User login
Cutis is a peer-reviewed clinical journal for the dermatologist, allergist, and general practitioner published monthly since 1965. Concise clinical articles present the practical side of dermatology, helping physicians to improve patient care. Cutis is referenced in Index Medicus/MEDLINE and is written and edited by industry leaders.
ass lick
assault rifle
balls
ballsac
black jack
bleach
Boko Haram
bondage
causas
cheap
child abuse
cocaine
compulsive behaviors
cost of miracles
cunt
Daech
display network stats
drug paraphernalia
explosion
fart
fda and death
fda AND warn
fda AND warning
fda AND warns
feom
fuck
gambling
gfc
gun
human trafficking
humira AND expensive
illegal
ISIL
ISIS
Islamic caliphate
Islamic state
madvocate
masturbation
mixed martial arts
MMA
molestation
national rifle association
NRA
nsfw
nuccitelli
pedophile
pedophilia
poker
porn
porn
pornography
psychedelic drug
recreational drug
sex slave rings
shit
slot machine
snort
substance abuse
terrorism
terrorist
texarkana
Texas hold 'em
UFC
section[contains(@class, 'nav-hidden')]
section[contains(@class, 'nav-hidden active')
A peer-reviewed, indexed journal for dermatologists with original research, image quizzes, cases and reviews, and columns.
Allergic Contact Dermatitis With Sparing of Exposed Psoriasis Plaques
To the Editor:
Allergic contact dermatitis (ACD) is a delayed-type hypersensitivity reaction against antigens to which the skin’s immune system was previously sensitized. The initial sensitization requires penetration of the antigen through the stratum corneum. Thus, the ability of a particle to cause ACD is related to its molecular structure and size, lipophilicity, and protein-binding affinity, as well as the dose and duration of exposure.1 Psoriasis typically presents as well-demarcated areas of skin that may be erythematous, indurated, and scaly to variable degrees. Histologically, psoriasis plaques are characterized by epidermal hyperplasia in the presence of a T-cell infiltrate and neutrophilic microabscesses. We report a case of a patient with plaque-type psoriasis who experienced ACD with sparing of exposed psoriatic plaques.
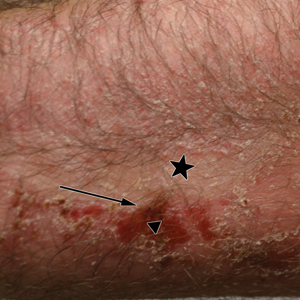
A 45-year-old man with a 5-year history of generalized moderate to severe psoriasis undergoing therapy with ustekinumab 45 mg subcutaneously once every 12 weeks presented to the emergency department with intensely erythematous, pruritic, vesicular lesions on the trunk, arms, and legs within 24 hours of exposure to poison oak while hiking. The patient reported pruritus, pain, and swelling of the affected areas. On physical examination, he was afebrile. Widespread erythematous vesicular lesions were noted on the face, trunk, arms, and legs, sparing the well-demarcated scaly psoriatic plaques on the arms and legs (Figure). The patient was given intravenous fluids and intravenous diphenhydramine. After responding to initial treatment, the patient was discharged with ibuprofen and a tapering dose of oral prednisone from 60 mg 5 times daily, to 40 mg 5 times daily, to 20 mg 5 times daily over 15 days.
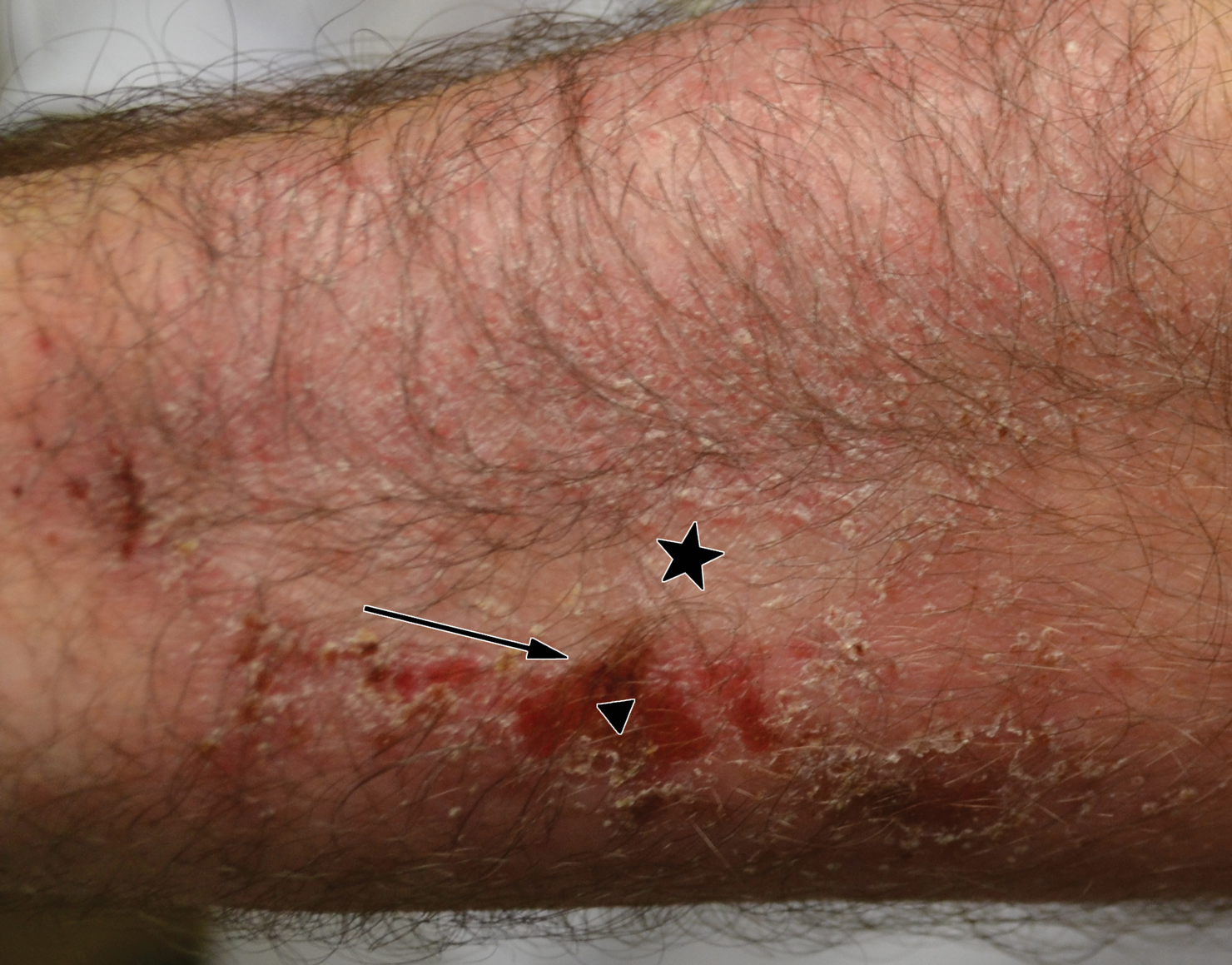
star), with a linear border demarcating the ACD lesion and the unaffected psoriatic plaque (black arrow).
Allergic contact dermatitis occurs after sensitization to environmental allergens or haptens. Clinically, ACD is characterized by pruritic, erythematous, vesicular papules and plaques. The predominant effector cells in ACD are CD8+ T cells, along with contributions from helper T cells (TH2). Together, these cell types produce an environment enriched in IFN-γ, IL-2, IL-4, IL-10, IL-17, and tumor necrosis factor α.2 Ultimately, the ACD response induces keratinocyte apoptosis via cytotoxic effects.3,4
Plaque psoriasis is a chronic, immune-mediated, inflammatory disease that presents clinically as erythematous well-demarcated plaques with a micaceous scale. The immunologic environment of psoriasis plaques is characterized by infiltration of CD4+ TH17 cells and elevated levels of IL-17, IL-23, tumor necrosis factor α, and IL-1β, which induce keratinocyte hyperproliferation through a complex mechanism resulting in hyperkeratosis composed of orthokeratosis and parakeratosis, a neutrophilic infiltrate, and Munro microabscesses.5
The predominant effector cells and the final effects on keratinocyte survival are divergent in psoriasis and ACD. The possibly antagonistic relationship between these immunologic processes is further supported by epidemiologic studies demonstrating a decreased incidence of ACD in patients with psoriasis.6,7
Our patient demonstrated a typical ACD reaction in response to exposure to urushiol, the allergen present in poison oak, in areas unaffected by psoriasis plaques. Interestingly, the patient displayed this response even while undergoing therapy with ustekinumab, a fully humanized antibody that binds IL-12 and IL-23 and ultimately downregulates TH17 cell-mediated release of IL-17 in the treatment of psoriasis. Although IL-17 also has been implicated in ACD, the lack of inhibition of ACD with ustekinumab treatment was previously demonstrated in a small retrospective study, indicating a potentially different source of IL-17 in ACD.8
Our patient did not demonstrate a typical ACD response in areas of active psoriasis plaques. This phenomenon was of great interest to us. It is possible that the presence of hyperkeratosis, manifested clinically as scaling, served as a mechanical barrier preventing the diffusion and exposure of cutaneous immune cells to urushiol. On the other hand, it is possible that the immunologic environment of the active psoriasis plaque was altered in such a way that it did not demonstrate the typical response to allergen exposure.
We hypothesize that the lack of a typical ACD response at sites of psoriatic plaques in our patient may be attributed to the intensity and duration of exposure to the allergen. Quaranta et al9 reported a typical ACD clinical response and a mixed immunohistologic response to nickel patch testing at sites of active plaques in nickel-sensitized psoriasis patients. Patch testing involves 48 hours of direct contact with an allergen, while our patient experienced an estimated 8 to 10 hours of exposure to the allergen prior to removal via washing. Supporting this line of reasoning, a proportion of patients who are responsive to nickel patch testing do not exhibit clinical symptoms in response to casual nickel exposure.10 Although a physical barrier effect due to hyperkeratosis may have contributed to the lack of ACD response in sites of psoriasis plaques in our patient, it remains possible that a more limited duration of exposure to the allergen is not sufficient to overcome the native immunologic milieu of the psoriasis plaque and induce the immunologic cascade resulting in ACD. Further research into the potentially antagonistic relationship of psoriasis and ACD should be performed to elucidate the interaction between these two common conditions.
- Kimber I, Basketter DA, Gerberick GF, et al. Allergic contact dermatitis. Int Immunopharmacol. 2002;2:201-211.
- Vocanson M, Hennino A, Cluzel-Tailhardat M, et al. CD8+ T cells are effector cells of contact dermatitis to common skin allergens in mice. J Invest Dermatol. 2006;126:815-820.
- Akiba H, Kehren J, Ducluzeau MT, et al. Skin inflammation during contact hypersensitivity is mediated by early recruitment of CD8+ T cytotoxic 1 cells inducing keratinocyte apoptosis. J Immunol. 2002;168:3079-3087.
- Trautmann A, Akdis M, Kleemann D, et al. T cell-mediated Fas-induced keratinocyte apoptosis plays a key pathogenetic role in eczematous dermatitis. J Clin Invest. 2000;106:25-35.
- Lynde CW, Poulin Y, Vender R, et al. Interleukin 17A: toward a new understanding of psoriasis pathogenesis. J Am Acad Dermatol. 2014;71:141-150.
- Bangsgaard N, Engkilde K, Thyssen JP, et al. Inverse relationship between contact allergy and psoriasis: results from a patient- and a population-based study. Br J Dermatol. 2009;161:1119-1123.
- Henseler T, Christophers E. Disease concomitance in psoriasis. J Am Acad Dermatol. 1995;32:982-986.
- Bangsgaard N, Zachariae C, Menne T, et al. Lack of effect of ustekinumab in treatment of allergic contact dermatitis. Contact Dermatitis. 2011;65:227-230.
- Quaranta M, Eyerich S, Knapp B, et al. Allergic contact dermatitis in psoriasis patients: typical, delayed, and non-interacting. PLoS One. 2014;9:e101814.
- Kimber I, Basketter DA, Gerberick GF, et al. Allergic contact dermatitis. Int Immunopharmacol. 2002;2:201-211.
To the Editor:
Allergic contact dermatitis (ACD) is a delayed-type hypersensitivity reaction against antigens to which the skin’s immune system was previously sensitized. The initial sensitization requires penetration of the antigen through the stratum corneum. Thus, the ability of a particle to cause ACD is related to its molecular structure and size, lipophilicity, and protein-binding affinity, as well as the dose and duration of exposure.1 Psoriasis typically presents as well-demarcated areas of skin that may be erythematous, indurated, and scaly to variable degrees. Histologically, psoriasis plaques are characterized by epidermal hyperplasia in the presence of a T-cell infiltrate and neutrophilic microabscesses. We report a case of a patient with plaque-type psoriasis who experienced ACD with sparing of exposed psoriatic plaques.
A 45-year-old man with a 5-year history of generalized moderate to severe psoriasis undergoing therapy with ustekinumab 45 mg subcutaneously once every 12 weeks presented to the emergency department with intensely erythematous, pruritic, vesicular lesions on the trunk, arms, and legs within 24 hours of exposure to poison oak while hiking. The patient reported pruritus, pain, and swelling of the affected areas. On physical examination, he was afebrile. Widespread erythematous vesicular lesions were noted on the face, trunk, arms, and legs, sparing the well-demarcated scaly psoriatic plaques on the arms and legs (Figure). The patient was given intravenous fluids and intravenous diphenhydramine. After responding to initial treatment, the patient was discharged with ibuprofen and a tapering dose of oral prednisone from 60 mg 5 times daily, to 40 mg 5 times daily, to 20 mg 5 times daily over 15 days.
star), with a linear border demarcating the ACD lesion and the unaffected psoriatic plaque (black arrow).
Allergic contact dermatitis occurs after sensitization to environmental allergens or haptens. Clinically, ACD is characterized by pruritic, erythematous, vesicular papules and plaques. The predominant effector cells in ACD are CD8+ T cells, along with contributions from helper T cells (TH2). Together, these cell types produce an environment enriched in IFN-γ, IL-2, IL-4, IL-10, IL-17, and tumor necrosis factor α.2 Ultimately, the ACD response induces keratinocyte apoptosis via cytotoxic effects.3,4
Plaque psoriasis is a chronic, immune-mediated, inflammatory disease that presents clinically as erythematous well-demarcated plaques with a micaceous scale. The immunologic environment of psoriasis plaques is characterized by infiltration of CD4+ TH17 cells and elevated levels of IL-17, IL-23, tumor necrosis factor α, and IL-1β, which induce keratinocyte hyperproliferation through a complex mechanism resulting in hyperkeratosis composed of orthokeratosis and parakeratosis, a neutrophilic infiltrate, and Munro microabscesses.5
The predominant effector cells and the final effects on keratinocyte survival are divergent in psoriasis and ACD. The possibly antagonistic relationship between these immunologic processes is further supported by epidemiologic studies demonstrating a decreased incidence of ACD in patients with psoriasis.6,7
Our patient demonstrated a typical ACD reaction in response to exposure to urushiol, the allergen present in poison oak, in areas unaffected by psoriasis plaques. Interestingly, the patient displayed this response even while undergoing therapy with ustekinumab, a fully humanized antibody that binds IL-12 and IL-23 and ultimately downregulates TH17 cell-mediated release of IL-17 in the treatment of psoriasis. Although IL-17 also has been implicated in ACD, the lack of inhibition of ACD with ustekinumab treatment was previously demonstrated in a small retrospective study, indicating a potentially different source of IL-17 in ACD.8
Our patient did not demonstrate a typical ACD response in areas of active psoriasis plaques. This phenomenon was of great interest to us. It is possible that the presence of hyperkeratosis, manifested clinically as scaling, served as a mechanical barrier preventing the diffusion and exposure of cutaneous immune cells to urushiol. On the other hand, it is possible that the immunologic environment of the active psoriasis plaque was altered in such a way that it did not demonstrate the typical response to allergen exposure.
We hypothesize that the lack of a typical ACD response at sites of psoriatic plaques in our patient may be attributed to the intensity and duration of exposure to the allergen. Quaranta et al9 reported a typical ACD clinical response and a mixed immunohistologic response to nickel patch testing at sites of active plaques in nickel-sensitized psoriasis patients. Patch testing involves 48 hours of direct contact with an allergen, while our patient experienced an estimated 8 to 10 hours of exposure to the allergen prior to removal via washing. Supporting this line of reasoning, a proportion of patients who are responsive to nickel patch testing do not exhibit clinical symptoms in response to casual nickel exposure.10 Although a physical barrier effect due to hyperkeratosis may have contributed to the lack of ACD response in sites of psoriasis plaques in our patient, it remains possible that a more limited duration of exposure to the allergen is not sufficient to overcome the native immunologic milieu of the psoriasis plaque and induce the immunologic cascade resulting in ACD. Further research into the potentially antagonistic relationship of psoriasis and ACD should be performed to elucidate the interaction between these two common conditions.
To the Editor:
Allergic contact dermatitis (ACD) is a delayed-type hypersensitivity reaction against antigens to which the skin’s immune system was previously sensitized. The initial sensitization requires penetration of the antigen through the stratum corneum. Thus, the ability of a particle to cause ACD is related to its molecular structure and size, lipophilicity, and protein-binding affinity, as well as the dose and duration of exposure.1 Psoriasis typically presents as well-demarcated areas of skin that may be erythematous, indurated, and scaly to variable degrees. Histologically, psoriasis plaques are characterized by epidermal hyperplasia in the presence of a T-cell infiltrate and neutrophilic microabscesses. We report a case of a patient with plaque-type psoriasis who experienced ACD with sparing of exposed psoriatic plaques.
A 45-year-old man with a 5-year history of generalized moderate to severe psoriasis undergoing therapy with ustekinumab 45 mg subcutaneously once every 12 weeks presented to the emergency department with intensely erythematous, pruritic, vesicular lesions on the trunk, arms, and legs within 24 hours of exposure to poison oak while hiking. The patient reported pruritus, pain, and swelling of the affected areas. On physical examination, he was afebrile. Widespread erythematous vesicular lesions were noted on the face, trunk, arms, and legs, sparing the well-demarcated scaly psoriatic plaques on the arms and legs (Figure). The patient was given intravenous fluids and intravenous diphenhydramine. After responding to initial treatment, the patient was discharged with ibuprofen and a tapering dose of oral prednisone from 60 mg 5 times daily, to 40 mg 5 times daily, to 20 mg 5 times daily over 15 days.
star), with a linear border demarcating the ACD lesion and the unaffected psoriatic plaque (black arrow).
Allergic contact dermatitis occurs after sensitization to environmental allergens or haptens. Clinically, ACD is characterized by pruritic, erythematous, vesicular papules and plaques. The predominant effector cells in ACD are CD8+ T cells, along with contributions from helper T cells (TH2). Together, these cell types produce an environment enriched in IFN-γ, IL-2, IL-4, IL-10, IL-17, and tumor necrosis factor α.2 Ultimately, the ACD response induces keratinocyte apoptosis via cytotoxic effects.3,4
Plaque psoriasis is a chronic, immune-mediated, inflammatory disease that presents clinically as erythematous well-demarcated plaques with a micaceous scale. The immunologic environment of psoriasis plaques is characterized by infiltration of CD4+ TH17 cells and elevated levels of IL-17, IL-23, tumor necrosis factor α, and IL-1β, which induce keratinocyte hyperproliferation through a complex mechanism resulting in hyperkeratosis composed of orthokeratosis and parakeratosis, a neutrophilic infiltrate, and Munro microabscesses.5
The predominant effector cells and the final effects on keratinocyte survival are divergent in psoriasis and ACD. The possibly antagonistic relationship between these immunologic processes is further supported by epidemiologic studies demonstrating a decreased incidence of ACD in patients with psoriasis.6,7
Our patient demonstrated a typical ACD reaction in response to exposure to urushiol, the allergen present in poison oak, in areas unaffected by psoriasis plaques. Interestingly, the patient displayed this response even while undergoing therapy with ustekinumab, a fully humanized antibody that binds IL-12 and IL-23 and ultimately downregulates TH17 cell-mediated release of IL-17 in the treatment of psoriasis. Although IL-17 also has been implicated in ACD, the lack of inhibition of ACD with ustekinumab treatment was previously demonstrated in a small retrospective study, indicating a potentially different source of IL-17 in ACD.8
Our patient did not demonstrate a typical ACD response in areas of active psoriasis plaques. This phenomenon was of great interest to us. It is possible that the presence of hyperkeratosis, manifested clinically as scaling, served as a mechanical barrier preventing the diffusion and exposure of cutaneous immune cells to urushiol. On the other hand, it is possible that the immunologic environment of the active psoriasis plaque was altered in such a way that it did not demonstrate the typical response to allergen exposure.
We hypothesize that the lack of a typical ACD response at sites of psoriatic plaques in our patient may be attributed to the intensity and duration of exposure to the allergen. Quaranta et al9 reported a typical ACD clinical response and a mixed immunohistologic response to nickel patch testing at sites of active plaques in nickel-sensitized psoriasis patients. Patch testing involves 48 hours of direct contact with an allergen, while our patient experienced an estimated 8 to 10 hours of exposure to the allergen prior to removal via washing. Supporting this line of reasoning, a proportion of patients who are responsive to nickel patch testing do not exhibit clinical symptoms in response to casual nickel exposure.10 Although a physical barrier effect due to hyperkeratosis may have contributed to the lack of ACD response in sites of psoriasis plaques in our patient, it remains possible that a more limited duration of exposure to the allergen is not sufficient to overcome the native immunologic milieu of the psoriasis plaque and induce the immunologic cascade resulting in ACD. Further research into the potentially antagonistic relationship of psoriasis and ACD should be performed to elucidate the interaction between these two common conditions.
- Kimber I, Basketter DA, Gerberick GF, et al. Allergic contact dermatitis. Int Immunopharmacol. 2002;2:201-211.
- Vocanson M, Hennino A, Cluzel-Tailhardat M, et al. CD8+ T cells are effector cells of contact dermatitis to common skin allergens in mice. J Invest Dermatol. 2006;126:815-820.
- Akiba H, Kehren J, Ducluzeau MT, et al. Skin inflammation during contact hypersensitivity is mediated by early recruitment of CD8+ T cytotoxic 1 cells inducing keratinocyte apoptosis. J Immunol. 2002;168:3079-3087.
- Trautmann A, Akdis M, Kleemann D, et al. T cell-mediated Fas-induced keratinocyte apoptosis plays a key pathogenetic role in eczematous dermatitis. J Clin Invest. 2000;106:25-35.
- Lynde CW, Poulin Y, Vender R, et al. Interleukin 17A: toward a new understanding of psoriasis pathogenesis. J Am Acad Dermatol. 2014;71:141-150.
- Bangsgaard N, Engkilde K, Thyssen JP, et al. Inverse relationship between contact allergy and psoriasis: results from a patient- and a population-based study. Br J Dermatol. 2009;161:1119-1123.
- Henseler T, Christophers E. Disease concomitance in psoriasis. J Am Acad Dermatol. 1995;32:982-986.
- Bangsgaard N, Zachariae C, Menne T, et al. Lack of effect of ustekinumab in treatment of allergic contact dermatitis. Contact Dermatitis. 2011;65:227-230.
- Quaranta M, Eyerich S, Knapp B, et al. Allergic contact dermatitis in psoriasis patients: typical, delayed, and non-interacting. PLoS One. 2014;9:e101814.
- Kimber I, Basketter DA, Gerberick GF, et al. Allergic contact dermatitis. Int Immunopharmacol. 2002;2:201-211.
- Kimber I, Basketter DA, Gerberick GF, et al. Allergic contact dermatitis. Int Immunopharmacol. 2002;2:201-211.
- Vocanson M, Hennino A, Cluzel-Tailhardat M, et al. CD8+ T cells are effector cells of contact dermatitis to common skin allergens in mice. J Invest Dermatol. 2006;126:815-820.
- Akiba H, Kehren J, Ducluzeau MT, et al. Skin inflammation during contact hypersensitivity is mediated by early recruitment of CD8+ T cytotoxic 1 cells inducing keratinocyte apoptosis. J Immunol. 2002;168:3079-3087.
- Trautmann A, Akdis M, Kleemann D, et al. T cell-mediated Fas-induced keratinocyte apoptosis plays a key pathogenetic role in eczematous dermatitis. J Clin Invest. 2000;106:25-35.
- Lynde CW, Poulin Y, Vender R, et al. Interleukin 17A: toward a new understanding of psoriasis pathogenesis. J Am Acad Dermatol. 2014;71:141-150.
- Bangsgaard N, Engkilde K, Thyssen JP, et al. Inverse relationship between contact allergy and psoriasis: results from a patient- and a population-based study. Br J Dermatol. 2009;161:1119-1123.
- Henseler T, Christophers E. Disease concomitance in psoriasis. J Am Acad Dermatol. 1995;32:982-986.
- Bangsgaard N, Zachariae C, Menne T, et al. Lack of effect of ustekinumab in treatment of allergic contact dermatitis. Contact Dermatitis. 2011;65:227-230.
- Quaranta M, Eyerich S, Knapp B, et al. Allergic contact dermatitis in psoriasis patients: typical, delayed, and non-interacting. PLoS One. 2014;9:e101814.
- Kimber I, Basketter DA, Gerberick GF, et al. Allergic contact dermatitis. Int Immunopharmacol. 2002;2:201-211.
Practice Points
- Patients with plaque-type psoriasis who experience allergic contact dermatitis (ACD) may present with sparing of exposed psoriatic plaques.
- The divergent immunologic milieus present in ACD and psoriasis likely underly the decreased incidence of ACD in patients with psoriasis.
Generalized Granuloma Annulare Responsive to Narrowband UVB
To the Editor:
Granuloma annulare (GA) is a common dermatosis that usually presents with dermal papules and annular plaques in a symmetric distribution.1 The etiology is unknown, but a delayed-type hypersensitivity reaction is the favored pathogenesis. Several systemic associations have been reported with generalized GA including diabetes mellitus, hyperlipidemia, autoimmune thyroiditis, rheumatoid arthritis, and lymphoproliferative malignancies, as well as other malignancies and viral infections such as human immunodeficiency virus and hepatitis C. Localized GA often is self-limiting, but generalized disease can be chronic and progressive. Although asymptomatic in most cases, the lesions can be cosmetically bothersome, and many patients desire treatment. There are few well-controlled studies of treatment, and most are limited to case reports and series. A review of GA treatment noted only 3 randomized studies: 2 relating to photodynamic therapy and 1 to cryosurgery. Well-accepted therapies, such as topical and intralesional corticosteroids, antimalarials, immunosuppressants, antibiotics, and phototherapy, are substantiated by lesser-quality evidence.1 Phototherapy has been studied for the treatment of GA and other disorders with altered dermal matrix deposition for which there are limited effective treatment options. UV irradiation promotes degradation of structural components of the dermis and inhibition of collagen production.2 Granuloma annulare generally is resistant to therapy. We report a case of generalized GA of long duration that responded well to phototherapy with narrowband UVB (NB-UVB).
A 60-year-old woman presented with generalized GA of 4 years’ duration that was confirmed on biopsy on 2 occasions (Figure 1). The lesions were asymptomatic but disfiguring and consisted of extensive pink, thin, annular plaques and papules on the torso, arms, and legs (Figure 2A). Apart from mild depression for which she was being treated with paroxetine and trazodone, she was otherwise healthy without evidence of thyroid disease, hyperlipidemia, or diabetes mellitus. Prior treatments for GA had included tapering courses of prednisone (up to 30 mg/d, tapered by 5 mg every 4 days) and betamethasone dipropionate cream 0.05%. She was started on NB-UVB therapy 5 times weekly in incremental doses with no adjuvant therapy. After 100 treatments, there was notable improvement with lesions becoming paler and flatter, with some involuting completely (Figure 2B). The frequency of treatment was reduced to 3 times weekly with continued improvement. An NB-UVB device was used containing 48 TL 100W/01-FS72 lamps with a mean irradiance of 2.9 mW/cm2. Her starting dose was 90 mJ/cm2. The cumulative dose after 100 treatments was 35,600 mJ/cm2. Apart from occasional mild erythema, there were no adverse effects.
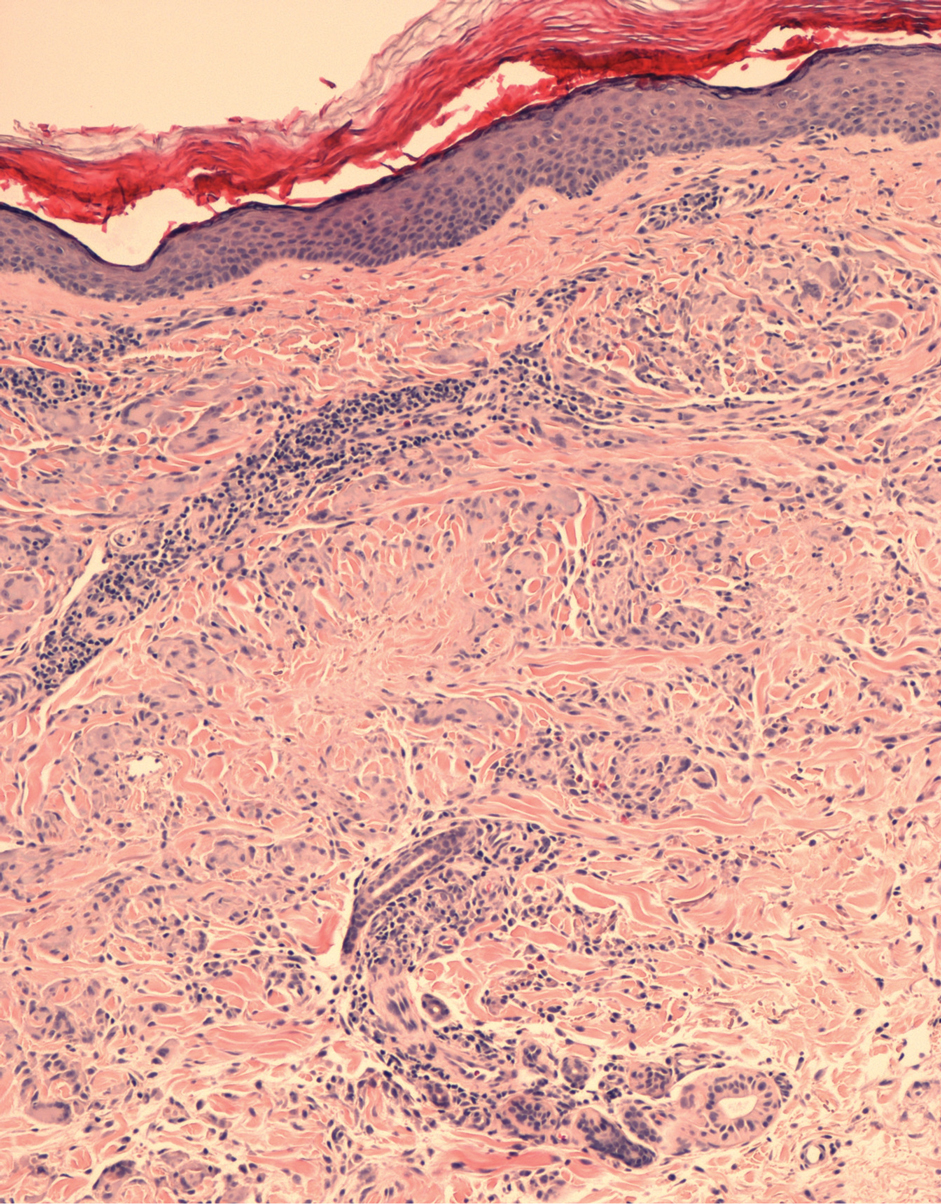

Inui et al3 described the successful treatment of generalized GA with NB-UVB. A retrospective review of NB-UVB for vitiligo, pruritus, and inflammatory dermatoses included 2 cases of generalized GA that were noted to have only a minimal to mild improvement.4 Most reports relating to phototherapy of GA have focused on psoralen plus UVA (PUVA). A retrospective study of 33 patients treated with systemic PUVA showed improvement in two-thirds of patients.5 Older studies showed systemic PUVA was effective in 1 patient after 53 treatments6 and in 4 patients using a high-dose protocol7; topical PUVA was effective in 4 patients after an average of 26 treatments.8 Psoralen plus UVA bath was reported as an effective treatment of generalized GA in a child.9 UVA1 phototherapy provided good or excellent results in half of patients (10/20) studied with generalized GA; however, discontinuation of treatment resulted in early recurrence of disease.10 In general, NB-UVB has been preferred over PUVA and UVA1 due to long-term safety, tolerability, and access. Although further clinical trials are needed, our report suggests that NB-UVB could be a useful modality in generalized GA.
