User login
Pertuzumab plus high-dose trastuzumab shows activity in HER-2-positive breast cancer
Key clinical point: In patients with human epidermal growth factor 2 (HER2)-positive metastatic breast cancer and progressive brain metastases despite radiotherapy, pertuzumab in combination with high-dose trastuzumab shows modest central nervous system (CNS) response and good clinical benefit.
Major finding: The confirmed CNS objective response rate was 11% with all partial responses. The clinical benefit rate at 4 months was 68% and at 6 months was 51%. The grade 3-4 adverse event rate was 44%. There were no new safety signals.
Study details: A phase 2 PATRICIA study evaluated pertuzumab in combination with high-dose trastuzumab in 39 patients with HER2-positive metastatic breast cancer and progressive brain metastases despite radiotherapy.
Disclosures: The study was sponsored by F. Hoffmann-La Roche/ Genentech. The authors received consulting/advisory fees, research funding, royalties, and travel/accommodation expenses from various sources. Dr. Fung, Dr. Cheng, and Dr. Kirschbrown reported employment by, stocks, and other ownership interests in Genentech/Roche.
Source: Lin NU et al. J Clin Oncol. 2021 May 4. doi: 10.1200/JCO.20.02822.
Key clinical point: In patients with human epidermal growth factor 2 (HER2)-positive metastatic breast cancer and progressive brain metastases despite radiotherapy, pertuzumab in combination with high-dose trastuzumab shows modest central nervous system (CNS) response and good clinical benefit.
Major finding: The confirmed CNS objective response rate was 11% with all partial responses. The clinical benefit rate at 4 months was 68% and at 6 months was 51%. The grade 3-4 adverse event rate was 44%. There were no new safety signals.
Study details: A phase 2 PATRICIA study evaluated pertuzumab in combination with high-dose trastuzumab in 39 patients with HER2-positive metastatic breast cancer and progressive brain metastases despite radiotherapy.
Disclosures: The study was sponsored by F. Hoffmann-La Roche/ Genentech. The authors received consulting/advisory fees, research funding, royalties, and travel/accommodation expenses from various sources. Dr. Fung, Dr. Cheng, and Dr. Kirschbrown reported employment by, stocks, and other ownership interests in Genentech/Roche.
Source: Lin NU et al. J Clin Oncol. 2021 May 4. doi: 10.1200/JCO.20.02822.
Key clinical point: In patients with human epidermal growth factor 2 (HER2)-positive metastatic breast cancer and progressive brain metastases despite radiotherapy, pertuzumab in combination with high-dose trastuzumab shows modest central nervous system (CNS) response and good clinical benefit.
Major finding: The confirmed CNS objective response rate was 11% with all partial responses. The clinical benefit rate at 4 months was 68% and at 6 months was 51%. The grade 3-4 adverse event rate was 44%. There were no new safety signals.
Study details: A phase 2 PATRICIA study evaluated pertuzumab in combination with high-dose trastuzumab in 39 patients with HER2-positive metastatic breast cancer and progressive brain metastases despite radiotherapy.
Disclosures: The study was sponsored by F. Hoffmann-La Roche/ Genentech. The authors received consulting/advisory fees, research funding, royalties, and travel/accommodation expenses from various sources. Dr. Fung, Dr. Cheng, and Dr. Kirschbrown reported employment by, stocks, and other ownership interests in Genentech/Roche.
Source: Lin NU et al. J Clin Oncol. 2021 May 4. doi: 10.1200/JCO.20.02822.
Immediate breast reconstruction delays radiotherapy and increases complication
Key clinical point: Immediate reconstruction after neoadjuvant chemotherapy in women with T4 breast cancer is associated with higher odds for complication resulting in unplanned reoperations and a longer time to postmastectomy radiotherapy.
Major finding: Unplanned reoperations for complications were more common after immediate vs. delayed and no reconstruction (22% vs. 7.3% and 4.4%, respectively; P less than .001). The median time to initiation of postmastectomy radiotherapy was longer after immediate reconstruction vs. delayed and no reconstruction (60 days vs. 42 days and 49 days, respectively; P less than .001). The median time to the first recurrence was 18 months and was not significantly different between the groups (P = .13).
Study details: A retrospective study of 269 consecutive women with stage T4 breast cancer treated with surgery, chemotherapy, and radiotherapy between 2007 and 2019.
Disclosure: This study was supported in part by the National Institutes of Health. Dr. Morrow received speaking honoraria from Roche. The other authors did not disclose any conflicts of interest.
Source: Pawloski KR et al. J Am Coll Surg. 2021 Apr 24. doi: 10.1016/j.jamcollsurg.2021.04.016.
Key clinical point: Immediate reconstruction after neoadjuvant chemotherapy in women with T4 breast cancer is associated with higher odds for complication resulting in unplanned reoperations and a longer time to postmastectomy radiotherapy.
Major finding: Unplanned reoperations for complications were more common after immediate vs. delayed and no reconstruction (22% vs. 7.3% and 4.4%, respectively; P less than .001). The median time to initiation of postmastectomy radiotherapy was longer after immediate reconstruction vs. delayed and no reconstruction (60 days vs. 42 days and 49 days, respectively; P less than .001). The median time to the first recurrence was 18 months and was not significantly different between the groups (P = .13).
Study details: A retrospective study of 269 consecutive women with stage T4 breast cancer treated with surgery, chemotherapy, and radiotherapy between 2007 and 2019.
Disclosure: This study was supported in part by the National Institutes of Health. Dr. Morrow received speaking honoraria from Roche. The other authors did not disclose any conflicts of interest.
Source: Pawloski KR et al. J Am Coll Surg. 2021 Apr 24. doi: 10.1016/j.jamcollsurg.2021.04.016.
Key clinical point: Immediate reconstruction after neoadjuvant chemotherapy in women with T4 breast cancer is associated with higher odds for complication resulting in unplanned reoperations and a longer time to postmastectomy radiotherapy.
Major finding: Unplanned reoperations for complications were more common after immediate vs. delayed and no reconstruction (22% vs. 7.3% and 4.4%, respectively; P less than .001). The median time to initiation of postmastectomy radiotherapy was longer after immediate reconstruction vs. delayed and no reconstruction (60 days vs. 42 days and 49 days, respectively; P less than .001). The median time to the first recurrence was 18 months and was not significantly different between the groups (P = .13).
Study details: A retrospective study of 269 consecutive women with stage T4 breast cancer treated with surgery, chemotherapy, and radiotherapy between 2007 and 2019.
Disclosure: This study was supported in part by the National Institutes of Health. Dr. Morrow received speaking honoraria from Roche. The other authors did not disclose any conflicts of interest.
Source: Pawloski KR et al. J Am Coll Surg. 2021 Apr 24. doi: 10.1016/j.jamcollsurg.2021.04.016.
Breast cancer: Routine scans can identify heart disease
Key clinical point: The presence and extent of coronary artery calcium (CAC), as automatically quantified on routinely performed computed tomography scans, are associated with cardiovascular and coronary artery diseases.
Major finding: The risk for cardiovascular disease increased with a higher CAC score (adjusted hazard ratio, 1.8, 2.1, and 3.4 for CAC scores 11-100, 101-400, and greater than 400, respectively). The risk for coronary artery disease also increased with an increase in CAC score (adjusted hazard ratio, 2.8, 4.3, and 7.8 for CAC scores 11-100, 101-400, and greater than 400, respectively).
Study details: A multicenter cohort study of 15,915 patients with breast cancer who received radiotherapy between 2005 and 2016. The CAC scores were automatically extracted from computed tomography scans using a deep learning algorithm.
Disclosure: This study was funded by the Dutch Cancer Society. The authors received grants, lecture fees, and research support from various sources. Dr. Leiner and Dr. Isgum owned shares of Quantib-U BV and/or a patent with royalties planned. No other conflicts of interest were reported.
Source: Gal R et al. JAMA Oncol. 2021 May 6. doi: 10.1001/jamaoncol.2021.1144.
Key clinical point: The presence and extent of coronary artery calcium (CAC), as automatically quantified on routinely performed computed tomography scans, are associated with cardiovascular and coronary artery diseases.
Major finding: The risk for cardiovascular disease increased with a higher CAC score (adjusted hazard ratio, 1.8, 2.1, and 3.4 for CAC scores 11-100, 101-400, and greater than 400, respectively). The risk for coronary artery disease also increased with an increase in CAC score (adjusted hazard ratio, 2.8, 4.3, and 7.8 for CAC scores 11-100, 101-400, and greater than 400, respectively).
Study details: A multicenter cohort study of 15,915 patients with breast cancer who received radiotherapy between 2005 and 2016. The CAC scores were automatically extracted from computed tomography scans using a deep learning algorithm.
Disclosure: This study was funded by the Dutch Cancer Society. The authors received grants, lecture fees, and research support from various sources. Dr. Leiner and Dr. Isgum owned shares of Quantib-U BV and/or a patent with royalties planned. No other conflicts of interest were reported.
Source: Gal R et al. JAMA Oncol. 2021 May 6. doi: 10.1001/jamaoncol.2021.1144.
Key clinical point: The presence and extent of coronary artery calcium (CAC), as automatically quantified on routinely performed computed tomography scans, are associated with cardiovascular and coronary artery diseases.
Major finding: The risk for cardiovascular disease increased with a higher CAC score (adjusted hazard ratio, 1.8, 2.1, and 3.4 for CAC scores 11-100, 101-400, and greater than 400, respectively). The risk for coronary artery disease also increased with an increase in CAC score (adjusted hazard ratio, 2.8, 4.3, and 7.8 for CAC scores 11-100, 101-400, and greater than 400, respectively).
Study details: A multicenter cohort study of 15,915 patients with breast cancer who received radiotherapy between 2005 and 2016. The CAC scores were automatically extracted from computed tomography scans using a deep learning algorithm.
Disclosure: This study was funded by the Dutch Cancer Society. The authors received grants, lecture fees, and research support from various sources. Dr. Leiner and Dr. Isgum owned shares of Quantib-U BV and/or a patent with royalties planned. No other conflicts of interest were reported.
Source: Gal R et al. JAMA Oncol. 2021 May 6. doi: 10.1001/jamaoncol.2021.1144.
Breast-conserving surgery tops mastectomy
Key clinical point: Breast-conserving surgery (BCS) with radiotherapy yields superior survival vs. mastectomy in patients with early-stage breast cancer.
Major finding: At a median follow-up of 6.28 years, mastectomy without radiotherapy vs. BCS with radiotherapy was associated with worse overall survival (OS; hazard ratio [HR], 1.79; P less than .001) and breast cancer-specific survival (BCSS; HR, 1.66; P less than .001). Mastectomy with radiotherapy also showed lower OS (HR, 1.24; P less than .001) and BCSS (HR, 1.28; P=.001) vs. BCS and radiotherapy.
Study details: A cohort study of 48,986 patients with primary invasive T1-2 N0-2 breast cancer who underwent breast surgery between 2008 and 2017.
Disclosure: This work was funded by the Swedish Breast Cancer Association. Dr de Boniface was supported by an investigator award from the Swedish Cancer Society, and Ms Johansson was supported by a research grant from the Swedish Research Council. The authors did not declare any conflict of interest.
Source: de Boniface J et al. JAMA Surg. 2021 May 5. doi: 10.1001/jamasurg.2021.1438.
Key clinical point: Breast-conserving surgery (BCS) with radiotherapy yields superior survival vs. mastectomy in patients with early-stage breast cancer.
Major finding: At a median follow-up of 6.28 years, mastectomy without radiotherapy vs. BCS with radiotherapy was associated with worse overall survival (OS; hazard ratio [HR], 1.79; P less than .001) and breast cancer-specific survival (BCSS; HR, 1.66; P less than .001). Mastectomy with radiotherapy also showed lower OS (HR, 1.24; P less than .001) and BCSS (HR, 1.28; P=.001) vs. BCS and radiotherapy.
Study details: A cohort study of 48,986 patients with primary invasive T1-2 N0-2 breast cancer who underwent breast surgery between 2008 and 2017.
Disclosure: This work was funded by the Swedish Breast Cancer Association. Dr de Boniface was supported by an investigator award from the Swedish Cancer Society, and Ms Johansson was supported by a research grant from the Swedish Research Council. The authors did not declare any conflict of interest.
