User login
Cutis is a peer-reviewed clinical journal for the dermatologist, allergist, and general practitioner published monthly since 1965. Concise clinical articles present the practical side of dermatology, helping physicians to improve patient care. Cutis is referenced in Index Medicus/MEDLINE and is written and edited by industry leaders.
ass lick
assault rifle
balls
ballsac
black jack
bleach
Boko Haram
bondage
causas
cheap
child abuse
cocaine
compulsive behaviors
cost of miracles
cunt
Daech
display network stats
drug paraphernalia
explosion
fart
fda and death
fda AND warn
fda AND warning
fda AND warns
feom
fuck
gambling
gfc
gun
human trafficking
humira AND expensive
illegal
ISIL
ISIS
Islamic caliphate
Islamic state
madvocate
masturbation
mixed martial arts
MMA
molestation
national rifle association
NRA
nsfw
nuccitelli
pedophile
pedophilia
poker
porn
porn
pornography
psychedelic drug
recreational drug
sex slave rings
shit
slot machine
snort
substance abuse
terrorism
terrorist
texarkana
Texas hold 'em
UFC
section[contains(@class, 'nav-hidden')]
section[contains(@class, 'nav-hidden active')
A peer-reviewed, indexed journal for dermatologists with original research, image quizzes, cases and reviews, and columns.
Pseudoepitheliomatous Hyperplasia Arising From Purple Tattoo Pigment
To the Editor:
Pseudoepitheliomatous hyperplasia (PEH) is an uncommon type of reactive epidermal proliferation that can occur from a variety of causes, including an underlying infection, inflammation, neoplastic condition, or trauma induced from tattooing.1 Diagnosis can be challenging and requires clinicopathologic correlation, as PEH can mimic malignancy on histopathology.2-4 Histologically, PEH shows irregular hyperplasia of the epidermis and adnexal epithelium, elongation of the rete ridges, and extension of the reactive proliferation into the dermis. Absence of cytologic atypia is key to the diagnosis of PEH, helping to distinguish it from squamous cell carcinoma and keratoacanthoma. Clinically, patients typically present with well-demarcated, erythematous, scaly plaques or nodules in reactive areas, which can be symptomatically pruritic.
A 48-year-old woman presented with scaly and crusted verrucous plaques of 2 months’ duration that were isolated to the areas of purple pigment within a tattoo on the right lower leg. The patient reported pruritus in the affected areas that occurred immediately after obtaining the tattoo, which was her first and only tattoo. She denied any pertinent medical history, including an absence of immunosuppression and autoimmune or chronic inflammatory diseases.
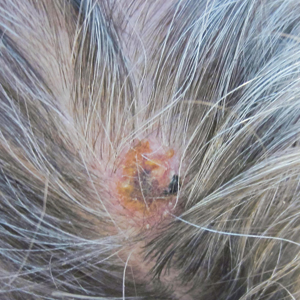
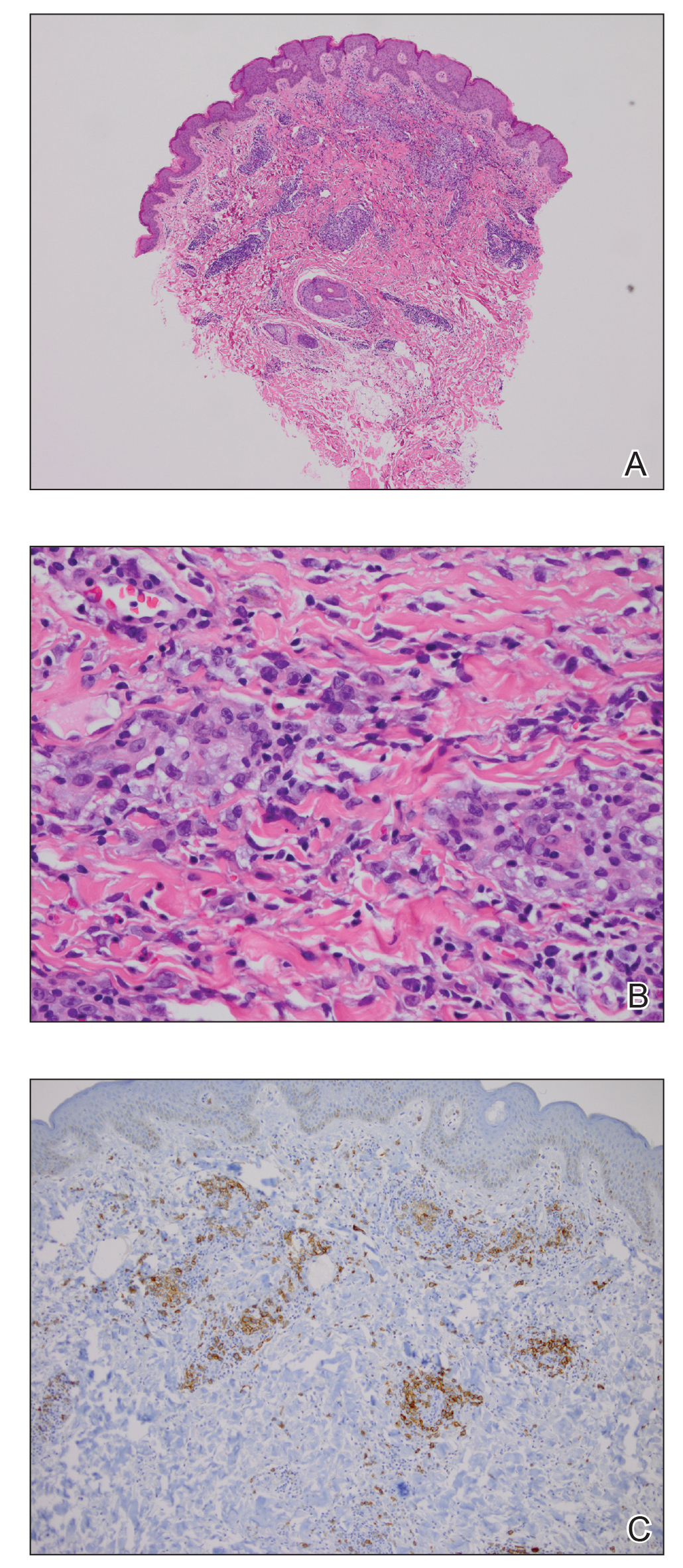
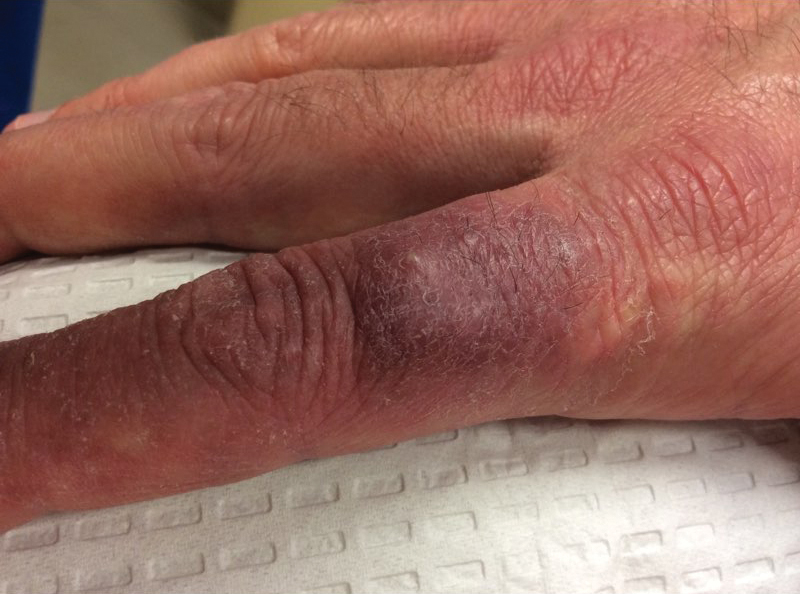
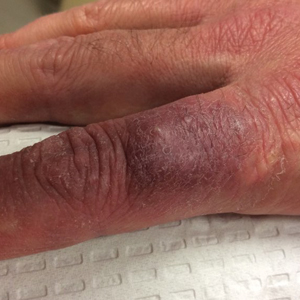
Physical examination revealed scaly and crusted plaques isolated to areas of purple tattoo pigment (Figure 1). Areas of red, green, black, and blue pigmentation within the tattoo were uninvolved. With the initial suspicion of allergic contact dermatitis, two 6-mm punch biopsies were taken from adjacent linear plaques on the right leg for histology and tissue culture. Histopathologic evaluation revealed dermal tattoo pigment with overlying PEH and was negative for signs of infection (Figure 2). Infectious stains such as periodic acid–Schiff, Grocott-Gomori methenamine-silver, and Gram stains were performed and found to be negative. In addition, culture for mycobacteria came back negative. Prurigo was on the differential; however, histopathologic changes were more compatible with a PEH reaction to the tattoo.


Upon diagnosis, the patient was treated with clobetasol ointment 0.05% under occlusion for 1 month without reported improvement. The patient subsequently elected to undergo treatment with intralesional triamcinolone 5 mg/mL to all areas of PEH, except the areas immediately surrounding the healing biopsy sites. Twice-daily application of tacrolimus ointment 0.1% to all affected areas also was initiated. At follow-up 1 month later, she reported symptomatic relief of pruritus with a notable reduction in the thickness of the plaques in all treated areas (Figure 3). A second course of intralesional triamcinolone 5 mg/mL was performed. No additional plaques appeared during the treatment course, and the patient reported high satisfaction with the final result that was achieved.
An increase in the popularity of tattooing has led to more reports of various tattoo skin reactions.4-6 The differential diagnosis is broad for tattoo reactions and includes granulomatous inflammation, sarcoidosis, psoriasis (Köbner phenomenon), allergic contact dermatitis, lichen planus, morphealike reactions, squamous cell carcinoma, and keratoacanthoma,5 which makes clinicopathologic correlation essential for accurate diagnosis. Our case demonstrated the characteristic epithelial hyperplasia in the absence of cytologic atypia. In addition, the presence of mixed dermal inflammation histologically was noted in our patient.

Pseudoepitheliomatous hyperplasia development from a tattoo in areas of both mercury-based and non–mercury-based red pigment is a known association.7-9 Balfour et al10 also reported a case of PEH occurring secondary to manganese-based purple pigment. Because few cases have been reported, the epidemiology for PEH currently is unknown. Treatment of this condition primarily is anecdotal, with prior cases showing success with topical or intralesional steroids.5,7 As with any steroid-based treatment, we recommend less aggressive treatments initially with close follow-up and adaptation as needed to minimize adverse effects such as unwanted atrophy. Some success has been reported with the use of the Q-switched Nd:YAG laser in the setting of a PEH tattoo reaction.5 Similar to other tattoo reactions, surgical removal can be considered with failure of more conservative treatment methods and focal involvement.
We report an unusual case of PEH occurring secondary to purple tattoo pigment. Our report also demonstrates the clinical and symptomatic improvement of PEH that can be achieved through the use of intralesional corticosteroid therapy. Our patient represents a case of PEH reactive to tattooing with purple ink. Further research to elucidate the precise pathogenesis of PEH tattoo reactions would be helpful in identifying high-risk patients and determining the most efficacious treatments.
- Meani RE, Nixon RL, O’Keefe R, et al. Pseudoepitheliomatous hyperplasia secondary to allergic contact dermatitis to Grevillea Robyn Gordon. Australas J Dermatol. 2017;58:E8-E10.
- Chakrabarti S, Chakrabarti P, Agrawal D, et al. Pseudoepitheliomatous hyperplasia: a clinical entity mistaken for squamous cell carcinoma. J Cutan Aesthet Surg. 2014;7:232.
- Kluger N. Issues with keratoacanthoma, pseudoepitheliomatous hyperplasia and squamous cell carcinoma within tattoos: a clinical point of view. J Cutan Pathol. 2009;37:812-813.
- Zayour M, Lazova R. Pseudoepitheliomatous hyperplasia: a review. Am J Dermatopathol. 2011;33:112-126.
- Bassi A, Campolmi P, Cannarozzo G, et al. Tattoo-associated skin reaction: the importance of an early diagnosis and proper treatment [published online July 23, 2014]. Biomed Res Int. 2014;2014:354608.
- Serup J. Diagnostic tools for doctors’ evaluation of tattoo complications. Curr Probl Dermatol. 2017;52:42-57.
- Kazlouskaya V, Junkins-Hopkins JM. Pseudoepitheliomatous hyperplasia in a red pigment tattoo: a separate entity or hypertrophic lichen planus-like reaction? J Clin Aesthet Dermatol. 2015;8:48-52.
- Kluger N, Durand L, Minier-Thoumin C, et al. Pseudoepitheliomatous epidermal hyperplasia in tattoos: report of three cases. Am J Clin Dermatol. 2008;9:337-340.
- Cui W, McGregor DH, Stark SP, et al. Pseudoepitheliomatous hyperplasia—an unusual reaction following tattoo: report of a case and review of the literature. Int J Dermatol. 2007;46:743-745.
- Balfour E, Olhoffer I, Leffell D, et al. Massive pseudoepitheliomatous hyperplasia: an unusual reaction to a tattoo. Am J Dermatopathol. 2003;25:338-340.
To the Editor:
Pseudoepitheliomatous hyperplasia (PEH) is an uncommon type of reactive epidermal proliferation that can occur from a variety of causes, including an underlying infection, inflammation, neoplastic condition, or trauma induced from tattooing.1 Diagnosis can be challenging and requires clinicopathologic correlation, as PEH can mimic malignancy on histopathology.2-4 Histologically, PEH shows irregular hyperplasia of the epidermis and adnexal epithelium, elongation of the rete ridges, and extension of the reactive proliferation into the dermis. Absence of cytologic atypia is key to the diagnosis of PEH, helping to distinguish it from squamous cell carcinoma and keratoacanthoma. Clinically, patients typically present with well-demarcated, erythematous, scaly plaques or nodules in reactive areas, which can be symptomatically pruritic.
A 48-year-old woman presented with scaly and crusted verrucous plaques of 2 months’ duration that were isolated to the areas of purple pigment within a tattoo on the right lower leg. The patient reported pruritus in the affected areas that occurred immediately after obtaining the tattoo, which was her first and only tattoo. She denied any pertinent medical history, including an absence of immunosuppression and autoimmune or chronic inflammatory diseases.
Physical examination revealed scaly and crusted plaques isolated to areas of purple tattoo pigment (Figure 1). Areas of red, green, black, and blue pigmentation within the tattoo were uninvolved. With the initial suspicion of allergic contact dermatitis, two 6-mm punch biopsies were taken from adjacent linear plaques on the right leg for histology and tissue culture. Histopathologic evaluation revealed dermal tattoo pigment with overlying PEH and was negative for signs of infection (Figure 2). Infectious stains such as periodic acid–Schiff, Grocott-Gomori methenamine-silver, and Gram stains were performed and found to be negative. In addition, culture for mycobacteria came back negative. Prurigo was on the differential; however, histopathologic changes were more compatible with a PEH reaction to the tattoo.
Upon diagnosis, the patient was treated with clobetasol ointment 0.05% under occlusion for 1 month without reported improvement. The patient subsequently elected to undergo treatment with intralesional triamcinolone 5 mg/mL to all areas of PEH, except the areas immediately surrounding the healing biopsy sites. Twice-daily application of tacrolimus ointment 0.1% to all affected areas also was initiated. At follow-up 1 month later, she reported symptomatic relief of pruritus with a notable reduction in the thickness of the plaques in all treated areas (Figure 3). A second course of intralesional triamcinolone 5 mg/mL was performed. No additional plaques appeared during the treatment course, and the patient reported high satisfaction with the final result that was achieved.
An increase in the popularity of tattooing has led to more reports of various tattoo skin reactions.4-6 The differential diagnosis is broad for tattoo reactions and includes granulomatous inflammation, sarcoidosis, psoriasis (Köbner phenomenon), allergic contact dermatitis, lichen planus, morphealike reactions, squamous cell carcinoma, and keratoacanthoma,5 which makes clinicopathologic correlation essential for accurate diagnosis. Our case demonstrated the characteristic epithelial hyperplasia in the absence of cytologic atypia. In addition, the presence of mixed dermal inflammation histologically was noted in our patient.
Pseudoepitheliomatous hyperplasia development from a tattoo in areas of both mercury-based and non–mercury-based red pigment is a known association.7-9 Balfour et al10 also reported a case of PEH occurring secondary to manganese-based purple pigment. Because few cases have been reported, the epidemiology for PEH currently is unknown. Treatment of this condition primarily is anecdotal, with prior cases showing success with topical or intralesional steroids.5,7 As with any steroid-based treatment, we recommend less aggressive treatments initially with close follow-up and adaptation as needed to minimize adverse effects such as unwanted atrophy. Some success has been reported with the use of the Q-switched Nd:YAG laser in the setting of a PEH tattoo reaction.5 Similar to other tattoo reactions, surgical removal can be considered with failure of more conservative treatment methods and focal involvement.
We report an unusual case of PEH occurring secondary to purple tattoo pigment. Our report also demonstrates the clinical and symptomatic improvement of PEH that can be achieved through the use of intralesional corticosteroid therapy. Our patient represents a case of PEH reactive to tattooing with purple ink. Further research to elucidate the precise pathogenesis of PEH tattoo reactions would be helpful in identifying high-risk patients and determining the most efficacious treatments.
To the Editor:
Pseudoepitheliomatous hyperplasia (PEH) is an uncommon type of reactive epidermal proliferation that can occur from a variety of causes, including an underlying infection, inflammation, neoplastic condition, or trauma induced from tattooing.1 Diagnosis can be challenging and requires clinicopathologic correlation, as PEH can mimic malignancy on histopathology.2-4 Histologically, PEH shows irregular hyperplasia of the epidermis and adnexal epithelium, elongation of the rete ridges, and extension of the reactive proliferation into the dermis. Absence of cytologic atypia is key to the diagnosis of PEH, helping to distinguish it from squamous cell carcinoma and keratoacanthoma. Clinically, patients typically present with well-demarcated, erythematous, scaly plaques or nodules in reactive areas, which can be symptomatically pruritic.
A 48-year-old woman presented with scaly and crusted verrucous plaques of 2 months’ duration that were isolated to the areas of purple pigment within a tattoo on the right lower leg. The patient reported pruritus in the affected areas that occurred immediately after obtaining the tattoo, which was her first and only tattoo. She denied any pertinent medical history, including an absence of immunosuppression and autoimmune or chronic inflammatory diseases.
Physical examination revealed scaly and crusted plaques isolated to areas of purple tattoo pigment (Figure 1). Areas of red, green, black, and blue pigmentation within the tattoo were uninvolved. With the initial suspicion of allergic contact dermatitis, two 6-mm punch biopsies were taken from adjacent linear plaques on the right leg for histology and tissue culture. Histopathologic evaluation revealed dermal tattoo pigment with overlying PEH and was negative for signs of infection (Figure 2). Infectious stains such as periodic acid–Schiff, Grocott-Gomori methenamine-silver, and Gram stains were performed and found to be negative. In addition, culture for mycobacteria came back negative. Prurigo was on the differential; however, histopathologic changes were more compatible with a PEH reaction to the tattoo.
Upon diagnosis, the patient was treated with clobetasol ointment 0.05% under occlusion for 1 month without reported improvement. The patient subsequently elected to undergo treatment with intralesional triamcinolone 5 mg/mL to all areas of PEH, except the areas immediately surrounding the healing biopsy sites. Twice-daily application of tacrolimus ointment 0.1% to all affected areas also was initiated. At follow-up 1 month later, she reported symptomatic relief of pruritus with a notable reduction in the thickness of the plaques in all treated areas (Figure 3). A second course of intralesional triamcinolone 5 mg/mL was performed. No additional plaques appeared during the treatment course, and the patient reported high satisfaction with the final result that was achieved.
An increase in the popularity of tattooing has led to more reports of various tattoo skin reactions.4-6 The differential diagnosis is broad for tattoo reactions and includes granulomatous inflammation, sarcoidosis, psoriasis (Köbner phenomenon), allergic contact dermatitis, lichen planus, morphealike reactions, squamous cell carcinoma, and keratoacanthoma,5 which makes clinicopathologic correlation essential for accurate diagnosis. Our case demonstrated the characteristic epithelial hyperplasia in the absence of cytologic atypia. In addition, the presence of mixed dermal inflammation histologically was noted in our patient.
Pseudoepitheliomatous hyperplasia development from a tattoo in areas of both mercury-based and non–mercury-based red pigment is a known association.7-9 Balfour et al10 also reported a case of PEH occurring secondary to manganese-based purple pigment. Because few cases have been reported, the epidemiology for PEH currently is unknown. Treatment of this condition primarily is anecdotal, with prior cases showing success with topical or intralesional steroids.5,7 As with any steroid-based treatment, we recommend less aggressive treatments initially with close follow-up and adaptation as needed to minimize adverse effects such as unwanted atrophy. Some success has been reported with the use of the Q-switched Nd:YAG laser in the setting of a PEH tattoo reaction.5 Similar to other tattoo reactions, surgical removal can be considered with failure of more conservative treatment methods and focal involvement.
We report an unusual case of PEH occurring secondary to purple tattoo pigment. Our report also demonstrates the clinical and symptomatic improvement of PEH that can be achieved through the use of intralesional corticosteroid therapy. Our patient represents a case of PEH reactive to tattooing with purple ink. Further research to elucidate the precise pathogenesis of PEH tattoo reactions would be helpful in identifying high-risk patients and determining the most efficacious treatments.
- Meani RE, Nixon RL, O’Keefe R, et al. Pseudoepitheliomatous hyperplasia secondary to allergic contact dermatitis to Grevillea Robyn Gordon. Australas J Dermatol. 2017;58:E8-E10.
- Chakrabarti S, Chakrabarti P, Agrawal D, et al. Pseudoepitheliomatous hyperplasia: a clinical entity mistaken for squamous cell carcinoma. J Cutan Aesthet Surg. 2014;7:232.
- Kluger N. Issues with keratoacanthoma, pseudoepitheliomatous hyperplasia and squamous cell carcinoma within tattoos: a clinical point of view. J Cutan Pathol. 2009;37:812-813.
- Zayour M, Lazova R. Pseudoepitheliomatous hyperplasia: a review. Am J Dermatopathol. 2011;33:112-126.
- Bassi A, Campolmi P, Cannarozzo G, et al. Tattoo-associated skin reaction: the importance of an early diagnosis and proper treatment [published online July 23, 2014]. Biomed Res Int. 2014;2014:354608.
- Serup J. Diagnostic tools for doctors’ evaluation of tattoo complications. Curr Probl Dermatol. 2017;52:42-57.
- Kazlouskaya V, Junkins-Hopkins JM. Pseudoepitheliomatous hyperplasia in a red pigment tattoo: a separate entity or hypertrophic lichen planus-like reaction? J Clin Aesthet Dermatol. 2015;8:48-52.
- Kluger N, Durand L, Minier-Thoumin C, et al. Pseudoepitheliomatous epidermal hyperplasia in tattoos: report of three cases. Am J Clin Dermatol. 2008;9:337-340.
- Cui W, McGregor DH, Stark SP, et al. Pseudoepitheliomatous hyperplasia—an unusual reaction following tattoo: report of a case and review of the literature. Int J Dermatol. 2007;46:743-745.
- Balfour E, Olhoffer I, Leffell D, et al. Massive pseudoepitheliomatous hyperplasia: an unusual reaction to a tattoo. Am J Dermatopathol. 2003;25:338-340.
- Meani RE, Nixon RL, O’Keefe R, et al. Pseudoepitheliomatous hyperplasia secondary to allergic contact dermatitis to Grevillea Robyn Gordon. Australas J Dermatol. 2017;58:E8-E10.
- Chakrabarti S, Chakrabarti P, Agrawal D, et al. Pseudoepitheliomatous hyperplasia: a clinical entity mistaken for squamous cell carcinoma. J Cutan Aesthet Surg. 2014;7:232.
- Kluger N. Issues with keratoacanthoma, pseudoepitheliomatous hyperplasia and squamous cell carcinoma within tattoos: a clinical point of view. J Cutan Pathol. 2009;37:812-813.
- Zayour M, Lazova R. Pseudoepitheliomatous hyperplasia: a review. Am J Dermatopathol. 2011;33:112-126.
- Bassi A, Campolmi P, Cannarozzo G, et al. Tattoo-associated skin reaction: the importance of an early diagnosis and proper treatment [published online July 23, 2014]. Biomed Res Int. 2014;2014:354608.
- Serup J. Diagnostic tools for doctors’ evaluation of tattoo complications. Curr Probl Dermatol. 2017;52:42-57.
- Kazlouskaya V, Junkins-Hopkins JM. Pseudoepitheliomatous hyperplasia in a red pigment tattoo: a separate entity or hypertrophic lichen planus-like reaction? J Clin Aesthet Dermatol. 2015;8:48-52.
- Kluger N, Durand L, Minier-Thoumin C, et al. Pseudoepitheliomatous epidermal hyperplasia in tattoos: report of three cases. Am J Clin Dermatol. 2008;9:337-340.
- Cui W, McGregor DH, Stark SP, et al. Pseudoepitheliomatous hyperplasia—an unusual reaction following tattoo: report of a case and review of the literature. Int J Dermatol. 2007;46:743-745.
- Balfour E, Olhoffer I, Leffell D, et al. Massive pseudoepitheliomatous hyperplasia: an unusual reaction to a tattoo. Am J Dermatopathol. 2003;25:338-340.
Practice Points
- Pseudoepitheliomatous hyperplasia (PEH) is a rare benign condition that can arise in response to multiple underlying triggers such as tattoo pigment.
- Histopathologic evaluation is essential for diagnosis and shows characteristic hyperplasia of the epidermis.
- Clinicians should consider intralesional steroids in the treatment of PEH once atypical mycobacterial and deep fungal infections have been ruled out.

Lichen Planopilaris in a Patient Treated With Bexarotene for Lymphomatoid Papulosis
To the Editor:
Lymphomatoid papulosis is a rare chronic skin disorder characterized by recurrent, self-healing crops of papulonodular eruptions, often resembling cutaneous T-cell lymphoma.1 Oral bexarotene, a retinoid X receptor–selective retinoid, can be used to control the disease.2,3 Lichen planopilaris (LPP) is a type of cicatricial alopecia characterized by irreversible hair loss, perifollicular inflammation, and follicular hyperkeratosis, commonly affecting the scalp vertex in adults.4 We report a case of a patient with lymphomatoid papulosis who was treated with bexarotene and subsequently developed LPP. We also discuss a proposed mechanism by which bexarotene may have influenced the onset of LPP.
A 35-year-old woman who was previously healthy initially presented with recurrent pruritic papular eruptions on the flank, axillae, and groin of several months’ duration. The lesions appeared as 2-mm, flat-topped, violaceous papules. The patient had no known drug allergies, no medical or family history of skin disease, and was only taking 3000 mg/d of omega-3 fatty acids (fish oil). Histopathologic examination of a biopsy specimen from the inner thigh showed enlarged, atypical, dermal lymphocytes that were CD30+ (Figure 1). These findings were consistent with lymphomatoid papulosis. As she had undergone tubal ligation several years prior, she was prescribed oral bexarotene 300 mg once daily in addition to triamcinolone cream 0.1% twice daily, as needed. Symptoms were well controlled on this regimen.
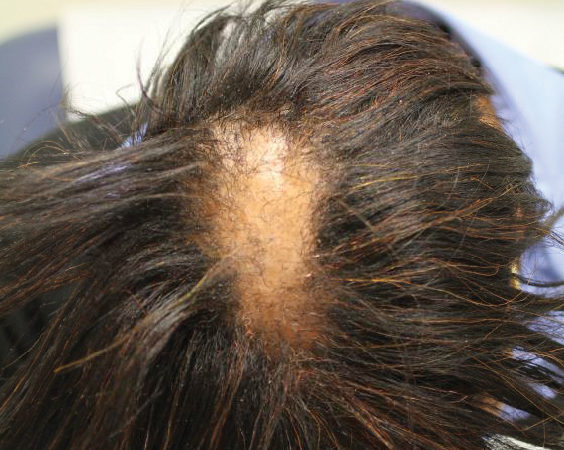
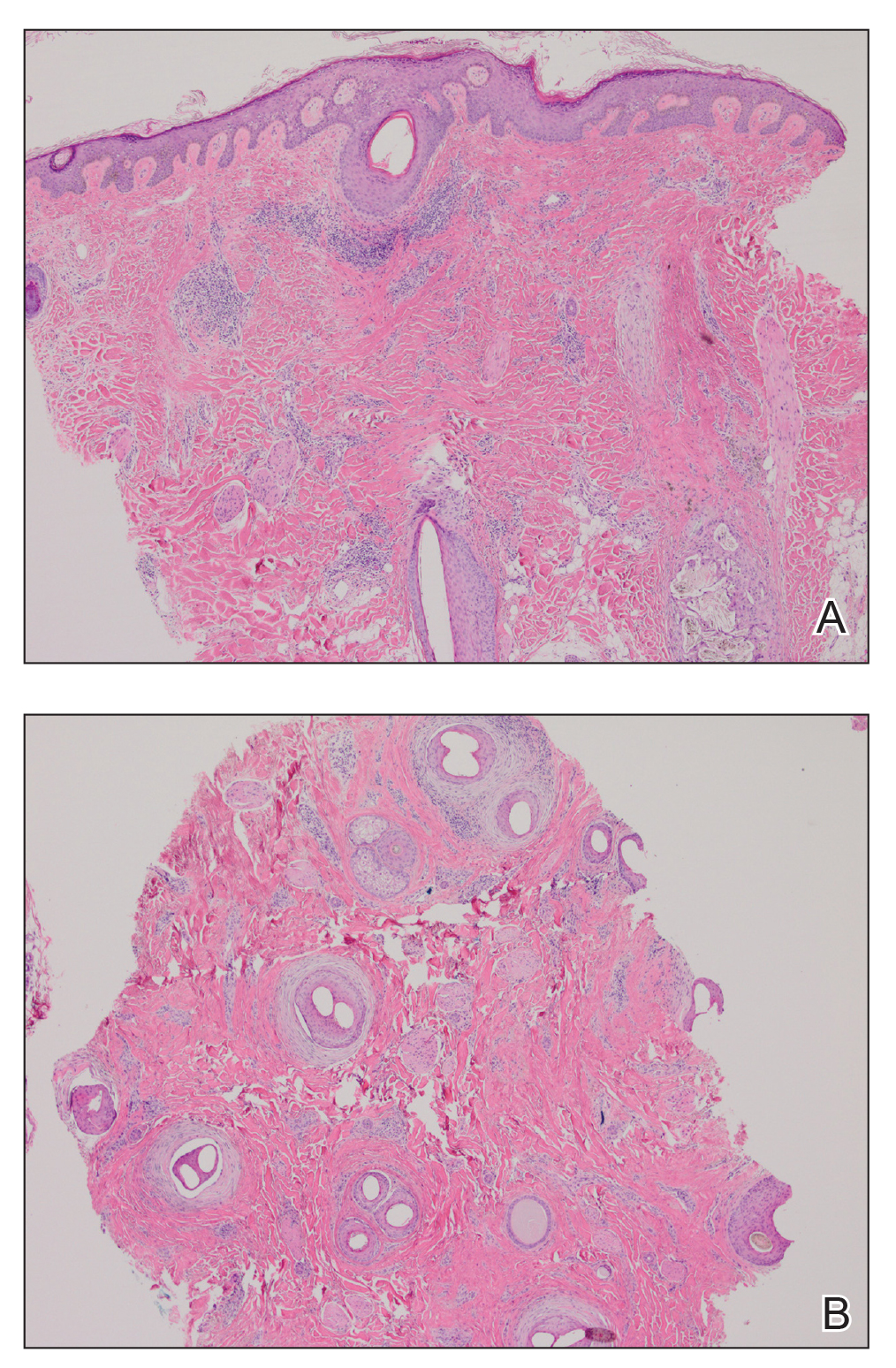
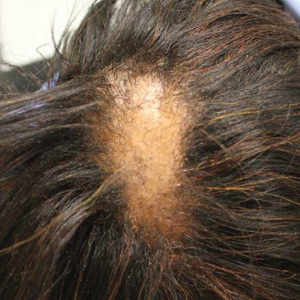
Six months later the patient returned, presenting with a new central patch of scarring alopecia on the vertex of the scalp (Figure 2). Adjacent to the area of hair loss were areas of prominent perifollicular scale that were slightly violaceous in color. Two 4-mm punch biopsies of the scalp showed dermal scarring with perifollicular lamellar fibrosis surrounded by a rim of lymphoplasmacytic inflammation (Figure 3). Sebaceous glands were found to be reduced in number. These findings were consistent with cicatricial alopecia, which was further classified as LPP in conjunction with the clinical findings. No CD30+ lymphocytes were identified in these specimens.
