 3
A 51-year-old woman with a history of papillary carcinoma and extensive ductal carcinoma in situ presented with persistent hair loss for 3.5 years following chemotherapy for recurrent breast cancer. After her initial diagnosis in the left breast, she received cyclophosphamide, methotrexate, and 5-fluorouracil but did not receive endocrine therapy. Her hair thinned during chemotherapy but returned to normal density within 1 year. She had a recurrence of the cancer in the right breast 14 years later and received 6 cycles of chemotherapy with cyclophosphamide and docetaxel followed by radiation therapy. After this course, her hair loss incompletely recovered. One year after chemotherapy, she underwent bilateral salpingo-oophorectomy and started anastrozole. Three months later, she noticed increased shedding and progressive thinning of the hair. Physical examination revealed diffuse thinning that was most pronounced over the crown. She also experienced lateral thinning of the eyebrows, decreased eyelashes, and dystrophic fingernails. Fluocinonide solution was discontinued by the patient due to scalp burning. She had a brother with bitemporal recession. Her medical history was notable for Hashimoto thyroiditis, vitamin D deficiency, and peripheral neuropathy. Her TSH occasionally was elevated, and she was intermittently on levothyroxine; however, her free T4 was maintained within reference range on all records. Her medications at the time of evaluation were anastrozole and gabapentin. Biopsies taken from the right and left temporal scalp revealed decreased follicle density with a majority of follicles in anagen, scattered miniaturized follicles, and a mild perivascular and perifollicular lymphoid infiltrate. Mild dermal fibrosis was present without evidence of frank scarring (Figure 2). She declined treatment, and there was no change in her condition over 3 years of follow-up.
Comment
Classification of Chemotherapy-Induced Hair Loss
Chemotherapy-induced alopecia is typically an anagen effluvium that is reversed within 6 months following the final course of chemotherapy. When incomplete regrowth persists, the patient is considered to have PCIA.1 The pathophysiology of PCIA is unclear.
Traditional grading for chemotherapy-induced alopecia does not account for the patterns of loss seen in PCIA, of which the most common appears to be a female pattern with accentuated hair loss in androgen-dependent regions of the scalp.18 Other patterns include a diffuse type with body hair loss, patchy alopecia, and complete alopecia with or without body hair loss (Table).3-8 Whether these patterns all can be attributed to chemotherapy remains to be explored.
Breast Cancer Therapies Causing PCIA
The main agents thought to be responsible for PCIA in breast cancer patients are taxanes. The role of endocrine therapies has not been well explored. Trastuzumab lacks several of the common side effects of chemotherapy due to its specificity for the HER2/neu receptor and has not been found to increase the rate of hair loss when combined with standard chemotherapy.19,20 Although radiation therapy has the potential to damage hair follicles, and a dose-dependent relationship has been described for temporary and permanent alopecia at irradiated sites, permanent alopecia predominantly has been reported with cranial radiation used in the treatment of intracranial malignancies.21 The role of radiation therapy of the breasts in PCIA is unclear, as its inclusion in therapy has not been consistently reported in the literature.
Docetaxel is known to cause chemotherapy-induced alopecia, with an 83.4% incidence in phase 2 trials; however, it also appears to be related to PCIA.20 A PubMed search of articles indexed for MEDLINE was performed using the terms permanent chemotherapy induced alopecia, chemotherapy, docetaxel, endocrine therapies, hair loss, alopecia, and breast cancer. More than 400 cases of PCIA related to chemotherapy in breast cancer patients have been reported in the literature from a combination of case reports/series, retrospective surveys, and at least one prospective study. Data from some of the more detailed reports (n=52) are summarized in the Table. In the single-center, 3-year prospective study of women given adjuvant taxane-based or non–taxane-based chemotherapy, those who received taxane therapy were more likely to develop PCIA (odds ratio, 8.01).9
All 3 of our patients received taxanes. Interestingly, patient 3 underwent 2 rounds of chemotherapy 14 years apart and experienced full regrowth of the hair after the first course of taxane-free chemotherapy but experienced persistent hair loss following docetaxel treatment. Adjuvant endocrine therapies also may contribute to PCIA. A review of the side effects of endocrine therapies revealed an incidence of alopecia that was higher than expected; tamoxifen was the greatest offender. Additionally, using endocrine treatments in combination was found to have a synergistic effect on alopecia.18 Adjuvant endocrine therapy was used in patients 2 and 3. Although endocrine therapies appear to have a milder effect on hair loss compared to chemotherapy, these medications are continued for a longer duration, potentially contributing to the severity of hair loss and prolonging the time to regrowth.
Furthermore, endocrine therapies used in breast cancer treatment decrease estrogen levels or antagonize estrogen receptors, creating an environment of relative hyperandrogenism that may contribute to FPHL in genetically susceptible women.18 Although taxanes may cause irreversible hair loss in these patients, the action of endocrine therapies on the remaining hair follicles may affect the typical female pattern seen clinically. Patients 2 and 3 who presented with FPHL received adjuvant endocrine therapies and had positive family history, while patient 1 did not. Of note, patient 3 experienced worsening hair loss following the addition of anastrozole, which suggests a contribution of endocrine therapy to her PCIA. Our limited cases do not allow for evaluation of a worsened outcome with the combination of taxanes and endocrine therapies; however, we suggest further evaluation for a synergistic effect that may be contributing to PCIA.
Conclusion
Permanent alopecia in breast cancer patients appears to be a true potential adverse effect of taxanes and endocrine therapies, and it is important to characterize it appropriately so that its mechanism can be understood and appropriate treatment and counseling can take place. Although it may not influence clinical decision-making, patients should be informed that hair loss with chemotherapy can be permanent. Treatment with scalp cooling can reduce the risk for severe chemotherapy-induced alopecia, but it is unclear if it reduces risk for PCIA.12,15 Topical or oral minoxidil may be helpful in the treatment of PCIA once it has developed.7,8,15,22 Better characterization of these cases may elucidate risk factors for developing permanent alopecia, allowing for more appropriate risk stratification, counseling, and treatment.
- Dorr VJ. A practitioner’s guide to cancer-related alopecia. Semin Oncol. 1998;25:562-570.
- Machado M, Moreb JS, Khan SA. Six cases of permanent alopecia after various conditioning regimens commonly used in hematopoietic stem cell transplantation. Bone Marrow Transplant. 2007;40:979-982.
- Tallon B, Blanchard E, Goldberg LJ. Permanent chemotherapy-induced alopecia: case report and review of the literature. J Am Acad Dermatol. 2010;63:333-336.
- Miteva M, Misciali C, Fanti PA, et al. Permanent alopecia after systemic chemotherapy: a clinicopathological study of 10 cases. Am J Dermatopathol. 2011;33:345-350.
- Prevezas C, Matard B, Pinquier L, et al. Irreversible and severe alopecia following docetaxel or paclitaxel cytotoxic therapy for breast cancer. Br J Dermatol. 2009;160:883-885.
- Masidonski P, Mahon SM. Permanent alopecia in women being treated for breast cancer. Clin J Oncol Nurs. 2009;13:13-14.
- Kluger N, Jacot W, Frouin E, et al. Permanent scalp alopecia related to breast cancer chemotherapy by sequential fluorouracil/epirubicin/cyclophosphamide (FEC) and docetaxel: a prospective study of 20 patients. Ann Oncol. 2012;23:2879-2884.
- Fonia A, Cota C, Setterfield JF, et al. Permanent alopecia in patients with breast cancer after taxane chemotherapy and adjuvant hormonal therapy: clinicopathologic findings in a cohort of 10 patients. J Am Acad Dermatol. 2017;76:948-957.
- Kang D, Kim IR, Choi EK, et al. Permanent chemotherapy-induced alopecia in patients with breast cancer: a 3-year prospective cohort study [published online August 17, 2018]. Oncologist. 2019;24:414-420.
- Chan J, Adderley H, Alameddine M, et al. Permanent hair loss associated with taxane chemotherapy use in breast cancer: a retrospective survey at two tertiary UK cancer centres [published online December 22, 2020]. Eur J Cancer Care (Engl). doi:10.1111/ecc.13395
- Bourgeois H, Denis F, Kerbrat P, et al. Long term persistent alopecia and suboptimal hair regrowth after adjuvant chemotherapy for breast cancer: alert for an emerging side effect: ALOPERS Observatory. Cancer Res. 2009;69(24 suppl). doi:10.1158/0008-5472.SABCS-09-3174
- Bertrand M, Mailliez A, Vercambre S, et al. Permanent chemotherapy induced alopecia in early breast cancer patients after (neo)adjuvant chemotherapy: long term follow up. Cancer Res. 2013;73(24 suppl). doi:10.1158/0008-5472.SABCS13-P3-09-15
- Kim S, Park HS, Kim JY, et al. Irreversible chemotherapy-induced alopecia in breast cancer patient. Cancer Res. 2016;76(4 suppl). doi:10.1158/1538-7445.SABCS15-P1-15-04
- Thorp NJ, Swift F, Arundell D, et al. Long term hair loss in patients with early breast cancer receiving docetaxel chemotherapy. Cancer Res. 2015;75(9 suppl). doi:10.1158/1538-7445.SABCS14-P5-17-04
- Freites-Martinez A, Shapiro J, van den Hurk C, et al. Hair disorders in cancer survivors. J Am Acad Dermatol. 2019;80:1199-1213.
- Freites-Martinez A, Chan D, Sibaud V, et al. Assessment of quality of life and treatment outcomes of patients with persistent postchemotherapy alopecia. JAMA Dermatol. 2019;155:724-728.
- Sinclair R, Jolley D, Mallari R, et al. The reliability of horizontally sectioned scalp biopsies in the diagnosis of chronic diffuse telogen hair loss in women. J Am Acad Dermatol. 2004;51:189-199.
- Saggar V, Wu S, Dickler MN, et al. Alopecia with endocrine therapies in patients with cancer. Oncologist. 2013;18:1126-1134.
- Yeager CE, Olsen EA. Treatment of chemotherapy-induced alopecia. Dermatol Ther. 2011;24:432-442.
- Baselga J. Clinical trials of single-agent trastuzumab (Herceptin). Semin Oncol. 2000;27(5 suppl 9):20-26.
- Lawenda BD, Gagne HM, Gierga DP, et al. Permanent alopecia after cranial irradiation: dose-response relationship. Int J Radiat Oncol Biol Phys. 2004;60:879-887.
- Yang X, Thai KE. Treatment of permanent chemotherapy-induced alopecia with low dose oral minoxidil [published online May 13, 2015]. Australas J Dermatol. 2016;57:E130-E132.
- Dorr VJ. A practitioner’s guide to cancer-related alopecia. Semin Oncol. 1998;25:562-570.
- Machado M, Moreb JS, Khan SA. Six cases of permanent alopecia after various conditioning regimens commonly used in hematopoietic stem cell transplantation. Bone Marrow Transplant. 2007;40:979-982.
- Tallon B, Blanchard E, Goldberg LJ. Permanent chemotherapy-induced alopecia: case report and review of the literature. J Am Acad Dermatol. 2010;63:333-336.
- Miteva M, Misciali C, Fanti PA, et al. Permanent alopecia after systemic chemotherapy: a clinicopathological study of 10 cases. Am J Dermatopathol. 2011;33:345-350.
- Prevezas C, Matard B, Pinquier L, et al. Irreversible and severe alopecia following docetaxel or paclitaxel cytotoxic therapy for breast cancer. Br J Dermatol. 2009;160:883-885.
- Masidonski P, Mahon SM. Permanent alopecia in women being treated for breast cancer. Clin J Oncol Nurs. 2009;13:13-14.
- Kluger N, Jacot W, Frouin E, et al. Permanent scalp alopecia related to breast cancer chemotherapy by sequential fluorouracil/epirubicin/cyclophosphamide (FEC) and docetaxel: a prospective study of 20 patients. Ann Oncol. 2012;23:2879-2884.
- Fonia A, Cota C, Setterfield JF, et al. Permanent alopecia in patients with breast cancer after taxane chemotherapy and adjuvant hormonal therapy: clinicopathologic findings in a cohort of 10 patients. J Am Acad Dermatol. 2017;76:948-957.
- Kang D, Kim IR, Choi EK, et al. Permanent chemotherapy-induced alopecia in patients with breast cancer: a 3-year prospective cohort study [published online August 17, 2018]. Oncologist. 2019;24:414-420.
- Chan J, Adderley H, Alameddine M, et al. Permanent hair loss associated with taxane chemotherapy use in breast cancer: a retrospective survey at two tertiary UK cancer centres [published online December 22, 2020]. Eur J Cancer Care (Engl). doi:10.1111/ecc.13395
- Bourgeois H, Denis F, Kerbrat P, et al. Long term persistent alopecia and suboptimal hair regrowth after adjuvant chemotherapy for breast cancer: alert for an emerging side effect: ALOPERS Observatory. Cancer Res. 2009;69(24 suppl). doi:10.1158/0008-5472.SABCS-09-3174
- Bertrand M, Mailliez A, Vercambre S, et al. Permanent chemotherapy induced alopecia in early breast cancer patients after (neo)adjuvant chemotherapy: long term follow up. Cancer Res. 2013;73(24 suppl). doi:10.1158/0008-5472.SABCS13-P3-09-15
- Kim S, Park HS, Kim JY, et al. Irreversible chemotherapy-induced alopecia in breast cancer patient. Cancer Res. 2016;76(4 suppl). doi:10.1158/1538-7445.SABCS15-P1-15-04
- Thorp NJ, Swift F, Arundell D, et al. Long term hair loss in patients with early breast cancer receiving docetaxel chemotherapy. Cancer Res. 2015;75(9 suppl). doi:10.1158/1538-7445.SABCS14-P5-17-04
- Freites-Martinez A, Shapiro J, van den Hurk C, et al. Hair disorders in cancer survivors. J Am Acad Dermatol. 2019;80:1199-1213.
- Freites-Martinez A, Chan D, Sibaud V, et al. Assessment of quality of life and treatment outcomes of patients with persistent postchemotherapy alopecia. JAMA Dermatol. 2019;155:724-728.
- Sinclair R, Jolley D, Mallari R, et al. The reliability of horizontally sectioned scalp biopsies in the diagnosis of chronic diffuse telogen hair loss in women. J Am Acad Dermatol. 2004;51:189-199.
- Saggar V, Wu S, Dickler MN, et al. Alopecia with endocrine therapies in patients with cancer. Oncologist. 2013;18:1126-1134.
- Yeager CE, Olsen EA. Treatment of chemotherapy-induced alopecia. Dermatol Ther. 2011;24:432-442.
- Baselga J. Clinical trials of single-agent trastuzumab (Herceptin). Semin Oncol. 2000;27(5 suppl 9):20-26.
- Lawenda BD, Gagne HM, Gierga DP, et al. Permanent alopecia after cranial irradiation: dose-response relationship. Int J Radiat Oncol Biol Phys. 2004;60:879-887.
- Yang X, Thai KE. Treatment of permanent chemotherapy-induced alopecia with low dose oral minoxidil [published online May 13, 2015]. Australas J Dermatol. 2016;57:E130-E132.
Practice Points
- Permanent chemotherapy-induced alopecia (PCIA) is defined as hair loss that persists beyond 6 months after treatment with chemotherapy. It may be complicated by the addition of endocrine therapies.
- Patients and clinicians should be aware that PCIA can occur and appears to be a higher risk with taxane therapy.
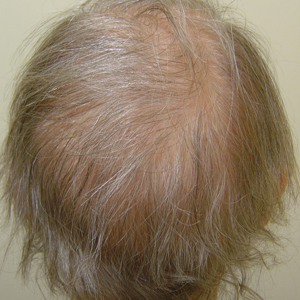
Wax Stripping and Isotretinoin Treatment: A Warning Not to Be Missed
To the Editor:
Oral isotretinoin is a widely used treatment modality in dermatologic practice that is highly effective for severe and recalcitrant acne vulgaris in addition to other conditions. Its use is accompanied by a variety of side effects that are mainly mucocutaneous. These dose-dependent side effects are experienced by almost all patients treated with this medication.1
A generally healthy 14-year-old adolescent girl presented with severe widespread erosions located in a linear pattern corresponding to areas of wax depilation on the shins and thighs (Figure). Approximately 5 months prior, the patient started oral isotretinoin 40 mg daily for severe and recalcitrant acne vulgaris. She was not taking other medications. After 4 months of treatment, during which the acne lesions improved and the patient experienced only mild xerosis and cheilitis, the dosage was increased to 60 mg daily. Three weeks later, the patient underwent wax depilation, which resulted in the erosions.
Oral isotretinoin treatment leads to structural and functional changes to the skin, related to epidermal dyscohesion and sebo-suppression. Although these changes may not be clinically evident in all patients, they still make the skin much more sensitive to external mechanical stimuli.1 Wax depilation commonly is used for treating excess hair on the body. Because it exerts remarkable mechanical stress on the epidermis, it may lead to epidermal stripping in patients taking isotretinoin, manifesting as widespread erosions and resulting in notable patient distress.
Dermatologists typically advise patients to avoid wax epilation while being treated with isotretinoin; however, some patients do not adhere to this recommendation. Also, there are dermatologists who are not aware of this potential side effect. In one survey (N=54), only 4% of consulting dermatologists were aware of this complication.2 A PubMed search of articles indexed for MEDLINE using the terms isotretinoin and wax revealed that this severe side effect with isotretinoin has been reported only 4 times in the medical literature.2-5 The fact that wax epilation should be avoided during isotretinoin treatment previously was not included in the prescribing information. It currently is included in the isotretinoin prescribing information6 with an indication not to perform wax depilation for 6 months after stopping treatment. This case should serve as a reminder to avoid wax depilation during isotretinoin treatment.
- Del Rosso JQ. Clinical relevance of skin barrier changes associated with the use of oral isotretinoin: the importance of barrier repair therapy in patient management. J Drugs Dermatol. 2013;12:626-631.
Woollons A, Price ML. Roaccutane and wax epilation: a cautionary tale. Br J Dermatol. 1997;137:839-840. - Egido Romo M. Isotretinoin and wax epilation. Br J Dermatol. 1991;124:393.
- Holmes SC, Thomson J. Isotretinoin and skin fragility. Br J Dermatol. 1995;132:165.
- Turel-Ermertcan A, Sahin MT, Yurtman D, et al. Inappropriate treatments at beauty centers: a case report of burns caused by hot wax stripping. J Dermatol. 2004;31:854-855.
- Accutane. Package insert. Roche; 2008.
To the Editor:
Oral isotretinoin is a widely used treatment modality in dermatologic practice that is highly effective for severe and recalcitrant acne vulgaris in addition to other conditions. Its use is accompanied by a variety of side effects that are mainly mucocutaneous. These dose-dependent side effects are experienced by almost all patients treated with this medication.1
A generally healthy 14-year-old adolescent girl presented with severe widespread erosions located in a linear pattern corresponding to areas of wax depilation on the shins and thighs (Figure). Approximately 5 months prior, the patient started oral isotretinoin 40 mg daily for severe and recalcitrant acne vulgaris. She was not taking other medications. After 4 months of treatment, during which the acne lesions improved and the patient experienced only mild xerosis and cheilitis, the dosage was increased to 60 mg daily. Three weeks later, the patient underwent wax depilation, which resulted in the erosions.
Oral isotretinoin treatment leads to structural and functional changes to the skin, related to epidermal dyscohesion and sebo-suppression. Although these changes may not be clinically evident in all patients, they still make the skin much more sensitive to external mechanical stimuli.1 Wax depilation commonly is used for treating excess hair on the body. Because it exerts remarkable mechanical stress on the epidermis, it may lead to epidermal stripping in patients taking isotretinoin, manifesting as widespread erosions and resulting in notable patient distress.
Dermatologists typically advise patients to avoid wax epilation while being treated with isotretinoin; however, some patients do not adhere to this recommendation. Also, there are dermatologists who are not aware of this potential side effect. In one survey (N=54), only 4% of consulting dermatologists were aware of this complication.2 A PubMed search of articles indexed for MEDLINE using the terms isotretinoin and wax revealed that this severe side effect with isotretinoin has been reported only 4 times in the medical literature.2-5 The fact that wax epilation should be avoided during isotretinoin treatment previously was not included in the prescribing information. It currently is included in the isotretinoin prescribing information6 with an indication not to perform wax depilation for 6 months after stopping treatment. This case should serve as a reminder to avoid wax depilation during isotretinoin treatment.