Source: de Boniface J et al. JAMA Surg. 2021 May 5. doi: 10.1001/jamasurg.2021.1438.
Key clinical point: Breast-conserving surgery (BCS) with radiotherapy yields superior survival vs. mastectomy in patients with early-stage breast cancer.
Major finding: At a median follow-up of 6.28 years, mastectomy without radiotherapy vs. BCS with radiotherapy was associated with worse overall survival (OS; hazard ratio [HR], 1.79; P less than .001) and breast cancer-specific survival (BCSS; HR, 1.66; P less than .001). Mastectomy with radiotherapy also showed lower OS (HR, 1.24; P less than .001) and BCSS (HR, 1.28; P=.001) vs. BCS and radiotherapy.
Study details: A cohort study of 48,986 patients with primary invasive T1-2 N0-2 breast cancer who underwent breast surgery between 2008 and 2017.
Disclosure: This work was funded by the Swedish Breast Cancer Association. Dr de Boniface was supported by an investigator award from the Swedish Cancer Society, and Ms Johansson was supported by a research grant from the Swedish Research Council. The authors did not declare any conflict of interest.
Source: de Boniface J et al. JAMA Surg. 2021 May 5. doi: 10.1001/jamasurg.2021.1438.
Clinical Edge Journal Scan Commentary: Breast Cancer June 2021

Adjuvant T-DM1 is recommended for patients with HER2-positive early breast cancer with residual disease after neoadjuvant therapy based on phase 3 results. The KATHERINE study found a significant reduction for the risk of recurrence and death with adjuvant T-DM1 vs trastuzumab. Subgroup analyses from KATHERINE showed similar benefits with T-DM1 irrespective of type of neoadjuvant regimen. Furthermore, T-DM1 appeared to benefit small node-negative tumors and particularly those tumors considered high-risk (Mamounas). In the phase 3 TRAIN-2 study, similar pCR rates (68% vs 67%), as well as 3 year event-free (94% vs 93%) and overall (98% vs 98%) survival, were observed for non-anthracycline and anthracycline-containing regimens. These findings highlight the broad applicability of T-DM1 in the adjuvant setting, the rationale to support de-escalation and omission of anthracyclines for HER2-positive tumors, and the importance of tailoring therapy based on response.
Brain metastases occur in up to 50% of patients with HER2-positive metastatic breast cancer (MBC). Effective therapies for this population represent an unmet clinical need. The phase 2 PATRICIA study demonstrated activity of pertuzumab plus high-dose trastuzumab (6mg/kg weekly) for patients with HER2-positive MBC and central nervous system (CNS) progression after radiotherapy. In 37 patients evaluable for efficacy, the CNS objective response rate was 11%, clinical benefit rate at 4 and 6 months was 68% and 51%, respectively, and 2 patients had stable disease for over 2 years (Lin). Data supports the role of other HER2-targeted therapies for CNS disease including T-DM1, neratinib, and tucatinib. Among 291 patients with brain metastases in HER2CLIMB, the combination of tucatinib, capecitabine, and trastuzumab improved median overall survival (18 vs 12 months) and CNS progression-free survival (9.9 vs 4.2 months), compared with capecitabine plus trastuzumab. Further investigation exploring other novel therapy combinations and biomarkers will help further improve outcomes for these patients.
Adjuvant endocrine therapy decreases the risk for recurrence and improves survival for women diagnosed with HR-positive breast cancer. For young women, endocrine therapy options include tamoxifen, as well as ovarian suppression plus tamoxifen, or an aromatase inhibitor. Recommended duration of therapy is at least 5 years and can extend to 10 years. These treatments and duration may present challenges related to childbearing attempts and raise fertility concerns among young women. In the Young Women’s Breast Cancer Study, among 643 women aged 40 years or younger and diagnosed with early stage HR-positive breast cancer, one-third reported fertility concerns impacting endocrine therapy decisions. Those who reported fertility concerns were more likely to exhibit non-initiation or non-persistence to endocrine therapy (40% vs 20%). Among women with fertility concerns, 7% did not initiate endocrine therapy, and 33% were non-persistent over 5 years (Sella). It is essential to integrate early oncofertility dialogue to help achieve optimal endocrine therapy and address family planning goals.
Studies have shown sugar-sweetened beverages (SSB) increase the risk for insulin resistance, diabetes, and heart disease. In a subsample of Women’s Health Initiative participants, higher levels of insulin resistance were shown to be linked to an increased incidence of breast cancer and all-cause mortality after breast cancer. Researchers found that among 8,863 women diagnosed with early breast cancer, those who consumed SSB after diagnosis had higher breast cancer-specific mortality and all-cause mortality. Additionally, replacing SSB with coffee, tea or water was linked to a decrease in mortality (Farvid). These findings support discussion of lifestyle and dietary behaviors in the survivorship setting, as these modifiable risk factors can potentially have significant health implications.
References:
van der Voort A, van Ramshorst MS, van Werkhoven ED, et al. Three-year follow-up of neoadjuvant chemotherapy with or without anthracyclines in the presence of dual HER2-blockade for HER2-positive breast cancer (TRAIN-2): A randomized phase III trial. J Clin Oncol. 2020;38S:ASCO #501.
Lin NU, Borges V, Anders C, et al. Intracranial efficacy and survival with tucatinib plus trastuzumab and capecitabine for previously treated HER2-positive breast cancer with brain metastases in the HER2CLIMB trial. J Clin Oncol. 2020;38(23):2610-2619.
Pan K, Chlebowski RT, Mortimer JE, et al. Insulin resistance and breast cancer incidence and mortality in postmenopausal women in the Women's Health Initiative. Cancer. 2020;126(16):3638-3647.
Adjuvant T-DM1 is recommended for patients with HER2-positive early breast cancer with residual disease after neoadjuvant therapy based on phase 3 results. The KATHERINE study found a significant reduction for the risk of recurrence and death with adjuvant T-DM1 vs trastuzumab. Subgroup analyses from KATHERINE showed similar benefits with T-DM1 irrespective of type of neoadjuvant regimen. Furthermore, T-DM1 appeared to benefit small node-negative tumors and particularly those tumors considered high-risk (Mamounas). In the phase 3 TRAIN-2 study, similar pCR rates (68% vs 67%), as well as 3 year event-free (94% vs 93%) and overall (98% vs 98%) survival, were observed for non-anthracycline and anthracycline-containing regimens. These findings highlight the broad applicability of T-DM1 in the adjuvant setting, the rationale to support de-escalation and omission of anthracyclines for HER2-positive tumors, and the importance of tailoring therapy based on response.
Brain metastases occur in up to 50% of patients with HER2-positive metastatic breast cancer (MBC). Effective therapies for this population represent an unmet clinical need. The phase 2 PATRICIA study demonstrated activity of pertuzumab plus high-dose trastuzumab (6mg/kg weekly) for patients with HER2-positive MBC and central nervous system (CNS) progression after radiotherapy. In 37 patients evaluable for efficacy, the CNS objective response rate was 11%, clinical benefit rate at 4 and 6 months was 68% and 51%, respectively, and 2 patients had stable disease for over 2 years (Lin). Data supports the role of other HER2-targeted therapies for CNS disease including T-DM1, neratinib, and tucatinib. Among 291 patients with brain metastases in HER2CLIMB, the combination of tucatinib, capecitabine, and trastuzumab improved median overall survival (18 vs 12 months) and CNS progression-free survival (9.9 vs 4.2 months), compared with capecitabine plus trastuzumab. Further investigation exploring other novel therapy combinations and biomarkers will help further improve outcomes for these patients.
Adjuvant endocrine therapy decreases the risk for recurrence and improves survival for women diagnosed with HR-positive breast cancer. For young women, endocrine therapy options include tamoxifen, as well as ovarian suppression plus tamoxifen, or an aromatase inhibitor. Recommended duration of therapy is at least 5 years and can extend to 10 years. These treatments and duration may present challenges related to childbearing attempts and raise fertility concerns among young women. In the Young Women’s Breast Cancer Study, among 643 women aged 40 years or younger and diagnosed with early stage HR-positive breast cancer, one-third reported fertility concerns impacting endocrine therapy decisions. Those who reported fertility concerns were more likely to exhibit non-initiation or non-persistence to endocrine therapy (40% vs 20%). Among women with fertility concerns, 7% did not initiate endocrine therapy, and 33% were non-persistent over 5 years (Sella). It is essential to integrate early oncofertility dialogue to help achieve optimal endocrine therapy and address family planning goals.
Studies have shown sugar-sweetened beverages (SSB) increase the risk for insulin resistance, diabetes, and heart disease. In a subsample of Women’s Health Initiative participants, higher levels of insulin resistance were shown to be linked to an increased incidence of breast cancer and all-cause mortality after breast cancer. Researchers found that among 8,863 women diagnosed with early breast cancer, those who consumed SSB after diagnosis had higher breast cancer-specific mortality and all-cause mortality. Additionally, replacing SSB with coffee, tea or water was linked to a decrease in mortality (Farvid). These findings support discussion of lifestyle and dietary behaviors in the survivorship setting, as these modifiable risk factors can potentially have significant health implications.
References:
van der Voort A, van Ramshorst MS, van Werkhoven ED, et al. Three-year follow-up of neoadjuvant chemotherapy with or without anthracyclines in the presence of dual HER2-blockade for HER2-positive breast cancer (TRAIN-2): A randomized phase III trial. J Clin Oncol. 2020;38S:ASCO #501.
Lin NU, Borges V, Anders C, et al. Intracranial efficacy and survival with tucatinib plus trastuzumab and capecitabine for previously treated HER2-positive breast cancer with brain metastases in the HER2CLIMB trial. J Clin Oncol. 2020;38(23):2610-2619.
Pan K, Chlebowski RT, Mortimer JE, et al. Insulin resistance and breast cancer incidence and mortality in postmenopausal women in the Women's Health Initiative. Cancer. 2020;126(16):3638-3647.
Adjuvant T-DM1 is recommended for patients with HER2-positive early breast cancer with residual disease after neoadjuvant therapy based on phase 3 results. The KATHERINE study found a significant reduction for the risk of recurrence and death with adjuvant T-DM1 vs trastuzumab. Subgroup analyses from KATHERINE showed similar benefits with T-DM1 irrespective of type of neoadjuvant regimen. Furthermore, T-DM1 appeared to benefit small node-negative tumors and particularly those tumors considered high-risk (Mamounas). In the phase 3 TRAIN-2 study, similar pCR rates (68% vs 67%), as well as 3 year event-free (94% vs 93%) and overall (98% vs 98%) survival, were observed for non-anthracycline and anthracycline-containing regimens. These findings highlight the broad applicability of T-DM1 in the adjuvant setting, the rationale to support de-escalation and omission of anthracyclines for HER2-positive tumors, and the importance of tailoring therapy based on response.
Brain metastases occur in up to 50% of patients with HER2-positive metastatic breast cancer (MBC). Effective therapies for this population represent an unmet clinical need. The phase 2 PATRICIA study demonstrated activity of pertuzumab plus high-dose trastuzumab (6mg/kg weekly) for patients with HER2-positive MBC and central nervous system (CNS) progression after radiotherapy. In 37 patients evaluable for efficacy, the CNS objective response rate was 11%, clinical benefit rate at 4 and 6 months was 68% and 51%, respectively, and 2 patients had stable disease for over 2 years (Lin). Data supports the role of other HER2-targeted therapies for CNS disease including T-DM1, neratinib, and tucatinib. Among 291 patients with brain metastases in HER2CLIMB, the combination of tucatinib, capecitabine, and trastuzumab improved median overall survival (18 vs 12 months) and CNS progression-free survival (9.9 vs 4.2 months), compared with capecitabine plus trastuzumab. Further investigation exploring other novel therapy combinations and biomarkers will help further improve outcomes for these patients.
