User login
October 2017 Digital Edition
Click here to access the October 2017 Digital Edition.
Table of Contents
- A Mission for Graduate Medical Education at VA
- Trends in Hysterectomy Rates and Approaches in the VA
- A Severe Case of Paliperidone Palmitate-Induced Parkinsonism: Opportunities for Improvement
- A Case of Streptococcus pyogenes Sepsis of Possible Oral Origin
- Cannabinoid Hyperemesis Syndrome
- Improving Care and Reducing Length of Stay in Patients Undergoing Total Knee Replacement
- Restoring Function in Veterans With Complex Chronic Pain
- Bearing the Standard
- FDA Updates
- NOVA Updates

Click here to access the October 2017 Digital Edition.
Table of Contents
- A Mission for Graduate Medical Education at VA
- Trends in Hysterectomy Rates and Approaches in the VA
- A Severe Case of Paliperidone Palmitate-Induced Parkinsonism: Opportunities for Improvement
- A Case of Streptococcus pyogenes Sepsis of Possible Oral Origin
- Cannabinoid Hyperemesis Syndrome
- Improving Care and Reducing Length of Stay in Patients Undergoing Total Knee Replacement
- Restoring Function in Veterans With Complex Chronic Pain
- Bearing the Standard
- FDA Updates
- NOVA Updates
Click here to access the October 2017 Digital Edition.
Table of Contents
- A Mission for Graduate Medical Education at VA
- Trends in Hysterectomy Rates and Approaches in the VA
- A Severe Case of Paliperidone Palmitate-Induced Parkinsonism: Opportunities for Improvement
- A Case of Streptococcus pyogenes Sepsis of Possible Oral Origin
- Cannabinoid Hyperemesis Syndrome
- Improving Care and Reducing Length of Stay in Patients Undergoing Total Knee Replacement
- Restoring Function in Veterans With Complex Chronic Pain
- Bearing the Standard
- FDA Updates
- NOVA Updates

Prescribing antipsychotics in geriatric patients: Focus on schizophrenia and bipolar disorder
Antipsychotics are FDA-approved as a primary treatment for schizophrenia and bipolar disorder and as adjunctive therapy for major depressive disorder. In the United States, approximately 26% of antipsychotic prescriptions written for these indications are for individuals age >65.1 Additionally, antipsychotics are widely used to treat behavioral symptoms associated with dementia.1 The rapid expansion of the use of second-generation antipsychotics (SGAs), in particular, has been driven in part by their lower risk for extrapyramidal symptoms (EPS) compared with first-generation antipsychotics (FGAs).1 However, a growing body of data indicates that all antipsychotics have a range of adverse effects in older patients. This focus is critical in light of demographic trends—in the next 10 to 15 years, the population age >60 will grow 3.5 times more rapidly than the general population.2
In this context, psychiatrists need information on the relative risks of antipsychotics for older patients. This 3-part series summarizes findings and recommendations on safety and tolerability when prescribing antipsychotics in older individuals with chronic psychotic disorders, such as schizophrenia, bipolar disorder, depression, and dementia. This review aims to:
- briefly summarize the major studies and analyses relevant to older patients with these diagnoses
- provide a summative opinion on safety and tolerability issues in these older adults
- highlight the gaps in the evidence base and areas that need additional research.
Part 1 focuses on older adults with schizophrenia or bipolar disorder. Subsequent articles will focus on prescribing antipsychotics to older adults with depression and those with dementia.

Schizophrenia

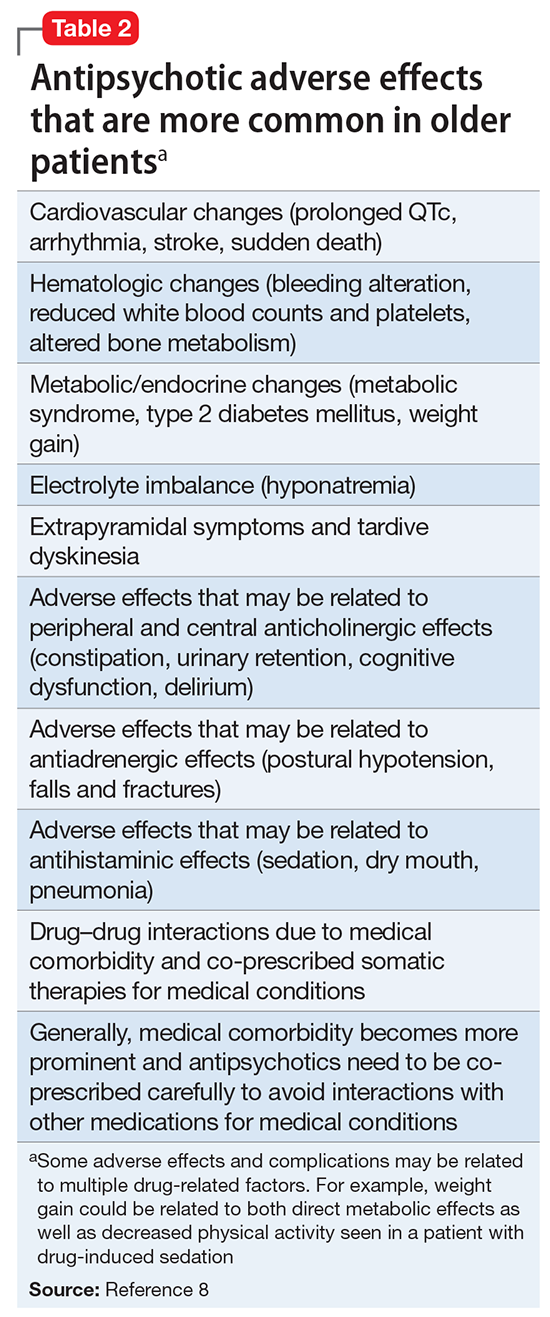
Summary of benefits, place in treatment armamentarium. Individuals with schizophrenia have a shorter life expectancy than that of the general population mostly as a result of suicide and comorbid physical illnesses,3 but the number of patients with schizophrenia age >55 will double over the next 2 decades.4 With aging, both positive and negative symptoms may be a focus of treatment (Table 1).5,6 Antipsychotics are a first-line treatment for older patients with schizophrenia with few medication alternatives.7 Safety risks associated with antipsychotics in older people span a broad spectrum (Table 2).8
A 6-week prospective RCT evaluated paliperidone extended-release vs placebo in 114 older adults (age ≥65 years; mean age, 70 years) with schizophrenia.14 There was an optional 24-week extension of open-label treatment with paliperidone. Mean daily dose of paliperidone was 8.4 mg. Efficacy measures did not show consistent statistically significant differences between treatment groups. Discontinuation rates were similar between paliperidone (7%) vs placebo (8%). Serious adverse events occurred in 3% of paliperidone-treated vs 8% of placebo-treated patients. Elevated prolactin levels occurred in one-half of paliperidone-treated patients. There were no prolactin or glucose treatment-related adverse events or significant mean changes in body weight for either paliperidone-treated or placebo-treated patients. Safety findings in the 24-week, open-label extension group were consistent with the RCT results.
Howanitz et al15 conducted a 12-week, prospective RCT that compared clozapine (mean dose, 300 mg/d) with chlorpromazine (mean dose, 600 mg/d) in 42 older adults (mean age, 67 years) with schizophrenia. Drop-out rate prior to 5 weeks was 19% and similar between groups. Common adverse effects included sialorrhea, hematologic abnormalities, sedation, tachycardia, EPS, and weight gain. Although both drugs were effective, more patients taking clozapine had tachycardia and weight gain, while more chlorpromazine patients reported sedation.
There have been other, less rigorous studies.7,8 Most of these studies evaluated risperidone and olanzapine, and most were conducted in “younger” geriatric patients (age <75 years). Although patients who participate in clinical trials may be healthier than “typical” patients, adverse effects such as EPS, sedation, and weight gain were still relatively common in these studies.
Other clinical data. A major consideration in treating older adults with schizophrenia is balancing the need to administer an antipsychotic dose high enough to alleviate psychotic symptoms while minimizing dose-dependent adverse effects. There is a U-shaped relationship between age and vulnerability to antipsychotic adverse effects,16,17 wherein adverse effects are highest at younger and older ages. Evidence supports using the lowest effective antipsychotic dose for geriatric patients with schizophrenia. Positive emission tomography (PET) studies suggest that older patients develop EPS with lower doses despite lower receptor occupancy.17,18 A recent study of 35 older patients (mean age, 60.1 years) with schizophrenia obtained PET, clinical measures, and blood pharmacokinetic measures before and after reduction of risperidone or olanzapine doses.18 A ≥40% reduction in dose was associated with reduced adverse effects, particularly EPS and elevation of prolactin levels. Moreover, the therapeutic window of striatal D2/D3 receptor occupancy appeared to be 50% to 60% in these older patients, compared with 65% to 80% in younger patients.
Long-term risks of antipsychotic treatment across the lifespan are less clear, with evidence suggesting both lower and higher mortality risk.19,20 It is difficult to fully disentangle the long-term risks of antipsychotics from the cumulative effects of lifestyle and comorbidity among individuals who have lived with schizophrenia for decades. Large naturalistic studies that include substantial numbers of older people with schizophrenia might be a way to elicit more information on long-term safety. The Schizophrenia Outpatient Health Outcome (SOHO) study was a large naturalistic trial that recruited >10,000 individuals with schizophrenia in 10 European countries.21 Although the SOHO study found differences between antipsychotics and adverse effects, such as EPS, weight gain, and sexual dysfunction, because the mean age of these patients was approximately 40 years and the follow-up period was only 3 years, it is difficult to draw conclusions that could be relevant to older individuals who have had schizophrenia for decades.
Bipolar Disorder

Clinical trials: Bipolar depression. A post hoc, secondary analysis of two 8-week, double-blind, randomized, placebo-controlled studies in bipolar depression compared 2 dosages of quetiapine (300 mg/d and 600 mg/d) with placebo in mixed-age patients.31 In a subgroup of 72 patients, ages 55 to 65, remission occurred more often with quetiapine than with placebo. Study discontinuation rates were similar between older people and younger people (age <55 years): quetiapine, 300 mg/d, 29.2%; quetiapine, 600 mg/d, 48.1%; and placebo, 29.6% in older adults, compared with 37.1%, 45.8%, and 38.1%, respectively, in younger adults. In all patients, the most common reason for discontinuation was adverse events with quetiapine and lack of efficacy for placebo. Adverse event rates were similar in older and younger adults. Dry mouth and dizziness were more common in older adults. Proportions of adults experiencing clinically significant weight gain (≥7% of body weight) were 5.3%, 8.3%, and 0% in older adults receiving quetiapine, 300 mg/d, quetiapine, 600 mg/d, and placebo, respectively, compared with 7.2%, 10.1%, and 2.6% in younger adults. EPS and treatment-emergent mania were minimal.
A secondary analysis of mixed-age, RCTs examined response in older adults (age ≥55 years) with bipolar I depression who received lurasidone as monotherapy or adjunctive therapy.32 In the monotherapy study, these patients were randomized to 6 weeks of lurasidone 20 to 60 mg/d, lurasidone 80 to 120 mg/d, or placebo. In the adjunctive therapy study, they were randomized to lurasidone 20 to 120 mg/d or placebo with either lithium or valproate. There were 83 older adults (17.1% of the sample) in the monotherapy study and 53 (15.6%) in the adjunctive therapy study. Mean improvement in depression was significantly higher for both doses of lurasidone monotherapy than placebo. Adjunctive lurasidone was not associated with statistically significant improvement vs placebo. The most frequent adverse events in older patients on lurasidone monotherapy 20 to 60 mg/d or 80 to 120 mg/d were nausea (18.5% and 9.7%, respectively) and somnolence (11.1% and 0%, respectively). Akathisia (9.7%) and insomnia (9.7%) were the most common adverse events in the group receiving 80 to 120 mg/d, with the rate of akathisia exhibiting a dose-related increase. Weight change with lurasidone was similar to placebo, and there were no clinically meaningful group changes in vital signs, electrocardiography, or laboratory parameters.
A small (N = 20) open study found improvement in older adults with bipolar depression with aripiprazole (mean dose, 10.3 mg/d).33 Adverse effects included restlessness and weight gain (n = 3, 9% each), sedation (n = 2, 10%), and drooling and diarrhea/loose stools (n = 1, 5% each). In another small study (N = 15) using asenapine (mean dose, 11.2 mg/d) in mainly older bipolar patients with depression, the most common adverse effects were gastrointestinal (GI) discomfort (n = 5, 33%) and restlessness, tremors, cognitive difficulties, and sluggishness (n = 2, 13% each).34
Clinical trials: Bipolar mania. Researchers conducted a pooled analysis of two 12-week randomized trials comparing quetiapine with placebo in a mixed-age sample with bipolar mania.35 In a subgroup of 59 older patients (mean age, 62.9 years), manic symptoms improved significantly more with quetiapine (modal dose, 550 mg/d) than with placebo. Adverse effects reported by >10% of older patients were dry mouth, somnolence, postural hypotension, insomnia, weight gain, and dizziness. Insomnia was reported by >10% of patients receiving placebo.
In a case series of 11 elderly patients with mania receiving asenapine, Baruch et al36 reported a 63% remission rate. One patient discontinued the study because of a new rash, 1 discontinued after developing peripheral edema, and 3 patients reported mild sedation.
Beyer et al37 reported on a post hoc analysis of 94 older adults (mean age, 57.1 years; range, 50.1 to 74.8 years) with acute bipolar mania receiving olanzapine (n = 47), divalproex (n = 31), or placebo (n = 16) in a pooled olanzapine clinical trials database. Patients receiving olanzapine or divalproex had improvement in mania; those receiving placebo did not improve. Safety findings were comparable with reports in younger patients with mania.
Other clinical data. Adverse effects found in mixed-age samples using secondary analyses of clinical trials need to be interpreted with caution because these types of studies usually exclude individuals with significant medical comorbidity. Medical burden, cognitive impairment, or concomitant medications generally necessitate slower drug titration and lower total daily dosing. For example, a secondary analysis of the U.S. National Institute of Health-funded Systematic Treatment Enhancement Program for Bipolar Disorder study, which had broader inclusion criteria than most clinical trials, reported that, although recovery rates in older adults with bipolar disorder were fairly good (78.5%), lower doses of risperidone were used in older vs younger patients.38
Clinical considerations
Interpretation of the relative risks of antipsychotics in older people must be tempered by the caveat that there is limited high-quality data (Table 4). Antipsychotics are the first-line therapy for older patients with schizophrenia, although their use is supported by a small number of prospective RCTs. SGAs are preferred because of their lower propensity to cause EPS and other motor adverse effects. Older persons with schizophrenia have an EPS threshold lower than younger patients and determining the lowest effective dosage may minimize EPS and cognitive adverse effects. As individuals with long-standing schizophrenia get older, their antipsychotic dosages may need to be reduced, and clinicians need to monitor for adverse effects that are more common among older people, such as tardive dyskinesia and metabolic abnormalities. In healthy, “younger” geriatric patients, monitoring for adverse effects may be similar to monitoring of younger patients. Patients who are older or frail may need more frequent assessment.

Like older adults with schizophrenia, geriatric patients with bipolar disorder have reduced drug tolerability and experience more adverse effects than younger patients. There are no prospective controlled studies that evaluated using antipsychotics in older patients with bipolar disorder. In older bipolar patients, the most problematic adverse effects of antipsychotics are akathisia, parkinsonism, other EPS, sedation and dizziness (which may increase fall risk), and GI discomfort. A key tolerability and safety consideration when treating older adults with bipolar disorder is the role of antipsychotics in relation to the use of lithium and mood stabilizers. Some studies have suggested that lithium has neuroprotective effects when used long-term; however, at least 1 report suggested that long-term antipsychotic treatment may be associated with neurodegeneration.39
The literature does not provide strong evidence on the many clinical variations that we see in routine practice settings, such as combinations of drug treatments or drugs prescribed to patients with specific comorbid conditions. There is a need for large cohort studies that monitor treatment course, medical comorbidity, and prognosis. Additionally, well-designed clinical trials such as the DART-AD, which investigated longer-term trajectories of people with dementia taking antipsychotics, should serve as a model for the type of research that is needed to better understand outcome variability among older people with chronic psychotic or bipolar disorders.40
1. Alexander GC, Gallagher SA, Mascola A, et al. Increasing off-label use of antipsychotic medications in the United States, 1995-2008. Pharmacoepidemiol Drug Saf. 2011;20(2):177-184.
2. United Nations, Department of Economic and Social Affairs, Population Division. World population ageing: 1950-2050. http://www.un.org/esa/population/publications/worldageing19502050. Accessed September 1, 2017.
3. Lawrence D, Kisely S, Pais J. The epidemiology of excess mortality in people with mental illness. Can J Psychiatry. 2010;55(12):752-760.
4. Cohen CI, Vahia I, Reyes P, et al. Focus on geriatric psychiatry: schizophrenia in later life: clinical symptoms and social well-being. Psychiatr Serv. 2008;59(3):232-234.
5. Jeste DV, Barak Y, Madhusoodanan S, et al. International multisite double-blind trial of the atypical antipsychotics risperidone and olanzapine in 175 elderly patients with chronic schizophrenia. Am J Geriatr Psychiatry. 2003;11(6):638-647.
6. Kalache SM, Mulsant BH, Davies SJ, et al. The impact of aging, cognition, and symptoms on functional competence in individuals with schizophrenia across the lifespan. Schizophr Bull. 2015;41(2):374-381.
7. Suzuki T, Remington G, Uchida H, et al. Management of schizophrenia in late life with antipsychotic medications: a qualitative review. Drugs Aging. 2011;28(12):961-980.
8. Mulsant BH, Pollock BG. Psychopharmacology. In: David C. Steffens DC, Blazer DG, Thakur ME (eds). The American Psychiatric Publishing Textbook of Geriatric Psychiatry, 5th Edition. Arlington, VA: American Psychiatric Publishing; 2015:527-587.
9. Cohen CI, Meesters PD, Zhao J. New perspectives on schizophrenia in later life: implications for treatment, policy, and research. Lancet Psychiatry. 2015;2(4):340-350.
10. Marriott RG, Neil W, Waddingham S. Antipsychotic medication for elderly people with schizophrenia. Cochrane Database Syst Rev. 2006;(1):CD005580.
11. Essali A, Ali G. Antipsychotic drug treatment for elderly people with late-onset schizophrenia. Cochrane Database Syst Rev. 2012(2):CD004162.
12. Scott J, Greenwald BS, Kramer E, et al. Atypical (second generation) antipsychotic treatment response in very late-onset schizophrenia-like psychosis. Int Psychogeriatr. 2011;23(5):742-748.
13. Rado J, Janicak PG. Pharmacological and clinical profile of recently approved second-generation antipsychotics: implications for treatment of schizophrenia in older patients. Drugs Aging. 2012;29(10):783-791.
14. Tzimos A, Samokhvalov V, Kramer M, et al. Safety and tolerability of oral paliperidone extended-release tablets in elderly patients with schizophrenia: a double-blind, placebo-controlled study with six-month open-label extension. Am J Geriatr Psychiatry. 2008;16(1):31-43.
15. Howanitz E, Pardo M, Smelson DA, et al. The efficacy and safety of clozapine versus chlorpromazine in geriatric schizophrenia. J Clin Psychiatry. 1999;60(1):41-44.
16. Sproule BA, Lake J, Mamo DC, et al. Are antipsychotic prescribing patterns different in older and younger adults?: a survey of 1357 psychiatric inpatients in Toronto. Can J Psychiatry. 2010;55(4):248-254.
17. Uchida H, Suzuki T, Mamo DC, et al. Effects of age and age of onset on prescribed antipsychotic dose in schizophrenia spectrum disorders: a survey of 1,418 patients in Japan. Am J Geriatr Psychiatry. 2008;16(7):584-593.
18. Graff-Guerrero A, Rajji TK, Mulsant BH, et al. Evaluation of antipsychotic dose reduction in late-life schizophrenia: a prospective dopamine D2/3 occupancy study. JAMA Psychiatry. 2015;72(9):927-934.
19. Khan A, Schwartz K, Stern C, et al. Mortality risk in patients with schizophrenia participating in premarketing atypical antipsychotic clinical trials. J Clin Psychiatry. 2007;68(12):1828-1833.
20. Weinmann S, Read J, Aderhold V. Influence of antipsychotics on mortality in schizophrenia: a systematic review. Schizophr Res. 2009;113(1):1-11.
21. Novick D, Haro JM, Perrin E, et al. Tolerability of outpatient antipsychotic treatment: 36-month results from the European Schizophrenia Outpatient Health Outcomes (SOHO) study. Eur Neuropsychopharmacol. 2009;19(8):542-550.
22. Sajatovic M, Blow FC, Ignacio RV, et al. Age-related modifiers of clinical presentation and health service use among veterans with bipolar disorder. Psychiatr Serv. 2004;55(9):1014-1021.
23. Jeste DV, Alexopoulos GS, Bartels SJ, et al. Consensus statement on the upcoming crisis in geriatric mental health: research agenda for the next 2 decades. Arch Gen Psychiatry. 1999;56(9):848-853.
24. Sajatovic M, Chen P. Geriatric bipolar disorder. Psychiatr Clin North Am. 2011;34(2):319-333,vii.
25. Sajatovic M, Strejilevich SA, Gildengers AG, et al. A report on older-age bipolar disorder from the International Society for Bipolar Disorders Task Force. Bipolar Disord. 2015;17(7):689-704.
26. Lala SV, Sajatovic M. Medical and psychiatric comorbidities among elderly individuals with bipolar disorder: a literature review. J Geriatr Psychiatry Neurol. 2012;25(1):20-25.
27. Dols A, Rhebergen D, Beekman A, et al. Psychiatric and medical comorbidities: results from a bipolar elderly cohort study. Am J Geriatr Psychiatry. 2014;22(11):1066-1074.
28. Pillarella J, Higashi A, Alexander GC, et al. Trends in use of second-generation antipsychotics for treatment of bipolar disorder in the United States, 1998-2009. Psychiatr Serv. 2012;63(1):83-86.
29. De Fruyt J, Deschepper E, Audenaert K, et al. Second generation antipsychotics in the treatment of bipolar depression: a systematic review and meta-analysis. J Psychopharmacol. 2012;26(5):603-617.
30. Nivoli AM, Murru A, Goikolea JM, et al. New treatment guidelines for acute bipolar mania: a critical review. J Affect Disord. 2012;140(2):125-141.
31. Sajatovic M, Paulsson B. Quetiapine for the treatment of depressive episodes in adults aged 55 to 65 years with bipolar disorder. Paper presented at: American Association of Geriatric Psychiatry Annual Meeting; 2007; New Orleans, LA.
32. Sajatovic M, Forester B, Tsai J, et al. Efficacy and safety of lurasidone in older adults with bipolar depression: analysis of two double-blind, placebo-controlled studies. Paper presented at: American College of Neuropsychopharmacology (ACNP) 53rd Annual Meeting; 2014; Phoenix, AZ.
33. Sajatovic M, Coconcea N, Ignacio RV, et al. Aripiprazole therapy in 20 older adults with bipolar disorder: a 12-week, open-label trial. J Clin Psychiatry. 2008;69(1):41-46.
34. Sajatovic M, Dines P, Fuentes-Casiano E, et al. Asenapine in the treatment of older adults with bipolar disorder. Int J Geriatr Psychiatry. 2015;30(7):710-719.
35. Sajatovic M, Calabrese JR, Mullen J. Quetiapine for the treatment of bipolar mania in older adults. Bipolar Disord. 2008;10(6):662-671.
36. Baruch Y, Tadger S, Plopski I, et al. Asenapine for elderly bipolar manic patients. J Affect Disord. 2013;145(1):130-132.
37. Beyer JL, Siegal A, Kennedy JS. Olanzapine, divalproex and placebo treatment, non-head to head comparisons of older adults acute mania. Paper presented at: 10th Congress of the International Psychogeriatric Association; 2001; Nice, France.
38. Al Jurdi RK, Marangell LB, Petersen NJ, et al. Prescription patterns of psychotropic medications in elderly compared with younger participants who achieved a “recovered” status in the systematic treatment enhancement program for bipolar disorder. Am J Geriatr Psychiatry. 2008;16(11):922-933.
39. Gildengers AG, Chung KH, Huang SH, et al. Neuroprogressive effects of lifetime illness duration in older adults with bipolar disorder. Bipolar Disord. 2014;16(6):617-623.
40. Ballard C, Lana MM, Theodoulou M, et al. A randomised, blinded, placebo-controlled trial in dementia patients continuing or stopping neuroleptics (the DART-AD trial). PLoS Med. 2008;5(4):e76.
Antipsychotics are FDA-approved as a primary treatment for schizophrenia and bipolar disorder and as adjunctive therapy for major depressive disorder. In the United States, approximately 26% of antipsychotic prescriptions written for these indications are for individuals age >65.1 Additionally, antipsychotics are widely used to treat behavioral symptoms associated with dementia.1 The rapid expansion of the use of second-generation antipsychotics (SGAs), in particular, has been driven in part by their lower risk for extrapyramidal symptoms (EPS) compared with first-generation antipsychotics (FGAs).1 However, a growing body of data indicates that all antipsychotics have a range of adverse effects in older patients. This focus is critical in light of demographic trends—in the next 10 to 15 years, the population age >60 will grow 3.5 times more rapidly than the general population.2
In this context, psychiatrists need information on the relative risks of antipsychotics for older patients. This 3-part series summarizes findings and recommendations on safety and tolerability when prescribing antipsychotics in older individuals with chronic psychotic disorders, such as schizophrenia, bipolar disorder, depression, and dementia. This review aims to:
- briefly summarize the major studies and analyses relevant to older patients with these diagnoses
- provide a summative opinion on safety and tolerability issues in these older adults
- highlight the gaps in the evidence base and areas that need additional research.
Part 1 focuses on older adults with schizophrenia or bipolar disorder. Subsequent articles will focus on prescribing antipsychotics to older adults with depression and those with dementia.
Schizophrenia
Summary of benefits, place in treatment armamentarium. Individuals with schizophrenia have a shorter life expectancy than that of the general population mostly as a result of suicide and comorbid physical illnesses,3 but the number of patients with schizophrenia age >55 will double over the next 2 decades.4 With aging, both positive and negative symptoms may be a focus of treatment (Table 1).5,6 Antipsychotics are a first-line treatment for older patients with schizophrenia with few medication alternatives.7 Safety risks associated with antipsychotics in older people span a broad spectrum (Table 2).8
A 6-week prospective RCT evaluated paliperidone extended-release vs placebo in 114 older adults (age ≥65 years; mean age, 70 years) with schizophrenia.14 There was an optional 24-week extension of open-label treatment with paliperidone. Mean daily dose of paliperidone was 8.4 mg. Efficacy measures did not show consistent statistically significant differences between treatment groups. Discontinuation rates were similar between paliperidone (7%) vs placebo (8%). Serious adverse events occurred in 3% of paliperidone-treated vs 8% of placebo-treated patients. Elevated prolactin levels occurred in one-half of paliperidone-treated patients. There were no prolactin or glucose treatment-related adverse events or significant mean changes in body weight for either paliperidone-treated or placebo-treated patients. Safety findings in the 24-week, open-label extension group were consistent with the RCT results.
Howanitz et al15 conducted a 12-week, prospective RCT that compared clozapine (mean dose, 300 mg/d) with chlorpromazine (mean dose, 600 mg/d) in 42 older adults (mean age, 67 years) with schizophrenia. Drop-out rate prior to 5 weeks was 19% and similar between groups. Common adverse effects included sialorrhea, hematologic abnormalities, sedation, tachycardia, EPS, and weight gain. Although both drugs were effective, more patients taking clozapine had tachycardia and weight gain, while more chlorpromazine patients reported sedation.
There have been other, less rigorous studies.7,8 Most of these studies evaluated risperidone and olanzapine, and most were conducted in “younger” geriatric patients (age <75 years). Although patients who participate in clinical trials may be healthier than “typical” patients, adverse effects such as EPS, sedation, and weight gain were still relatively common in these studies.
Other clinical data. A major consideration in treating older adults with schizophrenia is balancing the need to administer an antipsychotic dose high enough to alleviate psychotic symptoms while minimizing dose-dependent adverse effects. There is a U-shaped relationship between age and vulnerability to antipsychotic adverse effects,16,17 wherein adverse effects are highest at younger and older ages. Evidence supports using the lowest effective antipsychotic dose for geriatric patients with schizophrenia. Positive emission tomography (PET) studies suggest that older patients develop EPS with lower doses despite lower receptor occupancy.17,18 A recent study of 35 older patients (mean age, 60.1 years) with schizophrenia obtained PET, clinical measures, and blood pharmacokinetic measures before and after reduction of risperidone or olanzapine doses.18 A ≥40% reduction in dose was associated with reduced adverse effects, particularly EPS and elevation of prolactin levels. Moreover, the therapeutic window of striatal D2/D3 receptor occupancy appeared to be 50% to 60% in these older patients, compared with 65% to 80% in younger patients.
Long-term risks of antipsychotic treatment across the lifespan are less clear, with evidence suggesting both lower and higher mortality risk.19,20 It is difficult to fully disentangle the long-term risks of antipsychotics from the cumulative effects of lifestyle and comorbidity among individuals who have lived with schizophrenia for decades. Large naturalistic studies that include substantial numbers of older people with schizophrenia might be a way to elicit more information on long-term safety. The Schizophrenia Outpatient Health Outcome (SOHO) study was a large naturalistic trial that recruited >10,000 individuals with schizophrenia in 10 European countries.21 Although the SOHO study found differences between antipsychotics and adverse effects, such as EPS, weight gain, and sexual dysfunction, because the mean age of these patients was approximately 40 years and the follow-up period was only 3 years, it is difficult to draw conclusions that could be relevant to older individuals who have had schizophrenia for decades.
Bipolar Disorder
Clinical trials: Bipolar depression. A post hoc, secondary analysis of two 8-week, double-blind, randomized, placebo-controlled studies in bipolar depression compared 2 dosages of quetiapine (300 mg/d and 600 mg/d) with placebo in mixed-age patients.31 In a subgroup of 72 patients, ages 55 to 65, remission occurred more often with quetiapine than with placebo. Study discontinuation rates were similar between older people and younger people (age <55 years): quetiapine, 300 mg/d, 29.2%; quetiapine, 600 mg/d, 48.1%; and placebo, 29.6% in older adults, compared with 37.1%, 45.8%, and 38.1%, respectively, in younger adults. In all patients, the most common reason for discontinuation was adverse events with quetiapine and lack of efficacy for placebo. Adverse event rates were similar in older and younger adults. Dry mouth and dizziness were more common in older adults. Proportions of adults experiencing clinically significant weight gain (≥7% of body weight) were 5.3%, 8.3%, and 0% in older adults receiving quetiapine, 300 mg/d, quetiapine, 600 mg/d, and placebo, respectively, compared with 7.2%, 10.1%, and 2.6% in younger adults. EPS and treatment-emergent mania were minimal.
A secondary analysis of mixed-age, RCTs examined response in older adults (age ≥55 years) with bipolar I depression who received lurasidone as monotherapy or adjunctive therapy.32 In the monotherapy study, these patients were randomized to 6 weeks of lurasidone 20 to 60 mg/d, lurasidone 80 to 120 mg/d, or placebo. In the adjunctive therapy study, they were randomized to lurasidone 20 to 120 mg/d or placebo with either lithium or valproate. There were 83 older adults (17.1% of the sample) in the monotherapy study and 53 (15.6%) in the adjunctive therapy study. Mean improvement in depression was significantly higher for both doses of lurasidone monotherapy than placebo. Adjunctive lurasidone was not associated with statistically significant improvement vs placebo. The most frequent adverse events in older patients on lurasidone monotherapy 20 to 60 mg/d or 80 to 120 mg/d were nausea (18.5% and 9.7%, respectively) and somnolence (11.1% and 0%, respectively). Akathisia (9.7%) and insomnia (9.7%) were the most common adverse events in the group receiving 80 to 120 mg/d, with the rate of akathisia exhibiting a dose-related increase. Weight change with lurasidone was similar to placebo, and there were no clinically meaningful group changes in vital signs, electrocardiography, or laboratory parameters.
A small (N = 20) open study found improvement in older adults with bipolar depression with aripiprazole (mean dose, 10.3 mg/d).33 Adverse effects included restlessness and weight gain (n = 3, 9% each), sedation (n = 2, 10%), and drooling and diarrhea/loose stools (n = 1, 5% each). In another small study (N = 15) using asenapine (mean dose, 11.2 mg/d) in mainly older bipolar patients with depression, the most common adverse effects were gastrointestinal (GI) discomfort (n = 5, 33%) and restlessness, tremors, cognitive difficulties, and sluggishness (n = 2, 13% each).34
Clinical trials: Bipolar mania. Researchers conducted a pooled analysis of two 12-week randomized trials comparing quetiapine with placebo in a mixed-age sample with bipolar mania.35 In a subgroup of 59 older patients (mean age, 62.9 years), manic symptoms improved significantly more with quetiapine (modal dose, 550 mg/d) than with placebo. Adverse effects reported by >10% of older patients were dry mouth, somnolence, postural hypotension, insomnia, weight gain, and dizziness. Insomnia was reported by >10% of patients receiving placebo.
In a case series of 11 elderly patients with mania receiving asenapine, Baruch et al36 reported a 63% remission rate. One patient discontinued the study because of a new rash, 1 discontinued after developing peripheral edema, and 3 patients reported mild sedation.
Beyer et al37 reported on a post hoc analysis of 94 older adults (mean age, 57.1 years; range, 50.1 to 74.8 years) with acute bipolar mania receiving olanzapine (n = 47), divalproex (n = 31), or placebo (n = 16) in a pooled olanzapine clinical trials database. Patients receiving olanzapine or divalproex had improvement in mania; those receiving placebo did not improve. Safety findings were comparable with reports in younger patients with mania.
Other clinical data. Adverse effects found in mixed-age samples using secondary analyses of clinical trials need to be interpreted with caution because these types of studies usually exclude individuals with significant medical comorbidity. Medical burden, cognitive impairment, or concomitant medications generally necessitate slower drug titration and lower total daily dosing. For example, a secondary analysis of the U.S. National Institute of Health-funded Systematic Treatment Enhancement Program for Bipolar Disorder study, which had broader inclusion criteria than most clinical trials, reported that, although recovery rates in older adults with bipolar disorder were fairly good (78.5%), lower doses of risperidone were used in older vs younger patients.38
Clinical considerations
Interpretation of the relative risks of antipsychotics in older people must be tempered by the caveat that there is limited high-quality data (Table 4). Antipsychotics are the first-line therapy for older patients with schizophrenia, although their use is supported by a small number of prospective RCTs. SGAs are preferred because of their lower propensity to cause EPS and other motor adverse effects. Older persons with schizophrenia have an EPS threshold lower than younger patients and determining the lowest effective dosage may minimize EPS and cognitive adverse effects. As individuals with long-standing schizophrenia get older, their antipsychotic dosages may need to be reduced, and clinicians need to monitor for adverse effects that are more common among older people, such as tardive dyskinesia and metabolic abnormalities. In healthy, “younger” geriatric patients, monitoring for adverse effects may be similar to monitoring of younger patients. Patients who are older or frail may need more frequent assessment.
