User login
Cutis is a peer-reviewed clinical journal for the dermatologist, allergist, and general practitioner published monthly since 1965. Concise clinical articles present the practical side of dermatology, helping physicians to improve patient care. Cutis is referenced in Index Medicus/MEDLINE and is written and edited by industry leaders.
ass lick
assault rifle
balls
ballsac
black jack
bleach
Boko Haram
bondage
causas
cheap
child abuse
cocaine
compulsive behaviors
cost of miracles
cunt
Daech
display network stats
drug paraphernalia
explosion
fart
fda and death
fda AND warn
fda AND warning
fda AND warns
feom
fuck
gambling
gfc
gun
human trafficking
humira AND expensive
illegal
ISIL
ISIS
Islamic caliphate
Islamic state
madvocate
masturbation
mixed martial arts
MMA
molestation
national rifle association
NRA
nsfw
nuccitelli
pedophile
pedophilia
poker
porn
porn
pornography
psychedelic drug
recreational drug
sex slave rings
shit
slot machine
snort
substance abuse
terrorism
terrorist
texarkana
Texas hold 'em
UFC
section[contains(@class, 'nav-hidden')]
section[contains(@class, 'nav-hidden active')
A peer-reviewed, indexed journal for dermatologists with original research, image quizzes, cases and reviews, and columns.
Epidermolysis Bullosa Acquisita in Association With Mantle Cell Lymphoma
To the Editor:
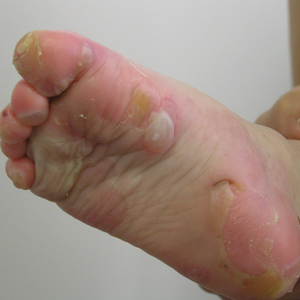
A 46-year-old man presented with multiple tense bullae and denuded patches on the palms (Figure 1A) and soles (Figure 1B). The blisters first appeared 2 months prior to presentation, shortly after he was diagnosed with stage IVB mantle cell lymphoma, and waxed and waned in intensity since then. He denied antecedent trauma or friction and reported that all sites were painful. He had no family or personal history of blistering disorders.
 and multiple bullae on the sole (B).")
The mantle cell lymphoma initially was treated with 4 cycles of R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone) chemotherapy more than 2.5 years prior to the current presentation, which resulted in partial remission, followed by R-ICE (rituximab, ifosfamide, carboplatin, etoposide) therapy as well as autologous stem cell transplantation; complete remission was achieved. His recovery was complicated by a necrotic small bowel leading to resection. Eighteen months following the second course of chemotherapy, a mass was noted on the neck; biopsy performed by an outside dermatologist revealed mantle cell lymphoma.
Punch biopsy revealed a subepidermal bulla. Six weeks later, biopsy of a newly developed hand lesion performed at our office revealed a subepidermal cleft with minimal dermal infiltrate (Figure 2). Direct immunofluorescence was negative for immunoglobulin and complement deposition. Porphyrin elevation was not detected with a 24-hour urine assay. New lesions were drained and injected with triamcinolone, which appeared to hasten healing.
.")
Mantle cell lymphoma is a distinct lymphoproliferative disorder of B cells that represents less than 7% of non-Hodgkin lymphoma cases.1 The tumor cells originate in the mantle zone of the lymph nodes. Most patients present with advanced disease involving lymph nodes and other organs. The disease is characterized by male predominance and an aggressive course with a median overall survival of less than 5 years.1
Epidermolysis bullosa acquisita is a rare blistering disease that usually develops in adulthood. It is a subepidermal disorder characterized by the appearance of fragile tense bullae. Epidermolysis bullosa acquisita can be divided into 2 subtypes: inflammatory and mechanobullous (classic EBA).2 Inflammatory EBA presents similarly to bullous pemphigoid and other subepithelial autoimmune blistering diseases. Vesiculobullous lesions predominate on the trunk and extremities and often are accompanied by intense pruritus. The less common mechanobullous noninflammatory subtype, illustrated in our case, presents in trauma-prone areas with skin fragility and tense noninflamed vesicles and bullae that rupture leaving erosions. Associated findings may include milia and scarring. Lesions appear in areas exposed to friction and trauma such as the hands, feet, elbows, knees, and lower back. The differential diagnosis includes dystrophic epidermolysis bullosa, porphyria cutanea tarda, and pseudoporphyria. Dystrophic epidermolysis bullosa is ruled out by family history and disease onset at birth. The lesions of porphyria cutanea tarda and pseudoporphyria occur on sun-exposed areas; porphyrin levels are elevated in the former. Direct immunofluorescence of a perilesional EBA site usually reveals IgG deposition.3 Negative direct immunofluorescence in our case could have resulted from technical error, sample location, or response to systemic immunosuppressive treatment.4
Epidermolysis bullosa acquisita is caused by autoantibodies against type VII collagen.2,3 After the autoantibodies bind, a complement cascade reaction is activated, leading to deposition of C3a and C5a, which recruit leukocytes and mast cells. The anchoring fibrils in the basement membrane zones of the skin and mucosa are disrupted.5,6 Injection of anti–type VII collagen antibodies into mice induces a blistering disease resembling EBA.7 In a study of 14 patients with EBA, disease severity was correlated to levels of anticollagen autoantibodies measured by enzyme-linked immunosorbent assay.8
Epidermolysis bullosa acquisita has been linked to Crohn disease and approximately 30% of EBA cases occur in patients with this disease.9,10 Two case reports document an association with multiple myeloma.11,12 Treatment often proves challenging and unsatisfactory; valid controlled clinical trials are impossible given the paucity of cases. Successful therapeutic outcomes have been reported with oral prednisone,13 colchicine,14 cyclosporine,15 dapsone,16 and rituximab.17 Our patient received 2 separate courses of rituximab as part of chemotherapy for mantle cell lymphoma without measurable improvement. He was lost to follow-up after recurrence of the lymphoma and we learned from his wife that he had died.
- Hitz F, Bargetzi M, Cogliatti S, et al. Diagnosis and treatment of mantle cell lymphoma. Swiss Med Wkly. 2013;143:w13868.
- Ludwig RJ. Clinical presentation, pathogenesis, diagnosis, and treatment of epidermolysis bullosa acquisita. ISRN Dermatol. 2013;2013:812029.
- Gupta R, Woodley DT, Chen M. Epidermolysis bullosa acquisita. Clin Dermatol. 2012;30:60-69.
- Mutasim DF, Adams BB. Immunofluorescence in dermatology. J Am Acad Dermatol. 2001;45:803-822.
- Woodley DT, Briggaman RA, O’Keefe EJ. Identification of the skin basement-membrane autoantigen in epidermolysis bullosa acquisita. N Engl J Med. 1984;310:1007-1013.
- Hashimoto T, Ishii N, Ohata C, et al. Pathogenesis of epidermolysis bullosa acquisita, an autoimmune subepidermal bullous disease. J Pathol. 2012;228:1-7.
- Sitaru C, Chiriac MT, Mihai S, et al. Induction of complement-fixing autoantibodies against type VII collagen results in subepidermal blistering in mice. J Immunol. 2006;177:3461-3468.
- Marzano AV, Cozzani E, Fanoni D, et al. Diagnosis and disease severity assessment of epidermolysis bullosa acquisita by ELISA for anti-type VII collagen autoantibodies: an Italian multicentre study. Br J Dermatol. 2013;168:80-84.
- Chen M, O’Toole EA, Sanghavi J, et al. The epidermolysis bullosa acquisita antigen (type VII collagen) is present in human colon and patients with Crohn’s disease have autoantibodies to type VII collagen. J Invest Dermatol. 2002;118:1059-1064.
- Reddy H, Shipman AR, Wojnarowska F. Epidermolysis bullosa acquisita and inflammatory bowel disease: a review of the literature. Clin Exp Dermatol. 2013;38:225-229.
- Radfar L, Fatahzadeh M, Shahamat Y, et al. Paraneoplastic epidermolysis bullosa acquisita associated with multiple myeloma. Spec Care Dentist. 2006;26:159-163.
- Engineer L, Dow EC, Braverman IM, et al. Epidermolysis bullosa acquisita and multiple myeloma. J Am Acad Dermatol. 2002;47:943-946.
- Ishii N, Hamada T, Dainichi T, et al. Epidermolysis bullosa acquisita: what’s new? J Dermatol. 2010;37:220-230.
- Megahed M, Scharffetter-Kochanek K. Epidermolysis bullosa acquisita—successful treatment with colchicine. Arch Dermatol Res. 1994;286:35-46.
- Khatri ML, Benghazeil M, Shafi M. Epidermolysis bullosa acquisita responsive to cyclosporin therapy. J Eur Acad Dermatol Venereol. 2001;15:182-184.
- Hughes AP, Callen JP. Epidermolysis bullosa acquisita responsive to dapsone therapy. J Cutan Med Surg. 2001;5:397-399.
- Kim JH, Lee SE, Kim SC. Successful treatment of epidermolysis bullosa acquisita with rituximab therapy. J Dermatol. 2012;39:477-479.
To the Editor:
A 46-year-old man presented with multiple tense bullae and denuded patches on the palms (Figure 1A) and soles (Figure 1B). The blisters first appeared 2 months prior to presentation, shortly after he was diagnosed with stage IVB mantle cell lymphoma, and waxed and waned in intensity since then. He denied antecedent trauma or friction and reported that all sites were painful. He had no family or personal history of blistering disorders.
The mantle cell lymphoma initially was treated with 4 cycles of R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone) chemotherapy more than 2.5 years prior to the current presentation, which resulted in partial remission, followed by R-ICE (rituximab, ifosfamide, carboplatin, etoposide) therapy as well as autologous stem cell transplantation; complete remission was achieved. His recovery was complicated by a necrotic small bowel leading to resection. Eighteen months following the second course of chemotherapy, a mass was noted on the neck; biopsy performed by an outside dermatologist revealed mantle cell lymphoma.
Punch biopsy revealed a subepidermal bulla. Six weeks later, biopsy of a newly developed hand lesion performed at our office revealed a subepidermal cleft with minimal dermal infiltrate (Figure 2). Direct immunofluorescence was negative for immunoglobulin and complement deposition. Porphyrin elevation was not detected with a 24-hour urine assay. New lesions were drained and injected with triamcinolone, which appeared to hasten healing.
Mantle cell lymphoma is a distinct lymphoproliferative disorder of B cells that represents less than 7% of non-Hodgkin lymphoma cases.1 The tumor cells originate in the mantle zone of the lymph nodes. Most patients present with advanced disease involving lymph nodes and other organs. The disease is characterized by male predominance and an aggressive course with a median overall survival of less than 5 years.1
Epidermolysis bullosa acquisita is a rare blistering disease that usually develops in adulthood. It is a subepidermal disorder characterized by the appearance of fragile tense bullae. Epidermolysis bullosa acquisita can be divided into 2 subtypes: inflammatory and mechanobullous (classic EBA).2 Inflammatory EBA presents similarly to bullous pemphigoid and other subepithelial autoimmune blistering diseases. Vesiculobullous lesions predominate on the trunk and extremities and often are accompanied by intense pruritus. The less common mechanobullous noninflammatory subtype, illustrated in our case, presents in trauma-prone areas with skin fragility and tense noninflamed vesicles and bullae that rupture leaving erosions. Associated findings may include milia and scarring. Lesions appear in areas exposed to friction and trauma such as the hands, feet, elbows, knees, and lower back. The differential diagnosis includes dystrophic epidermolysis bullosa, porphyria cutanea tarda, and pseudoporphyria. Dystrophic epidermolysis bullosa is ruled out by family history and disease onset at birth. The lesions of porphyria cutanea tarda and pseudoporphyria occur on sun-exposed areas; porphyrin levels are elevated in the former. Direct immunofluorescence of a perilesional EBA site usually reveals IgG deposition.3 Negative direct immunofluorescence in our case could have resulted from technical error, sample location, or response to systemic immunosuppressive treatment.4
Epidermolysis bullosa acquisita is caused by autoantibodies against type VII collagen.2,3 After the autoantibodies bind, a complement cascade reaction is activated, leading to deposition of C3a and C5a, which recruit leukocytes and mast cells. The anchoring fibrils in the basement membrane zones of the skin and mucosa are disrupted.5,6 Injection of anti–type VII collagen antibodies into mice induces a blistering disease resembling EBA.7 In a study of 14 patients with EBA, disease severity was correlated to levels of anticollagen autoantibodies measured by enzyme-linked immunosorbent assay.8
Epidermolysis bullosa acquisita has been linked to Crohn disease and approximately 30% of EBA cases occur in patients with this disease.9,10 Two case reports document an association with multiple myeloma.11,12 Treatment often proves challenging and unsatisfactory; valid controlled clinical trials are impossible given the paucity of cases. Successful therapeutic outcomes have been reported with oral prednisone,13 colchicine,14 cyclosporine,15 dapsone,16 and rituximab.17 Our patient received 2 separate courses of rituximab as part of chemotherapy for mantle cell lymphoma without measurable improvement. He was lost to follow-up after recurrence of the lymphoma and we learned from his wife that he had died.
To the Editor:
A 46-year-old man presented with multiple tense bullae and denuded patches on the palms (Figure 1A) and soles (Figure 1B). The blisters first appeared 2 months prior to presentation, shortly after he was diagnosed with stage IVB mantle cell lymphoma, and waxed and waned in intensity since then. He denied antecedent trauma or friction and reported that all sites were painful. He had no family or personal history of blistering disorders.
The mantle cell lymphoma initially was treated with 4 cycles of R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone) chemotherapy more than 2.5 years prior to the current presentation, which resulted in partial remission, followed by R-ICE (rituximab, ifosfamide, carboplatin, etoposide) therapy as well as autologous stem cell transplantation; complete remission was achieved. His recovery was complicated by a necrotic small bowel leading to resection. Eighteen months following the second course of chemotherapy, a mass was noted on the neck; biopsy performed by an outside dermatologist revealed mantle cell lymphoma.
Punch biopsy revealed a subepidermal bulla. Six weeks later, biopsy of a newly developed hand lesion performed at our office revealed a subepidermal cleft with minimal dermal infiltrate (Figure 2). Direct immunofluorescence was negative for immunoglobulin and complement deposition. Porphyrin elevation was not detected with a 24-hour urine assay. New lesions were drained and injected with triamcinolone, which appeared to hasten healing.
Mantle cell lymphoma is a distinct lymphoproliferative disorder of B cells that represents less than 7% of non-Hodgkin lymphoma cases.1 The tumor cells originate in the mantle zone of the lymph nodes. Most patients present with advanced disease involving lymph nodes and other organs. The disease is characterized by male predominance and an aggressive course with a median overall survival of less than 5 years.1
Epidermolysis bullosa acquisita is a rare blistering disease that usually develops in adulthood. It is a subepidermal disorder characterized by the appearance of fragile tense bullae. Epidermolysis bullosa acquisita can be divided into 2 subtypes: inflammatory and mechanobullous (classic EBA).2 Inflammatory EBA presents similarly to bullous pemphigoid and other subepithelial autoimmune blistering diseases. Vesiculobullous lesions predominate on the trunk and extremities and often are accompanied by intense pruritus. The less common mechanobullous noninflammatory subtype, illustrated in our case, presents in trauma-prone areas with skin fragility and tense noninflamed vesicles and bullae that rupture leaving erosions. Associated findings may include milia and scarring. Lesions appear in areas exposed to friction and trauma such as the hands, feet, elbows, knees, and lower back. The differential diagnosis includes dystrophic epidermolysis bullosa, porphyria cutanea tarda, and pseudoporphyria. Dystrophic epidermolysis bullosa is ruled out by family history and disease onset at birth. The lesions of porphyria cutanea tarda and pseudoporphyria occur on sun-exposed areas; porphyrin levels are elevated in the former. Direct immunofluorescence of a perilesional EBA site usually reveals IgG deposition.3 Negative direct immunofluorescence in our case could have resulted from technical error, sample location, or response to systemic immunosuppressive treatment.4
Epidermolysis bullosa acquisita is caused by autoantibodies against type VII collagen.2,3 After the autoantibodies bind, a complement cascade reaction is activated, leading to deposition of C3a and C5a, which recruit leukocytes and mast cells. The anchoring fibrils in the basement membrane zones of the skin and mucosa are disrupted.5,6 Injection of anti–type VII collagen antibodies into mice induces a blistering disease resembling EBA.7 In a study of 14 patients with EBA, disease severity was correlated to levels of anticollagen autoantibodies measured by enzyme-linked immunosorbent assay.8
Epidermolysis bullosa acquisita has been linked to Crohn disease and approximately 30% of EBA cases occur in patients with this disease.9,10 Two case reports document an association with multiple myeloma.11,12 Treatment often proves challenging and unsatisfactory; valid controlled clinical trials are impossible given the paucity of cases. Successful therapeutic outcomes have been reported with oral prednisone,13 colchicine,14 cyclosporine,15 dapsone,16 and rituximab.17 Our patient received 2 separate courses of rituximab as part of chemotherapy for mantle cell lymphoma without measurable improvement. He was lost to follow-up after recurrence of the lymphoma and we learned from his wife that he had died.
- Hitz F, Bargetzi M, Cogliatti S, et al. Diagnosis and treatment of mantle cell lymphoma. Swiss Med Wkly. 2013;143:w13868.
- Ludwig RJ. Clinical presentation, pathogenesis, diagnosis, and treatment of epidermolysis bullosa acquisita. ISRN Dermatol. 2013;2013:812029.
- Gupta R, Woodley DT, Chen M. Epidermolysis bullosa acquisita. Clin Dermatol. 2012;30:60-69.
- Mutasim DF, Adams BB. Immunofluorescence in dermatology. J Am Acad Dermatol. 2001;45:803-822.
- Woodley DT, Briggaman RA, O’Keefe EJ. Identification of the skin basement-membrane autoantigen in epidermolysis bullosa acquisita. N Engl J Med. 1984;310:1007-1013.
- Hashimoto T, Ishii N, Ohata C, et al. Pathogenesis of epidermolysis bullosa acquisita, an autoimmune subepidermal bullous disease. J Pathol. 2012;228:1-7.
- Sitaru C, Chiriac MT, Mihai S, et al. Induction of complement-fixing autoantibodies against type VII collagen results in subepidermal blistering in mice. J Immunol. 2006;177:3461-3468.
- Marzano AV, Cozzani E, Fanoni D, et al. Diagnosis and disease severity assessment of epidermolysis bullosa acquisita by ELISA for anti-type VII collagen autoantibodies: an Italian multicentre study. Br J Dermatol. 2013;168:80-84.
- Chen M, O’Toole EA, Sanghavi J, et al. The epidermolysis bullosa acquisita antigen (type VII collagen) is present in human colon and patients with Crohn’s disease have autoantibodies to type VII collagen. J Invest Dermatol. 2002;118:1059-1064.
- Reddy H, Shipman AR, Wojnarowska F. Epidermolysis bullosa acquisita and inflammatory bowel disease: a review of the literature. Clin Exp Dermatol. 2013;38:225-229.
- Radfar L, Fatahzadeh M, Shahamat Y, et al. Paraneoplastic epidermolysis bullosa acquisita associated with multiple myeloma. Spec Care Dentist. 2006;26:159-163.
- Engineer L, Dow EC, Braverman IM, et al. Epidermolysis bullosa acquisita and multiple myeloma. J Am Acad Dermatol. 2002;47:943-946.
- Ishii N, Hamada T, Dainichi T, et al. Epidermolysis bullosa acquisita: what’s new? J Dermatol. 2010;37:220-230.
- Megahed M, Scharffetter-Kochanek K. Epidermolysis bullosa acquisita—successful treatment with colchicine. Arch Dermatol Res. 1994;286:35-46.
- Khatri ML, Benghazeil M, Shafi M. Epidermolysis bullosa acquisita responsive to cyclosporin therapy. J Eur Acad Dermatol Venereol. 2001;15:182-184.
- Hughes AP, Callen JP. Epidermolysis bullosa acquisita responsive to dapsone therapy. J Cutan Med Surg. 2001;5:397-399.
- Kim JH, Lee SE, Kim SC. Successful treatment of epidermolysis bullosa acquisita with rituximab therapy. J Dermatol. 2012;39:477-479.
- Hitz F, Bargetzi M, Cogliatti S, et al. Diagnosis and treatment of mantle cell lymphoma. Swiss Med Wkly. 2013;143:w13868.
- Ludwig RJ. Clinical presentation, pathogenesis, diagnosis, and treatment of epidermolysis bullosa acquisita. ISRN Dermatol. 2013;2013:812029.
- Gupta R, Woodley DT, Chen M. Epidermolysis bullosa acquisita. Clin Dermatol. 2012;30:60-69.
- Mutasim DF, Adams BB. Immunofluorescence in dermatology. J Am Acad Dermatol. 2001;45:803-822.
- Woodley DT, Briggaman RA, O’Keefe EJ. Identification of the skin basement-membrane autoantigen in epidermolysis bullosa acquisita. N Engl J Med. 1984;310:1007-1013.
- Hashimoto T, Ishii N, Ohata C, et al. Pathogenesis of epidermolysis bullosa acquisita, an autoimmune subepidermal bullous disease. J Pathol. 2012;228:1-7.
- Sitaru C, Chiriac MT, Mihai S, et al. Induction of complement-fixing autoantibodies against type VII collagen results in subepidermal blistering in mice. J Immunol. 2006;177:3461-3468.
- Marzano AV, Cozzani E, Fanoni D, et al. Diagnosis and disease severity assessment of epidermolysis bullosa acquisita by ELISA for anti-type VII collagen autoantibodies: an Italian multicentre study. Br J Dermatol. 2013;168:80-84.
- Chen M, O’Toole EA, Sanghavi J, et al. The epidermolysis bullosa acquisita antigen (type VII collagen) is present in human colon and patients with Crohn’s disease have autoantibodies to type VII collagen. J Invest Dermatol. 2002;118:1059-1064.
- Reddy H, Shipman AR, Wojnarowska F. Epidermolysis bullosa acquisita and inflammatory bowel disease: a review of the literature. Clin Exp Dermatol. 2013;38:225-229.
- Radfar L, Fatahzadeh M, Shahamat Y, et al. Paraneoplastic epidermolysis bullosa acquisita associated with multiple myeloma. Spec Care Dentist. 2006;26:159-163.
- Engineer L, Dow EC, Braverman IM, et al. Epidermolysis bullosa acquisita and multiple myeloma. J Am Acad Dermatol. 2002;47:943-946.
- Ishii N, Hamada T, Dainichi T, et al. Epidermolysis bullosa acquisita: what’s new? J Dermatol. 2010;37:220-230.
