This article reviews recommendations and evidence concerning current anticoagulant management for venous thromboembolism and perioperative care, with an emphasis on individualizing treatment for real-world patients.
TREATING ACUTE VENOUS THROMBOEMBOLISM
Case 1: Deep vein thrombosis in an otherwise healthy man
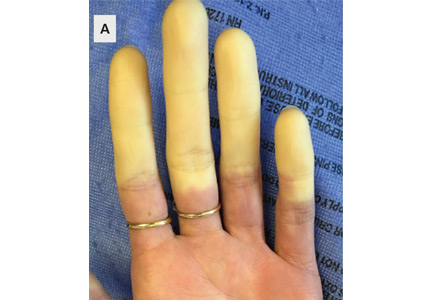
A 40-year-old man presents with 7 days of progressive right leg swelling. He has no antecedent risk factors for deep vein thrombosis or other medical problems. Venous ultrasonography reveals an iliofemoral deep vein thrombosis. How should he be managed?
Outpatient treatment with low-molecular-weight heparin for 4 to 6 days plus warfarin
Outpatient treatment with a direct oral anticoagulant, ie, apixaban, dabigatran (which requires 4 to 6 days of initial treatment with low-molecular-weight heparin), or rivaroxaban
Catheter-directed thrombolysis followed by low-molecular-weight heparin, then warfarin or a direct oral anticoagulant
Inpatient intravenous heparin for 7 to 10 days, then warfarin or a direct oral anticoagulant
All of these are acceptable for managing acute venous thromboembolism, but the clinician’s role is to identify which treatment is most appropriate for an individual patient.
Deep vein thrombosis is not a single condition
Multiple guidelines exist to help decide on a management strategy. Those of the American College of Chest Physicians (ACCP)1 are used most often.
That said, guidelines are established for “average” patients, so it is important to look beyond guidelines and individualize management. Venous thromboembolism is not a single entity; it has a myriad of clinical presentations that could call for different treatments. Most patients have submassive deep vein thrombosis or pulmonary embolism, which is not limb-threatening nor associated with hemodynamic instability. It can also differ in terms of etiology and can be unprovoked (or idiopathic), cancer-related, catheter-associated, or provoked by surgery or immobility.
Deep vein thrombosis has a wide spectrum of presentations. It can involve the veins of the calf only, or it can involve the femoral and iliac veins and other locations including the splanchnic veins, the cerebral sinuses, and upper extremities. Pulmonary embolism can be massive (defined as being associated with hemodynamic instability or impending respiratory failure) or submassive. Similarly, patients differ in terms of baseline medical conditions, mobility, and lifestyle. Anticoagulant management decisions should take all these factors into account.
Consider clot location
Our patient with iliofemoral deep vein thrombosis is best managed differently than a more typical patient with less extensive thrombosis that would involve the popliteal or femoral vein segments, or both. A clot that involves the iliac vein is more likely to lead to postthrombotic chronic pain and swelling as the lack of venous outflow bypass channels to circumvent the clot location creates higher venous pressure within the affected leg. Therefore, for our patient, catheter-directed thrombolysis is an option that should be considered.
Catheter-directed thrombolysis trials
According to the “open-vein hypothesis,” quickly eliminating the thrombus and restoring unobstructed venous flow may mitigate the risk not only of recurrent thrombosis, but also of postthrombotic syndrome, which is often not given much consideration acutely but can cause significant, life-altering chronic disability.
The “valve-integrity hypothesis” is also important; it considers whether lytic therapy may help prevent damage to such valves in an attempt to mitigate the amount of venous hypertension.
Thus, catheter-directed thrombolysis offers theoretical benefits, and recent trials have assessed it against standard anticoagulation treatments.
The CaVenT trial (Catheter-Directed Venous Thrombolysis),2 conducted in Norway, randomized 209 patients with midfemoral to iliac deep vein thrombosis to conventional treatment (anticoagulation alone) or anticoagulation plus catheter-directed thrombolysis. At 2 years, postthrombotic syndrome had occurred in 41% of the catheter-directed thrombolysis group compared with 56% of the conventional treatment group (P = .047). At 5 years, the difference widened to 43% vs 71% (P < .01, number needed to treat = 4).3 Despite the superiority of lytic therapy, the incidence of postthrombotic syndrome remained high in patients who received this treatment.
The ATTRACT trial (Acute Venous Thrombosis: Thrombus Removal With Adjunctive Catheter-Directed Thrombolysis),4 a US multicenter, open-label, assessor-blind study, randomized 698 patients with femoral or more-proximal deep vein thrombosis to either standard care (anticoagulant therapy and graduated elastic compression stockings) or standard care plus catheter-directed thrombolysis. In preliminary results presented at the Society of Interventional Radiology meeting in March 2017, although no difference was found in the primary outcome (postthrombotic syndrome at 24 months), catheter-directed thrombolysis for iliofemoral deep vein thrombosis led to a 25% reduction in moderate to severe postthrombotic syndrome.
Although it is too early to draw conclusions before publication of the ATTRACT study, the preliminary results highlight the need to individualize treatment and to be selective about using catheter-directed thrombolysis. The trials provide reassurance that catheter-directed lysis is a reasonable and safe intervention when performed by physicians experienced in the procedure. The risk of major bleeding appears to be low (about 2%) and that for intracranial hemorrhage even lower (< 0.5%).
Catheter-directed thrombolysis is appropriate in some cases
The 2016 ACCP guidelines1 recommend anticoagulant therapy alone over catheter-directed thrombolysis for patients with acute proximal deep vein thrombosis of the leg. However, it is a grade 2C (weak) recommendation.
They provide no specific recommendation as to the clinical indications for catheter-directed thrombolysis, but identify patients who would be most likely to benefit, ie, those who have:
Iliofemoral deep vein thrombosis
Symptoms for less than 14 days
Good functional status
Life expectancy of more than 1 year
Low risk of bleeding.
Our patient satisfies these criteria, suggesting that catheter-directed thrombolysis is a reasonable option for him.
Timing is important. Catheter-directed lysis is more likely to be beneficial if used before fibrin deposits form and stiffen the venous valves, causing irreversible damage that leads to postthrombotic syndrome.
Role of direct oral anticoagulants
The availability of direct oral anticoagulants has generated interest in defining their therapeutic role in patients with venous thromboembolism.
In a meta-analysis5 of major trials comparing direct oral anticoagulants and vitamin K antagonists such as warfarin, no significant difference was found for the risk of recurrent venous thromboembolism or venous thromboembolism-related deaths. However, fewer patients experienced major bleeding with direct oral anticoagulants (relative risk 0.61, P = .002). Although significant, the absolute risk reduction was small; the incidence of major bleeding was 1.1% with direct oral anticoagulants vs 1.8% with vitamin K antagonists.
The main advantage of direct oral anticoagulants is greater convenience for the patient.
The 2016 ACCP guidelines1 on the treatment of venous thrombosis and pulmonary embolism are summarized in Table 1. They suggest using direct oral anticoagulants rather than vitamin K antagonists to manage venous thromboembolism, but this is a weak (ie, grade 2B) recommendation, likely because the net clinical benefit of direct oral anticoagulants over vitamin K antagonists is modest.
WHICH PATIENTS ON WARFARIN NEED BRIDGING PREOPERATIVELY?
Many patients still take warfarin, particularly those with atrial fibrillation, a mechanical heart valve, or venous thromboembolism. In many countries, warfarin remains the dominant anticoagulant for stroke prevention. Whether these patients need heparin during the period of perioperative warfarin interruption is a frequently encountered scenario that, until recently, was controversial. Recent studies have helped to inform the need for heparin bridging in many of these patients.
Case 2: An elderly woman on warfarin facing cancer surgery
A 75-year-old woman weighing 65 kg is scheduled for elective colon resection for incidentally found colon cancer. She is taking warfarin for atrial fibrillation. She also has hypertension and diabetes and had a transient ischemic attack 10 years ago.
