User login
Cutis is a peer-reviewed clinical journal for the dermatologist, allergist, and general practitioner published monthly since 1965. Concise clinical articles present the practical side of dermatology, helping physicians to improve patient care. Cutis is referenced in Index Medicus/MEDLINE and is written and edited by industry leaders.
ass lick
assault rifle
balls
ballsac
black jack
bleach
Boko Haram
bondage
causas
cheap
child abuse
cocaine
compulsive behaviors
cost of miracles
cunt
Daech
display network stats
drug paraphernalia
explosion
fart
fda and death
fda AND warn
fda AND warning
fda AND warns
feom
fuck
gambling
gfc
gun
human trafficking
humira AND expensive
illegal
ISIL
ISIS
Islamic caliphate
Islamic state
madvocate
masturbation
mixed martial arts
MMA
molestation
national rifle association
NRA
nsfw
nuccitelli
pedophile
pedophilia
poker
porn
porn
pornography
psychedelic drug
recreational drug
sex slave rings
shit
slot machine
snort
substance abuse
terrorism
terrorist
texarkana
Texas hold 'em
UFC
section[contains(@class, 'nav-hidden')]
section[contains(@class, 'nav-hidden active')
A peer-reviewed, indexed journal for dermatologists with original research, image quizzes, cases and reviews, and columns.
MRSA in Dermatology Inpatients With a Vesiculobullous Disorder
Methicillin, cloxacillin, flucloxacillin, and cefoxitin are stable, penicillinase-producing β-lactam antibiotics; Staphylococcus aureus strains resistant to these agents are designated as methicillin-resistant S aureus (MRSA). Based on genotypic and phenotypic differences there are 2 strains of MRSA: hospital acquired and community acquired.
The potential for nosocomial transmission and the limited number of antibiotics available to treat MRSA are problematic. Moreover, MRSA has emerged worldwide as a major nosocomial pathogen that contributes to morbidity and mortality. Methicillin-resistant S aureus infection in vesiculobullous disorders such as pemphigus vulgaris (PV) and toxic epidermal necrolysis (TEN) is known to contribute to mortality.1
The reported prevalence of MRSA in India ranges from 12% to 38.44%.2-4 We frequently encounter MRSA in dermatology inpatients, especially those with a vesiculobullous disorder. The primary objective of this study was to determine the prevalence of MRSA in dermatology inpatients with a vesiculobullous disorder; the secondary objective was to determine if MRSA contributes to mortality.
Materials and Methods
A 1-year prospective, cross-sectional, descriptive study was conducted in a tertiary-care center. The study population included all dermatology inpatients with a vesiculobullous disorder. Patients with a vesiculobullous disorder secondary to a primary viral or bacterial disorder were excluded. Permission to conduct the study was granted by the institution’s Human Ethics Committee.
All patients underwent a detailed history and clinical examination. Routine hematology testing, urinalysis, measurement of the blood glucose level, and other investigations relevant to the vesiculobullous disorder were performed. Special investigations were Gram staining, culture, and susceptibility testing of material from a nasal swab and a swab of a representative skin lesion.
Detection of MRSA
Skin lesions were thoroughly cleaned with sterile normal saline. Specimens of pus were drawn with a sterile swab for Gram staining, culture, and susceptibility testing and were analyzed in the institution’s microbiology department. A direct colony suspension (equivalent to McFarland Standard No. 0.5) was inoculated on a Mueller-Hinton agar plate, incorporating cefoxitin, linezolid, vancomycin, amikacin, and rifampicin supplemented with sodium chloride 2% and incubated at 37°C for 24 hours. Staphylococcus aureus colonies were identified by their smooth, convex, shiny, and opaque appearance with a golden yellow pigment, as well as by coagulase positivity, mannitol fermentation, and production of phosphatase.
Methicillin-resistant S aureus was defined as an isolate having a minimum inhibitory concentration of more than 2 μg/mL of cefoxitin; a methicillin-sensitive S aureus isolate was defined as having a minimum inhibitory concentration of less than or equal to 2 μg/mL of cefoxitin. Specimens showing moderate to heavy growth of MRSA were included in the study. For specimens showing mild growth, testing was repeated; if no growth was seen on repeat testing, results were interpreted as negative.
Data were collected and analyzed for frequency and percentage; P<.05 was considered significant.
Results
The number of patients analyzed in the study period was 43. Table 1 shows their salient demographic characteristics, clinical features, and findings of the investigation. The youngest patient was aged 13 years; the oldest was aged 80 years. The male to female ratio was 0.65 to 1. The most common primary lesion was a combined vesicle and bulla (34 patients [79.1%]); the most common secondary lesion was a combination of erosion with crusting (22 patients [51.2%]).
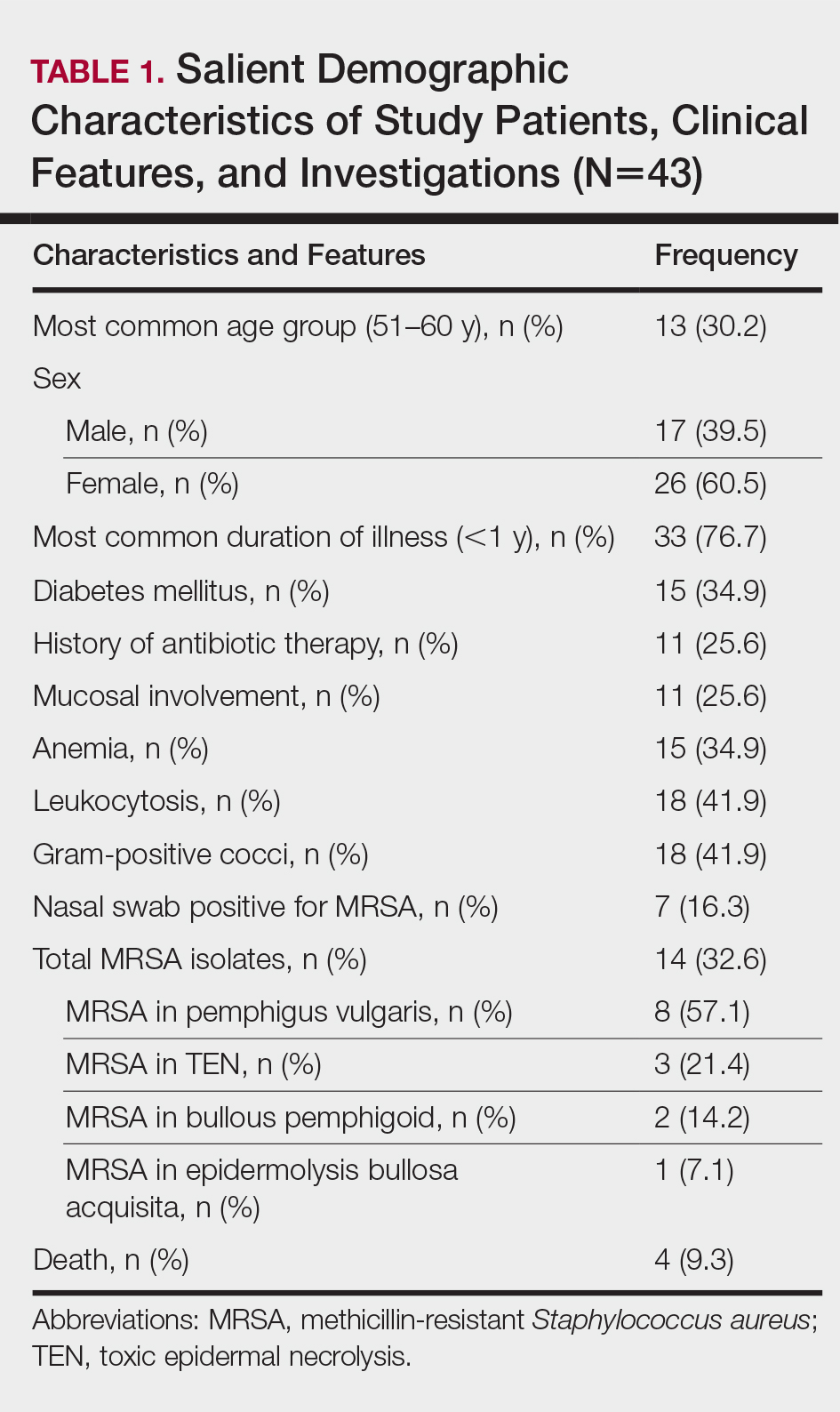
Table 2 lists the types of vesiculobullous disorders seen in this study. Pemphigus vulgaris was the most common (21 patients [48.8%])(Figure 1). Drug-induced vesiculobullous disorders (eg, TEN) were noted in 11 patients (25.6%)(Figure 2).


Table 2 also lists pathogens cultured in the study group. There were 24 bacterial isolates, of which S aureus accounted for 22 (91.7%). Methicillin-resistant S aureus was cultured in 14 patients (32.6%); culture was sterile in 19 patients (44.2%).
Among the 22 cultured staphylococcal species, MRSA accounted for 14 (63.6%) and constituted 58.3% (14/24) of all bacterial isolates. The nasal swab for MRSA was positive in 4 PV patients (9.3%), 2 TEN patients (4.6%), and 1 bullous pemphigoid patient (2.3%). Methicillin-resistant S aureus was most commonly cultured in PV patients (8/14 [57.1%]).
All MRSA strains (100%) were sensitive to vancomycin and linezolid; 34 (79.1%) were sensitive to amikacin. Additionally, 100% of MRSA strains were resistant to oxacillin, cloxacillin, and cefoxitin.
Three patients with PV (7.0%) and 1 patient with TEN (2.3%) died during the course of the study; only 1 death (2.3%) occurred in a patient who had a positive MRSA culture.
Comment
In this 1-year study, we tested and followed 43 patients with autoimmune and drug-induced vesiculobullous disorders. Vesiculobullous disorders in dermatology inpatients are a cause of great concern. When lesions rupture, they leave behind a large area of erosion that forms a nidus of bacterial colonization; often, these bacteria cause severe infection, including septicemia, and result in death.5 Moreover, autoimmune bullous disorders usually require a prolonged hospital stay and powerful immunosuppressive drugs, which contributes to bacterial infection, especially MRSA.6
The age of patients in this study ranged from 13 to 80 years; most patients were in the 6th decade, a pattern seen in studies worldwide.5 In a study by Kanwar and De,7 however, most cases were aged 20 to 40 years.7 In our study, there was a female preponderance (male to female ratio of 0.65 to 1).
Studies have shown that the duration of illness in vesiculobullous disorder is directly associated with MRSA infection. However, in our study with MRSA detected in 14 patients, most patients had a duration of illness less than 1 year (statistically insignificant [P>.05]), a finding similar to Shafi et al.8
The symptomatic nature of these diseases, their unsightly appearance, and mucous-membrane involvement of vesiculobullous disorders prompts these patients to present to the hospital early. However, a prolonged hospital stay by patients with an autoimmune vesiculobullous disorders sets the stage for MRSA colonization.
In this study, diabetes mellitus (DM) was seen in 15 patients (34.9%); 5 of them had MRSA infection (statistically insignificant [P>.05]). Diabetes mellitus contributing to sepsis and MRSA infection, which in turn contributes to morbidity and mortality, has been well-documented.2,4,9
Methicillin-resistant S aureus in this study was isolated most often from blisters and erosions. Vesiculobullous disorders and drug reactions (eg, Stevens-Johnson syndrome, TEN) are characterized by blisters that rupture to form erosions and crusting, which form fissures in the epidermal barrier function that are nidi for colonization by microbes, especially S aureus and MRSA in particular; later, these bacteria can enter dermal vessels and then the bloodstream, leading to septicemia.10
The prevalence of MRSA in this study was 32.6% (14/43), which is high compared to other studies.2-4 Pemphigus vulgaris was the most common disorder infected by MRSA in this study (57.1% [8/14] of MRSA isolates)(Table 1), a finding that reveals that the incidence of MRSA is high among staphylococcal isolates in vesiculobullous disorders. However, the high incidence of MRSA in this study could be a reflection of the number of patients with a severe and chronic vesiculobullous disorder, such as PV, and serious drug reactions such as TEN referred to our tertiary-care center, where we get a large number of patients affected by autoimmune and drug-induced vesiculobullous disorders. Similar findings have been reported by Stryjewski et al.11
A high prevalence of MRSA in a dermatology unit has grave consequences, contributing to morbidity and mortality in particular among patients with a vesiculobullous disorder. Immunosuppressive therapy and comorbidities such as DM contribute to MRSA colonization in vesiculobullous disorders.12 Overcrowding and poor sterilization techniques in public hospitals in India may contribute to the high prevalence of MRSA seen in hospital units.
Patients with a vesiculobullous disorder who are chronic nasal carriers of MRSA are at risk for cutaneous MRSA infection, which in turn can lead to MRSA septicemia and an elevated risk of death. In this study, however, a nasal swab was positive for MRSA in only 7 patients. One patient with MRSA colonization died, which was statistically insignificant (P=1).
In this study, all MRSA strains (100%) were resistant to first-line antibiotics, such as oxacillin, cloxacillin, and cefoxitin; all strains were susceptible to vancomycin and linezolid.
Conclusion
Our study shows that MRSA is becoming the prominent pathogen in nosocomial infections, especially in bedridden patients, which has grave implications. The use of a prophylactic S aureus conjugate vaccine in patients with a chronic vesiculobullous disorder might be justified in the future.15 We found a high prevalence (32.6%) of MRSA in vesiculobullous disorders, no relationship between DM and MRSA colonization, PV was the most common disorder complicated by MRSA, no relationship between nasal colonization and MRSA infection, no relationship between death during the study period and MRSA infection, 100% of MRSA strains were susceptible to vancomycin and linezolid, and 79.1% of MRSA strains were susceptible to amikacin.
- Nair SP. A retrospective study of mortality of pemphigus patients in a tertiary care hospital. Indian J Dermatol Venereol Leprol. 2013;79:706-709.
- Sachdev D, Amladi S, Natarj G, et al. An outbreak of methicillin-resistant Staphylococcus aureus (MRSA) infection in dermatology inpatients. Indian J Dermatol Venereol Leprol. 2003;69:377-380.
- Vijayamohan N, Nair SP. A study of the prevalence of methicillin-resistant Staphylococcus aureus in dermatology inpatients. Indian Dermatol Online J. 2014;5:441-445.
- Malhotra SK, Malhotra S, Dhaliwal GS, et al. Bacterial study of pyodermas in a tertiary care dermatological center. Indian J Dermatol. 2012;57:358-361.
- Valencia IC, Kirsner RS, Kerdel FA. Microbiological evaluation of skin wounds: alarming trends towards antibiotic resistance in an inpatient dermatology service during a 10-year period. J Am Acad Dermatol. 2004;50:845-849.
- Lehman JS, Murell DF, Camilleri MJ, et al. Infection and infection prevention in patients treated with immunosuppressive medications for autoimmune bullous disorders. Dermatol Clin. 2011;29:591-598.
- Kanwar AJ, De D. Pemphigus in India. Indian J Dermatol Venereol Leprol. 2011;77:439-449.
- Shafi M, Khatri ML, Mashima M, et al. Pemphigus: a clinical study of 109 cases from Tripoli, Libya. Indian J Dermatol Venereol Leprol. 1994;60:140-143.
- Torres K, Sampathkumar P. Predictors of methicillin-resistant Staphylococcus aureus colonization at hospital admission. Am J Infect Control. 2013;41:1043-1047.
- Miller LG, Quan C, Shay A, et al. A prospective investigation of outcomes after hospital discharge for endemic, community-acquired methicillin-resistant Staphylococcus aureus skin infection. Clin Infect Dis. 2007;44:483-492.
- Stryjewski M, Chambers HF. Skin and soft-tissue infections caused by community-acquired methicillin-resistant Staphylococcus aureus. Clin Infect Dis. 2008;46(suppl 5):S368-S377.
- Mutasim DF. Management of autoimmune bullous diseases: pharmacology and therapeutics. J Am Acad Dermatol. 2004;51:859-877.
- Cohen PR. Community-acquired methicillin-resistant Staphylococcus aureus skin infections: a review of epidemiology, clinical features, management, and prevention. Int J Dermatol. 2007;46:1-11.
- Elston DM. Methicillin-sensitive and methicillin-resistant Staphylococcus aureus: management principles and selection of antibiotic therapy. Dermatol Clin. 2007;25:157-164.
- Shinefield H, Black S, Fattom A, et al. Use of a Staphylococcus aureus conjugate vaccine in patients receiving hemodialysis. N Engl J Med. 2001;346:491-496.
Methicillin, cloxacillin, flucloxacillin, and cefoxitin are stable, penicillinase-producing β-lactam antibiotics; Staphylococcus aureus strains resistant to these agents are designated as methicillin-resistant S aureus (MRSA). Based on genotypic and phenotypic differences there are 2 strains of MRSA: hospital acquired and community acquired.
The potential for nosocomial transmission and the limited number of antibiotics available to treat MRSA are problematic. Moreover, MRSA has emerged worldwide as a major nosocomial pathogen that contributes to morbidity and mortality. Methicillin-resistant S aureus infection in vesiculobullous disorders such as pemphigus vulgaris (PV) and toxic epidermal necrolysis (TEN) is known to contribute to mortality.1
The reported prevalence of MRSA in India ranges from 12% to 38.44%.2-4 We frequently encounter MRSA in dermatology inpatients, especially those with a vesiculobullous disorder. The primary objective of this study was to determine the prevalence of MRSA in dermatology inpatients with a vesiculobullous disorder; the secondary objective was to determine if MRSA contributes to mortality.
Materials and Methods
A 1-year prospective, cross-sectional, descriptive study was conducted in a tertiary-care center. The study population included all dermatology inpatients with a vesiculobullous disorder. Patients with a vesiculobullous disorder secondary to a primary viral or bacterial disorder were excluded. Permission to conduct the study was granted by the institution’s Human Ethics Committee.
All patients underwent a detailed history and clinical examination. Routine hematology testing, urinalysis, measurement of the blood glucose level, and other investigations relevant to the vesiculobullous disorder were performed. Special investigations were Gram staining, culture, and susceptibility testing of material from a nasal swab and a swab of a representative skin lesion.
Detection of MRSA
Skin lesions were thoroughly cleaned with sterile normal saline. Specimens of pus were drawn with a sterile swab for Gram staining, culture, and susceptibility testing and were analyzed in the institution’s microbiology department. A direct colony suspension (equivalent to McFarland Standard No. 0.5) was inoculated on a Mueller-Hinton agar plate, incorporating cefoxitin, linezolid, vancomycin, amikacin, and rifampicin supplemented with sodium chloride 2% and incubated at 37°C for 24 hours. Staphylococcus aureus colonies were identified by their smooth, convex, shiny, and opaque appearance with a golden yellow pigment, as well as by coagulase positivity, mannitol fermentation, and production of phosphatase.
Methicillin-resistant S aureus was defined as an isolate having a minimum inhibitory concentration of more than 2 μg/mL of cefoxitin; a methicillin-sensitive S aureus isolate was defined as having a minimum inhibitory concentration of less than or equal to 2 μg/mL of cefoxitin. Specimens showing moderate to heavy growth of MRSA were included in the study. For specimens showing mild growth, testing was repeated; if no growth was seen on repeat testing, results were interpreted as negative.
Data were collected and analyzed for frequency and percentage; P<.05 was considered significant.
Results
The number of patients analyzed in the study period was 43. Table 1 shows their salient demographic characteristics, clinical features, and findings of the investigation. The youngest patient was aged 13 years; the oldest was aged 80 years. The male to female ratio was 0.65 to 1. The most common primary lesion was a combined vesicle and bulla (34 patients [79.1%]); the most common secondary lesion was a combination of erosion with crusting (22 patients [51.2%]).
Table 2 lists the types of vesiculobullous disorders seen in this study. Pemphigus vulgaris was the most common (21 patients [48.8%])(Figure 1). Drug-induced vesiculobullous disorders (eg, TEN) were noted in 11 patients (25.6%)(Figure 2).
Table 2 also lists pathogens cultured in the study group. There were 24 bacterial isolates, of which S aureus accounted for 22 (91.7%). Methicillin-resistant S aureus was cultured in 14 patients (32.6%); culture was sterile in 19 patients (44.2%).
Among the 22 cultured staphylococcal species, MRSA accounted for 14 (63.6%) and constituted 58.3% (14/24) of all bacterial isolates. The nasal swab for MRSA was positive in 4 PV patients (9.3%), 2 TEN patients (4.6%), and 1 bullous pemphigoid patient (2.3%). Methicillin-resistant S aureus was most commonly cultured in PV patients (8/14 [57.1%]).
All MRSA strains (100%) were sensitive to vancomycin and linezolid; 34 (79.1%) were sensitive to amikacin. Additionally, 100% of MRSA strains were resistant to oxacillin, cloxacillin, and cefoxitin.
Three patients with PV (7.0%) and 1 patient with TEN (2.3%) died during the course of the study; only 1 death (2.3%) occurred in a patient who had a positive MRSA culture.
Comment
In this 1-year study, we tested and followed 43 patients with autoimmune and drug-induced vesiculobullous disorders. Vesiculobullous disorders in dermatology inpatients are a cause of great concern. When lesions rupture, they leave behind a large area of erosion that forms a nidus of bacterial colonization; often, these bacteria cause severe infection, including septicemia, and result in death.5 Moreover, autoimmune bullous disorders usually require a prolonged hospital stay and powerful immunosuppressive drugs, which contributes to bacterial infection, especially MRSA.6
The age of patients in this study ranged from 13 to 80 years; most patients were in the 6th decade, a pattern seen in studies worldwide.5 In a study by Kanwar and De,7 however, most cases were aged 20 to 40 years.7 In our study, there was a female preponderance (male to female ratio of 0.65 to 1).
Studies have shown that the duration of illness in vesiculobullous disorder is directly associated with MRSA infection. However, in our study with MRSA detected in 14 patients, most patients had a duration of illness less than 1 year (statistically insignificant [P>.05]), a finding similar to Shafi et al.8
The symptomatic nature of these diseases, their unsightly appearance, and mucous-membrane involvement of vesiculobullous disorders prompts these patients to present to the hospital early. However, a prolonged hospital stay by patients with an autoimmune vesiculobullous disorders sets the stage for MRSA colonization.
In this study, diabetes mellitus (DM) was seen in 15 patients (34.9%); 5 of them had MRSA infection (statistically insignificant [P>.05]). Diabetes mellitus contributing to sepsis and MRSA infection, which in turn contributes to morbidity and mortality, has been well-documented.2,4,9
Methicillin-resistant S aureus in this study was isolated most often from blisters and erosions. Vesiculobullous disorders and drug reactions (eg, Stevens-Johnson syndrome, TEN) are characterized by blisters that rupture to form erosions and crusting, which form fissures in the epidermal barrier function that are nidi for colonization by microbes, especially S aureus and MRSA in particular; later, these bacteria can enter dermal vessels and then the bloodstream, leading to septicemia.10
The prevalence of MRSA in this study was 32.6% (14/43), which is high compared to other studies.2-4 Pemphigus vulgaris was the most common disorder infected by MRSA in this study (57.1% [8/14] of MRSA isolates)(Table 1), a finding that reveals that the incidence of MRSA is high among staphylococcal isolates in vesiculobullous disorders. However, the high incidence of MRSA in this study could be a reflection of the number of patients with a severe and chronic vesiculobullous disorder, such as PV, and serious drug reactions such as TEN referred to our tertiary-care center, where we get a large number of patients affected by autoimmune and drug-induced vesiculobullous disorders. Similar findings have been reported by Stryjewski et al.11
A high prevalence of MRSA in a dermatology unit has grave consequences, contributing to morbidity and mortality in particular among patients with a vesiculobullous disorder. Immunosuppressive therapy and comorbidities such as DM contribute to MRSA colonization in vesiculobullous disorders.12 Overcrowding and poor sterilization techniques in public hospitals in India may contribute to the high prevalence of MRSA seen in hospital units.
Patients with a vesiculobullous disorder who are chronic nasal carriers of MRSA are at risk for cutaneous MRSA infection, which in turn can lead to MRSA septicemia and an elevated risk of death. In this study, however, a nasal swab was positive for MRSA in only 7 patients. One patient with MRSA colonization died, which was statistically insignificant (P=1).
In this study, all MRSA strains (100%) were resistant to first-line antibiotics, such as oxacillin, cloxacillin, and cefoxitin; all strains were susceptible to vancomycin and linezolid.
Conclusion
Our study shows that MRSA is becoming the prominent pathogen in nosocomial infections, especially in bedridden patients, which has grave implications. The use of a prophylactic S aureus conjugate vaccine in patients with a chronic vesiculobullous disorder might be justified in the future.15 We found a high prevalence (32.6%) of MRSA in vesiculobullous disorders, no relationship between DM and MRSA colonization, PV was the most common disorder complicated by MRSA, no relationship between nasal colonization and MRSA infection, no relationship between death during the study period and MRSA infection, 100% of MRSA strains were susceptible to vancomycin and linezolid, and 79.1% of MRSA strains were susceptible to amikacin.
Methicillin, cloxacillin, flucloxacillin, and cefoxitin are stable, penicillinase-producing β-lactam antibiotics; Staphylococcus aureus strains resistant to these agents are designated as methicillin-resistant S aureus (MRSA). Based on genotypic and phenotypic differences there are 2 strains of MRSA: hospital acquired and community acquired.
The potential for nosocomial transmission and the limited number of antibiotics available to treat MRSA are problematic. Moreover, MRSA has emerged worldwide as a major nosocomial pathogen that contributes to morbidity and mortality. Methicillin-resistant S aureus infection in vesiculobullous disorders such as pemphigus vulgaris (PV) and toxic epidermal necrolysis (TEN) is known to contribute to mortality.1
The reported prevalence of MRSA in India ranges from 12% to 38.44%.2-4 We frequently encounter MRSA in dermatology inpatients, especially those with a vesiculobullous disorder. The primary objective of this study was to determine the prevalence of MRSA in dermatology inpatients with a vesiculobullous disorder; the secondary objective was to determine if MRSA contributes to mortality.
Materials and Methods
A 1-year prospective, cross-sectional, descriptive study was conducted in a tertiary-care center. The study population included all dermatology inpatients with a vesiculobullous disorder. Patients with a vesiculobullous disorder secondary to a primary viral or bacterial disorder were excluded. Permission to conduct the study was granted by the institution’s Human Ethics Committee.
All patients underwent a detailed history and clinical examination. Routine hematology testing, urinalysis, measurement of the blood glucose level, and other investigations relevant to the vesiculobullous disorder were performed. Special investigations were Gram staining, culture, and susceptibility testing of material from a nasal swab and a swab of a representative skin lesion.
Detection of MRSA
Skin lesions were thoroughly cleaned with sterile normal saline. Specimens of pus were drawn with a sterile swab for Gram staining, culture, and susceptibility testing and were analyzed in the institution’s microbiology department. A direct colony suspension (equivalent to McFarland Standard No. 0.5) was inoculated on a Mueller-Hinton agar plate, incorporating cefoxitin, linezolid, vancomycin, amikacin, and rifampicin supplemented with sodium chloride 2% and incubated at 37°C for 24 hours. Staphylococcus aureus colonies were identified by their smooth, convex, shiny, and opaque appearance with a golden yellow pigment, as well as by coagulase positivity, mannitol fermentation, and production of phosphatase.
Methicillin-resistant S aureus was defined as an isolate having a minimum inhibitory concentration of more than 2 μg/mL of cefoxitin; a methicillin-sensitive S aureus isolate was defined as having a minimum inhibitory concentration of less than or equal to 2 μg/mL of cefoxitin. Specimens showing moderate to heavy growth of MRSA were included in the study. For specimens showing mild growth, testing was repeated; if no growth was seen on repeat testing, results were interpreted as negative.
Data were collected and analyzed for frequency and percentage; P<.05 was considered significant.
Results
The number of patients analyzed in the study period was 43. Table 1 shows their salient demographic characteristics, clinical features, and findings of the investigation. The youngest patient was aged 13 years; the oldest was aged 80 years. The male to female ratio was 0.65 to 1. The most common primary lesion was a combined vesicle and bulla (34 patients [79.1%]); the most common secondary lesion was a combination of erosion with crusting (22 patients [51.2%]).
Table 2 lists the types of vesiculobullous disorders seen in this study. Pemphigus vulgaris was the most common (21 patients [48.8%])(Figure 1). Drug-induced vesiculobullous disorders (eg, TEN) were noted in 11 patients (25.6%)(Figure 2).
Table 2 also lists pathogens cultured in the study group. There were 24 bacterial isolates, of which S aureus accounted for 22 (91.7%). Methicillin-resistant S aureus was cultured in 14 patients (32.6%); culture was sterile in 19 patients (44.2%).
Among the 22 cultured staphylococcal species, MRSA accounted for 14 (63.6%) and constituted 58.3% (14/24) of all bacterial isolates. The nasal swab for MRSA was positive in 4 PV patients (9.3%), 2 TEN patients (4.6%), and 1 bullous pemphigoid patient (2.3%). Methicillin-resistant S aureus was most commonly cultured in PV patients (8/14 [57.1%]).
All MRSA strains (100%) were sensitive to vancomycin and linezolid; 34 (79.1%) were sensitive to amikacin. Additionally, 100% of MRSA strains were resistant to oxacillin, cloxacillin, and cefoxitin.
Three patients with PV (7.0%) and 1 patient with TEN (2.3%) died during the course of the study; only 1 death (2.3%) occurred in a patient who had a positive MRSA culture.
Comment
In this 1-year study, we tested and followed 43 patients with autoimmune and drug-induced vesiculobullous disorders. Vesiculobullous disorders in dermatology inpatients are a cause of great concern. When lesions rupture, they leave behind a large area of erosion that forms a nidus of bacterial colonization; often, these bacteria cause severe infection, including septicemia, and result in death.5 Moreover, autoimmune bullous disorders usually require a prolonged hospital stay and powerful immunosuppressive drugs, which contributes to bacterial infection, especially MRSA.6
The age of patients in this study ranged from 13 to 80 years; most patients were in the 6th decade, a pattern seen in studies worldwide.5 In a study by Kanwar and De,7 however, most cases were aged 20 to 40 years.7 In our study, there was a female preponderance (male to female ratio of 0.65 to 1).
Studies have shown that the duration of illness in vesiculobullous disorder is directly associated with MRSA infection. However, in our study with MRSA detected in 14 patients, most patients had a duration of illness less than 1 year (statistically insignificant [P>.05]), a finding similar to Shafi et al.8
The symptomatic nature of these diseases, their unsightly appearance, and mucous-membrane involvement of vesiculobullous disorders prompts these patients to present to the hospital early. However, a prolonged hospital stay by patients with an autoimmune vesiculobullous disorders sets the stage for MRSA colonization.
In this study, diabetes mellitus (DM) was seen in 15 patients (34.9%); 5 of them had MRSA infection (statistically insignificant [P>.05]). Diabetes mellitus contributing to sepsis and MRSA infection, which in turn contributes to morbidity and mortality, has been well-documented.2,4,9
Methicillin-resistant S aureus in this study was isolated most often from blisters and erosions. Vesiculobullous disorders and drug reactions (eg, Stevens-Johnson syndrome, TEN) are characterized by blisters that rupture to form erosions and crusting, which form fissures in the epidermal barrier function that are nidi for colonization by microbes, especially S aureus and MRSA in particular; later, these bacteria can enter dermal vessels and then the bloodstream, leading to septicemia.10
The prevalence of MRSA in this study was 32.6% (14/43), which is high compared to other studies.2-4 Pemphigus vulgaris was the most common disorder infected by MRSA in this study (57.1% [8/14] of MRSA isolates)(Table 1), a finding that reveals that the incidence of MRSA is high among staphylococcal isolates in vesiculobullous disorders. However, the high incidence of MRSA in this study could be a reflection of the number of patients with a severe and chronic vesiculobullous disorder, such as PV, and serious drug reactions such as TEN referred to our tertiary-care center, where we get a large number of patients affected by autoimmune and drug-induced vesiculobullous disorders. Similar findings have been reported by Stryjewski et al.11
A high prevalence of MRSA in a dermatology unit has grave consequences, contributing to morbidity and mortality in particular among patients with a vesiculobullous disorder. Immunosuppressive therapy and comorbidities such as DM contribute to MRSA colonization in vesiculobullous disorders.12 Overcrowding and poor sterilization techniques in public hospitals in India may contribute to the high prevalence of MRSA seen in hospital units.
Patients with a vesiculobullous disorder who are chronic nasal carriers of MRSA are at risk for cutaneous MRSA infection, which in turn can lead to MRSA septicemia and an elevated risk of death. In this study, however, a nasal swab was positive for MRSA in only 7 patients. One patient with MRSA colonization died, which was statistically insignificant (P=1).
In this study, all MRSA strains (100%) were resistant to first-line antibiotics, such as oxacillin, cloxacillin, and cefoxitin; all strains were susceptible to vancomycin and linezolid.
Conclusion
Our study shows that MRSA is becoming the prominent pathogen in nosocomial infections, especially in bedridden patients, which has grave implications. The use of a prophylactic S aureus conjugate vaccine in patients with a chronic vesiculobullous disorder might be justified in the future.15 We found a high prevalence (32.6%) of MRSA in vesiculobullous disorders, no relationship between DM and MRSA colonization, PV was the most common disorder complicated by MRSA, no relationship between nasal colonization and MRSA infection, no relationship between death during the study period and MRSA infection, 100% of MRSA strains were susceptible to vancomycin and linezolid, and 79.1% of MRSA strains were susceptible to amikacin.
- Nair SP. A retrospective study of mortality of pemphigus patients in a tertiary care hospital. Indian J Dermatol Venereol Leprol. 2013;79:706-709.
- Sachdev D, Amladi S, Natarj G, et al. An outbreak of methicillin-resistant Staphylococcus aureus (MRSA) infection in dermatology inpatients. Indian J Dermatol Venereol Leprol. 2003;69:377-380.
- Vijayamohan N, Nair SP. A study of the prevalence of methicillin-resistant Staphylococcus aureus in dermatology inpatients. Indian Dermatol Online J. 2014;5:441-445.
- Malhotra SK, Malhotra S, Dhaliwal GS, et al. Bacterial study of pyodermas in a tertiary care dermatological center. Indian J Dermatol. 2012;57:358-361.
- Valencia IC, Kirsner RS, Kerdel FA. Microbiological evaluation of skin wounds: alarming trends towards antibiotic resistance in an inpatient dermatology service during a 10-year period. J Am Acad Dermatol. 2004;50:845-849.
- Lehman JS, Murell DF, Camilleri MJ, et al. Infection and infection prevention in patients treated with immunosuppressive medications for autoimmune bullous disorders. Dermatol Clin. 2011;29:591-598.
- Kanwar AJ, De D. Pemphigus in India. Indian J Dermatol Venereol Leprol. 2011;77:439-449.
- Shafi M, Khatri ML, Mashima M, et al. Pemphigus: a clinical study of 109 cases from Tripoli, Libya. Indian J Dermatol Venereol Leprol. 1994;60:140-143.
- Torres K, Sampathkumar P. Predictors of methicillin-resistant Staphylococcus aureus colonization at hospital admission. Am J Infect Control. 2013;41:1043-1047.
- Miller LG, Quan C, Shay A, et al. A prospective investigation of outcomes after hospital discharge for endemic, community-acquired methicillin-resistant Staphylococcus aureus skin infection. Clin Infect Dis. 2007;44:483-492.