- Thornsberry LA, English JC III. Etiology, diagnosis, and therapeutic management of granuloma annulare: an update. Am J Clin Dermatol. 2013;14:279-290.
- Fisher GJ, Wang ZQ, Datta SC, et al. Pathophysiology of premature skin aging induced by ultraviolet light. N Engl J Med. 1997;337:1419-1428.
- Inui S, Nishida Y, Itami S, et al. Disseminated granuloma annulare responsive to narrowband ultraviolet B therapy. J Am Acad Dermatol. 2005;53:532-533.
- Samson Yashar S, Gielczyk R, Scherschun L, et al. Narrow-band ultraviolet B treatment for vitiligo, pruritus, and inflammatory dermatoses. Photodermatol Photoimmunol Photomed. 2003;19:164-168.
- Browne F, Turner D, Goulden V. Psoralen and ultraviolet A in the treatment of granuloma annulare. Photodermatol Photoimmunol Photomed. 2011;27:81-84.
- Setterfield J, Huilgol SC, Black MM. Generalised granuloma annulare successfully treated with PUVA. Clin Exp Dermatol. 1999;24:458-460.
- Munchenberger S, Schopf E, Simon JC. Phototherapy with UVA-1 for generalized granuloma annulare. Arch Dermatol. 1997;133:1605.
- Grundmann-Kollmann M, Ochsendorf FR, Zollner TM, et al. Cream psoralen plus ultraviolet A therapy for granuloma annulare. Br J Dermatol. 2001;144:996-999.
- Batchelor R, Clark S. Clearance of generalized popular umbilicated granuloma annulare in a child with bath PUVA therapy. Pediatr Dermatol. 2006;23:72-74.
- Schnopp C, Tzaneva S, Mempel M, et al. UVA1 phototherapy for disseminated granuloma annulare. Photodermatol Photoimmunol Photomed. 2005;21:68-71.
To the Editor:
Granuloma annulare (GA) is a common dermatosis that usually presents with dermal papules and annular plaques in a symmetric distribution.1 The etiology is unknown, but a delayed-type hypersensitivity reaction is the favored pathogenesis. Several systemic associations have been reported with generalized GA including diabetes mellitus, hyperlipidemia, autoimmune thyroiditis, rheumatoid arthritis, and lymphoproliferative malignancies, as well as other malignancies and viral infections such as human immunodeficiency virus and hepatitis C. Localized GA often is self-limiting, but generalized disease can be chronic and progressive. Although asymptomatic in most cases, the lesions can be cosmetically bothersome, and many patients desire treatment. There are few well-controlled studies of treatment, and most are limited to case reports and series. A review of GA treatment noted only 3 randomized studies: 2 relating to photodynamic therapy and 1 to cryosurgery. Well-accepted therapies, such as topical and intralesional corticosteroids, antimalarials, immunosuppressants, antibiotics, and phototherapy, are substantiated by lesser-quality evidence.1 Phototherapy has been studied for the treatment of GA and other disorders with altered dermal matrix deposition for which there are limited effective treatment options. UV irradiation promotes degradation of structural components of the dermis and inhibition of collagen production.2 Granuloma annulare generally is resistant to therapy. We report a case of generalized GA of long duration that responded well to phototherapy with narrowband UVB (NB-UVB).
A 60-year-old woman presented with generalized GA of 4 years’ duration that was confirmed on biopsy on 2 occasions (Figure 1). The lesions were asymptomatic but disfiguring and consisted of extensive pink, thin, annular plaques and papules on the torso, arms, and legs (Figure 2A). Apart from mild depression for which she was being treated with paroxetine and trazodone, she was otherwise healthy without evidence of thyroid disease, hyperlipidemia, or diabetes mellitus. Prior treatments for GA had included tapering courses of prednisone (up to 30 mg/d, tapered by 5 mg every 4 days) and betamethasone dipropionate cream 0.05%. She was started on NB-UVB therapy 5 times weekly in incremental doses with no adjuvant therapy. After 100 treatments, there was notable improvement with lesions becoming paler and flatter, with some involuting completely (Figure 2B). The frequency of treatment was reduced to 3 times weekly with continued improvement. An NB-UVB device was used containing 48 TL 100W/01-FS72 lamps with a mean irradiance of 2.9 mW/cm2. Her starting dose was 90 mJ/cm2. The cumulative dose after 100 treatments was 35,600 mJ/cm2. Apart from occasional mild erythema, there were no adverse effects.
Inui et al3 described the successful treatment of generalized GA with NB-UVB. A retrospective review of NB-UVB for vitiligo, pruritus, and inflammatory dermatoses included 2 cases of generalized GA that were noted to have only a minimal to mild improvement.4 Most reports relating to phototherapy of GA have focused on psoralen plus UVA (PUVA). A retrospective study of 33 patients treated with systemic PUVA showed improvement in two-thirds of patients.5 Older studies showed systemic PUVA was effective in 1 patient after 53 treatments6 and in 4 patients using a high-dose protocol7; topical PUVA was effective in 4 patients after an average of 26 treatments.8 Psoralen plus UVA bath was reported as an effective treatment of generalized GA in a child.9 UVA1 phototherapy provided good or excellent results in half of patients (10/20) studied with generalized GA; however, discontinuation of treatment resulted in early recurrence of disease.10 In general, NB-UVB has been preferred over PUVA and UVA1 due to long-term safety, tolerability, and access. Although further clinical trials are needed, our report suggests that NB-UVB could be a useful modality in generalized GA.
To the Editor:
Granuloma annulare (GA) is a common dermatosis that usually presents with dermal papules and annular plaques in a symmetric distribution.1 The etiology is unknown, but a delayed-type hypersensitivity reaction is the favored pathogenesis. Several systemic associations have been reported with generalized GA including diabetes mellitus, hyperlipidemia, autoimmune thyroiditis, rheumatoid arthritis, and lymphoproliferative malignancies, as well as other malignancies and viral infections such as human immunodeficiency virus and hepatitis C. Localized GA often is self-limiting, but generalized disease can be chronic and progressive. Although asymptomatic in most cases, the lesions can be cosmetically bothersome, and many patients desire treatment. There are few well-controlled studies of treatment, and most are limited to case reports and series. A review of GA treatment noted only 3 randomized studies: 2 relating to photodynamic therapy and 1 to cryosurgery. Well-accepted therapies, such as topical and intralesional corticosteroids, antimalarials, immunosuppressants, antibiotics, and phototherapy, are substantiated by lesser-quality evidence.1 Phototherapy has been studied for the treatment of GA and other disorders with altered dermal matrix deposition for which there are limited effective treatment options. UV irradiation promotes degradation of structural components of the dermis and inhibition of collagen production.2 Granuloma annulare generally is resistant to therapy. We report a case of generalized GA of long duration that responded well to phototherapy with narrowband UVB (NB-UVB).
A 60-year-old woman presented with generalized GA of 4 years’ duration that was confirmed on biopsy on 2 occasions (Figure 1). The lesions were asymptomatic but disfiguring and consisted of extensive pink, thin, annular plaques and papules on the torso, arms, and legs (Figure 2A). Apart from mild depression for which she was being treated with paroxetine and trazodone, she was otherwise healthy without evidence of thyroid disease, hyperlipidemia, or diabetes mellitus. Prior treatments for GA had included tapering courses of prednisone (up to 30 mg/d, tapered by 5 mg every 4 days) and betamethasone dipropionate cream 0.05%. She was started on NB-UVB therapy 5 times weekly in incremental doses with no adjuvant therapy. After 100 treatments, there was notable improvement with lesions becoming paler and flatter, with some involuting completely (Figure 2B). The frequency of treatment was reduced to 3 times weekly with continued improvement. An NB-UVB device was used containing 48 TL 100W/01-FS72 lamps with a mean irradiance of 2.9 mW/cm2. Her starting dose was 90 mJ/cm2. The cumulative dose after 100 treatments was 35,600 mJ/cm2. Apart from occasional mild erythema, there were no adverse effects.
Inui et al3 described the successful treatment of generalized GA with NB-UVB. A retrospective review of NB-UVB for vitiligo, pruritus, and inflammatory dermatoses included 2 cases of generalized GA that were noted to have only a minimal to mild improvement.4 Most reports relating to phototherapy of GA have focused on psoralen plus UVA (PUVA). A retrospective study of 33 patients treated with systemic PUVA showed improvement in two-thirds of patients.5 Older studies showed systemic PUVA was effective in 1 patient after 53 treatments6 and in 4 patients using a high-dose protocol7; topical PUVA was effective in 4 patients after an average of 26 treatments.8 Psoralen plus UVA bath was reported as an effective treatment of generalized GA in a child.9 UVA1 phototherapy provided good or excellent results in half of patients (10/20) studied with generalized GA; however, discontinuation of treatment resulted in early recurrence of disease.10 In general, NB-UVB has been preferred over PUVA and UVA1 due to long-term safety, tolerability, and access. Although further clinical trials are needed, our report suggests that NB-UVB could be a useful modality in generalized GA.
- Thornsberry LA, English JC III. Etiology, diagnosis, and therapeutic management of granuloma annulare: an update. Am J Clin Dermatol. 2013;14:279-290.
- Fisher GJ, Wang ZQ, Datta SC, et al. Pathophysiology of premature skin aging induced by ultraviolet light. N Engl J Med. 1997;337:1419-1428.
- Inui S, Nishida Y, Itami S, et al. Disseminated granuloma annulare responsive to narrowband ultraviolet B therapy. J Am Acad Dermatol. 2005;53:532-533.
- Samson Yashar S, Gielczyk R, Scherschun L, et al. Narrow-band ultraviolet B treatment for vitiligo, pruritus, and inflammatory dermatoses. Photodermatol Photoimmunol Photomed. 2003;19:164-168.
- Browne F, Turner D, Goulden V. Psoralen and ultraviolet A in the treatment of granuloma annulare. Photodermatol Photoimmunol Photomed. 2011;27:81-84.
- Setterfield J, Huilgol SC, Black MM. Generalised granuloma annulare successfully treated with PUVA. Clin Exp Dermatol. 1999;24:458-460.
- Munchenberger S, Schopf E, Simon JC. Phototherapy with UVA-1 for generalized granuloma annulare. Arch Dermatol. 1997;133:1605.
- Grundmann-Kollmann M, Ochsendorf FR, Zollner TM, et al. Cream psoralen plus ultraviolet A therapy for granuloma annulare. Br J Dermatol. 2001;144:996-999.
- Batchelor R, Clark S. Clearance of generalized popular umbilicated granuloma annulare in a child with bath PUVA therapy. Pediatr Dermatol. 2006;23:72-74.
- Schnopp C, Tzaneva S, Mempel M, et al. UVA1 phototherapy for disseminated granuloma annulare. Photodermatol Photoimmunol Photomed. 2005;21:68-71.
- Thornsberry LA, English JC III. Etiology, diagnosis, and therapeutic management of granuloma annulare: an update. Am J Clin Dermatol. 2013;14:279-290.
- Fisher GJ, Wang ZQ, Datta SC, et al. Pathophysiology of premature skin aging induced by ultraviolet light. N Engl J Med. 1997;337:1419-1428.
- Inui S, Nishida Y, Itami S, et al. Disseminated granuloma annulare responsive to narrowband ultraviolet B therapy. J Am Acad Dermatol. 2005;53:532-533.
- Samson Yashar S, Gielczyk R, Scherschun L, et al. Narrow-band ultraviolet B treatment for vitiligo, pruritus, and inflammatory dermatoses. Photodermatol Photoimmunol Photomed. 2003;19:164-168.
- Browne F, Turner D, Goulden V. Psoralen and ultraviolet A in the treatment of granuloma annulare. Photodermatol Photoimmunol Photomed. 2011;27:81-84.
- Setterfield J, Huilgol SC, Black MM. Generalised granuloma annulare successfully treated with PUVA. Clin Exp Dermatol. 1999;24:458-460.
- Munchenberger S, Schopf E, Simon JC. Phototherapy with UVA-1 for generalized granuloma annulare. Arch Dermatol. 1997;133:1605.
- Grundmann-Kollmann M, Ochsendorf FR, Zollner TM, et al. Cream psoralen plus ultraviolet A therapy for granuloma annulare. Br J Dermatol. 2001;144:996-999.
- Batchelor R, Clark S. Clearance of generalized popular umbilicated granuloma annulare in a child with bath PUVA therapy. Pediatr Dermatol. 2006;23:72-74.
- Schnopp C, Tzaneva S, Mempel M, et al. UVA1 phototherapy for disseminated granuloma annulare. Photodermatol Photoimmunol Photomed. 2005;21:68-71.
Practice Points
- The generalized variant of granuloma annulare (GA) can be persistent, sometimes lasting years to decades; treatment is not always effective.
- The safety profile and tolerability of narrowband UVB phototherapy make it a suitable treatment option for generalized GA.
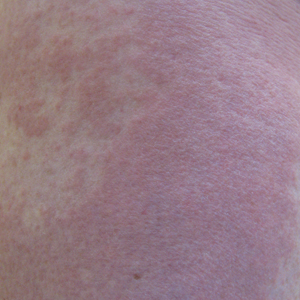
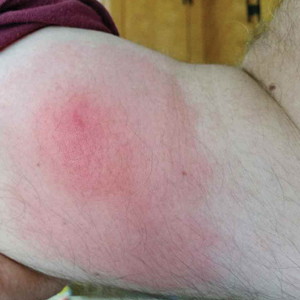
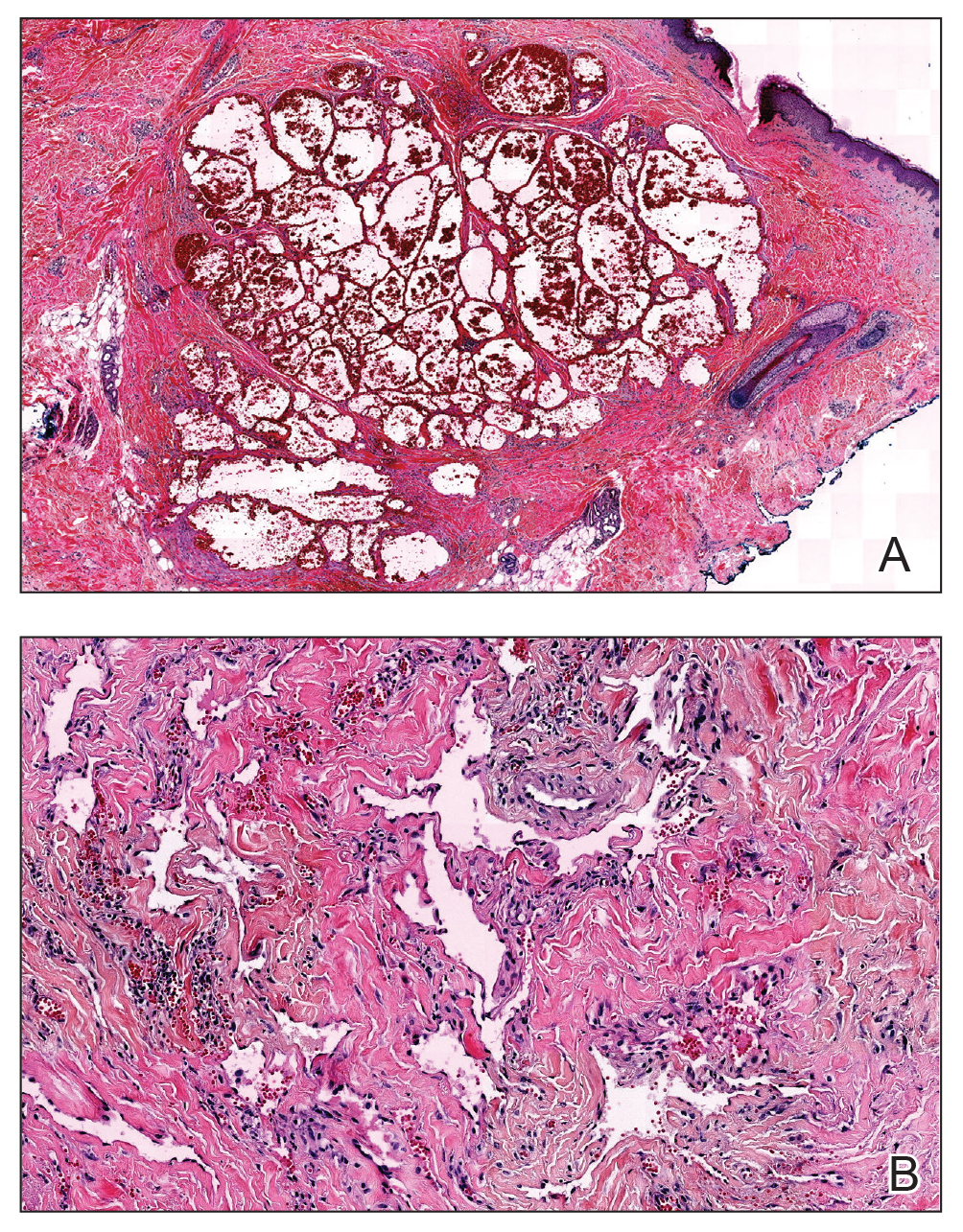
Recurrent Pruritic Multifocal Erythematous Rash
The Diagnosis: Wells Syndrome
Histopathologic examination of the biopsy demonstrated overlying acanthosis, focal spongiosis, and exocytosis. There also was proliferation and thickening of superficial capillaries and papillary fibrosis (Figure, A). There was a mixed interstitial and perivascular inflammatory infiltrate consisting of lymphocytes, histiocytes, plasma cells, and eosinophils (Figure, A and B). Occasional flame figures were identified (Figure, C).

Wells syndrome, also known as eosinophilic cellulitis, was first described in 1971 by Wells1 as a recurrent granulomatous dermatitis with eosinophilia. Rarely reported worldwide, this chronic relapsing condition is characterized by a pronounced eosinophilic infiltrate of the dermis resembling urticaria or cellulitis.2 The exact etiology has not been elucidated; however, links to certain medications, vaccines, exaggerated arthropod reactions, infections, and malignancies have been documented.3
Wells syndrome is a diagnosis of exclusion and lacks a predictable dermatologic presentation, thereby mandating focused clinical follow-up as well as correlation with histopathology findings. Although the classic histologic hallmark of Wells syndrome is scattered flame figures, this finding is not specific and can be found in other hypereosinophilic conditions.2 Clinical manifestations most often consist of 2 distinct phases: an initial painful burning or pruritic sensation, followed by the development of erythematous and edematous dermal plaques that may heal with slight hyperpigmentation over 4 to 8 weeks. A case series of 19 patients demonstrated variants of Wells syndrome, with an annular granuloma-like appearance found primarily in adults and the signature plaque-type appearance predominating in children.4
Acute urticaria is characterized by pruritic erythematous wheals secondary to a histamine-mediated response brought on by a variety of triggers, typically allergic and self-resolving within 24 hours. When such lesions last longer than 24 hours, biopsy should be performed to exclude urticarial vasculitis, which is characterized by a burning or painful sensation rather than pruritis, in addition to dermal neutrophilia and perivascular infiltrate on histology. Erythema migrans of Lyme disease begins at the site of a tick bite, evolving from a red macule to an expanding targetoid lesion and typically is not pruritic. Infectious cellulitis presents with warm, tender, and poorly defined erythematous patches; can progress rapidly; and is accompanied by systemic symptoms such as fevers, malaise, and lymphadenopathy.
Best evidence favors the use of moderate- to high-dose corticosteroids as first-line treatment.5 The use of tumor necrosis factor blockers, various immunomodulating agents, and combination therapy with levocetirizine and hydroxyzine have demonstrated variable levels of efficacy, albeit often followed by high rates of relapse with drug discontinuation.6
- Wells GC. Recurrent granulomatous dermatitis with eosinophilia. Trans St Johns Hosp Dermatol Soc. 1971;57:46-56.
- Aberer W, Konrad K, Wolff K. Wells' syndrome is a distinctive disease entity and not a histologic diagnosis. J Am Acad Dermatol. 1988;18:105-114.
- Kaufmann D, Pichler W, Beer JH. Severe episode of high fever with rash, lymphadenopathy, neutropenia, and eosinophilia after minocycline therapy for acne. Arch Intern Med. 1994;154:1983-1984.
- Caputo R, Marzano AV, Vezzoli P, et al. Wells syndrome in adults and children: a report of 19 cases. Arch Dermatol. 2006;142:1157-1161.
- Ferreli C, Pinna AL, Atzori L, et al. Eosinophilic cellulitis (Well's syndrome): a new case description. J Eur Acad Dermatol Venereol. 1999;13:41-45.
- Cormerais M, Poizeau F, Darrieux L, et al. Wells' syndrome mimicking facial cellulitis: a report of two cases. Case Rep Dermatol. 2015;7:117-122.
The Diagnosis: Wells Syndrome
Histopathologic examination of the biopsy demonstrated overlying acanthosis, focal spongiosis, and exocytosis. There also was proliferation and thickening of superficial capillaries and papillary fibrosis (Figure, A). There was a mixed interstitial and perivascular inflammatory infiltrate consisting of lymphocytes, histiocytes, plasma cells, and eosinophils (Figure, A and B). Occasional flame figures were identified (Figure, C).
Wells syndrome, also known as eosinophilic cellulitis, was first described in 1971 by Wells1 as a recurrent granulomatous dermatitis with eosinophilia. Rarely reported worldwide, this chronic relapsing condition is characterized by a pronounced eosinophilic infiltrate of the dermis resembling urticaria or cellulitis.2 The exact etiology has not been elucidated; however, links to certain medications, vaccines, exaggerated arthropod reactions, infections, and malignancies have been documented.3
Wells syndrome is a diagnosis of exclusion and lacks a predictable dermatologic presentation, thereby mandating focused clinical follow-up as well as correlation with histopathology findings. Although the classic histologic hallmark of Wells syndrome is scattered flame figures, this finding is not specific and can be found in other hypereosinophilic conditions.2 Clinical manifestations most often consist of 2 distinct phases: an initial painful burning or pruritic sensation, followed by the development of erythematous and edematous dermal plaques that may heal with slight hyperpigmentation over 4 to 8 weeks. A case series of 19 patients demonstrated variants of Wells syndrome, with an annular granuloma-like appearance found primarily in adults and the signature plaque-type appearance predominating in children.4
Acute urticaria is characterized by pruritic erythematous wheals secondary to a histamine-mediated response brought on by a variety of triggers, typically allergic and self-resolving within 24 hours. When such lesions last longer than 24 hours, biopsy should be performed to exclude urticarial vasculitis, which is characterized by a burning or painful sensation rather than pruritis, in addition to dermal neutrophilia and perivascular infiltrate on histology. Erythema migrans of Lyme disease begins at the site of a tick bite, evolving from a red macule to an expanding targetoid lesion and typically is not pruritic. Infectious cellulitis presents with warm, tender, and poorly defined erythematous patches; can progress rapidly; and is accompanied by systemic symptoms such as fevers, malaise, and lymphadenopathy.
Best evidence favors the use of moderate- to high-dose corticosteroids as first-line treatment.5 The use of tumor necrosis factor blockers, various immunomodulating agents, and combination therapy with levocetirizine and hydroxyzine have demonstrated variable levels of efficacy, albeit often followed by high rates of relapse with drug discontinuation.6
The Diagnosis: Wells Syndrome
Histopathologic examination of the biopsy demonstrated overlying acanthosis, focal spongiosis, and exocytosis. There also was proliferation and thickening of superficial capillaries and papillary fibrosis (Figure, A). There was a mixed interstitial and perivascular inflammatory infiltrate consisting of lymphocytes, histiocytes, plasma cells, and eosinophils (Figure, A and B). Occasional flame figures were identified (Figure, C).
Wells syndrome, also known as eosinophilic cellulitis, was first described in 1971 by Wells1 as a recurrent granulomatous dermatitis with eosinophilia. Rarely reported worldwide, this chronic relapsing condition is characterized by a pronounced eosinophilic infiltrate of the dermis resembling urticaria or cellulitis.2 The exact etiology has not been elucidated; however, links to certain medications, vaccines, exaggerated arthropod reactions, infections, and malignancies have been documented.3
Wells syndrome is a diagnosis of exclusion and lacks a predictable dermatologic presentation, thereby mandating focused clinical follow-up as well as correlation with histopathology findings. Although the classic histologic hallmark of Wells syndrome is scattered flame figures, this finding is not specific and can be found in other hypereosinophilic conditions.2 Clinical manifestations most often consist of 2 distinct phases: an initial painful burning or pruritic sensation, followed by the development of erythematous and edematous dermal plaques that may heal with slight hyperpigmentation over 4 to 8 weeks. A case series of 19 patients demonstrated variants of Wells syndrome, with an annular granuloma-like appearance found primarily in adults and the signature plaque-type appearance predominating in children.4
Acute urticaria is characterized by pruritic erythematous wheals secondary to a histamine-mediated response brought on by a variety of triggers, typically allergic and self-resolving within 24 hours. When such lesions last longer than 24 hours, biopsy should be performed to exclude urticarial vasculitis, which is characterized by a burning or painful sensation rather than pruritis, in addition to dermal neutrophilia and perivascular infiltrate on histology. Erythema migrans of Lyme disease begins at the site of a tick bite, evolving from a red macule to an expanding targetoid lesion and typically is not pruritic. Infectious cellulitis presents with warm, tender, and poorly defined erythematous patches; can progress rapidly; and is accompanied by systemic symptoms such as fevers, malaise, and lymphadenopathy.
Best evidence favors the use of moderate- to high-dose corticosteroids as first-line treatment.5 The use of tumor necrosis factor blockers, various immunomodulating agents, and combination therapy with levocetirizine and hydroxyzine have demonstrated variable levels of efficacy, albeit often followed by high rates of relapse with drug discontinuation.6
- Wells GC. Recurrent granulomatous dermatitis with eosinophilia. Trans St Johns Hosp Dermatol Soc. 1971;57:46-56.
- Aberer W, Konrad K, Wolff K. Wells' syndrome is a distinctive disease entity and not a histologic diagnosis. J Am Acad Dermatol. 1988;18:105-114.
- Kaufmann D, Pichler W, Beer JH. Severe episode of high fever with rash, lymphadenopathy, neutropenia, and eosinophilia after minocycline therapy for acne. Arch Intern Med. 1994;154:1983-1984.
- Caputo R, Marzano AV, Vezzoli P, et al. Wells syndrome in adults and children: a report of 19 cases. Arch Dermatol. 2006;142:1157-1161.
- Ferreli C, Pinna AL, Atzori L, et al. Eosinophilic cellulitis (Well's syndrome): a new case description. J Eur Acad Dermatol Venereol. 1999;13:41-45.
- Cormerais M, Poizeau F, Darrieux L, et al. Wells' syndrome mimicking facial cellulitis: a report of two cases. Case Rep Dermatol. 2015;7:117-122.
- Wells GC. Recurrent granulomatous dermatitis with eosinophilia. Trans St Johns Hosp Dermatol Soc. 1971;57:46-56.
- Aberer W, Konrad K, Wolff K. Wells' syndrome is a distinctive disease entity and not a histologic diagnosis. J Am Acad Dermatol. 1988;18:105-114.
- Kaufmann D, Pichler W, Beer JH. Severe episode of high fever with rash, lymphadenopathy, neutropenia, and eosinophilia after minocycline therapy for acne. Arch Intern Med. 1994;154:1983-1984.
- Caputo R, Marzano AV, Vezzoli P, et al. Wells syndrome in adults and children: a report of 19 cases. Arch Dermatol. 2006;142:1157-1161.
- Ferreli C, Pinna AL, Atzori L, et al. Eosinophilic cellulitis (Well's syndrome): a new case description. J Eur Acad Dermatol Venereol. 1999;13:41-45.
- Cormerais M, Poizeau F, Darrieux L, et al. Wells' syndrome mimicking facial cellulitis: a report of two cases. Case Rep Dermatol. 2015;7:117-122.
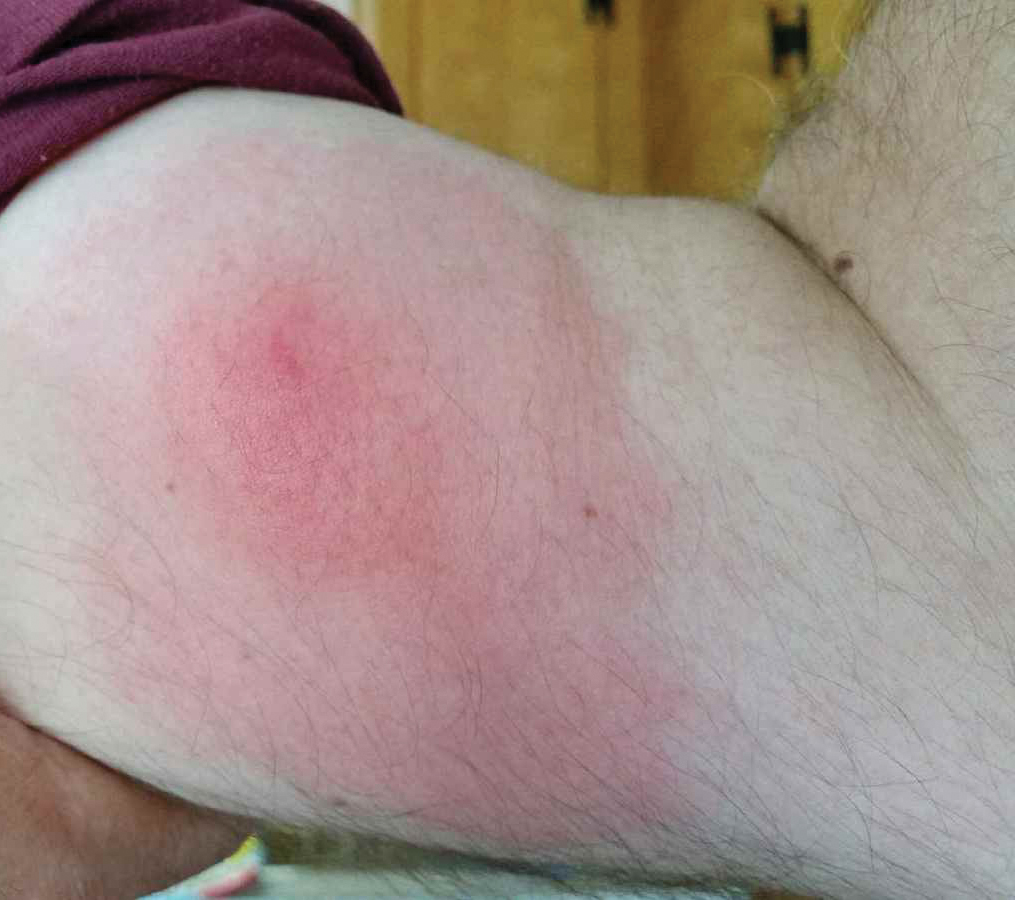
A 60-year-old man with a history of hyperlipidemia developed acute onset of an intensely pruritic and painful burning rash on the dorsal aspect of the left forearm of 8 days' duration. The patient described the rash as red and warm. It measured 2 cm at inception and peaked at 12 cm 6 months later when the patient presented. These symptoms resolved without therapeutic intervention.