Like older adults with schizophrenia, geriatric patients with bipolar disorder have reduced drug tolerability and experience more adverse effects than younger patients. There are no prospective controlled studies that evaluated using antipsychotics in older patients with bipolar disorder. In older bipolar patients, the most problematic adverse effects of antipsychotics are akathisia, parkinsonism, other EPS, sedation and dizziness (which may increase fall risk), and GI discomfort. A key tolerability and safety consideration when treating older adults with bipolar disorder is the role of antipsychotics in relation to the use of lithium and mood stabilizers. Some studies have suggested that lithium has neuroprotective effects when used long-term; however, at least 1 report suggested that long-term antipsychotic treatment may be associated with neurodegeneration.39
The literature does not provide strong evidence on the many clinical variations that we see in routine practice settings, such as combinations of drug treatments or drugs prescribed to patients with specific comorbid conditions. There is a need for large cohort studies that monitor treatment course, medical comorbidity, and prognosis. Additionally, well-designed clinical trials such as the DART-AD, which investigated longer-term trajectories of people with dementia taking antipsychotics, should serve as a model for the type of research that is needed to better understand outcome variability among older people with chronic psychotic or bipolar disorders.40
Antipsychotics are FDA-approved as a primary treatment for schizophrenia and bipolar disorder and as adjunctive therapy for major depressive disorder. In the United States, approximately 26% of antipsychotic prescriptions written for these indications are for individuals age >65.1 Additionally, antipsychotics are widely used to treat behavioral symptoms associated with dementia.1 The rapid expansion of the use of second-generation antipsychotics (SGAs), in particular, has been driven in part by their lower risk for extrapyramidal symptoms (EPS) compared with first-generation antipsychotics (FGAs).1 However, a growing body of data indicates that all antipsychotics have a range of adverse effects in older patients. This focus is critical in light of demographic trends—in the next 10 to 15 years, the population age >60 will grow 3.5 times more rapidly than the general population.2
In this context, psychiatrists need information on the relative risks of antipsychotics for older patients. This 3-part series summarizes findings and recommendations on safety and tolerability when prescribing antipsychotics in older individuals with chronic psychotic disorders, such as schizophrenia, bipolar disorder, depression, and dementia. This review aims to:
- briefly summarize the major studies and analyses relevant to older patients with these diagnoses
- provide a summative opinion on safety and tolerability issues in these older adults
- highlight the gaps in the evidence base and areas that need additional research.
Part 1 focuses on older adults with schizophrenia or bipolar disorder. Subsequent articles will focus on prescribing antipsychotics to older adults with depression and those with dementia.
Schizophrenia
Summary of benefits, place in treatment armamentarium. Individuals with schizophrenia have a shorter life expectancy than that of the general population mostly as a result of suicide and comorbid physical illnesses,3 but the number of patients with schizophrenia age >55 will double over the next 2 decades.4 With aging, both positive and negative symptoms may be a focus of treatment (Table 1).5,6 Antipsychotics are a first-line treatment for older patients with schizophrenia with few medication alternatives.7 Safety risks associated with antipsychotics in older people span a broad spectrum (Table 2).8
A 6-week prospective RCT evaluated paliperidone extended-release vs placebo in 114 older adults (age ≥65 years; mean age, 70 years) with schizophrenia.14 There was an optional 24-week extension of open-label treatment with paliperidone. Mean daily dose of paliperidone was 8.4 mg. Efficacy measures did not show consistent statistically significant differences between treatment groups. Discontinuation rates were similar between paliperidone (7%) vs placebo (8%). Serious adverse events occurred in 3% of paliperidone-treated vs 8% of placebo-treated patients. Elevated prolactin levels occurred in one-half of paliperidone-treated patients. There were no prolactin or glucose treatment-related adverse events or significant mean changes in body weight for either paliperidone-treated or placebo-treated patients. Safety findings in the 24-week, open-label extension group were consistent with the RCT results.
Howanitz et al15 conducted a 12-week, prospective RCT that compared clozapine (mean dose, 300 mg/d) with chlorpromazine (mean dose, 600 mg/d) in 42 older adults (mean age, 67 years) with schizophrenia. Drop-out rate prior to 5 weeks was 19% and similar between groups. Common adverse effects included sialorrhea, hematologic abnormalities, sedation, tachycardia, EPS, and weight gain. Although both drugs were effective, more patients taking clozapine had tachycardia and weight gain, while more chlorpromazine patients reported sedation.
There have been other, less rigorous studies.7,8 Most of these studies evaluated risperidone and olanzapine, and most were conducted in “younger” geriatric patients (age <75 years). Although patients who participate in clinical trials may be healthier than “typical” patients, adverse effects such as EPS, sedation, and weight gain were still relatively common in these studies.
Other clinical data. A major consideration in treating older adults with schizophrenia is balancing the need to administer an antipsychotic dose high enough to alleviate psychotic symptoms while minimizing dose-dependent adverse effects. There is a U-shaped relationship between age and vulnerability to antipsychotic adverse effects,16,17 wherein adverse effects are highest at younger and older ages. Evidence supports using the lowest effective antipsychotic dose for geriatric patients with schizophrenia. Positive emission tomography (PET) studies suggest that older patients develop EPS with lower doses despite lower receptor occupancy.17,18 A recent study of 35 older patients (mean age, 60.1 years) with schizophrenia obtained PET, clinical measures, and blood pharmacokinetic measures before and after reduction of risperidone or olanzapine doses.18 A ≥40% reduction in dose was associated with reduced adverse effects, particularly EPS and elevation of prolactin levels. Moreover, the therapeutic window of striatal D2/D3 receptor occupancy appeared to be 50% to 60% in these older patients, compared with 65% to 80% in younger patients.
Long-term risks of antipsychotic treatment across the lifespan are less clear, with evidence suggesting both lower and higher mortality risk.19,20 It is difficult to fully disentangle the long-term risks of antipsychotics from the cumulative effects of lifestyle and comorbidity among individuals who have lived with schizophrenia for decades. Large naturalistic studies that include substantial numbers of older people with schizophrenia might be a way to elicit more information on long-term safety. The Schizophrenia Outpatient Health Outcome (SOHO) study was a large naturalistic trial that recruited >10,000 individuals with schizophrenia in 10 European countries.21 Although the SOHO study found differences between antipsychotics and adverse effects, such as EPS, weight gain, and sexual dysfunction, because the mean age of these patients was approximately 40 years and the follow-up period was only 3 years, it is difficult to draw conclusions that could be relevant to older individuals who have had schizophrenia for decades.
Bipolar Disorder
Clinical trials: Bipolar depression. A post hoc, secondary analysis of two 8-week, double-blind, randomized, placebo-controlled studies in bipolar depression compared 2 dosages of quetiapine (300 mg/d and 600 mg/d) with placebo in mixed-age patients.31 In a subgroup of 72 patients, ages 55 to 65, remission occurred more often with quetiapine than with placebo. Study discontinuation rates were similar between older people and younger people (age <55 years): quetiapine, 300 mg/d, 29.2%; quetiapine, 600 mg/d, 48.1%; and placebo, 29.6% in older adults, compared with 37.1%, 45.8%, and 38.1%, respectively, in younger adults. In all patients, the most common reason for discontinuation was adverse events with quetiapine and lack of efficacy for placebo. Adverse event rates were similar in older and younger adults. Dry mouth and dizziness were more common in older adults. Proportions of adults experiencing clinically significant weight gain (≥7% of body weight) were 5.3%, 8.3%, and 0% in older adults receiving quetiapine, 300 mg/d, quetiapine, 600 mg/d, and placebo, respectively, compared with 7.2%, 10.1%, and 2.6% in younger adults. EPS and treatment-emergent mania were minimal.
A secondary analysis of mixed-age, RCTs examined response in older adults (age ≥55 years) with bipolar I depression who received lurasidone as monotherapy or adjunctive therapy.32 In the monotherapy study, these patients were randomized to 6 weeks of lurasidone 20 to 60 mg/d, lurasidone 80 to 120 mg/d, or placebo. In the adjunctive therapy study, they were randomized to lurasidone 20 to 120 mg/d or placebo with either lithium or valproate. There were 83 older adults (17.1% of the sample) in the monotherapy study and 53 (15.6%) in the adjunctive therapy study. Mean improvement in depression was significantly higher for both doses of lurasidone monotherapy than placebo. Adjunctive lurasidone was not associated with statistically significant improvement vs placebo. The most frequent adverse events in older patients on lurasidone monotherapy 20 to 60 mg/d or 80 to 120 mg/d were nausea (18.5% and 9.7%, respectively) and somnolence (11.1% and 0%, respectively). Akathisia (9.7%) and insomnia (9.7%) were the most common adverse events in the group receiving 80 to 120 mg/d, with the rate of akathisia exhibiting a dose-related increase. Weight change with lurasidone was similar to placebo, and there were no clinically meaningful group changes in vital signs, electrocardiography, or laboratory parameters.
A small (N = 20) open study found improvement in older adults with bipolar depression with aripiprazole (mean dose, 10.3 mg/d).33 Adverse effects included restlessness and weight gain (n = 3, 9% each), sedation (n = 2, 10%), and drooling and diarrhea/loose stools (n = 1, 5% each). In another small study (N = 15) using asenapine (mean dose, 11.2 mg/d) in mainly older bipolar patients with depression, the most common adverse effects were gastrointestinal (GI) discomfort (n = 5, 33%) and restlessness, tremors, cognitive difficulties, and sluggishness (n = 2, 13% each).34
Clinical trials: Bipolar mania. Researchers conducted a pooled analysis of two 12-week randomized trials comparing quetiapine with placebo in a mixed-age sample with bipolar mania.35 In a subgroup of 59 older patients (mean age, 62.9 years), manic symptoms improved significantly more with quetiapine (modal dose, 550 mg/d) than with placebo. Adverse effects reported by >10% of older patients were dry mouth, somnolence, postural hypotension, insomnia, weight gain, and dizziness. Insomnia was reported by >10% of patients receiving placebo.
In a case series of 11 elderly patients with mania receiving asenapine, Baruch et al36 reported a 63% remission rate. One patient discontinued the study because of a new rash, 1 discontinued after developing peripheral edema, and 3 patients reported mild sedation.
Beyer et al37 reported on a post hoc analysis of 94 older adults (mean age, 57.1 years; range, 50.1 to 74.8 years) with acute bipolar mania receiving olanzapine (n = 47), divalproex (n = 31), or placebo (n = 16) in a pooled olanzapine clinical trials database. Patients receiving olanzapine or divalproex had improvement in mania; those receiving placebo did not improve. Safety findings were comparable with reports in younger patients with mania.
Other clinical data. Adverse effects found in mixed-age samples using secondary analyses of clinical trials need to be interpreted with caution because these types of studies usually exclude individuals with significant medical comorbidity. Medical burden, cognitive impairment, or concomitant medications generally necessitate slower drug titration and lower total daily dosing. For example, a secondary analysis of the U.S. National Institute of Health-funded Systematic Treatment Enhancement Program for Bipolar Disorder study, which had broader inclusion criteria than most clinical trials, reported that, although recovery rates in older adults with bipolar disorder were fairly good (78.5%), lower doses of risperidone were used in older vs younger patients.38
Clinical considerations
Interpretation of the relative risks of antipsychotics in older people must be tempered by the caveat that there is limited high-quality data (Table 4). Antipsychotics are the first-line therapy for older patients with schizophrenia, although their use is supported by a small number of prospective RCTs. SGAs are preferred because of their lower propensity to cause EPS and other motor adverse effects. Older persons with schizophrenia have an EPS threshold lower than younger patients and determining the lowest effective dosage may minimize EPS and cognitive adverse effects. As individuals with long-standing schizophrenia get older, their antipsychotic dosages may need to be reduced, and clinicians need to monitor for adverse effects that are more common among older people, such as tardive dyskinesia and metabolic abnormalities. In healthy, “younger” geriatric patients, monitoring for adverse effects may be similar to monitoring of younger patients. Patients who are older or frail may need more frequent assessment.
Like older adults with schizophrenia, geriatric patients with bipolar disorder have reduced drug tolerability and experience more adverse effects than younger patients. There are no prospective controlled studies that evaluated using antipsychotics in older patients with bipolar disorder. In older bipolar patients, the most problematic adverse effects of antipsychotics are akathisia, parkinsonism, other EPS, sedation and dizziness (which may increase fall risk), and GI discomfort. A key tolerability and safety consideration when treating older adults with bipolar disorder is the role of antipsychotics in relation to the use of lithium and mood stabilizers. Some studies have suggested that lithium has neuroprotective effects when used long-term; however, at least 1 report suggested that long-term antipsychotic treatment may be associated with neurodegeneration.39
The literature does not provide strong evidence on the many clinical variations that we see in routine practice settings, such as combinations of drug treatments or drugs prescribed to patients with specific comorbid conditions. There is a need for large cohort studies that monitor treatment course, medical comorbidity, and prognosis. Additionally, well-designed clinical trials such as the DART-AD, which investigated longer-term trajectories of people with dementia taking antipsychotics, should serve as a model for the type of research that is needed to better understand outcome variability among older people with chronic psychotic or bipolar disorders.40
1. Alexander GC, Gallagher SA, Mascola A, et al. Increasing off-label use of antipsychotic medications in the United States, 1995-2008. Pharmacoepidemiol Drug Saf. 2011;20(2):177-184.
2. United Nations, Department of Economic and Social Affairs, Population Division. World population ageing: 1950-2050. http://www.un.org/esa/population/publications/worldageing19502050. Accessed September 1, 2017.
3. Lawrence D, Kisely S, Pais J. The epidemiology of excess mortality in people with mental illness. Can J Psychiatry. 2010;55(12):752-760.
4. Cohen CI, Vahia I, Reyes P, et al. Focus on geriatric psychiatry: schizophrenia in later life: clinical symptoms and social well-being. Psychiatr Serv. 2008;59(3):232-234.
5. Jeste DV, Barak Y, Madhusoodanan S, et al. International multisite double-blind trial of the atypical antipsychotics risperidone and olanzapine in 175 elderly patients with chronic schizophrenia. Am J Geriatr Psychiatry. 2003;11(6):638-647.
6. Kalache SM, Mulsant BH, Davies SJ, et al. The impact of aging, cognition, and symptoms on functional competence in individuals with schizophrenia across the lifespan. Schizophr Bull. 2015;41(2):374-381.
7. Suzuki T, Remington G, Uchida H, et al. Management of schizophrenia in late life with antipsychotic medications: a qualitative review. Drugs Aging. 2011;28(12):961-980.
8. Mulsant BH, Pollock BG. Psychopharmacology. In: David C. Steffens DC, Blazer DG, Thakur ME (eds). The American Psychiatric Publishing Textbook of Geriatric Psychiatry, 5th Edition. Arlington, VA: American Psychiatric Publishing; 2015:527-587.
9. Cohen CI, Meesters PD, Zhao J. New perspectives on schizophrenia in later life: implications for treatment, policy, and research. Lancet Psychiatry. 2015;2(4):340-350.
10. Marriott RG, Neil W, Waddingham S. Antipsychotic medication for elderly people with schizophrenia. Cochrane Database Syst Rev. 2006;(1):CD005580.
11. Essali A, Ali G. Antipsychotic drug treatment for elderly people with late-onset schizophrenia. Cochrane Database Syst Rev. 2012(2):CD004162.
12. Scott J, Greenwald BS, Kramer E, et al. Atypical (second generation) antipsychotic treatment response in very late-onset schizophrenia-like psychosis. Int Psychogeriatr. 2011;23(5):742-748.
13. Rado J, Janicak PG. Pharmacological and clinical profile of recently approved second-generation antipsychotics: implications for treatment of schizophrenia in older patients. Drugs Aging. 2012;29(10):783-791.
14. Tzimos A, Samokhvalov V, Kramer M, et al. Safety and tolerability of oral paliperidone extended-release tablets in elderly patients with schizophrenia: a double-blind, placebo-controlled study with six-month open-label extension. Am J Geriatr Psychiatry. 2008;16(1):31-43.
15. Howanitz E, Pardo M, Smelson DA, et al. The efficacy and safety of clozapine versus chlorpromazine in geriatric schizophrenia. J Clin Psychiatry. 1999;60(1):41-44.
16. Sproule BA, Lake J, Mamo DC, et al. Are antipsychotic prescribing patterns different in older and younger adults?: a survey of 1357 psychiatric inpatients in Toronto. Can J Psychiatry. 2010;55(4):248-254.
17. Uchida H, Suzuki T, Mamo DC, et al. Effects of age and age of onset on prescribed antipsychotic dose in schizophrenia spectrum disorders: a survey of 1,418 patients in Japan. Am J Geriatr Psychiatry. 2008;16(7):584-593.
18. Graff-Guerrero A, Rajji TK, Mulsant BH, et al. Evaluation of antipsychotic dose reduction in late-life schizophrenia: a prospective dopamine D2/3 occupancy study. JAMA Psychiatry. 2015;72(9):927-934.
19. Khan A, Schwartz K, Stern C, et al. Mortality risk in patients with schizophrenia participating in premarketing atypical antipsychotic clinical trials. J Clin Psychiatry. 2007;68(12):1828-1833.
20. Weinmann S, Read J, Aderhold V. Influence of antipsychotics on mortality in schizophrenia: a systematic review. Schizophr Res. 2009;113(1):1-11.
21. Novick D, Haro JM, Perrin E, et al. Tolerability of outpatient antipsychotic treatment: 36-month results from the European Schizophrenia Outpatient Health Outcomes (SOHO) study. Eur Neuropsychopharmacol. 2009;19(8):542-550.
22. Sajatovic M, Blow FC, Ignacio RV, et al. Age-related modifiers of clinical presentation and health service use among veterans with bipolar disorder. Psychiatr Serv. 2004;55(9):1014-1021.
23. Jeste DV, Alexopoulos GS, Bartels SJ, et al. Consensus statement on the upcoming crisis in geriatric mental health: research agenda for the next 2 decades. Arch Gen Psychiatry. 1999;56(9):848-853.
24. Sajatovic M, Chen P. Geriatric bipolar disorder. Psychiatr Clin North Am. 2011;34(2):319-333,vii.
25. Sajatovic M, Strejilevich SA, Gildengers AG, et al. A report on older-age bipolar disorder from the International Society for Bipolar Disorders Task Force. Bipolar Disord. 2015;17(7):689-704.
26. Lala SV, Sajatovic M. Medical and psychiatric comorbidities among elderly individuals with bipolar disorder: a literature review. J Geriatr Psychiatry Neurol. 2012;25(1):20-25.
27. Dols A, Rhebergen D, Beekman A, et al. Psychiatric and medical comorbidities: results from a bipolar elderly cohort study. Am J Geriatr Psychiatry. 2014;22(11):1066-1074.
28. Pillarella J, Higashi A, Alexander GC, et al. Trends in use of second-generation antipsychotics for treatment of bipolar disorder in the United States, 1998-2009. Psychiatr Serv. 2012;63(1):83-86.
29. De Fruyt J, Deschepper E, Audenaert K, et al. Second generation antipsychotics in the treatment of bipolar depression: a systematic review and meta-analysis. J Psychopharmacol. 2012;26(5):603-617.
30. Nivoli AM, Murru A, Goikolea JM, et al. New treatment guidelines for acute bipolar mania: a critical review. J Affect Disord. 2012;140(2):125-141.
31. Sajatovic M, Paulsson B. Quetiapine for the treatment of depressive episodes in adults aged 55 to 65 years with bipolar disorder. Paper presented at: American Association of Geriatric Psychiatry Annual Meeting; 2007; New Orleans, LA.
32. Sajatovic M, Forester B, Tsai J, et al. Efficacy and safety of lurasidone in older adults with bipolar depression: analysis of two double-blind, placebo-controlled studies. Paper presented at: American College of Neuropsychopharmacology (ACNP) 53rd Annual Meeting; 2014; Phoenix, AZ.
33. Sajatovic M, Coconcea N, Ignacio RV, et al. Aripiprazole therapy in 20 older adults with bipolar disorder: a 12-week, open-label trial. J Clin Psychiatry. 2008;69(1):41-46.
34. Sajatovic M, Dines P, Fuentes-Casiano E, et al. Asenapine in the treatment of older adults with bipolar disorder. Int J Geriatr Psychiatry. 2015;30(7):710-719.
35. Sajatovic M, Calabrese JR, Mullen J. Quetiapine for the treatment of bipolar mania in older adults. Bipolar Disord. 2008;10(6):662-671.
36. Baruch Y, Tadger S, Plopski I, et al. Asenapine for elderly bipolar manic patients. J Affect Disord. 2013;145(1):130-132.
37. Beyer JL, Siegal A, Kennedy JS. Olanzapine, divalproex and placebo treatment, non-head to head comparisons of older adults acute mania. Paper presented at: 10th Congress of the International Psychogeriatric Association; 2001; Nice, France.
38. Al Jurdi RK, Marangell LB, Petersen NJ, et al. Prescription patterns of psychotropic medications in elderly compared with younger participants who achieved a “recovered” status in the systematic treatment enhancement program for bipolar disorder. Am J Geriatr Psychiatry. 2008;16(11):922-933.
39. Gildengers AG, Chung KH, Huang SH, et al. Neuroprogressive effects of lifetime illness duration in older adults with bipolar disorder. Bipolar Disord. 2014;16(6):617-623.
40. Ballard C, Lana MM, Theodoulou M, et al. A randomised, blinded, placebo-controlled trial in dementia patients continuing or stopping neuroleptics (the DART-AD trial). PLoS Med. 2008;5(4):e76.
1. Alexander GC, Gallagher SA, Mascola A, et al. Increasing off-label use of antipsychotic medications in the United States, 1995-2008. Pharmacoepidemiol Drug Saf. 2011;20(2):177-184.
2. United Nations, Department of Economic and Social Affairs, Population Division. World population ageing: 1950-2050. http://www.un.org/esa/population/publications/worldageing19502050. Accessed September 1, 2017.
3. Lawrence D, Kisely S, Pais J. The epidemiology of excess mortality in people with mental illness. Can J Psychiatry. 2010;55(12):752-760.
4. Cohen CI, Vahia I, Reyes P, et al. Focus on geriatric psychiatry: schizophrenia in later life: clinical symptoms and social well-being. Psychiatr Serv. 2008;59(3):232-234.
5. Jeste DV, Barak Y, Madhusoodanan S, et al. International multisite double-blind trial of the atypical antipsychotics risperidone and olanzapine in 175 elderly patients with chronic schizophrenia. Am J Geriatr Psychiatry. 2003;11(6):638-647.
6. Kalache SM, Mulsant BH, Davies SJ, et al. The impact of aging, cognition, and symptoms on functional competence in individuals with schizophrenia across the lifespan. Schizophr Bull. 2015;41(2):374-381.
7. Suzuki T, Remington G, Uchida H, et al. Management of schizophrenia in late life with antipsychotic medications: a qualitative review. Drugs Aging. 2011;28(12):961-980.
8. Mulsant BH, Pollock BG. Psychopharmacology. In: David C. Steffens DC, Blazer DG, Thakur ME (eds). The American Psychiatric Publishing Textbook of Geriatric Psychiatry, 5th Edition. Arlington, VA: American Psychiatric Publishing; 2015:527-587.
9. Cohen CI, Meesters PD, Zhao J. New perspectives on schizophrenia in later life: implications for treatment, policy, and research. Lancet Psychiatry. 2015;2(4):340-350.
10. Marriott RG, Neil W, Waddingham S. Antipsychotic medication for elderly people with schizophrenia. Cochrane Database Syst Rev. 2006;(1):CD005580.
11. Essali A, Ali G. Antipsychotic drug treatment for elderly people with late-onset schizophrenia. Cochrane Database Syst Rev. 2012(2):CD004162.
12. Scott J, Greenwald BS, Kramer E, et al. Atypical (second generation) antipsychotic treatment response in very late-onset schizophrenia-like psychosis. Int Psychogeriatr. 2011;23(5):742-748.
13. Rado J, Janicak PG. Pharmacological and clinical profile of recently approved second-generation antipsychotics: implications for treatment of schizophrenia in older patients. Drugs Aging. 2012;29(10):783-791.
14. Tzimos A, Samokhvalov V, Kramer M, et al. Safety and tolerability of oral paliperidone extended-release tablets in elderly patients with schizophrenia: a double-blind, placebo-controlled study with six-month open-label extension. Am J Geriatr Psychiatry. 2008;16(1):31-43.
15. Howanitz E, Pardo M, Smelson DA, et al. The efficacy and safety of clozapine versus chlorpromazine in geriatric schizophrenia. J Clin Psychiatry. 1999;60(1):41-44.
16. Sproule BA, Lake J, Mamo DC, et al. Are antipsychotic prescribing patterns different in older and younger adults?: a survey of 1357 psychiatric inpatients in Toronto. Can J Psychiatry. 2010;55(4):248-254.
17. Uchida H, Suzuki T, Mamo DC, et al. Effects of age and age of onset on prescribed antipsychotic dose in schizophrenia spectrum disorders: a survey of 1,418 patients in Japan. Am J Geriatr Psychiatry. 2008;16(7):584-593.
18. Graff-Guerrero A, Rajji TK, Mulsant BH, et al. Evaluation of antipsychotic dose reduction in late-life schizophrenia: a prospective dopamine D2/3 occupancy study. JAMA Psychiatry. 2015;72(9):927-934.
19. Khan A, Schwartz K, Stern C, et al. Mortality risk in patients with schizophrenia participating in premarketing atypical antipsychotic clinical trials. J Clin Psychiatry. 2007;68(12):1828-1833.
20. Weinmann S, Read J, Aderhold V. Influence of antipsychotics on mortality in schizophrenia: a systematic review. Schizophr Res. 2009;113(1):1-11.
21. Novick D, Haro JM, Perrin E, et al. Tolerability of outpatient antipsychotic treatment: 36-month results from the European Schizophrenia Outpatient Health Outcomes (SOHO) study. Eur Neuropsychopharmacol. 2009;19(8):542-550.
22. Sajatovic M, Blow FC, Ignacio RV, et al. Age-related modifiers of clinical presentation and health service use among veterans with bipolar disorder. Psychiatr Serv. 2004;55(9):1014-1021.
23. Jeste DV, Alexopoulos GS, Bartels SJ, et al. Consensus statement on the upcoming crisis in geriatric mental health: research agenda for the next 2 decades. Arch Gen Psychiatry. 1999;56(9):848-853.
24. Sajatovic M, Chen P. Geriatric bipolar disorder. Psychiatr Clin North Am. 2011;34(2):319-333,vii.
25. Sajatovic M, Strejilevich SA, Gildengers AG, et al. A report on older-age bipolar disorder from the International Society for Bipolar Disorders Task Force. Bipolar Disord. 2015;17(7):689-704.
26. Lala SV, Sajatovic M. Medical and psychiatric comorbidities among elderly individuals with bipolar disorder: a literature review. J Geriatr Psychiatry Neurol. 2012;25(1):20-25.
27. Dols A, Rhebergen D, Beekman A, et al. Psychiatric and medical comorbidities: results from a bipolar elderly cohort study. Am J Geriatr Psychiatry. 2014;22(11):1066-1074.
28. Pillarella J, Higashi A, Alexander GC, et al. Trends in use of second-generation antipsychotics for treatment of bipolar disorder in the United States, 1998-2009. Psychiatr Serv. 2012;63(1):83-86.
29. De Fruyt J, Deschepper E, Audenaert K, et al. Second generation antipsychotics in the treatment of bipolar depression: a systematic review and meta-analysis. J Psychopharmacol. 2012;26(5):603-617.
30. Nivoli AM, Murru A, Goikolea JM, et al. New treatment guidelines for acute bipolar mania: a critical review. J Affect Disord. 2012;140(2):125-141.
31. Sajatovic M, Paulsson B. Quetiapine for the treatment of depressive episodes in adults aged 55 to 65 years with bipolar disorder. Paper presented at: American Association of Geriatric Psychiatry Annual Meeting; 2007; New Orleans, LA.
32. Sajatovic M, Forester B, Tsai J, et al. Efficacy and safety of lurasidone in older adults with bipolar depression: analysis of two double-blind, placebo-controlled studies. Paper presented at: American College of Neuropsychopharmacology (ACNP) 53rd Annual Meeting; 2014; Phoenix, AZ.
33. Sajatovic M, Coconcea N, Ignacio RV, et al. Aripiprazole therapy in 20 older adults with bipolar disorder: a 12-week, open-label trial. J Clin Psychiatry. 2008;69(1):41-46.
34. Sajatovic M, Dines P, Fuentes-Casiano E, et al. Asenapine in the treatment of older adults with bipolar disorder. Int J Geriatr Psychiatry. 2015;30(7):710-719.
35. Sajatovic M, Calabrese JR, Mullen J. Quetiapine for the treatment of bipolar mania in older adults. Bipolar Disord. 2008;10(6):662-671.
36. Baruch Y, Tadger S, Plopski I, et al. Asenapine for elderly bipolar manic patients. J Affect Disord. 2013;145(1):130-132.
37. Beyer JL, Siegal A, Kennedy JS. Olanzapine, divalproex and placebo treatment, non-head to head comparisons of older adults acute mania. Paper presented at: 10th Congress of the International Psychogeriatric Association; 2001; Nice, France.
38. Al Jurdi RK, Marangell LB, Petersen NJ, et al. Prescription patterns of psychotropic medications in elderly compared with younger participants who achieved a “recovered” status in the systematic treatment enhancement program for bipolar disorder. Am J Geriatr Psychiatry. 2008;16(11):922-933.
39. Gildengers AG, Chung KH, Huang SH, et al. Neuroprogressive effects of lifetime illness duration in older adults with bipolar disorder. Bipolar Disord. 2014;16(6):617-623.
40. Ballard C, Lana MM, Theodoulou M, et al. A randomised, blinded, placebo-controlled trial in dementia patients continuing or stopping neuroleptics (the DART-AD trial). PLoS Med. 2008;5(4):e76.

Improving the recognition of borderline personality disorder
Borderline personality disorder (BPD) is associated with impaired psychosocial functioning,1-4 reduced health-related quality of life,5 high utilization of services,6,7 and excess mortality.8-10 Although BPD occurs in up to 40% of psychiatric inpatients11 and 10% of outpatients,12 it is underrecognized.13-15 Often, patients with BPD do not receive an accurate diagnosis until ≥10 years after initially seeking treatment.16 The treatment and clinical implications of failing to recognize BPD include overprescribing medication and underutilizing empirically effective psychotherapies.14
This review summarizes studies of the underdiagnosis of BPD in routine clinical practice, describes which patients should be screened, and reviews alternative approaches to screening.
Underrecognition of BPD
The Rhode Island Methods to Improve Diagnostic Assessment and Services (MIDAS) project is an ongoing clinical research study involving the integration of research assessment methods into routine clinical practice.17 In an early report from the MIDAS project, BPD diagnoses derived from structured and unstructured clinical interviews were compared between 2 groups of psychiatric outpatients in the same practice.15 Individuals in the structured interview cohort were 35 times more often diagnosed with BPD than individuals evaluated with an unstructured clinical interview. Importantly, when the information from the structured interview was presented to the clinicians, BPD was more likely to be diagnosed clinically.
Other studies13,16 also found that the rate of diagnosing BPD was higher when the diagnosis was based on a semi-structured diagnostic interview compared with an unstructured clinical interview, and that clinicians were reluctant to diagnose BPD during their routine intake diagnostic evaluation.
Clinicians, however, do not use semi-structured interviews in their practice, and they also do not tend to diagnose personality disorders (PDs) based on direct questioning, as they typically would when assessing a symptom-based disorder such as depression or anxiety. Rather, clinicians report that they rely on longitudinal observations to diagnose PDs.18 However, the results from the MIDAS project were inconsistent with clinicians’ reports. When clinicians were presented with the results of the semi-structured interview, they usually would diagnose BPD, even though it was the initial evaluation. If clinicians actually relied on longitudinal observations and considered information based on the direct question approach of research interviews to be irrelevant or invalid, then the results from the semi-structured interview should not have influenced the rate at which they diagnosed BPD. This suggests that the primary issue in diagnosing PDs is not the need for longitudinal observation but rather the need for more information, and that there is a role for screening questionnaires.
One potential criticism of studies demonstrating underrecognition of BPD in clinical practice is that patients typically were interviewed when they presented for treatment, when most were depressed or anxious. The possible pathologizing effects of psychiatric state on personality have been known for years.19 However, a large body of literature examining the treatment, prognostic, familial, and biological correlates of PDs supports the validity of diagnosing PDs in this manner. Moreover, from a clinical perspective, the sooner a clinician is aware of the presence of BPD, the more likely this information can be used for treatment planning.
Who should be screened for BPD?
BPD is underrecognized and underdiagnosed because patients with BPD often also have comorbid mood, anxiety, or substance use disorders.20,21 The symptoms associated with these disorders are typically the chief concern of patients with undiagnosed BPD who present for treatment. Patients with BPD rarely present for an intake evaluation and state that they are struggling with abandonment fears, chronic feelings of emptiness, or an identity disturbance. If patients identified these problems as their chief concerns, BPD would be easier to recognize.
Although several studies have documented the frequency of BPD in patients with a specific psychiatric diagnosis such as major depressive disorder (MDD) or attention-deficit/hyperactivity disorder,22-26 the MIDAS project examined the frequency of BPD in patients with various diagnoses and evaluated which disorders were associated with a significantly increased rate of BPD.27 The highest rate of BPD was found in patients with bipolar disorder. Approximately 25% of patients with bipolar II disorder and one-third of those with bipolar I disorder were diagnosed with BPD; these rates were significantly higher than the rate of BPD in patients without these disorders (Table 127). The rate of BPD was second highest in patients with a principal diagnosis of posttraumatic stress disorder (PTSD) and MDD; however, the rate of BPD in these patients was not significantly elevated compared with patients who did not have these principal diagnoses. Three disorders were associated with a significantly lower rate of BPD: adjustment disorder, dysthymic disorder, and generalized anxiety disorder.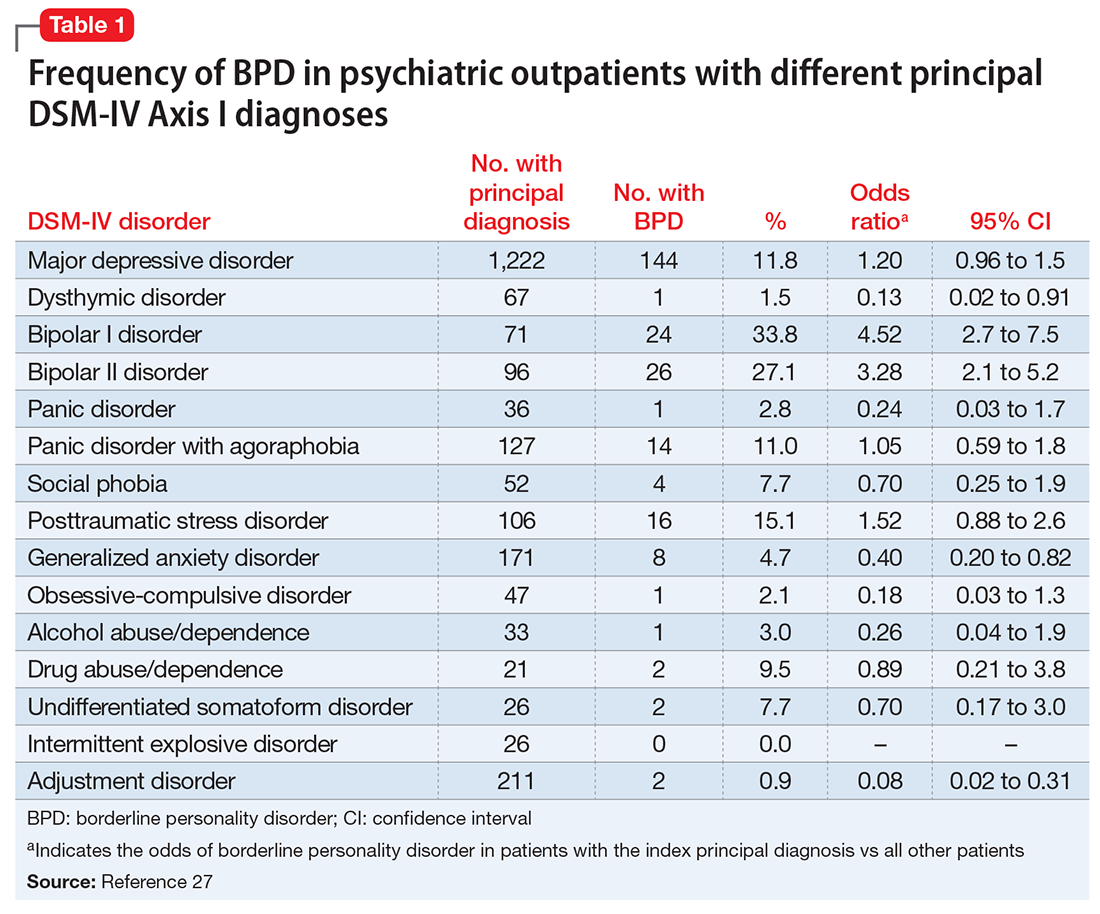
It would be easy to recommend screening for BPD in all psychiatric patients. However, that is not feasible or practical. In making screening recommendations, absolute risk should be considered more important than relative risk. Clinicians should screen for BPD in patients presenting to a general psychiatric outpatient practice with a principal diagnosis of MDD, bipolar disorder, PTSD, or panic disorder with agoraphobia. That is, I recommend screening for BPD in patients with a principal diagnosis in which the prevalence of BPD is ≥10% (Table 127).