To the Editor:
Oral isotretinoin is a widely used treatment modality in dermatologic practice that is highly effective for severe and recalcitrant acne vulgaris in addition to other conditions. Its use is accompanied by a variety of side effects that are mainly mucocutaneous. These dose-dependent side effects are experienced by almost all patients treated with this medication.1
A generally healthy 14-year-old adolescent girl presented with severe widespread erosions located in a linear pattern corresponding to areas of wax depilation on the shins and thighs (Figure). Approximately 5 months prior, the patient started oral isotretinoin 40 mg daily for severe and recalcitrant acne vulgaris. She was not taking other medications. After 4 months of treatment, during which the acne lesions improved and the patient experienced only mild xerosis and cheilitis, the dosage was increased to 60 mg daily. Three weeks later, the patient underwent wax depilation, which resulted in the erosions.
Oral isotretinoin treatment leads to structural and functional changes to the skin, related to epidermal dyscohesion and sebo-suppression. Although these changes may not be clinically evident in all patients, they still make the skin much more sensitive to external mechanical stimuli.1 Wax depilation commonly is used for treating excess hair on the body. Because it exerts remarkable mechanical stress on the epidermis, it may lead to epidermal stripping in patients taking isotretinoin, manifesting as widespread erosions and resulting in notable patient distress.
Dermatologists typically advise patients to avoid wax epilation while being treated with isotretinoin; however, some patients do not adhere to this recommendation. Also, there are dermatologists who are not aware of this potential side effect. In one survey (N=54), only 4% of consulting dermatologists were aware of this complication.2 A PubMed search of articles indexed for MEDLINE using the terms isotretinoin and wax revealed that this severe side effect with isotretinoin has been reported only 4 times in the medical literature.2-5 The fact that wax epilation should be avoided during isotretinoin treatment previously was not included in the prescribing information. It currently is included in the isotretinoin prescribing information6 with an indication not to perform wax depilation for 6 months after stopping treatment. This case should serve as a reminder to avoid wax depilation during isotretinoin treatment.
- Del Rosso JQ. Clinical relevance of skin barrier changes associated with the use of oral isotretinoin: the importance of barrier repair therapy in patient management. J Drugs Dermatol. 2013;12:626-631.
Woollons A, Price ML. Roaccutane and wax epilation: a cautionary tale. Br J Dermatol. 1997;137:839-840. - Egido Romo M. Isotretinoin and wax epilation. Br J Dermatol. 1991;124:393.
- Holmes SC, Thomson J. Isotretinoin and skin fragility. Br J Dermatol. 1995;132:165.
- Turel-Ermertcan A, Sahin MT, Yurtman D, et al. Inappropriate treatments at beauty centers: a case report of burns caused by hot wax stripping. J Dermatol. 2004;31:854-855.
- Accutane. Package insert. Roche; 2008.
- Del Rosso JQ. Clinical relevance of skin barrier changes associated with the use of oral isotretinoin: the importance of barrier repair therapy in patient management. J Drugs Dermatol. 2013;12:626-631.
Woollons A, Price ML. Roaccutane and wax epilation: a cautionary tale. Br J Dermatol. 1997;137:839-840. - Egido Romo M. Isotretinoin and wax epilation. Br J Dermatol. 1991;124:393.
- Holmes SC, Thomson J. Isotretinoin and skin fragility. Br J Dermatol. 1995;132:165.
- Turel-Ermertcan A, Sahin MT, Yurtman D, et al. Inappropriate treatments at beauty centers: a case report of burns caused by hot wax stripping. J Dermatol. 2004;31:854-855.
- Accutane. Package insert. Roche; 2008.
Practice Points
- Oral isotretinoin treatment leads to structural and functional changes to the skin, making it much more sensitive to external mechanical stimuli.
- Wax depilation may lead to epidermal stripping in patients taking isotretinoin and therefore should be avoided in these patients.
Crusted Papules on the Bilateral Helices and Lobules
The Diagnosis: Kikuchi-Fujimoto Disease
A skin biopsy from the left helix was obtained. Histopathologic examination revealed a vacuolar interface reaction with marked papillary dermal edema and a patchy perijunctional lymphocytic infiltrate. The dermis was free of increased mucin (Figure 1). Immunohistochemical staining for CD56 and Epstein-Barr virus (EBV)–encoded small nuclear RNA chromogenic in situ hybridization were negative. Laboratory workup was remarkable for elevated transaminases and inflammatory markers (eg, C-reactive protein, erythrocyte sedimentation rate) but negative for rheumatologic markers (eg, antinuclear antibodies, antineutrophil cytoplasmic antibodies, myeloperoxidase antibodies, serine protease IgG). An extensive infectious workup was unrevealing. Computed tomography highlighted prominent lymphadenopathy throughout the cervical and supraclavicular chains and a large necrotic lymph node in the porta hepatis (Figure 2). Right neck lymph node aspiration revealed necrotizing lymphadenitis in a background of histiocytes and mixed lymphocytes. Coupling the clinical presentation and histomorphology with imaging, a diagnosis of Kikuchi-Fujimoto disease (KD) was rendered.
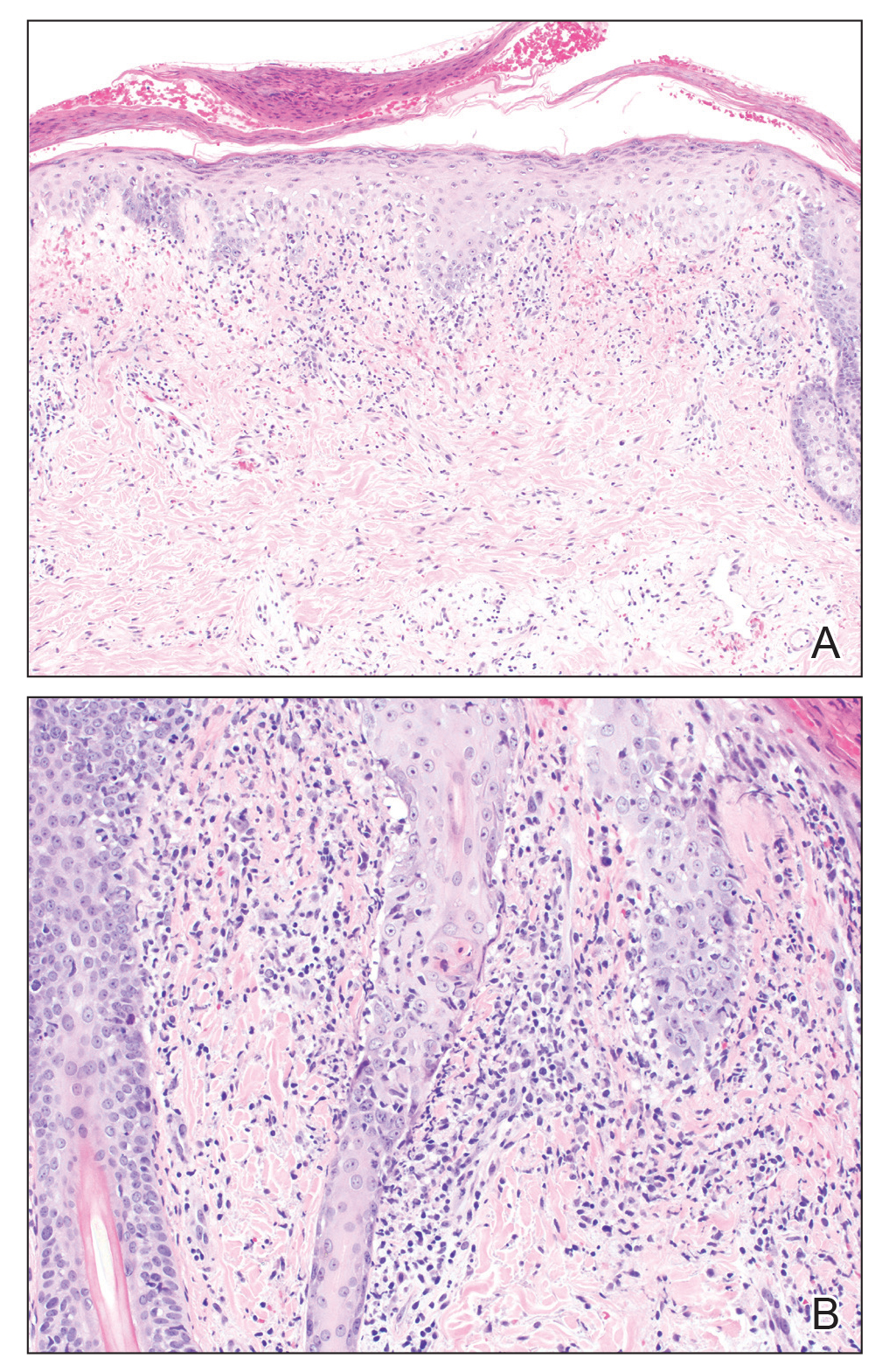

Kikuchi-Fujimoto disease is a rare illness of unknown etiology characterized by cervical lymphadenopathy and fever. Originally described in Japan, KD affects all racial and ethnic groups1,2 but more commonly is seen in women and patients younger than 40 years.3 It can be associated with systemic lupus erythematosus (SLE) and other autoimmune diseases (eg, relapsing polychondritis, adult-onset Still disease),3 and lymphoma.4 Multiple infections have been implicated in the pathogenesis of KD, including EBV and other human herpesviruses; HIV; human T-cell leukemia virus type 1; dengue virus; parvovirus B19; and Yersinia enterocolitica, Bartonella, Brucella, and Toxoplasma infections.3,5,6
Kikuchi-Fujimoto disease classically presents with fever and cervical lymphadenopathy. In a retrospective review of 244 patients with KD, the 3 most common manifestations included lymphadenopathy, fever, and rash.7 A diagnosis of KD is rendered based on clinical presentation and lymph node histopathologic findings of paracortical necrosis and florid histiocytic infiltrate.1
The cutaneous manifestations of KD are heterogeneous yet mostly transient. Cutaneous involvement is reported in 16.6% to 40% of patients.3,5,6 Common cutaneous manifestations include erythematous macules, papules, patches, and plaques; erosions, nodules, and bullae less commonly can occur.6 A variety of cutaneous manifestations have been reported in KD, including lesions mimicking pigmented purpuric dermatoses, vasculitis, Sweet syndrome, drug eruptions, and viral exanthems.6 Signs and symptoms of KD usually resolve within 1 to 4 months. Although there are no established treatments for this disease, patients with severe or persistent symptoms can be treated with steroids or hydroxychloroquine. Recurrences after treatment have been reported.8
Systemic lupus erythematosus is a multiorgan disease with protean manifestations. Cutaneous manifestations of SLE include malar erythema and discoid, annular, and papulosquamous lesions. Histopathologic patterns frequently observed in cutaneous lesions associated with SLE include interface dermatitis with perivascular infiltrates, dermal mucin, and plasmacytoid dendritic cells (marked by CD123 staining); these findings were notably absent in our case.6
Lupus vulgaris is a form of cutaneous tuberculosis that results from reactivation of Mycobacterium tuberculosis in tubercles formed during preceding hematogenous dissemination. The head and neck region is the most common location, particularly the nose, cheeks, and earlobes. Small, brown-red, soft papules coalesce into gelatinous plaques, demonstrating a characteristic apple jelly appearance on diascopy. Other clinical manifestations include the plaque/plane, hypertrophic/tumorlike, and ulcerative/scarring forms.9 Delayed-type hypersensitivity testing by tuberculin skin test, interferon-gamma release assay, or polymerase chain reaction–based assays can detect Mycobacterium tuberculosis. Histopathology shows well-formed granulomas surrounded by chronic inflammatory cells and central necrosis.
Hydroa vacciniforme–like (HV-like) eruption is a rare photosensitive disorder characterized by vesiculopapules on sun-exposed areas. Hydroa vacciniforme–like eruptions rarely have been reported to progress to EBVassociated malignant lymphoma.10 Unlike typical hydroa vacciniforme, which resolves by early adulthood, HV-like eruptions can become more severe with age and are associated with systemic manifestations, including fevers, lymphadenopathy, and liver damage. Histopathologic examination reveals a dense infiltrate of atypical T lymphocytes or natural killer cells (CD56+), which stain positive for EBV-encoded small nuclear RNA,10 in contrast to the patchy perijunctional lymphocytic infiltrate seen in KD.
This case highlights the protean cutaneous manifestations of a rare rheumatologic entity. It demonstrates the importance of a full systemic workup when considering an enigmatic disease. Our patient was started on prednisone 20 mg and hydroxychloroquine 200 mg daily. Within 24 hours, the fevers and rash both improved.
- Turner RR, Martin J, Dorfman RF. Necrotizing lymphadenitis. a study of 30 cases. Am J Surg Pathol. 1983;7:115-123.
- Dorfman RF, Berry GJ. Kikuchi’s histiocytic necrotizing lymphadenitis: an analysis of 108 cases with emphasis on differential diagnosis. Semin Diagn Pathol. 1988;5:329-345.
- Atwater AR, Longley BJ, Aughenbaugh WD. Kikuchi’s disease: case report and systematic review of cutaneous and histopathologic presentations. J Am Acad Dermatol. 2008;59:130-136.
- Yoshino T, Mannami T, Ichimura K, et al. Two cases of histiocytic necrotizing lymphadenitis (Kikuchi-Fujimoto’s disease) following diffuse large B-cell lymphoma. Hum Pathol. 2000;31:1328-1331.
- Yen A, Fearneyhough P, Raimer SS, et al. EBV-associated Kikuchi’s histiocytic necrotizing lymphadenitis with cutaneous manifestations. J Am Acad Dermatol. 1997;36:342-346.
- Kim JH, Kim YB, In SI, et al. The cutaneous lesions of Kikuchi’s disease: a comprehensive analysis of 16 cases based on the clinicopathologic, immunohistochemical, and immunofluorescence studies with an emphasis on the differential diagnosis. Hum Pathol. 2010;41:1245-1254.
- Kucukardali Y, Solmazgul E, Kunter E, et al. Kikuchi-Fujimoto Disease: analysis of 244 cases. Clin Rheumatol. 2007;26:50-54.
- Smith KG, Becker GJ, Busmanis I. Recurrent Kikuchi’s disease. Lancet. 1992;340:124.
- Macgregor R. Cutaneous tuberculosis. Clin Dermatol. 1995;13:245-255.
- Iwatsuki K, Ohtsuka M, Harada H, et al. Clinicopathologic manifestations of Epstein-Barr virus–associated cutaneous lymphoproliferative disorders. Arch Dermatol. 1997;133:1081-1086.
The Diagnosis: Kikuchi-Fujimoto Disease
A skin biopsy from the left helix was obtained. Histopathologic examination revealed a vacuolar interface reaction with marked papillary dermal edema and a patchy perijunctional lymphocytic infiltrate. The dermis was free of increased mucin (Figure 1). Immunohistochemical staining for CD56 and Epstein-Barr virus (EBV)–encoded small nuclear RNA chromogenic in situ hybridization were negative. Laboratory workup was remarkable for elevated transaminases and inflammatory markers (eg, C-reactive protein, erythrocyte sedimentation rate) but negative for rheumatologic markers (eg, antinuclear antibodies, antineutrophil cytoplasmic antibodies, myeloperoxidase antibodies, serine protease IgG). An extensive infectious workup was unrevealing. Computed tomography highlighted prominent lymphadenopathy throughout the cervical and supraclavicular chains and a large necrotic lymph node in the porta hepatis (Figure 2). Right neck lymph node aspiration revealed necrotizing lymphadenitis in a background of histiocytes and mixed lymphocytes. Coupling the clinical presentation and histomorphology with imaging, a diagnosis of Kikuchi-Fujimoto disease (KD) was rendered.
Kikuchi-Fujimoto disease is a rare illness of unknown etiology characterized by cervical lymphadenopathy and fever. Originally described in Japan, KD affects all racial and ethnic groups1,2 but more commonly is seen in women and patients younger than 40 years.3 It can be associated with systemic lupus erythematosus (SLE) and other autoimmune diseases (eg, relapsing polychondritis, adult-onset Still disease),3 and lymphoma.4 Multiple infections have been implicated in the pathogenesis of KD, including EBV and other human herpesviruses; HIV; human T-cell leukemia virus type 1; dengue virus; parvovirus B19; and Yersinia enterocolitica, Bartonella, Brucella, and Toxoplasma infections.3,5,6
Kikuchi-Fujimoto disease classically presents with fever and cervical lymphadenopathy. In a retrospective review of 244 patients with KD, the 3 most common manifestations included lymphadenopathy, fever, and rash.7 A diagnosis of KD is rendered based on clinical presentation and lymph node histopathologic findings of paracortical necrosis and florid histiocytic infiltrate.1
The cutaneous manifestations of KD are heterogeneous yet mostly transient. Cutaneous involvement is reported in 16.6% to 40% of patients.3,5,6 Common cutaneous manifestations include erythematous macules, papules, patches, and plaques; erosions, nodules, and bullae less commonly can occur.6 A variety of cutaneous manifestations have been reported in KD, including lesions mimicking pigmented purpuric dermatoses, vasculitis, Sweet syndrome, drug eruptions, and viral exanthems.6 Signs and symptoms of KD usually resolve within 1 to 4 months. Although there are no established treatments for this disease, patients with severe or persistent symptoms can be treated with steroids or hydroxychloroquine. Recurrences after treatment have been reported.8
Systemic lupus erythematosus is a multiorgan disease with protean manifestations. Cutaneous manifestations of SLE include malar erythema and discoid, annular, and papulosquamous lesions. Histopathologic patterns frequently observed in cutaneous lesions associated with SLE include interface dermatitis with perivascular infiltrates, dermal mucin, and plasmacytoid dendritic cells (marked by CD123 staining); these findings were notably absent in our case.6
Lupus vulgaris is a form of cutaneous tuberculosis that results from reactivation of Mycobacterium tuberculosis in tubercles formed during preceding hematogenous dissemination. The head and neck region is the most common location, particularly the nose, cheeks, and earlobes. Small, brown-red, soft papules coalesce into gelatinous plaques, demonstrating a characteristic apple jelly appearance on diascopy. Other clinical manifestations include the plaque/plane, hypertrophic/tumorlike, and ulcerative/scarring forms.9 Delayed-type hypersensitivity testing by tuberculin skin test, interferon-gamma release assay, or polymerase chain reaction–based assays can detect Mycobacterium tuberculosis. Histopathology shows well-formed granulomas surrounded by chronic inflammatory cells and central necrosis.
Hydroa vacciniforme–like (HV-like) eruption is a rare photosensitive disorder characterized by vesiculopapules on sun-exposed areas. Hydroa vacciniforme–like eruptions rarely have been reported to progress to EBVassociated malignant lymphoma.10 Unlike typical hydroa vacciniforme, which resolves by early adulthood, HV-like eruptions can become more severe with age and are associated with systemic manifestations, including fevers, lymphadenopathy, and liver damage. Histopathologic examination reveals a dense infiltrate of atypical T lymphocytes or natural killer cells (CD56+), which stain positive for EBV-encoded small nuclear RNA,10 in contrast to the patchy perijunctional lymphocytic infiltrate seen in KD.
This case highlights the protean cutaneous manifestations of a rare rheumatologic entity. It demonstrates the importance of a full systemic workup when considering an enigmatic disease. Our patient was started on prednisone 20 mg and hydroxychloroquine 200 mg daily. Within 24 hours, the fevers and rash both improved.
The Diagnosis: Kikuchi-Fujimoto Disease
A skin biopsy from the left helix was obtained. Histopathologic examination revealed a vacuolar interface reaction with marked papillary dermal edema and a patchy perijunctional lymphocytic infiltrate. The dermis was free of increased mucin (Figure 1). Immunohistochemical staining for CD56 and Epstein-Barr virus (EBV)–encoded small nuclear RNA chromogenic in situ hybridization were negative. Laboratory workup was remarkable for elevated transaminases and inflammatory markers (eg, C-reactive protein, erythrocyte sedimentation rate) but negative for rheumatologic markers (eg, antinuclear antibodies, antineutrophil cytoplasmic antibodies, myeloperoxidase antibodies, serine protease IgG). An extensive infectious workup was unrevealing. Computed tomography highlighted prominent lymphadenopathy throughout the cervical and supraclavicular chains and a large necrotic lymph node in the porta hepatis (Figure 2). Right neck lymph node aspiration revealed necrotizing lymphadenitis in a background of histiocytes and mixed lymphocytes. Coupling the clinical presentation and histomorphology with imaging, a diagnosis of Kikuchi-Fujimoto disease (KD) was rendered.