Adjuvant endocrine therapy decreases the risk for recurrence and improves survival for women diagnosed with HR-positive breast cancer. For young women, endocrine therapy options include tamoxifen, as well as ovarian suppression plus tamoxifen, or an aromatase inhibitor. Recommended duration of therapy is at least 5 years and can extend to 10 years. These treatments and duration may present challenges related to childbearing attempts and raise fertility concerns among young women. In the Young Women’s Breast Cancer Study, among 643 women aged 40 years or younger and diagnosed with early stage HR-positive breast cancer, one-third reported fertility concerns impacting endocrine therapy decisions. Those who reported fertility concerns were more likely to exhibit non-initiation or non-persistence to endocrine therapy (40% vs 20%). Among women with fertility concerns, 7% did not initiate endocrine therapy, and 33% were non-persistent over 5 years (Sella). It is essential to integrate early oncofertility dialogue to help achieve optimal endocrine therapy and address family planning goals.
Studies have shown sugar-sweetened beverages (SSB) increase the risk for insulin resistance, diabetes, and heart disease. In a subsample of Women’s Health Initiative participants, higher levels of insulin resistance were shown to be linked to an increased incidence of breast cancer and all-cause mortality after breast cancer. Researchers found that among 8,863 women diagnosed with early breast cancer, those who consumed SSB after diagnosis had higher breast cancer-specific mortality and all-cause mortality. Additionally, replacing SSB with coffee, tea or water was linked to a decrease in mortality (Farvid). These findings support discussion of lifestyle and dietary behaviors in the survivorship setting, as these modifiable risk factors can potentially have significant health implications.
References:
van der Voort A, van Ramshorst MS, van Werkhoven ED, et al. Three-year follow-up of neoadjuvant chemotherapy with or without anthracyclines in the presence of dual HER2-blockade for HER2-positive breast cancer (TRAIN-2): A randomized phase III trial. J Clin Oncol. 2020;38S:ASCO #501.
Lin NU, Borges V, Anders C, et al. Intracranial efficacy and survival with tucatinib plus trastuzumab and capecitabine for previously treated HER2-positive breast cancer with brain metastases in the HER2CLIMB trial. J Clin Oncol. 2020;38(23):2610-2619.
Pan K, Chlebowski RT, Mortimer JE, et al. Insulin resistance and breast cancer incidence and mortality in postmenopausal women in the Women's Health Initiative. Cancer. 2020;126(16):3638-3647.
Hospital medicine leaders offer tips for gender equity
When Marisha Burden, MD, division head of hospital medicine at the University of Colorado at Denver, Aurora, would go to medical conferences, it seemed as if very few women were giving talks. She wondered if she could be wrong.
“I started doing my own assessments at every conference I would go to, just to make sure I wasn’t biased in my own belief system,” she said in a session at SHM Converge 2021, the annual conference of the Society of Hospital Medicine.
She wasn’t wrong.
In 2015, only 35% of all speakers at the SHM annual conference were women, and only 23% of the plenary speakers were women. In the years after that, when the society put out open calls for speakers, the numbers of women who spoke increased substantially, to 47% overall and 45% of plenary speakers.
The results – part of the SPEAK UP study Dr. Burden led in 2020 – show how gender disparity can be improved with a systematic process that is designed to improve it. The results of the study also showed that as the percentages of female speakers increased, the attendee ratings of the sessions did, too.
“You can do these things, and the quality of your conference doesn’t get negatively impacted – and in this case, actually improved,” Dr. Burden said.
That study marked progress toward leveling a traditionally uneven playing field when it comes to men and women in medicine, and the panelists in the session called on the field to use a variety of tools and strategies to continue toward something closer to equality.
Sara Spilseth, MD, MBA, chief of staff at Regions Hospital, in St. Paul, Minn., said it’s well established that although almost 50% of medical school students are women, the percentage shrinks each step from faculty to full professor to dean – of which only 16% are women. She referred to what’s known as the “leaky pipe.”
In what Dr. Spilseth said was one of her favorite studies, researchers in 2015 found that only 13% of clinical department leaders at the top 50 U.S. medical schools were women – they were outnumbered by the percentage of department leaders with mustaches, at 19%, even though mustaches are dwindling in popularity.
“Why does this exist? Why did we end up like this?” Part of the problem is a “respect gap,” she said, pointing to a study on the tendency of women to use the formal title of “doctor” when introducing male colleagues, whereas men who introduce women use that title less than half the time.
The COVID-19 pandemic has only made these disparities worse. Women are responsible for childcare much more frequently than men, Dr. Burden said, although the pandemic has brought caregiving duties to the forefront.
Dr. Spilseth said mentoring can help women navigate the workplace so as to help overcome these disparities. At Regions, the mentoring program is robust.
“Even before a new hire steps foot in the hospital, we have established them with a mentor,” she said. Sponsoring – the “ability of someone with political capital to use it to help colleagues” – can also help boost women’s careers, she said.
Her hospital also has a Women in Medicine Cooperative, which provides a way for women to talk about common struggles and to network.
Flexible work opportunities – working in transitional care units, being a physician advisor, and doing research – can all help boost a career as well, Dr. Spilseth said.
She said that at the University of Colorado, leaders set out to reach salary equity in a year and a half – and “it was a painful, painful process.” They found that different people held different beliefs about how people were paid, which led to a lot of unnecessary stress as they tried to construct a fairer system.
“On the back end of having done that, while it was a rough year and half, it has saved so much time – and I think built a culture of trust and transparency,” she said.
Recruiting in a more thoughtful way can also have a big impact, Dr. Spilseth said. The manner in which people are told about opportunities could exclude people without intending to.
“Are you casting a wide net?” she asked.
Adia Ross, MD, MHA, chief medical officer at Duke Regional Hospital, Durham, N.C., said that even in the face of obvious disparities, women can take steps on their own to boost their careers. She encouraged taking on “stretch assignments,” a project or task that is a bit beyond one’s current comfort level or level of experience or knowledge. “It can be a little scary, and sometimes there are bumps along the way,” she said.
All of these measures, though incremental, are the way to make bigger change, she said. “We want to take small steps but big strides forward.”
A version of this article first appeared on Medscape.com.
When Marisha Burden, MD, division head of hospital medicine at the University of Colorado at Denver, Aurora, would go to medical conferences, it seemed as if very few women were giving talks. She wondered if she could be wrong.
“I started doing my own assessments at every conference I would go to, just to make sure I wasn’t biased in my own belief system,” she said in a session at SHM Converge 2021, the annual conference of the Society of Hospital Medicine.
She wasn’t wrong.
In 2015, only 35% of all speakers at the SHM annual conference were women, and only 23% of the plenary speakers were women. In the years after that, when the society put out open calls for speakers, the numbers of women who spoke increased substantially, to 47% overall and 45% of plenary speakers.
The results – part of the SPEAK UP study Dr. Burden led in 2020 – show how gender disparity can be improved with a systematic process that is designed to improve it. The results of the study also showed that as the percentages of female speakers increased, the attendee ratings of the sessions did, too.
“You can do these things, and the quality of your conference doesn’t get negatively impacted – and in this case, actually improved,” Dr. Burden said.
That study marked progress toward leveling a traditionally uneven playing field when it comes to men and women in medicine, and the panelists in the session called on the field to use a variety of tools and strategies to continue toward something closer to equality.
Sara Spilseth, MD, MBA, chief of staff at Regions Hospital, in St. Paul, Minn., said it’s well established that although almost 50% of medical school students are women, the percentage shrinks each step from faculty to full professor to dean – of which only 16% are women. She referred to what’s known as the “leaky pipe.”
In what Dr. Spilseth said was one of her favorite studies, researchers in 2015 found that only 13% of clinical department leaders at the top 50 U.S. medical schools were women – they were outnumbered by the percentage of department leaders with mustaches, at 19%, even though mustaches are dwindling in popularity.
“Why does this exist? Why did we end up like this?” Part of the problem is a “respect gap,” she said, pointing to a study on the tendency of women to use the formal title of “doctor” when introducing male colleagues, whereas men who introduce women use that title less than half the time.
The COVID-19 pandemic has only made these disparities worse. Women are responsible for childcare much more frequently than men, Dr. Burden said, although the pandemic has brought caregiving duties to the forefront.
Dr. Spilseth said mentoring can help women navigate the workplace so as to help overcome these disparities. At Regions, the mentoring program is robust.
“Even before a new hire steps foot in the hospital, we have established them with a mentor,” she said. Sponsoring – the “ability of someone with political capital to use it to help colleagues” – can also help boost women’s careers, she said.
Her hospital also has a Women in Medicine Cooperative, which provides a way for women to talk about common struggles and to network.
Flexible work opportunities – working in transitional care units, being a physician advisor, and doing research – can all help boost a career as well, Dr. Spilseth said.
She said that at the University of Colorado, leaders set out to reach salary equity in a year and a half – and “it was a painful, painful process.” They found that different people held different beliefs about how people were paid, which led to a lot of unnecessary stress as they tried to construct a fairer system.
“On the back end of having done that, while it was a rough year and half, it has saved so much time – and I think built a culture of trust and transparency,” she said.
Recruiting in a more thoughtful way can also have a big impact, Dr. Spilseth said. The manner in which people are told about opportunities could exclude people without intending to.
“Are you casting a wide net?” she asked.
Adia Ross, MD, MHA, chief medical officer at Duke Regional Hospital, Durham, N.C., said that even in the face of obvious disparities, women can take steps on their own to boost their careers. She encouraged taking on “stretch assignments,” a project or task that is a bit beyond one’s current comfort level or level of experience or knowledge. “It can be a little scary, and sometimes there are bumps along the way,” she said.
All of these measures, though incremental, are the way to make bigger change, she said. “We want to take small steps but big strides forward.”
A version of this article first appeared on Medscape.com.
When Marisha Burden, MD, division head of hospital medicine at the University of Colorado at Denver, Aurora, would go to medical conferences, it seemed as if very few women were giving talks. She wondered if she could be wrong.
“I started doing my own assessments at every conference I would go to, just to make sure I wasn’t biased in my own belief system,” she said in a session at SHM Converge 2021, the annual conference of the Society of Hospital Medicine.
She wasn’t wrong.
In 2015, only 35% of all speakers at the SHM annual conference were women, and only 23% of the plenary speakers were women. In the years after that, when the society put out open calls for speakers, the numbers of women who spoke increased substantially, to 47% overall and 45% of plenary speakers.
The results – part of the SPEAK UP study Dr. Burden led in 2020 – show how gender disparity can be improved with a systematic process that is designed to improve it. The results of the study also showed that as the percentages of female speakers increased, the attendee ratings of the sessions did, too.
“You can do these things, and the quality of your conference doesn’t get negatively impacted – and in this case, actually improved,” Dr. Burden said.
That study marked progress toward leveling a traditionally uneven playing field when it comes to men and women in medicine, and the panelists in the session called on the field to use a variety of tools and strategies to continue toward something closer to equality.
Sara Spilseth, MD, MBA, chief of staff at Regions Hospital, in St. Paul, Minn., said it’s well established that although almost 50% of medical school students are women, the percentage shrinks each step from faculty to full professor to dean – of which only 16% are women. She referred to what’s known as the “leaky pipe.”
In what Dr. Spilseth said was one of her favorite studies, researchers in 2015 found that only 13% of clinical department leaders at the top 50 U.S. medical schools were women – they were outnumbered by the percentage of department leaders with mustaches, at 19%, even though mustaches are dwindling in popularity.
“Why does this exist? Why did we end up like this?” Part of the problem is a “respect gap,” she said, pointing to a study on the tendency of women to use the formal title of “doctor” when introducing male colleagues, whereas men who introduce women use that title less than half the time.
The COVID-19 pandemic has only made these disparities worse. Women are responsible for childcare much more frequently than men, Dr. Burden said, although the pandemic has brought caregiving duties to the forefront.
Dr. Spilseth said mentoring can help women navigate the workplace so as to help overcome these disparities. At Regions, the mentoring program is robust.
“Even before a new hire steps foot in the hospital, we have established them with a mentor,” she said. Sponsoring – the “ability of someone with political capital to use it to help colleagues” – can also help boost women’s careers, she said.
Her hospital also has a Women in Medicine Cooperative, which provides a way for women to talk about common struggles and to network.
Flexible work opportunities – working in transitional care units, being a physician advisor, and doing research – can all help boost a career as well, Dr. Spilseth said.
She said that at the University of Colorado, leaders set out to reach salary equity in a year and a half – and “it was a painful, painful process.” They found that different people held different beliefs about how people were paid, which led to a lot of unnecessary stress as they tried to construct a fairer system.
