User login
Bringing you the latest news, research and reviews, exclusive interviews, podcasts, quizzes, and more.
Phase 1 CAR T trial for NHL launches in Cleveland
University Hospitals Seidman Cancer Center in Cleveland has launched a phase 1 clinical trial to study the safety of CAR T therapy for non-Hodgkin lymphoma.
The trial will enroll 12-15 adult patients with non-Hodgkin lymphoma who have not responded to standard therapies, according to a statement from University Hospitals Seidman Cancer Center.
The principal investigator for the trial will be Paolo Caimi, MD, of UH Seidman and Case Western Reserve University.
UH Seidman, affiliated with Case Western Reserve University, is one of a handful of centers that has the ability to manufacture the CAR T cells from the patient’s own genetically modified T cells on site in the shared Case Western Reserve University National Center for Regenerative Medicine and the UH Seidman Cellular Therapy Laboratory, saving time for patients.
“Having the ability to make cells on-site means there will be a shorter turnaround time in having the cells available for the patient, compared to shipping them off-site,” said Dr. Caimi in the press statement.
University Hospitals Seidman Cancer Center in Cleveland has launched a phase 1 clinical trial to study the safety of CAR T therapy for non-Hodgkin lymphoma.
The trial will enroll 12-15 adult patients with non-Hodgkin lymphoma who have not responded to standard therapies, according to a statement from University Hospitals Seidman Cancer Center.
The principal investigator for the trial will be Paolo Caimi, MD, of UH Seidman and Case Western Reserve University.
UH Seidman, affiliated with Case Western Reserve University, is one of a handful of centers that has the ability to manufacture the CAR T cells from the patient’s own genetically modified T cells on site in the shared Case Western Reserve University National Center for Regenerative Medicine and the UH Seidman Cellular Therapy Laboratory, saving time for patients.
“Having the ability to make cells on-site means there will be a shorter turnaround time in having the cells available for the patient, compared to shipping them off-site,” said Dr. Caimi in the press statement.
University Hospitals Seidman Cancer Center in Cleveland has launched a phase 1 clinical trial to study the safety of CAR T therapy for non-Hodgkin lymphoma.
The trial will enroll 12-15 adult patients with non-Hodgkin lymphoma who have not responded to standard therapies, according to a statement from University Hospitals Seidman Cancer Center.
The principal investigator for the trial will be Paolo Caimi, MD, of UH Seidman and Case Western Reserve University.
UH Seidman, affiliated with Case Western Reserve University, is one of a handful of centers that has the ability to manufacture the CAR T cells from the patient’s own genetically modified T cells on site in the shared Case Western Reserve University National Center for Regenerative Medicine and the UH Seidman Cellular Therapy Laboratory, saving time for patients.
“Having the ability to make cells on-site means there will be a shorter turnaround time in having the cells available for the patient, compared to shipping them off-site,” said Dr. Caimi in the press statement.
Key clinical point: A phase 1 trial of CAR T therapy is enrolling adult patients with NHL who have not responded to standard therapies.
Major finding: The trial site has the ability to manufacture the cells on site, saving patients time.
Study details: A phase 1 trial to evaluate safety.
Disclosures: The study will be funded by University Hospitals Seidman Cancer Center.
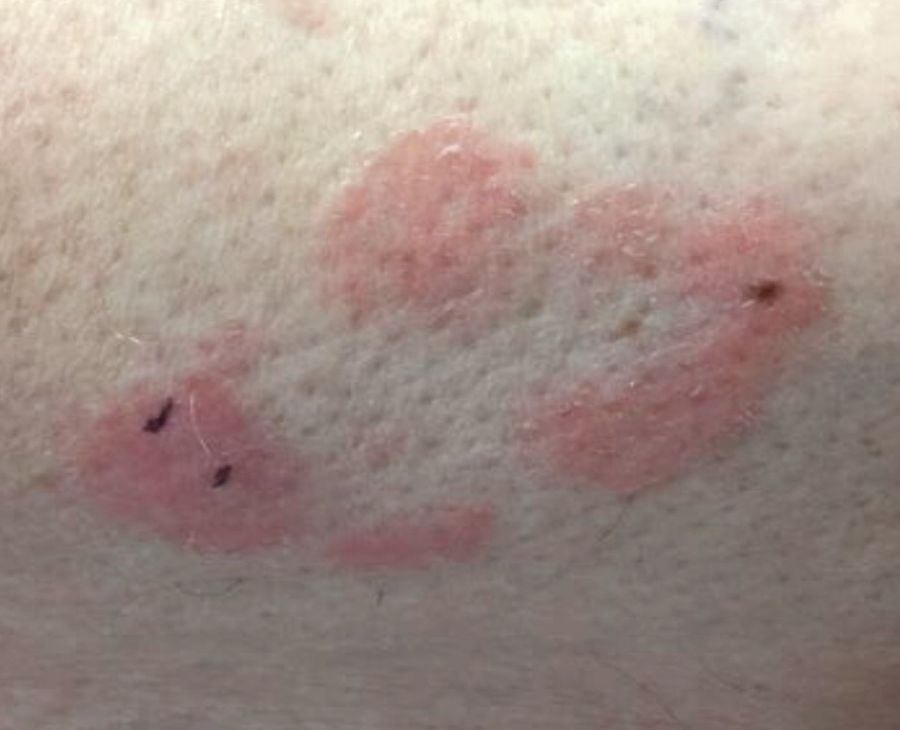
Make The Diagnosis - September 2018
Some have postulated an infectious agent as the cause. Atopic dermatitis may confer an increased risk because of the chronic stimulation of T cells. Males are more commonly affected than females by a 2:1 ratio. A worse prognosis is associated with advanced age. Children and adolescents may be affected as well.
With mycosis fungoides, there are three main types of skin lesions: patch, plaque, and tumor. Patients will progress from patch to plaque to tumor stage in classic MF. Often, lesions begin as scaly, erythematous patches that resemble eczema. Because of the nonspecific nature of early lesions, the median duration from the onset of skin lesions to the diagnosis of MF is 4-6 years. Patch stage lesions may be pruritic or asymptomatic. Commonly, they present in non–sun-exposed areas, such as the buttocks. Annular, infiltrated, red-brown or violaceous plaques can develop, which represent malignant T-cell infiltration. Many patients never progress past the plaque stage. Tumor stage MF is more aggressive, with nodules that may undergo necrosis and ulceration.
The leukemic form of MF is Sézary syndrome. Patients present with pruritic erythroderma and lymphadenopathy. Nail dystrophy, scaling of palms and soles, and alopecia may be present. A peripheral blood smear reveals Sézary cells, which are large, hyperconvoluted lymphocytes. The count of Sézary cells is usually greater than 1000 cells/mm3.
Histology of early lesions may not be diagnostic for CTCL. Often, biopsies will be read as eczematous or psoriasiform for years before the diagnosis of MF is made. Classically, epidermotropism (single-cell exocytosis of lymphocytes into the epidermis) is present. Advanced stages may show a dense infiltrate of lymphocytes in the dermis. Groups of lymphocytes in the epidermis form Pautrier’s microabscesses. Mycosis cells may exhibit cerebriform nuclei. Neoplastic cells in MF are CD3+, CD4+, CD45RO+, CD8–. Tissue can be sent for T-cell gene rearrangement polymerase chain reaction. The presence of monoclonal T-cell gene receptor rearrangements can aid in the diagnosis of MF.
Treatment includes topical steroids, mechlorethamine (nitrogen mustard) or bexarotene gel, PUVA therapy, and narrow-band UVB light for limited and/or patch disease. Localized radiotherapy can be used for more resistant lesions. Topical therapies are preferred in the early stages in MF. Systemic treatments for patients who do not respond to local therapy, or in more advanced disease include methotrexate, interferon-alpha, oral bexarotene, denileukin diftitox, and combination chemotherapy. Photopheresis is reserved for erythrodermic disease.

This case and photo were submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice in Aventura, Fla. More diagnostic cases are available at edermatologynews.com. To submit a case for possible publication, send an email to dermnews@mdedge.com.
Some have postulated an infectious agent as the cause. Atopic dermatitis may confer an increased risk because of the chronic stimulation of T cells. Males are more commonly affected than females by a 2:1 ratio. A worse prognosis is associated with advanced age. Children and adolescents may be affected as well.
With mycosis fungoides, there are three main types of skin lesions: patch, plaque, and tumor. Patients will progress from patch to plaque to tumor stage in classic MF. Often, lesions begin as scaly, erythematous patches that resemble eczema. Because of the nonspecific nature of early lesions, the median duration from the onset of skin lesions to the diagnosis of MF is 4-6 years. Patch stage lesions may be pruritic or asymptomatic. Commonly, they present in non–sun-exposed areas, such as the buttocks. Annular, infiltrated, red-brown or violaceous plaques can develop, which represent malignant T-cell infiltration. Many patients never progress past the plaque stage. Tumor stage MF is more aggressive, with nodules that may undergo necrosis and ulceration.
The leukemic form of MF is Sézary syndrome. Patients present with pruritic erythroderma and lymphadenopathy. Nail dystrophy, scaling of palms and soles, and alopecia may be present. A peripheral blood smear reveals Sézary cells, which are large, hyperconvoluted lymphocytes. The count of Sézary cells is usually greater than 1000 cells/mm3.
Histology of early lesions may not be diagnostic for CTCL. Often, biopsies will be read as eczematous or psoriasiform for years before the diagnosis of MF is made. Classically, epidermotropism (single-cell exocytosis of lymphocytes into the epidermis) is present. Advanced stages may show a dense infiltrate of lymphocytes in the dermis. Groups of lymphocytes in the epidermis form Pautrier’s microabscesses. Mycosis cells may exhibit cerebriform nuclei. Neoplastic cells in MF are CD3+, CD4+, CD45RO+, CD8–. Tissue can be sent for T-cell gene rearrangement polymerase chain reaction. The presence of monoclonal T-cell gene receptor rearrangements can aid in the diagnosis of MF.
Treatment includes topical steroids, mechlorethamine (nitrogen mustard) or bexarotene gel, PUVA therapy, and narrow-band UVB light for limited and/or patch disease. Localized radiotherapy can be used for more resistant lesions. Topical therapies are preferred in the early stages in MF. Systemic treatments for patients who do not respond to local therapy, or in more advanced disease include methotrexate, interferon-alpha, oral bexarotene, denileukin diftitox, and combination chemotherapy. Photopheresis is reserved for erythrodermic disease.
This case and photo were submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice in Aventura, Fla. More diagnostic cases are available at edermatologynews.com. To submit a case for possible publication, send an email to dermnews@mdedge.com.
Some have postulated an infectious agent as the cause. Atopic dermatitis may confer an increased risk because of the chronic stimulation of T cells. Males are more commonly affected than females by a 2:1 ratio. A worse prognosis is associated with advanced age. Children and adolescents may be affected as well.