Kikuchi-Fujimoto disease is a rare illness of unknown etiology characterized by cervical lymphadenopathy and fever. Originally described in Japan, KD affects all racial and ethnic groups1,2 but more commonly is seen in women and patients younger than 40 years.3 It can be associated with systemic lupus erythematosus (SLE) and other autoimmune diseases (eg, relapsing polychondritis, adult-onset Still disease),3 and lymphoma.4 Multiple infections have been implicated in the pathogenesis of KD, including EBV and other human herpesviruses; HIV; human T-cell leukemia virus type 1; dengue virus; parvovirus B19; and Yersinia enterocolitica, Bartonella, Brucella, and Toxoplasma infections.3,5,6
Kikuchi-Fujimoto disease classically presents with fever and cervical lymphadenopathy. In a retrospective review of 244 patients with KD, the 3 most common manifestations included lymphadenopathy, fever, and rash.7 A diagnosis of KD is rendered based on clinical presentation and lymph node histopathologic findings of paracortical necrosis and florid histiocytic infiltrate.1
The cutaneous manifestations of KD are heterogeneous yet mostly transient. Cutaneous involvement is reported in 16.6% to 40% of patients.3,5,6 Common cutaneous manifestations include erythematous macules, papules, patches, and plaques; erosions, nodules, and bullae less commonly can occur.6 A variety of cutaneous manifestations have been reported in KD, including lesions mimicking pigmented purpuric dermatoses, vasculitis, Sweet syndrome, drug eruptions, and viral exanthems.6 Signs and symptoms of KD usually resolve within 1 to 4 months. Although there are no established treatments for this disease, patients with severe or persistent symptoms can be treated with steroids or hydroxychloroquine. Recurrences after treatment have been reported.8
Systemic lupus erythematosus is a multiorgan disease with protean manifestations. Cutaneous manifestations of SLE include malar erythema and discoid, annular, and papulosquamous lesions. Histopathologic patterns frequently observed in cutaneous lesions associated with SLE include interface dermatitis with perivascular infiltrates, dermal mucin, and plasmacytoid dendritic cells (marked by CD123 staining); these findings were notably absent in our case.6
Lupus vulgaris is a form of cutaneous tuberculosis that results from reactivation of Mycobacterium tuberculosis in tubercles formed during preceding hematogenous dissemination. The head and neck region is the most common location, particularly the nose, cheeks, and earlobes. Small, brown-red, soft papules coalesce into gelatinous plaques, demonstrating a characteristic apple jelly appearance on diascopy. Other clinical manifestations include the plaque/plane, hypertrophic/tumorlike, and ulcerative/scarring forms.9 Delayed-type hypersensitivity testing by tuberculin skin test, interferon-gamma release assay, or polymerase chain reaction–based assays can detect Mycobacterium tuberculosis. Histopathology shows well-formed granulomas surrounded by chronic inflammatory cells and central necrosis.
Hydroa vacciniforme–like (HV-like) eruption is a rare photosensitive disorder characterized by vesiculopapules on sun-exposed areas. Hydroa vacciniforme–like eruptions rarely have been reported to progress to EBVassociated malignant lymphoma.10 Unlike typical hydroa vacciniforme, which resolves by early adulthood, HV-like eruptions can become more severe with age and are associated with systemic manifestations, including fevers, lymphadenopathy, and liver damage. Histopathologic examination reveals a dense infiltrate of atypical T lymphocytes or natural killer cells (CD56+), which stain positive for EBV-encoded small nuclear RNA,10 in contrast to the patchy perijunctional lymphocytic infiltrate seen in KD.
This case highlights the protean cutaneous manifestations of a rare rheumatologic entity. It demonstrates the importance of a full systemic workup when considering an enigmatic disease. Our patient was started on prednisone 20 mg and hydroxychloroquine 200 mg daily. Within 24 hours, the fevers and rash both improved.
- Turner RR, Martin J, Dorfman RF. Necrotizing lymphadenitis. a study of 30 cases. Am J Surg Pathol. 1983;7:115-123.
- Dorfman RF, Berry GJ. Kikuchi’s histiocytic necrotizing lymphadenitis: an analysis of 108 cases with emphasis on differential diagnosis. Semin Diagn Pathol. 1988;5:329-345.
- Atwater AR, Longley BJ, Aughenbaugh WD. Kikuchi’s disease: case report and systematic review of cutaneous and histopathologic presentations. J Am Acad Dermatol. 2008;59:130-136.
- Yoshino T, Mannami T, Ichimura K, et al. Two cases of histiocytic necrotizing lymphadenitis (Kikuchi-Fujimoto’s disease) following diffuse large B-cell lymphoma. Hum Pathol. 2000;31:1328-1331.
- Yen A, Fearneyhough P, Raimer SS, et al. EBV-associated Kikuchi’s histiocytic necrotizing lymphadenitis with cutaneous manifestations. J Am Acad Dermatol. 1997;36:342-346.
- Kim JH, Kim YB, In SI, et al. The cutaneous lesions of Kikuchi’s disease: a comprehensive analysis of 16 cases based on the clinicopathologic, immunohistochemical, and immunofluorescence studies with an emphasis on the differential diagnosis. Hum Pathol. 2010;41:1245-1254.
- Kucukardali Y, Solmazgul E, Kunter E, et al. Kikuchi-Fujimoto Disease: analysis of 244 cases. Clin Rheumatol. 2007;26:50-54.
- Smith KG, Becker GJ, Busmanis I. Recurrent Kikuchi’s disease. Lancet. 1992;340:124.
- Macgregor R. Cutaneous tuberculosis. Clin Dermatol. 1995;13:245-255.
- Iwatsuki K, Ohtsuka M, Harada H, et al. Clinicopathologic manifestations of Epstein-Barr virus–associated cutaneous lymphoproliferative disorders. Arch Dermatol. 1997;133:1081-1086.
- Turner RR, Martin J, Dorfman RF. Necrotizing lymphadenitis. a study of 30 cases. Am J Surg Pathol. 1983;7:115-123.
- Dorfman RF, Berry GJ. Kikuchi’s histiocytic necrotizing lymphadenitis: an analysis of 108 cases with emphasis on differential diagnosis. Semin Diagn Pathol. 1988;5:329-345.
- Atwater AR, Longley BJ, Aughenbaugh WD. Kikuchi’s disease: case report and systematic review of cutaneous and histopathologic presentations. J Am Acad Dermatol. 2008;59:130-136.
- Yoshino T, Mannami T, Ichimura K, et al. Two cases of histiocytic necrotizing lymphadenitis (Kikuchi-Fujimoto’s disease) following diffuse large B-cell lymphoma. Hum Pathol. 2000;31:1328-1331.
- Yen A, Fearneyhough P, Raimer SS, et al. EBV-associated Kikuchi’s histiocytic necrotizing lymphadenitis with cutaneous manifestations. J Am Acad Dermatol. 1997;36:342-346.
- Kim JH, Kim YB, In SI, et al. The cutaneous lesions of Kikuchi’s disease: a comprehensive analysis of 16 cases based on the clinicopathologic, immunohistochemical, and immunofluorescence studies with an emphasis on the differential diagnosis. Hum Pathol. 2010;41:1245-1254.
- Kucukardali Y, Solmazgul E, Kunter E, et al. Kikuchi-Fujimoto Disease: analysis of 244 cases. Clin Rheumatol. 2007;26:50-54.
- Smith KG, Becker GJ, Busmanis I. Recurrent Kikuchi’s disease. Lancet. 1992;340:124.
- Macgregor R. Cutaneous tuberculosis. Clin Dermatol. 1995;13:245-255.
- Iwatsuki K, Ohtsuka M, Harada H, et al. Clinicopathologic manifestations of Epstein-Barr virus–associated cutaneous lymphoproliferative disorders. Arch Dermatol. 1997;133:1081-1086.
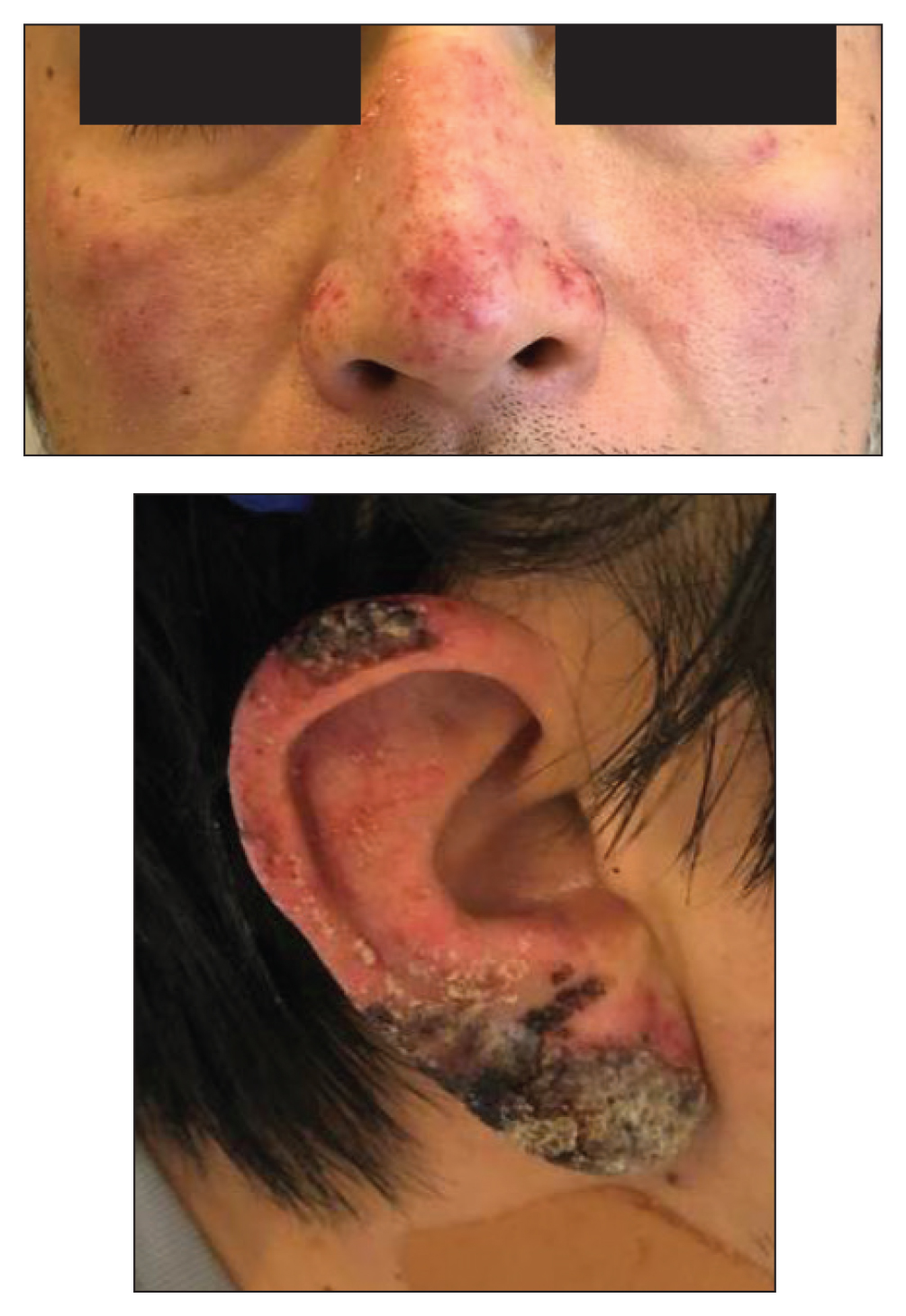
A healthy 42-year-old Japanese man presented with painful lymphadenopathy and fevers of 1 month’s duration as well as a pruritic rash and bilateral ear redness and crusting of 1 week’s duration. He initially was seen at an outside facility and was treated with antibiotics and supportive care for cervical adenitis. During clinical evaluation, he denied joint pain, photosensitivity, and oral lesions. His medical and family history were noncontributory. Although he reported recent travel to multiple countries, he denied exposure to animals, ticks, or sick individuals. Physical examination revealed erythematous blanching papules on the nose and cheeks (top) as well as crusted papules coalescing into plaques on the bilateral helices and lobules (bottom).
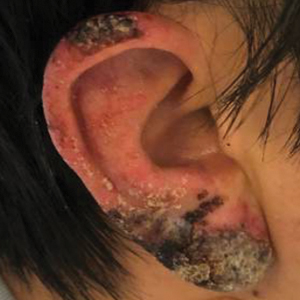
Cutaneous Cholesterol Embolization to the Lower Trunk: An Underrecognized Presentation
To the Editor:
A 65-year-old man with severe atherosclerotic disease developed multiple painful eschars on the lower abdomen, thighs, sacrum, and perineum. He initially presented with myocardial ischemia and claudication and underwent 3 cardiac catheterizations as well as stenting of the superficial femoral artery. Within 2 weeks, he developed exquisitely tender nodules on the lower abdomen, clinically presumed to be sites of enoxaparin injections. These lesions gradually expanded and ulcerated to involve the sacrum, buttock, perineum, and upper thighs (Figure 1). Two punch biopsies from ulcerated skin taken 10 days apart demonstrated necrosis of skin and subcutaneous fat without evidence of vasculitis, vasculopathy, emboli, or notable inflammation despite examination of multiple levels of all submitted tissue. A definitive cause for the ulcerations remained elusive with development of new lesions. A third incisional biopsy of a newly developed, nonulcerated, subcutaneous nodule performed 8 weeks after presentation revealed multiple cholesterol emboli (Figure 2). He was treated with warfarin and clopidogrel bisulfate as well as local wound care. The lesions slowly resolved over the next 4 to 6 months.
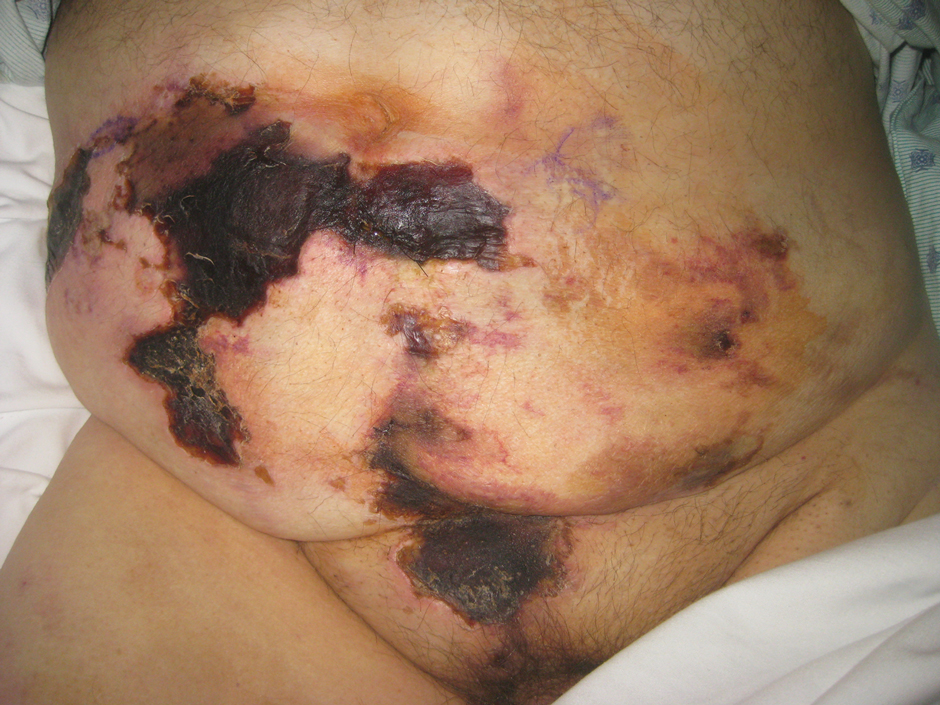
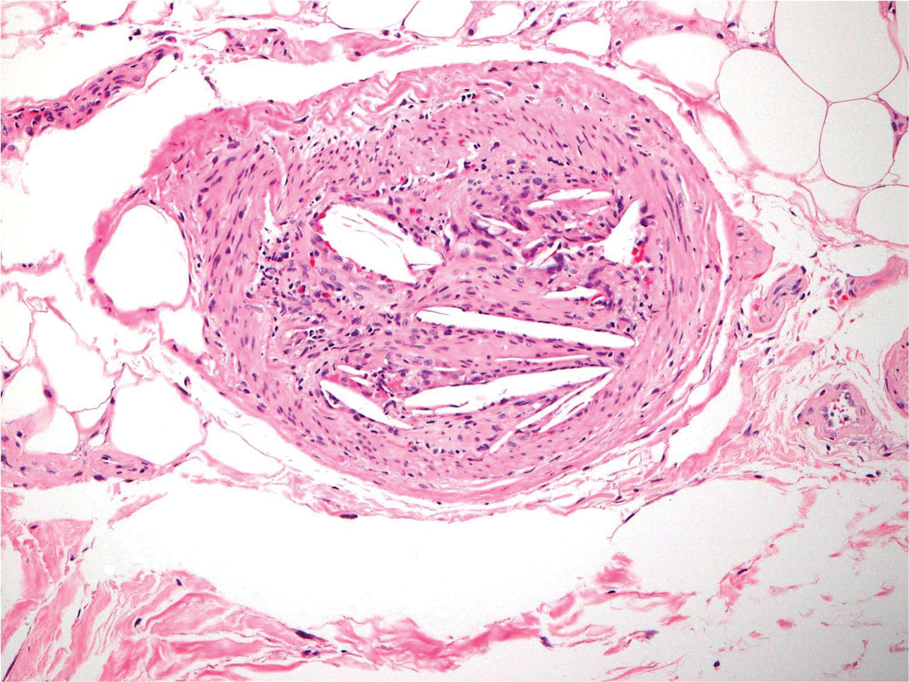
Cholesterol embolization syndrome occurs when disrupted atherosclerotic plaques embolize from large proximal arteries to more distal arterioles, resulting in ischemic damage to 1 or more organ systems.1 It can occur spontaneously but often is a consequence of thrombolytic therapy, anticoagulation, and angioinvasive procedures.2,3 Cutaneous manifestations include livedo reticularis, retiform purpura, nodules, and gangrene. Although livedo reticularis may extend from the legs to the trunk, gangrenous lesions predominantly involve the distal digits.
This case illustrates the challenge in diagnosis of cholesterol emboli, both clinically and histologically. Cutaneous lesions are morphologically variable and often occur with systemic manifestations, mimicking numerous conditions.1 Lower extremity involvement is a well-known occurrence in cholesterol embolization (ie, blue toe syndrome); however, periumbilical and lumbosacral lesions have not been emphasized in the dermatologic or peripheral vascular literature. Our patient’s initial diagnosis was enoxaparin necrosis at abdominal injection sites; however, this unusual distribution of lesions was ultimately determined to be the consequence of cholesterol embolization from the inferior epigastric and superficial external pudendal arteries at the time of stenting of the superficial femoral artery. Proximal truncal involvement should be recognized as an atypical but important cutaneous manifestation to facilitate timely diagnosis and treatment.4,5
Our patient’s course also highlights the potential need for multiple biopsies. Although the gold standard for diagnosis is histologic confirmation, a negative biopsy does not always exclude cholesterol emboli, and one should have a low threshold to perform additional biopsies in the appropriate clinical setting.
- Fine MJ, Kapoor W, Falanga V. Cholesterol crystal embolization: a review of 221 cases in the English literature. Angiology. 1987;38:769-784.
- Fukumoto Y, Tsutsui H, Tsuchihashi M, et al. The incidence and risk factors of cholesterol embolization syndrome, a complication of cardiac catheterization: a prospective study. J Am Coll Cardiol. 2003;42:211-216.
- Karalis DG, Chandrasekaran K, Victor MF, et al. Recognition and embolic potential of intraaortic atherosclerotic debris. J Am Coll Cardiol. 1991;17:73.
- Zaytsev P, Miller K, Pellettiere EV. Cutaneous cholesterol emboli with infarction clinically mimicking heparin necrosis—a case report. Angiology. 1986;37:471-476.
- Erdim M, Tezel E, Biskin N. A case of skin necrosis as a result of cholesterol crystal embolisation. J Plast Reconstr Aesthet Surg. 2006;59:429-432.
To the Editor:
A 65-year-old man with severe atherosclerotic disease developed multiple painful eschars on the lower abdomen, thighs, sacrum, and perineum. He initially presented with myocardial ischemia and claudication and underwent 3 cardiac catheterizations as well as stenting of the superficial femoral artery. Within 2 weeks, he developed exquisitely tender nodules on the lower abdomen, clinically presumed to be sites of enoxaparin injections. These lesions gradually expanded and ulcerated to involve the sacrum, buttock, perineum, and upper thighs (Figure 1). Two punch biopsies from ulcerated skin taken 10 days apart demonstrated necrosis of skin and subcutaneous fat without evidence of vasculitis, vasculopathy, emboli, or notable inflammation despite examination of multiple levels of all submitted tissue. A definitive cause for the ulcerations remained elusive with development of new lesions. A third incisional biopsy of a newly developed, nonulcerated, subcutaneous nodule performed 8 weeks after presentation revealed multiple cholesterol emboli (Figure 2). He was treated with warfarin and clopidogrel bisulfate as well as local wound care. The lesions slowly resolved over the next 4 to 6 months.
