User login
Cutis is a peer-reviewed clinical journal for the dermatologist, allergist, and general practitioner published monthly since 1965. Concise clinical articles present the practical side of dermatology, helping physicians to improve patient care. Cutis is referenced in Index Medicus/MEDLINE and is written and edited by industry leaders.
ass lick
assault rifle
balls
ballsac
black jack
bleach
Boko Haram
bondage
causas
cheap
child abuse
cocaine
compulsive behaviors
cost of miracles
cunt
Daech
display network stats
drug paraphernalia
explosion
fart
fda and death
fda AND warn
fda AND warning
fda AND warns
feom
fuck
gambling
gfc
gun
human trafficking
humira AND expensive
illegal
ISIL
ISIS
Islamic caliphate
Islamic state
madvocate
masturbation
mixed martial arts
MMA
molestation
national rifle association
NRA
nsfw
nuccitelli
pedophile
pedophilia
poker
porn
porn
pornography
psychedelic drug
recreational drug
sex slave rings
shit
slot machine
snort
substance abuse
terrorism
terrorist
texarkana
Texas hold 'em
UFC
section[contains(@class, 'nav-hidden')]
section[contains(@class, 'nav-hidden active')
A peer-reviewed, indexed journal for dermatologists with original research, image quizzes, cases and reviews, and columns.
Multiple Subcutaneous Dermoid Cysts
To the Editor:
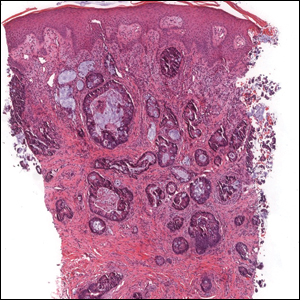
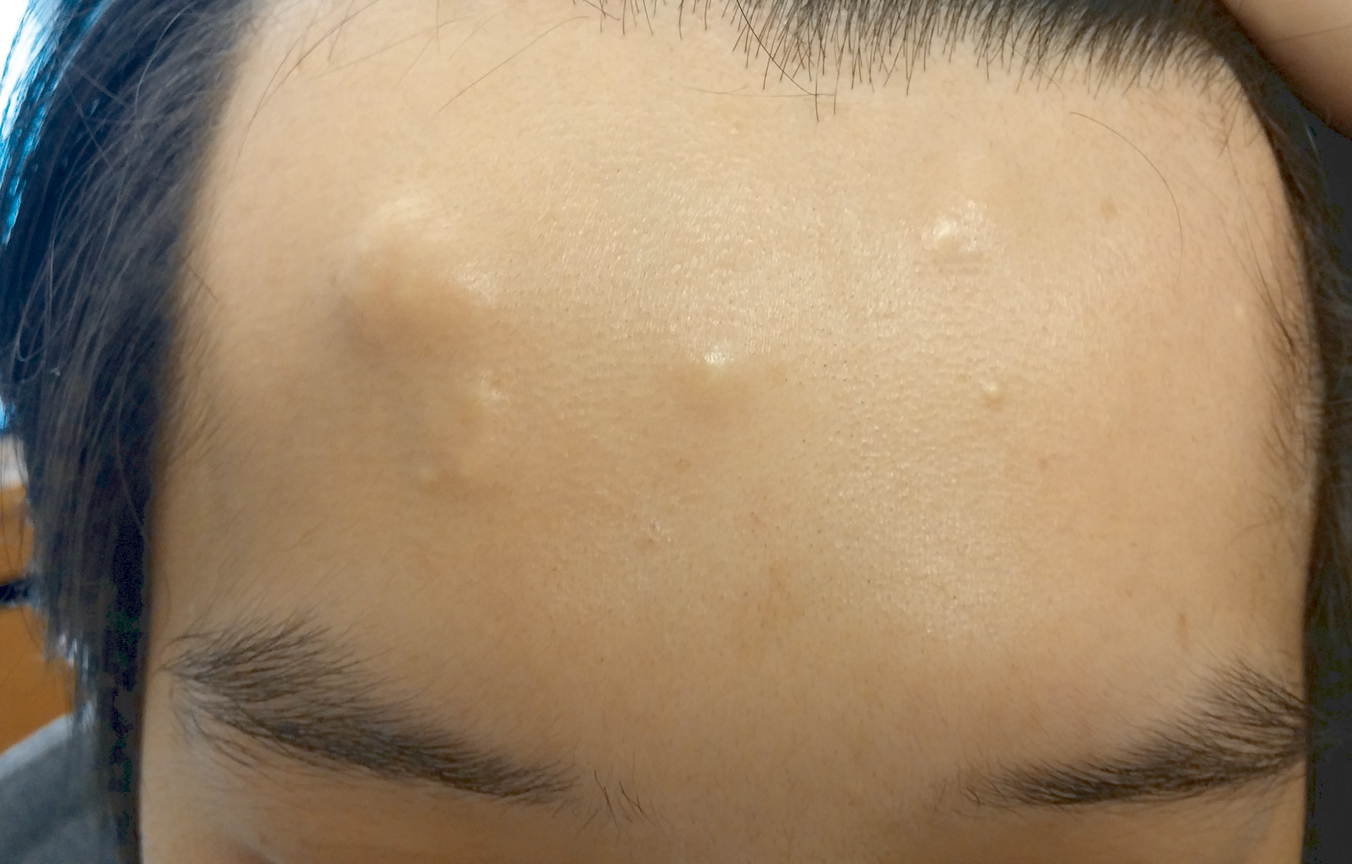
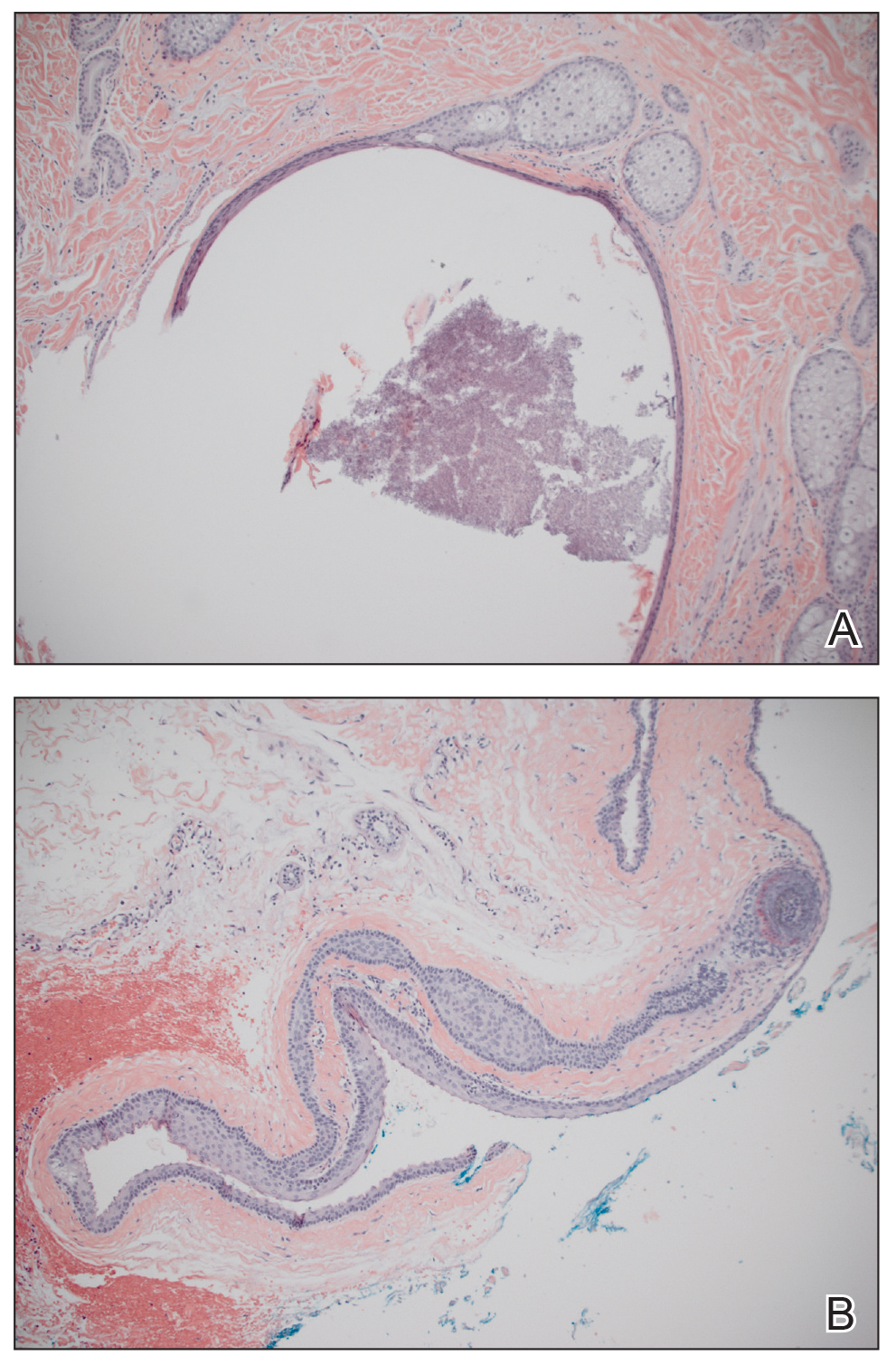
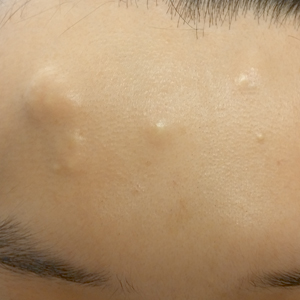
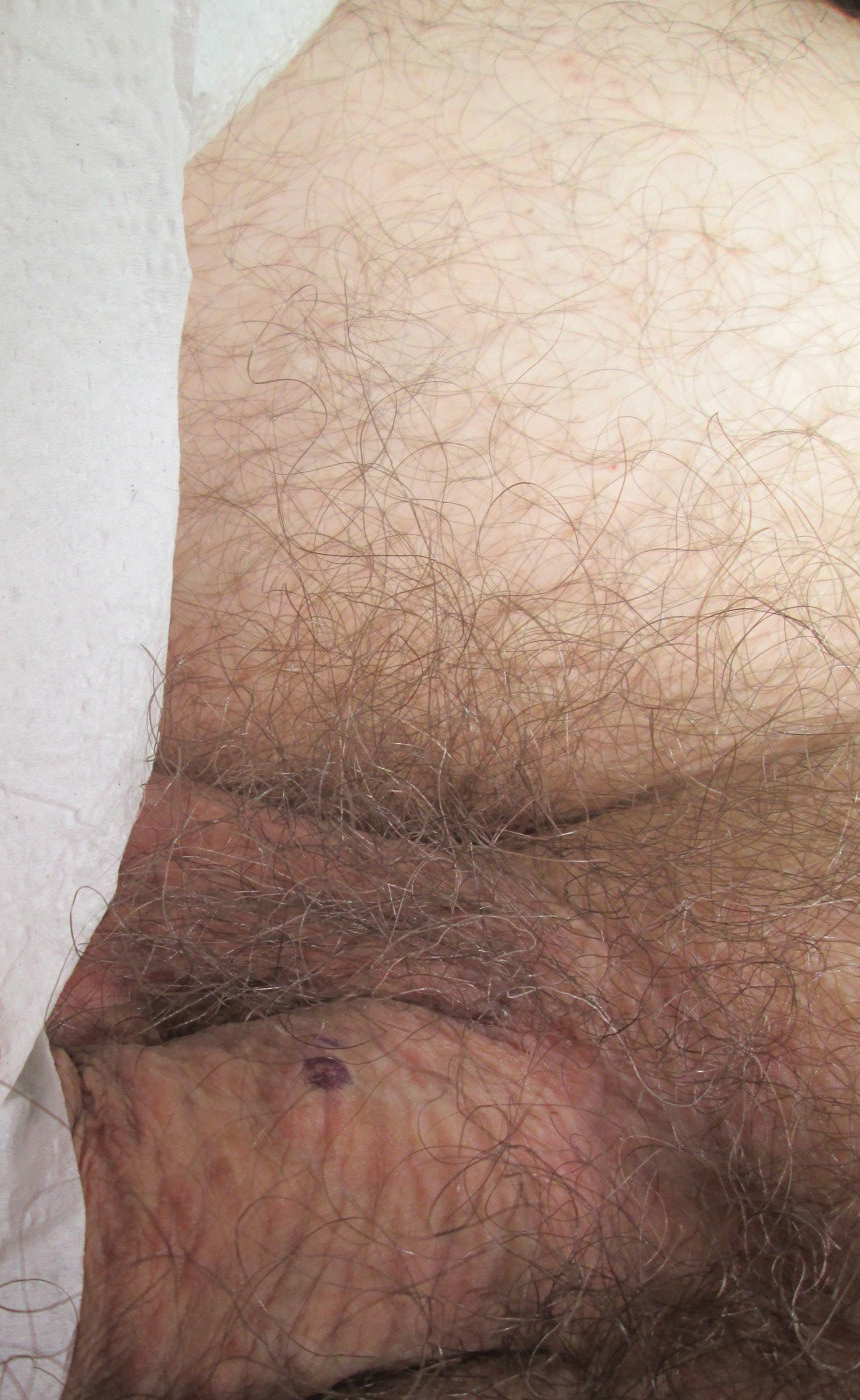
A 30-year-old man with no notable medical history presented to the dermatology clinic with multiple subcutaneous nodules on the forehead of 5 years’ duration. He reported no history of forehead trauma or manipulation of the lesions, and there was no accompanying pruritis, pain, erythema, or purulent discharge. There was no family history of skin or gastrointestinal tract tumors. On physical examination, the patient had 5 firm, flesh-colored to yellow nodules measuring approximately 0.2 to 1.5 cm in diameter without central punctae scattered over the central forehead (Figure 1). Due to cosmetic concerns, the patient elected to pursue surgical excision of the lesions, which occurred over several office visits. During surgical excision, the lesions were found to be smooth, encapsulated, and mobile, and they were excised without surgical complication. Histopathologic examination showed subcutaneous cysts lined by squamous epithelium with associated sebaceous glands (Figure 2A) and hair follicles in the cyst lumen (Figure 2B). These findings confirmed the diagnosis of multiple subcutaneous dermoid cysts.
Dermoid cysts are relatively uncommon, benign tumors consisting of tissue derived from ectodermal and mesodermal germ cell layers. Dermoid cysts may be distinguished from teratomas, which may contain tissues derived from all 3 germ cell layers and typically consist of types of tissues foreign to the site of origin, such as dental, thyroid, gastrointestinal, or neural tissue.1,2 The majority of dermoid cysts are congenitally developed along the lines of embryologic fusion due to an error in the division of the ectoderm and mesoderm3,4; however, some dermoid cysts may be acquired from epidermal elements being traumatically implanted into the dermis.5
Our patient’s presentation with multiple dermoid cysts was atypical, as dermoid cysts are almost always solitary tumors. A similar case was reported in a 41-year-old man who developed multiple dermoid cysts on the forehead over a 20-year period.This patient also was otherwise healthy, denied prior trauma to the forehead, and reported no family history of skin or gastrointestinal tract tumors.5
Another unusual feature in our case was the location of the dermoid cysts on the central forehead. The most common location for dermoid cysts is the lateral third of the eyebrows (47%–70% of cases).1,4,6-10 These cysts occur because of sequestration of the surface ectoderm during fusion along the naso-optic groove.2 Dermoid cysts also have been noted in other anatomical areas such as the scalp, nose, anterior neck, and trunk.6
Dermoid cysts tend to be small, round, smooth, and slowly growing until sudden enlargement prompts surgical evaluation.4,6 During surgical excision, they often are fixed to the underlying bone but also may be freely mobile, as in our patient.6 Histopathologic examination reveals a stratified squamous epithelium with associated adnexal structures such as sebaceous glands or hair follicles.1 Smooth muscle fibers, prominent vascular stroma, small nerves, and collagen and elastic fibers also may be found within the lumen of dermoid cysts.2
In some cases, dermoid cysts may be invasive and carry the risk of bony erosion, intracranial extension, osteomyelitis, meningitis, or cerebral abscess. Imaging studies sometimes are needed to rule out intracranial or intraspinal extension, particularly for midline dermoid cysts.6 The standard of treatment for dermoid cysts is surgical excision and complete enucleation without disruption of the cyst wall; however, invasive dermoid cysts may require endoscopic excision, orbitotomy, or craniotomy.4,6
- Brownstein MH, Helwig EB. Subcutaneous dermoid cysts. Arch Dermatol. 1973;107:237-239.
- Smirniotopoulos JG, Chiechi MV. Teratomas, dermoids, and epidermoids of the head and neck. Radiographics. 1995;15:1437-1455.
- Pryor SG, Lewis JE, Weaver AL, et al. Pediatric dermoid cysts of the head and neck. Otolaryngol Head Neck Surg. 2005;132:938-942.
- Yamaki T, Higuchi R, Sasaki K, et al. Multiple dermoid cysts on the forehead. case report. Scand J Plast Reconstr Surg Hand Surg. 1996;30:321-324.
- Prior A, Anania P, Pacetti M, et al. Dermoid and epidermoid cysts of scalp: case series of 234 consecutive patients. World Neurosurg. 2018;120:119-124.
- Orozco-Covarrubias L, Lara-Carpio R, Saez-De-Ocariz M, et al. Dermoid cysts: a report of 75 pediatric patients. Pediatr Dermatol. 2013;30:706-711.
- Al-Khateeb TH, Al-Masri NM, Al-Zoubi F. Cutaneous cysts of the head and neck. J Oral Maxillofac Surg. 2009;67:52-57.
- McAvoy JM, Zuckerbraun L. Dermoid cysts of the head and neck in children. Arch Otolaryngol. 1976;102:529-531.
- Taylor BW, Erich JB, Dockerty MB. Dermoids of the head and neck. Minnesota Med. 1966;49:1535-1540.
- Golden BA, Zide MF. Cutaneous cysts of the head and neck. J Oral Maxillofac Surg. 2005;63:1613-1619.
To the Editor:
A 30-year-old man with no notable medical history presented to the dermatology clinic with multiple subcutaneous nodules on the forehead of 5 years’ duration. He reported no history of forehead trauma or manipulation of the lesions, and there was no accompanying pruritis, pain, erythema, or purulent discharge. There was no family history of skin or gastrointestinal tract tumors. On physical examination, the patient had 5 firm, flesh-colored to yellow nodules measuring approximately 0.2 to 1.5 cm in diameter without central punctae scattered over the central forehead (Figure 1). Due to cosmetic concerns, the patient elected to pursue surgical excision of the lesions, which occurred over several office visits. During surgical excision, the lesions were found to be smooth, encapsulated, and mobile, and they were excised without surgical complication. Histopathologic examination showed subcutaneous cysts lined by squamous epithelium with associated sebaceous glands (Figure 2A) and hair follicles in the cyst lumen (Figure 2B). These findings confirmed the diagnosis of multiple subcutaneous dermoid cysts.
Dermoid cysts are relatively uncommon, benign tumors consisting of tissue derived from ectodermal and mesodermal germ cell layers. Dermoid cysts may be distinguished from teratomas, which may contain tissues derived from all 3 germ cell layers and typically consist of types of tissues foreign to the site of origin, such as dental, thyroid, gastrointestinal, or neural tissue.1,2 The majority of dermoid cysts are congenitally developed along the lines of embryologic fusion due to an error in the division of the ectoderm and mesoderm3,4; however, some dermoid cysts may be acquired from epidermal elements being traumatically implanted into the dermis.5
Our patient’s presentation with multiple dermoid cysts was atypical, as dermoid cysts are almost always solitary tumors. A similar case was reported in a 41-year-old man who developed multiple dermoid cysts on the forehead over a 20-year period.This patient also was otherwise healthy, denied prior trauma to the forehead, and reported no family history of skin or gastrointestinal tract tumors.5
Another unusual feature in our case was the location of the dermoid cysts on the central forehead. The most common location for dermoid cysts is the lateral third of the eyebrows (47%–70% of cases).1,4,6-10 These cysts occur because of sequestration of the surface ectoderm during fusion along the naso-optic groove.2 Dermoid cysts also have been noted in other anatomical areas such as the scalp, nose, anterior neck, and trunk.6
Dermoid cysts tend to be small, round, smooth, and slowly growing until sudden enlargement prompts surgical evaluation.4,6 During surgical excision, they often are fixed to the underlying bone but also may be freely mobile, as in our patient.6 Histopathologic examination reveals a stratified squamous epithelium with associated adnexal structures such as sebaceous glands or hair follicles.1 Smooth muscle fibers, prominent vascular stroma, small nerves, and collagen and elastic fibers also may be found within the lumen of dermoid cysts.2
In some cases, dermoid cysts may be invasive and carry the risk of bony erosion, intracranial extension, osteomyelitis, meningitis, or cerebral abscess. Imaging studies sometimes are needed to rule out intracranial or intraspinal extension, particularly for midline dermoid cysts.6 The standard of treatment for dermoid cysts is surgical excision and complete enucleation without disruption of the cyst wall; however, invasive dermoid cysts may require endoscopic excision, orbitotomy, or craniotomy.4,6
To the Editor:
A 30-year-old man with no notable medical history presented to the dermatology clinic with multiple subcutaneous nodules on the forehead of 5 years’ duration. He reported no history of forehead trauma or manipulation of the lesions, and there was no accompanying pruritis, pain, erythema, or purulent discharge. There was no family history of skin or gastrointestinal tract tumors. On physical examination, the patient had 5 firm, flesh-colored to yellow nodules measuring approximately 0.2 to 1.5 cm in diameter without central punctae scattered over the central forehead (Figure 1). Due to cosmetic concerns, the patient elected to pursue surgical excision of the lesions, which occurred over several office visits. During surgical excision, the lesions were found to be smooth, encapsulated, and mobile, and they were excised without surgical complication. Histopathologic examination showed subcutaneous cysts lined by squamous epithelium with associated sebaceous glands (Figure 2A) and hair follicles in the cyst lumen (Figure 2B). These findings confirmed the diagnosis of multiple subcutaneous dermoid cysts.
Dermoid cysts are relatively uncommon, benign tumors consisting of tissue derived from ectodermal and mesodermal germ cell layers. Dermoid cysts may be distinguished from teratomas, which may contain tissues derived from all 3 germ cell layers and typically consist of types of tissues foreign to the site of origin, such as dental, thyroid, gastrointestinal, or neural tissue.1,2 The majority of dermoid cysts are congenitally developed along the lines of embryologic fusion due to an error in the division of the ectoderm and mesoderm3,4; however, some dermoid cysts may be acquired from epidermal elements being traumatically implanted into the dermis.5
Our patient’s presentation with multiple dermoid cysts was atypical, as dermoid cysts are almost always solitary tumors. A similar case was reported in a 41-year-old man who developed multiple dermoid cysts on the forehead over a 20-year period.This patient also was otherwise healthy, denied prior trauma to the forehead, and reported no family history of skin or gastrointestinal tract tumors.5
Another unusual feature in our case was the location of the dermoid cysts on the central forehead. The most common location for dermoid cysts is the lateral third of the eyebrows (47%–70% of cases).1,4,6-10 These cysts occur because of sequestration of the surface ectoderm during fusion along the naso-optic groove.2 Dermoid cysts also have been noted in other anatomical areas such as the scalp, nose, anterior neck, and trunk.6
Dermoid cysts tend to be small, round, smooth, and slowly growing until sudden enlargement prompts surgical evaluation.4,6 During surgical excision, they often are fixed to the underlying bone but also may be freely mobile, as in our patient.6 Histopathologic examination reveals a stratified squamous epithelium with associated adnexal structures such as sebaceous glands or hair follicles.1 Smooth muscle fibers, prominent vascular stroma, small nerves, and collagen and elastic fibers also may be found within the lumen of dermoid cysts.2
In some cases, dermoid cysts may be invasive and carry the risk of bony erosion, intracranial extension, osteomyelitis, meningitis, or cerebral abscess. Imaging studies sometimes are needed to rule out intracranial or intraspinal extension, particularly for midline dermoid cysts.6 The standard of treatment for dermoid cysts is surgical excision and complete enucleation without disruption of the cyst wall; however, invasive dermoid cysts may require endoscopic excision, orbitotomy, or craniotomy.4,6
- Brownstein MH, Helwig EB. Subcutaneous dermoid cysts. Arch Dermatol. 1973;107:237-239.
- Smirniotopoulos JG, Chiechi MV. Teratomas, dermoids, and epidermoids of the head and neck. Radiographics. 1995;15:1437-1455.
- Pryor SG, Lewis JE, Weaver AL, et al. Pediatric dermoid cysts of the head and neck. Otolaryngol Head Neck Surg. 2005;132:938-942.
- Yamaki T, Higuchi R, Sasaki K, et al. Multiple dermoid cysts on the forehead. case report. Scand J Plast Reconstr Surg Hand Surg. 1996;30:321-324.
- Prior A, Anania P, Pacetti M, et al. Dermoid and epidermoid cysts of scalp: case series of 234 consecutive patients. World Neurosurg. 2018;120:119-124.
- Orozco-Covarrubias L, Lara-Carpio R, Saez-De-Ocariz M, et al. Dermoid cysts: a report of 75 pediatric patients. Pediatr Dermatol. 2013;30:706-711.
- Al-Khateeb TH, Al-Masri NM, Al-Zoubi F. Cutaneous cysts of the head and neck. J Oral Maxillofac Surg. 2009;67:52-57.
- McAvoy JM, Zuckerbraun L. Dermoid cysts of the head and neck in children. Arch Otolaryngol. 1976;102:529-531.
- Taylor BW, Erich JB, Dockerty MB. Dermoids of the head and neck. Minnesota Med. 1966;49:1535-1540.
- Golden BA, Zide MF. Cutaneous cysts of the head and neck. J Oral Maxillofac Surg. 2005;63:1613-1619.
- Brownstein MH, Helwig EB. Subcutaneous dermoid cysts. Arch Dermatol. 1973;107:237-239.
- Smirniotopoulos JG, Chiechi MV. Teratomas, dermoids, and epidermoids of the head and neck. Radiographics. 1995;15:1437-1455.
- Pryor SG, Lewis JE, Weaver AL, et al. Pediatric dermoid cysts of the head and neck. Otolaryngol Head Neck Surg. 2005;132:938-942.
- Yamaki T, Higuchi R, Sasaki K, et al. Multiple dermoid cysts on the forehead. case report. Scand J Plast Reconstr Surg Hand Surg. 1996;30:321-324.
- Prior A, Anania P, Pacetti M, et al. Dermoid and epidermoid cysts of scalp: case series of 234 consecutive patients. World Neurosurg. 2018;120:119-124.
- Orozco-Covarrubias L, Lara-Carpio R, Saez-De-Ocariz M, et al. Dermoid cysts: a report of 75 pediatric patients. Pediatr Dermatol. 2013;30:706-711.
- Al-Khateeb TH, Al-Masri NM, Al-Zoubi F. Cutaneous cysts of the head and neck. J Oral Maxillofac Surg. 2009;67:52-57.
- McAvoy JM, Zuckerbraun L. Dermoid cysts of the head and neck in children. Arch Otolaryngol. 1976;102:529-531.
- Taylor BW, Erich JB, Dockerty MB. Dermoids of the head and neck. Minnesota Med. 1966;49:1535-1540.
- Golden BA, Zide MF. Cutaneous cysts of the head and neck. J Oral Maxillofac Surg. 2005;63:1613-1619.
Practice Points
- The majority of dermoid cysts are congenital; however, they may be acquired from traumatic implantation of epidermal elements into the dermis.
- The most common location for dermoid cysts is the lateral third of the eyebrows; however, they also may occur on the mid forehead, scalp, nose, anterior neck, and trunk.
- Imaging studies may be needed to rule out intracranial or intraspinal extension of dermoid cysts, particularly for those presenting in the midline.
