User login
Cutis is a peer-reviewed clinical journal for the dermatologist, allergist, and general practitioner published monthly since 1965. Concise clinical articles present the practical side of dermatology, helping physicians to improve patient care. Cutis is referenced in Index Medicus/MEDLINE and is written and edited by industry leaders.
ass lick
assault rifle
balls
ballsac
black jack
bleach
Boko Haram
bondage
causas
cheap
child abuse
cocaine
compulsive behaviors
cost of miracles
cunt
Daech
display network stats
drug paraphernalia
explosion
fart
fda and death
fda AND warn
fda AND warning
fda AND warns
feom
fuck
gambling
gfc
gun
human trafficking
humira AND expensive
illegal
ISIL
ISIS
Islamic caliphate
Islamic state
madvocate
masturbation
mixed martial arts
MMA
molestation
national rifle association
NRA
nsfw
nuccitelli
pedophile
pedophilia
poker
porn
porn
pornography
psychedelic drug
recreational drug
sex slave rings
shit
slot machine
snort
substance abuse
terrorism
terrorist
texarkana
Texas hold 'em
UFC
section[contains(@class, 'nav-hidden')]
section[contains(@class, 'nav-hidden active')
A peer-reviewed, indexed journal for dermatologists with original research, image quizzes, cases and reviews, and columns.
Reducing the Cost of Dermatology Residency Applications: An Applicant’s Perspective
Another Match Day is approaching. Students find themselves paying more each year to apply to one of the most competitive fields, while program directors struggle to sort through hundreds of stellar applications to invite a handful of candidates for interviews. Estimates place the cost of the application process at $5 million in total for all medical school seniors, or roughly $10,000 per applicant.1 Approximately 60% of these costs occur during the interview process.1,2 In an era in which students routinely graduate medical school with hundreds of thousands of dollars of debt, these costs must be addressed as soon as possible.
This problem is not unique to dermatology; otolaryngology, another especially competitive field, has considered various changes to the match process based on applicants’ feedback.3 As an applicant during the 2018-2019 match cycle for dermatology, I offer 2 solutions that are a starting point aimed at streamlining the application process for both applicants and program directors: regional interview coordination and a cap on the number of residency applications.
Regional Interview Coordination
Regional interview coordination would reduce travel costs and facilitate greater predictability in scheduling clinical rotations. In the current climate, it is not uncommon for applicants to make multiple cross-country round trips in the same week, especially given that the interview season for dermatology, including interviews for preliminary programs, now ranges from mid-October to early February. Although affluent applicants may not be concerned with financial costs of frequent travel, all applicants face travel inconveniences that could be mitigated through regional coordination. For example, an applicant invited to multiple interviews in the New York City area could reserve a room in a single hotel over a period of several days. During each interview day, he/she could travel back and forth from that accommodation to each institution without needing to bring luggage, worrying about reaching the airport on time, or missing a pre-interview dinner at a program in a faraway city.
Given the amount of coordination required among programs, it may lead to more positive working relationships among regional dermatology programs. One limitation of this approach is that competitive programs may be unwilling to cooperate. If even one program deviates from the interview time frame, it reduces the incentive for others to participate. Programs must be willing to sacrifice short-term autonomy in interview scheduling for their long-term shared interest in reducing the application burden for students, which is known as a commitment problem in game theory, and could be addressed through joint decision-making that incorporates the time frame preferences of all programs as well as binding commitments on interview dates that are decided before the process begins.4 Another limitation is that inclement weather could affect all regional programs simultaneously. In this case, offering interviews via video conference for affected students may be a solution.
Capping the Number of Applications
A second method of reducing interview costs would be capping the number of applications. Although matched seniors applied to a median of 72 programs, the Association of American Medical Colleges suggests that dermatology applicants can maximize their return on investment (ie, ratio of interviews to applications) by sending 35 to 55 applications depending on US Medical Licensing Examination scores. Attending more than 10 interviews does not meaningfully improve the chance of matching.5,6
Programs have limited capacity for interviews and must judiciously allocate invitations based solely on the information provided through the Electronic Residency Application Service (ERAS). Given the competitiveness of dermatology, applicants usually will accept every interview invitation. Therefore, applicants who are not genuinely interested in a program may crowd out others who are interested. In a survey of otolaryngology applicants (N=150), 90.6% of respondents admitted applying to programs in which they had no specific interest, simply to increase their chance of matching.3 Capping application numbers would force students to apply more selectively and enable residencies to gauge students’ true interest more effectively. In contrast to regional interview coordination, this policy change would be easy to enforce. It also may be popular; nearly two-thirds of otolaryngology applicants agreed to a hypothetical cap on residency applications to reduce the burden on students and programs.3
An alternative to a hard cap on applications could be restructuring the ERAS application fee to incentivize students to apply to fewer programs. For example, a flat fee might cover application numbers up to the point of diminishing returns, after which the price per application could increase exponentially. This approach would have a similar effect of a hard cap and cause many students to apply to fewer programs; however, one notable drawback is that highly affluent applicants would simply absorb the extra cost and still gain a competitive advantage in applying to more programs, which might further decrease the number of lower-income individuals successfully matching into dermatology.
A benefit of decreased application numbers to program directors would be giving them more time to conduct a holistic review of applicants, rather than attempting to weed out candidates through arbitrary cutoffs for US Medical Licensing Examination scores or Alpha Omega Alpha Honor Medical Society membership. The ERAS could allow applicants the option of stating preferences for geographic regions, desired fellowships, areas of research interest, and other intangible metrics. Selection committees could filter their candidate search by different variables and then look at each candidate holistically.
Limitations of capping application numbers include the risk that such a cap would harm less-competitive applicants while failing to address the primary cost drivers (ie, travel costs). The specific cap number would be controversial and may need to be adjusted higher for special cases such as couples matching and international applicants, thus making a cap seem arbitrary.
Final Thoughts
The dermatology residency match can be streamlined to the benefit of both applicants and selection committees. Regional interview coordination would reduce both financial and logistical barriers for applicants but may be difficult to enforce without cooperation from multiple programs. Capping the number of applications, either through a hard cap or an increased financial barrier, would be relatively easy to enforce and might empower selection committees to conduct more detailed, holistic reviews of applicants; however, certain types of applicants may find the application limits detrimental to their chances of matching. These policy recommendations are meant to be a starting point for discussion. Streamlining the application process is critical to improving the diversity of dermatology residencies.
- Mansouri B, Walker GD, Mitchell J, et al. The cost of applying to dermatology residency: 2014 data estimates. J Am Acad Dermatol. 2016;74:754-756.
- Tichy AL, Peng DH, Lane AT. Applying for dermatology residency is difficult and expensive. J Am Acad Dermatol. 2012;66:696-697.
- Ward M, Pingree C, Laury AM, et al. Applicant perspectives on the otolaryngology residency application process. JAMA Otolaryngol Head Neck Surg. 2017;143:782-787.
- North DC. Institutions and credible commitment. J Inst Theor Econ. 1993;149:11-23.
- Association of American Medical Colleges. Apply smart: data to consider when applying to residency. https://students-residents.aamc.org/applying-residency/filteredresult/apply-smart-data-consider-when-applying-residency/. Accessed November 12, 2019.
- Charting Outcomes in the Match: Characteristics of U.S. Allopathic Seniors Who Matched to Their Preferred Specialty in the 2018 Main Residency Match. 2nd ed. Washington, DC: National Resident Matching Program; July 2018. https://www.nrmp.org/wp-content/uploads/2018/06/Charting-Outcomes-in-the-Match-2018-Seniors.pdf. Accessed November 11, 2019.
Another Match Day is approaching. Students find themselves paying more each year to apply to one of the most competitive fields, while program directors struggle to sort through hundreds of stellar applications to invite a handful of candidates for interviews. Estimates place the cost of the application process at $5 million in total for all medical school seniors, or roughly $10,000 per applicant.1 Approximately 60% of these costs occur during the interview process.1,2 In an era in which students routinely graduate medical school with hundreds of thousands of dollars of debt, these costs must be addressed as soon as possible.
This problem is not unique to dermatology; otolaryngology, another especially competitive field, has considered various changes to the match process based on applicants’ feedback.3 As an applicant during the 2018-2019 match cycle for dermatology, I offer 2 solutions that are a starting point aimed at streamlining the application process for both applicants and program directors: regional interview coordination and a cap on the number of residency applications.
Regional Interview Coordination
Regional interview coordination would reduce travel costs and facilitate greater predictability in scheduling clinical rotations. In the current climate, it is not uncommon for applicants to make multiple cross-country round trips in the same week, especially given that the interview season for dermatology, including interviews for preliminary programs, now ranges from mid-October to early February. Although affluent applicants may not be concerned with financial costs of frequent travel, all applicants face travel inconveniences that could be mitigated through regional coordination. For example, an applicant invited to multiple interviews in the New York City area could reserve a room in a single hotel over a period of several days. During each interview day, he/she could travel back and forth from that accommodation to each institution without needing to bring luggage, worrying about reaching the airport on time, or missing a pre-interview dinner at a program in a faraway city.
Given the amount of coordination required among programs, it may lead to more positive working relationships among regional dermatology programs. One limitation of this approach is that competitive programs may be unwilling to cooperate. If even one program deviates from the interview time frame, it reduces the incentive for others to participate. Programs must be willing to sacrifice short-term autonomy in interview scheduling for their long-term shared interest in reducing the application burden for students, which is known as a commitment problem in game theory, and could be addressed through joint decision-making that incorporates the time frame preferences of all programs as well as binding commitments on interview dates that are decided before the process begins.4 Another limitation is that inclement weather could affect all regional programs simultaneously. In this case, offering interviews via video conference for affected students may be a solution.
Capping the Number of Applications
A second method of reducing interview costs would be capping the number of applications. Although matched seniors applied to a median of 72 programs, the Association of American Medical Colleges suggests that dermatology applicants can maximize their return on investment (ie, ratio of interviews to applications) by sending 35 to 55 applications depending on US Medical Licensing Examination scores. Attending more than 10 interviews does not meaningfully improve the chance of matching.5,6
Programs have limited capacity for interviews and must judiciously allocate invitations based solely on the information provided through the Electronic Residency Application Service (ERAS). Given the competitiveness of dermatology, applicants usually will accept every interview invitation. Therefore, applicants who are not genuinely interested in a program may crowd out others who are interested. In a survey of otolaryngology applicants (N=150), 90.6% of respondents admitted applying to programs in which they had no specific interest, simply to increase their chance of matching.3 Capping application numbers would force students to apply more selectively and enable residencies to gauge students’ true interest more effectively. In contrast to regional interview coordination, this policy change would be easy to enforce. It also may be popular; nearly two-thirds of otolaryngology applicants agreed to a hypothetical cap on residency applications to reduce the burden on students and programs.3
An alternative to a hard cap on applications could be restructuring the ERAS application fee to incentivize students to apply to fewer programs. For example, a flat fee might cover application numbers up to the point of diminishing returns, after which the price per application could increase exponentially. This approach would have a similar effect of a hard cap and cause many students to apply to fewer programs; however, one notable drawback is that highly affluent applicants would simply absorb the extra cost and still gain a competitive advantage in applying to more programs, which might further decrease the number of lower-income individuals successfully matching into dermatology.
A benefit of decreased application numbers to program directors would be giving them more time to conduct a holistic review of applicants, rather than attempting to weed out candidates through arbitrary cutoffs for US Medical Licensing Examination scores or Alpha Omega Alpha Honor Medical Society membership. The ERAS could allow applicants the option of stating preferences for geographic regions, desired fellowships, areas of research interest, and other intangible metrics. Selection committees could filter their candidate search by different variables and then look at each candidate holistically.
Limitations of capping application numbers include the risk that such a cap would harm less-competitive applicants while failing to address the primary cost drivers (ie, travel costs). The specific cap number would be controversial and may need to be adjusted higher for special cases such as couples matching and international applicants, thus making a cap seem arbitrary.
Final Thoughts
The dermatology residency match can be streamlined to the benefit of both applicants and selection committees. Regional interview coordination would reduce both financial and logistical barriers for applicants but may be difficult to enforce without cooperation from multiple programs. Capping the number of applications, either through a hard cap or an increased financial barrier, would be relatively easy to enforce and might empower selection committees to conduct more detailed, holistic reviews of applicants; however, certain types of applicants may find the application limits detrimental to their chances of matching. These policy recommendations are meant to be a starting point for discussion. Streamlining the application process is critical to improving the diversity of dermatology residencies.
Another Match Day is approaching. Students find themselves paying more each year to apply to one of the most competitive fields, while program directors struggle to sort through hundreds of stellar applications to invite a handful of candidates for interviews. Estimates place the cost of the application process at $5 million in total for all medical school seniors, or roughly $10,000 per applicant.1 Approximately 60% of these costs occur during the interview process.1,2 In an era in which students routinely graduate medical school with hundreds of thousands of dollars of debt, these costs must be addressed as soon as possible.
This problem is not unique to dermatology; otolaryngology, another especially competitive field, has considered various changes to the match process based on applicants’ feedback.3 As an applicant during the 2018-2019 match cycle for dermatology, I offer 2 solutions that are a starting point aimed at streamlining the application process for both applicants and program directors: regional interview coordination and a cap on the number of residency applications.
Regional Interview Coordination
Regional interview coordination would reduce travel costs and facilitate greater predictability in scheduling clinical rotations. In the current climate, it is not uncommon for applicants to make multiple cross-country round trips in the same week, especially given that the interview season for dermatology, including interviews for preliminary programs, now ranges from mid-October to early February. Although affluent applicants may not be concerned with financial costs of frequent travel, all applicants face travel inconveniences that could be mitigated through regional coordination. For example, an applicant invited to multiple interviews in the New York City area could reserve a room in a single hotel over a period of several days. During each interview day, he/she could travel back and forth from that accommodation to each institution without needing to bring luggage, worrying about reaching the airport on time, or missing a pre-interview dinner at a program in a faraway city.
Given the amount of coordination required among programs, it may lead to more positive working relationships among regional dermatology programs. One limitation of this approach is that competitive programs may be unwilling to cooperate. If even one program deviates from the interview time frame, it reduces the incentive for others to participate. Programs must be willing to sacrifice short-term autonomy in interview scheduling for their long-term shared interest in reducing the application burden for students, which is known as a commitment problem in game theory, and could be addressed through joint decision-making that incorporates the time frame preferences of all programs as well as binding commitments on interview dates that are decided before the process begins.4 Another limitation is that inclement weather could affect all regional programs simultaneously. In this case, offering interviews via video conference for affected students may be a solution.
Capping the Number of Applications
A second method of reducing interview costs would be capping the number of applications. Although matched seniors applied to a median of 72 programs, the Association of American Medical Colleges suggests that dermatology applicants can maximize their return on investment (ie, ratio of interviews to applications) by sending 35 to 55 applications depending on US Medical Licensing Examination scores. Attending more than 10 interviews does not meaningfully improve the chance of matching.5,6
Programs have limited capacity for interviews and must judiciously allocate invitations based solely on the information provided through the Electronic Residency Application Service (ERAS). Given the competitiveness of dermatology, applicants usually will accept every interview invitation. Therefore, applicants who are not genuinely interested in a program may crowd out others who are interested. In a survey of otolaryngology applicants (N=150), 90.6% of respondents admitted applying to programs in which they had no specific interest, simply to increase their chance of matching.3 Capping application numbers would force students to apply more selectively and enable residencies to gauge students’ true interest more effectively. In contrast to regional interview coordination, this policy change would be easy to enforce. It also may be popular; nearly two-thirds of otolaryngology applicants agreed to a hypothetical cap on residency applications to reduce the burden on students and programs.3
An alternative to a hard cap on applications could be restructuring the ERAS application fee to incentivize students to apply to fewer programs. For example, a flat fee might cover application numbers up to the point of diminishing returns, after which the price per application could increase exponentially. This approach would have a similar effect of a hard cap and cause many students to apply to fewer programs; however, one notable drawback is that highly affluent applicants would simply absorb the extra cost and still gain a competitive advantage in applying to more programs, which might further decrease the number of lower-income individuals successfully matching into dermatology.
A benefit of decreased application numbers to program directors would be giving them more time to conduct a holistic review of applicants, rather than attempting to weed out candidates through arbitrary cutoffs for US Medical Licensing Examination scores or Alpha Omega Alpha Honor Medical Society membership. The ERAS could allow applicants the option of stating preferences for geographic regions, desired fellowships, areas of research interest, and other intangible metrics. Selection committees could filter their candidate search by different variables and then look at each candidate holistically.
Limitations of capping application numbers include the risk that such a cap would harm less-competitive applicants while failing to address the primary cost drivers (ie, travel costs). The specific cap number would be controversial and may need to be adjusted higher for special cases such as couples matching and international applicants, thus making a cap seem arbitrary.
Final Thoughts
The dermatology residency match can be streamlined to the benefit of both applicants and selection committees. Regional interview coordination would reduce both financial and logistical barriers for applicants but may be difficult to enforce without cooperation from multiple programs. Capping the number of applications, either through a hard cap or an increased financial barrier, would be relatively easy to enforce and might empower selection committees to conduct more detailed, holistic reviews of applicants; however, certain types of applicants may find the application limits detrimental to their chances of matching. These policy recommendations are meant to be a starting point for discussion. Streamlining the application process is critical to improving the diversity of dermatology residencies.
- Mansouri B, Walker GD, Mitchell J, et al. The cost of applying to dermatology residency: 2014 data estimates. J Am Acad Dermatol. 2016;74:754-756.
- Tichy AL, Peng DH, Lane AT. Applying for dermatology residency is difficult and expensive. J Am Acad Dermatol. 2012;66:696-697.
- Ward M, Pingree C, Laury AM, et al. Applicant perspectives on the otolaryngology residency application process. JAMA Otolaryngol Head Neck Surg. 2017;143:782-787.
- North DC. Institutions and credible commitment. J Inst Theor Econ. 1993;149:11-23.
- Association of American Medical Colleges. Apply smart: data to consider when applying to residency. https://students-residents.aamc.org/applying-residency/filteredresult/apply-smart-data-consider-when-applying-residency/. Accessed November 12, 2019.
- Charting Outcomes in the Match: Characteristics of U.S. Allopathic Seniors Who Matched to Their Preferred Specialty in the 2018 Main Residency Match. 2nd ed. Washington, DC: National Resident Matching Program; July 2018. https://www.nrmp.org/wp-content/uploads/2018/06/Charting-Outcomes-in-the-Match-2018-Seniors.pdf. Accessed November 11, 2019.
- Mansouri B, Walker GD, Mitchell J, et al. The cost of applying to dermatology residency: 2014 data estimates. J Am Acad Dermatol. 2016;74:754-756.
- Tichy AL, Peng DH, Lane AT. Applying for dermatology residency is difficult and expensive. J Am Acad Dermatol. 2012;66:696-697.
- Ward M, Pingree C, Laury AM, et al. Applicant perspectives on the otolaryngology residency application process. JAMA Otolaryngol Head Neck Surg. 2017;143:782-787.
- North DC. Institutions and credible commitment. J Inst Theor Econ. 1993;149:11-23.
- Association of American Medical Colleges. Apply smart: data to consider when applying to residency. https://students-residents.aamc.org/applying-residency/filteredresult/apply-smart-data-consider-when-applying-residency/. Accessed November 12, 2019.
- Charting Outcomes in the Match: Characteristics of U.S. Allopathic Seniors Who Matched to Their Preferred Specialty in the 2018 Main Residency Match. 2nd ed. Washington, DC: National Resident Matching Program; July 2018. https://www.nrmp.org/wp-content/uploads/2018/06/Charting-Outcomes-in-the-Match-2018-Seniors.pdf. Accessed November 11, 2019.

Pyoderma Gangrenosum Developing After Chest Tube Placement in a Patient With Chronic Lymphocytic Leukemia
Diagnosis of a neutrophilic dermatosis, such as pyoderma gangrenosum (PG), often is challenging at onset because it can be impossible to distinguish clinically and histopathologically from acute infection in an immunosuppressed patient, necessitating a detailed history as well as correlation pathology with microbial tissue cultures. The dermatologist’s ability to distinguish a neutrophilic dermatosis from active infection is of paramount importance because the decision to treat with surgical debridement, in addition to an antibiotic regimen, can have grave consequences in the misdiagnosed patient.
Pyoderma gangrenosum is a neutrophilic dermatosis histologically characterized by a pandermal neutrophilic infiltrate without evidence of an infectious cause or true vasculitis. It is classically associated with inflammatory bowel disease or an underlying hematologic malignancy. Pyoderma gangrenosum in the setting of chronic lymphocytic leukemia (CLL) is rare, with as few as 4 cases having been described in the literature and only 1 case of PG developing after a surgical procedure.1-4 We present a case of PG occurring at a chest tube site in a patient with CLL. We highlight the challenges and therapeutic importance of arriving at the correct diagnosis.
Case Report
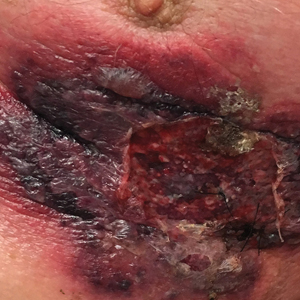
An 87-year-old man with a history of refractory CLL was admitted to the hospital with pneumonia and pleural effusion requiring chest tube placement (left). His most recent therapeutic regimen for CLL was rituximab and bendamustine, which was administered 9 days prior to admission. After removal of the chest tube, an erythematous plaque with central necrosis surrounding the chest tube site developed (Figure 1A). During this time period, the patient had documented intermittent fevers, leukopenia, and neutropenia. Serial blood cultures yielded no growth. Because the patient was on broad-spectrum antibiotic coverage, dermatology was consulted for possible angioinvasive fungal infection.
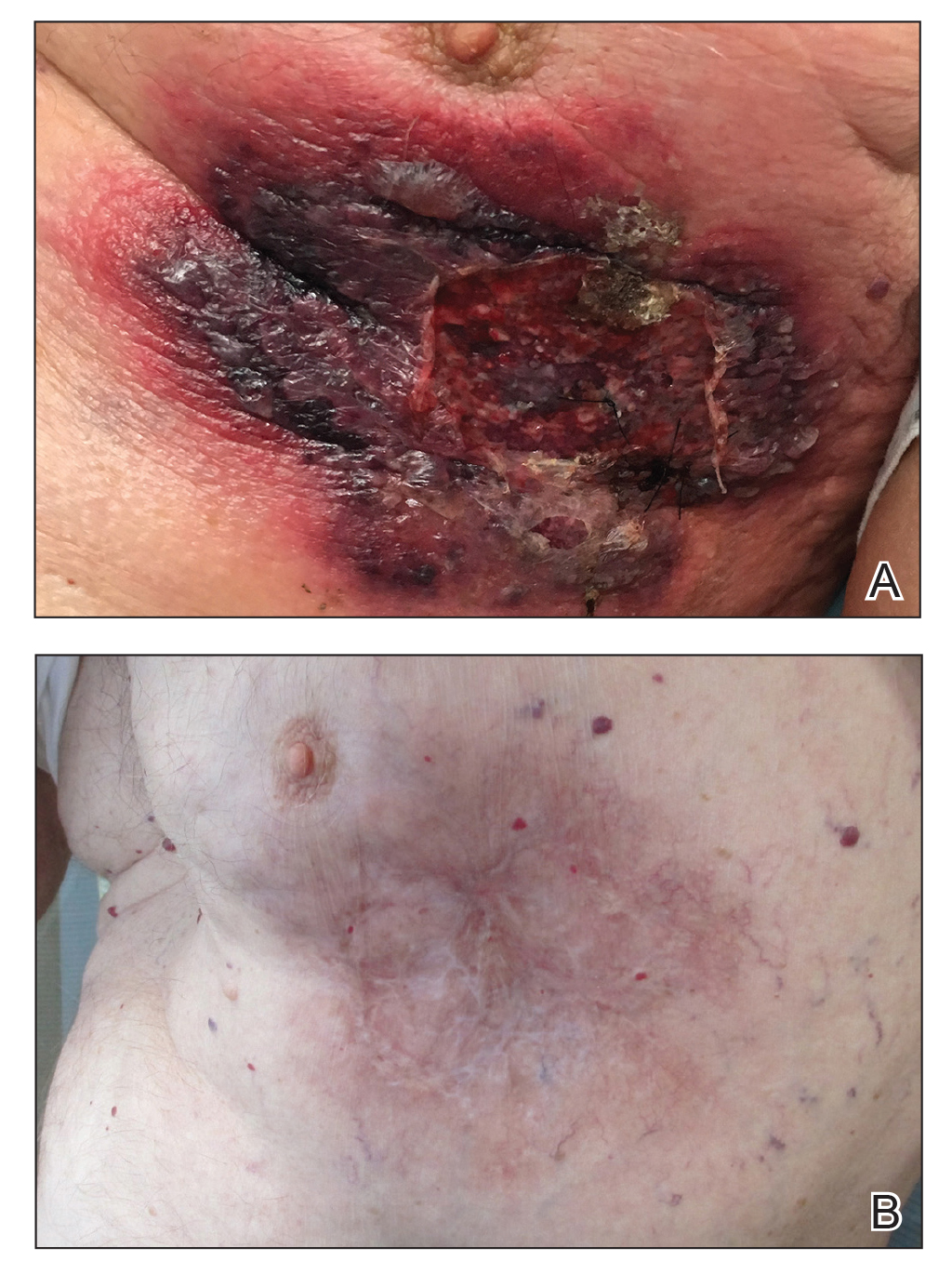
Physical examination revealed an indurated, erythematous-violaceous, targetoid, well-defined, ulcerated plaque with central necrosis on the left side of the chest. Notably, we observed an isolated bulla with an erythematous base within the right antecubital fossa at the site of intravenous placement, suggesting pathergy.