- Megahed M, Scharffetter-Kochanek K. Epidermolysis bullosa acquisita—successful treatment with colchicine. Arch Dermatol Res. 1994;286:35-46.
- Khatri ML, Benghazeil M, Shafi M. Epidermolysis bullosa acquisita responsive to cyclosporin therapy. J Eur Acad Dermatol Venereol. 2001;15:182-184.
- Hughes AP, Callen JP. Epidermolysis bullosa acquisita responsive to dapsone therapy. J Cutan Med Surg. 2001;5:397-399.
- Kim JH, Lee SE, Kim SC. Successful treatment of epidermolysis bullosa acquisita with rituximab therapy. J Dermatol. 2012;39:477-479.
Practice Points
- Epidermolysis bullosa acquisita (EBA) is an uncommon blistering disorder and few cases have been associated with malignancy.
- Diagnosis of EBA is challenging and requires exclusion of other blistering diseases.
Slow-growing, Asymptomatic, Annular Plaques on the Bilateral Palms
The Diagnosis: Circumscribed Palmar Hypokeratosis
Circumscribed palmar hypokeratosis is a rare, benign, acquired dermatosis that was first described by Pérez et al1 in 2002 and is characterized by annular plaques with an atrophic center and hyperkeratotic edges. Classically, the lesions present on the thenar and hypothenar eminences of the palms.2 The condition predominantly affects women (4:1 ratio), with a mean age of onset of 65 years.3
Although the pathogenesis of circumscribed palmar hypokeratosis is unknown, local trauma generally is considered to be the causative factor. Other hypotheses include human papillomaviruses 4 and 6 infection and primary abnormal keratinization in the epidermis.3 Immunohistochemical studies have demonstrated increased expression of keratin 16 and Ki-67 in cutaneous lesions, which is postulated to be responsible for keratinocyte fragility associated with epidermal hyperproliferation. Other reported cases have shown diminished keratin 9, keratin 2e, and connexin 26 expression, which normally are abundant in the acral epidermis. Abnormal expression of antigens associated with epidermal proliferation and differentiation also have been reported,3 suggesting that there is an altered regulation of the cutaneous desquamation process.
Histologically, circumscribed palmar hypokeratosis is characterized by an abrupt reduction in the stratum corneum (Figure), forming a step between the lesion and the perilesional normal skin.2,3 The clinical appearance of erythema is due to visualization of dermal blood circulation in the area of corneal thinning and is not a result of vasodilation. The dermis is uninvolved, and inflammation is absent. The differential diagnosis includes psoriasis, Bowen disease, porokeratosis, and dermatophytosis.3
(H&E, original magnification ×4). No notable inflammation was evident in the dermis (B)(H&E, original magnification ×10).")
Circumscribed palmar hypokeratosis is a chronic condition, and there are no known reports of development of malignancy. Treatment is not required but may include cryotherapy; topical therapy with corticosteroids, retinoids, urea, and calcipotriene; and photodynamic therapy. Circumscribed hypokeratosis should be included in the differential diagnosis of palmar lesions.
- Pérez A, Rütten A, Gold R, et al. Circumscribed palmar or plantar hypokeratosis: a distinctive epidermal malformation of the palms or soles. J Am Acad Dermatol. 2002;47:21-27.
- Mitkov M, Balagula Y, Lockshin B. Case report: circumscribed plantar hypokeratosis. Int J Dermatol. 2015;54:E203-E205.
- Rocha L, Nico M. Circumscribed palmoplantar hypokeratosis: report of two Brazilian cases. An Bras Dermatol. 2013;88:623-626.
The Diagnosis: Circumscribed Palmar Hypokeratosis
Circumscribed palmar hypokeratosis is a rare, benign, acquired dermatosis that was first described by Pérez et al1 in 2002 and is characterized by annular plaques with an atrophic center and hyperkeratotic edges. Classically, the lesions present on the thenar and hypothenar eminences of the palms.2 The condition predominantly affects women (4:1 ratio), with a mean age of onset of 65 years.3
Although the pathogenesis of circumscribed palmar hypokeratosis is unknown, local trauma generally is considered to be the causative factor. Other hypotheses include human papillomaviruses 4 and 6 infection and primary abnormal keratinization in the epidermis.3 Immunohistochemical studies have demonstrated increased expression of keratin 16 and Ki-67 in cutaneous lesions, which is postulated to be responsible for keratinocyte fragility associated with epidermal hyperproliferation. Other reported cases have shown diminished keratin 9, keratin 2e, and connexin 26 expression, which normally are abundant in the acral epidermis. Abnormal expression of antigens associated with epidermal proliferation and differentiation also have been reported,3 suggesting that there is an altered regulation of the cutaneous desquamation process.
Histologically, circumscribed palmar hypokeratosis is characterized by an abrupt reduction in the stratum corneum (Figure), forming a step between the lesion and the perilesional normal skin.2,3 The clinical appearance of erythema is due to visualization of dermal blood circulation in the area of corneal thinning and is not a result of vasodilation. The dermis is uninvolved, and inflammation is absent. The differential diagnosis includes psoriasis, Bowen disease, porokeratosis, and dermatophytosis.3
Circumscribed palmar hypokeratosis is a chronic condition, and there are no known reports of development of malignancy. Treatment is not required but may include cryotherapy; topical therapy with corticosteroids, retinoids, urea, and calcipotriene; and photodynamic therapy. Circumscribed hypokeratosis should be included in the differential diagnosis of palmar lesions.
The Diagnosis: Circumscribed Palmar Hypokeratosis
Circumscribed palmar hypokeratosis is a rare, benign, acquired dermatosis that was first described by Pérez et al1 in 2002 and is characterized by annular plaques with an atrophic center and hyperkeratotic edges. Classically, the lesions present on the thenar and hypothenar eminences of the palms.2 The condition predominantly affects women (4:1 ratio), with a mean age of onset of 65 years.3
Although the pathogenesis of circumscribed palmar hypokeratosis is unknown, local trauma generally is considered to be the causative factor. Other hypotheses include human papillomaviruses 4 and 6 infection and primary abnormal keratinization in the epidermis.3 Immunohistochemical studies have demonstrated increased expression of keratin 16 and Ki-67 in cutaneous lesions, which is postulated to be responsible for keratinocyte fragility associated with epidermal hyperproliferation. Other reported cases have shown diminished keratin 9, keratin 2e, and connexin 26 expression, which normally are abundant in the acral epidermis. Abnormal expression of antigens associated with epidermal proliferation and differentiation also have been reported,3 suggesting that there is an altered regulation of the cutaneous desquamation process.
Histologically, circumscribed palmar hypokeratosis is characterized by an abrupt reduction in the stratum corneum (Figure), forming a step between the lesion and the perilesional normal skin.2,3 The clinical appearance of erythema is due to visualization of dermal blood circulation in the area of corneal thinning and is not a result of vasodilation. The dermis is uninvolved, and inflammation is absent. The differential diagnosis includes psoriasis, Bowen disease, porokeratosis, and dermatophytosis.3
Circumscribed palmar hypokeratosis is a chronic condition, and there are no known reports of development of malignancy. Treatment is not required but may include cryotherapy; topical therapy with corticosteroids, retinoids, urea, and calcipotriene; and photodynamic therapy. Circumscribed hypokeratosis should be included in the differential diagnosis of palmar lesions.
- Pérez A, Rütten A, Gold R, et al. Circumscribed palmar or plantar hypokeratosis: a distinctive epidermal malformation of the palms or soles. J Am Acad Dermatol. 2002;47:21-27.
- Mitkov M, Balagula Y, Lockshin B. Case report: circumscribed plantar hypokeratosis. Int J Dermatol. 2015;54:E203-E205.
- Rocha L, Nico M. Circumscribed palmoplantar hypokeratosis: report of two Brazilian cases. An Bras Dermatol. 2013;88:623-626.
- Pérez A, Rütten A, Gold R, et al. Circumscribed palmar or plantar hypokeratosis: a distinctive epidermal malformation of the palms or soles. J Am Acad Dermatol. 2002;47:21-27.
- Mitkov M, Balagula Y, Lockshin B. Case report: circumscribed plantar hypokeratosis. Int J Dermatol. 2015;54:E203-E205.
- Rocha L, Nico M. Circumscribed palmoplantar hypokeratosis: report of two Brazilian cases. An Bras Dermatol. 2013;88:623-626.
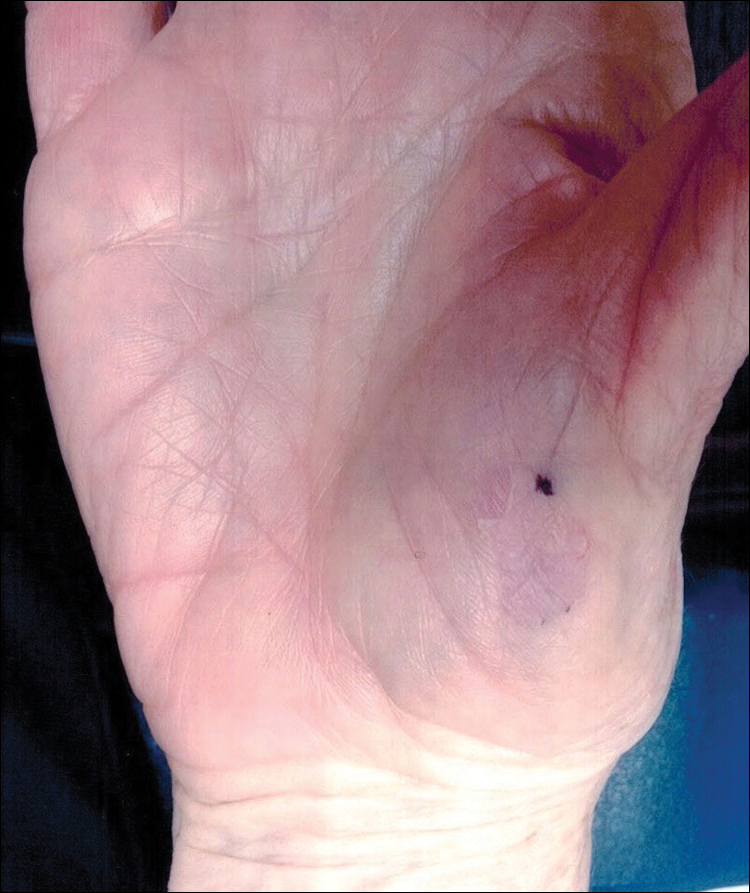
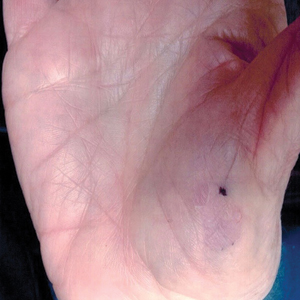
A 77-year-old woman presented with slow-growing, asymptomatic, annular plaques on the bilateral palms of many years' duration. There was no history of trauma or local infection. Prior treatment with over-the-counter creams was unsuccessful. A 3-mm punch biopsy of the lesion on the right palm was performed.
New Guidelines for Nonmelanoma Skin Cancer: What You Need to Know


Painful Nonhealing Vulvar and Perianal Erosions
The Diagnosis: Cutaneous Crohn Disease
A punch biopsy of the vulvar skin revealed epidermal hyperplasia with moderate spongiosis and exocytosis of lymphocytes and neutrophils in the epidermis. A brisk mixed inflammatory infiltrate of epithelioid histiocytes, multinucleate foreign body-type giant cells, lymphocytes, plasma cells, neutrophils, and eosinophils in a granulomatous pattern also were present in the dermis (Figure). Periodic acid-Schiff and acid-fast bacillus stains were negative. Given the history of Crohn disease (CD) and the characteristic dermal noncaseating granulomas on histology, the patient was diagnosed with cutaneous CD.
(H&E, original magnification ×4) and mixed inflammatory granulomas (B)(H&E, original magnification ×40).")
Although the patient was offered a topical corticosteroid, she deferred topical therapy. Given the lack of response to adalimumab, the gastroenterology department switched the patient to a treatment of infliximab 5 mg/kg every 8 weeks. Azathioprine was discontinued and the patient was switched to intramuscular methotrexate 25 mg/mL weekly. Slow reepithelialization of the vulvar and perianal erosions occurred on this regimen.
Although CD has numerous cutaneous features, cutaneous CD, also known as metastatic CD, is the rarest cutaneous manifestation of CD.1 This disease process is characterized by noncaseating granulomatous cutaneous lesions that are not contiguous with the affected gastrointestinal tract.2 The pathogenesis of cutaneous CD is unknown. Young adults tend to be more predisposed to developing cutaneous CD, likely due to the age distribution of CD.3
Cutaneous CD commonly presents in patients with a well-established history of gastrointestinal CD but occasionally can be the presenting sign of CD.1 The most common sites of involvement are the legs, vulva, penis, trunk, face, and intertriginous areas. Cutaneous CD findings can be divided into 2 subgroups: genital and nongenital lesions. Genital findings involve ulceration, erythema, edema, and fissuring of the vulva, labia, clitoris, scrotum, penis, and perineum. Nongenital cutaneous manifestations include ulcers; erythematous papules, plaques, and nodules; abscesslike lesions; and lichenoid papules.4,5 The severity of cutaneous lesions does not correlate to the severity of gastrointestinal disease; however, colon involvement is more common in patients with cutaneous CD.6
Histologically, cutaneous CD presents as noncaseating granulomatous inflammation in the papillary and reticular dermis. These granulomas consist of epithelioid histiocytes and multinucleated giant cells with a lymphocytic infiltrate.5
Given the rarity of cutaneous CD, treatment approach is based on anecdotal evidence from case reports and case series. For a single lesion or localized disease, topical superpotent or intralesional steroids are recommended for initial therapy.3 Oral metronidazole also is an effective treatment and can be combined with topical or intralesional steroids.7 For disseminated disease, systemic corticosteroids have shown efficacy.3 Other reported treatment options include oral corticosteroids, sulfasalazine, azathioprine, 6-mercaptopurine, infliximab, and adalimumab. If monotherapy fails, combination therapy may be needed. Surgical debridement may be attempted if medical therapy fails but is complicated by wound dehiscence and disease recurrence.3
Although genital ulcers can be a presentation of Behçet disease and genital herpes infection, genital nodules and plaques are not typical for these 2 diseases. Also, the patient did not have oral ulcers, which is a common feature of Behçet disease. Genital sarcoidosis is extremely rare, and cutaneous CD was more likely given the patient's medical history. Finally, Jacquet dermatitis is more common in children, and patients with this condition typically have history of fecal and urinary incontinence.
- Teixeira M, Machado S, Lago P, et al. Cutaneous Crohn's disease. Int J Dermatol. 2006;45:1074-1076.
- Stingeni L, Neve D, Bassotti G, et al. Cutaneous Crohn's disease successfully treated with adalimumab [published online Sep 15, 2015]. J Eur Acad Dermatol Venerol. 2016;30:E72-E74.
- Kurtzman DJ, Jones T, Fangru L, et al. Metastatic Crohn's disease: a review and approach to therapy. J Am Acad Dermatol. 2014;71:804-813.
- Hagen JW, Swoger JM, Grandinetti LM. Cutaneous manifestations of Crohn disease. Dermatol Clin. 2015;33:417-431.
- Palamaras I, El-Jabbour J, Pietropaolo N, et al. Metastatic Crohn's disease: a review [published online June 19, 2008]. J Eur Acad Dermatol Venereol. 2008;22:1033-1043.
- Thrash B, Patel M, Shah KR, et al. Cutaneous manifestations of gastrointestinal disease, part II. J Am Acad Dermatol. 2013;68:211.e1-211.e33.
- Abide JM. Metastatic Crohn disease: clearance with metronidazole. J Am Acad Dermatol. 2011;64:448-449.
The Diagnosis: Cutaneous Crohn Disease
A punch biopsy of the vulvar skin revealed epidermal hyperplasia with moderate spongiosis and exocytosis of lymphocytes and neutrophils in the epidermis. A brisk mixed inflammatory infiltrate of epithelioid histiocytes, multinucleate foreign body-type giant cells, lymphocytes, plasma cells, neutrophils, and eosinophils in a granulomatous pattern also were present in the dermis (Figure). Periodic acid-Schiff and acid-fast bacillus stains were negative. Given the history of Crohn disease (CD) and the characteristic dermal noncaseating granulomas on histology, the patient was diagnosed with cutaneous CD.
Although the patient was offered a topical corticosteroid, she deferred topical therapy. Given the lack of response to adalimumab, the gastroenterology department switched the patient to a treatment of infliximab 5 mg/kg every 8 weeks. Azathioprine was discontinued and the patient was switched to intramuscular methotrexate 25 mg/mL weekly. Slow reepithelialization of the vulvar and perianal erosions occurred on this regimen.
Although CD has numerous cutaneous features, cutaneous CD, also known as metastatic CD, is the rarest cutaneous manifestation of CD.1 This disease process is characterized by noncaseating granulomatous cutaneous lesions that are not contiguous with the affected gastrointestinal tract.2 The pathogenesis of cutaneous CD is unknown. Young adults tend to be more predisposed to developing cutaneous CD, likely due to the age distribution of CD.3
Cutaneous CD commonly presents in patients with a well-established history of gastrointestinal CD but occasionally can be the presenting sign of CD.1 The most common sites of involvement are the legs, vulva, penis, trunk, face, and intertriginous areas. Cutaneous CD findings can be divided into 2 subgroups: genital and nongenital lesions. Genital findings involve ulceration, erythema, edema, and fissuring of the vulva, labia, clitoris, scrotum, penis, and perineum. Nongenital cutaneous manifestations include ulcers; erythematous papules, plaques, and nodules; abscesslike lesions; and lichenoid papules.4,5 The severity of cutaneous lesions does not correlate to the severity of gastrointestinal disease; however, colon involvement is more common in patients with cutaneous CD.6
Histologically, cutaneous CD presents as noncaseating granulomatous inflammation in the papillary and reticular dermis. These granulomas consist of epithelioid histiocytes and multinucleated giant cells with a lymphocytic infiltrate.5
Given the rarity of cutaneous CD, treatment approach is based on anecdotal evidence from case reports and case series. For a single lesion or localized disease, topical superpotent or intralesional steroids are recommended for initial therapy.3 Oral metronidazole also is an effective treatment and can be combined with topical or intralesional steroids.7 For disseminated disease, systemic corticosteroids have shown efficacy.3 Other reported treatment options include oral corticosteroids, sulfasalazine, azathioprine, 6-mercaptopurine, infliximab, and adalimumab. If monotherapy fails, combination therapy may be needed. Surgical debridement may be attempted if medical therapy fails but is complicated by wound dehiscence and disease recurrence.3
Although genital ulcers can be a presentation of Behçet disease and genital herpes infection, genital nodules and plaques are not typical for these 2 diseases. Also, the patient did not have oral ulcers, which is a common feature of Behçet disease. Genital sarcoidosis is extremely rare, and cutaneous CD was more likely given the patient's medical history. Finally, Jacquet dermatitis is more common in children, and patients with this condition typically have history of fecal and urinary incontinence.
The Diagnosis: Cutaneous Crohn Disease
A punch biopsy of the vulvar skin revealed epidermal hyperplasia with moderate spongiosis and exocytosis of lymphocytes and neutrophils in the epidermis. A brisk mixed inflammatory infiltrate of epithelioid histiocytes, multinucleate foreign body-type giant cells, lymphocytes, plasma cells, neutrophils, and eosinophils in a granulomatous pattern also were present in the dermis (Figure). Periodic acid-Schiff and acid-fast bacillus stains were negative. Given the history of Crohn disease (CD) and the characteristic dermal noncaseating granulomas on histology, the patient was diagnosed with cutaneous CD.
Although the patient was offered a topical corticosteroid, she deferred topical therapy. Given the lack of response to adalimumab, the gastroenterology department switched the patient to a treatment of infliximab 5 mg/kg every 8 weeks. Azathioprine was discontinued and the patient was switched to intramuscular methotrexate 25 mg/mL weekly. Slow reepithelialization of the vulvar and perianal erosions occurred on this regimen.
Although CD has numerous cutaneous features, cutaneous CD, also known as metastatic CD, is the rarest cutaneous manifestation of CD.1 This disease process is characterized by noncaseating granulomatous cutaneous lesions that are not contiguous with the affected gastrointestinal tract.2 The pathogenesis of cutaneous CD is unknown. Young adults tend to be more predisposed to developing cutaneous CD, likely due to the age distribution of CD.3
Cutaneous CD commonly presents in patients with a well-established history of gastrointestinal CD but occasionally can be the presenting sign of CD.1 The most common sites of involvement are the legs, vulva, penis, trunk, face, and intertriginous areas. Cutaneous CD findings can be divided into 2 subgroups: genital and nongenital lesions. Genital findings involve ulceration, erythema, edema, and fissuring of the vulva, labia, clitoris, scrotum, penis, and perineum. Nongenital cutaneous manifestations include ulcers; erythematous papules, plaques, and nodules; abscesslike lesions; and lichenoid papules.4,5 The severity of cutaneous lesions does not correlate to the severity of gastrointestinal disease; however, colon involvement is more common in patients with cutaneous CD.6
Histologically, cutaneous CD presents as noncaseating granulomatous inflammation in the papillary and reticular dermis. These granulomas consist of epithelioid histiocytes and multinucleated giant cells with a lymphocytic infiltrate.5
Given the rarity of cutaneous CD, treatment approach is based on anecdotal evidence from case reports and case series. For a single lesion or localized disease, topical superpotent or intralesional steroids are recommended for initial therapy.3 Oral metronidazole also is an effective treatment and can be combined with topical or intralesional steroids.7 For disseminated disease, systemic corticosteroids have shown efficacy.3 Other reported treatment options include oral corticosteroids, sulfasalazine, azathioprine, 6-mercaptopurine, infliximab, and adalimumab. If monotherapy fails, combination therapy may be needed. Surgical debridement may be attempted if medical therapy fails but is complicated by wound dehiscence and disease recurrence.3
Although genital ulcers can be a presentation of Behçet disease and genital herpes infection, genital nodules and plaques are not typical for these 2 diseases. Also, the patient did not have oral ulcers, which is a common feature of Behçet disease. Genital sarcoidosis is extremely rare, and cutaneous CD was more likely given the patient's medical history. Finally, Jacquet dermatitis is more common in children, and patients with this condition typically have history of fecal and urinary incontinence.
- Teixeira M, Machado S, Lago P, et al. Cutaneous Crohn's disease. Int J Dermatol. 2006;45:1074-1076.