Violaceous Nodules on the Hard Palate
The Diagnosis: Kaposi Sarcoma
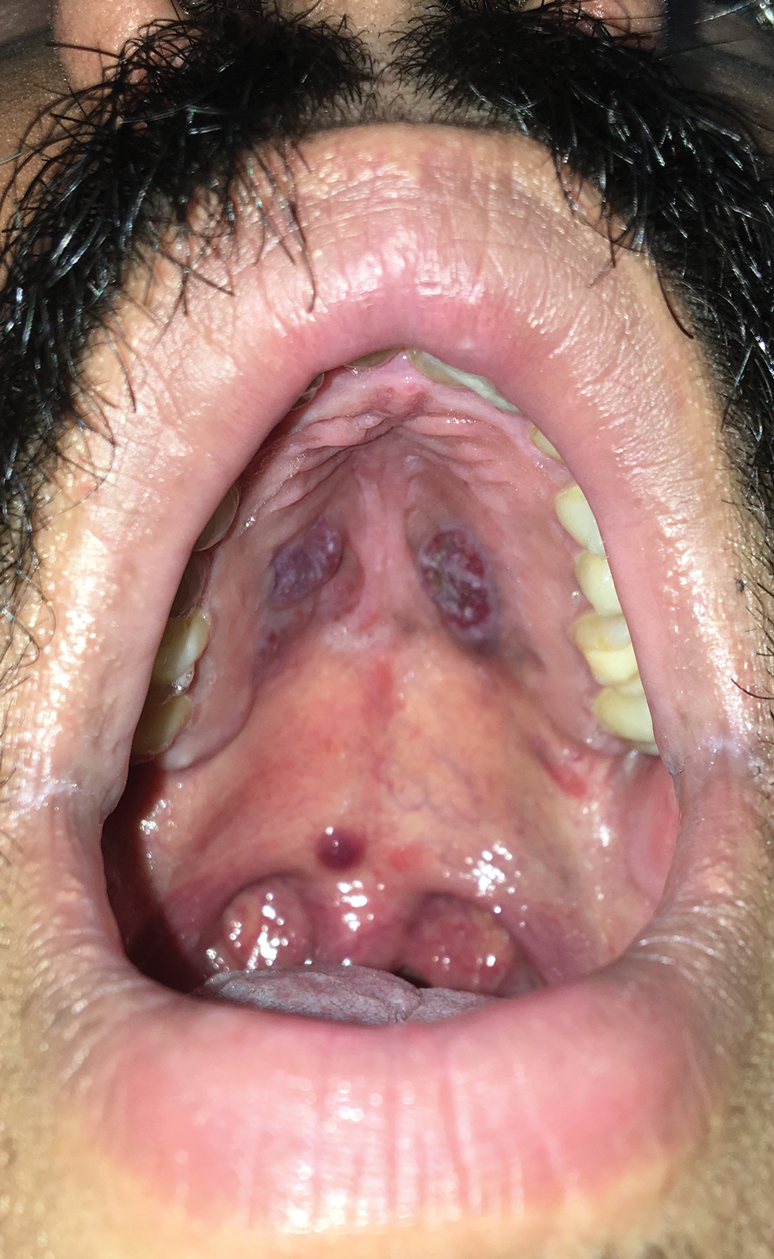
A 4-mm punch biopsy from the border of an ulcerated nodular lesion on the hard palate demonstrated diffusely distributed spindle cells, cleftlike microvascularity with extravasated erythrocytes, and widespread human herpesvirus 8 immunoreactivity on histopathology (Figure 1). Serologic tests were positive for human immunodeficiency virus (HIV) infection; HIV RNA was 14,584 IU/mL and the CD4 count was 254/mm3. The patient was diagnosed with Kaposi sarcoma (KS) and referred to the infectious disease department for initiation of antiviral therapy. Marked regression was detected after 6 months of highly active antiretroviral therapy (HAART) without any additional treatment (Figure 2).
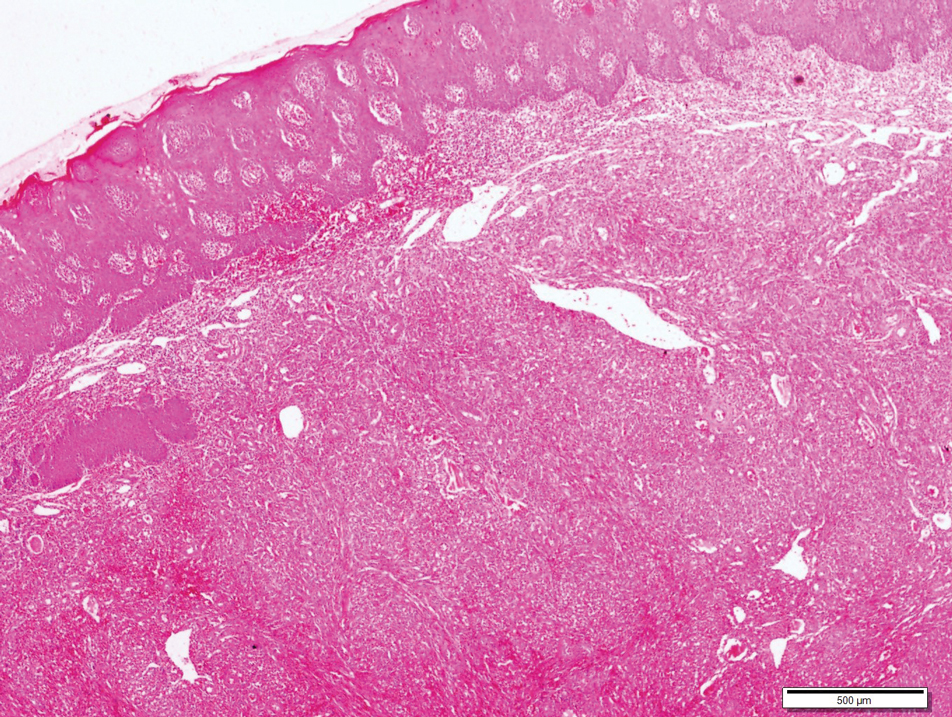
widespread human herpesvirus 8 immunoreactivity (H&E, original magnification ×4).
Kaposi sarcoma is a human herpesvirus 8-associated angioproliferative disorder with low-grade malignant potential. There are 4 well-known clinical types: classic, endemic, iatrogenic, and AIDS associated.1 Involvement of the oral cavity may be seen in all types but mostly is associated with the AIDS-associated type, which also could be a signal for undiagnosed asymptomatic HIV infection.2 Oral KS most often affects the hard and soft palate, gingiva, and dorsal tongue, with plaques or tumors ranging from nonpigmented to brownish red or violaceous. AIDS-associated KS is known to be related to cytokine expression, which is induced by HIV infection causing immune dysregulation by altering the expression of cytokines, including IL-1, tumor necrosis factor α, and IL-6.1 An in vitro study showed that cytokines secrete a number of angiogenic growth factors that, along with HIV proteins, induce and proliferate cells to become sarcoma cells. Integrins and the apoptosis process also are important in proliferation and neovascularization of KS tumor cells.3
Bacillary angiomatosis (BA) is a rare manifestation of infection caused by Bartonella species, which leads to vasoproliferative lesions of the skin and other organs. Bacillary angiomatosis affects individuals with advanced HIV or other immunocompromised individuals and may clinically mimic KS, which is similarly characterized by red-purple papules, nodules, or plaques. Differentiating BA from KS largely depends on histopathologic examination, with BA demonstrating protuberant endothelial cells surrounded by clumps of bacilli that are visible on Warthin-Starry silver stain.
Lymphangioma is a benign hamartomatous hyperplasia of the lymphatic vessels. The majority of lymphangiomas are superficial, but a few may extend deeply into the connective tissue. Intraoral lymphangiomas occur more frequently on the dorsum of the tongue, followed by the palate, buccal mucosa, gingiva, and lips. They may be differentiated with their soft quality, pebblelike surface, and translucent vesicles.
Malignant tumors of the oral cavity are rare, representing only 5% of tumors occurring in the body.4 Among malignant tumors of the oral cavity, squamous cell carcinomas are the most frequent type (90%-98%), and lymphomas and melanoma are the most outstanding among the remaining 2% to 10%. Both for lymphoma and mucosal melanoma, the most common sites of involvement are the soft tissues of the oral cavity, palatal mucosa, gingiva, tongue, cheeks, floor of the mouth, and lips.4 Although mucosal melanoma lesions usually are characterized by pigmented and ulcerated lesions, amelanotic variants also should be kept in mind. Histopathologic examination is mandatory for diagnosis.
Intralesional chemotherapy with vinblastine or bleomycin, radiotherapy, electrochemotherapy, systemic antiretroviral therapy (ie, HAART), and chemotherapy with daunorubicin and pegylated liposomal doxorubicin are the main treatment options.5,6 The immune system activator role of HAART leads to an increased CD4 count and reduces HIV proteins, which helps induction of the proliferation and neovascularization of KS tumor cells.3 This effect may help resolution of KS with localized involvement and allows physicians to utilize HAART without any other additional local and systemic chemotherapy treatment.
- Fatahzadeh M, Schwartz RA. Oral Kaposi's sarcoma: a review and update. Int J Dermatol. 2013;52:666-672.
- Martorano LM, Cannella JD, Lloyd JR. Mucocutaneous presentation of Kaposi sarcoma in an asymptomatic human immunodeficiency virus-positive man. Cutis. 2015;95:E19-E22.
- Stebbing J, Portsmouth S, Gazzard B. How does HAART lead to the resolution of Kaposi's sarcoma? J Antimicrobial Chemother. 2003;51:1095-1098.
- Guevara-Canales JO, Morales-Vadillo R, Sacsaquispe-Contreras SJ, et al. Malignant lymphoma of the oral cavity and the maxillofacial region: overall survivalprognostic factors. Med Oral Patol Oral Cir Bucal. 2013;18:E619-E626.
- Donato V, Guarnaccia R, Dognini J, et al. Radiation therapy in the treatment of HIV-related Kaposi's sarcoma. Anticancer Res. 2013;33:2153-2157.
- Gbabe OF, Okwundu CI, Dedicoat M, et al. Treatment of severe or progressive Kaposi's sarcoma in HIV-infected adults. Cochrane Database Syst Rev. 2014:CD003256.
The Diagnosis: Kaposi Sarcoma
A 4-mm punch biopsy from the border of an ulcerated nodular lesion on the hard palate demonstrated diffusely distributed spindle cells, cleftlike microvascularity with extravasated erythrocytes, and widespread human herpesvirus 8 immunoreactivity on histopathology (Figure 1). Serologic tests were positive for human immunodeficiency virus (HIV) infection; HIV RNA was 14,584 IU/mL and the CD4 count was 254/mm3. The patient was diagnosed with Kaposi sarcoma (KS) and referred to the infectious disease department for initiation of antiviral therapy. Marked regression was detected after 6 months of highly active antiretroviral therapy (HAART) without any additional treatment (Figure 2).
widespread human herpesvirus 8 immunoreactivity (H&E, original magnification ×4).
Kaposi sarcoma is a human herpesvirus 8-associated angioproliferative disorder with low-grade malignant potential. There are 4 well-known clinical types: classic, endemic, iatrogenic, and AIDS associated.1 Involvement of the oral cavity may be seen in all types but mostly is associated with the AIDS-associated type, which also could be a signal for undiagnosed asymptomatic HIV infection.2 Oral KS most often affects the hard and soft palate, gingiva, and dorsal tongue, with plaques or tumors ranging from nonpigmented to brownish red or violaceous. AIDS-associated KS is known to be related to cytokine expression, which is induced by HIV infection causing immune dysregulation by altering the expression of cytokines, including IL-1, tumor necrosis factor α, and IL-6.1 An in vitro study showed that cytokines secrete a number of angiogenic growth factors that, along with HIV proteins, induce and proliferate cells to become sarcoma cells. Integrins and the apoptosis process also are important in proliferation and neovascularization of KS tumor cells.3
Bacillary angiomatosis (BA) is a rare manifestation of infection caused by Bartonella species, which leads to vasoproliferative lesions of the skin and other organs. Bacillary angiomatosis affects individuals with advanced HIV or other immunocompromised individuals and may clinically mimic KS, which is similarly characterized by red-purple papules, nodules, or plaques. Differentiating BA from KS largely depends on histopathologic examination, with BA demonstrating protuberant endothelial cells surrounded by clumps of bacilli that are visible on Warthin-Starry silver stain.
Lymphangioma is a benign hamartomatous hyperplasia of the lymphatic vessels. The majority of lymphangiomas are superficial, but a few may extend deeply into the connective tissue. Intraoral lymphangiomas occur more frequently on the dorsum of the tongue, followed by the palate, buccal mucosa, gingiva, and lips. They may be differentiated with their soft quality, pebblelike surface, and translucent vesicles.
Malignant tumors of the oral cavity are rare, representing only 5% of tumors occurring in the body.4 Among malignant tumors of the oral cavity, squamous cell carcinomas are the most frequent type (90%-98%), and lymphomas and melanoma are the most outstanding among the remaining 2% to 10%. Both for lymphoma and mucosal melanoma, the most common sites of involvement are the soft tissues of the oral cavity, palatal mucosa, gingiva, tongue, cheeks, floor of the mouth, and lips.4 Although mucosal melanoma lesions usually are characterized by pigmented and ulcerated lesions, amelanotic variants also should be kept in mind. Histopathologic examination is mandatory for diagnosis.
Intralesional chemotherapy with vinblastine or bleomycin, radiotherapy, electrochemotherapy, systemic antiretroviral therapy (ie, HAART), and chemotherapy with daunorubicin and pegylated liposomal doxorubicin are the main treatment options.5,6 The immune system activator role of HAART leads to an increased CD4 count and reduces HIV proteins, which helps induction of the proliferation and neovascularization of KS tumor cells.3 This effect may help resolution of KS with localized involvement and allows physicians to utilize HAART without any other additional local and systemic chemotherapy treatment.
The Diagnosis: Kaposi Sarcoma
A 4-mm punch biopsy from the border of an ulcerated nodular lesion on the hard palate demonstrated diffusely distributed spindle cells, cleftlike microvascularity with extravasated erythrocytes, and widespread human herpesvirus 8 immunoreactivity on histopathology (Figure 1). Serologic tests were positive for human immunodeficiency virus (HIV) infection; HIV RNA was 14,584 IU/mL and the CD4 count was 254/mm3. The patient was diagnosed with Kaposi sarcoma (KS) and referred to the infectious disease department for initiation of antiviral therapy. Marked regression was detected after 6 months of highly active antiretroviral therapy (HAART) without any additional treatment (Figure 2).
widespread human herpesvirus 8 immunoreactivity (H&E, original magnification ×4).
Kaposi sarcoma is a human herpesvirus 8-associated angioproliferative disorder with low-grade malignant potential. There are 4 well-known clinical types: classic, endemic, iatrogenic, and AIDS associated.1 Involvement of the oral cavity may be seen in all types but mostly is associated with the AIDS-associated type, which also could be a signal for undiagnosed asymptomatic HIV infection.2 Oral KS most often affects the hard and soft palate, gingiva, and dorsal tongue, with plaques or tumors ranging from nonpigmented to brownish red or violaceous. AIDS-associated KS is known to be related to cytokine expression, which is induced by HIV infection causing immune dysregulation by altering the expression of cytokines, including IL-1, tumor necrosis factor α, and IL-6.1 An in vitro study showed that cytokines secrete a number of angiogenic growth factors that, along with HIV proteins, induce and proliferate cells to become sarcoma cells. Integrins and the apoptosis process also are important in proliferation and neovascularization of KS tumor cells.3
Bacillary angiomatosis (BA) is a rare manifestation of infection caused by Bartonella species, which leads to vasoproliferative lesions of the skin and other organs. Bacillary angiomatosis affects individuals with advanced HIV or other immunocompromised individuals and may clinically mimic KS, which is similarly characterized by red-purple papules, nodules, or plaques. Differentiating BA from KS largely depends on histopathologic examination, with BA demonstrating protuberant endothelial cells surrounded by clumps of bacilli that are visible on Warthin-Starry silver stain.
Lymphangioma is a benign hamartomatous hyperplasia of the lymphatic vessels. The majority of lymphangiomas are superficial, but a few may extend deeply into the connective tissue. Intraoral lymphangiomas occur more frequently on the dorsum of the tongue, followed by the palate, buccal mucosa, gingiva, and lips. They may be differentiated with their soft quality, pebblelike surface, and translucent vesicles.
Malignant tumors of the oral cavity are rare, representing only 5% of tumors occurring in the body.4 Among malignant tumors of the oral cavity, squamous cell carcinomas are the most frequent type (90%-98%), and lymphomas and melanoma are the most outstanding among the remaining 2% to 10%. Both for lymphoma and mucosal melanoma, the most common sites of involvement are the soft tissues of the oral cavity, palatal mucosa, gingiva, tongue, cheeks, floor of the mouth, and lips.4 Although mucosal melanoma lesions usually are characterized by pigmented and ulcerated lesions, amelanotic variants also should be kept in mind. Histopathologic examination is mandatory for diagnosis.
Intralesional chemotherapy with vinblastine or bleomycin, radiotherapy, electrochemotherapy, systemic antiretroviral therapy (ie, HAART), and chemotherapy with daunorubicin and pegylated liposomal doxorubicin are the main treatment options.5,6 The immune system activator role of HAART leads to an increased CD4 count and reduces HIV proteins, which helps induction of the proliferation and neovascularization of KS tumor cells.3 This effect may help resolution of KS with localized involvement and allows physicians to utilize HAART without any other additional local and systemic chemotherapy treatment.
- Fatahzadeh M, Schwartz RA. Oral Kaposi's sarcoma: a review and update. Int J Dermatol. 2013;52:666-672.
- Martorano LM, Cannella JD, Lloyd JR. Mucocutaneous presentation of Kaposi sarcoma in an asymptomatic human immunodeficiency virus-positive man. Cutis. 2015;95:E19-E22.
- Stebbing J, Portsmouth S, Gazzard B. How does HAART lead to the resolution of Kaposi's sarcoma? J Antimicrobial Chemother. 2003;51:1095-1098.
- Guevara-Canales JO, Morales-Vadillo R, Sacsaquispe-Contreras SJ, et al. Malignant lymphoma of the oral cavity and the maxillofacial region: overall survivalprognostic factors. Med Oral Patol Oral Cir Bucal. 2013;18:E619-E626.
- Donato V, Guarnaccia R, Dognini J, et al. Radiation therapy in the treatment of HIV-related Kaposi's sarcoma. Anticancer Res. 2013;33:2153-2157.
- Gbabe OF, Okwundu CI, Dedicoat M, et al. Treatment of severe or progressive Kaposi's sarcoma in HIV-infected adults. Cochrane Database Syst Rev. 2014:CD003256.
- Fatahzadeh M, Schwartz RA. Oral Kaposi's sarcoma: a review and update. Int J Dermatol. 2013;52:666-672.
- Martorano LM, Cannella JD, Lloyd JR. Mucocutaneous presentation of Kaposi sarcoma in an asymptomatic human immunodeficiency virus-positive man. Cutis. 2015;95:E19-E22.
- Stebbing J, Portsmouth S, Gazzard B. How does HAART lead to the resolution of Kaposi's sarcoma? J Antimicrobial Chemother. 2003;51:1095-1098.
- Guevara-Canales JO, Morales-Vadillo R, Sacsaquispe-Contreras SJ, et al. Malignant lymphoma of the oral cavity and the maxillofacial region: overall survivalprognostic factors. Med Oral Patol Oral Cir Bucal. 2013;18:E619-E626.
- Donato V, Guarnaccia R, Dognini J, et al. Radiation therapy in the treatment of HIV-related Kaposi's sarcoma. Anticancer Res. 2013;33:2153-2157.
- Gbabe OF, Okwundu CI, Dedicoat M, et al. Treatment of severe or progressive Kaposi's sarcoma in HIV-infected adults. Cochrane Database Syst Rev. 2014:CD003256.
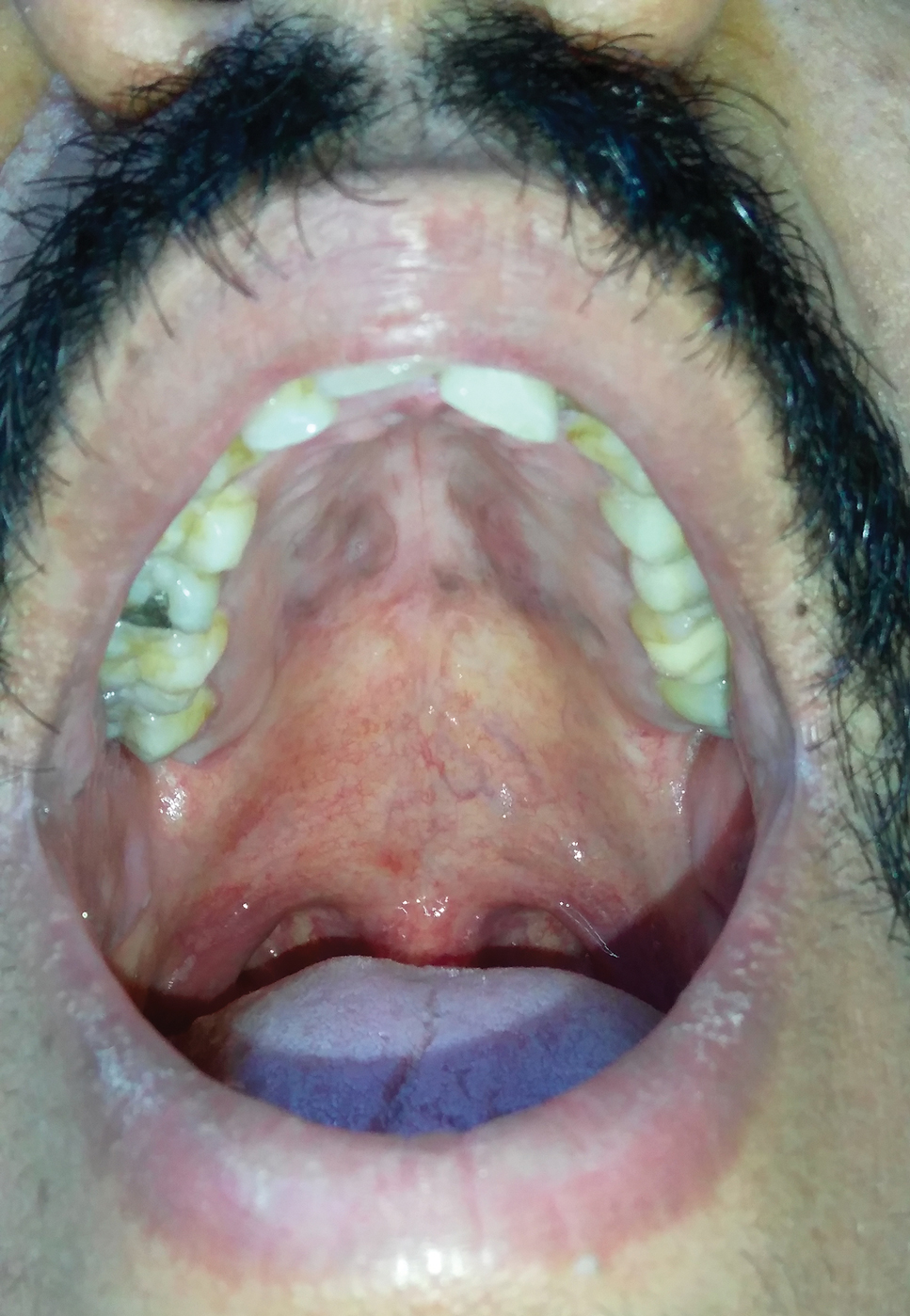
A 30-year-old man presented to our outpatient clinic with rapidly growing, ulcerated, violaceous lesions on the hard palate of 4 months' duration. Physical examination revealed approximately 2.0×1.5-cm, centrally ulcerated, violaceous, nodular lesions on the hard palate, as well as a 4-mm pinkish papular lesion on the soft palate.
Multiple Eruptive Syringomas on the Penis
To the Editor:
Syringomas are small, benign, asymptomatic eccrine or apocrine tumors that present as multiple discrete flesh-colored papules. They are more common in females than males.1 The etiology of eruptive syringomas is unclear, though an inflammatory process has been implicated in the abnormal proliferation of sweat glands.2 However, a minority of tumors have been known to have an autosomal-dominant mode of transmission. Multiple or eruptive syringomas are associated with Down syndrome, Marfan syndrome, Ehlers-Danlos syndrome, and Blau syndrome.3 The clear cell variant has been found to be associated with diabetes mellitus.4 Syringomas most commonly appear on the lower eyelids, upper cheeks, neck, and upper chest; presentation on the penis is rare.5 We report a case of multiple eruptive syringomas located exclusively on the penis mimicking a sexually transmitted condition.
A 53-year-old man who was otherwise healthy presented with multiple flesh-colored papules on the penis that initially began to develop 30 years prior, but increased crops of lesions appeared 4 to 6 weeks prior to presentation. The patient described the lesions as rashlike, nonpruritic, and sensitive to the touch. He denied any discharge, oozing, crusting, or bleeding from the lesions. He did not report any high-risk sexual behaviors and stated that he was in a monogamous relationship with his wife. He had a medical history of molluscum contagiosum that was diagnosed and treated with cryotherapy 30 years prior; however, he did not have a history of any other sexually transmitted diseases. He also did not have a history of diabetes mellitus or thyroid disease.
Physical examination revealed multiple pink papules on the dorsal and ventral shaft of the penis, measuring 2 to 4 mm in diameter, with koebnerization (Figure 1). Based on clinical examination, the differential included condyloma, inflamed seborrheic keratosis, bowenoid papulosis, atypical molluscum contagiosum, or lichen planus. Consequently, a punch biopsy of the penile shaft was performed and histopathologic examination revealed proliferation of ducts focally that were tadpole shaped and embedded in a sclerotic stroma. The lining of the ducts was composed of cuboidal cells, some with clear cell change. The microscopic findings were consistent with penile syringomas (Figure 2). Laboratory results revealed the patient was negative for human immunodeficiency virus, hepatitis B, hepatitis C, and syphilis. The patient was given topical hydrocortisone butyrate and tacrolimus for symptomatic treatment. He declined further aggressive treatment.

Due to the rarity of syringomas on the penis, presentation of these benign eccrine tumors can be commonly mistaken for lichen planus, molluscum contagiosum, genital warts, or bowenoid papulosis.5 The characteristic histopathology of syringomas consists of multiple, small, tadpole or paisley tie–shaped ducts within an eosinophilic stroma. Often, the findings can be histologically confused with desmoplastic trichoepithelioma, morpheaform basal cell carcinoma, and microcystic adnexal carcinoma. Although the histopathology of our patient’s biopsy showed clear cell change, the patient did not report a history of diabetes mellitus, which is a disease that can be associated with the clear cell variant of syringoma. Because syringomas are benign tumors, treatment is not medically necessary unless the lesions are symptomatic. Treatment often is regarded as challenging, as lesions often recur and scarring is a consideration. Possible treatments for removal of the benign papules include surgical excision, electrodesiccation and curettage, shave removal, chemical peels, liquid nitrogen cryotherapy, and CO2 laser vaporization.6
To prevent misdiagnosis and unnecessary treatment, it is important to have syringomas as part of the differential diagnosis when patients present with multiple small flesh-colored papules on the penis. The lesions should be biopsied for accurate diagnosis and to provide reassurance to patients who usually come in for evaluation for fear of having acquired a sexually transmitted disease.
- Yalisove B, Stolar EEH, Williams CM. Multiple penile papules. syringoma. Arch Dermatol. 1987;123:1391-1396.
- Cohen PR, Tschen JA, Rapini RP. Penile syringoma: reports and review of patients with syringoma located on the penis. J Clin Aesthet Dermatol. 2013;6:38-42.
- Yoshimi N, Kurokawa I, Kakuno A, et al. Case of generalized eruptive clear cell syringoma with diabetes mellitus. J Dermatol. 2012;39:744-745.
- Petersson F, Mjornberg PA, Kazakov DV, et al. Eruptive syringoma of the penis. a report of 2 cases and a review of the literature. Am J Dermatopathol. 2009;31:436-438.
- Wu CY. Multifocal penile syringoma masquerading as genital warts. Clin Exp Dermatol. 2009;34:e290-e291.
- Lipshutz RL, Kantor GR, Vonderheid EC. Multiple penile syringomas mimicking verrucae. Int J Dermatol. 1991;30:69.