- Stryjewski M, Chambers HF. Skin and soft-tissue infections caused by community-acquired methicillin-resistant Staphylococcus aureus. Clin Infect Dis. 2008;46(suppl 5):S368-S377.
- Mutasim DF. Management of autoimmune bullous diseases: pharmacology and therapeutics. J Am Acad Dermatol. 2004;51:859-877.
- Cohen PR. Community-acquired methicillin-resistant Staphylococcus aureus skin infections: a review of epidemiology, clinical features, management, and prevention. Int J Dermatol. 2007;46:1-11.
- Elston DM. Methicillin-sensitive and methicillin-resistant Staphylococcus aureus: management principles and selection of antibiotic therapy. Dermatol Clin. 2007;25:157-164.
- Shinefield H, Black S, Fattom A, et al. Use of a Staphylococcus aureus conjugate vaccine in patients receiving hemodialysis. N Engl J Med. 2001;346:491-496.
- Nair SP. A retrospective study of mortality of pemphigus patients in a tertiary care hospital. Indian J Dermatol Venereol Leprol. 2013;79:706-709.
- Sachdev D, Amladi S, Natarj G, et al. An outbreak of methicillin-resistant Staphylococcus aureus (MRSA) infection in dermatology inpatients. Indian J Dermatol Venereol Leprol. 2003;69:377-380.
- Vijayamohan N, Nair SP. A study of the prevalence of methicillin-resistant Staphylococcus aureus in dermatology inpatients. Indian Dermatol Online J. 2014;5:441-445.
- Malhotra SK, Malhotra S, Dhaliwal GS, et al. Bacterial study of pyodermas in a tertiary care dermatological center. Indian J Dermatol. 2012;57:358-361.
- Valencia IC, Kirsner RS, Kerdel FA. Microbiological evaluation of skin wounds: alarming trends towards antibiotic resistance in an inpatient dermatology service during a 10-year period. J Am Acad Dermatol. 2004;50:845-849.
- Lehman JS, Murell DF, Camilleri MJ, et al. Infection and infection prevention in patients treated with immunosuppressive medications for autoimmune bullous disorders. Dermatol Clin. 2011;29:591-598.
- Kanwar AJ, De D. Pemphigus in India. Indian J Dermatol Venereol Leprol. 2011;77:439-449.
- Shafi M, Khatri ML, Mashima M, et al. Pemphigus: a clinical study of 109 cases from Tripoli, Libya. Indian J Dermatol Venereol Leprol. 1994;60:140-143.
- Torres K, Sampathkumar P. Predictors of methicillin-resistant Staphylococcus aureus colonization at hospital admission. Am J Infect Control. 2013;41:1043-1047.
- Miller LG, Quan C, Shay A, et al. A prospective investigation of outcomes after hospital discharge for endemic, community-acquired methicillin-resistant Staphylococcus aureus skin infection. Clin Infect Dis. 2007;44:483-492.
- Stryjewski M, Chambers HF. Skin and soft-tissue infections caused by community-acquired methicillin-resistant Staphylococcus aureus. Clin Infect Dis. 2008;46(suppl 5):S368-S377.
- Mutasim DF. Management of autoimmune bullous diseases: pharmacology and therapeutics. J Am Acad Dermatol. 2004;51:859-877.
- Cohen PR. Community-acquired methicillin-resistant Staphylococcus aureus skin infections: a review of epidemiology, clinical features, management, and prevention. Int J Dermatol. 2007;46:1-11.
- Elston DM. Methicillin-sensitive and methicillin-resistant Staphylococcus aureus: management principles and selection of antibiotic therapy. Dermatol Clin. 2007;25:157-164.
- Shinefield H, Black S, Fattom A, et al. Use of a Staphylococcus aureus conjugate vaccine in patients receiving hemodialysis. N Engl J Med. 2001;346:491-496.
Practice Points
- Methicillin-resistant Staphylococcus aureus (MRSA) infection in vesiculobullous disorders such as pemphigus vulgaris and toxic epidermal necrolysis is known to contribute to an increase in disease-related mortality.
- Methicillin-resistant S aureus is becoming the prominent pathogen in nosocomial infections, especially in bedridden patients.
- The prevalence of MRSA in vesiculobullous disorders is high; pemphigus vulgaris is the most common vesiculobullous disorder complicated by MRSA.
- Early diagnosis of MRSA helps reduce morbidity and mortality and improves the patient’s prognosis.
Sweet Syndrome With Aseptic Splenic Abscesses and Multiple Myeloma
To the Editor:
An 84-year-old man was admitted to the hospital with 5 erythematous cutaneous nodules of several days’ duration on the legs ranging in size from 1.0 to 1.5 cm. Upon admission, the patient also had a chest radiograph suspicious for pneumonia. The patient had received sulfamethoxazole/trimethoprim for a urinary tract infection as an outpatient 5 days prior to presentation, but he stopped the medication due to the appearance of the cutaneous nodules. Of note, the patient also reported unintentional weight loss of 15 pounds over the last few months.
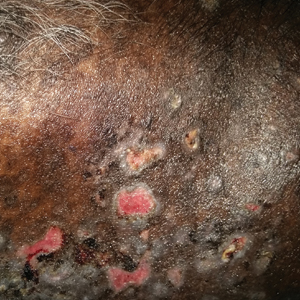
New nodules had developed at a rate of 1 to 2 lesions daily in the 3 days prior to presentation and continued to develop after admission to the hospital. The nodules appeared as tender, erythematous lesions that evolved to form pustules and developed overlying crusts in later stages (Figure 1). They were limited to the arms and legs, primarily involving the lower legs. There was no evidence of oral or ocular involvement. A hemoglobin count of 10.9 g/dL (reference range, 14.0–17.5 g/dL), white blood cell count of 8.8×109/L (reference range, 4.5–11.0×109/L), and erythrocyte sedimentation rate of 69 mm/h (reference range, 0–20 mm/h) were noted on admission.
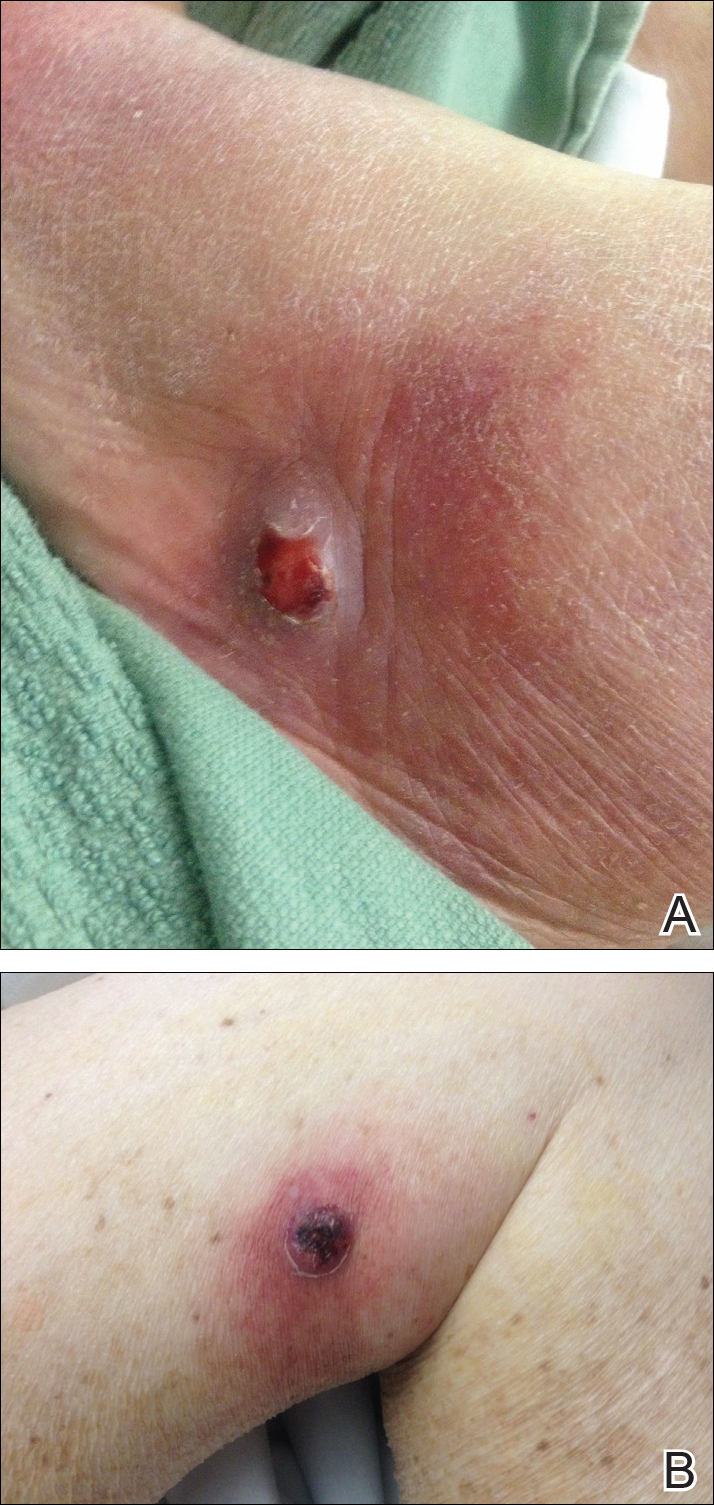
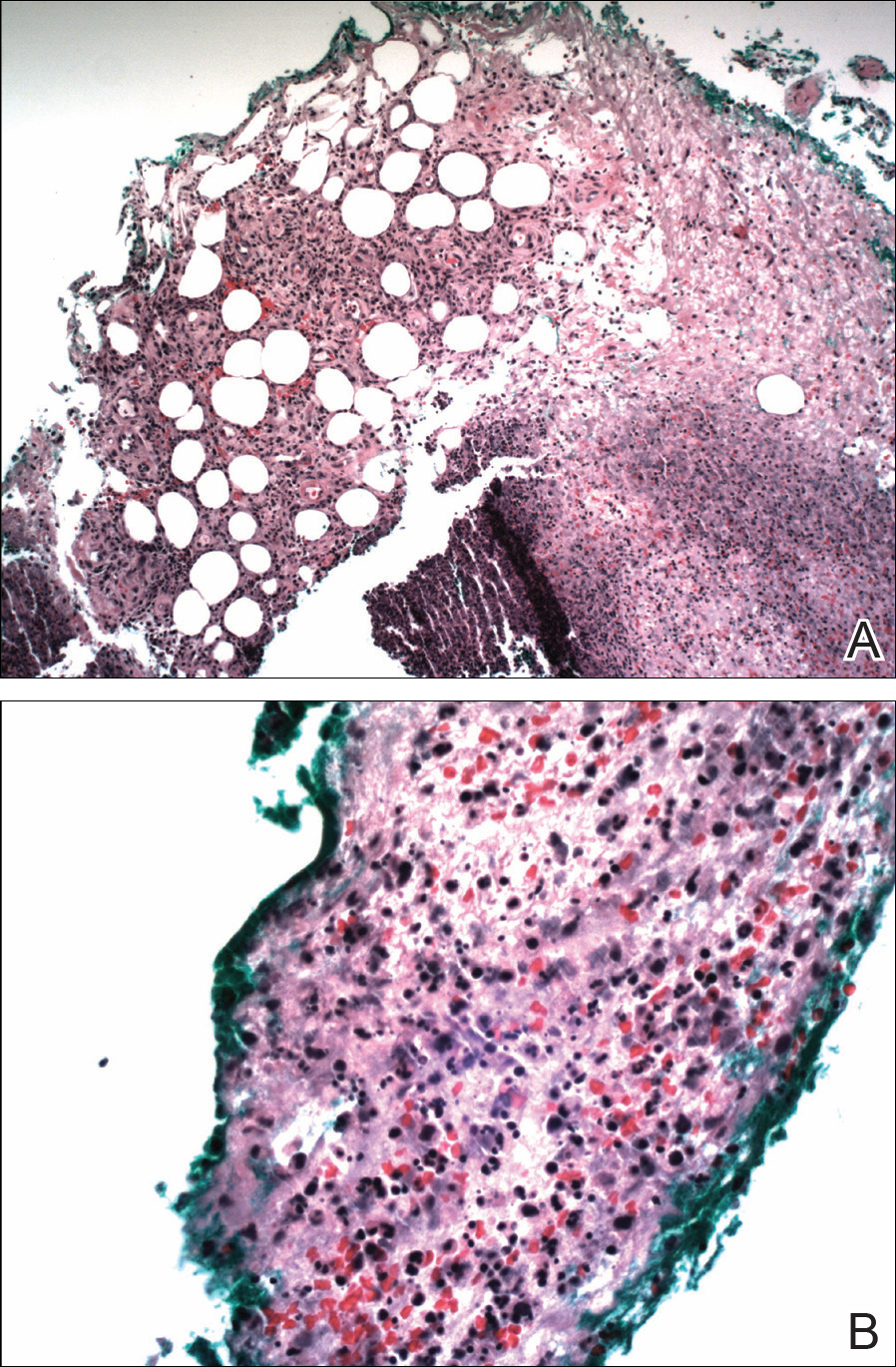
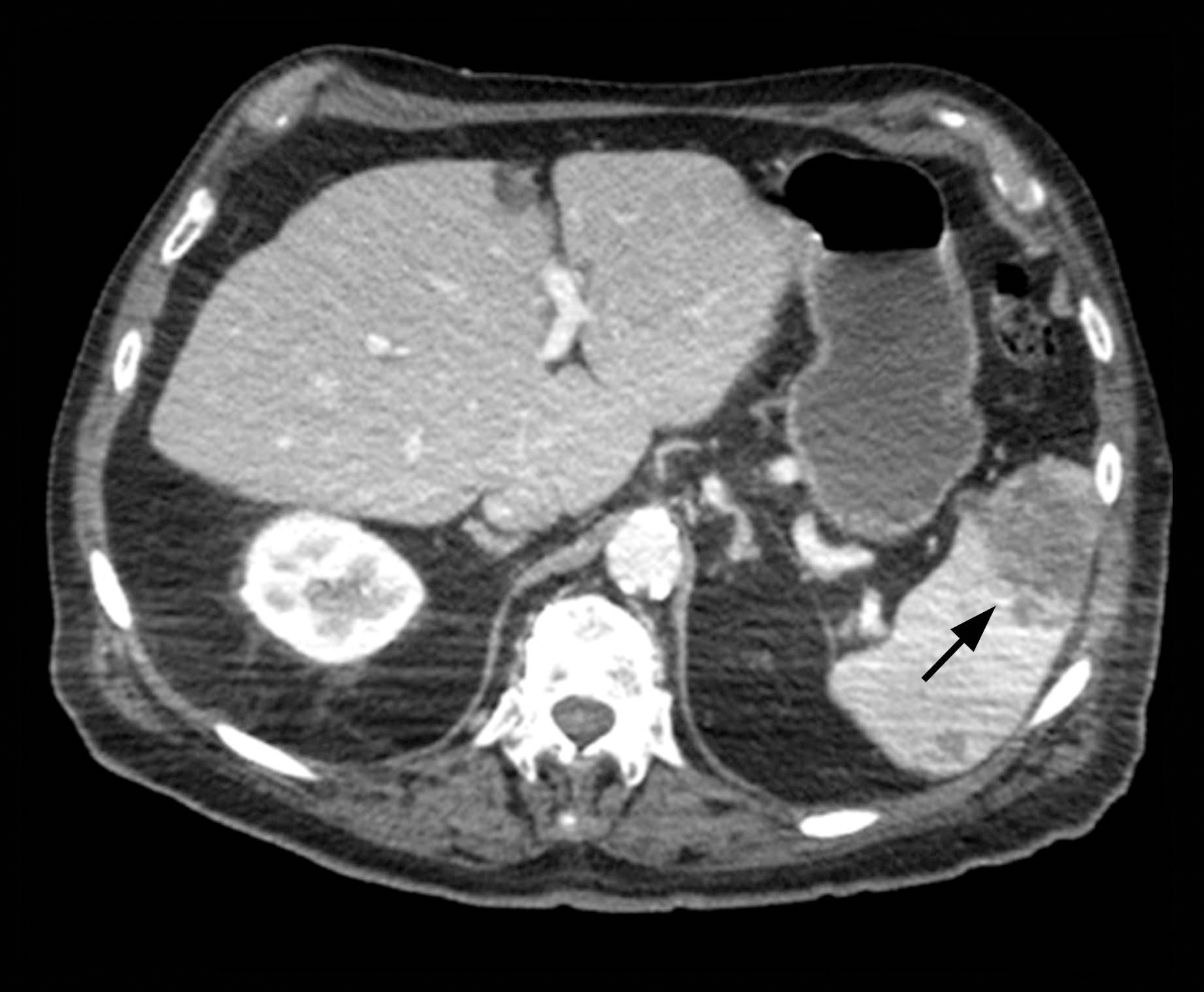
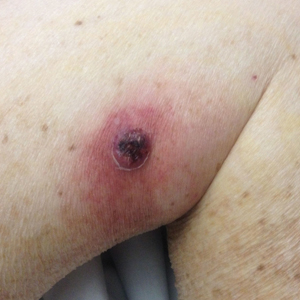
The patient was started on ceftriaxone and azithromycin for suspected pneumonia. The differential diagnosis for the cutaneous nodules included lymphoma, acid-fast bacilli (AFB) infection, deep fungal infection, pyoderma gangrenosum, Sweet syndrome (SS), panniculitis, erythema elevatum diutinum, and polyarteritis nodosa. A punch biopsy of a nodule on the left foot was performed. Histopathology demonstrated a neutrophilic panniculitis (Figure 2) with an epidermal abscess. No vasculitis was identified, and periodic acid–Schiff and AFB staining of the skin biopsy were negative. These findings were consistent with SS. Computed tomography scans of the chest, abdomen, and pelvis, which were completed early in the course of hospitalization due to concern for underlying malignancy, revealed pericardial and pleural effusions as well as cystic lesions in the lungs, spleen, kidneys, and prostate, with the largest lesion on the spleen measuring 5.6×4.8 cm (Figure 3). Computed tomography scanning was negative for areas of consolidation in the lungs. A splenic biopsy was performed by an interventional radiologist during the patient's hospitalization that identified an aseptic, neutrophilic process. Fungal, bacterial, and AFB cultures of the splenic tissue and cystic contents were negative. Bilateral pleural effusions also were identified, and a thoracentesis was performed. The pleural fluid indicated rare mesothelial cells in the background of acute inflammation with no growth of the bacterial, fungal, or AFB cultures.
Due to the association of hematologic malignances with SS, a bone marrow biopsy was performed, which revealed multiple myeloma. Serum protein electrophoresis demonstrated monoclonal gammopathy of κ light chains. During the course of his hospitalization, new skin lesions continued to develop on the hands, face, and trunk. The patient was discharged from the hospital shortly after diagnosis to receive outpatient treatment for multiple myeloma with lenalidomide and dexamethasone. Upon follow-up with the patient’s family via telephone 3 weeks into treatment, his son confirmed that the nodules were resolving.
Our case could be consistent with either drug-induced or malignancy-associated SS. Sweet syndrome initially was described in 1964 in 8 female patients with leukocytosis and cutaneous plaques infiltrated by neutrophils.1 The skin lesions typically are red and painful, ranging in size from 0.5 cm to 12.0 cm, and can last weeks to years if not treated.2 Variations of skin lesions include bullous and pustular morphologies.3
Diagnostic criteria for SS have been established.4 Both of the major criteria must be met as well as 2 of 4 minor criteria. Major criteria include abrupt onset of tender erythematous plaques and nodules; secondly, a dense neutrophilic infiltrate without evidence of leukocytoclastic vasculitis must be seen on histopathology. Minor criteria include pyrexia, association with underlying condition (malignancy, pregnancy, drug exposure, inflammatory disorder), responsiveness to systemic steroids, and abnormal laboratory values (erythrocyte sedimentation rate, white blood cell count, C-reactive protein, neutrophilia).4
Sweet syndrome can be divided into 3 classifications: classical or idiopathic, drug-induced, or malignancy-associated.4 Classical SS most commonly is seen in middle-aged women after an upper respiratory or gastrointestinal infection. Drug-induced SS most often is associated with granulocyte-stimulating factor colony therapy4; however, it has been associated with use of trimethoprim/sulfamethoxazole.5 Malignancy-associated SS most commonly is seen in individuals with hematologic malignancy, specifically acute myeloid leukemia. Although its association with multiple myeloma is not as frequent, cases of malignancy-associated SS identifying this association have been reported.6,7 Mucosal involvement in the form of aphthouslike lesions more frequently is seen in malignancy-associated SS.8 Differing from classical SS, which has a female predilection of around 4:1, the malignancy-associated disorder has a 1:1 female-to-male ratio.4
In the majority of cases of SS, the neutrophilic infiltrate is in the papillary and upper reticular dermis; however, if the neutrophilic infiltrate is predominately in the subcutaneous tissue (known as subcutaneous SS), there is a strong association with malignancy.9 The histopathology in our case demonstrated a neutrophilic infiltrate in the subcutaneous tissue.
Fever is the most common systemic manifestation of SS and is present in 54% to 65% of patients.8,10 Besides the skin, the most common site affected is the eye, with 13% to 75% of patients reporting ocular involvement, usually conjunctivitis.4,10 Although infrequent, extracutaneous SS has been identified in the bones, central nervous system, kidneys, heart, liver, spleen, lungs, ears, eyes, and intestines.4 A case of SS with splenic involvement in the form of sterile abscesses also was reported.11 This case was related to parvovirus B19.
Sweet syndrome is a condition characterized by tender, erythematous cutaneous lesions with histopathology demonstrating neutrophilic infiltrate in the absence of vasculitis. We report a case of suspected extracutaneous SS in the form of splenic cysts in a patient whose SS was associated with malignancy and/or drug ingestion.
- Sweet RD. An acute febrile neutrophilic dermatosis. Br J Dermatol. 1964;76:349-356.
- Cohen PR, Kurzrock R. Sweet’s syndrome and malignancy. Am J Med. 1987;82:1220-1226.
- Cohen PR, Kurzrock R. Sweet’s syndrome revisited: a review of disease concepts. Int J Dermatol. 2002;41:182-184.
- Cohen PR. Sweet’s syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:34.
- Walker DC, Cohen PR. Trimethoprim-sulfamethoxazole-associated acute febrile neutrophilic dermatosis: case report and review of drug-induced Sweet’s syndrome. J Am Acad Dermatol. 1996;34:918-923.
- Belhadjali H, Chaabane S, Njim L, et al. Sweet’s syndrome associated with multiple myeloma. Acta Dermatovenerol Alp Pannonica Adriat. 2008;17:31-33.
- Bayer-Garner IB, Cottler-Fox M, Smoller BR. Sweet syndrome in multiple myeloma: a series of six cases. J Cutan Pathol. 2003;30:261-264.
- Fett DL, Gibson LE, Su WP. Sweet’s syndrome: systemic signs and symptoms and associated disorders. Mayo Clin Proc. 1995;70:234-240.
- von den Driesch P. Sweet’s syndrome (acute febrile neutrophilic dermatosis). J Am Acad Dermatol. 1994;31:535-556; quiz 557-560.
- Neoh CY, Tan AW, Ng SK. Sweet’s syndrome: a spectrum of unusual clinical presentation and associations. Br J Dermatol. 2007;156:480-485.
- Fortna RR, Toporcer M, Elder DE, et al. A case of sweet syndrome with spleen and lymph node involvement preceded by parvovirus B19 infection, and review of the literature on extracutaneous Sweet syndrome. Am J Dermatopathol. 2010;32:621-627.
To the Editor:
An 84-year-old man was admitted to the hospital with 5 erythematous cutaneous nodules of several days’ duration on the legs ranging in size from 1.0 to 1.5 cm. Upon admission, the patient also had a chest radiograph suspicious for pneumonia. The patient had received sulfamethoxazole/trimethoprim for a urinary tract infection as an outpatient 5 days prior to presentation, but he stopped the medication due to the appearance of the cutaneous nodules. Of note, the patient also reported unintentional weight loss of 15 pounds over the last few months.
New nodules had developed at a rate of 1 to 2 lesions daily in the 3 days prior to presentation and continued to develop after admission to the hospital. The nodules appeared as tender, erythematous lesions that evolved to form pustules and developed overlying crusts in later stages (Figure 1). They were limited to the arms and legs, primarily involving the lower legs. There was no evidence of oral or ocular involvement. A hemoglobin count of 10.9 g/dL (reference range, 14.0–17.5 g/dL), white blood cell count of 8.8×109/L (reference range, 4.5–11.0×109/L), and erythrocyte sedimentation rate of 69 mm/h (reference range, 0–20 mm/h) were noted on admission.
The patient was started on ceftriaxone and azithromycin for suspected pneumonia. The differential diagnosis for the cutaneous nodules included lymphoma, acid-fast bacilli (AFB) infection, deep fungal infection, pyoderma gangrenosum, Sweet syndrome (SS), panniculitis, erythema elevatum diutinum, and polyarteritis nodosa. A punch biopsy of a nodule on the left foot was performed. Histopathology demonstrated a neutrophilic panniculitis (Figure 2) with an epidermal abscess. No vasculitis was identified, and periodic acid–Schiff and AFB staining of the skin biopsy were negative. These findings were consistent with SS. Computed tomography scans of the chest, abdomen, and pelvis, which were completed early in the course of hospitalization due to concern for underlying malignancy, revealed pericardial and pleural effusions as well as cystic lesions in the lungs, spleen, kidneys, and prostate, with the largest lesion on the spleen measuring 5.6×4.8 cm (Figure 3). Computed tomography scanning was negative for areas of consolidation in the lungs. A splenic biopsy was performed by an interventional radiologist during the patient's hospitalization that identified an aseptic, neutrophilic process. Fungal, bacterial, and AFB cultures of the splenic tissue and cystic contents were negative. Bilateral pleural effusions also were identified, and a thoracentesis was performed. The pleural fluid indicated rare mesothelial cells in the background of acute inflammation with no growth of the bacterial, fungal, or AFB cultures.
Due to the association of hematologic malignances with SS, a bone marrow biopsy was performed, which revealed multiple myeloma. Serum protein electrophoresis demonstrated monoclonal gammopathy of κ light chains. During the course of his hospitalization, new skin lesions continued to develop on the hands, face, and trunk. The patient was discharged from the hospital shortly after diagnosis to receive outpatient treatment for multiple myeloma with lenalidomide and dexamethasone. Upon follow-up with the patient’s family via telephone 3 weeks into treatment, his son confirmed that the nodules were resolving.
Our case could be consistent with either drug-induced or malignancy-associated SS. Sweet syndrome initially was described in 1964 in 8 female patients with leukocytosis and cutaneous plaques infiltrated by neutrophils.1 The skin lesions typically are red and painful, ranging in size from 0.5 cm to 12.0 cm, and can last weeks to years if not treated.2 Variations of skin lesions include bullous and pustular morphologies.3
Diagnostic criteria for SS have been established.4 Both of the major criteria must be met as well as 2 of 4 minor criteria. Major criteria include abrupt onset of tender erythematous plaques and nodules; secondly, a dense neutrophilic infiltrate without evidence of leukocytoclastic vasculitis must be seen on histopathology. Minor criteria include pyrexia, association with underlying condition (malignancy, pregnancy, drug exposure, inflammatory disorder), responsiveness to systemic steroids, and abnormal laboratory values (erythrocyte sedimentation rate, white blood cell count, C-reactive protein, neutrophilia).4
Sweet syndrome can be divided into 3 classifications: classical or idiopathic, drug-induced, or malignancy-associated.4 Classical SS most commonly is seen in middle-aged women after an upper respiratory or gastrointestinal infection. Drug-induced SS most often is associated with granulocyte-stimulating factor colony therapy4; however, it has been associated with use of trimethoprim/sulfamethoxazole.5 Malignancy-associated SS most commonly is seen in individuals with hematologic malignancy, specifically acute myeloid leukemia. Although its association with multiple myeloma is not as frequent, cases of malignancy-associated SS identifying this association have been reported.6,7 Mucosal involvement in the form of aphthouslike lesions more frequently is seen in malignancy-associated SS.8 Differing from classical SS, which has a female predilection of around 4:1, the malignancy-associated disorder has a 1:1 female-to-male ratio.4
In the majority of cases of SS, the neutrophilic infiltrate is in the papillary and upper reticular dermis; however, if the neutrophilic infiltrate is predominately in the subcutaneous tissue (known as subcutaneous SS), there is a strong association with malignancy.9 The histopathology in our case demonstrated a neutrophilic infiltrate in the subcutaneous tissue.
Fever is the most common systemic manifestation of SS and is present in 54% to 65% of patients.8,10 Besides the skin, the most common site affected is the eye, with 13% to 75% of patients reporting ocular involvement, usually conjunctivitis.4,10 Although infrequent, extracutaneous SS has been identified in the bones, central nervous system, kidneys, heart, liver, spleen, lungs, ears, eyes, and intestines.4 A case of SS with splenic involvement in the form of sterile abscesses also was reported.11 This case was related to parvovirus B19.
Sweet syndrome is a condition characterized by tender, erythematous cutaneous lesions with histopathology demonstrating neutrophilic infiltrate in the absence of vasculitis. We report a case of suspected extracutaneous SS in the form of splenic cysts in a patient whose SS was associated with malignancy and/or drug ingestion.
To the Editor:
An 84-year-old man was admitted to the hospital with 5 erythematous cutaneous nodules of several days’ duration on the legs ranging in size from 1.0 to 1.5 cm. Upon admission, the patient also had a chest radiograph suspicious for pneumonia. The patient had received sulfamethoxazole/trimethoprim for a urinary tract infection as an outpatient 5 days prior to presentation, but he stopped the medication due to the appearance of the cutaneous nodules. Of note, the patient also reported unintentional weight loss of 15 pounds over the last few months.
New nodules had developed at a rate of 1 to 2 lesions daily in the 3 days prior to presentation and continued to develop after admission to the hospital. The nodules appeared as tender, erythematous lesions that evolved to form pustules and developed overlying crusts in later stages (Figure 1). They were limited to the arms and legs, primarily involving the lower legs. There was no evidence of oral or ocular involvement. A hemoglobin count of 10.9 g/dL (reference range, 14.0–17.5 g/dL), white blood cell count of 8.8×109/L (reference range, 4.5–11.0×109/L), and erythrocyte sedimentation rate of 69 mm/h (reference range, 0–20 mm/h) were noted on admission.
The patient was started on ceftriaxone and azithromycin for suspected pneumonia. The differential diagnosis for the cutaneous nodules included lymphoma, acid-fast bacilli (AFB) infection, deep fungal infection, pyoderma gangrenosum, Sweet syndrome (SS), panniculitis, erythema elevatum diutinum, and polyarteritis nodosa. A punch biopsy of a nodule on the left foot was performed. Histopathology demonstrated a neutrophilic panniculitis (Figure 2) with an epidermal abscess. No vasculitis was identified, and periodic acid–Schiff and AFB staining of the skin biopsy were negative. These findings were consistent with SS. Computed tomography scans of the chest, abdomen, and pelvis, which were completed early in the course of hospitalization due to concern for underlying malignancy, revealed pericardial and pleural effusions as well as cystic lesions in the lungs, spleen, kidneys, and prostate, with the largest lesion on the spleen measuring 5.6×4.8 cm (Figure 3). Computed tomography scanning was negative for areas of consolidation in the lungs. A splenic biopsy was performed by an interventional radiologist during the patient's hospitalization that identified an aseptic, neutrophilic process. Fungal, bacterial, and AFB cultures of the splenic tissue and cystic contents were negative. Bilateral pleural effusions also were identified, and a thoracentesis was performed. The pleural fluid indicated rare mesothelial cells in the background of acute inflammation with no growth of the bacterial, fungal, or AFB cultures.
Due to the association of hematologic malignances with SS, a bone marrow biopsy was performed, which revealed multiple myeloma. Serum protein electrophoresis demonstrated monoclonal gammopathy of κ light chains. During the course of his hospitalization, new skin lesions continued to develop on the hands, face, and trunk. The patient was discharged from the hospital shortly after diagnosis to receive outpatient treatment for multiple myeloma with lenalidomide and dexamethasone. Upon follow-up with the patient’s family via telephone 3 weeks into treatment, his son confirmed that the nodules were resolving.
Our case could be consistent with either drug-induced or malignancy-associated SS. Sweet syndrome initially was described in 1964 in 8 female patients with leukocytosis and cutaneous plaques infiltrated by neutrophils.1 The skin lesions typically are red and painful, ranging in size from 0.5 cm to 12.0 cm, and can last weeks to years if not treated.2 Variations of skin lesions include bullous and pustular morphologies.3
Diagnostic criteria for SS have been established.4 Both of the major criteria must be met as well as 2 of 4 minor criteria. Major criteria include abrupt onset of tender erythematous plaques and nodules; secondly, a dense neutrophilic infiltrate without evidence of leukocytoclastic vasculitis must be seen on histopathology. Minor criteria include pyrexia, association with underlying condition (malignancy, pregnancy, drug exposure, inflammatory disorder), responsiveness to systemic steroids, and abnormal laboratory values (erythrocyte sedimentation rate, white blood cell count, C-reactive protein, neutrophilia).4
Sweet syndrome can be divided into 3 classifications: classical or idiopathic, drug-induced, or malignancy-associated.4 Classical SS most commonly is seen in middle-aged women after an upper respiratory or gastrointestinal infection. Drug-induced SS most often is associated with granulocyte-stimulating factor colony therapy4; however, it has been associated with use of trimethoprim/sulfamethoxazole.5 Malignancy-associated SS most commonly is seen in individuals with hematologic malignancy, specifically acute myeloid leukemia. Although its association with multiple myeloma is not as frequent, cases of malignancy-associated SS identifying this association have been reported.6,7 Mucosal involvement in the form of aphthouslike lesions more frequently is seen in malignancy-associated SS.8 Differing from classical SS, which has a female predilection of around 4:1, the malignancy-associated disorder has a 1:1 female-to-male ratio.4
In the majority of cases of SS, the neutrophilic infiltrate is in the papillary and upper reticular dermis; however, if the neutrophilic infiltrate is predominately in the subcutaneous tissue (known as subcutaneous SS), there is a strong association with malignancy.9 The histopathology in our case demonstrated a neutrophilic infiltrate in the subcutaneous tissue.
Fever is the most common systemic manifestation of SS and is present in 54% to 65% of patients.8,10 Besides the skin, the most common site affected is the eye, with 13% to 75% of patients reporting ocular involvement, usually conjunctivitis.4,10 Although infrequent, extracutaneous SS has been identified in the bones, central nervous system, kidneys, heart, liver, spleen, lungs, ears, eyes, and intestines.4 A case of SS with splenic involvement in the form of sterile abscesses also was reported.11 This case was related to parvovirus B19.
Sweet syndrome is a condition characterized by tender, erythematous cutaneous lesions with histopathology demonstrating neutrophilic infiltrate in the absence of vasculitis. We report a case of suspected extracutaneous SS in the form of splenic cysts in a patient whose SS was associated with malignancy and/or drug ingestion.
- Sweet RD. An acute febrile neutrophilic dermatosis. Br J Dermatol. 1964;76:349-356.
- Cohen PR, Kurzrock R. Sweet’s syndrome and malignancy. Am J Med. 1987;82:1220-1226.
- Cohen PR, Kurzrock R. Sweet’s syndrome revisited: a review of disease concepts. Int J Dermatol. 2002;41:182-184.
- Cohen PR. Sweet’s syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:34.
- Walker DC, Cohen PR. Trimethoprim-sulfamethoxazole-associated acute febrile neutrophilic dermatosis: case report and review of drug-induced Sweet’s syndrome. J Am Acad Dermatol. 1996;34:918-923.
