User login
Cutis is a peer-reviewed clinical journal for the dermatologist, allergist, and general practitioner published monthly since 1965. Concise clinical articles present the practical side of dermatology, helping physicians to improve patient care. Cutis is referenced in Index Medicus/MEDLINE and is written and edited by industry leaders.
ass lick
assault rifle
balls
ballsac
black jack
bleach
Boko Haram
bondage
causas
cheap
child abuse
cocaine
compulsive behaviors
cost of miracles
cunt
Daech
display network stats
drug paraphernalia
explosion
fart
fda and death
fda AND warn
fda AND warning
fda AND warns
feom
fuck
gambling
gfc
gun
human trafficking
humira AND expensive
illegal
ISIL
ISIS
Islamic caliphate
Islamic state
madvocate
masturbation
mixed martial arts
MMA
molestation
national rifle association
NRA
nsfw
nuccitelli
pedophile
pedophilia
poker
porn
porn
pornography
psychedelic drug
recreational drug
sex slave rings
shit
slot machine
snort
substance abuse
terrorism
terrorist
texarkana
Texas hold 'em
UFC
section[contains(@class, 'nav-hidden')]
section[contains(@class, 'nav-hidden active')
A peer-reviewed, indexed journal for dermatologists with original research, image quizzes, cases and reviews, and columns.
Localized Acanthosis Nigricans at the Site of Repetitive Insulin Injections
To the Editor:
Acanthosis nigricans (AN) is characterized by asymptomatic, hyperpigmented, velvety plaques that can occur anywhere on the body but most often present on the skin of the neck, axillae, groin, and other body folds.1-12 Although there are 5 subtypes, benign AN is the most common and is related to insulin resistance.1-4 Insulin can bind to insulinlike growth factor 1 (IGF-1) on keratinocytes, stimulating their proliferation. In type 2 diabetes mellitus, endogenous insulin levels are high enough to bind IGF-1 and activate keratinocytes, leading to the development of AN. Insulin injections have been associated with cutaneous side effects including lipoatrophy, lipohypertrophy, and postinflammatory hyperpigmentation.3 Acanthosis nigricans at insulin injection sites is a rare clinical condition.
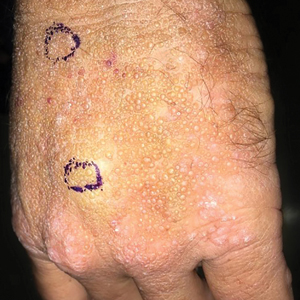
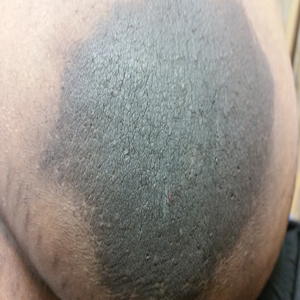
A 64-year-old man presented for evaluation of a growing asymptomatic lesion on the abdomen of 6 years’ duration. He had a 17-year history of type 2 diabetes mellitus treated with insulin injections for 14 years after failing oral hypoglycemic agents. The patient reported injecting at the same site on the abdomen for the last 14 years. Physical examination revealed a lichenified, hyperpigmented, verrucous plaque on the lower abdomen under the umbilicus (Figure 1). No similar skin lesions were observed elsewhere on the body. A punch biopsy of the plaque showed hyperkeratosis, papillomatosis, acanthosis, and hyperpigmentation, which was consistent with AN (Figure 2). The patient was instructed to rotate injection sites and avoid the affected area. Over-the-counter ammonium lactate cream applied twice daily to the affected site also was recommended. After 2 months of treatment with this regimen, the patient reported softening and lightening of the lesion on the abdomen.

A PubMed search of articles indexed for MEDLINE for all English-language studies with human participants using the terms acanthosis nigricans and insulin injections yielded 20 results. Of them, 13 discussed localized AN at insulin injection sites: 12 case reports (Table)1-12 and 1 observational study in a group of diabetic patients.13
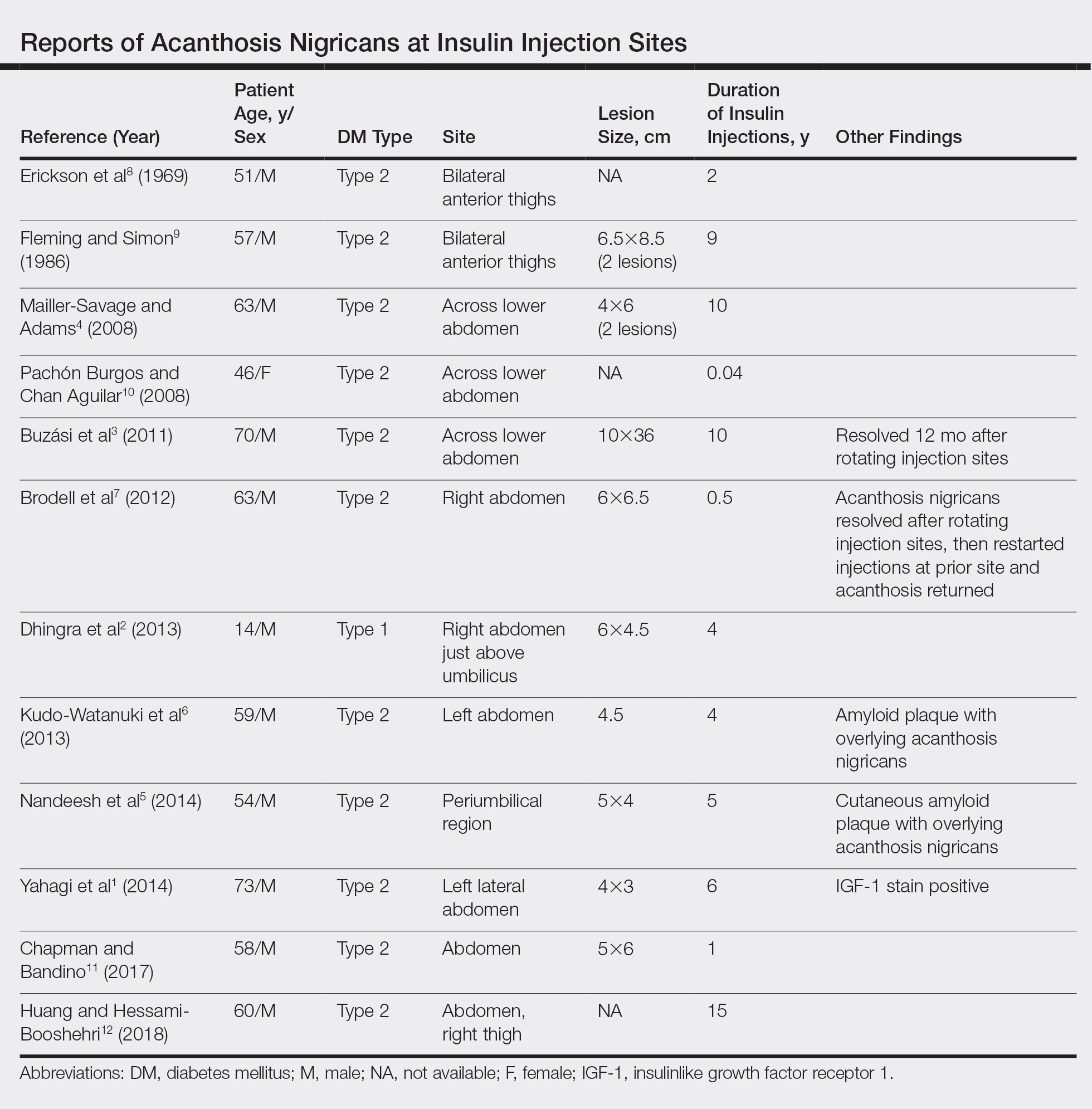
In the observational study, 500 diabetic patients were examined for insulin injection-site dermatoses and only 2 had localized injection-site AN. No other information was provided for these 2 patients.13 In the 12 published case reports,1-12 all patients did not rotate sites for their insulin injections and repeatedly injected into the affected area. The abdomen was the most commonly affected site, seen in 83% (10/12) of cases, while 25% (3/12) involved the thighs. All but 1 patient had type 2 diabetes mellitus. In 2 patients, “amyloid” was noted on the biopsy report in addition to changes consistent with AN. At least 2 patients had clearance after rotating injection sites.3,12
It has been suggested that localized AN at insulin injection sites develops through similar mechanisms as benign AN. Contributing factors to the development of benign AN may be IGF-1, fibroblast growth factor, and epidermal growth factor.1-3 Insulin is similar in structure to IGF-1 and can bind to IGF-1 receptors at high enough concentrations. Insulinlike growth factor 1 receptors are present on keratinocytes and fibroblasts, which proliferate upon activation, leading to the development of AN.1-3 Localized AN is thought to occur when repetitive insulin at the injection site saturates IGF-1 receptors on local keratinocytes.
Based on our patient and prior reports in the literature, localized AN is an uncommon cutaneous complication of insulin injections. Physicians should ask about repetitive insulin injections in the same site when localized AN occurs in patients with diabetes mellitus on insulin therapy. They also should discuss the importance of rotating sites for insulin adminstration to prevent the development of cutaneous complications including AN.
- Yahagi E, Mabuchi T, Nuruki H, et al. Case of exogenous insulin-derived acanthosis nigricans caused by insulin injections. Tokai J Exp Clin Med. 2014;39:5-9.
- Dhingra M, Garg G, Gupta M, et al. Exogenous insulin-derived acanthosis nigricans: could it be a cause of increased insulin requirement? Dermatol Online J. 2013;19:9.
- Buzási K, Sápi Z, Jermendy G. Acanthosis nigricans as a local cutaneous side effect of repeated human insulin injections. Diabetes Res Clin Pract. 2011;94:E34-E36.
- Mailler-Savage EA, Adams BB. Exogenous insulin-derived acanthosis nigricans. Arch Dermatol. 2008;144:126-127.
- Nandeesh BN, Rajalakshmi T, Shubha B. Cutaneous amyloidosis and insulin with coexistence of acanthosis nigricans. Indian J Pathol Microbiol. 2014;57:127-129.
- Kudo-Watanuki S, Kurihara E, Yamamoto K, et al. Coexistence of insulin-derived amyloidosis and an overlying acanthosis nigricans-like lesion at the site of insulin injection. Clin Exp Dermatol. 2013;38:25-29.
- Brodell JD Jr, Cannella JD, Helms SE. Case report: acanthosis nigricans resulting from repetitive same-site insulin injections. J Drugs Dermatol. 2012;11:E85-E87.
- Erickson L, Lipschutz DE, Wrigley W, et al. A peculiar cutaneous reaction to repeated injections of insulin. JAMA. 1969;209:934-935.
- Fleming MG, Simon SI. Cutaneous insulin reaction resembling acanthosis nigricans. Arch Dermatol. 1986;122:1054-1056.
- Pachon Burgos A, Chan Aguilar MP. Visual vignette. hyperpigmented hyperkeratotic cutaneous insulin reaction that resembles acanthosis nigricans with lipohypertrophy. Endocr Pract. 2008;14:514.
- Chapman SE, Bandino JP. A verrucous plaque on the abdomen: challenge. Am J Dermatopathol. 2017;39:E163.
- Huang Y, Hessami-Booshehri M. Acanthosis nigricans at sites of insulin injection in a man with diabetes. CMAJ. 2018;190:E1390.
- Sawatkar GU, Kanwar AJ, Dogra S, et al. Spectrum of cutaneous manifestations of type 1 diabetes mellitus in 500 South Asian patients. Br J Dermatol. 2014;171:1402-1406.
To the Editor:
Acanthosis nigricans (AN) is characterized by asymptomatic, hyperpigmented, velvety plaques that can occur anywhere on the body but most often present on the skin of the neck, axillae, groin, and other body folds.1-12 Although there are 5 subtypes, benign AN is the most common and is related to insulin resistance.1-4 Insulin can bind to insulinlike growth factor 1 (IGF-1) on keratinocytes, stimulating their proliferation. In type 2 diabetes mellitus, endogenous insulin levels are high enough to bind IGF-1 and activate keratinocytes, leading to the development of AN. Insulin injections have been associated with cutaneous side effects including lipoatrophy, lipohypertrophy, and postinflammatory hyperpigmentation.3 Acanthosis nigricans at insulin injection sites is a rare clinical condition.
A 64-year-old man presented for evaluation of a growing asymptomatic lesion on the abdomen of 6 years’ duration. He had a 17-year history of type 2 diabetes mellitus treated with insulin injections for 14 years after failing oral hypoglycemic agents. The patient reported injecting at the same site on the abdomen for the last 14 years. Physical examination revealed a lichenified, hyperpigmented, verrucous plaque on the lower abdomen under the umbilicus (Figure 1). No similar skin lesions were observed elsewhere on the body. A punch biopsy of the plaque showed hyperkeratosis, papillomatosis, acanthosis, and hyperpigmentation, which was consistent with AN (Figure 2). The patient was instructed to rotate injection sites and avoid the affected area. Over-the-counter ammonium lactate cream applied twice daily to the affected site also was recommended. After 2 months of treatment with this regimen, the patient reported softening and lightening of the lesion on the abdomen.
A PubMed search of articles indexed for MEDLINE for all English-language studies with human participants using the terms acanthosis nigricans and insulin injections yielded 20 results. Of them, 13 discussed localized AN at insulin injection sites: 12 case reports (Table)1-12 and 1 observational study in a group of diabetic patients.13
In the observational study, 500 diabetic patients were examined for insulin injection-site dermatoses and only 2 had localized injection-site AN. No other information was provided for these 2 patients.13 In the 12 published case reports,1-12 all patients did not rotate sites for their insulin injections and repeatedly injected into the affected area. The abdomen was the most commonly affected site, seen in 83% (10/12) of cases, while 25% (3/12) involved the thighs. All but 1 patient had type 2 diabetes mellitus. In 2 patients, “amyloid” was noted on the biopsy report in addition to changes consistent with AN. At least 2 patients had clearance after rotating injection sites.3,12
It has been suggested that localized AN at insulin injection sites develops through similar mechanisms as benign AN. Contributing factors to the development of benign AN may be IGF-1, fibroblast growth factor, and epidermal growth factor.1-3 Insulin is similar in structure to IGF-1 and can bind to IGF-1 receptors at high enough concentrations. Insulinlike growth factor 1 receptors are present on keratinocytes and fibroblasts, which proliferate upon activation, leading to the development of AN.1-3 Localized AN is thought to occur when repetitive insulin at the injection site saturates IGF-1 receptors on local keratinocytes.
Based on our patient and prior reports in the literature, localized AN is an uncommon cutaneous complication of insulin injections. Physicians should ask about repetitive insulin injections in the same site when localized AN occurs in patients with diabetes mellitus on insulin therapy. They also should discuss the importance of rotating sites for insulin adminstration to prevent the development of cutaneous complications including AN.
To the Editor:
Acanthosis nigricans (AN) is characterized by asymptomatic, hyperpigmented, velvety plaques that can occur anywhere on the body but most often present on the skin of the neck, axillae, groin, and other body folds.1-12 Although there are 5 subtypes, benign AN is the most common and is related to insulin resistance.1-4 Insulin can bind to insulinlike growth factor 1 (IGF-1) on keratinocytes, stimulating their proliferation. In type 2 diabetes mellitus, endogenous insulin levels are high enough to bind IGF-1 and activate keratinocytes, leading to the development of AN. Insulin injections have been associated with cutaneous side effects including lipoatrophy, lipohypertrophy, and postinflammatory hyperpigmentation.3 Acanthosis nigricans at insulin injection sites is a rare clinical condition.
A 64-year-old man presented for evaluation of a growing asymptomatic lesion on the abdomen of 6 years’ duration. He had a 17-year history of type 2 diabetes mellitus treated with insulin injections for 14 years after failing oral hypoglycemic agents. The patient reported injecting at the same site on the abdomen for the last 14 years. Physical examination revealed a lichenified, hyperpigmented, verrucous plaque on the lower abdomen under the umbilicus (Figure 1). No similar skin lesions were observed elsewhere on the body. A punch biopsy of the plaque showed hyperkeratosis, papillomatosis, acanthosis, and hyperpigmentation, which was consistent with AN (Figure 2). The patient was instructed to rotate injection sites and avoid the affected area. Over-the-counter ammonium lactate cream applied twice daily to the affected site also was recommended. After 2 months of treatment with this regimen, the patient reported softening and lightening of the lesion on the abdomen.
A PubMed search of articles indexed for MEDLINE for all English-language studies with human participants using the terms acanthosis nigricans and insulin injections yielded 20 results. Of them, 13 discussed localized AN at insulin injection sites: 12 case reports (Table)1-12 and 1 observational study in a group of diabetic patients.13
In the observational study, 500 diabetic patients were examined for insulin injection-site dermatoses and only 2 had localized injection-site AN. No other information was provided for these 2 patients.13 In the 12 published case reports,1-12 all patients did not rotate sites for their insulin injections and repeatedly injected into the affected area. The abdomen was the most commonly affected site, seen in 83% (10/12) of cases, while 25% (3/12) involved the thighs. All but 1 patient had type 2 diabetes mellitus. In 2 patients, “amyloid” was noted on the biopsy report in addition to changes consistent with AN. At least 2 patients had clearance after rotating injection sites.3,12
It has been suggested that localized AN at insulin injection sites develops through similar mechanisms as benign AN. Contributing factors to the development of benign AN may be IGF-1, fibroblast growth factor, and epidermal growth factor.1-3 Insulin is similar in structure to IGF-1 and can bind to IGF-1 receptors at high enough concentrations. Insulinlike growth factor 1 receptors are present on keratinocytes and fibroblasts, which proliferate upon activation, leading to the development of AN.1-3 Localized AN is thought to occur when repetitive insulin at the injection site saturates IGF-1 receptors on local keratinocytes.
Based on our patient and prior reports in the literature, localized AN is an uncommon cutaneous complication of insulin injections. Physicians should ask about repetitive insulin injections in the same site when localized AN occurs in patients with diabetes mellitus on insulin therapy. They also should discuss the importance of rotating sites for insulin adminstration to prevent the development of cutaneous complications including AN.
- Yahagi E, Mabuchi T, Nuruki H, et al. Case of exogenous insulin-derived acanthosis nigricans caused by insulin injections. Tokai J Exp Clin Med. 2014;39:5-9.
- Dhingra M, Garg G, Gupta M, et al. Exogenous insulin-derived acanthosis nigricans: could it be a cause of increased insulin requirement? Dermatol Online J. 2013;19:9.
- Buzási K, Sápi Z, Jermendy G. Acanthosis nigricans as a local cutaneous side effect of repeated human insulin injections. Diabetes Res Clin Pract. 2011;94:E34-E36.
- Mailler-Savage EA, Adams BB. Exogenous insulin-derived acanthosis nigricans. Arch Dermatol. 2008;144:126-127.
- Nandeesh BN, Rajalakshmi T, Shubha B. Cutaneous amyloidosis and insulin with coexistence of acanthosis nigricans. Indian J Pathol Microbiol. 2014;57:127-129.
- Kudo-Watanuki S, Kurihara E, Yamamoto K, et al. Coexistence of insulin-derived amyloidosis and an overlying acanthosis nigricans-like lesion at the site of insulin injection. Clin Exp Dermatol. 2013;38:25-29.
- Brodell JD Jr, Cannella JD, Helms SE. Case report: acanthosis nigricans resulting from repetitive same-site insulin injections. J Drugs Dermatol. 2012;11:E85-E87.
- Erickson L, Lipschutz DE, Wrigley W, et al. A peculiar cutaneous reaction to repeated injections of insulin. JAMA. 1969;209:934-935.
- Fleming MG, Simon SI. Cutaneous insulin reaction resembling acanthosis nigricans. Arch Dermatol. 1986;122:1054-1056.
- Pachon Burgos A, Chan Aguilar MP. Visual vignette. hyperpigmented hyperkeratotic cutaneous insulin reaction that resembles acanthosis nigricans with lipohypertrophy. Endocr Pract. 2008;14:514.
- Chapman SE, Bandino JP. A verrucous plaque on the abdomen: challenge. Am J Dermatopathol. 2017;39:E163.
- Huang Y, Hessami-Booshehri M. Acanthosis nigricans at sites of insulin injection in a man with diabetes. CMAJ. 2018;190:E1390.
- Sawatkar GU, Kanwar AJ, Dogra S, et al. Spectrum of cutaneous manifestations of type 1 diabetes mellitus in 500 South Asian patients. Br J Dermatol. 2014;171:1402-1406.
- Yahagi E, Mabuchi T, Nuruki H, et al. Case of exogenous insulin-derived acanthosis nigricans caused by insulin injections. Tokai J Exp Clin Med. 2014;39:5-9.
- Dhingra M, Garg G, Gupta M, et al. Exogenous insulin-derived acanthosis nigricans: could it be a cause of increased insulin requirement? Dermatol Online J. 2013;19:9.
- Buzási K, Sápi Z, Jermendy G. Acanthosis nigricans as a local cutaneous side effect of repeated human insulin injections. Diabetes Res Clin Pract. 2011;94:E34-E36.
- Mailler-Savage EA, Adams BB. Exogenous insulin-derived acanthosis nigricans. Arch Dermatol. 2008;144:126-127.
- Nandeesh BN, Rajalakshmi T, Shubha B. Cutaneous amyloidosis and insulin with coexistence of acanthosis nigricans. Indian J Pathol Microbiol. 2014;57:127-129.
- Kudo-Watanuki S, Kurihara E, Yamamoto K, et al. Coexistence of insulin-derived amyloidosis and an overlying acanthosis nigricans-like lesion at the site of insulin injection. Clin Exp Dermatol. 2013;38:25-29.
- Brodell JD Jr, Cannella JD, Helms SE. Case report: acanthosis nigricans resulting from repetitive same-site insulin injections. J Drugs Dermatol. 2012;11:E85-E87.
- Erickson L, Lipschutz DE, Wrigley W, et al. A peculiar cutaneous reaction to repeated injections of insulin. JAMA. 1969;209:934-935.
- Fleming MG, Simon SI. Cutaneous insulin reaction resembling acanthosis nigricans. Arch Dermatol. 1986;122:1054-1056.
- Pachon Burgos A, Chan Aguilar MP. Visual vignette. hyperpigmented hyperkeratotic cutaneous insulin reaction that resembles acanthosis nigricans with lipohypertrophy. Endocr Pract. 2008;14:514.
- Chapman SE, Bandino JP. A verrucous plaque on the abdomen: challenge. Am J Dermatopathol. 2017;39:E163.
- Huang Y, Hessami-Booshehri M. Acanthosis nigricans at sites of insulin injection in a man with diabetes. CMAJ. 2018;190:E1390.
- Sawatkar GU, Kanwar AJ, Dogra S, et al. Spectrum of cutaneous manifestations of type 1 diabetes mellitus in 500 South Asian patients. Br J Dermatol. 2014;171:1402-1406.
Practice Points
- Benign acanthosis nigricans (AN) is most often related to insulin resistance and presents as asymptomatic, hyperpigmented, velvety plaques on the neck, axillae, groin, and other body folds.
- Benign AN related to insulin resistance occurs when insulin binds to insulinlike growth factor 1 on keratinocytes and stimulates proliferations.
- Although insulin injections have been associated with several cutaneous side effects, including lipoatrophy, lipohypertrophy, and postinflammatory hyperpigmentation, localized AN is an uncommonly reported cutaneous adverse effect.
Not So Classic, Classic Kaposi Sarcoma
To the Editor:
Kaposi sarcoma (KS) is a lymphatic endothelial cell neoplasm that frequently presents as multiple vascular cutaneous and mucosal nodules.1 The classic KS variant typically is described as the presentation of KS in otherwise healthy elderly men of Jewish or Mediterranean descent.2 We present 2 cases of classic KS presenting in Mexican women living in Los Angeles County, California, with atypical clinical features.
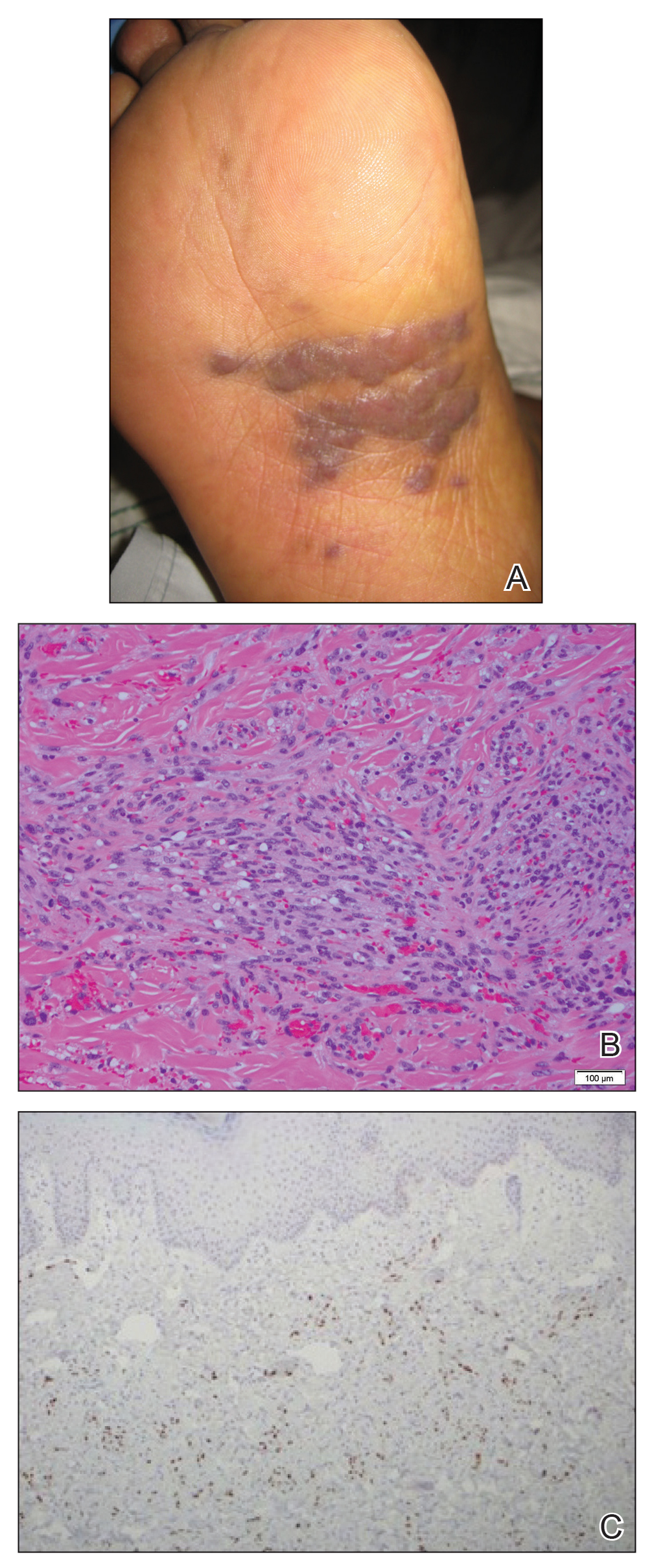
A 65-year-old woman of Mexican descent presented to the dermatology clinic for evaluation of an asymptomatic growth on the right ventral forearm of 4 months’ duration. Biopsy of the lesion demonstrated a spindle cell proliferation suspicious for KS. Physical examination several months following the initial biopsy revealed a mildly indurated scar on the right ventral forearm with an adjacent faintly erythematous papule (Figure 1A). Repeat biopsy revealed a dermal spindle cell proliferation suggestive of KS (Figures 1B and 1C). S-100 and cytokeratin stains were negative, but a latent nuclear antigen 1 stain for human herpesvirus 8 was positive. Human immunodeficiency virus screening also was negative. Given the isolated findings, the oncology service determined that observation alone was the most appropriate management. Over time, the patient developed several similar scattered erythematous papules, and treatment with imiquimod cream 5% was initiated for 1 month without improvement. She was subsequently given a trial of alitretinoin ointment 0.1% twice daily. The lesions improved, and she continues to be well controlled on this topical therapy alone.