To the Editor:
Syringomas are small, benign, asymptomatic eccrine or apocrine tumors that present as multiple discrete flesh-colored papules. They are more common in females than males.1 The etiology of eruptive syringomas is unclear, though an inflammatory process has been implicated in the abnormal proliferation of sweat glands.2 However, a minority of tumors have been known to have an autosomal-dominant mode of transmission. Multiple or eruptive syringomas are associated with Down syndrome, Marfan syndrome, Ehlers-Danlos syndrome, and Blau syndrome.3 The clear cell variant has been found to be associated with diabetes mellitus.4 Syringomas most commonly appear on the lower eyelids, upper cheeks, neck, and upper chest; presentation on the penis is rare.5 We report a case of multiple eruptive syringomas located exclusively on the penis mimicking a sexually transmitted condition.
A 53-year-old man who was otherwise healthy presented with multiple flesh-colored papules on the penis that initially began to develop 30 years prior, but increased crops of lesions appeared 4 to 6 weeks prior to presentation. The patient described the lesions as rashlike, nonpruritic, and sensitive to the touch. He denied any discharge, oozing, crusting, or bleeding from the lesions. He did not report any high-risk sexual behaviors and stated that he was in a monogamous relationship with his wife. He had a medical history of molluscum contagiosum that was diagnosed and treated with cryotherapy 30 years prior; however, he did not have a history of any other sexually transmitted diseases. He also did not have a history of diabetes mellitus or thyroid disease.
Physical examination revealed multiple pink papules on the dorsal and ventral shaft of the penis, measuring 2 to 4 mm in diameter, with koebnerization (Figure 1). Based on clinical examination, the differential included condyloma, inflamed seborrheic keratosis, bowenoid papulosis, atypical molluscum contagiosum, or lichen planus. Consequently, a punch biopsy of the penile shaft was performed and histopathologic examination revealed proliferation of ducts focally that were tadpole shaped and embedded in a sclerotic stroma. The lining of the ducts was composed of cuboidal cells, some with clear cell change. The microscopic findings were consistent with penile syringomas (Figure 2). Laboratory results revealed the patient was negative for human immunodeficiency virus, hepatitis B, hepatitis C, and syphilis. The patient was given topical hydrocortisone butyrate and tacrolimus for symptomatic treatment. He declined further aggressive treatment.
Due to the rarity of syringomas on the penis, presentation of these benign eccrine tumors can be commonly mistaken for lichen planus, molluscum contagiosum, genital warts, or bowenoid papulosis.5 The characteristic histopathology of syringomas consists of multiple, small, tadpole or paisley tie–shaped ducts within an eosinophilic stroma. Often, the findings can be histologically confused with desmoplastic trichoepithelioma, morpheaform basal cell carcinoma, and microcystic adnexal carcinoma. Although the histopathology of our patient’s biopsy showed clear cell change, the patient did not report a history of diabetes mellitus, which is a disease that can be associated with the clear cell variant of syringoma. Because syringomas are benign tumors, treatment is not medically necessary unless the lesions are symptomatic. Treatment often is regarded as challenging, as lesions often recur and scarring is a consideration. Possible treatments for removal of the benign papules include surgical excision, electrodesiccation and curettage, shave removal, chemical peels, liquid nitrogen cryotherapy, and CO2 laser vaporization.6
To prevent misdiagnosis and unnecessary treatment, it is important to have syringomas as part of the differential diagnosis when patients present with multiple small flesh-colored papules on the penis. The lesions should be biopsied for accurate diagnosis and to provide reassurance to patients who usually come in for evaluation for fear of having acquired a sexually transmitted disease.
To the Editor:
Syringomas are small, benign, asymptomatic eccrine or apocrine tumors that present as multiple discrete flesh-colored papules. They are more common in females than males.1 The etiology of eruptive syringomas is unclear, though an inflammatory process has been implicated in the abnormal proliferation of sweat glands.2 However, a minority of tumors have been known to have an autosomal-dominant mode of transmission. Multiple or eruptive syringomas are associated with Down syndrome, Marfan syndrome, Ehlers-Danlos syndrome, and Blau syndrome.3 The clear cell variant has been found to be associated with diabetes mellitus.4 Syringomas most commonly appear on the lower eyelids, upper cheeks, neck, and upper chest; presentation on the penis is rare.5 We report a case of multiple eruptive syringomas located exclusively on the penis mimicking a sexually transmitted condition.
A 53-year-old man who was otherwise healthy presented with multiple flesh-colored papules on the penis that initially began to develop 30 years prior, but increased crops of lesions appeared 4 to 6 weeks prior to presentation. The patient described the lesions as rashlike, nonpruritic, and sensitive to the touch. He denied any discharge, oozing, crusting, or bleeding from the lesions. He did not report any high-risk sexual behaviors and stated that he was in a monogamous relationship with his wife. He had a medical history of molluscum contagiosum that was diagnosed and treated with cryotherapy 30 years prior; however, he did not have a history of any other sexually transmitted diseases. He also did not have a history of diabetes mellitus or thyroid disease.
Physical examination revealed multiple pink papules on the dorsal and ventral shaft of the penis, measuring 2 to 4 mm in diameter, with koebnerization (Figure 1). Based on clinical examination, the differential included condyloma, inflamed seborrheic keratosis, bowenoid papulosis, atypical molluscum contagiosum, or lichen planus. Consequently, a punch biopsy of the penile shaft was performed and histopathologic examination revealed proliferation of ducts focally that were tadpole shaped and embedded in a sclerotic stroma. The lining of the ducts was composed of cuboidal cells, some with clear cell change. The microscopic findings were consistent with penile syringomas (Figure 2). Laboratory results revealed the patient was negative for human immunodeficiency virus, hepatitis B, hepatitis C, and syphilis. The patient was given topical hydrocortisone butyrate and tacrolimus for symptomatic treatment. He declined further aggressive treatment.
Due to the rarity of syringomas on the penis, presentation of these benign eccrine tumors can be commonly mistaken for lichen planus, molluscum contagiosum, genital warts, or bowenoid papulosis.5 The characteristic histopathology of syringomas consists of multiple, small, tadpole or paisley tie–shaped ducts within an eosinophilic stroma. Often, the findings can be histologically confused with desmoplastic trichoepithelioma, morpheaform basal cell carcinoma, and microcystic adnexal carcinoma. Although the histopathology of our patient’s biopsy showed clear cell change, the patient did not report a history of diabetes mellitus, which is a disease that can be associated with the clear cell variant of syringoma. Because syringomas are benign tumors, treatment is not medically necessary unless the lesions are symptomatic. Treatment often is regarded as challenging, as lesions often recur and scarring is a consideration. Possible treatments for removal of the benign papules include surgical excision, electrodesiccation and curettage, shave removal, chemical peels, liquid nitrogen cryotherapy, and CO2 laser vaporization.6
To prevent misdiagnosis and unnecessary treatment, it is important to have syringomas as part of the differential diagnosis when patients present with multiple small flesh-colored papules on the penis. The lesions should be biopsied for accurate diagnosis and to provide reassurance to patients who usually come in for evaluation for fear of having acquired a sexually transmitted disease.
- Yalisove B, Stolar EEH, Williams CM. Multiple penile papules. syringoma. Arch Dermatol. 1987;123:1391-1396.
- Cohen PR, Tschen JA, Rapini RP. Penile syringoma: reports and review of patients with syringoma located on the penis. J Clin Aesthet Dermatol. 2013;6:38-42.
- Yoshimi N, Kurokawa I, Kakuno A, et al. Case of generalized eruptive clear cell syringoma with diabetes mellitus. J Dermatol. 2012;39:744-745.
- Petersson F, Mjornberg PA, Kazakov DV, et al. Eruptive syringoma of the penis. a report of 2 cases and a review of the literature. Am J Dermatopathol. 2009;31:436-438.
- Wu CY. Multifocal penile syringoma masquerading as genital warts. Clin Exp Dermatol. 2009;34:e290-e291.
- Lipshutz RL, Kantor GR, Vonderheid EC. Multiple penile syringomas mimicking verrucae. Int J Dermatol. 1991;30:69.
- Yalisove B, Stolar EEH, Williams CM. Multiple penile papules. syringoma. Arch Dermatol. 1987;123:1391-1396.
- Cohen PR, Tschen JA, Rapini RP. Penile syringoma: reports and review of patients with syringoma located on the penis. J Clin Aesthet Dermatol. 2013;6:38-42.
- Yoshimi N, Kurokawa I, Kakuno A, et al. Case of generalized eruptive clear cell syringoma with diabetes mellitus. J Dermatol. 2012;39:744-745.
- Petersson F, Mjornberg PA, Kazakov DV, et al. Eruptive syringoma of the penis. a report of 2 cases and a review of the literature. Am J Dermatopathol. 2009;31:436-438.
- Wu CY. Multifocal penile syringoma masquerading as genital warts. Clin Exp Dermatol. 2009;34:e290-e291.
- Lipshutz RL, Kantor GR, Vonderheid EC. Multiple penile syringomas mimicking verrucae. Int J Dermatol. 1991;30:69.
Practice Points
- Penile syringoma can mimic sexually transmitted disease such as condyloma acuminatum or molluscum contagiosum.
- Penile syringomas can be long-standing and require biopsy to differentiate from other conditions.

Infographic: Hyperhidrosis Survey Results
"Doctor, Do I Need a Skin Check?"
What does your patient need to know at the first visit?
A patient may be scheduled for a total-body skin examination (TBSE) through several routes: primary care referral, continued cancer screening for an at-risk patient or patient transfer, or patient-directed scheduling for general screening regardless of risk factors. At the patient's first visit, it is imperative that the course of the appointment is smooth and predictable for patient comfort and for a thorough and effective examination. The nurse initially solicits salient medical history, particularly personal and family history of skin cancer, current medications, and any acute concerns. The nurse then prepares the patient for the logistics of the TBSE, namely to undress, don a gown that ties and opens in the back, and be seated on the examination table. When I enter the room, the conversation commences with me seated across from the patient, reviewing specifics about his/her history and risk factors. Then the TBSE is executed from head to toe.
Do you broadly recommend TBSE?
Firstly, TBSE is a safe clinical tool, supported by data outlining a lack of notable patient morbidity during the examination, including psychosocial factors, and it is generally well-received by patients (Risica et al). In 2016, the US Preventative Services Task Force (USPSTF) outlined its recommendations regarding screening for skin cancer, concluding that there is insufficient evidence to broadly recommend TBSE. Unfortunately, USPSTF findings amassed data from all types of screenings, including those by nondermatologists, and did not extract specialty-specific benefits and risks to patients. The recommendation also did not outline the influence of TBSE on morbidity and mortality for at-risk groups. The guidelines target primary care practice trends; therefore, specialty societies such as the American Academy of Dermatology issued statements following the USPSTF recommendation outlining these salient clarifications, namely that TBSE detects melanoma and keratinocyte carcinomas earlier than in patients who are not screened. Randomized controlled trials to prove this observation are lacking, particularly because of the ethics of withholding screening from a prospective study group. However, in 2017, Johnson et al outlined the best available survival data in concert with the USPSTF statement to arrive at the most beneficial screening recommendations for patients, specifically targeting risk groups--those with a history of skin cancer, immunosuppression, indoor tanning and/or many blistering sunburns, and several other genetic parameters--for at least annual TBSE.
The technique and reproducibility of TBSE also are not standardized, though they seem to have been endearingly apprenticed but variably implemented through generations of dermatology residents going forward into practice. As it is, depending on patient body surface area, mobility, willingness to disrobe, and adornments (eg, tattoos, hair appliances), multiple factors can restrict full view of a patient's skin. Recently, Helm et al proposed standardizing the TBSE sequence to minimize omitted areas of the body, which may become an imperative tool for streamlined resident teaching and optimal screening encounters.
How do you keep patients compliant with TBSE?
During and following TBSE, I typically outline any lesions of concern and plan for further testing, screening, and behavioral prevention strategies. Frequency of TBSE and importance of compliance are discussed during the visit and reinforced at checkout where the appointment templates are established a year in advance for those with skin cancer. Further, for those with melanoma, their appointment slots are given priority status so that any cancellations or delays are rescheduled preferentially. Particularly during the discussion about TBSE frequency, I emphasize the comparison and importance of this visit akin to other recommended screenings, such as mammograms and colonoscopies, and that we, as dermatologists, are part of their cancer surveillance team.
What do you do if patients refuse your recommendations?
Some patients refuse a gown or removal of certain clothing items (eg, undergarments, socks, wigs). Some patients defer a yearly TBSE upon checkout and schedule an appointment only when a lesion of concern arises. My advice is not to shame patients and to take advantage of as much as the patient is able and comfortable to show us and be present for, welcoming that we have the opportunity to take care of them and screen for cancer in any capacity. In underserved or limited budget practice regions, lesion-directed examination vs TBSE may be the only screening method utilized and may even attract more patients to a screening facility (Hoorens et al).
In the opposite corner are those patients who deem the recommended TBSE interval as too infrequent, which poses a delicate dilemma. In my opinion, these situations present another cohort of risks. Namely, the patient may become (or continue to be) overly fixated on the small details of every skin lesion, and in my experience, they tend to develop the habit of expecting at least 1 biopsy at each visit, typically of a lesion of their choosing. Depending on the validity of this expectation vs my clinical examination, it can lead to a difficult discussion with the patient about oversampling lesions and the potential for many scars, copious reexcisions for ambiguous lesion pathology, and a trend away from prudent clinical care. In addition, multiple visits incur more patient co-pays and time away from school, work, or home. To ease the patient's mind, I advise to call our office for a more acute visit if there is a lesion of concern; I additionally recommend taking a smartphone photograph of a concerning lesion and monitoring it for changes or sending the photograph to our patient portal messaging system so we can evaluate its acuity.
What take-home advice do you give to patients?
As the visit ends, I further explain that home self-examination or examination by a partner between visits is intuitively a valuable screening adjunct for skin cancer. In 2018, the USPSTF recommended behavioral skin cancer prevention counseling and self-examination only for younger-age cohorts with fair skin (6 months to 24 years), but its utility in specialty practice must be qualified. The American Academy of Dermatology Association subsequently issued a statement to support safe sun-protective practices and diligent self-screening for changing lesions, as earlier detection and management of skin cancer can lead to decreased morbidity and mortality from these neoplasms.
Resources for Patients
American Academy of Dermatology's SPOT Skin Cancer
Centers for Disease Control and Prevention: What Screening Tests Are There?
Suggested Readings
AAD statement on USPSTF recommendation on skin cancer screening. Schaumburg, IL: American Academy of Dermatology; July 26, 2016. https://www.aad.org/media/news-releases/aad-statement-on-uspstf. Accessed April 26, 2019.
AADA responds to USPSTF recommendation on skin cancer prevention counseling. Rosemont, IL: American Academy of Dermatology Association; March 20, 2018. https://www.aad.org/media/news-releases/skin-cancer-prevention-counseling. Accessed April 26, 2019.
Helm MF, Hallock KK, Bisbee E, et al. Optimizing the total body skin exam: an observational cohort study [published online February 15, 2019]. J Am Acad Dermatol. doi:10.1016/j.jaad.2019.02.028.
Hoorens I, Vossaert K, Pil L, et al. Total-body examination vs lesion-directed skin cancer screening. JAMA Dermatol. 2016;152:27-34.
Johnson MM, Leachman SA, Aspinwall LG, et al. Skin cancer screening: recommendations for data-driven screening guidelines and a review of the US Preventive Services Task Force controversy. Melanoma Manag. 2017;4:13-37.
Risica PM, Matthews NH, Dionne L, et al. Psychosocial consequences of skin cancer screening. Prev Med Rep. 2018;10:310-316.
US Preventive Services Task Force, Bibbins-Domingo K, Grossman DC, et al. Screening for skin cancer: US Preventive Services Task Force recommendation statement. JAMA. 2016;316:429-435.
US Preventive Services Task Force, Grossman DC, Curry SJ, et al. Behavioral counseling to prevent skin cancer: US Preventive Services Task Force recommendation statement. JAMA. 2018;319:1134-1142.
What does your patient need to know at the first visit?
A patient may be scheduled for a total-body skin examination (TBSE) through several routes: primary care referral, continued cancer screening for an at-risk patient or patient transfer, or patient-directed scheduling for general screening regardless of risk factors. At the patient's first visit, it is imperative that the course of the appointment is smooth and predictable for patient comfort and for a thorough and effective examination. The nurse initially solicits salient medical history, particularly personal and family history of skin cancer, current medications, and any acute concerns. The nurse then prepares the patient for the logistics of the TBSE, namely to undress, don a gown that ties and opens in the back, and be seated on the examination table. When I enter the room, the conversation commences with me seated across from the patient, reviewing specifics about his/her history and risk factors. Then the TBSE is executed from head to toe.
Do you broadly recommend TBSE?
Firstly, TBSE is a safe clinical tool, supported by data outlining a lack of notable patient morbidity during the examination, including psychosocial factors, and it is generally well-received by patients (Risica et al). In 2016, the US Preventative Services Task Force (USPSTF) outlined its recommendations regarding screening for skin cancer, concluding that there is insufficient evidence to broadly recommend TBSE. Unfortunately, USPSTF findings amassed data from all types of screenings, including those by nondermatologists, and did not extract specialty-specific benefits and risks to patients. The recommendation also did not outline the influence of TBSE on morbidity and mortality for at-risk groups. The guidelines target primary care practice trends; therefore, specialty societies such as the American Academy of Dermatology issued statements following the USPSTF recommendation outlining these salient clarifications, namely that TBSE detects melanoma and keratinocyte carcinomas earlier than in patients who are not screened. Randomized controlled trials to prove this observation are lacking, particularly because of the ethics of withholding screening from a prospective study group. However, in 2017, Johnson et al outlined the best available survival data in concert with the USPSTF statement to arrive at the most beneficial screening recommendations for patients, specifically targeting risk groups--those with a history of skin cancer, immunosuppression, indoor tanning and/or many blistering sunburns, and several other genetic parameters--for at least annual TBSE.
The technique and reproducibility of TBSE also are not standardized, though they seem to have been endearingly apprenticed but variably implemented through generations of dermatology residents going forward into practice. As it is, depending on patient body surface area, mobility, willingness to disrobe, and adornments (eg, tattoos, hair appliances), multiple factors can restrict full view of a patient's skin. Recently, Helm et al proposed standardizing the TBSE sequence to minimize omitted areas of the body, which may become an imperative tool for streamlined resident teaching and optimal screening encounters.
How do you keep patients compliant with TBSE?
During and following TBSE, I typically outline any lesions of concern and plan for further testing, screening, and behavioral prevention strategies. Frequency of TBSE and importance of compliance are discussed during the visit and reinforced at checkout where the appointment templates are established a year in advance for those with skin cancer. Further, for those with melanoma, their appointment slots are given priority status so that any cancellations or delays are rescheduled preferentially. Particularly during the discussion about TBSE frequency, I emphasize the comparison and importance of this visit akin to other recommended screenings, such as mammograms and colonoscopies, and that we, as dermatologists, are part of their cancer surveillance team.
What do you do if patients refuse your recommendations?
Some patients refuse a gown or removal of certain clothing items (eg, undergarments, socks, wigs). Some patients defer a yearly TBSE upon checkout and schedule an appointment only when a lesion of concern arises. My advice is not to shame patients and to take advantage of as much as the patient is able and comfortable to show us and be present for, welcoming that we have the opportunity to take care of them and screen for cancer in any capacity. In underserved or limited budget practice regions, lesion-directed examination vs TBSE may be the only screening method utilized and may even attract more patients to a screening facility (Hoorens et al).
In the opposite corner are those patients who deem the recommended TBSE interval as too infrequent, which poses a delicate dilemma. In my opinion, these situations present another cohort of risks. Namely, the patient may become (or continue to be) overly fixated on the small details of every skin lesion, and in my experience, they tend to develop the habit of expecting at least 1 biopsy at each visit, typically of a lesion of their choosing. Depending on the validity of this expectation vs my clinical examination, it can lead to a difficult discussion with the patient about oversampling lesions and the potential for many scars, copious reexcisions for ambiguous lesion pathology, and a trend away from prudent clinical care. In addition, multiple visits incur more patient co-pays and time away from school, work, or home. To ease the patient's mind, I advise to call our office for a more acute visit if there is a lesion of concern; I additionally recommend taking a smartphone photograph of a concerning lesion and monitoring it for changes or sending the photograph to our patient portal messaging system so we can evaluate its acuity.
What take-home advice do you give to patients?
As the visit ends, I further explain that home self-examination or examination by a partner between visits is intuitively a valuable screening adjunct for skin cancer. In 2018, the USPSTF recommended behavioral skin cancer prevention counseling and self-examination only for younger-age cohorts with fair skin (6 months to 24 years), but its utility in specialty practice must be qualified. The American Academy of Dermatology Association subsequently issued a statement to support safe sun-protective practices and diligent self-screening for changing lesions, as earlier detection and management of skin cancer can lead to decreased morbidity and mortality from these neoplasms.
Resources for Patients
American Academy of Dermatology's SPOT Skin Cancer
Centers for Disease Control and Prevention: What Screening Tests Are There?
What does your patient need to know at the first visit?
A patient may be scheduled for a total-body skin examination (TBSE) through several routes: primary care referral, continued cancer screening for an at-risk patient or patient transfer, or patient-directed scheduling for general screening regardless of risk factors. At the patient's first visit, it is imperative that the course of the appointment is smooth and predictable for patient comfort and for a thorough and effective examination. The nurse initially solicits salient medical history, particularly personal and family history of skin cancer, current medications, and any acute concerns. The nurse then prepares the patient for the logistics of the TBSE, namely to undress, don a gown that ties and opens in the back, and be seated on the examination table. When I enter the room, the conversation commences with me seated across from the patient, reviewing specifics about his/her history and risk factors. Then the TBSE is executed from head to toe.
Do you broadly recommend TBSE?
Firstly, TBSE is a safe clinical tool, supported by data outlining a lack of notable patient morbidity during the examination, including psychosocial factors, and it is generally well-received by patients (Risica et al). In 2016, the US Preventative Services Task Force (USPSTF) outlined its recommendations regarding screening for skin cancer, concluding that there is insufficient evidence to broadly recommend TBSE. Unfortunately, USPSTF findings amassed data from all types of screenings, including those by nondermatologists, and did not extract specialty-specific benefits and risks to patients. The recommendation also did not outline the influence of TBSE on morbidity and mortality for at-risk groups. The guidelines target primary care practice trends; therefore, specialty societies such as the American Academy of Dermatology issued statements following the USPSTF recommendation outlining these salient clarifications, namely that TBSE detects melanoma and keratinocyte carcinomas earlier than in patients who are not screened. Randomized controlled trials to prove this observation are lacking, particularly because of the ethics of withholding screening from a prospective study group. However, in 2017, Johnson et al outlined the best available survival data in concert with the USPSTF statement to arrive at the most beneficial screening recommendations for patients, specifically targeting risk groups--those with a history of skin cancer, immunosuppression, indoor tanning and/or many blistering sunburns, and several other genetic parameters--for at least annual TBSE.
The technique and reproducibility of TBSE also are not standardized, though they seem to have been endearingly apprenticed but variably implemented through generations of dermatology residents going forward into practice. As it is, depending on patient body surface area, mobility, willingness to disrobe, and adornments (eg, tattoos, hair appliances), multiple factors can restrict full view of a patient's skin. Recently, Helm et al proposed standardizing the TBSE sequence to minimize omitted areas of the body, which may become an imperative tool for streamlined resident teaching and optimal screening encounters.
How do you keep patients compliant with TBSE?
During and following TBSE, I typically outline any lesions of concern and plan for further testing, screening, and behavioral prevention strategies. Frequency of TBSE and importance of compliance are discussed during the visit and reinforced at checkout where the appointment templates are established a year in advance for those with skin cancer. Further, for those with melanoma, their appointment slots are given priority status so that any cancellations or delays are rescheduled preferentially. Particularly during the discussion about TBSE frequency, I emphasize the comparison and importance of this visit akin to other recommended screenings, such as mammograms and colonoscopies, and that we, as dermatologists, are part of their cancer surveillance team.
What do you do if patients refuse your recommendations?
Some patients refuse a gown or removal of certain clothing items (eg, undergarments, socks, wigs). Some patients defer a yearly TBSE upon checkout and schedule an appointment only when a lesion of concern arises. My advice is not to shame patients and to take advantage of as much as the patient is able and comfortable to show us and be present for, welcoming that we have the opportunity to take care of them and screen for cancer in any capacity. In underserved or limited budget practice regions, lesion-directed examination vs TBSE may be the only screening method utilized and may even attract more patients to a screening facility (Hoorens et al).
In the opposite corner are those patients who deem the recommended TBSE interval as too infrequent, which poses a delicate dilemma. In my opinion, these situations present another cohort of risks. Namely, the patient may become (or continue to be) overly fixated on the small details of every skin lesion, and in my experience, they tend to develop the habit of expecting at least 1 biopsy at each visit, typically of a lesion of their choosing. Depending on the validity of this expectation vs my clinical examination, it can lead to a difficult discussion with the patient about oversampling lesions and the potential for many scars, copious reexcisions for ambiguous lesion pathology, and a trend away from prudent clinical care. In addition, multiple visits incur more patient co-pays and time away from school, work, or home. To ease the patient's mind, I advise to call our office for a more acute visit if there is a lesion of concern; I additionally recommend taking a smartphone photograph of a concerning lesion and monitoring it for changes or sending the photograph to our patient portal messaging system so we can evaluate its acuity.
What take-home advice do you give to patients?
As the visit ends, I further explain that home self-examination or examination by a partner between visits is intuitively a valuable screening adjunct for skin cancer. In 2018, the USPSTF recommended behavioral skin cancer prevention counseling and self-examination only for younger-age cohorts with fair skin (6 months to 24 years), but its utility in specialty practice must be qualified. The American Academy of Dermatology Association subsequently issued a statement to support safe sun-protective practices and diligent self-screening for changing lesions, as earlier detection and management of skin cancer can lead to decreased morbidity and mortality from these neoplasms.
Resources for Patients
American Academy of Dermatology's SPOT Skin Cancer
Centers for Disease Control and Prevention: What Screening Tests Are There?
Suggested Readings
AAD statement on USPSTF recommendation on skin cancer screening. Schaumburg, IL: American Academy of Dermatology; July 26, 2016. https://www.aad.org/media/news-releases/aad-statement-on-uspstf. Accessed April 26, 2019.
AADA responds to USPSTF recommendation on skin cancer prevention counseling. Rosemont, IL: American Academy of Dermatology Association; March 20, 2018. https://www.aad.org/media/news-releases/skin-cancer-prevention-counseling. Accessed April 26, 2019.
Helm MF, Hallock KK, Bisbee E, et al. Optimizing the total body skin exam: an observational cohort study [published online February 15, 2019]. J Am Acad Dermatol. doi:10.1016/j.jaad.2019.02.028.
Hoorens I, Vossaert K, Pil L, et al. Total-body examination vs lesion-directed skin cancer screening. JAMA Dermatol. 2016;152:27-34.
Johnson MM, Leachman SA, Aspinwall LG, et al. Skin cancer screening: recommendations for data-driven screening guidelines and a review of the US Preventive Services Task Force controversy. Melanoma Manag. 2017;4:13-37.
Risica PM, Matthews NH, Dionne L, et al. Psychosocial consequences of skin cancer screening. Prev Med Rep. 2018;10:310-316.
US Preventive Services Task Force, Bibbins-Domingo K, Grossman DC, et al. Screening for skin cancer: US Preventive Services Task Force recommendation statement. JAMA. 2016;316:429-435.
US Preventive Services Task Force, Grossman DC, Curry SJ, et al. Behavioral counseling to prevent skin cancer: US Preventive Services Task Force recommendation statement. JAMA. 2018;319:1134-1142.
Suggested Readings
AAD statement on USPSTF recommendation on skin cancer screening. Schaumburg, IL: American Academy of Dermatology; July 26, 2016. https://www.aad.org/media/news-releases/aad-statement-on-uspstf. Accessed April 26, 2019.
AADA responds to USPSTF recommendation on skin cancer prevention counseling. Rosemont, IL: American Academy of Dermatology Association; March 20, 2018. https://www.aad.org/media/news-releases/skin-cancer-prevention-counseling. Accessed April 26, 2019.