One doctor told her she needs to be assessed for heparin bridging, but another told her she does not need bridging.
The default management should be not to bridge patients who have atrial fibrillation, but to consider bridging in selected patients, such as those with recent stroke or transient ischemic attack or a prior thromboembolic event during warfarin interruption. However, decisions about bridging should not be made on the basis of the CHADS2 score alone. For the patient described here, I would recommend not bridging.
Complex factors contribute to stroke risk
Stroke risk for patients with atrial fibrillation can be quickly estimated with the CHADS2 score, based on:
Congestive heart failure (1 point)
Hypertension (1 point)
Age at least 75 (1 point)
Diabetes (1 point)
Stroke or transient ischemic attack (2 points).
Our patient has a score of 5, corresponding to an annual adjusted stroke risk of 12.5%. Whether her transient ischemic attack of 10 years ago is comparable in significance to a recent stroke is debatable and highlights a weakness of clinical prediction rules. Moreover, such prediction scores were developed to estimate the long-term risk of stroke if anticoagulants are not given, and they have not been assessed in a perioperative setting where there is short-term interruption of anticoagulants. Also, the perioperative milieu is associated with additional factors not captured in these clinical prediction rules that may affect the risk of stroke.
Thus, the risk of perioperative stroke likely involves the interplay of multiple factors, including the type of surgery the patient is undergoing. Some factors may be mitigated:
Rebound hypercoagulability after stopping an oral anticoagulant can be prevented by intraoperative blood pressure and volume control
Elevated biochemical factors (eg, D-dimer, B-type natriuretic peptide, troponin) may be lowered with perioperative aspirin therapy
Lipid and genetic factors may be mitigated with perioperative statin use.
Can heparin bridging also mitigate the risk?
Bridging in patients with atrial fibrillation
Most patients who are taking warfarin are doing so because of atrial fibrillation, so most evidence about perioperative bridging was developed in such patients.
The BRIDGE trial (Bridging Anticoagulation in Patients Who Require Temporary Interruption of Warfarin Therapy for an Elective Invasive Procedure or Surgery)6 was the first randomized controlled trial to compare a bridging and no-bridging strategy for patients with atrial fibrillation who required warfarin interruption for elective surgery. Nearly 2,000 patients were given either low-molecular-weight heparin or placebo starting 3 days before until 24 hours before a procedure, and then for 5 to 10 days afterwards. For all patients, warfarin was stopped 5 days before the procedure and was resumed within 24 hours afterwards.
A no-bridging strategy was noninferior to bridging: the risk of perioperative arterial thromboembolism was 0.4% without bridging vs 0.3% with bridging (P = .01 for noninferiority). In addition, a no-bridging strategy conferred a lower risk of major bleeding than bridging: 1.3% vs 3.2% (relative risk 0.41, P = .005 for superiority).
Although the difference in absolute bleeding risk was small, bleeding rates were lower than those seen outside of clinical trials, as the bridging protocol used in BRIDGE was designed to minimize the risk of bleeding. Also, although only 5% of patients had a CHADS2 score of 5 or 6, such patients are infrequent in clinical practice, and BRIDGE did include a considerable proportion (17%) of patients with a prior stroke or transient ischemic attack who would be considered at high risk.
Other evidence about heparin bridging is derived from observational studies, more than 10 of which have been conducted. In general, they have found that not bridging is associated with low rates of arterial thromboembolism (< 0.5%) and that bridging is associated with high rates of major bleeding (4%–7%).7–12
Bridging in patients with a mechanical heart valve
Warfarin is the only anticoagulant option for patients who have a mechanical heart valve. No randomized controlled trials have evaluated the benefits of perioperative bridging vs no bridging in this setting.
Observational (cohort) studies suggest that the risk of perioperative arterial thromboembolism is similar with or without bridging anticoagulation, although most patients studied were bridged and those not bridged were considered at low risk (eg, with a bileaflet aortic valve and no additional risk factors).13 However, without stronger evidence from randomized controlled trials, bridging should be the default management for patients with a mechanical heart valve. In our practice, we bridge most patients who have a mechanical heart valve unless they are considered to be at low risk, such as those who have a bileaflet aortic valve.
Bridging in patients with prior venous thromboembolism
Even less evidence is available for periprocedural management of patients who have a history of venous thromboembolism. No randomized controlled trials exist evaluating bridging vs no bridging. In 1 cohort study in which more than 90% of patients had had thromboembolism more than 3 months before the procedure, the rate of recurrent venous thromboembolism without bridging was less than 0.5%.14
It is reasonable to bridge patients who need anticoagulant interruption within 3 months of diagnosis of a deep vein thrombosis or pulmonary embolism, and to consider using a temporary inferior vena cava filter for patients who have had a clot who need treatment interruption during the initial 3 to 4 weeks after diagnosis.
Practice guidelines: Perioperative anticoagulation
The ACCP,15 the American College of Cardiology,16 and the American Heart Association17 have published guidelines for perioperative management of antithrombotic therapy. Despite a paucity of evidence from randomized trials, there are sufficient data to inform clinical management. Some guidelines are complex. A simplified algorithm has been proposed that considers the type of procedure, the CHADS2 score, whether the patient has a mechanical heart valve, and whether there has been a recent venous thromboembolic event.18
Guidance for preoperative and postoperative bridging for patients taking warfarin is summarized in Table 2.
CARDIAC PROCEDURES
For patients facing a procedure to implant an implantable cardioverter-defibrillator (ICD) or pacemaker, a procedure-specific concern is the avoidance of pocket hematoma.
Patients on warfarin: Do not bridge
The BRUISE CONTROL-1 trial (Bridge or Continue Coumadin for Device Surgery Randomized Controlled Trial)19 randomized patients undergoing pacemaker or ICD implantation to either continued anticoagulation therapy and not bridging (ie, continued warfarin so long as the international normalized ratio was < 3) vs conventional bridging treatment (ie, stopping warfarin and bridging with low-molecular-weight heparin). A clinically significant device-pocket hematoma occurred in 3.5% of the continued-warfarin group vs 16.0% in the heparin-bridging group (P < .001). Thromboembolic complications were rare, and rates did not differ between the 2 groups.
Results of the BRUISE CONTROL-1 trial serve as a caution to at least not be too aggressive with bridging. The study design involved resuming heparin 24 hours after surgery, which is perhaps more aggressive than standard practice. In our practice, we wait at least 24 hours to reinstate heparin after minor surgery, and 48 to 72 hours after surgery with higher bleeding risk.
These results are perhaps not surprising if one considers how carefully surgeons try to control bleeding during surgery for patients taking anticoagulants. For patients who are not on an anticoagulant, small bleeding may be less of a concern during a procedure. When high doses of heparin are introduced soon after surgery, small concerns during surgery may become big problems afterward.
Based on these results, it is reasonable to undertake device implantation without interruption of a vitamin K antagonist such as warfarin.
Patients on direct oral anticoagulants: The jury is still out
The similar BRUISE CONTROL-2 trial is currently under way, comparing interruption vs continuation of dabigatran for patients undergoing cardiac device surgery.
In Europe, surgeons are less concerned than those in the United States about operating while a patient is on anticoagulant therapy. But the safety of this practice is not backed by strong evidence.
Direct oral anticoagulants: Consider pharmacokinetics
Direct oral anticoagulants are potent and fast-acting, with a peak effect 1 to 3 hours after intake. This rapid anticoagulant action is similar to that of bridging with low-molecular-weight heparin, and caution is needed when administering direct oral anticoagulants, especially after major surgery or surgery with a high bleeding risk.
Frost et al20 compared the pharmacokinetics of apixaban (with twice-daily dosing) and rivaroxaban (once-daily dosing) and found that peak anticoagulant activity is faster and higher with rivaroxaban. This is important, because many patients will take their anticoagulant first thing in the morning. Consequently, if patients require any kind of procedure (including dental), they should skip the morning dose of the direct oral anticoagulant to avoid having the procedure done during the peak anticoagulant effect, and they should either not take that day’s dose or defer the dose until the evening after the procedure.