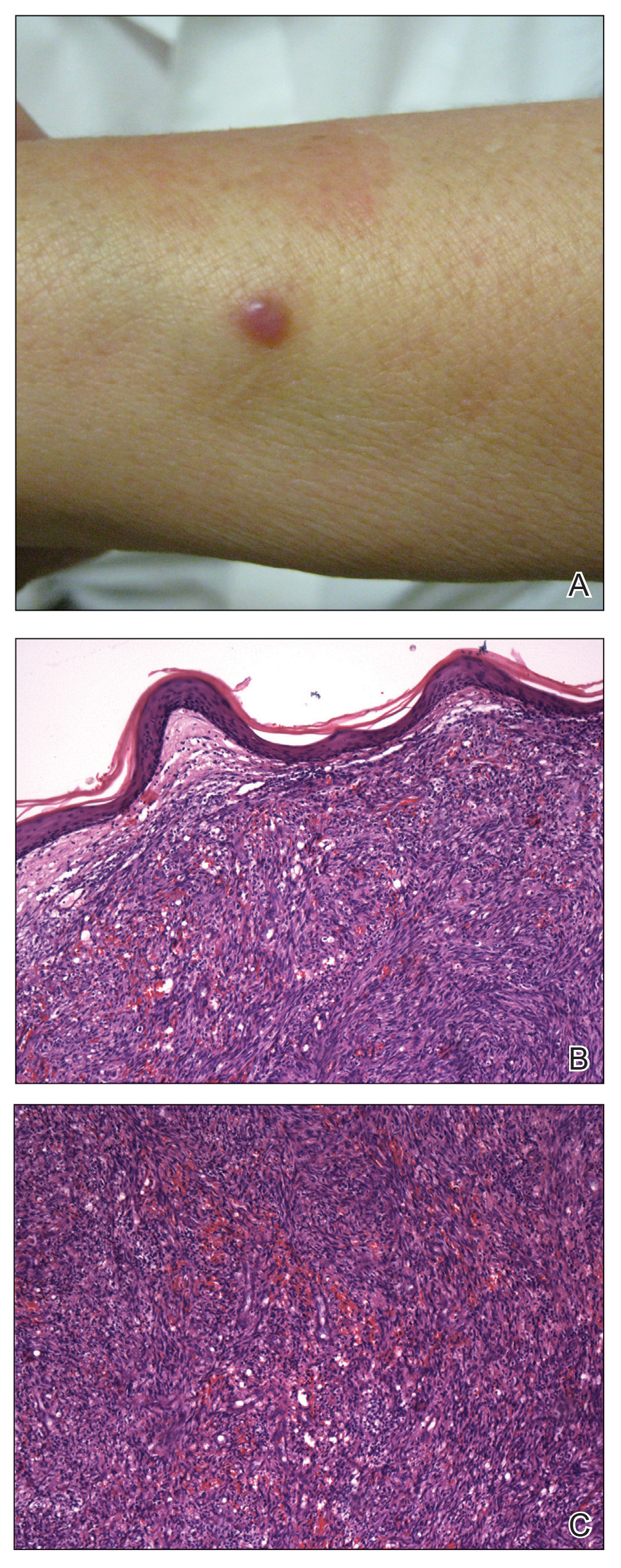
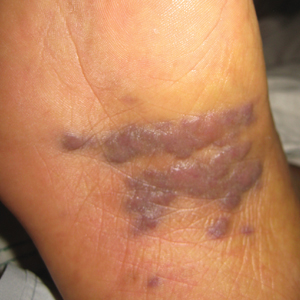
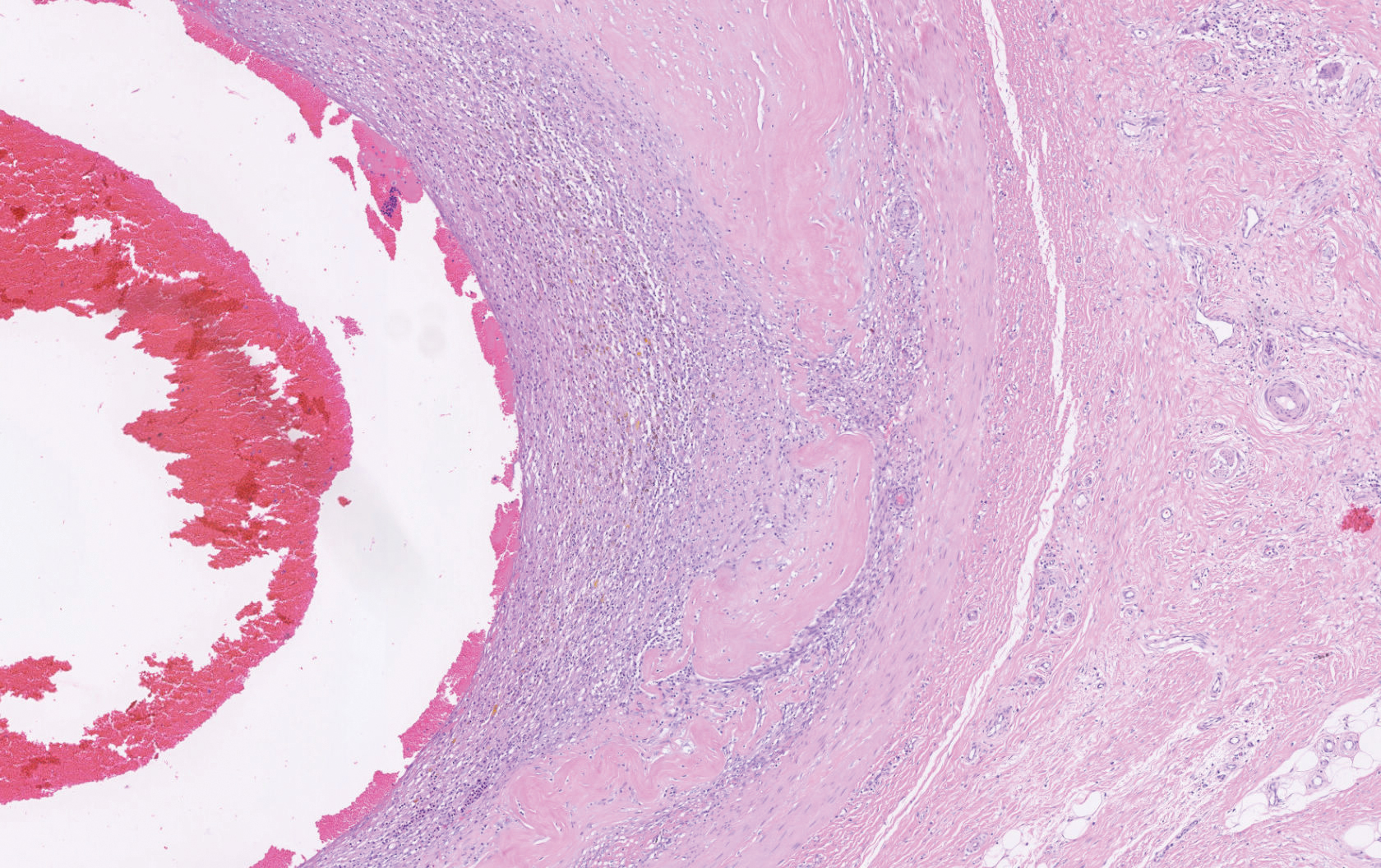
A 62-year-old woman of Mexican descent with end-stage cryptogenic cirrhosis was admitted to the hospital for evaluation of transplant candidacy. Dermatology was consulted to assess a 5×3-cm, asymptomatic, solitary, violaceous plaque on the right plantar foot (Figure 2A) with no palpable lymph nodes. Biopsy revealed a dermal proliferation of slit-like vascular channels infiltrating through the collagen and surrounding preexisting vascular spaces and adnexal structures (Figure 2B). Extravasation of erythrocytes and plasma cells also was appreciated. Latent nuclear antigen 1 staining showed strong nuclear positivity consistent with KS (Figure 2C), and a human immunodeficiency virus test and workup for underlying immunosuppression were negative. The patient had no history of treatment with immunosuppressive medications. Further workup revealed involvement of the lymphatic system. The patient was removed from the transplant list and was not a candidate for chemotherapy due to liver failure.
Kaposi sarcoma is a vascular neoplasm associated with human herpesvirus 8. It typically presents as erythematous to violaceous papules and plaques on the extremities.1 At least 10 morphologic variants of KS have been identified.3 The indolent classic variant of KS most commonly is found in immunocompetent individuals and has been reported to primarily affect elderly men of Jewish or Mediterranean descent.
Epidemiologic analyses of this disease in the South American population are rare. More than 250 cases have been published from South American countries with the largest series published from patients in Argentina, Peru, and Colombia.4 The incidence of classic KS in Peru is 2.54 per 10,000 individuals,5 and the disease has been diagnosed in 1 of 1000 malignant neoplasms at the National Cancer Institute of Colombia.6
A search of the Armed Forces Institute of Pathology Soft Tissue Pathology registry for classic KS patients (1980-2000) showed that 18% of 438 cases of classic KS were diagnosed in South American Hispanics,7 a percentage that nearly approximates the proportion of patients with KS of Mediterranean descent. Interestingly, in an analysis conducted within Los Angeles County, classic KS was most frequently diagnosed in Jewish, Eastern European–born men who were 55 years or older, followed by European-born women. In this study, white, Spanish, and black populations were diagnosed with classic KS with equal frequency.8
Our 2 cases of classic KS from Los Angeles County had several notable features. Both patients were women from Mexico, a demographic not previously associated with classic KS,8 and they did not have risk factors commonly associated with classic KS. We emphasize that classic KS likely is underreported and understudied in the Mexican and South American populations with need for further epidemiological and clinical analyses.
- Kaposi M. Idiopathisches multiples Pigmentsarkom der Haut. Arch Dermatol Syphilol. 1872;4:265-272.
- Akasbi Y, Awada A, Arifi S, et al. Non-HIV Kaposi’s sarcoma: a review and therapeutic perspectives. Bull Cancer. 2012;99:92-99.
- Schwartz RA. Kaposi’s sarcoma: an update. J Surg Oncol. 2004;87:146-151.
- Mohanna S, Maco V, Bravo F, et al. Epidemiology and clinical characteristics of classic Kaposi’s sarcoma, seroprevalence, and variants of human herpesvirus 8 in South America: a critical review of an old disease. Int J Infect Dis. 2005;9:2392-2350.
- Mohanna S, Ferrufino JC, Sanchez J, et al. Epidemiological and clinical characteristics of classic Kaposi’s sarcoma in Peru. J Am Acad Dermatol. 2005;53:435-441.
- García A, Olivella F, Valderrama S, et al. Kaposi’s sarcoma in Colombia. Cancer. 1989;64:2393-2398.
- Hiatt KM, Nelson AM, Lichy JH, et al. Classic Kaposi Sarcoma in the United States over the last two decades: a clinicopathologic and molecular study of 438 non-HIV related Kaposi Sarcoma patients with comparison to HIV-related Kaposi Sarcoma. Mod Pathol. 2008;21:572-582.
- Ross RK, Casagrande JT, Dworsky RL, et al. Kaposi’s sarcoma in Los Angeles, California. J Natl Cancer Inst. 1985;75:1011-1015.
To the Editor:
Kaposi sarcoma (KS) is a lymphatic endothelial cell neoplasm that frequently presents as multiple vascular cutaneous and mucosal nodules.1 The classic KS variant typically is described as the presentation of KS in otherwise healthy elderly men of Jewish or Mediterranean descent.2 We present 2 cases of classic KS presenting in Mexican women living in Los Angeles County, California, with atypical clinical features.
A 65-year-old woman of Mexican descent presented to the dermatology clinic for evaluation of an asymptomatic growth on the right ventral forearm of 4 months’ duration. Biopsy of the lesion demonstrated a spindle cell proliferation suspicious for KS. Physical examination several months following the initial biopsy revealed a mildly indurated scar on the right ventral forearm with an adjacent faintly erythematous papule (Figure 1A). Repeat biopsy revealed a dermal spindle cell proliferation suggestive of KS (Figures 1B and 1C). S-100 and cytokeratin stains were negative, but a latent nuclear antigen 1 stain for human herpesvirus 8 was positive. Human immunodeficiency virus screening also was negative. Given the isolated findings, the oncology service determined that observation alone was the most appropriate management. Over time, the patient developed several similar scattered erythematous papules, and treatment with imiquimod cream 5% was initiated for 1 month without improvement. She was subsequently given a trial of alitretinoin ointment 0.1% twice daily. The lesions improved, and she continues to be well controlled on this topical therapy alone.
A 62-year-old woman of Mexican descent with end-stage cryptogenic cirrhosis was admitted to the hospital for evaluation of transplant candidacy. Dermatology was consulted to assess a 5×3-cm, asymptomatic, solitary, violaceous plaque on the right plantar foot (Figure 2A) with no palpable lymph nodes. Biopsy revealed a dermal proliferation of slit-like vascular channels infiltrating through the collagen and surrounding preexisting vascular spaces and adnexal structures (Figure 2B). Extravasation of erythrocytes and plasma cells also was appreciated. Latent nuclear antigen 1 staining showed strong nuclear positivity consistent with KS (Figure 2C), and a human immunodeficiency virus test and workup for underlying immunosuppression were negative. The patient had no history of treatment with immunosuppressive medications. Further workup revealed involvement of the lymphatic system. The patient was removed from the transplant list and was not a candidate for chemotherapy due to liver failure.
Kaposi sarcoma is a vascular neoplasm associated with human herpesvirus 8. It typically presents as erythematous to violaceous papules and plaques on the extremities.1 At least 10 morphologic variants of KS have been identified.3 The indolent classic variant of KS most commonly is found in immunocompetent individuals and has been reported to primarily affect elderly men of Jewish or Mediterranean descent.
Epidemiologic analyses of this disease in the South American population are rare. More than 250 cases have been published from South American countries with the largest series published from patients in Argentina, Peru, and Colombia.4 The incidence of classic KS in Peru is 2.54 per 10,000 individuals,5 and the disease has been diagnosed in 1 of 1000 malignant neoplasms at the National Cancer Institute of Colombia.6
A search of the Armed Forces Institute of Pathology Soft Tissue Pathology registry for classic KS patients (1980-2000) showed that 18% of 438 cases of classic KS were diagnosed in South American Hispanics,7 a percentage that nearly approximates the proportion of patients with KS of Mediterranean descent. Interestingly, in an analysis conducted within Los Angeles County, classic KS was most frequently diagnosed in Jewish, Eastern European–born men who were 55 years or older, followed by European-born women. In this study, white, Spanish, and black populations were diagnosed with classic KS with equal frequency.8
Our 2 cases of classic KS from Los Angeles County had several notable features. Both patients were women from Mexico, a demographic not previously associated with classic KS,8 and they did not have risk factors commonly associated with classic KS. We emphasize that classic KS likely is underreported and understudied in the Mexican and South American populations with need for further epidemiological and clinical analyses.
To the Editor:
Kaposi sarcoma (KS) is a lymphatic endothelial cell neoplasm that frequently presents as multiple vascular cutaneous and mucosal nodules.1 The classic KS variant typically is described as the presentation of KS in otherwise healthy elderly men of Jewish or Mediterranean descent.2 We present 2 cases of classic KS presenting in Mexican women living in Los Angeles County, California, with atypical clinical features.
A 65-year-old woman of Mexican descent presented to the dermatology clinic for evaluation of an asymptomatic growth on the right ventral forearm of 4 months’ duration. Biopsy of the lesion demonstrated a spindle cell proliferation suspicious for KS. Physical examination several months following the initial biopsy revealed a mildly indurated scar on the right ventral forearm with an adjacent faintly erythematous papule (Figure 1A). Repeat biopsy revealed a dermal spindle cell proliferation suggestive of KS (Figures 1B and 1C). S-100 and cytokeratin stains were negative, but a latent nuclear antigen 1 stain for human herpesvirus 8 was positive. Human immunodeficiency virus screening also was negative. Given the isolated findings, the oncology service determined that observation alone was the most appropriate management. Over time, the patient developed several similar scattered erythematous papules, and treatment with imiquimod cream 5% was initiated for 1 month without improvement. She was subsequently given a trial of alitretinoin ointment 0.1% twice daily. The lesions improved, and she continues to be well controlled on this topical therapy alone.
A 62-year-old woman of Mexican descent with end-stage cryptogenic cirrhosis was admitted to the hospital for evaluation of transplant candidacy. Dermatology was consulted to assess a 5×3-cm, asymptomatic, solitary, violaceous plaque on the right plantar foot (Figure 2A) with no palpable lymph nodes. Biopsy revealed a dermal proliferation of slit-like vascular channels infiltrating through the collagen and surrounding preexisting vascular spaces and adnexal structures (Figure 2B). Extravasation of erythrocytes and plasma cells also was appreciated. Latent nuclear antigen 1 staining showed strong nuclear positivity consistent with KS (Figure 2C), and a human immunodeficiency virus test and workup for underlying immunosuppression were negative. The patient had no history of treatment with immunosuppressive medications. Further workup revealed involvement of the lymphatic system. The patient was removed from the transplant list and was not a candidate for chemotherapy due to liver failure.
Kaposi sarcoma is a vascular neoplasm associated with human herpesvirus 8. It typically presents as erythematous to violaceous papules and plaques on the extremities.1 At least 10 morphologic variants of KS have been identified.3 The indolent classic variant of KS most commonly is found in immunocompetent individuals and has been reported to primarily affect elderly men of Jewish or Mediterranean descent.
Epidemiologic analyses of this disease in the South American population are rare. More than 250 cases have been published from South American countries with the largest series published from patients in Argentina, Peru, and Colombia.4 The incidence of classic KS in Peru is 2.54 per 10,000 individuals,5 and the disease has been diagnosed in 1 of 1000 malignant neoplasms at the National Cancer Institute of Colombia.6
A search of the Armed Forces Institute of Pathology Soft Tissue Pathology registry for classic KS patients (1980-2000) showed that 18% of 438 cases of classic KS were diagnosed in South American Hispanics,7 a percentage that nearly approximates the proportion of patients with KS of Mediterranean descent. Interestingly, in an analysis conducted within Los Angeles County, classic KS was most frequently diagnosed in Jewish, Eastern European–born men who were 55 years or older, followed by European-born women. In this study, white, Spanish, and black populations were diagnosed with classic KS with equal frequency.8
Our 2 cases of classic KS from Los Angeles County had several notable features. Both patients were women from Mexico, a demographic not previously associated with classic KS,8 and they did not have risk factors commonly associated with classic KS. We emphasize that classic KS likely is underreported and understudied in the Mexican and South American populations with need for further epidemiological and clinical analyses.
- Kaposi M. Idiopathisches multiples Pigmentsarkom der Haut. Arch Dermatol Syphilol. 1872;4:265-272.
- Akasbi Y, Awada A, Arifi S, et al. Non-HIV Kaposi’s sarcoma: a review and therapeutic perspectives. Bull Cancer. 2012;99:92-99.
- Schwartz RA. Kaposi’s sarcoma: an update. J Surg Oncol. 2004;87:146-151.
- Mohanna S, Maco V, Bravo F, et al. Epidemiology and clinical characteristics of classic Kaposi’s sarcoma, seroprevalence, and variants of human herpesvirus 8 in South America: a critical review of an old disease. Int J Infect Dis. 2005;9:2392-2350.
- Mohanna S, Ferrufino JC, Sanchez J, et al. Epidemiological and clinical characteristics of classic Kaposi’s sarcoma in Peru. J Am Acad Dermatol. 2005;53:435-441.
- García A, Olivella F, Valderrama S, et al. Kaposi’s sarcoma in Colombia. Cancer. 1989;64:2393-2398.
- Hiatt KM, Nelson AM, Lichy JH, et al. Classic Kaposi Sarcoma in the United States over the last two decades: a clinicopathologic and molecular study of 438 non-HIV related Kaposi Sarcoma patients with comparison to HIV-related Kaposi Sarcoma. Mod Pathol. 2008;21:572-582.
- Ross RK, Casagrande JT, Dworsky RL, et al. Kaposi’s sarcoma in Los Angeles, California. J Natl Cancer Inst. 1985;75:1011-1015.
- Kaposi M. Idiopathisches multiples Pigmentsarkom der Haut. Arch Dermatol Syphilol. 1872;4:265-272.
- Akasbi Y, Awada A, Arifi S, et al. Non-HIV Kaposi’s sarcoma: a review and therapeutic perspectives. Bull Cancer. 2012;99:92-99.
- Schwartz RA. Kaposi’s sarcoma: an update. J Surg Oncol. 2004;87:146-151.
- Mohanna S, Maco V, Bravo F, et al. Epidemiology and clinical characteristics of classic Kaposi’s sarcoma, seroprevalence, and variants of human herpesvirus 8 in South America: a critical review of an old disease. Int J Infect Dis. 2005;9:2392-2350.
- Mohanna S, Ferrufino JC, Sanchez J, et al. Epidemiological and clinical characteristics of classic Kaposi’s sarcoma in Peru. J Am Acad Dermatol. 2005;53:435-441.
- García A, Olivella F, Valderrama S, et al. Kaposi’s sarcoma in Colombia. Cancer. 1989;64:2393-2398.
- Hiatt KM, Nelson AM, Lichy JH, et al. Classic Kaposi Sarcoma in the United States over the last two decades: a clinicopathologic and molecular study of 438 non-HIV related Kaposi Sarcoma patients with comparison to HIV-related Kaposi Sarcoma. Mod Pathol. 2008;21:572-582.
- Ross RK, Casagrande JT, Dworsky RL, et al. Kaposi’s sarcoma in Los Angeles, California. J Natl Cancer Inst. 1985;75:1011-1015.
Practice Points
- Classic Kaposi sarcoma is an indolent neoplasm that can be diagnosed in immunocompetent females of Mexican descent.
- Common diagnoses appearing in skin of color may appear morphologically disparate, and a high index of suspicion is needed for correct diagnosis.