A brief review of screening statistics
Screening tests for most psychiatric disorders are based on multi-item scales in which a total score is computed from a sum of item scores, and a cutoff point is established to determine who does and does not screen positive on the test. However, sensitivity, specificity, and positive and negative predictive values are not invariant properties of a screening test with a continuous score distribution. Rather, the performance statistics of a scale can be altered by changing the threshold score to distinguish cases from non-cases. When the screening threshold is lowered, sensitivity increases and specificity decreases.
For screening, a broad net needs to be cast so that all (or almost all) cases are included. Therefore, the cutoff score should be set low to prioritize the sensitivity of the instrument. A screening scale also should have high negative predictive value so that the clinician can be confident that patients who screen negative on the test do not have the disorder.
Screening questionnaires for BPD
Several questionnaires have been developed to screen for PDs (Table 228-35). Some screen for each of the DSM PDs,28,36-42 and some screen more broadly for the presence or absence of any PD.29,43,44 The most commonly studied self-report scale for BPD is the McLean Screening Instrument for Borderline Personality Disorder (MSI-BPD),30 a 10-item self-report scale derived from a subset of questions from the BPD module of a semi-structured diagnostic interview.
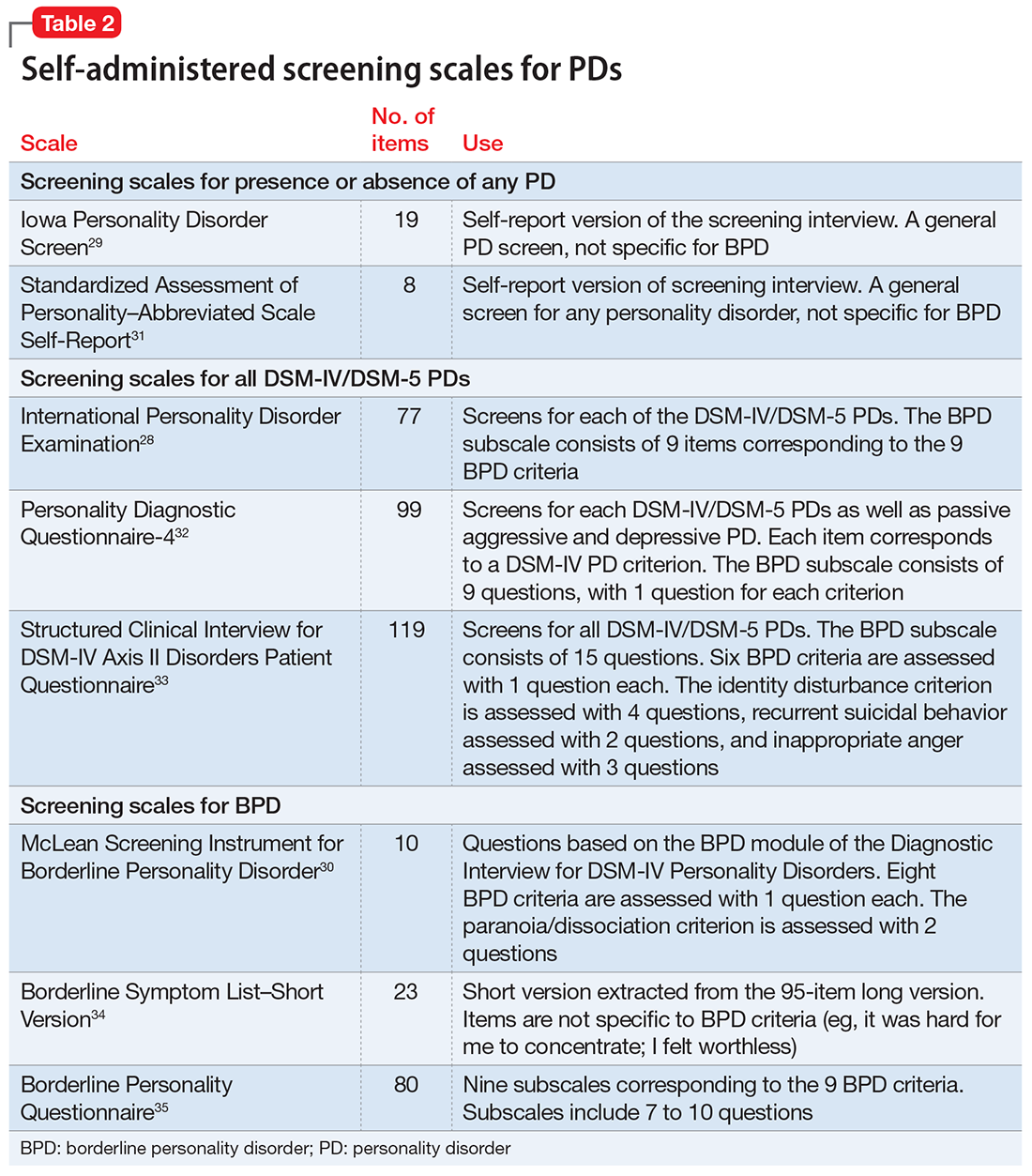
The initial validation study30 found that the optimal cutoff score was 7, which resulted in a sensitivity of 81% and specificity of 89%. Three studies have evaluated the scale in adolescents and young adults,45-47 and 3 studies examined the scale in adult outpatients.48-50 Across all 6 studies, at the optimal cutoff scores determined in each study, the sensitivity of the MSI-BPD ranged from 68% to 94% (mean, 80%) and the specificity ranged from 66% to 80% (mean, 72%).
Problems with screening questionnaires. Although screening scales have been developed for many psychiatric disorders, they have not been widely used in mental health settings. In a previous commentary, I argued that the conceptual justification for using self-report screening scales for single disorders in psychiatric settings is questionable.51 Another problem with screening scales is their potential misuse as case-finding instruments. In the literature on bipolar disorder screening, several researchers misconstrued a positive screen to indicate caseness.51 Although this is not a problem with the screening measures or the selection of a cutoff score, caution must be taken to not confuse screening with diagnosis.52
Screening for BPD as part of your diagnostic interview
An alternative approach to using self-administered questionnaires for screening is for clinicians to include questions in their evaluation as part of a psychiatric review of systems. When conducting a diagnostic interview, clinicians typically screen for disorders that are comorbid with the principal diagnosis by asking about the comorbid disorders’ necessary features or “gate criteria.” For example, in a patient with a principal diagnosis of MDD, the clinician would inquire about the presence of panic attacks, excessive worry, or substance use to screen for the presence of panic disorder, generalized anxiety disorder, or a substance use disorder. In contrast, for polythetically defined disorders such as BPD, there is no single gate criterion, because the disorder is diagnosed based on the presence of at least 5 of 9 criteria and no single one of these criteria is required to be present to establish the diagnosis.
As part of the MIDAS project, the psychometric properties of the BPD criteria were examined to determine if it was possible to identify 1 or 2 criteria that could serve as gate criteria to screen for the disorder. If the sensitivity of 1 criterion or a combination of 2 BPD criteria was sufficiently high (ie, >90%), then the assessment of this criterion (or these criteria) could be included in a psychiatric review of systems, thus potentially improving the detection of BPD. Researchers hypothesized that affective instability, considered first by Linehan53 and later by other theorists54 to be of central importance to the clinical manifestations of BPD, could function as a gate criterion. In the sample of 3,674 psychiatric outpatients who were evaluated with a semi-structured interview, the sensitivity of the affective instability criterion was 92.8%, and the negative predictive value of the criterion was 99%.
Identifying a single BPD criterion that is present in the vast majority of patients diagnosed with BPD will allow clinicians to follow their usual clinical practice when conducting a psychiatric review of systems and inquire about the gate criteria of various disorders. Several studies have found that >90% of patients with BPD report affective instability. However, this does not mean that the diagnosis of BPD can be abbreviated to an assessment of the presence or absence of affective instability. Many patients who screen positive will not have BPD when a more definitive diagnostic evaluation is conducted. In the case of BPD, the more costly definitive diagnostic procedure simply entails inquiry of the other diagnostic criteria.
1. Bellino S, Patria L, Paradiso E, et al. Major depression in patients with borderline personality disorder: a clinical investigation. Can J Psychiatry. 2005;50(4):234-238.
2. Skodol AE, Gunderson JG, McGlashan TH, et al. Functional impairment in patients with schizotypal, borderline, avoidant, or obsessive-compulsive personality disorder. Am J Psychiatry. 2002;159(2):276-283.
3. Gunderson JG, Stout RL, McGlashan TH, et al. Ten-year course of borderline personality disorder: psychopathology and function from the Collaborative Longitudinal Personality Disorders study. Arch Gen Psychiatry. 2011;68(8):827-837.
4. Zanarini MC, Jacoby RJ, Frankenburg FR, et al. The 10-year course of social security disability income reported by patients with borderline personality disorder and axis II comparison subjects. J Pers Disord. 2009;23(4):346-356.
5. Grant BF, Chou SP, Goldstein RB, et al. Prevalence, correlates, disability, and comorbidity of DSM-IV borderline personality disorder: results from the Wave 2 National Epidemiologic Survey on Alcohol and Related Conditions. J Clin Psychiatry. 2008;69(4):533-545.
6. Bender DS, Dolan RT, Skodol AE, et al. Treatment utilization by patients with personality disorders. Am J Psychiatry. 2001;158(2):295-302.
7. Zanarini MC, Frankenburg FR, Hennen J, et al. Mental health service utilization by borderline personality disorder patients and Axis II comparison subjects followed prospectively for 6 years. J Clin Psychiatry. 2004;65(1):28-36.
8. Pompili M, Girardi P, Ruberto A, et al. Suicide in borderline personality disorder: a meta-analysis. Nord J Psychiatry. 2005;59(5):319-324.
9. Oldham JM. Borderline personality disorder and suicidality. Am J Psychiatry. 2006;163(1):20-26.
10. Black DW, Blum N, Pfohl B, et al. Suicidal behavior in borderline personality disorder: prevalence, risk factors, prediction, and prevention. J Pers Disord. 2004;18(3):226-239.
11. Marinangeli M, Butti G, Scinto A, et al. Patterns of comorbidity among DSM-III-R personality disorders. Psychopathology. 2000;33(2):69-74.
12. Zimmerman M, Rothschild L, Chelminski I. The frequency of DSM-IV personality disorders in psychiatric outpatients. Am J Psychiatry. 2005;162(10):1911-1918.
13. Comtois KA, Carmel A. Borderline personality disorder and high utilization of inpatient psychiatric hospitalization: concordance between research and clinical diagnosis. J Behav Health Servi Res. 2016;43(2):272-280.
14. Paris J, Black DW. Borderline personality disorder and bipolar disorder: what is the difference and why does it matter? J Nerv Ment Dis. 2015;203(1):3-7.
15. Zimmerman M, Mattia JI. Differences between clinical and research practice in diagnosing borderline personality disorder. Am J Psychiatry. 1999;156(10):1570-1574.
16. Magnavita JJ, Levy KN, Critchfield KL, et al. Ethical considerations in treatment of personality dysfunction: using evidence, principles, and clinical judgment. Professional Psychology: Research and Practice. 2010;41(1):64-74.
17. Zimmerman M. A review of 20 years of research on overdiagnosis and underdiagnosis in the Rhode Island Methods to Improve Diagnostic Assessment and Services (MIDAS) Project. Can J Psychiatry. 2016;61(2):71-79.
18. Westen D. Divergences between clinical and research methods for assessing personality disorders: implications for research and the evolution of axis II. Am J Psychiatry. 1997;154(7):895-903.
19. Zimmerman M. Diagnosing personality disorders: a review of issues and research methods. Arch Gen Psychiatry. 1994;51(3):225-245.
20. Zanarini MC, Gunderson JG, Frankenberg FR. Axis I phenomenology of borderline personality disorder. Compr Psychiatry. 1989;30(2):149-156.
21. Zimmerman M, Mattia JI. Axis I diagnostic comorbidity and borderline personality disorder. Compr Psychiatry. 1999;40(4):245-252.
22. Gunderson JG, Morey LC, Stout RL, et al. Major depressive disorder and borderline personality disorder revisited: longitudinal interactions. J Clin Psychiatry. 2004;65(8):1049-1056.
23. Bayes AJ, Parker GB. Clinical vs. DSM diagnosis of bipolar disorder, borderline personality disorder and their co-occurrence. Acta Psychiatr Scand. 2016;135(3):259-265.
24. Carpenter RW, Wood PK, Trull TJ. Comorbidity of borderline personality disorder and lifetime substance use disorders in a nationally representative sample. J Pers Disord. 2016;30(3):336-350.
25. Trull TJ, Sher KJ, Minks-Brown C, et al. Borderline personality disorder and substance use disorders: a review and integration. Clin Psychol Rev. 2000;20(2):235-253.
26. Matthies SD, Philipsen A. Common ground in attention deficit hyperactivity disorder (ADHD) and borderline personality disorder (BPD)-review of recent findings. Borderline Personal Disord Emot Dysregul. 2014;1:3.
27. Zimmerman M, Chelminski I, Dalrymple K, et al. Principal diagnoses in psychiatric outpatients with borderline personality disorder: implications for screening recommendations. Ann Clin Psychiatry. 2017;29(1):54-60.
28. Magallón-Neri EM, Forns M, Canalda G, et al. Usefulness of the International Personality Disorder Examination Screening Questionnaire for borderline and impulsive personality pathology in adolescents. Compr Psychiatry. 2013;54(3):301-308.
29. Germans S, Van Heck GL, Langbehn DR, et al. The Iowa Personality Disorder Screen. Eur J Psychol Assess. 2010;26(1):11-18.
30. Zanarini MC, Vujanovic AA, Parachini EA, et al. A screening measure for BPD: the McLean Screening Instrument for Borderline Personality Disorder (MSI-BPD). J Pers Disord. 2003;17(6):568-573.
31. Germans S, Van Heck GL, Hodiamont PP. Results of the search for personality disorder screening tools: clinical implications. J Clin Psychiatry. 2012;73(2):165-173.
32. Hyler SE. Personality diagnostic questionnaire-4. New York, NY: New York State Psychiatric Institute; 1994.
33. First MB, Spitzer RL, Gibbon M, et al. Structured Clinical Interview for DSM-IV Axis II Disorders - Patient edition (SCID-I/P, version 2.0). New York, NY: Biometrics Research Department, New York State Psychiatric Institute; 1995.
34. Bohus M, Kleindienst N, Limberger MF, et al. The short version of the Borderline Symptom List (BSL-23): development and initial data on psychometric properties. Psychopathology. 2009;42(1):32-39.
35. Poreh AM, Rawlings D, Claridge G, et al. The BPQ: a scale for the assessment of boderline personality based on DSM-IV criteria. J Pers Disord. 2006;20(3):247-260.
36. Ekselius L, Lindstrom E, von Knorring L, et al. SCID II interviews and the SCID Screen questionnaire as diagnostic tools for personality disorders in DSM-III-R. Acta Psychiatr Scand. 1994;90(2):120-123.
37. Hyler SE, Skodol AE, Oldham JM, et al. Validity of the Personality Diagnostic Questionnaire-Revised: a replication in an outpatient sample. Compr Psychiatry. 1992;33(2):73-77.
38. Davison S, Leese M, Taylor PJ. Examination of the screening properties of the personality diagnostic questionnaire 4+ (PDQ-4+) in a prison population. J Pers Disord. 2001;15(2):180-194.
39. Jacobsberg L, Perry S, Frances A. Diagnostic agreement between the SCID-II screening questionnaire and the Personality Disorder Examination. J Pers Assess. 1995;65(3):428-433.
40. Germans S, Van Heck GL, Masthoff ED, et al. Diagnostic efficiency among psychiatric outpatients of a self-report version of a subset of screen items of the Structured Clinical Interview for DSM-IV-TR Personality Disorders (SCID-II). Psychol Assess. 2010;22(4):945-952.
41. Lloyd C, Overall JE, Click M Jr. Screening for borderline personality disorders with the MMPI-168. J Clin Psychol. 1983;39(5):722-726.
42. Neal LA, Fox C, Carroll N, et al. Development and validation of a computerized screening test for personality disorders in DSM-III-R. Acta Psychiatr Scand. 1997;95(4):351-356.
43. Germans S, Van Heck GL, Moran P, et al. The Self-Report Standardized Assessment of Personality-abbreviated Scale: preliminary results of a brief screening test for personality disorders. Pers Ment Health. 2008;2(2):70-76.
44. Moran P, Leese M, Lee T, et al. Standardized Assessment of Personality - Abbreviated Scale (SAPAS): preliminary validation of a brief screen for personality disorder. Br J Psychiatry. 2003;183:228-232.
45. Chanen AM, Jovev M, Djaja D, et al. Screening for borderline personality disorder in outpatient youth. J Pers Disord. 2008;22(4):353-364.
46. van Alebeek A, van der Heijden PT, Hessels C, et al. Comparison of three questionnaires to screen for borderline personality disorder in adolescents and young adults. Eur J Psychol Assess. 2017:33;123-128.
47. Noblin JL, Venta A, Sharp C. The validity of the MSI-BPD among inpatient adolescents. Assessment. 2014;21(2):210-217.
48. Kröger C, Vonau M, Kliem S, et al. Emotion dysregulation as a core feature of borderline personality disorder: comparison of the discriminatory ability of two self-rating measures. Psychopathology. 2011;44(4):253-260.
49. Soler J, Domínguez-Clav E, García-Rizo C, et al. Validation of the Spanish version of the McLean Screening Instrument for Borderline Personality Disorder. Rev Psiquiatr Salud Ment. 2016;9(4):195-202.
50. Melartin T, Häkkinen M, Koivisto M, et al. Screening of psychiatric outpatients for borderline personality disorder with the McLean Screening Instrument for Borderline Personality Disorder (MSI-BPD). Nord J Psychiatry. 2009;63(6):475-479.
51. Zimmerman M. Misuse of the Mood Disorders Questionnaire as a case-finding measure and a critique of the concept of using a screening scale for bipolar disorder in psychiatric practice. Bipolar Disord. 2012;14(2):127-134.
52. Zimmerman M. Screening for bipolar disorder: confusion between case-finding and screening. Psychother Psychosom. 2014;83(5):259-262.
53. Linehan MM. Cognitive-behavioral treatment of borderline personality disorder. New York, NY: Guilford Press; 1993.
54. Koenigsberg HW, Harvey PD, Mitropoulou V, et al. Are the interpersonal and identity disturbances in the borderline personality disorder criteria linked to the traits of affective instability and impulsivity? J Pers Disord. 2001;15(4):358-370.
55. Grilo CM, Becker DF, Anez LM, et al. Diagnostic efficiency of DSM-IV criteria for borderline personality disorder: an evaluation in Hispanic men and women with substance use disorders. J Consult Clin Psychol. 2004;72(1):126-131.
56. Korfine L, Hooley JM. Detecting individuals with borderline personality disorder in the community: an ascertainment strategy and comparison with a hospital sample. J Pers Disord. 2009;23(1):62-75.
57. Leppänen V, Lindeman S, Arntz A, et al. Preliminary evaluation of psychometric properties of the Finnish Borderline Personality Disorder Severity Index: Oulu-BPD-Study. Nord J Psychiatry. 2013;67(5):312-319.
58. Pfohl B, Coryell W, Zimmerman M, et al. DSM-III personality disorders: diagnostic overlap and internal consistency of individual DSM-III criteria. Compr Psychiatry. 1986;27(1):22-34.
59. Reich J. Criteria for diagnosing DSM-III borderline personality disorder. Ann Clin Psychiatry. 1990;2(3):189-197.
60. Nurnberg HG, Raskin M, Levine PE, et al. Hierarchy of DSM-III-R criteria efficiency for the diagnosis of borderline personality disorder. J Pers Disord. 1991;5(3):211-224.
61. Farmer RF, Chapman AL. Evaluation of DSM-IV personality disorder criteria as assessed by the structured clinical interview for DSM-IV personality disorders. Compr Psychiatry. 2002;43(4):285-300.
62. Grilo CM, McGlashan TH, Morey LC, et al. Internal consistency, intercriterion overlap and diagnostic efficiency of criteria sets for DSM-IV schizotypal, borderline, avoidant and obsessive-compulsive personality disorders. Acta Psychiatr Scand. 2001;104(4):264-272.
63. Grilo CM, Sanislow CA, Skodol AE, et al. Longitudinal diagnostic efficiency of DSM-IV criteria for borderline personality disorder: a 2-year prospective study. Can J Psychiatry. 2007;52(6):357-362.
Borderline personality disorder (BPD) is associated with impaired psychosocial functioning,1-4 reduced health-related quality of life,5 high utilization of services,6,7 and excess mortality.8-10 Although BPD occurs in up to 40% of psychiatric inpatients11 and 10% of outpatients,12 it is underrecognized.13-15 Often, patients with BPD do not receive an accurate diagnosis until ≥10 years after initially seeking treatment.16 The treatment and clinical implications of failing to recognize BPD include overprescribing medication and underutilizing empirically effective psychotherapies.14
This review summarizes studies of the underdiagnosis of BPD in routine clinical practice, describes which patients should be screened, and reviews alternative approaches to screening.
Underrecognition of BPD
The Rhode Island Methods to Improve Diagnostic Assessment and Services (MIDAS) project is an ongoing clinical research study involving the integration of research assessment methods into routine clinical practice.17 In an early report from the MIDAS project, BPD diagnoses derived from structured and unstructured clinical interviews were compared between 2 groups of psychiatric outpatients in the same practice.15 Individuals in the structured interview cohort were 35 times more often diagnosed with BPD than individuals evaluated with an unstructured clinical interview. Importantly, when the information from the structured interview was presented to the clinicians, BPD was more likely to be diagnosed clinically.
Other studies13,16 also found that the rate of diagnosing BPD was higher when the diagnosis was based on a semi-structured diagnostic interview compared with an unstructured clinical interview, and that clinicians were reluctant to diagnose BPD during their routine intake diagnostic evaluation.
Clinicians, however, do not use semi-structured interviews in their practice, and they also do not tend to diagnose personality disorders (PDs) based on direct questioning, as they typically would when assessing a symptom-based disorder such as depression or anxiety. Rather, clinicians report that they rely on longitudinal observations to diagnose PDs.18 However, the results from the MIDAS project were inconsistent with clinicians’ reports. When clinicians were presented with the results of the semi-structured interview, they usually would diagnose BPD, even though it was the initial evaluation. If clinicians actually relied on longitudinal observations and considered information based on the direct question approach of research interviews to be irrelevant or invalid, then the results from the semi-structured interview should not have influenced the rate at which they diagnosed BPD. This suggests that the primary issue in diagnosing PDs is not the need for longitudinal observation but rather the need for more information, and that there is a role for screening questionnaires.
One potential criticism of studies demonstrating underrecognition of BPD in clinical practice is that patients typically were interviewed when they presented for treatment, when most were depressed or anxious. The possible pathologizing effects of psychiatric state on personality have been known for years.19 However, a large body of literature examining the treatment, prognostic, familial, and biological correlates of PDs supports the validity of diagnosing PDs in this manner. Moreover, from a clinical perspective, the sooner a clinician is aware of the presence of BPD, the more likely this information can be used for treatment planning.
Who should be screened for BPD?
BPD is underrecognized and underdiagnosed because patients with BPD often also have comorbid mood, anxiety, or substance use disorders.20,21 The symptoms associated with these disorders are typically the chief concern of patients with undiagnosed BPD who present for treatment. Patients with BPD rarely present for an intake evaluation and state that they are struggling with abandonment fears, chronic feelings of emptiness, or an identity disturbance. If patients identified these problems as their chief concerns, BPD would be easier to recognize.
Although several studies have documented the frequency of BPD in patients with a specific psychiatric diagnosis such as major depressive disorder (MDD) or attention-deficit/hyperactivity disorder,22-26 the MIDAS project examined the frequency of BPD in patients with various diagnoses and evaluated which disorders were associated with a significantly increased rate of BPD.27 The highest rate of BPD was found in patients with bipolar disorder. Approximately 25% of patients with bipolar II disorder and one-third of those with bipolar I disorder were diagnosed with BPD; these rates were significantly higher than the rate of BPD in patients without these disorders (Table 127). The rate of BPD was second highest in patients with a principal diagnosis of posttraumatic stress disorder (PTSD) and MDD; however, the rate of BPD in these patients was not significantly elevated compared with patients who did not have these principal diagnoses. Three disorders were associated with a significantly lower rate of BPD: adjustment disorder, dysthymic disorder, and generalized anxiety disorder.
It would be easy to recommend screening for BPD in all psychiatric patients. However, that is not feasible or practical. In making screening recommendations, absolute risk should be considered more important than relative risk. Clinicians should screen for BPD in patients presenting to a general psychiatric outpatient practice with a principal diagnosis of MDD, bipolar disorder, PTSD, or panic disorder with agoraphobia. That is, I recommend screening for BPD in patients with a principal diagnosis in which the prevalence of BPD is ≥10% (Table 127).
A brief review of screening statistics
Screening tests for most psychiatric disorders are based on multi-item scales in which a total score is computed from a sum of item scores, and a cutoff point is established to determine who does and does not screen positive on the test. However, sensitivity, specificity, and positive and negative predictive values are not invariant properties of a screening test with a continuous score distribution. Rather, the performance statistics of a scale can be altered by changing the threshold score to distinguish cases from non-cases. When the screening threshold is lowered, sensitivity increases and specificity decreases.
For screening, a broad net needs to be cast so that all (or almost all) cases are included. Therefore, the cutoff score should be set low to prioritize the sensitivity of the instrument. A screening scale also should have high negative predictive value so that the clinician can be confident that patients who screen negative on the test do not have the disorder.
Screening questionnaires for BPD
Several questionnaires have been developed to screen for PDs (Table 228-35). Some screen for each of the DSM PDs,28,36-42 and some screen more broadly for the presence or absence of any PD.29,43,44 The most commonly studied self-report scale for BPD is the McLean Screening Instrument for Borderline Personality Disorder (MSI-BPD),30 a 10-item self-report scale derived from a subset of questions from the BPD module of a semi-structured diagnostic interview.
The initial validation study30 found that the optimal cutoff score was 7, which resulted in a sensitivity of 81% and specificity of 89%. Three studies have evaluated the scale in adolescents and young adults,45-47 and 3 studies examined the scale in adult outpatients.48-50 Across all 6 studies, at the optimal cutoff scores determined in each study, the sensitivity of the MSI-BPD ranged from 68% to 94% (mean, 80%) and the specificity ranged from 66% to 80% (mean, 72%).
Problems with screening questionnaires. Although screening scales have been developed for many psychiatric disorders, they have not been widely used in mental health settings. In a previous commentary, I argued that the conceptual justification for using self-report screening scales for single disorders in psychiatric settings is questionable.51 Another problem with screening scales is their potential misuse as case-finding instruments. In the literature on bipolar disorder screening, several researchers misconstrued a positive screen to indicate caseness.51 Although this is not a problem with the screening measures or the selection of a cutoff score, caution must be taken to not confuse screening with diagnosis.52
Screening for BPD as part of your diagnostic interview
An alternative approach to using self-administered questionnaires for screening is for clinicians to include questions in their evaluation as part of a psychiatric review of systems. When conducting a diagnostic interview, clinicians typically screen for disorders that are comorbid with the principal diagnosis by asking about the comorbid disorders’ necessary features or “gate criteria.” For example, in a patient with a principal diagnosis of MDD, the clinician would inquire about the presence of panic attacks, excessive worry, or substance use to screen for the presence of panic disorder, generalized anxiety disorder, or a substance use disorder. In contrast, for polythetically defined disorders such as BPD, there is no single gate criterion, because the disorder is diagnosed based on the presence of at least 5 of 9 criteria and no single one of these criteria is required to be present to establish the diagnosis.
As part of the MIDAS project, the psychometric properties of the BPD criteria were examined to determine if it was possible to identify 1 or 2 criteria that could serve as gate criteria to screen for the disorder. If the sensitivity of 1 criterion or a combination of 2 BPD criteria was sufficiently high (ie, >90%), then the assessment of this criterion (or these criteria) could be included in a psychiatric review of systems, thus potentially improving the detection of BPD. Researchers hypothesized that affective instability, considered first by Linehan53 and later by other theorists54 to be of central importance to the clinical manifestations of BPD, could function as a gate criterion. In the sample of 3,674 psychiatric outpatients who were evaluated with a semi-structured interview, the sensitivity of the affective instability criterion was 92.8%, and the negative predictive value of the criterion was 99%.
Identifying a single BPD criterion that is present in the vast majority of patients diagnosed with BPD will allow clinicians to follow their usual clinical practice when conducting a psychiatric review of systems and inquire about the gate criteria of various disorders. Several studies have found that >90% of patients with BPD report affective instability. However, this does not mean that the diagnosis of BPD can be abbreviated to an assessment of the presence or absence of affective instability. Many patients who screen positive will not have BPD when a more definitive diagnostic evaluation is conducted. In the case of BPD, the more costly definitive diagnostic procedure simply entails inquiry of the other diagnostic criteria.
Borderline personality disorder (BPD) is associated with impaired psychosocial functioning,1-4 reduced health-related quality of life,5 high utilization of services,6,7 and excess mortality.8-10 Although BPD occurs in up to 40% of psychiatric inpatients11 and 10% of outpatients,12 it is underrecognized.13-15 Often, patients with BPD do not receive an accurate diagnosis until ≥10 years after initially seeking treatment.16 The treatment and clinical implications of failing to recognize BPD include overprescribing medication and underutilizing empirically effective psychotherapies.14
This review summarizes studies of the underdiagnosis of BPD in routine clinical practice, describes which patients should be screened, and reviews alternative approaches to screening.
Underrecognition of BPD
The Rhode Island Methods to Improve Diagnostic Assessment and Services (MIDAS) project is an ongoing clinical research study involving the integration of research assessment methods into routine clinical practice.17 In an early report from the MIDAS project, BPD diagnoses derived from structured and unstructured clinical interviews were compared between 2 groups of psychiatric outpatients in the same practice.15 Individuals in the structured interview cohort were 35 times more often diagnosed with BPD than individuals evaluated with an unstructured clinical interview. Importantly, when the information from the structured interview was presented to the clinicians, BPD was more likely to be diagnosed clinically.
Other studies13,16 also found that the rate of diagnosing BPD was higher when the diagnosis was based on a semi-structured diagnostic interview compared with an unstructured clinical interview, and that clinicians were reluctant to diagnose BPD during their routine intake diagnostic evaluation.
Clinicians, however, do not use semi-structured interviews in their practice, and they also do not tend to diagnose personality disorders (PDs) based on direct questioning, as they typically would when assessing a symptom-based disorder such as depression or anxiety. Rather, clinicians report that they rely on longitudinal observations to diagnose PDs.18 However, the results from the MIDAS project were inconsistent with clinicians’ reports. When clinicians were presented with the results of the semi-structured interview, they usually would diagnose BPD, even though it was the initial evaluation. If clinicians actually relied on longitudinal observations and considered information based on the direct question approach of research interviews to be irrelevant or invalid, then the results from the semi-structured interview should not have influenced the rate at which they diagnosed BPD. This suggests that the primary issue in diagnosing PDs is not the need for longitudinal observation but rather the need for more information, and that there is a role for screening questionnaires.
One potential criticism of studies demonstrating underrecognition of BPD in clinical practice is that patients typically were interviewed when they presented for treatment, when most were depressed or anxious. The possible pathologizing effects of psychiatric state on personality have been known for years.19 However, a large body of literature examining the treatment, prognostic, familial, and biological correlates of PDs supports the validity of diagnosing PDs in this manner. Moreover, from a clinical perspective, the sooner a clinician is aware of the presence of BPD, the more likely this information can be used for treatment planning.
Who should be screened for BPD?
BPD is underrecognized and underdiagnosed because patients with BPD often also have comorbid mood, anxiety, or substance use disorders.20,21 The symptoms associated with these disorders are typically the chief concern of patients with undiagnosed BPD who present for treatment. Patients with BPD rarely present for an intake evaluation and state that they are struggling with abandonment fears, chronic feelings of emptiness, or an identity disturbance. If patients identified these problems as their chief concerns, BPD would be easier to recognize.
Although several studies have documented the frequency of BPD in patients with a specific psychiatric diagnosis such as major depressive disorder (MDD) or attention-deficit/hyperactivity disorder,22-26 the MIDAS project examined the frequency of BPD in patients with various diagnoses and evaluated which disorders were associated with a significantly increased rate of BPD.27 The highest rate of BPD was found in patients with bipolar disorder. Approximately 25% of patients with bipolar II disorder and one-third of those with bipolar I disorder were diagnosed with BPD; these rates were significantly higher than the rate of BPD in patients without these disorders (Table 127). The rate of BPD was second highest in patients with a principal diagnosis of posttraumatic stress disorder (PTSD) and MDD; however, the rate of BPD in these patients was not significantly elevated compared with patients who did not have these principal diagnoses. Three disorders were associated with a significantly lower rate of BPD: adjustment disorder, dysthymic disorder, and generalized anxiety disorder.
It would be easy to recommend screening for BPD in all psychiatric patients. However, that is not feasible or practical. In making screening recommendations, absolute risk should be considered more important than relative risk. Clinicians should screen for BPD in patients presenting to a general psychiatric outpatient practice with a principal diagnosis of MDD, bipolar disorder, PTSD, or panic disorder with agoraphobia. That is, I recommend screening for BPD in patients with a principal diagnosis in which the prevalence of BPD is ≥10% (Table 127).