Cholesterol embolization syndrome occurs when disrupted atherosclerotic plaques embolize from large proximal arteries to more distal arterioles, resulting in ischemic damage to 1 or more organ systems.1 It can occur spontaneously but often is a consequence of thrombolytic therapy, anticoagulation, and angioinvasive procedures.2,3 Cutaneous manifestations include livedo reticularis, retiform purpura, nodules, and gangrene. Although livedo reticularis may extend from the legs to the trunk, gangrenous lesions predominantly involve the distal digits.
This case illustrates the challenge in diagnosis of cholesterol emboli, both clinically and histologically. Cutaneous lesions are morphologically variable and often occur with systemic manifestations, mimicking numerous conditions.1 Lower extremity involvement is a well-known occurrence in cholesterol embolization (ie, blue toe syndrome); however, periumbilical and lumbosacral lesions have not been emphasized in the dermatologic or peripheral vascular literature. Our patient’s initial diagnosis was enoxaparin necrosis at abdominal injection sites; however, this unusual distribution of lesions was ultimately determined to be the consequence of cholesterol embolization from the inferior epigastric and superficial external pudendal arteries at the time of stenting of the superficial femoral artery. Proximal truncal involvement should be recognized as an atypical but important cutaneous manifestation to facilitate timely diagnosis and treatment.4,5
Our patient’s course also highlights the potential need for multiple biopsies. Although the gold standard for diagnosis is histologic confirmation, a negative biopsy does not always exclude cholesterol emboli, and one should have a low threshold to perform additional biopsies in the appropriate clinical setting.
To the Editor:
A 65-year-old man with severe atherosclerotic disease developed multiple painful eschars on the lower abdomen, thighs, sacrum, and perineum. He initially presented with myocardial ischemia and claudication and underwent 3 cardiac catheterizations as well as stenting of the superficial femoral artery. Within 2 weeks, he developed exquisitely tender nodules on the lower abdomen, clinically presumed to be sites of enoxaparin injections. These lesions gradually expanded and ulcerated to involve the sacrum, buttock, perineum, and upper thighs (Figure 1). Two punch biopsies from ulcerated skin taken 10 days apart demonstrated necrosis of skin and subcutaneous fat without evidence of vasculitis, vasculopathy, emboli, or notable inflammation despite examination of multiple levels of all submitted tissue. A definitive cause for the ulcerations remained elusive with development of new lesions. A third incisional biopsy of a newly developed, nonulcerated, subcutaneous nodule performed 8 weeks after presentation revealed multiple cholesterol emboli (Figure 2). He was treated with warfarin and clopidogrel bisulfate as well as local wound care. The lesions slowly resolved over the next 4 to 6 months.
Cholesterol embolization syndrome occurs when disrupted atherosclerotic plaques embolize from large proximal arteries to more distal arterioles, resulting in ischemic damage to 1 or more organ systems.1 It can occur spontaneously but often is a consequence of thrombolytic therapy, anticoagulation, and angioinvasive procedures.2,3 Cutaneous manifestations include livedo reticularis, retiform purpura, nodules, and gangrene. Although livedo reticularis may extend from the legs to the trunk, gangrenous lesions predominantly involve the distal digits.
This case illustrates the challenge in diagnosis of cholesterol emboli, both clinically and histologically. Cutaneous lesions are morphologically variable and often occur with systemic manifestations, mimicking numerous conditions.1 Lower extremity involvement is a well-known occurrence in cholesterol embolization (ie, blue toe syndrome); however, periumbilical and lumbosacral lesions have not been emphasized in the dermatologic or peripheral vascular literature. Our patient’s initial diagnosis was enoxaparin necrosis at abdominal injection sites; however, this unusual distribution of lesions was ultimately determined to be the consequence of cholesterol embolization from the inferior epigastric and superficial external pudendal arteries at the time of stenting of the superficial femoral artery. Proximal truncal involvement should be recognized as an atypical but important cutaneous manifestation to facilitate timely diagnosis and treatment.4,5
Our patient’s course also highlights the potential need for multiple biopsies. Although the gold standard for diagnosis is histologic confirmation, a negative biopsy does not always exclude cholesterol emboli, and one should have a low threshold to perform additional biopsies in the appropriate clinical setting.
- Fine MJ, Kapoor W, Falanga V. Cholesterol crystal embolization: a review of 221 cases in the English literature. Angiology. 1987;38:769-784.
- Fukumoto Y, Tsutsui H, Tsuchihashi M, et al. The incidence and risk factors of cholesterol embolization syndrome, a complication of cardiac catheterization: a prospective study. J Am Coll Cardiol. 2003;42:211-216.
- Karalis DG, Chandrasekaran K, Victor MF, et al. Recognition and embolic potential of intraaortic atherosclerotic debris. J Am Coll Cardiol. 1991;17:73.
- Zaytsev P, Miller K, Pellettiere EV. Cutaneous cholesterol emboli with infarction clinically mimicking heparin necrosis—a case report. Angiology. 1986;37:471-476.
- Erdim M, Tezel E, Biskin N. A case of skin necrosis as a result of cholesterol crystal embolisation. J Plast Reconstr Aesthet Surg. 2006;59:429-432.
- Fine MJ, Kapoor W, Falanga V. Cholesterol crystal embolization: a review of 221 cases in the English literature. Angiology. 1987;38:769-784.
- Fukumoto Y, Tsutsui H, Tsuchihashi M, et al. The incidence and risk factors of cholesterol embolization syndrome, a complication of cardiac catheterization: a prospective study. J Am Coll Cardiol. 2003;42:211-216.
- Karalis DG, Chandrasekaran K, Victor MF, et al. Recognition and embolic potential of intraaortic atherosclerotic debris. J Am Coll Cardiol. 1991;17:73.
- Zaytsev P, Miller K, Pellettiere EV. Cutaneous cholesterol emboli with infarction clinically mimicking heparin necrosis—a case report. Angiology. 1986;37:471-476.
- Erdim M, Tezel E, Biskin N. A case of skin necrosis as a result of cholesterol crystal embolisation. J Plast Reconstr Aesthet Surg. 2006;59:429-432.
Practice Points
- Cholesterol embolization may occur in proximal locations, and index of suspicion should be high in patients who are at risk.
- Several biopsies may be necessary to make a diagnosis of cholesterol emboli.

Candida Esophagitis Associated With Adalimumab for Hidradenitis Suppurativa
To the Editor:
Hidradenitis suppurativa (HS) is a chronic inflammatory disease characterized by the development of painful abscesses, fistulous tracts, and scars. It most commonly affects the apocrine gland–bearing areas of the body such as the axillary, inguinal, and anogenital regions. With a prevalence of approximately 1%, HS can lead to notable morbidity.1 The pathogenesis is thought to be due to occlusion of terminal hair follicles that subsequently stimulates release of proinflammatory cytokines from nearby keratinocytes. The mechanism of initial occlusion is not well understood but may be due to friction or trauma. An inflammatory mechanism of disease also has been hypothesized; however, the exact cytokine profile is not known. Treatment of HS consists of several different modalities, including oral retinoids, antibiotics, antiandrogenic therapy, and surgery.1,2 Adalimumab is a well-known biologic that has been approved by the US Food and Drug Administration for the treatment of HS.
Adalimumab is a human monoclonal antibody against tumor necrosis factor (TNF) α and is thought to improve HS by several mechanisms. Inhibition of TNF-α and other proinflammatory cytokines found in inflammatory lesions and apocrine glands directly decreases the severity of lesion size and the frequency of recurrence.3 Adalimumab also is thought to downregulate expression of keratin 6 and prevent the hyperkeratinization seen in HS.4 Additionally, TNF-α inhibition decreases production of IL-1, which has been shown to cause hypercornification of follicles and perpetuate HS pathogenesis.5
A 41-year-old woman with a history of endometriosis, adenomyosis, polycystic ovary syndrome, interstitial cystitis, asthma, fibromyalgia, depression, and Hashimoto thyroiditis presented to our dermatology clinic with active draining lesions and sinus tracts in the perivaginal area that were consistent with HS, which initially was treated with doxycycline 100 mg twice daily. She experienced minimal improvement of the HS lesions at 2-month follow-up.
Due to disease severity, adalimumab was started. The patient received a loading dose of 4 injections totaling 160 mg and 80 mg on day 15, followed by a maintenance dose of 40 mg/0.4 mL weekly. The patient reported substantial improvement of pain, and complete resolution of active lesions was noted on physical examination after 4 weeks of treatment with adalimumab.
Six weeks after adalimumab was started, the patient developed severe dysphagia. She was evaluated by a gastroenterologist and underwent endoscopy (Figure), which led to a diagnosis of esophageal candidiasis. Adalimumab was discontinued immediately thereafter. The patient started treatment with nystatin oral rinse 4 times daily and oral fluconazole 200 mg daily. The candidiasis resolved within 2 weeks; however, she experienced recurrence of HS with draining lesions in the perivaginal area approximately 8 weeks after discontinuation of adalimumab. The patient requested to restart adalimumab treatment despite the recent history of esophagitis. Adalimumab 40 mg/0.4 mL weekly was restarted along with oral fluconazole 200 mg twice weekly and nystatin oral rinse 4 times daily. This regimen resulted in complete resolution of HS symptoms within 6 weeks with no recurrence of esophageal candidiasis during 6 months of follow-up.
Although the side effect of Candida esophagitis associated with adalimumab treatment in our patient may be logical given the medication’s mechanism of action and side-effect profile, this case warrants additional attention. An increase in fungal infections occurs from treatment with adalimumab because TNF-α is involved in many immune regulatory steps that counteract infection. Candida typically activates the innate immune system through macrophages via pathogen-associated molecular pattern stimulation, subsequently stimulating the release of inflammatory cytokines such as TNF-α. The cellular immune system also is activated. Helper T cells (TH1) release TNF-α along with other proinflammatory cytokines to increase phagocytosis in polymorphonuclear cells and macrophages.6 Thus, inhibition of TNF-α compromises innate and cellular immunity, thereby increasing susceptibility to fungal organisms.
A PubMed search of articles indexed for MEDLINE using the terms Candida, candidiasis, esophageal, adalimumab, anti-TNF, and TNF revealed no reports of esophageal candidiasis in patients receiving adalimumab or any of the TNF inhibitors. Candida laryngitis was reported in a patient receiving adalimumab for treatment of rheumatoid arthritis.7 Other studies have demonstrated an incidence of mucocutaneous candidiasis, most notably oropharyngeal and vaginal candidiasis.8-10 One study found that anti-TNF medications were associated with an increased risk for candidiasis by a hazard ratio of 2.7 in patients with Crohn disease.8 Other studies have shown that the highest incidence of fungal infection is seen with the use of infliximab, while adalimumab is associated with lower rates of fungal infection.9,10 Although it is known that anti-TNF therapy predisposes patients to fungal infection, the dose of medication known to preclude the highest risk has not been studied. Furthermore, most studies assess rates of Candida infection in individuals receiving anti-TNF therapy in addition to several other immunosuppressant agents (ie, corticosteroids), which confounds the interpretation of results. Additional studies assessing rates of Candida and other opportunistic infections associated with use of adalimumab alone are needed to better guide clinical practices in dermatology.
Patients receiving adalimumab for dermatologic or other conditions should be closely monitored for opportunistic infections. Although immunomodulatory medications offer promising therapeutic benefits in patients with HS, larger studies regarding treatment with anti-TNF agents in HS are warranted to prevent complications from treatment and promote long-term efficacy and safety.
- Kurayev A, Ashkar H, Saraiya A, et al. Hidradenitis suppurativa: review of the pathogenesis and treatment. J Drugs Dermatol. 2016;15:1107-1022.
- Rambhatla PV, Lim HW, Hamzavi I. A systematic review of treatments for hidradenitis suppurativa. Arch Dermatol. 2012;148:439-446.
- van der Zee HH, de Ruiter L, van den Broecke DG, et al. Elevated levels of tumour necrosis factor (TNF)-alpha, interleukin (IL)-1beta and IL-10 in hidradenitis suppurativa skin: a rationale for targeting TNF-alpha and IL-1beta. Br J Dermatol. 2011;164:1292-1298.
- Shuja F, Chan CS, Rosen T. Biologic drugs for the treatment of hidradenitis suppurativa: an evidence-based review. Dermatol Clin. 2010;28:511-521, 523-514.
- Kutsch CL, Norris DA, Arend WP. Tumor necrosis factor-alpha induces interleukin-1 alpha and interleukin-1 receptor antagonist production by cultured human keratinocytes. J Invest Dermatol. 1993;101:79-85.
- Senet JM. Risk factors and physiopathology of candidiasis. Rev Iberoam Micol. 1997;14:6-13.
- Kobak S, Yilmaz H, Guclu O, et al. Severe candida laryngitis in a patient with rheumatoid arthritis treated with adalimumab. Eur J Rheumatol. 2014;1:167-169.
- Marehbian J, Arrighi HM, Hass S, et al. Adverse events associated with common therapy regimens for moderate-to-severe Crohn’s disease. Am J Gastroenterol. 2009;104:2524-2533.
- Tsiodras S, Samonis G, Boumpas DT, et al. Fungal infections complicating tumor necrosis factor alpha blockade therapy. Mayo Clin Proc. 2008;83:181-194.
- Aikawa NE, Rosa DT, Del Negro GM, et al. Systemic and localized infection by Candida species in patients with rheumatic diseases receiving anti-TNF therapy [in Portuguese]. Rev Bras Reumatol. doi:10.1016/j.rbr.2015.03.010
To the Editor:
Hidradenitis suppurativa (HS) is a chronic inflammatory disease characterized by the development of painful abscesses, fistulous tracts, and scars. It most commonly affects the apocrine gland–bearing areas of the body such as the axillary, inguinal, and anogenital regions. With a prevalence of approximately 1%, HS can lead to notable morbidity.1 The pathogenesis is thought to be due to occlusion of terminal hair follicles that subsequently stimulates release of proinflammatory cytokines from nearby keratinocytes. The mechanism of initial occlusion is not well understood but may be due to friction or trauma. An inflammatory mechanism of disease also has been hypothesized; however, the exact cytokine profile is not known. Treatment of HS consists of several different modalities, including oral retinoids, antibiotics, antiandrogenic therapy, and surgery.1,2 Adalimumab is a well-known biologic that has been approved by the US Food and Drug Administration for the treatment of HS.
Adalimumab is a human monoclonal antibody against tumor necrosis factor (TNF) α and is thought to improve HS by several mechanisms. Inhibition of TNF-α and other proinflammatory cytokines found in inflammatory lesions and apocrine glands directly decreases the severity of lesion size and the frequency of recurrence.3 Adalimumab also is thought to downregulate expression of keratin 6 and prevent the hyperkeratinization seen in HS.4 Additionally, TNF-α inhibition decreases production of IL-1, which has been shown to cause hypercornification of follicles and perpetuate HS pathogenesis.5
A 41-year-old woman with a history of endometriosis, adenomyosis, polycystic ovary syndrome, interstitial cystitis, asthma, fibromyalgia, depression, and Hashimoto thyroiditis presented to our dermatology clinic with active draining lesions and sinus tracts in the perivaginal area that were consistent with HS, which initially was treated with doxycycline 100 mg twice daily. She experienced minimal improvement of the HS lesions at 2-month follow-up.
Due to disease severity, adalimumab was started. The patient received a loading dose of 4 injections totaling 160 mg and 80 mg on day 15, followed by a maintenance dose of 40 mg/0.4 mL weekly. The patient reported substantial improvement of pain, and complete resolution of active lesions was noted on physical examination after 4 weeks of treatment with adalimumab.
Six weeks after adalimumab was started, the patient developed severe dysphagia. She was evaluated by a gastroenterologist and underwent endoscopy (Figure), which led to a diagnosis of esophageal candidiasis. Adalimumab was discontinued immediately thereafter. The patient started treatment with nystatin oral rinse 4 times daily and oral fluconazole 200 mg daily. The candidiasis resolved within 2 weeks; however, she experienced recurrence of HS with draining lesions in the perivaginal area approximately 8 weeks after discontinuation of adalimumab. The patient requested to restart adalimumab treatment despite the recent history of esophagitis. Adalimumab 40 mg/0.4 mL weekly was restarted along with oral fluconazole 200 mg twice weekly and nystatin oral rinse 4 times daily. This regimen resulted in complete resolution of HS symptoms within 6 weeks with no recurrence of esophageal candidiasis during 6 months of follow-up.
Although the side effect of Candida esophagitis associated with adalimumab treatment in our patient may be logical given the medication’s mechanism of action and side-effect profile, this case warrants additional attention. An increase in fungal infections occurs from treatment with adalimumab because TNF-α is involved in many immune regulatory steps that counteract infection. Candida typically activates the innate immune system through macrophages via pathogen-associated molecular pattern stimulation, subsequently stimulating the release of inflammatory cytokines such as TNF-α. The cellular immune system also is activated. Helper T cells (TH1) release TNF-α along with other proinflammatory cytokines to increase phagocytosis in polymorphonuclear cells and macrophages.6 Thus, inhibition of TNF-α compromises innate and cellular immunity, thereby increasing susceptibility to fungal organisms.
A PubMed search of articles indexed for MEDLINE using the terms Candida, candidiasis, esophageal, adalimumab, anti-TNF, and TNF revealed no reports of esophageal candidiasis in patients receiving adalimumab or any of the TNF inhibitors. Candida laryngitis was reported in a patient receiving adalimumab for treatment of rheumatoid arthritis.7 Other studies have demonstrated an incidence of mucocutaneous candidiasis, most notably oropharyngeal and vaginal candidiasis.8-10 One study found that anti-TNF medications were associated with an increased risk for candidiasis by a hazard ratio of 2.7 in patients with Crohn disease.8 Other studies have shown that the highest incidence of fungal infection is seen with the use of infliximab, while adalimumab is associated with lower rates of fungal infection.9,10 Although it is known that anti-TNF therapy predisposes patients to fungal infection, the dose of medication known to preclude the highest risk has not been studied. Furthermore, most studies assess rates of Candida infection in individuals receiving anti-TNF therapy in addition to several other immunosuppressant agents (ie, corticosteroids), which confounds the interpretation of results. Additional studies assessing rates of Candida and other opportunistic infections associated with use of adalimumab alone are needed to better guide clinical practices in dermatology.
Patients receiving adalimumab for dermatologic or other conditions should be closely monitored for opportunistic infections. Although immunomodulatory medications offer promising therapeutic benefits in patients with HS, larger studies regarding treatment with anti-TNF agents in HS are warranted to prevent complications from treatment and promote long-term efficacy and safety.
To the Editor:
Hidradenitis suppurativa (HS) is a chronic inflammatory disease characterized by the development of painful abscesses, fistulous tracts, and scars. It most commonly affects the apocrine gland–bearing areas of the body such as the axillary, inguinal, and anogenital regions. With a prevalence of approximately 1%, HS can lead to notable morbidity.1 The pathogenesis is thought to be due to occlusion of terminal hair follicles that subsequently stimulates release of proinflammatory cytokines from nearby keratinocytes. The mechanism of initial occlusion is not well understood but may be due to friction or trauma. An inflammatory mechanism of disease also has been hypothesized; however, the exact cytokine profile is not known. Treatment of HS consists of several different modalities, including oral retinoids, antibiotics, antiandrogenic therapy, and surgery.1,2 Adalimumab is a well-known biologic that has been approved by the US Food and Drug Administration for the treatment of HS.
Adalimumab is a human monoclonal antibody against tumor necrosis factor (TNF) α and is thought to improve HS by several mechanisms. Inhibition of TNF-α and other proinflammatory cytokines found in inflammatory lesions and apocrine glands directly decreases the severity of lesion size and the frequency of recurrence.3 Adalimumab also is thought to downregulate expression of keratin 6 and prevent the hyperkeratinization seen in HS.4 Additionally, TNF-α inhibition decreases production of IL-1, which has been shown to cause hypercornification of follicles and perpetuate HS pathogenesis.5
A 41-year-old woman with a history of endometriosis, adenomyosis, polycystic ovary syndrome, interstitial cystitis, asthma, fibromyalgia, depression, and Hashimoto thyroiditis presented to our dermatology clinic with active draining lesions and sinus tracts in the perivaginal area that were consistent with HS, which initially was treated with doxycycline 100 mg twice daily. She experienced minimal improvement of the HS lesions at 2-month follow-up.
Due to disease severity, adalimumab was started. The patient received a loading dose of 4 injections totaling 160 mg and 80 mg on day 15, followed by a maintenance dose of 40 mg/0.4 mL weekly. The patient reported substantial improvement of pain, and complete resolution of active lesions was noted on physical examination after 4 weeks of treatment with adalimumab.