“On the back end of having done that, while it was a rough year and half, it has saved so much time – and I think built a culture of trust and transparency,” she said.
Recruiting in a more thoughtful way can also have a big impact, Dr. Spilseth said. The manner in which people are told about opportunities could exclude people without intending to.
“Are you casting a wide net?” she asked.
Adia Ross, MD, MHA, chief medical officer at Duke Regional Hospital, Durham, N.C., said that even in the face of obvious disparities, women can take steps on their own to boost their careers. She encouraged taking on “stretch assignments,” a project or task that is a bit beyond one’s current comfort level or level of experience or knowledge. “It can be a little scary, and sometimes there are bumps along the way,” she said.
All of these measures, though incremental, are the way to make bigger change, she said. “We want to take small steps but big strides forward.”
A version of this article first appeared on Medscape.com.
FROM SHM CONVERGE 2021
FDA approves secukinumab in psoriasis patients age six and older
The who are candidates for systemic therapy or phototherapy. The expanded indication marks the first time the drug has been available for a pediatric population in the United States.

Children with plaque psoriasis are often undertreated because of fear of side effects of therapies, according to Kelly M. Cordoro, MD, professor of dermatology and pediatrics at the University of California, San Francisco. “Now, more and more medicines are being tested for safety and efficacy in children, and we no longer have to rely on adult studies to inform treatment choices for children,” Dr. Cordoro told this news organization.
The FDA approval of secukinumab for children aged 6 and older with moderate to severe psoriasis “is a welcome addition to the therapeutic toolbox for pediatric psoriasis,” she said. “We’ve entered an era where severe pediatric psoriasis has become a condition that can be adequately controlled with minimal risk and with the convenience of intermittent injections. This has changed the playing field for these children and their families completely. Given the potential short- and long-term negative impact of chronic inflammation on the body of a growing child, we now have approved treatments that can safely offset the risks of undertreated severe psoriasis on the functional and psychological health of the child.”
The approved pediatric dosing for secukinumab is 75 mg or 150 mg depending on the child’s weight at the time of dosing, and it is administered by subcutaneous injection every 4 weeks after an initial loading regimen. According to a press release from Novartis, the FDA approval came on the heels of two phase 3 studies that evaluated the use of secukinumab in children aged 6 to younger than 18 years with plaque psoriasis. The first was a 52-week, randomized, double-blind, placebo- and active-controlled study which included 162 children 6 years of age and older with severe plaque psoriasis. The doses evaluated were 75 mg for children who weighed less than 50 kg and 150 mg for those 50 kg or greater.
At week 12, the Psoriasis Area Severity Index (PASI)-75 response was 55% among children in the 75-mg dosing group vs. 10% in the placebo group and 86% in the 150-mg dosing group vs. 19% in the placebo group.
Meanwhile, the Investigator’s Global Assessment modified 2011 (IGA) “clear” response was achieved in 32% of children in the 75-mg dosing group vs. 5% in the placebo group and in 81% of children in the 150-mg dosing group vs. 5% in the placebo group. An IGA “almost clear” skin response was achieved in 81% of children in the 75-mg dosing group vs. 5% in the placebo group.
The second phase 3 study was a randomized open-label, 208-week trial of 84 subjects 6 years of age and older with moderate to severe plaque psoriasis. According to the Novartis press release, the safety profile reported in both trials was consistent with the safety profile reported in adult plaque psoriasis trials and no new safety signals were observed. The updated prescribing information for secukinumab can be found here.
“When considering treatment with a systemic agent such as a biologic, it is important to consider objective measures of severity, such as extent of disease and involvement of joints but also subjective indicators of severity such as impact beyond the skin on psychological well-being,” Dr. Cordoro said in the interview. “Kids with psoriasis in visible locations may socially isolate themselves due to embarrassment or bullying. Therefore, the impact of moderate to severe psoriasis not only on overall health but on self-esteem and identity formation can be significant, and therefore adequately treating children of all ages to prevent the downstream negative consequences of childhood psoriasis is critical.”
Dr. Cordoro reported having no financial disclosures.
The who are candidates for systemic therapy or phototherapy. The expanded indication marks the first time the drug has been available for a pediatric population in the United States.
Children with plaque psoriasis are often undertreated because of fear of side effects of therapies, according to Kelly M. Cordoro, MD, professor of dermatology and pediatrics at the University of California, San Francisco. “Now, more and more medicines are being tested for safety and efficacy in children, and we no longer have to rely on adult studies to inform treatment choices for children,” Dr. Cordoro told this news organization.
The FDA approval of secukinumab for children aged 6 and older with moderate to severe psoriasis “is a welcome addition to the therapeutic toolbox for pediatric psoriasis,” she said. “We’ve entered an era where severe pediatric psoriasis has become a condition that can be adequately controlled with minimal risk and with the convenience of intermittent injections. This has changed the playing field for these children and their families completely. Given the potential short- and long-term negative impact of chronic inflammation on the body of a growing child, we now have approved treatments that can safely offset the risks of undertreated severe psoriasis on the functional and psychological health of the child.”
The approved pediatric dosing for secukinumab is 75 mg or 150 mg depending on the child’s weight at the time of dosing, and it is administered by subcutaneous injection every 4 weeks after an initial loading regimen. According to a press release from Novartis, the FDA approval came on the heels of two phase 3 studies that evaluated the use of secukinumab in children aged 6 to younger than 18 years with plaque psoriasis. The first was a 52-week, randomized, double-blind, placebo- and active-controlled study which included 162 children 6 years of age and older with severe plaque psoriasis. The doses evaluated were 75 mg for children who weighed less than 50 kg and 150 mg for those 50 kg or greater.
At week 12, the Psoriasis Area Severity Index (PASI)-75 response was 55% among children in the 75-mg dosing group vs. 10% in the placebo group and 86% in the 150-mg dosing group vs. 19% in the placebo group.
Meanwhile, the Investigator’s Global Assessment modified 2011 (IGA) “clear” response was achieved in 32% of children in the 75-mg dosing group vs. 5% in the placebo group and in 81% of children in the 150-mg dosing group vs. 5% in the placebo group. An IGA “almost clear” skin response was achieved in 81% of children in the 75-mg dosing group vs. 5% in the placebo group.
The second phase 3 study was a randomized open-label, 208-week trial of 84 subjects 6 years of age and older with moderate to severe plaque psoriasis. According to the Novartis press release, the safety profile reported in both trials was consistent with the safety profile reported in adult plaque psoriasis trials and no new safety signals were observed. The updated prescribing information for secukinumab can be found here.
“When considering treatment with a systemic agent such as a biologic, it is important to consider objective measures of severity, such as extent of disease and involvement of joints but also subjective indicators of severity such as impact beyond the skin on psychological well-being,” Dr. Cordoro said in the interview. “Kids with psoriasis in visible locations may socially isolate themselves due to embarrassment or bullying. Therefore, the impact of moderate to severe psoriasis not only on overall health but on self-esteem and identity formation can be significant, and therefore adequately treating children of all ages to prevent the downstream negative consequences of childhood psoriasis is critical.”
Dr. Cordoro reported having no financial disclosures.
The who are candidates for systemic therapy or phototherapy. The expanded indication marks the first time the drug has been available for a pediatric population in the United States.
Children with plaque psoriasis are often undertreated because of fear of side effects of therapies, according to Kelly M. Cordoro, MD, professor of dermatology and pediatrics at the University of California, San Francisco. “Now, more and more medicines are being tested for safety and efficacy in children, and we no longer have to rely on adult studies to inform treatment choices for children,” Dr. Cordoro told this news organization.
The FDA approval of secukinumab for children aged 6 and older with moderate to severe psoriasis “is a welcome addition to the therapeutic toolbox for pediatric psoriasis,” she said. “We’ve entered an era where severe pediatric psoriasis has become a condition that can be adequately controlled with minimal risk and with the convenience of intermittent injections. This has changed the playing field for these children and their families completely. Given the potential short- and long-term negative impact of chronic inflammation on the body of a growing child, we now have approved treatments that can safely offset the risks of undertreated severe psoriasis on the functional and psychological health of the child.”
The approved pediatric dosing for secukinumab is 75 mg or 150 mg depending on the child’s weight at the time of dosing, and it is administered by subcutaneous injection every 4 weeks after an initial loading regimen. According to a press release from Novartis, the FDA approval came on the heels of two phase 3 studies that evaluated the use of secukinumab in children aged 6 to younger than 18 years with plaque psoriasis. The first was a 52-week, randomized, double-blind, placebo- and active-controlled study which included 162 children 6 years of age and older with severe plaque psoriasis. The doses evaluated were 75 mg for children who weighed less than 50 kg and 150 mg for those 50 kg or greater.
At week 12, the Psoriasis Area Severity Index (PASI)-75 response was 55% among children in the 75-mg dosing group vs. 10% in the placebo group and 86% in the 150-mg dosing group vs. 19% in the placebo group.
Meanwhile, the Investigator’s Global Assessment modified 2011 (IGA) “clear” response was achieved in 32% of children in the 75-mg dosing group vs. 5% in the placebo group and in 81% of children in the 150-mg dosing group vs. 5% in the placebo group. An IGA “almost clear” skin response was achieved in 81% of children in the 75-mg dosing group vs. 5% in the placebo group.
The second phase 3 study was a randomized open-label, 208-week trial of 84 subjects 6 years of age and older with moderate to severe plaque psoriasis. According to the Novartis press release, the safety profile reported in both trials was consistent with the safety profile reported in adult plaque psoriasis trials and no new safety signals were observed. The updated prescribing information for secukinumab can be found here.
“When considering treatment with a systemic agent such as a biologic, it is important to consider objective measures of severity, such as extent of disease and involvement of joints but also subjective indicators of severity such as impact beyond the skin on psychological well-being,” Dr. Cordoro said in the interview. “Kids with psoriasis in visible locations may socially isolate themselves due to embarrassment or bullying. Therefore, the impact of moderate to severe psoriasis not only on overall health but on self-esteem and identity formation can be significant, and therefore adequately treating children of all ages to prevent the downstream negative consequences of childhood psoriasis is critical.”
Dr. Cordoro reported having no financial disclosures.
Phacomatosis Pigmentokeratotica Associated With Raynaud Phenomenon, Segmental Nevi, Hyperhidrosis, and Scoliosis
To the Editor:
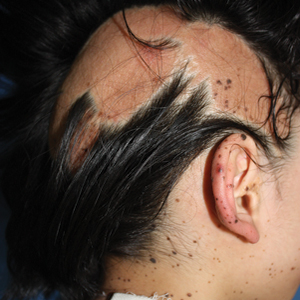
Phacomatosis pigmentokeratotica (PPK) is a rare epidermal nevus syndrome complicated by multiple extracutaneous anomalies, including skeletal defects and neurologic anomalies. Less common associations include lateral curvature of the spine and hyperhidrosis. We present a patient with PPK and unilateral Raynaud phenomenon in addition to a segmental distribution of melanocytic nevi, hyperhidrosis, and scoliosis.
A 9-year-old girl was born with a yellow-orange alopecic plaque on the right side of the scalp (Figure 1). There also were 2 large, irregularly pigmented patches localized on the right side of the upper back and buttock. Over 3 years, numerous papular nevi developed within these pigmented patches and were diagnosed as speckled lentiginous nevi (Figure 2). In addition, numerous nevi of various sizes affected the right face, right shoulder, right arm (Figure 3), and right neck and were clearly demarcated along the midline. Several nevi also were noted within the nevus sebaceous on the right scalp. These skin lesions expanded progressively with age. At 6 years of age, she was diagnosed with hyperhidrosis of the right half of the body, which was most pronounced on the face. Raynaud phenomenon restricted to the right hand also was noted (Figure 4). Upon cold exposure, the digits become pale white, cold, and numb; then blue; and finally red. She lacked other features of connective tissue disease, and autoantibody testing was negative. She also was noted to have an abnormal lateral curvature of the spine (scoliosis). Auditory, ocular, and neurologic examinations were normal. Cranial and cerebral magnetic resonance imaging showed no central nervous system abnormalities. Her family history was negative for nevus spilus, nevus sebaceous, and neurofibromatosis. The clinical findings in our patient led to the diagnosis of PPK.