Helm MF, Hallock KK, Bisbee E, et al. Optimizing the total body skin exam: an observational cohort study [published online February 15, 2019]. J Am Acad Dermatol. doi:10.1016/j.jaad.2019.02.028.
Hoorens I, Vossaert K, Pil L, et al. Total-body examination vs lesion-directed skin cancer screening. JAMA Dermatol. 2016;152:27-34.
Johnson MM, Leachman SA, Aspinwall LG, et al. Skin cancer screening: recommendations for data-driven screening guidelines and a review of the US Preventive Services Task Force controversy. Melanoma Manag. 2017;4:13-37.
Risica PM, Matthews NH, Dionne L, et al. Psychosocial consequences of skin cancer screening. Prev Med Rep. 2018;10:310-316.
US Preventive Services Task Force, Bibbins-Domingo K, Grossman DC, et al. Screening for skin cancer: US Preventive Services Task Force recommendation statement. JAMA. 2016;316:429-435.
US Preventive Services Task Force, Grossman DC, Curry SJ, et al. Behavioral counseling to prevent skin cancer: US Preventive Services Task Force recommendation statement. JAMA. 2018;319:1134-1142.
Cystic Scalp Lesion
The Diagnosis: Merkel Cell Carcinoma
An excisional biopsy revealed that the dermis was mostly replaced by a malignant neoplastic infiltrate morphologically resembling small cell carcinoma (Figure 1). The cells had uniform hyperchromatic nuclei with fairly even chromatin and generally inconspicuous nucleoli. There was a tendency for smudgy artifacts at the periphery of the infiltrate, and the cells had relatively scant cytoplasm with slight streaming. Occasional apoptotic forms were present. Immunohistochemistry showed strong dotlike staining with cytokeratin 20 and moderate positivity with synaptophysin and chromogranin A (Figure 2). Unusually, there also was weak staining in a few tumor cells with thyroid transcription factor 1, a marker usually indicative of small cell carcinoma of the lungs that typically is negative in Merkel cell carcinoma (MCC). A second thyroid transcription factor 1 monoclonal antibody used in a double immunostain for lung adenocarcinomas was completely negative. This second antibody is more specific but less sensitive than the stand-alone version. The skin biopsy results confirmed the diagnosis of MCC. Given the patient's frailty and comorbidities, wide local excision was not performed and the patient was referred to radiation oncology. He died several months later from metastatic MCC.
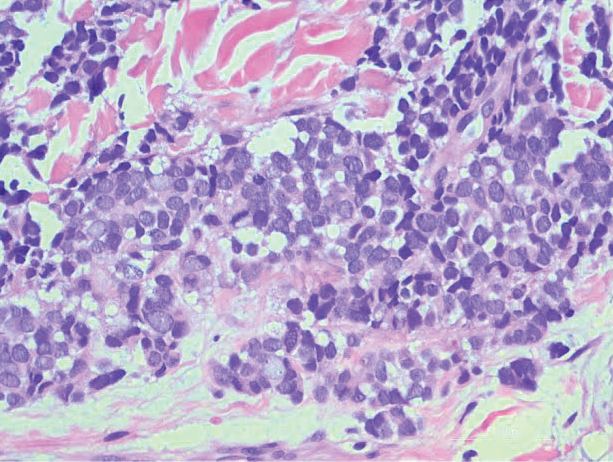
dermis was mostly replaced by a malignant neoplastic infiltrate morphologically resembling small cell carcinoma. The cells had uniform hyperchromatic nuclei with fairly even chromatin and generally inconspicuous nucleoli (H&E, original magnification ×200).
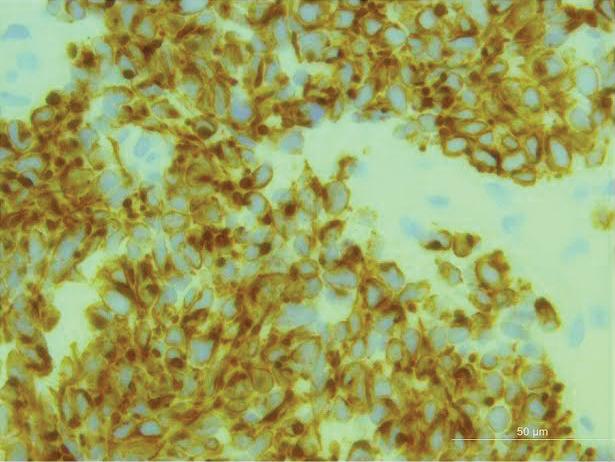
Merkel cell carcinoma (original magnification ×200).
Merkel cell carcinoma is an uncommon skin malignancy that can be easily mistaken for other conditions if the clinician is not familiar with its typical presentation. It most commonly is found on the head and neck in elderly individuals, most often aged 60 to 80 years,1 with a notable history of sun exposure and/or immunosuppression. It is an aggressive skin cancer that originally was thought to be due to pathogenic changes of Merkel cells,2 which are specialized touch receptors located at the dermoepidermal junction of the skin; however, newer evidence has suggested that MCC arises from malignant changes to skin stem cells.3 It shares more characteristics with extracutaneous neuroendocrine tumors and is more aptly labeled by pathologists as a primary neuroendocrine carcinoma of the skin.4
The frequency of MCC is highest in Australia, likely due to intense sun exposure, where the age-adjusted incidence rate reported in Queensland was 1.6 per 100,000 individuals from 2006 to 2010.5 The lowest incidence rates were reported in Finland (0.11 and 0.12 per 100,000 males and females, respectively)6 and Denmark (2.2 cases per million person-years).7 The clinical features of MCC are summarized by the mnemonic AEIOU: asymptomatic/lack of tenderness, expanding rapidly, immune suppression, older than 50 years, UV-exposed site on a person with fair skin.8 In a 2008 study of 195 patients, 89% of primary MCC lesions met 3 or more criteria, 32% met 4 or more criteria, and 7% met all 5 criteria.8
The classic presentation of MCC is a pink-red to violaceous nodule on the head or neck in an elderly patient, but there is a need to maintain suspicion of malignancy when examining a presumed infected cystic lesion, especially when a round of antibiotics has not ameliorated the symptoms. According to Heath et al,8 of 106 patients treated for MCC, 56% of first clinical impressions were benign. A PubMed and Scopus search was performed with the MeSH headings Merkel cell carcinoma +/- presentation to uncover similar unusual presentations between 1970 and the present day. Merkel cell carcinoma has been misdiagnosed as seemingly benign lesions including lipoma,9 allergic contact dermatitis,10 and atheroma.11 The differential diagnosis of MCC also includes cysts, amelanotic melanoma, basal cell carcinoma, dermatofibrosarcoma protuberans, squamous cell carcinoma, fungal kerion, leiomyosarcoma, neurothekeoma, abscesses, and cutaneous lymphoma.
Merkel cell polyomavirus has been implicated in the malignant transformation of MCC. It is a small, human, nonenveloped, double-stranded DNA virus1 and is found in approximately 70% to 80% of MCC cases.12 Merkel cell polyomavirus is a respiratory tract pathogen that is acquired by immunocompetent infants; it integrates itself into the host's genome and then enters a long latency period to later reactivate in immunocompromised adults.13
Wide local excision down to fascia is the mainstay of treatment of MCC, with recommended margins of 1 to 2 cm.14 Mohs micrographic surgery also can be considered.15 Similar to other neuroendocrine tumors, MCC is considered a radiosensitive tumor; radiation likely improves local control and is recommended in early-stage disease.16,17 It also has been described as the sole treatment modality in patients who are not candidates for surgery. The role of chemotherapy is more controversial, as responses do not appear to be long-lasting but should be considered in patients with advanced disease.14,18 There have been major advances in immunotherapy with the recent approvals of avelumab, an anti-PD-L1 inhibitor,19 and pembrolizumab,20 an anti-PD-1 inhibitor, for metastatic MCC. Clinical trials for MCC using kinase inhibitors and somatostatin analogues currently are ongoing.21
Several studies have demonstrated high rates of occult nodal disease in clinically node-negative patients, which has led to widespread use of sentinel lymph node biopsies.22,23 A sentinel lymph node biopsy is recommended at the time of surgery to aid with treatment decisions and prognosis.24
Merkel cell carcinoma is highly aggressive, and more than one-third of patients die from their disease, making it twice as lethal as melanoma. Overall survival rates remain low (5-year overall survival, 0%-18%) for advanced disease.5 Unfortunately, progression to metastasis is common and most often occurs within 2 years of diagnosis.17,25 Follow-up after treatment of MCC is crucial, with the 2019 National Comprehensive Cancer Network (NCCN) guidelines suggesting a physical examination with complete skin and complete lymph node examination every 3 to 6 months for 3 years and every 6 to 12 months thereafter.15
This case is an important reminder to include MCC in the differential diagnosis of presumed infected cysts, particularly on sun-exposed sites in elderly patients, as our patient was treated with antibiotics twice without improvement. An infected cyst with a lack of response to antibiotics should alert clinicians to the potential of malignancy.
- Sourvinos G, Mammas IN, Spandidos GA. 2015 Merkel cell polyoma virus infections in childhood. Arch Virol. 2015;160:887-892.
- Sibley RK, Rosai J, Foucar E, et al. Neuroendocrine (Merkel cell) carcinoma of the skin. a histologic and ultrastructural study of two cases. Am J Surg Pathol. 1980;4:211-221.
- Tilling T, Moll I. Which are the cells of origin in Merkel cell carcinoma? J Skin Cancer. 2012;2012:1-7.
- Succaria F, Radfar A, Bhawan J. Merkel cell carcinoma (primary neuroendocrine carcinoma of skin) mimicking basal cell carcinoma with review of different histopathologic features. Am J Dermatopathol. 2014;36:160-166.
- Youlden DR, Soyer HP, Youl PH, et al. Incidence and survival for Merkel cell carcinoma in Queensland, Australia, 1993-2010. JAMA Dermatol. 2014;150:864-872.
- Kukko H, Böhling T, Koljonen V, et al. Merkel cell carcinoma--a population-based epidemiological study in Finland with a clinical series of 181 cases. Eur J Cancer. 2012;48:737-742.
- Kaae J, Hansen AV, Biggar RJ, et al. Merkel cell carcinoma: incidence, mortality, and risk of other cancers. J Natl Cancer Inst. 2010;102:793-801.
- Heath M, Jaimes N, Lamos B, et al. Clinical characteristics of Merkel cell carcinoma at diagnosis of 195 patients: the AEIOU features. J Am Acad Dermatol. 2008;59:375-381.
- Sarma DP, Heagley DE, Chalupa J, et al. An unusual clinical presentation of Merkel cell carcinoma: a case report. Case Rep Med. 2010;2010:905414.
- Craven E, Alexandroff A, Liu JK, et al. Merkel cell carcinoma mistaken for allergic contact dermatitis. BMJ. 2015;351:h4635.
- Kinoshita A, Hoashi T, Okazaki S, et al. Atypical case of Merkel cell carcinoma difficult to diagnose clinically. J Dermatol. 2017;44:E158-E159.
- Donepudi S, DeConti LC, Samlowski WE. Recent advances in the understanding of the genetics, etiology, and treatment of Merkel cell carcinoma. Semin Oncol. 2012;39:163-172.
- Abedi Kiasari B, Vallely PJ, Klapper PE. Merkel cell polyoma virus DNA in immunocompetent and immunocompromised patients with respiratory disease. J Med Virol. 2011;83:2220-2224.
- Tai P. A practical update of surgical management of Merkel cell carcinoma of the skin. ISRN Surg. 2013;2013:850797.
- National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines): Merkel Cell Carcinoma. Version 2.2019. Fort Washington, PA: National Comprehensive Cancer Network; 2019.
- Jabbour J. Merkel cell carcinoma: assessing the effect of wide local excision, lymph node dissection, and radiotherapy on recurrence and survival in early-stage disease--results from a review of 82 consecutive cases diagnosed between 1992 and 2004. Ann Surg Oncol. 2007;14:1943-1952.
- Medina-Franco H, Urist MM, Fiveash J, et al. Multimodality treatment of Merkel cell carcinoma: case series and literature review of 1024 cases. Ann Surg Oncol. 2001;8:204-208.
- Akhtar S, Oza KK, Wright J. Merkel cell carcinoma: report of 10 cases and review of the literature. J Am Acad Dermatol 2000;43:755-767.
- Palla AR, Doll D. Immunotherapy in Merkel cell carcinoma: role of avelumab. Immunotargets Ther. 2018;7:15-19.
- FDA approves pembrolizumab for Merkel cell carcinoma. US Food & Drug Administration website. http://www.fda.gov/Drugs/Information OnDrugs/ApprovedDrugs/ucm628867.htm. Published December 19, 2018. Accessed April 23, 2019.
- Schadendorff D, Lebbé C, zur Hausen A, et al. Merkel cell carcinoma: epidemiology, prognosis, therapy, and unmet medical needs. Eur J Cancer. 2017;71:53-69.
- Schwartz JL, Griffith KA, Lowe L, et al. Features predicting sentinel lymph node positivity in Merkel cell carcinoma. J Clin Oncol. 2011;29:1036-1041.
- Kachare SD, Wong JH, Vohra NA, et al. Sentinel lymph node biopsy is associated with improved survival in Merkel cell carcinoma. Ann Surg Oncol. 2014;21:1624-1630.
- Gupta SG, Wang LC, Penas LC, et al. Sentinel lymph node biopsy for evaluation and treatment of patients with Merkel cell carcinoma: the Dana-Farber experience and meta-analysis of the literature. Arch Dermatol. 2006;142:685-690.
- Bajetta E, Celio L, Platania M, et al. Single-institution series of early-stage Merkel cell carcinoma: long-term outcomes in 95 patients managed with surgery alone. Ann Surg Oncol. 2009;16:2985-2993.
The Diagnosis: Merkel Cell Carcinoma
An excisional biopsy revealed that the dermis was mostly replaced by a malignant neoplastic infiltrate morphologically resembling small cell carcinoma (Figure 1). The cells had uniform hyperchromatic nuclei with fairly even chromatin and generally inconspicuous nucleoli. There was a tendency for smudgy artifacts at the periphery of the infiltrate, and the cells had relatively scant cytoplasm with slight streaming. Occasional apoptotic forms were present. Immunohistochemistry showed strong dotlike staining with cytokeratin 20 and moderate positivity with synaptophysin and chromogranin A (Figure 2). Unusually, there also was weak staining in a few tumor cells with thyroid transcription factor 1, a marker usually indicative of small cell carcinoma of the lungs that typically is negative in Merkel cell carcinoma (MCC). A second thyroid transcription factor 1 monoclonal antibody used in a double immunostain for lung adenocarcinomas was completely negative. This second antibody is more specific but less sensitive than the stand-alone version. The skin biopsy results confirmed the diagnosis of MCC. Given the patient's frailty and comorbidities, wide local excision was not performed and the patient was referred to radiation oncology. He died several months later from metastatic MCC.
dermis was mostly replaced by a malignant neoplastic infiltrate morphologically resembling small cell carcinoma. The cells had uniform hyperchromatic nuclei with fairly even chromatin and generally inconspicuous nucleoli (H&E, original magnification ×200).
Merkel cell carcinoma (original magnification ×200).
Merkel cell carcinoma is an uncommon skin malignancy that can be easily mistaken for other conditions if the clinician is not familiar with its typical presentation. It most commonly is found on the head and neck in elderly individuals, most often aged 60 to 80 years,1 with a notable history of sun exposure and/or immunosuppression. It is an aggressive skin cancer that originally was thought to be due to pathogenic changes of Merkel cells,2 which are specialized touch receptors located at the dermoepidermal junction of the skin; however, newer evidence has suggested that MCC arises from malignant changes to skin stem cells.3 It shares more characteristics with extracutaneous neuroendocrine tumors and is more aptly labeled by pathologists as a primary neuroendocrine carcinoma of the skin.4
The frequency of MCC is highest in Australia, likely due to intense sun exposure, where the age-adjusted incidence rate reported in Queensland was 1.6 per 100,000 individuals from 2006 to 2010.5 The lowest incidence rates were reported in Finland (0.11 and 0.12 per 100,000 males and females, respectively)6 and Denmark (2.2 cases per million person-years).7 The clinical features of MCC are summarized by the mnemonic AEIOU: asymptomatic/lack of tenderness, expanding rapidly, immune suppression, older than 50 years, UV-exposed site on a person with fair skin.8 In a 2008 study of 195 patients, 89% of primary MCC lesions met 3 or more criteria, 32% met 4 or more criteria, and 7% met all 5 criteria.8
The classic presentation of MCC is a pink-red to violaceous nodule on the head or neck in an elderly patient, but there is a need to maintain suspicion of malignancy when examining a presumed infected cystic lesion, especially when a round of antibiotics has not ameliorated the symptoms. According to Heath et al,8 of 106 patients treated for MCC, 56% of first clinical impressions were benign. A PubMed and Scopus search was performed with the MeSH headings Merkel cell carcinoma +/- presentation to uncover similar unusual presentations between 1970 and the present day. Merkel cell carcinoma has been misdiagnosed as seemingly benign lesions including lipoma,9 allergic contact dermatitis,10 and atheroma.11 The differential diagnosis of MCC also includes cysts, amelanotic melanoma, basal cell carcinoma, dermatofibrosarcoma protuberans, squamous cell carcinoma, fungal kerion, leiomyosarcoma, neurothekeoma, abscesses, and cutaneous lymphoma.
Merkel cell polyomavirus has been implicated in the malignant transformation of MCC. It is a small, human, nonenveloped, double-stranded DNA virus1 and is found in approximately 70% to 80% of MCC cases.12 Merkel cell polyomavirus is a respiratory tract pathogen that is acquired by immunocompetent infants; it integrates itself into the host's genome and then enters a long latency period to later reactivate in immunocompromised adults.13
Wide local excision down to fascia is the mainstay of treatment of MCC, with recommended margins of 1 to 2 cm.14 Mohs micrographic surgery also can be considered.15 Similar to other neuroendocrine tumors, MCC is considered a radiosensitive tumor; radiation likely improves local control and is recommended in early-stage disease.16,17 It also has been described as the sole treatment modality in patients who are not candidates for surgery. The role of chemotherapy is more controversial, as responses do not appear to be long-lasting but should be considered in patients with advanced disease.14,18 There have been major advances in immunotherapy with the recent approvals of avelumab, an anti-PD-L1 inhibitor,19 and pembrolizumab,20 an anti-PD-1 inhibitor, for metastatic MCC. Clinical trials for MCC using kinase inhibitors and somatostatin analogues currently are ongoing.21
Several studies have demonstrated high rates of occult nodal disease in clinically node-negative patients, which has led to widespread use of sentinel lymph node biopsies.22,23 A sentinel lymph node biopsy is recommended at the time of surgery to aid with treatment decisions and prognosis.24
Merkel cell carcinoma is highly aggressive, and more than one-third of patients die from their disease, making it twice as lethal as melanoma. Overall survival rates remain low (5-year overall survival, 0%-18%) for advanced disease.5 Unfortunately, progression to metastasis is common and most often occurs within 2 years of diagnosis.17,25 Follow-up after treatment of MCC is crucial, with the 2019 National Comprehensive Cancer Network (NCCN) guidelines suggesting a physical examination with complete skin and complete lymph node examination every 3 to 6 months for 3 years and every 6 to 12 months thereafter.15
This case is an important reminder to include MCC in the differential diagnosis of presumed infected cysts, particularly on sun-exposed sites in elderly patients, as our patient was treated with antibiotics twice without improvement. An infected cyst with a lack of response to antibiotics should alert clinicians to the potential of malignancy.
The Diagnosis: Merkel Cell Carcinoma
An excisional biopsy revealed that the dermis was mostly replaced by a malignant neoplastic infiltrate morphologically resembling small cell carcinoma (Figure 1). The cells had uniform hyperchromatic nuclei with fairly even chromatin and generally inconspicuous nucleoli. There was a tendency for smudgy artifacts at the periphery of the infiltrate, and the cells had relatively scant cytoplasm with slight streaming. Occasional apoptotic forms were present. Immunohistochemistry showed strong dotlike staining with cytokeratin 20 and moderate positivity with synaptophysin and chromogranin A (Figure 2). Unusually, there also was weak staining in a few tumor cells with thyroid transcription factor 1, a marker usually indicative of small cell carcinoma of the lungs that typically is negative in Merkel cell carcinoma (MCC). A second thyroid transcription factor 1 monoclonal antibody used in a double immunostain for lung adenocarcinomas was completely negative. This second antibody is more specific but less sensitive than the stand-alone version. The skin biopsy results confirmed the diagnosis of MCC. Given the patient's frailty and comorbidities, wide local excision was not performed and the patient was referred to radiation oncology. He died several months later from metastatic MCC.
dermis was mostly replaced by a malignant neoplastic infiltrate morphologically resembling small cell carcinoma. The cells had uniform hyperchromatic nuclei with fairly even chromatin and generally inconspicuous nucleoli (H&E, original magnification ×200).
Merkel cell carcinoma (original magnification ×200).
Merkel cell carcinoma is an uncommon skin malignancy that can be easily mistaken for other conditions if the clinician is not familiar with its typical presentation. It most commonly is found on the head and neck in elderly individuals, most often aged 60 to 80 years,1 with a notable history of sun exposure and/or immunosuppression. It is an aggressive skin cancer that originally was thought to be due to pathogenic changes of Merkel cells,2 which are specialized touch receptors located at the dermoepidermal junction of the skin; however, newer evidence has suggested that MCC arises from malignant changes to skin stem cells.3 It shares more characteristics with extracutaneous neuroendocrine tumors and is more aptly labeled by pathologists as a primary neuroendocrine carcinoma of the skin.4
The frequency of MCC is highest in Australia, likely due to intense sun exposure, where the age-adjusted incidence rate reported in Queensland was 1.6 per 100,000 individuals from 2006 to 2010.5 The lowest incidence rates were reported in Finland (0.11 and 0.12 per 100,000 males and females, respectively)6 and Denmark (2.2 cases per million person-years).7 The clinical features of MCC are summarized by the mnemonic AEIOU: asymptomatic/lack of tenderness, expanding rapidly, immune suppression, older than 50 years, UV-exposed site on a person with fair skin.8 In a 2008 study of 195 patients, 89% of primary MCC lesions met 3 or more criteria, 32% met 4 or more criteria, and 7% met all 5 criteria.8
The classic presentation of MCC is a pink-red to violaceous nodule on the head or neck in an elderly patient, but there is a need to maintain suspicion of malignancy when examining a presumed infected cystic lesion, especially when a round of antibiotics has not ameliorated the symptoms. According to Heath et al,8 of 106 patients treated for MCC, 56% of first clinical impressions were benign. A PubMed and Scopus search was performed with the MeSH headings Merkel cell carcinoma +/- presentation to uncover similar unusual presentations between 1970 and the present day. Merkel cell carcinoma has been misdiagnosed as seemingly benign lesions including lipoma,9 allergic contact dermatitis,10 and atheroma.11 The differential diagnosis of MCC also includes cysts, amelanotic melanoma, basal cell carcinoma, dermatofibrosarcoma protuberans, squamous cell carcinoma, fungal kerion, leiomyosarcoma, neurothekeoma, abscesses, and cutaneous lymphoma.
Merkel cell polyomavirus has been implicated in the malignant transformation of MCC. It is a small, human, nonenveloped, double-stranded DNA virus1 and is found in approximately 70% to 80% of MCC cases.12 Merkel cell polyomavirus is a respiratory tract pathogen that is acquired by immunocompetent infants; it integrates itself into the host's genome and then enters a long latency period to later reactivate in immunocompromised adults.13
Wide local excision down to fascia is the mainstay of treatment of MCC, with recommended margins of 1 to 2 cm.14 Mohs micrographic surgery also can be considered.15 Similar to other neuroendocrine tumors, MCC is considered a radiosensitive tumor; radiation likely improves local control and is recommended in early-stage disease.16,17 It also has been described as the sole treatment modality in patients who are not candidates for surgery. The role of chemotherapy is more controversial, as responses do not appear to be long-lasting but should be considered in patients with advanced disease.14,18 There have been major advances in immunotherapy with the recent approvals of avelumab, an anti-PD-L1 inhibitor,19 and pembrolizumab,20 an anti-PD-1 inhibitor, for metastatic MCC. Clinical trials for MCC using kinase inhibitors and somatostatin analogues currently are ongoing.21
Several studies have demonstrated high rates of occult nodal disease in clinically node-negative patients, which has led to widespread use of sentinel lymph node biopsies.22,23 A sentinel lymph node biopsy is recommended at the time of surgery to aid with treatment decisions and prognosis.24
Merkel cell carcinoma is highly aggressive, and more than one-third of patients die from their disease, making it twice as lethal as melanoma. Overall survival rates remain low (5-year overall survival, 0%-18%) for advanced disease.5 Unfortunately, progression to metastasis is common and most often occurs within 2 years of diagnosis.17,25 Follow-up after treatment of MCC is crucial, with the 2019 National Comprehensive Cancer Network (NCCN) guidelines suggesting a physical examination with complete skin and complete lymph node examination every 3 to 6 months for 3 years and every 6 to 12 months thereafter.15
This case is an important reminder to include MCC in the differential diagnosis of presumed infected cysts, particularly on sun-exposed sites in elderly patients, as our patient was treated with antibiotics twice without improvement. An infected cyst with a lack of response to antibiotics should alert clinicians to the potential of malignancy.
- Sourvinos G, Mammas IN, Spandidos GA. 2015 Merkel cell polyoma virus infections in childhood. Arch Virol. 2015;160:887-892.
- Sibley RK, Rosai J, Foucar E, et al. Neuroendocrine (Merkel cell) carcinoma of the skin. a histologic and ultrastructural study of two cases. Am J Surg Pathol. 1980;4:211-221.
- Tilling T, Moll I. Which are the cells of origin in Merkel cell carcinoma? J Skin Cancer. 2012;2012:1-7.
- Succaria F, Radfar A, Bhawan J. Merkel cell carcinoma (primary neuroendocrine carcinoma of skin) mimicking basal cell carcinoma with review of different histopathologic features. Am J Dermatopathol. 2014;36:160-166.
- Youlden DR, Soyer HP, Youl PH, et al. Incidence and survival for Merkel cell carcinoma in Queensland, Australia, 1993-2010. JAMA Dermatol. 2014;150:864-872.
- Kukko H, Böhling T, Koljonen V, et al. Merkel cell carcinoma--a population-based epidemiological study in Finland with a clinical series of 181 cases. Eur J Cancer. 2012;48:737-742.
- Kaae J, Hansen AV, Biggar RJ, et al. Merkel cell carcinoma: incidence, mortality, and risk of other cancers. J Natl Cancer Inst. 2010;102:793-801.
- Heath M, Jaimes N, Lamos B, et al. Clinical characteristics of Merkel cell carcinoma at diagnosis of 195 patients: the AEIOU features. J Am Acad Dermatol. 2008;59:375-381.
- Sarma DP, Heagley DE, Chalupa J, et al. An unusual clinical presentation of Merkel cell carcinoma: a case report. Case Rep Med. 2010;2010:905414.
- Craven E, Alexandroff A, Liu JK, et al. Merkel cell carcinoma mistaken for allergic contact dermatitis. BMJ. 2015;351:h4635.
- Kinoshita A, Hoashi T, Okazaki S, et al. Atypical case of Merkel cell carcinoma difficult to diagnose clinically. J Dermatol. 2017;44:E158-E159.
- Donepudi S, DeConti LC, Samlowski WE. Recent advances in the understanding of the genetics, etiology, and treatment of Merkel cell carcinoma. Semin Oncol. 2012;39:163-172.
- Abedi Kiasari B, Vallely PJ, Klapper PE. Merkel cell polyoma virus DNA in immunocompetent and immunocompromised patients with respiratory disease. J Med Virol. 2011;83:2220-2224.
- Tai P. A practical update of surgical management of Merkel cell carcinoma of the skin. ISRN Surg. 2013;2013:850797.
- National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines): Merkel Cell Carcinoma. Version 2.2019. Fort Washington, PA: National Comprehensive Cancer Network; 2019.