Six weeks after adalimumab was started, the patient developed severe dysphagia. She was evaluated by a gastroenterologist and underwent endoscopy (Figure), which led to a diagnosis of esophageal candidiasis. Adalimumab was discontinued immediately thereafter. The patient started treatment with nystatin oral rinse 4 times daily and oral fluconazole 200 mg daily. The candidiasis resolved within 2 weeks; however, she experienced recurrence of HS with draining lesions in the perivaginal area approximately 8 weeks after discontinuation of adalimumab. The patient requested to restart adalimumab treatment despite the recent history of esophagitis. Adalimumab 40 mg/0.4 mL weekly was restarted along with oral fluconazole 200 mg twice weekly and nystatin oral rinse 4 times daily. This regimen resulted in complete resolution of HS symptoms within 6 weeks with no recurrence of esophageal candidiasis during 6 months of follow-up.
Although the side effect of Candida esophagitis associated with adalimumab treatment in our patient may be logical given the medication’s mechanism of action and side-effect profile, this case warrants additional attention. An increase in fungal infections occurs from treatment with adalimumab because TNF-α is involved in many immune regulatory steps that counteract infection. Candida typically activates the innate immune system through macrophages via pathogen-associated molecular pattern stimulation, subsequently stimulating the release of inflammatory cytokines such as TNF-α. The cellular immune system also is activated. Helper T cells (TH1) release TNF-α along with other proinflammatory cytokines to increase phagocytosis in polymorphonuclear cells and macrophages.6 Thus, inhibition of TNF-α compromises innate and cellular immunity, thereby increasing susceptibility to fungal organisms.
A PubMed search of articles indexed for MEDLINE using the terms Candida, candidiasis, esophageal, adalimumab, anti-TNF, and TNF revealed no reports of esophageal candidiasis in patients receiving adalimumab or any of the TNF inhibitors. Candida laryngitis was reported in a patient receiving adalimumab for treatment of rheumatoid arthritis.7 Other studies have demonstrated an incidence of mucocutaneous candidiasis, most notably oropharyngeal and vaginal candidiasis.8-10 One study found that anti-TNF medications were associated with an increased risk for candidiasis by a hazard ratio of 2.7 in patients with Crohn disease.8 Other studies have shown that the highest incidence of fungal infection is seen with the use of infliximab, while adalimumab is associated with lower rates of fungal infection.9,10 Although it is known that anti-TNF therapy predisposes patients to fungal infection, the dose of medication known to preclude the highest risk has not been studied. Furthermore, most studies assess rates of Candida infection in individuals receiving anti-TNF therapy in addition to several other immunosuppressant agents (ie, corticosteroids), which confounds the interpretation of results. Additional studies assessing rates of Candida and other opportunistic infections associated with use of adalimumab alone are needed to better guide clinical practices in dermatology.
Patients receiving adalimumab for dermatologic or other conditions should be closely monitored for opportunistic infections. Although immunomodulatory medications offer promising therapeutic benefits in patients with HS, larger studies regarding treatment with anti-TNF agents in HS are warranted to prevent complications from treatment and promote long-term efficacy and safety.
- Kurayev A, Ashkar H, Saraiya A, et al. Hidradenitis suppurativa: review of the pathogenesis and treatment. J Drugs Dermatol. 2016;15:1107-1022.
- Rambhatla PV, Lim HW, Hamzavi I. A systematic review of treatments for hidradenitis suppurativa. Arch Dermatol. 2012;148:439-446.
- van der Zee HH, de Ruiter L, van den Broecke DG, et al. Elevated levels of tumour necrosis factor (TNF)-alpha, interleukin (IL)-1beta and IL-10 in hidradenitis suppurativa skin: a rationale for targeting TNF-alpha and IL-1beta. Br J Dermatol. 2011;164:1292-1298.
- Shuja F, Chan CS, Rosen T. Biologic drugs for the treatment of hidradenitis suppurativa: an evidence-based review. Dermatol Clin. 2010;28:511-521, 523-514.
- Kutsch CL, Norris DA, Arend WP. Tumor necrosis factor-alpha induces interleukin-1 alpha and interleukin-1 receptor antagonist production by cultured human keratinocytes. J Invest Dermatol. 1993;101:79-85.
- Senet JM. Risk factors and physiopathology of candidiasis. Rev Iberoam Micol. 1997;14:6-13.
- Kobak S, Yilmaz H, Guclu O, et al. Severe candida laryngitis in a patient with rheumatoid arthritis treated with adalimumab. Eur J Rheumatol. 2014;1:167-169.
- Marehbian J, Arrighi HM, Hass S, et al. Adverse events associated with common therapy regimens for moderate-to-severe Crohn’s disease. Am J Gastroenterol. 2009;104:2524-2533.
- Tsiodras S, Samonis G, Boumpas DT, et al. Fungal infections complicating tumor necrosis factor alpha blockade therapy. Mayo Clin Proc. 2008;83:181-194.
- Aikawa NE, Rosa DT, Del Negro GM, et al. Systemic and localized infection by Candida species in patients with rheumatic diseases receiving anti-TNF therapy [in Portuguese]. Rev Bras Reumatol. doi:10.1016/j.rbr.2015.03.010
- Kurayev A, Ashkar H, Saraiya A, et al. Hidradenitis suppurativa: review of the pathogenesis and treatment. J Drugs Dermatol. 2016;15:1107-1022.
- Rambhatla PV, Lim HW, Hamzavi I. A systematic review of treatments for hidradenitis suppurativa. Arch Dermatol. 2012;148:439-446.
- van der Zee HH, de Ruiter L, van den Broecke DG, et al. Elevated levels of tumour necrosis factor (TNF)-alpha, interleukin (IL)-1beta and IL-10 in hidradenitis suppurativa skin: a rationale for targeting TNF-alpha and IL-1beta. Br J Dermatol. 2011;164:1292-1298.
- Shuja F, Chan CS, Rosen T. Biologic drugs for the treatment of hidradenitis suppurativa: an evidence-based review. Dermatol Clin. 2010;28:511-521, 523-514.
- Kutsch CL, Norris DA, Arend WP. Tumor necrosis factor-alpha induces interleukin-1 alpha and interleukin-1 receptor antagonist production by cultured human keratinocytes. J Invest Dermatol. 1993;101:79-85.
- Senet JM. Risk factors and physiopathology of candidiasis. Rev Iberoam Micol. 1997;14:6-13.
- Kobak S, Yilmaz H, Guclu O, et al. Severe candida laryngitis in a patient with rheumatoid arthritis treated with adalimumab. Eur J Rheumatol. 2014;1:167-169.
- Marehbian J, Arrighi HM, Hass S, et al. Adverse events associated with common therapy regimens for moderate-to-severe Crohn’s disease. Am J Gastroenterol. 2009;104:2524-2533.
- Tsiodras S, Samonis G, Boumpas DT, et al. Fungal infections complicating tumor necrosis factor alpha blockade therapy. Mayo Clin Proc. 2008;83:181-194.
- Aikawa NE, Rosa DT, Del Negro GM, et al. Systemic and localized infection by Candida species in patients with rheumatic diseases receiving anti-TNF therapy [in Portuguese]. Rev Bras Reumatol. doi:10.1016/j.rbr.2015.03.010
Practice Points
- Adalimumab is an effective treatment for patients with hidradenitis suppurativa.
- There is risk for opportunistic infections with adalimumab, and patients should be monitored closely.
Treatment of Generalized Pustular Psoriasis of Pregnancy With Infliximab
Generalized pustular psoriasis of pregnancy (GPPP), formerly known as impetigo herpetiformis, is a rare dermatosis that causes maternal and fetal morbidity and mortality. It is characterized by widespread, circular, erythematous plaques with pustules at the periphery.1 Conventional first-line treatment includes systemic corticosteroids and cyclosporine. The National Psoriasis Foundation Medical Board also has included infliximab among the first-line treatment options for GPPP.2 Herein, we report a case of GPPP treated with infliximab at 30 weeks’ gestation and during the postpartum period.
Case Report
A 22-year-old woman was admitted to our inpatient clinic at 20 weeks’ gestation in her second pregnancy for evaluation of cutaneous eruptions covering the entire body. The lesions first appeared 3 to 4 days prior to her admission and dramatically progressed. She had a history of psoriasis vulgaris diagnosed during her first pregnancy 2 years prior that was treated with topical steroids throughout the pregnancy and methotrexate during lactation for a total of 11 months. She then was started on cyclosporine, which she used for 6 months due to ineffectiveness of the methotrexate, but she stopped treatment 4 months before the second pregnancy.
At the current presentation, physical examination revealed erythroderma and widespread pustules on the chest, abdomen, arms, and legs, including the intertriginous regions, that tended to coalesce and form lakes of pus over an erythematous base (Figure 1). The mucosae were normal. She exhibited a low blood pressure (85/50 mmHg) and high body temperature (102 °F [38.9 °C]). Routine laboratory examination revealed anemia and a normal leukocyte count. Her erythrocyte sedimentation rate (57 mm/h [reference range, <20 mm/h]) and C-reactive protein level (102 mg/L [reference range, <6 mg/L]) were elevated, whereas total calcium (8.11 mg/dL [reference range, 8.2–10.6 mg/dL]) and albumin (3.15 g/dL [reference range, >4.0 g/dL]) levels were low.
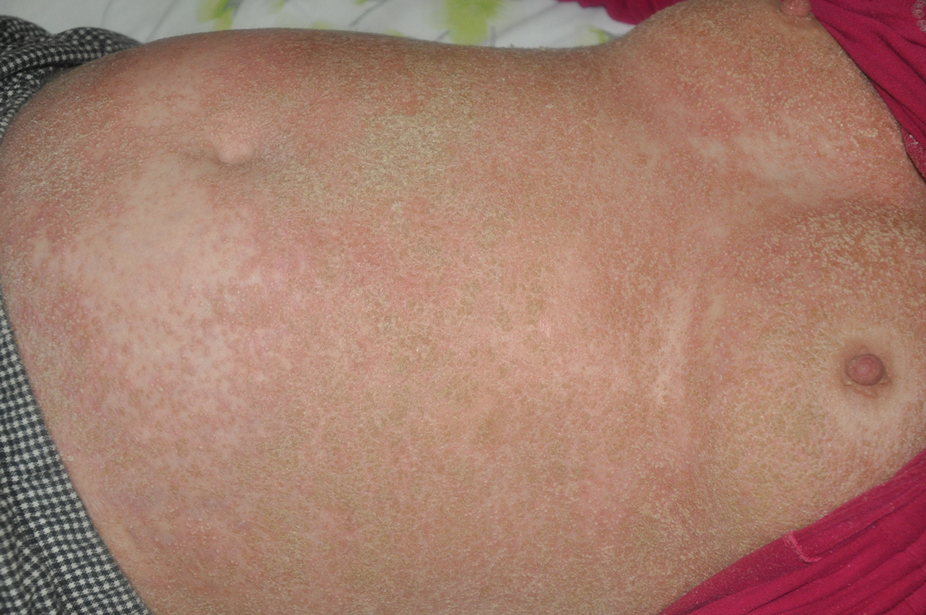
Empirical intravenous piperacillin/tazobactam was started due to hypotension, high fever, and elevated C-reactive protein levels; however, treatment was stopped after 4 days when microbiological cultures taken from blood and pustules revealed no bacterial growth, and therefore the fever was assumed to be caused by erythroderma. A skin biopsy before the start of topical and systemic treatment revealed changes consistent with GPPP.
Because her disease was extensive, systemic methylprednisolone 1.5 mg/kg once daily was started, and the dose was increased up to 2.5 mg/kg once daily on the tenth day of treatment to control new crops of eruptions. The dose was tapered to 2 mg/kg once daily when the lesions subsided 4 weeks into the treatment. The patient was discharged after 7 weeks at 27 weeks’ gestation.
Twelve days later, the patient was readmitted to the clinic in an erythrodermic state. The lesions were not controlled with increased doses of systemic corticosteroids. Treatment with cyclosporine was considered, but the patient refused; thus, infliximab treatment was planned. Isoniazid 300 mg once daily was started due to a risk of latent Mycobacterium tuberculosis infection revealed by a tuberculosis blood test. Other evaluations revealed no contraindications, and an infusion of infliximab 300 mg (5 mg/kg) was administered at 30 weeks’ gestation. There was visible improvement in the erythroderma and pustular lesions within the same day of treatment, and the lesions were completely cleared within 2 days of the infusion. The methylprednisolone dose was reduced to 1.5 mg/kg once daily.
Three days after treatment with infliximab, lesions with yellow encrustation appeared in the perioral region and on the oral mucosa and left ear. She was diagnosed with an oral herpes infection. Oral valacyclovir 1 g twice daily and topical mupirocin were started and the lesions subsided within 1 week. Twelve days after the infliximab infusion, new pustular lesions appeared, and a second infusion of infliximab was administered 13 days after the first, which cleared all lesions within 48 hours.
The patient’s methylprednisolone dose was tapered and stopped prior to delivery at 34 weeks’ gestation—2 weeks after the second dose of infliximab—as she did not have any new skin eruptions. A third infliximab infusion that normally would have occurred 4 weeks after the second treatment was postponed for a Cesarean section scheduled at 36 weeks’ gestation due to suspected intrauterine growth retardation. The patient stayed at the hospital until delivery without any new skin lesions. The gross and histopathologic examination of the placenta was normal. The neonate weighed 4.8 lb at birth and had neonatal jaundice that resolved spontaneously within 10 days but was otherwise healthy.
The patient returned to the clinic 3 weeks postpartum with a few pustules on erythematous plaques on the chest, abdomen, and back. At this time, she received a third infusion of infliximab 8 weeks after the second dose. For the past 5 years, the patient has been undergoing infliximab maintenance treatment, which she receives at the hospital every 8 weeks with excellent response. She has had no further pregnancies to date.
Comment
Generalized pustular psoriasis of pregnancy is a rare condition that typically occurs in the third trimester but also can start in the first and second trimesters. It may result in maternal and fetal morbidity by causing fluid and electrolyte imbalance and/or placental insufficiency, resulting in an increased risk for fetal abnormalities, stillbirth, and neonatal death.3 In subsequent pregnancies, GPPP has been observed to recur at an earlier gestational age with a more severe presentation.1,3
Generalized pustular psoriasis of pregnancy usually involves an eruption that begins symmetrically in the intertriginous areas and spreads to the rest of the body. The lesions present as erythematous annular plaques with pustules on the periphery and desquamation in the center due to older pustules.1,3 The mucous membranes also may be involved with erosive and exfoliative plaques, and there may be nail involvement. Patients often present with systemic symptoms such as fever, malaise, diarrhea, and vomiting.1 Laboratory investigations may reveal neutrophilic leukocytosis, high erythrocyte sedimentation rate, hypocalcemia, and hypoalbuminemia.4 Cultures from blood and pustules show no bacterial growth. A skin biopsy is helpful in diagnosis, with features similar to generalized pustular psoriasis, demonstrating spongiform pustules containing neutrophils, lymphocytic and neutrophilic infiltrates in the papillary dermis, and negative direct immunofluorescence.3
The differential diagnosis of GPPP includes subcorneal pustular dermatosis, dermatitis herpetiformis, herpes gestationis, impetigo, and acute generalized exanthematous pustulosis.1,3 Due to concerns of fetal implications, treatment options in GPPP are somewhat limited; however, the condition requires treatment because it may result in unfavorable pregnancy outcomes. Topical corticosteroids may be an option for limited disease.5,6 Systemic corticosteroids (eg, prednisone 60–80 mg/d) were previously considered as first-line agents, although they have shown limited efficacy in our case as well as in other case reports.7 Their ineffectiveness and risk for flare-up after dose tapering should be kept in mind when starting GPPP patients on systemic corticosteroids. Systemic cyclosporine (2–3 mg/kg/d) may be added to increase the efficacy of systemic steroids, which was done in several cases in literature.1,6,8 Although cyclosporine has been classified as a pregnancy category C drug, an analysis of pregnancy outcomes of 629 renal transplant patients revealed no association with adverse pregnancy outcomes compared to the general population and no increase in fetal malformations.9 Therefore, cyclosporine is a safe treatment option and was classified as a first-line drug for GPPP in a 2012 review by the National Psoriasis Foundation Medical Board.2 Narrowband UVB also has been reported to be used for the treatment of GPPP.10 Methotrexate and retinoids have been used in cases with lesions that persisted postpartum.1
Anti–tumor necrosis factor (TNF) α agents are another effective option for treatment of GPPP. Anti-TNF agents are classified as pregnancy category B due to results showing that anti-mouse TNF-α monoclonal antibodies did not cause embryotoxicity or teratogenicity in pregnant mice.11 Although Carter et al12 published a review of US Food and Drug Administration data on pregnant women receiving anti-TNF treatment and concluded that these agents were associated with the VACTERL group of malformations (vertebral defects, anal atresia, cardiac defect, tracheoesophageal fistula with esophageal atresia, cardiac defects, renal and limb anomalies), no such association was found in further studies. A 2014 study showed no difference in the rate of major malformations in infants born to women who were treated with anti-TNF drugs compared to the disease-matched group not treated with these agents and pregnant women counselled for nonteratogenic exposure.13 The same study detected an increase in preterm and low-birth-weight deliveries and suggested this might be caused by the increased severity of disease in patients requiring anti-TNF medication. The British Society of Rheumatology Biologics Register published data on pregnancy outcomes in 130 rheumatoid arthritis patients who had been exposed to anti-TNF agents.14 The results suggested an increased rate of spontaneous abortions in women exposed to anti-TNF treatment around the time of conception, especially in those taking these medications together with methotrexate or leflunomide; however, results also indicated that disease activity may have had an impact on the rate of spontaneous abortions in these patients. In a 2013 review of 462 women with inflammatory bowel disease who had been exposed to anti-TNF agents during pregnancy, the investigators concluded that pregnancy outcomes and the rate of congenital anomalies did not significantly differ from other inflammatory bowel disease patients not receiving anti-TNF drugs or the general population.15
In 2012, the National Board of the National Psoriasis Foundation put infliximab amongst the first-line treatment modalities for GPPP.2 In one case of GPPP in which the eruption persisted after delivery, the patient was treated with infliximab 7 weeks postpartum due to failure to control the disease with prednisolone 60 mg daily and cyclosporine 7.5 mg/kg daily. Unlike our patient, this patient was only started on an infliximab regimen after delivery.16 In another case reported in 2010, the patient was started on infliximab during the postpartum period of her first pregnancy following a pustular flare of previously diagnosed plaque psoriasis (not a generalized pustular psoriasis, as in our case).17 As a good response was obtained, infliximab treatment was continued in the patient throughout her second pregnancy.
Our case is unique in that infliximab was started during pregnancy because of intractable disease leading to systemic symptoms. Our patient showed an excellent response to infliximab after a 10-week disease course with repeated flare-ups and impairment to her overall condition. Delivery occurred at 36 weeks’ gestation due to suspected intrauterine growth retardation; however, the neonate was born with a 5-minute APGAR score of 10 and required no special medical care, which suggests that the low birth weight was constitutional due to the patient’s small frame (her height was 4 ft 11 in). The breast milk of patients with inflammatory bowel disease has been detected to contain very small amounts of infliximab (101 ng/mL, about 1/200 of the therapeutic blood level).18 Considering the large molecular weight of this agent and possible proteolysis in the stomach and intestines, infliximab is unlikely to affect the neonate.15 Thus, we encouraged our patient to breastfeed her baby. A case of fatal disseminated Bacille-Calmette-Guérin infection in an infant whose mother received infliximab treatment during pregnancy has been reported.19 It has been suggested that live vaccines should be avoided in neonates exposed to anti-TNF agents at least for the first 6 months of life or until the agent is no longer detectable in their blood.15 We therefore informed our patient’s family practitioner about this data.
Conclusion
We report a case of infliximab treatment for GPPP that was continued during the postpartum period. Infliximab was an effective treatment option in our patient with no detected serious adverse events and may be considered in other cases of GPPP that are not responsive to systemic steroids. However, further studies are warranted to evaluate the safety and efficacy of infliximab treatment for GPPP and psoriasis in pregnancy.
- Lerhoff S, Pomeranz MK. Specific dermatoses of pregnancy and their treatment. Dermatol Ther. 2013;26:274-284.
- Robinson A, Van Voorhees AS, Hsu S, et al. Treatment of pustular psoriasis: from the Medical Board of the National Psoriasis Foundation. J Am Acad Dermatol. 2012;67:279-288.
- Oumeish OY, Parish JL. Impetigo herpetiformis. Clin Dermatol. 2006;24:101-104.
- Gao QQ, Xi MR, Yao Q. Impetigo herpetiformis during pregnancy: a case report and literature review. Dermatology. 2013;226:35-40.
- Bae YS, Van Voorhees AS, Hsu S, et al. Review of treatment options for psoriasis in pregnant or lactating women: from the Medical Board of the National Psoriasis Foundation. J Am Acad Dermatol. 2012;67:459-477.