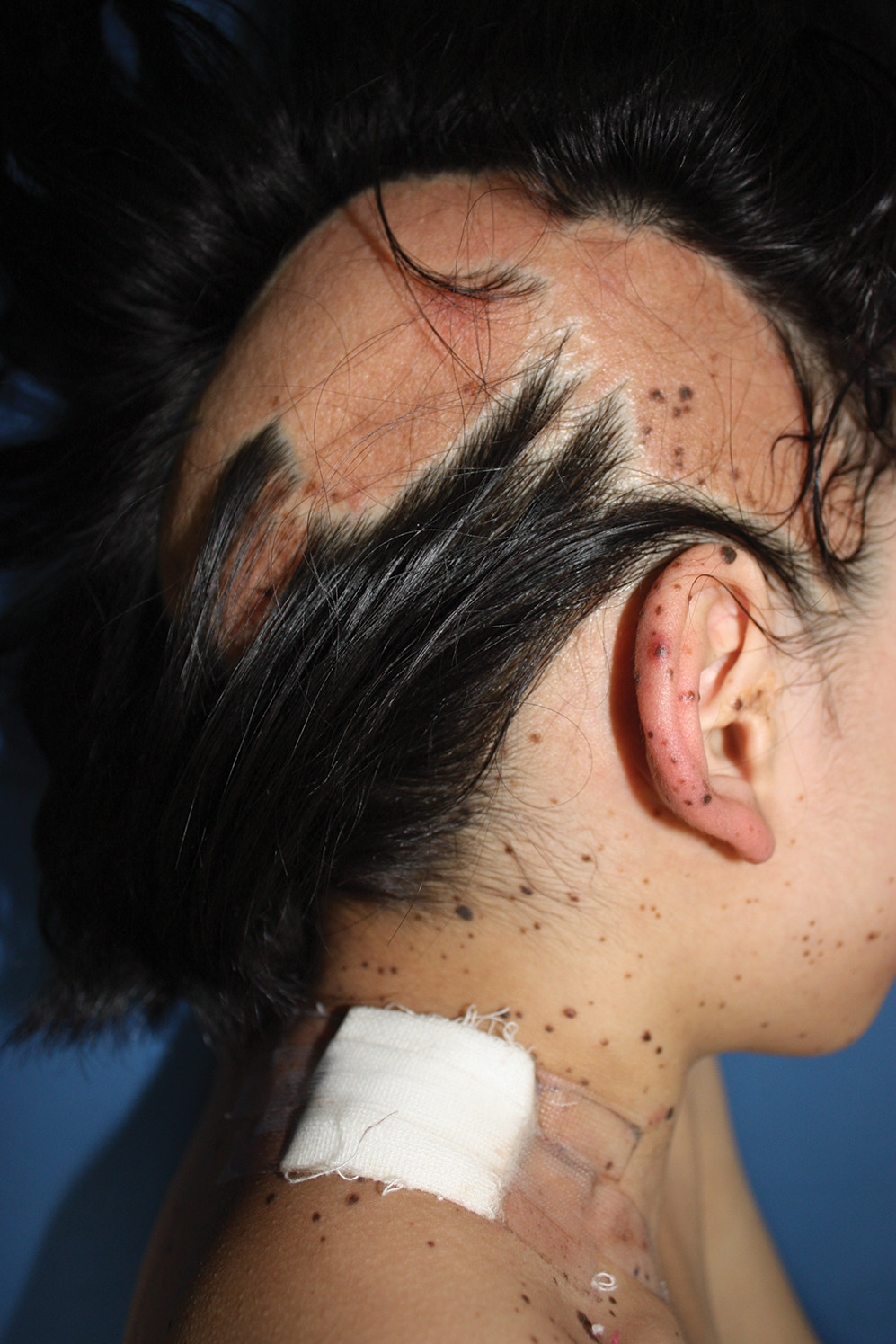
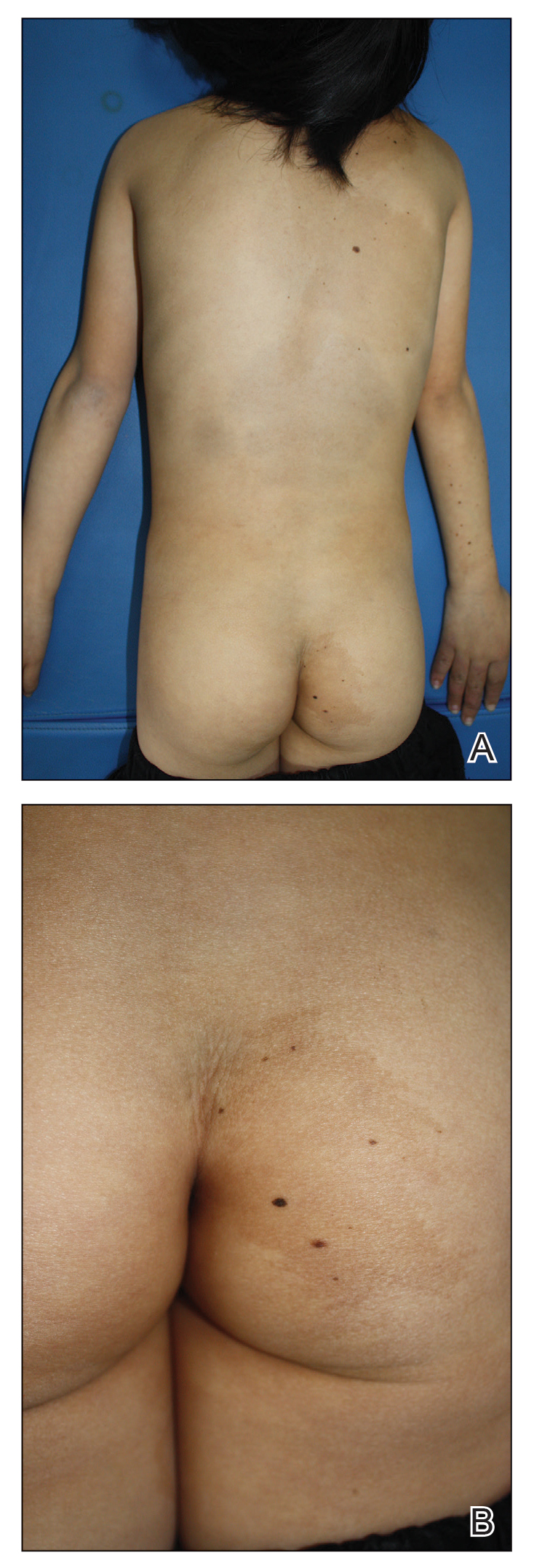
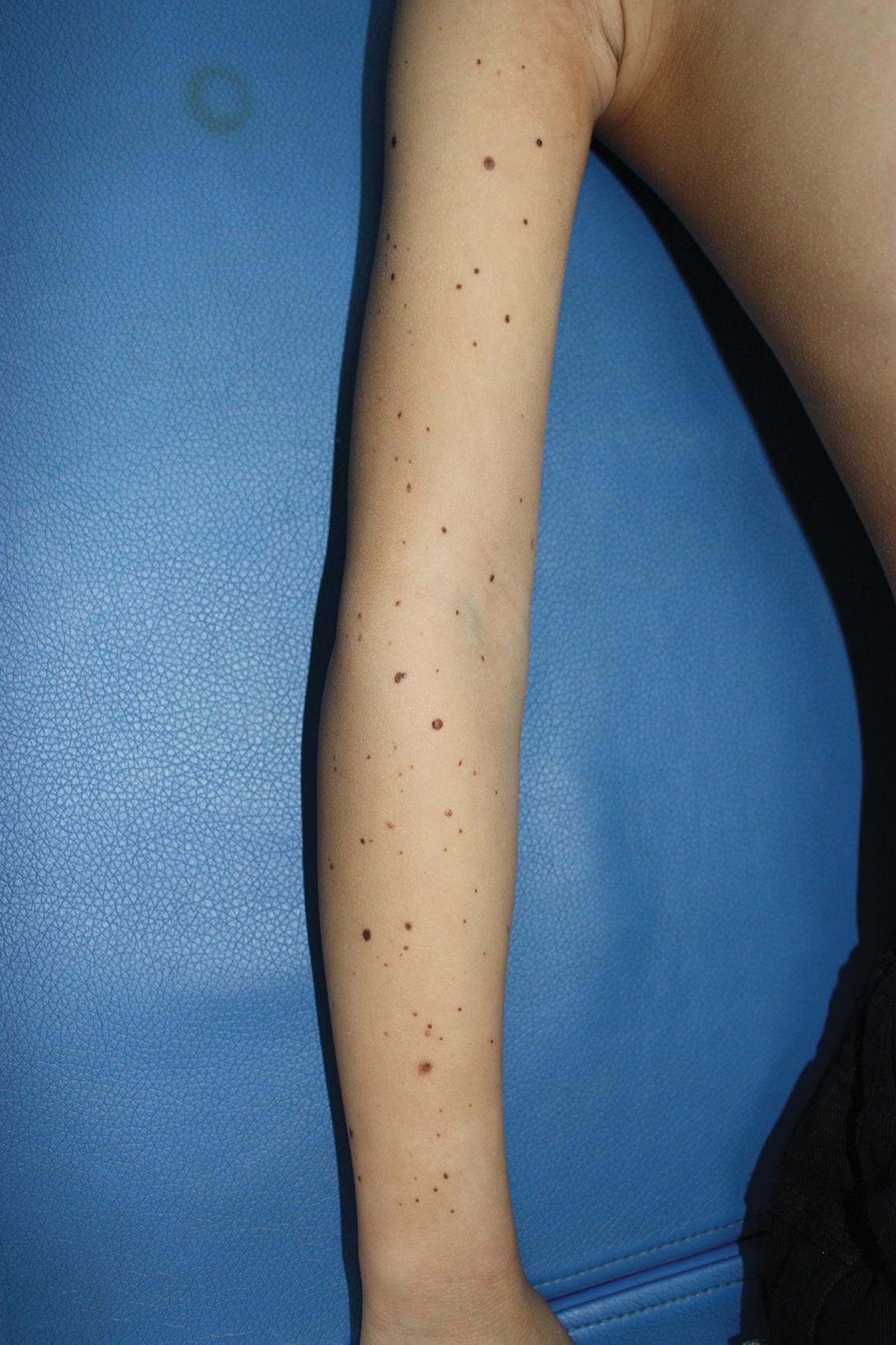
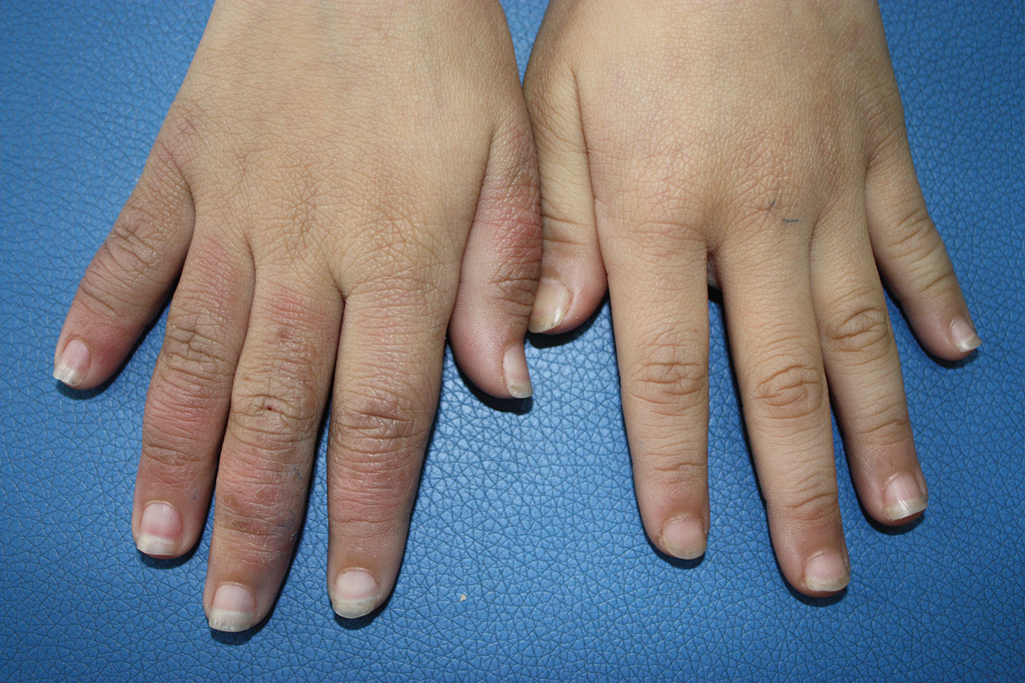
Phacomatosis pigmentokeratotica is a distinctive epidermal nevus syndrome characterized by the coexistence of a speckled lentiginous nevus, also known as a nevus spilus, and a nevus sebaceous1; PPK frequently is complicated by skeletal, ophthalmic, or neurologic abnormalities.2 Most cases reported are sporadic, and a postzygotic mosaic HRas proto-oncogene, GTPase, HRAS, mutation has been demonstrated in some patients and may contribute to the phenotype of PPK.3,4
Other anomalies have included ichthyosislike diffuse hyperkeratosis, laxity of the hands, pelvic hypoplasia, glaucoma, psychomotor retardation, and hypophosphatemic rickets. These patients also should be monitored for the development of malignant neoplasms within the nevus sebaceous.5 Segmental hyperhidrosis may be seen in association with the nevus spilus component.2
Raynaud phenomenon involving only the right hand was a unique finding in our patient. In 3 years of follow-up, our patient developed no evidence of connective tissue disease or other systemic illness. We speculate that Raynaud phenomenon of the right hand along with hyperhidrosis of the right side of the body could be a result of dysfunction of the autonomic nervous system. We propose that Raynaud phenomenon represents an unusual manifestation of PPK and may broaden the spectrum of extracutaneous anomalies associated with the disease. The finding of segmental nevi outside of the confines of the nevus spilus was another unusual manifestation of mosaicism.