Chronic Diarrhea in an Adolescent Girl With a Genetic Skin Condition
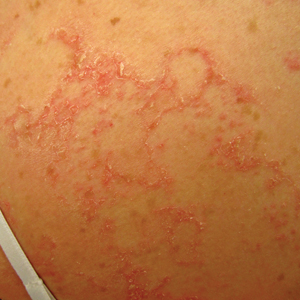
The Diagnosis: Netherton Syndrome
Netherton syndrome (NS) is a rare autosomal-recessive disorder characterized by a clinical triad of ichthyosis linearis circumflexa; atopic diathesis; and hair shaft abnormalities, most classically trichorrhexis invaginata.1 Netherton syndrome is caused by a loss-of-function mutation in the serine peptidase inhibitor Kazal-type gene, SPINK5, which encodes LEKTI proteins and is found in all stratified epithelia as well as the thymus.2 A lack of functional LEKTI leads to the activation of a cascade of allergy and inflammation as well as uncontrolled proteolytic activity in the stratum corneum, which causes increased desquamation.1
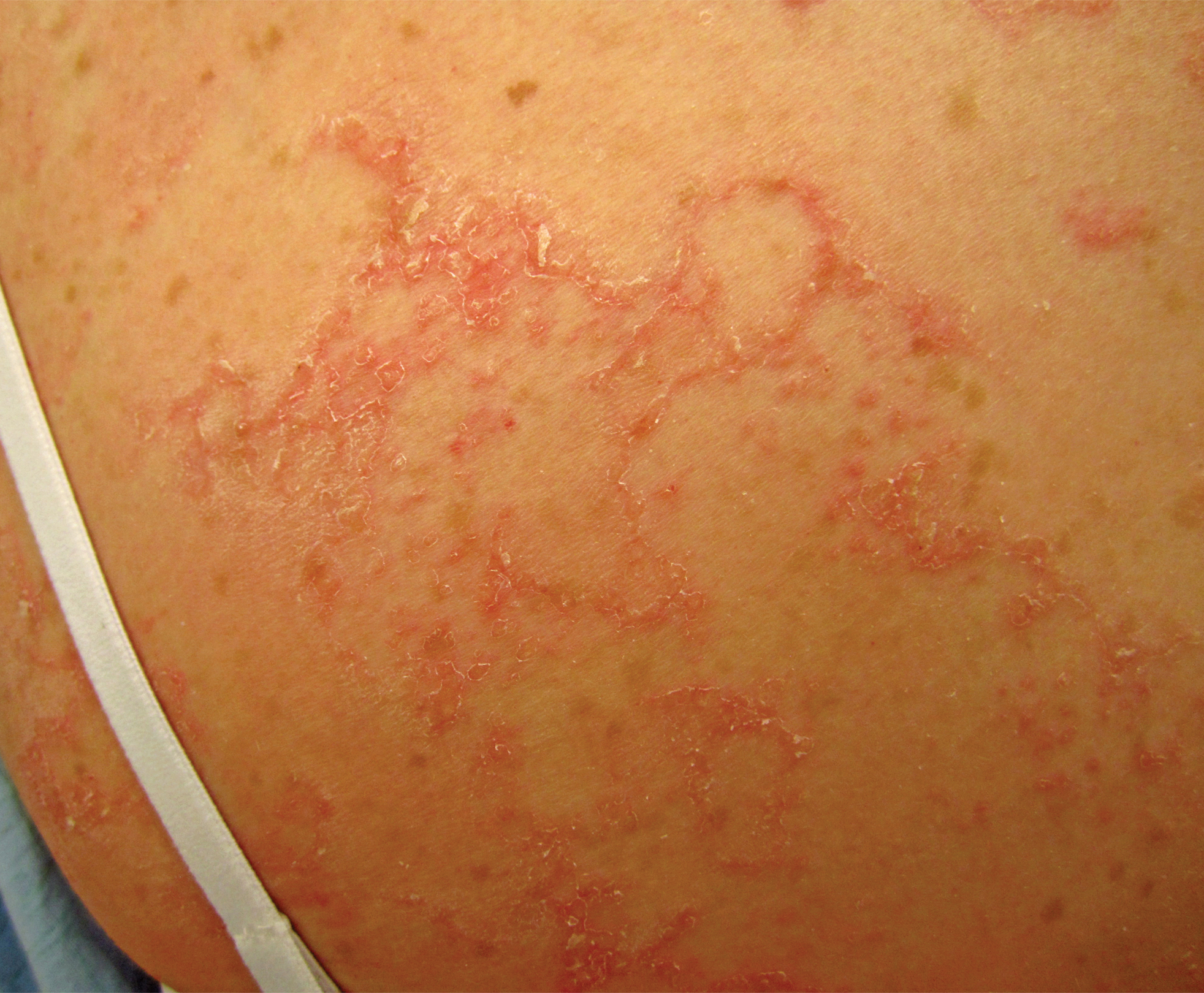
Netherton syndrome presents with serpiginous or circinate scaling plaques with double-edged scale referred to as ichthyosis linearis circumflexa (quiz image). Skin plaques are intensely pruritic and migratory with fluctuating severity. Alternately, patients may have a generalized scaling erythroderma. Infants are at an especially high risk for recurrent infections, sepsis, hypernatremic dehydration, and failure to thrive.2
Netherton syndrome often gradually improves over time, though adults with NS usually have intensely pruritic, localized patches of redness, scaling, or ichthyosis linearis circumflexa. Lichenification and eczematous plaques of the popliteal and antecubital fossae also are common.1 Therapeutic options for NS include emollients, topical steroids, phototherapy, and intravenous immunoglobulin for severe cases.3 Because there is skin barrier dysfunction in NS, supratherapeutic serum levels of tacrolimus following topical application have been reported.4 Topical pimecrolimus has been demonstrated as an effective and safer application.5 Trichorrhexis invaginata (also known as bamboo hair) of the hair and eyebrows is a pathognomonic finding in NS, involving invagination of the distal hair shaft into the proximal shaft on light microscopy examination.1
Histopathology is variable and nonspecific with psoriasiform hyperplasia as the most frequent finding. Other histologic findings include incomplete keratinization of the epidermis, incomplete cornification with a severely reduced granular layer, and mild to moderate inflammatory dermal infiltrate.6 LEKTI immunostaining is confirmatory and shows the reduction or complete absence of LEKTI in the granular layer and inner root sheath of follicles.1 Patchy LEKTI staining would be suggestive of atopic dermatitis and psoriasis instead of NS.2
Atopic manifestations include angioedema, urticaria, and anaphylaxis, as well as chronic diarrhea or vomiting due to food allergies.1 Elevated IgE levels for staple foods (eg, milk, wheat), elevated total serum IgE, and eosinophilia frequently are seen.7 Biopsy of the esophagus and colon likely would show mucosal eosinophilia.7,8 Elimination of major food triggers through specific serum IgE testing and oral allergen desensitization can lead to the reduction of digestive symptoms.9 Cisapride and omeprazole are effective treatments for gastroesophageal reflux and poor feeding.8 Biopsy of the intestines in this patient likely would not have shown total villous atrophy, which is rare and primarily reported in infants with NS who have failure to thrive.10 There is a limited association between NS and intestinal metaplasia, intraepithelial lymphocytes, and bacterial overgrowth.
The primary morphology of dyskeratosis follicularis includes keratotic papules developing in sebaceous areas of the skin rather than scaly serpiginous plaques as seen in NS. Elastosis perforans serpiginosa is a perforating disorder seen in the context of several genetic conditions. It has a serpiginous appearance but, unlike NS, tends to be localized and features keratotic papules rather than patches with scale. Erythema marginatum is an uncommon feature of rheumatic fever and appears as pink annular macules and tends not to be pruritic. Subacute cutaneous lupus does feature scaly annular and serpiginous plaques but features trailing scale without the double-edge appearance of NS and is acquired rather than genetic.
- Hovnanian A. Netherton syndrome: skin inflammation and allergy by loss of protease inhibition. Cell Tissue Res. 2013;351:289-300.
- Bitoun E, Micheloni A, Lamant L, et al. LEKTI proteolytic processing in human primary keratinocytes, tissue distribution and defective expression in Netherton syndrome. Human Mol Genet. 2003;12:2417-2430.
- Yan AC, Honig PJ, Ming ME, et al. The safety and efficacy of pimecrolimus, 1%, cream for the treatment of Netherton syndrome: results from an exploratory study. Arch Dermatol. 2010;146:57-62.
- Shah KN, Yan AC. Low but detectable serum levels of tacrolimus seen with the use of very dilute, extemporaneously compounded formulations of tacrolimus ointment in the treatment of patients with Netherton syndrome. Arch Dermatol. 2006;142:1362-1363.
- Yan AC, Honig PJ, Ming ME, et al. The safety and efficacy of pimecrolimus, 1%, cream for the treatment of Netherton syndrome: results from an exploratory study. Arch Dermatol. 2010;146:57-62.
- Leclerc-Mercier S, Bodemer C, Furio L, et al. Skin biopsy in Netherton syndrome: a histological review of a large series and new findings. Am J Dermatopathol. 2016;38:83-91.
- Pauluel-Marmont C, Bellon N, Barbet P, et al. Eosinophilic esophagitis and colonic mucosal eosinophilia in Netherton syndrome. J Allergy Clin Immunol. 2017;139:2003-2005.e1.
- Hannula-Jouppi K, Laasanen SL, Heikkila H, et al. IgE allergen component-based profiling and atopic manifestations in patients with Netherton syndrome. J Allergy Clin Immunol. 2014;134:985-988.
- Kagalwalla AF, Sentongo TA, Ritz S, et al. Effect of six-food elimination diet on clinical and histologic outcomes in eosinophilic esophagitis. Clin Gastroenterol Hepatol. 2006;4:1097-1102.
- Judge MR, Morgan G , Harper JI. A clinical and immunological study of Netherton's syndrome. Br J Dermatol. 1994;131:615-21.
The Diagnosis: Netherton Syndrome
Netherton syndrome (NS) is a rare autosomal-recessive disorder characterized by a clinical triad of ichthyosis linearis circumflexa; atopic diathesis; and hair shaft abnormalities, most classically trichorrhexis invaginata.1 Netherton syndrome is caused by a loss-of-function mutation in the serine peptidase inhibitor Kazal-type gene, SPINK5, which encodes LEKTI proteins and is found in all stratified epithelia as well as the thymus.2 A lack of functional LEKTI leads to the activation of a cascade of allergy and inflammation as well as uncontrolled proteolytic activity in the stratum corneum, which causes increased desquamation.1
Netherton syndrome presents with serpiginous or circinate scaling plaques with double-edged scale referred to as ichthyosis linearis circumflexa (quiz image). Skin plaques are intensely pruritic and migratory with fluctuating severity. Alternately, patients may have a generalized scaling erythroderma. Infants are at an especially high risk for recurrent infections, sepsis, hypernatremic dehydration, and failure to thrive.2
Netherton syndrome often gradually improves over time, though adults with NS usually have intensely pruritic, localized patches of redness, scaling, or ichthyosis linearis circumflexa. Lichenification and eczematous plaques of the popliteal and antecubital fossae also are common.1 Therapeutic options for NS include emollients, topical steroids, phototherapy, and intravenous immunoglobulin for severe cases.3 Because there is skin barrier dysfunction in NS, supratherapeutic serum levels of tacrolimus following topical application have been reported.4 Topical pimecrolimus has been demonstrated as an effective and safer application.5 Trichorrhexis invaginata (also known as bamboo hair) of the hair and eyebrows is a pathognomonic finding in NS, involving invagination of the distal hair shaft into the proximal shaft on light microscopy examination.1
Histopathology is variable and nonspecific with psoriasiform hyperplasia as the most frequent finding. Other histologic findings include incomplete keratinization of the epidermis, incomplete cornification with a severely reduced granular layer, and mild to moderate inflammatory dermal infiltrate.6 LEKTI immunostaining is confirmatory and shows the reduction or complete absence of LEKTI in the granular layer and inner root sheath of follicles.1 Patchy LEKTI staining would be suggestive of atopic dermatitis and psoriasis instead of NS.2
Atopic manifestations include angioedema, urticaria, and anaphylaxis, as well as chronic diarrhea or vomiting due to food allergies.1 Elevated IgE levels for staple foods (eg, milk, wheat), elevated total serum IgE, and eosinophilia frequently are seen.7 Biopsy of the esophagus and colon likely would show mucosal eosinophilia.7,8 Elimination of major food triggers through specific serum IgE testing and oral allergen desensitization can lead to the reduction of digestive symptoms.9 Cisapride and omeprazole are effective treatments for gastroesophageal reflux and poor feeding.8 Biopsy of the intestines in this patient likely would not have shown total villous atrophy, which is rare and primarily reported in infants with NS who have failure to thrive.10 There is a limited association between NS and intestinal metaplasia, intraepithelial lymphocytes, and bacterial overgrowth.
The primary morphology of dyskeratosis follicularis includes keratotic papules developing in sebaceous areas of the skin rather than scaly serpiginous plaques as seen in NS. Elastosis perforans serpiginosa is a perforating disorder seen in the context of several genetic conditions. It has a serpiginous appearance but, unlike NS, tends to be localized and features keratotic papules rather than patches with scale. Erythema marginatum is an uncommon feature of rheumatic fever and appears as pink annular macules and tends not to be pruritic. Subacute cutaneous lupus does feature scaly annular and serpiginous plaques but features trailing scale without the double-edge appearance of NS and is acquired rather than genetic.
The Diagnosis: Netherton Syndrome
Netherton syndrome (NS) is a rare autosomal-recessive disorder characterized by a clinical triad of ichthyosis linearis circumflexa; atopic diathesis; and hair shaft abnormalities, most classically trichorrhexis invaginata.1 Netherton syndrome is caused by a loss-of-function mutation in the serine peptidase inhibitor Kazal-type gene, SPINK5, which encodes LEKTI proteins and is found in all stratified epithelia as well as the thymus.2 A lack of functional LEKTI leads to the activation of a cascade of allergy and inflammation as well as uncontrolled proteolytic activity in the stratum corneum, which causes increased desquamation.1
Netherton syndrome presents with serpiginous or circinate scaling plaques with double-edged scale referred to as ichthyosis linearis circumflexa (quiz image). Skin plaques are intensely pruritic and migratory with fluctuating severity. Alternately, patients may have a generalized scaling erythroderma. Infants are at an especially high risk for recurrent infections, sepsis, hypernatremic dehydration, and failure to thrive.2
Netherton syndrome often gradually improves over time, though adults with NS usually have intensely pruritic, localized patches of redness, scaling, or ichthyosis linearis circumflexa. Lichenification and eczematous plaques of the popliteal and antecubital fossae also are common.1 Therapeutic options for NS include emollients, topical steroids, phototherapy, and intravenous immunoglobulin for severe cases.3 Because there is skin barrier dysfunction in NS, supratherapeutic serum levels of tacrolimus following topical application have been reported.4 Topical pimecrolimus has been demonstrated as an effective and safer application.5 Trichorrhexis invaginata (also known as bamboo hair) of the hair and eyebrows is a pathognomonic finding in NS, involving invagination of the distal hair shaft into the proximal shaft on light microscopy examination.1
Histopathology is variable and nonspecific with psoriasiform hyperplasia as the most frequent finding. Other histologic findings include incomplete keratinization of the epidermis, incomplete cornification with a severely reduced granular layer, and mild to moderate inflammatory dermal infiltrate.6 LEKTI immunostaining is confirmatory and shows the reduction or complete absence of LEKTI in the granular layer and inner root sheath of follicles.1 Patchy LEKTI staining would be suggestive of atopic dermatitis and psoriasis instead of NS.2
Atopic manifestations include angioedema, urticaria, and anaphylaxis, as well as chronic diarrhea or vomiting due to food allergies.1 Elevated IgE levels for staple foods (eg, milk, wheat), elevated total serum IgE, and eosinophilia frequently are seen.7 Biopsy of the esophagus and colon likely would show mucosal eosinophilia.7,8 Elimination of major food triggers through specific serum IgE testing and oral allergen desensitization can lead to the reduction of digestive symptoms.9 Cisapride and omeprazole are effective treatments for gastroesophageal reflux and poor feeding.8 Biopsy of the intestines in this patient likely would not have shown total villous atrophy, which is rare and primarily reported in infants with NS who have failure to thrive.10 There is a limited association between NS and intestinal metaplasia, intraepithelial lymphocytes, and bacterial overgrowth.
The primary morphology of dyskeratosis follicularis includes keratotic papules developing in sebaceous areas of the skin rather than scaly serpiginous plaques as seen in NS. Elastosis perforans serpiginosa is a perforating disorder seen in the context of several genetic conditions. It has a serpiginous appearance but, unlike NS, tends to be localized and features keratotic papules rather than patches with scale. Erythema marginatum is an uncommon feature of rheumatic fever and appears as pink annular macules and tends not to be pruritic. Subacute cutaneous lupus does feature scaly annular and serpiginous plaques but features trailing scale without the double-edge appearance of NS and is acquired rather than genetic.
- Hovnanian A. Netherton syndrome: skin inflammation and allergy by loss of protease inhibition. Cell Tissue Res. 2013;351:289-300.
- Bitoun E, Micheloni A, Lamant L, et al. LEKTI proteolytic processing in human primary keratinocytes, tissue distribution and defective expression in Netherton syndrome. Human Mol Genet. 2003;12:2417-2430.
- Yan AC, Honig PJ, Ming ME, et al. The safety and efficacy of pimecrolimus, 1%, cream for the treatment of Netherton syndrome: results from an exploratory study. Arch Dermatol. 2010;146:57-62.
- Shah KN, Yan AC. Low but detectable serum levels of tacrolimus seen with the use of very dilute, extemporaneously compounded formulations of tacrolimus ointment in the treatment of patients with Netherton syndrome. Arch Dermatol. 2006;142:1362-1363.
- Yan AC, Honig PJ, Ming ME, et al. The safety and efficacy of pimecrolimus, 1%, cream for the treatment of Netherton syndrome: results from an exploratory study. Arch Dermatol. 2010;146:57-62.
- Leclerc-Mercier S, Bodemer C, Furio L, et al. Skin biopsy in Netherton syndrome: a histological review of a large series and new findings. Am J Dermatopathol. 2016;38:83-91.
- Pauluel-Marmont C, Bellon N, Barbet P, et al. Eosinophilic esophagitis and colonic mucosal eosinophilia in Netherton syndrome. J Allergy Clin Immunol. 2017;139:2003-2005.e1.
- Hannula-Jouppi K, Laasanen SL, Heikkila H, et al. IgE allergen component-based profiling and atopic manifestations in patients with Netherton syndrome. J Allergy Clin Immunol. 2014;134:985-988.
- Kagalwalla AF, Sentongo TA, Ritz S, et al. Effect of six-food elimination diet on clinical and histologic outcomes in eosinophilic esophagitis. Clin Gastroenterol Hepatol. 2006;4:1097-1102.
- Judge MR, Morgan G , Harper JI. A clinical and immunological study of Netherton's syndrome. Br J Dermatol. 1994;131:615-21.
- Hovnanian A. Netherton syndrome: skin inflammation and allergy by loss of protease inhibition. Cell Tissue Res. 2013;351:289-300.
- Bitoun E, Micheloni A, Lamant L, et al. LEKTI proteolytic processing in human primary keratinocytes, tissue distribution and defective expression in Netherton syndrome. Human Mol Genet. 2003;12:2417-2430.
- Yan AC, Honig PJ, Ming ME, et al. The safety and efficacy of pimecrolimus, 1%, cream for the treatment of Netherton syndrome: results from an exploratory study. Arch Dermatol. 2010;146:57-62.
- Shah KN, Yan AC. Low but detectable serum levels of tacrolimus seen with the use of very dilute, extemporaneously compounded formulations of tacrolimus ointment in the treatment of patients with Netherton syndrome. Arch Dermatol. 2006;142:1362-1363.
- Yan AC, Honig PJ, Ming ME, et al. The safety and efficacy of pimecrolimus, 1%, cream for the treatment of Netherton syndrome: results from an exploratory study. Arch Dermatol. 2010;146:57-62.
- Leclerc-Mercier S, Bodemer C, Furio L, et al. Skin biopsy in Netherton syndrome: a histological review of a large series and new findings. Am J Dermatopathol. 2016;38:83-91.
- Pauluel-Marmont C, Bellon N, Barbet P, et al. Eosinophilic esophagitis and colonic mucosal eosinophilia in Netherton syndrome. J Allergy Clin Immunol. 2017;139:2003-2005.e1.
- Hannula-Jouppi K, Laasanen SL, Heikkila H, et al. IgE allergen component-based profiling and atopic manifestations in patients with Netherton syndrome. J Allergy Clin Immunol. 2014;134:985-988.
- Kagalwalla AF, Sentongo TA, Ritz S, et al. Effect of six-food elimination diet on clinical and histologic outcomes in eosinophilic esophagitis. Clin Gastroenterol Hepatol. 2006;4:1097-1102.
- Judge MR, Morgan G , Harper JI. A clinical and immunological study of Netherton's syndrome. Br J Dermatol. 1994;131:615-21.
A 17-year-old adolescent girl visited our clinic to establish care for her genetic skin condition. She exhibited red scaly plaques and patches over much of the body surface area consistent with atopic dermatitis but also had areas on the trunk with serpiginous red plaques with scale on the leading and trailing edges. She also noted fragile hair with sparse eyebrows. The patient reported that she had experienced chronic diarrhea and abdominal pain since childhood. She asked if it could be related to her genetic condition.
Unilateral Vesicular Eruption in a Neonate
The Diagnosis: Incontinentia Pigmenti
The patient was diagnosed clinically with the vesicular stage of incontinentia pigmenti (IP), a rare, X-linked dominant neuroectodermal dysplasia that usually is lethal in males. The genetic mutation has been identified in the IKBKG gene (inhibitor of nuclear factor κB; formally NEMO), which leads to a truncated and defective nuclear factor κB. Female infants survive and display characteristic findings on examination due to X-inactivation leading to mosaicism.1 Worldwide, there are approximately 27.6 new cases of IP per year. Although it is heritable, the majority (65%-75%) of cases are due to sporadic mutations, with the remaining minority (25%-35%) representing familial disease.1
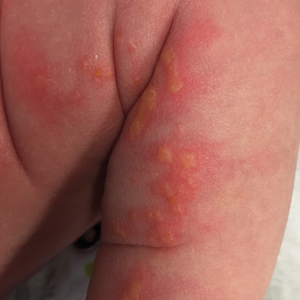
Cutaneous findings of IP classically progress through 4 stages, though individual patients often do not develop the characteristic lesions of each of the 4 stages. The vesicular stage (stage 1) presented in our patient (quiz image). This stage presents within 2 weeks of birth in 90% of patients and typically disappears when the patient is approximately 4 months of age.1-3 Although the clinical presentation is striking, it is essential to rule out herpes simplex virus infection, which can mimic vesicular IP. Localized herpes simplex virus is most commonly seen in clusters on the scalp and often is not present at birth. Alternatively, IP is most often seen on the extremities in bands or whorls of distribution along Blaschko lines,4 as in this patient.
Stage 2 (the verrucous stage) presents with verrucous papules or pustules in a similar blaschkoid distribution. Areas previously involved in stage 1 are not always the same areas affected in stage 2. Approximately 70% of patients develop stage 2 lesions, usually at 2 to 6 weeks of age.1-3 Erythema toxicum neonatorum presents in the first week of life with pustules often on the trunk or extremities, but these lesions are not confined to Blaschko lines, differentiating it from IP.4
The third stage (hyperpigmented stage) lends the disease its name and occurs in 90% to 95% of patients with IP. Linear and whorled hyperpigmentation develops in early infancy and can either persist or fade by adolescence.1 Pustules and hyperpigmentation in transient neonatal pustular melanosis may be similar to this stage of IP, but the distribution is more variable and progression to other lesions is not seen.5
The fourth and final stage is the hypopigmented stage, whereby blaschkoid linear and whorled lines of hypopigmentation with or without both atrophy and alopecia develop in 75% of patients. This is the last finding, beginning in adolescence and often persisting into adulthood.1 Goltz syndrome is another X-linked dominant disorder with features similar to IP. Verrucous and atrophic lesions along Blaschko lines are reminiscent of the second and fourth stages of IP but are differentiated in Goltz syndrome because they present concurrently rather than in sequential stages such as IP. Similar extracutaneous organs are affected such as the eyes, teeth, and nails; however, Goltz syndrome may be associated with more distinguishing systemic signs such as sweating and skeletal abnormalities.6
Given its unique appearance, physicians usually diagnose IP clinically after identification of characteristic linear lesions along the lines of Blaschko in an infant or neonate. Skin biopsy is confirmatory, which would differ depending on the stage of disease biopsied. The vesicular stage is characterized by eosinophilic spongiosis and is differentiated from other items on the histologic differential diagnosis by the presence of dyskeratosis.7 Genetic testing is available and should be performed along with a physical examination of the mother for counseling purposes.1
Proper diagnosis is critical because of the potential multisystem nature of the disease with implications for longitudinal care and prognosis in patients. As in other neurocutaneous disease, IP can affect the hair, nails, teeth, central nervous system, and eyes. All IP patients receive a referral to ophthalmology at the time of diagnosis for a dilated fundus examination, with repeat examinations every several months initially--every 3 months for a year, every 6 months from 1 to 3 years of age--and annually thereafter. Dental evaluation should occur at 6 months of age or whenever the first tooth erupts.1 Mental retardation, seizures, and developmental delay can occur and usually are evident in the first year of life. Patients should have developmental milestones closely monitored and be referred to appropriate specialists if signs or symptoms develop consistent with neurologic involvement.1
- Greene-Roethke C. Incontinentia pigmenti: a summary review of this rare ectodermal dysplasia with neurologic manifestations, including treatment protocols. J Pediatr Health Care. 2017;31:e45-e52.
- Shah KN. Incontinentia pigmenti clinical presentation. Medscape. https://emedicine.medscape.com/article/1114205-clinical. Updated March 5, 2019. Accessed August 2, 2019.
- Poziomczyk CS, Recuero JK, Bringhenti L, et al. Incontinentia pigmenti. An Bras Dermatol. 2014;89:23-36.
- Mathes E, Howard RM. Vesicular, pustular, and bullous lesions in the newborn and infant. UpToDate. https://www.uptodate.com/contents/vesicular-pustular-and-bullous-lesions-in-the-newborn-and-infant. Updated December 3, 2018. Accessed February 20, 2020.
- Ghosh S. Neonatal pustular dermatosis: an overview. Indian J Dermatol. 2015;60:211.
- Temple IK, MacDowall P, Baraitser M, et al. Focal dermal hypoplasia (Goltz syndrome). J Med Genet. 1990;27:180-187.
- Ferringer T. Genodermatoses. In: Elston D, Ferringer T, Ko CJ, et al, eds. Dermatology. 2nd ed. Philadelphia, PA: Elsevier Saunders; 2014:208-213.
The Diagnosis: Incontinentia Pigmenti
The patient was diagnosed clinically with the vesicular stage of incontinentia pigmenti (IP), a rare, X-linked dominant neuroectodermal dysplasia that usually is lethal in males. The genetic mutation has been identified in the IKBKG gene (inhibitor of nuclear factor κB; formally NEMO), which leads to a truncated and defective nuclear factor κB. Female infants survive and display characteristic findings on examination due to X-inactivation leading to mosaicism.1 Worldwide, there are approximately 27.6 new cases of IP per year. Although it is heritable, the majority (65%-75%) of cases are due to sporadic mutations, with the remaining minority (25%-35%) representing familial disease.1
Cutaneous findings of IP classically progress through 4 stages, though individual patients often do not develop the characteristic lesions of each of the 4 stages. The vesicular stage (stage 1) presented in our patient (quiz image). This stage presents within 2 weeks of birth in 90% of patients and typically disappears when the patient is approximately 4 months of age.1-3 Although the clinical presentation is striking, it is essential to rule out herpes simplex virus infection, which can mimic vesicular IP. Localized herpes simplex virus is most commonly seen in clusters on the scalp and often is not present at birth. Alternatively, IP is most often seen on the extremities in bands or whorls of distribution along Blaschko lines,4 as in this patient.