- Jabbour J. Merkel cell carcinoma: assessing the effect of wide local excision, lymph node dissection, and radiotherapy on recurrence and survival in early-stage disease--results from a review of 82 consecutive cases diagnosed between 1992 and 2004. Ann Surg Oncol. 2007;14:1943-1952.
- Medina-Franco H, Urist MM, Fiveash J, et al. Multimodality treatment of Merkel cell carcinoma: case series and literature review of 1024 cases. Ann Surg Oncol. 2001;8:204-208.
- Akhtar S, Oza KK, Wright J. Merkel cell carcinoma: report of 10 cases and review of the literature. J Am Acad Dermatol 2000;43:755-767.
- Palla AR, Doll D. Immunotherapy in Merkel cell carcinoma: role of avelumab. Immunotargets Ther. 2018;7:15-19.
- FDA approves pembrolizumab for Merkel cell carcinoma. US Food & Drug Administration website. http://www.fda.gov/Drugs/Information OnDrugs/ApprovedDrugs/ucm628867.htm. Published December 19, 2018. Accessed April 23, 2019.
- Schadendorff D, Lebbé C, zur Hausen A, et al. Merkel cell carcinoma: epidemiology, prognosis, therapy, and unmet medical needs. Eur J Cancer. 2017;71:53-69.
- Schwartz JL, Griffith KA, Lowe L, et al. Features predicting sentinel lymph node positivity in Merkel cell carcinoma. J Clin Oncol. 2011;29:1036-1041.
- Kachare SD, Wong JH, Vohra NA, et al. Sentinel lymph node biopsy is associated with improved survival in Merkel cell carcinoma. Ann Surg Oncol. 2014;21:1624-1630.
- Gupta SG, Wang LC, Penas LC, et al. Sentinel lymph node biopsy for evaluation and treatment of patients with Merkel cell carcinoma: the Dana-Farber experience and meta-analysis of the literature. Arch Dermatol. 2006;142:685-690.
- Bajetta E, Celio L, Platania M, et al. Single-institution series of early-stage Merkel cell carcinoma: long-term outcomes in 95 patients managed with surgery alone. Ann Surg Oncol. 2009;16:2985-2993.
- Sourvinos G, Mammas IN, Spandidos GA. 2015 Merkel cell polyoma virus infections in childhood. Arch Virol. 2015;160:887-892.
- Sibley RK, Rosai J, Foucar E, et al. Neuroendocrine (Merkel cell) carcinoma of the skin. a histologic and ultrastructural study of two cases. Am J Surg Pathol. 1980;4:211-221.
- Tilling T, Moll I. Which are the cells of origin in Merkel cell carcinoma? J Skin Cancer. 2012;2012:1-7.
- Succaria F, Radfar A, Bhawan J. Merkel cell carcinoma (primary neuroendocrine carcinoma of skin) mimicking basal cell carcinoma with review of different histopathologic features. Am J Dermatopathol. 2014;36:160-166.
- Youlden DR, Soyer HP, Youl PH, et al. Incidence and survival for Merkel cell carcinoma in Queensland, Australia, 1993-2010. JAMA Dermatol. 2014;150:864-872.
- Kukko H, Böhling T, Koljonen V, et al. Merkel cell carcinoma--a population-based epidemiological study in Finland with a clinical series of 181 cases. Eur J Cancer. 2012;48:737-742.
- Kaae J, Hansen AV, Biggar RJ, et al. Merkel cell carcinoma: incidence, mortality, and risk of other cancers. J Natl Cancer Inst. 2010;102:793-801.
- Heath M, Jaimes N, Lamos B, et al. Clinical characteristics of Merkel cell carcinoma at diagnosis of 195 patients: the AEIOU features. J Am Acad Dermatol. 2008;59:375-381.
- Sarma DP, Heagley DE, Chalupa J, et al. An unusual clinical presentation of Merkel cell carcinoma: a case report. Case Rep Med. 2010;2010:905414.
- Craven E, Alexandroff A, Liu JK, et al. Merkel cell carcinoma mistaken for allergic contact dermatitis. BMJ. 2015;351:h4635.
- Kinoshita A, Hoashi T, Okazaki S, et al. Atypical case of Merkel cell carcinoma difficult to diagnose clinically. J Dermatol. 2017;44:E158-E159.
- Donepudi S, DeConti LC, Samlowski WE. Recent advances in the understanding of the genetics, etiology, and treatment of Merkel cell carcinoma. Semin Oncol. 2012;39:163-172.
- Abedi Kiasari B, Vallely PJ, Klapper PE. Merkel cell polyoma virus DNA in immunocompetent and immunocompromised patients with respiratory disease. J Med Virol. 2011;83:2220-2224.
- Tai P. A practical update of surgical management of Merkel cell carcinoma of the skin. ISRN Surg. 2013;2013:850797.
- National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines): Merkel Cell Carcinoma. Version 2.2019. Fort Washington, PA: National Comprehensive Cancer Network; 2019.
- Jabbour J. Merkel cell carcinoma: assessing the effect of wide local excision, lymph node dissection, and radiotherapy on recurrence and survival in early-stage disease--results from a review of 82 consecutive cases diagnosed between 1992 and 2004. Ann Surg Oncol. 2007;14:1943-1952.
- Medina-Franco H, Urist MM, Fiveash J, et al. Multimodality treatment of Merkel cell carcinoma: case series and literature review of 1024 cases. Ann Surg Oncol. 2001;8:204-208.
- Akhtar S, Oza KK, Wright J. Merkel cell carcinoma: report of 10 cases and review of the literature. J Am Acad Dermatol 2000;43:755-767.
- Palla AR, Doll D. Immunotherapy in Merkel cell carcinoma: role of avelumab. Immunotargets Ther. 2018;7:15-19.
- FDA approves pembrolizumab for Merkel cell carcinoma. US Food & Drug Administration website. http://www.fda.gov/Drugs/Information OnDrugs/ApprovedDrugs/ucm628867.htm. Published December 19, 2018. Accessed April 23, 2019.
- Schadendorff D, Lebbé C, zur Hausen A, et al. Merkel cell carcinoma: epidemiology, prognosis, therapy, and unmet medical needs. Eur J Cancer. 2017;71:53-69.
- Schwartz JL, Griffith KA, Lowe L, et al. Features predicting sentinel lymph node positivity in Merkel cell carcinoma. J Clin Oncol. 2011;29:1036-1041.
- Kachare SD, Wong JH, Vohra NA, et al. Sentinel lymph node biopsy is associated with improved survival in Merkel cell carcinoma. Ann Surg Oncol. 2014;21:1624-1630.
- Gupta SG, Wang LC, Penas LC, et al. Sentinel lymph node biopsy for evaluation and treatment of patients with Merkel cell carcinoma: the Dana-Farber experience and meta-analysis of the literature. Arch Dermatol. 2006;142:685-690.
- Bajetta E, Celio L, Platania M, et al. Single-institution series of early-stage Merkel cell carcinoma: long-term outcomes in 95 patients managed with surgery alone. Ann Surg Oncol. 2009;16:2985-2993.

A frail 85-year-old man presented to the emergency department for treatment of a 4.0.2 ×2.5-cm, erythematous, tender nodule on the scalp. The area was increasingly painful with persistent throbbing, which led to sleep disruption. The nodule did not express any material and was not aspirated or surgically treated. The lesion had been present for 1 to 2 years and was small and stable in size until it grew rapidly in the 6 weeks prior to presentation. The patient initially presented to his general practitioner during this period of rapid growth and was diagnosed with an infected sebaceous cyst that was treated with a course of oral cephalexin without improvement. Bacterial or fungal cultures were not performed. No other similar lesions were present, but there was 1 palpable lymph node in the right posterior cervical chain. At the time of presentation to the emergency department, the patient felt well and denied weight loss, night sweats, or fevers. He was given a dose of intravenous cefazolin by the emergency physician and then was referred to surgery for management of an infected sebaceous cyst.
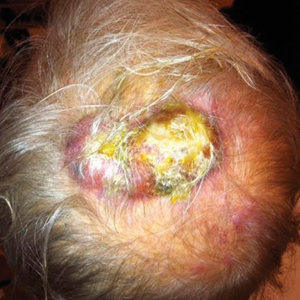
Apremilast and Phototherapy for Treatment of Psoriasis in a Patient With Human Immunodeficiency Virus
To the Editor:
A 50-year old man with Fitzpatrick skin type IV, human immunodeficiency virus (HIV), fatty liver disease, and moderate psoriasis (10% body surface area [BSA] affected) currently treated with clobetasol spray and calcitriol ointment presented with persistent psoriatic lesions on the trunk, arms, legs, and buttocks. His CD4 count was 460 and his HIV RNA count was 48 copies/mL on polymerase chain reaction 2 months prior to the current presentation. He had been undergoing phototherapy 3 times weekly for the last 5 months for treatment of psoriasis.
At the current presentation, he was started on an apremilast starter pack with the dosage titrated from 10 mg to 30 mg over the course of 1 week. He was maintained on a dose of 30 mg twice daily after 1 week and continued clobetasol spray, calcitriol ointment, and phototherapy 3 times weekly with the intent to reduce the frequency after adequate control of psoriasis was achieved. After 3 months of treatment, the affected BSA was 0%. He continued apremilast, and phototherapy was reduced to once weekly. Phototherapy was discontinued after 7 months of concomitant treatment with apremilast after clearance was maintained. It was reinitiated twice weekly after a mild flare (3% BSA affected). After 20 total months of treatment, the patient was no longer able to afford apremilast treatment and presented with a severe psoriasis flare (40% BSA affected). He was switched to acitretin with a plan to apply for apremilast financial assistance programs.
Psoriasis treatment in the HIV population poses a challenge given the immunosuppressed state of these patients, the risk of reactivation of latent infections, and the refractory nature of psoriasis in the setting of HIV. Two of the authors (S.P.R. and J.J.W.) previously reported a case of moderate to severe psoriasis in a patient with HIV and hepatitis C who demonstrated treatment success with apremilast until it was discontinued due to financial implications.1 Currently, apremilast is not widely used to treat psoriasis in the HIV population. The National Psoriasis Foundation 2010 guidelines recommended UV light therapy for treatment of moderate to severe psoriasis in HIV-positive patients, with oral retinoids as the second-line treatment.2 There remains a need for updated guidelines on the use of systemic agents for psoriasis treatment in the HIV population.
Apremilast, a phosphodiesterase 4 inhibitor, is an oral therapy that restores the balance of proinflammatory and anti-inflammatory cytokines by inhibiting inflammatory cytokine (eg, tumor necrosis factor α, IFN-γ, IL-2, IL-12, IL-23) secretion and stimulating anti-inflammatory cytokine (eg, IL-6, IL-10) production. In 2015, the phase 3 ESTEEM 13 and ESTEEM 24 trials demonstrated the efficacy of apremilast 30 mg twice daily for treatment of psoriasis. In both trials, the psoriasis area and severity index 75 response rate at week 16 was significantly
Use of other systemic agents such as tumor necrosis factor α inhibitors and ustekinumab has been reported in HIV-positive patients.5-7 There is no current data on IL-17 and IL-23 inhibitors. Acitretin generally is recommended as a second-line agent in HIV patients given its lack of immunosuppression2; however, methotrexate and cyclosporine should be avoided given the risk of opportunistic infections.8
Apremilast is a promising therapy with a favorable safety profile that should be considered as an adjuvant treatment to first-line agents such as phototherapy in HIV-positive patients. Apremilast has been successfully used in an HIV patient with a concomitant chronic hepatitis C infection.1 Systemic medications such as apremilast should be managed in coordination with infectious disease specialists with close monitoring of CD4 levels and viral loads as well as prophylactic agents.
- Reddy SP, Shah VV, Wu JJ. Apremilast for a psoriasis patient with HIV and hepatitis C. J Eur Acad Dermatol Venereol. 2017;31:e481-e482.
- Menon K, Van Voorhees AS, Bebo BF Jr, et al. Psoriasis in patients with HIV infection: from the medical board of the National Psoriasis Foundation [published online July 31, 2009]. J Am Acad Dermatol. 2010;62:291-299.
- Papp K, Reich K, Leonardi CL, et al. Apremilast, an oral phosphodiesterase 4 (PDE4) inhibitor, in patients with moderate to severe plaque psoriasis: results of a phase III, randomized, controlled trial (Efficacy and Safety Trial Evaluating the Effects of Apremilast in Psoriasis [ESTEEM] 1). J Am Acad Dermatol. 2015;73:37-49.
- Paul C, Cather J, Gooderham M, et al. Efficacy and safety of apremilast, an oral phosphodiesterase 4 inhibitor, in patients with moderate-to-severe plaque psoriasis over 52 weeks: a phase III, randomized controlled trial (ESTEEM 2). Br J Dermatol. 2015;173:1387-1399.
- Lindsey SF, Weiss J, Lee ES, et al. Treatment of severe psoriasis and psoriatic arthritis with adalimumab in an HIV-positive patient. J Drugs Dermatol. 2014;13:869-871.
- Saeki H, Ito T, Hayashi M, et al. Successful treatment of ustekinumab in a severe psoriasis patient with human immunodeficiency virus infection. J Eur Acad Dermatol Venereol. 2015;29:1653-1655.
- Paparizos V, Rallis E, Kirsten L, et al. Ustekinumab for the treatment of HIV psoriasis. J Dermatolog Treat. 2012;23:398-399.
- Kaushik SB, Lebwohl MG. Psoriasis: which therapy for which patient: focus on special populations and chronic infections [published online July 11, 2018]. J Am Acad Dermatol. 2019;80:43-53.
To the Editor:
A 50-year old man with Fitzpatrick skin type IV, human immunodeficiency virus (HIV), fatty liver disease, and moderate psoriasis (10% body surface area [BSA] affected) currently treated with clobetasol spray and calcitriol ointment presented with persistent psoriatic lesions on the trunk, arms, legs, and buttocks. His CD4 count was 460 and his HIV RNA count was 48 copies/mL on polymerase chain reaction 2 months prior to the current presentation. He had been undergoing phototherapy 3 times weekly for the last 5 months for treatment of psoriasis.
At the current presentation, he was started on an apremilast starter pack with the dosage titrated from 10 mg to 30 mg over the course of 1 week. He was maintained on a dose of 30 mg twice daily after 1 week and continued clobetasol spray, calcitriol ointment, and phototherapy 3 times weekly with the intent to reduce the frequency after adequate control of psoriasis was achieved. After 3 months of treatment, the affected BSA was 0%. He continued apremilast, and phototherapy was reduced to once weekly. Phototherapy was discontinued after 7 months of concomitant treatment with apremilast after clearance was maintained. It was reinitiated twice weekly after a mild flare (3% BSA affected). After 20 total months of treatment, the patient was no longer able to afford apremilast treatment and presented with a severe psoriasis flare (40% BSA affected). He was switched to acitretin with a plan to apply for apremilast financial assistance programs.
Psoriasis treatment in the HIV population poses a challenge given the immunosuppressed state of these patients, the risk of reactivation of latent infections, and the refractory nature of psoriasis in the setting of HIV. Two of the authors (S.P.R. and J.J.W.) previously reported a case of moderate to severe psoriasis in a patient with HIV and hepatitis C who demonstrated treatment success with apremilast until it was discontinued due to financial implications.1 Currently, apremilast is not widely used to treat psoriasis in the HIV population. The National Psoriasis Foundation 2010 guidelines recommended UV light therapy for treatment of moderate to severe psoriasis in HIV-positive patients, with oral retinoids as the second-line treatment.2 There remains a need for updated guidelines on the use of systemic agents for psoriasis treatment in the HIV population.
Apremilast, a phosphodiesterase 4 inhibitor, is an oral therapy that restores the balance of proinflammatory and anti-inflammatory cytokines by inhibiting inflammatory cytokine (eg, tumor necrosis factor α, IFN-γ, IL-2, IL-12, IL-23) secretion and stimulating anti-inflammatory cytokine (eg, IL-6, IL-10) production. In 2015, the phase 3 ESTEEM 13 and ESTEEM 24 trials demonstrated the efficacy of apremilast 30 mg twice daily for treatment of psoriasis. In both trials, the psoriasis area and severity index 75 response rate at week 16 was significantly
Use of other systemic agents such as tumor necrosis factor α inhibitors and ustekinumab has been reported in HIV-positive patients.5-7 There is no current data on IL-17 and IL-23 inhibitors. Acitretin generally is recommended as a second-line agent in HIV patients given its lack of immunosuppression2; however, methotrexate and cyclosporine should be avoided given the risk of opportunistic infections.8
Apremilast is a promising therapy with a favorable safety profile that should be considered as an adjuvant treatment to first-line agents such as phototherapy in HIV-positive patients. Apremilast has been successfully used in an HIV patient with a concomitant chronic hepatitis C infection.1 Systemic medications such as apremilast should be managed in coordination with infectious disease specialists with close monitoring of CD4 levels and viral loads as well as prophylactic agents.
To the Editor:
A 50-year old man with Fitzpatrick skin type IV, human immunodeficiency virus (HIV), fatty liver disease, and moderate psoriasis (10% body surface area [BSA] affected) currently treated with clobetasol spray and calcitriol ointment presented with persistent psoriatic lesions on the trunk, arms, legs, and buttocks. His CD4 count was 460 and his HIV RNA count was 48 copies/mL on polymerase chain reaction 2 months prior to the current presentation. He had been undergoing phototherapy 3 times weekly for the last 5 months for treatment of psoriasis.
At the current presentation, he was started on an apremilast starter pack with the dosage titrated from 10 mg to 30 mg over the course of 1 week. He was maintained on a dose of 30 mg twice daily after 1 week and continued clobetasol spray, calcitriol ointment, and phototherapy 3 times weekly with the intent to reduce the frequency after adequate control of psoriasis was achieved. After 3 months of treatment, the affected BSA was 0%. He continued apremilast, and phototherapy was reduced to once weekly. Phototherapy was discontinued after 7 months of concomitant treatment with apremilast after clearance was maintained. It was reinitiated twice weekly after a mild flare (3% BSA affected). After 20 total months of treatment, the patient was no longer able to afford apremilast treatment and presented with a severe psoriasis flare (40% BSA affected). He was switched to acitretin with a plan to apply for apremilast financial assistance programs.
Psoriasis treatment in the HIV population poses a challenge given the immunosuppressed state of these patients, the risk of reactivation of latent infections, and the refractory nature of psoriasis in the setting of HIV. Two of the authors (S.P.R. and J.J.W.) previously reported a case of moderate to severe psoriasis in a patient with HIV and hepatitis C who demonstrated treatment success with apremilast until it was discontinued due to financial implications.1 Currently, apremilast is not widely used to treat psoriasis in the HIV population. The National Psoriasis Foundation 2010 guidelines recommended UV light therapy for treatment of moderate to severe psoriasis in HIV-positive patients, with oral retinoids as the second-line treatment.2 There remains a need for updated guidelines on the use of systemic agents for psoriasis treatment in the HIV population.
Apremilast, a phosphodiesterase 4 inhibitor, is an oral therapy that restores the balance of proinflammatory and anti-inflammatory cytokines by inhibiting inflammatory cytokine (eg, tumor necrosis factor α, IFN-γ, IL-2, IL-12, IL-23) secretion and stimulating anti-inflammatory cytokine (eg, IL-6, IL-10) production. In 2015, the phase 3 ESTEEM 13 and ESTEEM 24 trials demonstrated the efficacy of apremilast 30 mg twice daily for treatment of psoriasis. In both trials, the psoriasis area and severity index 75 response rate at week 16 was significantly
Use of other systemic agents such as tumor necrosis factor α inhibitors and ustekinumab has been reported in HIV-positive patients.5-7 There is no current data on IL-17 and IL-23 inhibitors. Acitretin generally is recommended as a second-line agent in HIV patients given its lack of immunosuppression2; however, methotrexate and cyclosporine should be avoided given the risk of opportunistic infections.8
Apremilast is a promising therapy with a favorable safety profile that should be considered as an adjuvant treatment to first-line agents such as phototherapy in HIV-positive patients. Apremilast has been successfully used in an HIV patient with a concomitant chronic hepatitis C infection.1 Systemic medications such as apremilast should be managed in coordination with infectious disease specialists with close monitoring of CD4 levels and viral loads as well as prophylactic agents.
- Reddy SP, Shah VV, Wu JJ. Apremilast for a psoriasis patient with HIV and hepatitis C. J Eur Acad Dermatol Venereol. 2017;31:e481-e482.
- Menon K, Van Voorhees AS, Bebo BF Jr, et al. Psoriasis in patients with HIV infection: from the medical board of the National Psoriasis Foundation [published online July 31, 2009]. J Am Acad Dermatol. 2010;62:291-299.
- Papp K, Reich K, Leonardi CL, et al. Apremilast, an oral phosphodiesterase 4 (PDE4) inhibitor, in patients with moderate to severe plaque psoriasis: results of a phase III, randomized, controlled trial (Efficacy and Safety Trial Evaluating the Effects of Apremilast in Psoriasis [ESTEEM] 1). J Am Acad Dermatol. 2015;73:37-49.
- Paul C, Cather J, Gooderham M, et al. Efficacy and safety of apremilast, an oral phosphodiesterase 4 inhibitor, in patients with moderate-to-severe plaque psoriasis over 52 weeks: a phase III, randomized controlled trial (ESTEEM 2). Br J Dermatol. 2015;173:1387-1399.
- Lindsey SF, Weiss J, Lee ES, et al. Treatment of severe psoriasis and psoriatic arthritis with adalimumab in an HIV-positive patient. J Drugs Dermatol. 2014;13:869-871.
- Saeki H, Ito T, Hayashi M, et al. Successful treatment of ustekinumab in a severe psoriasis patient with human immunodeficiency virus infection. J Eur Acad Dermatol Venereol. 2015;29:1653-1655.
- Paparizos V, Rallis E, Kirsten L, et al. Ustekinumab for the treatment of HIV psoriasis. J Dermatolog Treat. 2012;23:398-399.
- Kaushik SB, Lebwohl MG. Psoriasis: which therapy for which patient: focus on special populations and chronic infections [published online July 11, 2018]. J Am Acad Dermatol. 2019;80:43-53.
- Reddy SP, Shah VV, Wu JJ. Apremilast for a psoriasis patient with HIV and hepatitis C. J Eur Acad Dermatol Venereol. 2017;31:e481-e482.
- Menon K, Van Voorhees AS, Bebo BF Jr, et al. Psoriasis in patients with HIV infection: from the medical board of the National Psoriasis Foundation [published online July 31, 2009]. J Am Acad Dermatol. 2010;62:291-299.
- Papp K, Reich K, Leonardi CL, et al. Apremilast, an oral phosphodiesterase 4 (PDE4) inhibitor, in patients with moderate to severe plaque psoriasis: results of a phase III, randomized, controlled trial (Efficacy and Safety Trial Evaluating the Effects of Apremilast in Psoriasis [ESTEEM] 1). J Am Acad Dermatol. 2015;73:37-49.
- Paul C, Cather J, Gooderham M, et al. Efficacy and safety of apremilast, an oral phosphodiesterase 4 inhibitor, in patients with moderate-to-severe plaque psoriasis over 52 weeks: a phase III, randomized controlled trial (ESTEEM 2). Br J Dermatol. 2015;173:1387-1399.
- Lindsey SF, Weiss J, Lee ES, et al. Treatment of severe psoriasis and psoriatic arthritis with adalimumab in an HIV-positive patient. J Drugs Dermatol. 2014;13:869-871.
- Saeki H, Ito T, Hayashi M, et al. Successful treatment of ustekinumab in a severe psoriasis patient with human immunodeficiency virus infection. J Eur Acad Dermatol Venereol. 2015;29:1653-1655.
- Paparizos V, Rallis E, Kirsten L, et al. Ustekinumab for the treatment of HIV psoriasis. J Dermatolog Treat. 2012;23:398-399.
- Kaushik SB, Lebwohl MG. Psoriasis: which therapy for which patient: focus on special populations and chronic infections [published online July 11, 2018]. J Am Acad Dermatol. 2019;80:43-53.
Practice Point
- Apremilast may be considered as a first-line therapy in the human immunodeficiency virus population due to decreased immunosuppression.
Netherton Syndrome: An Atypical Presentation
To the Editor:
Netherton syndrome (NS) is a rare autosomal-recessive ichthyosiform disease.1 The incidence is estimated to be 1 in 200,000 individuals.2 Netherton syndrome presents with generalized erythroderma and scaling, characteristic hair shaft abnormalities, and dysregulation of the immune system. Treatment is largely symptomatic and includes fragrance-free emollients, keratolytics, tretinoin, and corticosteroids, either alone or in combination. We report a case of NS in a man with congenital erythroderma, pili torti, and elevated IgE levels.
A 23-year-old man presented with generalized scaly skin that was present since birth. He was the first child born of nonconsanguineous parents. His medical history was suggestive of atopic diatheses such as allergic rhinitis and recurrent urticaria. The patient was of thin build and had widespread erythematous, annular, and polycyclic scaly lesions (Figure 1A), some with characteristic double-edged scale (Figure 1B). The skin was dry due to anhidrosis that was present since birth. Flexural lichenification was present at the cubital fossa of both arms. Scalp hairs were easily pluckable and had generalized thinning of hair density. Hair mount examination showed characteristic features of both trichorrhexis invaginata (Figure 2A) and pili torti (Figure 2B).
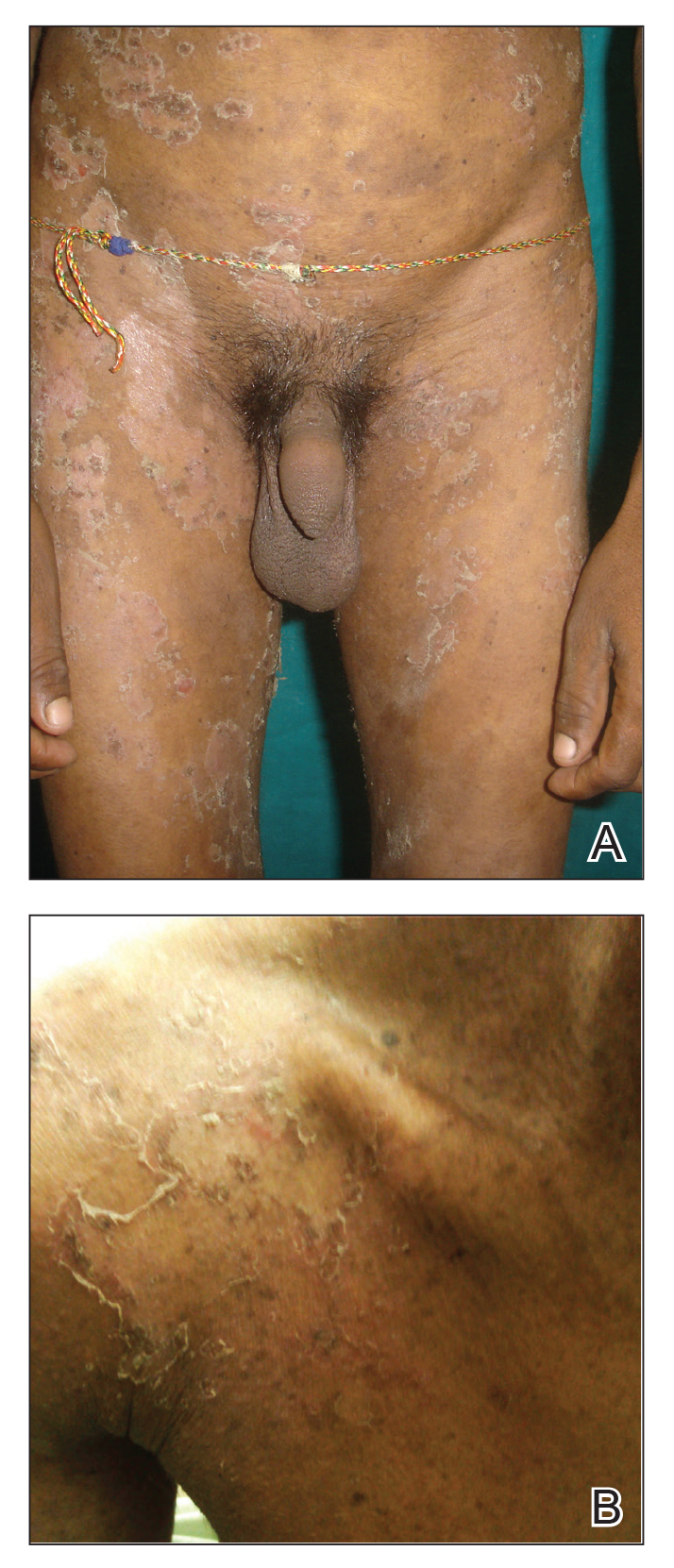

hair shaft known as bamboo hair or trichorrhexis invaginata. B, Features of pili torti; the hair
shaft twisted at irregular intervals.