Baseline fasting triglycerides were 123 mg/dL (desirable: <150 mg/dL; borderline: 150–199 mg/dL; high: ≥200 mg/dL) and were stable over the first 4 months on bexarotene. After 5 months of therapy, the triglycerides increased to a high of 255 mg/dL, which corresponded with the onset of LPP. She was treated for the hypertriglyceridemia with omega-3 fatty acids (fish oil), and subsequent triglyceride levels have normalized and been stable. Her alopecia has not progressed but is persistent. She continues to have central hypothyroidism due to bexarotene and is on levothyroxine. The lymphomatoid papulosis also remains stable with no signs of progression to cutaneous T-cell lymphoma.
Although the exact mechanism of LPP is not fully understood, studies have suggested that cellular lipid metabolism may be responsible for the inflammation of the pilosebaceous unit.4-11 Hyperlipidemia is the most common side effect of oral bexarotene, typically occurring within the first 2 to 4 weeks of treatment.3,12 Considering the insights into the role of lipid regulation on LPP pathogenesis, it is reasonable to suspect that the dyslipidemia caused by bexarotene may have triggered the onset of LPP in our patient. The patient’s lipid values mostly remained within reference range throughout the course of treatment, though she did have elevation of triglycerides around the onset of LPP. Dyslipidemia has been reported in patients with lichen planus but not in patients with LPP. One case-control study showed no dyslipidemia in patients with LPP, but the triglyceride levels were not tracked over time and patients had varying durations since onset of disease at presentation.9-11,13 In our case, we were fortunate to have this information, and it may suggest an interaction between lipid dysregulation and the development of LPP. It would be interesting to explore this further in a larger patient population and to evaluate if control of dyslipidemia reduces progression of disease as it appears to have done for our patient.
- Karp DL, Horn TD. Lymphomatoid papulosis. J Am Acad Dermatol. 1994;30:379-395; quiz 396-398.
- Krathen RA, Ward S, Duvic M. Bexarotene is a new treatment option for lymphomatoid papulosis. Dermatology. 2003;206:142-147.
- Targretin (bexarotene) capsule [package insert]. St. Petersburg, FL: Cardinal Health; 2003. http://dailymed.nlm.nih.gov/dailymed/lookup.cfm?setid=63656f64-e240-4855-8df9-ca1655863735. Accessed April 9, 2020.
- Assouly P, Reygagne P. Lichen planopilaris: update on diagnosis and treatment. Semin Cutan Med Surg. 2009;28:3-10.
- Dogra S, Sarangal R. What’s new in cicatricial alopecia? Indian J Dermatol Venereol Leprol. 2013;79:576-90.
- Zheng Y, Eilertsen KJ, Ge L, et al. Scd1 is expressed in sebaceous glands and is disrupted in the asebia mouse. Nat Genet. 1999;23:268-270.
- Sundberg JP, Boggess D, Sundberg BA, et al. Asebia-2J (Scd1(ab2J)): a new allele and a model for scarring alopecia. Am J Pathol. 2000;156:2067-2075.
- Karnik P, Tekeste Z, McCormick TS, et al. Hair follicle stem cell-specific PPARgamma deletion causes scarring alopecia. J Invest Dermatol. 2009;129:1243-157.
- López-Jornet P, Camacho-Alonso F, Rodríguez-Martínes MA. Alterations in serum lipid profile patterns in oral lichen planus: a cross-sectional study. Am J Clin Dermatol. 2012;13:399-404.
- Arias-Santiago S, Buendía-Eisman A, Aneiros-Fernández J, et al. Lipid levels in patients with lichen planus: a case-control study. J Eur Acad Dermatol Venereol. 2011;25:1398-1401.
- Dreiher J, Shapiro J, Cohen AD. Lichen planus and dyslipidaemia: a case-control study. Br J Dermatol. 2009;161:626-629.
- de Vries-van der Weij J, de Haan W, Hu L, et al. Bexarotene induces dyslipidemia by increased very low-density lipoprotein production and cholesteryl ester transfer protein-mediated reduction of high-density lipoprotein. Endocrinology. 2009;150:2368-2375.
- Conic RRZ, Piliang M, Bergfeld W, et al. Association of lichen planopilaris with dyslipidemia. JAMA Dermatol. 2018;154:1088-1089.
To the Editor:
Lymphomatoid papulosis is a rare chronic skin disorder characterized by recurrent, self-healing crops of papulonodular eruptions, often resembling cutaneous T-cell lymphoma.1 Oral bexarotene, a retinoid X receptor–selective retinoid, can be used to control the disease.2,3 Lichen planopilaris (LPP) is a type of cicatricial alopecia characterized by irreversible hair loss, perifollicular inflammation, and follicular hyperkeratosis, commonly affecting the scalp vertex in adults.4 We report a case of a patient with lymphomatoid papulosis who was treated with bexarotene and subsequently developed LPP. We also discuss a proposed mechanism by which bexarotene may have influenced the onset of LPP.
A 35-year-old woman who was previously healthy initially presented with recurrent pruritic papular eruptions on the flank, axillae, and groin of several months’ duration. The lesions appeared as 2-mm, flat-topped, violaceous papules. The patient had no known drug allergies, no medical or family history of skin disease, and was only taking 3000 mg/d of omega-3 fatty acids (fish oil). Histopathologic examination of a biopsy specimen from the inner thigh showed enlarged, atypical, dermal lymphocytes that were CD30+ (Figure 1). These findings were consistent with lymphomatoid papulosis. As she had undergone tubal ligation several years prior, she was prescribed oral bexarotene 300 mg once daily in addition to triamcinolone cream 0.1% twice daily, as needed. Symptoms were well controlled on this regimen.
Six months later the patient returned, presenting with a new central patch of scarring alopecia on the vertex of the scalp (Figure 2). Adjacent to the area of hair loss were areas of prominent perifollicular scale that were slightly violaceous in color. Two 4-mm punch biopsies of the scalp showed dermal scarring with perifollicular lamellar fibrosis surrounded by a rim of lymphoplasmacytic inflammation (Figure 3). Sebaceous glands were found to be reduced in number. These findings were consistent with cicatricial alopecia, which was further classified as LPP in conjunction with the clinical findings. No CD30+ lymphocytes were identified in these specimens.
Baseline fasting triglycerides were 123 mg/dL (desirable: <150 mg/dL; borderline: 150–199 mg/dL; high: ≥200 mg/dL) and were stable over the first 4 months on bexarotene. After 5 months of therapy, the triglycerides increased to a high of 255 mg/dL, which corresponded with the onset of LPP. She was treated for the hypertriglyceridemia with omega-3 fatty acids (fish oil), and subsequent triglyceride levels have normalized and been stable. Her alopecia has not progressed but is persistent. She continues to have central hypothyroidism due to bexarotene and is on levothyroxine. The lymphomatoid papulosis also remains stable with no signs of progression to cutaneous T-cell lymphoma.
Although the exact mechanism of LPP is not fully understood, studies have suggested that cellular lipid metabolism may be responsible for the inflammation of the pilosebaceous unit.4-11 Hyperlipidemia is the most common side effect of oral bexarotene, typically occurring within the first 2 to 4 weeks of treatment.3,12 Considering the insights into the role of lipid regulation on LPP pathogenesis, it is reasonable to suspect that the dyslipidemia caused by bexarotene may have triggered the onset of LPP in our patient. The patient’s lipid values mostly remained within reference range throughout the course of treatment, though she did have elevation of triglycerides around the onset of LPP. Dyslipidemia has been reported in patients with lichen planus but not in patients with LPP. One case-control study showed no dyslipidemia in patients with LPP, but the triglyceride levels were not tracked over time and patients had varying durations since onset of disease at presentation.9-11,13 In our case, we were fortunate to have this information, and it may suggest an interaction between lipid dysregulation and the development of LPP. It would be interesting to explore this further in a larger patient population and to evaluate if control of dyslipidemia reduces progression of disease as it appears to have done for our patient.
To the Editor:
Lymphomatoid papulosis is a rare chronic skin disorder characterized by recurrent, self-healing crops of papulonodular eruptions, often resembling cutaneous T-cell lymphoma.1 Oral bexarotene, a retinoid X receptor–selective retinoid, can be used to control the disease.2,3 Lichen planopilaris (LPP) is a type of cicatricial alopecia characterized by irreversible hair loss, perifollicular inflammation, and follicular hyperkeratosis, commonly affecting the scalp vertex in adults.4 We report a case of a patient with lymphomatoid papulosis who was treated with bexarotene and subsequently developed LPP. We also discuss a proposed mechanism by which bexarotene may have influenced the onset of LPP.
A 35-year-old woman who was previously healthy initially presented with recurrent pruritic papular eruptions on the flank, axillae, and groin of several months’ duration. The lesions appeared as 2-mm, flat-topped, violaceous papules. The patient had no known drug allergies, no medical or family history of skin disease, and was only taking 3000 mg/d of omega-3 fatty acids (fish oil). Histopathologic examination of a biopsy specimen from the inner thigh showed enlarged, atypical, dermal lymphocytes that were CD30+ (Figure 1). These findings were consistent with lymphomatoid papulosis. As she had undergone tubal ligation several years prior, she was prescribed oral bexarotene 300 mg once daily in addition to triamcinolone cream 0.1% twice daily, as needed. Symptoms were well controlled on this regimen.
Six months later the patient returned, presenting with a new central patch of scarring alopecia on the vertex of the scalp (Figure 2). Adjacent to the area of hair loss were areas of prominent perifollicular scale that were slightly violaceous in color. Two 4-mm punch biopsies of the scalp showed dermal scarring with perifollicular lamellar fibrosis surrounded by a rim of lymphoplasmacytic inflammation (Figure 3). Sebaceous glands were found to be reduced in number. These findings were consistent with cicatricial alopecia, which was further classified as LPP in conjunction with the clinical findings. No CD30+ lymphocytes were identified in these specimens.
Baseline fasting triglycerides were 123 mg/dL (desirable: <150 mg/dL; borderline: 150–199 mg/dL; high: ≥200 mg/dL) and were stable over the first 4 months on bexarotene. After 5 months of therapy, the triglycerides increased to a high of 255 mg/dL, which corresponded with the onset of LPP. She was treated for the hypertriglyceridemia with omega-3 fatty acids (fish oil), and subsequent triglyceride levels have normalized and been stable. Her alopecia has not progressed but is persistent. She continues to have central hypothyroidism due to bexarotene and is on levothyroxine. The lymphomatoid papulosis also remains stable with no signs of progression to cutaneous T-cell lymphoma.
Although the exact mechanism of LPP is not fully understood, studies have suggested that cellular lipid metabolism may be responsible for the inflammation of the pilosebaceous unit.4-11 Hyperlipidemia is the most common side effect of oral bexarotene, typically occurring within the first 2 to 4 weeks of treatment.3,12 Considering the insights into the role of lipid regulation on LPP pathogenesis, it is reasonable to suspect that the dyslipidemia caused by bexarotene may have triggered the onset of LPP in our patient. The patient’s lipid values mostly remained within reference range throughout the course of treatment, though she did have elevation of triglycerides around the onset of LPP. Dyslipidemia has been reported in patients with lichen planus but not in patients with LPP. One case-control study showed no dyslipidemia in patients with LPP, but the triglyceride levels were not tracked over time and patients had varying durations since onset of disease at presentation.9-11,13 In our case, we were fortunate to have this information, and it may suggest an interaction between lipid dysregulation and the development of LPP. It would be interesting to explore this further in a larger patient population and to evaluate if control of dyslipidemia reduces progression of disease as it appears to have done for our patient.
- Karp DL, Horn TD. Lymphomatoid papulosis. J Am Acad Dermatol. 1994;30:379-395; quiz 396-398.
- Krathen RA, Ward S, Duvic M. Bexarotene is a new treatment option for lymphomatoid papulosis. Dermatology. 2003;206:142-147.
- Targretin (bexarotene) capsule [package insert]. St. Petersburg, FL: Cardinal Health; 2003. http://dailymed.nlm.nih.gov/dailymed/lookup.cfm?setid=63656f64-e240-4855-8df9-ca1655863735. Accessed April 9, 2020.
- Assouly P, Reygagne P. Lichen planopilaris: update on diagnosis and treatment. Semin Cutan Med Surg. 2009;28:3-10.
- Dogra S, Sarangal R. What’s new in cicatricial alopecia? Indian J Dermatol Venereol Leprol. 2013;79:576-90.
- Zheng Y, Eilertsen KJ, Ge L, et al. Scd1 is expressed in sebaceous glands and is disrupted in the asebia mouse. Nat Genet. 1999;23:268-270.
- Sundberg JP, Boggess D, Sundberg BA, et al. Asebia-2J (Scd1(ab2J)): a new allele and a model for scarring alopecia. Am J Pathol. 2000;156:2067-2075.
- Karnik P, Tekeste Z, McCormick TS, et al. Hair follicle stem cell-specific PPARgamma deletion causes scarring alopecia. J Invest Dermatol. 2009;129:1243-157.
- López-Jornet P, Camacho-Alonso F, Rodríguez-Martínes MA. Alterations in serum lipid profile patterns in oral lichen planus: a cross-sectional study. Am J Clin Dermatol. 2012;13:399-404.
- Arias-Santiago S, Buendía-Eisman A, Aneiros-Fernández J, et al. Lipid levels in patients with lichen planus: a case-control study. J Eur Acad Dermatol Venereol. 2011;25:1398-1401.
- Dreiher J, Shapiro J, Cohen AD. Lichen planus and dyslipidaemia: a case-control study. Br J Dermatol. 2009;161:626-629.
- de Vries-van der Weij J, de Haan W, Hu L, et al. Bexarotene induces dyslipidemia by increased very low-density lipoprotein production and cholesteryl ester transfer protein-mediated reduction of high-density lipoprotein. Endocrinology. 2009;150:2368-2375.
- Conic RRZ, Piliang M, Bergfeld W, et al. Association of lichen planopilaris with dyslipidemia. JAMA Dermatol. 2018;154:1088-1089.
- Karp DL, Horn TD. Lymphomatoid papulosis. J Am Acad Dermatol. 1994;30:379-395; quiz 396-398.
- Krathen RA, Ward S, Duvic M. Bexarotene is a new treatment option for lymphomatoid papulosis. Dermatology. 2003;206:142-147.
- Targretin (bexarotene) capsule [package insert]. St. Petersburg, FL: Cardinal Health; 2003. http://dailymed.nlm.nih.gov/dailymed/lookup.cfm?setid=63656f64-e240-4855-8df9-ca1655863735. Accessed April 9, 2020.
- Assouly P, Reygagne P. Lichen planopilaris: update on diagnosis and treatment. Semin Cutan Med Surg. 2009;28:3-10.
- Dogra S, Sarangal R. What’s new in cicatricial alopecia? Indian J Dermatol Venereol Leprol. 2013;79:576-90.
- Zheng Y, Eilertsen KJ, Ge L, et al. Scd1 is expressed in sebaceous glands and is disrupted in the asebia mouse. Nat Genet. 1999;23:268-270.
- Sundberg JP, Boggess D, Sundberg BA, et al. Asebia-2J (Scd1(ab2J)): a new allele and a model for scarring alopecia. Am J Pathol. 2000;156:2067-2075.
- Karnik P, Tekeste Z, McCormick TS, et al. Hair follicle stem cell-specific PPARgamma deletion causes scarring alopecia. J Invest Dermatol. 2009;129:1243-157.
- López-Jornet P, Camacho-Alonso F, Rodríguez-Martínes MA. Alterations in serum lipid profile patterns in oral lichen planus: a cross-sectional study. Am J Clin Dermatol. 2012;13:399-404.
- Arias-Santiago S, Buendía-Eisman A, Aneiros-Fernández J, et al. Lipid levels in patients with lichen planus: a case-control study. J Eur Acad Dermatol Venereol. 2011;25:1398-1401.
- Dreiher J, Shapiro J, Cohen AD. Lichen planus and dyslipidaemia: a case-control study. Br J Dermatol. 2009;161:626-629.
- de Vries-van der Weij J, de Haan W, Hu L, et al. Bexarotene induces dyslipidemia by increased very low-density lipoprotein production and cholesteryl ester transfer protein-mediated reduction of high-density lipoprotein. Endocrinology. 2009;150:2368-2375.
- Conic RRZ, Piliang M, Bergfeld W, et al. Association of lichen planopilaris with dyslipidemia. JAMA Dermatol. 2018;154:1088-1089.
Practice Points
- Oral retinoids may be associated with development of lichen planopilaris (LPP).
- Hypertriglyceridemia may be associated with onset of LPP.
Pruritic Papules on the Face and Chest
The Diagnosis: Eosinophilic Folliculitis
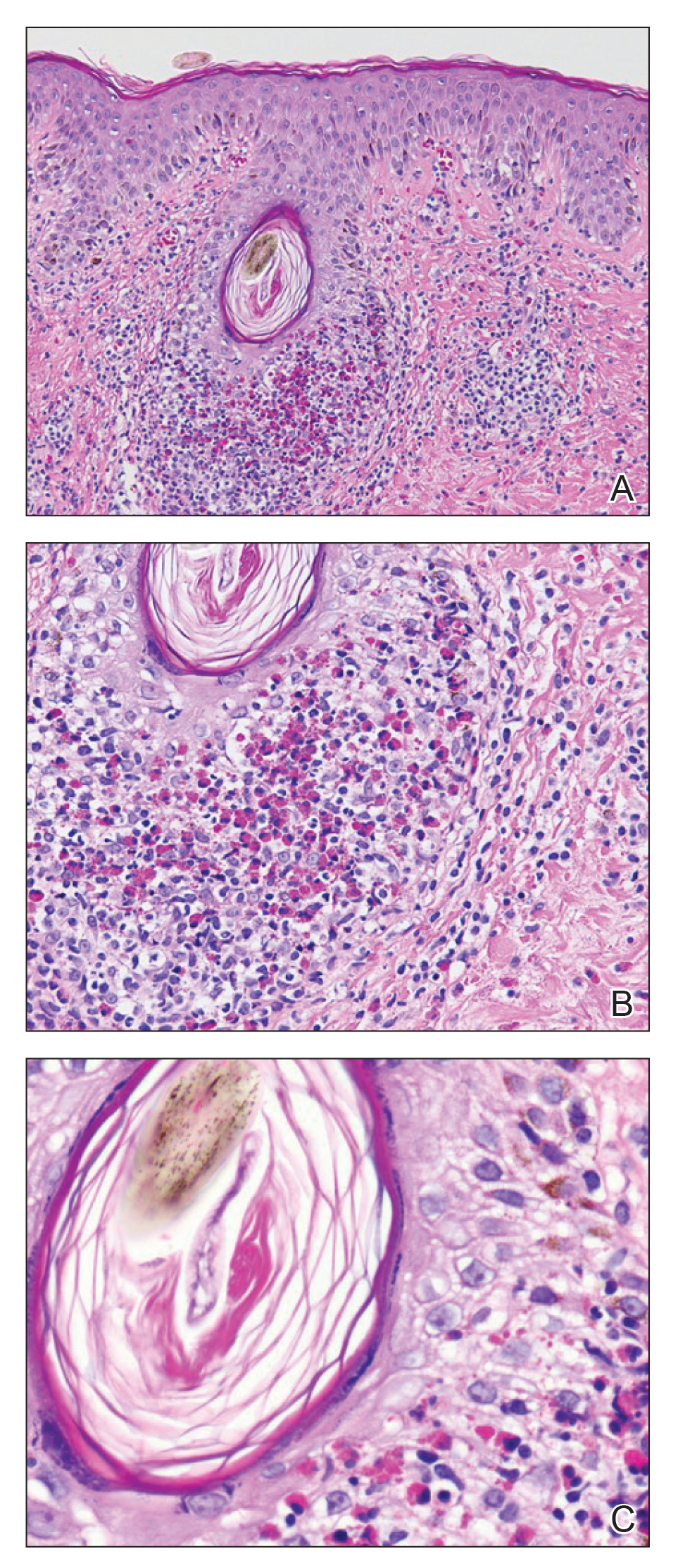
A shave biopsy specimen of an intact pustule on the left side of the chest was obtained. Histopathologic examination revealed follicular inflammation with copious eosinophils (Figure, A and B). Based on the histopathology and clinical presentation, a diagnosis of human immunodeficiency virus (HIV)-associated eosinophilic folliculitis (EF) was made.
The patient was started on triamcinolone ointment 0.1% twice daily to active lesions, oral cetirizine 10 mg in the morning, and oral hydroxyzine 25 mg at bedtime. Laboratory evaluation at the time of diagnosis showed eosinophilia with a peripheral blood eosinophil count of 0.5 K/μL (reference range, 0.03–0.48 K/μL).
Human immunodeficiency virus-associated EF is a pruritic follicular eruption that occurs in HIV-positive individuals with advanced disease. Clinically, it is characterized by intermittent, urticarial, red or flesh-colored, 2- to 5-mm papules with sparse pustules involving the head, neck, arms, and upper trunk.1,2 The cardinal clinical feature of the disorder is intense pruritus, with overlying crusts and excoriations present on physical examination.3
Patients usually have a CD4 count of less than 250 cells/mm3.2,3 Patients with HIV can develop an exacerbation of EF in the first 3 to 6 months after initiating antiretroviral therapy. This clinical pattern is believed to be due to the reconstituted immune system and increased circulation of inflammatory cells.4 Peripheral eosinophilia and elevated serum IgE levels are found in 25% to 50% of patients with HIV-associated EF.2,3
Clinically, the differential diagnosis of intensely pruritic papules with excoriations should include scabies.3 Other diagnoses to consider include opportunistic infections and papular urticaria.5 Acne vulgaris and Demodex folliculitis also may present with lesions similar to HIV-associated EF; however, these lesions tend not to be as intensely pruritic.1,5
The etiology of HIV-associated EF is unknown.3 One proposed mechanism involves a hypersensitivity reaction to Pityrosporum or Demodex mite fragments, as evidenced by studies that found fragments of these microorganisms in biopsied lesions of HIV-associated EF.3,6 In our patient's histopathology, it was noted that the afflicted hair follicle held a single Demodex mite (Figure, C).
The histopathology is characterized by a perifollicular inflammatory infiltrate of eosinophils and CD8+ lymphocytes with areas of sebaceous lysis.3,6 Spongiosis of the follicular epithelium is seen in early lesions of HIV-associated EF.6
The first-line treatment of HIV-associated EF includes antiretroviral therapy with topical steroids and antihistamines. Human immunodeficiency virus-associated EF improves as CD4 helper T-cell counts rise above 250 cells/mm3 with continued antiretroviral therapy, though it initially can cause a flare of the condition.4 High-potency steroids and antihistamines are added during this period to treat the severe pruritus.1,7 In particular, daily cetirizine has been shown to be effective, which may be due to its ability to block eosinophil migration in addition to H1-receptor antagonist properties.3,7
Various alternative therapies have been described in case reports and case series; however, there have been no controlled studies comparing therapies. Phototherapy with UVB light 3 times weekly for 3 to 6 weeks has been effective and curative in recalcitrant cases.7 Other frequently used treatments include oral metronidazole, oral itraconazole, and permethrin cream 5%. The effectiveness of the latter 2 treatments is believed to be related to the proposed role of Pityrosporum and Demodex in the pathogenesis.3
Acknowledgment
The authors thank Garth Fraga, MD (Kansas City, Kansas), for his help compiling the histopathological images and their diagnostic descriptions.
- Parker SR, Parker DC, McCall CO. Eosinophilic folliculitis in HIV-infected women: case series and review. Am J Clin Dermatol. 2006;7:193-200.
- Rosenthal D, LeBoit PE, Klumpp L, et al. Human immunodeficiency virus-associated eosinophilic folliculitis. a unique dermatosis associated with advanced human immunodeficiency virus infection. Arch Dermatol. 1991;127:206-209.
- Fearfield LA, Rowe A, Francis N, et al. Itchy folliculitis and human immunodeficiency virus infection: clinicopathological and immunological features, pathogenesis, and treatment. Br J Dermatol. 1999;141:3-11.
- Rajendran PM, Dolev JC, Heaphy MR, et al. Eosinophilic folliculitis: before and after the introduction of antiretroviral therapy. Arch Dermatol. 2005;141:1227-1231.
- Nervi SJ, Schwartz RA, Dmochowski M. Eosinophilic pustular folliculitis: a 40 year retrospect. J Am Acad Dermatol. 2006;55:285-289.
- McCalmont TH, Altemus D, Maurer T, et al. Eosinophilic folliculitis: the histological spectrum. Am J Dermatopathol. 1995;17:439-446.
- Ellis E, Scheinfeld N. Eosinophilic pustular folliculitis: a comprehensive review of treatment options. Am J Clin Dermatol. 2004;5:189-197.
The Diagnosis: Eosinophilic Folliculitis
A shave biopsy specimen of an intact pustule on the left side of the chest was obtained. Histopathologic examination revealed follicular inflammation with copious eosinophils (Figure, A and B). Based on the histopathology and clinical presentation, a diagnosis of human immunodeficiency virus (HIV)-associated eosinophilic folliculitis (EF) was made.
The patient was started on triamcinolone ointment 0.1% twice daily to active lesions, oral cetirizine 10 mg in the morning, and oral hydroxyzine 25 mg at bedtime. Laboratory evaluation at the time of diagnosis showed eosinophilia with a peripheral blood eosinophil count of 0.5 K/μL (reference range, 0.03–0.48 K/μL).
Human immunodeficiency virus-associated EF is a pruritic follicular eruption that occurs in HIV-positive individuals with advanced disease. Clinically, it is characterized by intermittent, urticarial, red or flesh-colored, 2- to 5-mm papules with sparse pustules involving the head, neck, arms, and upper trunk.1,2 The cardinal clinical feature of the disorder is intense pruritus, with overlying crusts and excoriations present on physical examination.3
Patients usually have a CD4 count of less than 250 cells/mm3.2,3 Patients with HIV can develop an exacerbation of EF in the first 3 to 6 months after initiating antiretroviral therapy. This clinical pattern is believed to be due to the reconstituted immune system and increased circulation of inflammatory cells.4 Peripheral eosinophilia and elevated serum IgE levels are found in 25% to 50% of patients with HIV-associated EF.2,3
Clinically, the differential diagnosis of intensely pruritic papules with excoriations should include scabies.3 Other diagnoses to consider include opportunistic infections and papular urticaria.5 Acne vulgaris and Demodex folliculitis also may present with lesions similar to HIV-associated EF; however, these lesions tend not to be as intensely pruritic.1,5
The etiology of HIV-associated EF is unknown.3 One proposed mechanism involves a hypersensitivity reaction to Pityrosporum or Demodex mite fragments, as evidenced by studies that found fragments of these microorganisms in biopsied lesions of HIV-associated EF.3,6 In our patient's histopathology, it was noted that the afflicted hair follicle held a single Demodex mite (Figure, C).
The histopathology is characterized by a perifollicular inflammatory infiltrate of eosinophils and CD8+ lymphocytes with areas of sebaceous lysis.3,6 Spongiosis of the follicular epithelium is seen in early lesions of HIV-associated EF.6
The first-line treatment of HIV-associated EF includes antiretroviral therapy with topical steroids and antihistamines. Human immunodeficiency virus-associated EF improves as CD4 helper T-cell counts rise above 250 cells/mm3 with continued antiretroviral therapy, though it initially can cause a flare of the condition.4 High-potency steroids and antihistamines are added during this period to treat the severe pruritus.1,7 In particular, daily cetirizine has been shown to be effective, which may be due to its ability to block eosinophil migration in addition to H1-receptor antagonist properties.3,7
Various alternative therapies have been described in case reports and case series; however, there have been no controlled studies comparing therapies. Phototherapy with UVB light 3 times weekly for 3 to 6 weeks has been effective and curative in recalcitrant cases.7 Other frequently used treatments include oral metronidazole, oral itraconazole, and permethrin cream 5%. The effectiveness of the latter 2 treatments is believed to be related to the proposed role of Pityrosporum and Demodex in the pathogenesis.3
Acknowledgment
The authors thank Garth Fraga, MD (Kansas City, Kansas), for his help compiling the histopathological images and their diagnostic descriptions.
The Diagnosis: Eosinophilic Folliculitis
A shave biopsy specimen of an intact pustule on the left side of the chest was obtained. Histopathologic examination revealed follicular inflammation with copious eosinophils (Figure, A and B). Based on the histopathology and clinical presentation, a diagnosis of human immunodeficiency virus (HIV)-associated eosinophilic folliculitis (EF) was made.
The patient was started on triamcinolone ointment 0.1% twice daily to active lesions, oral cetirizine 10 mg in the morning, and oral hydroxyzine 25 mg at bedtime. Laboratory evaluation at the time of diagnosis showed eosinophilia with a peripheral blood eosinophil count of 0.5 K/μL (reference range, 0.03–0.48 K/μL).
Human immunodeficiency virus-associated EF is a pruritic follicular eruption that occurs in HIV-positive individuals with advanced disease. Clinically, it is characterized by intermittent, urticarial, red or flesh-colored, 2- to 5-mm papules with sparse pustules involving the head, neck, arms, and upper trunk.1,2 The cardinal clinical feature of the disorder is intense pruritus, with overlying crusts and excoriations present on physical examination.3
Patients usually have a CD4 count of less than 250 cells/mm3.2,3 Patients with HIV can develop an exacerbation of EF in the first 3 to 6 months after initiating antiretroviral therapy. This clinical pattern is believed to be due to the reconstituted immune system and increased circulation of inflammatory cells.4 Peripheral eosinophilia and elevated serum IgE levels are found in 25% to 50% of patients with HIV-associated EF.2,3
Clinically, the differential diagnosis of intensely pruritic papules with excoriations should include scabies.3 Other diagnoses to consider include opportunistic infections and papular urticaria.5 Acne vulgaris and Demodex folliculitis also may present with lesions similar to HIV-associated EF; however, these lesions tend not to be as intensely pruritic.1,5
The etiology of HIV-associated EF is unknown.3 One proposed mechanism involves a hypersensitivity reaction to Pityrosporum or Demodex mite fragments, as evidenced by studies that found fragments of these microorganisms in biopsied lesions of HIV-associated EF.3,6 In our patient's histopathology, it was noted that the afflicted hair follicle held a single Demodex mite (Figure, C).
The histopathology is characterized by a perifollicular inflammatory infiltrate of eosinophils and CD8+ lymphocytes with areas of sebaceous lysis.3,6 Spongiosis of the follicular epithelium is seen in early lesions of HIV-associated EF.6
The first-line treatment of HIV-associated EF includes antiretroviral therapy with topical steroids and antihistamines. Human immunodeficiency virus-associated EF improves as CD4 helper T-cell counts rise above 250 cells/mm3 with continued antiretroviral therapy, though it initially can cause a flare of the condition.4 High-potency steroids and antihistamines are added during this period to treat the severe pruritus.1,7 In particular, daily cetirizine has been shown to be effective, which may be due to its ability to block eosinophil migration in addition to H1-receptor antagonist properties.3,7
Various alternative therapies have been described in case reports and case series; however, there have been no controlled studies comparing therapies. Phototherapy with UVB light 3 times weekly for 3 to 6 weeks has been effective and curative in recalcitrant cases.7 Other frequently used treatments include oral metronidazole, oral itraconazole, and permethrin cream 5%. The effectiveness of the latter 2 treatments is believed to be related to the proposed role of Pityrosporum and Demodex in the pathogenesis.3
Acknowledgment
The authors thank Garth Fraga, MD (Kansas City, Kansas), for his help compiling the histopathological images and their diagnostic descriptions.
- Parker SR, Parker DC, McCall CO. Eosinophilic folliculitis in HIV-infected women: case series and review. Am J Clin Dermatol. 2006;7:193-200.
- Rosenthal D, LeBoit PE, Klumpp L, et al. Human immunodeficiency virus-associated eosinophilic folliculitis. a unique dermatosis associated with advanced human immunodeficiency virus infection. Arch Dermatol. 1991;127:206-209.
- Fearfield LA, Rowe A, Francis N, et al. Itchy folliculitis and human immunodeficiency virus infection: clinicopathological and immunological features, pathogenesis, and treatment. Br J Dermatol. 1999;141:3-11.
- Rajendran PM, Dolev JC, Heaphy MR, et al. Eosinophilic folliculitis: before and after the introduction of antiretroviral therapy. Arch Dermatol. 2005;141:1227-1231.
- Nervi SJ, Schwartz RA, Dmochowski M. Eosinophilic pustular folliculitis: a 40 year retrospect. J Am Acad Dermatol. 2006;55:285-289.
