User login
Cutis is a peer-reviewed clinical journal for the dermatologist, allergist, and general practitioner published monthly since 1965. Concise clinical articles present the practical side of dermatology, helping physicians to improve patient care. Cutis is referenced in Index Medicus/MEDLINE and is written and edited by industry leaders.
ass lick
assault rifle
balls
ballsac
black jack
bleach
Boko Haram
bondage
causas
cheap
child abuse
cocaine
compulsive behaviors
cost of miracles
cunt
Daech
display network stats
drug paraphernalia
explosion
fart
fda and death
fda AND warn
fda AND warning
fda AND warns
feom
fuck
gambling
gfc
gun
human trafficking
humira AND expensive
illegal
ISIL
ISIS
Islamic caliphate
Islamic state
madvocate
masturbation
mixed martial arts
MMA
molestation
national rifle association
NRA
nsfw
nuccitelli
pedophile
pedophilia
poker
porn
porn
pornography
psychedelic drug
recreational drug
sex slave rings
shit
slot machine
snort
substance abuse
terrorism
terrorist
texarkana
Texas hold 'em
UFC
section[contains(@class, 'nav-hidden')]
section[contains(@class, 'nav-hidden active')
A peer-reviewed, indexed journal for dermatologists with original research, image quizzes, cases and reviews, and columns.
Trends in Nail Services May Cause Dermatitis: Not Your Mother’s Nail Polish
In 2017, consumers spent an average of $8.53 billion on nail services.1 This booming industry is set to grow to more than $15.5 billion by 2024.2 Nail polishes and other nail cosmetic trends can present new exposures for consumers, including chemicals that can elicit allergic contact dermatitis. In this article, we discuss new nail trends and their associated allergens, the acrylates.
Tosylamide/Formaldehyde Resin
Traditionally, the most widely recognized nail polish allergen has been tosylamide/formaldehyde resin (TSFR). However, there now are many touted TSFR-free nail polishes on the market, and the rate of positive reactions to this chemical has been declining in recent years. The North American Contact Dermatitis Group reported a positive reaction rate of 1.3% from 2005 through 2006,3 and rates decreased to 0.9% from 2015 through 2016.4 An Australian study demonstrated a similar reduction in positive reaction rates to nail polish chemicals, with only 0.7% of patients reacting to TSFR from 2014 to 2016 and 0% in 2017. It is theorized that this reduction occurred from replacing TSFR in traditional nail polishes with other chemicals such as polyester resins and cellulose acetate butyrate.5
Acrylate-Based Nail Treatments
Consumers recently have been gravitating toward acrylate-based nail treatments vs traditional nail polishes for a variety of reasons. Often referred to as gels, dips, or shellac, acrylate-based nail treatments represent a hot new trend in nail cosmetics. These manicures are resistant to chipping and scratches, creating a like-new look that lasts for weeks after application. The long-lasting nature of acrylate-based nail polishes has made them wildly popular with consumers.
Traditional acrylic nails consist of a powder polymer mixed with a liquid monomer, which polymerizes when a catalyst is added.6 The procedure is time consuming and can take up to 2 hours for application. In contrast, the newer gel manicure can be completed faster and includes application of acrylate-based nail polish, including a base coat, 2 coats of color, and a top coat. Exposure to either a light-emitting diode (30–60 seconds) or UVA (2 minutes) lamp is necessary after each coat is applied for polymerization (Figure 1).6 This long-lasting, semipermanent manicure typically is what patients are referring to when they say they have “gel nails.”
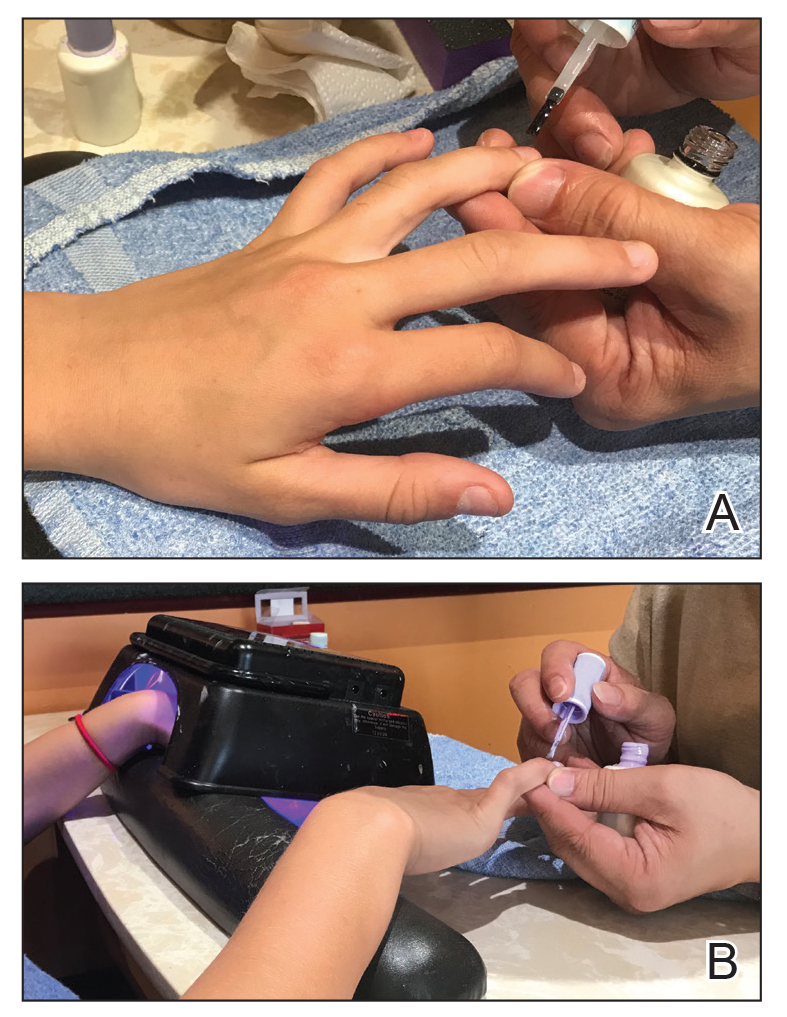
Gel dipping powders (referred to as dips) are another long-lasting acrylate-based nail treatment. This type of polish uses ethyl cyanoacrylate, a slightly different acrylate (yes, that IS super glue). After the nail is prepared, a base polish is applied to three-quarters of the nail and it is dipped into a natural color dip powder. The base polish is then applied to the entire nail, followed by a dip into the polish color of choice. This process is completed twice, followed by shaping and application of a top coat (Figure 2).
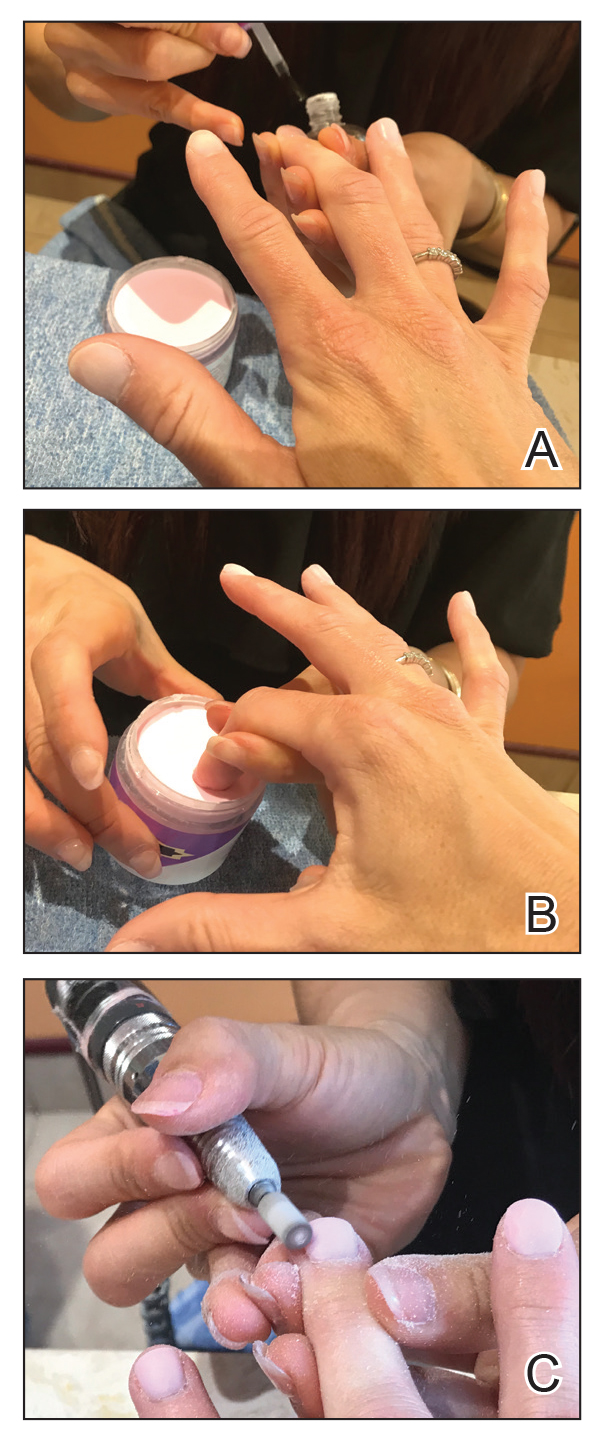
base coat. B, Application of dip powder to gel polish. Note the entire
distal finger and nail are dipped into the powder. C, Shaping of the
nail after the second coat of color is applied.
Finally, there are nail wraps, which are similar to stickers placed over or extending the nail plate. The wraps can be made from linen, silk, vinyl, or other material. Ethyl cyanoacrylate and isopropyl-2-cyanoacrylates have been identified in nail wrap adhesive.7 The heated product is directly applied to the prepared nail, and the excess wrap is filed off. Additional nail polish and a top coat usually are applied to finish the nail. Many of these products are available for in-salon use as well as online purchase and home application by consumers.
Acrylate Allergy
Patients who are allergic to acrylates can present with different patterns of dermatitis. Although the majority of patients present with dermatitis on the hands, fingers, or wrists, up to 10% may only have facial and neck dermatitis.8 Less commonly, the abdomen and thighs can be involved.6,8 Nail technicians most commonly present with pulpitis with cutaneous fissures.8 Other symptoms can include subungual hyperkeratosis, onycholysis, and nail dystrophy. Paresthesia, urticaria, and upper respiratory tract symptoms can occur but are less common.6,8
Acrylate allergy typically is the result of sensitization to the acrylate monomers. In theory, gel nail acrylate materials are polymerized following exposure to a light-emitting diode or UVA lamp; however, there likely is some incomplete polymerization, which can increase the risk for development of allergy. Allergen exposure can occur due to incorrect application of the light source; inadvertent monomer exposure, which occurs when nail technicians wipe extra acrylate off of a client’s finger(s); or inadvertent application of acrylate monomers to objects in the nail technician’s work environment.6,8
Several acrylate nail allergens have been reported. Many studies have identified 2-hydroxyethyl methacrylate (HEMA) as the most common nail acrylate allergen.8,9 At least one study identified 2-hydroxypropyl methacrylate as the most common, with HEMA in second place.6 Other reported acrylate allergens have included ethylene glycol dimethacrylate, triethylene glycol dimethacrylate, methyl methacrylate, ethyl cyanoacrylate, 1,4-butanediol diacrylate, hydroxypropyl acrylate, and 2-hydroxyethyl acrylate.8,9
The American Contact Dermatitis Society Core Allergen Series and the North American Contact Dermatitis Group screening series currently include HEMA, methyl methacrylate, ethyl acrylate, ethyl cyanoacrylate, and TSFR.4,10 Of note, acrylates are not included in the thin-layer rapid use epicutaneous (T.R.U.E.) patch test (SmartPractice), so they will be missed if this series is used.11 In the setting of suspected nail acrylate allergy, some authors recommend initial screening with HEMA and ethyl cyanoacrylate, with extended acrylate testing if both are negative.8
Upon patch testing with an acrylate series, patients frequently react to 2 or more acrylates and the reactions can be strong (++) or extreme (+++), which may represent cosensitization or cross-sensitization.8 The likelihood of cross-reactivity between acrylates is not clear, though it has been postulated that it is theoretically possible.6
An important pearl for patch testers using the chamber method is proper storage of acrylate allergens and assembly of trays prior to patch testing. Similar to all haptens, manufacturers recommend that acrylates should be stored in a refrigerator, but some authors suggest that acrylates should be stored in the freezer.12 Acrylates are volatile chemicals and rapidly degrade when exposed to air. A methyl methacrylate preparation loaded into an inert quadrate (IQ) chamber and stored at room temperature showed a nearly undetectable amount of any residual methyl methacrylate 24 hours later. Refrigeration of allergens in chambers slowed but did not stop eventual degradation, with nearly all acrylate preparations reaching an undetectable level of allergen by day 8.13 Acrylates, along with other volatile allergens, should only be loaded into chambers immediately prior to placement on the patient.
Allergy Prevention
Prevention of nail acrylate allergy among consumers is simple: avoid contact with the offending allergen. Acrylate spillover (ie, applying the acrylate onto the skin) and direct contact with objects and working surfaces contaminated with acrylate-based nail products should be avoided.8 Avoidance is more complicated for nail technicians, but it is thought that nitrile gloves allow for the best dexterity and allergen avoidance when acrylate exposure is brief.14 Allowable exposure times with nitrile gloves may be 15 to 30 minutes. After this times passes, a glove change is required to avoid exposure.14 Wearing nitrile gloves for longer than 15 to 30 minutes will result in cutaneous exposure and risk for dermatitis in sensitized patients. If longer wear is desired, one option includes cutting the fingertips off of Silver Shield/4H gloves (Honeywell Safety Products USA, Inc), applying them to the distal fingers, and wearing a standard nitrile glove over top, known as the finger stall technique.6 In one study, this technique was recommended to nail technicians with acrylate allergy. A telephone survey conducted 4 to 43 months later confirmed that 36% (8/22) of participants were using the technique without symptoms. In this same study, 73% (16/22) had continued working as nail technicians.6
Acrylates are used for other medical purposes, including dental procedures, orthopedic procedures, surgical glues, wound dressings, and contact and intraocular lenses. They also have additional cosmetic applications, including eyelash and hair extensions.8 Therefore, it is vital that patients disclose any history of acrylate allergy to both their medical and cosmetic providers.
Our Final Interpretation
Acrylate allergy has become increasingly common, and long-lasting nail treatments often are the culprit. Whether through gels, dips, or shellac, repeated exposure to acrylates through nail treatments can increase the risk for allergy. The T.R.U.E. test alone will not make the diagnosis, as acrylates are not present in this patch test system. It is important to remind your allergic patients that acrylates are present in other compounds used for medical and cosmetic purposes. Avoidance is key, and for allergic patients who love to bedazzle their nails, we suggest less-permanent, acrylate-free nail polishes as alternatives.
- 2017-2018 industry statistics highlights. Nails Magazine. http://files.nailsmag.com/handouts/nabb2017-18stats-lr.pdf. Accessed May 17, 2019.
- Nail polish market size worth $15.55 billion by 2024. Grand View Research website. https://www.grandviewresearch.com/press-release/global-nail-polish-market. Published October 2017. Accessed May 17, 2019.
- Zug KA, Warshaw EM, Fowler JF, et al. Patch-test results of the North American Contact Dermatitis Group 2005-2006. Dermatitis. 2009;20:149-160.
- DeKoven J, Warshaw EM, Zug KA, et al. North American Contact Dermatitis Group patch test results: 2015-2016. Dermatitis. 2018;29:297-309.
- Lee S, Maor D, Palmer A, et al. Declining prevalence of allergic contact dermatitis caused by tosylamide/formaldehyde in nail polish. Contact Dermatitis. 2018;79:184-185.
- Gatica-Ortega ME, Pastor-Nieto MA, Mercader-García P, et al. Allergic contact dermatitis caused by (meth)acrylates in long-lasting nail polish: are we facing a new epidemic in the beauty industry? Contact Dermatitis. 2017;7:360-366.
- Fitzgerald DA, Bhaggoe R, English JS. Contact sensitivity to cyanoacrylate nail-adhesive with dermatitis at remote sites. Contact Dermatitis. 1995;32:175-176.
- Goncalo M, Pinho A, Agner T et al. Allergic contact dermatitis caused by nail acrylates in Europe. an EECDRG study. Contact Dermatitis. 2017;78:254-260.
- Fisch A, Hamnerius N, Isaksson M. Dermatitis and occupational (meth)acrylate contact allergy in nail technicians—a 10-year study [published online January 14, 2019]. Contact Dermatitis. doi:10.1111/cod.13216.
- Schalock PC, Dunnick CA, Nedorost S, et al. American Contact Dermatitis Society core allergen series: 2017 update. Dermatitis. 2017;28:141-143.
- T.R.U.E. TEST ready-to-use patch test panels. Smart Practice website. https://www.smartpractice.com/shop/wa/category?cn=T.R.U.E.-TEST%C2%AE-Ready-to-Use-Patch-Test-Panels&id=508222&m=SPA. Accessed May 17, 2019.
- Good AT, Bruze M, Zimerson E, et al. Variation in allergen content over time of acrylates/methylacrylates in patch test preparations. Br J Dermatol. 2011;164:116-124.
- Goon A, Bruze M, Zimerson E, et al. Variation in allergen content over time of acrylates/methacrylates in patch test preparations. Br J Dermatol. 2011;164:116-124.
- Morgado F, Batista M, Gonçalo M. Short exposures and glove protection against (meth)acrylates in nail beauticians—thoughts on a rising concern [published online January 17, 2019]. Contact Dermatitis. doi:10.1111/cod.13222.
In 2017, consumers spent an average of $8.53 billion on nail services.1 This booming industry is set to grow to more than $15.5 billion by 2024.2 Nail polishes and other nail cosmetic trends can present new exposures for consumers, including chemicals that can elicit allergic contact dermatitis. In this article, we discuss new nail trends and their associated allergens, the acrylates.
Tosylamide/Formaldehyde Resin
Traditionally, the most widely recognized nail polish allergen has been tosylamide/formaldehyde resin (TSFR). However, there now are many touted TSFR-free nail polishes on the market, and the rate of positive reactions to this chemical has been declining in recent years. The North American Contact Dermatitis Group reported a positive reaction rate of 1.3% from 2005 through 2006,3 and rates decreased to 0.9% from 2015 through 2016.4 An Australian study demonstrated a similar reduction in positive reaction rates to nail polish chemicals, with only 0.7% of patients reacting to TSFR from 2014 to 2016 and 0% in 2017. It is theorized that this reduction occurred from replacing TSFR in traditional nail polishes with other chemicals such as polyester resins and cellulose acetate butyrate.5
Acrylate-Based Nail Treatments
Consumers recently have been gravitating toward acrylate-based nail treatments vs traditional nail polishes for a variety of reasons. Often referred to as gels, dips, or shellac, acrylate-based nail treatments represent a hot new trend in nail cosmetics. These manicures are resistant to chipping and scratches, creating a like-new look that lasts for weeks after application. The long-lasting nature of acrylate-based nail polishes has made them wildly popular with consumers.
Traditional acrylic nails consist of a powder polymer mixed with a liquid monomer, which polymerizes when a catalyst is added.6 The procedure is time consuming and can take up to 2 hours for application. In contrast, the newer gel manicure can be completed faster and includes application of acrylate-based nail polish, including a base coat, 2 coats of color, and a top coat. Exposure to either a light-emitting diode (30–60 seconds) or UVA (2 minutes) lamp is necessary after each coat is applied for polymerization (Figure 1).6 This long-lasting, semipermanent manicure typically is what patients are referring to when they say they have “gel nails.”
Gel dipping powders (referred to as dips) are another long-lasting acrylate-based nail treatment. This type of polish uses ethyl cyanoacrylate, a slightly different acrylate (yes, that IS super glue). After the nail is prepared, a base polish is applied to three-quarters of the nail and it is dipped into a natural color dip powder. The base polish is then applied to the entire nail, followed by a dip into the polish color of choice. This process is completed twice, followed by shaping and application of a top coat (Figure 2).
base coat. B, Application of dip powder to gel polish. Note the entire
distal finger and nail are dipped into the powder. C, Shaping of the
nail after the second coat of color is applied.
Finally, there are nail wraps, which are similar to stickers placed over or extending the nail plate. The wraps can be made from linen, silk, vinyl, or other material. Ethyl cyanoacrylate and isopropyl-2-cyanoacrylates have been identified in nail wrap adhesive.7 The heated product is directly applied to the prepared nail, and the excess wrap is filed off. Additional nail polish and a top coat usually are applied to finish the nail. Many of these products are available for in-salon use as well as online purchase and home application by consumers.
Acrylate Allergy
Patients who are allergic to acrylates can present with different patterns of dermatitis. Although the majority of patients present with dermatitis on the hands, fingers, or wrists, up to 10% may only have facial and neck dermatitis.8 Less commonly, the abdomen and thighs can be involved.6,8 Nail technicians most commonly present with pulpitis with cutaneous fissures.8 Other symptoms can include subungual hyperkeratosis, onycholysis, and nail dystrophy. Paresthesia, urticaria, and upper respiratory tract symptoms can occur but are less common.6,8
Acrylate allergy typically is the result of sensitization to the acrylate monomers. In theory, gel nail acrylate materials are polymerized following exposure to a light-emitting diode or UVA lamp; however, there likely is some incomplete polymerization, which can increase the risk for development of allergy. Allergen exposure can occur due to incorrect application of the light source; inadvertent monomer exposure, which occurs when nail technicians wipe extra acrylate off of a client’s finger(s); or inadvertent application of acrylate monomers to objects in the nail technician’s work environment.6,8
Several acrylate nail allergens have been reported. Many studies have identified 2-hydroxyethyl methacrylate (HEMA) as the most common nail acrylate allergen.8,9 At least one study identified 2-hydroxypropyl methacrylate as the most common, with HEMA in second place.6 Other reported acrylate allergens have included ethylene glycol dimethacrylate, triethylene glycol dimethacrylate, methyl methacrylate, ethyl cyanoacrylate, 1,4-butanediol diacrylate, hydroxypropyl acrylate, and 2-hydroxyethyl acrylate.8,9
The American Contact Dermatitis Society Core Allergen Series and the North American Contact Dermatitis Group screening series currently include HEMA, methyl methacrylate, ethyl acrylate, ethyl cyanoacrylate, and TSFR.4,10 Of note, acrylates are not included in the thin-layer rapid use epicutaneous (T.R.U.E.) patch test (SmartPractice), so they will be missed if this series is used.11 In the setting of suspected nail acrylate allergy, some authors recommend initial screening with HEMA and ethyl cyanoacrylate, with extended acrylate testing if both are negative.8
Upon patch testing with an acrylate series, patients frequently react to 2 or more acrylates and the reactions can be strong (++) or extreme (+++), which may represent cosensitization or cross-sensitization.8 The likelihood of cross-reactivity between acrylates is not clear, though it has been postulated that it is theoretically possible.6
An important pearl for patch testers using the chamber method is proper storage of acrylate allergens and assembly of trays prior to patch testing. Similar to all haptens, manufacturers recommend that acrylates should be stored in a refrigerator, but some authors suggest that acrylates should be stored in the freezer.12 Acrylates are volatile chemicals and rapidly degrade when exposed to air. A methyl methacrylate preparation loaded into an inert quadrate (IQ) chamber and stored at room temperature showed a nearly undetectable amount of any residual methyl methacrylate 24 hours later. Refrigeration of allergens in chambers slowed but did not stop eventual degradation, with nearly all acrylate preparations reaching an undetectable level of allergen by day 8.13 Acrylates, along with other volatile allergens, should only be loaded into chambers immediately prior to placement on the patient.
Allergy Prevention
Prevention of nail acrylate allergy among consumers is simple: avoid contact with the offending allergen. Acrylate spillover (ie, applying the acrylate onto the skin) and direct contact with objects and working surfaces contaminated with acrylate-based nail products should be avoided.8 Avoidance is more complicated for nail technicians, but it is thought that nitrile gloves allow for the best dexterity and allergen avoidance when acrylate exposure is brief.14 Allowable exposure times with nitrile gloves may be 15 to 30 minutes. After this times passes, a glove change is required to avoid exposure.14 Wearing nitrile gloves for longer than 15 to 30 minutes will result in cutaneous exposure and risk for dermatitis in sensitized patients. If longer wear is desired, one option includes cutting the fingertips off of Silver Shield/4H gloves (Honeywell Safety Products USA, Inc), applying them to the distal fingers, and wearing a standard nitrile glove over top, known as the finger stall technique.6 In one study, this technique was recommended to nail technicians with acrylate allergy. A telephone survey conducted 4 to 43 months later confirmed that 36% (8/22) of participants were using the technique without symptoms. In this same study, 73% (16/22) had continued working as nail technicians.6
Acrylates are used for other medical purposes, including dental procedures, orthopedic procedures, surgical glues, wound dressings, and contact and intraocular lenses. They also have additional cosmetic applications, including eyelash and hair extensions.8 Therefore, it is vital that patients disclose any history of acrylate allergy to both their medical and cosmetic providers.
Our Final Interpretation
Acrylate allergy has become increasingly common, and long-lasting nail treatments often are the culprit. Whether through gels, dips, or shellac, repeated exposure to acrylates through nail treatments can increase the risk for allergy. The T.R.U.E. test alone will not make the diagnosis, as acrylates are not present in this patch test system. It is important to remind your allergic patients that acrylates are present in other compounds used for medical and cosmetic purposes. Avoidance is key, and for allergic patients who love to bedazzle their nails, we suggest less-permanent, acrylate-free nail polishes as alternatives.
In 2017, consumers spent an average of $8.53 billion on nail services.1 This booming industry is set to grow to more than $15.5 billion by 2024.2 Nail polishes and other nail cosmetic trends can present new exposures for consumers, including chemicals that can elicit allergic contact dermatitis. In this article, we discuss new nail trends and their associated allergens, the acrylates.
Tosylamide/Formaldehyde Resin
Traditionally, the most widely recognized nail polish allergen has been tosylamide/formaldehyde resin (TSFR). However, there now are many touted TSFR-free nail polishes on the market, and the rate of positive reactions to this chemical has been declining in recent years. The North American Contact Dermatitis Group reported a positive reaction rate of 1.3% from 2005 through 2006,3 and rates decreased to 0.9% from 2015 through 2016.4 An Australian study demonstrated a similar reduction in positive reaction rates to nail polish chemicals, with only 0.7% of patients reacting to TSFR from 2014 to 2016 and 0% in 2017. It is theorized that this reduction occurred from replacing TSFR in traditional nail polishes with other chemicals such as polyester resins and cellulose acetate butyrate.5
Acrylate-Based Nail Treatments
Consumers recently have been gravitating toward acrylate-based nail treatments vs traditional nail polishes for a variety of reasons. Often referred to as gels, dips, or shellac, acrylate-based nail treatments represent a hot new trend in nail cosmetics. These manicures are resistant to chipping and scratches, creating a like-new look that lasts for weeks after application. The long-lasting nature of acrylate-based nail polishes has made them wildly popular with consumers.
Traditional acrylic nails consist of a powder polymer mixed with a liquid monomer, which polymerizes when a catalyst is added.6 The procedure is time consuming and can take up to 2 hours for application. In contrast, the newer gel manicure can be completed faster and includes application of acrylate-based nail polish, including a base coat, 2 coats of color, and a top coat. Exposure to either a light-emitting diode (30–60 seconds) or UVA (2 minutes) lamp is necessary after each coat is applied for polymerization (Figure 1).6 This long-lasting, semipermanent manicure typically is what patients are referring to when they say they have “gel nails.”
Gel dipping powders (referred to as dips) are another long-lasting acrylate-based nail treatment. This type of polish uses ethyl cyanoacrylate, a slightly different acrylate (yes, that IS super glue). After the nail is prepared, a base polish is applied to three-quarters of the nail and it is dipped into a natural color dip powder. The base polish is then applied to the entire nail, followed by a dip into the polish color of choice. This process is completed twice, followed by shaping and application of a top coat (Figure 2).
base coat. B, Application of dip powder to gel polish. Note the entire
distal finger and nail are dipped into the powder. C, Shaping of the
nail after the second coat of color is applied.
Finally, there are nail wraps, which are similar to stickers placed over or extending the nail plate. The wraps can be made from linen, silk, vinyl, or other material. Ethyl cyanoacrylate and isopropyl-2-cyanoacrylates have been identified in nail wrap adhesive.7 The heated product is directly applied to the prepared nail, and the excess wrap is filed off. Additional nail polish and a top coat usually are applied to finish the nail. Many of these products are available for in-salon use as well as online purchase and home application by consumers.
Acrylate Allergy
Patients who are allergic to acrylates can present with different patterns of dermatitis. Although the majority of patients present with dermatitis on the hands, fingers, or wrists, up to 10% may only have facial and neck dermatitis.8 Less commonly, the abdomen and thighs can be involved.6,8 Nail technicians most commonly present with pulpitis with cutaneous fissures.8 Other symptoms can include subungual hyperkeratosis, onycholysis, and nail dystrophy. Paresthesia, urticaria, and upper respiratory tract symptoms can occur but are less common.6,8
Acrylate allergy typically is the result of sensitization to the acrylate monomers. In theory, gel nail acrylate materials are polymerized following exposure to a light-emitting diode or UVA lamp; however, there likely is some incomplete polymerization, which can increase the risk for development of allergy. Allergen exposure can occur due to incorrect application of the light source; inadvertent monomer exposure, which occurs when nail technicians wipe extra acrylate off of a client’s finger(s); or inadvertent application of acrylate monomers to objects in the nail technician’s work environment.6,8
Several acrylate nail allergens have been reported. Many studies have identified 2-hydroxyethyl methacrylate (HEMA) as the most common nail acrylate allergen.8,9 At least one study identified 2-hydroxypropyl methacrylate as the most common, with HEMA in second place.6 Other reported acrylate allergens have included ethylene glycol dimethacrylate, triethylene glycol dimethacrylate, methyl methacrylate, ethyl cyanoacrylate, 1,4-butanediol diacrylate, hydroxypropyl acrylate, and 2-hydroxyethyl acrylate.8,9
The American Contact Dermatitis Society Core Allergen Series and the North American Contact Dermatitis Group screening series currently include HEMA, methyl methacrylate, ethyl acrylate, ethyl cyanoacrylate, and TSFR.4,10 Of note, acrylates are not included in the thin-layer rapid use epicutaneous (T.R.U.E.) patch test (SmartPractice), so they will be missed if this series is used.11 In the setting of suspected nail acrylate allergy, some authors recommend initial screening with HEMA and ethyl cyanoacrylate, with extended acrylate testing if both are negative.8
Upon patch testing with an acrylate series, patients frequently react to 2 or more acrylates and the reactions can be strong (++) or extreme (+++), which may represent cosensitization or cross-sensitization.8 The likelihood of cross-reactivity between acrylates is not clear, though it has been postulated that it is theoretically possible.6
An important pearl for patch testers using the chamber method is proper storage of acrylate allergens and assembly of trays prior to patch testing. Similar to all haptens, manufacturers recommend that acrylates should be stored in a refrigerator, but some authors suggest that acrylates should be stored in the freezer.12 Acrylates are volatile chemicals and rapidly degrade when exposed to air. A methyl methacrylate preparation loaded into an inert quadrate (IQ) chamber and stored at room temperature showed a nearly undetectable amount of any residual methyl methacrylate 24 hours later. Refrigeration of allergens in chambers slowed but did not stop eventual degradation, with nearly all acrylate preparations reaching an undetectable level of allergen by day 8.13 Acrylates, along with other volatile allergens, should only be loaded into chambers immediately prior to placement on the patient.
Allergy Prevention
Prevention of nail acrylate allergy among consumers is simple: avoid contact with the offending allergen. Acrylate spillover (ie, applying the acrylate onto the skin) and direct contact with objects and working surfaces contaminated with acrylate-based nail products should be avoided.8 Avoidance is more complicated for nail technicians, but it is thought that nitrile gloves allow for the best dexterity and allergen avoidance when acrylate exposure is brief.14 Allowable exposure times with nitrile gloves may be 15 to 30 minutes. After this times passes, a glove change is required to avoid exposure.14 Wearing nitrile gloves for longer than 15 to 30 minutes will result in cutaneous exposure and risk for dermatitis in sensitized patients. If longer wear is desired, one option includes cutting the fingertips off of Silver Shield/4H gloves (Honeywell Safety Products USA, Inc), applying them to the distal fingers, and wearing a standard nitrile glove over top, known as the finger stall technique.6 In one study, this technique was recommended to nail technicians with acrylate allergy. A telephone survey conducted 4 to 43 months later confirmed that 36% (8/22) of participants were using the technique without symptoms. In this same study, 73% (16/22) had continued working as nail technicians.6
Acrylates are used for other medical purposes, including dental procedures, orthopedic procedures, surgical glues, wound dressings, and contact and intraocular lenses. They also have additional cosmetic applications, including eyelash and hair extensions.8 Therefore, it is vital that patients disclose any history of acrylate allergy to both their medical and cosmetic providers.
Our Final Interpretation
Acrylate allergy has become increasingly common, and long-lasting nail treatments often are the culprit. Whether through gels, dips, or shellac, repeated exposure to acrylates through nail treatments can increase the risk for allergy. The T.R.U.E. test alone will not make the diagnosis, as acrylates are not present in this patch test system. It is important to remind your allergic patients that acrylates are present in other compounds used for medical and cosmetic purposes. Avoidance is key, and for allergic patients who love to bedazzle their nails, we suggest less-permanent, acrylate-free nail polishes as alternatives.
- 2017-2018 industry statistics highlights. Nails Magazine. http://files.nailsmag.com/handouts/nabb2017-18stats-lr.pdf. Accessed May 17, 2019.
- Nail polish market size worth $15.55 billion by 2024. Grand View Research website. https://www.grandviewresearch.com/press-release/global-nail-polish-market. Published October 2017. Accessed May 17, 2019.
- Zug KA, Warshaw EM, Fowler JF, et al. Patch-test results of the North American Contact Dermatitis Group 2005-2006. Dermatitis. 2009;20:149-160.
- DeKoven J, Warshaw EM, Zug KA, et al. North American Contact Dermatitis Group patch test results: 2015-2016. Dermatitis. 2018;29:297-309.
- Lee S, Maor D, Palmer A, et al. Declining prevalence of allergic contact dermatitis caused by tosylamide/formaldehyde in nail polish. Contact Dermatitis. 2018;79:184-185.
- Gatica-Ortega ME, Pastor-Nieto MA, Mercader-García P, et al. Allergic contact dermatitis caused by (meth)acrylates in long-lasting nail polish: are we facing a new epidemic in the beauty industry? Contact Dermatitis. 2017;7:360-366.
- Fitzgerald DA, Bhaggoe R, English JS. Contact sensitivity to cyanoacrylate nail-adhesive with dermatitis at remote sites. Contact Dermatitis. 1995;32:175-176.
- Goncalo M, Pinho A, Agner T et al. Allergic contact dermatitis caused by nail acrylates in Europe. an EECDRG study. Contact Dermatitis. 2017;78:254-260.
- Fisch A, Hamnerius N, Isaksson M. Dermatitis and occupational (meth)acrylate contact allergy in nail technicians—a 10-year study [published online January 14, 2019]. Contact Dermatitis. doi:10.1111/cod.13216.
- Schalock PC, Dunnick CA, Nedorost S, et al. American Contact Dermatitis Society core allergen series: 2017 update. Dermatitis. 2017;28:141-143.
- T.R.U.E. TEST ready-to-use patch test panels. Smart Practice website. https://www.smartpractice.com/shop/wa/category?cn=T.R.U.E.-TEST%C2%AE-Ready-to-Use-Patch-Test-Panels&id=508222&m=SPA. Accessed May 17, 2019.
- Good AT, Bruze M, Zimerson E, et al. Variation in allergen content over time of acrylates/methylacrylates in patch test preparations. Br J Dermatol. 2011;164:116-124.
- Goon A, Bruze M, Zimerson E, et al. Variation in allergen content over time of acrylates/methacrylates in patch test preparations. Br J Dermatol. 2011;164:116-124.
- Morgado F, Batista M, Gonçalo M. Short exposures and glove protection against (meth)acrylates in nail beauticians—thoughts on a rising concern [published online January 17, 2019]. Contact Dermatitis. doi:10.1111/cod.13222.
- 2017-2018 industry statistics highlights. Nails Magazine. http://files.nailsmag.com/handouts/nabb2017-18stats-lr.pdf. Accessed May 17, 2019.
- Nail polish market size worth $15.55 billion by 2024. Grand View Research website. https://www.grandviewresearch.com/press-release/global-nail-polish-market. Published October 2017. Accessed May 17, 2019.
- Zug KA, Warshaw EM, Fowler JF, et al. Patch-test results of the North American Contact Dermatitis Group 2005-2006. Dermatitis. 2009;20:149-160.
- DeKoven J, Warshaw EM, Zug KA, et al. North American Contact Dermatitis Group patch test results: 2015-2016. Dermatitis. 2018;29:297-309.
- Lee S, Maor D, Palmer A, et al. Declining prevalence of allergic contact dermatitis caused by tosylamide/formaldehyde in nail polish. Contact Dermatitis. 2018;79:184-185.
- Gatica-Ortega ME, Pastor-Nieto MA, Mercader-García P, et al. Allergic contact dermatitis caused by (meth)acrylates in long-lasting nail polish: are we facing a new epidemic in the beauty industry? Contact Dermatitis. 2017;7:360-366.
- Fitzgerald DA, Bhaggoe R, English JS. Contact sensitivity to cyanoacrylate nail-adhesive with dermatitis at remote sites. Contact Dermatitis. 1995;32:175-176.
- Goncalo M, Pinho A, Agner T et al. Allergic contact dermatitis caused by nail acrylates in Europe. an EECDRG study. Contact Dermatitis. 2017;78:254-260.
- Fisch A, Hamnerius N, Isaksson M. Dermatitis and occupational (meth)acrylate contact allergy in nail technicians—a 10-year study [published online January 14, 2019]. Contact Dermatitis. doi:10.1111/cod.13216.
- Schalock PC, Dunnick CA, Nedorost S, et al. American Contact Dermatitis Society core allergen series: 2017 update. Dermatitis. 2017;28:141-143.
- T.R.U.E. TEST ready-to-use patch test panels. Smart Practice website. https://www.smartpractice.com/shop/wa/category?cn=T.R.U.E.-TEST%C2%AE-Ready-to-Use-Patch-Test-Panels&id=508222&m=SPA. Accessed May 17, 2019.
- Good AT, Bruze M, Zimerson E, et al. Variation in allergen content over time of acrylates/methylacrylates in patch test preparations. Br J Dermatol. 2011;164:116-124.
- Goon A, Bruze M, Zimerson E, et al. Variation in allergen content over time of acrylates/methacrylates in patch test preparations. Br J Dermatol. 2011;164:116-124.
- Morgado F, Batista M, Gonçalo M. Short exposures and glove protection against (meth)acrylates in nail beauticians—thoughts on a rising concern [published online January 17, 2019]. Contact Dermatitis. doi:10.1111/cod.13222.
Practice Points
- Changing trends in nail services mean new exposures for consumers. Traditional nail polish has been replaced by semipermanent nail polish, which contains acrylates.
- Acrylates are a common cause of allergic contact dermatitis from nail polish. Acrylates can be found in gel, dip, and shellac nail polishes, among others.
- Patch testing with 2-hydroxyethyl methacrylate and ethyl cyanoacrylate can screen many patients for allergy due to nail services.

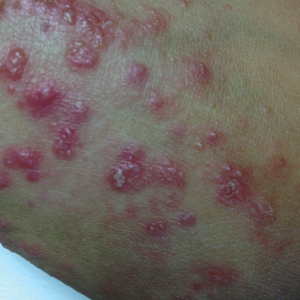
Pustular Tinea Id Reaction
To the Editor:
A 17-year-old adolescent girl presented to the dermatology clinic with a tender pruritic rash on the left wrist that was spreading to the bilateral arms and legs of several years’ duration. An area of a prior biopsy on the left wrist was healing well with use of petroleum jelly and halcinonide cream. The patient denied any constitutional symptoms.
Physical examination revealed numerous erythematous papules coalescing into plaques on the bilateral anterior and posterior arms and legs, including some erythematous macules and papules on the palms and soles. The original area of involvement on the left dorsal medial wrist demonstrated a background of erythema with overlying peripheral scaling and resolving violaceous to erythematous papules with signs of serosanguineous crusting (Figure 1). Scattered perifollicular erythema was present on the posterior aspects of the bilateral thighs and arms (Figure 2). Baseline complete blood cell count and complete metabolic panel were within reference range.