Potassium hydroxide mount from a lesion was negative for fungal elements. Complete hematologic workup showed moderate anemia at 8.0 g/dL (reference range, 8.0–10.9 g/dL) and peripheral eosinophilia at 12% (reference range, 0%–6%). His IgE level was markedly elevated at6331 IU/mL (reference range, 150–1000 IU/mL) when tested with fully automated bidirectionally interfaced chemiluminescent immunoassay. Histopathologic examination of a lesion biopsy showed psoriasiform epidermal hyperplasia, papillomatosis, and acanthosis, consistent with ichthyosis linearis circumflexa (ILC)(Figure 3). Clinicopathologic correlation led to a diagnosis of ILC, trichorrhexis invaginata/pili torti, and atopic diathesis, which is a constellation of disorders related to NS.
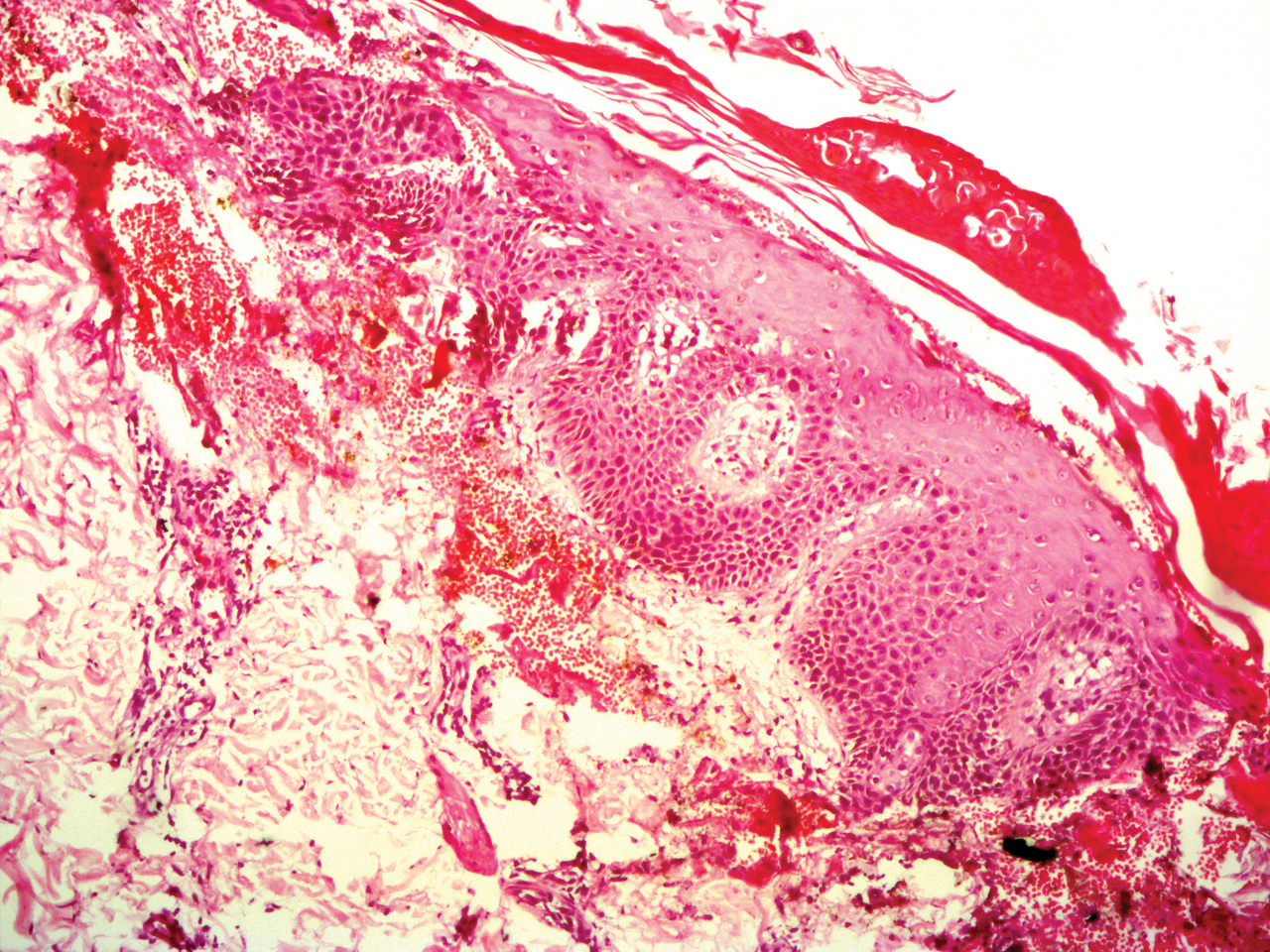
We prescribed oral acitretin 25 mg once daily and instructed the patient to apply petroleum jelly; however, the patient returned after 2 weeks due to aggravation of the skin condition with increased scaling and redness. Because the patient showed signs of acute skin failure and erythroderma, we stopped acitretin treatment and managed his condition conservatively with the application of petroleum jelly.
Netherton syndrome is caused by mutation of the SPINK5 gene, serine protease inhibitor Kazal type 5; the corresponding gene is located on the long arm of chromosome 5.3 The gene encodes a serine protease inhibitor proprotein LEKTI (lymphoepithelial Kazal type inhibitor).4 The product of the gene is thought to be necessary for epidermal cell growth and differentiation. The classic clinical triad of NS includes ichthyosiform dermatosis with double-edged scale, hair shaft abnormalities, and atopy or elevated IgE levels.5 Generalized (congenital) erythroderma usually becomes evident at birth or shortly thereafter. Half of patients develop lesions of ILC on the trunk and limbs during childhood.6 A typical ILC lesion is characterized by an erythematous scaly patch that may be annular or polycyclic with double-edged scale at the advancing border. The ability to sweat is impaired, which may cause episodes of hyperpyrexia, especially during humid weather. Patients with hyperpyrexia may be incorrectly diagnosed with bacterial infection and treated with antipyretic drugs or a prolonged course of antibiotics. Trichorrhexis invaginata, also referred to as bamboo hair or ball-and-socket defect, is the pathognomonic hair shaft abnormality seen in NS.7 Other hair shaft abnormalities in this syndrome include trichorrhexis nodosa and pili torti.8 Our patient had hair shaft abnormalities of trichorrhexis invaginata and pili torti, which are rare findings. The third component of this syndrome is atopy, which generally manifests as angioedema, urticaria, allergic rhinitis, peripheral eosinophilia, atopic dermatitis–like skin lesions, asthma, and elevated IgE levels.9
Treatment with emollients, topical steroids, tacrolimus, and psoralen plus UVA does not elicit a satisfactory response. The Table highlights the clinical features and management of NS.
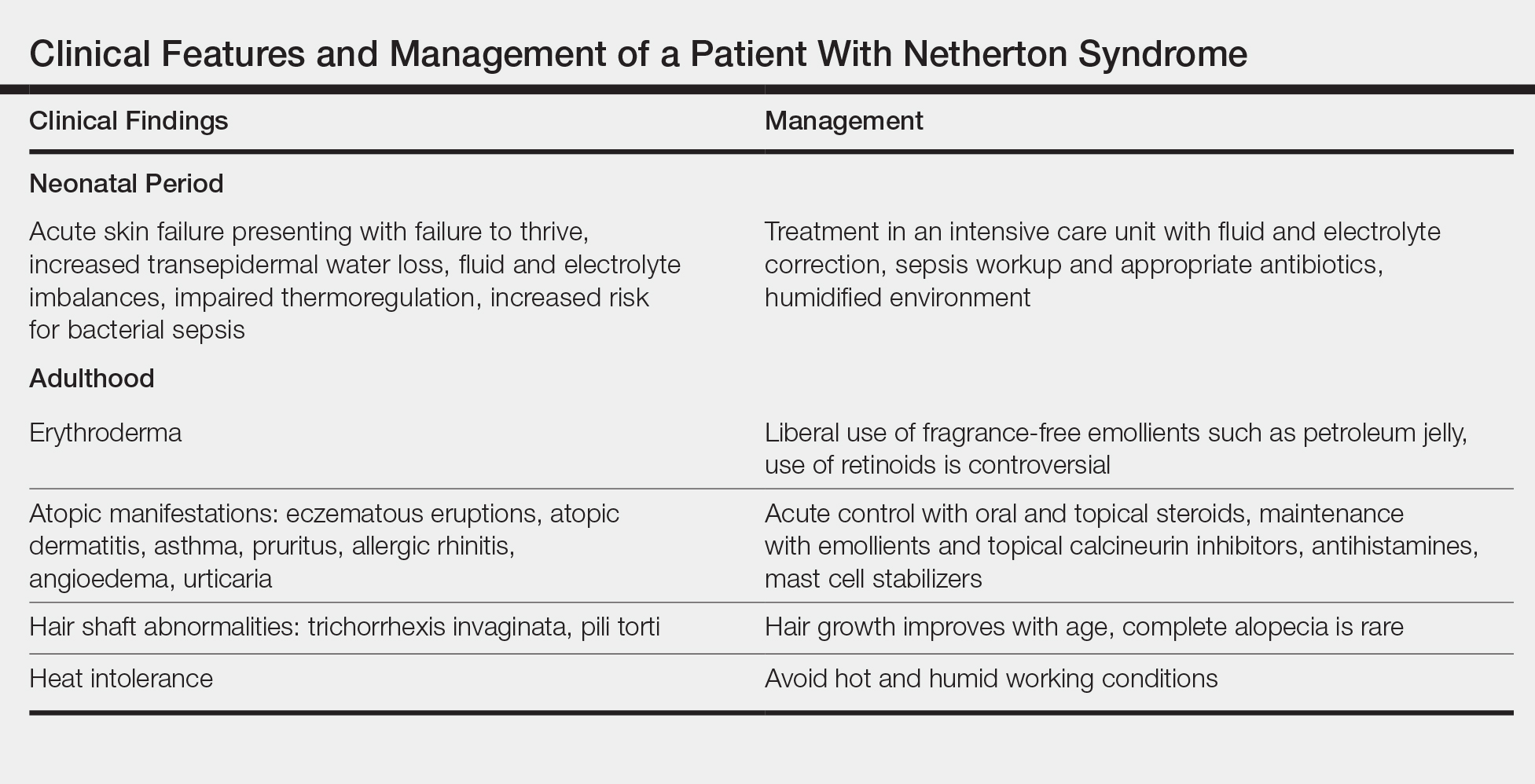
Generally, systemic retinoid therapy is helpful in cases of erythrodermic ichthyosis, but a unique feature of NS is that erythroderma may worsen with systemic retinoid therapy, as retinoids aggravate atopic dermatitis by worsening existing xerosis.4 Our case highlights the rare association of trichorrhexis invaginata with pili torti as well as acitretin treatment worsening our patient’s condition. This paradoxical effect of retinoid therapy further confirmed the diagnosis of NS.
- Suhaila O, Muzhirah A. Netherton syndrome: a case report. Malaysian J Pediatr Child Health. 2010;16:26.
- Emre S, Metin A, Demirseren D, et al. Two siblings with Netherton syndrome. Turk J Med Sci. 2010;40:819-823.
- Chavanas S, Bodemer C, Rochat A, et al. Mutations in SPINK5, encoding a serine protease inhibitor, cause Netherton syndrome. Nat Genet. 2000;25:141-142.
- Judge MR, Mclean WH, Munro CS. Disorders of keratinization. In: Burns T, Breathnach S, Cox N, et al, eds. Rook’s Textbook of Dermatology. 8th ed. Singapore: Wiley-Blackwell; 2010:19.1-19.122.
- Greene SL, Muller SA. Netherton’s syndrome. report of a case and review of the literature. J Am Acad Dermatol. 1985;13:329-337.
- Khan I-U, Chaudhary R. Netherton’s syndrome, an uncommon genodermatosis. J Pakistan Assoc Dermatol. 2006;16.
- Boskabadi H, Maamouri G, Mafinejad S. Netherton syndrome, a case report and review of literature. Iran J Pediatr. 2013;23:611-612.
- Hurwitz S. Hereditary skin disorders: the genodermatoses. In: Hurwitz, ed. Clinical Pediatric Dermatology. Philadelphia, PA: WB Saunders; 1993:173.
- Judge MR, McLean WH, Munro CS. Disorders of keratinization. In: Burns T, Breathnach S, Cox N, et al, eds. Rook’s Textbook of Dermatology. 7th ed. Vol 2. Oxford, England: Blackwell Science; 2004:34.35.
To the Editor:
Netherton syndrome (NS) is a rare autosomal-recessive ichthyosiform disease.1 The incidence is estimated to be 1 in 200,000 individuals.2 Netherton syndrome presents with generalized erythroderma and scaling, characteristic hair shaft abnormalities, and dysregulation of the immune system. Treatment is largely symptomatic and includes fragrance-free emollients, keratolytics, tretinoin, and corticosteroids, either alone or in combination. We report a case of NS in a man with congenital erythroderma, pili torti, and elevated IgE levels.
A 23-year-old man presented with generalized scaly skin that was present since birth. He was the first child born of nonconsanguineous parents. His medical history was suggestive of atopic diatheses such as allergic rhinitis and recurrent urticaria. The patient was of thin build and had widespread erythematous, annular, and polycyclic scaly lesions (Figure 1A), some with characteristic double-edged scale (Figure 1B). The skin was dry due to anhidrosis that was present since birth. Flexural lichenification was present at the cubital fossa of both arms. Scalp hairs were easily pluckable and had generalized thinning of hair density. Hair mount examination showed characteristic features of both trichorrhexis invaginata (Figure 2A) and pili torti (Figure 2B).
hair shaft known as bamboo hair or trichorrhexis invaginata. B, Features of pili torti; the hair
shaft twisted at irregular intervals.
Potassium hydroxide mount from a lesion was negative for fungal elements. Complete hematologic workup showed moderate anemia at 8.0 g/dL (reference range, 8.0–10.9 g/dL) and peripheral eosinophilia at 12% (reference range, 0%–6%). His IgE level was markedly elevated at6331 IU/mL (reference range, 150–1000 IU/mL) when tested with fully automated bidirectionally interfaced chemiluminescent immunoassay. Histopathologic examination of a lesion biopsy showed psoriasiform epidermal hyperplasia, papillomatosis, and acanthosis, consistent with ichthyosis linearis circumflexa (ILC)(Figure 3). Clinicopathologic correlation led to a diagnosis of ILC, trichorrhexis invaginata/pili torti, and atopic diathesis, which is a constellation of disorders related to NS.
We prescribed oral acitretin 25 mg once daily and instructed the patient to apply petroleum jelly; however, the patient returned after 2 weeks due to aggravation of the skin condition with increased scaling and redness. Because the patient showed signs of acute skin failure and erythroderma, we stopped acitretin treatment and managed his condition conservatively with the application of petroleum jelly.
Netherton syndrome is caused by mutation of the SPINK5 gene, serine protease inhibitor Kazal type 5; the corresponding gene is located on the long arm of chromosome 5.3 The gene encodes a serine protease inhibitor proprotein LEKTI (lymphoepithelial Kazal type inhibitor).4 The product of the gene is thought to be necessary for epidermal cell growth and differentiation. The classic clinical triad of NS includes ichthyosiform dermatosis with double-edged scale, hair shaft abnormalities, and atopy or elevated IgE levels.5 Generalized (congenital) erythroderma usually becomes evident at birth or shortly thereafter. Half of patients develop lesions of ILC on the trunk and limbs during childhood.6 A typical ILC lesion is characterized by an erythematous scaly patch that may be annular or polycyclic with double-edged scale at the advancing border. The ability to sweat is impaired, which may cause episodes of hyperpyrexia, especially during humid weather. Patients with hyperpyrexia may be incorrectly diagnosed with bacterial infection and treated with antipyretic drugs or a prolonged course of antibiotics. Trichorrhexis invaginata, also referred to as bamboo hair or ball-and-socket defect, is the pathognomonic hair shaft abnormality seen in NS.7 Other hair shaft abnormalities in this syndrome include trichorrhexis nodosa and pili torti.8 Our patient had hair shaft abnormalities of trichorrhexis invaginata and pili torti, which are rare findings. The third component of this syndrome is atopy, which generally manifests as angioedema, urticaria, allergic rhinitis, peripheral eosinophilia, atopic dermatitis–like skin lesions, asthma, and elevated IgE levels.9
Treatment with emollients, topical steroids, tacrolimus, and psoralen plus UVA does not elicit a satisfactory response. The Table highlights the clinical features and management of NS.
Generally, systemic retinoid therapy is helpful in cases of erythrodermic ichthyosis, but a unique feature of NS is that erythroderma may worsen with systemic retinoid therapy, as retinoids aggravate atopic dermatitis by worsening existing xerosis.4 Our case highlights the rare association of trichorrhexis invaginata with pili torti as well as acitretin treatment worsening our patient’s condition. This paradoxical effect of retinoid therapy further confirmed the diagnosis of NS.
To the Editor:
Netherton syndrome (NS) is a rare autosomal-recessive ichthyosiform disease.1 The incidence is estimated to be 1 in 200,000 individuals.2 Netherton syndrome presents with generalized erythroderma and scaling, characteristic hair shaft abnormalities, and dysregulation of the immune system. Treatment is largely symptomatic and includes fragrance-free emollients, keratolytics, tretinoin, and corticosteroids, either alone or in combination. We report a case of NS in a man with congenital erythroderma, pili torti, and elevated IgE levels.
A 23-year-old man presented with generalized scaly skin that was present since birth. He was the first child born of nonconsanguineous parents. His medical history was suggestive of atopic diatheses such as allergic rhinitis and recurrent urticaria. The patient was of thin build and had widespread erythematous, annular, and polycyclic scaly lesions (Figure 1A), some with characteristic double-edged scale (Figure 1B). The skin was dry due to anhidrosis that was present since birth. Flexural lichenification was present at the cubital fossa of both arms. Scalp hairs were easily pluckable and had generalized thinning of hair density. Hair mount examination showed characteristic features of both trichorrhexis invaginata (Figure 2A) and pili torti (Figure 2B).
hair shaft known as bamboo hair or trichorrhexis invaginata. B, Features of pili torti; the hair
shaft twisted at irregular intervals.
Potassium hydroxide mount from a lesion was negative for fungal elements. Complete hematologic workup showed moderate anemia at 8.0 g/dL (reference range, 8.0–10.9 g/dL) and peripheral eosinophilia at 12% (reference range, 0%–6%). His IgE level was markedly elevated at6331 IU/mL (reference range, 150–1000 IU/mL) when tested with fully automated bidirectionally interfaced chemiluminescent immunoassay. Histopathologic examination of a lesion biopsy showed psoriasiform epidermal hyperplasia, papillomatosis, and acanthosis, consistent with ichthyosis linearis circumflexa (ILC)(Figure 3). Clinicopathologic correlation led to a diagnosis of ILC, trichorrhexis invaginata/pili torti, and atopic diathesis, which is a constellation of disorders related to NS.
We prescribed oral acitretin 25 mg once daily and instructed the patient to apply petroleum jelly; however, the patient returned after 2 weeks due to aggravation of the skin condition with increased scaling and redness. Because the patient showed signs of acute skin failure and erythroderma, we stopped acitretin treatment and managed his condition conservatively with the application of petroleum jelly.
Netherton syndrome is caused by mutation of the SPINK5 gene, serine protease inhibitor Kazal type 5; the corresponding gene is located on the long arm of chromosome 5.3 The gene encodes a serine protease inhibitor proprotein LEKTI (lymphoepithelial Kazal type inhibitor).4 The product of the gene is thought to be necessary for epidermal cell growth and differentiation. The classic clinical triad of NS includes ichthyosiform dermatosis with double-edged scale, hair shaft abnormalities, and atopy or elevated IgE levels.5 Generalized (congenital) erythroderma usually becomes evident at birth or shortly thereafter. Half of patients develop lesions of ILC on the trunk and limbs during childhood.6 A typical ILC lesion is characterized by an erythematous scaly patch that may be annular or polycyclic with double-edged scale at the advancing border. The ability to sweat is impaired, which may cause episodes of hyperpyrexia, especially during humid weather. Patients with hyperpyrexia may be incorrectly diagnosed with bacterial infection and treated with antipyretic drugs or a prolonged course of antibiotics. Trichorrhexis invaginata, also referred to as bamboo hair or ball-and-socket defect, is the pathognomonic hair shaft abnormality seen in NS.7 Other hair shaft abnormalities in this syndrome include trichorrhexis nodosa and pili torti.8 Our patient had hair shaft abnormalities of trichorrhexis invaginata and pili torti, which are rare findings. The third component of this syndrome is atopy, which generally manifests as angioedema, urticaria, allergic rhinitis, peripheral eosinophilia, atopic dermatitis–like skin lesions, asthma, and elevated IgE levels.9
Treatment with emollients, topical steroids, tacrolimus, and psoralen plus UVA does not elicit a satisfactory response. The Table highlights the clinical features and management of NS.
Generally, systemic retinoid therapy is helpful in cases of erythrodermic ichthyosis, but a unique feature of NS is that erythroderma may worsen with systemic retinoid therapy, as retinoids aggravate atopic dermatitis by worsening existing xerosis.4 Our case highlights the rare association of trichorrhexis invaginata with pili torti as well as acitretin treatment worsening our patient’s condition. This paradoxical effect of retinoid therapy further confirmed the diagnosis of NS.
- Suhaila O, Muzhirah A. Netherton syndrome: a case report. Malaysian J Pediatr Child Health. 2010;16:26.
- Emre S, Metin A, Demirseren D, et al. Two siblings with Netherton syndrome. Turk J Med Sci. 2010;40:819-823.
- Chavanas S, Bodemer C, Rochat A, et al. Mutations in SPINK5, encoding a serine protease inhibitor, cause Netherton syndrome. Nat Genet. 2000;25:141-142.
- Judge MR, Mclean WH, Munro CS. Disorders of keratinization. In: Burns T, Breathnach S, Cox N, et al, eds. Rook’s Textbook of Dermatology. 8th ed. Singapore: Wiley-Blackwell; 2010:19.1-19.122.
- Greene SL, Muller SA. Netherton’s syndrome. report of a case and review of the literature. J Am Acad Dermatol. 1985;13:329-337.
- Khan I-U, Chaudhary R. Netherton’s syndrome, an uncommon genodermatosis. J Pakistan Assoc Dermatol. 2006;16.
- Boskabadi H, Maamouri G, Mafinejad S. Netherton syndrome, a case report and review of literature. Iran J Pediatr. 2013;23:611-612.
- Hurwitz S. Hereditary skin disorders: the genodermatoses. In: Hurwitz, ed. Clinical Pediatric Dermatology. Philadelphia, PA: WB Saunders; 1993:173.
- Judge MR, McLean WH, Munro CS. Disorders of keratinization. In: Burns T, Breathnach S, Cox N, et al, eds. Rook’s Textbook of Dermatology. 7th ed. Vol 2. Oxford, England: Blackwell Science; 2004:34.35.
- Suhaila O, Muzhirah A. Netherton syndrome: a case report. Malaysian J Pediatr Child Health. 2010;16:26.
- Emre S, Metin A, Demirseren D, et al. Two siblings with Netherton syndrome. Turk J Med Sci. 2010;40:819-823.
- Chavanas S, Bodemer C, Rochat A, et al. Mutations in SPINK5, encoding a serine protease inhibitor, cause Netherton syndrome. Nat Genet. 2000;25:141-142.
- Judge MR, Mclean WH, Munro CS. Disorders of keratinization. In: Burns T, Breathnach S, Cox N, et al, eds. Rook’s Textbook of Dermatology. 8th ed. Singapore: Wiley-Blackwell; 2010:19.1-19.122.
- Greene SL, Muller SA. Netherton’s syndrome. report of a case and review of the literature. J Am Acad Dermatol. 1985;13:329-337.
- Khan I-U, Chaudhary R. Netherton’s syndrome, an uncommon genodermatosis. J Pakistan Assoc Dermatol. 2006;16.
- Boskabadi H, Maamouri G, Mafinejad S. Netherton syndrome, a case report and review of literature. Iran J Pediatr. 2013;23:611-612.
- Hurwitz S. Hereditary skin disorders: the genodermatoses. In: Hurwitz, ed. Clinical Pediatric Dermatology. Philadelphia, PA: WB Saunders; 1993:173.
- Judge MR, McLean WH, Munro CS. Disorders of keratinization. In: Burns T, Breathnach S, Cox N, et al, eds. Rook’s Textbook of Dermatology. 7th ed. Vol 2. Oxford, England: Blackwell Science; 2004:34.35.
Practice Points
- Netherton syndrome is characterized by generalized erythroderma and scaling, hair shaft abnormalities, and dysregulation of the immune system.
- Treatment is largely symptomatic and includes fragrance-free emollients, keratolytics, tretinoin, and corticosteroids, either alone or in combination.
Adult-Onset Asymmetrical Lipomatosis
To the Editor:
An 85-year-old woman presented with extra growth of subcutaneous fat at the left anterior infradiaphragm that expanded circumferentially to the left back over the last 4 years. Two years prior to the current presentation, the left thigh became visibly thicker than the right. Diffuse subtle lipomatosis affecting the ipsilateral face, neck, arms, calf, and foot was noted at that time. Additionally, the patient had hyperlipidemia, gastroesophageal reflux disease, osteoporosis, and scoliosis, all beginning in her late 60s. She reported no alcohol or tobacco use and was taking rosuvastatin, esomeprazole, calcium, vitamin D, and glucosamine. There was no reported family history of asymmetric growth or bony deformities, and her children were healthy.
On physical examination, the lipomatosis affected the entire left side, most prominently around the abdomen, back, and thighs. The affected side was nontender and nonpruritic; there was no atrophy of the unaffected side (Figure). Maximum thigh circumference was 55.1 cm on the affected side and 52.6 cm on the unaffected side. There were no differences in power, reflex, or sensation between the 2 sides, and no hyperhidrosis or vascular malformations were present. Laboratory investigations, including complete blood cell count, complete metabolic panel, lipids, and thyroid-stimulating and sex hormone panels all were within reference range.
Enzi et al1 reported 2 women who developed asymmetrical lipomatosis between the ages of 13 and 20 years. Acquired asymmetrical lipomatosis should be differentiated from the asymmetrical overgrowth diagnosed in neonates and infants.
Proteus syndrome (PS) is a progressive disease involving a combination of overgrowth in a mosaic distribution, connective tissue and epidermal nevi, ovarian cysts, parotid gland tumor, dysregulated adipose tissue, lymphovascular malformation, and certain facial phenotypes.2,3 The average age of onset is 6 to 18 months, and half of cases present at birth.3,4 Hemihyperplasia-multiple lipomatosis syndrome (HHML) describes a mild and nonprogressive variant that does not satisfy the diagnostic criteria of PS; it typically is diagnosed at birth.5 One case of mild and delayed-onset PS was described in a woman who started developing signs at 15 years of age.6 In comparison, asymmetrical lipomatosis and scoliosis were the only abnormal clinical signs present in our patient, and the lipomatosis developed diffusely, as opposed to the typical mosaic distribution found in PS and HHML. Scoliosis can be found in PS and HHML secondary to hemihypertrophy of vertebra or infiltrative intraspinal lipomatosis.7,8 Our patient’s scoliosis was diagnosed more than 10 years prior to the onset of lipomatosis, likely representing degenerative joint disease.9
Prior reported cases of asymmetrical lipomatosis did not describe treatment.1 Ultrasound-guided or conventional liposuction and lipectomy are mainstream therapies for multiple symmetrical lipomatosis, an acquired lipomatosis typically affecting alcoholics in the fourth decade of life. However, recurrence rates are high for surgical treatment of unencapsulated lipomatosis, likely due to incomplete removal of the adipose tissue.10 Alternative treatments found in case reports, including oral salbutamol, mesotherapy using phosphatidylcholine, and fenofibrate (200 mg/d), require further study.11-13 Our patient was not aesthetically bothered by her lipomatosis; therefore, imaging and treatment options were not pursued. In conclusion, we report a patient with acquired asymmetrical lipomatosis with onset in late adulthood, unique from the existing syndromes of asymmetrical hemihyperplasia.1,14
- Enzi G, Digito M, Enzi GB, et al. Asymmetrical lipomatosis: report of two cases. Postgrad Med J. 1985;61:797-800.