- Happle R, Hoffmann R, Restano L, et al. Phacomatosis pigmentokeratotica: a melanocytic-epidermal twin nevus syndrome. Am J Med Genet. 1996;65:363-365.
- Happle R. The group of epidermal nevus syndromes part I. well defined phenotypes. J Am Acad Dermatol. 2010;63:1-22, 23-24.
- Groesser L, Herschberger E, Sagrera A, et al. Phacomatosis pigmentokeratotica is caused by a postzygotic HRAS mutation in a multipotent progenitor cell. J Invest Dermatol. 2013;133:1998-2003.
- Martin RJ, Arefi M, Splitt M, et al. Phacomatosis pigmentokeratotica and precocious puberty associated with HRAS mutation. Br J Dermatol. 2018;178:289-291.
- Chu GY, Wu CY. Phacomatosis pigmentokeratotica: a follow-up report with fatal outcome. Acta Derm Venereol. 2014;94:467-468.
To the Editor:
Phacomatosis pigmentokeratotica (PPK) is a rare epidermal nevus syndrome complicated by multiple extracutaneous anomalies, including skeletal defects and neurologic anomalies. Less common associations include lateral curvature of the spine and hyperhidrosis. We present a patient with PPK and unilateral Raynaud phenomenon in addition to a segmental distribution of melanocytic nevi, hyperhidrosis, and scoliosis.
A 9-year-old girl was born with a yellow-orange alopecic plaque on the right side of the scalp (Figure 1). There also were 2 large, irregularly pigmented patches localized on the right side of the upper back and buttock. Over 3 years, numerous papular nevi developed within these pigmented patches and were diagnosed as speckled lentiginous nevi (Figure 2). In addition, numerous nevi of various sizes affected the right face, right shoulder, right arm (Figure 3), and right neck and were clearly demarcated along the midline. Several nevi also were noted within the nevus sebaceous on the right scalp. These skin lesions expanded progressively with age. At 6 years of age, she was diagnosed with hyperhidrosis of the right half of the body, which was most pronounced on the face. Raynaud phenomenon restricted to the right hand also was noted (Figure 4). Upon cold exposure, the digits become pale white, cold, and numb; then blue; and finally red. She lacked other features of connective tissue disease, and autoantibody testing was negative. She also was noted to have an abnormal lateral curvature of the spine (scoliosis). Auditory, ocular, and neurologic examinations were normal. Cranial and cerebral magnetic resonance imaging showed no central nervous system abnormalities. Her family history was negative for nevus spilus, nevus sebaceous, and neurofibromatosis. The clinical findings in our patient led to the diagnosis of PPK.
Phacomatosis pigmentokeratotica is a distinctive epidermal nevus syndrome characterized by the coexistence of a speckled lentiginous nevus, also known as a nevus spilus, and a nevus sebaceous1; PPK frequently is complicated by skeletal, ophthalmic, or neurologic abnormalities.2 Most cases reported are sporadic, and a postzygotic mosaic HRas proto-oncogene, GTPase, HRAS, mutation has been demonstrated in some patients and may contribute to the phenotype of PPK.3,4
Other anomalies have included ichthyosislike diffuse hyperkeratosis, laxity of the hands, pelvic hypoplasia, glaucoma, psychomotor retardation, and hypophosphatemic rickets. These patients also should be monitored for the development of malignant neoplasms within the nevus sebaceous.5 Segmental hyperhidrosis may be seen in association with the nevus spilus component.2
Raynaud phenomenon involving only the right hand was a unique finding in our patient. In 3 years of follow-up, our patient developed no evidence of connective tissue disease or other systemic illness. We speculate that Raynaud phenomenon of the right hand along with hyperhidrosis of the right side of the body could be a result of dysfunction of the autonomic nervous system. We propose that Raynaud phenomenon represents an unusual manifestation of PPK and may broaden the spectrum of extracutaneous anomalies associated with the disease. The finding of segmental nevi outside of the confines of the nevus spilus was another unusual manifestation of mosaicism.
To the Editor:
Phacomatosis pigmentokeratotica (PPK) is a rare epidermal nevus syndrome complicated by multiple extracutaneous anomalies, including skeletal defects and neurologic anomalies. Less common associations include lateral curvature of the spine and hyperhidrosis. We present a patient with PPK and unilateral Raynaud phenomenon in addition to a segmental distribution of melanocytic nevi, hyperhidrosis, and scoliosis.
A 9-year-old girl was born with a yellow-orange alopecic plaque on the right side of the scalp (Figure 1). There also were 2 large, irregularly pigmented patches localized on the right side of the upper back and buttock. Over 3 years, numerous papular nevi developed within these pigmented patches and were diagnosed as speckled lentiginous nevi (Figure 2). In addition, numerous nevi of various sizes affected the right face, right shoulder, right arm (Figure 3), and right neck and were clearly demarcated along the midline. Several nevi also were noted within the nevus sebaceous on the right scalp. These skin lesions expanded progressively with age. At 6 years of age, she was diagnosed with hyperhidrosis of the right half of the body, which was most pronounced on the face. Raynaud phenomenon restricted to the right hand also was noted (Figure 4). Upon cold exposure, the digits become pale white, cold, and numb; then blue; and finally red. She lacked other features of connective tissue disease, and autoantibody testing was negative. She also was noted to have an abnormal lateral curvature of the spine (scoliosis). Auditory, ocular, and neurologic examinations were normal. Cranial and cerebral magnetic resonance imaging showed no central nervous system abnormalities. Her family history was negative for nevus spilus, nevus sebaceous, and neurofibromatosis. The clinical findings in our patient led to the diagnosis of PPK.
Phacomatosis pigmentokeratotica is a distinctive epidermal nevus syndrome characterized by the coexistence of a speckled lentiginous nevus, also known as a nevus spilus, and a nevus sebaceous1; PPK frequently is complicated by skeletal, ophthalmic, or neurologic abnormalities.2 Most cases reported are sporadic, and a postzygotic mosaic HRas proto-oncogene, GTPase, HRAS, mutation has been demonstrated in some patients and may contribute to the phenotype of PPK.3,4
Other anomalies have included ichthyosislike diffuse hyperkeratosis, laxity of the hands, pelvic hypoplasia, glaucoma, psychomotor retardation, and hypophosphatemic rickets. These patients also should be monitored for the development of malignant neoplasms within the nevus sebaceous.5 Segmental hyperhidrosis may be seen in association with the nevus spilus component.2
Raynaud phenomenon involving only the right hand was a unique finding in our patient. In 3 years of follow-up, our patient developed no evidence of connective tissue disease or other systemic illness. We speculate that Raynaud phenomenon of the right hand along with hyperhidrosis of the right side of the body could be a result of dysfunction of the autonomic nervous system. We propose that Raynaud phenomenon represents an unusual manifestation of PPK and may broaden the spectrum of extracutaneous anomalies associated with the disease. The finding of segmental nevi outside of the confines of the nevus spilus was another unusual manifestation of mosaicism.
- Happle R, Hoffmann R, Restano L, et al. Phacomatosis pigmentokeratotica: a melanocytic-epidermal twin nevus syndrome. Am J Med Genet. 1996;65:363-365.
- Happle R. The group of epidermal nevus syndromes part I. well defined phenotypes. J Am Acad Dermatol. 2010;63:1-22, 23-24.
- Groesser L, Herschberger E, Sagrera A, et al. Phacomatosis pigmentokeratotica is caused by a postzygotic HRAS mutation in a multipotent progenitor cell. J Invest Dermatol. 2013;133:1998-2003.
- Martin RJ, Arefi M, Splitt M, et al. Phacomatosis pigmentokeratotica and precocious puberty associated with HRAS mutation. Br J Dermatol. 2018;178:289-291.
- Chu GY, Wu CY. Phacomatosis pigmentokeratotica: a follow-up report with fatal outcome. Acta Derm Venereol. 2014;94:467-468.
- Happle R, Hoffmann R, Restano L, et al. Phacomatosis pigmentokeratotica: a melanocytic-epidermal twin nevus syndrome. Am J Med Genet. 1996;65:363-365.
- Happle R. The group of epidermal nevus syndromes part I. well defined phenotypes. J Am Acad Dermatol. 2010;63:1-22, 23-24.
- Groesser L, Herschberger E, Sagrera A, et al. Phacomatosis pigmentokeratotica is caused by a postzygotic HRAS mutation in a multipotent progenitor cell. J Invest Dermatol. 2013;133:1998-2003.
- Martin RJ, Arefi M, Splitt M, et al. Phacomatosis pigmentokeratotica and precocious puberty associated with HRAS mutation. Br J Dermatol. 2018;178:289-291.
- Chu GY, Wu CY. Phacomatosis pigmentokeratotica: a follow-up report with fatal outcome. Acta Derm Venereol. 2014;94:467-468.
Practice Points
- Phacomatosis pigmentokeratotica (PPK) is characterized by the coexistence of speckled lentiginous nevus and nevus sebaceous.
- Raynaud phenomenon may be an unreported association with PPK.
FDA approves diagnostic device for autism spectrum disorder
The Food and Drug Administration has approved marketing for a device that will help diagnose autism spectrum disorder (ASD) in children between the ages of 18 months and 5 years old who exhibit potential symptoms.
Cognoa ASD Diagnosis Aid is a machine learning–based software program that receives information from parents or caregivers, video analysts, and health care providers to assist physicians in evaluating whether a child is at risk of having autism.
Autism is a developmental disorder that can cause social, communication, and behavioral challenges, according to the Centers for Disease Control and Prevention. The disorder affects about 1 in 54 children. The disorder is difficult to diagnose because there isn’t a medical test to diagnose the it. Instead, physicians have to look at a child’s developmental history and behavior to make a diagnosis.
Many children are not diagnosed with ASD until later in childhood, which in some cases delays treatment and early intervention. ASD may be detected as early as 18 months, but the average age of diagnosis for ASD is 4.3 years, according to the FDA.
“[ASD] can delay a child’s physical, cognitive, and social development, including motor skill development, learning, communication, and interacting with others. The earlier ASD can be diagnosed, the more quickly intervention strategies and appropriate therapies can begin,” Jeff Shuren, MD, JD, director of the FDA’s Center for Devices and Radiological Health, said in a statement. “Today’s marketing authorization provides a new tool for helping diagnose children with ASD.”