With mycosis fungoides, there are three main types of skin lesions: patch, plaque, and tumor. Patients will progress from patch to plaque to tumor stage in classic MF. Often, lesions begin as scaly, erythematous patches that resemble eczema. Because of the nonspecific nature of early lesions, the median duration from the onset of skin lesions to the diagnosis of MF is 4-6 years. Patch stage lesions may be pruritic or asymptomatic. Commonly, they present in non–sun-exposed areas, such as the buttocks. Annular, infiltrated, red-brown or violaceous plaques can develop, which represent malignant T-cell infiltration. Many patients never progress past the plaque stage. Tumor stage MF is more aggressive, with nodules that may undergo necrosis and ulceration.
The leukemic form of MF is Sézary syndrome. Patients present with pruritic erythroderma and lymphadenopathy. Nail dystrophy, scaling of palms and soles, and alopecia may be present. A peripheral blood smear reveals Sézary cells, which are large, hyperconvoluted lymphocytes. The count of Sézary cells is usually greater than 1000 cells/mm3.
Histology of early lesions may not be diagnostic for CTCL. Often, biopsies will be read as eczematous or psoriasiform for years before the diagnosis of MF is made. Classically, epidermotropism (single-cell exocytosis of lymphocytes into the epidermis) is present. Advanced stages may show a dense infiltrate of lymphocytes in the dermis. Groups of lymphocytes in the epidermis form Pautrier’s microabscesses. Mycosis cells may exhibit cerebriform nuclei. Neoplastic cells in MF are CD3+, CD4+, CD45RO+, CD8–. Tissue can be sent for T-cell gene rearrangement polymerase chain reaction. The presence of monoclonal T-cell gene receptor rearrangements can aid in the diagnosis of MF.
Treatment includes topical steroids, mechlorethamine (nitrogen mustard) or bexarotene gel, PUVA therapy, and narrow-band UVB light for limited and/or patch disease. Localized radiotherapy can be used for more resistant lesions. Topical therapies are preferred in the early stages in MF. Systemic treatments for patients who do not respond to local therapy, or in more advanced disease include methotrexate, interferon-alpha, oral bexarotene, denileukin diftitox, and combination chemotherapy. Photopheresis is reserved for erythrodermic disease.
This case and photo were submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice in Aventura, Fla. More diagnostic cases are available at edermatologynews.com. To submit a case for possible publication, send an email to dermnews@mdedge.com.
Autotransplant is linked to higher AML, MDS risk
Patients undergoing autologous hematopoietic cell transplantation for lymphoma or plasma cell myeloma have 10-100 times the risk of acute myeloid leukemia (AML) or myelodysplastic syndrome (MDS) seen in the general population, according to a retrospective cohort study.
The elevated risk also exceeds that of similar patients largely untreated with autotransplant.
Exposure to DNA-damaging drugs and ionizing radiation – both used in autotransplant – is known to increase risk of these treatment-related myeloid neoplasms, according to Tomas Radivoyevitch, PhD, of the Cleveland Clinic Foundation and his colleagues. Concern about this complication has been growing as long-term survivorship after transplant improves.
The investigators analyzed data reported to the Center for International Blood and Marrow Transplant Research. Analyses were based on 9,028 patients undergoing autotransplant during 1995-2010 for Hodgkin lymphoma (916 patients), non-Hodgkin lymphoma (3,546 patients), or plasma cell myeloma (4,566 patients). Their median duration of follow-up was 90 months, 110 months, and 97 months, respectively.
Overall, 3.7% of the cohort developed AML or MDS after their transplant. More aggressive transplantation protocols increased the likelihood of this outcome: Risk was higher for patients with Hodgkin lymphoma who received conditioning with total body radiation versus chemotherapy alone (hazard ratio, 4.0); patients with non-Hodgkin lymphoma who received conditioning with total body radiation (HR, 1.7) or with busulfan and melphalan or cyclophosphamide (HR, 1.8) versus the BEAM regimen; patients with non-Hodgkin lymphoma or plasma cell myeloma who received three or more lines of chemotherapy versus just one line (HR, 1.9 and 1.8, respectively); and patients with non-Hodgkin lymphoma who underwent transplantation in 2005-2010 versus 1995-1999 (HR, 2.1).
Patients reported to Surveillance, Epidemiology and End Results (SEER) database with the same lymphoma and plasma cell myeloma diagnoses, few of whom underwent autotransplant, had risks of AML and MDS that were 5-10 times higher than the background level in the population. But the study autotransplant cohort had a risk of AML that was 10-50 times higher, and a relative risk of MDS that was roughly 100 times higher than the background level.
“These increases may be related to exposure to high doses of DNA-damaging drugs given for the autotransplant, but this hypothesis can only be tested in a prospective study,” Dr. Radivoyevitch and his coinvestigators wrote.
The reason for the greater elevation of MDS risk, compared with AML risk, is unknown. “One possible explanation is that many cases of MDS evolve to AML, and that earlier diagnosis from increased posttransplant surveillance resulted in a deficiency of AML,” they wrote. “A second is based on steeper MDS versus AML incidences versus age … and the possibility that transplantation recipient marrow ages (i.e., marrow biological ages) are perhaps decades older than calendar ages.”
The Center for International Blood and Marrow Transplant Research is supported by several U.S. government agencies and numerous pharmaceutical companies. The authors reported that they had no relevant conflicts of interest.
SOURCE: Radivoyevitch T et al. Leuk Res. 2018 Jul 19. pii: S0145-2126(18)30160-7.
Patients undergoing autologous hematopoietic cell transplantation for lymphoma or plasma cell myeloma have 10-100 times the risk of acute myeloid leukemia (AML) or myelodysplastic syndrome (MDS) seen in the general population, according to a retrospective cohort study.
The elevated risk also exceeds that of similar patients largely untreated with autotransplant.
Exposure to DNA-damaging drugs and ionizing radiation – both used in autotransplant – is known to increase risk of these treatment-related myeloid neoplasms, according to Tomas Radivoyevitch, PhD, of the Cleveland Clinic Foundation and his colleagues. Concern about this complication has been growing as long-term survivorship after transplant improves.
The investigators analyzed data reported to the Center for International Blood and Marrow Transplant Research. Analyses were based on 9,028 patients undergoing autotransplant during 1995-2010 for Hodgkin lymphoma (916 patients), non-Hodgkin lymphoma (3,546 patients), or plasma cell myeloma (4,566 patients). Their median duration of follow-up was 90 months, 110 months, and 97 months, respectively.
Overall, 3.7% of the cohort developed AML or MDS after their transplant. More aggressive transplantation protocols increased the likelihood of this outcome: Risk was higher for patients with Hodgkin lymphoma who received conditioning with total body radiation versus chemotherapy alone (hazard ratio, 4.0); patients with non-Hodgkin lymphoma who received conditioning with total body radiation (HR, 1.7) or with busulfan and melphalan or cyclophosphamide (HR, 1.8) versus the BEAM regimen; patients with non-Hodgkin lymphoma or plasma cell myeloma who received three or more lines of chemotherapy versus just one line (HR, 1.9 and 1.8, respectively); and patients with non-Hodgkin lymphoma who underwent transplantation in 2005-2010 versus 1995-1999 (HR, 2.1).
Patients reported to Surveillance, Epidemiology and End Results (SEER) database with the same lymphoma and plasma cell myeloma diagnoses, few of whom underwent autotransplant, had risks of AML and MDS that were 5-10 times higher than the background level in the population. But the study autotransplant cohort had a risk of AML that was 10-50 times higher, and a relative risk of MDS that was roughly 100 times higher than the background level.
“These increases may be related to exposure to high doses of DNA-damaging drugs given for the autotransplant, but this hypothesis can only be tested in a prospective study,” Dr. Radivoyevitch and his coinvestigators wrote.
The reason for the greater elevation of MDS risk, compared with AML risk, is unknown. “One possible explanation is that many cases of MDS evolve to AML, and that earlier diagnosis from increased posttransplant surveillance resulted in a deficiency of AML,” they wrote. “A second is based on steeper MDS versus AML incidences versus age … and the possibility that transplantation recipient marrow ages (i.e., marrow biological ages) are perhaps decades older than calendar ages.”
The Center for International Blood and Marrow Transplant Research is supported by several U.S. government agencies and numerous pharmaceutical companies. The authors reported that they had no relevant conflicts of interest.
SOURCE: Radivoyevitch T et al. Leuk Res. 2018 Jul 19. pii: S0145-2126(18)30160-7.
Patients undergoing autologous hematopoietic cell transplantation for lymphoma or plasma cell myeloma have 10-100 times the risk of acute myeloid leukemia (AML) or myelodysplastic syndrome (MDS) seen in the general population, according to a retrospective cohort study.
The elevated risk also exceeds that of similar patients largely untreated with autotransplant.
Exposure to DNA-damaging drugs and ionizing radiation – both used in autotransplant – is known to increase risk of these treatment-related myeloid neoplasms, according to Tomas Radivoyevitch, PhD, of the Cleveland Clinic Foundation and his colleagues. Concern about this complication has been growing as long-term survivorship after transplant improves.
The investigators analyzed data reported to the Center for International Blood and Marrow Transplant Research. Analyses were based on 9,028 patients undergoing autotransplant during 1995-2010 for Hodgkin lymphoma (916 patients), non-Hodgkin lymphoma (3,546 patients), or plasma cell myeloma (4,566 patients). Their median duration of follow-up was 90 months, 110 months, and 97 months, respectively.
Overall, 3.7% of the cohort developed AML or MDS after their transplant. More aggressive transplantation protocols increased the likelihood of this outcome: Risk was higher for patients with Hodgkin lymphoma who received conditioning with total body radiation versus chemotherapy alone (hazard ratio, 4.0); patients with non-Hodgkin lymphoma who received conditioning with total body radiation (HR, 1.7) or with busulfan and melphalan or cyclophosphamide (HR, 1.8) versus the BEAM regimen; patients with non-Hodgkin lymphoma or plasma cell myeloma who received three or more lines of chemotherapy versus just one line (HR, 1.9 and 1.8, respectively); and patients with non-Hodgkin lymphoma who underwent transplantation in 2005-2010 versus 1995-1999 (HR, 2.1).
Patients reported to Surveillance, Epidemiology and End Results (SEER) database with the same lymphoma and plasma cell myeloma diagnoses, few of whom underwent autotransplant, had risks of AML and MDS that were 5-10 times higher than the background level in the population. But the study autotransplant cohort had a risk of AML that was 10-50 times higher, and a relative risk of MDS that was roughly 100 times higher than the background level.
“These increases may be related to exposure to high doses of DNA-damaging drugs given for the autotransplant, but this hypothesis can only be tested in a prospective study,” Dr. Radivoyevitch and his coinvestigators wrote.
The reason for the greater elevation of MDS risk, compared with AML risk, is unknown. “One possible explanation is that many cases of MDS evolve to AML, and that earlier diagnosis from increased posttransplant surveillance resulted in a deficiency of AML,” they wrote. “A second is based on steeper MDS versus AML incidences versus age … and the possibility that transplantation recipient marrow ages (i.e., marrow biological ages) are perhaps decades older than calendar ages.”