- Belhadjali H, Chaabane S, Njim L, et al. Sweet’s syndrome associated with multiple myeloma. Acta Dermatovenerol Alp Pannonica Adriat. 2008;17:31-33.
- Bayer-Garner IB, Cottler-Fox M, Smoller BR. Sweet syndrome in multiple myeloma: a series of six cases. J Cutan Pathol. 2003;30:261-264.
- Fett DL, Gibson LE, Su WP. Sweet’s syndrome: systemic signs and symptoms and associated disorders. Mayo Clin Proc. 1995;70:234-240.
- von den Driesch P. Sweet’s syndrome (acute febrile neutrophilic dermatosis). J Am Acad Dermatol. 1994;31:535-556; quiz 557-560.
- Neoh CY, Tan AW, Ng SK. Sweet’s syndrome: a spectrum of unusual clinical presentation and associations. Br J Dermatol. 2007;156:480-485.
- Fortna RR, Toporcer M, Elder DE, et al. A case of sweet syndrome with spleen and lymph node involvement preceded by parvovirus B19 infection, and review of the literature on extracutaneous Sweet syndrome. Am J Dermatopathol. 2010;32:621-627.
- Sweet RD. An acute febrile neutrophilic dermatosis. Br J Dermatol. 1964;76:349-356.
- Cohen PR, Kurzrock R. Sweet’s syndrome and malignancy. Am J Med. 1987;82:1220-1226.
- Cohen PR, Kurzrock R. Sweet’s syndrome revisited: a review of disease concepts. Int J Dermatol. 2002;41:182-184.
- Cohen PR. Sweet’s syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:34.
- Walker DC, Cohen PR. Trimethoprim-sulfamethoxazole-associated acute febrile neutrophilic dermatosis: case report and review of drug-induced Sweet’s syndrome. J Am Acad Dermatol. 1996;34:918-923.
- Belhadjali H, Chaabane S, Njim L, et al. Sweet’s syndrome associated with multiple myeloma. Acta Dermatovenerol Alp Pannonica Adriat. 2008;17:31-33.
- Bayer-Garner IB, Cottler-Fox M, Smoller BR. Sweet syndrome in multiple myeloma: a series of six cases. J Cutan Pathol. 2003;30:261-264.
- Fett DL, Gibson LE, Su WP. Sweet’s syndrome: systemic signs and symptoms and associated disorders. Mayo Clin Proc. 1995;70:234-240.
- von den Driesch P. Sweet’s syndrome (acute febrile neutrophilic dermatosis). J Am Acad Dermatol. 1994;31:535-556; quiz 557-560.
- Neoh CY, Tan AW, Ng SK. Sweet’s syndrome: a spectrum of unusual clinical presentation and associations. Br J Dermatol. 2007;156:480-485.
- Fortna RR, Toporcer M, Elder DE, et al. A case of sweet syndrome with spleen and lymph node involvement preceded by parvovirus B19 infection, and review of the literature on extracutaneous Sweet syndrome. Am J Dermatopathol. 2010;32:621-627.
Practice Points
- Sweet syndrome (SS), also known as acute febrile neutrophilic dermatosis, is an inflammatory process characterized by a diffuse dermal neutrophilic infiltrate in the absence of vasculitis.
- A diagnosis of SS warrants further investigation due to its association with malignancy, especially hematologic malignancy.
- Other organs in SS also may have aseptic involvement.
Eosinophilic Pustular Folliculitis With Underlying Mantle Cell Lymphoma
Eosinophilic pustular folliculitis (EPF) was originally described in 1965 and has since evolved into 3 distinct subtypes: classic, immunosuppressed (IS), and infantile types. Immunosuppressed EPF can be further subdivided into human immunodeficiency virus (HIV) associated (IS-HIV) and non-HIV associated. Human immunodeficiency virus–seronegative cases have been associated with underlying malignancies (IS-heme) or chronic immunosuppression, such as that seen in transplant patients.
Case Report
A 52-year-old man with a medical history limited to prostate adenocarcinoma treated with a robotic prostatectomy presented with a pruritic red rash on the face, neck, shoulders, and chest of 1 month’s duration. The patient previously completed a course of azithromycin 250 mg, intramuscular triamcinolone, and oral prednisone with only minor improvement. Physical examination demonstrated multiple pink folliculocentric papules and pustules scattered on the head (Figure 1A), neck, and chest (Figure 1B), as well as edematous pink papules and plaques on the forehead (Figures 1C and 1D). The palms, soles, and oral mucosa were clear.
, neck, and chest (B), as well as edematous pink papules and plaques on the forehead (C and D).")
Initial biopsy of the right side of the chest was nonspecific and most consistent with a reaction to an arthropod bite. The patient was started on oral doxycycline 100 mg twice daily for 2 weeks. With no improvement seen, additional biopsies were obtained from the left side of the chest and forehead. The biopsy of the chest showed ruptured folliculitis with evidence of acute and chronic inflammation. The biopsy of the forehead demonstrated eosinophilic follicular spongiosis with intrafollicular Langerhans cell microgranulomas along with abundant eosinophils adjacent to follicles, consistent with EPF (Figure 2). Serum HIV testing was negative. Serum white blood cell count was normal at 6400/µL (reference range, 4500–11,000/µL) with mild elevation of eosinophils (8%). The remaining complete blood cell count and comprehensive metabolic panel were within reference range. The patient was subsequently started on oral indomethacin 25 mg twice daily and triamcinolone cream 0.1%. Within a few days he experienced initial improvement in his symptoms of pruritus and diminution in the number of inflammatory follicular papules.
(H&E, original magnifications ×10 and ×20).")
Approximately 1 month after presentation, he began to experience symptoms of dysphagia and fatigue. In addition, tonsillar hypertrophy and palpable neck and axillary lymphadenopathy were present. Computed tomography of the neck, chest, and abdomen showed diffuse lymphadenopathy. Full-body positron emission tomography–computed tomography demonstrated extensive metabolically active lymphoma in multiple nodal groups above and below the diaphragm. There also was lymphomatous involvement of the spleen. An axillary lymph node biopsy was diagnostic for mantle cell lymphoma (CD4:CD8, 1:1; CD45 negative; CD20 positive; CD5 positive). He
Comment
Subtypes of EPF
Eosinophilic pustular folliculitis was first described in a Japanese female presenting with folliculocentric pustules distributed on the face, torso, and arms.1 This noninfectious eosinophilic infiltration of hair follicles predominantly seen in the Japanese population is now regarded as the classic form. Three distinct subtypes of EPF now exist, including the originally described classic variant (Ofuji disease), an IS variant, and a rare infantile form.1
All 3 subtypes of EPF are more commonly seen in men than women. The classic form has a peak incidence between the third and fourth decades of life. It presents as chronic annular papules and sterile pustules exhibiting peripheral extension, with individual lesions lasting for approximately 7 to 10 days with frequent relapses. The face is the most common area of involvement, followed by the trunk, extremities, and more rarely the palmoplantar surfaces. Concomitant leukocytosis with eosinophilia is seen in up to 35% of patients.1 The infantile type represents the rarest EPF form. The average age of onset is 5 months, with most cases resolving by 14 months of age.1
Clinically, EPF is characterized by recurrent papules and pustules predominantly on the scalp without annular or polycyclic ring formation, as seen in the classic type. The palms and soles may be involved, which can clinically mimic infantile acropustulosis and scabies infection. Most patients exhibit a concomitant peripheral eosinophilia.1,2
In the late 1980s, the IS variant of EPF was recognized in HIV-positive (IS-HIV) and HIV-negative malignancy-associated (IS-heme) populations.1,3 This newly characterized form differs morphologically and biologically from the classic and infantile subtypes. The IS subtype has a unique presentation including intensely pruritic, discrete, erythematous, follicular papules with palmoplantar sparing and infrequent annular or circinate plaque forms.1 Frequently, with the IS-HIV form, CD4+ T-cell counts are below 300 cells/mL, and 25% to 50% of patients have lymphopenia with eosinophilia.3 Highly active antiretroviral therapy has been associated with EPF resolution in HIV-positive individuals; however, it also has been shown to induce transient EPF during the first 3 to 6 months of initiation.1,3,4
Unlike the IS-HIV form, the IS-heme form has occurred solely in males and is predominantly associated with hematologic malignancies (eg, non-Hodgkin lymphoma, acute lymphoblastic leukemia, acute myeloid leukemia, myelodysplastic syndrome) 30 to 90 days following bone marrow transplant, peripheral blood stem cell transplant, or chemotherapy treatment.5,6 Unlike the chronic and persistent IS-HIV form, prior cases of IS-heme EPF have been predominantly self-limited. Interestingly, only 2 reported cases of EPF have occurred prior to the diagnosis of malignancy including B-cell leukemia and myelodysplastic syndrome.5
Histopathology
All 3 identified forms of EPF histopathologically show acute and chronic lymphoeosinophilic infiltrate concentrated at the follicular isthmus, which can lead to follicular destruction. Scattered mononuclear cells, eosinophils, and neutrophils are found within the pilar outer root sheath, sebaceous glands, and ducts. Approximately 40% of cases demonstrate follicular mucinosis.1 Histopathology of lesional palmar skin in classic-type EPF demonstrates intraepidermal pustule formation with abundant eosinophils and neutrophils adjacent to the acrosyringium.7,8
Pathogenesis
Although the pathophysiology of EPF is largely unknown, it is thought to represent a helper T cell (TH2) response involving IL-4, IL-5, and IL-13 cytokines.9 Chemoattractant receptor homologous molecule 2, which is expressed on eosinophils and lymphocytes, is believed to play a role in the pruritus, edema, and inflammatory response seen adjacent to pilosebaceous units in EPF.10 Moreover, immunohistochemical and flow cytometry analysis has revealed a prevalence of prostaglandin D2 within the perisebocyte infiltrate in EPF.9 Prostaglandin D2 induces eotaxin-3 production within sebocytes via peroxisome proliferator-activated receptor γ, which enhances chemoattraction of eosinophils. This pathogenesis represents a prostaglandin-based mechanism and potentially explains the efficacy of indomethacin treatment of EPF through its cyclooxygenase inhibition and reduction of chemoattractant receptor homologous molecule 2 expression.9-11
Treatment
Multiple therapeutic modalities have been reported for the treatment of EPF. For all 3 subtypes, moderate- to high-potency topical corticosteroids are considered first-line therapy. UVB phototherapy 2 to 3 times weekly remains the gold standard, given its consistent efficacy.1,12 Indomethacin (50–75 mg daily) remains first-line treatment of classic EPF.4,12 Previously reported cases of classic EPF and IS-EPF have responded well to oral prednisone (1 mg/kg daily).12,13 In a retrospective review of EPF treatment data, the following treatments also have been reported to be successful: psoralen plus UVA, oral cetirizine (20–40 mg daily, particularly for IS-EPF cases), metronidazole (250 mg 3 times daily), minocycline (150 mg daily), itraconazole (200–400 mg daily, dapsone (50–200 mg daily), systemic retinoids, tacrolimus ointment 0.1%, and permethrin cream.4,12
Malignancy
Although the entity of IS-heme EPF is rare, the morphology and treatment are unique and can potentially unmask an underlying hematologic malignancy. In patients with EPF and associated malignancy, such as our patient, a differential diagnosis to consider is eosinophilic dermatosis of hematologic malignancy (EDHM). Eosinophilic dermatosis of hematologic malignancy is most commonly associated with chronic lymphocytic leukemia and can be differentiated from EPF clinically, histopathologically, and by treatment response. Eosinophilic dermatosis of hematologic malignancy clinically presents with nonspecific papules, pustules, and/or vesicles on the head, trunk, and extremities. On histopathology, EDHM shows a superficial and deep perivascular and interstitial lymphoeosinophilic infiltration. Furthermore, EDHM patients typically exhibit a poor treatment response to oral indomethacin.14
Conclusion
Eosinophilic pustular folliculitis is a noninfectious folliculocentric process comprised of 3 distinct types. The histopathology shows follicular spongiosis with increased eosinophils. The pathogenesis is most likely related to a multifactorial immune system dysregulation involving TH2 T cells, prostaglandin D2, and eotaxin-3. The treatment of EPF may involve topical corticosteroids, UVB phototherapy, or most notably oral indomethacin. In patients with EPF and malignancy, EDHM is a differential diagnosis to consider. Our case serves as a reminder that rare eosinophilic dermatoses may represent manifestations of underlying hematopoietic malignancy and, when investigated early, can lead to appropriate life-saving treatment.
- Nervi J, Stephen. Eosinophilic pustular folliculitis: a 40 year retrospect. J Am Acad Dermatol. 2006;55:285-289.
- Hernández-Martín Á, Nuño-González A, Colmenero I, et al. Eosinophilic pustular folliculitis of infancy: a series of 15 cases and review of the literature [published online July 21, 2012]. J Am Acad Dermatol. 2013;68:150-155.
- Soepr
ono F, Schinella R. Eosinophilic pustular folliculitis in patients with acquired immunodeficiency syndrome. report of three cases. J Am Acad Dermatol. 1986;14:1020-1022. - Katoh
M, Nomura T, Miyachi Y, et al. Eosinophilic pustular folliculitis: a review of the Japanese published works. J Dermatol. 2013;40:15-20. - Keida
T, Hayashi N, Kawashima M. Eosinophilic pustular folliculitis following autologous peripheral blood stem-cell transplant. J Dermatol. 2004;31:21-26. - Goiriz R, Gul-Millán G, Peñas PF, et al. Eosinophilic folliculitis following allogeneic peripheral blood stem cell transplantation: case report and review. J Cutan Pathol. 2007;34(suppl 1):33-36.
- Satoh T, Ikeda H, Yokozeki H. Acrosyringeal involvement of palmoplantar lesions of eosinophilic pustular folliculitis. Acta Derm Venereol. 2013;93:99.
- Tsuboi H, Wakita K, Fujimura T, et al. Acral variant of eosinophilic pustular folliculitis (Ofuji’s disease). Clin Exp Dermatol. 2003;28:321-324.
- Nakahig
ashi K, Doi H, Otsuka A, et al. PGD2 induces eotaxin-3 via PPARgamma from sebocytes: a possible pathogenesis of eosinophilic pustular folliculitis. J Allergy Clin Immunol. 2012;129:536-543. - Satoh
T, Shimura C, Miyagishi C, et al. Indomethacin-induced reduction in CRTH2 in eosinophilic pustular folliculitis (Ofuji’s disease): a proposed mechanism of action. Acta Derm Venereol. 2010;90:18-22. - Hagiwara A, Fujimura T, Furudate S, et al. Induction of CD163(+)M2 macrophages in the lesional skin of eosinophilic pustular folliculitis. Acta Derm Venereol. 2014;94:104-106.
- Ellis
E, Scheinfeld N. Eosinophilic pustular folliculitis: a comprehensive review of treatment options. Am J Clin Dermatol. 2004;5:189-197. - Bull R
H, Harland CA, Fallowfield ME, et al. Eosinophilic folliculitis: a self-limiting illness in patients being treated for haematological malignancy. Br J Dermatol. 1993;129:178-182. - Farber M, Forgia S, Sahu J, et al. Eosinophilic dermatosis of hematologic malignancy. J Cutan Pathol. 2012;39:690-695.
Eosinophilic pustular folliculitis (EPF) was originally described in 1965 and has since evolved into 3 distinct subtypes: classic, immunosuppressed (IS), and infantile types. Immunosuppressed EPF can be further subdivided into human immunodeficiency virus (HIV) associated (IS-HIV) and non-HIV associated. Human immunodeficiency virus–seronegative cases have been associated with underlying malignancies (IS-heme) or chronic immunosuppression, such as that seen in transplant patients.
Case Report
A 52-year-old man with a medical history limited to prostate adenocarcinoma treated with a robotic prostatectomy presented with a pruritic red rash on the face, neck, shoulders, and chest of 1 month’s duration. The patient previously completed a course of azithromycin 250 mg, intramuscular triamcinolone, and oral prednisone with only minor improvement. Physical examination demonstrated multiple pink folliculocentric papules and pustules scattered on the head (Figure 1A), neck, and chest (Figure 1B), as well as edematous pink papules and plaques on the forehead (Figures 1C and 1D). The palms, soles, and oral mucosa were clear.
Initial biopsy of the right side of the chest was nonspecific and most consistent with a reaction to an arthropod bite. The patient was started on oral doxycycline 100 mg twice daily for 2 weeks. With no improvement seen, additional biopsies were obtained from the left side of the chest and forehead. The biopsy of the chest showed ruptured folliculitis with evidence of acute and chronic inflammation. The biopsy of the forehead demonstrated eosinophilic follicular spongiosis with intrafollicular Langerhans cell microgranulomas along with abundant eosinophils adjacent to follicles, consistent with EPF (Figure 2). Serum HIV testing was negative. Serum white blood cell count was normal at 6400/µL (reference range, 4500–11,000/µL) with mild elevation of eosinophils (8%). The remaining complete blood cell count and comprehensive metabolic panel were within reference range. The patient was subsequently started on oral indomethacin 25 mg twice daily and triamcinolone cream 0.1%. Within a few days he experienced initial improvement in his symptoms of pruritus and diminution in the number of inflammatory follicular papules.
Approximately 1 month after presentation, he began to experience symptoms of dysphagia and fatigue. In addition, tonsillar hypertrophy and palpable neck and axillary lymphadenopathy were present. Computed tomography of the neck, chest, and abdomen showed diffuse lymphadenopathy. Full-body positron emission tomography–computed tomography demonstrated extensive metabolically active lymphoma in multiple nodal groups above and below the diaphragm. There also was lymphomatous involvement of the spleen. An axillary lymph node biopsy was diagnostic for mantle cell lymphoma (CD4:CD8, 1:1; CD45 negative; CD20 positive; CD5 positive). He
Comment
Subtypes of EPF
Eosinophilic pustular folliculitis was first described in a Japanese female presenting with folliculocentric pustules distributed on the face, torso, and arms.1 This noninfectious eosinophilic infiltration of hair follicles predominantly seen in the Japanese population is now regarded as the classic form. Three distinct subtypes of EPF now exist, including the originally described classic variant (Ofuji disease), an IS variant, and a rare infantile form.1
All 3 subtypes of EPF are more commonly seen in men than women. The classic form has a peak incidence between the third and fourth decades of life. It presents as chronic annular papules and sterile pustules exhibiting peripheral extension, with individual lesions lasting for approximately 7 to 10 days with frequent relapses. The face is the most common area of involvement, followed by the trunk, extremities, and more rarely the palmoplantar surfaces. Concomitant leukocytosis with eosinophilia is seen in up to 35% of patients.1 The infantile type represents the rarest EPF form. The average age of onset is 5 months, with most cases resolving by 14 months of age.1
Clinically, EPF is characterized by recurrent papules and pustules predominantly on the scalp without annular or polycyclic ring formation, as seen in the classic type. The palms and soles may be involved, which can clinically mimic infantile acropustulosis and scabies infection. Most patients exhibit a concomitant peripheral eosinophilia.1,2
In the late 1980s, the IS variant of EPF was recognized in HIV-positive (IS-HIV) and HIV-negative malignancy-associated (IS-heme) populations.1,3 This newly characterized form differs morphologically and biologically from the classic and infantile subtypes. The IS subtype has a unique presentation including intensely pruritic, discrete, erythematous, follicular papules with palmoplantar sparing and infrequent annular or circinate plaque forms.1 Frequently, with the IS-HIV form, CD4+ T-cell counts are below 300 cells/mL, and 25% to 50% of patients have lymphopenia with eosinophilia.3 Highly active antiretroviral therapy has been associated with EPF resolution in HIV-positive individuals; however, it also has been shown to induce transient EPF during the first 3 to 6 months of initiation.1,3,4
Unlike the IS-HIV form, the IS-heme form has occurred solely in males and is predominantly associated with hematologic malignancies (eg, non-Hodgkin lymphoma, acute lymphoblastic leukemia, acute myeloid leukemia, myelodysplastic syndrome) 30 to 90 days following bone marrow transplant, peripheral blood stem cell transplant, or chemotherapy treatment.5,6 Unlike the chronic and persistent IS-HIV form, prior cases of IS-heme EPF have been predominantly self-limited. Interestingly, only 2 reported cases of EPF have occurred prior to the diagnosis of malignancy including B-cell leukemia and myelodysplastic syndrome.5
Histopathology
All 3 identified forms of EPF histopathologically show acute and chronic lymphoeosinophilic infiltrate concentrated at the follicular isthmus, which can lead to follicular destruction. Scattered mononuclear cells, eosinophils, and neutrophils are found within the pilar outer root sheath, sebaceous glands, and ducts. Approximately 40% of cases demonstrate follicular mucinosis.1 Histopathology of lesional palmar skin in classic-type EPF demonstrates intraepidermal pustule formation with abundant eosinophils and neutrophils adjacent to the acrosyringium.7,8
Pathogenesis
Although the pathophysiology of EPF is largely unknown, it is thought to represent a helper T cell (TH2) response involving IL-4, IL-5, and IL-13 cytokines.9 Chemoattractant receptor homologous molecule 2, which is expressed on eosinophils and lymphocytes, is believed to play a role in the pruritus, edema, and inflammatory response seen adjacent to pilosebaceous units in EPF.10 Moreover, immunohistochemical and flow cytometry analysis has revealed a prevalence of prostaglandin D2 within the perisebocyte infiltrate in EPF.9 Prostaglandin D2 induces eotaxin-3 production within sebocytes via peroxisome proliferator-activated receptor γ, which enhances chemoattraction of eosinophils. This pathogenesis represents a prostaglandin-based mechanism and potentially explains the efficacy of indomethacin treatment of EPF through its cyclooxygenase inhibition and reduction of chemoattractant receptor homologous molecule 2 expression.9-11
Treatment
Multiple therapeutic modalities have been reported for the treatment of EPF. For all 3 subtypes, moderate- to high-potency topical corticosteroids are considered first-line therapy. UVB phototherapy 2 to 3 times weekly remains the gold standard, given its consistent efficacy.1,12 Indomethacin (50–75 mg daily) remains first-line treatment of classic EPF.4,12 Previously reported cases of classic EPF and IS-EPF have responded well to oral prednisone (1 mg/kg daily).12,13 In a retrospective review of EPF treatment data, the following treatments also have been reported to be successful: psoralen plus UVA, oral cetirizine (20–40 mg daily, particularly for IS-EPF cases), metronidazole (250 mg 3 times daily), minocycline (150 mg daily), itraconazole (200–400 mg daily, dapsone (50–200 mg daily), systemic retinoids, tacrolimus ointment 0.1%, and permethrin cream.4,12
Malignancy
Although the entity of IS-heme EPF is rare, the morphology and treatment are unique and can potentially unmask an underlying hematologic malignancy. In patients with EPF and associated malignancy, such as our patient, a differential diagnosis to consider is eosinophilic dermatosis of hematologic malignancy (EDHM). Eosinophilic dermatosis of hematologic malignancy is most commonly associated with chronic lymphocytic leukemia and can be differentiated from EPF clinically, histopathologically, and by treatment response. Eosinophilic dermatosis of hematologic malignancy clinically presents with nonspecific papules, pustules, and/or vesicles on the head, trunk, and extremities. On histopathology, EDHM shows a superficial and deep perivascular and interstitial lymphoeosinophilic infiltration. Furthermore, EDHM patients typically exhibit a poor treatment response to oral indomethacin.14
Conclusion
Eosinophilic pustular folliculitis is a noninfectious folliculocentric process comprised of 3 distinct types. The histopathology shows follicular spongiosis with increased eosinophils. The pathogenesis is most likely related to a multifactorial immune system dysregulation involving TH2 T cells, prostaglandin D2, and eotaxin-3. The treatment of EPF may involve topical corticosteroids, UVB phototherapy, or most notably oral indomethacin. In patients with EPF and malignancy, EDHM is a differential diagnosis to consider. Our case serves as a reminder that rare eosinophilic dermatoses may represent manifestations of underlying hematopoietic malignancy and, when investigated early, can lead to appropriate life-saving treatment.
Eosinophilic pustular folliculitis (EPF) was originally described in 1965 and has since evolved into 3 distinct subtypes: classic, immunosuppressed (IS), and infantile types. Immunosuppressed EPF can be further subdivided into human immunodeficiency virus (HIV) associated (IS-HIV) and non-HIV associated. Human immunodeficiency virus–seronegative cases have been associated with underlying malignancies (IS-heme) or chronic immunosuppression, such as that seen in transplant patients.
Case Report
A 52-year-old man with a medical history limited to prostate adenocarcinoma treated with a robotic prostatectomy presented with a pruritic red rash on the face, neck, shoulders, and chest of 1 month’s duration. The patient previously completed a course of azithromycin 250 mg, intramuscular triamcinolone, and oral prednisone with only minor improvement. Physical examination demonstrated multiple pink folliculocentric papules and pustules scattered on the head (Figure 1A), neck, and chest (Figure 1B), as well as edematous pink papules and plaques on the forehead (Figures 1C and 1D). The palms, soles, and oral mucosa were clear.
Initial biopsy of the right side of the chest was nonspecific and most consistent with a reaction to an arthropod bite. The patient was started on oral doxycycline 100 mg twice daily for 2 weeks. With no improvement seen, additional biopsies were obtained from the left side of the chest and forehead. The biopsy of the chest showed ruptured folliculitis with evidence of acute and chronic inflammation. The biopsy of the forehead demonstrated eosinophilic follicular spongiosis with intrafollicular Langerhans cell microgranulomas along with abundant eosinophils adjacent to follicles, consistent with EPF (Figure 2). Serum HIV testing was negative. Serum white blood cell count was normal at 6400/µL (reference range, 4500–11,000/µL) with mild elevation of eosinophils (8%). The remaining complete blood cell count and comprehensive metabolic panel were within reference range. The patient was subsequently started on oral indomethacin 25 mg twice daily and triamcinolone cream 0.1%. Within a few days he experienced initial improvement in his symptoms of pruritus and diminution in the number of inflammatory follicular papules.
Approximately 1 month after presentation, he began to experience symptoms of dysphagia and fatigue. In addition, tonsillar hypertrophy and palpable neck and axillary lymphadenopathy were present. Computed tomography of the neck, chest, and abdomen showed diffuse lymphadenopathy. Full-body positron emission tomography–computed tomography demonstrated extensive metabolically active lymphoma in multiple nodal groups above and below the diaphragm. There also was lymphomatous involvement of the spleen. An axillary lymph node biopsy was diagnostic for mantle cell lymphoma (CD4:CD8, 1:1; CD45 negative; CD20 positive; CD5 positive). He
Comment
Subtypes of EPF
Eosinophilic pustular folliculitis was first described in a Japanese female presenting with folliculocentric pustules distributed on the face, torso, and arms.1 This noninfectious eosinophilic infiltration of hair follicles predominantly seen in the Japanese population is now regarded as the classic form. Three distinct subtypes of EPF now exist, including the originally described classic variant (Ofuji disease), an IS variant, and a rare infantile form.1
All 3 subtypes of EPF are more commonly seen in men than women. The classic form has a peak incidence between the third and fourth decades of life. It presents as chronic annular papules and sterile pustules exhibiting peripheral extension, with individual lesions lasting for approximately 7 to 10 days with frequent relapses. The face is the most common area of involvement, followed by the trunk, extremities, and more rarely the palmoplantar surfaces. Concomitant leukocytosis with eosinophilia is seen in up to 35% of patients.1 The infantile type represents the rarest EPF form. The average age of onset is 5 months, with most cases resolving by 14 months of age.1
Clinically, EPF is characterized by recurrent papules and pustules predominantly on the scalp without annular or polycyclic ring formation, as seen in the classic type. The palms and soles may be involved, which can clinically mimic infantile acropustulosis and scabies infection. Most patients exhibit a concomitant peripheral eosinophilia.1,2
In the late 1980s, the IS variant of EPF was recognized in HIV-positive (IS-HIV) and HIV-negative malignancy-associated (IS-heme) populations.1,3 This newly characterized form differs morphologically and biologically from the classic and infantile subtypes. The IS subtype has a unique presentation including intensely pruritic, discrete, erythematous, follicular papules with palmoplantar sparing and infrequent annular or circinate plaque forms.1 Frequently, with the IS-HIV form, CD4+ T-cell counts are below 300 cells/mL, and 25% to 50% of patients have lymphopenia with eosinophilia.3 Highly active antiretroviral therapy has been associated with EPF resolution in HIV-positive individuals; however, it also has been shown to induce transient EPF during the first 3 to 6 months of initiation.1,3,4
Unlike the IS-HIV form, the IS-heme form has occurred solely in males and is predominantly associated with hematologic malignancies (eg, non-Hodgkin lymphoma, acute lymphoblastic leukemia, acute myeloid leukemia, myelodysplastic syndrome) 30 to 90 days following bone marrow transplant, peripheral blood stem cell transplant, or chemotherapy treatment.5,6 Unlike the chronic and persistent IS-HIV form, prior cases of IS-heme EPF have been predominantly self-limited. Interestingly, only 2 reported cases of EPF have occurred prior to the diagnosis of malignancy including B-cell leukemia and myelodysplastic syndrome.5
Histopathology
All 3 identified forms of EPF histopathologically show acute and chronic lymphoeosinophilic infiltrate concentrated at the follicular isthmus, which can lead to follicular destruction. Scattered mononuclear cells, eosinophils, and neutrophils are found within the pilar outer root sheath, sebaceous glands, and ducts. Approximately 40% of cases demonstrate follicular mucinosis.1 Histopathology of lesional palmar skin in classic-type EPF demonstrates intraepidermal pustule formation with abundant eosinophils and neutrophils adjacent to the acrosyringium.7,8
Pathogenesis
Although the pathophysiology of EPF is largely unknown, it is thought to represent a helper T cell (TH2) response involving IL-4, IL-5, and IL-13 cytokines.9 Chemoattractant receptor homologous molecule 2, which is expressed on eosinophils and lymphocytes, is believed to play a role in the pruritus, edema, and inflammatory response seen adjacent to pilosebaceous units in EPF.10 Moreover, immunohistochemical and flow cytometry analysis has revealed a prevalence of prostaglandin D2 within the perisebocyte infiltrate in EPF.9 Prostaglandin D2 induces eotaxin-3 production within sebocytes via peroxisome proliferator-activated receptor γ, which enhances chemoattraction of eosinophils. This pathogenesis represents a prostaglandin-based mechanism and potentially explains the efficacy of indomethacin treatment of EPF through its cyclooxygenase inhibition and reduction of chemoattractant receptor homologous molecule 2 expression.9-11
Treatment
Multiple therapeutic modalities have been reported for the treatment of EPF. For all 3 subtypes, moderate- to high-potency topical corticosteroids are considered first-line therapy. UVB phototherapy 2 to 3 times weekly remains the gold standard, given its consistent efficacy.1,12 Indomethacin (50–75 mg daily) remains first-line treatment of classic EPF.4,12 Previously reported cases of classic EPF and IS-EPF have responded well to oral prednisone (1 mg/kg daily).12,13 In a retrospective review of EPF treatment data, the following treatments also have been reported to be successful: psoralen plus UVA, oral cetirizine (20–40 mg daily, particularly for IS-EPF cases), metronidazole (250 mg 3 times daily), minocycline (150 mg daily), itraconazole (200–400 mg daily, dapsone (50–200 mg daily), systemic retinoids, tacrolimus ointment 0.1%, and permethrin cream.4,12
Malignancy
Although the entity of IS-heme EPF is rare, the morphology and treatment are unique and can potentially unmask an underlying hematologic malignancy. In patients with EPF and associated malignancy, such as our patient, a differential diagnosis to consider is eosinophilic dermatosis of hematologic malignancy (EDHM). Eosinophilic dermatosis of hematologic malignancy is most commonly associated with chronic lymphocytic leukemia and can be differentiated from EPF clinically, histopathologically, and by treatment response. Eosinophilic dermatosis of hematologic malignancy clinically presents with nonspecific papules, pustules, and/or vesicles on the head, trunk, and extremities. On histopathology, EDHM shows a superficial and deep perivascular and interstitial lymphoeosinophilic infiltration. Furthermore, EDHM patients typically exhibit a poor treatment response to oral indomethacin.14
Conclusion
Eosinophilic pustular folliculitis is a noninfectious folliculocentric process comprised of 3 distinct types. The histopathology shows follicular spongiosis with increased eosinophils. The pathogenesis is most likely related to a multifactorial immune system dysregulation involving TH2 T cells, prostaglandin D2, and eotaxin-3. The treatment of EPF may involve topical corticosteroids, UVB phototherapy, or most notably oral indomethacin. In patients with EPF and malignancy, EDHM is a differential diagnosis to consider. Our case serves as a reminder that rare eosinophilic dermatoses may represent manifestations of underlying hematopoietic malignancy and, when investigated early, can lead to appropriate life-saving treatment.
- Nervi J, Stephen. Eosinophilic pustular folliculitis: a 40 year retrospect. J Am Acad Dermatol. 2006;55:285-289.
- Hernández-Martín Á, Nuño-González A, Colmenero I, et al. Eosinophilic pustular folliculitis of infancy: a series of 15 cases and review of the literature [published online July 21, 2012]. J Am Acad Dermatol. 2013;68:150-155.
- Soepr
ono F, Schinella R. Eosinophilic pustular folliculitis in patients with acquired immunodeficiency syndrome. report of three cases. J Am Acad Dermatol. 1986;14:1020-1022. - Katoh
M, Nomura T, Miyachi Y, et al. Eosinophilic pustular folliculitis: a review of the Japanese published works. J Dermatol. 2013;40:15-20. - Keida
T, Hayashi N, Kawashima M. Eosinophilic pustular folliculitis following autologous peripheral blood stem-cell transplant. J Dermatol. 2004;31:21-26. - Goiriz R, Gul-Millán G, Peñas PF, et al. Eosinophilic folliculitis following allogeneic peripheral blood stem cell transplantation: case report and review. J Cutan Pathol. 2007;34(suppl 1):33-36.
- Satoh T, Ikeda H, Yokozeki H. Acrosyringeal involvement of palmoplantar lesions of eosinophilic pustular folliculitis. Acta Derm Venereol. 2013;93:99.
- Tsuboi H, Wakita K, Fujimura T, et al. Acral variant of eosinophilic pustular folliculitis (Ofuji’s disease). Clin Exp Dermatol. 2003;28:321-324.
- Nakahig
ashi K, Doi H, Otsuka A, et al. PGD2 induces eotaxin-3 via PPARgamma from sebocytes: a possible pathogenesis of eosinophilic pustular folliculitis. J Allergy Clin Immunol. 2012;129:536-543. - Satoh
T, Shimura C, Miyagishi C, et al. Indomethacin-induced reduction in CRTH2 in eosinophilic pustular folliculitis (Ofuji’s disease): a proposed mechanism of action. Acta Derm Venereol. 2010;90:18-22. - Hagiwara A, Fujimura T, Furudate S, et al. Induction of CD163(+)M2 macrophages in the lesional skin of eosinophilic pustular folliculitis. Acta Derm Venereol. 2014;94:104-106.
- Ellis
E, Scheinfeld N. Eosinophilic pustular folliculitis: a comprehensive review of treatment options. Am J Clin Dermatol. 2004;5:189-197. - Bull R
H, Harland CA, Fallowfield ME, et al. Eosinophilic folliculitis: a self-limiting illness in patients being treated for haematological malignancy. Br J Dermatol. 1993;129:178-182. - Farber M, Forgia S, Sahu J, et al. Eosinophilic dermatosis of hematologic malignancy. J Cutan Pathol. 2012;39:690-695.