The safety and efficacy of the Cognoa ASD Diagnosis Aid was assessed in a study of 425 patients between the ages of 18 months and 5 years old. For the study, researchers compared the diagnostic assessments made by the device to those made by a panel of clinical experts who used the current standard ASD diagnostic process. The device diagnosed 32% of the children with either a “Positive for ASD” or a “Negative for ASD” result. Researchers found that the device matched the panel’s conclusions for 81% of the patients who received a positive diagnosis. For those who received a negative diagnosis, the device matched the panel’s conclusions for 98% of the patients. In addition, the device made an accurate ASD determination in 98.4% of patients with the condition and in 78.9% of patients without the condition.
Cognoa ASD Diagnosis Aid has three main components. One component includes a mobile app for caregivers to answer questions about the child’s behavioral problems and to upload videos of the child. The next component is a video analysis portal for specialists to view and analyze uploaded videos of patients. Another component is a portal for health care providers that allows them to enter answers to preloaded questions about behavior problems, track the information provided by parents, and review a report of the results.
After the machine learning–based device processes the information provided by parents and health care providers, it reports either a positive or a negative diagnosis. If there is insufficient information to make either a positive or a negative diagnosis, the ASD Diagnostic AID will report that no result can be generated.
Some of the risks associated with this device include misdiagnosis and delayed diagnosis of ASD because of a false-positive or false-negative result, or when no result is generated. Researchers said a false-positive result occurred in 15 out of 303 study subjects without ASD and a false-negative result occurred in 1 out of 122 study subjects with ASD.
The FDA emphasized that the device is indicated to aid physicians in the process of diagnosing ASD in children. This means it shouldn’t be treated as a standalone diagnostic device, but as an adjunct to the diagnostic process.
The Food and Drug Administration has approved marketing for a device that will help diagnose autism spectrum disorder (ASD) in children between the ages of 18 months and 5 years old who exhibit potential symptoms.
Cognoa ASD Diagnosis Aid is a machine learning–based software program that receives information from parents or caregivers, video analysts, and health care providers to assist physicians in evaluating whether a child is at risk of having autism.
Autism is a developmental disorder that can cause social, communication, and behavioral challenges, according to the Centers for Disease Control and Prevention. The disorder affects about 1 in 54 children. The disorder is difficult to diagnose because there isn’t a medical test to diagnose the it. Instead, physicians have to look at a child’s developmental history and behavior to make a diagnosis.
Many children are not diagnosed with ASD until later in childhood, which in some cases delays treatment and early intervention. ASD may be detected as early as 18 months, but the average age of diagnosis for ASD is 4.3 years, according to the FDA.
“[ASD] can delay a child’s physical, cognitive, and social development, including motor skill development, learning, communication, and interacting with others. The earlier ASD can be diagnosed, the more quickly intervention strategies and appropriate therapies can begin,” Jeff Shuren, MD, JD, director of the FDA’s Center for Devices and Radiological Health, said in a statement. “Today’s marketing authorization provides a new tool for helping diagnose children with ASD.”
The safety and efficacy of the Cognoa ASD Diagnosis Aid was assessed in a study of 425 patients between the ages of 18 months and 5 years old. For the study, researchers compared the diagnostic assessments made by the device to those made by a panel of clinical experts who used the current standard ASD diagnostic process. The device diagnosed 32% of the children with either a “Positive for ASD” or a “Negative for ASD” result. Researchers found that the device matched the panel’s conclusions for 81% of the patients who received a positive diagnosis. For those who received a negative diagnosis, the device matched the panel’s conclusions for 98% of the patients. In addition, the device made an accurate ASD determination in 98.4% of patients with the condition and in 78.9% of patients without the condition.
Cognoa ASD Diagnosis Aid has three main components. One component includes a mobile app for caregivers to answer questions about the child’s behavioral problems and to upload videos of the child. The next component is a video analysis portal for specialists to view and analyze uploaded videos of patients. Another component is a portal for health care providers that allows them to enter answers to preloaded questions about behavior problems, track the information provided by parents, and review a report of the results.
After the machine learning–based device processes the information provided by parents and health care providers, it reports either a positive or a negative diagnosis. If there is insufficient information to make either a positive or a negative diagnosis, the ASD Diagnostic AID will report that no result can be generated.
Some of the risks associated with this device include misdiagnosis and delayed diagnosis of ASD because of a false-positive or false-negative result, or when no result is generated. Researchers said a false-positive result occurred in 15 out of 303 study subjects without ASD and a false-negative result occurred in 1 out of 122 study subjects with ASD.
The FDA emphasized that the device is indicated to aid physicians in the process of diagnosing ASD in children. This means it shouldn’t be treated as a standalone diagnostic device, but as an adjunct to the diagnostic process.
The Food and Drug Administration has approved marketing for a device that will help diagnose autism spectrum disorder (ASD) in children between the ages of 18 months and 5 years old who exhibit potential symptoms.
Cognoa ASD Diagnosis Aid is a machine learning–based software program that receives information from parents or caregivers, video analysts, and health care providers to assist physicians in evaluating whether a child is at risk of having autism.
Autism is a developmental disorder that can cause social, communication, and behavioral challenges, according to the Centers for Disease Control and Prevention. The disorder affects about 1 in 54 children. The disorder is difficult to diagnose because there isn’t a medical test to diagnose the it. Instead, physicians have to look at a child’s developmental history and behavior to make a diagnosis.
Many children are not diagnosed with ASD until later in childhood, which in some cases delays treatment and early intervention. ASD may be detected as early as 18 months, but the average age of diagnosis for ASD is 4.3 years, according to the FDA.
“[ASD] can delay a child’s physical, cognitive, and social development, including motor skill development, learning, communication, and interacting with others. The earlier ASD can be diagnosed, the more quickly intervention strategies and appropriate therapies can begin,” Jeff Shuren, MD, JD, director of the FDA’s Center for Devices and Radiological Health, said in a statement. “Today’s marketing authorization provides a new tool for helping diagnose children with ASD.”
The safety and efficacy of the Cognoa ASD Diagnosis Aid was assessed in a study of 425 patients between the ages of 18 months and 5 years old. For the study, researchers compared the diagnostic assessments made by the device to those made by a panel of clinical experts who used the current standard ASD diagnostic process. The device diagnosed 32% of the children with either a “Positive for ASD” or a “Negative for ASD” result. Researchers found that the device matched the panel’s conclusions for 81% of the patients who received a positive diagnosis. For those who received a negative diagnosis, the device matched the panel’s conclusions for 98% of the patients. In addition, the device made an accurate ASD determination in 98.4% of patients with the condition and in 78.9% of patients without the condition.
Cognoa ASD Diagnosis Aid has three main components. One component includes a mobile app for caregivers to answer questions about the child’s behavioral problems and to upload videos of the child. The next component is a video analysis portal for specialists to view and analyze uploaded videos of patients. Another component is a portal for health care providers that allows them to enter answers to preloaded questions about behavior problems, track the information provided by parents, and review a report of the results.
After the machine learning–based device processes the information provided by parents and health care providers, it reports either a positive or a negative diagnosis. If there is insufficient information to make either a positive or a negative diagnosis, the ASD Diagnostic AID will report that no result can be generated.
Some of the risks associated with this device include misdiagnosis and delayed diagnosis of ASD because of a false-positive or false-negative result, or when no result is generated. Researchers said a false-positive result occurred in 15 out of 303 study subjects without ASD and a false-negative result occurred in 1 out of 122 study subjects with ASD.
The FDA emphasized that the device is indicated to aid physicians in the process of diagnosing ASD in children. This means it shouldn’t be treated as a standalone diagnostic device, but as an adjunct to the diagnostic process.
The Power of a Multidisciplinary Tumor Board: Managing Unresectable and/or High-Risk Skin Cancers
Multidisciplinary tumor boards are composed of providers from many fields who deliver coordinated care for patients with unresectable and high-risk skin cancers. Providers who comprise the tumor board often are radiation oncologists, hematologists/oncologists, general surgeons, dermatologists, dermatologic surgeons, and pathologists. The benefit of having a tumor board is that each patient is evaluated simultaneously by a group of physicians from various specialties who bring diverse perspectives that will contribute to the overall treatment plan. The cases often encompass high-risk tumors including unresectable basal cell carcinomas or invasive melanomas. By combining knowledge from each specialty in a team approach, the tumor board can effectively and holistically develop a care plan for each patient.
For the tumor board at the Warren Alpert Medical School of Brown University (Providence, Rhode Island), we often prepare a presentation with comprehensive details about the patient and tumor. During the presentation, we also propose a treatment plan prior to describing each patient at the weekly conference and amend the plans during the discussion. Tumor boards also provide a consulting role to the community and hospital providers in which patients are being referred by their primary provider and are seeking a second opinion or guidance.
In many ways, the tumor board is a multidisciplinary approach for patient advocacy in the form of treatment. These physicians meet on a regular basis to check on the patient’s progress and continually reevaluate how to have discussions about the patient’s care. There are many reasons why it is important to refer patients to a multidisciplinary tumor board.
Improved Workup and Diagnosis
One of the values of a tumor board is that it allows for patient data to be collected and assembled in a way that tells a story. The specialist from each field can then discuss and weigh the benefits and risks for each diagnostic test that should be performed for the workup in each patient. Physicians who refer their patients to the tumor board use their recommendations to both confirm the diagnosis and shift their treatment plans, depending on the information presented during the meeting.1 There may be a change in the tumor type, decision to refer for surgery, cancer staging, and list of viable options, especially after reviewing pathology and imaging.2 The discussion of the treatment plan may consider not only surgical considerations but also the patient’s quality of life. At times, noninvasive interventions are more appropriate and align with the patient’s goals of care. In addition, during the tumor board clinic there may be new tumors that are identified and biopsied, providing increased diagnosis and surveillance for patients who may have a higher risk for developing skin cancer.
Education for Residents and Providers
The multidisciplinary tumor board not only helps patients but also educates both residents and providers on the evidence-based therapeutic management of high-risk tumors.2 Research literature on cutaneous oncology is dynamic, and the weekly tumor board meetings help providers stay informed about the best and most effective treatments for their patients.3 In addition to the attending specialists, participants of the tumor board also may include residents, medical students, medical assistance staff, nurses, physician assistants, and fellows. Furthermore, the recommendations given by the tumor board serve to educate both the patient and the provider who referred them to the tumor board. Although we have access to excellent dermatology textbooks as residents, the most impactful educational experience is seeing the patients in tumor board clinic and participating in the immensely educational discussions at the weekly conferences. Through this experience, I have learned that treatment plans should be personalized to the patient. There are many factors to take into consideration when deciphering what the best course of treatment will be for a patient. Sometimes the best option is Mohs micrographic surgery, while other times it may be scheduling several sessions of palliative radiation oncology. Treatment depends on the individual patient and their condition.
Coordination of Care
During a week that I was on call, I was consulted to biopsy a patient with a giant hemorrhagic basal cell carcinoma that caused substantial cheek and nose distortion as well as anemia secondary to acute blood loss. The patient not only did not have a dermatologist but also did not have a primary care physician given he had not had contact with the health care system in more than 30 years. The reason for him not seeking care was multifactorial, but the approach to his care became multidisciplinary. We sought to connect him with the right providers to help him in any way that we could. We presented him at our multidisciplinary tumor board and started him on sonedigib, a medication that binds to and inhibits the smoothened protein.4 Through the tumor board, we were able to establish sustained contact with the patient. The tumor board created effective communication between providers to get him the referrals that he needed for dermatology, pathology, radiation oncology, hematology/oncology, and otolaryngology. The discussions centered around being cognizant of the patient’s apprehension with the health care system as well as providing medical and surgical treatment that would help his quality of life. We built a consensus on what the best plan was for the patient and his family. This consensus would have been more difficult had it not been for the combined specialties of the tumor board. In general, studies have shown that weekly tumor boards have resulted in decreased mortality rates for patients with advanced cancers.5
Final Thoughts
The multidisciplinary tumor board is a powerful resource for hospitals and the greater medical community. At these weekly conferences you realize there may still be hope that begins at the line where your expertise ends. It represents a team of providers who compassionately refuse to give up on patients when they are the last refuge.
- Foster TJ, Bouchard-Fortier A, Olivotto IA, et al. Effect of multidisciplinary case conferences on physician decision making: breast diagnostic rounds. Cureus. 2016;8:E895.