The Center for International Blood and Marrow Transplant Research is supported by several U.S. government agencies and numerous pharmaceutical companies. The authors reported that they had no relevant conflicts of interest.
SOURCE: Radivoyevitch T et al. Leuk Res. 2018 Jul 19. pii: S0145-2126(18)30160-7.
FROM LEUKEMIA RESEARCH
Key clinical point:
Major finding: Patients undergoing autologous hematopoietic cell transplantation have risks for AML and MDS that are 10-100 times higher than those of the general population.
Study details: A retrospective cohort study of 9,028 patients undergoing hematopoietic cell autotransplant during 1995-2010 for Hodgkin lymphoma, non-Hodgkin lymphoma, or plasma cell myeloma.
Disclosures: The Center for International Blood and Marrow Transplant Research is supported by U.S. government agencies and numerous pharmaceutical companies. The authors reported that they have no relevant conflicts of interest.
Source: Radivoyevitch T et al. Leuk Res. 2018 Jul 19. pii: S0145-2126(18)30160-7.
FDA approves biologic for mycosis fungoides, Sézary syndrome
The Food and Drug Administration has approved mogamulizumab-kpkc (Poteligeo) for the treatment of adults with relapsed or refractory mycosis fungoides (MF) or Sézary syndrome (SS) who have received at least one prior systemic therapy.

Mogamulizumab is a humanized monoclonal antibody directed against CC chemokine receptor 4 (CCR4). It is the first biologic agent targeting CCR4 to be approved for patients in the United States.
Mogamulizumab is expected to be commercially available in the fourth quarter of 2018.
The FDA previously granted mogamulizumab breakthrough therapy and orphan drug designations, as well as priority review.
The approval is supported by the phase 3 MAVORIC trial. Results from this trial were presented at the 10th Annual T-cell Lymphoma Forum in February 2018.
MAVORIC enrolled 372 adults with histologically confirmed MF or SS who had failed at least one systemic therapy. They were randomized to receive mogamulizumab at 1.0 mg/kg (weekly for the first 4-week cycle and then every 2 weeks) or vorinostat at 400 mg daily. Patients were treated until disease progression or unacceptable toxicity. Those receiving vorinostat could cross over to mogamulizumab if they progressed or experienced intolerable toxicity. Baseline characteristics were similar between the treatment arms. The study’s primary endpoint was progression-free survival. The median progression-free survival was 7.7 months with mogamulizumab and 3.1 months with vorinostat (hazard ratio, 0.53; P less than .0001).
The global overall response rate was 28% (52/189) in the mogamulizumab arm and 5% (9/186) in the vorinostat arm (P less than .0001). For patients with MF, the ORR was 21% with mogamulizumab and 7% with vorinostat; for patients with SS, the ORR was 37% and 2%, respectively. After crossover, the ORR in the mogamulizumab arm was 30% (41/136).
The median duration of response (DOR) was 14 months in the mogamulizumab arm and 9 months in the vorinostat arm. For MF patients, the median DOR was 13 months with mogamulizumab and 9 months with vorinostat; for SS patients, the median DOR was 17 months and 7 months, respectively.
The most common treatment-emergent adverse events (AEs), which occurred in at least 20% of patients in either arm (mogamulizumab and vorinostat, respectively), included the following:
- Infusion-related reactions (33.2% vs. 0.5%).
- Drug eruptions (23.9% vs. 0.5%).
- Diarrhea (23.4% vs. 61.8%).
- Nausea (15.2% vs. 42.5%).
- Thrombocytopenia (11.4% vs. 30.6%).
- Dysgeusia (3.3% vs. 28.0%).
- Increased blood creatinine (3.3% vs. 28.0%).
- Decreased appetite (7.6% vs. 24.7%).
There were no grade 4 AEs in the mogamulizumab arm. Grade 3 AEs in mogamulizumab recipients included drug eruptions (n = 8), infusion-related reactions (n = 3), fatigue (n = 3), decreased appetite (n = 2), nausea (n = 1), pyrexia (n = 1), and diarrhea (n = 1).
The drug is marketed by Kyowa Kirin.
The Food and Drug Administration has approved mogamulizumab-kpkc (Poteligeo) for the treatment of adults with relapsed or refractory mycosis fungoides (MF) or Sézary syndrome (SS) who have received at least one prior systemic therapy.
Mogamulizumab is a humanized monoclonal antibody directed against CC chemokine receptor 4 (CCR4). It is the first biologic agent targeting CCR4 to be approved for patients in the United States.
Mogamulizumab is expected to be commercially available in the fourth quarter of 2018.
The FDA previously granted mogamulizumab breakthrough therapy and orphan drug designations, as well as priority review.
The approval is supported by the phase 3 MAVORIC trial. Results from this trial were presented at the 10th Annual T-cell Lymphoma Forum in February 2018.
MAVORIC enrolled 372 adults with histologically confirmed MF or SS who had failed at least one systemic therapy. They were randomized to receive mogamulizumab at 1.0 mg/kg (weekly for the first 4-week cycle and then every 2 weeks) or vorinostat at 400 mg daily. Patients were treated until disease progression or unacceptable toxicity. Those receiving vorinostat could cross over to mogamulizumab if they progressed or experienced intolerable toxicity. Baseline characteristics were similar between the treatment arms. The study’s primary endpoint was progression-free survival. The median progression-free survival was 7.7 months with mogamulizumab and 3.1 months with vorinostat (hazard ratio, 0.53; P less than .0001).
The global overall response rate was 28% (52/189) in the mogamulizumab arm and 5% (9/186) in the vorinostat arm (P less than .0001). For patients with MF, the ORR was 21% with mogamulizumab and 7% with vorinostat; for patients with SS, the ORR was 37% and 2%, respectively. After crossover, the ORR in the mogamulizumab arm was 30% (41/136).
The median duration of response (DOR) was 14 months in the mogamulizumab arm and 9 months in the vorinostat arm. For MF patients, the median DOR was 13 months with mogamulizumab and 9 months with vorinostat; for SS patients, the median DOR was 17 months and 7 months, respectively.
The most common treatment-emergent adverse events (AEs), which occurred in at least 20% of patients in either arm (mogamulizumab and vorinostat, respectively), included the following:
- Infusion-related reactions (33.2% vs. 0.5%).
- Drug eruptions (23.9% vs. 0.5%).
- Diarrhea (23.4% vs. 61.8%).
- Nausea (15.2% vs. 42.5%).
- Thrombocytopenia (11.4% vs. 30.6%).
- Dysgeusia (3.3% vs. 28.0%).
- Increased blood creatinine (3.3% vs. 28.0%).
- Decreased appetite (7.6% vs. 24.7%).
There were no grade 4 AEs in the mogamulizumab arm. Grade 3 AEs in mogamulizumab recipients included drug eruptions (n = 8), infusion-related reactions (n = 3), fatigue (n = 3), decreased appetite (n = 2), nausea (n = 1), pyrexia (n = 1), and diarrhea (n = 1).
The drug is marketed by Kyowa Kirin.
The Food and Drug Administration has approved mogamulizumab-kpkc (Poteligeo) for the treatment of adults with relapsed or refractory mycosis fungoides (MF) or Sézary syndrome (SS) who have received at least one prior systemic therapy.
Mogamulizumab is a humanized monoclonal antibody directed against CC chemokine receptor 4 (CCR4). It is the first biologic agent targeting CCR4 to be approved for patients in the United States.
Mogamulizumab is expected to be commercially available in the fourth quarter of 2018.
The FDA previously granted mogamulizumab breakthrough therapy and orphan drug designations, as well as priority review.
The approval is supported by the phase 3 MAVORIC trial. Results from this trial were presented at the 10th Annual T-cell Lymphoma Forum in February 2018.
MAVORIC enrolled 372 adults with histologically confirmed MF or SS who had failed at least one systemic therapy. They were randomized to receive mogamulizumab at 1.0 mg/kg (weekly for the first 4-week cycle and then every 2 weeks) or vorinostat at 400 mg daily. Patients were treated until disease progression or unacceptable toxicity. Those receiving vorinostat could cross over to mogamulizumab if they progressed or experienced intolerable toxicity. Baseline characteristics were similar between the treatment arms. The study’s primary endpoint was progression-free survival. The median progression-free survival was 7.7 months with mogamulizumab and 3.1 months with vorinostat (hazard ratio, 0.53; P less than .0001).
The global overall response rate was 28% (52/189) in the mogamulizumab arm and 5% (9/186) in the vorinostat arm (P less than .0001). For patients with MF, the ORR was 21% with mogamulizumab and 7% with vorinostat; for patients with SS, the ORR was 37% and 2%, respectively. After crossover, the ORR in the mogamulizumab arm was 30% (41/136).
The median duration of response (DOR) was 14 months in the mogamulizumab arm and 9 months in the vorinostat arm. For MF patients, the median DOR was 13 months with mogamulizumab and 9 months with vorinostat; for SS patients, the median DOR was 17 months and 7 months, respectively.
The most common treatment-emergent adverse events (AEs), which occurred in at least 20% of patients in either arm (mogamulizumab and vorinostat, respectively), included the following:
- Infusion-related reactions (33.2% vs. 0.5%).
- Drug eruptions (23.9% vs. 0.5%).
- Diarrhea (23.4% vs. 61.8%).
- Nausea (15.2% vs. 42.5%).
- Thrombocytopenia (11.4% vs. 30.6%).
- Dysgeusia (3.3% vs. 28.0%).
- Increased blood creatinine (3.3% vs. 28.0%).
- Decreased appetite (7.6% vs. 24.7%).
There were no grade 4 AEs in the mogamulizumab arm. Grade 3 AEs in mogamulizumab recipients included drug eruptions (n = 8), infusion-related reactions (n = 3), fatigue (n = 3), decreased appetite (n = 2), nausea (n = 1), pyrexia (n = 1), and diarrhea (n = 1).
The drug is marketed by Kyowa Kirin.
Increased B-cell lymphoma risk with JAK1/2 inhibitors
Patients with myeloproliferative neoplasms treated with Janus-kinase (JAK) 1/2 inhibitors may be at significantly increased risk of aggressive B cell non-Hodgkin lymphomas, according to a study published in Blood.
A retrospective cohort study of 626 Viennese patients with myeloproliferative neoplasms – 69 of whom were treated with JAK1/2 inhibitors – found that 4 of the 69 patients (5.8%) developed aggressive B-cell lymphoma, compared with just 2 patients (0.36%) in the rest of the group. This represented a significant, 16-fold higher risk of aggressive B cell lymphoma associated with JAK1/2 inhibitor therapy (P = .0017).
The lymphoma was diagnosed within 13-35 months of starting JAK1/2 inhibitors. In three patients, the disease was in the bone marrow and peripheral blood, one patient had it in mammary tissue, and another had it in mucosal tissue. All four lymphomas showed positive MYC and p53 staining.