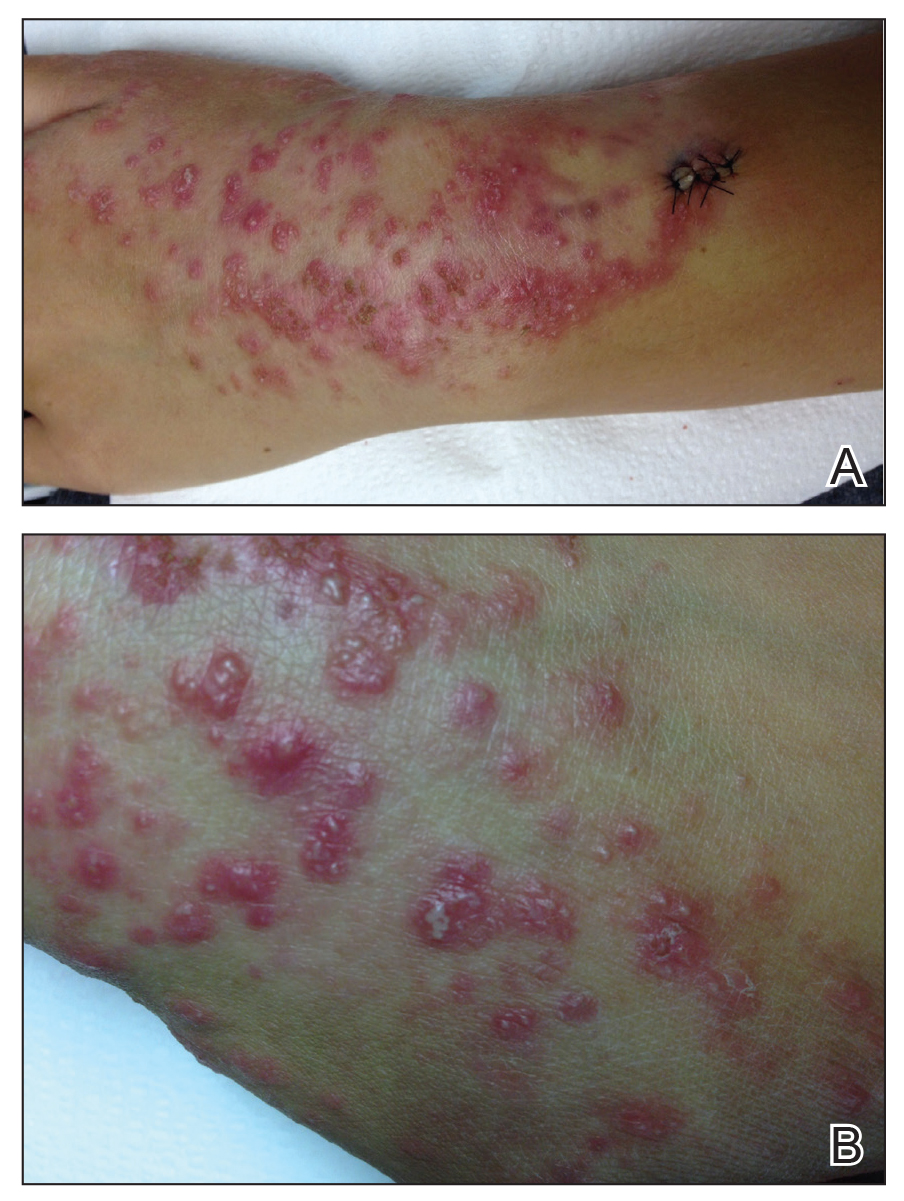
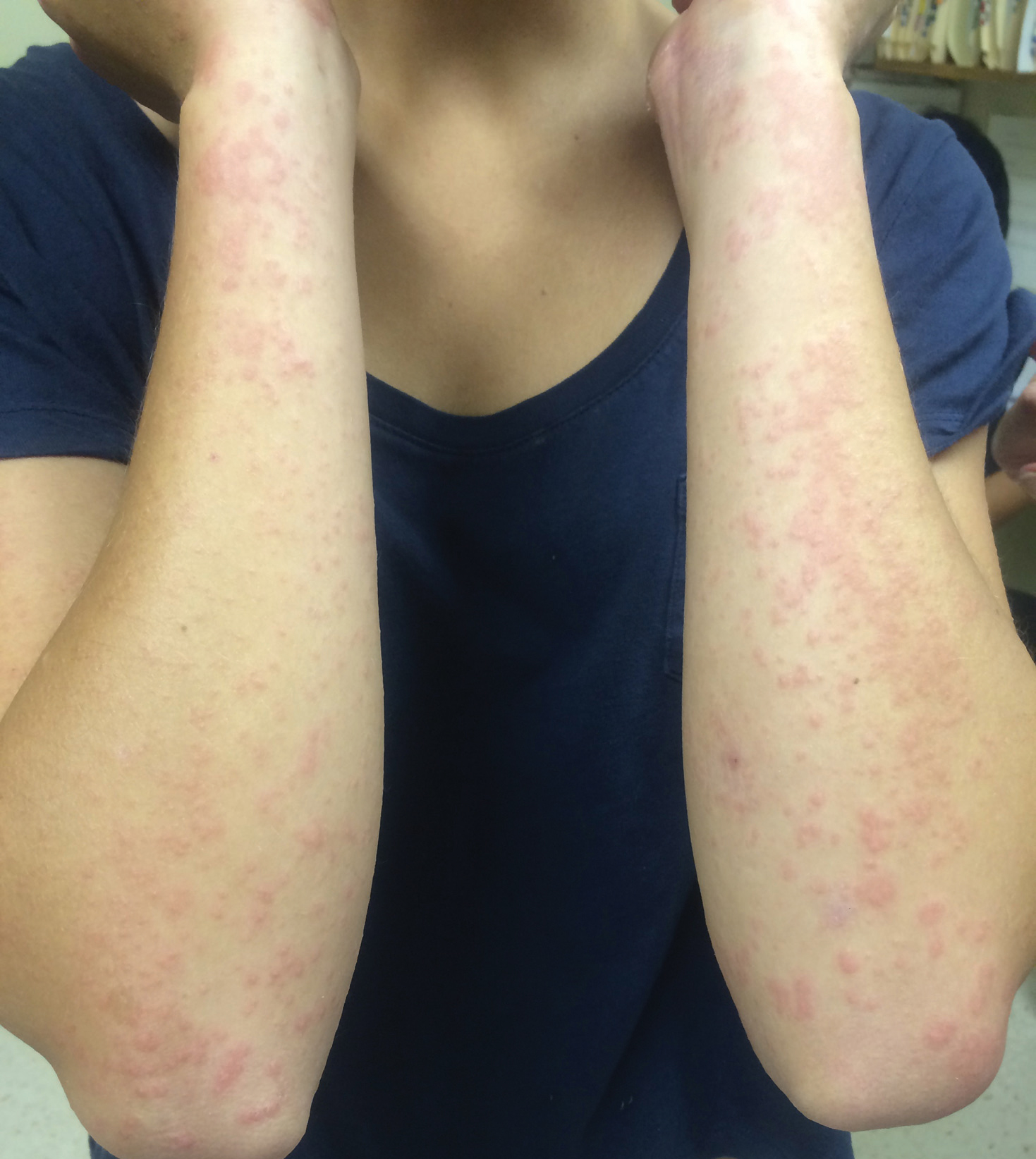
Clinical histopathology showed evidence of a pustular superficial dermatophyte infection, and Grocott-Gomori methenamine-silver stain demonstrated numerous fungal hyphae within subcorneal pustules, indicating pustular tinea. Based on the clinicopathologic correlation, the initial presentation was diagnosed as pustular tinea of the entire left wrist, followed by a generalized id reaction 1 week later.
The patient was prescribed oral terbinafine 250 mg once daily to treat the diffuse involvement of the pustular tinea as well as once-daily oral cetirizine, once-daily oral diphenhydramine, a topical emollient, and a topical nonsteroidal antipruritic gel.
Tinea is a superficial fungal infection commonly caused by the dermatophytes Epidermophyton, Trichophyton, and Microsporum. It has a variety of clinical presentations based on the anatomic location, including tinea capitis (hair/scalp), tinea pedis (feet), tinea corporis (face/trunk/extremities), tinea cruris (groin), and tinea unguium (nails).1 Tinea infections occur in the stratum corneum, hair, and nails, thriving on dead keratin in these areas.2 Tinea corporis usually appears as an erythematous ring-shaped lesion with a scaly border, but atypical cases presenting with vesicles, pustules, and bullae also have been reported.3 Additionally, secondary eruptions called id reactions, or autoeczematization, can present in the setting of dermatophyte infections. Such outbreaks may be due to a delayed hypersensitivity reaction to the fungal antigens. Id reactions can manifest in many forms of tinea with patients generally exhibiting pruritic papulovesicular lesions that can present far from the site of origin.4
Patients with id reactions can have atypical and varied presentations. In a case of id reaction due to tinea corporis, a patient presented with vesicles and pustules that grew in number and coalesced to form annular lesions.5 A case of an id reaction caused by tinea pedis also noted the presence of pustules, which are atypical in this form of tinea.6 In another case of tinea pedis, a generalized id reaction was noted, illustrating that such eruptions do not necessarily appear at the original site of infection.7 Additionally, in a rare presentation of tinea invading the nares, a patient developed an erythema multiforme id reaction.8 Id reactions also were noted in 14 patients with refractory otitis externa, illustrating the ability of this fungal infection to persist and infect distant locations.9
Because the differential diagnoses for tinea infection are extensive, pathology or laboratory confirmation is necessary for diagnosis, and potassium hydroxide preparation often is used to diagnose dermatophyte infections.1,2 Additionally, the possibility of a hypersensitivity drug rash should remain in the differential if the patient received allergy-inducing medications prior to the outbreak, which may in turn complicate the diagnosis.
Tinea infections typically can be treated with topical antifungals such as terbinafine, butenafine,1 and luliconazole10; however, more involved cases may require oral antifungal treatment.1 Systemic treatment of tinea corporis includes itraconazole, terbinafine, and fluconazole,11 all of which exhibit fewer side effects and greater efficacy when compared to griseofulvin.12-15
Treatment of id reactions centers on the proper clearance of the dermatophyte infection, and treatment with oral antifungals generally is sufficient. In the cases of id reaction in patients with refractory otitis, some success was achieved with treatment involving immunotherapy with dermatophyte and dust mite allergen extracts coupled with a yeast elimination diet.9 In acute id reactions, topical corticosteroids and antipruritic agents can be applied.4 Rarely, systemic glucocorticoids are required, such as in cases in which the id reaction persists despite proper treatment of the primary infection.16
- Ely JW, Rosenfeld S, Seabury Stone M. Diagnosis and management of tinea infections. Am Fam Physician. 2014;90:702-710.
- Habif TP. Clinical Dermatology: A Color Guide to Diagnosis and Therapy. 5th ed. Hanover, NH: Elsevier, Inc; 2010.
- Ziemer M, Seyfarth F, Elsner P, et al. Atypical manifestations of tinea corporis. Mycoses. 2007;50(suppl 2):31-35.
- Cheng N, Rucker Wright D, Cohen BA. Dermatophytid in tinea capitis: rarely reported common phenomenon with clinical implications [published online July 4, 2011]. Pediatrics. 2011;128:e453-e457.
- Ohno S, Tanabe H, Kawasaki M, et al. Tinea corporis with acute inflammation caused by Trichophyton tonsurans. J Dermatol. 2008;35:590-593.
- Hirschmann JV, Raugi GJ. Pustular tinea pedis. J Am Acad Dermatol. 2000;42:132-133.
- Iglesias ME, España A, Idoate MA, et al. Generalized skin reaction following tinea pedis (dermatophytids). J Dermatol. 1994;21:31-34.
- Atzori L, Pau M, Aste M. Erythema multiforme ID reaction in atypical dermatophytosis: a case report. J Eur Acad Dermatol Venereol. 2003;17:699-701.
- Derebery J, Berliner KI. Foot and ear disease—the dermatophytid reaction in otology. Laryngoscope. 1996;106(2 Pt 1):181-186.
- Khanna D, Bharti S. Luliconazole for the treatment of fungal infections: an evidence-based review. Core Evid. 2014;9:113-124.
- Korting HC, Schöllmann C. The significance of itraconazole for treatment of fungal infections of skin, nails and mucous membranes. J Dtsch Dermatol Ges. 2009;7:11-20.
- Goldstein AO, Goldstein BG. Dermatophyte (tinea) infections. UpToDate website. https://www.uptodate.com/contents/dermatophyte-tinea-infections. Updated December 28, 2018. Accessed April 24, 2019.
- Cole GW, Stricklin G. A comparison of a new oral antifungal, terbinafine, with griseofulvin as therapy for tinea corporis. Arch Dermatol. 1989;125:1537.
- Panagiotidou D, Kousidou T, Chaidemenos G, et al. A comparison of itraconazole and griseofulvin in the treatment of tinea corporis and tinea cruris: a double-blind study. J Int Med Res. 1992;20:392-400.
- Faergemann J, Mörk NJ, Haglund A, et al. A multicentre (double-blind) comparative study to assess the safety and efficacy of fluconazole and griseofulvin in the treatment of tinea corporis and tinea cruris. Br J Dermatol. 1997;136:575-577.
- Ilkit M, Durdu M, Karakas M. Cutaneous id reactions: a comprehensive review of clinical manifestations, epidemiology, etiology, and management. Crit Rev Microbiol. 2012;38:191-202.
To the Editor:
A 17-year-old adolescent girl presented to the dermatology clinic with a tender pruritic rash on the left wrist that was spreading to the bilateral arms and legs of several years’ duration. An area of a prior biopsy on the left wrist was healing well with use of petroleum jelly and halcinonide cream. The patient denied any constitutional symptoms.
Physical examination revealed numerous erythematous papules coalescing into plaques on the bilateral anterior and posterior arms and legs, including some erythematous macules and papules on the palms and soles. The original area of involvement on the left dorsal medial wrist demonstrated a background of erythema with overlying peripheral scaling and resolving violaceous to erythematous papules with signs of serosanguineous crusting (Figure 1). Scattered perifollicular erythema was present on the posterior aspects of the bilateral thighs and arms (Figure 2). Baseline complete blood cell count and complete metabolic panel were within reference range.
Clinical histopathology showed evidence of a pustular superficial dermatophyte infection, and Grocott-Gomori methenamine-silver stain demonstrated numerous fungal hyphae within subcorneal pustules, indicating pustular tinea. Based on the clinicopathologic correlation, the initial presentation was diagnosed as pustular tinea of the entire left wrist, followed by a generalized id reaction 1 week later.
The patient was prescribed oral terbinafine 250 mg once daily to treat the diffuse involvement of the pustular tinea as well as once-daily oral cetirizine, once-daily oral diphenhydramine, a topical emollient, and a topical nonsteroidal antipruritic gel.
Tinea is a superficial fungal infection commonly caused by the dermatophytes Epidermophyton, Trichophyton, and Microsporum. It has a variety of clinical presentations based on the anatomic location, including tinea capitis (hair/scalp), tinea pedis (feet), tinea corporis (face/trunk/extremities), tinea cruris (groin), and tinea unguium (nails).1 Tinea infections occur in the stratum corneum, hair, and nails, thriving on dead keratin in these areas.2 Tinea corporis usually appears as an erythematous ring-shaped lesion with a scaly border, but atypical cases presenting with vesicles, pustules, and bullae also have been reported.3 Additionally, secondary eruptions called id reactions, or autoeczematization, can present in the setting of dermatophyte infections. Such outbreaks may be due to a delayed hypersensitivity reaction to the fungal antigens. Id reactions can manifest in many forms of tinea with patients generally exhibiting pruritic papulovesicular lesions that can present far from the site of origin.4
Patients with id reactions can have atypical and varied presentations. In a case of id reaction due to tinea corporis, a patient presented with vesicles and pustules that grew in number and coalesced to form annular lesions.5 A case of an id reaction caused by tinea pedis also noted the presence of pustules, which are atypical in this form of tinea.6 In another case of tinea pedis, a generalized id reaction was noted, illustrating that such eruptions do not necessarily appear at the original site of infection.7 Additionally, in a rare presentation of tinea invading the nares, a patient developed an erythema multiforme id reaction.8 Id reactions also were noted in 14 patients with refractory otitis externa, illustrating the ability of this fungal infection to persist and infect distant locations.9
Because the differential diagnoses for tinea infection are extensive, pathology or laboratory confirmation is necessary for diagnosis, and potassium hydroxide preparation often is used to diagnose dermatophyte infections.1,2 Additionally, the possibility of a hypersensitivity drug rash should remain in the differential if the patient received allergy-inducing medications prior to the outbreak, which may in turn complicate the diagnosis.
Tinea infections typically can be treated with topical antifungals such as terbinafine, butenafine,1 and luliconazole10; however, more involved cases may require oral antifungal treatment.1 Systemic treatment of tinea corporis includes itraconazole, terbinafine, and fluconazole,11 all of which exhibit fewer side effects and greater efficacy when compared to griseofulvin.12-15
Treatment of id reactions centers on the proper clearance of the dermatophyte infection, and treatment with oral antifungals generally is sufficient. In the cases of id reaction in patients with refractory otitis, some success was achieved with treatment involving immunotherapy with dermatophyte and dust mite allergen extracts coupled with a yeast elimination diet.9 In acute id reactions, topical corticosteroids and antipruritic agents can be applied.4 Rarely, systemic glucocorticoids are required, such as in cases in which the id reaction persists despite proper treatment of the primary infection.16
To the Editor:
A 17-year-old adolescent girl presented to the dermatology clinic with a tender pruritic rash on the left wrist that was spreading to the bilateral arms and legs of several years’ duration. An area of a prior biopsy on the left wrist was healing well with use of petroleum jelly and halcinonide cream. The patient denied any constitutional symptoms.
Physical examination revealed numerous erythematous papules coalescing into plaques on the bilateral anterior and posterior arms and legs, including some erythematous macules and papules on the palms and soles. The original area of involvement on the left dorsal medial wrist demonstrated a background of erythema with overlying peripheral scaling and resolving violaceous to erythematous papules with signs of serosanguineous crusting (Figure 1). Scattered perifollicular erythema was present on the posterior aspects of the bilateral thighs and arms (Figure 2). Baseline complete blood cell count and complete metabolic panel were within reference range.
Clinical histopathology showed evidence of a pustular superficial dermatophyte infection, and Grocott-Gomori methenamine-silver stain demonstrated numerous fungal hyphae within subcorneal pustules, indicating pustular tinea. Based on the clinicopathologic correlation, the initial presentation was diagnosed as pustular tinea of the entire left wrist, followed by a generalized id reaction 1 week later.
The patient was prescribed oral terbinafine 250 mg once daily to treat the diffuse involvement of the pustular tinea as well as once-daily oral cetirizine, once-daily oral diphenhydramine, a topical emollient, and a topical nonsteroidal antipruritic gel.
Tinea is a superficial fungal infection commonly caused by the dermatophytes Epidermophyton, Trichophyton, and Microsporum. It has a variety of clinical presentations based on the anatomic location, including tinea capitis (hair/scalp), tinea pedis (feet), tinea corporis (face/trunk/extremities), tinea cruris (groin), and tinea unguium (nails).1 Tinea infections occur in the stratum corneum, hair, and nails, thriving on dead keratin in these areas.2 Tinea corporis usually appears as an erythematous ring-shaped lesion with a scaly border, but atypical cases presenting with vesicles, pustules, and bullae also have been reported.3 Additionally, secondary eruptions called id reactions, or autoeczematization, can present in the setting of dermatophyte infections. Such outbreaks may be due to a delayed hypersensitivity reaction to the fungal antigens. Id reactions can manifest in many forms of tinea with patients generally exhibiting pruritic papulovesicular lesions that can present far from the site of origin.4
Patients with id reactions can have atypical and varied presentations. In a case of id reaction due to tinea corporis, a patient presented with vesicles and pustules that grew in number and coalesced to form annular lesions.5 A case of an id reaction caused by tinea pedis also noted the presence of pustules, which are atypical in this form of tinea.6 In another case of tinea pedis, a generalized id reaction was noted, illustrating that such eruptions do not necessarily appear at the original site of infection.7 Additionally, in a rare presentation of tinea invading the nares, a patient developed an erythema multiforme id reaction.8 Id reactions also were noted in 14 patients with refractory otitis externa, illustrating the ability of this fungal infection to persist and infect distant locations.9
Because the differential diagnoses for tinea infection are extensive, pathology or laboratory confirmation is necessary for diagnosis, and potassium hydroxide preparation often is used to diagnose dermatophyte infections.1,2 Additionally, the possibility of a hypersensitivity drug rash should remain in the differential if the patient received allergy-inducing medications prior to the outbreak, which may in turn complicate the diagnosis.
Tinea infections typically can be treated with topical antifungals such as terbinafine, butenafine,1 and luliconazole10; however, more involved cases may require oral antifungal treatment.1 Systemic treatment of tinea corporis includes itraconazole, terbinafine, and fluconazole,11 all of which exhibit fewer side effects and greater efficacy when compared to griseofulvin.12-15
Treatment of id reactions centers on the proper clearance of the dermatophyte infection, and treatment with oral antifungals generally is sufficient. In the cases of id reaction in patients with refractory otitis, some success was achieved with treatment involving immunotherapy with dermatophyte and dust mite allergen extracts coupled with a yeast elimination diet.9 In acute id reactions, topical corticosteroids and antipruritic agents can be applied.4 Rarely, systemic glucocorticoids are required, such as in cases in which the id reaction persists despite proper treatment of the primary infection.16
- Ely JW, Rosenfeld S, Seabury Stone M. Diagnosis and management of tinea infections. Am Fam Physician. 2014;90:702-710.
- Habif TP. Clinical Dermatology: A Color Guide to Diagnosis and Therapy. 5th ed. Hanover, NH: Elsevier, Inc; 2010.
- Ziemer M, Seyfarth F, Elsner P, et al. Atypical manifestations of tinea corporis. Mycoses. 2007;50(suppl 2):31-35.
- Cheng N, Rucker Wright D, Cohen BA. Dermatophytid in tinea capitis: rarely reported common phenomenon with clinical implications [published online July 4, 2011]. Pediatrics. 2011;128:e453-e457.
- Ohno S, Tanabe H, Kawasaki M, et al. Tinea corporis with acute inflammation caused by Trichophyton tonsurans. J Dermatol. 2008;35:590-593.
- Hirschmann JV, Raugi GJ. Pustular tinea pedis. J Am Acad Dermatol. 2000;42:132-133.
- Iglesias ME, España A, Idoate MA, et al. Generalized skin reaction following tinea pedis (dermatophytids). J Dermatol. 1994;21:31-34.
- Atzori L, Pau M, Aste M. Erythema multiforme ID reaction in atypical dermatophytosis: a case report. J Eur Acad Dermatol Venereol. 2003;17:699-701.
- Derebery J, Berliner KI. Foot and ear disease—the dermatophytid reaction in otology. Laryngoscope. 1996;106(2 Pt 1):181-186.
- Khanna D, Bharti S. Luliconazole for the treatment of fungal infections: an evidence-based review. Core Evid. 2014;9:113-124.
- Korting HC, Schöllmann C. The significance of itraconazole for treatment of fungal infections of skin, nails and mucous membranes. J Dtsch Dermatol Ges. 2009;7:11-20.
- Goldstein AO, Goldstein BG. Dermatophyte (tinea) infections. UpToDate website. https://www.uptodate.com/contents/dermatophyte-tinea-infections. Updated December 28, 2018. Accessed April 24, 2019.
- Cole GW, Stricklin G. A comparison of a new oral antifungal, terbinafine, with griseofulvin as therapy for tinea corporis. Arch Dermatol. 1989;125:1537.
- Panagiotidou D, Kousidou T, Chaidemenos G, et al. A comparison of itraconazole and griseofulvin in the treatment of tinea corporis and tinea cruris: a double-blind study. J Int Med Res. 1992;20:392-400.
- Faergemann J, Mörk NJ, Haglund A, et al. A multicentre (double-blind) comparative study to assess the safety and efficacy of fluconazole and griseofulvin in the treatment of tinea corporis and tinea cruris. Br J Dermatol. 1997;136:575-577.
- Ilkit M, Durdu M, Karakas M. Cutaneous id reactions: a comprehensive review of clinical manifestations, epidemiology, etiology, and management. Crit Rev Microbiol. 2012;38:191-202.
- Ely JW, Rosenfeld S, Seabury Stone M. Diagnosis and management of tinea infections. Am Fam Physician. 2014;90:702-710.
- Habif TP. Clinical Dermatology: A Color Guide to Diagnosis and Therapy. 5th ed. Hanover, NH: Elsevier, Inc; 2010.
- Ziemer M, Seyfarth F, Elsner P, et al. Atypical manifestations of tinea corporis. Mycoses. 2007;50(suppl 2):31-35.
- Cheng N, Rucker Wright D, Cohen BA. Dermatophytid in tinea capitis: rarely reported common phenomenon with clinical implications [published online July 4, 2011]. Pediatrics. 2011;128:e453-e457.
- Ohno S, Tanabe H, Kawasaki M, et al. Tinea corporis with acute inflammation caused by Trichophyton tonsurans. J Dermatol. 2008;35:590-593.
- Hirschmann JV, Raugi GJ. Pustular tinea pedis. J Am Acad Dermatol. 2000;42:132-133.
- Iglesias ME, España A, Idoate MA, et al. Generalized skin reaction following tinea pedis (dermatophytids). J Dermatol. 1994;21:31-34.
- Atzori L, Pau M, Aste M. Erythema multiforme ID reaction in atypical dermatophytosis: a case report. J Eur Acad Dermatol Venereol. 2003;17:699-701.
- Derebery J, Berliner KI. Foot and ear disease—the dermatophytid reaction in otology. Laryngoscope. 1996;106(2 Pt 1):181-186.
- Khanna D, Bharti S. Luliconazole for the treatment of fungal infections: an evidence-based review. Core Evid. 2014;9:113-124.
- Korting HC, Schöllmann C. The significance of itraconazole for treatment of fungal infections of skin, nails and mucous membranes. J Dtsch Dermatol Ges. 2009;7:11-20.
- Goldstein AO, Goldstein BG. Dermatophyte (tinea) infections. UpToDate website. https://www.uptodate.com/contents/dermatophyte-tinea-infections. Updated December 28, 2018. Accessed April 24, 2019.
- Cole GW, Stricklin G. A comparison of a new oral antifungal, terbinafine, with griseofulvin as therapy for tinea corporis. Arch Dermatol. 1989;125:1537.
- Panagiotidou D, Kousidou T, Chaidemenos G, et al. A comparison of itraconazole and griseofulvin in the treatment of tinea corporis and tinea cruris: a double-blind study. J Int Med Res. 1992;20:392-400.
- Faergemann J, Mörk NJ, Haglund A, et al. A multicentre (double-blind) comparative study to assess the safety and efficacy of fluconazole and griseofulvin in the treatment of tinea corporis and tinea cruris. Br J Dermatol. 1997;136:575-577.
- Ilkit M, Durdu M, Karakas M. Cutaneous id reactions: a comprehensive review of clinical manifestations, epidemiology, etiology, and management. Crit Rev Microbiol. 2012;38:191-202.
Practice Points
• Id reactions, or autoeczematization, can occur secondary to dermatophyte infections, possibly due to a hypersensitivity reaction to the fungus. These eruptions can occur in many forms of tinea and in a variety of clinical presentations.
• Treatment is based on clearance of the original dermatophyte infection.
Dupilumab for Treatment of Severe Atopic Dermatitis in a Heart Transplant Recipient
To the Editor:
Solid-organ transplant recipients can develop a range of dermatologic consequences due to chronic immunosuppression, including frequent skin infections and malignancies. Atopic dermatitis (AD) and psoriasis are relatively rare in this population because many immunosuppressive therapies, such as mycophenolate mofetil and tacrolimus, also are used to treat inflammatory dermatoses.1 In a large renal transplant population, the prevalence of AD was 1.3%.2 The pathogenesis of posttransplantation AD is poorly understood, and standard treatment regimens have not been defined. Dupilumab is a novel biologic medication that has demonstrated efficacy in the treatment of AD.3 Reports of dupilumab use for AD management in solid-organ transplant recipients are limited in the literature.
A 29-year-old woman with a history of a heart transplant 4 years prior presented to our dermatology clinic with an itchy rash over the entire body. Since the transplant, she had been on long-term immunosuppression with prednisone, mycophenolate mofetil, and tacrolimus. The rash appeared after she switched from brand-name to generic versions of the medications. Physical examination revealed erythematous scaly plaques on the lateral face, back, chest, arms, and legs covering approximately 10% of the body surface area. The patient’s total serum IgE level was elevated at 711,500 µg/L (reference range, 0–1500 µg/L). Outside biopsies revealed changes consistent with spongiotic dermatitis, and patch testing performed by an outside physician was positive for sensitivity to the preservative bronopol.
The patient was switched back to brand-name tacrolimus, but the rash did not improve. Topical steroids, phototherapy, and omalizumab were ineffective. The itching was primarily managed with desoximetasone spray, mometasone cream, and loratidine. With approval from the patient’s transplant team outside of our hospital system, she was started on dupilumab 300 mg once every 14 days. Complete clearance of the rash was noted within 3 months of treatment. Besides bilateral conjunctivitis, which was treated with ophthalmic prednisolone and moxifloxacin solutions, dupilumab was well tolerated. No issues related to immunosuppressant levels or graft-related issues, including rejection, were reported at 6-, 12-, and 18-month follow-up visits.
Atopic dermatitis is characterized by activation of type 2 immune responses, skin barrier defects, and increased Staphylococcus aureus colonization.4 A potential mechanism for the development of AD in transplant recipients relates to their use of tacrolimus for chronic immunosuppression. Tacrolimus increases intestinal permeability and therefore allows greater absorption of allergens. This influx of allergens promotes hypersensitivity reactions, resulting in elevated IgE levels and eosinophilia. Tacrolimus also facilitates predominance of helper T cells (TH2 cytokines) through selective inhibition of the TH1 cytokine IL-2.5
Dupilumab is a human monoclonal antibody that blocks IL-4 and IL-13, which are key drivers of TH2-mediated inflammation. In addition to downregulation of inflammatory mediators, dupilumab also increases production of epidermal barrier proteins, resulting in skin repair. It has demonstrated rapid, dose-dependent efficacy in patients with moderate to severe AD.6 Dupilumab boasts a good safety profile with no increase in risk for skin infections compared to placebo6; however, its safety has not yet been verified in transplant recipients.
Our case is notable for the severity of the patient’s AD despite considerable immunosuppression with transplant medications. Development of AD was associated with a switch from brand-name to generic drugs, which is not commonly reported. Her condition was refractory to a litany of treatments prior to a trial of dupilumab. The rapid clearance observed with this novel biologic medication highlights its potential to provide relief to patients who have particularly tenacious cases of AD. Prior to starting dupilumab, we do recommend more extensive laboratory testing in immunosuppressed patients including transplant recipients and patients with human immunodeficiency virus. We illustrate that a history of solid-organ transplant need not exclude patients from consideration for dupilumab therapy.
- Savoia P, Cavaliere G, Zavattaro E, et al. Inflammatory cutaneous diseases in renal transplant recipients [published online August 19, 2016]. Int J Mol Sci. doi:10.3390/ijms17081362.
- Lally A, Casabonne D, Imko-Walczuk B, et al. Prevalence of benign cutaneous disease among Oxford renal transplant recipients. J Eur Acad Dermatol Venereol. 2011;25:462-470.
- Beck L, Thaci D, Hamilton JD, et al. Dupilumab treatment in adults with moderate-to-severe atopic dermatitis. N Engl J Med. 2014;371:130-139.
- Simpson EL, Bieber T, Guttman-Yassky E, et al; SOLO 1 and SOLO 2 Investigators. Two phase 3 trials of dupilumab versus placebo in atopic dermatitis. N Engl J Med. 2016;375:2335-2348.
- Machura E, Chodór B, Kleszyk M, et al. Atopic allergy and chronic inflammation of the oral mucosa in a 3-year-old boy after heart transplantation—diagnostic and therapeutic difficulties. Kardiochir Torakochirurgia Pol. 2015;12:176-180.
- Beck L, Thaci D, Hamilton JD, et al. Dupilumab treatment in adults with moderate-to-severe atopic dermatitis. N Engl J Med. 2014;371:130-139.
To the Editor:
Solid-organ transplant recipients can develop a range of dermatologic consequences due to chronic immunosuppression, including frequent skin infections and malignancies. Atopic dermatitis (AD) and psoriasis are relatively rare in this population because many immunosuppressive therapies, such as mycophenolate mofetil and tacrolimus, also are used to treat inflammatory dermatoses.1 In a large renal transplant population, the prevalence of AD was 1.3%.2 The pathogenesis of posttransplantation AD is poorly understood, and standard treatment regimens have not been defined. Dupilumab is a novel biologic medication that has demonstrated efficacy in the treatment of AD.3 Reports of dupilumab use for AD management in solid-organ transplant recipients are limited in the literature.
A 29-year-old woman with a history of a heart transplant 4 years prior presented to our dermatology clinic with an itchy rash over the entire body. Since the transplant, she had been on long-term immunosuppression with prednisone, mycophenolate mofetil, and tacrolimus. The rash appeared after she switched from brand-name to generic versions of the medications. Physical examination revealed erythematous scaly plaques on the lateral face, back, chest, arms, and legs covering approximately 10% of the body surface area. The patient’s total serum IgE level was elevated at 711,500 µg/L (reference range, 0–1500 µg/L). Outside biopsies revealed changes consistent with spongiotic dermatitis, and patch testing performed by an outside physician was positive for sensitivity to the preservative bronopol.
The patient was switched back to brand-name tacrolimus, but the rash did not improve. Topical steroids, phototherapy, and omalizumab were ineffective. The itching was primarily managed with desoximetasone spray, mometasone cream, and loratidine. With approval from the patient’s transplant team outside of our hospital system, she was started on dupilumab 300 mg once every 14 days. Complete clearance of the rash was noted within 3 months of treatment. Besides bilateral conjunctivitis, which was treated with ophthalmic prednisolone and moxifloxacin solutions, dupilumab was well tolerated. No issues related to immunosuppressant levels or graft-related issues, including rejection, were reported at 6-, 12-, and 18-month follow-up visits.
Atopic dermatitis is characterized by activation of type 2 immune responses, skin barrier defects, and increased Staphylococcus aureus colonization.4 A potential mechanism for the development of AD in transplant recipients relates to their use of tacrolimus for chronic immunosuppression. Tacrolimus increases intestinal permeability and therefore allows greater absorption of allergens. This influx of allergens promotes hypersensitivity reactions, resulting in elevated IgE levels and eosinophilia. Tacrolimus also facilitates predominance of helper T cells (TH2 cytokines) through selective inhibition of the TH1 cytokine IL-2.5
Dupilumab is a human monoclonal antibody that blocks IL-4 and IL-13, which are key drivers of TH2-mediated inflammation. In addition to downregulation of inflammatory mediators, dupilumab also increases production of epidermal barrier proteins, resulting in skin repair. It has demonstrated rapid, dose-dependent efficacy in patients with moderate to severe AD.6 Dupilumab boasts a good safety profile with no increase in risk for skin infections compared to placebo6; however, its safety has not yet been verified in transplant recipients.
Our case is notable for the severity of the patient’s AD despite considerable immunosuppression with transplant medications. Development of AD was associated with a switch from brand-name to generic drugs, which is not commonly reported. Her condition was refractory to a litany of treatments prior to a trial of dupilumab. The rapid clearance observed with this novel biologic medication highlights its potential to provide relief to patients who have particularly tenacious cases of AD. Prior to starting dupilumab, we do recommend more extensive laboratory testing in immunosuppressed patients including transplant recipients and patients with human immunodeficiency virus. We illustrate that a history of solid-organ transplant need not exclude patients from consideration for dupilumab therapy.
To the Editor:
Solid-organ transplant recipients can develop a range of dermatologic consequences due to chronic immunosuppression, including frequent skin infections and malignancies. Atopic dermatitis (AD) and psoriasis are relatively rare in this population because many immunosuppressive therapies, such as mycophenolate mofetil and tacrolimus, also are used to treat inflammatory dermatoses.1 In a large renal transplant population, the prevalence of AD was 1.3%.2 The pathogenesis of posttransplantation AD is poorly understood, and standard treatment regimens have not been defined. Dupilumab is a novel biologic medication that has demonstrated efficacy in the treatment of AD.3 Reports of dupilumab use for AD management in solid-organ transplant recipients are limited in the literature.
A 29-year-old woman with a history of a heart transplant 4 years prior presented to our dermatology clinic with an itchy rash over the entire body. Since the transplant, she had been on long-term immunosuppression with prednisone, mycophenolate mofetil, and tacrolimus. The rash appeared after she switched from brand-name to generic versions of the medications. Physical examination revealed erythematous scaly plaques on the lateral face, back, chest, arms, and legs covering approximately 10% of the body surface area. The patient’s total serum IgE level was elevated at 711,500 µg/L (reference range, 0–1500 µg/L). Outside biopsies revealed changes consistent with spongiotic dermatitis, and patch testing performed by an outside physician was positive for sensitivity to the preservative bronopol.
The patient was switched back to brand-name tacrolimus, but the rash did not improve. Topical steroids, phototherapy, and omalizumab were ineffective. The itching was primarily managed with desoximetasone spray, mometasone cream, and loratidine. With approval from the patient’s transplant team outside of our hospital system, she was started on dupilumab 300 mg once every 14 days. Complete clearance of the rash was noted within 3 months of treatment. Besides bilateral conjunctivitis, which was treated with ophthalmic prednisolone and moxifloxacin solutions, dupilumab was well tolerated. No issues related to immunosuppressant levels or graft-related issues, including rejection, were reported at 6-, 12-, and 18-month follow-up visits.
Atopic dermatitis is characterized by activation of type 2 immune responses, skin barrier defects, and increased Staphylococcus aureus colonization.4 A potential mechanism for the development of AD in transplant recipients relates to their use of tacrolimus for chronic immunosuppression. Tacrolimus increases intestinal permeability and therefore allows greater absorption of allergens. This influx of allergens promotes hypersensitivity reactions, resulting in elevated IgE levels and eosinophilia. Tacrolimus also facilitates predominance of helper T cells (TH2 cytokines) through selective inhibition of the TH1 cytokine IL-2.5
Dupilumab is a human monoclonal antibody that blocks IL-4 and IL-13, which are key drivers of TH2-mediated inflammation. In addition to downregulation of inflammatory mediators, dupilumab also increases production of epidermal barrier proteins, resulting in skin repair. It has demonstrated rapid, dose-dependent efficacy in patients with moderate to severe AD.6 Dupilumab boasts a good safety profile with no increase in risk for skin infections compared to placebo6; however, its safety has not yet been verified in transplant recipients.
Our case is notable for the severity of the patient’s AD despite considerable immunosuppression with transplant medications. Development of AD was associated with a switch from brand-name to generic drugs, which is not commonly reported. Her condition was refractory to a litany of treatments prior to a trial of dupilumab. The rapid clearance observed with this novel biologic medication highlights its potential to provide relief to patients who have particularly tenacious cases of AD. Prior to starting dupilumab, we do recommend more extensive laboratory testing in immunosuppressed patients including transplant recipients and patients with human immunodeficiency virus. We illustrate that a history of solid-organ transplant need not exclude patients from consideration for dupilumab therapy.
- Savoia P, Cavaliere G, Zavattaro E, et al. Inflammatory cutaneous diseases in renal transplant recipients [published online August 19, 2016]. Int J Mol Sci. doi:10.3390/ijms17081362.
- Lally A, Casabonne D, Imko-Walczuk B, et al. Prevalence of benign cutaneous disease among Oxford renal transplant recipients. J Eur Acad Dermatol Venereol. 2011;25:462-470.
- Beck L, Thaci D, Hamilton JD, et al. Dupilumab treatment in adults with moderate-to-severe atopic dermatitis. N Engl J Med. 2014;371:130-139.
- Simpson EL, Bieber T, Guttman-Yassky E, et al; SOLO 1 and SOLO 2 Investigators. Two phase 3 trials of dupilumab versus placebo in atopic dermatitis. N Engl J Med. 2016;375:2335-2348.
- Machura E, Chodór B, Kleszyk M, et al. Atopic allergy and chronic inflammation of the oral mucosa in a 3-year-old boy after heart transplantation—diagnostic and therapeutic difficulties. Kardiochir Torakochirurgia Pol. 2015;12:176-180.
- Beck L, Thaci D, Hamilton JD, et al. Dupilumab treatment in adults with moderate-to-severe atopic dermatitis. N Engl J Med. 2014;371:130-139.
- Savoia P, Cavaliere G, Zavattaro E, et al. Inflammatory cutaneous diseases in renal transplant recipients [published online August 19, 2016]. Int J Mol Sci. doi:10.3390/ijms17081362.
- Lally A, Casabonne D, Imko-Walczuk B, et al. Prevalence of benign cutaneous disease among Oxford renal transplant recipients. J Eur Acad Dermatol Venereol. 2011;25:462-470.
- Beck L, Thaci D, Hamilton JD, et al. Dupilumab treatment in adults with moderate-to-severe atopic dermatitis. N Engl J Med. 2014;371:130-139.
- Simpson EL, Bieber T, Guttman-Yassky E, et al; SOLO 1 and SOLO 2 Investigators. Two phase 3 trials of dupilumab versus placebo in atopic dermatitis. N Engl J Med. 2016;375:2335-2348.
- Machura E, Chodór B, Kleszyk M, et al. Atopic allergy and chronic inflammation of the oral mucosa in a 3-year-old boy after heart transplantation—diagnostic and therapeutic difficulties. Kardiochir Torakochirurgia Pol. 2015;12:176-180.
- Beck L, Thaci D, Hamilton JD, et al. Dupilumab treatment in adults with moderate-to-severe atopic dermatitis. N Engl J Med. 2014;371:130-139.
Practice Points
- Chronic tacrolimus use in solid-organ transplant recipients may increase intestinal permeability to allergens and is a potential cause for development of atopic dermatitis (AD).
- Dupilumab has the potential to provide relief from particularly tenacious cases of AD.
- History of solid-organ transplant should not be cause for exclusion from consideration for dupilumab therapy.
Linear Vulvar Lesions
The Diagnosis: Vestibular Papillomatosis
Vestibular papillomatosis (VP), the female equivalent of pearly penile papules, is characterized by multiple papules in a linear array on the labia minora and is considered a normal anatomic variant. It typically presents as monomorphous, soft, flesh-colored, filiform papules that are distributed in a symmetric fashion. In women, the papules present as linear arrays on the inner aspects of the labia minora, whereas in men, they present in a circumferential array along the sulcus of the glans penis.1 Lesions often are asymptomatic but may cause itching, burning, and dyspareunia.2 Previously believed to be associated with human papillomavirus infection,3 VP is now considered a noninfectious condition. Biopsy reveals parakeratosis and perinuclear vacuolization in the absence of true koilocytes.4,5 Dermoscopy and reflectance confocal microscopy have been used to differentiate VP from clinically similar lesions (eg, condyloma acuminatum).6,7 The prevalence of this condition is not well established; however, one study found VP in 1% of women attending genitourinary medicine clinics.3
Condyloma acuminatum, known colloquially as genital warts, is a human papillomavirus infection. Lesions tend to be painless and firm and are distributed asymmetrically with a cauliflowerlike appearance.1 Condyloma latum, found in secondary syphilis, is characterized by papules that are pale, smooth, flat topped, and moist.8 Molluscum contagiosum is an infection caused by a poxvirus presenting with flesh-colored, dome-shaped papules with central umbilication.9 The lesions of papulosquamous lichen planus are violaceous polygonal papules that affect the clitoral hood and labia minora and may cause pruritus. The cause of lichen planus is unknown; however, clinically similar lesions may occur in a lichenoid drug eruption due to certain medications.