- Stingeni L, Neve D, Bassotti G, et al. Cutaneous Crohn's disease successfully treated with adalimumab [published online Sep 15, 2015]. J Eur Acad Dermatol Venerol. 2016;30:E72-E74.
- Kurtzman DJ, Jones T, Fangru L, et al. Metastatic Crohn's disease: a review and approach to therapy. J Am Acad Dermatol. 2014;71:804-813.
- Hagen JW, Swoger JM, Grandinetti LM. Cutaneous manifestations of Crohn disease. Dermatol Clin. 2015;33:417-431.
- Palamaras I, El-Jabbour J, Pietropaolo N, et al. Metastatic Crohn's disease: a review [published online June 19, 2008]. J Eur Acad Dermatol Venereol. 2008;22:1033-1043.
- Thrash B, Patel M, Shah KR, et al. Cutaneous manifestations of gastrointestinal disease, part II. J Am Acad Dermatol. 2013;68:211.e1-211.e33.
- Abide JM. Metastatic Crohn disease: clearance with metronidazole. J Am Acad Dermatol. 2011;64:448-449.
- Teixeira M, Machado S, Lago P, et al. Cutaneous Crohn's disease. Int J Dermatol. 2006;45:1074-1076.
- Stingeni L, Neve D, Bassotti G, et al. Cutaneous Crohn's disease successfully treated with adalimumab [published online Sep 15, 2015]. J Eur Acad Dermatol Venerol. 2016;30:E72-E74.
- Kurtzman DJ, Jones T, Fangru L, et al. Metastatic Crohn's disease: a review and approach to therapy. J Am Acad Dermatol. 2014;71:804-813.
- Hagen JW, Swoger JM, Grandinetti LM. Cutaneous manifestations of Crohn disease. Dermatol Clin. 2015;33:417-431.
- Palamaras I, El-Jabbour J, Pietropaolo N, et al. Metastatic Crohn's disease: a review [published online June 19, 2008]. J Eur Acad Dermatol Venereol. 2008;22:1033-1043.
- Thrash B, Patel M, Shah KR, et al. Cutaneous manifestations of gastrointestinal disease, part II. J Am Acad Dermatol. 2013;68:211.e1-211.e33.
- Abide JM. Metastatic Crohn disease: clearance with metronidazole. J Am Acad Dermatol. 2011;64:448-449.
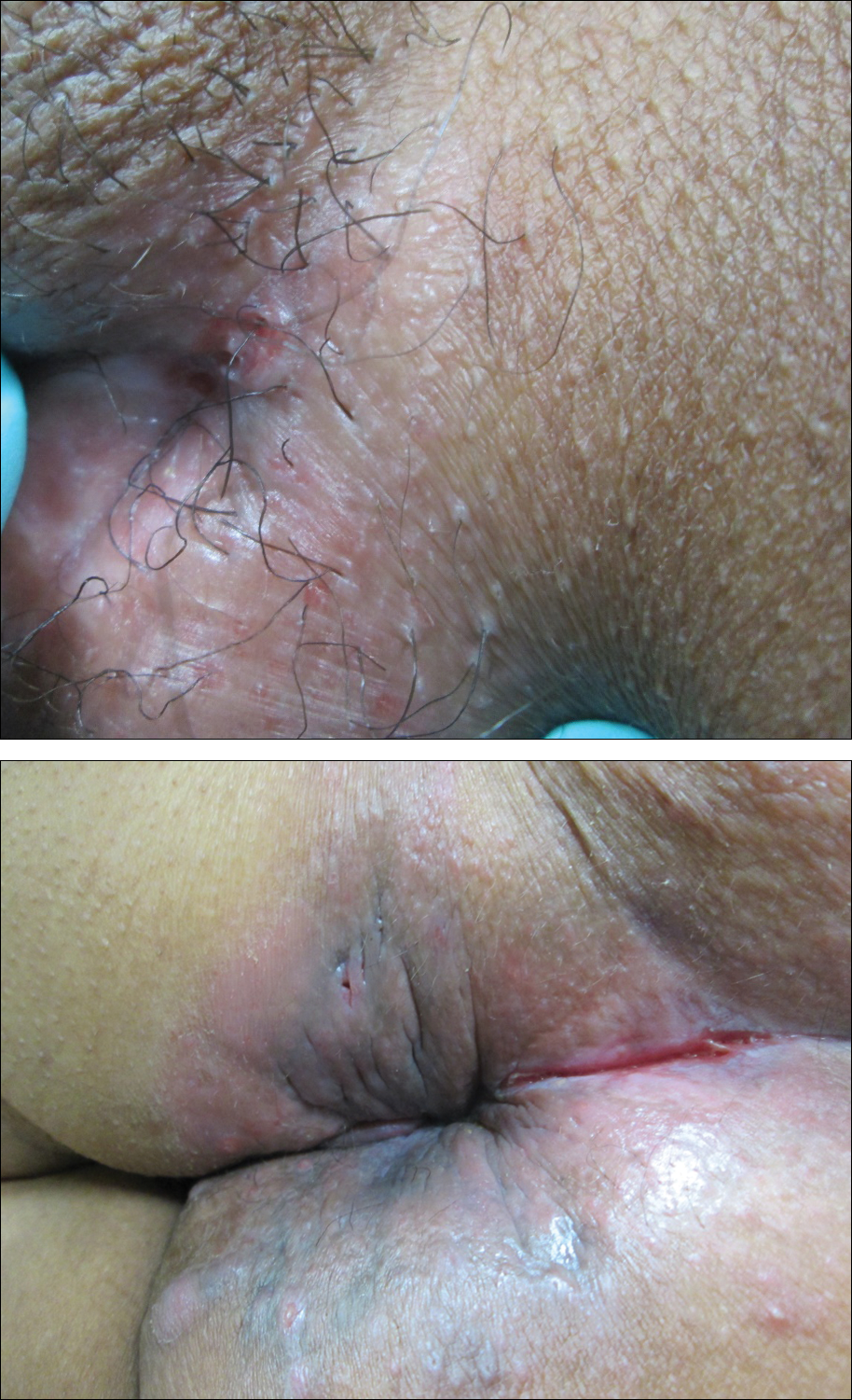
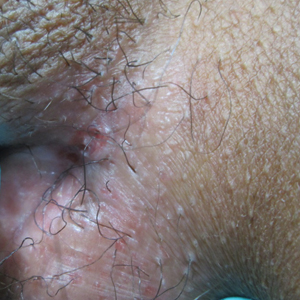
A 38-year-old woman with a history of Crohn disease presented with painful nonhealing vulvar and perianal erosions of 6 months' duration. The erosions developed 4 months after discontinuing adalimumab for a planned surgery. During this time, the patient also had an exacerbation of Crohn colitis and developed an anal fistula. Prior to this break in adalimumab, the patient's Crohn disease was well controlled on adalimumab 40 mg every 2 weeks, azathioprine 100 mg daily, and mesalamine 4.8 g daily. Despite restarting adalimumab and therapy with multiple antibiotics (ie, metronidazole, ciprofloxacin), the erosions persisted. On physical examination erythematous plaques and nodules were present at the vulvar (top) and perianal (bottom) skin. In addition, well-demarcated erosions measuring 20 mm and 80 mm were present on the vulvar and perianal skin, respectively. Human immunodeficiency virus screening and rapid plasma reagin were negative.
Polypoid Melanoma: An Aggressive Variant of Nodular Melanoma
To the Editor:
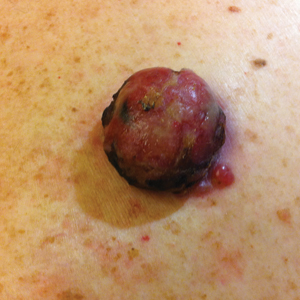
An 81-year-old man presented with a nodular polypoid lesion that developed on a flat lesion on the back of 2 years’ duration. The lesion grew progressively over the course of 3 months prior to presentation. The patient had a history of melanoma in situ on the forehead that was treated with conventional surgery with clear surgical margins 6 years prior to the current presentation.
On physical examination the patient had a 4×2-cm ulcerated polypoid lesion on the back. The lesion was pink with a pigmented base. Additionally, 2 pink papules with superficial telangiectases were observed around the main lesion (Figure 1).
The gross section showed an exophytic tumor largely growing above the skin surface (Figure 2). Histopathologic analysis revealed an ulcerated lesion consisting of confluent nest and sheets of epithelioid and spindle atypical cells with numerous mitotic figures and necrotic foci (Figure 3). The thickness of the lesion was 2200 µm, and the mitotic count was 28 mitoses/mm2. There also was peritumoral vascular invasion and satellite metastasis within the perilesional hypodermis measuring 0.4 mm. Immunohistochemistry staining for S-100, human melanoma black 45 (HMB-45)(Figure 4), and Melan-A was positive in neoplastic cells.


.")
 immunostain (original magnification ×200).")
The dissemination study revealed multiple mediastinal and axillary lymphadenopathies and lesions with metastatic appearance in the brain, liver, pancreas, and muscle, together with peritoneal carcinomatosis. The patient was lost to follow-up and did not follow coadjuvant therapy with interferon alfa.
Polypoid melanoma initially was described as a type of melanoma characterized by an exophytic growth in which most of the tumor is located on the cutaneous surface, together with ulceration.1 It usually occurs in patients aged 20 to 39 years,2 and the reported incidence ranges from 1.9% to 43.3%.1 It more commonly affects mucosae, including the upper respiratory tract, esophagus, and vagina. Polypoid melanoma has a rapid progression and a poor prognosis.3 Polypoid melanoma involving the skin primarily affects the back and has a 5-year survival rate of 32% to 42%.4 Poor prognosis has been attributed to the high risk for vascular embolization under the lesion.5 Histologically, there is marked cell atypia with nuclear and cellular pleomorphism and a high mitotic count. The tumor rarely involves the reticular dermis.1,2
Polypoid melanomas are rare; however, reported frequency rates cover a wide range. These frequency rates may be due to the definition of polypoid melanoma used by the pathologist issuing the report. One of the most accepted definitions at present is a pigmented macule that progresses in months with a rapid vertical growth, invading the epidermis and the papillary dermis.2 The differential diagnosis includes pyogenic granuloma, squamous cell carcinoma, basal cell carcinoma, soft tissue sarcomas, and hemangioma.
Although our patient had a history of melanoma and the polypoid lesion developed from a flat lesion, he was late to seek medical care. The diagnosis of melanoma is made on increasingly smaller lesions with better prognosis, but there still are reports of larger melanomas. This case highlights the role dermatologists serve in the education of patients on their diagnoses and risk factors so that we may be able to diagnose non–life-threatening small lesions. It is important to remember this morphologic variety of melanoma and highlight its rapid progression and poor prognosis.
- Knezević F, Duancić V, Sitić S, et al. Histological types of polypoid cutaneous melanoma II. Coll Antropol. 2007;31:1049-1053.
- Dini M, Quercioli F, Caldarella V, et al. Head and neck polypoid melanoma. J Craniofac Surg. 2012;23:E23-E25.
- Plotnick H, Rachmaninoff N, VandenBerg HJ Jr. Polypoid melanoma: a virulent variant of nodular melanoma. report of three cases and literature review. J Am Acad Dermatol. 1990;23(5, pt 1):880-884.
- Manci EA, Balch CM, Murad TM, et al. Polypoid melanoma, a virulent variant of the nodular growth pattern. Am J Clin Pathol. 1981;75:810-815.
- De Giorgi V, Massi D, Gerlini G, et al. Immediate local and regional recurrence after the excision of a polypoid melanoma: tumor dormancy or tumor activation? Dermatol Surg. 2003;29:664-667.
To the Editor:
An 81-year-old man presented with a nodular polypoid lesion that developed on a flat lesion on the back of 2 years’ duration. The lesion grew progressively over the course of 3 months prior to presentation. The patient had a history of melanoma in situ on the forehead that was treated with conventional surgery with clear surgical margins 6 years prior to the current presentation.
On physical examination the patient had a 4×2-cm ulcerated polypoid lesion on the back. The lesion was pink with a pigmented base. Additionally, 2 pink papules with superficial telangiectases were observed around the main lesion (Figure 1).
The gross section showed an exophytic tumor largely growing above the skin surface (Figure 2). Histopathologic analysis revealed an ulcerated lesion consisting of confluent nest and sheets of epithelioid and spindle atypical cells with numerous mitotic figures and necrotic foci (Figure 3). The thickness of the lesion was 2200 µm, and the mitotic count was 28 mitoses/mm2. There also was peritumoral vascular invasion and satellite metastasis within the perilesional hypodermis measuring 0.4 mm. Immunohistochemistry staining for S-100, human melanoma black 45 (HMB-45)(Figure 4), and Melan-A was positive in neoplastic cells.
The dissemination study revealed multiple mediastinal and axillary lymphadenopathies and lesions with metastatic appearance in the brain, liver, pancreas, and muscle, together with peritoneal carcinomatosis. The patient was lost to follow-up and did not follow coadjuvant therapy with interferon alfa.
Polypoid melanoma initially was described as a type of melanoma characterized by an exophytic growth in which most of the tumor is located on the cutaneous surface, together with ulceration.1 It usually occurs in patients aged 20 to 39 years,2 and the reported incidence ranges from 1.9% to 43.3%.1 It more commonly affects mucosae, including the upper respiratory tract, esophagus, and vagina. Polypoid melanoma has a rapid progression and a poor prognosis.3 Polypoid melanoma involving the skin primarily affects the back and has a 5-year survival rate of 32% to 42%.4 Poor prognosis has been attributed to the high risk for vascular embolization under the lesion.5 Histologically, there is marked cell atypia with nuclear and cellular pleomorphism and a high mitotic count. The tumor rarely involves the reticular dermis.1,2
Polypoid melanomas are rare; however, reported frequency rates cover a wide range. These frequency rates may be due to the definition of polypoid melanoma used by the pathologist issuing the report. One of the most accepted definitions at present is a pigmented macule that progresses in months with a rapid vertical growth, invading the epidermis and the papillary dermis.2 The differential diagnosis includes pyogenic granuloma, squamous cell carcinoma, basal cell carcinoma, soft tissue sarcomas, and hemangioma.
Although our patient had a history of melanoma and the polypoid lesion developed from a flat lesion, he was late to seek medical care. The diagnosis of melanoma is made on increasingly smaller lesions with better prognosis, but there still are reports of larger melanomas. This case highlights the role dermatologists serve in the education of patients on their diagnoses and risk factors so that we may be able to diagnose non–life-threatening small lesions. It is important to remember this morphologic variety of melanoma and highlight its rapid progression and poor prognosis.
To the Editor:
An 81-year-old man presented with a nodular polypoid lesion that developed on a flat lesion on the back of 2 years’ duration. The lesion grew progressively over the course of 3 months prior to presentation. The patient had a history of melanoma in situ on the forehead that was treated with conventional surgery with clear surgical margins 6 years prior to the current presentation.
On physical examination the patient had a 4×2-cm ulcerated polypoid lesion on the back. The lesion was pink with a pigmented base. Additionally, 2 pink papules with superficial telangiectases were observed around the main lesion (Figure 1).
The gross section showed an exophytic tumor largely growing above the skin surface (Figure 2). Histopathologic analysis revealed an ulcerated lesion consisting of confluent nest and sheets of epithelioid and spindle atypical cells with numerous mitotic figures and necrotic foci (Figure 3). The thickness of the lesion was 2200 µm, and the mitotic count was 28 mitoses/mm2. There also was peritumoral vascular invasion and satellite metastasis within the perilesional hypodermis measuring 0.4 mm. Immunohistochemistry staining for S-100, human melanoma black 45 (HMB-45)(Figure 4), and Melan-A was positive in neoplastic cells.
The dissemination study revealed multiple mediastinal and axillary lymphadenopathies and lesions with metastatic appearance in the brain, liver, pancreas, and muscle, together with peritoneal carcinomatosis. The patient was lost to follow-up and did not follow coadjuvant therapy with interferon alfa.
Polypoid melanoma initially was described as a type of melanoma characterized by an exophytic growth in which most of the tumor is located on the cutaneous surface, together with ulceration.1 It usually occurs in patients aged 20 to 39 years,2 and the reported incidence ranges from 1.9% to 43.3%.1 It more commonly affects mucosae, including the upper respiratory tract, esophagus, and vagina. Polypoid melanoma has a rapid progression and a poor prognosis.3 Polypoid melanoma involving the skin primarily affects the back and has a 5-year survival rate of 32% to 42%.4 Poor prognosis has been attributed to the high risk for vascular embolization under the lesion.5 Histologically, there is marked cell atypia with nuclear and cellular pleomorphism and a high mitotic count. The tumor rarely involves the reticular dermis.1,2
Polypoid melanomas are rare; however, reported frequency rates cover a wide range. These frequency rates may be due to the definition of polypoid melanoma used by the pathologist issuing the report. One of the most accepted definitions at present is a pigmented macule that progresses in months with a rapid vertical growth, invading the epidermis and the papillary dermis.2 The differential diagnosis includes pyogenic granuloma, squamous cell carcinoma, basal cell carcinoma, soft tissue sarcomas, and hemangioma.
Although our patient had a history of melanoma and the polypoid lesion developed from a flat lesion, he was late to seek medical care. The diagnosis of melanoma is made on increasingly smaller lesions with better prognosis, but there still are reports of larger melanomas. This case highlights the role dermatologists serve in the education of patients on their diagnoses and risk factors so that we may be able to diagnose non–life-threatening small lesions. It is important to remember this morphologic variety of melanoma and highlight its rapid progression and poor prognosis.
- Knezević F, Duancić V, Sitić S, et al. Histological types of polypoid cutaneous melanoma II. Coll Antropol. 2007;31:1049-1053.
- Dini M, Quercioli F, Caldarella V, et al. Head and neck polypoid melanoma. J Craniofac Surg. 2012;23:E23-E25.
- Plotnick H, Rachmaninoff N, VandenBerg HJ Jr. Polypoid melanoma: a virulent variant of nodular melanoma. report of three cases and literature review. J Am Acad Dermatol. 1990;23(5, pt 1):880-884.
- Manci EA, Balch CM, Murad TM, et al. Polypoid melanoma, a virulent variant of the nodular growth pattern. Am J Clin Pathol. 1981;75:810-815.
- De Giorgi V, Massi D, Gerlini G, et al. Immediate local and regional recurrence after the excision of a polypoid melanoma: tumor dormancy or tumor activation? Dermatol Surg. 2003;29:664-667.
- Knezević F, Duancić V, Sitić S, et al. Histological types of polypoid cutaneous melanoma II. Coll Antropol. 2007;31:1049-1053.
- Dini M, Quercioli F, Caldarella V, et al. Head and neck polypoid melanoma. J Craniofac Surg. 2012;23:E23-E25.
- Plotnick H, Rachmaninoff N, VandenBerg HJ Jr. Polypoid melanoma: a virulent variant of nodular melanoma. report of three cases and literature review. J Am Acad Dermatol. 1990;23(5, pt 1):880-884.
- Manci EA, Balch CM, Murad TM, et al. Polypoid melanoma, a virulent variant of the nodular growth pattern. Am J Clin Pathol. 1981;75:810-815.
- De Giorgi V, Massi D, Gerlini G, et al. Immediate local and regional recurrence after the excision of a polypoid melanoma: tumor dormancy or tumor activation? Dermatol Surg. 2003;29:664-667.
Practice Points
- The differential diagnosis of polypoid melanoma includes pyogenic granuloma and squamous cell carcinoma.
- Polypoid melanoma has a poor prognosis because of its thickness and ulceration at the time of diagnosis and the risk of vascular embolization.
Scaly Annular and Concentric Plaques
The Diagnosis: Annular Psoriasis
Because the patient's history was nonconcordant with the clinical appearance, a 4-mm punch biopsy was performed from a lesion on the left hip. Hematoxylin and eosin-stained sections demonstrated mild irregular acanthosis of the epidermis with discrete mounds of parakeratin (Figure 1A). Higher power revealed numerous neutrophils entrapped within focal scale crusts (Figure 1B). Periodic acid-Schiff stain for fungus demonstrated no hyphal elements or yeast forms in the stratum corneum. These histopathology findings were consistent with the diagnosis of annular psoriasis.
(H&E, original magnification ×4) with neutrophils entrapped in thescale (B)(H&E, original magnification ×20).")
The manifestation of psoriasis may take many forms, ranging from classic plaques to pustular eruptions--either annular or generalized--and erythroderma. Primarily annular plaque-type psoriasis without pustules, however, remains an uncommon finding.1 Psoriatic plaques may become annular or arcuate with central clearing from partial treatment with topical medications, though our patient reported annular plaques prior to any treatment. His presentation was notably different than annular pustular psoriasis in that there were no pustules in the leading edge, and there was no trailing scale, which is typical of annular pustular psoriasis.
Topical triamcinolone prescribed at the initial presentation to the dermatology department helped with pruritus, but due to the large body surface area involved, methotrexate later was initiated. After a 10-mg test dose of methotrexate and titration to 15 mg weekly, dramatic improvement in the rash was noted after 8 weeks. As the rash resolved, only faint hyperpigmented patches remained (Figure 2).

Erythema gyratum repens is a rare paraneoplastic syndrome that presents with annular scaly plaques with concentric circles with a wood grain-like appearance. The borders can advance up to 1 cm daily and show nonspecific findings on histopathology.2 Due to the observation that approximately 80% of cases of erythema gyratum repens were associated with an underlying malignancy, most often of the lung,3 this diagnosis was entertained given our patient's clinical presentation.
Erythema annulare centrifugum (EAC) historically has been divided into 2 forms: superficial and deep.4 Both present with slowly expanding, annular, pink plaques. Superficial EAC demonstrates parakeratosis and trailing scale and has not been proven to be associated with other systemic diseases, while deep EAC has infiltrated borders without scale, and many cases of EAC may represent annular forms of tumid lupus.4 Inflammatory cells may cuff vessels tightly, resulting in so-called coat sleeve infiltrate in superficial EAC. Along with trailing scale, this finding suggests the diagnosis. It has been argued that EAC is not an entity on its own and should prompt evaluation for lupus erythematosus, dermatitis, hypersensitivity to tinea pedis, and Lyme disease in appropriate circumstances.5
Tinea corporis always should be considered when evaluating annular scaly plaques with central clearing. Diagnosis and treatment are straightforward when hyphae are found on microscopy of skin scrapings or seen on periodic acid-Schiff stains of formalin-fixed tissue. Tinea imbricata presents with an interesting morphology and appears more ornate or cerebriform than tinea corporis caused by Trichophyton rubrum. It is caused by infection with Trichophyton circumscriptum and occurs in certain regions in the South Pacific, Southeast Asia, and Central and South America, making the diagnosis within the United States unlikely for a patient who has not traveled to these areas.6
Erythema chronicum migrans is diagnostic of Lyme disease infection with Borrelia burgdorferi, and solitary lesions occur surrounding the site of a tick bite in the majority of patients. Only 20% of patients will develop multiple lesions consistent with erythema chronicum migrans due to multiple tick bites, spirochetemia, or lymphatic spread.7 Up to one-third of patients are unaware that they were bitten by a tick. In endemic areas, this diagnosis must be entertained in any patient presenting with an annular rash, as treatment may prevent notable morbidity.