MANAGING SURGERY FOR PATIENTS ON A DIRECT ORAL ANTICOAGULANT
Case 3: An elderly woman on apixaban facing surgery
Let us imagine that our previous patient takes apixaban instead of warfarin. She is 75 years old, has atrial fibrillation, and is about to undergo elective colon resection for cancer. One doctor advises her to simply stop apixaban for 2 days, while another says she should go off apixaban for 5 days and will need bridging. Which plan is best?
In the perioperative setting, our goal is to interrupt patients’ anticoagulant therapy for the shortest time that results in no residual anticoagulant effect at the time of the procedure.
The European Society of Regional Anaesthesia and Pain Therapy and the American Society of Regional Anesthesia and Pain Medicine21 recommend an extended period of interruption of direct oral anticoagulants (Table 3)
They further recommend that if the risk of venous thromboembolism is high, low-molecular-weight heparin bridging should be done while stopping the direct oral anticoagulant, with the heparin discontinued 24 hours before the procedure. This recommendation seems counterintuitive, as it is advising replacing a short-acting anticoagulant with low-molecular-weight heparin, another short-acting anticoagulant.
The guidelines committee was unable to provide strength and grading of their recommendations, as too few well-designed studies are available to support them. The doctor in case 3 who advised stopping apixaban for 5 days and bridging is following the guidelines, but without much evidence to support this strategy.
Is bridging needed during interruption of a direct oral anticoagulant?
There are no randomized, controlled trials of bridging vs no bridging in patients taking direct oral anticoagulants. Substudies exist of patients taking these drugs for atrial fibrillation who had treatment interrupted for procedures, but the studies did not randomize bridging vs no bridging, nor were bridging regimens standardized. Three of the four atrial fibrillation trials had a blinded design (warfarin vs direct oral anticoagulants), making perioperative management difficult, as physicians did not know the pharmacokinetics of the drugs their patients were taking.22–24
We used the database from the Randomized Evaluation of Long-Term Anticoagulation Therapy (RE-LY) trial22 to evaluate bridging in patients taking either warfarin or dabigatran. With an open-label study design (the blinding was only for the 110 mg and 150 mg dabigatran doses), clinicians were aware of whether patients were receiving warfarin or dabigatran, thereby facilitating perioperative management. Among dabigatran-treated patients, those who were bridged had significantly more major bleeding than those not bridged (6.5% vs 1.8%, P < .001), with no difference between the groups for stroke or systemic embolism. Although it is not a randomized controlled trial, it does provide evidence that bridging may not be advisable for patients taking a direct oral anticoagulant.
The 2017 American College of Cardiology guidelines25 conclude that parenteral bridging is not indicated for direct oral anticoagulants. Although this is not based on strong evidence, the guidance appears reasonable according to the evidence at hand.
The 2017 American Heart Association Guidelines16 recommend a somewhat complex approach based on periprocedural bleeding risk and thromboembolic risk.
How long to interrupt direct oral anticoagulants?
Table 4 shows a simplified approach to interrupting direct oral anticoagulants that we use in Canada. The approach takes into account the type of surgery and kidney function for patients taking dabigatran, a drug that depends more on renal clearance than the other direct oral anticoagulants do.26
Evidence for this approach comes from a prospective cohort study27 of 541 patients being treated with dabigatran who were having an elective surgery or invasive procedure. Patients received standard perioperative management, with the timing of the last dabigatran dose before the procedure (24 hours, 48 hours, or 96 hours) based on the bleeding risk of surgery and the patient’s creatinine clearance. Dabigatran was resumed 24 to 72 hours after the procedure. No heparin bridging was done. Patients were followed for up to 30 days postoperatively. The results were favorable with few complications: one transient ischemic attack (0.2%), 10 major bleeding episodes (1.8%), and 28 minor bleeding episodes (5.2%).
A subgroup of 181 patients in this study28 had a plasma sample drawn just before surgery, allowing the investigators to assess the level of coagulation factors after dabigatran interruption. Results were as follows:
93% had a normal prothrombin time
80% had a normal activated partial thromboplastin time
33% had a normal thrombin time
81% had a normal dilute thrombin time.
The dilute thrombin time is considered the most reliable test of the anticoagulant effect of dabigatran but is not widely available. The activated partial thromboplastin time can provide a more widely used coagulation test to assess (in a less precise manner) whether there is an anticoagulant effect of dabigatran present, and more sensitive activated partial thromboplastin time assays can be used to better detect any residual dabigatran effect.
Dabigatran levels were also measured. Although 66% of patients had low drug levels just before surgery, the others still had substantial dabigatran on board. The fact that bleeding event rates were so low in this study despite the presence of dabigatran in many patients raises the question of whether having some drug on board is a good predictor of bleeding risk.
An interruption protocol with a longer interruption interval—12 to 14 hours longer than in the previous study (3 days for high-bleed risk procedures, 2 days for low-bleed risk procedures)—brought the activated partial thromboplastin time and dilute thrombin time to normal levels for 100% of patients with the protocol for high-bleeding-risk surgery. This study was based on small numbers and its interruption strategy needs further investigation.29
Case 3 continued
Based on the current empiric evidence, we recommend interrupting direct oral anticoagulants for 2 days (or approximately a 60-hour interval between the last dose and surgery) for this 75-year-old woman who is taking apixaban (Table 5). This interruption interval corresponds to 5 elimination half-lives for apixaban, which should result in little to no residual anticoagulant and will facilitate major surgery and, if indicated, neuraxial anesthesia.
The PAUSE study (NCT02228798), a multicenter, prospective cohort study, is designed to establish a safe, standardized protocol for the perioperative management of patients with atrial fibrillation taking dabigatran, rivaroxaban, or apixaban and will include 3,300 patients.
PATIENTS WITH A CORONARY STENT WHO NEED SURGERY
Case 4: A woman with a stent facing surgery
A 70-year-old woman needs breast cancer resection. She has coronary artery disease and had a drug-eluting stent placed 5 months ago after elective cardiac catheterization. She also has hypertension, obesity, and type 2 diabetes. Her medications include an angiotensin II receptor blocker, hydrochlorothiazide, insulin, and an oral hypoglycemic. She is also taking aspirin 81 mg daily and ticagrelor (a P2Y12 receptor antagonist) 90 mg twice daily.
Her cardiologist is concerned that stopping antiplatelet therapy could trigger acute stent thrombosis, which has a 50% or higher mortality rate.
Should she stop taking aspirin before surgery? What about the ticagrelor?
Is aspirin safe during surgery?
Evidence concerning aspirin during surgery comes from Perioperative Ischemic Evaluation 2 (POISE-2), a double-blind, randomized controlled trial.30 Patients who had known cardiovascular disease or risk factors for cardiovascular disease and were about to undergo noncardiac surgery were stratified according to whether they had been taking aspirin before the study (patients taking aspirin within 72 hours of the surgery were excluded from randomization). Participants in each group were randomized to take either aspirin or placebo just before surgery. The primary outcome was the combined rate of death or nonfatal myocardial infarction 30 days after randomization.
The study found no differences in the primary end point between the two groups. However, major bleeding occurred significantly more often in the aspirin group (4.6% vs 3.8%, hazard ratio 1.2, 95% confidence interval 1.0–1.5).
Moreover, only 4% of the patients in this trial had a cardiac stent. The trial excluded patients who had had a bare-metal stent placed within 6 weeks or a drug-eluting stent placed within 1 year, so it does not help us answer whether aspirin should be stopped for our current patient.
Is surgery safe for patients with stents?
The safety of undergoing surgery with a stent was investigated in a large US Veterans Administration retrospective cohort study.31 More than 20,000 patients with stents who underwent noncardiac surgery within 2 years of stent placement were compared with a control group of more than 41,000 patients with stents who did not undergo surgery. Patients were matched by stent type and cardiac risk factors at the time of stent placement.
