User login
Cutis is a peer-reviewed clinical journal for the dermatologist, allergist, and general practitioner published monthly since 1965. Concise clinical articles present the practical side of dermatology, helping physicians to improve patient care. Cutis is referenced in Index Medicus/MEDLINE and is written and edited by industry leaders.
ass lick
assault rifle
balls
ballsac
black jack
bleach
Boko Haram
bondage
causas
cheap
child abuse
cocaine
compulsive behaviors
cost of miracles
cunt
Daech
display network stats
drug paraphernalia
explosion
fart
fda and death
fda AND warn
fda AND warning
fda AND warns
feom
fuck
gambling
gfc
gun
human trafficking
humira AND expensive
illegal
ISIL
ISIS
Islamic caliphate
Islamic state
madvocate
masturbation
mixed martial arts
MMA
molestation
national rifle association
NRA
nsfw
nuccitelli
pedophile
pedophilia
poker
porn
porn
pornography
psychedelic drug
recreational drug
sex slave rings
shit
slot machine
snort
substance abuse
terrorism
terrorist
texarkana
Texas hold 'em
UFC
section[contains(@class, 'nav-hidden')]
section[contains(@class, 'nav-hidden active')
A peer-reviewed, indexed journal for dermatologists with original research, image quizzes, cases and reviews, and columns.
Chondrodermatitis Nodularis Helicis in an Adolescent Boy: Not Just for Old Men
Chondrodermatitis nodularis helicis (CNH) is a chronic painful or crusted, 4- to 6-mm, solitary nodule, primarily on the upper part of the ear (most commonly on the right side). The presence of pain, which increases the likelihood that a person will seek treatment, clinically distinguishes CNH from other cutaneous tumors in the differential diagnosis that produce painless ulceration.
It is roughly 5 times more prevalent in males (72.9%),1 with an average age of onset of 65 years.2 However, CNH has been reported in females3 and rarely in individuals younger than 20 years. According to a PubMed search of articles indexed for MEDLINE and a Google Scholar search using the terms chrondodermatitis nodularis helices child, only 6 cases of CNH have been reported in the pediatric population.4-8 The youngest reported case was a 9-month-old infant.8 Including the present case, males and females in the pediatric population are equally affected; 4 patients had an underlying dermatomyositis,7 rheumatoid nodule,8 or systemic disease, including systemic lupus erythematosus and Beckwith-Wiedemann syndrome.5,9 Chronic intermittent pressure from headwear was the etiologic agent in the remaining cases.4 Recognizing that CNH can occur in young patients and can be associated with underlying autoimmune disease helps direct management and avoid overly invasive treatment.
Case Report
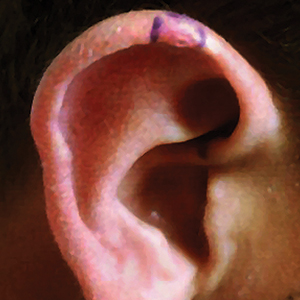
A 17-year-old adolescent boy presented with a painful ulcerated papule on the right upper helix of 3 months’ duration (Figure 1). The patient habitually slept on the right side, pressed a cell phone to that ear, and wore a tight-fitting visor while lifeguarding, which, along with solar damage, all may have contributed to the disease process. He was otherwise in good health, without a history of underlying systemic disease. Given the patient’s extensive occupational sun exposure, biopsy of the lesion was taken under the impression of CNH vs squamous cell carcinoma or basal cell carcinoma.

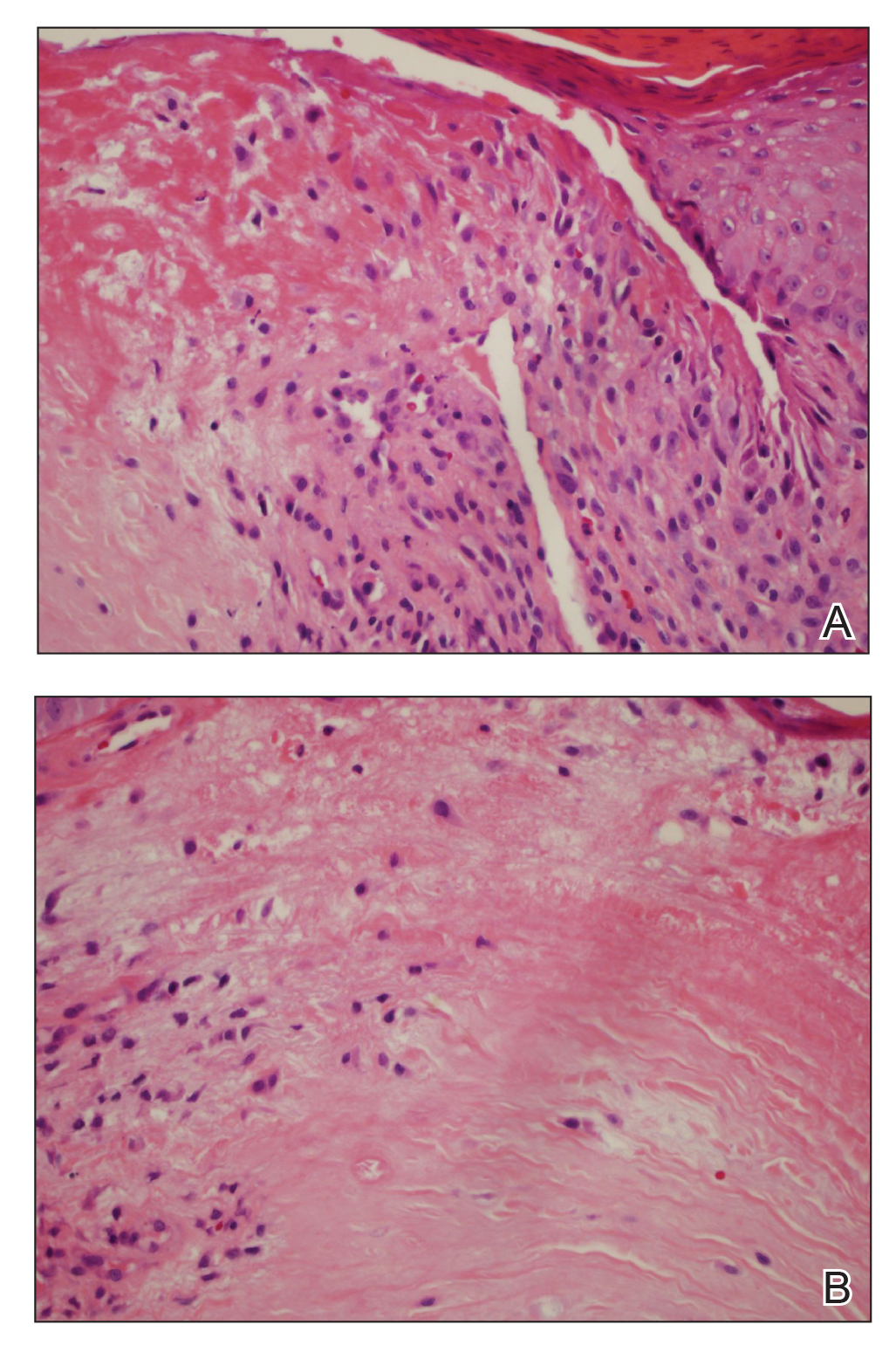
Histopathologic analysis revealed a central area of ulceration with edematous degenerated dermal collagen and overlying inflammatory crust, characteristic of CNH (Figure 2A). Biopsy in this patient demonstrated classic histopathologic findings of CNH, including a central area of epidermal ulceration capped by an inflammatory crust and an underlying edematous degenerated dermal collagen (Figure 2B).
Following biopsy, the patient was advised of this diagnosis and recommended to avoid applying pressure to the area with cell phones or hats or when sleeping to prevent recurrence. At 3-month follow-up, no residual lesion remained.
Comment
Pathogenesis
The exact cause of CNH is unknown but is probably the result of prolonged and excessive pressure on the ear that leads to ischemic injury to cartilage and skin. The external location of CNH, lack of bony support, and exquisitely thin padding or insulation in the form of subcutaneous tissue make the small dermal blood vessels supplying the outer ear vulnerable to compression. Dermal inflammation; edema; and necrosis from trauma, cold, or actinic damage also can help initiate CNH. This disruption of blood perfusion to the external ear also inhibits the ear’s ability to heal. A cycle of pressure from objects such as a pillow or cell phone, followed by inadequate healing, leads to secondary perichondritis and remodeling of perichondrial arterioles, which is demonstrated histologically by the presence of perichondrial fibrous thickening, mild chronic inflammation, collagen degeneration, hyalinization, and rarely necrosis or calcification. Healed lesions often show dermal fibrosis overlying perichondrium.
Repeated pressure can lead to vascular changes, but underlying vascular disease also can predispose a person to CNH at a younger age. A striking case of bilateral CNH was reported in an 8-year-old girl with a known history of dermatomyositis.7 Furthermore, in 24 patients with CNH (mean age, 43 years), Magro et al9 observed an association between CNH and collagen vascular disease, scleroderma, hypertension, thyroid disease, and heart disease, with a higher incidence of any of these medical problems in younger patients. Therefore, screening all patients presenting with CNH, particularly those younger than their fourth decade, for underlying vasculopathy and an autoimmune connective tissue disorder is advised.9
Other findings of CNH reported in the literature include loss of elastic fibers in the central area of degenerated dermal collagen and nerve hyperplasia, which might account for pain.6 Many of the biopsies in cases of CNH reported in the literature also demonstrate perichondrial fibrous thickening, mild chronic inflammation, and degenerative changes in collagen, including hyalinization and rarely necrosis and calcification. Skin at the periphery of the lesion usually contains granulation tissue, with a mild to moderate inflammatory infiltrate and dilated vessels extending beyond the lesion.2
Genetics might play a role in the disorder, which is suggested by the occurrence of CNH in monozygotic twins10 and in an otherwise healthy 16-year-old adolescent girl with CNH of the right ear who screened negative for underlying connective tissue disease—serologic tests included antinuclear antibody, anti-Sm, anti-SCL-70, anti-Ro, anti-La, and rheumatoid factor—but who had a family history of a maternal grandmother with CNH, also on the right side.6
In the present case, there was no family history or signs and symptoms of underlying systemic disease at the time of diagnosis. The social history revealed excessive occupational sun exposure, habitually wearing a tight visor, and frequent cell phone use, all of which might have contributed to CNH.
Management
Medical management is geared toward relieving pressure at the site of the lesion, which was accomplished by use of an off-loading, ring-shaped, foam pillow at night in a 9-month-old girl with CNH, in which the smaller of her 2 left-sided lesions completely resolved by 6-month follow-up.8 However, it often is difficult to achieve adequate relief of pressure because of the patient’s preference for holding a cell phone to a particular ear or unconscious sleeping habits that perpetuate lesions. There are many creative physical interventions to offload aggravating pressure from the area during sleep. A prosthesis can be fashioned by cutting a hole from the center of a bath sponge and securing it with a headband,11 or a crescentic or rectangular piece of self-adhering foam sponge can be applied to the non–hair-bearing postauricular scalp during sleep.12 Topical antibiotics might relieve pain caused by secondary infection.
Surgical intervention, with or without placement of a full-thickness skin graft, is the mainstay of therapy. Excision was performed in 3 previously reported pediatric cases, with no recurrence reported at 6- to 24-month follow-up. Other treatments employed to varying effect include topical and intralesional steroids, collagen injection, cryotherapy, nitroglycerin paste 2% twice daily,13 and electrodesiccation and curettage.14 In adults, if full resolution is desired, multiple surgeries might be required to remove underlying protuberant cartilage; however, this strategy is not without risk of complication, including formation of adjacent cartilaginous nodules that can become site(s) of CNH recurrence due to a change in pressure points.
Conclusion
Although uncommon, CNH can present on the ears of young patients. A causal link between underlying vasculopathy and CNH has yet to be determined, but the association discovered by Magro et al9 merits obtaining a more detailed rheumatologic history and examination, followed by serologic testing (if indicated). Once the diagnosis of CNH is determined, patient education is paramount to prevent recurrence. Increased awareness of habits that inflict persistent repetitive trauma or pressure to the site—from sleeping patterns to cell phone use—will help to extinguish the behavior and therefore the lesion.
- Rex J, Rivera M, Bielsa I, et al. Narrow elliptical skin excision and cartilage shaving for treatment of chondrodermatitis nodularis. Dermatol Surg. 2006;32:400-404.
- Wettlé C, Keller F, Will F, et al. Chondrodermatitis nodularis chronical helicis: a descriptive study of 99 patients [in French]. Ann Dermatol Venereol. 2013;140:687-692.
- Oelzner S, Elsner P. Bilateral chondrodermatitis nodularis chronica helicis on the free border of the helix in a woman. J Am Acad Dermatol. 2003;49:720-722.
- Grigoryants V, Qureshi H, Patterson J, et al. Pediatric chondrodermatitis nodularis helicis. J Craniofac Surg. 2007;18:228-231.
- Fix WC, Cornejo C, Duffy KA, et al. Pediatric chondrodermatitis nodularis helicis (CNH) in a child with Beckwith-Wiedemann syndrome (BWS). Pediatr Dermatol. 2019;36:388-390.
- Rogers NE, Farris PK, Wang AR. Juvenile chondrodermatitis nodularis helicis: case report and literature review. Pediatr Dermatol. 2003;20:488-490.
- Sasaki T, Nishizawa H, Sugita Y. Chondrodermatitis nodularis helicis in childhood dermatomyositis. Br J Dermatol. 1999;141:363-365.
- Tsai TH, Lin YC, Chen HC. Infantile chondrodermatitis nodularis. Pediatr Dermatol. 2007;24:337-339.
- Magro CM, Frambach GE, Crowson AN. Chondrodermatitis nodularis helicis as a marker of internal disease associated with microvascular injury. J Cutan Pathol. 2005;32:329-333.
- Chan HP, Neuhaus IM, Maibach HI. Chondrodermatitis nodularis chronica helicis in monozygotic twins. Clin Exp Dermatol. 2009;34:358-359.
- Moncrieff M, Sassoon EM. Effective treatment of chondrodermatitis nodularis chronica helicis using a conservative approach. Br J Dermatol. 2004;150:892-894.
- Travelute CR. Self-adhering foam: a simple method for pressure relief during sleep in patients with chondrodermatitis nodularis helicis. Dermatol Surg. 2013;39:317-319.
- Flynn V, Chisholm C, Grimwood R. Topical nitroglycerin: a promising treatment option for chondrodermatitis nodularis helicis. J Am Acad Dermatol. 2011;65:531-536.
- Kromann N, Høyer H, Reymann F. Chondrodermatitis nodularis chronica helicis treated with curettage and electrocauterization: follow-up of a 15-year material. Acta Derm Venereol. 1983;63:85-87.
Chondrodermatitis nodularis helicis (CNH) is a chronic painful or crusted, 4- to 6-mm, solitary nodule, primarily on the upper part of the ear (most commonly on the right side). The presence of pain, which increases the likelihood that a person will seek treatment, clinically distinguishes CNH from other cutaneous tumors in the differential diagnosis that produce painless ulceration.
It is roughly 5 times more prevalent in males (72.9%),1 with an average age of onset of 65 years.2 However, CNH has been reported in females3 and rarely in individuals younger than 20 years. According to a PubMed search of articles indexed for MEDLINE and a Google Scholar search using the terms chrondodermatitis nodularis helices child, only 6 cases of CNH have been reported in the pediatric population.4-8 The youngest reported case was a 9-month-old infant.8 Including the present case, males and females in the pediatric population are equally affected; 4 patients had an underlying dermatomyositis,7 rheumatoid nodule,8 or systemic disease, including systemic lupus erythematosus and Beckwith-Wiedemann syndrome.5,9 Chronic intermittent pressure from headwear was the etiologic agent in the remaining cases.4 Recognizing that CNH can occur in young patients and can be associated with underlying autoimmune disease helps direct management and avoid overly invasive treatment.
Case Report
A 17-year-old adolescent boy presented with a painful ulcerated papule on the right upper helix of 3 months’ duration (Figure 1). The patient habitually slept on the right side, pressed a cell phone to that ear, and wore a tight-fitting visor while lifeguarding, which, along with solar damage, all may have contributed to the disease process. He was otherwise in good health, without a history of underlying systemic disease. Given the patient’s extensive occupational sun exposure, biopsy of the lesion was taken under the impression of CNH vs squamous cell carcinoma or basal cell carcinoma.
Histopathologic analysis revealed a central area of ulceration with edematous degenerated dermal collagen and overlying inflammatory crust, characteristic of CNH (Figure 2A). Biopsy in this patient demonstrated classic histopathologic findings of CNH, including a central area of epidermal ulceration capped by an inflammatory crust and an underlying edematous degenerated dermal collagen (Figure 2B).
Following biopsy, the patient was advised of this diagnosis and recommended to avoid applying pressure to the area with cell phones or hats or when sleeping to prevent recurrence. At 3-month follow-up, no residual lesion remained.
Comment
Pathogenesis
The exact cause of CNH is unknown but is probably the result of prolonged and excessive pressure on the ear that leads to ischemic injury to cartilage and skin. The external location of CNH, lack of bony support, and exquisitely thin padding or insulation in the form of subcutaneous tissue make the small dermal blood vessels supplying the outer ear vulnerable to compression. Dermal inflammation; edema; and necrosis from trauma, cold, or actinic damage also can help initiate CNH. This disruption of blood perfusion to the external ear also inhibits the ear’s ability to heal. A cycle of pressure from objects such as a pillow or cell phone, followed by inadequate healing, leads to secondary perichondritis and remodeling of perichondrial arterioles, which is demonstrated histologically by the presence of perichondrial fibrous thickening, mild chronic inflammation, collagen degeneration, hyalinization, and rarely necrosis or calcification. Healed lesions often show dermal fibrosis overlying perichondrium.
Repeated pressure can lead to vascular changes, but underlying vascular disease also can predispose a person to CNH at a younger age. A striking case of bilateral CNH was reported in an 8-year-old girl with a known history of dermatomyositis.7 Furthermore, in 24 patients with CNH (mean age, 43 years), Magro et al9 observed an association between CNH and collagen vascular disease, scleroderma, hypertension, thyroid disease, and heart disease, with a higher incidence of any of these medical problems in younger patients. Therefore, screening all patients presenting with CNH, particularly those younger than their fourth decade, for underlying vasculopathy and an autoimmune connective tissue disorder is advised.9
Other findings of CNH reported in the literature include loss of elastic fibers in the central area of degenerated dermal collagen and nerve hyperplasia, which might account for pain.6 Many of the biopsies in cases of CNH reported in the literature also demonstrate perichondrial fibrous thickening, mild chronic inflammation, and degenerative changes in collagen, including hyalinization and rarely necrosis and calcification. Skin at the periphery of the lesion usually contains granulation tissue, with a mild to moderate inflammatory infiltrate and dilated vessels extending beyond the lesion.2
Genetics might play a role in the disorder, which is suggested by the occurrence of CNH in monozygotic twins10 and in an otherwise healthy 16-year-old adolescent girl with CNH of the right ear who screened negative for underlying connective tissue disease—serologic tests included antinuclear antibody, anti-Sm, anti-SCL-70, anti-Ro, anti-La, and rheumatoid factor—but who had a family history of a maternal grandmother with CNH, also on the right side.6
In the present case, there was no family history or signs and symptoms of underlying systemic disease at the time of diagnosis. The social history revealed excessive occupational sun exposure, habitually wearing a tight visor, and frequent cell phone use, all of which might have contributed to CNH.
Management
Medical management is geared toward relieving pressure at the site of the lesion, which was accomplished by use of an off-loading, ring-shaped, foam pillow at night in a 9-month-old girl with CNH, in which the smaller of her 2 left-sided lesions completely resolved by 6-month follow-up.8 However, it often is difficult to achieve adequate relief of pressure because of the patient’s preference for holding a cell phone to a particular ear or unconscious sleeping habits that perpetuate lesions. There are many creative physical interventions to offload aggravating pressure from the area during sleep. A prosthesis can be fashioned by cutting a hole from the center of a bath sponge and securing it with a headband,11 or a crescentic or rectangular piece of self-adhering foam sponge can be applied to the non–hair-bearing postauricular scalp during sleep.12 Topical antibiotics might relieve pain caused by secondary infection.
Surgical intervention, with or without placement of a full-thickness skin graft, is the mainstay of therapy. Excision was performed in 3 previously reported pediatric cases, with no recurrence reported at 6- to 24-month follow-up. Other treatments employed to varying effect include topical and intralesional steroids, collagen injection, cryotherapy, nitroglycerin paste 2% twice daily,13 and electrodesiccation and curettage.14 In adults, if full resolution is desired, multiple surgeries might be required to remove underlying protuberant cartilage; however, this strategy is not without risk of complication, including formation of adjacent cartilaginous nodules that can become site(s) of CNH recurrence due to a change in pressure points.
Conclusion
Although uncommon, CNH can present on the ears of young patients. A causal link between underlying vasculopathy and CNH has yet to be determined, but the association discovered by Magro et al9 merits obtaining a more detailed rheumatologic history and examination, followed by serologic testing (if indicated). Once the diagnosis of CNH is determined, patient education is paramount to prevent recurrence. Increased awareness of habits that inflict persistent repetitive trauma or pressure to the site—from sleeping patterns to cell phone use—will help to extinguish the behavior and therefore the lesion.
Chondrodermatitis nodularis helicis (CNH) is a chronic painful or crusted, 4- to 6-mm, solitary nodule, primarily on the upper part of the ear (most commonly on the right side). The presence of pain, which increases the likelihood that a person will seek treatment, clinically distinguishes CNH from other cutaneous tumors in the differential diagnosis that produce painless ulceration.
It is roughly 5 times more prevalent in males (72.9%),1 with an average age of onset of 65 years.2 However, CNH has been reported in females3 and rarely in individuals younger than 20 years. According to a PubMed search of articles indexed for MEDLINE and a Google Scholar search using the terms chrondodermatitis nodularis helices child, only 6 cases of CNH have been reported in the pediatric population.4-8 The youngest reported case was a 9-month-old infant.8 Including the present case, males and females in the pediatric population are equally affected; 4 patients had an underlying dermatomyositis,7 rheumatoid nodule,8 or systemic disease, including systemic lupus erythematosus and Beckwith-Wiedemann syndrome.5,9 Chronic intermittent pressure from headwear was the etiologic agent in the remaining cases.4 Recognizing that CNH can occur in young patients and can be associated with underlying autoimmune disease helps direct management and avoid overly invasive treatment.
Case Report
A 17-year-old adolescent boy presented with a painful ulcerated papule on the right upper helix of 3 months’ duration (Figure 1). The patient habitually slept on the right side, pressed a cell phone to that ear, and wore a tight-fitting visor while lifeguarding, which, along with solar damage, all may have contributed to the disease process. He was otherwise in good health, without a history of underlying systemic disease. Given the patient’s extensive occupational sun exposure, biopsy of the lesion was taken under the impression of CNH vs squamous cell carcinoma or basal cell carcinoma.
Histopathologic analysis revealed a central area of ulceration with edematous degenerated dermal collagen and overlying inflammatory crust, characteristic of CNH (Figure 2A). Biopsy in this patient demonstrated classic histopathologic findings of CNH, including a central area of epidermal ulceration capped by an inflammatory crust and an underlying edematous degenerated dermal collagen (Figure 2B).
Following biopsy, the patient was advised of this diagnosis and recommended to avoid applying pressure to the area with cell phones or hats or when sleeping to prevent recurrence. At 3-month follow-up, no residual lesion remained.
Comment
Pathogenesis
The exact cause of CNH is unknown but is probably the result of prolonged and excessive pressure on the ear that leads to ischemic injury to cartilage and skin. The external location of CNH, lack of bony support, and exquisitely thin padding or insulation in the form of subcutaneous tissue make the small dermal blood vessels supplying the outer ear vulnerable to compression. Dermal inflammation; edema; and necrosis from trauma, cold, or actinic damage also can help initiate CNH. This disruption of blood perfusion to the external ear also inhibits the ear’s ability to heal. A cycle of pressure from objects such as a pillow or cell phone, followed by inadequate healing, leads to secondary perichondritis and remodeling of perichondrial arterioles, which is demonstrated histologically by the presence of perichondrial fibrous thickening, mild chronic inflammation, collagen degeneration, hyalinization, and rarely necrosis or calcification. Healed lesions often show dermal fibrosis overlying perichondrium.
Repeated pressure can lead to vascular changes, but underlying vascular disease also can predispose a person to CNH at a younger age. A striking case of bilateral CNH was reported in an 8-year-old girl with a known history of dermatomyositis.7 Furthermore, in 24 patients with CNH (mean age, 43 years), Magro et al9 observed an association between CNH and collagen vascular disease, scleroderma, hypertension, thyroid disease, and heart disease, with a higher incidence of any of these medical problems in younger patients. Therefore, screening all patients presenting with CNH, particularly those younger than their fourth decade, for underlying vasculopathy and an autoimmune connective tissue disorder is advised.9
Other findings of CNH reported in the literature include loss of elastic fibers in the central area of degenerated dermal collagen and nerve hyperplasia, which might account for pain.6 Many of the biopsies in cases of CNH reported in the literature also demonstrate perichondrial fibrous thickening, mild chronic inflammation, and degenerative changes in collagen, including hyalinization and rarely necrosis and calcification. Skin at the periphery of the lesion usually contains granulation tissue, with a mild to moderate inflammatory infiltrate and dilated vessels extending beyond the lesion.2
Genetics might play a role in the disorder, which is suggested by the occurrence of CNH in monozygotic twins10 and in an otherwise healthy 16-year-old adolescent girl with CNH of the right ear who screened negative for underlying connective tissue disease—serologic tests included antinuclear antibody, anti-Sm, anti-SCL-70, anti-Ro, anti-La, and rheumatoid factor—but who had a family history of a maternal grandmother with CNH, also on the right side.6
In the present case, there was no family history or signs and symptoms of underlying systemic disease at the time of diagnosis. The social history revealed excessive occupational sun exposure, habitually wearing a tight visor, and frequent cell phone use, all of which might have contributed to CNH.
Management
Medical management is geared toward relieving pressure at the site of the lesion, which was accomplished by use of an off-loading, ring-shaped, foam pillow at night in a 9-month-old girl with CNH, in which the smaller of her 2 left-sided lesions completely resolved by 6-month follow-up.8 However, it often is difficult to achieve adequate relief of pressure because of the patient’s preference for holding a cell phone to a particular ear or unconscious sleeping habits that perpetuate lesions. There are many creative physical interventions to offload aggravating pressure from the area during sleep. A prosthesis can be fashioned by cutting a hole from the center of a bath sponge and securing it with a headband,11 or a crescentic or rectangular piece of self-adhering foam sponge can be applied to the non–hair-bearing postauricular scalp during sleep.12 Topical antibiotics might relieve pain caused by secondary infection.
Surgical intervention, with or without placement of a full-thickness skin graft, is the mainstay of therapy. Excision was performed in 3 previously reported pediatric cases, with no recurrence reported at 6- to 24-month follow-up. Other treatments employed to varying effect include topical and intralesional steroids, collagen injection, cryotherapy, nitroglycerin paste 2% twice daily,13 and electrodesiccation and curettage.14 In adults, if full resolution is desired, multiple surgeries might be required to remove underlying protuberant cartilage; however, this strategy is not without risk of complication, including formation of adjacent cartilaginous nodules that can become site(s) of CNH recurrence due to a change in pressure points.
Conclusion
Although uncommon, CNH can present on the ears of young patients. A causal link between underlying vasculopathy and CNH has yet to be determined, but the association discovered by Magro et al9 merits obtaining a more detailed rheumatologic history and examination, followed by serologic testing (if indicated). Once the diagnosis of CNH is determined, patient education is paramount to prevent recurrence. Increased awareness of habits that inflict persistent repetitive trauma or pressure to the site—from sleeping patterns to cell phone use—will help to extinguish the behavior and therefore the lesion.
- Rex J, Rivera M, Bielsa I, et al. Narrow elliptical skin excision and cartilage shaving for treatment of chondrodermatitis nodularis. Dermatol Surg. 2006;32:400-404.
- Wettlé C, Keller F, Will F, et al. Chondrodermatitis nodularis chronical helicis: a descriptive study of 99 patients [in French]. Ann Dermatol Venereol. 2013;140:687-692.
- Oelzner S, Elsner P. Bilateral chondrodermatitis nodularis chronica helicis on the free border of the helix in a woman. J Am Acad Dermatol. 2003;49:720-722.
- Grigoryants V, Qureshi H, Patterson J, et al. Pediatric chondrodermatitis nodularis helicis. J Craniofac Surg. 2007;18:228-231.
- Fix WC, Cornejo C, Duffy KA, et al. Pediatric chondrodermatitis nodularis helicis (CNH) in a child with Beckwith-Wiedemann syndrome (BWS). Pediatr Dermatol. 2019;36:388-390.
- Rogers NE, Farris PK, Wang AR. Juvenile chondrodermatitis nodularis helicis: case report and literature review. Pediatr Dermatol. 2003;20:488-490.
- Sasaki T, Nishizawa H, Sugita Y. Chondrodermatitis nodularis helicis in childhood dermatomyositis. Br J Dermatol. 1999;141:363-365.
- Tsai TH, Lin YC, Chen HC. Infantile chondrodermatitis nodularis. Pediatr Dermatol. 2007;24:337-339.
- Magro CM, Frambach GE, Crowson AN. Chondrodermatitis nodularis helicis as a marker of internal disease associated with microvascular injury. J Cutan Pathol. 2005;32:329-333.
- Chan HP, Neuhaus IM, Maibach HI. Chondrodermatitis nodularis chronica helicis in monozygotic twins. Clin Exp Dermatol. 2009;34:358-359.
- Moncrieff M, Sassoon EM. Effective treatment of chondrodermatitis nodularis chronica helicis using a conservative approach. Br J Dermatol. 2004;150:892-894.
- Travelute CR. Self-adhering foam: a simple method for pressure relief during sleep in patients with chondrodermatitis nodularis helicis. Dermatol Surg. 2013;39:317-319.
- Flynn V, Chisholm C, Grimwood R. Topical nitroglycerin: a promising treatment option for chondrodermatitis nodularis helicis. J Am Acad Dermatol. 2011;65:531-536.
- Kromann N, Høyer H, Reymann F. Chondrodermatitis nodularis chronica helicis treated with curettage and electrocauterization: follow-up of a 15-year material. Acta Derm Venereol. 1983;63:85-87.
- Rex J, Rivera M, Bielsa I, et al. Narrow elliptical skin excision and cartilage shaving for treatment of chondrodermatitis nodularis. Dermatol Surg. 2006;32:400-404.
- Wettlé C, Keller F, Will F, et al. Chondrodermatitis nodularis chronical helicis: a descriptive study of 99 patients [in French]. Ann Dermatol Venereol. 2013;140:687-692.
- Oelzner S, Elsner P. Bilateral chondrodermatitis nodularis chronica helicis on the free border of the helix in a woman. J Am Acad Dermatol. 2003;49:720-722.
- Grigoryants V, Qureshi H, Patterson J, et al. Pediatric chondrodermatitis nodularis helicis. J Craniofac Surg. 2007;18:228-231.
- Fix WC, Cornejo C, Duffy KA, et al. Pediatric chondrodermatitis nodularis helicis (CNH) in a child with Beckwith-Wiedemann syndrome (BWS). Pediatr Dermatol. 2019;36:388-390.
- Rogers NE, Farris PK, Wang AR. Juvenile chondrodermatitis nodularis helicis: case report and literature review. Pediatr Dermatol. 2003;20:488-490.
- Sasaki T, Nishizawa H, Sugita Y. Chondrodermatitis nodularis helicis in childhood dermatomyositis. Br J Dermatol. 1999;141:363-365.
- Tsai TH, Lin YC, Chen HC. Infantile chondrodermatitis nodularis. Pediatr Dermatol. 2007;24:337-339.
- Magro CM, Frambach GE, Crowson AN. Chondrodermatitis nodularis helicis as a marker of internal disease associated with microvascular injury. J Cutan Pathol. 2005;32:329-333.
- Chan HP, Neuhaus IM, Maibach HI. Chondrodermatitis nodularis chronica helicis in monozygotic twins. Clin Exp Dermatol. 2009;34:358-359.
- Moncrieff M, Sassoon EM. Effective treatment of chondrodermatitis nodularis chronica helicis using a conservative approach. Br J Dermatol. 2004;150:892-894.
- Travelute CR. Self-adhering foam: a simple method for pressure relief during sleep in patients with chondrodermatitis nodularis helicis. Dermatol Surg. 2013;39:317-319.
- Flynn V, Chisholm C, Grimwood R. Topical nitroglycerin: a promising treatment option for chondrodermatitis nodularis helicis. J Am Acad Dermatol. 2011;65:531-536.
- Kromann N, Høyer H, Reymann F. Chondrodermatitis nodularis chronica helicis treated with curettage and electrocauterization: follow-up of a 15-year material. Acta Derm Venereol. 1983;63:85-87.
Practice Points
- Chondrodermatitis nodularis helicis should be in the differential for nodular lesions on the ears of adolescents, as societal shifts in behavior have altered the epidemiology of this condition such that it is no longer exclusive to the geriatric population.
- Make sure to get a thorough history of potential pressure triggers when evaluating nodules on the ears of adolescents.
Product News November 2019
Aklief Cream Topical Retinoid Approved for Acne Vulgaris
Galderma Laboratories, LP, announces US Food and Drug Administration approval of Aklief (trifarotene) Cream 0.005% for the treatment of acne vulgaris in patients 9 years and older. Trifarotene is a retinoid that selectively targets retinoic acid receptor γ. Aklief Cream treats both facial and truncal acne. Aklief Cream is expected to be available in the United States in November 2019 in a 45-g pump. For more information, visit www.galderma.com.
Altreno Lotion Now Available in a 20-g Tube for Dermatologist Dispensing
Ortho Dermatologics launches a 20-g tube of Altreno (tretinoin) Lotion 0.05% for dermatologists to dispense in their offices. Offering the product in the physician’s office helps ensure that patients will be ready to begin their acne regimen, increasing patient compliance. Altreno Lotion is approved for the treatment of acne vulgaris in patients 9 years and older. It provides efficacy and tolerability in a once-daily dosing regimen. For more information, visit www.altrenohcp.com.
Amzeeq Topical Minocycline Approved for Acne
Foamix Pharmaceuticals Ltd receives US Food and Drug Administration approval of Amzeeq (minocycline) Foam 4% for the treatment of moderate to severe acne vulgaris in patients 9 years and older. Foamix’s proprietary Molecule Stabilizing Technology is used to effectively deliver minocycline—a broad-spectrum antibiotic—in a foam-based vehicle for once-daily application. Amzeeq is expected to be available for prescribing in January 2020. For more information, visit www.foamix.com.
FDA Clears Protego Antimicrobial Wound Dressing
Turn Therapeutics, Inc, receives US Food and Drug Administration clearance of Protego antimicrobial wound dressing for acute and chronic wound management. Protego wound dressings are single-use, sterile, antimicrobial gauze dressings impregnated with Hexagen, a proprietary petrolatum-based wound care emulsion. Protego offers patients the utility of traditional petrolatum-saturated gauze dressings with the added benefit of broad-spectrum antimicrobial protection against bacteria, fungi, and yeasts. For more information, visit www.turntherapeutics.com.
Skin Cancer Foundation Champions for Change Gala Raises More Than $700,000
The Skin Cancer Foundation held its 23rd annual Champions for Change Gala on October 17, 2019. The foundation’s signature fundraising event raised more than $700,000 to support educational campaigns, community programs, and research initiatives. More than 400 guests attended the event at The Plaza Hotel in New York, New York. The event was emceed by comedian Tom Kelly, and President Dr. Deborah S. Sarnoff reflected on the 40th birthday of the foundation, reinforcing the goal “to change behaviors and save lives.” For more information, visit www.
If you would like your product included in Product News, please email a press release to the Editorial Office at cutis@mdedge.com.
Aklief Cream Topical Retinoid Approved for Acne Vulgaris
Galderma Laboratories, LP, announces US Food and Drug Administration approval of Aklief (trifarotene) Cream 0.005% for the treatment of acne vulgaris in patients 9 years and older. Trifarotene is a retinoid that selectively targets retinoic acid receptor γ. Aklief Cream treats both facial and truncal acne. Aklief Cream is expected to be available in the United States in November 2019 in a 45-g pump. For more information, visit www.galderma.com.
Altreno Lotion Now Available in a 20-g Tube for Dermatologist Dispensing
Ortho Dermatologics launches a 20-g tube of Altreno (tretinoin) Lotion 0.05% for dermatologists to dispense in their offices. Offering the product in the physician’s office helps ensure that patients will be ready to begin their acne regimen, increasing patient compliance. Altreno Lotion is approved for the treatment of acne vulgaris in patients 9 years and older. It provides efficacy and tolerability in a once-daily dosing regimen. For more information, visit www.altrenohcp.com.
Amzeeq Topical Minocycline Approved for Acne
Foamix Pharmaceuticals Ltd receives US Food and Drug Administration approval of Amzeeq (minocycline) Foam 4% for the treatment of moderate to severe acne vulgaris in patients 9 years and older. Foamix’s proprietary Molecule Stabilizing Technology is used to effectively deliver minocycline—a broad-spectrum antibiotic—in a foam-based vehicle for once-daily application. Amzeeq is expected to be available for prescribing in January 2020. For more information, visit www.foamix.com.
FDA Clears Protego Antimicrobial Wound Dressing
Turn Therapeutics, Inc, receives US Food and Drug Administration clearance of Protego antimicrobial wound dressing for acute and chronic wound management. Protego wound dressings are single-use, sterile, antimicrobial gauze dressings impregnated with Hexagen, a proprietary petrolatum-based wound care emulsion. Protego offers patients the utility of traditional petrolatum-saturated gauze dressings with the added benefit of broad-spectrum antimicrobial protection against bacteria, fungi, and yeasts. For more information, visit www.turntherapeutics.com.
Skin Cancer Foundation Champions for Change Gala Raises More Than $700,000
The Skin Cancer Foundation held its 23rd annual Champions for Change Gala on October 17, 2019. The foundation’s signature fundraising event raised more than $700,000 to support educational campaigns, community programs, and research initiatives. More than 400 guests attended the event at The Plaza Hotel in New York, New York. The event was emceed by comedian Tom Kelly, and President Dr. Deborah S. Sarnoff reflected on the 40th birthday of the foundation, reinforcing the goal “to change behaviors and save lives.” For more information, visit www.
If you would like your product included in Product News, please email a press release to the Editorial Office at cutis@mdedge.com.
Aklief Cream Topical Retinoid Approved for Acne Vulgaris
Galderma Laboratories, LP, announces US Food and Drug Administration approval of Aklief (trifarotene) Cream 0.005% for the treatment of acne vulgaris in patients 9 years and older. Trifarotene is a retinoid that selectively targets retinoic acid receptor γ. Aklief Cream treats both facial and truncal acne. Aklief Cream is expected to be available in the United States in November 2019 in a 45-g pump. For more information, visit www.galderma.com.
Altreno Lotion Now Available in a 20-g Tube for Dermatologist Dispensing
Ortho Dermatologics launches a 20-g tube of Altreno (tretinoin) Lotion 0.05% for dermatologists to dispense in their offices. Offering the product in the physician’s office helps ensure that patients will be ready to begin their acne regimen, increasing patient compliance. Altreno Lotion is approved for the treatment of acne vulgaris in patients 9 years and older. It provides efficacy and tolerability in a once-daily dosing regimen. For more information, visit www.altrenohcp.com.
Amzeeq Topical Minocycline Approved for Acne
Foamix Pharmaceuticals Ltd receives US Food and Drug Administration approval of Amzeeq (minocycline) Foam 4% for the treatment of moderate to severe acne vulgaris in patients 9 years and older. Foamix’s proprietary Molecule Stabilizing Technology is used to effectively deliver minocycline—a broad-spectrum antibiotic—in a foam-based vehicle for once-daily application. Amzeeq is expected to be available for prescribing in January 2020. For more information, visit www.foamix.com.
FDA Clears Protego Antimicrobial Wound Dressing
Turn Therapeutics, Inc, receives US Food and Drug Administration clearance of Protego antimicrobial wound dressing for acute and chronic wound management. Protego wound dressings are single-use, sterile, antimicrobial gauze dressings impregnated with Hexagen, a proprietary petrolatum-based wound care emulsion. Protego offers patients the utility of traditional petrolatum-saturated gauze dressings with the added benefit of broad-spectrum antimicrobial protection against bacteria, fungi, and yeasts. For more information, visit www.turntherapeutics.com.
Skin Cancer Foundation Champions for Change Gala Raises More Than $700,000
The Skin Cancer Foundation held its 23rd annual Champions for Change Gala on October 17, 2019. The foundation’s signature fundraising event raised more than $700,000 to support educational campaigns, community programs, and research initiatives. More than 400 guests attended the event at The Plaza Hotel in New York, New York. The event was emceed by comedian Tom Kelly, and President Dr. Deborah S. Sarnoff reflected on the 40th birthday of the foundation, reinforcing the goal “to change behaviors and save lives.” For more information, visit www.
If you would like your product included in Product News, please email a press release to the Editorial Office at cutis@mdedge.com.
Yellow-Brown Ulcerated Papule in a Newborn
The Diagnosis: Congenital Self-healing Reticulohistiocytosis
Biopsy of a representative lesion from this patient was consistent with congenital self-healing reticulohistiocytosis, as shown in the Figure. Characteristic Langerhans cells were present in the dermis that stained CD1a positive, S-100 positive, and CD68 negative to confirm the diagnosis.
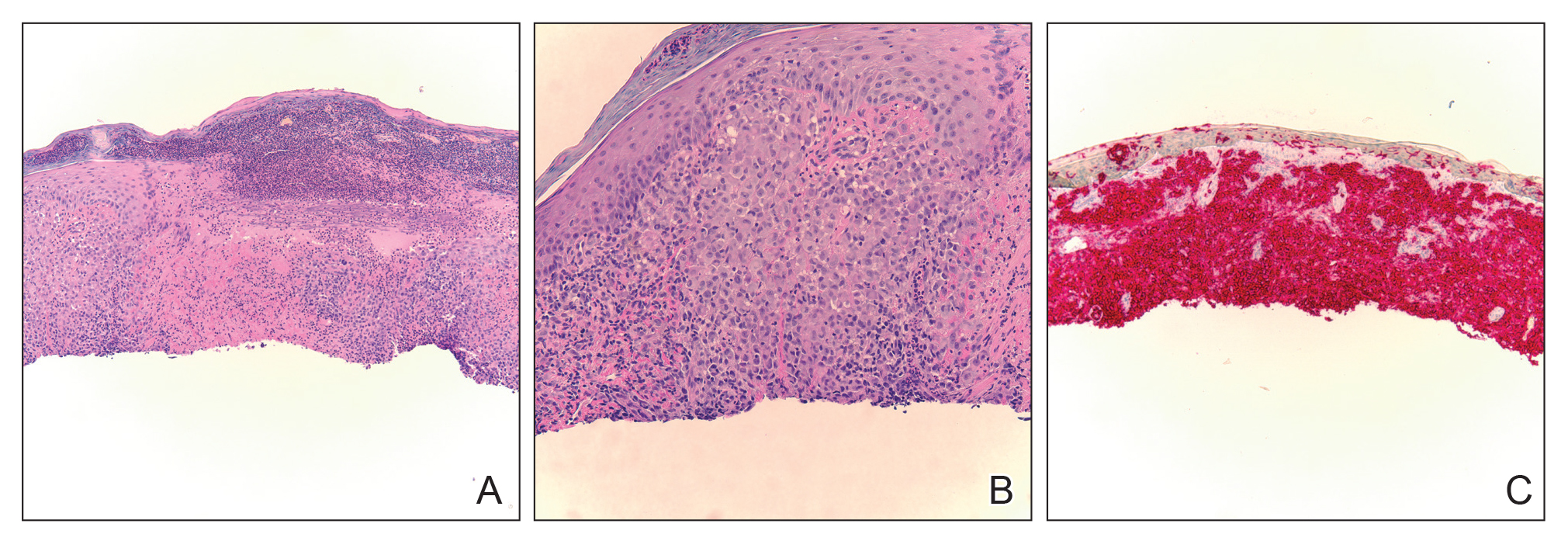
Congenital self-healing reticulohistiocytosis, or Hashimoto-Pritzker syndrome, is a rare benign form of Langerhans cell histiocytosis. It is twice as common in males than females and typically noted at birth or early during the neonatal period. Lesions may present as pink, firm, asymptomatic papulonodular lesions that often ulcerate with possible residual hypopigmentation or hyperpigmentation.1 The differential diagnosis includes congenital infectious and hematologic diseases typically associated with blueberry muffin baby. Thus, varicella, cytomegalovirus, syphilis, toxoplasmosis, rubella, neuroblastoma, leukemia cutis, and extramedullary hematopoiesis, among others, may be considered. Juvenile xanthogranuloma or urticaria pigmentosa also enter the differential diagnosis given the yellow-brown appearance. As a clonal proliferation of Langerhans cells, pathology reveals lesions that stain positive for CD1a and S-100.2
Although typically absent, evaluation for systemic involvement is warranted, which may be an early presentation of multisystem Langerhans cell histiocytosis. Continued monitoring is recommended given the risk of relapse and associated mortality. Our patient continues to do well. He will continue to be followed by our team and hematology/oncology during early childhood.
The treatment of congenital self-healing reticulohistiocytosis may include conservative monitoring, topical steroids, topical nitrogen mustard, tacrolimus, or psoralen plus UVA.3 Surgical excision may be considered for large lesions.
- Parimi LR, You J, Hong L, et al. Congenital self-healing reticulohistiocytosis with spontaneous regression. An Bras Dermatol. 2017;92:553-555.
- Chen AJ, Jarrett P, Macfarlane S. Congenital self-healing reticulohistiocytosis: the need for investigation. Australas J Dermatol. 2016;57:76-77.
- Gothwal S, Gupta AK, Choudhary R. Congenital self healing Langerhans cell histiocytosis. Indian J Pediatr. 2018;85:316-317.
The Diagnosis: Congenital Self-healing Reticulohistiocytosis
Biopsy of a representative lesion from this patient was consistent with congenital self-healing reticulohistiocytosis, as shown in the Figure. Characteristic Langerhans cells were present in the dermis that stained CD1a positive, S-100 positive, and CD68 negative to confirm the diagnosis.
Congenital self-healing reticulohistiocytosis, or Hashimoto-Pritzker syndrome, is a rare benign form of Langerhans cell histiocytosis. It is twice as common in males than females and typically noted at birth or early during the neonatal period. Lesions may present as pink, firm, asymptomatic papulonodular lesions that often ulcerate with possible residual hypopigmentation or hyperpigmentation.1 The differential diagnosis includes congenital infectious and hematologic diseases typically associated with blueberry muffin baby. Thus, varicella, cytomegalovirus, syphilis, toxoplasmosis, rubella, neuroblastoma, leukemia cutis, and extramedullary hematopoiesis, among others, may be considered. Juvenile xanthogranuloma or urticaria pigmentosa also enter the differential diagnosis given the yellow-brown appearance. As a clonal proliferation of Langerhans cells, pathology reveals lesions that stain positive for CD1a and S-100.2
Although typically absent, evaluation for systemic involvement is warranted, which may be an early presentation of multisystem Langerhans cell histiocytosis. Continued monitoring is recommended given the risk of relapse and associated mortality. Our patient continues to do well. He will continue to be followed by our team and hematology/oncology during early childhood.
The treatment of congenital self-healing reticulohistiocytosis may include conservative monitoring, topical steroids, topical nitrogen mustard, tacrolimus, or psoralen plus UVA.3 Surgical excision may be considered for large lesions.
The Diagnosis: Congenital Self-healing Reticulohistiocytosis
Biopsy of a representative lesion from this patient was consistent with congenital self-healing reticulohistiocytosis, as shown in the Figure. Characteristic Langerhans cells were present in the dermis that stained CD1a positive, S-100 positive, and CD68 negative to confirm the diagnosis.
Congenital self-healing reticulohistiocytosis, or Hashimoto-Pritzker syndrome, is a rare benign form of Langerhans cell histiocytosis. It is twice as common in males than females and typically noted at birth or early during the neonatal period. Lesions may present as pink, firm, asymptomatic papulonodular lesions that often ulcerate with possible residual hypopigmentation or hyperpigmentation.1 The differential diagnosis includes congenital infectious and hematologic diseases typically associated with blueberry muffin baby. Thus, varicella, cytomegalovirus, syphilis, toxoplasmosis, rubella, neuroblastoma, leukemia cutis, and extramedullary hematopoiesis, among others, may be considered. Juvenile xanthogranuloma or urticaria pigmentosa also enter the differential diagnosis given the yellow-brown appearance. As a clonal proliferation of Langerhans cells, pathology reveals lesions that stain positive for CD1a and S-100.2
Although typically absent, evaluation for systemic involvement is warranted, which may be an early presentation of multisystem Langerhans cell histiocytosis. Continued monitoring is recommended given the risk of relapse and associated mortality. Our patient continues to do well. He will continue to be followed by our team and hematology/oncology during early childhood.
The treatment of congenital self-healing reticulohistiocytosis may include conservative monitoring, topical steroids, topical nitrogen mustard, tacrolimus, or psoralen plus UVA.3 Surgical excision may be considered for large lesions.
- Parimi LR, You J, Hong L, et al. Congenital self-healing reticulohistiocytosis with spontaneous regression. An Bras Dermatol. 2017;92:553-555.
- Chen AJ, Jarrett P, Macfarlane S. Congenital self-healing reticulohistiocytosis: the need for investigation. Australas J Dermatol. 2016;57:76-77.
- Gothwal S, Gupta AK, Choudhary R. Congenital self healing Langerhans cell histiocytosis. Indian J Pediatr. 2018;85:316-317.