- Guill C, Hoang M, Carder K. Primary annular plaque-type psoriasis. Pediatr Dermatol. 2005;22:15-18.
- Boyd A, Neldner K, Menter A. Erythema gyratum repens: a paraneoplastic eruption. J Am Acad Dermatol. 1992;26:757-762.
- Kawakami T, Saito R. Erythema gyratum repens unassociated with underlying malignancy. J Dermatol. 1995;22:587-589.
- Weyers W, Diaz-Cascajo C, Weyers I. Erythema annulare centrifugum: results of a clinicopathologic study of 73 patients. Am J Dermatopathol. 2003;25:451-462.
- Ziemer M, Eisendle K, Zelger B. New concepts on erythema annulare centrifugum: a clinical reaction pattern that does notrepresent a specific clinicopathological entity. Br J Dermatol. 2009;160:119-126.
- Bonifaz A, Vázquez-González D. Tinea imbricata in the Americas. Curr Opin Infect Dis. 2011;24:106-111.
- Müllegger R, Glatz M. Skin manifestations of Lyme borreliosis: diagnosis and management. Am J Clin Dermatol. 2008;9:355-368.
The Diagnosis: Annular Psoriasis
Because the patient's history was nonconcordant with the clinical appearance, a 4-mm punch biopsy was performed from a lesion on the left hip. Hematoxylin and eosin-stained sections demonstrated mild irregular acanthosis of the epidermis with discrete mounds of parakeratin (Figure 1A). Higher power revealed numerous neutrophils entrapped within focal scale crusts (Figure 1B). Periodic acid-Schiff stain for fungus demonstrated no hyphal elements or yeast forms in the stratum corneum. These histopathology findings were consistent with the diagnosis of annular psoriasis.
The manifestation of psoriasis may take many forms, ranging from classic plaques to pustular eruptions--either annular or generalized--and erythroderma. Primarily annular plaque-type psoriasis without pustules, however, remains an uncommon finding.1 Psoriatic plaques may become annular or arcuate with central clearing from partial treatment with topical medications, though our patient reported annular plaques prior to any treatment. His presentation was notably different than annular pustular psoriasis in that there were no pustules in the leading edge, and there was no trailing scale, which is typical of annular pustular psoriasis.
Topical triamcinolone prescribed at the initial presentation to the dermatology department helped with pruritus, but due to the large body surface area involved, methotrexate later was initiated. After a 10-mg test dose of methotrexate and titration to 15 mg weekly, dramatic improvement in the rash was noted after 8 weeks. As the rash resolved, only faint hyperpigmented patches remained (Figure 2).
Erythema gyratum repens is a rare paraneoplastic syndrome that presents with annular scaly plaques with concentric circles with a wood grain-like appearance. The borders can advance up to 1 cm daily and show nonspecific findings on histopathology.2 Due to the observation that approximately 80% of cases of erythema gyratum repens were associated with an underlying malignancy, most often of the lung,3 this diagnosis was entertained given our patient's clinical presentation.
Erythema annulare centrifugum (EAC) historically has been divided into 2 forms: superficial and deep.4 Both present with slowly expanding, annular, pink plaques. Superficial EAC demonstrates parakeratosis and trailing scale and has not been proven to be associated with other systemic diseases, while deep EAC has infiltrated borders without scale, and many cases of EAC may represent annular forms of tumid lupus.4 Inflammatory cells may cuff vessels tightly, resulting in so-called coat sleeve infiltrate in superficial EAC. Along with trailing scale, this finding suggests the diagnosis. It has been argued that EAC is not an entity on its own and should prompt evaluation for lupus erythematosus, dermatitis, hypersensitivity to tinea pedis, and Lyme disease in appropriate circumstances.5
Tinea corporis always should be considered when evaluating annular scaly plaques with central clearing. Diagnosis and treatment are straightforward when hyphae are found on microscopy of skin scrapings or seen on periodic acid-Schiff stains of formalin-fixed tissue. Tinea imbricata presents with an interesting morphology and appears more ornate or cerebriform than tinea corporis caused by Trichophyton rubrum. It is caused by infection with Trichophyton circumscriptum and occurs in certain regions in the South Pacific, Southeast Asia, and Central and South America, making the diagnosis within the United States unlikely for a patient who has not traveled to these areas.6
Erythema chronicum migrans is diagnostic of Lyme disease infection with Borrelia burgdorferi, and solitary lesions occur surrounding the site of a tick bite in the majority of patients. Only 20% of patients will develop multiple lesions consistent with erythema chronicum migrans due to multiple tick bites, spirochetemia, or lymphatic spread.7 Up to one-third of patients are unaware that they were bitten by a tick. In endemic areas, this diagnosis must be entertained in any patient presenting with an annular rash, as treatment may prevent notable morbidity.
The Diagnosis: Annular Psoriasis
Because the patient's history was nonconcordant with the clinical appearance, a 4-mm punch biopsy was performed from a lesion on the left hip. Hematoxylin and eosin-stained sections demonstrated mild irregular acanthosis of the epidermis with discrete mounds of parakeratin (Figure 1A). Higher power revealed numerous neutrophils entrapped within focal scale crusts (Figure 1B). Periodic acid-Schiff stain for fungus demonstrated no hyphal elements or yeast forms in the stratum corneum. These histopathology findings were consistent with the diagnosis of annular psoriasis.
The manifestation of psoriasis may take many forms, ranging from classic plaques to pustular eruptions--either annular or generalized--and erythroderma. Primarily annular plaque-type psoriasis without pustules, however, remains an uncommon finding.1 Psoriatic plaques may become annular or arcuate with central clearing from partial treatment with topical medications, though our patient reported annular plaques prior to any treatment. His presentation was notably different than annular pustular psoriasis in that there were no pustules in the leading edge, and there was no trailing scale, which is typical of annular pustular psoriasis.
Topical triamcinolone prescribed at the initial presentation to the dermatology department helped with pruritus, but due to the large body surface area involved, methotrexate later was initiated. After a 10-mg test dose of methotrexate and titration to 15 mg weekly, dramatic improvement in the rash was noted after 8 weeks. As the rash resolved, only faint hyperpigmented patches remained (Figure 2).
Erythema gyratum repens is a rare paraneoplastic syndrome that presents with annular scaly plaques with concentric circles with a wood grain-like appearance. The borders can advance up to 1 cm daily and show nonspecific findings on histopathology.2 Due to the observation that approximately 80% of cases of erythema gyratum repens were associated with an underlying malignancy, most often of the lung,3 this diagnosis was entertained given our patient's clinical presentation.
Erythema annulare centrifugum (EAC) historically has been divided into 2 forms: superficial and deep.4 Both present with slowly expanding, annular, pink plaques. Superficial EAC demonstrates parakeratosis and trailing scale and has not been proven to be associated with other systemic diseases, while deep EAC has infiltrated borders without scale, and many cases of EAC may represent annular forms of tumid lupus.4 Inflammatory cells may cuff vessels tightly, resulting in so-called coat sleeve infiltrate in superficial EAC. Along with trailing scale, this finding suggests the diagnosis. It has been argued that EAC is not an entity on its own and should prompt evaluation for lupus erythematosus, dermatitis, hypersensitivity to tinea pedis, and Lyme disease in appropriate circumstances.5
Tinea corporis always should be considered when evaluating annular scaly plaques with central clearing. Diagnosis and treatment are straightforward when hyphae are found on microscopy of skin scrapings or seen on periodic acid-Schiff stains of formalin-fixed tissue. Tinea imbricata presents with an interesting morphology and appears more ornate or cerebriform than tinea corporis caused by Trichophyton rubrum. It is caused by infection with Trichophyton circumscriptum and occurs in certain regions in the South Pacific, Southeast Asia, and Central and South America, making the diagnosis within the United States unlikely for a patient who has not traveled to these areas.6
Erythema chronicum migrans is diagnostic of Lyme disease infection with Borrelia burgdorferi, and solitary lesions occur surrounding the site of a tick bite in the majority of patients. Only 20% of patients will develop multiple lesions consistent with erythema chronicum migrans due to multiple tick bites, spirochetemia, or lymphatic spread.7 Up to one-third of patients are unaware that they were bitten by a tick. In endemic areas, this diagnosis must be entertained in any patient presenting with an annular rash, as treatment may prevent notable morbidity.
- Guill C, Hoang M, Carder K. Primary annular plaque-type psoriasis. Pediatr Dermatol. 2005;22:15-18.
- Boyd A, Neldner K, Menter A. Erythema gyratum repens: a paraneoplastic eruption. J Am Acad Dermatol. 1992;26:757-762.
- Kawakami T, Saito R. Erythema gyratum repens unassociated with underlying malignancy. J Dermatol. 1995;22:587-589.
- Weyers W, Diaz-Cascajo C, Weyers I. Erythema annulare centrifugum: results of a clinicopathologic study of 73 patients. Am J Dermatopathol. 2003;25:451-462.
- Ziemer M, Eisendle K, Zelger B. New concepts on erythema annulare centrifugum: a clinical reaction pattern that does notrepresent a specific clinicopathological entity. Br J Dermatol. 2009;160:119-126.
- Bonifaz A, Vázquez-González D. Tinea imbricata in the Americas. Curr Opin Infect Dis. 2011;24:106-111.
- Müllegger R, Glatz M. Skin manifestations of Lyme borreliosis: diagnosis and management. Am J Clin Dermatol. 2008;9:355-368.
- Guill C, Hoang M, Carder K. Primary annular plaque-type psoriasis. Pediatr Dermatol. 2005;22:15-18.
- Boyd A, Neldner K, Menter A. Erythema gyratum repens: a paraneoplastic eruption. J Am Acad Dermatol. 1992;26:757-762.
- Kawakami T, Saito R. Erythema gyratum repens unassociated with underlying malignancy. J Dermatol. 1995;22:587-589.
- Weyers W, Diaz-Cascajo C, Weyers I. Erythema annulare centrifugum: results of a clinicopathologic study of 73 patients. Am J Dermatopathol. 2003;25:451-462.
- Ziemer M, Eisendle K, Zelger B. New concepts on erythema annulare centrifugum: a clinical reaction pattern that does notrepresent a specific clinicopathological entity. Br J Dermatol. 2009;160:119-126.
- Bonifaz A, Vázquez-González D. Tinea imbricata in the Americas. Curr Opin Infect Dis. 2011;24:106-111.
- Müllegger R, Glatz M. Skin manifestations of Lyme borreliosis: diagnosis and management. Am J Clin Dermatol. 2008;9:355-368.
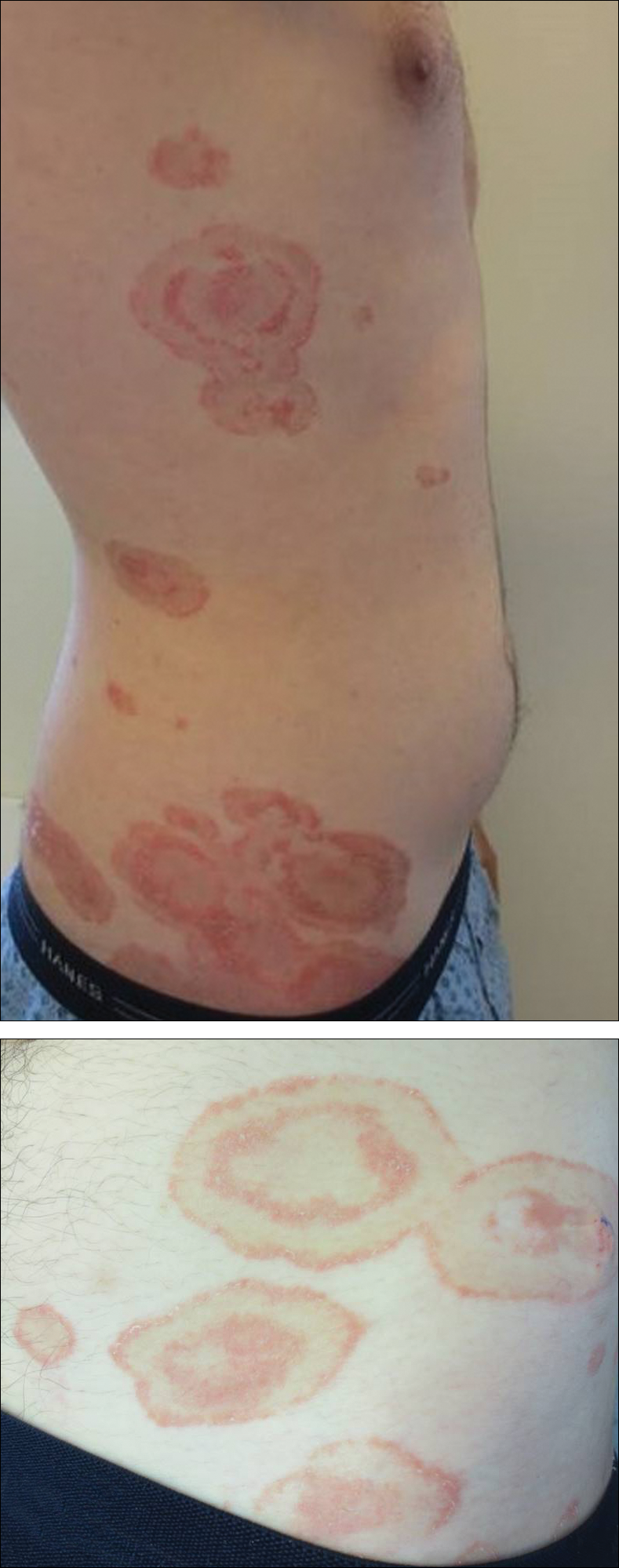
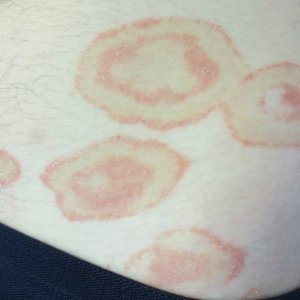
A healthy 23-year-old man presented for evaluation of an enlarging annular pruritic rash of 1.5 years' duration. Treatment with ciclopirox cream 0.77%, calcipotriene cream 0.005%, tacrolimus ointment 0.1%, fluticasone cream 0.05%, and halobetasol cream 0.05% prescribed by an outside physician provided only modest temporary improvement. The patient reported no history of travel outside of western New York, camping, tick bites, or medications. He denied any joint swelling or morning stiffness. Physical examination revealed multiple 4- to 6-cm pink, annular, scaly plaques with central clearing on the abdomen (top) and thighs. A few 1-cm pink scaly patches were present on the back (bottom), and few 2- to 3-mm pink scaly papules were noted on the extensor aspects of the elbows and forearms. A potassium hydroxide examination revealed no hyphal elements or yeast forms.
Idiopathic Eruptive Macular Pigmentation With Papillomatosis
To the Editor:
A 13-year-old white adolescent girl presented with asymptomatic discrete hyperpigmented papules on the chest, back, arms, and upper legs of 7 months’ duration. The patient otherwise was in good health; her weight and height were on the 40th percentile on growth curves and she had no history of any medications. Treatments for the skin condition prescribed by outside dermatologists included minocycline 75 mg twice daily for 2 months, lactic acid lotion 12% daily, and ketoconazole 400 mg administered twice 1 week apart.
Physical examination revealed more than 50 scattered hyperpigmented papules on the chest, back, arms, and upper legs ranging in size from 2 to 3.5 cm (Figure 1). Stroking of lesions failed to elicit Darier sign. A potassium hydroxide preparation and fungal culture were negative for pathogenic fungal organisms. The plasma insulin level was within reference range. A punch biopsy from the abdomen was obtained and sent for histopathologic examination. Histopathology showed mild hyperkeratosis, subtle papillomatosis, and interanastomosing acanthosis comprising squamoid cells with mild basilar hyperpigmentation (Figure 2). Sparse superficial perivascular lymphocytic infiltrate and increased pigmentation was seen in the basal layer. The dermis showed a few scattered dermal melanophages. A periodic acid–Schiff with diastase stain was negative. Giemsa and Leder stains highlighted a normal number and distribution of mast cells. Based on the histologic findings, the patient was diagnosed with idiopathic eruptive macular pigmentation (IEMP).

.")
Idiopathic eruptive macular pigmentation is a rare condition that was described in 1978 by Degos et al.1 Sanz de Galdeano et al2 established the following diagnostic criteria: (1) eruption of brownish black, nonconfluent, asymptomatic macules involving the trunk, neck, and proximal arms and legs in children or adolescents; (2) absence of preceding inflammatory lesions; (3) no prior drug exposure; (4) basal cell layer hyperpigmentation of the epidermis and prominent dermal melanophages without visible basal layer damage or lichenoid inflammatory infiltrate; and (5) normal mast cell count.
Idiopathic eruptive macular pigmentation with papillomatosis (IEMPwP) is a variant of IEMP.3 It is undecided if IEMP and IEMPwP are variants of the same entity or distinct conditions. Until a clear etiology of these entities is established, we prefer to separate them on purely morphologic grounds. Marcoux et al4 labeled IEMPwP as a variant of acanthosis nigricans. Although morphologically the 2 conditions appear similar, our patient’s plasma insulin level essentially ruled out acanthosis nigricans.
Idiopathic eruptive macular pigmentation is a rare condition with the majority of cases reported in the Asian population with some reports in white, Hispanic, and black individuals.5 Idiopathic eruptive macular pigmentation with papillomatosis was reported by Joshi3 in 2007 in 9 Indian children with the classic findings of IEMP along with a velvety rash that correlated with papillomatosis. Diagnosis of IEMPwP is important, as the disease generally is self-limited and resolves over the course of a few weeks to a few years.
- Degos R, Civatte J, Belaïch S. Idiopathic eruptive macular pigmentation (author’s transl)[in French]. Ann Dermatol Venereol. 1978;105:177-182.
- Sanz de Galdeano C, Léauté-Labrèze C, Bioulac-Sage P, et al. Idiopathic eruptive macular pigmentation: report of five patients. Pediatr Dermatol. 1996;13:274-277.
- Joshi R. Idiopathic eruptive macular pigmentation with papillomatosis: report of nine cases. Indian J Dermatol Venereol Leprol. 2007;73:402-405.
- Marcoux DA, Durán-McKinster C, Baselga E. Pigmentary abnormalities. In: Schachner LA, Hansen RC, eds. Pediatric Dermatology. Philadelphia, PA: Mosby; 2011:700-746.
- Torres-Romero LF, Lisle A, Waxman L. Asymptomatic hyperpigmented macules and patches on the trunk. Am J Dermatopathol. 2015;37:546, 586.
To the Editor:
A 13-year-old white adolescent girl presented with asymptomatic discrete hyperpigmented papules on the chest, back, arms, and upper legs of 7 months’ duration. The patient otherwise was in good health; her weight and height were on the 40th percentile on growth curves and she had no history of any medications. Treatments for the skin condition prescribed by outside dermatologists included minocycline 75 mg twice daily for 2 months, lactic acid lotion 12% daily, and ketoconazole 400 mg administered twice 1 week apart.
Physical examination revealed more than 50 scattered hyperpigmented papules on the chest, back, arms, and upper legs ranging in size from 2 to 3.5 cm (Figure 1). Stroking of lesions failed to elicit Darier sign. A potassium hydroxide preparation and fungal culture were negative for pathogenic fungal organisms. The plasma insulin level was within reference range. A punch biopsy from the abdomen was obtained and sent for histopathologic examination. Histopathology showed mild hyperkeratosis, subtle papillomatosis, and interanastomosing acanthosis comprising squamoid cells with mild basilar hyperpigmentation (Figure 2). Sparse superficial perivascular lymphocytic infiltrate and increased pigmentation was seen in the basal layer. The dermis showed a few scattered dermal melanophages. A periodic acid–Schiff with diastase stain was negative. Giemsa and Leder stains highlighted a normal number and distribution of mast cells. Based on the histologic findings, the patient was diagnosed with idiopathic eruptive macular pigmentation (IEMP).
Idiopathic eruptive macular pigmentation is a rare condition that was described in 1978 by Degos et al.1 Sanz de Galdeano et al2 established the following diagnostic criteria: (1) eruption of brownish black, nonconfluent, asymptomatic macules involving the trunk, neck, and proximal arms and legs in children or adolescents; (2) absence of preceding inflammatory lesions; (3) no prior drug exposure; (4) basal cell layer hyperpigmentation of the epidermis and prominent dermal melanophages without visible basal layer damage or lichenoid inflammatory infiltrate; and (5) normal mast cell count.
Idiopathic eruptive macular pigmentation with papillomatosis (IEMPwP) is a variant of IEMP.3 It is undecided if IEMP and IEMPwP are variants of the same entity or distinct conditions. Until a clear etiology of these entities is established, we prefer to separate them on purely morphologic grounds. Marcoux et al4 labeled IEMPwP as a variant of acanthosis nigricans. Although morphologically the 2 conditions appear similar, our patient’s plasma insulin level essentially ruled out acanthosis nigricans.
Idiopathic eruptive macular pigmentation is a rare condition with the majority of cases reported in the Asian population with some reports in white, Hispanic, and black individuals.5 Idiopathic eruptive macular pigmentation with papillomatosis was reported by Joshi3 in 2007 in 9 Indian children with the classic findings of IEMP along with a velvety rash that correlated with papillomatosis. Diagnosis of IEMPwP is important, as the disease generally is self-limited and resolves over the course of a few weeks to a few years.
To the Editor:
A 13-year-old white adolescent girl presented with asymptomatic discrete hyperpigmented papules on the chest, back, arms, and upper legs of 7 months’ duration. The patient otherwise was in good health; her weight and height were on the 40th percentile on growth curves and she had no history of any medications. Treatments for the skin condition prescribed by outside dermatologists included minocycline 75 mg twice daily for 2 months, lactic acid lotion 12% daily, and ketoconazole 400 mg administered twice 1 week apart.