- Shaw CJ, Wu P, Sriemevan A. First trimester impetigo herpetiformis in multiparous female successfully treated with oral cyclosporine [published May 12, 2011]. BMJ Case Rep. doi:10.1136/bcr.02.2011.3915
- Hazarika D. Generalized pustular psoriasis of pregnancy successfully treated with cyclosporine. Indian J Dermatol Venereol Leprol. 2009;75:638.
- Luan L, Han S, Zhang Z, et al. Personal treatment experience for severe generalized pustular psoriasis of pregnancy: two case reports. Dermatol Ther. 2014;27:174-177.
- Lamarque V, Leleu MF, Monka C, et al. Analysis of 629 pregnancy outcomes in transplant recipients treated with Sandimmun. Transplant Proc. 1997;29:2480.
- Bozdag K, Ozturk S, Ermete M. A case of recurrent impetigo herpetiformis treated with systemic corticosteroids and narrowband UVB. Cutan Ocul Toxicol. 2012;31:67-69.
- Treacy G. Using an analogous monoclonal antibody to evaluate the reproductive and chronic toxicity potential for a humanized anti-TNF alpha monoclonal antibody. Hum Exp Toxicol. 2000;19:226-228.
- Carter JD, Ladhani A, Ricca LR, et al. A safety assessment of tumor necrosis factor antagonists during pregnancy: a review of the Food and Drug Administration database. J Rheumatol. 2009;36:635-641.
- Diav-Citrin O, Otcheretianski-Volodarsky A, Shechtman S, et al. Pregnancy outcome following gestational exposure to TNF-alpha-inhibitors: a prospective, comparative, observational study. Reprod Toxicol. 2014;43:78-84.
- Verstappen SM, King Y, Watson KD, et al. Anti-TNF therapies and pregnancy: outcome of 130 pregnancies in the British Society for Rheumatology Biologics Register. Ann Rheum Dis. 2011;70:823-826.
- Gisbert JP, Chaparro M. Safety of anti-TNF agents during pregnancy and breastfeeding in women with inflammatory bowel disease. Am J Gastroenterol. 2013;108:1426-1438.
- Sheth N, Greenblatt DT, Acland K, et al. Generalized pustular psoriasis of pregnancy treated with infliximab. Clin Exp Dermatol. 2009;34:521-522.
- Puig L, Barco D, Alomar A. Treatment of psoriasis with anti-TNF drugs during pregnancy: case report and review of the literature. Dermatology. 2010;220:71-76.
- Ben-Horin S, Yavzori M, Kopylov U, et al. Detection of infliximab in breast milk of nursing mothers with inflammatory bowel disease. J Crohns Colitis. 2011;5:555-558.
- Cheent K, Nolan J, Shariq S, et al. Case report: fatal case of disseminated BCG infection in an infant born to a mother taking infliximab for Crohn’s disease. J Crohns Colitis. 2010;4:603-605.
Generalized pustular psoriasis of pregnancy (GPPP), formerly known as impetigo herpetiformis, is a rare dermatosis that causes maternal and fetal morbidity and mortality. It is characterized by widespread, circular, erythematous plaques with pustules at the periphery.1 Conventional first-line treatment includes systemic corticosteroids and cyclosporine. The National Psoriasis Foundation Medical Board also has included infliximab among the first-line treatment options for GPPP.2 Herein, we report a case of GPPP treated with infliximab at 30 weeks’ gestation and during the postpartum period.
Case Report
A 22-year-old woman was admitted to our inpatient clinic at 20 weeks’ gestation in her second pregnancy for evaluation of cutaneous eruptions covering the entire body. The lesions first appeared 3 to 4 days prior to her admission and dramatically progressed. She had a history of psoriasis vulgaris diagnosed during her first pregnancy 2 years prior that was treated with topical steroids throughout the pregnancy and methotrexate during lactation for a total of 11 months. She then was started on cyclosporine, which she used for 6 months due to ineffectiveness of the methotrexate, but she stopped treatment 4 months before the second pregnancy.
At the current presentation, physical examination revealed erythroderma and widespread pustules on the chest, abdomen, arms, and legs, including the intertriginous regions, that tended to coalesce and form lakes of pus over an erythematous base (Figure 1). The mucosae were normal. She exhibited a low blood pressure (85/50 mmHg) and high body temperature (102 °F [38.9 °C]). Routine laboratory examination revealed anemia and a normal leukocyte count. Her erythrocyte sedimentation rate (57 mm/h [reference range, <20 mm/h]) and C-reactive protein level (102 mg/L [reference range, <6 mg/L]) were elevated, whereas total calcium (8.11 mg/dL [reference range, 8.2–10.6 mg/dL]) and albumin (3.15 g/dL [reference range, >4.0 g/dL]) levels were low.
Empirical intravenous piperacillin/tazobactam was started due to hypotension, high fever, and elevated C-reactive protein levels; however, treatment was stopped after 4 days when microbiological cultures taken from blood and pustules revealed no bacterial growth, and therefore the fever was assumed to be caused by erythroderma. A skin biopsy before the start of topical and systemic treatment revealed changes consistent with GPPP.
Because her disease was extensive, systemic methylprednisolone 1.5 mg/kg once daily was started, and the dose was increased up to 2.5 mg/kg once daily on the tenth day of treatment to control new crops of eruptions. The dose was tapered to 2 mg/kg once daily when the lesions subsided 4 weeks into the treatment. The patient was discharged after 7 weeks at 27 weeks’ gestation.
Twelve days later, the patient was readmitted to the clinic in an erythrodermic state. The lesions were not controlled with increased doses of systemic corticosteroids. Treatment with cyclosporine was considered, but the patient refused; thus, infliximab treatment was planned. Isoniazid 300 mg once daily was started due to a risk of latent Mycobacterium tuberculosis infection revealed by a tuberculosis blood test. Other evaluations revealed no contraindications, and an infusion of infliximab 300 mg (5 mg/kg) was administered at 30 weeks’ gestation. There was visible improvement in the erythroderma and pustular lesions within the same day of treatment, and the lesions were completely cleared within 2 days of the infusion. The methylprednisolone dose was reduced to 1.5 mg/kg once daily.
Three days after treatment with infliximab, lesions with yellow encrustation appeared in the perioral region and on the oral mucosa and left ear. She was diagnosed with an oral herpes infection. Oral valacyclovir 1 g twice daily and topical mupirocin were started and the lesions subsided within 1 week. Twelve days after the infliximab infusion, new pustular lesions appeared, and a second infusion of infliximab was administered 13 days after the first, which cleared all lesions within 48 hours.
The patient’s methylprednisolone dose was tapered and stopped prior to delivery at 34 weeks’ gestation—2 weeks after the second dose of infliximab—as she did not have any new skin eruptions. A third infliximab infusion that normally would have occurred 4 weeks after the second treatment was postponed for a Cesarean section scheduled at 36 weeks’ gestation due to suspected intrauterine growth retardation. The patient stayed at the hospital until delivery without any new skin lesions. The gross and histopathologic examination of the placenta was normal. The neonate weighed 4.8 lb at birth and had neonatal jaundice that resolved spontaneously within 10 days but was otherwise healthy.
The patient returned to the clinic 3 weeks postpartum with a few pustules on erythematous plaques on the chest, abdomen, and back. At this time, she received a third infusion of infliximab 8 weeks after the second dose. For the past 5 years, the patient has been undergoing infliximab maintenance treatment, which she receives at the hospital every 8 weeks with excellent response. She has had no further pregnancies to date.
Comment
Generalized pustular psoriasis of pregnancy is a rare condition that typically occurs in the third trimester but also can start in the first and second trimesters. It may result in maternal and fetal morbidity by causing fluid and electrolyte imbalance and/or placental insufficiency, resulting in an increased risk for fetal abnormalities, stillbirth, and neonatal death.3 In subsequent pregnancies, GPPP has been observed to recur at an earlier gestational age with a more severe presentation.1,3
Generalized pustular psoriasis of pregnancy usually involves an eruption that begins symmetrically in the intertriginous areas and spreads to the rest of the body. The lesions present as erythematous annular plaques with pustules on the periphery and desquamation in the center due to older pustules.1,3 The mucous membranes also may be involved with erosive and exfoliative plaques, and there may be nail involvement. Patients often present with systemic symptoms such as fever, malaise, diarrhea, and vomiting.1 Laboratory investigations may reveal neutrophilic leukocytosis, high erythrocyte sedimentation rate, hypocalcemia, and hypoalbuminemia.4 Cultures from blood and pustules show no bacterial growth. A skin biopsy is helpful in diagnosis, with features similar to generalized pustular psoriasis, demonstrating spongiform pustules containing neutrophils, lymphocytic and neutrophilic infiltrates in the papillary dermis, and negative direct immunofluorescence.3
The differential diagnosis of GPPP includes subcorneal pustular dermatosis, dermatitis herpetiformis, herpes gestationis, impetigo, and acute generalized exanthematous pustulosis.1,3 Due to concerns of fetal implications, treatment options in GPPP are somewhat limited; however, the condition requires treatment because it may result in unfavorable pregnancy outcomes. Topical corticosteroids may be an option for limited disease.5,6 Systemic corticosteroids (eg, prednisone 60–80 mg/d) were previously considered as first-line agents, although they have shown limited efficacy in our case as well as in other case reports.7 Their ineffectiveness and risk for flare-up after dose tapering should be kept in mind when starting GPPP patients on systemic corticosteroids. Systemic cyclosporine (2–3 mg/kg/d) may be added to increase the efficacy of systemic steroids, which was done in several cases in literature.1,6,8 Although cyclosporine has been classified as a pregnancy category C drug, an analysis of pregnancy outcomes of 629 renal transplant patients revealed no association with adverse pregnancy outcomes compared to the general population and no increase in fetal malformations.9 Therefore, cyclosporine is a safe treatment option and was classified as a first-line drug for GPPP in a 2012 review by the National Psoriasis Foundation Medical Board.2 Narrowband UVB also has been reported to be used for the treatment of GPPP.10 Methotrexate and retinoids have been used in cases with lesions that persisted postpartum.1
Anti–tumor necrosis factor (TNF) α agents are another effective option for treatment of GPPP. Anti-TNF agents are classified as pregnancy category B due to results showing that anti-mouse TNF-α monoclonal antibodies did not cause embryotoxicity or teratogenicity in pregnant mice.11 Although Carter et al12 published a review of US Food and Drug Administration data on pregnant women receiving anti-TNF treatment and concluded that these agents were associated with the VACTERL group of malformations (vertebral defects, anal atresia, cardiac defect, tracheoesophageal fistula with esophageal atresia, cardiac defects, renal and limb anomalies), no such association was found in further studies. A 2014 study showed no difference in the rate of major malformations in infants born to women who were treated with anti-TNF drugs compared to the disease-matched group not treated with these agents and pregnant women counselled for nonteratogenic exposure.13 The same study detected an increase in preterm and low-birth-weight deliveries and suggested this might be caused by the increased severity of disease in patients requiring anti-TNF medication. The British Society of Rheumatology Biologics Register published data on pregnancy outcomes in 130 rheumatoid arthritis patients who had been exposed to anti-TNF agents.14 The results suggested an increased rate of spontaneous abortions in women exposed to anti-TNF treatment around the time of conception, especially in those taking these medications together with methotrexate or leflunomide; however, results also indicated that disease activity may have had an impact on the rate of spontaneous abortions in these patients. In a 2013 review of 462 women with inflammatory bowel disease who had been exposed to anti-TNF agents during pregnancy, the investigators concluded that pregnancy outcomes and the rate of congenital anomalies did not significantly differ from other inflammatory bowel disease patients not receiving anti-TNF drugs or the general population.15
In 2012, the National Board of the National Psoriasis Foundation put infliximab amongst the first-line treatment modalities for GPPP.2 In one case of GPPP in which the eruption persisted after delivery, the patient was treated with infliximab 7 weeks postpartum due to failure to control the disease with prednisolone 60 mg daily and cyclosporine 7.5 mg/kg daily. Unlike our patient, this patient was only started on an infliximab regimen after delivery.16 In another case reported in 2010, the patient was started on infliximab during the postpartum period of her first pregnancy following a pustular flare of previously diagnosed plaque psoriasis (not a generalized pustular psoriasis, as in our case).17 As a good response was obtained, infliximab treatment was continued in the patient throughout her second pregnancy.
Our case is unique in that infliximab was started during pregnancy because of intractable disease leading to systemic symptoms. Our patient showed an excellent response to infliximab after a 10-week disease course with repeated flare-ups and impairment to her overall condition. Delivery occurred at 36 weeks’ gestation due to suspected intrauterine growth retardation; however, the neonate was born with a 5-minute APGAR score of 10 and required no special medical care, which suggests that the low birth weight was constitutional due to the patient’s small frame (her height was 4 ft 11 in). The breast milk of patients with inflammatory bowel disease has been detected to contain very small amounts of infliximab (101 ng/mL, about 1/200 of the therapeutic blood level).18 Considering the large molecular weight of this agent and possible proteolysis in the stomach and intestines, infliximab is unlikely to affect the neonate.15 Thus, we encouraged our patient to breastfeed her baby. A case of fatal disseminated Bacille-Calmette-Guérin infection in an infant whose mother received infliximab treatment during pregnancy has been reported.19 It has been suggested that live vaccines should be avoided in neonates exposed to anti-TNF agents at least for the first 6 months of life or until the agent is no longer detectable in their blood.15 We therefore informed our patient’s family practitioner about this data.
Conclusion
We report a case of infliximab treatment for GPPP that was continued during the postpartum period. Infliximab was an effective treatment option in our patient with no detected serious adverse events and may be considered in other cases of GPPP that are not responsive to systemic steroids. However, further studies are warranted to evaluate the safety and efficacy of infliximab treatment for GPPP and psoriasis in pregnancy.
Generalized pustular psoriasis of pregnancy (GPPP), formerly known as impetigo herpetiformis, is a rare dermatosis that causes maternal and fetal morbidity and mortality. It is characterized by widespread, circular, erythematous plaques with pustules at the periphery.1 Conventional first-line treatment includes systemic corticosteroids and cyclosporine. The National Psoriasis Foundation Medical Board also has included infliximab among the first-line treatment options for GPPP.2 Herein, we report a case of GPPP treated with infliximab at 30 weeks’ gestation and during the postpartum period.
Case Report
A 22-year-old woman was admitted to our inpatient clinic at 20 weeks’ gestation in her second pregnancy for evaluation of cutaneous eruptions covering the entire body. The lesions first appeared 3 to 4 days prior to her admission and dramatically progressed. She had a history of psoriasis vulgaris diagnosed during her first pregnancy 2 years prior that was treated with topical steroids throughout the pregnancy and methotrexate during lactation for a total of 11 months. She then was started on cyclosporine, which she used for 6 months due to ineffectiveness of the methotrexate, but she stopped treatment 4 months before the second pregnancy.
At the current presentation, physical examination revealed erythroderma and widespread pustules on the chest, abdomen, arms, and legs, including the intertriginous regions, that tended to coalesce and form lakes of pus over an erythematous base (Figure 1). The mucosae were normal. She exhibited a low blood pressure (85/50 mmHg) and high body temperature (102 °F [38.9 °C]). Routine laboratory examination revealed anemia and a normal leukocyte count. Her erythrocyte sedimentation rate (57 mm/h [reference range, <20 mm/h]) and C-reactive protein level (102 mg/L [reference range, <6 mg/L]) were elevated, whereas total calcium (8.11 mg/dL [reference range, 8.2–10.6 mg/dL]) and albumin (3.15 g/dL [reference range, >4.0 g/dL]) levels were low.
Empirical intravenous piperacillin/tazobactam was started due to hypotension, high fever, and elevated C-reactive protein levels; however, treatment was stopped after 4 days when microbiological cultures taken from blood and pustules revealed no bacterial growth, and therefore the fever was assumed to be caused by erythroderma. A skin biopsy before the start of topical and systemic treatment revealed changes consistent with GPPP.
Because her disease was extensive, systemic methylprednisolone 1.5 mg/kg once daily was started, and the dose was increased up to 2.5 mg/kg once daily on the tenth day of treatment to control new crops of eruptions. The dose was tapered to 2 mg/kg once daily when the lesions subsided 4 weeks into the treatment. The patient was discharged after 7 weeks at 27 weeks’ gestation.
Twelve days later, the patient was readmitted to the clinic in an erythrodermic state. The lesions were not controlled with increased doses of systemic corticosteroids. Treatment with cyclosporine was considered, but the patient refused; thus, infliximab treatment was planned. Isoniazid 300 mg once daily was started due to a risk of latent Mycobacterium tuberculosis infection revealed by a tuberculosis blood test. Other evaluations revealed no contraindications, and an infusion of infliximab 300 mg (5 mg/kg) was administered at 30 weeks’ gestation. There was visible improvement in the erythroderma and pustular lesions within the same day of treatment, and the lesions were completely cleared within 2 days of the infusion. The methylprednisolone dose was reduced to 1.5 mg/kg once daily.
Three days after treatment with infliximab, lesions with yellow encrustation appeared in the perioral region and on the oral mucosa and left ear. She was diagnosed with an oral herpes infection. Oral valacyclovir 1 g twice daily and topical mupirocin were started and the lesions subsided within 1 week. Twelve days after the infliximab infusion, new pustular lesions appeared, and a second infusion of infliximab was administered 13 days after the first, which cleared all lesions within 48 hours.
The patient’s methylprednisolone dose was tapered and stopped prior to delivery at 34 weeks’ gestation—2 weeks after the second dose of infliximab—as she did not have any new skin eruptions. A third infliximab infusion that normally would have occurred 4 weeks after the second treatment was postponed for a Cesarean section scheduled at 36 weeks’ gestation due to suspected intrauterine growth retardation. The patient stayed at the hospital until delivery without any new skin lesions. The gross and histopathologic examination of the placenta was normal. The neonate weighed 4.8 lb at birth and had neonatal jaundice that resolved spontaneously within 10 days but was otherwise healthy.
The patient returned to the clinic 3 weeks postpartum with a few pustules on erythematous plaques on the chest, abdomen, and back. At this time, she received a third infusion of infliximab 8 weeks after the second dose. For the past 5 years, the patient has been undergoing infliximab maintenance treatment, which she receives at the hospital every 8 weeks with excellent response. She has had no further pregnancies to date.
Comment
Generalized pustular psoriasis of pregnancy is a rare condition that typically occurs in the third trimester but also can start in the first and second trimesters. It may result in maternal and fetal morbidity by causing fluid and electrolyte imbalance and/or placental insufficiency, resulting in an increased risk for fetal abnormalities, stillbirth, and neonatal death.3 In subsequent pregnancies, GPPP has been observed to recur at an earlier gestational age with a more severe presentation.1,3
Generalized pustular psoriasis of pregnancy usually involves an eruption that begins symmetrically in the intertriginous areas and spreads to the rest of the body. The lesions present as erythematous annular plaques with pustules on the periphery and desquamation in the center due to older pustules.1,3 The mucous membranes also may be involved with erosive and exfoliative plaques, and there may be nail involvement. Patients often present with systemic symptoms such as fever, malaise, diarrhea, and vomiting.1 Laboratory investigations may reveal neutrophilic leukocytosis, high erythrocyte sedimentation rate, hypocalcemia, and hypoalbuminemia.4 Cultures from blood and pustules show no bacterial growth. A skin biopsy is helpful in diagnosis, with features similar to generalized pustular psoriasis, demonstrating spongiform pustules containing neutrophils, lymphocytic and neutrophilic infiltrates in the papillary dermis, and negative direct immunofluorescence.3
The differential diagnosis of GPPP includes subcorneal pustular dermatosis, dermatitis herpetiformis, herpes gestationis, impetigo, and acute generalized exanthematous pustulosis.1,3 Due to concerns of fetal implications, treatment options in GPPP are somewhat limited; however, the condition requires treatment because it may result in unfavorable pregnancy outcomes. Topical corticosteroids may be an option for limited disease.5,6 Systemic corticosteroids (eg, prednisone 60–80 mg/d) were previously considered as first-line agents, although they have shown limited efficacy in our case as well as in other case reports.7 Their ineffectiveness and risk for flare-up after dose tapering should be kept in mind when starting GPPP patients on systemic corticosteroids. Systemic cyclosporine (2–3 mg/kg/d) may be added to increase the efficacy of systemic steroids, which was done in several cases in literature.1,6,8 Although cyclosporine has been classified as a pregnancy category C drug, an analysis of pregnancy outcomes of 629 renal transplant patients revealed no association with adverse pregnancy outcomes compared to the general population and no increase in fetal malformations.9 Therefore, cyclosporine is a safe treatment option and was classified as a first-line drug for GPPP in a 2012 review by the National Psoriasis Foundation Medical Board.2 Narrowband UVB also has been reported to be used for the treatment of GPPP.10 Methotrexate and retinoids have been used in cases with lesions that persisted postpartum.1
Anti–tumor necrosis factor (TNF) α agents are another effective option for treatment of GPPP. Anti-TNF agents are classified as pregnancy category B due to results showing that anti-mouse TNF-α monoclonal antibodies did not cause embryotoxicity or teratogenicity in pregnant mice.11 Although Carter et al12 published a review of US Food and Drug Administration data on pregnant women receiving anti-TNF treatment and concluded that these agents were associated with the VACTERL group of malformations (vertebral defects, anal atresia, cardiac defect, tracheoesophageal fistula with esophageal atresia, cardiac defects, renal and limb anomalies), no such association was found in further studies. A 2014 study showed no difference in the rate of major malformations in infants born to women who were treated with anti-TNF drugs compared to the disease-matched group not treated with these agents and pregnant women counselled for nonteratogenic exposure.13 The same study detected an increase in preterm and low-birth-weight deliveries and suggested this might be caused by the increased severity of disease in patients requiring anti-TNF medication. The British Society of Rheumatology Biologics Register published data on pregnancy outcomes in 130 rheumatoid arthritis patients who had been exposed to anti-TNF agents.14 The results suggested an increased rate of spontaneous abortions in women exposed to anti-TNF treatment around the time of conception, especially in those taking these medications together with methotrexate or leflunomide; however, results also indicated that disease activity may have had an impact on the rate of spontaneous abortions in these patients. In a 2013 review of 462 women with inflammatory bowel disease who had been exposed to anti-TNF agents during pregnancy, the investigators concluded that pregnancy outcomes and the rate of congenital anomalies did not significantly differ from other inflammatory bowel disease patients not receiving anti-TNF drugs or the general population.15
In 2012, the National Board of the National Psoriasis Foundation put infliximab amongst the first-line treatment modalities for GPPP.2 In one case of GPPP in which the eruption persisted after delivery, the patient was treated with infliximab 7 weeks postpartum due to failure to control the disease with prednisolone 60 mg daily and cyclosporine 7.5 mg/kg daily. Unlike our patient, this patient was only started on an infliximab regimen after delivery.16 In another case reported in 2010, the patient was started on infliximab during the postpartum period of her first pregnancy following a pustular flare of previously diagnosed plaque psoriasis (not a generalized pustular psoriasis, as in our case).17 As a good response was obtained, infliximab treatment was continued in the patient throughout her second pregnancy.