Over the ensuing 6 months, he experienced 13 self-limited episodes of erythematous indurated cutaneous streaks, usually with proximal migration on the arms along with involvement of the posterior thorax and right leg. Five months prior to the onset of the initial rash, the patient had discontinued ezetimibe to treat hyperlipidemia due to swelling of the lips and tongue. He also reported that he regularly hunted in upstate Pennsylvania but reported no history of arthropod or animal bites. The patient did not take prescription or over-the-counter medications, and he denied the presence of fever, night sweats, fatigue, adenopathy, anorexia, weight loss, diarrhea, joint pain or swelling, or illicit drug use. Lyme titers, complete blood cell count, erythrocyte sedimentation rate, and comprehensive metabolic panel were within reference range. A punch biopsy was performed.
Multiple Atypical Vascular Lesions Following Breast-Conserving Surgery and Radiation
Atypical vascular lesions (AVLs) are rare flesh-colored, erythematous, or violaceous macules, patches, papules, or plaques that may occur following adjuvant radiation in breast cancer patients who have undergone conservative lumpectomy.1,2 They range in size from 1 mm to 6 cm and are most often confined to the radiation field. Presentation occurs 1 to 20 years following radiation, though the lesions most often present within 5 years.1,2 Although generally considered benign, 2 of 29 cases of AVLs progressed to angiosarcoma over a 5-year follow-up period in a retrospective clinicopathologic study.1
Atypical vascular lesions show considerable histologic and clinical overlap with radiation-induced angiosarcomas (RIAs), making differentiation between the two challenging.3,4 Mentzel et al5 compared benign, atypical, and malignant postradiation vascular lesions with nonradiation-associated angiosarcomas and found that RIAs were highly variable histopathologically, ranging from well differentiated to poorly differentiated, with atypia ranging from mild to severe. Radiation-induced angiosarcomas could be distinguished from AVLs and nonradiation-associated angiosarcomas by their oncogene amplification and protein expression profiles. Most strikingly, they found amplification of the MYC oncogene by fluorescence in situ hybridization in the nucleus of almost all the RIA cells, which was not seen in AVLs or nonradiation-associated angiosarcomas. Similarly, they found positive nuclear staining for MYC protein by immunohistochemistry in the nucleus of almost all cases of RIA but not in AVL or nonradiation-associated angiosarcomas, making MYC staining a useful diagnostic marker.5 In contrast, a study by Patton et al1 concluded that AVLs demonstrate morphologic patterns and clinical outcomes that suggest they are precursors of angiosarcoma rather than just markers of risk.
Atypical vascular lesions and RIAs usually follow a total radiation dose of 40 to 50 Gy, but RIAs typically are diagnosed later (approximately 10 years following exposure).6,7 Although RIAs are rare, they are known to be aggressive and often high grade, with a median survival of less than 5 years.6,7 Survival is poor even with radical surgical treatment.8 We present a patient with at least 29 AVLs following breast-conserving surgery and radiation and suggest the need for increased awareness of the elevated risk for RIA in patients with numerous benign AVLs.
Case Report
A 43-year-old woman with a history of breast cancer who underwent breast-conserving lumpectomy and adjuvant radiation presented to dermatology upon referral from surgical oncology for multiple lesions on the right breast (Figure 1). Seven years prior to presentation she was diagnosed with grade 3 poorly differentiated invasive ductal carcinoma with lobular features in the right breast that was positive for human epidermal growth factor receptor 2 but negative for estrogen or progesterone receptors. She was given neoadjuvant treatment with trastuzumab, docetaxel, and carboplatin prior to conservation lumpectomy with adjuvant radiation. She received a total dose of 50.4 Gy in 28 fractions of 1.8 Gy each over 1 month, with a final boost of 10 Gy in 5 fractions of 2 Gy, each with local skin irritation as the only concern posttreatment.

She initially presented to dermatology approximately 3 years after radiotherapy (5 years prior to current presentation) with lesions on the breast that had been present for 6 to 9 months. Physical examination showed 2 firm, painless, 4- to 5-mm papules on the right upper breast. The patient was reassured that the lesions were not suspicious for malignancy; however, 3 years later she presented to surgical oncology with 8 bluish papules or macules (all approximately 4 mm in diameter) on the right breast. These lesions were biopsied and examined by 2 institutions. Pathology of the initial punch biopsy favored a diagnosis of AVLs, though the possibility of RIA could not be ruled out without a complete excisional biopsy. Two excisional biopsies a month later were again consistent with AVLs. In all cases, the lesions were negative for MYC protein. The patient was again reassured but referred to dermatology for a second opinion.
At the current presentation, physical examination showed at least 29 subcutaneous nodules on the right breast ranging in color from pink to deep blue to flesh colored with others more superficially hyperpigmented, possibly secondary to prior biopsy, and measuring 2 to 8 mm in diameter. Histopathologic examination of the biopsy specimens showed a vascular proliferation extending from the dermis into the subcutaneous tissue comprised of dilated and cavernous vascular channels lined by a single layer of endothelial cells with minimal cytologic atypia (Figure 2). There were focal areas of anastomosing slitlike vascular spaces dissecting dermal collagen. No features of malignancy, such as nuclear crowding, multilayering, or increased mitotic activity, were evident. Immunohistochemical studies for MYC protein were negative. The overall morphologic features and immunoprofile were felt to be most consistent with postradiation AVLs.
At the time, surgical oncology felt that the risk of radical mastectomy outweighed the risk of angiosarcoma due to the absence of frank angiosarcoma and the patient’s notable comorbidities, including diabetes mellitus, cerebrovascular disease, peripheral vascular disease, and smoking; however, after reviewing the literature and considering the difficulty of following such a large number of lesions, the dermatology team brought the patient’s case to the multidisciplinary cutaneous tumor board at the University of Massachusetts (Worcester, Massachusetts). In consensus, the tumor board recommended radical mastectomy despite the comorbidities, given her young age and the potential risk for malignant transformation of any one of the numerous AVLs to angiosarcoma.
Postmastectomy pathology showed multiple scattered foci of AVLs ranging from 1.5 to 4 mm in the dermis, similar to those seen on prior biopsies, with no frank evidence of RIA. At 3-year follow-up, the patient has had no recurrence of AVLs or findings suggestive of RIA. There were no reported complications.
Comment
Conservative breast cancer surgery and radiotherapy are becoming more prevalent for breast cancer treatment, thus the number of patients likely to present with AVLs has increased. These patients are at risk for transformation to RIAs.6 It is important for clinicians to be aware of the diagnosis of both AVLs and RIAs and their management given their more frequent presentation. In most cases, one or a few AVLs are present, and excision is the treatment of choice. In a retrospective study by Brenn and Fletcher3 examining 16 patients with AVLs and 26 patients with RIA, the majority of cases of AVL had a single lesion and the maximum number of AVLs was 4. One patient in their study had 30 AVLs (each 3–4 mm in diameter), and she was diagnosed with RIA.3 Our patient—with at least 29 identifiable AVL lesions—was felt to be at considerable risk for developing RIA, as the only other case reported with this many AVLs developed RIA.1 Given the large number of lesions, it was neither feasible to excise each one individually nor monitor all of them for malignant transformation.
Our case demonstrates the important role dermatologists may play in orchestrating care by a multispecialty team including oncology, radiation oncology, surgery, and plastic surgery. In our patient, a close examination of the literature by the dermatology team led to recognition of the potentially elevated risk for malignant transformation. The dermatology team also brought the case for review at the tumor board.
Although future studies are required to determine the relationship between AVL burden and the risk for progression to RIA, it is clear that a multidisciplinary approach and careful consideration of the current literature can prevent unnecessary morbidity and mortality for patients with this increasingly common problem.
- Patton KT, Deyrup AT, Weiss SW. Atypical vascular lesions after surgery and radiation of the breast: a clinicopathologic study of 32 cases analyzing histologic heterogeneity and association with angiosarcoma. Am J Surg Pathol. 2008;32:943-950.
- Mandrell J, Mehta S, McClure S. Atypical vascular lesion of the breast. J Am Acad Dermatol. 2010;63:337-340.
- Brenn T, Fletcher CD. Radiation-associated cutaneous atypical vascular lesions and angiosarcoma: clinicopathologic analysis of 42 cases. Am J Surg Pathol. 2005;29:983-996.
- Losch A, Chilek KD, Zirwas MJ. Post-radiation atypical vascular proliferation mimicking angiosarcoma eight months following breast-conserving therapy for breast carcinoma. J Clin Aesthet Dermatol. 2011;4:47-48.
- Mentzel T, Schildhaus HU, Palmedo G, et al. Postradiation cutaneous angiosarcoma after treatment of breast carcinoma is characterized by MYC amplification in contrast to atypical vascular lesions after radiotherapy and control cases: clinicopathological, immunohistochemical and molecular analysis of 66 cases. Mod Pathol. 2012;25:75-85.
- Tahir M, Hendry P, Baird L, et al. Radiation induced angiosarcoma a sequela of radiotherapy for breast cancer following conservative surgery. Int Semin Surg Oncol. 2006;3:26.
- Hillenbrand T, Menge F, Hohenberger P, et al. Primary and secondary angiosarcomas: a comparative single-center analysis. Clin Sarcoma Res. 2015;5:14.
- Seinen JM, Styring E, Verstappen V, et al. Radiation-associated angiosarcoma after breast cancer: high recurrence rate and poor survival despite surgical treatment with R0 resection. Ann Surg Oncol. 2012;19:2700-2706.
Atypical vascular lesions (AVLs) are rare flesh-colored, erythematous, or violaceous macules, patches, papules, or plaques that may occur following adjuvant radiation in breast cancer patients who have undergone conservative lumpectomy.1,2 They range in size from 1 mm to 6 cm and are most often confined to the radiation field. Presentation occurs 1 to 20 years following radiation, though the lesions most often present within 5 years.1,2 Although generally considered benign, 2 of 29 cases of AVLs progressed to angiosarcoma over a 5-year follow-up period in a retrospective clinicopathologic study.1
Atypical vascular lesions show considerable histologic and clinical overlap with radiation-induced angiosarcomas (RIAs), making differentiation between the two challenging.3,4 Mentzel et al5 compared benign, atypical, and malignant postradiation vascular lesions with nonradiation-associated angiosarcomas and found that RIAs were highly variable histopathologically, ranging from well differentiated to poorly differentiated, with atypia ranging from mild to severe. Radiation-induced angiosarcomas could be distinguished from AVLs and nonradiation-associated angiosarcomas by their oncogene amplification and protein expression profiles. Most strikingly, they found amplification of the MYC oncogene by fluorescence in situ hybridization in the nucleus of almost all the RIA cells, which was not seen in AVLs or nonradiation-associated angiosarcomas. Similarly, they found positive nuclear staining for MYC protein by immunohistochemistry in the nucleus of almost all cases of RIA but not in AVL or nonradiation-associated angiosarcomas, making MYC staining a useful diagnostic marker.5 In contrast, a study by Patton et al1 concluded that AVLs demonstrate morphologic patterns and clinical outcomes that suggest they are precursors of angiosarcoma rather than just markers of risk.
Atypical vascular lesions and RIAs usually follow a total radiation dose of 40 to 50 Gy, but RIAs typically are diagnosed later (approximately 10 years following exposure).6,7 Although RIAs are rare, they are known to be aggressive and often high grade, with a median survival of less than 5 years.6,7 Survival is poor even with radical surgical treatment.8 We present a patient with at least 29 AVLs following breast-conserving surgery and radiation and suggest the need for increased awareness of the elevated risk for RIA in patients with numerous benign AVLs.
Case Report
A 43-year-old woman with a history of breast cancer who underwent breast-conserving lumpectomy and adjuvant radiation presented to dermatology upon referral from surgical oncology for multiple lesions on the right breast (Figure 1). Seven years prior to presentation she was diagnosed with grade 3 poorly differentiated invasive ductal carcinoma with lobular features in the right breast that was positive for human epidermal growth factor receptor 2 but negative for estrogen or progesterone receptors. She was given neoadjuvant treatment with trastuzumab, docetaxel, and carboplatin prior to conservation lumpectomy with adjuvant radiation. She received a total dose of 50.4 Gy in 28 fractions of 1.8 Gy each over 1 month, with a final boost of 10 Gy in 5 fractions of 2 Gy, each with local skin irritation as the only concern posttreatment.
She initially presented to dermatology approximately 3 years after radiotherapy (5 years prior to current presentation) with lesions on the breast that had been present for 6 to 9 months. Physical examination showed 2 firm, painless, 4- to 5-mm papules on the right upper breast. The patient was reassured that the lesions were not suspicious for malignancy; however, 3 years later she presented to surgical oncology with 8 bluish papules or macules (all approximately 4 mm in diameter) on the right breast. These lesions were biopsied and examined by 2 institutions. Pathology of the initial punch biopsy favored a diagnosis of AVLs, though the possibility of RIA could not be ruled out without a complete excisional biopsy. Two excisional biopsies a month later were again consistent with AVLs. In all cases, the lesions were negative for MYC protein. The patient was again reassured but referred to dermatology for a second opinion.
At the current presentation, physical examination showed at least 29 subcutaneous nodules on the right breast ranging in color from pink to deep blue to flesh colored with others more superficially hyperpigmented, possibly secondary to prior biopsy, and measuring 2 to 8 mm in diameter. Histopathologic examination of the biopsy specimens showed a vascular proliferation extending from the dermis into the subcutaneous tissue comprised of dilated and cavernous vascular channels lined by a single layer of endothelial cells with minimal cytologic atypia (Figure 2). There were focal areas of anastomosing slitlike vascular spaces dissecting dermal collagen. No features of malignancy, such as nuclear crowding, multilayering, or increased mitotic activity, were evident. Immunohistochemical studies for MYC protein were negative. The overall morphologic features and immunoprofile were felt to be most consistent with postradiation AVLs.
At the time, surgical oncology felt that the risk of radical mastectomy outweighed the risk of angiosarcoma due to the absence of frank angiosarcoma and the patient’s notable comorbidities, including diabetes mellitus, cerebrovascular disease, peripheral vascular disease, and smoking; however, after reviewing the literature and considering the difficulty of following such a large number of lesions, the dermatology team brought the patient’s case to the multidisciplinary cutaneous tumor board at the University of Massachusetts (Worcester, Massachusetts). In consensus, the tumor board recommended radical mastectomy despite the comorbidities, given her young age and the potential risk for malignant transformation of any one of the numerous AVLs to angiosarcoma.
Postmastectomy pathology showed multiple scattered foci of AVLs ranging from 1.5 to 4 mm in the dermis, similar to those seen on prior biopsies, with no frank evidence of RIA. At 3-year follow-up, the patient has had no recurrence of AVLs or findings suggestive of RIA. There were no reported complications.
Comment
Conservative breast cancer surgery and radiotherapy are becoming more prevalent for breast cancer treatment, thus the number of patients likely to present with AVLs has increased. These patients are at risk for transformation to RIAs.6 It is important for clinicians to be aware of the diagnosis of both AVLs and RIAs and their management given their more frequent presentation. In most cases, one or a few AVLs are present, and excision is the treatment of choice. In a retrospective study by Brenn and Fletcher3 examining 16 patients with AVLs and 26 patients with RIA, the majority of cases of AVL had a single lesion and the maximum number of AVLs was 4. One patient in their study had 30 AVLs (each 3–4 mm in diameter), and she was diagnosed with RIA.3 Our patient—with at least 29 identifiable AVL lesions—was felt to be at considerable risk for developing RIA, as the only other case reported with this many AVLs developed RIA.1 Given the large number of lesions, it was neither feasible to excise each one individually nor monitor all of them for malignant transformation.
Our case demonstrates the important role dermatologists may play in orchestrating care by a multispecialty team including oncology, radiation oncology, surgery, and plastic surgery. In our patient, a close examination of the literature by the dermatology team led to recognition of the potentially elevated risk for malignant transformation. The dermatology team also brought the case for review at the tumor board.
Although future studies are required to determine the relationship between AVL burden and the risk for progression to RIA, it is clear that a multidisciplinary approach and careful consideration of the current literature can prevent unnecessary morbidity and mortality for patients with this increasingly common problem.
Atypical vascular lesions (AVLs) are rare flesh-colored, erythematous, or violaceous macules, patches, papules, or plaques that may occur following adjuvant radiation in breast cancer patients who have undergone conservative lumpectomy.1,2 They range in size from 1 mm to 6 cm and are most often confined to the radiation field. Presentation occurs 1 to 20 years following radiation, though the lesions most often present within 5 years.1,2 Although generally considered benign, 2 of 29 cases of AVLs progressed to angiosarcoma over a 5-year follow-up period in a retrospective clinicopathologic study.1
Atypical vascular lesions show considerable histologic and clinical overlap with radiation-induced angiosarcomas (RIAs), making differentiation between the two challenging.3,4 Mentzel et al5 compared benign, atypical, and malignant postradiation vascular lesions with nonradiation-associated angiosarcomas and found that RIAs were highly variable histopathologically, ranging from well differentiated to poorly differentiated, with atypia ranging from mild to severe. Radiation-induced angiosarcomas could be distinguished from AVLs and nonradiation-associated angiosarcomas by their oncogene amplification and protein expression profiles. Most strikingly, they found amplification of the MYC oncogene by fluorescence in situ hybridization in the nucleus of almost all the RIA cells, which was not seen in AVLs or nonradiation-associated angiosarcomas. Similarly, they found positive nuclear staining for MYC protein by immunohistochemistry in the nucleus of almost all cases of RIA but not in AVL or nonradiation-associated angiosarcomas, making MYC staining a useful diagnostic marker.5 In contrast, a study by Patton et al1 concluded that AVLs demonstrate morphologic patterns and clinical outcomes that suggest they are precursors of angiosarcoma rather than just markers of risk.
Atypical vascular lesions and RIAs usually follow a total radiation dose of 40 to 50 Gy, but RIAs typically are diagnosed later (approximately 10 years following exposure).6,7 Although RIAs are rare, they are known to be aggressive and often high grade, with a median survival of less than 5 years.6,7 Survival is poor even with radical surgical treatment.8 We present a patient with at least 29 AVLs following breast-conserving surgery and radiation and suggest the need for increased awareness of the elevated risk for RIA in patients with numerous benign AVLs.
Case Report
A 43-year-old woman with a history of breast cancer who underwent breast-conserving lumpectomy and adjuvant radiation presented to dermatology upon referral from surgical oncology for multiple lesions on the right breast (Figure 1). Seven years prior to presentation she was diagnosed with grade 3 poorly differentiated invasive ductal carcinoma with lobular features in the right breast that was positive for human epidermal growth factor receptor 2 but negative for estrogen or progesterone receptors. She was given neoadjuvant treatment with trastuzumab, docetaxel, and carboplatin prior to conservation lumpectomy with adjuvant radiation. She received a total dose of 50.4 Gy in 28 fractions of 1.8 Gy each over 1 month, with a final boost of 10 Gy in 5 fractions of 2 Gy, each with local skin irritation as the only concern posttreatment.
She initially presented to dermatology approximately 3 years after radiotherapy (5 years prior to current presentation) with lesions on the breast that had been present for 6 to 9 months. Physical examination showed 2 firm, painless, 4- to 5-mm papules on the right upper breast. The patient was reassured that the lesions were not suspicious for malignancy; however, 3 years later she presented to surgical oncology with 8 bluish papules or macules (all approximately 4 mm in diameter) on the right breast. These lesions were biopsied and examined by 2 institutions. Pathology of the initial punch biopsy favored a diagnosis of AVLs, though the possibility of RIA could not be ruled out without a complete excisional biopsy. Two excisional biopsies a month later were again consistent with AVLs. In all cases, the lesions were negative for MYC protein. The patient was again reassured but referred to dermatology for a second opinion.
At the current presentation, physical examination showed at least 29 subcutaneous nodules on the right breast ranging in color from pink to deep blue to flesh colored with others more superficially hyperpigmented, possibly secondary to prior biopsy, and measuring 2 to 8 mm in diameter. Histopathologic examination of the biopsy specimens showed a vascular proliferation extending from the dermis into the subcutaneous tissue comprised of dilated and cavernous vascular channels lined by a single layer of endothelial cells with minimal cytologic atypia (Figure 2). There were focal areas of anastomosing slitlike vascular spaces dissecting dermal collagen. No features of malignancy, such as nuclear crowding, multilayering, or increased mitotic activity, were evident. Immunohistochemical studies for MYC protein were negative. The overall morphologic features and immunoprofile were felt to be most consistent with postradiation AVLs.
At the time, surgical oncology felt that the risk of radical mastectomy outweighed the risk of angiosarcoma due to the absence of frank angiosarcoma and the patient’s notable comorbidities, including diabetes mellitus, cerebrovascular disease, peripheral vascular disease, and smoking; however, after reviewing the literature and considering the difficulty of following such a large number of lesions, the dermatology team brought the patient’s case to the multidisciplinary cutaneous tumor board at the University of Massachusetts (Worcester, Massachusetts). In consensus, the tumor board recommended radical mastectomy despite the comorbidities, given her young age and the potential risk for malignant transformation of any one of the numerous AVLs to angiosarcoma.
Postmastectomy pathology showed multiple scattered foci of AVLs ranging from 1.5 to 4 mm in the dermis, similar to those seen on prior biopsies, with no frank evidence of RIA. At 3-year follow-up, the patient has had no recurrence of AVLs or findings suggestive of RIA. There were no reported complications.
Comment
Conservative breast cancer surgery and radiotherapy are becoming more prevalent for breast cancer treatment, thus the number of patients likely to present with AVLs has increased. These patients are at risk for transformation to RIAs.6 It is important for clinicians to be aware of the diagnosis of both AVLs and RIAs and their management given their more frequent presentation. In most cases, one or a few AVLs are present, and excision is the treatment of choice. In a retrospective study by Brenn and Fletcher3 examining 16 patients with AVLs and 26 patients with RIA, the majority of cases of AVL had a single lesion and the maximum number of AVLs was 4. One patient in their study had 30 AVLs (each 3–4 mm in diameter), and she was diagnosed with RIA.3 Our patient—with at least 29 identifiable AVL lesions—was felt to be at considerable risk for developing RIA, as the only other case reported with this many AVLs developed RIA.1 Given the large number of lesions, it was neither feasible to excise each one individually nor monitor all of them for malignant transformation.
Our case demonstrates the important role dermatologists may play in orchestrating care by a multispecialty team including oncology, radiation oncology, surgery, and plastic surgery. In our patient, a close examination of the literature by the dermatology team led to recognition of the potentially elevated risk for malignant transformation. The dermatology team also brought the case for review at the tumor board.
Although future studies are required to determine the relationship between AVL burden and the risk for progression to RIA, it is clear that a multidisciplinary approach and careful consideration of the current literature can prevent unnecessary morbidity and mortality for patients with this increasingly common problem.
- Patton KT, Deyrup AT, Weiss SW. Atypical vascular lesions after surgery and radiation of the breast: a clinicopathologic study of 32 cases analyzing histologic heterogeneity and association with angiosarcoma. Am J Surg Pathol. 2008;32:943-950.
- Mandrell J, Mehta S, McClure S. Atypical vascular lesion of the breast. J Am Acad Dermatol. 2010;63:337-340.
- Brenn T, Fletcher CD. Radiation-associated cutaneous atypical vascular lesions and angiosarcoma: clinicopathologic analysis of 42 cases. Am J Surg Pathol. 2005;29:983-996.
- Losch A, Chilek KD, Zirwas MJ. Post-radiation atypical vascular proliferation mimicking angiosarcoma eight months following breast-conserving therapy for breast carcinoma. J Clin Aesthet Dermatol. 2011;4:47-48.
- Mentzel T, Schildhaus HU, Palmedo G, et al. Postradiation cutaneous angiosarcoma after treatment of breast carcinoma is characterized by MYC amplification in contrast to atypical vascular lesions after radiotherapy and control cases: clinicopathological, immunohistochemical and molecular analysis of 66 cases. Mod Pathol. 2012;25:75-85.
- Tahir M, Hendry P, Baird L, et al. Radiation induced angiosarcoma a sequela of radiotherapy for breast cancer following conservative surgery. Int Semin Surg Oncol. 2006;3:26.
- Hillenbrand T, Menge F, Hohenberger P, et al. Primary and secondary angiosarcomas: a comparative single-center analysis. Clin Sarcoma Res. 2015;5:14.
- Seinen JM, Styring E, Verstappen V, et al. Radiation-associated angiosarcoma after breast cancer: high recurrence rate and poor survival despite surgical treatment with R0 resection. Ann Surg Oncol. 2012;19:2700-2706.
- Patton KT, Deyrup AT, Weiss SW. Atypical vascular lesions after surgery and radiation of the breast: a clinicopathologic study of 32 cases analyzing histologic heterogeneity and association with angiosarcoma. Am J Surg Pathol. 2008;32:943-950.
- Mandrell J, Mehta S, McClure S. Atypical vascular lesion of the breast. J Am Acad Dermatol. 2010;63:337-340.
- Brenn T, Fletcher CD. Radiation-associated cutaneous atypical vascular lesions and angiosarcoma: clinicopathologic analysis of 42 cases. Am J Surg Pathol. 2005;29:983-996.
- Losch A, Chilek KD, Zirwas MJ. Post-radiation atypical vascular proliferation mimicking angiosarcoma eight months following breast-conserving therapy for breast carcinoma. J Clin Aesthet Dermatol. 2011;4:47-48.
- Mentzel T, Schildhaus HU, Palmedo G, et al. Postradiation cutaneous angiosarcoma after treatment of breast carcinoma is characterized by MYC amplification in contrast to atypical vascular lesions after radiotherapy and control cases: clinicopathological, immunohistochemical and molecular analysis of 66 cases. Mod Pathol. 2012;25:75-85.
- Tahir M, Hendry P, Baird L, et al. Radiation induced angiosarcoma a sequela of radiotherapy for breast cancer following conservative surgery. Int Semin Surg Oncol. 2006;3:26.
- Hillenbrand T, Menge F, Hohenberger P, et al. Primary and secondary angiosarcomas: a comparative single-center analysis. Clin Sarcoma Res. 2015;5:14.
- Seinen JM, Styring E, Verstappen V, et al. Radiation-associated angiosarcoma after breast cancer: high recurrence rate and poor survival despite surgical treatment with R0 resection. Ann Surg Oncol. 2012;19:2700-2706.
Practice Points
- Atypical vascular lesions (AVLs) of the breast have been reported in breast cancer patients following radiation treatment.
- Conservative breast cancer surgery and radiotherapy are becoming more prevalent for breast cancer treatment, thus the number of patients likely to present with AVLs has increased.
- Differentiation between AVLs and radiation-induced angiosarcomas (RIAs) can be challenging due to considerable histologic and clinical overlap; therefore, it is important for clinicians to be aware of the diagnosis and management of both AVLs and RIAs.
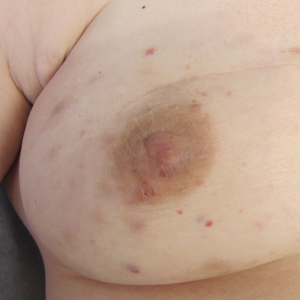
Rapidly Growing Cutaneous Nodules on the Scalp
The Diagnosis: B-Cell Acute Lymphoblastic Leukemia
A 4-mm punch biopsy of one of the scalp lesions showed a diffuse infiltrate of intermediately sized cells with variably mature chromatin and irregular nuclear contours, consistent with a neoplastic process. Numerous mitotic figures were present, indicating a high proliferation rate (Figure 1). At that time there was no evidence of systemic involvement. A repeat biopsy with concurrent bone marrow biopsy was scheduled 10 days after the patient's initial presentation for further classification. Laboratory studies at that time revealed leukocytosis with elevated neutrophils and lymphocytes as well as a high absolute blast count.
On immunohistochemical staining, the neoplastic cells were positive for CD45, which indicated the neoplasm was hematopoietic, as well as CD10 and the B-cell antigens PAX-5 and CD79a. The cells were negative for CD20, which also is a B-cell marker, but this marker is only expressed in approximately half of pediatric acute lymphoblastic leukemia (ALL) cases with B-cell precursor origin.1 Markers that typically are expressed in B-cell acute lymphoblastic leukemia (B-ALL)--CD34 and terminal deoxynucleotidyl transferase--were both negative. These results were somewhat contradictory, and the differential remained open to both B-ALL and mature B-cell lymphoma. A bone marrow biopsy showed approximately 65% blasts or leukemic cells (Figure 2). Flow cytometry showed the cells were positive for CD10, CD19, weak CD79a, and variable lambda surface antigen expression. The cells were negative for expression of CD20, CD34, terminal deoxynucleotidyl transferase, myeloid antigens, and CD3. Ultimately, the morphology and immunophenotype were most consistent with a diagnosis of B-ALL. Fluorescence in situ hybridization revealed mixed lineage leukemia, MLL, gene rearrangements. 
In general, when considering the differential diagnosis of superficial nodules, 5 elements are helpful to consider: the number of nodules (single vs multiple); the location; and the presence or absence of tenderness, pigmentation or erythema, and firmness.2 Our patient had multiple nodules on the scalp, which were erythematous to slightly purple and firm. The differential diagnosis can be categorized into malignant; infectious; and benign inflammatory, vascular, and fibrous tumors.