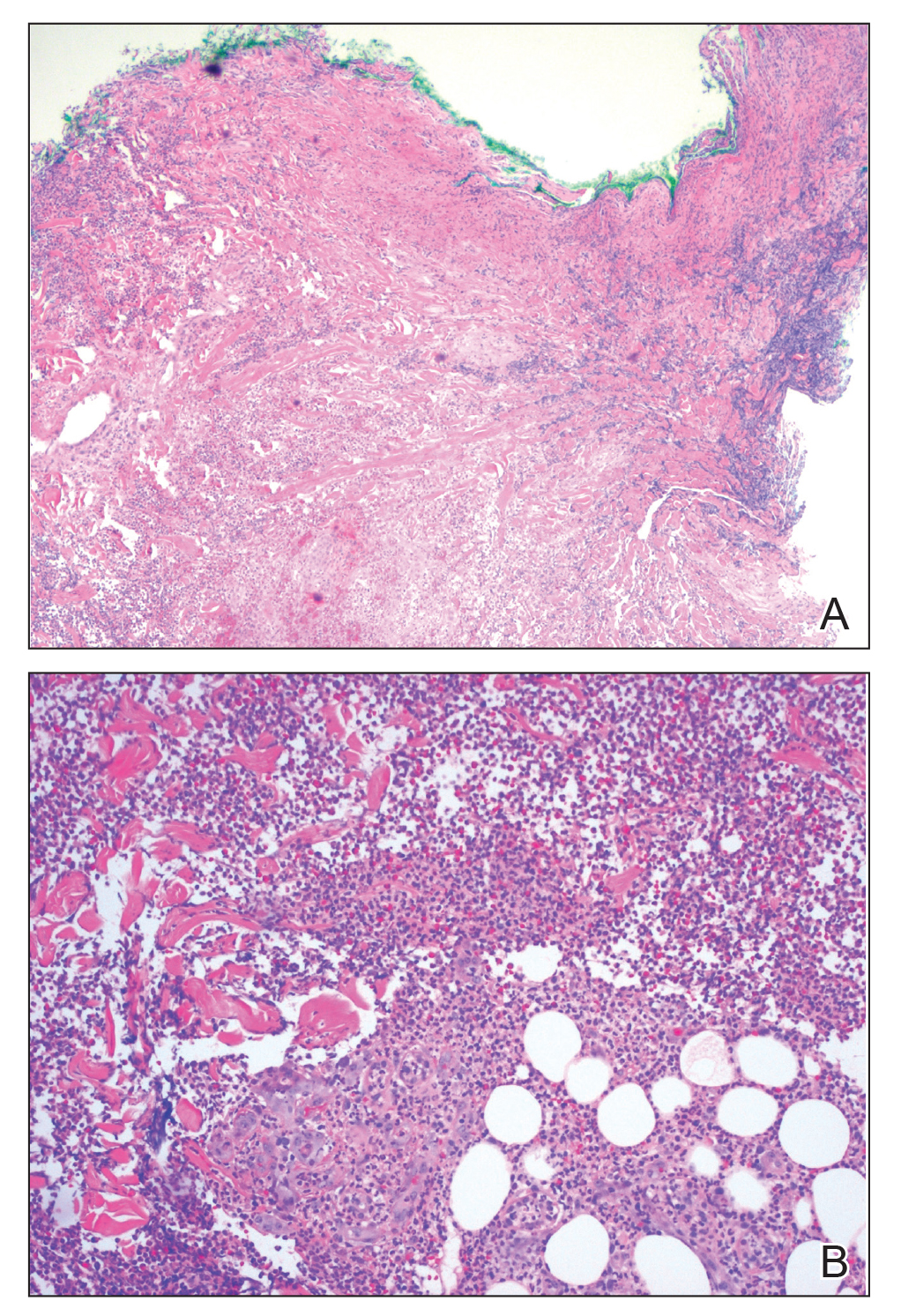
Multiple punch biopsies revealed an ulcer with an underlying dense neutrophilic infiltrate within the dermis and subcutaneous tissues (Figure 2). Grocott-Gomori methenamine-silver, periodic acid–Schiff, and acid-fast bacillus stains were all negative for organisms. Tissue cultures for bacterial, fungal, and acid-fast bacilli revealed no growth. Due to the rapidly expanding nature of the plaque and the possibility of infection despite negative microbial stains and cultures, the patient was scheduled for surgical debridement by the surgical team.
Opportunely, after thoughtful consideration of the clinical history, histopathology, and negative tissue cultures, we made a diagnosis of PG, a condition that would have been further exacerbated by debridement and unimproved with antibiotics. Based on our recommendation, the patient received immunosuppressive treatment with prednisone 60 mg/d and triamcinolone ointment 0.1%. He experienced immediate clinical improvement, allowing him to be discharged to the care of dermatology as an outpatient. He continued to receive a monthly rituximab infusion. We intentionally tapered the patient’s prednisone dosage slowly over 4 months and photodocumented steady improvement with eventual resolution of the PG (Figure 1B).
Comment
Pathogenesis of PG
Pyoderma gangrenosum lies in the spectrum of neutrophilic dermatoses, which are characterized histologically by a pandermal neutrophilic infiltrate without evidence of an infectious cause or true vasculitis. Clinically, PG typically presents as a steadily expanding ulceration with an undermined or slightly raised border, and often is associated with the pathergy phenomenon. Historically, PG is classically linked to inflammatory bowel disease; however, association with underlying malignancy, including acute myelogenous leukemia, chronic myelogenous leukemia, myeloma, and myeloid metaplasia, also has been described.5
Pathogenesis of CLL
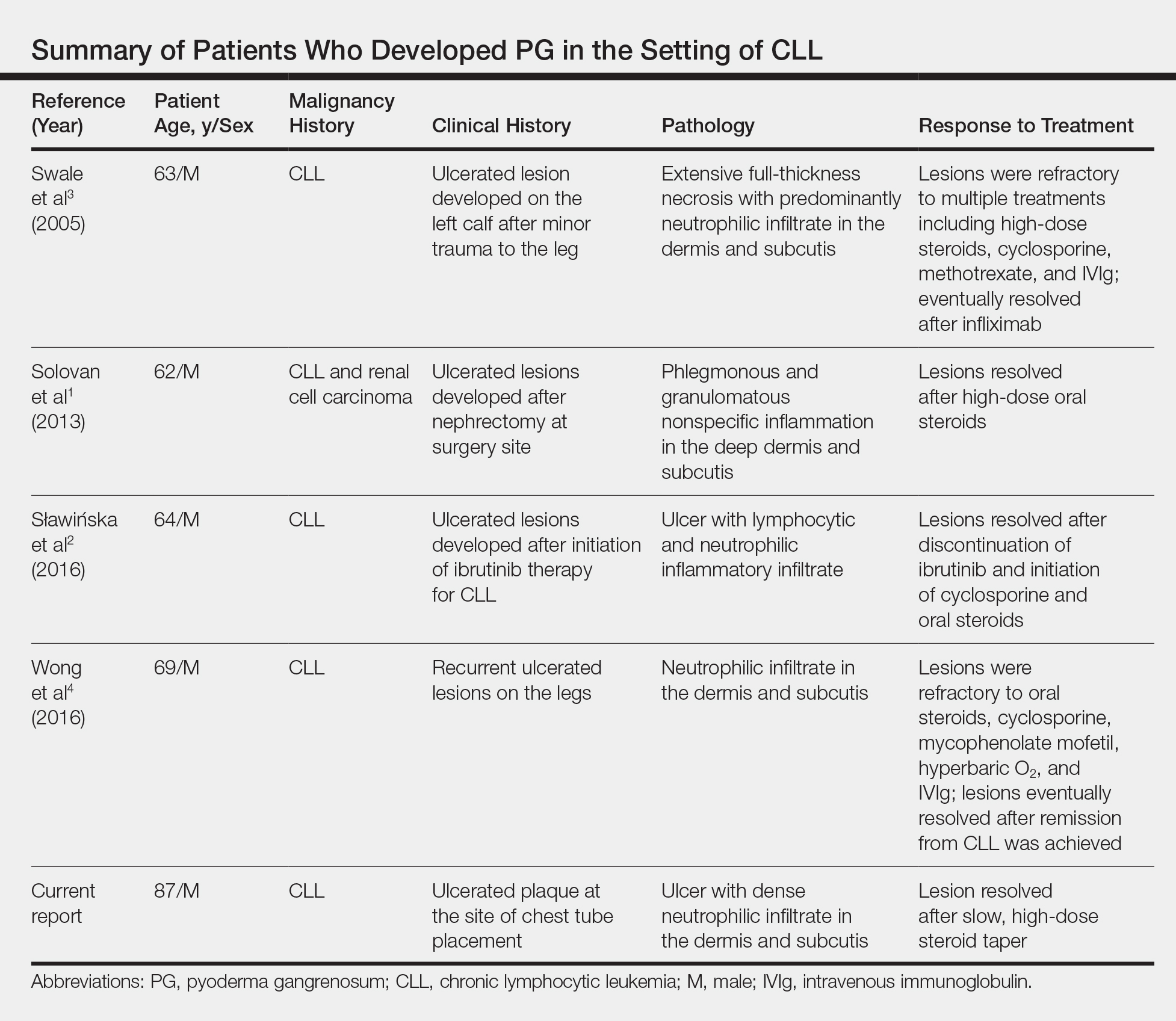
Chronic lymphocytic leukemia represents the most prevalent form of leukemia in US adults, with the second highest annual incidence.6 Cutaneous findings are seen in 25% of patients with CLL, varying from leukemia cutis to secondary findings such as vasculitis, purpura, generalized pruritus, exfoliative erythroderma, paraneoplastic pemphigus, infections, and rarely neutrophilic dermatoses.7 According to a PubMed search of articles indexed for MEDLINE using the term pyoderma gangrenosum in CLL, only 4 cases of PG occurring in the setting of CLL exist in the literature, with 1 case demonstrating development after a surgical procedure, making ours the second such case (Table).1-4
Diagnosis
Making the diagnosis of a neutrophilic dermatosis such as PG or Sweet syndrome (SS) in the hospital setting is not only difficult but also imperative, considering that the counterdiagnosis more often is an infectious process. The distinction between individual neutrophilic dermatoses is less crucial at the onset because the initial treatment is the same.
Sweet syndrome is classically the most challenging entity within the spectrum to differentiate from PG. However, our case outlines several key distinguishing features:
• The lesion in classic PG is a rapidly expanding ulceration with undermined borders, whereas SS is less commonly associated with ulceration and instead classically presents with multiple edematous papules that progress to juicy plaques.8
• The pathergy phenomenon has been reported in SS, though it is more commonly associated with PG.9
• In reported cases of SS that were related to cutaneous trauma, lesions developed outside the area of trauma and there was documented leukocytosis and neutrophilia.10-14
• Although leukocytosis is part of the minor diagnostic criteria for SS, it is not required for the diagnosis of PG. Considering that our patient had ulcerated lesions, lesions only at the site of trauma, and leukopenia with intermittent neutropenia, the diagnosis was consistent with PG.
The primary value of early recognition and diagnosis of PG lies in the physician’s ability to distinguish PG from an infectious process, which can be challenging in an immunosuppressed patient with an underlying hematologic malignancy.
Conclusion
This case report represents our experience in arriving at the correct diagnosis of PG in a febrile neutrophilic patient with CLL. In the case of PG in a complicated patient, it is critical to initiate appropriate treatment and avoid inappropriate therapies. Aggressive surgical debridement could have resulted in a fatal outcome for our patient, highlighting the need for dermatologists to raise physician awareness of this challenging disease.
Acknowledgments
The authors acknowledge the contributions of Sarah Shalin, MD, PhD; Nikhil Meena, MD; and Aditya Chada, MD (all from Little Rock, Arkansas), for excellent patient care.
- Solovan C, Smiszek R, Wickenhauser C, et al. Postoperative pyoderma gangrenosum in association with renal cell carcinoma and chronic lymphocytic leukemia. Infect Dis Ther. 2013;2:75-80.
- Sławińska M, Barańska-Rybak W, Sobjanek M, et al. Ibrutinib-induced pyoderma gangrenosum. Pol Arch Med Wewn. 2016;126:710-711.
- Swale VJ, Saha M, Kapur N, et al. Pyoderma gangrenosum outside the context of inflammatory bowel disease treated successfully with infliximab. Clin Exp Dermatol. 2005;30:134-136.
- Wong SM, McComish J, Douglass J, et al. Rare skin manifestations successfully treated with primary B-cell chronic lymphocytic leukemia treatment. J Cutan Pathol. 2016;43:552-555.
- Jockenhöfer F, Herberger K, Schaller J, et al. Tricenter analysis of cofactors and comorbidity in patients with pyoderma gangrenosum. J Dtsch Dermatol Ges. 2016;14:1023-1030.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7-30. 7.
- Robak E, Robak T. Skin lesions in chronic lymphocytic leukemia. Leuk Lymphoma. 2007;48:855-865.
- Beasley JM, Sluzevich JC. A recurrent vesiculobullous eruption on the head, trunk, and extremities. Bullous Sweet’s syndrome. Int J Dermatol. 2016;55:149-150.
- Awan F, Hamadani M, Devine S. Paraneoplastic Sweet’s syndrome and the pathergy phenomenon. Ann Hematol. 2007;86:613-614.
- de Moya MA, Wong JT, Kroshinsky D, et al. Case records of the Massachusetts General Hospital. Case 28-2012. A 30-year-old woman with shock and abdominal-wall necrosis after cesarean section. N Engl J Med. 2012;367:1046-1057.
- Minocha R, Sebaratnam DF, Choi JY. Sweet’s syndrome following surgery: cutaneous trauma as a possible aetiological co-factor in neutrophilic dermatoses. Australas J Dermatol. 2015;56:
e74-e76. - Phua YS, Al-Ani SA, She RB, et al. Sweet’s syndrome triggered by scalding: a case study and review of the literature. Burns. 2010;36:e49-e52.
- Schwarz RE, Quinn MA, Molina A. Acute postoperative dermatosis at the site of the electrocautery pad: sweet diagnosis of a burning issue. Surg Today. 2000;30:207-209.
- Tan AW, Tan HH, Lim PL. Bullous Sweet’s syndrome following influenza vaccination in a HIV-infected patient. Int J Dermatol. 2006;45:1254-1255.
Diagnosis of a neutrophilic dermatosis, such as pyoderma gangrenosum (PG), often is challenging at onset because it can be impossible to distinguish clinically and histopathologically from acute infection in an immunosuppressed patient, necessitating a detailed history as well as correlation pathology with microbial tissue cultures. The dermatologist’s ability to distinguish a neutrophilic dermatosis from active infection is of paramount importance because the decision to treat with surgical debridement, in addition to an antibiotic regimen, can have grave consequences in the misdiagnosed patient.
Pyoderma gangrenosum is a neutrophilic dermatosis histologically characterized by a pandermal neutrophilic infiltrate without evidence of an infectious cause or true vasculitis. It is classically associated with inflammatory bowel disease or an underlying hematologic malignancy. Pyoderma gangrenosum in the setting of chronic lymphocytic leukemia (CLL) is rare, with as few as 4 cases having been described in the literature and only 1 case of PG developing after a surgical procedure.1-4 We present a case of PG occurring at a chest tube site in a patient with CLL. We highlight the challenges and therapeutic importance of arriving at the correct diagnosis.
Case Report
An 87-year-old man with a history of refractory CLL was admitted to the hospital with pneumonia and pleural effusion requiring chest tube placement (left). His most recent therapeutic regimen for CLL was rituximab and bendamustine, which was administered 9 days prior to admission. After removal of the chest tube, an erythematous plaque with central necrosis surrounding the chest tube site developed (Figure 1A). During this time period, the patient had documented intermittent fevers, leukopenia, and neutropenia. Serial blood cultures yielded no growth. Because the patient was on broad-spectrum antibiotic coverage, dermatology was consulted for possible angioinvasive fungal infection.
Physical examination revealed an indurated, erythematous-violaceous, targetoid, well-defined, ulcerated plaque with central necrosis on the left side of the chest. Notably, we observed an isolated bulla with an erythematous base within the right antecubital fossa at the site of intravenous placement, suggesting pathergy.
Multiple punch biopsies revealed an ulcer with an underlying dense neutrophilic infiltrate within the dermis and subcutaneous tissues (Figure 2). Grocott-Gomori methenamine-silver, periodic acid–Schiff, and acid-fast bacillus stains were all negative for organisms. Tissue cultures for bacterial, fungal, and acid-fast bacilli revealed no growth. Due to the rapidly expanding nature of the plaque and the possibility of infection despite negative microbial stains and cultures, the patient was scheduled for surgical debridement by the surgical team.
Opportunely, after thoughtful consideration of the clinical history, histopathology, and negative tissue cultures, we made a diagnosis of PG, a condition that would have been further exacerbated by debridement and unimproved with antibiotics. Based on our recommendation, the patient received immunosuppressive treatment with prednisone 60 mg/d and triamcinolone ointment 0.1%. He experienced immediate clinical improvement, allowing him to be discharged to the care of dermatology as an outpatient. He continued to receive a monthly rituximab infusion. We intentionally tapered the patient’s prednisone dosage slowly over 4 months and photodocumented steady improvement with eventual resolution of the PG (Figure 1B).
Comment
Pathogenesis of PG
Pyoderma gangrenosum lies in the spectrum of neutrophilic dermatoses, which are characterized histologically by a pandermal neutrophilic infiltrate without evidence of an infectious cause or true vasculitis. Clinically, PG typically presents as a steadily expanding ulceration with an undermined or slightly raised border, and often is associated with the pathergy phenomenon. Historically, PG is classically linked to inflammatory bowel disease; however, association with underlying malignancy, including acute myelogenous leukemia, chronic myelogenous leukemia, myeloma, and myeloid metaplasia, also has been described.5
Pathogenesis of CLL
Chronic lymphocytic leukemia represents the most prevalent form of leukemia in US adults, with the second highest annual incidence.6 Cutaneous findings are seen in 25% of patients with CLL, varying from leukemia cutis to secondary findings such as vasculitis, purpura, generalized pruritus, exfoliative erythroderma, paraneoplastic pemphigus, infections, and rarely neutrophilic dermatoses.7 According to a PubMed search of articles indexed for MEDLINE using the term pyoderma gangrenosum in CLL, only 4 cases of PG occurring in the setting of CLL exist in the literature, with 1 case demonstrating development after a surgical procedure, making ours the second such case (Table).1-4
Diagnosis
Making the diagnosis of a neutrophilic dermatosis such as PG or Sweet syndrome (SS) in the hospital setting is not only difficult but also imperative, considering that the counterdiagnosis more often is an infectious process. The distinction between individual neutrophilic dermatoses is less crucial at the onset because the initial treatment is the same.
Sweet syndrome is classically the most challenging entity within the spectrum to differentiate from PG. However, our case outlines several key distinguishing features:
• The lesion in classic PG is a rapidly expanding ulceration with undermined borders, whereas SS is less commonly associated with ulceration and instead classically presents with multiple edematous papules that progress to juicy plaques.8
• The pathergy phenomenon has been reported in SS, though it is more commonly associated with PG.9
• In reported cases of SS that were related to cutaneous trauma, lesions developed outside the area of trauma and there was documented leukocytosis and neutrophilia.10-14
• Although leukocytosis is part of the minor diagnostic criteria for SS, it is not required for the diagnosis of PG. Considering that our patient had ulcerated lesions, lesions only at the site of trauma, and leukopenia with intermittent neutropenia, the diagnosis was consistent with PG.
The primary value of early recognition and diagnosis of PG lies in the physician’s ability to distinguish PG from an infectious process, which can be challenging in an immunosuppressed patient with an underlying hematologic malignancy.
Conclusion
This case report represents our experience in arriving at the correct diagnosis of PG in a febrile neutrophilic patient with CLL. In the case of PG in a complicated patient, it is critical to initiate appropriate treatment and avoid inappropriate therapies. Aggressive surgical debridement could have resulted in a fatal outcome for our patient, highlighting the need for dermatologists to raise physician awareness of this challenging disease.
Acknowledgments
The authors acknowledge the contributions of Sarah Shalin, MD, PhD; Nikhil Meena, MD; and Aditya Chada, MD (all from Little Rock, Arkansas), for excellent patient care.
Diagnosis of a neutrophilic dermatosis, such as pyoderma gangrenosum (PG), often is challenging at onset because it can be impossible to distinguish clinically and histopathologically from acute infection in an immunosuppressed patient, necessitating a detailed history as well as correlation pathology with microbial tissue cultures. The dermatologist’s ability to distinguish a neutrophilic dermatosis from active infection is of paramount importance because the decision to treat with surgical debridement, in addition to an antibiotic regimen, can have grave consequences in the misdiagnosed patient.
Pyoderma gangrenosum is a neutrophilic dermatosis histologically characterized by a pandermal neutrophilic infiltrate without evidence of an infectious cause or true vasculitis. It is classically associated with inflammatory bowel disease or an underlying hematologic malignancy. Pyoderma gangrenosum in the setting of chronic lymphocytic leukemia (CLL) is rare, with as few as 4 cases having been described in the literature and only 1 case of PG developing after a surgical procedure.1-4 We present a case of PG occurring at a chest tube site in a patient with CLL. We highlight the challenges and therapeutic importance of arriving at the correct diagnosis.
Case Report
An 87-year-old man with a history of refractory CLL was admitted to the hospital with pneumonia and pleural effusion requiring chest tube placement (left). His most recent therapeutic regimen for CLL was rituximab and bendamustine, which was administered 9 days prior to admission. After removal of the chest tube, an erythematous plaque with central necrosis surrounding the chest tube site developed (Figure 1A). During this time period, the patient had documented intermittent fevers, leukopenia, and neutropenia. Serial blood cultures yielded no growth. Because the patient was on broad-spectrum antibiotic coverage, dermatology was consulted for possible angioinvasive fungal infection.
Physical examination revealed an indurated, erythematous-violaceous, targetoid, well-defined, ulcerated plaque with central necrosis on the left side of the chest. Notably, we observed an isolated bulla with an erythematous base within the right antecubital fossa at the site of intravenous placement, suggesting pathergy.
Multiple punch biopsies revealed an ulcer with an underlying dense neutrophilic infiltrate within the dermis and subcutaneous tissues (Figure 2). Grocott-Gomori methenamine-silver, periodic acid–Schiff, and acid-fast bacillus stains were all negative for organisms. Tissue cultures for bacterial, fungal, and acid-fast bacilli revealed no growth. Due to the rapidly expanding nature of the plaque and the possibility of infection despite negative microbial stains and cultures, the patient was scheduled for surgical debridement by the surgical team.
Opportunely, after thoughtful consideration of the clinical history, histopathology, and negative tissue cultures, we made a diagnosis of PG, a condition that would have been further exacerbated by debridement and unimproved with antibiotics. Based on our recommendation, the patient received immunosuppressive treatment with prednisone 60 mg/d and triamcinolone ointment 0.1%. He experienced immediate clinical improvement, allowing him to be discharged to the care of dermatology as an outpatient. He continued to receive a monthly rituximab infusion. We intentionally tapered the patient’s prednisone dosage slowly over 4 months and photodocumented steady improvement with eventual resolution of the PG (Figure 1B).
Comment
Pathogenesis of PG
Pyoderma gangrenosum lies in the spectrum of neutrophilic dermatoses, which are characterized histologically by a pandermal neutrophilic infiltrate without evidence of an infectious cause or true vasculitis. Clinically, PG typically presents as a steadily expanding ulceration with an undermined or slightly raised border, and often is associated with the pathergy phenomenon. Historically, PG is classically linked to inflammatory bowel disease; however, association with underlying malignancy, including acute myelogenous leukemia, chronic myelogenous leukemia, myeloma, and myeloid metaplasia, also has been described.5
Pathogenesis of CLL
Chronic lymphocytic leukemia represents the most prevalent form of leukemia in US adults, with the second highest annual incidence.6 Cutaneous findings are seen in 25% of patients with CLL, varying from leukemia cutis to secondary findings such as vasculitis, purpura, generalized pruritus, exfoliative erythroderma, paraneoplastic pemphigus, infections, and rarely neutrophilic dermatoses.7 According to a PubMed search of articles indexed for MEDLINE using the term pyoderma gangrenosum in CLL, only 4 cases of PG occurring in the setting of CLL exist in the literature, with 1 case demonstrating development after a surgical procedure, making ours the second such case (Table).1-4
Diagnosis
Making the diagnosis of a neutrophilic dermatosis such as PG or Sweet syndrome (SS) in the hospital setting is not only difficult but also imperative, considering that the counterdiagnosis more often is an infectious process. The distinction between individual neutrophilic dermatoses is less crucial at the onset because the initial treatment is the same.
Sweet syndrome is classically the most challenging entity within the spectrum to differentiate from PG. However, our case outlines several key distinguishing features:
• The lesion in classic PG is a rapidly expanding ulceration with undermined borders, whereas SS is less commonly associated with ulceration and instead classically presents with multiple edematous papules that progress to juicy plaques.8
• The pathergy phenomenon has been reported in SS, though it is more commonly associated with PG.9
• In reported cases of SS that were related to cutaneous trauma, lesions developed outside the area of trauma and there was documented leukocytosis and neutrophilia.10-14
• Although leukocytosis is part of the minor diagnostic criteria for SS, it is not required for the diagnosis of PG. Considering that our patient had ulcerated lesions, lesions only at the site of trauma, and leukopenia with intermittent neutropenia, the diagnosis was consistent with PG.
The primary value of early recognition and diagnosis of PG lies in the physician’s ability to distinguish PG from an infectious process, which can be challenging in an immunosuppressed patient with an underlying hematologic malignancy.
Conclusion
This case report represents our experience in arriving at the correct diagnosis of PG in a febrile neutrophilic patient with CLL. In the case of PG in a complicated patient, it is critical to initiate appropriate treatment and avoid inappropriate therapies. Aggressive surgical debridement could have resulted in a fatal outcome for our patient, highlighting the need for dermatologists to raise physician awareness of this challenging disease.
Acknowledgments
The authors acknowledge the contributions of Sarah Shalin, MD, PhD; Nikhil Meena, MD; and Aditya Chada, MD (all from Little Rock, Arkansas), for excellent patient care.
- Solovan C, Smiszek R, Wickenhauser C, et al. Postoperative pyoderma gangrenosum in association with renal cell carcinoma and chronic lymphocytic leukemia. Infect Dis Ther. 2013;2:75-80.
- Sławińska M, Barańska-Rybak W, Sobjanek M, et al. Ibrutinib-induced pyoderma gangrenosum. Pol Arch Med Wewn. 2016;126:710-711.
- Swale VJ, Saha M, Kapur N, et al. Pyoderma gangrenosum outside the context of inflammatory bowel disease treated successfully with infliximab. Clin Exp Dermatol. 2005;30:134-136.
- Wong SM, McComish J, Douglass J, et al. Rare skin manifestations successfully treated with primary B-cell chronic lymphocytic leukemia treatment. J Cutan Pathol. 2016;43:552-555.
- Jockenhöfer F, Herberger K, Schaller J, et al. Tricenter analysis of cofactors and comorbidity in patients with pyoderma gangrenosum. J Dtsch Dermatol Ges. 2016;14:1023-1030.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7-30. 7.
- Robak E, Robak T. Skin lesions in chronic lymphocytic leukemia. Leuk Lymphoma. 2007;48:855-865.