Our case is unique in that infliximab was started during pregnancy because of intractable disease leading to systemic symptoms. Our patient showed an excellent response to infliximab after a 10-week disease course with repeated flare-ups and impairment to her overall condition. Delivery occurred at 36 weeks’ gestation due to suspected intrauterine growth retardation; however, the neonate was born with a 5-minute APGAR score of 10 and required no special medical care, which suggests that the low birth weight was constitutional due to the patient’s small frame (her height was 4 ft 11 in). The breast milk of patients with inflammatory bowel disease has been detected to contain very small amounts of infliximab (101 ng/mL, about 1/200 of the therapeutic blood level).18 Considering the large molecular weight of this agent and possible proteolysis in the stomach and intestines, infliximab is unlikely to affect the neonate.15 Thus, we encouraged our patient to breastfeed her baby. A case of fatal disseminated Bacille-Calmette-Guérin infection in an infant whose mother received infliximab treatment during pregnancy has been reported.19 It has been suggested that live vaccines should be avoided in neonates exposed to anti-TNF agents at least for the first 6 months of life or until the agent is no longer detectable in their blood.15 We therefore informed our patient’s family practitioner about this data.
Conclusion
We report a case of infliximab treatment for GPPP that was continued during the postpartum period. Infliximab was an effective treatment option in our patient with no detected serious adverse events and may be considered in other cases of GPPP that are not responsive to systemic steroids. However, further studies are warranted to evaluate the safety and efficacy of infliximab treatment for GPPP and psoriasis in pregnancy.
- Lerhoff S, Pomeranz MK. Specific dermatoses of pregnancy and their treatment. Dermatol Ther. 2013;26:274-284.
- Robinson A, Van Voorhees AS, Hsu S, et al. Treatment of pustular psoriasis: from the Medical Board of the National Psoriasis Foundation. J Am Acad Dermatol. 2012;67:279-288.
- Oumeish OY, Parish JL. Impetigo herpetiformis. Clin Dermatol. 2006;24:101-104.
- Gao QQ, Xi MR, Yao Q. Impetigo herpetiformis during pregnancy: a case report and literature review. Dermatology. 2013;226:35-40.
- Bae YS, Van Voorhees AS, Hsu S, et al. Review of treatment options for psoriasis in pregnant or lactating women: from the Medical Board of the National Psoriasis Foundation. J Am Acad Dermatol. 2012;67:459-477.
- Shaw CJ, Wu P, Sriemevan A. First trimester impetigo herpetiformis in multiparous female successfully treated with oral cyclosporine [published May 12, 2011]. BMJ Case Rep. doi:10.1136/bcr.02.2011.3915
- Hazarika D. Generalized pustular psoriasis of pregnancy successfully treated with cyclosporine. Indian J Dermatol Venereol Leprol. 2009;75:638.
- Luan L, Han S, Zhang Z, et al. Personal treatment experience for severe generalized pustular psoriasis of pregnancy: two case reports. Dermatol Ther. 2014;27:174-177.
- Lamarque V, Leleu MF, Monka C, et al. Analysis of 629 pregnancy outcomes in transplant recipients treated with Sandimmun. Transplant Proc. 1997;29:2480.
- Bozdag K, Ozturk S, Ermete M. A case of recurrent impetigo herpetiformis treated with systemic corticosteroids and narrowband UVB. Cutan Ocul Toxicol. 2012;31:67-69.
- Treacy G. Using an analogous monoclonal antibody to evaluate the reproductive and chronic toxicity potential for a humanized anti-TNF alpha monoclonal antibody. Hum Exp Toxicol. 2000;19:226-228.
- Carter JD, Ladhani A, Ricca LR, et al. A safety assessment of tumor necrosis factor antagonists during pregnancy: a review of the Food and Drug Administration database. J Rheumatol. 2009;36:635-641.
- Diav-Citrin O, Otcheretianski-Volodarsky A, Shechtman S, et al. Pregnancy outcome following gestational exposure to TNF-alpha-inhibitors: a prospective, comparative, observational study. Reprod Toxicol. 2014;43:78-84.
- Verstappen SM, King Y, Watson KD, et al. Anti-TNF therapies and pregnancy: outcome of 130 pregnancies in the British Society for Rheumatology Biologics Register. Ann Rheum Dis. 2011;70:823-826.
- Gisbert JP, Chaparro M. Safety of anti-TNF agents during pregnancy and breastfeeding in women with inflammatory bowel disease. Am J Gastroenterol. 2013;108:1426-1438.
- Sheth N, Greenblatt DT, Acland K, et al. Generalized pustular psoriasis of pregnancy treated with infliximab. Clin Exp Dermatol. 2009;34:521-522.
- Puig L, Barco D, Alomar A. Treatment of psoriasis with anti-TNF drugs during pregnancy: case report and review of the literature. Dermatology. 2010;220:71-76.
- Ben-Horin S, Yavzori M, Kopylov U, et al. Detection of infliximab in breast milk of nursing mothers with inflammatory bowel disease. J Crohns Colitis. 2011;5:555-558.
- Cheent K, Nolan J, Shariq S, et al. Case report: fatal case of disseminated BCG infection in an infant born to a mother taking infliximab for Crohn’s disease. J Crohns Colitis. 2010;4:603-605.
- Lerhoff S, Pomeranz MK. Specific dermatoses of pregnancy and their treatment. Dermatol Ther. 2013;26:274-284.
- Robinson A, Van Voorhees AS, Hsu S, et al. Treatment of pustular psoriasis: from the Medical Board of the National Psoriasis Foundation. J Am Acad Dermatol. 2012;67:279-288.
- Oumeish OY, Parish JL. Impetigo herpetiformis. Clin Dermatol. 2006;24:101-104.
- Gao QQ, Xi MR, Yao Q. Impetigo herpetiformis during pregnancy: a case report and literature review. Dermatology. 2013;226:35-40.
- Bae YS, Van Voorhees AS, Hsu S, et al. Review of treatment options for psoriasis in pregnant or lactating women: from the Medical Board of the National Psoriasis Foundation. J Am Acad Dermatol. 2012;67:459-477.
- Shaw CJ, Wu P, Sriemevan A. First trimester impetigo herpetiformis in multiparous female successfully treated with oral cyclosporine [published May 12, 2011]. BMJ Case Rep. doi:10.1136/bcr.02.2011.3915
- Hazarika D. Generalized pustular psoriasis of pregnancy successfully treated with cyclosporine. Indian J Dermatol Venereol Leprol. 2009;75:638.
- Luan L, Han S, Zhang Z, et al. Personal treatment experience for severe generalized pustular psoriasis of pregnancy: two case reports. Dermatol Ther. 2014;27:174-177.
- Lamarque V, Leleu MF, Monka C, et al. Analysis of 629 pregnancy outcomes in transplant recipients treated with Sandimmun. Transplant Proc. 1997;29:2480.
- Bozdag K, Ozturk S, Ermete M. A case of recurrent impetigo herpetiformis treated with systemic corticosteroids and narrowband UVB. Cutan Ocul Toxicol. 2012;31:67-69.
- Treacy G. Using an analogous monoclonal antibody to evaluate the reproductive and chronic toxicity potential for a humanized anti-TNF alpha monoclonal antibody. Hum Exp Toxicol. 2000;19:226-228.
- Carter JD, Ladhani A, Ricca LR, et al. A safety assessment of tumor necrosis factor antagonists during pregnancy: a review of the Food and Drug Administration database. J Rheumatol. 2009;36:635-641.
- Diav-Citrin O, Otcheretianski-Volodarsky A, Shechtman S, et al. Pregnancy outcome following gestational exposure to TNF-alpha-inhibitors: a prospective, comparative, observational study. Reprod Toxicol. 2014;43:78-84.
- Verstappen SM, King Y, Watson KD, et al. Anti-TNF therapies and pregnancy: outcome of 130 pregnancies in the British Society for Rheumatology Biologics Register. Ann Rheum Dis. 2011;70:823-826.
- Gisbert JP, Chaparro M. Safety of anti-TNF agents during pregnancy and breastfeeding in women with inflammatory bowel disease. Am J Gastroenterol. 2013;108:1426-1438.
- Sheth N, Greenblatt DT, Acland K, et al. Generalized pustular psoriasis of pregnancy treated with infliximab. Clin Exp Dermatol. 2009;34:521-522.
- Puig L, Barco D, Alomar A. Treatment of psoriasis with anti-TNF drugs during pregnancy: case report and review of the literature. Dermatology. 2010;220:71-76.
- Ben-Horin S, Yavzori M, Kopylov U, et al. Detection of infliximab in breast milk of nursing mothers with inflammatory bowel disease. J Crohns Colitis. 2011;5:555-558.
- Cheent K, Nolan J, Shariq S, et al. Case report: fatal case of disseminated BCG infection in an infant born to a mother taking infliximab for Crohn’s disease. J Crohns Colitis. 2010;4:603-605.
Practice Points
- Generalized pustular psoriasis of pregnancy (GPPP) is a rare and severe condition that may lead to complications in both the mother and the fetus. Effective treatment with low impact on the fetus is essential.
- Infliximab, among other biologic agents, may be considered for the rapid clearing of skin lesions in GPPP.
New Approaches in Managing Cellulite: EXPERT INSIGHTS

Progressive Telangiectatic Rash
The Diagnosis: Cutaneous Collagenous Vasculopathy
Cutaneous collagenous vasculopathy (CCV) is an idiopathic microangiopathy of the small vessels in the superficial dermis. A condition first identified by Salama and Rosenthal1 in 2000, CCV likely is underreported, as its clinical mimickers are not routinely biopsied.2 It presents as asymptomatic telangiectatic macules, initially on the lower extremities and often spreading to the trunk. Cutaneous collagenous vasculopathy often is seen in middle-aged adults, and most patients have comorbidities such as hypertension, diabetes mellitus, or cardiovascular disease. The exact etiology of this disease is unknown.3,4
Histopathologically, CCV is characterized by dilated superficial vessels with thickened eosinophilic walls. The eosinophilic material is composed of hyalinized type IV collagen, which is periodic acid-Schiff positive and diastase resistant (Figure 1).3,4 Stains for amyloid are negative.
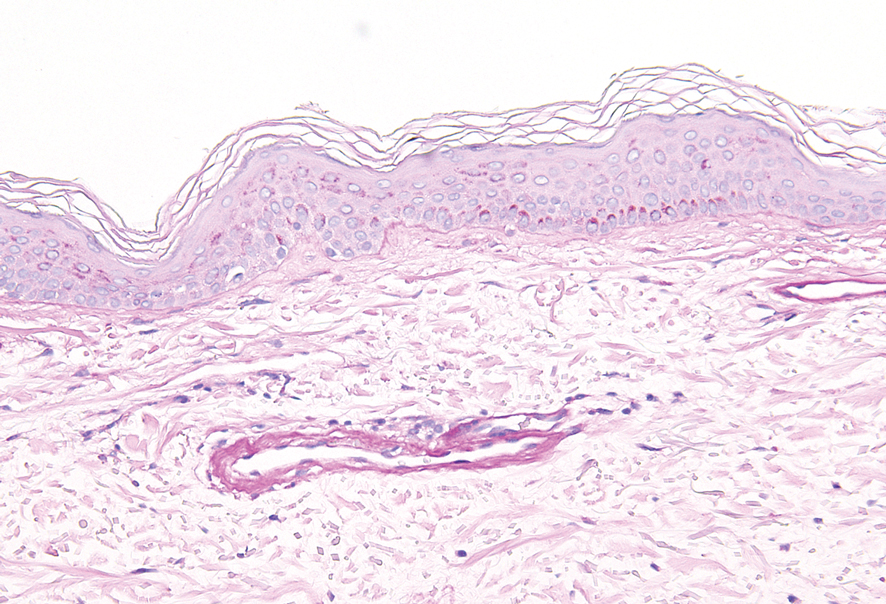
Generalized essential telangiectasia (GET) is a condition that presents with symmetric, blanchable, erythematous telangiectases.5 These lesions can occur alone or can accompany systemic diseases. Similar to CCV, the telangiectases tend to begin on the legs before gradually spreading to the trunk; however, this process more often is seen in females and occurs at an earlier age. Unlike CCV, GET can occur on mucosal surfaces, with cases of conjunctival and oral involvement reported.6 Generalized essential telangiectasia usually is a diagnosis of exclusion.7,8 It is thought that many CCV lesions have been misclassified clinically as GET, which highlights the importance of biopsy. Microscopically, GET is distinct from CCV in that the superficial dermis lacks thick-walled vessels.5,7 Although usually not associated with systemic diseases or progressive morbidity, treatment options are limited.8
Livedoid vasculopathy, also known as atrophie blanche, is caused by fibrin thrombi occlusion of dermal vessels. Clinically, patients have recurrent telangiectatic papules and painful ulcers on the lower extremities that gradually heal, leaving behind white stellate scars. It is caused by an underlying prothrombotic state with a superimposed inflammatory response.9 Livedoid vasculopathy primarily affects middle-aged women, and many patients have comorbidities such as scleroderma or systemic lupus erythematosus. Histologically, the epidermis often is ulcerated, and thrombi are visualized within small vessels. Eosinophilic fibrinoid material is deposited in vessel walls, including but not confined to vessels at the base of the epidermal ulcer (Figure 2). The fibrinoid material is periodic acid-Schiff positive and diastase resistant and can be highlighted with immunofluorescence, which may help to distinguish this entity from CCV.1,9 As the disease progresses, vessels are diffusely hyalinized, and there is epidermal atrophy and dermal sclerosis. Treatment options include antiplatelet and fibrinolytic drugs with a multidisciplinary approach to resolve pain and scarring.9
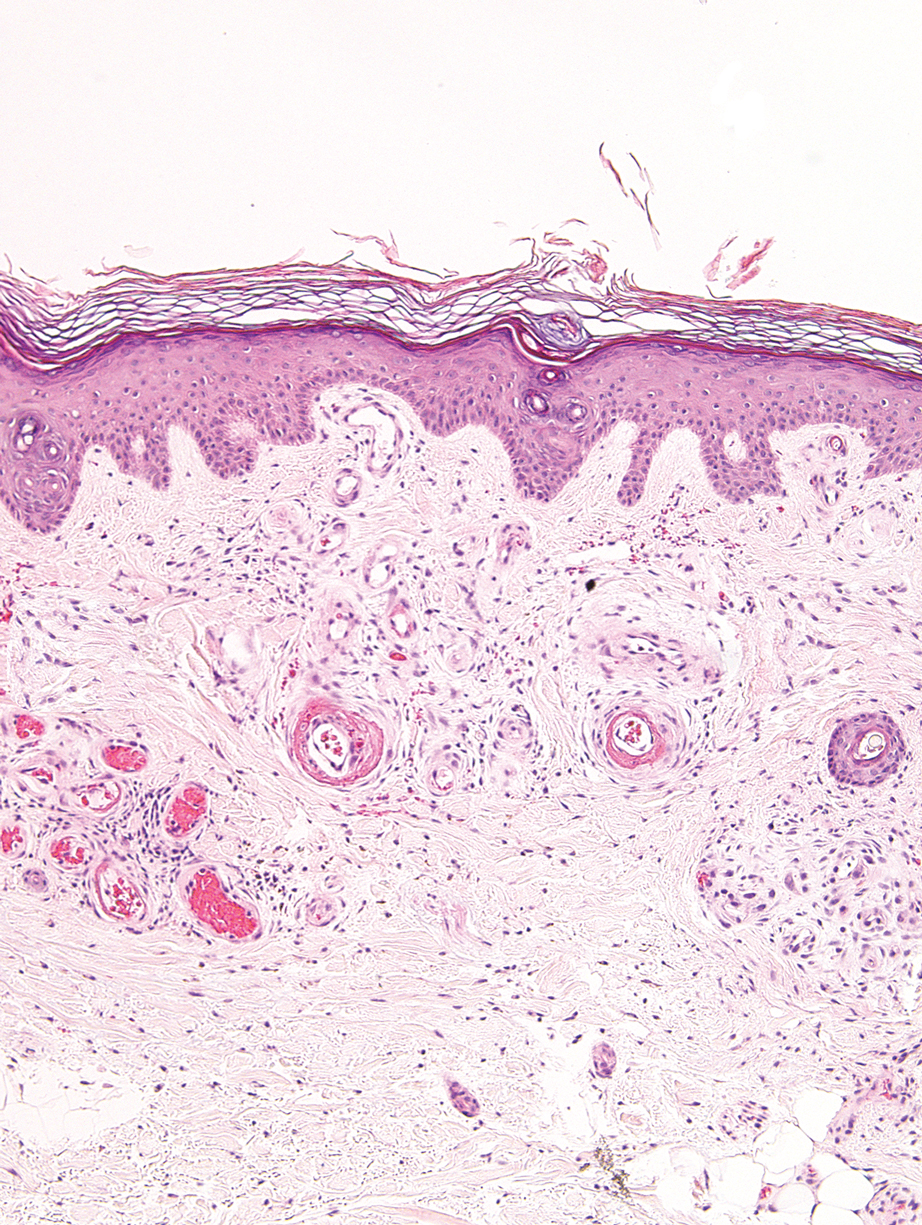
Primary systemic amyloidosis is a rare condition, and cutaneous manifestations are seen in approximately one-third of affected individuals. Amyloid deposition results in waxy papules that predominantly affect the face and periorbital areas but also may occur on the neck, flexural areas, and genitalia.5 Because the amyloid deposits also can be found within vessel walls, hemorrhagic lesions may occur. Microscopically, amorphous eosinophilic material can be found within the vessel walls, similar to CCV (Figure 3A); however, when stained with Congo red, cutaneous amyloidosis shows waxy red-orange material involving the vessel walls and exhibits apple green birefringence under polarization (Figure 3B).10 Amyloid also will be negative for type IV collagen, fibronectin, and laminin, whereas CCV will be positive.5
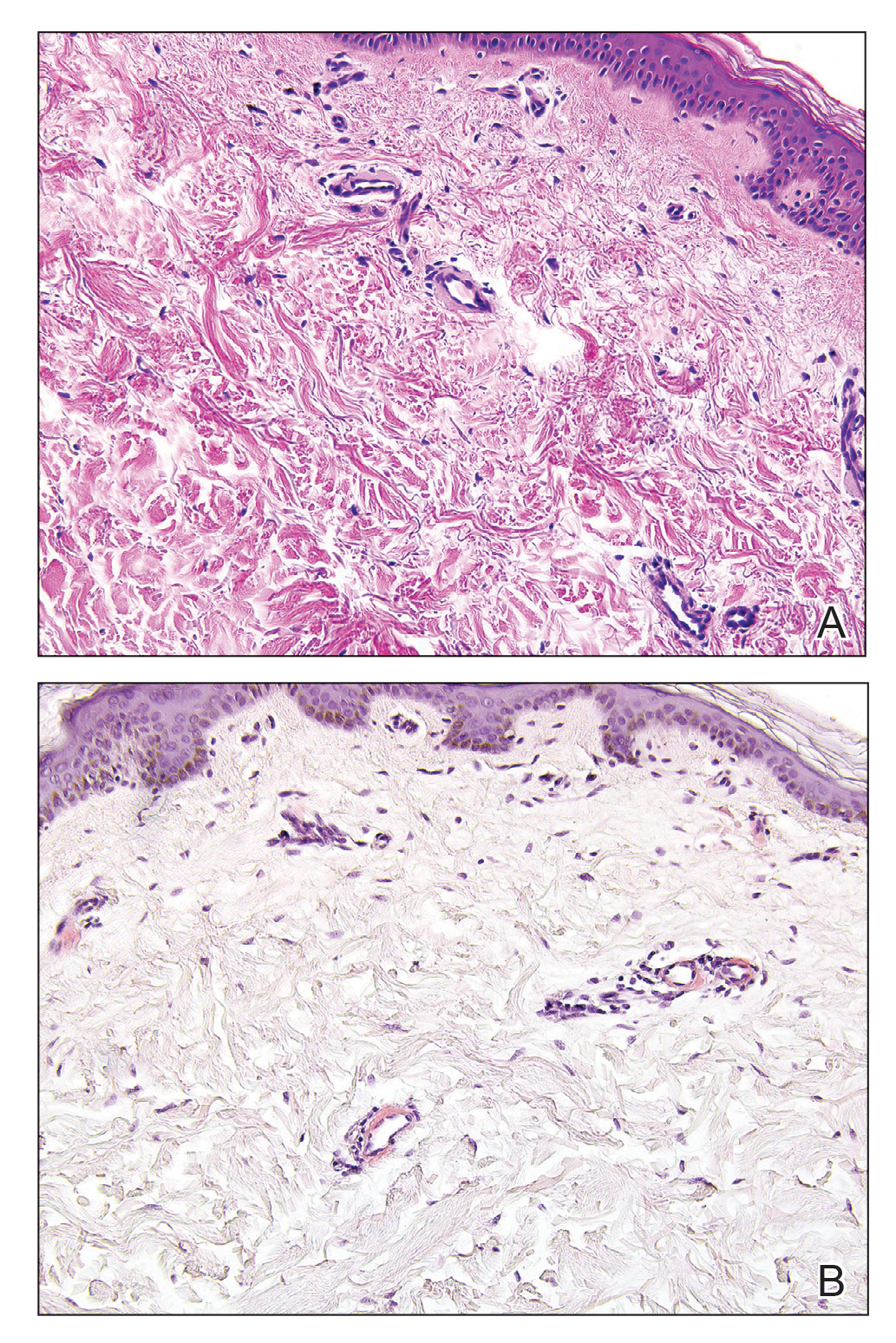
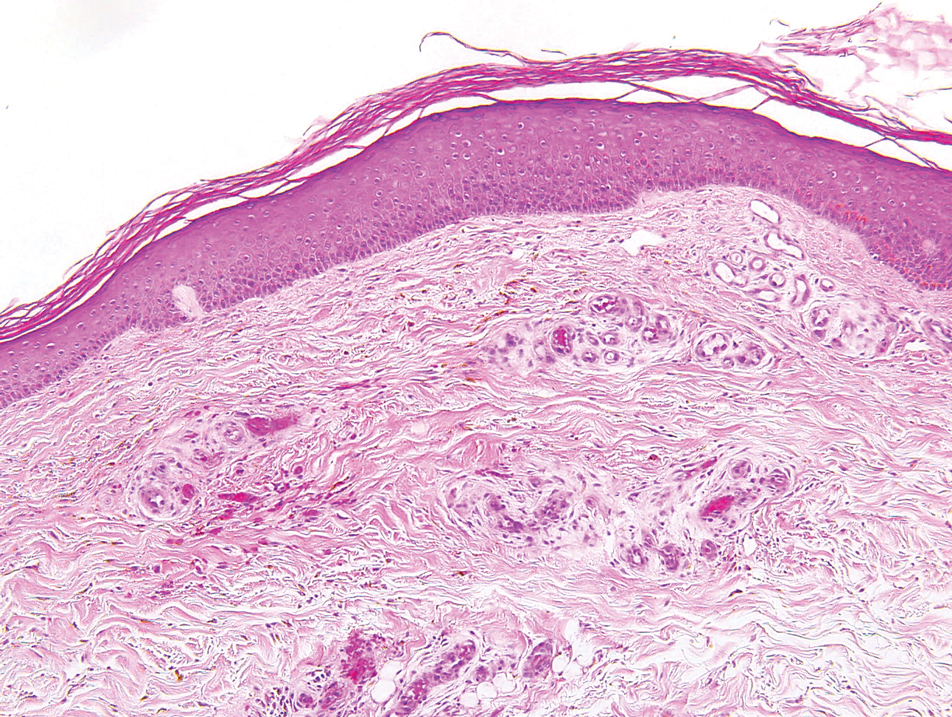
- Salama S, Rosenthal D. Cutaneous collagenous vasculopathy with generalized telangiectasia: an immunohistochemical and ultrastructural study. J Cutan Pathol. 2000;27:40-48.