Physical examination revealed more than 50 scattered hyperpigmented papules on the chest, back, arms, and upper legs ranging in size from 2 to 3.5 cm (Figure 1). Stroking of lesions failed to elicit Darier sign. A potassium hydroxide preparation and fungal culture were negative for pathogenic fungal organisms. The plasma insulin level was within reference range. A punch biopsy from the abdomen was obtained and sent for histopathologic examination. Histopathology showed mild hyperkeratosis, subtle papillomatosis, and interanastomosing acanthosis comprising squamoid cells with mild basilar hyperpigmentation (Figure 2). Sparse superficial perivascular lymphocytic infiltrate and increased pigmentation was seen in the basal layer. The dermis showed a few scattered dermal melanophages. A periodic acid–Schiff with diastase stain was negative. Giemsa and Leder stains highlighted a normal number and distribution of mast cells. Based on the histologic findings, the patient was diagnosed with idiopathic eruptive macular pigmentation (IEMP).
Idiopathic eruptive macular pigmentation is a rare condition that was described in 1978 by Degos et al.1 Sanz de Galdeano et al2 established the following diagnostic criteria: (1) eruption of brownish black, nonconfluent, asymptomatic macules involving the trunk, neck, and proximal arms and legs in children or adolescents; (2) absence of preceding inflammatory lesions; (3) no prior drug exposure; (4) basal cell layer hyperpigmentation of the epidermis and prominent dermal melanophages without visible basal layer damage or lichenoid inflammatory infiltrate; and (5) normal mast cell count.
Idiopathic eruptive macular pigmentation with papillomatosis (IEMPwP) is a variant of IEMP.3 It is undecided if IEMP and IEMPwP are variants of the same entity or distinct conditions. Until a clear etiology of these entities is established, we prefer to separate them on purely morphologic grounds. Marcoux et al4 labeled IEMPwP as a variant of acanthosis nigricans. Although morphologically the 2 conditions appear similar, our patient’s plasma insulin level essentially ruled out acanthosis nigricans.
Idiopathic eruptive macular pigmentation is a rare condition with the majority of cases reported in the Asian population with some reports in white, Hispanic, and black individuals.5 Idiopathic eruptive macular pigmentation with papillomatosis was reported by Joshi3 in 2007 in 9 Indian children with the classic findings of IEMP along with a velvety rash that correlated with papillomatosis. Diagnosis of IEMPwP is important, as the disease generally is self-limited and resolves over the course of a few weeks to a few years.
- Degos R, Civatte J, Belaïch S. Idiopathic eruptive macular pigmentation (author’s transl)[in French]. Ann Dermatol Venereol. 1978;105:177-182.
- Sanz de Galdeano C, Léauté-Labrèze C, Bioulac-Sage P, et al. Idiopathic eruptive macular pigmentation: report of five patients. Pediatr Dermatol. 1996;13:274-277.
- Joshi R. Idiopathic eruptive macular pigmentation with papillomatosis: report of nine cases. Indian J Dermatol Venereol Leprol. 2007;73:402-405.
- Marcoux DA, Durán-McKinster C, Baselga E. Pigmentary abnormalities. In: Schachner LA, Hansen RC, eds. Pediatric Dermatology. Philadelphia, PA: Mosby; 2011:700-746.
- Torres-Romero LF, Lisle A, Waxman L. Asymptomatic hyperpigmented macules and patches on the trunk. Am J Dermatopathol. 2015;37:546, 586.
- Degos R, Civatte J, Belaïch S. Idiopathic eruptive macular pigmentation (author’s transl)[in French]. Ann Dermatol Venereol. 1978;105:177-182.
- Sanz de Galdeano C, Léauté-Labrèze C, Bioulac-Sage P, et al. Idiopathic eruptive macular pigmentation: report of five patients. Pediatr Dermatol. 1996;13:274-277.
- Joshi R. Idiopathic eruptive macular pigmentation with papillomatosis: report of nine cases. Indian J Dermatol Venereol Leprol. 2007;73:402-405.
- Marcoux DA, Durán-McKinster C, Baselga E. Pigmentary abnormalities. In: Schachner LA, Hansen RC, eds. Pediatric Dermatology. Philadelphia, PA: Mosby; 2011:700-746.
- Torres-Romero LF, Lisle A, Waxman L. Asymptomatic hyperpigmented macules and patches on the trunk. Am J Dermatopathol. 2015;37:546, 586.
Practice Points
- Idiopathic eruptive macular pigmentation with papillomatosis is a rare disorder that most frequently affects children and young adults.
- Idiopathic eruptive macular pigmentation with papillomatosis is characterized by asymptomatic, brownish, hyperpigmented macules involving the neck, trunk, arms, and legs.
- The disorder is important to consider in the differential diagnosis of asymptomatic pigmentary disorders to avoid unnecessary treatment because the disease is self-limiting and resolves over weeks to years.
Debunking Atopic Dermatitis Myths: Should You Use Systemic Therapy?
Myth: Because atopic dermatitis is skin-deep, systemic therapy is unnecessary.
Although atopic dermatitis (AD) primarily is known as a skin condition, recent research has indicated that it may be the start of the “atopic march” leading to the development of 1 or more other atopic conditions with multiorgan involvement. In infancy AD can progress to asthma and allergic rhinitis. Adult AD can be accompanied by systemic diseases such as inflammatory bowel disease, nephritic syndrome, and others. There also is a link between impairment of epidermal barrier function and disturbed skin microbiome in patients with AD. Therefore, systemic therapy may be warranted; the question is when should you use systemic therapy?
Most AD patients have mild to moderate disease that responds well to emollients and avoidance of disease triggers and other skin irritants. However, many AD patients experience a more severe disease course that does not respond adequately to topical therapy. For these patients, systemic therapy is a viable treatment option to improve quality of life (QOL), prevent flares, and control skin inflammation and other AD symptoms.
In 2017 an expert panel of the International Eczema Council proposed an algorithm to be used to determine if systemic therapy is warranted in patients with AD. Dermatologists must consider disease severity, impact on QOL, and risks and benefits of systemic therapies. Before starting systemic therapy, the panel recommends the following:
- Consider alternate or concomitant diagnoses
- Avoid triggers
- Optimize topical therapy
- Ensure adequate patient/caregiver education
- Treat coexistent infection
- Assess QOL
- Consider phototherapy
The American Academy of Dermatology established Guidelines of Care for the Management of AD in 2014, which provide recommendations for the most efficacious systemic agents.
Armed with these guidelines, dermatologists can work with patients to determine the most appropriate treatment course for this condition that is more than skin-deep.
Expert Commentary
Atopic dermatitis is a skin barrier abnormality that causes inflammatory skin disease and an inflammatory disorder triggering abnormal barrier. Whether we choose the outside-in or inside-out approach, it is clear that there is a systemic inflammation associated with skin disease. It is true that children respond well to barrier repair and topical therapy in many settings, as do many adults. However, chronic skin inflammation is not in isolation, triggering mucosal barrier changes allowing for more sensitization to foods and respiratory allergens as well as systemic inflammation in adults. Despite the utility of systemic steroids, the side effects generally outweigh benefit. On the other hand, phototherapy and systemic agents can clear skin and induce remissions and improved QOL. The AAD guidelines were reported before US Food and Drug Administration approval of newer agents such as dupilumab, leaving it up to the dermatologist to find the niche for this first biologic agent for AD.
—Nanette B. Silverberg, MD
Suggested Readings
Darlenski R, Kazandjieva J, Hristakieva E, et al. Atopic dermatitis as a systemic disease [published online November 22, 2013]. Clin Dermatol. 2014;32:409-413.
Sidbury R, Davis DM, Cohen DE, et al; American Academy of Dermatology. Guidelines of care for the management of atopic dermatitis: section 3. management and treatment with phototherapy and systemic agents [published online May 9, 2014]. J Am Acad Dermatol. 2014;71:327-349.
Simpson EL, Bruin-Weller M, Flohr C, et al. When does atopic dermatitis warrant systemic therapy? recommendations from an expert panel of the International Eczema Council [published online August 10, 2017]. J Am Acad Dermatol. 2017;77:623-633.
Thomas CL, Fernández-Peñas P. The microbiome and atopic eczema: more than skin deep [published online January 28, 2016]. Australas J Dermatol. 2017;58:18-24.
Myth: Because atopic dermatitis is skin-deep, systemic therapy is unnecessary.
Although atopic dermatitis (AD) primarily is known as a skin condition, recent research has indicated that it may be the start of the “atopic march” leading to the development of 1 or more other atopic conditions with multiorgan involvement. In infancy AD can progress to asthma and allergic rhinitis. Adult AD can be accompanied by systemic diseases such as inflammatory bowel disease, nephritic syndrome, and others. There also is a link between impairment of epidermal barrier function and disturbed skin microbiome in patients with AD. Therefore, systemic therapy may be warranted; the question is when should you use systemic therapy?
Most AD patients have mild to moderate disease that responds well to emollients and avoidance of disease triggers and other skin irritants. However, many AD patients experience a more severe disease course that does not respond adequately to topical therapy. For these patients, systemic therapy is a viable treatment option to improve quality of life (QOL), prevent flares, and control skin inflammation and other AD symptoms.
In 2017 an expert panel of the International Eczema Council proposed an algorithm to be used to determine if systemic therapy is warranted in patients with AD. Dermatologists must consider disease severity, impact on QOL, and risks and benefits of systemic therapies. Before starting systemic therapy, the panel recommends the following:
- Consider alternate or concomitant diagnoses
- Avoid triggers
- Optimize topical therapy
- Ensure adequate patient/caregiver education
- Treat coexistent infection
- Assess QOL
- Consider phototherapy
The American Academy of Dermatology established Guidelines of Care for the Management of AD in 2014, which provide recommendations for the most efficacious systemic agents.
Armed with these guidelines, dermatologists can work with patients to determine the most appropriate treatment course for this condition that is more than skin-deep.
Expert Commentary
Atopic dermatitis is a skin barrier abnormality that causes inflammatory skin disease and an inflammatory disorder triggering abnormal barrier. Whether we choose the outside-in or inside-out approach, it is clear that there is a systemic inflammation associated with skin disease. It is true that children respond well to barrier repair and topical therapy in many settings, as do many adults. However, chronic skin inflammation is not in isolation, triggering mucosal barrier changes allowing for more sensitization to foods and respiratory allergens as well as systemic inflammation in adults. Despite the utility of systemic steroids, the side effects generally outweigh benefit. On the other hand, phototherapy and systemic agents can clear skin and induce remissions and improved QOL. The AAD guidelines were reported before US Food and Drug Administration approval of newer agents such as dupilumab, leaving it up to the dermatologist to find the niche for this first biologic agent for AD.
—Nanette B. Silverberg, MD
Suggested Readings
Darlenski R, Kazandjieva J, Hristakieva E, et al. Atopic dermatitis as a systemic disease [published online November 22, 2013]. Clin Dermatol. 2014;32:409-413.
Sidbury R, Davis DM, Cohen DE, et al; American Academy of Dermatology. Guidelines of care for the management of atopic dermatitis: section 3. management and treatment with phototherapy and systemic agents [published online May 9, 2014]. J Am Acad Dermatol. 2014;71:327-349.
Simpson EL, Bruin-Weller M, Flohr C, et al. When does atopic dermatitis warrant systemic therapy? recommendations from an expert panel of the International Eczema Council [published online August 10, 2017]. J Am Acad Dermatol. 2017;77:623-633.
Thomas CL, Fernández-Peñas P. The microbiome and atopic eczema: more than skin deep [published online January 28, 2016]. Australas J Dermatol. 2017;58:18-24.
Myth: Because atopic dermatitis is skin-deep, systemic therapy is unnecessary.
Although atopic dermatitis (AD) primarily is known as a skin condition, recent research has indicated that it may be the start of the “atopic march” leading to the development of 1 or more other atopic conditions with multiorgan involvement. In infancy AD can progress to asthma and allergic rhinitis. Adult AD can be accompanied by systemic diseases such as inflammatory bowel disease, nephritic syndrome, and others. There also is a link between impairment of epidermal barrier function and disturbed skin microbiome in patients with AD. Therefore, systemic therapy may be warranted; the question is when should you use systemic therapy?
Most AD patients have mild to moderate disease that responds well to emollients and avoidance of disease triggers and other skin irritants. However, many AD patients experience a more severe disease course that does not respond adequately to topical therapy. For these patients, systemic therapy is a viable treatment option to improve quality of life (QOL), prevent flares, and control skin inflammation and other AD symptoms.
In 2017 an expert panel of the International Eczema Council proposed an algorithm to be used to determine if systemic therapy is warranted in patients with AD. Dermatologists must consider disease severity, impact on QOL, and risks and benefits of systemic therapies. Before starting systemic therapy, the panel recommends the following:
- Consider alternate or concomitant diagnoses
- Avoid triggers
- Optimize topical therapy
- Ensure adequate patient/caregiver education
- Treat coexistent infection
- Assess QOL
- Consider phototherapy
The American Academy of Dermatology established Guidelines of Care for the Management of AD in 2014, which provide recommendations for the most efficacious systemic agents.
Armed with these guidelines, dermatologists can work with patients to determine the most appropriate treatment course for this condition that is more than skin-deep.
Expert Commentary
Atopic dermatitis is a skin barrier abnormality that causes inflammatory skin disease and an inflammatory disorder triggering abnormal barrier. Whether we choose the outside-in or inside-out approach, it is clear that there is a systemic inflammation associated with skin disease. It is true that children respond well to barrier repair and topical therapy in many settings, as do many adults. However, chronic skin inflammation is not in isolation, triggering mucosal barrier changes allowing for more sensitization to foods and respiratory allergens as well as systemic inflammation in adults. Despite the utility of systemic steroids, the side effects generally outweigh benefit. On the other hand, phototherapy and systemic agents can clear skin and induce remissions and improved QOL. The AAD guidelines were reported before US Food and Drug Administration approval of newer agents such as dupilumab, leaving it up to the dermatologist to find the niche for this first biologic agent for AD.
—Nanette B. Silverberg, MD
Suggested Readings
Darlenski R, Kazandjieva J, Hristakieva E, et al. Atopic dermatitis as a systemic disease [published online November 22, 2013]. Clin Dermatol. 2014;32:409-413.
Sidbury R, Davis DM, Cohen DE, et al; American Academy of Dermatology. Guidelines of care for the management of atopic dermatitis: section 3. management and treatment with phototherapy and systemic agents [published online May 9, 2014]. J Am Acad Dermatol. 2014;71:327-349.
Simpson EL, Bruin-Weller M, Flohr C, et al. When does atopic dermatitis warrant systemic therapy? recommendations from an expert panel of the International Eczema Council [published online August 10, 2017]. J Am Acad Dermatol. 2017;77:623-633.
Thomas CL, Fernández-Peñas P. The microbiome and atopic eczema: more than skin deep [published online January 28, 2016]. Australas J Dermatol. 2017;58:18-24.

Imiquimod-Induced Hypopigmentation Following Treatment of Periungual Verruca Vulgaris
Imiquimod is derived from the imidazoquinoline family and works by activating both innate and adaptive immune pathways. Imiquimod binds to toll-like receptor 7 located on monocytes, macrophages, and dendritic cells,1 which allows nuclear factor κβ light chain enhancer of activated B cells to induce production of proinflammatory cytokines, including IFN-α and tumor necrosis factor α, as well as IL-1, IL-6, IL-8, IL-10, and IL-12.2 These proinflammatory cytokines play a role in the innate immunity, triggering upregulation of the adaptive immune pathway and activating type 1 helper T cells, cytotoxic T cells, and natural killer cells. These cells have antiviral and antitumoral effects that lend to their significance in coordinating innate and adaptive immune mechanisms.3 More specifically, imiquimod enhances dendritic cell migration to regional lymph nodes and induces apoptosis via activation of proapoptotic B-cell lymphoma 2 proteins.1,2 Imiquimod has been approved by the US Food and Drug Administration (FDA) to treat external genitalia and perianal condyloma acuminata, actinic keratoses (AKs), and superficial basal cell carcinoma (BCC). It often is used off label for antiviral or antitumoral therapy in Bowen disease, squamous cell carcinoma, lentigo maligna, vulvar intraepithelial neoplasia, molluscum contagiosum, common warts, and leishmaniasis.1,2 Imiquimod is generally well tolerated; erythema and irritation at the application site are the most common side effects, with pigmentary change being less common.
Case Report
A 51-year-old man with a medical history of vitamin D deficiency, vitamin B12 deficiency, tinea pedis, and BCC presented with periungual verruca vulgaris on the right fifth digit and left thumb (Figure 1). The patient was prescribed imiquimod cream 5% to be applied 3 times weekly for 3 months. At 5-month follow-up the patient reported new-onset vitiligolike patches of depigmentation on the hands and feet that abruptly began 3 months after initiating treatment with imiquimod. On examination he had several depigmented patches with well-defined irregular borders on the bilateral dorsal hands and right foot as well as the right elbow (Figure 2). There was no personal or family history of vitiligo, thyroid disease, or autoimmune disease. Thyroid function studies and autoimmune panel were unremarkable. The patient also denied applying imiquimod to areas other than the periungual region of the right fifth digit and left thumb. He declined a biopsy of the lesions and was given a prescription for tacrolimus ointment 0.1% for twice-daily application. At 3-month follow-up the depigmented patches had spread. The patient is currently on 5-fluorouracil cream 5%. Despite loss of pigmentation, the periungual verruca vulgaris has persisted as well as depigmentation.

 and the right elbow (B).")
Comment
Imiquimod therapy is commonly used to treat conditions for which an antiviral or antitumor immune response is necessary for treatment and full resolution of skin conditions. It can yield positive results in conditions that are difficult to treat, such as periungual verruca vulgaris.4 The most common adverse effects of imiquimod include localized inflammation and application-site reactions. Pigment changes, though less common, also have been reported. From 1997 to 2003, 1257 cases of imiquimod adverse effects were reported to the FDA. There were 68 reported cases of pigmentary change, of which 51 documented vitiligo, hypopigmentation, or depigmentation. The others reported hyperpigmentation following imiquimod use.4 The imiquimod package insert lists application-site hypopigmentation as a possible adverse effect.5 Imiquimod-induced hypopigmentation and depigmentation have been reported in the peer-reviewed literature.4,6-14 Pigment loss has been reported in imiquimod treatment of condyloma acuminata, superficial BCC, nodular BCC, and extramammary Paget disease.6-8 Duration of therapy to onset of pigment loss ranged from 7 to 28 weeks.9 Imiquimod dosing varied among reported cases, ranging from 3 times weekly to daily application. Interestingly, hypopigmentation or depigmentation are not commonly associated with imiquimod use for the treatment of AKs, which Burnett and Kouba9 proposed may be due to the twice weekly imiquimod dosing regimen recommended by the FDA for the treatment of AK (below the minimum threshold for pigment loss). Our patient applied imiquimod cream 5% to periungual verruca vulgaris 3 times weekly for 3 months and may have developed vitiligolike depigmentation because he met this theoretical dosage threshold. Further research is necessary to confirm a dosage-related threshold for the development of depigmentation. Imiquimod-induced pigment loss has mainly been limited to the site of application.
Depigmentation was limited to the application site the majority of the time; however, depigmentation at adjacent sites has been reported.10 This finding was consistent with the proposed notion that cytokines induced by imiquimod have localized paracrine activity.11 Our patient was unique in that his depigmentation was present at the site of application, adjacent to the site of application, and at distant sites. He applied imiquimod only to the periungual area of the right fifth digit and left thumb but experienced depigmentation at several other sites. Although it is possible that our patient unintentionally spread imiquimod on the distant sites, it is less likely that the application would have been sufficient to cause depigmentation. Although systemic absorption of topical medications varies depending on multiple factors, the systemic absorption of imiquimod is minimal with mild systemic side effects reported, including headache, myalgia, and influenzalike symptoms.5 Thus, it is possible that our patient developed distant vitiligolike depigmentation as a systemic side effect of imiquimod therapy. Although our patient declined to have a biopsy performed, Gowda et al15 reported biopsy-proven vitiligo, demonstrating the absence of melanin and melanocytes following the use of imiquimod.
Several mechanisms have been proposed for imiquimod-induced depigmentation. For example, imiquimod may induce melanocyte apoptosis by increasing the levels of several proinflammatory and proapoptotic cytokines.16 Imiquimod-induced melanocyte apoptosis appears to involve elevated caspase-3, decreased B-cell lymphoma 2, altered mitogen-activated protein kinase expression, and ubiquitin-mediated proteolysis.13,17 Additionally, increased levels of IL-6 appear to increase melanocyte-binding molecules and increase melanocyte-leukocyte interactions. Another proposed theory targets toll-like receptor 7 on melanocytes that are acted on directly by imiquimod.11,17 In contrast, development of vitiligo following trauma (Koebner phenomenon) is not uncommon, and the immune effects induced by imiquimod may mimic those seen with trauma.14 Further research is needed to elucidate the mechanism by which imiquimod causes vitiligolike depigmentation.
Unfortunately, the depigmentation seen with imiquimod generally is permanent. Stefanaki et al10 showed repigmentation on cessation of imiquimod use. Our patient’s depigmentation remains unchanged despite treatment with tacrolimus ointment. Although it is possible for vitiligo to occur de novo without obvious inciting event or laboratory abnormality, the timeline and number of other cases in the literature make ours highly suspect for imiquimod-induced depigmentation.
Conclusion
Imiquimod is a commonly used immune-enhancing medication with an increasing list of off-label uses. Prior to prescribing imiquimod for a benign skin condition, clinicians should be cognizant of the potential for localized or possibly even distant depigmentation. We report a case of distant depigmentation following the use of imiquimod for periungual verruca vulgaris.
- Ganjian S, Ourian AJ, Shamtoub G, et al. Off-label indications for imiquimod. Dermatol Online J. 2009;15:4.
- Skinner RB Jr. Imiquimod. Dermatol Clin. 2003;21:291-300.
- Murphy K, Travers P, Walport M. Innate immunity. In: Murphy K, Travers P, Walport M, eds. Janeway’s Immunobiology. 7th ed. New York, NY: Garland Science. 2008:39-108.
- Brown T, Zirvi M, Cotsarelis G, et al. Vitiligo-like hypopigmentation associated with imiquimod treatment of genital warts. J Am Acad Dermatol. 2005;52:715-716.
- Aldara [package insert]. Bristol, TN: Graceway Pharmaceuticals, LLC; 2007.
- Kwon HH, Cho KH. Induction of vitiligo-like hypopigmentation after imiquimod treatment of extramammary Paget’s disease. Ann Dermatol. 2012;24:482-484.