- Beasley JM, Sluzevich JC. A recurrent vesiculobullous eruption on the head, trunk, and extremities. Bullous Sweet’s syndrome. Int J Dermatol. 2016;55:149-150.
- Awan F, Hamadani M, Devine S. Paraneoplastic Sweet’s syndrome and the pathergy phenomenon. Ann Hematol. 2007;86:613-614.
- de Moya MA, Wong JT, Kroshinsky D, et al. Case records of the Massachusetts General Hospital. Case 28-2012. A 30-year-old woman with shock and abdominal-wall necrosis after cesarean section. N Engl J Med. 2012;367:1046-1057.
- Minocha R, Sebaratnam DF, Choi JY. Sweet’s syndrome following surgery: cutaneous trauma as a possible aetiological co-factor in neutrophilic dermatoses. Australas J Dermatol. 2015;56:
e74-e76. - Phua YS, Al-Ani SA, She RB, et al. Sweet’s syndrome triggered by scalding: a case study and review of the literature. Burns. 2010;36:e49-e52.
- Schwarz RE, Quinn MA, Molina A. Acute postoperative dermatosis at the site of the electrocautery pad: sweet diagnosis of a burning issue. Surg Today. 2000;30:207-209.
- Tan AW, Tan HH, Lim PL. Bullous Sweet’s syndrome following influenza vaccination in a HIV-infected patient. Int J Dermatol. 2006;45:1254-1255.
- Solovan C, Smiszek R, Wickenhauser C, et al. Postoperative pyoderma gangrenosum in association with renal cell carcinoma and chronic lymphocytic leukemia. Infect Dis Ther. 2013;2:75-80.
- Sławińska M, Barańska-Rybak W, Sobjanek M, et al. Ibrutinib-induced pyoderma gangrenosum. Pol Arch Med Wewn. 2016;126:710-711.
- Swale VJ, Saha M, Kapur N, et al. Pyoderma gangrenosum outside the context of inflammatory bowel disease treated successfully with infliximab. Clin Exp Dermatol. 2005;30:134-136.
- Wong SM, McComish J, Douglass J, et al. Rare skin manifestations successfully treated with primary B-cell chronic lymphocytic leukemia treatment. J Cutan Pathol. 2016;43:552-555.
- Jockenhöfer F, Herberger K, Schaller J, et al. Tricenter analysis of cofactors and comorbidity in patients with pyoderma gangrenosum. J Dtsch Dermatol Ges. 2016;14:1023-1030.
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7-30. 7.
- Robak E, Robak T. Skin lesions in chronic lymphocytic leukemia. Leuk Lymphoma. 2007;48:855-865.
- Beasley JM, Sluzevich JC. A recurrent vesiculobullous eruption on the head, trunk, and extremities. Bullous Sweet’s syndrome. Int J Dermatol. 2016;55:149-150.
- Awan F, Hamadani M, Devine S. Paraneoplastic Sweet’s syndrome and the pathergy phenomenon. Ann Hematol. 2007;86:613-614.
- de Moya MA, Wong JT, Kroshinsky D, et al. Case records of the Massachusetts General Hospital. Case 28-2012. A 30-year-old woman with shock and abdominal-wall necrosis after cesarean section. N Engl J Med. 2012;367:1046-1057.
- Minocha R, Sebaratnam DF, Choi JY. Sweet’s syndrome following surgery: cutaneous trauma as a possible aetiological co-factor in neutrophilic dermatoses. Australas J Dermatol. 2015;56:
e74-e76. - Phua YS, Al-Ani SA, She RB, et al. Sweet’s syndrome triggered by scalding: a case study and review of the literature. Burns. 2010;36:e49-e52.
- Schwarz RE, Quinn MA, Molina A. Acute postoperative dermatosis at the site of the electrocautery pad: sweet diagnosis of a burning issue. Surg Today. 2000;30:207-209.
- Tan AW, Tan HH, Lim PL. Bullous Sweet’s syndrome following influenza vaccination in a HIV-infected patient. Int J Dermatol. 2006;45:1254-1255.
Practice Points
- The primary value of early recognition and diagnosis of pyoderma gangrenosum (PG) lies in the physician’s ability to distinguish PG from an infectious process.
- Surgical debridement would further exacerbate PG, making proper diagnosis of a neutrophilic dermatosis of paramount importance to avoid treatments that could have grave consequences in the misdiagnosed patient.
- Cutaneous findings are seen in one-quarter of patients with chronic lymphocytic leukemia.
- Pyoderma gangrenosum is commonly associated with inflammatory bowel disease but also can be seen in many hematologic malignancies. Physicians should be aware of this association to ensure these patients are diagnosed properly.
Don’t Forget These 5 Things When Treating Hidradenitis Suppurativa
Hidradenitis suppurativa (HS) is a common and debilitating inflammatory disorder of the pilosebaceous unit that presents with recurrent scarring inflammatory nodules and sinus tracts in the intertriginous folds of the body. It is a complex condition that requires multimodal management to address the medical, surgical, and psychosocial needs of affected patients. However, it can be difficult to coordinate all that goes into HS management beyond the standard therapeutic ladder of topical and oral antimicrobials, intralesional corticosteroids, biologics, and surgery. In this article, I will outline 5 important aspects of HS treatment that often are overlooked.
Talk About Pathophysiology
Patients with HS often have limited understanding of their condition. One common misperception is that HS is an infectious disease and that disease activity is associated with poor hygiene.1 Dispelling this myth may help patients avoid unnecessary hygiene practices, decrease perceived stigma, and enhance your therapeutic alliance.
The current model of HS pathophysiology implicates an aberrant inflammatory response to the cutaneous bacterial microbiome, which leads to follicular occlusion and then rupture of debris and bacteria into the surrounding dermis. Immune cells and inflammatory mediators such as nuclear factor κB and tumor necrosis factor α respond to the disruption. Chronic lesions develop due to tissue repair with scarring and re-epithelialization.2,3 Although most patients probably are not interested in the esoteric details, I typically make a point of explaining to patients that HS is a chronic inflammatory disease and provide reassurance that it is not a sign of poor hygiene.
Counsel on Smoking Cessation
Most HS patients use tobacco. As many as 75% of HS patients are active smokers and another 10% to 15% are former smokers. Although there is mixed evidence that disease activity correlates with smoking status, the Hidradenitis Suppurativa Foundation in the United States and Canada concluded in the 2019 North American Clinical Management Guidelines for Hidradenitis Suppurativa that due to the overall health risks of smoking, we should recommend cessation to our patients.4
Laser Hair Removal Works
Don’t forget about laser hair removal! Evidence from randomized controlled trials supports the use of the Nd:YAG laser in the treatment of HS. Treat the entire affected anatomic area and use stacked double pulses on active nodules (typical settings: 10-mm spot size; 10-millisecond pulse duration and 35–50 J/cm2 in Fitzpatrick skin types I–III; 20-millisecond pulse duration and 25–40 J/cm2 in Fitzpatrick skin types IV–VI).4 Especially if it is covered by your patient’s insurance, Nd:YAG is a great adjunctive treatment to consider. The guidelines also recommend long-pulsed alexandrite and diode lasers as well as intense pulsed light, all of which result in follicular destruction, though these treatments have less supporting evidence.4
Have a Plan for Flares
Intralesional injection of triamcinolone is a mainstay of HS treatment and provides patients with rapid relief of symptoms during a flare.5 One case series found that there was a notable decrease in pain, size, and drainage after just 1 day of treatment with intralesional triamcinolone 10 mg/mL (0.2–2.0 mL).6
Intralesional steroid injection is a great tool for quieting an active disease flare while simultaneously instating ongoing treatment for preventive management. However, even when disease control is optimized, patients may still experience intermittent flares of disease. For some patients, it may be appropriate to have a plan in place for a return to clinic during the beginning of a flare to obtain intralesional steroids. The ability to come in on short notice may help avoid visits to the emergency department and urgent care where your patients may receive treatments such as short courses of antibiotics or incision and drainage that may deviate from your overall treatment plan.
Consider Childbearing Status
Don’t forget to consider childbearing plans and childbearing potential when treating female patients with HS. Pregnancy is a frequent consideration in HS patients, as HS affects 3 to 4 times more women than men and typically presents after puberty (second or third decades of life). Many of the medications in the HS armamentarium are contraindicated in pregnancy including tetracyclines, retinoids, and hormonal agents. Surgery should be avoided in pregnant patients whenever possible, particularly in the first trimester. Relatively safe options include topical antibiotics such as clindamycin and metronidazole, as well as tumor necrosis factor α inhibitors, which are classified as category B in pregnancy.5
Before making treatment decisions in pregnant and breastfeeding patients, consult the US Food and Drug Administration recommendations. Perng et al7 reviewed current management strategies for HS in pregnant and breastfeeding women, and their review article in the Journal of the American Academy of Dermatology is an excellent resource.
Final Thoughts
Comprehensive management of HS may include a combination of medication and procedures, lifestyle modification, management of comorbidities, and social support. Formulating a good treatment plan may be a challenge but can drastically improve your patient’s quality of life.
- What is hidradenitis suppurativa? Hidradenitis Suppurativa Foundation website. https://www.hs-foundation.org/what-is-hs. Accessed October 9, 2019.
- Frew JW, Hawkes JE, Krueger JG. Topical, systemic and biologic therapies in hidradenitis suppurativa: pathogenic insights by examining therapeutic mechanisms. Ther Adv Chronic Dis. 2019;10:2040622319830646. doi:10.1177/2040622319830646
- Lacarrubba F, Musumeci ML, Nasca MR, et al. Double-ended pseudocomedones in hidradenitis suppurativa: clinical, dermoscopic, and histopathological correlation. Acta Derm Venereol. 2017;97:763-764.
- Alikhan A, Sayed C, Alavi A, et al. North American Clinical Management Guidelines for Hidradenitis Suppurativa: a publication from the United States and Canadian Hidradenitis Suppurativa Foundations. part I: diagnosis, evaluation, and the use of complementary and procedural management. J Am Acad Dermatol. 2019;81:76-90.
- Alikhan A, Sayed C, Alavi A, et al. North American Clinical Management Guidelines for Hidradenitis Suppurativa: a publication from the United States and Canadian Hidradenitis Suppurativa Foundations. part II: topical, intralesional, and systemic medical management. J Am Acad Dermatol. 2019;81:91-101.
- Riis PT, Boer J, Prens EP, et al. Intralesional triamcinolone for flares of hidradenitis suppurativa (HS): a case series. J Am Acad Dermatol. 2016;75:1151-1155.
- Perng P, Zampella JG, Okoye GA. Management of hidradenitis suppurativa in pregnancy. J Am Acad Dermatol. 2017;76:979-989.
Hidradenitis suppurativa (HS) is a common and debilitating inflammatory disorder of the pilosebaceous unit that presents with recurrent scarring inflammatory nodules and sinus tracts in the intertriginous folds of the body. It is a complex condition that requires multimodal management to address the medical, surgical, and psychosocial needs of affected patients. However, it can be difficult to coordinate all that goes into HS management beyond the standard therapeutic ladder of topical and oral antimicrobials, intralesional corticosteroids, biologics, and surgery. In this article, I will outline 5 important aspects of HS treatment that often are overlooked.
Talk About Pathophysiology
Patients with HS often have limited understanding of their condition. One common misperception is that HS is an infectious disease and that disease activity is associated with poor hygiene.1 Dispelling this myth may help patients avoid unnecessary hygiene practices, decrease perceived stigma, and enhance your therapeutic alliance.
The current model of HS pathophysiology implicates an aberrant inflammatory response to the cutaneous bacterial microbiome, which leads to follicular occlusion and then rupture of debris and bacteria into the surrounding dermis. Immune cells and inflammatory mediators such as nuclear factor κB and tumor necrosis factor α respond to the disruption. Chronic lesions develop due to tissue repair with scarring and re-epithelialization.2,3 Although most patients probably are not interested in the esoteric details, I typically make a point of explaining to patients that HS is a chronic inflammatory disease and provide reassurance that it is not a sign of poor hygiene.
Counsel on Smoking Cessation
Most HS patients use tobacco. As many as 75% of HS patients are active smokers and another 10% to 15% are former smokers. Although there is mixed evidence that disease activity correlates with smoking status, the Hidradenitis Suppurativa Foundation in the United States and Canada concluded in the 2019 North American Clinical Management Guidelines for Hidradenitis Suppurativa that due to the overall health risks of smoking, we should recommend cessation to our patients.4
Laser Hair Removal Works
Don’t forget about laser hair removal! Evidence from randomized controlled trials supports the use of the Nd:YAG laser in the treatment of HS. Treat the entire affected anatomic area and use stacked double pulses on active nodules (typical settings: 10-mm spot size; 10-millisecond pulse duration and 35–50 J/cm2 in Fitzpatrick skin types I–III; 20-millisecond pulse duration and 25–40 J/cm2 in Fitzpatrick skin types IV–VI).4 Especially if it is covered by your patient’s insurance, Nd:YAG is a great adjunctive treatment to consider. The guidelines also recommend long-pulsed alexandrite and diode lasers as well as intense pulsed light, all of which result in follicular destruction, though these treatments have less supporting evidence.4
Have a Plan for Flares
Intralesional injection of triamcinolone is a mainstay of HS treatment and provides patients with rapid relief of symptoms during a flare.5 One case series found that there was a notable decrease in pain, size, and drainage after just 1 day of treatment with intralesional triamcinolone 10 mg/mL (0.2–2.0 mL).6
Intralesional steroid injection is a great tool for quieting an active disease flare while simultaneously instating ongoing treatment for preventive management. However, even when disease control is optimized, patients may still experience intermittent flares of disease. For some patients, it may be appropriate to have a plan in place for a return to clinic during the beginning of a flare to obtain intralesional steroids. The ability to come in on short notice may help avoid visits to the emergency department and urgent care where your patients may receive treatments such as short courses of antibiotics or incision and drainage that may deviate from your overall treatment plan.
Consider Childbearing Status
Don’t forget to consider childbearing plans and childbearing potential when treating female patients with HS. Pregnancy is a frequent consideration in HS patients, as HS affects 3 to 4 times more women than men and typically presents after puberty (second or third decades of life). Many of the medications in the HS armamentarium are contraindicated in pregnancy including tetracyclines, retinoids, and hormonal agents. Surgery should be avoided in pregnant patients whenever possible, particularly in the first trimester. Relatively safe options include topical antibiotics such as clindamycin and metronidazole, as well as tumor necrosis factor α inhibitors, which are classified as category B in pregnancy.5
Before making treatment decisions in pregnant and breastfeeding patients, consult the US Food and Drug Administration recommendations. Perng et al7 reviewed current management strategies for HS in pregnant and breastfeeding women, and their review article in the Journal of the American Academy of Dermatology is an excellent resource.
Final Thoughts
Comprehensive management of HS may include a combination of medication and procedures, lifestyle modification, management of comorbidities, and social support. Formulating a good treatment plan may be a challenge but can drastically improve your patient’s quality of life.
Hidradenitis suppurativa (HS) is a common and debilitating inflammatory disorder of the pilosebaceous unit that presents with recurrent scarring inflammatory nodules and sinus tracts in the intertriginous folds of the body. It is a complex condition that requires multimodal management to address the medical, surgical, and psychosocial needs of affected patients. However, it can be difficult to coordinate all that goes into HS management beyond the standard therapeutic ladder of topical and oral antimicrobials, intralesional corticosteroids, biologics, and surgery. In this article, I will outline 5 important aspects of HS treatment that often are overlooked.
Talk About Pathophysiology
Patients with HS often have limited understanding of their condition. One common misperception is that HS is an infectious disease and that disease activity is associated with poor hygiene.1 Dispelling this myth may help patients avoid unnecessary hygiene practices, decrease perceived stigma, and enhance your therapeutic alliance.
The current model of HS pathophysiology implicates an aberrant inflammatory response to the cutaneous bacterial microbiome, which leads to follicular occlusion and then rupture of debris and bacteria into the surrounding dermis. Immune cells and inflammatory mediators such as nuclear factor κB and tumor necrosis factor α respond to the disruption. Chronic lesions develop due to tissue repair with scarring and re-epithelialization.2,3 Although most patients probably are not interested in the esoteric details, I typically make a point of explaining to patients that HS is a chronic inflammatory disease and provide reassurance that it is not a sign of poor hygiene.
Counsel on Smoking Cessation
Most HS patients use tobacco. As many as 75% of HS patients are active smokers and another 10% to 15% are former smokers. Although there is mixed evidence that disease activity correlates with smoking status, the Hidradenitis Suppurativa Foundation in the United States and Canada concluded in the 2019 North American Clinical Management Guidelines for Hidradenitis Suppurativa that due to the overall health risks of smoking, we should recommend cessation to our patients.4
Laser Hair Removal Works
Don’t forget about laser hair removal! Evidence from randomized controlled trials supports the use of the Nd:YAG laser in the treatment of HS. Treat the entire affected anatomic area and use stacked double pulses on active nodules (typical settings: 10-mm spot size; 10-millisecond pulse duration and 35–50 J/cm2 in Fitzpatrick skin types I–III; 20-millisecond pulse duration and 25–40 J/cm2 in Fitzpatrick skin types IV–VI).4 Especially if it is covered by your patient’s insurance, Nd:YAG is a great adjunctive treatment to consider. The guidelines also recommend long-pulsed alexandrite and diode lasers as well as intense pulsed light, all of which result in follicular destruction, though these treatments have less supporting evidence.4
Have a Plan for Flares
Intralesional injection of triamcinolone is a mainstay of HS treatment and provides patients with rapid relief of symptoms during a flare.5 One case series found that there was a notable decrease in pain, size, and drainage after just 1 day of treatment with intralesional triamcinolone 10 mg/mL (0.2–2.0 mL).6
Intralesional steroid injection is a great tool for quieting an active disease flare while simultaneously instating ongoing treatment for preventive management. However, even when disease control is optimized, patients may still experience intermittent flares of disease. For some patients, it may be appropriate to have a plan in place for a return to clinic during the beginning of a flare to obtain intralesional steroids. The ability to come in on short notice may help avoid visits to the emergency department and urgent care where your patients may receive treatments such as short courses of antibiotics or incision and drainage that may deviate from your overall treatment plan.
Consider Childbearing Status
Don’t forget to consider childbearing plans and childbearing potential when treating female patients with HS. Pregnancy is a frequent consideration in HS patients, as HS affects 3 to 4 times more women than men and typically presents after puberty (second or third decades of life). Many of the medications in the HS armamentarium are contraindicated in pregnancy including tetracyclines, retinoids, and hormonal agents. Surgery should be avoided in pregnant patients whenever possible, particularly in the first trimester. Relatively safe options include topical antibiotics such as clindamycin and metronidazole, as well as tumor necrosis factor α inhibitors, which are classified as category B in pregnancy.5
Before making treatment decisions in pregnant and breastfeeding patients, consult the US Food and Drug Administration recommendations. Perng et al7 reviewed current management strategies for HS in pregnant and breastfeeding women, and their review article in the Journal of the American Academy of Dermatology is an excellent resource.
Final Thoughts
Comprehensive management of HS may include a combination of medication and procedures, lifestyle modification, management of comorbidities, and social support. Formulating a good treatment plan may be a challenge but can drastically improve your patient’s quality of life.
- What is hidradenitis suppurativa? Hidradenitis Suppurativa Foundation website. https://www.hs-foundation.org/what-is-hs. Accessed October 9, 2019.
- Frew JW, Hawkes JE, Krueger JG. Topical, systemic and biologic therapies in hidradenitis suppurativa: pathogenic insights by examining therapeutic mechanisms. Ther Adv Chronic Dis. 2019;10:2040622319830646. doi:10.1177/2040622319830646
- Lacarrubba F, Musumeci ML, Nasca MR, et al. Double-ended pseudocomedones in hidradenitis suppurativa: clinical, dermoscopic, and histopathological correlation. Acta Derm Venereol. 2017;97:763-764.
- Alikhan A, Sayed C, Alavi A, et al. North American Clinical Management Guidelines for Hidradenitis Suppurativa: a publication from the United States and Canadian Hidradenitis Suppurativa Foundations. part I: diagnosis, evaluation, and the use of complementary and procedural management. J Am Acad Dermatol. 2019;81:76-90.
- Alikhan A, Sayed C, Alavi A, et al. North American Clinical Management Guidelines for Hidradenitis Suppurativa: a publication from the United States and Canadian Hidradenitis Suppurativa Foundations. part II: topical, intralesional, and systemic medical management. J Am Acad Dermatol. 2019;81:91-101.
- Riis PT, Boer J, Prens EP, et al. Intralesional triamcinolone for flares of hidradenitis suppurativa (HS): a case series. J Am Acad Dermatol. 2016;75:1151-1155.
- Perng P, Zampella JG, Okoye GA. Management of hidradenitis suppurativa in pregnancy. J Am Acad Dermatol. 2017;76:979-989.
- What is hidradenitis suppurativa? Hidradenitis Suppurativa Foundation website. https://www.hs-foundation.org/what-is-hs. Accessed October 9, 2019.
- Frew JW, Hawkes JE, Krueger JG. Topical, systemic and biologic therapies in hidradenitis suppurativa: pathogenic insights by examining therapeutic mechanisms. Ther Adv Chronic Dis. 2019;10:2040622319830646. doi:10.1177/2040622319830646
- Lacarrubba F, Musumeci ML, Nasca MR, et al. Double-ended pseudocomedones in hidradenitis suppurativa: clinical, dermoscopic, and histopathological correlation. Acta Derm Venereol. 2017;97:763-764.
- Alikhan A, Sayed C, Alavi A, et al. North American Clinical Management Guidelines for Hidradenitis Suppurativa: a publication from the United States and Canadian Hidradenitis Suppurativa Foundations. part I: diagnosis, evaluation, and the use of complementary and procedural management. J Am Acad Dermatol. 2019;81:76-90.
- Alikhan A, Sayed C, Alavi A, et al. North American Clinical Management Guidelines for Hidradenitis Suppurativa: a publication from the United States and Canadian Hidradenitis Suppurativa Foundations. part II: topical, intralesional, and systemic medical management. J Am Acad Dermatol. 2019;81:91-101.
- Riis PT, Boer J, Prens EP, et al. Intralesional triamcinolone for flares of hidradenitis suppurativa (HS): a case series. J Am Acad Dermatol. 2016;75:1151-1155.
- Perng P, Zampella JG, Okoye GA. Management of hidradenitis suppurativa in pregnancy. J Am Acad Dermatol. 2017;76:979-989.
Resident Pearls
- Medical treatment of hidradenitis suppurativa (HS) can be relatively straightforward, but optimal comprehensive management is multifaceted.
- Educate patients about pathophysiology, counsel on smoking cessation, remember laser hair removal, consider an ongoing plan for addressing flares, and think about childbearing status when treating HS patients.

Kaposi Sarcoma in a Patient With Postpolio Syndrome
Kaposi sarcoma (KS) is a low-grade vascular tumor that is rare among the general US population, with an incidence rate of less than 1 per 100,000.1 The tumor is more common among certain groups of individuals due to geographic differences in the prevalence of KS-associated herpesvirus (also referred to as human herpesvirus 8) as well as host immune factors.2 Kaposi sarcoma often is defined by the patient's predisposing characteristics yielding the following distinct epidemiologic subtypes: (1) classic KS is a rare disease affecting older men of Mediterranean descent; (2) African KS is an endemic cancer with male predominance in sub-Saharan Africa; (3) AIDS-associated KS is an often aggressive AIDS-defining illness; and (4) iatrogenic KS occurs in patients on immunosuppressive therapy.3 When evaluating a patient without any of these risk factors, the clinical suspicion for KS may be low. We report a patient with postpolio syndrome (PPS) who presented with KS of the right leg, ankle, and foot.
A 77-year-old man with a distant history of paralytic poliomyelitis presented for an annual skin examination with concern for a new lesion on the right ankle. The patient had a history of PPS primarily affecting the right leg. Physical examination revealed residual weakness in an atrophic right lower extremity with a mottled appearance and mild pitting edema to the knee. Two red, dome-shaped, vascular papules were appreciated on the medial aspect of the right ankle (Figure 1), and a shave biopsy of the larger papule was performed. Microscopic examination of the biopsy specimen was consistent with KS (Figure 2). This patient had no history of human immunodeficiency virus or immunosuppressive therapy and was not of Mediterranean descent.