- Parimi LR, You J, Hong L, et al. Congenital self-healing reticulohistiocytosis with spontaneous regression. An Bras Dermatol. 2017;92:553-555.
- Chen AJ, Jarrett P, Macfarlane S. Congenital self-healing reticulohistiocytosis: the need for investigation. Australas J Dermatol. 2016;57:76-77.
- Gothwal S, Gupta AK, Choudhary R. Congenital self healing Langerhans cell histiocytosis. Indian J Pediatr. 2018;85:316-317.
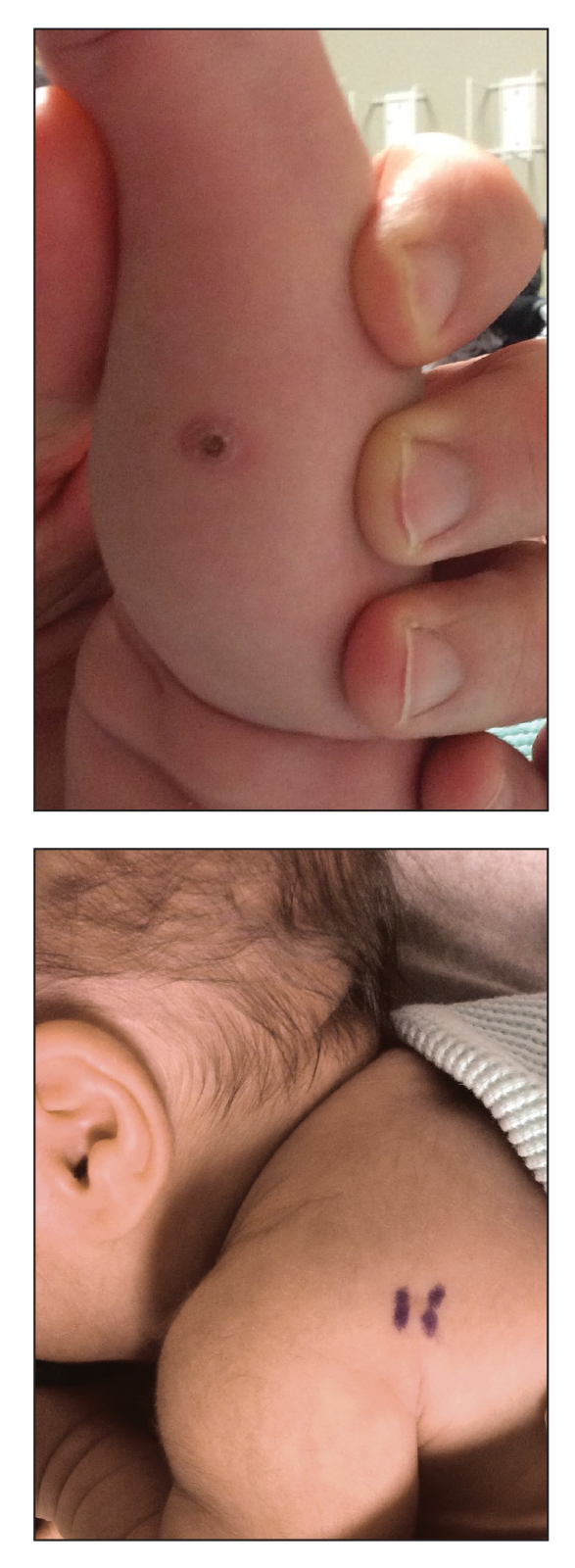
An 18-day-old infant boy presented with yellow-brown ulcerated papules on the left posterior leg (top) and left posterior shoulder (bottom). He was born via spontaneous vaginal delivery at 33 1/7 weeks' gestation complicated by preeclampsia. The lesions were unchanged during the infant's stay in the neonatal intensive care unit. However, his mother noted that one lesion crusted once home without an increase in size. His fraternal twin sister did not have any similar lesions. Jaundice and hepatosplenomegaly were absent. He was referred to hematology/oncology. A complete blood cell count revealed nonconcerning slight anemia. Liver function tests, coagulation studies, a chest radiograph, and a skeletal survey were ordered.
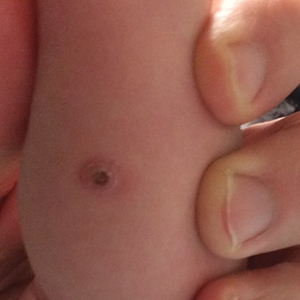
Solitary Papule on the Nose
The Diagnosis: Sclerosing Perineurioma
Sclerosing perineurioma, first described in 1997 by Fetsch and Miettinen,1 is a subtype of perineurioma with a strong predilection for the fingers and palms of young adults. Rare cases involving extra-acral sites including the forearm, elbow, axilla, back, neck, lower leg, thigh, knee, lips, nose, and mouth have been reported.2-4 Perineurioma is a relatively uncommon and benign peripheral nerve sheath tumor with exclusive perineurial differentiation.5 Perineurioma is divided into intraneural and extraneural types; the latter are further subclassified into soft tissue, sclerosing, reticular, and plexiform types. Other rare forms include the sclerosing, Pacinian corpuscle-like perineurioma, lipomatous perineurioma, perineurioma with xanthomatous areas, and perineurioma with granular cells.6,7
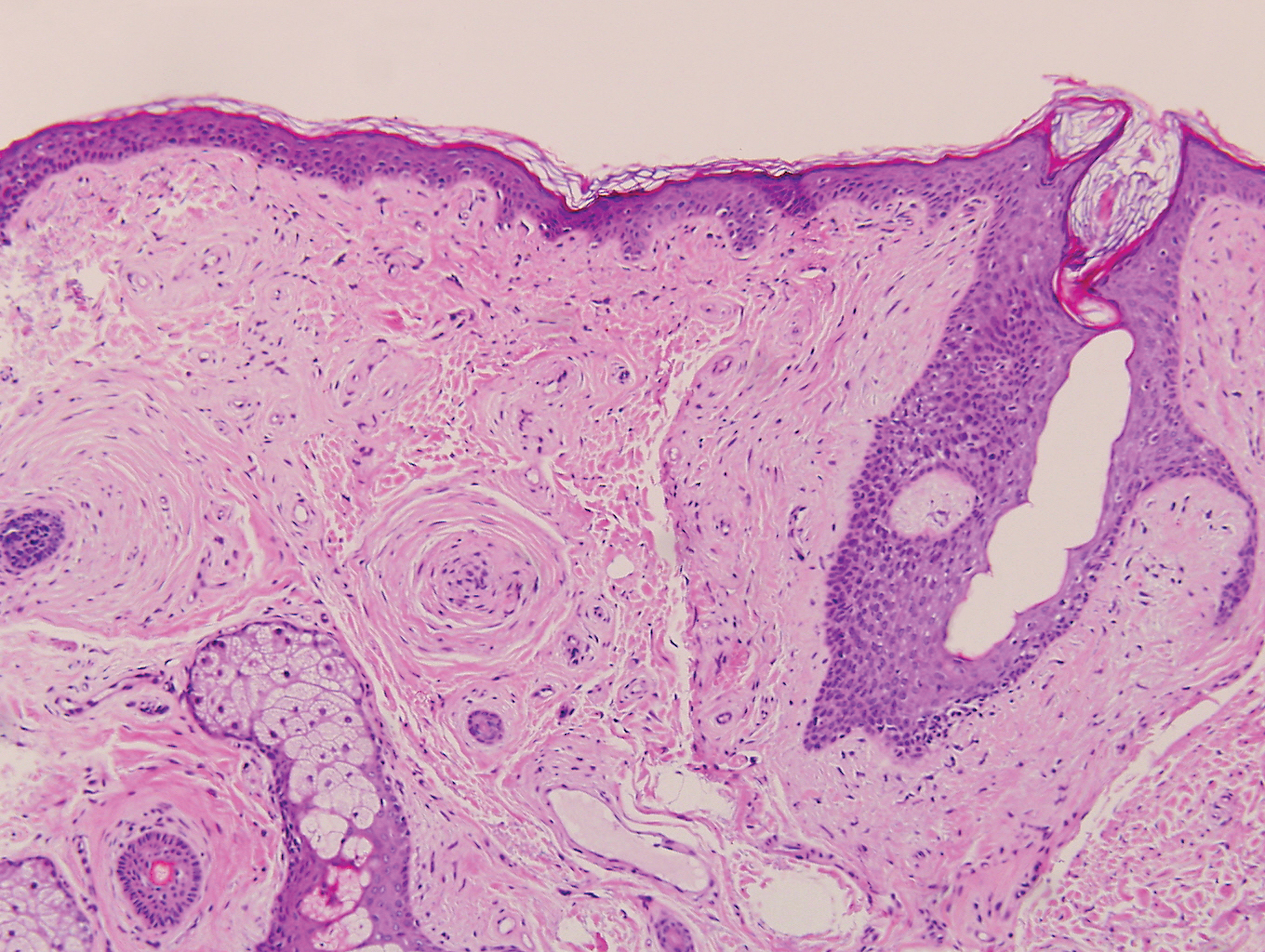
Clinically, sclerosing perineurioma usually presents as a solitary lesion; however, rare cases of multiple lesions have been reported.8 Our patient presented with a solitary papule on the nose. Histopathologically, sclerosing perineurioma demonstrates slender spindle cells in a whorled growth pattern (onion skin) embedded in a hyalinized, lamellar, and dense collagenous stroma with intervening cleftlike spaces. Immunohistochemically, the spindle cells of our case stained positive for epithelial membrane antigen (quiz images). Other positive immunostains for perineurioma include claudin-1 and glucose transporter 1 (GLUT1). Perineurioma lacks expression of S-100 but can express CD34.2 As a benign tumor, the prognosis of sclerosing perineurioma is excellent. Complete local excision is considered curative.1
Angiofibroma, also known as fibrous papule, is a common and benign lesion located primarily on or in close proximity to the nose.9 Angiofibromas can be associated with genodermatoses such as tuberous sclerosis, multiple endocrine neoplasia type 1, or Birt-Hogg-Dubé syndrome. When angiofibromas involve the penis, they are called pearly penile papules. Ungual angiofibroma, also known as Koenen tumor, occurs underneath the nail.10-12 Both facial angiofibromas (>3) and ungual angiofibromas (>2) are independent major criteria for tuberous sclerosis.13 Clinically, angiofibroma presents as a small, dome-shaped, pink papule arising on the lower portion of the nose or nearby area of the central face. Histopathologically, angiofibromas classically demonstrate increased dilated vessels and fibrosis in the dermis. Stellate, plump, spindle-shaped, and multinucleated cells can be seen in the collagenous stroma. The collagen fibers around hair follicles are arranged concentrically, resulting in an onion skin-like appearance. The epidermal rete ridges can be effaced (Figure 1). Increased numbers of single-unit melanocytes along the dermoepidermal junction can be seen in some cases. Immunohistochemically, a variable number of spindled and multinucleated cells in the dermis stain with factor XIIIa. There are at least 7 histologic variants of angiofibroma including hypercellular, pigmented, inflammatory, pleomorphic, clear cell, granular cell, and epithelioid.9,14
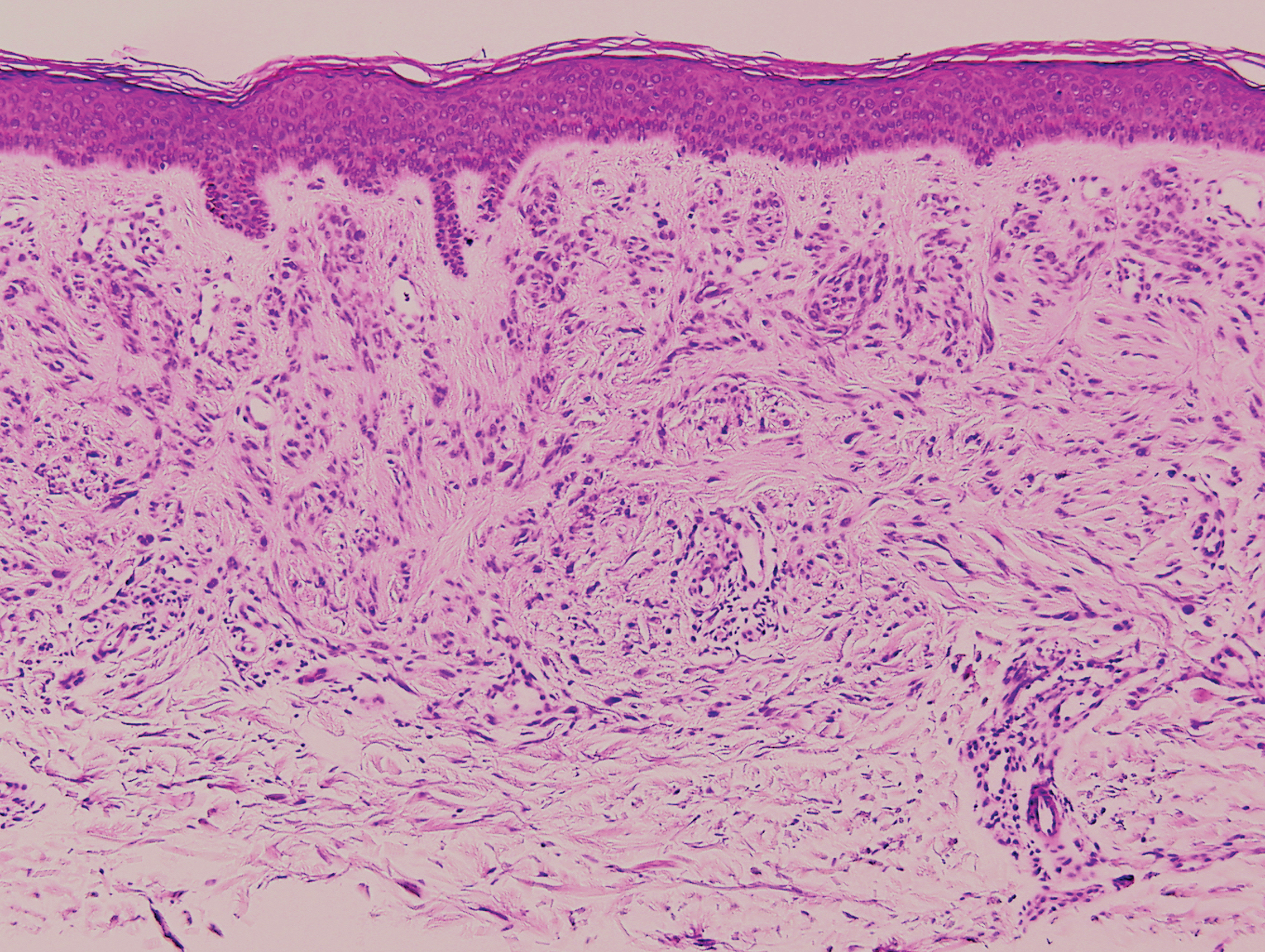
Desmoplastic nevus (DN) is a benign melanocytic neoplasm characterized by predominantly spindle-shaped nevus cells embedded within a fibrotic stroma. Although it can resemble a Spitz nevus, it is recognized as a distinct entity.15-17 Clinically, DN presents as a small and flesh-colored, erythematous or slightly pigmented papule or nodule that usually occurs on the arms and legs of young adults. Histopathologically, DN demonstrates a dermal-based proliferation of spindled melanocytes embedded in a dense collagenous stroma with sparse or absent melanin deposition. The collagen bundles often show artifactual clefts and onion skin-like accentuation around vessels. Melanocytes may be epithelioid (Figure 2).16 Immunohistochemically, DN expresses melanocytic markers such as S-100, Melan-A, and human melanoma black 45, but epithelial membrane antigen is negative. Human melanoma black 45 demonstrates maturation with stronger staining in superficial portions of the lesion and diminution of staining with increasing dermal depth.18 Many other melanocytic tumors share histologic similarities to DN including blue nevus and desmoplastic melanoma.17,19,20
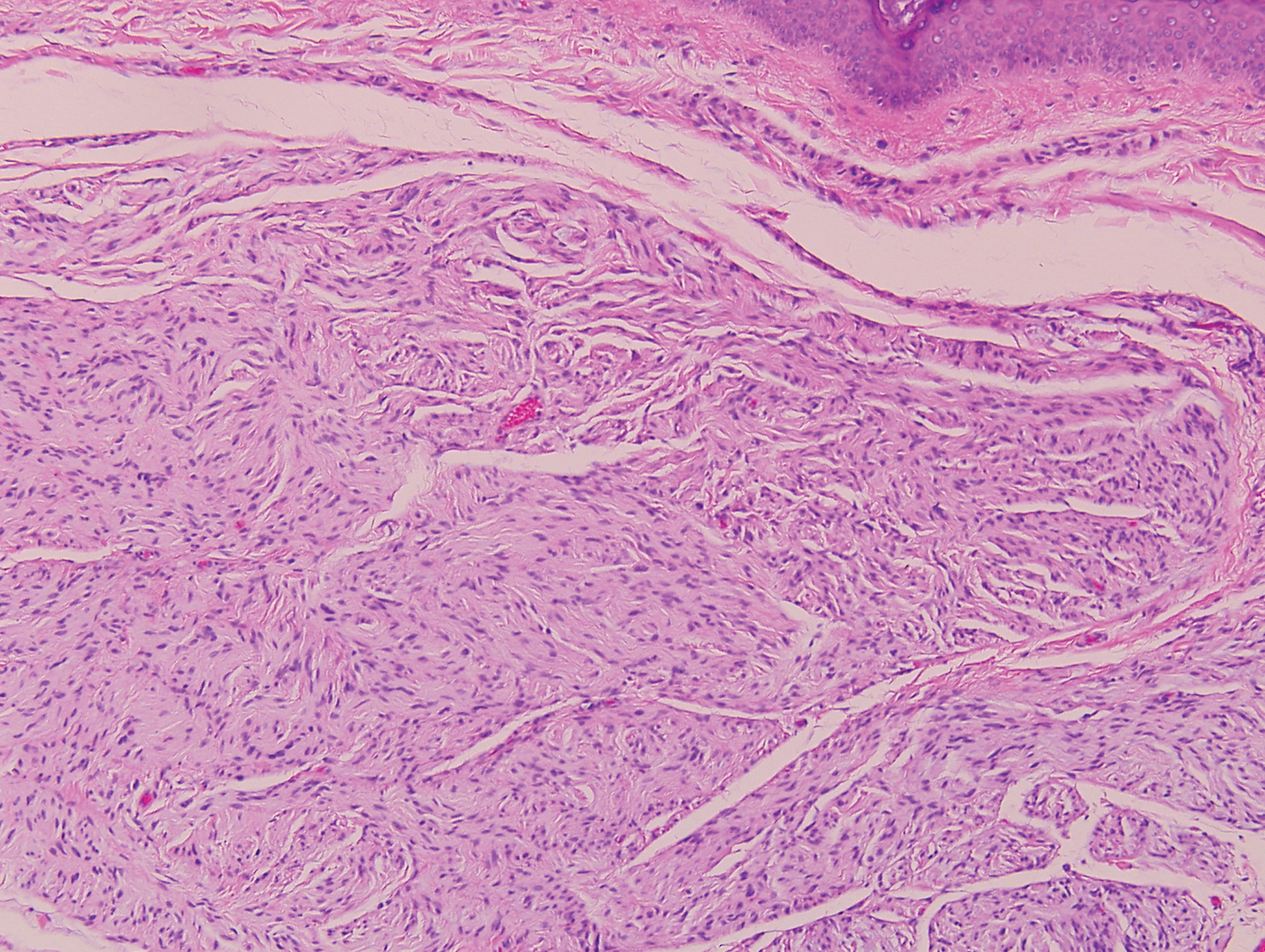
Palisaded encapsulated neuroma, also referred to as solitary circumscribed neuroma, was first described by Reed et al21 in 1972. It is a benign and solitary, firm, dome-shaped, flesh-colored papule that occurs in middle-aged adults, predominately near mucocutaneous junctions of the face. Other locations include the oral mucosa, eyelid, nasal fossa, shoulder, arm, hand, foot, and glans penis.22,23 Histopathologically, palisaded encapsulated neuroma demonstrates a solitary, well-circumscribed, partially encapsulated, intradermal nodule composed of interweaving fascicles of spindle cells with prominent clefts (Figure 3). Rarely, palisaded encapsulated neuroma may have a plexiform or multinodular architecture.24 Immunohistochemically, tumor cells stain positively for S-100 protein, type IV collagen, and vimentin. The capsule, composed of perineural cells, stains positive for epithelial membrane antigen. A neurofilament stain will highlight axons within the tumor.24,25 Currently, palisaded encapsulated neuroma does not have a well-established link to known neurocutaneous or inherited syndromes. Excision is curative with a low risk of recurrence.26
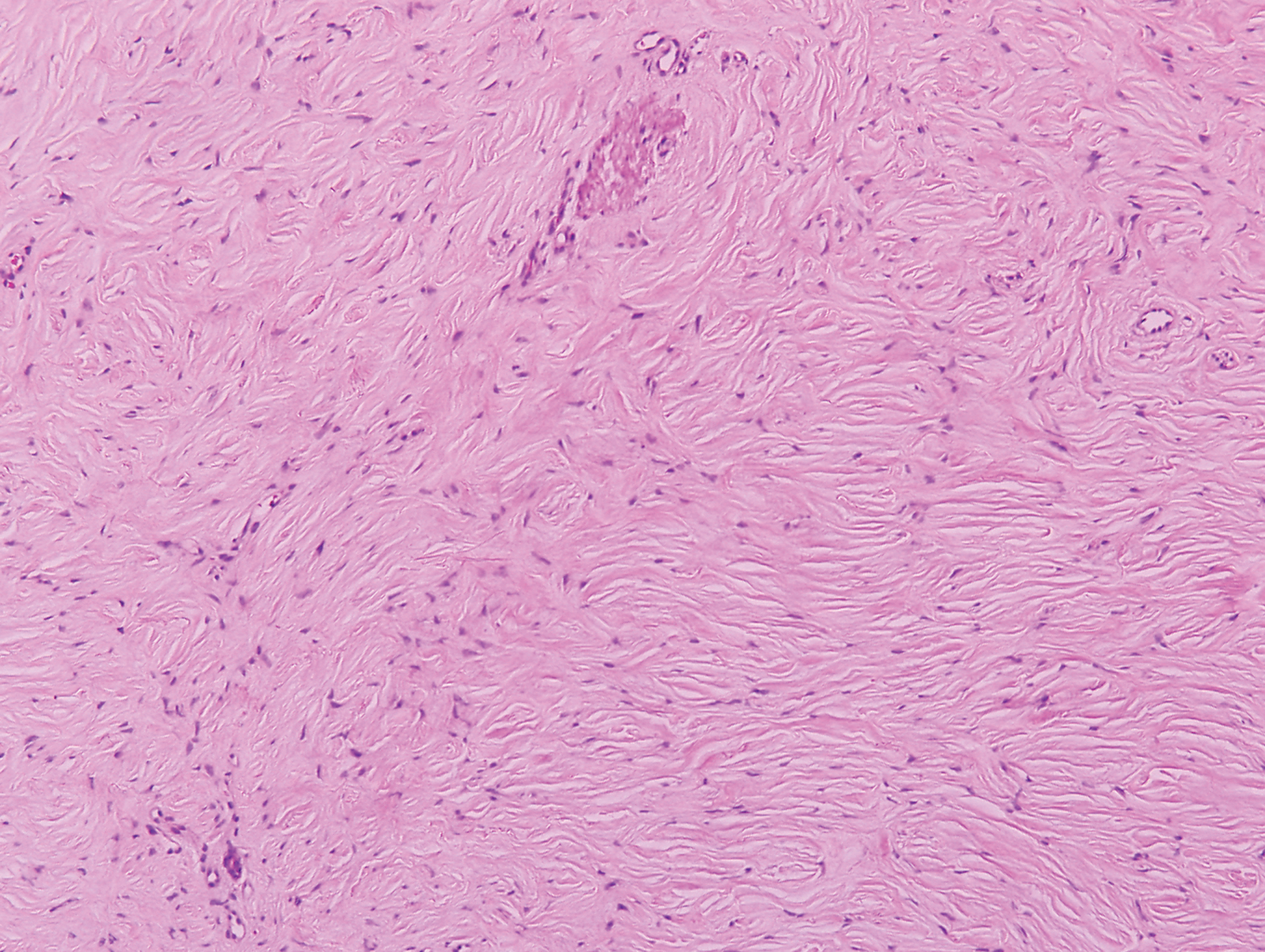
Sclerotic fibromas (SFs) were first reported by Weary et al27 as multiple tumors involving the tongues of patients with Cowden syndrome. Sporadic or solitary SFs of the skin in patients without Cowden syndrome have been reported, and both multiple and solitary SFs present with similar pathologic changes.28-30 Clinically, the solitary variant manifests as a well-demarcated, flesh-colored to erythematous, waxy papule or nodule with no site or sex predilection.30,31 Histologically, SF demonstrates a well-demarcated, nonencapsulated dermal nodule composed of hypocellular and sclerotic collagen bundles with scattered spindled cells and prominent clefts resembling Vincent van Gogh's Starry Night or plywood (Figure 4). Immunohistochemically, the spindled cells strongly express CD34. Factor XIIIa and markers of melanocytic, neural, and muscular differentiation are negative. When rendering a diagnosis in a patient with multiple SFs, a comment regarding the possibility of Cowden syndrome should be mentioned.32
- Fetsch JF, Miettinen M. Sclerosing perineurioma: a clinicopathologic study of 19 cases of a distinctive soft tissue lesion with a predilection for the fingers and palms of young adults. Am J Surg Pathol. 1997;21:1433-1442.
- Fox MD, Gleason BC, Thomas AB, et al. Extra-acral cutaneous/soft tissue sclerosing perineurioma: an under-recognized entity in the differential of CD34-positive cutaneous neoplasms. J Cutan Pathol. 2010;37:1053-1056.
- Erstine EM, Ko JS, Rubin BP, et al. Broadening the anatomic landscape of sclerosing perineurioma: a series of 5 cases in nonacral sites. Am J Dermatopathol. 2017;39:679-681.
- Senghore N, Cunliffe D, Watt-Smith S, et al. Extraneural perineurioma of the face: an unusual cutaneous presentation of an uncommon tumour. Br J Oral Maxillofac Surg. 2001;39:315-319.
- Lazarus SS, Trombetta LD. Ultrastructural identification of a benign perineurial cell tumor. Cancer. 1978;41:1823-1829.
- Macarenco RS, Cury-Martins J. Extra-acral cutaneous sclerosing perineurioma with CD34 fingerprint pattern. J Cutan Pathol. 2017;44:388-392.
- Santos-Briz A, Godoy E, Canueto J, et al. Cutaneous intraneural perineurioma: a case report. Am J Dermatopathol. 2013;35:E45-E48.
- Rubin AI, Yassaee M, Johnson W, et al. Multiple cutaneous sclerosing perineuriomas: an extensive presentation with involvement of the bilateral upper extremities. J Cutan Pathol. 2009;36(suppl 1):60-65.
- Damman J, Biswas A. Fibrous papule: a histopathologic review. Am J Dermatopathol. 2018;40:551-560.
- Macri A, Tanner LS. Cutaneous angiofibroma. StatPearls. https://www.statpearls.com/kb/viewarticle/17566/. Updated January 24, 2019. Accessed October 21, 2019.
- Darling TN, Skarulis MC, Steinberg SM, et al. Multiple facial angiofibromas and collagenomas in patients with multiple endocrine neoplasia type 1. Arch Dermatol. 1997;133:853-857.
- Schaffer JV, Gohara MA, McNiff JM, et al. Multiple facial angiofibromas: a cutaneous manifestation of Birt-Hogg-Dube syndrome. J Am Acad Dermatol. 2005;53:S108-S111.
- Northrup H, Krueger DA; International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol. 2013;49:243-254.
- Bansal C, Stewart D, Li A, et al. Histologic variants of fibrous papule. J Cutan Pathol. 2005;32:424-428.
- Harris GR, Shea CR, Horenstein MG, et al. Desmoplastic (sclerotic) nevus: an underrecognized entity that resembles dermatofibroma and desmoplastic melanoma. Am J Surg Pathol. 1999;23:786-794.
- Ferrara G, Brasiello M, Annese P, et al. Desmoplastic nevus: clinicopathologic keynotes. Am J Dermatopathol. 2009;31:718-722.
- Sherrill AM, Crespo G, Prakash AV, et al. Desmoplastic nevus: an entity distinct from Spitz nevus and blue nevus. Am J Dermatopathol. 2011;33:35-39.
- Kucher C, Zhang PJ, Pasha T, et al. Expression of Melan-A and Ki-67 in desmoplastic melanoma and desmoplastic nevi. Am J Dermatopathol. 2004;26:452-457.
- Sidiropoulos M, Sholl LM, Obregon R, et al. Desmoplastic nevus of chronically sun-damaged skin: an entity to be distinguished from desmoplastic melanoma. Am J Dermatopathol. 2014;36:629-634.
- Kiuru M, Patel RM, Busam KJ. Desmoplastic melanocytic nevi with lymphocytic aggregates. J Cutan Pathol. 2012;39:940-944.
- Reed RJ, Fine RM, Meltzer HD. Palisaded, encapsulated neuromas of the skin. Arch Dermatol. 1972;106:865-870.
- Newman MD, Milgraum S. Palisaded encapsulated neuroma (PEN): an often misdiagnosed neural tumor. Dermatol Online J. 2008;14:12.
- Beutler B, Cohen PR. Palisaded encapsulated neuroma of the trunk: a case report and review of palisaded encapsulated neuroma. Cureus. 2016;8:E726.
- Jokinen CH, Ragsdale BD, Argenyi ZB. Expanding the clinicopathologic spectrum of palisaded encapsulated neuroma. J Cutan Pathol. 2010;37:43-48.
- Argenyi ZB. Immunohistochemical characterization of palisaded, encapsulated neuroma. J Cutan Pathol. 1990;17:329-335.
- Batra J, Ramesh V, Molpariya A, et al. Palisaded encapsulated neuroma: an unusual presentation. Indian Dermatol Online J. 2018;9:262-264.
- Weary PE, Gorlin RJ, Gentry WC Jr, et al. Multiple hamartoma syndrome (Cowden's disease). Arch Dermatol. 1972;106:682-690.
- Mahmood MN, Salama ME, Chaffins M, et al. Solitary sclerotic fibroma of skin: a possible link with pleomorphic fibroma with immunophenotypic expression for O13 (CD99) and CD34. J Cutan Pathol. 2003;30:631-636.
- Nakashima K, Yamada N, Adachi K, et al. Solitary sclerotic fibroma of the skin: morphological characterization of the 'plywood-like pattern'. J Cutan Pathol. 2008;35(suppl 1):74-79.
- Rapini RP, Golitz LE. Sclerotic fibromas of the skin. J Am Acad Dermatol. 1989;20:266-271.
- Abbas O, Ghosn S, Bahhady R, et al. Solitary sclerotic fibroma on the scalp of a young girl: reactive sclerosis pattern? J Dermatol. 2010;37:575-577.
- Hanft VN, Shea CR, McNutt NS, et al. Expression of CD34 in sclerotic ("plywood") fibromas. Am J Dermatopathol. 2000;22:17-21.
The Diagnosis: Sclerosing Perineurioma
Sclerosing perineurioma, first described in 1997 by Fetsch and Miettinen,1 is a subtype of perineurioma with a strong predilection for the fingers and palms of young adults. Rare cases involving extra-acral sites including the forearm, elbow, axilla, back, neck, lower leg, thigh, knee, lips, nose, and mouth have been reported.2-4 Perineurioma is a relatively uncommon and benign peripheral nerve sheath tumor with exclusive perineurial differentiation.5 Perineurioma is divided into intraneural and extraneural types; the latter are further subclassified into soft tissue, sclerosing, reticular, and plexiform types. Other rare forms include the sclerosing, Pacinian corpuscle-like perineurioma, lipomatous perineurioma, perineurioma with xanthomatous areas, and perineurioma with granular cells.6,7
Clinically, sclerosing perineurioma usually presents as a solitary lesion; however, rare cases of multiple lesions have been reported.8 Our patient presented with a solitary papule on the nose. Histopathologically, sclerosing perineurioma demonstrates slender spindle cells in a whorled growth pattern (onion skin) embedded in a hyalinized, lamellar, and dense collagenous stroma with intervening cleftlike spaces. Immunohistochemically, the spindle cells of our case stained positive for epithelial membrane antigen (quiz images). Other positive immunostains for perineurioma include claudin-1 and glucose transporter 1 (GLUT1). Perineurioma lacks expression of S-100 but can express CD34.2 As a benign tumor, the prognosis of sclerosing perineurioma is excellent. Complete local excision is considered curative.1
Angiofibroma, also known as fibrous papule, is a common and benign lesion located primarily on or in close proximity to the nose.9 Angiofibromas can be associated with genodermatoses such as tuberous sclerosis, multiple endocrine neoplasia type 1, or Birt-Hogg-Dubé syndrome. When angiofibromas involve the penis, they are called pearly penile papules. Ungual angiofibroma, also known as Koenen tumor, occurs underneath the nail.10-12 Both facial angiofibromas (>3) and ungual angiofibromas (>2) are independent major criteria for tuberous sclerosis.13 Clinically, angiofibroma presents as a small, dome-shaped, pink papule arising on the lower portion of the nose or nearby area of the central face. Histopathologically, angiofibromas classically demonstrate increased dilated vessels and fibrosis in the dermis. Stellate, plump, spindle-shaped, and multinucleated cells can be seen in the collagenous stroma. The collagen fibers around hair follicles are arranged concentrically, resulting in an onion skin-like appearance. The epidermal rete ridges can be effaced (Figure 1). Increased numbers of single-unit melanocytes along the dermoepidermal junction can be seen in some cases. Immunohistochemically, a variable number of spindled and multinucleated cells in the dermis stain with factor XIIIa. There are at least 7 histologic variants of angiofibroma including hypercellular, pigmented, inflammatory, pleomorphic, clear cell, granular cell, and epithelioid.9,14
Desmoplastic nevus (DN) is a benign melanocytic neoplasm characterized by predominantly spindle-shaped nevus cells embedded within a fibrotic stroma. Although it can resemble a Spitz nevus, it is recognized as a distinct entity.15-17 Clinically, DN presents as a small and flesh-colored, erythematous or slightly pigmented papule or nodule that usually occurs on the arms and legs of young adults. Histopathologically, DN demonstrates a dermal-based proliferation of spindled melanocytes embedded in a dense collagenous stroma with sparse or absent melanin deposition. The collagen bundles often show artifactual clefts and onion skin-like accentuation around vessels. Melanocytes may be epithelioid (Figure 2).16 Immunohistochemically, DN expresses melanocytic markers such as S-100, Melan-A, and human melanoma black 45, but epithelial membrane antigen is negative. Human melanoma black 45 demonstrates maturation with stronger staining in superficial portions of the lesion and diminution of staining with increasing dermal depth.18 Many other melanocytic tumors share histologic similarities to DN including blue nevus and desmoplastic melanoma.17,19,20
Palisaded encapsulated neuroma, also referred to as solitary circumscribed neuroma, was first described by Reed et al21 in 1972. It is a benign and solitary, firm, dome-shaped, flesh-colored papule that occurs in middle-aged adults, predominately near mucocutaneous junctions of the face. Other locations include the oral mucosa, eyelid, nasal fossa, shoulder, arm, hand, foot, and glans penis.22,23 Histopathologically, palisaded encapsulated neuroma demonstrates a solitary, well-circumscribed, partially encapsulated, intradermal nodule composed of interweaving fascicles of spindle cells with prominent clefts (Figure 3). Rarely, palisaded encapsulated neuroma may have a plexiform or multinodular architecture.24 Immunohistochemically, tumor cells stain positively for S-100 protein, type IV collagen, and vimentin. The capsule, composed of perineural cells, stains positive for epithelial membrane antigen. A neurofilament stain will highlight axons within the tumor.24,25 Currently, palisaded encapsulated neuroma does not have a well-established link to known neurocutaneous or inherited syndromes. Excision is curative with a low risk of recurrence.26
Sclerotic fibromas (SFs) were first reported by Weary et al27 as multiple tumors involving the tongues of patients with Cowden syndrome. Sporadic or solitary SFs of the skin in patients without Cowden syndrome have been reported, and both multiple and solitary SFs present with similar pathologic changes.28-30 Clinically, the solitary variant manifests as a well-demarcated, flesh-colored to erythematous, waxy papule or nodule with no site or sex predilection.30,31 Histologically, SF demonstrates a well-demarcated, nonencapsulated dermal nodule composed of hypocellular and sclerotic collagen bundles with scattered spindled cells and prominent clefts resembling Vincent van Gogh's Starry Night or plywood (Figure 4). Immunohistochemically, the spindled cells strongly express CD34. Factor XIIIa and markers of melanocytic, neural, and muscular differentiation are negative. When rendering a diagnosis in a patient with multiple SFs, a comment regarding the possibility of Cowden syndrome should be mentioned.32
The Diagnosis: Sclerosing Perineurioma
Sclerosing perineurioma, first described in 1997 by Fetsch and Miettinen,1 is a subtype of perineurioma with a strong predilection for the fingers and palms of young adults. Rare cases involving extra-acral sites including the forearm, elbow, axilla, back, neck, lower leg, thigh, knee, lips, nose, and mouth have been reported.2-4 Perineurioma is a relatively uncommon and benign peripheral nerve sheath tumor with exclusive perineurial differentiation.5 Perineurioma is divided into intraneural and extraneural types; the latter are further subclassified into soft tissue, sclerosing, reticular, and plexiform types. Other rare forms include the sclerosing, Pacinian corpuscle-like perineurioma, lipomatous perineurioma, perineurioma with xanthomatous areas, and perineurioma with granular cells.6,7
Clinically, sclerosing perineurioma usually presents as a solitary lesion; however, rare cases of multiple lesions have been reported.8 Our patient presented with a solitary papule on the nose. Histopathologically, sclerosing perineurioma demonstrates slender spindle cells in a whorled growth pattern (onion skin) embedded in a hyalinized, lamellar, and dense collagenous stroma with intervening cleftlike spaces. Immunohistochemically, the spindle cells of our case stained positive for epithelial membrane antigen (quiz images). Other positive immunostains for perineurioma include claudin-1 and glucose transporter 1 (GLUT1). Perineurioma lacks expression of S-100 but can express CD34.2 As a benign tumor, the prognosis of sclerosing perineurioma is excellent. Complete local excision is considered curative.1
Angiofibroma, also known as fibrous papule, is a common and benign lesion located primarily on or in close proximity to the nose.9 Angiofibromas can be associated with genodermatoses such as tuberous sclerosis, multiple endocrine neoplasia type 1, or Birt-Hogg-Dubé syndrome. When angiofibromas involve the penis, they are called pearly penile papules. Ungual angiofibroma, also known as Koenen tumor, occurs underneath the nail.10-12 Both facial angiofibromas (>3) and ungual angiofibromas (>2) are independent major criteria for tuberous sclerosis.13 Clinically, angiofibroma presents as a small, dome-shaped, pink papule arising on the lower portion of the nose or nearby area of the central face. Histopathologically, angiofibromas classically demonstrate increased dilated vessels and fibrosis in the dermis. Stellate, plump, spindle-shaped, and multinucleated cells can be seen in the collagenous stroma. The collagen fibers around hair follicles are arranged concentrically, resulting in an onion skin-like appearance. The epidermal rete ridges can be effaced (Figure 1). Increased numbers of single-unit melanocytes along the dermoepidermal junction can be seen in some cases. Immunohistochemically, a variable number of spindled and multinucleated cells in the dermis stain with factor XIIIa. There are at least 7 histologic variants of angiofibroma including hypercellular, pigmented, inflammatory, pleomorphic, clear cell, granular cell, and epithelioid.9,14
Desmoplastic nevus (DN) is a benign melanocytic neoplasm characterized by predominantly spindle-shaped nevus cells embedded within a fibrotic stroma. Although it can resemble a Spitz nevus, it is recognized as a distinct entity.15-17 Clinically, DN presents as a small and flesh-colored, erythematous or slightly pigmented papule or nodule that usually occurs on the arms and legs of young adults. Histopathologically, DN demonstrates a dermal-based proliferation of spindled melanocytes embedded in a dense collagenous stroma with sparse or absent melanin deposition. The collagen bundles often show artifactual clefts and onion skin-like accentuation around vessels. Melanocytes may be epithelioid (Figure 2).16 Immunohistochemically, DN expresses melanocytic markers such as S-100, Melan-A, and human melanoma black 45, but epithelial membrane antigen is negative. Human melanoma black 45 demonstrates maturation with stronger staining in superficial portions of the lesion and diminution of staining with increasing dermal depth.18 Many other melanocytic tumors share histologic similarities to DN including blue nevus and desmoplastic melanoma.17,19,20
Palisaded encapsulated neuroma, also referred to as solitary circumscribed neuroma, was first described by Reed et al21 in 1972. It is a benign and solitary, firm, dome-shaped, flesh-colored papule that occurs in middle-aged adults, predominately near mucocutaneous junctions of the face. Other locations include the oral mucosa, eyelid, nasal fossa, shoulder, arm, hand, foot, and glans penis.22,23 Histopathologically, palisaded encapsulated neuroma demonstrates a solitary, well-circumscribed, partially encapsulated, intradermal nodule composed of interweaving fascicles of spindle cells with prominent clefts (Figure 3). Rarely, palisaded encapsulated neuroma may have a plexiform or multinodular architecture.24 Immunohistochemically, tumor cells stain positively for S-100 protein, type IV collagen, and vimentin. The capsule, composed of perineural cells, stains positive for epithelial membrane antigen. A neurofilament stain will highlight axons within the tumor.24,25 Currently, palisaded encapsulated neuroma does not have a well-established link to known neurocutaneous or inherited syndromes. Excision is curative with a low risk of recurrence.26
Sclerotic fibromas (SFs) were first reported by Weary et al27 as multiple tumors involving the tongues of patients with Cowden syndrome. Sporadic or solitary SFs of the skin in patients without Cowden syndrome have been reported, and both multiple and solitary SFs present with similar pathologic changes.28-30 Clinically, the solitary variant manifests as a well-demarcated, flesh-colored to erythematous, waxy papule or nodule with no site or sex predilection.30,31 Histologically, SF demonstrates a well-demarcated, nonencapsulated dermal nodule composed of hypocellular and sclerotic collagen bundles with scattered spindled cells and prominent clefts resembling Vincent van Gogh's Starry Night or plywood (Figure 4). Immunohistochemically, the spindled cells strongly express CD34. Factor XIIIa and markers of melanocytic, neural, and muscular differentiation are negative. When rendering a diagnosis in a patient with multiple SFs, a comment regarding the possibility of Cowden syndrome should be mentioned.32
- Fetsch JF, Miettinen M. Sclerosing perineurioma: a clinicopathologic study of 19 cases of a distinctive soft tissue lesion with a predilection for the fingers and palms of young adults. Am J Surg Pathol. 1997;21:1433-1442.
- Fox MD, Gleason BC, Thomas AB, et al. Extra-acral cutaneous/soft tissue sclerosing perineurioma: an under-recognized entity in the differential of CD34-positive cutaneous neoplasms. J Cutan Pathol. 2010;37:1053-1056.
- Erstine EM, Ko JS, Rubin BP, et al. Broadening the anatomic landscape of sclerosing perineurioma: a series of 5 cases in nonacral sites. Am J Dermatopathol. 2017;39:679-681.
- Senghore N, Cunliffe D, Watt-Smith S, et al. Extraneural perineurioma of the face: an unusual cutaneous presentation of an uncommon tumour. Br J Oral Maxillofac Surg. 2001;39:315-319.
- Lazarus SS, Trombetta LD. Ultrastructural identification of a benign perineurial cell tumor. Cancer. 1978;41:1823-1829.
- Macarenco RS, Cury-Martins J. Extra-acral cutaneous sclerosing perineurioma with CD34 fingerprint pattern. J Cutan Pathol. 2017;44:388-392.
- Santos-Briz A, Godoy E, Canueto J, et al. Cutaneous intraneural perineurioma: a case report. Am J Dermatopathol. 2013;35:E45-E48.
- Rubin AI, Yassaee M, Johnson W, et al. Multiple cutaneous sclerosing perineuriomas: an extensive presentation with involvement of the bilateral upper extremities. J Cutan Pathol. 2009;36(suppl 1):60-65.
- Damman J, Biswas A. Fibrous papule: a histopathologic review. Am J Dermatopathol. 2018;40:551-560.
- Macri A, Tanner LS. Cutaneous angiofibroma. StatPearls. https://www.statpearls.com/kb/viewarticle/17566/. Updated January 24, 2019. Accessed October 21, 2019.
- Darling TN, Skarulis MC, Steinberg SM, et al. Multiple facial angiofibromas and collagenomas in patients with multiple endocrine neoplasia type 1. Arch Dermatol. 1997;133:853-857.
- Schaffer JV, Gohara MA, McNiff JM, et al. Multiple facial angiofibromas: a cutaneous manifestation of Birt-Hogg-Dube syndrome. J Am Acad Dermatol. 2005;53:S108-S111.
- Northrup H, Krueger DA; International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol. 2013;49:243-254.
- Bansal C, Stewart D, Li A, et al. Histologic variants of fibrous papule. J Cutan Pathol. 2005;32:424-428.
- Harris GR, Shea CR, Horenstein MG, et al. Desmoplastic (sclerotic) nevus: an underrecognized entity that resembles dermatofibroma and desmoplastic melanoma. Am J Surg Pathol. 1999;23:786-794.
- Ferrara G, Brasiello M, Annese P, et al. Desmoplastic nevus: clinicopathologic keynotes. Am J Dermatopathol. 2009;31:718-722.
- Sherrill AM, Crespo G, Prakash AV, et al. Desmoplastic nevus: an entity distinct from Spitz nevus and blue nevus. Am J Dermatopathol. 2011;33:35-39.
- Kucher C, Zhang PJ, Pasha T, et al. Expression of Melan-A and Ki-67 in desmoplastic melanoma and desmoplastic nevi. Am J Dermatopathol. 2004;26:452-457.
- Sidiropoulos M, Sholl LM, Obregon R, et al. Desmoplastic nevus of chronically sun-damaged skin: an entity to be distinguished from desmoplastic melanoma. Am J Dermatopathol. 2014;36:629-634.
- Kiuru M, Patel RM, Busam KJ. Desmoplastic melanocytic nevi with lymphocytic aggregates. J Cutan Pathol. 2012;39:940-944.
- Reed RJ, Fine RM, Meltzer HD. Palisaded, encapsulated neuromas of the skin. Arch Dermatol. 1972;106:865-870.
- Newman MD, Milgraum S. Palisaded encapsulated neuroma (PEN): an often misdiagnosed neural tumor. Dermatol Online J. 2008;14:12.
- Beutler B, Cohen PR. Palisaded encapsulated neuroma of the trunk: a case report and review of palisaded encapsulated neuroma. Cureus. 2016;8:E726.
- Jokinen CH, Ragsdale BD, Argenyi ZB. Expanding the clinicopathologic spectrum of palisaded encapsulated neuroma. J Cutan Pathol. 2010;37:43-48.
- Argenyi ZB. Immunohistochemical characterization of palisaded, encapsulated neuroma. J Cutan Pathol. 1990;17:329-335.
- Batra J, Ramesh V, Molpariya A, et al. Palisaded encapsulated neuroma: an unusual presentation. Indian Dermatol Online J. 2018;9:262-264.
- Weary PE, Gorlin RJ, Gentry WC Jr, et al. Multiple hamartoma syndrome (Cowden's disease). Arch Dermatol. 1972;106:682-690.
- Mahmood MN, Salama ME, Chaffins M, et al. Solitary sclerotic fibroma of skin: a possible link with pleomorphic fibroma with immunophenotypic expression for O13 (CD99) and CD34. J Cutan Pathol. 2003;30:631-636.
- Nakashima K, Yamada N, Adachi K, et al. Solitary sclerotic fibroma of the skin: morphological characterization of the 'plywood-like pattern'. J Cutan Pathol. 2008;35(suppl 1):74-79.
- Rapini RP, Golitz LE. Sclerotic fibromas of the skin. J Am Acad Dermatol. 1989;20:266-271.
- Abbas O, Ghosn S, Bahhady R, et al. Solitary sclerotic fibroma on the scalp of a young girl: reactive sclerosis pattern? J Dermatol. 2010;37:575-577.
- Hanft VN, Shea CR, McNutt NS, et al. Expression of CD34 in sclerotic ("plywood") fibromas. Am J Dermatopathol. 2000;22:17-21.
- Fetsch JF, Miettinen M. Sclerosing perineurioma: a clinicopathologic study of 19 cases of a distinctive soft tissue lesion with a predilection for the fingers and palms of young adults. Am J Surg Pathol. 1997;21:1433-1442.
- Fox MD, Gleason BC, Thomas AB, et al. Extra-acral cutaneous/soft tissue sclerosing perineurioma: an under-recognized entity in the differential of CD34-positive cutaneous neoplasms. J Cutan Pathol. 2010;37:1053-1056.
- Erstine EM, Ko JS, Rubin BP, et al. Broadening the anatomic landscape of sclerosing perineurioma: a series of 5 cases in nonacral sites. Am J Dermatopathol. 2017;39:679-681.
- Senghore N, Cunliffe D, Watt-Smith S, et al. Extraneural perineurioma of the face: an unusual cutaneous presentation of an uncommon tumour. Br J Oral Maxillofac Surg. 2001;39:315-319.
- Lazarus SS, Trombetta LD. Ultrastructural identification of a benign perineurial cell tumor. Cancer. 1978;41:1823-1829.
- Macarenco RS, Cury-Martins J. Extra-acral cutaneous sclerosing perineurioma with CD34 fingerprint pattern. J Cutan Pathol. 2017;44:388-392.