- Nervi J, Stephen. Eosinophilic pustular folliculitis: a 40 year retrospect. J Am Acad Dermatol. 2006;55:285-289.
- Hernández-Martín Á, Nuño-González A, Colmenero I, et al. Eosinophilic pustular folliculitis of infancy: a series of 15 cases and review of the literature [published online July 21, 2012]. J Am Acad Dermatol. 2013;68:150-155.
- Soepr
ono F, Schinella R. Eosinophilic pustular folliculitis in patients with acquired immunodeficiency syndrome. report of three cases. J Am Acad Dermatol. 1986;14:1020-1022. - Katoh
M, Nomura T, Miyachi Y, et al. Eosinophilic pustular folliculitis: a review of the Japanese published works. J Dermatol. 2013;40:15-20. - Keida
T, Hayashi N, Kawashima M. Eosinophilic pustular folliculitis following autologous peripheral blood stem-cell transplant. J Dermatol. 2004;31:21-26. - Goiriz R, Gul-Millán G, Peñas PF, et al. Eosinophilic folliculitis following allogeneic peripheral blood stem cell transplantation: case report and review. J Cutan Pathol. 2007;34(suppl 1):33-36.
- Satoh T, Ikeda H, Yokozeki H. Acrosyringeal involvement of palmoplantar lesions of eosinophilic pustular folliculitis. Acta Derm Venereol. 2013;93:99.
- Tsuboi H, Wakita K, Fujimura T, et al. Acral variant of eosinophilic pustular folliculitis (Ofuji’s disease). Clin Exp Dermatol. 2003;28:321-324.
- Nakahig
ashi K, Doi H, Otsuka A, et al. PGD2 induces eotaxin-3 via PPARgamma from sebocytes: a possible pathogenesis of eosinophilic pustular folliculitis. J Allergy Clin Immunol. 2012;129:536-543. - Satoh
T, Shimura C, Miyagishi C, et al. Indomethacin-induced reduction in CRTH2 in eosinophilic pustular folliculitis (Ofuji’s disease): a proposed mechanism of action. Acta Derm Venereol. 2010;90:18-22. - Hagiwara A, Fujimura T, Furudate S, et al. Induction of CD163(+)M2 macrophages in the lesional skin of eosinophilic pustular folliculitis. Acta Derm Venereol. 2014;94:104-106.
- Ellis
E, Scheinfeld N. Eosinophilic pustular folliculitis: a comprehensive review of treatment options. Am J Clin Dermatol. 2004;5:189-197. - Bull R
H, Harland CA, Fallowfield ME, et al. Eosinophilic folliculitis: a self-limiting illness in patients being treated for haematological malignancy. Br J Dermatol. 1993;129:178-182. - Farber M, Forgia S, Sahu J, et al. Eosinophilic dermatosis of hematologic malignancy. J Cutan Pathol. 2012;39:690-695.
Practice Points
- Recalcitrant folliculocentric papules and pustules involving the head, trunk, arms, and legs should raise suspicion of possible eosinophilic pustular folliculitis (EPF).
- Underlying hematopoietic malignancy may be associated with cases of EPF.
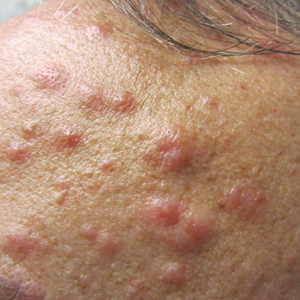
Acrodermatitis Enteropathica From Zinc-Deficient Total Parenteral Nutrition
Case Report
A 54-year-old woman presented with a pruritic and slightly painful skin eruption that began perinasally and progressed over 1 week to involve the labial commissures, finger webs, dorsal surfaces of the feet, heels, and bilateral gluteal folds. In addition, the eruption involved the left thigh at the donor site of a prior skin graft. She received no relief after an intramuscular steroid injection and hydrocortisone cream 1% prescribed by a primary care physician who diagnosed the rash as poison ivy contact dermatitis despite no exposure to plants. Review of systems was negative and she denied any new medication use. Her medical history was notable for extensive mesenteric injury secondary to a motor vehicle accident. She subsequently had multiple enterocutaneous fistulas that resulted in a complete small bowel enterectomy 10 months prior to presentation, which caused her to become dependent on total parenteral nutrition (TPN).
Physical examination revealed sharply demarcated, erythematous, scaly plaques perinasally, periorally, and on the bilateral gluteal folds (Figure 1). There were sharply demarcated, erythematous, scaly plaques on the right and left finger webs, dorsal surface of the right foot, and left upper thigh. Hemorrhagic bullae were appreciated on the left finger webs. Large flaccid bullae were present on the bilateral heels and dorsum of the right foot (Figure 2).

 and dorsum of the right foot (B).")
Suspecting a diagnosis of acrodermatitis enteropathica (AE), laboratory testing included a serum zinc level, which was 42 µg/dL (reference range, 70–130 µg/dL). The copper and selenium levels also were low with values of 71 µg/dL (reference range, 80–155 µg/dL) and 31 µg/dL (reference range, 79–326 µg/dL), respectively. No additional vitamin or mineral deficiencies were discovered. A complete blood cell count and comprehensive metabolic panel were performed and showed no abnormalities other than a mildly elevated sodium level of 147 mEq/L (reference range, 136–142 mEq/L).
A punch biopsy was performed. Histopathology revealed subcorneal neutrophils and neutrophilic crust, mild spongiosis, and a dense upper dermal mixed neutrophilic and lymphohistiocytic infiltrate. The specimen also exhibited mild intercellular edema and prominent capillaries (Figure 3).

After further investigation, the company providing the patient’s TPN confirmed that zinc had been removed several weeks prior to the onset of symptoms due to a critical national shortage of trace element additives. Zinc was supplemented at 15 mg daily to the TPN solution. Three days later a skin examination revealed dramatic changes with notable improvement of the finger web plaques and complete resolution of the facial lesions. The plaques and bullae on the lower extremities also had resolved (Figure 4).

Comment
Background
Acrodermatitis enteropathica is a rare autosomal-recessive disorder of zinc metabolism characterized by skin lesions predominantly distributed in acral and periorificial sites as well as alopecia and diarrhea. Acrodermatitis enteropathica was first described by Brandt1 in 1936 and later characterized by Danbolt and Closs2 in 1942 as a unique and often fatal disease of unknown etiology. More than 30 years later, the link between zinc deficiency and AE was illustrated by Moynahan3 who demonstrated clinical improvement with zinc supplementation. It was not until 2002 that the molecular pathogenesis of hypozincemia in patients with inherited AE was described. Küry et al4 identified a mutation in the SLC39A4 gene responsible for encoding the Zip4 protein, a zinc transporter found on enterocytes, particularly in the proximal small intestine.5,6 Classically, patients with inherited AE are children who present within days of birth or days to weeks after being weaned from breast milk to cow’s milk. The zinc in bovine milk is less bioavailable than breast milk, though both have similar total zinc concentrations, which results in the decreased plasma zinc levels seen in children with inherited AE.5-8 Occasionally, children present before weaning due to decreased maternal mammary zinc secretion (lactogenic AE).9,10
Clinical Presentation
Similar clinical findings are seen in patients with noninherited forms of zinc deficiency known as acquired AE. Acquired zinc deficiency may be broadly categorized as being from inadequate intake, deficient absorption, excess demand, or overexcretion.8 Such disturbances of zinc balance are most frequently seen in patients with restrictive diets, anorexia nervosa, intestinal bypass procedures, Crohn disease, pancreatic insufficiency, alcoholism, human immunodeficiency virus, and extensive cutaneous burns. Premature infants, mothers who are breastfeeding, and those dependent on TPN are at risk for developing acquired zinc deficiency.7-9,11
Differentiating Characteristics
Both acquired and inherited AE present as erythematous or pink eczematous scaly plaques with the variable presence of vesicular or bullous lesions involving periorificial, acral, and anogenital regions. Early manifestations of AE may include angular cheilitis and paronychia. Alopecia and diarrhea are characteristics of later disease. In fact, the complete triad of dermatitis, alopecia, and diarrhea is seen in only 20% of cases.7Without treatment, patients may develop blepharitis, conjunctivitis, photophobia, irritability, anorexia, apathy, growth retardation, hypogonadism, hypogeusia, and mental slowing. Skin lesions frequently become secondarily infected with Candida albicans and/or bacteria.5,7,11
Histopathology
Histopathologic examination of skin biopsy specimens from AE lesions demonstrates nonspecific findings similar to other deficiency dermatoses, such as pellagra and glucagonoma-associated necrolytic migratory erythema. Histology typically reveals cytoplasmic pallor with vacuolization and ballooning degeneration of keratinocytes, followed by confluent keratinocyte necrosis within the stratum granulosum and stratum spinosum of the epidermis.5 Confluent parakeratosis with hypogranulosis variably associated with neutrophil crust also is seen. Scattered dyskeratotic keratinocytes may be found within all levels of the epidermis. In resolving or chronic AE lesions, psoriasiform hyperplasia is prevalent, though necrolysis may be minimal or absent.5,11
Diagnosis
Evaluation includes measurement of plasma zinc levels. Zinc levels less than 50 µg/dL are suggestive but not diagnostic of AE.5 Although plasma zinc measurement is the most useful indicator of zinc status, its utility in assessing the true total body store of zinc is limited. Plasma zinc is tightly regulated and only represents 0.1% of body stores.5,6 Additionally, zinc levels may decrease in proinflammatory states.12 Beyond zinc measurement, evaluation of alkaline phosphatase, a zinc-dependent enzyme, can provide useful diagnostic information.5,6
Zinc and TPN
Patients on TPN are at a unique risk for developing zinc and other nutritional deficiencies. Because the daily recommended dietary allowance for zinc is low (8 mg daily for adult women and 11 mg daily for adult men)5 and the element is found in a wide variety of foods, maintaining adequate zinc levels is easily achieved in healthy individuals with normal diets. Kay et al13 described 4 patients on parenteral nutrition who developed hypozincemia and an AE-like syndrome within weeks of TPN induction. The authors described rapid and drastic clinical improvement after initiating zinc supplementation, accentuating the importance of including zinc as a component of TPN.13,14 Brazin et al15 also reported a case of an AE-like syndrome from zinc-deficient hyperalimentation in a patient receiving TPN for short bowel syndrome. Chun et al16 described another case of acquired AE in a patient on TPN for acute pancreatitis. Both cases demonstrated prompt improvement of skin lesions after treatment with zinc supplementation. Other nutrient deficiencies may reveal themselves through similar dermatologic manifestations. For example, cases of scaly dermatitis secondary to the development of essential fatty acid deficiency from TPN formulations lacking adequate quantities of linoleic acid have been reported.Similar to our case, the resolution of skin lesions was seen after TPN was supplemented with the deficient nutrient.17 These cases exemplify the importance in considering deficiency dermatoses in the TPN-dependent patient population.
Conclusion
In our case, the development of skin lesions directly coincided with a recent removal of zinc from the patient’s TPN, which provided us with a unique opportunity to observe the causal relationship between decreased zinc intake and the development of clinical signs of acquired AE. This association was further elucidated by laboratory confirmation of low serum zinc levels and rapid improvement in all skin lesions after zinc supplementation was initiated.
- Brandt T. Dermatitis in children with disturbances of general condition and absorption of food. Acta Derm Venereol. 1936;17:513-537.
- Danbolt N, Closs K. Acrodermatitis enteropathica. Acta Derm Venereol. 1942;23:127-169.
- Moynahan E. Acrodermatitis enteropathica: a lethal inherited human zinc deficiency disorder. Lancet. 1974;2:299-400.
- Küry S, Dréno B, Bézieau S, et al. Identification of SLC39A4, a gene involved in acrodermatitis enteropathica. Nat Genet. 2002;31:238-240.
- Maverakis E, Fung MA, Lynch PJ, et al. Acrodermatitis enteropathica and an overview of zinc metabolism. J Am Acad Dermatol. 2007;56:116-124.
- Thrash B, Patel M, Shah KR, et al. Cutaneous manifestations of gastrointestinal disease: part II. J Am Acad Dermatol. 2013;68:211.e1-211.e33; quiz 244-246.
- Perafán-Riveros C, França LF, Alves AC, et al. Acrodermatitis enteropathica: case report and review of the literature. Pediatr Dermatol. 2002;19:426-431.
- Kumar P, Ranjan NR, Mondal AK. Zinc and skin: a brief summary. Dermatol Online J. 2012;18:1.
- Saritha M, Gupta D, Chandrashekar L, et al. Acquired zinc deficiency in an adult female. Indian J Dermatol. 2012;57:492-494.
- Neldner K, Hambidge K, Walravens P. Acrodermatitis enteropathica.Int J Dermatol. 1978;17:380-387.
- Gehrig K, Dinulos J. Acrodermatitis due to nutritional deficiency. Curr Opin Pediatr. 2010;22:107-112.
- Liuzzi JP, Lichten LA, Rivera S, et al. Interleukin-6 regulates the zinc transporter Zip14 in liver and contributes to hypozincemia of the acute-phase response. Proct Natl Acad Sci U S A. 2005;102:6843-6848.
- Kay RG, Tasman-Jones C, Pybus J, et al. A syndrome of acute zinc deficiency during total parenteral nutrition in man. Ann Surg. 1976;183:331-340.
- Jeejeebhoy K. Zinc: an essential trace element for parenteral nutrition. Gastroenterology. 2009;137(5 suppl):S7-S12.
- Brazin SA, Johnson WT, Abramson LJ. The acrodermatitis enteropathica-like syndrome. Arch Dermatol. 1979;115:597-599.
- Chun JH, Baek JH, Chung NG. Development of bullous acrodermatitis enteropathica during the course of chemotherapy for acute lymphocytic leukemia. Ann Dermatol. 2011;23(suppl 3):S326-S328.
- Roongpisuthipong W, Phanachet P, Roongpisuthipong C, et al. Essential fatty acid deficiency while a patient receiving fat regimen total parenteral nutrition [published June 14, 2012]. BMJ Case Rep. doi:10.1136/bcr.07.2011.4475.
Case Report
A 54-year-old woman presented with a pruritic and slightly painful skin eruption that began perinasally and progressed over 1 week to involve the labial commissures, finger webs, dorsal surfaces of the feet, heels, and bilateral gluteal folds. In addition, the eruption involved the left thigh at the donor site of a prior skin graft. She received no relief after an intramuscular steroid injection and hydrocortisone cream 1% prescribed by a primary care physician who diagnosed the rash as poison ivy contact dermatitis despite no exposure to plants. Review of systems was negative and she denied any new medication use. Her medical history was notable for extensive mesenteric injury secondary to a motor vehicle accident. She subsequently had multiple enterocutaneous fistulas that resulted in a complete small bowel enterectomy 10 months prior to presentation, which caused her to become dependent on total parenteral nutrition (TPN).
Physical examination revealed sharply demarcated, erythematous, scaly plaques perinasally, periorally, and on the bilateral gluteal folds (Figure 1). There were sharply demarcated, erythematous, scaly plaques on the right and left finger webs, dorsal surface of the right foot, and left upper thigh. Hemorrhagic bullae were appreciated on the left finger webs. Large flaccid bullae were present on the bilateral heels and dorsum of the right foot (Figure 2).
Suspecting a diagnosis of acrodermatitis enteropathica (AE), laboratory testing included a serum zinc level, which was 42 µg/dL (reference range, 70–130 µg/dL). The copper and selenium levels also were low with values of 71 µg/dL (reference range, 80–155 µg/dL) and 31 µg/dL (reference range, 79–326 µg/dL), respectively. No additional vitamin or mineral deficiencies were discovered. A complete blood cell count and comprehensive metabolic panel were performed and showed no abnormalities other than a mildly elevated sodium level of 147 mEq/L (reference range, 136–142 mEq/L).
A punch biopsy was performed. Histopathology revealed subcorneal neutrophils and neutrophilic crust, mild spongiosis, and a dense upper dermal mixed neutrophilic and lymphohistiocytic infiltrate. The specimen also exhibited mild intercellular edema and prominent capillaries (Figure 3).
After further investigation, the company providing the patient’s TPN confirmed that zinc had been removed several weeks prior to the onset of symptoms due to a critical national shortage of trace element additives. Zinc was supplemented at 15 mg daily to the TPN solution. Three days later a skin examination revealed dramatic changes with notable improvement of the finger web plaques and complete resolution of the facial lesions. The plaques and bullae on the lower extremities also had resolved (Figure 4).
Comment
Background
Acrodermatitis enteropathica is a rare autosomal-recessive disorder of zinc metabolism characterized by skin lesions predominantly distributed in acral and periorificial sites as well as alopecia and diarrhea. Acrodermatitis enteropathica was first described by Brandt1 in 1936 and later characterized by Danbolt and Closs2 in 1942 as a unique and often fatal disease of unknown etiology. More than 30 years later, the link between zinc deficiency and AE was illustrated by Moynahan3 who demonstrated clinical improvement with zinc supplementation. It was not until 2002 that the molecular pathogenesis of hypozincemia in patients with inherited AE was described. Küry et al4 identified a mutation in the SLC39A4 gene responsible for encoding the Zip4 protein, a zinc transporter found on enterocytes, particularly in the proximal small intestine.5,6 Classically, patients with inherited AE are children who present within days of birth or days to weeks after being weaned from breast milk to cow’s milk. The zinc in bovine milk is less bioavailable than breast milk, though both have similar total zinc concentrations, which results in the decreased plasma zinc levels seen in children with inherited AE.5-8 Occasionally, children present before weaning due to decreased maternal mammary zinc secretion (lactogenic AE).9,10
Clinical Presentation
Similar clinical findings are seen in patients with noninherited forms of zinc deficiency known as acquired AE. Acquired zinc deficiency may be broadly categorized as being from inadequate intake, deficient absorption, excess demand, or overexcretion.8 Such disturbances of zinc balance are most frequently seen in patients with restrictive diets, anorexia nervosa, intestinal bypass procedures, Crohn disease, pancreatic insufficiency, alcoholism, human immunodeficiency virus, and extensive cutaneous burns. Premature infants, mothers who are breastfeeding, and those dependent on TPN are at risk for developing acquired zinc deficiency.7-9,11
Differentiating Characteristics
Both acquired and inherited AE present as erythematous or pink eczematous scaly plaques with the variable presence of vesicular or bullous lesions involving periorificial, acral, and anogenital regions. Early manifestations of AE may include angular cheilitis and paronychia. Alopecia and diarrhea are characteristics of later disease. In fact, the complete triad of dermatitis, alopecia, and diarrhea is seen in only 20% of cases.7Without treatment, patients may develop blepharitis, conjunctivitis, photophobia, irritability, anorexia, apathy, growth retardation, hypogonadism, hypogeusia, and mental slowing. Skin lesions frequently become secondarily infected with Candida albicans and/or bacteria.5,7,11
Histopathology
Histopathologic examination of skin biopsy specimens from AE lesions demonstrates nonspecific findings similar to other deficiency dermatoses, such as pellagra and glucagonoma-associated necrolytic migratory erythema. Histology typically reveals cytoplasmic pallor with vacuolization and ballooning degeneration of keratinocytes, followed by confluent keratinocyte necrosis within the stratum granulosum and stratum spinosum of the epidermis.5 Confluent parakeratosis with hypogranulosis variably associated with neutrophil crust also is seen. Scattered dyskeratotic keratinocytes may be found within all levels of the epidermis. In resolving or chronic AE lesions, psoriasiform hyperplasia is prevalent, though necrolysis may be minimal or absent.5,11
Diagnosis
Evaluation includes measurement of plasma zinc levels. Zinc levels less than 50 µg/dL are suggestive but not diagnostic of AE.5 Although plasma zinc measurement is the most useful indicator of zinc status, its utility in assessing the true total body store of zinc is limited. Plasma zinc is tightly regulated and only represents 0.1% of body stores.5,6 Additionally, zinc levels may decrease in proinflammatory states.12 Beyond zinc measurement, evaluation of alkaline phosphatase, a zinc-dependent enzyme, can provide useful diagnostic information.5,6
Zinc and TPN
Patients on TPN are at a unique risk for developing zinc and other nutritional deficiencies. Because the daily recommended dietary allowance for zinc is low (8 mg daily for adult women and 11 mg daily for adult men)5 and the element is found in a wide variety of foods, maintaining adequate zinc levels is easily achieved in healthy individuals with normal diets. Kay et al13 described 4 patients on parenteral nutrition who developed hypozincemia and an AE-like syndrome within weeks of TPN induction. The authors described rapid and drastic clinical improvement after initiating zinc supplementation, accentuating the importance of including zinc as a component of TPN.13,14 Brazin et al15 also reported a case of an AE-like syndrome from zinc-deficient hyperalimentation in a patient receiving TPN for short bowel syndrome. Chun et al16 described another case of acquired AE in a patient on TPN for acute pancreatitis. Both cases demonstrated prompt improvement of skin lesions after treatment with zinc supplementation. Other nutrient deficiencies may reveal themselves through similar dermatologic manifestations. For example, cases of scaly dermatitis secondary to the development of essential fatty acid deficiency from TPN formulations lacking adequate quantities of linoleic acid have been reported.Similar to our case, the resolution of skin lesions was seen after TPN was supplemented with the deficient nutrient.17 These cases exemplify the importance in considering deficiency dermatoses in the TPN-dependent patient population.
Conclusion
In our case, the development of skin lesions directly coincided with a recent removal of zinc from the patient’s TPN, which provided us with a unique opportunity to observe the causal relationship between decreased zinc intake and the development of clinical signs of acquired AE. This association was further elucidated by laboratory confirmation of low serum zinc levels and rapid improvement in all skin lesions after zinc supplementation was initiated.
Case Report
A 54-year-old woman presented with a pruritic and slightly painful skin eruption that began perinasally and progressed over 1 week to involve the labial commissures, finger webs, dorsal surfaces of the feet, heels, and bilateral gluteal folds. In addition, the eruption involved the left thigh at the donor site of a prior skin graft. She received no relief after an intramuscular steroid injection and hydrocortisone cream 1% prescribed by a primary care physician who diagnosed the rash as poison ivy contact dermatitis despite no exposure to plants. Review of systems was negative and she denied any new medication use. Her medical history was notable for extensive mesenteric injury secondary to a motor vehicle accident. She subsequently had multiple enterocutaneous fistulas that resulted in a complete small bowel enterectomy 10 months prior to presentation, which caused her to become dependent on total parenteral nutrition (TPN).
Physical examination revealed sharply demarcated, erythematous, scaly plaques perinasally, periorally, and on the bilateral gluteal folds (Figure 1). There were sharply demarcated, erythematous, scaly plaques on the right and left finger webs, dorsal surface of the right foot, and left upper thigh. Hemorrhagic bullae were appreciated on the left finger webs. Large flaccid bullae were present on the bilateral heels and dorsum of the right foot (Figure 2).
Suspecting a diagnosis of acrodermatitis enteropathica (AE), laboratory testing included a serum zinc level, which was 42 µg/dL (reference range, 70–130 µg/dL). The copper and selenium levels also were low with values of 71 µg/dL (reference range, 80–155 µg/dL) and 31 µg/dL (reference range, 79–326 µg/dL), respectively. No additional vitamin or mineral deficiencies were discovered. A complete blood cell count and comprehensive metabolic panel were performed and showed no abnormalities other than a mildly elevated sodium level of 147 mEq/L (reference range, 136–142 mEq/L).
A punch biopsy was performed. Histopathology revealed subcorneal neutrophils and neutrophilic crust, mild spongiosis, and a dense upper dermal mixed neutrophilic and lymphohistiocytic infiltrate. The specimen also exhibited mild intercellular edema and prominent capillaries (Figure 3).
After further investigation, the company providing the patient’s TPN confirmed that zinc had been removed several weeks prior to the onset of symptoms due to a critical national shortage of trace element additives. Zinc was supplemented at 15 mg daily to the TPN solution. Three days later a skin examination revealed dramatic changes with notable improvement of the finger web plaques and complete resolution of the facial lesions. The plaques and bullae on the lower extremities also had resolved (Figure 4).
Comment
Background
Acrodermatitis enteropathica is a rare autosomal-recessive disorder of zinc metabolism characterized by skin lesions predominantly distributed in acral and periorificial sites as well as alopecia and diarrhea. Acrodermatitis enteropathica was first described by Brandt1 in 1936 and later characterized by Danbolt and Closs2 in 1942 as a unique and often fatal disease of unknown etiology. More than 30 years later, the link between zinc deficiency and AE was illustrated by Moynahan3 who demonstrated clinical improvement with zinc supplementation. It was not until 2002 that the molecular pathogenesis of hypozincemia in patients with inherited AE was described. Küry et al4 identified a mutation in the SLC39A4 gene responsible for encoding the Zip4 protein, a zinc transporter found on enterocytes, particularly in the proximal small intestine.5,6 Classically, patients with inherited AE are children who present within days of birth or days to weeks after being weaned from breast milk to cow’s milk. The zinc in bovine milk is less bioavailable than breast milk, though both have similar total zinc concentrations, which results in the decreased plasma zinc levels seen in children with inherited AE.5-8 Occasionally, children present before weaning due to decreased maternal mammary zinc secretion (lactogenic AE).9,10
Clinical Presentation
Similar clinical findings are seen in patients with noninherited forms of zinc deficiency known as acquired AE. Acquired zinc deficiency may be broadly categorized as being from inadequate intake, deficient absorption, excess demand, or overexcretion.8 Such disturbances of zinc balance are most frequently seen in patients with restrictive diets, anorexia nervosa, intestinal bypass procedures, Crohn disease, pancreatic insufficiency, alcoholism, human immunodeficiency virus, and extensive cutaneous burns. Premature infants, mothers who are breastfeeding, and those dependent on TPN are at risk for developing acquired zinc deficiency.7-9,11
Differentiating Characteristics
Both acquired and inherited AE present as erythematous or pink eczematous scaly plaques with the variable presence of vesicular or bullous lesions involving periorificial, acral, and anogenital regions. Early manifestations of AE may include angular cheilitis and paronychia. Alopecia and diarrhea are characteristics of later disease. In fact, the complete triad of dermatitis, alopecia, and diarrhea is seen in only 20% of cases.7Without treatment, patients may develop blepharitis, conjunctivitis, photophobia, irritability, anorexia, apathy, growth retardation, hypogonadism, hypogeusia, and mental slowing. Skin lesions frequently become secondarily infected with Candida albicans and/or bacteria.5,7,11
Histopathology
Histopathologic examination of skin biopsy specimens from AE lesions demonstrates nonspecific findings similar to other deficiency dermatoses, such as pellagra and glucagonoma-associated necrolytic migratory erythema. Histology typically reveals cytoplasmic pallor with vacuolization and ballooning degeneration of keratinocytes, followed by confluent keratinocyte necrosis within the stratum granulosum and stratum spinosum of the epidermis.5 Confluent parakeratosis with hypogranulosis variably associated with neutrophil crust also is seen. Scattered dyskeratotic keratinocytes may be found within all levels of the epidermis. In resolving or chronic AE lesions, psoriasiform hyperplasia is prevalent, though necrolysis may be minimal or absent.5,11
Diagnosis
Evaluation includes measurement of plasma zinc levels. Zinc levels less than 50 µg/dL are suggestive but not diagnostic of AE.5 Although plasma zinc measurement is the most useful indicator of zinc status, its utility in assessing the true total body store of zinc is limited. Plasma zinc is tightly regulated and only represents 0.1% of body stores.5,6 Additionally, zinc levels may decrease in proinflammatory states.12 Beyond zinc measurement, evaluation of alkaline phosphatase, a zinc-dependent enzyme, can provide useful diagnostic information.5,6
Zinc and TPN
Patients on TPN are at a unique risk for developing zinc and other nutritional deficiencies. Because the daily recommended dietary allowance for zinc is low (8 mg daily for adult women and 11 mg daily for adult men)5 and the element is found in a wide variety of foods, maintaining adequate zinc levels is easily achieved in healthy individuals with normal diets. Kay et al13 described 4 patients on parenteral nutrition who developed hypozincemia and an AE-like syndrome within weeks of TPN induction. The authors described rapid and drastic clinical improvement after initiating zinc supplementation, accentuating the importance of including zinc as a component of TPN.13,14 Brazin et al15 also reported a case of an AE-like syndrome from zinc-deficient hyperalimentation in a patient receiving TPN for short bowel syndrome. Chun et al16 described another case of acquired AE in a patient on TPN for acute pancreatitis. Both cases demonstrated prompt improvement of skin lesions after treatment with zinc supplementation. Other nutrient deficiencies may reveal themselves through similar dermatologic manifestations. For example, cases of scaly dermatitis secondary to the development of essential fatty acid deficiency from TPN formulations lacking adequate quantities of linoleic acid have been reported.Similar to our case, the resolution of skin lesions was seen after TPN was supplemented with the deficient nutrient.17 These cases exemplify the importance in considering deficiency dermatoses in the TPN-dependent patient population.
Conclusion
In our case, the development of skin lesions directly coincided with a recent removal of zinc from the patient’s TPN, which provided us with a unique opportunity to observe the causal relationship between decreased zinc intake and the development of clinical signs of acquired AE. This association was further elucidated by laboratory confirmation of low serum zinc levels and rapid improvement in all skin lesions after zinc supplementation was initiated.
- Brandt T. Dermatitis in children with disturbances of general condition and absorption of food. Acta Derm Venereol. 1936;17:513-537.
- Danbolt N, Closs K. Acrodermatitis enteropathica. Acta Derm Venereol. 1942;23:127-169.
- Moynahan E. Acrodermatitis enteropathica: a lethal inherited human zinc deficiency disorder. Lancet. 1974;2:299-400.
- Küry S, Dréno B, Bézieau S, et al. Identification of SLC39A4, a gene involved in acrodermatitis enteropathica. Nat Genet. 2002;31:238-240.
- Maverakis E, Fung MA, Lynch PJ, et al. Acrodermatitis enteropathica and an overview of zinc metabolism. J Am Acad Dermatol. 2007;56:116-124.
- Thrash B, Patel M, Shah KR, et al. Cutaneous manifestations of gastrointestinal disease: part II. J Am Acad Dermatol. 2013;68:211.e1-211.e33; quiz 244-246.
- Perafán-Riveros C, França LF, Alves AC, et al. Acrodermatitis enteropathica: case report and review of the literature. Pediatr Dermatol. 2002;19:426-431.
- Kumar P, Ranjan NR, Mondal AK. Zinc and skin: a brief summary. Dermatol Online J. 2012;18:1.
- Saritha M, Gupta D, Chandrashekar L, et al. Acquired zinc deficiency in an adult female. Indian J Dermatol. 2012;57:492-494.
- Neldner K, Hambidge K, Walravens P. Acrodermatitis enteropathica.Int J Dermatol. 1978;17:380-387.
- Gehrig K, Dinulos J. Acrodermatitis due to nutritional deficiency. Curr Opin Pediatr. 2010;22:107-112.
- Liuzzi JP, Lichten LA, Rivera S, et al. Interleukin-6 regulates the zinc transporter Zip14 in liver and contributes to hypozincemia of the acute-phase response. Proct Natl Acad Sci U S A. 2005;102:6843-6848.
- Kay RG, Tasman-Jones C, Pybus J, et al. A syndrome of acute zinc deficiency during total parenteral nutrition in man. Ann Surg. 1976;183:331-340.
- Jeejeebhoy K. Zinc: an essential trace element for parenteral nutrition. Gastroenterology. 2009;137(5 suppl):S7-S12.
- Brazin SA, Johnson WT, Abramson LJ. The acrodermatitis enteropathica-like syndrome. Arch Dermatol. 1979;115:597-599.
- Chun JH, Baek JH, Chung NG. Development of bullous acrodermatitis enteropathica during the course of chemotherapy for acute lymphocytic leukemia. Ann Dermatol. 2011;23(suppl 3):S326-S328.
- Roongpisuthipong W, Phanachet P, Roongpisuthipong C, et al. Essential fatty acid deficiency while a patient receiving fat regimen total parenteral nutrition [published June 14, 2012]. BMJ Case Rep. doi:10.1136/bcr.07.2011.4475.
- Brandt T. Dermatitis in children with disturbances of general condition and absorption of food. Acta Derm Venereol. 1936;17:513-537.
- Danbolt N, Closs K. Acrodermatitis enteropathica. Acta Derm Venereol. 1942;23:127-169.
- Moynahan E. Acrodermatitis enteropathica: a lethal inherited human zinc deficiency disorder. Lancet. 1974;2:299-400.
- Küry S, Dréno B, Bézieau S, et al. Identification of SLC39A4, a gene involved in acrodermatitis enteropathica. Nat Genet. 2002;31:238-240.
- Maverakis E, Fung MA, Lynch PJ, et al. Acrodermatitis enteropathica and an overview of zinc metabolism. J Am Acad Dermatol. 2007;56:116-124.
- Thrash B, Patel M, Shah KR, et al. Cutaneous manifestations of gastrointestinal disease: part II. J Am Acad Dermatol. 2013;68:211.e1-211.e33; quiz 244-246.
- Perafán-Riveros C, França LF, Alves AC, et al. Acrodermatitis enteropathica: case report and review of the literature. Pediatr Dermatol. 2002;19:426-431.
- Kumar P, Ranjan NR, Mondal AK. Zinc and skin: a brief summary. Dermatol Online J. 2012;18:1.
- Saritha M, Gupta D, Chandrashekar L, et al. Acquired zinc deficiency in an adult female. Indian J Dermatol. 2012;57:492-494.
- Neldner K, Hambidge K, Walravens P. Acrodermatitis enteropathica.Int J Dermatol. 1978;17:380-387.
- Gehrig K, Dinulos J. Acrodermatitis due to nutritional deficiency. Curr Opin Pediatr. 2010;22:107-112.
- Liuzzi JP, Lichten LA, Rivera S, et al. Interleukin-6 regulates the zinc transporter Zip14 in liver and contributes to hypozincemia of the acute-phase response. Proct Natl Acad Sci U S A. 2005;102:6843-6848.
- Kay RG, Tasman-Jones C, Pybus J, et al. A syndrome of acute zinc deficiency during total parenteral nutrition in man. Ann Surg. 1976;183:331-340.
- Jeejeebhoy K. Zinc: an essential trace element for parenteral nutrition. Gastroenterology. 2009;137(5 suppl):S7-S12.
- Brazin SA, Johnson WT, Abramson LJ. The acrodermatitis enteropathica-like syndrome. Arch Dermatol. 1979;115:597-599.
- Chun JH, Baek JH, Chung NG. Development of bullous acrodermatitis enteropathica during the course of chemotherapy for acute lymphocytic leukemia. Ann Dermatol. 2011;23(suppl 3):S326-S328.
- Roongpisuthipong W, Phanachet P, Roongpisuthipong C, et al. Essential fatty acid deficiency while a patient receiving fat regimen total parenteral nutrition [published June 14, 2012]. BMJ Case Rep. doi:10.1136/bcr.07.2011.4475.
Practice Points
- Acrodermatitis enteropathica (AE) may be acquired or due to a rare autosomal-recessive disorder of zinc absorption.
- Hereditary AE typically becomes symptomatic during infancy, while acquired AE may develop during hypozincemia in patients of any age.
- Both acquired and hereditary AE improve with zinc supplementation.