- McCalmont TH, Altemus D, Maurer T, et al. Eosinophilic folliculitis: the histological spectrum. Am J Dermatopathol. 1995;17:439-446.
- Ellis E, Scheinfeld N. Eosinophilic pustular folliculitis: a comprehensive review of treatment options. Am J Clin Dermatol. 2004;5:189-197.
- Parker SR, Parker DC, McCall CO. Eosinophilic folliculitis in HIV-infected women: case series and review. Am J Clin Dermatol. 2006;7:193-200.
- Rosenthal D, LeBoit PE, Klumpp L, et al. Human immunodeficiency virus-associated eosinophilic folliculitis. a unique dermatosis associated with advanced human immunodeficiency virus infection. Arch Dermatol. 1991;127:206-209.
- Fearfield LA, Rowe A, Francis N, et al. Itchy folliculitis and human immunodeficiency virus infection: clinicopathological and immunological features, pathogenesis, and treatment. Br J Dermatol. 1999;141:3-11.
- Rajendran PM, Dolev JC, Heaphy MR, et al. Eosinophilic folliculitis: before and after the introduction of antiretroviral therapy. Arch Dermatol. 2005;141:1227-1231.
- Nervi SJ, Schwartz RA, Dmochowski M. Eosinophilic pustular folliculitis: a 40 year retrospect. J Am Acad Dermatol. 2006;55:285-289.
- McCalmont TH, Altemus D, Maurer T, et al. Eosinophilic folliculitis: the histological spectrum. Am J Dermatopathol. 1995;17:439-446.
- Ellis E, Scheinfeld N. Eosinophilic pustular folliculitis: a comprehensive review of treatment options. Am J Clin Dermatol. 2004;5:189-197.

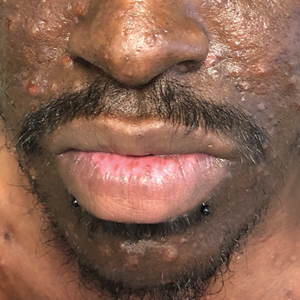
A 31-year-old man presented with a severely pruritic rash of 2 weeks' duration. Physical examination revealed numerous urticarial papules and rare erythematous pustules over the face (top), upper chest (bottom), and proximal arms; most lesions were excoriated. Additionally, there were numerous hyperpigmented papules with central hypopigmentation on the upper chest and arms. The lower half of the body was spared. His medical history was notable for human immunodeficiency virus/AIDS with a prior episode of Pneumocystis pneumonia. He had been noncompliant with antiretroviral therapy for the last 2 years but restarted therapy 3 weeks prior to presentation. Laboratory test results revealed a CD4 cell count of 13 cells/mm3 (reference range, 500-1500 cells/mm3) with a viral load of 179 copies/mL (reference range, undetectable).
Dusky Pink Nodular Plaque on the Finger
The Diagnosis: Majocchi Granuloma
Majocchi granuloma (MG) is a dermatophytic infection that reveals hyphal elements within the cornified cells of follicles and most commonly is caused by Trichophyton rubrum. However, occasionally other Trichophyton, Trichosporon, and Aspergillus species are involved.1
There typically are 2 forms of MG: (1) the small perifollicular papular form that usually is localized to the dermis and occurs in immunocompetent individuals, and (2) a deep form featuring subcutaneous plaques and nodules that generally occur on the hair-bearing surfaces in immunosuppressed hosts.2 Majocchi granuloma also commonly occurs from the use of potent topical steroids on unsuspected tinea.3
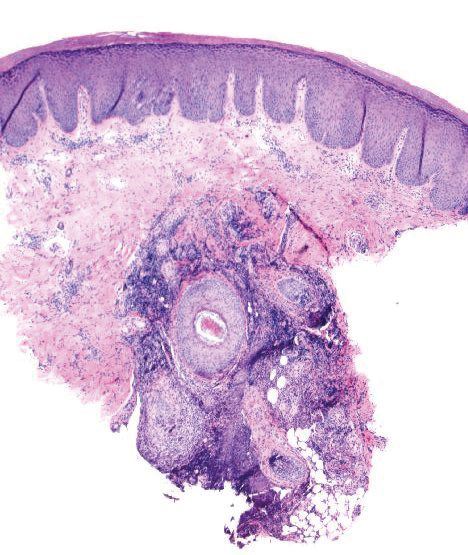
Histopathologically, MG generally presents as granulomatous inflammation with perifollicular neutrophilic infiltration. This polymorphonuclear cell infiltrate was visible clinically as a single pustule overlying the nodular plaque, a clue appreciable only on close inspection. Histopathologic examination revealed segmented branching filaments present within cornified elements of a follicle (Figure). Notably, potassium hydroxide (KOH) preparations are unreliable diagnostic aids in MG, as evidenced by the 2 negative KOH preparations in this case. According to Chou and Hsu,4 because KOH preparation can only detect fungi located in the stratum corneum, the result may be negative for MG due to deeper invasion of the fungi into the dermal follicular component. In fact, KOH preparations of MG may reveal no hyphae in 23.3% of cases.2
The initiating factor in MG is not entirely known but is thought to be physical trauma that either directly or indirectly leads to follicle disruption and passive introduction of the organism into the dermis (eg, traumatic implantation via gardening or other recreational activities).2 Other proposed mechanisms include the presentation of the membrane-associated ATP-binding cassette transporter on the surface of T rubrum.1 Dermatophytes evade the host immune system through a variety of mechanisms: (1) cell wall glycoproteins, (2) release of anti-inflammatory cytokines, and (3) generation of immunosuppressive regulatory T cells.1
Collectively, the clinical and histopathologic findings distinguish MG from other cutaneous conditions. Sporotrichosis, a granulomatous infection caused by Sporothrix schenckii, typically is found in tropical regions of the world and often is associated with floriculture.5 Sporotrichosis initially presents in a subcutaneous papulonodular form, but unlike MG, it later ulcerates and progresses along adjacent lymphatic chains.5 Pathology of sporotrichosis exhibits pseudoepitheliomatous hyperplasia with granulomas, possible foci of suppuration, and yeastlike forms called cigar bodies. Chromoblastomycosis clinically is defined by tumorlike lesions on the skin including verrucous, nodular, or scarlike plaques and typically is associated with traumatic injury and implantation of the microorganism. Histologically, chromoblastomycosis demonstrates pseudoepitheliomatous hyperplasia with granulomas and characteristic darkly pigmented, thick-walled sclerotic cells called Medlar bodies.6Mycobacterium marinum is one cause of nontuberculous mycobacterial skin infections in humans. Clinically, M marinum is associated with improper hygiene techniques and contact with fish tanks and other aqueous environments. Mycobacterium marinum can present histopathologically as early neutrophilic infiltration or late dermal granulomatous inflammation.7 Acid-fast bacilli typically are scant, leaving the diagnosis best secured via polymerase chain reaction assay. Nodular Kaposi sarcoma (KS) can present as a dusky nodular plaque on an acral surface but typically is seen in patients with underlying human immunodeficiency virus/AIDS or other immunosuppressive conditions. The pathology for KS shows a proliferation of human herpes virus 8-positive spindle cells with slitlike spaces containing red blood cells instead of granulomatous inflammation.
Treatment regimens with topical corticosteroids can exacerbate the infection due to local suppression of cell-mediated immunity.8 In these scenarios, fungal infection is suspected, and systemic antifungals such as ketoconazole; itraconazole; or terbinafine, which has become the mainstay, are prescribed. Resolution of the infection with these medications usually is seen after 4 weeks.2
A diagnosis of MG can be elusive and often may take multiple visits. Clinicians should note that MG could demonstrate repeated false-negative KOH preparations; therefore, these tests should not be relied on as the sole determination of a diagnosis. Although chromoblastomycosis, sporotrichosis, nodular KS, and infection with M marinum may all present as nodular plaques with granulomatous pathology, a follicular pustule may be a clinical clue to MG, as its mimics typically lack folliculocentric neutrophils.
- Tirado-Sánchez A, Ponce-Olivera RM, Bonifaz A. Majocchi's granuloma (dermatophytic granuloma): updated therapeutic options. Curr Fungal Infect Rep. 2015;9:204-212.
- Ilkit M, Durdu M, Karakas¸ M. Majocchi's granuloma: a symptom complex caused by fungal pathogens. Med Mycol. 2012;50:449-457.
- Schwartz RA, Janniger CK. Majocchi granuloma. Medscape website. https://emedicine.medscape.com/article/1092601-overview. Updated May 14, 2019. Accessed April 13, 2020.
- Chou WY, Hsu CJ. A case report of Majocchi's granuloma associated with combined therapy of topical steroids and adalimumab. Medicine (Baltimore). 2016;95:E2245.
- Barros MB, de Almeida Paes R, Schubach AO. Sporothrix schenckii and sporotrichosis. Clin Microbiol Rev. 2011;24:633-654.
- Guarner J, Brandt ME. Histopathologic diagnosis of fungal infections in the 21st century. Clin Microbiol Rev. 2011;24:247-280.
- Slany M, Jezek P, Bodnarova M. Fish tank granuloma caused by Mycobacterium marinum in two aquarists: two case reports. Biomed Res Int. 2013;2013:161329.
- Coondoo A, Phiske M, Verma S, et al. Side-effects of topical steroids: a long overdue revisit. Indian Dermatol Online J. 2014;5:416-425.
The Diagnosis: Majocchi Granuloma
Majocchi granuloma (MG) is a dermatophytic infection that reveals hyphal elements within the cornified cells of follicles and most commonly is caused by Trichophyton rubrum. However, occasionally other Trichophyton, Trichosporon, and Aspergillus species are involved.1
There typically are 2 forms of MG: (1) the small perifollicular papular form that usually is localized to the dermis and occurs in immunocompetent individuals, and (2) a deep form featuring subcutaneous plaques and nodules that generally occur on the hair-bearing surfaces in immunosuppressed hosts.2 Majocchi granuloma also commonly occurs from the use of potent topical steroids on unsuspected tinea.3
Histopathologically, MG generally presents as granulomatous inflammation with perifollicular neutrophilic infiltration. This polymorphonuclear cell infiltrate was visible clinically as a single pustule overlying the nodular plaque, a clue appreciable only on close inspection. Histopathologic examination revealed segmented branching filaments present within cornified elements of a follicle (Figure). Notably, potassium hydroxide (KOH) preparations are unreliable diagnostic aids in MG, as evidenced by the 2 negative KOH preparations in this case. According to Chou and Hsu,4 because KOH preparation can only detect fungi located in the stratum corneum, the result may be negative for MG due to deeper invasion of the fungi into the dermal follicular component. In fact, KOH preparations of MG may reveal no hyphae in 23.3% of cases.2
The initiating factor in MG is not entirely known but is thought to be physical trauma that either directly or indirectly leads to follicle disruption and passive introduction of the organism into the dermis (eg, traumatic implantation via gardening or other recreational activities).2 Other proposed mechanisms include the presentation of the membrane-associated ATP-binding cassette transporter on the surface of T rubrum.1 Dermatophytes evade the host immune system through a variety of mechanisms: (1) cell wall glycoproteins, (2) release of anti-inflammatory cytokines, and (3) generation of immunosuppressive regulatory T cells.1
Collectively, the clinical and histopathologic findings distinguish MG from other cutaneous conditions. Sporotrichosis, a granulomatous infection caused by Sporothrix schenckii, typically is found in tropical regions of the world and often is associated with floriculture.5 Sporotrichosis initially presents in a subcutaneous papulonodular form, but unlike MG, it later ulcerates and progresses along adjacent lymphatic chains.5 Pathology of sporotrichosis exhibits pseudoepitheliomatous hyperplasia with granulomas, possible foci of suppuration, and yeastlike forms called cigar bodies. Chromoblastomycosis clinically is defined by tumorlike lesions on the skin including verrucous, nodular, or scarlike plaques and typically is associated with traumatic injury and implantation of the microorganism. Histologically, chromoblastomycosis demonstrates pseudoepitheliomatous hyperplasia with granulomas and characteristic darkly pigmented, thick-walled sclerotic cells called Medlar bodies.6Mycobacterium marinum is one cause of nontuberculous mycobacterial skin infections in humans. Clinically, M marinum is associated with improper hygiene techniques and contact with fish tanks and other aqueous environments. Mycobacterium marinum can present histopathologically as early neutrophilic infiltration or late dermal granulomatous inflammation.7 Acid-fast bacilli typically are scant, leaving the diagnosis best secured via polymerase chain reaction assay. Nodular Kaposi sarcoma (KS) can present as a dusky nodular plaque on an acral surface but typically is seen in patients with underlying human immunodeficiency virus/AIDS or other immunosuppressive conditions. The pathology for KS shows a proliferation of human herpes virus 8-positive spindle cells with slitlike spaces containing red blood cells instead of granulomatous inflammation.
Treatment regimens with topical corticosteroids can exacerbate the infection due to local suppression of cell-mediated immunity.8 In these scenarios, fungal infection is suspected, and systemic antifungals such as ketoconazole; itraconazole; or terbinafine, which has become the mainstay, are prescribed. Resolution of the infection with these medications usually is seen after 4 weeks.2
A diagnosis of MG can be elusive and often may take multiple visits. Clinicians should note that MG could demonstrate repeated false-negative KOH preparations; therefore, these tests should not be relied on as the sole determination of a diagnosis. Although chromoblastomycosis, sporotrichosis, nodular KS, and infection with M marinum may all present as nodular plaques with granulomatous pathology, a follicular pustule may be a clinical clue to MG, as its mimics typically lack folliculocentric neutrophils.
The Diagnosis: Majocchi Granuloma
Majocchi granuloma (MG) is a dermatophytic infection that reveals hyphal elements within the cornified cells of follicles and most commonly is caused by Trichophyton rubrum. However, occasionally other Trichophyton, Trichosporon, and Aspergillus species are involved.1
There typically are 2 forms of MG: (1) the small perifollicular papular form that usually is localized to the dermis and occurs in immunocompetent individuals, and (2) a deep form featuring subcutaneous plaques and nodules that generally occur on the hair-bearing surfaces in immunosuppressed hosts.2 Majocchi granuloma also commonly occurs from the use of potent topical steroids on unsuspected tinea.3
Histopathologically, MG generally presents as granulomatous inflammation with perifollicular neutrophilic infiltration. This polymorphonuclear cell infiltrate was visible clinically as a single pustule overlying the nodular plaque, a clue appreciable only on close inspection. Histopathologic examination revealed segmented branching filaments present within cornified elements of a follicle (Figure). Notably, potassium hydroxide (KOH) preparations are unreliable diagnostic aids in MG, as evidenced by the 2 negative KOH preparations in this case. According to Chou and Hsu,4 because KOH preparation can only detect fungi located in the stratum corneum, the result may be negative for MG due to deeper invasion of the fungi into the dermal follicular component. In fact, KOH preparations of MG may reveal no hyphae in 23.3% of cases.2
The initiating factor in MG is not entirely known but is thought to be physical trauma that either directly or indirectly leads to follicle disruption and passive introduction of the organism into the dermis (eg, traumatic implantation via gardening or other recreational activities).2 Other proposed mechanisms include the presentation of the membrane-associated ATP-binding cassette transporter on the surface of T rubrum.1 Dermatophytes evade the host immune system through a variety of mechanisms: (1) cell wall glycoproteins, (2) release of anti-inflammatory cytokines, and (3) generation of immunosuppressive regulatory T cells.1
Collectively, the clinical and histopathologic findings distinguish MG from other cutaneous conditions. Sporotrichosis, a granulomatous infection caused by Sporothrix schenckii, typically is found in tropical regions of the world and often is associated with floriculture.5 Sporotrichosis initially presents in a subcutaneous papulonodular form, but unlike MG, it later ulcerates and progresses along adjacent lymphatic chains.5 Pathology of sporotrichosis exhibits pseudoepitheliomatous hyperplasia with granulomas, possible foci of suppuration, and yeastlike forms called cigar bodies. Chromoblastomycosis clinically is defined by tumorlike lesions on the skin including verrucous, nodular, or scarlike plaques and typically is associated with traumatic injury and implantation of the microorganism. Histologically, chromoblastomycosis demonstrates pseudoepitheliomatous hyperplasia with granulomas and characteristic darkly pigmented, thick-walled sclerotic cells called Medlar bodies.6Mycobacterium marinum is one cause of nontuberculous mycobacterial skin infections in humans. Clinically, M marinum is associated with improper hygiene techniques and contact with fish tanks and other aqueous environments. Mycobacterium marinum can present histopathologically as early neutrophilic infiltration or late dermal granulomatous inflammation.7 Acid-fast bacilli typically are scant, leaving the diagnosis best secured via polymerase chain reaction assay. Nodular Kaposi sarcoma (KS) can present as a dusky nodular plaque on an acral surface but typically is seen in patients with underlying human immunodeficiency virus/AIDS or other immunosuppressive conditions. The pathology for KS shows a proliferation of human herpes virus 8-positive spindle cells with slitlike spaces containing red blood cells instead of granulomatous inflammation.
Treatment regimens with topical corticosteroids can exacerbate the infection due to local suppression of cell-mediated immunity.8 In these scenarios, fungal infection is suspected, and systemic antifungals such as ketoconazole; itraconazole; or terbinafine, which has become the mainstay, are prescribed. Resolution of the infection with these medications usually is seen after 4 weeks.2
A diagnosis of MG can be elusive and often may take multiple visits. Clinicians should note that MG could demonstrate repeated false-negative KOH preparations; therefore, these tests should not be relied on as the sole determination of a diagnosis. Although chromoblastomycosis, sporotrichosis, nodular KS, and infection with M marinum may all present as nodular plaques with granulomatous pathology, a follicular pustule may be a clinical clue to MG, as its mimics typically lack folliculocentric neutrophils.
- Tirado-Sánchez A, Ponce-Olivera RM, Bonifaz A. Majocchi's granuloma (dermatophytic granuloma): updated therapeutic options. Curr Fungal Infect Rep. 2015;9:204-212.
- Ilkit M, Durdu M, Karakas¸ M. Majocchi's granuloma: a symptom complex caused by fungal pathogens. Med Mycol. 2012;50:449-457.
- Schwartz RA, Janniger CK. Majocchi granuloma. Medscape website. https://emedicine.medscape.com/article/1092601-overview. Updated May 14, 2019. Accessed April 13, 2020.
- Chou WY, Hsu CJ. A case report of Majocchi's granuloma associated with combined therapy of topical steroids and adalimumab. Medicine (Baltimore). 2016;95:E2245.
- Barros MB, de Almeida Paes R, Schubach AO. Sporothrix schenckii and sporotrichosis. Clin Microbiol Rev. 2011;24:633-654.
- Guarner J, Brandt ME. Histopathologic diagnosis of fungal infections in the 21st century. Clin Microbiol Rev. 2011;24:247-280.
- Slany M, Jezek P, Bodnarova M. Fish tank granuloma caused by Mycobacterium marinum in two aquarists: two case reports. Biomed Res Int. 2013;2013:161329.
- Coondoo A, Phiske M, Verma S, et al. Side-effects of topical steroids: a long overdue revisit. Indian Dermatol Online J. 2014;5:416-425.
- Tirado-Sánchez A, Ponce-Olivera RM, Bonifaz A. Majocchi's granuloma (dermatophytic granuloma): updated therapeutic options. Curr Fungal Infect Rep. 2015;9:204-212.
- Ilkit M, Durdu M, Karakas¸ M. Majocchi's granuloma: a symptom complex caused by fungal pathogens. Med Mycol. 2012;50:449-457.
- Schwartz RA, Janniger CK. Majocchi granuloma. Medscape website. https://emedicine.medscape.com/article/1092601-overview. Updated May 14, 2019. Accessed April 13, 2020.
- Chou WY, Hsu CJ. A case report of Majocchi's granuloma associated with combined therapy of topical steroids and adalimumab. Medicine (Baltimore). 2016;95:E2245.
- Barros MB, de Almeida Paes R, Schubach AO. Sporothrix schenckii and sporotrichosis. Clin Microbiol Rev. 2011;24:633-654.
- Guarner J, Brandt ME. Histopathologic diagnosis of fungal infections in the 21st century. Clin Microbiol Rev. 2011;24:247-280.
- Slany M, Jezek P, Bodnarova M. Fish tank granuloma caused by Mycobacterium marinum in two aquarists: two case reports. Biomed Res Int. 2013;2013:161329.
- Coondoo A, Phiske M, Verma S, et al. Side-effects of topical steroids: a long overdue revisit. Indian Dermatol Online J. 2014;5:416-425.
A 38-year-old man presented with a persistent pruritic nodular plaque on the proximal right index finger of 4 months' duration. He reported pruning roses in the garden but denied any trauma. The patient previously had been treated by another clinician with fluocinonide cream 0.05%, clobetasol cream 0.05%, intramuscular methylprednisolone 40 mg, and oral doxycycline hyclate 100 mg with no improvement. Two potassium hydroxide preparations were performed as well as a bacterial culture and sensitivity, with all results returning as negative. Physical examination revealed a 2-cm pink to purple, scaly, nodular plaque on the right index finger. A punch biopsy was obtained for histopathology with hematoxylin and eosin stain.
The DNA Mismatch Repair System in Sebaceous Tumors: An Update on the Genetics and Workup of Muir-Torre Syndrome
It is well known by now that tumor formation is driven by accumulation of numerous genetic and epigenetic mutations. Human cells are equipped with an apparatus called the DNA mismatch repair (MMR) system that corrects errors during replication.1 If these genes are themselves mutated, cells then start accumulating mutations in other genes, including oncogenes and tumor suppressor genes, which results in the development of sustained proliferative signaling pathways, evasion of growth suppression, resistance to cell death, and the potential for invasion and metastasis.2
Gene mutations in DNA MMR have been detected in several tumors, such as sebaceous tumors,3 colorectal adenocarcinomas,4 keratoacanthomas,5 and other visceral malignancies.6 Sebaceous tumors are rare in the general population; however, they are common in patients with inherited or acquired mutations in MMR genes.5 These patients also have been found to have other visceral malignancies such as colorectal adenocarcinomas and breast, lung, and central nervous system (CNS) tumors.7 This observation was made in the 1960s, and patients were referred to as having Muir-Torre syndrome (MTS).8 This article serves to briefly describe the DNA MMR system and its implication in sebaceous tumors as well as discuss the recent recommendations for screening for MTS in patients presenting with sebaceous tumors.
The DNA MMR System
Mismatch repair proteins are responsible for detecting and repairing errors during cell division, especially in microsatellite regions.9 Microsatellites are common and widely distributed DNA motifs consisting of repeated nucleotide sequences that normally account for 3% of the genome.10 Mutations in MMR result in insertion or deletion of nucleotides in these DNA motifs, making them either abnormally long or short, referred to as microsatellite instability (MSI), which results in downstream cumulative accumulation of mutations in oncogenes and tumor suppressor genes, and thus carcinogenesis.9
There are 7 human MMR proteins: MLH1, MLH3, MSH2, MSH3, MSH6, PMS1, and PMS2. These proteins are highly conserved across different living species.11 Loss of MMR proteins can be due to a mutation in the coding sequence of the gene or due to epigenetic hypermethylation of the gene promoter.12 These alterations can be inherited or acquired and in most cases result in MSI.
When assessing for MSI, tumor genomes can be divided into 3 subtypes: high-level and low-level MSI and stable microsatellites.13 Tumors with high-level MSI respond better to treatment and show a better prognosis than those with low-level MSI or stable microsatellites,14 which is thought to be due to tumor-induced immune activation. Microsatellite instability results in the generation of frameshift peptides that are immunogenic and induce tumor-specific immune responses.15 Several research laboratories have artificially synthesized frameshift peptides as vaccines and have successfully used them as targets for immune therapy as a way for preventing and treating malignancies.16
Sebaceous Tumors in MTS
A typical example of tumors that arise from mutations in the DNA MMR system is seen in MTS,a rare inherited genetic syndrome that predisposes patients to sebaceous neoplasms, keratoacanthomas, and visceral malignancies.17 It was first described as an autosomal-dominant condition in patients who have at least 1 sebaceous tumor and 1 visceral malignancy, with or without keratoacanthomas. It was then later characterized as a skin variant of Lynch syndrome, also known as hereditary nonpolyposis colorectal cancer syndrome.18
Sebaceous tumors are the hallmark of MTS. Although sebaceous hyperplasia is common in the general population, sebaceous tumors are rare outside the context of MTS. There are 3 types of sebaceous tumors with distinct pathologic features: adenoma, epithelioma, and carcinoma.19 Sebaceous adenomas and epitheliomas are benign growths; however, sebaceous carcinomas can be aggressive and have metastatic potential.20 Because it is difficult to clinically distinguish carcinomas from the benign sebaceous growths, biopsy of a large, changing, or ulcerated lesion is important in these patients to rule out a sebaceous carcinoma. Other aggressive skin tumors can develop in MTS, such as rapidly growing keratoacanthomas and basal cell carcinomas with sebaceous differentiation.21
Types of MTS
For most cases, MTS is characterized by germline mutations in DNA MMR genes. The most common mutation involves MSH2 (MutS Homolog 2)—found in approximately 90% of patients—followed by MLH1 (MutL Homolog 1)—found in approximately 10% of patients.22 Other MMR genes such as MSH6 (MutS Homolog 6), PMS2 (PMS1 homolog 2, mismatch repair system component), and MLH3 (MutL Homolog 3) less commonly are reported in MTS. There is a subset of patients who lose MSH2 or MLH1 expression due to promoter hypermethylation rather than a germline mutation. Methylation results in biallelic inactivation of the gene and loss of expression.23
A new subtype of MTS has been identified that demonstrates an autosomal-recessive pattern of inheritance and is referred to as MTS type 2 (autosomal-recessive colorectal adenomatous polyposis).24 In contrast to the classic MTS type 1, MTS type 2 exhibits microsatellite stability. Recent molecular analyses revealed that type 2 is due to a mutation in a base excision repair gene called MUTYH (mutY DNA glycosylase).25 These patients are likely to develop hundreds of polyps at an early age.
Muir-Torre syndrome also can occur sporadically without inheriting a germline mutation, which has been reported in a transplant patient from de novo somatic mutations or promoter hypermethylation.26 A case report of a renal transplant patient showed that switching from tacrolimus to sirolimus halted the appearance of new sebaceous neoplasms, which suggests that patients with MTS who undergo organ transplantation should potentially avoid tacrolimus and be put on sirolimus instead.27
Visceral Malignancies in MTS
Apart from frequent skin examinations, MTS patients should have frequent and rigorous visceral malignancy screening. Patients most commonly develop colorectal adenocarcinoma, especially in the proximal parts of the colon.28 In addition, they can develop numerous premalignant tumors, especially in MTS type 2. Other common tumors include endometrial, ovarian, genitourinary, hepatobiliary, breast, lung, hematopoietic, and CNS malignancies.29
Studies showed that specific loss of certain MMR proteins predispose patients to different types of visceral malignancies.30-32 For example, loss of MSH2 predisposes patients to development of extracolonic tumors, while loss of MLH1 more strongly is associated with development of colorectal adenocarcinoma.30 Patients with MSH2 also are at risk for development of CNS tumors, while patients with MLH1 mutations have never been reported to develop CNS tumors.31 Patients with loss of PMS2 have the lowest risk for development of any visceral malignancy.32
Diagnosing MTS
Let us consider a scenario whereby a dermatologist biopsied a solitary lesion and it came back as a sebaceous tumor. What would be the next step to establish a diagnosis of MTS?
Sebaceous tumors are rare outside the context of MTS. Therefore, patients presenting with a solitary sebaceous tumor should be worked up for MTS, as there are implications for further cancer screening. One helpful clue that can affect the pretest probability for MTS diagnosis is location of the tumor. A sebaceous tumor inferior to the neck most likely is associated with MTS. On the other hand, tumors on the head and neck can be spontaneous or associated with MTS.33 Another helpful tool is the Mayo score, a risk score for MTS in patients with sebaceous tumors.34 The score is established by adding up points, with 1 point given to each of the following: age of onset of a sebaceous tumor less than 60 years, personal history of visceral malignancy, and family history of Lynch syndrome–related visceral malignancy. Two points are given if the patient has 2 or more sebaceous tumors. The score ranges from 0 to 5. A risk score of 2 or more has a sensitivity of 100% and specificity of 81% for predicting a germline mutation in MMR genes.34
Testing for loss of MMR proteins is performed using immunohistochemistry (IHC) as well as microsatellite gene analysis on the biopsied tumor. There is no need to perform another biopsy, as these tests can be performed on the paraffin-embedded formalin fixed tissue. Immunohistochemistry testing looks for loss of expression of one of the MMR proteins. Staining usually is performed for MSH2, MSH6, and MLH1, as the combination offers a sensitivity of 81% and a positive predictive value of 100%.23,35,36
If IHC shows loss of MMR proteins, then MSI gene analysis should be performed as a confirmatory test by using MSI gene locus assays, which utilize 5 markers of mononucleotide and dinucleotide repeats. If the genome is positive for 2 of 5 of these markers, then the patient most likely has MTS.13
One caveat for IHC analysis is that there is a subset of patients who develop a solitary sebaceous tumor due to a sporadic loss of MMR protein without having MTS. These tumors also exhibit BRAF (B-Raf proto-oncogene, serine/threonine kinase) mutations or loss of p16, features that distinguish these tumors from those developed in MTS.37 As such, in a patient with a low Mayo score who developed a solitary sebaceous tumor that showed loss of MMR protein on IHC without evidence of MSI, it is reasonable to perform IHC for BRAF and p16 to avoid inaccurate diagnosis of MTS.
Another caveat is that standard MSI analysis will not detect MSI in tumors with loss of MSH6 because the markers used in the MSI analysis do not detect MSI caused by MSH6 loss. For these patients, MSI analysis using a panel composed of mononucleotides alone (pentaplex assay) should be performed in lieu of the standard panel.38
It is important to note that these molecular tests are not helpful for patients with MTS type 2, as the sebaceous tumors maintain MMR proteins and have microsatellite stability. As such, if MTS is highly suspected based on the Mayo score (either personal history of malignancy or strong family history) but the IHC and MSI analysis are negative, then referral to a geneticist for identification for MUTYH gene mutation is a reasonable next step. These patients with high Mayo scores should still be managed as MTS patients and should be screened for visceral malignancies despite lack of confirmatory tests.
Final Thoughts
Dermatologists should be highly suspicious of MTS when they diagnose sebaceous tumors. Making a diagnosis of MTS notably affects patients’ primary care. Patients with MTS should have annual skin examinations, neurologic examinations, colonoscopies starting at the age of 18 years, and surveillance for breast and pelvic cancers in women (by annual transvaginal ultrasound and endometrial aspirations) or for prostate and testicular cancers in men.17,39,40 Other tests to be ordered annually include complete blood cell count with differential and urinalysis.19
- Yamamoto H, Imai K. An updated review of microsatellite instability in the era of next-generation sequencing and precision medicine. Semin Oncol. 2019;46:261-270.
- Tamura K, Kaneda M, Futagawa M, et al. Genetic and genomic basis of the mismatch repair system involved in Lynch syndrome. Int J Clin Oncol. 2019;24:999-1011.
- Shiki M, Hida T, Sugano K, et al. Muir-Torre syndrome caused by exonic deletion of MLH1 due to homologous recombination. Eur J Dermatol. 2017;27:54-58.
- Büttner R, Friedrichs N. Hereditary colon cancer in Lynch syndrome/HNPCC syndrome in Germany. Pathologe. 2019;40:584-591.
- Kuwabara K, Suzuki O, Chika N, et al. Prevalence and molecular characteristics of DNA mismatch repair protein-deficient sebaceous neoplasms and keratoacanthomas in a Japanese hospital-based population. Jpn J Clin Oncol. 2018;48:514-521.
- Burris CKH, Rodriguez ME, Raven ML, et al. Muir-torre syndrome: the importance of a detailed family history. Case Rep Ophthalmol. 2019;10:180-185.
- Walsh MD, Jayasekara H, Huang A, et al. Clinico-pathological predictors of mismatch repair deficiency in sebaceous neoplasia: a large case series from a single Australian private pathology service. Australas J Dermatol. 2019;60:126-133.
- Georgeson P, Walsh MD, Clendenning M, et al. Tumor mutational signatures in sebaceous skin lesions from individuals with Lynch syndrome. Mol Genet Genomic Med. 2019;7:E00781.
- Hsieh P, Yamane K. DNA mismatch repair: molecular mechanism, cancer, and ageing. Mech Ageing Dev. 2008;129:391-407.
- Li YC, Korol AB, Fahima T, et al. Microsatellites within genes: structure, function, and evolution [published online February 12, 2004]. Mol Biol Evol. 2004;21:991-1007.
- Ellegren H. Microsatellites: simple sequences with complex evolution. Nat Rev Genet. 2004;5:435-445.