- Santos-Briz A, Godoy E, Canueto J, et al. Cutaneous intraneural perineurioma: a case report. Am J Dermatopathol. 2013;35:E45-E48.
- Rubin AI, Yassaee M, Johnson W, et al. Multiple cutaneous sclerosing perineuriomas: an extensive presentation with involvement of the bilateral upper extremities. J Cutan Pathol. 2009;36(suppl 1):60-65.
- Damman J, Biswas A. Fibrous papule: a histopathologic review. Am J Dermatopathol. 2018;40:551-560.
- Macri A, Tanner LS. Cutaneous angiofibroma. StatPearls. https://www.statpearls.com/kb/viewarticle/17566/. Updated January 24, 2019. Accessed October 21, 2019.
- Darling TN, Skarulis MC, Steinberg SM, et al. Multiple facial angiofibromas and collagenomas in patients with multiple endocrine neoplasia type 1. Arch Dermatol. 1997;133:853-857.
- Schaffer JV, Gohara MA, McNiff JM, et al. Multiple facial angiofibromas: a cutaneous manifestation of Birt-Hogg-Dube syndrome. J Am Acad Dermatol. 2005;53:S108-S111.
- Northrup H, Krueger DA; International Tuberous Sclerosis Complex Consensus Group. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol. 2013;49:243-254.
- Bansal C, Stewart D, Li A, et al. Histologic variants of fibrous papule. J Cutan Pathol. 2005;32:424-428.
- Harris GR, Shea CR, Horenstein MG, et al. Desmoplastic (sclerotic) nevus: an underrecognized entity that resembles dermatofibroma and desmoplastic melanoma. Am J Surg Pathol. 1999;23:786-794.
- Ferrara G, Brasiello M, Annese P, et al. Desmoplastic nevus: clinicopathologic keynotes. Am J Dermatopathol. 2009;31:718-722.
- Sherrill AM, Crespo G, Prakash AV, et al. Desmoplastic nevus: an entity distinct from Spitz nevus and blue nevus. Am J Dermatopathol. 2011;33:35-39.
- Kucher C, Zhang PJ, Pasha T, et al. Expression of Melan-A and Ki-67 in desmoplastic melanoma and desmoplastic nevi. Am J Dermatopathol. 2004;26:452-457.
- Sidiropoulos M, Sholl LM, Obregon R, et al. Desmoplastic nevus of chronically sun-damaged skin: an entity to be distinguished from desmoplastic melanoma. Am J Dermatopathol. 2014;36:629-634.
- Kiuru M, Patel RM, Busam KJ. Desmoplastic melanocytic nevi with lymphocytic aggregates. J Cutan Pathol. 2012;39:940-944.
- Reed RJ, Fine RM, Meltzer HD. Palisaded, encapsulated neuromas of the skin. Arch Dermatol. 1972;106:865-870.
- Newman MD, Milgraum S. Palisaded encapsulated neuroma (PEN): an often misdiagnosed neural tumor. Dermatol Online J. 2008;14:12.
- Beutler B, Cohen PR. Palisaded encapsulated neuroma of the trunk: a case report and review of palisaded encapsulated neuroma. Cureus. 2016;8:E726.
- Jokinen CH, Ragsdale BD, Argenyi ZB. Expanding the clinicopathologic spectrum of palisaded encapsulated neuroma. J Cutan Pathol. 2010;37:43-48.
- Argenyi ZB. Immunohistochemical characterization of palisaded, encapsulated neuroma. J Cutan Pathol. 1990;17:329-335.
- Batra J, Ramesh V, Molpariya A, et al. Palisaded encapsulated neuroma: an unusual presentation. Indian Dermatol Online J. 2018;9:262-264.
- Weary PE, Gorlin RJ, Gentry WC Jr, et al. Multiple hamartoma syndrome (Cowden's disease). Arch Dermatol. 1972;106:682-690.
- Mahmood MN, Salama ME, Chaffins M, et al. Solitary sclerotic fibroma of skin: a possible link with pleomorphic fibroma with immunophenotypic expression for O13 (CD99) and CD34. J Cutan Pathol. 2003;30:631-636.
- Nakashima K, Yamada N, Adachi K, et al. Solitary sclerotic fibroma of the skin: morphological characterization of the 'plywood-like pattern'. J Cutan Pathol. 2008;35(suppl 1):74-79.
- Rapini RP, Golitz LE. Sclerotic fibromas of the skin. J Am Acad Dermatol. 1989;20:266-271.
- Abbas O, Ghosn S, Bahhady R, et al. Solitary sclerotic fibroma on the scalp of a young girl: reactive sclerosis pattern? J Dermatol. 2010;37:575-577.
- Hanft VN, Shea CR, McNutt NS, et al. Expression of CD34 in sclerotic ("plywood") fibromas. Am J Dermatopathol. 2000;22:17-21.
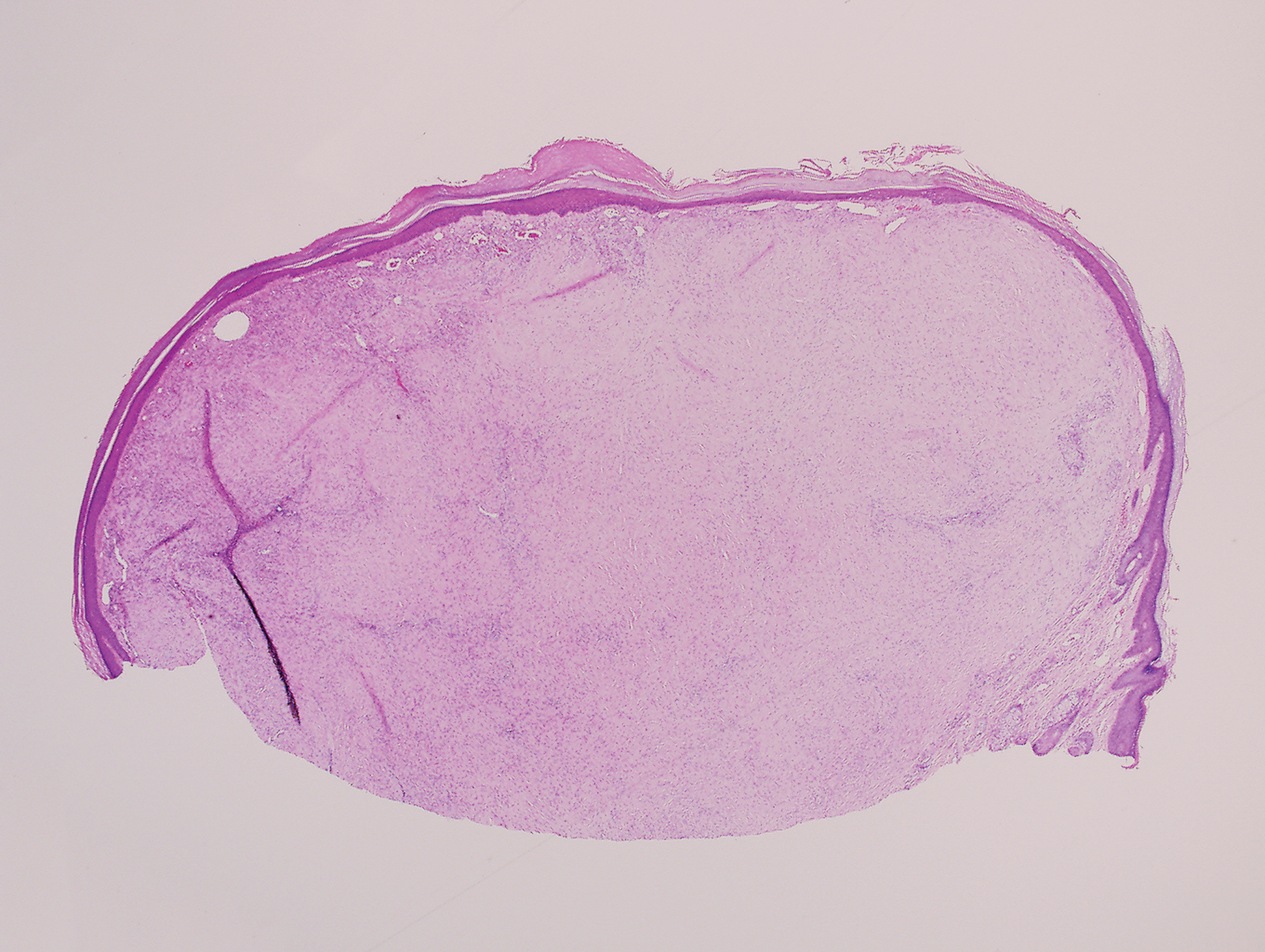
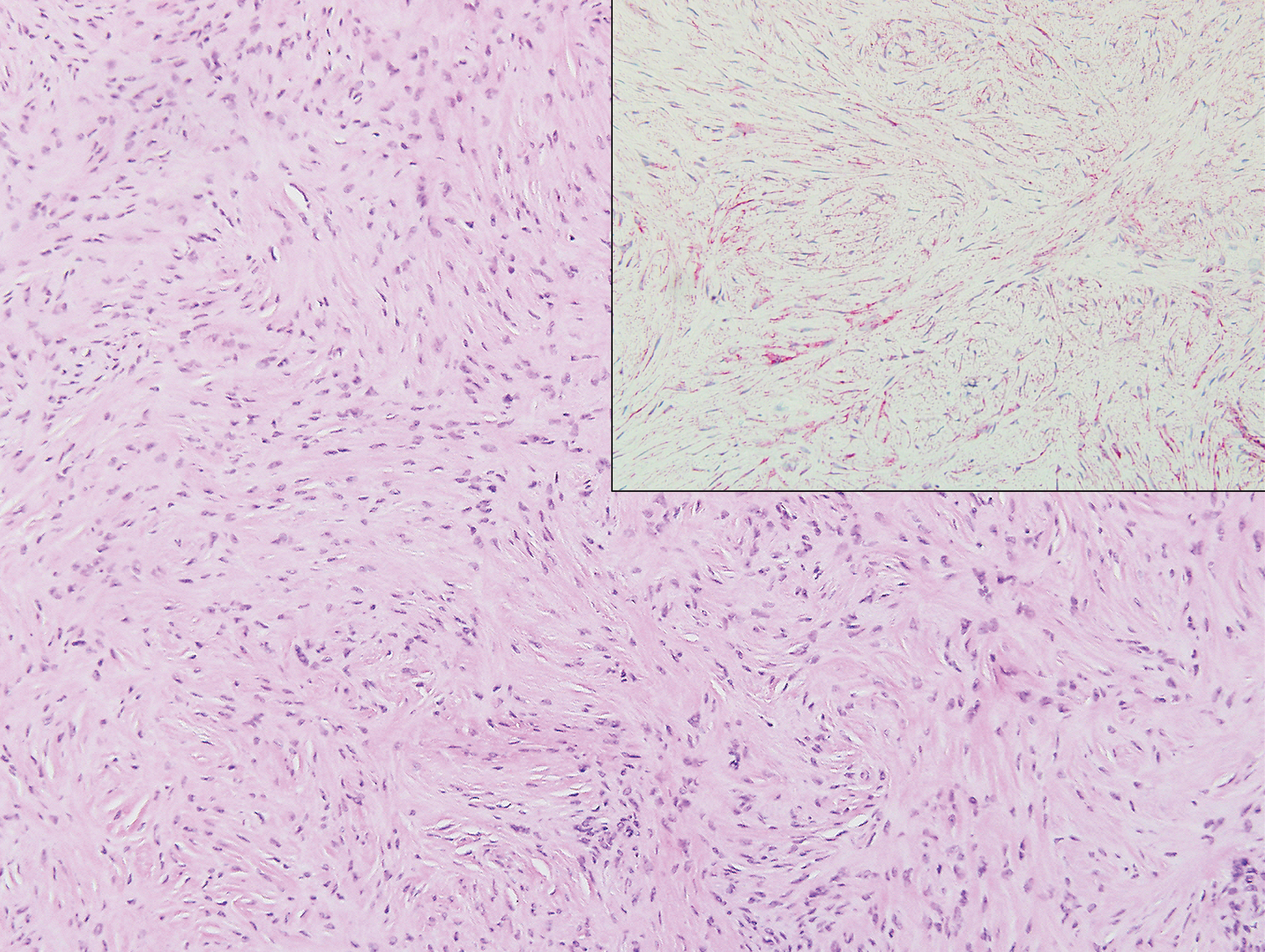
A 25-year-old man presented with a flesh-colored papule on the left side of the nose of 2 years' duration.
Systemic Medications Linked to an Increased Risk for Skin Malignancy
Dermatologists are increasingly called on to evaluate patients with complex medical problems who are often taking many medications. Over the last several decades, many new drugs that target molecular pathways in carcinogenesis and the inflammatory immune system have been developed. Increased skin cancer risk has been reported in association with BRAF inhibitors, sonic hedgehog–inhibiting agents, Janus kinase (JAK) inhibitors, and phosphodiesterase 5 (PDE-5) inhibitors. We review the literature and data regarding the significance and strength of these associations and the molecular pathways by which these medications promote cutaneous tumorigenesis. The association of skin cancer with drugs that either induce photosensitivity—nonsteroidal anti-inflammatory drugs, antibiotics (eg, tetracyclines, fluoroquinolones, trimethoprim-sulfamethoxazole), voriconazole, thiazides—or suppress the immune system—certain biologics (eg, anti–tumor necrosis factor agents), calcineurin inhibitors, thiopurines, methotrexate, cyclosporine—is well known and is therefore not reviewed in this discussion.
BRAF Inhibitors
The mitogen-activated protein kinase (MAPK) pathway (also known as the RAS/RAF/MAPK signaling pathway) is important in growth factor–receptor signaling and plays a key role in cell differentiation, survival, and proliferation. Activating mutations in this pathway allow cells to grow and proliferate in a growth factor–independent manner. Twenty percent of human cancers harbor a mutation in the RAS oncogene, an upstream mediator of the pathway.1 Activating mutations in BRAF, a serine/threonine kinase, predominate in cutaneous melanoma and also have been found in 40% to 70% of papillary thyroid malignancies, 10% to 20% of cholangiocarcinomas, and 5% to 20% of colorectal carcinomas. The most common BRAF mutation in cutaneous melanoma is V600E, which involves a glutamic acid for valine substitution at codon 600. This mutation activates BRAF 500-fold and is present in approximately 50% of melanomas.1,2
Vemurafenib, a selective BRAF inhibitor, was approved by the US Food and Drug Administration (FDA) for the treatment of metastatic melanoma in the United States in 2011. Phase 3 trial data demonstrated that vemurafenib resulted in improved survival and decreased risk for disease progression compared to dacarbazine, the former best treatment.3 During phase 1 testing, it became apparent that vemurafenib treatment was associated with a 31% increased risk for squamous cell carcinoma (SCC), most commonly well-differentiated SCC, and keratoacanthomas (KAs).4 This association was confirmed in phase 2 and 3 studies, though the incidence was lower. McArthur et al5 reported a 19% incidence of cutaneous SCC with extended follow-up analysis of the phase 3 trial. Dabrafenib, another BRAF inhibitor, has been similarly associated with increasing the risk for SCC and KA.
In one study, the mean time to development of SCC after initiating vemurafenib therapy was 10 weeks, with lesions reported as early as 3 weeks. Most patients had clinical signs of chronically sun damaged skin; however, a history of SCC was present in only 17%. Most lesions (63%) were characterized as KAs.6
The mechanism for BRAF inhibitor–induced squamoproliferative growth is due to paradoxical activation of the MAPK pathway in cells with wild-type BRAF that harbor upstream-activating mutations in RAS or tyrosine kinase receptors.7 In the presence of a BRAF inhibitor, inactivated BRAF forms heterodimers with wild-type CRAF (a BRAF-CRAF heterodimer). The heterodimer forms a complex with the mutant RAS that leads to transactivation of the CRAF molecule,8,9 resulting in a paradoxical increase in MAPK signaling and consequent ERK phosphorylation and activation through CRAF signaling. RAS, particularly HRAS, mutations have been found in 60% of all vemurafenib-associated SCCs and KAs. For this reason, it is thought that vemurafenib potentiates tumorigenesis in subclinical lesions harboring upstream MAPK pathway mutations as opposed to inducing de novo lesions.6
Because BRAF inhibitors are remarkably efficacious in the treatment of metastatic melanomas harboring the V600E BRAF mutation, there are no restrictions on their use, despite the known increased risk for SCC. Squamous cell carcinomas tend to be low grade, and all tumors that developed in phase 1 to 3 trials were treated with simple excision. The development of SCC did not necessitate interruption of treatment. Furthermore, the addition of MEK inhibition to BRAF inhibitor therapy reduces the risk for SCC from 19% to 7%.7,10,11
In addition to SCC, second primary melanomas (SPMs) have been reported in patients treated with BRAF inhibitors. It has been shown that these melanomas occur in melanocytes with wild-type BRAF. It has been postulated that some of these tumors occur in cells that harbor upstream mutations in RAS, whereas others might result from alternate signaling through non-RAF oncogenic pathways.9,12
Zimmer et al1 reported 12 SPMs in 11 patients treated with BRAF inhibitor therapy. They reported a median delay of 8 weeks (range, 4–27 weeks) for SPM development. Tumors were detected in early stages; 1 tumor harbored an NRAS mutation.1
Dalle et al13 reported 25 SPMs in 120 vemurafenib-treated patients. Median delay in SPM development was 14 weeks (range, 4–42 weeks). All tumors were thin, ranging from in situ to 0.45-mm thick. Wild-type BRAF was detected in the 21 melanomas sampled; 1 lesion showed mutated NRAS.13
The exact incidence of SPM in the setting of BRAF inhibition is thought to be at least 10-fold less than SCC and KA.2 Patients on BRAF inhibitor therapy should have routine full-body skin examinations, given the increased risk for SPM and SCC.
Another drug belonging to the tyrosine kinase inhibitor family, sorafenib, is used in the treatment of solid tumors, particularly hepatocellular and renal cell carcinomas, and also has been associated with development of cutaneous SCC and KAs.14 Sorafenib is a multiple tyrosine kinase inhibitor that also inhibits the RAF serine/threonine kinases. Similar to vemurafenib and dabrafenib, SCCs and KAs associated with sorafenib tend to arise in patients with chronic actinic damage during the first 2 months of treatment. It has been hypothesized that inhibition of RAF kinases is pathogenic in inducing SCCs because these lesions have not been reported with sunitinib, another multiple tyrosine kinase inhibitor that lacks the ability to inhibit serine/threonine kinases.15,16 Although SCCs and KAs associated with sorafenib tend to be low grade, it is reasonable to consider sunitinib or an alternative tyrosine kinase inhibitor in patients who develop multiple SCCs while taking sorafenib.16
Sonic Hedgehog–Inhibiting Agents
Vismodegib, the first small molecule inhibitor of the signaling protein smoothened, gained FDA approval for the treatment of metastatic or locally advanced basal cell carcinoma (BCC) in 2012. A second agent with an identical mechanism of action, sonidegib, was approved by the FDA for locally advanced BCC in 2015. Approximately 90% of BCCs contain mutations in the sonic hedgehog pathway, which lead to constitutive smoothened activation and uncontrolled cell proliferation.17 The development of smoothened inhibitors introduced a much-needed treatment for inoperable or metastatic BCC,17,18 though long-term utility is limited by drug resistance with extended use in this patient population.19,20 Several case reports have documented the emergence of KA21 and cutaneous SCC following vismodegib treatment of advanced or metastatic BCC.22-24 A larger case-control study by Mohan et al25 showed that patients with BCC treated with vismodegib had an increased risk for non-BCC malignancy (hazard ratio [HR]=6.37), most of which were cutaneous SCC (HR=8.12).
The mechanism by which selective inhibition of smoothened leads to cutaneous SCC is unclear. A study found that patients on vismodegib who developed SCC within the original BCC site had elevated ERK levels within tumor tissue, suggesting that the RAS/RAF/MAPK pathway can become upregulated during hedgehog inhibition.26 Other studies looking at hedgehog inhibition in medulloblastoma models also have shown activated RAS/RAF/MAPK pathways.25 These findings suggest that tumors under smoothened inhibition might be able to bypass the sonic hedgehog pathway and continue to grow by upregulating alternative growth pathways, such as RAS/RAF/MAPK.25,26
The incidence of cutaneous SCC following vismodegib treatment is unknown. Chang and Oro27 examined BCC tumor regrowth from secondary (acquired) resistance to vismodegib and noted that lesions recurred within 1 cm of the original tumor 21% of the time. Although none of the 12 patients whose tumors regrew during treatment were reported to have developed SCC, several demonstrated different BCC subtypes than the pretreatment specimen. The authors proposed that regrowth of BCC was due to upregulated alternative pathways allowing tumors to bypass smoothened inhibition, which is similar to the proposed mechanism for SCC development in vismodegib patients.27
Prospective studies are needed to confirm the link between vismodegib and cutaneous SCC; establish the incidence of SCC development; and identify any pretreatment factors, tumor characteristics, or treatment details (eg, dosage, duration) that might contribute to SCC development. Furthermore, because Mohan et al25 observed that vismodegib-treated patients were less likely to develop SCC in situ than controls, it is unknown if these tumors are more aggressive than traditional SCC. At this point, careful surveillance and regular full-body skin examinations are advised for patients on vismodegib for treatment of advanced BCC.
JAK Inhibitors
Another class of medications potentially associated with increased development of nonmelanoma skin cancer (NMSC) is the JAK inhibitors (also known as jakinibs). Many proinflammatory signaling pathways converge on the JAK family of enzymes—JAK1, JAK2, JAK3, and TYK2. These enzymes operate in cytokine signal transduction by phosphorylating activated cytokine receptors, which allows for recruitment and activation by means of phosphorylation of transcription factors collectively known as signal transducers and activators of transcription (STATs). Phosphorylated STATs dimerize and translocate to the nucleus, acting as direct transcription promoters. Janus kinase inhibitors modulate the immune response by reducing the effect of interleukin and interferon signaling.
Ruxolitinib, a JAK1/JAK2 inhibitor, was the first JAK inhibitor approved by the FDA and is indicated for the treatment of myelofibrosis and polycythemia vera. Additionally, oral and topical JAK inhibitors have shown efficacy in the treatment of psoriasis, rheumatoid arthritis, alopecia areata, vitiligo, and pruritus from atopic dermatitis.28
The JAK-STAT pathway is complex, and the biological activity of the pathway is both proinflammatory and pro–cell survival and proliferation. Because signaling through the pathway can increase angiogenesis and inhibit apoptosis, inhibition of this pathway has been exploited for the treatment of some tumors. However, inhibition of interferon and proinflammatory interleukin signaling also can potentially promote tumor growth by means of inhibition of downstream cytotoxic T-cell signaling, theoretically increasing the risk for NMSC. A study examining the 5-year efficacy of ruxolitinib in myelofibrosis patients (COMFORT-II trial) found that 17.1% of patients developed NMSC compared to only 2.7% of those on the best available therapy. After adjustment by patient exposure, the NMSC rate was still doubled for ruxolitinib-treated patients compared to controls (6.1/100 patient-years and 3.0/100 patient-years, respectively).29 Eighty-week follow-up of the phase 3 clinical trial of ruxolitinib for the treatment of polycythemia vera also noted an increased incidence of NMSC, albeit a more conservative increase. Patients randomized to the ruxolitinib treatment group developed NMSC at a rate of 4.4/100 patient-years, whereas the rate for controls treated with best available therapy was 2.7/100 patient-years.30 In contrast, 5-year follow-up of the COMFORT-I trial, also examining the efficacy of ruxolitinib in myelofibrosis, showed no increased risk for NMSC between ruxolitinib-treated patients and placebo (2.7/100 patient-years and 3.9/100 patient-years, respectively).31
A 2017 case series described 5 patients with myelofibrosis who developed multiple skin cancers with aggressive features while receiving ruxolitinib.32 Duration of ruxolitinib therapy ranged from 4 months to 4 years; 3 patients had a history of hydroxyurea exposure, and only 1 patient had a history of NMSC. High-risk cutaneous SCC, undifferentiated pleomorphic sarcoma, and lentigo maligna melanoma (Breslow thickness, 0.45 mm) were among the tumors reported in this series. Although no definitive conclusion can be made regarding the causality of JAK inhibitors in promoting these tumors, the association warrants further investigation. Clinicians should be aware that ruxolitinib might amplify the risk for NMSC in patients with pre-existing genetic or exposure-related susceptibility. Interruption of drug therapy may be necessary in managing patients who develop an aggressive tumor.32
In contrast, tofacitinib, which specifically inhibits JAK3, carries very low risk, if any, for NMSC when used for the treatment of psoriasis and rheumatoid arthritis. Results from 2 phase 3 trials analyzing the efficacy of tofacitinib in psoriasis demonstrated that only 2 of 1486 patients treated developed NMSC compared to none in the control group.33 Furthermore, analysis of NMSC across the tofacitinib rheumatoid arthritis clinical program, which included a total of 15,103 patient-years of exposure, demonstrated that the overall NMSC incidence was 0.55 for every 100 patient-years. Of note, the risk in patients receiving high-dose treatment (10 mg vs 5 mg) was nearly doubled in long-term follow-up studies (0.79/100 patient-years and 0.41/100 patient-years, respectively). Overall, the study concluded that treatment with tofacitinib presents no greater increased risk for NMSC than treatment with tumor necrosis factor inhibitors.33
PDE-5 Inhibitors
Phosphodiesterase 5 inhibitors, such as sildenafil citrate, have been widely prescribed for the treatment of erectile dysfunction. Studies have shown that BRAF-activated melanomas, which occur in approximately 50% to 70% of melanomas, also result in reduced PDE-5 expression.34-36 In these melanomas, downregulation of PDE-5 results in increased intracellular calcium,36 which has been shown to induce melanoma invasion.36,37 Given this similarity in molecular pathway between BRAF-activated melanomas and PDE-5 inhibitors, there has been increased concern that PDE-5 inhibitors might be associated with an increased risk for melanoma.
In 2014, Li et al38 published a retrospective analysis suggesting an association with sildenafil and an increased risk for melanoma. Their study utilized the Health Professionals Follow-up Study to identify a statistically significant elevation in the risk for invasive melanoma with both recent sildenafil use (multivariate-adjusted HR=2.24) and use at any time (HR=1.92). These results controlled for confounding variables, such as presence of major chronic disease, use of other erectile dysfunction treatments, family history of melanoma, history of sun exposure, and UV index of the patient’s residence. Notably, the study also found that sildenafil did not affect the incidence of BCC or SCC.38
In 2015, Loeb et al39 also examined the potential association between PDE-5 inhibitors and melanoma. Review of several Swedish drug and cancer registries allowed for analysis of melanoma risk and PDE-5 inhibitor use, based on number of prescriptions filled and type of PDE-5 inhibitor prescribed. Their analysis showed that men developing melanoma were more likely than nonmelanoma controls to have taken a PDE-5 inhibitor (11% vs 8%). In a subgroup analysis, however, statistical significance was shown for men with only a single prescription filled (34% of cases; P<.05), whereas the difference for men with multiple filled prescriptions did not meet statistical significance. Furthermore, the study did not find increased risk with longer-acting tadalafil and vardenafil (odds ratio [OR]=1.16) compared to sildenafil (OR=1.14). Last, use of PDE-5 inhibitors was only associated with stage 0 (OR=1.49) and stage I (OR=1.21) tumors, not with stages II to IV (OR=0.83) tumors. Although there was a statistically significant association between PDE-5 inhibitors and malignant melanoma (P<.05), the subgroup analysis findings pointed away from a causal relationship and likely toward a confounding of variable(s).39
A 2016 study by Lian et al40 looked at the risk for melanoma in a cohort of patients diagnosed with erectile dysfunction. No association between PDE-5 inhibitors and melanoma risk was shown when comparing patients who received a PDE-5 inhibitor and those who did not receive a PDE-5 inhibitor. However, secondary analysis did show that melanoma risk was increased among patients receiving more pills (34%) and prescriptions (30%). The authors concluded that there was no association between PDE-5 inhibitor use and overall increased risk for melanoma, and the increased risk associated with a greater number of pills and prescriptions would require further study.40
In contrast, a 2017 meta-analysis by Tang et al41 of 5 studies (3 of which were the aforementioned trials38-40) concluded that use of PDE-5 inhibitors was associated with a small but significantly increased risk for melanoma (OR=1.12) and BCC (OR=1.14) but not SCC. Furthermore, the study found no evidence of dosage-dependent association between PDE-5 inhibitor use and melanoma risk.41
Overall, clinical studies have been inconclusive in determining the risk for melanoma in the setting of PDE-5 inhibitor use. Studies showing an increased rate of melanoma within patient cohorts receiving PDE-5 inhibitors are limited; results might be affected by confounding variables. However, given the similarity in mechanism between PDE-5 inhibitors and HRAS-activated melanomas, it is reasonable to continue research into this potential association.
Conclusion
Since the turn of the century, drugs targeting cell-signaling pathways have been developed to treat inflammatory, oncologic, and immune conditions. The role of immunosuppressants in promoting skin cancer is well established and supported by a vast literature base. However, associations are less clear with newer immunomodulatory and antineoplastic medications. Skin cancer has been reported in association with BRAF inhibitors, sonic hedgehog–inhibiting agents, JAK inhibitors, and PDE-5 inhibitors. In the case of JAK and PDE-5 inhibitors, the increased risk for melanoma and NMSC is somewhat inconclusive; risk is more firmly established for BRAF inhibitors and smoothened inhibitors. For the antineoplastic agents reviewed, the therapeutic effect of cancer regression is well documented, and benefits of continued therapy outweigh the increased risk for skin cancer promotion in nearly all cases. The value of early detection has been well documented for skin malignancy; therefore, increased skin surveillance and prompt management of suspicious lesions should be a priority for physicians treating patients undergoing therapy with these medications
- Zimmer L, Hillen U, Livingstone E, et al. Atypical melanocytic proliferations and new primary melanoma in patients with advanced melanoma undergoing selective BRAF inhibition. J Clin Oncol. 2012;30:2375-2383.
- Long GV, Menzies AM, Nagrial AM, et al. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J Clin Oncol. 2011;29:1239-1246.
- Chapman PB, Hauschild A, Robert C, et al; BRIM-3 Study Group. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507-2516.
- Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809-819.
- McArthur GA, Chapman PB, Robert C, et al. Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM-3): extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol. 2014;15:323-332.
- Su F, Viros A, Milagre C, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med. 2012;366:207-215.
- Carlos G, Anforth R, Clements A, et al. Cutaneous toxic effects of BRAF inhibitors alone and in combination with MEK inhibitors for metastatic melanoma. JAMA Dermatol. 2015;151:1103-1109.
- Poulikakos PI, Zhang C, Bollag G, et al. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464:427-430.
- Ryan MB, Der CJ, Wang-Gillam A, et al. Targeting RAS-mutant cancers: is ERK the key? Trends Cancer. 2015;1:183-198.
- Long GV, Stroyakovskiy D, Gogas H, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med. 2014;371:1877-1888.
- Robert C, Karaszewska B, Schachter J, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015;372:30-39.
- Holderfield M, Nagel TE, Stuart DD. Mechanism and consequence of RAF kinase activation by small-molecule inhibitors. Br J Cancer. 2014;111:640-645.
- Dalle S, Poulalhon N, Debarbieux S, et al. Tracking of second primary melanomas in vemurafenib-treated patients. JAMA Dermatol. 2013;149:488-490.
- Williams VL, Cohen PR, Stewart DJ. Sorafenib-induced premalignant and malignant skin lesions. Int J Dermatol. 2011;50:396-402.
- Arnault JP, Wechsler J, Escudier B, et al. Keratoacanthomas and squamous cell carcinomas in patients receiving sorafenib. J Clin Oncol. 2009;27:e59-e61.
- Smith KJ, Haley H, Hamza S, et al. Eruptive keratoacanthoma-type squamous cell carcinomas in patients taking sorafenib for the treatment of solid tumors. Dermatol Surg. 2009;35:1766-1770.
- Sekulic A, Migden MR, Oro AE, et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N Engl J Med. 2012;366:2171-2179.
- Demirci H, Worden F, Nelson CC, et al. Efficacy of vismodegib (Erivedge) for basal cell carcinoma involving the orbit and periocular area. Ophthalmic Plast Reconstr Surg. 2015;31:463-466.
- Atwood SX, Sarin KY, Whitson RJ, et al. Smoothened variants explain the majority of drug resistance in basal cell carcinoma. Cancer Cell. 2015;27:342-353.
- Ridky TW, Cotsarelis G. Vismodegib resistance in basal cell carcinoma: not a smooth fit. Cancer Cell. 2015;27:315-316.
- Aasi S, Silkiss R, Tang JY, et al. New onset of keratoacanthomas after vismodegib treatment for locally advanced basal cell carcinomas: a report of 2 cases. JAMA Dermatol. 2013;149:242-243.
- Orouji A, Goerdt S, Utikal J, et al. Multiple highly and moderately differentiated squamous cell carcinomas of the skin during vismodegib treatment of inoperable basal cell carcinoma. Br J Dermatol. 2014;171:431-433.
- Iarrobino A, Messina JL, Kudchadkar R, et al. Emergence of a squamous cell carcinoma phenotype following treatment of metastatic basal cell carcinoma with vismodegib. J Am Acad Dermatol. 2013;69:e33-e34.
- Saintes C, Saint-Jean M, Brocard A, et al. Development of squamous cell carcinoma into basal cell carcinoma under treatment with vismodegib. J Eur Acad Dermatol Venereol. 2015;29:1006-1009.
- Mohan SV, Chang J, Li S, et al. Increased risk of cutaneous squamous cell carcinoma after vismodegib therapy for basal cell carcinoma. JAMA Dermatol. 2016;152:527-532.
- Zhao X, Ponomaryov T, Ornell KJ, et al. RAS/MAPK activation drives resistance to Smo inhibition, metastasis, and tumor evolution in Shh pathway-dependent tumors. Cancer Res. 2015;75:3623-3635.
- Chang AL, Oro AE. Initial assessment of tumor regrowth after vismodegib in advanced basal cell carcinoma. Arch Dermatol. 2012;148:1324-1325.
- Damsky W, King BA. JAK inhibitors in dermatology: the promise of a new drug class. J Am Acad Dermatol. 2017;76:736-744.
- Harrison CN, Vannucchi AM, Kiladjian JJ, et al. Long-term findings from COMFORT-II, a phase 3 study of ruxolitinib vs best available therapy for myelofibrosis. Leukemia. 2016;30:1701-1707.
- Verstovsek S, Vannucchi AM, Griesshammer M, et al. Ruxolitinib versus best available therapy in patients with polycythemia vera: 80-week follow-up from the RESPONSE trial. Haematologica. 2016;101:821-829.
- Verstovsek S, Mesa RA, Gotlib J, et al; COMFORT-I investigators. Long-term treatment with ruxolitinib for patients with myelofibrosis: 5-year update from the randomized, double-blind, placebo-controlled, phase 3 COMFORT-I trial. J Hematol Oncol. 2017;10:55.
- Blechman AB, Cabell CE, Weinberger CH, et al. Aggressive skin cancers occurring in patients treated with the Janus kinase inhibitor ruxolitinib. J Drugs Dermatol. 2017;16:508-511.
- Papp KA, Menter MA, Abe M, et al; OPT Pivotal 1 and OPT Pivotal 2 investigators. Tofacitinib, an oral Janus kinase inhibitor, for the treatment of chronic plaque psoriasis: results from two randomized, placebo-controlled, phase III trials. Br J Dermatol. 2015;173:949-961.
- Wellbrock C, Karasarides M, Marais R. The RAF proteins take centre stage. Nat Rev Mol Cell Biol. 2004;5:875-885.
- Gray-Schopfer V, Wellbrock C, Marais R. Melanoma biology and new targeted therapy. Nature. 2007;445:851-857.
- Arozarena I, Sanchez-Laorden B, Packer L, et al. Oncogenic BRAF induces melanoma cell invasion by downregulating the cGMP-specific phosphodiesterase PDE5A. Cancer Cell. 2011;19:45-57.
- Houslay MD. Hard times for oncogenic BRAF-expressing melanoma cells. Cancer Cell. 2011;19:3-4.
- Li WQ, Qureshi AA, Robinson KC, et al. Sildenafil use and increased risk of incident melanoma in US men: a prospective cohort study. JAMA Intern Med. 2014;174:964-970.
- Loeb S, Folkvaljon Y, Lambe M, et al. Use of phosphodiesterase type 5 inhibitors for erectile dysfunction and risk of malignant melanoma. JAMA. 2015;313:2449-2455.
- Lian Y, Yin H, Pollak MN, et al. Phosphodiesterase type 5 inhibitors and the risk of melanoma skin cancer. Eur Urol. 2016;70:808-815.
- Tang H, Wu W, Fu S, et al. Phosphodiesterase type 5 inhibitors and risk of melanoma: a meta-analysis. J Am Acad Dermatol. 2017;77:480.e9-488.e9.
Dermatologists are increasingly called on to evaluate patients with complex medical problems who are often taking many medications. Over the last several decades, many new drugs that target molecular pathways in carcinogenesis and the inflammatory immune system have been developed. Increased skin cancer risk has been reported in association with BRAF inhibitors, sonic hedgehog–inhibiting agents, Janus kinase (JAK) inhibitors, and phosphodiesterase 5 (PDE-5) inhibitors. We review the literature and data regarding the significance and strength of these associations and the molecular pathways by which these medications promote cutaneous tumorigenesis. The association of skin cancer with drugs that either induce photosensitivity—nonsteroidal anti-inflammatory drugs, antibiotics (eg, tetracyclines, fluoroquinolones, trimethoprim-sulfamethoxazole), voriconazole, thiazides—or suppress the immune system—certain biologics (eg, anti–tumor necrosis factor agents), calcineurin inhibitors, thiopurines, methotrexate, cyclosporine—is well known and is therefore not reviewed in this discussion.
BRAF Inhibitors
The mitogen-activated protein kinase (MAPK) pathway (also known as the RAS/RAF/MAPK signaling pathway) is important in growth factor–receptor signaling and plays a key role in cell differentiation, survival, and proliferation. Activating mutations in this pathway allow cells to grow and proliferate in a growth factor–independent manner. Twenty percent of human cancers harbor a mutation in the RAS oncogene, an upstream mediator of the pathway.1 Activating mutations in BRAF, a serine/threonine kinase, predominate in cutaneous melanoma and also have been found in 40% to 70% of papillary thyroid malignancies, 10% to 20% of cholangiocarcinomas, and 5% to 20% of colorectal carcinomas. The most common BRAF mutation in cutaneous melanoma is V600E, which involves a glutamic acid for valine substitution at codon 600. This mutation activates BRAF 500-fold and is present in approximately 50% of melanomas.1,2
Vemurafenib, a selective BRAF inhibitor, was approved by the US Food and Drug Administration (FDA) for the treatment of metastatic melanoma in the United States in 2011. Phase 3 trial data demonstrated that vemurafenib resulted in improved survival and decreased risk for disease progression compared to dacarbazine, the former best treatment.3 During phase 1 testing, it became apparent that vemurafenib treatment was associated with a 31% increased risk for squamous cell carcinoma (SCC), most commonly well-differentiated SCC, and keratoacanthomas (KAs).4 This association was confirmed in phase 2 and 3 studies, though the incidence was lower. McArthur et al5 reported a 19% incidence of cutaneous SCC with extended follow-up analysis of the phase 3 trial. Dabrafenib, another BRAF inhibitor, has been similarly associated with increasing the risk for SCC and KA.
In one study, the mean time to development of SCC after initiating vemurafenib therapy was 10 weeks, with lesions reported as early as 3 weeks. Most patients had clinical signs of chronically sun damaged skin; however, a history of SCC was present in only 17%. Most lesions (63%) were characterized as KAs.6
The mechanism for BRAF inhibitor–induced squamoproliferative growth is due to paradoxical activation of the MAPK pathway in cells with wild-type BRAF that harbor upstream-activating mutations in RAS or tyrosine kinase receptors.7 In the presence of a BRAF inhibitor, inactivated BRAF forms heterodimers with wild-type CRAF (a BRAF-CRAF heterodimer). The heterodimer forms a complex with the mutant RAS that leads to transactivation of the CRAF molecule,8,9 resulting in a paradoxical increase in MAPK signaling and consequent ERK phosphorylation and activation through CRAF signaling. RAS, particularly HRAS, mutations have been found in 60% of all vemurafenib-associated SCCs and KAs. For this reason, it is thought that vemurafenib potentiates tumorigenesis in subclinical lesions harboring upstream MAPK pathway mutations as opposed to inducing de novo lesions.6
Because BRAF inhibitors are remarkably efficacious in the treatment of metastatic melanomas harboring the V600E BRAF mutation, there are no restrictions on their use, despite the known increased risk for SCC. Squamous cell carcinomas tend to be low grade, and all tumors that developed in phase 1 to 3 trials were treated with simple excision. The development of SCC did not necessitate interruption of treatment. Furthermore, the addition of MEK inhibition to BRAF inhibitor therapy reduces the risk for SCC from 19% to 7%.7,10,11
In addition to SCC, second primary melanomas (SPMs) have been reported in patients treated with BRAF inhibitors. It has been shown that these melanomas occur in melanocytes with wild-type BRAF. It has been postulated that some of these tumors occur in cells that harbor upstream mutations in RAS, whereas others might result from alternate signaling through non-RAF oncogenic pathways.9,12
Zimmer et al1 reported 12 SPMs in 11 patients treated with BRAF inhibitor therapy. They reported a median delay of 8 weeks (range, 4–27 weeks) for SPM development. Tumors were detected in early stages; 1 tumor harbored an NRAS mutation.1
Dalle et al13 reported 25 SPMs in 120 vemurafenib-treated patients. Median delay in SPM development was 14 weeks (range, 4–42 weeks). All tumors were thin, ranging from in situ to 0.45-mm thick. Wild-type BRAF was detected in the 21 melanomas sampled; 1 lesion showed mutated NRAS.13
The exact incidence of SPM in the setting of BRAF inhibition is thought to be at least 10-fold less than SCC and KA.2 Patients on BRAF inhibitor therapy should have routine full-body skin examinations, given the increased risk for SPM and SCC.
Another drug belonging to the tyrosine kinase inhibitor family, sorafenib, is used in the treatment of solid tumors, particularly hepatocellular and renal cell carcinomas, and also has been associated with development of cutaneous SCC and KAs.14 Sorafenib is a multiple tyrosine kinase inhibitor that also inhibits the RAF serine/threonine kinases. Similar to vemurafenib and dabrafenib, SCCs and KAs associated with sorafenib tend to arise in patients with chronic actinic damage during the first 2 months of treatment. It has been hypothesized that inhibition of RAF kinases is pathogenic in inducing SCCs because these lesions have not been reported with sunitinib, another multiple tyrosine kinase inhibitor that lacks the ability to inhibit serine/threonine kinases.15,16 Although SCCs and KAs associated with sorafenib tend to be low grade, it is reasonable to consider sunitinib or an alternative tyrosine kinase inhibitor in patients who develop multiple SCCs while taking sorafenib.16
Sonic Hedgehog–Inhibiting Agents
Vismodegib, the first small molecule inhibitor of the signaling protein smoothened, gained FDA approval for the treatment of metastatic or locally advanced basal cell carcinoma (BCC) in 2012. A second agent with an identical mechanism of action, sonidegib, was approved by the FDA for locally advanced BCC in 2015. Approximately 90% of BCCs contain mutations in the sonic hedgehog pathway, which lead to constitutive smoothened activation and uncontrolled cell proliferation.17 The development of smoothened inhibitors introduced a much-needed treatment for inoperable or metastatic BCC,17,18 though long-term utility is limited by drug resistance with extended use in this patient population.19,20 Several case reports have documented the emergence of KA21 and cutaneous SCC following vismodegib treatment of advanced or metastatic BCC.22-24 A larger case-control study by Mohan et al25 showed that patients with BCC treated with vismodegib had an increased risk for non-BCC malignancy (hazard ratio [HR]=6.37), most of which were cutaneous SCC (HR=8.12).
The mechanism by which selective inhibition of smoothened leads to cutaneous SCC is unclear. A study found that patients on vismodegib who developed SCC within the original BCC site had elevated ERK levels within tumor tissue, suggesting that the RAS/RAF/MAPK pathway can become upregulated during hedgehog inhibition.26 Other studies looking at hedgehog inhibition in medulloblastoma models also have shown activated RAS/RAF/MAPK pathways.25 These findings suggest that tumors under smoothened inhibition might be able to bypass the sonic hedgehog pathway and continue to grow by upregulating alternative growth pathways, such as RAS/RAF/MAPK.25,26
The incidence of cutaneous SCC following vismodegib treatment is unknown. Chang and Oro27 examined BCC tumor regrowth from secondary (acquired) resistance to vismodegib and noted that lesions recurred within 1 cm of the original tumor 21% of the time. Although none of the 12 patients whose tumors regrew during treatment were reported to have developed SCC, several demonstrated different BCC subtypes than the pretreatment specimen. The authors proposed that regrowth of BCC was due to upregulated alternative pathways allowing tumors to bypass smoothened inhibition, which is similar to the proposed mechanism for SCC development in vismodegib patients.27
Prospective studies are needed to confirm the link between vismodegib and cutaneous SCC; establish the incidence of SCC development; and identify any pretreatment factors, tumor characteristics, or treatment details (eg, dosage, duration) that might contribute to SCC development. Furthermore, because Mohan et al25 observed that vismodegib-treated patients were less likely to develop SCC in situ than controls, it is unknown if these tumors are more aggressive than traditional SCC. At this point, careful surveillance and regular full-body skin examinations are advised for patients on vismodegib for treatment of advanced BCC.
JAK Inhibitors
Another class of medications potentially associated with increased development of nonmelanoma skin cancer (NMSC) is the JAK inhibitors (also known as jakinibs). Many proinflammatory signaling pathways converge on the JAK family of enzymes—JAK1, JAK2, JAK3, and TYK2. These enzymes operate in cytokine signal transduction by phosphorylating activated cytokine receptors, which allows for recruitment and activation by means of phosphorylation of transcription factors collectively known as signal transducers and activators of transcription (STATs). Phosphorylated STATs dimerize and translocate to the nucleus, acting as direct transcription promoters. Janus kinase inhibitors modulate the immune response by reducing the effect of interleukin and interferon signaling.
Ruxolitinib, a JAK1/JAK2 inhibitor, was the first JAK inhibitor approved by the FDA and is indicated for the treatment of myelofibrosis and polycythemia vera. Additionally, oral and topical JAK inhibitors have shown efficacy in the treatment of psoriasis, rheumatoid arthritis, alopecia areata, vitiligo, and pruritus from atopic dermatitis.28
The JAK-STAT pathway is complex, and the biological activity of the pathway is both proinflammatory and pro–cell survival and proliferation. Because signaling through the pathway can increase angiogenesis and inhibit apoptosis, inhibition of this pathway has been exploited for the treatment of some tumors. However, inhibition of interferon and proinflammatory interleukin signaling also can potentially promote tumor growth by means of inhibition of downstream cytotoxic T-cell signaling, theoretically increasing the risk for NMSC. A study examining the 5-year efficacy of ruxolitinib in myelofibrosis patients (COMFORT-II trial) found that 17.1% of patients developed NMSC compared to only 2.7% of those on the best available therapy. After adjustment by patient exposure, the NMSC rate was still doubled for ruxolitinib-treated patients compared to controls (6.1/100 patient-years and 3.0/100 patient-years, respectively).29 Eighty-week follow-up of the phase 3 clinical trial of ruxolitinib for the treatment of polycythemia vera also noted an increased incidence of NMSC, albeit a more conservative increase. Patients randomized to the ruxolitinib treatment group developed NMSC at a rate of 4.4/100 patient-years, whereas the rate for controls treated with best available therapy was 2.7/100 patient-years.30 In contrast, 5-year follow-up of the COMFORT-I trial, also examining the efficacy of ruxolitinib in myelofibrosis, showed no increased risk for NMSC between ruxolitinib-treated patients and placebo (2.7/100 patient-years and 3.9/100 patient-years, respectively).31
A 2017 case series described 5 patients with myelofibrosis who developed multiple skin cancers with aggressive features while receiving ruxolitinib.32 Duration of ruxolitinib therapy ranged from 4 months to 4 years; 3 patients had a history of hydroxyurea exposure, and only 1 patient had a history of NMSC. High-risk cutaneous SCC, undifferentiated pleomorphic sarcoma, and lentigo maligna melanoma (Breslow thickness, 0.45 mm) were among the tumors reported in this series. Although no definitive conclusion can be made regarding the causality of JAK inhibitors in promoting these tumors, the association warrants further investigation. Clinicians should be aware that ruxolitinib might amplify the risk for NMSC in patients with pre-existing genetic or exposure-related susceptibility. Interruption of drug therapy may be necessary in managing patients who develop an aggressive tumor.32
In contrast, tofacitinib, which specifically inhibits JAK3, carries very low risk, if any, for NMSC when used for the treatment of psoriasis and rheumatoid arthritis. Results from 2 phase 3 trials analyzing the efficacy of tofacitinib in psoriasis demonstrated that only 2 of 1486 patients treated developed NMSC compared to none in the control group.33 Furthermore, analysis of NMSC across the tofacitinib rheumatoid arthritis clinical program, which included a total of 15,103 patient-years of exposure, demonstrated that the overall NMSC incidence was 0.55 for every 100 patient-years. Of note, the risk in patients receiving high-dose treatment (10 mg vs 5 mg) was nearly doubled in long-term follow-up studies (0.79/100 patient-years and 0.41/100 patient-years, respectively). Overall, the study concluded that treatment with tofacitinib presents no greater increased risk for NMSC than treatment with tumor necrosis factor inhibitors.33
PDE-5 Inhibitors
Phosphodiesterase 5 inhibitors, such as sildenafil citrate, have been widely prescribed for the treatment of erectile dysfunction. Studies have shown that BRAF-activated melanomas, which occur in approximately 50% to 70% of melanomas, also result in reduced PDE-5 expression.34-36 In these melanomas, downregulation of PDE-5 results in increased intracellular calcium,36 which has been shown to induce melanoma invasion.36,37 Given this similarity in molecular pathway between BRAF-activated melanomas and PDE-5 inhibitors, there has been increased concern that PDE-5 inhibitors might be associated with an increased risk for melanoma.