A brief review of screening statistics
Screening tests for most psychiatric disorders are based on multi-item scales in which a total score is computed from a sum of item scores, and a cutoff point is established to determine who does and does not screen positive on the test. However, sensitivity, specificity, and positive and negative predictive values are not invariant properties of a screening test with a continuous score distribution. Rather, the performance statistics of a scale can be altered by changing the threshold score to distinguish cases from non-cases. When the screening threshold is lowered, sensitivity increases and specificity decreases.
For screening, a broad net needs to be cast so that all (or almost all) cases are included. Therefore, the cutoff score should be set low to prioritize the sensitivity of the instrument. A screening scale also should have high negative predictive value so that the clinician can be confident that patients who screen negative on the test do not have the disorder.
Screening questionnaires for BPD
Several questionnaires have been developed to screen for PDs (Table 228-35). Some screen for each of the DSM PDs,28,36-42 and some screen more broadly for the presence or absence of any PD.29,43,44 The most commonly studied self-report scale for BPD is the McLean Screening Instrument for Borderline Personality Disorder (MSI-BPD),30 a 10-item self-report scale derived from a subset of questions from the BPD module of a semi-structured diagnostic interview.
The initial validation study30 found that the optimal cutoff score was 7, which resulted in a sensitivity of 81% and specificity of 89%. Three studies have evaluated the scale in adolescents and young adults,45-47 and 3 studies examined the scale in adult outpatients.48-50 Across all 6 studies, at the optimal cutoff scores determined in each study, the sensitivity of the MSI-BPD ranged from 68% to 94% (mean, 80%) and the specificity ranged from 66% to 80% (mean, 72%).
Problems with screening questionnaires. Although screening scales have been developed for many psychiatric disorders, they have not been widely used in mental health settings. In a previous commentary, I argued that the conceptual justification for using self-report screening scales for single disorders in psychiatric settings is questionable.51 Another problem with screening scales is their potential misuse as case-finding instruments. In the literature on bipolar disorder screening, several researchers misconstrued a positive screen to indicate caseness.51 Although this is not a problem with the screening measures or the selection of a cutoff score, caution must be taken to not confuse screening with diagnosis.52
Screening for BPD as part of your diagnostic interview
An alternative approach to using self-administered questionnaires for screening is for clinicians to include questions in their evaluation as part of a psychiatric review of systems. When conducting a diagnostic interview, clinicians typically screen for disorders that are comorbid with the principal diagnosis by asking about the comorbid disorders’ necessary features or “gate criteria.” For example, in a patient with a principal diagnosis of MDD, the clinician would inquire about the presence of panic attacks, excessive worry, or substance use to screen for the presence of panic disorder, generalized anxiety disorder, or a substance use disorder. In contrast, for polythetically defined disorders such as BPD, there is no single gate criterion, because the disorder is diagnosed based on the presence of at least 5 of 9 criteria and no single one of these criteria is required to be present to establish the diagnosis.
As part of the MIDAS project, the psychometric properties of the BPD criteria were examined to determine if it was possible to identify 1 or 2 criteria that could serve as gate criteria to screen for the disorder. If the sensitivity of 1 criterion or a combination of 2 BPD criteria was sufficiently high (ie, >90%), then the assessment of this criterion (or these criteria) could be included in a psychiatric review of systems, thus potentially improving the detection of BPD. Researchers hypothesized that affective instability, considered first by Linehan53 and later by other theorists54 to be of central importance to the clinical manifestations of BPD, could function as a gate criterion. In the sample of 3,674 psychiatric outpatients who were evaluated with a semi-structured interview, the sensitivity of the affective instability criterion was 92.8%, and the negative predictive value of the criterion was 99%.
Identifying a single BPD criterion that is present in the vast majority of patients diagnosed with BPD will allow clinicians to follow their usual clinical practice when conducting a psychiatric review of systems and inquire about the gate criteria of various disorders. Several studies have found that >90% of patients with BPD report affective instability. However, this does not mean that the diagnosis of BPD can be abbreviated to an assessment of the presence or absence of affective instability. Many patients who screen positive will not have BPD when a more definitive diagnostic evaluation is conducted. In the case of BPD, the more costly definitive diagnostic procedure simply entails inquiry of the other diagnostic criteria.
1. Bellino S, Patria L, Paradiso E, et al. Major depression in patients with borderline personality disorder: a clinical investigation. Can J Psychiatry. 2005;50(4):234-238.
2. Skodol AE, Gunderson JG, McGlashan TH, et al. Functional impairment in patients with schizotypal, borderline, avoidant, or obsessive-compulsive personality disorder. Am J Psychiatry. 2002;159(2):276-283.
3. Gunderson JG, Stout RL, McGlashan TH, et al. Ten-year course of borderline personality disorder: psychopathology and function from the Collaborative Longitudinal Personality Disorders study. Arch Gen Psychiatry. 2011;68(8):827-837.
4. Zanarini MC, Jacoby RJ, Frankenburg FR, et al. The 10-year course of social security disability income reported by patients with borderline personality disorder and axis II comparison subjects. J Pers Disord. 2009;23(4):346-356.
5. Grant BF, Chou SP, Goldstein RB, et al. Prevalence, correlates, disability, and comorbidity of DSM-IV borderline personality disorder: results from the Wave 2 National Epidemiologic Survey on Alcohol and Related Conditions. J Clin Psychiatry. 2008;69(4):533-545.
6. Bender DS, Dolan RT, Skodol AE, et al. Treatment utilization by patients with personality disorders. Am J Psychiatry. 2001;158(2):295-302.
7. Zanarini MC, Frankenburg FR, Hennen J, et al. Mental health service utilization by borderline personality disorder patients and Axis II comparison subjects followed prospectively for 6 years. J Clin Psychiatry. 2004;65(1):28-36.
8. Pompili M, Girardi P, Ruberto A, et al. Suicide in borderline personality disorder: a meta-analysis. Nord J Psychiatry. 2005;59(5):319-324.
9. Oldham JM. Borderline personality disorder and suicidality. Am J Psychiatry. 2006;163(1):20-26.
10. Black DW, Blum N, Pfohl B, et al. Suicidal behavior in borderline personality disorder: prevalence, risk factors, prediction, and prevention. J Pers Disord. 2004;18(3):226-239.
11. Marinangeli M, Butti G, Scinto A, et al. Patterns of comorbidity among DSM-III-R personality disorders. Psychopathology. 2000;33(2):69-74.
12. Zimmerman M, Rothschild L, Chelminski I. The frequency of DSM-IV personality disorders in psychiatric outpatients. Am J Psychiatry. 2005;162(10):1911-1918.
13. Comtois KA, Carmel A. Borderline personality disorder and high utilization of inpatient psychiatric hospitalization: concordance between research and clinical diagnosis. J Behav Health Servi Res. 2016;43(2):272-280.
14. Paris J, Black DW. Borderline personality disorder and bipolar disorder: what is the difference and why does it matter? J Nerv Ment Dis. 2015;203(1):3-7.
15. Zimmerman M, Mattia JI. Differences between clinical and research practice in diagnosing borderline personality disorder. Am J Psychiatry. 1999;156(10):1570-1574.
16. Magnavita JJ, Levy KN, Critchfield KL, et al. Ethical considerations in treatment of personality dysfunction: using evidence, principles, and clinical judgment. Professional Psychology: Research and Practice. 2010;41(1):64-74.
17. Zimmerman M. A review of 20 years of research on overdiagnosis and underdiagnosis in the Rhode Island Methods to Improve Diagnostic Assessment and Services (MIDAS) Project. Can J Psychiatry. 2016;61(2):71-79.
18. Westen D. Divergences between clinical and research methods for assessing personality disorders: implications for research and the evolution of axis II. Am J Psychiatry. 1997;154(7):895-903.
19. Zimmerman M. Diagnosing personality disorders: a review of issues and research methods. Arch Gen Psychiatry. 1994;51(3):225-245.
20. Zanarini MC, Gunderson JG, Frankenberg FR. Axis I phenomenology of borderline personality disorder. Compr Psychiatry. 1989;30(2):149-156.
21. Zimmerman M, Mattia JI. Axis I diagnostic comorbidity and borderline personality disorder. Compr Psychiatry. 1999;40(4):245-252.
22. Gunderson JG, Morey LC, Stout RL, et al. Major depressive disorder and borderline personality disorder revisited: longitudinal interactions. J Clin Psychiatry. 2004;65(8):1049-1056.
23. Bayes AJ, Parker GB. Clinical vs. DSM diagnosis of bipolar disorder, borderline personality disorder and their co-occurrence. Acta Psychiatr Scand. 2016;135(3):259-265.
24. Carpenter RW, Wood PK, Trull TJ. Comorbidity of borderline personality disorder and lifetime substance use disorders in a nationally representative sample. J Pers Disord. 2016;30(3):336-350.
25. Trull TJ, Sher KJ, Minks-Brown C, et al. Borderline personality disorder and substance use disorders: a review and integration. Clin Psychol Rev. 2000;20(2):235-253.
26. Matthies SD, Philipsen A. Common ground in attention deficit hyperactivity disorder (ADHD) and borderline personality disorder (BPD)-review of recent findings. Borderline Personal Disord Emot Dysregul. 2014;1:3.
27. Zimmerman M, Chelminski I, Dalrymple K, et al. Principal diagnoses in psychiatric outpatients with borderline personality disorder: implications for screening recommendations. Ann Clin Psychiatry. 2017;29(1):54-60.
28. Magallón-Neri EM, Forns M, Canalda G, et al. Usefulness of the International Personality Disorder Examination Screening Questionnaire for borderline and impulsive personality pathology in adolescents. Compr Psychiatry. 2013;54(3):301-308.
29. Germans S, Van Heck GL, Langbehn DR, et al. The Iowa Personality Disorder Screen. Eur J Psychol Assess. 2010;26(1):11-18.
30. Zanarini MC, Vujanovic AA, Parachini EA, et al. A screening measure for BPD: the McLean Screening Instrument for Borderline Personality Disorder (MSI-BPD). J Pers Disord. 2003;17(6):568-573.
31. Germans S, Van Heck GL, Hodiamont PP. Results of the search for personality disorder screening tools: clinical implications. J Clin Psychiatry. 2012;73(2):165-173.
32. Hyler SE. Personality diagnostic questionnaire-4. New York, NY: New York State Psychiatric Institute; 1994.
33. First MB, Spitzer RL, Gibbon M, et al. Structured Clinical Interview for DSM-IV Axis II Disorders - Patient edition (SCID-I/P, version 2.0). New York, NY: Biometrics Research Department, New York State Psychiatric Institute; 1995.
34. Bohus M, Kleindienst N, Limberger MF, et al. The short version of the Borderline Symptom List (BSL-23): development and initial data on psychometric properties. Psychopathology. 2009;42(1):32-39.
35. Poreh AM, Rawlings D, Claridge G, et al. The BPQ: a scale for the assessment of boderline personality based on DSM-IV criteria. J Pers Disord. 2006;20(3):247-260.
36. Ekselius L, Lindstrom E, von Knorring L, et al. SCID II interviews and the SCID Screen questionnaire as diagnostic tools for personality disorders in DSM-III-R. Acta Psychiatr Scand. 1994;90(2):120-123.
37. Hyler SE, Skodol AE, Oldham JM, et al. Validity of the Personality Diagnostic Questionnaire-Revised: a replication in an outpatient sample. Compr Psychiatry. 1992;33(2):73-77.
38. Davison S, Leese M, Taylor PJ. Examination of the screening properties of the personality diagnostic questionnaire 4+ (PDQ-4+) in a prison population. J Pers Disord. 2001;15(2):180-194.
39. Jacobsberg L, Perry S, Frances A. Diagnostic agreement between the SCID-II screening questionnaire and the Personality Disorder Examination. J Pers Assess. 1995;65(3):428-433.
40. Germans S, Van Heck GL, Masthoff ED, et al. Diagnostic efficiency among psychiatric outpatients of a self-report version of a subset of screen items of the Structured Clinical Interview for DSM-IV-TR Personality Disorders (SCID-II). Psychol Assess. 2010;22(4):945-952.
41. Lloyd C, Overall JE, Click M Jr. Screening for borderline personality disorders with the MMPI-168. J Clin Psychol. 1983;39(5):722-726.
42. Neal LA, Fox C, Carroll N, et al. Development and validation of a computerized screening test for personality disorders in DSM-III-R. Acta Psychiatr Scand. 1997;95(4):351-356.
43. Germans S, Van Heck GL, Moran P, et al. The Self-Report Standardized Assessment of Personality-abbreviated Scale: preliminary results of a brief screening test for personality disorders. Pers Ment Health. 2008;2(2):70-76.
44. Moran P, Leese M, Lee T, et al. Standardized Assessment of Personality - Abbreviated Scale (SAPAS): preliminary validation of a brief screen for personality disorder. Br J Psychiatry. 2003;183:228-232.
45. Chanen AM, Jovev M, Djaja D, et al. Screening for borderline personality disorder in outpatient youth. J Pers Disord. 2008;22(4):353-364.
46. van Alebeek A, van der Heijden PT, Hessels C, et al. Comparison of three questionnaires to screen for borderline personality disorder in adolescents and young adults. Eur J Psychol Assess. 2017:33;123-128.
47. Noblin JL, Venta A, Sharp C. The validity of the MSI-BPD among inpatient adolescents. Assessment. 2014;21(2):210-217.
48. Kröger C, Vonau M, Kliem S, et al. Emotion dysregulation as a core feature of borderline personality disorder: comparison of the discriminatory ability of two self-rating measures. Psychopathology. 2011;44(4):253-260.
49. Soler J, Domínguez-Clav E, García-Rizo C, et al. Validation of the Spanish version of the McLean Screening Instrument for Borderline Personality Disorder. Rev Psiquiatr Salud Ment. 2016;9(4):195-202.
50. Melartin T, Häkkinen M, Koivisto M, et al. Screening of psychiatric outpatients for borderline personality disorder with the McLean Screening Instrument for Borderline Personality Disorder (MSI-BPD). Nord J Psychiatry. 2009;63(6):475-479.
51. Zimmerman M. Misuse of the Mood Disorders Questionnaire as a case-finding measure and a critique of the concept of using a screening scale for bipolar disorder in psychiatric practice. Bipolar Disord. 2012;14(2):127-134.
52. Zimmerman M. Screening for bipolar disorder: confusion between case-finding and screening. Psychother Psychosom. 2014;83(5):259-262.
53. Linehan MM. Cognitive-behavioral treatment of borderline personality disorder. New York, NY: Guilford Press; 1993.
54. Koenigsberg HW, Harvey PD, Mitropoulou V, et al. Are the interpersonal and identity disturbances in the borderline personality disorder criteria linked to the traits of affective instability and impulsivity? J Pers Disord. 2001;15(4):358-370.
55. Grilo CM, Becker DF, Anez LM, et al. Diagnostic efficiency of DSM-IV criteria for borderline personality disorder: an evaluation in Hispanic men and women with substance use disorders. J Consult Clin Psychol. 2004;72(1):126-131.
56. Korfine L, Hooley JM. Detecting individuals with borderline personality disorder in the community: an ascertainment strategy and comparison with a hospital sample. J Pers Disord. 2009;23(1):62-75.
57. Leppänen V, Lindeman S, Arntz A, et al. Preliminary evaluation of psychometric properties of the Finnish Borderline Personality Disorder Severity Index: Oulu-BPD-Study. Nord J Psychiatry. 2013;67(5):312-319.
58. Pfohl B, Coryell W, Zimmerman M, et al. DSM-III personality disorders: diagnostic overlap and internal consistency of individual DSM-III criteria. Compr Psychiatry. 1986;27(1):22-34.
59. Reich J. Criteria for diagnosing DSM-III borderline personality disorder. Ann Clin Psychiatry. 1990;2(3):189-197.
60. Nurnberg HG, Raskin M, Levine PE, et al. Hierarchy of DSM-III-R criteria efficiency for the diagnosis of borderline personality disorder. J Pers Disord. 1991;5(3):211-224.
61. Farmer RF, Chapman AL. Evaluation of DSM-IV personality disorder criteria as assessed by the structured clinical interview for DSM-IV personality disorders. Compr Psychiatry. 2002;43(4):285-300.
62. Grilo CM, McGlashan TH, Morey LC, et al. Internal consistency, intercriterion overlap and diagnostic efficiency of criteria sets for DSM-IV schizotypal, borderline, avoidant and obsessive-compulsive personality disorders. Acta Psychiatr Scand. 2001;104(4):264-272.
63. Grilo CM, Sanislow CA, Skodol AE, et al. Longitudinal diagnostic efficiency of DSM-IV criteria for borderline personality disorder: a 2-year prospective study. Can J Psychiatry. 2007;52(6):357-362.
1. Bellino S, Patria L, Paradiso E, et al. Major depression in patients with borderline personality disorder: a clinical investigation. Can J Psychiatry. 2005;50(4):234-238.
2. Skodol AE, Gunderson JG, McGlashan TH, et al. Functional impairment in patients with schizotypal, borderline, avoidant, or obsessive-compulsive personality disorder. Am J Psychiatry. 2002;159(2):276-283.
3. Gunderson JG, Stout RL, McGlashan TH, et al. Ten-year course of borderline personality disorder: psychopathology and function from the Collaborative Longitudinal Personality Disorders study. Arch Gen Psychiatry. 2011;68(8):827-837.
4. Zanarini MC, Jacoby RJ, Frankenburg FR, et al. The 10-year course of social security disability income reported by patients with borderline personality disorder and axis II comparison subjects. J Pers Disord. 2009;23(4):346-356.
5. Grant BF, Chou SP, Goldstein RB, et al. Prevalence, correlates, disability, and comorbidity of DSM-IV borderline personality disorder: results from the Wave 2 National Epidemiologic Survey on Alcohol and Related Conditions. J Clin Psychiatry. 2008;69(4):533-545.
6. Bender DS, Dolan RT, Skodol AE, et al. Treatment utilization by patients with personality disorders. Am J Psychiatry. 2001;158(2):295-302.
7. Zanarini MC, Frankenburg FR, Hennen J, et al. Mental health service utilization by borderline personality disorder patients and Axis II comparison subjects followed prospectively for 6 years. J Clin Psychiatry. 2004;65(1):28-36.
8. Pompili M, Girardi P, Ruberto A, et al. Suicide in borderline personality disorder: a meta-analysis. Nord J Psychiatry. 2005;59(5):319-324.
9. Oldham JM. Borderline personality disorder and suicidality. Am J Psychiatry. 2006;163(1):20-26.
10. Black DW, Blum N, Pfohl B, et al. Suicidal behavior in borderline personality disorder: prevalence, risk factors, prediction, and prevention. J Pers Disord. 2004;18(3):226-239.
11. Marinangeli M, Butti G, Scinto A, et al. Patterns of comorbidity among DSM-III-R personality disorders. Psychopathology. 2000;33(2):69-74.
12. Zimmerman M, Rothschild L, Chelminski I. The frequency of DSM-IV personality disorders in psychiatric outpatients. Am J Psychiatry. 2005;162(10):1911-1918.
13. Comtois KA, Carmel A. Borderline personality disorder and high utilization of inpatient psychiatric hospitalization: concordance between research and clinical diagnosis. J Behav Health Servi Res. 2016;43(2):272-280.
14. Paris J, Black DW. Borderline personality disorder and bipolar disorder: what is the difference and why does it matter? J Nerv Ment Dis. 2015;203(1):3-7.
15. Zimmerman M, Mattia JI. Differences between clinical and research practice in diagnosing borderline personality disorder. Am J Psychiatry. 1999;156(10):1570-1574.
16. Magnavita JJ, Levy KN, Critchfield KL, et al. Ethical considerations in treatment of personality dysfunction: using evidence, principles, and clinical judgment. Professional Psychology: Research and Practice. 2010;41(1):64-74.
17. Zimmerman M. A review of 20 years of research on overdiagnosis and underdiagnosis in the Rhode Island Methods to Improve Diagnostic Assessment and Services (MIDAS) Project. Can J Psychiatry. 2016;61(2):71-79.
18. Westen D. Divergences between clinical and research methods for assessing personality disorders: implications for research and the evolution of axis II. Am J Psychiatry. 1997;154(7):895-903.
19. Zimmerman M. Diagnosing personality disorders: a review of issues and research methods. Arch Gen Psychiatry. 1994;51(3):225-245.
20. Zanarini MC, Gunderson JG, Frankenberg FR. Axis I phenomenology of borderline personality disorder. Compr Psychiatry. 1989;30(2):149-156.
21. Zimmerman M, Mattia JI. Axis I diagnostic comorbidity and borderline personality disorder. Compr Psychiatry. 1999;40(4):245-252.
22. Gunderson JG, Morey LC, Stout RL, et al. Major depressive disorder and borderline personality disorder revisited: longitudinal interactions. J Clin Psychiatry. 2004;65(8):1049-1056.
23. Bayes AJ, Parker GB. Clinical vs. DSM diagnosis of bipolar disorder, borderline personality disorder and their co-occurrence. Acta Psychiatr Scand. 2016;135(3):259-265.
24. Carpenter RW, Wood PK, Trull TJ. Comorbidity of borderline personality disorder and lifetime substance use disorders in a nationally representative sample. J Pers Disord. 2016;30(3):336-350.
25. Trull TJ, Sher KJ, Minks-Brown C, et al. Borderline personality disorder and substance use disorders: a review and integration. Clin Psychol Rev. 2000;20(2):235-253.
26. Matthies SD, Philipsen A. Common ground in attention deficit hyperactivity disorder (ADHD) and borderline personality disorder (BPD)-review of recent findings. Borderline Personal Disord Emot Dysregul. 2014;1:3.
27. Zimmerman M, Chelminski I, Dalrymple K, et al. Principal diagnoses in psychiatric outpatients with borderline personality disorder: implications for screening recommendations. Ann Clin Psychiatry. 2017;29(1):54-60.
28. Magallón-Neri EM, Forns M, Canalda G, et al. Usefulness of the International Personality Disorder Examination Screening Questionnaire for borderline and impulsive personality pathology in adolescents. Compr Psychiatry. 2013;54(3):301-308.
29. Germans S, Van Heck GL, Langbehn DR, et al. The Iowa Personality Disorder Screen. Eur J Psychol Assess. 2010;26(1):11-18.
30. Zanarini MC, Vujanovic AA, Parachini EA, et al. A screening measure for BPD: the McLean Screening Instrument for Borderline Personality Disorder (MSI-BPD). J Pers Disord. 2003;17(6):568-573.
31. Germans S, Van Heck GL, Hodiamont PP. Results of the search for personality disorder screening tools: clinical implications. J Clin Psychiatry. 2012;73(2):165-173.
32. Hyler SE. Personality diagnostic questionnaire-4. New York, NY: New York State Psychiatric Institute; 1994.
33. First MB, Spitzer RL, Gibbon M, et al. Structured Clinical Interview for DSM-IV Axis II Disorders - Patient edition (SCID-I/P, version 2.0). New York, NY: Biometrics Research Department, New York State Psychiatric Institute; 1995.
34. Bohus M, Kleindienst N, Limberger MF, et al. The short version of the Borderline Symptom List (BSL-23): development and initial data on psychometric properties. Psychopathology. 2009;42(1):32-39.
35. Poreh AM, Rawlings D, Claridge G, et al. The BPQ: a scale for the assessment of boderline personality based on DSM-IV criteria. J Pers Disord. 2006;20(3):247-260.
36. Ekselius L, Lindstrom E, von Knorring L, et al. SCID II interviews and the SCID Screen questionnaire as diagnostic tools for personality disorders in DSM-III-R. Acta Psychiatr Scand. 1994;90(2):120-123.
37. Hyler SE, Skodol AE, Oldham JM, et al. Validity of the Personality Diagnostic Questionnaire-Revised: a replication in an outpatient sample. Compr Psychiatry. 1992;33(2):73-77.
38. Davison S, Leese M, Taylor PJ. Examination of the screening properties of the personality diagnostic questionnaire 4+ (PDQ-4+) in a prison population. J Pers Disord. 2001;15(2):180-194.
39. Jacobsberg L, Perry S, Frances A. Diagnostic agreement between the SCID-II screening questionnaire and the Personality Disorder Examination. J Pers Assess. 1995;65(3):428-433.
40. Germans S, Van Heck GL, Masthoff ED, et al. Diagnostic efficiency among psychiatric outpatients of a self-report version of a subset of screen items of the Structured Clinical Interview for DSM-IV-TR Personality Disorders (SCID-II). Psychol Assess. 2010;22(4):945-952.
41. Lloyd C, Overall JE, Click M Jr. Screening for borderline personality disorders with the MMPI-168. J Clin Psychol. 1983;39(5):722-726.
42. Neal LA, Fox C, Carroll N, et al. Development and validation of a computerized screening test for personality disorders in DSM-III-R. Acta Psychiatr Scand. 1997;95(4):351-356.
43. Germans S, Van Heck GL, Moran P, et al. The Self-Report Standardized Assessment of Personality-abbreviated Scale: preliminary results of a brief screening test for personality disorders. Pers Ment Health. 2008;2(2):70-76.
44. Moran P, Leese M, Lee T, et al. Standardized Assessment of Personality - Abbreviated Scale (SAPAS): preliminary validation of a brief screen for personality disorder. Br J Psychiatry. 2003;183:228-232.
45. Chanen AM, Jovev M, Djaja D, et al. Screening for borderline personality disorder in outpatient youth. J Pers Disord. 2008;22(4):353-364.
46. van Alebeek A, van der Heijden PT, Hessels C, et al. Comparison of three questionnaires to screen for borderline personality disorder in adolescents and young adults. Eur J Psychol Assess. 2017:33;123-128.
47. Noblin JL, Venta A, Sharp C. The validity of the MSI-BPD among inpatient adolescents. Assessment. 2014;21(2):210-217.
48. Kröger C, Vonau M, Kliem S, et al. Emotion dysregulation as a core feature of borderline personality disorder: comparison of the discriminatory ability of two self-rating measures. Psychopathology. 2011;44(4):253-260.
49. Soler J, Domínguez-Clav E, García-Rizo C, et al. Validation of the Spanish version of the McLean Screening Instrument for Borderline Personality Disorder. Rev Psiquiatr Salud Ment. 2016;9(4):195-202.
50. Melartin T, Häkkinen M, Koivisto M, et al. Screening of psychiatric outpatients for borderline personality disorder with the McLean Screening Instrument for Borderline Personality Disorder (MSI-BPD). Nord J Psychiatry. 2009;63(6):475-479.
51. Zimmerman M. Misuse of the Mood Disorders Questionnaire as a case-finding measure and a critique of the concept of using a screening scale for bipolar disorder in psychiatric practice. Bipolar Disord. 2012;14(2):127-134.
52. Zimmerman M. Screening for bipolar disorder: confusion between case-finding and screening. Psychother Psychosom. 2014;83(5):259-262.
53. Linehan MM. Cognitive-behavioral treatment of borderline personality disorder. New York, NY: Guilford Press; 1993.
54. Koenigsberg HW, Harvey PD, Mitropoulou V, et al. Are the interpersonal and identity disturbances in the borderline personality disorder criteria linked to the traits of affective instability and impulsivity? J Pers Disord. 2001;15(4):358-370.
55. Grilo CM, Becker DF, Anez LM, et al. Diagnostic efficiency of DSM-IV criteria for borderline personality disorder: an evaluation in Hispanic men and women with substance use disorders. J Consult Clin Psychol. 2004;72(1):126-131.
56. Korfine L, Hooley JM. Detecting individuals with borderline personality disorder in the community: an ascertainment strategy and comparison with a hospital sample. J Pers Disord. 2009;23(1):62-75.
57. Leppänen V, Lindeman S, Arntz A, et al. Preliminary evaluation of psychometric properties of the Finnish Borderline Personality Disorder Severity Index: Oulu-BPD-Study. Nord J Psychiatry. 2013;67(5):312-319.
58. Pfohl B, Coryell W, Zimmerman M, et al. DSM-III personality disorders: diagnostic overlap and internal consistency of individual DSM-III criteria. Compr Psychiatry. 1986;27(1):22-34.
59. Reich J. Criteria for diagnosing DSM-III borderline personality disorder. Ann Clin Psychiatry. 1990;2(3):189-197.
60. Nurnberg HG, Raskin M, Levine PE, et al. Hierarchy of DSM-III-R criteria efficiency for the diagnosis of borderline personality disorder. J Pers Disord. 1991;5(3):211-224.
61. Farmer RF, Chapman AL. Evaluation of DSM-IV personality disorder criteria as assessed by the structured clinical interview for DSM-IV personality disorders. Compr Psychiatry. 2002;43(4):285-300.
62. Grilo CM, McGlashan TH, Morey LC, et al. Internal consistency, intercriterion overlap and diagnostic efficiency of criteria sets for DSM-IV schizotypal, borderline, avoidant and obsessive-compulsive personality disorders. Acta Psychiatr Scand. 2001;104(4):264-272.
63. Grilo CM, Sanislow CA, Skodol AE, et al. Longitudinal diagnostic efficiency of DSM-IV criteria for borderline personality disorder: a 2-year prospective study. Can J Psychiatry. 2007;52(6):357-362.

Screening for psychiatric disorders

Beyond DSM-5: Clinical and biologic features shared by major psychiatric syndromes
It does not adequately inform psychiatric practitioners about the many clinical and biologic features shared across the various diagnostic categories. It does not do justice to the galloping advances in the neurobiology of psychiatric brain disorders and the wealth of potential biomarkers that will eventually endow psychiatry with an objective and ultimately more valid, not just reliable, diagnostic model that is compatible with a future of precision medicine.
The Research Domain Criteria (RDoC) Project1 is a valiant attempt to transcend the DSM’s “Chinese menu” approach to diagnosis. It was championed by the former director of the National Institute of Mental Health (NIMH), who used his authority to encourage investigators applying for federal grants to employ the RDoC principles in their research programs. Who does not recall the awkward moment, a few weeks before the official baptism of DSM-5 as psychiatry’s latest diagnostic Bible in May 2013? The NIMH director’s unflattering portrayal of the incipient DSM-5 was a well-publicized shot across the bow. The kerfuffle was later resolved, but its effects linger among clinical researchers who relentlessly hope for neuroscience advances to translate into a more objective diagnostic approach to psychiatric diagnoses. The neurobiologic foundations of psychopathology are bound to guide us to a more valid set of diagnostic categories, yet the pace remains painfully slow.
However, the copious advances in brain research are providing other dividends beyond a better diagnostic forest. Many intriguing insights are emerging about the connectedness among major psychiatric “trees,” including schizophrenia, bipolar disorders, and major depressive disorder. The following are examples of neurobiologic, clinical, and treatment commonalities across those psychotic and mood disorders.
Shared neurobiology
Progressive brain tissue loss/neurodegeneration. Numerous studies have established that abnormal neuroplasticity is a common theme during psychotic, manic, and depressive episodes. These findings have demonstrated that the more recurrent the episodes, the more prominent the atrophy in either overall brain volume or specific brain regions, especially in the hippocampus, prefrontal cortex, and cerebellum as measured on MRI.
White matter pathology. Multiple studies have reported loss of myelin integrity in psychotic and mood disorders. Abnormalities are detected by using diffusion tensor imaging and measuring anisotropy and diffusivity of water flow in white matter traits. White matter pathology can be associated with intra- and inter-hemispheric disconnectivity and impairment of brain functional integration that may contribute to positive, negative, and cognitive symptoms.
Neuroinflammation. Acute psychotic and mood episodes have been shown to be associated with significant elevation in inflammatory cytokines in CSF and serum, including interleukins (such as interleukin-6), tumor necrosis factor-alpha, interferon gamma, and C-reactive protein. Those inflammatory biomarkers subside when the acute episodes are treated. It is believed that activation of the microglia leads to release of proinflammatory cytokines.
Mitochondrial dysfunction. Many studies document various dysfunctions of the mitochondria in schizophrenia, bipolar disorders, and major depressive disorder. The consequences include oxidative stress due to a decrease in the antioxidant glutathione, produced in the mitochondria, which is vital for neutralizing the reactive oxygen and nitrogen species referred to as free radicals. There is a substantial increase of free radicals during acute psychotic and mood episodes, which contributes to neurodegeneration.
Glutamate pathway abnormalities. A large body of literature has focused on the glutamate N-methyl-
Gene/environment interaction. Neurogenetic advances have demonstrated some shared genes among schizophrenia, bipolar disorders, and major depressive disorder (such as the CACNA1C gene).2 Also, environmental factors, such as severe childhood maltreatment, lead to high rates of psychosis and mood disorders in adulthood. Risk genes in schizophrenia and mood disorders are likely to be overexpressed with adverse environmental factors and epigenetics.
Shortened telomeres. Patients with psychotic and mood disorders have been reported to have shorter telomeres—proteins that cap the end of chromosomes and shorten with repeated cycles of mitosis and aging—at a younger age, predicting early senescence and mortality. Telomere shortening is associated with multiple factors, including chronic stress, smoking, poor diet, obesity, infections, inflammation, and free radicals, all shared by major psychiatric disorders.
Genetic heterogeneity. Schizophrenia, bipolar disorders, and major depressive disorder are associated with complex genetics (eg, risk genes, mutations, and copy number variants) and various perinatal complications (eg, infections, gestational diabetes, vitamin D deficiency, hypoxia at delivery), which makes them highly heterogeneous syndromes, comprised of hundreds of biotypes. There are many established endophenotypes that a future diagnostic system should adopt.
Elevated cortisol levels. Increased serum cortisol levels are found in depression and schizophrenia related to HPA axis dysregulation as well as life stress. Hypercortisolemia can contribute to neurodegeneration as well as to multiple systemic medical disorders often encountered in mood and psychotic disorders.
Shared clinical features
Psychotic and mood disorders share several key clinical features, including:
- cognitive deficits
- substance use disorders (especially Cannabis and alcohol) as a common comorbidity
- increased suicide risk
- high prevalence of smoking
- premature mortality, by 10 to 20 years
- anxiety as a common comorbidity
- elevated cardiometabolic risk factors, even before pharmacotherapy
- recurrent relapses lead to treatment resistance
- various degrees of fixed false beliefs (delusions)
- perceptional aberrations (various types of hallucinations)
- response to dopamine-serotonin antagonists (atypical antipsychotics) as monotherapy or adjunctive therapy.
While it is fair to say that a diagnostic manual like DSM-5 should focus on the diagnosis of individual psychiatric diseases and syndromes, it is also reasonable to say that focusing primarily on clinical features does not do justice to the biologic complexities of psychiatric disorders and the importance of including biomarkers to increase the validity of psychopathological categories. The shared neurobiologic and clinical features across major psychiatric syndromes, such as schizophrenia, bipolar disorders, and depression, indicate how multifaceted psychiatric diagnosis can be. The same approach is applicable to other psychiatric syndromes, such as anxiety, personality disorders, attention-deficit/hyperactivity disorder, or dementia. Our field should move firmly and steadily toward a diagnostic schema that incorporates ongoing breakthroughs in psychiatric neuroscience as soon as they are replicated.
If psychopathology is a forest, then DSM-5 is a simplistic depiction of each tree’s structure as roots, a trunk, branches, and leaves. Psychiatry needs to move to a far more sophisticated perspective of each tree as an amazingly complex, dynamic, and evolving organism, designed genetically but continuously influenced by its environment. Psychiatry also should keep an eye on the entire forest and detect distinctive patterns as well as idiosyncratic or shared features among the trees. Major insights will ensue about the etiology, course, and management of each diagnostic tree or the mosaic of related trees.