Vestibular papillomatosis typically does not require treatment, except in symptomatic cases. To date, limited studies have reported variable treatment success utilizing destructive techniques such as CO2 laser or topical application of 5-fluorouracil or trichloroacetic acid.10
The lesions on our patient's left medial labia minora were successfully treated with low-voltage (3.0 V) electrodesiccation. Following local anesthesia with 1% lidocaine, each papule was gently electrodesiccated utilizing a standard hyfrecation electrode tip to a light gray discoloration. Postprocedural care involved only twice-daily cleansing with a gentle soap and application of petrolatum. The patient tolerated the procedure well and was satisfied with the cosmetic and functional results. She subsequently underwent treatment of the lesions on the right labia minora with equivalent treatment success.
- Moyal-Barracco M, Leibowitch M, Orth G. Vestibular papillae of the vulva. lack of evidence for human papillomavirus etiology. Arch Dermatol. 1990;126:1594-1598.
- Strand A, Wilander E, Zehbe I, et al. Vulvar papillomatosis, aceto-white lesions, and normal-looking vulvar mucosa evaluated by microscopy and human papillomavirus analysis. Gynecol Obstet Invest. 1995;40:265-270.
- Welch JM, Nayagam M, Parry G, et al. What is vestibular papillomatosis? a study of its prevalence, aetiology and natural history. Br J Obstet Gynaecol. 1993;100:939-942.
- Wilkinson EJ, Guerrero E, Daniel R, et al. Vulvar vestibulitis is rarely associated with human papillomavirus infection types 6, 11, 16, or 18. Int J Gynecol Pathol. 1993;12:344-349.
- Beznos G, Coates V, Focchi J, et al. Biomolecular study of the correlation between papillomatosis of the vulvar vestibule in adolescents and human papillomavirus. ScientificWorldJournal. 2006;6:628-636.
- Kim SH, Seo SH, Ko HC, et al. The use of dermatoscopy to differentiate vestibular papillae, a normal variant of the female external genitalia, from condyloma acuminata. J Am Acad Dermatol. 2009;60:353-355.
- Ozkur E, Falay T, Turgut Erdemir AV, et al. Vestibular papillomatosis: an important differential diagnosis of vulvar papillomas. Dermatol Online J. 2016;22. pii:13030/qt7933q377
- Chang GJ, Welton ML. Human papillomavirus, condylomata acuminata, and anal neoplasia. Clin Colon Rectal Surg. 2004;17:221-230.
- Lynch PJ, Moyal-Barracco M, Bogliatto F, et al. 2006 ISSVD classification of vulvar dermatoses: pathologic subsets and their clinical correlates. J Reprod Med. 2007;52:3-9.
- Bergeron C, Ferenczy A, Richart RM, et al. Micropapillomatosis labialis appears unrelated to human papillomavirus. Obstet Gynecol. 1990;76:281-286.
The Diagnosis: Vestibular Papillomatosis
Vestibular papillomatosis (VP), the female equivalent of pearly penile papules, is characterized by multiple papules in a linear array on the labia minora and is considered a normal anatomic variant. It typically presents as monomorphous, soft, flesh-colored, filiform papules that are distributed in a symmetric fashion. In women, the papules present as linear arrays on the inner aspects of the labia minora, whereas in men, they present in a circumferential array along the sulcus of the glans penis.1 Lesions often are asymptomatic but may cause itching, burning, and dyspareunia.2 Previously believed to be associated with human papillomavirus infection,3 VP is now considered a noninfectious condition. Biopsy reveals parakeratosis and perinuclear vacuolization in the absence of true koilocytes.4,5 Dermoscopy and reflectance confocal microscopy have been used to differentiate VP from clinically similar lesions (eg, condyloma acuminatum).6,7 The prevalence of this condition is not well established; however, one study found VP in 1% of women attending genitourinary medicine clinics.3
Condyloma acuminatum, known colloquially as genital warts, is a human papillomavirus infection. Lesions tend to be painless and firm and are distributed asymmetrically with a cauliflowerlike appearance.1 Condyloma latum, found in secondary syphilis, is characterized by papules that are pale, smooth, flat topped, and moist.8 Molluscum contagiosum is an infection caused by a poxvirus presenting with flesh-colored, dome-shaped papules with central umbilication.9 The lesions of papulosquamous lichen planus are violaceous polygonal papules that affect the clitoral hood and labia minora and may cause pruritus. The cause of lichen planus is unknown; however, clinically similar lesions may occur in a lichenoid drug eruption due to certain medications.
Vestibular papillomatosis typically does not require treatment, except in symptomatic cases. To date, limited studies have reported variable treatment success utilizing destructive techniques such as CO2 laser or topical application of 5-fluorouracil or trichloroacetic acid.10
The lesions on our patient's left medial labia minora were successfully treated with low-voltage (3.0 V) electrodesiccation. Following local anesthesia with 1% lidocaine, each papule was gently electrodesiccated utilizing a standard hyfrecation electrode tip to a light gray discoloration. Postprocedural care involved only twice-daily cleansing with a gentle soap and application of petrolatum. The patient tolerated the procedure well and was satisfied with the cosmetic and functional results. She subsequently underwent treatment of the lesions on the right labia minora with equivalent treatment success.
The Diagnosis: Vestibular Papillomatosis
Vestibular papillomatosis (VP), the female equivalent of pearly penile papules, is characterized by multiple papules in a linear array on the labia minora and is considered a normal anatomic variant. It typically presents as monomorphous, soft, flesh-colored, filiform papules that are distributed in a symmetric fashion. In women, the papules present as linear arrays on the inner aspects of the labia minora, whereas in men, they present in a circumferential array along the sulcus of the glans penis.1 Lesions often are asymptomatic but may cause itching, burning, and dyspareunia.2 Previously believed to be associated with human papillomavirus infection,3 VP is now considered a noninfectious condition. Biopsy reveals parakeratosis and perinuclear vacuolization in the absence of true koilocytes.4,5 Dermoscopy and reflectance confocal microscopy have been used to differentiate VP from clinically similar lesions (eg, condyloma acuminatum).6,7 The prevalence of this condition is not well established; however, one study found VP in 1% of women attending genitourinary medicine clinics.3
Condyloma acuminatum, known colloquially as genital warts, is a human papillomavirus infection. Lesions tend to be painless and firm and are distributed asymmetrically with a cauliflowerlike appearance.1 Condyloma latum, found in secondary syphilis, is characterized by papules that are pale, smooth, flat topped, and moist.8 Molluscum contagiosum is an infection caused by a poxvirus presenting with flesh-colored, dome-shaped papules with central umbilication.9 The lesions of papulosquamous lichen planus are violaceous polygonal papules that affect the clitoral hood and labia minora and may cause pruritus. The cause of lichen planus is unknown; however, clinically similar lesions may occur in a lichenoid drug eruption due to certain medications.
Vestibular papillomatosis typically does not require treatment, except in symptomatic cases. To date, limited studies have reported variable treatment success utilizing destructive techniques such as CO2 laser or topical application of 5-fluorouracil or trichloroacetic acid.10
The lesions on our patient's left medial labia minora were successfully treated with low-voltage (3.0 V) electrodesiccation. Following local anesthesia with 1% lidocaine, each papule was gently electrodesiccated utilizing a standard hyfrecation electrode tip to a light gray discoloration. Postprocedural care involved only twice-daily cleansing with a gentle soap and application of petrolatum. The patient tolerated the procedure well and was satisfied with the cosmetic and functional results. She subsequently underwent treatment of the lesions on the right labia minora with equivalent treatment success.
- Moyal-Barracco M, Leibowitch M, Orth G. Vestibular papillae of the vulva. lack of evidence for human papillomavirus etiology. Arch Dermatol. 1990;126:1594-1598.
- Strand A, Wilander E, Zehbe I, et al. Vulvar papillomatosis, aceto-white lesions, and normal-looking vulvar mucosa evaluated by microscopy and human papillomavirus analysis. Gynecol Obstet Invest. 1995;40:265-270.
- Welch JM, Nayagam M, Parry G, et al. What is vestibular papillomatosis? a study of its prevalence, aetiology and natural history. Br J Obstet Gynaecol. 1993;100:939-942.
- Wilkinson EJ, Guerrero E, Daniel R, et al. Vulvar vestibulitis is rarely associated with human papillomavirus infection types 6, 11, 16, or 18. Int J Gynecol Pathol. 1993;12:344-349.
- Beznos G, Coates V, Focchi J, et al. Biomolecular study of the correlation between papillomatosis of the vulvar vestibule in adolescents and human papillomavirus. ScientificWorldJournal. 2006;6:628-636.
- Kim SH, Seo SH, Ko HC, et al. The use of dermatoscopy to differentiate vestibular papillae, a normal variant of the female external genitalia, from condyloma acuminata. J Am Acad Dermatol. 2009;60:353-355.
- Ozkur E, Falay T, Turgut Erdemir AV, et al. Vestibular papillomatosis: an important differential diagnosis of vulvar papillomas. Dermatol Online J. 2016;22. pii:13030/qt7933q377
- Chang GJ, Welton ML. Human papillomavirus, condylomata acuminata, and anal neoplasia. Clin Colon Rectal Surg. 2004;17:221-230.
- Lynch PJ, Moyal-Barracco M, Bogliatto F, et al. 2006 ISSVD classification of vulvar dermatoses: pathologic subsets and their clinical correlates. J Reprod Med. 2007;52:3-9.
- Bergeron C, Ferenczy A, Richart RM, et al. Micropapillomatosis labialis appears unrelated to human papillomavirus. Obstet Gynecol. 1990;76:281-286.
- Moyal-Barracco M, Leibowitch M, Orth G. Vestibular papillae of the vulva. lack of evidence for human papillomavirus etiology. Arch Dermatol. 1990;126:1594-1598.
- Strand A, Wilander E, Zehbe I, et al. Vulvar papillomatosis, aceto-white lesions, and normal-looking vulvar mucosa evaluated by microscopy and human papillomavirus analysis. Gynecol Obstet Invest. 1995;40:265-270.
- Welch JM, Nayagam M, Parry G, et al. What is vestibular papillomatosis? a study of its prevalence, aetiology and natural history. Br J Obstet Gynaecol. 1993;100:939-942.
- Wilkinson EJ, Guerrero E, Daniel R, et al. Vulvar vestibulitis is rarely associated with human papillomavirus infection types 6, 11, 16, or 18. Int J Gynecol Pathol. 1993;12:344-349.
- Beznos G, Coates V, Focchi J, et al. Biomolecular study of the correlation between papillomatosis of the vulvar vestibule in adolescents and human papillomavirus. ScientificWorldJournal. 2006;6:628-636.
- Kim SH, Seo SH, Ko HC, et al. The use of dermatoscopy to differentiate vestibular papillae, a normal variant of the female external genitalia, from condyloma acuminata. J Am Acad Dermatol. 2009;60:353-355.
- Ozkur E, Falay T, Turgut Erdemir AV, et al. Vestibular papillomatosis: an important differential diagnosis of vulvar papillomas. Dermatol Online J. 2016;22. pii:13030/qt7933q377
- Chang GJ, Welton ML. Human papillomavirus, condylomata acuminata, and anal neoplasia. Clin Colon Rectal Surg. 2004;17:221-230.
- Lynch PJ, Moyal-Barracco M, Bogliatto F, et al. 2006 ISSVD classification of vulvar dermatoses: pathologic subsets and their clinical correlates. J Reprod Med. 2007;52:3-9.
- Bergeron C, Ferenczy A, Richart RM, et al. Micropapillomatosis labialis appears unrelated to human papillomavirus. Obstet Gynecol. 1990;76:281-286.
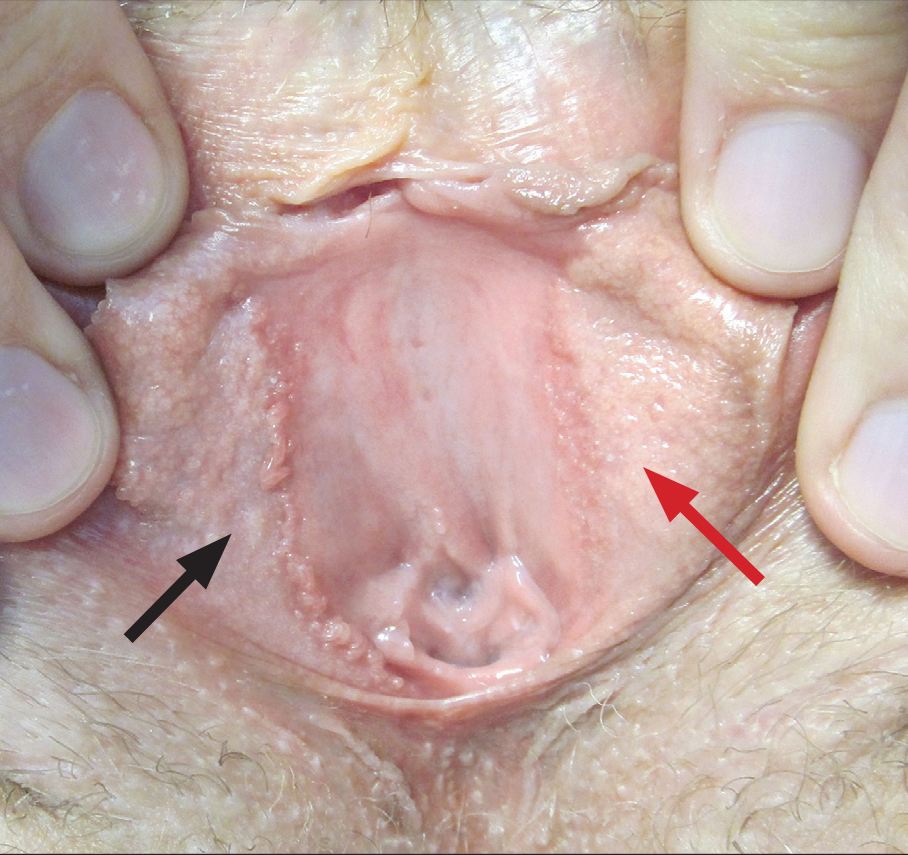
A 30-year-old woman with congenital absence of the uterus presented to dermatology for a second opinion of vulvar lesions that were first noted during adolescence. The patient reported that the lesions had not changed and were painful during sexual intercourse. The lesions were otherwise asymptomatic, and she had no additional relevant medical history or family history of similar lesions. She denied any history of sexually transmitted infections. Physical examination revealed multiple, soft, flesh-colored, 1- to 2-mm, discrete and coalescing, filiform papules distributed symmetrically in a linear array on the inner aspect of the bilateral medial labia minora. The rest of the mucocutaneous examination was normal.
The lesions on the left medial labia minora were treated with low-voltage (3.0 V) electrodesiccation following local anesthesia with 1% lidocaine (red arrow), while the lesions on the right medial labia minora were left untreated (black arrow). The clinical image shows the left labia minora approximately 1 month after treatment; the papules on the right labia minora were unchanged from the prior examination.
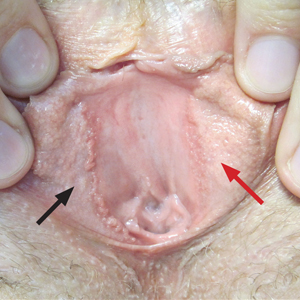
Melanocytic Matrical Carcinoma in a Solid-Organ Transplant Recipient
To the Editor:
A 68-year-old white man presented with a firm, gradually enlarging, mildly tender, grayish black papule with central ulceration on the left dorsal wrist of 4 months’ duration (Figure 1). His relevant medical history included multiple basal cell carcinomas (BCCs) and squamous cell carcinomas, as well as a single-lung transplant 2 years prior, for which he was on chronic immunosuppressive therapy with azathioprine, everolimus, tacrolimus, and prednisone. The clinical differential diagnosis included pigmented BCC, malignant melanoma, and ulcerated squamous cell carcinoma.
Histologic examination of the lesion (Figure 2) demonstrated irregular nodules of basaloid tumor cells with rounded nuclei, visible nucleoli, and scant cytoplasm involving the dermis. The tumor produced abrupt matrical-type keratinization, forming ghost cells. The lesion also contained frequent mitotic figures, apoptotic cells, focal areas of necrosis, and abundant melanin pigment. Admixed throughout the lesion were pigmented and dendritic melanocytic cells. The overlying epidermis was focally ulcerated with an adjacent localized connection between the tumor and the epidermis. Keratinocyte atypia was found in the surrounding epidermis, which contained melanophages, solar elastosis, and scattered chronic inflammatory cells. An immunohistochemical study (Figure 3) for tyrosinase demonstrated abundant admixed melanocytic cells. β-Catenin expression was shown in both nuclear and cytoplasmic distributions, and there was focal labeling on BerEP4 staining.
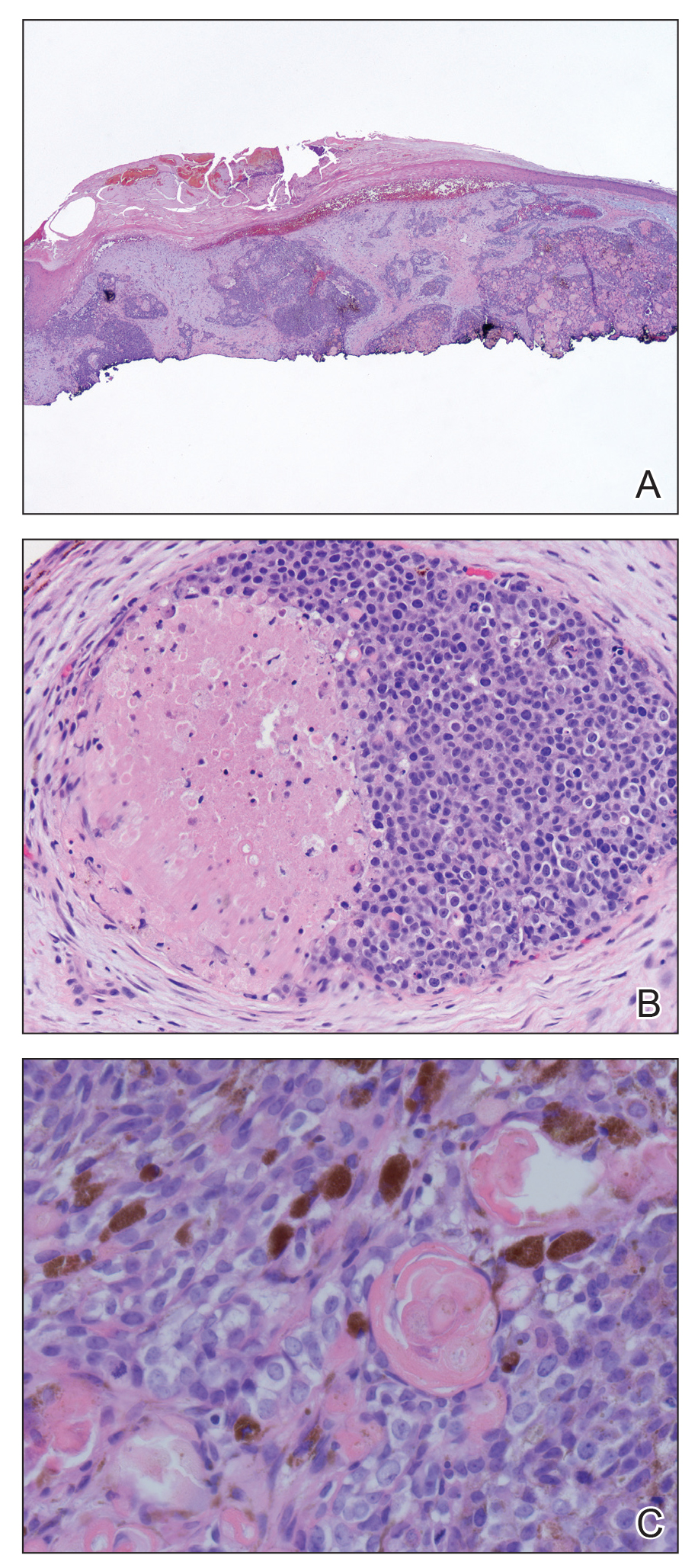
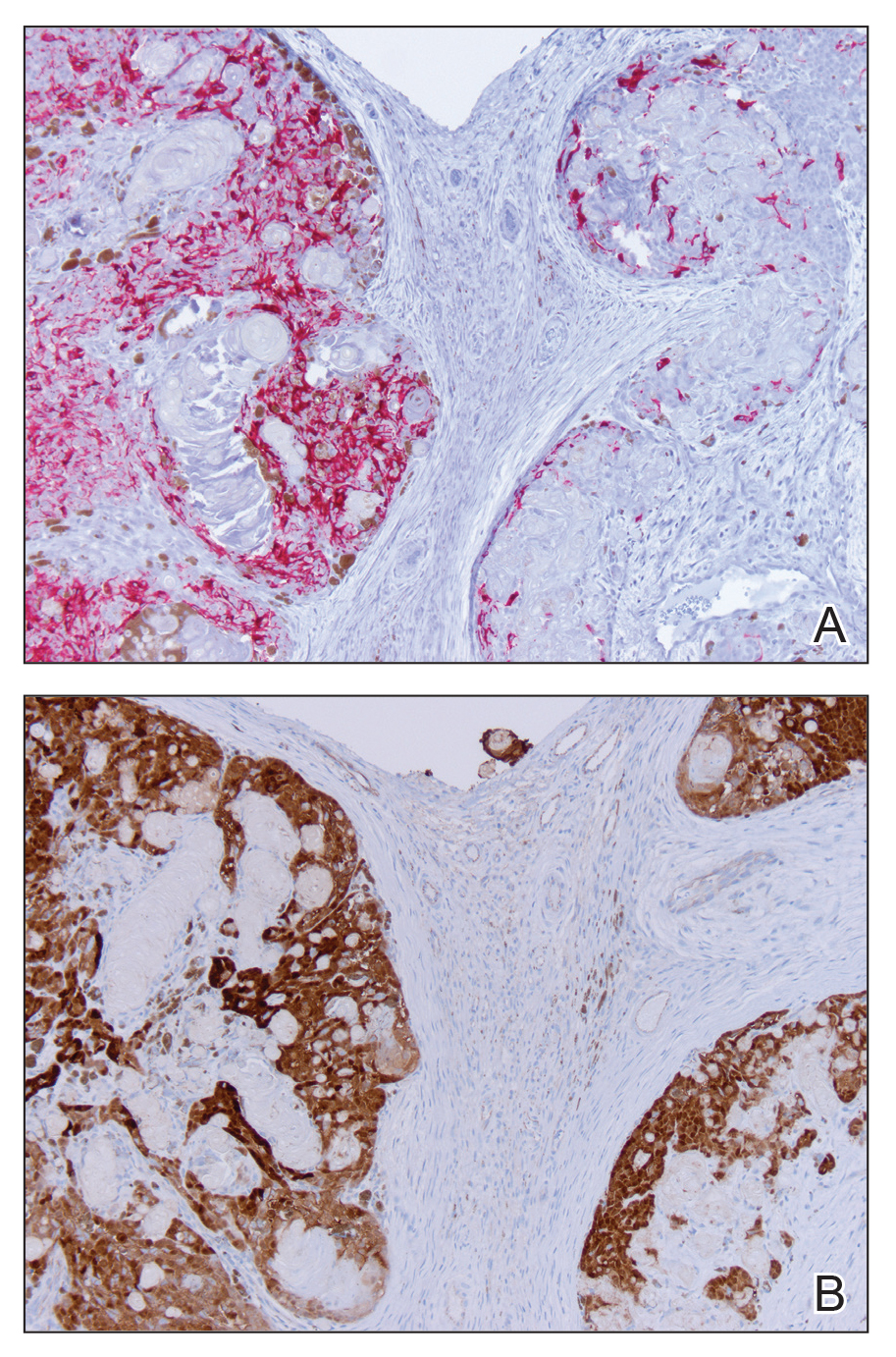
The lesion was subsequently treated with wide local excision. The patient has not had recurrence to date.
Melanocytic matricoma (MM), a rare adnexal tumor, was first described in 1999 by Carlson et al.1 A PubMed search of articles indexed for MEDLINE using the terms melanocytic and matricoma yielded 24 reported cases in the English-language literature.1-17 It consists of an admixed population of basaloid matrical and supramatrical cells, ghost cells, and dendritic melanocytes in a well-circumscribed dermal nodule, typically without epidermal or adnexal connection. In comparison to the more commonly described pilomatricoma, which can be uncommonly pigmented, MM typically has only focal areas of ghost cells and lacks cystic architecture.1,9,10,18 A granulomatous reaction to keratinaceous debris is variably present.1,9,10 Histologically, the scattered dendritic melanocytes are classically benign, but cases demonstrating melanocyte atypia have been reported.10,13 Melanocytic matricoma appears most commonly as a black or gray papule on sun-damaged skin in older men and tends not to recur following complete excision; thus, MM is considered to be a clinically benign neoplasm. Given the demographics and distribution of the lesions, exposure to UV radiation is thought to play a contributory role in the pathogenesis.2,10,19 Melanocytic matricoma is believed to recapitulate the hair follicle in the anagen phase, where there is close interplay between matrical keratinocytes and melanocytes prior to cessation of melanogenesis during the catagen phase.5,6,8,20,21 Evidence demonstrating highly conserved β-catenin and downstream lymphoid enhancer binding factor 1 (LEF1) expression, as well as pleckstrin homology-like domain, family A, member 1 (PHLDA1) expression (as a marker for follicular stem cells), points to constitutive activity in the Wnt signaling pathway in follicular stem cells of the bulge area as a major agent of tumorigenesis.12
Melanocytic matrical carcinoma, also known as malignant MM or matrical carcinoma with melanocytic hyperplasia, may be considered the malignant counterpart to MM.22 A PubMed search of articles indexed for MEDLINE using the terms melanocytic matrical carcinoma, malignant melanocytic matricoma, and matrical carcinoma with melanocytic hyperplasia, with review of references to identify additional citations, yielded 13 reported cases of MMC in the English-language literature (Table).19,22-30 As with MM, MMC is a biphasic tumor with basaloid matrical and supramatrical cells; focal areas of ghost cells; and admixed, banal-appearing dendritic melanocytes. However, the basaloid component also demonstrates nuclear atypia, mitoses, occasional ulceration, and variably poor circumscription. Clinically these lesions can mimic pigmented BCC, malignant melanoma, or other malignant adnexal tumors.25 Their natural history is unknown due to few reported cases, but they can be correlated with matrical carcinomas, which were first described by Weedon et al31 in 1980. A summary of more than 130 cases of matrical carcinomas in the English-language literature found that MMCs have high rates of local recurrence and metastasize in approximately 13% of cases. Wide local excision demonstrated lower rates of recurrence than simple excision (23% vs 83%), but there were insufficient cases to determine the incidence following Mohs micrographic surgery.32 Melanocytic matrical carcinomas also demonstrate mutations in the β-catenin pathway,pointing to a similar pathogenesis as their benign counterparts or perhaps direct malignant transformation.25,33,34
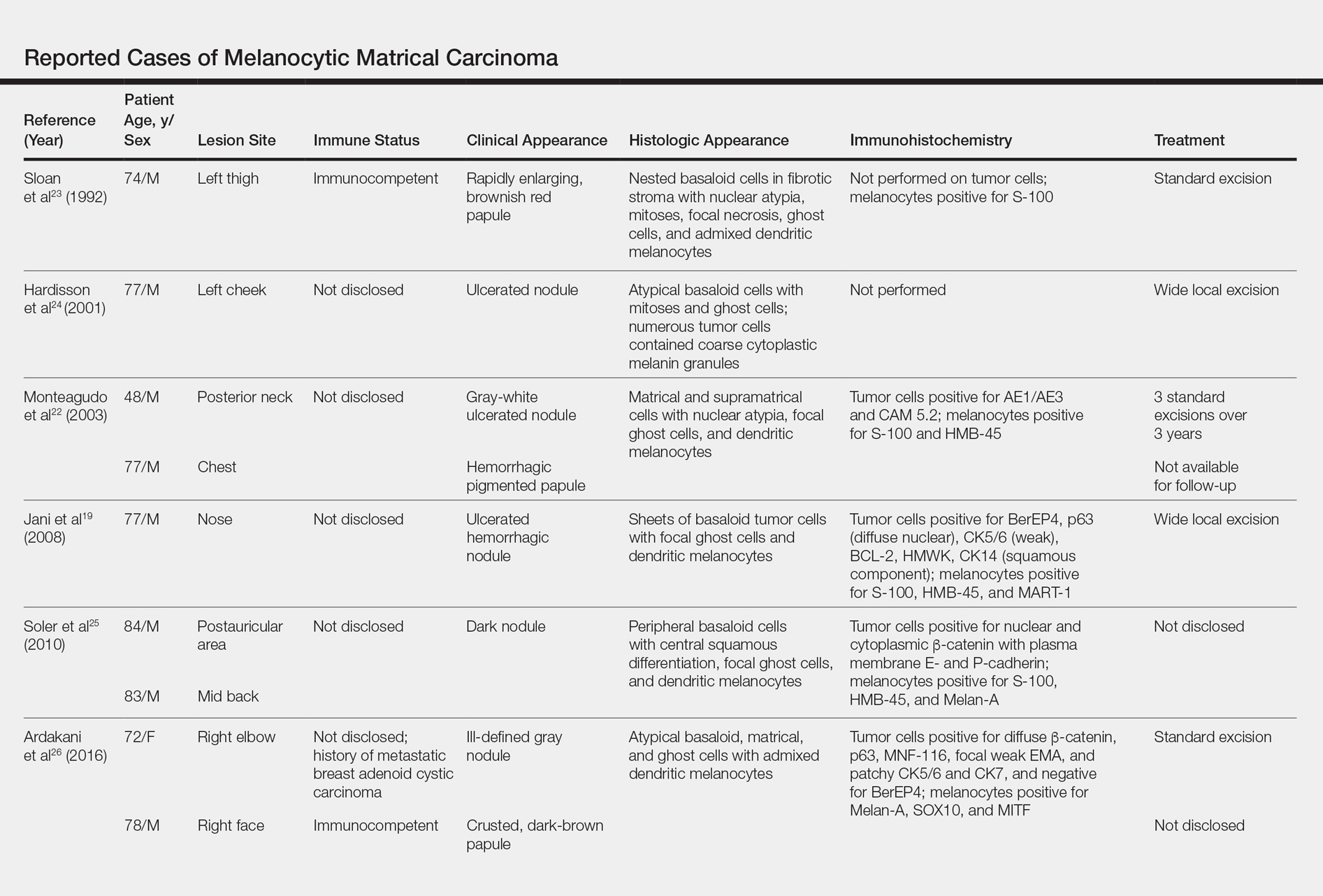
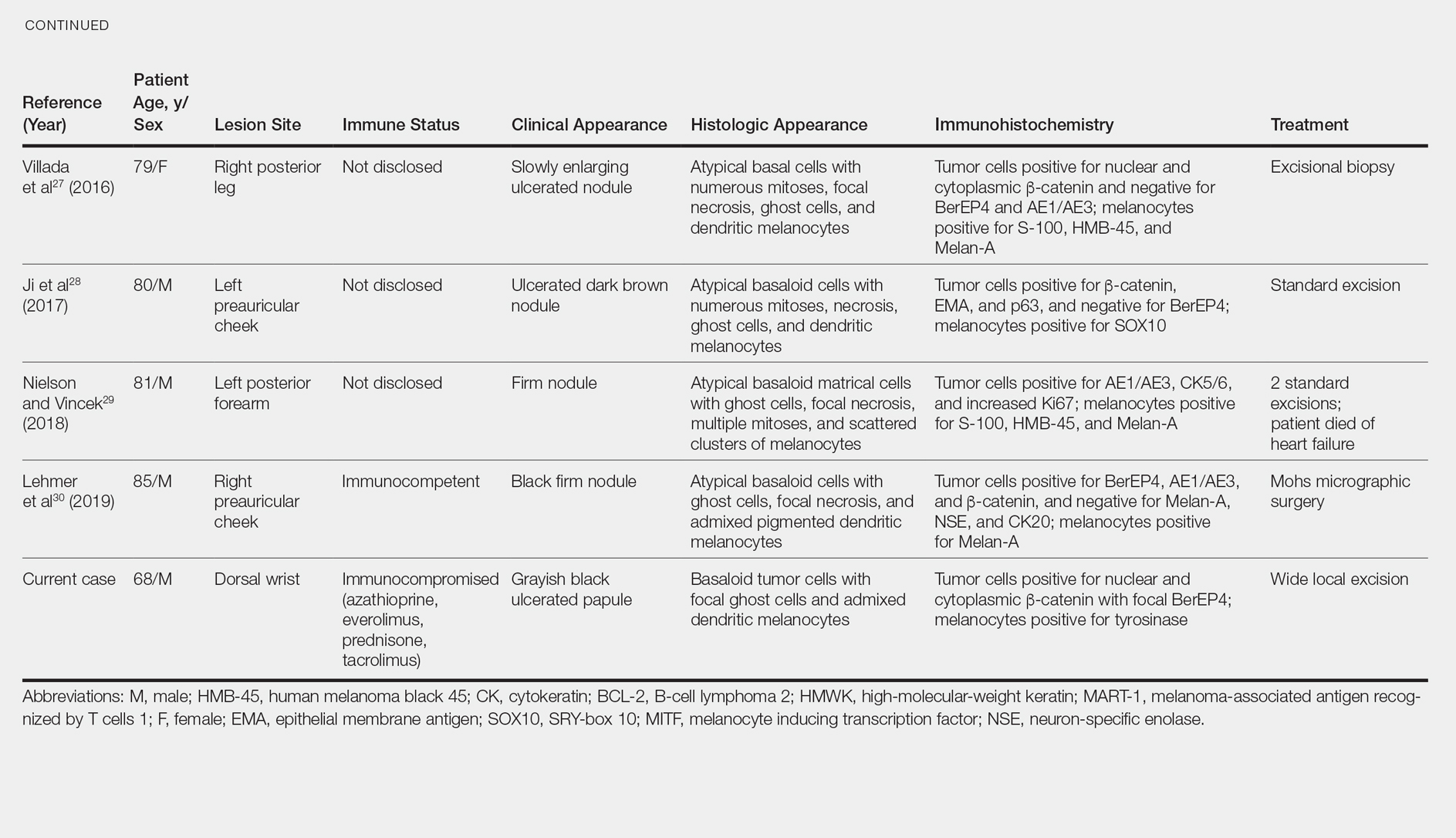
A subset of MMCs are combined cutaneous tumors (CCTs) consisting of epithelial neoplasms in close association with malignant melanocytes. Two of the more common variants include dermal squamomelanocytic tumors, a term first used by Pool et al,35 and malignant basomelanocytic tumors, as named by Erickson et al,36 but trichoblastomelanomas and other types have been documented.37 Although CCTs typically occur in the same patient populations as MMCs, namely elderly white men with chronically sun-damaged skin,they exhibit several important distinctions.37-39 By definition, CCTs have a malignant melanocytic component, whereas melanocytes are nonneoplastic in MMCs. The pathogenesis may differ as well. Various mechanisms for the close association of epithelial tumors and melanoma have been proposed, including field cancerization, tumor collision, tumor-tumor metastases, tumor colonization, and others, though CCTs likely arise through combinations of these processes depending upon their subtype.37-39 Paracrine signaling may play an important role in the pathogenesis of both tumors.5,6,8,38 As with MMCs, the prognosis of CCTs is limited by relatively few reported cases. Despite advanced Breslow depths in many cases, these tumors display more indolent behavior suggestive of melanoma in situ rather than invasive melanoma, perhaps due to dependence upon epithelial paracrine factors.37,39-42
Solid-organ transplant recipients have higher rates of more aggressive malignancies, of which skin cancer is the most common.43-49 Squamous cell carcinoma of the skin accounts for 95% of cutaneous malignancies in this population and occurs at approximately 65 times the rate of the general population.50 The risk of other skin cancers also is increased, though less dramatically, including BCC (10-fold increased risk) and melanoma (2- to 8-fold increased risk).46,50-53 The cause likely is multifactorial, including older age, history of skin cancer pretransplant, more than 5 years posttransplant, male sex, and incrementally as Fitzpatrick skin type decreases from VI to I.54-56 Immunosuppressive therapy also plays a role in tumorigenesis. Azathioprine metabolites have specifically been implicated in UVA radiation–induced promutagenic oxidative damage to DNA.57 Other studies have found no significant differences in the type of immunosuppressant used but instead have correlated rates of skin cancer to overall immunosuppression.48,55,58 Lung transplant recipients in particular demonstrate high rates of cutaneous malignancy, likely due in part to the necessity of more potent immunosuppressive regimens. Nearly one-third of patients develop a cutaneous malignancy by 5 years and nearly half by 10 years posttransplant.55
We report a rare case of MMC in a solid-organ transplant recipient. We hypothesize that the combination of UV radiation exposure–induced photodamage acquired pretransplant in addition to an aggressive immunosuppressive regimen with azathioprine and other agents posttransplant contributed to the development of this patient’s rare malignancy. Although rare, these tumors should remain in the differential diagnosis of clinicians and pathologists caring for this unique patient population.
- Carlson JA, Healy K, Slominski A, et al. Melanocytic matricoma: a report of two cases of a new entity. Am J Dermatopathol. 1999;21:344-349.
- Rizzardi C, Brollo A, Colonna A, et al. A tumor with composite pilo-folliculosebaceous differentiation harboring a recently described new entity—melanocytic matricoma. Am J Dermatopathol. 2002;24:493-497.
- Williams CM, Bozner P, Oliveri CV, et al. Melanocytic matricoma: case confirmation of a recently described entity. J Cutan Pathol. 2003;30:275-278.
- Horenstein MG, Kahn AG. Pathologic quiz case: a 69-year-old man with a brown-black facial papule. melanocytic matricoma. Arch Pathol Lab Med. 2004;128:e163-e164.
- Soler AP, Burchette JL, Bellet JS, et al. Cell adhesion protein expression in melanocytic matricoma. J Cutan Pathol. 2007;34:456-460.
- Islam MN, Bhattacharyya I, Proper SA, et al. Melanocytic matricoma: a distinctive clinicopathologic entity. Dermatol Surg. 2007;33:857-863.
- Monteagudo B, Requena L, Used-Aznar MM, et al. Melanocytic matricoma. Actas Dermosifiliogr. 2008;99:573-582.
- Cartaginese F, Sidoni A. Melanocytic matricoma. report of a further case with clinicopathological and immunohistochemical findings, differential diagnosis and review of the literature. Histol Histopathol. 2010;25:713-717.
- Tallon B, Cerroni L. Where pigmented pilomatricoma and melanocytic matricoma collide. Am J Dermatopathol. 2010;32:769-773.
- Zussman J, Sheth S, Ra SH, et al. Melanocytic matricoma with melanocytic atypia: report of a unique case and review of the literature. Am J Dermatopathol. 2011;33:508-512.
- Tanboon J, Manonukul J, Pattanaprichakul P. Melanocytic matricoma: two cases of a rare entity in women. J Cutan Pathol. 2014;41:775-782.
- Battistella M, Carlson JA, Oslo A, et al. Skin tumors with matrical differentiation: lessons from hair keratins, beta-catenin and PHLDA-1 expression. J Cutan Pathol. 2014;41:427-436.
- Barrado-Solis N, Moles-Poveda P, Roca-Estelles MJ, et al. Melanocytic matricoma with melanocytic atypia: report of a new case [published online February 11, 2015]. J Eur Acad Dermatol Venereol. 2016;30:859-860.
- Pagliarello C, Stanganelli I, Ricci R, et al. A pinkish-blue exophytic nodule on the arm of an elderly man: a quiz. melanocytic matricoma. Acta Derm Venereol. 2017;97:1261-1262.
- Winslow CY, Camacho I, Nousari CH. Melanocytic matricoma with consumption of the epidermis: an atypical histologic attribute or a malignant variant? Am J Dermatopathol. 2017;39:907-909.
- Sangiorgio V, Moneghini L, Tosi D, et al. A case of melanocytic matricoma with prominent mitotic activity and melanocytic hyperplasia. Int J Dermatol. 2018;57:e78-e81.
- Song J, Lu S, Wu Z. An unusual case of melanocytic matricoma in a young pregnant woman. Australas J Dermatol. 2019;60:140-141.
- Ishida M, Okabe H. Pigmented pilomatricoma: an underrecognized variant. Int J Clin Exp Pathol. 2013;6:1890-1893.
- Jani P, Chetty R, Ghazarian DM. An unusual composite pilomatrix carcinoma with intralesional melanocytes: differential diagnosis, immunohistochemical evaluation, and review of the literature. Am J Dermatopathol. 2008;30:174-177.
- Slominski A, Paus R. Melanogenesis is coupled to murine anagen: toward new concepts for the role of melanocytes and the regulation of melanogenesis in hair growth. J Invest Dermatol. 1993;101:90S-97S.
- De Berker D, Higgins CA, Jahada C, et al. Biology of hair and nails. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. China: Elsevier Saunders; 2012:1075-1092.
- Monteagudo C, Fernandez-Figueras MT, San Juan J, et al. Matrical carcinoma with prominent melanocytc hyperplasia (malignant melanocytic matricoma?). Am J Dermatopathol. 2003;25:485-489.
- Sloan JB, Sueki H, Jaworsky C. Pigmented malignant pilomatrixoma: report of a case and review of the literature. J Cutan Pathol. 1992;19:240-246.
- Hardisson D, Linares MD, Cuevas-Santos J, et al. Pilomatrix carcinoma: a clinicopathologic study of six cases and review of the literature. Am J Dermatopathol. 2001;23:394-401.
- Soler AP, Kindel SE, McCloskey G, et al. Cell-cell adhesion proteins in melanocytic pilomatrix carcinoma. Rare Tumors. 2010;2:e43-e45.
- Ardakani NM, Palmer DL, Wood BA. Malignant melanocytic matricoma: a report of 2 cases and review of the literature. Am J Dermatopathol. 2016;38:33-38.
- Villada G, Romagosa R, Miteva M, et al. Matrical carcinoma with melanocytic proliferation and prominent squamoid whorls. Am J Dermatopathol. 2016;38:e11-e14.