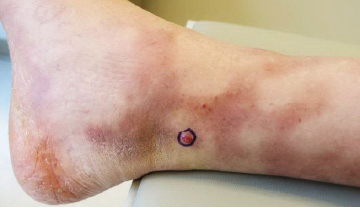
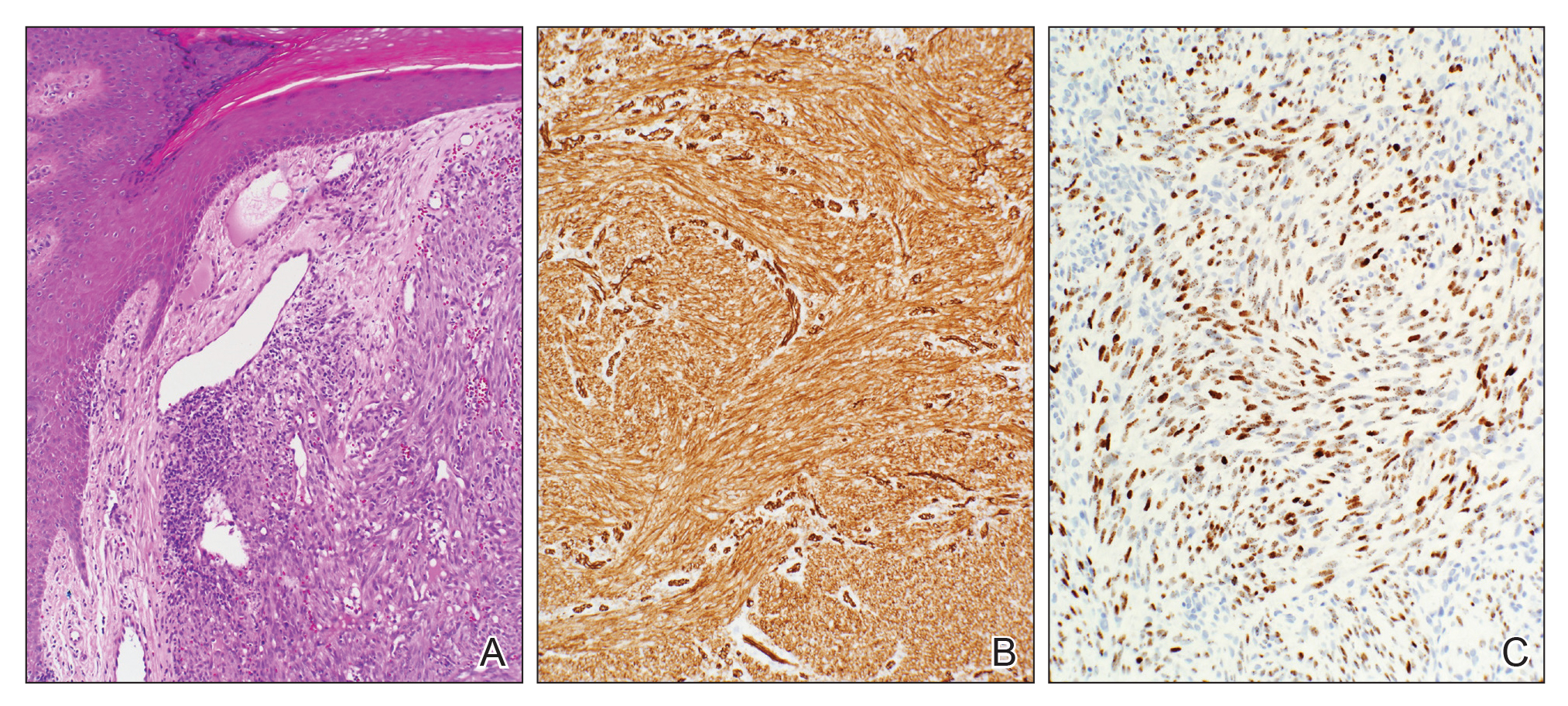
Because KS is a radiosensitive vascular neoplasm and radiation therapy (RT) alone can achieve local control,4 the patient was treated with 6 megaelectron-volt electron-beam RT. He received 30 Gy in 10 fractions to the affected area of the medial ankle. The patient tolerated RT well. Three weeks after completing treatment, he was found to have mild lichenification on the right medial ankle with no clinical evidence of disease. Four months later, he presented with multiple additional vascular papules on the right third toe and in the interdigital web space (Figure 3). Shave biopsy of one of these lesions was consistent with KS. Contrast computed tomography of the chest, abdomen, and pelvis was performed, revealing no evidence of metastatic disease. The patient was treated with 30 Gy in 15 fractions using opposed lateral 6 megaelectron-volt photon fields to the entire right lower extremity below the knee to treat all of the skin affected by the PPS. His posttreatment course was complicated by edema in the affected leg that resolved after daily pneumatic compression. He had no evidence of residual or recurrent disease 6 months after completing RT (Figure 4).
Cutaneous KS is a human herpesvirus 8-positive tumor of endothelial origin typically seen in older men of Mediterranean or African descent and among immunosuppressed patients.4 Our patient did not have any classic risk factors for KS, but his disease did arise in the setting of a right lower extremity that was notably affected by PPS. Postpolio syndrome is characterized by muscle atrophy due to denervation of the motor unit.5 Bruno et al6 found that such deficits in motor innervation could lead to impairments in venous outflow causing cutaneous venous congestion. Acroangiodermatitis clinically resembles KS but is a benign reactive vasoproliferative disorder and is well known to occur in the lower extremities as a sequela of chronic venous insufficiency.7 A case of bilateral lower extremity pseudo-KS was reported in a patient with notable PPS.8 A report of 2 patients describes KS arising in the setting of chronic venous insufficiency without any classic risk factors.9 Therefore, patients with PPS characterized by venous insufficiency may represent a population at increased risk for KS.
- Surveillance, Epidemiology, and End Results (SEER) Program. US Population Data--1969-2017. https://seer.cancer.gov/popdata/. Published January 2019. Accessed November 25, 2019.
- Uldrick TS, Whitby D. Update on KSHV epidemiology, kaposi sarcoma pathogenesis, and treatment of saposi sarcoma. Cancer Lett. 2011;305:150-162.
- Schwartz RA, Micali G, Nasca MR, et al. Kaposi sarcoma: a continuing conundrum. J Am Acad Dermatol. 2008;59:179-206.
- Arnold HL, Odom RB, James WD, et al. Andrews' Diseases of the Skin: Clinical Dermatology. Philadelphia, PA: Saunders; 1990.
- Boyer FV, Tiffreau V, Rapin A, et al. Post-polio syndrome: pathophysiological hypotheses, diagnosis criteria, drug therapy. Ann Phys Rehabil Med. 2010;53:34-41.
- Bruno RL, Johnson JC, Berman WS. Vasomotor abnormalities as post-polio sequelae: functional and clinical implications. Orthopedics. 1985;8:865-869.
- Palmer B, Xia Y, Cho S, Lewis FS. Acroangiodermatitis secondary to chronic venous insufficiency. Cutis. 2010;86:239-240.
- Rotbart G. Kaposi's disease and venous insufficiency. Phlebologie. 1978;31:439-443.
- Que SK, DeFelice T, Abdulla FR, et al. Non-HIV-related kaposi sarcoma in 2 Hispanic patients arising in the setting of chronic venous insufficiency. Cutis. 2015;95:E30-E33.
Kaposi sarcoma (KS) is a low-grade vascular tumor that is rare among the general US population, with an incidence rate of less than 1 per 100,000.1 The tumor is more common among certain groups of individuals due to geographic differences in the prevalence of KS-associated herpesvirus (also referred to as human herpesvirus 8) as well as host immune factors.2 Kaposi sarcoma often is defined by the patient's predisposing characteristics yielding the following distinct epidemiologic subtypes: (1) classic KS is a rare disease affecting older men of Mediterranean descent; (2) African KS is an endemic cancer with male predominance in sub-Saharan Africa; (3) AIDS-associated KS is an often aggressive AIDS-defining illness; and (4) iatrogenic KS occurs in patients on immunosuppressive therapy.3 When evaluating a patient without any of these risk factors, the clinical suspicion for KS may be low. We report a patient with postpolio syndrome (PPS) who presented with KS of the right leg, ankle, and foot.
A 77-year-old man with a distant history of paralytic poliomyelitis presented for an annual skin examination with concern for a new lesion on the right ankle. The patient had a history of PPS primarily affecting the right leg. Physical examination revealed residual weakness in an atrophic right lower extremity with a mottled appearance and mild pitting edema to the knee. Two red, dome-shaped, vascular papules were appreciated on the medial aspect of the right ankle (Figure 1), and a shave biopsy of the larger papule was performed. Microscopic examination of the biopsy specimen was consistent with KS (Figure 2). This patient had no history of human immunodeficiency virus or immunosuppressive therapy and was not of Mediterranean descent.
Because KS is a radiosensitive vascular neoplasm and radiation therapy (RT) alone can achieve local control,4 the patient was treated with 6 megaelectron-volt electron-beam RT. He received 30 Gy in 10 fractions to the affected area of the medial ankle. The patient tolerated RT well. Three weeks after completing treatment, he was found to have mild lichenification on the right medial ankle with no clinical evidence of disease. Four months later, he presented with multiple additional vascular papules on the right third toe and in the interdigital web space (Figure 3). Shave biopsy of one of these lesions was consistent with KS. Contrast computed tomography of the chest, abdomen, and pelvis was performed, revealing no evidence of metastatic disease. The patient was treated with 30 Gy in 15 fractions using opposed lateral 6 megaelectron-volt photon fields to the entire right lower extremity below the knee to treat all of the skin affected by the PPS. His posttreatment course was complicated by edema in the affected leg that resolved after daily pneumatic compression. He had no evidence of residual or recurrent disease 6 months after completing RT (Figure 4).
Cutaneous KS is a human herpesvirus 8-positive tumor of endothelial origin typically seen in older men of Mediterranean or African descent and among immunosuppressed patients.4 Our patient did not have any classic risk factors for KS, but his disease did arise in the setting of a right lower extremity that was notably affected by PPS. Postpolio syndrome is characterized by muscle atrophy due to denervation of the motor unit.5 Bruno et al6 found that such deficits in motor innervation could lead to impairments in venous outflow causing cutaneous venous congestion. Acroangiodermatitis clinically resembles KS but is a benign reactive vasoproliferative disorder and is well known to occur in the lower extremities as a sequela of chronic venous insufficiency.7 A case of bilateral lower extremity pseudo-KS was reported in a patient with notable PPS.8 A report of 2 patients describes KS arising in the setting of chronic venous insufficiency without any classic risk factors.9 Therefore, patients with PPS characterized by venous insufficiency may represent a population at increased risk for KS.
Kaposi sarcoma (KS) is a low-grade vascular tumor that is rare among the general US population, with an incidence rate of less than 1 per 100,000.1 The tumor is more common among certain groups of individuals due to geographic differences in the prevalence of KS-associated herpesvirus (also referred to as human herpesvirus 8) as well as host immune factors.2 Kaposi sarcoma often is defined by the patient's predisposing characteristics yielding the following distinct epidemiologic subtypes: (1) classic KS is a rare disease affecting older men of Mediterranean descent; (2) African KS is an endemic cancer with male predominance in sub-Saharan Africa; (3) AIDS-associated KS is an often aggressive AIDS-defining illness; and (4) iatrogenic KS occurs in patients on immunosuppressive therapy.3 When evaluating a patient without any of these risk factors, the clinical suspicion for KS may be low. We report a patient with postpolio syndrome (PPS) who presented with KS of the right leg, ankle, and foot.
A 77-year-old man with a distant history of paralytic poliomyelitis presented for an annual skin examination with concern for a new lesion on the right ankle. The patient had a history of PPS primarily affecting the right leg. Physical examination revealed residual weakness in an atrophic right lower extremity with a mottled appearance and mild pitting edema to the knee. Two red, dome-shaped, vascular papules were appreciated on the medial aspect of the right ankle (Figure 1), and a shave biopsy of the larger papule was performed. Microscopic examination of the biopsy specimen was consistent with KS (Figure 2). This patient had no history of human immunodeficiency virus or immunosuppressive therapy and was not of Mediterranean descent.
Because KS is a radiosensitive vascular neoplasm and radiation therapy (RT) alone can achieve local control,4 the patient was treated with 6 megaelectron-volt electron-beam RT. He received 30 Gy in 10 fractions to the affected area of the medial ankle. The patient tolerated RT well. Three weeks after completing treatment, he was found to have mild lichenification on the right medial ankle with no clinical evidence of disease. Four months later, he presented with multiple additional vascular papules on the right third toe and in the interdigital web space (Figure 3). Shave biopsy of one of these lesions was consistent with KS. Contrast computed tomography of the chest, abdomen, and pelvis was performed, revealing no evidence of metastatic disease. The patient was treated with 30 Gy in 15 fractions using opposed lateral 6 megaelectron-volt photon fields to the entire right lower extremity below the knee to treat all of the skin affected by the PPS. His posttreatment course was complicated by edema in the affected leg that resolved after daily pneumatic compression. He had no evidence of residual or recurrent disease 6 months after completing RT (Figure 4).
Cutaneous KS is a human herpesvirus 8-positive tumor of endothelial origin typically seen in older men of Mediterranean or African descent and among immunosuppressed patients.4 Our patient did not have any classic risk factors for KS, but his disease did arise in the setting of a right lower extremity that was notably affected by PPS. Postpolio syndrome is characterized by muscle atrophy due to denervation of the motor unit.5 Bruno et al6 found that such deficits in motor innervation could lead to impairments in venous outflow causing cutaneous venous congestion. Acroangiodermatitis clinically resembles KS but is a benign reactive vasoproliferative disorder and is well known to occur in the lower extremities as a sequela of chronic venous insufficiency.7 A case of bilateral lower extremity pseudo-KS was reported in a patient with notable PPS.8 A report of 2 patients describes KS arising in the setting of chronic venous insufficiency without any classic risk factors.9 Therefore, patients with PPS characterized by venous insufficiency may represent a population at increased risk for KS.
- Surveillance, Epidemiology, and End Results (SEER) Program. US Population Data--1969-2017. https://seer.cancer.gov/popdata/. Published January 2019. Accessed November 25, 2019.
- Uldrick TS, Whitby D. Update on KSHV epidemiology, kaposi sarcoma pathogenesis, and treatment of saposi sarcoma. Cancer Lett. 2011;305:150-162.
- Schwartz RA, Micali G, Nasca MR, et al. Kaposi sarcoma: a continuing conundrum. J Am Acad Dermatol. 2008;59:179-206.
- Arnold HL, Odom RB, James WD, et al. Andrews' Diseases of the Skin: Clinical Dermatology. Philadelphia, PA: Saunders; 1990.
- Boyer FV, Tiffreau V, Rapin A, et al. Post-polio syndrome: pathophysiological hypotheses, diagnosis criteria, drug therapy. Ann Phys Rehabil Med. 2010;53:34-41.
- Bruno RL, Johnson JC, Berman WS. Vasomotor abnormalities as post-polio sequelae: functional and clinical implications. Orthopedics. 1985;8:865-869.
- Palmer B, Xia Y, Cho S, Lewis FS. Acroangiodermatitis secondary to chronic venous insufficiency. Cutis. 2010;86:239-240.
- Rotbart G. Kaposi's disease and venous insufficiency. Phlebologie. 1978;31:439-443.
- Que SK, DeFelice T, Abdulla FR, et al. Non-HIV-related kaposi sarcoma in 2 Hispanic patients arising in the setting of chronic venous insufficiency. Cutis. 2015;95:E30-E33.
- Surveillance, Epidemiology, and End Results (SEER) Program. US Population Data--1969-2017. https://seer.cancer.gov/popdata/. Published January 2019. Accessed November 25, 2019.
- Uldrick TS, Whitby D. Update on KSHV epidemiology, kaposi sarcoma pathogenesis, and treatment of saposi sarcoma. Cancer Lett. 2011;305:150-162.
- Schwartz RA, Micali G, Nasca MR, et al. Kaposi sarcoma: a continuing conundrum. J Am Acad Dermatol. 2008;59:179-206.
- Arnold HL, Odom RB, James WD, et al. Andrews' Diseases of the Skin: Clinical Dermatology. Philadelphia, PA: Saunders; 1990.
- Boyer FV, Tiffreau V, Rapin A, et al. Post-polio syndrome: pathophysiological hypotheses, diagnosis criteria, drug therapy. Ann Phys Rehabil Med. 2010;53:34-41.
- Bruno RL, Johnson JC, Berman WS. Vasomotor abnormalities as post-polio sequelae: functional and clinical implications. Orthopedics. 1985;8:865-869.
- Palmer B, Xia Y, Cho S, Lewis FS. Acroangiodermatitis secondary to chronic venous insufficiency. Cutis. 2010;86:239-240.
- Rotbart G. Kaposi's disease and venous insufficiency. Phlebologie. 1978;31:439-443.
- Que SK, DeFelice T, Abdulla FR, et al. Non-HIV-related kaposi sarcoma in 2 Hispanic patients arising in the setting of chronic venous insufficiency. Cutis. 2015;95:E30-E33.
Practice Points
- Cutaneous Kaposi sarcoma (KS) is a human herpesvirus 8–positive tumor of endothelial origin typically seen in older men of Mediterranean or African descent and among immunosuppressed patients.
- In addition, patients with postpolio syndrome characterized by venous insufficiency may represent a population at increased risk for KS.
- Kaposi sarcoma is a radiosensitive vascular neoplasm, and radiation therapy can achieve local control.
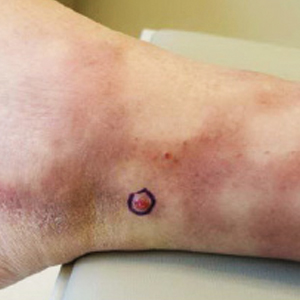
Multiple Facial Papules
The Diagnosis: Birt-Hogg-Dubé Syndrome
Histopathologic examination revealed a collection of bland spindle cells with perifollicular fibrosis consistent with a fibrofolliculoma, confirming the diagnosis of Birt-Hogg-Dubé syndrome (Figure). Cosmetic treatment with ablative therapy was offered, but the patient declined.
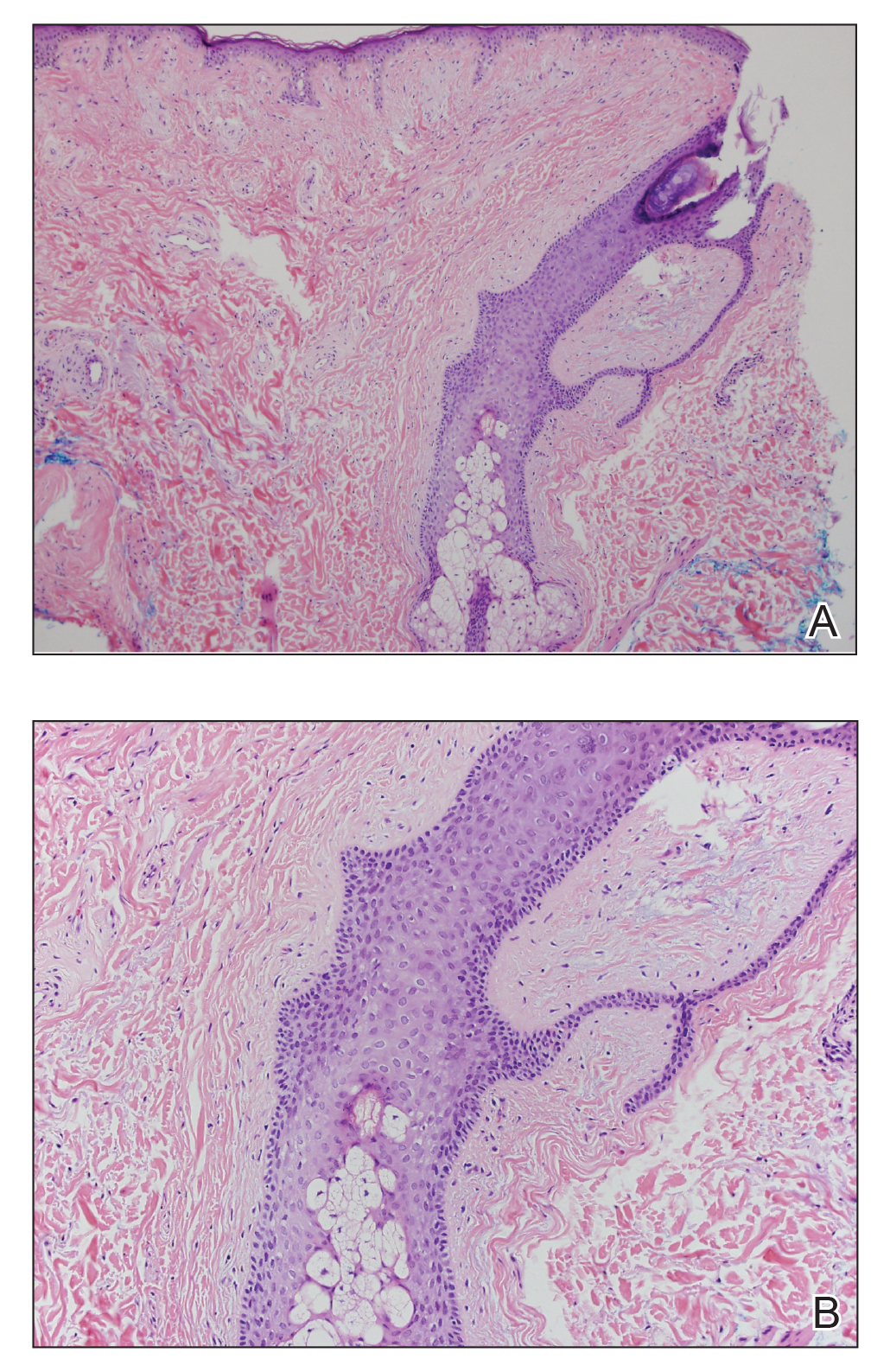
Birt-Hogg-Dubé syndrome is an autosomal-dominant genodermatosis caused by a loss-of-function mutation in the folliculin gene, FLCN, on chromosome arm 17p11.2.1 Cutaneous findings include benign follicular hamartomas, such as fibrofolliculomas and trichodiscomas. Angiofibromas, perifollicular fibromas, oral papillomas, and acrochordons also can be present.1 Cutaneous lesions usually appear on the head and neck in the third decade of life.
Patients with Birt-Hogg-Dubé syndrome are at an increased risk for pneumothorax and renal cancer, specifically hybrid oncocytic-chromophobe renal cell carcinomas.2 In a study of 89 patients with a FLCN mutation, 90% (80/89) of patients had cutaneous lesions, 84% (34/89) had pulmonary cysts, and 34% (30/89) had kidney tumors. Affected individuals were at a higher risk for pneumothorax and kidney tumors if there was a family history of these tumors.2
Proposed diagnostic criteria include any 1 of the following: 2 or more skin lesions clinically consistent with fibrofolliculomas and 1 histologically confirmed fibrofolliculoma; multiple bilateral pulmonary cysts in the basilar lung with or without pneumothorax before 40 years of age; bilateral multifocal chromophobe renal carcinomas or hybrid oncocytic tumors; combination of cutaneous, pulmonary, or renal manifestation in the patient and family; or a FLCN mutation.3
Current recommendations for the workup of a patient with Birt-Hogg-Dubé syndrome include referral to genetic counseling for the patient and family, a baseline computed tomography of the chest to evaluate for pulmonary cysts, and gadolinium-enhanced abdominal magnetic resonance imaging starting at 20 years of age and repeated every 3 to 4 years to screen for renal tumors.1 Pulmonary function tests can be considered if the patient is symptomatic or has a high cyst burden. Patients should be advised against smoking and scuba diving.
The differential diagnosis of multiple facial papules includes Cowden syndrome, tuberous sclerosis, Brooke-Spiegler syndrome, and Muir-Torre syndrome. Cowden syndrome is caused by a mutation in the protein tyrosine phosphatase gene, PTEN.4 The characteristic cutaneous findings on the face are trichilemmomas, which appear as flesh-colored papules that may have a verrucous surface.
Tuberous sclerosis is caused by mutations in hamartin (TSC1) or tuberin (TSC2). Angiofibromas are most commonly found on the face and appear as flesh-colored to red-brown papules. Fibrous plaques, periungual fibromas, gingival fibromas, hypopigmented macules, and connective tissue nevi also are found in tuberous sclerosis.5
Brooke-Spiegler syndrome is caused by a mutation in the CYLD lysine 63 deubiquitinase gene, CYLD. Trichoepitheliomas, cylindromas, and spiradenomas are caused by the CYLD mutation and appear on the head and neck. Trichoepitheliomas are flesh-colored to pink papules found on the face, often concentrated in the nasolabial folds.6 Cylindromas and spiradenomas are flesh-colored to pink papules or nodules most commonly found on the scalp.6
Muir-Torre syndrome is caused by a mutation in DNA mismatch repair genes MSH2 and/or MLH1.7 Sebaceous neoplasms, including sebaceous adenomas, sebaceomas, and less frequently sebaceous carcinomas, are characteristic cutaneous findings and appear as pink to yellow papules commonly found on the head and neck.
Careful history taking, physical examination, and histopathologic analysis are important in recognizing the features of Birt-Hogg-Dubé syndrome. Accurate and timely diagnosis is essential for the appropriate care of patients and their families, given the syndrome's systemic implications.
- Gupta N, Sunwoo BY, Kotloff RM. Birt-Hogg-Dubé syndrome. Clin Chest Med. 2016;37:475-486.
- Toro JR, Wei MH, Glenn GM, et al. BHD mutations, clinical and molecular genetic investigations of Birt-Hogg-Dubé syndrome: a new series of 50 families and a review of published reports. J Med Genet. 2008;45:321-331.
- Schmidt LS, Linehan WM. Molecular genetics and clinical features of Birt-Hogg-Dubé syndrome. Nat Rev Urol. 2015;12:558-569.
- Marsh D, Kum JB, Lunetta KL, et al. PTEN mutation spectrum and genotype-phenotype correlations in Bannayan-Riley-Ruvalcaba syndrome suggest a single entity with Cowden syndrome. Hum Mol Genet. 1999;8:1461-1472.
- Wataya-Kaneda M, Uemura M, Fujita K, et al. Tuberous sclerosis complex: recent advances in manifestations and therapy. Int J Urol. 2017;24:681-691.
- Kazakov DV. Brooke-Spiegler syndrome and phenotypic variants: an update. Head Neck Pathol. 2016;10:125-130.
- Mahalingam M. MSH6, Past and present and Muir-Torre syndrome--connecting the dots. Am J Dermatopathol. 2017;39:239-249.
The Diagnosis: Birt-Hogg-Dubé Syndrome
Histopathologic examination revealed a collection of bland spindle cells with perifollicular fibrosis consistent with a fibrofolliculoma, confirming the diagnosis of Birt-Hogg-Dubé syndrome (Figure). Cosmetic treatment with ablative therapy was offered, but the patient declined.
Birt-Hogg-Dubé syndrome is an autosomal-dominant genodermatosis caused by a loss-of-function mutation in the folliculin gene, FLCN, on chromosome arm 17p11.2.1 Cutaneous findings include benign follicular hamartomas, such as fibrofolliculomas and trichodiscomas. Angiofibromas, perifollicular fibromas, oral papillomas, and acrochordons also can be present.1 Cutaneous lesions usually appear on the head and neck in the third decade of life.
Patients with Birt-Hogg-Dubé syndrome are at an increased risk for pneumothorax and renal cancer, specifically hybrid oncocytic-chromophobe renal cell carcinomas.2 In a study of 89 patients with a FLCN mutation, 90% (80/89) of patients had cutaneous lesions, 84% (34/89) had pulmonary cysts, and 34% (30/89) had kidney tumors. Affected individuals were at a higher risk for pneumothorax and kidney tumors if there was a family history of these tumors.2
Proposed diagnostic criteria include any 1 of the following: 2 or more skin lesions clinically consistent with fibrofolliculomas and 1 histologically confirmed fibrofolliculoma; multiple bilateral pulmonary cysts in the basilar lung with or without pneumothorax before 40 years of age; bilateral multifocal chromophobe renal carcinomas or hybrid oncocytic tumors; combination of cutaneous, pulmonary, or renal manifestation in the patient and family; or a FLCN mutation.3
Current recommendations for the workup of a patient with Birt-Hogg-Dubé syndrome include referral to genetic counseling for the patient and family, a baseline computed tomography of the chest to evaluate for pulmonary cysts, and gadolinium-enhanced abdominal magnetic resonance imaging starting at 20 years of age and repeated every 3 to 4 years to screen for renal tumors.1 Pulmonary function tests can be considered if the patient is symptomatic or has a high cyst burden. Patients should be advised against smoking and scuba diving.