All four patients had been treated with ruxolitinib, one was also treated with fedratinib, and three of the four had been pretreated with alkylating agents.
Meanwhile, a second retrospective cohort study in Paris of 929 patients with myeloproliferative neoplasms, reported in the same paper, found that 3.51% of those treated with ruxolitinib developed lymphoma, compared with 0.23% of conventionally-treated patients.
Using archived bone marrow samples from 54 of the 69 patients treated with JAK1/2 inhibitors, researchers discovered that 15.9% of them – including three of the B-cell lymphoma patients (the fourth was not tested) – had a preexisting B cell clone. This was present as early as 47-70 months before the lymphoma diagnosis.
“In patients, the clonal B-cell population was present as long as 6 years before overt lymphoma and preceded JAK1/2 inhibition which offers the opportunity to determine patients at risk,” wrote Edit Porpaczy, MD, of the Comprehensive Cancer Center at the Medical University of Vienna, and her coauthors. “Targeted inhibition of JAK-STAT signaling appears to be required to trigger the appearance of the B-cell clone as other treatments eliminating the myeloid cell load in men do not exert a comparable effect.”
In the Viennese cohort, three of the lymphomas were aggressive CD19+ B-cell type, and the fourth was a nonspecified high-grade B-cell lymphoma.
Researchers also looked at the effects of JAK1/2 inhibition in STAT1-/- mice, and found that two-thirds developed a spontaneous myeloid hyperplasia with the concomitant presence of aberrant B-cells.
“Upon STAT1-deficiency myeloid hyperplasia is paralleled by the occurrence of a malignant B-cell clone, which evolves into disease upon bone-marrow transplantation and gives rise to a leukemic lymphoma phenotype,” the authors wrote.
The study was supported by the Austrian Science Fund, the Anniversary Fund of the Austrian National Bank and the WWTF Precision Medicine Program. Several authors reported support, funding or advisory board positions with the pharmaceutical industry.
SOURCE: Porpaczy E et al. Blood. 2018 Jun 14. doi: 10.1182/blood-2017-10-810739.
Patients with myeloproliferative neoplasms treated with Janus-kinase (JAK) 1/2 inhibitors may be at significantly increased risk of aggressive B cell non-Hodgkin lymphomas, according to a study published in Blood.
A retrospective cohort study of 626 Viennese patients with myeloproliferative neoplasms – 69 of whom were treated with JAK1/2 inhibitors – found that 4 of the 69 patients (5.8%) developed aggressive B-cell lymphoma, compared with just 2 patients (0.36%) in the rest of the group. This represented a significant, 16-fold higher risk of aggressive B cell lymphoma associated with JAK1/2 inhibitor therapy (P = .0017).
The lymphoma was diagnosed within 13-35 months of starting JAK1/2 inhibitors. In three patients, the disease was in the bone marrow and peripheral blood, one patient had it in mammary tissue, and another had it in mucosal tissue. All four lymphomas showed positive MYC and p53 staining.
All four patients had been treated with ruxolitinib, one was also treated with fedratinib, and three of the four had been pretreated with alkylating agents.
Meanwhile, a second retrospective cohort study in Paris of 929 patients with myeloproliferative neoplasms, reported in the same paper, found that 3.51% of those treated with ruxolitinib developed lymphoma, compared with 0.23% of conventionally-treated patients.
Using archived bone marrow samples from 54 of the 69 patients treated with JAK1/2 inhibitors, researchers discovered that 15.9% of them – including three of the B-cell lymphoma patients (the fourth was not tested) – had a preexisting B cell clone. This was present as early as 47-70 months before the lymphoma diagnosis.
“In patients, the clonal B-cell population was present as long as 6 years before overt lymphoma and preceded JAK1/2 inhibition which offers the opportunity to determine patients at risk,” wrote Edit Porpaczy, MD, of the Comprehensive Cancer Center at the Medical University of Vienna, and her coauthors. “Targeted inhibition of JAK-STAT signaling appears to be required to trigger the appearance of the B-cell clone as other treatments eliminating the myeloid cell load in men do not exert a comparable effect.”
In the Viennese cohort, three of the lymphomas were aggressive CD19+ B-cell type, and the fourth was a nonspecified high-grade B-cell lymphoma.
Researchers also looked at the effects of JAK1/2 inhibition in STAT1-/- mice, and found that two-thirds developed a spontaneous myeloid hyperplasia with the concomitant presence of aberrant B-cells.
“Upon STAT1-deficiency myeloid hyperplasia is paralleled by the occurrence of a malignant B-cell clone, which evolves into disease upon bone-marrow transplantation and gives rise to a leukemic lymphoma phenotype,” the authors wrote.
The study was supported by the Austrian Science Fund, the Anniversary Fund of the Austrian National Bank and the WWTF Precision Medicine Program. Several authors reported support, funding or advisory board positions with the pharmaceutical industry.
SOURCE: Porpaczy E et al. Blood. 2018 Jun 14. doi: 10.1182/blood-2017-10-810739.
Patients with myeloproliferative neoplasms treated with Janus-kinase (JAK) 1/2 inhibitors may be at significantly increased risk of aggressive B cell non-Hodgkin lymphomas, according to a study published in Blood.
A retrospective cohort study of 626 Viennese patients with myeloproliferative neoplasms – 69 of whom were treated with JAK1/2 inhibitors – found that 4 of the 69 patients (5.8%) developed aggressive B-cell lymphoma, compared with just 2 patients (0.36%) in the rest of the group. This represented a significant, 16-fold higher risk of aggressive B cell lymphoma associated with JAK1/2 inhibitor therapy (P = .0017).
The lymphoma was diagnosed within 13-35 months of starting JAK1/2 inhibitors. In three patients, the disease was in the bone marrow and peripheral blood, one patient had it in mammary tissue, and another had it in mucosal tissue. All four lymphomas showed positive MYC and p53 staining.
All four patients had been treated with ruxolitinib, one was also treated with fedratinib, and three of the four had been pretreated with alkylating agents.
Meanwhile, a second retrospective cohort study in Paris of 929 patients with myeloproliferative neoplasms, reported in the same paper, found that 3.51% of those treated with ruxolitinib developed lymphoma, compared with 0.23% of conventionally-treated patients.
Using archived bone marrow samples from 54 of the 69 patients treated with JAK1/2 inhibitors, researchers discovered that 15.9% of them – including three of the B-cell lymphoma patients (the fourth was not tested) – had a preexisting B cell clone. This was present as early as 47-70 months before the lymphoma diagnosis.
“In patients, the clonal B-cell population was present as long as 6 years before overt lymphoma and preceded JAK1/2 inhibition which offers the opportunity to determine patients at risk,” wrote Edit Porpaczy, MD, of the Comprehensive Cancer Center at the Medical University of Vienna, and her coauthors. “Targeted inhibition of JAK-STAT signaling appears to be required to trigger the appearance of the B-cell clone as other treatments eliminating the myeloid cell load in men do not exert a comparable effect.”
In the Viennese cohort, three of the lymphomas were aggressive CD19+ B-cell type, and the fourth was a nonspecified high-grade B-cell lymphoma.
Researchers also looked at the effects of JAK1/2 inhibition in STAT1-/- mice, and found that two-thirds developed a spontaneous myeloid hyperplasia with the concomitant presence of aberrant B-cells.
“Upon STAT1-deficiency myeloid hyperplasia is paralleled by the occurrence of a malignant B-cell clone, which evolves into disease upon bone-marrow transplantation and gives rise to a leukemic lymphoma phenotype,” the authors wrote.
The study was supported by the Austrian Science Fund, the Anniversary Fund of the Austrian National Bank and the WWTF Precision Medicine Program. Several authors reported support, funding or advisory board positions with the pharmaceutical industry.
SOURCE: Porpaczy E et al. Blood. 2018 Jun 14. doi: 10.1182/blood-2017-10-810739.
FROM BLOOD
Key clinical point:
Major finding: Patients with myeloproliferative neoplasms treated with JAK1/2 inhibitors have a 16-fold higher incidence of lymphoma.
Study details: A retrospective cohort study of 626 patients with myeloproliferative neoplasms.
Disclosures: The study was supported by the Austrian Science Fund, the Anniversary Fund of the Austrian National Bank, and the WWTF Precision Medicine Program. Several authors reported support, funding, or advisory board positions with the pharmaceutical industry.
Source: Porpaczy E et al. Blood. 2018 Jun 14. doi: 10.1182/blood-2017-10-810739.
New chronic lymphocytic leukemia guidelines from the UK
Fludarabine, cyclophosphamide, and rituximab are recommended as initial therapy for patients with chronic lymphocytic leukemia who do not have TP53 disruption, according to new guidelines from the British Society for Haematology.
The guidelines update the 2012 recommendations on chronic lymphocytic leukemia (CLL) to include “significant” developments in treatment. They were published in the British Journal of Haematology.
Anna H. Schuh, MD, of the department of oncology at the University of Oxford (England), and her coauthors noted that, while these guidelines apply to treatments available outside clinical trials, wherever possible patients with CLL should be treated within the clinical trial setting.
While recommending fludarabine, cyclophosphamide, and rituximab as first-line therapy, the guideline authors acknowledged that the combination of bendamustine and rituximab is an acceptable alternative for patients who could not take the triple therapy because of comorbidities such as advanced age, renal impairment, or issues with marrow capacity.
Similarly, less-fit patients could also be considered for chlorambucil-obinutuzumab or chlorambucil-ofatumumab combinations.
All patients diagnosed with CLL should be tested for TP53 deletions and mutations before each line of therapy, the guideline committee recommended. TP53 disruption makes chemoimmunotherapy ineffective because of either a deletion of chromosome 17p or a mutation in the TP53 gene. However, there is compelling evidence for the efficacy of ibrutinib in these patients, or idelalisib and rituximab for those with cardiac disease or receiving vitamin K antagonists.
With respect to maintenance therapy, the guidelines noted that this was not routinely recommended in CLL as “it is unclear to what extent the progression-free survival benefit is offset by long-term toxicity.”
Patients who are refractory to chemoimmunotherapy, who have relapsed, or who cannot be retreated with chemoimmunotherapy should be treated with idelalisib with rituximab or ibrutinib monotherapy, the guidelines suggested.
“Deciding whether ibrutinib or idelalisib with rituximab is most appropriate for an individual patient depends on a range of factors, including toxicity profile and convenience of delivery,” the authors wrote. However, they noted that the value of adding bendamustine to either option was unclear as research had not shown significant, associated gains in median progression-free survival.
Allogeneic stem cell transplantation should be considered as a treatment option for patients who have either failed chemotherapy, have a TP53 disruption and have not responded to B-cell receptor signaling pathway inhibitors such as ibrutinib, or have a Richter transformation.
The guidelines also addressed the issue of autoimmune cytopenias, which occur in 5%-10% of patients with CLL and can actually precede the diagnosis of CLL in about 9% of cases.