- Ji C, Zhang Y, Heller P, et al. Melanocytic matrical carcinoma mimicking melanoma. Am J Dermatopathol. 2017;39:903-906.
- Nielson CB, Vincek V. Malignant melanocytic matricoma and criteria for malignancy. Open J Pathol. 2018;8:94-100.
- Lehmer L, Carly SK, de Feraudy S. Matrical carcinoma with melanocytic hyperplasia mimicking nodular melanoma in an elderly Mexican male. J Cutan Pathol. 2019;46:442-446.
- Weedon D, Bell J, Mayze J. Matrical carcinoma of the skin. J Cutan Pathol. 1980;7:39-42.
- Herrmann JL, Allan A, Trapp KM, et al. Pilomatrix carcinoma: 13 new cases and review of the literature with emphasis on predictors of metastasis. J Am Acad Dermatol. 2014;71:38-43.
- Lazar AJ, Calonje E, Grayson W, et al. Pilomatrix carcinomas contain mutations in CTNNB1, the gene encoding beta-catenin. J Cutan Pathol. 2005;32:148-157.
- Hassanein AM, Glanz SM. Beta-catenin expression in benign and malignant pilomatrix neoplasms. Br J Dermatol. 2004;150:511-516.
- Pool SE, Manieei F, Clark WH Jr, et al. Dermal squamo-melanocytic tumor: a unique biphenotypic neoplasm of uncertain biological potential. Hum Pathol. 1999;30:525-529.
- Erickson LA, Myers JL, Mihm MC, et al. Malignant basomelanocytic tumor manifesting as metastatic melanoma. Am J Surg Pathol. 2004;28:1393-1396.
- Amin SM, Cooper C, Yelamos O, et al. Combined cutaneous tumors with a melanoma component: a clinical, histologic, and molecular study. J Am Acad Dermatol. 2015;73:451-460.
- Miteva M, Herschthal D, Ricotti C, et al. A rare case of a cutaneous squamomelanocytic tumor: revisiting the histogenesis of combined neoplasms. Am J Dermatopathol. 2009;31:599-603.
- Satter EK, Metcalf J, Lountzis N, et al. Tumors composed of malignant epithelial and melanocytic populations: a case series and review of the literature. J Cutan Pathol. 2009;36:211-219.
- Pouryazdanparast P, Yu L, Johnson T, et al. An unusual squamo-melanocytic tumor of uncertain biologic behavior: a variant of melanoma? Am J Dermatopathol. 2009;31:457-461.
- Burkhalter A, White W. Malignant melanoma in situ colonizing basal cell carcinoma: a simulator of invasive melanoma. Am J Dermatopathol. 1997;19:303-307.
- Papa G, Grandi G, Pascone M. Collision tumor of malignant skin cancers: a case of melanoma in basal cell carcinoma. Pathol Res Pract. 2006;202:691-694.
- Miao Y, Everly JJ, Gross TG, et al. De novo cancers arising in organ transplant recipients are associated with adverse outcomes compared with the general population. Transplantation. 2009;87:1347-1359.
- Bouwes Bavinck JN, Hardie DR, Green A, et al. The risk of skin cancer in renal transplant recipients in Queensland, Australia. a follow-up study. Transplantation. 1996;61:715-721.
- Berg D, Otley CC. Skin cancer in organ transplant recipients: epidemiology, pathogenesis, and management. J Am Acad Dermatol. 2002;47:1-17.
- Zwald FO, Brown M. Skin cancer in solid organ transplant recipients: advances in therapy and management: part I. epidemiology of skin cancer in solid organ transplant recipients. J Am Acad Dermatol. 2011;65:253-261.
- Zwald FO, Brown M. Skin cancer in solid organ transplant recipients: advances in therapy and management: part II. management of skin cancer in solid organ transplant recipients. J Am Acad Dermatol. 2011;65:263-273.
- DePry JL, Reed KB, Cook-Harris RH, et al. Iatrogenic immunosuppression and cutaneous malignancy. Clin Dermatol. 2011;29:602-613.
- Tessari G, Girolomoni G. Nonmelanoma skin cancer in solid organ transplant recipients: update on epidemiology, risk factors, and management. Dermatol Surg. 2012;38:1622-1630.
- Jensen P, Hansen S, Møller B, et al. Skin cancer in kidney and heart transplant recipients and different long-term immunosuppressive therapy regimens. J Am Acad Dermatol. 1999;40:177-186.
- Kasiske BL, Snyder JJ, Gilbertson DT, et al. Cancer after kidney transplantation in the United States. Am J Transplant. 2004;4:905-913.
- Hollenbeak CS, Todd MM, Billingsley EM, et al. Increased incidence of melanoma in renal transplantation recipients. Cancer. 2005;104:1962-1967.
- Le Mire L, Hollowood K, Gray D, et al. Melanomas in renal transplant recipients. Br J Dermatol. 2006;154:472-477.
- Gogia R, Binstock M, Hirose R, et al. Fitzpatrick skin phototype is an independent predictor of squamous cell carcinoma risk after solid organ transplantation. J Am Acad Dermatol. 2013;68:585-591.
- Rashtak S, Dierkhising RA, Kremers WK, et al. Incidence and risk factors for skin cancer following lung transplantation. J Am Acad Dermatol. 2015;72:92-98.
- Ruiz DE, Luzuriaga AM, Hsieh C. Yearly burden of skin cancer in non-Caucasian and Caucasian solid-organ transplant recipients. J Clin Aesthet Dermatol. 2015;8:16-19.
- Perrett CM, Walker SL, O’Donovan P, et al. Azathioprine treatment photosensitizes human skin to ultraviolet A radiation. Br J Dermatol. 2008;159:198-204.
- Abou Ayache R, Thierry A, Bridoux F, et al. Long-term maintenance of calcineurin inhibitor monotherapy reduces the risk for squamous cell carcinomas after kidney transplantation compared with bi- or tritherapy. Transplant Proc. 2007;39:2592-2594.
To the Editor:
A 68-year-old white man presented with a firm, gradually enlarging, mildly tender, grayish black papule with central ulceration on the left dorsal wrist of 4 months’ duration (Figure 1). His relevant medical history included multiple basal cell carcinomas (BCCs) and squamous cell carcinomas, as well as a single-lung transplant 2 years prior, for which he was on chronic immunosuppressive therapy with azathioprine, everolimus, tacrolimus, and prednisone. The clinical differential diagnosis included pigmented BCC, malignant melanoma, and ulcerated squamous cell carcinoma.
Histologic examination of the lesion (Figure 2) demonstrated irregular nodules of basaloid tumor cells with rounded nuclei, visible nucleoli, and scant cytoplasm involving the dermis. The tumor produced abrupt matrical-type keratinization, forming ghost cells. The lesion also contained frequent mitotic figures, apoptotic cells, focal areas of necrosis, and abundant melanin pigment. Admixed throughout the lesion were pigmented and dendritic melanocytic cells. The overlying epidermis was focally ulcerated with an adjacent localized connection between the tumor and the epidermis. Keratinocyte atypia was found in the surrounding epidermis, which contained melanophages, solar elastosis, and scattered chronic inflammatory cells. An immunohistochemical study (Figure 3) for tyrosinase demonstrated abundant admixed melanocytic cells. β-Catenin expression was shown in both nuclear and cytoplasmic distributions, and there was focal labeling on BerEP4 staining.
The lesion was subsequently treated with wide local excision. The patient has not had recurrence to date.
Melanocytic matricoma (MM), a rare adnexal tumor, was first described in 1999 by Carlson et al.1 A PubMed search of articles indexed for MEDLINE using the terms melanocytic and matricoma yielded 24 reported cases in the English-language literature.1-17 It consists of an admixed population of basaloid matrical and supramatrical cells, ghost cells, and dendritic melanocytes in a well-circumscribed dermal nodule, typically without epidermal or adnexal connection. In comparison to the more commonly described pilomatricoma, which can be uncommonly pigmented, MM typically has only focal areas of ghost cells and lacks cystic architecture.1,9,10,18 A granulomatous reaction to keratinaceous debris is variably present.1,9,10 Histologically, the scattered dendritic melanocytes are classically benign, but cases demonstrating melanocyte atypia have been reported.10,13 Melanocytic matricoma appears most commonly as a black or gray papule on sun-damaged skin in older men and tends not to recur following complete excision; thus, MM is considered to be a clinically benign neoplasm. Given the demographics and distribution of the lesions, exposure to UV radiation is thought to play a contributory role in the pathogenesis.2,10,19 Melanocytic matricoma is believed to recapitulate the hair follicle in the anagen phase, where there is close interplay between matrical keratinocytes and melanocytes prior to cessation of melanogenesis during the catagen phase.5,6,8,20,21 Evidence demonstrating highly conserved β-catenin and downstream lymphoid enhancer binding factor 1 (LEF1) expression, as well as pleckstrin homology-like domain, family A, member 1 (PHLDA1) expression (as a marker for follicular stem cells), points to constitutive activity in the Wnt signaling pathway in follicular stem cells of the bulge area as a major agent of tumorigenesis.12
Melanocytic matrical carcinoma, also known as malignant MM or matrical carcinoma with melanocytic hyperplasia, may be considered the malignant counterpart to MM.22 A PubMed search of articles indexed for MEDLINE using the terms melanocytic matrical carcinoma, malignant melanocytic matricoma, and matrical carcinoma with melanocytic hyperplasia, with review of references to identify additional citations, yielded 13 reported cases of MMC in the English-language literature (Table).19,22-30 As with MM, MMC is a biphasic tumor with basaloid matrical and supramatrical cells; focal areas of ghost cells; and admixed, banal-appearing dendritic melanocytes. However, the basaloid component also demonstrates nuclear atypia, mitoses, occasional ulceration, and variably poor circumscription. Clinically these lesions can mimic pigmented BCC, malignant melanoma, or other malignant adnexal tumors.25 Their natural history is unknown due to few reported cases, but they can be correlated with matrical carcinomas, which were first described by Weedon et al31 in 1980. A summary of more than 130 cases of matrical carcinomas in the English-language literature found that MMCs have high rates of local recurrence and metastasize in approximately 13% of cases. Wide local excision demonstrated lower rates of recurrence than simple excision (23% vs 83%), but there were insufficient cases to determine the incidence following Mohs micrographic surgery.32 Melanocytic matrical carcinomas also demonstrate mutations in the β-catenin pathway,pointing to a similar pathogenesis as their benign counterparts or perhaps direct malignant transformation.25,33,34
A subset of MMCs are combined cutaneous tumors (CCTs) consisting of epithelial neoplasms in close association with malignant melanocytes. Two of the more common variants include dermal squamomelanocytic tumors, a term first used by Pool et al,35 and malignant basomelanocytic tumors, as named by Erickson et al,36 but trichoblastomelanomas and other types have been documented.37 Although CCTs typically occur in the same patient populations as MMCs, namely elderly white men with chronically sun-damaged skin,they exhibit several important distinctions.37-39 By definition, CCTs have a malignant melanocytic component, whereas melanocytes are nonneoplastic in MMCs. The pathogenesis may differ as well. Various mechanisms for the close association of epithelial tumors and melanoma have been proposed, including field cancerization, tumor collision, tumor-tumor metastases, tumor colonization, and others, though CCTs likely arise through combinations of these processes depending upon their subtype.37-39 Paracrine signaling may play an important role in the pathogenesis of both tumors.5,6,8,38 As with MMCs, the prognosis of CCTs is limited by relatively few reported cases. Despite advanced Breslow depths in many cases, these tumors display more indolent behavior suggestive of melanoma in situ rather than invasive melanoma, perhaps due to dependence upon epithelial paracrine factors.37,39-42
Solid-organ transplant recipients have higher rates of more aggressive malignancies, of which skin cancer is the most common.43-49 Squamous cell carcinoma of the skin accounts for 95% of cutaneous malignancies in this population and occurs at approximately 65 times the rate of the general population.50 The risk of other skin cancers also is increased, though less dramatically, including BCC (10-fold increased risk) and melanoma (2- to 8-fold increased risk).46,50-53 The cause likely is multifactorial, including older age, history of skin cancer pretransplant, more than 5 years posttransplant, male sex, and incrementally as Fitzpatrick skin type decreases from VI to I.54-56 Immunosuppressive therapy also plays a role in tumorigenesis. Azathioprine metabolites have specifically been implicated in UVA radiation–induced promutagenic oxidative damage to DNA.57 Other studies have found no significant differences in the type of immunosuppressant used but instead have correlated rates of skin cancer to overall immunosuppression.48,55,58 Lung transplant recipients in particular demonstrate high rates of cutaneous malignancy, likely due in part to the necessity of more potent immunosuppressive regimens. Nearly one-third of patients develop a cutaneous malignancy by 5 years and nearly half by 10 years posttransplant.55
We report a rare case of MMC in a solid-organ transplant recipient. We hypothesize that the combination of UV radiation exposure–induced photodamage acquired pretransplant in addition to an aggressive immunosuppressive regimen with azathioprine and other agents posttransplant contributed to the development of this patient’s rare malignancy. Although rare, these tumors should remain in the differential diagnosis of clinicians and pathologists caring for this unique patient population.
To the Editor:
A 68-year-old white man presented with a firm, gradually enlarging, mildly tender, grayish black papule with central ulceration on the left dorsal wrist of 4 months’ duration (Figure 1). His relevant medical history included multiple basal cell carcinomas (BCCs) and squamous cell carcinomas, as well as a single-lung transplant 2 years prior, for which he was on chronic immunosuppressive therapy with azathioprine, everolimus, tacrolimus, and prednisone. The clinical differential diagnosis included pigmented BCC, malignant melanoma, and ulcerated squamous cell carcinoma.
Histologic examination of the lesion (Figure 2) demonstrated irregular nodules of basaloid tumor cells with rounded nuclei, visible nucleoli, and scant cytoplasm involving the dermis. The tumor produced abrupt matrical-type keratinization, forming ghost cells. The lesion also contained frequent mitotic figures, apoptotic cells, focal areas of necrosis, and abundant melanin pigment. Admixed throughout the lesion were pigmented and dendritic melanocytic cells. The overlying epidermis was focally ulcerated with an adjacent localized connection between the tumor and the epidermis. Keratinocyte atypia was found in the surrounding epidermis, which contained melanophages, solar elastosis, and scattered chronic inflammatory cells. An immunohistochemical study (Figure 3) for tyrosinase demonstrated abundant admixed melanocytic cells. β-Catenin expression was shown in both nuclear and cytoplasmic distributions, and there was focal labeling on BerEP4 staining.
The lesion was subsequently treated with wide local excision. The patient has not had recurrence to date.
Melanocytic matricoma (MM), a rare adnexal tumor, was first described in 1999 by Carlson et al.1 A PubMed search of articles indexed for MEDLINE using the terms melanocytic and matricoma yielded 24 reported cases in the English-language literature.1-17 It consists of an admixed population of basaloid matrical and supramatrical cells, ghost cells, and dendritic melanocytes in a well-circumscribed dermal nodule, typically without epidermal or adnexal connection. In comparison to the more commonly described pilomatricoma, which can be uncommonly pigmented, MM typically has only focal areas of ghost cells and lacks cystic architecture.1,9,10,18 A granulomatous reaction to keratinaceous debris is variably present.1,9,10 Histologically, the scattered dendritic melanocytes are classically benign, but cases demonstrating melanocyte atypia have been reported.10,13 Melanocytic matricoma appears most commonly as a black or gray papule on sun-damaged skin in older men and tends not to recur following complete excision; thus, MM is considered to be a clinically benign neoplasm. Given the demographics and distribution of the lesions, exposure to UV radiation is thought to play a contributory role in the pathogenesis.2,10,19 Melanocytic matricoma is believed to recapitulate the hair follicle in the anagen phase, where there is close interplay between matrical keratinocytes and melanocytes prior to cessation of melanogenesis during the catagen phase.5,6,8,20,21 Evidence demonstrating highly conserved β-catenin and downstream lymphoid enhancer binding factor 1 (LEF1) expression, as well as pleckstrin homology-like domain, family A, member 1 (PHLDA1) expression (as a marker for follicular stem cells), points to constitutive activity in the Wnt signaling pathway in follicular stem cells of the bulge area as a major agent of tumorigenesis.12
Melanocytic matrical carcinoma, also known as malignant MM or matrical carcinoma with melanocytic hyperplasia, may be considered the malignant counterpart to MM.22 A PubMed search of articles indexed for MEDLINE using the terms melanocytic matrical carcinoma, malignant melanocytic matricoma, and matrical carcinoma with melanocytic hyperplasia, with review of references to identify additional citations, yielded 13 reported cases of MMC in the English-language literature (Table).19,22-30 As with MM, MMC is a biphasic tumor with basaloid matrical and supramatrical cells; focal areas of ghost cells; and admixed, banal-appearing dendritic melanocytes. However, the basaloid component also demonstrates nuclear atypia, mitoses, occasional ulceration, and variably poor circumscription. Clinically these lesions can mimic pigmented BCC, malignant melanoma, or other malignant adnexal tumors.25 Their natural history is unknown due to few reported cases, but they can be correlated with matrical carcinomas, which were first described by Weedon et al31 in 1980. A summary of more than 130 cases of matrical carcinomas in the English-language literature found that MMCs have high rates of local recurrence and metastasize in approximately 13% of cases. Wide local excision demonstrated lower rates of recurrence than simple excision (23% vs 83%), but there were insufficient cases to determine the incidence following Mohs micrographic surgery.32 Melanocytic matrical carcinomas also demonstrate mutations in the β-catenin pathway,pointing to a similar pathogenesis as their benign counterparts or perhaps direct malignant transformation.25,33,34
A subset of MMCs are combined cutaneous tumors (CCTs) consisting of epithelial neoplasms in close association with malignant melanocytes. Two of the more common variants include dermal squamomelanocytic tumors, a term first used by Pool et al,35 and malignant basomelanocytic tumors, as named by Erickson et al,36 but trichoblastomelanomas and other types have been documented.37 Although CCTs typically occur in the same patient populations as MMCs, namely elderly white men with chronically sun-damaged skin,they exhibit several important distinctions.37-39 By definition, CCTs have a malignant melanocytic component, whereas melanocytes are nonneoplastic in MMCs. The pathogenesis may differ as well. Various mechanisms for the close association of epithelial tumors and melanoma have been proposed, including field cancerization, tumor collision, tumor-tumor metastases, tumor colonization, and others, though CCTs likely arise through combinations of these processes depending upon their subtype.37-39 Paracrine signaling may play an important role in the pathogenesis of both tumors.5,6,8,38 As with MMCs, the prognosis of CCTs is limited by relatively few reported cases. Despite advanced Breslow depths in many cases, these tumors display more indolent behavior suggestive of melanoma in situ rather than invasive melanoma, perhaps due to dependence upon epithelial paracrine factors.37,39-42
Solid-organ transplant recipients have higher rates of more aggressive malignancies, of which skin cancer is the most common.43-49 Squamous cell carcinoma of the skin accounts for 95% of cutaneous malignancies in this population and occurs at approximately 65 times the rate of the general population.50 The risk of other skin cancers also is increased, though less dramatically, including BCC (10-fold increased risk) and melanoma (2- to 8-fold increased risk).46,50-53 The cause likely is multifactorial, including older age, history of skin cancer pretransplant, more than 5 years posttransplant, male sex, and incrementally as Fitzpatrick skin type decreases from VI to I.54-56 Immunosuppressive therapy also plays a role in tumorigenesis. Azathioprine metabolites have specifically been implicated in UVA radiation–induced promutagenic oxidative damage to DNA.57 Other studies have found no significant differences in the type of immunosuppressant used but instead have correlated rates of skin cancer to overall immunosuppression.48,55,58 Lung transplant recipients in particular demonstrate high rates of cutaneous malignancy, likely due in part to the necessity of more potent immunosuppressive regimens. Nearly one-third of patients develop a cutaneous malignancy by 5 years and nearly half by 10 years posttransplant.55
We report a rare case of MMC in a solid-organ transplant recipient. We hypothesize that the combination of UV radiation exposure–induced photodamage acquired pretransplant in addition to an aggressive immunosuppressive regimen with azathioprine and other agents posttransplant contributed to the development of this patient’s rare malignancy. Although rare, these tumors should remain in the differential diagnosis of clinicians and pathologists caring for this unique patient population.
- Carlson JA, Healy K, Slominski A, et al. Melanocytic matricoma: a report of two cases of a new entity. Am J Dermatopathol. 1999;21:344-349.
- Rizzardi C, Brollo A, Colonna A, et al. A tumor with composite pilo-folliculosebaceous differentiation harboring a recently described new entity—melanocytic matricoma. Am J Dermatopathol. 2002;24:493-497.
- Williams CM, Bozner P, Oliveri CV, et al. Melanocytic matricoma: case confirmation of a recently described entity. J Cutan Pathol. 2003;30:275-278.
- Horenstein MG, Kahn AG. Pathologic quiz case: a 69-year-old man with a brown-black facial papule. melanocytic matricoma. Arch Pathol Lab Med. 2004;128:e163-e164.
- Soler AP, Burchette JL, Bellet JS, et al. Cell adhesion protein expression in melanocytic matricoma. J Cutan Pathol. 2007;34:456-460.
- Islam MN, Bhattacharyya I, Proper SA, et al. Melanocytic matricoma: a distinctive clinicopathologic entity. Dermatol Surg. 2007;33:857-863.
- Monteagudo B, Requena L, Used-Aznar MM, et al. Melanocytic matricoma. Actas Dermosifiliogr. 2008;99:573-582.
- Cartaginese F, Sidoni A. Melanocytic matricoma. report of a further case with clinicopathological and immunohistochemical findings, differential diagnosis and review of the literature. Histol Histopathol. 2010;25:713-717.
- Tallon B, Cerroni L. Where pigmented pilomatricoma and melanocytic matricoma collide. Am J Dermatopathol. 2010;32:769-773.
- Zussman J, Sheth S, Ra SH, et al. Melanocytic matricoma with melanocytic atypia: report of a unique case and review of the literature. Am J Dermatopathol. 2011;33:508-512.
- Tanboon J, Manonukul J, Pattanaprichakul P. Melanocytic matricoma: two cases of a rare entity in women. J Cutan Pathol. 2014;41:775-782.
- Battistella M, Carlson JA, Oslo A, et al. Skin tumors with matrical differentiation: lessons from hair keratins, beta-catenin and PHLDA-1 expression. J Cutan Pathol. 2014;41:427-436.
- Barrado-Solis N, Moles-Poveda P, Roca-Estelles MJ, et al. Melanocytic matricoma with melanocytic atypia: report of a new case [published online February 11, 2015]. J Eur Acad Dermatol Venereol. 2016;30:859-860.
- Pagliarello C, Stanganelli I, Ricci R, et al. A pinkish-blue exophytic nodule on the arm of an elderly man: a quiz. melanocytic matricoma. Acta Derm Venereol. 2017;97:1261-1262.
- Winslow CY, Camacho I, Nousari CH. Melanocytic matricoma with consumption of the epidermis: an atypical histologic attribute or a malignant variant? Am J Dermatopathol. 2017;39:907-909.
- Sangiorgio V, Moneghini L, Tosi D, et al. A case of melanocytic matricoma with prominent mitotic activity and melanocytic hyperplasia. Int J Dermatol. 2018;57:e78-e81.
- Song J, Lu S, Wu Z. An unusual case of melanocytic matricoma in a young pregnant woman. Australas J Dermatol. 2019;60:140-141.
- Ishida M, Okabe H. Pigmented pilomatricoma: an underrecognized variant. Int J Clin Exp Pathol. 2013;6:1890-1893.
- Jani P, Chetty R, Ghazarian DM. An unusual composite pilomatrix carcinoma with intralesional melanocytes: differential diagnosis, immunohistochemical evaluation, and review of the literature. Am J Dermatopathol. 2008;30:174-177.
- Slominski A, Paus R. Melanogenesis is coupled to murine anagen: toward new concepts for the role of melanocytes and the regulation of melanogenesis in hair growth. J Invest Dermatol. 1993;101:90S-97S.
- De Berker D, Higgins CA, Jahada C, et al. Biology of hair and nails. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. China: Elsevier Saunders; 2012:1075-1092.
- Monteagudo C, Fernandez-Figueras MT, San Juan J, et al. Matrical carcinoma with prominent melanocytc hyperplasia (malignant melanocytic matricoma?). Am J Dermatopathol. 2003;25:485-489.
- Sloan JB, Sueki H, Jaworsky C. Pigmented malignant pilomatrixoma: report of a case and review of the literature. J Cutan Pathol. 1992;19:240-246.
- Hardisson D, Linares MD, Cuevas-Santos J, et al. Pilomatrix carcinoma: a clinicopathologic study of six cases and review of the literature. Am J Dermatopathol. 2001;23:394-401.
- Soler AP, Kindel SE, McCloskey G, et al. Cell-cell adhesion proteins in melanocytic pilomatrix carcinoma. Rare Tumors. 2010;2:e43-e45.
- Ardakani NM, Palmer DL, Wood BA. Malignant melanocytic matricoma: a report of 2 cases and review of the literature. Am J Dermatopathol. 2016;38:33-38.
- Villada G, Romagosa R, Miteva M, et al. Matrical carcinoma with melanocytic proliferation and prominent squamoid whorls. Am J Dermatopathol. 2016;38:e11-e14.
- Ji C, Zhang Y, Heller P, et al. Melanocytic matrical carcinoma mimicking melanoma. Am J Dermatopathol. 2017;39:903-906.
- Nielson CB, Vincek V. Malignant melanocytic matricoma and criteria for malignancy. Open J Pathol. 2018;8:94-100.
- Lehmer L, Carly SK, de Feraudy S. Matrical carcinoma with melanocytic hyperplasia mimicking nodular melanoma in an elderly Mexican male. J Cutan Pathol. 2019;46:442-446.
- Weedon D, Bell J, Mayze J. Matrical carcinoma of the skin. J Cutan Pathol. 1980;7:39-42.
- Herrmann JL, Allan A, Trapp KM, et al. Pilomatrix carcinoma: 13 new cases and review of the literature with emphasis on predictors of metastasis. J Am Acad Dermatol. 2014;71:38-43.
- Lazar AJ, Calonje E, Grayson W, et al. Pilomatrix carcinomas contain mutations in CTNNB1, the gene encoding beta-catenin. J Cutan Pathol. 2005;32:148-157.
- Hassanein AM, Glanz SM. Beta-catenin expression in benign and malignant pilomatrix neoplasms. Br J Dermatol. 2004;150:511-516.
- Pool SE, Manieei F, Clark WH Jr, et al. Dermal squamo-melanocytic tumor: a unique biphenotypic neoplasm of uncertain biological potential. Hum Pathol. 1999;30:525-529.
- Erickson LA, Myers JL, Mihm MC, et al. Malignant basomelanocytic tumor manifesting as metastatic melanoma. Am J Surg Pathol. 2004;28:1393-1396.
- Amin SM, Cooper C, Yelamos O, et al. Combined cutaneous tumors with a melanoma component: a clinical, histologic, and molecular study. J Am Acad Dermatol. 2015;73:451-460.
- Miteva M, Herschthal D, Ricotti C, et al. A rare case of a cutaneous squamomelanocytic tumor: revisiting the histogenesis of combined neoplasms. Am J Dermatopathol. 2009;31:599-603.
- Satter EK, Metcalf J, Lountzis N, et al. Tumors composed of malignant epithelial and melanocytic populations: a case series and review of the literature. J Cutan Pathol. 2009;36:211-219.
- Pouryazdanparast P, Yu L, Johnson T, et al. An unusual squamo-melanocytic tumor of uncertain biologic behavior: a variant of melanoma? Am J Dermatopathol. 2009;31:457-461.
- Burkhalter A, White W. Malignant melanoma in situ colonizing basal cell carcinoma: a simulator of invasive melanoma. Am J Dermatopathol. 1997;19:303-307.
- Papa G, Grandi G, Pascone M. Collision tumor of malignant skin cancers: a case of melanoma in basal cell carcinoma. Pathol Res Pract. 2006;202:691-694.
- Miao Y, Everly JJ, Gross TG, et al. De novo cancers arising in organ transplant recipients are associated with adverse outcomes compared with the general population. Transplantation. 2009;87:1347-1359.
- Bouwes Bavinck JN, Hardie DR, Green A, et al. The risk of skin cancer in renal transplant recipients in Queensland, Australia. a follow-up study. Transplantation. 1996;61:715-721.
- Berg D, Otley CC. Skin cancer in organ transplant recipients: epidemiology, pathogenesis, and management. J Am Acad Dermatol. 2002;47:1-17.
- Zwald FO, Brown M. Skin cancer in solid organ transplant recipients: advances in therapy and management: part I. epidemiology of skin cancer in solid organ transplant recipients. J Am Acad Dermatol. 2011;65:253-261.
- Zwald FO, Brown M. Skin cancer in solid organ transplant recipients: advances in therapy and management: part II. management of skin cancer in solid organ transplant recipients. J Am Acad Dermatol. 2011;65:263-273.
- DePry JL, Reed KB, Cook-Harris RH, et al. Iatrogenic immunosuppression and cutaneous malignancy. Clin Dermatol. 2011;29:602-613.
- Tessari G, Girolomoni G. Nonmelanoma skin cancer in solid organ transplant recipients: update on epidemiology, risk factors, and management. Dermatol Surg. 2012;38:1622-1630.
- Jensen P, Hansen S, Møller B, et al. Skin cancer in kidney and heart transplant recipients and different long-term immunosuppressive therapy regimens. J Am Acad Dermatol. 1999;40:177-186.
- Kasiske BL, Snyder JJ, Gilbertson DT, et al. Cancer after kidney transplantation in the United States. Am J Transplant. 2004;4:905-913.
- Hollenbeak CS, Todd MM, Billingsley EM, et al. Increased incidence of melanoma in renal transplantation recipients. Cancer. 2005;104:1962-1967.
- Le Mire L, Hollowood K, Gray D, et al. Melanomas in renal transplant recipients. Br J Dermatol. 2006;154:472-477.
- Gogia R, Binstock M, Hirose R, et al. Fitzpatrick skin phototype is an independent predictor of squamous cell carcinoma risk after solid organ transplantation. J Am Acad Dermatol. 2013;68:585-591.
- Rashtak S, Dierkhising RA, Kremers WK, et al. Incidence and risk factors for skin cancer following lung transplantation. J Am Acad Dermatol. 2015;72:92-98.
- Ruiz DE, Luzuriaga AM, Hsieh C. Yearly burden of skin cancer in non-Caucasian and Caucasian solid-organ transplant recipients. J Clin Aesthet Dermatol. 2015;8:16-19.
- Perrett CM, Walker SL, O’Donovan P, et al. Azathioprine treatment photosensitizes human skin to ultraviolet A radiation. Br J Dermatol. 2008;159:198-204.
- Abou Ayache R, Thierry A, Bridoux F, et al. Long-term maintenance of calcineurin inhibitor monotherapy reduces the risk for squamous cell carcinomas after kidney transplantation compared with bi- or tritherapy. Transplant Proc. 2007;39:2592-2594.
- Carlson JA, Healy K, Slominski A, et al. Melanocytic matricoma: a report of two cases of a new entity. Am J Dermatopathol. 1999;21:344-349.
- Rizzardi C, Brollo A, Colonna A, et al. A tumor with composite pilo-folliculosebaceous differentiation harboring a recently described new entity—melanocytic matricoma. Am J Dermatopathol. 2002;24:493-497.
- Williams CM, Bozner P, Oliveri CV, et al. Melanocytic matricoma: case confirmation of a recently described entity. J Cutan Pathol. 2003;30:275-278.
- Horenstein MG, Kahn AG. Pathologic quiz case: a 69-year-old man with a brown-black facial papule. melanocytic matricoma. Arch Pathol Lab Med. 2004;128:e163-e164.
- Soler AP, Burchette JL, Bellet JS, et al. Cell adhesion protein expression in melanocytic matricoma. J Cutan Pathol. 2007;34:456-460.
- Islam MN, Bhattacharyya I, Proper SA, et al. Melanocytic matricoma: a distinctive clinicopathologic entity. Dermatol Surg. 2007;33:857-863.
- Monteagudo B, Requena L, Used-Aznar MM, et al. Melanocytic matricoma. Actas Dermosifiliogr. 2008;99:573-582.
- Cartaginese F, Sidoni A. Melanocytic matricoma. report of a further case with clinicopathological and immunohistochemical findings, differential diagnosis and review of the literature. Histol Histopathol. 2010;25:713-717.
- Tallon B, Cerroni L. Where pigmented pilomatricoma and melanocytic matricoma collide. Am J Dermatopathol. 2010;32:769-773.
- Zussman J, Sheth S, Ra SH, et al. Melanocytic matricoma with melanocytic atypia: report of a unique case and review of the literature. Am J Dermatopathol. 2011;33:508-512.
- Tanboon J, Manonukul J, Pattanaprichakul P. Melanocytic matricoma: two cases of a rare entity in women. J Cutan Pathol. 2014;41:775-782.
- Battistella M, Carlson JA, Oslo A, et al. Skin tumors with matrical differentiation: lessons from hair keratins, beta-catenin and PHLDA-1 expression. J Cutan Pathol. 2014;41:427-436.
- Barrado-Solis N, Moles-Poveda P, Roca-Estelles MJ, et al. Melanocytic matricoma with melanocytic atypia: report of a new case [published online February 11, 2015]. J Eur Acad Dermatol Venereol. 2016;30:859-860.
- Pagliarello C, Stanganelli I, Ricci R, et al. A pinkish-blue exophytic nodule on the arm of an elderly man: a quiz. melanocytic matricoma. Acta Derm Venereol. 2017;97:1261-1262.
- Winslow CY, Camacho I, Nousari CH. Melanocytic matricoma with consumption of the epidermis: an atypical histologic attribute or a malignant variant? Am J Dermatopathol. 2017;39:907-909.
- Sangiorgio V, Moneghini L, Tosi D, et al. A case of melanocytic matricoma with prominent mitotic activity and melanocytic hyperplasia. Int J Dermatol. 2018;57:e78-e81.
- Song J, Lu S, Wu Z. An unusual case of melanocytic matricoma in a young pregnant woman. Australas J Dermatol. 2019;60:140-141.
- Ishida M, Okabe H. Pigmented pilomatricoma: an underrecognized variant. Int J Clin Exp Pathol. 2013;6:1890-1893.
- Jani P, Chetty R, Ghazarian DM. An unusual composite pilomatrix carcinoma with intralesional melanocytes: differential diagnosis, immunohistochemical evaluation, and review of the literature. Am J Dermatopathol. 2008;30:174-177.
- Slominski A, Paus R. Melanogenesis is coupled to murine anagen: toward new concepts for the role of melanocytes and the regulation of melanogenesis in hair growth. J Invest Dermatol. 1993;101:90S-97S.
- De Berker D, Higgins CA, Jahada C, et al. Biology of hair and nails. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. China: Elsevier Saunders; 2012:1075-1092.
- Monteagudo C, Fernandez-Figueras MT, San Juan J, et al. Matrical carcinoma with prominent melanocytc hyperplasia (malignant melanocytic matricoma?). Am J Dermatopathol. 2003;25:485-489.
- Sloan JB, Sueki H, Jaworsky C. Pigmented malignant pilomatrixoma: report of a case and review of the literature. J Cutan Pathol. 1992;19:240-246.
- Hardisson D, Linares MD, Cuevas-Santos J, et al. Pilomatrix carcinoma: a clinicopathologic study of six cases and review of the literature. Am J Dermatopathol. 2001;23:394-401.
- Soler AP, Kindel SE, McCloskey G, et al. Cell-cell adhesion proteins in melanocytic pilomatrix carcinoma. Rare Tumors. 2010;2:e43-e45.
- Ardakani NM, Palmer DL, Wood BA. Malignant melanocytic matricoma: a report of 2 cases and review of the literature. Am J Dermatopathol. 2016;38:33-38.
- Villada G, Romagosa R, Miteva M, et al. Matrical carcinoma with melanocytic proliferation and prominent squamoid whorls. Am J Dermatopathol. 2016;38:e11-e14.
- Ji C, Zhang Y, Heller P, et al. Melanocytic matrical carcinoma mimicking melanoma. Am J Dermatopathol. 2017;39:903-906.
- Nielson CB, Vincek V. Malignant melanocytic matricoma and criteria for malignancy. Open J Pathol. 2018;8:94-100.
- Lehmer L, Carly SK, de Feraudy S. Matrical carcinoma with melanocytic hyperplasia mimicking nodular melanoma in an elderly Mexican male. J Cutan Pathol. 2019;46:442-446.
- Weedon D, Bell J, Mayze J. Matrical carcinoma of the skin. J Cutan Pathol. 1980;7:39-42.
- Herrmann JL, Allan A, Trapp KM, et al. Pilomatrix carcinoma: 13 new cases and review of the literature with emphasis on predictors of metastasis. J Am Acad Dermatol. 2014;71:38-43.
- Lazar AJ, Calonje E, Grayson W, et al. Pilomatrix carcinomas contain mutations in CTNNB1, the gene encoding beta-catenin. J Cutan Pathol. 2005;32:148-157.
- Hassanein AM, Glanz SM. Beta-catenin expression in benign and malignant pilomatrix neoplasms. Br J Dermatol. 2004;150:511-516.
- Pool SE, Manieei F, Clark WH Jr, et al. Dermal squamo-melanocytic tumor: a unique biphenotypic neoplasm of uncertain biological potential. Hum Pathol. 1999;30:525-529.
- Erickson LA, Myers JL, Mihm MC, et al. Malignant basomelanocytic tumor manifesting as metastatic melanoma. Am J Surg Pathol. 2004;28:1393-1396.
- Amin SM, Cooper C, Yelamos O, et al. Combined cutaneous tumors with a melanoma component: a clinical, histologic, and molecular study. J Am Acad Dermatol. 2015;73:451-460.
- Miteva M, Herschthal D, Ricotti C, et al. A rare case of a cutaneous squamomelanocytic tumor: revisiting the histogenesis of combined neoplasms. Am J Dermatopathol. 2009;31:599-603.
- Satter EK, Metcalf J, Lountzis N, et al. Tumors composed of malignant epithelial and melanocytic populations: a case series and review of the literature. J Cutan Pathol. 2009;36:211-219.
- Pouryazdanparast P, Yu L, Johnson T, et al. An unusual squamo-melanocytic tumor of uncertain biologic behavior: a variant of melanoma? Am J Dermatopathol. 2009;31:457-461.
- Burkhalter A, White W. Malignant melanoma in situ colonizing basal cell carcinoma: a simulator of invasive melanoma. Am J Dermatopathol. 1997;19:303-307.
- Papa G, Grandi G, Pascone M. Collision tumor of malignant skin cancers: a case of melanoma in basal cell carcinoma. Pathol Res Pract. 2006;202:691-694.
- Miao Y, Everly JJ, Gross TG, et al. De novo cancers arising in organ transplant recipients are associated with adverse outcomes compared with the general population. Transplantation. 2009;87:1347-1359.
- Bouwes Bavinck JN, Hardie DR, Green A, et al. The risk of skin cancer in renal transplant recipients in Queensland, Australia. a follow-up study. Transplantation. 1996;61:715-721.
- Berg D, Otley CC. Skin cancer in organ transplant recipients: epidemiology, pathogenesis, and management. J Am Acad Dermatol. 2002;47:1-17.
- Zwald FO, Brown M. Skin cancer in solid organ transplant recipients: advances in therapy and management: part I. epidemiology of skin cancer in solid organ transplant recipients. J Am Acad Dermatol. 2011;65:253-261.
- Zwald FO, Brown M. Skin cancer in solid organ transplant recipients: advances in therapy and management: part II. management of skin cancer in solid organ transplant recipients. J Am Acad Dermatol. 2011;65:263-273.
- DePry JL, Reed KB, Cook-Harris RH, et al. Iatrogenic immunosuppression and cutaneous malignancy. Clin Dermatol. 2011;29:602-613.
- Tessari G, Girolomoni G. Nonmelanoma skin cancer in solid organ transplant recipients: update on epidemiology, risk factors, and management. Dermatol Surg. 2012;38:1622-1630.
- Jensen P, Hansen S, Møller B, et al. Skin cancer in kidney and heart transplant recipients and different long-term immunosuppressive therapy regimens. J Am Acad Dermatol. 1999;40:177-186.
- Kasiske BL, Snyder JJ, Gilbertson DT, et al. Cancer after kidney transplantation in the United States. Am J Transplant. 2004;4:905-913.
- Hollenbeak CS, Todd MM, Billingsley EM, et al. Increased incidence of melanoma in renal transplantation recipients. Cancer. 2005;104:1962-1967.
- Le Mire L, Hollowood K, Gray D, et al. Melanomas in renal transplant recipients. Br J Dermatol. 2006;154:472-477.
- Gogia R, Binstock M, Hirose R, et al. Fitzpatrick skin phototype is an independent predictor of squamous cell carcinoma risk after solid organ transplantation. J Am Acad Dermatol. 2013;68:585-591.
- Rashtak S, Dierkhising RA, Kremers WK, et al. Incidence and risk factors for skin cancer following lung transplantation. J Am Acad Dermatol. 2015;72:92-98.
- Ruiz DE, Luzuriaga AM, Hsieh C. Yearly burden of skin cancer in non-Caucasian and Caucasian solid-organ transplant recipients. J Clin Aesthet Dermatol. 2015;8:16-19.
- Perrett CM, Walker SL, O’Donovan P, et al. Azathioprine treatment photosensitizes human skin to ultraviolet A radiation. Br J Dermatol. 2008;159:198-204.
- Abou Ayache R, Thierry A, Bridoux F, et al. Long-term maintenance of calcineurin inhibitor monotherapy reduces the risk for squamous cell carcinomas after kidney transplantation compared with bi- or tritherapy. Transplant Proc. 2007;39:2592-2594.
Practice Points
- Melanocytic matrical carcinoma (MMC) is an extremely rare adnexal malignancy that can present as a hyperpigmented papule with or without ulceration.
- Histologically, the lesion resembles a matrical carcinoma with admixed, banal-appearing dendritic melanocytes.
- Solid-organ transplant recipients are at an increased risk of cutaneous malignancies, including rare cancers such as MMC, and these neoplasms should remain in the clinician’s differential diagnosis.