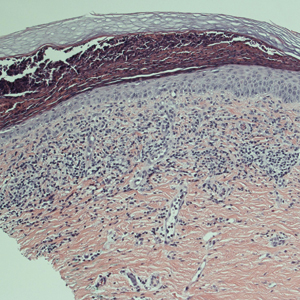
Uncommon Presentation of Chromoblastomycosis
Case Report
A 25-year-old man who was a dairy farmer in Ahmednagar, Maharashtra, India, presented with a history of slowly growing, occasionally itchy lesions on both cheeks of 20 years’ duration. Most of the right cheek was covered by a well-defined, lobulated, gray-brown verrucous mass with a cerebriform surface (Figure 1). The left cheek was covered with a gray-brown infiltrated plaque surrounded by brown-tinged monomorphic papules.

Routine investigations were normal at presentation. Tests for purified protein derivative (tuberculin) and antibodies to human immunodeficiency virus were negative. Magnetic resonance imaging of the head showed soft tissue thickening with ulcerations involving the skin, subcutaneous tissue, and underlying facial muscles of the right cheek.
On histopathology, a hematoxylin and eosin–stained section showed hyperkeratosis, parakeratosis, pseudoepitheliomatous hyperplasia, and follicular plugs in the epidermis, as well as a mixed cellular infiltrate with Langhans giant cells and sclerotic bodies in the dermis (Figure 2). Periodic acid–Schiff and methenamine silver special stains revealed sclerotic bodies.
 in the dermis (H&E, original magnification ×100).")
Fungal culture on Sabouraud dextrose agar at 25°C and 37°C grew olive green, rugose, velvety, leathery colonies within 48 hours, with pigmentation front and reverse (Figure 3). A panfungal polymerase chain reaction assay was positive. Direct microscopic examination of a 10% potassium hydroxide mount of the colonies showed mycelia with dematiaceous septate hyphae (Figure 4), apical branching, branching conidiophores, elliptical conidia in long chains, and pathognomonic round yeastlike bodies resembling copper pennies known as sclerotic cells (also called muriform cells and medlar bodies).1,2 The causative organism was identified as Cladosporium carrionii. A final diagnosis of chromoblastomycosis was made.
 grew olive green, rugose, velvety, leathery colonies within 48 hours.")
.")
After 2 months of treatment with oral itraconazole 400 mg daily, there was no notable clinical improvement and fungal elements were still seen on culture. Four treatment cycles of intravenous liposomal amphotericin B 50 mg daily (1 mg/kg daily) for 15 days followed by itraconazole 200 mg daily for another 15 days caused substantial reduction and flattening of the lesion on the right side and resolution of the lesions on the left side. Healing was accompanied by central erythema and depigmentation (Figure 5). With a suspicion of continuing C carrionii activity on the right cheek, intralesional liposomal amphotericin B 0.2 mL (in a dilution of 5 mg in 1 mL) was given weekly in the peripheral hyperpigmented raised margin, which resulted in further flattening and reduction in tissue resistance. Fungal elements were absent on repeat biopsy and culture after 4 weeks.

Six months after negative culture, further cosmetic correction of the scar on the right cheek was performed with a patterned full-thickness graft for the upper half and excision with approximation of the edges for the lower half (Figure 6). Cultures have been negative for the last 20 months; as of this writing, there has been no recurrence of lesions.

Comment
Distribution
Chromoblastomycosis, also known as chromomycosis and verrucous dermatitis,3 is a chronic subcutaneous mycosis found in tropical and subtropical regions.3,4 It is caused by traumatic inoculation of any of several members of a specific group of dematiaceous fungi through the skin.2,3 Common causative organisms include Fonsecaea pedrosoi, C carrionii, Fonsecaea compacta, and Phialophora verrucosa, all of which are saprophytes in soil and plants. Fonsecaea pedrosoi is the most common causative agent worldwide (70%–90% of cases).2Cladosporium carrionii tends to be the predominant pathogen isolated in patients who present in drier climates, with F pedrosoi in humid forests.1-4
In India, chromoblastomycosis has been reported from the sub-Himalayan belt and western and eastern coasts.1,5 Our patient resided in Ahmednagar, Maharashtra, India, which has a predominantly hot and dry climate. The history might include vegetational trauma, such as a thorn prick. Time between inoculation and development of disease is believed to be years.
Clinical Presentation
Chromoblastomycosis is characterized by a slowly enlarging lesion at the site of inoculation. Five morphological variants are known: nodular, tumoral, verrucous, plaque, and cicatricial; verrucous and nodular types are most common.3,4
The disease is limited to the skin and subcutaneous tissue, growing in extent rather than in depth and not directly invading muscle or bone.4 Lymphatic and hematogenous dissemination can occur.3,4 Secondary bacterial infection is common. The most common affected site is the lower limb, especially the foot.1,3 The upper limb and rarely the ear, trunk, face, and breast can be affected.
Diagnosis
Routine laboratory investigations are usually within reference range. Diagnosis is made by histopathological and mycological studies. Preferably, scrapings or biopsy material are taken from lesions that are covered with what is described as “black dots” (an area of transepidermal elimination of the fungus) where there is a better diagnostic yield.2-4 Routine histopathology shows hyperkeratosis, pseudoepitheliomatous hyperplasia of the epidermis, a mixed granulomatous neutrophil response with multinucleated giant cells and neutrophil abscesses, refractile fungal spores, typical sclerotic cells around abscesses or granulomas, and a dense fibrous response in the dermis and subcutaneous tissue.
Extensive fibrosis, coupled with a chronic inflammatory infiltrate and increased susceptibility to secondary infection, leads to obstruction of lymphatic flow and lymphedema below the affected site.2-4 Periodic acid–Schiff and Gomori methenamine silver stains confirm the presence of fungus. Direct microscopic examination of a 10% potassium hydroxide mount of scrapings reveals spherical, thick-walled, darkly pigmented, multiseptate sclerotic cells known as medlar bodies, copper pennies, and muriform cells that are pathognomonic for chromoblastomycosis.1-4Cladosporium carrionii culture on Sabouraud dextrose agar at 37°C shows olive green, dark, rugose, smooth, hairy, leathery or velvety colonies with pigmentation front and reverse. Direct microscopic examination of the colonies shows dematiaceous septate hyphae and sparsely branching conidiophores bearing ellipsoidal, smooth-walled conidia in long acropetal chains.1,4
Treatment
Treatment options for chromoblastomycosis can be divided into antifungal agents and physical methods.Antifungal agents include itraconazole (200–400 mg daily),3 terbinafine (250–500 mg daily),3 5-fluorocytosine (100–150 mg/kg daily),3 amphotericin B (intravenous/intralesional), and others (eg, fluconazole, ketoconazole, posaconazole [800 mg daily],6,7 potassium iodide, voriconazole). Physical methods include CO2 laser, cryosurgery, local heat therapy, Mohs micrographic surgery, and standard surgery.3 There is no evidence-based treatment protocol. Itraconazole and terbinafine are considered drugs of first choice1,8; however, combination therapy is the best option.9
- Ajanta S, Naba KH, Deepak G. Chromoblastomycosis in sub-tropical regions of India. Mycopathologia. 2010;169:381-386.
- Ameen M. Chromoblastomycosis: clinical presentation and management. Clin Exp Dermatol. 2009;34:849-854.
- Flavio QT, Phillippe E, Maigualida PB, et al. Chromoblastomycosis: an overview of clinical manifestations, diagnosis and treatment. Med Mycol. 2009;47:3-15.
- López Martínez R, Méndez Tovar LJ. Chromoblastomycosis. Clin Dermatol. 2007;25:188-194.
- Pradhan SV, Talwar OP, Ghosh A, et al. Chromoblastomycosis in Nepal: a study of 13 cases. Indian J Dermatol Venereol Leprol. 2007;73:176-178.
- Krzys´ciak PM, Pindycka-Piaszczys´ska M, Piaszczys´ski M. Chromoblastomycosis [published online October 22, 2014]. Postepy Dermatol Alergol. 2014;31:310-321.
- Negroni R, Tobón A, Bustamante B, et al. Posaconazole treatment of refractory eumycetoma and chromoblastomycosis. Rev Inst Med Trop Sao Paulo. 2005;47:339-346.
- Mohanty L, Mohanty P, Padhi T, et al. Verrucous growth on leg. Indian J Dermatol Venereol Leprol. 2006;72:399-400.
- Najafzadeh MJ, Rezusta A, Cameo MI, et al. Successful treatment of chromoblastomycosis of 36 years duration caused by Fonsecaea monophora. Med Mycol. 2010;48:390-393.
Case Report
A 25-year-old man who was a dairy farmer in Ahmednagar, Maharashtra, India, presented with a history of slowly growing, occasionally itchy lesions on both cheeks of 20 years’ duration. Most of the right cheek was covered by a well-defined, lobulated, gray-brown verrucous mass with a cerebriform surface (Figure 1). The left cheek was covered with a gray-brown infiltrated plaque surrounded by brown-tinged monomorphic papules.
Routine investigations were normal at presentation. Tests for purified protein derivative (tuberculin) and antibodies to human immunodeficiency virus were negative. Magnetic resonance imaging of the head showed soft tissue thickening with ulcerations involving the skin, subcutaneous tissue, and underlying facial muscles of the right cheek.
On histopathology, a hematoxylin and eosin–stained section showed hyperkeratosis, parakeratosis, pseudoepitheliomatous hyperplasia, and follicular plugs in the epidermis, as well as a mixed cellular infiltrate with Langhans giant cells and sclerotic bodies in the dermis (Figure 2). Periodic acid–Schiff and methenamine silver special stains revealed sclerotic bodies.
Fungal culture on Sabouraud dextrose agar at 25°C and 37°C grew olive green, rugose, velvety, leathery colonies within 48 hours, with pigmentation front and reverse (Figure 3). A panfungal polymerase chain reaction assay was positive. Direct microscopic examination of a 10% potassium hydroxide mount of the colonies showed mycelia with dematiaceous septate hyphae (Figure 4), apical branching, branching conidiophores, elliptical conidia in long chains, and pathognomonic round yeastlike bodies resembling copper pennies known as sclerotic cells (also called muriform cells and medlar bodies).1,2 The causative organism was identified as Cladosporium carrionii. A final diagnosis of chromoblastomycosis was made.
After 2 months of treatment with oral itraconazole 400 mg daily, there was no notable clinical improvement and fungal elements were still seen on culture. Four treatment cycles of intravenous liposomal amphotericin B 50 mg daily (1 mg/kg daily) for 15 days followed by itraconazole 200 mg daily for another 15 days caused substantial reduction and flattening of the lesion on the right side and resolution of the lesions on the left side. Healing was accompanied by central erythema and depigmentation (Figure 5). With a suspicion of continuing C carrionii activity on the right cheek, intralesional liposomal amphotericin B 0.2 mL (in a dilution of 5 mg in 1 mL) was given weekly in the peripheral hyperpigmented raised margin, which resulted in further flattening and reduction in tissue resistance. Fungal elements were absent on repeat biopsy and culture after 4 weeks.
Six months after negative culture, further cosmetic correction of the scar on the right cheek was performed with a patterned full-thickness graft for the upper half and excision with approximation of the edges for the lower half (Figure 6). Cultures have been negative for the last 20 months; as of this writing, there has been no recurrence of lesions.
Comment
Distribution
Chromoblastomycosis, also known as chromomycosis and verrucous dermatitis,3 is a chronic subcutaneous mycosis found in tropical and subtropical regions.3,4 It is caused by traumatic inoculation of any of several members of a specific group of dematiaceous fungi through the skin.2,3 Common causative organisms include Fonsecaea pedrosoi, C carrionii, Fonsecaea compacta, and Phialophora verrucosa, all of which are saprophytes in soil and plants. Fonsecaea pedrosoi is the most common causative agent worldwide (70%–90% of cases).2Cladosporium carrionii tends to be the predominant pathogen isolated in patients who present in drier climates, with F pedrosoi in humid forests.1-4
In India, chromoblastomycosis has been reported from the sub-Himalayan belt and western and eastern coasts.1,5 Our patient resided in Ahmednagar, Maharashtra, India, which has a predominantly hot and dry climate. The history might include vegetational trauma, such as a thorn prick. Time between inoculation and development of disease is believed to be years.
Clinical Presentation
Chromoblastomycosis is characterized by a slowly enlarging lesion at the site of inoculation. Five morphological variants are known: nodular, tumoral, verrucous, plaque, and cicatricial; verrucous and nodular types are most common.3,4
The disease is limited to the skin and subcutaneous tissue, growing in extent rather than in depth and not directly invading muscle or bone.4 Lymphatic and hematogenous dissemination can occur.3,4 Secondary bacterial infection is common. The most common affected site is the lower limb, especially the foot.1,3 The upper limb and rarely the ear, trunk, face, and breast can be affected.
Diagnosis
Routine laboratory investigations are usually within reference range. Diagnosis is made by histopathological and mycological studies. Preferably, scrapings or biopsy material are taken from lesions that are covered with what is described as “black dots” (an area of transepidermal elimination of the fungus) where there is a better diagnostic yield.2-4 Routine histopathology shows hyperkeratosis, pseudoepitheliomatous hyperplasia of the epidermis, a mixed granulomatous neutrophil response with multinucleated giant cells and neutrophil abscesses, refractile fungal spores, typical sclerotic cells around abscesses or granulomas, and a dense fibrous response in the dermis and subcutaneous tissue.
Extensive fibrosis, coupled with a chronic inflammatory infiltrate and increased susceptibility to secondary infection, leads to obstruction of lymphatic flow and lymphedema below the affected site.2-4 Periodic acid–Schiff and Gomori methenamine silver stains confirm the presence of fungus. Direct microscopic examination of a 10% potassium hydroxide mount of scrapings reveals spherical, thick-walled, darkly pigmented, multiseptate sclerotic cells known as medlar bodies, copper pennies, and muriform cells that are pathognomonic for chromoblastomycosis.1-4Cladosporium carrionii culture on Sabouraud dextrose agar at 37°C shows olive green, dark, rugose, smooth, hairy, leathery or velvety colonies with pigmentation front and reverse. Direct microscopic examination of the colonies shows dematiaceous septate hyphae and sparsely branching conidiophores bearing ellipsoidal, smooth-walled conidia in long acropetal chains.1,4
Treatment
Treatment options for chromoblastomycosis can be divided into antifungal agents and physical methods.Antifungal agents include itraconazole (200–400 mg daily),3 terbinafine (250–500 mg daily),3 5-fluorocytosine (100–150 mg/kg daily),3 amphotericin B (intravenous/intralesional), and others (eg, fluconazole, ketoconazole, posaconazole [800 mg daily],6,7 potassium iodide, voriconazole). Physical methods include CO2 laser, cryosurgery, local heat therapy, Mohs micrographic surgery, and standard surgery.3 There is no evidence-based treatment protocol. Itraconazole and terbinafine are considered drugs of first choice1,8; however, combination therapy is the best option.9
Case Report
A 25-year-old man who was a dairy farmer in Ahmednagar, Maharashtra, India, presented with a history of slowly growing, occasionally itchy lesions on both cheeks of 20 years’ duration. Most of the right cheek was covered by a well-defined, lobulated, gray-brown verrucous mass with a cerebriform surface (Figure 1). The left cheek was covered with a gray-brown infiltrated plaque surrounded by brown-tinged monomorphic papules.
Routine investigations were normal at presentation. Tests for purified protein derivative (tuberculin) and antibodies to human immunodeficiency virus were negative. Magnetic resonance imaging of the head showed soft tissue thickening with ulcerations involving the skin, subcutaneous tissue, and underlying facial muscles of the right cheek.
On histopathology, a hematoxylin and eosin–stained section showed hyperkeratosis, parakeratosis, pseudoepitheliomatous hyperplasia, and follicular plugs in the epidermis, as well as a mixed cellular infiltrate with Langhans giant cells and sclerotic bodies in the dermis (Figure 2). Periodic acid–Schiff and methenamine silver special stains revealed sclerotic bodies.
Fungal culture on Sabouraud dextrose agar at 25°C and 37°C grew olive green, rugose, velvety, leathery colonies within 48 hours, with pigmentation front and reverse (Figure 3). A panfungal polymerase chain reaction assay was positive. Direct microscopic examination of a 10% potassium hydroxide mount of the colonies showed mycelia with dematiaceous septate hyphae (Figure 4), apical branching, branching conidiophores, elliptical conidia in long chains, and pathognomonic round yeastlike bodies resembling copper pennies known as sclerotic cells (also called muriform cells and medlar bodies).1,2 The causative organism was identified as Cladosporium carrionii. A final diagnosis of chromoblastomycosis was made.
After 2 months of treatment with oral itraconazole 400 mg daily, there was no notable clinical improvement and fungal elements were still seen on culture. Four treatment cycles of intravenous liposomal amphotericin B 50 mg daily (1 mg/kg daily) for 15 days followed by itraconazole 200 mg daily for another 15 days caused substantial reduction and flattening of the lesion on the right side and resolution of the lesions on the left side. Healing was accompanied by central erythema and depigmentation (Figure 5). With a suspicion of continuing C carrionii activity on the right cheek, intralesional liposomal amphotericin B 0.2 mL (in a dilution of 5 mg in 1 mL) was given weekly in the peripheral hyperpigmented raised margin, which resulted in further flattening and reduction in tissue resistance. Fungal elements were absent on repeat biopsy and culture after 4 weeks.
Six months after negative culture, further cosmetic correction of the scar on the right cheek was performed with a patterned full-thickness graft for the upper half and excision with approximation of the edges for the lower half (Figure 6). Cultures have been negative for the last 20 months; as of this writing, there has been no recurrence of lesions.
Comment
Distribution
Chromoblastomycosis, also known as chromomycosis and verrucous dermatitis,3 is a chronic subcutaneous mycosis found in tropical and subtropical regions.3,4 It is caused by traumatic inoculation of any of several members of a specific group of dematiaceous fungi through the skin.2,3 Common causative organisms include Fonsecaea pedrosoi, C carrionii, Fonsecaea compacta, and Phialophora verrucosa, all of which are saprophytes in soil and plants. Fonsecaea pedrosoi is the most common causative agent worldwide (70%–90% of cases).2Cladosporium carrionii tends to be the predominant pathogen isolated in patients who present in drier climates, with F pedrosoi in humid forests.1-4
In India, chromoblastomycosis has been reported from the sub-Himalayan belt and western and eastern coasts.1,5 Our patient resided in Ahmednagar, Maharashtra, India, which has a predominantly hot and dry climate. The history might include vegetational trauma, such as a thorn prick. Time between inoculation and development of disease is believed to be years.
Clinical Presentation
Chromoblastomycosis is characterized by a slowly enlarging lesion at the site of inoculation. Five morphological variants are known: nodular, tumoral, verrucous, plaque, and cicatricial; verrucous and nodular types are most common.3,4
The disease is limited to the skin and subcutaneous tissue, growing in extent rather than in depth and not directly invading muscle or bone.4 Lymphatic and hematogenous dissemination can occur.3,4 Secondary bacterial infection is common. The most common affected site is the lower limb, especially the foot.1,3 The upper limb and rarely the ear, trunk, face, and breast can be affected.
Diagnosis
Routine laboratory investigations are usually within reference range. Diagnosis is made by histopathological and mycological studies. Preferably, scrapings or biopsy material are taken from lesions that are covered with what is described as “black dots” (an area of transepidermal elimination of the fungus) where there is a better diagnostic yield.2-4 Routine histopathology shows hyperkeratosis, pseudoepitheliomatous hyperplasia of the epidermis, a mixed granulomatous neutrophil response with multinucleated giant cells and neutrophil abscesses, refractile fungal spores, typical sclerotic cells around abscesses or granulomas, and a dense fibrous response in the dermis and subcutaneous tissue.
Extensive fibrosis, coupled with a chronic inflammatory infiltrate and increased susceptibility to secondary infection, leads to obstruction of lymphatic flow and lymphedema below the affected site.2-4 Periodic acid–Schiff and Gomori methenamine silver stains confirm the presence of fungus. Direct microscopic examination of a 10% potassium hydroxide mount of scrapings reveals spherical, thick-walled, darkly pigmented, multiseptate sclerotic cells known as medlar bodies, copper pennies, and muriform cells that are pathognomonic for chromoblastomycosis.1-4Cladosporium carrionii culture on Sabouraud dextrose agar at 37°C shows olive green, dark, rugose, smooth, hairy, leathery or velvety colonies with pigmentation front and reverse. Direct microscopic examination of the colonies shows dematiaceous septate hyphae and sparsely branching conidiophores bearing ellipsoidal, smooth-walled conidia in long acropetal chains.1,4
Treatment
Treatment options for chromoblastomycosis can be divided into antifungal agents and physical methods.Antifungal agents include itraconazole (200–400 mg daily),3 terbinafine (250–500 mg daily),3 5-fluorocytosine (100–150 mg/kg daily),3 amphotericin B (intravenous/intralesional), and others (eg, fluconazole, ketoconazole, posaconazole [800 mg daily],6,7 potassium iodide, voriconazole). Physical methods include CO2 laser, cryosurgery, local heat therapy, Mohs micrographic surgery, and standard surgery.3 There is no evidence-based treatment protocol. Itraconazole and terbinafine are considered drugs of first choice1,8; however, combination therapy is the best option.9
- Ajanta S, Naba KH, Deepak G. Chromoblastomycosis in sub-tropical regions of India. Mycopathologia. 2010;169:381-386.
- Ameen M. Chromoblastomycosis: clinical presentation and management. Clin Exp Dermatol. 2009;34:849-854.
- Flavio QT, Phillippe E, Maigualida PB, et al. Chromoblastomycosis: an overview of clinical manifestations, diagnosis and treatment. Med Mycol. 2009;47:3-15.
- López Martínez R, Méndez Tovar LJ. Chromoblastomycosis. Clin Dermatol. 2007;25:188-194.
- Pradhan SV, Talwar OP, Ghosh A, et al. Chromoblastomycosis in Nepal: a study of 13 cases. Indian J Dermatol Venereol Leprol. 2007;73:176-178.
- Krzys´ciak PM, Pindycka-Piaszczys´ska M, Piaszczys´ski M. Chromoblastomycosis [published online October 22, 2014]. Postepy Dermatol Alergol. 2014;31:310-321.
- Negroni R, Tobón A, Bustamante B, et al. Posaconazole treatment of refractory eumycetoma and chromoblastomycosis. Rev Inst Med Trop Sao Paulo. 2005;47:339-346.
- Mohanty L, Mohanty P, Padhi T, et al. Verrucous growth on leg. Indian J Dermatol Venereol Leprol. 2006;72:399-400.
- Najafzadeh MJ, Rezusta A, Cameo MI, et al. Successful treatment of chromoblastomycosis of 36 years duration caused by Fonsecaea monophora. Med Mycol. 2010;48:390-393.
- Ajanta S, Naba KH, Deepak G. Chromoblastomycosis in sub-tropical regions of India. Mycopathologia. 2010;169:381-386.
- Ameen M. Chromoblastomycosis: clinical presentation and management. Clin Exp Dermatol. 2009;34:849-854.
- Flavio QT, Phillippe E, Maigualida PB, et al. Chromoblastomycosis: an overview of clinical manifestations, diagnosis and treatment. Med Mycol. 2009;47:3-15.
- López Martínez R, Méndez Tovar LJ. Chromoblastomycosis. Clin Dermatol. 2007;25:188-194.
- Pradhan SV, Talwar OP, Ghosh A, et al. Chromoblastomycosis in Nepal: a study of 13 cases. Indian J Dermatol Venereol Leprol. 2007;73:176-178.
- Krzys´ciak PM, Pindycka-Piaszczys´ska M, Piaszczys´ski M. Chromoblastomycosis [published online October 22, 2014]. Postepy Dermatol Alergol. 2014;31:310-321.
- Negroni R, Tobón A, Bustamante B, et al. Posaconazole treatment of refractory eumycetoma and chromoblastomycosis. Rev Inst Med Trop Sao Paulo. 2005;47:339-346.
- Mohanty L, Mohanty P, Padhi T, et al. Verrucous growth on leg. Indian J Dermatol Venereol Leprol. 2006;72:399-400.
- Najafzadeh MJ, Rezusta A, Cameo MI, et al. Successful treatment of chromoblastomycosis of 36 years duration caused by Fonsecaea monophora. Med Mycol. 2010;48:390-393.
Practice Points
- Chromoblastomycosis is limited to skin and subcutaneous tissue, most commonly of the lower limb, especially the foot; it does not directly invade muscle or bone. Secondary bacterial infection is common.
- Chromoblastomycosis is a therapeutic challenge due to its recalcitrant nature. Itraconazole and terbinafine are considered drugs of first choice, but consensus and evidence are lacking on a standard of care.
Delayed Cutaneous Reactions to Iodinated Contrast
Case Report
A 67-year-old woman with a history of allergic rhinitis presented in the spring with a pruritic eruption of 2 days’ duration that began on the abdomen and spread to the chest, back, and bilateral arms. Six days prior to the onset of the symptoms she underwent computed tomography (CT) of the abdomen and pelvis to evaluate abdominal pain and peripheral eosinophilia. Two iodinated contrast (IC) agents were used: intravenous iohexol and oral diatrizoate meglumine–diatrizoate sodium. The eruption was not preceded by fever, malaise, sore throat, rhinorrhea, cough, arthralgia, headache, diarrhea, or new medication or supplement use. The patient denied any history of drug allergy or cutaneous eruptions. Her current medications, which she had been taking long-term, were aspirin, lisinopril, diltiazem, levothyroxine, esomeprazole, paroxetine, gabapentin, and diphenhydramine.
Physical examination was notable for erythematous, blanchable, nontender macules coalescing into patches on the trunk and bilateral arms (Figure). There was slight erythema on the nasolabial folds and ears. The mucosal surfaces and distal legs were clear. The patient was afebrile. Her white blood cell count was 12.5×109/L with 32.3% eosinophils (baseline: white blood cell count, 14.8×109/L; 22% eosinophils)(reference range, 4.8–10.8×109/L; 1%–4% eosinophils). Her comprehensive metabolic panel was within reference range. The human immunodeficiency virus 1/2 antibody immunoassay was nonreactive.
 and left arm (B).")
The patient was diagnosed with an exanthematous eruption caused by IC and was treated with oral hydroxyzine and triamcinolone acetonide cream 0.1%. The eruption resolved within 2 weeks without recurrence at 3-month follow-up.
Comment
Del
Clinical Presentation of Delayed Reactions
Most delayed cutaneous reactions to IC present as exanthematous eruptions in the week following a contrast-enhanced CT scan or coronary angiogram.2,12 The reactions tend to resolve within 2 weeks of onset, and the treatment is largely supportive with antihistamines and/or corticosteroids for the associated pruritus.2,5,6 In a study of 98 patients with a history of delayed reactions to IC, delayed-onset urticaria and angioedema also have been reported with incidence rates of 19% and 24%, respectively.2 Other reactions are less common. In the same study, 7% of patients developed palpable purpura; acute generalized exanthematous pustulosis; bullous, flexural, or psoriasislike exanthema; exfoliative eruptions; or purpura and a maculopapular eruption combined with eosinophilia.2 There also have been reports of IC-induced erythema multiforme,3 fixed drug eruptions,10,11 symmetrical drug-related intertriginous and flexural exanthema,13 cutaneous vasculitis,14 drug reactions with eosinophilia and systemic symptoms,15 Stevens-Johnson syndrome/TEN,7,8,16,17 and iododerma.18
IC Agents
Virtually all delayed cutaneous reactions to IC reportedly are due to intravascular rather than oral agents,1,2,19 with the exception of iododerma18 and 1 reported case of TEN.17 Intravenous iohexol was most likely the offending drug in our case. In a prospective cohort study of 539 patients undergoing CT scans, the absolute risk for developing a delayed cutaneous reaction (defined as rash, itching, or skin redness or swelling) to intravascular iohexol was 9.4%.20 Randomized, double-blind studies have found that the risk for delayed cutaneous eruptions is similar among various types of IC, except for iodixanol, which confers a higher risk.5,6,21
Risk Factors
Interestingly, analyses have shown that delayed reactions to IC are more common in atopic patients and during high pollen season.22 Our patient displayed these risk factors, as she had allergic rhinitis and presented for evaluation in late spring when local pollen counts were high. Additionally, patients who develop delayed reactions to IC are notably more likely than controls to have a history of other cutaneous drug reactions, serum creatinine levels greater than 2.0 mg/dL (reference range, 0.6–1.2 mg/dL),3 or history of treatment with recombinant interleukin 2.19
Patients with a history of delayed reactions to IC are not at increased risk for immediate reactions and vice versa.2,3 This finding is consistent with the evidence that delayed and immediate reactions to IC are mechanistically unrelated.23 Additionally, seafood allergy is not a risk factor for either immediate or delayed reactions to IC, despite a common misconception among physicians and patients because seafood is iodinated.24,25
Reexposure to IC
Patients who have had delayed cutaneous reactions to IC are at risk for similar eruptions upon reexposure. Although the reactions are believed to be cell mediated, skin testing with IC is not sensitive enough to reliably identify tolerable alternatives.12 Consequently, gadolinium-based agents have been recommended in patients with a history of reactions to IC if additional contrast-enhanced studies are needed.13,26 Iodinated and gadolinium-based contrast agents do not cross-react, and gadolinium is compatible with studies other than magnetic resonance imaging.1,27
Premedication
Despite the absence of cross-reactivity, the American College of Radiology considers patients with hypersensitivity reactions to IC to be at increased risk for reactions to gadolinium but does not make specific recommendations regarding premedication given the dearth of data.1 As a result, premedication may be considered prior to gadolinium administration depending on the severity of the delayed reaction to IC. Additionally, premedication may be beneficial in cases in which gadolinium is contraindicated and IC must be reused. In a retrospective study, all patients with suspected delayed reactions to IC tolerated IC or gadolinium contrast when pretreated with corticosteroids with or without antihistamines.28 Regimens with corticosteroids and either cyclosporine or intravenous immunoglobulin also have prevented the recurrence of IC-induced exanthematous eruptions and Stevens-Johnson syndrome.29,30 Despite these reports, delayed cutaneous reactions to IC have recurred in other patients receiving corticosteroids, antihistamines, and/or cyclosporine for premedication or concurrent treatment of an underlying condition.16,29-31
Conclusion
It is important for dermatologists to recognize IC as a cause of delayed drug reactions. Current awareness is limited, and as a result, patients often are reexposed to the offending contrast agents unsuspectingly. In addition to diagnosing these eruptions, dermatologists may help prevent their recurrence if future contrast-enhanced studies are required by recommending gadolinium-based agents and/or premedication.
- Cohan RH, Davenport MS, Dillman JR, et al; ACR Committee on Drugs and Contrast Media. ACR Manual on Contrast Media. 9th ed. Reston, VA: American College of Radiology; 2013.
- Brockow K, Romano A, Aberer W, et al; European Network of Drug Allergy and the EAACI Interest Group on Drug Hypersensitivity. Skin testing in patients with hypersensitivity reactions to iodinated contrast media—a European multicenter study. Allergy. 2009;64:234-241.
Hosoya T, Yamaguchi K, Akutsu T, et al. Delayed adverse reactions to iodinated contrast media and their risk factors. Radiat Med. 2000;18:39-45. - Rydberg J, Charles J, Aspelin P. Frequency of late allergy-like adverse reactions following injection of intravascular non-ionic contrast media: a retrospective study comparing a non-ionic monomeric contrast medium with a non-ionic dimeric contrast medium. Acta Radiol. 1998;39:219-222.
- Sutton AG, Finn P, Grech ED, et al. Early and late reactions after the use of iopamidol 340, ioxaglate 320, and iodixanol 320 in cardiac catheterization. Am Heart J. 2001;141:677-683.
- Sutton AG, Finn P, Campbell PG, et al. Early and late reactions following the use of iopamidol 340, iomeprol 350 and iodixanol 320 in cardiac catheterization. J Invasive Cardiol. 2003;15:133-138.
- Brown M, Yowler C, Brandt C. Recurrent toxic epidermal necrolysis secondary to iopromide contrast. J Burn Care Res. 2013;34:E53-E56.
- Rosado A, Canto G, Veleiro B, et al. Toxic epidermal necrolysis after repeated injections of iohexol. AJR Am J Roentgenol. 2001;176:262-263.
- Peterson A, Katzberg RW, Fung MA, et al. Acute generalized exanthematous pustulosis as a delayed dermatotoxic reaction to IV-administered nonionic contrast media. AJR Am J Roentgenol. 2006;187:W198-W201.
- Good AE, Novak E, Sonda LP III. Fixed eruption and fever after urography. South Med J. 1980;73:948-949.
- Benson PM, Giblin WJ, Douglas DM. Transient, nonpigmenting fixed drug eruption caused by radiopaque contrast media. J Am Acad Dermatol. 1990;23(2, pt 2):379-381.
- Torres MJ, Gomez F, Doña I, et al. Diagnostic evaluation of patients with nonimmediate cutaneous hypersensitivity reactions to iodinated contrast media. Allergy. 2012;67:929-935.
- Scherer K, Harr T, Bach S, et al. The role of iodine in hypersensitivity reactions to radio contrast media. Clin Exp Allergy. 2010;40:468-475.
- Reynolds NJ, Wallington TB, Burton JL. Hydralazine predisposes to acute cutaneous vasculitis following urography with iopamidol. Br J Dermatol. 1993;129:82-85.
- Belhadjali H, Bouzgarrou L, Youssef M, et al. DRESS syndrome induced by sodium meglumine ioxitalamate. Allergy. 2008;63:786-787.
- Baldwin BT, Lien MH, Khan H, et al. Case of fatal toxic epidermal necrolysis due to cardiac catheterization dye. J Drugs Dermatol. 2010;9:837-840.
- Schmidt BJ, Foley WD, Bohorfoush AG. Toxic epidermal necrolysis related to oral administration of diluted diatrizoate meglumine and diatrizoate sodium. AJR Am J Roentgenol. 1998;171:1215-1216.
- Young AL, Grossman ME. Acute iododerma secondary to iodinated contrast media. Br J Dermatol. 2014;170:1377-1379.
- Choyke PL, Miller DL, Lotze MT, et al. Delayed reactions to contrast media after interleukin-2 immunotherapy. Radiology. 1992;183:111-114.
- Loh S, Bagheri S, Katzberg RW, et al. Delayed adverse reaction to contrast-enhanced CT: a prospective single-center study comparison to control group without enhancement. Radiology. 2010;255:764-771.
- Bertrand P, Delhommais A, Alison D, et al. Immediate and delayed tolerance of iohexol and ioxaglate in lower limb phlebography: a double-blind comparative study in humans. Acad Radiol. 1995;2:683-686.
- Munechika H, Hiramatsu Y, Kudo S, et al. A prospective survey of delayed adverse reactions to iohexol in urography and computed tomography. Eur Radiol. 2003;13:185-194.
- Guéant-Rodriguez RM, Romano A, Barbaud A, et al. Hypersensitivity reactions to iodinated contrast media. Curr Pharm Des. 2006;12:3359-3372.
- H
uang SW. Seafood and iodine: an analysis of a medical myth. Allergy Asthma Proc. 2005;26:468-469. - B
aig M, Farag A, Sajid J, et al. Shellfish allergy and relation to iodinated contrast media: United Kingdom survey. World J Cardiol. 2014;6:107-111. - B
öhm I, Schild HH. A practical guide to diagnose lesser-known immediate and delayed contrast media-induced adverse cutaneous reactions. Eur Radiol. 2006;16:1570-1579. - Ose K, Doue T, Zen K, et al. “Gadolinium” as an alternative to iodinated contrast media for X-ray angiography in patients with severe allergy. Circ J. 2005;69:507-509.