In patients where autoimmune cytopenia is the dominant clinical feature, they should be treated with corticosteroids, intravenous immunoglobulin, or rituximab. However, for patients where the cytopenia is triggered by CLL therapy, the guidelines recommended halting treatment and beginning immunosuppression.
The guideline development was supported by the British Society for Haematology. The UK CLL Forum is a registered charity that receives funding from a number of pharmaceutical companies.
SOURCE: Schuh AH et al. Br J Haematol. 2018 Jul 15. doi: 10.1111/bjh.15460.
Fludarabine, cyclophosphamide, and rituximab are recommended as initial therapy for patients with chronic lymphocytic leukemia who do not have TP53 disruption, according to new guidelines from the British Society for Haematology.
The guidelines update the 2012 recommendations on chronic lymphocytic leukemia (CLL) to include “significant” developments in treatment. They were published in the British Journal of Haematology.
Anna H. Schuh, MD, of the department of oncology at the University of Oxford (England), and her coauthors noted that, while these guidelines apply to treatments available outside clinical trials, wherever possible patients with CLL should be treated within the clinical trial setting.
While recommending fludarabine, cyclophosphamide, and rituximab as first-line therapy, the guideline authors acknowledged that the combination of bendamustine and rituximab is an acceptable alternative for patients who could not take the triple therapy because of comorbidities such as advanced age, renal impairment, or issues with marrow capacity.
Similarly, less-fit patients could also be considered for chlorambucil-obinutuzumab or chlorambucil-ofatumumab combinations.
All patients diagnosed with CLL should be tested for TP53 deletions and mutations before each line of therapy, the guideline committee recommended. TP53 disruption makes chemoimmunotherapy ineffective because of either a deletion of chromosome 17p or a mutation in the TP53 gene. However, there is compelling evidence for the efficacy of ibrutinib in these patients, or idelalisib and rituximab for those with cardiac disease or receiving vitamin K antagonists.
With respect to maintenance therapy, the guidelines noted that this was not routinely recommended in CLL as “it is unclear to what extent the progression-free survival benefit is offset by long-term toxicity.”
Patients who are refractory to chemoimmunotherapy, who have relapsed, or who cannot be retreated with chemoimmunotherapy should be treated with idelalisib with rituximab or ibrutinib monotherapy, the guidelines suggested.
“Deciding whether ibrutinib or idelalisib with rituximab is most appropriate for an individual patient depends on a range of factors, including toxicity profile and convenience of delivery,” the authors wrote. However, they noted that the value of adding bendamustine to either option was unclear as research had not shown significant, associated gains in median progression-free survival.
Allogeneic stem cell transplantation should be considered as a treatment option for patients who have either failed chemotherapy, have a TP53 disruption and have not responded to B-cell receptor signaling pathway inhibitors such as ibrutinib, or have a Richter transformation.
The guidelines also addressed the issue of autoimmune cytopenias, which occur in 5%-10% of patients with CLL and can actually precede the diagnosis of CLL in about 9% of cases.
In patients where autoimmune cytopenia is the dominant clinical feature, they should be treated with corticosteroids, intravenous immunoglobulin, or rituximab. However, for patients where the cytopenia is triggered by CLL therapy, the guidelines recommended halting treatment and beginning immunosuppression.
The guideline development was supported by the British Society for Haematology. The UK CLL Forum is a registered charity that receives funding from a number of pharmaceutical companies.
SOURCE: Schuh AH et al. Br J Haematol. 2018 Jul 15. doi: 10.1111/bjh.15460.
Fludarabine, cyclophosphamide, and rituximab are recommended as initial therapy for patients with chronic lymphocytic leukemia who do not have TP53 disruption, according to new guidelines from the British Society for Haematology.
The guidelines update the 2012 recommendations on chronic lymphocytic leukemia (CLL) to include “significant” developments in treatment. They were published in the British Journal of Haematology.
Anna H. Schuh, MD, of the department of oncology at the University of Oxford (England), and her coauthors noted that, while these guidelines apply to treatments available outside clinical trials, wherever possible patients with CLL should be treated within the clinical trial setting.
While recommending fludarabine, cyclophosphamide, and rituximab as first-line therapy, the guideline authors acknowledged that the combination of bendamustine and rituximab is an acceptable alternative for patients who could not take the triple therapy because of comorbidities such as advanced age, renal impairment, or issues with marrow capacity.
Similarly, less-fit patients could also be considered for chlorambucil-obinutuzumab or chlorambucil-ofatumumab combinations.
All patients diagnosed with CLL should be tested for TP53 deletions and mutations before each line of therapy, the guideline committee recommended. TP53 disruption makes chemoimmunotherapy ineffective because of either a deletion of chromosome 17p or a mutation in the TP53 gene. However, there is compelling evidence for the efficacy of ibrutinib in these patients, or idelalisib and rituximab for those with cardiac disease or receiving vitamin K antagonists.
With respect to maintenance therapy, the guidelines noted that this was not routinely recommended in CLL as “it is unclear to what extent the progression-free survival benefit is offset by long-term toxicity.”
Patients who are refractory to chemoimmunotherapy, who have relapsed, or who cannot be retreated with chemoimmunotherapy should be treated with idelalisib with rituximab or ibrutinib monotherapy, the guidelines suggested.
“Deciding whether ibrutinib or idelalisib with rituximab is most appropriate for an individual patient depends on a range of factors, including toxicity profile and convenience of delivery,” the authors wrote. However, they noted that the value of adding bendamustine to either option was unclear as research had not shown significant, associated gains in median progression-free survival.
Allogeneic stem cell transplantation should be considered as a treatment option for patients who have either failed chemotherapy, have a TP53 disruption and have not responded to B-cell receptor signaling pathway inhibitors such as ibrutinib, or have a Richter transformation.
The guidelines also addressed the issue of autoimmune cytopenias, which occur in 5%-10% of patients with CLL and can actually precede the diagnosis of CLL in about 9% of cases.
In patients where autoimmune cytopenia is the dominant clinical feature, they should be treated with corticosteroids, intravenous immunoglobulin, or rituximab. However, for patients where the cytopenia is triggered by CLL therapy, the guidelines recommended halting treatment and beginning immunosuppression.
The guideline development was supported by the British Society for Haematology. The UK CLL Forum is a registered charity that receives funding from a number of pharmaceutical companies.
SOURCE: Schuh AH et al. Br J Haematol. 2018 Jul 15. doi: 10.1111/bjh.15460.
FROM THE BRITISH JOURNAL OF HAEMATOLOGY
Key clinical point:
Major finding: All patients diagnosed with CLL should be tested for TP53 disruption.
Study details: A guideline developed by the British Society for Haematology offering recommendations for CLL treatment outside clinical trials.
Disclosures: The guideline development was supported by the British Society for Haematology. The UK CLL Forum is a registered charity that receives funding from a number of pharmaceutical companies.
Source: Schuh AH et al. Br J Haematol. 2018 Jul 15. doi: 10.1111/bjh.15460.
Insurance is a matter of life or death for lymphoma patients
Having health insurance can mean the difference between life and death for patients with follicular lymphoma, suggest results of a study showing that patients with private health insurance had nearly twofold better survival outcomes than patients without insurance or those who were covered by Medicare or Medicaid.

A review of records on more than 43,000 patients with follicular lymphoma (FL) in a national cancer registry showed that, compared with patients under age 65 with private insurance, the hazard ratios (HR) for death among patients in the same age bracket with either no insurance, Medicaid, or Medicare were, respectively, 1.96, 1.83, and 1.96 (P less than .0001 for each comparison).
“Our study finds that insurance status contributes to survival disparities in FL. Future studies on outcomes in FL should include insurance status as an important predictor,” Christopher R. Flowers, MD, of Emory University in Atlanta and his colleagues wrote in Blood.
“Further research on prognosis for FL should examine the impact of public policy, such as the passage of the [Affordable Care Act], on FL outcomes, as well as examine other factors that influence access to care, such as individual-level socioeconomic status, regular primary care visits, access to prescription medications, and care affordability,” they added.
The investigators noted that earlier research found that patients with Medicaid or no insurance were more likely than privately-insured patients to be diagnosed with cancers at advanced stages, and that some patients with aggressive non-Hodgkin lymphomas have been shown to have insurance-related disparities in treatments and outcomes.
To see whether the same could be true for patients with indolent-histology lymphomas such as FL, they extracted data from the National Cancer Database, a nationwide hospital-based cancer registry sponsored jointly by the American College of Surgeons and the American Cancer Society.
They identified a total of 43,648 patients aged 18 years or older who were diagnosed with FL from 2004 through 2014. They looked at both patients 18-64 years and those 65 years and older to account for changes in insurance with Medicare eligibility.
Overall survival among patients younger than age 65 was significantly worse for patients with public insurance (Medicaid or Medicare) or no insurance in Cox proportional hazard models controlling for available data on sociodemographic factors and prognostic indicators.
However, compared with patients aged 65 and older with private insurance, only patients with Medicare as their sole source of insurance had significantly worse overall survival (HR, 1.28; P less than .0001).
Patients who were uninsured or had Medicaid were more likely than others to have lower socioeconomic status, present with advanced-stage disease, have systemic symptoms, and have multiple comorbidities that persisted after controlling for known sociodemographic and prognostic factors.
The investigators found that, among patients under age 65, those with a comorbidity score of 1 had an HR for death of 1.71, compared with patients with no comorbidities, and that patients with a score of 2 or greater had a HR of 3.1 (P less than .0001 for each comparison).
“The findings of the study indicate that improving access to affordable, quality health care may reduce disparities in survival for those currently lacking coverage,” the investigators wrote.
The study was supported by Emory University, the National Institutes of Health, and the National Center for Advancing Translational Sciences. Dr. Flowers reported financial relationships with AbbVie, Spectrum, Celgene, and several other companies. The other authors reported having nothing to disclose.
SOURCE: Goldstein JS et al. Blood. 2018 Jul 24. doi: 10.1182/blood-2018-03-839035.
Having health insurance can mean the difference between life and death for patients with follicular lymphoma, suggest results of a study showing that patients with private health insurance had nearly twofold better survival outcomes than patients without insurance or those who were covered by Medicare or Medicaid.
A review of records on more than 43,000 patients with follicular lymphoma (FL) in a national cancer registry showed that, compared with patients under age 65 with private insurance, the hazard ratios (HR) for death among patients in the same age bracket with either no insurance, Medicaid, or Medicare were, respectively, 1.96, 1.83, and 1.96 (P less than .0001 for each comparison).
“Our study finds that insurance status contributes to survival disparities in FL. Future studies on outcomes in FL should include insurance status as an important predictor,” Christopher R. Flowers, MD, of Emory University in Atlanta and his colleagues wrote in Blood.
“Further research on prognosis for FL should examine the impact of public policy, such as the passage of the [Affordable Care Act], on FL outcomes, as well as examine other factors that influence access to care, such as individual-level socioeconomic status, regular primary care visits, access to prescription medications, and care affordability,” they added.