In 2014, Li et al38 published a retrospective analysis suggesting an association with sildenafil and an increased risk for melanoma. Their study utilized the Health Professionals Follow-up Study to identify a statistically significant elevation in the risk for invasive melanoma with both recent sildenafil use (multivariate-adjusted HR=2.24) and use at any time (HR=1.92). These results controlled for confounding variables, such as presence of major chronic disease, use of other erectile dysfunction treatments, family history of melanoma, history of sun exposure, and UV index of the patient’s residence. Notably, the study also found that sildenafil did not affect the incidence of BCC or SCC.38
In 2015, Loeb et al39 also examined the potential association between PDE-5 inhibitors and melanoma. Review of several Swedish drug and cancer registries allowed for analysis of melanoma risk and PDE-5 inhibitor use, based on number of prescriptions filled and type of PDE-5 inhibitor prescribed. Their analysis showed that men developing melanoma were more likely than nonmelanoma controls to have taken a PDE-5 inhibitor (11% vs 8%). In a subgroup analysis, however, statistical significance was shown for men with only a single prescription filled (34% of cases; P<.05), whereas the difference for men with multiple filled prescriptions did not meet statistical significance. Furthermore, the study did not find increased risk with longer-acting tadalafil and vardenafil (odds ratio [OR]=1.16) compared to sildenafil (OR=1.14). Last, use of PDE-5 inhibitors was only associated with stage 0 (OR=1.49) and stage I (OR=1.21) tumors, not with stages II to IV (OR=0.83) tumors. Although there was a statistically significant association between PDE-5 inhibitors and malignant melanoma (P<.05), the subgroup analysis findings pointed away from a causal relationship and likely toward a confounding of variable(s).39
A 2016 study by Lian et al40 looked at the risk for melanoma in a cohort of patients diagnosed with erectile dysfunction. No association between PDE-5 inhibitors and melanoma risk was shown when comparing patients who received a PDE-5 inhibitor and those who did not receive a PDE-5 inhibitor. However, secondary analysis did show that melanoma risk was increased among patients receiving more pills (34%) and prescriptions (30%). The authors concluded that there was no association between PDE-5 inhibitor use and overall increased risk for melanoma, and the increased risk associated with a greater number of pills and prescriptions would require further study.40
In contrast, a 2017 meta-analysis by Tang et al41 of 5 studies (3 of which were the aforementioned trials38-40) concluded that use of PDE-5 inhibitors was associated with a small but significantly increased risk for melanoma (OR=1.12) and BCC (OR=1.14) but not SCC. Furthermore, the study found no evidence of dosage-dependent association between PDE-5 inhibitor use and melanoma risk.41
Overall, clinical studies have been inconclusive in determining the risk for melanoma in the setting of PDE-5 inhibitor use. Studies showing an increased rate of melanoma within patient cohorts receiving PDE-5 inhibitors are limited; results might be affected by confounding variables. However, given the similarity in mechanism between PDE-5 inhibitors and HRAS-activated melanomas, it is reasonable to continue research into this potential association.
Conclusion
Since the turn of the century, drugs targeting cell-signaling pathways have been developed to treat inflammatory, oncologic, and immune conditions. The role of immunosuppressants in promoting skin cancer is well established and supported by a vast literature base. However, associations are less clear with newer immunomodulatory and antineoplastic medications. Skin cancer has been reported in association with BRAF inhibitors, sonic hedgehog–inhibiting agents, JAK inhibitors, and PDE-5 inhibitors. In the case of JAK and PDE-5 inhibitors, the increased risk for melanoma and NMSC is somewhat inconclusive; risk is more firmly established for BRAF inhibitors and smoothened inhibitors. For the antineoplastic agents reviewed, the therapeutic effect of cancer regression is well documented, and benefits of continued therapy outweigh the increased risk for skin cancer promotion in nearly all cases. The value of early detection has been well documented for skin malignancy; therefore, increased skin surveillance and prompt management of suspicious lesions should be a priority for physicians treating patients undergoing therapy with these medications
Dermatologists are increasingly called on to evaluate patients with complex medical problems who are often taking many medications. Over the last several decades, many new drugs that target molecular pathways in carcinogenesis and the inflammatory immune system have been developed. Increased skin cancer risk has been reported in association with BRAF inhibitors, sonic hedgehog–inhibiting agents, Janus kinase (JAK) inhibitors, and phosphodiesterase 5 (PDE-5) inhibitors. We review the literature and data regarding the significance and strength of these associations and the molecular pathways by which these medications promote cutaneous tumorigenesis. The association of skin cancer with drugs that either induce photosensitivity—nonsteroidal anti-inflammatory drugs, antibiotics (eg, tetracyclines, fluoroquinolones, trimethoprim-sulfamethoxazole), voriconazole, thiazides—or suppress the immune system—certain biologics (eg, anti–tumor necrosis factor agents), calcineurin inhibitors, thiopurines, methotrexate, cyclosporine—is well known and is therefore not reviewed in this discussion.
BRAF Inhibitors
The mitogen-activated protein kinase (MAPK) pathway (also known as the RAS/RAF/MAPK signaling pathway) is important in growth factor–receptor signaling and plays a key role in cell differentiation, survival, and proliferation. Activating mutations in this pathway allow cells to grow and proliferate in a growth factor–independent manner. Twenty percent of human cancers harbor a mutation in the RAS oncogene, an upstream mediator of the pathway.1 Activating mutations in BRAF, a serine/threonine kinase, predominate in cutaneous melanoma and also have been found in 40% to 70% of papillary thyroid malignancies, 10% to 20% of cholangiocarcinomas, and 5% to 20% of colorectal carcinomas. The most common BRAF mutation in cutaneous melanoma is V600E, which involves a glutamic acid for valine substitution at codon 600. This mutation activates BRAF 500-fold and is present in approximately 50% of melanomas.1,2
Vemurafenib, a selective BRAF inhibitor, was approved by the US Food and Drug Administration (FDA) for the treatment of metastatic melanoma in the United States in 2011. Phase 3 trial data demonstrated that vemurafenib resulted in improved survival and decreased risk for disease progression compared to dacarbazine, the former best treatment.3 During phase 1 testing, it became apparent that vemurafenib treatment was associated with a 31% increased risk for squamous cell carcinoma (SCC), most commonly well-differentiated SCC, and keratoacanthomas (KAs).4 This association was confirmed in phase 2 and 3 studies, though the incidence was lower. McArthur et al5 reported a 19% incidence of cutaneous SCC with extended follow-up analysis of the phase 3 trial. Dabrafenib, another BRAF inhibitor, has been similarly associated with increasing the risk for SCC and KA.
In one study, the mean time to development of SCC after initiating vemurafenib therapy was 10 weeks, with lesions reported as early as 3 weeks. Most patients had clinical signs of chronically sun damaged skin; however, a history of SCC was present in only 17%. Most lesions (63%) were characterized as KAs.6
The mechanism for BRAF inhibitor–induced squamoproliferative growth is due to paradoxical activation of the MAPK pathway in cells with wild-type BRAF that harbor upstream-activating mutations in RAS or tyrosine kinase receptors.7 In the presence of a BRAF inhibitor, inactivated BRAF forms heterodimers with wild-type CRAF (a BRAF-CRAF heterodimer). The heterodimer forms a complex with the mutant RAS that leads to transactivation of the CRAF molecule,8,9 resulting in a paradoxical increase in MAPK signaling and consequent ERK phosphorylation and activation through CRAF signaling. RAS, particularly HRAS, mutations have been found in 60% of all vemurafenib-associated SCCs and KAs. For this reason, it is thought that vemurafenib potentiates tumorigenesis in subclinical lesions harboring upstream MAPK pathway mutations as opposed to inducing de novo lesions.6
Because BRAF inhibitors are remarkably efficacious in the treatment of metastatic melanomas harboring the V600E BRAF mutation, there are no restrictions on their use, despite the known increased risk for SCC. Squamous cell carcinomas tend to be low grade, and all tumors that developed in phase 1 to 3 trials were treated with simple excision. The development of SCC did not necessitate interruption of treatment. Furthermore, the addition of MEK inhibition to BRAF inhibitor therapy reduces the risk for SCC from 19% to 7%.7,10,11
In addition to SCC, second primary melanomas (SPMs) have been reported in patients treated with BRAF inhibitors. It has been shown that these melanomas occur in melanocytes with wild-type BRAF. It has been postulated that some of these tumors occur in cells that harbor upstream mutations in RAS, whereas others might result from alternate signaling through non-RAF oncogenic pathways.9,12
Zimmer et al1 reported 12 SPMs in 11 patients treated with BRAF inhibitor therapy. They reported a median delay of 8 weeks (range, 4–27 weeks) for SPM development. Tumors were detected in early stages; 1 tumor harbored an NRAS mutation.1
Dalle et al13 reported 25 SPMs in 120 vemurafenib-treated patients. Median delay in SPM development was 14 weeks (range, 4–42 weeks). All tumors were thin, ranging from in situ to 0.45-mm thick. Wild-type BRAF was detected in the 21 melanomas sampled; 1 lesion showed mutated NRAS.13
The exact incidence of SPM in the setting of BRAF inhibition is thought to be at least 10-fold less than SCC and KA.2 Patients on BRAF inhibitor therapy should have routine full-body skin examinations, given the increased risk for SPM and SCC.
Another drug belonging to the tyrosine kinase inhibitor family, sorafenib, is used in the treatment of solid tumors, particularly hepatocellular and renal cell carcinomas, and also has been associated with development of cutaneous SCC and KAs.14 Sorafenib is a multiple tyrosine kinase inhibitor that also inhibits the RAF serine/threonine kinases. Similar to vemurafenib and dabrafenib, SCCs and KAs associated with sorafenib tend to arise in patients with chronic actinic damage during the first 2 months of treatment. It has been hypothesized that inhibition of RAF kinases is pathogenic in inducing SCCs because these lesions have not been reported with sunitinib, another multiple tyrosine kinase inhibitor that lacks the ability to inhibit serine/threonine kinases.15,16 Although SCCs and KAs associated with sorafenib tend to be low grade, it is reasonable to consider sunitinib or an alternative tyrosine kinase inhibitor in patients who develop multiple SCCs while taking sorafenib.16
Sonic Hedgehog–Inhibiting Agents
Vismodegib, the first small molecule inhibitor of the signaling protein smoothened, gained FDA approval for the treatment of metastatic or locally advanced basal cell carcinoma (BCC) in 2012. A second agent with an identical mechanism of action, sonidegib, was approved by the FDA for locally advanced BCC in 2015. Approximately 90% of BCCs contain mutations in the sonic hedgehog pathway, which lead to constitutive smoothened activation and uncontrolled cell proliferation.17 The development of smoothened inhibitors introduced a much-needed treatment for inoperable or metastatic BCC,17,18 though long-term utility is limited by drug resistance with extended use in this patient population.19,20 Several case reports have documented the emergence of KA21 and cutaneous SCC following vismodegib treatment of advanced or metastatic BCC.22-24 A larger case-control study by Mohan et al25 showed that patients with BCC treated with vismodegib had an increased risk for non-BCC malignancy (hazard ratio [HR]=6.37), most of which were cutaneous SCC (HR=8.12).
The mechanism by which selective inhibition of smoothened leads to cutaneous SCC is unclear. A study found that patients on vismodegib who developed SCC within the original BCC site had elevated ERK levels within tumor tissue, suggesting that the RAS/RAF/MAPK pathway can become upregulated during hedgehog inhibition.26 Other studies looking at hedgehog inhibition in medulloblastoma models also have shown activated RAS/RAF/MAPK pathways.25 These findings suggest that tumors under smoothened inhibition might be able to bypass the sonic hedgehog pathway and continue to grow by upregulating alternative growth pathways, such as RAS/RAF/MAPK.25,26
The incidence of cutaneous SCC following vismodegib treatment is unknown. Chang and Oro27 examined BCC tumor regrowth from secondary (acquired) resistance to vismodegib and noted that lesions recurred within 1 cm of the original tumor 21% of the time. Although none of the 12 patients whose tumors regrew during treatment were reported to have developed SCC, several demonstrated different BCC subtypes than the pretreatment specimen. The authors proposed that regrowth of BCC was due to upregulated alternative pathways allowing tumors to bypass smoothened inhibition, which is similar to the proposed mechanism for SCC development in vismodegib patients.27
Prospective studies are needed to confirm the link between vismodegib and cutaneous SCC; establish the incidence of SCC development; and identify any pretreatment factors, tumor characteristics, or treatment details (eg, dosage, duration) that might contribute to SCC development. Furthermore, because Mohan et al25 observed that vismodegib-treated patients were less likely to develop SCC in situ than controls, it is unknown if these tumors are more aggressive than traditional SCC. At this point, careful surveillance and regular full-body skin examinations are advised for patients on vismodegib for treatment of advanced BCC.
JAK Inhibitors
Another class of medications potentially associated with increased development of nonmelanoma skin cancer (NMSC) is the JAK inhibitors (also known as jakinibs). Many proinflammatory signaling pathways converge on the JAK family of enzymes—JAK1, JAK2, JAK3, and TYK2. These enzymes operate in cytokine signal transduction by phosphorylating activated cytokine receptors, which allows for recruitment and activation by means of phosphorylation of transcription factors collectively known as signal transducers and activators of transcription (STATs). Phosphorylated STATs dimerize and translocate to the nucleus, acting as direct transcription promoters. Janus kinase inhibitors modulate the immune response by reducing the effect of interleukin and interferon signaling.
Ruxolitinib, a JAK1/JAK2 inhibitor, was the first JAK inhibitor approved by the FDA and is indicated for the treatment of myelofibrosis and polycythemia vera. Additionally, oral and topical JAK inhibitors have shown efficacy in the treatment of psoriasis, rheumatoid arthritis, alopecia areata, vitiligo, and pruritus from atopic dermatitis.28
The JAK-STAT pathway is complex, and the biological activity of the pathway is both proinflammatory and pro–cell survival and proliferation. Because signaling through the pathway can increase angiogenesis and inhibit apoptosis, inhibition of this pathway has been exploited for the treatment of some tumors. However, inhibition of interferon and proinflammatory interleukin signaling also can potentially promote tumor growth by means of inhibition of downstream cytotoxic T-cell signaling, theoretically increasing the risk for NMSC. A study examining the 5-year efficacy of ruxolitinib in myelofibrosis patients (COMFORT-II trial) found that 17.1% of patients developed NMSC compared to only 2.7% of those on the best available therapy. After adjustment by patient exposure, the NMSC rate was still doubled for ruxolitinib-treated patients compared to controls (6.1/100 patient-years and 3.0/100 patient-years, respectively).29 Eighty-week follow-up of the phase 3 clinical trial of ruxolitinib for the treatment of polycythemia vera also noted an increased incidence of NMSC, albeit a more conservative increase. Patients randomized to the ruxolitinib treatment group developed NMSC at a rate of 4.4/100 patient-years, whereas the rate for controls treated with best available therapy was 2.7/100 patient-years.30 In contrast, 5-year follow-up of the COMFORT-I trial, also examining the efficacy of ruxolitinib in myelofibrosis, showed no increased risk for NMSC between ruxolitinib-treated patients and placebo (2.7/100 patient-years and 3.9/100 patient-years, respectively).31
A 2017 case series described 5 patients with myelofibrosis who developed multiple skin cancers with aggressive features while receiving ruxolitinib.32 Duration of ruxolitinib therapy ranged from 4 months to 4 years; 3 patients had a history of hydroxyurea exposure, and only 1 patient had a history of NMSC. High-risk cutaneous SCC, undifferentiated pleomorphic sarcoma, and lentigo maligna melanoma (Breslow thickness, 0.45 mm) were among the tumors reported in this series. Although no definitive conclusion can be made regarding the causality of JAK inhibitors in promoting these tumors, the association warrants further investigation. Clinicians should be aware that ruxolitinib might amplify the risk for NMSC in patients with pre-existing genetic or exposure-related susceptibility. Interruption of drug therapy may be necessary in managing patients who develop an aggressive tumor.32
In contrast, tofacitinib, which specifically inhibits JAK3, carries very low risk, if any, for NMSC when used for the treatment of psoriasis and rheumatoid arthritis. Results from 2 phase 3 trials analyzing the efficacy of tofacitinib in psoriasis demonstrated that only 2 of 1486 patients treated developed NMSC compared to none in the control group.33 Furthermore, analysis of NMSC across the tofacitinib rheumatoid arthritis clinical program, which included a total of 15,103 patient-years of exposure, demonstrated that the overall NMSC incidence was 0.55 for every 100 patient-years. Of note, the risk in patients receiving high-dose treatment (10 mg vs 5 mg) was nearly doubled in long-term follow-up studies (0.79/100 patient-years and 0.41/100 patient-years, respectively). Overall, the study concluded that treatment with tofacitinib presents no greater increased risk for NMSC than treatment with tumor necrosis factor inhibitors.33
PDE-5 Inhibitors
Phosphodiesterase 5 inhibitors, such as sildenafil citrate, have been widely prescribed for the treatment of erectile dysfunction. Studies have shown that BRAF-activated melanomas, which occur in approximately 50% to 70% of melanomas, also result in reduced PDE-5 expression.34-36 In these melanomas, downregulation of PDE-5 results in increased intracellular calcium,36 which has been shown to induce melanoma invasion.36,37 Given this similarity in molecular pathway between BRAF-activated melanomas and PDE-5 inhibitors, there has been increased concern that PDE-5 inhibitors might be associated with an increased risk for melanoma.
In 2014, Li et al38 published a retrospective analysis suggesting an association with sildenafil and an increased risk for melanoma. Their study utilized the Health Professionals Follow-up Study to identify a statistically significant elevation in the risk for invasive melanoma with both recent sildenafil use (multivariate-adjusted HR=2.24) and use at any time (HR=1.92). These results controlled for confounding variables, such as presence of major chronic disease, use of other erectile dysfunction treatments, family history of melanoma, history of sun exposure, and UV index of the patient’s residence. Notably, the study also found that sildenafil did not affect the incidence of BCC or SCC.38
In 2015, Loeb et al39 also examined the potential association between PDE-5 inhibitors and melanoma. Review of several Swedish drug and cancer registries allowed for analysis of melanoma risk and PDE-5 inhibitor use, based on number of prescriptions filled and type of PDE-5 inhibitor prescribed. Their analysis showed that men developing melanoma were more likely than nonmelanoma controls to have taken a PDE-5 inhibitor (11% vs 8%). In a subgroup analysis, however, statistical significance was shown for men with only a single prescription filled (34% of cases; P<.05), whereas the difference for men with multiple filled prescriptions did not meet statistical significance. Furthermore, the study did not find increased risk with longer-acting tadalafil and vardenafil (odds ratio [OR]=1.16) compared to sildenafil (OR=1.14). Last, use of PDE-5 inhibitors was only associated with stage 0 (OR=1.49) and stage I (OR=1.21) tumors, not with stages II to IV (OR=0.83) tumors. Although there was a statistically significant association between PDE-5 inhibitors and malignant melanoma (P<.05), the subgroup analysis findings pointed away from a causal relationship and likely toward a confounding of variable(s).39
A 2016 study by Lian et al40 looked at the risk for melanoma in a cohort of patients diagnosed with erectile dysfunction. No association between PDE-5 inhibitors and melanoma risk was shown when comparing patients who received a PDE-5 inhibitor and those who did not receive a PDE-5 inhibitor. However, secondary analysis did show that melanoma risk was increased among patients receiving more pills (34%) and prescriptions (30%). The authors concluded that there was no association between PDE-5 inhibitor use and overall increased risk for melanoma, and the increased risk associated with a greater number of pills and prescriptions would require further study.40
In contrast, a 2017 meta-analysis by Tang et al41 of 5 studies (3 of which were the aforementioned trials38-40) concluded that use of PDE-5 inhibitors was associated with a small but significantly increased risk for melanoma (OR=1.12) and BCC (OR=1.14) but not SCC. Furthermore, the study found no evidence of dosage-dependent association between PDE-5 inhibitor use and melanoma risk.41
Overall, clinical studies have been inconclusive in determining the risk for melanoma in the setting of PDE-5 inhibitor use. Studies showing an increased rate of melanoma within patient cohorts receiving PDE-5 inhibitors are limited; results might be affected by confounding variables. However, given the similarity in mechanism between PDE-5 inhibitors and HRAS-activated melanomas, it is reasonable to continue research into this potential association.
Conclusion
Since the turn of the century, drugs targeting cell-signaling pathways have been developed to treat inflammatory, oncologic, and immune conditions. The role of immunosuppressants in promoting skin cancer is well established and supported by a vast literature base. However, associations are less clear with newer immunomodulatory and antineoplastic medications. Skin cancer has been reported in association with BRAF inhibitors, sonic hedgehog–inhibiting agents, JAK inhibitors, and PDE-5 inhibitors. In the case of JAK and PDE-5 inhibitors, the increased risk for melanoma and NMSC is somewhat inconclusive; risk is more firmly established for BRAF inhibitors and smoothened inhibitors. For the antineoplastic agents reviewed, the therapeutic effect of cancer regression is well documented, and benefits of continued therapy outweigh the increased risk for skin cancer promotion in nearly all cases. The value of early detection has been well documented for skin malignancy; therefore, increased skin surveillance and prompt management of suspicious lesions should be a priority for physicians treating patients undergoing therapy with these medications
- Zimmer L, Hillen U, Livingstone E, et al. Atypical melanocytic proliferations and new primary melanoma in patients with advanced melanoma undergoing selective BRAF inhibition. J Clin Oncol. 2012;30:2375-2383.
- Long GV, Menzies AM, Nagrial AM, et al. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J Clin Oncol. 2011;29:1239-1246.
- Chapman PB, Hauschild A, Robert C, et al; BRIM-3 Study Group. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507-2516.
- Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809-819.
- McArthur GA, Chapman PB, Robert C, et al. Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM-3): extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol. 2014;15:323-332.
- Su F, Viros A, Milagre C, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med. 2012;366:207-215.
- Carlos G, Anforth R, Clements A, et al. Cutaneous toxic effects of BRAF inhibitors alone and in combination with MEK inhibitors for metastatic melanoma. JAMA Dermatol. 2015;151:1103-1109.
- Poulikakos PI, Zhang C, Bollag G, et al. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464:427-430.
- Ryan MB, Der CJ, Wang-Gillam A, et al. Targeting RAS-mutant cancers: is ERK the key? Trends Cancer. 2015;1:183-198.
- Long GV, Stroyakovskiy D, Gogas H, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med. 2014;371:1877-1888.
- Robert C, Karaszewska B, Schachter J, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015;372:30-39.
- Holderfield M, Nagel TE, Stuart DD. Mechanism and consequence of RAF kinase activation by small-molecule inhibitors. Br J Cancer. 2014;111:640-645.
- Dalle S, Poulalhon N, Debarbieux S, et al. Tracking of second primary melanomas in vemurafenib-treated patients. JAMA Dermatol. 2013;149:488-490.
- Williams VL, Cohen PR, Stewart DJ. Sorafenib-induced premalignant and malignant skin lesions. Int J Dermatol. 2011;50:396-402.
- Arnault JP, Wechsler J, Escudier B, et al. Keratoacanthomas and squamous cell carcinomas in patients receiving sorafenib. J Clin Oncol. 2009;27:e59-e61.
- Smith KJ, Haley H, Hamza S, et al. Eruptive keratoacanthoma-type squamous cell carcinomas in patients taking sorafenib for the treatment of solid tumors. Dermatol Surg. 2009;35:1766-1770.
- Sekulic A, Migden MR, Oro AE, et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N Engl J Med. 2012;366:2171-2179.
- Demirci H, Worden F, Nelson CC, et al. Efficacy of vismodegib (Erivedge) for basal cell carcinoma involving the orbit and periocular area. Ophthalmic Plast Reconstr Surg. 2015;31:463-466.
- Atwood SX, Sarin KY, Whitson RJ, et al. Smoothened variants explain the majority of drug resistance in basal cell carcinoma. Cancer Cell. 2015;27:342-353.
- Ridky TW, Cotsarelis G. Vismodegib resistance in basal cell carcinoma: not a smooth fit. Cancer Cell. 2015;27:315-316.
- Aasi S, Silkiss R, Tang JY, et al. New onset of keratoacanthomas after vismodegib treatment for locally advanced basal cell carcinomas: a report of 2 cases. JAMA Dermatol. 2013;149:242-243.
- Orouji A, Goerdt S, Utikal J, et al. Multiple highly and moderately differentiated squamous cell carcinomas of the skin during vismodegib treatment of inoperable basal cell carcinoma. Br J Dermatol. 2014;171:431-433.
- Iarrobino A, Messina JL, Kudchadkar R, et al. Emergence of a squamous cell carcinoma phenotype following treatment of metastatic basal cell carcinoma with vismodegib. J Am Acad Dermatol. 2013;69:e33-e34.
- Saintes C, Saint-Jean M, Brocard A, et al. Development of squamous cell carcinoma into basal cell carcinoma under treatment with vismodegib. J Eur Acad Dermatol Venereol. 2015;29:1006-1009.
- Mohan SV, Chang J, Li S, et al. Increased risk of cutaneous squamous cell carcinoma after vismodegib therapy for basal cell carcinoma. JAMA Dermatol. 2016;152:527-532.
- Zhao X, Ponomaryov T, Ornell KJ, et al. RAS/MAPK activation drives resistance to Smo inhibition, metastasis, and tumor evolution in Shh pathway-dependent tumors. Cancer Res. 2015;75:3623-3635.
- Chang AL, Oro AE. Initial assessment of tumor regrowth after vismodegib in advanced basal cell carcinoma. Arch Dermatol. 2012;148:1324-1325.
- Damsky W, King BA. JAK inhibitors in dermatology: the promise of a new drug class. J Am Acad Dermatol. 2017;76:736-744.
- Harrison CN, Vannucchi AM, Kiladjian JJ, et al. Long-term findings from COMFORT-II, a phase 3 study of ruxolitinib vs best available therapy for myelofibrosis. Leukemia. 2016;30:1701-1707.
- Verstovsek S, Vannucchi AM, Griesshammer M, et al. Ruxolitinib versus best available therapy in patients with polycythemia vera: 80-week follow-up from the RESPONSE trial. Haematologica. 2016;101:821-829.
- Verstovsek S, Mesa RA, Gotlib J, et al; COMFORT-I investigators. Long-term treatment with ruxolitinib for patients with myelofibrosis: 5-year update from the randomized, double-blind, placebo-controlled, phase 3 COMFORT-I trial. J Hematol Oncol. 2017;10:55.
- Blechman AB, Cabell CE, Weinberger CH, et al. Aggressive skin cancers occurring in patients treated with the Janus kinase inhibitor ruxolitinib. J Drugs Dermatol. 2017;16:508-511.
- Papp KA, Menter MA, Abe M, et al; OPT Pivotal 1 and OPT Pivotal 2 investigators. Tofacitinib, an oral Janus kinase inhibitor, for the treatment of chronic plaque psoriasis: results from two randomized, placebo-controlled, phase III trials. Br J Dermatol. 2015;173:949-961.
- Wellbrock C, Karasarides M, Marais R. The RAF proteins take centre stage. Nat Rev Mol Cell Biol. 2004;5:875-885.
- Gray-Schopfer V, Wellbrock C, Marais R. Melanoma biology and new targeted therapy. Nature. 2007;445:851-857.
- Arozarena I, Sanchez-Laorden B, Packer L, et al. Oncogenic BRAF induces melanoma cell invasion by downregulating the cGMP-specific phosphodiesterase PDE5A. Cancer Cell. 2011;19:45-57.
- Houslay MD. Hard times for oncogenic BRAF-expressing melanoma cells. Cancer Cell. 2011;19:3-4.
- Li WQ, Qureshi AA, Robinson KC, et al. Sildenafil use and increased risk of incident melanoma in US men: a prospective cohort study. JAMA Intern Med. 2014;174:964-970.
- Loeb S, Folkvaljon Y, Lambe M, et al. Use of phosphodiesterase type 5 inhibitors for erectile dysfunction and risk of malignant melanoma. JAMA. 2015;313:2449-2455.
- Lian Y, Yin H, Pollak MN, et al. Phosphodiesterase type 5 inhibitors and the risk of melanoma skin cancer. Eur Urol. 2016;70:808-815.
- Tang H, Wu W, Fu S, et al. Phosphodiesterase type 5 inhibitors and risk of melanoma: a meta-analysis. J Am Acad Dermatol. 2017;77:480.e9-488.e9.
- Zimmer L, Hillen U, Livingstone E, et al. Atypical melanocytic proliferations and new primary melanoma in patients with advanced melanoma undergoing selective BRAF inhibition. J Clin Oncol. 2012;30:2375-2383.
- Long GV, Menzies AM, Nagrial AM, et al. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J Clin Oncol. 2011;29:1239-1246.
- Chapman PB, Hauschild A, Robert C, et al; BRIM-3 Study Group. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507-2516.
- Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809-819.
- McArthur GA, Chapman PB, Robert C, et al. Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM-3): extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol. 2014;15:323-332.
- Su F, Viros A, Milagre C, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med. 2012;366:207-215.
- Carlos G, Anforth R, Clements A, et al. Cutaneous toxic effects of BRAF inhibitors alone and in combination with MEK inhibitors for metastatic melanoma. JAMA Dermatol. 2015;151:1103-1109.
- Poulikakos PI, Zhang C, Bollag G, et al. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464:427-430.
- Ryan MB, Der CJ, Wang-Gillam A, et al. Targeting RAS-mutant cancers: is ERK the key? Trends Cancer. 2015;1:183-198.
- Long GV, Stroyakovskiy D, Gogas H, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med. 2014;371:1877-1888.
- Robert C, Karaszewska B, Schachter J, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015;372:30-39.
- Holderfield M, Nagel TE, Stuart DD. Mechanism and consequence of RAF kinase activation by small-molecule inhibitors. Br J Cancer. 2014;111:640-645.
- Dalle S, Poulalhon N, Debarbieux S, et al. Tracking of second primary melanomas in vemurafenib-treated patients. JAMA Dermatol. 2013;149:488-490.
- Williams VL, Cohen PR, Stewart DJ. Sorafenib-induced premalignant and malignant skin lesions. Int J Dermatol. 2011;50:396-402.
- Arnault JP, Wechsler J, Escudier B, et al. Keratoacanthomas and squamous cell carcinomas in patients receiving sorafenib. J Clin Oncol. 2009;27:e59-e61.
- Smith KJ, Haley H, Hamza S, et al. Eruptive keratoacanthoma-type squamous cell carcinomas in patients taking sorafenib for the treatment of solid tumors. Dermatol Surg. 2009;35:1766-1770.
- Sekulic A, Migden MR, Oro AE, et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N Engl J Med. 2012;366:2171-2179.
- Demirci H, Worden F, Nelson CC, et al. Efficacy of vismodegib (Erivedge) for basal cell carcinoma involving the orbit and periocular area. Ophthalmic Plast Reconstr Surg. 2015;31:463-466.
- Atwood SX, Sarin KY, Whitson RJ, et al. Smoothened variants explain the majority of drug resistance in basal cell carcinoma. Cancer Cell. 2015;27:342-353.
- Ridky TW, Cotsarelis G. Vismodegib resistance in basal cell carcinoma: not a smooth fit. Cancer Cell. 2015;27:315-316.
- Aasi S, Silkiss R, Tang JY, et al. New onset of keratoacanthomas after vismodegib treatment for locally advanced basal cell carcinomas: a report of 2 cases. JAMA Dermatol. 2013;149:242-243.
- Orouji A, Goerdt S, Utikal J, et al. Multiple highly and moderately differentiated squamous cell carcinomas of the skin during vismodegib treatment of inoperable basal cell carcinoma. Br J Dermatol. 2014;171:431-433.
- Iarrobino A, Messina JL, Kudchadkar R, et al. Emergence of a squamous cell carcinoma phenotype following treatment of metastatic basal cell carcinoma with vismodegib. J Am Acad Dermatol. 2013;69:e33-e34.
- Saintes C, Saint-Jean M, Brocard A, et al. Development of squamous cell carcinoma into basal cell carcinoma under treatment with vismodegib. J Eur Acad Dermatol Venereol. 2015;29:1006-1009.
- Mohan SV, Chang J, Li S, et al. Increased risk of cutaneous squamous cell carcinoma after vismodegib therapy for basal cell carcinoma. JAMA Dermatol. 2016;152:527-532.
- Zhao X, Ponomaryov T, Ornell KJ, et al. RAS/MAPK activation drives resistance to Smo inhibition, metastasis, and tumor evolution in Shh pathway-dependent tumors. Cancer Res. 2015;75:3623-3635.
- Chang AL, Oro AE. Initial assessment of tumor regrowth after vismodegib in advanced basal cell carcinoma. Arch Dermatol. 2012;148:1324-1325.
- Damsky W, King BA. JAK inhibitors in dermatology: the promise of a new drug class. J Am Acad Dermatol. 2017;76:736-744.
- Harrison CN, Vannucchi AM, Kiladjian JJ, et al. Long-term findings from COMFORT-II, a phase 3 study of ruxolitinib vs best available therapy for myelofibrosis. Leukemia. 2016;30:1701-1707.
- Verstovsek S, Vannucchi AM, Griesshammer M, et al. Ruxolitinib versus best available therapy in patients with polycythemia vera: 80-week follow-up from the RESPONSE trial. Haematologica. 2016;101:821-829.
- Verstovsek S, Mesa RA, Gotlib J, et al; COMFORT-I investigators. Long-term treatment with ruxolitinib for patients with myelofibrosis: 5-year update from the randomized, double-blind, placebo-controlled, phase 3 COMFORT-I trial. J Hematol Oncol. 2017;10:55.
- Blechman AB, Cabell CE, Weinberger CH, et al. Aggressive skin cancers occurring in patients treated with the Janus kinase inhibitor ruxolitinib. J Drugs Dermatol. 2017;16:508-511.
- Papp KA, Menter MA, Abe M, et al; OPT Pivotal 1 and OPT Pivotal 2 investigators. Tofacitinib, an oral Janus kinase inhibitor, for the treatment of chronic plaque psoriasis: results from two randomized, placebo-controlled, phase III trials. Br J Dermatol. 2015;173:949-961.
- Wellbrock C, Karasarides M, Marais R. The RAF proteins take centre stage. Nat Rev Mol Cell Biol. 2004;5:875-885.
- Gray-Schopfer V, Wellbrock C, Marais R. Melanoma biology and new targeted therapy. Nature. 2007;445:851-857.
- Arozarena I, Sanchez-Laorden B, Packer L, et al. Oncogenic BRAF induces melanoma cell invasion by downregulating the cGMP-specific phosphodiesterase PDE5A. Cancer Cell. 2011;19:45-57.
- Houslay MD. Hard times for oncogenic BRAF-expressing melanoma cells. Cancer Cell. 2011;19:3-4.
- Li WQ, Qureshi AA, Robinson KC, et al. Sildenafil use and increased risk of incident melanoma in US men: a prospective cohort study. JAMA Intern Med. 2014;174:964-970.
- Loeb S, Folkvaljon Y, Lambe M, et al. Use of phosphodiesterase type 5 inhibitors for erectile dysfunction and risk of malignant melanoma. JAMA. 2015;313:2449-2455.
- Lian Y, Yin H, Pollak MN, et al. Phosphodiesterase type 5 inhibitors and the risk of melanoma skin cancer. Eur Urol. 2016;70:808-815.
- Tang H, Wu W, Fu S, et al. Phosphodiesterase type 5 inhibitors and risk of melanoma: a meta-analysis. J Am Acad Dermatol. 2017;77:480.e9-488.e9.
Practice Points
- Patients should be educated about the increased risk for skin malignancy while undergoing treatment with BRAF inhibitors, sonic hedgehog–inhibiting agents, Janus kinase (JAK) inhibitors, and phosphodiesterase 5 (PDE-5) inhibitors.
- For BRAF inhibitors, sonic hedgehog–inhibiting agents, and JAK inhibitors, the increased risk for skin cancer warrants regular surveillance; however, given the indications for these medications, many patients will already be receiving regular skin screenings.
- The association between PDE-5 inhibitors and melanoma as well as nonmelanoma skin cancer remains questionable, and increased skin surveillance is not recommended at this time, unless patients have other risk factors for cutaneous malignancy.

Systemic Epstein-Barr Virus–Positive T-cell Lymphoma of Childhood
Case Report
A 7-year-old Chinese boy presented with multiple painful oral and tongue ulcers of 2 weeks’ duration as well as acute onset of moderate to high fever (highest temperature, 39.3°C) for 5 days. The fever was reported to have run a relapsing course, accompanied by rigors but without convulsions or cognitive changes. At times, the patient had nasal congestion, nasal discharge, and cough. He also had a transient eruption on the back and hands as well as an indurated red nodule on the left forearm.
Before the patient was hospitalized, antibiotic therapy was administered by other physicians, but the condition of fever and oral ulcers did not improve. After the patient was hospitalized, new tender nodules emerged on the scalp, buttocks, and lower extremities. New ulcers also appeared on the palate.
History
Two months earlier, the patient had presented with a painful perioral skin ulcer that resolved after being treated as contagious eczema. Another dermatologist previously had considered a diagnosis of hand-foot-and-mouth disease.
The patient was born by normal spontaneous vaginal delivery, without abnormality. He was breastfed; feeding, growth, and the developmental history showed no abnormality. He was the family’s eldest child, with a healthy brother and sister. There was no history of familial illness. He received bacillus Calmette-Guérin and poliomyelitis vaccines after birth; the rest of the vaccine history was unclear. There was no history of immunologic abnormality.
Physical Examination
A 1.5×1.5-cm, warm, red nodule with a central black crust was noted on the left forearm (Figure 1A). Several similar lesions were noted on the buttocks, scalp, and lower extremities. Multiple ulcers, as large as 1 cm, were present on the tongue, palate, and left angle of the mouth (Figure 1B). The pharynx was congested, and the tonsils were mildly enlarged. Multiple enlarged, movable, nontender lymph nodes could be palpated in the cervical basins, axillae, and groin. No purpura or ecchymosis was detected.
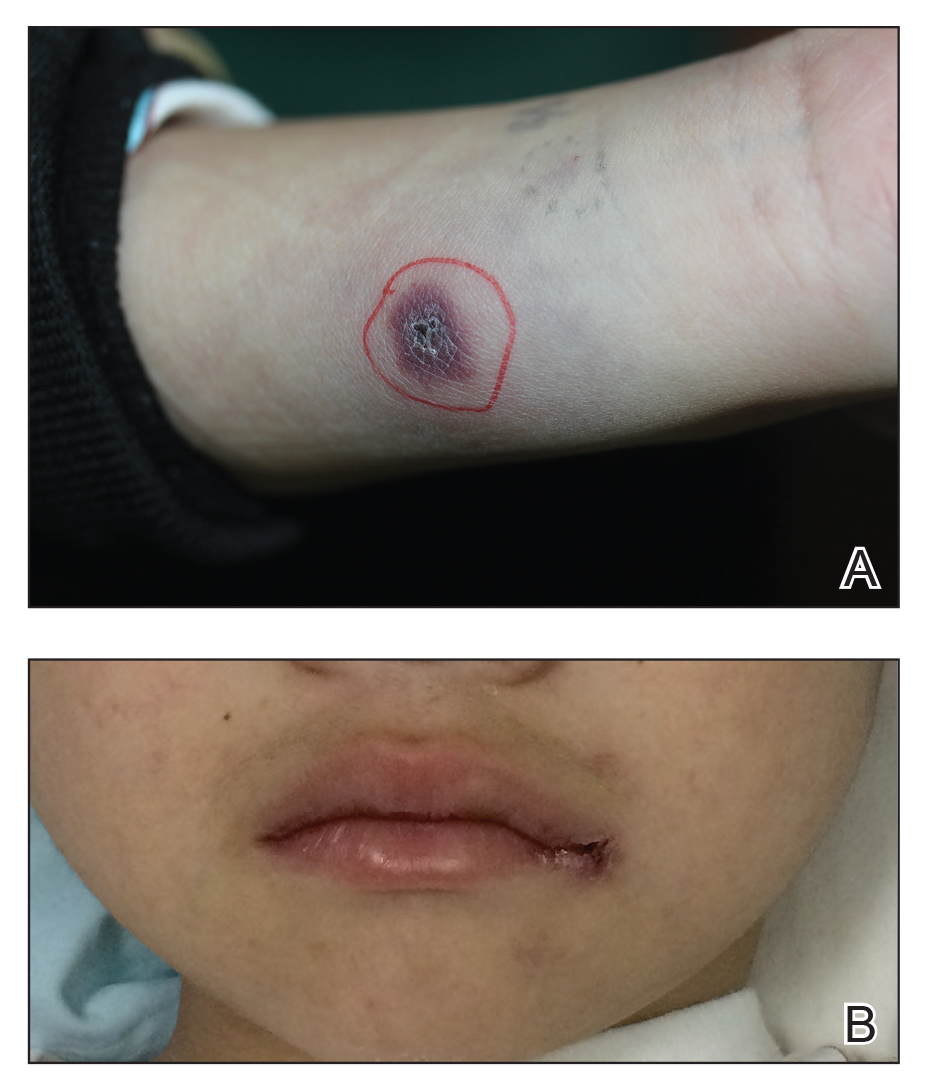
Laboratory Results
Laboratory testing revealed a normal total white blood cell count (4.26×109/L [reference range, 4.0–12.0×109/L]), with normal neutrophils (1.36×109/L [reference range, 1.32–7.90×109/L]), lymphocytes (2.77×109/L [reference range, 1.20–6.00×109/L]), and monocytes (0.13×109/L [reference range, 0.08–0.80×109/L]); a mildly decreased hemoglobin level (115 g/L [reference range, 120–160 g/L]); a normal platelet count (102×109/L [reference range, 100–380×109/L]); an elevated lactate dehydrogenase level (614 U/L [reference range, 110–330 U/L]); an elevated α-hydroxybutyrate dehydrogenase level (483 U/L [reference range, 120–270 U/L]); elevated prothrombin time (15.3 s [reference range, 9–14 s]); elevated activated partial thromboplastin time (59.8 s [reference range, 20.6–39.6 s]); and an elevated D-dimer level (1.51 mg/L [reference range, <0.73 mg/L]). In addition, autoantibody testing revealed a positive antinuclear antibody titer of 1:320 and a strong positive anti–Ro-52 level.
The peripheral blood lymphocyte classification demonstrated a prominent elevated percentage of T lymphocytes, with predominantly CD8+ cells (CD3, 94.87%; CD8, 71.57%; CD4, 24.98%; CD4:CD8 ratio, 0.35) and a diminished percentage of B lymphocytes and natural killer (NK) cells. Epstein-Barr virus (EBV) antibody testing was positive for anti–viral capsid antigen (VCA) IgG and negative for anti-VCA IgM.
Smears of the ulcer on the tongue demonstrated gram-positive cocci, gram-negative bacilli, and diplococci. Culture of sputum showed methicillin-resistant Staphylococcus aureus. Inspection for acid-fast bacilli in sputum yielded negative results 3 times. A purified protein derivative skin test for Mycobacterium tuberculosis infection was negative.
Imaging and Other Studies
Computed tomography of the chest and abdomen demonstrated 2 nodular opacities on the lower right lung; spotted opacities on the upper right lung; floccular opacities on the rest area of the lung; mild pleural effusion; enlargement of lymph nodes on the mediastinum, the bilateral hilum of the lung, and mesentery; and hepatosplenomegaly. Electrocardiography showed sinus tachycardia. Nasal cavity endoscopy showed sinusitis. Fundus examination showed vasculopathy of the left retina. A colonoscopy showed normal mucosa.
Histopathology
Biopsy of the nodule on the left arm showed dense, superficial to deep perivascular, periadnexal, perineural, and panniculitislike lymphoid infiltrates, as well as a sparse interstitial infiltrate with irregular and pleomorphic medium to large nuclei. Lymphoid cells showed mild epidermotropism, with tagging to the basal layer. Some vessel walls were infiltrated by similar cells (Figure 2). Infiltrative atypical lymphoid cells expressed CD3 and CD7 and were mostly CD8+, with a few CD4+ cells and most cells negative for CD5, CD20, CD30, CD56, and anaplastic lymphoma kinase. Cytotoxic markers granzyme B and T-cell intracellular antigen protein 1 were scattered positive. Immunostaining for Ki-67 protein highlighted an increased proliferative rate of 80% in malignant cells. In situ hybridization for EBV-encoded RNA (EBER) demonstrated EBV-positive atypical lymphoid cells (Figure 3). Analysis for T-cell receptor (TCR) γ gene rearrangement revealed a monoclonal pattern. Bone marrow aspirate showed proliferation of the 3 cell lines. The percentage of T lymphocytes was increased (20% of all nucleated cells). No hemophagocytic activity was found.
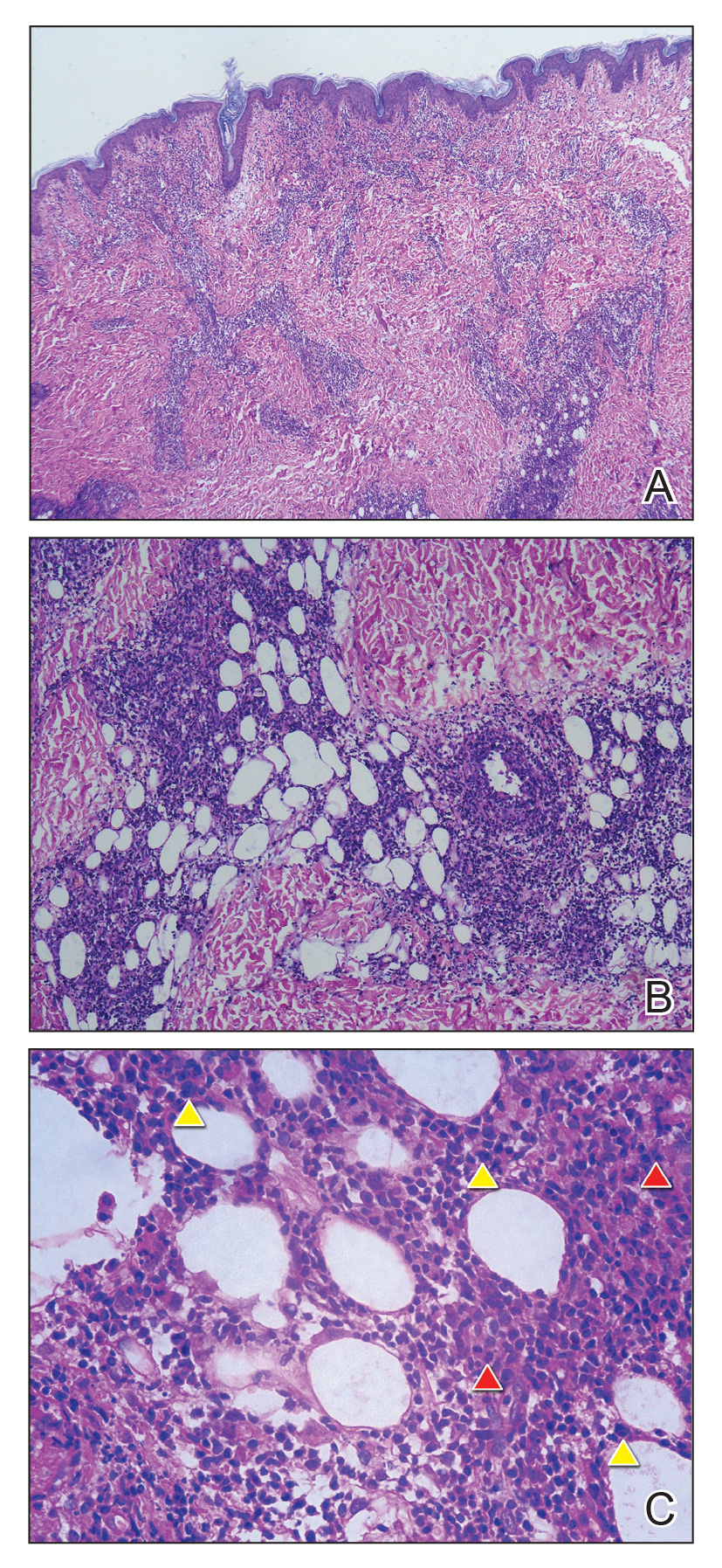
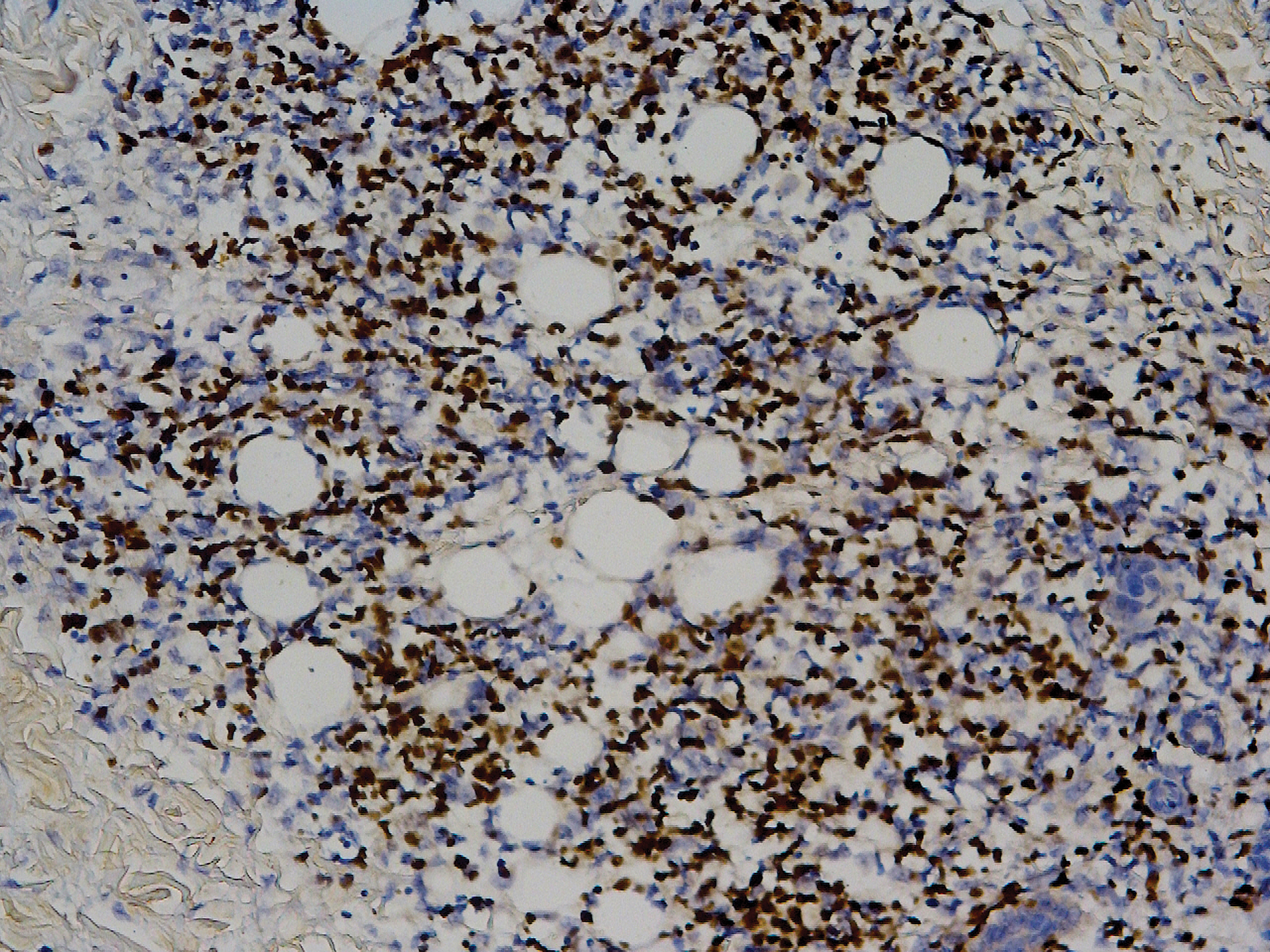
Diagnosis
A diagnosis of systemic EBV-positive T-cell lymphoma was made. Before the final diagnosis was made, the patient was treated by rheumatologists with antibiotics, antiviral drugs, nonsteroidal anti-inflammatory drugs, and other symptomatic treatments. Following antibiotic therapy, a sputum culture reverted to normal flora, the coagulation index (ie, prothrombin time, activated partial thromboplastin time) returned to normal, and the D-dimer level decreased to 1.19 mg/L.