The risk of an adverse cardiac event in both the surgical and nonsurgical cohorts was highest in the initial 6 weeks after stent placement and plateaued 6 months after stent placement, when the risk difference between the surgical and nonsurgical groups leveled off to 1%.
The risk of a major adverse cardiac event postoperatively was much more dependent on the timing of stent placement in complex and inpatient surgeries. For outpatient surgeries, the risk of a major cardiac event was very low and the timing of stent placement did not matter.
A Danish observational study32 compared more than 4,000 patients with drug-eluting stents having surgery to more than 20,000 matched controls without coronary heart disease having similar surgery. The risk of myocardial infarction or cardiac death was much higher for patients undergoing surgery within 1 month after drug-eluting stent placement compared with controls without heart disease and patients with stent placement longer than 1 month before surgery.
Our practice is to continue aspirin for surgery in patients with coronary stents regardless of the timing of placement. Although there is a small increased risk of bleeding, this must be balanced against thrombotic risk. We typically stop clopidogrel 5 to 7 days before surgery and ticagrelor 3 to 5 days before surgery. We may decide to give platelets before very-high-risk surgery (eg, intracranial, spinal) if there is a decision to continue both antiplatelet drugs—for example, in a patient who recently received a drug-eluting stent (ie, within 3 months). It is essential to involve the cardiologist and surgeon in these decisions.
BOTTOM LINE
Navigating the anticoagulant landscape in 2017 is complex. Doctors should review professional society guidelines while considering the strength of evidence on which they are based and tailor management to individual patient characteristics. Table 6 summarizes the management recommendations reviewed in this article.
References
Kearon C, Aki EA, Ornelas J, et al. Antithrombotic therapy for VTE disease: CHEST guideline and expert panel report. Chest 2016; 149:315–352.
Enden T, Haig Y, Klow NE, et al; CaVenT Study Group. Long-term outcome after additional catheter-directed thrombolysis versus standard treatment for acute iliofemoral deep vein thrombosis (the CaVenT study): a randomised controlled trial. Lancet 2012; 379:31–38.
Haig Y, Enden T, Grotta O, et al; CaVenT Study Group. Post-thrombotic syndrome after catheter-directed thrombolysis for deep vein thrombosis (CaVenT): 5-year follow-up results of an open-label, randomized controlled trial. Lancet Haematol 2016; 3:e64–e71.
Vedantham S, Goldhaber SZ, Kahn SR, et al. Rationale and design of the ATTRACT Study: a multicenter randomized trial to evaluate pharmacomechanical catheter-directed thrombolysis for the prevention of postthrombotic syndrome in patients with proximal deep vein thrombosis. Am Heart J 2013; 165:523–530.
Van Es N, Coppens M, Schulman S, Middeldorp S, Buller HR. Direct oral anticoagulants compared with vitamin K antagonists for acute venous thromboembolism: evidence from phase 3 trials. Blood 2014; 124:1968–1975.
Douketis JD, Spyropoulos AC, Kaatz S, et al; BRIDGE Investigators. Perioperative bridging anticoagulation in patients with atrial fibrillation. N Engl J Med 2015; 373:823–833.
Douketis J, Johnson JA, Turpie AG. Low-molecular-weight heparin as bridging anticoagulation during interruption of warfarin: assessment of a standardized periprocedural anticoagulation regimen. Arch Intern Med 2004; 164:1319–1326.
Dunn AS, Spyropoulos AC, Turpie AG. Bridging therapy in patients on long-term oral anticoagulants who require surgery: the Prospective Peri-operative Enoxaparin Cohort Trial (PROSPECT). J Thromb Haemost 2007; 5:2211–2218.
Kovacs MJ, Kearon C, Rodger M, et al. Single-arm study of bridging therapy with low-molecular-weight heparin for patients at risk of arterial embolism who require temporary interruption of warfarin. Circulation 2004; 110:1658–1663.
Spyropoulos AC, Turpie AG, Dunn AS, et al; REGIMEN Investigators. Clinical outcomes with unfractionated heparin or low-molecular-weight heparin as bridging therapy in patients on long-term oral anticoagulants: the REGIMEN registry. J Thromb Haemost 2006; 4:1246–1252.
Douketis JD, Woods K, Foster GA, Crowther MA. Bridging anticoagulation with low-molecular-weight heparin after interruption of warfarin therapy is associated with a residual anticoagulant effect prior to surgery. Thromb Haemost 2005; 94:528–531.
Schulman S, Hwang HG, Eikelboom JW, Kearon C, Pai M, Delaney J. Loading dose vs. maintenance dose of warfarin for reinitiation after invasive procedures: a randomized trial. J Thromb Haemost 2014; 12:1254-1259.
Siegal D, Yudin J, Kaatz S, Douketis JD, Lim W, Spyropoulos AC. Periprocedural heparin bridging in patients receiving vitamin K antagonists: systematic review and meta-analysis of bleeding and thromboembolic rates. Circulation 2012; 126:1630–1639.
Skeith L, Taylor J, Lazo-Langner A, Kovacs MJ. Conservative perioperative anticoagulation management in patients with chronic venous thromboembolic disease: a cohort study. J Thromb Haemost 2012; 10:2298–2304.
Douketis JD, Spyropoulos AC, Spencer FA, et al. Perioperative management of antithrombotic therapy: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest 2012; 141(2 suppl):e326S–e350S.
Doherty JU, Gluckman TJ, Hucker WJ, et al. 2017 ACC expert consensus decision pathway for periprocedural management of anticoagulation in patients with nonvalvular atrial fibrillation: a report of the American College of Cardiology Clinical Expert Consensus Document Task Force. J Am Coll Cardiol 2017; 69:871–898.
Raval AN, Cigarroa JE, Chung MK, et al; American Heart Association Clinical Pharmacology Subcommittee of the Acute Cardiac Care and General Cardiology Committee of the Council on Clinical Cardiology; Council on Cardiovascular Disease in the Young; and Council on Quality of Care and Outcomes Research. Management of patients on non-vitamin K antagonist oral anticoagulants in the acute care and periprocedural setting: a scientific statement from the American Heart Association. Circulation 2017; 135:e604–e633.
Tafur A, Douketis J. Perioperative anticoagulant management in patients with atrial fibrillation: practical implications of recent clinical trials. Pol Arch Med Wewn 2015; 125:666–671.
Birnie DH, Healey JS, Wells GA, et al: BRUISE CONTROL Investigators. Pacemaker or defibrillator surgery without interruption of anticoagulation. N Engl J Med 2013; 368:2084–2093.
Frost C, Song Y, Barrett YC, et al. A randomized direct comparison of the pharmacokinetics and pharmacodynamics of apixaban and rivaroxaban. Clin Pharmacol 2014; 6:179–187.
Narouze S, Benzon HT, Provenzano DA, et al. Interventional spine and pain procedures in patients on antiplatelet and anticoagulant medications: guidelines from the American Society of Regional Anesthesia and Pain Medicine, the European Society of Regional Anesthesia and Pain Therapy, the American Academy of Pain Medicine, the International Neuromodulation Society, the North American Neuromodulation Society, and the World institute of Pain. Reg Anesth Pain Med 2015; 40:182–212.
Douketis JD, Healey JS, Brueckmann M, et al. Perioperative bridging anticoagulation during dabigatran or warfarin interruption among patients who had an elective surgery or procedure. Substudy of the RE-LY trial. Thromb Haemost 2015; 113:625–632.
Steinberg BA, Peterson ED, Kim S, et al; Outcomes Registry for Better Informed Treatment of Atrial Fibrillation Investigators and Patients. Use and outcomes associated with bridging during anticoagulation interruptions in patients with atrial fibrillation: findings from the Outcomes Registry for Better Informed Treatment of Atrial Fibrillation (ORBIT-AF). Circulation 2015; 131:488–494.
Garcia D, Alexander JH, Wallentin L, et al. Management and clinical outcomes in patients treated with apixaban vs warfarin undergoing procedures. Blood 2014; 124:3692–3698.