Stage 2 (the verrucous stage) presents with verrucous papules or pustules in a similar blaschkoid distribution. Areas previously involved in stage 1 are not always the same areas affected in stage 2. Approximately 70% of patients develop stage 2 lesions, usually at 2 to 6 weeks of age.1-3 Erythema toxicum neonatorum presents in the first week of life with pustules often on the trunk or extremities, but these lesions are not confined to Blaschko lines, differentiating it from IP.4
The third stage (hyperpigmented stage) lends the disease its name and occurs in 90% to 95% of patients with IP. Linear and whorled hyperpigmentation develops in early infancy and can either persist or fade by adolescence.1 Pustules and hyperpigmentation in transient neonatal pustular melanosis may be similar to this stage of IP, but the distribution is more variable and progression to other lesions is not seen.5
The fourth and final stage is the hypopigmented stage, whereby blaschkoid linear and whorled lines of hypopigmentation with or without both atrophy and alopecia develop in 75% of patients. This is the last finding, beginning in adolescence and often persisting into adulthood.1 Goltz syndrome is another X-linked dominant disorder with features similar to IP. Verrucous and atrophic lesions along Blaschko lines are reminiscent of the second and fourth stages of IP but are differentiated in Goltz syndrome because they present concurrently rather than in sequential stages such as IP. Similar extracutaneous organs are affected such as the eyes, teeth, and nails; however, Goltz syndrome may be associated with more distinguishing systemic signs such as sweating and skeletal abnormalities.6
Given its unique appearance, physicians usually diagnose IP clinically after identification of characteristic linear lesions along the lines of Blaschko in an infant or neonate. Skin biopsy is confirmatory, which would differ depending on the stage of disease biopsied. The vesicular stage is characterized by eosinophilic spongiosis and is differentiated from other items on the histologic differential diagnosis by the presence of dyskeratosis.7 Genetic testing is available and should be performed along with a physical examination of the mother for counseling purposes.1
Proper diagnosis is critical because of the potential multisystem nature of the disease with implications for longitudinal care and prognosis in patients. As in other neurocutaneous disease, IP can affect the hair, nails, teeth, central nervous system, and eyes. All IP patients receive a referral to ophthalmology at the time of diagnosis for a dilated fundus examination, with repeat examinations every several months initially--every 3 months for a year, every 6 months from 1 to 3 years of age--and annually thereafter. Dental evaluation should occur at 6 months of age or whenever the first tooth erupts.1 Mental retardation, seizures, and developmental delay can occur and usually are evident in the first year of life. Patients should have developmental milestones closely monitored and be referred to appropriate specialists if signs or symptoms develop consistent with neurologic involvement.1
The Diagnosis: Incontinentia Pigmenti
The patient was diagnosed clinically with the vesicular stage of incontinentia pigmenti (IP), a rare, X-linked dominant neuroectodermal dysplasia that usually is lethal in males. The genetic mutation has been identified in the IKBKG gene (inhibitor of nuclear factor κB; formally NEMO), which leads to a truncated and defective nuclear factor κB. Female infants survive and display characteristic findings on examination due to X-inactivation leading to mosaicism.1 Worldwide, there are approximately 27.6 new cases of IP per year. Although it is heritable, the majority (65%-75%) of cases are due to sporadic mutations, with the remaining minority (25%-35%) representing familial disease.1
Cutaneous findings of IP classically progress through 4 stages, though individual patients often do not develop the characteristic lesions of each of the 4 stages. The vesicular stage (stage 1) presented in our patient (quiz image). This stage presents within 2 weeks of birth in 90% of patients and typically disappears when the patient is approximately 4 months of age.1-3 Although the clinical presentation is striking, it is essential to rule out herpes simplex virus infection, which can mimic vesicular IP. Localized herpes simplex virus is most commonly seen in clusters on the scalp and often is not present at birth. Alternatively, IP is most often seen on the extremities in bands or whorls of distribution along Blaschko lines,4 as in this patient.
Stage 2 (the verrucous stage) presents with verrucous papules or pustules in a similar blaschkoid distribution. Areas previously involved in stage 1 are not always the same areas affected in stage 2. Approximately 70% of patients develop stage 2 lesions, usually at 2 to 6 weeks of age.1-3 Erythema toxicum neonatorum presents in the first week of life with pustules often on the trunk or extremities, but these lesions are not confined to Blaschko lines, differentiating it from IP.4
The third stage (hyperpigmented stage) lends the disease its name and occurs in 90% to 95% of patients with IP. Linear and whorled hyperpigmentation develops in early infancy and can either persist or fade by adolescence.1 Pustules and hyperpigmentation in transient neonatal pustular melanosis may be similar to this stage of IP, but the distribution is more variable and progression to other lesions is not seen.5
The fourth and final stage is the hypopigmented stage, whereby blaschkoid linear and whorled lines of hypopigmentation with or without both atrophy and alopecia develop in 75% of patients. This is the last finding, beginning in adolescence and often persisting into adulthood.1 Goltz syndrome is another X-linked dominant disorder with features similar to IP. Verrucous and atrophic lesions along Blaschko lines are reminiscent of the second and fourth stages of IP but are differentiated in Goltz syndrome because they present concurrently rather than in sequential stages such as IP. Similar extracutaneous organs are affected such as the eyes, teeth, and nails; however, Goltz syndrome may be associated with more distinguishing systemic signs such as sweating and skeletal abnormalities.6
Given its unique appearance, physicians usually diagnose IP clinically after identification of characteristic linear lesions along the lines of Blaschko in an infant or neonate. Skin biopsy is confirmatory, which would differ depending on the stage of disease biopsied. The vesicular stage is characterized by eosinophilic spongiosis and is differentiated from other items on the histologic differential diagnosis by the presence of dyskeratosis.7 Genetic testing is available and should be performed along with a physical examination of the mother for counseling purposes.1
Proper diagnosis is critical because of the potential multisystem nature of the disease with implications for longitudinal care and prognosis in patients. As in other neurocutaneous disease, IP can affect the hair, nails, teeth, central nervous system, and eyes. All IP patients receive a referral to ophthalmology at the time of diagnosis for a dilated fundus examination, with repeat examinations every several months initially--every 3 months for a year, every 6 months from 1 to 3 years of age--and annually thereafter. Dental evaluation should occur at 6 months of age or whenever the first tooth erupts.1 Mental retardation, seizures, and developmental delay can occur and usually are evident in the first year of life. Patients should have developmental milestones closely monitored and be referred to appropriate specialists if signs or symptoms develop consistent with neurologic involvement.1
- Greene-Roethke C. Incontinentia pigmenti: a summary review of this rare ectodermal dysplasia with neurologic manifestations, including treatment protocols. J Pediatr Health Care. 2017;31:e45-e52.
- Shah KN. Incontinentia pigmenti clinical presentation. Medscape. https://emedicine.medscape.com/article/1114205-clinical. Updated March 5, 2019. Accessed August 2, 2019.
- Poziomczyk CS, Recuero JK, Bringhenti L, et al. Incontinentia pigmenti. An Bras Dermatol. 2014;89:23-36.
- Mathes E, Howard RM. Vesicular, pustular, and bullous lesions in the newborn and infant. UpToDate. https://www.uptodate.com/contents/vesicular-pustular-and-bullous-lesions-in-the-newborn-and-infant. Updated December 3, 2018. Accessed February 20, 2020.
- Ghosh S. Neonatal pustular dermatosis: an overview. Indian J Dermatol. 2015;60:211.
- Temple IK, MacDowall P, Baraitser M, et al. Focal dermal hypoplasia (Goltz syndrome). J Med Genet. 1990;27:180-187.
- Ferringer T. Genodermatoses. In: Elston D, Ferringer T, Ko CJ, et al, eds. Dermatology. 2nd ed. Philadelphia, PA: Elsevier Saunders; 2014:208-213.
- Greene-Roethke C. Incontinentia pigmenti: a summary review of this rare ectodermal dysplasia with neurologic manifestations, including treatment protocols. J Pediatr Health Care. 2017;31:e45-e52.
- Shah KN. Incontinentia pigmenti clinical presentation. Medscape. https://emedicine.medscape.com/article/1114205-clinical. Updated March 5, 2019. Accessed August 2, 2019.
- Poziomczyk CS, Recuero JK, Bringhenti L, et al. Incontinentia pigmenti. An Bras Dermatol. 2014;89:23-36.
- Mathes E, Howard RM. Vesicular, pustular, and bullous lesions in the newborn and infant. UpToDate. https://www.uptodate.com/contents/vesicular-pustular-and-bullous-lesions-in-the-newborn-and-infant. Updated December 3, 2018. Accessed February 20, 2020.
- Ghosh S. Neonatal pustular dermatosis: an overview. Indian J Dermatol. 2015;60:211.
- Temple IK, MacDowall P, Baraitser M, et al. Focal dermal hypoplasia (Goltz syndrome). J Med Genet. 1990;27:180-187.
- Ferringer T. Genodermatoses. In: Elston D, Ferringer T, Ko CJ, et al, eds. Dermatology. 2nd ed. Philadelphia, PA: Elsevier Saunders; 2014:208-213.

A 4-day-old female neonate presented to the dermatology clinic with a vesicular eruption on the left leg of 1 day's duration. The eruption was asymptomatic without any extracutaneous findings. This term infant was born without complication, and the mother denied any symptoms consistent with herpes simplex virus infection. Physical examination revealed yellow-red vesicles on an erythematous base in a blaschkoid distribution on the left leg. The rest of the examination was unremarkable. Herpes simplex virus polymerase chain reaction testing was negative.
CUTIS Celebrates 55 Years
When the first issue of Cutis was published in February 1965:
- Alopecia was featured on the cover
- Eugene F. Traub, MD, was Chief Editor, and John T. McCarthy, MD, was Assistant Chief Editor
- The cost of a year's subscription was $10
- The editorial objective was to bring readers "in simple and concise form the latest in diagnosis, prognosis and treatment" with articles "dealing with common dermatoses or those rarer diseases of great interest"
- From the Consultant's Corner answered the question: Is diet actually important in the treatment of acne vulgaris?
To our loyal readers, contributors, and Editorial Board members, thank you for continuing to turn to Cutis for the latest in diagnosis, prognosis, and treatment.
To our new readers, we hope you find our articles in simple and concise form relevant to your practice.
To our resident readers, you are entering one of the most rewarding specialties in medicine—dermatology.
Access past issues of Cutis online.
When the first issue of Cutis was published in February 1965:
- Alopecia was featured on the cover
- Eugene F. Traub, MD, was Chief Editor, and John T. McCarthy, MD, was Assistant Chief Editor
- The cost of a year's subscription was $10
- The editorial objective was to bring readers "in simple and concise form the latest in diagnosis, prognosis and treatment" with articles "dealing with common dermatoses or those rarer diseases of great interest"
- From the Consultant's Corner answered the question: Is diet actually important in the treatment of acne vulgaris?
To our loyal readers, contributors, and Editorial Board members, thank you for continuing to turn to Cutis for the latest in diagnosis, prognosis, and treatment.
To our new readers, we hope you find our articles in simple and concise form relevant to your practice.
To our resident readers, you are entering one of the most rewarding specialties in medicine—dermatology.
Access past issues of Cutis online.
When the first issue of Cutis was published in February 1965:
- Alopecia was featured on the cover
- Eugene F. Traub, MD, was Chief Editor, and John T. McCarthy, MD, was Assistant Chief Editor
- The cost of a year's subscription was $10
- The editorial objective was to bring readers "in simple and concise form the latest in diagnosis, prognosis and treatment" with articles "dealing with common dermatoses or those rarer diseases of great interest"
- From the Consultant's Corner answered the question: Is diet actually important in the treatment of acne vulgaris?
To our loyal readers, contributors, and Editorial Board members, thank you for continuing to turn to Cutis for the latest in diagnosis, prognosis, and treatment.
To our new readers, we hope you find our articles in simple and concise form relevant to your practice.
To our resident readers, you are entering one of the most rewarding specialties in medicine—dermatology.
Access past issues of Cutis online.
Bleeding Hand Mass in an Older Man
The Diagnosis: Epithelioid Angiosarcoma
Histopathology showed a large soft-tissue neoplasm with extensive hemorrhage (Figure 1). The epithelioid angiosarcoma (EA) consisted mostly of irregular slit-shaped vessels lined by sheets of atypical endothelial cells (Figure 2). At higher-power magnification, the cellular atypia was prominent and diffuse (Figure 3). Immunostaining of the tumor cells showed positive uptake for CD31, confirming vascular origin (Figure 4). Other vascular markers, including CD34 and factor VIII, as well as nuclear positivity for the erythroblast transformation-specific transcription factor gene, ERG, can be demonstrated by EA. Irregular, smooth muscle actin-positive spindle cells are distributed around some of the vessels. The human herpesvirus 8 stain is negative.
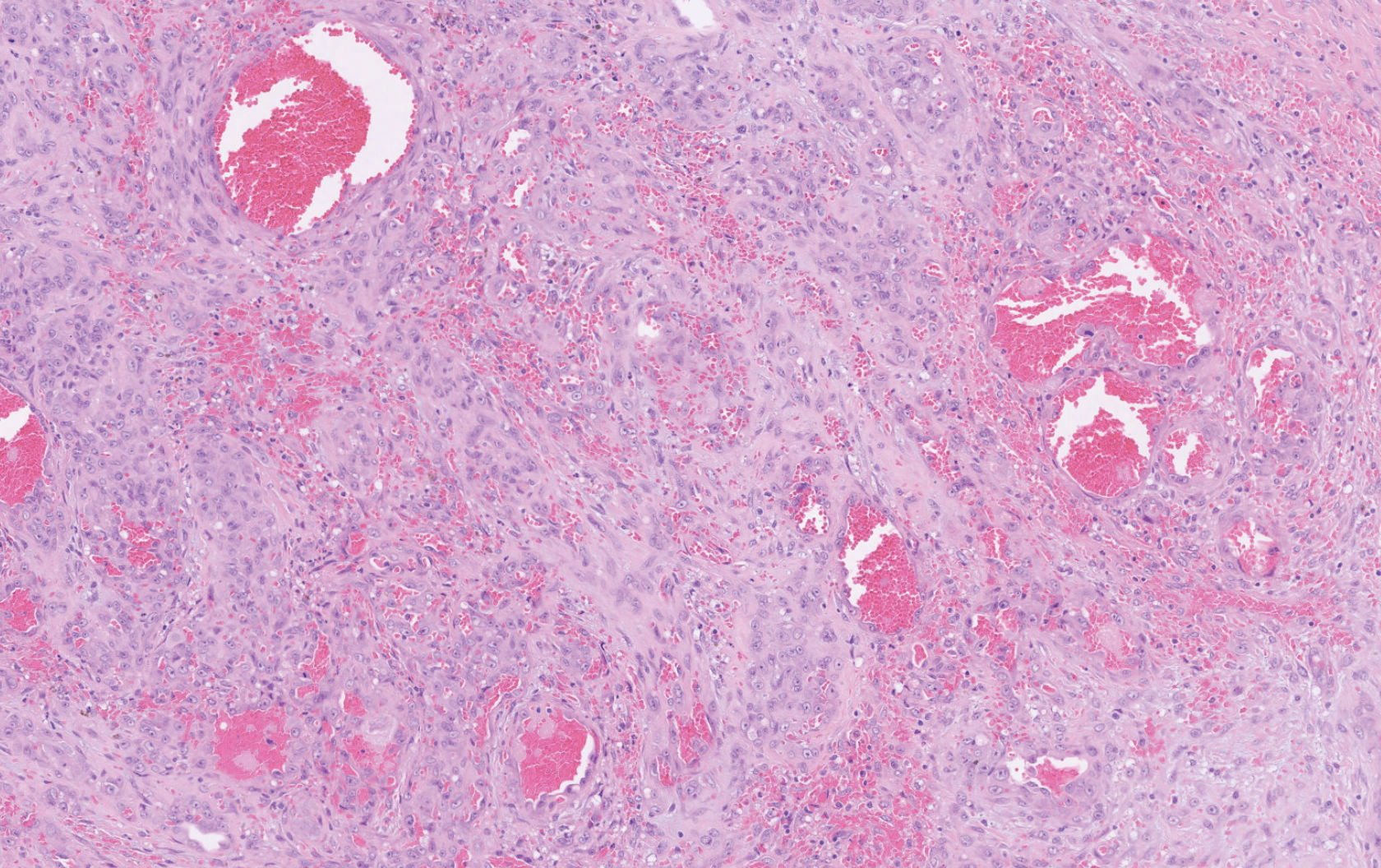
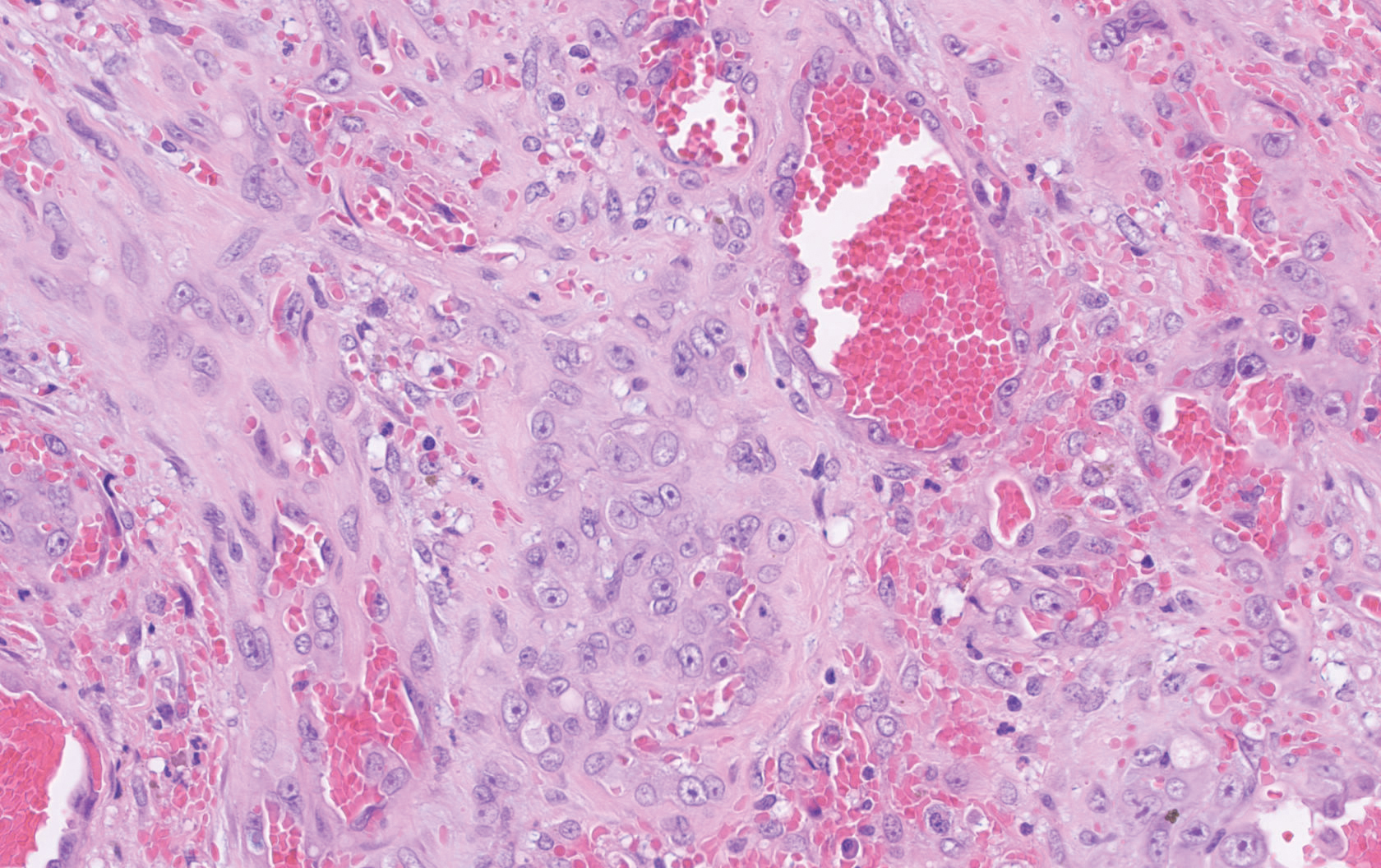

Compared to classic angiosarcomas, EAs have a predilection for the extremities rather than the head and scalp. Histopathologically, the cells are epithelioid and are strongly positive for vimentin and CD31, in addition to factor VIII, friend leukemia integration 1 transcription factor, and CD34.1,2 In contrast, epithelioid sarcomas more typically are seen in younger adults and less likely to be CD31 positive.3 An epithelioid hemangioendothelioma is more focal in cellular atypia and forms small nests and trabeculae rather than sheets of atypical cells. Melanoma cells stain positive for human melanoma black 45, Melan-A, and S-100 but not for CD31.3 Glomangiosarcomas typically stain positive for smooth muscle actin and muscle-specific actin.4
Epithelioid angiosarcomas are rare and aggressive malignancies of endothelial origin.3 They are more prevalent in men and have a peak incidence in the seventh decade of life. They most commonly occur in the deep soft tissues of the extremities but have been reported to form in a variety of primary sites, including the skin, bones, thyroid, and adrenal glands.3
Tumors tend to be highly aggressive and demonstrate early nodal and solid organ metastases.3 Our case demonstrated the aggressive nature of this high-grade malignancy by showing neoplastic invasion through a vascular wall. Within 2 to 3 years of diagnosis, 50% of patients die of the disease, and the 5-year survival rate is estimated to be 12% to 20%.3,5 The etiology remains unknown, but EA has been linked to prior exposure to toxic chemicals, irradiation, or Thorotrast contrast media, and it may arise in the setting of arteriovenous fistulae and chronic lymphedema.6
Although radiation therapy often is utilized, surgery is the primary treatment modality.5 Even with wide excision, local recurrence is common. Tumor size is one of the most important prognostic features, with a worse prognosis for tumors larger than 5 cm. Evidence suggests that paclitaxel-based chemotherapeutic regimens may improve survival, and a combination of paclitaxel and sorafenib has been reported to induce remission in metastatic angiosarcoma of parietal EA.5 Currently, no standardized treatment regimen for this condition exists.
Acknowledgment
The authors thank Amanda Marsch, MD (Chicago, Illinois), for obtaining outside pathology consultation.
- Suchak R, Thway K, Zelger B, et al. Primary cutaneous epithelioid angiosarcoma: a clinicopathologic study of 13 cases of a rare neoplasm occurring outside the setting of conventional angiosarcomas and with predilection for the limbs. Am J Surg Pathol. 2011;35:60-69.
- Prescott RJ, Banerjee SS, Eyden BP, et al. Cutaneous epithelioid angiosarcoma: a clinicopathological study of four cases. Histopathology. 1994;25:421-429.
- Hart J, Mandavilli S. Epithelioid angiosarcoma: a brief diagnostic review and differential diagnosis. Arch Pathol Lab Med. 2011;135:268-272.
- Maselli AM, Jambhekar AV, Hunter JG. Glomangiosarcoma arising from a prior biopsy site. Plast Reconstr Surg Glob Open. 2017;5:e1219.
- Donghi D, Dummer R, Cozzio A. Complete remission in a patient with multifocal metastatic cutaneous angiosarcoma with a combination of paclitaxel and sorafenib. Br J Dermatol. 2010;162:697-699.
- Wu J, Li X, Liu X. Epithelioid angiosarcoma: a clinicopathological study of 16 Chinese cases. Int J Clin Exp Pathol. 2015;8:3901-3909.
The Diagnosis: Epithelioid Angiosarcoma
Histopathology showed a large soft-tissue neoplasm with extensive hemorrhage (Figure 1). The epithelioid angiosarcoma (EA) consisted mostly of irregular slit-shaped vessels lined by sheets of atypical endothelial cells (Figure 2). At higher-power magnification, the cellular atypia was prominent and diffuse (Figure 3). Immunostaining of the tumor cells showed positive uptake for CD31, confirming vascular origin (Figure 4). Other vascular markers, including CD34 and factor VIII, as well as nuclear positivity for the erythroblast transformation-specific transcription factor gene, ERG, can be demonstrated by EA. Irregular, smooth muscle actin-positive spindle cells are distributed around some of the vessels. The human herpesvirus 8 stain is negative.
Compared to classic angiosarcomas, EAs have a predilection for the extremities rather than the head and scalp. Histopathologically, the cells are epithelioid and are strongly positive for vimentin and CD31, in addition to factor VIII, friend leukemia integration 1 transcription factor, and CD34.1,2 In contrast, epithelioid sarcomas more typically are seen in younger adults and less likely to be CD31 positive.3 An epithelioid hemangioendothelioma is more focal in cellular atypia and forms small nests and trabeculae rather than sheets of atypical cells. Melanoma cells stain positive for human melanoma black 45, Melan-A, and S-100 but not for CD31.3 Glomangiosarcomas typically stain positive for smooth muscle actin and muscle-specific actin.4
Epithelioid angiosarcomas are rare and aggressive malignancies of endothelial origin.3 They are more prevalent in men and have a peak incidence in the seventh decade of life. They most commonly occur in the deep soft tissues of the extremities but have been reported to form in a variety of primary sites, including the skin, bones, thyroid, and adrenal glands.3
Tumors tend to be highly aggressive and demonstrate early nodal and solid organ metastases.3 Our case demonstrated the aggressive nature of this high-grade malignancy by showing neoplastic invasion through a vascular wall. Within 2 to 3 years of diagnosis, 50% of patients die of the disease, and the 5-year survival rate is estimated to be 12% to 20%.3,5 The etiology remains unknown, but EA has been linked to prior exposure to toxic chemicals, irradiation, or Thorotrast contrast media, and it may arise in the setting of arteriovenous fistulae and chronic lymphedema.6
Although radiation therapy often is utilized, surgery is the primary treatment modality.5 Even with wide excision, local recurrence is common. Tumor size is one of the most important prognostic features, with a worse prognosis for tumors larger than 5 cm. Evidence suggests that paclitaxel-based chemotherapeutic regimens may improve survival, and a combination of paclitaxel and sorafenib has been reported to induce remission in metastatic angiosarcoma of parietal EA.5 Currently, no standardized treatment regimen for this condition exists.
Acknowledgment
The authors thank Amanda Marsch, MD (Chicago, Illinois), for obtaining outside pathology consultation.
The Diagnosis: Epithelioid Angiosarcoma
Histopathology showed a large soft-tissue neoplasm with extensive hemorrhage (Figure 1). The epithelioid angiosarcoma (EA) consisted mostly of irregular slit-shaped vessels lined by sheets of atypical endothelial cells (Figure 2). At higher-power magnification, the cellular atypia was prominent and diffuse (Figure 3). Immunostaining of the tumor cells showed positive uptake for CD31, confirming vascular origin (Figure 4). Other vascular markers, including CD34 and factor VIII, as well as nuclear positivity for the erythroblast transformation-specific transcription factor gene, ERG, can be demonstrated by EA. Irregular, smooth muscle actin-positive spindle cells are distributed around some of the vessels. The human herpesvirus 8 stain is negative.