- Biesecker LG, Happle R, Mulliken JB, et al. Proteus syndrome: diagnostic criteria, differential diagnosis, and patient evaluation. Am J Med Genet. 1999;84:389-395.
- Biesecker L. The challenges of Proteus syndrome: diagnosis and management. Eur J Hum Genet. 2006;14:1151-1157.
- Cohen MM Jr. Proteus syndrome: an update. Am J Med Genet C Semin Med Genet. 2005;137C:38-52.
- Biesecker LG, Peters KF, Darling TN, et al. Clinical differentiation between Proteus syndrome and hemihyperplasia: description of a distinct form of hemihyperplasia. Am J Med Genet. 1998;79:311-318.
- Luo S, Feng Y, Zheng Y, et al. Mild and delayed-onset Proteus syndrome. Eur J Dermatol. 2007;17:172-173.
- Takebayashi T, Yamashita T, Yokogushi K, et al. Scoliosis in Proteus syndrome: case report. Spine. 2001;26:E395-E398.
- Schulte TL, Liljenqvist U, Görgens H, et al. Hemihyperplasia-multiple lipomatosis syndrome (HHML): a challenge in spinal care. Acta Orthop Belg. 2008;74:714-719.
- Robin GC, Span Y, Steinberg R, et al. Scoliosis in the elderly: a follow-up study. Spine. 1982;7:355-359.
- Brea-García B, Cameselle-Teijeiro J, Couto-González I, et al. Madelung’s disease: comorbidities, fatty mass distribution, and response to treatment of 22 patients. Aesthet Plast Surg. 2013;37:409-416.
- Hasegawa T, Matsukura T, Ikeda S. Mesotherapy for benign symmetric lipomatosis. Aesthet Plast Surg. 2010;34:153-156.
- Zeitler H, Ulrich-Merzenich G, Richter DF, et al. Multiple benign symmetric lipomatosis—a differential diagnosis of obesity. is there a rationale for fibrate treatment? Obes Surg. 2008;18:1354-1356.
- Leung N, Gaer J, Beggs D, et al. Multiple symmetric lipomatosis (Launois‐Bensaude syndrome): effect of oral salbutamol. Clin Endocrinol. 1987;27:601-606.
- Craiglow BG, Ko CJ, Antaya RJ. Two cases of hemihyperplasia-multiple lipomatosis syndrome and review of asymmetric hemihyperplasia syndromes. Pediatr Dermatol. 2014;31:507-510.
To the Editor:
An 85-year-old woman presented with extra growth of subcutaneous fat at the left anterior infradiaphragm that expanded circumferentially to the left back over the last 4 years. Two years prior to the current presentation, the left thigh became visibly thicker than the right. Diffuse subtle lipomatosis affecting the ipsilateral face, neck, arms, calf, and foot was noted at that time. Additionally, the patient had hyperlipidemia, gastroesophageal reflux disease, osteoporosis, and scoliosis, all beginning in her late 60s. She reported no alcohol or tobacco use and was taking rosuvastatin, esomeprazole, calcium, vitamin D, and glucosamine. There was no reported family history of asymmetric growth or bony deformities, and her children were healthy.
On physical examination, the lipomatosis affected the entire left side, most prominently around the abdomen, back, and thighs. The affected side was nontender and nonpruritic; there was no atrophy of the unaffected side (Figure). Maximum thigh circumference was 55.1 cm on the affected side and 52.6 cm on the unaffected side. There were no differences in power, reflex, or sensation between the 2 sides, and no hyperhidrosis or vascular malformations were present. Laboratory investigations, including complete blood cell count, complete metabolic panel, lipids, and thyroid-stimulating and sex hormone panels all were within reference range.
Enzi et al1 reported 2 women who developed asymmetrical lipomatosis between the ages of 13 and 20 years. Acquired asymmetrical lipomatosis should be differentiated from the asymmetrical overgrowth diagnosed in neonates and infants.
Proteus syndrome (PS) is a progressive disease involving a combination of overgrowth in a mosaic distribution, connective tissue and epidermal nevi, ovarian cysts, parotid gland tumor, dysregulated adipose tissue, lymphovascular malformation, and certain facial phenotypes.2,3 The average age of onset is 6 to 18 months, and half of cases present at birth.3,4 Hemihyperplasia-multiple lipomatosis syndrome (HHML) describes a mild and nonprogressive variant that does not satisfy the diagnostic criteria of PS; it typically is diagnosed at birth.5 One case of mild and delayed-onset PS was described in a woman who started developing signs at 15 years of age.6 In comparison, asymmetrical lipomatosis and scoliosis were the only abnormal clinical signs present in our patient, and the lipomatosis developed diffusely, as opposed to the typical mosaic distribution found in PS and HHML. Scoliosis can be found in PS and HHML secondary to hemihypertrophy of vertebra or infiltrative intraspinal lipomatosis.7,8 Our patient’s scoliosis was diagnosed more than 10 years prior to the onset of lipomatosis, likely representing degenerative joint disease.9
Prior reported cases of asymmetrical lipomatosis did not describe treatment.1 Ultrasound-guided or conventional liposuction and lipectomy are mainstream therapies for multiple symmetrical lipomatosis, an acquired lipomatosis typically affecting alcoholics in the fourth decade of life. However, recurrence rates are high for surgical treatment of unencapsulated lipomatosis, likely due to incomplete removal of the adipose tissue.10 Alternative treatments found in case reports, including oral salbutamol, mesotherapy using phosphatidylcholine, and fenofibrate (200 mg/d), require further study.11-13 Our patient was not aesthetically bothered by her lipomatosis; therefore, imaging and treatment options were not pursued. In conclusion, we report a patient with acquired asymmetrical lipomatosis with onset in late adulthood, unique from the existing syndromes of asymmetrical hemihyperplasia.1,14
To the Editor:
An 85-year-old woman presented with extra growth of subcutaneous fat at the left anterior infradiaphragm that expanded circumferentially to the left back over the last 4 years. Two years prior to the current presentation, the left thigh became visibly thicker than the right. Diffuse subtle lipomatosis affecting the ipsilateral face, neck, arms, calf, and foot was noted at that time. Additionally, the patient had hyperlipidemia, gastroesophageal reflux disease, osteoporosis, and scoliosis, all beginning in her late 60s. She reported no alcohol or tobacco use and was taking rosuvastatin, esomeprazole, calcium, vitamin D, and glucosamine. There was no reported family history of asymmetric growth or bony deformities, and her children were healthy.
On physical examination, the lipomatosis affected the entire left side, most prominently around the abdomen, back, and thighs. The affected side was nontender and nonpruritic; there was no atrophy of the unaffected side (Figure). Maximum thigh circumference was 55.1 cm on the affected side and 52.6 cm on the unaffected side. There were no differences in power, reflex, or sensation between the 2 sides, and no hyperhidrosis or vascular malformations were present. Laboratory investigations, including complete blood cell count, complete metabolic panel, lipids, and thyroid-stimulating and sex hormone panels all were within reference range.
Enzi et al1 reported 2 women who developed asymmetrical lipomatosis between the ages of 13 and 20 years. Acquired asymmetrical lipomatosis should be differentiated from the asymmetrical overgrowth diagnosed in neonates and infants.
Proteus syndrome (PS) is a progressive disease involving a combination of overgrowth in a mosaic distribution, connective tissue and epidermal nevi, ovarian cysts, parotid gland tumor, dysregulated adipose tissue, lymphovascular malformation, and certain facial phenotypes.2,3 The average age of onset is 6 to 18 months, and half of cases present at birth.3,4 Hemihyperplasia-multiple lipomatosis syndrome (HHML) describes a mild and nonprogressive variant that does not satisfy the diagnostic criteria of PS; it typically is diagnosed at birth.5 One case of mild and delayed-onset PS was described in a woman who started developing signs at 15 years of age.6 In comparison, asymmetrical lipomatosis and scoliosis were the only abnormal clinical signs present in our patient, and the lipomatosis developed diffusely, as opposed to the typical mosaic distribution found in PS and HHML. Scoliosis can be found in PS and HHML secondary to hemihypertrophy of vertebra or infiltrative intraspinal lipomatosis.7,8 Our patient’s scoliosis was diagnosed more than 10 years prior to the onset of lipomatosis, likely representing degenerative joint disease.9
Prior reported cases of asymmetrical lipomatosis did not describe treatment.1 Ultrasound-guided or conventional liposuction and lipectomy are mainstream therapies for multiple symmetrical lipomatosis, an acquired lipomatosis typically affecting alcoholics in the fourth decade of life. However, recurrence rates are high for surgical treatment of unencapsulated lipomatosis, likely due to incomplete removal of the adipose tissue.10 Alternative treatments found in case reports, including oral salbutamol, mesotherapy using phosphatidylcholine, and fenofibrate (200 mg/d), require further study.11-13 Our patient was not aesthetically bothered by her lipomatosis; therefore, imaging and treatment options were not pursued. In conclusion, we report a patient with acquired asymmetrical lipomatosis with onset in late adulthood, unique from the existing syndromes of asymmetrical hemihyperplasia.1,14
- Enzi G, Digito M, Enzi GB, et al. Asymmetrical lipomatosis: report of two cases. Postgrad Med J. 1985;61:797-800.
- Biesecker LG, Happle R, Mulliken JB, et al. Proteus syndrome: diagnostic criteria, differential diagnosis, and patient evaluation. Am J Med Genet. 1999;84:389-395.
- Biesecker L. The challenges of Proteus syndrome: diagnosis and management. Eur J Hum Genet. 2006;14:1151-1157.
- Cohen MM Jr. Proteus syndrome: an update. Am J Med Genet C Semin Med Genet. 2005;137C:38-52.
- Biesecker LG, Peters KF, Darling TN, et al. Clinical differentiation between Proteus syndrome and hemihyperplasia: description of a distinct form of hemihyperplasia. Am J Med Genet. 1998;79:311-318.
- Luo S, Feng Y, Zheng Y, et al. Mild and delayed-onset Proteus syndrome. Eur J Dermatol. 2007;17:172-173.
- Takebayashi T, Yamashita T, Yokogushi K, et al. Scoliosis in Proteus syndrome: case report. Spine. 2001;26:E395-E398.
- Schulte TL, Liljenqvist U, Görgens H, et al. Hemihyperplasia-multiple lipomatosis syndrome (HHML): a challenge in spinal care. Acta Orthop Belg. 2008;74:714-719.
- Robin GC, Span Y, Steinberg R, et al. Scoliosis in the elderly: a follow-up study. Spine. 1982;7:355-359.
- Brea-García B, Cameselle-Teijeiro J, Couto-González I, et al. Madelung’s disease: comorbidities, fatty mass distribution, and response to treatment of 22 patients. Aesthet Plast Surg. 2013;37:409-416.
- Hasegawa T, Matsukura T, Ikeda S. Mesotherapy for benign symmetric lipomatosis. Aesthet Plast Surg. 2010;34:153-156.
- Zeitler H, Ulrich-Merzenich G, Richter DF, et al. Multiple benign symmetric lipomatosis—a differential diagnosis of obesity. is there a rationale for fibrate treatment? Obes Surg. 2008;18:1354-1356.
- Leung N, Gaer J, Beggs D, et al. Multiple symmetric lipomatosis (Launois‐Bensaude syndrome): effect of oral salbutamol. Clin Endocrinol. 1987;27:601-606.
- Craiglow BG, Ko CJ, Antaya RJ. Two cases of hemihyperplasia-multiple lipomatosis syndrome and review of asymmetric hemihyperplasia syndromes. Pediatr Dermatol. 2014;31:507-510.
- Enzi G, Digito M, Enzi GB, et al. Asymmetrical lipomatosis: report of two cases. Postgrad Med J. 1985;61:797-800.
- Biesecker LG, Happle R, Mulliken JB, et al. Proteus syndrome: diagnostic criteria, differential diagnosis, and patient evaluation. Am J Med Genet. 1999;84:389-395.
- Biesecker L. The challenges of Proteus syndrome: diagnosis and management. Eur J Hum Genet. 2006;14:1151-1157.
- Cohen MM Jr. Proteus syndrome: an update. Am J Med Genet C Semin Med Genet. 2005;137C:38-52.
- Biesecker LG, Peters KF, Darling TN, et al. Clinical differentiation between Proteus syndrome and hemihyperplasia: description of a distinct form of hemihyperplasia. Am J Med Genet. 1998;79:311-318.
- Luo S, Feng Y, Zheng Y, et al. Mild and delayed-onset Proteus syndrome. Eur J Dermatol. 2007;17:172-173.
- Takebayashi T, Yamashita T, Yokogushi K, et al. Scoliosis in Proteus syndrome: case report. Spine. 2001;26:E395-E398.
- Schulte TL, Liljenqvist U, Görgens H, et al. Hemihyperplasia-multiple lipomatosis syndrome (HHML): a challenge in spinal care. Acta Orthop Belg. 2008;74:714-719.
- Robin GC, Span Y, Steinberg R, et al. Scoliosis in the elderly: a follow-up study. Spine. 1982;7:355-359.
- Brea-García B, Cameselle-Teijeiro J, Couto-González I, et al. Madelung’s disease: comorbidities, fatty mass distribution, and response to treatment of 22 patients. Aesthet Plast Surg. 2013;37:409-416.
- Hasegawa T, Matsukura T, Ikeda S. Mesotherapy for benign symmetric lipomatosis. Aesthet Plast Surg. 2010;34:153-156.
- Zeitler H, Ulrich-Merzenich G, Richter DF, et al. Multiple benign symmetric lipomatosis—a differential diagnosis of obesity. is there a rationale for fibrate treatment? Obes Surg. 2008;18:1354-1356.
- Leung N, Gaer J, Beggs D, et al. Multiple symmetric lipomatosis (Launois‐Bensaude syndrome): effect of oral salbutamol. Clin Endocrinol. 1987;27:601-606.
- Craiglow BG, Ko CJ, Antaya RJ. Two cases of hemihyperplasia-multiple lipomatosis syndrome and review of asymmetric hemihyperplasia syndromes. Pediatr Dermatol. 2014;31:507-510.
Practice Points
- Acquired asymmetrical lipomatosis is a rare condition that can develop at any age; it should be differentiated from existing syndromes of asymmetrical hemihyperplasia.
- Acquired asymmetrical lipomatosis is a clinical diagnosis with no laboratory changes. If the patient is clinically stable and asymptomatic, no further investigation or management is required.
Enlarging Nodule on the Thigh
The Diagnosis: Metastatic Adenocarcinoma of the Colon
Cutaneous adenocarcinomas are uncommon, whether they present as a primary lesion or metastatic disease. In our patient, the histologic findings and immunohistochemical staining pattern were consistent with metastatic adenocarcinoma of the colon, an uncommon clinical presentation.
Colonic adenocarcinoma can cause cutaneous metastasis in 3% of cases. The most common sites of metastases include the abdomen, chest, and back.1 On histologic examination, hematoxylin and eosin (H&E)-stained sections of cutaneous metastatic adenocarcinoma illustrate a malignant gland-forming neoplasm in the dermis with luminal mucin and necrotic debris (quiz image). The glands are lined by tall columnar epithelial cells with hyperchromatic nuclei. Alternatively, poorly differentiated morphology can be seen with fewer glands and more infiltrating nests of tumor cells.2 Immunohistochemically, colonic adenocarcinoma typically is negative for cytokeratin (CK) 7 and positive for CK20 and caudal type homeobox transcription factor 2 (CDX-2).3
Primary cutaneous mucinous carcinoma is characterized by islands of neoplastic cells floating in pools of mucin (Figure 1). It may be indistinguishable from metastatic mucinous carcinomas of the colon or breast. Immunohistochemistry can be helpful in differentiating metastatic breast vs colon carcinoma. Cytokeratin 7, GATA binding protein 3, gross cystic disease fluid protein 15, and estrogen receptor will be positive in carcinomas of the breast and will be negative in colonic adenocarcinomas.4-6 Furthermore, lesional cells in metastatic adenocarcinoma of the colon are positive for CDX-2 and CK20, while those in metastatic carcinoma of the breast are negative.2 Immunohistochemistry also can differentiate primary cutaneous carcinoma from metastatic adenocarcinoma. When used in combination, p63 and podoplanin (D2-40) offer a highly sensitive and specific indicator of a primary cutaneous neoplasm, as both demonstrate either focal or diffuse positivity in this setting. In contrast, these stains typically are negative in metastatic adenocarcinomas of the skin.7
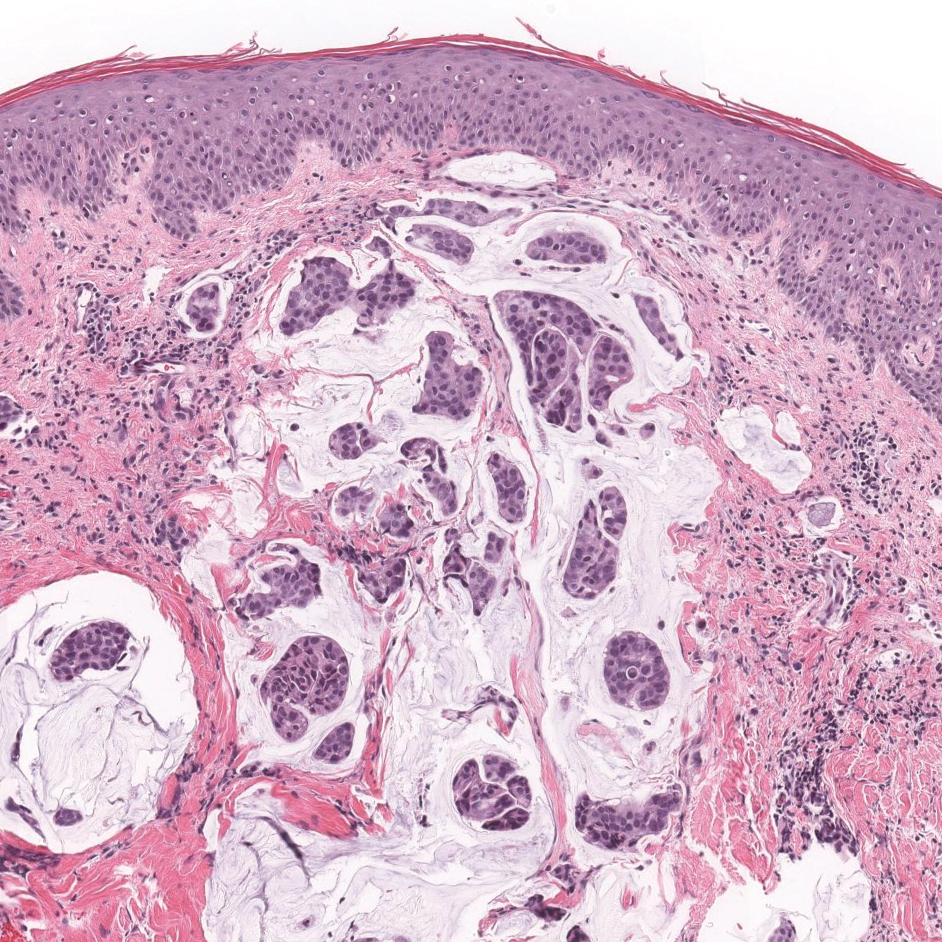
Endometriosis affects 1% to 2% of all reproductive-age females, of which extrapelvic manifestations account for only 0.5% to 1.0% of cases.8 Histologically, extrapelvic endometriosis is characterized by the triad of endometrial-type glands, endometrial stroma, and hemorrhage or hemosiderin deposition (Figure 2). The glands can enlarge and demonstrate architectural distortion with partial lack of polarity. These features initially can be concerning for adenocarcinoma, but on closer examination, nuclear morphology is regular and mitoses are absent.8,9 The diagnosis usually can be rendered with H&E alone; however, immunohistochemical stains for CD10 and estrogen receptor can highlight the endometrial stroma.10 Furthermore, endometrial glands will stain positive for paired box gene 8 (PAX8), a marker that is not expressed within the gastrointestinal tract and associated malignancies.11
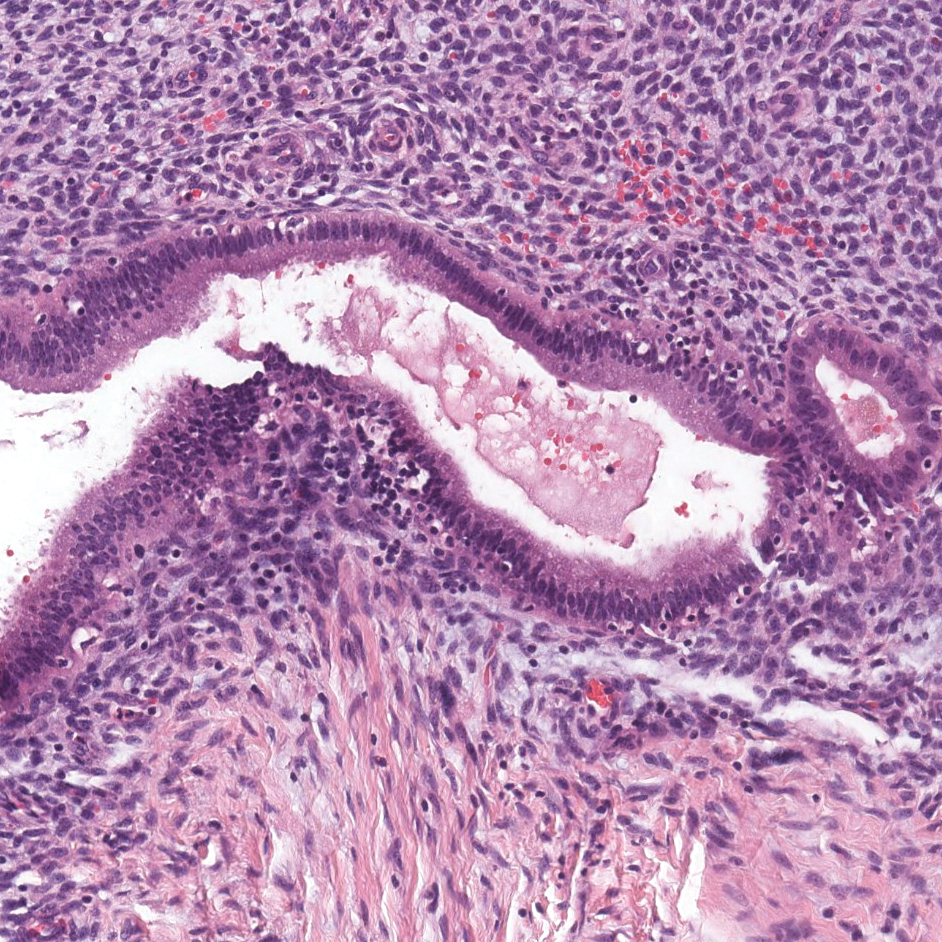
cytoplasm (H&E, original magnification ×100).
Primary cutaneous angiosarcoma may mimic adenocarcinoma, as the endothelial-lined vessels can be confused as malignant glands (Figure 3). Angiosarcoma often is seen in 1 of 3 clinical presentations: the head and neck of elderly patients, postradiation treatment, and chronic lymphedema.12,13 Regardless of the location, the disease carries a poor prognosis, with a 5-year survival rate of 12% following initial diagnosis.13 Angiosarcoma is characterized by malignant endothelial cells dissecting through the dermis. Although the histology can be deceptively bland in some cases, the neoplasm most commonly demonstrates notable atypia with a multilayered endothelium and occasional intravascular atypical cells ("fish in the creek appearance").13,14 There can be frequent mitoses, and the atypical cells may show intracytoplasmic lumina containing red blood cells. The lesional cells are positive for endothelial markers such as erythroblast transformation specific related gene (ERG), CD31, CD34, and friend leukemia integration factor 1 (FLI-1).15,16
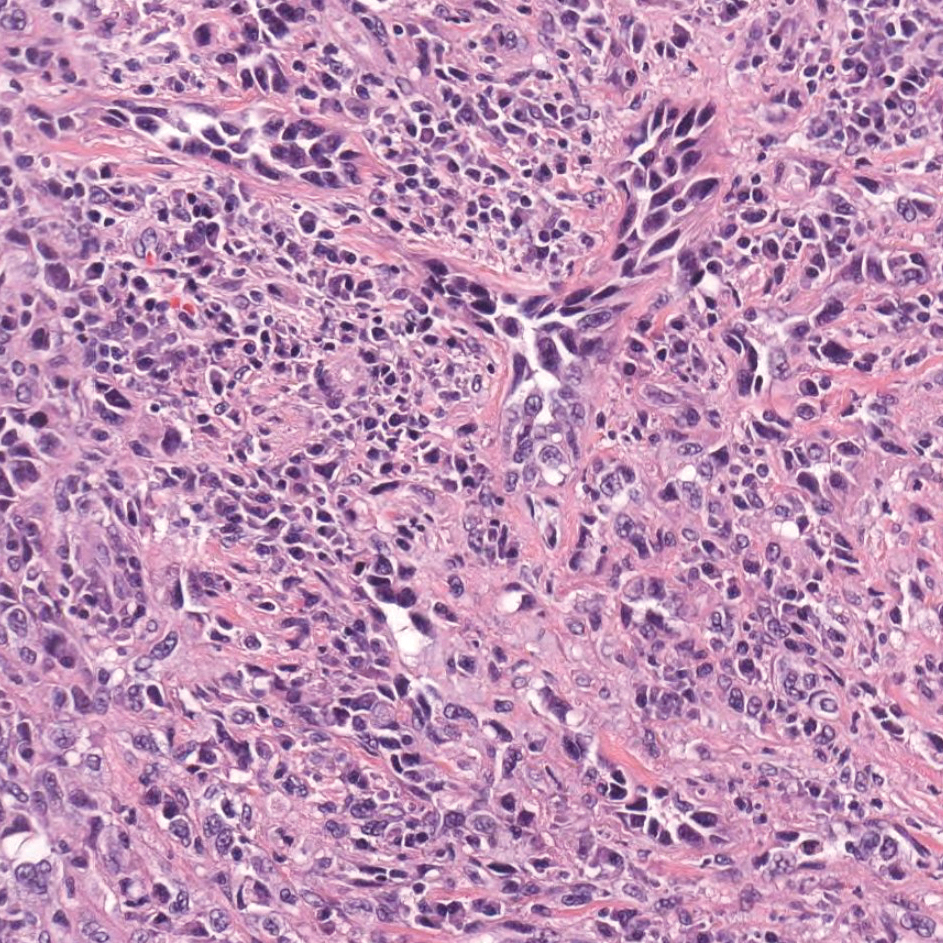
Breast cancer also can cause cutaneous metastases in approximately 20% of cases, with the most common presenting site being the anterior chest wall.17 Macroscopically, these lesions appear most commonly as painless nodules but also as telangiectatic, erysipeloid, fibrotic, and alopecic lesions.17-19 The histologic findings from H&E-stained sections of a cutaneous metastasis of breast cancer are variable and depend on the specific tumor subtype (eg, ductal, lobular, mucinous). However, the classic histologic presentation is that of nests and cords of malignant epithelial cells with variable gland formation. Often, tumor cells infiltrate in a single-file fashion (Figure 4).17 Although inflammatory breast carcinoma is a strictly clinical diagnosis, the presence of tumor cells in the lymphovascular spaces is a histologic clue to this diagnosis. Immunohistochemically, GATA binding protein 3 is helpful in identifying both hormone receptor-positive and -negative breast cancer subtypes that have metastasized.20
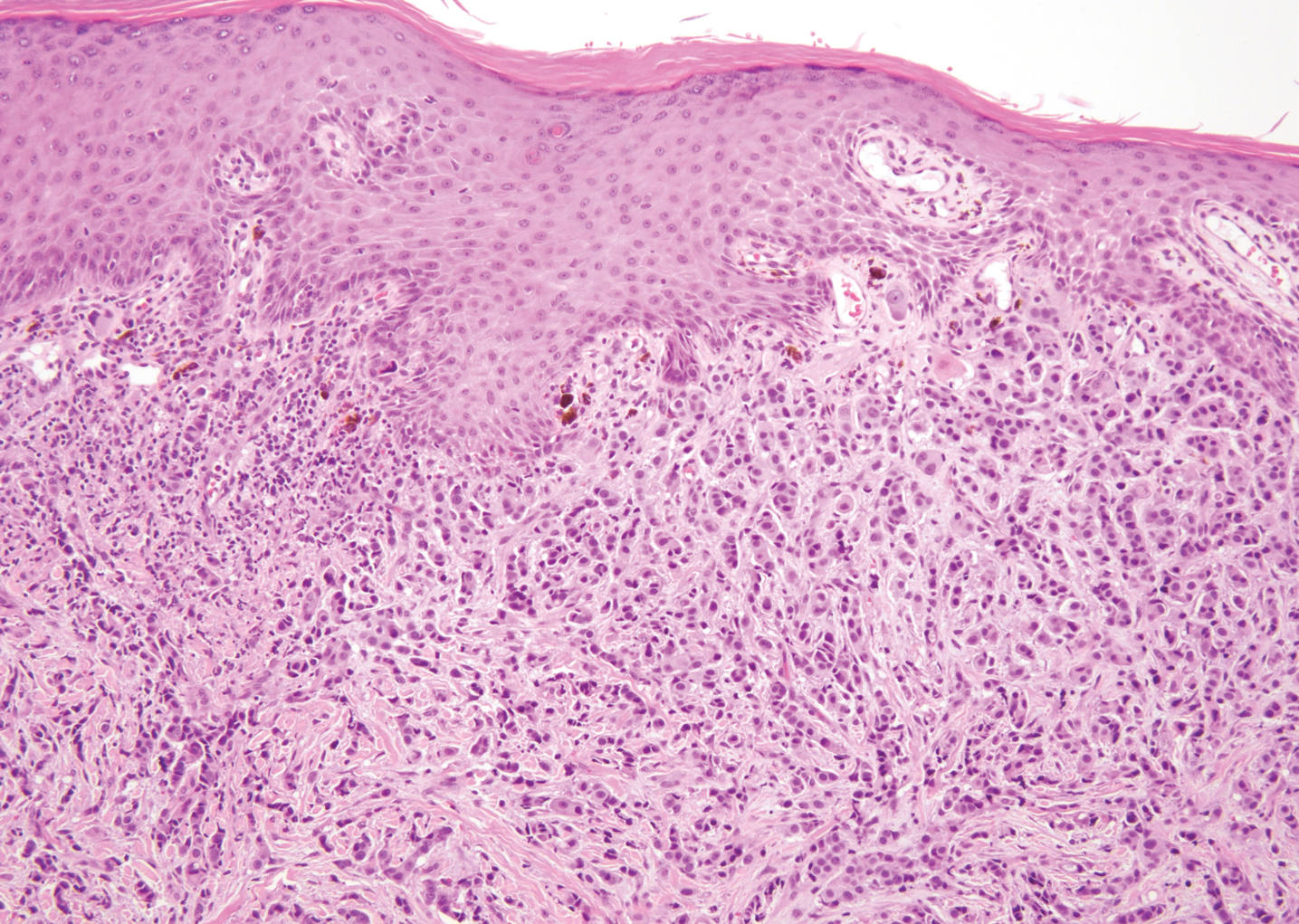
Within the histologic differential diagnoses, the most useful tool to diagnose metastatic adenocarcinoma of the colon often is a thorough clinical history. In the absence of a clinical history of adenocarcinoma, immunohistochemistry can be a useful adjunct to aid in the correct characterization and classification of a malignant gland-forming tumor.2,3,6
- Lookingbill DP, Spangler N, Helm KF. Cutaneous metastases in patients with metastatic carcinoma: a retrospective study of 4020 patients. J Am Acad Dermatol. 1993;29:228-236.