The investigators noted that earlier research found that patients with Medicaid or no insurance were more likely than privately-insured patients to be diagnosed with cancers at advanced stages, and that some patients with aggressive non-Hodgkin lymphomas have been shown to have insurance-related disparities in treatments and outcomes.
To see whether the same could be true for patients with indolent-histology lymphomas such as FL, they extracted data from the National Cancer Database, a nationwide hospital-based cancer registry sponsored jointly by the American College of Surgeons and the American Cancer Society.
They identified a total of 43,648 patients aged 18 years or older who were diagnosed with FL from 2004 through 2014. They looked at both patients 18-64 years and those 65 years and older to account for changes in insurance with Medicare eligibility.
Overall survival among patients younger than age 65 was significantly worse for patients with public insurance (Medicaid or Medicare) or no insurance in Cox proportional hazard models controlling for available data on sociodemographic factors and prognostic indicators.
However, compared with patients aged 65 and older with private insurance, only patients with Medicare as their sole source of insurance had significantly worse overall survival (HR, 1.28; P less than .0001).
Patients who were uninsured or had Medicaid were more likely than others to have lower socioeconomic status, present with advanced-stage disease, have systemic symptoms, and have multiple comorbidities that persisted after controlling for known sociodemographic and prognostic factors.
The investigators found that, among patients under age 65, those with a comorbidity score of 1 had an HR for death of 1.71, compared with patients with no comorbidities, and that patients with a score of 2 or greater had a HR of 3.1 (P less than .0001 for each comparison).
“The findings of the study indicate that improving access to affordable, quality health care may reduce disparities in survival for those currently lacking coverage,” the investigators wrote.
The study was supported by Emory University, the National Institutes of Health, and the National Center for Advancing Translational Sciences. Dr. Flowers reported financial relationships with AbbVie, Spectrum, Celgene, and several other companies. The other authors reported having nothing to disclose.
SOURCE: Goldstein JS et al. Blood. 2018 Jul 24. doi: 10.1182/blood-2018-03-839035.
Having health insurance can mean the difference between life and death for patients with follicular lymphoma, suggest results of a study showing that patients with private health insurance had nearly twofold better survival outcomes than patients without insurance or those who were covered by Medicare or Medicaid.
A review of records on more than 43,000 patients with follicular lymphoma (FL) in a national cancer registry showed that, compared with patients under age 65 with private insurance, the hazard ratios (HR) for death among patients in the same age bracket with either no insurance, Medicaid, or Medicare were, respectively, 1.96, 1.83, and 1.96 (P less than .0001 for each comparison).
“Our study finds that insurance status contributes to survival disparities in FL. Future studies on outcomes in FL should include insurance status as an important predictor,” Christopher R. Flowers, MD, of Emory University in Atlanta and his colleagues wrote in Blood.
“Further research on prognosis for FL should examine the impact of public policy, such as the passage of the [Affordable Care Act], on FL outcomes, as well as examine other factors that influence access to care, such as individual-level socioeconomic status, regular primary care visits, access to prescription medications, and care affordability,” they added.
The investigators noted that earlier research found that patients with Medicaid or no insurance were more likely than privately-insured patients to be diagnosed with cancers at advanced stages, and that some patients with aggressive non-Hodgkin lymphomas have been shown to have insurance-related disparities in treatments and outcomes.
To see whether the same could be true for patients with indolent-histology lymphomas such as FL, they extracted data from the National Cancer Database, a nationwide hospital-based cancer registry sponsored jointly by the American College of Surgeons and the American Cancer Society.
They identified a total of 43,648 patients aged 18 years or older who were diagnosed with FL from 2004 through 2014. They looked at both patients 18-64 years and those 65 years and older to account for changes in insurance with Medicare eligibility.
Overall survival among patients younger than age 65 was significantly worse for patients with public insurance (Medicaid or Medicare) or no insurance in Cox proportional hazard models controlling for available data on sociodemographic factors and prognostic indicators.
However, compared with patients aged 65 and older with private insurance, only patients with Medicare as their sole source of insurance had significantly worse overall survival (HR, 1.28; P less than .0001).
Patients who were uninsured or had Medicaid were more likely than others to have lower socioeconomic status, present with advanced-stage disease, have systemic symptoms, and have multiple comorbidities that persisted after controlling for known sociodemographic and prognostic factors.
The investigators found that, among patients under age 65, those with a comorbidity score of 1 had an HR for death of 1.71, compared with patients with no comorbidities, and that patients with a score of 2 or greater had a HR of 3.1 (P less than .0001 for each comparison).
“The findings of the study indicate that improving access to affordable, quality health care may reduce disparities in survival for those currently lacking coverage,” the investigators wrote.
The study was supported by Emory University, the National Institutes of Health, and the National Center for Advancing Translational Sciences. Dr. Flowers reported financial relationships with AbbVie, Spectrum, Celgene, and several other companies. The other authors reported having nothing to disclose.
SOURCE: Goldstein JS et al. Blood. 2018 Jul 24. doi: 10.1182/blood-2018-03-839035.
FROM BLOOD
Key clinical point:
Major finding: The risk for death among patients under age 65 with no insurance, Medicaid, or Medicare was nearly twice that of similar patients with private health insurance.
Study details: Review of data on 43,648 patients with follicular lymphoma in the National Cancer Database.
Disclosures: The study was supported by Emory University, the National Institutes of Health, and the National Center for Advancing Translational Sciences. Dr. Flowers reported financial relationships with AbbVie, Spectrum, Celgene, and several other companies. The other authors reported having nothing to disclose.
Source: Goldstein JS et al. Blood. 2018 Jul 24. doi: 10.1182/blood-2018-03-839035.
PET/CT accurately predicts MCL stage
Bone marrow involvement in mantle cell lymphoma could be assessed using just 18fluorodeoxyglucose (FDG)–PET/CT, according to findings from a small, retrospective study published in Clinical Lymphoma, Myeloma & Leukemia.
Rustain Morgan, MD, of the University of Colorado, Aurora, and his colleagues found that, at a certain threshold of bone marrow voxels in standard uptake value (SUV), there was 100% sensitivity and 80% specificity in determining bone marrow involvement in mantle cell lymphoma (MCL).
Currently, National Comprehensive Cancer Network guidelines call for bone marrow biopsy and whole body FDG PET/CT scan to complete an initial diagnosis of MCL.
“One of the most important factors for correct staging is the identification of bone marrow involvement, occurring in approximately 55% of patients with MCL, which classifies patients as advanced stage. However, accurate analysis of bone marrow involvement can be challenging due to sampling error,” the researchers wrote. “While bone marrow biopsy remains the gold standard, it is not a perfect standard given unilateral variability.”
In previous studies, FDG PET/CT was not considered sensitive enough to detect gastrointestinal or bone marrow involvement. However, these earlier studies used SUV maximum or mean or a visual assessment of the bone marrow activity, compared with hepatic uptake. To address this issue, the researchers developed a new method of examining SUV distribution throughout the pelvic bones by analyzing thousands of bone marrow voxels within the bilateral iliacs.
During the developmental phase, an institutional dataset of 11 patients with MCL was used to define the voxel-based analysis. These patients had undergone both unilateral iliac bone marrow biopsy and FDG PET/CT at the initial diagnosis. Then, FDG PET/CT scans from another 12 patients with MCL from a different institution were used to validate the developmental phase findings. Finally, a control group of 5 people with no known malignancy were referred for FDG PET/CT pulmonary nodule evaluation.
“The hypothesis of the study was that, if the bone marrow was involved by lymphoma, then there would be a small increase in the SUV of each voxel, reflecting involvement by the lymphoma. In order to capture such changes, we analyzed the percent of total voxels in SUV ranging from 0.75 to 1.20, in increments of 0.05, as this is where the greatest divergence was visually identified,” the researchers wrote. “The goal was to identify if a percentage of voxels at a set SUV could detect lymphomatous involvement.”
The researchers identified 10 candidate thresholds in the developmental phase; 4 of these performed better than the others in the validation phase. Using those thresholds, 10 of the 12 patients in the validation cohort could be correctly staged using FDG PET/CT.
Further analysis identified a single threshold that performed best: If greater than 38% of the voxels (averaging 1,734 voxels) demonstrated an SUV of less than 0.95, the sensitivity was 100% and the specificity was 80%.
The researchers acknowledged that the findings are limited because of the study’s small sample size and said the results should be validated in a larger trial.
There was no external funding for the study and the researchers reported having no financial disclosures.
SOURCE: Morgan R et al. Clin Lymphoma Myeloma Leuk. 2018 Jul 4. doi: 10.1016/j.clml.2018.06.024.
Bone marrow involvement in mantle cell lymphoma could be assessed using just 18fluorodeoxyglucose (FDG)–PET/CT, according to findings from a small, retrospective study published in Clinical Lymphoma, Myeloma & Leukemia.
Rustain Morgan, MD, of the University of Colorado, Aurora, and his colleagues found that, at a certain threshold of bone marrow voxels in standard uptake value (SUV), there was 100% sensitivity and 80% specificity in determining bone marrow involvement in mantle cell lymphoma (MCL).
Currently, National Comprehensive Cancer Network guidelines call for bone marrow biopsy and whole body FDG PET/CT scan to complete an initial diagnosis of MCL.
“One of the most important factors for correct staging is the identification of bone marrow involvement, occurring in approximately 55% of patients with MCL, which classifies patients as advanced stage. However, accurate analysis of bone marrow involvement can be challenging due to sampling error,” the researchers wrote. “While bone marrow biopsy remains the gold standard, it is not a perfect standard given unilateral variability.”
In previous studies, FDG PET/CT was not considered sensitive enough to detect gastrointestinal or bone marrow involvement. However, these earlier studies used SUV maximum or mean or a visual assessment of the bone marrow activity, compared with hepatic uptake. To address this issue, the researchers developed a new method of examining SUV distribution throughout the pelvic bones by analyzing thousands of bone marrow voxels within the bilateral iliacs.
During the developmental phase, an institutional dataset of 11 patients with MCL was used to define the voxel-based analysis. These patients had undergone both unilateral iliac bone marrow biopsy and FDG PET/CT at the initial diagnosis. Then, FDG PET/CT scans from another 12 patients with MCL from a different institution were used to validate the developmental phase findings. Finally, a control group of 5 people with no known malignancy were referred for FDG PET/CT pulmonary nodule evaluation.
“The hypothesis of the study was that, if the bone marrow was involved by lymphoma, then there would be a small increase in the SUV of each voxel, reflecting involvement by the lymphoma. In order to capture such changes, we analyzed the percent of total voxels in SUV ranging from 0.75 to 1.20, in increments of 0.05, as this is where the greatest divergence was visually identified,” the researchers wrote. “The goal was to identify if a percentage of voxels at a set SUV could detect lymphomatous involvement.”