Compared to classic angiosarcomas, EAs have a predilection for the extremities rather than the head and scalp. Histopathologically, the cells are epithelioid and are strongly positive for vimentin and CD31, in addition to factor VIII, friend leukemia integration 1 transcription factor, and CD34.1,2 In contrast, epithelioid sarcomas more typically are seen in younger adults and less likely to be CD31 positive.3 An epithelioid hemangioendothelioma is more focal in cellular atypia and forms small nests and trabeculae rather than sheets of atypical cells. Melanoma cells stain positive for human melanoma black 45, Melan-A, and S-100 but not for CD31.3 Glomangiosarcomas typically stain positive for smooth muscle actin and muscle-specific actin.4
Epithelioid angiosarcomas are rare and aggressive malignancies of endothelial origin.3 They are more prevalent in men and have a peak incidence in the seventh decade of life. They most commonly occur in the deep soft tissues of the extremities but have been reported to form in a variety of primary sites, including the skin, bones, thyroid, and adrenal glands.3
Tumors tend to be highly aggressive and demonstrate early nodal and solid organ metastases.3 Our case demonstrated the aggressive nature of this high-grade malignancy by showing neoplastic invasion through a vascular wall. Within 2 to 3 years of diagnosis, 50% of patients die of the disease, and the 5-year survival rate is estimated to be 12% to 20%.3,5 The etiology remains unknown, but EA has been linked to prior exposure to toxic chemicals, irradiation, or Thorotrast contrast media, and it may arise in the setting of arteriovenous fistulae and chronic lymphedema.6
Although radiation therapy often is utilized, surgery is the primary treatment modality.5 Even with wide excision, local recurrence is common. Tumor size is one of the most important prognostic features, with a worse prognosis for tumors larger than 5 cm. Evidence suggests that paclitaxel-based chemotherapeutic regimens may improve survival, and a combination of paclitaxel and sorafenib has been reported to induce remission in metastatic angiosarcoma of parietal EA.5 Currently, no standardized treatment regimen for this condition exists.
Acknowledgment
The authors thank Amanda Marsch, MD (Chicago, Illinois), for obtaining outside pathology consultation.
- Suchak R, Thway K, Zelger B, et al. Primary cutaneous epithelioid angiosarcoma: a clinicopathologic study of 13 cases of a rare neoplasm occurring outside the setting of conventional angiosarcomas and with predilection for the limbs. Am J Surg Pathol. 2011;35:60-69.
- Prescott RJ, Banerjee SS, Eyden BP, et al. Cutaneous epithelioid angiosarcoma: a clinicopathological study of four cases. Histopathology. 1994;25:421-429.
- Hart J, Mandavilli S. Epithelioid angiosarcoma: a brief diagnostic review and differential diagnosis. Arch Pathol Lab Med. 2011;135:268-272.
- Maselli AM, Jambhekar AV, Hunter JG. Glomangiosarcoma arising from a prior biopsy site. Plast Reconstr Surg Glob Open. 2017;5:e1219.
- Donghi D, Dummer R, Cozzio A. Complete remission in a patient with multifocal metastatic cutaneous angiosarcoma with a combination of paclitaxel and sorafenib. Br J Dermatol. 2010;162:697-699.
- Wu J, Li X, Liu X. Epithelioid angiosarcoma: a clinicopathological study of 16 Chinese cases. Int J Clin Exp Pathol. 2015;8:3901-3909.
- Suchak R, Thway K, Zelger B, et al. Primary cutaneous epithelioid angiosarcoma: a clinicopathologic study of 13 cases of a rare neoplasm occurring outside the setting of conventional angiosarcomas and with predilection for the limbs. Am J Surg Pathol. 2011;35:60-69.
- Prescott RJ, Banerjee SS, Eyden BP, et al. Cutaneous epithelioid angiosarcoma: a clinicopathological study of four cases. Histopathology. 1994;25:421-429.
- Hart J, Mandavilli S. Epithelioid angiosarcoma: a brief diagnostic review and differential diagnosis. Arch Pathol Lab Med. 2011;135:268-272.
- Maselli AM, Jambhekar AV, Hunter JG. Glomangiosarcoma arising from a prior biopsy site. Plast Reconstr Surg Glob Open. 2017;5:e1219.
- Donghi D, Dummer R, Cozzio A. Complete remission in a patient with multifocal metastatic cutaneous angiosarcoma with a combination of paclitaxel and sorafenib. Br J Dermatol. 2010;162:697-699.
- Wu J, Li X, Liu X. Epithelioid angiosarcoma: a clinicopathological study of 16 Chinese cases. Int J Clin Exp Pathol. 2015;8:3901-3909.
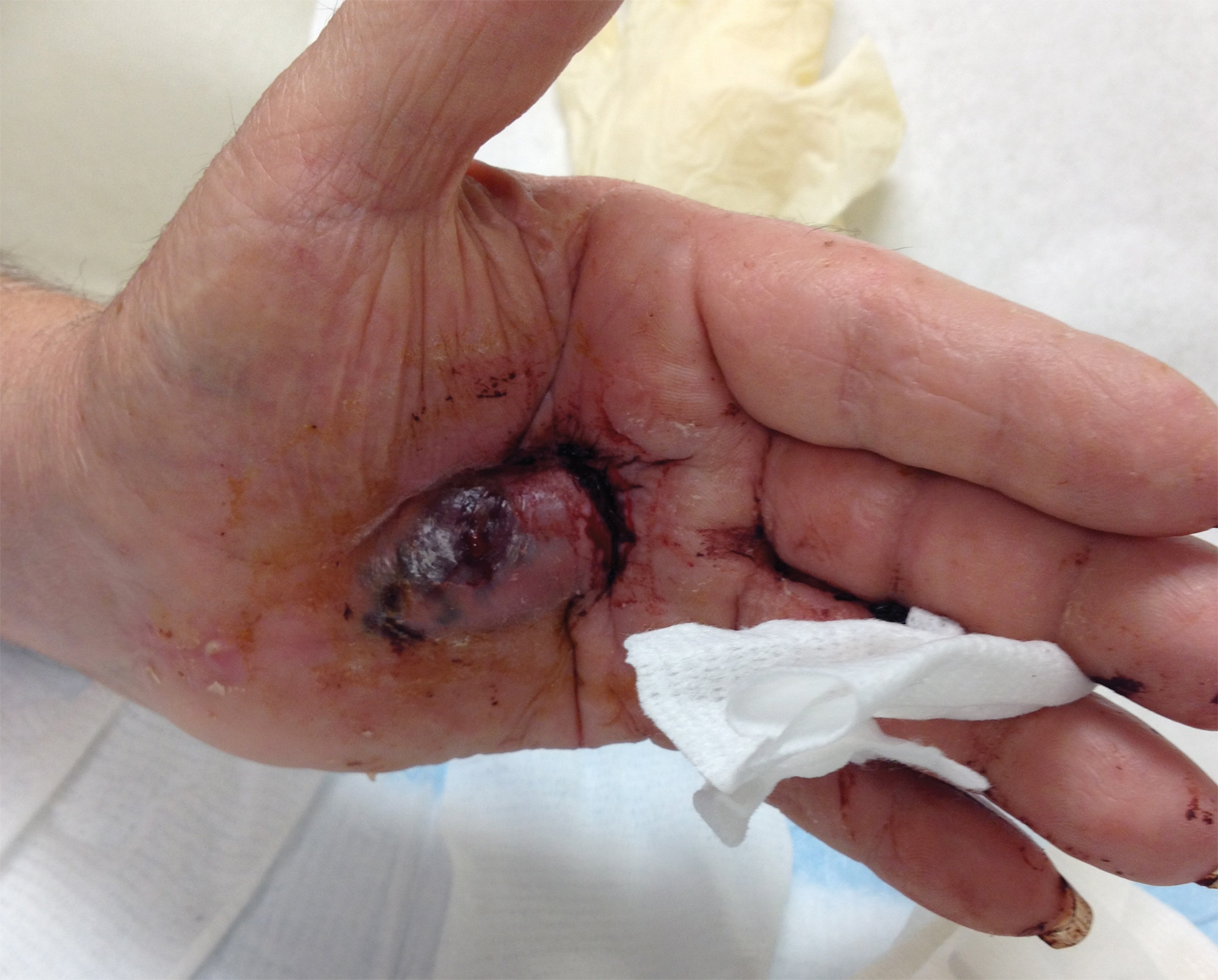
A 72-year-old man presented for evaluation of a mass on the left hand that continued to grow over the last few months and eventually bled. The patient first noticed a small firm lump on the palm approximately 1 year prior to presentation, and it was originally diagnosed as a Dupuytren contracture by his primary care physician. Months later, the lesion grew and began to bleed. Magnetic resonance imaging showed large hematomas of the hand with areas of nodular enhancement. The mass was located between the third and fourth proximal phalanges and abutted the extensor tendon. Complete excision yielded a definitive diagnosis.
Broadly Distributed Vascular Macules in a Pediatric Patient
The Diagnosis: Capillary Malformation-Arteriovenous Malformation Syndrome
Capillary malformation-arteriovenous malformation (CM-AVM) was suspected, and a sample of the patient's blood was sent for a diagnostic genetic workup. DNA sequencing evaluated the following 5 genes that have been implicated in telangiectasia or AVM disorders: ACVRL1 (activin A receptorlike type 1), ENG (endoglin), GDF2 (growth differentiation factor 2), RASA1 (RAS p21 protein activator 1), and SMAD4 (SMAD family member 4). The patient was found to be heterozygous for a known pathogenic splice-site mutation in the RASA1 gene, consistent with a diagnosis of CM-AVM.
Capillary malformation-arteriovenous malformation presents with multiple small cutaneous CMs and associated arteriovenous fistulas as well as high-flow AVMs located in the soft tissues, bones, or central nervous system (CNS). Occasionally, the cutaneous CMs are surrounded by a blanched halo.1 Because of the potential for CNS involvement in CM-AVM, our patient was further evaluated with spine and brain magnetic resonance imaging (MRI). The brain MRI revealed 2 right occipital pole and fusiform gyral AVMs (Figure). No vascular abnormalities were found in the spine. The patient was referred to interventional neuroradiology to assess the feasibility of ablation to reduce the risk for complications, including intracranial hemorrhage.
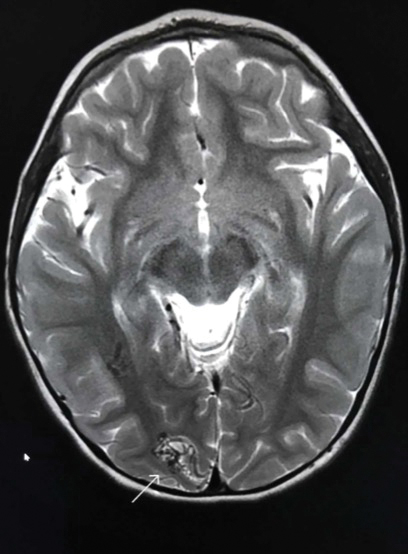
Compared to other well-established congenital vascular disorders, CM-AVM has only recently been described in the literature. It was first reported by Eerola and colleagues2 in 2003. They studied several families with CMs and identified heterozygous inactivating RASA1 mutations in 6 families manifesting atypical CMs that were multiple small, round to oval, and pinkish red.2
It has been estimated that RASA1 mutations contribute to 68% of CM-AVM cases. Another gene--EPHB4 (EPH receptor B4)--has been implicated in patients with RASA1-negative disease. Two separate subtypes for patients with CM-AVM have been described: (1) CM-AVM type 1 for patients with RASA1 mutations, and (2) CM-AVM type 2 for those with EPHB4 mutations.3
Both CM-AVM types are characterized by small multifocal CMs and an increased risk for CNS fast-flow vascular malformations.4 It has been suggested that there are morphologic differences between the cutaneous manifestations of the 2 types. For example, one group stated Bier spots are more frequently observed in CM-AVM type 2. This same group suggested telangiectases seen primarily on the lips but also in the perioral region and on the upper thorax were seen in CM-AVM type 2 but not in CM-AVM type 1.4 In our patient, it is plausible that the pinpoint red macules on the lips and oral mucosa could be confused for telangiectases (quiz image [bottom]). At this time, we do not feel that there is sufficient evidence to clinically distinguish between CM-AVM types 1 and 2.
Central nervous system involvement seems to be more common in patients with CM-AVM type 1 (10%) than those with CM-AVM type 2 (3%).1,4 Of the 2 CM-AVM type 2 patients found to have intracranial AVMs in one study, both were found to have vein of Galen aneurysmal malformations (VGAMs).4 The study examining CNS involvement in CM-AVM type 1 did not comment on the percentage of VGAMs seen in all patients.1 However, in the retrospective component of the study, the authors reported that in 161 patients with CM-AVM type 1, 24 AVMs were observed, 6 of which were intracranial. Half of these intracranial AVMs were at the vein of Galen, demonstrating that VGAMs are seen in both types of CM-AVM.1 Further study is necessary to better characterize potential phenotypic differences between the 2 forms of CM-AVM.
Overall, the annual risk for hemorrhage associated with brain AVMs is approximately 2% per year.5 Because the morbidity and mortality of undiagnosed CNS malformations is high, it is recommended that patients with both types of CM-AVM undergo spine and brain MRI evaluation. If CNS malformations are identified, patients should be referred to interventional neuroradiology to assess the feasibility of ablation.
It is unclear if patients who initially screen negative for AVMs will go on to develop these fast-flow lesions later. We have noted that new CMs develop over time in our patients. Therefore, it does not seem far-fetched to hypothesize that AVMs of CNS are similarly dynamic. Ultimately, we recommend ongoing screening for brain and spinal AVMs at regular intervals, determined by discussions of risks and benefits between the treating team and patient/family.
It is important to distinguish CM-AVM from hereditary hemorrhagic telangiectasia (HHT), as the distinction affects patient management. Unlike the AVMs found in HHT, AVMs in CM-AVM seldom are found in the lungs or liver.1 Thus, asymptomatic patients with HHT, but not CM-AVM, often are screened for pulmonary AVMs.
The diagnosis of HHT is based on the following 4 findings: spontaneous and recurrent epistaxis; multiple mucocutaneous telangiectasia at characteristic sites, including the lips, oral cavity, fingers, and nose; visceral involvement, such as gastrointestinal, pulmonary, cerebral, or hepatic AVMs; and a first-degree relative with the disorder. Three of the criteria are required for diagnosis.
Notably, the lesions seen in HHT and CM-AVM are morphologically different. Our patient did have 1-mm red macules on the lower lip that had clinical features overlapping with telangiectases, but other cutaneous findings including the presence of red macules and small patches, some with blanched halos, were clearly characteristic of CMs, not telangiectases.6 Furthermore, our patient did not have a personal history of epistaxis or a family history of any affected first-degree relatives. Finally, individuals with HHT tend to develop symptoms later in life compared to patients with CM-AVM, starting with epistaxis at 12 years of age.6
Patients with Henoch-Schönlein purpura also present in childhood but typically demonstrate palpable purpura and acute abdominal pain. Patients with Klippel-Trenaunay syndrome present with CM and venous malformation but also typically display limb overgrowth. Most patients with Klippel-Trenaunay syndrome are born with a port-wine stain.
Diffuse neonatal hemangiomatosis is characterized by multiple progressive, rapidly growing cutaneous hemangiomas associated with widespread visceral hemangiomas in the liver, lungs, gastrointestinal tract, brain, and meninges. Our patient's macules were much more slowly progressive.
- Revencu N, Boon LM, Mendola A, et al. RASA1 mutations and associated phenotypes in 68 families with capillary malformation-arteriovenous malformation. Hum Mutat. 2013;34:1632-1641.
- Eerola I, Boon LM, Mulliken JB, et al. Capillary malformation-arteriovenous malformation, a new clinical and genetic disorder caused by RASA1 mutations. Am J Hum Genet. 2003;73:1240-1249.
- Yu J, Streicher JL, Medne L, et al. EPHB4 mutation implicated in capillary malformation-arteriovenous malformation syndrome: a case report. Pediatr Dermatol. 2017;34:227-230.
- Amyere M, Revencu N, Helaers R, et al. Germline loss-of-function mutations in EPHB4 cause a second form of capillary malformation-arteriovenous malformation (CM-AVM2) deregulating RAS-MAPK signaling. Circulation. 2017;136:1037-1048.
- Mohr JP, Parides MK, Stapf C, et al. Medical management with or without interventional therapy for unruptured brain arteriovenous malformations (ARUBA): a multicentre, non-blinded, randomised trial. Lancet. 2014;383:614-621.
- Edwards LR, Blechman AB, Zlotoff BJ. RASA1 mutation in a family with capillary malformation-arteriovenous malformation syndrome: a discussion of the differential diagnosis. Pediatr Dermatol. 2017;35:e9-e12.
The Diagnosis: Capillary Malformation-Arteriovenous Malformation Syndrome
Capillary malformation-arteriovenous malformation (CM-AVM) was suspected, and a sample of the patient's blood was sent for a diagnostic genetic workup. DNA sequencing evaluated the following 5 genes that have been implicated in telangiectasia or AVM disorders: ACVRL1 (activin A receptorlike type 1), ENG (endoglin), GDF2 (growth differentiation factor 2), RASA1 (RAS p21 protein activator 1), and SMAD4 (SMAD family member 4). The patient was found to be heterozygous for a known pathogenic splice-site mutation in the RASA1 gene, consistent with a diagnosis of CM-AVM.
Capillary malformation-arteriovenous malformation presents with multiple small cutaneous CMs and associated arteriovenous fistulas as well as high-flow AVMs located in the soft tissues, bones, or central nervous system (CNS). Occasionally, the cutaneous CMs are surrounded by a blanched halo.1 Because of the potential for CNS involvement in CM-AVM, our patient was further evaluated with spine and brain magnetic resonance imaging (MRI). The brain MRI revealed 2 right occipital pole and fusiform gyral AVMs (Figure). No vascular abnormalities were found in the spine. The patient was referred to interventional neuroradiology to assess the feasibility of ablation to reduce the risk for complications, including intracranial hemorrhage.
Compared to other well-established congenital vascular disorders, CM-AVM has only recently been described in the literature. It was first reported by Eerola and colleagues2 in 2003. They studied several families with CMs and identified heterozygous inactivating RASA1 mutations in 6 families manifesting atypical CMs that were multiple small, round to oval, and pinkish red.2
It has been estimated that RASA1 mutations contribute to 68% of CM-AVM cases. Another gene--EPHB4 (EPH receptor B4)--has been implicated in patients with RASA1-negative disease. Two separate subtypes for patients with CM-AVM have been described: (1) CM-AVM type 1 for patients with RASA1 mutations, and (2) CM-AVM type 2 for those with EPHB4 mutations.3
Both CM-AVM types are characterized by small multifocal CMs and an increased risk for CNS fast-flow vascular malformations.4 It has been suggested that there are morphologic differences between the cutaneous manifestations of the 2 types. For example, one group stated Bier spots are more frequently observed in CM-AVM type 2. This same group suggested telangiectases seen primarily on the lips but also in the perioral region and on the upper thorax were seen in CM-AVM type 2 but not in CM-AVM type 1.4 In our patient, it is plausible that the pinpoint red macules on the lips and oral mucosa could be confused for telangiectases (quiz image [bottom]). At this time, we do not feel that there is sufficient evidence to clinically distinguish between CM-AVM types 1 and 2.
Central nervous system involvement seems to be more common in patients with CM-AVM type 1 (10%) than those with CM-AVM type 2 (3%).1,4 Of the 2 CM-AVM type 2 patients found to have intracranial AVMs in one study, both were found to have vein of Galen aneurysmal malformations (VGAMs).4 The study examining CNS involvement in CM-AVM type 1 did not comment on the percentage of VGAMs seen in all patients.1 However, in the retrospective component of the study, the authors reported that in 161 patients with CM-AVM type 1, 24 AVMs were observed, 6 of which were intracranial. Half of these intracranial AVMs were at the vein of Galen, demonstrating that VGAMs are seen in both types of CM-AVM.1 Further study is necessary to better characterize potential phenotypic differences between the 2 forms of CM-AVM.
Overall, the annual risk for hemorrhage associated with brain AVMs is approximately 2% per year.5 Because the morbidity and mortality of undiagnosed CNS malformations is high, it is recommended that patients with both types of CM-AVM undergo spine and brain MRI evaluation. If CNS malformations are identified, patients should be referred to interventional neuroradiology to assess the feasibility of ablation.
It is unclear if patients who initially screen negative for AVMs will go on to develop these fast-flow lesions later. We have noted that new CMs develop over time in our patients. Therefore, it does not seem far-fetched to hypothesize that AVMs of CNS are similarly dynamic. Ultimately, we recommend ongoing screening for brain and spinal AVMs at regular intervals, determined by discussions of risks and benefits between the treating team and patient/family.
It is important to distinguish CM-AVM from hereditary hemorrhagic telangiectasia (HHT), as the distinction affects patient management. Unlike the AVMs found in HHT, AVMs in CM-AVM seldom are found in the lungs or liver.1 Thus, asymptomatic patients with HHT, but not CM-AVM, often are screened for pulmonary AVMs.
The diagnosis of HHT is based on the following 4 findings: spontaneous and recurrent epistaxis; multiple mucocutaneous telangiectasia at characteristic sites, including the lips, oral cavity, fingers, and nose; visceral involvement, such as gastrointestinal, pulmonary, cerebral, or hepatic AVMs; and a first-degree relative with the disorder. Three of the criteria are required for diagnosis.
Notably, the lesions seen in HHT and CM-AVM are morphologically different. Our patient did have 1-mm red macules on the lower lip that had clinical features overlapping with telangiectases, but other cutaneous findings including the presence of red macules and small patches, some with blanched halos, were clearly characteristic of CMs, not telangiectases.6 Furthermore, our patient did not have a personal history of epistaxis or a family history of any affected first-degree relatives. Finally, individuals with HHT tend to develop symptoms later in life compared to patients with CM-AVM, starting with epistaxis at 12 years of age.6
Patients with Henoch-Schönlein purpura also present in childhood but typically demonstrate palpable purpura and acute abdominal pain. Patients with Klippel-Trenaunay syndrome present with CM and venous malformation but also typically display limb overgrowth. Most patients with Klippel-Trenaunay syndrome are born with a port-wine stain.
Diffuse neonatal hemangiomatosis is characterized by multiple progressive, rapidly growing cutaneous hemangiomas associated with widespread visceral hemangiomas in the liver, lungs, gastrointestinal tract, brain, and meninges. Our patient's macules were much more slowly progressive.
The Diagnosis: Capillary Malformation-Arteriovenous Malformation Syndrome
Capillary malformation-arteriovenous malformation (CM-AVM) was suspected, and a sample of the patient's blood was sent for a diagnostic genetic workup. DNA sequencing evaluated the following 5 genes that have been implicated in telangiectasia or AVM disorders: ACVRL1 (activin A receptorlike type 1), ENG (endoglin), GDF2 (growth differentiation factor 2), RASA1 (RAS p21 protein activator 1), and SMAD4 (SMAD family member 4). The patient was found to be heterozygous for a known pathogenic splice-site mutation in the RASA1 gene, consistent with a diagnosis of CM-AVM.
Capillary malformation-arteriovenous malformation presents with multiple small cutaneous CMs and associated arteriovenous fistulas as well as high-flow AVMs located in the soft tissues, bones, or central nervous system (CNS). Occasionally, the cutaneous CMs are surrounded by a blanched halo.1 Because of the potential for CNS involvement in CM-AVM, our patient was further evaluated with spine and brain magnetic resonance imaging (MRI). The brain MRI revealed 2 right occipital pole and fusiform gyral AVMs (Figure). No vascular abnormalities were found in the spine. The patient was referred to interventional neuroradiology to assess the feasibility of ablation to reduce the risk for complications, including intracranial hemorrhage.
Compared to other well-established congenital vascular disorders, CM-AVM has only recently been described in the literature. It was first reported by Eerola and colleagues2 in 2003. They studied several families with CMs and identified heterozygous inactivating RASA1 mutations in 6 families manifesting atypical CMs that were multiple small, round to oval, and pinkish red.2
It has been estimated that RASA1 mutations contribute to 68% of CM-AVM cases. Another gene--EPHB4 (EPH receptor B4)--has been implicated in patients with RASA1-negative disease. Two separate subtypes for patients with CM-AVM have been described: (1) CM-AVM type 1 for patients with RASA1 mutations, and (2) CM-AVM type 2 for those with EPHB4 mutations.3
Both CM-AVM types are characterized by small multifocal CMs and an increased risk for CNS fast-flow vascular malformations.4 It has been suggested that there are morphologic differences between the cutaneous manifestations of the 2 types. For example, one group stated Bier spots are more frequently observed in CM-AVM type 2. This same group suggested telangiectases seen primarily on the lips but also in the perioral region and on the upper thorax were seen in CM-AVM type 2 but not in CM-AVM type 1.4 In our patient, it is plausible that the pinpoint red macules on the lips and oral mucosa could be confused for telangiectases (quiz image [bottom]). At this time, we do not feel that there is sufficient evidence to clinically distinguish between CM-AVM types 1 and 2.
Central nervous system involvement seems to be more common in patients with CM-AVM type 1 (10%) than those with CM-AVM type 2 (3%).1,4 Of the 2 CM-AVM type 2 patients found to have intracranial AVMs in one study, both were found to have vein of Galen aneurysmal malformations (VGAMs).4 The study examining CNS involvement in CM-AVM type 1 did not comment on the percentage of VGAMs seen in all patients.1 However, in the retrospective component of the study, the authors reported that in 161 patients with CM-AVM type 1, 24 AVMs were observed, 6 of which were intracranial. Half of these intracranial AVMs were at the vein of Galen, demonstrating that VGAMs are seen in both types of CM-AVM.1 Further study is necessary to better characterize potential phenotypic differences between the 2 forms of CM-AVM.