Doherty JU, Gluckman TJ, Hucker WJ, et al. 2017 ACC expert consensus decision pathway for periprocedural management of anticoagulation in patients with nonvalvular atrial fibrillation: a report of the American College of Cardiology Clinical Expert Consensus Document Task Force. J Am Coll Cardiol 2017; 69:871–898.
Schulman S, Carrier M, Lee AY, et al; Periop Dabigatran Study Group. Perioperative management of dabigatran: a prospective cohort study. Circulation 2015; 132:167–173.
Douketis JD, Wang G, Chan N, et al. Effect of standardized perioperative dabigatran interruption on the residual anticoagulation effect at the time of surgery or procedure. J Thromb Haemost 2016; 14:89–97.
Douketis JD, Syed S, Schulman S. Periprocedural management of direct oral anticoagulants: comment on the 2015 American Society of Regional Anesthesia and Pain Medicine guidelines. Reg Anesth Pain Med 2016; 41:127–129.
Devereaux PJ, Mrkobrada M, Sessler DI, et al; POISE-2 Investigators. Aspirin in patients undergoing noncardiac surgery. N Engl J Med 2014; 370:1494–1503.
Holcomb CN, Graham LA, Richman JS, et al. The incremental risk of noncardiac surgery on adverse cardiac events following coronary stenting. J Am Coll Cardiol 2014; 64:2730–2739.
Egholm G, Kristensen SD, Thim T, et al. Risk associated with surgery within 12 months after coronary drug-eluting stent implantation. J Am Coll Cardiol 2016; 68:2622–2632.
James D. Douketis, MD, FRCP(C), FACP, FCCP Professor of Medicine, McMaster University, Hamilton, ON, Canada; Chair, American College of Physicians Practice Guidelines in Perioperative Management of Antithrombotic Therapy
Address: James D. Douketis, MD, St Joseph’s Healthcare Hamilton, Room F-544, 50 Charlton Ave E, Hamilton, ON, Canada L8N 4A6; jdouket@mcmaster.ca
This article is based on an edited transcript from a Heart and Vascular Institute Grand Rounds presentation at Cleveland Clinic. It was approved by the author but not peer-reviewed.
James D. Douketis, MD, FRCP(C), FACP, FCCP Professor of Medicine, McMaster University, Hamilton, ON, Canada; Chair, American College of Physicians Practice Guidelines in Perioperative Management of Antithrombotic Therapy
Address: James D. Douketis, MD, St Joseph’s Healthcare Hamilton, Room F-544, 50 Charlton Ave E, Hamilton, ON, Canada L8N 4A6; jdouket@mcmaster.ca
This article is based on an edited transcript from a Heart and Vascular Institute Grand Rounds presentation at Cleveland Clinic. It was approved by the author but not peer-reviewed.
Author and Disclosure Information
James D. Douketis, MD, FRCP(C), FACP, FCCP Professor of Medicine, McMaster University, Hamilton, ON, Canada; Chair, American College of Physicians Practice Guidelines in Perioperative Management of Antithrombotic Therapy
Address: James D. Douketis, MD, St Joseph’s Healthcare Hamilton, Room F-544, 50 Charlton Ave E, Hamilton, ON, Canada L8N 4A6; jdouket@mcmaster.ca
This article is based on an edited transcript from a Heart and Vascular Institute Grand Rounds presentation at Cleveland Clinic. It was approved by the author but not peer-reviewed.
This article reviews recommendations and evidence concerning current anticoagulant management for venous thromboembolism and perioperative care, with an emphasis on individualizing treatment for real-world patients.
TREATING ACUTE VENOUS THROMBOEMBOLISM
Case 1: Deep vein thrombosis in an otherwise healthy man
A 40-year-old man presents with 7 days of progressive right leg swelling. He has no antecedent risk factors for deep vein thrombosis or other medical problems. Venous ultrasonography reveals an iliofemoral deep vein thrombosis. How should he be managed?
Outpatient treatment with low-molecular-weight heparin for 4 to 6 days plus warfarin
Outpatient treatment with a direct oral anticoagulant, ie, apixaban, dabigatran (which requires 4 to 6 days of initial treatment with low-molecular-weight heparin), or rivaroxaban
Catheter-directed thrombolysis followed by low-molecular-weight heparin, then warfarin or a direct oral anticoagulant
Inpatient intravenous heparin for 7 to 10 days, then warfarin or a direct oral anticoagulant
All of these are acceptable for managing acute venous thromboembolism, but the clinician’s role is to identify which treatment is most appropriate for an individual patient.
Deep vein thrombosis is not a single condition
Multiple guidelines exist to help decide on a management strategy. Those of the American College of Chest Physicians (ACCP)1 are used most often.
That said, guidelines are established for “average” patients, so it is important to look beyond guidelines and individualize management. Venous thromboembolism is not a single entity; it has a myriad of clinical presentations that could call for different treatments. Most patients have submassive deep vein thrombosis or pulmonary embolism, which is not limb-threatening nor associated with hemodynamic instability. It can also differ in terms of etiology and can be unprovoked (or idiopathic), cancer-related, catheter-associated, or provoked by surgery or immobility.
Deep vein thrombosis has a wide spectrum of presentations. It can involve the veins of the calf only, or it can involve the femoral and iliac veins and other locations including the splanchnic veins, the cerebral sinuses, and upper extremities. Pulmonary embolism can be massive (defined as being associated with hemodynamic instability or impending respiratory failure) or submassive. Similarly, patients differ in terms of baseline medical conditions, mobility, and lifestyle. Anticoagulant management decisions should take all these factors into account.
Consider clot location
Our patient with iliofemoral deep vein thrombosis is best managed differently than a more typical patient with less extensive thrombosis that would involve the popliteal or femoral vein segments, or both. A clot that involves the iliac vein is more likely to lead to postthrombotic chronic pain and swelling as the lack of venous outflow bypass channels to circumvent the clot location creates higher venous pressure within the affected leg. Therefore, for our patient, catheter-directed thrombolysis is an option that should be considered.
Catheter-directed thrombolysis trials
According to the “open-vein hypothesis,” quickly eliminating the thrombus and restoring unobstructed venous flow may mitigate the risk not only of recurrent thrombosis, but also of postthrombotic syndrome, which is often not given much consideration acutely but can cause significant, life-altering chronic disability.
The “valve-integrity hypothesis” is also important; it considers whether lytic therapy may help prevent damage to such valves in an attempt to mitigate the amount of venous hypertension.
Thus, catheter-directed thrombolysis offers theoretical benefits, and recent trials have assessed it against standard anticoagulation treatments.
The CaVenT trial (Catheter-Directed Venous Thrombolysis),2 conducted in Norway, randomized 209 patients with midfemoral to iliac deep vein thrombosis to conventional treatment (anticoagulation alone) or anticoagulation plus catheter-directed thrombolysis. At 2 years, postthrombotic syndrome had occurred in 41% of the catheter-directed thrombolysis group compared with 56% of the conventional treatment group (P = .047). At 5 years, the difference widened to 43% vs 71% (P < .01, number needed to treat = 4).3 Despite the superiority of lytic therapy, the incidence of postthrombotic syndrome remained high in patients who received this treatment.
The ATTRACT trial (Acute Venous Thrombosis: Thrombus Removal With Adjunctive Catheter-Directed Thrombolysis),4 a US multicenter, open-label, assessor-blind study, randomized 698 patients with femoral or more-proximal deep vein thrombosis to either standard care (anticoagulant therapy and graduated elastic compression stockings) or standard care plus catheter-directed thrombolysis. In preliminary results presented at the Society of Interventional Radiology meeting in March 2017, although no difference was found in the primary outcome (postthrombotic syndrome at 24 months), catheter-directed thrombolysis for iliofemoral deep vein thrombosis led to a 25% reduction in moderate to severe postthrombotic syndrome.