- Mendonca CO, Yates VM. Permanent facial hypopigmentation following treatment with imiquimod. Clin Exp Dermatol. 2006;31:721-722.
- Zhang R, Zhu W. Genital vitiligo following use of imiquimod 5% cream. Indian J Dermatol. 2011;56:335-336.
- Burnett CT, Kouba DJ. Imiquimod-induced depigmentation: report of two cases and review of the literature. Dermatol Surg. 2012;38:1872-1875.
- Stefanaki C, Nicolaidou E, Hadjivassiliou M. Imiquimod-induced vitiligo in a patient with genital warts. J Eur Acad Dermatol Venereol. 2006;20:755-756.
- Al-Dujaili Z, Hsu S. Imiquimod-induced vitiligo. Dermatol Online J. 2007;13:10.
- Mashiah J, Brenner S. Possible mechanisms in the induction of vitiligo-like hypopigmentation by topical imiquimod. Clin Exp Dermatol. 2007;33:74-76.
- Grahovac M, Ehmann LM, Flaig M, et al. Giant basal cell carcinoma. Improvement and vitiligo-like hypopigmentation after intermittent treatment with 5% imiquimod. Acta Dermatovenerol Croat. 2012;20:275-278.
- Serrão VV, Páris FR, Feio AB. Genital vitiligo-like depigmentation following use of imiquimod 5% cream. Eur J Dermatol. 2008;18:342-343.
- Gowda S, Tillman DK, Fitzpatrick JE, et al. Imiquimod-induced vitiligo after treatment of nodular basal cell carcinoma. J Cutan Pathol. 2009;36:878-881.
- Kim CH, Ahn JH, Kang SU, et al. Imiquimod induces apoptosis of human melanocytes. Arch Dermatol Res. 2010;302:301-306.
- Eapen BR. Vitiligo, psoriasis, and imiquimod: fitting all into the same pathway. Indian J Dermatol Venereol Leprol. 2008;74:169.
Imiquimod is derived from the imidazoquinoline family and works by activating both innate and adaptive immune pathways. Imiquimod binds to toll-like receptor 7 located on monocytes, macrophages, and dendritic cells,1 which allows nuclear factor κβ light chain enhancer of activated B cells to induce production of proinflammatory cytokines, including IFN-α and tumor necrosis factor α, as well as IL-1, IL-6, IL-8, IL-10, and IL-12.2 These proinflammatory cytokines play a role in the innate immunity, triggering upregulation of the adaptive immune pathway and activating type 1 helper T cells, cytotoxic T cells, and natural killer cells. These cells have antiviral and antitumoral effects that lend to their significance in coordinating innate and adaptive immune mechanisms.3 More specifically, imiquimod enhances dendritic cell migration to regional lymph nodes and induces apoptosis via activation of proapoptotic B-cell lymphoma 2 proteins.1,2 Imiquimod has been approved by the US Food and Drug Administration (FDA) to treat external genitalia and perianal condyloma acuminata, actinic keratoses (AKs), and superficial basal cell carcinoma (BCC). It often is used off label for antiviral or antitumoral therapy in Bowen disease, squamous cell carcinoma, lentigo maligna, vulvar intraepithelial neoplasia, molluscum contagiosum, common warts, and leishmaniasis.1,2 Imiquimod is generally well tolerated; erythema and irritation at the application site are the most common side effects, with pigmentary change being less common.
Case Report
A 51-year-old man with a medical history of vitamin D deficiency, vitamin B12 deficiency, tinea pedis, and BCC presented with periungual verruca vulgaris on the right fifth digit and left thumb (Figure 1). The patient was prescribed imiquimod cream 5% to be applied 3 times weekly for 3 months. At 5-month follow-up the patient reported new-onset vitiligolike patches of depigmentation on the hands and feet that abruptly began 3 months after initiating treatment with imiquimod. On examination he had several depigmented patches with well-defined irregular borders on the bilateral dorsal hands and right foot as well as the right elbow (Figure 2). There was no personal or family history of vitiligo, thyroid disease, or autoimmune disease. Thyroid function studies and autoimmune panel were unremarkable. The patient also denied applying imiquimod to areas other than the periungual region of the right fifth digit and left thumb. He declined a biopsy of the lesions and was given a prescription for tacrolimus ointment 0.1% for twice-daily application. At 3-month follow-up the depigmented patches had spread. The patient is currently on 5-fluorouracil cream 5%. Despite loss of pigmentation, the periungual verruca vulgaris has persisted as well as depigmentation.
Comment
Imiquimod therapy is commonly used to treat conditions for which an antiviral or antitumor immune response is necessary for treatment and full resolution of skin conditions. It can yield positive results in conditions that are difficult to treat, such as periungual verruca vulgaris.4 The most common adverse effects of imiquimod include localized inflammation and application-site reactions. Pigment changes, though less common, also have been reported. From 1997 to 2003, 1257 cases of imiquimod adverse effects were reported to the FDA. There were 68 reported cases of pigmentary change, of which 51 documented vitiligo, hypopigmentation, or depigmentation. The others reported hyperpigmentation following imiquimod use.4 The imiquimod package insert lists application-site hypopigmentation as a possible adverse effect.5 Imiquimod-induced hypopigmentation and depigmentation have been reported in the peer-reviewed literature.4,6-14 Pigment loss has been reported in imiquimod treatment of condyloma acuminata, superficial BCC, nodular BCC, and extramammary Paget disease.6-8 Duration of therapy to onset of pigment loss ranged from 7 to 28 weeks.9 Imiquimod dosing varied among reported cases, ranging from 3 times weekly to daily application. Interestingly, hypopigmentation or depigmentation are not commonly associated with imiquimod use for the treatment of AKs, which Burnett and Kouba9 proposed may be due to the twice weekly imiquimod dosing regimen recommended by the FDA for the treatment of AK (below the minimum threshold for pigment loss). Our patient applied imiquimod cream 5% to periungual verruca vulgaris 3 times weekly for 3 months and may have developed vitiligolike depigmentation because he met this theoretical dosage threshold. Further research is necessary to confirm a dosage-related threshold for the development of depigmentation. Imiquimod-induced pigment loss has mainly been limited to the site of application.
Depigmentation was limited to the application site the majority of the time; however, depigmentation at adjacent sites has been reported.10 This finding was consistent with the proposed notion that cytokines induced by imiquimod have localized paracrine activity.11 Our patient was unique in that his depigmentation was present at the site of application, adjacent to the site of application, and at distant sites. He applied imiquimod only to the periungual area of the right fifth digit and left thumb but experienced depigmentation at several other sites. Although it is possible that our patient unintentionally spread imiquimod on the distant sites, it is less likely that the application would have been sufficient to cause depigmentation. Although systemic absorption of topical medications varies depending on multiple factors, the systemic absorption of imiquimod is minimal with mild systemic side effects reported, including headache, myalgia, and influenzalike symptoms.5 Thus, it is possible that our patient developed distant vitiligolike depigmentation as a systemic side effect of imiquimod therapy. Although our patient declined to have a biopsy performed, Gowda et al15 reported biopsy-proven vitiligo, demonstrating the absence of melanin and melanocytes following the use of imiquimod.
Several mechanisms have been proposed for imiquimod-induced depigmentation. For example, imiquimod may induce melanocyte apoptosis by increasing the levels of several proinflammatory and proapoptotic cytokines.16 Imiquimod-induced melanocyte apoptosis appears to involve elevated caspase-3, decreased B-cell lymphoma 2, altered mitogen-activated protein kinase expression, and ubiquitin-mediated proteolysis.13,17 Additionally, increased levels of IL-6 appear to increase melanocyte-binding molecules and increase melanocyte-leukocyte interactions. Another proposed theory targets toll-like receptor 7 on melanocytes that are acted on directly by imiquimod.11,17 In contrast, development of vitiligo following trauma (Koebner phenomenon) is not uncommon, and the immune effects induced by imiquimod may mimic those seen with trauma.14 Further research is needed to elucidate the mechanism by which imiquimod causes vitiligolike depigmentation.
Unfortunately, the depigmentation seen with imiquimod generally is permanent. Stefanaki et al10 showed repigmentation on cessation of imiquimod use. Our patient’s depigmentation remains unchanged despite treatment with tacrolimus ointment. Although it is possible for vitiligo to occur de novo without obvious inciting event or laboratory abnormality, the timeline and number of other cases in the literature make ours highly suspect for imiquimod-induced depigmentation.
Conclusion
Imiquimod is a commonly used immune-enhancing medication with an increasing list of off-label uses. Prior to prescribing imiquimod for a benign skin condition, clinicians should be cognizant of the potential for localized or possibly even distant depigmentation. We report a case of distant depigmentation following the use of imiquimod for periungual verruca vulgaris.
Imiquimod is derived from the imidazoquinoline family and works by activating both innate and adaptive immune pathways. Imiquimod binds to toll-like receptor 7 located on monocytes, macrophages, and dendritic cells,1 which allows nuclear factor κβ light chain enhancer of activated B cells to induce production of proinflammatory cytokines, including IFN-α and tumor necrosis factor α, as well as IL-1, IL-6, IL-8, IL-10, and IL-12.2 These proinflammatory cytokines play a role in the innate immunity, triggering upregulation of the adaptive immune pathway and activating type 1 helper T cells, cytotoxic T cells, and natural killer cells. These cells have antiviral and antitumoral effects that lend to their significance in coordinating innate and adaptive immune mechanisms.3 More specifically, imiquimod enhances dendritic cell migration to regional lymph nodes and induces apoptosis via activation of proapoptotic B-cell lymphoma 2 proteins.1,2 Imiquimod has been approved by the US Food and Drug Administration (FDA) to treat external genitalia and perianal condyloma acuminata, actinic keratoses (AKs), and superficial basal cell carcinoma (BCC). It often is used off label for antiviral or antitumoral therapy in Bowen disease, squamous cell carcinoma, lentigo maligna, vulvar intraepithelial neoplasia, molluscum contagiosum, common warts, and leishmaniasis.1,2 Imiquimod is generally well tolerated; erythema and irritation at the application site are the most common side effects, with pigmentary change being less common.
Case Report
A 51-year-old man with a medical history of vitamin D deficiency, vitamin B12 deficiency, tinea pedis, and BCC presented with periungual verruca vulgaris on the right fifth digit and left thumb (Figure 1). The patient was prescribed imiquimod cream 5% to be applied 3 times weekly for 3 months. At 5-month follow-up the patient reported new-onset vitiligolike patches of depigmentation on the hands and feet that abruptly began 3 months after initiating treatment with imiquimod. On examination he had several depigmented patches with well-defined irregular borders on the bilateral dorsal hands and right foot as well as the right elbow (Figure 2). There was no personal or family history of vitiligo, thyroid disease, or autoimmune disease. Thyroid function studies and autoimmune panel were unremarkable. The patient also denied applying imiquimod to areas other than the periungual region of the right fifth digit and left thumb. He declined a biopsy of the lesions and was given a prescription for tacrolimus ointment 0.1% for twice-daily application. At 3-month follow-up the depigmented patches had spread. The patient is currently on 5-fluorouracil cream 5%. Despite loss of pigmentation, the periungual verruca vulgaris has persisted as well as depigmentation.
Comment
Imiquimod therapy is commonly used to treat conditions for which an antiviral or antitumor immune response is necessary for treatment and full resolution of skin conditions. It can yield positive results in conditions that are difficult to treat, such as periungual verruca vulgaris.4 The most common adverse effects of imiquimod include localized inflammation and application-site reactions. Pigment changes, though less common, also have been reported. From 1997 to 2003, 1257 cases of imiquimod adverse effects were reported to the FDA. There were 68 reported cases of pigmentary change, of which 51 documented vitiligo, hypopigmentation, or depigmentation. The others reported hyperpigmentation following imiquimod use.4 The imiquimod package insert lists application-site hypopigmentation as a possible adverse effect.5 Imiquimod-induced hypopigmentation and depigmentation have been reported in the peer-reviewed literature.4,6-14 Pigment loss has been reported in imiquimod treatment of condyloma acuminata, superficial BCC, nodular BCC, and extramammary Paget disease.6-8 Duration of therapy to onset of pigment loss ranged from 7 to 28 weeks.9 Imiquimod dosing varied among reported cases, ranging from 3 times weekly to daily application. Interestingly, hypopigmentation or depigmentation are not commonly associated with imiquimod use for the treatment of AKs, which Burnett and Kouba9 proposed may be due to the twice weekly imiquimod dosing regimen recommended by the FDA for the treatment of AK (below the minimum threshold for pigment loss). Our patient applied imiquimod cream 5% to periungual verruca vulgaris 3 times weekly for 3 months and may have developed vitiligolike depigmentation because he met this theoretical dosage threshold. Further research is necessary to confirm a dosage-related threshold for the development of depigmentation. Imiquimod-induced pigment loss has mainly been limited to the site of application.
Depigmentation was limited to the application site the majority of the time; however, depigmentation at adjacent sites has been reported.10 This finding was consistent with the proposed notion that cytokines induced by imiquimod have localized paracrine activity.11 Our patient was unique in that his depigmentation was present at the site of application, adjacent to the site of application, and at distant sites. He applied imiquimod only to the periungual area of the right fifth digit and left thumb but experienced depigmentation at several other sites. Although it is possible that our patient unintentionally spread imiquimod on the distant sites, it is less likely that the application would have been sufficient to cause depigmentation. Although systemic absorption of topical medications varies depending on multiple factors, the systemic absorption of imiquimod is minimal with mild systemic side effects reported, including headache, myalgia, and influenzalike symptoms.5 Thus, it is possible that our patient developed distant vitiligolike depigmentation as a systemic side effect of imiquimod therapy. Although our patient declined to have a biopsy performed, Gowda et al15 reported biopsy-proven vitiligo, demonstrating the absence of melanin and melanocytes following the use of imiquimod.
Several mechanisms have been proposed for imiquimod-induced depigmentation. For example, imiquimod may induce melanocyte apoptosis by increasing the levels of several proinflammatory and proapoptotic cytokines.16 Imiquimod-induced melanocyte apoptosis appears to involve elevated caspase-3, decreased B-cell lymphoma 2, altered mitogen-activated protein kinase expression, and ubiquitin-mediated proteolysis.13,17 Additionally, increased levels of IL-6 appear to increase melanocyte-binding molecules and increase melanocyte-leukocyte interactions. Another proposed theory targets toll-like receptor 7 on melanocytes that are acted on directly by imiquimod.11,17 In contrast, development of vitiligo following trauma (Koebner phenomenon) is not uncommon, and the immune effects induced by imiquimod may mimic those seen with trauma.14 Further research is needed to elucidate the mechanism by which imiquimod causes vitiligolike depigmentation.
Unfortunately, the depigmentation seen with imiquimod generally is permanent. Stefanaki et al10 showed repigmentation on cessation of imiquimod use. Our patient’s depigmentation remains unchanged despite treatment with tacrolimus ointment. Although it is possible for vitiligo to occur de novo without obvious inciting event or laboratory abnormality, the timeline and number of other cases in the literature make ours highly suspect for imiquimod-induced depigmentation.
Conclusion
Imiquimod is a commonly used immune-enhancing medication with an increasing list of off-label uses. Prior to prescribing imiquimod for a benign skin condition, clinicians should be cognizant of the potential for localized or possibly even distant depigmentation. We report a case of distant depigmentation following the use of imiquimod for periungual verruca vulgaris.
- Ganjian S, Ourian AJ, Shamtoub G, et al. Off-label indications for imiquimod. Dermatol Online J. 2009;15:4.
- Skinner RB Jr. Imiquimod. Dermatol Clin. 2003;21:291-300.
- Murphy K, Travers P, Walport M. Innate immunity. In: Murphy K, Travers P, Walport M, eds. Janeway’s Immunobiology. 7th ed. New York, NY: Garland Science. 2008:39-108.
- Brown T, Zirvi M, Cotsarelis G, et al. Vitiligo-like hypopigmentation associated with imiquimod treatment of genital warts. J Am Acad Dermatol. 2005;52:715-716.
- Aldara [package insert]. Bristol, TN: Graceway Pharmaceuticals, LLC; 2007.
- Kwon HH, Cho KH. Induction of vitiligo-like hypopigmentation after imiquimod treatment of extramammary Paget’s disease. Ann Dermatol. 2012;24:482-484.
- Mendonca CO, Yates VM. Permanent facial hypopigmentation following treatment with imiquimod. Clin Exp Dermatol. 2006;31:721-722.
- Zhang R, Zhu W. Genital vitiligo following use of imiquimod 5% cream. Indian J Dermatol. 2011;56:335-336.
- Burnett CT, Kouba DJ. Imiquimod-induced depigmentation: report of two cases and review of the literature. Dermatol Surg. 2012;38:1872-1875.
- Stefanaki C, Nicolaidou E, Hadjivassiliou M. Imiquimod-induced vitiligo in a patient with genital warts. J Eur Acad Dermatol Venereol. 2006;20:755-756.
- Al-Dujaili Z, Hsu S. Imiquimod-induced vitiligo. Dermatol Online J. 2007;13:10.
- Mashiah J, Brenner S. Possible mechanisms in the induction of vitiligo-like hypopigmentation by topical imiquimod. Clin Exp Dermatol. 2007;33:74-76.
- Grahovac M, Ehmann LM, Flaig M, et al. Giant basal cell carcinoma. Improvement and vitiligo-like hypopigmentation after intermittent treatment with 5% imiquimod. Acta Dermatovenerol Croat. 2012;20:275-278.
- Serrão VV, Páris FR, Feio AB. Genital vitiligo-like depigmentation following use of imiquimod 5% cream. Eur J Dermatol. 2008;18:342-343.
- Gowda S, Tillman DK, Fitzpatrick JE, et al. Imiquimod-induced vitiligo after treatment of nodular basal cell carcinoma. J Cutan Pathol. 2009;36:878-881.
- Kim CH, Ahn JH, Kang SU, et al. Imiquimod induces apoptosis of human melanocytes. Arch Dermatol Res. 2010;302:301-306.
- Eapen BR. Vitiligo, psoriasis, and imiquimod: fitting all into the same pathway. Indian J Dermatol Venereol Leprol. 2008;74:169.
- Ganjian S, Ourian AJ, Shamtoub G, et al. Off-label indications for imiquimod. Dermatol Online J. 2009;15:4.
- Skinner RB Jr. Imiquimod. Dermatol Clin. 2003;21:291-300.
- Murphy K, Travers P, Walport M. Innate immunity. In: Murphy K, Travers P, Walport M, eds. Janeway’s Immunobiology. 7th ed. New York, NY: Garland Science. 2008:39-108.
- Brown T, Zirvi M, Cotsarelis G, et al. Vitiligo-like hypopigmentation associated with imiquimod treatment of genital warts. J Am Acad Dermatol. 2005;52:715-716.
- Aldara [package insert]. Bristol, TN: Graceway Pharmaceuticals, LLC; 2007.
- Kwon HH, Cho KH. Induction of vitiligo-like hypopigmentation after imiquimod treatment of extramammary Paget’s disease. Ann Dermatol. 2012;24:482-484.
- Mendonca CO, Yates VM. Permanent facial hypopigmentation following treatment with imiquimod. Clin Exp Dermatol. 2006;31:721-722.
- Zhang R, Zhu W. Genital vitiligo following use of imiquimod 5% cream. Indian J Dermatol. 2011;56:335-336.
- Burnett CT, Kouba DJ. Imiquimod-induced depigmentation: report of two cases and review of the literature. Dermatol Surg. 2012;38:1872-1875.
- Stefanaki C, Nicolaidou E, Hadjivassiliou M. Imiquimod-induced vitiligo in a patient with genital warts. J Eur Acad Dermatol Venereol. 2006;20:755-756.
- Al-Dujaili Z, Hsu S. Imiquimod-induced vitiligo. Dermatol Online J. 2007;13:10.
- Mashiah J, Brenner S. Possible mechanisms in the induction of vitiligo-like hypopigmentation by topical imiquimod. Clin Exp Dermatol. 2007;33:74-76.
- Grahovac M, Ehmann LM, Flaig M, et al. Giant basal cell carcinoma. Improvement and vitiligo-like hypopigmentation after intermittent treatment with 5% imiquimod. Acta Dermatovenerol Croat. 2012;20:275-278.
- Serrão VV, Páris FR, Feio AB. Genital vitiligo-like depigmentation following use of imiquimod 5% cream. Eur J Dermatol. 2008;18:342-343.
- Gowda S, Tillman DK, Fitzpatrick JE, et al. Imiquimod-induced vitiligo after treatment of nodular basal cell carcinoma. J Cutan Pathol. 2009;36:878-881.
- Kim CH, Ahn JH, Kang SU, et al. Imiquimod induces apoptosis of human melanocytes. Arch Dermatol Res. 2010;302:301-306.
- Eapen BR. Vitiligo, psoriasis, and imiquimod: fitting all into the same pathway. Indian J Dermatol Venereol Leprol. 2008;74:169.
Practice Points
- Imiquimod commonly is used off label to treat viral and neoplastic processes.
- Clinicians should be aware of the potential for dyspigmentation or depigmentation as a side effect from treatment.
Unusual Presentation of Erythema Elevatum Diutinum With Underlying Hepatitis B Infection
Erythema elevatum diutinum (EED) manifests on a clinicopathologic spectrum of chronic cutaneous small vessel vasculitis. The lesions typically present as persistent, symmetric, firm, red to purple papules or nodules on the extensor arms and dorsal hands.1,2 Underlying infectious, malignant, or autoimmune processes are commonly associated with the disease, notably Streptococcus infection and IgA monoclonal gammopathy.2,3 Hepatitis virus also is often implicated in association with EED. Cases of EED have been seen with concomitant human immunodeficiency virus (HIV) infection.4-6 We report a case of EED presenting in various stages of evolution associated with underlying hepatitis B infection alone.
Case Report
A 57-year-old man originally presented to an outpatient dermatology practice with a nodular, painful, episodic rash on the trunk and upper and lower extremities. A biopsy revealed leukocytoclastic vasculitis (LCV) with prominent eosinophils. At the time, the skin findings were believed to be a manifestation of drug hypersensitivity, likely to opioid use. The patient was lost to follow-up.
Seven years later, the patient was admitted to the hospital with new-onset burning and stinging red nodules on the dorsum of the hands and persistence of the original episodic rash over the lower legs and bilateral flanks. In the interim, he was briefly treated with an oral prednisone taper and topical corticosteroids including triamcinolone cream 0.1% and clobetasol cream 0.05% without improvement.