- Kumar V, Robbins SL. Robbins Basic Pathology. 8th ed. Philadelphia, PA: Saunders/Elsevier; 2007.
- Taliano RJ, LeGolvan M, Resnick MB. Immunohistochemistry of colorectal carcinoma: current practice and evolving applications. Hum Pathol. 2013;44:151-163.
- Kamalpour L, Brindise RT, Nodzenski M, et al. Primary cutaneous mucinous carcinoma: a systematic review and meta-analysis of outcomes after surgery. JAMA Dermatol. 2014;150:380-384.
- Roshan MH, Tambo A, Pace NP. The role of testosterone in colorectal carcinoma: pathomechanisms and open questions. EPMA J. 2016;7:22.
- Mazoujian G, Pinkus GS, Davis S, et al. Immunohistochemistry of a gross cystic disease fluid protein (GCDFP-15) of the breast. a marker of apocrine epithelium and breast carcinomas with apocrine features. Am J Pathol. 1983;110:105-112.
- Plaza JA, Ortega PF, Stockman DL, et al. Value of p63 and podoplanin (D2-40) immunoreactivity in the distinction between primary cutaneous tumors and adenocarcinomas metastatic to the skin: a clinicopathologic and immunohistochemical study of 79 cases. J Cutan Pathol. 2010;37:403-410.
- Machairiotis N, Stylianaki A, Dryllis G, et al. Extrapelvic endometriosis: a rare entity or an under diagnosed condition? Diagn Pathol. 2013;8:194.
- Chen H, Luo Q, Liu S, et al. Rectal mucosal endometriosis primarily misinterpreted as adenocarcinoma: a case report and review of literature. Int J Clin Exp Pathol. 2015;8:5902-5907.
- Terada S, Miyata Y, Nakazawa H, et al. Immunohistochemical analysis of an ectopic endometriosis in the uterine round ligament. Diagn Pathol. 2006;1:27.
- Yemelyanova A, Gown AM, Wu LS, et al. PAX8 expression in uterine adenocarcinomas and mesonephric proliferations. Int J Gynecol Pathol. 2014;33:492-499.
- Farid M, Ong WS, Lee MJ, et al. Cutaneous versus non-cutaneous angiosarcoma: clinicopathologic features and treatment outcomes in 60 patients at a single Asian cancer centre. Oncology. 2013;85:182-190.
- Requena C, Sendra E, Llombart B, et al. Cutaneous angiosarcoma: clinical and pathology study of 16 cases. Actas Dermosifiliogr. 2017;108:457-465.
- Schmidt AP, Tjarks BJ, Lynch DW. Gone fishing: a unique histologic pattern in cutaneous angiosarcoma. Cutis. 2018;101:270-272.
- Sullivan HC, Edgar MA, Cohen C, et al. The utility of ERG, CD31 and CD34 in the cytological diagnosis of angiosarcoma: an analysis of 25 cases. J Clin Pathol. 2015;68:44-50.
- Rossi S, Orvieto E, Furlanetto A, et al. Utility of the immunohistochemical detection of FLI-1 expression in round cell and vascular neoplasm using a monoclonal antibody. Mod Pathol. 2004;17:547-552.
- Tan AR. Cutaneous manifestations of breast cancer. Semin Oncol. 2016;43:331-334.
- Schwartz RA, Wiederkehr M, Lambert WC. Secondary mucinous carcinoma of the skin: metastatic breast cancer. Dermatol Surg. 2004;30(2, pt 1):234-235.
- Mallon E, Dawber RP. Alopecia neoplastica without alopecia: a unique presentation of breast carcinoma scalp metastasis. J Am Acad Dermatol. 1994;31(2, pt 2):319-321.
- Braxton DR, Cohen C, Siddiqui MT. Utility of GATA3 immunohistochemistry for diagnosis of metastatic breast carcinoma in cytology specimens. Diagn Cytopathol. 2015;43:271-277.
The Diagnosis: Metastatic Adenocarcinoma of the Colon
Cutaneous adenocarcinomas are uncommon, whether they present as a primary lesion or metastatic disease. In our patient, the histologic findings and immunohistochemical staining pattern were consistent with metastatic adenocarcinoma of the colon, an uncommon clinical presentation.
Colonic adenocarcinoma can cause cutaneous metastasis in 3% of cases. The most common sites of metastases include the abdomen, chest, and back.1 On histologic examination, hematoxylin and eosin (H&E)-stained sections of cutaneous metastatic adenocarcinoma illustrate a malignant gland-forming neoplasm in the dermis with luminal mucin and necrotic debris (quiz image). The glands are lined by tall columnar epithelial cells with hyperchromatic nuclei. Alternatively, poorly differentiated morphology can be seen with fewer glands and more infiltrating nests of tumor cells.2 Immunohistochemically, colonic adenocarcinoma typically is negative for cytokeratin (CK) 7 and positive for CK20 and caudal type homeobox transcription factor 2 (CDX-2).3
Primary cutaneous mucinous carcinoma is characterized by islands of neoplastic cells floating in pools of mucin (Figure 1). It may be indistinguishable from metastatic mucinous carcinomas of the colon or breast. Immunohistochemistry can be helpful in differentiating metastatic breast vs colon carcinoma. Cytokeratin 7, GATA binding protein 3, gross cystic disease fluid protein 15, and estrogen receptor will be positive in carcinomas of the breast and will be negative in colonic adenocarcinomas.4-6 Furthermore, lesional cells in metastatic adenocarcinoma of the colon are positive for CDX-2 and CK20, while those in metastatic carcinoma of the breast are negative.2 Immunohistochemistry also can differentiate primary cutaneous carcinoma from metastatic adenocarcinoma. When used in combination, p63 and podoplanin (D2-40) offer a highly sensitive and specific indicator of a primary cutaneous neoplasm, as both demonstrate either focal or diffuse positivity in this setting. In contrast, these stains typically are negative in metastatic adenocarcinomas of the skin.7
Endometriosis affects 1% to 2% of all reproductive-age females, of which extrapelvic manifestations account for only 0.5% to 1.0% of cases.8 Histologically, extrapelvic endometriosis is characterized by the triad of endometrial-type glands, endometrial stroma, and hemorrhage or hemosiderin deposition (Figure 2). The glands can enlarge and demonstrate architectural distortion with partial lack of polarity. These features initially can be concerning for adenocarcinoma, but on closer examination, nuclear morphology is regular and mitoses are absent.8,9 The diagnosis usually can be rendered with H&E alone; however, immunohistochemical stains for CD10 and estrogen receptor can highlight the endometrial stroma.10 Furthermore, endometrial glands will stain positive for paired box gene 8 (PAX8), a marker that is not expressed within the gastrointestinal tract and associated malignancies.11
cytoplasm (H&E, original magnification ×100).
Primary cutaneous angiosarcoma may mimic adenocarcinoma, as the endothelial-lined vessels can be confused as malignant glands (Figure 3). Angiosarcoma often is seen in 1 of 3 clinical presentations: the head and neck of elderly patients, postradiation treatment, and chronic lymphedema.12,13 Regardless of the location, the disease carries a poor prognosis, with a 5-year survival rate of 12% following initial diagnosis.13 Angiosarcoma is characterized by malignant endothelial cells dissecting through the dermis. Although the histology can be deceptively bland in some cases, the neoplasm most commonly demonstrates notable atypia with a multilayered endothelium and occasional intravascular atypical cells ("fish in the creek appearance").13,14 There can be frequent mitoses, and the atypical cells may show intracytoplasmic lumina containing red blood cells. The lesional cells are positive for endothelial markers such as erythroblast transformation specific related gene (ERG), CD31, CD34, and friend leukemia integration factor 1 (FLI-1).15,16
Breast cancer also can cause cutaneous metastases in approximately 20% of cases, with the most common presenting site being the anterior chest wall.17 Macroscopically, these lesions appear most commonly as painless nodules but also as telangiectatic, erysipeloid, fibrotic, and alopecic lesions.17-19 The histologic findings from H&E-stained sections of a cutaneous metastasis of breast cancer are variable and depend on the specific tumor subtype (eg, ductal, lobular, mucinous). However, the classic histologic presentation is that of nests and cords of malignant epithelial cells with variable gland formation. Often, tumor cells infiltrate in a single-file fashion (Figure 4).17 Although inflammatory breast carcinoma is a strictly clinical diagnosis, the presence of tumor cells in the lymphovascular spaces is a histologic clue to this diagnosis. Immunohistochemically, GATA binding protein 3 is helpful in identifying both hormone receptor-positive and -negative breast cancer subtypes that have metastasized.20
Within the histologic differential diagnoses, the most useful tool to diagnose metastatic adenocarcinoma of the colon often is a thorough clinical history. In the absence of a clinical history of adenocarcinoma, immunohistochemistry can be a useful adjunct to aid in the correct characterization and classification of a malignant gland-forming tumor.2,3,6
The Diagnosis: Metastatic Adenocarcinoma of the Colon
Cutaneous adenocarcinomas are uncommon, whether they present as a primary lesion or metastatic disease. In our patient, the histologic findings and immunohistochemical staining pattern were consistent with metastatic adenocarcinoma of the colon, an uncommon clinical presentation.
Colonic adenocarcinoma can cause cutaneous metastasis in 3% of cases. The most common sites of metastases include the abdomen, chest, and back.1 On histologic examination, hematoxylin and eosin (H&E)-stained sections of cutaneous metastatic adenocarcinoma illustrate a malignant gland-forming neoplasm in the dermis with luminal mucin and necrotic debris (quiz image). The glands are lined by tall columnar epithelial cells with hyperchromatic nuclei. Alternatively, poorly differentiated morphology can be seen with fewer glands and more infiltrating nests of tumor cells.2 Immunohistochemically, colonic adenocarcinoma typically is negative for cytokeratin (CK) 7 and positive for CK20 and caudal type homeobox transcription factor 2 (CDX-2).3
Primary cutaneous mucinous carcinoma is characterized by islands of neoplastic cells floating in pools of mucin (Figure 1). It may be indistinguishable from metastatic mucinous carcinomas of the colon or breast. Immunohistochemistry can be helpful in differentiating metastatic breast vs colon carcinoma. Cytokeratin 7, GATA binding protein 3, gross cystic disease fluid protein 15, and estrogen receptor will be positive in carcinomas of the breast and will be negative in colonic adenocarcinomas.4-6 Furthermore, lesional cells in metastatic adenocarcinoma of the colon are positive for CDX-2 and CK20, while those in metastatic carcinoma of the breast are negative.2 Immunohistochemistry also can differentiate primary cutaneous carcinoma from metastatic adenocarcinoma. When used in combination, p63 and podoplanin (D2-40) offer a highly sensitive and specific indicator of a primary cutaneous neoplasm, as both demonstrate either focal or diffuse positivity in this setting. In contrast, these stains typically are negative in metastatic adenocarcinomas of the skin.7
Endometriosis affects 1% to 2% of all reproductive-age females, of which extrapelvic manifestations account for only 0.5% to 1.0% of cases.8 Histologically, extrapelvic endometriosis is characterized by the triad of endometrial-type glands, endometrial stroma, and hemorrhage or hemosiderin deposition (Figure 2). The glands can enlarge and demonstrate architectural distortion with partial lack of polarity. These features initially can be concerning for adenocarcinoma, but on closer examination, nuclear morphology is regular and mitoses are absent.8,9 The diagnosis usually can be rendered with H&E alone; however, immunohistochemical stains for CD10 and estrogen receptor can highlight the endometrial stroma.10 Furthermore, endometrial glands will stain positive for paired box gene 8 (PAX8), a marker that is not expressed within the gastrointestinal tract and associated malignancies.11
cytoplasm (H&E, original magnification ×100).
Primary cutaneous angiosarcoma may mimic adenocarcinoma, as the endothelial-lined vessels can be confused as malignant glands (Figure 3). Angiosarcoma often is seen in 1 of 3 clinical presentations: the head and neck of elderly patients, postradiation treatment, and chronic lymphedema.12,13 Regardless of the location, the disease carries a poor prognosis, with a 5-year survival rate of 12% following initial diagnosis.13 Angiosarcoma is characterized by malignant endothelial cells dissecting through the dermis. Although the histology can be deceptively bland in some cases, the neoplasm most commonly demonstrates notable atypia with a multilayered endothelium and occasional intravascular atypical cells ("fish in the creek appearance").13,14 There can be frequent mitoses, and the atypical cells may show intracytoplasmic lumina containing red blood cells. The lesional cells are positive for endothelial markers such as erythroblast transformation specific related gene (ERG), CD31, CD34, and friend leukemia integration factor 1 (FLI-1).15,16
Breast cancer also can cause cutaneous metastases in approximately 20% of cases, with the most common presenting site being the anterior chest wall.17 Macroscopically, these lesions appear most commonly as painless nodules but also as telangiectatic, erysipeloid, fibrotic, and alopecic lesions.17-19 The histologic findings from H&E-stained sections of a cutaneous metastasis of breast cancer are variable and depend on the specific tumor subtype (eg, ductal, lobular, mucinous). However, the classic histologic presentation is that of nests and cords of malignant epithelial cells with variable gland formation. Often, tumor cells infiltrate in a single-file fashion (Figure 4).17 Although inflammatory breast carcinoma is a strictly clinical diagnosis, the presence of tumor cells in the lymphovascular spaces is a histologic clue to this diagnosis. Immunohistochemically, GATA binding protein 3 is helpful in identifying both hormone receptor-positive and -negative breast cancer subtypes that have metastasized.20
Within the histologic differential diagnoses, the most useful tool to diagnose metastatic adenocarcinoma of the colon often is a thorough clinical history. In the absence of a clinical history of adenocarcinoma, immunohistochemistry can be a useful adjunct to aid in the correct characterization and classification of a malignant gland-forming tumor.2,3,6
- Lookingbill DP, Spangler N, Helm KF. Cutaneous metastases in patients with metastatic carcinoma: a retrospective study of 4020 patients. J Am Acad Dermatol. 1993;29:228-236.
- Kumar V, Robbins SL. Robbins Basic Pathology. 8th ed. Philadelphia, PA: Saunders/Elsevier; 2007.
- Taliano RJ, LeGolvan M, Resnick MB. Immunohistochemistry of colorectal carcinoma: current practice and evolving applications. Hum Pathol. 2013;44:151-163.
- Kamalpour L, Brindise RT, Nodzenski M, et al. Primary cutaneous mucinous carcinoma: a systematic review and meta-analysis of outcomes after surgery. JAMA Dermatol. 2014;150:380-384.
- Roshan MH, Tambo A, Pace NP. The role of testosterone in colorectal carcinoma: pathomechanisms and open questions. EPMA J. 2016;7:22.
- Mazoujian G, Pinkus GS, Davis S, et al. Immunohistochemistry of a gross cystic disease fluid protein (GCDFP-15) of the breast. a marker of apocrine epithelium and breast carcinomas with apocrine features. Am J Pathol. 1983;110:105-112.
- Plaza JA, Ortega PF, Stockman DL, et al. Value of p63 and podoplanin (D2-40) immunoreactivity in the distinction between primary cutaneous tumors and adenocarcinomas metastatic to the skin: a clinicopathologic and immunohistochemical study of 79 cases. J Cutan Pathol. 2010;37:403-410.
- Machairiotis N, Stylianaki A, Dryllis G, et al. Extrapelvic endometriosis: a rare entity or an under diagnosed condition? Diagn Pathol. 2013;8:194.
- Chen H, Luo Q, Liu S, et al. Rectal mucosal endometriosis primarily misinterpreted as adenocarcinoma: a case report and review of literature. Int J Clin Exp Pathol. 2015;8:5902-5907.
- Terada S, Miyata Y, Nakazawa H, et al. Immunohistochemical analysis of an ectopic endometriosis in the uterine round ligament. Diagn Pathol. 2006;1:27.
- Yemelyanova A, Gown AM, Wu LS, et al. PAX8 expression in uterine adenocarcinomas and mesonephric proliferations. Int J Gynecol Pathol. 2014;33:492-499.
- Farid M, Ong WS, Lee MJ, et al. Cutaneous versus non-cutaneous angiosarcoma: clinicopathologic features and treatment outcomes in 60 patients at a single Asian cancer centre. Oncology. 2013;85:182-190.
- Requena C, Sendra E, Llombart B, et al. Cutaneous angiosarcoma: clinical and pathology study of 16 cases. Actas Dermosifiliogr. 2017;108:457-465.
- Schmidt AP, Tjarks BJ, Lynch DW. Gone fishing: a unique histologic pattern in cutaneous angiosarcoma. Cutis. 2018;101:270-272.
- Sullivan HC, Edgar MA, Cohen C, et al. The utility of ERG, CD31 and CD34 in the cytological diagnosis of angiosarcoma: an analysis of 25 cases. J Clin Pathol. 2015;68:44-50.
- Rossi S, Orvieto E, Furlanetto A, et al. Utility of the immunohistochemical detection of FLI-1 expression in round cell and vascular neoplasm using a monoclonal antibody. Mod Pathol. 2004;17:547-552.
- Tan AR. Cutaneous manifestations of breast cancer. Semin Oncol. 2016;43:331-334.
- Schwartz RA, Wiederkehr M, Lambert WC. Secondary mucinous carcinoma of the skin: metastatic breast cancer. Dermatol Surg. 2004;30(2, pt 1):234-235.
- Mallon E, Dawber RP. Alopecia neoplastica without alopecia: a unique presentation of breast carcinoma scalp metastasis. J Am Acad Dermatol. 1994;31(2, pt 2):319-321.
- Braxton DR, Cohen C, Siddiqui MT. Utility of GATA3 immunohistochemistry for diagnosis of metastatic breast carcinoma in cytology specimens. Diagn Cytopathol. 2015;43:271-277.
- Lookingbill DP, Spangler N, Helm KF. Cutaneous metastases in patients with metastatic carcinoma: a retrospective study of 4020 patients. J Am Acad Dermatol. 1993;29:228-236.
- Kumar V, Robbins SL. Robbins Basic Pathology. 8th ed. Philadelphia, PA: Saunders/Elsevier; 2007.
- Taliano RJ, LeGolvan M, Resnick MB. Immunohistochemistry of colorectal carcinoma: current practice and evolving applications. Hum Pathol. 2013;44:151-163.
- Kamalpour L, Brindise RT, Nodzenski M, et al. Primary cutaneous mucinous carcinoma: a systematic review and meta-analysis of outcomes after surgery. JAMA Dermatol. 2014;150:380-384.
- Roshan MH, Tambo A, Pace NP. The role of testosterone in colorectal carcinoma: pathomechanisms and open questions. EPMA J. 2016;7:22.
- Mazoujian G, Pinkus GS, Davis S, et al. Immunohistochemistry of a gross cystic disease fluid protein (GCDFP-15) of the breast. a marker of apocrine epithelium and breast carcinomas with apocrine features. Am J Pathol. 1983;110:105-112.
- Plaza JA, Ortega PF, Stockman DL, et al. Value of p63 and podoplanin (D2-40) immunoreactivity in the distinction between primary cutaneous tumors and adenocarcinomas metastatic to the skin: a clinicopathologic and immunohistochemical study of 79 cases. J Cutan Pathol. 2010;37:403-410.
- Machairiotis N, Stylianaki A, Dryllis G, et al. Extrapelvic endometriosis: a rare entity or an under diagnosed condition? Diagn Pathol. 2013;8:194.
- Chen H, Luo Q, Liu S, et al. Rectal mucosal endometriosis primarily misinterpreted as adenocarcinoma: a case report and review of literature. Int J Clin Exp Pathol. 2015;8:5902-5907.
- Terada S, Miyata Y, Nakazawa H, et al. Immunohistochemical analysis of an ectopic endometriosis in the uterine round ligament. Diagn Pathol. 2006;1:27.
- Yemelyanova A, Gown AM, Wu LS, et al. PAX8 expression in uterine adenocarcinomas and mesonephric proliferations. Int J Gynecol Pathol. 2014;33:492-499.
- Farid M, Ong WS, Lee MJ, et al. Cutaneous versus non-cutaneous angiosarcoma: clinicopathologic features and treatment outcomes in 60 patients at a single Asian cancer centre. Oncology. 2013;85:182-190.
- Requena C, Sendra E, Llombart B, et al. Cutaneous angiosarcoma: clinical and pathology study of 16 cases. Actas Dermosifiliogr. 2017;108:457-465.
- Schmidt AP, Tjarks BJ, Lynch DW. Gone fishing: a unique histologic pattern in cutaneous angiosarcoma. Cutis. 2018;101:270-272.
- Sullivan HC, Edgar MA, Cohen C, et al. The utility of ERG, CD31 and CD34 in the cytological diagnosis of angiosarcoma: an analysis of 25 cases. J Clin Pathol. 2015;68:44-50.
- Rossi S, Orvieto E, Furlanetto A, et al. Utility of the immunohistochemical detection of FLI-1 expression in round cell and vascular neoplasm using a monoclonal antibody. Mod Pathol. 2004;17:547-552.
- Tan AR. Cutaneous manifestations of breast cancer. Semin Oncol. 2016;43:331-334.
- Schwartz RA, Wiederkehr M, Lambert WC. Secondary mucinous carcinoma of the skin: metastatic breast cancer. Dermatol Surg. 2004;30(2, pt 1):234-235.
- Mallon E, Dawber RP. Alopecia neoplastica without alopecia: a unique presentation of breast carcinoma scalp metastasis. J Am Acad Dermatol. 1994;31(2, pt 2):319-321.
- Braxton DR, Cohen C, Siddiqui MT. Utility of GATA3 immunohistochemistry for diagnosis of metastatic breast carcinoma in cytology specimens. Diagn Cytopathol. 2015;43:271-277.
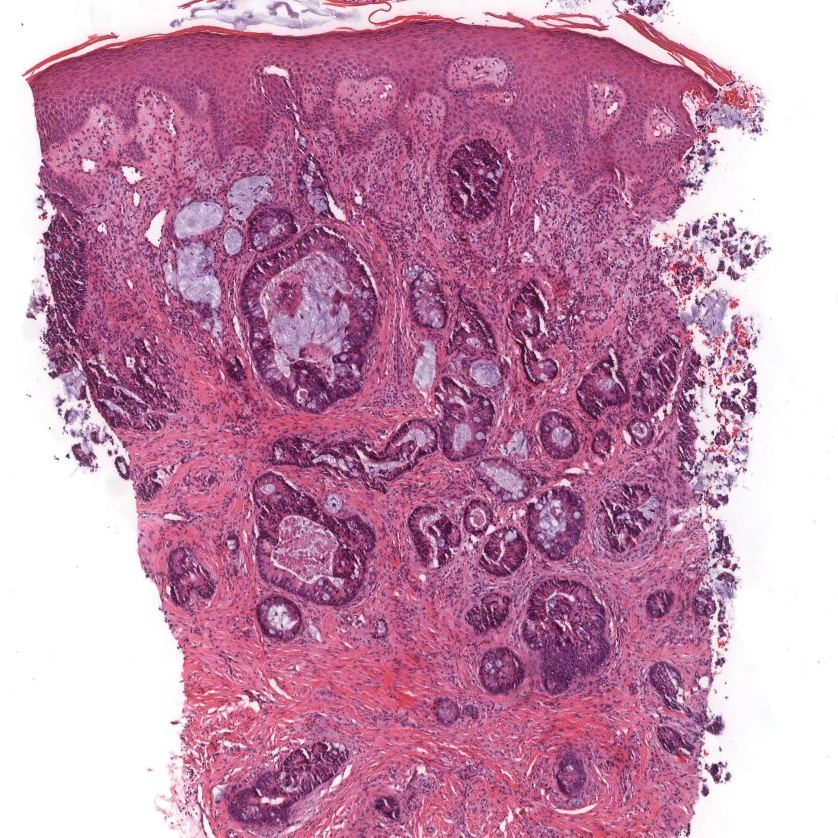
A 68-year-old patient presented with an enlarging flesh-colored nodule on the thigh that was positive for cytokeratin 20 and negative for cytokeratin 7.