Although it is too early to draw conclusions before publication of the ATTRACT study, the preliminary results highlight the need to individualize treatment and to be selective about using catheter-directed thrombolysis. The trials provide reassurance that catheter-directed lysis is a reasonable and safe intervention when performed by physicians experienced in the procedure. The risk of major bleeding appears to be low (about 2%) and that for intracranial hemorrhage even lower (< 0.5%).
Catheter-directed thrombolysis is appropriate in some cases
The 2016 ACCP guidelines1 recommend anticoagulant therapy alone over catheter-directed thrombolysis for patients with acute proximal deep vein thrombosis of the leg. However, it is a grade 2C (weak) recommendation.
They provide no specific recommendation as to the clinical indications for catheter-directed thrombolysis, but identify patients who would be most likely to benefit, ie, those who have:
Iliofemoral deep vein thrombosis
Symptoms for less than 14 days
Good functional status
Life expectancy of more than 1 year
Low risk of bleeding.
Our patient satisfies these criteria, suggesting that catheter-directed thrombolysis is a reasonable option for him.
Timing is important. Catheter-directed lysis is more likely to be beneficial if used before fibrin deposits form and stiffen the venous valves, causing irreversible damage that leads to postthrombotic syndrome.
Role of direct oral anticoagulants
The availability of direct oral anticoagulants has generated interest in defining their therapeutic role in patients with venous thromboembolism.
In a meta-analysis5 of major trials comparing direct oral anticoagulants and vitamin K antagonists such as warfarin, no significant difference was found for the risk of recurrent venous thromboembolism or venous thromboembolism-related deaths. However, fewer patients experienced major bleeding with direct oral anticoagulants (relative risk 0.61, P = .002). Although significant, the absolute risk reduction was small; the incidence of major bleeding was 1.1% with direct oral anticoagulants vs 1.8% with vitamin K antagonists.
The main advantage of direct oral anticoagulants is greater convenience for the patient.
The 2016 ACCP guidelines1 on the treatment of venous thrombosis and pulmonary embolism are summarized in Table 1. They suggest using direct oral anticoagulants rather than vitamin K antagonists to manage venous thromboembolism, but this is a weak (ie, grade 2B) recommendation, likely because the net clinical benefit of direct oral anticoagulants over vitamin K antagonists is modest.
WHICH PATIENTS ON WARFARIN NEED BRIDGING PREOPERATIVELY?
Many patients still take warfarin, particularly those with atrial fibrillation, a mechanical heart valve, or venous thromboembolism. In many countries, warfarin remains the dominant anticoagulant for stroke prevention. Whether these patients need heparin during the period of perioperative warfarin interruption is a frequently encountered scenario that, until recently, was controversial. Recent studies have helped to inform the need for heparin bridging in many of these patients.
Case 2: An elderly woman on warfarin facing cancer surgery
A 75-year-old woman weighing 65 kg is scheduled for elective colon resection for incidentally found colon cancer. She is taking warfarin for atrial fibrillation. She also has hypertension and diabetes and had a transient ischemic attack 10 years ago.
One doctor told her she needs to be assessed for heparin bridging, but another told her she does not need bridging.
The default management should be not to bridge patients who have atrial fibrillation, but to consider bridging in selected patients, such as those with recent stroke or transient ischemic attack or a prior thromboembolic event during warfarin interruption. However, decisions about bridging should not be made on the basis of the CHADS2 score alone. For the patient described here, I would recommend not bridging.
Complex factors contribute to stroke risk
Stroke risk for patients with atrial fibrillation can be quickly estimated with the CHADS2 score, based on:
Congestive heart failure (1 point)
Hypertension (1 point)
Age at least 75 (1 point)
Diabetes (1 point)
Stroke or transient ischemic attack (2 points).
Our patient has a score of 5, corresponding to an annual adjusted stroke risk of 12.5%. Whether her transient ischemic attack of 10 years ago is comparable in significance to a recent stroke is debatable and highlights a weakness of clinical prediction rules. Moreover, such prediction scores were developed to estimate the long-term risk of stroke if anticoagulants are not given, and they have not been assessed in a perioperative setting where there is short-term interruption of anticoagulants. Also, the perioperative milieu is associated with additional factors not captured in these clinical prediction rules that may affect the risk of stroke.
Thus, the risk of perioperative stroke likely involves the interplay of multiple factors, including the type of surgery the patient is undergoing. Some factors may be mitigated:
Rebound hypercoagulability after stopping an oral anticoagulant can be prevented by intraoperative blood pressure and volume control
Elevated biochemical factors (eg, D-dimer, B-type natriuretic peptide, troponin) may be lowered with perioperative aspirin therapy
Lipid and genetic factors may be mitigated with perioperative statin use.
Can heparin bridging also mitigate the risk?
Bridging in patients with atrial fibrillation
Most patients who are taking warfarin are doing so because of atrial fibrillation, so most evidence about perioperative bridging was developed in such patients.
The BRIDGE trial (Bridging Anticoagulation in Patients Who Require Temporary Interruption of Warfarin Therapy for an Elective Invasive Procedure or Surgery)6 was the first randomized controlled trial to compare a bridging and no-bridging strategy for patients with atrial fibrillation who required warfarin interruption for elective surgery. Nearly 2,000 patients were given either low-molecular-weight heparin or placebo starting 3 days before until 24 hours before a procedure, and then for 5 to 10 days afterwards. For all patients, warfarin was stopped 5 days before the procedure and was resumed within 24 hours afterwards.
A no-bridging strategy was noninferior to bridging: the risk of perioperative arterial thromboembolism was 0.4% without bridging vs 0.3% with bridging (P = .01 for noninferiority). In addition, a no-bridging strategy conferred a lower risk of major bleeding than bridging: 1.3% vs 3.2% (relative risk 0.41, P = .005 for superiority).
Although the difference in absolute bleeding risk was small, bleeding rates were lower than those seen outside of clinical trials, as the bridging protocol used in BRIDGE was designed to minimize the risk of bleeding. Also, although only 5% of patients had a CHADS2 score of 5 or 6, such patients are infrequent in clinical practice, and BRIDGE did include a considerable proportion (17%) of patients with a prior stroke or transient ischemic attack who would be considered at high risk.
Other evidence about heparin bridging is derived from observational studies, more than 10 of which have been conducted. In general, they have found that not bridging is associated with low rates of arterial thromboembolism (< 0.5%) and that bridging is associated with high rates of major bleeding (4%–7%).7–12
Bridging in patients with a mechanical heart valve
Warfarin is the only anticoagulant option for patients who have a mechanical heart valve. No randomized controlled trials have evaluated the benefits of perioperative bridging vs no bridging in this setting.
Observational (cohort) studies suggest that the risk of perioperative arterial thromboembolism is similar with or without bridging anticoagulation, although most patients studied were bridged and those not bridged were considered at low risk (eg, with a bileaflet aortic valve and no additional risk factors).13 However, without stronger evidence from randomized controlled trials, bridging should be the default management for patients with a mechanical heart valve. In our practice, we bridge most patients who have a mechanical heart valve unless they are considered to be at low risk, such as those who have a bileaflet aortic valve.
Bridging in patients with prior venous thromboembolism
Even less evidence is available for periprocedural management of patients who have a history of venous thromboembolism. No randomized controlled trials exist evaluating bridging vs no bridging. In 1 cohort study in which more than 90% of patients had had thromboembolism more than 3 months before the procedure, the rate of recurrent venous thromboembolism without bridging was less than 0.5%.14
It is reasonable to bridge patients who need anticoagulant interruption within 3 months of diagnosis of a deep vein thrombosis or pulmonary embolism, and to consider using a temporary inferior vena cava filter for patients who have had a clot who need treatment interruption during the initial 3 to 4 weeks after diagnosis.
Practice guidelines: Perioperative anticoagulation
The ACCP,15 the American College of Cardiology,16 and the American Heart Association17 have published guidelines for perioperative management of antithrombotic therapy. Despite a paucity of evidence from randomized trials, there are sufficient data to inform clinical management. Some guidelines are complex. A simplified algorithm has been proposed that considers the type of procedure, the CHADS2 score, whether the patient has a mechanical heart valve, and whether there has been a recent venous thromboembolic event.18
Guidance for preoperative and postoperative bridging for patients taking warfarin is summarized in Table 2.