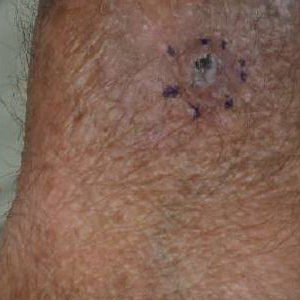
Vandetanib Photoinduced Cutaneous Toxicities
Vandetanib is a once-daily oral multikinase inhibitor that targets the rearranged during transfection (RET) tyrosine kinase, vascular endothelial growth factor receptor, and epidermal growth factor receptor. It has shown efficacy at doses of 300 mg daily in the treatment of progressive medullary thyroid cancer and has shown promise in non–small cell lung cancer and breast cancer. Vandetanib’s toxicity profile includes QT prolongation, diarrhea, and rash.1-3 Cutaneous involvement has been described in the literature as a photodistributed drug reaction with both erythema multiforme (EM) and Stevens-Johnson syndrome (SJS)–like eruptions, phototoxicity, and photoallergy (Table).4-12 Photoinduction is the common thread, but various mechanisms have been proposed, including drug deposition within the dermis and direct toxicity to keratinocytes; however, an understanding of the varied presentation is lacking.
We present 3 cases of vandetanib photoinduced cutaneous toxicities and review the literature on this novel kinase inhibitor. This discussion highlights the spectrum of photosensitivity reactions to vandetanib among patients with varying histologic and clinical presentations.
Case Reports
Patient 1A
74-year-old woman with a history of recurrent metastatic squamous cell carcinoma of the cervix and Fitzpatrick skin type III presented with erythematous, well-demarcated, photodistributed, eczematous papules that were coalescing into plaques on the scalp, hands, and face. The rash appeared sharply demarcated at the wrists bilaterally and principally involved the dorsal sun-exposed areas of her hands (Figure 1). The rash also involved the face and the V of the neck with sharp demarcation. Two weeks prior to onset, she initiated a phase 1 trial of oral vandetanib 100 mg twice daily and oral everolimus 5 mg daily. She did not recall practicing sun protection or experiencing increased sun exposure after starting that trial. The patient demonstrated symptom improvement with desonide cream, hydrocortisone cream 2.5%, and over-the-counter analgesic cream while continuing with the study drugs. However, she developed new, warm, painful papules on the hands and face. Phototesting and biopsy were not performed, and the etiology of the photosensitivity was unknown.
The patient was counseled about regular sun protection and was prescribed triamcinolone cream 0.1% for the arms and hydrocortisone cream 2.5% for the affected facial areas. Therapy with vandetanib and everolimus was continued without dose reduction or further cutaneous eruptions.
Patient 2
A 54-year-old man with a history of progressive medullary thyroid carcinoma and Fitzpatrick skin type II presented with erythematous, well-demarcated, photodistributed, edematous plaques and bullae of the head and neck, bilateral dorsal hands, and bilateral palms of 2 weeks’ duration. The rash spared the upper back and chest with a well-demarcated border (Figure 2A). There were ulcerations and erosions at the base of the neck and the dorsal hands (Figure 2B). He also had conjunctivitis but uninvolved oral and genital mucosae.
Two weeks before the rash appeared, oral vandetanib 300 mg daily was initiated. The patient initially noted some dry skin, which progressed to an eruption involving the face and neck and later the hands with palmar blistering and desquamation. Medication cessation for 1 month led to moderate improvement of the rash on the face and neck. He had not been practicing sun protection but did wear a baseball cap when outside. The patient did not recall an incidence of increased sun exposure. He underwent a skin biopsy of the right dorsal hand, which revealed interface dermatitis with dyskeratosis and subepidermal and intraepidermal bullae (Figure 3). The biopsy findings were most consistent with a phototoxic eruption. Phototesting was not performed.

The patient then initiated sun-protective measures, a prednisone taper, and high-potency steroid ointments. As he tapered his prednisone, he noted continued improvement in the rash. His disease progressed, however, and he did not restart vandetanib.
Patient 3
A 73-year-old man with a history of metastatic lung carcinoma and Fitzpatrick skin type II presented with a rash on the scalp, face, and arms of 2.5 weeks’ duration. There was sharp demarcation at the edges of sun-exposed skin, and no bullae were noted (Figure 4). Prior to presentation, the patient started a 4-week phase 1 trial with vandetanib 300 mg daily and everolimus 10 mg daily. He did not recall any episodes of increased sun exposure. A punch biopsy of the arm showed an interface dermatitis suggestive of a phototoxic reaction. Phototesting was not performed to further clarify if there was a diminished minimal erythema dose with UVA or UVB radiation. Both drugs were discontinued, strict photoprotection was practiced, and triamcinolone cream 0.1% was initiated with resolution of rash. Vandetanib and everolimus were resumed at initial doses with strict photoprotection, and the rash has not recurred.

Comment
Adverse Events Associated With Vandetanib
Vandetanib is a novel multikinase inhibitor that targets RET tyrosine kinase, vascular endothelial growth factor receptor, and epidermal growth factor receptor.1,2 It currently is approved by the US Food and Drug Administration for the treatment of progressive medullary thyroid cancer and is being used in clinical trials for non–small cell lung cancer, glioma, advanced biliary tract cancer, breast cancer, and other advanced solid malignancies. Frequently reported adverse events (AEs) include QT prolongation, diarrhea, and rash.1-3 In a large phase 3 trial, 45% of patients had a rash; of these, 4% were grade 3 and above.3 The most common reasons for dose decrease or cessation were diarrhea and rash (1% and 1.3%, respectively).13 Outside of a trial setting, 75% (45/60) of patients in one French study reported a cutaneous AE, with photosensitivity noted in 22% (13/60). Thus, cutaneous reactions tend to be a common occurrence for patients on this drug, requiring diligent dermatologic examinations.14 In one meta-analysis comprising 9 studies with a total of 2961 patients, the incidence of all-grade rash was 46.1% (95% CI, 40.6%-51.8%), and it was concluded that vandetanib has the highest association of all-grade rash among the anti–vascular endothelial growth factor tyrosine kinase inhibitors. In this meta-analysis, the specific diagnosis of AEs was not further classified.15 In another cohort of vandetanib-treated patients, as many as 37% (28/63) of patients had photosensitivity, with no clarification of the etiology.16
Photoallergic vs Phototoxic Reactions
Photosensitivity reactions are cutaneous reactions that occur from UV light exposure, typically in conjunction with a photosensitizing agent. Photosensitivity reactions can be further classified into phototoxic and photoallergic reactions, which can be distinguished by histopathologic evaluation and history. Although phototoxic reactions will cause keratinocyte necrosis similar to a sunburn, photoallergic reactions will cause epidermal spongiosis similar to allergic contact dermatitis or eczema. Also, phototoxic reactions appear within 1 to 2 days of UV exposure and often are painful, whereas photoallergic reactions can be delayed for 2 to 3 weeks and usually are pruritic. Photosensitivity reactions related to vandetanib have been reported and are summarized in the Table.4-12
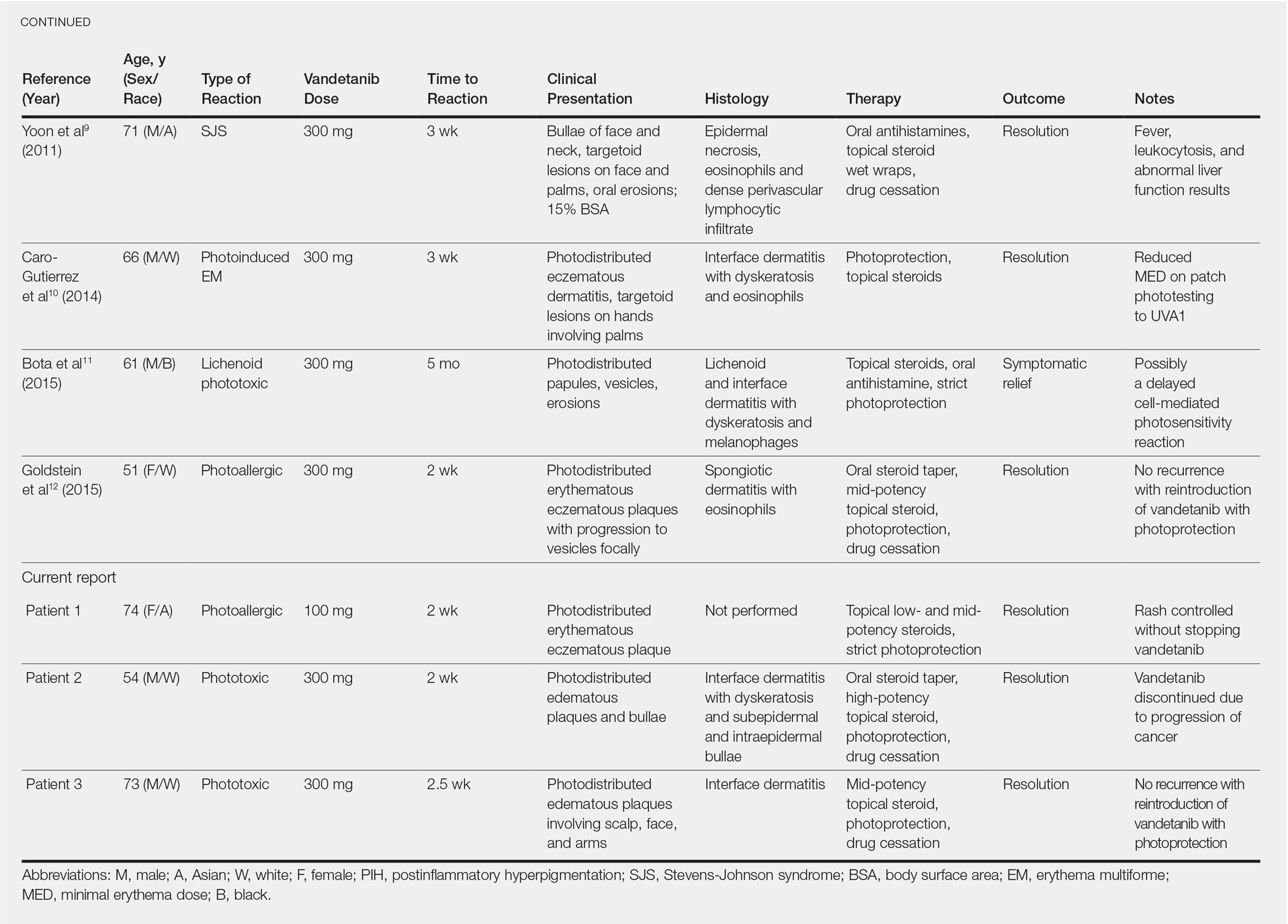
Although reported cutaneous reactions to vandetanib thus far in the literature were reported as photoinduced reactions, there have been isolated case reports of other eruptions including cutaneous pigmentation5 and one case of SJS.9 According to a PubMed search of articles indexed for MEDLINE using the terms vandetanib and rash, we found that there are a variety of clinical findings, but most of the reported photosensitivity cases were phototoxic. Fava et al7 and Goldstein et al12 both reported 1 photoallergic reaction each, plus patient 1 in our case series was noted to have a photoallergic reaction. Phototoxic reactions were reported in 4 patients (including our patient 2) who had dyskeratotic keratinocytes and vacuolar degeneration of the basal layer on histopathology.4,8 Fava et al7 described a lichenoid infiltrate with spongiosis consistent with a photoallergic reaction, but Chang et al4 and Bota et al11 described a lichenoid infiltrate with dyskeratotic cells. Also, Giacchero et al16 described a photosensitivity reaction in 28 of 63 patients. Although only 6 patients had biopsies performed, the range of photosensitivity reactions was demonstrated with lichenoid, dyskeratotic, and spongiotic reactions. However, the cases were not further defined as photoallergic or phototoxic.16 Vandetanib also has been associated with cutaneous blue pigmentation after likely phototoxic reactions. Pigment changes occurred after photosensitivity, but the clinical presentation of photosensitivity was not further characterized.5,16
Classic Drug Eruptions
Two patients were described as having classic drug eruptions—EM10 and SJS9—in photodistributed locations. Histologically, these entities are identical to phototoxic reactions, resulting in epidermal necrosis and an interface dermatitis, but the presence of targetoid lesions on the palms prompted the diagnosis of photodistributed EM and SJS in both cases.9,10 Unique to the SJS case was oral involvement.9
Distinguishing between a phototoxic reaction and photodistributed EM or SJS may be inconsequential if both can be prevented with photoprotection. Rechallenging patients with vandetanib while practicing photoprotection would help to clarify the mechanism, though this course is not always practical.
Mechanism of Action
As seen in our case series, cutaneous reactions occurred only on sun-exposed surfaces, and patients presented with sharp cutoff points that spared non–sun-exposed areas. Although clinically organized as a subtype of photosensitivity, the phototoxicity mechanism of action is considered a direct toxic effect on keratinocytes, which explains the histopathologic finding of dyskeratotic cells and the clinical spectrum of sunburn reaction, phototoxic EM, and SJS. UVA1 induces 2 photoproducts of vandetanib via a UVA1-mediated debromination process,17 but these photoproducts are not responsible for epidermal dyskeratosis.18 It was subsequently demonstrated that keratinocyte death was induced by apoptosis through photoinduced DNA cleavage and the formation of an aryl radical, which can induce further DNA damage.18 Caro-Gutierrez et al10 demonstrated a lowered minimal erythema dose in their patient with vandetanib-induced phototoxic EM.
Conversely, photoallergic reactions are considered immune-mediated delayed-type hypersensitivity reactions.4,7,11 Although the mechanism of a photoallergic reaction remains unclear, it is possible that vandetanib or a metabolite (in susceptible patients) induces an immune-mediated delayed-type hypersensitivity reaction with repeated exposure to the compound, which may explain the varied timing of photoallergic onset, including the events featured in the Bota et al11 case that occurred several months after drug initiation.
Conclusion
Considering the high prevalence of cutaneous AEs, especially varied photosensitivity reactions, these cases emphasize the importance of sun protection to help prevent dose reduction or drug cessation among patients taking vandetanib therapy.
- Carlomagno F, Vitagliano D, Guida T, et al. ZD6474, an orally available inhibitor of KDR tyrosine kinase activity, efficiently blocks oncogenic RET kinases. Cancer Res. 2002;62:7284-7290.
- Wedge SR, Ogilvie DJ, Dukes M, et al. ZD6474 inhibits vascular endothelial growth factor signaling, angiogenesis, and tumor growth following oral administration. Cancer Res. 2002;62:4645-4655.
- Wells SA Jr, Robinson BG, Gagel RF, et al. Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: a randomized, double-blind phase III trial. J Clin Oncol. 2012;30:134-141.
- Chang CH, Chang JW, Hui CY, et al. Severe photosensitivity reaction to vandetanib. J Clin Oncol. 2009;27:E114-E115.
- Kong HH, Fine HA, Stern JB, et al. Cutaneous pigmentation after photosensitivity induced by vandetanib therapy. Arch Dermatol. 2009;145:923-925.
- Brooks S, Linehan WM, Srinivasan R, et al. Successful laser treatment of vandetanib-associated cutaneous pigmentation. Arch Dermatol. 2011;147:364-365.
- Fava P, Quaglino P, Fierro MT, et al. Therapeutic hotline. a rare vandetanib-induced photo-allergic drug eruption. Dermatol Ther. 2010;23:553-555.
- Son YM, Roh JY, Cho EK, et al. Photosensitivity reactions to vandetanib: redevelopment after sequential treatment with docetaxel. Ann Dermatol. 2011;23(suppl 3):S314-S318.
- Yoon J, Oh CW, Kim CY. Stevens-Johnson syndrome induced by vandetanib. Ann Dermatol. 2011;23(suppl 3):S343-S345.
- Caro-Gutierrez D, Floristan Muruzabal MU, Gomez de la Fuente E, et al. Photo-induced erythema multiforme associated with vandetanib administration. J Am Acad Dermatol. 2014;71:E142-E144.11.
- Bota J, Harvey V, Ferguson C, et al. A rare case of late-onset lichenoid photodermatitis after vandetanib therapy. JAAD Case Rep. 2015;1:141-143.
- Goldstein J, Patel AB, Curry JL, et al. Photoallergic reaction in a patient receiving vandetanib for metastatic follicular thyroid carcinoma: a case report. BMC Dermatol. 2015;15:2.
- Thornton K, Kim G, Maher VE, et al. Vandetanib for the treatment of symptomatic or progressive medullary thyroid cancer in patients with unresectable locally advanced or metastatic disease: US Food and Drug Administration drug approval summary. Clin Cancer Res. 2012;18:3722-3730.
- Chougnet CN, Borget I, Leboulleux S, et al. Vandetanib for the treatment of advanced medullary thyroid cancer outside a clinical trial: results from a French cohort. Thyroid. 2015;25:386-391.
- Rosen AC, Wu S, Damse A, et al. Risk of rash in cancer patients treated with vandetanib: systematic review and meta-analysis. J Clin Endocrinol Metab. 2012;97:1125-1133.
- Giacchero D, Ramacciotti C, Arnault JP, et al. A new spectrum of skin toxic effects associated with the multikinase inhibitor vandetanib. Arch Dermatol. 2012;148:1418-1420.
- Dall’acqua S, Vedaldi D, Salvador A. Isolation and structure elucidation of the main UV-A photoproducts of vandetanib. J Pharm Biomed Anal. 2013;84:196-200.
- Salvador A, Vedaldi D, Brun P, et al. Vandetanib-induced phototoxicity in human keratinocytes NCTC-2544. Toxicol In Vitro. 2014;28:803-811.
Vandetanib is a once-daily oral multikinase inhibitor that targets the rearranged during transfection (RET) tyrosine kinase, vascular endothelial growth factor receptor, and epidermal growth factor receptor. It has shown efficacy at doses of 300 mg daily in the treatment of progressive medullary thyroid cancer and has shown promise in non–small cell lung cancer and breast cancer. Vandetanib’s toxicity profile includes QT prolongation, diarrhea, and rash.1-3 Cutaneous involvement has been described in the literature as a photodistributed drug reaction with both erythema multiforme (EM) and Stevens-Johnson syndrome (SJS)–like eruptions, phototoxicity, and photoallergy (Table).4-12 Photoinduction is the common thread, but various mechanisms have been proposed, including drug deposition within the dermis and direct toxicity to keratinocytes; however, an understanding of the varied presentation is lacking.
We present 3 cases of vandetanib photoinduced cutaneous toxicities and review the literature on this novel kinase inhibitor. This discussion highlights the spectrum of photosensitivity reactions to vandetanib among patients with varying histologic and clinical presentations.
Case Reports
Patient 1A
74-year-old woman with a history of recurrent metastatic squamous cell carcinoma of the cervix and Fitzpatrick skin type III presented with erythematous, well-demarcated, photodistributed, eczematous papules that were coalescing into plaques on the scalp, hands, and face. The rash appeared sharply demarcated at the wrists bilaterally and principally involved the dorsal sun-exposed areas of her hands (Figure 1). The rash also involved the face and the V of the neck with sharp demarcation. Two weeks prior to onset, she initiated a phase 1 trial of oral vandetanib 100 mg twice daily and oral everolimus 5 mg daily. She did not recall practicing sun protection or experiencing increased sun exposure after starting that trial. The patient demonstrated symptom improvement with desonide cream, hydrocortisone cream 2.5%, and over-the-counter analgesic cream while continuing with the study drugs. However, she developed new, warm, painful papules on the hands and face. Phototesting and biopsy were not performed, and the etiology of the photosensitivity was unknown.
The patient was counseled about regular sun protection and was prescribed triamcinolone cream 0.1% for the arms and hydrocortisone cream 2.5% for the affected facial areas. Therapy with vandetanib and everolimus was continued without dose reduction or further cutaneous eruptions.
Patient 2
A 54-year-old man with a history of progressive medullary thyroid carcinoma and Fitzpatrick skin type II presented with erythematous, well-demarcated, photodistributed, edematous plaques and bullae of the head and neck, bilateral dorsal hands, and bilateral palms of 2 weeks’ duration. The rash spared the upper back and chest with a well-demarcated border (Figure 2A). There were ulcerations and erosions at the base of the neck and the dorsal hands (Figure 2B). He also had conjunctivitis but uninvolved oral and genital mucosae.
Two weeks before the rash appeared, oral vandetanib 300 mg daily was initiated. The patient initially noted some dry skin, which progressed to an eruption involving the face and neck and later the hands with palmar blistering and desquamation. Medication cessation for 1 month led to moderate improvement of the rash on the face and neck. He had not been practicing sun protection but did wear a baseball cap when outside. The patient did not recall an incidence of increased sun exposure. He underwent a skin biopsy of the right dorsal hand, which revealed interface dermatitis with dyskeratosis and subepidermal and intraepidermal bullae (Figure 3). The biopsy findings were most consistent with a phototoxic eruption. Phototesting was not performed.
The patient then initiated sun-protective measures, a prednisone taper, and high-potency steroid ointments. As he tapered his prednisone, he noted continued improvement in the rash. His disease progressed, however, and he did not restart vandetanib.
Patient 3
A 73-year-old man with a history of metastatic lung carcinoma and Fitzpatrick skin type II presented with a rash on the scalp, face, and arms of 2.5 weeks’ duration. There was sharp demarcation at the edges of sun-exposed skin, and no bullae were noted (Figure 4). Prior to presentation, the patient started a 4-week phase 1 trial with vandetanib 300 mg daily and everolimus 10 mg daily. He did not recall any episodes of increased sun exposure. A punch biopsy of the arm showed an interface dermatitis suggestive of a phototoxic reaction. Phototesting was not performed to further clarify if there was a diminished minimal erythema dose with UVA or UVB radiation. Both drugs were discontinued, strict photoprotection was practiced, and triamcinolone cream 0.1% was initiated with resolution of rash. Vandetanib and everolimus were resumed at initial doses with strict photoprotection, and the rash has not recurred.
Comment
Adverse Events Associated With Vandetanib
Vandetanib is a novel multikinase inhibitor that targets RET tyrosine kinase, vascular endothelial growth factor receptor, and epidermal growth factor receptor.1,2 It currently is approved by the US Food and Drug Administration for the treatment of progressive medullary thyroid cancer and is being used in clinical trials for non–small cell lung cancer, glioma, advanced biliary tract cancer, breast cancer, and other advanced solid malignancies. Frequently reported adverse events (AEs) include QT prolongation, diarrhea, and rash.1-3 In a large phase 3 trial, 45% of patients had a rash; of these, 4% were grade 3 and above.3 The most common reasons for dose decrease or cessation were diarrhea and rash (1% and 1.3%, respectively).13 Outside of a trial setting, 75% (45/60) of patients in one French study reported a cutaneous AE, with photosensitivity noted in 22% (13/60). Thus, cutaneous reactions tend to be a common occurrence for patients on this drug, requiring diligent dermatologic examinations.14 In one meta-analysis comprising 9 studies with a total of 2961 patients, the incidence of all-grade rash was 46.1% (95% CI, 40.6%-51.8%), and it was concluded that vandetanib has the highest association of all-grade rash among the anti–vascular endothelial growth factor tyrosine kinase inhibitors. In this meta-analysis, the specific diagnosis of AEs was not further classified.15 In another cohort of vandetanib-treated patients, as many as 37% (28/63) of patients had photosensitivity, with no clarification of the etiology.16
Photoallergic vs Phototoxic Reactions
Photosensitivity reactions are cutaneous reactions that occur from UV light exposure, typically in conjunction with a photosensitizing agent. Photosensitivity reactions can be further classified into phototoxic and photoallergic reactions, which can be distinguished by histopathologic evaluation and history. Although phototoxic reactions will cause keratinocyte necrosis similar to a sunburn, photoallergic reactions will cause epidermal spongiosis similar to allergic contact dermatitis or eczema. Also, phototoxic reactions appear within 1 to 2 days of UV exposure and often are painful, whereas photoallergic reactions can be delayed for 2 to 3 weeks and usually are pruritic. Photosensitivity reactions related to vandetanib have been reported and are summarized in the Table.4-12
Although reported cutaneous reactions to vandetanib thus far in the literature were reported as photoinduced reactions, there have been isolated case reports of other eruptions including cutaneous pigmentation5 and one case of SJS.9 According to a PubMed search of articles indexed for MEDLINE using the terms vandetanib and rash, we found that there are a variety of clinical findings, but most of the reported photosensitivity cases were phototoxic. Fava et al7 and Goldstein et al12 both reported 1 photoallergic reaction each, plus patient 1 in our case series was noted to have a photoallergic reaction. Phototoxic reactions were reported in 4 patients (including our patient 2) who had dyskeratotic keratinocytes and vacuolar degeneration of the basal layer on histopathology.4,8 Fava et al7 described a lichenoid infiltrate with spongiosis consistent with a photoallergic reaction, but Chang et al4 and Bota et al11 described a lichenoid infiltrate with dyskeratotic cells. Also, Giacchero et al16 described a photosensitivity reaction in 28 of 63 patients. Although only 6 patients had biopsies performed, the range of photosensitivity reactions was demonstrated with lichenoid, dyskeratotic, and spongiotic reactions. However, the cases were not further defined as photoallergic or phototoxic.16 Vandetanib also has been associated with cutaneous blue pigmentation after likely phototoxic reactions. Pigment changes occurred after photosensitivity, but the clinical presentation of photosensitivity was not further characterized.5,16
Classic Drug Eruptions
Two patients were described as having classic drug eruptions—EM10 and SJS9—in photodistributed locations. Histologically, these entities are identical to phototoxic reactions, resulting in epidermal necrosis and an interface dermatitis, but the presence of targetoid lesions on the palms prompted the diagnosis of photodistributed EM and SJS in both cases.9,10 Unique to the SJS case was oral involvement.9
Distinguishing between a phototoxic reaction and photodistributed EM or SJS may be inconsequential if both can be prevented with photoprotection. Rechallenging patients with vandetanib while practicing photoprotection would help to clarify the mechanism, though this course is not always practical.
Mechanism of Action
As seen in our case series, cutaneous reactions occurred only on sun-exposed surfaces, and patients presented with sharp cutoff points that spared non–sun-exposed areas. Although clinically organized as a subtype of photosensitivity, the phototoxicity mechanism of action is considered a direct toxic effect on keratinocytes, which explains the histopathologic finding of dyskeratotic cells and the clinical spectrum of sunburn reaction, phototoxic EM, and SJS. UVA1 induces 2 photoproducts of vandetanib via a UVA1-mediated debromination process,17 but these photoproducts are not responsible for epidermal dyskeratosis.18 It was subsequently demonstrated that keratinocyte death was induced by apoptosis through photoinduced DNA cleavage and the formation of an aryl radical, which can induce further DNA damage.18 Caro-Gutierrez et al10 demonstrated a lowered minimal erythema dose in their patient with vandetanib-induced phototoxic EM.
Conversely, photoallergic reactions are considered immune-mediated delayed-type hypersensitivity reactions.4,7,11 Although the mechanism of a photoallergic reaction remains unclear, it is possible that vandetanib or a metabolite (in susceptible patients) induces an immune-mediated delayed-type hypersensitivity reaction with repeated exposure to the compound, which may explain the varied timing of photoallergic onset, including the events featured in the Bota et al11 case that occurred several months after drug initiation.
Conclusion
Considering the high prevalence of cutaneous AEs, especially varied photosensitivity reactions, these cases emphasize the importance of sun protection to help prevent dose reduction or drug cessation among patients taking vandetanib therapy.
Vandetanib is a once-daily oral multikinase inhibitor that targets the rearranged during transfection (RET) tyrosine kinase, vascular endothelial growth factor receptor, and epidermal growth factor receptor. It has shown efficacy at doses of 300 mg daily in the treatment of progressive medullary thyroid cancer and has shown promise in non–small cell lung cancer and breast cancer. Vandetanib’s toxicity profile includes QT prolongation, diarrhea, and rash.1-3 Cutaneous involvement has been described in the literature as a photodistributed drug reaction with both erythema multiforme (EM) and Stevens-Johnson syndrome (SJS)–like eruptions, phototoxicity, and photoallergy (Table).4-12 Photoinduction is the common thread, but various mechanisms have been proposed, including drug deposition within the dermis and direct toxicity to keratinocytes; however, an understanding of the varied presentation is lacking.
We present 3 cases of vandetanib photoinduced cutaneous toxicities and review the literature on this novel kinase inhibitor. This discussion highlights the spectrum of photosensitivity reactions to vandetanib among patients with varying histologic and clinical presentations.
Case Reports
Patient 1A
74-year-old woman with a history of recurrent metastatic squamous cell carcinoma of the cervix and Fitzpatrick skin type III presented with erythematous, well-demarcated, photodistributed, eczematous papules that were coalescing into plaques on the scalp, hands, and face. The rash appeared sharply demarcated at the wrists bilaterally and principally involved the dorsal sun-exposed areas of her hands (Figure 1). The rash also involved the face and the V of the neck with sharp demarcation. Two weeks prior to onset, she initiated a phase 1 trial of oral vandetanib 100 mg twice daily and oral everolimus 5 mg daily. She did not recall practicing sun protection or experiencing increased sun exposure after starting that trial. The patient demonstrated symptom improvement with desonide cream, hydrocortisone cream 2.5%, and over-the-counter analgesic cream while continuing with the study drugs. However, she developed new, warm, painful papules on the hands and face. Phototesting and biopsy were not performed, and the etiology of the photosensitivity was unknown.
The patient was counseled about regular sun protection and was prescribed triamcinolone cream 0.1% for the arms and hydrocortisone cream 2.5% for the affected facial areas. Therapy with vandetanib and everolimus was continued without dose reduction or further cutaneous eruptions.
Patient 2
A 54-year-old man with a history of progressive medullary thyroid carcinoma and Fitzpatrick skin type II presented with erythematous, well-demarcated, photodistributed, edematous plaques and bullae of the head and neck, bilateral dorsal hands, and bilateral palms of 2 weeks’ duration. The rash spared the upper back and chest with a well-demarcated border (Figure 2A). There were ulcerations and erosions at the base of the neck and the dorsal hands (Figure 2B). He also had conjunctivitis but uninvolved oral and genital mucosae.
Two weeks before the rash appeared, oral vandetanib 300 mg daily was initiated. The patient initially noted some dry skin, which progressed to an eruption involving the face and neck and later the hands with palmar blistering and desquamation. Medication cessation for 1 month led to moderate improvement of the rash on the face and neck. He had not been practicing sun protection but did wear a baseball cap when outside. The patient did not recall an incidence of increased sun exposure. He underwent a skin biopsy of the right dorsal hand, which revealed interface dermatitis with dyskeratosis and subepidermal and intraepidermal bullae (Figure 3). The biopsy findings were most consistent with a phototoxic eruption. Phototesting was not performed.
The patient then initiated sun-protective measures, a prednisone taper, and high-potency steroid ointments. As he tapered his prednisone, he noted continued improvement in the rash. His disease progressed, however, and he did not restart vandetanib.
Patient 3
A 73-year-old man with a history of metastatic lung carcinoma and Fitzpatrick skin type II presented with a rash on the scalp, face, and arms of 2.5 weeks’ duration. There was sharp demarcation at the edges of sun-exposed skin, and no bullae were noted (Figure 4). Prior to presentation, the patient started a 4-week phase 1 trial with vandetanib 300 mg daily and everolimus 10 mg daily. He did not recall any episodes of increased sun exposure. A punch biopsy of the arm showed an interface dermatitis suggestive of a phototoxic reaction. Phototesting was not performed to further clarify if there was a diminished minimal erythema dose with UVA or UVB radiation. Both drugs were discontinued, strict photoprotection was practiced, and triamcinolone cream 0.1% was initiated with resolution of rash. Vandetanib and everolimus were resumed at initial doses with strict photoprotection, and the rash has not recurred.
Comment
Adverse Events Associated With Vandetanib
Vandetanib is a novel multikinase inhibitor that targets RET tyrosine kinase, vascular endothelial growth factor receptor, and epidermal growth factor receptor.1,2 It currently is approved by the US Food and Drug Administration for the treatment of progressive medullary thyroid cancer and is being used in clinical trials for non–small cell lung cancer, glioma, advanced biliary tract cancer, breast cancer, and other advanced solid malignancies. Frequently reported adverse events (AEs) include QT prolongation, diarrhea, and rash.1-3 In a large phase 3 trial, 45% of patients had a rash; of these, 4% were grade 3 and above.3 The most common reasons for dose decrease or cessation were diarrhea and rash (1% and 1.3%, respectively).13 Outside of a trial setting, 75% (45/60) of patients in one French study reported a cutaneous AE, with photosensitivity noted in 22% (13/60). Thus, cutaneous reactions tend to be a common occurrence for patients on this drug, requiring diligent dermatologic examinations.14 In one meta-analysis comprising 9 studies with a total of 2961 patients, the incidence of all-grade rash was 46.1% (95% CI, 40.6%-51.8%), and it was concluded that vandetanib has the highest association of all-grade rash among the anti–vascular endothelial growth factor tyrosine kinase inhibitors. In this meta-analysis, the specific diagnosis of AEs was not further classified.15 In another cohort of vandetanib-treated patients, as many as 37% (28/63) of patients had photosensitivity, with no clarification of the etiology.16
Photoallergic vs Phototoxic Reactions
Photosensitivity reactions are cutaneous reactions that occur from UV light exposure, typically in conjunction with a photosensitizing agent. Photosensitivity reactions can be further classified into phototoxic and photoallergic reactions, which can be distinguished by histopathologic evaluation and history. Although phototoxic reactions will cause keratinocyte necrosis similar to a sunburn, photoallergic reactions will cause epidermal spongiosis similar to allergic contact dermatitis or eczema. Also, phototoxic reactions appear within 1 to 2 days of UV exposure and often are painful, whereas photoallergic reactions can be delayed for 2 to 3 weeks and usually are pruritic. Photosensitivity reactions related to vandetanib have been reported and are summarized in the Table.4-12
Although reported cutaneous reactions to vandetanib thus far in the literature were reported as photoinduced reactions, there have been isolated case reports of other eruptions including cutaneous pigmentation5 and one case of SJS.9 According to a PubMed search of articles indexed for MEDLINE using the terms vandetanib and rash, we found that there are a variety of clinical findings, but most of the reported photosensitivity cases were phototoxic. Fava et al7 and Goldstein et al12 both reported 1 photoallergic reaction each, plus patient 1 in our case series was noted to have a photoallergic reaction. Phototoxic reactions were reported in 4 patients (including our patient 2) who had dyskeratotic keratinocytes and vacuolar degeneration of the basal layer on histopathology.4,8 Fava et al7 described a lichenoid infiltrate with spongiosis consistent with a photoallergic reaction, but Chang et al4 and Bota et al11 described a lichenoid infiltrate with dyskeratotic cells. Also, Giacchero et al16 described a photosensitivity reaction in 28 of 63 patients. Although only 6 patients had biopsies performed, the range of photosensitivity reactions was demonstrated with lichenoid, dyskeratotic, and spongiotic reactions. However, the cases were not further defined as photoallergic or phototoxic.16 Vandetanib also has been associated with cutaneous blue pigmentation after likely phototoxic reactions. Pigment changes occurred after photosensitivity, but the clinical presentation of photosensitivity was not further characterized.5,16
Classic Drug Eruptions
Two patients were described as having classic drug eruptions—EM10 and SJS9—in photodistributed locations. Histologically, these entities are identical to phototoxic reactions, resulting in epidermal necrosis and an interface dermatitis, but the presence of targetoid lesions on the palms prompted the diagnosis of photodistributed EM and SJS in both cases.9,10 Unique to the SJS case was oral involvement.9
Distinguishing between a phototoxic reaction and photodistributed EM or SJS may be inconsequential if both can be prevented with photoprotection. Rechallenging patients with vandetanib while practicing photoprotection would help to clarify the mechanism, though this course is not always practical.
Mechanism of Action
As seen in our case series, cutaneous reactions occurred only on sun-exposed surfaces, and patients presented with sharp cutoff points that spared non–sun-exposed areas. Although clinically organized as a subtype of photosensitivity, the phototoxicity mechanism of action is considered a direct toxic effect on keratinocytes, which explains the histopathologic finding of dyskeratotic cells and the clinical spectrum of sunburn reaction, phototoxic EM, and SJS. UVA1 induces 2 photoproducts of vandetanib via a UVA1-mediated debromination process,17 but these photoproducts are not responsible for epidermal dyskeratosis.18 It was subsequently demonstrated that keratinocyte death was induced by apoptosis through photoinduced DNA cleavage and the formation of an aryl radical, which can induce further DNA damage.18 Caro-Gutierrez et al10 demonstrated a lowered minimal erythema dose in their patient with vandetanib-induced phototoxic EM.
Conversely, photoallergic reactions are considered immune-mediated delayed-type hypersensitivity reactions.4,7,11 Although the mechanism of a photoallergic reaction remains unclear, it is possible that vandetanib or a metabolite (in susceptible patients) induces an immune-mediated delayed-type hypersensitivity reaction with repeated exposure to the compound, which may explain the varied timing of photoallergic onset, including the events featured in the Bota et al11 case that occurred several months after drug initiation.
Conclusion
Considering the high prevalence of cutaneous AEs, especially varied photosensitivity reactions, these cases emphasize the importance of sun protection to help prevent dose reduction or drug cessation among patients taking vandetanib therapy.
- Carlomagno F, Vitagliano D, Guida T, et al. ZD6474, an orally available inhibitor of KDR tyrosine kinase activity, efficiently blocks oncogenic RET kinases. Cancer Res. 2002;62:7284-7290.
- Wedge SR, Ogilvie DJ, Dukes M, et al. ZD6474 inhibits vascular endothelial growth factor signaling, angiogenesis, and tumor growth following oral administration. Cancer Res. 2002;62:4645-4655.
- Wells SA Jr, Robinson BG, Gagel RF, et al. Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: a randomized, double-blind phase III trial. J Clin Oncol. 2012;30:134-141.
- Chang CH, Chang JW, Hui CY, et al. Severe photosensitivity reaction to vandetanib. J Clin Oncol. 2009;27:E114-E115.
- Kong HH, Fine HA, Stern JB, et al. Cutaneous pigmentation after photosensitivity induced by vandetanib therapy. Arch Dermatol. 2009;145:923-925.
- Brooks S, Linehan WM, Srinivasan R, et al. Successful laser treatment of vandetanib-associated cutaneous pigmentation. Arch Dermatol. 2011;147:364-365.
- Fava P, Quaglino P, Fierro MT, et al. Therapeutic hotline. a rare vandetanib-induced photo-allergic drug eruption. Dermatol Ther. 2010;23:553-555.
- Son YM, Roh JY, Cho EK, et al. Photosensitivity reactions to vandetanib: redevelopment after sequential treatment with docetaxel. Ann Dermatol. 2011;23(suppl 3):S314-S318.
- Yoon J, Oh CW, Kim CY. Stevens-Johnson syndrome induced by vandetanib. Ann Dermatol. 2011;23(suppl 3):S343-S345.
- Caro-Gutierrez D, Floristan Muruzabal MU, Gomez de la Fuente E, et al. Photo-induced erythema multiforme associated with vandetanib administration. J Am Acad Dermatol. 2014;71:E142-E144.11.
- Bota J, Harvey V, Ferguson C, et al. A rare case of late-onset lichenoid photodermatitis after vandetanib therapy. JAAD Case Rep. 2015;1:141-143.
- Goldstein J, Patel AB, Curry JL, et al. Photoallergic reaction in a patient receiving vandetanib for metastatic follicular thyroid carcinoma: a case report. BMC Dermatol. 2015;15:2.
- Thornton K, Kim G, Maher VE, et al. Vandetanib for the treatment of symptomatic or progressive medullary thyroid cancer in patients with unresectable locally advanced or metastatic disease: US Food and Drug Administration drug approval summary. Clin Cancer Res. 2012;18:3722-3730.
- Chougnet CN, Borget I, Leboulleux S, et al. Vandetanib for the treatment of advanced medullary thyroid cancer outside a clinical trial: results from a French cohort. Thyroid. 2015;25:386-391.
- Rosen AC, Wu S, Damse A, et al. Risk of rash in cancer patients treated with vandetanib: systematic review and meta-analysis. J Clin Endocrinol Metab. 2012;97:1125-1133.
- Giacchero D, Ramacciotti C, Arnault JP, et al. A new spectrum of skin toxic effects associated with the multikinase inhibitor vandetanib. Arch Dermatol. 2012;148:1418-1420.
- Dall’acqua S, Vedaldi D, Salvador A. Isolation and structure elucidation of the main UV-A photoproducts of vandetanib. J Pharm Biomed Anal. 2013;84:196-200.
- Salvador A, Vedaldi D, Brun P, et al. Vandetanib-induced phototoxicity in human keratinocytes NCTC-2544. Toxicol In Vitro. 2014;28:803-811.
- Carlomagno F, Vitagliano D, Guida T, et al. ZD6474, an orally available inhibitor of KDR tyrosine kinase activity, efficiently blocks oncogenic RET kinases. Cancer Res. 2002;62:7284-7290.
- Wedge SR, Ogilvie DJ, Dukes M, et al. ZD6474 inhibits vascular endothelial growth factor signaling, angiogenesis, and tumor growth following oral administration. Cancer Res. 2002;62:4645-4655.
- Wells SA Jr, Robinson BG, Gagel RF, et al. Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: a randomized, double-blind phase III trial. J Clin Oncol. 2012;30:134-141.
- Chang CH, Chang JW, Hui CY, et al. Severe photosensitivity reaction to vandetanib. J Clin Oncol. 2009;27:E114-E115.
- Kong HH, Fine HA, Stern JB, et al. Cutaneous pigmentation after photosensitivity induced by vandetanib therapy. Arch Dermatol. 2009;145:923-925.
- Brooks S, Linehan WM, Srinivasan R, et al. Successful laser treatment of vandetanib-associated cutaneous pigmentation. Arch Dermatol. 2011;147:364-365.
- Fava P, Quaglino P, Fierro MT, et al. Therapeutic hotline. a rare vandetanib-induced photo-allergic drug eruption. Dermatol Ther. 2010;23:553-555.
- Son YM, Roh JY, Cho EK, et al. Photosensitivity reactions to vandetanib: redevelopment after sequential treatment with docetaxel. Ann Dermatol. 2011;23(suppl 3):S314-S318.
- Yoon J, Oh CW, Kim CY. Stevens-Johnson syndrome induced by vandetanib. Ann Dermatol. 2011;23(suppl 3):S343-S345.