Potential oncologic processes include leukemia cutis, lymphoblastic leukemia/lymphoma, Langerhans cell histiocytosis, and rhabdomyosarcoma. Initial laboratory test results were reassuring. Infectious processes in the differential include deep fungal infections such as coccidioidomycosis and nontuberculous mycobacterial infections. Coccidioidomycosis was the most likely to cause skin lesions or masses in our patient; however, it was considered less likely because the patient's family had not traveled or been exposed to an endemic area.3
Benign tumors in the differential include deep hemangioma, which was deemed less likely in our patient because most hemangiomas reach 80% of their maximum size by 5 months of age.4 Another possible benign tumor is infantile myofibromatosis, which is rare but is the most common fibrous tumor of infancy.5
Early-onset childhood sarcoidosis also has been shown to produce multiple nontender firm nodules.2 This process was considered unlikely in our patient because not only is the disease relatively rare in the pediatric population, but most reported childhood cases have occurred in patients aged 13 to 15 years.6 Additionally, no uveitis or arthritis was observed in this case.
Ultimately, histopathology and bone marrow biopsy were necessary to determine the diagnosis of B-ALL. Although uncommon, cutaneous involvement can be an early sign of ALL in children.7 Thus, neoplastic etiologies should be considered in the workup of cutaneous nodules in children, especially when these nodules are hard, rapidly growing, ulcerated, fixed, and/or vascular.8 Once the diagnosis is established, initial workup of ALL in children should include complete blood cell count with manual differential, prothrombin time, partial thromboplastin time, electrolytes, uric acid, and renal and liver function tests. Often, baseline viral titers such as cytomegalovirus, Epstein-Barr virus, human immunodeficiency virus, hepatitis B virus, and varicella-zoster virus also are included. Patients are risk stratified to the appropriate level of treatment based on tumor immunophenotype, cytogenetic findings, patient age, white blood cell count at the time of diagnosis, and response to initial therapy. Treatment typically is comprised of a multidrug regimen divided into several phases--induction, consolidation, and maintenance--as well as therapy directed to the central nervous system. Treatment protocols usually take 2 to 3 years to complete.
Our patient was treated with 1 dose of intrathecal methotrexate before starting the Interfant-06 protocol with a 7-day methylprednisolone prophase. The patient's nodules shrank over time and were no longer present after 14 days of treatment.
- Dworzak MN, Schumich A, Printz D, et al. CD20 up-regulation in pediatric B-cell precursor acute lymphoblastic leukemia during induction treatment: setting the stage for anti-CD20 directed immunotherapy. Blood. 2008;112:3982-3988.
- Whelan JP, Zembowicz A. Case records of the Massachusetts General Hospital. case 19-2006. a 22-month-old boy with the rapid growth of subcutaneous nodules. N Engl J Med. 2006;354:2697-2704.
- Malo J, Luraschi-Monjagatta C, Wolk DM, et al. Update on the diagnosis of pulmonary coccidioidomycosis. Ann Am Thorac Soc. 2014;11:243-253.
- Chang LC, Haggstrom AN, Drolet BA, et al. Growth characteristics of infantile hemangiomas: implications for management. Pediatrics. 2008;122:360-367.
- Schurr P, Moulsdale W. Infantile myofibroma. Adv Neonatal Care. 2008;8:13-20.
- Shetty AK, Gedalia A. Childhood sarcoidosis: a rare but fascinating disorder. Pediatr Rheumatol Online J. 2008;6:16.
- Millot F, Robert A, Bertrand Y, et al. Cutaneous involvement in children with acute lymphoblastic leukemia or lymphoblastic lymphoma. The Children's Leukemia Cooperative Group of the European Organization of Research and Treatment of Cancer (EORTC). Pediatrics. 1997;100:60-64.
- Fogelson S, Dohil M. Papular and nodular skin lesions in children. Semin Plast Surg. 2006;20:180-191.
The Diagnosis: B-Cell Acute Lymphoblastic Leukemia
A 4-mm punch biopsy of one of the scalp lesions showed a diffuse infiltrate of intermediately sized cells with variably mature chromatin and irregular nuclear contours, consistent with a neoplastic process. Numerous mitotic figures were present, indicating a high proliferation rate (Figure 1). At that time there was no evidence of systemic involvement. A repeat biopsy with concurrent bone marrow biopsy was scheduled 10 days after the patient's initial presentation for further classification. Laboratory studies at that time revealed leukocytosis with elevated neutrophils and lymphocytes as well as a high absolute blast count.
On immunohistochemical staining, the neoplastic cells were positive for CD45, which indicated the neoplasm was hematopoietic, as well as CD10 and the B-cell antigens PAX-5 and CD79a. The cells were negative for CD20, which also is a B-cell marker, but this marker is only expressed in approximately half of pediatric acute lymphoblastic leukemia (ALL) cases with B-cell precursor origin.1 Markers that typically are expressed in B-cell acute lymphoblastic leukemia (B-ALL)--CD34 and terminal deoxynucleotidyl transferase--were both negative. These results were somewhat contradictory, and the differential remained open to both B-ALL and mature B-cell lymphoma. A bone marrow biopsy showed approximately 65% blasts or leukemic cells (Figure 2). Flow cytometry showed the cells were positive for CD10, CD19, weak CD79a, and variable lambda surface antigen expression. The cells were negative for expression of CD20, CD34, terminal deoxynucleotidyl transferase, myeloid antigens, and CD3. Ultimately, the morphology and immunophenotype were most consistent with a diagnosis of B-ALL. Fluorescence in situ hybridization revealed mixed lineage leukemia, MLL, gene rearrangements.
In general, when considering the differential diagnosis of superficial nodules, 5 elements are helpful to consider: the number of nodules (single vs multiple); the location; and the presence or absence of tenderness, pigmentation or erythema, and firmness.2 Our patient had multiple nodules on the scalp, which were erythematous to slightly purple and firm. The differential diagnosis can be categorized into malignant; infectious; and benign inflammatory, vascular, and fibrous tumors.
Potential oncologic processes include leukemia cutis, lymphoblastic leukemia/lymphoma, Langerhans cell histiocytosis, and rhabdomyosarcoma. Initial laboratory test results were reassuring. Infectious processes in the differential include deep fungal infections such as coccidioidomycosis and nontuberculous mycobacterial infections. Coccidioidomycosis was the most likely to cause skin lesions or masses in our patient; however, it was considered less likely because the patient's family had not traveled or been exposed to an endemic area.3
Benign tumors in the differential include deep hemangioma, which was deemed less likely in our patient because most hemangiomas reach 80% of their maximum size by 5 months of age.4 Another possible benign tumor is infantile myofibromatosis, which is rare but is the most common fibrous tumor of infancy.5
Early-onset childhood sarcoidosis also has been shown to produce multiple nontender firm nodules.2 This process was considered unlikely in our patient because not only is the disease relatively rare in the pediatric population, but most reported childhood cases have occurred in patients aged 13 to 15 years.6 Additionally, no uveitis or arthritis was observed in this case.
Ultimately, histopathology and bone marrow biopsy were necessary to determine the diagnosis of B-ALL. Although uncommon, cutaneous involvement can be an early sign of ALL in children.7 Thus, neoplastic etiologies should be considered in the workup of cutaneous nodules in children, especially when these nodules are hard, rapidly growing, ulcerated, fixed, and/or vascular.8 Once the diagnosis is established, initial workup of ALL in children should include complete blood cell count with manual differential, prothrombin time, partial thromboplastin time, electrolytes, uric acid, and renal and liver function tests. Often, baseline viral titers such as cytomegalovirus, Epstein-Barr virus, human immunodeficiency virus, hepatitis B virus, and varicella-zoster virus also are included. Patients are risk stratified to the appropriate level of treatment based on tumor immunophenotype, cytogenetic findings, patient age, white blood cell count at the time of diagnosis, and response to initial therapy. Treatment typically is comprised of a multidrug regimen divided into several phases--induction, consolidation, and maintenance--as well as therapy directed to the central nervous system. Treatment protocols usually take 2 to 3 years to complete.
Our patient was treated with 1 dose of intrathecal methotrexate before starting the Interfant-06 protocol with a 7-day methylprednisolone prophase. The patient's nodules shrank over time and were no longer present after 14 days of treatment.
The Diagnosis: B-Cell Acute Lymphoblastic Leukemia
A 4-mm punch biopsy of one of the scalp lesions showed a diffuse infiltrate of intermediately sized cells with variably mature chromatin and irregular nuclear contours, consistent with a neoplastic process. Numerous mitotic figures were present, indicating a high proliferation rate (Figure 1). At that time there was no evidence of systemic involvement. A repeat biopsy with concurrent bone marrow biopsy was scheduled 10 days after the patient's initial presentation for further classification. Laboratory studies at that time revealed leukocytosis with elevated neutrophils and lymphocytes as well as a high absolute blast count.
On immunohistochemical staining, the neoplastic cells were positive for CD45, which indicated the neoplasm was hematopoietic, as well as CD10 and the B-cell antigens PAX-5 and CD79a. The cells were negative for CD20, which also is a B-cell marker, but this marker is only expressed in approximately half of pediatric acute lymphoblastic leukemia (ALL) cases with B-cell precursor origin.1 Markers that typically are expressed in B-cell acute lymphoblastic leukemia (B-ALL)--CD34 and terminal deoxynucleotidyl transferase--were both negative. These results were somewhat contradictory, and the differential remained open to both B-ALL and mature B-cell lymphoma. A bone marrow biopsy showed approximately 65% blasts or leukemic cells (Figure 2). Flow cytometry showed the cells were positive for CD10, CD19, weak CD79a, and variable lambda surface antigen expression. The cells were negative for expression of CD20, CD34, terminal deoxynucleotidyl transferase, myeloid antigens, and CD3. Ultimately, the morphology and immunophenotype were most consistent with a diagnosis of B-ALL. Fluorescence in situ hybridization revealed mixed lineage leukemia, MLL, gene rearrangements.
In general, when considering the differential diagnosis of superficial nodules, 5 elements are helpful to consider: the number of nodules (single vs multiple); the location; and the presence or absence of tenderness, pigmentation or erythema, and firmness.2 Our patient had multiple nodules on the scalp, which were erythematous to slightly purple and firm. The differential diagnosis can be categorized into malignant; infectious; and benign inflammatory, vascular, and fibrous tumors.
Potential oncologic processes include leukemia cutis, lymphoblastic leukemia/lymphoma, Langerhans cell histiocytosis, and rhabdomyosarcoma. Initial laboratory test results were reassuring. Infectious processes in the differential include deep fungal infections such as coccidioidomycosis and nontuberculous mycobacterial infections. Coccidioidomycosis was the most likely to cause skin lesions or masses in our patient; however, it was considered less likely because the patient's family had not traveled or been exposed to an endemic area.3
Benign tumors in the differential include deep hemangioma, which was deemed less likely in our patient because most hemangiomas reach 80% of their maximum size by 5 months of age.4 Another possible benign tumor is infantile myofibromatosis, which is rare but is the most common fibrous tumor of infancy.5
Early-onset childhood sarcoidosis also has been shown to produce multiple nontender firm nodules.2 This process was considered unlikely in our patient because not only is the disease relatively rare in the pediatric population, but most reported childhood cases have occurred in patients aged 13 to 15 years.6 Additionally, no uveitis or arthritis was observed in this case.
Ultimately, histopathology and bone marrow biopsy were necessary to determine the diagnosis of B-ALL. Although uncommon, cutaneous involvement can be an early sign of ALL in children.7 Thus, neoplastic etiologies should be considered in the workup of cutaneous nodules in children, especially when these nodules are hard, rapidly growing, ulcerated, fixed, and/or vascular.8 Once the diagnosis is established, initial workup of ALL in children should include complete blood cell count with manual differential, prothrombin time, partial thromboplastin time, electrolytes, uric acid, and renal and liver function tests. Often, baseline viral titers such as cytomegalovirus, Epstein-Barr virus, human immunodeficiency virus, hepatitis B virus, and varicella-zoster virus also are included. Patients are risk stratified to the appropriate level of treatment based on tumor immunophenotype, cytogenetic findings, patient age, white blood cell count at the time of diagnosis, and response to initial therapy. Treatment typically is comprised of a multidrug regimen divided into several phases--induction, consolidation, and maintenance--as well as therapy directed to the central nervous system. Treatment protocols usually take 2 to 3 years to complete.
Our patient was treated with 1 dose of intrathecal methotrexate before starting the Interfant-06 protocol with a 7-day methylprednisolone prophase. The patient's nodules shrank over time and were no longer present after 14 days of treatment.
- Dworzak MN, Schumich A, Printz D, et al. CD20 up-regulation in pediatric B-cell precursor acute lymphoblastic leukemia during induction treatment: setting the stage for anti-CD20 directed immunotherapy. Blood. 2008;112:3982-3988.
- Whelan JP, Zembowicz A. Case records of the Massachusetts General Hospital. case 19-2006. a 22-month-old boy with the rapid growth of subcutaneous nodules. N Engl J Med. 2006;354:2697-2704.
- Malo J, Luraschi-Monjagatta C, Wolk DM, et al. Update on the diagnosis of pulmonary coccidioidomycosis. Ann Am Thorac Soc. 2014;11:243-253.
- Chang LC, Haggstrom AN, Drolet BA, et al. Growth characteristics of infantile hemangiomas: implications for management. Pediatrics. 2008;122:360-367.
- Schurr P, Moulsdale W. Infantile myofibroma. Adv Neonatal Care. 2008;8:13-20.
- Shetty AK, Gedalia A. Childhood sarcoidosis: a rare but fascinating disorder. Pediatr Rheumatol Online J. 2008;6:16.
- Millot F, Robert A, Bertrand Y, et al. Cutaneous involvement in children with acute lymphoblastic leukemia or lymphoblastic lymphoma. The Children's Leukemia Cooperative Group of the European Organization of Research and Treatment of Cancer (EORTC). Pediatrics. 1997;100:60-64.
- Fogelson S, Dohil M. Papular and nodular skin lesions in children. Semin Plast Surg. 2006;20:180-191.
- Dworzak MN, Schumich A, Printz D, et al. CD20 up-regulation in pediatric B-cell precursor acute lymphoblastic leukemia during induction treatment: setting the stage for anti-CD20 directed immunotherapy. Blood. 2008;112:3982-3988.
- Whelan JP, Zembowicz A. Case records of the Massachusetts General Hospital. case 19-2006. a 22-month-old boy with the rapid growth of subcutaneous nodules. N Engl J Med. 2006;354:2697-2704.
- Malo J, Luraschi-Monjagatta C, Wolk DM, et al. Update on the diagnosis of pulmonary coccidioidomycosis. Ann Am Thorac Soc. 2014;11:243-253.
- Chang LC, Haggstrom AN, Drolet BA, et al. Growth characteristics of infantile hemangiomas: implications for management. Pediatrics. 2008;122:360-367.
- Schurr P, Moulsdale W. Infantile myofibroma. Adv Neonatal Care. 2008;8:13-20.
- Shetty AK, Gedalia A. Childhood sarcoidosis: a rare but fascinating disorder. Pediatr Rheumatol Online J. 2008;6:16.
- Millot F, Robert A, Bertrand Y, et al. Cutaneous involvement in children with acute lymphoblastic leukemia or lymphoblastic lymphoma. The Children's Leukemia Cooperative Group of the European Organization of Research and Treatment of Cancer (EORTC). Pediatrics. 1997;100:60-64.
- Fogelson S, Dohil M. Papular and nodular skin lesions in children. Semin Plast Surg. 2006;20:180-191.
An 8-month-old infant girl presented with rapidly growing cutaneous nodules on the scalp of 1 month's duration. Her parents reported that she disliked lying flat but was otherwise growing and developing normally. Nondiagnostic ultrasonography of the head and brain had been performed as well as a skull radiograph, which found no evidence of lytic lesions. On physical examination, 3 erythematous to violaceous, subcutaneous, firm, fixed nodules were observed on the scalp. Notable cervical lymphadenopathy with several distinct, fixed, firm, subcutaneous nodules in the postauricular lymph chains also were noted. The patient had no pertinent medical history and was born via normal spontaneous vaginal delivery to healthy parents. The remainder of the physical examination and review of systems was negative.
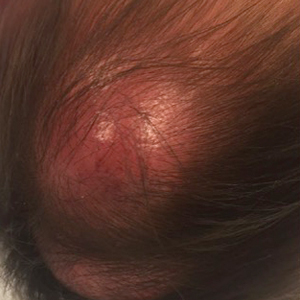
Bullous Systemic Lupus Erythematosus Successfully Treated With Rituximab
Bullous systemic lupus erythematosus (BSLE) is a rare cutaneous presentation of systemic lupus erythematosus (SLE).1 Although 59% to 85% of SLE patients develop skin-related symptoms, fewer than 5% of SLE patients develop BSLE.1-3 This acquired autoimmune bullous disease, characterized by subepidermal bullae with a neutrophilic infiltrate on histopathology, is precipitated by autoantibodies to type VII collagen. Bullae can appear on both cutaneous and mucosal surfaces but tend to favor the trunk, upper extremities, neck, face, and vermilion border.3
Our case of an 18-year-old black woman with BSLE was originally reported in 2011.4 We update the case to illustrate the heterogeneous presentation of BSLE in a single patient and to expand on the role of rituximab in this disease.
Case Report
An 18-year-old black woman presented with a vesicular eruption of 3 weeks’ duration that started on the trunk and buttocks and progressed to involve the face, oral mucosa, and posterior auricular area. The vesicular eruption was accompanied by fatigue, arthralgia, and myalgia.
Physical examination revealed multiple tense, fluid-filled vesicles, measuring roughly 2 to 3 mm in diameter, over the cheeks, chin, postauricular area, vermilion border, oral mucosa, and left side of the neck and shoulder. Resolved lesions on the trunk and buttocks were marked by superficial crust and postinflammatory hyperpigmentation. Scarring was absent.
Laboratory analysis demonstrated hemolytic anemia with a positive direct antiglobulin test, hypocomplementemia, and an elevated erythrocyte sedimentation rate. Antinuclear antibody testing was positive (titer, 1:640).
Biopsies were taken from the left cheek for hematoxylin and eosin (H&E) staining and direct immunofluorescence (DIF), which revealed subepidermal clefting, few neutrophils, and notable mucin deposition. Direct immunofluorescence showed a broad deposition of IgG, IgA, and IgM, as well as C3 in a ribbonlike pattern at the dermoepidermal junction.
A diagnosis of SLE with BSLE was made. The patient initially was treated with prednisone, hydroxychloroquine, mycophenolate mofetil, and intravenous immunoglobulin, but the cutaneous disease persisted. The bullous eruption resolved with 2 infusions of rituximab (1000 mg) spaced 2 weeks apart.
The patient was in remission on 5 mg of prednisone for 2 years following the initial course of rituximab. However, she developed a flare of SLE, with fatigue, arthralgia, hypocomplementemia, and recurrence of BSLE with tense bullae on the face and lips. The flare resolved with prednisone and a single infusion of rituximab (1000 mg). She was then maintained on hydroxychloroquine (200 mg/d).
Three years later (5 years after the initial presentation), the patient presented with pruritic erythematous papulovesicles on the bilateral extensor elbows and right knee (Figure 1). The clinical appearance suggested dermatitis herpetiformis (DH).
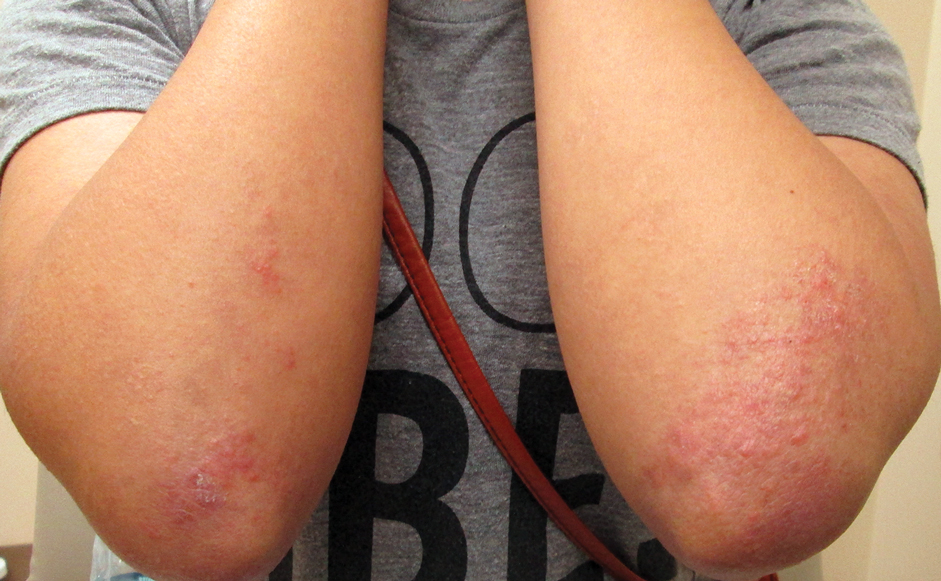
Punch biopsies were obtained from the right elbow for H&E and DIF testing; the H&E-stained specimen showed lichenoid dermatitis with prominent dermal mucin, consistent with cutaneous lupus erythematosus. Direct immunofluorescence showed prominent linear IgG, linear IgA, and granular IgM along the basement membrane, which were identical to DIF findings of the original eruption.
Further laboratory testing revealed hypocomplementemia, anemia of chronic disease (hemoglobin, 8.4 g/dL [reference range, 14.0–17.5 g/dL]), and an elevated erythrocyte sedimentation rate. Given the clinical appearance of the vesicles, DIF findings, and the corresponding SLE flare, a diagnosis of BSLE was made. Because of the systemic symptoms, skin findings, and laboratory results, azathioprine was started. The cutaneous symptoms were treated and resolved with the addition of triamcinolone ointment 0.1% twice daily.
Six months later, the patient presented to our facility with fatigue, arthralgia, and numerous erythematous papules coalescing into a large plaque on the left upper arm (Figure 2). Biopsy showed interface dermatitis with numerous neutrophils and early vesiculation, consistent with BSLE (Figure 3). She underwent another course of 2 infusions of rituximab (1000 mg) administered 2 weeks apart, with resolution of cutaneous and systemic disease.
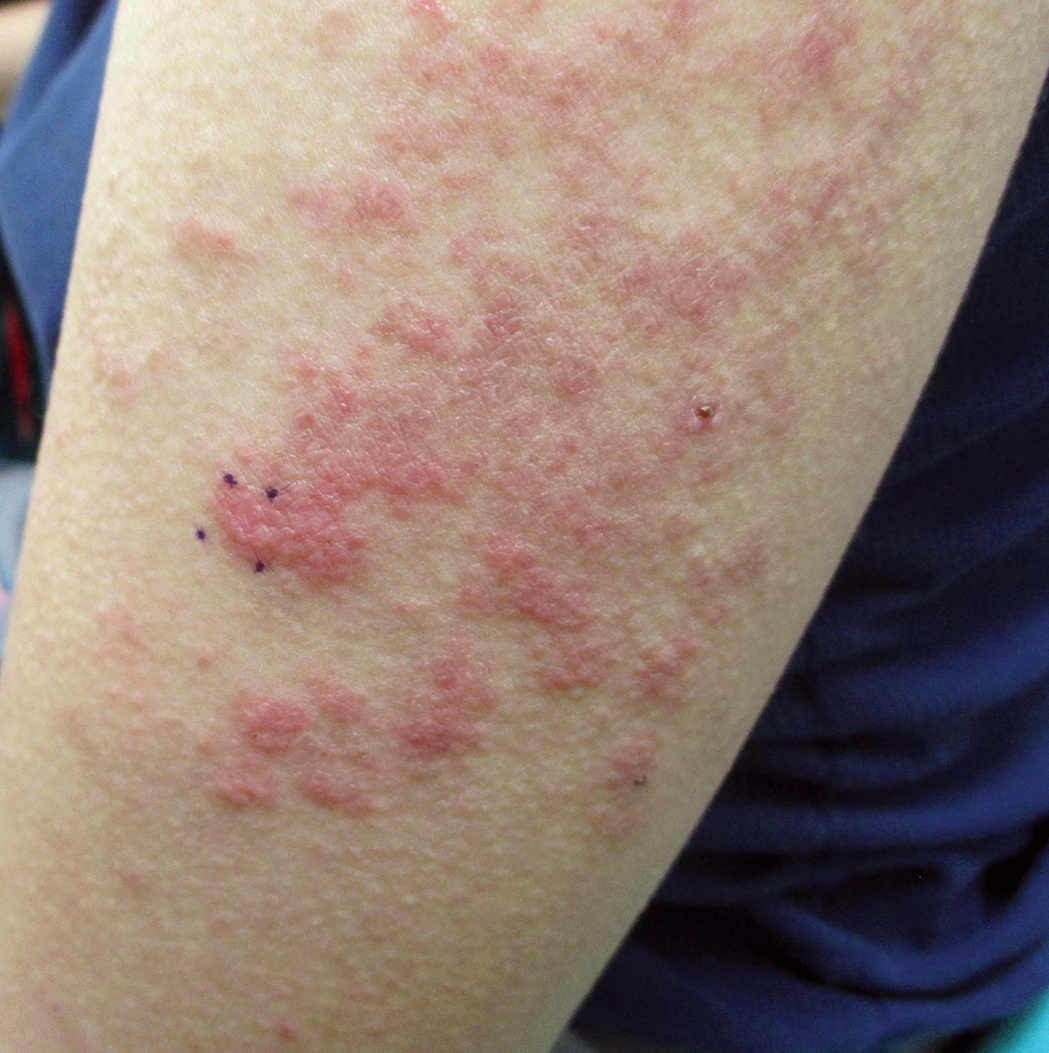
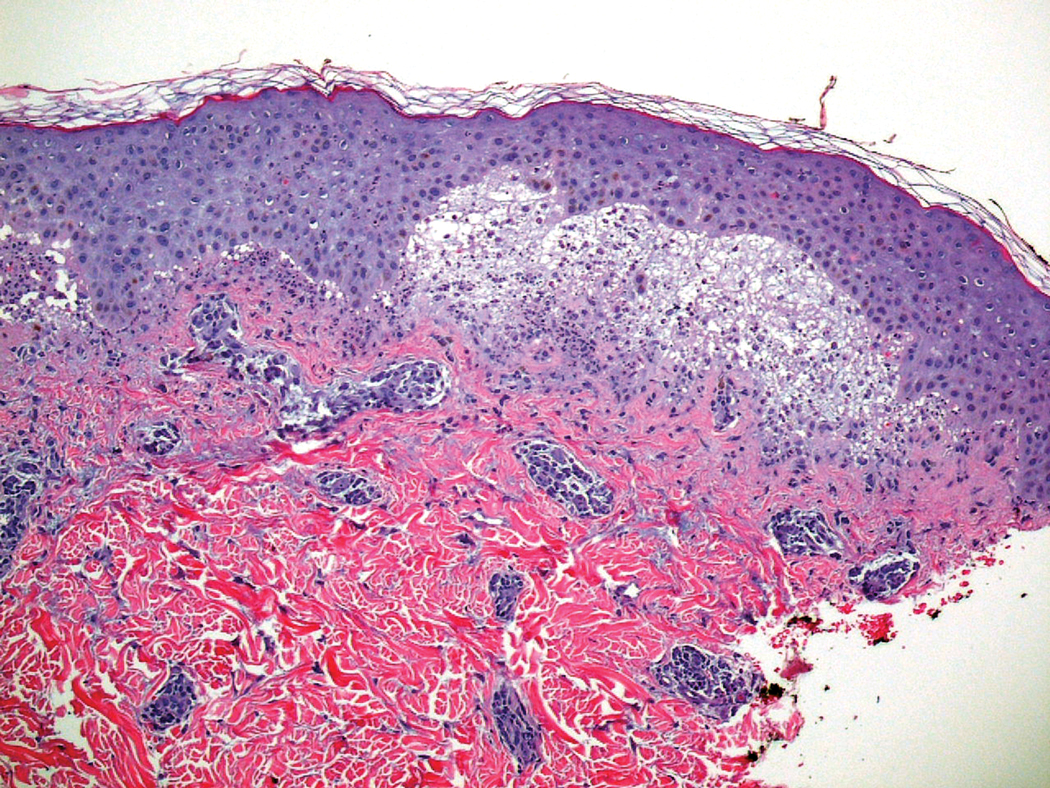
Comment
Diagnosis of BSLE
Bullous systemic lupus erythematosus is a rare cutaneous complication of SLE. It typically affects young black women in the second to fourth decades of life.1 It is a heterogeneous disorder with several clinical variants reported in the literature, and it can be mistaken for bullous pemphigoid, epidermolysis bullosa acquisita (EBA), linear IgA bullous dermatosis, and DH.1-3 Despite its varying clinical phenotypes, BSLE is associated with autoantibodies to the EBA antigen, type VII collagen.1
Current diagnostic criteria for BSLE, revised in 1995,5 include the following: (1) a diagnosis of SLE, based on criteria outlined by the American College of Rheumatology6; (2) vesicles or bullae, or both, involving but not limited to sun-exposed skin; (3) histopathologic features similar to DH; (4) DIF with IgG or IgM, or both, and IgA at the basement membrane zone; and (5) indirect immunofluorescence testing for circulating autoantibodies against the basement membrane zone, using the salt-split skin technique.
Clinical Presentation of BSLE
The classic phenotype associated with BSLE is similar to our patient’s original eruption, with tense bullae favoring the upper trunk and healing without scarring. The extensor surfaces typically are spared. Another presentation of BSLE is an EBA-like phenotype, with bullae on acral and extensor surfaces that heal with scarring. The EBA-like phenotype usually is more difficult to control. Lesions appearing clinically similar to DH have been reported, either as DH associated with SLE (later postulated to have been BSLE) or as herpetiform BSLE.1,4,7-10
Histopathology of BSLE
The typical histologic appearance of BSLE is similar to DH or linear IgA bullous dermatosis, with a predominantly neutrophilic inflammatory infiltrate in the upper dermis and a subepidermal split. Direct immunofluorescence shows broad deposition of IgG along the basement membrane zone (93% of cases; 60% of which are linear and 40% are granular), with approximately 70% of cases showing positive IgA or IgM, or both, at the basement membrane zone. Indirect immunofluorescence performed on 1 M NaCl salt-split skin showed staining on the dermal side of the split, similar to EBA.11
Treatment Options
Rapid clinical response has been reported with dapsone, usually in combination with other immunosuppresants.1,2 A subset of patients does not respond to dapsone, however, as was the case in our patient who tried dapsone early in the disease course but was not effective. Other therapies including azathioprine, cyclophosphamide, mycophenolate mofetil, and antimalarials have been used with some success.3
Rituximab, an anti-CD20 monoclonal antibody, has been used off label to treat BSLE cases that are resistant to dapsone, corticosteroids, and other immunosuppressants.12 Rituximab functions by depleting CD20+ B cells, thus altering the production of autoantibodies and, in the case of BSLE, reducing the concentration of circulating anti–type VII collagen antibodies. Rituximab was approved by the US Food and Drug Administration in 1997 for the treatment of non–Hodgkin lymphoma and later for chronic lymphocytic leukemia, rheumatoid arthritis, granulomatosis with polyangiitis (Wegener granulomatosis), and microscopic polyangiitis.12 Off-label administration of rituximab to treat autoimmune bullous dermatoses has been increasing, and the drug is now approved by the US Food and Drug Administration to treat pemphigus vulgaris (as of June 2018).13
In 2011, Alsanafi et al12 reported successful treatment of BSLE with rituximab in a 61-year-old black woman who had rapid clearance of skin lesions. Our patient had rapid resolution of cutaneous disease with rituximab after the second infusion in a 2-infusion regimen. Interestingly, rituximab is the only agent that has reliably resulted in resolution of our patient’s cutaneous and systemic disease during multiple episodes.