The patient’s parents refused to accept chemotherapy for him. Instead, they chose herbal therapy only; 5 months later, they reported that all of his symptoms had resolved; however, the disease suddenly relapsed after another 7 months, with multiple skin nodules and fever. The patient died, even with chemotherapy in another hospital.
Comment
Prevalence and Presentation
Epstein-Barr virus is a ubiquitous γ-herpesvirus with tropism for B cells, affecting more than 90% of the adult population worldwide. In addition to infecting B cells, EBV is capable of infecting T and NK cells, leading to various EBV-related lymphoproliferative disorders (LPDs). The frequency and clinical presentation of infection varies based on the type of EBV-infected cells and the state of host immunity.1-3
Primary infection usually is asymptomatic and occurs early in life; when symptomatic, the disease usually presents as infectious mononucleosis (IM), characterized by polyclonal expansion of infected B cells and subsequent cytotoxic T-cell response. A diagnosis of EBV infection can be made by testing for specific IgM and IgG antibodies against VCA, early antigens, and EBV nuclear antigen proteins.3,4
Associated LPDs
Although most symptoms associated with IM resolve within weeks or months, persistent or recurrent IM-like symptoms or even lasting disease occasionally occur, particularly in children and young adults. This complication is known as chronic active EBV infection (CAEBV), frequently associated with EBV-infected T-cell or NK-cell proliferation, especially in East Asian populations.3,5
Epstein-Barr virus–positive T-cell and NK-cell LPDs of childhood include CAEBV infection of T-cell and NK-cell types and systemic EBV-positive T-cell lymphoma of childhood. The former includes hydroa vacciniforme–like LPD and severe mosquito bite allergy.3
Systemic EBV-Positive T-cell Lymphoma of Childhood
This entity occurs not only in children but also in adolescents and young adults. A fulminant illness characterized by clonal proliferation of EBV-infected cytotoxic T cells, it can develop shortly after primary EBV infection or is linked to CAEBV infection. The disorder is rare and has a racial predilection for Asian (ie, Japanese, Chinese, Korean) populations and indigenous populations of Mexico and Central and South America.6-8
Complications
Systemic EBV-positive T-cell lymphoma of childhood is often complicated by hemophagocytic syndrome, coagulopathy, sepsis, and multiorgan failure. Other signs and symptoms include high fever, rash, jaundice, diarrhea, pancytopenia, and hepatosplenomegaly. The liver, spleen, lymph nodes, and bone marrow are commonly involved, and the disease can involve skin, the heart, and the lungs.9,10
Diagnosis
When systemic EBV-positive T-cell lymphoma occurs shortly after IM, serology shows low or absent anti-VCA IgM and positive anti-VCA IgG. Infiltrating T cells usually are small and lack cytologic atypia; however, cases with pleomorphic, medium to large lymphoid cells, irregular nuclei, and frequent mitoses have been described. Hemophagocytosis can be seen in the liver, spleen, and bone marrow.3,11
The most typical phenotype of systemic EBV-positive T-cell lymphoma is CD2+CD3+CD8+CD20−CD56−, with expression of the cytotoxic granules known as T-cell intracellular antigen 1 and granzyme B. Rare cases of CD4+ and mixed CD4+/CD8+ phenotypes have been described, usually in the setting of CAEBV infection.3,12 Neoplastic cells have monoclonally rearranged TCR-γ genes and consistent EBER positivity with in situ hybridization.13 A final diagnosis is based on a comprehensive analysis of clinical, morphological, immunohistochemical, and molecular biological aspects.
Clinical Course and Prognosis
Most patients with systemic EBV-positive T-cell lymphoma have an aggressive clinical course with high mortality. In a few cases, patients were reported to respond to a regimen of etoposide and dexamethasone, followed by allogeneic hematopoietic stem cell transplantation.3
In recognition of the aggressive clinical behavior and desire to clearly distinguish systemic EBV-positive T-cell lymphoma from CAEBV infection, the older term systemic EBV-positive T-cell LPD of childhood, which had been introduced in 2008 to the World Health Organization classification, was changed to systemic EBV-positive T-cell lymphoma of childhood in the revised 2016 World Health Organization classification.6,12 However, Kim et al14 reported a case with excellent response to corticosteroid administration, suggesting that systemic EBV-positive T-cell lymphoma of childhood may be more heterogeneous in terms of prognosis.
Our patient presented with acute IM-like symptoms, including high fever, tonsillar enlargement, lymphadenopathy, and hepatosplenomegaly, as well as uncommon oral ulcers and skin lesions, including indurated nodules. Histopathologic changes in the skin nodule, proliferation in bone marrow, immunohistochemical phenotype, and positivity of EBER and TCR-γ monoclonal rearrangement were all consistent with systemic EBV-positive T-cell lymphoma of childhood. The patient was positive for VCA IgG and negative for VCA IgM, compatible with systemic EBV-positive T-cell lymphoma of childhood occurring shortly after IM. Neither pancytopenia, hemophagocytic syndrome, nor multiorgan failure occurred during the course.
Differential Diagnosis
It is important to distinguish IM from systemic EBV-positive T-cell lymphoma of childhood and CAEBV infection. Detection of anti–VCA IgM in the early stage, its disappearance during the clinical course, and appearance of anti-EBV–determined nuclear antigen is useful to distinguish IM from the neoplasms, as systemic EBV-positive T-cell lymphoma of childhood is negative for anti-EBV–determined nuclear antigen. Carefully following the clinical course also is important.3,15
Epstein-Barr virus–associated hemophagocytic lymphohistiocytosis can occur in association with systemic EBV-positive T-cell lymphoma of childhood and might represent a continuum of disease rather than distinct entities.14 The most useful marker for differentiating EBV-associated hemophagocytic lymphohistiocytosis and systemic EBV-positive T-cell lymphoma of childhood is an abnormal karyotype rather than molecular clonality.16
Outcome
Mortality risk in EBV-associated T-cell and NK-cell LPD is not primarily dependent on whether the lesion has progressed to lymphoma but instead is related to associated complications.17
Conclusion
Although systemic EBV-positive T-cell lymphoma of childhood is a rare disorder and has race predilection, dermatologists should be aware due to the aggressive clinical source and poor prognosis. Histopathology and in situ hybridization for EBER and TCR gene rearrangements are critical for final diagnosis. Although rare cases can show temporary resolution, the final outcome of this disease is not optimistic.
- Ameli F, Ghafourian F, Masir N. Systematic Epstein-Barr virus-positive T-cell lymphoproliferative disease presenting as a persistent fever and cough: a case report. J Med Case Rep. 2014;8:288.
- Kim HJ, Ko YH, Kim JE, et al. Epstein-Barr virus-associated lympho-proliferative disorders: review and update on 2016 WHO classification. J Pathol Transl Med. 2017;51:352-358.
- Dojcinov SD, Fend F, Quintanilla-Martinez L. EBV-positive lymphoproliferations of B- T- and NK-cell derivation in non-immunocompromised hosts [published online March 7, 2018]. Pathogens. doi:10.3390/pathogens7010028.
- Luzuriaga K, Sullivan JL. Infectious mononucleosis. N Engl J Med. 2010;362:1993-2000.
- Cohen JI, Kimura H, Nakamura S, et al. Epstein-Barr virus-associated lymphoproliferative disease in non-immunocompromised hosts: a status report and summary of an international meeting, 8-9 September 2008. Ann Oncol. 2009;20:1472-1482.
- Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127:2375-2390.
- Kim WY, Montes-Mojarro IA, Fend F, et al. Epstein-Barr virus-associated T and NK-cell lymphoproliferative diseases. Front Pediatr. 2019;7:71.
- Hong M, Ko YH, Yoo KH, et al. EBV-positive T/NK-cell lymphoproliferative disease of childhood. Korean J Pathol. 2013;47:137-147.
- Quintanilla-Martinez L, Kumar S, Fend F, et al. Fulminant EBV(+) T-cell lymphoproliferative disorder following acute/chronic EBV infection: a distinct clinicopathologic syndrome. Blood. 2000;96:443-451.
- Chen G, Chen L, Qin X, et al. Systemic Epstein-Barr virus positive T-cell lymphoproliferative disease of childhood with hemophagocytic syndrome. Int J Clin Exp Pathol. 2014;7:7110-7113.
- Grywalska E, Rolinski J. Epstein-Barr virus-associated lymphomas. Semin Oncol. 2015;42:291-303.
- Huang W, Lv N, Ying J, et al. Clinicopathological characteristics of four cases of EBV positive T-cell lymphoproliferative disorders of childhood in China. Int J Clin Exp Pathol. 2014;7:4991-4999.
- Tabanelli V, Agostinelli C, Sabattini E, et al. Systemic Epstein-Barr-virus-positive T cell lymphoproliferative childhood disease in a 22-year-old Caucasian man: a case report and review of the literature. J Med Case Rep. 2011;5:218.
- Kim DH, Kim M, Kim Y, et al. Systemic Epstein-Barr virus-positive T-cell lymphoproliferative disease of childhood with good response to steroid therapy. J Pediatr Hematol Oncol. 2017;39:e497-e500.
- Arai A, Yamaguchi T, Komatsu H, et al. Infectious mononucleosis accompanied by clonal proliferation of EBV-infected cells and infection of CD8-positive cells. Int J Hematol. 2014;99:671-675.
- Smith MC, Cohen DN, Greig B, et al. The ambiguous boundary between EBV-related hemophagocytic lymphohistiocytosis and systemic EBV-driven T cell lymphoproliferative disorder. Int J Clin Exp Pathol. 2014;7:5738-5749.
- Paik JH, Choe JY, Kim H, et al. Clinicopathological categorization of Epstein-Barr virus-positive T/NK-cell lymphoproliferative disease: an analysis of 42 cases with an emphasis on prognostic implications. Leuk Lymphoma. 2017;58:53-63.
Case Report
A 7-year-old Chinese boy presented with multiple painful oral and tongue ulcers of 2 weeks’ duration as well as acute onset of moderate to high fever (highest temperature, 39.3°C) for 5 days. The fever was reported to have run a relapsing course, accompanied by rigors but without convulsions or cognitive changes. At times, the patient had nasal congestion, nasal discharge, and cough. He also had a transient eruption on the back and hands as well as an indurated red nodule on the left forearm.
Before the patient was hospitalized, antibiotic therapy was administered by other physicians, but the condition of fever and oral ulcers did not improve. After the patient was hospitalized, new tender nodules emerged on the scalp, buttocks, and lower extremities. New ulcers also appeared on the palate.
History
Two months earlier, the patient had presented with a painful perioral skin ulcer that resolved after being treated as contagious eczema. Another dermatologist previously had considered a diagnosis of hand-foot-and-mouth disease.
The patient was born by normal spontaneous vaginal delivery, without abnormality. He was breastfed; feeding, growth, and the developmental history showed no abnormality. He was the family’s eldest child, with a healthy brother and sister. There was no history of familial illness. He received bacillus Calmette-Guérin and poliomyelitis vaccines after birth; the rest of the vaccine history was unclear. There was no history of immunologic abnormality.
Physical Examination
A 1.5×1.5-cm, warm, red nodule with a central black crust was noted on the left forearm (Figure 1A). Several similar lesions were noted on the buttocks, scalp, and lower extremities. Multiple ulcers, as large as 1 cm, were present on the tongue, palate, and left angle of the mouth (Figure 1B). The pharynx was congested, and the tonsils were mildly enlarged. Multiple enlarged, movable, nontender lymph nodes could be palpated in the cervical basins, axillae, and groin. No purpura or ecchymosis was detected.
Laboratory Results
Laboratory testing revealed a normal total white blood cell count (4.26×109/L [reference range, 4.0–12.0×109/L]), with normal neutrophils (1.36×109/L [reference range, 1.32–7.90×109/L]), lymphocytes (2.77×109/L [reference range, 1.20–6.00×109/L]), and monocytes (0.13×109/L [reference range, 0.08–0.80×109/L]); a mildly decreased hemoglobin level (115 g/L [reference range, 120–160 g/L]); a normal platelet count (102×109/L [reference range, 100–380×109/L]); an elevated lactate dehydrogenase level (614 U/L [reference range, 110–330 U/L]); an elevated α-hydroxybutyrate dehydrogenase level (483 U/L [reference range, 120–270 U/L]); elevated prothrombin time (15.3 s [reference range, 9–14 s]); elevated activated partial thromboplastin time (59.8 s [reference range, 20.6–39.6 s]); and an elevated D-dimer level (1.51 mg/L [reference range, <0.73 mg/L]). In addition, autoantibody testing revealed a positive antinuclear antibody titer of 1:320 and a strong positive anti–Ro-52 level.
The peripheral blood lymphocyte classification demonstrated a prominent elevated percentage of T lymphocytes, with predominantly CD8+ cells (CD3, 94.87%; CD8, 71.57%; CD4, 24.98%; CD4:CD8 ratio, 0.35) and a diminished percentage of B lymphocytes and natural killer (NK) cells. Epstein-Barr virus (EBV) antibody testing was positive for anti–viral capsid antigen (VCA) IgG and negative for anti-VCA IgM.
Smears of the ulcer on the tongue demonstrated gram-positive cocci, gram-negative bacilli, and diplococci. Culture of sputum showed methicillin-resistant Staphylococcus aureus. Inspection for acid-fast bacilli in sputum yielded negative results 3 times. A purified protein derivative skin test for Mycobacterium tuberculosis infection was negative.
Imaging and Other Studies
Computed tomography of the chest and abdomen demonstrated 2 nodular opacities on the lower right lung; spotted opacities on the upper right lung; floccular opacities on the rest area of the lung; mild pleural effusion; enlargement of lymph nodes on the mediastinum, the bilateral hilum of the lung, and mesentery; and hepatosplenomegaly. Electrocardiography showed sinus tachycardia. Nasal cavity endoscopy showed sinusitis. Fundus examination showed vasculopathy of the left retina. A colonoscopy showed normal mucosa.
Histopathology
Biopsy of the nodule on the left arm showed dense, superficial to deep perivascular, periadnexal, perineural, and panniculitislike lymphoid infiltrates, as well as a sparse interstitial infiltrate with irregular and pleomorphic medium to large nuclei. Lymphoid cells showed mild epidermotropism, with tagging to the basal layer. Some vessel walls were infiltrated by similar cells (Figure 2). Infiltrative atypical lymphoid cells expressed CD3 and CD7 and were mostly CD8+, with a few CD4+ cells and most cells negative for CD5, CD20, CD30, CD56, and anaplastic lymphoma kinase. Cytotoxic markers granzyme B and T-cell intracellular antigen protein 1 were scattered positive. Immunostaining for Ki-67 protein highlighted an increased proliferative rate of 80% in malignant cells. In situ hybridization for EBV-encoded RNA (EBER) demonstrated EBV-positive atypical lymphoid cells (Figure 3). Analysis for T-cell receptor (TCR) γ gene rearrangement revealed a monoclonal pattern. Bone marrow aspirate showed proliferation of the 3 cell lines. The percentage of T lymphocytes was increased (20% of all nucleated cells). No hemophagocytic activity was found.
Diagnosis
A diagnosis of systemic EBV-positive T-cell lymphoma was made. Before the final diagnosis was made, the patient was treated by rheumatologists with antibiotics, antiviral drugs, nonsteroidal anti-inflammatory drugs, and other symptomatic treatments. Following antibiotic therapy, a sputum culture reverted to normal flora, the coagulation index (ie, prothrombin time, activated partial thromboplastin time) returned to normal, and the D-dimer level decreased to 1.19 mg/L.
The patient’s parents refused to accept chemotherapy for him. Instead, they chose herbal therapy only; 5 months later, they reported that all of his symptoms had resolved; however, the disease suddenly relapsed after another 7 months, with multiple skin nodules and fever. The patient died, even with chemotherapy in another hospital.
Comment
Prevalence and Presentation
Epstein-Barr virus is a ubiquitous γ-herpesvirus with tropism for B cells, affecting more than 90% of the adult population worldwide. In addition to infecting B cells, EBV is capable of infecting T and NK cells, leading to various EBV-related lymphoproliferative disorders (LPDs). The frequency and clinical presentation of infection varies based on the type of EBV-infected cells and the state of host immunity.1-3
Primary infection usually is asymptomatic and occurs early in life; when symptomatic, the disease usually presents as infectious mononucleosis (IM), characterized by polyclonal expansion of infected B cells and subsequent cytotoxic T-cell response. A diagnosis of EBV infection can be made by testing for specific IgM and IgG antibodies against VCA, early antigens, and EBV nuclear antigen proteins.3,4
Associated LPDs
Although most symptoms associated with IM resolve within weeks or months, persistent or recurrent IM-like symptoms or even lasting disease occasionally occur, particularly in children and young adults. This complication is known as chronic active EBV infection (CAEBV), frequently associated with EBV-infected T-cell or NK-cell proliferation, especially in East Asian populations.3,5
Epstein-Barr virus–positive T-cell and NK-cell LPDs of childhood include CAEBV infection of T-cell and NK-cell types and systemic EBV-positive T-cell lymphoma of childhood. The former includes hydroa vacciniforme–like LPD and severe mosquito bite allergy.3
Systemic EBV-Positive T-cell Lymphoma of Childhood
This entity occurs not only in children but also in adolescents and young adults. A fulminant illness characterized by clonal proliferation of EBV-infected cytotoxic T cells, it can develop shortly after primary EBV infection or is linked to CAEBV infection. The disorder is rare and has a racial predilection for Asian (ie, Japanese, Chinese, Korean) populations and indigenous populations of Mexico and Central and South America.6-8
Complications
Systemic EBV-positive T-cell lymphoma of childhood is often complicated by hemophagocytic syndrome, coagulopathy, sepsis, and multiorgan failure. Other signs and symptoms include high fever, rash, jaundice, diarrhea, pancytopenia, and hepatosplenomegaly. The liver, spleen, lymph nodes, and bone marrow are commonly involved, and the disease can involve skin, the heart, and the lungs.9,10
Diagnosis
When systemic EBV-positive T-cell lymphoma occurs shortly after IM, serology shows low or absent anti-VCA IgM and positive anti-VCA IgG. Infiltrating T cells usually are small and lack cytologic atypia; however, cases with pleomorphic, medium to large lymphoid cells, irregular nuclei, and frequent mitoses have been described. Hemophagocytosis can be seen in the liver, spleen, and bone marrow.3,11
The most typical phenotype of systemic EBV-positive T-cell lymphoma is CD2+CD3+CD8+CD20−CD56−, with expression of the cytotoxic granules known as T-cell intracellular antigen 1 and granzyme B. Rare cases of CD4+ and mixed CD4+/CD8+ phenotypes have been described, usually in the setting of CAEBV infection.3,12 Neoplastic cells have monoclonally rearranged TCR-γ genes and consistent EBER positivity with in situ hybridization.13 A final diagnosis is based on a comprehensive analysis of clinical, morphological, immunohistochemical, and molecular biological aspects.
Clinical Course and Prognosis
Most patients with systemic EBV-positive T-cell lymphoma have an aggressive clinical course with high mortality. In a few cases, patients were reported to respond to a regimen of etoposide and dexamethasone, followed by allogeneic hematopoietic stem cell transplantation.3
In recognition of the aggressive clinical behavior and desire to clearly distinguish systemic EBV-positive T-cell lymphoma from CAEBV infection, the older term systemic EBV-positive T-cell LPD of childhood, which had been introduced in 2008 to the World Health Organization classification, was changed to systemic EBV-positive T-cell lymphoma of childhood in the revised 2016 World Health Organization classification.6,12 However, Kim et al14 reported a case with excellent response to corticosteroid administration, suggesting that systemic EBV-positive T-cell lymphoma of childhood may be more heterogeneous in terms of prognosis.
Our patient presented with acute IM-like symptoms, including high fever, tonsillar enlargement, lymphadenopathy, and hepatosplenomegaly, as well as uncommon oral ulcers and skin lesions, including indurated nodules. Histopathologic changes in the skin nodule, proliferation in bone marrow, immunohistochemical phenotype, and positivity of EBER and TCR-γ monoclonal rearrangement were all consistent with systemic EBV-positive T-cell lymphoma of childhood. The patient was positive for VCA IgG and negative for VCA IgM, compatible with systemic EBV-positive T-cell lymphoma of childhood occurring shortly after IM. Neither pancytopenia, hemophagocytic syndrome, nor multiorgan failure occurred during the course.
Differential Diagnosis
It is important to distinguish IM from systemic EBV-positive T-cell lymphoma of childhood and CAEBV infection. Detection of anti–VCA IgM in the early stage, its disappearance during the clinical course, and appearance of anti-EBV–determined nuclear antigen is useful to distinguish IM from the neoplasms, as systemic EBV-positive T-cell lymphoma of childhood is negative for anti-EBV–determined nuclear antigen. Carefully following the clinical course also is important.3,15
Epstein-Barr virus–associated hemophagocytic lymphohistiocytosis can occur in association with systemic EBV-positive T-cell lymphoma of childhood and might represent a continuum of disease rather than distinct entities.14 The most useful marker for differentiating EBV-associated hemophagocytic lymphohistiocytosis and systemic EBV-positive T-cell lymphoma of childhood is an abnormal karyotype rather than molecular clonality.16
Outcome
Mortality risk in EBV-associated T-cell and NK-cell LPD is not primarily dependent on whether the lesion has progressed to lymphoma but instead is related to associated complications.17
Conclusion
Although systemic EBV-positive T-cell lymphoma of childhood is a rare disorder and has race predilection, dermatologists should be aware due to the aggressive clinical source and poor prognosis. Histopathology and in situ hybridization for EBER and TCR gene rearrangements are critical for final diagnosis. Although rare cases can show temporary resolution, the final outcome of this disease is not optimistic.
Case Report
A 7-year-old Chinese boy presented with multiple painful oral and tongue ulcers of 2 weeks’ duration as well as acute onset of moderate to high fever (highest temperature, 39.3°C) for 5 days. The fever was reported to have run a relapsing course, accompanied by rigors but without convulsions or cognitive changes. At times, the patient had nasal congestion, nasal discharge, and cough. He also had a transient eruption on the back and hands as well as an indurated red nodule on the left forearm.
Before the patient was hospitalized, antibiotic therapy was administered by other physicians, but the condition of fever and oral ulcers did not improve. After the patient was hospitalized, new tender nodules emerged on the scalp, buttocks, and lower extremities. New ulcers also appeared on the palate.
History
Two months earlier, the patient had presented with a painful perioral skin ulcer that resolved after being treated as contagious eczema. Another dermatologist previously had considered a diagnosis of hand-foot-and-mouth disease.
The patient was born by normal spontaneous vaginal delivery, without abnormality. He was breastfed; feeding, growth, and the developmental history showed no abnormality. He was the family’s eldest child, with a healthy brother and sister. There was no history of familial illness. He received bacillus Calmette-Guérin and poliomyelitis vaccines after birth; the rest of the vaccine history was unclear. There was no history of immunologic abnormality.
Physical Examination
A 1.5×1.5-cm, warm, red nodule with a central black crust was noted on the left forearm (Figure 1A). Several similar lesions were noted on the buttocks, scalp, and lower extremities. Multiple ulcers, as large as 1 cm, were present on the tongue, palate, and left angle of the mouth (Figure 1B). The pharynx was congested, and the tonsils were mildly enlarged. Multiple enlarged, movable, nontender lymph nodes could be palpated in the cervical basins, axillae, and groin. No purpura or ecchymosis was detected.
Laboratory Results
Laboratory testing revealed a normal total white blood cell count (4.26×109/L [reference range, 4.0–12.0×109/L]), with normal neutrophils (1.36×109/L [reference range, 1.32–7.90×109/L]), lymphocytes (2.77×109/L [reference range, 1.20–6.00×109/L]), and monocytes (0.13×109/L [reference range, 0.08–0.80×109/L]); a mildly decreased hemoglobin level (115 g/L [reference range, 120–160 g/L]); a normal platelet count (102×109/L [reference range, 100–380×109/L]); an elevated lactate dehydrogenase level (614 U/L [reference range, 110–330 U/L]); an elevated α-hydroxybutyrate dehydrogenase level (483 U/L [reference range, 120–270 U/L]); elevated prothrombin time (15.3 s [reference range, 9–14 s]); elevated activated partial thromboplastin time (59.8 s [reference range, 20.6–39.6 s]); and an elevated D-dimer level (1.51 mg/L [reference range, <0.73 mg/L]). In addition, autoantibody testing revealed a positive antinuclear antibody titer of 1:320 and a strong positive anti–Ro-52 level.
The peripheral blood lymphocyte classification demonstrated a prominent elevated percentage of T lymphocytes, with predominantly CD8+ cells (CD3, 94.87%; CD8, 71.57%; CD4, 24.98%; CD4:CD8 ratio, 0.35) and a diminished percentage of B lymphocytes and natural killer (NK) cells. Epstein-Barr virus (EBV) antibody testing was positive for anti–viral capsid antigen (VCA) IgG and negative for anti-VCA IgM.
Smears of the ulcer on the tongue demonstrated gram-positive cocci, gram-negative bacilli, and diplococci. Culture of sputum showed methicillin-resistant Staphylococcus aureus. Inspection for acid-fast bacilli in sputum yielded negative results 3 times. A purified protein derivative skin test for Mycobacterium tuberculosis infection was negative.
Imaging and Other Studies
Computed tomography of the chest and abdomen demonstrated 2 nodular opacities on the lower right lung; spotted opacities on the upper right lung; floccular opacities on the rest area of the lung; mild pleural effusion; enlargement of lymph nodes on the mediastinum, the bilateral hilum of the lung, and mesentery; and hepatosplenomegaly. Electrocardiography showed sinus tachycardia. Nasal cavity endoscopy showed sinusitis. Fundus examination showed vasculopathy of the left retina. A colonoscopy showed normal mucosa.
Histopathology
Biopsy of the nodule on the left arm showed dense, superficial to deep perivascular, periadnexal, perineural, and panniculitislike lymphoid infiltrates, as well as a sparse interstitial infiltrate with irregular and pleomorphic medium to large nuclei. Lymphoid cells showed mild epidermotropism, with tagging to the basal layer. Some vessel walls were infiltrated by similar cells (Figure 2). Infiltrative atypical lymphoid cells expressed CD3 and CD7 and were mostly CD8+, with a few CD4+ cells and most cells negative for CD5, CD20, CD30, CD56, and anaplastic lymphoma kinase. Cytotoxic markers granzyme B and T-cell intracellular antigen protein 1 were scattered positive. Immunostaining for Ki-67 protein highlighted an increased proliferative rate of 80% in malignant cells. In situ hybridization for EBV-encoded RNA (EBER) demonstrated EBV-positive atypical lymphoid cells (Figure 3). Analysis for T-cell receptor (TCR) γ gene rearrangement revealed a monoclonal pattern. Bone marrow aspirate showed proliferation of the 3 cell lines. The percentage of T lymphocytes was increased (20% of all nucleated cells). No hemophagocytic activity was found.
Diagnosis
A diagnosis of systemic EBV-positive T-cell lymphoma was made. Before the final diagnosis was made, the patient was treated by rheumatologists with antibiotics, antiviral drugs, nonsteroidal anti-inflammatory drugs, and other symptomatic treatments. Following antibiotic therapy, a sputum culture reverted to normal flora, the coagulation index (ie, prothrombin time, activated partial thromboplastin time) returned to normal, and the D-dimer level decreased to 1.19 mg/L.
The patient’s parents refused to accept chemotherapy for him. Instead, they chose herbal therapy only; 5 months later, they reported that all of his symptoms had resolved; however, the disease suddenly relapsed after another 7 months, with multiple skin nodules and fever. The patient died, even with chemotherapy in another hospital.
Comment
Prevalence and Presentation
Epstein-Barr virus is a ubiquitous γ-herpesvirus with tropism for B cells, affecting more than 90% of the adult population worldwide. In addition to infecting B cells, EBV is capable of infecting T and NK cells, leading to various EBV-related lymphoproliferative disorders (LPDs). The frequency and clinical presentation of infection varies based on the type of EBV-infected cells and the state of host immunity.1-3
Primary infection usually is asymptomatic and occurs early in life; when symptomatic, the disease usually presents as infectious mononucleosis (IM), characterized by polyclonal expansion of infected B cells and subsequent cytotoxic T-cell response. A diagnosis of EBV infection can be made by testing for specific IgM and IgG antibodies against VCA, early antigens, and EBV nuclear antigen proteins.3,4
Associated LPDs
Although most symptoms associated with IM resolve within weeks or months, persistent or recurrent IM-like symptoms or even lasting disease occasionally occur, particularly in children and young adults. This complication is known as chronic active EBV infection (CAEBV), frequently associated with EBV-infected T-cell or NK-cell proliferation, especially in East Asian populations.3,5
Epstein-Barr virus–positive T-cell and NK-cell LPDs of childhood include CAEBV infection of T-cell and NK-cell types and systemic EBV-positive T-cell lymphoma of childhood. The former includes hydroa vacciniforme–like LPD and severe mosquito bite allergy.3
Systemic EBV-Positive T-cell Lymphoma of Childhood
This entity occurs not only in children but also in adolescents and young adults. A fulminant illness characterized by clonal proliferation of EBV-infected cytotoxic T cells, it can develop shortly after primary EBV infection or is linked to CAEBV infection. The disorder is rare and has a racial predilection for Asian (ie, Japanese, Chinese, Korean) populations and indigenous populations of Mexico and Central and South America.6-8
Complications
Systemic EBV-positive T-cell lymphoma of childhood is often complicated by hemophagocytic syndrome, coagulopathy, sepsis, and multiorgan failure. Other signs and symptoms include high fever, rash, jaundice, diarrhea, pancytopenia, and hepatosplenomegaly. The liver, spleen, lymph nodes, and bone marrow are commonly involved, and the disease can involve skin, the heart, and the lungs.9,10
Diagnosis
When systemic EBV-positive T-cell lymphoma occurs shortly after IM, serology shows low or absent anti-VCA IgM and positive anti-VCA IgG. Infiltrating T cells usually are small and lack cytologic atypia; however, cases with pleomorphic, medium to large lymphoid cells, irregular nuclei, and frequent mitoses have been described. Hemophagocytosis can be seen in the liver, spleen, and bone marrow.3,11
The most typical phenotype of systemic EBV-positive T-cell lymphoma is CD2+CD3+CD8+CD20−CD56−, with expression of the cytotoxic granules known as T-cell intracellular antigen 1 and granzyme B. Rare cases of CD4+ and mixed CD4+/CD8+ phenotypes have been described, usually in the setting of CAEBV infection.3,12 Neoplastic cells have monoclonally rearranged TCR-γ genes and consistent EBER positivity with in situ hybridization.13 A final diagnosis is based on a comprehensive analysis of clinical, morphological, immunohistochemical, and molecular biological aspects.
Clinical Course and Prognosis
Most patients with systemic EBV-positive T-cell lymphoma have an aggressive clinical course with high mortality. In a few cases, patients were reported to respond to a regimen of etoposide and dexamethasone, followed by allogeneic hematopoietic stem cell transplantation.3
In recognition of the aggressive clinical behavior and desire to clearly distinguish systemic EBV-positive T-cell lymphoma from CAEBV infection, the older term systemic EBV-positive T-cell LPD of childhood, which had been introduced in 2008 to the World Health Organization classification, was changed to systemic EBV-positive T-cell lymphoma of childhood in the revised 2016 World Health Organization classification.6,12 However, Kim et al14 reported a case with excellent response to corticosteroid administration, suggesting that systemic EBV-positive T-cell lymphoma of childhood may be more heterogeneous in terms of prognosis.
Our patient presented with acute IM-like symptoms, including high fever, tonsillar enlargement, lymphadenopathy, and hepatosplenomegaly, as well as uncommon oral ulcers and skin lesions, including indurated nodules. Histopathologic changes in the skin nodule, proliferation in bone marrow, immunohistochemical phenotype, and positivity of EBER and TCR-γ monoclonal rearrangement were all consistent with systemic EBV-positive T-cell lymphoma of childhood. The patient was positive for VCA IgG and negative for VCA IgM, compatible with systemic EBV-positive T-cell lymphoma of childhood occurring shortly after IM. Neither pancytopenia, hemophagocytic syndrome, nor multiorgan failure occurred during the course.
Differential Diagnosis
It is important to distinguish IM from systemic EBV-positive T-cell lymphoma of childhood and CAEBV infection. Detection of anti–VCA IgM in the early stage, its disappearance during the clinical course, and appearance of anti-EBV–determined nuclear antigen is useful to distinguish IM from the neoplasms, as systemic EBV-positive T-cell lymphoma of childhood is negative for anti-EBV–determined nuclear antigen. Carefully following the clinical course also is important.3,15
Epstein-Barr virus–associated hemophagocytic lymphohistiocytosis can occur in association with systemic EBV-positive T-cell lymphoma of childhood and might represent a continuum of disease rather than distinct entities.14 The most useful marker for differentiating EBV-associated hemophagocytic lymphohistiocytosis and systemic EBV-positive T-cell lymphoma of childhood is an abnormal karyotype rather than molecular clonality.16
Outcome
Mortality risk in EBV-associated T-cell and NK-cell LPD is not primarily dependent on whether the lesion has progressed to lymphoma but instead is related to associated complications.17
Conclusion
Although systemic EBV-positive T-cell lymphoma of childhood is a rare disorder and has race predilection, dermatologists should be aware due to the aggressive clinical source and poor prognosis. Histopathology and in situ hybridization for EBER and TCR gene rearrangements are critical for final diagnosis. Although rare cases can show temporary resolution, the final outcome of this disease is not optimistic.
- Ameli F, Ghafourian F, Masir N. Systematic Epstein-Barr virus-positive T-cell lymphoproliferative disease presenting as a persistent fever and cough: a case report. J Med Case Rep. 2014;8:288.
- Kim HJ, Ko YH, Kim JE, et al. Epstein-Barr virus-associated lympho-proliferative disorders: review and update on 2016 WHO classification. J Pathol Transl Med. 2017;51:352-358.
- Dojcinov SD, Fend F, Quintanilla-Martinez L. EBV-positive lymphoproliferations of B- T- and NK-cell derivation in non-immunocompromised hosts [published online March 7, 2018]. Pathogens. doi:10.3390/pathogens7010028.
- Luzuriaga K, Sullivan JL. Infectious mononucleosis. N Engl J Med. 2010;362:1993-2000.
- Cohen JI, Kimura H, Nakamura S, et al. Epstein-Barr virus-associated lymphoproliferative disease in non-immunocompromised hosts: a status report and summary of an international meeting, 8-9 September 2008. Ann Oncol. 2009;20:1472-1482.
- Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127:2375-2390.
- Kim WY, Montes-Mojarro IA, Fend F, et al. Epstein-Barr virus-associated T and NK-cell lymphoproliferative diseases. Front Pediatr. 2019;7:71.
- Hong M, Ko YH, Yoo KH, et al. EBV-positive T/NK-cell lymphoproliferative disease of childhood. Korean J Pathol. 2013;47:137-147.
- Quintanilla-Martinez L, Kumar S, Fend F, et al. Fulminant EBV(+) T-cell lymphoproliferative disorder following acute/chronic EBV infection: a distinct clinicopathologic syndrome. Blood. 2000;96:443-451.
- Chen G, Chen L, Qin X, et al. Systemic Epstein-Barr virus positive T-cell lymphoproliferative disease of childhood with hemophagocytic syndrome. Int J Clin Exp Pathol. 2014;7:7110-7113.
- Grywalska E, Rolinski J. Epstein-Barr virus-associated lymphomas. Semin Oncol. 2015;42:291-303.
- Huang W, Lv N, Ying J, et al. Clinicopathological characteristics of four cases of EBV positive T-cell lymphoproliferative disorders of childhood in China. Int J Clin Exp Pathol. 2014;7:4991-4999.
- Tabanelli V, Agostinelli C, Sabattini E, et al. Systemic Epstein-Barr-virus-positive T cell lymphoproliferative childhood disease in a 22-year-old Caucasian man: a case report and review of the literature. J Med Case Rep. 2011;5:218.
- Kim DH, Kim M, Kim Y, et al. Systemic Epstein-Barr virus-positive T-cell lymphoproliferative disease of childhood with good response to steroid therapy. J Pediatr Hematol Oncol. 2017;39:e497-e500.
- Arai A, Yamaguchi T, Komatsu H, et al. Infectious mononucleosis accompanied by clonal proliferation of EBV-infected cells and infection of CD8-positive cells. Int J Hematol. 2014;99:671-675.
- Smith MC, Cohen DN, Greig B, et al. The ambiguous boundary between EBV-related hemophagocytic lymphohistiocytosis and systemic EBV-driven T cell lymphoproliferative disorder. Int J Clin Exp Pathol. 2014;7:5738-5749.
- Paik JH, Choe JY, Kim H, et al. Clinicopathological categorization of Epstein-Barr virus-positive T/NK-cell lymphoproliferative disease: an analysis of 42 cases with an emphasis on prognostic implications. Leuk Lymphoma. 2017;58:53-63.
- Ameli F, Ghafourian F, Masir N. Systematic Epstein-Barr virus-positive T-cell lymphoproliferative disease presenting as a persistent fever and cough: a case report. J Med Case Rep. 2014;8:288.
- Kim HJ, Ko YH, Kim JE, et al. Epstein-Barr virus-associated lympho-proliferative disorders: review and update on 2016 WHO classification. J Pathol Transl Med. 2017;51:352-358.
- Dojcinov SD, Fend F, Quintanilla-Martinez L. EBV-positive lymphoproliferations of B- T- and NK-cell derivation in non-immunocompromised hosts [published online March 7, 2018]. Pathogens. doi:10.3390/pathogens7010028.
- Luzuriaga K, Sullivan JL. Infectious mononucleosis. N Engl J Med. 2010;362:1993-2000.
- Cohen JI, Kimura H, Nakamura S, et al. Epstein-Barr virus-associated lymphoproliferative disease in non-immunocompromised hosts: a status report and summary of an international meeting, 8-9 September 2008. Ann Oncol. 2009;20:1472-1482.
- Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127:2375-2390.
- Kim WY, Montes-Mojarro IA, Fend F, et al. Epstein-Barr virus-associated T and NK-cell lymphoproliferative diseases. Front Pediatr. 2019;7:71.
- Hong M, Ko YH, Yoo KH, et al. EBV-positive T/NK-cell lymphoproliferative disease of childhood. Korean J Pathol. 2013;47:137-147.
- Quintanilla-Martinez L, Kumar S, Fend F, et al. Fulminant EBV(+) T-cell lymphoproliferative disorder following acute/chronic EBV infection: a distinct clinicopathologic syndrome. Blood. 2000;96:443-451.
- Chen G, Chen L, Qin X, et al. Systemic Epstein-Barr virus positive T-cell lymphoproliferative disease of childhood with hemophagocytic syndrome. Int J Clin Exp Pathol. 2014;7:7110-7113.
- Grywalska E, Rolinski J. Epstein-Barr virus-associated lymphomas. Semin Oncol. 2015;42:291-303.
- Huang W, Lv N, Ying J, et al. Clinicopathological characteristics of four cases of EBV positive T-cell lymphoproliferative disorders of childhood in China. Int J Clin Exp Pathol. 2014;7:4991-4999.
- Tabanelli V, Agostinelli C, Sabattini E, et al. Systemic Epstein-Barr-virus-positive T cell lymphoproliferative childhood disease in a 22-year-old Caucasian man: a case report and review of the literature. J Med Case Rep. 2011;5:218.
- Kim DH, Kim M, Kim Y, et al. Systemic Epstein-Barr virus-positive T-cell lymphoproliferative disease of childhood with good response to steroid therapy. J Pediatr Hematol Oncol. 2017;39:e497-e500.
- Arai A, Yamaguchi T, Komatsu H, et al. Infectious mononucleosis accompanied by clonal proliferation of EBV-infected cells and infection of CD8-positive cells. Int J Hematol. 2014;99:671-675.
- Smith MC, Cohen DN, Greig B, et al. The ambiguous boundary between EBV-related hemophagocytic lymphohistiocytosis and systemic EBV-driven T cell lymphoproliferative disorder. Int J Clin Exp Pathol. 2014;7:5738-5749.
- Paik JH, Choe JY, Kim H, et al. Clinicopathological categorization of Epstein-Barr virus-positive T/NK-cell lymphoproliferative disease: an analysis of 42 cases with an emphasis on prognostic implications. Leuk Lymphoma. 2017;58:53-63.
Practice Points
- Systemic Epstein-Barr virus (EBV)–positive T-cell lymphoma of childhood is a fulminant illness with a predilection for Asians and indigenous populations from Mexico and Central and South America. In most patients, the disease has an aggressive clinical course with high mortality.
- The disease often is complicated by hemophagocytic syndrome, coagulopathy, sepsis, and multiorgan failure. When these severe complications are absent, the prognosis might be better.
- In situ hybridization for EBV-encoded RNA and for T-cell receptor gene rearrangements is an important tool to establish the diagnosis as well as for treatment options and predicting the prognosis.
Seborrhea Herpeticum: Cutaneous Herpes Simplex Virus Infection Within Infantile Seborrheic Dermatitis
Classically, eczema herpeticum is associated with atopic dermatitis (AD), but it also has been previously reported in the setting of pemphigus vulgaris, Darier disease, ichthyosis vulgaris, burns, psoriasis, and irritant contact dermatitis.1,2 Descriptions of cutaneous herpes simplex virus (HSV) in the setting of seborrheic dermatitis are lacking.
Case Report
A 2-month-old infant boy who was otherwise healthy presented to the emergency department with a new rash on the scalp. Initially there were a few clusters of small fluid-filled lesions that evolved over several days into diffuse clusters covering the scalp and extending onto the forehead and upper chest (Figure). The patient’s medical history was notable for infantile seborrheic dermatitis and a family history of AD. His grandmother, who was his primary caretaker, had a recent history of herpes labialis.
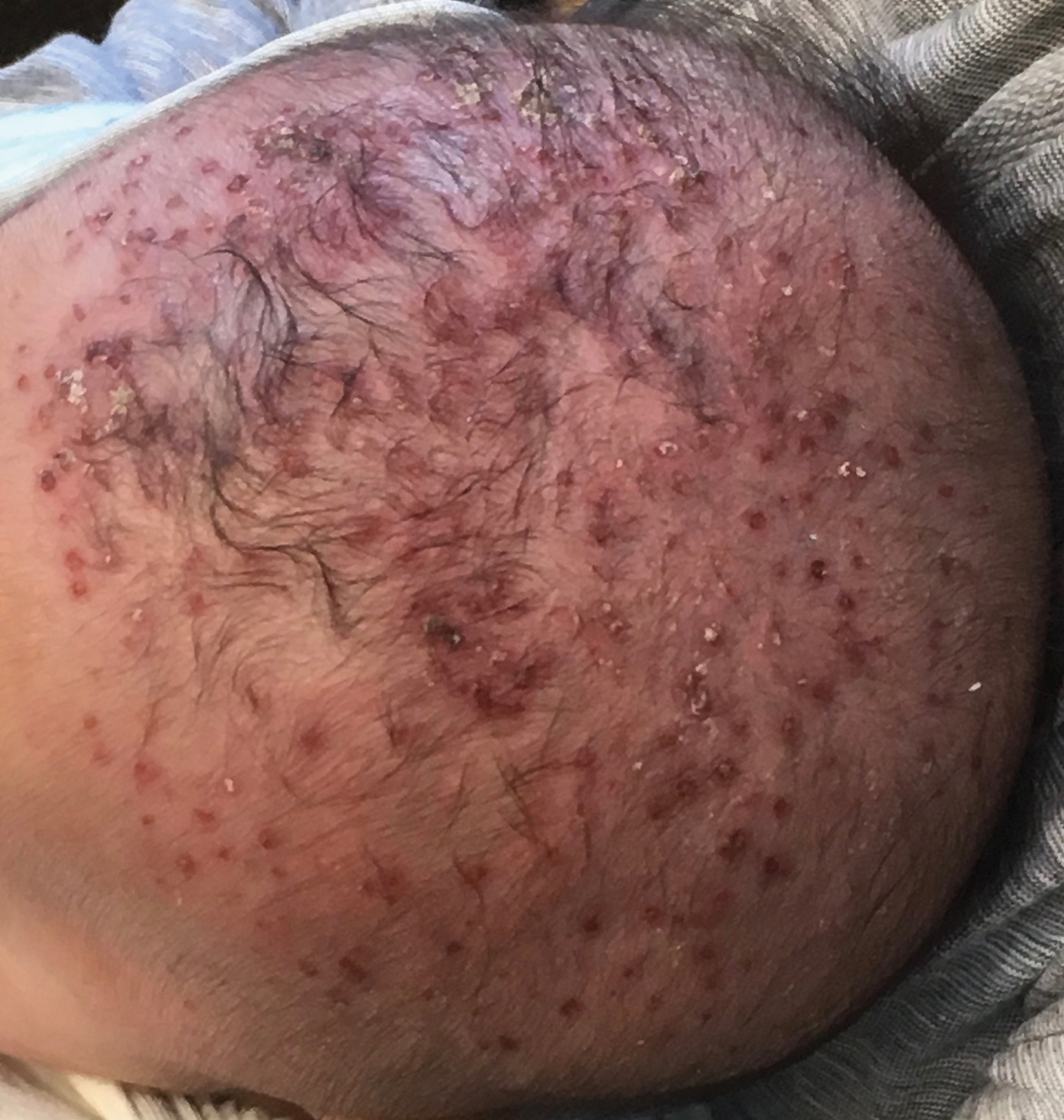
Physical examination revealed numerous discrete, erythematous, and punched-out erosions diffusely on the scalp. There were fewer similar erosions on the forehead and upper chest. There were no oral or periocular lesions. There were no areas of lichenification or eczematous plaques on the remainder of the trunk or extremities. Laboratory testing was positive for HSV type 1 polymerase chain reaction and positive for HSV type 1 viral culture. Liver enzymes were elevated with alanine aminotransferase at 107 U/L (reference range, 7–52 U/L) and aspartate aminotransferase at 94 U/L (reference range, 13–39 U/L).
The patient was admitted to the hospital and was treated by the dermatology and infectious disease services. Intravenous acyclovir 60 mg/kg daily was administered for 3 days until all lesions had crusted over. On the day of discharge, the patient was transitioned to oral valacyclovir 20 mg/kg daily for 7 days with resolution. One month later he developed a recurrence that was within his existing seborrheic dermatitis. After a repeat 7-day course of oral valacyclovir 20 mg/kg daily, he was placed on prophylaxis therapy of oral acyclovir 10 mg/kg daily. Gentle skin care precautions also were recommended.
Comment
Eczema herpeticum refers to disseminated cutaneous infection with HSV types 1 or 2 in the setting of underlying dermatosis.2 Although it is classically associated with AD, it has been reported in a number of other chronic skin disorders and can lead to serious complications, including hepatitis, keratoconjunctivitis, and meningitis. In those with AD who develop HSV, presentation may occur in active dermatitis locations because of skin barrier disruption, which may lead to increased susceptibility to viral infection.3
Herpes simplex virus in a background of seborrheic dermatitis has not been well described. Although the pathogenesis of seborrheic dermatitis has not been fully reported, several gene mutations and protein deficiencies have been identified in patients and animal models that are associated with immune response or epidermal differentiation.4 Therefore, it is possible that, as with AD, a disruption in the skin barrier increases susceptibility to viral infection.