The differential diagnosis of multiple facial papules includes Cowden syndrome, tuberous sclerosis, Brooke-Spiegler syndrome, and Muir-Torre syndrome. Cowden syndrome is caused by a mutation in the protein tyrosine phosphatase gene, PTEN.4 The characteristic cutaneous findings on the face are trichilemmomas, which appear as flesh-colored papules that may have a verrucous surface.
Tuberous sclerosis is caused by mutations in hamartin (TSC1) or tuberin (TSC2). Angiofibromas are most commonly found on the face and appear as flesh-colored to red-brown papules. Fibrous plaques, periungual fibromas, gingival fibromas, hypopigmented macules, and connective tissue nevi also are found in tuberous sclerosis.5
Brooke-Spiegler syndrome is caused by a mutation in the CYLD lysine 63 deubiquitinase gene, CYLD. Trichoepitheliomas, cylindromas, and spiradenomas are caused by the CYLD mutation and appear on the head and neck. Trichoepitheliomas are flesh-colored to pink papules found on the face, often concentrated in the nasolabial folds.6 Cylindromas and spiradenomas are flesh-colored to pink papules or nodules most commonly found on the scalp.6
Muir-Torre syndrome is caused by a mutation in DNA mismatch repair genes MSH2 and/or MLH1.7 Sebaceous neoplasms, including sebaceous adenomas, sebaceomas, and less frequently sebaceous carcinomas, are characteristic cutaneous findings and appear as pink to yellow papules commonly found on the head and neck.
Careful history taking, physical examination, and histopathologic analysis are important in recognizing the features of Birt-Hogg-Dubé syndrome. Accurate and timely diagnosis is essential for the appropriate care of patients and their families, given the syndrome's systemic implications.
The Diagnosis: Birt-Hogg-Dubé Syndrome
Histopathologic examination revealed a collection of bland spindle cells with perifollicular fibrosis consistent with a fibrofolliculoma, confirming the diagnosis of Birt-Hogg-Dubé syndrome (Figure). Cosmetic treatment with ablative therapy was offered, but the patient declined.
Birt-Hogg-Dubé syndrome is an autosomal-dominant genodermatosis caused by a loss-of-function mutation in the folliculin gene, FLCN, on chromosome arm 17p11.2.1 Cutaneous findings include benign follicular hamartomas, such as fibrofolliculomas and trichodiscomas. Angiofibromas, perifollicular fibromas, oral papillomas, and acrochordons also can be present.1 Cutaneous lesions usually appear on the head and neck in the third decade of life.
Patients with Birt-Hogg-Dubé syndrome are at an increased risk for pneumothorax and renal cancer, specifically hybrid oncocytic-chromophobe renal cell carcinomas.2 In a study of 89 patients with a FLCN mutation, 90% (80/89) of patients had cutaneous lesions, 84% (34/89) had pulmonary cysts, and 34% (30/89) had kidney tumors. Affected individuals were at a higher risk for pneumothorax and kidney tumors if there was a family history of these tumors.2
Proposed diagnostic criteria include any 1 of the following: 2 or more skin lesions clinically consistent with fibrofolliculomas and 1 histologically confirmed fibrofolliculoma; multiple bilateral pulmonary cysts in the basilar lung with or without pneumothorax before 40 years of age; bilateral multifocal chromophobe renal carcinomas or hybrid oncocytic tumors; combination of cutaneous, pulmonary, or renal manifestation in the patient and family; or a FLCN mutation.3
Current recommendations for the workup of a patient with Birt-Hogg-Dubé syndrome include referral to genetic counseling for the patient and family, a baseline computed tomography of the chest to evaluate for pulmonary cysts, and gadolinium-enhanced abdominal magnetic resonance imaging starting at 20 years of age and repeated every 3 to 4 years to screen for renal tumors.1 Pulmonary function tests can be considered if the patient is symptomatic or has a high cyst burden. Patients should be advised against smoking and scuba diving.
The differential diagnosis of multiple facial papules includes Cowden syndrome, tuberous sclerosis, Brooke-Spiegler syndrome, and Muir-Torre syndrome. Cowden syndrome is caused by a mutation in the protein tyrosine phosphatase gene, PTEN.4 The characteristic cutaneous findings on the face are trichilemmomas, which appear as flesh-colored papules that may have a verrucous surface.
Tuberous sclerosis is caused by mutations in hamartin (TSC1) or tuberin (TSC2). Angiofibromas are most commonly found on the face and appear as flesh-colored to red-brown papules. Fibrous plaques, periungual fibromas, gingival fibromas, hypopigmented macules, and connective tissue nevi also are found in tuberous sclerosis.5
Brooke-Spiegler syndrome is caused by a mutation in the CYLD lysine 63 deubiquitinase gene, CYLD. Trichoepitheliomas, cylindromas, and spiradenomas are caused by the CYLD mutation and appear on the head and neck. Trichoepitheliomas are flesh-colored to pink papules found on the face, often concentrated in the nasolabial folds.6 Cylindromas and spiradenomas are flesh-colored to pink papules or nodules most commonly found on the scalp.6
Muir-Torre syndrome is caused by a mutation in DNA mismatch repair genes MSH2 and/or MLH1.7 Sebaceous neoplasms, including sebaceous adenomas, sebaceomas, and less frequently sebaceous carcinomas, are characteristic cutaneous findings and appear as pink to yellow papules commonly found on the head and neck.
Careful history taking, physical examination, and histopathologic analysis are important in recognizing the features of Birt-Hogg-Dubé syndrome. Accurate and timely diagnosis is essential for the appropriate care of patients and their families, given the syndrome's systemic implications.
- Gupta N, Sunwoo BY, Kotloff RM. Birt-Hogg-Dubé syndrome. Clin Chest Med. 2016;37:475-486.
- Toro JR, Wei MH, Glenn GM, et al. BHD mutations, clinical and molecular genetic investigations of Birt-Hogg-Dubé syndrome: a new series of 50 families and a review of published reports. J Med Genet. 2008;45:321-331.
- Schmidt LS, Linehan WM. Molecular genetics and clinical features of Birt-Hogg-Dubé syndrome. Nat Rev Urol. 2015;12:558-569.
- Marsh D, Kum JB, Lunetta KL, et al. PTEN mutation spectrum and genotype-phenotype correlations in Bannayan-Riley-Ruvalcaba syndrome suggest a single entity with Cowden syndrome. Hum Mol Genet. 1999;8:1461-1472.
- Wataya-Kaneda M, Uemura M, Fujita K, et al. Tuberous sclerosis complex: recent advances in manifestations and therapy. Int J Urol. 2017;24:681-691.
- Kazakov DV. Brooke-Spiegler syndrome and phenotypic variants: an update. Head Neck Pathol. 2016;10:125-130.
- Mahalingam M. MSH6, Past and present and Muir-Torre syndrome--connecting the dots. Am J Dermatopathol. 2017;39:239-249.
- Gupta N, Sunwoo BY, Kotloff RM. Birt-Hogg-Dubé syndrome. Clin Chest Med. 2016;37:475-486.
- Toro JR, Wei MH, Glenn GM, et al. BHD mutations, clinical and molecular genetic investigations of Birt-Hogg-Dubé syndrome: a new series of 50 families and a review of published reports. J Med Genet. 2008;45:321-331.
- Schmidt LS, Linehan WM. Molecular genetics and clinical features of Birt-Hogg-Dubé syndrome. Nat Rev Urol. 2015;12:558-569.
- Marsh D, Kum JB, Lunetta KL, et al. PTEN mutation spectrum and genotype-phenotype correlations in Bannayan-Riley-Ruvalcaba syndrome suggest a single entity with Cowden syndrome. Hum Mol Genet. 1999;8:1461-1472.
- Wataya-Kaneda M, Uemura M, Fujita K, et al. Tuberous sclerosis complex: recent advances in manifestations and therapy. Int J Urol. 2017;24:681-691.
- Kazakov DV. Brooke-Spiegler syndrome and phenotypic variants: an update. Head Neck Pathol. 2016;10:125-130.
- Mahalingam M. MSH6, Past and present and Muir-Torre syndrome--connecting the dots. Am J Dermatopathol. 2017;39:239-249.

A 50-year-old man presented with facial papules on the cheeks that had appeared approximately 1.5 years prior and gradually spread over the face and neck. They were occasionally pruritic but otherwise were asymptomatic. His mother and brother reportedly had similar clinical findings. Family history was notable for a maternal uncle who had died in his 30s of an unknown type of renal cancer. Physical examination revealed innumerable white-gray papules that measured 1 to 5 mm and were scattered across the face and neck. Punch biopsies were obtained. Computed tomography of the chest showed multiple bibasilar pulmonary cysts. Magnetic resonance imaging was negative for renal tumors.
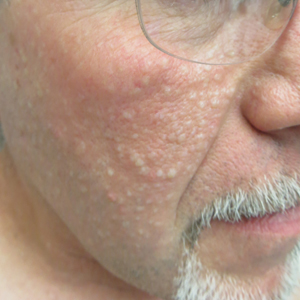
New Plaques Arising at Site of Previously Excised Basal Cell Carcinoma
The Diagnosis: Actinic Comedonal Plaque
Histopathologic examination showed multiple small, keratin-filled cystic spaces in the superficial dermis lined by stratified squamous epithelium that keratinized with a granular layer with surrounding solar elastosis (Figure 1). These findings were consistent with an actinic comedonal plaque (ACP), a rare variant of Favre-Racouchot syndrome (FRS). Due to other cases of invasive squamous cell carcinomas developing within these lesions1 and the patient's history of basal cell carcinoma in the area, it was important to rule out malignancy and further confirm the diagnosis. Thus, an additional biopsy was obtained, which revealed no sign of cellular atypia. The patient was bothered by the appearance and texture of the lesion, so he elected to pursue treatment. Because of the lesion's moderate size and location, surgical excision was not recommended. He was treated with cryosurgery followed by tretinoin gel 0.025% for 3 months (Figure 2). At 1-year follow-up, there was no recurrence and an acceptable cosmetic outcome.
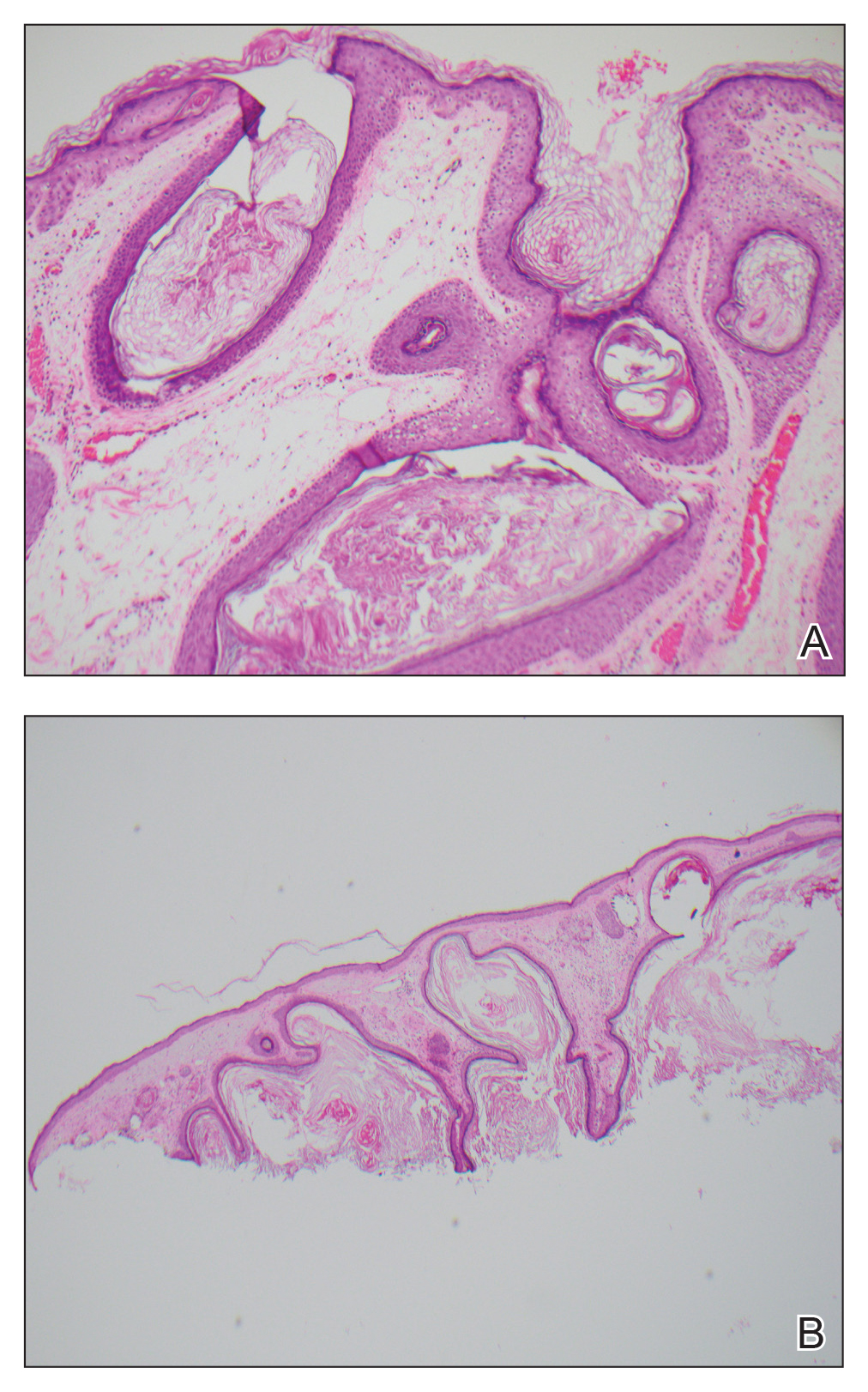
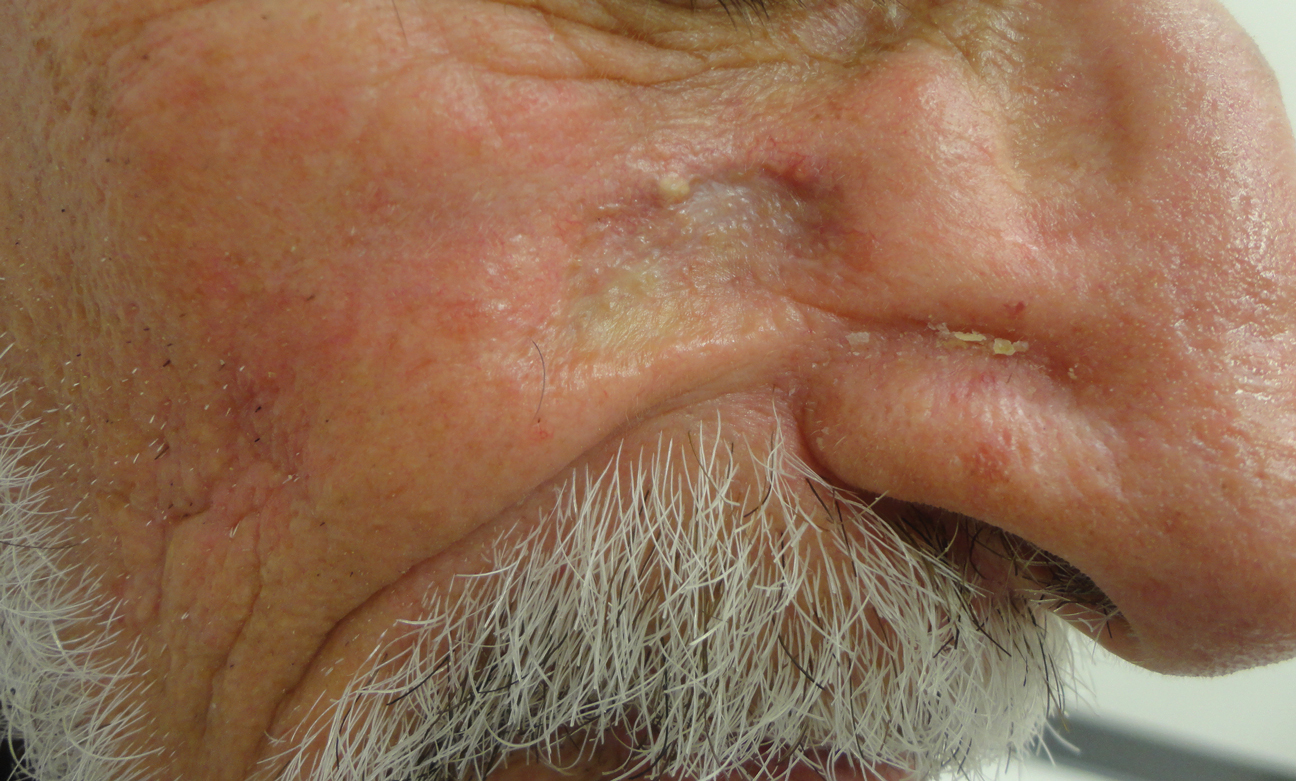
Actinic comedonal plaques are characterized by cysts, comedones, and papules that form well-defined yellow plaques on the skin after chronic sun exposure.2 First described in 1980 by Eastern and Martin,3 these lesions are most often found on the neck, thorax, nasal dorsum, helix, and forearms; they often are mistaken for basal cell carcinomas, chondrodermatitis nodularis chronica helicis, and amyloidosis.2,4
Similar to the cases described by Eastern and Martin,3 our patient presented with a solitary 2.8×1.6-cm yellowish and bumpy plaque that was growing on chronically sun-exposed skin of the medial right cheek. The patient's history of smoking and basal cell carcinoma removal led to the following differential diagnoses: recurrent malignancy, tumors of dermal appendages, and xanthelasmalike deposition within a scar, among others.
Histologic examination demonstrated keratin-filled cystic spaces in the superficial dermis, along with hyperkeratosis and solar elastosis. These findings were consistent with the original histologic descriptions of ACP that were cited as a rare ectopic form of FRS.5 Leeuwis-Fedorovich et al1 described multifocal squamous cell carcinomas arising in a FRS lesion in 2 cases. Additional biopsy was performed in our patient to rule out this possibility.
Aside from its role in dermatologic malignancy, exposure to UV light can lead to dilation of the sebaceous gland infundibulum and formation of comedones in various locations.2 Histologically, lesions of ACP feature dilated, keratin-filled follicles within a matrix of amorphous damaged collagen in the middle and lower dermis, along with elastosis and basophilic degeneration with fragmentation of collagen bundles in the upper dermis coated with flattened epidermis.2,3
Several clinical diagnoses were considered in the differential for our patient. In contrast to our patient's solitary lesion, classic FRS--nodular elastosis with cysts and comedones--is characterized by a diffuse yellowish hue of large open black comedones that are symmetrically distributed on the temporal and periorbital areas.6
Xanthoma has several clinical presentations. Plane xanthomas typically develop in skin folds, especially in palmar creases, while xanthelasma typically consists of yellow soft plaques on the eyelids and periorbital skin. Histologically, xanthomas contain lipid-laden macrophages, which were absent in our patient.7
Cutaneous amyloidosis can be characterized as macular, papular, and nodular. Nodular, the rarest subtype, commonly manifests on the face.8 However, its characteristic histologic features include atrophic epidermal changes with an amorphous, eosinophilic-appearing dermis due to amyloid deposition. These findings were absent in our case.
Recurrent neoplasm would be in the differential diagnosis of any solitary nodule arising within a previously treated site of malignancy, which was excluded by histologic examination of our patient.7
Topical tretinoin has been demonstrated as effective treatment of both FRS and ACP.2,9 Other effective treatment modalities include cryotherapy and photoprotection.9 For our patient, a combination of cryosurgery and topical tretinoin resulted in depression of the plaque and a good cosmetic outcome.
Acknowledgments
We thank the patient for granting permission to publish this article. We also are indebted to Morgan L. Wilson, MD (Springfield, Illinois), for his expert dermatopathologic evaluation in this case. He has received no compensation for his contribution.
- Leeuwis-Fedorovich NE, Starink M, van der Wal AC. Multifocal squamous cell carcinoma arising in a Favre-Racouchot lesion—report of two cases and review of the literature. J Dermatol Case Rep. 2015;9:103-106.
- Cardoso F, Nakandakari S, Zattar GA, et al. Actinic comedonal plaquevariant of Favre-Racouchot syndrome: report of two cases. An Bras Dermatol. 2015;90(suppl 1):185-187.
- Eastern JS, Martin S. Actinic comedonal plaque. J Am Acad Dermatol. 1980;3:633-636.
- Hauptman G, Kopf A, Rabinovitz HS, et al. The actinic comedonal plaque. Cutis. 1997;60:145-146.
- John SM, Hamm H. Actinic comedonal plaque—a rare ectopic form of the Favre-Racouchot syndrome. Clin Exp Dermatol. 1993;18:256-258.
- Sonthalia S, Arora R, Chhabra N, et al. Favre-Racouchot syndrome. Indian Dermatol Online J. 2014;5(suppl 2):S128-S129.
- Elder DE, Elenitsas R, Murphy GF, et al. Benign pigmented lesions and malignant melanoma. In: Elder DE, Elenitsas R, Johnson BL Jr, et al, eds. Lever’s Histopathology of the Skin. 10th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2009:699-790.
- Lee DY, Kim YJ, Lee JY, et al. Primary localized cutaneous nodular amyloidosis following local trauma. Ann Dermatol. 2011;23:515-518.
- Patterson WM, Fox MD, Schwartz RA. Favre-Racouchot disease. Int J Dermatol. 2004;43:167-169.
The Diagnosis: Actinic Comedonal Plaque
Histopathologic examination showed multiple small, keratin-filled cystic spaces in the superficial dermis lined by stratified squamous epithelium that keratinized with a granular layer with surrounding solar elastosis (Figure 1). These findings were consistent with an actinic comedonal plaque (ACP), a rare variant of Favre-Racouchot syndrome (FRS). Due to other cases of invasive squamous cell carcinomas developing within these lesions1 and the patient's history of basal cell carcinoma in the area, it was important to rule out malignancy and further confirm the diagnosis. Thus, an additional biopsy was obtained, which revealed no sign of cellular atypia. The patient was bothered by the appearance and texture of the lesion, so he elected to pursue treatment. Because of the lesion's moderate size and location, surgical excision was not recommended. He was treated with cryosurgery followed by tretinoin gel 0.025% for 3 months (Figure 2). At 1-year follow-up, there was no recurrence and an acceptable cosmetic outcome.
Actinic comedonal plaques are characterized by cysts, comedones, and papules that form well-defined yellow plaques on the skin after chronic sun exposure.2 First described in 1980 by Eastern and Martin,3 these lesions are most often found on the neck, thorax, nasal dorsum, helix, and forearms; they often are mistaken for basal cell carcinomas, chondrodermatitis nodularis chronica helicis, and amyloidosis.2,4
Similar to the cases described by Eastern and Martin,3 our patient presented with a solitary 2.8×1.6-cm yellowish and bumpy plaque that was growing on chronically sun-exposed skin of the medial right cheek. The patient's history of smoking and basal cell carcinoma removal led to the following differential diagnoses: recurrent malignancy, tumors of dermal appendages, and xanthelasmalike deposition within a scar, among others.
Histologic examination demonstrated keratin-filled cystic spaces in the superficial dermis, along with hyperkeratosis and solar elastosis. These findings were consistent with the original histologic descriptions of ACP that were cited as a rare ectopic form of FRS.5 Leeuwis-Fedorovich et al1 described multifocal squamous cell carcinomas arising in a FRS lesion in 2 cases. Additional biopsy was performed in our patient to rule out this possibility.
Aside from its role in dermatologic malignancy, exposure to UV light can lead to dilation of the sebaceous gland infundibulum and formation of comedones in various locations.2 Histologically, lesions of ACP feature dilated, keratin-filled follicles within a matrix of amorphous damaged collagen in the middle and lower dermis, along with elastosis and basophilic degeneration with fragmentation of collagen bundles in the upper dermis coated with flattened epidermis.2,3
Several clinical diagnoses were considered in the differential for our patient. In contrast to our patient's solitary lesion, classic FRS--nodular elastosis with cysts and comedones--is characterized by a diffuse yellowish hue of large open black comedones that are symmetrically distributed on the temporal and periorbital areas.6
Xanthoma has several clinical presentations. Plane xanthomas typically develop in skin folds, especially in palmar creases, while xanthelasma typically consists of yellow soft plaques on the eyelids and periorbital skin. Histologically, xanthomas contain lipid-laden macrophages, which were absent in our patient.7
Cutaneous amyloidosis can be characterized as macular, papular, and nodular. Nodular, the rarest subtype, commonly manifests on the face.8 However, its characteristic histologic features include atrophic epidermal changes with an amorphous, eosinophilic-appearing dermis due to amyloid deposition. These findings were absent in our case.
Recurrent neoplasm would be in the differential diagnosis of any solitary nodule arising within a previously treated site of malignancy, which was excluded by histologic examination of our patient.7
Topical tretinoin has been demonstrated as effective treatment of both FRS and ACP.2,9 Other effective treatment modalities include cryotherapy and photoprotection.9 For our patient, a combination of cryosurgery and topical tretinoin resulted in depression of the plaque and a good cosmetic outcome.
Acknowledgments
We thank the patient for granting permission to publish this article. We also are indebted to Morgan L. Wilson, MD (Springfield, Illinois), for his expert dermatopathologic evaluation in this case. He has received no compensation for his contribution.
The Diagnosis: Actinic Comedonal Plaque
Histopathologic examination showed multiple small, keratin-filled cystic spaces in the superficial dermis lined by stratified squamous epithelium that keratinized with a granular layer with surrounding solar elastosis (Figure 1). These findings were consistent with an actinic comedonal plaque (ACP), a rare variant of Favre-Racouchot syndrome (FRS). Due to other cases of invasive squamous cell carcinomas developing within these lesions1 and the patient's history of basal cell carcinoma in the area, it was important to rule out malignancy and further confirm the diagnosis. Thus, an additional biopsy was obtained, which revealed no sign of cellular atypia. The patient was bothered by the appearance and texture of the lesion, so he elected to pursue treatment. Because of the lesion's moderate size and location, surgical excision was not recommended. He was treated with cryosurgery followed by tretinoin gel 0.025% for 3 months (Figure 2). At 1-year follow-up, there was no recurrence and an acceptable cosmetic outcome.