The researchers identified 10 candidate thresholds in the developmental phase; 4 of these performed better than the others in the validation phase. Using those thresholds, 10 of the 12 patients in the validation cohort could be correctly staged using FDG PET/CT.
Further analysis identified a single threshold that performed best: If greater than 38% of the voxels (averaging 1,734 voxels) demonstrated an SUV of less than 0.95, the sensitivity was 100% and the specificity was 80%.
The researchers acknowledged that the findings are limited because of the study’s small sample size and said the results should be validated in a larger trial.
There was no external funding for the study and the researchers reported having no financial disclosures.
SOURCE: Morgan R et al. Clin Lymphoma Myeloma Leuk. 2018 Jul 4. doi: 10.1016/j.clml.2018.06.024.
Bone marrow involvement in mantle cell lymphoma could be assessed using just 18fluorodeoxyglucose (FDG)–PET/CT, according to findings from a small, retrospective study published in Clinical Lymphoma, Myeloma & Leukemia.
Rustain Morgan, MD, of the University of Colorado, Aurora, and his colleagues found that, at a certain threshold of bone marrow voxels in standard uptake value (SUV), there was 100% sensitivity and 80% specificity in determining bone marrow involvement in mantle cell lymphoma (MCL).
Currently, National Comprehensive Cancer Network guidelines call for bone marrow biopsy and whole body FDG PET/CT scan to complete an initial diagnosis of MCL.
“One of the most important factors for correct staging is the identification of bone marrow involvement, occurring in approximately 55% of patients with MCL, which classifies patients as advanced stage. However, accurate analysis of bone marrow involvement can be challenging due to sampling error,” the researchers wrote. “While bone marrow biopsy remains the gold standard, it is not a perfect standard given unilateral variability.”
In previous studies, FDG PET/CT was not considered sensitive enough to detect gastrointestinal or bone marrow involvement. However, these earlier studies used SUV maximum or mean or a visual assessment of the bone marrow activity, compared with hepatic uptake. To address this issue, the researchers developed a new method of examining SUV distribution throughout the pelvic bones by analyzing thousands of bone marrow voxels within the bilateral iliacs.
During the developmental phase, an institutional dataset of 11 patients with MCL was used to define the voxel-based analysis. These patients had undergone both unilateral iliac bone marrow biopsy and FDG PET/CT at the initial diagnosis. Then, FDG PET/CT scans from another 12 patients with MCL from a different institution were used to validate the developmental phase findings. Finally, a control group of 5 people with no known malignancy were referred for FDG PET/CT pulmonary nodule evaluation.
“The hypothesis of the study was that, if the bone marrow was involved by lymphoma, then there would be a small increase in the SUV of each voxel, reflecting involvement by the lymphoma. In order to capture such changes, we analyzed the percent of total voxels in SUV ranging from 0.75 to 1.20, in increments of 0.05, as this is where the greatest divergence was visually identified,” the researchers wrote. “The goal was to identify if a percentage of voxels at a set SUV could detect lymphomatous involvement.”
The researchers identified 10 candidate thresholds in the developmental phase; 4 of these performed better than the others in the validation phase. Using those thresholds, 10 of the 12 patients in the validation cohort could be correctly staged using FDG PET/CT.
Further analysis identified a single threshold that performed best: If greater than 38% of the voxels (averaging 1,734 voxels) demonstrated an SUV of less than 0.95, the sensitivity was 100% and the specificity was 80%.
The researchers acknowledged that the findings are limited because of the study’s small sample size and said the results should be validated in a larger trial.
There was no external funding for the study and the researchers reported having no financial disclosures.
SOURCE: Morgan R et al. Clin Lymphoma Myeloma Leuk. 2018 Jul 4. doi: 10.1016/j.clml.2018.06.024.
REPORTING FROM CLINICAL LYMPHOMA, MYELOMA & LEUKEMIA
Key clinical point:
Major finding: If greater than 38% of the voxels demonstrated an standard uptake value of less than 0.95, there was a sensitivity of 100% and a specificity of 80%.
Study details: A retrospective cohort study of 23 patients with mantle cell leukemia and 5 controls.
Disclosures: There was no external funding for the study and the researchers reported having no financial disclosures.
Source: Morgan R et al. Clin Lymphoma Myeloma Leuk. 2018 Jul 4. doi: 10.1016/j.clml.2018.06.024.
Isavuconazole resolved invasive fungal disease in patients on ibrutinib
Treatment with isavuconazole resolved or substantially improved invasive fungal disease among seven of eight patients receiving concomitant ibrutinib, according to the results of a small two-center study.
The combination “was well-tolerated overall,” wrote Kaelyn C. Cummins of Brigham and Women’s Hospital, together with her associates there and at the Dana-Farber Cancer Institute, Boston. Their letter to the editor was published in Leukemia & Lymphoma.
Although ibrutinib is considered less immunosuppressive than conventional chemotherapy, it has been tied to invasive fungal infections, even in seemingly low-risk patients. The preferred treatment, voriconazole, is a very strong inhibitor of cytochrome P450 systems, of which ibrutinib is a substrate. For this study, the researchers queried the pharmacy databases of their institutions to identify adults who received concomitant isavuconazole (200 mg per day) and ibrutinib between 2015 and 2018. Drug exposures were confirmed by medical record review.
Four patients experienced clinical and radiologic resolution of invasive aspergillosis, fusariosis, mucormycosis, or phaeohyphomycosis. Another three had clinical and radiologic improvement of confirmed or probable aspergillosis or histoplasmosis. One of these patients underwent five debridements for central nervous system invasive aspergillosis but had 8 months of clinical improvement between debridements. This patient’s fungal isolate remained susceptible to isavuconazole throughout treatment. The patient who did not respond at all to isavuconazole had invasive aspergillosis with recurrent brain abscesses. The fungal isolate remained susceptible to isavuconazole, and the patient switched to long-term voriconazole therapy after stopping ibrutinib.
Several adverse events occurred while patients were on concomitant therapy. One patient developed paroxysmal atrial fibrillation that persisted after stopping ibrutinib. Another had worsening of preexisting thrombocytopenia. Among four patients with electrocardiogram data, two had transient QTc prolongation. No patient died within 12 weeks of starting concomitant therapy. Two patients eventually died after their cancer progressed.
The median age of the patients was 60 years (range, 38-76 years). Five were men. Six had chronic lymphocytic leukemia (CLL) and two had marginal zone lymphoma. Two CLL patients were on ibrutinib monotherapy, two also received rituximab, one also received umbralisib, and one also received obinutuzumab. One patient with marginal zone lymphoma was on ibrutinib monotherapy, and the other received concomitant rituximab, gemcitabine, dexamethasone, and cisplatin.
Researchers should study the mechanisms by which [Bruton’s tyrosine kinase] inhibitors might increase susceptibility to fungal infections among patients with lymphoma or CLL, said Ms. Cummins and her associates. Because the CYP3A enzyme system also metabolizes PI3K and BCL-2 inhibitors, their results “could be more broadly applicable.”
Ms. Cummins had no disclosures.
SOURCE: Cummins KC et al. Leuk. Lymphoma 2018 Jul 24. doi: 10.1080/10428194.2018.1485913.
Treatment with isavuconazole resolved or substantially improved invasive fungal disease among seven of eight patients receiving concomitant ibrutinib, according to the results of a small two-center study.
The combination “was well-tolerated overall,” wrote Kaelyn C. Cummins of Brigham and Women’s Hospital, together with her associates there and at the Dana-Farber Cancer Institute, Boston. Their letter to the editor was published in Leukemia & Lymphoma.
Although ibrutinib is considered less immunosuppressive than conventional chemotherapy, it has been tied to invasive fungal infections, even in seemingly low-risk patients. The preferred treatment, voriconazole, is a very strong inhibitor of cytochrome P450 systems, of which ibrutinib is a substrate. For this study, the researchers queried the pharmacy databases of their institutions to identify adults who received concomitant isavuconazole (200 mg per day) and ibrutinib between 2015 and 2018. Drug exposures were confirmed by medical record review.
Four patients experienced clinical and radiologic resolution of invasive aspergillosis, fusariosis, mucormycosis, or phaeohyphomycosis. Another three had clinical and radiologic improvement of confirmed or probable aspergillosis or histoplasmosis. One of these patients underwent five debridements for central nervous system invasive aspergillosis but had 8 months of clinical improvement between debridements. This patient’s fungal isolate remained susceptible to isavuconazole throughout treatment. The patient who did not respond at all to isavuconazole had invasive aspergillosis with recurrent brain abscesses. The fungal isolate remained susceptible to isavuconazole, and the patient switched to long-term voriconazole therapy after stopping ibrutinib.
Several adverse events occurred while patients were on concomitant therapy. One patient developed paroxysmal atrial fibrillation that persisted after stopping ibrutinib. Another had worsening of preexisting thrombocytopenia. Among four patients with electrocardiogram data, two had transient QTc prolongation. No patient died within 12 weeks of starting concomitant therapy. Two patients eventually died after their cancer progressed.
The median age of the patients was 60 years (range, 38-76 years). Five were men. Six had chronic lymphocytic leukemia (CLL) and two had marginal zone lymphoma. Two CLL patients were on ibrutinib monotherapy, two also received rituximab, one also received umbralisib, and one also received obinutuzumab. One patient with marginal zone lymphoma was on ibrutinib monotherapy, and the other received concomitant rituximab, gemcitabine, dexamethasone, and cisplatin.
Researchers should study the mechanisms by which [Bruton’s tyrosine kinase] inhibitors might increase susceptibility to fungal infections among patients with lymphoma or CLL, said Ms. Cummins and her associates. Because the CYP3A enzyme system also metabolizes PI3K and BCL-2 inhibitors, their results “could be more broadly applicable.”
Ms. Cummins had no disclosures.
SOURCE: Cummins KC et al. Leuk. Lymphoma 2018 Jul 24. doi: 10.1080/10428194.2018.1485913.
Treatment with isavuconazole resolved or substantially improved invasive fungal disease among seven of eight patients receiving concomitant ibrutinib, according to the results of a small two-center study.
The combination “was well-tolerated overall,” wrote Kaelyn C. Cummins of Brigham and Women’s Hospital, together with her associates there and at the Dana-Farber Cancer Institute, Boston. Their letter to the editor was published in Leukemia & Lymphoma.
Although ibrutinib is considered less immunosuppressive than conventional chemotherapy, it has been tied to invasive fungal infections, even in seemingly low-risk patients. The preferred treatment, voriconazole, is a very strong inhibitor of cytochrome P450 systems, of which ibrutinib is a substrate. For this study, the researchers queried the pharmacy databases of their institutions to identify adults who received concomitant isavuconazole (200 mg per day) and ibrutinib between 2015 and 2018. Drug exposures were confirmed by medical record review.