It also has been suggested that infantile seborrheic dermatitis and AD represent the same spectrum of disease.5 Given our patient’s family history of AD, it is possible his presentation represents early underlying AD. Providers should be aware that cutaneous HSV can be confined to a seborrheic distribution and may represent underlying epidermal dysfunction secondary to seborrheic dermatitis.
- Wheeler CE, Abele DC. Eczema herpeticum, primary and recurrent. Arch Dermatol. 1966;93:162-173.
- Santmyire-Rosenberger BR, Nigra TP. Psoriasis herpeticum: three cases of Kaposi’s varicelliform eruption in psoriasis. J Am Acad Dermatol. 2005;53:52-56.
- Wollenberg A, Wetzel S, Burgdorf WH, et al. Viral infections in atopic dermatitis: pathogenic aspects and clinical management. J Allergy Clin Immunol. 2003;112:667-674.
- Karakadze M, Hirt P, Wikramanayake T. The genetic basis of seborrhoeic dermatitis: a review. J Eur Acad Dermatol Venereol. 2017;32:529-536.
- Alexopoulos A, Kakourou T, Orfanou I, et al. Retrospective analysis of the relationship between infantile seborrheic dermatitis and atopic dermatitis. Pediatr Dermatol. 2013;31:125-130.
Classically, eczema herpeticum is associated with atopic dermatitis (AD), but it also has been previously reported in the setting of pemphigus vulgaris, Darier disease, ichthyosis vulgaris, burns, psoriasis, and irritant contact dermatitis.1,2 Descriptions of cutaneous herpes simplex virus (HSV) in the setting of seborrheic dermatitis are lacking.
Case Report
A 2-month-old infant boy who was otherwise healthy presented to the emergency department with a new rash on the scalp. Initially there were a few clusters of small fluid-filled lesions that evolved over several days into diffuse clusters covering the scalp and extending onto the forehead and upper chest (Figure). The patient’s medical history was notable for infantile seborrheic dermatitis and a family history of AD. His grandmother, who was his primary caretaker, had a recent history of herpes labialis.
Physical examination revealed numerous discrete, erythematous, and punched-out erosions diffusely on the scalp. There were fewer similar erosions on the forehead and upper chest. There were no oral or periocular lesions. There were no areas of lichenification or eczematous plaques on the remainder of the trunk or extremities. Laboratory testing was positive for HSV type 1 polymerase chain reaction and positive for HSV type 1 viral culture. Liver enzymes were elevated with alanine aminotransferase at 107 U/L (reference range, 7–52 U/L) and aspartate aminotransferase at 94 U/L (reference range, 13–39 U/L).
The patient was admitted to the hospital and was treated by the dermatology and infectious disease services. Intravenous acyclovir 60 mg/kg daily was administered for 3 days until all lesions had crusted over. On the day of discharge, the patient was transitioned to oral valacyclovir 20 mg/kg daily for 7 days with resolution. One month later he developed a recurrence that was within his existing seborrheic dermatitis. After a repeat 7-day course of oral valacyclovir 20 mg/kg daily, he was placed on prophylaxis therapy of oral acyclovir 10 mg/kg daily. Gentle skin care precautions also were recommended.
Comment
Eczema herpeticum refers to disseminated cutaneous infection with HSV types 1 or 2 in the setting of underlying dermatosis.2 Although it is classically associated with AD, it has been reported in a number of other chronic skin disorders and can lead to serious complications, including hepatitis, keratoconjunctivitis, and meningitis. In those with AD who develop HSV, presentation may occur in active dermatitis locations because of skin barrier disruption, which may lead to increased susceptibility to viral infection.3
Herpes simplex virus in a background of seborrheic dermatitis has not been well described. Although the pathogenesis of seborrheic dermatitis has not been fully reported, several gene mutations and protein deficiencies have been identified in patients and animal models that are associated with immune response or epidermal differentiation.4 Therefore, it is possible that, as with AD, a disruption in the skin barrier increases susceptibility to viral infection.
It also has been suggested that infantile seborrheic dermatitis and AD represent the same spectrum of disease.5 Given our patient’s family history of AD, it is possible his presentation represents early underlying AD. Providers should be aware that cutaneous HSV can be confined to a seborrheic distribution and may represent underlying epidermal dysfunction secondary to seborrheic dermatitis.
Classically, eczema herpeticum is associated with atopic dermatitis (AD), but it also has been previously reported in the setting of pemphigus vulgaris, Darier disease, ichthyosis vulgaris, burns, psoriasis, and irritant contact dermatitis.1,2 Descriptions of cutaneous herpes simplex virus (HSV) in the setting of seborrheic dermatitis are lacking.
Case Report
A 2-month-old infant boy who was otherwise healthy presented to the emergency department with a new rash on the scalp. Initially there were a few clusters of small fluid-filled lesions that evolved over several days into diffuse clusters covering the scalp and extending onto the forehead and upper chest (Figure). The patient’s medical history was notable for infantile seborrheic dermatitis and a family history of AD. His grandmother, who was his primary caretaker, had a recent history of herpes labialis.
Physical examination revealed numerous discrete, erythematous, and punched-out erosions diffusely on the scalp. There were fewer similar erosions on the forehead and upper chest. There were no oral or periocular lesions. There were no areas of lichenification or eczematous plaques on the remainder of the trunk or extremities. Laboratory testing was positive for HSV type 1 polymerase chain reaction and positive for HSV type 1 viral culture. Liver enzymes were elevated with alanine aminotransferase at 107 U/L (reference range, 7–52 U/L) and aspartate aminotransferase at 94 U/L (reference range, 13–39 U/L).
The patient was admitted to the hospital and was treated by the dermatology and infectious disease services. Intravenous acyclovir 60 mg/kg daily was administered for 3 days until all lesions had crusted over. On the day of discharge, the patient was transitioned to oral valacyclovir 20 mg/kg daily for 7 days with resolution. One month later he developed a recurrence that was within his existing seborrheic dermatitis. After a repeat 7-day course of oral valacyclovir 20 mg/kg daily, he was placed on prophylaxis therapy of oral acyclovir 10 mg/kg daily. Gentle skin care precautions also were recommended.
Comment
Eczema herpeticum refers to disseminated cutaneous infection with HSV types 1 or 2 in the setting of underlying dermatosis.2 Although it is classically associated with AD, it has been reported in a number of other chronic skin disorders and can lead to serious complications, including hepatitis, keratoconjunctivitis, and meningitis. In those with AD who develop HSV, presentation may occur in active dermatitis locations because of skin barrier disruption, which may lead to increased susceptibility to viral infection.3
Herpes simplex virus in a background of seborrheic dermatitis has not been well described. Although the pathogenesis of seborrheic dermatitis has not been fully reported, several gene mutations and protein deficiencies have been identified in patients and animal models that are associated with immune response or epidermal differentiation.4 Therefore, it is possible that, as with AD, a disruption in the skin barrier increases susceptibility to viral infection.
It also has been suggested that infantile seborrheic dermatitis and AD represent the same spectrum of disease.5 Given our patient’s family history of AD, it is possible his presentation represents early underlying AD. Providers should be aware that cutaneous HSV can be confined to a seborrheic distribution and may represent underlying epidermal dysfunction secondary to seborrheic dermatitis.
- Wheeler CE, Abele DC. Eczema herpeticum, primary and recurrent. Arch Dermatol. 1966;93:162-173.
- Santmyire-Rosenberger BR, Nigra TP. Psoriasis herpeticum: three cases of Kaposi’s varicelliform eruption in psoriasis. J Am Acad Dermatol. 2005;53:52-56.
- Wollenberg A, Wetzel S, Burgdorf WH, et al. Viral infections in atopic dermatitis: pathogenic aspects and clinical management. J Allergy Clin Immunol. 2003;112:667-674.
- Karakadze M, Hirt P, Wikramanayake T. The genetic basis of seborrhoeic dermatitis: a review. J Eur Acad Dermatol Venereol. 2017;32:529-536.
- Alexopoulos A, Kakourou T, Orfanou I, et al. Retrospective analysis of the relationship between infantile seborrheic dermatitis and atopic dermatitis. Pediatr Dermatol. 2013;31:125-130.
- Wheeler CE, Abele DC. Eczema herpeticum, primary and recurrent. Arch Dermatol. 1966;93:162-173.
- Santmyire-Rosenberger BR, Nigra TP. Psoriasis herpeticum: three cases of Kaposi’s varicelliform eruption in psoriasis. J Am Acad Dermatol. 2005;53:52-56.
- Wollenberg A, Wetzel S, Burgdorf WH, et al. Viral infections in atopic dermatitis: pathogenic aspects and clinical management. J Allergy Clin Immunol. 2003;112:667-674.
- Karakadze M, Hirt P, Wikramanayake T. The genetic basis of seborrhoeic dermatitis: a review. J Eur Acad Dermatol Venereol. 2017;32:529-536.
- Alexopoulos A, Kakourou T, Orfanou I, et al. Retrospective analysis of the relationship between infantile seborrheic dermatitis and atopic dermatitis. Pediatr Dermatol. 2013;31:125-130.
Practice Points
- Cutaneous herpes simplex virus may present in a seborrheic distribution within infantile seborrheic dermatitis, suggesting underlying dysfunction secondary to seborrheic dermatitis.
- Treatment of seborrhea herpeticum involves antiviral therapy to treat the secondary viral infection and gentle skin care precautions for the primary condition.
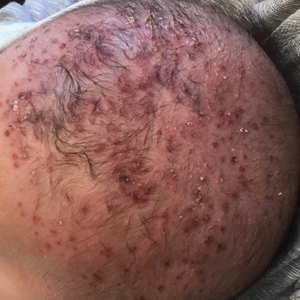
Pediatric Molluscum: An Update
Molluscum contagiosum virus (MCV) infection causes the cutaneous lesions we call molluscum. Molluscum has become common in the last 30 years. Deciding the best course of therapy requires some fundamental understanding about how MCV relates to the following factors: epidemiology, childhood immunity and vaccination, clinical features, comorbidities, and quality of life. Treatment depends on many factors, including presence or absence of atopic dermatitis (AD) and/or pruritus, other symptoms, cosmetic location, and the child’s concern about the lesions. Therapeutics include destructive and immunologic therapies, the latter geared toward increasing immune response.
Epidemiology
Molluscum contagiosum virus is the solo member of the Molluscipoxvirus genus. Infection with MCV causes benign growth or tumors in the skin (ie, molluscum). The infection is slow to clear because the virus reduces the host’s immunity.1,2 Molluscum contagiosum virus is a double-stranded DNA virus that affects keratinocytes and genetically carries the tools for its own replication (ie, DNA-dependent RNA polymerase). The virus has a few subtypes—I/Ia, II, III, and IV—with MCV-I predominating in children and healthy humans and MCV-II in patients with human immunodeficiency virus.1,2 Typing is experimental and is not standardly performed in clinical practice. Molluscum contagiosum virus produces a variety of factors that block the host’s immune response, prolonging infection and preventing erythema and inflammatory response.3
Molluscum contagiosum virus is transmitted through skin-to-skin contact and fomites, including shared towels, bathtubs, spas, bath sponges, and pool equipment.2,4,5 Transmission from household contact and bathing together has been noted in pediatric patients with MCV. Based on the data it can be posited that the lesions are softer when wet and more readily release viral particles or fomites, and fomites may be left on surfaces, especially when a child is wet.6,7 Propensity for infection occurs in patients with AD and in immunosuppressed hosts, including children with human immunodeficiency virus and iatrogenic immunosuppression caused by chemotherapy.1,2,8 Contact sports can increase the risk of transmission, and outbreaks have occurred in pools,5,9 day-care facilities,10 and sports settings.11 Cases of congenital and vertically transmitted molluscum have been documented.12,13 Sexual transmission of MCV may be seen in adolescents who are sexually active. Although child-to-child transmission can occur in the groin area from shared equipment, transmission via sexual abuse also is possible.14 Bargman15 has mentioned the isolated genital location and lack of contact with other infected children as concerning features. Latency of new lesion appearance is anywhere from 1 to 50 days from the date of inoculation; therefore, new lesions are possible and expected even after therapy has been effective in eradicating visible lesions.10 Although clearance has been reported in 6 to 12 months, one pediatric study demonstrated 70% clearance by 1.5 years, suggesting the disease often is more prolonged.16 One-third of children will experience signs of inflammation, such as pruritus and/or erythema. Rare side effects include bacterial superinfection and hypersensitivity.2
One Dutch study from 1994, the largest database survey of children to date, cited a 17% cumulative incidence of molluscum in children by reviewing the data from 103 general practices.17 In a survey and review of molluscum by Braue et al,18 annual rates in populations vary but seem to maximize at approximately 6% to 7%. Sturt et al19 reviewed the prevalence in the indigenous West Sepik section of New Guinea and noted annual incidence rates of 6% in children younger than 10 years (range, 1.8%–10.9%). Epidemics occur and can produce large numbers of cases in a short time period.18 The cumulative prevalence in early childhood may be as high as 22%, as Sturt et al19 observed in children younger than 10 years.
Rising incidence and therefore rising lifetime prevalence appear to have been an issue in the last few decades. Data from the Indian Health Service have demonstrated increases in MCV in Native American children between 2001 and 2005.20 In adults, the data support a steady increase of molluscum from 1988-2007, with a 3-fold increase from 1988-1997 to 1998-2007 in a Spanish study.21 Better population-based data are needed.
Childhood Immunity and Vaccination
Sequence homology between MC133L, a protein of MCV, with vaccinia virus suggests overlapping genes.22 Therefore, it is conceptually possible that the rise in incidence of MCV since the 1980s relates to the loss of herd immunity to variola due to lack of vaccination for smallpox, which has not been offered in the United States since 1972.23 Childhood immunity to MCV varies among studies, but it appears that children do develop antibodies to molluscum in the setting of forming an immune response. Because the rise in molluscum incidence began after the smallpox vaccine was discontinued, the factors appear related; however, the scientific data do not support the theory of a relationship. Mitchell24 has shown that a patient can develop antibodies in response to ground molluscum bodies inoculated into the skin; however, vaccination against molluscum and natural infection do not appear to produce antibodies that would cross-react and protect against other poxviruses, including vaccinia or fowl pox infections.25 Cell-mediated immunity also is required to clear MCV and may account for the inflammatory appearance of lesions as they resolve.26
Demonstrated factors that account for the rise in MCV incidence, aside from alterations in vaccination practices, include spread through sports,9 swimming,11 and AD,7 which have become more commonplace in the United States in the last few decades, supporting the theory that they may be the cause of the increase in childhood MCV infections. Another cause may be the ability of MCV to create factors that stem host immune response.1
Clinical Features
Molluscum lesions have a typical appearance of pearly papules with a central dell. These lesions are lighter to flesh colored and measure 1 to 3 mm.2,4,5 The lesions cluster in the axillae and extremities and average from 10 to 20 per child.6 Lesions clear spontaneously, but new ones will continue to form until immunity is developed. Specific clinical appearances of lesions that are not pearly papules are not infrequent. Table 1 contains a short list of the manifold clinical appearances of molluscum lesions in children.1,2,7,27-35 In particular, certain clinical appearances should be considered. In small children, head and neck lesions resembling milia are not uncommon. Giant or wartlike lesions can appear on the head, neck, or gluteal region in children and are clinical mimics of condyloma or other warts (Figure 1). Giant lesions also can grow in the subcutaneous space and mimic a cyst or abscess.27 Erosive lesions mimicking eczema vaccinatum can be seen (Figure 2), but dermoscopy may demonstrate central dells in some lesions. Other viral processes mimicked include Gianotti Crosti–like lesions (Figure 3) that appear when a papular id reaction forms over the extremities or a localized version in the axilla, mimicking unilateral laterothoracic exanthema.2,36,37 Hypersensitivity reactions are commonly noted with clearance and can be papular or demonstrate swelling and erythema, termed the beginning-of-the-end sign.38

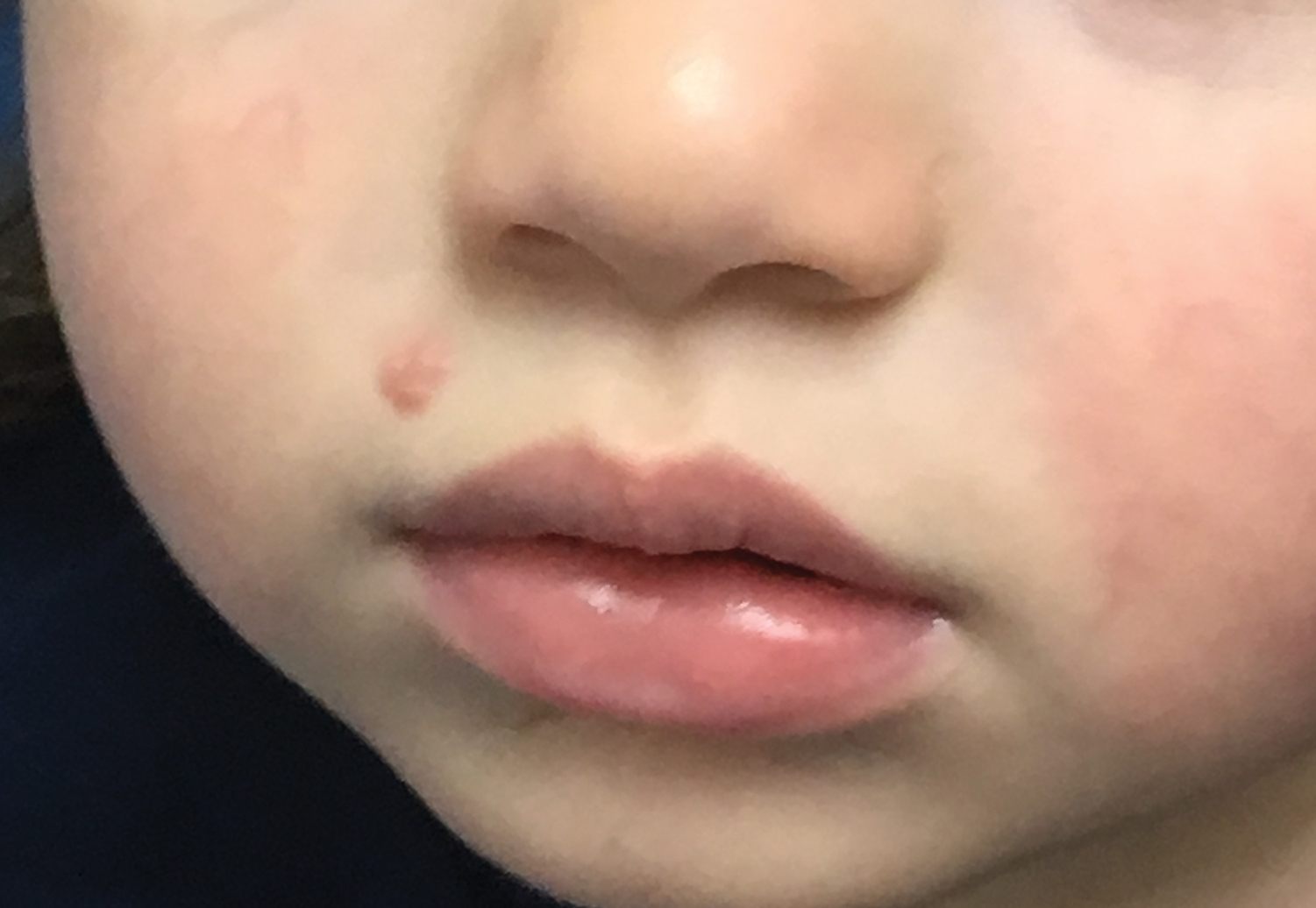

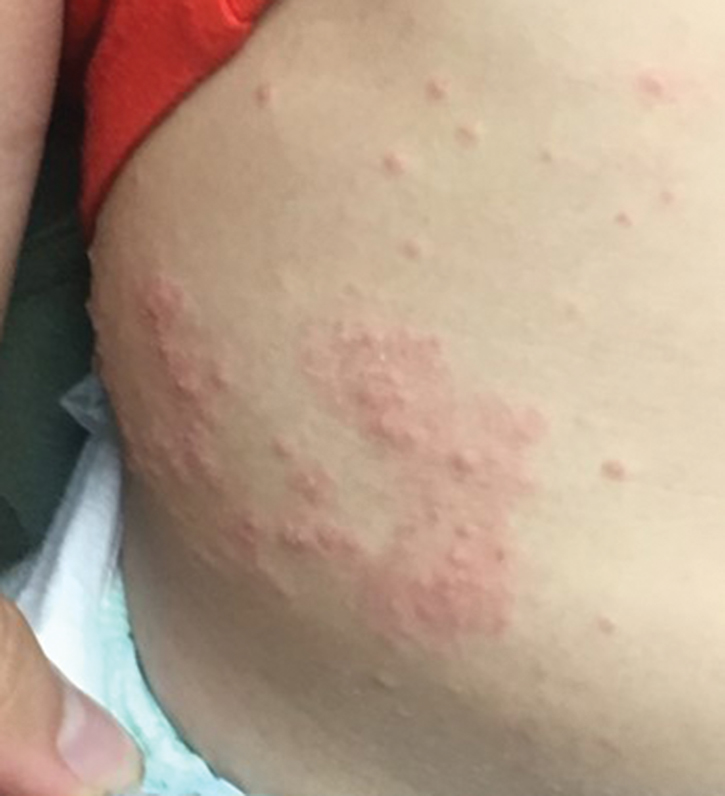
Pruritus, erythema, and swelling can occur with clearance but do not appear in all patients. Addressing pruritus is important to prevent disease spread, as patients are likely to inoculate other areas of the skin with virus when they scratch, and lesion number is reduced with dermatitis interventions.36
Comorbidities
Molluscum lesions can occur in any child; however, the impaired immunologic status and skin barrier in patients with AD is ripe for the extensive spread of lesions that is associated with higher lesion count.36 Children with molluscum infection can experience new-onset dermatitis or triggering of AD flares, especially on the extremities, such as the antecubital and popliteal regions.7 A study of children with MCV infection demonstrated that treatment of active dermatitis reduced spread. The authors mentioned autoinoculation as the mechanism; however, these data also suggest supporting barrier state as a factor in disease spread.36 Superinfection can occur prior to6 or after therapy for lesions,37 but it is unclear if this relates to the underlying atopic diathesis. Children with molluscum have been described to have warts, psoriasis, family history of atopy, diabetes mellitus, and pityriasis alba,7 while immunosuppression of any kind is associated with molluscum and high lesion count or prolonged disease in childhood.1,2
Quality of Life
Children with molluscum who have higher lesion counts appear to be at risk for severe effects on their quality of life. Approximately 10% of children with MCV infection have been documented to have severe impairments on quality of life.39 In my practice, quality of life in children with MCV appears to be affected by many factors (Table 2).7,18,39

Treatments
Proper Skin Care and Treatment of AD
Therapy for AD and/or pruritus appears to limit lesion number in children with MCV and rashes or itch.7,36 I recommend barrier repair agents, including emollients and syndet bar cleansers, to prevent small breaks in the skin that occur with xerosis and AD and that increase itch and risk of spread. Therapy for AD and molluscum dermatitis is similar and overlapping. There is always a concern about the spread of MCV when using topical calcineurin inhibitors. I, therefore, focus the dermatitis therapeutics on topical corticosteroid–based care.6,40
Prevention of Spread
Prevention of spread begins with hygiene interventions. Cobathing is common in children with MCV and should be held off when possible. It is important for the child with MCV to avoid sharing bath towels and equipment23 and having bare skin come in contact with mats in sports. I request that children with MCV wear bathing suits that cover the areas affected.
Reassurance
The most important therapy is reassurance.41 Many parents/guardians are truly unaware that the MCV infection can last for more than a year and therefore worry over normal disease course. When counseled as to the benign course of illness and given instructions on proper skin care, the parent/guardian of a child with MCV will often opt against therapy of uncomplicated cases. On the other hand, there are medical reasons for treatment, and they support the need for intervention (Table 3). Seventy percent of lesions resolve in 1.5 years; however, of the residual infections, some may last as long as 4 years.16 It is not recommended to stop children from attending school because of MCV.

Interventional Therapy
Therapeutics of MCV include destructive therapies in office (ie, cantharidin, cryotherapy, curettage, trichloroacetic acid, and glycolic acid) and at-home therapies (ie, topical retinoids, nitric oxide releasers)(eTable).2,5,6,42-58 When there are many lesions or spread is noted, immunotherapies can be used, including topical imiquimod, oral cimetidine, and intralesional Candida antigen.2,4,7 Pulsed dye laser cuts off the lesion vascular supply, while cidofovir is directly antiviral both topically and systemically, the latter reserved for severe cases in immunosuppressed adults.59 Head-to-head studies of cantharidin, curettage, topical peeling agents, and imiquimod demonstrated better satisfaction and fewer office visits with topical anesthetic and curettage on the first visit. Side effects were greatest for salicylic acid and glycolic acid; therefore, these agents are less desirable.42
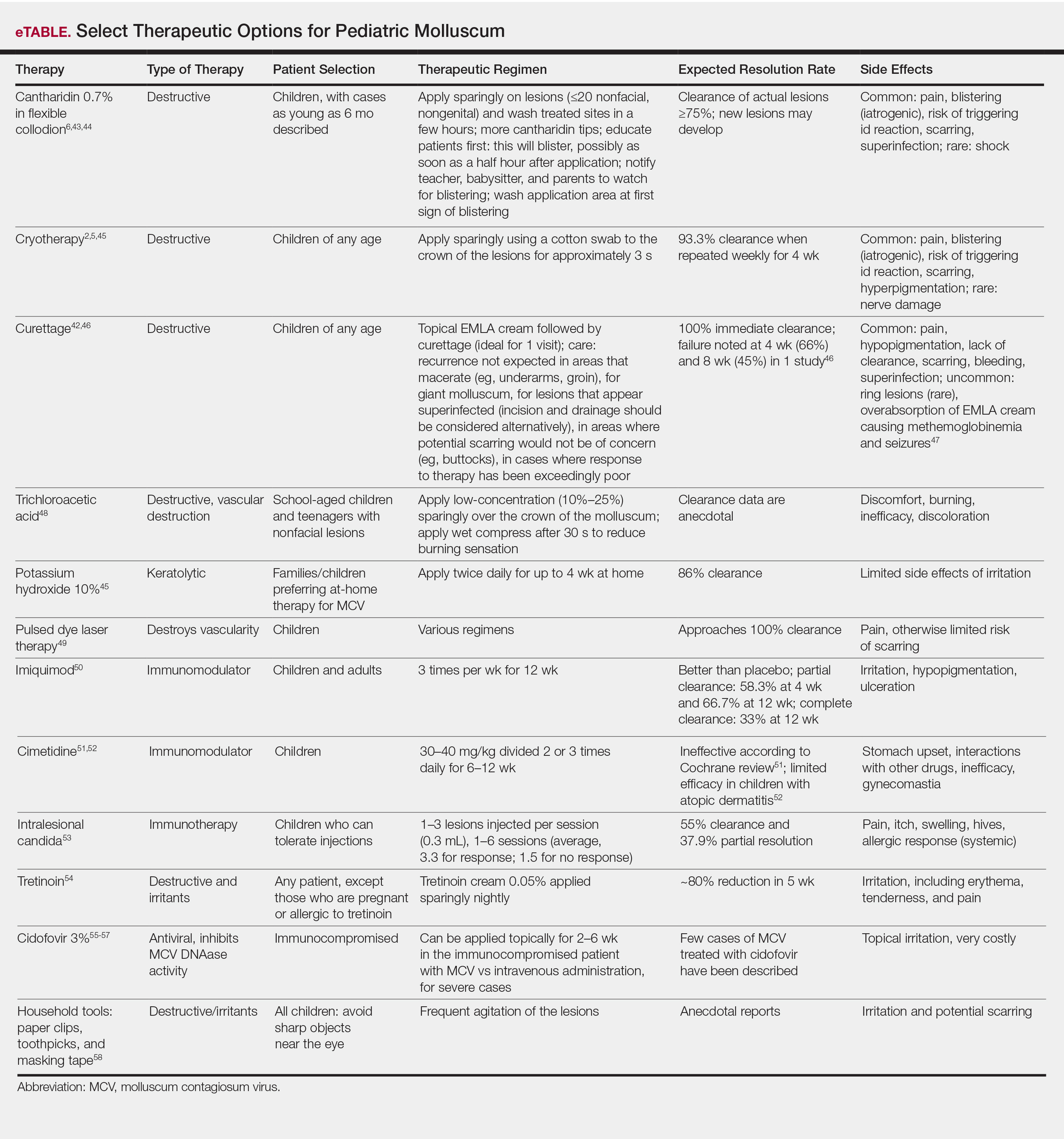
Conclusion
Molluscum is a cutaneous viral infection that is common in children and has associated morbidities, including AD, pruritus, poor quality of life in some cases, and risk of contagion. Addressing the disease includes understanding its natural history and explaining it to parents/guardians. Therapeutics can be offered in cases where need is demonstrated, such as with lesions that spread and cause discomfort. Choice of therapeutics depends on the practitioner’s experience, the child’s clinical appearance, availability of therapy, and review of options with the parents/guardians. When avoidance of intervention is desired, barrier enhancement and treatment of symptomatic dermatitis are still beneficial, as are household (eg, not sharing towels) and activity (eg, adhesive bandages over active lesions) interventions to reduce transmission.
- Shisler JL. Immune evasion strategies of molluscum contagiosum virus. Adv Virus Res. 2015;92:201-252.
- Brown J, Janniger CK, Schwartz RA, et al. Childhood molluscum contagiosum. Int J Dermatol. 2006;45:93-99.
- Moss B, Shisler JL, Xiang Y, et al. Immune-defense molecules of molluscum contagiosum virus, a human poxvirus. Trends Microbiol. 2000;8:473-477.
- Silverberg NB. Warts and molluscum in children. Adv Dermatol. 2004;20:23-73.
- Choong KY, Roberts LJ. Molluscum contagiosum, swimming and bathing: a clinical analysis. Australas J Dermatol. 1999;40:89-92.
- Silverberg NB, Sidbury R, Mancini AJ. Childhood molluscum contagiosum: experience with cantharidin therapy in 300 patients. J Am Acad Dermatol. 2000;43:503-507.
- Silverberg NB. Molluscum contagiosum virus infection can trigger atopic dermatitis disease onset or flare. Cutis. 2018;102:191-194.
- Ajithkumar VT, Sasidharanpillai S, Muhammed K, et al. Disseminated molluscum contagiosum following chemotherapy: a therapeutic challenge. Indian J Dermatol Venereol Leprol. 2017;83:516.
- Oren B, Wende SO. An outbreak of molluscum contagiosum in a kibbutz. Infection. 1991;19:159-161.
- Molluscum contagiosum. Healthy Children website. https://www.healthychildren.org/English/health-issues/conditions/skin/Pages/Molluscum-Contagiosum.aspx. Updated November 21, 2015. Accessed October 16, 2019.
- Peterson AR, Nash E, Anderson BJ. Infectious disease in contact sports. Sports Health. 2019;11:47-58.
- Connell CO, Oranje A, Van Gysel D, et al. Congenital molluscum contagiosum: report of four cases and review of the literature. Pediatr Dermatol. 2008;25:553-556.
- Luke JD, Silverberg NB. Vertically transmitted molluscum contagiosum infection. Pediatrics. 2010;125:E423-E425.
- Mendiratta V, Agarwal S, Chander R. Reappraisal of sexually transmitted infections in children: a hospital-based study from an urban area. Indian J Sex Transm Dis AIDS. 2014;35:25-28.
- Bargman H. Genital molluscum contagiosum in children: evidence of sexual abuse? CMAJ. 1986;135:432-433.
- Basdag H, Rainer BM, Cohen BA. Molluscum contagiosum: to treat or not to treat? experience with 170 children in an outpatient clinic setting in the northeastern United States. Pediatr Dermatol. 2015;32:353-357.
- Koning S, Bruijnzeels MA, van Suijlekom-Smit LW, et al. Molluscum contagiosum in Dutch general practice. Br J Gen Pract. 1994;44:417-419.
- Braue A, Ross G, Varigos G, et al. Epidemiology and impact of childhood molluscum contagiosum: a case series and critical review of the literature. Pediatr Dermatol. 2005;22:287-294.
- Sturt RJ, Muller HK, Francis GD. Molluscum contagiosum in villages of the West Sepik District of New Guinea. Med J Aust. 1971;2:751-754.
- Reynolds MG, Homan RC, Yorita Christensen KL, et al. The incidence of molluscum contagiosum among American Indians and Alaska Natives. PLoS One. 2009;4:e5255.
- Villa L, Varela JA, Otero L, et al. Molluscum contagiosum: a 20-year study in a sexually transmitted infections unit. Sex Transm Dis. 2010;37:423-424.
- Watanabe T, Morikawa S, Suzuki K, et al. Two major antigenic polypeptides of molluscum contagiosum virus. J Infect Dis. 1998;177:284-292.
- Vaccine basics. Centers for Disease Control and Prevention website. https://www.cdc.gov/smallpox/vaccine-basics/index.html. Updated July 12, 2017. Accessed October 16, 2019.
- Mitchell JC. Observations on the virus of molluscum contagiosum. Br J Exp Pathol. 1953;34:44-49.
- Konya J, Thompson CH. Molluscum contagiosum virus: antibody responses in patients with clinical lesions and its sero-epidemiology in a representative Australian population. J Infect Dis. 1999;179:701-704.
- Steffen C, Markman JA. Spontaneous disappearance of molluscum contagiosum. Arch Dermatol. 1980;116:923-924.
- Uzuncakmak TK, Kuru BC, Zemheri EI, et al. Isolated giant molluscum contagiosum mimicking epidermoid cyst. Dermatol Pract Concept. 2016;6:71-73.
- Persechino S, Abruzzese C, Caperchi C, et al. Condyloma acuminata and mollusca contagiosa: a giant manifestation in a patient with lupus. Skinmed. 2014;12:310-311.
- Kim SK, Do JE, Kang HY, et al. Giant molluscum contagiosum of immunocompetent children occurring on the anogenital area. Eur J Dermatol. 2007;17:537-538.
- Alam MS, Shrirao N. Giant molluscum contagiosum presenting as lid neoplasm in an immunocompetent child. Dermatol Online J. 2016;22. pii:13030/qt56v567gn.
- Krishnamurthy J, Nagappa DK. The cytology of molluscum contagiosum mimicking skin adnexal tumor. J Cytol. 2010;27:74-75.
- Baek YS, Oh CH, Song HJ, et al. Asymmetrical periflexural exanthem of childhood with concurrence of molluscum contagiosum infection. Clin Exp Dermatol. 2011;36:676-677.
- Lee HJ, Kwon JA, Kim JW. Erythema multiforme-like molluscum dermatitis. Acta Derm Venereol. 2002;82:217-218.
- Lee YB, Choi HJ, Park HJ, et al. Two cases of erythema multiforme associated with molluscum contagiosum. Int J Dermatol. 2009;48:659-660.
- Vasily DB, Bhatia SG. Erythema annulare centrifugum and molluscum contagiosum.
- Berger EM, Orlow SJ, Patel RR, et al. Experience with molluscum contagiosum and associated inflammatory reactions in a pediatric dermatology practice: the bump that rashes. Arch Dermatol. 2012;148:1257-1264.
- Groner A, Laing-Grayman D, Silverberg NB. Outpatient pediatric community-acquired methicillin-resistant Staphylococcus aureus: a polymorphous clinical disease. Cutis. 2008;81:115-122.
- Butala N, Siegfried E, Weissler A. Molluscum BOTE sign: a predictor of imminent resolution. Pediatrics. 2013;131:E1650-E1653.
- Olsen JR, Gallagher J, Finlay AY, et al. Time to resolution and effect on quality of life of molluscum contagiosum in children in the UK: a prospective community cohort study. Lancet Infect Dis. 2015;15:190-195.
- Goksugur N, Ozbostanci B, Goksugur SB. Molluscum contagiosum infection associated with pimecrolimus use in pityriasis alba. Pediatr Dermatol. 2007;24:E63-E65.
- Lee R, Schwartz RA. Pediatric molluscum contagiosum: reflections on the last challenging poxvirus infection, part 1. Cutis. 2010;86:230-236.
- Hanna D, Hatami A, Powell J, et al. A prospective randomized trial comparing the efficacy and adverse effects of four recognized treatments of molluscum contagiosum in children. Pediatr Dermatol. 2006;23:574-579.
- Coloe Dosal J, Stewart PW, Lin JA, et al. Cantharidin for the treatment of molluscum contagiosum: a prospective, double-blinded, placebo-controlled trial. Pediatr Dermatol. 2014;31:440-449.
- Vakharia PP, Chopra R, Silverberg NB, et al. Efficacy and safety of topical cantharidin treatment for molluscum contagiosum and warts: a systematic review. Am J Clin Dermatol. 2018;19:791-803.
- Handjani F, Behazin E, Sadati MS. Comparison of 10% potassium hydroxide solution versus cryotherapy in the treatment of molluscum contagiosum: an open randomized clinical trial. J Dermatolog Treat. 2014;25:249-250.
- Simonart T, De Maertelaer V. Curettage treatment for molluscum contagiosum: a follow-up survey study. Br J Dermatol. 2008;159:1144-1147.
- Cho YS, Chung BY, Park CW, et al. Seizures and methemoglobinemia after topical application of eutectic mixture of lidocaine and prilocaine on a 3.5-year-old child with molluscum contagiosum and atopic dermatitis. Pediatr Dermatol. 2016;33:E284-E285.
- Bard S, Shiman MI, Bellman B, et al. Treatment of facial molluscum contagiosum with trichloroacetic acid. Pediatr Dermatol. 2009;26:425-426.
- Griffith RD, Yazdani Abyaneh MA, Falto-Aizpurua L, et al. Pulsed dye laser therapy for molluscum contagiosum: a systematic review. J Drugs Dermatol. 2014;13:1349-1352.
- Theos AU, Cummins R, Silverberg NB, et al. Effectiveness of imiquimod cream 5% for treating childhood molluscum contagiosum in a double-blind, randomized pilot trial. Cutis. 2004;74:134-138, 141-142.
- van der Wouden JC, Menke J, Gajadin S, et al. Interventions for cutaneous molluscum contagiosum. Cochrane Database Syst Rev. 2006:CD004767.
- Cunningham BB, Paller AS, Garzon M. Inefficacy of oral cimetidine for nonatopic children with molluscum contagiosum. Pediatr Dermatol. 1998;15:71-72.
- Enns LL, Evans MS. Intralesional immunotherapy with Candida antigen for the treatment of molluscum contagiosum in children. Pediatr Dermatol. 2011;28:254-258.
- Rajouria EA, Amatya A, Karn D. Comparative study of 5% potassium hydroxide solution versus 0.05% tretinoin cream for molluscum contagiosum in children. Kathmandu Univ Med J (KUMJ). 2011;9:291-294.
- Briand S, Milpied B, Navas D, et al. 1% topical cidofovir used as last alternative to treat viral infections. J Eur Acad Dermatol Venereol. 2008;22:249-250.
- Zabawski EJ Jr, Cockerell CJ. Topical cidofovir for molluscum contagiosum in children. Pediatr Dermatol. 1999;16:414-415.
- Watanabe T. Cidofovir diphosphate inhibits molluscum contagiosum virus DNA polymerase activity. J Invest Dermatol. 2008;128:1327-1329.
- Lindau MS, Munar MY. Use of duct tape occlusion in the treatment of recurrent molluscum contagiosum. Pediatr Dermatol. 2004;21:609.
- Silverberg N. Pediatric molluscum contagiosum: optimal treatment strategies. Paediatr Drugs. 2003;5:505-512.
Molluscum contagiosum virus (MCV) infection causes the cutaneous lesions we call molluscum. Molluscum has become common in the last 30 years. Deciding the best course of therapy requires some fundamental understanding about how MCV relates to the following factors: epidemiology, childhood immunity and vaccination, clinical features, comorbidities, and quality of life. Treatment depends on many factors, including presence or absence of atopic dermatitis (AD) and/or pruritus, other symptoms, cosmetic location, and the child’s concern about the lesions. Therapeutics include destructive and immunologic therapies, the latter geared toward increasing immune response.
Epidemiology
Molluscum contagiosum virus is the solo member of the Molluscipoxvirus genus. Infection with MCV causes benign growth or tumors in the skin (ie, molluscum). The infection is slow to clear because the virus reduces the host’s immunity.1,2 Molluscum contagiosum virus is a double-stranded DNA virus that affects keratinocytes and genetically carries the tools for its own replication (ie, DNA-dependent RNA polymerase). The virus has a few subtypes—I/Ia, II, III, and IV—with MCV-I predominating in children and healthy humans and MCV-II in patients with human immunodeficiency virus.1,2 Typing is experimental and is not standardly performed in clinical practice. Molluscum contagiosum virus produces a variety of factors that block the host’s immune response, prolonging infection and preventing erythema and inflammatory response.3
Molluscum contagiosum virus is transmitted through skin-to-skin contact and fomites, including shared towels, bathtubs, spas, bath sponges, and pool equipment.2,4,5 Transmission from household contact and bathing together has been noted in pediatric patients with MCV. Based on the data it can be posited that the lesions are softer when wet and more readily release viral particles or fomites, and fomites may be left on surfaces, especially when a child is wet.6,7 Propensity for infection occurs in patients with AD and in immunosuppressed hosts, including children with human immunodeficiency virus and iatrogenic immunosuppression caused by chemotherapy.1,2,8 Contact sports can increase the risk of transmission, and outbreaks have occurred in pools,5,9 day-care facilities,10 and sports settings.11 Cases of congenital and vertically transmitted molluscum have been documented.12,13 Sexual transmission of MCV may be seen in adolescents who are sexually active. Although child-to-child transmission can occur in the groin area from shared equipment, transmission via sexual abuse also is possible.14 Bargman15 has mentioned the isolated genital location and lack of contact with other infected children as concerning features. Latency of new lesion appearance is anywhere from 1 to 50 days from the date of inoculation; therefore, new lesions are possible and expected even after therapy has been effective in eradicating visible lesions.10 Although clearance has been reported in 6 to 12 months, one pediatric study demonstrated 70% clearance by 1.5 years, suggesting the disease often is more prolonged.16 One-third of children will experience signs of inflammation, such as pruritus and/or erythema. Rare side effects include bacterial superinfection and hypersensitivity.2
One Dutch study from 1994, the largest database survey of children to date, cited a 17% cumulative incidence of molluscum in children by reviewing the data from 103 general practices.17 In a survey and review of molluscum by Braue et al,18 annual rates in populations vary but seem to maximize at approximately 6% to 7%. Sturt et al19 reviewed the prevalence in the indigenous West Sepik section of New Guinea and noted annual incidence rates of 6% in children younger than 10 years (range, 1.8%–10.9%). Epidemics occur and can produce large numbers of cases in a short time period.18 The cumulative prevalence in early childhood may be as high as 22%, as Sturt et al19 observed in children younger than 10 years.
Rising incidence and therefore rising lifetime prevalence appear to have been an issue in the last few decades. Data from the Indian Health Service have demonstrated increases in MCV in Native American children between 2001 and 2005.20 In adults, the data support a steady increase of molluscum from 1988-2007, with a 3-fold increase from 1988-1997 to 1998-2007 in a Spanish study.21 Better population-based data are needed.
Childhood Immunity and Vaccination
Sequence homology between MC133L, a protein of MCV, with vaccinia virus suggests overlapping genes.22 Therefore, it is conceptually possible that the rise in incidence of MCV since the 1980s relates to the loss of herd immunity to variola due to lack of vaccination for smallpox, which has not been offered in the United States since 1972.23 Childhood immunity to MCV varies among studies, but it appears that children do develop antibodies to molluscum in the setting of forming an immune response. Because the rise in molluscum incidence began after the smallpox vaccine was discontinued, the factors appear related; however, the scientific data do not support the theory of a relationship. Mitchell24 has shown that a patient can develop antibodies in response to ground molluscum bodies inoculated into the skin; however, vaccination against molluscum and natural infection do not appear to produce antibodies that would cross-react and protect against other poxviruses, including vaccinia or fowl pox infections.25 Cell-mediated immunity also is required to clear MCV and may account for the inflammatory appearance of lesions as they resolve.26
Demonstrated factors that account for the rise in MCV incidence, aside from alterations in vaccination practices, include spread through sports,9 swimming,11 and AD,7 which have become more commonplace in the United States in the last few decades, supporting the theory that they may be the cause of the increase in childhood MCV infections. Another cause may be the ability of MCV to create factors that stem host immune response.1
Clinical Features
Molluscum lesions have a typical appearance of pearly papules with a central dell. These lesions are lighter to flesh colored and measure 1 to 3 mm.2,4,5 The lesions cluster in the axillae and extremities and average from 10 to 20 per child.6 Lesions clear spontaneously, but new ones will continue to form until immunity is developed. Specific clinical appearances of lesions that are not pearly papules are not infrequent. Table 1 contains a short list of the manifold clinical appearances of molluscum lesions in children.1,2,7,27-35 In particular, certain clinical appearances should be considered. In small children, head and neck lesions resembling milia are not uncommon. Giant or wartlike lesions can appear on the head, neck, or gluteal region in children and are clinical mimics of condyloma or other warts (Figure 1). Giant lesions also can grow in the subcutaneous space and mimic a cyst or abscess.27 Erosive lesions mimicking eczema vaccinatum can be seen (Figure 2), but dermoscopy may demonstrate central dells in some lesions. Other viral processes mimicked include Gianotti Crosti–like lesions (Figure 3) that appear when a papular id reaction forms over the extremities or a localized version in the axilla, mimicking unilateral laterothoracic exanthema.2,36,37 Hypersensitivity reactions are commonly noted with clearance and can be papular or demonstrate swelling and erythema, termed the beginning-of-the-end sign.38
Pruritus, erythema, and swelling can occur with clearance but do not appear in all patients. Addressing pruritus is important to prevent disease spread, as patients are likely to inoculate other areas of the skin with virus when they scratch, and lesion number is reduced with dermatitis interventions.36
Comorbidities
Molluscum lesions can occur in any child; however, the impaired immunologic status and skin barrier in patients with AD is ripe for the extensive spread of lesions that is associated with higher lesion count.36 Children with molluscum infection can experience new-onset dermatitis or triggering of AD flares, especially on the extremities, such as the antecubital and popliteal regions.7 A study of children with MCV infection demonstrated that treatment of active dermatitis reduced spread. The authors mentioned autoinoculation as the mechanism; however, these data also suggest supporting barrier state as a factor in disease spread.36 Superinfection can occur prior to6 or after therapy for lesions,37 but it is unclear if this relates to the underlying atopic diathesis. Children with molluscum have been described to have warts, psoriasis, family history of atopy, diabetes mellitus, and pityriasis alba,7 while immunosuppression of any kind is associated with molluscum and high lesion count or prolonged disease in childhood.1,2
Quality of Life
Children with molluscum who have higher lesion counts appear to be at risk for severe effects on their quality of life. Approximately 10% of children with MCV infection have been documented to have severe impairments on quality of life.39 In my practice, quality of life in children with MCV appears to be affected by many factors (Table 2).7,18,39
Treatments
Proper Skin Care and Treatment of AD
Therapy for AD and/or pruritus appears to limit lesion number in children with MCV and rashes or itch.7,36 I recommend barrier repair agents, including emollients and syndet bar cleansers, to prevent small breaks in the skin that occur with xerosis and AD and that increase itch and risk of spread. Therapy for AD and molluscum dermatitis is similar and overlapping. There is always a concern about the spread of MCV when using topical calcineurin inhibitors. I, therefore, focus the dermatitis therapeutics on topical corticosteroid–based care.6,40
Prevention of Spread
Prevention of spread begins with hygiene interventions. Cobathing is common in children with MCV and should be held off when possible. It is important for the child with MCV to avoid sharing bath towels and equipment23 and having bare skin come in contact with mats in sports. I request that children with MCV wear bathing suits that cover the areas affected.
Reassurance
The most important therapy is reassurance.41 Many parents/guardians are truly unaware that the MCV infection can last for more than a year and therefore worry over normal disease course. When counseled as to the benign course of illness and given instructions on proper skin care, the parent/guardian of a child with MCV will often opt against therapy of uncomplicated cases. On the other hand, there are medical reasons for treatment, and they support the need for intervention (Table 3). Seventy percent of lesions resolve in 1.5 years; however, of the residual infections, some may last as long as 4 years.16 It is not recommended to stop children from attending school because of MCV.
Interventional Therapy
Therapeutics of MCV include destructive therapies in office (ie, cantharidin, cryotherapy, curettage, trichloroacetic acid, and glycolic acid) and at-home therapies (ie, topical retinoids, nitric oxide releasers)(eTable).2,5,6,42-58 When there are many lesions or spread is noted, immunotherapies can be used, including topical imiquimod, oral cimetidine, and intralesional Candida antigen.2,4,7 Pulsed dye laser cuts off the lesion vascular supply, while cidofovir is directly antiviral both topically and systemically, the latter reserved for severe cases in immunosuppressed adults.59 Head-to-head studies of cantharidin, curettage, topical peeling agents, and imiquimod demonstrated better satisfaction and fewer office visits with topical anesthetic and curettage on the first visit. Side effects were greatest for salicylic acid and glycolic acid; therefore, these agents are less desirable.42
Conclusion
Molluscum is a cutaneous viral infection that is common in children and has associated morbidities, including AD, pruritus, poor quality of life in some cases, and risk of contagion. Addressing the disease includes understanding its natural history and explaining it to parents/guardians. Therapeutics can be offered in cases where need is demonstrated, such as with lesions that spread and cause discomfort. Choice of therapeutics depends on the practitioner’s experience, the child’s clinical appearance, availability of therapy, and review of options with the parents/guardians. When avoidance of intervention is desired, barrier enhancement and treatment of symptomatic dermatitis are still beneficial, as are household (eg, not sharing towels) and activity (eg, adhesive bandages over active lesions) interventions to reduce transmission.