Actinic comedonal plaques are characterized by cysts, comedones, and papules that form well-defined yellow plaques on the skin after chronic sun exposure.2 First described in 1980 by Eastern and Martin,3 these lesions are most often found on the neck, thorax, nasal dorsum, helix, and forearms; they often are mistaken for basal cell carcinomas, chondrodermatitis nodularis chronica helicis, and amyloidosis.2,4
Similar to the cases described by Eastern and Martin,3 our patient presented with a solitary 2.8×1.6-cm yellowish and bumpy plaque that was growing on chronically sun-exposed skin of the medial right cheek. The patient's history of smoking and basal cell carcinoma removal led to the following differential diagnoses: recurrent malignancy, tumors of dermal appendages, and xanthelasmalike deposition within a scar, among others.
Histologic examination demonstrated keratin-filled cystic spaces in the superficial dermis, along with hyperkeratosis and solar elastosis. These findings were consistent with the original histologic descriptions of ACP that were cited as a rare ectopic form of FRS.5 Leeuwis-Fedorovich et al1 described multifocal squamous cell carcinomas arising in a FRS lesion in 2 cases. Additional biopsy was performed in our patient to rule out this possibility.
Aside from its role in dermatologic malignancy, exposure to UV light can lead to dilation of the sebaceous gland infundibulum and formation of comedones in various locations.2 Histologically, lesions of ACP feature dilated, keratin-filled follicles within a matrix of amorphous damaged collagen in the middle and lower dermis, along with elastosis and basophilic degeneration with fragmentation of collagen bundles in the upper dermis coated with flattened epidermis.2,3
Several clinical diagnoses were considered in the differential for our patient. In contrast to our patient's solitary lesion, classic FRS--nodular elastosis with cysts and comedones--is characterized by a diffuse yellowish hue of large open black comedones that are symmetrically distributed on the temporal and periorbital areas.6
Xanthoma has several clinical presentations. Plane xanthomas typically develop in skin folds, especially in palmar creases, while xanthelasma typically consists of yellow soft plaques on the eyelids and periorbital skin. Histologically, xanthomas contain lipid-laden macrophages, which were absent in our patient.7
Cutaneous amyloidosis can be characterized as macular, papular, and nodular. Nodular, the rarest subtype, commonly manifests on the face.8 However, its characteristic histologic features include atrophic epidermal changes with an amorphous, eosinophilic-appearing dermis due to amyloid deposition. These findings were absent in our case.
Recurrent neoplasm would be in the differential diagnosis of any solitary nodule arising within a previously treated site of malignancy, which was excluded by histologic examination of our patient.7
Topical tretinoin has been demonstrated as effective treatment of both FRS and ACP.2,9 Other effective treatment modalities include cryotherapy and photoprotection.9 For our patient, a combination of cryosurgery and topical tretinoin resulted in depression of the plaque and a good cosmetic outcome.
Acknowledgments
We thank the patient for granting permission to publish this article. We also are indebted to Morgan L. Wilson, MD (Springfield, Illinois), for his expert dermatopathologic evaluation in this case. He has received no compensation for his contribution.
- Leeuwis-Fedorovich NE, Starink M, van der Wal AC. Multifocal squamous cell carcinoma arising in a Favre-Racouchot lesion—report of two cases and review of the literature. J Dermatol Case Rep. 2015;9:103-106.
- Cardoso F, Nakandakari S, Zattar GA, et al. Actinic comedonal plaquevariant of Favre-Racouchot syndrome: report of two cases. An Bras Dermatol. 2015;90(suppl 1):185-187.
- Eastern JS, Martin S. Actinic comedonal plaque. J Am Acad Dermatol. 1980;3:633-636.
- Hauptman G, Kopf A, Rabinovitz HS, et al. The actinic comedonal plaque. Cutis. 1997;60:145-146.
- John SM, Hamm H. Actinic comedonal plaque—a rare ectopic form of the Favre-Racouchot syndrome. Clin Exp Dermatol. 1993;18:256-258.
- Sonthalia S, Arora R, Chhabra N, et al. Favre-Racouchot syndrome. Indian Dermatol Online J. 2014;5(suppl 2):S128-S129.
- Elder DE, Elenitsas R, Murphy GF, et al. Benign pigmented lesions and malignant melanoma. In: Elder DE, Elenitsas R, Johnson BL Jr, et al, eds. Lever’s Histopathology of the Skin. 10th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2009:699-790.
- Lee DY, Kim YJ, Lee JY, et al. Primary localized cutaneous nodular amyloidosis following local trauma. Ann Dermatol. 2011;23:515-518.
- Patterson WM, Fox MD, Schwartz RA. Favre-Racouchot disease. Int J Dermatol. 2004;43:167-169.
- Leeuwis-Fedorovich NE, Starink M, van der Wal AC. Multifocal squamous cell carcinoma arising in a Favre-Racouchot lesion—report of two cases and review of the literature. J Dermatol Case Rep. 2015;9:103-106.
- Cardoso F, Nakandakari S, Zattar GA, et al. Actinic comedonal plaquevariant of Favre-Racouchot syndrome: report of two cases. An Bras Dermatol. 2015;90(suppl 1):185-187.
- Eastern JS, Martin S. Actinic comedonal plaque. J Am Acad Dermatol. 1980;3:633-636.
- Hauptman G, Kopf A, Rabinovitz HS, et al. The actinic comedonal plaque. Cutis. 1997;60:145-146.
- John SM, Hamm H. Actinic comedonal plaque—a rare ectopic form of the Favre-Racouchot syndrome. Clin Exp Dermatol. 1993;18:256-258.
- Sonthalia S, Arora R, Chhabra N, et al. Favre-Racouchot syndrome. Indian Dermatol Online J. 2014;5(suppl 2):S128-S129.
- Elder DE, Elenitsas R, Murphy GF, et al. Benign pigmented lesions and malignant melanoma. In: Elder DE, Elenitsas R, Johnson BL Jr, et al, eds. Lever’s Histopathology of the Skin. 10th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2009:699-790.
- Lee DY, Kim YJ, Lee JY, et al. Primary localized cutaneous nodular amyloidosis following local trauma. Ann Dermatol. 2011;23:515-518.
- Patterson WM, Fox MD, Schwartz RA. Favre-Racouchot disease. Int J Dermatol. 2004;43:167-169.
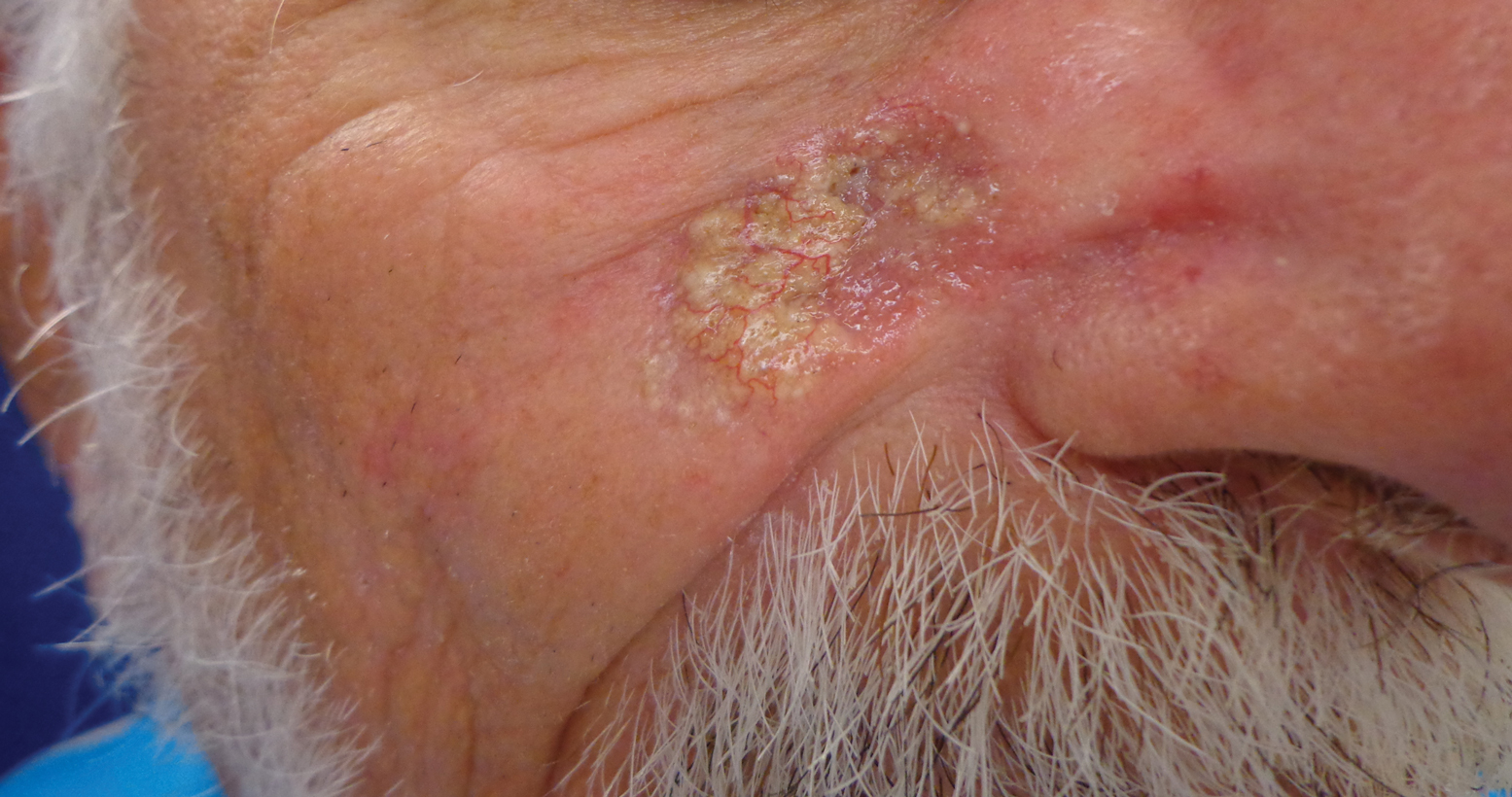
A 79-year-old man presented to dermatology with an enlarging bump on the right cheek. He reported a history of basal cell carcinoma on the medial right cheek that was removed 15 years prior with a resultant scar. Over the last 6 months, the area became more red and bumpy, and the lesion increased in size but was otherwise asymptomatic. The patient reported no history of other dermatologic conditions or skin cancer aside from the prior basal cell carcinoma. His medical history was notable for hypertension and hyperlipidemia. He had a history of smoking for many years (only recently quit) with extensive sun exposure in his lifetime as an outdoor worker. Physical examination revealed a 2.8.2 ×1.6-cm, pink, telangiectatic, and slightly depressed plaque, with the surface consisting of multiple white to yellowish coalescing papules. As part of the dermatologic workup, 4-mm punch and shave biopsies were obtained.
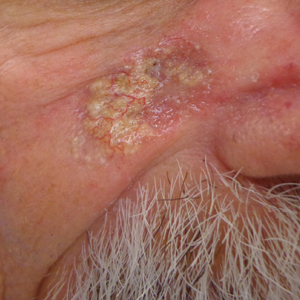
Papulonecrotic Tuberculid Secondary to Mycobacterium avium Complex
To the Editor:
Papulonecrotic tuberculid (PNT) is a cutaneous hypersensitivity reaction to antigenic components of Mycobacterium species, most commonly Mycobacterium tuberculosis. According to a PubMed search of articles indexed for MEDLINE using the terms papulonecrotic tuberculid, Mycobacterium avium complex, and Mycobacterium, only 1 case of PNT secondary to infection with Mycobacterium avium complex (MAC) has been reported.1,2 Papulonecrotic tuberculid classically presents with symmetrical, dusky red papules with necrosis on the extremities.3 Patients may or may not have associated symptoms of fever and weight loss. It is diagnosed through skin biopsy as well as identification of a distant source of mycobacterial infection. Papulonecrotic tuberculid is considered a reactive process to a distant site of mycobacterial infection, and skin lesions contain few, if any, mycobacteria.4
A 65-year-old man was admitted to the hospital for expedited workup of chronic fevers, 20-lb weight loss, and night sweats of 8 months’ duration. He had a medical history of myelodysplastic syndrome and autoimmune hemolytic anemia. During hospitalization, positron emission tomography revealed multilevel vertebral lytic and sclerotic lesions. Subsequent T10 vertebral biopsy showed necrotizing granulomatous inflammation with extensive necrosis and acid-fast bacilli–positive organisms. The patient was empirically started on rifampicin, isoniazid, pyrazinamide, ethambutol, and pyridoxine for presumed M tuberculosis and placed on respiratory isolation.
Dermatology was consulted for a recurrent tender rash on the bilateral upper and lower extremities of 5 years’ duration. Physical examination revealed numerous erythematous papulonecrotic lesions in various states of healing on the bilateral upper and lower extremities (Figure 1). Three years prior to the current presentation, 2 lesions were biopsied and demonstrated leukocytoclastic vasculitis with neutrophilic panniculitis and vasculopathy. A presumptive diagnosis of Sweet syndrome was made given the history of myelodysplastic syndrome, though an infectious etiology could not be ruled out at that time. Concurrently, the patient was diagnosed with autoimmune hemolytic anemia and was started on prednisone. Initially, the skin lesions improved with prednisone but never fully resolved; however, as the dosage of oral steroids decreased, the skin lesions worsened and presented in larger numbers with more frequency. The patient was titrated down to prednisone 5 mg daily with no additional treatment of the skin lesions at that time.
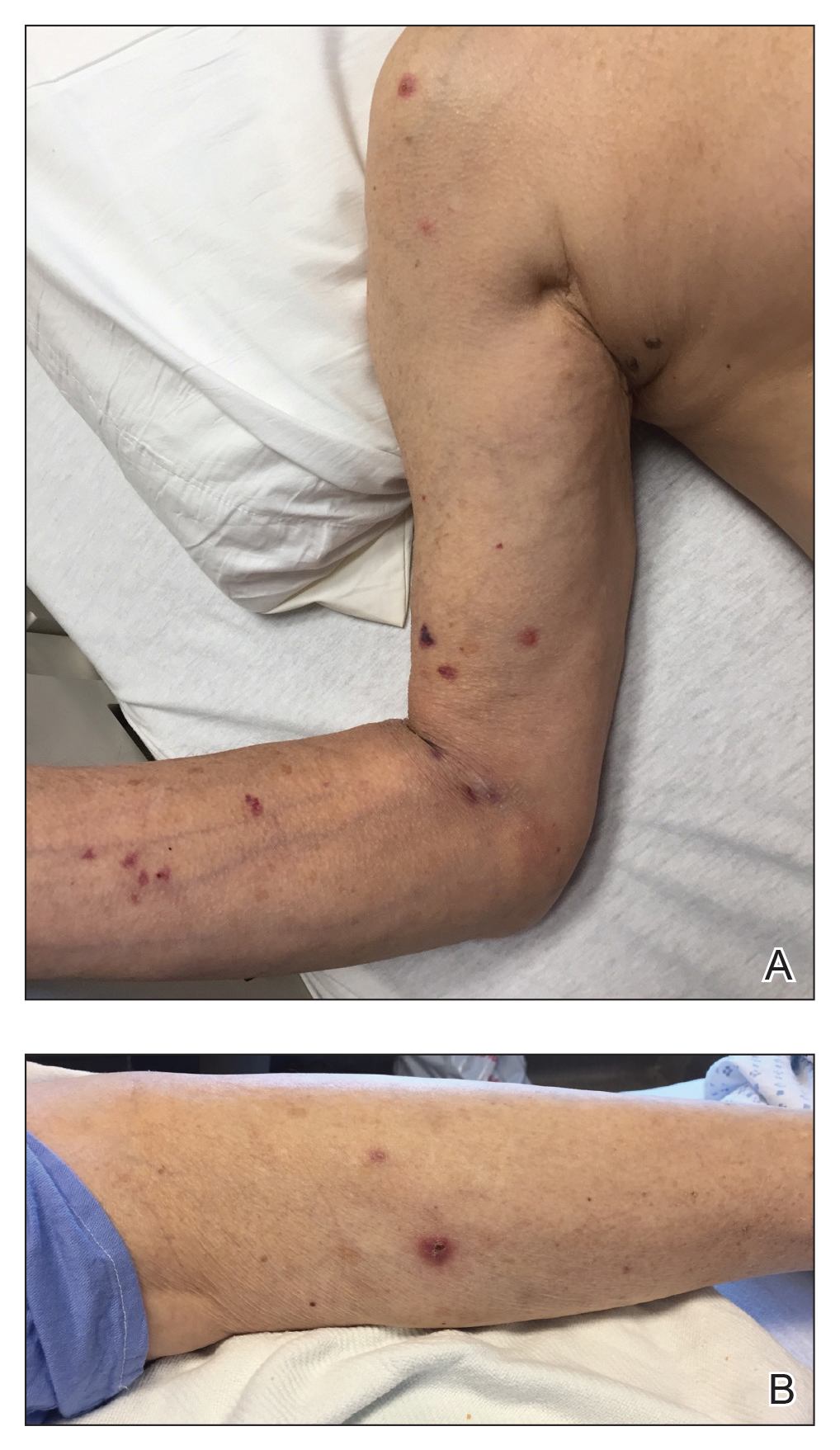
During the current hospitalization, 2 additional biopsies were taken from the arm for routine histopathology and tissue culture. Dermatopathology revealed robust neutrophilic and granulomatous inflammation as well as remarkable necrosis with a few mycobacteria identified on acid-fast and Fite stains (Figure 2). Tissue culture was negative. Additionally, the patient’s spinal biopsy was sent for polymerase chain reaction analysis for Mycobacterium typing, which confirmed MAC. The patient was diagnosed with Pott disease, a mycobacterial infection of the spine, as well as cutaneous papulonecrotic tuberculid secondary to MAC.
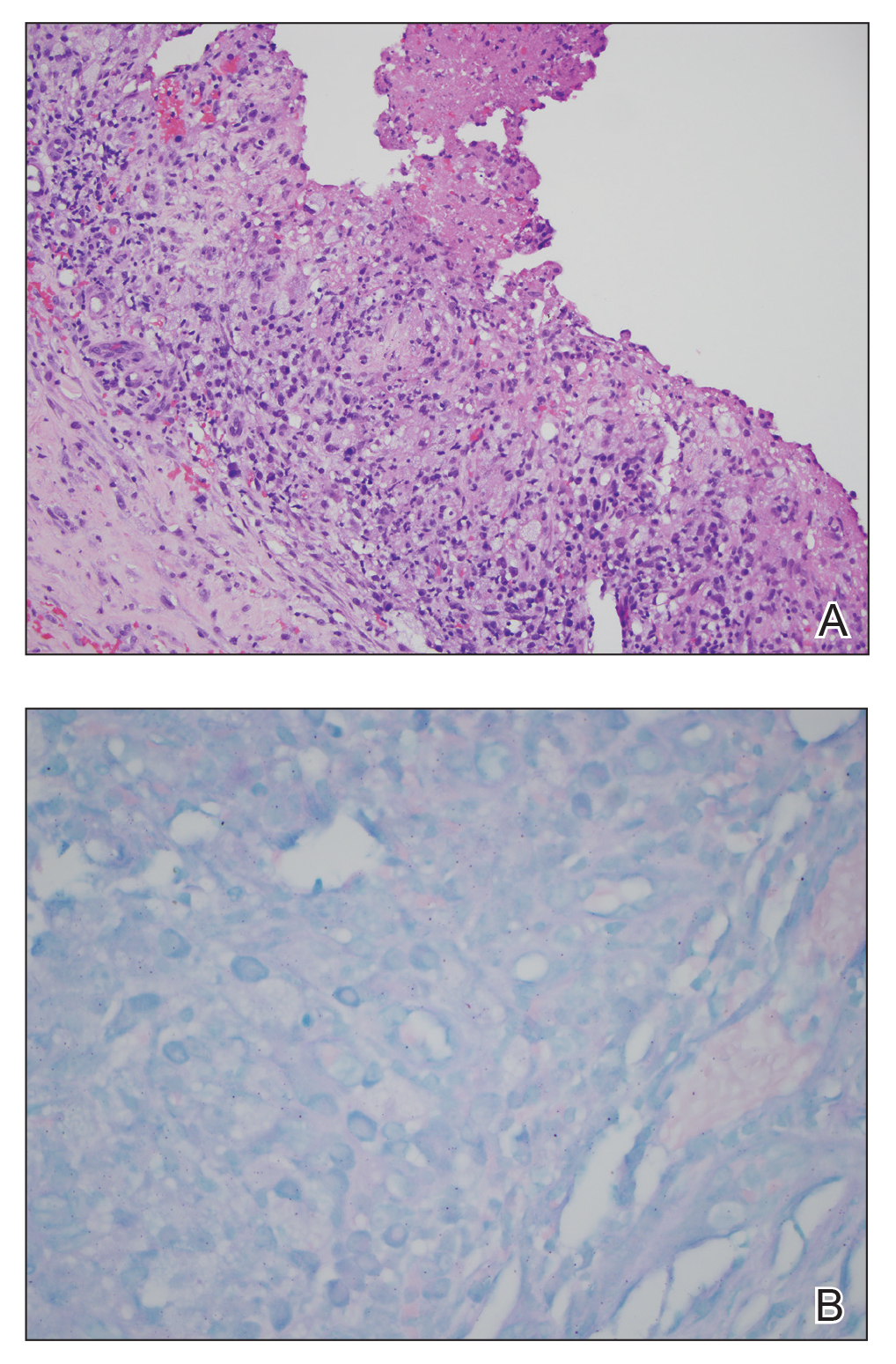
Papulonecrotic tuberculid is the rarest form of cutaneous tuberculosis infection and rarely has been reported in connection to MAC.1 This condition is considered a hypersensitivity reaction that occurs in response to antigenic components of mycobacteria.4 Patients with PNT typically present with recurrent crops of painful papulonecrotic lesions distributed on the extremities. Histopathology in PNT classically reveals necrosis, notable inflammatory infiltrate, and lack of observed organisms.5 Diagnosis often is made through skin biopsy, though histopathology varies based on lesion maturity.4 Early lesions often reveal leukocytoclastic vasculitis, whereas late lesions usually demonstrate granulomatous inflammation.4 Mycobacterium avium complex is difficult to culture, as it is a slow-growing, fastidious bacterium and therefore polymerase chain reaction genotyping is useful for bacterial classification.6
Disseminated MAC infection also was on the differential for our patient; however, we felt it was less likely than PNT for several reasons. First, disseminated infection rarely presents with cutaneous involvement and is associated with pulmonary involvement in 90% of cases.7-9 Second, the granuloma formation noted on our patient’s skin biopsy was not typical for disseminated MAC but is well described in cases of PNT.4,8,9 Finally, in the rare cases in which cutaneous involvement has occurred with disseminated mycobacterial infections, skin biopsies typically revealed numerous Mycobacterium organisms.8,10 In contrast, skin lesions associated with PNT usually reveal few, if any, organisms, as was seen with our patient.2
The patient’s initial biopsies also supported a diagnosis of PNT, as early lesions of PNT typically show leukocytoclastic vasculitis. His response to low and high doses of prednisone also fit well with a PNT diagnosis. In fact, a case of PNT secondary to Mycobacterium bovis similarly showed an improvement in the rash with high-dose steroids but progression with lower doses.11 It is possible that our patient’s response to steroids complicated the diagnosis of his rash.
The treatment of PNT is clearance of the underlying infection. Macrolide antibiotics, such as clarithromycin and azithromycin, have the best efficacy against MAC, in combination with ethambutol and/or rifabutin.6,12 Treatment duration should be 1 year. Amikacin or streptomycin may be added to this regimen during early treatment.6 Mycobacterium avium complex is resistant to many antibiotics, including typical antituberculosis drugs, and sensitivities should be identified at the onset of treatment.11,12
Albeit rare, clinicians should be aware of PNT secondary to MAC or other mycobacterial infections. Because this condition is difficult to diagnose with varying histologic findings and often negative tissue cultures, a high index of suspicion is necessary when a patient presents with recurrent papulonecrotic lesions, especially in immunocompromised hosts and patients with exposure to mycobacteria.
- Williams JT, Pulitzer DR, DeVillez RL. Papulonecrotic tuberculid secondary to disseminated Mycobacterium avium complex. Int J Dermatol. 1994;33:109-112.
- Jordaan HF, Schneider JW. Papulonecrotic tuberculid. Int J Dermatol. 1995;34:217-219.
- Scollard DM, Dacso MM, Abad-Venida ML. Tuberculosis and leprosy: classical granulomatous diseases in the twenty-first century. Dermatol Clin. 2015;33:541-562.
- Kim GW, Park HJ, Kim HS, et al. Simultaneous occurrence of papulonecrotic tuberculid and erythema induratum in a patient with pulmonary tuberculosis. Pediatr Dermatol. 2013;30:256-259.
- Spelta K, Diniz LM. Cutaneous tuberculosis: a 26-year retrospective study in an endemic area. Rev Inst Med Trop Sao Paulo. 2016;58:49.
- Griffith DE, Aksamit T, Brown-Elliott BA, et al. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. 2007;175:367-416.
- Dyer J, Weiss J, Steiner WS, et al. Primary cutaneous Mycobacterium avium complex infection following squamous cell carcinoma excision. Cutis. 2016;98:E8-E11.
- Kollipara R, Richards K, Tschen J, et al. Disseminated Mycobacterium avium complex with cutaneous lesions. J Cutan Med Surg. 2016;20:272-274.
- Endly DC, Ackerman LS. Disseminated cutaneous Mycobacterium avium complex in a person with AIDS. Dermatol Online J. 2014;20:22616.
- Li JJ, Beresford R, Fyfe J, et al. Clinical and histopathological features of cutaneous nontuberculous mycobacterial infection: a review of 13 cases. J Cutan Pathol. 2017;44:433-443.
- Iden DL, Rogers RS 3rd, Schroeter AL. Papulonecrotic tuberculid secondary to Mycobacterium bovis. Arch Dermatol. 1978;114:564-566.
- Wong NM, Sun LK, Lau PY. Spinal infection caused by Mycobacterium avium complex in a patient with no acquired immune deficiency syndrome: a case report. J Orthop Surg (Hong Kong). 2008;16:359-363.