On examination deep red to violaceous discrete nodules and plaques with overlying hyperkeratosis involving all distal and proximal interphalangeal joints of the hands and extensor elbows were seen (Figure 1A). On the bilateral posterior arms (Figure 1B), anterior legs, and periumbilical area were deeply erythematous papules and plaques with background hyperpigmentation. Across his lower back and bilateral flanks were erythematous papules with central hemorrhagic crusting (Figure 1C).
Pertinent laboratory findings included a positive hepatitis B surface antigen with hepatitis B DNA value 4,313,876 IU/mL and a hepatitis B virus quantitative polymerase chain reaction value of 6.64 U. The etiology was suspected to be intravenous drug abuse; however, the patient denied recreational drug use.
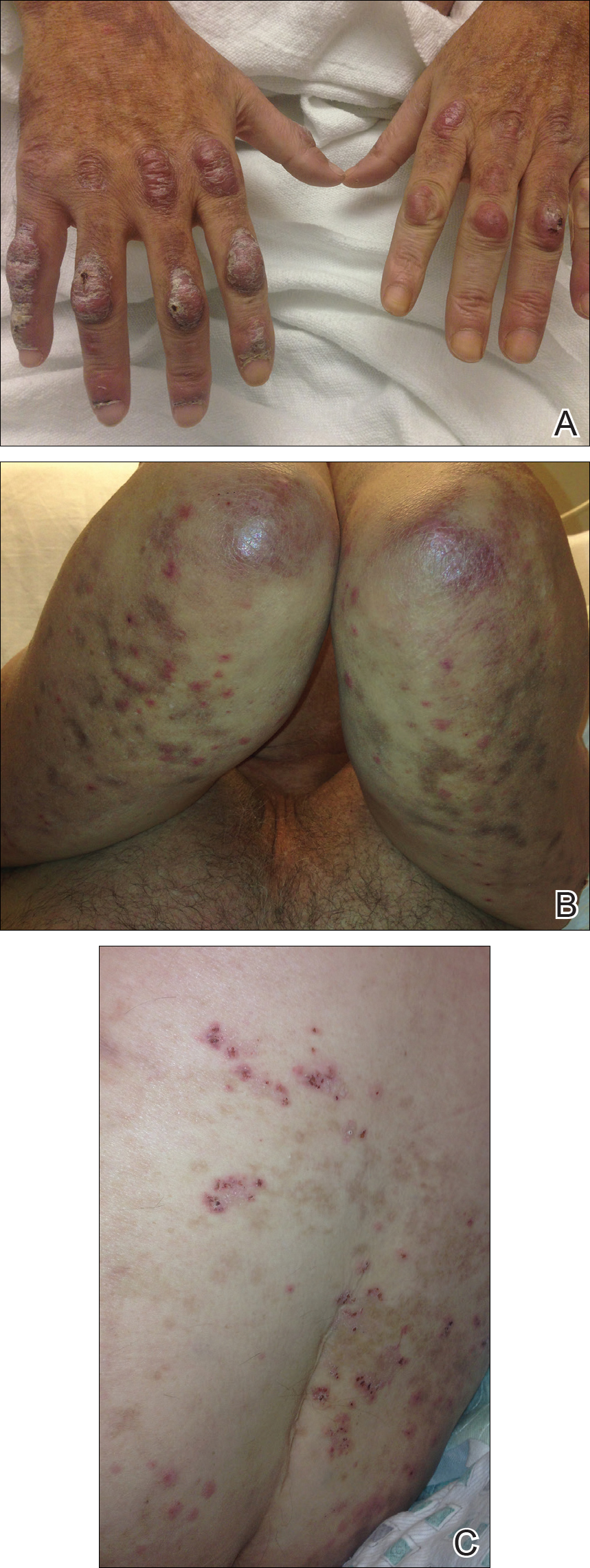
An additional infectious workup was negative for hepatitis C, streptococcus, syphilis, tuberculosis, and HIV. A complete blood cell count, complete metabolic panel, urinalysis, complement, cryoglobulins, and serum protein electrophoresis were within reference range. Autoimmune serologies were negative including antinuclear antibody, rheumatoid factor, anti-Sjögren syndrome–related antigen A and B, anticyclic citrullinated peptide, anti-Smith, and antineutrophilic cytoplasmic antibodies. Peripheral blood immunophenotyping, lactate dehydrogenase, quantitative immunoglobulins, and age-appropriate cancer screens did not demonstrate evidence for malignancy underlying the disease. Bilateral hand radiographs showed mild periostitis of the proximal phalanges without obvious erosions.
Three 4-mm punch biopsies were performed from the left fifth digit, left posterior arm, and left flank. Tissue of the left fifth digit showed an intradermal vascular proliferation with a concentric pattern resembling onion skin in a background of increased fibrosis. The blood vessels showed focal fibrinoid necrosis (Figure 2A). The biopsy of the left posterior arm showed an intradermal vascular proliferation with an associated mild acute and chronic perivascular inflammation (Figure 2B). The left flank biopsy showed LCV with focal epidermal necrosis (Figure 2C).
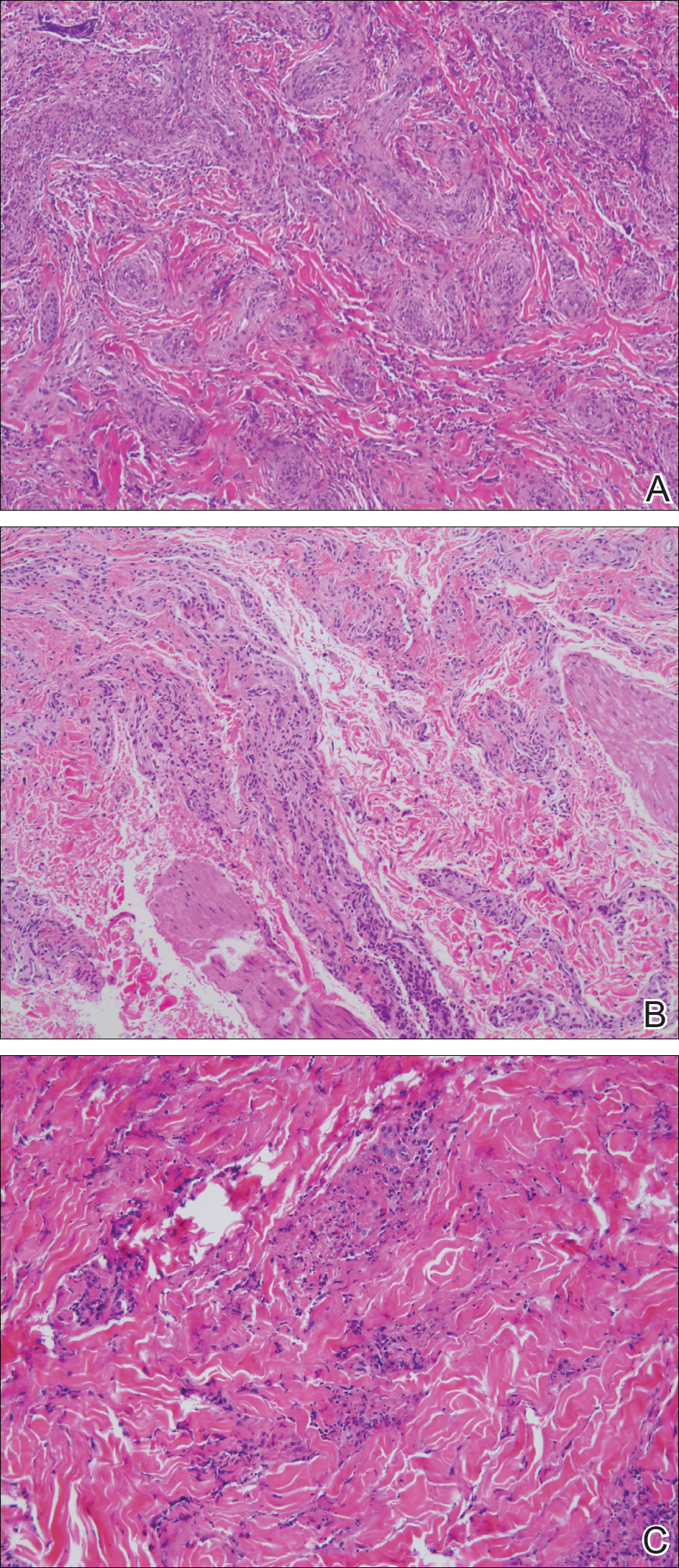
The constellation of clinical findings together with the histopathologic changes represented EED in various stages of evolution. The patient was started on dapsone 100 mg daily and referred to the infectious disease service for treatment of chronic hepatitis B; however, he was subsequently lost to follow-up.
Comment
Overview of EED
Erythema elevatum diutinum represents a rare form of chronic cutaneous small vessel vasculitis. Originally described by Hutchinson7 and Bury8 as symmetric purpuric nodules of the skin, it was later named by Crocker and Williams9 in 1894. The disease classically presents as firm, fixed, red-brown to violaceous papules, plaques, and nodules affecting the extensor upper or lower extremities.1 Lesions are most commonly found symmetrically overlying joints of the hands, feet, elbows, and knees, as well as the Achilles tendon and buttocks.3 Less common locations include the palms and soles, face,10,11 trunk,12 and periauricular region.1 Although they are typically asymptomatic, sensations such as burning, stinging, and pruritus have been noted.1 Our patient was unique because in addition to typical lesions of EED, he presented with crusted papules on the flanks and violaceous papules of the lower legs and periumbilicus.
Etiology
Originally associated with Streptococcus as isolated from EED lesions,3,13 additional infectious etiologies include viral hepatitis,4-6 human herpesvirus 6,14 and rarely HIV.1,15 Hepatitis B and C are well known to be associated with EED, with only rare reports in patients with concomitant HIV infection. Erythema elevatum diutinum also has been described in relationship to myeloproliferative disorders and hematologic malignancies such as IgA myeloma,16 non-Hodgkin lymphoma,17 chronic lymphocytic leukemia,18 and hypergammaglobulinemia.19 In a study of 13 patients with EED, 4 had associated underlying IgA monoclonal gammopathy.2 Autoimmune conditions such as rheumatoid arthritis,20 ulcerative colitis,21 relapsing polychondritis,22 and systemic lupus erythematosus23 also have been implicated.
Pathogenesis
Although the precise pathogenesis of EED remains unknown, it has been suggested that a complement cascade initiated by immune-complex deposition in postcapillary venules induces an LCV.24,25 Chronic antigenic exposure or high antibody levels26 in the face of infections, autoimmune disease, or malignancy may incite this immune-complex reaction. Skin lesions seen in association with hepatitis reflect circulating immune-complex deposition in vessel walls causing destruction. It has been postulated that the duration of immune complexemia may be sufficient to account for the differences in the type of vascular injury seen in acute versus chronic infection.27
Histopathology
Erythema elevatum diutinum may present on a histopathologic spectrum of LCV, as manifested in our patient. Early lesions show predominantly polymorphonuclear cells with nuclear dust pattern in a wedge-shaped infiltrate with fibrin deposition in the superficial and mid dermis.2,3 Later lesions show vasculitis in addition to dermal aggregates of lymphocytes, neutrophils, fibrosis, and areas of granulation tissue. The fibrosis may be dense and comprised of fibroblasts and myofibroblasts.28 Newly formed vessels within the granulation tissue have been postulated to be more susceptible to immune-complex deposition, thus potentiating the process.1,29
Management
Spontaneous resolution of EED may occur, albeit after a prolonged and recurrent course of up to 5 to 10 years.30 Treatment of the underlying cause, when identified, remains paramount. First-line therapy includes dapsone, shown to be effective in reducing lesion size to complete resolution in 80% of the 47 cases reviewed by Momen et al.31 Dapsone monotherapy tends to be less effective in treating nodular lesions associated with HIV-positivity, likely due to the extensive fibrosis.4,31 Combination therapy with dapsone and a sulfonamide,32 niacinamide and tetracycline,33 colchicine,34 or surgical excision35 may be necessary in more resistant cases.
Conclusion
Our case exemplifies the clinical histologic spectrum that EED can present. The constellation of clinical findings was histologically confirmed to be manifestations of the disease in various stages of evolution. When typical lesions of EED present along with cutaneous findings in less common locations, performing multiple biopsies can be helpful. The clinician should retain a high index of suspicion for an underlying etiology and perform a complete workup for infection, malignancy, or autoimmune disease.
- Gibson LE, el-Azhary RA. Erythema elevatum diutinum. Clin Dermatol. 2000;18:295-299.
- Yiannias JA, el-Azhary RA, Gibson LE. Erythema elevatum diutinum: a clinical and histopathologic study of 13 patients. J Am Acad Dermatol. 1992;26:38-44.
- Wilkinson SM, English JS, Smith NP, et al. Erythema elevatum diutinum: a clinicopathological study. Clin Exp Dermatol. 1992;17:87-93.
- Fakheri A, Gupta SM, White SM, et al. Erythema elevatum diutinum in a patient with human immunodeficiency virus. Cutis. 2001;68:41-42, 55.
- Kim H. Erythema elevatum diutinum in an HIV-positive patient. J Drugs Dermatol. 2003;2:411-412.
- Revenga F, Vera A, Muñoz A, et al. Erythema elevatum diutinum and AIDS: are they related? Clin Exp Dermatol. 1997;22:250-251.
- Hutchinson J. On two remarkable cases of symmetrieal purple congestion of the skin in patches, with induration. Br J Dermatol. 1888;1:10-15.
- Bury JS. A case of erythema with remarkable nodular thickening and induration of the skin associated with intermittent albuminuria. Illustrated Medical News. 1889;3:145-149.
- Crocker HR, Williams C. Erythema elevatum diutinum. Br J Dermatol. 1894;6:33-38.
- Barzegar M, Davatchi CC, Akhyani M, et al. An atypical presentation of erythema elevatum diutinum involving palms and soles. Int J Dermatol. 2009;48:73-75.
- Futei Y, Konohana I. A case of erythema elevatum diutinum associated with B-cell lymphoma: a rare distribution involving palms, soles and nails. Br J Dermatol. 2000;142:116-119.
- Ben-Zvi GT, Bardsley V, Burrows NP. An atypical distribution of erythema elevatum diutinum. Clin Exp Dermatol. 2014;39:269-270.
- Weidman FD, Besancon JH. Erythema elevatum diutinum. role of streptococci, and relationship to other rheumatic dermatoses. Arch Dermatol Syphilol. 1929;20:593-620.
- Drago F, Semino M, Rampini P, et al. Erythema elevatum diutinum in a patient with human herpesvirus 6 infection. Acta Derm Venereol. 1999;79:91-92.
- Muratori S, Carrera C, Gorani A, et al. Erythema elevatum diutinum and HIV infection: a report of five cases. Br J Dermatol. 1999;141:335-338.
- Archimandritis AJ, Fertakis A, Alegakis G, et al. Erythema elevatum diutinum and IgA myeloma: an interesting association. Br Med J. 1977;2:613-614.
- Hatzitolios A, Tzellos TG, Savopoulos C, et al. Erythema elevatum diutinum with rare distribution as a first clinical sign of non-Hodgkin’s lymphoma: a novel association? J Dermatol. 2008;35:297-300.
- Delaporte E, Alfandari S, Fenaux P, et al. Erythema elevatum diutinum and chronic lymphocytic leukaemia. Clin Exp Dermatol. 1994;19:188-189.
- Miyagawa S, Kitamura W, Morita K, et al. Association of hyperimmunoglobulinaemia D syndrome with erythema elevatum diutinum. Br J Dermatol. 1993;128:572-574.
- Collier PM, Neill SM, Branfoot AC, et al. Erythema elevatum diutinum—a solitary lesion in a patient with rheumatoid arthritis. Clin Exp Dermatol. 1990;15:394-395.
- Buahene K, Hudson M, Mowat A, et al. Erythema elevatum diutinum—an unusual association with ulcerative colitis. Clin Exp Dermatol. 1991;16:204-206.
- Bernard P, Bedane C, Delrous JL, et al. Erythema elevatum diutinum in a patient with relapsing polychondritis. J Am Acad Dermatol. 1992;26:312-315.
- Hancox JG, Wallace CA, Sangueza OP, et al. Erythema elevatum diutinum associated with lupus panniculitis in a patient with discoid lesions of chronic cutaneous lupus erythematosus. J Am Acad Dermatol. 2004;50:652-653.
- Haber H. Erythema elevatum diutinum. Br J Dermatol. 1955;67:121-145.
- Katz SI, Gallin JL, Hertz KC, et al. Erythema elevatum diutinum: skin and systemic manifestations, immunologic studies, and successful treatment with dapsone. Medicine (Baltimore). 1977;56:443-455.
- Walker KD, Badame AJ. Erythema elevatum diutinum in a patient with Crohn’s disease. J Am Acad Dermatol. 1990;22:948-952.
- Popp JW, Harrist T, Dienstag JL, et al. Cutaneous vasculitis associated with acute and chronic hepatitis. Arch Intern Med. 1981;141:623-629.
- Lee AY, Nakagawa H, Nogita T, et al. Erythema elevatum diutinum: an ultrastructural case study. J Cutan Pathol. 1989;16:211-217.
- LeBoit PE, Yen TS, Wintroub B. The evolution of lesions in erythema elevatum diutinum. Am J Dermatopathol. 1986;8:392-402.
- Soubeiran E, Wacker J, Hausser I, et al. Erythema elevatum diutinum with unusual clinical appearance. J Dtsch Dermatol Ges. 2008;6:303-305.
- Momen SE, Jorizzo J, Al-Niaimi F. Erythema elevatum diutinum: a review of presentation and treatment. J Eur Acad Dermatol Venereol. 2014;28:1594-1602.
- Vollum DI. Erythema elevatum diutinum—vesicular lesions and sulfone response. Br J Dermatol. 1968;80:178-183.
- Kohler IK, Lorincz AL. Erythema elevatum diutinum treated with niacinamide and tetracycline. Arch Dermatol. 1980;116:693-695.
- Henriksson R, Hofor PA, Hörngvist R. Erythema elevatum diutinum—a case successfully treated with colchicine. Clin Exp Dermatol. 1989;14:451-453.
- Zacaron LH, Gonçalves JC, Curty VM, et al. Clinical and surgical therapeutic approach in erythema elevatum diutinum—case report. An Bras Dermatol. 2013;88(6, suppl 1):15-18.
Erythema elevatum diutinum (EED) manifests on a clinicopathologic spectrum of chronic cutaneous small vessel vasculitis. The lesions typically present as persistent, symmetric, firm, red to purple papules or nodules on the extensor arms and dorsal hands.1,2 Underlying infectious, malignant, or autoimmune processes are commonly associated with the disease, notably Streptococcus infection and IgA monoclonal gammopathy.2,3 Hepatitis virus also is often implicated in association with EED. Cases of EED have been seen with concomitant human immunodeficiency virus (HIV) infection.4-6 We report a case of EED presenting in various stages of evolution associated with underlying hepatitis B infection alone.
Case Report
A 57-year-old man originally presented to an outpatient dermatology practice with a nodular, painful, episodic rash on the trunk and upper and lower extremities. A biopsy revealed leukocytoclastic vasculitis (LCV) with prominent eosinophils. At the time, the skin findings were believed to be a manifestation of drug hypersensitivity, likely to opioid use. The patient was lost to follow-up.
Seven years later, the patient was admitted to the hospital with new-onset burning and stinging red nodules on the dorsum of the hands and persistence of the original episodic rash over the lower legs and bilateral flanks. In the interim, he was briefly treated with an oral prednisone taper and topical corticosteroids including triamcinolone cream 0.1% and clobetasol cream 0.05% without improvement.
On examination deep red to violaceous discrete nodules and plaques with overlying hyperkeratosis involving all distal and proximal interphalangeal joints of the hands and extensor elbows were seen (Figure 1A). On the bilateral posterior arms (Figure 1B), anterior legs, and periumbilical area were deeply erythematous papules and plaques with background hyperpigmentation. Across his lower back and bilateral flanks were erythematous papules with central hemorrhagic crusting (Figure 1C).
Pertinent laboratory findings included a positive hepatitis B surface antigen with hepatitis B DNA value 4,313,876 IU/mL and a hepatitis B virus quantitative polymerase chain reaction value of 6.64 U. The etiology was suspected to be intravenous drug abuse; however, the patient denied recreational drug use.
An additional infectious workup was negative for hepatitis C, streptococcus, syphilis, tuberculosis, and HIV. A complete blood cell count, complete metabolic panel, urinalysis, complement, cryoglobulins, and serum protein electrophoresis were within reference range. Autoimmune serologies were negative including antinuclear antibody, rheumatoid factor, anti-Sjögren syndrome–related antigen A and B, anticyclic citrullinated peptide, anti-Smith, and antineutrophilic cytoplasmic antibodies. Peripheral blood immunophenotyping, lactate dehydrogenase, quantitative immunoglobulins, and age-appropriate cancer screens did not demonstrate evidence for malignancy underlying the disease. Bilateral hand radiographs showed mild periostitis of the proximal phalanges without obvious erosions.
Three 4-mm punch biopsies were performed from the left fifth digit, left posterior arm, and left flank. Tissue of the left fifth digit showed an intradermal vascular proliferation with a concentric pattern resembling onion skin in a background of increased fibrosis. The blood vessels showed focal fibrinoid necrosis (Figure 2A). The biopsy of the left posterior arm showed an intradermal vascular proliferation with an associated mild acute and chronic perivascular inflammation (Figure 2B). The left flank biopsy showed LCV with focal epidermal necrosis (Figure 2C).
The constellation of clinical findings together with the histopathologic changes represented EED in various stages of evolution. The patient was started on dapsone 100 mg daily and referred to the infectious disease service for treatment of chronic hepatitis B; however, he was subsequently lost to follow-up.
Comment
Overview of EED
Erythema elevatum diutinum represents a rare form of chronic cutaneous small vessel vasculitis. Originally described by Hutchinson7 and Bury8 as symmetric purpuric nodules of the skin, it was later named by Crocker and Williams9 in 1894. The disease classically presents as firm, fixed, red-brown to violaceous papules, plaques, and nodules affecting the extensor upper or lower extremities.1 Lesions are most commonly found symmetrically overlying joints of the hands, feet, elbows, and knees, as well as the Achilles tendon and buttocks.3 Less common locations include the palms and soles, face,10,11 trunk,12 and periauricular region.1 Although they are typically asymptomatic, sensations such as burning, stinging, and pruritus have been noted.1 Our patient was unique because in addition to typical lesions of EED, he presented with crusted papules on the flanks and violaceous papules of the lower legs and periumbilicus.