1. Insel TR. The NIMH Research Domain Criteria (RDoC) Project: precision medicine for psychiatry. Am J Psychiatry. 2014;171(4):395-397.
2. Nasrallah HA. Pleiotropy of psychiatric disorders will reinvent DSM. Current Psychiatry. 2013;12(4):6-7.
It does not adequately inform psychiatric practitioners about the many clinical and biologic features shared across the various diagnostic categories. It does not do justice to the galloping advances in the neurobiology of psychiatric brain disorders and the wealth of potential biomarkers that will eventually endow psychiatry with an objective and ultimately more valid, not just reliable, diagnostic model that is compatible with a future of precision medicine.
The Research Domain Criteria (RDoC) Project1 is a valiant attempt to transcend the DSM’s “Chinese menu” approach to diagnosis. It was championed by the former director of the National Institute of Mental Health (NIMH), who used his authority to encourage investigators applying for federal grants to employ the RDoC principles in their research programs. Who does not recall the awkward moment, a few weeks before the official baptism of DSM-5 as psychiatry’s latest diagnostic Bible in May 2013? The NIMH director’s unflattering portrayal of the incipient DSM-5 was a well-publicized shot across the bow. The kerfuffle was later resolved, but its effects linger among clinical researchers who relentlessly hope for neuroscience advances to translate into a more objective diagnostic approach to psychiatric diagnoses. The neurobiologic foundations of psychopathology are bound to guide us to a more valid set of diagnostic categories, yet the pace remains painfully slow.
However, the copious advances in brain research are providing other dividends beyond a better diagnostic forest. Many intriguing insights are emerging about the connectedness among major psychiatric “trees,” including schizophrenia, bipolar disorders, and major depressive disorder. The following are examples of neurobiologic, clinical, and treatment commonalities across those psychotic and mood disorders.
Shared neurobiology
Progressive brain tissue loss/neurodegeneration. Numerous studies have established that abnormal neuroplasticity is a common theme during psychotic, manic, and depressive episodes. These findings have demonstrated that the more recurrent the episodes, the more prominent the atrophy in either overall brain volume or specific brain regions, especially in the hippocampus, prefrontal cortex, and cerebellum as measured on MRI.
White matter pathology. Multiple studies have reported loss of myelin integrity in psychotic and mood disorders. Abnormalities are detected by using diffusion tensor imaging and measuring anisotropy and diffusivity of water flow in white matter traits. White matter pathology can be associated with intra- and inter-hemispheric disconnectivity and impairment of brain functional integration that may contribute to positive, negative, and cognitive symptoms.
Neuroinflammation. Acute psychotic and mood episodes have been shown to be associated with significant elevation in inflammatory cytokines in CSF and serum, including interleukins (such as interleukin-6), tumor necrosis factor-alpha, interferon gamma, and C-reactive protein. Those inflammatory biomarkers subside when the acute episodes are treated. It is believed that activation of the microglia leads to release of proinflammatory cytokines.
Mitochondrial dysfunction. Many studies document various dysfunctions of the mitochondria in schizophrenia, bipolar disorders, and major depressive disorder. The consequences include oxidative stress due to a decrease in the antioxidant glutathione, produced in the mitochondria, which is vital for neutralizing the reactive oxygen and nitrogen species referred to as free radicals. There is a substantial increase of free radicals during acute psychotic and mood episodes, which contributes to neurodegeneration.
Glutamate pathway abnormalities. A large body of literature has focused on the glutamate N-methyl-
Gene/environment interaction. Neurogenetic advances have demonstrated some shared genes among schizophrenia, bipolar disorders, and major depressive disorder (such as the CACNA1C gene).2 Also, environmental factors, such as severe childhood maltreatment, lead to high rates of psychosis and mood disorders in adulthood. Risk genes in schizophrenia and mood disorders are likely to be overexpressed with adverse environmental factors and epigenetics.
Shortened telomeres. Patients with psychotic and mood disorders have been reported to have shorter telomeres—proteins that cap the end of chromosomes and shorten with repeated cycles of mitosis and aging—at a younger age, predicting early senescence and mortality. Telomere shortening is associated with multiple factors, including chronic stress, smoking, poor diet, obesity, infections, inflammation, and free radicals, all shared by major psychiatric disorders.
Genetic heterogeneity. Schizophrenia, bipolar disorders, and major depressive disorder are associated with complex genetics (eg, risk genes, mutations, and copy number variants) and various perinatal complications (eg, infections, gestational diabetes, vitamin D deficiency, hypoxia at delivery), which makes them highly heterogeneous syndromes, comprised of hundreds of biotypes. There are many established endophenotypes that a future diagnostic system should adopt.
Elevated cortisol levels. Increased serum cortisol levels are found in depression and schizophrenia related to HPA axis dysregulation as well as life stress. Hypercortisolemia can contribute to neurodegeneration as well as to multiple systemic medical disorders often encountered in mood and psychotic disorders.
Shared clinical features
Psychotic and mood disorders share several key clinical features, including:
- cognitive deficits
- substance use disorders (especially Cannabis and alcohol) as a common comorbidity
- increased suicide risk
- high prevalence of smoking
- premature mortality, by 10 to 20 years
- anxiety as a common comorbidity
- elevated cardiometabolic risk factors, even before pharmacotherapy
- recurrent relapses lead to treatment resistance
- various degrees of fixed false beliefs (delusions)
- perceptional aberrations (various types of hallucinations)
- response to dopamine-serotonin antagonists (atypical antipsychotics) as monotherapy or adjunctive therapy.
While it is fair to say that a diagnostic manual like DSM-5 should focus on the diagnosis of individual psychiatric diseases and syndromes, it is also reasonable to say that focusing primarily on clinical features does not do justice to the biologic complexities of psychiatric disorders and the importance of including biomarkers to increase the validity of psychopathological categories. The shared neurobiologic and clinical features across major psychiatric syndromes, such as schizophrenia, bipolar disorders, and depression, indicate how multifaceted psychiatric diagnosis can be. The same approach is applicable to other psychiatric syndromes, such as anxiety, personality disorders, attention-deficit/hyperactivity disorder, or dementia. Our field should move firmly and steadily toward a diagnostic schema that incorporates ongoing breakthroughs in psychiatric neuroscience as soon as they are replicated.
If psychopathology is a forest, then DSM-5 is a simplistic depiction of each tree’s structure as roots, a trunk, branches, and leaves. Psychiatry needs to move to a far more sophisticated perspective of each tree as an amazingly complex, dynamic, and evolving organism, designed genetically but continuously influenced by its environment. Psychiatry also should keep an eye on the entire forest and detect distinctive patterns as well as idiosyncratic or shared features among the trees. Major insights will ensue about the etiology, course, and management of each diagnostic tree or the mosaic of related trees.
It does not adequately inform psychiatric practitioners about the many clinical and biologic features shared across the various diagnostic categories. It does not do justice to the galloping advances in the neurobiology of psychiatric brain disorders and the wealth of potential biomarkers that will eventually endow psychiatry with an objective and ultimately more valid, not just reliable, diagnostic model that is compatible with a future of precision medicine.
The Research Domain Criteria (RDoC) Project1 is a valiant attempt to transcend the DSM’s “Chinese menu” approach to diagnosis. It was championed by the former director of the National Institute of Mental Health (NIMH), who used his authority to encourage investigators applying for federal grants to employ the RDoC principles in their research programs. Who does not recall the awkward moment, a few weeks before the official baptism of DSM-5 as psychiatry’s latest diagnostic Bible in May 2013? The NIMH director’s unflattering portrayal of the incipient DSM-5 was a well-publicized shot across the bow. The kerfuffle was later resolved, but its effects linger among clinical researchers who relentlessly hope for neuroscience advances to translate into a more objective diagnostic approach to psychiatric diagnoses. The neurobiologic foundations of psychopathology are bound to guide us to a more valid set of diagnostic categories, yet the pace remains painfully slow.
However, the copious advances in brain research are providing other dividends beyond a better diagnostic forest. Many intriguing insights are emerging about the connectedness among major psychiatric “trees,” including schizophrenia, bipolar disorders, and major depressive disorder. The following are examples of neurobiologic, clinical, and treatment commonalities across those psychotic and mood disorders.
Shared neurobiology
Progressive brain tissue loss/neurodegeneration. Numerous studies have established that abnormal neuroplasticity is a common theme during psychotic, manic, and depressive episodes. These findings have demonstrated that the more recurrent the episodes, the more prominent the atrophy in either overall brain volume or specific brain regions, especially in the hippocampus, prefrontal cortex, and cerebellum as measured on MRI.
White matter pathology. Multiple studies have reported loss of myelin integrity in psychotic and mood disorders. Abnormalities are detected by using diffusion tensor imaging and measuring anisotropy and diffusivity of water flow in white matter traits. White matter pathology can be associated with intra- and inter-hemispheric disconnectivity and impairment of brain functional integration that may contribute to positive, negative, and cognitive symptoms.
Neuroinflammation. Acute psychotic and mood episodes have been shown to be associated with significant elevation in inflammatory cytokines in CSF and serum, including interleukins (such as interleukin-6), tumor necrosis factor-alpha, interferon gamma, and C-reactive protein. Those inflammatory biomarkers subside when the acute episodes are treated. It is believed that activation of the microglia leads to release of proinflammatory cytokines.
Mitochondrial dysfunction. Many studies document various dysfunctions of the mitochondria in schizophrenia, bipolar disorders, and major depressive disorder. The consequences include oxidative stress due to a decrease in the antioxidant glutathione, produced in the mitochondria, which is vital for neutralizing the reactive oxygen and nitrogen species referred to as free radicals. There is a substantial increase of free radicals during acute psychotic and mood episodes, which contributes to neurodegeneration.
Glutamate pathway abnormalities. A large body of literature has focused on the glutamate N-methyl-
Gene/environment interaction. Neurogenetic advances have demonstrated some shared genes among schizophrenia, bipolar disorders, and major depressive disorder (such as the CACNA1C gene).2 Also, environmental factors, such as severe childhood maltreatment, lead to high rates of psychosis and mood disorders in adulthood. Risk genes in schizophrenia and mood disorders are likely to be overexpressed with adverse environmental factors and epigenetics.
Shortened telomeres. Patients with psychotic and mood disorders have been reported to have shorter telomeres—proteins that cap the end of chromosomes and shorten with repeated cycles of mitosis and aging—at a younger age, predicting early senescence and mortality. Telomere shortening is associated with multiple factors, including chronic stress, smoking, poor diet, obesity, infections, inflammation, and free radicals, all shared by major psychiatric disorders.
Genetic heterogeneity. Schizophrenia, bipolar disorders, and major depressive disorder are associated with complex genetics (eg, risk genes, mutations, and copy number variants) and various perinatal complications (eg, infections, gestational diabetes, vitamin D deficiency, hypoxia at delivery), which makes them highly heterogeneous syndromes, comprised of hundreds of biotypes. There are many established endophenotypes that a future diagnostic system should adopt.
Elevated cortisol levels. Increased serum cortisol levels are found in depression and schizophrenia related to HPA axis dysregulation as well as life stress. Hypercortisolemia can contribute to neurodegeneration as well as to multiple systemic medical disorders often encountered in mood and psychotic disorders.
Shared clinical features
Psychotic and mood disorders share several key clinical features, including:
- cognitive deficits
- substance use disorders (especially Cannabis and alcohol) as a common comorbidity
- increased suicide risk
- high prevalence of smoking
- premature mortality, by 10 to 20 years
- anxiety as a common comorbidity
- elevated cardiometabolic risk factors, even before pharmacotherapy
- recurrent relapses lead to treatment resistance
- various degrees of fixed false beliefs (delusions)
- perceptional aberrations (various types of hallucinations)
- response to dopamine-serotonin antagonists (atypical antipsychotics) as monotherapy or adjunctive therapy.
While it is fair to say that a diagnostic manual like DSM-5 should focus on the diagnosis of individual psychiatric diseases and syndromes, it is also reasonable to say that focusing primarily on clinical features does not do justice to the biologic complexities of psychiatric disorders and the importance of including biomarkers to increase the validity of psychopathological categories. The shared neurobiologic and clinical features across major psychiatric syndromes, such as schizophrenia, bipolar disorders, and depression, indicate how multifaceted psychiatric diagnosis can be. The same approach is applicable to other psychiatric syndromes, such as anxiety, personality disorders, attention-deficit/hyperactivity disorder, or dementia. Our field should move firmly and steadily toward a diagnostic schema that incorporates ongoing breakthroughs in psychiatric neuroscience as soon as they are replicated.
If psychopathology is a forest, then DSM-5 is a simplistic depiction of each tree’s structure as roots, a trunk, branches, and leaves. Psychiatry needs to move to a far more sophisticated perspective of each tree as an amazingly complex, dynamic, and evolving organism, designed genetically but continuously influenced by its environment. Psychiatry also should keep an eye on the entire forest and detect distinctive patterns as well as idiosyncratic or shared features among the trees. Major insights will ensue about the etiology, course, and management of each diagnostic tree or the mosaic of related trees.
1. Insel TR. The NIMH Research Domain Criteria (RDoC) Project: precision medicine for psychiatry. Am J Psychiatry. 2014;171(4):395-397.
2. Nasrallah HA. Pleiotropy of psychiatric disorders will reinvent DSM. Current Psychiatry. 2013;12(4):6-7.
1. Insel TR. The NIMH Research Domain Criteria (RDoC) Project: precision medicine for psychiatry. Am J Psychiatry. 2014;171(4):395-397.
2. Nasrallah HA. Pleiotropy of psychiatric disorders will reinvent DSM. Current Psychiatry. 2013;12(4):6-7.

Cannabis use disorder

Deutetrabenazine for tardive dyskinesia
Compared with first-generation antipsychotics, second-generation antipsychotics (SGAs) have a lower risk for extrapyramidal symptoms. Yet tardive dyskinesia (TD) remains a concern because of the widespread use of SGAs for multiple indications.1 Prior to April 2017, clinicians had no FDA-approved TD treatment options. The most widely used agent worldwide, tetrabenazine, had positive efficacy data in TD trials over the past 45 years but was not available in the United States until 2008, and its sole indication was for chorea associated with Huntington’s disease.2 Moreover, the use of tetrabenazine involved slow titration, multiple daily dosing, cytochrome P450 (CYP) 2D6 genotyping for doses >50 mg/d, and tolerability issues.
Tetrabenazine is an inhibitor of vesicular monoamine transport type 2 (VMAT2), a transport protein located almost exclusively in the CNS whose role is to place monoamine neurotransmitters (dopamine, serotonin, norepinephrine) into presynaptic vesicles. By decreasing dopamine transport into these presynaptic vesicles, synaptic dopamine release is lessened, thus reducing postsynaptic dopamine D2 receptor activity and the severity of dyskinetic movements.1
In 2 pivotal 12-week clinical trials, deutetrabenazine significantly reduced TD severity as measured by Abnormal Involuntary Movement Scale (AIMS) scores (see Efficacy).6,7
Clinical implications
TD remains a substantial public health concern due to the increasing use of antipsychotics for mood and other disorders beyond the initial indications for schizophrenia.1 Although exposure to dopamine D2antagonism results in postsynaptic receptor upregulation and supersensitivity that underlies the development of dyskinesia, this process is often rapidly reversible in animal models.1 The persistence of TD symptoms in up to 80% of patients after dopamine receptor blocking agents (DRBAs) are stopped has led to hypotheses that the underlying pathophysiology of TD is also a problem with neuroplasticity. Aside from DRBA exposure, environmental factors (eg, oxidative stress) and genetic predisposition might contribute to TD risk.1
Before 2017, only 1 medication (branched-chain amino acids) had been FDA-approved for treating TD in the United States, and only a few existing medications (clonazepam, amantadine, and ginkgo biloba extract [EGb-761]) had positive results from controlled trials, most with small effect sizes.8 Moreover, there was only 1 controlled trial each for clonazepam and EGb-761.1 A branched-chain amino acid preparation received FDA approval for managing TD in male patients, but is no longer commercially available, except from compounding pharmacies.9
Tetrabenazine was developed in the mid-1950s to avoid orthostasis and sedation associated with reserpine.10 Both reserpine and tetrabenazine proved effective for TD,11 but tetrabenazine lacked reserpine’s peripheral adverse effects. However, the kinetics of tetrabenazine necessitated multiple daily doses, and CYP2D6 genotyping was required for doses >50 mg/d.2
Receptor blocking. The mechanism that distinguishes the clinical profiles of reserpine and tetrabenazine relates to their differential properties at VMAT.12 VMAT exists in 2 forms (VMAT1 and VMAT2) that vary in distribution, with VMAT1 expressed mainly in the peripheral nervous system and VMAT2 expressed mainly in monoaminergic cells of the CNS.13 Tetrabenazine is a specific and reversible VMAT2 inhibitor, whereas reserpine is an irreversible and nonselective antagonist of VMAT1 and VMAT2. It is reserpine’s VMAT1 inhibition that results in peripheral adverse effects such as orthostasis. Tetrabenazine is rapidly and extensively converted into 2 isomers, alpha-dihydrotetrabenazine (α-DHTBZ) and beta-dihydrotetrabenazine (β-DHTBZ), both of which are metabolized by CYP2D6, with a role for CYP3A4 in α-DHTBZ metabolism.1 These DHTBZ metabolites have a short half-life when generated from oral tetrabenazine, a feature that necessitates multiple daily dosing; moreover, the existence of 2D6 polymorphisms led to FDA-mandated CYP2D6 genotyping for tetrabenazine doses >50 mg/d when it was approved for Huntington’s chorea. The concern is that 2D6 poor metabolizers will have excessive exposure to the VMAT2 effects of DHTBZ, resulting in sedation, akathisia, parkinsonism, and mood symptoms.2
How deuterium impacts medication kinetics. Deuterium is a naturally occurring, stable, nontoxic isotope of hydrogen. Humans have 5 g of deuterium in their body at any time, mostly in the form of heavy water (D2O).14 When deuterium is used to replace selected hydrogen atoms, the resulting molecule will have similar configuration and receptor-binding properties but markedly different kinetics. Because the carbon–deuterium covalent bond requires 8 times more energy to break than a carbon–hydrogen bond, the half-life is prolonged.15 Utilizing this knowledge, a deuterated form of tetrabenazine, deutetrabenazine, was synthesized with such a purpose in mind. While the active metabolites of deutetrabenazine retain the VMAT2 affinity of non-deuterated tetrabenazine, the substitution of deuterium for hydrogen at specific positions slows the breakdown of metabolites, resulting in sustained duration of action, greater active drug exposure, and less impact of 2D6 genotype on drug exposure, thus eliminating the need for genotyping, unless one wants to exceed 36 mg/d.
Deutetrabenazine was first studied in Huntington’s chorea in a 13-week, double-blind, placebo-controlled, parallel-group study (N = 90).4 The maximum daily deutetrabenazine dose was 48 mg, but reduced to 36 mg in those taking strong CYP2D6 inhibitors (bupropion, fluoxetine, or paroxetine). Blinded 2D6 genotyping was performed, but there was no dose modification required based on 2D6 genotype. There was a 36.4% reduction in total maximal chorea score for deutetrabenazine compared with 14.4% for placebo (P < .001).4 Importantly, adverse effects were comparable between both groups, with 1 drop-out in the deutetrabenazine arm vs 2 in the placebo arm. The only adverse event occurring in ≥5% of deutetrabenazine participants and at a rate ≥2 times that of placebo was somnolence: 11.1% for deutetrabenazine vs 4.4% for placebo.4 The mean deutetrabenazine daily dose at the end of the treatment period was 39.7 ± 9.3 mg, and for those with impaired CYP2D6 function (poor metabolizers or those taking strong CYP2D6 inhibiting medications), the mean daily dose was 34.8 mg ± 3.8 mg.4
Use in tardive dyskinesia. The recommended starting dosage for TD treatment is 6 mg, twice daily with food. The dose may be increased at weekly intervals in increments of 6 mg/d to a maximum recommended daily dosage of 48 mg.5 The maximum daily dose is 36 mg (18 mg, twice daily) in patients receiving strong CYP2D6 inhibitors or who are 2D6 poor metabolizers.5
Deutetrabenazine has not been studied in those with moderate or severe hepatic impairment, and its use is contraindicated in these patients.5 No clinical studies have been conducted to assess the effect of renal impairment on the pharmacokinetics of deutetrabenazine.5
Pharmacologic profile, adverse reactions
When the data from the two 12-week, phase 3 placebo-controlled studies were pooled, the most common adverse reactions occurring in >3% of deutetrabenazine patients and greater than placebo were nasopharyngeal symptoms (4% vs 2% placebo) and insomnia (4% vs 1% placebo).5 Importantly, in neither TD study were there clinically significant changes in rating scales for depression, suicidal ideation and behavior, or parkinsonism. There also were no clinically significant changes in measures of schizophrenia symptoms. The mean QT prolongation for a single 24 mg dose of deutetrabenazine in healthy volunteers was 4.5 milliseconds, with the upper bound of the double-sided 90% confidence interval reaching 6.5 milliseconds.5 For tetrabenazine, single 50 mg doses administered to volunteers resulted in mean QT prolongation of 8 milliseconds.5 In patients requiring deutetrabenazine doses >24 mg/d who are taking other medications known to prolong QTc, assess the QTc interval before and after increasing the dose of deutetrabenazine or other medications that are known to prolong QTc.5
How it works
Tetrabenazine is the only agent that has demonstrated significant efficacy for TD management, but its use involves slow titration, multiple daily dosing, CYP2D6 genotyping for doses >50 mg/d, and tolerability issues. For example, the most common adverse effects in the pivotal tetrabenazine Huntington’s disease trial were sedation/somnolence (tetrabenazine 31% vs 3% for placebo), insomnia (tetrabenazine 22% vs 0% for placebo), depression (tetrabenazine 19% vs 0% for placebo), fatigue (tetrabenazine 22% vs 13% for placebo), and akathisia (tetrabenazine 19% vs 0% for placebo).2 For comparison, the only adverse event occurring in ≥5% of deutetrabenazine participants and at a rate ≥2 times that of placebo in the pivotal Huntington’s disease trial was somnolence (11.1% for deutetrabenazine vs 4.4% for placebo).4
Pharmacokinetics
Deutetrabenazine has 80% oral bioavailability, and is rapidly converted to its active metabolites after oral dosing (Table 2).5 Linear dose dependence of Cmax and area under the curve (AUC) was observed for the active metabolites following single or multiple doses of deutetrabenazine (6 to 24 mg and 7.5 to 22.5 mg, twice daily).15 Cmax of deuterated α-DHTBZ and β-DHTBZ is reached within 3 to 4 hours after dosing, with a steady state ratio of 3:1 for the α-DHTBZ vs the β-DHTBZ form. Food had no effect on AUC, but did increase Cmax by 50%.5
Deutetrabenazine is metabolized through carbonyl reductase enzymes to its active metabolites, and these are further metabolized through multiple CYP pathways, predominantly 2D6 and to a lesser extent 3A4. The effect of CYP2D6 inhibition on the pharmacokinetics of deutetrabenazine and its α-DHTBZ and β-DHTBZ metabolites was studied in 24 healthy participants following a single 22.5 mg dose of deutetrabenazine given after 8 days of administration of the strong CYP2D6 inhibitor paroxetine, 20 mg/d. In the presence of paroxetine, systemic exposure (AUC) of α-DHTBZ was 1.9-fold higher and β-DHTBZ was 6.5-fold higher, resulting in an approximately 3-fold increase in AUC for total (α+β)-DHTBZ, with corresponding increases in mean half-life of approximately 1.5-fold and 2.7-fold, respectively.5 Neither deutetrabenazine or its metabolites are inhibitors or inducers of major CYP enzymes. Aside from VMAT2, the results of in vitro studies suggest that deutetrabenazine and its active metabolites are unlikely to inhibit most major drug transporters at clinically relevant concentrations.
Efficacy
Efficacy was established in two 12-week, double-blind, placebo-controlled trials of adult patients with TD (ages 18 to 80).6,7 Eligible participants had:
- TD diagnosis for ≥3 months before screening and a history of DRBA treatment for ≥3 months (≥1 month if age ≥60)
- Total AIMS motor score ≥6 (items 1 to 7) at both screening and baseline, verified by a blinded central rater at screening via central video rating
- Patients with an underlying psychiatric illness had to be stable. Psychoactive medication use, including antipsychotics, was allowed if stable for ≥30 days before screening (antidepressants, ≥45 days).
Exclusion criteria included treatment with tetrabenazine, reserpine, α-methyl-p-tyrosine, strong anticholinergic medications, dopamine antagonizing antiemetics (eg, metoclopramide, prochlorperazine, promethazine), dopamine agonists, levodopa, stimulants, or a monoamine oxidase inhibitor (MAOI) within 30 days of screening or baseline, or treatment with botulinum toxin within 3 months of screening; and presence of a neurologic condition that could confound TD assessments, serious untreated or undertreated psychiatric illness, or unstable medical illness. Patients with a history of or active suicidal ideation or behavior within 6 months of screening or score ≥11 on the depression subscale of the Hospital Anxiety and Depression Scale were excluded. Those participants with Fridericia-corrected QT interval values >450 milliseconds in men, >460 milliseconds in women, or >480 milliseconds in patients with a right bundle branch block on electrocardiography at screening also were excluded.
The flexible-dose TD study was performed in 117 participants randomized in a 1:1 manner to deutetrabenazine or placebo, both administered twice daily, titrated to optimal dosage (12 to 48 mg/d) over 6 weeks, and then administered at that dose for another 6 weeks.7 The population demographics were: mean age, 54.6 ± 10.3 years, 52.1% female, 69.2% white, and 80.3% receiving ongoing dopamine antagonists, with a mean TD duration of 74.7 ± 81.5 months. Sixty-eight percent had schizophrenia spectrum disorders, and 30% had mood disorders. The primary outcome was change in total AIMS score (items 1 to 7) assessed by central, independent raters. The mean baseline AIMS score for items 1 to 7 was 9.6 ± 3.9, with 82.9% of participants with baseline AIMS scores ≥6. Study treatment retention was high: placebo 88.1%, deutetrabenazine 89.7%.7 There was a mean 3 point decrease in AIMS score for deutetrabenazine compared with 1.4 for placebo (P = .019). Among those with baseline AIMS scores ≥6, there was a 3.4 point decrease in AIMS scores for deutetrabenazine compared with a 1.9 point decrease for placebo (P = .027). The only adverse effects that occurred in ≥5% of deutetrabenazine participants and at a rate ≥2 times the rate in placebo were insomnia (deutetrabenazine 6.9% vs placebo 1.7%) and akathisia (deutetrabenazine 5.2% vs placebo 0%).
The fixed-dose TD study was performed in 293 participants randomized in 1:1:1:1 manner to 1 of 3 fixed doses of deutetrabenazine (12 mg/d, 24 mg/d, or 36 mg/d) or placebo, both administered twice daily.6 The starting dose of deutetrabenazine was 6 mg twice daily. During the dose escalation period (through Week 4), the dose of study drug was increased weekly in increments of 6 mg/d until the randomized dose was achieved. Patients continued to receive the dose they were assigned to over a maintenance period of 8 weeks.6 The population demographics were: mean age, 56.4 ± 11.3 years, 55% female, 79% white, 76% receiving ongoing dopamine antagonists, with a mean TD duration of 67.2 ± 66 months. Sixty percent had schizophrenia spectrum disorders, and 36% had mood disorders. The primary outcome was change in AIMS total score (items 1 to 7) assessed by central, independent raters. The mean AIMS score at baseline was 9.5 ± 2.7 in the placebo group, and for deutetrabenazine: 9.6 ± 2.4 in the 12 mg/d group, 9.4 ± 2.9 in the 24 mg/d group, and 10.1 ± 3.2 in the 36 mg/d group. The 24 mg/d and 36 mg/d doses significantly reduced AIMS scores from baseline vs placebo: 36 mg: −3.3 (0.42) vs −1.4 (0.41) (P = .001); 24 mg: −3.2 (0.45) vs −1.4 (0.41) (P = .003). Study treatment retention rates were high: placebo 90.5%, deutetrabenazine 88%. Across all doses, only 1 adverse effect occurred in ≥5% of deutetrabenazine participants: headache (5% deutetrabenazine vs 6% placebo). At the highest dose, 36 mg/d, the only adverse effects that occurred in ≥5% of participants were diarrhea (7% deutetrabenazine vs 3% placebo) and headache (7% deutetrabenazine vs 6% placebo).
Outcome. In the flexible-dose study (mean dose 38.8 ± 7.92 mg/d), the deutetrabenazine arm experienced a mean 30% reduction in AIMS scores from baseline at the Week 12 endpoint. Compared with placebo, the mean reduction in AIMS scores (standard error) was: −3.0 (0.45) deutetrabenazine vs −1.6 (0.46) placebo (P = .019).7 For the fixed-dose study, the 24 mg/d and 36 mg/d doses significantly reduced AIMS scores from baseline vs placebo: 36 mg: −3.3 (0.42) vs −1.4 (0.41) (P = .001); 24 mg: −3.2 (0.45) vs −1.4 (0.41) (P = .003). In addition to these mean changes from baseline, 35% of the 24 mg/d group and 33% of the 36 mg/d group demonstrated ≥50% reduction in AIMS scores.6
Tolerability
In the 2 phase 3 trials, there were no adverse effects occurring with an incidence ≥5% and at least twice the rate of placebo.5 Discontinuations because of adverse events were low in both pivotal studies across all treatment groups: 3.4% for placebo vs 1.7% for deutetrabenazine in the flexible-dose trial,7 and 3% for placebo vs 4% for deutetrabenazine in the fixed-dose study.6 In neither trial were there clinically significant changes in ratings of depression, suicidality, parkinsonism, or schizophrenia symptoms. The mean QT prolongation in healthy volunteers is described above.
Clinical considerations
Unique properties. Deutetrabenazine utilizes the greater bond strength of the carbon–deuterium bond to slow CYP metabolism, resulting in prolonged duration of action that is well tolerated, and provides significant efficacy.
Why Rx? The reasons to prescribe deutetrabenazine for TD patients include:
- only 1 of 2 agents with FDA approval for TD
- fewer tolerability issues than with tetrabenazine
- lower sedation rates in TD trials than with valbenazine
- no signal for effects on mood parameters or rates of parkinsonism when used for TD.
Dosing
The recommended starting dosage of deutetrabenazine is 6 mg twice daily taken with food, increasing by 6 mg/d weekly as needed, with a maximum dose of 48 mg/d or 36 mg/d in those taking strong CYP2D6 inhibitors or who are 2D6 poor metabolizers. Deutetrabenazine is contraindicated in patients with hepatic impairment (as determined by Child-Pugh criteria16). There are no data in patients with renal impairment. The combined efficacy and tolerability of dosages >48 mg/d has not been evaluated. Overdoses of tetrabenazine ranging from 100 to 1,000 mg have been reported in the literature and were associated with acute dystonia, oculogyric crisis, nausea and vomiting, sweating, sedation, hypotension, confusion, diarrhea, hallucinations, rubor, and tremor.5
Contraindications
When used for TD, deutetrabenazine is contraindicated for patients taking reserpine, tetrabenazine, valbenazine, or MAOIs, and for patients with hepatic impairment. As with most medications, there are no data on deutetrabenazine use in pregnant women; however, oral administration of deutetrabenazine (5, 10, or 30 mg/kg/d) or tetrabenazine (30 mg/kg/d) to pregnant rats during organogenesis had no clear effect on embryofetal development. The highest dose tested was 6 times the maximum recommended human dose of 48 mg/d on a body surface area (mg/m2) basis. There are no data on the presence of deutetrabenazine or its metabolites in human milk, the effects on the breastfed infant, or the effects of the drug on milk production.
1. Meyer JM. Forgotten but not gone: new developments in the understanding and treatment of tardive dyskinesia. CNS Spectr. 2016;21(S1):13-24.
2. Jankovic J, Clarence-Smith K. Tetrabenazine for the treatment of chorea and other hyperkinetic movement disorders. Expert Rev Neurother. 2011;11(11):1509-1523.
3. Meyer JM. Valbenazine for tardive dyskinesia. Current Psychiatry. 2017;16(5):40-46.
4. Huntington Study Group; Frank S, Testa CM, Stamler D, et al. Effect of deutetrabenazine on chorea among patients with Huntington disease: a randomized clinical trial. JAMA. 2016;316(1):40-50.
5. Austedo [package insert]. North Wales, PA: Teva Pharmaceuticals USA, Inc.; 2017.
6. Anderson KE, Stamler D, Davis MD, et al. Deutetrabenazine for treatment of involuntary movements in patients with tardive dyskinesia (AIM-TD): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Psychiatry. 2017;4(8):595-604.
7. Fernandez HH, Factor SA, Hauser RA, et al. Randomized controlled trial of deutetrabenazine for tardive dyskinesia: the ARM-TD study. Neurology. 2017;88(21):2003-2010.
8. Bhidayasiri R, Fahn S, Weiner WJ, et al. Evidence-based guideline: treatment of tardive syndromes: report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. 2013;81(5):463-469.
9. Richardson MA, Small AM, Read LL, et al. Branched chain amino acid treatment of tardive dyskinesia in children and adolescents. J Clin Psychiatry. 2004;65(1):92-96.
10. Quinn GP, Shore PA, Brodie BB. Biochemical and pharmacological studies of RO 1-9569 (tetrabenazine), a nonindole tranquilizing agent with reserpine-like effects. J Pharmacol Exp Ther. 1959;127:103-109.
11. Kazamatsuri H, Chien C, Cole JO. Treatment of tardive dyskinesia. I. Clinical efficacy of a dopamine-depleting agent, tetrabenazine. Arch Gen Psychiatry. 1972;27(1):95-99.
12. Scherman D, Weber MJ. Characterization of the vesicular monoamine transporter in cultured rat sympathetic neurons: persistence upon induction of cholinergic phenotypic traits. Dev Biol. 1987;119(1):68-74.
13. Erickson JD, Schafer MK, Bonner TI, et al. Distinct pharmacological properties and distribution in neurons and endocrine cells of two isoforms of the human vesicular monoamine transporter. Proc Natl Acad Sci U S A. 1996;93(10):5166-5171.
14. Kushner DJ, Baker A, Dunstall TG. Pharmacological uses and perspectives of heavy water and deuterated compounds. Can J Physiol Pharmacol. 1999;77(2):79-88.
15. United States Securities and Exchange Commission. Form S-1 Registration Statement of Auspex Pharmaceuticals, Inc. https://www.sec.gov/Archives/edgar/data/1454189/000119312513481239/d627086ds1.htm. Published December 20, 2013. Accessed July 1, 2016.
16. Cholongitas E, Papatheodoridis GV, Vangeli M, et al. Systematic review: the model for end-stage liver disease—should it replace Child-Pugh’s classification for assessing prognosis in cirrhosis? Aliment Pharmacol Ther. 2005;22(11-12):1079-1089.
Compared with first-generation antipsychotics, second-generation antipsychotics (SGAs) have a lower risk for extrapyramidal symptoms. Yet tardive dyskinesia (TD) remains a concern because of the widespread use of SGAs for multiple indications.1 Prior to April 2017, clinicians had no FDA-approved TD treatment options. The most widely used agent worldwide, tetrabenazine, had positive efficacy data in TD trials over the past 45 years but was not available in the United States until 2008, and its sole indication was for chorea associated with Huntington’s disease.2 Moreover, the use of tetrabenazine involved slow titration, multiple daily dosing, cytochrome P450 (CYP) 2D6 genotyping for doses >50 mg/d, and tolerability issues.