- Ji
ngu A, Fukuda J, Taketomi-Takahashi A, et al. Breakthrough reactions of iodinated and gadolinium contrast media after oral steroid premedication protocol. BMC Med Imaging. 2014;14:34. - Ro
mano A, Artesani MC, Andriolo M, et al. Effective prophylactic protocol in delayed hypersensitivity to contrast media: report of a case involving lymphocyte transformation studies with different compounds. Radiology. 2002;225:466-470. - He
bert AA, Bogle MA. Intravenous immunoglobulin prophylaxis for recurrent Stevens-Johnson syndrome. J Am Acad Dermatol. 2004;50:286-288. - Ha
sdenteufel F, Waton J, Cordebar V, et al. Delayed hypersensitivity reactions caused by iodixanol: an assessment of cross-reactivity in 22 patients. J Allergy Clin Immunol. 2011;128:1356-1357.
Case Report
A 67-year-old woman with a history of allergic rhinitis presented in the spring with a pruritic eruption of 2 days’ duration that began on the abdomen and spread to the chest, back, and bilateral arms. Six days prior to the onset of the symptoms she underwent computed tomography (CT) of the abdomen and pelvis to evaluate abdominal pain and peripheral eosinophilia. Two iodinated contrast (IC) agents were used: intravenous iohexol and oral diatrizoate meglumine–diatrizoate sodium. The eruption was not preceded by fever, malaise, sore throat, rhinorrhea, cough, arthralgia, headache, diarrhea, or new medication or supplement use. The patient denied any history of drug allergy or cutaneous eruptions. Her current medications, which she had been taking long-term, were aspirin, lisinopril, diltiazem, levothyroxine, esomeprazole, paroxetine, gabapentin, and diphenhydramine.
Physical examination was notable for erythematous, blanchable, nontender macules coalescing into patches on the trunk and bilateral arms (Figure). There was slight erythema on the nasolabial folds and ears. The mucosal surfaces and distal legs were clear. The patient was afebrile. Her white blood cell count was 12.5×109/L with 32.3% eosinophils (baseline: white blood cell count, 14.8×109/L; 22% eosinophils)(reference range, 4.8–10.8×109/L; 1%–4% eosinophils). Her comprehensive metabolic panel was within reference range. The human immunodeficiency virus 1/2 antibody immunoassay was nonreactive.
The patient was diagnosed with an exanthematous eruption caused by IC and was treated with oral hydroxyzine and triamcinolone acetonide cream 0.1%. The eruption resolved within 2 weeks without recurrence at 3-month follow-up.
Comment
Del
Clinical Presentation of Delayed Reactions
Most delayed cutaneous reactions to IC present as exanthematous eruptions in the week following a contrast-enhanced CT scan or coronary angiogram.2,12 The reactions tend to resolve within 2 weeks of onset, and the treatment is largely supportive with antihistamines and/or corticosteroids for the associated pruritus.2,5,6 In a study of 98 patients with a history of delayed reactions to IC, delayed-onset urticaria and angioedema also have been reported with incidence rates of 19% and 24%, respectively.2 Other reactions are less common. In the same study, 7% of patients developed palpable purpura; acute generalized exanthematous pustulosis; bullous, flexural, or psoriasislike exanthema; exfoliative eruptions; or purpura and a maculopapular eruption combined with eosinophilia.2 There also have been reports of IC-induced erythema multiforme,3 fixed drug eruptions,10,11 symmetrical drug-related intertriginous and flexural exanthema,13 cutaneous vasculitis,14 drug reactions with eosinophilia and systemic symptoms,15 Stevens-Johnson syndrome/TEN,7,8,16,17 and iododerma.18
IC Agents
Virtually all delayed cutaneous reactions to IC reportedly are due to intravascular rather than oral agents,1,2,19 with the exception of iododerma18 and 1 reported case of TEN.17 Intravenous iohexol was most likely the offending drug in our case. In a prospective cohort study of 539 patients undergoing CT scans, the absolute risk for developing a delayed cutaneous reaction (defined as rash, itching, or skin redness or swelling) to intravascular iohexol was 9.4%.20 Randomized, double-blind studies have found that the risk for delayed cutaneous eruptions is similar among various types of IC, except for iodixanol, which confers a higher risk.5,6,21
Risk Factors
Interestingly, analyses have shown that delayed reactions to IC are more common in atopic patients and during high pollen season.22 Our patient displayed these risk factors, as she had allergic rhinitis and presented for evaluation in late spring when local pollen counts were high. Additionally, patients who develop delayed reactions to IC are notably more likely than controls to have a history of other cutaneous drug reactions, serum creatinine levels greater than 2.0 mg/dL (reference range, 0.6–1.2 mg/dL),3 or history of treatment with recombinant interleukin 2.19
Patients with a history of delayed reactions to IC are not at increased risk for immediate reactions and vice versa.2,3 This finding is consistent with the evidence that delayed and immediate reactions to IC are mechanistically unrelated.23 Additionally, seafood allergy is not a risk factor for either immediate or delayed reactions to IC, despite a common misconception among physicians and patients because seafood is iodinated.24,25
Reexposure to IC
Patients who have had delayed cutaneous reactions to IC are at risk for similar eruptions upon reexposure. Although the reactions are believed to be cell mediated, skin testing with IC is not sensitive enough to reliably identify tolerable alternatives.12 Consequently, gadolinium-based agents have been recommended in patients with a history of reactions to IC if additional contrast-enhanced studies are needed.13,26 Iodinated and gadolinium-based contrast agents do not cross-react, and gadolinium is compatible with studies other than magnetic resonance imaging.1,27
Premedication
Despite the absence of cross-reactivity, the American College of Radiology considers patients with hypersensitivity reactions to IC to be at increased risk for reactions to gadolinium but does not make specific recommendations regarding premedication given the dearth of data.1 As a result, premedication may be considered prior to gadolinium administration depending on the severity of the delayed reaction to IC. Additionally, premedication may be beneficial in cases in which gadolinium is contraindicated and IC must be reused. In a retrospective study, all patients with suspected delayed reactions to IC tolerated IC or gadolinium contrast when pretreated with corticosteroids with or without antihistamines.28 Regimens with corticosteroids and either cyclosporine or intravenous immunoglobulin also have prevented the recurrence of IC-induced exanthematous eruptions and Stevens-Johnson syndrome.29,30 Despite these reports, delayed cutaneous reactions to IC have recurred in other patients receiving corticosteroids, antihistamines, and/or cyclosporine for premedication or concurrent treatment of an underlying condition.16,29-31
Conclusion
It is important for dermatologists to recognize IC as a cause of delayed drug reactions. Current awareness is limited, and as a result, patients often are reexposed to the offending contrast agents unsuspectingly. In addition to diagnosing these eruptions, dermatologists may help prevent their recurrence if future contrast-enhanced studies are required by recommending gadolinium-based agents and/or premedication.
Case Report
A 67-year-old woman with a history of allergic rhinitis presented in the spring with a pruritic eruption of 2 days’ duration that began on the abdomen and spread to the chest, back, and bilateral arms. Six days prior to the onset of the symptoms she underwent computed tomography (CT) of the abdomen and pelvis to evaluate abdominal pain and peripheral eosinophilia. Two iodinated contrast (IC) agents were used: intravenous iohexol and oral diatrizoate meglumine–diatrizoate sodium. The eruption was not preceded by fever, malaise, sore throat, rhinorrhea, cough, arthralgia, headache, diarrhea, or new medication or supplement use. The patient denied any history of drug allergy or cutaneous eruptions. Her current medications, which she had been taking long-term, were aspirin, lisinopril, diltiazem, levothyroxine, esomeprazole, paroxetine, gabapentin, and diphenhydramine.
Physical examination was notable for erythematous, blanchable, nontender macules coalescing into patches on the trunk and bilateral arms (Figure). There was slight erythema on the nasolabial folds and ears. The mucosal surfaces and distal legs were clear. The patient was afebrile. Her white blood cell count was 12.5×109/L with 32.3% eosinophils (baseline: white blood cell count, 14.8×109/L; 22% eosinophils)(reference range, 4.8–10.8×109/L; 1%–4% eosinophils). Her comprehensive metabolic panel was within reference range. The human immunodeficiency virus 1/2 antibody immunoassay was nonreactive.
The patient was diagnosed with an exanthematous eruption caused by IC and was treated with oral hydroxyzine and triamcinolone acetonide cream 0.1%. The eruption resolved within 2 weeks without recurrence at 3-month follow-up.
Comment
Del
Clinical Presentation of Delayed Reactions
Most delayed cutaneous reactions to IC present as exanthematous eruptions in the week following a contrast-enhanced CT scan or coronary angiogram.2,12 The reactions tend to resolve within 2 weeks of onset, and the treatment is largely supportive with antihistamines and/or corticosteroids for the associated pruritus.2,5,6 In a study of 98 patients with a history of delayed reactions to IC, delayed-onset urticaria and angioedema also have been reported with incidence rates of 19% and 24%, respectively.2 Other reactions are less common. In the same study, 7% of patients developed palpable purpura; acute generalized exanthematous pustulosis; bullous, flexural, or psoriasislike exanthema; exfoliative eruptions; or purpura and a maculopapular eruption combined with eosinophilia.2 There also have been reports of IC-induced erythema multiforme,3 fixed drug eruptions,10,11 symmetrical drug-related intertriginous and flexural exanthema,13 cutaneous vasculitis,14 drug reactions with eosinophilia and systemic symptoms,15 Stevens-Johnson syndrome/TEN,7,8,16,17 and iododerma.18
IC Agents
Virtually all delayed cutaneous reactions to IC reportedly are due to intravascular rather than oral agents,1,2,19 with the exception of iododerma18 and 1 reported case of TEN.17 Intravenous iohexol was most likely the offending drug in our case. In a prospective cohort study of 539 patients undergoing CT scans, the absolute risk for developing a delayed cutaneous reaction (defined as rash, itching, or skin redness or swelling) to intravascular iohexol was 9.4%.20 Randomized, double-blind studies have found that the risk for delayed cutaneous eruptions is similar among various types of IC, except for iodixanol, which confers a higher risk.5,6,21
Risk Factors
Interestingly, analyses have shown that delayed reactions to IC are more common in atopic patients and during high pollen season.22 Our patient displayed these risk factors, as she had allergic rhinitis and presented for evaluation in late spring when local pollen counts were high. Additionally, patients who develop delayed reactions to IC are notably more likely than controls to have a history of other cutaneous drug reactions, serum creatinine levels greater than 2.0 mg/dL (reference range, 0.6–1.2 mg/dL),3 or history of treatment with recombinant interleukin 2.19
Patients with a history of delayed reactions to IC are not at increased risk for immediate reactions and vice versa.2,3 This finding is consistent with the evidence that delayed and immediate reactions to IC are mechanistically unrelated.23 Additionally, seafood allergy is not a risk factor for either immediate or delayed reactions to IC, despite a common misconception among physicians and patients because seafood is iodinated.24,25
Reexposure to IC
Patients who have had delayed cutaneous reactions to IC are at risk for similar eruptions upon reexposure. Although the reactions are believed to be cell mediated, skin testing with IC is not sensitive enough to reliably identify tolerable alternatives.12 Consequently, gadolinium-based agents have been recommended in patients with a history of reactions to IC if additional contrast-enhanced studies are needed.13,26 Iodinated and gadolinium-based contrast agents do not cross-react, and gadolinium is compatible with studies other than magnetic resonance imaging.1,27
Premedication
Despite the absence of cross-reactivity, the American College of Radiology considers patients with hypersensitivity reactions to IC to be at increased risk for reactions to gadolinium but does not make specific recommendations regarding premedication given the dearth of data.1 As a result, premedication may be considered prior to gadolinium administration depending on the severity of the delayed reaction to IC. Additionally, premedication may be beneficial in cases in which gadolinium is contraindicated and IC must be reused. In a retrospective study, all patients with suspected delayed reactions to IC tolerated IC or gadolinium contrast when pretreated with corticosteroids with or without antihistamines.28 Regimens with corticosteroids and either cyclosporine or intravenous immunoglobulin also have prevented the recurrence of IC-induced exanthematous eruptions and Stevens-Johnson syndrome.29,30 Despite these reports, delayed cutaneous reactions to IC have recurred in other patients receiving corticosteroids, antihistamines, and/or cyclosporine for premedication or concurrent treatment of an underlying condition.16,29-31
Conclusion
It is important for dermatologists to recognize IC as a cause of delayed drug reactions. Current awareness is limited, and as a result, patients often are reexposed to the offending contrast agents unsuspectingly. In addition to diagnosing these eruptions, dermatologists may help prevent their recurrence if future contrast-enhanced studies are required by recommending gadolinium-based agents and/or premedication.
- Cohan RH, Davenport MS, Dillman JR, et al; ACR Committee on Drugs and Contrast Media. ACR Manual on Contrast Media. 9th ed. Reston, VA: American College of Radiology; 2013.
- Brockow K, Romano A, Aberer W, et al; European Network of Drug Allergy and the EAACI Interest Group on Drug Hypersensitivity. Skin testing in patients with hypersensitivity reactions to iodinated contrast media—a European multicenter study. Allergy. 2009;64:234-241.
Hosoya T, Yamaguchi K, Akutsu T, et al. Delayed adverse reactions to iodinated contrast media and their risk factors. Radiat Med. 2000;18:39-45. - Rydberg J, Charles J, Aspelin P. Frequency of late allergy-like adverse reactions following injection of intravascular non-ionic contrast media: a retrospective study comparing a non-ionic monomeric contrast medium with a non-ionic dimeric contrast medium. Acta Radiol. 1998;39:219-222.
- Sutton AG, Finn P, Grech ED, et al. Early and late reactions after the use of iopamidol 340, ioxaglate 320, and iodixanol 320 in cardiac catheterization. Am Heart J. 2001;141:677-683.
- Sutton AG, Finn P, Campbell PG, et al. Early and late reactions following the use of iopamidol 340, iomeprol 350 and iodixanol 320 in cardiac catheterization. J Invasive Cardiol. 2003;15:133-138.
- Brown M, Yowler C, Brandt C. Recurrent toxic epidermal necrolysis secondary to iopromide contrast. J Burn Care Res. 2013;34:E53-E56.
- Rosado A, Canto G, Veleiro B, et al. Toxic epidermal necrolysis after repeated injections of iohexol. AJR Am J Roentgenol. 2001;176:262-263.
- Peterson A, Katzberg RW, Fung MA, et al. Acute generalized exanthematous pustulosis as a delayed dermatotoxic reaction to IV-administered nonionic contrast media. AJR Am J Roentgenol. 2006;187:W198-W201.
- Good AE, Novak E, Sonda LP III. Fixed eruption and fever after urography. South Med J. 1980;73:948-949.
- Benson PM, Giblin WJ, Douglas DM. Transient, nonpigmenting fixed drug eruption caused by radiopaque contrast media. J Am Acad Dermatol. 1990;23(2, pt 2):379-381.
- Torres MJ, Gomez F, Doña I, et al. Diagnostic evaluation of patients with nonimmediate cutaneous hypersensitivity reactions to iodinated contrast media. Allergy. 2012;67:929-935.
- Scherer K, Harr T, Bach S, et al. The role of iodine in hypersensitivity reactions to radio contrast media. Clin Exp Allergy. 2010;40:468-475.
- Reynolds NJ, Wallington TB, Burton JL. Hydralazine predisposes to acute cutaneous vasculitis following urography with iopamidol. Br J Dermatol. 1993;129:82-85.
- Belhadjali H, Bouzgarrou L, Youssef M, et al. DRESS syndrome induced by sodium meglumine ioxitalamate. Allergy. 2008;63:786-787.
- Baldwin BT, Lien MH, Khan H, et al. Case of fatal toxic epidermal necrolysis due to cardiac catheterization dye. J Drugs Dermatol. 2010;9:837-840.
- Schmidt BJ, Foley WD, Bohorfoush AG. Toxic epidermal necrolysis related to oral administration of diluted diatrizoate meglumine and diatrizoate sodium. AJR Am J Roentgenol. 1998;171:1215-1216.
- Young AL, Grossman ME. Acute iododerma secondary to iodinated contrast media. Br J Dermatol. 2014;170:1377-1379.
- Choyke PL, Miller DL, Lotze MT, et al. Delayed reactions to contrast media after interleukin-2 immunotherapy. Radiology. 1992;183:111-114.
- Loh S, Bagheri S, Katzberg RW, et al. Delayed adverse reaction to contrast-enhanced CT: a prospective single-center study comparison to control group without enhancement. Radiology. 2010;255:764-771.
- Bertrand P, Delhommais A, Alison D, et al. Immediate and delayed tolerance of iohexol and ioxaglate in lower limb phlebography: a double-blind comparative study in humans. Acad Radiol. 1995;2:683-686.
- Munechika H, Hiramatsu Y, Kudo S, et al. A prospective survey of delayed adverse reactions to iohexol in urography and computed tomography. Eur Radiol. 2003;13:185-194.
- Guéant-Rodriguez RM, Romano A, Barbaud A, et al. Hypersensitivity reactions to iodinated contrast media. Curr Pharm Des. 2006;12:3359-3372.
- H
uang SW. Seafood and iodine: an analysis of a medical myth. Allergy Asthma Proc. 2005;26:468-469. - B
aig M, Farag A, Sajid J, et al. Shellfish allergy and relation to iodinated contrast media: United Kingdom survey. World J Cardiol. 2014;6:107-111. - B
öhm I, Schild HH. A practical guide to diagnose lesser-known immediate and delayed contrast media-induced adverse cutaneous reactions. Eur Radiol. 2006;16:1570-1579. - Ose K, Doue T, Zen K, et al. “Gadolinium” as an alternative to iodinated contrast media for X-ray angiography in patients with severe allergy. Circ J. 2005;69:507-509.
- Ji
ngu A, Fukuda J, Taketomi-Takahashi A, et al. Breakthrough reactions of iodinated and gadolinium contrast media after oral steroid premedication protocol. BMC Med Imaging. 2014;14:34. - Ro
mano A, Artesani MC, Andriolo M, et al. Effective prophylactic protocol in delayed hypersensitivity to contrast media: report of a case involving lymphocyte transformation studies with different compounds. Radiology. 2002;225:466-470. - He
bert AA, Bogle MA. Intravenous immunoglobulin prophylaxis for recurrent Stevens-Johnson syndrome. J Am Acad Dermatol. 2004;50:286-288. - Ha
sdenteufel F, Waton J, Cordebar V, et al. Delayed hypersensitivity reactions caused by iodixanol: an assessment of cross-reactivity in 22 patients. J Allergy Clin Immunol. 2011;128:1356-1357.
- Cohan RH, Davenport MS, Dillman JR, et al; ACR Committee on Drugs and Contrast Media. ACR Manual on Contrast Media. 9th ed. Reston, VA: American College of Radiology; 2013.
- Brockow K, Romano A, Aberer W, et al; European Network of Drug Allergy and the EAACI Interest Group on Drug Hypersensitivity. Skin testing in patients with hypersensitivity reactions to iodinated contrast media—a European multicenter study. Allergy. 2009;64:234-241.
Hosoya T, Yamaguchi K, Akutsu T, et al. Delayed adverse reactions to iodinated contrast media and their risk factors. Radiat Med. 2000;18:39-45. - Rydberg J, Charles J, Aspelin P. Frequency of late allergy-like adverse reactions following injection of intravascular non-ionic contrast media: a retrospective study comparing a non-ionic monomeric contrast medium with a non-ionic dimeric contrast medium. Acta Radiol. 1998;39:219-222.
- Sutton AG, Finn P, Grech ED, et al. Early and late reactions after the use of iopamidol 340, ioxaglate 320, and iodixanol 320 in cardiac catheterization. Am Heart J. 2001;141:677-683.
- Sutton AG, Finn P, Campbell PG, et al. Early and late reactions following the use of iopamidol 340, iomeprol 350 and iodixanol 320 in cardiac catheterization. J Invasive Cardiol. 2003;15:133-138.
- Brown M, Yowler C, Brandt C. Recurrent toxic epidermal necrolysis secondary to iopromide contrast. J Burn Care Res. 2013;34:E53-E56.
- Rosado A, Canto G, Veleiro B, et al. Toxic epidermal necrolysis after repeated injections of iohexol. AJR Am J Roentgenol. 2001;176:262-263.
- Peterson A, Katzberg RW, Fung MA, et al. Acute generalized exanthematous pustulosis as a delayed dermatotoxic reaction to IV-administered nonionic contrast media. AJR Am J Roentgenol. 2006;187:W198-W201.
- Good AE, Novak E, Sonda LP III. Fixed eruption and fever after urography. South Med J. 1980;73:948-949.
- Benson PM, Giblin WJ, Douglas DM. Transient, nonpigmenting fixed drug eruption caused by radiopaque contrast media. J Am Acad Dermatol. 1990;23(2, pt 2):379-381.
- Torres MJ, Gomez F, Doña I, et al. Diagnostic evaluation of patients with nonimmediate cutaneous hypersensitivity reactions to iodinated contrast media. Allergy. 2012;67:929-935.
- Scherer K, Harr T, Bach S, et al. The role of iodine in hypersensitivity reactions to radio contrast media. Clin Exp Allergy. 2010;40:468-475.
- Reynolds NJ, Wallington TB, Burton JL. Hydralazine predisposes to acute cutaneous vasculitis following urography with iopamidol. Br J Dermatol. 1993;129:82-85.
- Belhadjali H, Bouzgarrou L, Youssef M, et al. DRESS syndrome induced by sodium meglumine ioxitalamate. Allergy. 2008;63:786-787.
- Baldwin BT, Lien MH, Khan H, et al. Case of fatal toxic epidermal necrolysis due to cardiac catheterization dye. J Drugs Dermatol. 2010;9:837-840.
- Schmidt BJ, Foley WD, Bohorfoush AG. Toxic epidermal necrolysis related to oral administration of diluted diatrizoate meglumine and diatrizoate sodium. AJR Am J Roentgenol. 1998;171:1215-1216.
- Young AL, Grossman ME. Acute iododerma secondary to iodinated contrast media. Br J Dermatol. 2014;170:1377-1379.
- Choyke PL, Miller DL, Lotze MT, et al. Delayed reactions to contrast media after interleukin-2 immunotherapy. Radiology. 1992;183:111-114.
- Loh S, Bagheri S, Katzberg RW, et al. Delayed adverse reaction to contrast-enhanced CT: a prospective single-center study comparison to control group without enhancement. Radiology. 2010;255:764-771.
- Bertrand P, Delhommais A, Alison D, et al. Immediate and delayed tolerance of iohexol and ioxaglate in lower limb phlebography: a double-blind comparative study in humans. Acad Radiol. 1995;2:683-686.
- Munechika H, Hiramatsu Y, Kudo S, et al. A prospective survey of delayed adverse reactions to iohexol in urography and computed tomography. Eur Radiol. 2003;13:185-194.
- Guéant-Rodriguez RM, Romano A, Barbaud A, et al. Hypersensitivity reactions to iodinated contrast media. Curr Pharm Des. 2006;12:3359-3372.
- H
uang SW. Seafood and iodine: an analysis of a medical myth. Allergy Asthma Proc. 2005;26:468-469. - B
aig M, Farag A, Sajid J, et al. Shellfish allergy and relation to iodinated contrast media: United Kingdom survey. World J Cardiol. 2014;6:107-111. - B
öhm I, Schild HH. A practical guide to diagnose lesser-known immediate and delayed contrast media-induced adverse cutaneous reactions. Eur Radiol. 2006;16:1570-1579. - Ose K, Doue T, Zen K, et al. “Gadolinium” as an alternative to iodinated contrast media for X-ray angiography in patients with severe allergy. Circ J. 2005;69:507-509.
- Ji
ngu A, Fukuda J, Taketomi-Takahashi A, et al. Breakthrough reactions of iodinated and gadolinium contrast media after oral steroid premedication protocol. BMC Med Imaging. 2014;14:34. - Ro
mano A, Artesani MC, Andriolo M, et al. Effective prophylactic protocol in delayed hypersensitivity to contrast media: report of a case involving lymphocyte transformation studies with different compounds. Radiology. 2002;225:466-470. - He
bert AA, Bogle MA. Intravenous immunoglobulin prophylaxis for recurrent Stevens-Johnson syndrome. J Am Acad Dermatol. 2004;50:286-288. - Ha
sdenteufel F, Waton J, Cordebar V, et al. Delayed hypersensitivity reactions caused by iodixanol: an assessment of cross-reactivity in 22 patients. J Allergy Clin Immunol. 2011;128:1356-1357.
Practice Points
- Delayed cutaneous reactions to iodinated contrast (IC) are common, but patients frequently are misdiagnosed and inadvertently readministered the offending agent.
- The most common IC-induced delayed reactions are self-limited exanthematous eruptions that develop within 1 week of exposure.
- Risk factors for delayed reactions to IC include atopy, contrast exposure during high pollen season, use of the agent iodixanol, a history of other cutaneous drug eruptions, elevated serum creatinine levels, and treatment with recombinant interleukin 2.
- Dermatologists can help prevent recurrent reactions in patients who require repeated exposure to IC by recommending gadolinium-based contrast agents and/or premedication.
Unusual Presentation of Ectopic Extramammary Paget Disease
Extramammary Paget disease (EMPD) is a malignant epithelial tumor that most commonly affects the anogenital region and less frequently arises in the axillae. Most cases occur in locations where apocrine glands predominate.1 Few cases of EMPD arising in nonapocrine-bearing regions, or ectopic EMPD, have been reported.2 We describe a case of primary ectopic EMPD with an infiltrative growth pattern arising on the back of a 67-year-old Thai man.
Case Report
A 67-year-old Thai man presented to the dermatology clinic for evaluation of a persistent rash on the right lower back of approximately 30 years’ duration. He reported that the eruption had started out as a small coin-shaped area but had slowly increased in size. Over the last 2 years, the area had grown more rapidly and became pruritic. His medical history was remarkable for hypertension treated with losartan, but he was otherwise healthy. He had no history of cancer or gastrointestinal tract or genitourinary symptoms, and he had no recent fever, weight loss, or night sweats.
On physical examination a well-demarcated, asymmetric, erythematous to brown plaque was noted on the right lower back. The plaque was surfaced by scale and contained a central hyperkeratotic papule (Figure 1). The skin examination was otherwise unremarkable. The patient had no lymphadenopathy.
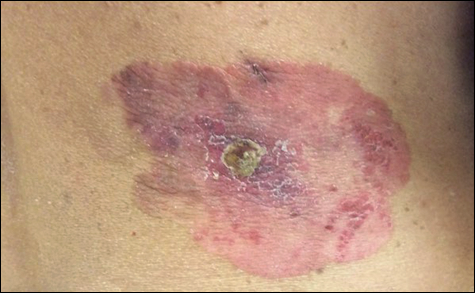
Two punch biopsies were performed. On low power, acanthosis and hyperkeratosis of the epidermis were noted. The epidermis contained a proliferation of large (tumor) cells with pleomorphic nuclei, prominent nucleoli, and abundant pale to clear cytoplasm. The cells were present singularly as well as in clusters and were most prominent along the basal layer but many were also seen extending to more superficial levels of the epidermis (Figure 2A). In one biopsy, the tumor cells were found in the dermis with an infiltrative growth pattern (Figure 2B). Immunohistochemistry (IHC) studies for cytokeratin 7 (Figure 3A and 3B) and carcinoembryonic antigen (Figure 3C) labeled the tumor cells. An IHC study for gross cystic disease fluid protein 15 labeled some of the tumor cells. Immunohistochemistry studies for S-100, human melanoma black 45 (HMB-45), p16, and renal cell carcinoma did not label the tumor cells. An IHC study for MIB-1 labeled many of the tumor cells, indicating a notably increased mitotic index. The patient was diagnosed with ectopic EMPD. He underwent an endoscopy, colonoscopy, and cystoscopy, all of which were normal.
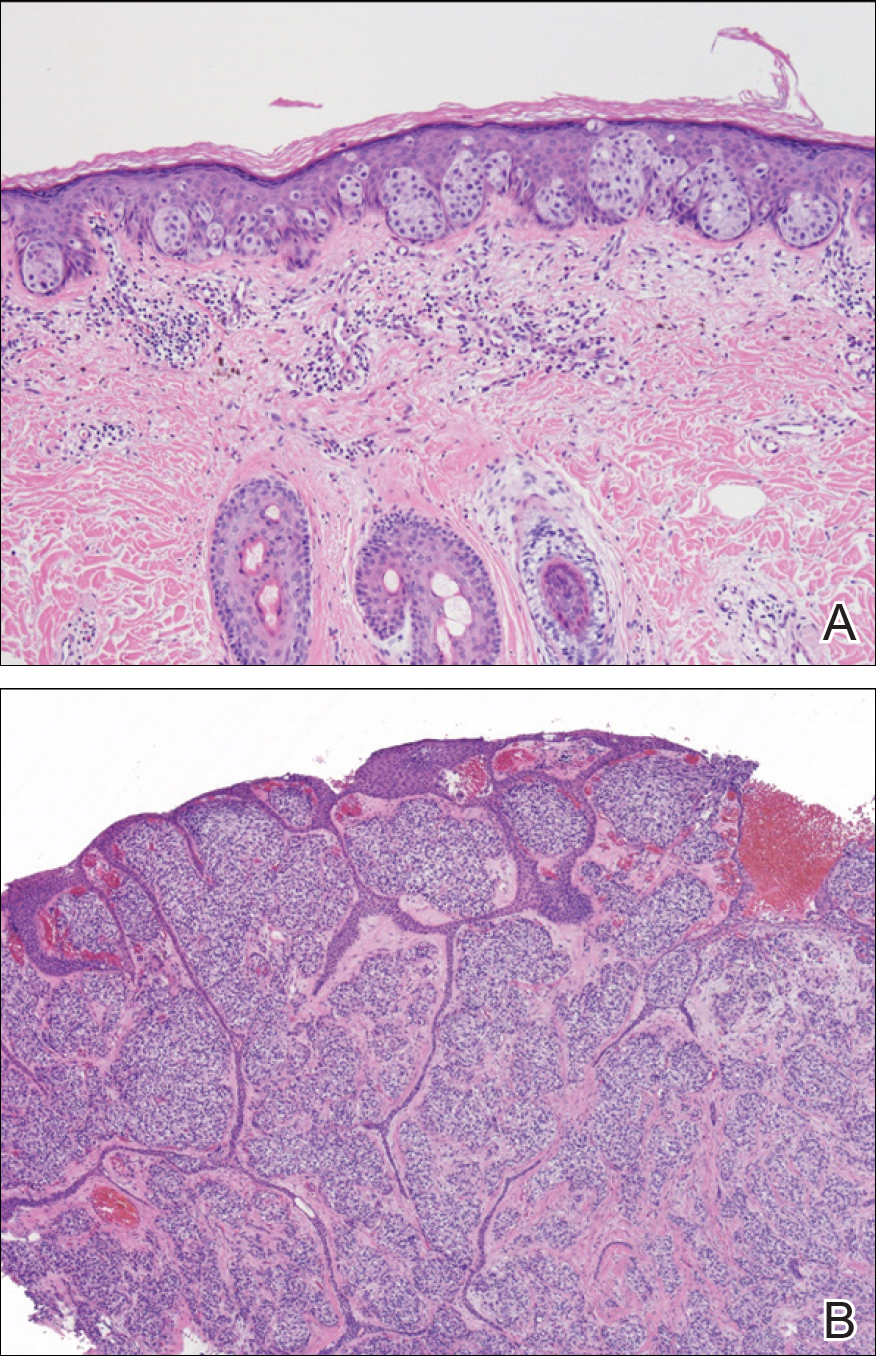
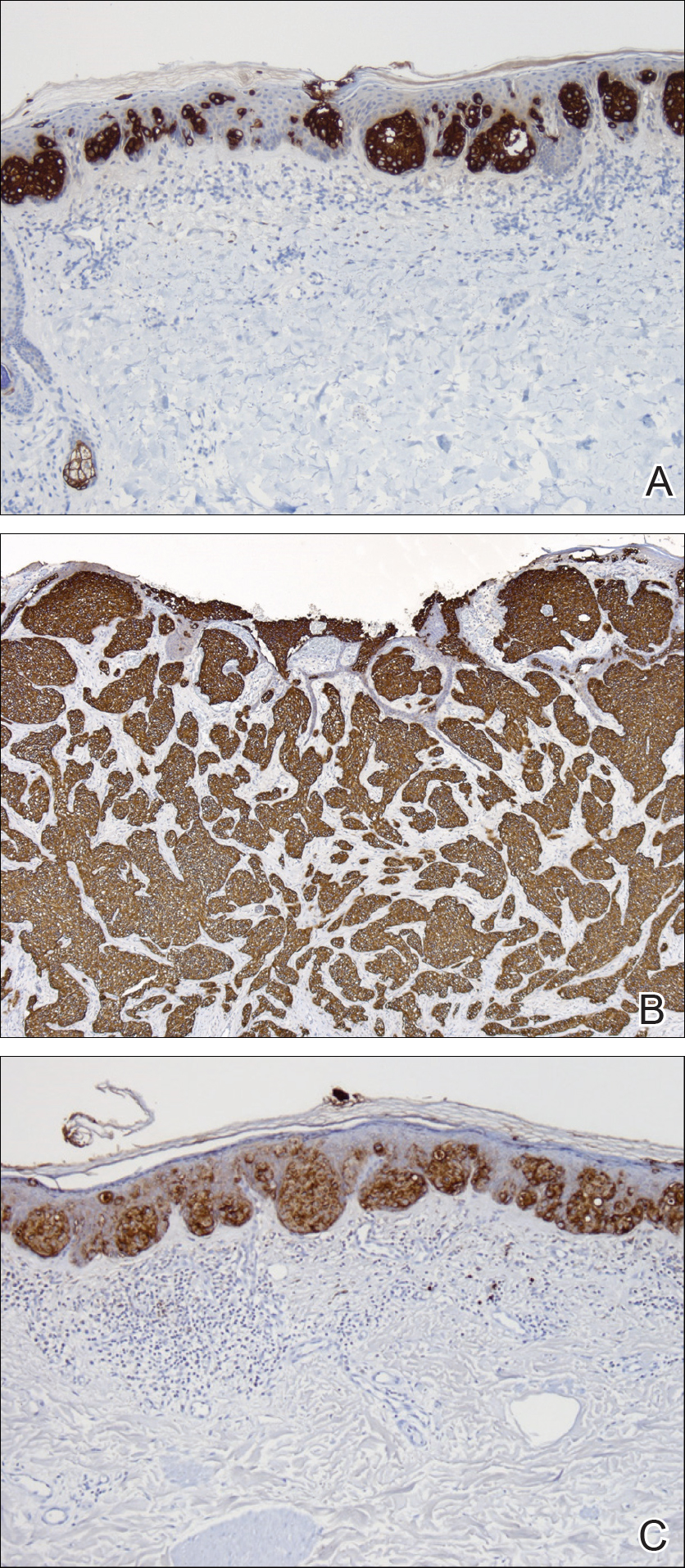
Comment
Extramammary Paget disease is a malignant tumor typically found in apocrine-rich areas of the skin, particularly the anogenital skin. It is categorized as primary or secondary EMPD. Primary EMPD arises as an intraepithelial adenocarcinoma, with the Toker cell as the cell of origin.3 Secondary EMPD represents a cutaneous extension of an underlying malignancy (eg, colorectal, urothelial, prostatic, gynecologic).4
Ectopic EMPD arises in nonapocrine-bearing areas, specifically the nongerminative milk line. A review of the literature using Google Scholar and the search term ectopic extramammary Paget disease showed that there have been at least 30 cases of ectopic EMPD reported. Older men are more commonly affected, with a mean age at diagnosis of approximately 68 years. Although the tumor is most commonly seen on the trunk, cases on the head, arms, and legs have been reported.5
This tumor is most frequently seen in Asian individuals, as in our patient, with a ratio of approximately 3:1.5 Interestingly, triple or quadruple EMPD was reported in 68 Japanese patients but rarely has been reported outside of Japan.6 It is thought that some germinative apocrine-differentiating cells might preferentially exist on the trunk of Asians, leading to an increased incidence of EMPD in this population5; however, the exact reason for this racial preference is not completely understood, and more studies are needed to investigate this association.