CARDIAC PROCEDURES
For patients facing a procedure to implant an implantable cardioverter-defibrillator (ICD) or pacemaker, a procedure-specific concern is the avoidance of pocket hematoma.
Patients on warfarin: Do not bridge
The BRUISE CONTROL-1 trial (Bridge or Continue Coumadin for Device Surgery Randomized Controlled Trial)19 randomized patients undergoing pacemaker or ICD implantation to either continued anticoagulation therapy and not bridging (ie, continued warfarin so long as the international normalized ratio was < 3) vs conventional bridging treatment (ie, stopping warfarin and bridging with low-molecular-weight heparin). A clinically significant device-pocket hematoma occurred in 3.5% of the continued-warfarin group vs 16.0% in the heparin-bridging group (P < .001). Thromboembolic complications were rare, and rates did not differ between the 2 groups.
Results of the BRUISE CONTROL-1 trial serve as a caution to at least not be too aggressive with bridging. The study design involved resuming heparin 24 hours after surgery, which is perhaps more aggressive than standard practice. In our practice, we wait at least 24 hours to reinstate heparin after minor surgery, and 48 to 72 hours after surgery with higher bleeding risk.
These results are perhaps not surprising if one considers how carefully surgeons try to control bleeding during surgery for patients taking anticoagulants. For patients who are not on an anticoagulant, small bleeding may be less of a concern during a procedure. When high doses of heparin are introduced soon after surgery, small concerns during surgery may become big problems afterward.
Based on these results, it is reasonable to undertake device implantation without interruption of a vitamin K antagonist such as warfarin.
Patients on direct oral anticoagulants: The jury is still out
The similar BRUISE CONTROL-2 trial is currently under way, comparing interruption vs continuation of dabigatran for patients undergoing cardiac device surgery.
In Europe, surgeons are less concerned than those in the United States about operating while a patient is on anticoagulant therapy. But the safety of this practice is not backed by strong evidence.
Direct oral anticoagulants: Consider pharmacokinetics
Direct oral anticoagulants are potent and fast-acting, with a peak effect 1 to 3 hours after intake. This rapid anticoagulant action is similar to that of bridging with low-molecular-weight heparin, and caution is needed when administering direct oral anticoagulants, especially after major surgery or surgery with a high bleeding risk.
Frost et al20 compared the pharmacokinetics of apixaban (with twice-daily dosing) and rivaroxaban (once-daily dosing) and found that peak anticoagulant activity is faster and higher with rivaroxaban. This is important, because many patients will take their anticoagulant first thing in the morning. Consequently, if patients require any kind of procedure (including dental), they should skip the morning dose of the direct oral anticoagulant to avoid having the procedure done during the peak anticoagulant effect, and they should either not take that day’s dose or defer the dose until the evening after the procedure.
MANAGING SURGERY FOR PATIENTS ON A DIRECT ORAL ANTICOAGULANT
Case 3: An elderly woman on apixaban facing surgery
Let us imagine that our previous patient takes apixaban instead of warfarin. She is 75 years old, has atrial fibrillation, and is about to undergo elective colon resection for cancer. One doctor advises her to simply stop apixaban for 2 days, while another says she should go off apixaban for 5 days and will need bridging. Which plan is best?
In the perioperative setting, our goal is to interrupt patients’ anticoagulant therapy for the shortest time that results in no residual anticoagulant effect at the time of the procedure.
The European Society of Regional Anaesthesia and Pain Therapy and the American Society of Regional Anesthesia and Pain Medicine21 recommend an extended period of interruption of direct oral anticoagulants (Table 3)
They further recommend that if the risk of venous thromboembolism is high, low-molecular-weight heparin bridging should be done while stopping the direct oral anticoagulant, with the heparin discontinued 24 hours before the procedure. This recommendation seems counterintuitive, as it is advising replacing a short-acting anticoagulant with low-molecular-weight heparin, another short-acting anticoagulant.
The guidelines committee was unable to provide strength and grading of their recommendations, as too few well-designed studies are available to support them. The doctor in case 3 who advised stopping apixaban for 5 days and bridging is following the guidelines, but without much evidence to support this strategy.
Is bridging needed during interruption of a direct oral anticoagulant?
There are no randomized, controlled trials of bridging vs no bridging in patients taking direct oral anticoagulants. Substudies exist of patients taking these drugs for atrial fibrillation who had treatment interrupted for procedures, but the studies did not randomize bridging vs no bridging, nor were bridging regimens standardized. Three of the four atrial fibrillation trials had a blinded design (warfarin vs direct oral anticoagulants), making perioperative management difficult, as physicians did not know the pharmacokinetics of the drugs their patients were taking.22–24
We used the database from the Randomized Evaluation of Long-Term Anticoagulation Therapy (RE-LY) trial22 to evaluate bridging in patients taking either warfarin or dabigatran. With an open-label study design (the blinding was only for the 110 mg and 150 mg dabigatran doses), clinicians were aware of whether patients were receiving warfarin or dabigatran, thereby facilitating perioperative management. Among dabigatran-treated patients, those who were bridged had significantly more major bleeding than those not bridged (6.5% vs 1.8%, P < .001), with no difference between the groups for stroke or systemic embolism. Although it is not a randomized controlled trial, it does provide evidence that bridging may not be advisable for patients taking a direct oral anticoagulant.
The 2017 American College of Cardiology guidelines25 conclude that parenteral bridging is not indicated for direct oral anticoagulants. Although this is not based on strong evidence, the guidance appears reasonable according to the evidence at hand.
The 2017 American Heart Association Guidelines16 recommend a somewhat complex approach based on periprocedural bleeding risk and thromboembolic risk.
How long to interrupt direct oral anticoagulants?
Table 4 shows a simplified approach to interrupting direct oral anticoagulants that we use in Canada. The approach takes into account the type of surgery and kidney function for patients taking dabigatran, a drug that depends more on renal clearance than the other direct oral anticoagulants do.26
Evidence for this approach comes from a prospective cohort study27 of 541 patients being treated with dabigatran who were having an elective surgery or invasive procedure. Patients received standard perioperative management, with the timing of the last dabigatran dose before the procedure (24 hours, 48 hours, or 96 hours) based on the bleeding risk of surgery and the patient’s creatinine clearance. Dabigatran was resumed 24 to 72 hours after the procedure. No heparin bridging was done. Patients were followed for up to 30 days postoperatively. The results were favorable with few complications: one transient ischemic attack (0.2%), 10 major bleeding episodes (1.8%), and 28 minor bleeding episodes (5.2%).
A subgroup of 181 patients in this study28 had a plasma sample drawn just before surgery, allowing the investigators to assess the level of coagulation factors after dabigatran interruption. Results were as follows:
93% had a normal prothrombin time
80% had a normal activated partial thromboplastin time
33% had a normal thrombin time
81% had a normal dilute thrombin time.
The dilute thrombin time is considered the most reliable test of the anticoagulant effect of dabigatran but is not widely available. The activated partial thromboplastin time can provide a more widely used coagulation test to assess (in a less precise manner) whether there is an anticoagulant effect of dabigatran present, and more sensitive activated partial thromboplastin time assays can be used to better detect any residual dabigatran effect.
Dabigatran levels were also measured. Although 66% of patients had low drug levels just before surgery, the others still had substantial dabigatran on board. The fact that bleeding event rates were so low in this study despite the presence of dabigatran in many patients raises the question of whether having some drug on board is a good predictor of bleeding risk.