To the Editor:
Papulonecrotic tuberculid (PNT) is a cutaneous hypersensitivity reaction to antigenic components of Mycobacterium species, most commonly Mycobacterium tuberculosis. According to a PubMed search of articles indexed for MEDLINE using the terms papulonecrotic tuberculid, Mycobacterium avium complex, and Mycobacterium, only 1 case of PNT secondary to infection with Mycobacterium avium complex (MAC) has been reported.1,2 Papulonecrotic tuberculid classically presents with symmetrical, dusky red papules with necrosis on the extremities.3 Patients may or may not have associated symptoms of fever and weight loss. It is diagnosed through skin biopsy as well as identification of a distant source of mycobacterial infection. Papulonecrotic tuberculid is considered a reactive process to a distant site of mycobacterial infection, and skin lesions contain few, if any, mycobacteria.4
A 65-year-old man was admitted to the hospital for expedited workup of chronic fevers, 20-lb weight loss, and night sweats of 8 months’ duration. He had a medical history of myelodysplastic syndrome and autoimmune hemolytic anemia. During hospitalization, positron emission tomography revealed multilevel vertebral lytic and sclerotic lesions. Subsequent T10 vertebral biopsy showed necrotizing granulomatous inflammation with extensive necrosis and acid-fast bacilli–positive organisms. The patient was empirically started on rifampicin, isoniazid, pyrazinamide, ethambutol, and pyridoxine for presumed M tuberculosis and placed on respiratory isolation.
Dermatology was consulted for a recurrent tender rash on the bilateral upper and lower extremities of 5 years’ duration. Physical examination revealed numerous erythematous papulonecrotic lesions in various states of healing on the bilateral upper and lower extremities (Figure 1). Three years prior to the current presentation, 2 lesions were biopsied and demonstrated leukocytoclastic vasculitis with neutrophilic panniculitis and vasculopathy. A presumptive diagnosis of Sweet syndrome was made given the history of myelodysplastic syndrome, though an infectious etiology could not be ruled out at that time. Concurrently, the patient was diagnosed with autoimmune hemolytic anemia and was started on prednisone. Initially, the skin lesions improved with prednisone but never fully resolved; however, as the dosage of oral steroids decreased, the skin lesions worsened and presented in larger numbers with more frequency. The patient was titrated down to prednisone 5 mg daily with no additional treatment of the skin lesions at that time.
During the current hospitalization, 2 additional biopsies were taken from the arm for routine histopathology and tissue culture. Dermatopathology revealed robust neutrophilic and granulomatous inflammation as well as remarkable necrosis with a few mycobacteria identified on acid-fast and Fite stains (Figure 2). Tissue culture was negative. Additionally, the patient’s spinal biopsy was sent for polymerase chain reaction analysis for Mycobacterium typing, which confirmed MAC. The patient was diagnosed with Pott disease, a mycobacterial infection of the spine, as well as cutaneous papulonecrotic tuberculid secondary to MAC.
Papulonecrotic tuberculid is the rarest form of cutaneous tuberculosis infection and rarely has been reported in connection to MAC.1 This condition is considered a hypersensitivity reaction that occurs in response to antigenic components of mycobacteria.4 Patients with PNT typically present with recurrent crops of painful papulonecrotic lesions distributed on the extremities. Histopathology in PNT classically reveals necrosis, notable inflammatory infiltrate, and lack of observed organisms.5 Diagnosis often is made through skin biopsy, though histopathology varies based on lesion maturity.4 Early lesions often reveal leukocytoclastic vasculitis, whereas late lesions usually demonstrate granulomatous inflammation.4 Mycobacterium avium complex is difficult to culture, as it is a slow-growing, fastidious bacterium and therefore polymerase chain reaction genotyping is useful for bacterial classification.6
Disseminated MAC infection also was on the differential for our patient; however, we felt it was less likely than PNT for several reasons. First, disseminated infection rarely presents with cutaneous involvement and is associated with pulmonary involvement in 90% of cases.7-9 Second, the granuloma formation noted on our patient’s skin biopsy was not typical for disseminated MAC but is well described in cases of PNT.4,8,9 Finally, in the rare cases in which cutaneous involvement has occurred with disseminated mycobacterial infections, skin biopsies typically revealed numerous Mycobacterium organisms.8,10 In contrast, skin lesions associated with PNT usually reveal few, if any, organisms, as was seen with our patient.2
The patient’s initial biopsies also supported a diagnosis of PNT, as early lesions of PNT typically show leukocytoclastic vasculitis. His response to low and high doses of prednisone also fit well with a PNT diagnosis. In fact, a case of PNT secondary to Mycobacterium bovis similarly showed an improvement in the rash with high-dose steroids but progression with lower doses.11 It is possible that our patient’s response to steroids complicated the diagnosis of his rash.
The treatment of PNT is clearance of the underlying infection. Macrolide antibiotics, such as clarithromycin and azithromycin, have the best efficacy against MAC, in combination with ethambutol and/or rifabutin.6,12 Treatment duration should be 1 year. Amikacin or streptomycin may be added to this regimen during early treatment.6 Mycobacterium avium complex is resistant to many antibiotics, including typical antituberculosis drugs, and sensitivities should be identified at the onset of treatment.11,12
Albeit rare, clinicians should be aware of PNT secondary to MAC or other mycobacterial infections. Because this condition is difficult to diagnose with varying histologic findings and often negative tissue cultures, a high index of suspicion is necessary when a patient presents with recurrent papulonecrotic lesions, especially in immunocompromised hosts and patients with exposure to mycobacteria.
To the Editor:
Papulonecrotic tuberculid (PNT) is a cutaneous hypersensitivity reaction to antigenic components of Mycobacterium species, most commonly Mycobacterium tuberculosis. According to a PubMed search of articles indexed for MEDLINE using the terms papulonecrotic tuberculid, Mycobacterium avium complex, and Mycobacterium, only 1 case of PNT secondary to infection with Mycobacterium avium complex (MAC) has been reported.1,2 Papulonecrotic tuberculid classically presents with symmetrical, dusky red papules with necrosis on the extremities.3 Patients may or may not have associated symptoms of fever and weight loss. It is diagnosed through skin biopsy as well as identification of a distant source of mycobacterial infection. Papulonecrotic tuberculid is considered a reactive process to a distant site of mycobacterial infection, and skin lesions contain few, if any, mycobacteria.4
A 65-year-old man was admitted to the hospital for expedited workup of chronic fevers, 20-lb weight loss, and night sweats of 8 months’ duration. He had a medical history of myelodysplastic syndrome and autoimmune hemolytic anemia. During hospitalization, positron emission tomography revealed multilevel vertebral lytic and sclerotic lesions. Subsequent T10 vertebral biopsy showed necrotizing granulomatous inflammation with extensive necrosis and acid-fast bacilli–positive organisms. The patient was empirically started on rifampicin, isoniazid, pyrazinamide, ethambutol, and pyridoxine for presumed M tuberculosis and placed on respiratory isolation.
Dermatology was consulted for a recurrent tender rash on the bilateral upper and lower extremities of 5 years’ duration. Physical examination revealed numerous erythematous papulonecrotic lesions in various states of healing on the bilateral upper and lower extremities (Figure 1). Three years prior to the current presentation, 2 lesions were biopsied and demonstrated leukocytoclastic vasculitis with neutrophilic panniculitis and vasculopathy. A presumptive diagnosis of Sweet syndrome was made given the history of myelodysplastic syndrome, though an infectious etiology could not be ruled out at that time. Concurrently, the patient was diagnosed with autoimmune hemolytic anemia and was started on prednisone. Initially, the skin lesions improved with prednisone but never fully resolved; however, as the dosage of oral steroids decreased, the skin lesions worsened and presented in larger numbers with more frequency. The patient was titrated down to prednisone 5 mg daily with no additional treatment of the skin lesions at that time.
During the current hospitalization, 2 additional biopsies were taken from the arm for routine histopathology and tissue culture. Dermatopathology revealed robust neutrophilic and granulomatous inflammation as well as remarkable necrosis with a few mycobacteria identified on acid-fast and Fite stains (Figure 2). Tissue culture was negative. Additionally, the patient’s spinal biopsy was sent for polymerase chain reaction analysis for Mycobacterium typing, which confirmed MAC. The patient was diagnosed with Pott disease, a mycobacterial infection of the spine, as well as cutaneous papulonecrotic tuberculid secondary to MAC.
Papulonecrotic tuberculid is the rarest form of cutaneous tuberculosis infection and rarely has been reported in connection to MAC.1 This condition is considered a hypersensitivity reaction that occurs in response to antigenic components of mycobacteria.4 Patients with PNT typically present with recurrent crops of painful papulonecrotic lesions distributed on the extremities. Histopathology in PNT classically reveals necrosis, notable inflammatory infiltrate, and lack of observed organisms.5 Diagnosis often is made through skin biopsy, though histopathology varies based on lesion maturity.4 Early lesions often reveal leukocytoclastic vasculitis, whereas late lesions usually demonstrate granulomatous inflammation.4 Mycobacterium avium complex is difficult to culture, as it is a slow-growing, fastidious bacterium and therefore polymerase chain reaction genotyping is useful for bacterial classification.6
Disseminated MAC infection also was on the differential for our patient; however, we felt it was less likely than PNT for several reasons. First, disseminated infection rarely presents with cutaneous involvement and is associated with pulmonary involvement in 90% of cases.7-9 Second, the granuloma formation noted on our patient’s skin biopsy was not typical for disseminated MAC but is well described in cases of PNT.4,8,9 Finally, in the rare cases in which cutaneous involvement has occurred with disseminated mycobacterial infections, skin biopsies typically revealed numerous Mycobacterium organisms.8,10 In contrast, skin lesions associated with PNT usually reveal few, if any, organisms, as was seen with our patient.2
The patient’s initial biopsies also supported a diagnosis of PNT, as early lesions of PNT typically show leukocytoclastic vasculitis. His response to low and high doses of prednisone also fit well with a PNT diagnosis. In fact, a case of PNT secondary to Mycobacterium bovis similarly showed an improvement in the rash with high-dose steroids but progression with lower doses.11 It is possible that our patient’s response to steroids complicated the diagnosis of his rash.
The treatment of PNT is clearance of the underlying infection. Macrolide antibiotics, such as clarithromycin and azithromycin, have the best efficacy against MAC, in combination with ethambutol and/or rifabutin.6,12 Treatment duration should be 1 year. Amikacin or streptomycin may be added to this regimen during early treatment.6 Mycobacterium avium complex is resistant to many antibiotics, including typical antituberculosis drugs, and sensitivities should be identified at the onset of treatment.11,12
Albeit rare, clinicians should be aware of PNT secondary to MAC or other mycobacterial infections. Because this condition is difficult to diagnose with varying histologic findings and often negative tissue cultures, a high index of suspicion is necessary when a patient presents with recurrent papulonecrotic lesions, especially in immunocompromised hosts and patients with exposure to mycobacteria.
- Williams JT, Pulitzer DR, DeVillez RL. Papulonecrotic tuberculid secondary to disseminated Mycobacterium avium complex. Int J Dermatol. 1994;33:109-112.
- Jordaan HF, Schneider JW. Papulonecrotic tuberculid. Int J Dermatol. 1995;34:217-219.
- Scollard DM, Dacso MM, Abad-Venida ML. Tuberculosis and leprosy: classical granulomatous diseases in the twenty-first century. Dermatol Clin. 2015;33:541-562.
- Kim GW, Park HJ, Kim HS, et al. Simultaneous occurrence of papulonecrotic tuberculid and erythema induratum in a patient with pulmonary tuberculosis. Pediatr Dermatol. 2013;30:256-259.
- Spelta K, Diniz LM. Cutaneous tuberculosis: a 26-year retrospective study in an endemic area. Rev Inst Med Trop Sao Paulo. 2016;58:49.
- Griffith DE, Aksamit T, Brown-Elliott BA, et al. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. 2007;175:367-416.
- Dyer J, Weiss J, Steiner WS, et al. Primary cutaneous Mycobacterium avium complex infection following squamous cell carcinoma excision. Cutis. 2016;98:E8-E11.
- Kollipara R, Richards K, Tschen J, et al. Disseminated Mycobacterium avium complex with cutaneous lesions. J Cutan Med Surg. 2016;20:272-274.
- Endly DC, Ackerman LS. Disseminated cutaneous Mycobacterium avium complex in a person with AIDS. Dermatol Online J. 2014;20:22616.
- Li JJ, Beresford R, Fyfe J, et al. Clinical and histopathological features of cutaneous nontuberculous mycobacterial infection: a review of 13 cases. J Cutan Pathol. 2017;44:433-443.
- Iden DL, Rogers RS 3rd, Schroeter AL. Papulonecrotic tuberculid secondary to Mycobacterium bovis. Arch Dermatol. 1978;114:564-566.
- Wong NM, Sun LK, Lau PY. Spinal infection caused by Mycobacterium avium complex in a patient with no acquired immune deficiency syndrome: a case report. J Orthop Surg (Hong Kong). 2008;16:359-363.
- Williams JT, Pulitzer DR, DeVillez RL. Papulonecrotic tuberculid secondary to disseminated Mycobacterium avium complex. Int J Dermatol. 1994;33:109-112.
- Jordaan HF, Schneider JW. Papulonecrotic tuberculid. Int J Dermatol. 1995;34:217-219.
- Scollard DM, Dacso MM, Abad-Venida ML. Tuberculosis and leprosy: classical granulomatous diseases in the twenty-first century. Dermatol Clin. 2015;33:541-562.
- Kim GW, Park HJ, Kim HS, et al. Simultaneous occurrence of papulonecrotic tuberculid and erythema induratum in a patient with pulmonary tuberculosis. Pediatr Dermatol. 2013;30:256-259.
- Spelta K, Diniz LM. Cutaneous tuberculosis: a 26-year retrospective study in an endemic area. Rev Inst Med Trop Sao Paulo. 2016;58:49.
- Griffith DE, Aksamit T, Brown-Elliott BA, et al. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. 2007;175:367-416.
- Dyer J, Weiss J, Steiner WS, et al. Primary cutaneous Mycobacterium avium complex infection following squamous cell carcinoma excision. Cutis. 2016;98:E8-E11.
- Kollipara R, Richards K, Tschen J, et al. Disseminated Mycobacterium avium complex with cutaneous lesions. J Cutan Med Surg. 2016;20:272-274.
- Endly DC, Ackerman LS. Disseminated cutaneous Mycobacterium avium complex in a person with AIDS. Dermatol Online J. 2014;20:22616.
- Li JJ, Beresford R, Fyfe J, et al. Clinical and histopathological features of cutaneous nontuberculous mycobacterial infection: a review of 13 cases. J Cutan Pathol. 2017;44:433-443.
- Iden DL, Rogers RS 3rd, Schroeter AL. Papulonecrotic tuberculid secondary to Mycobacterium bovis. Arch Dermatol. 1978;114:564-566.
- Wong NM, Sun LK, Lau PY. Spinal infection caused by Mycobacterium avium complex in a patient with no acquired immune deficiency syndrome: a case report. J Orthop Surg (Hong Kong). 2008;16:359-363.
Practice Points
- Papulonecrotic tuberculid (PNT) is a hypersensitivity reaction that presents with reddish papules with central necrosis on the extremities.
- Early PNT histopathology shows leukocytoclastic vasculitis. Later lesions demonstrate granulomatous inflammation on histopathology.
- Mycobacterium avium is difficult to culture; therefore, if you suspect it, we recommend polymerase chain reaction genotyping for bacterial classification.
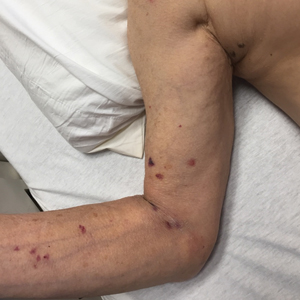
Antecedent Chronic Lymphocytic Leukemia May Be Associated With More Aggressive Mycosis Fungoides
To the Editor:
Mycosis fungoides (MF) is the most common form of primary cutaneous T-cell lymphoma. It has been associated with increased risk for other visceral and hematologic malignancies.1 Chronic lymphocytic leukemia (CLL) is one of the most common hematologic malignancies. In the United States, a patient’s lifetime risk for CLL is 0.6%. Chronic lymphocytic leukemia often is diagnosed as an incidental finding and typically is not detrimental to a patient’s health. Six cases of MF with antecedent or concomitant CLL were identified in a cohort of patients treated at the University of Minnesota (Minneapolis, Minnesota) from 2005 to 2017 (Table).
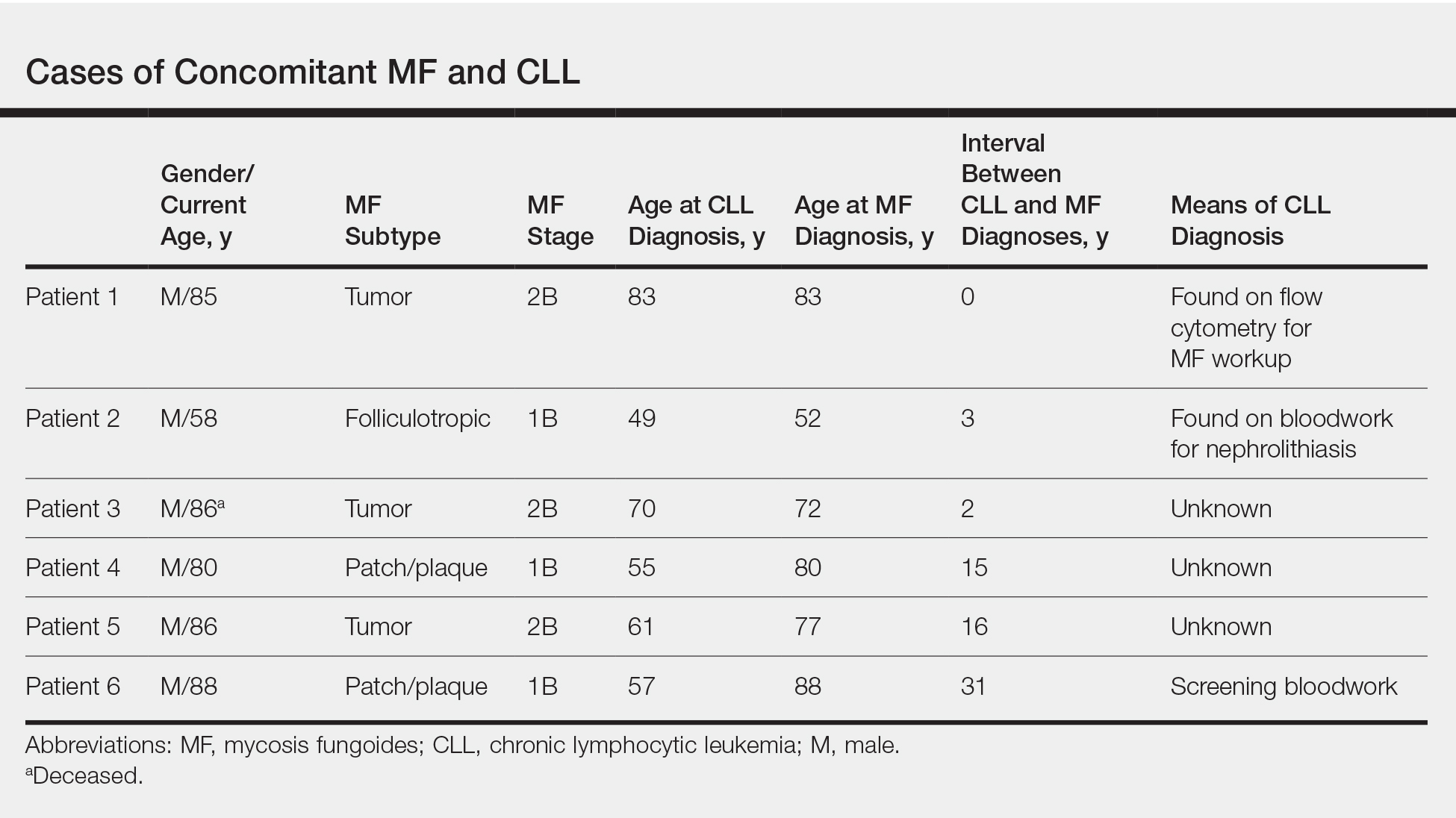
All 6 patients were male, with a mean age of 80.5 years. The mean age at CLL diagnosis was 62.5 years, while the mean age at MF diagnosis was 75.3 years. Three patients were younger than 60 years when their CLL was diagnosed: 49, 55, and 57 years. Notably, 4 patients had more aggressive types of MF: 3 with tumor-stage disease, and 1 with folliculotropic MF. Five patients were diagnosed with CLL before their MF was diagnosed (mean, 13.4 years prior; range, 3–31 years), and 1 was diagnosed as part of the initial MF workup.
Given the frequency of both MF and CLL, the co-occurrence of these diseases is not surprising, as other case reports and a larger case series have described the relationship between MF and malignancy.2 It is possible that CLL patients are more likely to be diagnosed with MF because of their regular hematology/oncology follow-up; however, none of our patients were referred from hematology/oncology to dermatology. Alternatively, patients with MF may be more likely to be diagnosed with CLL because of repeated bloodwork performed for diagnosis and screening, which occurred in only 1 of 6 cases. Most of the other patients were diagnosed with MF more than a decade after being diagnosed with CLL.
Does having CLL make patients more likely to develop MF? It is known that patients with CLL may experience immunodeficiency secondary to immune dysregulation, making them more susceptible to infection and secondary malignancies.3 Of our 6 cases, 4 had aggressive or advanced forms of MF, which is similar to the findings of Chang et al.2 In their report, of 8 patients with MF, 2 had tumor-stage disease and 2 had erythrodermic MF. They determined that these patients had worse overall survival.2 Our data corroborate the finding that patients with CLL may develop more severe MF, which leads to the conclusion that patients diagnosed with CLL before, concomitantly, or after their diagnosis of MF should be closely monitored. It is notable that patients with more advanced disease tend to be older at the time of diagnosis and that patients who are diagnosed at 57 years or older have been found to have worse disease-specific survival.4,5
This report is limited by the small sample size (6 cases), but it serves to draw attention to the phenomenon of co-occurrence of MF and CLL, and the concern that patients with CLL may develop more aggressive MF.
- Huang KP, Weinstock MA, Clarke CA, et al. Second lymphomas and other malignant neoplasms in patients with mycosis fungoides and Sézary syndrome. Arch Dermatol. 2007;143:45-50.
- Chang MB, Weaver AL, Brewer JD. Cutaneous T-cell lymphoma in patients with chronic lymphocytic leukemia: clinical characteristics, temporal relationships, and survival data in a series of 14 patients at Mayo Clinic. Int J Dermatol. 2014;53:966-970.
- Hamblin AD, Hamblin TJ. The immunodeficiency of chronic lymphocytic leukaemia. Br Med Bull. 2008;87:49-62.
- Kim YH, Liu HL, Mraz-Gernhard S, et al. Long-term outcome of 525 patients with mycosis fungoides and Sezary syndrome: clinical prognostic factors and risk for disease progression. Arch Dermatol. 2003;139:857-866.
- Agar NS, Wedgeworth E, Crichton S, et al. Survival outcomes and prognostic factors in mycosis fungoides/Sézary syndrome: validation of the revised International Society for Cutaneous Lymphomas/European Organisation for Research and Treatment of Cancer staging proposal. J Clin Oncol. 2010;28:4730-4739.
To the Editor:
Mycosis fungoides (MF) is the most common form of primary cutaneous T-cell lymphoma. It has been associated with increased risk for other visceral and hematologic malignancies.1 Chronic lymphocytic leukemia (CLL) is one of the most common hematologic malignancies. In the United States, a patient’s lifetime risk for CLL is 0.6%. Chronic lymphocytic leukemia often is diagnosed as an incidental finding and typically is not detrimental to a patient’s health. Six cases of MF with antecedent or concomitant CLL were identified in a cohort of patients treated at the University of Minnesota (Minneapolis, Minnesota) from 2005 to 2017 (Table).
All 6 patients were male, with a mean age of 80.5 years. The mean age at CLL diagnosis was 62.5 years, while the mean age at MF diagnosis was 75.3 years. Three patients were younger than 60 years when their CLL was diagnosed: 49, 55, and 57 years. Notably, 4 patients had more aggressive types of MF: 3 with tumor-stage disease, and 1 with folliculotropic MF. Five patients were diagnosed with CLL before their MF was diagnosed (mean, 13.4 years prior; range, 3–31 years), and 1 was diagnosed as part of the initial MF workup.
Given the frequency of both MF and CLL, the co-occurrence of these diseases is not surprising, as other case reports and a larger case series have described the relationship between MF and malignancy.2 It is possible that CLL patients are more likely to be diagnosed with MF because of their regular hematology/oncology follow-up; however, none of our patients were referred from hematology/oncology to dermatology. Alternatively, patients with MF may be more likely to be diagnosed with CLL because of repeated bloodwork performed for diagnosis and screening, which occurred in only 1 of 6 cases. Most of the other patients were diagnosed with MF more than a decade after being diagnosed with CLL.
Does having CLL make patients more likely to develop MF? It is known that patients with CLL may experience immunodeficiency secondary to immune dysregulation, making them more susceptible to infection and secondary malignancies.3 Of our 6 cases, 4 had aggressive or advanced forms of MF, which is similar to the findings of Chang et al.2 In their report, of 8 patients with MF, 2 had tumor-stage disease and 2 had erythrodermic MF. They determined that these patients had worse overall survival.2 Our data corroborate the finding that patients with CLL may develop more severe MF, which leads to the conclusion that patients diagnosed with CLL before, concomitantly, or after their diagnosis of MF should be closely monitored. It is notable that patients with more advanced disease tend to be older at the time of diagnosis and that patients who are diagnosed at 57 years or older have been found to have worse disease-specific survival.4,5
This report is limited by the small sample size (6 cases), but it serves to draw attention to the phenomenon of co-occurrence of MF and CLL, and the concern that patients with CLL may develop more aggressive MF.
To the Editor:
Mycosis fungoides (MF) is the most common form of primary cutaneous T-cell lymphoma. It has been associated with increased risk for other visceral and hematologic malignancies.1 Chronic lymphocytic leukemia (CLL) is one of the most common hematologic malignancies. In the United States, a patient’s lifetime risk for CLL is 0.6%. Chronic lymphocytic leukemia often is diagnosed as an incidental finding and typically is not detrimental to a patient’s health. Six cases of MF with antecedent or concomitant CLL were identified in a cohort of patients treated at the University of Minnesota (Minneapolis, Minnesota) from 2005 to 2017 (Table).
All 6 patients were male, with a mean age of 80.5 years. The mean age at CLL diagnosis was 62.5 years, while the mean age at MF diagnosis was 75.3 years. Three patients were younger than 60 years when their CLL was diagnosed: 49, 55, and 57 years. Notably, 4 patients had more aggressive types of MF: 3 with tumor-stage disease, and 1 with folliculotropic MF. Five patients were diagnosed with CLL before their MF was diagnosed (mean, 13.4 years prior; range, 3–31 years), and 1 was diagnosed as part of the initial MF workup.