Tetrabenazine is an inhibitor of vesicular monoamine transport type 2 (VMAT2), a transport protein located almost exclusively in the CNS whose role is to place monoamine neurotransmitters (dopamine, serotonin, norepinephrine) into presynaptic vesicles. By decreasing dopamine transport into these presynaptic vesicles, synaptic dopamine release is lessened, thus reducing postsynaptic dopamine D2 receptor activity and the severity of dyskinetic movements.1
In 2 pivotal 12-week clinical trials, deutetrabenazine significantly reduced TD severity as measured by Abnormal Involuntary Movement Scale (AIMS) scores (see Efficacy).6,7
Clinical implications
TD remains a substantial public health concern due to the increasing use of antipsychotics for mood and other disorders beyond the initial indications for schizophrenia.1 Although exposure to dopamine D2antagonism results in postsynaptic receptor upregulation and supersensitivity that underlies the development of dyskinesia, this process is often rapidly reversible in animal models.1 The persistence of TD symptoms in up to 80% of patients after dopamine receptor blocking agents (DRBAs) are stopped has led to hypotheses that the underlying pathophysiology of TD is also a problem with neuroplasticity. Aside from DRBA exposure, environmental factors (eg, oxidative stress) and genetic predisposition might contribute to TD risk.1
Before 2017, only 1 medication (branched-chain amino acids) had been FDA-approved for treating TD in the United States, and only a few existing medications (clonazepam, amantadine, and ginkgo biloba extract [EGb-761]) had positive results from controlled trials, most with small effect sizes.8 Moreover, there was only 1 controlled trial each for clonazepam and EGb-761.1 A branched-chain amino acid preparation received FDA approval for managing TD in male patients, but is no longer commercially available, except from compounding pharmacies.9
Tetrabenazine was developed in the mid-1950s to avoid orthostasis and sedation associated with reserpine.10 Both reserpine and tetrabenazine proved effective for TD,11 but tetrabenazine lacked reserpine’s peripheral adverse effects. However, the kinetics of tetrabenazine necessitated multiple daily doses, and CYP2D6 genotyping was required for doses >50 mg/d.2
Receptor blocking. The mechanism that distinguishes the clinical profiles of reserpine and tetrabenazine relates to their differential properties at VMAT.12 VMAT exists in 2 forms (VMAT1 and VMAT2) that vary in distribution, with VMAT1 expressed mainly in the peripheral nervous system and VMAT2 expressed mainly in monoaminergic cells of the CNS.13 Tetrabenazine is a specific and reversible VMAT2 inhibitor, whereas reserpine is an irreversible and nonselective antagonist of VMAT1 and VMAT2. It is reserpine’s VMAT1 inhibition that results in peripheral adverse effects such as orthostasis. Tetrabenazine is rapidly and extensively converted into 2 isomers, alpha-dihydrotetrabenazine (α-DHTBZ) and beta-dihydrotetrabenazine (β-DHTBZ), both of which are metabolized by CYP2D6, with a role for CYP3A4 in α-DHTBZ metabolism.1 These DHTBZ metabolites have a short half-life when generated from oral tetrabenazine, a feature that necessitates multiple daily dosing; moreover, the existence of 2D6 polymorphisms led to FDA-mandated CYP2D6 genotyping for tetrabenazine doses >50 mg/d when it was approved for Huntington’s chorea. The concern is that 2D6 poor metabolizers will have excessive exposure to the VMAT2 effects of DHTBZ, resulting in sedation, akathisia, parkinsonism, and mood symptoms.2
How deuterium impacts medication kinetics. Deuterium is a naturally occurring, stable, nontoxic isotope of hydrogen. Humans have 5 g of deuterium in their body at any time, mostly in the form of heavy water (D2O).14 When deuterium is used to replace selected hydrogen atoms, the resulting molecule will have similar configuration and receptor-binding properties but markedly different kinetics. Because the carbon–deuterium covalent bond requires 8 times more energy to break than a carbon–hydrogen bond, the half-life is prolonged.15 Utilizing this knowledge, a deuterated form of tetrabenazine, deutetrabenazine, was synthesized with such a purpose in mind. While the active metabolites of deutetrabenazine retain the VMAT2 affinity of non-deuterated tetrabenazine, the substitution of deuterium for hydrogen at specific positions slows the breakdown of metabolites, resulting in sustained duration of action, greater active drug exposure, and less impact of 2D6 genotype on drug exposure, thus eliminating the need for genotyping, unless one wants to exceed 36 mg/d.
Deutetrabenazine was first studied in Huntington’s chorea in a 13-week, double-blind, placebo-controlled, parallel-group study (N = 90).4 The maximum daily deutetrabenazine dose was 48 mg, but reduced to 36 mg in those taking strong CYP2D6 inhibitors (bupropion, fluoxetine, or paroxetine). Blinded 2D6 genotyping was performed, but there was no dose modification required based on 2D6 genotype. There was a 36.4% reduction in total maximal chorea score for deutetrabenazine compared with 14.4% for placebo (P < .001).4 Importantly, adverse effects were comparable between both groups, with 1 drop-out in the deutetrabenazine arm vs 2 in the placebo arm. The only adverse event occurring in ≥5% of deutetrabenazine participants and at a rate ≥2 times that of placebo was somnolence: 11.1% for deutetrabenazine vs 4.4% for placebo.4 The mean deutetrabenazine daily dose at the end of the treatment period was 39.7 ± 9.3 mg, and for those with impaired CYP2D6 function (poor metabolizers or those taking strong CYP2D6 inhibiting medications), the mean daily dose was 34.8 mg ± 3.8 mg.4
Use in tardive dyskinesia. The recommended starting dosage for TD treatment is 6 mg, twice daily with food. The dose may be increased at weekly intervals in increments of 6 mg/d to a maximum recommended daily dosage of 48 mg.5 The maximum daily dose is 36 mg (18 mg, twice daily) in patients receiving strong CYP2D6 inhibitors or who are 2D6 poor metabolizers.5
Deutetrabenazine has not been studied in those with moderate or severe hepatic impairment, and its use is contraindicated in these patients.5 No clinical studies have been conducted to assess the effect of renal impairment on the pharmacokinetics of deutetrabenazine.5
Pharmacologic profile, adverse reactions
When the data from the two 12-week, phase 3 placebo-controlled studies were pooled, the most common adverse reactions occurring in >3% of deutetrabenazine patients and greater than placebo were nasopharyngeal symptoms (4% vs 2% placebo) and insomnia (4% vs 1% placebo).5 Importantly, in neither TD study were there clinically significant changes in rating scales for depression, suicidal ideation and behavior, or parkinsonism. There also were no clinically significant changes in measures of schizophrenia symptoms. The mean QT prolongation for a single 24 mg dose of deutetrabenazine in healthy volunteers was 4.5 milliseconds, with the upper bound of the double-sided 90% confidence interval reaching 6.5 milliseconds.5 For tetrabenazine, single 50 mg doses administered to volunteers resulted in mean QT prolongation of 8 milliseconds.5 In patients requiring deutetrabenazine doses >24 mg/d who are taking other medications known to prolong QTc, assess the QTc interval before and after increasing the dose of deutetrabenazine or other medications that are known to prolong QTc.5
How it works
Tetrabenazine is the only agent that has demonstrated significant efficacy for TD management, but its use involves slow titration, multiple daily dosing, CYP2D6 genotyping for doses >50 mg/d, and tolerability issues. For example, the most common adverse effects in the pivotal tetrabenazine Huntington’s disease trial were sedation/somnolence (tetrabenazine 31% vs 3% for placebo), insomnia (tetrabenazine 22% vs 0% for placebo), depression (tetrabenazine 19% vs 0% for placebo), fatigue (tetrabenazine 22% vs 13% for placebo), and akathisia (tetrabenazine 19% vs 0% for placebo).2 For comparison, the only adverse event occurring in ≥5% of deutetrabenazine participants and at a rate ≥2 times that of placebo in the pivotal Huntington’s disease trial was somnolence (11.1% for deutetrabenazine vs 4.4% for placebo).4
Pharmacokinetics
Deutetrabenazine has 80% oral bioavailability, and is rapidly converted to its active metabolites after oral dosing (Table 2).5 Linear dose dependence of Cmax and area under the curve (AUC) was observed for the active metabolites following single or multiple doses of deutetrabenazine (6 to 24 mg and 7.5 to 22.5 mg, twice daily).15 Cmax of deuterated α-DHTBZ and β-DHTBZ is reached within 3 to 4 hours after dosing, with a steady state ratio of 3:1 for the α-DHTBZ vs the β-DHTBZ form. Food had no effect on AUC, but did increase Cmax by 50%.5
Deutetrabenazine is metabolized through carbonyl reductase enzymes to its active metabolites, and these are further metabolized through multiple CYP pathways, predominantly 2D6 and to a lesser extent 3A4. The effect of CYP2D6 inhibition on the pharmacokinetics of deutetrabenazine and its α-DHTBZ and β-DHTBZ metabolites was studied in 24 healthy participants following a single 22.5 mg dose of deutetrabenazine given after 8 days of administration of the strong CYP2D6 inhibitor paroxetine, 20 mg/d. In the presence of paroxetine, systemic exposure (AUC) of α-DHTBZ was 1.9-fold higher and β-DHTBZ was 6.5-fold higher, resulting in an approximately 3-fold increase in AUC for total (α+β)-DHTBZ, with corresponding increases in mean half-life of approximately 1.5-fold and 2.7-fold, respectively.5 Neither deutetrabenazine or its metabolites are inhibitors or inducers of major CYP enzymes. Aside from VMAT2, the results of in vitro studies suggest that deutetrabenazine and its active metabolites are unlikely to inhibit most major drug transporters at clinically relevant concentrations.
Efficacy
Efficacy was established in two 12-week, double-blind, placebo-controlled trials of adult patients with TD (ages 18 to 80).6,7 Eligible participants had:
- TD diagnosis for ≥3 months before screening and a history of DRBA treatment for ≥3 months (≥1 month if age ≥60)
- Total AIMS motor score ≥6 (items 1 to 7) at both screening and baseline, verified by a blinded central rater at screening via central video rating
- Patients with an underlying psychiatric illness had to be stable. Psychoactive medication use, including antipsychotics, was allowed if stable for ≥30 days before screening (antidepressants, ≥45 days).
Exclusion criteria included treatment with tetrabenazine, reserpine, α-methyl-p-tyrosine, strong anticholinergic medications, dopamine antagonizing antiemetics (eg, metoclopramide, prochlorperazine, promethazine), dopamine agonists, levodopa, stimulants, or a monoamine oxidase inhibitor (MAOI) within 30 days of screening or baseline, or treatment with botulinum toxin within 3 months of screening; and presence of a neurologic condition that could confound TD assessments, serious untreated or undertreated psychiatric illness, or unstable medical illness. Patients with a history of or active suicidal ideation or behavior within 6 months of screening or score ≥11 on the depression subscale of the Hospital Anxiety and Depression Scale were excluded. Those participants with Fridericia-corrected QT interval values >450 milliseconds in men, >460 milliseconds in women, or >480 milliseconds in patients with a right bundle branch block on electrocardiography at screening also were excluded.
The flexible-dose TD study was performed in 117 participants randomized in a 1:1 manner to deutetrabenazine or placebo, both administered twice daily, titrated to optimal dosage (12 to 48 mg/d) over 6 weeks, and then administered at that dose for another 6 weeks.7 The population demographics were: mean age, 54.6 ± 10.3 years, 52.1% female, 69.2% white, and 80.3% receiving ongoing dopamine antagonists, with a mean TD duration of 74.7 ± 81.5 months. Sixty-eight percent had schizophrenia spectrum disorders, and 30% had mood disorders. The primary outcome was change in total AIMS score (items 1 to 7) assessed by central, independent raters. The mean baseline AIMS score for items 1 to 7 was 9.6 ± 3.9, with 82.9% of participants with baseline AIMS scores ≥6. Study treatment retention was high: placebo 88.1%, deutetrabenazine 89.7%.7 There was a mean 3 point decrease in AIMS score for deutetrabenazine compared with 1.4 for placebo (P = .019). Among those with baseline AIMS scores ≥6, there was a 3.4 point decrease in AIMS scores for deutetrabenazine compared with a 1.9 point decrease for placebo (P = .027). The only adverse effects that occurred in ≥5% of deutetrabenazine participants and at a rate ≥2 times the rate in placebo were insomnia (deutetrabenazine 6.9% vs placebo 1.7%) and akathisia (deutetrabenazine 5.2% vs placebo 0%).
The fixed-dose TD study was performed in 293 participants randomized in 1:1:1:1 manner to 1 of 3 fixed doses of deutetrabenazine (12 mg/d, 24 mg/d, or 36 mg/d) or placebo, both administered twice daily.6 The starting dose of deutetrabenazine was 6 mg twice daily. During the dose escalation period (through Week 4), the dose of study drug was increased weekly in increments of 6 mg/d until the randomized dose was achieved. Patients continued to receive the dose they were assigned to over a maintenance period of 8 weeks.6 The population demographics were: mean age, 56.4 ± 11.3 years, 55% female, 79% white, 76% receiving ongoing dopamine antagonists, with a mean TD duration of 67.2 ± 66 months. Sixty percent had schizophrenia spectrum disorders, and 36% had mood disorders. The primary outcome was change in AIMS total score (items 1 to 7) assessed by central, independent raters. The mean AIMS score at baseline was 9.5 ± 2.7 in the placebo group, and for deutetrabenazine: 9.6 ± 2.4 in the 12 mg/d group, 9.4 ± 2.9 in the 24 mg/d group, and 10.1 ± 3.2 in the 36 mg/d group. The 24 mg/d and 36 mg/d doses significantly reduced AIMS scores from baseline vs placebo: 36 mg: −3.3 (0.42) vs −1.4 (0.41) (P = .001); 24 mg: −3.2 (0.45) vs −1.4 (0.41) (P = .003). Study treatment retention rates were high: placebo 90.5%, deutetrabenazine 88%. Across all doses, only 1 adverse effect occurred in ≥5% of deutetrabenazine participants: headache (5% deutetrabenazine vs 6% placebo). At the highest dose, 36 mg/d, the only adverse effects that occurred in ≥5% of participants were diarrhea (7% deutetrabenazine vs 3% placebo) and headache (7% deutetrabenazine vs 6% placebo).
Outcome. In the flexible-dose study (mean dose 38.8 ± 7.92 mg/d), the deutetrabenazine arm experienced a mean 30% reduction in AIMS scores from baseline at the Week 12 endpoint. Compared with placebo, the mean reduction in AIMS scores (standard error) was: −3.0 (0.45) deutetrabenazine vs −1.6 (0.46) placebo (P = .019).7 For the fixed-dose study, the 24 mg/d and 36 mg/d doses significantly reduced AIMS scores from baseline vs placebo: 36 mg: −3.3 (0.42) vs −1.4 (0.41) (P = .001); 24 mg: −3.2 (0.45) vs −1.4 (0.41) (P = .003). In addition to these mean changes from baseline, 35% of the 24 mg/d group and 33% of the 36 mg/d group demonstrated ≥50% reduction in AIMS scores.6
Tolerability
In the 2 phase 3 trials, there were no adverse effects occurring with an incidence ≥5% and at least twice the rate of placebo.5 Discontinuations because of adverse events were low in both pivotal studies across all treatment groups: 3.4% for placebo vs 1.7% for deutetrabenazine in the flexible-dose trial,7 and 3% for placebo vs 4% for deutetrabenazine in the fixed-dose study.6 In neither trial were there clinically significant changes in ratings of depression, suicidality, parkinsonism, or schizophrenia symptoms. The mean QT prolongation in healthy volunteers is described above.
Clinical considerations
Unique properties. Deutetrabenazine utilizes the greater bond strength of the carbon–deuterium bond to slow CYP metabolism, resulting in prolonged duration of action that is well tolerated, and provides significant efficacy.
Why Rx? The reasons to prescribe deutetrabenazine for TD patients include:
- only 1 of 2 agents with FDA approval for TD
- fewer tolerability issues than with tetrabenazine
- lower sedation rates in TD trials than with valbenazine
- no signal for effects on mood parameters or rates of parkinsonism when used for TD.
Dosing
The recommended starting dosage of deutetrabenazine is 6 mg twice daily taken with food, increasing by 6 mg/d weekly as needed, with a maximum dose of 48 mg/d or 36 mg/d in those taking strong CYP2D6 inhibitors or who are 2D6 poor metabolizers. Deutetrabenazine is contraindicated in patients with hepatic impairment (as determined by Child-Pugh criteria16). There are no data in patients with renal impairment. The combined efficacy and tolerability of dosages >48 mg/d has not been evaluated. Overdoses of tetrabenazine ranging from 100 to 1,000 mg have been reported in the literature and were associated with acute dystonia, oculogyric crisis, nausea and vomiting, sweating, sedation, hypotension, confusion, diarrhea, hallucinations, rubor, and tremor.5
Contraindications
When used for TD, deutetrabenazine is contraindicated for patients taking reserpine, tetrabenazine, valbenazine, or MAOIs, and for patients with hepatic impairment. As with most medications, there are no data on deutetrabenazine use in pregnant women; however, oral administration of deutetrabenazine (5, 10, or 30 mg/kg/d) or tetrabenazine (30 mg/kg/d) to pregnant rats during organogenesis had no clear effect on embryofetal development. The highest dose tested was 6 times the maximum recommended human dose of 48 mg/d on a body surface area (mg/m2) basis. There are no data on the presence of deutetrabenazine or its metabolites in human milk, the effects on the breastfed infant, or the effects of the drug on milk production.
Compared with first-generation antipsychotics, second-generation antipsychotics (SGAs) have a lower risk for extrapyramidal symptoms. Yet tardive dyskinesia (TD) remains a concern because of the widespread use of SGAs for multiple indications.1 Prior to April 2017, clinicians had no FDA-approved TD treatment options. The most widely used agent worldwide, tetrabenazine, had positive efficacy data in TD trials over the past 45 years but was not available in the United States until 2008, and its sole indication was for chorea associated with Huntington’s disease.2 Moreover, the use of tetrabenazine involved slow titration, multiple daily dosing, cytochrome P450 (CYP) 2D6 genotyping for doses >50 mg/d, and tolerability issues.
Tetrabenazine is an inhibitor of vesicular monoamine transport type 2 (VMAT2), a transport protein located almost exclusively in the CNS whose role is to place monoamine neurotransmitters (dopamine, serotonin, norepinephrine) into presynaptic vesicles. By decreasing dopamine transport into these presynaptic vesicles, synaptic dopamine release is lessened, thus reducing postsynaptic dopamine D2 receptor activity and the severity of dyskinetic movements.1
In 2 pivotal 12-week clinical trials, deutetrabenazine significantly reduced TD severity as measured by Abnormal Involuntary Movement Scale (AIMS) scores (see Efficacy).6,7
Clinical implications
TD remains a substantial public health concern due to the increasing use of antipsychotics for mood and other disorders beyond the initial indications for schizophrenia.1 Although exposure to dopamine D2antagonism results in postsynaptic receptor upregulation and supersensitivity that underlies the development of dyskinesia, this process is often rapidly reversible in animal models.1 The persistence of TD symptoms in up to 80% of patients after dopamine receptor blocking agents (DRBAs) are stopped has led to hypotheses that the underlying pathophysiology of TD is also a problem with neuroplasticity. Aside from DRBA exposure, environmental factors (eg, oxidative stress) and genetic predisposition might contribute to TD risk.1
Before 2017, only 1 medication (branched-chain amino acids) had been FDA-approved for treating TD in the United States, and only a few existing medications (clonazepam, amantadine, and ginkgo biloba extract [EGb-761]) had positive results from controlled trials, most with small effect sizes.8 Moreover, there was only 1 controlled trial each for clonazepam and EGb-761.1 A branched-chain amino acid preparation received FDA approval for managing TD in male patients, but is no longer commercially available, except from compounding pharmacies.9
Tetrabenazine was developed in the mid-1950s to avoid orthostasis and sedation associated with reserpine.10 Both reserpine and tetrabenazine proved effective for TD,11 but tetrabenazine lacked reserpine’s peripheral adverse effects. However, the kinetics of tetrabenazine necessitated multiple daily doses, and CYP2D6 genotyping was required for doses >50 mg/d.2
Receptor blocking. The mechanism that distinguishes the clinical profiles of reserpine and tetrabenazine relates to their differential properties at VMAT.12 VMAT exists in 2 forms (VMAT1 and VMAT2) that vary in distribution, with VMAT1 expressed mainly in the peripheral nervous system and VMAT2 expressed mainly in monoaminergic cells of the CNS.13 Tetrabenazine is a specific and reversible VMAT2 inhibitor, whereas reserpine is an irreversible and nonselective antagonist of VMAT1 and VMAT2. It is reserpine’s VMAT1 inhibition that results in peripheral adverse effects such as orthostasis. Tetrabenazine is rapidly and extensively converted into 2 isomers, alpha-dihydrotetrabenazine (α-DHTBZ) and beta-dihydrotetrabenazine (β-DHTBZ), both of which are metabolized by CYP2D6, with a role for CYP3A4 in α-DHTBZ metabolism.1 These DHTBZ metabolites have a short half-life when generated from oral tetrabenazine, a feature that necessitates multiple daily dosing; moreover, the existence of 2D6 polymorphisms led to FDA-mandated CYP2D6 genotyping for tetrabenazine doses >50 mg/d when it was approved for Huntington’s chorea. The concern is that 2D6 poor metabolizers will have excessive exposure to the VMAT2 effects of DHTBZ, resulting in sedation, akathisia, parkinsonism, and mood symptoms.2
How deuterium impacts medication kinetics. Deuterium is a naturally occurring, stable, nontoxic isotope of hydrogen. Humans have 5 g of deuterium in their body at any time, mostly in the form of heavy water (D2O).14 When deuterium is used to replace selected hydrogen atoms, the resulting molecule will have similar configuration and receptor-binding properties but markedly different kinetics. Because the carbon–deuterium covalent bond requires 8 times more energy to break than a carbon–hydrogen bond, the half-life is prolonged.15 Utilizing this knowledge, a deuterated form of tetrabenazine, deutetrabenazine, was synthesized with such a purpose in mind. While the active metabolites of deutetrabenazine retain the VMAT2 affinity of non-deuterated tetrabenazine, the substitution of deuterium for hydrogen at specific positions slows the breakdown of metabolites, resulting in sustained duration of action, greater active drug exposure, and less impact of 2D6 genotype on drug exposure, thus eliminating the need for genotyping, unless one wants to exceed 36 mg/d.
Deutetrabenazine was first studied in Huntington’s chorea in a 13-week, double-blind, placebo-controlled, parallel-group study (N = 90).4 The maximum daily deutetrabenazine dose was 48 mg, but reduced to 36 mg in those taking strong CYP2D6 inhibitors (bupropion, fluoxetine, or paroxetine). Blinded 2D6 genotyping was performed, but there was no dose modification required based on 2D6 genotype. There was a 36.4% reduction in total maximal chorea score for deutetrabenazine compared with 14.4% for placebo (P < .001).4 Importantly, adverse effects were comparable between both groups, with 1 drop-out in the deutetrabenazine arm vs 2 in the placebo arm. The only adverse event occurring in ≥5% of deutetrabenazine participants and at a rate ≥2 times that of placebo was somnolence: 11.1% for deutetrabenazine vs 4.4% for placebo.4 The mean deutetrabenazine daily dose at the end of the treatment period was 39.7 ± 9.3 mg, and for those with impaired CYP2D6 function (poor metabolizers or those taking strong CYP2D6 inhibiting medications), the mean daily dose was 34.8 mg ± 3.8 mg.4
Use in tardive dyskinesia. The recommended starting dosage for TD treatment is 6 mg, twice daily with food. The dose may be increased at weekly intervals in increments of 6 mg/d to a maximum recommended daily dosage of 48 mg.5 The maximum daily dose is 36 mg (18 mg, twice daily) in patients receiving strong CYP2D6 inhibitors or who are 2D6 poor metabolizers.5
Deutetrabenazine has not been studied in those with moderate or severe hepatic impairment, and its use is contraindicated in these patients.5 No clinical studies have been conducted to assess the effect of renal impairment on the pharmacokinetics of deutetrabenazine.5
Pharmacologic profile, adverse reactions
When the data from the two 12-week, phase 3 placebo-controlled studies were pooled, the most common adverse reactions occurring in >3% of deutetrabenazine patients and greater than placebo were nasopharyngeal symptoms (4% vs 2% placebo) and insomnia (4% vs 1% placebo).5 Importantly, in neither TD study were there clinically significant changes in rating scales for depression, suicidal ideation and behavior, or parkinsonism. There also were no clinically significant changes in measures of schizophrenia symptoms. The mean QT prolongation for a single 24 mg dose of deutetrabenazine in healthy volunteers was 4.5 milliseconds, with the upper bound of the double-sided 90% confidence interval reaching 6.5 milliseconds.5 For tetrabenazine, single 50 mg doses administered to volunteers resulted in mean QT prolongation of 8 milliseconds.5 In patients requiring deutetrabenazine doses >24 mg/d who are taking other medications known to prolong QTc, assess the QTc interval before and after increasing the dose of deutetrabenazine or other medications that are known to prolong QTc.5
How it works
Tetrabenazine is the only agent that has demonstrated significant efficacy for TD management, but its use involves slow titration, multiple daily dosing, CYP2D6 genotyping for doses >50 mg/d, and tolerability issues. For example, the most common adverse effects in the pivotal tetrabenazine Huntington’s disease trial were sedation/somnolence (tetrabenazine 31% vs 3% for placebo), insomnia (tetrabenazine 22% vs 0% for placebo), depression (tetrabenazine 19% vs 0% for placebo), fatigue (tetrabenazine 22% vs 13% for placebo), and akathisia (tetrabenazine 19% vs 0% for placebo).2 For comparison, the only adverse event occurring in ≥5% of deutetrabenazine participants and at a rate ≥2 times that of placebo in the pivotal Huntington’s disease trial was somnolence (11.1% for deutetrabenazine vs 4.4% for placebo).4
Pharmacokinetics
Deutetrabenazine has 80% oral bioavailability, and is rapidly converted to its active metabolites after oral dosing (Table 2).5 Linear dose dependence of Cmax and area under the curve (AUC) was observed for the active metabolites following single or multiple doses of deutetrabenazine (6 to 24 mg and 7.5 to 22.5 mg, twice daily).15 Cmax of deuterated α-DHTBZ and β-DHTBZ is reached within 3 to 4 hours after dosing, with a steady state ratio of 3:1 for the α-DHTBZ vs the β-DHTBZ form. Food had no effect on AUC, but did increase Cmax by 50%.5
Deutetrabenazine is metabolized through carbonyl reductase enzymes to its active metabolites, and these are further metabolized through multiple CYP pathways, predominantly 2D6 and to a lesser extent 3A4. The effect of CYP2D6 inhibition on the pharmacokinetics of deutetrabenazine and its α-DHTBZ and β-DHTBZ metabolites was studied in 24 healthy participants following a single 22.5 mg dose of deutetrabenazine given after 8 days of administration of the strong CYP2D6 inhibitor paroxetine, 20 mg/d. In the presence of paroxetine, systemic exposure (AUC) of α-DHTBZ was 1.9-fold higher and β-DHTBZ was 6.5-fold higher, resulting in an approximately 3-fold increase in AUC for total (α+β)-DHTBZ, with corresponding increases in mean half-life of approximately 1.5-fold and 2.7-fold, respectively.5 Neither deutetrabenazine or its metabolites are inhibitors or inducers of major CYP enzymes. Aside from VMAT2, the results of in vitro studies suggest that deutetrabenazine and its active metabolites are unlikely to inhibit most major drug transporters at clinically relevant concentrations.
Efficacy
Efficacy was established in two 12-week, double-blind, placebo-controlled trials of adult patients with TD (ages 18 to 80).6,7 Eligible participants had:
- TD diagnosis for ≥3 months before screening and a history of DRBA treatment for ≥3 months (≥1 month if age ≥60)
- Total AIMS motor score ≥6 (items 1 to 7) at both screening and baseline, verified by a blinded central rater at screening via central video rating
- Patients with an underlying psychiatric illness had to be stable. Psychoactive medication use, including antipsychotics, was allowed if stable for ≥30 days before screening (antidepressants, ≥45 days).
Exclusion criteria included treatment with tetrabenazine, reserpine, α-methyl-p-tyrosine, strong anticholinergic medications, dopamine antagonizing antiemetics (eg, metoclopramide, prochlorperazine, promethazine), dopamine agonists, levodopa, stimulants, or a monoamine oxidase inhibitor (MAOI) within 30 days of screening or baseline, or treatment with botulinum toxin within 3 months of screening; and presence of a neurologic condition that could confound TD assessments, serious untreated or undertreated psychiatric illness, or unstable medical illness. Patients with a history of or active suicidal ideation or behavior within 6 months of screening or score ≥11 on the depression subscale of the Hospital Anxiety and Depression Scale were excluded. Those participants with Fridericia-corrected QT interval values >450 milliseconds in men, >460 milliseconds in women, or >480 milliseconds in patients with a right bundle branch block on electrocardiography at screening also were excluded.
The flexible-dose TD study was performed in 117 participants randomized in a 1:1 manner to deutetrabenazine or placebo, both administered twice daily, titrated to optimal dosage (12 to 48 mg/d) over 6 weeks, and then administered at that dose for another 6 weeks.7 The population demographics were: mean age, 54.6 ± 10.3 years, 52.1% female, 69.2% white, and 80.3% receiving ongoing dopamine antagonists, with a mean TD duration of 74.7 ± 81.5 months. Sixty-eight percent had schizophrenia spectrum disorders, and 30% had mood disorders. The primary outcome was change in total AIMS score (items 1 to 7) assessed by central, independent raters. The mean baseline AIMS score for items 1 to 7 was 9.6 ± 3.9, with 82.9% of participants with baseline AIMS scores ≥6. Study treatment retention was high: placebo 88.1%, deutetrabenazine 89.7%.7 There was a mean 3 point decrease in AIMS score for deutetrabenazine compared with 1.4 for placebo (P = .019). Among those with baseline AIMS scores ≥6, there was a 3.4 point decrease in AIMS scores for deutetrabenazine compared with a 1.9 point decrease for placebo (P = .027). The only adverse effects that occurred in ≥5% of deutetrabenazine participants and at a rate ≥2 times the rate in placebo were insomnia (deutetrabenazine 6.9% vs placebo 1.7%) and akathisia (deutetrabenazine 5.2% vs placebo 0%).
The fixed-dose TD study was performed in 293 participants randomized in 1:1:1:1 manner to 1 of 3 fixed doses of deutetrabenazine (12 mg/d, 24 mg/d, or 36 mg/d) or placebo, both administered twice daily.6 The starting dose of deutetrabenazine was 6 mg twice daily. During the dose escalation period (through Week 4), the dose of study drug was increased weekly in increments of 6 mg/d until the randomized dose was achieved. Patients continued to receive the dose they were assigned to over a maintenance period of 8 weeks.6 The population demographics were: mean age, 56.4 ± 11.3 years, 55% female, 79% white, 76% receiving ongoing dopamine antagonists, with a mean TD duration of 67.2 ± 66 months. Sixty percent had schizophrenia spectrum disorders, and 36% had mood disorders. The primary outcome was change in AIMS total score (items 1 to 7) assessed by central, independent raters. The mean AIMS score at baseline was 9.5 ± 2.7 in the placebo group, and for deutetrabenazine: 9.6 ± 2.4 in the 12 mg/d group, 9.4 ± 2.9 in the 24 mg/d group, and 10.1 ± 3.2 in the 36 mg/d group. The 24 mg/d and 36 mg/d doses significantly reduced AIMS scores from baseline vs placebo: 36 mg: −3.3 (0.42) vs −1.4 (0.41) (P = .001); 24 mg: −3.2 (0.45) vs −1.4 (0.41) (P = .003). Study treatment retention rates were high: placebo 90.5%, deutetrabenazine 88%. Across all doses, only 1 adverse effect occurred in ≥5% of deutetrabenazine participants: headache (5% deutetrabenazine vs 6% placebo). At the highest dose, 36 mg/d, the only adverse effects that occurred in ≥5% of participants were diarrhea (7% deutetrabenazine vs 3% placebo) and headache (7% deutetrabenazine vs 6% placebo).
Outcome. In the flexible-dose study (mean dose 38.8 ± 7.92 mg/d), the deutetrabenazine arm experienced a mean 30% reduction in AIMS scores from baseline at the Week 12 endpoint. Compared with placebo, the mean reduction in AIMS scores (standard error) was: −3.0 (0.45) deutetrabenazine vs −1.6 (0.46) placebo (P = .019).7 For the fixed-dose study, the 24 mg/d and 36 mg/d doses significantly reduced AIMS scores from baseline vs placebo: 36 mg: −3.3 (0.42) vs −1.4 (0.41) (P = .001); 24 mg: −3.2 (0.45) vs −1.4 (0.41) (P = .003). In addition to these mean changes from baseline, 35% of the 24 mg/d group and 33% of the 36 mg/d group demonstrated ≥50% reduction in AIMS scores.6
Tolerability
In the 2 phase 3 trials, there were no adverse effects occurring with an incidence ≥5% and at least twice the rate of placebo.5 Discontinuations because of adverse events were low in both pivotal studies across all treatment groups: 3.4% for placebo vs 1.7% for deutetrabenazine in the flexible-dose trial,7 and 3% for placebo vs 4% for deutetrabenazine in the fixed-dose study.6 In neither trial were there clinically significant changes in ratings of depression, suicidality, parkinsonism, or schizophrenia symptoms. The mean QT prolongation in healthy volunteers is described above.
Clinical considerations
Unique properties. Deutetrabenazine utilizes the greater bond strength of the carbon–deuterium bond to slow CYP metabolism, resulting in prolonged duration of action that is well tolerated, and provides significant efficacy.
Why Rx? The reasons to prescribe deutetrabenazine for TD patients include:
- only 1 of 2 agents with FDA approval for TD
- fewer tolerability issues than with tetrabenazine
- lower sedation rates in TD trials than with valbenazine
- no signal for effects on mood parameters or rates of parkinsonism when used for TD.
Dosing
The recommended starting dosage of deutetrabenazine is 6 mg twice daily taken with food, increasing by 6 mg/d weekly as needed, with a maximum dose of 48 mg/d or 36 mg/d in those taking strong CYP2D6 inhibitors or who are 2D6 poor metabolizers. Deutetrabenazine is contraindicated in patients with hepatic impairment (as determined by Child-Pugh criteria16). There are no data in patients with renal impairment. The combined efficacy and tolerability of dosages >48 mg/d has not been evaluated. Overdoses of tetrabenazine ranging from 100 to 1,000 mg have been reported in the literature and were associated with acute dystonia, oculogyric crisis, nausea and vomiting, sweating, sedation, hypotension, confusion, diarrhea, hallucinations, rubor, and tremor.5
Contraindications
When used for TD, deutetrabenazine is contraindicated for patients taking reserpine, tetrabenazine, valbenazine, or MAOIs, and for patients with hepatic impairment. As with most medications, there are no data on deutetrabenazine use in pregnant women; however, oral administration of deutetrabenazine (5, 10, or 30 mg/kg/d) or tetrabenazine (30 mg/kg/d) to pregnant rats during organogenesis had no clear effect on embryofetal development. The highest dose tested was 6 times the maximum recommended human dose of 48 mg/d on a body surface area (mg/m2) basis. There are no data on the presence of deutetrabenazine or its metabolites in human milk, the effects on the breastfed infant, or the effects of the drug on milk production.