There is little information in the literature regarding the duration of response to rituximab in BSLE or its use in subsequent flares. Our patient relapsed at 2 years and again 3 years later (5 years after the initial presentation). The original cutaneous outbreak and subsequent relapse had classic clinical and histological findings for BSLE; however, the third cutaneous relapse was more similar to DH, given its distribution and appearance. However, the histopathologic findings were the same at the third relapse as they were at the initial presentation and not reflective of DH. We propose that our patient’s prior treatment with rituximab and ongoing immunosuppression at presentation contributed to the more atypical cutaneous findings observed late in the disease course.
Conclusion
We report this case to highlight the heterogeneity of BSLE, even in a single patient, and to report the time course of treatment with rituximab. Although BSLE is considered a rare cutaneous complication of SLE, it is important to note that BSLE also can present as the initial manifestation of SLE.7 As such, BSLE should always be included in the differential diagnosis for a patient presenting with a bullous eruption and symptoms that suggest SLE.
This case also illustrates the repeated use of rituximab for the treatment of BSLE over a 5-year period and justifies the need for larger population-based studies to demonstrate the efficacy of rituximab in BSLE.
- Contestable JJ, Edhegard KD, Meyerle JH. Bullous systemic lupus erythematosus: a review and update to diagnosis and treatment. Am J Clin Dermatol. 2014;15:517-524.
- Camisa C. Vesiculobullous systemic lupus erythematosus. a report of four cases. J Am Acad Dermatol. 1988;18(1, pt 1):93-100.
- Tincopa M, Puttgen KB, Sule S, et al. Bullous lupus: an unusual initial presentation of systemic lupus erythematosus in an adolescent girl. Pediatr Dermatol. 2010;27:373-376.
- Burke KR, Green BP, Meyerle J. Bullous lupus in an 18-year-old. Pediatr Dermatol. 2011;28:483.
- Yell JA, Allen J, Wojnarowska F, et al. Bullous systemic lupus erythematosus: revised criteria for diagnosis. Br J Dermatol. 1995;132:921-928.
- Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheumat. 1997;40:1725.
- Fujimoto W, Hamada T, Yamada J, et al. Bullous systemic lupus erythematosus as an initial manifestation of SLE. J Dermatol. 2005;32:1021-1027.
- Moncada B. Dermatitis herpetiformis in association with systemic lupus erythematosus. Arch Dermatol. 1974;109:723-725.
- Davies MG, Marks R, Waddington E. Simultaneous systemic lupus erythematosus and dermatitis herpetiformis. Arch Dermatol. 1976;112:1292-1294.
- Burrows N, Bhogal BS, Black MM, et al. Bullous eruption of systemic lupus erythematosus: a clinicopathological study of four cases. Br J Dermatol. 1993;128:332-338.
- Sebaratnam DF, Murrell DF. Bullous systemic lupus erythematosus. Dermatol Clin. 2011;29:649-653.
- Alsanafi S, Kovarik C, Mermelstein AL, et al. Rituximab in the treatment of bullous systemic lupus erythematosus. J Clin Rheumatol. 2011;17:142-144.
- Durable remission of pemphigus with a fixed-dose rituximab protocol. JAMA Dermatol. 2014;150:703-708.
Bullous systemic lupus erythematosus (BSLE) is a rare cutaneous presentation of systemic lupus erythematosus (SLE).1 Although 59% to 85% of SLE patients develop skin-related symptoms, fewer than 5% of SLE patients develop BSLE.1-3 This acquired autoimmune bullous disease, characterized by subepidermal bullae with a neutrophilic infiltrate on histopathology, is precipitated by autoantibodies to type VII collagen. Bullae can appear on both cutaneous and mucosal surfaces but tend to favor the trunk, upper extremities, neck, face, and vermilion border.3
Our case of an 18-year-old black woman with BSLE was originally reported in 2011.4 We update the case to illustrate the heterogeneous presentation of BSLE in a single patient and to expand on the role of rituximab in this disease.
Case Report
An 18-year-old black woman presented with a vesicular eruption of 3 weeks’ duration that started on the trunk and buttocks and progressed to involve the face, oral mucosa, and posterior auricular area. The vesicular eruption was accompanied by fatigue, arthralgia, and myalgia.
Physical examination revealed multiple tense, fluid-filled vesicles, measuring roughly 2 to 3 mm in diameter, over the cheeks, chin, postauricular area, vermilion border, oral mucosa, and left side of the neck and shoulder. Resolved lesions on the trunk and buttocks were marked by superficial crust and postinflammatory hyperpigmentation. Scarring was absent.
Laboratory analysis demonstrated hemolytic anemia with a positive direct antiglobulin test, hypocomplementemia, and an elevated erythrocyte sedimentation rate. Antinuclear antibody testing was positive (titer, 1:640).
Biopsies were taken from the left cheek for hematoxylin and eosin (H&E) staining and direct immunofluorescence (DIF), which revealed subepidermal clefting, few neutrophils, and notable mucin deposition. Direct immunofluorescence showed a broad deposition of IgG, IgA, and IgM, as well as C3 in a ribbonlike pattern at the dermoepidermal junction.
A diagnosis of SLE with BSLE was made. The patient initially was treated with prednisone, hydroxychloroquine, mycophenolate mofetil, and intravenous immunoglobulin, but the cutaneous disease persisted. The bullous eruption resolved with 2 infusions of rituximab (1000 mg) spaced 2 weeks apart.
The patient was in remission on 5 mg of prednisone for 2 years following the initial course of rituximab. However, she developed a flare of SLE, with fatigue, arthralgia, hypocomplementemia, and recurrence of BSLE with tense bullae on the face and lips. The flare resolved with prednisone and a single infusion of rituximab (1000 mg). She was then maintained on hydroxychloroquine (200 mg/d).
Three years later (5 years after the initial presentation), the patient presented with pruritic erythematous papulovesicles on the bilateral extensor elbows and right knee (Figure 1). The clinical appearance suggested dermatitis herpetiformis (DH).
Punch biopsies were obtained from the right elbow for H&E and DIF testing; the H&E-stained specimen showed lichenoid dermatitis with prominent dermal mucin, consistent with cutaneous lupus erythematosus. Direct immunofluorescence showed prominent linear IgG, linear IgA, and granular IgM along the basement membrane, which were identical to DIF findings of the original eruption.
Further laboratory testing revealed hypocomplementemia, anemia of chronic disease (hemoglobin, 8.4 g/dL [reference range, 14.0–17.5 g/dL]), and an elevated erythrocyte sedimentation rate. Given the clinical appearance of the vesicles, DIF findings, and the corresponding SLE flare, a diagnosis of BSLE was made. Because of the systemic symptoms, skin findings, and laboratory results, azathioprine was started. The cutaneous symptoms were treated and resolved with the addition of triamcinolone ointment 0.1% twice daily.
Six months later, the patient presented to our facility with fatigue, arthralgia, and numerous erythematous papules coalescing into a large plaque on the left upper arm (Figure 2). Biopsy showed interface dermatitis with numerous neutrophils and early vesiculation, consistent with BSLE (Figure 3). She underwent another course of 2 infusions of rituximab (1000 mg) administered 2 weeks apart, with resolution of cutaneous and systemic disease.
Comment
Diagnosis of BSLE
Bullous systemic lupus erythematosus is a rare cutaneous complication of SLE. It typically affects young black women in the second to fourth decades of life.1 It is a heterogeneous disorder with several clinical variants reported in the literature, and it can be mistaken for bullous pemphigoid, epidermolysis bullosa acquisita (EBA), linear IgA bullous dermatosis, and DH.1-3 Despite its varying clinical phenotypes, BSLE is associated with autoantibodies to the EBA antigen, type VII collagen.1
Current diagnostic criteria for BSLE, revised in 1995,5 include the following: (1) a diagnosis of SLE, based on criteria outlined by the American College of Rheumatology6; (2) vesicles or bullae, or both, involving but not limited to sun-exposed skin; (3) histopathologic features similar to DH; (4) DIF with IgG or IgM, or both, and IgA at the basement membrane zone; and (5) indirect immunofluorescence testing for circulating autoantibodies against the basement membrane zone, using the salt-split skin technique.
Clinical Presentation of BSLE
The classic phenotype associated with BSLE is similar to our patient’s original eruption, with tense bullae favoring the upper trunk and healing without scarring. The extensor surfaces typically are spared. Another presentation of BSLE is an EBA-like phenotype, with bullae on acral and extensor surfaces that heal with scarring. The EBA-like phenotype usually is more difficult to control. Lesions appearing clinically similar to DH have been reported, either as DH associated with SLE (later postulated to have been BSLE) or as herpetiform BSLE.1,4,7-10
Histopathology of BSLE
The typical histologic appearance of BSLE is similar to DH or linear IgA bullous dermatosis, with a predominantly neutrophilic inflammatory infiltrate in the upper dermis and a subepidermal split. Direct immunofluorescence shows broad deposition of IgG along the basement membrane zone (93% of cases; 60% of which are linear and 40% are granular), with approximately 70% of cases showing positive IgA or IgM, or both, at the basement membrane zone. Indirect immunofluorescence performed on 1 M NaCl salt-split skin showed staining on the dermal side of the split, similar to EBA.11
Treatment Options
Rapid clinical response has been reported with dapsone, usually in combination with other immunosuppresants.1,2 A subset of patients does not respond to dapsone, however, as was the case in our patient who tried dapsone early in the disease course but was not effective. Other therapies including azathioprine, cyclophosphamide, mycophenolate mofetil, and antimalarials have been used with some success.3
Rituximab, an anti-CD20 monoclonal antibody, has been used off label to treat BSLE cases that are resistant to dapsone, corticosteroids, and other immunosuppressants.12 Rituximab functions by depleting CD20+ B cells, thus altering the production of autoantibodies and, in the case of BSLE, reducing the concentration of circulating anti–type VII collagen antibodies. Rituximab was approved by the US Food and Drug Administration in 1997 for the treatment of non–Hodgkin lymphoma and later for chronic lymphocytic leukemia, rheumatoid arthritis, granulomatosis with polyangiitis (Wegener granulomatosis), and microscopic polyangiitis.12 Off-label administration of rituximab to treat autoimmune bullous dermatoses has been increasing, and the drug is now approved by the US Food and Drug Administration to treat pemphigus vulgaris (as of June 2018).13
In 2011, Alsanafi et al12 reported successful treatment of BSLE with rituximab in a 61-year-old black woman who had rapid clearance of skin lesions. Our patient had rapid resolution of cutaneous disease with rituximab after the second infusion in a 2-infusion regimen. Interestingly, rituximab is the only agent that has reliably resulted in resolution of our patient’s cutaneous and systemic disease during multiple episodes.
There is little information in the literature regarding the duration of response to rituximab in BSLE or its use in subsequent flares. Our patient relapsed at 2 years and again 3 years later (5 years after the initial presentation). The original cutaneous outbreak and subsequent relapse had classic clinical and histological findings for BSLE; however, the third cutaneous relapse was more similar to DH, given its distribution and appearance. However, the histopathologic findings were the same at the third relapse as they were at the initial presentation and not reflective of DH. We propose that our patient’s prior treatment with rituximab and ongoing immunosuppression at presentation contributed to the more atypical cutaneous findings observed late in the disease course.
Conclusion
We report this case to highlight the heterogeneity of BSLE, even in a single patient, and to report the time course of treatment with rituximab. Although BSLE is considered a rare cutaneous complication of SLE, it is important to note that BSLE also can present as the initial manifestation of SLE.7 As such, BSLE should always be included in the differential diagnosis for a patient presenting with a bullous eruption and symptoms that suggest SLE.
This case also illustrates the repeated use of rituximab for the treatment of BSLE over a 5-year period and justifies the need for larger population-based studies to demonstrate the efficacy of rituximab in BSLE.
Bullous systemic lupus erythematosus (BSLE) is a rare cutaneous presentation of systemic lupus erythematosus (SLE).1 Although 59% to 85% of SLE patients develop skin-related symptoms, fewer than 5% of SLE patients develop BSLE.1-3 This acquired autoimmune bullous disease, characterized by subepidermal bullae with a neutrophilic infiltrate on histopathology, is precipitated by autoantibodies to type VII collagen. Bullae can appear on both cutaneous and mucosal surfaces but tend to favor the trunk, upper extremities, neck, face, and vermilion border.3
Our case of an 18-year-old black woman with BSLE was originally reported in 2011.4 We update the case to illustrate the heterogeneous presentation of BSLE in a single patient and to expand on the role of rituximab in this disease.
Case Report
An 18-year-old black woman presented with a vesicular eruption of 3 weeks’ duration that started on the trunk and buttocks and progressed to involve the face, oral mucosa, and posterior auricular area. The vesicular eruption was accompanied by fatigue, arthralgia, and myalgia.
Physical examination revealed multiple tense, fluid-filled vesicles, measuring roughly 2 to 3 mm in diameter, over the cheeks, chin, postauricular area, vermilion border, oral mucosa, and left side of the neck and shoulder. Resolved lesions on the trunk and buttocks were marked by superficial crust and postinflammatory hyperpigmentation. Scarring was absent.
Laboratory analysis demonstrated hemolytic anemia with a positive direct antiglobulin test, hypocomplementemia, and an elevated erythrocyte sedimentation rate. Antinuclear antibody testing was positive (titer, 1:640).
Biopsies were taken from the left cheek for hematoxylin and eosin (H&E) staining and direct immunofluorescence (DIF), which revealed subepidermal clefting, few neutrophils, and notable mucin deposition. Direct immunofluorescence showed a broad deposition of IgG, IgA, and IgM, as well as C3 in a ribbonlike pattern at the dermoepidermal junction.
A diagnosis of SLE with BSLE was made. The patient initially was treated with prednisone, hydroxychloroquine, mycophenolate mofetil, and intravenous immunoglobulin, but the cutaneous disease persisted. The bullous eruption resolved with 2 infusions of rituximab (1000 mg) spaced 2 weeks apart.
The patient was in remission on 5 mg of prednisone for 2 years following the initial course of rituximab. However, she developed a flare of SLE, with fatigue, arthralgia, hypocomplementemia, and recurrence of BSLE with tense bullae on the face and lips. The flare resolved with prednisone and a single infusion of rituximab (1000 mg). She was then maintained on hydroxychloroquine (200 mg/d).
Three years later (5 years after the initial presentation), the patient presented with pruritic erythematous papulovesicles on the bilateral extensor elbows and right knee (Figure 1). The clinical appearance suggested dermatitis herpetiformis (DH).
Punch biopsies were obtained from the right elbow for H&E and DIF testing; the H&E-stained specimen showed lichenoid dermatitis with prominent dermal mucin, consistent with cutaneous lupus erythematosus. Direct immunofluorescence showed prominent linear IgG, linear IgA, and granular IgM along the basement membrane, which were identical to DIF findings of the original eruption.
Further laboratory testing revealed hypocomplementemia, anemia of chronic disease (hemoglobin, 8.4 g/dL [reference range, 14.0–17.5 g/dL]), and an elevated erythrocyte sedimentation rate. Given the clinical appearance of the vesicles, DIF findings, and the corresponding SLE flare, a diagnosis of BSLE was made. Because of the systemic symptoms, skin findings, and laboratory results, azathioprine was started. The cutaneous symptoms were treated and resolved with the addition of triamcinolone ointment 0.1% twice daily.
Six months later, the patient presented to our facility with fatigue, arthralgia, and numerous erythematous papules coalescing into a large plaque on the left upper arm (Figure 2). Biopsy showed interface dermatitis with numerous neutrophils and early vesiculation, consistent with BSLE (Figure 3). She underwent another course of 2 infusions of rituximab (1000 mg) administered 2 weeks apart, with resolution of cutaneous and systemic disease.
Comment
Diagnosis of BSLE
Bullous systemic lupus erythematosus is a rare cutaneous complication of SLE. It typically affects young black women in the second to fourth decades of life.1 It is a heterogeneous disorder with several clinical variants reported in the literature, and it can be mistaken for bullous pemphigoid, epidermolysis bullosa acquisita (EBA), linear IgA bullous dermatosis, and DH.1-3 Despite its varying clinical phenotypes, BSLE is associated with autoantibodies to the EBA antigen, type VII collagen.1
Current diagnostic criteria for BSLE, revised in 1995,5 include the following: (1) a diagnosis of SLE, based on criteria outlined by the American College of Rheumatology6; (2) vesicles or bullae, or both, involving but not limited to sun-exposed skin; (3) histopathologic features similar to DH; (4) DIF with IgG or IgM, or both, and IgA at the basement membrane zone; and (5) indirect immunofluorescence testing for circulating autoantibodies against the basement membrane zone, using the salt-split skin technique.
Clinical Presentation of BSLE
The classic phenotype associated with BSLE is similar to our patient’s original eruption, with tense bullae favoring the upper trunk and healing without scarring. The extensor surfaces typically are spared. Another presentation of BSLE is an EBA-like phenotype, with bullae on acral and extensor surfaces that heal with scarring. The EBA-like phenotype usually is more difficult to control. Lesions appearing clinically similar to DH have been reported, either as DH associated with SLE (later postulated to have been BSLE) or as herpetiform BSLE.1,4,7-10
Histopathology of BSLE
The typical histologic appearance of BSLE is similar to DH or linear IgA bullous dermatosis, with a predominantly neutrophilic inflammatory infiltrate in the upper dermis and a subepidermal split. Direct immunofluorescence shows broad deposition of IgG along the basement membrane zone (93% of cases; 60% of which are linear and 40% are granular), with approximately 70% of cases showing positive IgA or IgM, or both, at the basement membrane zone. Indirect immunofluorescence performed on 1 M NaCl salt-split skin showed staining on the dermal side of the split, similar to EBA.11
Treatment Options
Rapid clinical response has been reported with dapsone, usually in combination with other immunosuppresants.1,2 A subset of patients does not respond to dapsone, however, as was the case in our patient who tried dapsone early in the disease course but was not effective. Other therapies including azathioprine, cyclophosphamide, mycophenolate mofetil, and antimalarials have been used with some success.3
Rituximab, an anti-CD20 monoclonal antibody, has been used off label to treat BSLE cases that are resistant to dapsone, corticosteroids, and other immunosuppressants.12 Rituximab functions by depleting CD20+ B cells, thus altering the production of autoantibodies and, in the case of BSLE, reducing the concentration of circulating anti–type VII collagen antibodies. Rituximab was approved by the US Food and Drug Administration in 1997 for the treatment of non–Hodgkin lymphoma and later for chronic lymphocytic leukemia, rheumatoid arthritis, granulomatosis with polyangiitis (Wegener granulomatosis), and microscopic polyangiitis.12 Off-label administration of rituximab to treat autoimmune bullous dermatoses has been increasing, and the drug is now approved by the US Food and Drug Administration to treat pemphigus vulgaris (as of June 2018).13
In 2011, Alsanafi et al12 reported successful treatment of BSLE with rituximab in a 61-year-old black woman who had rapid clearance of skin lesions. Our patient had rapid resolution of cutaneous disease with rituximab after the second infusion in a 2-infusion regimen. Interestingly, rituximab is the only agent that has reliably resulted in resolution of our patient’s cutaneous and systemic disease during multiple episodes.
There is little information in the literature regarding the duration of response to rituximab in BSLE or its use in subsequent flares. Our patient relapsed at 2 years and again 3 years later (5 years after the initial presentation). The original cutaneous outbreak and subsequent relapse had classic clinical and histological findings for BSLE; however, the third cutaneous relapse was more similar to DH, given its distribution and appearance. However, the histopathologic findings were the same at the third relapse as they were at the initial presentation and not reflective of DH. We propose that our patient’s prior treatment with rituximab and ongoing immunosuppression at presentation contributed to the more atypical cutaneous findings observed late in the disease course.
Conclusion
We report this case to highlight the heterogeneity of BSLE, even in a single patient, and to report the time course of treatment with rituximab. Although BSLE is considered a rare cutaneous complication of SLE, it is important to note that BSLE also can present as the initial manifestation of SLE.7 As such, BSLE should always be included in the differential diagnosis for a patient presenting with a bullous eruption and symptoms that suggest SLE.
This case also illustrates the repeated use of rituximab for the treatment of BSLE over a 5-year period and justifies the need for larger population-based studies to demonstrate the efficacy of rituximab in BSLE.
- Contestable JJ, Edhegard KD, Meyerle JH. Bullous systemic lupus erythematosus: a review and update to diagnosis and treatment. Am J Clin Dermatol. 2014;15:517-524.
- Camisa C. Vesiculobullous systemic lupus erythematosus. a report of four cases. J Am Acad Dermatol. 1988;18(1, pt 1):93-100.
- Tincopa M, Puttgen KB, Sule S, et al. Bullous lupus: an unusual initial presentation of systemic lupus erythematosus in an adolescent girl. Pediatr Dermatol. 2010;27:373-376.
- Burke KR, Green BP, Meyerle J. Bullous lupus in an 18-year-old. Pediatr Dermatol. 2011;28:483.
- Yell JA, Allen J, Wojnarowska F, et al. Bullous systemic lupus erythematosus: revised criteria for diagnosis. Br J Dermatol. 1995;132:921-928.
- Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheumat. 1997;40:1725.
- Fujimoto W, Hamada T, Yamada J, et al. Bullous systemic lupus erythematosus as an initial manifestation of SLE. J Dermatol. 2005;32:1021-1027.
- Moncada B. Dermatitis herpetiformis in association with systemic lupus erythematosus. Arch Dermatol. 1974;109:723-725.
- Davies MG, Marks R, Waddington E. Simultaneous systemic lupus erythematosus and dermatitis herpetiformis. Arch Dermatol. 1976;112:1292-1294.
- Burrows N, Bhogal BS, Black MM, et al. Bullous eruption of systemic lupus erythematosus: a clinicopathological study of four cases. Br J Dermatol. 1993;128:332-338.
- Sebaratnam DF, Murrell DF. Bullous systemic lupus erythematosus. Dermatol Clin. 2011;29:649-653.
- Alsanafi S, Kovarik C, Mermelstein AL, et al. Rituximab in the treatment of bullous systemic lupus erythematosus. J Clin Rheumatol. 2011;17:142-144.
- Durable remission of pemphigus with a fixed-dose rituximab protocol. JAMA Dermatol. 2014;150:703-708.
- Contestable JJ, Edhegard KD, Meyerle JH. Bullous systemic lupus erythematosus: a review and update to diagnosis and treatment. Am J Clin Dermatol. 2014;15:517-524.
- Camisa C. Vesiculobullous systemic lupus erythematosus. a report of four cases. J Am Acad Dermatol. 1988;18(1, pt 1):93-100.
- Tincopa M, Puttgen KB, Sule S, et al. Bullous lupus: an unusual initial presentation of systemic lupus erythematosus in an adolescent girl. Pediatr Dermatol. 2010;27:373-376.
- Burke KR, Green BP, Meyerle J. Bullous lupus in an 18-year-old. Pediatr Dermatol. 2011;28:483.
- Yell JA, Allen J, Wojnarowska F, et al. Bullous systemic lupus erythematosus: revised criteria for diagnosis. Br J Dermatol. 1995;132:921-928.
- Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheumat. 1997;40:1725.
- Fujimoto W, Hamada T, Yamada J, et al. Bullous systemic lupus erythematosus as an initial manifestation of SLE. J Dermatol. 2005;32:1021-1027.
- Moncada B. Dermatitis herpetiformis in association with systemic lupus erythematosus. Arch Dermatol. 1974;109:723-725.
- Davies MG, Marks R, Waddington E. Simultaneous systemic lupus erythematosus and dermatitis herpetiformis. Arch Dermatol. 1976;112:1292-1294.
- Burrows N, Bhogal BS, Black MM, et al. Bullous eruption of systemic lupus erythematosus: a clinicopathological study of four cases. Br J Dermatol. 1993;128:332-338.
- Sebaratnam DF, Murrell DF. Bullous systemic lupus erythematosus. Dermatol Clin. 2011;29:649-653.
- Alsanafi S, Kovarik C, Mermelstein AL, et al. Rituximab in the treatment of bullous systemic lupus erythematosus. J Clin Rheumatol. 2011;17:142-144.
- Durable remission of pemphigus with a fixed-dose rituximab protocol. JAMA Dermatol. 2014;150:703-708.
Practice Points
- Bullous systemic lupus erythematosus (BSLE) can present with a waxing and waning course punctuated by flares.
- Different clinical presentations can occur over the disease course.
- Rituximab is a viable treatment option in BSLE.
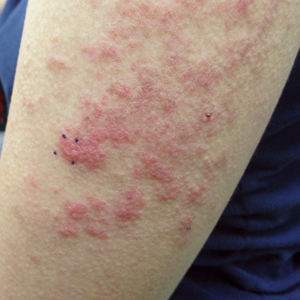
Acute Graft-vs-host Disease Following Liver Transplantation
Acute graft-vs-host disease (GVHD) is a T-cell mediated immunogenic response in which T lymphocytes from a donor regard host tissue as foreign and attack it in the setting of immunosuppression.1 The most common cause of acute GVHD is allogeneic stem cell transplantation, with solid-organ transplantation being a much less common cause.2 The incidence of acute GVHD following orthotopic liver transplantation (OLT) is 0.1%, as reported by the United Network for Organ Sharing, compared to an incidence of 40% to 60% in hematopoietic stem cell transplant recipients.3,4
Early recognition and treatment of acute GVHD following liver transplantation is imperative, as the mortality rate is 85% to 90%.2 We present a case of acute GVHD in a liver transplantation patient, with a focus on diagnostic criteria and comparison to acute GVHD following hematopoietic stem cell transplantation.
Case Report
A 68-year-old woman with a history of hepatitis C virus infection, hepatocellular carcinoma, and OLT 1 month prior presented to the hospital with fever and abdominal cellulitis in close proximity to the surgical site of 1 week’s duration. The patient was started on vancomycin and cefepime; pan cultures were performed.
At 10 days of hospitalization, the patient developed a pruritic, nontender, erythematous rash on the abdomen, with extension onto the chest and legs. The rash was associated with low-grade fever but not with diarrhea. Physical examination was notable for a few erythematous macules and scattered papules over the neck and chest and a large erythematous plaque with multiple ecchymoses over the lower abdomen (Figure 1A). Erythematous macules and papules coalescing into plaques were present on the lower back (Figure 1B) and proximal thighs. Oral, ocular, and genital lesions were absent.
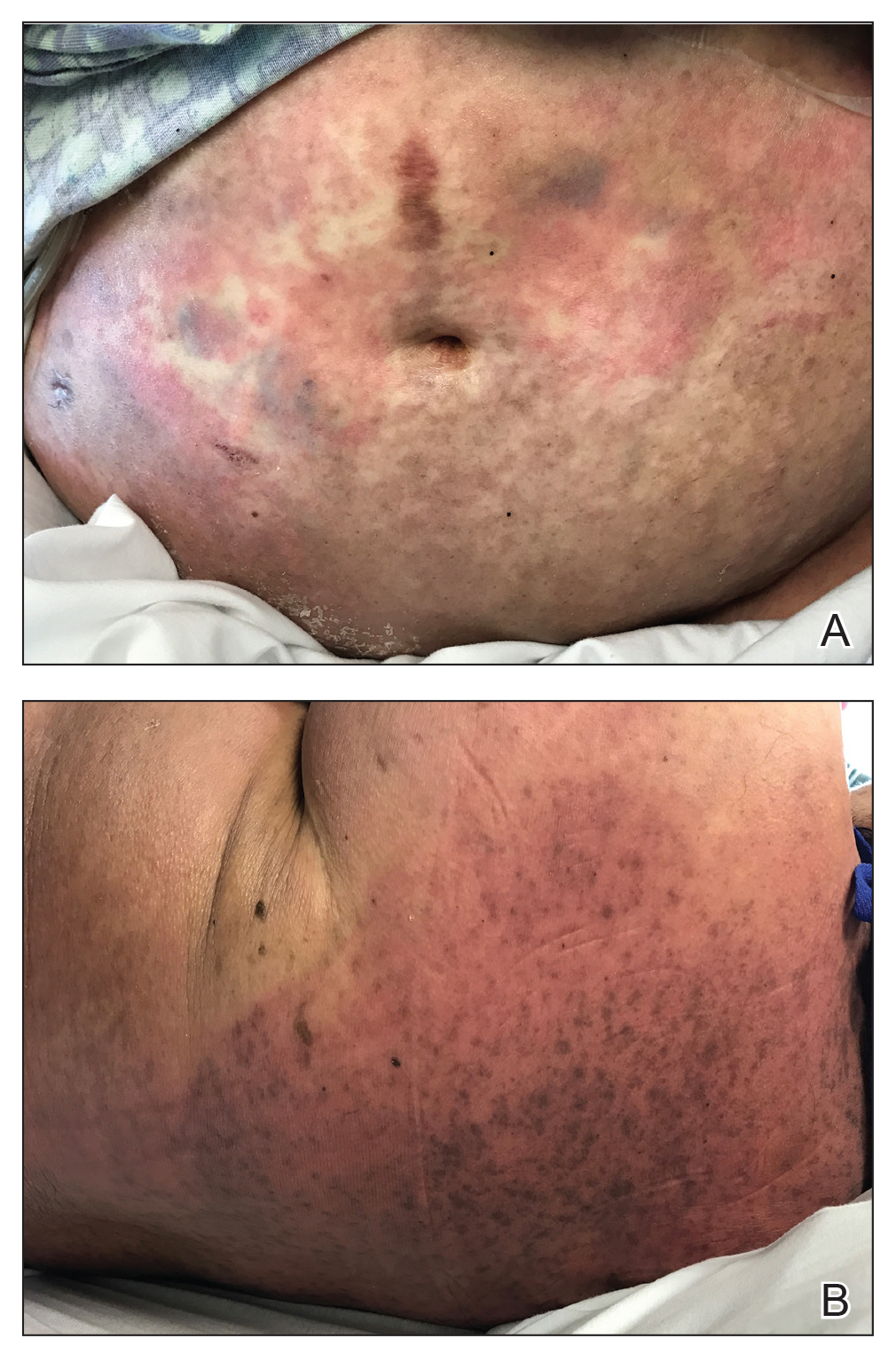
The differential diagnosis included drug reaction, viral infection, and acute GVHD. A skin biopsy was performed from the left side of the chest. Cefepime and vancomycin were discontinued; triamcinolone ointment 0.1% twice daily and antihistamines as needed for itching were started.
Over a 2-day period, the rash progressed to diffuse erythematous papules over the chest (Figure 2A) and bilateral arms (Figure 2B) including the palms. The patient also developed erythematous papules over the jawline and forehead as well as confluent erythematous plaques over the back with extension of the rash to involve the legs. She also had erythema and swelling bilaterally over the ears. She reported diarrhea. The low-grade fever resolved.