Given the frequency of both MF and CLL, the co-occurrence of these diseases is not surprising, as other case reports and a larger case series have described the relationship between MF and malignancy.2 It is possible that CLL patients are more likely to be diagnosed with MF because of their regular hematology/oncology follow-up; however, none of our patients were referred from hematology/oncology to dermatology. Alternatively, patients with MF may be more likely to be diagnosed with CLL because of repeated bloodwork performed for diagnosis and screening, which occurred in only 1 of 6 cases. Most of the other patients were diagnosed with MF more than a decade after being diagnosed with CLL.
Does having CLL make patients more likely to develop MF? It is known that patients with CLL may experience immunodeficiency secondary to immune dysregulation, making them more susceptible to infection and secondary malignancies.3 Of our 6 cases, 4 had aggressive or advanced forms of MF, which is similar to the findings of Chang et al.2 In their report, of 8 patients with MF, 2 had tumor-stage disease and 2 had erythrodermic MF. They determined that these patients had worse overall survival.2 Our data corroborate the finding that patients with CLL may develop more severe MF, which leads to the conclusion that patients diagnosed with CLL before, concomitantly, or after their diagnosis of MF should be closely monitored. It is notable that patients with more advanced disease tend to be older at the time of diagnosis and that patients who are diagnosed at 57 years or older have been found to have worse disease-specific survival.4,5
This report is limited by the small sample size (6 cases), but it serves to draw attention to the phenomenon of co-occurrence of MF and CLL, and the concern that patients with CLL may develop more aggressive MF.
- Huang KP, Weinstock MA, Clarke CA, et al. Second lymphomas and other malignant neoplasms in patients with mycosis fungoides and Sézary syndrome. Arch Dermatol. 2007;143:45-50.
- Chang MB, Weaver AL, Brewer JD. Cutaneous T-cell lymphoma in patients with chronic lymphocytic leukemia: clinical characteristics, temporal relationships, and survival data in a series of 14 patients at Mayo Clinic. Int J Dermatol. 2014;53:966-970.
- Hamblin AD, Hamblin TJ. The immunodeficiency of chronic lymphocytic leukaemia. Br Med Bull. 2008;87:49-62.
- Kim YH, Liu HL, Mraz-Gernhard S, et al. Long-term outcome of 525 patients with mycosis fungoides and Sezary syndrome: clinical prognostic factors and risk for disease progression. Arch Dermatol. 2003;139:857-866.
- Agar NS, Wedgeworth E, Crichton S, et al. Survival outcomes and prognostic factors in mycosis fungoides/Sézary syndrome: validation of the revised International Society for Cutaneous Lymphomas/European Organisation for Research and Treatment of Cancer staging proposal. J Clin Oncol. 2010;28:4730-4739.
- Huang KP, Weinstock MA, Clarke CA, et al. Second lymphomas and other malignant neoplasms in patients with mycosis fungoides and Sézary syndrome. Arch Dermatol. 2007;143:45-50.
- Chang MB, Weaver AL, Brewer JD. Cutaneous T-cell lymphoma in patients with chronic lymphocytic leukemia: clinical characteristics, temporal relationships, and survival data in a series of 14 patients at Mayo Clinic. Int J Dermatol. 2014;53:966-970.
- Hamblin AD, Hamblin TJ. The immunodeficiency of chronic lymphocytic leukaemia. Br Med Bull. 2008;87:49-62.
- Kim YH, Liu HL, Mraz-Gernhard S, et al. Long-term outcome of 525 patients with mycosis fungoides and Sezary syndrome: clinical prognostic factors and risk for disease progression. Arch Dermatol. 2003;139:857-866.
- Agar NS, Wedgeworth E, Crichton S, et al. Survival outcomes and prognostic factors in mycosis fungoides/Sézary syndrome: validation of the revised International Society for Cutaneous Lymphomas/European Organisation for Research and Treatment of Cancer staging proposal. J Clin Oncol. 2010;28:4730-4739.
Practice Points
- Patients with mycosis fungoides (MF) are at increased risk for second hematologic malignancies, including chronic lymphocytic leukemia (CLL).
- Anecdotal information suggests that patients with CLL prior to developing MF may have more severe phenotypes of MF.
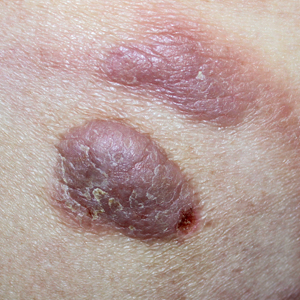
Unilateral Papules on the Face
The Diagnosis: Mosaic Tuberous Sclerosis
A punch biopsy of the facial lesion was stained with hematoxylin and eosin, which demonstrated spindled and stellate fibroblasts with dilated blood vessels (Figure), consistent with an angiofibroma. Given the clinical presentation and histologic findings, there was concern for a diagnosis of tuberous sclerosis (TSC). The patient was referred for genetic workup but tested negative for mutations of the TSC genes in the blood. Because the patient had only unilateral facial lesions, a possible cortical tuber, no other symptoms, and tested negative for TSC gene mutations, mosaic TSC was considered a likely diagnosis. Her facial lesions were treated with pulsed dye vascular laser therapy.
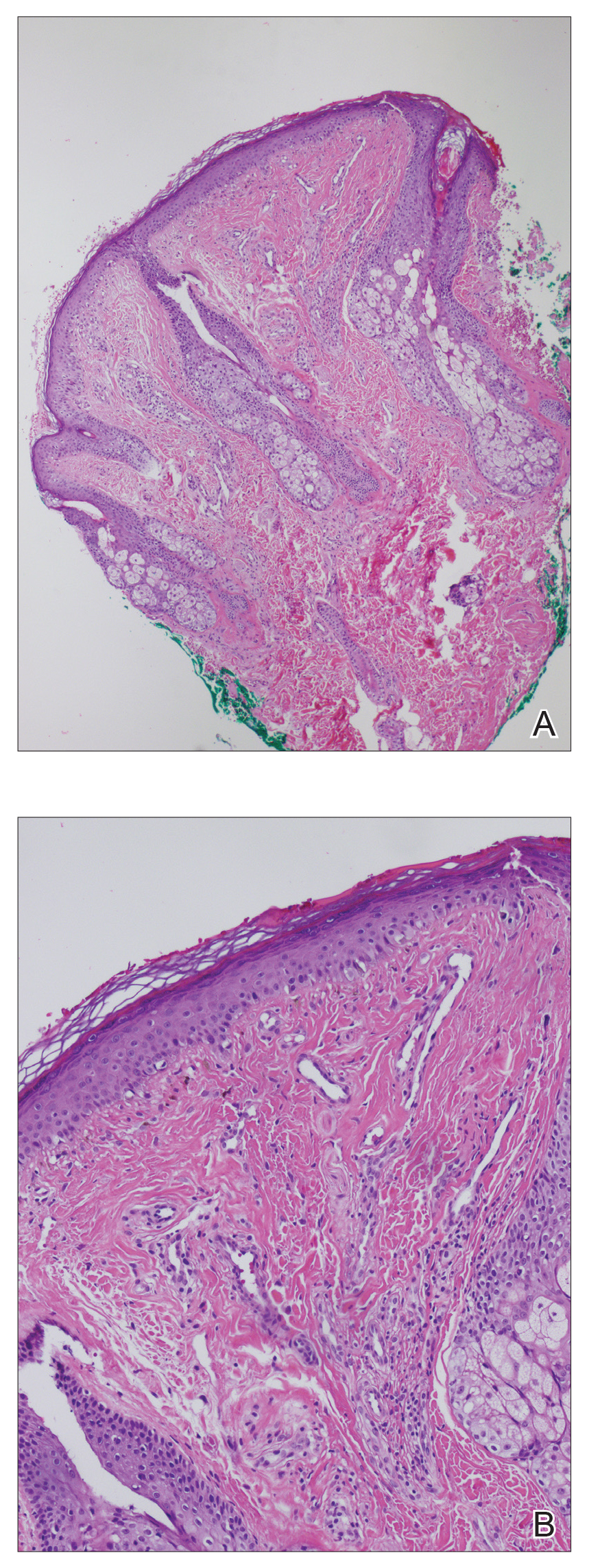
Tuberous sclerosis is an autosomal-dominant neurocutaneous disorder caused by inactivation of the genes TSC1 (encoding hamartin) and TSC2 (encoding tuberin). Mutation results in overactivation of the downstream mTOR (mammalian target of rapamycin) pathway, resulting in abnormal cellular proliferation and hamartomas. These benign tumors can be found in nearly every organ, most often in the central nervous system and skin, and they provide for a highly variable presentation of the disease.1
Tuberous sclerosis affects 1 in 6000 to 10,000 live births and has a prevalence of 1 in 20,000 individuals. Of these individuals, 75% carry sporadic mutations, and 75% to 90% eventually test positive for a TSC gene mutation.2 Genetic mosaicism has been reported in 28% of cases affected by large deletions1 and as few as 1% of cases involving small mutations.3
The dermatologic manifestation of mosaic TSC most often includes unilateral angiofibromas, whereas in nonmosaic cases, angiofibromas cover both cheeks, the forehead, and the eyelids. The other skin lesions of TSC--shagreen patches, forehead plaques, hypomelanotic macules, and ungual fibromas--are seen less frequently.4-6 Additionally, neurologic disease in mosaic patients is notably milder, with 57% of mosaic patients found to have epilepsy compared to 91% of nonmosaic patients.7 Our patient had both unilateral facial angiofibromas and a cortical lesion suspicious for a tuber, prompting a suspected diagnosis of mosaic TSC.
The methods of diagnosis outlined by the International Tuberous Sclerosis Complex Consensus Group pose a challenge in diagnosing mosaic TSC. The clinical criteria require 2 major (eg, multiple angiofibromas, angiomyolipomas, a shagreen patch) and 1 minor feature (eg, dental enamel pit, renal cyst).2 However, case reports detailing unilateral facial angiofibromas have described patients with isolated dermatologic findings.5,6 Further, it has been demonstrated that genetic studies in mosaic TSC can be unreliable depending on the tissue sampled.8 Thus, for patients who have mosaic TSC, establishing a definitive diagnosis is not always possible and may rely solely on the clinical picture.
Considering the differential diagnosis, benign cephalic histiocytosis usually would present with small red-brown macules and papules symmetrically located on the head and neck. The lesions occur at a younger age, usually in the first year or two of life. Fibrofolliculomas present as multiple whitish, slightly larger papules found on the central face. They are a marker for Birt-Hogg-Dubé syndrome, which also is associated with spontaneous pneumothorax.
Agminated means clustering or grouping of lesions. Agminated melanocytic nevi and agminated Spitz nevi are clusters of nevi. These lesions can vary in size and color. They may be elevated or flat. Melanocytic nevi usually are tan-brown or black. Spitz nevi may be pink or pigmented brown or black. To definitively differentiate between these 2 diagnoses and this patient's diagnosis of angiofibroma, a biopsy is needed.
- Curatolo P, Moavero R, Roberto D, et al. Genotype/phenotype correlations in tuberous sclerosis complex. Semin Pediatr Neurol. 2015;22:259-273.
- Northrup H, Krueger DA; International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol. 2013;49:243-254.
- Kwiatkowski DJ, Whittemore VH, Thiele EA. Tuberous Sclerosis Complex: Genes, Clinical Features and Therapeutics. Weinham, Germany: Wiley-Blackwell; 2011.
- Alshaiji JM, Spock CR, Connelly EA, et al. Facial angiofibromas in a mosaic pattern tuberous sclerosis: a case report. Dermatol Online J. 2012;18:8.
- Gutte R, Khopkar U. Unilateral multiple facial angiofibromas: a case report with brief review of literature. Indian J Dermatol. 2013;58:159.
- Silvestre JF, Bañuls J, Ramón R, et al. Unilateral multiple facial angiofibromas: a mosaic form of TSC. J Am Acad Dermatol. 2000;43(1, pt 1):127-129.
- Kozlowski P, Roberts P, Dabora S, et al. Identification of 54 large deletions/duplications in TSC1 and TSC2 using MLPA, and genotype-phenotype correlations. Hum Genet. 2007;121:389-400.
- Kwiatkowska J, Wigowska-Sowinska J, Napierala D, et al. Mosaicism in TSC as a potential cause of the failure of molecular diagnosis. N Engl J Med. 1999;340:703-707.
The Diagnosis: Mosaic Tuberous Sclerosis
A punch biopsy of the facial lesion was stained with hematoxylin and eosin, which demonstrated spindled and stellate fibroblasts with dilated blood vessels (Figure), consistent with an angiofibroma. Given the clinical presentation and histologic findings, there was concern for a diagnosis of tuberous sclerosis (TSC). The patient was referred for genetic workup but tested negative for mutations of the TSC genes in the blood. Because the patient had only unilateral facial lesions, a possible cortical tuber, no other symptoms, and tested negative for TSC gene mutations, mosaic TSC was considered a likely diagnosis. Her facial lesions were treated with pulsed dye vascular laser therapy.
Tuberous sclerosis is an autosomal-dominant neurocutaneous disorder caused by inactivation of the genes TSC1 (encoding hamartin) and TSC2 (encoding tuberin). Mutation results in overactivation of the downstream mTOR (mammalian target of rapamycin) pathway, resulting in abnormal cellular proliferation and hamartomas. These benign tumors can be found in nearly every organ, most often in the central nervous system and skin, and they provide for a highly variable presentation of the disease.1
Tuberous sclerosis affects 1 in 6000 to 10,000 live births and has a prevalence of 1 in 20,000 individuals. Of these individuals, 75% carry sporadic mutations, and 75% to 90% eventually test positive for a TSC gene mutation.2 Genetic mosaicism has been reported in 28% of cases affected by large deletions1 and as few as 1% of cases involving small mutations.3
The dermatologic manifestation of mosaic TSC most often includes unilateral angiofibromas, whereas in nonmosaic cases, angiofibromas cover both cheeks, the forehead, and the eyelids. The other skin lesions of TSC--shagreen patches, forehead plaques, hypomelanotic macules, and ungual fibromas--are seen less frequently.4-6 Additionally, neurologic disease in mosaic patients is notably milder, with 57% of mosaic patients found to have epilepsy compared to 91% of nonmosaic patients.7 Our patient had both unilateral facial angiofibromas and a cortical lesion suspicious for a tuber, prompting a suspected diagnosis of mosaic TSC.
The methods of diagnosis outlined by the International Tuberous Sclerosis Complex Consensus Group pose a challenge in diagnosing mosaic TSC. The clinical criteria require 2 major (eg, multiple angiofibromas, angiomyolipomas, a shagreen patch) and 1 minor feature (eg, dental enamel pit, renal cyst).2 However, case reports detailing unilateral facial angiofibromas have described patients with isolated dermatologic findings.5,6 Further, it has been demonstrated that genetic studies in mosaic TSC can be unreliable depending on the tissue sampled.8 Thus, for patients who have mosaic TSC, establishing a definitive diagnosis is not always possible and may rely solely on the clinical picture.
Considering the differential diagnosis, benign cephalic histiocytosis usually would present with small red-brown macules and papules symmetrically located on the head and neck. The lesions occur at a younger age, usually in the first year or two of life. Fibrofolliculomas present as multiple whitish, slightly larger papules found on the central face. They are a marker for Birt-Hogg-Dubé syndrome, which also is associated with spontaneous pneumothorax.
Agminated means clustering or grouping of lesions. Agminated melanocytic nevi and agminated Spitz nevi are clusters of nevi. These lesions can vary in size and color. They may be elevated or flat. Melanocytic nevi usually are tan-brown or black. Spitz nevi may be pink or pigmented brown or black. To definitively differentiate between these 2 diagnoses and this patient's diagnosis of angiofibroma, a biopsy is needed.
The Diagnosis: Mosaic Tuberous Sclerosis
A punch biopsy of the facial lesion was stained with hematoxylin and eosin, which demonstrated spindled and stellate fibroblasts with dilated blood vessels (Figure), consistent with an angiofibroma. Given the clinical presentation and histologic findings, there was concern for a diagnosis of tuberous sclerosis (TSC). The patient was referred for genetic workup but tested negative for mutations of the TSC genes in the blood. Because the patient had only unilateral facial lesions, a possible cortical tuber, no other symptoms, and tested negative for TSC gene mutations, mosaic TSC was considered a likely diagnosis. Her facial lesions were treated with pulsed dye vascular laser therapy.
Tuberous sclerosis is an autosomal-dominant neurocutaneous disorder caused by inactivation of the genes TSC1 (encoding hamartin) and TSC2 (encoding tuberin). Mutation results in overactivation of the downstream mTOR (mammalian target of rapamycin) pathway, resulting in abnormal cellular proliferation and hamartomas. These benign tumors can be found in nearly every organ, most often in the central nervous system and skin, and they provide for a highly variable presentation of the disease.1
Tuberous sclerosis affects 1 in 6000 to 10,000 live births and has a prevalence of 1 in 20,000 individuals. Of these individuals, 75% carry sporadic mutations, and 75% to 90% eventually test positive for a TSC gene mutation.2 Genetic mosaicism has been reported in 28% of cases affected by large deletions1 and as few as 1% of cases involving small mutations.3
The dermatologic manifestation of mosaic TSC most often includes unilateral angiofibromas, whereas in nonmosaic cases, angiofibromas cover both cheeks, the forehead, and the eyelids. The other skin lesions of TSC--shagreen patches, forehead plaques, hypomelanotic macules, and ungual fibromas--are seen less frequently.4-6 Additionally, neurologic disease in mosaic patients is notably milder, with 57% of mosaic patients found to have epilepsy compared to 91% of nonmosaic patients.7 Our patient had both unilateral facial angiofibromas and a cortical lesion suspicious for a tuber, prompting a suspected diagnosis of mosaic TSC.
The methods of diagnosis outlined by the International Tuberous Sclerosis Complex Consensus Group pose a challenge in diagnosing mosaic TSC. The clinical criteria require 2 major (eg, multiple angiofibromas, angiomyolipomas, a shagreen patch) and 1 minor feature (eg, dental enamel pit, renal cyst).2 However, case reports detailing unilateral facial angiofibromas have described patients with isolated dermatologic findings.5,6 Further, it has been demonstrated that genetic studies in mosaic TSC can be unreliable depending on the tissue sampled.8 Thus, for patients who have mosaic TSC, establishing a definitive diagnosis is not always possible and may rely solely on the clinical picture.
Considering the differential diagnosis, benign cephalic histiocytosis usually would present with small red-brown macules and papules symmetrically located on the head and neck. The lesions occur at a younger age, usually in the first year or two of life. Fibrofolliculomas present as multiple whitish, slightly larger papules found on the central face. They are a marker for Birt-Hogg-Dubé syndrome, which also is associated with spontaneous pneumothorax.
Agminated means clustering or grouping of lesions. Agminated melanocytic nevi and agminated Spitz nevi are clusters of nevi. These lesions can vary in size and color. They may be elevated or flat. Melanocytic nevi usually are tan-brown or black. Spitz nevi may be pink or pigmented brown or black. To definitively differentiate between these 2 diagnoses and this patient's diagnosis of angiofibroma, a biopsy is needed.
- Curatolo P, Moavero R, Roberto D, et al. Genotype/phenotype correlations in tuberous sclerosis complex. Semin Pediatr Neurol. 2015;22:259-273.
- Northrup H, Krueger DA; International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol. 2013;49:243-254.
- Kwiatkowski DJ, Whittemore VH, Thiele EA. Tuberous Sclerosis Complex: Genes, Clinical Features and Therapeutics. Weinham, Germany: Wiley-Blackwell; 2011.
- Alshaiji JM, Spock CR, Connelly EA, et al. Facial angiofibromas in a mosaic pattern tuberous sclerosis: a case report. Dermatol Online J. 2012;18:8.
- Gutte R, Khopkar U. Unilateral multiple facial angiofibromas: a case report with brief review of literature. Indian J Dermatol. 2013;58:159.
- Silvestre JF, Bañuls J, Ramón R, et al. Unilateral multiple facial angiofibromas: a mosaic form of TSC. J Am Acad Dermatol. 2000;43(1, pt 1):127-129.
- Kozlowski P, Roberts P, Dabora S, et al. Identification of 54 large deletions/duplications in TSC1 and TSC2 using MLPA, and genotype-phenotype correlations. Hum Genet. 2007;121:389-400.
- Kwiatkowska J, Wigowska-Sowinska J, Napierala D, et al. Mosaicism in TSC as a potential cause of the failure of molecular diagnosis. N Engl J Med. 1999;340:703-707.
- Curatolo P, Moavero R, Roberto D, et al. Genotype/phenotype correlations in tuberous sclerosis complex. Semin Pediatr Neurol. 2015;22:259-273.
- Northrup H, Krueger DA; International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol. 2013;49:243-254.
- Kwiatkowski DJ, Whittemore VH, Thiele EA. Tuberous Sclerosis Complex: Genes, Clinical Features and Therapeutics. Weinham, Germany: Wiley-Blackwell; 2011.
- Alshaiji JM, Spock CR, Connelly EA, et al. Facial angiofibromas in a mosaic pattern tuberous sclerosis: a case report. Dermatol Online J. 2012;18:8.
- Gutte R, Khopkar U. Unilateral multiple facial angiofibromas: a case report with brief review of literature. Indian J Dermatol. 2013;58:159.
- Silvestre JF, Bañuls J, Ramón R, et al. Unilateral multiple facial angiofibromas: a mosaic form of TSC. J Am Acad Dermatol. 2000;43(1, pt 1):127-129.
- Kozlowski P, Roberts P, Dabora S, et al. Identification of 54 large deletions/duplications in TSC1 and TSC2 using MLPA, and genotype-phenotype correlations. Hum Genet. 2007;121:389-400.
- Kwiatkowska J, Wigowska-Sowinska J, Napierala D, et al. Mosaicism in TSC as a potential cause of the failure of molecular diagnosis. N Engl J Med. 1999;340:703-707.
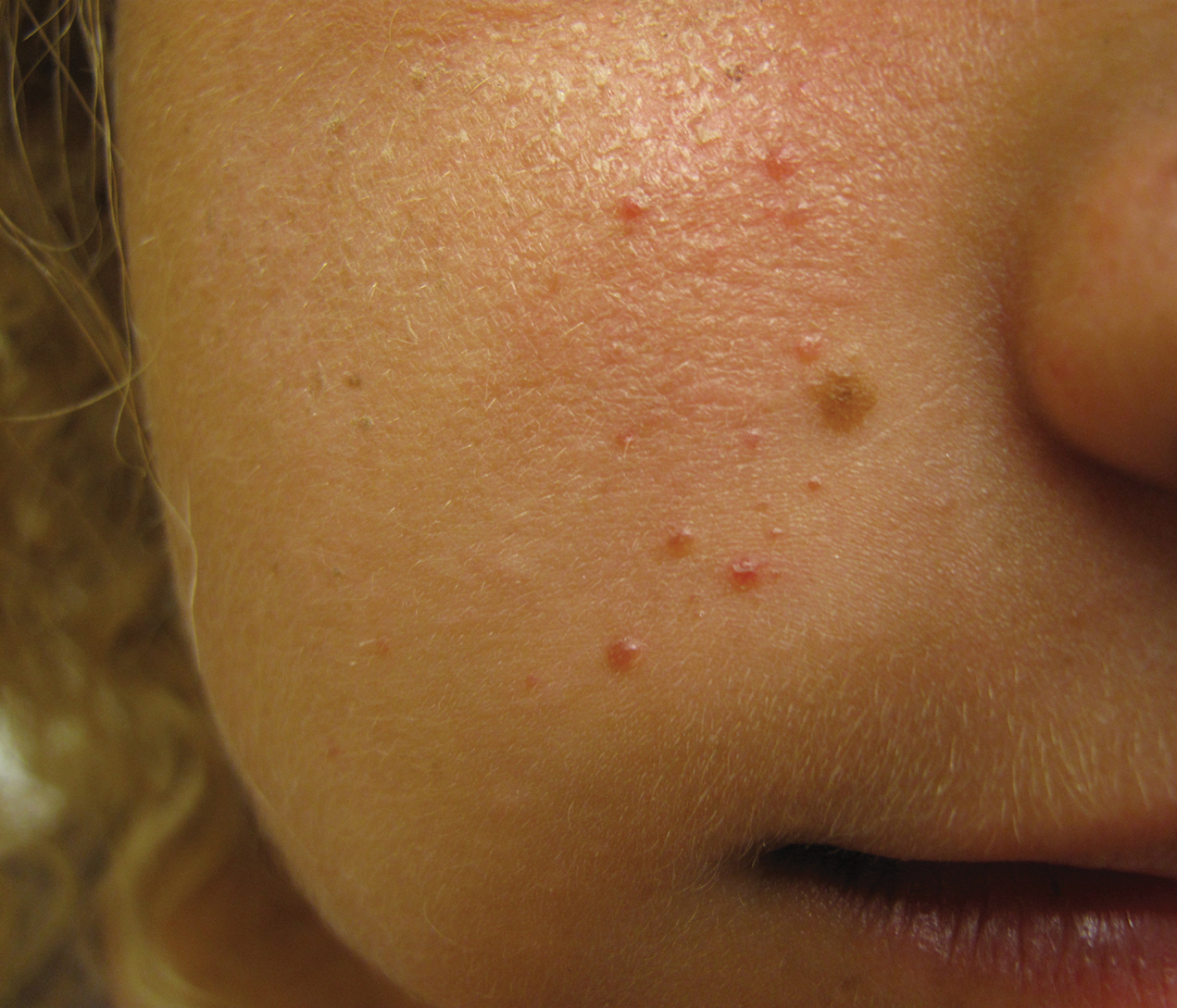
An 18-year-old woman presented with a progressive appearance of firm, red-brown, asymptomatic, 1- to 3-mm, dome-shaped papules on the right cheek that developed over the course of 2 years. She had 10 lesions that covered a 2.2 ×4-cm area on the right medial cheek. No similar-appearing lesions were detectable on a full-body skin examination, and no periungual tumors, café au lait macules, or shagreen patches were noted. A full-body skin examination using a Wood lamp revealed 1 small hypopigmented macule on the right second finger. The patient had a history of treatment-refractory migraines; magnetic resonance imaging 5 years prior to the current presentation revealed a nonspecific lesion in the left parietal gyrus. There was no personal or family history of seizures, cognitive delay, kidney disease, or ocular disease. Punch biopsy of a facial lesion was performed for histopathologic correlation.