1. Meyer JM. Forgotten but not gone: new developments in the understanding and treatment of tardive dyskinesia. CNS Spectr. 2016;21(S1):13-24.
2. Jankovic J, Clarence-Smith K. Tetrabenazine for the treatment of chorea and other hyperkinetic movement disorders. Expert Rev Neurother. 2011;11(11):1509-1523.
3. Meyer JM. Valbenazine for tardive dyskinesia. Current Psychiatry. 2017;16(5):40-46.
4. Huntington Study Group; Frank S, Testa CM, Stamler D, et al. Effect of deutetrabenazine on chorea among patients with Huntington disease: a randomized clinical trial. JAMA. 2016;316(1):40-50.
5. Austedo [package insert]. North Wales, PA: Teva Pharmaceuticals USA, Inc.; 2017.
6. Anderson KE, Stamler D, Davis MD, et al. Deutetrabenazine for treatment of involuntary movements in patients with tardive dyskinesia (AIM-TD): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Psychiatry. 2017;4(8):595-604.
7. Fernandez HH, Factor SA, Hauser RA, et al. Randomized controlled trial of deutetrabenazine for tardive dyskinesia: the ARM-TD study. Neurology. 2017;88(21):2003-2010.
8. Bhidayasiri R, Fahn S, Weiner WJ, et al. Evidence-based guideline: treatment of tardive syndromes: report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. 2013;81(5):463-469.
9. Richardson MA, Small AM, Read LL, et al. Branched chain amino acid treatment of tardive dyskinesia in children and adolescents. J Clin Psychiatry. 2004;65(1):92-96.
10. Quinn GP, Shore PA, Brodie BB. Biochemical and pharmacological studies of RO 1-9569 (tetrabenazine), a nonindole tranquilizing agent with reserpine-like effects. J Pharmacol Exp Ther. 1959;127:103-109.
11. Kazamatsuri H, Chien C, Cole JO. Treatment of tardive dyskinesia. I. Clinical efficacy of a dopamine-depleting agent, tetrabenazine. Arch Gen Psychiatry. 1972;27(1):95-99.
12. Scherman D, Weber MJ. Characterization of the vesicular monoamine transporter in cultured rat sympathetic neurons: persistence upon induction of cholinergic phenotypic traits. Dev Biol. 1987;119(1):68-74.
13. Erickson JD, Schafer MK, Bonner TI, et al. Distinct pharmacological properties and distribution in neurons and endocrine cells of two isoforms of the human vesicular monoamine transporter. Proc Natl Acad Sci U S A. 1996;93(10):5166-5171.
14. Kushner DJ, Baker A, Dunstall TG. Pharmacological uses and perspectives of heavy water and deuterated compounds. Can J Physiol Pharmacol. 1999;77(2):79-88.
15. United States Securities and Exchange Commission. Form S-1 Registration Statement of Auspex Pharmaceuticals, Inc. https://www.sec.gov/Archives/edgar/data/1454189/000119312513481239/d627086ds1.htm. Published December 20, 2013. Accessed July 1, 2016.
16. Cholongitas E, Papatheodoridis GV, Vangeli M, et al. Systematic review: the model for end-stage liver disease—should it replace Child-Pugh’s classification for assessing prognosis in cirrhosis? Aliment Pharmacol Ther. 2005;22(11-12):1079-1089.
1. Meyer JM. Forgotten but not gone: new developments in the understanding and treatment of tardive dyskinesia. CNS Spectr. 2016;21(S1):13-24.
2. Jankovic J, Clarence-Smith K. Tetrabenazine for the treatment of chorea and other hyperkinetic movement disorders. Expert Rev Neurother. 2011;11(11):1509-1523.
3. Meyer JM. Valbenazine for tardive dyskinesia. Current Psychiatry. 2017;16(5):40-46.
4. Huntington Study Group; Frank S, Testa CM, Stamler D, et al. Effect of deutetrabenazine on chorea among patients with Huntington disease: a randomized clinical trial. JAMA. 2016;316(1):40-50.
5. Austedo [package insert]. North Wales, PA: Teva Pharmaceuticals USA, Inc.; 2017.
6. Anderson KE, Stamler D, Davis MD, et al. Deutetrabenazine for treatment of involuntary movements in patients with tardive dyskinesia (AIM-TD): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Psychiatry. 2017;4(8):595-604.
7. Fernandez HH, Factor SA, Hauser RA, et al. Randomized controlled trial of deutetrabenazine for tardive dyskinesia: the ARM-TD study. Neurology. 2017;88(21):2003-2010.
8. Bhidayasiri R, Fahn S, Weiner WJ, et al. Evidence-based guideline: treatment of tardive syndromes: report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. 2013;81(5):463-469.
9. Richardson MA, Small AM, Read LL, et al. Branched chain amino acid treatment of tardive dyskinesia in children and adolescents. J Clin Psychiatry. 2004;65(1):92-96.
10. Quinn GP, Shore PA, Brodie BB. Biochemical and pharmacological studies of RO 1-9569 (tetrabenazine), a nonindole tranquilizing agent with reserpine-like effects. J Pharmacol Exp Ther. 1959;127:103-109.
11. Kazamatsuri H, Chien C, Cole JO. Treatment of tardive dyskinesia. I. Clinical efficacy of a dopamine-depleting agent, tetrabenazine. Arch Gen Psychiatry. 1972;27(1):95-99.
12. Scherman D, Weber MJ. Characterization of the vesicular monoamine transporter in cultured rat sympathetic neurons: persistence upon induction of cholinergic phenotypic traits. Dev Biol. 1987;119(1):68-74.
13. Erickson JD, Schafer MK, Bonner TI, et al. Distinct pharmacological properties and distribution in neurons and endocrine cells of two isoforms of the human vesicular monoamine transporter. Proc Natl Acad Sci U S A. 1996;93(10):5166-5171.
14. Kushner DJ, Baker A, Dunstall TG. Pharmacological uses and perspectives of heavy water and deuterated compounds. Can J Physiol Pharmacol. 1999;77(2):79-88.
15. United States Securities and Exchange Commission. Form S-1 Registration Statement of Auspex Pharmaceuticals, Inc. https://www.sec.gov/Archives/edgar/data/1454189/000119312513481239/d627086ds1.htm. Published December 20, 2013. Accessed July 1, 2016.
16. Cholongitas E, Papatheodoridis GV, Vangeli M, et al. Systematic review: the model for end-stage liver disease—should it replace Child-Pugh’s classification for assessing prognosis in cirrhosis? Aliment Pharmacol Ther. 2005;22(11-12):1079-1089.
When should you consider combining 2 long-acting injectable antipsychotics?
Ms. S, age 39, with a 15-year history of schizophrenia and severe paranoid delusions, is admitted after physically assaulting a staff member at a group home. She is receiving paliperidone palmitate, 234 mg every 4 weeks. This has reduced the severity of her symptoms, but she continues to have persistent delusions that affect her ability to accept redirection from staff. Ms. S frequently accuses staff and peers of sexual assault, says that she is pregnant, and does not adhere to treatment recommendations for laboratory monitoring because the “staff uses her blood for experiments.”
Ms. S frequently requires administration of oral and IM haloperidol, as needed, when she becomes aggressive with the staff. She has poor insight into her mental illness and does not believe that she needs medication. Ms. S has a long history of stopping her oral antipsychotic after a few days, reporting that it is “harming her baby.” Monotherapy has been tried with various long-acting injectable antipsychotics (LAIAs), but she still exhibits persistent delusions. The treatment team decides to add a second LAIA, haloperidol decanoate, 200 mg every 4 weeks, to her regimen.

Ladds et al.7 A 49-year-old woman with schizophrenia who was hospitalized for aggressive and bizarre behavior and had been institutionalized for 20 years stopped taking her medication regimen.7 She started taking 8-hour showers with bleach, talking incoherently, and believing that someone was poisoning her. She had poor response to oral risperidone monotherapy; however, 2 months after adding oral fluphenazine and benztropine to her regimen, her symptoms substantially improved (doses not reported). Because she had impaired insight into the need for daily medication, she was started on depot fluphenazine decanoate and risperidone microspheres (doses not reported) before discharge. No substantial adverse effects were noted with this regimen.
Wartelsteiner and Hofer.8 A man who had been diagnosed with paranoid schizophrenia at age 20 presented with thought blocking, incoherence, persecutory delusions, and uncontrolled self-damaging behavior.8 He had been admitted 27 times over 7 years; during this time he received many antipsychotic monotherapies and combination regimens. A total of 8 oral antipsychotics (including clozapine) and 5 LAIAs had been administered during these trials. He significantly improved with the combination of olanzapine and risperidone. Both medications were switched to LAIA formulations to address medication nonadherence. His symptoms remained stable with risperidone microspheres, 100 mg, and olanzapine pamoate, 300 mg, each administered every 2 weeks. He did not experience any adverse effects with this combination therapy.
Scangos et al.9 A 26-year-old Vietnamese man with schizophrenia and an extensive history of unprovoked, psychotically driven assaults was given multiple antipsychotics (including clozapine) during hospitalizations, and his medication regimen consistently included 2 antipsychotics. After contracting viral gastroenteritis, he refused oral medications and required short-acting IM administration of both haloperidol, 5 mg, twice a day, and olanzapine, 10 mg, twice a day. Because of concerns about continuing this regimen, he was switched to haloperidol decanoate (dose not reported) and olanzapine pamoate, 405 mg, administered once per month. The injections were scheduled to alternate so that the patient would receive 1 injection every 2 weeks. The patient’s assaultive behavior was significantly reduced, and no adverse effects were reported.
Ross and Fabian.11 An African American man, age 44, was receiving haloperidol decanoate, 400 mg every 2 weeks, and oral haloperidol, 20 mg/d.11 Because of residual symptoms, a history of nonadherence, and concerns about increasing the haloperidol decanoate dose or frequency, oral haloperidol was discontinued and paliperidone palmitate, 156 mg every 4 weeks, was started. The patient was able to transition into a step-down unit, and no adverse effects were reported.
What to consider before initiating dual LAIA treatment
Evaluate the frequency of administration, flexibility of dosing, administration site, adverse effects, and monitoring requirements of each LAIA (Table 212-19) to ensure the patient’s optimal tolerability of the regimen. Previous tolerability of each medication must be confirmed by evaluating the patient’s medication history or oral or IM administration of each agent prior to initiating the LAIA.
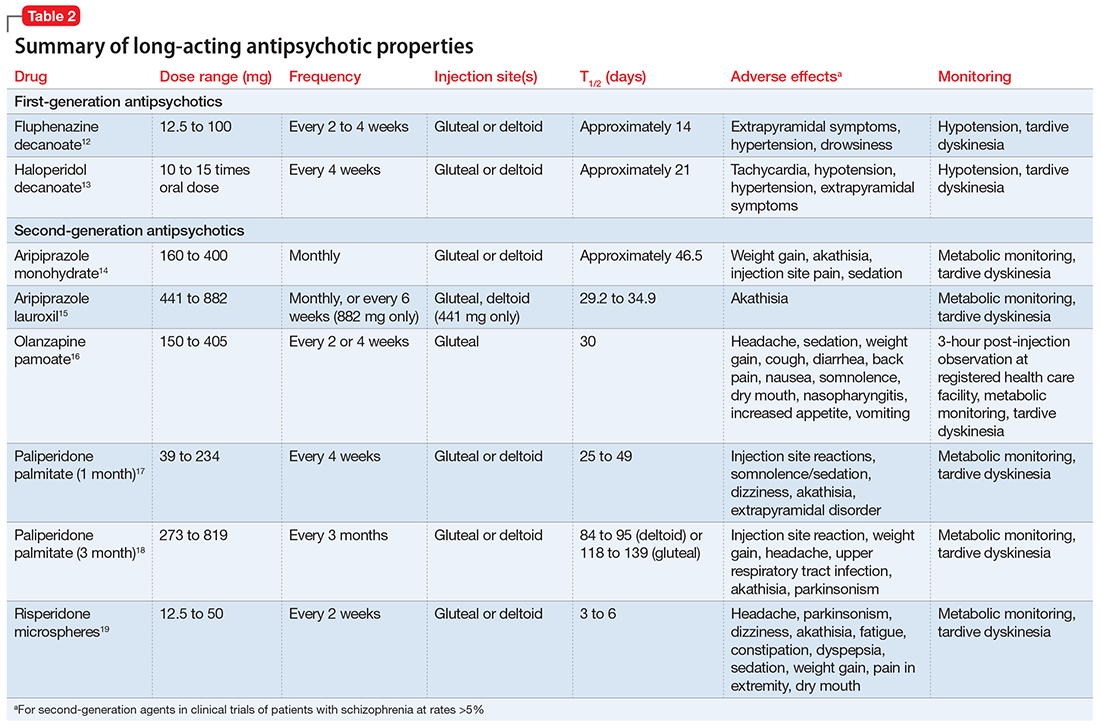
When choosing 2 agents that are each administered once every 4 weeks, consider administering the medications together every 4 weeks or alternating administration so that the patient receives an injection every 2 weeks. Receiving an injection once every 2 weeks might be beneficial for patients who need close follow-up or are more sensitive to injection site reactions, whereas a regimen of once every 4 weeks might be beneficial for patients who are more resistant to receiving the injections, so there is potentially less time spent agitated or anxious leading up to the date of the injection.
Use the lowest effective dose of each LAIA to limit adverse effects and improve tolerability of the regimen. Monitor patients closely for adverse reactions and discontinue the regimen as soon as possible if a severe adverse reaction occurs.
Cost may influence the decision to use 2 LAIAs. The majority of LAIAs in the United States are available only as branded formulations. Insurance companies may require prior authorization for the use of 2 LAIAs.
Although there are no treatment guidelines for combining 2 LAIAs, this practice has been used. A few case reports have described successful use of dual LAIA treatment, but one should consider the risk of the publication’s bias. Overall, the decision to use 2 LAIAs is difficult because there is lack of a large evidence base supporting the practice or direction from treatment guidelines. Because of this, dual LAIA treatment should not be used for most patients. In cases of treatment-resistant schizophrenia where clozapine is not an option and adherence is a concern, it is reasonable to consider this strategy on a case-by-case basis.
1. Kane J, Honigfeld G, Singer J, et al. Clozapine for the treatment-resistant schizophrenic. A double-blind comparison with chlorpromazine. Arch Gen Psychiatry. 1988;45(9):789-796.
2. Lehman A, Lieberman JA, Dixon LB, et al; American Psychiatric Association; Steering Committee on Practice Guidelines. Practice guideline for the treatment of patients with schizophrenia, second edition. Am J Psychiatry. 2004;161(suppl 2):1-56.
3. Hasan A, Falkai P, Wobrock T, et al; the WFSBP Task Force on Treatment Guidelines for Schizophrenia. World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for biological treatment of schizophrenia, part 1: update 2012 on the acute treatment of schizophrenia and management of treatment resistance. World J Biol Psychiatry. 2012;13(5):318-78.
4. Barnes TR; Schizophrenia Consensus Group of British Association for Psychopharmacology. Evidence-based guidelines for the pharmacological treatment of schizophrenia: recommendations from the British Association for Psychopharmacology. J Psychopharmacol. 2011;25(5):567-620. 5. Hasan A, Falkai P, Wobrock T, et al; WFSBP Task Force on Treatment Guidelines for Schizophrenia. World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for biological treatment of schizophrenia, part 2: update 2012 on the long-term treatment of schizophrenia and management of antipsychotic-induced side effects. World J Biol Psychiatry. 2013;14(1):2-44.
6. Kreyenbuhl J, Buchanan RW, Dickerson FB, et al; Schizophrenia Patient Outcomes Research Team (PORT). The Schizophrenia Patient Outcomes Research Team (PORT): updated treatment recommendations 2009. Schizophr Bull. 2010;36(1):94-103.
7. Ladds B, Cosme R, Rivera F. Concurrent use of two depot antipsychotic medications in schizophrenia. The Internet Journal of Psychiatry. 2009;1(1):1-3.
8. Wartelsteiner F, Hofer A. Treating schizophrenia with 2 long-acting injectable antipsychotic drugs: a case report. J Clin Psychopharmacol. 2015;35(4):474-475.
9. Scangos KW, Caton M, Newman WJ. Multiple long-acting injectable antipsychotics for treatment-resistant schizophrenia: case report. J Clin Psychopharmacol. 2016;36(3):283-285.
10. Yazdi K, Rosenleitner J, Pischinger B. Combination of two depot antipsychotic drugs [in German]. Nervenarzt. 2014;85(7):870-871.
11. Ross C, Fabian T. High dose haloperidol decanoate augmentation with paliperidone palmitate. Presented at: College of Psychiatric and Neurologic Pharmacists 16th Annual Meeting; April 21-24, 2013; Colorado Springs, CO.
12. Fluphenazine decanoate [package insert]. Schaumburg, IL: APP Pharmaceuticals, LLC; 2010.
13. Haloperidol decanoate [package insert]. Rockford, IL: Mylan; 2014.
14. Abilify Maintena [package insert]. Rockville, MD: Otsuka America Pharmaceutical, Inc.; 2016.
15. Aristada [package insert]. Waltham, MA: Alkermes; 2016.
16. Zyprexa Relprevv [package insert]. Indianapolis, IN: Lilly USA, LLC; 2016.
17. Invega Sustenna [package insert]. Titusville, NJ: Janssen Pharmaceuticals, Inc.; 2009.
18. Invega Trinza [package insert]. Titusville, NJ: Janssen Pharmaceuticals, Inc.; 2015.
19. Risperdal Consta [package insert]. Titusville, NJ: Janssen Pharmaceuticals, Inc.; 2007.
Ms. S, age 39, with a 15-year history of schizophrenia and severe paranoid delusions, is admitted after physically assaulting a staff member at a group home. She is receiving paliperidone palmitate, 234 mg every 4 weeks. This has reduced the severity of her symptoms, but she continues to have persistent delusions that affect her ability to accept redirection from staff. Ms. S frequently accuses staff and peers of sexual assault, says that she is pregnant, and does not adhere to treatment recommendations for laboratory monitoring because the “staff uses her blood for experiments.”
Ms. S frequently requires administration of oral and IM haloperidol, as needed, when she becomes aggressive with the staff. She has poor insight into her mental illness and does not believe that she needs medication. Ms. S has a long history of stopping her oral antipsychotic after a few days, reporting that it is “harming her baby.” Monotherapy has been tried with various long-acting injectable antipsychotics (LAIAs), but she still exhibits persistent delusions. The treatment team decides to add a second LAIA, haloperidol decanoate, 200 mg every 4 weeks, to her regimen.
Ladds et al.7 A 49-year-old woman with schizophrenia who was hospitalized for aggressive and bizarre behavior and had been institutionalized for 20 years stopped taking her medication regimen.7 She started taking 8-hour showers with bleach, talking incoherently, and believing that someone was poisoning her. She had poor response to oral risperidone monotherapy; however, 2 months after adding oral fluphenazine and benztropine to her regimen, her symptoms substantially improved (doses not reported). Because she had impaired insight into the need for daily medication, she was started on depot fluphenazine decanoate and risperidone microspheres (doses not reported) before discharge. No substantial adverse effects were noted with this regimen.
Wartelsteiner and Hofer.8 A man who had been diagnosed with paranoid schizophrenia at age 20 presented with thought blocking, incoherence, persecutory delusions, and uncontrolled self-damaging behavior.8 He had been admitted 27 times over 7 years; during this time he received many antipsychotic monotherapies and combination regimens. A total of 8 oral antipsychotics (including clozapine) and 5 LAIAs had been administered during these trials. He significantly improved with the combination of olanzapine and risperidone. Both medications were switched to LAIA formulations to address medication nonadherence. His symptoms remained stable with risperidone microspheres, 100 mg, and olanzapine pamoate, 300 mg, each administered every 2 weeks. He did not experience any adverse effects with this combination therapy.
Scangos et al.9 A 26-year-old Vietnamese man with schizophrenia and an extensive history of unprovoked, psychotically driven assaults was given multiple antipsychotics (including clozapine) during hospitalizations, and his medication regimen consistently included 2 antipsychotics. After contracting viral gastroenteritis, he refused oral medications and required short-acting IM administration of both haloperidol, 5 mg, twice a day, and olanzapine, 10 mg, twice a day. Because of concerns about continuing this regimen, he was switched to haloperidol decanoate (dose not reported) and olanzapine pamoate, 405 mg, administered once per month. The injections were scheduled to alternate so that the patient would receive 1 injection every 2 weeks. The patient’s assaultive behavior was significantly reduced, and no adverse effects were reported.
Ross and Fabian.11 An African American man, age 44, was receiving haloperidol decanoate, 400 mg every 2 weeks, and oral haloperidol, 20 mg/d.11 Because of residual symptoms, a history of nonadherence, and concerns about increasing the haloperidol decanoate dose or frequency, oral haloperidol was discontinued and paliperidone palmitate, 156 mg every 4 weeks, was started. The patient was able to transition into a step-down unit, and no adverse effects were reported.
What to consider before initiating dual LAIA treatment
Evaluate the frequency of administration, flexibility of dosing, administration site, adverse effects, and monitoring requirements of each LAIA (Table 212-19) to ensure the patient’s optimal tolerability of the regimen. Previous tolerability of each medication must be confirmed by evaluating the patient’s medication history or oral or IM administration of each agent prior to initiating the LAIA.
When choosing 2 agents that are each administered once every 4 weeks, consider administering the medications together every 4 weeks or alternating administration so that the patient receives an injection every 2 weeks. Receiving an injection once every 2 weeks might be beneficial for patients who need close follow-up or are more sensitive to injection site reactions, whereas a regimen of once every 4 weeks might be beneficial for patients who are more resistant to receiving the injections, so there is potentially less time spent agitated or anxious leading up to the date of the injection.
Use the lowest effective dose of each LAIA to limit adverse effects and improve tolerability of the regimen. Monitor patients closely for adverse reactions and discontinue the regimen as soon as possible if a severe adverse reaction occurs.
Cost may influence the decision to use 2 LAIAs. The majority of LAIAs in the United States are available only as branded formulations. Insurance companies may require prior authorization for the use of 2 LAIAs.
Although there are no treatment guidelines for combining 2 LAIAs, this practice has been used. A few case reports have described successful use of dual LAIA treatment, but one should consider the risk of the publication’s bias. Overall, the decision to use 2 LAIAs is difficult because there is lack of a large evidence base supporting the practice or direction from treatment guidelines. Because of this, dual LAIA treatment should not be used for most patients. In cases of treatment-resistant schizophrenia where clozapine is not an option and adherence is a concern, it is reasonable to consider this strategy on a case-by-case basis.
Ms. S, age 39, with a 15-year history of schizophrenia and severe paranoid delusions, is admitted after physically assaulting a staff member at a group home. She is receiving paliperidone palmitate, 234 mg every 4 weeks. This has reduced the severity of her symptoms, but she continues to have persistent delusions that affect her ability to accept redirection from staff. Ms. S frequently accuses staff and peers of sexual assault, says that she is pregnant, and does not adhere to treatment recommendations for laboratory monitoring because the “staff uses her blood for experiments.”
Ms. S frequently requires administration of oral and IM haloperidol, as needed, when she becomes aggressive with the staff. She has poor insight into her mental illness and does not believe that she needs medication. Ms. S has a long history of stopping her oral antipsychotic after a few days, reporting that it is “harming her baby.” Monotherapy has been tried with various long-acting injectable antipsychotics (LAIAs), but she still exhibits persistent delusions. The treatment team decides to add a second LAIA, haloperidol decanoate, 200 mg every 4 weeks, to her regimen.
Ladds et al.7 A 49-year-old woman with schizophrenia who was hospitalized for aggressive and bizarre behavior and had been institutionalized for 20 years stopped taking her medication regimen.7 She started taking 8-hour showers with bleach, talking incoherently, and believing that someone was poisoning her. She had poor response to oral risperidone monotherapy; however, 2 months after adding oral fluphenazine and benztropine to her regimen, her symptoms substantially improved (doses not reported). Because she had impaired insight into the need for daily medication, she was started on depot fluphenazine decanoate and risperidone microspheres (doses not reported) before discharge. No substantial adverse effects were noted with this regimen.
Wartelsteiner and Hofer.8 A man who had been diagnosed with paranoid schizophrenia at age 20 presented with thought blocking, incoherence, persecutory delusions, and uncontrolled self-damaging behavior.8 He had been admitted 27 times over 7 years; during this time he received many antipsychotic monotherapies and combination regimens. A total of 8 oral antipsychotics (including clozapine) and 5 LAIAs had been administered during these trials. He significantly improved with the combination of olanzapine and risperidone. Both medications were switched to LAIA formulations to address medication nonadherence. His symptoms remained stable with risperidone microspheres, 100 mg, and olanzapine pamoate, 300 mg, each administered every 2 weeks. He did not experience any adverse effects with this combination therapy.
Scangos et al.9 A 26-year-old Vietnamese man with schizophrenia and an extensive history of unprovoked, psychotically driven assaults was given multiple antipsychotics (including clozapine) during hospitalizations, and his medication regimen consistently included 2 antipsychotics. After contracting viral gastroenteritis, he refused oral medications and required short-acting IM administration of both haloperidol, 5 mg, twice a day, and olanzapine, 10 mg, twice a day. Because of concerns about continuing this regimen, he was switched to haloperidol decanoate (dose not reported) and olanzapine pamoate, 405 mg, administered once per month. The injections were scheduled to alternate so that the patient would receive 1 injection every 2 weeks. The patient’s assaultive behavior was significantly reduced, and no adverse effects were reported.
Ross and Fabian.11 An African American man, age 44, was receiving haloperidol decanoate, 400 mg every 2 weeks, and oral haloperidol, 20 mg/d.11 Because of residual symptoms, a history of nonadherence, and concerns about increasing the haloperidol decanoate dose or frequency, oral haloperidol was discontinued and paliperidone palmitate, 156 mg every 4 weeks, was started. The patient was able to transition into a step-down unit, and no adverse effects were reported.
What to consider before initiating dual LAIA treatment
Evaluate the frequency of administration, flexibility of dosing, administration site, adverse effects, and monitoring requirements of each LAIA (Table 212-19) to ensure the patient’s optimal tolerability of the regimen. Previous tolerability of each medication must be confirmed by evaluating the patient’s medication history or oral or IM administration of each agent prior to initiating the LAIA.
When choosing 2 agents that are each administered once every 4 weeks, consider administering the medications together every 4 weeks or alternating administration so that the patient receives an injection every 2 weeks. Receiving an injection once every 2 weeks might be beneficial for patients who need close follow-up or are more sensitive to injection site reactions, whereas a regimen of once every 4 weeks might be beneficial for patients who are more resistant to receiving the injections, so there is potentially less time spent agitated or anxious leading up to the date of the injection.
Use the lowest effective dose of each LAIA to limit adverse effects and improve tolerability of the regimen. Monitor patients closely for adverse reactions and discontinue the regimen as soon as possible if a severe adverse reaction occurs.
Cost may influence the decision to use 2 LAIAs. The majority of LAIAs in the United States are available only as branded formulations. Insurance companies may require prior authorization for the use of 2 LAIAs.
Although there are no treatment guidelines for combining 2 LAIAs, this practice has been used. A few case reports have described successful use of dual LAIA treatment, but one should consider the risk of the publication’s bias. Overall, the decision to use 2 LAIAs is difficult because there is lack of a large evidence base supporting the practice or direction from treatment guidelines. Because of this, dual LAIA treatment should not be used for most patients. In cases of treatment-resistant schizophrenia where clozapine is not an option and adherence is a concern, it is reasonable to consider this strategy on a case-by-case basis.
1. Kane J, Honigfeld G, Singer J, et al. Clozapine for the treatment-resistant schizophrenic. A double-blind comparison with chlorpromazine. Arch Gen Psychiatry. 1988;45(9):789-796.
2. Lehman A, Lieberman JA, Dixon LB, et al; American Psychiatric Association; Steering Committee on Practice Guidelines. Practice guideline for the treatment of patients with schizophrenia, second edition. Am J Psychiatry. 2004;161(suppl 2):1-56.
3. Hasan A, Falkai P, Wobrock T, et al; the WFSBP Task Force on Treatment Guidelines for Schizophrenia. World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for biological treatment of schizophrenia, part 1: update 2012 on the acute treatment of schizophrenia and management of treatment resistance. World J Biol Psychiatry. 2012;13(5):318-78.
4. Barnes TR; Schizophrenia Consensus Group of British Association for Psychopharmacology. Evidence-based guidelines for the pharmacological treatment of schizophrenia: recommendations from the British Association for Psychopharmacology. J Psychopharmacol. 2011;25(5):567-620. 5. Hasan A, Falkai P, Wobrock T, et al; WFSBP Task Force on Treatment Guidelines for Schizophrenia. World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for biological treatment of schizophrenia, part 2: update 2012 on the long-term treatment of schizophrenia and management of antipsychotic-induced side effects. World J Biol Psychiatry. 2013;14(1):2-44.
6. Kreyenbuhl J, Buchanan RW, Dickerson FB, et al; Schizophrenia Patient Outcomes Research Team (PORT). The Schizophrenia Patient Outcomes Research Team (PORT): updated treatment recommendations 2009. Schizophr Bull. 2010;36(1):94-103.
7. Ladds B, Cosme R, Rivera F. Concurrent use of two depot antipsychotic medications in schizophrenia. The Internet Journal of Psychiatry. 2009;1(1):1-3.
8. Wartelsteiner F, Hofer A. Treating schizophrenia with 2 long-acting injectable antipsychotic drugs: a case report. J Clin Psychopharmacol. 2015;35(4):474-475.
9. Scangos KW, Caton M, Newman WJ. Multiple long-acting injectable antipsychotics for treatment-resistant schizophrenia: case report. J Clin Psychopharmacol. 2016;36(3):283-285.
10. Yazdi K, Rosenleitner J, Pischinger B. Combination of two depot antipsychotic drugs [in German]. Nervenarzt. 2014;85(7):870-871.
11. Ross C, Fabian T. High dose haloperidol decanoate augmentation with paliperidone palmitate. Presented at: College of Psychiatric and Neurologic Pharmacists 16th Annual Meeting; April 21-24, 2013; Colorado Springs, CO.
12. Fluphenazine decanoate [package insert]. Schaumburg, IL: APP Pharmaceuticals, LLC; 2010.
13. Haloperidol decanoate [package insert]. Rockford, IL: Mylan; 2014.
14. Abilify Maintena [package insert]. Rockville, MD: Otsuka America Pharmaceutical, Inc.; 2016.
15. Aristada [package insert]. Waltham, MA: Alkermes; 2016.
16. Zyprexa Relprevv [package insert]. Indianapolis, IN: Lilly USA, LLC; 2016.
17. Invega Sustenna [package insert]. Titusville, NJ: Janssen Pharmaceuticals, Inc.; 2009.
18. Invega Trinza [package insert]. Titusville, NJ: Janssen Pharmaceuticals, Inc.; 2015.
19. Risperdal Consta [package insert]. Titusville, NJ: Janssen Pharmaceuticals, Inc.; 2007.
1. Kane J, Honigfeld G, Singer J, et al. Clozapine for the treatment-resistant schizophrenic. A double-blind comparison with chlorpromazine. Arch Gen Psychiatry. 1988;45(9):789-796.
2. Lehman A, Lieberman JA, Dixon LB, et al; American Psychiatric Association; Steering Committee on Practice Guidelines. Practice guideline for the treatment of patients with schizophrenia, second edition. Am J Psychiatry. 2004;161(suppl 2):1-56.
3. Hasan A, Falkai P, Wobrock T, et al; the WFSBP Task Force on Treatment Guidelines for Schizophrenia. World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for biological treatment of schizophrenia, part 1: update 2012 on the acute treatment of schizophrenia and management of treatment resistance. World J Biol Psychiatry. 2012;13(5):318-78.
4. Barnes TR; Schizophrenia Consensus Group of British Association for Psychopharmacology. Evidence-based guidelines for the pharmacological treatment of schizophrenia: recommendations from the British Association for Psychopharmacology. J Psychopharmacol. 2011;25(5):567-620. 5. Hasan A, Falkai P, Wobrock T, et al; WFSBP Task Force on Treatment Guidelines for Schizophrenia. World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for biological treatment of schizophrenia, part 2: update 2012 on the long-term treatment of schizophrenia and management of antipsychotic-induced side effects. World J Biol Psychiatry. 2013;14(1):2-44.
6. Kreyenbuhl J, Buchanan RW, Dickerson FB, et al; Schizophrenia Patient Outcomes Research Team (PORT). The Schizophrenia Patient Outcomes Research Team (PORT): updated treatment recommendations 2009. Schizophr Bull. 2010;36(1):94-103.
7. Ladds B, Cosme R, Rivera F. Concurrent use of two depot antipsychotic medications in schizophrenia. The Internet Journal of Psychiatry. 2009;1(1):1-3.
8. Wartelsteiner F, Hofer A. Treating schizophrenia with 2 long-acting injectable antipsychotic drugs: a case report. J Clin Psychopharmacol. 2015;35(4):474-475.
9. Scangos KW, Caton M, Newman WJ. Multiple long-acting injectable antipsychotics for treatment-resistant schizophrenia: case report. J Clin Psychopharmacol. 2016;36(3):283-285.
10. Yazdi K, Rosenleitner J, Pischinger B. Combination of two depot antipsychotic drugs [in German]. Nervenarzt. 2014;85(7):870-871.
11. Ross C, Fabian T. High dose haloperidol decanoate augmentation with paliperidone palmitate. Presented at: College of Psychiatric and Neurologic Pharmacists 16th Annual Meeting; April 21-24, 2013; Colorado Springs, CO.
12. Fluphenazine decanoate [package insert]. Schaumburg, IL: APP Pharmaceuticals, LLC; 2010.
13. Haloperidol decanoate [package insert]. Rockford, IL: Mylan; 2014.
14. Abilify Maintena [package insert]. Rockville, MD: Otsuka America Pharmaceutical, Inc.; 2016.
15. Aristada [package insert]. Waltham, MA: Alkermes; 2016.
16. Zyprexa Relprevv [package insert]. Indianapolis, IN: Lilly USA, LLC; 2016.
17. Invega Sustenna [package insert]. Titusville, NJ: Janssen Pharmaceuticals, Inc.; 2009.
18. Invega Trinza [package insert]. Titusville, NJ: Janssen Pharmaceuticals, Inc.; 2015.
19. Risperdal Consta [package insert]. Titusville, NJ: Janssen Pharmaceuticals, Inc.; 2007.
The girl who couldn’t stop stealing
CASE A lifelong habit
Ms. B, age 14, has diagnoses of attention-deficit/hyperactive disorder (ADHD) and oppositional defiant disorder, and is taking extended-release (ER) methylphenidate, 36 mg/d. Her mother brings her to the hospital with concerns that Ms. B has been stealing small objects, such as money, toys, and pencils from home, school, and her peers, even though she does not need them and her family can afford to buy them for her. Ms. B’s mother routinely searches her daughter when she leaves the house and when she returns and frequently finds things in Ms. B’s possession that do not belong to her.
The mother reports that Ms. B’s stealing has been a lifelong habit that worsened after Ms. B’s father died in a car accident last year.
Ms. B does not volunteer any information about her stealing. She is admitted to a partial hospitalization program for further evaluation and treatment.