- Bondier L, Tardieu M, Leveque P, et al. Cutaneous collagenous vasculopathy: report of two cases presenting as disseminated telangiectasias and review of the literature. Am J Dermatopathol. 2017;39:682-688.
- Sartori DS, Almeida HL Jr, Dorn TV, et al. Cutaneous collagenous vasculopathy: light and transmission electron microscopy. An Bras Dermatol. 2019;94:211-213.
- Brady BG, Ortleb M, Boyd AS, et al. Cutaneous collagenous vasculopathy. J Clin Aesthet Dermatol. 2015;8:49-52.
- Patterson JW, ed. Vascular tumors. Weedon's Skin Pathology. 4th ed. Churchill Livingstone/Elsevier; 2016:1069-1115.
- Knöpfel N, Martín-Santiago A, Saus C, et al. Extensive acquired telangiectasias: comparison of generalized essential telangiectasia and cutaneous collagenous vasculopathy. Actas Dermosifiliogr. 2017;108:E21-E26.
- Karimkhani C, Boyers LN, Olivere J, et al. Cutaneous collagenous vasculopathy. Cutis. 2019;103:E7-E8.
- McGrae JD, Winkelmann RK. Generalized essential telangiectasia: report of a clinical and histochemical study of 13 patients with acquired cutaneous lesions. JAMA. 1963;185:909-913.
- Vasudeva B, Neema S, Verma R. Livedoid vasculopathy: a review of pathogenesis and principles of management. Indian J Dermatol Venereol Leprol. 2016;82:478.
- Ko CJ, Barr RJ. Color--pink. In: Ko CJ, Barr RJ, eds. Dermatopathology: Diagnosis by First Impression. 3rd ed. Wiley; 2016:303-322.
- Clark ML, McGuinness AE, Vidal CI. Cutaneous collagenous vasculopathy: a unique case with positive direct immunofluorescence findings. Am J Dermatopathol. 2019;41:77-79.
The Diagnosis: Cutaneous Collagenous Vasculopathy
Cutaneous collagenous vasculopathy (CCV) is an idiopathic microangiopathy of the small vessels in the superficial dermis. A condition first identified by Salama and Rosenthal1 in 2000, CCV likely is underreported, as its clinical mimickers are not routinely biopsied.2 It presents as asymptomatic telangiectatic macules, initially on the lower extremities and often spreading to the trunk. Cutaneous collagenous vasculopathy often is seen in middle-aged adults, and most patients have comorbidities such as hypertension, diabetes mellitus, or cardiovascular disease. The exact etiology of this disease is unknown.3,4
Histopathologically, CCV is characterized by dilated superficial vessels with thickened eosinophilic walls. The eosinophilic material is composed of hyalinized type IV collagen, which is periodic acid-Schiff positive and diastase resistant (Figure 1).3,4 Stains for amyloid are negative.
Generalized essential telangiectasia (GET) is a condition that presents with symmetric, blanchable, erythematous telangiectases.5 These lesions can occur alone or can accompany systemic diseases. Similar to CCV, the telangiectases tend to begin on the legs before gradually spreading to the trunk; however, this process more often is seen in females and occurs at an earlier age. Unlike CCV, GET can occur on mucosal surfaces, with cases of conjunctival and oral involvement reported.6 Generalized essential telangiectasia usually is a diagnosis of exclusion.7,8 It is thought that many CCV lesions have been misclassified clinically as GET, which highlights the importance of biopsy. Microscopically, GET is distinct from CCV in that the superficial dermis lacks thick-walled vessels.5,7 Although usually not associated with systemic diseases or progressive morbidity, treatment options are limited.8
Livedoid vasculopathy, also known as atrophie blanche, is caused by fibrin thrombi occlusion of dermal vessels. Clinically, patients have recurrent telangiectatic papules and painful ulcers on the lower extremities that gradually heal, leaving behind white stellate scars. It is caused by an underlying prothrombotic state with a superimposed inflammatory response.9 Livedoid vasculopathy primarily affects middle-aged women, and many patients have comorbidities such as scleroderma or systemic lupus erythematosus. Histologically, the epidermis often is ulcerated, and thrombi are visualized within small vessels. Eosinophilic fibrinoid material is deposited in vessel walls, including but not confined to vessels at the base of the epidermal ulcer (Figure 2). The fibrinoid material is periodic acid-Schiff positive and diastase resistant and can be highlighted with immunofluorescence, which may help to distinguish this entity from CCV.1,9 As the disease progresses, vessels are diffusely hyalinized, and there is epidermal atrophy and dermal sclerosis. Treatment options include antiplatelet and fibrinolytic drugs with a multidisciplinary approach to resolve pain and scarring.9
Primary systemic amyloidosis is a rare condition, and cutaneous manifestations are seen in approximately one-third of affected individuals. Amyloid deposition results in waxy papules that predominantly affect the face and periorbital areas but also may occur on the neck, flexural areas, and genitalia.5 Because the amyloid deposits also can be found within vessel walls, hemorrhagic lesions may occur. Microscopically, amorphous eosinophilic material can be found within the vessel walls, similar to CCV (Figure 3A); however, when stained with Congo red, cutaneous amyloidosis shows waxy red-orange material involving the vessel walls and exhibits apple green birefringence under polarization (Figure 3B).10 Amyloid also will be negative for type IV collagen, fibronectin, and laminin, whereas CCV will be positive.5
The Diagnosis: Cutaneous Collagenous Vasculopathy
Cutaneous collagenous vasculopathy (CCV) is an idiopathic microangiopathy of the small vessels in the superficial dermis. A condition first identified by Salama and Rosenthal1 in 2000, CCV likely is underreported, as its clinical mimickers are not routinely biopsied.2 It presents as asymptomatic telangiectatic macules, initially on the lower extremities and often spreading to the trunk. Cutaneous collagenous vasculopathy often is seen in middle-aged adults, and most patients have comorbidities such as hypertension, diabetes mellitus, or cardiovascular disease. The exact etiology of this disease is unknown.3,4
Histopathologically, CCV is characterized by dilated superficial vessels with thickened eosinophilic walls. The eosinophilic material is composed of hyalinized type IV collagen, which is periodic acid-Schiff positive and diastase resistant (Figure 1).3,4 Stains for amyloid are negative.
Generalized essential telangiectasia (GET) is a condition that presents with symmetric, blanchable, erythematous telangiectases.5 These lesions can occur alone or can accompany systemic diseases. Similar to CCV, the telangiectases tend to begin on the legs before gradually spreading to the trunk; however, this process more often is seen in females and occurs at an earlier age. Unlike CCV, GET can occur on mucosal surfaces, with cases of conjunctival and oral involvement reported.6 Generalized essential telangiectasia usually is a diagnosis of exclusion.7,8 It is thought that many CCV lesions have been misclassified clinically as GET, which highlights the importance of biopsy. Microscopically, GET is distinct from CCV in that the superficial dermis lacks thick-walled vessels.5,7 Although usually not associated with systemic diseases or progressive morbidity, treatment options are limited.8
Livedoid vasculopathy, also known as atrophie blanche, is caused by fibrin thrombi occlusion of dermal vessels. Clinically, patients have recurrent telangiectatic papules and painful ulcers on the lower extremities that gradually heal, leaving behind white stellate scars. It is caused by an underlying prothrombotic state with a superimposed inflammatory response.9 Livedoid vasculopathy primarily affects middle-aged women, and many patients have comorbidities such as scleroderma or systemic lupus erythematosus. Histologically, the epidermis often is ulcerated, and thrombi are visualized within small vessels. Eosinophilic fibrinoid material is deposited in vessel walls, including but not confined to vessels at the base of the epidermal ulcer (Figure 2). The fibrinoid material is periodic acid-Schiff positive and diastase resistant and can be highlighted with immunofluorescence, which may help to distinguish this entity from CCV.1,9 As the disease progresses, vessels are diffusely hyalinized, and there is epidermal atrophy and dermal sclerosis. Treatment options include antiplatelet and fibrinolytic drugs with a multidisciplinary approach to resolve pain and scarring.9
Primary systemic amyloidosis is a rare condition, and cutaneous manifestations are seen in approximately one-third of affected individuals. Amyloid deposition results in waxy papules that predominantly affect the face and periorbital areas but also may occur on the neck, flexural areas, and genitalia.5 Because the amyloid deposits also can be found within vessel walls, hemorrhagic lesions may occur. Microscopically, amorphous eosinophilic material can be found within the vessel walls, similar to CCV (Figure 3A); however, when stained with Congo red, cutaneous amyloidosis shows waxy red-orange material involving the vessel walls and exhibits apple green birefringence under polarization (Figure 3B).10 Amyloid also will be negative for type IV collagen, fibronectin, and laminin, whereas CCV will be positive.5
- Salama S, Rosenthal D. Cutaneous collagenous vasculopathy with generalized telangiectasia: an immunohistochemical and ultrastructural study. J Cutan Pathol. 2000;27:40-48.
- Bondier L, Tardieu M, Leveque P, et al. Cutaneous collagenous vasculopathy: report of two cases presenting as disseminated telangiectasias and review of the literature. Am J Dermatopathol. 2017;39:682-688.
- Sartori DS, Almeida HL Jr, Dorn TV, et al. Cutaneous collagenous vasculopathy: light and transmission electron microscopy. An Bras Dermatol. 2019;94:211-213.
- Brady BG, Ortleb M, Boyd AS, et al. Cutaneous collagenous vasculopathy. J Clin Aesthet Dermatol. 2015;8:49-52.
- Patterson JW, ed. Vascular tumors. Weedon's Skin Pathology. 4th ed. Churchill Livingstone/Elsevier; 2016:1069-1115.
- Knöpfel N, Martín-Santiago A, Saus C, et al. Extensive acquired telangiectasias: comparison of generalized essential telangiectasia and cutaneous collagenous vasculopathy. Actas Dermosifiliogr. 2017;108:E21-E26.
- Karimkhani C, Boyers LN, Olivere J, et al. Cutaneous collagenous vasculopathy. Cutis. 2019;103:E7-E8.
- McGrae JD, Winkelmann RK. Generalized essential telangiectasia: report of a clinical and histochemical study of 13 patients with acquired cutaneous lesions. JAMA. 1963;185:909-913.
- Vasudeva B, Neema S, Verma R. Livedoid vasculopathy: a review of pathogenesis and principles of management. Indian J Dermatol Venereol Leprol. 2016;82:478.
- Ko CJ, Barr RJ. Color--pink. In: Ko CJ, Barr RJ, eds. Dermatopathology: Diagnosis by First Impression. 3rd ed. Wiley; 2016:303-322.
- Clark ML, McGuinness AE, Vidal CI. Cutaneous collagenous vasculopathy: a unique case with positive direct immunofluorescence findings. Am J Dermatopathol. 2019;41:77-79.
- Salama S, Rosenthal D. Cutaneous collagenous vasculopathy with generalized telangiectasia: an immunohistochemical and ultrastructural study. J Cutan Pathol. 2000;27:40-48.
- Bondier L, Tardieu M, Leveque P, et al. Cutaneous collagenous vasculopathy: report of two cases presenting as disseminated telangiectasias and review of the literature. Am J Dermatopathol. 2017;39:682-688.
- Sartori DS, Almeida HL Jr, Dorn TV, et al. Cutaneous collagenous vasculopathy: light and transmission electron microscopy. An Bras Dermatol. 2019;94:211-213.
- Brady BG, Ortleb M, Boyd AS, et al. Cutaneous collagenous vasculopathy. J Clin Aesthet Dermatol. 2015;8:49-52.
- Patterson JW, ed. Vascular tumors. Weedon's Skin Pathology. 4th ed. Churchill Livingstone/Elsevier; 2016:1069-1115.
- Knöpfel N, Martín-Santiago A, Saus C, et al. Extensive acquired telangiectasias: comparison of generalized essential telangiectasia and cutaneous collagenous vasculopathy. Actas Dermosifiliogr. 2017;108:E21-E26.
- Karimkhani C, Boyers LN, Olivere J, et al. Cutaneous collagenous vasculopathy. Cutis. 2019;103:E7-E8.
- McGrae JD, Winkelmann RK. Generalized essential telangiectasia: report of a clinical and histochemical study of 13 patients with acquired cutaneous lesions. JAMA. 1963;185:909-913.
- Vasudeva B, Neema S, Verma R. Livedoid vasculopathy: a review of pathogenesis and principles of management. Indian J Dermatol Venereol Leprol. 2016;82:478.
- Ko CJ, Barr RJ. Color--pink. In: Ko CJ, Barr RJ, eds. Dermatopathology: Diagnosis by First Impression. 3rd ed. Wiley; 2016:303-322.
- Clark ML, McGuinness AE, Vidal CI. Cutaneous collagenous vasculopathy: a unique case with positive direct immunofluorescence findings. Am J Dermatopathol. 2019;41:77-79.
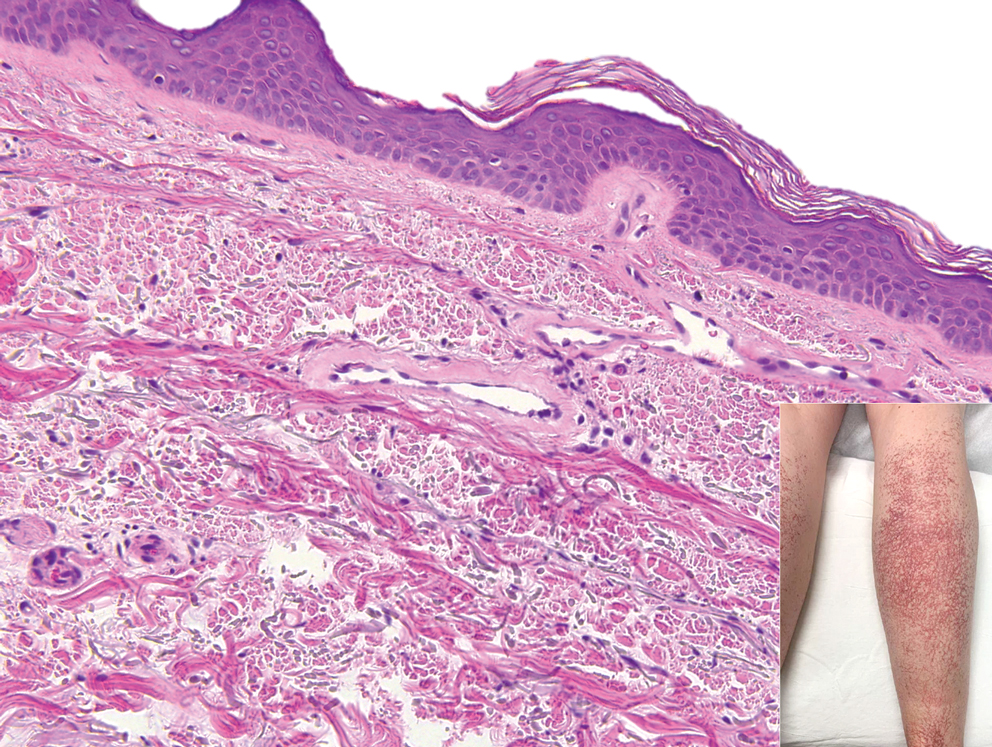
A 54-year-old woman presented with purple-red vessels on the lower legs of 15 years’ duration with gradual proximal progression to involve the thighs, breasts, and forearms. A punch biopsy of the inner thigh was obtained for histopathologic evaluation.