Overall, the annual risk for hemorrhage associated with brain AVMs is approximately 2% per year.5 Because the morbidity and mortality of undiagnosed CNS malformations is high, it is recommended that patients with both types of CM-AVM undergo spine and brain MRI evaluation. If CNS malformations are identified, patients should be referred to interventional neuroradiology to assess the feasibility of ablation.
It is unclear if patients who initially screen negative for AVMs will go on to develop these fast-flow lesions later. We have noted that new CMs develop over time in our patients. Therefore, it does not seem far-fetched to hypothesize that AVMs of CNS are similarly dynamic. Ultimately, we recommend ongoing screening for brain and spinal AVMs at regular intervals, determined by discussions of risks and benefits between the treating team and patient/family.
It is important to distinguish CM-AVM from hereditary hemorrhagic telangiectasia (HHT), as the distinction affects patient management. Unlike the AVMs found in HHT, AVMs in CM-AVM seldom are found in the lungs or liver.1 Thus, asymptomatic patients with HHT, but not CM-AVM, often are screened for pulmonary AVMs.
The diagnosis of HHT is based on the following 4 findings: spontaneous and recurrent epistaxis; multiple mucocutaneous telangiectasia at characteristic sites, including the lips, oral cavity, fingers, and nose; visceral involvement, such as gastrointestinal, pulmonary, cerebral, or hepatic AVMs; and a first-degree relative with the disorder. Three of the criteria are required for diagnosis.
Notably, the lesions seen in HHT and CM-AVM are morphologically different. Our patient did have 1-mm red macules on the lower lip that had clinical features overlapping with telangiectases, but other cutaneous findings including the presence of red macules and small patches, some with blanched halos, were clearly characteristic of CMs, not telangiectases.6 Furthermore, our patient did not have a personal history of epistaxis or a family history of any affected first-degree relatives. Finally, individuals with HHT tend to develop symptoms later in life compared to patients with CM-AVM, starting with epistaxis at 12 years of age.6
Patients with Henoch-Schönlein purpura also present in childhood but typically demonstrate palpable purpura and acute abdominal pain. Patients with Klippel-Trenaunay syndrome present with CM and venous malformation but also typically display limb overgrowth. Most patients with Klippel-Trenaunay syndrome are born with a port-wine stain.
Diffuse neonatal hemangiomatosis is characterized by multiple progressive, rapidly growing cutaneous hemangiomas associated with widespread visceral hemangiomas in the liver, lungs, gastrointestinal tract, brain, and meninges. Our patient's macules were much more slowly progressive.
- Revencu N, Boon LM, Mendola A, et al. RASA1 mutations and associated phenotypes in 68 families with capillary malformation-arteriovenous malformation. Hum Mutat. 2013;34:1632-1641.
- Eerola I, Boon LM, Mulliken JB, et al. Capillary malformation-arteriovenous malformation, a new clinical and genetic disorder caused by RASA1 mutations. Am J Hum Genet. 2003;73:1240-1249.
- Yu J, Streicher JL, Medne L, et al. EPHB4 mutation implicated in capillary malformation-arteriovenous malformation syndrome: a case report. Pediatr Dermatol. 2017;34:227-230.
- Amyere M, Revencu N, Helaers R, et al. Germline loss-of-function mutations in EPHB4 cause a second form of capillary malformation-arteriovenous malformation (CM-AVM2) deregulating RAS-MAPK signaling. Circulation. 2017;136:1037-1048.
- Mohr JP, Parides MK, Stapf C, et al. Medical management with or without interventional therapy for unruptured brain arteriovenous malformations (ARUBA): a multicentre, non-blinded, randomised trial. Lancet. 2014;383:614-621.
- Edwards LR, Blechman AB, Zlotoff BJ. RASA1 mutation in a family with capillary malformation-arteriovenous malformation syndrome: a discussion of the differential diagnosis. Pediatr Dermatol. 2017;35:e9-e12.
- Revencu N, Boon LM, Mendola A, et al. RASA1 mutations and associated phenotypes in 68 families with capillary malformation-arteriovenous malformation. Hum Mutat. 2013;34:1632-1641.
- Eerola I, Boon LM, Mulliken JB, et al. Capillary malformation-arteriovenous malformation, a new clinical and genetic disorder caused by RASA1 mutations. Am J Hum Genet. 2003;73:1240-1249.
- Yu J, Streicher JL, Medne L, et al. EPHB4 mutation implicated in capillary malformation-arteriovenous malformation syndrome: a case report. Pediatr Dermatol. 2017;34:227-230.
- Amyere M, Revencu N, Helaers R, et al. Germline loss-of-function mutations in EPHB4 cause a second form of capillary malformation-arteriovenous malformation (CM-AVM2) deregulating RAS-MAPK signaling. Circulation. 2017;136:1037-1048.
- Mohr JP, Parides MK, Stapf C, et al. Medical management with or without interventional therapy for unruptured brain arteriovenous malformations (ARUBA): a multicentre, non-blinded, randomised trial. Lancet. 2014;383:614-621.
- Edwards LR, Blechman AB, Zlotoff BJ. RASA1 mutation in a family with capillary malformation-arteriovenous malformation syndrome: a discussion of the differential diagnosis. Pediatr Dermatol. 2017;35:e9-e12.
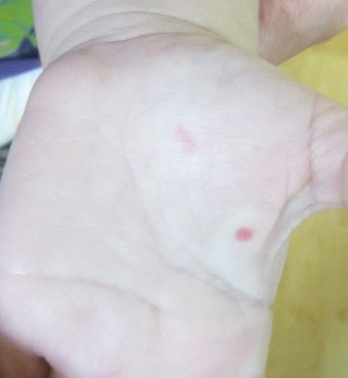
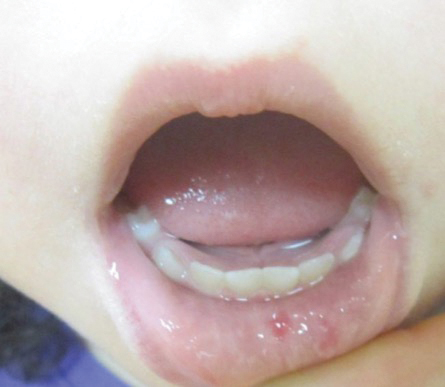
A 2-year-old girl presented with an erythematous macule on the left nasal sidewall that had been present since birth as well as other similar-appearing macules that had slowly evolved over the last 2 years. The patient was born via normal spontaneous vaginal delivery to healthy parents. She had 2 healthy siblings. Her parents reported that she was otherwise growing and developing normally. The patient had no history of epistaxis, and there was no family history of vascular anomalies. Physical examination revealed 2- to 6-mm vascular macules that blanched with pressure and filled quickly thereafter on the left nasal sidewall, upper (top) and lower extremities, and trunk. Some macules were surrounded by blanched halos. Several 1-mm red macules also were noted on the exterior and interior of the mucosal lower lip (bottom).
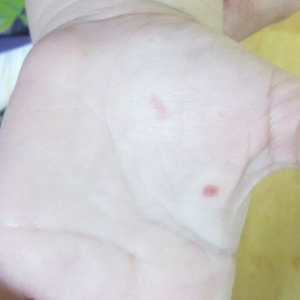
Product News February 2020
Arazlo Lotion Approved for Acne Vulgaris
Ortho Dermatologics announces US Food and Drug Administration approval of Arazlo (tazarotene) Lotion 0.045% for the topical treatment of acne vulgaris in patients 9 years and older. Arazlo provides the efficacy expected of tazarotene in a lotion formulation that helps minimize dryness and irritation historically associated with retinoids. Arazlo is expected to be available for prescription in the first half of 2020. For more information, visit https://ortho-dermatologics.com.
FDA Clears Lutronic LaseMD Ultra
Lutronic receives US Food and Drug Administration clearance of Lutronic LaseMD Ultra, a 1927-nm nonablative fractional laser to treat lentigos, solar lentigos, ephelides, actinic keratoses, and other benign pigmented lesions in all skin types. Lutronic LaseMD Ultra offers 20 W of power to treat multiple areas quickly and an advanced graphical user interface for customizable outcomes. For more information, visit https://us.aesthetic.lutronic.com.
18th World Congress on Cancers of the Skin
The 18th World Congress on Cancers of the Skin will be held in Buenos Aires, Argentina, from June 24 to June 27, 2020. The event is organized by Colegio Ibero Latinoamericano de Dermatología (CILAD) and cosponsored by The Skin Cancer Foundation. For more information, visit https://www.wccs2020.com/.
Wynzora Cream for Plaque Psoriasis
MC2 Therapeutics announces Wynzora Cream (calcipotriene 0.005% and betamethasone dipropionate 0.064%) has met its primary end point for its phase 3 trial in Europe in adult patients with plaque psoriasis. Wynzora Cream utilizes PAD Technology, which enables stability of both calcipotriene and betamethasone dipropionate in an aqueous cream
formulation. It offers patients with plaque psoriasis a once-daily topical treatment that can be quickly absorbed into the skin, is not greasy, and is convenient for routine use. The US Food and Drug Administration accepted the New Drug Application seeking marketing approval for Wynzora Cream. The combination of clinical efficacy, favorable safety profile, and high convenience of Wynzora Cream demonstrated in clinical trials hold promise to increase treatment adherence and overall patient satisfaction in the real-world setting. For more information, visit https://www.mc2therapeutics.com/.
If you would like your product included in Product News, please email a press release to the Editorial Office at cutis@mdedge.com.
Arazlo Lotion Approved for Acne Vulgaris
Ortho Dermatologics announces US Food and Drug Administration approval of Arazlo (tazarotene) Lotion 0.045% for the topical treatment of acne vulgaris in patients 9 years and older. Arazlo provides the efficacy expected of tazarotene in a lotion formulation that helps minimize dryness and irritation historically associated with retinoids. Arazlo is expected to be available for prescription in the first half of 2020. For more information, visit https://ortho-dermatologics.com.
FDA Clears Lutronic LaseMD Ultra
Lutronic receives US Food and Drug Administration clearance of Lutronic LaseMD Ultra, a 1927-nm nonablative fractional laser to treat lentigos, solar lentigos, ephelides, actinic keratoses, and other benign pigmented lesions in all skin types. Lutronic LaseMD Ultra offers 20 W of power to treat multiple areas quickly and an advanced graphical user interface for customizable outcomes. For more information, visit https://us.aesthetic.lutronic.com.
18th World Congress on Cancers of the Skin
The 18th World Congress on Cancers of the Skin will be held in Buenos Aires, Argentina, from June 24 to June 27, 2020. The event is organized by Colegio Ibero Latinoamericano de Dermatología (CILAD) and cosponsored by The Skin Cancer Foundation. For more information, visit https://www.wccs2020.com/.
Wynzora Cream for Plaque Psoriasis
MC2 Therapeutics announces Wynzora Cream (calcipotriene 0.005% and betamethasone dipropionate 0.064%) has met its primary end point for its phase 3 trial in Europe in adult patients with plaque psoriasis. Wynzora Cream utilizes PAD Technology, which enables stability of both calcipotriene and betamethasone dipropionate in an aqueous cream
formulation. It offers patients with plaque psoriasis a once-daily topical treatment that can be quickly absorbed into the skin, is not greasy, and is convenient for routine use. The US Food and Drug Administration accepted the New Drug Application seeking marketing approval for Wynzora Cream. The combination of clinical efficacy, favorable safety profile, and high convenience of Wynzora Cream demonstrated in clinical trials hold promise to increase treatment adherence and overall patient satisfaction in the real-world setting. For more information, visit https://www.mc2therapeutics.com/.
If you would like your product included in Product News, please email a press release to the Editorial Office at cutis@mdedge.com.
Arazlo Lotion Approved for Acne Vulgaris
Ortho Dermatologics announces US Food and Drug Administration approval of Arazlo (tazarotene) Lotion 0.045% for the topical treatment of acne vulgaris in patients 9 years and older. Arazlo provides the efficacy expected of tazarotene in a lotion formulation that helps minimize dryness and irritation historically associated with retinoids. Arazlo is expected to be available for prescription in the first half of 2020. For more information, visit https://ortho-dermatologics.com.
FDA Clears Lutronic LaseMD Ultra
Lutronic receives US Food and Drug Administration clearance of Lutronic LaseMD Ultra, a 1927-nm nonablative fractional laser to treat lentigos, solar lentigos, ephelides, actinic keratoses, and other benign pigmented lesions in all skin types. Lutronic LaseMD Ultra offers 20 W of power to treat multiple areas quickly and an advanced graphical user interface for customizable outcomes. For more information, visit https://us.aesthetic.lutronic.com.
18th World Congress on Cancers of the Skin
The 18th World Congress on Cancers of the Skin will be held in Buenos Aires, Argentina, from June 24 to June 27, 2020. The event is organized by Colegio Ibero Latinoamericano de Dermatología (CILAD) and cosponsored by The Skin Cancer Foundation. For more information, visit https://www.wccs2020.com/.
Wynzora Cream for Plaque Psoriasis
MC2 Therapeutics announces Wynzora Cream (calcipotriene 0.005% and betamethasone dipropionate 0.064%) has met its primary end point for its phase 3 trial in Europe in adult patients with plaque psoriasis. Wynzora Cream utilizes PAD Technology, which enables stability of both calcipotriene and betamethasone dipropionate in an aqueous cream
formulation. It offers patients with plaque psoriasis a once-daily topical treatment that can be quickly absorbed into the skin, is not greasy, and is convenient for routine use. The US Food and Drug Administration accepted the New Drug Application seeking marketing approval for Wynzora Cream. The combination of clinical efficacy, favorable safety profile, and high convenience of Wynzora Cream demonstrated in clinical trials hold promise to increase treatment adherence and overall patient satisfaction in the real-world setting. For more information, visit https://www.mc2therapeutics.com/.
If you would like your product included in Product News, please email a press release to the Editorial Office at cutis@mdedge.com.
Antineutrophil Cytoplasmic Antibody Vasculitis Induced by Hydralazine
To the Editor:
Hydralazine-induced antineutrophil cytoplasmic antibody vasculitis (HIAV) is a rare side effect that may develop in patients treated with hydralazine. Without early recognition and hydralazine cessation, patients often develop acute renal failure and pulmonary hemorrhage that may result in death. We present a case of HIAV.
A 67-year-old woman presented with progressive, tense, hemorrhagic, and necrotic bullae on both sides of the face and neck as well as the extremities of 2 weeks’ duration. She had a history of hypertension and a thyroid nodule after unilateral thyroid lobectomy. A review of symptoms was positive for worsening dyspnea and progressive generalized weakness. Noteworthy medications included amlodipine, metoprolol, levothyroxine, and oral hydralazine 75 mg 3 times daily for 13 months.
Bullae first appeared on the patient’s scalp and quickly progressed with a cephalocaudal pattern with a propensity for the eyes, nostrils, and labial mucosa (Figure 1). The tongue was covered by an eschar, and she had diffuse periorbital edema. Additionally, concentric purpuric patches were noted on the thighs and lower legs (Figure 2).
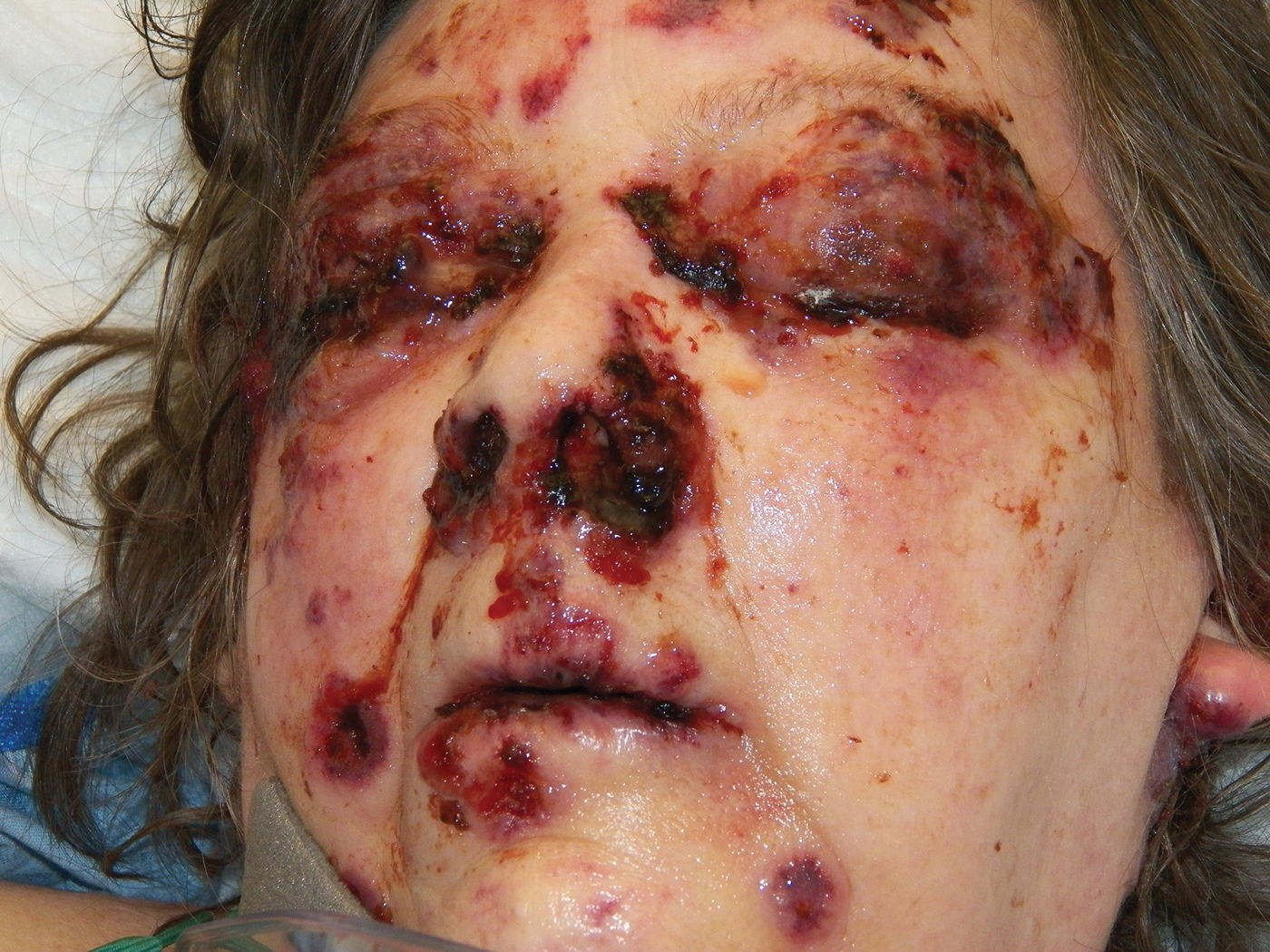
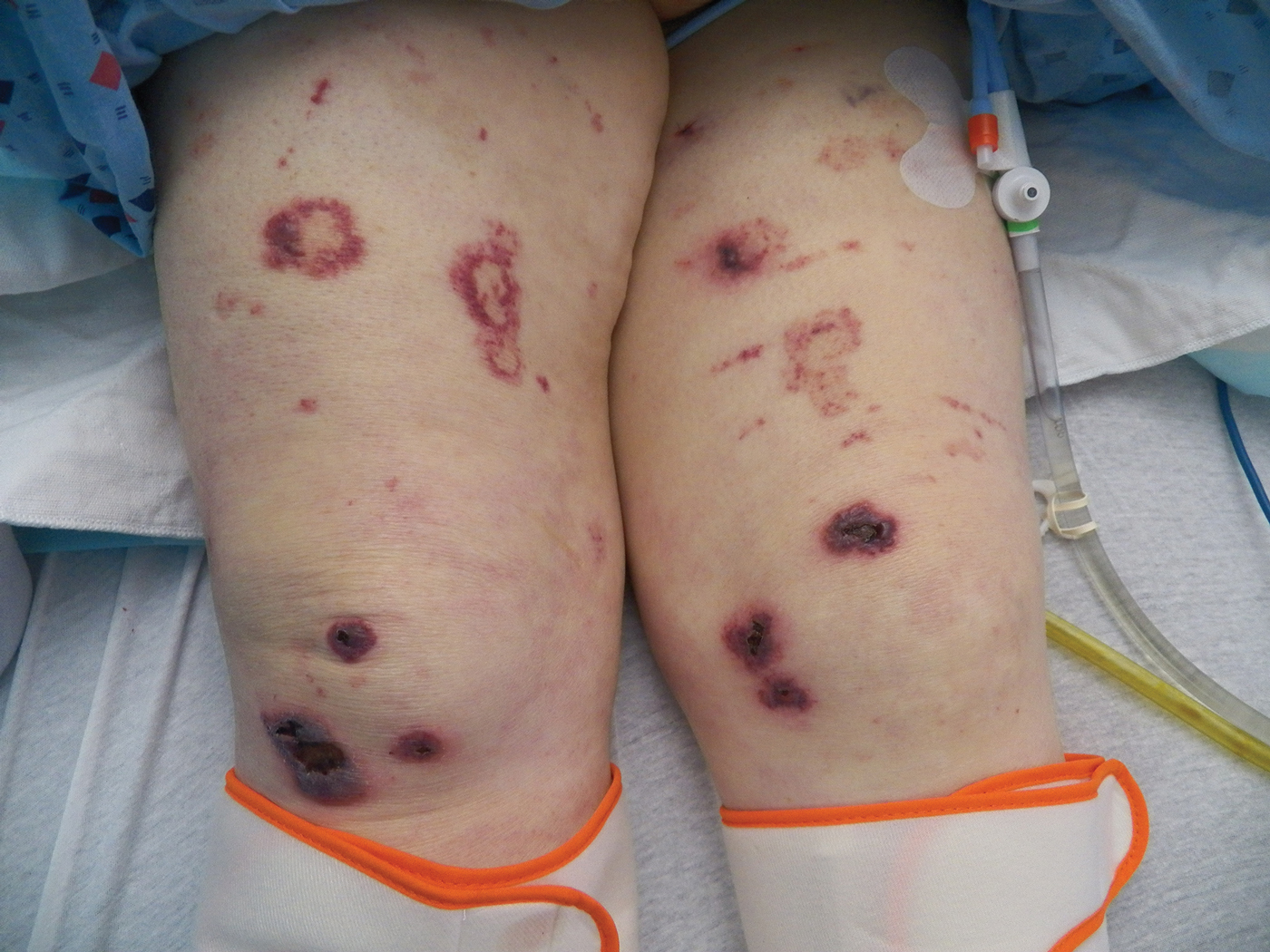
Pertinent laboratory findings included a positive antinuclear antibody titer of 1:320 and perinuclear antineutrophil cytoplasmic antibody (ANCA) titer of 1:160, along with an elevated serum creatinine level (2.31 mg/dL [reference range, 0.6–1.2 mg/dL]). Bilateral perihilar infiltrates with bilateral pleural effusions were noted on a chest radiograph.
While hospitalized, she developed pulmonary hemorrhages and a progressive decline in respiratory status. She subsequently was admitted to the medical intensive care unit. Aggressive support was administered, and several skin biopsy specimens were obtained along with an endobronchial biopsy of the right middle lobe.
Skin histopathology revealed a necrotic vasculitis (Figure 3). Direct immunofluorescence was not performed. Lung histopathology showed fragments of bronchial tissue with acute and chronic inflammation, focal necrosis, granulation tissue formation, edema, and squamous metaplasia. Together with the clinical history, these findings were consistent with HIAV.

Hydralazine was immediately discontinued, and the patient was started on 65 mg daily of intravenous methylprednisolone; methylprednisolone was later changed to oral prednisone 30 mg daily. Due to multiple organ involvement—lung and kidney—intravenous rituximab 375 mg/m2 every week for 4 weeks, per lymphoma protocol, was started. Within 2 weeks of beginning therapy, her renal function and respiratory status improved, and by week 4, the skin lesions had completely resolved. Although initially she did well on immunosuppressive therapy with resolution of all symptoms, the patient contracted Clostridium difficile–induced systemic inflammatory response syndrome after 5 weeks of therapy and died.
Hydralazine was first introduced in 1951 for adjunctive hypertension therapy due to its vasodilation effects.1-3 Since its introduction, it has been implicated in 2 important disease processes: HIAV and hydralazine-induced lupus.
Hydralazine-induced ANCA vasculitis was first documented in 1980; by 2011, multiple cases had been reported.1-7 Hydralazine-induced ANCA vasculitis has occurred in patients aged 11 to 79 years taking 50 to 300 mg daily. Symptom onset varies from 6 months to 14 years, with a mean exposure duration of 4.7 years and mean daily dose of 142 mg.1-7
Clinical manifestations range from less specific, such as fever, malaise, arthralgia, myalgia, and weight loss, to single tissue or organ involvement that may be fatal. The most frequent clinical features include kidney involvement (81%), cutaneous vasculitis (25%), arthralgia (24%), and pleuropulmonary involvement (19%). Cutaneous manifestations include but are not limited to palpable lower extremity purpura; morbilliform eruptions; and hemorrhagic blisters on the lower legs, arms, trunk, nasal septum, and uvula.1-4,8
The most commonly affected organ is the kidney, which commonly presents as hematuria, proteinuria, and elevated serum creatinine level. Histopathologically, patients most likely will have necrotizing and crescentic glomerulonephritis that is pauci-immune by immunofluorescence.7,9 The lungs are the next most commonly affected organ, with a classic presentation of cough, dyspnea, and hemoptysis in the setting of intra-alveolar hemorrhage.6,8 When both the kidneys and lungs are involved, the patient is said to have pulmonary-renal syndrome that is characterized by lung infiltrates or nodules with or without hemorrhage, hemoptysis, and pleuritis in the setting of glomerulonephritis.1,6
Clear data on incidence and prevalence of HIAV does not exist due to the rarity of the disease and the lack of prospective studies. To identify a clear incidence and prevalence, prospective longitudinal studies with larger cohorts along with better recognition and diagnosis are needed.2,8,10 A few predisposing risk factors have been identified, including older age, a cumulative dose of 100 g at the time of presentation, female sex, a history of thyroid disease, HLA-DR4 genotypes, slow hepatic acetylation, and the null gene for C4.1,3,5,9-11 Our patient was an older woman with a history of thyroid disease who had been taking oral hydralazine 75 mg 3 times daily for 13 months. During this 13-month duration, she had no dose adjustments.