Four patients experienced clinical and radiologic resolution of invasive aspergillosis, fusariosis, mucormycosis, or phaeohyphomycosis. Another three had clinical and radiologic improvement of confirmed or probable aspergillosis or histoplasmosis. One of these patients underwent five debridements for central nervous system invasive aspergillosis but had 8 months of clinical improvement between debridements. This patient’s fungal isolate remained susceptible to isavuconazole throughout treatment. The patient who did not respond at all to isavuconazole had invasive aspergillosis with recurrent brain abscesses. The fungal isolate remained susceptible to isavuconazole, and the patient switched to long-term voriconazole therapy after stopping ibrutinib.
Several adverse events occurred while patients were on concomitant therapy. One patient developed paroxysmal atrial fibrillation that persisted after stopping ibrutinib. Another had worsening of preexisting thrombocytopenia. Among four patients with electrocardiogram data, two had transient QTc prolongation. No patient died within 12 weeks of starting concomitant therapy. Two patients eventually died after their cancer progressed.
The median age of the patients was 60 years (range, 38-76 years). Five were men. Six had chronic lymphocytic leukemia (CLL) and two had marginal zone lymphoma. Two CLL patients were on ibrutinib monotherapy, two also received rituximab, one also received umbralisib, and one also received obinutuzumab. One patient with marginal zone lymphoma was on ibrutinib monotherapy, and the other received concomitant rituximab, gemcitabine, dexamethasone, and cisplatin.
Researchers should study the mechanisms by which [Bruton’s tyrosine kinase] inhibitors might increase susceptibility to fungal infections among patients with lymphoma or CLL, said Ms. Cummins and her associates. Because the CYP3A enzyme system also metabolizes PI3K and BCL-2 inhibitors, their results “could be more broadly applicable.”
Ms. Cummins had no disclosures.
SOURCE: Cummins KC et al. Leuk. Lymphoma 2018 Jul 24. doi: 10.1080/10428194.2018.1485913.
FROM LEUKEMIA & LYMPHOMA
Key clinical point: Treatment with isavuconazole resolved or substantially improved invasive fungal disease in patients receiving concomitant ibrutinib.
Major finding: Seven of eight patients experienced clinical and radiographic resolution or improvement. Adverse events of concomitant treatment included paroxysmal atrial fibrillation, worsening of baseline thrombocytopenia, and QTc interval prolongation.
Study details: Retrospective study at two centers.
Disclosures: The article did not include information on funding sources or conflicts of interests.
Source: Cummins KC. et al. Leuk. Lymphoma 2018 Jul 24. doi: 10.1080/10428194.2018.1485913.
Rituximab reduces risk of follicular lymphoma transformation
Rituximab-based chemotherapy can significantly reduce the risk of transformation of follicular lymphoma (FL) from an indolent to an aggressive histology, such as diffuse large B-cell lymphoma, results of a retrospective pooled analysis have suggested.
“Despite the intrinsic limitations related to the retrospective nature of our study, we confirmed that the cumulative hazard of histological transformation as a first event in follicular lymphoma can be reduced significantly by introducing rituximab to a backbone therapy. Moreover, our data also confirm that histological transformation still has an adverse effect on patient outcome, although it is less catastrophic than the pre-rituximab regimens,” they wrote in the Lancet Haematology.
These investigators, from 11 cooperative groups or institutions across Europe, pooled data on patients aged 18 years and older who had a histologically confirmed diagnosis of grade 1, 2, or 3a FL between Jan. 2, 1997, and Dec. 20, 2013.
They defined histologic transformation as a biopsy-proven aggressive lymphoma that occurred as a first event after first-line therapy.
Data on a total of 8,116 patients were available for analysis; 509 of these patients had had histologic transformations. After a median follow-up of 87 months, the 10-year cumulative hazard for all patients was 7.7%. The 10-year cumulative hazard – one of two primary endpoints – was 5.2% for patients who had received any rituximab versus 8.7% for those who did not, which translated into a hazard ratio of 0.73 (P = .004).
Among patients who received rituximab during induction only, the 10-year cumulative hazard was 5.9%, and it was 3.6% among those who received rituximab during induction and maintenance phases of treatment. This difference translated into a HR of 0.55 (P = .003).
The benefit of rituximab induction and maintenance – compared with induction only – held up in a multivariate analysis controlling for age at diagnosis, sex, FLIPI (Follicular Lymphoma International Prognostic Index) score, active surveillance vs. treatment, and FL grade (HR, 0.55; P = .016).
There were 287 deaths among the 509 patients with transformation, resulting in a 10-year survival after transformation of 32%.
The 5-year survival after transformation was 38% for patients who were not exposed to rituximab, 42% for patients who received induction rituximab, and 43% for those who received both induction and maintenance rituximab, but the differences between the three groups were not statistically significant.
“More comprehensive knowledge of the biological risk factors for follicular lymphoma transformation and the molecular pathways involved is likely to help clinicians make more accurate prognostic assessments and also inform the potential usefulness of novel drugs for the treatment of follicular lymphoma,” the researchers wrote.
The study was funded by the European Lymphoma Institute and other research groups. The researchers reported having no financial disclosures.
SOURCE: Federico M et al. Lancet Haematol. 2018 Jul 4. doi: 10.1016/S2352-3026(18)30090-5.
Rituximab-based chemotherapy can significantly reduce the risk of transformation of follicular lymphoma (FL) from an indolent to an aggressive histology, such as diffuse large B-cell lymphoma, results of a retrospective pooled analysis have suggested.
“Despite the intrinsic limitations related to the retrospective nature of our study, we confirmed that the cumulative hazard of histological transformation as a first event in follicular lymphoma can be reduced significantly by introducing rituximab to a backbone therapy. Moreover, our data also confirm that histological transformation still has an adverse effect on patient outcome, although it is less catastrophic than the pre-rituximab regimens,” they wrote in the Lancet Haematology.
These investigators, from 11 cooperative groups or institutions across Europe, pooled data on patients aged 18 years and older who had a histologically confirmed diagnosis of grade 1, 2, or 3a FL between Jan. 2, 1997, and Dec. 20, 2013.
They defined histologic transformation as a biopsy-proven aggressive lymphoma that occurred as a first event after first-line therapy.
Data on a total of 8,116 patients were available for analysis; 509 of these patients had had histologic transformations. After a median follow-up of 87 months, the 10-year cumulative hazard for all patients was 7.7%. The 10-year cumulative hazard – one of two primary endpoints – was 5.2% for patients who had received any rituximab versus 8.7% for those who did not, which translated into a hazard ratio of 0.73 (P = .004).
Among patients who received rituximab during induction only, the 10-year cumulative hazard was 5.9%, and it was 3.6% among those who received rituximab during induction and maintenance phases of treatment. This difference translated into a HR of 0.55 (P = .003).
The benefit of rituximab induction and maintenance – compared with induction only – held up in a multivariate analysis controlling for age at diagnosis, sex, FLIPI (Follicular Lymphoma International Prognostic Index) score, active surveillance vs. treatment, and FL grade (HR, 0.55; P = .016).
There were 287 deaths among the 509 patients with transformation, resulting in a 10-year survival after transformation of 32%.
The 5-year survival after transformation was 38% for patients who were not exposed to rituximab, 42% for patients who received induction rituximab, and 43% for those who received both induction and maintenance rituximab, but the differences between the three groups were not statistically significant.
“More comprehensive knowledge of the biological risk factors for follicular lymphoma transformation and the molecular pathways involved is likely to help clinicians make more accurate prognostic assessments and also inform the potential usefulness of novel drugs for the treatment of follicular lymphoma,” the researchers wrote.
The study was funded by the European Lymphoma Institute and other research groups. The researchers reported having no financial disclosures.
SOURCE: Federico M et al. Lancet Haematol. 2018 Jul 4. doi: 10.1016/S2352-3026(18)30090-5.
Rituximab-based chemotherapy can significantly reduce the risk of transformation of follicular lymphoma (FL) from an indolent to an aggressive histology, such as diffuse large B-cell lymphoma, results of a retrospective pooled analysis have suggested.
“Despite the intrinsic limitations related to the retrospective nature of our study, we confirmed that the cumulative hazard of histological transformation as a first event in follicular lymphoma can be reduced significantly by introducing rituximab to a backbone therapy. Moreover, our data also confirm that histological transformation still has an adverse effect on patient outcome, although it is less catastrophic than the pre-rituximab regimens,” they wrote in the Lancet Haematology.
These investigators, from 11 cooperative groups or institutions across Europe, pooled data on patients aged 18 years and older who had a histologically confirmed diagnosis of grade 1, 2, or 3a FL between Jan. 2, 1997, and Dec. 20, 2013.
They defined histologic transformation as a biopsy-proven aggressive lymphoma that occurred as a first event after first-line therapy.
Data on a total of 8,116 patients were available for analysis; 509 of these patients had had histologic transformations. After a median follow-up of 87 months, the 10-year cumulative hazard for all patients was 7.7%. The 10-year cumulative hazard – one of two primary endpoints – was 5.2% for patients who had received any rituximab versus 8.7% for those who did not, which translated into a hazard ratio of 0.73 (P = .004).
Among patients who received rituximab during induction only, the 10-year cumulative hazard was 5.9%, and it was 3.6% among those who received rituximab during induction and maintenance phases of treatment. This difference translated into a HR of 0.55 (P = .003).
The benefit of rituximab induction and maintenance – compared with induction only – held up in a multivariate analysis controlling for age at diagnosis, sex, FLIPI (Follicular Lymphoma International Prognostic Index) score, active surveillance vs. treatment, and FL grade (HR, 0.55; P = .016).
There were 287 deaths among the 509 patients with transformation, resulting in a 10-year survival after transformation of 32%.
The 5-year survival after transformation was 38% for patients who were not exposed to rituximab, 42% for patients who received induction rituximab, and 43% for those who received both induction and maintenance rituximab, but the differences between the three groups were not statistically significant.
“More comprehensive knowledge of the biological risk factors for follicular lymphoma transformation and the molecular pathways involved is likely to help clinicians make more accurate prognostic assessments and also inform the potential usefulness of novel drugs for the treatment of follicular lymphoma,” the researchers wrote.
The study was funded by the European Lymphoma Institute and other research groups. The researchers reported having no financial disclosures.
SOURCE: Federico M et al. Lancet Haematol. 2018 Jul 4. doi: 10.1016/S2352-3026(18)30090-5.
FROM THE LANCET HAEMATOLOGY
Key clinical point:
Major finding: The 10-year cumulative hazard of histologic transformation was 5.2% for patients who had received rituximab and 8.7% for those who had not.
Study details: Retrospective pooled analysis of 8,116 patients with FL, 509 of whom had transformation over a 10-year period.
Disclosures: The study was funded by Associazione Angela Serra per la Ricerca sul Cancro, European Lymphoma Institute, European Hematology Association Lymphoma Group, Fondazione Italiana Linfomi, and the Spanish Group of Lymphoma and Bone Marrow Transplantation. The researchers reported having no financial disclosures.
Source: Federico M et al. Lancet Haematol. 2018 Jul 4. doi: 10.1016/S2352-3026(18)30090-5.