Diagnosis of ectopic EMPD is made histologically. Tumor cells have abundant pale cytoplasm and large pleomorphic nuclei with prominent nucleoli. The cells are arranged in small groups or singly within the basal regions of the epidermis. In longstanding lesions, the entire thickness of the epidermis may be involved. Uncommonly, the tumor cells may invade the dermis, such as in our patient. On immunohistochemistry, the tumor cells stain positive for carcinoembryonic antigen, epithelial membrane antigen, and low-molecular-weight cytokeratins (eg, cytokeratin 7). Many of the tumor cells also express gross cystic disease fluid protein 15, which helps exclude cutaneous invasion of secondary EMPD.7-9 Cases of primary cutaneous apocrine carcinoma can have similar histologic and immunohistochemical findings to invasive EMPD, which further supports the possible apocrine derivation of Paget disease. In our patient, we considered the diagnosis of primary cutaneous apocrine adenocarcinoma with epidermotropism; however, we favored the diagnosis of ectopic EMPD with dermal invasion given the extensive epidermal-only involvement seen in one of the biopsies, which would be unusual for primary cutaneous apocrine adenocarcinoma.
Our patient had no identified underlying malignancy upon further workup; however, many cases of EMPD have been associated with an underlying malignancy.9-15 Several authors have reported a range of underlying malignancies associated with EMPD, with the incidence ranging from 11% to 45%.9-15 The location of the underlying internal malignancy appears to be closely related to the location of the EMPD.11 It is recommended that a thorough workup for internal malignancies be performed, including a full skin examination, lymph node examination, colonoscopy, cystoscopy, and gynecologic/prostate examination, among others.
No known differences in the prognosis or associated underlying malignancies between ectopic and ordinary EMPD have been reported; however, it has been noted that EMPD with invasion into the dermis does correlate with a more aggressive course and worse prognosis.8 Treatment includes surgical removal by Mohs micrographic surgery or wide local excision. Long-term follow-up is required since recurrences can be frequent.11-15
- Mazoujian G, Pinkus GS, Haagensen DE Jr. Extramammary Paget’s disease—evidence for an apocrine origin: an immunoperoxidase study of gross cystic disease fluid protein-15, carcinoembryonic antigen, and keratin proteins. Am J Surg Pathol. 1984;8:43-50.
- Saida T, Iwata M. “Ectopic” extramammary Paget’s disease affecting the lower anterior aspect of the chest. J Am Acad Dermatol. 1987;17(5, pt 2):910-913.
- Willman JH, Golitz LE, Fitzpatrick JE. Vulvar clear cells of Toker: precursors of extramammary Paget’s disease. Am J Dermatopathol. 2005;27:185-188.
- Lloyd J, Flanagan AM. Mammary and extramammary Paget’s disease.J Clin Pathol. 2000;53:742-749.
- Sawada Y, Bito T, Kabashima R, et al. Ectopic extramammary Paget’s disease: case report and literature review. Acta Derm Venereol. 2010;90:502-505.
- Abe S, Kabashima K, Nishio D, et al. Quadruple extramammary Paget’s disease. Acta Derm Venereol. 2007;87:80-81.
- Kanitakis J. Mammary and extramammary Paget’s disease. J Eur Acad Dermatol Venereol. 2007;21:581-590.
- Goldblum JR, Hart WR. Vulvar Paget’s disease: a clinicopathologic and immunohistochemical study of 19 cases. Am J Surg Pathol. 1997;21:1178-1187.
- Goldblum JR, Hart WR. Perianal Paget’s disease: a histologic and immunohistochemical study of 11 cases with and without associated rectal adenocarcinoma. Am J Surg Pathol. 1998;22:170-179.
- Shepherd V, Davidson EJ, Davies‐Humphreys J. Extramammary Paget’s disease. BJOG. 2005;112:273-279.
- Chanda JJ. Extramammary Paget’s disease: prognosis and relationship to internal malignancy. J Am Acad Dermatol. 1985;13:1009-1014.
- Besa P, Rich TA, Delclos L, et al. Extramammary Paget’s disease of the perineal skin: role of radiotherapy. Int J Radiat Oncol Biol Phys. 1992;24:73-78.
- Fanning J, Lambert HC, Hale TM, et al. Paget’s disease of the vulva: prevalence of associated vulvar adenocarcinoma, invasive Paget’s disease, and recurrence after surgical excision. Am J Obstet Gynecol. 1999;180:24-27.
- Parker LP, Parker JR, Bodurka-Bevers D, et al. Paget’s disease of the vulva: pathology, pattern of involvement, and prognosis. Gynecol Oncol. 2000;77:183-189.
- Marchesa P, Fazio VW, Oliart S, et al. Long-term outcome of patients with perianal Paget’s disease. Ann Surg Oncol. 1997;4:475-480.
Extramammary Paget disease (EMPD) is a malignant epithelial tumor that most commonly affects the anogenital region and less frequently arises in the axillae. Most cases occur in locations where apocrine glands predominate.1 Few cases of EMPD arising in nonapocrine-bearing regions, or ectopic EMPD, have been reported.2 We describe a case of primary ectopic EMPD with an infiltrative growth pattern arising on the back of a 67-year-old Thai man.
Case Report
A 67-year-old Thai man presented to the dermatology clinic for evaluation of a persistent rash on the right lower back of approximately 30 years’ duration. He reported that the eruption had started out as a small coin-shaped area but had slowly increased in size. Over the last 2 years, the area had grown more rapidly and became pruritic. His medical history was remarkable for hypertension treated with losartan, but he was otherwise healthy. He had no history of cancer or gastrointestinal tract or genitourinary symptoms, and he had no recent fever, weight loss, or night sweats.
On physical examination a well-demarcated, asymmetric, erythematous to brown plaque was noted on the right lower back. The plaque was surfaced by scale and contained a central hyperkeratotic papule (Figure 1). The skin examination was otherwise unremarkable. The patient had no lymphadenopathy.
Two punch biopsies were performed. On low power, acanthosis and hyperkeratosis of the epidermis were noted. The epidermis contained a proliferation of large (tumor) cells with pleomorphic nuclei, prominent nucleoli, and abundant pale to clear cytoplasm. The cells were present singularly as well as in clusters and were most prominent along the basal layer but many were also seen extending to more superficial levels of the epidermis (Figure 2A). In one biopsy, the tumor cells were found in the dermis with an infiltrative growth pattern (Figure 2B). Immunohistochemistry (IHC) studies for cytokeratin 7 (Figure 3A and 3B) and carcinoembryonic antigen (Figure 3C) labeled the tumor cells. An IHC study for gross cystic disease fluid protein 15 labeled some of the tumor cells. Immunohistochemistry studies for S-100, human melanoma black 45 (HMB-45), p16, and renal cell carcinoma did not label the tumor cells. An IHC study for MIB-1 labeled many of the tumor cells, indicating a notably increased mitotic index. The patient was diagnosed with ectopic EMPD. He underwent an endoscopy, colonoscopy, and cystoscopy, all of which were normal.
Comment
Extramammary Paget disease is a malignant tumor typically found in apocrine-rich areas of the skin, particularly the anogenital skin. It is categorized as primary or secondary EMPD. Primary EMPD arises as an intraepithelial adenocarcinoma, with the Toker cell as the cell of origin.3 Secondary EMPD represents a cutaneous extension of an underlying malignancy (eg, colorectal, urothelial, prostatic, gynecologic).4
Ectopic EMPD arises in nonapocrine-bearing areas, specifically the nongerminative milk line. A review of the literature using Google Scholar and the search term ectopic extramammary Paget disease showed that there have been at least 30 cases of ectopic EMPD reported. Older men are more commonly affected, with a mean age at diagnosis of approximately 68 years. Although the tumor is most commonly seen on the trunk, cases on the head, arms, and legs have been reported.5
This tumor is most frequently seen in Asian individuals, as in our patient, with a ratio of approximately 3:1.5 Interestingly, triple or quadruple EMPD was reported in 68 Japanese patients but rarely has been reported outside of Japan.6 It is thought that some germinative apocrine-differentiating cells might preferentially exist on the trunk of Asians, leading to an increased incidence of EMPD in this population5; however, the exact reason for this racial preference is not completely understood, and more studies are needed to investigate this association.
Diagnosis of ectopic EMPD is made histologically. Tumor cells have abundant pale cytoplasm and large pleomorphic nuclei with prominent nucleoli. The cells are arranged in small groups or singly within the basal regions of the epidermis. In longstanding lesions, the entire thickness of the epidermis may be involved. Uncommonly, the tumor cells may invade the dermis, such as in our patient. On immunohistochemistry, the tumor cells stain positive for carcinoembryonic antigen, epithelial membrane antigen, and low-molecular-weight cytokeratins (eg, cytokeratin 7). Many of the tumor cells also express gross cystic disease fluid protein 15, which helps exclude cutaneous invasion of secondary EMPD.7-9 Cases of primary cutaneous apocrine carcinoma can have similar histologic and immunohistochemical findings to invasive EMPD, which further supports the possible apocrine derivation of Paget disease. In our patient, we considered the diagnosis of primary cutaneous apocrine adenocarcinoma with epidermotropism; however, we favored the diagnosis of ectopic EMPD with dermal invasion given the extensive epidermal-only involvement seen in one of the biopsies, which would be unusual for primary cutaneous apocrine adenocarcinoma.
Our patient had no identified underlying malignancy upon further workup; however, many cases of EMPD have been associated with an underlying malignancy.9-15 Several authors have reported a range of underlying malignancies associated with EMPD, with the incidence ranging from 11% to 45%.9-15 The location of the underlying internal malignancy appears to be closely related to the location of the EMPD.11 It is recommended that a thorough workup for internal malignancies be performed, including a full skin examination, lymph node examination, colonoscopy, cystoscopy, and gynecologic/prostate examination, among others.
No known differences in the prognosis or associated underlying malignancies between ectopic and ordinary EMPD have been reported; however, it has been noted that EMPD with invasion into the dermis does correlate with a more aggressive course and worse prognosis.8 Treatment includes surgical removal by Mohs micrographic surgery or wide local excision. Long-term follow-up is required since recurrences can be frequent.11-15
Extramammary Paget disease (EMPD) is a malignant epithelial tumor that most commonly affects the anogenital region and less frequently arises in the axillae. Most cases occur in locations where apocrine glands predominate.1 Few cases of EMPD arising in nonapocrine-bearing regions, or ectopic EMPD, have been reported.2 We describe a case of primary ectopic EMPD with an infiltrative growth pattern arising on the back of a 67-year-old Thai man.
Case Report
A 67-year-old Thai man presented to the dermatology clinic for evaluation of a persistent rash on the right lower back of approximately 30 years’ duration. He reported that the eruption had started out as a small coin-shaped area but had slowly increased in size. Over the last 2 years, the area had grown more rapidly and became pruritic. His medical history was remarkable for hypertension treated with losartan, but he was otherwise healthy. He had no history of cancer or gastrointestinal tract or genitourinary symptoms, and he had no recent fever, weight loss, or night sweats.
On physical examination a well-demarcated, asymmetric, erythematous to brown plaque was noted on the right lower back. The plaque was surfaced by scale and contained a central hyperkeratotic papule (Figure 1). The skin examination was otherwise unremarkable. The patient had no lymphadenopathy.
Two punch biopsies were performed. On low power, acanthosis and hyperkeratosis of the epidermis were noted. The epidermis contained a proliferation of large (tumor) cells with pleomorphic nuclei, prominent nucleoli, and abundant pale to clear cytoplasm. The cells were present singularly as well as in clusters and were most prominent along the basal layer but many were also seen extending to more superficial levels of the epidermis (Figure 2A). In one biopsy, the tumor cells were found in the dermis with an infiltrative growth pattern (Figure 2B). Immunohistochemistry (IHC) studies for cytokeratin 7 (Figure 3A and 3B) and carcinoembryonic antigen (Figure 3C) labeled the tumor cells. An IHC study for gross cystic disease fluid protein 15 labeled some of the tumor cells. Immunohistochemistry studies for S-100, human melanoma black 45 (HMB-45), p16, and renal cell carcinoma did not label the tumor cells. An IHC study for MIB-1 labeled many of the tumor cells, indicating a notably increased mitotic index. The patient was diagnosed with ectopic EMPD. He underwent an endoscopy, colonoscopy, and cystoscopy, all of which were normal.
Comment
Extramammary Paget disease is a malignant tumor typically found in apocrine-rich areas of the skin, particularly the anogenital skin. It is categorized as primary or secondary EMPD. Primary EMPD arises as an intraepithelial adenocarcinoma, with the Toker cell as the cell of origin.3 Secondary EMPD represents a cutaneous extension of an underlying malignancy (eg, colorectal, urothelial, prostatic, gynecologic).4
Ectopic EMPD arises in nonapocrine-bearing areas, specifically the nongerminative milk line. A review of the literature using Google Scholar and the search term ectopic extramammary Paget disease showed that there have been at least 30 cases of ectopic EMPD reported. Older men are more commonly affected, with a mean age at diagnosis of approximately 68 years. Although the tumor is most commonly seen on the trunk, cases on the head, arms, and legs have been reported.5
This tumor is most frequently seen in Asian individuals, as in our patient, with a ratio of approximately 3:1.5 Interestingly, triple or quadruple EMPD was reported in 68 Japanese patients but rarely has been reported outside of Japan.6 It is thought that some germinative apocrine-differentiating cells might preferentially exist on the trunk of Asians, leading to an increased incidence of EMPD in this population5; however, the exact reason for this racial preference is not completely understood, and more studies are needed to investigate this association.
Diagnosis of ectopic EMPD is made histologically. Tumor cells have abundant pale cytoplasm and large pleomorphic nuclei with prominent nucleoli. The cells are arranged in small groups or singly within the basal regions of the epidermis. In longstanding lesions, the entire thickness of the epidermis may be involved. Uncommonly, the tumor cells may invade the dermis, such as in our patient. On immunohistochemistry, the tumor cells stain positive for carcinoembryonic antigen, epithelial membrane antigen, and low-molecular-weight cytokeratins (eg, cytokeratin 7). Many of the tumor cells also express gross cystic disease fluid protein 15, which helps exclude cutaneous invasion of secondary EMPD.7-9 Cases of primary cutaneous apocrine carcinoma can have similar histologic and immunohistochemical findings to invasive EMPD, which further supports the possible apocrine derivation of Paget disease. In our patient, we considered the diagnosis of primary cutaneous apocrine adenocarcinoma with epidermotropism; however, we favored the diagnosis of ectopic EMPD with dermal invasion given the extensive epidermal-only involvement seen in one of the biopsies, which would be unusual for primary cutaneous apocrine adenocarcinoma.
Our patient had no identified underlying malignancy upon further workup; however, many cases of EMPD have been associated with an underlying malignancy.9-15 Several authors have reported a range of underlying malignancies associated with EMPD, with the incidence ranging from 11% to 45%.9-15 The location of the underlying internal malignancy appears to be closely related to the location of the EMPD.11 It is recommended that a thorough workup for internal malignancies be performed, including a full skin examination, lymph node examination, colonoscopy, cystoscopy, and gynecologic/prostate examination, among others.
No known differences in the prognosis or associated underlying malignancies between ectopic and ordinary EMPD have been reported; however, it has been noted that EMPD with invasion into the dermis does correlate with a more aggressive course and worse prognosis.8 Treatment includes surgical removal by Mohs micrographic surgery or wide local excision. Long-term follow-up is required since recurrences can be frequent.11-15
- Mazoujian G, Pinkus GS, Haagensen DE Jr. Extramammary Paget’s disease—evidence for an apocrine origin: an immunoperoxidase study of gross cystic disease fluid protein-15, carcinoembryonic antigen, and keratin proteins. Am J Surg Pathol. 1984;8:43-50.
- Saida T, Iwata M. “Ectopic” extramammary Paget’s disease affecting the lower anterior aspect of the chest. J Am Acad Dermatol. 1987;17(5, pt 2):910-913.
- Willman JH, Golitz LE, Fitzpatrick JE. Vulvar clear cells of Toker: precursors of extramammary Paget’s disease. Am J Dermatopathol. 2005;27:185-188.
- Lloyd J, Flanagan AM. Mammary and extramammary Paget’s disease.J Clin Pathol. 2000;53:742-749.
- Sawada Y, Bito T, Kabashima R, et al. Ectopic extramammary Paget’s disease: case report and literature review. Acta Derm Venereol. 2010;90:502-505.
- Abe S, Kabashima K, Nishio D, et al. Quadruple extramammary Paget’s disease. Acta Derm Venereol. 2007;87:80-81.
- Kanitakis J. Mammary and extramammary Paget’s disease. J Eur Acad Dermatol Venereol. 2007;21:581-590.
- Goldblum JR, Hart WR. Vulvar Paget’s disease: a clinicopathologic and immunohistochemical study of 19 cases. Am J Surg Pathol. 1997;21:1178-1187.
- Goldblum JR, Hart WR. Perianal Paget’s disease: a histologic and immunohistochemical study of 11 cases with and without associated rectal adenocarcinoma. Am J Surg Pathol. 1998;22:170-179.
- Shepherd V, Davidson EJ, Davies‐Humphreys J. Extramammary Paget’s disease. BJOG. 2005;112:273-279.
- Chanda JJ. Extramammary Paget’s disease: prognosis and relationship to internal malignancy. J Am Acad Dermatol. 1985;13:1009-1014.
- Besa P, Rich TA, Delclos L, et al. Extramammary Paget’s disease of the perineal skin: role of radiotherapy. Int J Radiat Oncol Biol Phys. 1992;24:73-78.
- Fanning J, Lambert HC, Hale TM, et al. Paget’s disease of the vulva: prevalence of associated vulvar adenocarcinoma, invasive Paget’s disease, and recurrence after surgical excision. Am J Obstet Gynecol. 1999;180:24-27.
- Parker LP, Parker JR, Bodurka-Bevers D, et al. Paget’s disease of the vulva: pathology, pattern of involvement, and prognosis. Gynecol Oncol. 2000;77:183-189.
- Marchesa P, Fazio VW, Oliart S, et al. Long-term outcome of patients with perianal Paget’s disease. Ann Surg Oncol. 1997;4:475-480.
- Mazoujian G, Pinkus GS, Haagensen DE Jr. Extramammary Paget’s disease—evidence for an apocrine origin: an immunoperoxidase study of gross cystic disease fluid protein-15, carcinoembryonic antigen, and keratin proteins. Am J Surg Pathol. 1984;8:43-50.
- Saida T, Iwata M. “Ectopic” extramammary Paget’s disease affecting the lower anterior aspect of the chest. J Am Acad Dermatol. 1987;17(5, pt 2):910-913.
- Willman JH, Golitz LE, Fitzpatrick JE. Vulvar clear cells of Toker: precursors of extramammary Paget’s disease. Am J Dermatopathol. 2005;27:185-188.
- Lloyd J, Flanagan AM. Mammary and extramammary Paget’s disease.J Clin Pathol. 2000;53:742-749.
- Sawada Y, Bito T, Kabashima R, et al. Ectopic extramammary Paget’s disease: case report and literature review. Acta Derm Venereol. 2010;90:502-505.
- Abe S, Kabashima K, Nishio D, et al. Quadruple extramammary Paget’s disease. Acta Derm Venereol. 2007;87:80-81.
- Kanitakis J. Mammary and extramammary Paget’s disease. J Eur Acad Dermatol Venereol. 2007;21:581-590.
- Goldblum JR, Hart WR. Vulvar Paget’s disease: a clinicopathologic and immunohistochemical study of 19 cases. Am J Surg Pathol. 1997;21:1178-1187.
- Goldblum JR, Hart WR. Perianal Paget’s disease: a histologic and immunohistochemical study of 11 cases with and without associated rectal adenocarcinoma. Am J Surg Pathol. 1998;22:170-179.
- Shepherd V, Davidson EJ, Davies‐Humphreys J. Extramammary Paget’s disease. BJOG. 2005;112:273-279.
- Chanda JJ. Extramammary Paget’s disease: prognosis and relationship to internal malignancy. J Am Acad Dermatol. 1985;13:1009-1014.
- Besa P, Rich TA, Delclos L, et al. Extramammary Paget’s disease of the perineal skin: role of radiotherapy. Int J Radiat Oncol Biol Phys. 1992;24:73-78.
- Fanning J, Lambert HC, Hale TM, et al. Paget’s disease of the vulva: prevalence of associated vulvar adenocarcinoma, invasive Paget’s disease, and recurrence after surgical excision. Am J Obstet Gynecol. 1999;180:24-27.
- Parker LP, Parker JR, Bodurka-Bevers D, et al. Paget’s disease of the vulva: pathology, pattern of involvement, and prognosis. Gynecol Oncol. 2000;77:183-189.
- Marchesa P, Fazio VW, Oliart S, et al. Long-term outcome of patients with perianal Paget’s disease. Ann Surg Oncol. 1997;4:475-480.
Practice Points
- Ectopic extramammary Paget disease (EMPD) is a rare presentation of EMPD that is histologically identical to EMPD.
- Ectopic EMPD can be associated with underlying malignancy and therefore warrants a thorough workup.
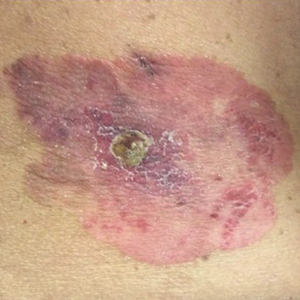
Atopic Dermatitis and Peanut Allergy Prevention: New Guidelines

Red-Brown Patches in the Groin
The Diagnosis: Erythrasma
Erythrasma usually involves intertriginous areas (eg, axillae, groin, inframammary area). Patients present with well-demarcated, minimally scaly, red-brown patches. The interdigital web space of the toes also can be involved with macerated white plaques, often with coexistent dermatophyte infection. Corynebacterium minutissimum, the bacteria responsible for erythrasma, produces coproporphyrin type III, which emits coral red fluorescence under Wood lamp examination.1 Bathing may result in removal of the porphyrin and result in a false-negative finding. Potassium hydroxide preparation of skin scrapings can show chains of bacilli. Biopsy appears relatively normal at low power but reveals compact orthokeratosis with coccobacilli and filamentous organisms in the superficial stratum corneum (quiz image). When not obvious on hematoxylin and eosin-stained sections, the organisms are Gram-positive and also are seen with periodic acid-Schiff (PAS) and methenamine silver stains. Unlike fungal hyphae, these organisms are thinner and nonrefractile. Inflammation typically is minimal. Due to the subtle histologic findings at low power, erythrasma is considered one of the invisible dermatoses.2 The differential diagnosis of these inconspicuous dermatoses that appear normal at first glance can be approached in a stepwise fashion starting in the stratum corneum, followed by the granular layer, basal layer, dermal papillae, dermal inflammatory cells, dermal connective tissue, and eccrine glands, and should consider each of the following diagnoses: candidiasis, dermatophytosis, ichthyosis vulgaris, vitiligo, macular amyloid, urticaria, telangiectasia macularis eruptiva perstans, connective tissue nevus, and argyria.2
Candidiasis, most commonly caused by Candida albicans, usually involves the oral cavity (eg, thrush, median rhomboid glossitis, angular cheilitis), intertriginous zones, nail fold (paronychia), genital areas (eg, vulvovaginitis, balanitis), and diaper area.3 The web space between the third and fourth fingers (erosio interdigitalis blastomycetica) can be involved in patients whose hands are frequently in water. Intertriginous candidiasis presents with bright red, sometimes erosive patches with satellite lesions. Spores and mycelia (filamentous forms) are noted on potassium hydroxide preparation of skin scrapings. Histologically, the epidermis often is acanthotic, mildly spongiotic, and contains groups of neutrophils in the superficial layers. The mnemonic device for diseases with clusters of neutrophils in the stratum corneum is PTICSS (psoriasis, tinea, impetigo, candida, seborrheic dermatitis, syphilis).2 Yeast, pseudohyphae, and even true hyphae can be seen in the stratum corneum with hematoxylin and eosin-stained sections and PAS. The filamentous forms tend to be vertically oriented in relation to the skin surface (Figure 1) compared to dermatophyte hyphae that tend to be parallel to the surface.2
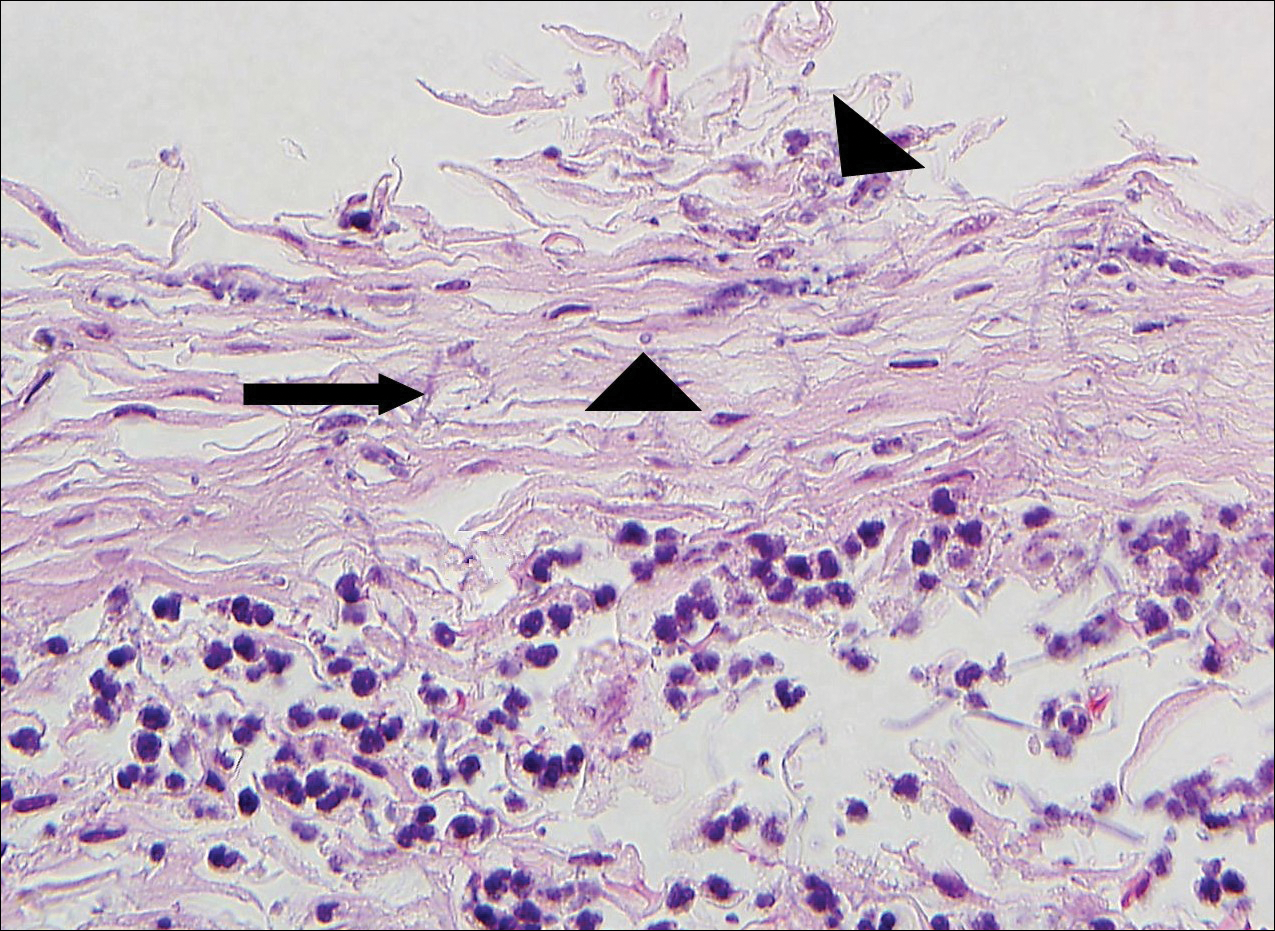
Pitted keratolysis is a superficial bacterial infection involving the soles of the feet. The classic clinical findings are shallow 1- to 2-mm pits in clusters that can coalesce on pressure-bearing areas. Hyperhidrosis, malodor, and maceration commonly are associated. Microscopic examination reveals clusters of small cocci and filamentous bacteria located in the dell or pit of a thick compact orthokeratotic stratum corneum of acral skin with no notable inflammatory infiltrate (Figure 2).2 Special stains such as Gram, methenamine silver, or PAS can assist in visualization of the organisms. Pitted keratolysis is caused by Dermatophilus congolensis and Kytococcus sedentarius (formerly Micrococcus sedentarius), which produce keratinolytic enzymes causing the defect in the stratum corneum.3
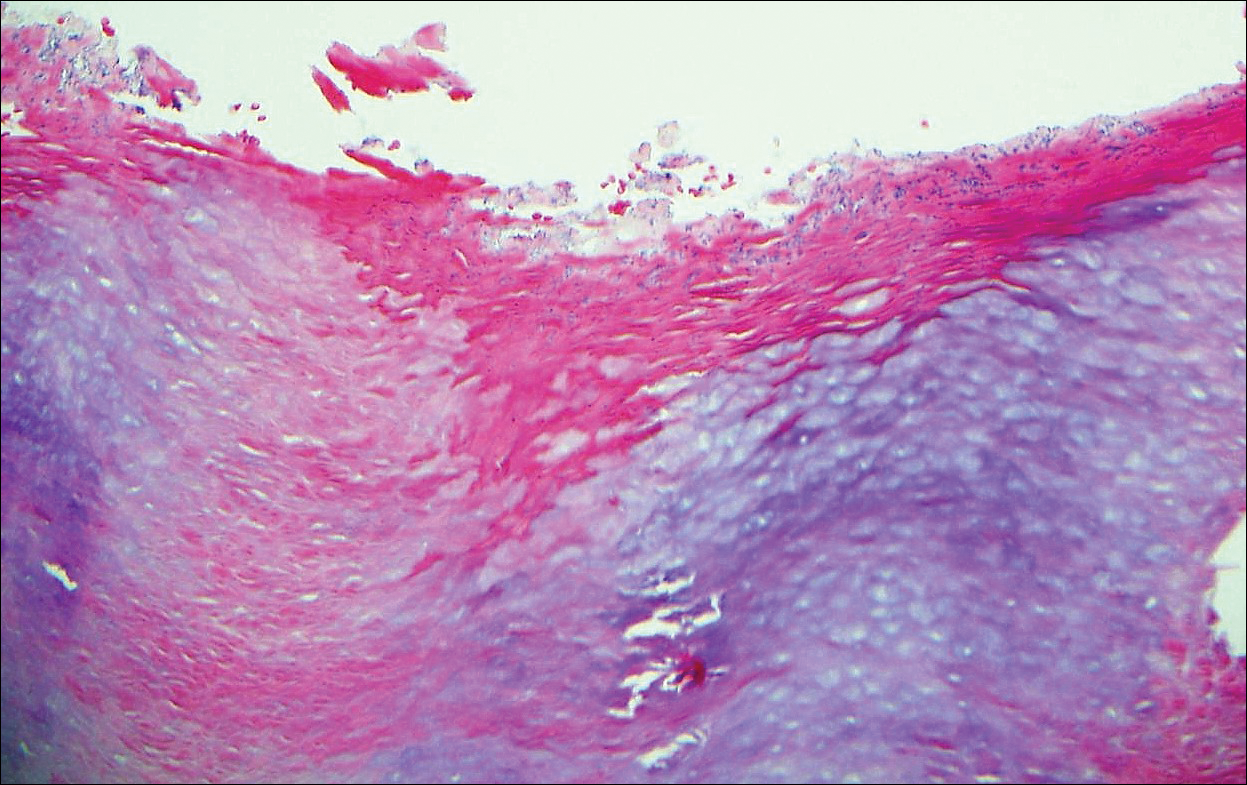
Tinea cruris, also known as jock itch and ringworm of the groin, presents with advancing pruritic, circinate, erythematous, scaling patches with central clearing on the inner thighs and crural folds. Similar to tinea pedis, Trichophyton rubrum is the most common dermatophyte to cause tinea cruris.4 Potassium hydroxide preparation of skin scrapings from the advancing border show fungal hyphae that cross the keratin cell borders. The histopathology of dermatophyte infections can be subtle and resemble normal skin before close inspection of the stratum corneum, which can show compact orthokeratosis, neutrophils, or "sandwich sign" where hyphae are sandwiched between an upper basket weave layer and a lower compact cornified layer (orthokeratotic or parakeratotic)(Figure 3).1 The presence of these patterns in the stratum corneum should result in performance of PAS to highlight obscure hyphae.
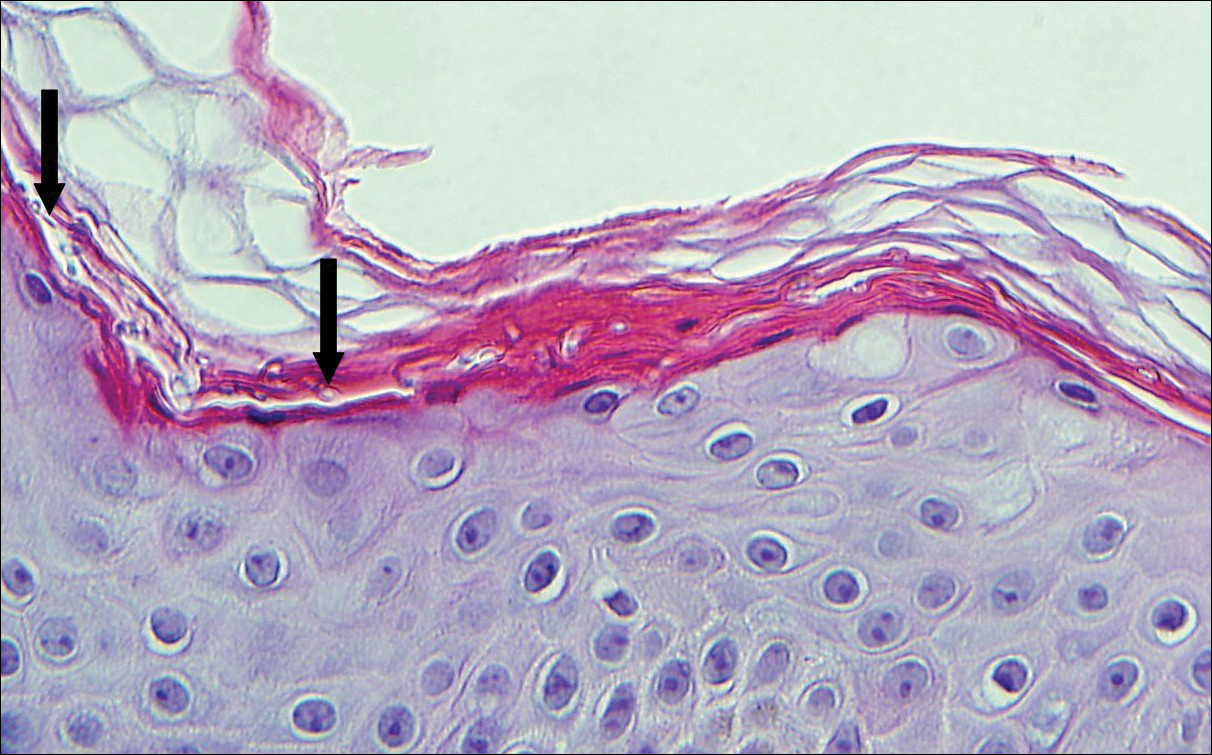
Tinea versicolor, also called pityriasis versicolor, usually presents with hypopigmented or less commonly hyperpigmented circular patches that coalesce on the upper trunk and shoulders. There is a fine fluffy scale that is most notable after scraping the skin for a potassium hydroxide preparation, which shows "spaghetti and meatballs" (hyphae and spores). Tinea versicolor typically is caused by the mycelial phase of the lipophilic yeast Malassezia globosae.3 Histologically, there are yeast and short septate hyphae scattered in a loose basket weave hyperkeratotic stratum corneum with minimal or no inflammation (Figure 4). On occasion, PAS is required for identification.