- Everett JN, Raymond VM, Dandapani M, et al. Screening for germline mismatch repair mutations following diagnosis of sebaceous neoplasm. JAMA Dermatol. 2014;150:1315-1321.
- Nojadeh JN, Sharif SB, Sakhinia E. Microsatellite instability in colorectal cancer. EXCLI J. 2018;17:159-168.
- Yang G, Zheng RY, Jin ZS. Correlations between microsatellite instability and the biological behaviour of tumours. J Cancer Res Clin Oncol. 2019;145:2891-2899.
- Garbe Y, Maletzki C, Linnebacher M. An MSI tumor specific frameshift mutation in a coding microsatellite of MSH3 encodes for HLA-A0201-restricted CD8+ cytotoxic T cell epitopes. PLoS One. 2011;6:E26517.
- Peng M, Mo Y, Wang Y, et al. Neoantigen vaccine: an emerging tumor immunotherapy. Mol Cancer. 2019;18:128.
- Rubay D, Ohanisian L, Bank MP, et al. Muir-Torre syndrome, a rare phenotype of hereditary nonpolyposis colorectal cancer with cutaneous manifestations. ACG Case Reports J. 2019;6:E00188.
- Velter C, Caussade P, Fricker JP, et al. Muir-Torre syndrome and Turcot syndrome [in French]. Ann Dermatol Venereol. 2017;144:525-529.
- John AM, Schwartz RA. Muir-Torre syndrome (MTS): an update and approach to diagnosis and management. J Am Acad Dermatol. 2016;74:558-566.
- Kibbi N, Worley B, Owen JL, et al. Sebaceous carcinoma: controversies and their evidence for clinical practice. Arch Dermatol Res. 2020;312:25-31.
- Marcoval J, Talavera-Belmonte A, Fornons-Servent R, et al. Cutaneous sebaceous tumours and Lynch syndrome: long-term follow-up of 60 patients. Clin Exp Dermatol. 2019;44:506-511.
- Roth RM, Haraldsdottir S, Hampel H, et al. Discordant mismatch repair protein immunoreactivity in Lynch syndrome-associated neoplasms: a recommendation for screening synchronous/metachronous neoplasms. Am J Clin Pathol. 2016;146:50-56.
- Westwood A, Glover A, Hutchins G, et al. Additional loss of MSH2 and MSH6 expression in sporadic deficient mismatch repair colorectal cancer due to MLH1 promoter hypermethylation. J Clin Pathol. 2019;72:443-447.
- Claes K, Dahan K, Tejpar S, et al. The genetics of familial adenomatous polyposis (FAP) and MutYH-associated polyposis (MAP). Acta Gastroenterol Belg. 2011;74:421-426.
- Sampson JR, Dolwani S, Jones S, et al. Autosomal recessive colorectal adenomatous polyposis due to inherited mutations of MYH. Lancet. 2003;362:39-41.
- Tomonari M, Shimada M, Nakada Y, et al. Muir-Torre syndrome: sebaceous carcinoma concurrent with colon cancer in a kidney transplant recipient; a case report. BMC Nephrol. 2019;20:394
- Levi Z, Hazazi R, Kedar-Barnes I, et al. Switching from tacrolimus to sirolimus halts the appearance of new sebaceous neoplasms in Muir-Torre syndrome. Am J Transplant. 2007;7:476-479.
- Mork ME, Rodriguez A, Taggart MW, et al. Identification of MSH2 inversion of exons 1–7 in clinical evaluation of families with suspected Lynch syndrome. Fam Cancer. 2017;16:357-361.
- Schwartz RA, Torre DP. The Muir-Torre syndrome: a 25-year retrospect. J Am Acad Dermatol. 1995;33:90-104.
- Chen W, Swanson BJ, Frankel WL. Molecular genetics of microsatellite-unstable colorectal cancer for pathologists. Diagn Pathol. 2017;12:24.
- Bansidhar BJ. Extracolonic manifestations of Lynch syndrome. Clin Colon Rectal Surg. 2012;25:103-110.
- Kato A, Sato N, Sugawara T, et al. Isolated loss of PMS2 immunohistochemical expression is frequently caused by heterogenous MLH1 promoter hypermethylation in Lynch syndrome screening for endometrial cancer patients. Am J Surg Pathol. 2016;40:770-776.
- Singh RS, Grayson W, Redston M, et al. Site and tumor type predicts DNA mismatch repair status in cutaneous sebaceous neoplasia. Am J Surg Pathol. 2008;32:936-942.
- Roberts ME, Riegert-Johnson DL, Thomas BC, et al. A clinical scoring system to identify patients with sebaceous neoplasms at risk for the Muir-Torre variant of Lynch syndrome [published online March 6, 2014]. Genet Med. 2014;16:711-716.
- Chhibber V, Dresser K, Mahalingam M. MSH-6: extending the reliability of immunohistochemistry as a screening tool in Muir-Torre syndrome. Mod Pathol. 2008;21:159-164.
- Orta L, Klimstra DS, Qin J, et al. Towards identification of hereditary DNA mismatch repair deficiency: sebaceous neoplasm warrants routine immunohistochemical screening regardless of patient’s age or other clinical characteristics. Am J Surg Pathol. 2009;33:934-944.
- Mathiak M, Rütten A, Mangold E, et al. Loss of DNA mismatch repair proteins in skin tumors from patients with Muir-Torre syndrome and MSH2 or MLH1 germline mutations: establishment of immunohistochemical analysis as a screening test. Am J Surg Pathol. 2002;26:338-343.
- Campanella NC, Berardinelli GN, Scapulatempo-Neto C, et al. Optimization of a pentaplex panel for MSI analysis without control DNA in a Brazilian population: correlation with ancestry markers. Eur J Hum Genet. 2014;22:875-880.
- Ponti G, Manfredini M, Tomasi A, et al. Muir-Torre Syndrome and founder mismatch repair gene mutations: a long gone historical genetic challenge. Gene. 2016;589:127-132.
- Ferreira I, Wiedemeyer K, Demetter P, et al. Update on the pathology, genetics and somatic landscape of sebaceous tumours [published online December 10, 2019]. Histopathology. doi:10.1111/his.14044
It is well known by now that tumor formation is driven by accumulation of numerous genetic and epigenetic mutations. Human cells are equipped with an apparatus called the DNA mismatch repair (MMR) system that corrects errors during replication.1 If these genes are themselves mutated, cells then start accumulating mutations in other genes, including oncogenes and tumor suppressor genes, which results in the development of sustained proliferative signaling pathways, evasion of growth suppression, resistance to cell death, and the potential for invasion and metastasis.2
Gene mutations in DNA MMR have been detected in several tumors, such as sebaceous tumors,3 colorectal adenocarcinomas,4 keratoacanthomas,5 and other visceral malignancies.6 Sebaceous tumors are rare in the general population; however, they are common in patients with inherited or acquired mutations in MMR genes.5 These patients also have been found to have other visceral malignancies such as colorectal adenocarcinomas and breast, lung, and central nervous system (CNS) tumors.7 This observation was made in the 1960s, and patients were referred to as having Muir-Torre syndrome (MTS).8 This article serves to briefly describe the DNA MMR system and its implication in sebaceous tumors as well as discuss the recent recommendations for screening for MTS in patients presenting with sebaceous tumors.
The DNA MMR System
Mismatch repair proteins are responsible for detecting and repairing errors during cell division, especially in microsatellite regions.9 Microsatellites are common and widely distributed DNA motifs consisting of repeated nucleotide sequences that normally account for 3% of the genome.10 Mutations in MMR result in insertion or deletion of nucleotides in these DNA motifs, making them either abnormally long or short, referred to as microsatellite instability (MSI), which results in downstream cumulative accumulation of mutations in oncogenes and tumor suppressor genes, and thus carcinogenesis.9
There are 7 human MMR proteins: MLH1, MLH3, MSH2, MSH3, MSH6, PMS1, and PMS2. These proteins are highly conserved across different living species.11 Loss of MMR proteins can be due to a mutation in the coding sequence of the gene or due to epigenetic hypermethylation of the gene promoter.12 These alterations can be inherited or acquired and in most cases result in MSI.
When assessing for MSI, tumor genomes can be divided into 3 subtypes: high-level and low-level MSI and stable microsatellites.13 Tumors with high-level MSI respond better to treatment and show a better prognosis than those with low-level MSI or stable microsatellites,14 which is thought to be due to tumor-induced immune activation. Microsatellite instability results in the generation of frameshift peptides that are immunogenic and induce tumor-specific immune responses.15 Several research laboratories have artificially synthesized frameshift peptides as vaccines and have successfully used them as targets for immune therapy as a way for preventing and treating malignancies.16
Sebaceous Tumors in MTS
A typical example of tumors that arise from mutations in the DNA MMR system is seen in MTS,a rare inherited genetic syndrome that predisposes patients to sebaceous neoplasms, keratoacanthomas, and visceral malignancies.17 It was first described as an autosomal-dominant condition in patients who have at least 1 sebaceous tumor and 1 visceral malignancy, with or without keratoacanthomas. It was then later characterized as a skin variant of Lynch syndrome, also known as hereditary nonpolyposis colorectal cancer syndrome.18
Sebaceous tumors are the hallmark of MTS. Although sebaceous hyperplasia is common in the general population, sebaceous tumors are rare outside the context of MTS. There are 3 types of sebaceous tumors with distinct pathologic features: adenoma, epithelioma, and carcinoma.19 Sebaceous adenomas and epitheliomas are benign growths; however, sebaceous carcinomas can be aggressive and have metastatic potential.20 Because it is difficult to clinically distinguish carcinomas from the benign sebaceous growths, biopsy of a large, changing, or ulcerated lesion is important in these patients to rule out a sebaceous carcinoma. Other aggressive skin tumors can develop in MTS, such as rapidly growing keratoacanthomas and basal cell carcinomas with sebaceous differentiation.21
Types of MTS
For most cases, MTS is characterized by germline mutations in DNA MMR genes. The most common mutation involves MSH2 (MutS Homolog 2)—found in approximately 90% of patients—followed by MLH1 (MutL Homolog 1)—found in approximately 10% of patients.22 Other MMR genes such as MSH6 (MutS Homolog 6), PMS2 (PMS1 homolog 2, mismatch repair system component), and MLH3 (MutL Homolog 3) less commonly are reported in MTS. There is a subset of patients who lose MSH2 or MLH1 expression due to promoter hypermethylation rather than a germline mutation. Methylation results in biallelic inactivation of the gene and loss of expression.23
A new subtype of MTS has been identified that demonstrates an autosomal-recessive pattern of inheritance and is referred to as MTS type 2 (autosomal-recessive colorectal adenomatous polyposis).24 In contrast to the classic MTS type 1, MTS type 2 exhibits microsatellite stability. Recent molecular analyses revealed that type 2 is due to a mutation in a base excision repair gene called MUTYH (mutY DNA glycosylase).25 These patients are likely to develop hundreds of polyps at an early age.
Muir-Torre syndrome also can occur sporadically without inheriting a germline mutation, which has been reported in a transplant patient from de novo somatic mutations or promoter hypermethylation.26 A case report of a renal transplant patient showed that switching from tacrolimus to sirolimus halted the appearance of new sebaceous neoplasms, which suggests that patients with MTS who undergo organ transplantation should potentially avoid tacrolimus and be put on sirolimus instead.27
Visceral Malignancies in MTS
Apart from frequent skin examinations, MTS patients should have frequent and rigorous visceral malignancy screening. Patients most commonly develop colorectal adenocarcinoma, especially in the proximal parts of the colon.28 In addition, they can develop numerous premalignant tumors, especially in MTS type 2. Other common tumors include endometrial, ovarian, genitourinary, hepatobiliary, breast, lung, hematopoietic, and CNS malignancies.29
Studies showed that specific loss of certain MMR proteins predispose patients to different types of visceral malignancies.30-32 For example, loss of MSH2 predisposes patients to development of extracolonic tumors, while loss of MLH1 more strongly is associated with development of colorectal adenocarcinoma.30 Patients with MSH2 also are at risk for development of CNS tumors, while patients with MLH1 mutations have never been reported to develop CNS tumors.31 Patients with loss of PMS2 have the lowest risk for development of any visceral malignancy.32
Diagnosing MTS
Let us consider a scenario whereby a dermatologist biopsied a solitary lesion and it came back as a sebaceous tumor. What would be the next step to establish a diagnosis of MTS?
Sebaceous tumors are rare outside the context of MTS. Therefore, patients presenting with a solitary sebaceous tumor should be worked up for MTS, as there are implications for further cancer screening. One helpful clue that can affect the pretest probability for MTS diagnosis is location of the tumor. A sebaceous tumor inferior to the neck most likely is associated with MTS. On the other hand, tumors on the head and neck can be spontaneous or associated with MTS.33 Another helpful tool is the Mayo score, a risk score for MTS in patients with sebaceous tumors.34 The score is established by adding up points, with 1 point given to each of the following: age of onset of a sebaceous tumor less than 60 years, personal history of visceral malignancy, and family history of Lynch syndrome–related visceral malignancy. Two points are given if the patient has 2 or more sebaceous tumors. The score ranges from 0 to 5. A risk score of 2 or more has a sensitivity of 100% and specificity of 81% for predicting a germline mutation in MMR genes.34
Testing for loss of MMR proteins is performed using immunohistochemistry (IHC) as well as microsatellite gene analysis on the biopsied tumor. There is no need to perform another biopsy, as these tests can be performed on the paraffin-embedded formalin fixed tissue. Immunohistochemistry testing looks for loss of expression of one of the MMR proteins. Staining usually is performed for MSH2, MSH6, and MLH1, as the combination offers a sensitivity of 81% and a positive predictive value of 100%.23,35,36
If IHC shows loss of MMR proteins, then MSI gene analysis should be performed as a confirmatory test by using MSI gene locus assays, which utilize 5 markers of mononucleotide and dinucleotide repeats. If the genome is positive for 2 of 5 of these markers, then the patient most likely has MTS.13
One caveat for IHC analysis is that there is a subset of patients who develop a solitary sebaceous tumor due to a sporadic loss of MMR protein without having MTS. These tumors also exhibit BRAF (B-Raf proto-oncogene, serine/threonine kinase) mutations or loss of p16, features that distinguish these tumors from those developed in MTS.37 As such, in a patient with a low Mayo score who developed a solitary sebaceous tumor that showed loss of MMR protein on IHC without evidence of MSI, it is reasonable to perform IHC for BRAF and p16 to avoid inaccurate diagnosis of MTS.
Another caveat is that standard MSI analysis will not detect MSI in tumors with loss of MSH6 because the markers used in the MSI analysis do not detect MSI caused by MSH6 loss. For these patients, MSI analysis using a panel composed of mononucleotides alone (pentaplex assay) should be performed in lieu of the standard panel.38
It is important to note that these molecular tests are not helpful for patients with MTS type 2, as the sebaceous tumors maintain MMR proteins and have microsatellite stability. As such, if MTS is highly suspected based on the Mayo score (either personal history of malignancy or strong family history) but the IHC and MSI analysis are negative, then referral to a geneticist for identification for MUTYH gene mutation is a reasonable next step. These patients with high Mayo scores should still be managed as MTS patients and should be screened for visceral malignancies despite lack of confirmatory tests.
Final Thoughts
Dermatologists should be highly suspicious of MTS when they diagnose sebaceous tumors. Making a diagnosis of MTS notably affects patients’ primary care. Patients with MTS should have annual skin examinations, neurologic examinations, colonoscopies starting at the age of 18 years, and surveillance for breast and pelvic cancers in women (by annual transvaginal ultrasound and endometrial aspirations) or for prostate and testicular cancers in men.17,39,40 Other tests to be ordered annually include complete blood cell count with differential and urinalysis.19
It is well known by now that tumor formation is driven by accumulation of numerous genetic and epigenetic mutations. Human cells are equipped with an apparatus called the DNA mismatch repair (MMR) system that corrects errors during replication.1 If these genes are themselves mutated, cells then start accumulating mutations in other genes, including oncogenes and tumor suppressor genes, which results in the development of sustained proliferative signaling pathways, evasion of growth suppression, resistance to cell death, and the potential for invasion and metastasis.2
Gene mutations in DNA MMR have been detected in several tumors, such as sebaceous tumors,3 colorectal adenocarcinomas,4 keratoacanthomas,5 and other visceral malignancies.6 Sebaceous tumors are rare in the general population; however, they are common in patients with inherited or acquired mutations in MMR genes.5 These patients also have been found to have other visceral malignancies such as colorectal adenocarcinomas and breast, lung, and central nervous system (CNS) tumors.7 This observation was made in the 1960s, and patients were referred to as having Muir-Torre syndrome (MTS).8 This article serves to briefly describe the DNA MMR system and its implication in sebaceous tumors as well as discuss the recent recommendations for screening for MTS in patients presenting with sebaceous tumors.
The DNA MMR System
Mismatch repair proteins are responsible for detecting and repairing errors during cell division, especially in microsatellite regions.9 Microsatellites are common and widely distributed DNA motifs consisting of repeated nucleotide sequences that normally account for 3% of the genome.10 Mutations in MMR result in insertion or deletion of nucleotides in these DNA motifs, making them either abnormally long or short, referred to as microsatellite instability (MSI), which results in downstream cumulative accumulation of mutations in oncogenes and tumor suppressor genes, and thus carcinogenesis.9
There are 7 human MMR proteins: MLH1, MLH3, MSH2, MSH3, MSH6, PMS1, and PMS2. These proteins are highly conserved across different living species.11 Loss of MMR proteins can be due to a mutation in the coding sequence of the gene or due to epigenetic hypermethylation of the gene promoter.12 These alterations can be inherited or acquired and in most cases result in MSI.
When assessing for MSI, tumor genomes can be divided into 3 subtypes: high-level and low-level MSI and stable microsatellites.13 Tumors with high-level MSI respond better to treatment and show a better prognosis than those with low-level MSI or stable microsatellites,14 which is thought to be due to tumor-induced immune activation. Microsatellite instability results in the generation of frameshift peptides that are immunogenic and induce tumor-specific immune responses.15 Several research laboratories have artificially synthesized frameshift peptides as vaccines and have successfully used them as targets for immune therapy as a way for preventing and treating malignancies.16
Sebaceous Tumors in MTS
A typical example of tumors that arise from mutations in the DNA MMR system is seen in MTS,a rare inherited genetic syndrome that predisposes patients to sebaceous neoplasms, keratoacanthomas, and visceral malignancies.17 It was first described as an autosomal-dominant condition in patients who have at least 1 sebaceous tumor and 1 visceral malignancy, with or without keratoacanthomas. It was then later characterized as a skin variant of Lynch syndrome, also known as hereditary nonpolyposis colorectal cancer syndrome.18
Sebaceous tumors are the hallmark of MTS. Although sebaceous hyperplasia is common in the general population, sebaceous tumors are rare outside the context of MTS. There are 3 types of sebaceous tumors with distinct pathologic features: adenoma, epithelioma, and carcinoma.19 Sebaceous adenomas and epitheliomas are benign growths; however, sebaceous carcinomas can be aggressive and have metastatic potential.20 Because it is difficult to clinically distinguish carcinomas from the benign sebaceous growths, biopsy of a large, changing, or ulcerated lesion is important in these patients to rule out a sebaceous carcinoma. Other aggressive skin tumors can develop in MTS, such as rapidly growing keratoacanthomas and basal cell carcinomas with sebaceous differentiation.21
Types of MTS
For most cases, MTS is characterized by germline mutations in DNA MMR genes. The most common mutation involves MSH2 (MutS Homolog 2)—found in approximately 90% of patients—followed by MLH1 (MutL Homolog 1)—found in approximately 10% of patients.22 Other MMR genes such as MSH6 (MutS Homolog 6), PMS2 (PMS1 homolog 2, mismatch repair system component), and MLH3 (MutL Homolog 3) less commonly are reported in MTS. There is a subset of patients who lose MSH2 or MLH1 expression due to promoter hypermethylation rather than a germline mutation. Methylation results in biallelic inactivation of the gene and loss of expression.23
A new subtype of MTS has been identified that demonstrates an autosomal-recessive pattern of inheritance and is referred to as MTS type 2 (autosomal-recessive colorectal adenomatous polyposis).24 In contrast to the classic MTS type 1, MTS type 2 exhibits microsatellite stability. Recent molecular analyses revealed that type 2 is due to a mutation in a base excision repair gene called MUTYH (mutY DNA glycosylase).25 These patients are likely to develop hundreds of polyps at an early age.
Muir-Torre syndrome also can occur sporadically without inheriting a germline mutation, which has been reported in a transplant patient from de novo somatic mutations or promoter hypermethylation.26 A case report of a renal transplant patient showed that switching from tacrolimus to sirolimus halted the appearance of new sebaceous neoplasms, which suggests that patients with MTS who undergo organ transplantation should potentially avoid tacrolimus and be put on sirolimus instead.27
Visceral Malignancies in MTS
Apart from frequent skin examinations, MTS patients should have frequent and rigorous visceral malignancy screening. Patients most commonly develop colorectal adenocarcinoma, especially in the proximal parts of the colon.28 In addition, they can develop numerous premalignant tumors, especially in MTS type 2. Other common tumors include endometrial, ovarian, genitourinary, hepatobiliary, breast, lung, hematopoietic, and CNS malignancies.29
Studies showed that specific loss of certain MMR proteins predispose patients to different types of visceral malignancies.30-32 For example, loss of MSH2 predisposes patients to development of extracolonic tumors, while loss of MLH1 more strongly is associated with development of colorectal adenocarcinoma.30 Patients with MSH2 also are at risk for development of CNS tumors, while patients with MLH1 mutations have never been reported to develop CNS tumors.31 Patients with loss of PMS2 have the lowest risk for development of any visceral malignancy.32
Diagnosing MTS
Let us consider a scenario whereby a dermatologist biopsied a solitary lesion and it came back as a sebaceous tumor. What would be the next step to establish a diagnosis of MTS?
Sebaceous tumors are rare outside the context of MTS. Therefore, patients presenting with a solitary sebaceous tumor should be worked up for MTS, as there are implications for further cancer screening. One helpful clue that can affect the pretest probability for MTS diagnosis is location of the tumor. A sebaceous tumor inferior to the neck most likely is associated with MTS. On the other hand, tumors on the head and neck can be spontaneous or associated with MTS.33 Another helpful tool is the Mayo score, a risk score for MTS in patients with sebaceous tumors.34 The score is established by adding up points, with 1 point given to each of the following: age of onset of a sebaceous tumor less than 60 years, personal history of visceral malignancy, and family history of Lynch syndrome–related visceral malignancy. Two points are given if the patient has 2 or more sebaceous tumors. The score ranges from 0 to 5. A risk score of 2 or more has a sensitivity of 100% and specificity of 81% for predicting a germline mutation in MMR genes.34
Testing for loss of MMR proteins is performed using immunohistochemistry (IHC) as well as microsatellite gene analysis on the biopsied tumor. There is no need to perform another biopsy, as these tests can be performed on the paraffin-embedded formalin fixed tissue. Immunohistochemistry testing looks for loss of expression of one of the MMR proteins. Staining usually is performed for MSH2, MSH6, and MLH1, as the combination offers a sensitivity of 81% and a positive predictive value of 100%.23,35,36
If IHC shows loss of MMR proteins, then MSI gene analysis should be performed as a confirmatory test by using MSI gene locus assays, which utilize 5 markers of mononucleotide and dinucleotide repeats. If the genome is positive for 2 of 5 of these markers, then the patient most likely has MTS.13
One caveat for IHC analysis is that there is a subset of patients who develop a solitary sebaceous tumor due to a sporadic loss of MMR protein without having MTS. These tumors also exhibit BRAF (B-Raf proto-oncogene, serine/threonine kinase) mutations or loss of p16, features that distinguish these tumors from those developed in MTS.37 As such, in a patient with a low Mayo score who developed a solitary sebaceous tumor that showed loss of MMR protein on IHC without evidence of MSI, it is reasonable to perform IHC for BRAF and p16 to avoid inaccurate diagnosis of MTS.
Another caveat is that standard MSI analysis will not detect MSI in tumors with loss of MSH6 because the markers used in the MSI analysis do not detect MSI caused by MSH6 loss. For these patients, MSI analysis using a panel composed of mononucleotides alone (pentaplex assay) should be performed in lieu of the standard panel.38
It is important to note that these molecular tests are not helpful for patients with MTS type 2, as the sebaceous tumors maintain MMR proteins and have microsatellite stability. As such, if MTS is highly suspected based on the Mayo score (either personal history of malignancy or strong family history) but the IHC and MSI analysis are negative, then referral to a geneticist for identification for MUTYH gene mutation is a reasonable next step. These patients with high Mayo scores should still be managed as MTS patients and should be screened for visceral malignancies despite lack of confirmatory tests.
Final Thoughts
Dermatologists should be highly suspicious of MTS when they diagnose sebaceous tumors. Making a diagnosis of MTS notably affects patients’ primary care. Patients with MTS should have annual skin examinations, neurologic examinations, colonoscopies starting at the age of 18 years, and surveillance for breast and pelvic cancers in women (by annual transvaginal ultrasound and endometrial aspirations) or for prostate and testicular cancers in men.17,39,40 Other tests to be ordered annually include complete blood cell count with differential and urinalysis.19
- Yamamoto H, Imai K. An updated review of microsatellite instability in the era of next-generation sequencing and precision medicine. Semin Oncol. 2019;46:261-270.
- Tamura K, Kaneda M, Futagawa M, et al. Genetic and genomic basis of the mismatch repair system involved in Lynch syndrome. Int J Clin Oncol. 2019;24:999-1011.
- Shiki M, Hida T, Sugano K, et al. Muir-Torre syndrome caused by exonic deletion of MLH1 due to homologous recombination. Eur J Dermatol. 2017;27:54-58.
- Büttner R, Friedrichs N. Hereditary colon cancer in Lynch syndrome/HNPCC syndrome in Germany. Pathologe. 2019;40:584-591.
- Kuwabara K, Suzuki O, Chika N, et al. Prevalence and molecular characteristics of DNA mismatch repair protein-deficient sebaceous neoplasms and keratoacanthomas in a Japanese hospital-based population. Jpn J Clin Oncol. 2018;48:514-521.
- Burris CKH, Rodriguez ME, Raven ML, et al. Muir-torre syndrome: the importance of a detailed family history. Case Rep Ophthalmol. 2019;10:180-185.
- Walsh MD, Jayasekara H, Huang A, et al. Clinico-pathological predictors of mismatch repair deficiency in sebaceous neoplasia: a large case series from a single Australian private pathology service. Australas J Dermatol. 2019;60:126-133.
- Georgeson P, Walsh MD, Clendenning M, et al. Tumor mutational signatures in sebaceous skin lesions from individuals with Lynch syndrome. Mol Genet Genomic Med. 2019;7:E00781.
- Hsieh P, Yamane K. DNA mismatch repair: molecular mechanism, cancer, and ageing. Mech Ageing Dev. 2008;129:391-407.
- Li YC, Korol AB, Fahima T, et al. Microsatellites within genes: structure, function, and evolution [published online February 12, 2004]. Mol Biol Evol. 2004;21:991-1007.
- Ellegren H. Microsatellites: simple sequences with complex evolution. Nat Rev Genet. 2004;5:435-445.
- Everett JN, Raymond VM, Dandapani M, et al. Screening for germline mismatch repair mutations following diagnosis of sebaceous neoplasm. JAMA Dermatol. 2014;150:1315-1321.
- Nojadeh JN, Sharif SB, Sakhinia E. Microsatellite instability in colorectal cancer. EXCLI J. 2018;17:159-168.
- Yang G, Zheng RY, Jin ZS. Correlations between microsatellite instability and the biological behaviour of tumours. J Cancer Res Clin Oncol. 2019;145:2891-2899.
- Garbe Y, Maletzki C, Linnebacher M. An MSI tumor specific frameshift mutation in a coding microsatellite of MSH3 encodes for HLA-A0201-restricted CD8+ cytotoxic T cell epitopes. PLoS One. 2011;6:E26517.
- Peng M, Mo Y, Wang Y, et al. Neoantigen vaccine: an emerging tumor immunotherapy. Mol Cancer. 2019;18:128.
- Rubay D, Ohanisian L, Bank MP, et al. Muir-Torre syndrome, a rare phenotype of hereditary nonpolyposis colorectal cancer with cutaneous manifestations. ACG Case Reports J. 2019;6:E00188.
- Velter C, Caussade P, Fricker JP, et al. Muir-Torre syndrome and Turcot syndrome [in French]. Ann Dermatol Venereol. 2017;144:525-529.
- John AM, Schwartz RA. Muir-Torre syndrome (MTS): an update and approach to diagnosis and management. J Am Acad Dermatol. 2016;74:558-566.
- Kibbi N, Worley B, Owen JL, et al. Sebaceous carcinoma: controversies and their evidence for clinical practice. Arch Dermatol Res. 2020;312:25-31.
- Marcoval J, Talavera-Belmonte A, Fornons-Servent R, et al. Cutaneous sebaceous tumours and Lynch syndrome: long-term follow-up of 60 patients. Clin Exp Dermatol. 2019;44:506-511.
- Roth RM, Haraldsdottir S, Hampel H, et al. Discordant mismatch repair protein immunoreactivity in Lynch syndrome-associated neoplasms: a recommendation for screening synchronous/metachronous neoplasms. Am J Clin Pathol. 2016;146:50-56.
- Westwood A, Glover A, Hutchins G, et al. Additional loss of MSH2 and MSH6 expression in sporadic deficient mismatch repair colorectal cancer due to MLH1 promoter hypermethylation. J Clin Pathol. 2019;72:443-447.
- Claes K, Dahan K, Tejpar S, et al. The genetics of familial adenomatous polyposis (FAP) and MutYH-associated polyposis (MAP). Acta Gastroenterol Belg. 2011;74:421-426.
- Sampson JR, Dolwani S, Jones S, et al. Autosomal recessive colorectal adenomatous polyposis due to inherited mutations of MYH. Lancet. 2003;362:39-41.
- Tomonari M, Shimada M, Nakada Y, et al. Muir-Torre syndrome: sebaceous carcinoma concurrent with colon cancer in a kidney transplant recipient; a case report. BMC Nephrol. 2019;20:394
- Levi Z, Hazazi R, Kedar-Barnes I, et al. Switching from tacrolimus to sirolimus halts the appearance of new sebaceous neoplasms in Muir-Torre syndrome. Am J Transplant. 2007;7:476-479.
- Mork ME, Rodriguez A, Taggart MW, et al. Identification of MSH2 inversion of exons 1–7 in clinical evaluation of families with suspected Lynch syndrome. Fam Cancer. 2017;16:357-361.
- Schwartz RA, Torre DP. The Muir-Torre syndrome: a 25-year retrospect. J Am Acad Dermatol. 1995;33:90-104.
- Chen W, Swanson BJ, Frankel WL. Molecular genetics of microsatellite-unstable colorectal cancer for pathologists. Diagn Pathol. 2017;12:24.
- Bansidhar BJ. Extracolonic manifestations of Lynch syndrome. Clin Colon Rectal Surg. 2012;25:103-110.
- Kato A, Sato N, Sugawara T, et al. Isolated loss of PMS2 immunohistochemical expression is frequently caused by heterogenous MLH1 promoter hypermethylation in Lynch syndrome screening for endometrial cancer patients. Am J Surg Pathol. 2016;40:770-776.
- Singh RS, Grayson W, Redston M, et al. Site and tumor type predicts DNA mismatch repair status in cutaneous sebaceous neoplasia. Am J Surg Pathol. 2008;32:936-942.
- Roberts ME, Riegert-Johnson DL, Thomas BC, et al. A clinical scoring system to identify patients with sebaceous neoplasms at risk for the Muir-Torre variant of Lynch syndrome [published online March 6, 2014]. Genet Med. 2014;16:711-716.
- Chhibber V, Dresser K, Mahalingam M. MSH-6: extending the reliability of immunohistochemistry as a screening tool in Muir-Torre syndrome. Mod Pathol. 2008;21:159-164.
- Orta L, Klimstra DS, Qin J, et al. Towards identification of hereditary DNA mismatch repair deficiency: sebaceous neoplasm warrants routine immunohistochemical screening regardless of patient’s age or other clinical characteristics. Am J Surg Pathol. 2009;33:934-944.
- Mathiak M, Rütten A, Mangold E, et al. Loss of DNA mismatch repair proteins in skin tumors from patients with Muir-Torre syndrome and MSH2 or MLH1 germline mutations: establishment of immunohistochemical analysis as a screening test. Am J Surg Pathol. 2002;26:338-343.