Molluscum contagiosum virus (MCV) infection causes the cutaneous lesions we call molluscum. Molluscum has become common in the last 30 years. Deciding the best course of therapy requires some fundamental understanding about how MCV relates to the following factors: epidemiology, childhood immunity and vaccination, clinical features, comorbidities, and quality of life. Treatment depends on many factors, including presence or absence of atopic dermatitis (AD) and/or pruritus, other symptoms, cosmetic location, and the child’s concern about the lesions. Therapeutics include destructive and immunologic therapies, the latter geared toward increasing immune response.
Epidemiology
Molluscum contagiosum virus is the solo member of the Molluscipoxvirus genus. Infection with MCV causes benign growth or tumors in the skin (ie, molluscum). The infection is slow to clear because the virus reduces the host’s immunity.1,2 Molluscum contagiosum virus is a double-stranded DNA virus that affects keratinocytes and genetically carries the tools for its own replication (ie, DNA-dependent RNA polymerase). The virus has a few subtypes—I/Ia, II, III, and IV—with MCV-I predominating in children and healthy humans and MCV-II in patients with human immunodeficiency virus.1,2 Typing is experimental and is not standardly performed in clinical practice. Molluscum contagiosum virus produces a variety of factors that block the host’s immune response, prolonging infection and preventing erythema and inflammatory response.3
Molluscum contagiosum virus is transmitted through skin-to-skin contact and fomites, including shared towels, bathtubs, spas, bath sponges, and pool equipment.2,4,5 Transmission from household contact and bathing together has been noted in pediatric patients with MCV. Based on the data it can be posited that the lesions are softer when wet and more readily release viral particles or fomites, and fomites may be left on surfaces, especially when a child is wet.6,7 Propensity for infection occurs in patients with AD and in immunosuppressed hosts, including children with human immunodeficiency virus and iatrogenic immunosuppression caused by chemotherapy.1,2,8 Contact sports can increase the risk of transmission, and outbreaks have occurred in pools,5,9 day-care facilities,10 and sports settings.11 Cases of congenital and vertically transmitted molluscum have been documented.12,13 Sexual transmission of MCV may be seen in adolescents who are sexually active. Although child-to-child transmission can occur in the groin area from shared equipment, transmission via sexual abuse also is possible.14 Bargman15 has mentioned the isolated genital location and lack of contact with other infected children as concerning features. Latency of new lesion appearance is anywhere from 1 to 50 days from the date of inoculation; therefore, new lesions are possible and expected even after therapy has been effective in eradicating visible lesions.10 Although clearance has been reported in 6 to 12 months, one pediatric study demonstrated 70% clearance by 1.5 years, suggesting the disease often is more prolonged.16 One-third of children will experience signs of inflammation, such as pruritus and/or erythema. Rare side effects include bacterial superinfection and hypersensitivity.2
One Dutch study from 1994, the largest database survey of children to date, cited a 17% cumulative incidence of molluscum in children by reviewing the data from 103 general practices.17 In a survey and review of molluscum by Braue et al,18 annual rates in populations vary but seem to maximize at approximately 6% to 7%. Sturt et al19 reviewed the prevalence in the indigenous West Sepik section of New Guinea and noted annual incidence rates of 6% in children younger than 10 years (range, 1.8%–10.9%). Epidemics occur and can produce large numbers of cases in a short time period.18 The cumulative prevalence in early childhood may be as high as 22%, as Sturt et al19 observed in children younger than 10 years.
Rising incidence and therefore rising lifetime prevalence appear to have been an issue in the last few decades. Data from the Indian Health Service have demonstrated increases in MCV in Native American children between 2001 and 2005.20 In adults, the data support a steady increase of molluscum from 1988-2007, with a 3-fold increase from 1988-1997 to 1998-2007 in a Spanish study.21 Better population-based data are needed.
Childhood Immunity and Vaccination
Sequence homology between MC133L, a protein of MCV, with vaccinia virus suggests overlapping genes.22 Therefore, it is conceptually possible that the rise in incidence of MCV since the 1980s relates to the loss of herd immunity to variola due to lack of vaccination for smallpox, which has not been offered in the United States since 1972.23 Childhood immunity to MCV varies among studies, but it appears that children do develop antibodies to molluscum in the setting of forming an immune response. Because the rise in molluscum incidence began after the smallpox vaccine was discontinued, the factors appear related; however, the scientific data do not support the theory of a relationship. Mitchell24 has shown that a patient can develop antibodies in response to ground molluscum bodies inoculated into the skin; however, vaccination against molluscum and natural infection do not appear to produce antibodies that would cross-react and protect against other poxviruses, including vaccinia or fowl pox infections.25 Cell-mediated immunity also is required to clear MCV and may account for the inflammatory appearance of lesions as they resolve.26
Demonstrated factors that account for the rise in MCV incidence, aside from alterations in vaccination practices, include spread through sports,9 swimming,11 and AD,7 which have become more commonplace in the United States in the last few decades, supporting the theory that they may be the cause of the increase in childhood MCV infections. Another cause may be the ability of MCV to create factors that stem host immune response.1
Clinical Features
Molluscum lesions have a typical appearance of pearly papules with a central dell. These lesions are lighter to flesh colored and measure 1 to 3 mm.2,4,5 The lesions cluster in the axillae and extremities and average from 10 to 20 per child.6 Lesions clear spontaneously, but new ones will continue to form until immunity is developed. Specific clinical appearances of lesions that are not pearly papules are not infrequent. Table 1 contains a short list of the manifold clinical appearances of molluscum lesions in children.1,2,7,27-35 In particular, certain clinical appearances should be considered. In small children, head and neck lesions resembling milia are not uncommon. Giant or wartlike lesions can appear on the head, neck, or gluteal region in children and are clinical mimics of condyloma or other warts (Figure 1). Giant lesions also can grow in the subcutaneous space and mimic a cyst or abscess.27 Erosive lesions mimicking eczema vaccinatum can be seen (Figure 2), but dermoscopy may demonstrate central dells in some lesions. Other viral processes mimicked include Gianotti Crosti–like lesions (Figure 3) that appear when a papular id reaction forms over the extremities or a localized version in the axilla, mimicking unilateral laterothoracic exanthema.2,36,37 Hypersensitivity reactions are commonly noted with clearance and can be papular or demonstrate swelling and erythema, termed the beginning-of-the-end sign.38
Pruritus, erythema, and swelling can occur with clearance but do not appear in all patients. Addressing pruritus is important to prevent disease spread, as patients are likely to inoculate other areas of the skin with virus when they scratch, and lesion number is reduced with dermatitis interventions.36
Comorbidities
Molluscum lesions can occur in any child; however, the impaired immunologic status and skin barrier in patients with AD is ripe for the extensive spread of lesions that is associated with higher lesion count.36 Children with molluscum infection can experience new-onset dermatitis or triggering of AD flares, especially on the extremities, such as the antecubital and popliteal regions.7 A study of children with MCV infection demonstrated that treatment of active dermatitis reduced spread. The authors mentioned autoinoculation as the mechanism; however, these data also suggest supporting barrier state as a factor in disease spread.36 Superinfection can occur prior to6 or after therapy for lesions,37 but it is unclear if this relates to the underlying atopic diathesis. Children with molluscum have been described to have warts, psoriasis, family history of atopy, diabetes mellitus, and pityriasis alba,7 while immunosuppression of any kind is associated with molluscum and high lesion count or prolonged disease in childhood.1,2
Quality of Life
Children with molluscum who have higher lesion counts appear to be at risk for severe effects on their quality of life. Approximately 10% of children with MCV infection have been documented to have severe impairments on quality of life.39 In my practice, quality of life in children with MCV appears to be affected by many factors (Table 2).7,18,39
Treatments
Proper Skin Care and Treatment of AD
Therapy for AD and/or pruritus appears to limit lesion number in children with MCV and rashes or itch.7,36 I recommend barrier repair agents, including emollients and syndet bar cleansers, to prevent small breaks in the skin that occur with xerosis and AD and that increase itch and risk of spread. Therapy for AD and molluscum dermatitis is similar and overlapping. There is always a concern about the spread of MCV when using topical calcineurin inhibitors. I, therefore, focus the dermatitis therapeutics on topical corticosteroid–based care.6,40
Prevention of Spread
Prevention of spread begins with hygiene interventions. Cobathing is common in children with MCV and should be held off when possible. It is important for the child with MCV to avoid sharing bath towels and equipment23 and having bare skin come in contact with mats in sports. I request that children with MCV wear bathing suits that cover the areas affected.
Reassurance
The most important therapy is reassurance.41 Many parents/guardians are truly unaware that the MCV infection can last for more than a year and therefore worry over normal disease course. When counseled as to the benign course of illness and given instructions on proper skin care, the parent/guardian of a child with MCV will often opt against therapy of uncomplicated cases. On the other hand, there are medical reasons for treatment, and they support the need for intervention (Table 3). Seventy percent of lesions resolve in 1.5 years; however, of the residual infections, some may last as long as 4 years.16 It is not recommended to stop children from attending school because of MCV.
Interventional Therapy
Therapeutics of MCV include destructive therapies in office (ie, cantharidin, cryotherapy, curettage, trichloroacetic acid, and glycolic acid) and at-home therapies (ie, topical retinoids, nitric oxide releasers)(eTable).2,5,6,42-58 When there are many lesions or spread is noted, immunotherapies can be used, including topical imiquimod, oral cimetidine, and intralesional Candida antigen.2,4,7 Pulsed dye laser cuts off the lesion vascular supply, while cidofovir is directly antiviral both topically and systemically, the latter reserved for severe cases in immunosuppressed adults.59 Head-to-head studies of cantharidin, curettage, topical peeling agents, and imiquimod demonstrated better satisfaction and fewer office visits with topical anesthetic and curettage on the first visit. Side effects were greatest for salicylic acid and glycolic acid; therefore, these agents are less desirable.42
Conclusion
Molluscum is a cutaneous viral infection that is common in children and has associated morbidities, including AD, pruritus, poor quality of life in some cases, and risk of contagion. Addressing the disease includes understanding its natural history and explaining it to parents/guardians. Therapeutics can be offered in cases where need is demonstrated, such as with lesions that spread and cause discomfort. Choice of therapeutics depends on the practitioner’s experience, the child’s clinical appearance, availability of therapy, and review of options with the parents/guardians. When avoidance of intervention is desired, barrier enhancement and treatment of symptomatic dermatitis are still beneficial, as are household (eg, not sharing towels) and activity (eg, adhesive bandages over active lesions) interventions to reduce transmission.
- Shisler JL. Immune evasion strategies of molluscum contagiosum virus. Adv Virus Res. 2015;92:201-252.
- Brown J, Janniger CK, Schwartz RA, et al. Childhood molluscum contagiosum. Int J Dermatol. 2006;45:93-99.
- Moss B, Shisler JL, Xiang Y, et al. Immune-defense molecules of molluscum contagiosum virus, a human poxvirus. Trends Microbiol. 2000;8:473-477.
- Silverberg NB. Warts and molluscum in children. Adv Dermatol. 2004;20:23-73.
- Choong KY, Roberts LJ. Molluscum contagiosum, swimming and bathing: a clinical analysis. Australas J Dermatol. 1999;40:89-92.
- Silverberg NB, Sidbury R, Mancini AJ. Childhood molluscum contagiosum: experience with cantharidin therapy in 300 patients. J Am Acad Dermatol. 2000;43:503-507.
- Silverberg NB. Molluscum contagiosum virus infection can trigger atopic dermatitis disease onset or flare. Cutis. 2018;102:191-194.
- Ajithkumar VT, Sasidharanpillai S, Muhammed K, et al. Disseminated molluscum contagiosum following chemotherapy: a therapeutic challenge. Indian J Dermatol Venereol Leprol. 2017;83:516.
- Oren B, Wende SO. An outbreak of molluscum contagiosum in a kibbutz. Infection. 1991;19:159-161.
- Molluscum contagiosum. Healthy Children website. https://www.healthychildren.org/English/health-issues/conditions/skin/Pages/Molluscum-Contagiosum.aspx. Updated November 21, 2015. Accessed October 16, 2019.
- Peterson AR, Nash E, Anderson BJ. Infectious disease in contact sports. Sports Health. 2019;11:47-58.
- Connell CO, Oranje A, Van Gysel D, et al. Congenital molluscum contagiosum: report of four cases and review of the literature. Pediatr Dermatol. 2008;25:553-556.
- Luke JD, Silverberg NB. Vertically transmitted molluscum contagiosum infection. Pediatrics. 2010;125:E423-E425.
- Mendiratta V, Agarwal S, Chander R. Reappraisal of sexually transmitted infections in children: a hospital-based study from an urban area. Indian J Sex Transm Dis AIDS. 2014;35:25-28.
- Bargman H. Genital molluscum contagiosum in children: evidence of sexual abuse? CMAJ. 1986;135:432-433.
- Basdag H, Rainer BM, Cohen BA. Molluscum contagiosum: to treat or not to treat? experience with 170 children in an outpatient clinic setting in the northeastern United States. Pediatr Dermatol. 2015;32:353-357.
- Koning S, Bruijnzeels MA, van Suijlekom-Smit LW, et al. Molluscum contagiosum in Dutch general practice. Br J Gen Pract. 1994;44:417-419.
- Braue A, Ross G, Varigos G, et al. Epidemiology and impact of childhood molluscum contagiosum: a case series and critical review of the literature. Pediatr Dermatol. 2005;22:287-294.
- Sturt RJ, Muller HK, Francis GD. Molluscum contagiosum in villages of the West Sepik District of New Guinea. Med J Aust. 1971;2:751-754.
- Reynolds MG, Homan RC, Yorita Christensen KL, et al. The incidence of molluscum contagiosum among American Indians and Alaska Natives. PLoS One. 2009;4:e5255.
- Villa L, Varela JA, Otero L, et al. Molluscum contagiosum: a 20-year study in a sexually transmitted infections unit. Sex Transm Dis. 2010;37:423-424.
- Watanabe T, Morikawa S, Suzuki K, et al. Two major antigenic polypeptides of molluscum contagiosum virus. J Infect Dis. 1998;177:284-292.
- Vaccine basics. Centers for Disease Control and Prevention website. https://www.cdc.gov/smallpox/vaccine-basics/index.html. Updated July 12, 2017. Accessed October 16, 2019.
- Mitchell JC. Observations on the virus of molluscum contagiosum. Br J Exp Pathol. 1953;34:44-49.
- Konya J, Thompson CH. Molluscum contagiosum virus: antibody responses in patients with clinical lesions and its sero-epidemiology in a representative Australian population. J Infect Dis. 1999;179:701-704.
- Steffen C, Markman JA. Spontaneous disappearance of molluscum contagiosum. Arch Dermatol. 1980;116:923-924.
- Uzuncakmak TK, Kuru BC, Zemheri EI, et al. Isolated giant molluscum contagiosum mimicking epidermoid cyst. Dermatol Pract Concept. 2016;6:71-73.
- Persechino S, Abruzzese C, Caperchi C, et al. Condyloma acuminata and mollusca contagiosa: a giant manifestation in a patient with lupus. Skinmed. 2014;12:310-311.
- Kim SK, Do JE, Kang HY, et al. Giant molluscum contagiosum of immunocompetent children occurring on the anogenital area. Eur J Dermatol. 2007;17:537-538.
- Alam MS, Shrirao N. Giant molluscum contagiosum presenting as lid neoplasm in an immunocompetent child. Dermatol Online J. 2016;22. pii:13030/qt56v567gn.
- Krishnamurthy J, Nagappa DK. The cytology of molluscum contagiosum mimicking skin adnexal tumor. J Cytol. 2010;27:74-75.
- Baek YS, Oh CH, Song HJ, et al. Asymmetrical periflexural exanthem of childhood with concurrence of molluscum contagiosum infection. Clin Exp Dermatol. 2011;36:676-677.
- Lee HJ, Kwon JA, Kim JW. Erythema multiforme-like molluscum dermatitis. Acta Derm Venereol. 2002;82:217-218.
- Lee YB, Choi HJ, Park HJ, et al. Two cases of erythema multiforme associated with molluscum contagiosum. Int J Dermatol. 2009;48:659-660.
- Vasily DB, Bhatia SG. Erythema annulare centrifugum and molluscum contagiosum.
- Berger EM, Orlow SJ, Patel RR, et al. Experience with molluscum contagiosum and associated inflammatory reactions in a pediatric dermatology practice: the bump that rashes. Arch Dermatol. 2012;148:1257-1264.
- Groner A, Laing-Grayman D, Silverberg NB. Outpatient pediatric community-acquired methicillin-resistant Staphylococcus aureus: a polymorphous clinical disease. Cutis. 2008;81:115-122.
- Butala N, Siegfried E, Weissler A. Molluscum BOTE sign: a predictor of imminent resolution. Pediatrics. 2013;131:E1650-E1653.
- Olsen JR, Gallagher J, Finlay AY, et al. Time to resolution and effect on quality of life of molluscum contagiosum in children in the UK: a prospective community cohort study. Lancet Infect Dis. 2015;15:190-195.
- Goksugur N, Ozbostanci B, Goksugur SB. Molluscum contagiosum infection associated with pimecrolimus use in pityriasis alba. Pediatr Dermatol. 2007;24:E63-E65.
- Lee R, Schwartz RA. Pediatric molluscum contagiosum: reflections on the last challenging poxvirus infection, part 1. Cutis. 2010;86:230-236.
- Hanna D, Hatami A, Powell J, et al. A prospective randomized trial comparing the efficacy and adverse effects of four recognized treatments of molluscum contagiosum in children. Pediatr Dermatol. 2006;23:574-579.
- Coloe Dosal J, Stewart PW, Lin JA, et al. Cantharidin for the treatment of molluscum contagiosum: a prospective, double-blinded, placebo-controlled trial. Pediatr Dermatol. 2014;31:440-449.
- Vakharia PP, Chopra R, Silverberg NB, et al. Efficacy and safety of topical cantharidin treatment for molluscum contagiosum and warts: a systematic review. Am J Clin Dermatol. 2018;19:791-803.
- Handjani F, Behazin E, Sadati MS. Comparison of 10% potassium hydroxide solution versus cryotherapy in the treatment of molluscum contagiosum: an open randomized clinical trial. J Dermatolog Treat. 2014;25:249-250.
- Simonart T, De Maertelaer V. Curettage treatment for molluscum contagiosum: a follow-up survey study. Br J Dermatol. 2008;159:1144-1147.
- Cho YS, Chung BY, Park CW, et al. Seizures and methemoglobinemia after topical application of eutectic mixture of lidocaine and prilocaine on a 3.5-year-old child with molluscum contagiosum and atopic dermatitis. Pediatr Dermatol. 2016;33:E284-E285.
- Bard S, Shiman MI, Bellman B, et al. Treatment of facial molluscum contagiosum with trichloroacetic acid. Pediatr Dermatol. 2009;26:425-426.
- Griffith RD, Yazdani Abyaneh MA, Falto-Aizpurua L, et al. Pulsed dye laser therapy for molluscum contagiosum: a systematic review. J Drugs Dermatol. 2014;13:1349-1352.
- Theos AU, Cummins R, Silverberg NB, et al. Effectiveness of imiquimod cream 5% for treating childhood molluscum contagiosum in a double-blind, randomized pilot trial. Cutis. 2004;74:134-138, 141-142.
- van der Wouden JC, Menke J, Gajadin S, et al. Interventions for cutaneous molluscum contagiosum. Cochrane Database Syst Rev. 2006:CD004767.
- Cunningham BB, Paller AS, Garzon M. Inefficacy of oral cimetidine for nonatopic children with molluscum contagiosum. Pediatr Dermatol. 1998;15:71-72.
- Enns LL, Evans MS. Intralesional immunotherapy with Candida antigen for the treatment of molluscum contagiosum in children. Pediatr Dermatol. 2011;28:254-258.
- Rajouria EA, Amatya A, Karn D. Comparative study of 5% potassium hydroxide solution versus 0.05% tretinoin cream for molluscum contagiosum in children. Kathmandu Univ Med J (KUMJ). 2011;9:291-294.
- Briand S, Milpied B, Navas D, et al. 1% topical cidofovir used as last alternative to treat viral infections. J Eur Acad Dermatol Venereol. 2008;22:249-250.
- Zabawski EJ Jr, Cockerell CJ. Topical cidofovir for molluscum contagiosum in children. Pediatr Dermatol. 1999;16:414-415.
- Watanabe T. Cidofovir diphosphate inhibits molluscum contagiosum virus DNA polymerase activity. J Invest Dermatol. 2008;128:1327-1329.
- Lindau MS, Munar MY. Use of duct tape occlusion in the treatment of recurrent molluscum contagiosum. Pediatr Dermatol. 2004;21:609.
- Silverberg N. Pediatric molluscum contagiosum: optimal treatment strategies. Paediatr Drugs. 2003;5:505-512.
- Shisler JL. Immune evasion strategies of molluscum contagiosum virus. Adv Virus Res. 2015;92:201-252.
- Brown J, Janniger CK, Schwartz RA, et al. Childhood molluscum contagiosum. Int J Dermatol. 2006;45:93-99.
- Moss B, Shisler JL, Xiang Y, et al. Immune-defense molecules of molluscum contagiosum virus, a human poxvirus. Trends Microbiol. 2000;8:473-477.
- Silverberg NB. Warts and molluscum in children. Adv Dermatol. 2004;20:23-73.
- Choong KY, Roberts LJ. Molluscum contagiosum, swimming and bathing: a clinical analysis. Australas J Dermatol. 1999;40:89-92.
- Silverberg NB, Sidbury R, Mancini AJ. Childhood molluscum contagiosum: experience with cantharidin therapy in 300 patients. J Am Acad Dermatol. 2000;43:503-507.
- Silverberg NB. Molluscum contagiosum virus infection can trigger atopic dermatitis disease onset or flare. Cutis. 2018;102:191-194.
- Ajithkumar VT, Sasidharanpillai S, Muhammed K, et al. Disseminated molluscum contagiosum following chemotherapy: a therapeutic challenge. Indian J Dermatol Venereol Leprol. 2017;83:516.
- Oren B, Wende SO. An outbreak of molluscum contagiosum in a kibbutz. Infection. 1991;19:159-161.
- Molluscum contagiosum. Healthy Children website. https://www.healthychildren.org/English/health-issues/conditions/skin/Pages/Molluscum-Contagiosum.aspx. Updated November 21, 2015. Accessed October 16, 2019.
- Peterson AR, Nash E, Anderson BJ. Infectious disease in contact sports. Sports Health. 2019;11:47-58.
- Connell CO, Oranje A, Van Gysel D, et al. Congenital molluscum contagiosum: report of four cases and review of the literature. Pediatr Dermatol. 2008;25:553-556.
- Luke JD, Silverberg NB. Vertically transmitted molluscum contagiosum infection. Pediatrics. 2010;125:E423-E425.
- Mendiratta V, Agarwal S, Chander R. Reappraisal of sexually transmitted infections in children: a hospital-based study from an urban area. Indian J Sex Transm Dis AIDS. 2014;35:25-28.
- Bargman H. Genital molluscum contagiosum in children: evidence of sexual abuse? CMAJ. 1986;135:432-433.
- Basdag H, Rainer BM, Cohen BA. Molluscum contagiosum: to treat or not to treat? experience with 170 children in an outpatient clinic setting in the northeastern United States. Pediatr Dermatol. 2015;32:353-357.
- Koning S, Bruijnzeels MA, van Suijlekom-Smit LW, et al. Molluscum contagiosum in Dutch general practice. Br J Gen Pract. 1994;44:417-419.
- Braue A, Ross G, Varigos G, et al. Epidemiology and impact of childhood molluscum contagiosum: a case series and critical review of the literature. Pediatr Dermatol. 2005;22:287-294.
- Sturt RJ, Muller HK, Francis GD. Molluscum contagiosum in villages of the West Sepik District of New Guinea. Med J Aust. 1971;2:751-754.
- Reynolds MG, Homan RC, Yorita Christensen KL, et al. The incidence of molluscum contagiosum among American Indians and Alaska Natives. PLoS One. 2009;4:e5255.
- Villa L, Varela JA, Otero L, et al. Molluscum contagiosum: a 20-year study in a sexually transmitted infections unit. Sex Transm Dis. 2010;37:423-424.
- Watanabe T, Morikawa S, Suzuki K, et al. Two major antigenic polypeptides of molluscum contagiosum virus. J Infect Dis. 1998;177:284-292.
- Vaccine basics. Centers for Disease Control and Prevention website. https://www.cdc.gov/smallpox/vaccine-basics/index.html. Updated July 12, 2017. Accessed October 16, 2019.
- Mitchell JC. Observations on the virus of molluscum contagiosum. Br J Exp Pathol. 1953;34:44-49.
- Konya J, Thompson CH. Molluscum contagiosum virus: antibody responses in patients with clinical lesions and its sero-epidemiology in a representative Australian population. J Infect Dis. 1999;179:701-704.
- Steffen C, Markman JA. Spontaneous disappearance of molluscum contagiosum. Arch Dermatol. 1980;116:923-924.
- Uzuncakmak TK, Kuru BC, Zemheri EI, et al. Isolated giant molluscum contagiosum mimicking epidermoid cyst. Dermatol Pract Concept. 2016;6:71-73.
- Persechino S, Abruzzese C, Caperchi C, et al. Condyloma acuminata and mollusca contagiosa: a giant manifestation in a patient with lupus. Skinmed. 2014;12:310-311.
- Kim SK, Do JE, Kang HY, et al. Giant molluscum contagiosum of immunocompetent children occurring on the anogenital area. Eur J Dermatol. 2007;17:537-538.
- Alam MS, Shrirao N. Giant molluscum contagiosum presenting as lid neoplasm in an immunocompetent child. Dermatol Online J. 2016;22. pii:13030/qt56v567gn.
- Krishnamurthy J, Nagappa DK. The cytology of molluscum contagiosum mimicking skin adnexal tumor. J Cytol. 2010;27:74-75.
- Baek YS, Oh CH, Song HJ, et al. Asymmetrical periflexural exanthem of childhood with concurrence of molluscum contagiosum infection. Clin Exp Dermatol. 2011;36:676-677.
- Lee HJ, Kwon JA, Kim JW. Erythema multiforme-like molluscum dermatitis. Acta Derm Venereol. 2002;82:217-218.
- Lee YB, Choi HJ, Park HJ, et al. Two cases of erythema multiforme associated with molluscum contagiosum. Int J Dermatol. 2009;48:659-660.
- Vasily DB, Bhatia SG. Erythema annulare centrifugum and molluscum contagiosum.
- Berger EM, Orlow SJ, Patel RR, et al. Experience with molluscum contagiosum and associated inflammatory reactions in a pediatric dermatology practice: the bump that rashes. Arch Dermatol. 2012;148:1257-1264.
- Groner A, Laing-Grayman D, Silverberg NB. Outpatient pediatric community-acquired methicillin-resistant Staphylococcus aureus: a polymorphous clinical disease. Cutis. 2008;81:115-122.
- Butala N, Siegfried E, Weissler A. Molluscum BOTE sign: a predictor of imminent resolution. Pediatrics. 2013;131:E1650-E1653.
- Olsen JR, Gallagher J, Finlay AY, et al. Time to resolution and effect on quality of life of molluscum contagiosum in children in the UK: a prospective community cohort study. Lancet Infect Dis. 2015;15:190-195.
- Goksugur N, Ozbostanci B, Goksugur SB. Molluscum contagiosum infection associated with pimecrolimus use in pityriasis alba. Pediatr Dermatol. 2007;24:E63-E65.
- Lee R, Schwartz RA. Pediatric molluscum contagiosum: reflections on the last challenging poxvirus infection, part 1. Cutis. 2010;86:230-236.
- Hanna D, Hatami A, Powell J, et al. A prospective randomized trial comparing the efficacy and adverse effects of four recognized treatments of molluscum contagiosum in children. Pediatr Dermatol. 2006;23:574-579.
- Coloe Dosal J, Stewart PW, Lin JA, et al. Cantharidin for the treatment of molluscum contagiosum: a prospective, double-blinded, placebo-controlled trial. Pediatr Dermatol. 2014;31:440-449.
- Vakharia PP, Chopra R, Silverberg NB, et al. Efficacy and safety of topical cantharidin treatment for molluscum contagiosum and warts: a systematic review. Am J Clin Dermatol. 2018;19:791-803.
- Handjani F, Behazin E, Sadati MS. Comparison of 10% potassium hydroxide solution versus cryotherapy in the treatment of molluscum contagiosum: an open randomized clinical trial. J Dermatolog Treat. 2014;25:249-250.
- Simonart T, De Maertelaer V. Curettage treatment for molluscum contagiosum: a follow-up survey study. Br J Dermatol. 2008;159:1144-1147.
- Cho YS, Chung BY, Park CW, et al. Seizures and methemoglobinemia after topical application of eutectic mixture of lidocaine and prilocaine on a 3.5-year-old child with molluscum contagiosum and atopic dermatitis. Pediatr Dermatol. 2016;33:E284-E285.
- Bard S, Shiman MI, Bellman B, et al. Treatment of facial molluscum contagiosum with trichloroacetic acid. Pediatr Dermatol. 2009;26:425-426.
- Griffith RD, Yazdani Abyaneh MA, Falto-Aizpurua L, et al. Pulsed dye laser therapy for molluscum contagiosum: a systematic review. J Drugs Dermatol. 2014;13:1349-1352.
- Theos AU, Cummins R, Silverberg NB, et al. Effectiveness of imiquimod cream 5% for treating childhood molluscum contagiosum in a double-blind, randomized pilot trial. Cutis. 2004;74:134-138, 141-142.
- van der Wouden JC, Menke J, Gajadin S, et al. Interventions for cutaneous molluscum contagiosum. Cochrane Database Syst Rev. 2006:CD004767.
- Cunningham BB, Paller AS, Garzon M. Inefficacy of oral cimetidine for nonatopic children with molluscum contagiosum. Pediatr Dermatol. 1998;15:71-72.
- Enns LL, Evans MS. Intralesional immunotherapy with Candida antigen for the treatment of molluscum contagiosum in children. Pediatr Dermatol. 2011;28:254-258.
- Rajouria EA, Amatya A, Karn D. Comparative study of 5% potassium hydroxide solution versus 0.05% tretinoin cream for molluscum contagiosum in children. Kathmandu Univ Med J (KUMJ). 2011;9:291-294.
- Briand S, Milpied B, Navas D, et al. 1% topical cidofovir used as last alternative to treat viral infections. J Eur Acad Dermatol Venereol. 2008;22:249-250.
- Zabawski EJ Jr, Cockerell CJ. Topical cidofovir for molluscum contagiosum in children. Pediatr Dermatol. 1999;16:414-415.
- Watanabe T. Cidofovir diphosphate inhibits molluscum contagiosum virus DNA polymerase activity. J Invest Dermatol. 2008;128:1327-1329.
- Lindau MS, Munar MY. Use of duct tape occlusion in the treatment of recurrent molluscum contagiosum. Pediatr Dermatol. 2004;21:609.
- Silverberg N. Pediatric molluscum contagiosum: optimal treatment strategies. Paediatr Drugs. 2003;5:505-512.
Practice Points
- Molluscum appears as pearly papules with a central dell (ie, umbilicated).
- Caused by a poxvirus, the disease is very contagious and transferred via skin-to-skin contact or fomites.
- One-third of children with molluscum will develop symptoms of local erythema, swelling, or pruritus.
- Diagnosis usually is clinical.
- Children are primarily managed through observation; however, cantharidin, cryotherapy, or curettage can be used for symptomatic or cosmetically concerning lesions.
Comment on “Intraoperative Electrosurgical Smoke During Outpatient Surgery: A Survey of Dermatologic Surgeon and Staff Preferences”
To the Editor:
We read with great interest the recent Cutis article by Golda et al,1 “Intraoperative Electrosurgical Smoke During Outpatient Surgery: A Survey of Dermatologic Surgeon and Staff Preferences.” We applaud the growing interest in the topic of dermatologist safety, as there are currently no established guidelines for precautions while performing surgical procedures. In 2018 we conducted a comprehensive review2 to characterize the specific risks, hazard reduction strategies available, and current use of surgical smoke safety techniques during surgery among dermatologists, and ultimately recommend guidance based on the current available evidence. To conduct this review, we collected data from 45 manuscripts in the dermatology, surgery, infectious disease, obstetrics, and cancer biology literature. Herein, we summarize key findings.2
Dermatologic surgeons, residents, staff, and patients are exposed to many infectious, inhalational, chemical, and mutagenic hazards when performing procedures that liberate smoke and plume. These risks are commonplace; however, they are particularly notable during ablative laser and laser hair removal procedures, which produce a heavy plume (averaging >100,000 particles/cm3). Brief periods of heavy plume exposure also are commonplace during electrosurgery.
Infectious particles in surgical plume have been extensively studied, and viral transmission has been demonstrated in animal studies. Human papillomavirus transmission appears to be the most prevalent risk. Surgical smoke has been shown to cause acute and chronic inhalational injury in rat and sheep studies.3-6
Additionally, chemicals with carcinogenic potential are present in surgical smoke and have been described.7,8 Chemicals in the greatest quantity include hydrocarbons, nitriles, fatty acids, and phenols. Although there have been no human studies on smoke carcinogenesis to date, surgical smoke has been shown to have carcinogenic properties in vitro.
Given these risks—both evidence based and theoretical—we believe that diligent hazard reduction strategies should be employed whenever possible. Surgical masks and high-efficiency particulate air respirators, such as N95 respirator masks, have been well studied and do provide smoke protection. High-efficiency particulate air masks can be worn when possible, especially during procedures that produce heavy plume, though surgical masks are capable of filtering most of the noxious chemicals in surgical smoke. It should be noted that proper fit with minimal air leak is the most important aspect of overall performance.
Smoke evacuators provide another level of protection. The physician should consider the evacuator’s filtration efficiency, capture velocity, and suction strength when evaluating overall performance. Furthermore, the smoke collection tip should be within 2 in of the surgical field to maximize efficacy. Maintenance for smoke evacuation systems should include regular (as defined by manufacturer instructions) flushing of the smoke evacuator lines.
Despite the risks of surgical smoke and the available options of minimizing these risks, the hazards of surgical smoke and the importance of protection are likely underemphasized. Many dermatologic surgeons do not use surgical masks or smoke evacuators in routine practice, according to several survey studies.9-11
It is important for the dermatologic community to consider effective ways of spreading awareness. We propose that surgical smoke safety be taught early in residency training. Additionally, smoke safety can be implemented into certification examinations. Access to masks and smoke evacuation devices are an important part of dermatology training. Accreditation Council for Graduate Medical Education funds should be appropriated to provide for such resources.
Finally, and perhaps most importantly, continued awareness should be established in the dermatology community via standardized guidelines and periodic updates in the dermatology literature and lectures at local and national conferences. Not until these strategies are implemented will surgical smoke protection be viewed as a necessary and important component of routine practice when performing dermatologic surgical procedures.
- Golda N, Merrill B, Neill B. Intraoperative electrosurgical smoke during outpatient surgery: a survey of dermatologic surgeon and staff preferences. Cutis. 2019;104:120-124.
- Georgesen C, Lipner SR. Surgical smoke: risk assessment and mitigation strategies. J Am Acad Dermatol. 2018;79:746-755.
- Wenig BL, Stenson KM, Wenig BM, et al. Effects of plume produced by the Nd:YAG laser and electrocautery on the respiratory system. Lasers Surg Med. 1993;13:242-245.
- Baggish MS, Elbakry M. The effects of laser smoke on the lungs of rats. Am J Obstet Gynecol. 1987;156:1260-1265.
- Baggish MS, Baltoyannis P, Sze E. Protection of the rat lung from the harmful effects of laser smoke. Lasers Surg Med. 1988;8:248-253.
- Freitag L, Chapman GA, Sielczak M, et al. Laser smoke effect on the bronchial system. Lasers Surg Med. 1987;7:283-288.
- Barrett WL, Garber SM. Surgical smoke: a review of the literature. Is this just a lot of hot air? Surg Endosc. 2003;17:979-987.
- Hensman C, Baty D, Willis RG, et al. Chemical composition of smoke produced by high-frequency electrosurgery in a closed gaseous environment. Surg Endosc. 1998;12:1017-1019.
- Edwards BE, Reiman RE. Results of a survey on current surgical smoke control practices. AORN J. 2008;87:739-749.
- Oganesyan G, Eimpunth S, Kim SS, et al. Surgical smoke in dermatologic surgery. Dermatol Surg. 2014;40:1373-1377.
- Chapman LW, Korta DZ, Lee PK, et al. Awareness of surgical smoke risks and assessment of safety practices during electrosurgery among US dermatology residents. JAMA Dermatol. 2017;153:467-468.
To the Editor:
We read with great interest the recent Cutis article by Golda et al,1 “Intraoperative Electrosurgical Smoke During Outpatient Surgery: A Survey of Dermatologic Surgeon and Staff Preferences.” We applaud the growing interest in the topic of dermatologist safety, as there are currently no established guidelines for precautions while performing surgical procedures. In 2018 we conducted a comprehensive review2 to characterize the specific risks, hazard reduction strategies available, and current use of surgical smoke safety techniques during surgery among dermatologists, and ultimately recommend guidance based on the current available evidence. To conduct this review, we collected data from 45 manuscripts in the dermatology, surgery, infectious disease, obstetrics, and cancer biology literature. Herein, we summarize key findings.2
Dermatologic surgeons, residents, staff, and patients are exposed to many infectious, inhalational, chemical, and mutagenic hazards when performing procedures that liberate smoke and plume. These risks are commonplace; however, they are particularly notable during ablative laser and laser hair removal procedures, which produce a heavy plume (averaging >100,000 particles/cm3). Brief periods of heavy plume exposure also are commonplace during electrosurgery.
Infectious particles in surgical plume have been extensively studied, and viral transmission has been demonstrated in animal studies. Human papillomavirus transmission appears to be the most prevalent risk. Surgical smoke has been shown to cause acute and chronic inhalational injury in rat and sheep studies.3-6
Additionally, chemicals with carcinogenic potential are present in surgical smoke and have been described.7,8 Chemicals in the greatest quantity include hydrocarbons, nitriles, fatty acids, and phenols. Although there have been no human studies on smoke carcinogenesis to date, surgical smoke has been shown to have carcinogenic properties in vitro.
Given these risks—both evidence based and theoretical—we believe that diligent hazard reduction strategies should be employed whenever possible. Surgical masks and high-efficiency particulate air respirators, such as N95 respirator masks, have been well studied and do provide smoke protection. High-efficiency particulate air masks can be worn when possible, especially during procedures that produce heavy plume, though surgical masks are capable of filtering most of the noxious chemicals in surgical smoke. It should be noted that proper fit with minimal air leak is the most important aspect of overall performance.
Smoke evacuators provide another level of protection. The physician should consider the evacuator’s filtration efficiency, capture velocity, and suction strength when evaluating overall performance. Furthermore, the smoke collection tip should be within 2 in of the surgical field to maximize efficacy. Maintenance for smoke evacuation systems should include regular (as defined by manufacturer instructions) flushing of the smoke evacuator lines.
Despite the risks of surgical smoke and the available options of minimizing these risks, the hazards of surgical smoke and the importance of protection are likely underemphasized. Many dermatologic surgeons do not use surgical masks or smoke evacuators in routine practice, according to several survey studies.9-11
It is important for the dermatologic community to consider effective ways of spreading awareness. We propose that surgical smoke safety be taught early in residency training. Additionally, smoke safety can be implemented into certification examinations. Access to masks and smoke evacuation devices are an important part of dermatology training. Accreditation Council for Graduate Medical Education funds should be appropriated to provide for such resources.
Finally, and perhaps most importantly, continued awareness should be established in the dermatology community via standardized guidelines and periodic updates in the dermatology literature and lectures at local and national conferences. Not until these strategies are implemented will surgical smoke protection be viewed as a necessary and important component of routine practice when performing dermatologic surgical procedures.
To the Editor:
We read with great interest the recent Cutis article by Golda et al,1 “Intraoperative Electrosurgical Smoke During Outpatient Surgery: A Survey of Dermatologic Surgeon and Staff Preferences.” We applaud the growing interest in the topic of dermatologist safety, as there are currently no established guidelines for precautions while performing surgical procedures. In 2018 we conducted a comprehensive review2 to characterize the specific risks, hazard reduction strategies available, and current use of surgical smoke safety techniques during surgery among dermatologists, and ultimately recommend guidance based on the current available evidence. To conduct this review, we collected data from 45 manuscripts in the dermatology, surgery, infectious disease, obstetrics, and cancer biology literature. Herein, we summarize key findings.2
Dermatologic surgeons, residents, staff, and patients are exposed to many infectious, inhalational, chemical, and mutagenic hazards when performing procedures that liberate smoke and plume. These risks are commonplace; however, they are particularly notable during ablative laser and laser hair removal procedures, which produce a heavy plume (averaging >100,000 particles/cm3). Brief periods of heavy plume exposure also are commonplace during electrosurgery.
Infectious particles in surgical plume have been extensively studied, and viral transmission has been demonstrated in animal studies. Human papillomavirus transmission appears to be the most prevalent risk. Surgical smoke has been shown to cause acute and chronic inhalational injury in rat and sheep studies.3-6
Additionally, chemicals with carcinogenic potential are present in surgical smoke and have been described.7,8 Chemicals in the greatest quantity include hydrocarbons, nitriles, fatty acids, and phenols. Although there have been no human studies on smoke carcinogenesis to date, surgical smoke has been shown to have carcinogenic properties in vitro.
Given these risks—both evidence based and theoretical—we believe that diligent hazard reduction strategies should be employed whenever possible. Surgical masks and high-efficiency particulate air respirators, such as N95 respirator masks, have been well studied and do provide smoke protection. High-efficiency particulate air masks can be worn when possible, especially during procedures that produce heavy plume, though surgical masks are capable of filtering most of the noxious chemicals in surgical smoke. It should be noted that proper fit with minimal air leak is the most important aspect of overall performance.
Smoke evacuators provide another level of protection. The physician should consider the evacuator’s filtration efficiency, capture velocity, and suction strength when evaluating overall performance. Furthermore, the smoke collection tip should be within 2 in of the surgical field to maximize efficacy. Maintenance for smoke evacuation systems should include regular (as defined by manufacturer instructions) flushing of the smoke evacuator lines.
Despite the risks of surgical smoke and the available options of minimizing these risks, the hazards of surgical smoke and the importance of protection are likely underemphasized. Many dermatologic surgeons do not use surgical masks or smoke evacuators in routine practice, according to several survey studies.9-11
It is important for the dermatologic community to consider effective ways of spreading awareness. We propose that surgical smoke safety be taught early in residency training. Additionally, smoke safety can be implemented into certification examinations. Access to masks and smoke evacuation devices are an important part of dermatology training. Accreditation Council for Graduate Medical Education funds should be appropriated to provide for such resources.
Finally, and perhaps most importantly, continued awareness should be established in the dermatology community via standardized guidelines and periodic updates in the dermatology literature and lectures at local and national conferences. Not until these strategies are implemented will surgical smoke protection be viewed as a necessary and important component of routine practice when performing dermatologic surgical procedures.
- Golda N, Merrill B, Neill B. Intraoperative electrosurgical smoke during outpatient surgery: a survey of dermatologic surgeon and staff preferences. Cutis. 2019;104:120-124.
- Georgesen C, Lipner SR. Surgical smoke: risk assessment and mitigation strategies. J Am Acad Dermatol. 2018;79:746-755.
- Wenig BL, Stenson KM, Wenig BM, et al. Effects of plume produced by the Nd:YAG laser and electrocautery on the respiratory system. Lasers Surg Med. 1993;13:242-245.
- Baggish MS, Elbakry M. The effects of laser smoke on the lungs of rats. Am J Obstet Gynecol. 1987;156:1260-1265.
- Baggish MS, Baltoyannis P, Sze E. Protection of the rat lung from the harmful effects of laser smoke. Lasers Surg Med. 1988;8:248-253.
- Freitag L, Chapman GA, Sielczak M, et al. Laser smoke effect on the bronchial system. Lasers Surg Med. 1987;7:283-288.
- Barrett WL, Garber SM. Surgical smoke: a review of the literature. Is this just a lot of hot air? Surg Endosc. 2003;17:979-987.
- Hensman C, Baty D, Willis RG, et al. Chemical composition of smoke produced by high-frequency electrosurgery in a closed gaseous environment. Surg Endosc. 1998;12:1017-1019.
- Edwards BE, Reiman RE. Results of a survey on current surgical smoke control practices. AORN J. 2008;87:739-749.
- Oganesyan G, Eimpunth S, Kim SS, et al. Surgical smoke in dermatologic surgery. Dermatol Surg. 2014;40:1373-1377.
- Chapman LW, Korta DZ, Lee PK, et al. Awareness of surgical smoke risks and assessment of safety practices during electrosurgery among US dermatology residents. JAMA Dermatol. 2017;153:467-468.
- Golda N, Merrill B, Neill B. Intraoperative electrosurgical smoke during outpatient surgery: a survey of dermatologic surgeon and staff preferences. Cutis. 2019;104:120-124.
- Georgesen C, Lipner SR. Surgical smoke: risk assessment and mitigation strategies. J Am Acad Dermatol. 2018;79:746-755.
- Wenig BL, Stenson KM, Wenig BM, et al. Effects of plume produced by the Nd:YAG laser and electrocautery on the respiratory system. Lasers Surg Med. 1993;13:242-245.
- Baggish MS, Elbakry M. The effects of laser smoke on the lungs of rats. Am J Obstet Gynecol. 1987;156:1260-1265.
- Baggish MS, Baltoyannis P, Sze E. Protection of the rat lung from the harmful effects of laser smoke. Lasers Surg Med. 1988;8:248-253.
- Freitag L, Chapman GA, Sielczak M, et al. Laser smoke effect on the bronchial system. Lasers Surg Med. 1987;7:283-288.
- Barrett WL, Garber SM. Surgical smoke: a review of the literature. Is this just a lot of hot air? Surg Endosc. 2003;17:979-987.
- Hensman C, Baty D, Willis RG, et al. Chemical composition of smoke produced by high-frequency electrosurgery in a closed gaseous environment. Surg Endosc. 1998;12:1017-1019.
- Edwards BE, Reiman RE. Results of a survey on current surgical smoke control practices. AORN J. 2008;87:739-749.
- Oganesyan G, Eimpunth S, Kim SS, et al. Surgical smoke in dermatologic surgery. Dermatol Surg. 2014;40:1373-1377.
- Chapman LW, Korta DZ, Lee PK, et al. Awareness of surgical smoke risks and assessment of safety practices during electrosurgery among US dermatology residents. JAMA Dermatol. 2017;153:467-468.
Hidradenitis Suppurativa for the Dermatologic Hospitalist
Hidradenitis suppurativa (HS) is a common chronic inflammatory skin disease characterized by purulent subcutaneous nodules, papules, abscesses, and fistula tracts that lead to scarring and fibrosis. Lesions develop primarily in the axilla, groin, and other intertriginous and hair-bearing areas.
The natural history of the disease is characterized by periods of disease flare, followed by periods of disease quiescence. Patients might have weeks or months of low disease activity but frequently develop multiple exacerbating episodes over the course of weeks or months. The condition primarily presents in adolescent and peripubescent years, continuing throughout adulthood. Some evidence suggests a bimodal disease distribution, with a second peak of incidence in middle-aged adults. Women and men are affected equally; however, the disease can be phenotypically different in men and women.
Patients frequently present in emergency and inpatient settings for evaluation because of the pain and severity of HS flares as well as associated systemic symptoms. Inpatient and emergency department (ED) care are unique opportunities for dermatologic hospitalist and dermatologic consultative services to educate other physicians about the condition and initiate aggressive treatments that are frequently necessary to control HS flares. This article aims to address best methods for treating HS in these settings.
Pathophysiology
Although the exact pathophysiology of the condition is unknown, HS is thought to begin with follicular occlusion with downstream inflammation mediating neutrophilic activity and scarring. Hyperplasia of the infundibular epithelium is observed on histology, and the resulting occlusion, contained keratin, and follicular rupture initiate robust downstream inflammation.1,2 Follicular occlusion might be initially androgen mediated3 or might occur in combination with friction4 and genetic or acquired factors involving Notch signaling. Although HS characteristically presents in areas of high apocrine density, apocrine glands are not thought to be the primary mediator of disease activity.5 IL-17, IL-23, tumor necrosis factor α, and IL-1β are implicated in the pathogenesis of HS, but it is unknown if these cytokines are the driving pathologic factor in HS or if they are merely secondary sequelae.6
Demographics and Prevalence in Hospitalized Patients
Although increasing treatment availability has brought more attention to HS, true prevalence is unknown. A prevalence of 1% has been reported in many European countries.7 Global prevalence has been more difficult to determine, with variable data suggesting a prevalence of 0.03% to 8%, depending on the population included.8 Most patients studied in a US-based claims database were aged 30 to 64 years, and the overall prevalence was 0.05%.9 Despite prevalence similar to psoriasis, utilization of high-cost emergency and inpatient admissions is notably higher among patients with HS. Recent claims data suggest that HS patients utilize the ED at a rate 3 times higher than psoriasis patients and are admitted as inpatients at a rate 5 times higher.10 Similar data suggest an associated increased cost of care for patients with HS vs other conditions, such as psoriasis, due to frequent ED and inpatient stays.11 Although HS frequently presents in the inpatient and emergency settings, there is little literature on best methods for managing patients in these settings.