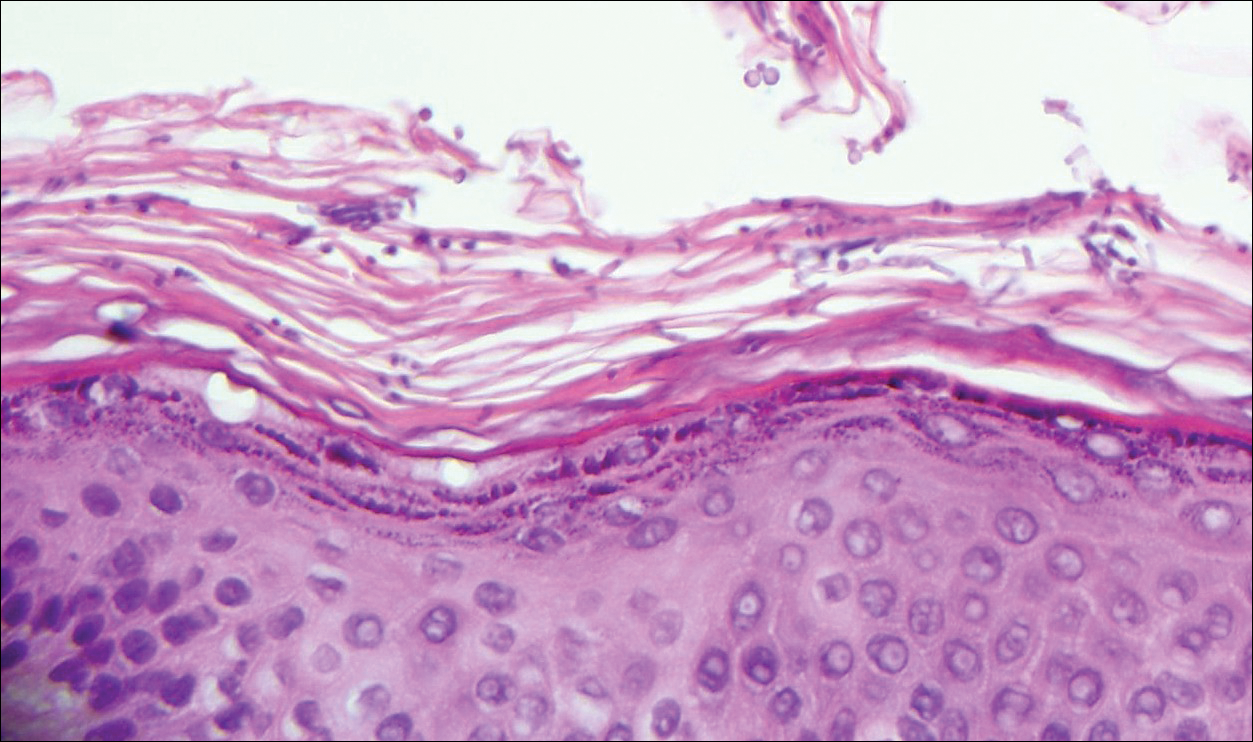
- Patterson JW, Hosler GA. Weedon's Skin Pathology. 4th ed. Philadelphia, PA: Churchill Livingstone/Elsevier; 2016.
- Elston DM, Ferringer T, eds. Dermatopathology. 2nd ed. Philadelphia, PA: Saunders Elsevier; 2014.
- Calonje E, McKee PH. McKee's Pathology of the Skin. 4th ed. Edinburgh, Scotland: Elsevier/Saunders; 2012.
- Bolognia JL, Shaffer JV, Cerroni L, eds. Dermatolology. 4th ed. China: Elsevier; 2018.
The Diagnosis: Erythrasma
Erythrasma usually involves intertriginous areas (eg, axillae, groin, inframammary area). Patients present with well-demarcated, minimally scaly, red-brown patches. The interdigital web space of the toes also can be involved with macerated white plaques, often with coexistent dermatophyte infection. Corynebacterium minutissimum, the bacteria responsible for erythrasma, produces coproporphyrin type III, which emits coral red fluorescence under Wood lamp examination.1 Bathing may result in removal of the porphyrin and result in a false-negative finding. Potassium hydroxide preparation of skin scrapings can show chains of bacilli. Biopsy appears relatively normal at low power but reveals compact orthokeratosis with coccobacilli and filamentous organisms in the superficial stratum corneum (quiz image). When not obvious on hematoxylin and eosin-stained sections, the organisms are Gram-positive and also are seen with periodic acid-Schiff (PAS) and methenamine silver stains. Unlike fungal hyphae, these organisms are thinner and nonrefractile. Inflammation typically is minimal. Due to the subtle histologic findings at low power, erythrasma is considered one of the invisible dermatoses.2 The differential diagnosis of these inconspicuous dermatoses that appear normal at first glance can be approached in a stepwise fashion starting in the stratum corneum, followed by the granular layer, basal layer, dermal papillae, dermal inflammatory cells, dermal connective tissue, and eccrine glands, and should consider each of the following diagnoses: candidiasis, dermatophytosis, ichthyosis vulgaris, vitiligo, macular amyloid, urticaria, telangiectasia macularis eruptiva perstans, connective tissue nevus, and argyria.2
Candidiasis, most commonly caused by Candida albicans, usually involves the oral cavity (eg, thrush, median rhomboid glossitis, angular cheilitis), intertriginous zones, nail fold (paronychia), genital areas (eg, vulvovaginitis, balanitis), and diaper area.3 The web space between the third and fourth fingers (erosio interdigitalis blastomycetica) can be involved in patients whose hands are frequently in water. Intertriginous candidiasis presents with bright red, sometimes erosive patches with satellite lesions. Spores and mycelia (filamentous forms) are noted on potassium hydroxide preparation of skin scrapings. Histologically, the epidermis often is acanthotic, mildly spongiotic, and contains groups of neutrophils in the superficial layers. The mnemonic device for diseases with clusters of neutrophils in the stratum corneum is PTICSS (psoriasis, tinea, impetigo, candida, seborrheic dermatitis, syphilis).2 Yeast, pseudohyphae, and even true hyphae can be seen in the stratum corneum with hematoxylin and eosin-stained sections and PAS. The filamentous forms tend to be vertically oriented in relation to the skin surface (Figure 1) compared to dermatophyte hyphae that tend to be parallel to the surface.2
Pitted keratolysis is a superficial bacterial infection involving the soles of the feet. The classic clinical findings are shallow 1- to 2-mm pits in clusters that can coalesce on pressure-bearing areas. Hyperhidrosis, malodor, and maceration commonly are associated. Microscopic examination reveals clusters of small cocci and filamentous bacteria located in the dell or pit of a thick compact orthokeratotic stratum corneum of acral skin with no notable inflammatory infiltrate (Figure 2).2 Special stains such as Gram, methenamine silver, or PAS can assist in visualization of the organisms. Pitted keratolysis is caused by Dermatophilus congolensis and Kytococcus sedentarius (formerly Micrococcus sedentarius), which produce keratinolytic enzymes causing the defect in the stratum corneum.3
Tinea cruris, also known as jock itch and ringworm of the groin, presents with advancing pruritic, circinate, erythematous, scaling patches with central clearing on the inner thighs and crural folds. Similar to tinea pedis, Trichophyton rubrum is the most common dermatophyte to cause tinea cruris.4 Potassium hydroxide preparation of skin scrapings from the advancing border show fungal hyphae that cross the keratin cell borders. The histopathology of dermatophyte infections can be subtle and resemble normal skin before close inspection of the stratum corneum, which can show compact orthokeratosis, neutrophils, or "sandwich sign" where hyphae are sandwiched between an upper basket weave layer and a lower compact cornified layer (orthokeratotic or parakeratotic)(Figure 3).1 The presence of these patterns in the stratum corneum should result in performance of PAS to highlight obscure hyphae.
Tinea versicolor, also called pityriasis versicolor, usually presents with hypopigmented or less commonly hyperpigmented circular patches that coalesce on the upper trunk and shoulders. There is a fine fluffy scale that is most notable after scraping the skin for a potassium hydroxide preparation, which shows "spaghetti and meatballs" (hyphae and spores). Tinea versicolor typically is caused by the mycelial phase of the lipophilic yeast Malassezia globosae.3 Histologically, there are yeast and short septate hyphae scattered in a loose basket weave hyperkeratotic stratum corneum with minimal or no inflammation (Figure 4). On occasion, PAS is required for identification.
The Diagnosis: Erythrasma
Erythrasma usually involves intertriginous areas (eg, axillae, groin, inframammary area). Patients present with well-demarcated, minimally scaly, red-brown patches. The interdigital web space of the toes also can be involved with macerated white plaques, often with coexistent dermatophyte infection. Corynebacterium minutissimum, the bacteria responsible for erythrasma, produces coproporphyrin type III, which emits coral red fluorescence under Wood lamp examination.1 Bathing may result in removal of the porphyrin and result in a false-negative finding. Potassium hydroxide preparation of skin scrapings can show chains of bacilli. Biopsy appears relatively normal at low power but reveals compact orthokeratosis with coccobacilli and filamentous organisms in the superficial stratum corneum (quiz image). When not obvious on hematoxylin and eosin-stained sections, the organisms are Gram-positive and also are seen with periodic acid-Schiff (PAS) and methenamine silver stains. Unlike fungal hyphae, these organisms are thinner and nonrefractile. Inflammation typically is minimal. Due to the subtle histologic findings at low power, erythrasma is considered one of the invisible dermatoses.2 The differential diagnosis of these inconspicuous dermatoses that appear normal at first glance can be approached in a stepwise fashion starting in the stratum corneum, followed by the granular layer, basal layer, dermal papillae, dermal inflammatory cells, dermal connective tissue, and eccrine glands, and should consider each of the following diagnoses: candidiasis, dermatophytosis, ichthyosis vulgaris, vitiligo, macular amyloid, urticaria, telangiectasia macularis eruptiva perstans, connective tissue nevus, and argyria.2
Candidiasis, most commonly caused by Candida albicans, usually involves the oral cavity (eg, thrush, median rhomboid glossitis, angular cheilitis), intertriginous zones, nail fold (paronychia), genital areas (eg, vulvovaginitis, balanitis), and diaper area.3 The web space between the third and fourth fingers (erosio interdigitalis blastomycetica) can be involved in patients whose hands are frequently in water. Intertriginous candidiasis presents with bright red, sometimes erosive patches with satellite lesions. Spores and mycelia (filamentous forms) are noted on potassium hydroxide preparation of skin scrapings. Histologically, the epidermis often is acanthotic, mildly spongiotic, and contains groups of neutrophils in the superficial layers. The mnemonic device for diseases with clusters of neutrophils in the stratum corneum is PTICSS (psoriasis, tinea, impetigo, candida, seborrheic dermatitis, syphilis).2 Yeast, pseudohyphae, and even true hyphae can be seen in the stratum corneum with hematoxylin and eosin-stained sections and PAS. The filamentous forms tend to be vertically oriented in relation to the skin surface (Figure 1) compared to dermatophyte hyphae that tend to be parallel to the surface.2
Pitted keratolysis is a superficial bacterial infection involving the soles of the feet. The classic clinical findings are shallow 1- to 2-mm pits in clusters that can coalesce on pressure-bearing areas. Hyperhidrosis, malodor, and maceration commonly are associated. Microscopic examination reveals clusters of small cocci and filamentous bacteria located in the dell or pit of a thick compact orthokeratotic stratum corneum of acral skin with no notable inflammatory infiltrate (Figure 2).2 Special stains such as Gram, methenamine silver, or PAS can assist in visualization of the organisms. Pitted keratolysis is caused by Dermatophilus congolensis and Kytococcus sedentarius (formerly Micrococcus sedentarius), which produce keratinolytic enzymes causing the defect in the stratum corneum.3
Tinea cruris, also known as jock itch and ringworm of the groin, presents with advancing pruritic, circinate, erythematous, scaling patches with central clearing on the inner thighs and crural folds. Similar to tinea pedis, Trichophyton rubrum is the most common dermatophyte to cause tinea cruris.4 Potassium hydroxide preparation of skin scrapings from the advancing border show fungal hyphae that cross the keratin cell borders. The histopathology of dermatophyte infections can be subtle and resemble normal skin before close inspection of the stratum corneum, which can show compact orthokeratosis, neutrophils, or "sandwich sign" where hyphae are sandwiched between an upper basket weave layer and a lower compact cornified layer (orthokeratotic or parakeratotic)(Figure 3).1 The presence of these patterns in the stratum corneum should result in performance of PAS to highlight obscure hyphae.
Tinea versicolor, also called pityriasis versicolor, usually presents with hypopigmented or less commonly hyperpigmented circular patches that coalesce on the upper trunk and shoulders. There is a fine fluffy scale that is most notable after scraping the skin for a potassium hydroxide preparation, which shows "spaghetti and meatballs" (hyphae and spores). Tinea versicolor typically is caused by the mycelial phase of the lipophilic yeast Malassezia globosae.3 Histologically, there are yeast and short septate hyphae scattered in a loose basket weave hyperkeratotic stratum corneum with minimal or no inflammation (Figure 4). On occasion, PAS is required for identification.
- Patterson JW, Hosler GA. Weedon's Skin Pathology. 4th ed. Philadelphia, PA: Churchill Livingstone/Elsevier; 2016.
- Elston DM, Ferringer T, eds. Dermatopathology. 2nd ed. Philadelphia, PA: Saunders Elsevier; 2014.
- Calonje E, McKee PH. McKee's Pathology of the Skin. 4th ed. Edinburgh, Scotland: Elsevier/Saunders; 2012.
- Bolognia JL, Shaffer JV, Cerroni L, eds. Dermatolology. 4th ed. China: Elsevier; 2018.
- Patterson JW, Hosler GA. Weedon's Skin Pathology. 4th ed. Philadelphia, PA: Churchill Livingstone/Elsevier; 2016.
- Elston DM, Ferringer T, eds. Dermatopathology. 2nd ed. Philadelphia, PA: Saunders Elsevier; 2014.
- Calonje E, McKee PH. McKee's Pathology of the Skin. 4th ed. Edinburgh, Scotland: Elsevier/Saunders; 2012.
- Bolognia JL, Shaffer JV, Cerroni L, eds. Dermatolology. 4th ed. China: Elsevier; 2018.

A 66-year-old man presented with reddish arciform patches in the inguinal area.
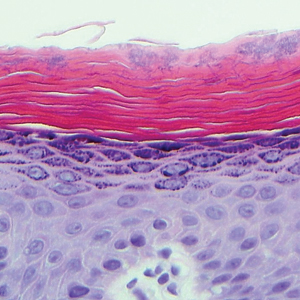
Red-Brown Plaque on the Leg
The Diagnosis: Wells Syndrome
A punch biopsy taken from the perimeter of the lesion demonstrated mild spongiosis overlying a dense nodular to diffuse infiltrate of lymphocytes, neutrophils, and numerous eosinophils, some involving underlying fat lobules (Figure, A and B). In some areas, eosinophilic degeneration of collagen bundles surrounded by a rim of histiocytes, "flame features," were observed (Figure C). The clinical and histological features were consistent with Wells syndrome (WS), also known as eosinophilic cellulitis. Given the localized mild nature of the disease, the patient was started on a midpotency topical corticosteroid.

Wells syndrome is a rare inflammatory condition characterized by clinical polymorphism, suggestive histologic findings, and a recurrent course.1,2 This condition is especially rare in children.3,4 Caputo et al1 described 7 variants in their case series of 19 patients: classic plaque-type variant (the most common clinical presentation in children); annular granuloma-like (the most common clinical presentation in adults); urticarialike; bullous; papulonodular; papulovesicular; and fixed drug eruption-like. Wells syndrome is thought to result from excess production of IL-5 in response to a hypersensitivity reaction to an exogenous or endogenous circulating antigen.3,4 Increased levels of IL-5 enhance eosinophil accumulation in the skin, degranulation, and subsequent tissue destruction.3,4 Reported triggers include insect bites, viral and bacterial infections, drug eruptions, recent vaccination, and paraphenylenediamine in henna tattoos.3-7 Additionally, WS has been reported in the setting of gastrointestinal pathologies, such as celiac disease and ulcerative colitis, and with asthma exacerbations.8,9 However, in half of pediatric cases, no trigger can be identified.7
Clinically, WS presents with pruritic, mildly tender plaques.7 Lesions may be localized or diffuse and range from mild annular or circinate plaques with infiltrated borders to cellulitic-appearing lesions that are occasionally associated with bullae.5,6 Patients often report prodromal symptoms of burning and pruritus.5,6 Lesions rapidly progress over 2 to 3 days, pass through a blue grayish discoloration phase, and gradually resolve over 2 to 8 weeks.5,6,10 Although patients generally heal without scarring, WS lesions have been described to resolve with atrophy and hyperpigmentation resembling morphea.5-7 Additionally, patients typically experience a relapsing remitting course over months to years with eventual spontaneous resolution.1,5 Patients also may experience systemic symptoms including fever, lymphadenopathy, and arthralgia, though they do not develop more widespread systemic manifestations.2,3,7
Diagnosis of WS is based on clinicopathologic correlation. Histopathology of WS lesions demonstrates 3 phases. The acute phase demonstrates edema of the superficial and mid dermis with a dense dermal eosinophilic infiltrate.1,6,10 The subacute granulomatous phase demonstrates flame figures in the dermis.1,2,6,7,10 Flame figures consist of palisading groups of eosinophils and histiocytes around a core of degenerating basophilic collagen bundles associated with major basic protein.1,2,6,7,10 Finally, in the resolution phase, eosinophils gradually disappear while histiocytes and giant cells persist, forming microgranulomas.1,2,10 Notably, no vasculitis is observed and direct immunofluorescence is negative.3,7 Although flame figures are suggestive of WS, they are not pathognomonic and are observed in other conditions including Churg-Strauss syndrome, parasitic and fungal infections, herpes gestationis, bullous pemphigoid, and follicular mucinosis.2,5
Wells syndrome is a self-resolving and benign condition.1,10 Physicians are recommended to gather a complete history including review of medications and vaccinations; a history of insect bites, infections, and asthma; laboratory workup consisting of a complete blood cell count with differential and stool samples for ova and parasites; and a skin biopsy if the diagnosis is unclear.7 Identification and treatment of underlying causes often results in resolution.6 Systemic corticosteroids frequently are used in both adult and pediatric patients, though practitioners should consider alternative treatments when recurrences occur to avoid steroid side effects.3,6 Midpotency topical corticosteroids present a safe alternative to systemic corticosteroids in the pediatric population, especially in cases of localized WS without systemic symptoms.3 Other medications reported in the literature include cyclosporine, dapsone, antimalarial medications, and azathioprine.6 Despite appropriate therapy, patients and physicians should anticipate recurrence over months to years.1,6
- Caputo R, Marzano AV, Vezzoli P, et al. Wells syndrome in adults and children: a report of 19 cases. Arch Dermatol. 2006;142:1157-1161.
- Smith SM, Kiracofe EA, Clark LN, et al. Idiopathic hypereosinophilic syndrome with cutaneous manifestations and flame figures: a spectrum of eosinophilic dermatoses whose features overlap with Wells' syndrome. Am J Dermatopathol. 2015;37:910-914.
- Gilliam AE, Bruckner AL, Howard RM, et al. Bullous "cellulitis" with eosinophilia: case report and review of Wells' syndrome in childhood. Pediatrics. 2005;116:E149-E155.
- Nacaroglu HT, Celegen M, Karkıner CS, et al. Eosinophilic cellulitis (Wells' syndrome) caused by a temporary henna tattoo. Postepy Dermatol Alergol. 2014;31:322-324.
- Heelan K, Ryan JF, Shear NH, et al. Wells syndrome (eosinophilic cellulitis): proposed diagnostic criteria and a literature review of the drug-induced variant. J Dermatol Case Rep. 2013;7:113-120.
- Sinno H, Lacroix JP, Lee J, et al. Diagnosis and management of eosinophilic cellulitis (Wells' syndrome): a case series and literature review. Can J Plast Surg. 2012;20:91-97.
- Cherng E, McClung AA, Rosenthal HM, et al. Wells' syndrome associated with parvovirus in a 5-year-old boy. Pediatr Dermatol. 2012;29:762-764.
- Eren M, Açikalin M. A case report of Wells' syndrome in a celiac patient. Turk J Gastroenterol. 2010;21:172-174.
- Cruz MJ, Mota A, Baudrier T, et al. Recurrent Wells' syndrome associated with allergic asthma exacerbation. Cutan Ocul Toxicol. 2012;31:154-156.
- Van der Straaten S, Wojciechowski M, Salgado R, et al. Eosinophilic cellulitis or Wells' syndrome in a 6-year-old child. Eur J Pediatr. 2006;165:197-198.
The Diagnosis: Wells Syndrome
A punch biopsy taken from the perimeter of the lesion demonstrated mild spongiosis overlying a dense nodular to diffuse infiltrate of lymphocytes, neutrophils, and numerous eosinophils, some involving underlying fat lobules (Figure, A and B). In some areas, eosinophilic degeneration of collagen bundles surrounded by a rim of histiocytes, "flame features," were observed (Figure C). The clinical and histological features were consistent with Wells syndrome (WS), also known as eosinophilic cellulitis. Given the localized mild nature of the disease, the patient was started on a midpotency topical corticosteroid.
Wells syndrome is a rare inflammatory condition characterized by clinical polymorphism, suggestive histologic findings, and a recurrent course.1,2 This condition is especially rare in children.3,4 Caputo et al1 described 7 variants in their case series of 19 patients: classic plaque-type variant (the most common clinical presentation in children); annular granuloma-like (the most common clinical presentation in adults); urticarialike; bullous; papulonodular; papulovesicular; and fixed drug eruption-like. Wells syndrome is thought to result from excess production of IL-5 in response to a hypersensitivity reaction to an exogenous or endogenous circulating antigen.3,4 Increased levels of IL-5 enhance eosinophil accumulation in the skin, degranulation, and subsequent tissue destruction.3,4 Reported triggers include insect bites, viral and bacterial infections, drug eruptions, recent vaccination, and paraphenylenediamine in henna tattoos.3-7 Additionally, WS has been reported in the setting of gastrointestinal pathologies, such as celiac disease and ulcerative colitis, and with asthma exacerbations.8,9 However, in half of pediatric cases, no trigger can be identified.7
Clinically, WS presents with pruritic, mildly tender plaques.7 Lesions may be localized or diffuse and range from mild annular or circinate plaques with infiltrated borders to cellulitic-appearing lesions that are occasionally associated with bullae.5,6 Patients often report prodromal symptoms of burning and pruritus.5,6 Lesions rapidly progress over 2 to 3 days, pass through a blue grayish discoloration phase, and gradually resolve over 2 to 8 weeks.5,6,10 Although patients generally heal without scarring, WS lesions have been described to resolve with atrophy and hyperpigmentation resembling morphea.5-7 Additionally, patients typically experience a relapsing remitting course over months to years with eventual spontaneous resolution.1,5 Patients also may experience systemic symptoms including fever, lymphadenopathy, and arthralgia, though they do not develop more widespread systemic manifestations.2,3,7
Diagnosis of WS is based on clinicopathologic correlation. Histopathology of WS lesions demonstrates 3 phases. The acute phase demonstrates edema of the superficial and mid dermis with a dense dermal eosinophilic infiltrate.1,6,10 The subacute granulomatous phase demonstrates flame figures in the dermis.1,2,6,7,10 Flame figures consist of palisading groups of eosinophils and histiocytes around a core of degenerating basophilic collagen bundles associated with major basic protein.1,2,6,7,10 Finally, in the resolution phase, eosinophils gradually disappear while histiocytes and giant cells persist, forming microgranulomas.1,2,10 Notably, no vasculitis is observed and direct immunofluorescence is negative.3,7 Although flame figures are suggestive of WS, they are not pathognomonic and are observed in other conditions including Churg-Strauss syndrome, parasitic and fungal infections, herpes gestationis, bullous pemphigoid, and follicular mucinosis.2,5
Wells syndrome is a self-resolving and benign condition.1,10 Physicians are recommended to gather a complete history including review of medications and vaccinations; a history of insect bites, infections, and asthma; laboratory workup consisting of a complete blood cell count with differential and stool samples for ova and parasites; and a skin biopsy if the diagnosis is unclear.7 Identification and treatment of underlying causes often results in resolution.6 Systemic corticosteroids frequently are used in both adult and pediatric patients, though practitioners should consider alternative treatments when recurrences occur to avoid steroid side effects.3,6 Midpotency topical corticosteroids present a safe alternative to systemic corticosteroids in the pediatric population, especially in cases of localized WS without systemic symptoms.3 Other medications reported in the literature include cyclosporine, dapsone, antimalarial medications, and azathioprine.6 Despite appropriate therapy, patients and physicians should anticipate recurrence over months to years.1,6
The Diagnosis: Wells Syndrome
A punch biopsy taken from the perimeter of the lesion demonstrated mild spongiosis overlying a dense nodular to diffuse infiltrate of lymphocytes, neutrophils, and numerous eosinophils, some involving underlying fat lobules (Figure, A and B). In some areas, eosinophilic degeneration of collagen bundles surrounded by a rim of histiocytes, "flame features," were observed (Figure C). The clinical and histological features were consistent with Wells syndrome (WS), also known as eosinophilic cellulitis. Given the localized mild nature of the disease, the patient was started on a midpotency topical corticosteroid.
Wells syndrome is a rare inflammatory condition characterized by clinical polymorphism, suggestive histologic findings, and a recurrent course.1,2 This condition is especially rare in children.3,4 Caputo et al1 described 7 variants in their case series of 19 patients: classic plaque-type variant (the most common clinical presentation in children); annular granuloma-like (the most common clinical presentation in adults); urticarialike; bullous; papulonodular; papulovesicular; and fixed drug eruption-like. Wells syndrome is thought to result from excess production of IL-5 in response to a hypersensitivity reaction to an exogenous or endogenous circulating antigen.3,4 Increased levels of IL-5 enhance eosinophil accumulation in the skin, degranulation, and subsequent tissue destruction.3,4 Reported triggers include insect bites, viral and bacterial infections, drug eruptions, recent vaccination, and paraphenylenediamine in henna tattoos.3-7 Additionally, WS has been reported in the setting of gastrointestinal pathologies, such as celiac disease and ulcerative colitis, and with asthma exacerbations.8,9 However, in half of pediatric cases, no trigger can be identified.7
Clinically, WS presents with pruritic, mildly tender plaques.7 Lesions may be localized or diffuse and range from mild annular or circinate plaques with infiltrated borders to cellulitic-appearing lesions that are occasionally associated with bullae.5,6 Patients often report prodromal symptoms of burning and pruritus.5,6 Lesions rapidly progress over 2 to 3 days, pass through a blue grayish discoloration phase, and gradually resolve over 2 to 8 weeks.5,6,10 Although patients generally heal without scarring, WS lesions have been described to resolve with atrophy and hyperpigmentation resembling morphea.5-7 Additionally, patients typically experience a relapsing remitting course over months to years with eventual spontaneous resolution.1,5 Patients also may experience systemic symptoms including fever, lymphadenopathy, and arthralgia, though they do not develop more widespread systemic manifestations.2,3,7
Diagnosis of WS is based on clinicopathologic correlation. Histopathology of WS lesions demonstrates 3 phases. The acute phase demonstrates edema of the superficial and mid dermis with a dense dermal eosinophilic infiltrate.1,6,10 The subacute granulomatous phase demonstrates flame figures in the dermis.1,2,6,7,10 Flame figures consist of palisading groups of eosinophils and histiocytes around a core of degenerating basophilic collagen bundles associated with major basic protein.1,2,6,7,10 Finally, in the resolution phase, eosinophils gradually disappear while histiocytes and giant cells persist, forming microgranulomas.1,2,10 Notably, no vasculitis is observed and direct immunofluorescence is negative.3,7 Although flame figures are suggestive of WS, they are not pathognomonic and are observed in other conditions including Churg-Strauss syndrome, parasitic and fungal infections, herpes gestationis, bullous pemphigoid, and follicular mucinosis.2,5
Wells syndrome is a self-resolving and benign condition.1,10 Physicians are recommended to gather a complete history including review of medications and vaccinations; a history of insect bites, infections, and asthma; laboratory workup consisting of a complete blood cell count with differential and stool samples for ova and parasites; and a skin biopsy if the diagnosis is unclear.7 Identification and treatment of underlying causes often results in resolution.6 Systemic corticosteroids frequently are used in both adult and pediatric patients, though practitioners should consider alternative treatments when recurrences occur to avoid steroid side effects.3,6 Midpotency topical corticosteroids present a safe alternative to systemic corticosteroids in the pediatric population, especially in cases of localized WS without systemic symptoms.3 Other medications reported in the literature include cyclosporine, dapsone, antimalarial medications, and azathioprine.6 Despite appropriate therapy, patients and physicians should anticipate recurrence over months to years.1,6
- Caputo R, Marzano AV, Vezzoli P, et al. Wells syndrome in adults and children: a report of 19 cases. Arch Dermatol. 2006;142:1157-1161.
- Smith SM, Kiracofe EA, Clark LN, et al. Idiopathic hypereosinophilic syndrome with cutaneous manifestations and flame figures: a spectrum of eosinophilic dermatoses whose features overlap with Wells' syndrome. Am J Dermatopathol. 2015;37:910-914.
- Gilliam AE, Bruckner AL, Howard RM, et al. Bullous "cellulitis" with eosinophilia: case report and review of Wells' syndrome in childhood. Pediatrics. 2005;116:E149-E155.
- Nacaroglu HT, Celegen M, Karkıner CS, et al. Eosinophilic cellulitis (Wells' syndrome) caused by a temporary henna tattoo. Postepy Dermatol Alergol. 2014;31:322-324.
- Heelan K, Ryan JF, Shear NH, et al. Wells syndrome (eosinophilic cellulitis): proposed diagnostic criteria and a literature review of the drug-induced variant. J Dermatol Case Rep. 2013;7:113-120.
- Sinno H, Lacroix JP, Lee J, et al. Diagnosis and management of eosinophilic cellulitis (Wells' syndrome): a case series and literature review. Can J Plast Surg. 2012;20:91-97.
- Cherng E, McClung AA, Rosenthal HM, et al. Wells' syndrome associated with parvovirus in a 5-year-old boy. Pediatr Dermatol. 2012;29:762-764.
- Eren M, Açikalin M. A case report of Wells' syndrome in a celiac patient. Turk J Gastroenterol. 2010;21:172-174.
- Cruz MJ, Mota A, Baudrier T, et al. Recurrent Wells' syndrome associated with allergic asthma exacerbation. Cutan Ocul Toxicol. 2012;31:154-156.
- Van der Straaten S, Wojciechowski M, Salgado R, et al. Eosinophilic cellulitis or Wells' syndrome in a 6-year-old child. Eur J Pediatr. 2006;165:197-198.
- Caputo R, Marzano AV, Vezzoli P, et al. Wells syndrome in adults and children: a report of 19 cases. Arch Dermatol. 2006;142:1157-1161.
- Smith SM, Kiracofe EA, Clark LN, et al. Idiopathic hypereosinophilic syndrome with cutaneous manifestations and flame figures: a spectrum of eosinophilic dermatoses whose features overlap with Wells' syndrome. Am J Dermatopathol. 2015;37:910-914.
- Gilliam AE, Bruckner AL, Howard RM, et al. Bullous "cellulitis" with eosinophilia: case report and review of Wells' syndrome in childhood. Pediatrics. 2005;116:E149-E155.
- Nacaroglu HT, Celegen M, Karkıner CS, et al. Eosinophilic cellulitis (Wells' syndrome) caused by a temporary henna tattoo. Postepy Dermatol Alergol. 2014;31:322-324.
- Heelan K, Ryan JF, Shear NH, et al. Wells syndrome (eosinophilic cellulitis): proposed diagnostic criteria and a literature review of the drug-induced variant. J Dermatol Case Rep. 2013;7:113-120.
- Sinno H, Lacroix JP, Lee J, et al. Diagnosis and management of eosinophilic cellulitis (Wells' syndrome): a case series and literature review. Can J Plast Surg. 2012;20:91-97.
- Cherng E, McClung AA, Rosenthal HM, et al. Wells' syndrome associated with parvovirus in a 5-year-old boy. Pediatr Dermatol. 2012;29:762-764.
- Eren M, Açikalin M. A case report of Wells' syndrome in a celiac patient. Turk J Gastroenterol. 2010;21:172-174.
- Cruz MJ, Mota A, Baudrier T, et al. Recurrent Wells' syndrome associated with allergic asthma exacerbation. Cutan Ocul Toxicol. 2012;31:154-156.
- Van der Straaten S, Wojciechowski M, Salgado R, et al. Eosinophilic cellulitis or Wells' syndrome in a 6-year-old child. Eur J Pediatr. 2006;165:197-198.
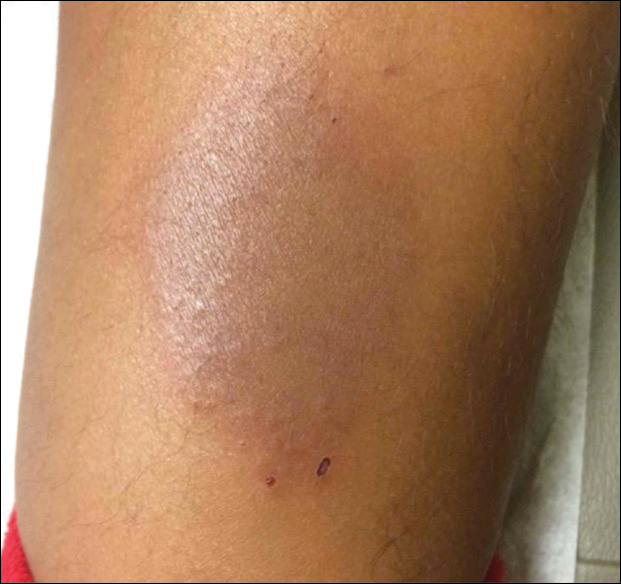
A healthy 7-year-old boy presented with an enlarging hyperpigmented plaque on the anterior aspect of the lower left leg of 2 months' duration. His mother reported onset following a mosquito bite. Clotrimazole was used without improvement. His mother denied recent travel, similar lesions in close contacts, fever, asthma, and arthralgia. Physical examination revealed a 5.2 ×3-cm nonscaly, red-brown, ovoid, thin plaque with a slightly raised border.