Currently, the pathomechanism for HIAV is unclear and may be multifactorial. There are 4 main theories2,8-10,12,13:
1. Hydralazine and its metabolites accumulate inside neutrophils, then subsequently bind and alter the configuration of myeloperoxidase (MPO). This alteration leads to spreading of the autoimmune response to other autoantigens, making neutrophil proteins (eg, elastase, lactoferrin, nuclear antigens) immunogenic.
2. Hydralazine binds MPO in neutrophils, creating cytotoxic products that induce neutrophil apoptosis. Neutrophil apoptosis without priming then results in ANCA antigen presence on the neutrophil cell membrane and the formation of MPO-ANCA. Myeloperoxidase-ANCA then binds to these membrane-bound antigens that cause self-perpetuating, constitutive activation through cross-linking with proteinase 3 or MPO and Fcγ receptors.
3. Activated neutrophils in the presence of hydrogen peroxidase release MPO that converts hydralazine into a cytotoxic product that is immunogenic for T cells that activate ANCA-producing B cells.
4. Histone H3 trimethyl Lys27 (H3K27me3) levels are perturbed in HIAV, which leads to aberrant gene silencing of proteinase 3 and MPO.In contrast, the demethylase Jumonji domain-containing protein 3 for the H3K27me3 histone is increased in patients without HIAV. Based on this data and the data showing a role for hydralazine in reversing epigenetic silencing of tumor suppressor genes in cancer cells,13 it has been proposed that hydralazine may reverse epigenetic silencing of proteinase 3 and MPO.
Diagnosing HIAV is still difficult because physicians do not recognize the drug as the etiologic agent, there is extensive variability in duration between starting the drug and onset of symptoms, and there often is a failure to order the appropriate laboratory and invasive tests needed for evaluation and diagnosis.3,5,8,10,12 Despite these difficulties, a set of criteria and practices for diagnosis are delineated in Table 1, with the key diagnostic feature being resolution with hydralazine cessation.1,5,7,8,12
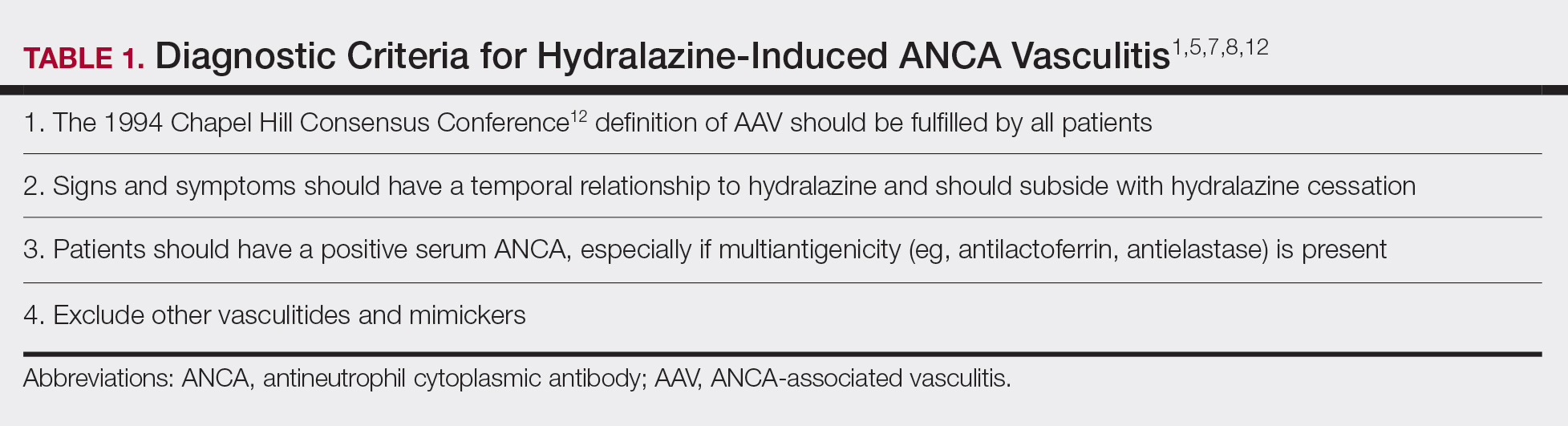
A comprehensive drug history from at least 6 months prior to presentation is essential. Biopsies also are strongly encouraged to confirm the presence of vasculitis and to determine its severity.8,12 If renal biopsies are performed, they typically show scant IgG, IgM, and C3 deposition that is characteristic of ANCA-positive pauci-immune glomerulonephritis. Compared to hydralazine-induced lupus, renal involvement in the setting of HIAV has a relative lack of immunoglobulin and complement deposition with histopathology and immunostaining.14
Laboratory test results including serum MPO-ANCA (perinuclear ANCA) with coexisting elastase and/or lactoferrin autoantibodies is characteristic of HIAV. Antinuclear antibody, antihistone, anti–double-stranded DNA, and antiphospholipid antibodies along with low complement levels also may be present.2,4,9,10,13,15 It is recommended that ANCA assays combine indirect immunofluorescence with antigen-specific enzyme-linked immunosorbent assay.8 With respect to its idiopathic counterpart, patients may only present with MPO-ANCA, while other aforementioned antibodies (eg, antihistone, anti–double-stranded DNA) are rarely found or are entirely absent.2,9 Patients with HIAV often have higher titers of MPO-ANCA.9,15 In hydralazine-induced lupus, patients rarely have MPO-ANCA.
When a diagnosis of HIAV is made, it cannot be confirmed until hydralazine is discontinued and the patient’s symptoms resolve. Therefore, it is both diagnostic and therapeutic to discontinue hydralazine when HIAV is suspected. If recognized when the patient is only presenting with nonspecific symptoms, simple hydralazine cessation may be all that is needed; however, because recognition and diagnosis of HIAV is difficult, most patients present when the disease is severe and has progressed to organ involvement.8-10
Treatment recommendations are highlighted in Table 2.8,9,12 Glucocorticoid therapy is believed to work by preventing T-cell and B-cell maturation needed to produce MPO-ANCA. Rituximab, on the other hand, is suspected to act by clearing the peripheral blood of MPO-ANCA B cells.12,16 Of note, patients with HIAV are different from their idiopathic counterparts because they usually need shorter courses of immunosuppressive therapy, long-term maintenance usually is unnecessary, and their prognosis generally is good if the offending agent is withdrawn.7-9,12 Once the appropriate therapy is instituted, vasculitic manifestations are expected to resolve 10 days to 8 months after hydralazine cessation; however, a response often is seen within 1 to 4 weeks after initiation of systemic treatment.4,8 Serum ANCA should be monitored, and there should be surveillance for the emergence of a chronic underlying vasculitis.8,12
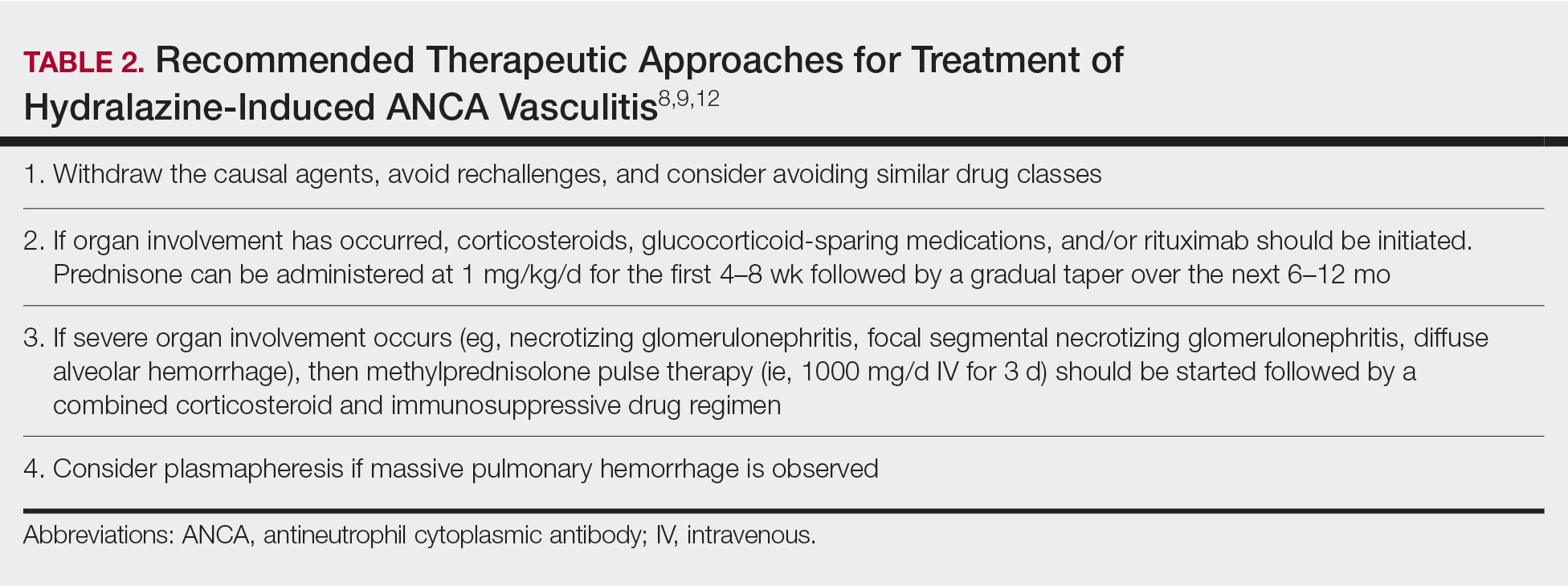
Our patient highlights the importance of identifying individuals at risk for HIAV. We seek to increase recognition of this entity, as it is not commonly seen in a dermatologic setting and is associated with high morbidity and mortality, as seen in our patient.
- Yokogawa N, Vivino FB. Hydralazine-induced autoimmune disease: comparison to idiopathic lupus and ANCA-positive vasculitis. Mod Rheumatol. 2009;19:338-347.
- Agarwal G, Sultan G, Werner SL, et al. Hydralazine induces myeloperoxidase and proteinase 3 anti-neutrophil cytoplasmic antibody vasculitis and leads to pulmonary renal syndrome. Case Rep Nephrol. 2014;2014:868590.
- Keasberry J, Frazier J, Isbel NM, et al. Hydralazine-induced anti-neutrophilic cytoplasmic antibody-positive renal vasculitis presenting with a vasculitic syndrome, acute nephritis and a puzzling skin rash: a case report. J Med Case Rep. 2013;7:20.
- ten Holder SM, Joy MS, Falk RJ. Cutaneous and systemic manifestations of drug-induced vasculitis. Ann Pharmacother. 2002;36:130-147.
- Namas R, Rubin B, Adwar W, et al. A challenging twist in pulmonary renal syndrome. Case Rep Rheumatol. 2014;2014:516362.
- Dobre M, Wish J, Negrea L. Hydralazine-induced ANCA-positive pauci-immune glomerulonephritis. Ren Fail. 2009;31:745-748.
- Hogan JJ, Markowitz GS, Radhakrishnan J. Drug-induced glomerular disease: immune-mediated injury. Clin J Am Soc Nephrol. 2015;10:1300-1310.
- Radic M, Martinovic Kaliterna D, Radic J. Drug-induced vasculitis: a clinical and pathological review. Neth J Med. 2012;70:12-17.
- Babar F, Posner JN, Obah EA. Hydralazine-induced pauci-immune glomerulonephritis: intriguing case series misleading diagnoses. J Community Hosp Intern Med Perspect. 2016;6:30632.
- Marina VP, Malhotra D, Kaw D. Hydralazine-induced ANCA vasculitis with pulmonary renal syndrome: a rare clinical presentation. Int Urol Nephrol. 2012;44:1907-1909.
- Magro CM. Associated ANCA positive vasculitis. The Dermatologist. 2015;23(7). http://www.the-dermatologist.com/content/associated-anca-positive-vasculitis. Accessed January 30, 2020.
- Gao Y, Zhao MH. Review article: Drug-induced anti-neutrophil cytoplasmic antibody-associated vasculitis. Nephrology (Carlton). 2009;14:33-41.
- Grau RG. Drug-induced vasculitis: new insights and a changing lineup of suspects. Curr Rheumatol Rep. 2015;17:71.
- Sangala N, Lee RW, Horsfield C, et al. Combined ANCA-associated vasculitis and lupus syndrome following prolonged use of hydralazine: a timely reminder of an old foe. Int Urol Nephrol. 2010;42:503-506.
- Choi HK, Merkel PA, Walker AM, et al. Drug-associated antineutrophil cytoplasmic antibody-positive vasculitis: prevalence among patients with high titers of antimyeloperoxidase antibodies. Arthritis Rheum. 2000;43:405-413.
- Coutinho AE, Chapman KE. The anti-inflammatory and immunosuppressive effects of glucocorticoids, recent developments and mechanistic insights. Mol Cell Endocrinol. 2011;335:2-13.
To the Editor:
Hydralazine-induced antineutrophil cytoplasmic antibody vasculitis (HIAV) is a rare side effect that may develop in patients treated with hydralazine. Without early recognition and hydralazine cessation, patients often develop acute renal failure and pulmonary hemorrhage that may result in death. We present a case of HIAV.
A 67-year-old woman presented with progressive, tense, hemorrhagic, and necrotic bullae on both sides of the face and neck as well as the extremities of 2 weeks’ duration. She had a history of hypertension and a thyroid nodule after unilateral thyroid lobectomy. A review of symptoms was positive for worsening dyspnea and progressive generalized weakness. Noteworthy medications included amlodipine, metoprolol, levothyroxine, and oral hydralazine 75 mg 3 times daily for 13 months.
Bullae first appeared on the patient’s scalp and quickly progressed with a cephalocaudal pattern with a propensity for the eyes, nostrils, and labial mucosa (Figure 1). The tongue was covered by an eschar, and she had diffuse periorbital edema. Additionally, concentric purpuric patches were noted on the thighs and lower legs (Figure 2).
Pertinent laboratory findings included a positive antinuclear antibody titer of 1:320 and perinuclear antineutrophil cytoplasmic antibody (ANCA) titer of 1:160, along with an elevated serum creatinine level (2.31 mg/dL [reference range, 0.6–1.2 mg/dL]). Bilateral perihilar infiltrates with bilateral pleural effusions were noted on a chest radiograph.
While hospitalized, she developed pulmonary hemorrhages and a progressive decline in respiratory status. She subsequently was admitted to the medical intensive care unit. Aggressive support was administered, and several skin biopsy specimens were obtained along with an endobronchial biopsy of the right middle lobe.
Skin histopathology revealed a necrotic vasculitis (Figure 3). Direct immunofluorescence was not performed. Lung histopathology showed fragments of bronchial tissue with acute and chronic inflammation, focal necrosis, granulation tissue formation, edema, and squamous metaplasia. Together with the clinical history, these findings were consistent with HIAV.
Hydralazine was immediately discontinued, and the patient was started on 65 mg daily of intravenous methylprednisolone; methylprednisolone was later changed to oral prednisone 30 mg daily. Due to multiple organ involvement—lung and kidney—intravenous rituximab 375 mg/m2 every week for 4 weeks, per lymphoma protocol, was started. Within 2 weeks of beginning therapy, her renal function and respiratory status improved, and by week 4, the skin lesions had completely resolved. Although initially she did well on immunosuppressive therapy with resolution of all symptoms, the patient contracted Clostridium difficile–induced systemic inflammatory response syndrome after 5 weeks of therapy and died.
Hydralazine was first introduced in 1951 for adjunctive hypertension therapy due to its vasodilation effects.1-3 Since its introduction, it has been implicated in 2 important disease processes: HIAV and hydralazine-induced lupus.
Hydralazine-induced ANCA vasculitis was first documented in 1980; by 2011, multiple cases had been reported.1-7 Hydralazine-induced ANCA vasculitis has occurred in patients aged 11 to 79 years taking 50 to 300 mg daily. Symptom onset varies from 6 months to 14 years, with a mean exposure duration of 4.7 years and mean daily dose of 142 mg.1-7
Clinical manifestations range from less specific, such as fever, malaise, arthralgia, myalgia, and weight loss, to single tissue or organ involvement that may be fatal. The most frequent clinical features include kidney involvement (81%), cutaneous vasculitis (25%), arthralgia (24%), and pleuropulmonary involvement (19%). Cutaneous manifestations include but are not limited to palpable lower extremity purpura; morbilliform eruptions; and hemorrhagic blisters on the lower legs, arms, trunk, nasal septum, and uvula.1-4,8
The most commonly affected organ is the kidney, which commonly presents as hematuria, proteinuria, and elevated serum creatinine level. Histopathologically, patients most likely will have necrotizing and crescentic glomerulonephritis that is pauci-immune by immunofluorescence.7,9 The lungs are the next most commonly affected organ, with a classic presentation of cough, dyspnea, and hemoptysis in the setting of intra-alveolar hemorrhage.6,8 When both the kidneys and lungs are involved, the patient is said to have pulmonary-renal syndrome that is characterized by lung infiltrates or nodules with or without hemorrhage, hemoptysis, and pleuritis in the setting of glomerulonephritis.1,6
Clear data on incidence and prevalence of HIAV does not exist due to the rarity of the disease and the lack of prospective studies. To identify a clear incidence and prevalence, prospective longitudinal studies with larger cohorts along with better recognition and diagnosis are needed.2,8,10 A few predisposing risk factors have been identified, including older age, a cumulative dose of 100 g at the time of presentation, female sex, a history of thyroid disease, HLA-DR4 genotypes, slow hepatic acetylation, and the null gene for C4.1,3,5,9-11 Our patient was an older woman with a history of thyroid disease who had been taking oral hydralazine 75 mg 3 times daily for 13 months. During this 13-month duration, she had no dose adjustments.
Currently, the pathomechanism for HIAV is unclear and may be multifactorial. There are 4 main theories2,8-10,12,13:
1. Hydralazine and its metabolites accumulate inside neutrophils, then subsequently bind and alter the configuration of myeloperoxidase (MPO). This alteration leads to spreading of the autoimmune response to other autoantigens, making neutrophil proteins (eg, elastase, lactoferrin, nuclear antigens) immunogenic.
2. Hydralazine binds MPO in neutrophils, creating cytotoxic products that induce neutrophil apoptosis. Neutrophil apoptosis without priming then results in ANCA antigen presence on the neutrophil cell membrane and the formation of MPO-ANCA. Myeloperoxidase-ANCA then binds to these membrane-bound antigens that cause self-perpetuating, constitutive activation through cross-linking with proteinase 3 or MPO and Fcγ receptors.
3. Activated neutrophils in the presence of hydrogen peroxidase release MPO that converts hydralazine into a cytotoxic product that is immunogenic for T cells that activate ANCA-producing B cells.
4. Histone H3 trimethyl Lys27 (H3K27me3) levels are perturbed in HIAV, which leads to aberrant gene silencing of proteinase 3 and MPO.In contrast, the demethylase Jumonji domain-containing protein 3 for the H3K27me3 histone is increased in patients without HIAV. Based on this data and the data showing a role for hydralazine in reversing epigenetic silencing of tumor suppressor genes in cancer cells,13 it has been proposed that hydralazine may reverse epigenetic silencing of proteinase 3 and MPO.
Diagnosing HIAV is still difficult because physicians do not recognize the drug as the etiologic agent, there is extensive variability in duration between starting the drug and onset of symptoms, and there often is a failure to order the appropriate laboratory and invasive tests needed for evaluation and diagnosis.3,5,8,10,12 Despite these difficulties, a set of criteria and practices for diagnosis are delineated in Table 1, with the key diagnostic feature being resolution with hydralazine cessation.1,5,7,8,12
A comprehensive drug history from at least 6 months prior to presentation is essential. Biopsies also are strongly encouraged to confirm the presence of vasculitis and to determine its severity.8,12 If renal biopsies are performed, they typically show scant IgG, IgM, and C3 deposition that is characteristic of ANCA-positive pauci-immune glomerulonephritis. Compared to hydralazine-induced lupus, renal involvement in the setting of HIAV has a relative lack of immunoglobulin and complement deposition with histopathology and immunostaining.14
Laboratory test results including serum MPO-ANCA (perinuclear ANCA) with coexisting elastase and/or lactoferrin autoantibodies is characteristic of HIAV. Antinuclear antibody, antihistone, anti–double-stranded DNA, and antiphospholipid antibodies along with low complement levels also may be present.2,4,9,10,13,15 It is recommended that ANCA assays combine indirect immunofluorescence with antigen-specific enzyme-linked immunosorbent assay.8 With respect to its idiopathic counterpart, patients may only present with MPO-ANCA, while other aforementioned antibodies (eg, antihistone, anti–double-stranded DNA) are rarely found or are entirely absent.2,9 Patients with HIAV often have higher titers of MPO-ANCA.9,15 In hydralazine-induced lupus, patients rarely have MPO-ANCA.
When a diagnosis of HIAV is made, it cannot be confirmed until hydralazine is discontinued and the patient’s symptoms resolve. Therefore, it is both diagnostic and therapeutic to discontinue hydralazine when HIAV is suspected. If recognized when the patient is only presenting with nonspecific symptoms, simple hydralazine cessation may be all that is needed; however, because recognition and diagnosis of HIAV is difficult, most patients present when the disease is severe and has progressed to organ involvement.8-10
Treatment recommendations are highlighted in Table 2.8,9,12 Glucocorticoid therapy is believed to work by preventing T-cell and B-cell maturation needed to produce MPO-ANCA. Rituximab, on the other hand, is suspected to act by clearing the peripheral blood of MPO-ANCA B cells.12,16 Of note, patients with HIAV are different from their idiopathic counterparts because they usually need shorter courses of immunosuppressive therapy, long-term maintenance usually is unnecessary, and their prognosis generally is good if the offending agent is withdrawn.7-9,12 Once the appropriate therapy is instituted, vasculitic manifestations are expected to resolve 10 days to 8 months after hydralazine cessation; however, a response often is seen within 1 to 4 weeks after initiation of systemic treatment.4,8 Serum ANCA should be monitored, and there should be surveillance for the emergence of a chronic underlying vasculitis.8,12
Our patient highlights the importance of identifying individuals at risk for HIAV. We seek to increase recognition of this entity, as it is not commonly seen in a dermatologic setting and is associated with high morbidity and mortality, as seen in our patient.
To the Editor:
Hydralazine-induced antineutrophil cytoplasmic antibody vasculitis (HIAV) is a rare side effect that may develop in patients treated with hydralazine. Without early recognition and hydralazine cessation, patients often develop acute renal failure and pulmonary hemorrhage that may result in death. We present a case of HIAV.
A 67-year-old woman presented with progressive, tense, hemorrhagic, and necrotic bullae on both sides of the face and neck as well as the extremities of 2 weeks’ duration. She had a history of hypertension and a thyroid nodule after unilateral thyroid lobectomy. A review of symptoms was positive for worsening dyspnea and progressive generalized weakness. Noteworthy medications included amlodipine, metoprolol, levothyroxine, and oral hydralazine 75 mg 3 times daily for 13 months.
Bullae first appeared on the patient’s scalp and quickly progressed with a cephalocaudal pattern with a propensity for the eyes, nostrils, and labial mucosa (Figure 1). The tongue was covered by an eschar, and she had diffuse periorbital edema. Additionally, concentric purpuric patches were noted on the thighs and lower legs (Figure 2).
Pertinent laboratory findings included a positive antinuclear antibody titer of 1:320 and perinuclear antineutrophil cytoplasmic antibody (ANCA) titer of 1:160, along with an elevated serum creatinine level (2.31 mg/dL [reference range, 0.6–1.2 mg/dL]). Bilateral perihilar infiltrates with bilateral pleural effusions were noted on a chest radiograph.
While hospitalized, she developed pulmonary hemorrhages and a progressive decline in respiratory status. She subsequently was admitted to the medical intensive care unit. Aggressive support was administered, and several skin biopsy specimens were obtained along with an endobronchial biopsy of the right middle lobe.
Skin histopathology revealed a necrotic vasculitis (Figure 3). Direct immunofluorescence was not performed. Lung histopathology showed fragments of bronchial tissue with acute and chronic inflammation, focal necrosis, granulation tissue formation, edema, and squamous metaplasia. Together with the clinical history, these findings were consistent with HIAV.
Hydralazine was immediately discontinued, and the patient was started on 65 mg daily of intravenous methylprednisolone; methylprednisolone was later changed to oral prednisone 30 mg daily. Due to multiple organ involvement—lung and kidney—intravenous rituximab 375 mg/m2 every week for 4 weeks, per lymphoma protocol, was started. Within 2 weeks of beginning therapy, her renal function and respiratory status improved, and by week 4, the skin lesions had completely resolved. Although initially she did well on immunosuppressive therapy with resolution of all symptoms, the patient contracted Clostridium difficile–induced systemic inflammatory response syndrome after 5 weeks of therapy and died.
Hydralazine was first introduced in 1951 for adjunctive hypertension therapy due to its vasodilation effects.1-3 Since its introduction, it has been implicated in 2 important disease processes: HIAV and hydralazine-induced lupus.
Hydralazine-induced ANCA vasculitis was first documented in 1980; by 2011, multiple cases had been reported.1-7 Hydralazine-induced ANCA vasculitis has occurred in patients aged 11 to 79 years taking 50 to 300 mg daily. Symptom onset varies from 6 months to 14 years, with a mean exposure duration of 4.7 years and mean daily dose of 142 mg.1-7
Clinical manifestations range from less specific, such as fever, malaise, arthralgia, myalgia, and weight loss, to single tissue or organ involvement that may be fatal. The most frequent clinical features include kidney involvement (81%), cutaneous vasculitis (25%), arthralgia (24%), and pleuropulmonary involvement (19%). Cutaneous manifestations include but are not limited to palpable lower extremity purpura; morbilliform eruptions; and hemorrhagic blisters on the lower legs, arms, trunk, nasal septum, and uvula.1-4,8
The most commonly affected organ is the kidney, which commonly presents as hematuria, proteinuria, and elevated serum creatinine level. Histopathologically, patients most likely will have necrotizing and crescentic glomerulonephritis that is pauci-immune by immunofluorescence.7,9 The lungs are the next most commonly affected organ, with a classic presentation of cough, dyspnea, and hemoptysis in the setting of intra-alveolar hemorrhage.6,8 When both the kidneys and lungs are involved, the patient is said to have pulmonary-renal syndrome that is characterized by lung infiltrates or nodules with or without hemorrhage, hemoptysis, and pleuritis in the setting of glomerulonephritis.1,6
Clear data on incidence and prevalence of HIAV does not exist due to the rarity of the disease and the lack of prospective studies. To identify a clear incidence and prevalence, prospective longitudinal studies with larger cohorts along with better recognition and diagnosis are needed.2,8,10 A few predisposing risk factors have been identified, including older age, a cumulative dose of 100 g at the time of presentation, female sex, a history of thyroid disease, HLA-DR4 genotypes, slow hepatic acetylation, and the null gene for C4.1,3,5,9-11 Our patient was an older woman with a history of thyroid disease who had been taking oral hydralazine 75 mg 3 times daily for 13 months. During this 13-month duration, she had no dose adjustments.