- Caro-Gutierrez D, Floristan Muruzabal MU, Gomez de la Fuente E, et al. Photo-induced erythema multiforme associated with vandetanib administration. J Am Acad Dermatol. 2014;71:E142-E144.11.
- Bota J, Harvey V, Ferguson C, et al. A rare case of late-onset lichenoid photodermatitis after vandetanib therapy. JAAD Case Rep. 2015;1:141-143.
- Goldstein J, Patel AB, Curry JL, et al. Photoallergic reaction in a patient receiving vandetanib for metastatic follicular thyroid carcinoma: a case report. BMC Dermatol. 2015;15:2.
- Thornton K, Kim G, Maher VE, et al. Vandetanib for the treatment of symptomatic or progressive medullary thyroid cancer in patients with unresectable locally advanced or metastatic disease: US Food and Drug Administration drug approval summary. Clin Cancer Res. 2012;18:3722-3730.
- Chougnet CN, Borget I, Leboulleux S, et al. Vandetanib for the treatment of advanced medullary thyroid cancer outside a clinical trial: results from a French cohort. Thyroid. 2015;25:386-391.
- Rosen AC, Wu S, Damse A, et al. Risk of rash in cancer patients treated with vandetanib: systematic review and meta-analysis. J Clin Endocrinol Metab. 2012;97:1125-1133.
- Giacchero D, Ramacciotti C, Arnault JP, et al. A new spectrum of skin toxic effects associated with the multikinase inhibitor vandetanib. Arch Dermatol. 2012;148:1418-1420.
- Dall’acqua S, Vedaldi D, Salvador A. Isolation and structure elucidation of the main UV-A photoproducts of vandetanib. J Pharm Biomed Anal. 2013;84:196-200.
- Salvador A, Vedaldi D, Brun P, et al. Vandetanib-induced phototoxicity in human keratinocytes NCTC-2544. Toxicol In Vitro. 2014;28:803-811.
Practice Points
- Vandetanib is a US Food and Drug Administration– approved once-daily oral multikinase inhibitor for patients with progressive medullary thyroid cancer with a high incidence of cutaneous toxicities including phototoxicity. Early recognition of such cutaneous toxicities leads to early intervention and may allow greater compliance with treatment.
- The most common toxicity is phototoxicity. Diligent interventions include photoprotection such as sunscreen, sun-protective clothing, and avoiding peak hours of sun exposure.
- Topical steroids as well as bland emollients are the mainstay of therapy for symptomatic lesions.
- Extensive cutaneous involvement may include blistering, pain, and pruritus and necessitate dose reduction or even drug cessation.
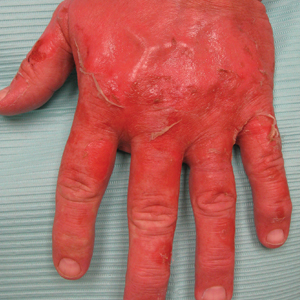
Asymptomatic, Slowly Enlarging Papule on the Nipple
The Diagnosis: Erosive Adenomatosis of the Nipple
Biopsy of the lesion revealed proliferative sections of glandular epithelium demonstrating apocrine differentiation, connecting to the epidermis and traversing throughout the entire dermis of the specimen (Figure). There were papillary projections of dilated ducts with a retained layer of myoepithelial cells surrounding the epithelial layers. Cytologic atypia was not appreciated. The patient was diagnosed with erosive adenomatosis of the nipple (EAN), also known as nipple adenoma. The lesion subsequently was treated and cleared with Mohs micrographic surgery (MMS). At 8-month follow-up there was no clinical recurrence of the lesion, and the patient was satisfied with the overall cosmetic appearance and conservation of the areola. The patient was followed clinically with annual breast examinations and mammography to monitor future recurrence.

Erosive adenomatosis of the nipple is an uncommon benign proliferative process of the lactiferous ducts of the nipple. Recognizing EAN is important because it resembles malignant breast diseases such as Paget disease of the nipple and invasive breast carcinoma. Due to these similarities, early cases of EAN have resulted in unnecessary mastectomies before the benignity of the condition was established.1 Accurate diagnosis is important to both the patient and the clinician for treatment planning as well as psychosocial consequences associated with the potential removal of this anatomically and cosmetically sensitive area.
Reviewing the literature on EAN is complicated by the variety of terms used to describe this condition, including but not limited to nipple adenoma, nipple duct adenoma, papillary adenoma of the nipple, and florid papillomatosis of the nipple. In 1955, Jones2 described EAN using the term florid papillomatosis of the nipple ducts. In 1962, Handley and Thackray1 argued that adenoma of the nipple was a more descriptive term because it more closely described the appearance of a sweat gland adenoma. They reasoned that adenoma of the nipple is a separate process from ductal papilloma due to the adenomatous proliferation into the nipple stroma rather than the lumen of the nipple ducts.1 The term adenoma of the nipple was further supported in 1965 by Taylor and Robertson.3 In 1959, Le Gal et al4 used the term erosive adenomatosis of the nipple to describe the erosive nature of nipple adenoma. The term nipple adenoma was published in the 2012 WHO Classification of Tumors of the Breast with 4 common histologic subtypes.5,6
Erosive adenomatosis of the nipple is clinically indistinguishable from Paget disease of the nipple, thus biopsy is essential for accurate diagnosis. In contrast to Paget disease, EAN tends to present in younger patients and progresses more slowly, and symptoms may be exacerbated around menstruation.1 Case reports demonstrate that patients may wait years before seeking medical attention for EAN.1,3,7,8 Presenting symptoms may include inflammation, crusting, nipple skin erosion, itching, and pain. Serous or sanguineous discharge from the lesions also is commonly reported. Palpation may reveal a small, hard, or elastic nodule within or underlying the nipple. In addition to Paget disease, EAN may resemble squamous cell carcinoma of the nipple, eczema, psoriasis, or a skin infection.6 Axillary lymphadenopathy is not present in the absence of a concomitant breast malignancy.8 On biopsy, nipple adenoma represents ductal proliferation of glandular structures within the stroma of the nipple that is well circumscribed but without borders. The erosive appearance of the lesion is produced by extensions of the glandular epithelium on the surface of the nipple.1,6 Specific to EAN is the presence of 2 cell types: an inner columnar epithelium and an outer cuboidal myoepithelium. These 2 cell types are present in normal lactiferous ducts; however, normal ducts are highly organized compared to EAN.9
After confirmation of EAN by nipple biopsy, complete surgical excision has been the gold standard for treatment, followed by reconstructive surgery.6 Handley and Thackray1 advocated for total excision of the nipple and areola with an underlying wedge of breast tissue to facilitate wound closure. More recently, successful alternative forms of treatment have been utilized to minimize disfiguring surgery. Alternative treatments include MMS,8 cryotherapy,10 and nipple splitting enucleation.6 Treatment with MMS has resulted in nipple sparing with the least amount of surface area sacrificed (1.1 cm2).9 Our case and prior case reports demonstrate that the tissue sparing potential of MMS is appropriate for the treatment of EAN, though traditionally it has been reserved for more malignant tumors. Preserving this sensitive area is both cosmetically and psychologically advantageous for the patient and thus should be considered when reviewing treatment options for EAN.
- Handley RS, Thackray AC. Adenoma of nipple. Br J Cancer. 1962;16:187-194.
- Jones DB. Florid papillomatosis of the nipple ducts. Cancer. 1955;8:315-319.
- Taylor HB, Robertson AG. Adenomas of the nipple. Cancer. 1965:18:995-1002.
- Le Gal Y, Gros CM, Bader P. Erosive adenomatosis of the nipple [in French]. Ann Anat Pathol (Paris). 1959;4:292-304.
- Eusebi V, Lester S. Tumours of the nipple. In: Lakhani SR, Ellis IO, Schnitt SJ, et al, eds. WHO Classification of Tumours of the Breast. Lyon, France: IARC; 2012.
- Spohn GP, Trotter SC, Tozbician G, et al. Nipple adenoma in a female patient presenting with persistent erythema of the right nipple skin: case report, review of the literature, clinical implications, and relevancy to health care providers who evaluate and treat patients with dermatologic conditions of the breast skin. BMC Dermatol. 2016;16:4.
- Kowal R, Miller CJ, Elenitsas R. Eroded patch on the nipple of a 57-year-old woman. Arch Dermatol. 2008;144:933-938.
- Van Mierlo PL, Geelen GM, Neumann HA. Mohs micrographic surgery for an erosive adenomatosis of the nipple. Dermatol Surg. 1998;24:681-683.
- Brankov N, Nino T, Hsiang D, et al. Utilizing Mohs surgery for tissue preservation in erosive adenomatosis of the nipple. Dermatol Surg. 2016;42:684-686.
- Kuflik EG. Erosive adenomatosis of the nipple treated with cryosurgery. J Am Acad Dermatol. 1998;38:270-271.
The Diagnosis: Erosive Adenomatosis of the Nipple
Biopsy of the lesion revealed proliferative sections of glandular epithelium demonstrating apocrine differentiation, connecting to the epidermis and traversing throughout the entire dermis of the specimen (Figure). There were papillary projections of dilated ducts with a retained layer of myoepithelial cells surrounding the epithelial layers. Cytologic atypia was not appreciated. The patient was diagnosed with erosive adenomatosis of the nipple (EAN), also known as nipple adenoma. The lesion subsequently was treated and cleared with Mohs micrographic surgery (MMS). At 8-month follow-up there was no clinical recurrence of the lesion, and the patient was satisfied with the overall cosmetic appearance and conservation of the areola. The patient was followed clinically with annual breast examinations and mammography to monitor future recurrence.
Erosive adenomatosis of the nipple is an uncommon benign proliferative process of the lactiferous ducts of the nipple. Recognizing EAN is important because it resembles malignant breast diseases such as Paget disease of the nipple and invasive breast carcinoma. Due to these similarities, early cases of EAN have resulted in unnecessary mastectomies before the benignity of the condition was established.1 Accurate diagnosis is important to both the patient and the clinician for treatment planning as well as psychosocial consequences associated with the potential removal of this anatomically and cosmetically sensitive area.
Reviewing the literature on EAN is complicated by the variety of terms used to describe this condition, including but not limited to nipple adenoma, nipple duct adenoma, papillary adenoma of the nipple, and florid papillomatosis of the nipple. In 1955, Jones2 described EAN using the term florid papillomatosis of the nipple ducts. In 1962, Handley and Thackray1 argued that adenoma of the nipple was a more descriptive term because it more closely described the appearance of a sweat gland adenoma. They reasoned that adenoma of the nipple is a separate process from ductal papilloma due to the adenomatous proliferation into the nipple stroma rather than the lumen of the nipple ducts.1 The term adenoma of the nipple was further supported in 1965 by Taylor and Robertson.3 In 1959, Le Gal et al4 used the term erosive adenomatosis of the nipple to describe the erosive nature of nipple adenoma. The term nipple adenoma was published in the 2012 WHO Classification of Tumors of the Breast with 4 common histologic subtypes.5,6
Erosive adenomatosis of the nipple is clinically indistinguishable from Paget disease of the nipple, thus biopsy is essential for accurate diagnosis. In contrast to Paget disease, EAN tends to present in younger patients and progresses more slowly, and symptoms may be exacerbated around menstruation.1 Case reports demonstrate that patients may wait years before seeking medical attention for EAN.1,3,7,8 Presenting symptoms may include inflammation, crusting, nipple skin erosion, itching, and pain. Serous or sanguineous discharge from the lesions also is commonly reported. Palpation may reveal a small, hard, or elastic nodule within or underlying the nipple. In addition to Paget disease, EAN may resemble squamous cell carcinoma of the nipple, eczema, psoriasis, or a skin infection.6 Axillary lymphadenopathy is not present in the absence of a concomitant breast malignancy.8 On biopsy, nipple adenoma represents ductal proliferation of glandular structures within the stroma of the nipple that is well circumscribed but without borders. The erosive appearance of the lesion is produced by extensions of the glandular epithelium on the surface of the nipple.1,6 Specific to EAN is the presence of 2 cell types: an inner columnar epithelium and an outer cuboidal myoepithelium. These 2 cell types are present in normal lactiferous ducts; however, normal ducts are highly organized compared to EAN.9
After confirmation of EAN by nipple biopsy, complete surgical excision has been the gold standard for treatment, followed by reconstructive surgery.6 Handley and Thackray1 advocated for total excision of the nipple and areola with an underlying wedge of breast tissue to facilitate wound closure. More recently, successful alternative forms of treatment have been utilized to minimize disfiguring surgery. Alternative treatments include MMS,8 cryotherapy,10 and nipple splitting enucleation.6 Treatment with MMS has resulted in nipple sparing with the least amount of surface area sacrificed (1.1 cm2).9 Our case and prior case reports demonstrate that the tissue sparing potential of MMS is appropriate for the treatment of EAN, though traditionally it has been reserved for more malignant tumors. Preserving this sensitive area is both cosmetically and psychologically advantageous for the patient and thus should be considered when reviewing treatment options for EAN.
The Diagnosis: Erosive Adenomatosis of the Nipple
Biopsy of the lesion revealed proliferative sections of glandular epithelium demonstrating apocrine differentiation, connecting to the epidermis and traversing throughout the entire dermis of the specimen (Figure). There were papillary projections of dilated ducts with a retained layer of myoepithelial cells surrounding the epithelial layers. Cytologic atypia was not appreciated. The patient was diagnosed with erosive adenomatosis of the nipple (EAN), also known as nipple adenoma. The lesion subsequently was treated and cleared with Mohs micrographic surgery (MMS). At 8-month follow-up there was no clinical recurrence of the lesion, and the patient was satisfied with the overall cosmetic appearance and conservation of the areola. The patient was followed clinically with annual breast examinations and mammography to monitor future recurrence.
Erosive adenomatosis of the nipple is an uncommon benign proliferative process of the lactiferous ducts of the nipple. Recognizing EAN is important because it resembles malignant breast diseases such as Paget disease of the nipple and invasive breast carcinoma. Due to these similarities, early cases of EAN have resulted in unnecessary mastectomies before the benignity of the condition was established.1 Accurate diagnosis is important to both the patient and the clinician for treatment planning as well as psychosocial consequences associated with the potential removal of this anatomically and cosmetically sensitive area.
Reviewing the literature on EAN is complicated by the variety of terms used to describe this condition, including but not limited to nipple adenoma, nipple duct adenoma, papillary adenoma of the nipple, and florid papillomatosis of the nipple. In 1955, Jones2 described EAN using the term florid papillomatosis of the nipple ducts. In 1962, Handley and Thackray1 argued that adenoma of the nipple was a more descriptive term because it more closely described the appearance of a sweat gland adenoma. They reasoned that adenoma of the nipple is a separate process from ductal papilloma due to the adenomatous proliferation into the nipple stroma rather than the lumen of the nipple ducts.1 The term adenoma of the nipple was further supported in 1965 by Taylor and Robertson.3 In 1959, Le Gal et al4 used the term erosive adenomatosis of the nipple to describe the erosive nature of nipple adenoma. The term nipple adenoma was published in the 2012 WHO Classification of Tumors of the Breast with 4 common histologic subtypes.5,6
Erosive adenomatosis of the nipple is clinically indistinguishable from Paget disease of the nipple, thus biopsy is essential for accurate diagnosis. In contrast to Paget disease, EAN tends to present in younger patients and progresses more slowly, and symptoms may be exacerbated around menstruation.1 Case reports demonstrate that patients may wait years before seeking medical attention for EAN.1,3,7,8 Presenting symptoms may include inflammation, crusting, nipple skin erosion, itching, and pain. Serous or sanguineous discharge from the lesions also is commonly reported. Palpation may reveal a small, hard, or elastic nodule within or underlying the nipple. In addition to Paget disease, EAN may resemble squamous cell carcinoma of the nipple, eczema, psoriasis, or a skin infection.6 Axillary lymphadenopathy is not present in the absence of a concomitant breast malignancy.8 On biopsy, nipple adenoma represents ductal proliferation of glandular structures within the stroma of the nipple that is well circumscribed but without borders. The erosive appearance of the lesion is produced by extensions of the glandular epithelium on the surface of the nipple.1,6 Specific to EAN is the presence of 2 cell types: an inner columnar epithelium and an outer cuboidal myoepithelium. These 2 cell types are present in normal lactiferous ducts; however, normal ducts are highly organized compared to EAN.9
After confirmation of EAN by nipple biopsy, complete surgical excision has been the gold standard for treatment, followed by reconstructive surgery.6 Handley and Thackray1 advocated for total excision of the nipple and areola with an underlying wedge of breast tissue to facilitate wound closure. More recently, successful alternative forms of treatment have been utilized to minimize disfiguring surgery. Alternative treatments include MMS,8 cryotherapy,10 and nipple splitting enucleation.6 Treatment with MMS has resulted in nipple sparing with the least amount of surface area sacrificed (1.1 cm2).9 Our case and prior case reports demonstrate that the tissue sparing potential of MMS is appropriate for the treatment of EAN, though traditionally it has been reserved for more malignant tumors. Preserving this sensitive area is both cosmetically and psychologically advantageous for the patient and thus should be considered when reviewing treatment options for EAN.
- Handley RS, Thackray AC. Adenoma of nipple. Br J Cancer. 1962;16:187-194.
- Jones DB. Florid papillomatosis of the nipple ducts. Cancer. 1955;8:315-319.
- Taylor HB, Robertson AG. Adenomas of the nipple. Cancer. 1965:18:995-1002.
- Le Gal Y, Gros CM, Bader P. Erosive adenomatosis of the nipple [in French]. Ann Anat Pathol (Paris). 1959;4:292-304.
- Eusebi V, Lester S. Tumours of the nipple. In: Lakhani SR, Ellis IO, Schnitt SJ, et al, eds. WHO Classification of Tumours of the Breast. Lyon, France: IARC; 2012.
- Spohn GP, Trotter SC, Tozbician G, et al. Nipple adenoma in a female patient presenting with persistent erythema of the right nipple skin: case report, review of the literature, clinical implications, and relevancy to health care providers who evaluate and treat patients with dermatologic conditions of the breast skin. BMC Dermatol. 2016;16:4.
- Kowal R, Miller CJ, Elenitsas R. Eroded patch on the nipple of a 57-year-old woman. Arch Dermatol. 2008;144:933-938.
- Van Mierlo PL, Geelen GM, Neumann HA. Mohs micrographic surgery for an erosive adenomatosis of the nipple. Dermatol Surg. 1998;24:681-683.
- Brankov N, Nino T, Hsiang D, et al. Utilizing Mohs surgery for tissue preservation in erosive adenomatosis of the nipple. Dermatol Surg. 2016;42:684-686.
- Kuflik EG. Erosive adenomatosis of the nipple treated with cryosurgery. J Am Acad Dermatol. 1998;38:270-271.
- Handley RS, Thackray AC. Adenoma of nipple. Br J Cancer. 1962;16:187-194.
- Jones DB. Florid papillomatosis of the nipple ducts. Cancer. 1955;8:315-319.
- Taylor HB, Robertson AG. Adenomas of the nipple. Cancer. 1965:18:995-1002.
- Le Gal Y, Gros CM, Bader P. Erosive adenomatosis of the nipple [in French]. Ann Anat Pathol (Paris). 1959;4:292-304.
- Eusebi V, Lester S. Tumours of the nipple. In: Lakhani SR, Ellis IO, Schnitt SJ, et al, eds. WHO Classification of Tumours of the Breast. Lyon, France: IARC; 2012.
- Spohn GP, Trotter SC, Tozbician G, et al. Nipple adenoma in a female patient presenting with persistent erythema of the right nipple skin: case report, review of the literature, clinical implications, and relevancy to health care providers who evaluate and treat patients with dermatologic conditions of the breast skin. BMC Dermatol. 2016;16:4.
- Kowal R, Miller CJ, Elenitsas R. Eroded patch on the nipple of a 57-year-old woman. Arch Dermatol. 2008;144:933-938.
- Van Mierlo PL, Geelen GM, Neumann HA. Mohs micrographic surgery for an erosive adenomatosis of the nipple. Dermatol Surg. 1998;24:681-683.
- Brankov N, Nino T, Hsiang D, et al. Utilizing Mohs surgery for tissue preservation in erosive adenomatosis of the nipple. Dermatol Surg. 2016;42:684-686.
- Kuflik EG. Erosive adenomatosis of the nipple treated with cryosurgery. J Am Acad Dermatol. 1998;38:270-271.
A 61-year-old woman presented with an asymptomatic, slowly enlarging, 9-mm, firm, red papule on the left nipple of 2 years' duration. She had no notable medical history, including a BI-RADS (Breast Imaging Reporting and Data System) mammogram score of 2 that was suggestive of benign findings 2 years prior. A repeat mammogram ordered by radiology and completed before presenting to dermatology had a BI-RADS score of 4, noting a concerning feature in the area of the lesion and prompting a biopsy.
Lambert-Eaton Myasthenic Syndrome and Merkel Cell Carcinoma
Merkel cell carcinoma (MCC) is an aggressive neuroendocrine malignancy of the skin that is thought to arise from neural crest cells. It has an estimated annual incidence of 0.6 per 100,000 individuals, typically occurs in the elderly population, and is most common in white males.1 The tumor presents as a rapidly growing, violaceous nodule in sun-exposed areas of the skin; early in the course, it can be mistaken for a benign entity such as an epidermal cyst.2 Merkel cell carcinoma has a propensity to spread to regional lymph nodes, and in some cases, it occurs in the absence of skin findings.3 Histologically, MCC is nearly indistinguishable from small cell lung carcinoma (SCLC).4 The overall prognosis for patients with MCC is poor and largely dependent on the stage at diagnosis. Patients with regional and distant metastases have a 5-year survival rate of 26% to 42% and 18%, respectively.3
Lambert-Eaton myasthenic syndrome (LEMS) is a paraneoplastic or autoimmune disorder of the neuromuscular junction that is found in 3% of cases of SCLC.4 Reported cases of LEMS in patients with MCC are exceedingly rare.5-8 We provide a full report and longitudinal clinical follow-up of a case that was briefly discussed by Simmons et al,8 and we review the literature regarding paraneoplastic syndromes associated with MCC and other extrapulmonary small cell carcinomas (EPSCCs).
Case Report
A 63-year-old man was evaluated in the neurology clinic due to difficulty walking, climbing stairs, and performing push-ups over the last month. Prior to the onset of symptoms, he was otherwise healthy, walking 3 miles daily; however, at presentation he required use of a cane. Leg weakness worsened as the day progressed. In addition, he reported constipation, urinary urgency, dry mouth, mild dysphagia, reduced sensation below the knees, and a nasal quality in his speech. He had no ptosis, diplopia, dysarthria, muscle cramps, myalgia, or facial weakness. He denied fevers, chills, and night sweats but did admit to an unintentional 10- to 15-lb weight loss over the preceding few months.
The neurologic examination revealed mild proximal upper extremity weakness in the bilateral shoulder abductors, infraspinatus, hip extensors, and hip flexors (Medical Research Council muscle scale grade 4). All deep tendon reflexes, except the Achilles reflex, were present. Despite subjective sensory concerns, objective examination of all sensory modalities was normal. Cranial nerve examination was normal, except for a slight nasal quality to his voice.
A qualitative assay was positive for the presence of P/Q-type voltage-gated calcium channel (VGCC) antibodies. Other laboratory studies were within reference range, including acetylcholine-receptor antibodies (blocking, binding, and modulating) and muscle-specific kinase antibodies.
Lumbar and cervical spine magnetic resonance imaging revealed multilevel neuroforaminal stenosis without spinal canal stenosis or myelopathy. Computed tomography (CT) of the chest was notable for 2 pathologically enlarged lymph nodes in the left axilla and no evidence of primary pulmonary malignancy. Nerve-conduction studies (NCSs) in conjunction with other clinical findings were consistent with the diagnosis of LEMS.
Ultrasound-guided biopsy of the enlarged axillary lymph nodes demonstrated sheets and nests of small round blue tumor cells with minimal cytoplasm, high mitotic rate, and foci of necrosis (Figure 1). The tumor cells were positive for pancytokeratin (Lu-5) and cytokeratin (CK) 20 in a perinuclear dotlike pattern (Figure 2), as well as for the neuroendocrine markers synaptophysin (Figure 3), chromogranin A, and CD56. The tumor cells showed no immunoreactivity for CK7, thyroid transcription factor 1, CD3, CD5, or CD20. Flow cytometry demonstrated low cellularity, low specimen viability, and no evidence of an abnormal B-cell population. These findings were consistent with the diagnosis of MCC.
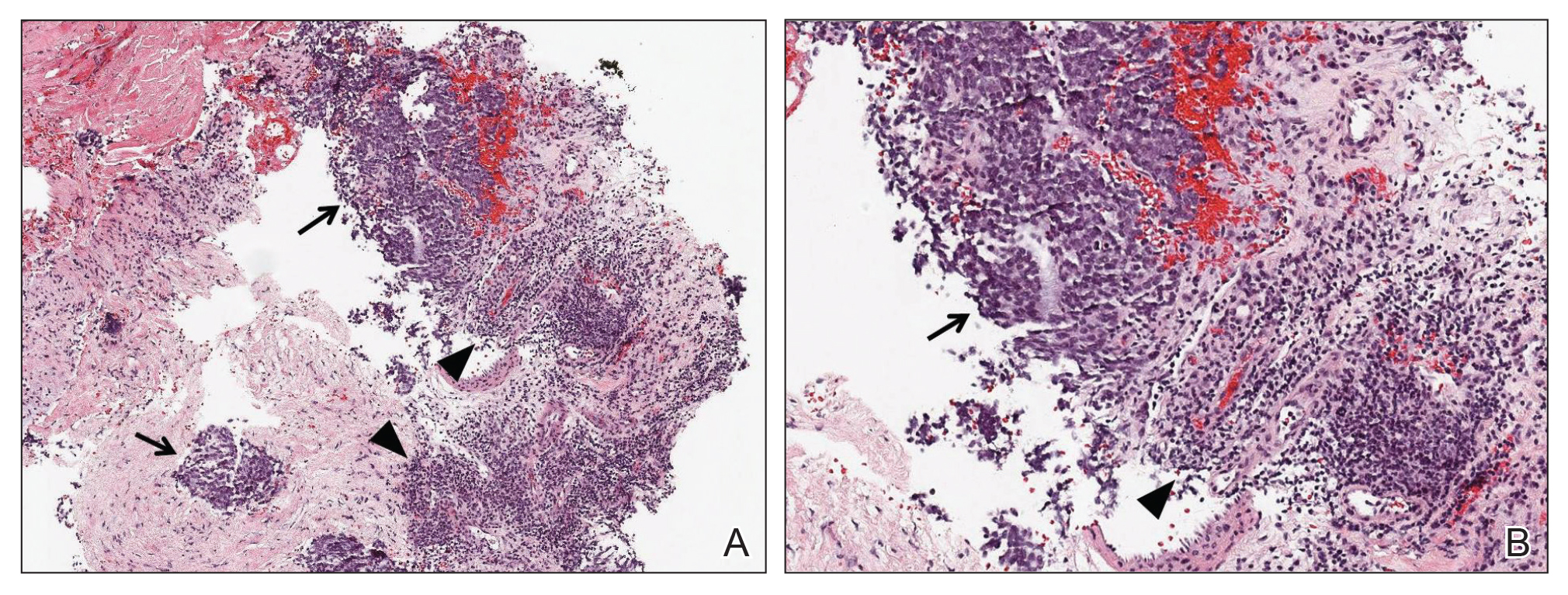
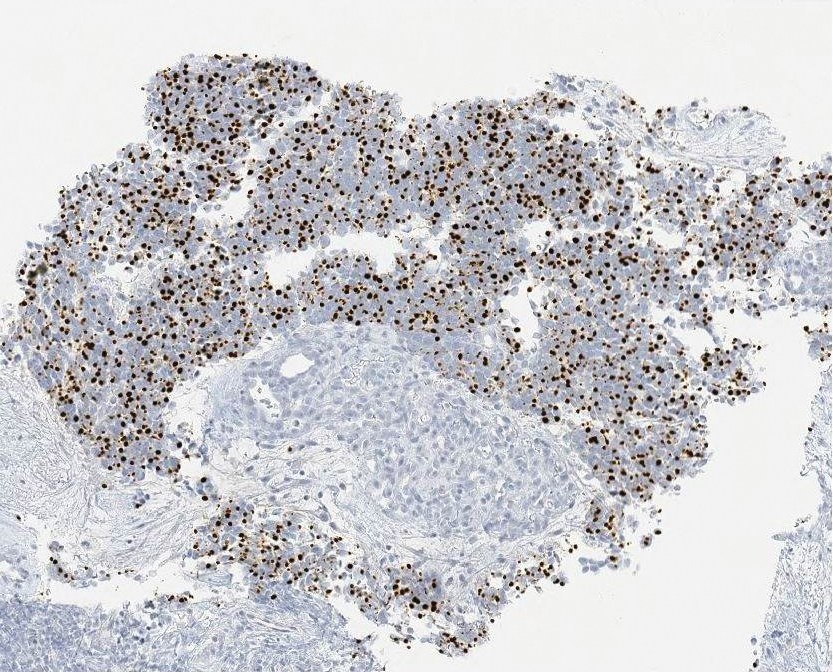
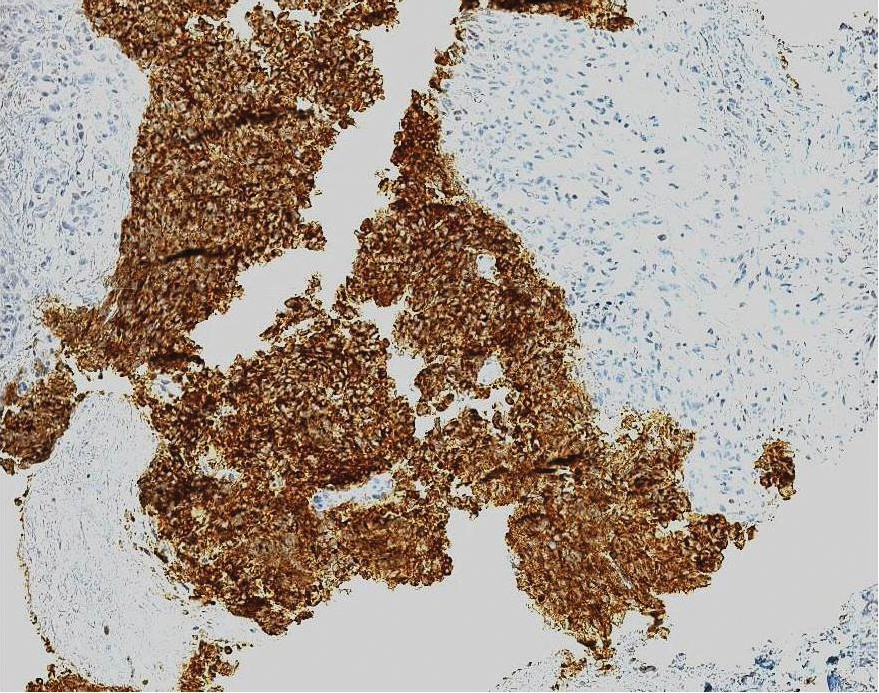
The patient underwent surgical excision of the involved lymph nodes. Four weeks after surgery, he reported dramatic improvement in strength, with complete resolution of the nasal speech, dysphagia, dry mouth, urinary retention, and constipation. Two months after surgery, his strength had normalized, except for slight persistent weakness in the bilateral shoulder abductors, trace weakness in the hip flexors, and a slight Trendelenburg gait. He was able to rise from a chair without using his arms and no longer required a cane for ambulation.
The patient underwent adjuvant radiation therapy after 2-month surgical follow-up with 5000-cGy radiation treatment to the left axillary region. Six months following primary definitive surgery and 4 months following adjuvant radiation therapy, he reported a 95% subjective return of physical strength. The patient was able to return to near-baseline physical activity. He continued to deny symptoms of dry mouth, incontinence, or constipation. Objectively, he had no focal neurologic deficits or weakness; no evidence of new skin lesions or lymphadenopathy was noted.
Comment
MCC vs SCLC
Merkel cell carcinoma is classified as a type of EPSCC. The histologic appearance of MCC is indistinguishable from SCLC. Both tumors are composed of uniform sheets of small round cells with a high nucleus to cytoplasm ratio, and both can express neuroendocrine markers, such as neuron-specific enolase, chromogranin A, and synaptophysin.9 Immunohistochemical positivity for CK20 and neurofilaments in combination with negative staining for thyroid transcription factor 1 and CK7 effectively differentiate MCC from SCLC.9 In addition, MCC often displays CK20 positivity in a perinuclear dotlike or punctate pattern, which is characteristic of this tumor.3,9,10 Negative immunohistochemical markers for B cells (CD20) and T cells (CD3) are important in excluding lymphoma.
LEMS Diagnosis
Lambert-Eaton myasthenic syndrome is a paraneoplastic or autoimmune disorder involving the neuromuscular junction. Autoantibodies to VGCC impair calcium influx into the presynaptic terminal, resulting in marked reduction of acetylcholine release into the synaptic cleft. The reduction in acetylcholine activity impairs production of muscle fiber action potentials, resulting in clinical weakness. The diagnosis of LEMS rests on clinical presentation, positive serology, and confirmatory neurophysiologic testing by NCS. Clinically, patients present with proximal weakness, hyporeflexia or areflexia, and autonomic dysfunction. Antibodies to P/Q-type VGCCs are found in 85% to 90% of cases of LEMS and are thought to play a direct causative role in the development of weakness.11 The finding of postexercise facilitation on motor NCS is the neurophysiologic hallmark and is highly specific for the diagnosis.
Approximately 50% to 60% of patients who present with LEMS have an underlying tumor, the vast majority of which are SCLC.11 There are a few reports of LEMS associated with other malignancies, including lymphoma; thymoma; neuroblastoma; and carcinoma of the breast, stomach, prostate, bladder, kidney, and gallbladder.12 Patients with nontumor or autoimmune LEMS tend to be younger, and there is no male predominance, as there is in paraneoplastic LEMS.13 Given the risk of underlying malignancy in LEMS, Titulaer et al14 proposed a screening protocol for patients presenting with LEMS, recommending initial primary screening using CT of the chest. If the CT scan is negative, total-body fludeoxyglucose positron emission tomography should be performed to assess for fludeoxyglucose avid lesions. If both initial studies are negative, routine follow-up with CT of the chest at 6-month intervals for a minimum of 2 to 4 years after the initial diagnosis of LEMS was recommended. An exception to this protocol was suggested to allow consideration to stop screening after the first 6-month follow-up chest CT for patients younger than 45 years who have never smoked and who have an HLA 8.1 haplotype for which nontumor LEMS would be a more probable diagnosis.14
In addition to a screening protocol, a validated prediction tool, the Dutch-English LEMS Tumor Association prediction score, was developed. It uses common signs and symptoms of LEMS and risk factors for SCLC to help guide the need for further screening.15
Paraneoplastic Syndromes Associated With MCC
Other paraneoplastic syndromes have been reported in association with MCC. A patient with brainstem encephalitis associated with MCC was reported in a trial of a novel immunotherapy for paraneoplastic neurologic syndromes.16,17 A syndrome of inappropriate antidiuretic hormone (SIADH) secretion was reported in a patient with N-type calcium channel antibodies.18 Two cases of paraneoplastic cerebellar degeneration have been reported; the first was associated with a novel 70-kD antibody,19 and the second was associated with the P/Q-type VGCC antibody.20 Anti-Hu antibodies have been found in a handful of reports of neurologic deterioration in patients with MCC. Hocar et al21 reported a severe necrotizing myopathy; Greenlee et al22 described a syndrome of progressive sensorimotor and autonomic neuropathy with encephalopathy; and Lopez et al23 described a constellation of vision changes, gait imbalance, and proximal weakness. Support for a pathophysiologic connection among these 3 cases is suggested by the finding of Hu antigen expression by MCC in 2 studies.24,25 Because MCC can present with occult lymph node involvement in the absence of primary cutaneous findings,3 there are more cases of paraneoplastic neurologic syndromes that were not recognized.
Extrapulmonary small cell carcinomas such as MCC are morphologically indistinguishable from their pulmonary counterparts and have been reported in most anatomic regions of the body, including gynecologic organs (eg, ovaries, cervix), genitourinary organs (eg, bladder, prostate), the gastrointestinal tract (eg, esophagus), skin (eg, MCC), and the head and neck region. Extrapulmonary small cell carcinoma is a rare entity, with the most common form found in the gynecologic tract, representing only 2% of gynecologic malignancies.26
Paraneoplastic syndromes of EPSCC are rare given the paucity of the malignancy. Several case reports discuss findings of SIADH in EPSCC of the cervix, as well as hypercalcemia, polyneuropathy, Cushing syndrome, limbic encephalitis, and peripheral neuropathy in EPSCC of the prostate.27,28 In contrast, SCLC has long been associated with paraneoplastic syndromes. Numerous case reports have been published describing SCLC-associated paraneoplastic syndromes to include hypercalcemia, Cushing syndrome, SIADH, vasoactive peptide production, cerebellar degeneration, limbic encephalitis, visceral plexopathy, autonomic dysfunction, and LEMS.29 As more cases of EPSCC with paraneoplastic syndromes are identified and reported, we might gain a better understanding of this interesting phenomenon.
Conclusion
Merkel cell carcinoma is an aggressive neuroendocrine malignancy associated with paraneoplastic neurologic syndromes, including LEMS. A thorough search for an underlying malignancy is highly recommended in patients with diagnosed LEMS without clear cause. Early identification and treatment of the primary tumor can lead to improvement of neurologic symptoms.
We present a case of LEMS with no clearly identifiable cause on presentation with later diagnosis of metastatic MCC of unknown primary origin. After surgical excision of affected lymph nodes and adjuvant radiation therapy, the patient had near-complete resolution of LEMS symptoms at 6-month follow-up, without additional findings of lymphadenopathy or skin lesions. Although this patient is not undergoing routine surveillance imaging to monitor for recurrence of MCC, a chest CT or positron emission tomography–CT for secondary screening would be considered if the patient experienced clinical symptoms consistent with LEMS.
In cases of LEMS without pulmonary malignancy, we recommend considering MCC in the differential diagnosis during the workup of an underlying malignancy
- Albores-Saavedra J, Batich K, Chable-Montero F, et al. Merkel cell carcinoma demographics, morphology, and survival based on 3870 cases: a population based study. J Cutan Pathol. 2010;37:20-27.
- Senchenkov A, Moran SL. Merkel cell carcinoma: diagnosis, management, and outcomes. Plast Reconstr Surg. 2013;131:E771-E778.
- Han SY, North JP, Canavan T, et al. Merkel cell carcinoma. Hematol Oncol Clin N Am. 2012;26:1351-1374.
- Vernino S. Paraneoplastic disorders affecting the neuromuscular junction or anterior horn cell. CONTINUUM Lifelong Learning in Neurology. 2009;15:132-146.
- Eggers SD, Salomao DR, Dinapoli RP, et al. Paraneoplastic and metastatic neurologic complications of Merkel cell carcinoma. Mayo Clin Proc. 2001;76:327-330.
- Siau RT, Morris A, Karoo RO. Surgery results in complete cure of Lambert-Eaton myasthenic syndrome in a patient with metastatic Merkel cell carcinoma. J Plast Reconstr Aesthet Surg. 2014;67:e162-e164.
- Bombelli F, Lispi L, Calabrò F, et al. Lambert-Eaton myasthenic syndrome associated to Merkel cell carcinoma: report of a case. Neurol Sci. 2015;36:1491-1492.
- Simmons DB, Duginski TM, McClean JC, et al. Lambert-eaton myasthenic syndrome and merkel cell carcinoma. Muscle Nerve. 2015;53:325-326.
- Bobos M, Hytiroglou P, Kostopoulos I, et al. Immunohistochemical distinction between Merkel cell carcinoma and small cell carcinoma of the lung. Am J Dermatopathol. 2006;28:99-104.
- Jensen K, Kohler S, Rouse RV. Cytokeratin staining in Merkel cell carcinoma: an immunohistochemical study of cytokeratins 5/6, 7, 17, and 20. Appl Immunohistochem Mol Morphol. 2000;8:310-315.
- Titulaer MJ, Lang B, Verschuuren JJ. Lambert-Eaton myasthenic syndrome: from clinical characteristics to therapeutic strategies. Lancet Neurol. 2011;10:1098-1107.
- Sanders DB. Lambert-Eaton myasthenic syndrome. In: Daroff R, Aminoff MJ, eds. Encyclopedia of the Neurological Sciences. Vol 2. New York, NY: Elsevier; 2009:819-822.
- Wirtz PW, Smallegange TM, Wintzen AR, et al. Differences in clinical features between the Lambert-Eaton myasthenic syndrome with and without cancer: an analysis of 227 published cases. Clin Neurol Neurosurg. 2002;104:359-363.
- Titulaer MJ, Wirtz PW, Willems LN, et al. Screening for small-cell lung cancer: a follow-up study of patients with Lambert-Eaton myasthenic syndrome. J Clin Oncol. 2008;26:4276-4281.
- Titulaer MJ, Maddison P, Sont JK, et al. Clinical Dutch-English Lambert-Eaton Myasthenic Syndrome (LEMS) Tumor Association prediction score accurately predicts small-cell lung cancer in the LEMS. J Clin Oncol. 2011;7:902-908.
- Cher LM, Hochberg FH, Teruya J, et al. Therapy for paraneoplastic neurologic syndromes in six patients with protein A column immunoadsorption. Cancer. 1995;75:1678-1683.
- Batchelor TT, Platten M, Hochberg FH. Immunoadsorption therapy for paraneoplastic syndromes. J Neurooncol. 1998;40:131-136.
- Blondin NA, Vortmeyer AO, Harel NY. Paraneoplastic syndrome of inappropriate antidiuretic hormone mimicking limbic encephalitis. Arch Neurol. 2011;68:1591-1594.
- Balegno S, Ceroni M, Corato M, et al. Antibodies to cerebellar nerve fibres in two patients with paraneoplastic cerebellar ataxia. Anticancer Res. 2005;25:3211-3214.
- Zhang C, Emery L, Lancaster E. Paraneoplastic cerebellar degeneration associated with noncutaneous Merkel cell carcinoma. Neurol Neuroimmunol Neuroinflamm. 2014;1:e17.