- Campanella NC, Berardinelli GN, Scapulatempo-Neto C, et al. Optimization of a pentaplex panel for MSI analysis without control DNA in a Brazilian population: correlation with ancestry markers. Eur J Hum Genet. 2014;22:875-880.
- Ponti G, Manfredini M, Tomasi A, et al. Muir-Torre Syndrome and founder mismatch repair gene mutations: a long gone historical genetic challenge. Gene. 2016;589:127-132.
- Ferreira I, Wiedemeyer K, Demetter P, et al. Update on the pathology, genetics and somatic landscape of sebaceous tumours [published online December 10, 2019]. Histopathology. doi:10.1111/his.14044
- Yamamoto H, Imai K. An updated review of microsatellite instability in the era of next-generation sequencing and precision medicine. Semin Oncol. 2019;46:261-270.
- Tamura K, Kaneda M, Futagawa M, et al. Genetic and genomic basis of the mismatch repair system involved in Lynch syndrome. Int J Clin Oncol. 2019;24:999-1011.
- Shiki M, Hida T, Sugano K, et al. Muir-Torre syndrome caused by exonic deletion of MLH1 due to homologous recombination. Eur J Dermatol. 2017;27:54-58.
- Büttner R, Friedrichs N. Hereditary colon cancer in Lynch syndrome/HNPCC syndrome in Germany. Pathologe. 2019;40:584-591.
- Kuwabara K, Suzuki O, Chika N, et al. Prevalence and molecular characteristics of DNA mismatch repair protein-deficient sebaceous neoplasms and keratoacanthomas in a Japanese hospital-based population. Jpn J Clin Oncol. 2018;48:514-521.
- Burris CKH, Rodriguez ME, Raven ML, et al. Muir-torre syndrome: the importance of a detailed family history. Case Rep Ophthalmol. 2019;10:180-185.
- Walsh MD, Jayasekara H, Huang A, et al. Clinico-pathological predictors of mismatch repair deficiency in sebaceous neoplasia: a large case series from a single Australian private pathology service. Australas J Dermatol. 2019;60:126-133.
- Georgeson P, Walsh MD, Clendenning M, et al. Tumor mutational signatures in sebaceous skin lesions from individuals with Lynch syndrome. Mol Genet Genomic Med. 2019;7:E00781.
- Hsieh P, Yamane K. DNA mismatch repair: molecular mechanism, cancer, and ageing. Mech Ageing Dev. 2008;129:391-407.
- Li YC, Korol AB, Fahima T, et al. Microsatellites within genes: structure, function, and evolution [published online February 12, 2004]. Mol Biol Evol. 2004;21:991-1007.
- Ellegren H. Microsatellites: simple sequences with complex evolution. Nat Rev Genet. 2004;5:435-445.
- Everett JN, Raymond VM, Dandapani M, et al. Screening for germline mismatch repair mutations following diagnosis of sebaceous neoplasm. JAMA Dermatol. 2014;150:1315-1321.
- Nojadeh JN, Sharif SB, Sakhinia E. Microsatellite instability in colorectal cancer. EXCLI J. 2018;17:159-168.
- Yang G, Zheng RY, Jin ZS. Correlations between microsatellite instability and the biological behaviour of tumours. J Cancer Res Clin Oncol. 2019;145:2891-2899.
- Garbe Y, Maletzki C, Linnebacher M. An MSI tumor specific frameshift mutation in a coding microsatellite of MSH3 encodes for HLA-A0201-restricted CD8+ cytotoxic T cell epitopes. PLoS One. 2011;6:E26517.
- Peng M, Mo Y, Wang Y, et al. Neoantigen vaccine: an emerging tumor immunotherapy. Mol Cancer. 2019;18:128.
- Rubay D, Ohanisian L, Bank MP, et al. Muir-Torre syndrome, a rare phenotype of hereditary nonpolyposis colorectal cancer with cutaneous manifestations. ACG Case Reports J. 2019;6:E00188.
- Velter C, Caussade P, Fricker JP, et al. Muir-Torre syndrome and Turcot syndrome [in French]. Ann Dermatol Venereol. 2017;144:525-529.
- John AM, Schwartz RA. Muir-Torre syndrome (MTS): an update and approach to diagnosis and management. J Am Acad Dermatol. 2016;74:558-566.
- Kibbi N, Worley B, Owen JL, et al. Sebaceous carcinoma: controversies and their evidence for clinical practice. Arch Dermatol Res. 2020;312:25-31.
- Marcoval J, Talavera-Belmonte A, Fornons-Servent R, et al. Cutaneous sebaceous tumours and Lynch syndrome: long-term follow-up of 60 patients. Clin Exp Dermatol. 2019;44:506-511.
- Roth RM, Haraldsdottir S, Hampel H, et al. Discordant mismatch repair protein immunoreactivity in Lynch syndrome-associated neoplasms: a recommendation for screening synchronous/metachronous neoplasms. Am J Clin Pathol. 2016;146:50-56.
- Westwood A, Glover A, Hutchins G, et al. Additional loss of MSH2 and MSH6 expression in sporadic deficient mismatch repair colorectal cancer due to MLH1 promoter hypermethylation. J Clin Pathol. 2019;72:443-447.
- Claes K, Dahan K, Tejpar S, et al. The genetics of familial adenomatous polyposis (FAP) and MutYH-associated polyposis (MAP). Acta Gastroenterol Belg. 2011;74:421-426.
- Sampson JR, Dolwani S, Jones S, et al. Autosomal recessive colorectal adenomatous polyposis due to inherited mutations of MYH. Lancet. 2003;362:39-41.
- Tomonari M, Shimada M, Nakada Y, et al. Muir-Torre syndrome: sebaceous carcinoma concurrent with colon cancer in a kidney transplant recipient; a case report. BMC Nephrol. 2019;20:394
- Levi Z, Hazazi R, Kedar-Barnes I, et al. Switching from tacrolimus to sirolimus halts the appearance of new sebaceous neoplasms in Muir-Torre syndrome. Am J Transplant. 2007;7:476-479.
- Mork ME, Rodriguez A, Taggart MW, et al. Identification of MSH2 inversion of exons 1–7 in clinical evaluation of families with suspected Lynch syndrome. Fam Cancer. 2017;16:357-361.
- Schwartz RA, Torre DP. The Muir-Torre syndrome: a 25-year retrospect. J Am Acad Dermatol. 1995;33:90-104.
- Chen W, Swanson BJ, Frankel WL. Molecular genetics of microsatellite-unstable colorectal cancer for pathologists. Diagn Pathol. 2017;12:24.
- Bansidhar BJ. Extracolonic manifestations of Lynch syndrome. Clin Colon Rectal Surg. 2012;25:103-110.
- Kato A, Sato N, Sugawara T, et al. Isolated loss of PMS2 immunohistochemical expression is frequently caused by heterogenous MLH1 promoter hypermethylation in Lynch syndrome screening for endometrial cancer patients. Am J Surg Pathol. 2016;40:770-776.
- Singh RS, Grayson W, Redston M, et al. Site and tumor type predicts DNA mismatch repair status in cutaneous sebaceous neoplasia. Am J Surg Pathol. 2008;32:936-942.
- Roberts ME, Riegert-Johnson DL, Thomas BC, et al. A clinical scoring system to identify patients with sebaceous neoplasms at risk for the Muir-Torre variant of Lynch syndrome [published online March 6, 2014]. Genet Med. 2014;16:711-716.
- Chhibber V, Dresser K, Mahalingam M. MSH-6: extending the reliability of immunohistochemistry as a screening tool in Muir-Torre syndrome. Mod Pathol. 2008;21:159-164.
- Orta L, Klimstra DS, Qin J, et al. Towards identification of hereditary DNA mismatch repair deficiency: sebaceous neoplasm warrants routine immunohistochemical screening regardless of patient’s age or other clinical characteristics. Am J Surg Pathol. 2009;33:934-944.
- Mathiak M, Rütten A, Mangold E, et al. Loss of DNA mismatch repair proteins in skin tumors from patients with Muir-Torre syndrome and MSH2 or MLH1 germline mutations: establishment of immunohistochemical analysis as a screening test. Am J Surg Pathol. 2002;26:338-343.
- Campanella NC, Berardinelli GN, Scapulatempo-Neto C, et al. Optimization of a pentaplex panel for MSI analysis without control DNA in a Brazilian population: correlation with ancestry markers. Eur J Hum Genet. 2014;22:875-880.
- Ponti G, Manfredini M, Tomasi A, et al. Muir-Torre Syndrome and founder mismatch repair gene mutations: a long gone historical genetic challenge. Gene. 2016;589:127-132.
- Ferreira I, Wiedemeyer K, Demetter P, et al. Update on the pathology, genetics and somatic landscape of sebaceous tumours [published online December 10, 2019]. Histopathology. doi:10.1111/his.14044
Resident Pearls
- When patients present with a solitary sebaceous tumor, there is a high likelihood they have Muir-Torre syndrome (MTS) and thus are at a high risk to develop visceral malignancies.
- It is important to perform further testing using immunohistochemistry for DNA mismatch repair proteins and microsatellite instability gene analysis in some cases to confirm the diagnosis of MTS and to perform the appropriate cancer screening tests.

Tuberous Sclerosis With Segmental Overgrowth
To the Editor:
A 3-year-old boy with a history of tuberous sclerosis presented to our clinic for evaluation of bumps on the second and third fingers of the left hand. Physical examination revealed firm rubbery nodules on the palmar third metacarpophalangeal joint extending to the palm and the lateral aspect of the distal third dorsal finger. There also was asymmetric overgrowth of the left second and third digits consistent with bony segmental overgrowth (Figure).
Tuberous sclerosis and overgrowth syndromes including Proteus syndrome have mutations that share a common pathway, namely the PI3K/AKT/mTOR (phosphoinositide 3-kinase/alpha serine/threonine-protein kinase/mammalian target of rapamycin) pathway.1 The mutations in tuberous sclerosis involve the loss of TSC1 (TSC complex subunit 1) on chromosome 9 or TSC2 (TSC complex subunit 2) on chromosome 16.2 The protein products of these genes, hamartin and tuberin, act together as a tumor suppressor complex.3 The inheritance pattern of tuberous sclerosis is autosomal dominant, though two-thirds of cases are due to de novo germline mutations.4 The second copy of the gene must be lost spontaneously in any particular cell for the deleterious effects of the disease to manifest. The mutation in overgrowth syndromes including Proteus syndrome involves the activation of AKT1 (AKT serine/threonine kinase 1) on chromosome 14. This mutation occurs in somatic cells as opposed to germ cells, as in tuberous sclerosis. This difference accounts for the mosaic expression of segmental overgrowth syndromes. This concept has been demonstrated in overgrowth syndromes such as Proteus syndrome, with cells from unaffected areas having different genetic makeup than those from affected tissues.5 These mutations, though different, result in the downstream effects of unchecked messenger RNA translation and dysregulated cellular growth.
In our patient, we hypothesized that a small proportion of his postfertilization somatic cells underwent a second de novo mutation in the AKT1 pathway, resulting in the bony overgrowth seen on the left hand. We suspected that this second mutation could be an activation of AKT1, the mutation seen in Proteus syndrome. Sequencing of the tissue may be performed in the future, especially if segmental overgrowth continues and necessitates therapy.
- Wu Y, Zhou BP. Kinases meet at TSC. Cell Res. 2007;17:971-973.
- Roach SE, Sparagana SP. Diagnosis of tuberous sclerosis complex. J Child Neurol. 2004;19:643-649.
- Barker KT, Houlston RS. Overgrowth syndromes: is dysfunctional PI3-kinase signaling a unifying mechanism? Eur J Hum Genet. 2003;11:665-670.
- Nothrup H, Koenig MK, Au KS. Tuberous sclerosis complex. GeneReviews. Seattle, WA: University of Washington; 1999.
- Lindhurst MJ, Parker VE, Payne F, et al. Mosaic overgrowth with fibroadipose hyperplasia is caused by somatic activating mutations in PIK3CA. Nat Genet. 2012;44:928-933.
To the Editor:
A 3-year-old boy with a history of tuberous sclerosis presented to our clinic for evaluation of bumps on the second and third fingers of the left hand. Physical examination revealed firm rubbery nodules on the palmar third metacarpophalangeal joint extending to the palm and the lateral aspect of the distal third dorsal finger. There also was asymmetric overgrowth of the left second and third digits consistent with bony segmental overgrowth (Figure).
Tuberous sclerosis and overgrowth syndromes including Proteus syndrome have mutations that share a common pathway, namely the PI3K/AKT/mTOR (phosphoinositide 3-kinase/alpha serine/threonine-protein kinase/mammalian target of rapamycin) pathway.1 The mutations in tuberous sclerosis involve the loss of TSC1 (TSC complex subunit 1) on chromosome 9 or TSC2 (TSC complex subunit 2) on chromosome 16.2 The protein products of these genes, hamartin and tuberin, act together as a tumor suppressor complex.3 The inheritance pattern of tuberous sclerosis is autosomal dominant, though two-thirds of cases are due to de novo germline mutations.4 The second copy of the gene must be lost spontaneously in any particular cell for the deleterious effects of the disease to manifest. The mutation in overgrowth syndromes including Proteus syndrome involves the activation of AKT1 (AKT serine/threonine kinase 1) on chromosome 14. This mutation occurs in somatic cells as opposed to germ cells, as in tuberous sclerosis. This difference accounts for the mosaic expression of segmental overgrowth syndromes. This concept has been demonstrated in overgrowth syndromes such as Proteus syndrome, with cells from unaffected areas having different genetic makeup than those from affected tissues.5 These mutations, though different, result in the downstream effects of unchecked messenger RNA translation and dysregulated cellular growth.
In our patient, we hypothesized that a small proportion of his postfertilization somatic cells underwent a second de novo mutation in the AKT1 pathway, resulting in the bony overgrowth seen on the left hand. We suspected that this second mutation could be an activation of AKT1, the mutation seen in Proteus syndrome. Sequencing of the tissue may be performed in the future, especially if segmental overgrowth continues and necessitates therapy.
To the Editor:
A 3-year-old boy with a history of tuberous sclerosis presented to our clinic for evaluation of bumps on the second and third fingers of the left hand. Physical examination revealed firm rubbery nodules on the palmar third metacarpophalangeal joint extending to the palm and the lateral aspect of the distal third dorsal finger. There also was asymmetric overgrowth of the left second and third digits consistent with bony segmental overgrowth (Figure).
Tuberous sclerosis and overgrowth syndromes including Proteus syndrome have mutations that share a common pathway, namely the PI3K/AKT/mTOR (phosphoinositide 3-kinase/alpha serine/threonine-protein kinase/mammalian target of rapamycin) pathway.1 The mutations in tuberous sclerosis involve the loss of TSC1 (TSC complex subunit 1) on chromosome 9 or TSC2 (TSC complex subunit 2) on chromosome 16.2 The protein products of these genes, hamartin and tuberin, act together as a tumor suppressor complex.3 The inheritance pattern of tuberous sclerosis is autosomal dominant, though two-thirds of cases are due to de novo germline mutations.4 The second copy of the gene must be lost spontaneously in any particular cell for the deleterious effects of the disease to manifest. The mutation in overgrowth syndromes including Proteus syndrome involves the activation of AKT1 (AKT serine/threonine kinase 1) on chromosome 14. This mutation occurs in somatic cells as opposed to germ cells, as in tuberous sclerosis. This difference accounts for the mosaic expression of segmental overgrowth syndromes. This concept has been demonstrated in overgrowth syndromes such as Proteus syndrome, with cells from unaffected areas having different genetic makeup than those from affected tissues.5 These mutations, though different, result in the downstream effects of unchecked messenger RNA translation and dysregulated cellular growth.
In our patient, we hypothesized that a small proportion of his postfertilization somatic cells underwent a second de novo mutation in the AKT1 pathway, resulting in the bony overgrowth seen on the left hand. We suspected that this second mutation could be an activation of AKT1, the mutation seen in Proteus syndrome. Sequencing of the tissue may be performed in the future, especially if segmental overgrowth continues and necessitates therapy.
- Wu Y, Zhou BP. Kinases meet at TSC. Cell Res. 2007;17:971-973.
- Roach SE, Sparagana SP. Diagnosis of tuberous sclerosis complex. J Child Neurol. 2004;19:643-649.
- Barker KT, Houlston RS. Overgrowth syndromes: is dysfunctional PI3-kinase signaling a unifying mechanism? Eur J Hum Genet. 2003;11:665-670.
- Nothrup H, Koenig MK, Au KS. Tuberous sclerosis complex. GeneReviews. Seattle, WA: University of Washington; 1999.
- Lindhurst MJ, Parker VE, Payne F, et al. Mosaic overgrowth with fibroadipose hyperplasia is caused by somatic activating mutations in PIK3CA. Nat Genet. 2012;44:928-933.
- Wu Y, Zhou BP. Kinases meet at TSC. Cell Res. 2007;17:971-973.
- Roach SE, Sparagana SP. Diagnosis of tuberous sclerosis complex. J Child Neurol. 2004;19:643-649.
- Barker KT, Houlston RS. Overgrowth syndromes: is dysfunctional PI3-kinase signaling a unifying mechanism? Eur J Hum Genet. 2003;11:665-670.
- Nothrup H, Koenig MK, Au KS. Tuberous sclerosis complex. GeneReviews. Seattle, WA: University of Washington; 1999.
- Lindhurst MJ, Parker VE, Payne F, et al. Mosaic overgrowth with fibroadipose hyperplasia is caused by somatic activating mutations in PIK3CA. Nat Genet. 2012;44:928-933.
Practice Points
- Tuberous sclerosis and Proteus syndrome share a common downstream effector pathway.
- For a patient to demonstrate features of both tuberous sclerosis and Proteus syndrome, he/she must have both a germline mutation (for tuberous sclerosis) as well as a postzygotic mutation (for Proteus syndrome) of this shared pathway.
Pruritic Eruption With Skinfold Sparing
The Diagnosis: Papuloerythroderma of Ofuji
The patient presented with a characteristic finding of skinfold sparing, known as the "deck-chair sign" (Figure 1).1 A repeat biopsy at our institution revealed a dermal perivascular and bandlike infiltrate with lymphocytes and occasional eosinophils (Figure 2). The epidermis showed mild spongiosis, lymphocytic exocytosis, and rare necrotic keratinocytes. A T-cell gene rearrangement assay was negative for a monoclonal population of T lymphocytes. Based on the clinical and histologic features, the diagnosis was most consistent with papuloerythroderma of Ofuji (PEO); however, a lymphoproliferative disorder needed to be excluded. Further workup included a peripheral smear, complete blood cell count with differential, comprehensive metabolic panel, IgE level, and hepatitis panel; all were normal, except for an elevated serum IgE level. Human immunodeficiency virus and age-appropriate malignancy screening were negative. The patient was prescribed betamethasone dipropionate cream 0.05% twice daily, which resulted in near-complete resolution of the rash and marked improvement in pruritus.
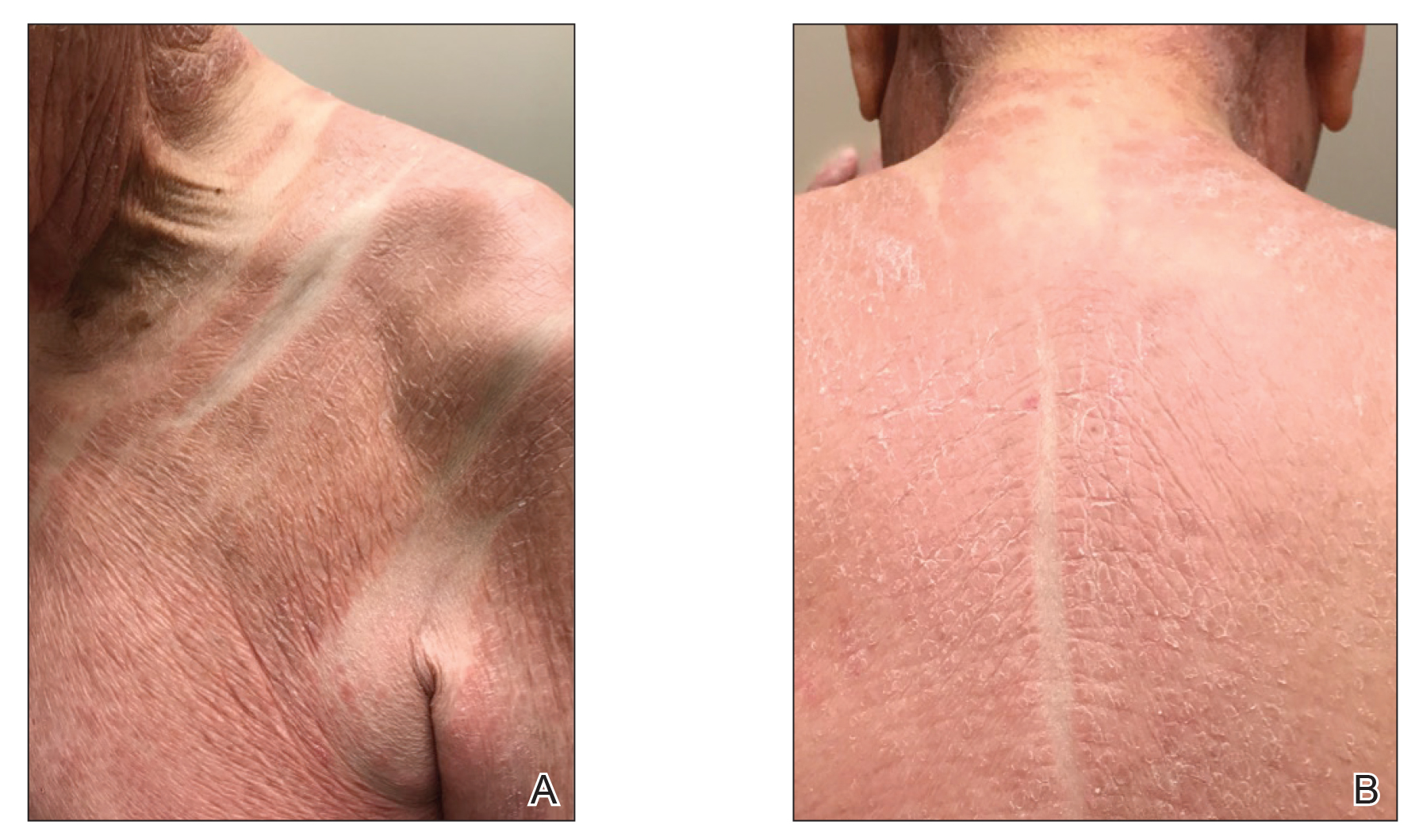
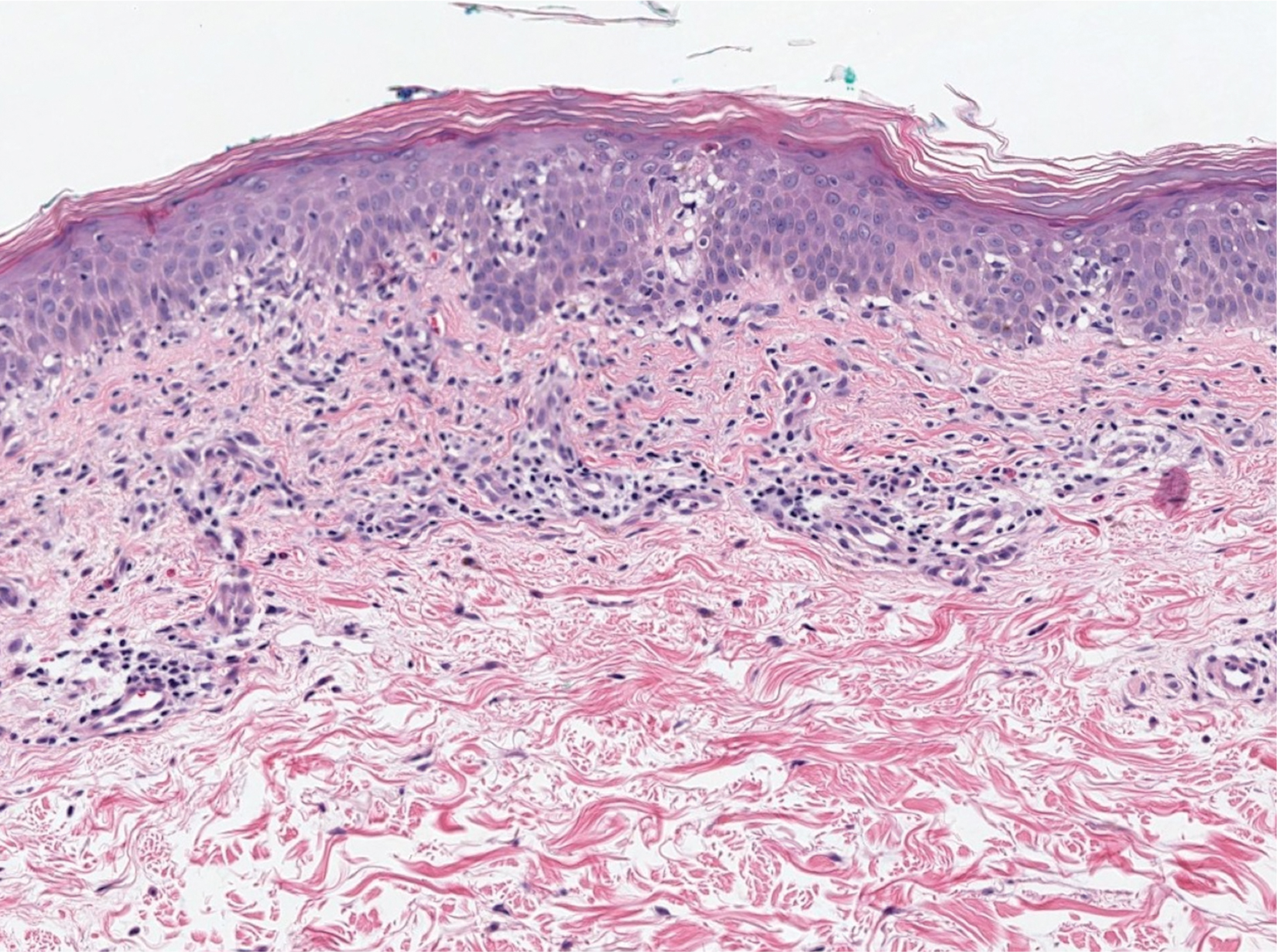
In 1984, PEO was described as an entity of generalized pruritic erythroderma characterized by flat-topped, red to brown, coalescing papules with sparing of the skinfolds, later coined the deck-chair sign.1,2 Papuloerythroderma of Ofuji commonly presents in elderly Asian males with a male to female ratio of 4:1.3 Papuloerythroderma of Ofuji is a T cell-mediated skin disease; however, the etiology of the signature rash remains unclear. One explanation includes circulating factors in the skin that elicit an inflammatory response, which does not occur in areas of external pressure.3 The deck-chair sign may occur more frequently in elderly individuals due to increased skin laxity, which creates crease lines that are spared from rubbing and excoriations.4
Although Ofuji et al2 initially reported 4 idiopathic cases, subsequent authors described PEO in association with other conditions, including cutaneous T-cell lymphoma (CTCL) and atopic diathesis, and infections, as well as secondary to medications. Some authors have suggested that PEO may be an early variant of mycosis fungoides; therefore, physicians should monitor patients closely.5-7 Maher et al6 commented that multiple causative factors including CTCL underlie the development of papuloerythroderma.
In a review of PEO, Torchia et al3 proposed diagnostic criteria and an etiologic classification to address whether PEO represents an independent entity or an unusual manifestation of other dermatoses. They established 4 categories of papuloerythroderma--primary, secondary, papuloerythrodermalike CTCL, and pseudopapuloerythroderma--and proposed that primary PEO is a diagnosis of exclusion. If no secondary association is found, they proposed 10 criteria for primary PEO: 5 major criteria include coalescing flat-topped papules, the deck-chair sign, pruritus, histopathologic exclusion of diseases such as CTCL, and a negative workup to exclude other causes.3 In 2018, Maher et al6 recommended workup to rule out cutaneous malignancy, including skin biopsy, flow cytometry, Sézary cell count, T-cell rearrangement, lactate dehydrogenase, and human T-cell lymphotropic virus 1. The 5 minor criteria proposed by Torchia et al3 include age older than 55 years, male sex, eosinophilia, elevated IgE level, and lymphopenia. Our patient fulfilled all 5 major criteria and 3 minor criteria; eosinophilia and lymphopenia were absent.
Clinically, PEO has been associated with the deck-chair sign, a pattern of selective sparing of skinfolds, including axillary, inguinal, submammary, and other flexural areas. Although the deck-chair sign was originally considered pathognomonic for PEO, this clinical pattern also has been observed in other entities, such as angioimmunoblastic lymphoma, cutaneous Waldenström macroglobulinemia, and acanthosis nigricans.5,8,9
Specific characteristics of the rash and certain clinical symptoms may help to distinguish the deck-chair sign of PEO from its other causes. Although malignant acanthosis nigricans may spare the skinfolds, lesions have a classic velvety thickening and brownish hyperpigmentation, which is not characteristic of the reddish brown, flat-topped papules of PEO.9 Pai et al5 described a patient with parthenium dermatitis presenting with the deck-chair sign that developed years after repeated exposure to the allergen. Our patient did not have a history of repeated episodes of allergic contact dermatitis. In addition, areas of sparing may mimic the appearance of pityriasis rubra pilaris. As in our patient, those with PEO generally lack the follicular hyperkeratotic papules, palmoplantar keratoderma, widespread orange-red erythema, and characteristic histopathologic finding of hyperkeratosis with alternating orthokeratosis and parakeratosis, allowing these entities to be easily distinguished in most instances.10
Histopathologically, primary PEO shows a nonspecific spongiotic dermatitis-like pattern characterized by slight epidermal hyperplasia with spongiosis and a predominantly perivascular dermal infiltrate with lymphocytes, histiocytes, and eosinophils.3 These histologic findings may at times show some overlap with CTCL, and therefore T-cell gene rearrangement and flow cytometry may be performed in those instances.6
Treatment includes the management of any underlying condition causing the papuloerythroderma.3,6 There are no large clinical trials of treatment options for primary PEO due to its rarity. Topical or systemic corticosteroids remain the mainstay of treatment.3 Alternative treatments used with variable success include phototherapy, interferon, etretinate, cyclosporine, and azathioprine.11 Allegue et al11 successfully used methotrexate to treat a patient with primary PEO and postulated that methotrexate may act through an immunosuppressive mechanism on activated T cells due to the involvement of helper T cells TH2 and TH22 in its pathogenesis.
Although the cutaneous manifestations of PEO may respond well to topical steroids, it is important to consider the possible presence of an underlying malignancy and other associated systemic conditions.
- Farthing CF, Staughton RC, Harper JI, et al. Papuloerythroderma--a further case with the 'deck chair sign.' Dermatologica. 1986;172:65-66.
- Ofuji S, Furukawa F, Miyachi Y, et al. Papuloerythroderma. Dermatologica. 1984;169:125-130.
- Torchia D, Miteva M, Hu S, et al. Papuloerythroderma 2009: two new cases and systematic review of the worldwide literature 25 years after its identification by Ofuji et al. Dermatology. 2010;220:311-320.
- Ochi H, Ang CC. Novel association of a papuloerythroderma of Ofuji phenotype with dermatitis herpetiformis. Int J Dermatol. 2018;57:856-857.
- Pai S, Shetty S, Rao R. Parthenium dermatitis with deck-chair sign. JAMA Dermatol. 2015;151:906-907.
- Maher AM, Ward CE, Glassman S, et al. The importance of excluding cutaneous T-cell lymphomas in patients with a working diagnosis of papuloerythroderma of Ofuji: a case series. Case Rep Dermatol. 2018;10:46-54.
- Grob JJ, Collet-Villette AM, Horchowski N, et al. Ofuji papuloerythroderma. report of a case with T cell skin lymphoma and discussion of the nature of this disease. J Am Acad Dermatol. 1989;20(5 pt 2):927-931.
- Ferran M, Gallardo F, Baena V, et al. The 'deck chair sign' in specific cutaneous involvement by angioimmunoblastic T cell lymphoma. Dermatology. 2006;213:50-52.
- Murao K, Sadamoto Y, Kubo Y, et al. Generalized malignant acanthosis nigricans with "deck-chair sign." Int J Dermatol. 2013;52:377-378.
- Regina G, Paramita L, Radiono S, et al. Papuloerythroderma of Ofuji in Indonesia: the first case report. JDVI. 2016;1:93-98.
- Allegue F, Fachal C, Gonzalez-Vilas D, et al. Papuloerythroderma of Ofuji successfully treated with methotrexate. Dermatol Ther. 2018;31:E12638.