- El Saghir NS, Charara RN, Kreidieh FY, et al. Global practice and efficiency of multidisciplinary tumor boards: results of an American Society of Clinical Oncology international survey. J Glob Oncol. 2015;1:57-64.
- Mori S, Navarrete-Dechent C, Petukhova TA, et al. Tumor board conferences for multidisciplinary skin cancer management: a survey of US cancer centers. J Natl Compr Canc Netw. 2018;16:1209-1215.
- Dummer R, Ascierto PA, Basset-Seguin N, et al. Sonidegib and vismodegib in the treatment of patients with locally advanced basal cell carcinoma: a joint expert opinion. J Eur Acad Dermatol Venereol. 2020;34:1944-1956.
- Kehl KL, Landrum MB, Kahn KL, et al. Tumor board participation among physicians caring for patients with lung or colorectal cancer. J Oncol Pract. 2015;11:E267-E278.
Multidisciplinary tumor boards are composed of providers from many fields who deliver coordinated care for patients with unresectable and high-risk skin cancers. Providers who comprise the tumor board often are radiation oncologists, hematologists/oncologists, general surgeons, dermatologists, dermatologic surgeons, and pathologists. The benefit of having a tumor board is that each patient is evaluated simultaneously by a group of physicians from various specialties who bring diverse perspectives that will contribute to the overall treatment plan. The cases often encompass high-risk tumors including unresectable basal cell carcinomas or invasive melanomas. By combining knowledge from each specialty in a team approach, the tumor board can effectively and holistically develop a care plan for each patient.
For the tumor board at the Warren Alpert Medical School of Brown University (Providence, Rhode Island), we often prepare a presentation with comprehensive details about the patient and tumor. During the presentation, we also propose a treatment plan prior to describing each patient at the weekly conference and amend the plans during the discussion. Tumor boards also provide a consulting role to the community and hospital providers in which patients are being referred by their primary provider and are seeking a second opinion or guidance.
In many ways, the tumor board is a multidisciplinary approach for patient advocacy in the form of treatment. These physicians meet on a regular basis to check on the patient’s progress and continually reevaluate how to have discussions about the patient’s care. There are many reasons why it is important to refer patients to a multidisciplinary tumor board.
Improved Workup and Diagnosis
One of the values of a tumor board is that it allows for patient data to be collected and assembled in a way that tells a story. The specialist from each field can then discuss and weigh the benefits and risks for each diagnostic test that should be performed for the workup in each patient. Physicians who refer their patients to the tumor board use their recommendations to both confirm the diagnosis and shift their treatment plans, depending on the information presented during the meeting.1 There may be a change in the tumor type, decision to refer for surgery, cancer staging, and list of viable options, especially after reviewing pathology and imaging.2 The discussion of the treatment plan may consider not only surgical considerations but also the patient’s quality of life. At times, noninvasive interventions are more appropriate and align with the patient’s goals of care. In addition, during the tumor board clinic there may be new tumors that are identified and biopsied, providing increased diagnosis and surveillance for patients who may have a higher risk for developing skin cancer.
Education for Residents and Providers
The multidisciplinary tumor board not only helps patients but also educates both residents and providers on the evidence-based therapeutic management of high-risk tumors.2 Research literature on cutaneous oncology is dynamic, and the weekly tumor board meetings help providers stay informed about the best and most effective treatments for their patients.3 In addition to the attending specialists, participants of the tumor board also may include residents, medical students, medical assistance staff, nurses, physician assistants, and fellows. Furthermore, the recommendations given by the tumor board serve to educate both the patient and the provider who referred them to the tumor board. Although we have access to excellent dermatology textbooks as residents, the most impactful educational experience is seeing the patients in tumor board clinic and participating in the immensely educational discussions at the weekly conferences. Through this experience, I have learned that treatment plans should be personalized to the patient. There are many factors to take into consideration when deciphering what the best course of treatment will be for a patient. Sometimes the best option is Mohs micrographic surgery, while other times it may be scheduling several sessions of palliative radiation oncology. Treatment depends on the individual patient and their condition.
Coordination of Care
During a week that I was on call, I was consulted to biopsy a patient with a giant hemorrhagic basal cell carcinoma that caused substantial cheek and nose distortion as well as anemia secondary to acute blood loss. The patient not only did not have a dermatologist but also did not have a primary care physician given he had not had contact with the health care system in more than 30 years. The reason for him not seeking care was multifactorial, but the approach to his care became multidisciplinary. We sought to connect him with the right providers to help him in any way that we could. We presented him at our multidisciplinary tumor board and started him on sonedigib, a medication that binds to and inhibits the smoothened protein.4 Through the tumor board, we were able to establish sustained contact with the patient. The tumor board created effective communication between providers to get him the referrals that he needed for dermatology, pathology, radiation oncology, hematology/oncology, and otolaryngology. The discussions centered around being cognizant of the patient’s apprehension with the health care system as well as providing medical and surgical treatment that would help his quality of life. We built a consensus on what the best plan was for the patient and his family. This consensus would have been more difficult had it not been for the combined specialties of the tumor board. In general, studies have shown that weekly tumor boards have resulted in decreased mortality rates for patients with advanced cancers.5
Final Thoughts
The multidisciplinary tumor board is a powerful resource for hospitals and the greater medical community. At these weekly conferences you realize there may still be hope that begins at the line where your expertise ends. It represents a team of providers who compassionately refuse to give up on patients when they are the last refuge.
Multidisciplinary tumor boards are composed of providers from many fields who deliver coordinated care for patients with unresectable and high-risk skin cancers. Providers who comprise the tumor board often are radiation oncologists, hematologists/oncologists, general surgeons, dermatologists, dermatologic surgeons, and pathologists. The benefit of having a tumor board is that each patient is evaluated simultaneously by a group of physicians from various specialties who bring diverse perspectives that will contribute to the overall treatment plan. The cases often encompass high-risk tumors including unresectable basal cell carcinomas or invasive melanomas. By combining knowledge from each specialty in a team approach, the tumor board can effectively and holistically develop a care plan for each patient.
For the tumor board at the Warren Alpert Medical School of Brown University (Providence, Rhode Island), we often prepare a presentation with comprehensive details about the patient and tumor. During the presentation, we also propose a treatment plan prior to describing each patient at the weekly conference and amend the plans during the discussion. Tumor boards also provide a consulting role to the community and hospital providers in which patients are being referred by their primary provider and are seeking a second opinion or guidance.
In many ways, the tumor board is a multidisciplinary approach for patient advocacy in the form of treatment. These physicians meet on a regular basis to check on the patient’s progress and continually reevaluate how to have discussions about the patient’s care. There are many reasons why it is important to refer patients to a multidisciplinary tumor board.
Improved Workup and Diagnosis
One of the values of a tumor board is that it allows for patient data to be collected and assembled in a way that tells a story. The specialist from each field can then discuss and weigh the benefits and risks for each diagnostic test that should be performed for the workup in each patient. Physicians who refer their patients to the tumor board use their recommendations to both confirm the diagnosis and shift their treatment plans, depending on the information presented during the meeting.1 There may be a change in the tumor type, decision to refer for surgery, cancer staging, and list of viable options, especially after reviewing pathology and imaging.2 The discussion of the treatment plan may consider not only surgical considerations but also the patient’s quality of life. At times, noninvasive interventions are more appropriate and align with the patient’s goals of care. In addition, during the tumor board clinic there may be new tumors that are identified and biopsied, providing increased diagnosis and surveillance for patients who may have a higher risk for developing skin cancer.
Education for Residents and Providers
The multidisciplinary tumor board not only helps patients but also educates both residents and providers on the evidence-based therapeutic management of high-risk tumors.2 Research literature on cutaneous oncology is dynamic, and the weekly tumor board meetings help providers stay informed about the best and most effective treatments for their patients.3 In addition to the attending specialists, participants of the tumor board also may include residents, medical students, medical assistance staff, nurses, physician assistants, and fellows. Furthermore, the recommendations given by the tumor board serve to educate both the patient and the provider who referred them to the tumor board. Although we have access to excellent dermatology textbooks as residents, the most impactful educational experience is seeing the patients in tumor board clinic and participating in the immensely educational discussions at the weekly conferences. Through this experience, I have learned that treatment plans should be personalized to the patient. There are many factors to take into consideration when deciphering what the best course of treatment will be for a patient. Sometimes the best option is Mohs micrographic surgery, while other times it may be scheduling several sessions of palliative radiation oncology. Treatment depends on the individual patient and their condition.
Coordination of Care
During a week that I was on call, I was consulted to biopsy a patient with a giant hemorrhagic basal cell carcinoma that caused substantial cheek and nose distortion as well as anemia secondary to acute blood loss. The patient not only did not have a dermatologist but also did not have a primary care physician given he had not had contact with the health care system in more than 30 years. The reason for him not seeking care was multifactorial, but the approach to his care became multidisciplinary. We sought to connect him with the right providers to help him in any way that we could. We presented him at our multidisciplinary tumor board and started him on sonedigib, a medication that binds to and inhibits the smoothened protein.4 Through the tumor board, we were able to establish sustained contact with the patient. The tumor board created effective communication between providers to get him the referrals that he needed for dermatology, pathology, radiation oncology, hematology/oncology, and otolaryngology. The discussions centered around being cognizant of the patient’s apprehension with the health care system as well as providing medical and surgical treatment that would help his quality of life. We built a consensus on what the best plan was for the patient and his family. This consensus would have been more difficult had it not been for the combined specialties of the tumor board. In general, studies have shown that weekly tumor boards have resulted in decreased mortality rates for patients with advanced cancers.5
Final Thoughts
The multidisciplinary tumor board is a powerful resource for hospitals and the greater medical community. At these weekly conferences you realize there may still be hope that begins at the line where your expertise ends. It represents a team of providers who compassionately refuse to give up on patients when they are the last refuge.
- Foster TJ, Bouchard-Fortier A, Olivotto IA, et al. Effect of multidisciplinary case conferences on physician decision making: breast diagnostic rounds. Cureus. 2016;8:E895.
- El Saghir NS, Charara RN, Kreidieh FY, et al. Global practice and efficiency of multidisciplinary tumor boards: results of an American Society of Clinical Oncology international survey. J Glob Oncol. 2015;1:57-64.
- Mori S, Navarrete-Dechent C, Petukhova TA, et al. Tumor board conferences for multidisciplinary skin cancer management: a survey of US cancer centers. J Natl Compr Canc Netw. 2018;16:1209-1215.
- Dummer R, Ascierto PA, Basset-Seguin N, et al. Sonidegib and vismodegib in the treatment of patients with locally advanced basal cell carcinoma: a joint expert opinion. J Eur Acad Dermatol Venereol. 2020;34:1944-1956.
- Kehl KL, Landrum MB, Kahn KL, et al. Tumor board participation among physicians caring for patients with lung or colorectal cancer. J Oncol Pract. 2015;11:E267-E278.
- Foster TJ, Bouchard-Fortier A, Olivotto IA, et al. Effect of multidisciplinary case conferences on physician decision making: breast diagnostic rounds. Cureus. 2016;8:E895.
- El Saghir NS, Charara RN, Kreidieh FY, et al. Global practice and efficiency of multidisciplinary tumor boards: results of an American Society of Clinical Oncology international survey. J Glob Oncol. 2015;1:57-64.
- Mori S, Navarrete-Dechent C, Petukhova TA, et al. Tumor board conferences for multidisciplinary skin cancer management: a survey of US cancer centers. J Natl Compr Canc Netw. 2018;16:1209-1215.
- Dummer R, Ascierto PA, Basset-Seguin N, et al. Sonidegib and vismodegib in the treatment of patients with locally advanced basal cell carcinoma: a joint expert opinion. J Eur Acad Dermatol Venereol. 2020;34:1944-1956.
- Kehl KL, Landrum MB, Kahn KL, et al. Tumor board participation among physicians caring for patients with lung or colorectal cancer. J Oncol Pract. 2015;11:E267-E278.
Resident Pearl
- Participating in a multidisciplinary tumor board allows residents to learn more about how to manage and treat high-risk skin cancers. The multidisciplinary team approach provides high-quality care for challenging patients.