Pearls for Inpatient and Emergency Evaluation and Management
Initial Evaluation
When dermatologic consultative services are asked to evaluate patients with HS, preliminary evaluation should reflect the acuity of the patient. Vital signs and toxicity should be reviewed to ensure that there is no evidence of severe infection necessitating critical or acute care.
History
History-taking should reflect assessment of the patient’s baseline disease, including date of initial onset; exacerbating factors, such as friction, smoking, pregnancy, and menses; and the current history of the patient’s flare. A history of antibiotics, immunosuppression, topical therapy, antiandrogen therapy, and vitamin A analog therapy also should be reviewed. If an initial diagnosis is made in the ED or inpatient setting, a family and personal history should focus on specific risk factors and disease associations, including inflammatory bowel disease,12 pilonidal cysts,13 polycystic ovary syndrome,14 and metabolic syndrome.15
Physical Examination
As with all dermatologic consultations, a full-body skin examination, with special attention to the axilla, inframammary skin, groin, buttocks, and perineum, should be undertaken. In addition to these common areas of disease progression, examination should focus on atypical sites for disease manifestation, including the posterior auricular scalp, skin folds in the pannus and back, and the beard area in men. Evaluation of axillary and gluteal hair should note features of folliculitis and hair removal, which can exacerbate HS. Examination also should include an investigation of cutaneous manifestations of comorbid conditions, including acanthosis nigricans, contiguous or metastatic cutaneous Crohn disease, erythema nodosum, and pilonidal cysts. Caution should be exercised when diagnosing pilonidal cysts, as isolated or evolving HS in the gluteal cleft often is misdiagnosed as a pilonidal cyst.
Laboratory Evaluation
Testing often is misleading in patients with HS, especially in the acute setting, because the condition is a chronic inflammatory process. The C-reactive protein level as well as the absolute white blood cell and neutrophil counts often are elevated, even in the absence of acute infection.16 In fact, although patients often are treated with intravenous antibiotics by inpatient and emergency teams in the setting of these 3 laboratory abnormalities, these findings often reflect disease activity, not frank infection. Fever, especially low-grade fever, also can reflect ongoing disease activity. Thrombocytosis and anemia also are anecdotally common, though these findings have not been reported specifically in the literature.
Bacterial Cultures
The role of lesional and perilesional bacterial cultures is controversial in HS. Prior studies have demonstrated that biofilm formation may be associated with the chronic inflammation seen in HS.17 However, most data to date suggest that infection is not the primary driver of HS disease flares, as demonstrated by the frequency of sterile cultures and the variable response of the disease to penicillin and related antibiotics.18
Imaging
Ultrasonography and magnetic resonance imaging can be conducted if there is concern about deeper abscesses that are not apparent on examination. When interpreted by nondermatologic practitioners, however, the findings of these modalities can result in unnecessary surgical intervention, given the concern for development of infectious abscess.19
Diagnosis
Many patients with HS experience a notable delay in time to diagnosis, living with symptoms for 7 years on average prior to being given a name for their condition.20 Often, patients seek ED care at initial presentation because lesions can present quickly and are associated with remarkable pain. Inpatient dermatologic evaluation can provide patients with definitive diagnosis, appropriate counseling that provides an overview of the natural history of the disease, lifestyle recommendations, and expedited connection to outpatient longitudinal care.
Diagnosis is made clinically by assessment for typical lesions, such as painful or tender papules, nodules, or abscesses in the axillae, inframammary region, groin, thighs, and perineal and perianal regions. Cordlike scarring often is seen in the absence of active inflammatory lesions.21 Double-headed open comedones and prominent follicular occlusion are seen in some phenotypes but are not required for diagnosis.22
Multiple scoring modalities are in use23; the Hurley staging system, initially developed for surgical staging, has become a commonly used method in the clinical setting24:
• Hurley stage I: isolated nodules or abscesses;
• Hurley stage II: widely separate lesions and sinus tracts or scarring are suggestive; and
• Hurley stage III: multiple lesions with near-diffuse involvement and formation of sinus tracts and scarring.
Other scoring modalities, such as the Hidradenitis Suppurativa Clinical Response (HiSCR), are more commonly used in the clinical trial setting and quantitatively capture lesion count improvement while the patient is being treated.25
Treatment
Evaluation in the ED might necessitate recommendations for inpatient admission. Dermatologic consultation can be helpful in providing ED physicians with context for interpretation of laboratory results and clinical findings. Specifically, dermatologic evaluation can help differentiate presentations consistent with a primary infection from a more common presentation of HS flaring and associated bacterial colonization. Indications for inpatient admission are severe pain; concern for systemic infection, including high fever or sepsis; and need for surgical intervention. Patients with severe disease who do not have a longitudinal care plan or who lack the ability to care for lesions at home also are candidates for inpatient admission, where they can receive more intensive nursing and wound care as well as outpatient logistical management.
Acute care should be aimed at treatments that work quickly and aggressively and have both anti-inflammatory and antimicrobial effects. Severe flares require aggressive initial treatment to ensure more long-term remission. Adalimumab, maintained at 40 mg/wk after a loading dose, is the mainstay of evidence-based treatment for moderate to severe HS in patients 12 years or older; however, this treatment might not be easy to initiate in the inpatient setting because of its cost and availability and the fact that it is not as fast acting as other therapies.26 For patients with severe disease flares, prednisone,27 infliximab,28 or cyclosporine29 can be used in combination with antimicrobial therapy in the inpatient setting to quickly control active flaring. Intravenous antimicrobial therapy might be necessary in severe disease and should include coverage of gram-positive30 and anaerobic organisms.31
Although management of acute flares is critical, especially for hospitalized patients, initiating longitudinal treatment modalities while the patient is an inpatient will help prevent future readmissions, facilitate better outcomes, and enable longer periods of disease-free progression. Specific treatments, stratified by disease severity, are listed in the Table.
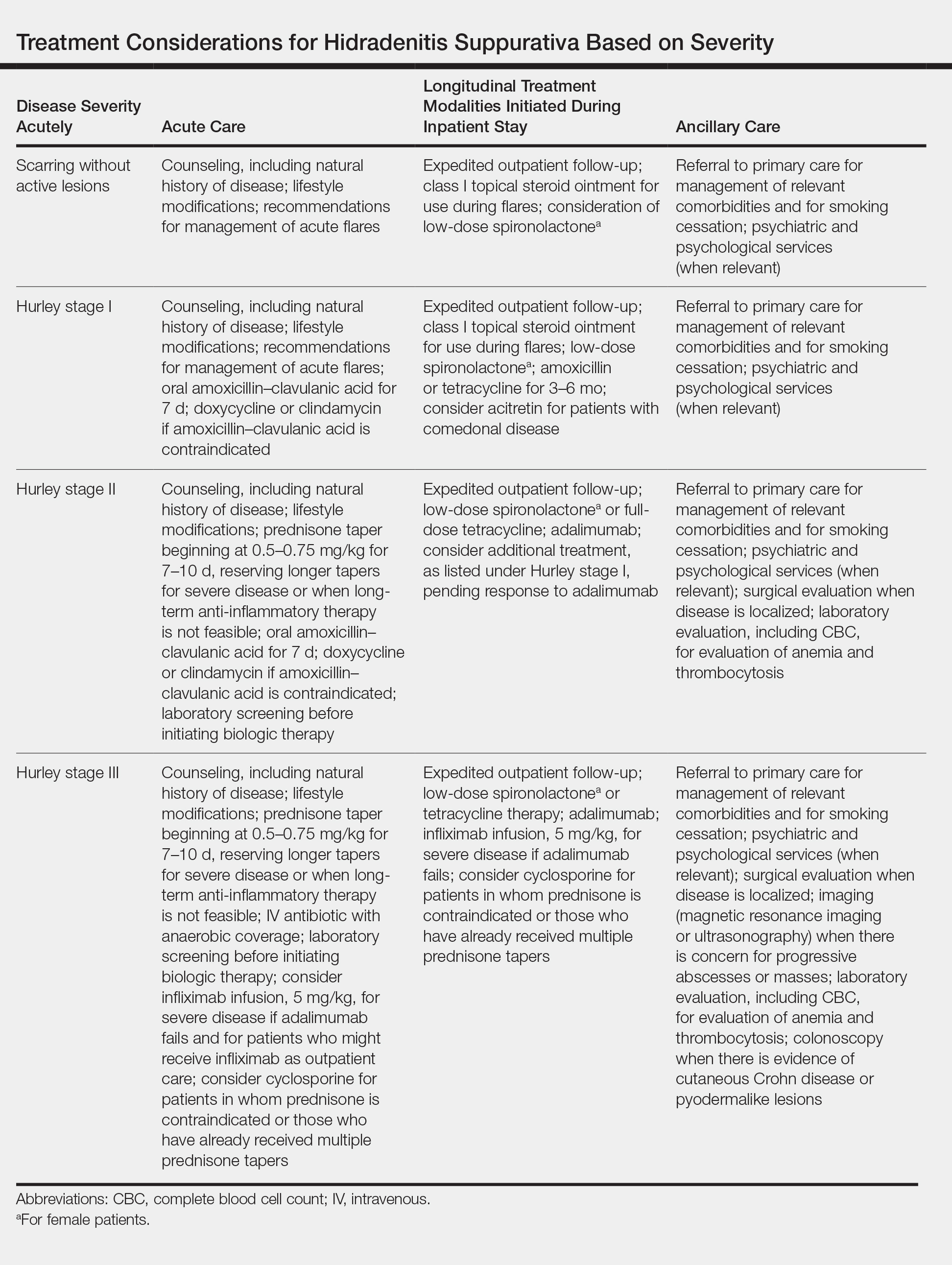
Postdischarge Lifestyle Modification
All disease management should include recommendations for lifestyle modification, including counseling on terminal hair removal (ie, avoid shaving, plucking, and waxing) and recommendations for daily and weekly decolonization with chlorhexidine or other antimicrobial soap, a weekly vinegar bath, and antiperspirant use in the groin and axilla. Avoiding tight clothes and humidity might also be helpful.
Other beneficial postdischarge strategies include smoking cessation and weight loss, which often are beneficial but difficult for many patients to achieve on their own; connecting patients with a primary care provider, which can facilitate better long-term outcomes; informing patients of the natural history of the disease and providing strategies for them to implement for acute flares to help avoid readmission and ED visits; and writing a “pill-in-pocket” prescription for a course of an antibiotic that provides good staphylococcal and anaerobic coverage, which can be helpful for patients who are prone to infrequent flares.
Lastly, appropriate postdischarge maintenance therapy also can be initiated during the inpatient stay, including maintenance antibiotic therapy, spironolactone32 for female patients, and acitretin33 for comedonal-predominant patients.
Final Thoughts
Hidradenitis suppurativa is a common dermatologic condition that frequently presents in emergency and inpatient settings, given its association with painful and acutely indurated lesions that often appear concerning for infection. Elevated inflammatory markers and fever are common in HS and are not necessarily suggestive of infection. As such, while antibiotics may be part of acute management of HS, care also should address the inflammatory component of the disease. Longitudinal outpatient care coordination with a dermatologist and primary care physician is imperative for limiting ED and inpatient care utilization.
- Jemec GB, Hansen U. Histology of hidradenitis suppurativa. J Am Acad Dermatol. 1996;34:994-999.
- Prens E, Deckers I. Pathophysiology of hidradenitis suppurativa: an update. J Am Acad Dermatol. 2015;73(suppl 1):S8-S11.
- Barth JH, Kealey T. Androgen metabolism by isolated human axillary apocrine glands in hidradenitis suppurativa. Br J Dermatol. 1991;125:304-308.
- de Winter K, van der Zee HH, Prens EP. Is mechanical stress an important pathogenic factor in hidradenitis suppurativa? Exp Dermatol. 2012;21:176-177.
- Yu CC, Cook MG. Hidradenitis suppurativa: a disease of follicular epithelium, rather than apocrine glands. Br J Dermatol. 1990;122:763-769.
- Deckers IE, van der Zee HH, Prens EP. Epidemiology of hidradenitis suppurativa: prevalence, pathogenesis, and factors associated with the development of HS. Curr Dermatol Rep. 2014;3:54-60.
- Revuz JE, Canoui-Poitrine F, Wolkenstein P, et al. Prevalence and factors associated with hidradenitis suppurativa: Results from two case-control studies. J Am Acad Dermatol. 2008;59:596-601.
- Jemec GE, Kimball AB. Hidradenitis suppurativa: epidemiology and scope of the problem. J Am Acad Dermatol. 2015;73(suppl 1):S4-S7.
- Cosmatos I, Matcho A, Weinstein R, et al. Analysis of patient claims data to determine the prevalence of hidradenitis suppurativa in the United States. J Am Acad Dermatol. 2013;68:412-419.
- Khalsa A, Liu G, Kirby JS. Increased utilization of emergency department and inpatient care by patients with hidradenitis suppurativa. J Am Acad Dermatol. 2015;73:609-614.
- Kirby JS, Miller JJ, Adams DR, et al. Health care utilization patterns and costs for patients with hidradenitis suppurativa. JAMA Dermatol. 2014;150:937-944.
- Deckers IE, Benhadou F, Koldijk MJ, et al. Inflammatory bowel disease is associated with hidradenitis suppurativa: results from a multicenter cross-sectional study. J Am Acad Dermatol. 2017;76:49-53.
- Benhadou F, Van der Zee HH, Pascual JC, et al. Pilonidal sinus disease: an intergluteal localization of hidradenitis suppurativa/acne inversa: a cross-sectional study among 2465 patients [published online March 27, 2019]. Br J Dermatol. doi:10.1111/bjd.17927.
- Garg A, Neuren E, Strunk A. Hidradenitis suppurativa is associated with polycystic ovary syndrome: a population-based analysis in the United States. J Invest Dermatol. 2018;138:1288-1292.
- Porter ML, Kimball AB. Comorbidities of hidradenitis suppurativa. Semin Cutan Med Surg. 2017;36:55-57.
- Hessam S, Sand M, Gambichler T, et al. Correlation of inflammatory serum markers with disease severity in patients with hidradenitis suppurativa (HS). J Am Acad Dermatol. 2015;73:998-1005.
- Ring HC, Bay L, Nilsson M, et al. Bacterial biofilm in chronic lesions of hidradenitis suppurativa. Br J Dermatol. 2017;176:993-1000.
- Yazdanyar S, Jemec GB. Hidradenitis suppurativa: a review of cause and treatment. Curr Opin Infect Dis. 2011;24:118-123.
- Wortsman X. Imaging of hidradenitis suppurativa. Dermatol Clin. 2016;34:59-68.
- Saunte DM, Boer J, Stratigos A, et al. Diagnostic delay in hidradenitis suppurativa is a global problem. Br J Dermatol. 2015;173:1546-1549.
- Revuz JE, Jemec GB. Diagnosing hidradenitis suppurativa. Dermatol Clin. 2016;34:1-5.
- Canoui-Poitrine F, Le Thuaut A, Revuz JE, et al. Identification of three hidradenitis suppurativa phenotypes: latent class analysis of a cross-sectional study. J Invest Dermatol. 2013;133:1506-1511.
- Porter ML, Kimball AB. Hidradenitis suppurativa scoring systems: can we choose just one? Cutis. 2017;99:156-157.
- Hurley HJ. Axillary hyperhidrosis, apocrine bromhidrosis, hidradenitis suppurativa, and familial benign pemphigus: surgical approach. In: Roenigk RK, Roenigk HH, Jr, eds. Dermatologic Surgery: Principles and Practice. New York, NY: Marcel Dekker, Inc; 1989:732-738.
- Kimball AB, Sobell JM, Zouboulis CC, et al. HiSCR (Hidradenitis Suppurativa Clinical Response): a novel clinical endpoint to evaluate therapeutic outcomes in patients with hidradenitis suppurativa from the placebo-controlled portion of a phase 2 adalimumab study. J Eur Acad Dermatol Venereol. 2016;30:989-994.
- Kimball AB, Okun MM, Williams DA, et al. Two phase 3 trials of adalimumab for hidradenitis suppurativa. N Engl J Med. 2016;375:422-434.
- Wong D, Walsh S, Alhusayen R. Low-dose systemic corticosteroid treatment for recalcitrant hidradenitis suppurativa. J Am Acad Dermatol. 2016;75:1059-1062.
- Sullivan TP, Welsh E, Kerdel FA. Infliximab for hidradenitis suppurativa. Br J Dermatol. 2003;149:1046-1049.
- Anderson MD, Zauli S, Bettoli V, et al. Cyclosporine treatment of severe hidradenitis suppurativa—a case series. J Dermatolog Treat. 2016;27:247-250.
- Ring HC, Riis Mikkelsen P, Miller IM, et al. The bacteriology of hidradenitis suppurativa: a systematic review. Exp Dermatol. 2015;24:727-731.
- Guet-Revillet H, Coignard-Biehler H, Jais JP, et al. Bacterial pathogens associated with hidradenitis suppurativa, France. Emerg Infect Dis. 2014;20:1990-1998.
- Golbari NM, Porter ML, Kimball AB. Antiandrogen therapy with spironolactone for the treatment of hidradenitis suppurativa. J Am Acad Dermatol. 2019;80:114-119.
- Matusiak L, Bieniek A, Szepietowski JC. Acitretin treatment for hidradenitis suppurativa: a prospective series of 17 patients. Br J Dermatol. 2014;171:170-174.
Hidradenitis suppurativa (HS) is a common chronic inflammatory skin disease characterized by purulent subcutaneous nodules, papules, abscesses, and fistula tracts that lead to scarring and fibrosis. Lesions develop primarily in the axilla, groin, and other intertriginous and hair-bearing areas.
The natural history of the disease is characterized by periods of disease flare, followed by periods of disease quiescence. Patients might have weeks or months of low disease activity but frequently develop multiple exacerbating episodes over the course of weeks or months. The condition primarily presents in adolescent and peripubescent years, continuing throughout adulthood. Some evidence suggests a bimodal disease distribution, with a second peak of incidence in middle-aged adults. Women and men are affected equally; however, the disease can be phenotypically different in men and women.
Patients frequently present in emergency and inpatient settings for evaluation because of the pain and severity of HS flares as well as associated systemic symptoms. Inpatient and emergency department (ED) care are unique opportunities for dermatologic hospitalist and dermatologic consultative services to educate other physicians about the condition and initiate aggressive treatments that are frequently necessary to control HS flares. This article aims to address best methods for treating HS in these settings.
Pathophysiology
Although the exact pathophysiology of the condition is unknown, HS is thought to begin with follicular occlusion with downstream inflammation mediating neutrophilic activity and scarring. Hyperplasia of the infundibular epithelium is observed on histology, and the resulting occlusion, contained keratin, and follicular rupture initiate robust downstream inflammation.1,2 Follicular occlusion might be initially androgen mediated3 or might occur in combination with friction4 and genetic or acquired factors involving Notch signaling. Although HS characteristically presents in areas of high apocrine density, apocrine glands are not thought to be the primary mediator of disease activity.5 IL-17, IL-23, tumor necrosis factor α, and IL-1β are implicated in the pathogenesis of HS, but it is unknown if these cytokines are the driving pathologic factor in HS or if they are merely secondary sequelae.6
Demographics and Prevalence in Hospitalized Patients
Although increasing treatment availability has brought more attention to HS, true prevalence is unknown. A prevalence of 1% has been reported in many European countries.7 Global prevalence has been more difficult to determine, with variable data suggesting a prevalence of 0.03% to 8%, depending on the population included.8 Most patients studied in a US-based claims database were aged 30 to 64 years, and the overall prevalence was 0.05%.9 Despite prevalence similar to psoriasis, utilization of high-cost emergency and inpatient admissions is notably higher among patients with HS. Recent claims data suggest that HS patients utilize the ED at a rate 3 times higher than psoriasis patients and are admitted as inpatients at a rate 5 times higher.10 Similar data suggest an associated increased cost of care for patients with HS vs other conditions, such as psoriasis, due to frequent ED and inpatient stays.11 Although HS frequently presents in the inpatient and emergency settings, there is little literature on best methods for managing patients in these settings.
Pearls for Inpatient and Emergency Evaluation and Management
Initial Evaluation
When dermatologic consultative services are asked to evaluate patients with HS, preliminary evaluation should reflect the acuity of the patient. Vital signs and toxicity should be reviewed to ensure that there is no evidence of severe infection necessitating critical or acute care.
History
History-taking should reflect assessment of the patient’s baseline disease, including date of initial onset; exacerbating factors, such as friction, smoking, pregnancy, and menses; and the current history of the patient’s flare. A history of antibiotics, immunosuppression, topical therapy, antiandrogen therapy, and vitamin A analog therapy also should be reviewed. If an initial diagnosis is made in the ED or inpatient setting, a family and personal history should focus on specific risk factors and disease associations, including inflammatory bowel disease,12 pilonidal cysts,13 polycystic ovary syndrome,14 and metabolic syndrome.15
Physical Examination
As with all dermatologic consultations, a full-body skin examination, with special attention to the axilla, inframammary skin, groin, buttocks, and perineum, should be undertaken. In addition to these common areas of disease progression, examination should focus on atypical sites for disease manifestation, including the posterior auricular scalp, skin folds in the pannus and back, and the beard area in men. Evaluation of axillary and gluteal hair should note features of folliculitis and hair removal, which can exacerbate HS. Examination also should include an investigation of cutaneous manifestations of comorbid conditions, including acanthosis nigricans, contiguous or metastatic cutaneous Crohn disease, erythema nodosum, and pilonidal cysts. Caution should be exercised when diagnosing pilonidal cysts, as isolated or evolving HS in the gluteal cleft often is misdiagnosed as a pilonidal cyst.
Laboratory Evaluation
Testing often is misleading in patients with HS, especially in the acute setting, because the condition is a chronic inflammatory process. The C-reactive protein level as well as the absolute white blood cell and neutrophil counts often are elevated, even in the absence of acute infection.16 In fact, although patients often are treated with intravenous antibiotics by inpatient and emergency teams in the setting of these 3 laboratory abnormalities, these findings often reflect disease activity, not frank infection. Fever, especially low-grade fever, also can reflect ongoing disease activity. Thrombocytosis and anemia also are anecdotally common, though these findings have not been reported specifically in the literature.
Bacterial Cultures
The role of lesional and perilesional bacterial cultures is controversial in HS. Prior studies have demonstrated that biofilm formation may be associated with the chronic inflammation seen in HS.17 However, most data to date suggest that infection is not the primary driver of HS disease flares, as demonstrated by the frequency of sterile cultures and the variable response of the disease to penicillin and related antibiotics.18
Imaging
Ultrasonography and magnetic resonance imaging can be conducted if there is concern about deeper abscesses that are not apparent on examination. When interpreted by nondermatologic practitioners, however, the findings of these modalities can result in unnecessary surgical intervention, given the concern for development of infectious abscess.19
Diagnosis
Many patients with HS experience a notable delay in time to diagnosis, living with symptoms for 7 years on average prior to being given a name for their condition.20 Often, patients seek ED care at initial presentation because lesions can present quickly and are associated with remarkable pain. Inpatient dermatologic evaluation can provide patients with definitive diagnosis, appropriate counseling that provides an overview of the natural history of the disease, lifestyle recommendations, and expedited connection to outpatient longitudinal care.
Diagnosis is made clinically by assessment for typical lesions, such as painful or tender papules, nodules, or abscesses in the axillae, inframammary region, groin, thighs, and perineal and perianal regions. Cordlike scarring often is seen in the absence of active inflammatory lesions.21 Double-headed open comedones and prominent follicular occlusion are seen in some phenotypes but are not required for diagnosis.22
Multiple scoring modalities are in use23; the Hurley staging system, initially developed for surgical staging, has become a commonly used method in the clinical setting24:
• Hurley stage I: isolated nodules or abscesses;
• Hurley stage II: widely separate lesions and sinus tracts or scarring are suggestive; and
• Hurley stage III: multiple lesions with near-diffuse involvement and formation of sinus tracts and scarring.
Other scoring modalities, such as the Hidradenitis Suppurativa Clinical Response (HiSCR), are more commonly used in the clinical trial setting and quantitatively capture lesion count improvement while the patient is being treated.25
Treatment
Evaluation in the ED might necessitate recommendations for inpatient admission. Dermatologic consultation can be helpful in providing ED physicians with context for interpretation of laboratory results and clinical findings. Specifically, dermatologic evaluation can help differentiate presentations consistent with a primary infection from a more common presentation of HS flaring and associated bacterial colonization. Indications for inpatient admission are severe pain; concern for systemic infection, including high fever or sepsis; and need for surgical intervention. Patients with severe disease who do not have a longitudinal care plan or who lack the ability to care for lesions at home also are candidates for inpatient admission, where they can receive more intensive nursing and wound care as well as outpatient logistical management.
Acute care should be aimed at treatments that work quickly and aggressively and have both anti-inflammatory and antimicrobial effects. Severe flares require aggressive initial treatment to ensure more long-term remission. Adalimumab, maintained at 40 mg/wk after a loading dose, is the mainstay of evidence-based treatment for moderate to severe HS in patients 12 years or older; however, this treatment might not be easy to initiate in the inpatient setting because of its cost and availability and the fact that it is not as fast acting as other therapies.26 For patients with severe disease flares, prednisone,27 infliximab,28 or cyclosporine29 can be used in combination with antimicrobial therapy in the inpatient setting to quickly control active flaring. Intravenous antimicrobial therapy might be necessary in severe disease and should include coverage of gram-positive30 and anaerobic organisms.31
Although management of acute flares is critical, especially for hospitalized patients, initiating longitudinal treatment modalities while the patient is an inpatient will help prevent future readmissions, facilitate better outcomes, and enable longer periods of disease-free progression. Specific treatments, stratified by disease severity, are listed in the Table.
Postdischarge Lifestyle Modification
All disease management should include recommendations for lifestyle modification, including counseling on terminal hair removal (ie, avoid shaving, plucking, and waxing) and recommendations for daily and weekly decolonization with chlorhexidine or other antimicrobial soap, a weekly vinegar bath, and antiperspirant use in the groin and axilla. Avoiding tight clothes and humidity might also be helpful.
Other beneficial postdischarge strategies include smoking cessation and weight loss, which often are beneficial but difficult for many patients to achieve on their own; connecting patients with a primary care provider, which can facilitate better long-term outcomes; informing patients of the natural history of the disease and providing strategies for them to implement for acute flares to help avoid readmission and ED visits; and writing a “pill-in-pocket” prescription for a course of an antibiotic that provides good staphylococcal and anaerobic coverage, which can be helpful for patients who are prone to infrequent flares.
Lastly, appropriate postdischarge maintenance therapy also can be initiated during the inpatient stay, including maintenance antibiotic therapy, spironolactone32 for female patients, and acitretin33 for comedonal-predominant patients.
Final Thoughts
Hidradenitis suppurativa is a common dermatologic condition that frequently presents in emergency and inpatient settings, given its association with painful and acutely indurated lesions that often appear concerning for infection. Elevated inflammatory markers and fever are common in HS and are not necessarily suggestive of infection. As such, while antibiotics may be part of acute management of HS, care also should address the inflammatory component of the disease. Longitudinal outpatient care coordination with a dermatologist and primary care physician is imperative for limiting ED and inpatient care utilization.
Hidradenitis suppurativa (HS) is a common chronic inflammatory skin disease characterized by purulent subcutaneous nodules, papules, abscesses, and fistula tracts that lead to scarring and fibrosis. Lesions develop primarily in the axilla, groin, and other intertriginous and hair-bearing areas.
The natural history of the disease is characterized by periods of disease flare, followed by periods of disease quiescence. Patients might have weeks or months of low disease activity but frequently develop multiple exacerbating episodes over the course of weeks or months. The condition primarily presents in adolescent and peripubescent years, continuing throughout adulthood. Some evidence suggests a bimodal disease distribution, with a second peak of incidence in middle-aged adults. Women and men are affected equally; however, the disease can be phenotypically different in men and women.
Patients frequently present in emergency and inpatient settings for evaluation because of the pain and severity of HS flares as well as associated systemic symptoms. Inpatient and emergency department (ED) care are unique opportunities for dermatologic hospitalist and dermatologic consultative services to educate other physicians about the condition and initiate aggressive treatments that are frequently necessary to control HS flares. This article aims to address best methods for treating HS in these settings.
Pathophysiology
Although the exact pathophysiology of the condition is unknown, HS is thought to begin with follicular occlusion with downstream inflammation mediating neutrophilic activity and scarring. Hyperplasia of the infundibular epithelium is observed on histology, and the resulting occlusion, contained keratin, and follicular rupture initiate robust downstream inflammation.1,2 Follicular occlusion might be initially androgen mediated3 or might occur in combination with friction4 and genetic or acquired factors involving Notch signaling. Although HS characteristically presents in areas of high apocrine density, apocrine glands are not thought to be the primary mediator of disease activity.5 IL-17, IL-23, tumor necrosis factor α, and IL-1β are implicated in the pathogenesis of HS, but it is unknown if these cytokines are the driving pathologic factor in HS or if they are merely secondary sequelae.6
Demographics and Prevalence in Hospitalized Patients
Although increasing treatment availability has brought more attention to HS, true prevalence is unknown. A prevalence of 1% has been reported in many European countries.7 Global prevalence has been more difficult to determine, with variable data suggesting a prevalence of 0.03% to 8%, depending on the population included.8 Most patients studied in a US-based claims database were aged 30 to 64 years, and the overall prevalence was 0.05%.9 Despite prevalence similar to psoriasis, utilization of high-cost emergency and inpatient admissions is notably higher among patients with HS. Recent claims data suggest that HS patients utilize the ED at a rate 3 times higher than psoriasis patients and are admitted as inpatients at a rate 5 times higher.10 Similar data suggest an associated increased cost of care for patients with HS vs other conditions, such as psoriasis, due to frequent ED and inpatient stays.11 Although HS frequently presents in the inpatient and emergency settings, there is little literature on best methods for managing patients in these settings.
Pearls for Inpatient and Emergency Evaluation and Management
Initial Evaluation
When dermatologic consultative services are asked to evaluate patients with HS, preliminary evaluation should reflect the acuity of the patient. Vital signs and toxicity should be reviewed to ensure that there is no evidence of severe infection necessitating critical or acute care.
History
History-taking should reflect assessment of the patient’s baseline disease, including date of initial onset; exacerbating factors, such as friction, smoking, pregnancy, and menses; and the current history of the patient’s flare. A history of antibiotics, immunosuppression, topical therapy, antiandrogen therapy, and vitamin A analog therapy also should be reviewed. If an initial diagnosis is made in the ED or inpatient setting, a family and personal history should focus on specific risk factors and disease associations, including inflammatory bowel disease,12 pilonidal cysts,13 polycystic ovary syndrome,14 and metabolic syndrome.15
Physical Examination
As with all dermatologic consultations, a full-body skin examination, with special attention to the axilla, inframammary skin, groin, buttocks, and perineum, should be undertaken. In addition to these common areas of disease progression, examination should focus on atypical sites for disease manifestation, including the posterior auricular scalp, skin folds in the pannus and back, and the beard area in men. Evaluation of axillary and gluteal hair should note features of folliculitis and hair removal, which can exacerbate HS. Examination also should include an investigation of cutaneous manifestations of comorbid conditions, including acanthosis nigricans, contiguous or metastatic cutaneous Crohn disease, erythema nodosum, and pilonidal cysts. Caution should be exercised when diagnosing pilonidal cysts, as isolated or evolving HS in the gluteal cleft often is misdiagnosed as a pilonidal cyst.
Laboratory Evaluation
Testing often is misleading in patients with HS, especially in the acute setting, because the condition is a chronic inflammatory process. The C-reactive protein level as well as the absolute white blood cell and neutrophil counts often are elevated, even in the absence of acute infection.16 In fact, although patients often are treated with intravenous antibiotics by inpatient and emergency teams in the setting of these 3 laboratory abnormalities, these findings often reflect disease activity, not frank infection. Fever, especially low-grade fever, also can reflect ongoing disease activity. Thrombocytosis and anemia also are anecdotally common, though these findings have not been reported specifically in the literature.
Bacterial Cultures
The role of lesional and perilesional bacterial cultures is controversial in HS. Prior studies have demonstrated that biofilm formation may be associated with the chronic inflammation seen in HS.17 However, most data to date suggest that infection is not the primary driver of HS disease flares, as demonstrated by the frequency of sterile cultures and the variable response of the disease to penicillin and related antibiotics.18
Imaging
Ultrasonography and magnetic resonance imaging can be conducted if there is concern about deeper abscesses that are not apparent on examination. When interpreted by nondermatologic practitioners, however, the findings of these modalities can result in unnecessary surgical intervention, given the concern for development of infectious abscess.19
Diagnosis
Many patients with HS experience a notable delay in time to diagnosis, living with symptoms for 7 years on average prior to being given a name for their condition.20 Often, patients seek ED care at initial presentation because lesions can present quickly and are associated with remarkable pain. Inpatient dermatologic evaluation can provide patients with definitive diagnosis, appropriate counseling that provides an overview of the natural history of the disease, lifestyle recommendations, and expedited connection to outpatient longitudinal care.
Diagnosis is made clinically by assessment for typical lesions, such as painful or tender papules, nodules, or abscesses in the axillae, inframammary region, groin, thighs, and perineal and perianal regions. Cordlike scarring often is seen in the absence of active inflammatory lesions.21 Double-headed open comedones and prominent follicular occlusion are seen in some phenotypes but are not required for diagnosis.22
Multiple scoring modalities are in use23; the Hurley staging system, initially developed for surgical staging, has become a commonly used method in the clinical setting24:
• Hurley stage I: isolated nodules or abscesses;
• Hurley stage II: widely separate lesions and sinus tracts or scarring are suggestive; and
• Hurley stage III: multiple lesions with near-diffuse involvement and formation of sinus tracts and scarring.
Other scoring modalities, such as the Hidradenitis Suppurativa Clinical Response (HiSCR), are more commonly used in the clinical trial setting and quantitatively capture lesion count improvement while the patient is being treated.25
Treatment
Evaluation in the ED might necessitate recommendations for inpatient admission. Dermatologic consultation can be helpful in providing ED physicians with context for interpretation of laboratory results and clinical findings. Specifically, dermatologic evaluation can help differentiate presentations consistent with a primary infection from a more common presentation of HS flaring and associated bacterial colonization. Indications for inpatient admission are severe pain; concern for systemic infection, including high fever or sepsis; and need for surgical intervention. Patients with severe disease who do not have a longitudinal care plan or who lack the ability to care for lesions at home also are candidates for inpatient admission, where they can receive more intensive nursing and wound care as well as outpatient logistical management.
Acute care should be aimed at treatments that work quickly and aggressively and have both anti-inflammatory and antimicrobial effects. Severe flares require aggressive initial treatment to ensure more long-term remission. Adalimumab, maintained at 40 mg/wk after a loading dose, is the mainstay of evidence-based treatment for moderate to severe HS in patients 12 years or older; however, this treatment might not be easy to initiate in the inpatient setting because of its cost and availability and the fact that it is not as fast acting as other therapies.26 For patients with severe disease flares, prednisone,27 infliximab,28 or cyclosporine29 can be used in combination with antimicrobial therapy in the inpatient setting to quickly control active flaring. Intravenous antimicrobial therapy might be necessary in severe disease and should include coverage of gram-positive30 and anaerobic organisms.31
Although management of acute flares is critical, especially for hospitalized patients, initiating longitudinal treatment modalities while the patient is an inpatient will help prevent future readmissions, facilitate better outcomes, and enable longer periods of disease-free progression. Specific treatments, stratified by disease severity, are listed in the Table.
Postdischarge Lifestyle Modification
All disease management should include recommendations for lifestyle modification, including counseling on terminal hair removal (ie, avoid shaving, plucking, and waxing) and recommendations for daily and weekly decolonization with chlorhexidine or other antimicrobial soap, a weekly vinegar bath, and antiperspirant use in the groin and axilla. Avoiding tight clothes and humidity might also be helpful.
Other beneficial postdischarge strategies include smoking cessation and weight loss, which often are beneficial but difficult for many patients to achieve on their own; connecting patients with a primary care provider, which can facilitate better long-term outcomes; informing patients of the natural history of the disease and providing strategies for them to implement for acute flares to help avoid readmission and ED visits; and writing a “pill-in-pocket” prescription for a course of an antibiotic that provides good staphylococcal and anaerobic coverage, which can be helpful for patients who are prone to infrequent flares.
Lastly, appropriate postdischarge maintenance therapy also can be initiated during the inpatient stay, including maintenance antibiotic therapy, spironolactone32 for female patients, and acitretin33 for comedonal-predominant patients.
Final Thoughts
Hidradenitis suppurativa is a common dermatologic condition that frequently presents in emergency and inpatient settings, given its association with painful and acutely indurated lesions that often appear concerning for infection. Elevated inflammatory markers and fever are common in HS and are not necessarily suggestive of infection. As such, while antibiotics may be part of acute management of HS, care also should address the inflammatory component of the disease. Longitudinal outpatient care coordination with a dermatologist and primary care physician is imperative for limiting ED and inpatient care utilization.
- Jemec GB, Hansen U. Histology of hidradenitis suppurativa. J Am Acad Dermatol. 1996;34:994-999.
- Prens E, Deckers I. Pathophysiology of hidradenitis suppurativa: an update. J Am Acad Dermatol. 2015;73(suppl 1):S8-S11.
- Barth JH, Kealey T. Androgen metabolism by isolated human axillary apocrine glands in hidradenitis suppurativa. Br J Dermatol. 1991;125:304-308.
- de Winter K, van der Zee HH, Prens EP. Is mechanical stress an important pathogenic factor in hidradenitis suppurativa? Exp Dermatol. 2012;21:176-177.
- Yu CC, Cook MG. Hidradenitis suppurativa: a disease of follicular epithelium, rather than apocrine glands. Br J Dermatol. 1990;122:763-769.
- Deckers IE, van der Zee HH, Prens EP. Epidemiology of hidradenitis suppurativa: prevalence, pathogenesis, and factors associated with the development of HS. Curr Dermatol Rep. 2014;3:54-60.
- Revuz JE, Canoui-Poitrine F, Wolkenstein P, et al. Prevalence and factors associated with hidradenitis suppurativa: Results from two case-control studies. J Am Acad Dermatol. 2008;59:596-601.
- Jemec GE, Kimball AB. Hidradenitis suppurativa: epidemiology and scope of the problem. J Am Acad Dermatol. 2015;73(suppl 1):S4-S7.
- Cosmatos I, Matcho A, Weinstein R, et al. Analysis of patient claims data to determine the prevalence of hidradenitis suppurativa in the United States. J Am Acad Dermatol. 2013;68:412-419.
- Khalsa A, Liu G, Kirby JS. Increased utilization of emergency department and inpatient care by patients with hidradenitis suppurativa. J Am Acad Dermatol. 2015;73:609-614.
- Kirby JS, Miller JJ, Adams DR, et al. Health care utilization patterns and costs for patients with hidradenitis suppurativa. JAMA Dermatol. 2014;150:937-944.
- Deckers IE, Benhadou F, Koldijk MJ, et al. Inflammatory bowel disease is associated with hidradenitis suppurativa: results from a multicenter cross-sectional study. J Am Acad Dermatol. 2017;76:49-53.
- Benhadou F, Van der Zee HH, Pascual JC, et al. Pilonidal sinus disease: an intergluteal localization of hidradenitis suppurativa/acne inversa: a cross-sectional study among 2465 patients [published online March 27, 2019]. Br J Dermatol. doi:10.1111/bjd.17927.
- Garg A, Neuren E, Strunk A. Hidradenitis suppurativa is associated with polycystic ovary syndrome: a population-based analysis in the United States. J Invest Dermatol. 2018;138:1288-1292.
- Porter ML, Kimball AB. Comorbidities of hidradenitis suppurativa. Semin Cutan Med Surg. 2017;36:55-57.
- Hessam S, Sand M, Gambichler T, et al. Correlation of inflammatory serum markers with disease severity in patients with hidradenitis suppurativa (HS). J Am Acad Dermatol. 2015;73:998-1005.
- Ring HC, Bay L, Nilsson M, et al. Bacterial biofilm in chronic lesions of hidradenitis suppurativa. Br J Dermatol. 2017;176:993-1000.
- Yazdanyar S, Jemec GB. Hidradenitis suppurativa: a review of cause and treatment. Curr Opin Infect Dis. 2011;24:118-123.
- Wortsman X. Imaging of hidradenitis suppurativa. Dermatol Clin. 2016;34:59-68.
- Saunte DM, Boer J, Stratigos A, et al. Diagnostic delay in hidradenitis suppurativa is a global problem. Br J Dermatol. 2015;173:1546-1549.
- Revuz JE, Jemec GB. Diagnosing hidradenitis suppurativa. Dermatol Clin. 2016;34:1-5.
- Canoui-Poitrine F, Le Thuaut A, Revuz JE, et al. Identification of three hidradenitis suppurativa phenotypes: latent class analysis of a cross-sectional study. J Invest Dermatol. 2013;133:1506-1511.
- Porter ML, Kimball AB. Hidradenitis suppurativa scoring systems: can we choose just one? Cutis. 2017;99:156-157.
- Hurley HJ. Axillary hyperhidrosis, apocrine bromhidrosis, hidradenitis suppurativa, and familial benign pemphigus: surgical approach. In: Roenigk RK, Roenigk HH, Jr, eds. Dermatologic Surgery: Principles and Practice. New York, NY: Marcel Dekker, Inc; 1989:732-738.
- Kimball AB, Sobell JM, Zouboulis CC, et al. HiSCR (Hidradenitis Suppurativa Clinical Response): a novel clinical endpoint to evaluate therapeutic outcomes in patients with hidradenitis suppurativa from the placebo-controlled portion of a phase 2 adalimumab study. J Eur Acad Dermatol Venereol. 2016;30:989-994.
- Kimball AB, Okun MM, Williams DA, et al. Two phase 3 trials of adalimumab for hidradenitis suppurativa. N Engl J Med. 2016;375:422-434.
- Wong D, Walsh S, Alhusayen R. Low-dose systemic corticosteroid treatment for recalcitrant hidradenitis suppurativa. J Am Acad Dermatol. 2016;75:1059-1062.
- Sullivan TP, Welsh E, Kerdel FA. Infliximab for hidradenitis suppurativa. Br J Dermatol. 2003;149:1046-1049.
- Anderson MD, Zauli S, Bettoli V, et al. Cyclosporine treatment of severe hidradenitis suppurativa—a case series. J Dermatolog Treat. 2016;27:247-250.
- Ring HC, Riis Mikkelsen P, Miller IM, et al. The bacteriology of hidradenitis suppurativa: a systematic review. Exp Dermatol. 2015;24:727-731.
- Guet-Revillet H, Coignard-Biehler H, Jais JP, et al. Bacterial pathogens associated with hidradenitis suppurativa, France. Emerg Infect Dis. 2014;20:1990-1998.
- Golbari NM, Porter ML, Kimball AB. Antiandrogen therapy with spironolactone for the treatment of hidradenitis suppurativa. J Am Acad Dermatol. 2019;80:114-119.
- Matusiak L, Bieniek A, Szepietowski JC. Acitretin treatment for hidradenitis suppurativa: a prospective series of 17 patients. Br J Dermatol. 2014;171:170-174.
- Jemec GB, Hansen U. Histology of hidradenitis suppurativa. J Am Acad Dermatol. 1996;34:994-999.
- Prens E, Deckers I. Pathophysiology of hidradenitis suppurativa: an update. J Am Acad Dermatol. 2015;73(suppl 1):S8-S11.
- Barth JH, Kealey T. Androgen metabolism by isolated human axillary apocrine glands in hidradenitis suppurativa. Br J Dermatol. 1991;125:304-308.
- de Winter K, van der Zee HH, Prens EP. Is mechanical stress an important pathogenic factor in hidradenitis suppurativa? Exp Dermatol. 2012;21:176-177.
- Yu CC, Cook MG. Hidradenitis suppurativa: a disease of follicular epithelium, rather than apocrine glands. Br J Dermatol. 1990;122:763-769.
- Deckers IE, van der Zee HH, Prens EP. Epidemiology of hidradenitis suppurativa: prevalence, pathogenesis, and factors associated with the development of HS. Curr Dermatol Rep. 2014;3:54-60.
- Revuz JE, Canoui-Poitrine F, Wolkenstein P, et al. Prevalence and factors associated with hidradenitis suppurativa: Results from two case-control studies. J Am Acad Dermatol. 2008;59:596-601.
- Jemec GE, Kimball AB. Hidradenitis suppurativa: epidemiology and scope of the problem. J Am Acad Dermatol. 2015;73(suppl 1):S4-S7.
- Cosmatos I, Matcho A, Weinstein R, et al. Analysis of patient claims data to determine the prevalence of hidradenitis suppurativa in the United States. J Am Acad Dermatol. 2013;68:412-419.
- Khalsa A, Liu G, Kirby JS. Increased utilization of emergency department and inpatient care by patients with hidradenitis suppurativa. J Am Acad Dermatol. 2015;73:609-614.
- Kirby JS, Miller JJ, Adams DR, et al. Health care utilization patterns and costs for patients with hidradenitis suppurativa. JAMA Dermatol. 2014;150:937-944.
- Deckers IE, Benhadou F, Koldijk MJ, et al. Inflammatory bowel disease is associated with hidradenitis suppurativa: results from a multicenter cross-sectional study. J Am Acad Dermatol. 2017;76:49-53.
- Benhadou F, Van der Zee HH, Pascual JC, et al. Pilonidal sinus disease: an intergluteal localization of hidradenitis suppurativa/acne inversa: a cross-sectional study among 2465 patients [published online March 27, 2019]. Br J Dermatol. doi:10.1111/bjd.17927.
- Garg A, Neuren E, Strunk A. Hidradenitis suppurativa is associated with polycystic ovary syndrome: a population-based analysis in the United States. J Invest Dermatol. 2018;138:1288-1292.
- Porter ML, Kimball AB. Comorbidities of hidradenitis suppurativa. Semin Cutan Med Surg. 2017;36:55-57.
- Hessam S, Sand M, Gambichler T, et al. Correlation of inflammatory serum markers with disease severity in patients with hidradenitis suppurativa (HS). J Am Acad Dermatol. 2015;73:998-1005.
- Ring HC, Bay L, Nilsson M, et al. Bacterial biofilm in chronic lesions of hidradenitis suppurativa. Br J Dermatol. 2017;176:993-1000.
- Yazdanyar S, Jemec GB. Hidradenitis suppurativa: a review of cause and treatment. Curr Opin Infect Dis. 2011;24:118-123.
- Wortsman X. Imaging of hidradenitis suppurativa. Dermatol Clin. 2016;34:59-68.
- Saunte DM, Boer J, Stratigos A, et al. Diagnostic delay in hidradenitis suppurativa is a global problem. Br J Dermatol. 2015;173:1546-1549.
- Revuz JE, Jemec GB. Diagnosing hidradenitis suppurativa. Dermatol Clin. 2016;34:1-5.
- Canoui-Poitrine F, Le Thuaut A, Revuz JE, et al. Identification of three hidradenitis suppurativa phenotypes: latent class analysis of a cross-sectional study. J Invest Dermatol. 2013;133:1506-1511.
- Porter ML, Kimball AB. Hidradenitis suppurativa scoring systems: can we choose just one? Cutis. 2017;99:156-157.
- Hurley HJ. Axillary hyperhidrosis, apocrine bromhidrosis, hidradenitis suppurativa, and familial benign pemphigus: surgical approach. In: Roenigk RK, Roenigk HH, Jr, eds. Dermatologic Surgery: Principles and Practice. New York, NY: Marcel Dekker, Inc; 1989:732-738.
- Kimball AB, Sobell JM, Zouboulis CC, et al. HiSCR (Hidradenitis Suppurativa Clinical Response): a novel clinical endpoint to evaluate therapeutic outcomes in patients with hidradenitis suppurativa from the placebo-controlled portion of a phase 2 adalimumab study. J Eur Acad Dermatol Venereol. 2016;30:989-994.
- Kimball AB, Okun MM, Williams DA, et al. Two phase 3 trials of adalimumab for hidradenitis suppurativa. N Engl J Med. 2016;375:422-434.
- Wong D, Walsh S, Alhusayen R. Low-dose systemic corticosteroid treatment for recalcitrant hidradenitis suppurativa. J Am Acad Dermatol. 2016;75:1059-1062.
- Sullivan TP, Welsh E, Kerdel FA. Infliximab for hidradenitis suppurativa. Br J Dermatol. 2003;149:1046-1049.
- Anderson MD, Zauli S, Bettoli V, et al. Cyclosporine treatment of severe hidradenitis suppurativa—a case series. J Dermatolog Treat. 2016;27:247-250.
- Ring HC, Riis Mikkelsen P, Miller IM, et al. The bacteriology of hidradenitis suppurativa: a systematic review. Exp Dermatol. 2015;24:727-731.
- Guet-Revillet H, Coignard-Biehler H, Jais JP, et al. Bacterial pathogens associated with hidradenitis suppurativa, France. Emerg Infect Dis. 2014;20:1990-1998.
- Golbari NM, Porter ML, Kimball AB. Antiandrogen therapy with spironolactone for the treatment of hidradenitis suppurativa. J Am Acad Dermatol. 2019;80:114-119.
- Matusiak L, Bieniek A, Szepietowski JC. Acitretin treatment for hidradenitis suppurativa: a prospective series of 17 patients. Br J Dermatol. 2014;171:170-174.
Practice Points
- Hidradenitis suppurativa (HS) is a common dermatologic condition that frequently presents in emergency and inpatient settings.
- Anemia, leukocytosis, neutrophilia, an elevated erythrocyte sedimentation rate, and an elevated C-reactive protein level are common markers of chronic inflammation in HS patients and might not signify infection.
- Acute management of HS should focus on anti-inflammatory and antibiotic regimens, with increasing severity dictating the need for more aggressive therapy.
- Longitudinal outpatient care coordination with a dermatologist and primary care physician is imperative for limiting emergency department and inpatient care utilization.