The Diagnosis: Papuloerythroderma of Ofuji
The patient presented with a characteristic finding of skinfold sparing, known as the "deck-chair sign" (Figure 1).1 A repeat biopsy at our institution revealed a dermal perivascular and bandlike infiltrate with lymphocytes and occasional eosinophils (Figure 2). The epidermis showed mild spongiosis, lymphocytic exocytosis, and rare necrotic keratinocytes. A T-cell gene rearrangement assay was negative for a monoclonal population of T lymphocytes. Based on the clinical and histologic features, the diagnosis was most consistent with papuloerythroderma of Ofuji (PEO); however, a lymphoproliferative disorder needed to be excluded. Further workup included a peripheral smear, complete blood cell count with differential, comprehensive metabolic panel, IgE level, and hepatitis panel; all were normal, except for an elevated serum IgE level. Human immunodeficiency virus and age-appropriate malignancy screening were negative. The patient was prescribed betamethasone dipropionate cream 0.05% twice daily, which resulted in near-complete resolution of the rash and marked improvement in pruritus.
In 1984, PEO was described as an entity of generalized pruritic erythroderma characterized by flat-topped, red to brown, coalescing papules with sparing of the skinfolds, later coined the deck-chair sign.1,2 Papuloerythroderma of Ofuji commonly presents in elderly Asian males with a male to female ratio of 4:1.3 Papuloerythroderma of Ofuji is a T cell-mediated skin disease; however, the etiology of the signature rash remains unclear. One explanation includes circulating factors in the skin that elicit an inflammatory response, which does not occur in areas of external pressure.3 The deck-chair sign may occur more frequently in elderly individuals due to increased skin laxity, which creates crease lines that are spared from rubbing and excoriations.4
Although Ofuji et al2 initially reported 4 idiopathic cases, subsequent authors described PEO in association with other conditions, including cutaneous T-cell lymphoma (CTCL) and atopic diathesis, and infections, as well as secondary to medications. Some authors have suggested that PEO may be an early variant of mycosis fungoides; therefore, physicians should monitor patients closely.5-7 Maher et al6 commented that multiple causative factors including CTCL underlie the development of papuloerythroderma.
In a review of PEO, Torchia et al3 proposed diagnostic criteria and an etiologic classification to address whether PEO represents an independent entity or an unusual manifestation of other dermatoses. They established 4 categories of papuloerythroderma--primary, secondary, papuloerythrodermalike CTCL, and pseudopapuloerythroderma--and proposed that primary PEO is a diagnosis of exclusion. If no secondary association is found, they proposed 10 criteria for primary PEO: 5 major criteria include coalescing flat-topped papules, the deck-chair sign, pruritus, histopathologic exclusion of diseases such as CTCL, and a negative workup to exclude other causes.3 In 2018, Maher et al6 recommended workup to rule out cutaneous malignancy, including skin biopsy, flow cytometry, Sézary cell count, T-cell rearrangement, lactate dehydrogenase, and human T-cell lymphotropic virus 1. The 5 minor criteria proposed by Torchia et al3 include age older than 55 years, male sex, eosinophilia, elevated IgE level, and lymphopenia. Our patient fulfilled all 5 major criteria and 3 minor criteria; eosinophilia and lymphopenia were absent.
Clinically, PEO has been associated with the deck-chair sign, a pattern of selective sparing of skinfolds, including axillary, inguinal, submammary, and other flexural areas. Although the deck-chair sign was originally considered pathognomonic for PEO, this clinical pattern also has been observed in other entities, such as angioimmunoblastic lymphoma, cutaneous Waldenström macroglobulinemia, and acanthosis nigricans.5,8,9
Specific characteristics of the rash and certain clinical symptoms may help to distinguish the deck-chair sign of PEO from its other causes. Although malignant acanthosis nigricans may spare the skinfolds, lesions have a classic velvety thickening and brownish hyperpigmentation, which is not characteristic of the reddish brown, flat-topped papules of PEO.9 Pai et al5 described a patient with parthenium dermatitis presenting with the deck-chair sign that developed years after repeated exposure to the allergen. Our patient did not have a history of repeated episodes of allergic contact dermatitis. In addition, areas of sparing may mimic the appearance of pityriasis rubra pilaris. As in our patient, those with PEO generally lack the follicular hyperkeratotic papules, palmoplantar keratoderma, widespread orange-red erythema, and characteristic histopathologic finding of hyperkeratosis with alternating orthokeratosis and parakeratosis, allowing these entities to be easily distinguished in most instances.10
Histopathologically, primary PEO shows a nonspecific spongiotic dermatitis-like pattern characterized by slight epidermal hyperplasia with spongiosis and a predominantly perivascular dermal infiltrate with lymphocytes, histiocytes, and eosinophils.3 These histologic findings may at times show some overlap with CTCL, and therefore T-cell gene rearrangement and flow cytometry may be performed in those instances.6
Treatment includes the management of any underlying condition causing the papuloerythroderma.3,6 There are no large clinical trials of treatment options for primary PEO due to its rarity. Topical or systemic corticosteroids remain the mainstay of treatment.3 Alternative treatments used with variable success include phototherapy, interferon, etretinate, cyclosporine, and azathioprine.11 Allegue et al11 successfully used methotrexate to treat a patient with primary PEO and postulated that methotrexate may act through an immunosuppressive mechanism on activated T cells due to the involvement of helper T cells TH2 and TH22 in its pathogenesis.
Although the cutaneous manifestations of PEO may respond well to topical steroids, it is important to consider the possible presence of an underlying malignancy and other associated systemic conditions.
The Diagnosis: Papuloerythroderma of Ofuji
The patient presented with a characteristic finding of skinfold sparing, known as the "deck-chair sign" (Figure 1).1 A repeat biopsy at our institution revealed a dermal perivascular and bandlike infiltrate with lymphocytes and occasional eosinophils (Figure 2). The epidermis showed mild spongiosis, lymphocytic exocytosis, and rare necrotic keratinocytes. A T-cell gene rearrangement assay was negative for a monoclonal population of T lymphocytes. Based on the clinical and histologic features, the diagnosis was most consistent with papuloerythroderma of Ofuji (PEO); however, a lymphoproliferative disorder needed to be excluded. Further workup included a peripheral smear, complete blood cell count with differential, comprehensive metabolic panel, IgE level, and hepatitis panel; all were normal, except for an elevated serum IgE level. Human immunodeficiency virus and age-appropriate malignancy screening were negative. The patient was prescribed betamethasone dipropionate cream 0.05% twice daily, which resulted in near-complete resolution of the rash and marked improvement in pruritus.
In 1984, PEO was described as an entity of generalized pruritic erythroderma characterized by flat-topped, red to brown, coalescing papules with sparing of the skinfolds, later coined the deck-chair sign.1,2 Papuloerythroderma of Ofuji commonly presents in elderly Asian males with a male to female ratio of 4:1.3 Papuloerythroderma of Ofuji is a T cell-mediated skin disease; however, the etiology of the signature rash remains unclear. One explanation includes circulating factors in the skin that elicit an inflammatory response, which does not occur in areas of external pressure.3 The deck-chair sign may occur more frequently in elderly individuals due to increased skin laxity, which creates crease lines that are spared from rubbing and excoriations.4
Although Ofuji et al2 initially reported 4 idiopathic cases, subsequent authors described PEO in association with other conditions, including cutaneous T-cell lymphoma (CTCL) and atopic diathesis, and infections, as well as secondary to medications. Some authors have suggested that PEO may be an early variant of mycosis fungoides; therefore, physicians should monitor patients closely.5-7 Maher et al6 commented that multiple causative factors including CTCL underlie the development of papuloerythroderma.
In a review of PEO, Torchia et al3 proposed diagnostic criteria and an etiologic classification to address whether PEO represents an independent entity or an unusual manifestation of other dermatoses. They established 4 categories of papuloerythroderma--primary, secondary, papuloerythrodermalike CTCL, and pseudopapuloerythroderma--and proposed that primary PEO is a diagnosis of exclusion. If no secondary association is found, they proposed 10 criteria for primary PEO: 5 major criteria include coalescing flat-topped papules, the deck-chair sign, pruritus, histopathologic exclusion of diseases such as CTCL, and a negative workup to exclude other causes.3 In 2018, Maher et al6 recommended workup to rule out cutaneous malignancy, including skin biopsy, flow cytometry, Sézary cell count, T-cell rearrangement, lactate dehydrogenase, and human T-cell lymphotropic virus 1. The 5 minor criteria proposed by Torchia et al3 include age older than 55 years, male sex, eosinophilia, elevated IgE level, and lymphopenia. Our patient fulfilled all 5 major criteria and 3 minor criteria; eosinophilia and lymphopenia were absent.
Clinically, PEO has been associated with the deck-chair sign, a pattern of selective sparing of skinfolds, including axillary, inguinal, submammary, and other flexural areas. Although the deck-chair sign was originally considered pathognomonic for PEO, this clinical pattern also has been observed in other entities, such as angioimmunoblastic lymphoma, cutaneous Waldenström macroglobulinemia, and acanthosis nigricans.5,8,9
Specific characteristics of the rash and certain clinical symptoms may help to distinguish the deck-chair sign of PEO from its other causes. Although malignant acanthosis nigricans may spare the skinfolds, lesions have a classic velvety thickening and brownish hyperpigmentation, which is not characteristic of the reddish brown, flat-topped papules of PEO.9 Pai et al5 described a patient with parthenium dermatitis presenting with the deck-chair sign that developed years after repeated exposure to the allergen. Our patient did not have a history of repeated episodes of allergic contact dermatitis. In addition, areas of sparing may mimic the appearance of pityriasis rubra pilaris. As in our patient, those with PEO generally lack the follicular hyperkeratotic papules, palmoplantar keratoderma, widespread orange-red erythema, and characteristic histopathologic finding of hyperkeratosis with alternating orthokeratosis and parakeratosis, allowing these entities to be easily distinguished in most instances.10
Histopathologically, primary PEO shows a nonspecific spongiotic dermatitis-like pattern characterized by slight epidermal hyperplasia with spongiosis and a predominantly perivascular dermal infiltrate with lymphocytes, histiocytes, and eosinophils.3 These histologic findings may at times show some overlap with CTCL, and therefore T-cell gene rearrangement and flow cytometry may be performed in those instances.6
Treatment includes the management of any underlying condition causing the papuloerythroderma.3,6 There are no large clinical trials of treatment options for primary PEO due to its rarity. Topical or systemic corticosteroids remain the mainstay of treatment.3 Alternative treatments used with variable success include phototherapy, interferon, etretinate, cyclosporine, and azathioprine.11 Allegue et al11 successfully used methotrexate to treat a patient with primary PEO and postulated that methotrexate may act through an immunosuppressive mechanism on activated T cells due to the involvement of helper T cells TH2 and TH22 in its pathogenesis.
Although the cutaneous manifestations of PEO may respond well to topical steroids, it is important to consider the possible presence of an underlying malignancy and other associated systemic conditions.
- Farthing CF, Staughton RC, Harper JI, et al. Papuloerythroderma--a further case with the 'deck chair sign.' Dermatologica. 1986;172:65-66.
- Ofuji S, Furukawa F, Miyachi Y, et al. Papuloerythroderma. Dermatologica. 1984;169:125-130.
- Torchia D, Miteva M, Hu S, et al. Papuloerythroderma 2009: two new cases and systematic review of the worldwide literature 25 years after its identification by Ofuji et al. Dermatology. 2010;220:311-320.
- Ochi H, Ang CC. Novel association of a papuloerythroderma of Ofuji phenotype with dermatitis herpetiformis. Int J Dermatol. 2018;57:856-857.
- Pai S, Shetty S, Rao R. Parthenium dermatitis with deck-chair sign. JAMA Dermatol. 2015;151:906-907.
- Maher AM, Ward CE, Glassman S, et al. The importance of excluding cutaneous T-cell lymphomas in patients with a working diagnosis of papuloerythroderma of Ofuji: a case series. Case Rep Dermatol. 2018;10:46-54.
- Grob JJ, Collet-Villette AM, Horchowski N, et al. Ofuji papuloerythroderma. report of a case with T cell skin lymphoma and discussion of the nature of this disease. J Am Acad Dermatol. 1989;20(5 pt 2):927-931.
- Ferran M, Gallardo F, Baena V, et al. The 'deck chair sign' in specific cutaneous involvement by angioimmunoblastic T cell lymphoma. Dermatology. 2006;213:50-52.
- Murao K, Sadamoto Y, Kubo Y, et al. Generalized malignant acanthosis nigricans with "deck-chair sign." Int J Dermatol. 2013;52:377-378.
- Regina G, Paramita L, Radiono S, et al. Papuloerythroderma of Ofuji in Indonesia: the first case report. JDVI. 2016;1:93-98.
- Allegue F, Fachal C, Gonzalez-Vilas D, et al. Papuloerythroderma of Ofuji successfully treated with methotrexate. Dermatol Ther. 2018;31:E12638.
- Farthing CF, Staughton RC, Harper JI, et al. Papuloerythroderma--a further case with the 'deck chair sign.' Dermatologica. 1986;172:65-66.
- Ofuji S, Furukawa F, Miyachi Y, et al. Papuloerythroderma. Dermatologica. 1984;169:125-130.
- Torchia D, Miteva M, Hu S, et al. Papuloerythroderma 2009: two new cases and systematic review of the worldwide literature 25 years after its identification by Ofuji et al. Dermatology. 2010;220:311-320.
- Ochi H, Ang CC. Novel association of a papuloerythroderma of Ofuji phenotype with dermatitis herpetiformis. Int J Dermatol. 2018;57:856-857.
- Pai S, Shetty S, Rao R. Parthenium dermatitis with deck-chair sign. JAMA Dermatol. 2015;151:906-907.
- Maher AM, Ward CE, Glassman S, et al. The importance of excluding cutaneous T-cell lymphomas in patients with a working diagnosis of papuloerythroderma of Ofuji: a case series. Case Rep Dermatol. 2018;10:46-54.
- Grob JJ, Collet-Villette AM, Horchowski N, et al. Ofuji papuloerythroderma. report of a case with T cell skin lymphoma and discussion of the nature of this disease. J Am Acad Dermatol. 1989;20(5 pt 2):927-931.
- Ferran M, Gallardo F, Baena V, et al. The 'deck chair sign' in specific cutaneous involvement by angioimmunoblastic T cell lymphoma. Dermatology. 2006;213:50-52.
- Murao K, Sadamoto Y, Kubo Y, et al. Generalized malignant acanthosis nigricans with "deck-chair sign." Int J Dermatol. 2013;52:377-378.
- Regina G, Paramita L, Radiono S, et al. Papuloerythroderma of Ofuji in Indonesia: the first case report. JDVI. 2016;1:93-98.
- Allegue F, Fachal C, Gonzalez-Vilas D, et al. Papuloerythroderma of Ofuji successfully treated with methotrexate. Dermatol Ther. 2018;31:E12638.
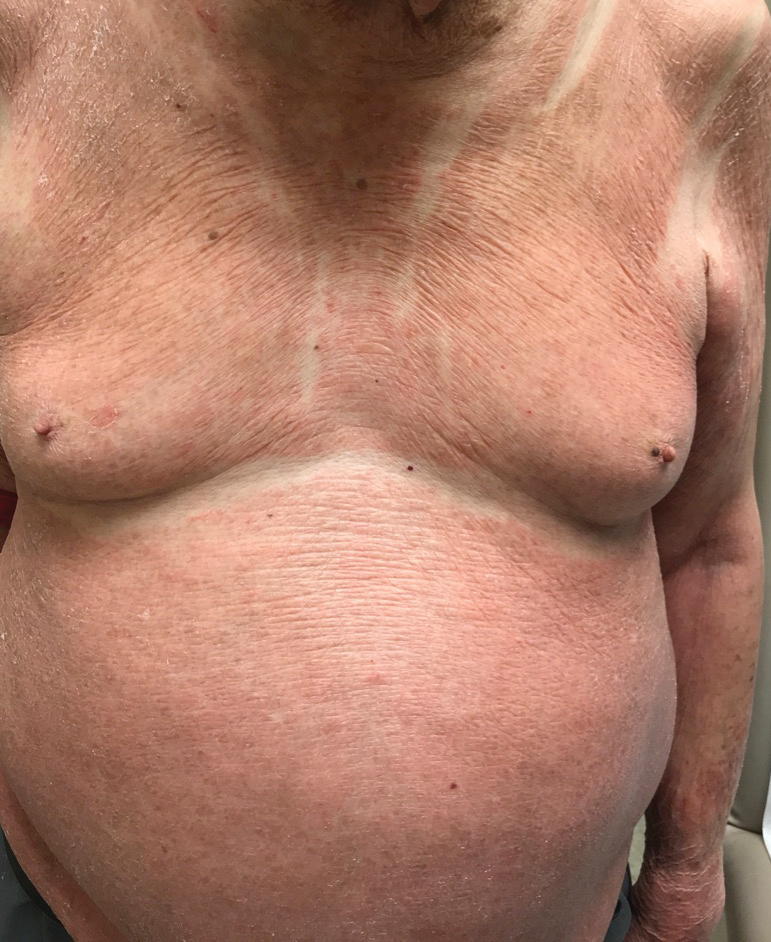
An 89-year-old Asian man presented with a generalized pruritic eruption of 2 months' duration. The rash started on the flanks and later spread to the arms and legs, abdomen, and back; the face and palms were spared. Physical examination revealed numerous erythematous papules coalescing into large scaly plaques on the trunk, arms, and legs. There were noticeable areas of sparing of the skinfolds, especially the axillary, inframammary, and inguinal folds, as well as the midline of the back. A biopsy performed by an outside physician showed findings consistent with a possible pityriasiform drug eruption; however, there were no recent changes in medication history.
Edema Affecting the Penis and Scrotum
The Diagnosis: Cutaneous Crohn Disease
Crohn disease (CD) is an inflammatory bowel disease that can involve any region of the gastrointestinal (GI) tract from the mouth to the anus but most commonly presents in the terminal ileum, colon, or small bowel with transmural inflammation, fistula formation, and knife-cut fissures among the frequently described findings. Extraintestinal manifestations may be found in the liver, eyes, and joints, with cutaneous extraintestinal manifestations occurring in up to one-third of patients.1
Crohn disease can be associated with multiple cutaneous findings, including erythema nodosum, pyoderma gangrenosum, aphthous ulcers, pyodermatitis-pyostomatitis vegetans, necrotizing vasculitis, and metastatic Crohn disease (MCD).2 Typical histopathologic findings seen in MCD such as noncaseating granulomatous inflammation in the papillary and reticular dermis, possibly extending to the subcutaneous fat, are not specific to MCD. Associated genital edema is thought to be a consequence of granulomatous inflammation of lymphatics. In one study reviewing specimens from 10 cases of CD, a mean of 46% of all granulomas identified on the slides (264 granulomas in total) were located proximal to lymphatic vessels, suggesting a common pathway for development of intestinal disease and genital edema.3 The differential diagnosis for penile and scrotal swelling is broad, and the diagnosis may be missed if attention is not given to the clinical history of the patient in addition to histopathologic findings.2
Skin changes in CD also can be separated into perianal disease and true metastatic disease--the former recognized when anal lesions appear associated with segmental involvement of the GI tract and the latter as ulceration of the skin separated from the GI tract by normal tissue.1 The term sarcoidal reaction often is used to describe histopathologic findings in cutaneous CD, as it refers to the noncaseating granulomas found in approximately 60% of all cases.4 Ultimately, the location of noncaseating granulomas within the dermis of our patient's biopsy, taken in conjunction with the clinical history and the lack of defining features for other potential etiologies (eg, polarizable material, organisms on special stains), led to the diagnosis of cutaneous CD.
Cutaneous manifestations of sarcoidosis most commonly occur as papules, plaques, and subcutaneous nodules predominantly on the face, upper back, arms, and legs. Although the histologic features of sarcoidosis are characterized by lymphocyte-poor noncaseating granulomas (Figure 1), these findings also can be seen as a consequence of multiple granulomatous causes.5,6 In a review of 48 cutaneous specimens from patients with sarcoidosis, the granulomas were found most frequently in the deep dermis (34/48 [70.8%]), with superficial dermis (21/48) and subcutaneous fat granulomas (20/48) each present in less than 50% of biopsies.5 Although less typical, cutaneous sarcoidosis also has been noted in the literature to present in the perianal and gluteal region, demonstrating dermal noncaseating granulomas on biopsy.7 One distinction in particular to be noted between sarcoid and CD is that sarcoid lesions in the skin rarely ulcerate, while the lesions of cutaneous CD often are ulcerated.4,6
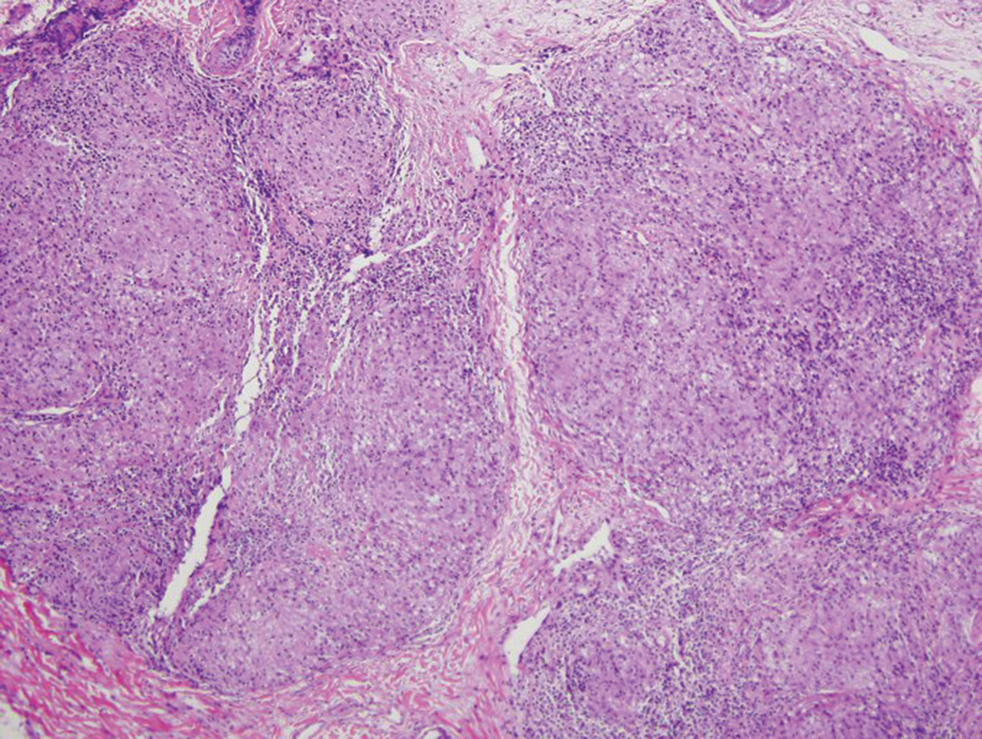
Lesions including abscesses in the groin may raise concern for hidradenitis suppurativa (HS), a disease of the apocrine gland-bearing skin. Typical lesions are tender subcutaneous erythematous nodules, cysts, and comedones that develop rapidly and may rupture to drain suppurative bloody discharge, subsequently healing with an atrophic scar.8 More persistent inflammation and rupture of nodules into the dermis may lead to formation of dermal tunnels with palpable cords and sinus tracts.8 Typical areas of disease involvement are in the axillae, inframammary folds, groin, or perigenital or perineal regions, with the diagnosis made on a combination of lesion morphology, location, and progression/recurrence frequency.9 Histologic examination of HS specimens can demonstrate a perifollicular lymphocytic infiltrate, with more advanced disease characterized by increased inflammatory cells, predominantly neutrophils, monocytes, and mast cells (Figure 2). The presence of granulomas in HS most often is of the foreign body type.9 Epithelioid granulomas noted in an area separate from inflammation in a patient with HS serve as a clue to be alert for systemic granulomatous disease.10
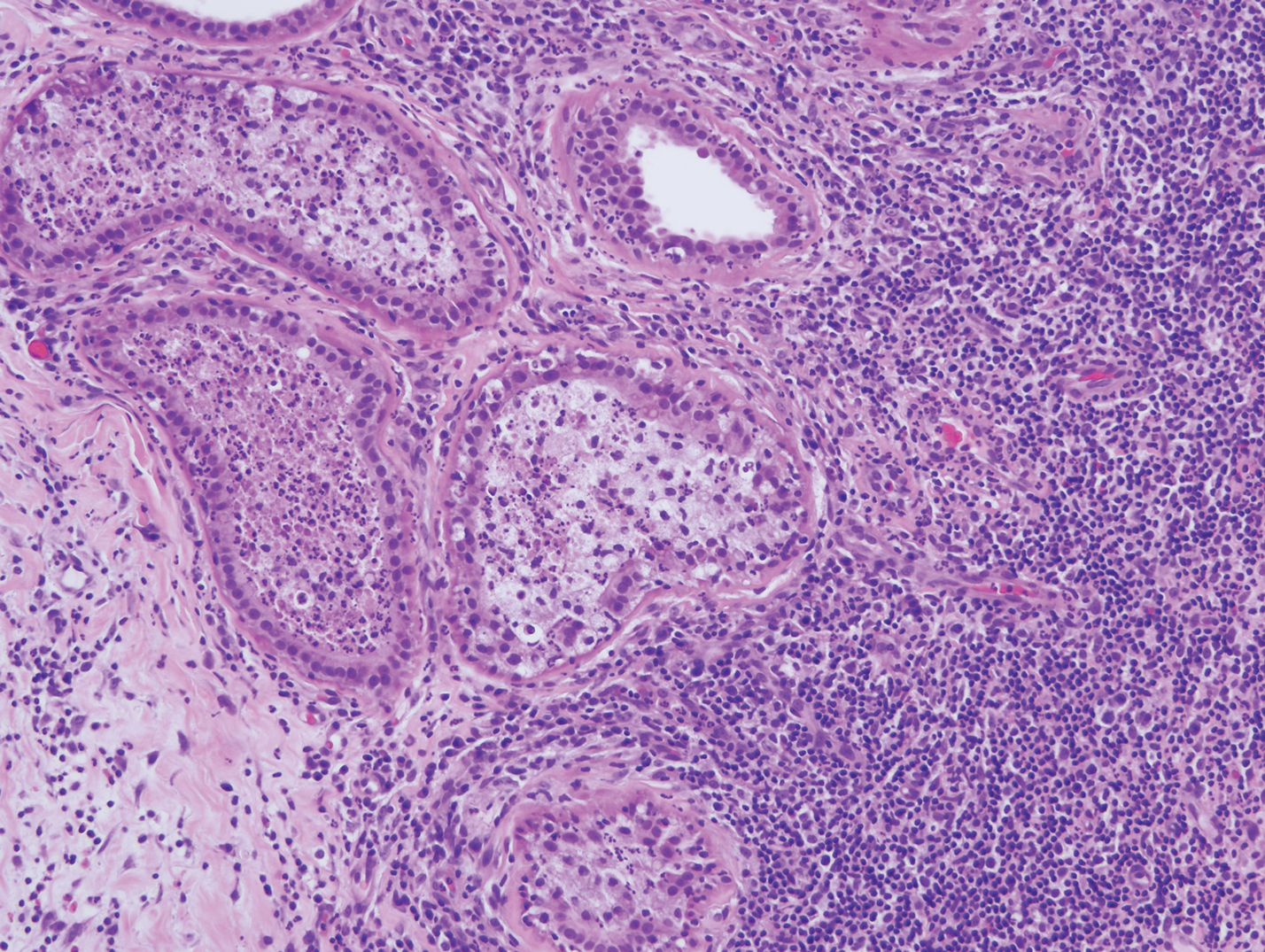
Mycosis fungoides is the most common primary cutaneous lymphoma to show a granulomatous infiltrate; the granuloma generally is sarcoidal, though other forms are described (Figure 3).11 Beyond these granulomatous foci, the key histopathologic feature of granulomatous mycosis fungoides (GMF) is diffuse dermal infiltration by atypical lymphoid cells. Epidermotropism and sparing of dermal nerves is the most critical finding in the diagnosis of GMF, especially in geographic regions where leprosy is endemic and high on the differential, as the conditions have histopathologic similarities.11,12 At the same time, lack of epidermotropism does not exclude the diagnosis of GMF.13 Clinically, GMF presentation is variable, but common findings include erythematous and hyperpigmented patches and plaques. Given the lack of clear clinical criteria, the diagnosis relies primarily on histopathologic features.11
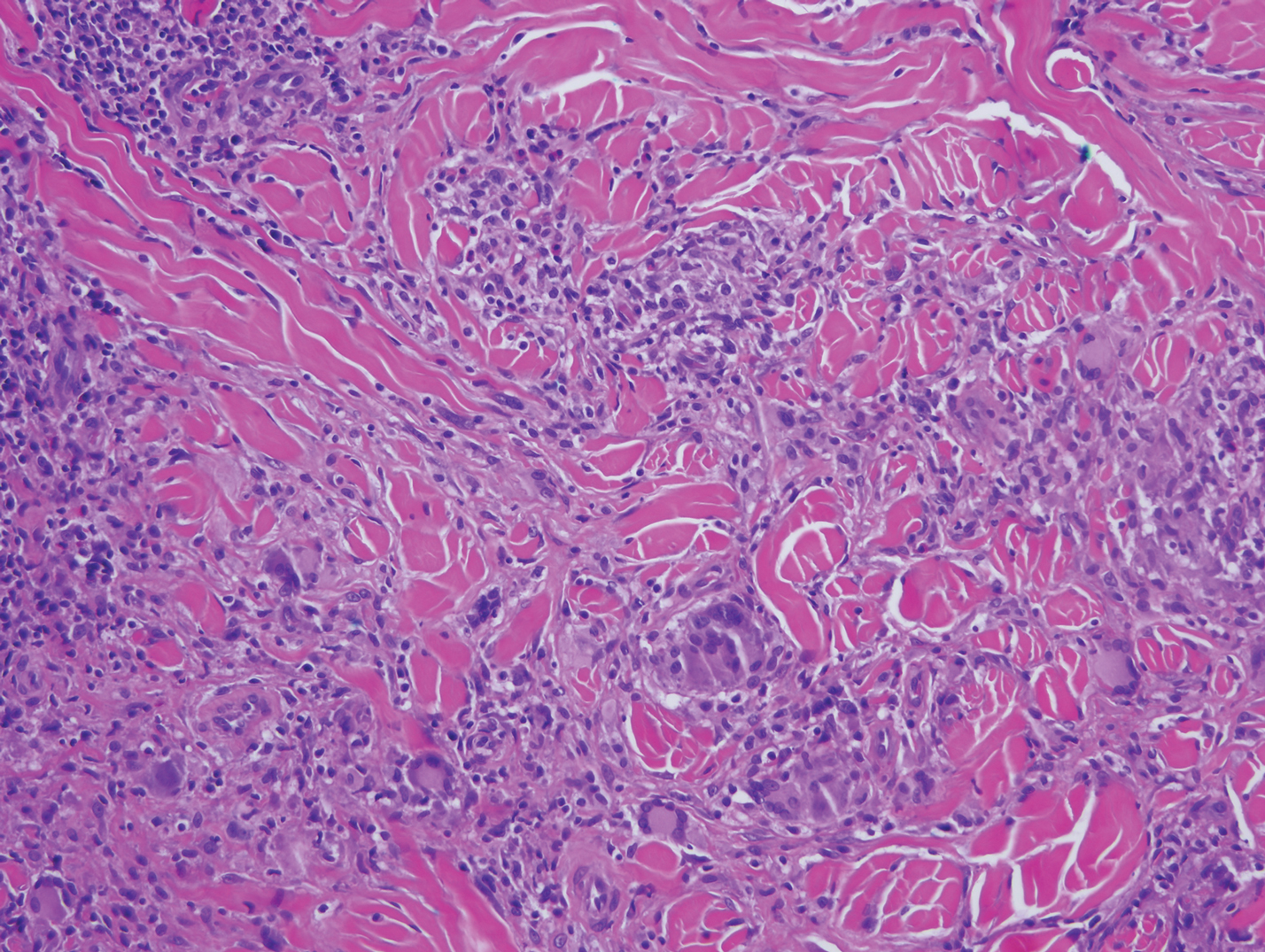
Mycobacterial skin and soft tissue infections may be attributed to both tuberculous and nontuberculous strains (atypical species).14 Clinical features range from small papules to large deformative plaques and ulcers.15 Histologic features also distinguish cutaneous tuberculosis (TB) from nontuberculous mycobacterial causes. Cutaneous TB shows caseous granulomas in the upper and mid dermis, while nontuberculous mycobacterial infections have more prominent neutrophil infiltration and interstitial granulomas (Figure 4).16
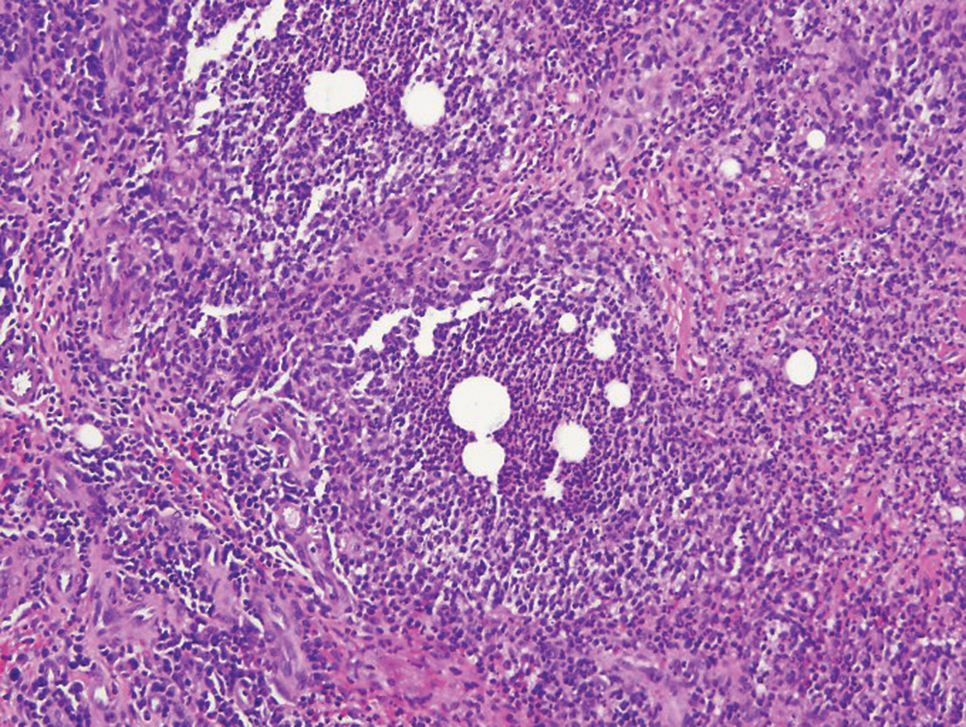
In cutaneous TB specifically, extrapulmonary manifestations may involve the skin in 1% to 1.5% of all TB cases, and although rare, ulcerative skin TB has been noted in one report as a nonhealing perianal ulcer that showed necrotizing granulomas on biopsy.17 Ultimately, diagnosis of cutaneous mycobacterial infection is confirmed with detection of acid-fast bacilli in the biopsy specimen.16
Diagnosis of cutaneous CD requires clinicopathologic correlation, as the clinical and histopathologic differential diagnoses of genital edema and noncaseating granulomas, respectively, are broad. Even though the clinical context was appropriate for cutaneous CD in this case, correct diagnosis required confirmatory histologic findings. Furthermore, taking multiple biopsies is prudent. In our patient, diagnostic findings only were present in the biopsy from the scrotum.