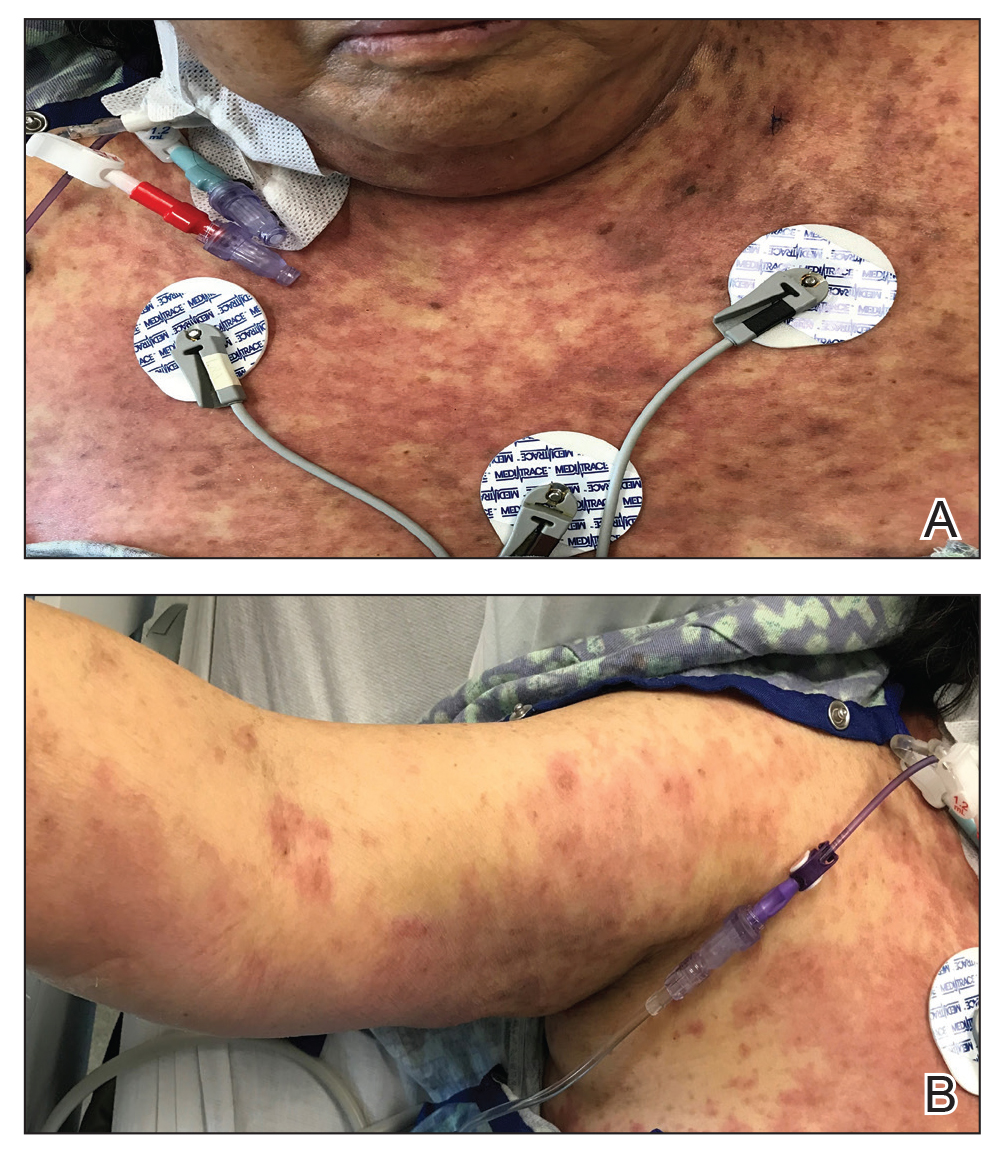
Laboratory review showed new-onset pancytopenia, normal liver function, and an elevated creatinine level of 2.3 mg/dL (reference range, 0.6–1.2 mg/dL), consistent with the patient’s baseline of stage 3 chronic kidney disease. Polymerase chain reaction analysis for cytomegalovirus was negative. Histology revealed vacuolar interface dermatitis with apoptotic keratinocytes, consistent with grade I GVHD (Figure 3). Duodenal biopsy revealed rare patchy glands with increased apoptosis, compatible with grade I GVHD.
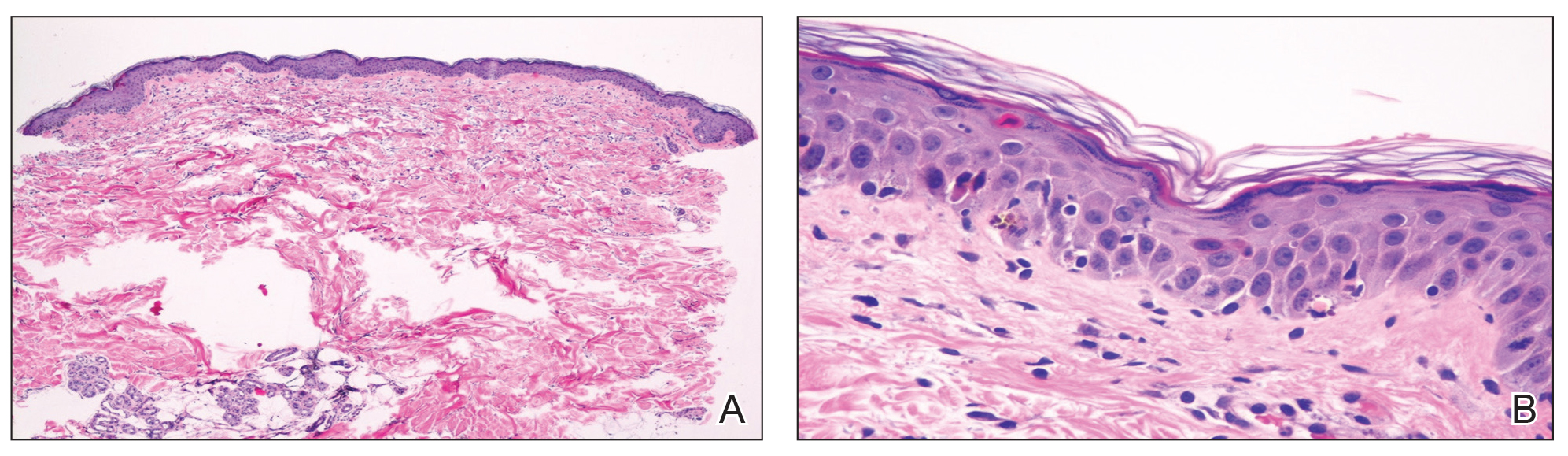
The patient was started on intravenous methylprednisolone 1 mg/kg for 3 days, then transitioned to an oral steroid taper, with improvement of the rash and other systemic symptoms.
Comment
GVHD Subtypes
The 2 types of GVHD are humoral and cellular.5 The humoral type results from ABO blood type incompatibility between donor and recipient and causes mild hemolytic anemia and fever. The cellular type is directed against major histocompatibility complexes and is associated with high morbidity and mortality.
Presentation of GVHD
Acute GVHD following OLT usually occurs 3 to 5 weeks after transplantation,6 as in our patient. Symptoms include rash, fever, pancytopenia, and diarrhea.2 Skin is the most commonly involved organ in acute GVHD; rash is the earliest manifestation.1 The rash can be asymptomatic or associated with pain and pruritus. Initial cutaneous manifestations include palmar erythema and erythematous to violaceous discoloration of the face and ears. A diffuse maculopapular rash can develop, involving the face, abdomen, and trunk. The rash may progress to formation of bullae or skin sloughing, resembling Stevens-Johnson syndrome or toxic epidermal necrolysis.1 The skin manifestation of acute GVHD following OLT is similar to hematopoietic stem cell transplantation (Table).7,8
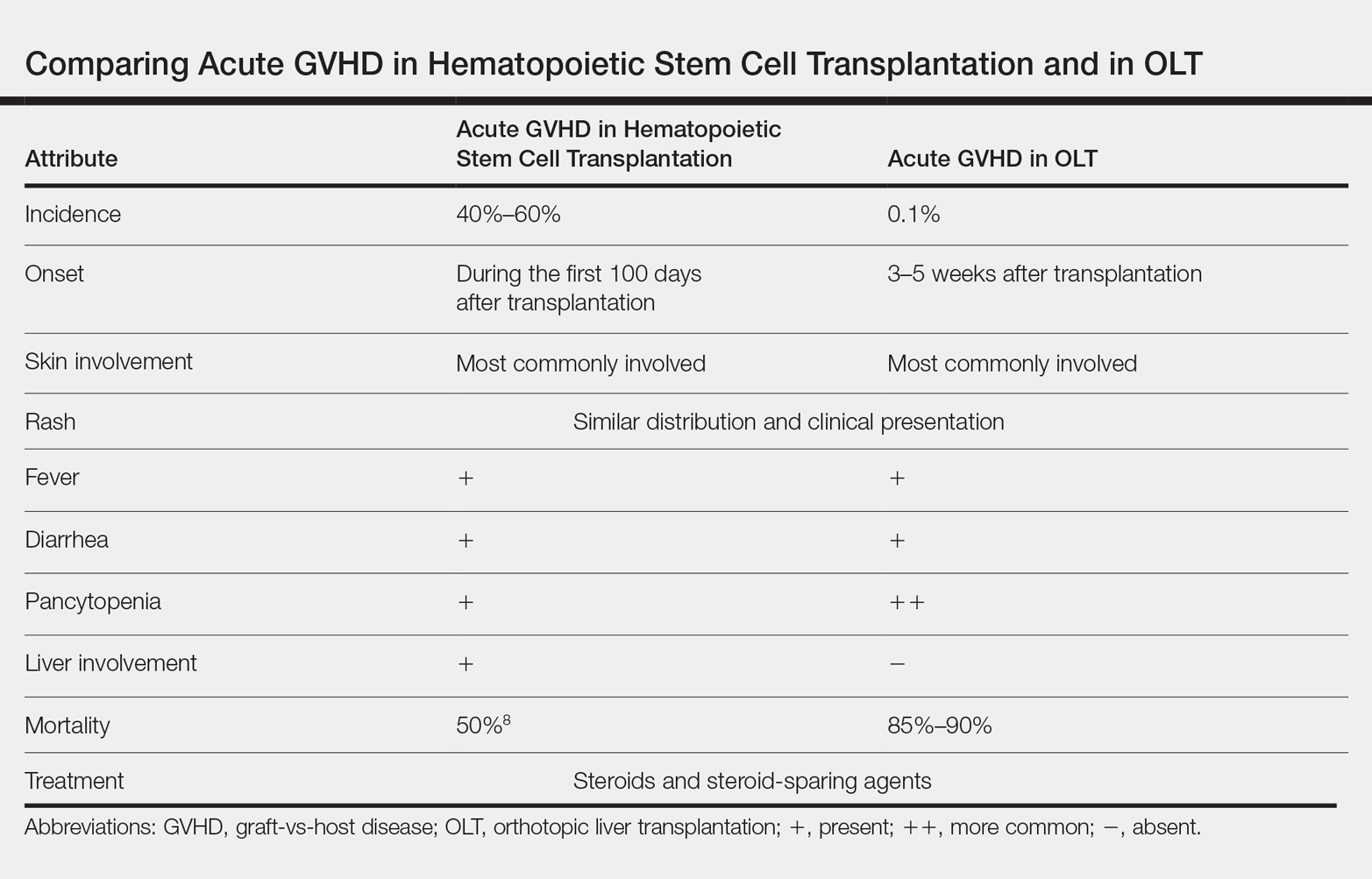
Pancytopenia is a common manifestation of GVHD following liver transplantation and is rarely seen following hematopoietic stem cell transplantation.7 Donor lymphocytes engraft and proliferate in the bone marrow, attacking recipient hematopoietic stem cells. It is important to note that more common causes of cytopenia following liver transplantation, including infection and drug-induced bone marrow suppression, should be ruled out before diagnosing acute GVHD.6
Acute GVHD can affect the gastrointestinal tract, causing diarrhea; however, other infectious and medication-induced causes of diarrhea also should be considered.6 In contrast to hematopoietic stem cell transplantation, in which the liver is usually involved,1 the liver is spared in acute GVHD following liver transplantation.5
Diagnosis of GVHD
The diagnosis of acute GVHD following liver transplantation can be challenging because the clinical manifestations can be caused by a drug reaction or viral infection, such as cytomegalovirus infection.2 Patients who are older than 50 years and glucose intolerant are at a higher risk of acute GVHD following OLT. The combination of younger donor age and the presence of an HLA class I match also increases the risk of acute GVHD.6 The diagnosis of acute GVHD is confirmed with biopsy of the skin or gastrointestinal tract.
Morbidity and Mortality of GVHD
Because of the high morbidity and mortality associated with acute GVHD following liver transplantation, early diagnosis and treatment are crucial.5 Death in patients with acute GVHD following OLT is mainly attributable to sepsis, multiorgan failure, and gastrointestinal tract bleeding.6 It remains unclear whether this high mortality is associated with delayed diagnosis due to nonspecific signs of acute GVHD following OLT or to the lack of appropriate treatment guidelines.6
Treatment Options
Because of the low incidence of acute GVHD following OLT, most treatment modalities are extrapolated from the literature on acute GVHD following stem cell transplantation.5 The most commonly used therapies include high-dose systemic steroids and anti–thymocyte globulin that attacks activated donor T cells.6 Other treatment modalities, including anti–tumor necrosis factor agents and antibodies to CD20, have been reported to be effective in steroid-refractory GVHD.2 The major drawback of systemic steroids is an increase in the risk for sepsis and infection; therefore, these patients should be diligently screened for infection and covered with antibiotics and antifungals. Extracorporeal photopheresis is another treatment modality that does not cause generalized immunosuppression but is not well studied in the setting of acute GVHD following OLT.6
Prevention
Acute GVHD following OLT can be prevented by eliminating donor T lymphocytes from the liver before transplantation. However, because the incidence of acute GVHD following OLT is very low, this approach is not routinely taken.2
Conclusion
Acute GVHD following liver transplantation is a rare complication; however, it has high mortality, necessitating further research regarding treatment and prevention. Early recognition and treatment of this condition can improve outcomes. Dermatologists should be familiar with the skin manifestations of acute GVHD following liver transplantation due to the rising number of cases of solid-organ transplantation.
- Hu SW, Cotliar J. Acute graft-versus-host disease following hematopoietic stem-cell transplantation. Dermatol Ther. 2011;24:411-423.
- Akbulut S, Yilmaz M, Yilmaz S. Graft-versus-host disease after liver transplantation: a comprehensive literature review. World J Gastroenterol. 2012;18:5240-5248.
- Taylor AL, Gibbs P, Bradley JA. Acute graft versus host disease following liver transplantation: the enemy within. Am J Transplant. 2004;4:466-474.
- Jagasia M, Arora M, Flowers ME, et al. Risk factor for acute GVHD and survival after hematopoietic cell transplantation. Blood. 2012;119:296-307.
- Kang WH, Hwang S, Song GW, et al. Acute graft-vs-host disease after liver transplantation: experience at a high-volume liver transplantation center in Korea. Transplant Proc. 2016;48:3368-3372.
- Murali AR, Chandra S, Stewart Z, et al. Graft versus host disease after liver transplantation in adults: a case series, review of literature, and an approach to management. Transplantation. 2016;100:2661-2670.
- Chaib E, Silva FD, Figueira ER, et al. Graft-versus-host disease after liver transplantation. Clinics (Sao Paulo). 2011;66:1115-1118.
- Barton-Burke M, Dwinell DM, Kafkas L, et al. Graft-versus-host disease: a complex long-term side effect of hematopoietic stem cell transplant. Oncology (Williston Park). 2008;22(11 Suppl Nurse Ed):31-45.
Acute graft-vs-host disease (GVHD) is a T-cell mediated immunogenic response in which T lymphocytes from a donor regard host tissue as foreign and attack it in the setting of immunosuppression.1 The most common cause of acute GVHD is allogeneic stem cell transplantation, with solid-organ transplantation being a much less common cause.2 The incidence of acute GVHD following orthotopic liver transplantation (OLT) is 0.1%, as reported by the United Network for Organ Sharing, compared to an incidence of 40% to 60% in hematopoietic stem cell transplant recipients.3,4
Early recognition and treatment of acute GVHD following liver transplantation is imperative, as the mortality rate is 85% to 90%.2 We present a case of acute GVHD in a liver transplantation patient, with a focus on diagnostic criteria and comparison to acute GVHD following hematopoietic stem cell transplantation.
Case Report
A 68-year-old woman with a history of hepatitis C virus infection, hepatocellular carcinoma, and OLT 1 month prior presented to the hospital with fever and abdominal cellulitis in close proximity to the surgical site of 1 week’s duration. The patient was started on vancomycin and cefepime; pan cultures were performed.
At 10 days of hospitalization, the patient developed a pruritic, nontender, erythematous rash on the abdomen, with extension onto the chest and legs. The rash was associated with low-grade fever but not with diarrhea. Physical examination was notable for a few erythematous macules and scattered papules over the neck and chest and a large erythematous plaque with multiple ecchymoses over the lower abdomen (Figure 1A). Erythematous macules and papules coalescing into plaques were present on the lower back (Figure 1B) and proximal thighs. Oral, ocular, and genital lesions were absent.
The differential diagnosis included drug reaction, viral infection, and acute GVHD. A skin biopsy was performed from the left side of the chest. Cefepime and vancomycin were discontinued; triamcinolone ointment 0.1% twice daily and antihistamines as needed for itching were started.
Over a 2-day period, the rash progressed to diffuse erythematous papules over the chest (Figure 2A) and bilateral arms (Figure 2B) including the palms. The patient also developed erythematous papules over the jawline and forehead as well as confluent erythematous plaques over the back with extension of the rash to involve the legs. She also had erythema and swelling bilaterally over the ears. She reported diarrhea. The low-grade fever resolved.
Laboratory review showed new-onset pancytopenia, normal liver function, and an elevated creatinine level of 2.3 mg/dL (reference range, 0.6–1.2 mg/dL), consistent with the patient’s baseline of stage 3 chronic kidney disease. Polymerase chain reaction analysis for cytomegalovirus was negative. Histology revealed vacuolar interface dermatitis with apoptotic keratinocytes, consistent with grade I GVHD (Figure 3). Duodenal biopsy revealed rare patchy glands with increased apoptosis, compatible with grade I GVHD.
The patient was started on intravenous methylprednisolone 1 mg/kg for 3 days, then transitioned to an oral steroid taper, with improvement of the rash and other systemic symptoms.
Comment
GVHD Subtypes
The 2 types of GVHD are humoral and cellular.5 The humoral type results from ABO blood type incompatibility between donor and recipient and causes mild hemolytic anemia and fever. The cellular type is directed against major histocompatibility complexes and is associated with high morbidity and mortality.
Presentation of GVHD
Acute GVHD following OLT usually occurs 3 to 5 weeks after transplantation,6 as in our patient. Symptoms include rash, fever, pancytopenia, and diarrhea.2 Skin is the most commonly involved organ in acute GVHD; rash is the earliest manifestation.1 The rash can be asymptomatic or associated with pain and pruritus. Initial cutaneous manifestations include palmar erythema and erythematous to violaceous discoloration of the face and ears. A diffuse maculopapular rash can develop, involving the face, abdomen, and trunk. The rash may progress to formation of bullae or skin sloughing, resembling Stevens-Johnson syndrome or toxic epidermal necrolysis.1 The skin manifestation of acute GVHD following OLT is similar to hematopoietic stem cell transplantation (Table).7,8
Pancytopenia is a common manifestation of GVHD following liver transplantation and is rarely seen following hematopoietic stem cell transplantation.7 Donor lymphocytes engraft and proliferate in the bone marrow, attacking recipient hematopoietic stem cells. It is important to note that more common causes of cytopenia following liver transplantation, including infection and drug-induced bone marrow suppression, should be ruled out before diagnosing acute GVHD.6
Acute GVHD can affect the gastrointestinal tract, causing diarrhea; however, other infectious and medication-induced causes of diarrhea also should be considered.6 In contrast to hematopoietic stem cell transplantation, in which the liver is usually involved,1 the liver is spared in acute GVHD following liver transplantation.5
Diagnosis of GVHD
The diagnosis of acute GVHD following liver transplantation can be challenging because the clinical manifestations can be caused by a drug reaction or viral infection, such as cytomegalovirus infection.2 Patients who are older than 50 years and glucose intolerant are at a higher risk of acute GVHD following OLT. The combination of younger donor age and the presence of an HLA class I match also increases the risk of acute GVHD.6 The diagnosis of acute GVHD is confirmed with biopsy of the skin or gastrointestinal tract.
Morbidity and Mortality of GVHD
Because of the high morbidity and mortality associated with acute GVHD following liver transplantation, early diagnosis and treatment are crucial.5 Death in patients with acute GVHD following OLT is mainly attributable to sepsis, multiorgan failure, and gastrointestinal tract bleeding.6 It remains unclear whether this high mortality is associated with delayed diagnosis due to nonspecific signs of acute GVHD following OLT or to the lack of appropriate treatment guidelines.6
Treatment Options
Because of the low incidence of acute GVHD following OLT, most treatment modalities are extrapolated from the literature on acute GVHD following stem cell transplantation.5 The most commonly used therapies include high-dose systemic steroids and anti–thymocyte globulin that attacks activated donor T cells.6 Other treatment modalities, including anti–tumor necrosis factor agents and antibodies to CD20, have been reported to be effective in steroid-refractory GVHD.2 The major drawback of systemic steroids is an increase in the risk for sepsis and infection; therefore, these patients should be diligently screened for infection and covered with antibiotics and antifungals. Extracorporeal photopheresis is another treatment modality that does not cause generalized immunosuppression but is not well studied in the setting of acute GVHD following OLT.6
Prevention
Acute GVHD following OLT can be prevented by eliminating donor T lymphocytes from the liver before transplantation. However, because the incidence of acute GVHD following OLT is very low, this approach is not routinely taken.2
Conclusion
Acute GVHD following liver transplantation is a rare complication; however, it has high mortality, necessitating further research regarding treatment and prevention. Early recognition and treatment of this condition can improve outcomes. Dermatologists should be familiar with the skin manifestations of acute GVHD following liver transplantation due to the rising number of cases of solid-organ transplantation.
Acute graft-vs-host disease (GVHD) is a T-cell mediated immunogenic response in which T lymphocytes from a donor regard host tissue as foreign and attack it in the setting of immunosuppression.1 The most common cause of acute GVHD is allogeneic stem cell transplantation, with solid-organ transplantation being a much less common cause.2 The incidence of acute GVHD following orthotopic liver transplantation (OLT) is 0.1%, as reported by the United Network for Organ Sharing, compared to an incidence of 40% to 60% in hematopoietic stem cell transplant recipients.3,4
Early recognition and treatment of acute GVHD following liver transplantation is imperative, as the mortality rate is 85% to 90%.2 We present a case of acute GVHD in a liver transplantation patient, with a focus on diagnostic criteria and comparison to acute GVHD following hematopoietic stem cell transplantation.
Case Report
A 68-year-old woman with a history of hepatitis C virus infection, hepatocellular carcinoma, and OLT 1 month prior presented to the hospital with fever and abdominal cellulitis in close proximity to the surgical site of 1 week’s duration. The patient was started on vancomycin and cefepime; pan cultures were performed.
At 10 days of hospitalization, the patient developed a pruritic, nontender, erythematous rash on the abdomen, with extension onto the chest and legs. The rash was associated with low-grade fever but not with diarrhea. Physical examination was notable for a few erythematous macules and scattered papules over the neck and chest and a large erythematous plaque with multiple ecchymoses over the lower abdomen (Figure 1A). Erythematous macules and papules coalescing into plaques were present on the lower back (Figure 1B) and proximal thighs. Oral, ocular, and genital lesions were absent.
The differential diagnosis included drug reaction, viral infection, and acute GVHD. A skin biopsy was performed from the left side of the chest. Cefepime and vancomycin were discontinued; triamcinolone ointment 0.1% twice daily and antihistamines as needed for itching were started.
Over a 2-day period, the rash progressed to diffuse erythematous papules over the chest (Figure 2A) and bilateral arms (Figure 2B) including the palms. The patient also developed erythematous papules over the jawline and forehead as well as confluent erythematous plaques over the back with extension of the rash to involve the legs. She also had erythema and swelling bilaterally over the ears. She reported diarrhea. The low-grade fever resolved.
Laboratory review showed new-onset pancytopenia, normal liver function, and an elevated creatinine level of 2.3 mg/dL (reference range, 0.6–1.2 mg/dL), consistent with the patient’s baseline of stage 3 chronic kidney disease. Polymerase chain reaction analysis for cytomegalovirus was negative. Histology revealed vacuolar interface dermatitis with apoptotic keratinocytes, consistent with grade I GVHD (Figure 3). Duodenal biopsy revealed rare patchy glands with increased apoptosis, compatible with grade I GVHD.
The patient was started on intravenous methylprednisolone 1 mg/kg for 3 days, then transitioned to an oral steroid taper, with improvement of the rash and other systemic symptoms.
Comment
GVHD Subtypes
The 2 types of GVHD are humoral and cellular.5 The humoral type results from ABO blood type incompatibility between donor and recipient and causes mild hemolytic anemia and fever. The cellular type is directed against major histocompatibility complexes and is associated with high morbidity and mortality.
Presentation of GVHD
Acute GVHD following OLT usually occurs 3 to 5 weeks after transplantation,6 as in our patient. Symptoms include rash, fever, pancytopenia, and diarrhea.2 Skin is the most commonly involved organ in acute GVHD; rash is the earliest manifestation.1 The rash can be asymptomatic or associated with pain and pruritus. Initial cutaneous manifestations include palmar erythema and erythematous to violaceous discoloration of the face and ears. A diffuse maculopapular rash can develop, involving the face, abdomen, and trunk. The rash may progress to formation of bullae or skin sloughing, resembling Stevens-Johnson syndrome or toxic epidermal necrolysis.1 The skin manifestation of acute GVHD following OLT is similar to hematopoietic stem cell transplantation (Table).7,8
Pancytopenia is a common manifestation of GVHD following liver transplantation and is rarely seen following hematopoietic stem cell transplantation.7 Donor lymphocytes engraft and proliferate in the bone marrow, attacking recipient hematopoietic stem cells. It is important to note that more common causes of cytopenia following liver transplantation, including infection and drug-induced bone marrow suppression, should be ruled out before diagnosing acute GVHD.6
Acute GVHD can affect the gastrointestinal tract, causing diarrhea; however, other infectious and medication-induced causes of diarrhea also should be considered.6 In contrast to hematopoietic stem cell transplantation, in which the liver is usually involved,1 the liver is spared in acute GVHD following liver transplantation.5
Diagnosis of GVHD
The diagnosis of acute GVHD following liver transplantation can be challenging because the clinical manifestations can be caused by a drug reaction or viral infection, such as cytomegalovirus infection.2 Patients who are older than 50 years and glucose intolerant are at a higher risk of acute GVHD following OLT. The combination of younger donor age and the presence of an HLA class I match also increases the risk of acute GVHD.6 The diagnosis of acute GVHD is confirmed with biopsy of the skin or gastrointestinal tract.
Morbidity and Mortality of GVHD
Because of the high morbidity and mortality associated with acute GVHD following liver transplantation, early diagnosis and treatment are crucial.5 Death in patients with acute GVHD following OLT is mainly attributable to sepsis, multiorgan failure, and gastrointestinal tract bleeding.6 It remains unclear whether this high mortality is associated with delayed diagnosis due to nonspecific signs of acute GVHD following OLT or to the lack of appropriate treatment guidelines.6
Treatment Options
Because of the low incidence of acute GVHD following OLT, most treatment modalities are extrapolated from the literature on acute GVHD following stem cell transplantation.5 The most commonly used therapies include high-dose systemic steroids and anti–thymocyte globulin that attacks activated donor T cells.6 Other treatment modalities, including anti–tumor necrosis factor agents and antibodies to CD20, have been reported to be effective in steroid-refractory GVHD.2 The major drawback of systemic steroids is an increase in the risk for sepsis and infection; therefore, these patients should be diligently screened for infection and covered with antibiotics and antifungals. Extracorporeal photopheresis is another treatment modality that does not cause generalized immunosuppression but is not well studied in the setting of acute GVHD following OLT.6
Prevention
Acute GVHD following OLT can be prevented by eliminating donor T lymphocytes from the liver before transplantation. However, because the incidence of acute GVHD following OLT is very low, this approach is not routinely taken.2
Conclusion
Acute GVHD following liver transplantation is a rare complication; however, it has high mortality, necessitating further research regarding treatment and prevention. Early recognition and treatment of this condition can improve outcomes. Dermatologists should be familiar with the skin manifestations of acute GVHD following liver transplantation due to the rising number of cases of solid-organ transplantation.
- Hu SW, Cotliar J. Acute graft-versus-host disease following hematopoietic stem-cell transplantation. Dermatol Ther. 2011;24:411-423.
- Akbulut S, Yilmaz M, Yilmaz S. Graft-versus-host disease after liver transplantation: a comprehensive literature review. World J Gastroenterol. 2012;18:5240-5248.
- Taylor AL, Gibbs P, Bradley JA. Acute graft versus host disease following liver transplantation: the enemy within. Am J Transplant. 2004;4:466-474.
- Jagasia M, Arora M, Flowers ME, et al. Risk factor for acute GVHD and survival after hematopoietic cell transplantation. Blood. 2012;119:296-307.
- Kang WH, Hwang S, Song GW, et al. Acute graft-vs-host disease after liver transplantation: experience at a high-volume liver transplantation center in Korea. Transplant Proc. 2016;48:3368-3372.
- Murali AR, Chandra S, Stewart Z, et al. Graft versus host disease after liver transplantation in adults: a case series, review of literature, and an approach to management. Transplantation. 2016;100:2661-2670.
- Chaib E, Silva FD, Figueira ER, et al. Graft-versus-host disease after liver transplantation. Clinics (Sao Paulo). 2011;66:1115-1118.
- Barton-Burke M, Dwinell DM, Kafkas L, et al. Graft-versus-host disease: a complex long-term side effect of hematopoietic stem cell transplant. Oncology (Williston Park). 2008;22(11 Suppl Nurse Ed):31-45.
- Hu SW, Cotliar J. Acute graft-versus-host disease following hematopoietic stem-cell transplantation. Dermatol Ther. 2011;24:411-423.
- Akbulut S, Yilmaz M, Yilmaz S. Graft-versus-host disease after liver transplantation: a comprehensive literature review. World J Gastroenterol. 2012;18:5240-5248.
- Taylor AL, Gibbs P, Bradley JA. Acute graft versus host disease following liver transplantation: the enemy within. Am J Transplant. 2004;4:466-474.
- Jagasia M, Arora M, Flowers ME, et al. Risk factor for acute GVHD and survival after hematopoietic cell transplantation. Blood. 2012;119:296-307.
- Kang WH, Hwang S, Song GW, et al. Acute graft-vs-host disease after liver transplantation: experience at a high-volume liver transplantation center in Korea. Transplant Proc. 2016;48:3368-3372.
- Murali AR, Chandra S, Stewart Z, et al. Graft versus host disease after liver transplantation in adults: a case series, review of literature, and an approach to management. Transplantation. 2016;100:2661-2670.