- Hocar O, Poszepczynska-Guigné E, Faye O, et al. Severe necrotizing myopathy subsequent to Merkel cell carcinoma. Ann Dermatol Venereol. 2011;138:130-134.
- Greenlee JE, Steffens JD, Clawson SA, et al. Anti-Hu antibodies in Merkel cell carcinoma. Ann Neurol. 2002;52:111-115.
- Lopez MC, Pericay C, Agustí M, et al. Merkel cell carcinoma associated with a paraneoplastic neurologic syndrome. Histopathology. 2004;44:628-629.
- Dalmau J, Furneaux HM, Cordon-Cardo C, et al. The expression of the Hu (paraneoplastic encephalomyelitis/sensory neuronopathy) antigen in human normal and tumor tissues. Am J Pathol. 1992;141:881-886.
- Gultekin SH, Rosai J, Demopoulos A, et al. Hu immunolabeling as a marker of neural and neuroendocrine differentiation in normal and neoplastic human tissues: assessment using a recombinant anti-Hu Fab fragment. Int J Surg Pathol. 2000;8:109-117.
- Zheng X, Liu D, Fallon JT, et al. Distinct genetic alterations in small cell carcinoma from different anatomic sites. Exp Hematol Oncol. 2015;4:2.
- Kim D, Yun H, Lee Y, et al. Small cell neuroendocrine carcinoma of the uterine cervix presenting with syndrome of inappropriate antidiuretic hormone secretion. Obstet Gynecol Sci. 2013;56:420-425.
- Venkatesh PK, Motwani B, Sherman N, et al. Metastatic pure small-cell carcinoma of prostate. Am J Med Sci. 2004;328:286-289.
- Kaltsas G, Androulakis II, de Herder WW, et al. Paraneoplastic syndromes secondary to neuroendocrine tumours. Endocr Relat Cancer. 2010;17:R173-R193.
Merkel cell carcinoma (MCC) is an aggressive neuroendocrine malignancy of the skin that is thought to arise from neural crest cells. It has an estimated annual incidence of 0.6 per 100,000 individuals, typically occurs in the elderly population, and is most common in white males.1 The tumor presents as a rapidly growing, violaceous nodule in sun-exposed areas of the skin; early in the course, it can be mistaken for a benign entity such as an epidermal cyst.2 Merkel cell carcinoma has a propensity to spread to regional lymph nodes, and in some cases, it occurs in the absence of skin findings.3 Histologically, MCC is nearly indistinguishable from small cell lung carcinoma (SCLC).4 The overall prognosis for patients with MCC is poor and largely dependent on the stage at diagnosis. Patients with regional and distant metastases have a 5-year survival rate of 26% to 42% and 18%, respectively.3
Lambert-Eaton myasthenic syndrome (LEMS) is a paraneoplastic or autoimmune disorder of the neuromuscular junction that is found in 3% of cases of SCLC.4 Reported cases of LEMS in patients with MCC are exceedingly rare.5-8 We provide a full report and longitudinal clinical follow-up of a case that was briefly discussed by Simmons et al,8 and we review the literature regarding paraneoplastic syndromes associated with MCC and other extrapulmonary small cell carcinomas (EPSCCs).
Case Report
A 63-year-old man was evaluated in the neurology clinic due to difficulty walking, climbing stairs, and performing push-ups over the last month. Prior to the onset of symptoms, he was otherwise healthy, walking 3 miles daily; however, at presentation he required use of a cane. Leg weakness worsened as the day progressed. In addition, he reported constipation, urinary urgency, dry mouth, mild dysphagia, reduced sensation below the knees, and a nasal quality in his speech. He had no ptosis, diplopia, dysarthria, muscle cramps, myalgia, or facial weakness. He denied fevers, chills, and night sweats but did admit to an unintentional 10- to 15-lb weight loss over the preceding few months.
The neurologic examination revealed mild proximal upper extremity weakness in the bilateral shoulder abductors, infraspinatus, hip extensors, and hip flexors (Medical Research Council muscle scale grade 4). All deep tendon reflexes, except the Achilles reflex, were present. Despite subjective sensory concerns, objective examination of all sensory modalities was normal. Cranial nerve examination was normal, except for a slight nasal quality to his voice.
A qualitative assay was positive for the presence of P/Q-type voltage-gated calcium channel (VGCC) antibodies. Other laboratory studies were within reference range, including acetylcholine-receptor antibodies (blocking, binding, and modulating) and muscle-specific kinase antibodies.
Lumbar and cervical spine magnetic resonance imaging revealed multilevel neuroforaminal stenosis without spinal canal stenosis or myelopathy. Computed tomography (CT) of the chest was notable for 2 pathologically enlarged lymph nodes in the left axilla and no evidence of primary pulmonary malignancy. Nerve-conduction studies (NCSs) in conjunction with other clinical findings were consistent with the diagnosis of LEMS.
Ultrasound-guided biopsy of the enlarged axillary lymph nodes demonstrated sheets and nests of small round blue tumor cells with minimal cytoplasm, high mitotic rate, and foci of necrosis (Figure 1). The tumor cells were positive for pancytokeratin (Lu-5) and cytokeratin (CK) 20 in a perinuclear dotlike pattern (Figure 2), as well as for the neuroendocrine markers synaptophysin (Figure 3), chromogranin A, and CD56. The tumor cells showed no immunoreactivity for CK7, thyroid transcription factor 1, CD3, CD5, or CD20. Flow cytometry demonstrated low cellularity, low specimen viability, and no evidence of an abnormal B-cell population. These findings were consistent with the diagnosis of MCC.
The patient underwent surgical excision of the involved lymph nodes. Four weeks after surgery, he reported dramatic improvement in strength, with complete resolution of the nasal speech, dysphagia, dry mouth, urinary retention, and constipation. Two months after surgery, his strength had normalized, except for slight persistent weakness in the bilateral shoulder abductors, trace weakness in the hip flexors, and a slight Trendelenburg gait. He was able to rise from a chair without using his arms and no longer required a cane for ambulation.
The patient underwent adjuvant radiation therapy after 2-month surgical follow-up with 5000-cGy radiation treatment to the left axillary region. Six months following primary definitive surgery and 4 months following adjuvant radiation therapy, he reported a 95% subjective return of physical strength. The patient was able to return to near-baseline physical activity. He continued to deny symptoms of dry mouth, incontinence, or constipation. Objectively, he had no focal neurologic deficits or weakness; no evidence of new skin lesions or lymphadenopathy was noted.
Comment
MCC vs SCLC
Merkel cell carcinoma is classified as a type of EPSCC. The histologic appearance of MCC is indistinguishable from SCLC. Both tumors are composed of uniform sheets of small round cells with a high nucleus to cytoplasm ratio, and both can express neuroendocrine markers, such as neuron-specific enolase, chromogranin A, and synaptophysin.9 Immunohistochemical positivity for CK20 and neurofilaments in combination with negative staining for thyroid transcription factor 1 and CK7 effectively differentiate MCC from SCLC.9 In addition, MCC often displays CK20 positivity in a perinuclear dotlike or punctate pattern, which is characteristic of this tumor.3,9,10 Negative immunohistochemical markers for B cells (CD20) and T cells (CD3) are important in excluding lymphoma.
LEMS Diagnosis
Lambert-Eaton myasthenic syndrome is a paraneoplastic or autoimmune disorder involving the neuromuscular junction. Autoantibodies to VGCC impair calcium influx into the presynaptic terminal, resulting in marked reduction of acetylcholine release into the synaptic cleft. The reduction in acetylcholine activity impairs production of muscle fiber action potentials, resulting in clinical weakness. The diagnosis of LEMS rests on clinical presentation, positive serology, and confirmatory neurophysiologic testing by NCS. Clinically, patients present with proximal weakness, hyporeflexia or areflexia, and autonomic dysfunction. Antibodies to P/Q-type VGCCs are found in 85% to 90% of cases of LEMS and are thought to play a direct causative role in the development of weakness.11 The finding of postexercise facilitation on motor NCS is the neurophysiologic hallmark and is highly specific for the diagnosis.
Approximately 50% to 60% of patients who present with LEMS have an underlying tumor, the vast majority of which are SCLC.11 There are a few reports of LEMS associated with other malignancies, including lymphoma; thymoma; neuroblastoma; and carcinoma of the breast, stomach, prostate, bladder, kidney, and gallbladder.12 Patients with nontumor or autoimmune LEMS tend to be younger, and there is no male predominance, as there is in paraneoplastic LEMS.13 Given the risk of underlying malignancy in LEMS, Titulaer et al14 proposed a screening protocol for patients presenting with LEMS, recommending initial primary screening using CT of the chest. If the CT scan is negative, total-body fludeoxyglucose positron emission tomography should be performed to assess for fludeoxyglucose avid lesions. If both initial studies are negative, routine follow-up with CT of the chest at 6-month intervals for a minimum of 2 to 4 years after the initial diagnosis of LEMS was recommended. An exception to this protocol was suggested to allow consideration to stop screening after the first 6-month follow-up chest CT for patients younger than 45 years who have never smoked and who have an HLA 8.1 haplotype for which nontumor LEMS would be a more probable diagnosis.14
In addition to a screening protocol, a validated prediction tool, the Dutch-English LEMS Tumor Association prediction score, was developed. It uses common signs and symptoms of LEMS and risk factors for SCLC to help guide the need for further screening.15
Paraneoplastic Syndromes Associated With MCC
Other paraneoplastic syndromes have been reported in association with MCC. A patient with brainstem encephalitis associated with MCC was reported in a trial of a novel immunotherapy for paraneoplastic neurologic syndromes.16,17 A syndrome of inappropriate antidiuretic hormone (SIADH) secretion was reported in a patient with N-type calcium channel antibodies.18 Two cases of paraneoplastic cerebellar degeneration have been reported; the first was associated with a novel 70-kD antibody,19 and the second was associated with the P/Q-type VGCC antibody.20 Anti-Hu antibodies have been found in a handful of reports of neurologic deterioration in patients with MCC. Hocar et al21 reported a severe necrotizing myopathy; Greenlee et al22 described a syndrome of progressive sensorimotor and autonomic neuropathy with encephalopathy; and Lopez et al23 described a constellation of vision changes, gait imbalance, and proximal weakness. Support for a pathophysiologic connection among these 3 cases is suggested by the finding of Hu antigen expression by MCC in 2 studies.24,25 Because MCC can present with occult lymph node involvement in the absence of primary cutaneous findings,3 there are more cases of paraneoplastic neurologic syndromes that were not recognized.
Extrapulmonary small cell carcinomas such as MCC are morphologically indistinguishable from their pulmonary counterparts and have been reported in most anatomic regions of the body, including gynecologic organs (eg, ovaries, cervix), genitourinary organs (eg, bladder, prostate), the gastrointestinal tract (eg, esophagus), skin (eg, MCC), and the head and neck region. Extrapulmonary small cell carcinoma is a rare entity, with the most common form found in the gynecologic tract, representing only 2% of gynecologic malignancies.26
Paraneoplastic syndromes of EPSCC are rare given the paucity of the malignancy. Several case reports discuss findings of SIADH in EPSCC of the cervix, as well as hypercalcemia, polyneuropathy, Cushing syndrome, limbic encephalitis, and peripheral neuropathy in EPSCC of the prostate.27,28 In contrast, SCLC has long been associated with paraneoplastic syndromes. Numerous case reports have been published describing SCLC-associated paraneoplastic syndromes to include hypercalcemia, Cushing syndrome, SIADH, vasoactive peptide production, cerebellar degeneration, limbic encephalitis, visceral plexopathy, autonomic dysfunction, and LEMS.29 As more cases of EPSCC with paraneoplastic syndromes are identified and reported, we might gain a better understanding of this interesting phenomenon.
Conclusion
Merkel cell carcinoma is an aggressive neuroendocrine malignancy associated with paraneoplastic neurologic syndromes, including LEMS. A thorough search for an underlying malignancy is highly recommended in patients with diagnosed LEMS without clear cause. Early identification and treatment of the primary tumor can lead to improvement of neurologic symptoms.
We present a case of LEMS with no clearly identifiable cause on presentation with later diagnosis of metastatic MCC of unknown primary origin. After surgical excision of affected lymph nodes and adjuvant radiation therapy, the patient had near-complete resolution of LEMS symptoms at 6-month follow-up, without additional findings of lymphadenopathy or skin lesions. Although this patient is not undergoing routine surveillance imaging to monitor for recurrence of MCC, a chest CT or positron emission tomography–CT for secondary screening would be considered if the patient experienced clinical symptoms consistent with LEMS.
In cases of LEMS without pulmonary malignancy, we recommend considering MCC in the differential diagnosis during the workup of an underlying malignancy
Merkel cell carcinoma (MCC) is an aggressive neuroendocrine malignancy of the skin that is thought to arise from neural crest cells. It has an estimated annual incidence of 0.6 per 100,000 individuals, typically occurs in the elderly population, and is most common in white males.1 The tumor presents as a rapidly growing, violaceous nodule in sun-exposed areas of the skin; early in the course, it can be mistaken for a benign entity such as an epidermal cyst.2 Merkel cell carcinoma has a propensity to spread to regional lymph nodes, and in some cases, it occurs in the absence of skin findings.3 Histologically, MCC is nearly indistinguishable from small cell lung carcinoma (SCLC).4 The overall prognosis for patients with MCC is poor and largely dependent on the stage at diagnosis. Patients with regional and distant metastases have a 5-year survival rate of 26% to 42% and 18%, respectively.3
Lambert-Eaton myasthenic syndrome (LEMS) is a paraneoplastic or autoimmune disorder of the neuromuscular junction that is found in 3% of cases of SCLC.4 Reported cases of LEMS in patients with MCC are exceedingly rare.5-8 We provide a full report and longitudinal clinical follow-up of a case that was briefly discussed by Simmons et al,8 and we review the literature regarding paraneoplastic syndromes associated with MCC and other extrapulmonary small cell carcinomas (EPSCCs).
Case Report
A 63-year-old man was evaluated in the neurology clinic due to difficulty walking, climbing stairs, and performing push-ups over the last month. Prior to the onset of symptoms, he was otherwise healthy, walking 3 miles daily; however, at presentation he required use of a cane. Leg weakness worsened as the day progressed. In addition, he reported constipation, urinary urgency, dry mouth, mild dysphagia, reduced sensation below the knees, and a nasal quality in his speech. He had no ptosis, diplopia, dysarthria, muscle cramps, myalgia, or facial weakness. He denied fevers, chills, and night sweats but did admit to an unintentional 10- to 15-lb weight loss over the preceding few months.
The neurologic examination revealed mild proximal upper extremity weakness in the bilateral shoulder abductors, infraspinatus, hip extensors, and hip flexors (Medical Research Council muscle scale grade 4). All deep tendon reflexes, except the Achilles reflex, were present. Despite subjective sensory concerns, objective examination of all sensory modalities was normal. Cranial nerve examination was normal, except for a slight nasal quality to his voice.
A qualitative assay was positive for the presence of P/Q-type voltage-gated calcium channel (VGCC) antibodies. Other laboratory studies were within reference range, including acetylcholine-receptor antibodies (blocking, binding, and modulating) and muscle-specific kinase antibodies.
Lumbar and cervical spine magnetic resonance imaging revealed multilevel neuroforaminal stenosis without spinal canal stenosis or myelopathy. Computed tomography (CT) of the chest was notable for 2 pathologically enlarged lymph nodes in the left axilla and no evidence of primary pulmonary malignancy. Nerve-conduction studies (NCSs) in conjunction with other clinical findings were consistent with the diagnosis of LEMS.
Ultrasound-guided biopsy of the enlarged axillary lymph nodes demonstrated sheets and nests of small round blue tumor cells with minimal cytoplasm, high mitotic rate, and foci of necrosis (Figure 1). The tumor cells were positive for pancytokeratin (Lu-5) and cytokeratin (CK) 20 in a perinuclear dotlike pattern (Figure 2), as well as for the neuroendocrine markers synaptophysin (Figure 3), chromogranin A, and CD56. The tumor cells showed no immunoreactivity for CK7, thyroid transcription factor 1, CD3, CD5, or CD20. Flow cytometry demonstrated low cellularity, low specimen viability, and no evidence of an abnormal B-cell population. These findings were consistent with the diagnosis of MCC.
The patient underwent surgical excision of the involved lymph nodes. Four weeks after surgery, he reported dramatic improvement in strength, with complete resolution of the nasal speech, dysphagia, dry mouth, urinary retention, and constipation. Two months after surgery, his strength had normalized, except for slight persistent weakness in the bilateral shoulder abductors, trace weakness in the hip flexors, and a slight Trendelenburg gait. He was able to rise from a chair without using his arms and no longer required a cane for ambulation.
The patient underwent adjuvant radiation therapy after 2-month surgical follow-up with 5000-cGy radiation treatment to the left axillary region. Six months following primary definitive surgery and 4 months following adjuvant radiation therapy, he reported a 95% subjective return of physical strength. The patient was able to return to near-baseline physical activity. He continued to deny symptoms of dry mouth, incontinence, or constipation. Objectively, he had no focal neurologic deficits or weakness; no evidence of new skin lesions or lymphadenopathy was noted.
Comment
MCC vs SCLC
Merkel cell carcinoma is classified as a type of EPSCC. The histologic appearance of MCC is indistinguishable from SCLC. Both tumors are composed of uniform sheets of small round cells with a high nucleus to cytoplasm ratio, and both can express neuroendocrine markers, such as neuron-specific enolase, chromogranin A, and synaptophysin.9 Immunohistochemical positivity for CK20 and neurofilaments in combination with negative staining for thyroid transcription factor 1 and CK7 effectively differentiate MCC from SCLC.9 In addition, MCC often displays CK20 positivity in a perinuclear dotlike or punctate pattern, which is characteristic of this tumor.3,9,10 Negative immunohistochemical markers for B cells (CD20) and T cells (CD3) are important in excluding lymphoma.
LEMS Diagnosis
Lambert-Eaton myasthenic syndrome is a paraneoplastic or autoimmune disorder involving the neuromuscular junction. Autoantibodies to VGCC impair calcium influx into the presynaptic terminal, resulting in marked reduction of acetylcholine release into the synaptic cleft. The reduction in acetylcholine activity impairs production of muscle fiber action potentials, resulting in clinical weakness. The diagnosis of LEMS rests on clinical presentation, positive serology, and confirmatory neurophysiologic testing by NCS. Clinically, patients present with proximal weakness, hyporeflexia or areflexia, and autonomic dysfunction. Antibodies to P/Q-type VGCCs are found in 85% to 90% of cases of LEMS and are thought to play a direct causative role in the development of weakness.11 The finding of postexercise facilitation on motor NCS is the neurophysiologic hallmark and is highly specific for the diagnosis.
Approximately 50% to 60% of patients who present with LEMS have an underlying tumor, the vast majority of which are SCLC.11 There are a few reports of LEMS associated with other malignancies, including lymphoma; thymoma; neuroblastoma; and carcinoma of the breast, stomach, prostate, bladder, kidney, and gallbladder.12 Patients with nontumor or autoimmune LEMS tend to be younger, and there is no male predominance, as there is in paraneoplastic LEMS.13 Given the risk of underlying malignancy in LEMS, Titulaer et al14 proposed a screening protocol for patients presenting with LEMS, recommending initial primary screening using CT of the chest. If the CT scan is negative, total-body fludeoxyglucose positron emission tomography should be performed to assess for fludeoxyglucose avid lesions. If both initial studies are negative, routine follow-up with CT of the chest at 6-month intervals for a minimum of 2 to 4 years after the initial diagnosis of LEMS was recommended. An exception to this protocol was suggested to allow consideration to stop screening after the first 6-month follow-up chest CT for patients younger than 45 years who have never smoked and who have an HLA 8.1 haplotype for which nontumor LEMS would be a more probable diagnosis.14
In addition to a screening protocol, a validated prediction tool, the Dutch-English LEMS Tumor Association prediction score, was developed. It uses common signs and symptoms of LEMS and risk factors for SCLC to help guide the need for further screening.15
Paraneoplastic Syndromes Associated With MCC
Other paraneoplastic syndromes have been reported in association with MCC. A patient with brainstem encephalitis associated with MCC was reported in a trial of a novel immunotherapy for paraneoplastic neurologic syndromes.16,17 A syndrome of inappropriate antidiuretic hormone (SIADH) secretion was reported in a patient with N-type calcium channel antibodies.18 Two cases of paraneoplastic cerebellar degeneration have been reported; the first was associated with a novel 70-kD antibody,19 and the second was associated with the P/Q-type VGCC antibody.20 Anti-Hu antibodies have been found in a handful of reports of neurologic deterioration in patients with MCC. Hocar et al21 reported a severe necrotizing myopathy; Greenlee et al22 described a syndrome of progressive sensorimotor and autonomic neuropathy with encephalopathy; and Lopez et al23 described a constellation of vision changes, gait imbalance, and proximal weakness. Support for a pathophysiologic connection among these 3 cases is suggested by the finding of Hu antigen expression by MCC in 2 studies.24,25 Because MCC can present with occult lymph node involvement in the absence of primary cutaneous findings,3 there are more cases of paraneoplastic neurologic syndromes that were not recognized.
Extrapulmonary small cell carcinomas such as MCC are morphologically indistinguishable from their pulmonary counterparts and have been reported in most anatomic regions of the body, including gynecologic organs (eg, ovaries, cervix), genitourinary organs (eg, bladder, prostate), the gastrointestinal tract (eg, esophagus), skin (eg, MCC), and the head and neck region. Extrapulmonary small cell carcinoma is a rare entity, with the most common form found in the gynecologic tract, representing only 2% of gynecologic malignancies.26
Paraneoplastic syndromes of EPSCC are rare given the paucity of the malignancy. Several case reports discuss findings of SIADH in EPSCC of the cervix, as well as hypercalcemia, polyneuropathy, Cushing syndrome, limbic encephalitis, and peripheral neuropathy in EPSCC of the prostate.27,28 In contrast, SCLC has long been associated with paraneoplastic syndromes. Numerous case reports have been published describing SCLC-associated paraneoplastic syndromes to include hypercalcemia, Cushing syndrome, SIADH, vasoactive peptide production, cerebellar degeneration, limbic encephalitis, visceral plexopathy, autonomic dysfunction, and LEMS.29 As more cases of EPSCC with paraneoplastic syndromes are identified and reported, we might gain a better understanding of this interesting phenomenon.
Conclusion
Merkel cell carcinoma is an aggressive neuroendocrine malignancy associated with paraneoplastic neurologic syndromes, including LEMS. A thorough search for an underlying malignancy is highly recommended in patients with diagnosed LEMS without clear cause. Early identification and treatment of the primary tumor can lead to improvement of neurologic symptoms.
We present a case of LEMS with no clearly identifiable cause on presentation with later diagnosis of metastatic MCC of unknown primary origin. After surgical excision of affected lymph nodes and adjuvant radiation therapy, the patient had near-complete resolution of LEMS symptoms at 6-month follow-up, without additional findings of lymphadenopathy or skin lesions. Although this patient is not undergoing routine surveillance imaging to monitor for recurrence of MCC, a chest CT or positron emission tomography–CT for secondary screening would be considered if the patient experienced clinical symptoms consistent with LEMS.
In cases of LEMS without pulmonary malignancy, we recommend considering MCC in the differential diagnosis during the workup of an underlying malignancy
- Albores-Saavedra J, Batich K, Chable-Montero F, et al. Merkel cell carcinoma demographics, morphology, and survival based on 3870 cases: a population based study. J Cutan Pathol. 2010;37:20-27.
- Senchenkov A, Moran SL. Merkel cell carcinoma: diagnosis, management, and outcomes. Plast Reconstr Surg. 2013;131:E771-E778.
- Han SY, North JP, Canavan T, et al. Merkel cell carcinoma. Hematol Oncol Clin N Am. 2012;26:1351-1374.
- Vernino S. Paraneoplastic disorders affecting the neuromuscular junction or anterior horn cell. CONTINUUM Lifelong Learning in Neurology. 2009;15:132-146.
- Eggers SD, Salomao DR, Dinapoli RP, et al. Paraneoplastic and metastatic neurologic complications of Merkel cell carcinoma. Mayo Clin Proc. 2001;76:327-330.
- Siau RT, Morris A, Karoo RO. Surgery results in complete cure of Lambert-Eaton myasthenic syndrome in a patient with metastatic Merkel cell carcinoma. J Plast Reconstr Aesthet Surg. 2014;67:e162-e164.
- Bombelli F, Lispi L, Calabrò F, et al. Lambert-Eaton myasthenic syndrome associated to Merkel cell carcinoma: report of a case. Neurol Sci. 2015;36:1491-1492.
- Simmons DB, Duginski TM, McClean JC, et al. Lambert-eaton myasthenic syndrome and merkel cell carcinoma. Muscle Nerve. 2015;53:325-326.
- Bobos M, Hytiroglou P, Kostopoulos I, et al. Immunohistochemical distinction between Merkel cell carcinoma and small cell carcinoma of the lung. Am J Dermatopathol. 2006;28:99-104.
- Jensen K, Kohler S, Rouse RV. Cytokeratin staining in Merkel cell carcinoma: an immunohistochemical study of cytokeratins 5/6, 7, 17, and 20. Appl Immunohistochem Mol Morphol. 2000;8:310-315.
- Titulaer MJ, Lang B, Verschuuren JJ. Lambert-Eaton myasthenic syndrome: from clinical characteristics to therapeutic strategies. Lancet Neurol. 2011;10:1098-1107.
- Sanders DB. Lambert-Eaton myasthenic syndrome. In: Daroff R, Aminoff MJ, eds. Encyclopedia of the Neurological Sciences. Vol 2. New York, NY: Elsevier; 2009:819-822.
- Wirtz PW, Smallegange TM, Wintzen AR, et al. Differences in clinical features between the Lambert-Eaton myasthenic syndrome with and without cancer: an analysis of 227 published cases. Clin Neurol Neurosurg. 2002;104:359-363.
- Titulaer MJ, Wirtz PW, Willems LN, et al. Screening for small-cell lung cancer: a follow-up study of patients with Lambert-Eaton myasthenic syndrome. J Clin Oncol. 2008;26:4276-4281.
- Titulaer MJ, Maddison P, Sont JK, et al. Clinical Dutch-English Lambert-Eaton Myasthenic Syndrome (LEMS) Tumor Association prediction score accurately predicts small-cell lung cancer in the LEMS. J Clin Oncol. 2011;7:902-908.
- Cher LM, Hochberg FH, Teruya J, et al. Therapy for paraneoplastic neurologic syndromes in six patients with protein A column immunoadsorption. Cancer. 1995;75:1678-1683.
- Batchelor TT, Platten M, Hochberg FH. Immunoadsorption therapy for paraneoplastic syndromes. J Neurooncol. 1998;40:131-136.
- Blondin NA, Vortmeyer AO, Harel NY. Paraneoplastic syndrome of inappropriate antidiuretic hormone mimicking limbic encephalitis. Arch Neurol. 2011;68:1591-1594.
- Balegno S, Ceroni M, Corato M, et al. Antibodies to cerebellar nerve fibres in two patients with paraneoplastic cerebellar ataxia. Anticancer Res. 2005;25:3211-3214.
- Zhang C, Emery L, Lancaster E. Paraneoplastic cerebellar degeneration associated with noncutaneous Merkel cell carcinoma. Neurol Neuroimmunol Neuroinflamm. 2014;1:e17.
- Hocar O, Poszepczynska-Guigné E, Faye O, et al. Severe necrotizing myopathy subsequent to Merkel cell carcinoma. Ann Dermatol Venereol. 2011;138:130-134.
- Greenlee JE, Steffens JD, Clawson SA, et al. Anti-Hu antibodies in Merkel cell carcinoma. Ann Neurol. 2002;52:111-115.
- Lopez MC, Pericay C, Agustí M, et al. Merkel cell carcinoma associated with a paraneoplastic neurologic syndrome. Histopathology. 2004;44:628-629.
- Dalmau J, Furneaux HM, Cordon-Cardo C, et al. The expression of the Hu (paraneoplastic encephalomyelitis/sensory neuronopathy) antigen in human normal and tumor tissues. Am J Pathol. 1992;141:881-886.
- Gultekin SH, Rosai J, Demopoulos A, et al. Hu immunolabeling as a marker of neural and neuroendocrine differentiation in normal and neoplastic human tissues: assessment using a recombinant anti-Hu Fab fragment. Int J Surg Pathol. 2000;8:109-117.
- Zheng X, Liu D, Fallon JT, et al. Distinct genetic alterations in small cell carcinoma from different anatomic sites. Exp Hematol Oncol. 2015;4:2.
- Kim D, Yun H, Lee Y, et al. Small cell neuroendocrine carcinoma of the uterine cervix presenting with syndrome of inappropriate antidiuretic hormone secretion. Obstet Gynecol Sci. 2013;56:420-425.
- Venkatesh PK, Motwani B, Sherman N, et al. Metastatic pure small-cell carcinoma of prostate. Am J Med Sci. 2004;328:286-289.
- Kaltsas G, Androulakis II, de Herder WW, et al. Paraneoplastic syndromes secondary to neuroendocrine tumours. Endocr Relat Cancer. 2010;17:R173-R193.
- Albores-Saavedra J, Batich K, Chable-Montero F, et al. Merkel cell carcinoma demographics, morphology, and survival based on 3870 cases: a population based study. J Cutan Pathol. 2010;37:20-27.
- Senchenkov A, Moran SL. Merkel cell carcinoma: diagnosis, management, and outcomes. Plast Reconstr Surg. 2013;131:E771-E778.
- Han SY, North JP, Canavan T, et al. Merkel cell carcinoma. Hematol Oncol Clin N Am. 2012;26:1351-1374.
- Vernino S. Paraneoplastic disorders affecting the neuromuscular junction or anterior horn cell. CONTINUUM Lifelong Learning in Neurology. 2009;15:132-146.
- Eggers SD, Salomao DR, Dinapoli RP, et al. Paraneoplastic and metastatic neurologic complications of Merkel cell carcinoma. Mayo Clin Proc. 2001;76:327-330.
- Siau RT, Morris A, Karoo RO. Surgery results in complete cure of Lambert-Eaton myasthenic syndrome in a patient with metastatic Merkel cell carcinoma. J Plast Reconstr Aesthet Surg. 2014;67:e162-e164.
- Bombelli F, Lispi L, Calabrò F, et al. Lambert-Eaton myasthenic syndrome associated to Merkel cell carcinoma: report of a case. Neurol Sci. 2015;36:1491-1492.
- Simmons DB, Duginski TM, McClean JC, et al. Lambert-eaton myasthenic syndrome and merkel cell carcinoma. Muscle Nerve. 2015;53:325-326.
- Bobos M, Hytiroglou P, Kostopoulos I, et al. Immunohistochemical distinction between Merkel cell carcinoma and small cell carcinoma of the lung. Am J Dermatopathol. 2006;28:99-104.
- Jensen K, Kohler S, Rouse RV. Cytokeratin staining in Merkel cell carcinoma: an immunohistochemical study of cytokeratins 5/6, 7, 17, and 20. Appl Immunohistochem Mol Morphol. 2000;8:310-315.
- Titulaer MJ, Lang B, Verschuuren JJ. Lambert-Eaton myasthenic syndrome: from clinical characteristics to therapeutic strategies. Lancet Neurol. 2011;10:1098-1107.
- Sanders DB. Lambert-Eaton myasthenic syndrome. In: Daroff R, Aminoff MJ, eds. Encyclopedia of the Neurological Sciences. Vol 2. New York, NY: Elsevier; 2009:819-822.
- Wirtz PW, Smallegange TM, Wintzen AR, et al. Differences in clinical features between the Lambert-Eaton myasthenic syndrome with and without cancer: an analysis of 227 published cases. Clin Neurol Neurosurg. 2002;104:359-363.
- Titulaer MJ, Wirtz PW, Willems LN, et al. Screening for small-cell lung cancer: a follow-up study of patients with Lambert-Eaton myasthenic syndrome. J Clin Oncol. 2008;26:4276-4281.
- Titulaer MJ, Maddison P, Sont JK, et al. Clinical Dutch-English Lambert-Eaton Myasthenic Syndrome (LEMS) Tumor Association prediction score accurately predicts small-cell lung cancer in the LEMS. J Clin Oncol. 2011;7:902-908.
- Cher LM, Hochberg FH, Teruya J, et al. Therapy for paraneoplastic neurologic syndromes in six patients with protein A column immunoadsorption. Cancer. 1995;75:1678-1683.
- Batchelor TT, Platten M, Hochberg FH. Immunoadsorption therapy for paraneoplastic syndromes. J Neurooncol. 1998;40:131-136.
- Blondin NA, Vortmeyer AO, Harel NY. Paraneoplastic syndrome of inappropriate antidiuretic hormone mimicking limbic encephalitis. Arch Neurol. 2011;68:1591-1594.
- Balegno S, Ceroni M, Corato M, et al. Antibodies to cerebellar nerve fibres in two patients with paraneoplastic cerebellar ataxia. Anticancer Res. 2005;25:3211-3214.
- Zhang C, Emery L, Lancaster E. Paraneoplastic cerebellar degeneration associated with noncutaneous Merkel cell carcinoma. Neurol Neuroimmunol Neuroinflamm. 2014;1:e17.
- Hocar O, Poszepczynska-Guigné E, Faye O, et al. Severe necrotizing myopathy subsequent to Merkel cell carcinoma. Ann Dermatol Venereol. 2011;138:130-134.
- Greenlee JE, Steffens JD, Clawson SA, et al. Anti-Hu antibodies in Merkel cell carcinoma. Ann Neurol. 2002;52:111-115.
- Lopez MC, Pericay C, Agustí M, et al. Merkel cell carcinoma associated with a paraneoplastic neurologic syndrome. Histopathology. 2004;44:628-629.
- Dalmau J, Furneaux HM, Cordon-Cardo C, et al. The expression of the Hu (paraneoplastic encephalomyelitis/sensory neuronopathy) antigen in human normal and tumor tissues. Am J Pathol. 1992;141:881-886.
- Gultekin SH, Rosai J, Demopoulos A, et al. Hu immunolabeling as a marker of neural and neuroendocrine differentiation in normal and neoplastic human tissues: assessment using a recombinant anti-Hu Fab fragment. Int J Surg Pathol. 2000;8:109-117.
- Zheng X, Liu D, Fallon JT, et al. Distinct genetic alterations in small cell carcinoma from different anatomic sites. Exp Hematol Oncol. 2015;4:2.
- Kim D, Yun H, Lee Y, et al. Small cell neuroendocrine carcinoma of the uterine cervix presenting with syndrome of inappropriate antidiuretic hormone secretion. Obstet Gynecol Sci. 2013;56:420-425.
- Venkatesh PK, Motwani B, Sherman N, et al. Metastatic pure small-cell carcinoma of prostate. Am J Med Sci. 2004;328:286-289.
- Kaltsas G, Androulakis II, de Herder WW, et al. Paraneoplastic syndromes secondary to neuroendocrine tumours. Endocr Relat Cancer. 2010;17:R173-R193.
Practice Points
- Approximately 50% to 60% of patients with Lambert-Eaton myasthenic syndrome (LEMS) have an underlying tumor, most commonly small cell lung carcinoma.
- A thorough search for an underlying malignancy is highly recommended in patients with diagnosed LEMS without clear cause; to this end, a screening protocol comprising computed tomography and total-body fludeoxyglucose positron emission tomography has been established.
- Because Merkel cell carcinoma (MCC) can present as occult lymph node involvement with primary cutaneous findings absent, it is recommended that MCC be considered in the differential diagnosis of an underlying malignancy in a LEMS patient.
- Early identification and treatment of the primary tumor can lead to improvement of neurologic symptoms.
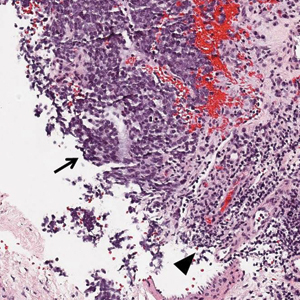
Graham-Little-Piccardi-Lassueur Syndrome
To the Editor:
A 56-year-old white woman with a history of melanoma and hypertension presented for evaluation of progressive hair loss of more than 1 year’s duration with associated pruritis. Scalp examination revealed diffuse erythema and scarring alopecia of the bilateral parietal and temporal regions. Physical examination also revealed nonscarring alopecia of the bilateral axillae, with associated thinning of the pubic hair, eyebrows, and eyelashes, as well as keratosis pilaris on the upper arms. Biopsy of the parietal scalp revealed mild scarring alopecia with isthmic fibroplasia consistent with early lichen planopilaris (LPP)(Figure). These histologic features combined with the patient’s clinical presentation were consistent with a diagnosis of Graham-Little-Piccardi-Lassueur syndrome (GLPL).
Graham-Little-Piccardi-Lassueur syndrome was first described by Piccardi in 1913.A second case was then described by Graham-Little in 1915 in a patient referred by Lassueur, resulting in the name it bears today.1,2 The condition presents most commonly in middle-aged white women and is characterized by a triad of cicatricial alopecia of the scalp, nonscarring alopecia of the axillae and/or groin, and a rough follicular eruption on the body and/or scalp. Symptoms may not be present simultaneously. In GLPL, scarring alopecia of the scalp often precedes follicular eruptions of the trunk, arms, and legs by as much as years,2 and the inverse also has been reported.1 The inflammatory lesions of the scalp eventually resolve spontaneously, but the hair loss is by definition irreversible.
This rare condition is considered one of the 3 clinical variants of LPP. Other variants include classic LPP, also known as follicular lichen planus, and frontal fibrosing alopecia.3 More recently, fibrosing alopecia in a pattern distribution has gained some popularity as a fourth variant of LPP.4 All variants of LPP, including GLPL, result in a scarring alopecia. The classic scalp finding is an erythematous to violaceous, perifollicular, hyperkeratotic scale at the base of the terminal hairs. The population of inflamed follicles spreads outward, leaving behind a round to oval, central, atrophic scar that often is devoid of follicles. Few hairs may persist within zones of alopecia at presentation; however, these hairs are affected by inflammation and also will likely shed. A hair pull test will be positive at the margins during active disease, consisting of mostly anagen hairs on trichogram examination.1,5 Patients may develop only a single foci of hair loss, but much more commonly, a patchy multifocal alopecia is noted.6 Sites often will coalesce. Onset of scalp alopecia may be insidious or fulminant.
The nonscarring alopecia of the axillae and groin may be described as subtle thinning to complete hair loss with no signs of atrophy or inflammation. Although not commonly reported, a case of nonscarring alopecia located on the shoulders has been seen.7
The follicular eruption that can be present on the trunk, arms, or legs in GLPL is most often but not limited to keratosis pilaris, as was seen in our patient. One reported case also described lichen spinulosus as a potential variant.8 Lichen planopilaris is separate from lichen planus (LP) because of its selective follicular involvement vs the nonselective mucocutaneous distribution of LP. The 2 processes also are histologically distinct; however, estimations have shown that more than 50% of patients with GLPL experience at least 1 episode of mucosal or cutaneous LP in their lifetime.9 Rarely, coexistence of GLPL and LP lesions has been described. One reported case of GLPL and concomitant hypertrophic LP could represent a severe form of the disease.9 Additionally, lichen planus pigmentosus, an uncommon variant of LP characterized by hyperpigmented brown macules in sun-exposed areas and flexural folds, was identified in a case report of an Asian woman with GLPL.10
As a general rule, the variants of LPP most commonly are seen in postmenopausal women aged 40 to 60 years; however, rare cases in a child and a teenager have been reported.11 The GLPL variant of LPP is reported up to 4 times more frequently in females.5 Pruritus and pain are inconsistent findings, and there are no systemic signs of illness. A case of androgen insensitivity syndrome associated with GLPL suggested a potential influence of hormones in LPP.12 Stress, vitamin A deficiency, and autoimmunity also have been proposed as triggers of GLPL.13 Furthermore, familial GLPL was described in a mother and daughter, though the association was uncertain.14 Our patient had no relevant family history.
Workups to reveal the etiology of GLPL have been inconclusive. Reports of laboratory testing including complete blood cell count, basic metabolic panel, liver function tests, testosterone and dehydroepiandrosterone levels, and chest radiograph have been normal.2 Additional workup for viral triggers also has been negative.15 A case series of 29 patients with LPP and its variants, including GLPL, revealed positive antinuclear antibodies in 10% of patients and a thyroid disorder in 24% of patients, with Hashimoto thyroiditis being the most prevalent in 7% of cases.16 There may be a strong association between the comorbidities of thyroid dysfunction and GLPL, as documented in other studies.10,17 A case-control study by Mesinkovska et al17 revealed a considerable increase in the prevalence of thyroid gland disease among patients with LPP vs controls. Human leukocyte antigen DR1 was found in a familial case of GLPL,4 and a case of GLPL following hepatitis B vaccination also has been described.18
Graham-Little-Piccardi-Lassueur syndrome most likely is a T-cell mediated autoimmune condition associated with one or multiple unknown keratinocyte antigens. Autoantibodies to the inner centromere protein were identified in a case that was positive on direct immunofluorescence, which may provide more insight into the disease pathophysiology.13 Interestingly, a study comparing the concentrations of inflammatory cells in LPP and traction alopecia found an elevation in the ratio of Langerhans cells to T lymphocytes within the follicular inflammatory infiltrate of LPP.19
Histologically, cicatricial alopecia of the scalp is characterized by an interface dermatitis and a lichenoid lymphocytic infiltrate of the isthmus and infundibulum of the hair follicle sparing the bulb (Figure). A follicular plug is present in the active border. The increased pressure from the keratinous plug from above and the pressure from the infiltrate from the sides has been proposed to decrease the blood supply to the follicle and result in its death.2 Late-stage disease is notable for fibrotic longitudinal tracks of the hair follicle, perifollicular lamellar fibrosis, and adjacent epidermal atrophy.20 Direct immunofluorescence in GLPL generally is negative. A trichogram performed in a 29-year-old woman with GLPL was normal, with 84% anagen, 2% catagen, and 14% telogen hairs. It was noted that 10% of the sampled hairs were classified as dystrophical dysplastic hairs.12 Despite the lack of fibrosis on physical examination in patients with GLPL, nonscarring alopecia of the axilla and groin may show follicular destruction on microscopic examination.1 The pathology of the papules present on the trunk and extremities—whether that of keratosis pilaris or lichen spinulosus—demonstrates similar hyperkeratosis, hypergranulosis, and follicular plugging with a possible superficial, perivascular, lymphocytic infiltrate.