- Hagen JW, Swoger JM, Grandinetti LM. Cutaneous manifestations of Crohn disease. Dermatol Clin. 2015;33:417-431.
- Barrick BJ, Tollefson MM, Schoch JJ, et al. Penile and scrotal swelling: an underrecognized presentation of Crohn's disease. Pediatr Dermatol. 2016;33:172-177.
- Mooney EE, Walker J, Hourihane DO. Relation of granulomas to lymphatic vessels in Crohn's disease. J Clin Pathol. 1995;48:335-338.
- Parks AG, Morson BC, Pegum JS. Crohn's disease with cutaneous involvement. Proc R Soc Med. 1965;58:241-242.
- García-Colmenero L, Sánchez-Schmidt JM, Barranco C, et al. The natural history of cutaneous sarcoidosis: clinical spectrum and histological analysis of 40 cases [published online October 18, 2018]. Int J Dermatol. 2019;58:178-184.
- Yoo SS, Mimouni D, Nikolskaia OV, et al. Clinicopathologic features of ulcerative-atrophic sarcoidosis. Int J Dermatol. 2004;43:108-112.
- Cohen GF, Wolfe CM. Recalcitrant diffuse cutaneous sarcoidosis with perianal involvement responding to adalimumab. J Drugs Dermatol. 2017;16:1305-1306.
- Hoffman LK, Ghias MH, Lowes MA. Pathophysiology of hidradenitis suppurativa. Semin Cutan Med Surg. 2017;36:47-54.
- Saunte DML, Jemec GBE. Hidradenitis suppurativa: advances in diagnosis and treatment. JAMA. 2017;318:2019-2032.
- Attanoos RL, Appleton MA, Hughes LE, et al. Granulomatous hidradenitis suppurativa and cutaneous Crohn's disease. Histopathology. 1993;23:111-115.
- Gutte R, Kharkar V, Mahajan S, et al. Granulomatous mycosis fungoides with hypohydrosis mimicking lepromatous leprosy. Indian J Dermatol Venerol Leprol. 2010;76:686-690.
- Pousa CM, Nery NS, Mann D, et al. Granulomatous mycosis fungoides--a diagnostic challenge. An Bras Dermatol. 2015;90:554-556.
- Kempf W, Ostheeren-Michaelis S, Paulli M, et al. Granulomatous mycosis fungoides and granulomatous slack skin: a multicenter study of the Cutaneous Lymphoma Histopathology Task Force Group of the European Organization for Research and Treatment of Cancer (EORTC). Arch Dermatol. 2008;144:1609-1617.
- van Mechelen M, van der Hilst J, Gyssens IC, et al. Mycobacterial skin and soft tissue infections: TB or not TB? Neth J Med. 2018;76:269-274.
- van Zyl L, du Plessis J, Viljoen J. Cutaneous tuberculosis overview and current treatment regimens. Tuberculosis (Edinb). 2015;95:629-638.
- De Maio F, Trecarichi EM, Visconti E, et al. Understanding cutaneous tuberculosis: two clinical cases. JMM Case Rep. 2016;3:E005070.
- Wu S, Wang W, Chen H, et al. Perianal ulcerative skin tuberculosis: a case report. Medicine (Baltimore). 2018;97:E10836.
The Diagnosis: Cutaneous Crohn Disease
Crohn disease (CD) is an inflammatory bowel disease that can involve any region of the gastrointestinal (GI) tract from the mouth to the anus but most commonly presents in the terminal ileum, colon, or small bowel with transmural inflammation, fistula formation, and knife-cut fissures among the frequently described findings. Extraintestinal manifestations may be found in the liver, eyes, and joints, with cutaneous extraintestinal manifestations occurring in up to one-third of patients.1
Crohn disease can be associated with multiple cutaneous findings, including erythema nodosum, pyoderma gangrenosum, aphthous ulcers, pyodermatitis-pyostomatitis vegetans, necrotizing vasculitis, and metastatic Crohn disease (MCD).2 Typical histopathologic findings seen in MCD such as noncaseating granulomatous inflammation in the papillary and reticular dermis, possibly extending to the subcutaneous fat, are not specific to MCD. Associated genital edema is thought to be a consequence of granulomatous inflammation of lymphatics. In one study reviewing specimens from 10 cases of CD, a mean of 46% of all granulomas identified on the slides (264 granulomas in total) were located proximal to lymphatic vessels, suggesting a common pathway for development of intestinal disease and genital edema.3 The differential diagnosis for penile and scrotal swelling is broad, and the diagnosis may be missed if attention is not given to the clinical history of the patient in addition to histopathologic findings.2
Skin changes in CD also can be separated into perianal disease and true metastatic disease--the former recognized when anal lesions appear associated with segmental involvement of the GI tract and the latter as ulceration of the skin separated from the GI tract by normal tissue.1 The term sarcoidal reaction often is used to describe histopathologic findings in cutaneous CD, as it refers to the noncaseating granulomas found in approximately 60% of all cases.4 Ultimately, the location of noncaseating granulomas within the dermis of our patient's biopsy, taken in conjunction with the clinical history and the lack of defining features for other potential etiologies (eg, polarizable material, organisms on special stains), led to the diagnosis of cutaneous CD.
Cutaneous manifestations of sarcoidosis most commonly occur as papules, plaques, and subcutaneous nodules predominantly on the face, upper back, arms, and legs. Although the histologic features of sarcoidosis are characterized by lymphocyte-poor noncaseating granulomas (Figure 1), these findings also can be seen as a consequence of multiple granulomatous causes.5,6 In a review of 48 cutaneous specimens from patients with sarcoidosis, the granulomas were found most frequently in the deep dermis (34/48 [70.8%]), with superficial dermis (21/48) and subcutaneous fat granulomas (20/48) each present in less than 50% of biopsies.5 Although less typical, cutaneous sarcoidosis also has been noted in the literature to present in the perianal and gluteal region, demonstrating dermal noncaseating granulomas on biopsy.7 One distinction in particular to be noted between sarcoid and CD is that sarcoid lesions in the skin rarely ulcerate, while the lesions of cutaneous CD often are ulcerated.4,6
Lesions including abscesses in the groin may raise concern for hidradenitis suppurativa (HS), a disease of the apocrine gland-bearing skin. Typical lesions are tender subcutaneous erythematous nodules, cysts, and comedones that develop rapidly and may rupture to drain suppurative bloody discharge, subsequently healing with an atrophic scar.8 More persistent inflammation and rupture of nodules into the dermis may lead to formation of dermal tunnels with palpable cords and sinus tracts.8 Typical areas of disease involvement are in the axillae, inframammary folds, groin, or perigenital or perineal regions, with the diagnosis made on a combination of lesion morphology, location, and progression/recurrence frequency.9 Histologic examination of HS specimens can demonstrate a perifollicular lymphocytic infiltrate, with more advanced disease characterized by increased inflammatory cells, predominantly neutrophils, monocytes, and mast cells (Figure 2). The presence of granulomas in HS most often is of the foreign body type.9 Epithelioid granulomas noted in an area separate from inflammation in a patient with HS serve as a clue to be alert for systemic granulomatous disease.10
Mycosis fungoides is the most common primary cutaneous lymphoma to show a granulomatous infiltrate; the granuloma generally is sarcoidal, though other forms are described (Figure 3).11 Beyond these granulomatous foci, the key histopathologic feature of granulomatous mycosis fungoides (GMF) is diffuse dermal infiltration by atypical lymphoid cells. Epidermotropism and sparing of dermal nerves is the most critical finding in the diagnosis of GMF, especially in geographic regions where leprosy is endemic and high on the differential, as the conditions have histopathologic similarities.11,12 At the same time, lack of epidermotropism does not exclude the diagnosis of GMF.13 Clinically, GMF presentation is variable, but common findings include erythematous and hyperpigmented patches and plaques. Given the lack of clear clinical criteria, the diagnosis relies primarily on histopathologic features.11
Mycobacterial skin and soft tissue infections may be attributed to both tuberculous and nontuberculous strains (atypical species).14 Clinical features range from small papules to large deformative plaques and ulcers.15 Histologic features also distinguish cutaneous tuberculosis (TB) from nontuberculous mycobacterial causes. Cutaneous TB shows caseous granulomas in the upper and mid dermis, while nontuberculous mycobacterial infections have more prominent neutrophil infiltration and interstitial granulomas (Figure 4).16
In cutaneous TB specifically, extrapulmonary manifestations may involve the skin in 1% to 1.5% of all TB cases, and although rare, ulcerative skin TB has been noted in one report as a nonhealing perianal ulcer that showed necrotizing granulomas on biopsy.17 Ultimately, diagnosis of cutaneous mycobacterial infection is confirmed with detection of acid-fast bacilli in the biopsy specimen.16
Diagnosis of cutaneous CD requires clinicopathologic correlation, as the clinical and histopathologic differential diagnoses of genital edema and noncaseating granulomas, respectively, are broad. Even though the clinical context was appropriate for cutaneous CD in this case, correct diagnosis required confirmatory histologic findings. Furthermore, taking multiple biopsies is prudent. In our patient, diagnostic findings only were present in the biopsy from the scrotum.
The Diagnosis: Cutaneous Crohn Disease
Crohn disease (CD) is an inflammatory bowel disease that can involve any region of the gastrointestinal (GI) tract from the mouth to the anus but most commonly presents in the terminal ileum, colon, or small bowel with transmural inflammation, fistula formation, and knife-cut fissures among the frequently described findings. Extraintestinal manifestations may be found in the liver, eyes, and joints, with cutaneous extraintestinal manifestations occurring in up to one-third of patients.1
Crohn disease can be associated with multiple cutaneous findings, including erythema nodosum, pyoderma gangrenosum, aphthous ulcers, pyodermatitis-pyostomatitis vegetans, necrotizing vasculitis, and metastatic Crohn disease (MCD).2 Typical histopathologic findings seen in MCD such as noncaseating granulomatous inflammation in the papillary and reticular dermis, possibly extending to the subcutaneous fat, are not specific to MCD. Associated genital edema is thought to be a consequence of granulomatous inflammation of lymphatics. In one study reviewing specimens from 10 cases of CD, a mean of 46% of all granulomas identified on the slides (264 granulomas in total) were located proximal to lymphatic vessels, suggesting a common pathway for development of intestinal disease and genital edema.3 The differential diagnosis for penile and scrotal swelling is broad, and the diagnosis may be missed if attention is not given to the clinical history of the patient in addition to histopathologic findings.2
Skin changes in CD also can be separated into perianal disease and true metastatic disease--the former recognized when anal lesions appear associated with segmental involvement of the GI tract and the latter as ulceration of the skin separated from the GI tract by normal tissue.1 The term sarcoidal reaction often is used to describe histopathologic findings in cutaneous CD, as it refers to the noncaseating granulomas found in approximately 60% of all cases.4 Ultimately, the location of noncaseating granulomas within the dermis of our patient's biopsy, taken in conjunction with the clinical history and the lack of defining features for other potential etiologies (eg, polarizable material, organisms on special stains), led to the diagnosis of cutaneous CD.
Cutaneous manifestations of sarcoidosis most commonly occur as papules, plaques, and subcutaneous nodules predominantly on the face, upper back, arms, and legs. Although the histologic features of sarcoidosis are characterized by lymphocyte-poor noncaseating granulomas (Figure 1), these findings also can be seen as a consequence of multiple granulomatous causes.5,6 In a review of 48 cutaneous specimens from patients with sarcoidosis, the granulomas were found most frequently in the deep dermis (34/48 [70.8%]), with superficial dermis (21/48) and subcutaneous fat granulomas (20/48) each present in less than 50% of biopsies.5 Although less typical, cutaneous sarcoidosis also has been noted in the literature to present in the perianal and gluteal region, demonstrating dermal noncaseating granulomas on biopsy.7 One distinction in particular to be noted between sarcoid and CD is that sarcoid lesions in the skin rarely ulcerate, while the lesions of cutaneous CD often are ulcerated.4,6
Lesions including abscesses in the groin may raise concern for hidradenitis suppurativa (HS), a disease of the apocrine gland-bearing skin. Typical lesions are tender subcutaneous erythematous nodules, cysts, and comedones that develop rapidly and may rupture to drain suppurative bloody discharge, subsequently healing with an atrophic scar.8 More persistent inflammation and rupture of nodules into the dermis may lead to formation of dermal tunnels with palpable cords and sinus tracts.8 Typical areas of disease involvement are in the axillae, inframammary folds, groin, or perigenital or perineal regions, with the diagnosis made on a combination of lesion morphology, location, and progression/recurrence frequency.9 Histologic examination of HS specimens can demonstrate a perifollicular lymphocytic infiltrate, with more advanced disease characterized by increased inflammatory cells, predominantly neutrophils, monocytes, and mast cells (Figure 2). The presence of granulomas in HS most often is of the foreign body type.9 Epithelioid granulomas noted in an area separate from inflammation in a patient with HS serve as a clue to be alert for systemic granulomatous disease.10
Mycosis fungoides is the most common primary cutaneous lymphoma to show a granulomatous infiltrate; the granuloma generally is sarcoidal, though other forms are described (Figure 3).11 Beyond these granulomatous foci, the key histopathologic feature of granulomatous mycosis fungoides (GMF) is diffuse dermal infiltration by atypical lymphoid cells. Epidermotropism and sparing of dermal nerves is the most critical finding in the diagnosis of GMF, especially in geographic regions where leprosy is endemic and high on the differential, as the conditions have histopathologic similarities.11,12 At the same time, lack of epidermotropism does not exclude the diagnosis of GMF.13 Clinically, GMF presentation is variable, but common findings include erythematous and hyperpigmented patches and plaques. Given the lack of clear clinical criteria, the diagnosis relies primarily on histopathologic features.11
Mycobacterial skin and soft tissue infections may be attributed to both tuberculous and nontuberculous strains (atypical species).14 Clinical features range from small papules to large deformative plaques and ulcers.15 Histologic features also distinguish cutaneous tuberculosis (TB) from nontuberculous mycobacterial causes. Cutaneous TB shows caseous granulomas in the upper and mid dermis, while nontuberculous mycobacterial infections have more prominent neutrophil infiltration and interstitial granulomas (Figure 4).16
In cutaneous TB specifically, extrapulmonary manifestations may involve the skin in 1% to 1.5% of all TB cases, and although rare, ulcerative skin TB has been noted in one report as a nonhealing perianal ulcer that showed necrotizing granulomas on biopsy.17 Ultimately, diagnosis of cutaneous mycobacterial infection is confirmed with detection of acid-fast bacilli in the biopsy specimen.16
Diagnosis of cutaneous CD requires clinicopathologic correlation, as the clinical and histopathologic differential diagnoses of genital edema and noncaseating granulomas, respectively, are broad. Even though the clinical context was appropriate for cutaneous CD in this case, correct diagnosis required confirmatory histologic findings. Furthermore, taking multiple biopsies is prudent. In our patient, diagnostic findings only were present in the biopsy from the scrotum.
- Hagen JW, Swoger JM, Grandinetti LM. Cutaneous manifestations of Crohn disease. Dermatol Clin. 2015;33:417-431.
- Barrick BJ, Tollefson MM, Schoch JJ, et al. Penile and scrotal swelling: an underrecognized presentation of Crohn's disease. Pediatr Dermatol. 2016;33:172-177.
- Mooney EE, Walker J, Hourihane DO. Relation of granulomas to lymphatic vessels in Crohn's disease. J Clin Pathol. 1995;48:335-338.
- Parks AG, Morson BC, Pegum JS. Crohn's disease with cutaneous involvement. Proc R Soc Med. 1965;58:241-242.
- García-Colmenero L, Sánchez-Schmidt JM, Barranco C, et al. The natural history of cutaneous sarcoidosis: clinical spectrum and histological analysis of 40 cases [published online October 18, 2018]. Int J Dermatol. 2019;58:178-184.
- Yoo SS, Mimouni D, Nikolskaia OV, et al. Clinicopathologic features of ulcerative-atrophic sarcoidosis. Int J Dermatol. 2004;43:108-112.
- Cohen GF, Wolfe CM. Recalcitrant diffuse cutaneous sarcoidosis with perianal involvement responding to adalimumab. J Drugs Dermatol. 2017;16:1305-1306.
- Hoffman LK, Ghias MH, Lowes MA. Pathophysiology of hidradenitis suppurativa. Semin Cutan Med Surg. 2017;36:47-54.
- Saunte DML, Jemec GBE. Hidradenitis suppurativa: advances in diagnosis and treatment. JAMA. 2017;318:2019-2032.
- Attanoos RL, Appleton MA, Hughes LE, et al. Granulomatous hidradenitis suppurativa and cutaneous Crohn's disease. Histopathology. 1993;23:111-115.
- Gutte R, Kharkar V, Mahajan S, et al. Granulomatous mycosis fungoides with hypohydrosis mimicking lepromatous leprosy. Indian J Dermatol Venerol Leprol. 2010;76:686-690.
- Pousa CM, Nery NS, Mann D, et al. Granulomatous mycosis fungoides--a diagnostic challenge. An Bras Dermatol. 2015;90:554-556.
- Kempf W, Ostheeren-Michaelis S, Paulli M, et al. Granulomatous mycosis fungoides and granulomatous slack skin: a multicenter study of the Cutaneous Lymphoma Histopathology Task Force Group of the European Organization for Research and Treatment of Cancer (EORTC). Arch Dermatol. 2008;144:1609-1617.
- van Mechelen M, van der Hilst J, Gyssens IC, et al. Mycobacterial skin and soft tissue infections: TB or not TB? Neth J Med. 2018;76:269-274.
- van Zyl L, du Plessis J, Viljoen J. Cutaneous tuberculosis overview and current treatment regimens. Tuberculosis (Edinb). 2015;95:629-638.
- De Maio F, Trecarichi EM, Visconti E, et al. Understanding cutaneous tuberculosis: two clinical cases. JMM Case Rep. 2016;3:E005070.
- Wu S, Wang W, Chen H, et al. Perianal ulcerative skin tuberculosis: a case report. Medicine (Baltimore). 2018;97:E10836.
- Hagen JW, Swoger JM, Grandinetti LM. Cutaneous manifestations of Crohn disease. Dermatol Clin. 2015;33:417-431.
- Barrick BJ, Tollefson MM, Schoch JJ, et al. Penile and scrotal swelling: an underrecognized presentation of Crohn's disease. Pediatr Dermatol. 2016;33:172-177.
- Mooney EE, Walker J, Hourihane DO. Relation of granulomas to lymphatic vessels in Crohn's disease. J Clin Pathol. 1995;48:335-338.
- Parks AG, Morson BC, Pegum JS. Crohn's disease with cutaneous involvement. Proc R Soc Med. 1965;58:241-242.
- García-Colmenero L, Sánchez-Schmidt JM, Barranco C, et al. The natural history of cutaneous sarcoidosis: clinical spectrum and histological analysis of 40 cases [published online October 18, 2018]. Int J Dermatol. 2019;58:178-184.
- Yoo SS, Mimouni D, Nikolskaia OV, et al. Clinicopathologic features of ulcerative-atrophic sarcoidosis. Int J Dermatol. 2004;43:108-112.
- Cohen GF, Wolfe CM. Recalcitrant diffuse cutaneous sarcoidosis with perianal involvement responding to adalimumab. J Drugs Dermatol. 2017;16:1305-1306.
- Hoffman LK, Ghias MH, Lowes MA. Pathophysiology of hidradenitis suppurativa. Semin Cutan Med Surg. 2017;36:47-54.
- Saunte DML, Jemec GBE. Hidradenitis suppurativa: advances in diagnosis and treatment. JAMA. 2017;318:2019-2032.
- Attanoos RL, Appleton MA, Hughes LE, et al. Granulomatous hidradenitis suppurativa and cutaneous Crohn's disease. Histopathology. 1993;23:111-115.
- Gutte R, Kharkar V, Mahajan S, et al. Granulomatous mycosis fungoides with hypohydrosis mimicking lepromatous leprosy. Indian J Dermatol Venerol Leprol. 2010;76:686-690.
- Pousa CM, Nery NS, Mann D, et al. Granulomatous mycosis fungoides--a diagnostic challenge. An Bras Dermatol. 2015;90:554-556.
- Kempf W, Ostheeren-Michaelis S, Paulli M, et al. Granulomatous mycosis fungoides and granulomatous slack skin: a multicenter study of the Cutaneous Lymphoma Histopathology Task Force Group of the European Organization for Research and Treatment of Cancer (EORTC). Arch Dermatol. 2008;144:1609-1617.
- van Mechelen M, van der Hilst J, Gyssens IC, et al. Mycobacterial skin and soft tissue infections: TB or not TB? Neth J Med. 2018;76:269-274.
- van Zyl L, du Plessis J, Viljoen J. Cutaneous tuberculosis overview and current treatment regimens. Tuberculosis (Edinb). 2015;95:629-638.
- De Maio F, Trecarichi EM, Visconti E, et al. Understanding cutaneous tuberculosis: two clinical cases. JMM Case Rep. 2016;3:E005070.
- Wu S, Wang W, Chen H, et al. Perianal ulcerative skin tuberculosis: a case report. Medicine (Baltimore). 2018;97:E10836.

A 44-year-old man presented for evaluation of self-described "skin ripping" on the penis with penile and scrotal edema of 1 year's duration. He had a history of bowel symptoms and anorectal fistula of 3 years' duration. Purulent penile drainage and inguinal lymphadenopathy were noted on physical examination. Excisional biopsies of the scrotum and penis were performed. Special stains for organisms were negative.
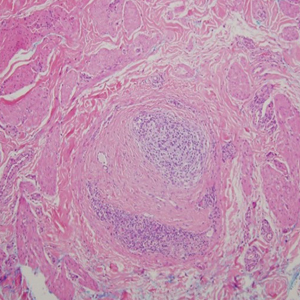
Is There an Association Between Hidradenitis Suppurativa and Fibromyalgia?
To the Editor:
Hidradenitis suppurativa (HS) is a chronic inflammatory condition that affects approximately 1% to 4% of the worldwide population and is 3 times more common in females than in males.1 The condition is characterized by painful inflamed nodules in apocrine gland–bearing regions that can progress to abscesses, sinus tracts, and/or scarring. Hidradenitis suppurativa is associated with intense pain, work disability, and poor quality of life.1
Recent evidence has suggested that HS is an autoimmune disease resulting from dysregulation of the γ-secretase/Notch pathway, leading to stimulation of the toll-like receptor–mediated innate immunity that contributes to occlusion and inflammation of the hair follicle. Additionally, elevated levels of proinflammatory cytokines such as tumor necrosis factor α and IL-17 are seen in HS lesions.2 The autoimmune nature of HS may account for its increased association with other autoimmune disorders such as thyroid disease and potentially with other unexplored conditions such as fibromyalgia.3
Fibromyalgia is a chronic pain condition that primarily affects females and is commonly associated with other autoimmune conditions.4 The primary objective of this retrospective study was to determine the prevalence of fibromyalgia in HS patients and assess if there is an association between HS disease severity and development of fibromyalgia.
We conducted a retrospective chart review of patients at Wake Forest Baptist Medical Center (Winston-Salem, North Carolina) who were 18 years and older and had a diagnosis of both HS and fibromyalgia from January 2008 to November 2018. The primary end point was the prevalence of fibromyalgia in the HS population. The secondary end point was the association of HS disease severity with the development of fibromyalgia. Hidradenitis disease severity was defined according to the number of body areas affected by HS: mild disease involved 1 body area, moderate disease involved 2 body areas, and severe disease involved 3 or more body areas. Patient age, sex, and race also were recorded.
A total of 1356 patients were seen during this time period for HS. The prevalence of fibromyalgia in the HS population was 3.2% (n=44). Ninety-five percent (42/44) of patients with HS and fibromyalgia were women; 22 (50%) patients had severe disease, 12 (27%) had moderate disease, 7 (16%) had mild disease, and 3 (7%) had an unknown number of affected body areas. Fifty-seven percent (25/44) of patients were diagnosed with HS prior to the diagnosis of fibromyalgia (Table).

In our study, the prevalence of fibromyalgia in HS patients was lower than the overall prevalence estimates of up to 6% in the United States.5 Although fibromyalgia is associated with other autoimmune conditions, it does not appear that fibromyalgia occurs more frequently in the HS population than the general population. A limitation of this study was that we only included academic outpatient clinic visits at one institution, which may not be representative of the entire HS population. Fibromyalgia was one of the many pain disorders in this population of patients. In this population of HS patients, many had pain issues with diagnose
- Smith MK, Nichlson CL, Parks-Miller A, et al. Hidradenitis suppurativa: an update on connecting the tracts. F1000Res. 2017;6:1272.
- Napolitano M, Megna M, Timoshchuk EA, et al. Hidradenitis suppurativa: from pathogenesis to diagnosis and treatment. Clin Cosmet Investig Dermatol. 2017;10:105-115.
- Miller IM, Vinding G, Sorensen HA, et al. Thyroid function in hidradenitis suppurativa: a population-based cross-sectional study from Denmark. Clin Exp Dermatol. 2018;43:899-905.
- Giacomelli C, Talarico R, Bombardieri S, et al. The interaction between autoimmune diseases and fibromyalgia: risk, disease course and management. Expert Rev Clin Immunol. 2013;9:1069-1076.
- Queiroz LP. Worldwide epidemiology of fibromyalgia. Curr Pain Headache Rep. 2013;17:356.
To the Editor:
Hidradenitis suppurativa (HS) is a chronic inflammatory condition that affects approximately 1% to 4% of the worldwide population and is 3 times more common in females than in males.1 The condition is characterized by painful inflamed nodules in apocrine gland–bearing regions that can progress to abscesses, sinus tracts, and/or scarring. Hidradenitis suppurativa is associated with intense pain, work disability, and poor quality of life.1
Recent evidence has suggested that HS is an autoimmune disease resulting from dysregulation of the γ-secretase/Notch pathway, leading to stimulation of the toll-like receptor–mediated innate immunity that contributes to occlusion and inflammation of the hair follicle. Additionally, elevated levels of proinflammatory cytokines such as tumor necrosis factor α and IL-17 are seen in HS lesions.2 The autoimmune nature of HS may account for its increased association with other autoimmune disorders such as thyroid disease and potentially with other unexplored conditions such as fibromyalgia.3
Fibromyalgia is a chronic pain condition that primarily affects females and is commonly associated with other autoimmune conditions.4 The primary objective of this retrospective study was to determine the prevalence of fibromyalgia in HS patients and assess if there is an association between HS disease severity and development of fibromyalgia.
We conducted a retrospective chart review of patients at Wake Forest Baptist Medical Center (Winston-Salem, North Carolina) who were 18 years and older and had a diagnosis of both HS and fibromyalgia from January 2008 to November 2018. The primary end point was the prevalence of fibromyalgia in the HS population. The secondary end point was the association of HS disease severity with the development of fibromyalgia. Hidradenitis disease severity was defined according to the number of body areas affected by HS: mild disease involved 1 body area, moderate disease involved 2 body areas, and severe disease involved 3 or more body areas. Patient age, sex, and race also were recorded.
A total of 1356 patients were seen during this time period for HS. The prevalence of fibromyalgia in the HS population was 3.2% (n=44). Ninety-five percent (42/44) of patients with HS and fibromyalgia were women; 22 (50%) patients had severe disease, 12 (27%) had moderate disease, 7 (16%) had mild disease, and 3 (7%) had an unknown number of affected body areas. Fifty-seven percent (25/44) of patients were diagnosed with HS prior to the diagnosis of fibromyalgia (Table).
In our study, the prevalence of fibromyalgia in HS patients was lower than the overall prevalence estimates of up to 6% in the United States.5 Although fibromyalgia is associated with other autoimmune conditions, it does not appear that fibromyalgia occurs more frequently in the HS population than the general population. A limitation of this study was that we only included academic outpatient clinic visits at one institution, which may not be representative of the entire HS population. Fibromyalgia was one of the many pain disorders in this population of patients. In this population of HS patients, many had pain issues with diagnose
To the Editor:
Hidradenitis suppurativa (HS) is a chronic inflammatory condition that affects approximately 1% to 4% of the worldwide population and is 3 times more common in females than in males.1 The condition is characterized by painful inflamed nodules in apocrine gland–bearing regions that can progress to abscesses, sinus tracts, and/or scarring. Hidradenitis suppurativa is associated with intense pain, work disability, and poor quality of life.1
Recent evidence has suggested that HS is an autoimmune disease resulting from dysregulation of the γ-secretase/Notch pathway, leading to stimulation of the toll-like receptor–mediated innate immunity that contributes to occlusion and inflammation of the hair follicle. Additionally, elevated levels of proinflammatory cytokines such as tumor necrosis factor α and IL-17 are seen in HS lesions.2 The autoimmune nature of HS may account for its increased association with other autoimmune disorders such as thyroid disease and potentially with other unexplored conditions such as fibromyalgia.3
Fibromyalgia is a chronic pain condition that primarily affects females and is commonly associated with other autoimmune conditions.4 The primary objective of this retrospective study was to determine the prevalence of fibromyalgia in HS patients and assess if there is an association between HS disease severity and development of fibromyalgia.
We conducted a retrospective chart review of patients at Wake Forest Baptist Medical Center (Winston-Salem, North Carolina) who were 18 years and older and had a diagnosis of both HS and fibromyalgia from January 2008 to November 2018. The primary end point was the prevalence of fibromyalgia in the HS population. The secondary end point was the association of HS disease severity with the development of fibromyalgia. Hidradenitis disease severity was defined according to the number of body areas affected by HS: mild disease involved 1 body area, moderate disease involved 2 body areas, and severe disease involved 3 or more body areas. Patient age, sex, and race also were recorded.