- Chaib E, Silva FD, Figueira ER, et al. Graft-versus-host disease after liver transplantation. Clinics (Sao Paulo). 2011;66:1115-1118.
- Barton-Burke M, Dwinell DM, Kafkas L, et al. Graft-versus-host disease: a complex long-term side effect of hematopoietic stem cell transplant. Oncology (Williston Park). 2008;22(11 Suppl Nurse Ed):31-45.
Practice Points
- Acute graft-vs-host disease (GVHD) is a T cell–mediated reaction in which donor T lymphocytes attack host tissue in the setting of immunosuppression.
- Acute GVHD is more common in allogeneic stem cell transplantation but can occur in the setting of solid organ transplantation.
- Symptoms of acute GVHD include rash with or without pruritus, fever, pancytopenia, and diarrhea.
- Early recognition and treatment with systemic steroids can improve mortality.
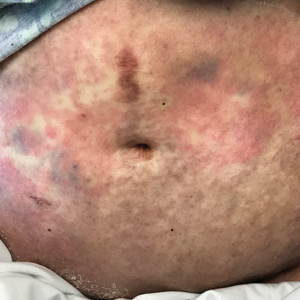
Surgical Dermatoethics for the Trainee
It is an uncomfortable and unavoidable reality as physicians that for every procedure we learn, there must be a first time we perform it. As with any type of skill, it takes practice to become proficient. The unique challenge in medicine is that the practice involves performing procedures on real patients. We cannot avoid the hands-on nature of the training process; we can, however, approach its ethical challenges mindfully. Herein, I will discuss some of the ethical considerations in providing care as a trainee and identify potential barriers to best practices, particularly as they relate to procedural dermatology.
Tell Patients You Are in Training
In every patient encounter, we must introduce ourselves as a trainee. The principle of right to the truth dictates that we are transparent about our level of training and do not misrepresent ourselves to our patients. A statement released by the American Medical Association (AMA) Council on Ethical and Judicial Affairs asserts that “[p]atients should be informed of the identity and training status of individuals involved in their care.”1
Although straightforward in theory, this mandate is not always simple in practice. With patients unfamiliar with the health care system, it could be more onerous to clearly communicate training status than simply introducing oneself as a resident. A study conducted in the emergency department at Vanderbilt University Hospital (Nashville, Tennessee) found that many patients and their family members (N=430) did not understand the various roles and responsibilities of physicians in the teaching hospital setting. For example, 30% believed an attending physician requires supervision by a resident, and an additional 17% of those surveyed were not sure.2 The AMA requests we “refrain from using terms that may be confusing when describing the training status of the students,”1 which evidently is audience specific. Thus, as with any type of patient education, a thorough introduction may require assessment of understanding.
Disclosure of Experience Level With a Particular Procedure
There is a clear professional expectation that we disclose to patients that we are in training; however, a universal standard does not exist for disclosure of our exact level of experience in a particular procedure. Do we need to tell patients if it is our first time performing a given procedure? What if it is our tenth? Multiple studies have found that patients want specifics. In one study of bariatric surgery patients (N=108), 93% felt that they should always be informed if it was the first time a trainee was performing a particular procedure.3 A study conducted in the emergency department setting (N=202) also found that the majority of patients thought they should be informed if a resident was performing a procedure for the first time, but the distribution differed by procedure (66% for suturing vs 82% for lumbar puncture).4
Despite these findings, this degree of specificity is not always discussed with patients and perhaps does not need to be. LaRosa and Grant-Kels5 analyzed a hypothetical scenario in which a dermatology resident is to perform his first excision under attending supervision and concluded that broad disclosure of training status would suffice in the given scenario, as it would not be necessary to state that it was his first time performing an excision. It is unclear if the same conclusion could be drawn for all procedures and levels of experience. Outcome data would help inform the analysis, but the available data are from other specialties including general surgery, gynecology, and urology. Some studies demonstrate an increased risk of adverse outcomes with trainee involvement in procedures such as bariatric surgery and emergency general surgery, but the data are mixed and may not be generalizable to dermatologic procedures.6-8
The appropriate level of detail to disclose regarding a physician’s experience may need to be assessed on a case-by-case basis, and the principles of informed consent can help. Informed consent requires understanding of the diagnosis, the treatment options including nonintervention, and the risks and benefits of each alternative. In obtaining informed consent, we must disclose “any facts which are necessary to form the basis of an intelligent consent by the patient to the proposed treatment.”9 Providers must determine what aspects of a trainee’s experience level are relevant to the risk-benefit analysis in a given set of circumstances. Surely, there is a large degree of subjectivity in this determination as data are limited, but information deemed relevant must be shared. Information that is inconsequential, on the other hand, may be omitted. It could even be argued that more detailed information, especially if it may cause anxiety, would be detrimental to share. For example, we would not list the chemical name of every preservative in every vaccine we recommend for children if there is no evidence of inflicting harm. If the information has not been shown to have clinical impact or affect safety concerns, the anxiety may be undue.
Withholding Information Can Violate Ethical Principles
We must be careful not to withhold details of our experience level with a particular procedure for the wrong reasons. It would be wrong, for example, to withhold information simply to avoid causing anxiety, which could be seen as an invocation of therapeutic privilege, a controversial practice of withholding important information that poses a psychological threat to the patient. A classic example is the physician who defers disclosure of a terminal diagnosis to preserve hope. Although therapeutic privilege theoretically promotes the principle of beneficence, it violates the principles of autonomy and right to truth and therefore generally is regarded as unethically paternalistic in modern medical ethics.9
Patients Can Refuse Trainee Participation
It also is unethical to withhold information to obtain consent and avoid refusal of our care. Refusal of trainee participation is not uncommon. In the aforementioned study of bariatric surgery patients, 92.4% supported their procedure being performed at a teaching hospital, but only 56% would consent to a resident assisting staff during the procedure. A mere 33% of those patients would consent to a resident primarily performing with staff assisting.3 Although the proportion of patients who refuse certainly depends on the type of procedure among other factors, it is a reality in any teaching environment. The training paradigm in medicine depends on being able to practice procedures with supervision before we are independent providers. If patients refuse our care, our training suffers. However, the AMA maintains that “[p]atients are free to choose from whom they receive treatment,”1 and we must respect this aspect of patient autonomy.
Final Thoughts
When it comes to the performance of procedures, there are a few basic principles to keep in mind to provide ethical care to our patients while we are in training. Although we must accept that a crucial part of learning dermatologic procedures is hands on with real patients, we also need to come prepared having learned what we can through reading and practice with cadavers or skin substitutes. Procedures we execute as residents should be performed with adequate supervision, and as we progress through residency, we should be given increased autonomy and graded responsibility to prepare us for independent practice at graduation. Although it is the responsibility of the attending physician to provide appropriate oversight for the resident’s level of training, we should feel empowered to ask for help and have the humility to know when we need it.
- Medical student involvement in patient care: report of the council on ethical and judicial affairs. Virtual Mentor. 2001;3. doi:10.1001/virtualmentor.2001.3.3.code1-0103.
- Santen S, Hemphill RR, Prough E, et al. Do patients understand their physician’s level of training? a survey of emergency department patients. Acad Med. 2004;79:139-143.
- McClellan JM, Nelson D, Porta CR, et al. Bariatric surgery patient perceptions and willingness to consent to resident participation. Surg Obes Relat Dis. 2016;12:1065-1071.
- Santen SA, Hemphill RR, McDonald MF, et al. Patients’ willingness to allow residents to learn to practice medical procedures. Acad Med. 2004;79:144-147.
- LaRosa C, Grant-Kels JM. See one, do one, teach one: the ethical dilemma of residents performing their first procedure on patients. J Am Acad Dermatol. 2016;75:845-848.
- Can MF. The trainee effect on early postoperative surgical outcomes: reflects the effect of resident involvement or hospital capacity to overcome complications? J Invest Surg. 2017;31:67-68.
- Goldberg I, Yang J, Park J, et al. Surgical trainee impact on bariatric surgery safety [published online November 13, 2018]. Surg Endosc. doi:10.1007/s00464-018-6587-0.
- Kasotakis G, Lakha A, Sarkar B, et al. Trainee participation is associated with adverse outcomes in emergency general surgery: an analysis of the National Surgical Quality Improvement Program database. Ann Surg. 2014;3:483-490.
- Richard C, Lajeunesse Y, Lussier MT. Therapeutic privilege: between the ethics of lying and the practice of truth. J Med Ethics. 2010;36:353-357.
It is an uncomfortable and unavoidable reality as physicians that for every procedure we learn, there must be a first time we perform it. As with any type of skill, it takes practice to become proficient. The unique challenge in medicine is that the practice involves performing procedures on real patients. We cannot avoid the hands-on nature of the training process; we can, however, approach its ethical challenges mindfully. Herein, I will discuss some of the ethical considerations in providing care as a trainee and identify potential barriers to best practices, particularly as they relate to procedural dermatology.
Tell Patients You Are in Training
In every patient encounter, we must introduce ourselves as a trainee. The principle of right to the truth dictates that we are transparent about our level of training and do not misrepresent ourselves to our patients. A statement released by the American Medical Association (AMA) Council on Ethical and Judicial Affairs asserts that “[p]atients should be informed of the identity and training status of individuals involved in their care.”1
Although straightforward in theory, this mandate is not always simple in practice. With patients unfamiliar with the health care system, it could be more onerous to clearly communicate training status than simply introducing oneself as a resident. A study conducted in the emergency department at Vanderbilt University Hospital (Nashville, Tennessee) found that many patients and their family members (N=430) did not understand the various roles and responsibilities of physicians in the teaching hospital setting. For example, 30% believed an attending physician requires supervision by a resident, and an additional 17% of those surveyed were not sure.2 The AMA requests we “refrain from using terms that may be confusing when describing the training status of the students,”1 which evidently is audience specific. Thus, as with any type of patient education, a thorough introduction may require assessment of understanding.
Disclosure of Experience Level With a Particular Procedure
There is a clear professional expectation that we disclose to patients that we are in training; however, a universal standard does not exist for disclosure of our exact level of experience in a particular procedure. Do we need to tell patients if it is our first time performing a given procedure? What if it is our tenth? Multiple studies have found that patients want specifics. In one study of bariatric surgery patients (N=108), 93% felt that they should always be informed if it was the first time a trainee was performing a particular procedure.3 A study conducted in the emergency department setting (N=202) also found that the majority of patients thought they should be informed if a resident was performing a procedure for the first time, but the distribution differed by procedure (66% for suturing vs 82% for lumbar puncture).4
Despite these findings, this degree of specificity is not always discussed with patients and perhaps does not need to be. LaRosa and Grant-Kels5 analyzed a hypothetical scenario in which a dermatology resident is to perform his first excision under attending supervision and concluded that broad disclosure of training status would suffice in the given scenario, as it would not be necessary to state that it was his first time performing an excision. It is unclear if the same conclusion could be drawn for all procedures and levels of experience. Outcome data would help inform the analysis, but the available data are from other specialties including general surgery, gynecology, and urology. Some studies demonstrate an increased risk of adverse outcomes with trainee involvement in procedures such as bariatric surgery and emergency general surgery, but the data are mixed and may not be generalizable to dermatologic procedures.6-8
The appropriate level of detail to disclose regarding a physician’s experience may need to be assessed on a case-by-case basis, and the principles of informed consent can help. Informed consent requires understanding of the diagnosis, the treatment options including nonintervention, and the risks and benefits of each alternative. In obtaining informed consent, we must disclose “any facts which are necessary to form the basis of an intelligent consent by the patient to the proposed treatment.”9 Providers must determine what aspects of a trainee’s experience level are relevant to the risk-benefit analysis in a given set of circumstances. Surely, there is a large degree of subjectivity in this determination as data are limited, but information deemed relevant must be shared. Information that is inconsequential, on the other hand, may be omitted. It could even be argued that more detailed information, especially if it may cause anxiety, would be detrimental to share. For example, we would not list the chemical name of every preservative in every vaccine we recommend for children if there is no evidence of inflicting harm. If the information has not been shown to have clinical impact or affect safety concerns, the anxiety may be undue.
Withholding Information Can Violate Ethical Principles
We must be careful not to withhold details of our experience level with a particular procedure for the wrong reasons. It would be wrong, for example, to withhold information simply to avoid causing anxiety, which could be seen as an invocation of therapeutic privilege, a controversial practice of withholding important information that poses a psychological threat to the patient. A classic example is the physician who defers disclosure of a terminal diagnosis to preserve hope. Although therapeutic privilege theoretically promotes the principle of beneficence, it violates the principles of autonomy and right to truth and therefore generally is regarded as unethically paternalistic in modern medical ethics.9
Patients Can Refuse Trainee Participation
It also is unethical to withhold information to obtain consent and avoid refusal of our care. Refusal of trainee participation is not uncommon. In the aforementioned study of bariatric surgery patients, 92.4% supported their procedure being performed at a teaching hospital, but only 56% would consent to a resident assisting staff during the procedure. A mere 33% of those patients would consent to a resident primarily performing with staff assisting.3 Although the proportion of patients who refuse certainly depends on the type of procedure among other factors, it is a reality in any teaching environment. The training paradigm in medicine depends on being able to practice procedures with supervision before we are independent providers. If patients refuse our care, our training suffers. However, the AMA maintains that “[p]atients are free to choose from whom they receive treatment,”1 and we must respect this aspect of patient autonomy.
Final Thoughts
When it comes to the performance of procedures, there are a few basic principles to keep in mind to provide ethical care to our patients while we are in training. Although we must accept that a crucial part of learning dermatologic procedures is hands on with real patients, we also need to come prepared having learned what we can through reading and practice with cadavers or skin substitutes. Procedures we execute as residents should be performed with adequate supervision, and as we progress through residency, we should be given increased autonomy and graded responsibility to prepare us for independent practice at graduation. Although it is the responsibility of the attending physician to provide appropriate oversight for the resident’s level of training, we should feel empowered to ask for help and have the humility to know when we need it.
It is an uncomfortable and unavoidable reality as physicians that for every procedure we learn, there must be a first time we perform it. As with any type of skill, it takes practice to become proficient. The unique challenge in medicine is that the practice involves performing procedures on real patients. We cannot avoid the hands-on nature of the training process; we can, however, approach its ethical challenges mindfully. Herein, I will discuss some of the ethical considerations in providing care as a trainee and identify potential barriers to best practices, particularly as they relate to procedural dermatology.
Tell Patients You Are in Training
In every patient encounter, we must introduce ourselves as a trainee. The principle of right to the truth dictates that we are transparent about our level of training and do not misrepresent ourselves to our patients. A statement released by the American Medical Association (AMA) Council on Ethical and Judicial Affairs asserts that “[p]atients should be informed of the identity and training status of individuals involved in their care.”1
Although straightforward in theory, this mandate is not always simple in practice. With patients unfamiliar with the health care system, it could be more onerous to clearly communicate training status than simply introducing oneself as a resident. A study conducted in the emergency department at Vanderbilt University Hospital (Nashville, Tennessee) found that many patients and their family members (N=430) did not understand the various roles and responsibilities of physicians in the teaching hospital setting. For example, 30% believed an attending physician requires supervision by a resident, and an additional 17% of those surveyed were not sure.2 The AMA requests we “refrain from using terms that may be confusing when describing the training status of the students,”1 which evidently is audience specific. Thus, as with any type of patient education, a thorough introduction may require assessment of understanding.
Disclosure of Experience Level With a Particular Procedure
There is a clear professional expectation that we disclose to patients that we are in training; however, a universal standard does not exist for disclosure of our exact level of experience in a particular procedure. Do we need to tell patients if it is our first time performing a given procedure? What if it is our tenth? Multiple studies have found that patients want specifics. In one study of bariatric surgery patients (N=108), 93% felt that they should always be informed if it was the first time a trainee was performing a particular procedure.3 A study conducted in the emergency department setting (N=202) also found that the majority of patients thought they should be informed if a resident was performing a procedure for the first time, but the distribution differed by procedure (66% for suturing vs 82% for lumbar puncture).4
Despite these findings, this degree of specificity is not always discussed with patients and perhaps does not need to be. LaRosa and Grant-Kels5 analyzed a hypothetical scenario in which a dermatology resident is to perform his first excision under attending supervision and concluded that broad disclosure of training status would suffice in the given scenario, as it would not be necessary to state that it was his first time performing an excision. It is unclear if the same conclusion could be drawn for all procedures and levels of experience. Outcome data would help inform the analysis, but the available data are from other specialties including general surgery, gynecology, and urology. Some studies demonstrate an increased risk of adverse outcomes with trainee involvement in procedures such as bariatric surgery and emergency general surgery, but the data are mixed and may not be generalizable to dermatologic procedures.6-8
The appropriate level of detail to disclose regarding a physician’s experience may need to be assessed on a case-by-case basis, and the principles of informed consent can help. Informed consent requires understanding of the diagnosis, the treatment options including nonintervention, and the risks and benefits of each alternative. In obtaining informed consent, we must disclose “any facts which are necessary to form the basis of an intelligent consent by the patient to the proposed treatment.”9 Providers must determine what aspects of a trainee’s experience level are relevant to the risk-benefit analysis in a given set of circumstances. Surely, there is a large degree of subjectivity in this determination as data are limited, but information deemed relevant must be shared. Information that is inconsequential, on the other hand, may be omitted. It could even be argued that more detailed information, especially if it may cause anxiety, would be detrimental to share. For example, we would not list the chemical name of every preservative in every vaccine we recommend for children if there is no evidence of inflicting harm. If the information has not been shown to have clinical impact or affect safety concerns, the anxiety may be undue.
Withholding Information Can Violate Ethical Principles
We must be careful not to withhold details of our experience level with a particular procedure for the wrong reasons. It would be wrong, for example, to withhold information simply to avoid causing anxiety, which could be seen as an invocation of therapeutic privilege, a controversial practice of withholding important information that poses a psychological threat to the patient. A classic example is the physician who defers disclosure of a terminal diagnosis to preserve hope. Although therapeutic privilege theoretically promotes the principle of beneficence, it violates the principles of autonomy and right to truth and therefore generally is regarded as unethically paternalistic in modern medical ethics.9
Patients Can Refuse Trainee Participation
It also is unethical to withhold information to obtain consent and avoid refusal of our care. Refusal of trainee participation is not uncommon. In the aforementioned study of bariatric surgery patients, 92.4% supported their procedure being performed at a teaching hospital, but only 56% would consent to a resident assisting staff during the procedure. A mere 33% of those patients would consent to a resident primarily performing with staff assisting.3 Although the proportion of patients who refuse certainly depends on the type of procedure among other factors, it is a reality in any teaching environment. The training paradigm in medicine depends on being able to practice procedures with supervision before we are independent providers. If patients refuse our care, our training suffers. However, the AMA maintains that “[p]atients are free to choose from whom they receive treatment,”1 and we must respect this aspect of patient autonomy.
Final Thoughts
When it comes to the performance of procedures, there are a few basic principles to keep in mind to provide ethical care to our patients while we are in training. Although we must accept that a crucial part of learning dermatologic procedures is hands on with real patients, we also need to come prepared having learned what we can through reading and practice with cadavers or skin substitutes. Procedures we execute as residents should be performed with adequate supervision, and as we progress through residency, we should be given increased autonomy and graded responsibility to prepare us for independent practice at graduation. Although it is the responsibility of the attending physician to provide appropriate oversight for the resident’s level of training, we should feel empowered to ask for help and have the humility to know when we need it.
- Medical student involvement in patient care: report of the council on ethical and judicial affairs. Virtual Mentor. 2001;3. doi:10.1001/virtualmentor.2001.3.3.code1-0103.
- Santen S, Hemphill RR, Prough E, et al. Do patients understand their physician’s level of training? a survey of emergency department patients. Acad Med. 2004;79:139-143.
- McClellan JM, Nelson D, Porta CR, et al. Bariatric surgery patient perceptions and willingness to consent to resident participation. Surg Obes Relat Dis. 2016;12:1065-1071.
- Santen SA, Hemphill RR, McDonald MF, et al. Patients’ willingness to allow residents to learn to practice medical procedures. Acad Med. 2004;79:144-147.
- LaRosa C, Grant-Kels JM. See one, do one, teach one: the ethical dilemma of residents performing their first procedure on patients. J Am Acad Dermatol. 2016;75:845-848.
- Can MF. The trainee effect on early postoperative surgical outcomes: reflects the effect of resident involvement or hospital capacity to overcome complications? J Invest Surg. 2017;31:67-68.
- Goldberg I, Yang J, Park J, et al. Surgical trainee impact on bariatric surgery safety [published online November 13, 2018]. Surg Endosc. doi:10.1007/s00464-018-6587-0.
- Kasotakis G, Lakha A, Sarkar B, et al. Trainee participation is associated with adverse outcomes in emergency general surgery: an analysis of the National Surgical Quality Improvement Program database. Ann Surg. 2014;3:483-490.
- Richard C, Lajeunesse Y, Lussier MT. Therapeutic privilege: between the ethics of lying and the practice of truth. J Med Ethics. 2010;36:353-357.
- Medical student involvement in patient care: report of the council on ethical and judicial affairs. Virtual Mentor. 2001;3. doi:10.1001/virtualmentor.2001.3.3.code1-0103.
- Santen S, Hemphill RR, Prough E, et al. Do patients understand their physician’s level of training? a survey of emergency department patients. Acad Med. 2004;79:139-143.
- McClellan JM, Nelson D, Porta CR, et al. Bariatric surgery patient perceptions and willingness to consent to resident participation. Surg Obes Relat Dis. 2016;12:1065-1071.
- Santen SA, Hemphill RR, McDonald MF, et al. Patients’ willingness to allow residents to learn to practice medical procedures. Acad Med. 2004;79:144-147.
- LaRosa C, Grant-Kels JM. See one, do one, teach one: the ethical dilemma of residents performing their first procedure on patients. J Am Acad Dermatol. 2016;75:845-848.
- Can MF. The trainee effect on early postoperative surgical outcomes: reflects the effect of resident involvement or hospital capacity to overcome complications? J Invest Surg. 2017;31:67-68.
- Goldberg I, Yang J, Park J, et al. Surgical trainee impact on bariatric surgery safety [published online November 13, 2018]. Surg Endosc. doi:10.1007/s00464-018-6587-0.
- Kasotakis G, Lakha A, Sarkar B, et al. Trainee participation is associated with adverse outcomes in emergency general surgery: an analysis of the National Surgical Quality Improvement Program database. Ann Surg. 2014;3:483-490.
- Richard C, Lajeunesse Y, Lussier MT. Therapeutic privilege: between the ethics of lying and the practice of truth. J Med Ethics. 2010;36:353-357.
Resident Pearl
- As residents, we must gain experience performing procedures on real patients to enter independent practice as proficient dermatologists. It is important to be mindful of the ethical challenges inherent to the hands-on training process and to understand the ethical principles that guide best practices.

Back to the Future: Integrating Technology to Improve Patient-Provider Interactions
The advent of electronic medical records (EMRs) is arguably the most important technological revolution in modern medicine. The transition from paper documentation to EMRs has improved organization of medical records, consolidating all physician notes, orders, consultations, laboratory test results, and radiologic studies into a single accessible location.1 However, this revolution has led to mixed consequences for patients, especially in the outpatient setting. The use of EMRs can facilitate questions, clarification, and discussion between patients and health care providers, prompted by the sections of the EMR. Unfortunately, patients too often encounter pressed-for-time, documentation-focused providers who may not even look up from the computer. Provider behaviors such as making eye contact, stopping typing during discussion of sensitive topics, and allowing patients to view the computer screen and using it as an educational tool are important for patients to have a positive care experience.2 We envision further integration of current and future technology to overcome the challenges of outpatient care. We use a hypothetical patient encounter to illustrate what the future may hold.
Hypothetical Patient Encounter
An established patient, Ms. PS, comes to the dermatology clinic for a follow-up appointment and walks into an examination room (Figure). Prior to entering the room, the provider, Dr. FT, reviews Ms. PS’s history via a dermatology-specific EMR and reads that Ms. PS has a 1.5-year history of psoriasis and is considering other therapeutic options.
Upon entering the room, Dr. FT tells Ms. PS that the visit is being recorded and transcribed. A large interactive screen is a key component of the examination room. A remote medical assistant is virtually present via video to transcribe and document the patient-provider interaction. There is potential for artificial intelligence to replace the remote medical assistant in the future. Wearable technology, including a smartwatch and Bluetooth headphones, allow the provider to record audio of the visit as well as through microphones on the interactive screen.
As the interaction begins, Ms. PS reports that her psoriasis is poorly controlled with her current regimen of topical steroids. Dr. FT inquires about Ms. PS’s current symptoms and psychosocial well-being. Dr. FT then performs a skin examination and is easily able to evaluate her skin vs prior visits, as clinical images from prior visits are automatically displayed on the interactive screen. Dr. FT also closely examines Ms. PS’s nails and conducts a joint examination, reminded by a notification on his wearable technology. After capturing clinical images of Ms. PS’s skin and nails with a secure EMR-connected tablet, Dr. FT briefly steps out of the room to allow Ms. PS to get dressed and feel more comfortable in the discussion to follow.
Once he reenters the examination room, Dr. FT initiates a discussion on next steps. Ms. PS’s pathology report and clinical images are displayed on the interactive screen, along with her most recent laboratory test results, which were completed prior to the visit in anticipation of changing therapies. Dr. FT presents Ms. PS with several evidence-based therapeutic options for psoriasis, and she expresses interest in methotrexate. Following the discussion, the remote medical assistant displays information about methotrexate on the interactive screen, including evidence for treatment of psoriasis, contraindications, laboratory monitoring requirements, and possible adverse effects for both the patient and provider to review together. Dr. FT reviews the laboratory test results displayed on the screen, specifically her transaminase levels, and confirms that methotrexate is an appropriate therapeutic option. After a full discussion of risks and benefits, Ms. PS chooses to initiate methotrexate treatment. Reminded by a notification on his wearable technology, Dr. FT follows evidence-based dosing guidelines and sends the prescription electronically to Ms. PS’s pharmacy, which concludes Ms. PS’s visit.
Analysis of the Patient Encounter
In this interaction, Dr. FT was able to fully engage with the patient, unencumbered by the demands of documentation. There were only a few instances when the provider looked at or touched the interactive screen. Furthermore, joint decision-making was optimized by allowing both the patient and provider to review diagnostic test results and current evidence-based therapeutic guidelines together through the interactive screen. Ms. PS goes home feeling satisfied that she received her provider’s complete attention and that they selected a therapeutic option supported by evidence. After the visit, the remote medial assistant’s transcript populates a patient note template, which Dr. FT reviews and amends to create the final note. Reducing the time required to write patient notes increases the speed at which Dr. FT can complete patient encounters and may improve clinic flow and productivity. In addition, a patient summary is generated from Dr. FT’s final note, with an emphasis on patient instructions, and is sent to Ms. PS.
Final Thoughts
Our proposed integration of currently available and future technology can help minimize documentation burdens on providers and improve patient-provider communication in the age of the EMR, thus optimizing patient satisfaction and outcomes.
- Evans RS. Electronic health records: then, now, and in the future. Yearb Med Inform. 2016;(suppl 1):S48-S61.
- Alkureishi MA, Lee WW, Lyons M, et al. Impact of electronic medical record use on the patient-doctor relationship and communication: a systematic review. J Gen Intern Med. 2016;31:548-560.
The advent of electronic medical records (EMRs) is arguably the most important technological revolution in modern medicine. The transition from paper documentation to EMRs has improved organization of medical records, consolidating all physician notes, orders, consultations, laboratory test results, and radiologic studies into a single accessible location.1 However, this revolution has led to mixed consequences for patients, especially in the outpatient setting. The use of EMRs can facilitate questions, clarification, and discussion between patients and health care providers, prompted by the sections of the EMR. Unfortunately, patients too often encounter pressed-for-time, documentation-focused providers who may not even look up from the computer. Provider behaviors such as making eye contact, stopping typing during discussion of sensitive topics, and allowing patients to view the computer screen and using it as an educational tool are important for patients to have a positive care experience.2 We envision further integration of current and future technology to overcome the challenges of outpatient care. We use a hypothetical patient encounter to illustrate what the future may hold.
Hypothetical Patient Encounter
An established patient, Ms. PS, comes to the dermatology clinic for a follow-up appointment and walks into an examination room (Figure). Prior to entering the room, the provider, Dr. FT, reviews Ms. PS’s history via a dermatology-specific EMR and reads that Ms. PS has a 1.5-year history of psoriasis and is considering other therapeutic options.
Upon entering the room, Dr. FT tells Ms. PS that the visit is being recorded and transcribed. A large interactive screen is a key component of the examination room. A remote medical assistant is virtually present via video to transcribe and document the patient-provider interaction. There is potential for artificial intelligence to replace the remote medical assistant in the future. Wearable technology, including a smartwatch and Bluetooth headphones, allow the provider to record audio of the visit as well as through microphones on the interactive screen.
As the interaction begins, Ms. PS reports that her psoriasis is poorly controlled with her current regimen of topical steroids. Dr. FT inquires about Ms. PS’s current symptoms and psychosocial well-being. Dr. FT then performs a skin examination and is easily able to evaluate her skin vs prior visits, as clinical images from prior visits are automatically displayed on the interactive screen. Dr. FT also closely examines Ms. PS’s nails and conducts a joint examination, reminded by a notification on his wearable technology. After capturing clinical images of Ms. PS’s skin and nails with a secure EMR-connected tablet, Dr. FT briefly steps out of the room to allow Ms. PS to get dressed and feel more comfortable in the discussion to follow.
Once he reenters the examination room, Dr. FT initiates a discussion on next steps. Ms. PS’s pathology report and clinical images are displayed on the interactive screen, along with her most recent laboratory test results, which were completed prior to the visit in anticipation of changing therapies. Dr. FT presents Ms. PS with several evidence-based therapeutic options for psoriasis, and she expresses interest in methotrexate. Following the discussion, the remote medical assistant displays information about methotrexate on the interactive screen, including evidence for treatment of psoriasis, contraindications, laboratory monitoring requirements, and possible adverse effects for both the patient and provider to review together. Dr. FT reviews the laboratory test results displayed on the screen, specifically her transaminase levels, and confirms that methotrexate is an appropriate therapeutic option. After a full discussion of risks and benefits, Ms. PS chooses to initiate methotrexate treatment. Reminded by a notification on his wearable technology, Dr. FT follows evidence-based dosing guidelines and sends the prescription electronically to Ms. PS’s pharmacy, which concludes Ms. PS’s visit.
Analysis of the Patient Encounter
In this interaction, Dr. FT was able to fully engage with the patient, unencumbered by the demands of documentation. There were only a few instances when the provider looked at or touched the interactive screen. Furthermore, joint decision-making was optimized by allowing both the patient and provider to review diagnostic test results and current evidence-based therapeutic guidelines together through the interactive screen. Ms. PS goes home feeling satisfied that she received her provider’s complete attention and that they selected a therapeutic option supported by evidence. After the visit, the remote medial assistant’s transcript populates a patient note template, which Dr. FT reviews and amends to create the final note. Reducing the time required to write patient notes increases the speed at which Dr. FT can complete patient encounters and may improve clinic flow and productivity. In addition, a patient summary is generated from Dr. FT’s final note, with an emphasis on patient instructions, and is sent to Ms. PS.