The differential diagnosis of GLPL includes other variants of LPP as well as discoid lupus erythematous (DLE), pseudopelade of Brocq, pityriasis rubra pilaris, sarcoidosis, acne keloidalis, central centrifugal scarring alopecia, follicular mucinosis, and folliculitis decalvans.14 Differentiation of LPP from DLE is difficult. Clinical clues include lack of central erythema and telangiectases within the lesions. Histologically, the lymphocytic dermatitis and folliculitis can be indistinguishable, but subtle findings suggesting DLE may be present, such as increased mucin in the reticular dermis, a focally thinned epidermis, and less severe dermal sclerosis when compared to cases of LPP.2 Direct immunofluorescence with IgG and C3 revealing linear granular deposits at the dermoepidermal junction is characteristic of DLE.20 Pseudopelade of Brocq is best thought of as an end-stage clinical pattern of hair loss in LPP rather than a separate condition. It is considered to be the end point of GLPL as well as DLE and others when the inflammation has subsided and the cicatricial alopecia is stable. For the duration of active disease, GLPL is classified as an unstable cicatricial alopecia that has a tendency to progress and recur periodically.20 Folliculitis decalvans also can mimic GLPL during a period when the pustules have resolved; however, a neutrophilic infiltrate will be present.
The goal of treatment in GLPL as well as other scarring alopecias is to stop the progression of hair loss. Early diagnosis is imperative if control is to be gained before considerable hair loss has occurred. Once follicular destruction has occurred as a result of the inflammation, there is minimal potential for hair rejuvenation.21 To date, treatment has been mostly fruitless, except in the management of keratosis pilaris that accompanies GLPL. First-line therapy often includes topical corticosteroids with or without intralesional corticosteroids. Systemic corticosteroids, retinoids, and psoralen plus UVA therapy also are frequently employed.1,2 Success in treating GLPL with cyclosporine A at a dosage of 4 mg/kg daily was described in several studies.1,2,15 Treatment resulted in reduction of perifollicular erythema and follicular hyperkeratotic papules as well as mild hair regrowth within the scarring patches.15 Nonetheless, cyclosporine A may prove useful in the initial inflammatory phase of GLPL. Consequently, cyclosporine A also is associated with a high relapse rate.1,2
Because the number of patients with GLPL is so few, therapy should mirror advances being made in treatments for other variants of LPP. More recent studies of LPP treatment with hydroxychloroquine showed opposing results, though the safety profile of this agent makes it an enticing treatment option.22,23 Tetracyclines showed improvement in 4 of 15 (26.7%) patients in a retrospective study by Spencer et al.24 Another retrospective study showed promising results with the potent 5-alpha reductase inhibitor dutasteride with 7 of 10 (70%) postmenopausal patients reporting stabilization over a mean duration of 28 months with no reported side effects.25 Antimalarial medications also have been implemented as adjunct therapies with mixed results.5 A case of a 26-year-old man with GLPL from South India showed systemic disease improvement following treatment with pulsed systemic steroids, isotretinoin, and anxiolytics.7 Chloroquine phosphate at a daily dose of 150 mg for 3 to 9 months yielded a transient response in one postmenopausal patient with frontal fibrosing alopecia.6 Stabilization of hair loss was achieved with a combination of hydroxychloroquine and doxycycline in a woman with GLPL who was previously unresponsive to tacrolimus ointment.10 Thalidomide showed early promise in an isolated report claiming successful treatment of LPP,26 but there is contradictory evidence, as thalidomide showed no benefit in a series of 4 patients with LPP.27
Peroxisome proliferator–activated receptor gamma (PPAR-γ), a transcription factor that regulates genes, is downregulated in LPP.28 Deletion of PPAR-γ within follicular stem cells in mice results in a phenotype similar to cicatricial alopecia. Data have supported the role of PPAR-γ in maintaining the pilosebaceous unit. A case report of pioglitazone (PPAR-γ agonist) therapy used at 15 mg daily for 8 months was successful in treating a patient with LPP.28 Further investigation must be conducted to evaluate these treatments since early attenuation of the disease process is crucial to the reduction of permanent hair loss.
Advances in the early recognition and successful treatment of GLPL are dependent on continued research in all variants of LPP. Randomized controlled trials are necessary to establish standard of care. Further studies should target the association of GLPL and other autoimmune phenomena. Moreover, research into the etiology will provide direction in understanding disease progression and outcome.
- Zegarska B, Kallas D, Schwartz RA, et al. Graham-Little syndrome. Acta Dermatovenerol Alp Pannonica Adriat. 2010;19:39-42.
- Assouly P, Reygagne P. Lichen planopilaris: update on diagnosis and treatment. Semin Cutan Med Surg. 2009;28:3-10.
- Olsen EA, Bergfield WF, Cotsarelis G, et al. Summary of North American Hair Research Society (NAHRS)–sponsored Workshop on Cicatricial Alopecia, Duke University Medical Center, February 10 and 11, 2001. J Am Acad Dermatol. 2003;48:103-110.
- Zinkernagel MS, Trueb RM. Fibrosing alopecia in a pattern distribution: patterned lichen planopilaris or androgenetic alopecia with a lichenoid tissue reaction pattern? Arch Dermatol. 2000;136:205-211.
- James WD, Berger TG, Elston DM. Andrews’ Diseases of the Skin: Clinical Dermatology. 12th ed. Philadelphia, PA: WB Saunders Company; 2016.
- Kossard S, Lee MS, Wilkinson B. Postmenopausal frontal fibrosing alopecia: a frontal variant of lichen planopilaris. J Am Acad Dermatol. 1997;36:59-66.
- Pai VV, Kikkeri NN, Sori T, et al. Graham-Little Piccardi Lassueur syndrome: an unusual variant of follicular lichen planus. Int J Trichology. 2011;3:28-30.
- Srivastava M, Mikkilineni R, Konstadt J. Lassueur-Graham-Little-Piccardi syndrome. Dermatol Online J. 2007;13:12.
- Brar BK, Khanna E, Mahajan BB. Graham Little Piccardi Lasseur syndrome: a rare case report with concomitant hypertrophic lichen planus. Int J Trichology. 2011;5:199-200.
- Vashi N, Newlove T, Chu J, et al. Graham-Little-Piccardi-Lassueur syndrome. Dermatol Online J. 2011;17:30.
- Chieregato C, Zini A, Barba A, et al. Lichen planopilaris: report of 30 cases and review of the literature. Int J Dermatol. 2003;42:342-345.
- Vega Gutierrez J, Miranda-Romera A, Perez Milan F, et al. Graham Little-Piccardi-Lassueur syndrome associated with androgen insensitivity syndrome (testicular feminization). J Eur Acad Dermatol Venereol. 2004;18:463-466.
- Rodríguez-Bayona B, Ruchaud S, Rodriguez C, et al. Autoantibodies against the chromosomal passenger protein INCENP found in a patient with Graham Little-Piccardi-Lassueur syndrome. J Autoimmune Dis. 2007;4:1.
- Viglizzo G, Verrini A, Rongioletti F. Familial Lassueur-Graham-Little-Piccardi syndrome. Dermatology. 2004;208:142-144.
- Bianchi L, Paro Vidolin A, Piemonte P, et al. Graham Little-Piccardi-Lassueur syndrome: effective treatment with cyclosporin A. Clin Exp Dermatol. 2001;26:518-520.
- Cevasco NC, Bergfeld WF, Remzi BK, et al. A case-series of 29 patients with lichen planopilaris: the Cleveland Clinic Foundation experience on evaluation, diagnosis, and treatment. J Am Acad Dermatol. 2007;57:47-53.
- Mesinkovska NA, Brankov N, Piliang M, et al. Association of lichen planopilaris with thyroid disease: a retrospective case-control study. J Am Acad Dermatol. 2014;70:889-892.
- Bardazzi F, Landi C, Orlandi C, et al. Graham Little-Piccardi-Lasseur syndrome following HBV vaccination. Acta Derm Venereol. 1999;79:93.
- Hutchens KA, Balfour EM, Smoller BR. Comparison between Langerhans cell concentration in lichen planopilaris and traction alopecia with possible immunologic implications. Am J Dermatopathol. 2011;33:277-280.
- Dogra S, Sarangal R. What’s new in cicatricial alopecia? Indian J Dermatol Venereol Leprol. 2013;79:576-590.
- Daoud MS, Pittelkow MR. Lichen planus. In: Wolff K, Goldsmith LA, Katz Si, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 7th ed. New York, NY: Mc Graw Hill; 2008:463-477.
- Donati A, Assouly P, Matard B, et al. Clinical and photographic assessment of lichen planopilaris treatment efficacy. J Am Acad Dermatol. 2011;64:597-599.
- Samrao A, Chew AL, Price V. Frontal fibrosing alopecia: a clinical review of 36 patients. Br J Dermatol. 2010;163:1296-1300.
- Spencer LA, Hawryluk EB, English JC. Lichen planopilaris: retrospective study and stepwise therapeutic approach. Arch Dermatol. 2009;145:333-334.
- Ladizinski B, Bazakas A, Selim MA, et al. Frontal fibrosing alopecia: a retrospective review of 19 patients seen at Duke University. J Am Acad Dermatol. 2013;68:749-755
- George SJ, Hsu SJ. Lichen planopilaris treated with thalidomide. J Am Acad Dermatol. 2001;45:965-966.
- Jouanique C, Reygagne P, Bachelez H, et al. Thalidomide is ineffective in the treatment of lichen planopilaris. J Am Acad Dermatol. 2004;51:480-481.
- Mirmirani P, Karnik P. Lichen planopilaris treated with a peroxisome proliferator–activated receptor γ agonist. Arch Dermatol. 2009;145:1363-1366.
To the Editor:
A 56-year-old white woman with a history of melanoma and hypertension presented for evaluation of progressive hair loss of more than 1 year’s duration with associated pruritis. Scalp examination revealed diffuse erythema and scarring alopecia of the bilateral parietal and temporal regions. Physical examination also revealed nonscarring alopecia of the bilateral axillae, with associated thinning of the pubic hair, eyebrows, and eyelashes, as well as keratosis pilaris on the upper arms. Biopsy of the parietal scalp revealed mild scarring alopecia with isthmic fibroplasia consistent with early lichen planopilaris (LPP)(Figure). These histologic features combined with the patient’s clinical presentation were consistent with a diagnosis of Graham-Little-Piccardi-Lassueur syndrome (GLPL).
Graham-Little-Piccardi-Lassueur syndrome was first described by Piccardi in 1913.A second case was then described by Graham-Little in 1915 in a patient referred by Lassueur, resulting in the name it bears today.1,2 The condition presents most commonly in middle-aged white women and is characterized by a triad of cicatricial alopecia of the scalp, nonscarring alopecia of the axillae and/or groin, and a rough follicular eruption on the body and/or scalp. Symptoms may not be present simultaneously. In GLPL, scarring alopecia of the scalp often precedes follicular eruptions of the trunk, arms, and legs by as much as years,2 and the inverse also has been reported.1 The inflammatory lesions of the scalp eventually resolve spontaneously, but the hair loss is by definition irreversible.
This rare condition is considered one of the 3 clinical variants of LPP. Other variants include classic LPP, also known as follicular lichen planus, and frontal fibrosing alopecia.3 More recently, fibrosing alopecia in a pattern distribution has gained some popularity as a fourth variant of LPP.4 All variants of LPP, including GLPL, result in a scarring alopecia. The classic scalp finding is an erythematous to violaceous, perifollicular, hyperkeratotic scale at the base of the terminal hairs. The population of inflamed follicles spreads outward, leaving behind a round to oval, central, atrophic scar that often is devoid of follicles. Few hairs may persist within zones of alopecia at presentation; however, these hairs are affected by inflammation and also will likely shed. A hair pull test will be positive at the margins during active disease, consisting of mostly anagen hairs on trichogram examination.1,5 Patients may develop only a single foci of hair loss, but much more commonly, a patchy multifocal alopecia is noted.6 Sites often will coalesce. Onset of scalp alopecia may be insidious or fulminant.
The nonscarring alopecia of the axillae and groin may be described as subtle thinning to complete hair loss with no signs of atrophy or inflammation. Although not commonly reported, a case of nonscarring alopecia located on the shoulders has been seen.7
The follicular eruption that can be present on the trunk, arms, or legs in GLPL is most often but not limited to keratosis pilaris, as was seen in our patient. One reported case also described lichen spinulosus as a potential variant.8 Lichen planopilaris is separate from lichen planus (LP) because of its selective follicular involvement vs the nonselective mucocutaneous distribution of LP. The 2 processes also are histologically distinct; however, estimations have shown that more than 50% of patients with GLPL experience at least 1 episode of mucosal or cutaneous LP in their lifetime.9 Rarely, coexistence of GLPL and LP lesions has been described. One reported case of GLPL and concomitant hypertrophic LP could represent a severe form of the disease.9 Additionally, lichen planus pigmentosus, an uncommon variant of LP characterized by hyperpigmented brown macules in sun-exposed areas and flexural folds, was identified in a case report of an Asian woman with GLPL.10
As a general rule, the variants of LPP most commonly are seen in postmenopausal women aged 40 to 60 years; however, rare cases in a child and a teenager have been reported.11 The GLPL variant of LPP is reported up to 4 times more frequently in females.5 Pruritus and pain are inconsistent findings, and there are no systemic signs of illness. A case of androgen insensitivity syndrome associated with GLPL suggested a potential influence of hormones in LPP.12 Stress, vitamin A deficiency, and autoimmunity also have been proposed as triggers of GLPL.13 Furthermore, familial GLPL was described in a mother and daughter, though the association was uncertain.14 Our patient had no relevant family history.
Workups to reveal the etiology of GLPL have been inconclusive. Reports of laboratory testing including complete blood cell count, basic metabolic panel, liver function tests, testosterone and dehydroepiandrosterone levels, and chest radiograph have been normal.2 Additional workup for viral triggers also has been negative.15 A case series of 29 patients with LPP and its variants, including GLPL, revealed positive antinuclear antibodies in 10% of patients and a thyroid disorder in 24% of patients, with Hashimoto thyroiditis being the most prevalent in 7% of cases.16 There may be a strong association between the comorbidities of thyroid dysfunction and GLPL, as documented in other studies.10,17 A case-control study by Mesinkovska et al17 revealed a considerable increase in the prevalence of thyroid gland disease among patients with LPP vs controls. Human leukocyte antigen DR1 was found in a familial case of GLPL,4 and a case of GLPL following hepatitis B vaccination also has been described.18
Graham-Little-Piccardi-Lassueur syndrome most likely is a T-cell mediated autoimmune condition associated with one or multiple unknown keratinocyte antigens. Autoantibodies to the inner centromere protein were identified in a case that was positive on direct immunofluorescence, which may provide more insight into the disease pathophysiology.13 Interestingly, a study comparing the concentrations of inflammatory cells in LPP and traction alopecia found an elevation in the ratio of Langerhans cells to T lymphocytes within the follicular inflammatory infiltrate of LPP.19
Histologically, cicatricial alopecia of the scalp is characterized by an interface dermatitis and a lichenoid lymphocytic infiltrate of the isthmus and infundibulum of the hair follicle sparing the bulb (Figure). A follicular plug is present in the active border. The increased pressure from the keratinous plug from above and the pressure from the infiltrate from the sides has been proposed to decrease the blood supply to the follicle and result in its death.2 Late-stage disease is notable for fibrotic longitudinal tracks of the hair follicle, perifollicular lamellar fibrosis, and adjacent epidermal atrophy.20 Direct immunofluorescence in GLPL generally is negative. A trichogram performed in a 29-year-old woman with GLPL was normal, with 84% anagen, 2% catagen, and 14% telogen hairs. It was noted that 10% of the sampled hairs were classified as dystrophical dysplastic hairs.12 Despite the lack of fibrosis on physical examination in patients with GLPL, nonscarring alopecia of the axilla and groin may show follicular destruction on microscopic examination.1 The pathology of the papules present on the trunk and extremities—whether that of keratosis pilaris or lichen spinulosus—demonstrates similar hyperkeratosis, hypergranulosis, and follicular plugging with a possible superficial, perivascular, lymphocytic infiltrate.
The differential diagnosis of GLPL includes other variants of LPP as well as discoid lupus erythematous (DLE), pseudopelade of Brocq, pityriasis rubra pilaris, sarcoidosis, acne keloidalis, central centrifugal scarring alopecia, follicular mucinosis, and folliculitis decalvans.14 Differentiation of LPP from DLE is difficult. Clinical clues include lack of central erythema and telangiectases within the lesions. Histologically, the lymphocytic dermatitis and folliculitis can be indistinguishable, but subtle findings suggesting DLE may be present, such as increased mucin in the reticular dermis, a focally thinned epidermis, and less severe dermal sclerosis when compared to cases of LPP.2 Direct immunofluorescence with IgG and C3 revealing linear granular deposits at the dermoepidermal junction is characteristic of DLE.20 Pseudopelade of Brocq is best thought of as an end-stage clinical pattern of hair loss in LPP rather than a separate condition. It is considered to be the end point of GLPL as well as DLE and others when the inflammation has subsided and the cicatricial alopecia is stable. For the duration of active disease, GLPL is classified as an unstable cicatricial alopecia that has a tendency to progress and recur periodically.20 Folliculitis decalvans also can mimic GLPL during a period when the pustules have resolved; however, a neutrophilic infiltrate will be present.
The goal of treatment in GLPL as well as other scarring alopecias is to stop the progression of hair loss. Early diagnosis is imperative if control is to be gained before considerable hair loss has occurred. Once follicular destruction has occurred as a result of the inflammation, there is minimal potential for hair rejuvenation.21 To date, treatment has been mostly fruitless, except in the management of keratosis pilaris that accompanies GLPL. First-line therapy often includes topical corticosteroids with or without intralesional corticosteroids. Systemic corticosteroids, retinoids, and psoralen plus UVA therapy also are frequently employed.1,2 Success in treating GLPL with cyclosporine A at a dosage of 4 mg/kg daily was described in several studies.1,2,15 Treatment resulted in reduction of perifollicular erythema and follicular hyperkeratotic papules as well as mild hair regrowth within the scarring patches.15 Nonetheless, cyclosporine A may prove useful in the initial inflammatory phase of GLPL. Consequently, cyclosporine A also is associated with a high relapse rate.1,2
Because the number of patients with GLPL is so few, therapy should mirror advances being made in treatments for other variants of LPP. More recent studies of LPP treatment with hydroxychloroquine showed opposing results, though the safety profile of this agent makes it an enticing treatment option.22,23 Tetracyclines showed improvement in 4 of 15 (26.7%) patients in a retrospective study by Spencer et al.24 Another retrospective study showed promising results with the potent 5-alpha reductase inhibitor dutasteride with 7 of 10 (70%) postmenopausal patients reporting stabilization over a mean duration of 28 months with no reported side effects.25 Antimalarial medications also have been implemented as adjunct therapies with mixed results.5 A case of a 26-year-old man with GLPL from South India showed systemic disease improvement following treatment with pulsed systemic steroids, isotretinoin, and anxiolytics.7 Chloroquine phosphate at a daily dose of 150 mg for 3 to 9 months yielded a transient response in one postmenopausal patient with frontal fibrosing alopecia.6 Stabilization of hair loss was achieved with a combination of hydroxychloroquine and doxycycline in a woman with GLPL who was previously unresponsive to tacrolimus ointment.10 Thalidomide showed early promise in an isolated report claiming successful treatment of LPP,26 but there is contradictory evidence, as thalidomide showed no benefit in a series of 4 patients with LPP.27
Peroxisome proliferator–activated receptor gamma (PPAR-γ), a transcription factor that regulates genes, is downregulated in LPP.28 Deletion of PPAR-γ within follicular stem cells in mice results in a phenotype similar to cicatricial alopecia. Data have supported the role of PPAR-γ in maintaining the pilosebaceous unit. A case report of pioglitazone (PPAR-γ agonist) therapy used at 15 mg daily for 8 months was successful in treating a patient with LPP.28 Further investigation must be conducted to evaluate these treatments since early attenuation of the disease process is crucial to the reduction of permanent hair loss.
Advances in the early recognition and successful treatment of GLPL are dependent on continued research in all variants of LPP. Randomized controlled trials are necessary to establish standard of care. Further studies should target the association of GLPL and other autoimmune phenomena. Moreover, research into the etiology will provide direction in understanding disease progression and outcome.
To the Editor:
A 56-year-old white woman with a history of melanoma and hypertension presented for evaluation of progressive hair loss of more than 1 year’s duration with associated pruritis. Scalp examination revealed diffuse erythema and scarring alopecia of the bilateral parietal and temporal regions. Physical examination also revealed nonscarring alopecia of the bilateral axillae, with associated thinning of the pubic hair, eyebrows, and eyelashes, as well as keratosis pilaris on the upper arms. Biopsy of the parietal scalp revealed mild scarring alopecia with isthmic fibroplasia consistent with early lichen planopilaris (LPP)(Figure). These histologic features combined with the patient’s clinical presentation were consistent with a diagnosis of Graham-Little-Piccardi-Lassueur syndrome (GLPL).
Graham-Little-Piccardi-Lassueur syndrome was first described by Piccardi in 1913.A second case was then described by Graham-Little in 1915 in a patient referred by Lassueur, resulting in the name it bears today.1,2 The condition presents most commonly in middle-aged white women and is characterized by a triad of cicatricial alopecia of the scalp, nonscarring alopecia of the axillae and/or groin, and a rough follicular eruption on the body and/or scalp. Symptoms may not be present simultaneously. In GLPL, scarring alopecia of the scalp often precedes follicular eruptions of the trunk, arms, and legs by as much as years,2 and the inverse also has been reported.1 The inflammatory lesions of the scalp eventually resolve spontaneously, but the hair loss is by definition irreversible.
This rare condition is considered one of the 3 clinical variants of LPP. Other variants include classic LPP, also known as follicular lichen planus, and frontal fibrosing alopecia.3 More recently, fibrosing alopecia in a pattern distribution has gained some popularity as a fourth variant of LPP.4 All variants of LPP, including GLPL, result in a scarring alopecia. The classic scalp finding is an erythematous to violaceous, perifollicular, hyperkeratotic scale at the base of the terminal hairs. The population of inflamed follicles spreads outward, leaving behind a round to oval, central, atrophic scar that often is devoid of follicles. Few hairs may persist within zones of alopecia at presentation; however, these hairs are affected by inflammation and also will likely shed. A hair pull test will be positive at the margins during active disease, consisting of mostly anagen hairs on trichogram examination.1,5 Patients may develop only a single foci of hair loss, but much more commonly, a patchy multifocal alopecia is noted.6 Sites often will coalesce. Onset of scalp alopecia may be insidious or fulminant.
The nonscarring alopecia of the axillae and groin may be described as subtle thinning to complete hair loss with no signs of atrophy or inflammation. Although not commonly reported, a case of nonscarring alopecia located on the shoulders has been seen.7
The follicular eruption that can be present on the trunk, arms, or legs in GLPL is most often but not limited to keratosis pilaris, as was seen in our patient. One reported case also described lichen spinulosus as a potential variant.8 Lichen planopilaris is separate from lichen planus (LP) because of its selective follicular involvement vs the nonselective mucocutaneous distribution of LP. The 2 processes also are histologically distinct; however, estimations have shown that more than 50% of patients with GLPL experience at least 1 episode of mucosal or cutaneous LP in their lifetime.9 Rarely, coexistence of GLPL and LP lesions has been described. One reported case of GLPL and concomitant hypertrophic LP could represent a severe form of the disease.9 Additionally, lichen planus pigmentosus, an uncommon variant of LP characterized by hyperpigmented brown macules in sun-exposed areas and flexural folds, was identified in a case report of an Asian woman with GLPL.10
As a general rule, the variants of LPP most commonly are seen in postmenopausal women aged 40 to 60 years; however, rare cases in a child and a teenager have been reported.11 The GLPL variant of LPP is reported up to 4 times more frequently in females.5 Pruritus and pain are inconsistent findings, and there are no systemic signs of illness. A case of androgen insensitivity syndrome associated with GLPL suggested a potential influence of hormones in LPP.12 Stress, vitamin A deficiency, and autoimmunity also have been proposed as triggers of GLPL.13 Furthermore, familial GLPL was described in a mother and daughter, though the association was uncertain.14 Our patient had no relevant family history.
Workups to reveal the etiology of GLPL have been inconclusive. Reports of laboratory testing including complete blood cell count, basic metabolic panel, liver function tests, testosterone and dehydroepiandrosterone levels, and chest radiograph have been normal.2 Additional workup for viral triggers also has been negative.15 A case series of 29 patients with LPP and its variants, including GLPL, revealed positive antinuclear antibodies in 10% of patients and a thyroid disorder in 24% of patients, with Hashimoto thyroiditis being the most prevalent in 7% of cases.16 There may be a strong association between the comorbidities of thyroid dysfunction and GLPL, as documented in other studies.10,17 A case-control study by Mesinkovska et al17 revealed a considerable increase in the prevalence of thyroid gland disease among patients with LPP vs controls. Human leukocyte antigen DR1 was found in a familial case of GLPL,4 and a case of GLPL following hepatitis B vaccination also has been described.18
Graham-Little-Piccardi-Lassueur syndrome most likely is a T-cell mediated autoimmune condition associated with one or multiple unknown keratinocyte antigens. Autoantibodies to the inner centromere protein were identified in a case that was positive on direct immunofluorescence, which may provide more insight into the disease pathophysiology.13 Interestingly, a study comparing the concentrations of inflammatory cells in LPP and traction alopecia found an elevation in the ratio of Langerhans cells to T lymphocytes within the follicular inflammatory infiltrate of LPP.19
Histologically, cicatricial alopecia of the scalp is characterized by an interface dermatitis and a lichenoid lymphocytic infiltrate of the isthmus and infundibulum of the hair follicle sparing the bulb (Figure). A follicular plug is present in the active border. The increased pressure from the keratinous plug from above and the pressure from the infiltrate from the sides has been proposed to decrease the blood supply to the follicle and result in its death.2 Late-stage disease is notable for fibrotic longitudinal tracks of the hair follicle, perifollicular lamellar fibrosis, and adjacent epidermal atrophy.20 Direct immunofluorescence in GLPL generally is negative. A trichogram performed in a 29-year-old woman with GLPL was normal, with 84% anagen, 2% catagen, and 14% telogen hairs. It was noted that 10% of the sampled hairs were classified as dystrophical dysplastic hairs.12 Despite the lack of fibrosis on physical examination in patients with GLPL, nonscarring alopecia of the axilla and groin may show follicular destruction on microscopic examination.1 The pathology of the papules present on the trunk and extremities—whether that of keratosis pilaris or lichen spinulosus—demonstrates similar hyperkeratosis, hypergranulosis, and follicular plugging with a possible superficial, perivascular, lymphocytic infiltrate.
The differential diagnosis of GLPL includes other variants of LPP as well as discoid lupus erythematous (DLE), pseudopelade of Brocq, pityriasis rubra pilaris, sarcoidosis, acne keloidalis, central centrifugal scarring alopecia, follicular mucinosis, and folliculitis decalvans.14 Differentiation of LPP from DLE is difficult. Clinical clues include lack of central erythema and telangiectases within the lesions. Histologically, the lymphocytic dermatitis and folliculitis can be indistinguishable, but subtle findings suggesting DLE may be present, such as increased mucin in the reticular dermis, a focally thinned epidermis, and less severe dermal sclerosis when compared to cases of LPP.2 Direct immunofluorescence with IgG and C3 revealing linear granular deposits at the dermoepidermal junction is characteristic of DLE.20 Pseudopelade of Brocq is best thought of as an end-stage clinical pattern of hair loss in LPP rather than a separate condition. It is considered to be the end point of GLPL as well as DLE and others when the inflammation has subsided and the cicatricial alopecia is stable. For the duration of active disease, GLPL is classified as an unstable cicatricial alopecia that has a tendency to progress and recur periodically.20 Folliculitis decalvans also can mimic GLPL during a period when the pustules have resolved; however, a neutrophilic infiltrate will be present.
The goal of treatment in GLPL as well as other scarring alopecias is to stop the progression of hair loss. Early diagnosis is imperative if control is to be gained before considerable hair loss has occurred. Once follicular destruction has occurred as a result of the inflammation, there is minimal potential for hair rejuvenation.21 To date, treatment has been mostly fruitless, except in the management of keratosis pilaris that accompanies GLPL. First-line therapy often includes topical corticosteroids with or without intralesional corticosteroids. Systemic corticosteroids, retinoids, and psoralen plus UVA therapy also are frequently employed.1,2 Success in treating GLPL with cyclosporine A at a dosage of 4 mg/kg daily was described in several studies.1,2,15 Treatment resulted in reduction of perifollicular erythema and follicular hyperkeratotic papules as well as mild hair regrowth within the scarring patches.15 Nonetheless, cyclosporine A may prove useful in the initial inflammatory phase of GLPL. Consequently, cyclosporine A also is associated with a high relapse rate.1,2
Because the number of patients with GLPL is so few, therapy should mirror advances being made in treatments for other variants of LPP. More recent studies of LPP treatment with hydroxychloroquine showed opposing results, though the safety profile of this agent makes it an enticing treatment option.22,23 Tetracyclines showed improvement in 4 of 15 (26.7%) patients in a retrospective study by Spencer et al.24 Another retrospective study showed promising results with the potent 5-alpha reductase inhibitor dutasteride with 7 of 10 (70%) postmenopausal patients reporting stabilization over a mean duration of 28 months with no reported side effects.25 Antimalarial medications also have been implemented as adjunct therapies with mixed results.5 A case of a 26-year-old man with GLPL from South India showed systemic disease improvement following treatment with pulsed systemic steroids, isotretinoin, and anxiolytics.7 Chloroquine phosphate at a daily dose of 150 mg for 3 to 9 months yielded a transient response in one postmenopausal patient with frontal fibrosing alopecia.6 Stabilization of hair loss was achieved with a combination of hydroxychloroquine and doxycycline in a woman with GLPL who was previously unresponsive to tacrolimus ointment.10 Thalidomide showed early promise in an isolated report claiming successful treatment of LPP,26 but there is contradictory evidence, as thalidomide showed no benefit in a series of 4 patients with LPP.27
Peroxisome proliferator–activated receptor gamma (PPAR-γ), a transcription factor that regulates genes, is downregulated in LPP.28 Deletion of PPAR-γ within follicular stem cells in mice results in a phenotype similar to cicatricial alopecia. Data have supported the role of PPAR-γ in maintaining the pilosebaceous unit. A case report of pioglitazone (PPAR-γ agonist) therapy used at 15 mg daily for 8 months was successful in treating a patient with LPP.28 Further investigation must be conducted to evaluate these treatments since early attenuation of the disease process is crucial to the reduction of permanent hair loss.
Advances in the early recognition and successful treatment of GLPL are dependent on continued research in all variants of LPP. Randomized controlled trials are necessary to establish standard of care. Further studies should target the association of GLPL and other autoimmune phenomena. Moreover, research into the etiology will provide direction in understanding disease progression and outcome.
- Zegarska B, Kallas D, Schwartz RA, et al. Graham-Little syndrome. Acta Dermatovenerol Alp Pannonica Adriat. 2010;19:39-42.
- Assouly P, Reygagne P. Lichen planopilaris: update on diagnosis and treatment. Semin Cutan Med Surg. 2009;28:3-10.
- Olsen EA, Bergfield WF, Cotsarelis G, et al. Summary of North American Hair Research Society (NAHRS)–sponsored Workshop on Cicatricial Alopecia, Duke University Medical Center, February 10 and 11, 2001. J Am Acad Dermatol. 2003;48:103-110.
- Zinkernagel MS, Trueb RM. Fibrosing alopecia in a pattern distribution: patterned lichen planopilaris or androgenetic alopecia with a lichenoid tissue reaction pattern? Arch Dermatol. 2000;136:205-211.
- James WD, Berger TG, Elston DM. Andrews’ Diseases of the Skin: Clinical Dermatology. 12th ed. Philadelphia, PA: WB Saunders Company; 2016.
- Kossard S, Lee MS, Wilkinson B. Postmenopausal frontal fibrosing alopecia: a frontal variant of lichen planopilaris. J Am Acad Dermatol. 1997;36:59-66.
- Pai VV, Kikkeri NN, Sori T, et al. Graham-Little Piccardi Lassueur syndrome: an unusual variant of follicular lichen planus. Int J Trichology. 2011;3:28-30.
- Srivastava M, Mikkilineni R, Konstadt J. Lassueur-Graham-Little-Piccardi syndrome. Dermatol Online J. 2007;13:12.
- Brar BK, Khanna E, Mahajan BB. Graham Little Piccardi Lasseur syndrome: a rare case report with concomitant hypertrophic lichen planus. Int J Trichology. 2011;5:199-200.
- Vashi N, Newlove T, Chu J, et al. Graham-Little-Piccardi-Lassueur syndrome. Dermatol Online J. 2011;17:30.
- Chieregato C, Zini A, Barba A, et al. Lichen planopilaris: report of 30 cases and review of the literature. Int J Dermatol. 2003;42:342-345.
- Vega Gutierrez J, Miranda-Romera A, Perez Milan F, et al. Graham Little-Piccardi-Lassueur syndrome associated with androgen insensitivity syndrome (testicular feminization). J Eur Acad Dermatol Venereol. 2004;18:463-466.
- Rodríguez-Bayona B, Ruchaud S, Rodriguez C, et al. Autoantibodies against the chromosomal passenger protein INCENP found in a patient with Graham Little-Piccardi-Lassueur syndrome. J Autoimmune Dis. 2007;4:1.
- Viglizzo G, Verrini A, Rongioletti F. Familial Lassueur-Graham-Little-Piccardi syndrome. Dermatology. 2004;208:142-144.
- Bianchi L, Paro Vidolin A, Piemonte P, et al. Graham Little-Piccardi-Lassueur syndrome: effective treatment with cyclosporin A. Clin Exp Dermatol. 2001;26:518-520.
- Cevasco NC, Bergfeld WF, Remzi BK, et al. A case-series of 29 patients with lichen planopilaris: the Cleveland Clinic Foundation experience on evaluation, diagnosis, and treatment. J Am Acad Dermatol. 2007;57:47-53.
- Mesinkovska NA, Brankov N, Piliang M, et al. Association of lichen planopilaris with thyroid disease: a retrospective case-control study. J Am Acad Dermatol. 2014;70:889-892.
- Bardazzi F, Landi C, Orlandi C, et al. Graham Little-Piccardi-Lasseur syndrome following HBV vaccination. Acta Derm Venereol. 1999;79:93.
- Hutchens KA, Balfour EM, Smoller BR. Comparison between Langerhans cell concentration in lichen planopilaris and traction alopecia with possible immunologic implications. Am J Dermatopathol. 2011;33:277-280.
- Dogra S, Sarangal R. What’s new in cicatricial alopecia? Indian J Dermatol Venereol Leprol. 2013;79:576-590.
- Daoud MS, Pittelkow MR. Lichen planus. In: Wolff K, Goldsmith LA, Katz Si, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 7th ed. New York, NY: Mc Graw Hill; 2008:463-477.
- Donati A, Assouly P, Matard B, et al. Clinical and photographic assessment of lichen planopilaris treatment efficacy. J Am Acad Dermatol. 2011;64:597-599.
- Samrao A, Chew AL, Price V. Frontal fibrosing alopecia: a clinical review of 36 patients. Br J Dermatol. 2010;163:1296-1300.
- Spencer LA, Hawryluk EB, English JC. Lichen planopilaris: retrospective study and stepwise therapeutic approach. Arch Dermatol. 2009;145:333-334.
- Ladizinski B, Bazakas A, Selim MA, et al. Frontal fibrosing alopecia: a retrospective review of 19 patients seen at Duke University. J Am Acad Dermatol. 2013;68:749-755
- George SJ, Hsu SJ. Lichen planopilaris treated with thalidomide. J Am Acad Dermatol. 2001;45:965-966.
- Jouanique C, Reygagne P, Bachelez H, et al. Thalidomide is ineffective in the treatment of lichen planopilaris. J Am Acad Dermatol. 2004;51:480-481.
- Mirmirani P, Karnik P. Lichen planopilaris treated with a peroxisome proliferator–activated receptor γ agonist. Arch Dermatol. 2009;145:1363-1366.
- Zegarska B, Kallas D, Schwartz RA, et al. Graham-Little syndrome. Acta Dermatovenerol Alp Pannonica Adriat. 2010;19:39-42.
- Assouly P, Reygagne P. Lichen planopilaris: update on diagnosis and treatment. Semin Cutan Med Surg. 2009;28:3-10.
- Olsen EA, Bergfield WF, Cotsarelis G, et al. Summary of North American Hair Research Society (NAHRS)–sponsored Workshop on Cicatricial Alopecia, Duke University Medical Center, February 10 and 11, 2001. J Am Acad Dermatol. 2003;48:103-110.
- Zinkernagel MS, Trueb RM. Fibrosing alopecia in a pattern distribution: patterned lichen planopilaris or androgenetic alopecia with a lichenoid tissue reaction pattern? Arch Dermatol. 2000;136:205-211.
- James WD, Berger TG, Elston DM. Andrews’ Diseases of the Skin: Clinical Dermatology. 12th ed. Philadelphia, PA: WB Saunders Company; 2016.
- Kossard S, Lee MS, Wilkinson B. Postmenopausal frontal fibrosing alopecia: a frontal variant of lichen planopilaris. J Am Acad Dermatol. 1997;36:59-66.
- Pai VV, Kikkeri NN, Sori T, et al. Graham-Little Piccardi Lassueur syndrome: an unusual variant of follicular lichen planus. Int J Trichology. 2011;3:28-30.
- Srivastava M, Mikkilineni R, Konstadt J. Lassueur-Graham-Little-Piccardi syndrome. Dermatol Online J. 2007;13:12.
- Brar BK, Khanna E, Mahajan BB. Graham Little Piccardi Lasseur syndrome: a rare case report with concomitant hypertrophic lichen planus. Int J Trichology. 2011;5:199-200.
- Vashi N, Newlove T, Chu J, et al. Graham-Little-Piccardi-Lassueur syndrome. Dermatol Online J. 2011;17:30.
- Chieregato C, Zini A, Barba A, et al. Lichen planopilaris: report of 30 cases and review of the literature. Int J Dermatol. 2003;42:342-345.
- Vega Gutierrez J, Miranda-Romera A, Perez Milan F, et al. Graham Little-Piccardi-Lassueur syndrome associated with androgen insensitivity syndrome (testicular feminization). J Eur Acad Dermatol Venereol. 2004;18:463-466.
- Rodríguez-Bayona B, Ruchaud S, Rodriguez C, et al. Autoantibodies against the chromosomal passenger protein INCENP found in a patient with Graham Little-Piccardi-Lassueur syndrome. J Autoimmune Dis. 2007;4:1.
- Viglizzo G, Verrini A, Rongioletti F. Familial Lassueur-Graham-Little-Piccardi syndrome. Dermatology. 2004;208:142-144.
- Bianchi L, Paro Vidolin A, Piemonte P, et al. Graham Little-Piccardi-Lassueur syndrome: effective treatment with cyclosporin A. Clin Exp Dermatol. 2001;26:518-520.
- Cevasco NC, Bergfeld WF, Remzi BK, et al. A case-series of 29 patients with lichen planopilaris: the Cleveland Clinic Foundation experience on evaluation, diagnosis, and treatment. J Am Acad Dermatol. 2007;57:47-53.
- Mesinkovska NA, Brankov N, Piliang M, et al. Association of lichen planopilaris with thyroid disease: a retrospective case-control study. J Am Acad Dermatol. 2014;70:889-892.
- Bardazzi F, Landi C, Orlandi C, et al. Graham Little-Piccardi-Lasseur syndrome following HBV vaccination. Acta Derm Venereol. 1999;79:93.
- Hutchens KA, Balfour EM, Smoller BR. Comparison between Langerhans cell concentration in lichen planopilaris and traction alopecia with possible immunologic implications. Am J Dermatopathol. 2011;33:277-280.
- Dogra S, Sarangal R. What’s new in cicatricial alopecia? Indian J Dermatol Venereol Leprol. 2013;79:576-590.
- Daoud MS, Pittelkow MR. Lichen planus. In: Wolff K, Goldsmith LA, Katz Si, et al, eds. Fitzpatrick’s Dermatology in General Medicine. 7th ed. New York, NY: Mc Graw Hill; 2008:463-477.
- Donati A, Assouly P, Matard B, et al. Clinical and photographic assessment of lichen planopilaris treatment efficacy. J Am Acad Dermatol. 2011;64:597-599.
- Samrao A, Chew AL, Price V. Frontal fibrosing alopecia: a clinical review of 36 patients. Br J Dermatol. 2010;163:1296-1300.
- Spencer LA, Hawryluk EB, English JC. Lichen planopilaris: retrospective study and stepwise therapeutic approach. Arch Dermatol. 2009;145:333-334.
- Ladizinski B, Bazakas A, Selim MA, et al. Frontal fibrosing alopecia: a retrospective review of 19 patients seen at Duke University. J Am Acad Dermatol. 2013;68:749-755
- George SJ, Hsu SJ. Lichen planopilaris treated with thalidomide. J Am Acad Dermatol. 2001;45:965-966.
- Jouanique C, Reygagne P, Bachelez H, et al. Thalidomide is ineffective in the treatment of lichen planopilaris. J Am Acad Dermatol. 2004;51:480-481.
- Mirmirani P, Karnik P. Lichen planopilaris treated with a peroxisome proliferator–activated receptor γ agonist. Arch Dermatol. 2009;145:1363-1366.
Practice Points
- Graham-Little-Piccardi-Lassueur syndrome (GLPL) is characterized by a triad of cicatricial alopecia of the scalp, nonscarring alopecia of the axillae and/or groin, and a rough follicular eruption on the body and/or scalp.
- Graham-Little-Piccardi-Lassueur syndrome is considered one of the 3 clinical variants of lichen planopilaris.
- Potential therapies for GLPL include hydroxychloroquine, cyclosporine, tetracyclines, and pioglitazone.