A total of 1356 patients were seen during this time period for HS. The prevalence of fibromyalgia in the HS population was 3.2% (n=44). Ninety-five percent (42/44) of patients with HS and fibromyalgia were women; 22 (50%) patients had severe disease, 12 (27%) had moderate disease, 7 (16%) had mild disease, and 3 (7%) had an unknown number of affected body areas. Fifty-seven percent (25/44) of patients were diagnosed with HS prior to the diagnosis of fibromyalgia (Table).
In our study, the prevalence of fibromyalgia in HS patients was lower than the overall prevalence estimates of up to 6% in the United States.5 Although fibromyalgia is associated with other autoimmune conditions, it does not appear that fibromyalgia occurs more frequently in the HS population than the general population. A limitation of this study was that we only included academic outpatient clinic visits at one institution, which may not be representative of the entire HS population. Fibromyalgia was one of the many pain disorders in this population of patients. In this population of HS patients, many had pain issues with diagnose
- Smith MK, Nichlson CL, Parks-Miller A, et al. Hidradenitis suppurativa: an update on connecting the tracts. F1000Res. 2017;6:1272.
- Napolitano M, Megna M, Timoshchuk EA, et al. Hidradenitis suppurativa: from pathogenesis to diagnosis and treatment. Clin Cosmet Investig Dermatol. 2017;10:105-115.
- Miller IM, Vinding G, Sorensen HA, et al. Thyroid function in hidradenitis suppurativa: a population-based cross-sectional study from Denmark. Clin Exp Dermatol. 2018;43:899-905.
- Giacomelli C, Talarico R, Bombardieri S, et al. The interaction between autoimmune diseases and fibromyalgia: risk, disease course and management. Expert Rev Clin Immunol. 2013;9:1069-1076.
- Queiroz LP. Worldwide epidemiology of fibromyalgia. Curr Pain Headache Rep. 2013;17:356.
- Smith MK, Nichlson CL, Parks-Miller A, et al. Hidradenitis suppurativa: an update on connecting the tracts. F1000Res. 2017;6:1272.
- Napolitano M, Megna M, Timoshchuk EA, et al. Hidradenitis suppurativa: from pathogenesis to diagnosis and treatment. Clin Cosmet Investig Dermatol. 2017;10:105-115.
- Miller IM, Vinding G, Sorensen HA, et al. Thyroid function in hidradenitis suppurativa: a population-based cross-sectional study from Denmark. Clin Exp Dermatol. 2018;43:899-905.
- Giacomelli C, Talarico R, Bombardieri S, et al. The interaction between autoimmune diseases and fibromyalgia: risk, disease course and management. Expert Rev Clin Immunol. 2013;9:1069-1076.
- Queiroz LP. Worldwide epidemiology of fibromyalgia. Curr Pain Headache Rep. 2013;17:356.
Practice Point
- Although fibromyalgia does not occur more frequently in hidradenitis suppurativa (HS) patients, it is important to recognize that HS patients can have comorbidities that should be addressed when possible to improve overall quality of life.
Cutaneous Metastases From Esophageal Adenocarcinoma on the Scalp
To the Editor:
A 59-year-old man presented with a lesion on the right frontal scalp of 4 months’ duration and a lesion on the left frontal scalp of 1 month’s duration. Both lesions were tender, bleeding, nonhealing, and growing in size. The patient reported no improvement with the use of triple antibiotic ointment. He denied any associated symptoms or trauma to the affected areas. He had a history of stage IV esophageal adenocarcinoma that initially had been surgically removed 6 years prior but metastasized to the lungs and bone. The patient subsequently underwent treatment with FOLFOX (folinic acid, fluorouracil, oxaliplatin), trastuzumab, and radiation therapy.
Physical examination revealed a hyperkeratotic pink nodule with a central erosion and crust on the right frontal scalp measuring 1.5×2 cm in diameter (Figure 1A). The left frontal scalp lesion was a smooth pearly papule measuring 5×5 mm in diameter (Figure 1B). The differential diagnosis included basal cell carcinoma, squamous cell carcinoma, and cutaneous metastases from esophageal adenocarcinoma. Shave biopsies were taken of both scalp lesions.
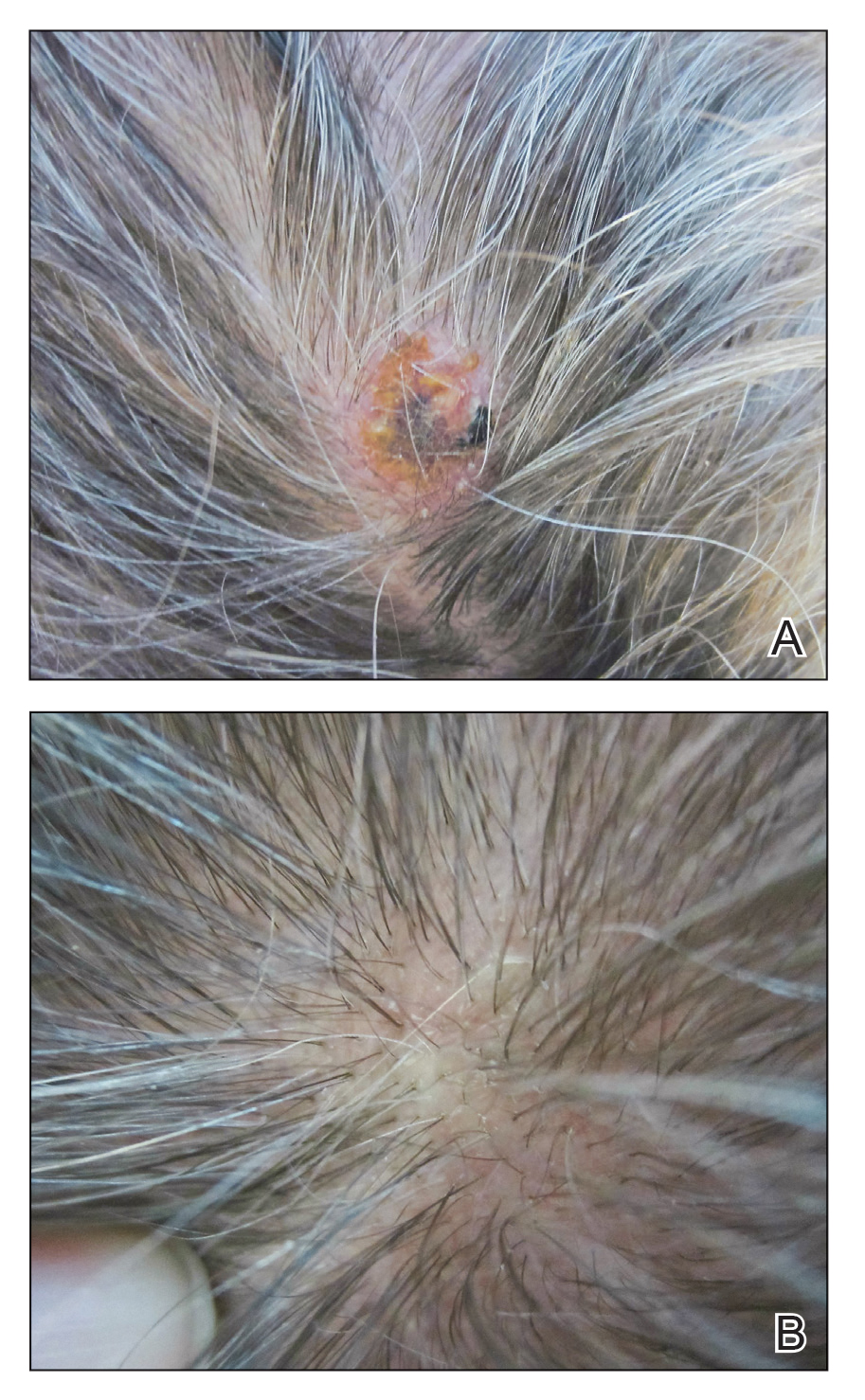
Histologic examination of both scalp lesions demonstrated a dermal gland-forming neoplasm with an infiltrative distribution that was comprised of irregular cribriform glands containing cellular debris (Figure 2). The cells of interest were enlarged and contained pleomorphic crowded nuclei that formed aberrant mitotic division figures. Both biopsies were positive for cytokeratin 7 and negative for cytokeratin 20 and CDX2. The final diagnosis for both scalp lesions was poorly differentiated adenocarcinoma, which was most suggestive of cutaneous metastases of the patient’s known esophageal adenocarcinoma. Given further metastasis, the patient was ultimately switched to ramucirumab and paclitaxel per oncology.
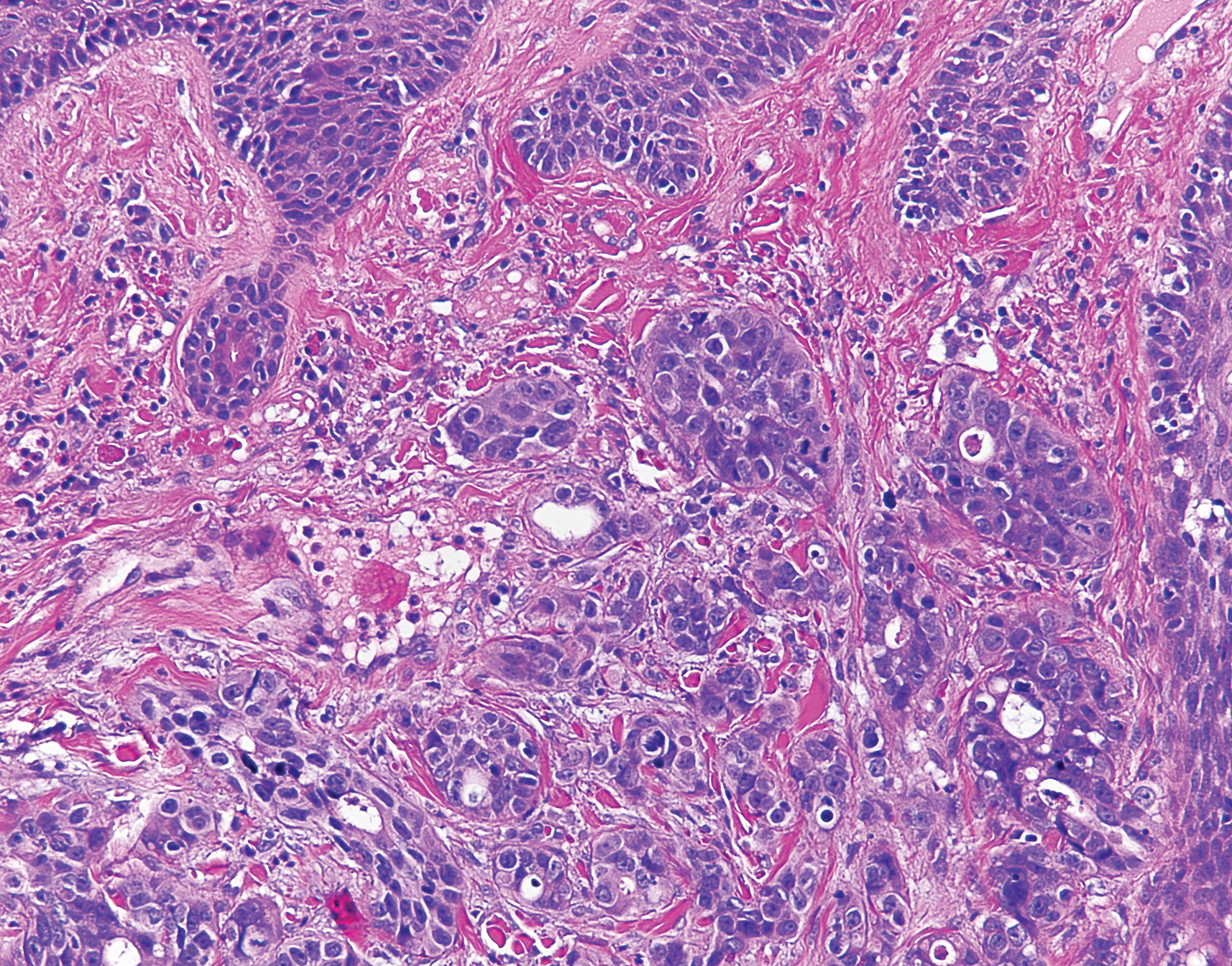
Esophageal carcinoma is the eighth most common cause of death related to cancer worldwide. Adenocarcinoma is the most prevalent histologic type of esophageal carcinoma, with an incidence as high as 5.69 per 100,000 individuals in the United States.1 Internal malignancies that lead to cutaneous metastases are not uncommon; however, the literature is limited on cutaneous scalp metastases from esophageal cancer. Cutaneous metastases secondary to internal malignancies present in less than 10% of overall cases; tend to derive from the breasts, lungs, and large bowel; and usually present in the sixth to seventh decades of life.2 Further, roughly 1% of all skin metastases originate from the esophagus.3 When there are cutaneous metastases to the scalp, they often arise from breast carcinomas and renal cell carcinomas.4,5 Rarely does esophageal cancer spread to the scalp.2,6-9 When cutaneous metastases originate from the esophagus, multiple cancers such as squamous cell carcinomas, mucoepidermoid carcinomas, small cell carcinomas, and adenocarcinomas can be the etiology of origin.10 Metastases originating from esophageal carcinomas frequently are diagnosed in the abdominal lymph nodes (45%), liver (35%), lungs (20%), cervical/supraclavicular lymph nodes (18%), bones (9%), adrenals (5%), peritoneum (2%), brain (2%), stomach (1%), pancreas (1%), pleura (1%), skin/body wall (1%), pericardium (1%), and spleen (1%).3 Additionally, multiple cutaneous scalp metastases from esophageal adenocarcinoma have been reported,7,9 as were seen in our case.
The clinical appearance of cutaneous scalp metastases has been described as inflammatory papules, indurated plaques, or nodules,2 which is consistent with our case, though the spectrum of presentation is admittedly broad. Histopathology of lesions characteristically shows prominent intraluminal necrotic cellular debris, which is common for adenocarcinomas of the gastrointestinal tract.7 However, utilizing immunohistochemical stains to detect specific antigens within tumor cells allows for better specificity of the tumor origin. More specifically, cytokeratin 7 and cytokeratin 20 are stained in esophageal metaplasia, such as Barrett esophagus, rather than in intestinal metaplasia inside the stomach.2,11 Therefore, discerning the location of the adenocarcinoma proves fruitful when using cytokeratin 7 and cytokeratin 20. Although CDX2 is an additional marker that can be used for gastrointestinal adenocarcinomas with decent sensitivity and specificity, it can still be expressed in mucinous ovarian carcinomas and urinary bladder adenocarcinomas.12 In our patient, the strong reactivity of cytokeratin 7 in addition to the characteristic morphology in both presenting biopsies was sufficient to make the diagnosis of cutaneous metastasis of esophageal adenocarcinoma to the scalp.
Our case highlights multiple cutaneous metastases of the scalp from a primary esophageal adenocarcinoma. Although cutaneous scalp metastasis of esophageal adenocarcinoma is rare, it is essential to provide a full-body skin examination, including the scalp, in patients with a history of esophageal cancer and to biopsy any suspicious nodules or plaques. The 1-year survival rate after diagnosis of esophageal carcinoma is less than 50%, and the 5-year survival rate is less than 10%.13 Identifying cutaneous metastasis of an esophageal adenocarcinoma can either change the staging of the cancer (if it was the first distant metastasis noted) or indicate an insufficient response to treatment in a patient with known metastatic disease, prompting a potential change in treatment.7
This case illustrates a rare site of metastasis of a fairly common cancer and highlights the histopathology and accompanying immunohistochemical stains that can be useful in diagnosis as well as the spectrum of its clinical presentation.
- Melhado R, Alderson D, Tucker O. The changing face of esophageal cancer. Cancers (Basel). 2010;2:1379-1404.
- Park JM, Kim DS, Oh SH, et al. A case of esophageal adenocarcinoma metastasized to the scalp [published online May 31, 2009]. Ann Dermatol. 2009;21:164-167.
- Quint LE, Hepburn LM, Francis IR, et al. Incidence and distribution of distant metastases from newly diagnosed esophageal carcinoma. Cancer. 1995;76:1120.
- Dobson C, Tagor V, Myint A, et al. Telangiectatic metastatic breast carcinoma in face and scalp mimicking cutaneous angiosarcoma. J Am Acad Dermatol. 2003;48:635-636.
- Riter H, Ghobrial I. Renal cell carcinoma with acrometastasis and scalp metastasis. Mayo Clin Proc. 2004;79:76.
- Roh EK, Nord R, Jukic DM. Scalp metastasis from esophageal adenocarcinoma. Cutis. 2006;77:106.
- Doumit G, Abouhassan W, Piliang M, et al. Scalp metastasis from esophageal adenocarcinoma: comparative histopathology dictates surgical approach. Ann Plast Surg. 2011;71:60-62.
- Roy AD, Sherparpa M, Prasad PR, et al. Scalp metastasis of gastro-esophageal junction adenocarcinoma: a rare occurrence. 2014;8:159-160.
- Stein R, Spencer J. Painful cutaneous metastases from esophageal carcinoma. Cutis. 2002;70:230.
- Schwartz RA. Cutaneous metastatic disease. J Am Acad Dermatol. 1995;33(2 pt 1):161-182.
- Ormsby AH, Goldblum JR, Rice TW, et al. Cytokeratin subsets can reliably distinguish Barrett’s esophagus from intestinal metaplasia of the stomach. Hum Pathol. 1999;30:288-294.
- Werling RW, Yaziji H, Bacchi CE, et al. CDX2, a highly sensitive and specific marker of adenocarcinomas of intestinal origin: an immunohistochemical survey of 476 primary and metastatic carcinomas. Am J Surg Pathol. 2003;27:303-310.
- Smith KJ, Williams J, Skelton H. Metastatic adenocarcinoma of the esophagus to the skin: new patterns of tumor recurrence and alternate treatments for palliation. J Cutan Pathol. 2001;28:425-431.
To the Editor:
A 59-year-old man presented with a lesion on the right frontal scalp of 4 months’ duration and a lesion on the left frontal scalp of 1 month’s duration. Both lesions were tender, bleeding, nonhealing, and growing in size. The patient reported no improvement with the use of triple antibiotic ointment. He denied any associated symptoms or trauma to the affected areas. He had a history of stage IV esophageal adenocarcinoma that initially had been surgically removed 6 years prior but metastasized to the lungs and bone. The patient subsequently underwent treatment with FOLFOX (folinic acid, fluorouracil, oxaliplatin), trastuzumab, and radiation therapy.
Physical examination revealed a hyperkeratotic pink nodule with a central erosion and crust on the right frontal scalp measuring 1.5×2 cm in diameter (Figure 1A). The left frontal scalp lesion was a smooth pearly papule measuring 5×5 mm in diameter (Figure 1B). The differential diagnosis included basal cell carcinoma, squamous cell carcinoma, and cutaneous metastases from esophageal adenocarcinoma. Shave biopsies were taken of both scalp lesions.
Histologic examination of both scalp lesions demonstrated a dermal gland-forming neoplasm with an infiltrative distribution that was comprised of irregular cribriform glands containing cellular debris (Figure 2). The cells of interest were enlarged and contained pleomorphic crowded nuclei that formed aberrant mitotic division figures. Both biopsies were positive for cytokeratin 7 and negative for cytokeratin 20 and CDX2. The final diagnosis for both scalp lesions was poorly differentiated adenocarcinoma, which was most suggestive of cutaneous metastases of the patient’s known esophageal adenocarcinoma. Given further metastasis, the patient was ultimately switched to ramucirumab and paclitaxel per oncology.
Esophageal carcinoma is the eighth most common cause of death related to cancer worldwide. Adenocarcinoma is the most prevalent histologic type of esophageal carcinoma, with an incidence as high as 5.69 per 100,000 individuals in the United States.1 Internal malignancies that lead to cutaneous metastases are not uncommon; however, the literature is limited on cutaneous scalp metastases from esophageal cancer. Cutaneous metastases secondary to internal malignancies present in less than 10% of overall cases; tend to derive from the breasts, lungs, and large bowel; and usually present in the sixth to seventh decades of life.2 Further, roughly 1% of all skin metastases originate from the esophagus.3 When there are cutaneous metastases to the scalp, they often arise from breast carcinomas and renal cell carcinomas.4,5 Rarely does esophageal cancer spread to the scalp.2,6-9 When cutaneous metastases originate from the esophagus, multiple cancers such as squamous cell carcinomas, mucoepidermoid carcinomas, small cell carcinomas, and adenocarcinomas can be the etiology of origin.10 Metastases originating from esophageal carcinomas frequently are diagnosed in the abdominal lymph nodes (45%), liver (35%), lungs (20%), cervical/supraclavicular lymph nodes (18%), bones (9%), adrenals (5%), peritoneum (2%), brain (2%), stomach (1%), pancreas (1%), pleura (1%), skin/body wall (1%), pericardium (1%), and spleen (1%).3 Additionally, multiple cutaneous scalp metastases from esophageal adenocarcinoma have been reported,7,9 as were seen in our case.
The clinical appearance of cutaneous scalp metastases has been described as inflammatory papules, indurated plaques, or nodules,2 which is consistent with our case, though the spectrum of presentation is admittedly broad. Histopathology of lesions characteristically shows prominent intraluminal necrotic cellular debris, which is common for adenocarcinomas of the gastrointestinal tract.7 However, utilizing immunohistochemical stains to detect specific antigens within tumor cells allows for better specificity of the tumor origin. More specifically, cytokeratin 7 and cytokeratin 20 are stained in esophageal metaplasia, such as Barrett esophagus, rather than in intestinal metaplasia inside the stomach.2,11 Therefore, discerning the location of the adenocarcinoma proves fruitful when using cytokeratin 7 and cytokeratin 20. Although CDX2 is an additional marker that can be used for gastrointestinal adenocarcinomas with decent sensitivity and specificity, it can still be expressed in mucinous ovarian carcinomas and urinary bladder adenocarcinomas.12 In our patient, the strong reactivity of cytokeratin 7 in addition to the characteristic morphology in both presenting biopsies was sufficient to make the diagnosis of cutaneous metastasis of esophageal adenocarcinoma to the scalp.
Our case highlights multiple cutaneous metastases of the scalp from a primary esophageal adenocarcinoma. Although cutaneous scalp metastasis of esophageal adenocarcinoma is rare, it is essential to provide a full-body skin examination, including the scalp, in patients with a history of esophageal cancer and to biopsy any suspicious nodules or plaques. The 1-year survival rate after diagnosis of esophageal carcinoma is less than 50%, and the 5-year survival rate is less than 10%.13 Identifying cutaneous metastasis of an esophageal adenocarcinoma can either change the staging of the cancer (if it was the first distant metastasis noted) or indicate an insufficient response to treatment in a patient with known metastatic disease, prompting a potential change in treatment.7
This case illustrates a rare site of metastasis of a fairly common cancer and highlights the histopathology and accompanying immunohistochemical stains that can be useful in diagnosis as well as the spectrum of its clinical presentation.
To the Editor:
A 59-year-old man presented with a lesion on the right frontal scalp of 4 months’ duration and a lesion on the left frontal scalp of 1 month’s duration. Both lesions were tender, bleeding, nonhealing, and growing in size. The patient reported no improvement with the use of triple antibiotic ointment. He denied any associated symptoms or trauma to the affected areas. He had a history of stage IV esophageal adenocarcinoma that initially had been surgically removed 6 years prior but metastasized to the lungs and bone. The patient subsequently underwent treatment with FOLFOX (folinic acid, fluorouracil, oxaliplatin), trastuzumab, and radiation therapy.
Physical examination revealed a hyperkeratotic pink nodule with a central erosion and crust on the right frontal scalp measuring 1.5×2 cm in diameter (Figure 1A). The left frontal scalp lesion was a smooth pearly papule measuring 5×5 mm in diameter (Figure 1B). The differential diagnosis included basal cell carcinoma, squamous cell carcinoma, and cutaneous metastases from esophageal adenocarcinoma. Shave biopsies were taken of both scalp lesions.
Histologic examination of both scalp lesions demonstrated a dermal gland-forming neoplasm with an infiltrative distribution that was comprised of irregular cribriform glands containing cellular debris (Figure 2). The cells of interest were enlarged and contained pleomorphic crowded nuclei that formed aberrant mitotic division figures. Both biopsies were positive for cytokeratin 7 and negative for cytokeratin 20 and CDX2. The final diagnosis for both scalp lesions was poorly differentiated adenocarcinoma, which was most suggestive of cutaneous metastases of the patient’s known esophageal adenocarcinoma. Given further metastasis, the patient was ultimately switched to ramucirumab and paclitaxel per oncology.
Esophageal carcinoma is the eighth most common cause of death related to cancer worldwide. Adenocarcinoma is the most prevalent histologic type of esophageal carcinoma, with an incidence as high as 5.69 per 100,000 individuals in the United States.1 Internal malignancies that lead to cutaneous metastases are not uncommon; however, the literature is limited on cutaneous scalp metastases from esophageal cancer. Cutaneous metastases secondary to internal malignancies present in less than 10% of overall cases; tend to derive from the breasts, lungs, and large bowel; and usually present in the sixth to seventh decades of life.2 Further, roughly 1% of all skin metastases originate from the esophagus.3 When there are cutaneous metastases to the scalp, they often arise from breast carcinomas and renal cell carcinomas.4,5 Rarely does esophageal cancer spread to the scalp.2,6-9 When cutaneous metastases originate from the esophagus, multiple cancers such as squamous cell carcinomas, mucoepidermoid carcinomas, small cell carcinomas, and adenocarcinomas can be the etiology of origin.10 Metastases originating from esophageal carcinomas frequently are diagnosed in the abdominal lymph nodes (45%), liver (35%), lungs (20%), cervical/supraclavicular lymph nodes (18%), bones (9%), adrenals (5%), peritoneum (2%), brain (2%), stomach (1%), pancreas (1%), pleura (1%), skin/body wall (1%), pericardium (1%), and spleen (1%).3 Additionally, multiple cutaneous scalp metastases from esophageal adenocarcinoma have been reported,7,9 as were seen in our case.
The clinical appearance of cutaneous scalp metastases has been described as inflammatory papules, indurated plaques, or nodules,2 which is consistent with our case, though the spectrum of presentation is admittedly broad. Histopathology of lesions characteristically shows prominent intraluminal necrotic cellular debris, which is common for adenocarcinomas of the gastrointestinal tract.7 However, utilizing immunohistochemical stains to detect specific antigens within tumor cells allows for better specificity of the tumor origin. More specifically, cytokeratin 7 and cytokeratin 20 are stained in esophageal metaplasia, such as Barrett esophagus, rather than in intestinal metaplasia inside the stomach.2,11 Therefore, discerning the location of the adenocarcinoma proves fruitful when using cytokeratin 7 and cytokeratin 20. Although CDX2 is an additional marker that can be used for gastrointestinal adenocarcinomas with decent sensitivity and specificity, it can still be expressed in mucinous ovarian carcinomas and urinary bladder adenocarcinomas.12 In our patient, the strong reactivity of cytokeratin 7 in addition to the characteristic morphology in both presenting biopsies was sufficient to make the diagnosis of cutaneous metastasis of esophageal adenocarcinoma to the scalp.
Our case highlights multiple cutaneous metastases of the scalp from a primary esophageal adenocarcinoma. Although cutaneous scalp metastasis of esophageal adenocarcinoma is rare, it is essential to provide a full-body skin examination, including the scalp, in patients with a history of esophageal cancer and to biopsy any suspicious nodules or plaques. The 1-year survival rate after diagnosis of esophageal carcinoma is less than 50%, and the 5-year survival rate is less than 10%.13 Identifying cutaneous metastasis of an esophageal adenocarcinoma can either change the staging of the cancer (if it was the first distant metastasis noted) or indicate an insufficient response to treatment in a patient with known metastatic disease, prompting a potential change in treatment.7
This case illustrates a rare site of metastasis of a fairly common cancer and highlights the histopathology and accompanying immunohistochemical stains that can be useful in diagnosis as well as the spectrum of its clinical presentation.
- Melhado R, Alderson D, Tucker O. The changing face of esophageal cancer. Cancers (Basel). 2010;2:1379-1404.
- Park JM, Kim DS, Oh SH, et al. A case of esophageal adenocarcinoma metastasized to the scalp [published online May 31, 2009]. Ann Dermatol. 2009;21:164-167.
- Quint LE, Hepburn LM, Francis IR, et al. Incidence and distribution of distant metastases from newly diagnosed esophageal carcinoma. Cancer. 1995;76:1120.
- Dobson C, Tagor V, Myint A, et al. Telangiectatic metastatic breast carcinoma in face and scalp mimicking cutaneous angiosarcoma. J Am Acad Dermatol. 2003;48:635-636.
- Riter H, Ghobrial I. Renal cell carcinoma with acrometastasis and scalp metastasis. Mayo Clin Proc. 2004;79:76.
- Roh EK, Nord R, Jukic DM. Scalp metastasis from esophageal adenocarcinoma. Cutis. 2006;77:106.
- Doumit G, Abouhassan W, Piliang M, et al. Scalp metastasis from esophageal adenocarcinoma: comparative histopathology dictates surgical approach. Ann Plast Surg. 2011;71:60-62.
- Roy AD, Sherparpa M, Prasad PR, et al. Scalp metastasis of gastro-esophageal junction adenocarcinoma: a rare occurrence. 2014;8:159-160.
- Stein R, Spencer J. Painful cutaneous metastases from esophageal carcinoma. Cutis. 2002;70:230.
- Schwartz RA. Cutaneous metastatic disease. J Am Acad Dermatol. 1995;33(2 pt 1):161-182.
- Ormsby AH, Goldblum JR, Rice TW, et al. Cytokeratin subsets can reliably distinguish Barrett’s esophagus from intestinal metaplasia of the stomach. Hum Pathol. 1999;30:288-294.
- Werling RW, Yaziji H, Bacchi CE, et al. CDX2, a highly sensitive and specific marker of adenocarcinomas of intestinal origin: an immunohistochemical survey of 476 primary and metastatic carcinomas. Am J Surg Pathol. 2003;27:303-310.
- Smith KJ, Williams J, Skelton H. Metastatic adenocarcinoma of the esophagus to the skin: new patterns of tumor recurrence and alternate treatments for palliation. J Cutan Pathol. 2001;28:425-431.
- Melhado R, Alderson D, Tucker O. The changing face of esophageal cancer. Cancers (Basel). 2010;2:1379-1404.
- Park JM, Kim DS, Oh SH, et al. A case of esophageal adenocarcinoma metastasized to the scalp [published online May 31, 2009]. Ann Dermatol. 2009;21:164-167.
- Quint LE, Hepburn LM, Francis IR, et al. Incidence and distribution of distant metastases from newly diagnosed esophageal carcinoma. Cancer. 1995;76:1120.
- Dobson C, Tagor V, Myint A, et al. Telangiectatic metastatic breast carcinoma in face and scalp mimicking cutaneous angiosarcoma. J Am Acad Dermatol. 2003;48:635-636.
- Riter H, Ghobrial I. Renal cell carcinoma with acrometastasis and scalp metastasis. Mayo Clin Proc. 2004;79:76.
- Roh EK, Nord R, Jukic DM. Scalp metastasis from esophageal adenocarcinoma. Cutis. 2006;77:106.
- Doumit G, Abouhassan W, Piliang M, et al. Scalp metastasis from esophageal adenocarcinoma: comparative histopathology dictates surgical approach. Ann Plast Surg. 2011;71:60-62.
- Roy AD, Sherparpa M, Prasad PR, et al. Scalp metastasis of gastro-esophageal junction adenocarcinoma: a rare occurrence. 2014;8:159-160.
- Stein R, Spencer J. Painful cutaneous metastases from esophageal carcinoma. Cutis. 2002;70:230.
- Schwartz RA. Cutaneous metastatic disease. J Am Acad Dermatol. 1995;33(2 pt 1):161-182.
- Ormsby AH, Goldblum JR, Rice TW, et al. Cytokeratin subsets can reliably distinguish Barrett’s esophagus from intestinal metaplasia of the stomach. Hum Pathol. 1999;30:288-294.
- Werling RW, Yaziji H, Bacchi CE, et al. CDX2, a highly sensitive and specific marker of adenocarcinomas of intestinal origin: an immunohistochemical survey of 476 primary and metastatic carcinomas. Am J Surg Pathol. 2003;27:303-310.
- Smith KJ, Williams J, Skelton H. Metastatic adenocarcinoma of the esophagus to the skin: new patterns of tumor recurrence and alternate treatments for palliation. J Cutan Pathol. 2001;28:425-431.
Practice Points
- In the setting of underlying esophageal adenocarcinoma, metastatic spread to the scalp should be considered in the differential diagnosis for any suspicious scalp lesions.
- Coupling histopathology with immunohistochemical stains may aid in the diagnosis for cutaneous metastasis of esophageal adenocarcinoma.