Etiology
Originally associated with Streptococcus as isolated from EED lesions,3,13 additional infectious etiologies include viral hepatitis,4-6 human herpesvirus 6,14 and rarely HIV.1,15 Hepatitis B and C are well known to be associated with EED, with only rare reports in patients with concomitant HIV infection. Erythema elevatum diutinum also has been described in relationship to myeloproliferative disorders and hematologic malignancies such as IgA myeloma,16 non-Hodgkin lymphoma,17 chronic lymphocytic leukemia,18 and hypergammaglobulinemia.19 In a study of 13 patients with EED, 4 had associated underlying IgA monoclonal gammopathy.2 Autoimmune conditions such as rheumatoid arthritis,20 ulcerative colitis,21 relapsing polychondritis,22 and systemic lupus erythematosus23 also have been implicated.
Pathogenesis
Although the precise pathogenesis of EED remains unknown, it has been suggested that a complement cascade initiated by immune-complex deposition in postcapillary venules induces an LCV.24,25 Chronic antigenic exposure or high antibody levels26 in the face of infections, autoimmune disease, or malignancy may incite this immune-complex reaction. Skin lesions seen in association with hepatitis reflect circulating immune-complex deposition in vessel walls causing destruction. It has been postulated that the duration of immune complexemia may be sufficient to account for the differences in the type of vascular injury seen in acute versus chronic infection.27
Histopathology
Erythema elevatum diutinum may present on a histopathologic spectrum of LCV, as manifested in our patient. Early lesions show predominantly polymorphonuclear cells with nuclear dust pattern in a wedge-shaped infiltrate with fibrin deposition in the superficial and mid dermis.2,3 Later lesions show vasculitis in addition to dermal aggregates of lymphocytes, neutrophils, fibrosis, and areas of granulation tissue. The fibrosis may be dense and comprised of fibroblasts and myofibroblasts.28 Newly formed vessels within the granulation tissue have been postulated to be more susceptible to immune-complex deposition, thus potentiating the process.1,29
Management
Spontaneous resolution of EED may occur, albeit after a prolonged and recurrent course of up to 5 to 10 years.30 Treatment of the underlying cause, when identified, remains paramount. First-line therapy includes dapsone, shown to be effective in reducing lesion size to complete resolution in 80% of the 47 cases reviewed by Momen et al.31 Dapsone monotherapy tends to be less effective in treating nodular lesions associated with HIV-positivity, likely due to the extensive fibrosis.4,31 Combination therapy with dapsone and a sulfonamide,32 niacinamide and tetracycline,33 colchicine,34 or surgical excision35 may be necessary in more resistant cases.
Conclusion
Our case exemplifies the clinical histologic spectrum that EED can present. The constellation of clinical findings was histologically confirmed to be manifestations of the disease in various stages of evolution. When typical lesions of EED present along with cutaneous findings in less common locations, performing multiple biopsies can be helpful. The clinician should retain a high index of suspicion for an underlying etiology and perform a complete workup for infection, malignancy, or autoimmune disease.
Erythema elevatum diutinum (EED) manifests on a clinicopathologic spectrum of chronic cutaneous small vessel vasculitis. The lesions typically present as persistent, symmetric, firm, red to purple papules or nodules on the extensor arms and dorsal hands.1,2 Underlying infectious, malignant, or autoimmune processes are commonly associated with the disease, notably Streptococcus infection and IgA monoclonal gammopathy.2,3 Hepatitis virus also is often implicated in association with EED. Cases of EED have been seen with concomitant human immunodeficiency virus (HIV) infection.4-6 We report a case of EED presenting in various stages of evolution associated with underlying hepatitis B infection alone.
Case Report
A 57-year-old man originally presented to an outpatient dermatology practice with a nodular, painful, episodic rash on the trunk and upper and lower extremities. A biopsy revealed leukocytoclastic vasculitis (LCV) with prominent eosinophils. At the time, the skin findings were believed to be a manifestation of drug hypersensitivity, likely to opioid use. The patient was lost to follow-up.
Seven years later, the patient was admitted to the hospital with new-onset burning and stinging red nodules on the dorsum of the hands and persistence of the original episodic rash over the lower legs and bilateral flanks. In the interim, he was briefly treated with an oral prednisone taper and topical corticosteroids including triamcinolone cream 0.1% and clobetasol cream 0.05% without improvement.
On examination deep red to violaceous discrete nodules and plaques with overlying hyperkeratosis involving all distal and proximal interphalangeal joints of the hands and extensor elbows were seen (Figure 1A). On the bilateral posterior arms (Figure 1B), anterior legs, and periumbilical area were deeply erythematous papules and plaques with background hyperpigmentation. Across his lower back and bilateral flanks were erythematous papules with central hemorrhagic crusting (Figure 1C).
Pertinent laboratory findings included a positive hepatitis B surface antigen with hepatitis B DNA value 4,313,876 IU/mL and a hepatitis B virus quantitative polymerase chain reaction value of 6.64 U. The etiology was suspected to be intravenous drug abuse; however, the patient denied recreational drug use.
An additional infectious workup was negative for hepatitis C, streptococcus, syphilis, tuberculosis, and HIV. A complete blood cell count, complete metabolic panel, urinalysis, complement, cryoglobulins, and serum protein electrophoresis were within reference range. Autoimmune serologies were negative including antinuclear antibody, rheumatoid factor, anti-Sjögren syndrome–related antigen A and B, anticyclic citrullinated peptide, anti-Smith, and antineutrophilic cytoplasmic antibodies. Peripheral blood immunophenotyping, lactate dehydrogenase, quantitative immunoglobulins, and age-appropriate cancer screens did not demonstrate evidence for malignancy underlying the disease. Bilateral hand radiographs showed mild periostitis of the proximal phalanges without obvious erosions.
Three 4-mm punch biopsies were performed from the left fifth digit, left posterior arm, and left flank. Tissue of the left fifth digit showed an intradermal vascular proliferation with a concentric pattern resembling onion skin in a background of increased fibrosis. The blood vessels showed focal fibrinoid necrosis (Figure 2A). The biopsy of the left posterior arm showed an intradermal vascular proliferation with an associated mild acute and chronic perivascular inflammation (Figure 2B). The left flank biopsy showed LCV with focal epidermal necrosis (Figure 2C).
The constellation of clinical findings together with the histopathologic changes represented EED in various stages of evolution. The patient was started on dapsone 100 mg daily and referred to the infectious disease service for treatment of chronic hepatitis B; however, he was subsequently lost to follow-up.
Comment
Overview of EED
Erythema elevatum diutinum represents a rare form of chronic cutaneous small vessel vasculitis. Originally described by Hutchinson7 and Bury8 as symmetric purpuric nodules of the skin, it was later named by Crocker and Williams9 in 1894. The disease classically presents as firm, fixed, red-brown to violaceous papules, plaques, and nodules affecting the extensor upper or lower extremities.1 Lesions are most commonly found symmetrically overlying joints of the hands, feet, elbows, and knees, as well as the Achilles tendon and buttocks.3 Less common locations include the palms and soles, face,10,11 trunk,12 and periauricular region.1 Although they are typically asymptomatic, sensations such as burning, stinging, and pruritus have been noted.1 Our patient was unique because in addition to typical lesions of EED, he presented with crusted papules on the flanks and violaceous papules of the lower legs and periumbilicus.
Etiology
Originally associated with Streptococcus as isolated from EED lesions,3,13 additional infectious etiologies include viral hepatitis,4-6 human herpesvirus 6,14 and rarely HIV.1,15 Hepatitis B and C are well known to be associated with EED, with only rare reports in patients with concomitant HIV infection. Erythema elevatum diutinum also has been described in relationship to myeloproliferative disorders and hematologic malignancies such as IgA myeloma,16 non-Hodgkin lymphoma,17 chronic lymphocytic leukemia,18 and hypergammaglobulinemia.19 In a study of 13 patients with EED, 4 had associated underlying IgA monoclonal gammopathy.2 Autoimmune conditions such as rheumatoid arthritis,20 ulcerative colitis,21 relapsing polychondritis,22 and systemic lupus erythematosus23 also have been implicated.
Pathogenesis
Although the precise pathogenesis of EED remains unknown, it has been suggested that a complement cascade initiated by immune-complex deposition in postcapillary venules induces an LCV.24,25 Chronic antigenic exposure or high antibody levels26 in the face of infections, autoimmune disease, or malignancy may incite this immune-complex reaction. Skin lesions seen in association with hepatitis reflect circulating immune-complex deposition in vessel walls causing destruction. It has been postulated that the duration of immune complexemia may be sufficient to account for the differences in the type of vascular injury seen in acute versus chronic infection.27
Histopathology
Erythema elevatum diutinum may present on a histopathologic spectrum of LCV, as manifested in our patient. Early lesions show predominantly polymorphonuclear cells with nuclear dust pattern in a wedge-shaped infiltrate with fibrin deposition in the superficial and mid dermis.2,3 Later lesions show vasculitis in addition to dermal aggregates of lymphocytes, neutrophils, fibrosis, and areas of granulation tissue. The fibrosis may be dense and comprised of fibroblasts and myofibroblasts.28 Newly formed vessels within the granulation tissue have been postulated to be more susceptible to immune-complex deposition, thus potentiating the process.1,29
Management
Spontaneous resolution of EED may occur, albeit after a prolonged and recurrent course of up to 5 to 10 years.30 Treatment of the underlying cause, when identified, remains paramount. First-line therapy includes dapsone, shown to be effective in reducing lesion size to complete resolution in 80% of the 47 cases reviewed by Momen et al.31 Dapsone monotherapy tends to be less effective in treating nodular lesions associated with HIV-positivity, likely due to the extensive fibrosis.4,31 Combination therapy with dapsone and a sulfonamide,32 niacinamide and tetracycline,33 colchicine,34 or surgical excision35 may be necessary in more resistant cases.
Conclusion
Our case exemplifies the clinical histologic spectrum that EED can present. The constellation of clinical findings was histologically confirmed to be manifestations of the disease in various stages of evolution. When typical lesions of EED present along with cutaneous findings in less common locations, performing multiple biopsies can be helpful. The clinician should retain a high index of suspicion for an underlying etiology and perform a complete workup for infection, malignancy, or autoimmune disease.
- Gibson LE, el-Azhary RA. Erythema elevatum diutinum. Clin Dermatol. 2000;18:295-299.
- Yiannias JA, el-Azhary RA, Gibson LE. Erythema elevatum diutinum: a clinical and histopathologic study of 13 patients. J Am Acad Dermatol. 1992;26:38-44.
- Wilkinson SM, English JS, Smith NP, et al. Erythema elevatum diutinum: a clinicopathological study. Clin Exp Dermatol. 1992;17:87-93.
- Fakheri A, Gupta SM, White SM, et al. Erythema elevatum diutinum in a patient with human immunodeficiency virus. Cutis. 2001;68:41-42, 55.
- Kim H. Erythema elevatum diutinum in an HIV-positive patient. J Drugs Dermatol. 2003;2:411-412.
- Revenga F, Vera A, Muñoz A, et al. Erythema elevatum diutinum and AIDS: are they related? Clin Exp Dermatol. 1997;22:250-251.
- Hutchinson J. On two remarkable cases of symmetrieal purple congestion of the skin in patches, with induration. Br J Dermatol. 1888;1:10-15.
- Bury JS. A case of erythema with remarkable nodular thickening and induration of the skin associated with intermittent albuminuria. Illustrated Medical News. 1889;3:145-149.
- Crocker HR, Williams C. Erythema elevatum diutinum. Br J Dermatol. 1894;6:33-38.
- Barzegar M, Davatchi CC, Akhyani M, et al. An atypical presentation of erythema elevatum diutinum involving palms and soles. Int J Dermatol. 2009;48:73-75.
- Futei Y, Konohana I. A case of erythema elevatum diutinum associated with B-cell lymphoma: a rare distribution involving palms, soles and nails. Br J Dermatol. 2000;142:116-119.
- Ben-Zvi GT, Bardsley V, Burrows NP. An atypical distribution of erythema elevatum diutinum. Clin Exp Dermatol. 2014;39:269-270.
- Weidman FD, Besancon JH. Erythema elevatum diutinum. role of streptococci, and relationship to other rheumatic dermatoses. Arch Dermatol Syphilol. 1929;20:593-620.
- Drago F, Semino M, Rampini P, et al. Erythema elevatum diutinum in a patient with human herpesvirus 6 infection. Acta Derm Venereol. 1999;79:91-92.
- Muratori S, Carrera C, Gorani A, et al. Erythema elevatum diutinum and HIV infection: a report of five cases. Br J Dermatol. 1999;141:335-338.
- Archimandritis AJ, Fertakis A, Alegakis G, et al. Erythema elevatum diutinum and IgA myeloma: an interesting association. Br Med J. 1977;2:613-614.
- Hatzitolios A, Tzellos TG, Savopoulos C, et al. Erythema elevatum diutinum with rare distribution as a first clinical sign of non-Hodgkin’s lymphoma: a novel association? J Dermatol. 2008;35:297-300.
- Delaporte E, Alfandari S, Fenaux P, et al. Erythema elevatum diutinum and chronic lymphocytic leukaemia. Clin Exp Dermatol. 1994;19:188-189.
- Miyagawa S, Kitamura W, Morita K, et al. Association of hyperimmunoglobulinaemia D syndrome with erythema elevatum diutinum. Br J Dermatol. 1993;128:572-574.
- Collier PM, Neill SM, Branfoot AC, et al. Erythema elevatum diutinum—a solitary lesion in a patient with rheumatoid arthritis. Clin Exp Dermatol. 1990;15:394-395.
- Buahene K, Hudson M, Mowat A, et al. Erythema elevatum diutinum—an unusual association with ulcerative colitis. Clin Exp Dermatol. 1991;16:204-206.
- Bernard P, Bedane C, Delrous JL, et al. Erythema elevatum diutinum in a patient with relapsing polychondritis. J Am Acad Dermatol. 1992;26:312-315.
- Hancox JG, Wallace CA, Sangueza OP, et al. Erythema elevatum diutinum associated with lupus panniculitis in a patient with discoid lesions of chronic cutaneous lupus erythematosus. J Am Acad Dermatol. 2004;50:652-653.
- Haber H. Erythema elevatum diutinum. Br J Dermatol. 1955;67:121-145.
- Katz SI, Gallin JL, Hertz KC, et al. Erythema elevatum diutinum: skin and systemic manifestations, immunologic studies, and successful treatment with dapsone. Medicine (Baltimore). 1977;56:443-455.
- Walker KD, Badame AJ. Erythema elevatum diutinum in a patient with Crohn’s disease. J Am Acad Dermatol. 1990;22:948-952.
- Popp JW, Harrist T, Dienstag JL, et al. Cutaneous vasculitis associated with acute and chronic hepatitis. Arch Intern Med. 1981;141:623-629.
- Lee AY, Nakagawa H, Nogita T, et al. Erythema elevatum diutinum: an ultrastructural case study. J Cutan Pathol. 1989;16:211-217.
- LeBoit PE, Yen TS, Wintroub B. The evolution of lesions in erythema elevatum diutinum. Am J Dermatopathol. 1986;8:392-402.
- Soubeiran E, Wacker J, Hausser I, et al. Erythema elevatum diutinum with unusual clinical appearance. J Dtsch Dermatol Ges. 2008;6:303-305.
- Momen SE, Jorizzo J, Al-Niaimi F. Erythema elevatum diutinum: a review of presentation and treatment. J Eur Acad Dermatol Venereol. 2014;28:1594-1602.
- Vollum DI. Erythema elevatum diutinum—vesicular lesions and sulfone response. Br J Dermatol. 1968;80:178-183.
- Kohler IK, Lorincz AL. Erythema elevatum diutinum treated with niacinamide and tetracycline. Arch Dermatol. 1980;116:693-695.
- Henriksson R, Hofor PA, Hörngvist R. Erythema elevatum diutinum—a case successfully treated with colchicine. Clin Exp Dermatol. 1989;14:451-453.
- Zacaron LH, Gonçalves JC, Curty VM, et al. Clinical and surgical therapeutic approach in erythema elevatum diutinum—case report. An Bras Dermatol. 2013;88(6, suppl 1):15-18.
- Gibson LE, el-Azhary RA. Erythema elevatum diutinum. Clin Dermatol. 2000;18:295-299.
- Yiannias JA, el-Azhary RA, Gibson LE. Erythema elevatum diutinum: a clinical and histopathologic study of 13 patients. J Am Acad Dermatol. 1992;26:38-44.
- Wilkinson SM, English JS, Smith NP, et al. Erythema elevatum diutinum: a clinicopathological study. Clin Exp Dermatol. 1992;17:87-93.
- Fakheri A, Gupta SM, White SM, et al. Erythema elevatum diutinum in a patient with human immunodeficiency virus. Cutis. 2001;68:41-42, 55.
- Kim H. Erythema elevatum diutinum in an HIV-positive patient. J Drugs Dermatol. 2003;2:411-412.
- Revenga F, Vera A, Muñoz A, et al. Erythema elevatum diutinum and AIDS: are they related? Clin Exp Dermatol. 1997;22:250-251.
- Hutchinson J. On two remarkable cases of symmetrieal purple congestion of the skin in patches, with induration. Br J Dermatol. 1888;1:10-15.
- Bury JS. A case of erythema with remarkable nodular thickening and induration of the skin associated with intermittent albuminuria. Illustrated Medical News. 1889;3:145-149.
- Crocker HR, Williams C. Erythema elevatum diutinum. Br J Dermatol. 1894;6:33-38.
- Barzegar M, Davatchi CC, Akhyani M, et al. An atypical presentation of erythema elevatum diutinum involving palms and soles. Int J Dermatol. 2009;48:73-75.
- Futei Y, Konohana I. A case of erythema elevatum diutinum associated with B-cell lymphoma: a rare distribution involving palms, soles and nails. Br J Dermatol. 2000;142:116-119.
- Ben-Zvi GT, Bardsley V, Burrows NP. An atypical distribution of erythema elevatum diutinum. Clin Exp Dermatol. 2014;39:269-270.
- Weidman FD, Besancon JH. Erythema elevatum diutinum. role of streptococci, and relationship to other rheumatic dermatoses. Arch Dermatol Syphilol. 1929;20:593-620.
- Drago F, Semino M, Rampini P, et al. Erythema elevatum diutinum in a patient with human herpesvirus 6 infection. Acta Derm Venereol. 1999;79:91-92.
- Muratori S, Carrera C, Gorani A, et al. Erythema elevatum diutinum and HIV infection: a report of five cases. Br J Dermatol. 1999;141:335-338.
- Archimandritis AJ, Fertakis A, Alegakis G, et al. Erythema elevatum diutinum and IgA myeloma: an interesting association. Br Med J. 1977;2:613-614.
- Hatzitolios A, Tzellos TG, Savopoulos C, et al. Erythema elevatum diutinum with rare distribution as a first clinical sign of non-Hodgkin’s lymphoma: a novel association? J Dermatol. 2008;35:297-300.
- Delaporte E, Alfandari S, Fenaux P, et al. Erythema elevatum diutinum and chronic lymphocytic leukaemia. Clin Exp Dermatol. 1994;19:188-189.
- Miyagawa S, Kitamura W, Morita K, et al. Association of hyperimmunoglobulinaemia D syndrome with erythema elevatum diutinum. Br J Dermatol. 1993;128:572-574.
- Collier PM, Neill SM, Branfoot AC, et al. Erythema elevatum diutinum—a solitary lesion in a patient with rheumatoid arthritis. Clin Exp Dermatol. 1990;15:394-395.
- Buahene K, Hudson M, Mowat A, et al. Erythema elevatum diutinum—an unusual association with ulcerative colitis. Clin Exp Dermatol. 1991;16:204-206.
- Bernard P, Bedane C, Delrous JL, et al. Erythema elevatum diutinum in a patient with relapsing polychondritis. J Am Acad Dermatol. 1992;26:312-315.
- Hancox JG, Wallace CA, Sangueza OP, et al. Erythema elevatum diutinum associated with lupus panniculitis in a patient with discoid lesions of chronic cutaneous lupus erythematosus. J Am Acad Dermatol. 2004;50:652-653.
- Haber H. Erythema elevatum diutinum. Br J Dermatol. 1955;67:121-145.
- Katz SI, Gallin JL, Hertz KC, et al. Erythema elevatum diutinum: skin and systemic manifestations, immunologic studies, and successful treatment with dapsone. Medicine (Baltimore). 1977;56:443-455.
- Walker KD, Badame AJ. Erythema elevatum diutinum in a patient with Crohn’s disease. J Am Acad Dermatol. 1990;22:948-952.
- Popp JW, Harrist T, Dienstag JL, et al. Cutaneous vasculitis associated with acute and chronic hepatitis. Arch Intern Med. 1981;141:623-629.
- Lee AY, Nakagawa H, Nogita T, et al. Erythema elevatum diutinum: an ultrastructural case study. J Cutan Pathol. 1989;16:211-217.
- LeBoit PE, Yen TS, Wintroub B. The evolution of lesions in erythema elevatum diutinum. Am J Dermatopathol. 1986;8:392-402.
- Soubeiran E, Wacker J, Hausser I, et al. Erythema elevatum diutinum with unusual clinical appearance. J Dtsch Dermatol Ges. 2008;6:303-305.
- Momen SE, Jorizzo J, Al-Niaimi F. Erythema elevatum diutinum: a review of presentation and treatment. J Eur Acad Dermatol Venereol. 2014;28:1594-1602.
- Vollum DI. Erythema elevatum diutinum—vesicular lesions and sulfone response. Br J Dermatol. 1968;80:178-183.
- Kohler IK, Lorincz AL. Erythema elevatum diutinum treated with niacinamide and tetracycline. Arch Dermatol. 1980;116:693-695.
- Henriksson R, Hofor PA, Hörngvist R. Erythema elevatum diutinum—a case successfully treated with colchicine. Clin Exp Dermatol. 1989;14:451-453.
- Zacaron LH, Gonçalves JC, Curty VM, et al. Clinical and surgical therapeutic approach in erythema elevatum diutinum—case report. An Bras Dermatol. 2013;88(6, suppl 1):15-18.
Practice Points
- Erythema elevatum diutinum (EED) often is associated with an underlying infectious process, including hepatitis B and hepatitis C, or a hematologic or autoimmune condition.
- If EED is suspected clinically, it may be beneficial to perform multiple biopsies from lesions at different stages of evolution to establish the diagnosis.
- First-line therapy includes treatment of any underlying condition and dapsone.