Currently, the pathomechanism for HIAV is unclear and may be multifactorial. There are 4 main theories2,8-10,12,13:
1. Hydralazine and its metabolites accumulate inside neutrophils, then subsequently bind and alter the configuration of myeloperoxidase (MPO). This alteration leads to spreading of the autoimmune response to other autoantigens, making neutrophil proteins (eg, elastase, lactoferrin, nuclear antigens) immunogenic.
2. Hydralazine binds MPO in neutrophils, creating cytotoxic products that induce neutrophil apoptosis. Neutrophil apoptosis without priming then results in ANCA antigen presence on the neutrophil cell membrane and the formation of MPO-ANCA. Myeloperoxidase-ANCA then binds to these membrane-bound antigens that cause self-perpetuating, constitutive activation through cross-linking with proteinase 3 or MPO and Fcγ receptors.
3. Activated neutrophils in the presence of hydrogen peroxidase release MPO that converts hydralazine into a cytotoxic product that is immunogenic for T cells that activate ANCA-producing B cells.
4. Histone H3 trimethyl Lys27 (H3K27me3) levels are perturbed in HIAV, which leads to aberrant gene silencing of proteinase 3 and MPO.In contrast, the demethylase Jumonji domain-containing protein 3 for the H3K27me3 histone is increased in patients without HIAV. Based on this data and the data showing a role for hydralazine in reversing epigenetic silencing of tumor suppressor genes in cancer cells,13 it has been proposed that hydralazine may reverse epigenetic silencing of proteinase 3 and MPO.
Diagnosing HIAV is still difficult because physicians do not recognize the drug as the etiologic agent, there is extensive variability in duration between starting the drug and onset of symptoms, and there often is a failure to order the appropriate laboratory and invasive tests needed for evaluation and diagnosis.3,5,8,10,12 Despite these difficulties, a set of criteria and practices for diagnosis are delineated in Table 1, with the key diagnostic feature being resolution with hydralazine cessation.1,5,7,8,12
A comprehensive drug history from at least 6 months prior to presentation is essential. Biopsies also are strongly encouraged to confirm the presence of vasculitis and to determine its severity.8,12 If renal biopsies are performed, they typically show scant IgG, IgM, and C3 deposition that is characteristic of ANCA-positive pauci-immune glomerulonephritis. Compared to hydralazine-induced lupus, renal involvement in the setting of HIAV has a relative lack of immunoglobulin and complement deposition with histopathology and immunostaining.14
Laboratory test results including serum MPO-ANCA (perinuclear ANCA) with coexisting elastase and/or lactoferrin autoantibodies is characteristic of HIAV. Antinuclear antibody, antihistone, anti–double-stranded DNA, and antiphospholipid antibodies along with low complement levels also may be present.2,4,9,10,13,15 It is recommended that ANCA assays combine indirect immunofluorescence with antigen-specific enzyme-linked immunosorbent assay.8 With respect to its idiopathic counterpart, patients may only present with MPO-ANCA, while other aforementioned antibodies (eg, antihistone, anti–double-stranded DNA) are rarely found or are entirely absent.2,9 Patients with HIAV often have higher titers of MPO-ANCA.9,15 In hydralazine-induced lupus, patients rarely have MPO-ANCA.
When a diagnosis of HIAV is made, it cannot be confirmed until hydralazine is discontinued and the patient’s symptoms resolve. Therefore, it is both diagnostic and therapeutic to discontinue hydralazine when HIAV is suspected. If recognized when the patient is only presenting with nonspecific symptoms, simple hydralazine cessation may be all that is needed; however, because recognition and diagnosis of HIAV is difficult, most patients present when the disease is severe and has progressed to organ involvement.8-10
Treatment recommendations are highlighted in Table 2.8,9,12 Glucocorticoid therapy is believed to work by preventing T-cell and B-cell maturation needed to produce MPO-ANCA. Rituximab, on the other hand, is suspected to act by clearing the peripheral blood of MPO-ANCA B cells.12,16 Of note, patients with HIAV are different from their idiopathic counterparts because they usually need shorter courses of immunosuppressive therapy, long-term maintenance usually is unnecessary, and their prognosis generally is good if the offending agent is withdrawn.7-9,12 Once the appropriate therapy is instituted, vasculitic manifestations are expected to resolve 10 days to 8 months after hydralazine cessation; however, a response often is seen within 1 to 4 weeks after initiation of systemic treatment.4,8 Serum ANCA should be monitored, and there should be surveillance for the emergence of a chronic underlying vasculitis.8,12
Our patient highlights the importance of identifying individuals at risk for HIAV. We seek to increase recognition of this entity, as it is not commonly seen in a dermatologic setting and is associated with high morbidity and mortality, as seen in our patient.
- Yokogawa N, Vivino FB. Hydralazine-induced autoimmune disease: comparison to idiopathic lupus and ANCA-positive vasculitis. Mod Rheumatol. 2009;19:338-347.
- Agarwal G, Sultan G, Werner SL, et al. Hydralazine induces myeloperoxidase and proteinase 3 anti-neutrophil cytoplasmic antibody vasculitis and leads to pulmonary renal syndrome. Case Rep Nephrol. 2014;2014:868590.
- Keasberry J, Frazier J, Isbel NM, et al. Hydralazine-induced anti-neutrophilic cytoplasmic antibody-positive renal vasculitis presenting with a vasculitic syndrome, acute nephritis and a puzzling skin rash: a case report. J Med Case Rep. 2013;7:20.
- ten Holder SM, Joy MS, Falk RJ. Cutaneous and systemic manifestations of drug-induced vasculitis. Ann Pharmacother. 2002;36:130-147.
- Namas R, Rubin B, Adwar W, et al. A challenging twist in pulmonary renal syndrome. Case Rep Rheumatol. 2014;2014:516362.
- Dobre M, Wish J, Negrea L. Hydralazine-induced ANCA-positive pauci-immune glomerulonephritis. Ren Fail. 2009;31:745-748.
- Hogan JJ, Markowitz GS, Radhakrishnan J. Drug-induced glomerular disease: immune-mediated injury. Clin J Am Soc Nephrol. 2015;10:1300-1310.
- Radic M, Martinovic Kaliterna D, Radic J. Drug-induced vasculitis: a clinical and pathological review. Neth J Med. 2012;70:12-17.
- Babar F, Posner JN, Obah EA. Hydralazine-induced pauci-immune glomerulonephritis: intriguing case series misleading diagnoses. J Community Hosp Intern Med Perspect. 2016;6:30632.
- Marina VP, Malhotra D, Kaw D. Hydralazine-induced ANCA vasculitis with pulmonary renal syndrome: a rare clinical presentation. Int Urol Nephrol. 2012;44:1907-1909.
- Magro CM. Associated ANCA positive vasculitis. The Dermatologist. 2015;23(7). http://www.the-dermatologist.com/content/associated-anca-positive-vasculitis. Accessed January 30, 2020.
- Gao Y, Zhao MH. Review article: Drug-induced anti-neutrophil cytoplasmic antibody-associated vasculitis. Nephrology (Carlton). 2009;14:33-41.
- Grau RG. Drug-induced vasculitis: new insights and a changing lineup of suspects. Curr Rheumatol Rep. 2015;17:71.
- Sangala N, Lee RW, Horsfield C, et al. Combined ANCA-associated vasculitis and lupus syndrome following prolonged use of hydralazine: a timely reminder of an old foe. Int Urol Nephrol. 2010;42:503-506.
- Choi HK, Merkel PA, Walker AM, et al. Drug-associated antineutrophil cytoplasmic antibody-positive vasculitis: prevalence among patients with high titers of antimyeloperoxidase antibodies. Arthritis Rheum. 2000;43:405-413.
- Coutinho AE, Chapman KE. The anti-inflammatory and immunosuppressive effects of glucocorticoids, recent developments and mechanistic insights. Mol Cell Endocrinol. 2011;335:2-13.
- Yokogawa N, Vivino FB. Hydralazine-induced autoimmune disease: comparison to idiopathic lupus and ANCA-positive vasculitis. Mod Rheumatol. 2009;19:338-347.
- Agarwal G, Sultan G, Werner SL, et al. Hydralazine induces myeloperoxidase and proteinase 3 anti-neutrophil cytoplasmic antibody vasculitis and leads to pulmonary renal syndrome. Case Rep Nephrol. 2014;2014:868590.
- Keasberry J, Frazier J, Isbel NM, et al. Hydralazine-induced anti-neutrophilic cytoplasmic antibody-positive renal vasculitis presenting with a vasculitic syndrome, acute nephritis and a puzzling skin rash: a case report. J Med Case Rep. 2013;7:20.
- ten Holder SM, Joy MS, Falk RJ. Cutaneous and systemic manifestations of drug-induced vasculitis. Ann Pharmacother. 2002;36:130-147.
- Namas R, Rubin B, Adwar W, et al. A challenging twist in pulmonary renal syndrome. Case Rep Rheumatol. 2014;2014:516362.
- Dobre M, Wish J, Negrea L. Hydralazine-induced ANCA-positive pauci-immune glomerulonephritis. Ren Fail. 2009;31:745-748.
- Hogan JJ, Markowitz GS, Radhakrishnan J. Drug-induced glomerular disease: immune-mediated injury. Clin J Am Soc Nephrol. 2015;10:1300-1310.
- Radic M, Martinovic Kaliterna D, Radic J. Drug-induced vasculitis: a clinical and pathological review. Neth J Med. 2012;70:12-17.
- Babar F, Posner JN, Obah EA. Hydralazine-induced pauci-immune glomerulonephritis: intriguing case series misleading diagnoses. J Community Hosp Intern Med Perspect. 2016;6:30632.
- Marina VP, Malhotra D, Kaw D. Hydralazine-induced ANCA vasculitis with pulmonary renal syndrome: a rare clinical presentation. Int Urol Nephrol. 2012;44:1907-1909.
- Magro CM. Associated ANCA positive vasculitis. The Dermatologist. 2015;23(7). http://www.the-dermatologist.com/content/associated-anca-positive-vasculitis. Accessed January 30, 2020.
- Gao Y, Zhao MH. Review article: Drug-induced anti-neutrophil cytoplasmic antibody-associated vasculitis. Nephrology (Carlton). 2009;14:33-41.
- Grau RG. Drug-induced vasculitis: new insights and a changing lineup of suspects. Curr Rheumatol Rep. 2015;17:71.
- Sangala N, Lee RW, Horsfield C, et al. Combined ANCA-associated vasculitis and lupus syndrome following prolonged use of hydralazine: a timely reminder of an old foe. Int Urol Nephrol. 2010;42:503-506.
- Choi HK, Merkel PA, Walker AM, et al. Drug-associated antineutrophil cytoplasmic antibody-positive vasculitis: prevalence among patients with high titers of antimyeloperoxidase antibodies. Arthritis Rheum. 2000;43:405-413.
- Coutinho AE, Chapman KE. The anti-inflammatory and immunosuppressive effects of glucocorticoids, recent developments and mechanistic insights. Mol Cell Endocrinol. 2011;335:2-13.
Practice Points
- Hydralazine-induced antineutrophil cytoplasmic antibody vasculitis (HIAV) is a rare side effect of hydralazine treatment and can have notable morbidity and mortality.
- Incidence and prevalence of HIAV is unclear due to its rarity, but risk factors that have been identified are older age, a cumulative dose of 100 g of hydralazine at the time of presentation, female sex, thyroid disease, HLA-DR4 genotypes, slow hepatic acetylation, and the null gene for C4.
- Symptoms of HIAV can include fever, malaise, arthralgia, weight loss, or even involvement of organs such as the kidneys and lungs.
- If recognized early, cessation of hydralazine and supportive therapy generally are sufficient; however, severe cases may need management with high-dose corticosteroids, rituximab, and even plasmapheresis.
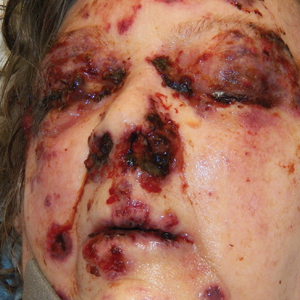
Smooth Papules on the Left Hand
The Diagnosis: Adult Colloid Milium
A 4-mm punch biopsy was performed and histopathologic evaluation revealed collections of amorphic eosinophilic material and fissures in the papillary dermis with sparing of the dermoepidermal junction, indicating adult colloid milium (Figure 1).

Adult colloid milium is an uncommon condition with grouped translucent to whitish papules that present on sun-exposed skin on the hands, face, neck, or ears in middle-aged adults.1 It has been associated with petrochemical exposure, tanning bed use, and excessive sun exposure. Our patient had a history of sun exposure, specifically to the left hand while driving. This condition is widely thought to be a result of photoinduced damage to elastic fibers and may potentially be a popular variant of severe solar elastosis.2 Due to vascular fragility, trauma to these locations often will result in hemorrhage into individual lesions, as observed in our patient (Figure 2).
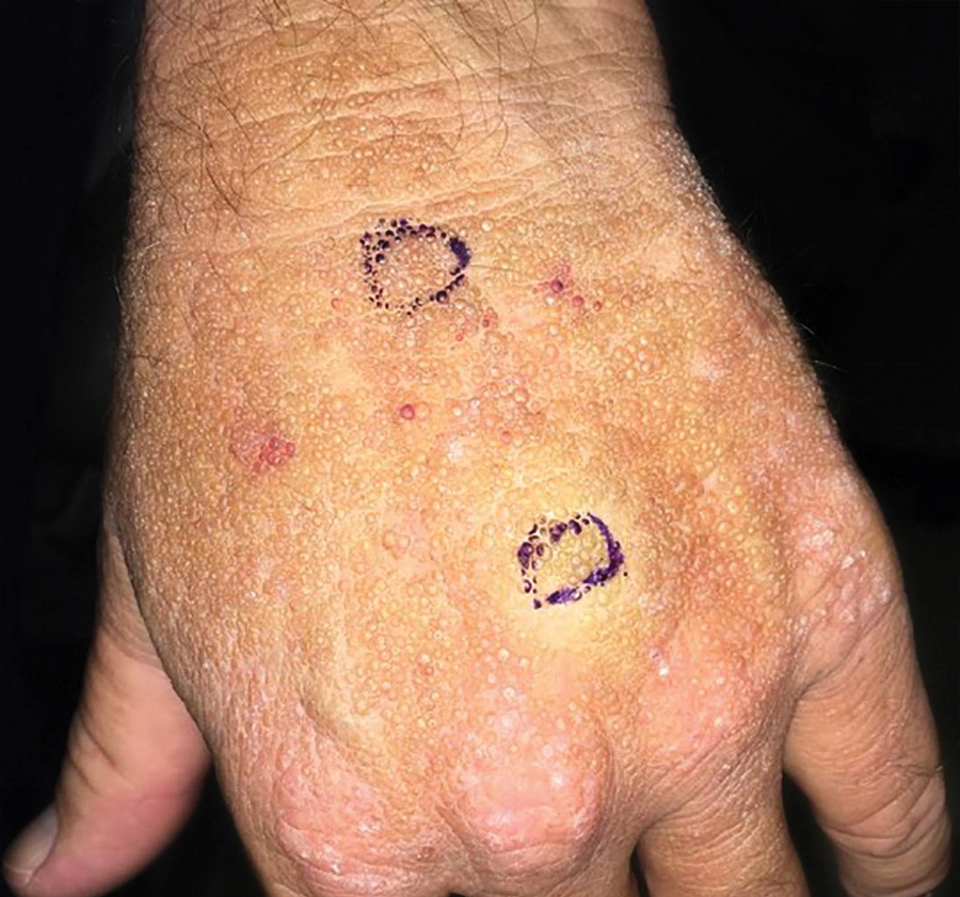
Adult colloid milium is diagnosed clinically and may mimic lichen or systemic amyloidosis, syringomas, lipoid proteinosis, molluscum contagiosum, steatocystoma multiplex, and sarcoidosis.2
Biopsy often is helpful in determining the diagnosis. Histopathology reveals amorphous eosinophilic deposits with fissures in the papillary dermis. These deposits are thought to be remnants of degenerated elastic fibers. Stains often are helpful, as the deposits are weakly apple-green birefringent on Congo red stain and are periodic acid-Schiff and thioflavin T positive. Laminin and type IV collagen stains are negative with adult colloid milium but are positive with amyloidosis and lipoid proteinosis.3 Electron microscopy also may help distinguish between amyloidosis and adult colloid milium, as these conditions may have a similar histologic appearance.
Treatment has not proven to be consistently helpful, as cryotherapy and dermabrasion have been the mainstay of treatment, often with disappointing results.4 Laser treatment has been shown to be of some benefit in treating these lesions.2
- Touart DM, Sau P. Cutaneous deposition diseases. part I. J Am Acad Dermatol. 1998;39(2, pt 1):149-171.
- Pourrabbani S, Marra DE, Iwasaki J, et al. Colloid milium: a review and update. J Drugs Dermatol. 2007;6:293-296.
- Calonje JE, Brenn T, Lazar A, et al. McKee's Pathology of the Skin. 4th ed. Philadelphia, PA: Saunders; 2012.
- Netscher DT, Sharma S, Kinner BM, et al. Adult-type colloid milium of hands and face successfully treated with dermabrasion. South Med J. 1996;89:1004-1007.
The Diagnosis: Adult Colloid Milium
A 4-mm punch biopsy was performed and histopathologic evaluation revealed collections of amorphic eosinophilic material and fissures in the papillary dermis with sparing of the dermoepidermal junction, indicating adult colloid milium (Figure 1).
Adult colloid milium is an uncommon condition with grouped translucent to whitish papules that present on sun-exposed skin on the hands, face, neck, or ears in middle-aged adults.1 It has been associated with petrochemical exposure, tanning bed use, and excessive sun exposure. Our patient had a history of sun exposure, specifically to the left hand while driving. This condition is widely thought to be a result of photoinduced damage to elastic fibers and may potentially be a popular variant of severe solar elastosis.2 Due to vascular fragility, trauma to these locations often will result in hemorrhage into individual lesions, as observed in our patient (Figure 2).
Adult colloid milium is diagnosed clinically and may mimic lichen or systemic amyloidosis, syringomas, lipoid proteinosis, molluscum contagiosum, steatocystoma multiplex, and sarcoidosis.2
Biopsy often is helpful in determining the diagnosis. Histopathology reveals amorphous eosinophilic deposits with fissures in the papillary dermis. These deposits are thought to be remnants of degenerated elastic fibers. Stains often are helpful, as the deposits are weakly apple-green birefringent on Congo red stain and are periodic acid-Schiff and thioflavin T positive. Laminin and type IV collagen stains are negative with adult colloid milium but are positive with amyloidosis and lipoid proteinosis.3 Electron microscopy also may help distinguish between amyloidosis and adult colloid milium, as these conditions may have a similar histologic appearance.
Treatment has not proven to be consistently helpful, as cryotherapy and dermabrasion have been the mainstay of treatment, often with disappointing results.4 Laser treatment has been shown to be of some benefit in treating these lesions.2
The Diagnosis: Adult Colloid Milium
A 4-mm punch biopsy was performed and histopathologic evaluation revealed collections of amorphic eosinophilic material and fissures in the papillary dermis with sparing of the dermoepidermal junction, indicating adult colloid milium (Figure 1).
Adult colloid milium is an uncommon condition with grouped translucent to whitish papules that present on sun-exposed skin on the hands, face, neck, or ears in middle-aged adults.1 It has been associated with petrochemical exposure, tanning bed use, and excessive sun exposure. Our patient had a history of sun exposure, specifically to the left hand while driving. This condition is widely thought to be a result of photoinduced damage to elastic fibers and may potentially be a popular variant of severe solar elastosis.2 Due to vascular fragility, trauma to these locations often will result in hemorrhage into individual lesions, as observed in our patient (Figure 2).
Adult colloid milium is diagnosed clinically and may mimic lichen or systemic amyloidosis, syringomas, lipoid proteinosis, molluscum contagiosum, steatocystoma multiplex, and sarcoidosis.2
Biopsy often is helpful in determining the diagnosis. Histopathology reveals amorphous eosinophilic deposits with fissures in the papillary dermis. These deposits are thought to be remnants of degenerated elastic fibers. Stains often are helpful, as the deposits are weakly apple-green birefringent on Congo red stain and are periodic acid-Schiff and thioflavin T positive. Laminin and type IV collagen stains are negative with adult colloid milium but are positive with amyloidosis and lipoid proteinosis.3 Electron microscopy also may help distinguish between amyloidosis and adult colloid milium, as these conditions may have a similar histologic appearance.
Treatment has not proven to be consistently helpful, as cryotherapy and dermabrasion have been the mainstay of treatment, often with disappointing results.4 Laser treatment has been shown to be of some benefit in treating these lesions.2
- Touart DM, Sau P. Cutaneous deposition diseases. part I. J Am Acad Dermatol. 1998;39(2, pt 1):149-171.
- Pourrabbani S, Marra DE, Iwasaki J, et al. Colloid milium: a review and update. J Drugs Dermatol. 2007;6:293-296.
- Calonje JE, Brenn T, Lazar A, et al. McKee's Pathology of the Skin. 4th ed. Philadelphia, PA: Saunders; 2012.
- Netscher DT, Sharma S, Kinner BM, et al. Adult-type colloid milium of hands and face successfully treated with dermabrasion. South Med J. 1996;89:1004-1007.
- Touart DM, Sau P. Cutaneous deposition diseases. part I. J Am Acad Dermatol. 1998;39(2, pt 1):149-171.
- Pourrabbani S, Marra DE, Iwasaki J, et al. Colloid milium: a review and update. J Drugs Dermatol. 2007;6:293-296.
- Calonje JE, Brenn T, Lazar A, et al. McKee's Pathology of the Skin. 4th ed. Philadelphia, PA: Saunders; 2012.
- Netscher DT, Sharma S, Kinner BM, et al. Adult-type colloid milium of hands and face successfully treated with dermabrasion. South Med J. 1996;89:1004-1007.
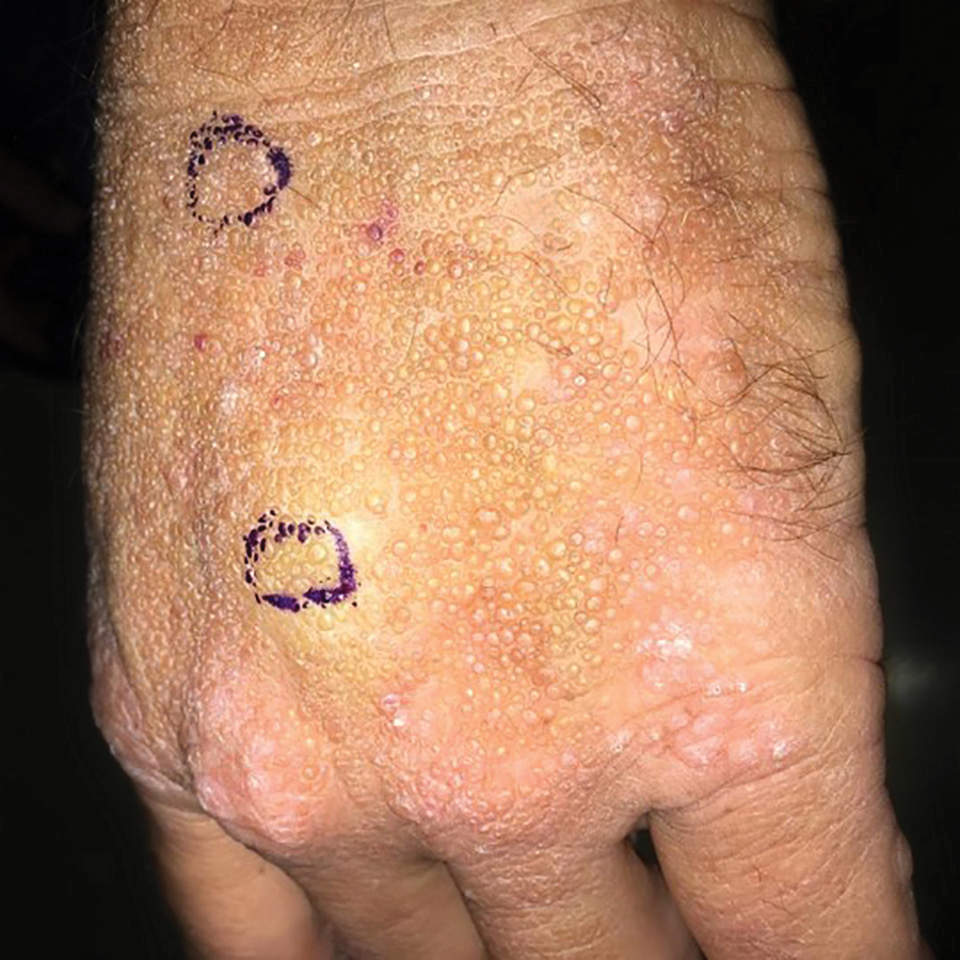
A 41-year-old man presented to the outpatient dermatology clinic with multiple smooth papules on the left hand of 7 years' duration. The papules had been steadily increasing in number, and the patient reported that they were frequently symptomatic with a burning itching sensation. Physical examination revealed multiple 1- to 3-mm, dome-shaped, translucent to flesh-colored papules on the left hand with a few scattered bright red papules. No similar lesions were present on the right hand or elsewhere on the body. He had a history of hypertension but was otherwise healthy with no other chronic medical conditions.