Final Thoughts
Our proposed integration of currently available and future technology can help minimize documentation burdens on providers and improve patient-provider communication in the age of the EMR, thus optimizing patient satisfaction and outcomes.
The advent of electronic medical records (EMRs) is arguably the most important technological revolution in modern medicine. The transition from paper documentation to EMRs has improved organization of medical records, consolidating all physician notes, orders, consultations, laboratory test results, and radiologic studies into a single accessible location.1 However, this revolution has led to mixed consequences for patients, especially in the outpatient setting. The use of EMRs can facilitate questions, clarification, and discussion between patients and health care providers, prompted by the sections of the EMR. Unfortunately, patients too often encounter pressed-for-time, documentation-focused providers who may not even look up from the computer. Provider behaviors such as making eye contact, stopping typing during discussion of sensitive topics, and allowing patients to view the computer screen and using it as an educational tool are important for patients to have a positive care experience.2 We envision further integration of current and future technology to overcome the challenges of outpatient care. We use a hypothetical patient encounter to illustrate what the future may hold.
Hypothetical Patient Encounter
An established patient, Ms. PS, comes to the dermatology clinic for a follow-up appointment and walks into an examination room (Figure). Prior to entering the room, the provider, Dr. FT, reviews Ms. PS’s history via a dermatology-specific EMR and reads that Ms. PS has a 1.5-year history of psoriasis and is considering other therapeutic options.
Upon entering the room, Dr. FT tells Ms. PS that the visit is being recorded and transcribed. A large interactive screen is a key component of the examination room. A remote medical assistant is virtually present via video to transcribe and document the patient-provider interaction. There is potential for artificial intelligence to replace the remote medical assistant in the future. Wearable technology, including a smartwatch and Bluetooth headphones, allow the provider to record audio of the visit as well as through microphones on the interactive screen.
As the interaction begins, Ms. PS reports that her psoriasis is poorly controlled with her current regimen of topical steroids. Dr. FT inquires about Ms. PS’s current symptoms and psychosocial well-being. Dr. FT then performs a skin examination and is easily able to evaluate her skin vs prior visits, as clinical images from prior visits are automatically displayed on the interactive screen. Dr. FT also closely examines Ms. PS’s nails and conducts a joint examination, reminded by a notification on his wearable technology. After capturing clinical images of Ms. PS’s skin and nails with a secure EMR-connected tablet, Dr. FT briefly steps out of the room to allow Ms. PS to get dressed and feel more comfortable in the discussion to follow.
Once he reenters the examination room, Dr. FT initiates a discussion on next steps. Ms. PS’s pathology report and clinical images are displayed on the interactive screen, along with her most recent laboratory test results, which were completed prior to the visit in anticipation of changing therapies. Dr. FT presents Ms. PS with several evidence-based therapeutic options for psoriasis, and she expresses interest in methotrexate. Following the discussion, the remote medical assistant displays information about methotrexate on the interactive screen, including evidence for treatment of psoriasis, contraindications, laboratory monitoring requirements, and possible adverse effects for both the patient and provider to review together. Dr. FT reviews the laboratory test results displayed on the screen, specifically her transaminase levels, and confirms that methotrexate is an appropriate therapeutic option. After a full discussion of risks and benefits, Ms. PS chooses to initiate methotrexate treatment. Reminded by a notification on his wearable technology, Dr. FT follows evidence-based dosing guidelines and sends the prescription electronically to Ms. PS’s pharmacy, which concludes Ms. PS’s visit.
Analysis of the Patient Encounter
In this interaction, Dr. FT was able to fully engage with the patient, unencumbered by the demands of documentation. There were only a few instances when the provider looked at or touched the interactive screen. Furthermore, joint decision-making was optimized by allowing both the patient and provider to review diagnostic test results and current evidence-based therapeutic guidelines together through the interactive screen. Ms. PS goes home feeling satisfied that she received her provider’s complete attention and that they selected a therapeutic option supported by evidence. After the visit, the remote medial assistant’s transcript populates a patient note template, which Dr. FT reviews and amends to create the final note. Reducing the time required to write patient notes increases the speed at which Dr. FT can complete patient encounters and may improve clinic flow and productivity. In addition, a patient summary is generated from Dr. FT’s final note, with an emphasis on patient instructions, and is sent to Ms. PS.
Final Thoughts
Our proposed integration of currently available and future technology can help minimize documentation burdens on providers and improve patient-provider communication in the age of the EMR, thus optimizing patient satisfaction and outcomes.
- Evans RS. Electronic health records: then, now, and in the future. Yearb Med Inform. 2016;(suppl 1):S48-S61.
- Alkureishi MA, Lee WW, Lyons M, et al. Impact of electronic medical record use on the patient-doctor relationship and communication: a systematic review. J Gen Intern Med. 2016;31:548-560.
- Evans RS. Electronic health records: then, now, and in the future. Yearb Med Inform. 2016;(suppl 1):S48-S61.
- Alkureishi MA, Lee WW, Lyons M, et al. Impact of electronic medical record use on the patient-doctor relationship and communication: a systematic review. J Gen Intern Med. 2016;31:548-560.
Practice Points
- Electronic medical records afford many benefits, but documentation burdens on health care providers can impede positive patient-provider interactions.
- Integration of current and future technology can shift the focus back to the patient and facilitate shared decision-making.

Painless Nodule on the Leg
The Diagnosis: Plasmablastic Lymphoma
Histopathologic examination revealed a diffuse dense proliferation of large, atypical, and pleomorphic mononuclear cells with prominent nucleoli and many mitotic figures representing plasmacytoid cells in the dermis (Figure). Immunostaining was positive for MUM-1 (marker of late-stage plasma cells and activated T cells) and BCL-2 (antiapoptotic marker). Fluorescent polymerase chain reaction was positive for clonal IgH gene arrangement, and fluorescence in situ hybridization was positive for C-MYC rearrangement in 94% of cells. Epstein-Barr encoding region in situ hybridization also was positive. Rare cells stained positive for T-cell markers. CD20, BCL-6, and CD30 immunostains were negative, suggesting that these cells were not B or T cells, though terminally differentiated B cells also can lack these markers. Bone marrow biopsy showed a similar staining pattern to the skin with 10% atypical plasmacytoid cells. Computed tomography of the left leg showed an enlargement of the semimembranosus muscle with internal areas of high density and heterogeneous enhancement. The patient underwent decompression of the left peroneal nerve. Biopsy showed a staining pattern similar to the right skin nodule and bone marrow, consistent with lymphoma.
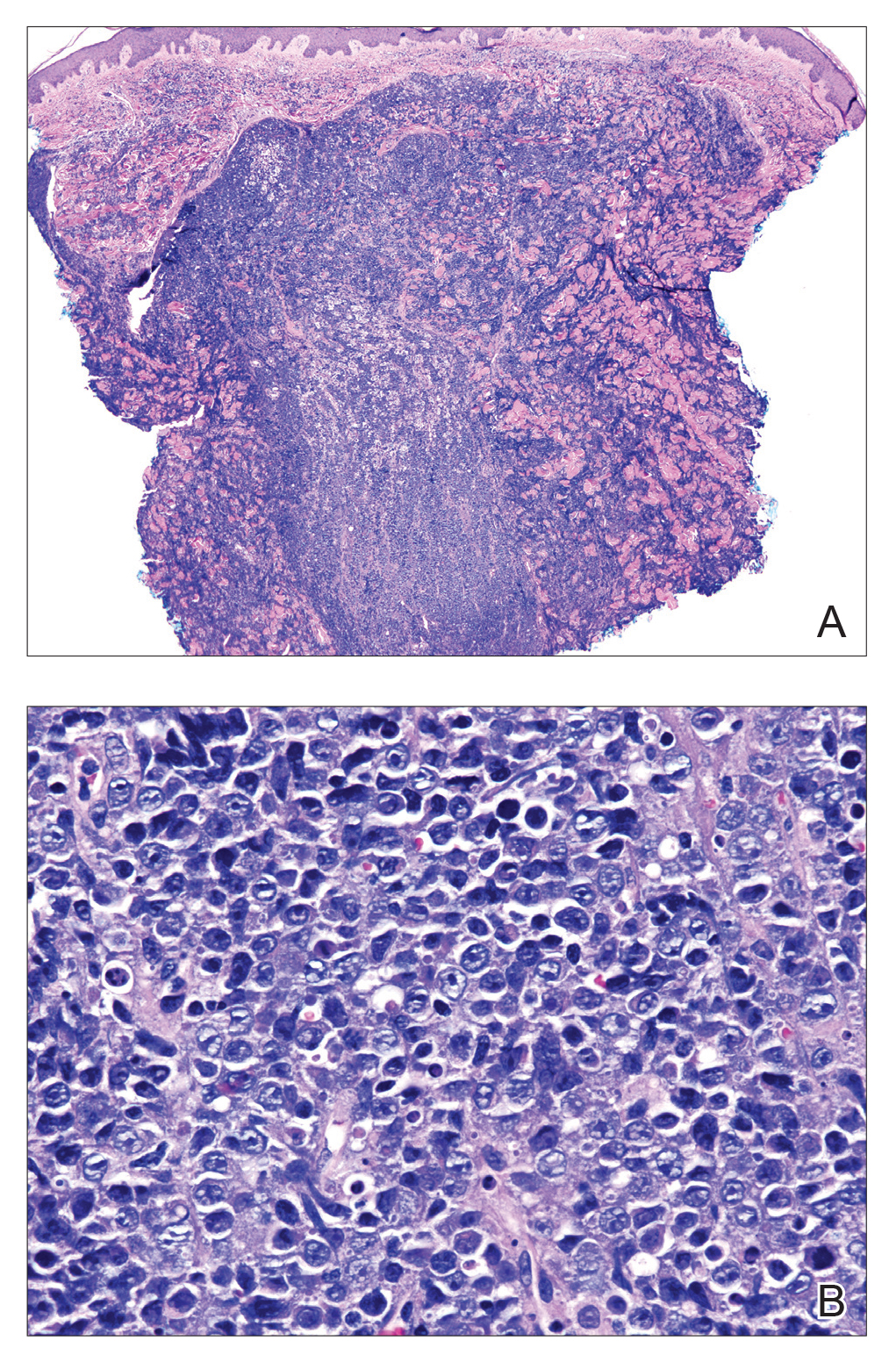
He was diagnosed with stage IV human immunodeficiency virus (HIV)-associated plasmablastic lymphoma (PBL) and received 6 cycles of R-EPOCH (rituximab, etoposide phosphate, prednisone, vincristine sulfate, cyclophosphamide, doxorubicin hydrochloride) without vincristine with intrathecal methotrexate, followed by 3 cycles of DHAP (dexamethasone, high dose Ara C, cisplatin) with bortezomib and daratumumab after relapse. Ultimately, he underwent autologous stem cell transplantation and was alive 13 months after diagnosis.
Plasmablastic lymphoma is a rare subtype of non-Hodgkin lymphoma that most commonly arises in the oral cavity of individuals with HIV.1 In addition to HIV infection, PBL also is seen in patients with other causes of immunodeficiency such as iatrogenic immunosuppression following solid organ transplantation.1 The typical disease presentation is an expanding mass in the oral cavity; however, 34% (52/151) of reported cases arose at extraoral primary sites, with a minority of cases confined to cutaneous sites with no systemic involvement.2 Cutaneous PBL presentations may include flesh-colored or purple, grouped or solitary nodules; an erythematous infiltrated plaque; or purple-red ulcerated nodules. The lesions usually are asymptomatic and located on the arms and legs.3
On histologic examination, PBL is characterized by a diffuse monomorphic lymphoid infiltrate that sometimes invades the surrounding soft tissue.4-6 The neoplastic cells have eccentric round nucleoli. Plasmablastic lymphoma characteristically displays a high proliferation index with many mitotic figures and signs of apoptosis.4-6 Definitive diagnosis requires immunohistochemical staining. Typical B-cell antigens (CD20) as well as CD45 are negative, while plasma cell markers such as CD38 are positive. Other B- and T-cell markers usually are negative.5,7 The pathogenesis of PBL is thought to be related to Epstein-Barr virus or human herpesvirus 8 infection. In a series of PBL cases, Epstein-Barr virus and human herpesvirus 8 was positive in 75% (97/129) and 17% (13/75) of tested cases, respectively.1
The prognosis for PBL is poor, with a median overall survival of 15 months and a 3-year survival rate of 25% in HIV-infected individuals.8 However, cutaneous PBL without systemic involvement has a considerably better prognosis, with only 1 of 12 cases resulting in death.2,3,9 Treatment of PBL depends on the extent of the disease. Cutaneous PBL can be treated with surgery and adjuvant radiation.3 Chemotherapy is required for patients with multiple lesions or systemic involvement. Current treatment regimens are similar to those used for other aggressive lymphomas such as CHOP (cyclophosphamide, doxorubicin, vincristine, prednisone).1 Transplant recipients should have their immunosuppression reduced, and HIV-infected patients should have their highly active antiretroviral therapy regimens optimized. Patients presenting with PBL without HIV should be tested for HIV, as PBL has previously been reported to be the presenting manifestation of HIV infection.10
The differential diagnosis for a rapidly expanding, vascular-appearing, red mass on the legs in an immunosuppressed individual includes abscess, malignancy, Kaposi sarcoma, Sweet syndrome, and tertiary syphilis.
Acknowledgment
We thank Sameera Husain, MD (New York, New York), for her assistance with histopathologic photographs and interpretation.
- Riedel DJ, Gonzalez-Cuyar LF, Zhao XF, et al. Plasmablastic lymphoma of the oral cavity: a rapidly progressive lymphoma associated with HIV infection. Lancet Infect Dis. 2008;8:261-267.
- Heiser D, Müller H, Kempf W, et al. Primary cutaneous plasmablastic lymphoma of the lower leg in an HIV-negative patient. J Am Acad Dermatol. 2012;67:E202-E205.
- Jambusaria A, Shafer D, Wu H, et al. Cutaneous plasmablastic lymphoma. J Am Acad Dermatol. 2008;58:676-678.
- Delecluse HJ, Anagnostopoulos I, Dallenbach F, et al. Plasmablastic lymphomas of the oral cavity: a new entity associated with the human immunodeficiency virus infection. Blood. 1997;89:1413-1420.
- Gaidano G, Cerri M, Capello D, et al. Molecular histogenesis of plasmablastic lymphoma of the oral cavity. Br J Haematol. 2002;119:622-628.
- Folk GS, Abbondanzo SL, Childers EL, et al. Plasmablastic lymphoma: a clinicopathologic correlation. Ann Diagn Pathol. 2006;10:8-12.
- Castillo JJ, Bibas M, Miranda RN. The biology and treatment of plasmablastic lymphoma. Blood. 2015;125:2323-2330.
- Castillo J, Pantanowitz L, Dezube BJ. HIV-associated plasmablastic lymphoma: lessons learned from 112 published cases. Am J Hematol. 2008;83:804-809.
- Horna P, Hamill JR, Sokol L, et al. Primary cutaneous plasmablastic lymphoma in an immunocompetent patient. J Am Acad Dermatol. 2013;69:E274-E276.
- Desai RS, Vanaki SS, Puranik RS, et al. Plasmablastic lymphoma presenting as a gingival growth in a previously undiagnosed HIV-positive patient: a case report. J Oral Maxillofac Surg. 2007;65:1358-1361.
The Diagnosis: Plasmablastic Lymphoma
Histopathologic examination revealed a diffuse dense proliferation of large, atypical, and pleomorphic mononuclear cells with prominent nucleoli and many mitotic figures representing plasmacytoid cells in the dermis (Figure). Immunostaining was positive for MUM-1 (marker of late-stage plasma cells and activated T cells) and BCL-2 (antiapoptotic marker). Fluorescent polymerase chain reaction was positive for clonal IgH gene arrangement, and fluorescence in situ hybridization was positive for C-MYC rearrangement in 94% of cells. Epstein-Barr encoding region in situ hybridization also was positive. Rare cells stained positive for T-cell markers. CD20, BCL-6, and CD30 immunostains were negative, suggesting that these cells were not B or T cells, though terminally differentiated B cells also can lack these markers. Bone marrow biopsy showed a similar staining pattern to the skin with 10% atypical plasmacytoid cells. Computed tomography of the left leg showed an enlargement of the semimembranosus muscle with internal areas of high density and heterogeneous enhancement. The patient underwent decompression of the left peroneal nerve. Biopsy showed a staining pattern similar to the right skin nodule and bone marrow, consistent with lymphoma.
He was diagnosed with stage IV human immunodeficiency virus (HIV)-associated plasmablastic lymphoma (PBL) and received 6 cycles of R-EPOCH (rituximab, etoposide phosphate, prednisone, vincristine sulfate, cyclophosphamide, doxorubicin hydrochloride) without vincristine with intrathecal methotrexate, followed by 3 cycles of DHAP (dexamethasone, high dose Ara C, cisplatin) with bortezomib and daratumumab after relapse. Ultimately, he underwent autologous stem cell transplantation and was alive 13 months after diagnosis.
Plasmablastic lymphoma is a rare subtype of non-Hodgkin lymphoma that most commonly arises in the oral cavity of individuals with HIV.1 In addition to HIV infection, PBL also is seen in patients with other causes of immunodeficiency such as iatrogenic immunosuppression following solid organ transplantation.1 The typical disease presentation is an expanding mass in the oral cavity; however, 34% (52/151) of reported cases arose at extraoral primary sites, with a minority of cases confined to cutaneous sites with no systemic involvement.2 Cutaneous PBL presentations may include flesh-colored or purple, grouped or solitary nodules; an erythematous infiltrated plaque; or purple-red ulcerated nodules. The lesions usually are asymptomatic and located on the arms and legs.3
On histologic examination, PBL is characterized by a diffuse monomorphic lymphoid infiltrate that sometimes invades the surrounding soft tissue.4-6 The neoplastic cells have eccentric round nucleoli. Plasmablastic lymphoma characteristically displays a high proliferation index with many mitotic figures and signs of apoptosis.4-6 Definitive diagnosis requires immunohistochemical staining. Typical B-cell antigens (CD20) as well as CD45 are negative, while plasma cell markers such as CD38 are positive. Other B- and T-cell markers usually are negative.5,7 The pathogenesis of PBL is thought to be related to Epstein-Barr virus or human herpesvirus 8 infection. In a series of PBL cases, Epstein-Barr virus and human herpesvirus 8 was positive in 75% (97/129) and 17% (13/75) of tested cases, respectively.1
The prognosis for PBL is poor, with a median overall survival of 15 months and a 3-year survival rate of 25% in HIV-infected individuals.8 However, cutaneous PBL without systemic involvement has a considerably better prognosis, with only 1 of 12 cases resulting in death.2,3,9 Treatment of PBL depends on the extent of the disease. Cutaneous PBL can be treated with surgery and adjuvant radiation.3 Chemotherapy is required for patients with multiple lesions or systemic involvement. Current treatment regimens are similar to those used for other aggressive lymphomas such as CHOP (cyclophosphamide, doxorubicin, vincristine, prednisone).1 Transplant recipients should have their immunosuppression reduced, and HIV-infected patients should have their highly active antiretroviral therapy regimens optimized. Patients presenting with PBL without HIV should be tested for HIV, as PBL has previously been reported to be the presenting manifestation of HIV infection.10
The differential diagnosis for a rapidly expanding, vascular-appearing, red mass on the legs in an immunosuppressed individual includes abscess, malignancy, Kaposi sarcoma, Sweet syndrome, and tertiary syphilis.
Acknowledgment
We thank Sameera Husain, MD (New York, New York), for her assistance with histopathologic photographs and interpretation.
The Diagnosis: Plasmablastic Lymphoma
Histopathologic examination revealed a diffuse dense proliferation of large, atypical, and pleomorphic mononuclear cells with prominent nucleoli and many mitotic figures representing plasmacytoid cells in the dermis (Figure). Immunostaining was positive for MUM-1 (marker of late-stage plasma cells and activated T cells) and BCL-2 (antiapoptotic marker). Fluorescent polymerase chain reaction was positive for clonal IgH gene arrangement, and fluorescence in situ hybridization was positive for C-MYC rearrangement in 94% of cells. Epstein-Barr encoding region in situ hybridization also was positive. Rare cells stained positive for T-cell markers. CD20, BCL-6, and CD30 immunostains were negative, suggesting that these cells were not B or T cells, though terminally differentiated B cells also can lack these markers. Bone marrow biopsy showed a similar staining pattern to the skin with 10% atypical plasmacytoid cells. Computed tomography of the left leg showed an enlargement of the semimembranosus muscle with internal areas of high density and heterogeneous enhancement. The patient underwent decompression of the left peroneal nerve. Biopsy showed a staining pattern similar to the right skin nodule and bone marrow, consistent with lymphoma.
He was diagnosed with stage IV human immunodeficiency virus (HIV)-associated plasmablastic lymphoma (PBL) and received 6 cycles of R-EPOCH (rituximab, etoposide phosphate, prednisone, vincristine sulfate, cyclophosphamide, doxorubicin hydrochloride) without vincristine with intrathecal methotrexate, followed by 3 cycles of DHAP (dexamethasone, high dose Ara C, cisplatin) with bortezomib and daratumumab after relapse. Ultimately, he underwent autologous stem cell transplantation and was alive 13 months after diagnosis.
Plasmablastic lymphoma is a rare subtype of non-Hodgkin lymphoma that most commonly arises in the oral cavity of individuals with HIV.1 In addition to HIV infection, PBL also is seen in patients with other causes of immunodeficiency such as iatrogenic immunosuppression following solid organ transplantation.1 The typical disease presentation is an expanding mass in the oral cavity; however, 34% (52/151) of reported cases arose at extraoral primary sites, with a minority of cases confined to cutaneous sites with no systemic involvement.2 Cutaneous PBL presentations may include flesh-colored or purple, grouped or solitary nodules; an erythematous infiltrated plaque; or purple-red ulcerated nodules. The lesions usually are asymptomatic and located on the arms and legs.3
On histologic examination, PBL is characterized by a diffuse monomorphic lymphoid infiltrate that sometimes invades the surrounding soft tissue.4-6 The neoplastic cells have eccentric round nucleoli. Plasmablastic lymphoma characteristically displays a high proliferation index with many mitotic figures and signs of apoptosis.4-6 Definitive diagnosis requires immunohistochemical staining. Typical B-cell antigens (CD20) as well as CD45 are negative, while plasma cell markers such as CD38 are positive. Other B- and T-cell markers usually are negative.5,7 The pathogenesis of PBL is thought to be related to Epstein-Barr virus or human herpesvirus 8 infection. In a series of PBL cases, Epstein-Barr virus and human herpesvirus 8 was positive in 75% (97/129) and 17% (13/75) of tested cases, respectively.1
The prognosis for PBL is poor, with a median overall survival of 15 months and a 3-year survival rate of 25% in HIV-infected individuals.8 However, cutaneous PBL without systemic involvement has a considerably better prognosis, with only 1 of 12 cases resulting in death.2,3,9 Treatment of PBL depends on the extent of the disease. Cutaneous PBL can be treated with surgery and adjuvant radiation.3 Chemotherapy is required for patients with multiple lesions or systemic involvement. Current treatment regimens are similar to those used for other aggressive lymphomas such as CHOP (cyclophosphamide, doxorubicin, vincristine, prednisone).1 Transplant recipients should have their immunosuppression reduced, and HIV-infected patients should have their highly active antiretroviral therapy regimens optimized. Patients presenting with PBL without HIV should be tested for HIV, as PBL has previously been reported to be the presenting manifestation of HIV infection.10
The differential diagnosis for a rapidly expanding, vascular-appearing, red mass on the legs in an immunosuppressed individual includes abscess, malignancy, Kaposi sarcoma, Sweet syndrome, and tertiary syphilis.
Acknowledgment
We thank Sameera Husain, MD (New York, New York), for her assistance with histopathologic photographs and interpretation.
- Riedel DJ, Gonzalez-Cuyar LF, Zhao XF, et al. Plasmablastic lymphoma of the oral cavity: a rapidly progressive lymphoma associated with HIV infection. Lancet Infect Dis. 2008;8:261-267.
- Heiser D, Müller H, Kempf W, et al. Primary cutaneous plasmablastic lymphoma of the lower leg in an HIV-negative patient. J Am Acad Dermatol. 2012;67:E202-E205.
- Jambusaria A, Shafer D, Wu H, et al. Cutaneous plasmablastic lymphoma. J Am Acad Dermatol. 2008;58:676-678.
- Delecluse HJ, Anagnostopoulos I, Dallenbach F, et al. Plasmablastic lymphomas of the oral cavity: a new entity associated with the human immunodeficiency virus infection. Blood. 1997;89:1413-1420.
- Gaidano G, Cerri M, Capello D, et al. Molecular histogenesis of plasmablastic lymphoma of the oral cavity. Br J Haematol. 2002;119:622-628.
- Folk GS, Abbondanzo SL, Childers EL, et al. Plasmablastic lymphoma: a clinicopathologic correlation. Ann Diagn Pathol. 2006;10:8-12.
- Castillo JJ, Bibas M, Miranda RN. The biology and treatment of plasmablastic lymphoma. Blood. 2015;125:2323-2330.
- Castillo J, Pantanowitz L, Dezube BJ. HIV-associated plasmablastic lymphoma: lessons learned from 112 published cases. Am J Hematol. 2008;83:804-809.
- Horna P, Hamill JR, Sokol L, et al. Primary cutaneous plasmablastic lymphoma in an immunocompetent patient. J Am Acad Dermatol. 2013;69:E274-E276.
- Desai RS, Vanaki SS, Puranik RS, et al. Plasmablastic lymphoma presenting as a gingival growth in a previously undiagnosed HIV-positive patient: a case report. J Oral Maxillofac Surg. 2007;65:1358-1361.
- Riedel DJ, Gonzalez-Cuyar LF, Zhao XF, et al. Plasmablastic lymphoma of the oral cavity: a rapidly progressive lymphoma associated with HIV infection. Lancet Infect Dis. 2008;8:261-267.
- Heiser D, Müller H, Kempf W, et al. Primary cutaneous plasmablastic lymphoma of the lower leg in an HIV-negative patient. J Am Acad Dermatol. 2012;67:E202-E205.
- Jambusaria A, Shafer D, Wu H, et al. Cutaneous plasmablastic lymphoma. J Am Acad Dermatol. 2008;58:676-678.
- Delecluse HJ, Anagnostopoulos I, Dallenbach F, et al. Plasmablastic lymphomas of the oral cavity: a new entity associated with the human immunodeficiency virus infection. Blood. 1997;89:1413-1420.
- Gaidano G, Cerri M, Capello D, et al. Molecular histogenesis of plasmablastic lymphoma of the oral cavity. Br J Haematol. 2002;119:622-628.
- Folk GS, Abbondanzo SL, Childers EL, et al. Plasmablastic lymphoma: a clinicopathologic correlation. Ann Diagn Pathol. 2006;10:8-12.
- Castillo JJ, Bibas M, Miranda RN. The biology and treatment of plasmablastic lymphoma. Blood. 2015;125:2323-2330.
- Castillo J, Pantanowitz L, Dezube BJ. HIV-associated plasmablastic lymphoma: lessons learned from 112 published cases. Am J Hematol. 2008;83:804-809.
- Horna P, Hamill JR, Sokol L, et al. Primary cutaneous plasmablastic lymphoma in an immunocompetent patient. J Am Acad Dermatol. 2013;69:E274-E276.
- Desai RS, Vanaki SS, Puranik RS, et al. Plasmablastic lymphoma presenting as a gingival growth in a previously undiagnosed HIV-positive patient: a case report. J Oral Maxillofac Surg. 2007;65:1358-1361.
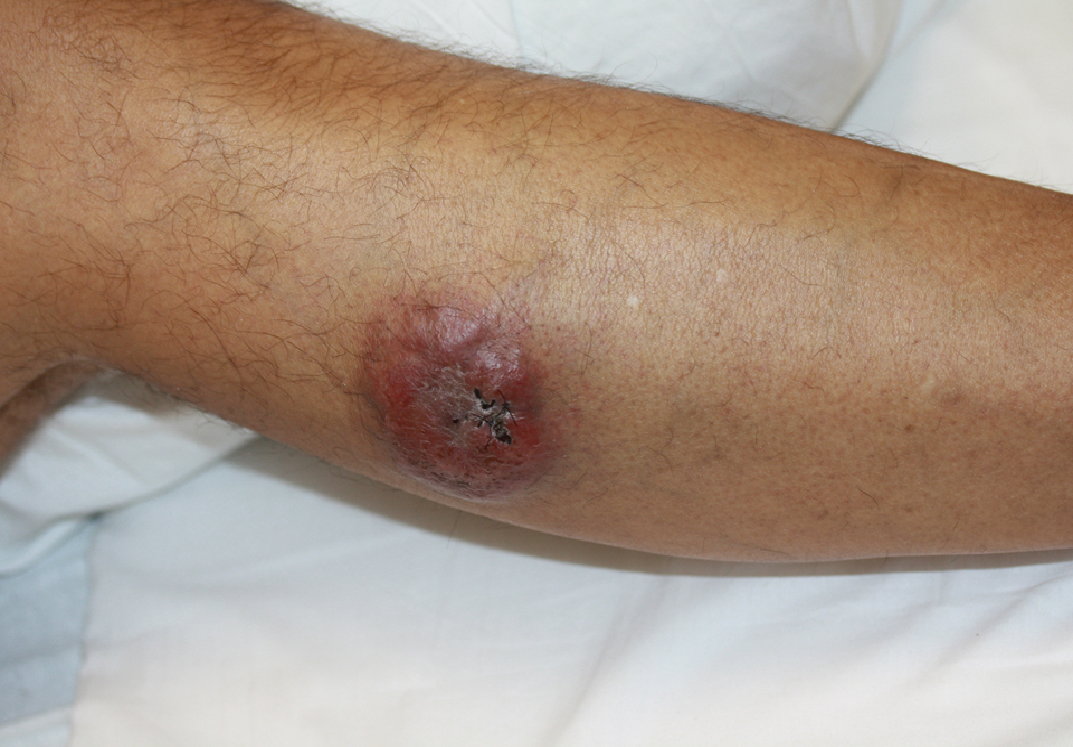
A 44-year-old man presented with numbness and a burning sensation of the left lateral leg and dorsal foot of 3 days' duration as well as a left foot drop of 1 day's duration. A painless red nodule on the right shin also developed over a 10-day period. He had been diagnosed with human immunodeficiency virus a year prior and reported compliance with antiretroviral therapy. There was a newly identified, well-demarcated, 6-cm, round, red-purple, flat-topped, nodular tumor with central depression on the right lateral shin. Ultrasonography of the nodule revealed a heterogeneous septate structure with increased vascularity. There was no regional or generalized lymphadenopathy. Laboratory values were notable for microcytic anemia. The white blood cell count was within reference range. Human immunodeficiency virus RNA viral load was elevated (3183 viral copies/mL [reference range, <20 viral copies/mL]). Two punch biopsies of the nodule were performed.