[polldaddy:9837962]
EVALUATION Continued stealing
A week later, Ms. B remains reluctant to talk about her stealing habit. However, once a therapeutic alliance is established, she reveals that she experiences increased anxiety before stealing and feels pleasure during the theft. Her methylphenidate ER dosage is increased to 54 mg/d in an attempt to address poor impulse control and subsequent stealing behavior. Her ADHD symptoms are controlled, and she does not exhibit poor impulse control in any situation other than stealing.
However, Ms. B continues to have poor insight and impaired judgment about her behavior. During treatment, Ms. B steals markers from the psychiatrist’s office, which later are found in her bag. When the staff convinces Ms. B to return the markers to the psychiatrist, she denies knowing how they got there. Behavioral interventions, including covert sensitization, systemic desensitization, positive reinforcement, body and bag search, and reminders, occur consistently as part of treatment, but have little effect on her symptoms.
The author’s observations
Risk-taking and novelty-seeking behaviors are common in adolescent patients. Impulsivity, instant reward-seeking behavior, and poor judgment can lead to stealing in this population, but this behavior is not necessarily indicative of kleptomania.
Kleptomania is the recurrent failure to resist impulses to steal objects.2 It differs from other forms of stealing in that the objects stolen by a patient with kleptomania are not needed for personal use or for their monetary value. Kleptomania usually begins in early adolescence, is found in about 0.5% of the general population, and is more common among females.3

There are 2 important theories to explain kleptomania:
- The psychoanalytical theory explains kleptomania as an immature defense against unconscious impulses, conflicts, and desires of destruction. By stealing, the individual protects the self from narcissistic injury and disintegration. The frantic search for objects helps to divert self-destructive aggressiveness and allows for the preservation of the self.4
- The biological model indicates that individuals with kleptomania have a significant deficit of white matter in inferior frontal regions and poor integrity of the tracts connecting the limbic system to the thalamus and to the prefrontal cortex.5 Reward system circuitry (ventral tegmental area–nucleus accumbens–orbital frontal cortex) is likely to be involved in impulse control disorders including kleptomania.6
Comorbidity. Kleptomania often is comorbid with substance use disorder (SUD), obsessive-compulsive disorder (OCD), and compulsive shopping, as well as depression, anxiety disorders, bulimia nervosa, and impulse control and conduct disorders.3,6
Kleptomania shares many characteristics with SUD, including continued engagement in a behavior despite negative consequences and the temporary reduction in urges after the behavior’s completion, followed by a return of the urge to steal. There also is a bidirectional relationship between OCD and kleptomania. Individuals with both disorders frequently engage in excessive and unnecessary rituals even when it is ego-dystonic. First-degree relatives of kleptomania patients have high rates of SUD and OCD.3
Serotonin, dopamine, and opioid pathways play a role in both kleptomania and other behavioral addictions.6 Clinicians should be cautious in treating comorbid disorders with stimulants. These agents may help patients with high impulsivity, but lead to disinhibition and worsen impulse control in patients with low impulsivity.7
TREATMENT Naltrexone
The psychiatrist discusses pharmacologic options to treat kleptomania with Ms. B and her mother. After considering the risks, benefits, adverse effects, and alternative treatments (including the option of no pharmacologic treatment), the mother consents and Ms. B assents to treatment with naltrexone, 25 mg/d. Before starting this medication, both the mother and Ms. B receive detailed psychoeducation describing naltrexone’s interactions with opioids. They are told that if Ms. B has a traumatic injury, they should inform the treatment team that she is taking naltrexone, which can acutely precipitate opiate withdrawal.
Before initiating pharmacotherapy, a comprehensive metabolic profile is obtained, and all values are within the normal range. After 1 week, naltrexone is increased to 50 mg/d. The medication is well tolerated, without any adverse effects.
[polldaddy:9837976]
The author’s observations
Behavioral interventions, such as covert sensitization and systemic desensitization, often are used to treat kleptomania.8 There are no FDA-approved medications for this condition. Opioid antagonists have been considered for the treatment of kleptomania.7
Mu-opioid receptors exist in highest concentrations in presynaptic neurons in the periaqueductal gray region and spinal cord and have high affinity for enkephalins and beta-endorphins. They also are involved in the reward and pleasure pathway. This neurocircuit is implicated in behavioral addiction.9
Naltrexone is an antagonist at μ-opioid receptors. It blocks the binding of endogenous and exogenous opioids at the receptors, particularly at the ventral tegmental area. By blocking the μ-receptor, naltrexone inhibits the processing of the reward and pleasure pathway involved in kleptomania. Naltrexone binds to these receptors, preventing the euphoric effects of behavioral addictions.10 This medication works best in conjunction with behavioral interventions.8
Naltrexone is a Schedule II drug. Use of naltrexone to treat kleptomania or other impulse control disorders is an off-label use of the medication. Naltrexone should not be prescribed to patients who are receiving opiates because it can cause acute opiate withdrawal.
Liver function tests should be monitored in all patients taking naltrexone. If liver function levels begin to rise, naltrexone should be discontinued. Naltrexone should be used with caution in patients with preexisting liver disease.11
OUTCOME Marked improvement
Ms. B’s K-SAS scores are evaluated 2 weeks after starting naltrexone. The results show a marked reduction in the urge to steal and in stealing behavior, and Ms. B’s mother reports no incidents of stealing in the previous week.
Ms. B is maintained on naltrexone, 50 mg/d, for 2 months. On repeated K-SAS scores, her mother rates Ms. B’s symptoms “very much improved” with “occasional” stealing. Ms. B is discharged from the intensive outpatient program.
1. Christianini AR, Conti MA, Hearst N, et al. Treating kleptomania: cross-cultural adaptation of the Kleptomania Symptom Assessment Scale and assessment of an outpatient program. Compr Psychiatry. 2015;56:289-294.
2. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
3. Talih FR. Kleptomania and potential exacerbating factors: a review and case report. Innov Clin Neurosci. 2011;8(10):35-39.
4. Cierpka M. Psychodynamics of neurotically-induced kleptomania [in German]. Psychiatr Prax. 1986;13(3):94-103.
5. Grant JE, Correia S, Brennan-Krohn T. White matter integrity in kleptomania: a pilot study. Psychiatry Res. 2006;147(2-3):233-237.
6. Grant JE, Odlaug BL, Kim SW. Kleptomania: clinical characteristics and relationship to substance use disorders. Am J Drug Alcohol Abuse. 2010;36(5):291-295.
7. Zack M, Poulos CX. Effects of the atypical stimulant modafinil on a brief gambling episode in pathological gamblers with high vs. low impulsivity. J Psychopharmacol. 2009;23(6):660-671.
8. Grant JE. Understanding and treating kleptomania: new models and new treatments. Isr J Psychiatry Relat Sci. 2006;43(2):81-87.
9. Potenza MN. Should addictive disorders include non-substance-related conditions? Addiction. 2006;101(suppl 1):142-151.
10. Grant JE, Kim SW. An open-label study of naltrexone in the treatment of kleptomania. J Clin Psychiatry. 2002;63(4):349-356.
11. Pfohl DN, Allen JI, Atkinson RL, et al. Naltrexone hydrochloride (Trexan): a review of serum transaminase elevations at high dosage. NIDA Res Monogr. 1986;67:66-72.
CASE A lifelong habit
Ms. B, age 14, has diagnoses of attention-deficit/hyperactive disorder (ADHD) and oppositional defiant disorder, and is taking extended-release (ER) methylphenidate, 36 mg/d. Her mother brings her to the hospital with concerns that Ms. B has been stealing small objects, such as money, toys, and pencils from home, school, and her peers, even though she does not need them and her family can afford to buy them for her. Ms. B’s mother routinely searches her daughter when she leaves the house and when she returns and frequently finds things in Ms. B’s possession that do not belong to her.
The mother reports that Ms. B’s stealing has been a lifelong habit that worsened after Ms. B’s father died in a car accident last year.
Ms. B does not volunteer any information about her stealing. She is admitted to a partial hospitalization program for further evaluation and treatment.
[polldaddy:9837962]
EVALUATION Continued stealing
A week later, Ms. B remains reluctant to talk about her stealing habit. However, once a therapeutic alliance is established, she reveals that she experiences increased anxiety before stealing and feels pleasure during the theft. Her methylphenidate ER dosage is increased to 54 mg/d in an attempt to address poor impulse control and subsequent stealing behavior. Her ADHD symptoms are controlled, and she does not exhibit poor impulse control in any situation other than stealing.
However, Ms. B continues to have poor insight and impaired judgment about her behavior. During treatment, Ms. B steals markers from the psychiatrist’s office, which later are found in her bag. When the staff convinces Ms. B to return the markers to the psychiatrist, she denies knowing how they got there. Behavioral interventions, including covert sensitization, systemic desensitization, positive reinforcement, body and bag search, and reminders, occur consistently as part of treatment, but have little effect on her symptoms.
The author’s observations
Risk-taking and novelty-seeking behaviors are common in adolescent patients. Impulsivity, instant reward-seeking behavior, and poor judgment can lead to stealing in this population, but this behavior is not necessarily indicative of kleptomania.
Kleptomania is the recurrent failure to resist impulses to steal objects.2 It differs from other forms of stealing in that the objects stolen by a patient with kleptomania are not needed for personal use or for their monetary value. Kleptomania usually begins in early adolescence, is found in about 0.5% of the general population, and is more common among females.3
There are 2 important theories to explain kleptomania:
- The psychoanalytical theory explains kleptomania as an immature defense against unconscious impulses, conflicts, and desires of destruction. By stealing, the individual protects the self from narcissistic injury and disintegration. The frantic search for objects helps to divert self-destructive aggressiveness and allows for the preservation of the self.4
- The biological model indicates that individuals with kleptomania have a significant deficit of white matter in inferior frontal regions and poor integrity of the tracts connecting the limbic system to the thalamus and to the prefrontal cortex.5 Reward system circuitry (ventral tegmental area–nucleus accumbens–orbital frontal cortex) is likely to be involved in impulse control disorders including kleptomania.6
Comorbidity. Kleptomania often is comorbid with substance use disorder (SUD), obsessive-compulsive disorder (OCD), and compulsive shopping, as well as depression, anxiety disorders, bulimia nervosa, and impulse control and conduct disorders.3,6
Kleptomania shares many characteristics with SUD, including continued engagement in a behavior despite negative consequences and the temporary reduction in urges after the behavior’s completion, followed by a return of the urge to steal. There also is a bidirectional relationship between OCD and kleptomania. Individuals with both disorders frequently engage in excessive and unnecessary rituals even when it is ego-dystonic. First-degree relatives of kleptomania patients have high rates of SUD and OCD.3
Serotonin, dopamine, and opioid pathways play a role in both kleptomania and other behavioral addictions.6 Clinicians should be cautious in treating comorbid disorders with stimulants. These agents may help patients with high impulsivity, but lead to disinhibition and worsen impulse control in patients with low impulsivity.7
TREATMENT Naltrexone
The psychiatrist discusses pharmacologic options to treat kleptomania with Ms. B and her mother. After considering the risks, benefits, adverse effects, and alternative treatments (including the option of no pharmacologic treatment), the mother consents and Ms. B assents to treatment with naltrexone, 25 mg/d. Before starting this medication, both the mother and Ms. B receive detailed psychoeducation describing naltrexone’s interactions with opioids. They are told that if Ms. B has a traumatic injury, they should inform the treatment team that she is taking naltrexone, which can acutely precipitate opiate withdrawal.
Before initiating pharmacotherapy, a comprehensive metabolic profile is obtained, and all values are within the normal range. After 1 week, naltrexone is increased to 50 mg/d. The medication is well tolerated, without any adverse effects.
[polldaddy:9837976]
The author’s observations
Behavioral interventions, such as covert sensitization and systemic desensitization, often are used to treat kleptomania.8 There are no FDA-approved medications for this condition. Opioid antagonists have been considered for the treatment of kleptomania.7
Mu-opioid receptors exist in highest concentrations in presynaptic neurons in the periaqueductal gray region and spinal cord and have high affinity for enkephalins and beta-endorphins. They also are involved in the reward and pleasure pathway. This neurocircuit is implicated in behavioral addiction.9
Naltrexone is an antagonist at μ-opioid receptors. It blocks the binding of endogenous and exogenous opioids at the receptors, particularly at the ventral tegmental area. By blocking the μ-receptor, naltrexone inhibits the processing of the reward and pleasure pathway involved in kleptomania. Naltrexone binds to these receptors, preventing the euphoric effects of behavioral addictions.10 This medication works best in conjunction with behavioral interventions.8
Naltrexone is a Schedule II drug. Use of naltrexone to treat kleptomania or other impulse control disorders is an off-label use of the medication. Naltrexone should not be prescribed to patients who are receiving opiates because it can cause acute opiate withdrawal.
Liver function tests should be monitored in all patients taking naltrexone. If liver function levels begin to rise, naltrexone should be discontinued. Naltrexone should be used with caution in patients with preexisting liver disease.11
OUTCOME Marked improvement
Ms. B’s K-SAS scores are evaluated 2 weeks after starting naltrexone. The results show a marked reduction in the urge to steal and in stealing behavior, and Ms. B’s mother reports no incidents of stealing in the previous week.
Ms. B is maintained on naltrexone, 50 mg/d, for 2 months. On repeated K-SAS scores, her mother rates Ms. B’s symptoms “very much improved” with “occasional” stealing. Ms. B is discharged from the intensive outpatient program.
CASE A lifelong habit
Ms. B, age 14, has diagnoses of attention-deficit/hyperactive disorder (ADHD) and oppositional defiant disorder, and is taking extended-release (ER) methylphenidate, 36 mg/d. Her mother brings her to the hospital with concerns that Ms. B has been stealing small objects, such as money, toys, and pencils from home, school, and her peers, even though she does not need them and her family can afford to buy them for her. Ms. B’s mother routinely searches her daughter when she leaves the house and when she returns and frequently finds things in Ms. B’s possession that do not belong to her.
The mother reports that Ms. B’s stealing has been a lifelong habit that worsened after Ms. B’s father died in a car accident last year.
Ms. B does not volunteer any information about her stealing. She is admitted to a partial hospitalization program for further evaluation and treatment.
[polldaddy:9837962]
EVALUATION Continued stealing
A week later, Ms. B remains reluctant to talk about her stealing habit. However, once a therapeutic alliance is established, she reveals that she experiences increased anxiety before stealing and feels pleasure during the theft. Her methylphenidate ER dosage is increased to 54 mg/d in an attempt to address poor impulse control and subsequent stealing behavior. Her ADHD symptoms are controlled, and she does not exhibit poor impulse control in any situation other than stealing.
However, Ms. B continues to have poor insight and impaired judgment about her behavior. During treatment, Ms. B steals markers from the psychiatrist’s office, which later are found in her bag. When the staff convinces Ms. B to return the markers to the psychiatrist, she denies knowing how they got there. Behavioral interventions, including covert sensitization, systemic desensitization, positive reinforcement, body and bag search, and reminders, occur consistently as part of treatment, but have little effect on her symptoms.
The author’s observations
Risk-taking and novelty-seeking behaviors are common in adolescent patients. Impulsivity, instant reward-seeking behavior, and poor judgment can lead to stealing in this population, but this behavior is not necessarily indicative of kleptomania.
Kleptomania is the recurrent failure to resist impulses to steal objects.2 It differs from other forms of stealing in that the objects stolen by a patient with kleptomania are not needed for personal use or for their monetary value. Kleptomania usually begins in early adolescence, is found in about 0.5% of the general population, and is more common among females.3
There are 2 important theories to explain kleptomania:
- The psychoanalytical theory explains kleptomania as an immature defense against unconscious impulses, conflicts, and desires of destruction. By stealing, the individual protects the self from narcissistic injury and disintegration. The frantic search for objects helps to divert self-destructive aggressiveness and allows for the preservation of the self.4
- The biological model indicates that individuals with kleptomania have a significant deficit of white matter in inferior frontal regions and poor integrity of the tracts connecting the limbic system to the thalamus and to the prefrontal cortex.5 Reward system circuitry (ventral tegmental area–nucleus accumbens–orbital frontal cortex) is likely to be involved in impulse control disorders including kleptomania.6
Comorbidity. Kleptomania often is comorbid with substance use disorder (SUD), obsessive-compulsive disorder (OCD), and compulsive shopping, as well as depression, anxiety disorders, bulimia nervosa, and impulse control and conduct disorders.3,6
Kleptomania shares many characteristics with SUD, including continued engagement in a behavior despite negative consequences and the temporary reduction in urges after the behavior’s completion, followed by a return of the urge to steal. There also is a bidirectional relationship between OCD and kleptomania. Individuals with both disorders frequently engage in excessive and unnecessary rituals even when it is ego-dystonic. First-degree relatives of kleptomania patients have high rates of SUD and OCD.3
Serotonin, dopamine, and opioid pathways play a role in both kleptomania and other behavioral addictions.6 Clinicians should be cautious in treating comorbid disorders with stimulants. These agents may help patients with high impulsivity, but lead to disinhibition and worsen impulse control in patients with low impulsivity.7
TREATMENT Naltrexone
The psychiatrist discusses pharmacologic options to treat kleptomania with Ms. B and her mother. After considering the risks, benefits, adverse effects, and alternative treatments (including the option of no pharmacologic treatment), the mother consents and Ms. B assents to treatment with naltrexone, 25 mg/d. Before starting this medication, both the mother and Ms. B receive detailed psychoeducation describing naltrexone’s interactions with opioids. They are told that if Ms. B has a traumatic injury, they should inform the treatment team that she is taking naltrexone, which can acutely precipitate opiate withdrawal.
Before initiating pharmacotherapy, a comprehensive metabolic profile is obtained, and all values are within the normal range. After 1 week, naltrexone is increased to 50 mg/d. The medication is well tolerated, without any adverse effects.
[polldaddy:9837976]
The author’s observations
Behavioral interventions, such as covert sensitization and systemic desensitization, often are used to treat kleptomania.8 There are no FDA-approved medications for this condition. Opioid antagonists have been considered for the treatment of kleptomania.7
Mu-opioid receptors exist in highest concentrations in presynaptic neurons in the periaqueductal gray region and spinal cord and have high affinity for enkephalins and beta-endorphins. They also are involved in the reward and pleasure pathway. This neurocircuit is implicated in behavioral addiction.9
Naltrexone is an antagonist at μ-opioid receptors. It blocks the binding of endogenous and exogenous opioids at the receptors, particularly at the ventral tegmental area. By blocking the μ-receptor, naltrexone inhibits the processing of the reward and pleasure pathway involved in kleptomania. Naltrexone binds to these receptors, preventing the euphoric effects of behavioral addictions.10 This medication works best in conjunction with behavioral interventions.8
Naltrexone is a Schedule II drug. Use of naltrexone to treat kleptomania or other impulse control disorders is an off-label use of the medication. Naltrexone should not be prescribed to patients who are receiving opiates because it can cause acute opiate withdrawal.
Liver function tests should be monitored in all patients taking naltrexone. If liver function levels begin to rise, naltrexone should be discontinued. Naltrexone should be used with caution in patients with preexisting liver disease.11
OUTCOME Marked improvement
Ms. B’s K-SAS scores are evaluated 2 weeks after starting naltrexone. The results show a marked reduction in the urge to steal and in stealing behavior, and Ms. B’s mother reports no incidents of stealing in the previous week.
Ms. B is maintained on naltrexone, 50 mg/d, for 2 months. On repeated K-SAS scores, her mother rates Ms. B’s symptoms “very much improved” with “occasional” stealing. Ms. B is discharged from the intensive outpatient program.
1. Christianini AR, Conti MA, Hearst N, et al. Treating kleptomania: cross-cultural adaptation of the Kleptomania Symptom Assessment Scale and assessment of an outpatient program. Compr Psychiatry. 2015;56:289-294.
2. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
3. Talih FR. Kleptomania and potential exacerbating factors: a review and case report. Innov Clin Neurosci. 2011;8(10):35-39.
4. Cierpka M. Psychodynamics of neurotically-induced kleptomania [in German]. Psychiatr Prax. 1986;13(3):94-103.
5. Grant JE, Correia S, Brennan-Krohn T. White matter integrity in kleptomania: a pilot study. Psychiatry Res. 2006;147(2-3):233-237.
6. Grant JE, Odlaug BL, Kim SW. Kleptomania: clinical characteristics and relationship to substance use disorders. Am J Drug Alcohol Abuse. 2010;36(5):291-295.
7. Zack M, Poulos CX. Effects of the atypical stimulant modafinil on a brief gambling episode in pathological gamblers with high vs. low impulsivity. J Psychopharmacol. 2009;23(6):660-671.
8. Grant JE. Understanding and treating kleptomania: new models and new treatments. Isr J Psychiatry Relat Sci. 2006;43(2):81-87.
9. Potenza MN. Should addictive disorders include non-substance-related conditions? Addiction. 2006;101(suppl 1):142-151.
10. Grant JE, Kim SW. An open-label study of naltrexone in the treatment of kleptomania. J Clin Psychiatry. 2002;63(4):349-356.
11. Pfohl DN, Allen JI, Atkinson RL, et al. Naltrexone hydrochloride (Trexan): a review of serum transaminase elevations at high dosage. NIDA Res Monogr. 1986;67:66-72.
1. Christianini AR, Conti MA, Hearst N, et al. Treating kleptomania: cross-cultural adaptation of the Kleptomania Symptom Assessment Scale and assessment of an outpatient program. Compr Psychiatry. 2015;56:289-294.
2. Diagnostic and statistical manual of mental disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
3. Talih FR. Kleptomania and potential exacerbating factors: a review and case report. Innov Clin Neurosci. 2011;8(10):35-39.
4. Cierpka M. Psychodynamics of neurotically-induced kleptomania [in German]. Psychiatr Prax. 1986;13(3):94-103.
5. Grant JE, Correia S, Brennan-Krohn T. White matter integrity in kleptomania: a pilot study. Psychiatry Res. 2006;147(2-3):233-237.
6. Grant JE, Odlaug BL, Kim SW. Kleptomania: clinical characteristics and relationship to substance use disorders. Am J Drug Alcohol Abuse. 2010;36(5):291-295.
7. Zack M, Poulos CX. Effects of the atypical stimulant modafinil on a brief gambling episode in pathological gamblers with high vs. low impulsivity. J Psychopharmacol. 2009;23(6):660-671.
8. Grant JE. Understanding and treating kleptomania: new models and new treatments. Isr J Psychiatry Relat Sci. 2006;43(2):81-87.
9. Potenza MN. Should addictive disorders include non-substance-related conditions? Addiction. 2006;101(suppl 1):142-151.
10. Grant JE, Kim SW. An open-label study of naltrexone in the treatment of kleptomania. J Clin Psychiatry. 2002;63(4):349-356.
11. Pfohl DN, Allen JI, Atkinson RL, et al. Naltrexone hydrochloride (Trexan): a review of serum transaminase elevations at high dosage. NIDA Res Monogr. 1986;67:66-72.
Physician impairment
Most physicians are likely familiar with guidelines relating to physician impairment, but they may not be aware that these guidelines typically conflict with the Americans with Disabilities Act (ADA), which protects all employees against unwarranted requests for mental health information or evaluations.
Under the ADA, employers cannot request mental health information from their employees or refer them for mental health evaluations without objective evidence showing that either the employee:
- is unable to perform essential job functions because of a mental health condition
- poses a high risk of substantial, imminent harm to himself (herself) or others in the workplace because of a mental health condition.1
Employers cannot rely on speculative evidence or generalizations about these conditions when making these determinations,1 and common mental disorders (eg, depressive disorders, anxiety disorders, attention-deficit/hyperactivity disorder, specific learning disorders, etc.) should almost never form the basis of such requests.2
In contrast, the American Medical Association (AMA) does not distinguish between the presence of a mental health condition and physician impairment,3,4 which may result in unwarranted requests and referrals for mental health evaluations. Some state laws on impairment, which all derive from AMA policies,5 even state outright that, “‘Impaired’ or ‘impairment’ means the presence of the diseases of alcoholism, drug abuse, or mental illness”6 and directly discriminate against physicians with these conditions.
State physician health programs (PHPs) also may describe impairment in problematic ways (eg, “Involvement in litigation against hospital”).7 Their descriptions also are overly inclusive in that they could be used to describe most physicians (N.D.L., J.W.B., unpublished data, 2017), and they rarely represent sufficient legal indications for a mental health evaluation under the ADA (N.D.L., J.W.B., unpublished data, 2017). Even the APA’s Clinical Guide to Psychiatric Ethics describes physician impairment as synonymous with mental illness.8
Requests for mental health information or evaluations not only can include referrals to state PHPs but also “suggestions” to see a psychologist, professional job coach, or any provider who may ask for mental health information. Under the ADA's guidelines, obtaining “voluntary” consent from an employee who could be fired for not cooperating does not change the involuntary nature of these requests.2,9
Employers who hire psychiatrists, physicians, and medical residents should comply with the ADA and disregard the AMA’s policies, state laws, PHPs, other institutional guidelines,10 and guidance from some articles published in
1. U.S. Equal Employment Opportunity Commission. EEOC enforcement guidance on the Americans with Disabilities Act and psychiatric disabilities. No. 915.002. http://www.eeoc.gov/policy/docs/psych.html. Updated March 9, 2009. Accessed July 20, 2017.
2. Lawson ND, Kalet AL. The administrative psychiatric evaluation. J Grad Med Educ. 2016;8(1):14-17.
3. American Medical Association. Physician impairment H-95.955: Drug Abuse. https://policy search.ama-assn.org/policyfinder/detail/physician%20impairment?uri=%2FAMADoc%2FHOD.xml-0-5334.xml. Updated 2009. Accessed April 20, 2017.
4. Myers MF, Gabbard GO. The physician as patient: a clinical handbook for mental health professionals. Arlington, VA: American Psychiatric Publishing, Inc.; 2008.
5. Sargent DA. The impaired physician movement: an interim report. Hosp Community Psychiatry. 1985;36(3):294-297.
6. Arkansas State Medical Board. Arkansas medical practices act and regulations. http://www.armedicalboard.org/professionals/pdf/mpa.pdf. Revised March 2017. Accessed July 11, 2017.
7. Oklahoma Health Professionals Program. Chemical dependency. https://www.okhpp.org/chemical-dependency. Accessed September 15, 2017.
8. Trockel M, Miller MN, Roberts LW. Clinician well-being and impairment. In: Roberts LW, ed. A clinical guide to psychiatric ethics. Arlington, VA: American Psychiatric Publishing, Inc.; 2016:223-236.
9. U.S. Equal Employment Opportunity Commission. Regulations under the Americans with Disabilities Act. Federal Register. https://www.gpo.gov/fdsys/pkg/FR-2016-05-17/pdf/2016-11558.pdf. Published May 17, 2016. Accessed August 2
10. Lawson ND. Comply with federal laws before checking institutional guidelines on resident referrals for psychiatric evaluations. J Grad Med Educ. In press.
11. Bright RP, Krahn L. Impaired physicians: how to recognize, when to report, and where to refer. Current Psychiatry. 2010;9(6):11-20.
12. Mossman D, Farrell HM. Physician impairment: when should you report? Current Psychiatry. 2011;10(9):67-71.
Most physicians are likely familiar with guidelines relating to physician impairment, but they may not be aware that these guidelines typically conflict with the Americans with Disabilities Act (ADA), which protects all employees against unwarranted requests for mental health information or evaluations.
Under the ADA, employers cannot request mental health information from their employees or refer them for mental health evaluations without objective evidence showing that either the employee:
- is unable to perform essential job functions because of a mental health condition
- poses a high risk of substantial, imminent harm to himself (herself) or others in the workplace because of a mental health condition.1
Employers cannot rely on speculative evidence or generalizations about these conditions when making these determinations,1 and common mental disorders (eg, depressive disorders, anxiety disorders, attention-deficit/hyperactivity disorder, specific learning disorders, etc.) should almost never form the basis of such requests.2
In contrast, the American Medical Association (AMA) does not distinguish between the presence of a mental health condition and physician impairment,3,4 which may result in unwarranted requests and referrals for mental health evaluations. Some state laws on impairment, which all derive from AMA policies,5 even state outright that, “‘Impaired’ or ‘impairment’ means the presence of the diseases of alcoholism, drug abuse, or mental illness”6 and directly discriminate against physicians with these conditions.
State physician health programs (PHPs) also may describe impairment in problematic ways (eg, “Involvement in litigation against hospital”).7 Their descriptions also are overly inclusive in that they could be used to describe most physicians (N.D.L., J.W.B., unpublished data, 2017), and they rarely represent sufficient legal indications for a mental health evaluation under the ADA (N.D.L., J.W.B., unpublished data, 2017). Even the APA’s Clinical Guide to Psychiatric Ethics describes physician impairment as synonymous with mental illness.8
Requests for mental health information or evaluations not only can include referrals to state PHPs but also “suggestions” to see a psychologist, professional job coach, or any provider who may ask for mental health information. Under the ADA's guidelines, obtaining “voluntary” consent from an employee who could be fired for not cooperating does not change the involuntary nature of these requests.2,9
Employers who hire psychiatrists, physicians, and medical residents should comply with the ADA and disregard the AMA’s policies, state laws, PHPs, other institutional guidelines,10 and guidance from some articles published in
Most physicians are likely familiar with guidelines relating to physician impairment, but they may not be aware that these guidelines typically conflict with the Americans with Disabilities Act (ADA), which protects all employees against unwarranted requests for mental health information or evaluations.
Under the ADA, employers cannot request mental health information from their employees or refer them for mental health evaluations without objective evidence showing that either the employee:
- is unable to perform essential job functions because of a mental health condition
- poses a high risk of substantial, imminent harm to himself (herself) or others in the workplace because of a mental health condition.1
Employers cannot rely on speculative evidence or generalizations about these conditions when making these determinations,1 and common mental disorders (eg, depressive disorders, anxiety disorders, attention-deficit/hyperactivity disorder, specific learning disorders, etc.) should almost never form the basis of such requests.2
In contrast, the American Medical Association (AMA) does not distinguish between the presence of a mental health condition and physician impairment,3,4 which may result in unwarranted requests and referrals for mental health evaluations. Some state laws on impairment, which all derive from AMA policies,5 even state outright that, “‘Impaired’ or ‘impairment’ means the presence of the diseases of alcoholism, drug abuse, or mental illness”6 and directly discriminate against physicians with these conditions.
State physician health programs (PHPs) also may describe impairment in problematic ways (eg, “Involvement in litigation against hospital”).7 Their descriptions also are overly inclusive in that they could be used to describe most physicians (N.D.L., J.W.B., unpublished data, 2017), and they rarely represent sufficient legal indications for a mental health evaluation under the ADA (N.D.L., J.W.B., unpublished data, 2017). Even the APA’s Clinical Guide to Psychiatric Ethics describes physician impairment as synonymous with mental illness.8
Requests for mental health information or evaluations not only can include referrals to state PHPs but also “suggestions” to see a psychologist, professional job coach, or any provider who may ask for mental health information. Under the ADA's guidelines, obtaining “voluntary” consent from an employee who could be fired for not cooperating does not change the involuntary nature of these requests.2,9
Employers who hire psychiatrists, physicians, and medical residents should comply with the ADA and disregard the AMA’s policies, state laws, PHPs, other institutional guidelines,10 and guidance from some articles published in
1. U.S. Equal Employment Opportunity Commission. EEOC enforcement guidance on the Americans with Disabilities Act and psychiatric disabilities. No. 915.002. http://www.eeoc.gov/policy/docs/psych.html. Updated March 9, 2009. Accessed July 20, 2017.
2. Lawson ND, Kalet AL. The administrative psychiatric evaluation. J Grad Med Educ. 2016;8(1):14-17.
3. American Medical Association. Physician impairment H-95.955: Drug Abuse. https://policy search.ama-assn.org/policyfinder/detail/physician%20impairment?uri=%2FAMADoc%2FHOD.xml-0-5334.xml. Updated 2009. Accessed April 20, 2017.
4. Myers MF, Gabbard GO. The physician as patient: a clinical handbook for mental health professionals. Arlington, VA: American Psychiatric Publishing, Inc.; 2008.
5. Sargent DA. The impaired physician movement: an interim report. Hosp Community Psychiatry. 1985;36(3):294-297.
6. Arkansas State Medical Board. Arkansas medical practices act and regulations. http://www.armedicalboard.org/professionals/pdf/mpa.pdf. Revised March 2017. Accessed July 11, 2017.
7. Oklahoma Health Professionals Program. Chemical dependency. https://www.okhpp.org/chemical-dependency. Accessed September 15, 2017.
8. Trockel M, Miller MN, Roberts LW. Clinician well-being and impairment. In: Roberts LW, ed. A clinical guide to psychiatric ethics. Arlington, VA: American Psychiatric Publishing, Inc.; 2016:223-236.
9. U.S. Equal Employment Opportunity Commission. Regulations under the Americans with Disabilities Act. Federal Register. https://www.gpo.gov/fdsys/pkg/FR-2016-05-17/pdf/2016-11558.pdf. Published May 17, 2016. Accessed August 2
10. Lawson ND. Comply with federal laws before checking institutional guidelines on resident referrals for psychiatric evaluations. J Grad Med Educ. In press.
11. Bright RP, Krahn L. Impaired physicians: how to recognize, when to report, and where to refer. Current Psychiatry. 2010;9(6):11-20.
12. Mossman D, Farrell HM. Physician impairment: when should you report? Current Psychiatry. 2011;10(9):67-71.
1. U.S. Equal Employment Opportunity Commission. EEOC enforcement guidance on the Americans with Disabilities Act and psychiatric disabilities. No. 915.002. http://www.eeoc.gov/policy/docs/psych.html. Updated March 9, 2009. Accessed July 20, 2017.
2. Lawson ND, Kalet AL. The administrative psychiatric evaluation. J Grad Med Educ. 2016;8(1):14-17.
3. American Medical Association. Physician impairment H-95.955: Drug Abuse. https://policy search.ama-assn.org/policyfinder/detail/physician%20impairment?uri=%2FAMADoc%2FHOD.xml-0-5334.xml. Updated 2009. Accessed April 20, 2017.
4. Myers MF, Gabbard GO. The physician as patient: a clinical handbook for mental health professionals. Arlington, VA: American Psychiatric Publishing, Inc.; 2008.
5. Sargent DA. The impaired physician movement: an interim report. Hosp Community Psychiatry. 1985;36(3):294-297.
6. Arkansas State Medical Board. Arkansas medical practices act and regulations. http://www.armedicalboard.org/professionals/pdf/mpa.pdf. Revised March 2017. Accessed July 11, 2017.
7. Oklahoma Health Professionals Program. Chemical dependency. https://www.okhpp.org/chemical-dependency. Accessed September 15, 2017.
8. Trockel M, Miller MN, Roberts LW. Clinician well-being and impairment. In: Roberts LW, ed. A clinical guide to psychiatric ethics. Arlington, VA: American Psychiatric Publishing, Inc.; 2016:223-236.
9. U.S. Equal Employment Opportunity Commission. Regulations under the Americans with Disabilities Act. Federal Register. https://www.gpo.gov/fdsys/pkg/FR-2016-05-17/pdf/2016-11558.pdf. Published May 17, 2016. Accessed August 2
10. Lawson ND. Comply with federal laws before checking institutional guidelines on resident referrals for psychiatric evaluations. J Grad Med Educ. In press.
11. Bright RP, Krahn L. Impaired physicians: how to recognize, when to report, and where to refer. Current Psychiatry. 2010;9(6):11-20.
12. Mossman D, Farrell HM. Physician impairment: when should you report? Current Psychiatry. 2011;10(9):67-71.