An interruption protocol with a longer interruption interval—12 to 14 hours longer than in the previous study (3 days for high-bleed risk procedures, 2 days for low-bleed risk procedures)—brought the activated partial thromboplastin time and dilute thrombin time to normal levels for 100% of patients with the protocol for high-bleeding-risk surgery. This study was based on small numbers and its interruption strategy needs further investigation.29
Case 3 continued
Based on the current empiric evidence, we recommend interrupting direct oral anticoagulants for 2 days (or approximately a 60-hour interval between the last dose and surgery) for this 75-year-old woman who is taking apixaban (Table 5). This interruption interval corresponds to 5 elimination half-lives for apixaban, which should result in little to no residual anticoagulant and will facilitate major surgery and, if indicated, neuraxial anesthesia.
The PAUSE study (NCT02228798), a multicenter, prospective cohort study, is designed to establish a safe, standardized protocol for the perioperative management of patients with atrial fibrillation taking dabigatran, rivaroxaban, or apixaban and will include 3,300 patients.
PATIENTS WITH A CORONARY STENT WHO NEED SURGERY
Case 4: A woman with a stent facing surgery
A 70-year-old woman needs breast cancer resection. She has coronary artery disease and had a drug-eluting stent placed 5 months ago after elective cardiac catheterization. She also has hypertension, obesity, and type 2 diabetes. Her medications include an angiotensin II receptor blocker, hydrochlorothiazide, insulin, and an oral hypoglycemic. She is also taking aspirin 81 mg daily and ticagrelor (a P2Y12 receptor antagonist) 90 mg twice daily.
Her cardiologist is concerned that stopping antiplatelet therapy could trigger acute stent thrombosis, which has a 50% or higher mortality rate.
Should she stop taking aspirin before surgery? What about the ticagrelor?
Is aspirin safe during surgery?
Evidence concerning aspirin during surgery comes from Perioperative Ischemic Evaluation 2 (POISE-2), a double-blind, randomized controlled trial.30 Patients who had known cardiovascular disease or risk factors for cardiovascular disease and were about to undergo noncardiac surgery were stratified according to whether they had been taking aspirin before the study (patients taking aspirin within 72 hours of the surgery were excluded from randomization). Participants in each group were randomized to take either aspirin or placebo just before surgery. The primary outcome was the combined rate of death or nonfatal myocardial infarction 30 days after randomization.
The study found no differences in the primary end point between the two groups. However, major bleeding occurred significantly more often in the aspirin group (4.6% vs 3.8%, hazard ratio 1.2, 95% confidence interval 1.0–1.5).
Moreover, only 4% of the patients in this trial had a cardiac stent. The trial excluded patients who had had a bare-metal stent placed within 6 weeks or a drug-eluting stent placed within 1 year, so it does not help us answer whether aspirin should be stopped for our current patient.
Is surgery safe for patients with stents?
The safety of undergoing surgery with a stent was investigated in a large US Veterans Administration retrospective cohort study.31 More than 20,000 patients with stents who underwent noncardiac surgery within 2 years of stent placement were compared with a control group of more than 41,000 patients with stents who did not undergo surgery. Patients were matched by stent type and cardiac risk factors at the time of stent placement.
The risk of an adverse cardiac event in both the surgical and nonsurgical cohorts was highest in the initial 6 weeks after stent placement and plateaued 6 months after stent placement, when the risk difference between the surgical and nonsurgical groups leveled off to 1%.
The risk of a major adverse cardiac event postoperatively was much more dependent on the timing of stent placement in complex and inpatient surgeries. For outpatient surgeries, the risk of a major cardiac event was very low and the timing of stent placement did not matter.
A Danish observational study32 compared more than 4,000 patients with drug-eluting stents having surgery to more than 20,000 matched controls without coronary heart disease having similar surgery. The risk of myocardial infarction or cardiac death was much higher for patients undergoing surgery within 1 month after drug-eluting stent placement compared with controls without heart disease and patients with stent placement longer than 1 month before surgery.
Our practice is to continue aspirin for surgery in patients with coronary stents regardless of the timing of placement. Although there is a small increased risk of bleeding, this must be balanced against thrombotic risk. We typically stop clopidogrel 5 to 7 days before surgery and ticagrelor 3 to 5 days before surgery. We may decide to give platelets before very-high-risk surgery (eg, intracranial, spinal) if there is a decision to continue both antiplatelet drugs—for example, in a patient who recently received a drug-eluting stent (ie, within 3 months). It is essential to involve the cardiologist and surgeon in these decisions.
BOTTOM LINE
Navigating the anticoagulant landscape in 2017 is complex. Doctors should review professional society guidelines while considering the strength of evidence on which they are based and tailor management to individual patient characteristics. Table 6 summarizes the management recommendations reviewed in this article.
This article reviews recommendations and evidence concerning current anticoagulant management for venous thromboembolism and perioperative care, with an emphasis on individualizing treatment for real-world patients.
TREATING ACUTE VENOUS THROMBOEMBOLISM
Case 1: Deep vein thrombosis in an otherwise healthy man
A 40-year-old man presents with 7 days of progressive right leg swelling. He has no antecedent risk factors for deep vein thrombosis or other medical problems. Venous ultrasonography reveals an iliofemoral deep vein thrombosis. How should he be managed?
Outpatient treatment with low-molecular-weight heparin for 4 to 6 days plus warfarin
Outpatient treatment with a direct oral anticoagulant, ie, apixaban, dabigatran (which requires 4 to 6 days of initial treatment with low-molecular-weight heparin), or rivaroxaban
Catheter-directed thrombolysis followed by low-molecular-weight heparin, then warfarin or a direct oral anticoagulant
Inpatient intravenous heparin for 7 to 10 days, then warfarin or a direct oral anticoagulant
All of these are acceptable for managing acute venous thromboembolism, but the clinician’s role is to identify which treatment is most appropriate for an individual patient.
Deep vein thrombosis is not a single condition
Multiple guidelines exist to help decide on a management strategy. Those of the American College of Chest Physicians (ACCP)1 are used most often.
That said, guidelines are established for “average” patients, so it is important to look beyond guidelines and individualize management. Venous thromboembolism is not a single entity; it has a myriad of clinical presentations that could call for different treatments. Most patients have submassive deep vein thrombosis or pulmonary embolism, which is not limb-threatening nor associated with hemodynamic instability. It can also differ in terms of etiology and can be unprovoked (or idiopathic), cancer-related, catheter-associated, or provoked by surgery or immobility.
Deep vein thrombosis has a wide spectrum of presentations. It can involve the veins of the calf only, or it can involve the femoral and iliac veins and other locations including the splanchnic veins, the cerebral sinuses, and upper extremities. Pulmonary embolism can be massive (defined as being associated with hemodynamic instability or impending respiratory failure) or submassive. Similarly, patients differ in terms of baseline medical conditions, mobility, and lifestyle. Anticoagulant management decisions should take all these factors into account.
Consider clot location
Our patient with iliofemoral deep vein thrombosis is best managed differently than a more typical patient with less extensive thrombosis that would involve the popliteal or femoral vein segments, or both. A clot that involves the iliac vein is more likely to lead to postthrombotic chronic pain and swelling as the lack of venous outflow bypass channels to circumvent the clot location creates higher venous pressure within the affected leg. Therefore, for our patient, catheter-directed thrombolysis is an option that should be considered.
Catheter-directed thrombolysis trials
According to the “open-vein hypothesis,” quickly eliminating the thrombus and restoring unobstructed venous flow may mitigate the risk not only of recurrent thrombosis, but also of postthrombotic syndrome, which is often not given much consideration acutely but can cause significant, life-altering chronic disability.
The “valve-integrity hypothesis” is also important; it considers whether lytic therapy may help prevent damage to such valves in an attempt to mitigate the amount of venous hypertension.
Thus, catheter-directed thrombolysis offers theoretical benefits, and recent trials have assessed it against standard anticoagulation treatments.
The CaVenT trial (Catheter-Directed Venous Thrombolysis),2 conducted in Norway, randomized 209 patients with midfemoral to iliac deep vein thrombosis to conventional treatment (anticoagulation alone) or anticoagulation plus catheter-directed thrombolysis. At 2 years, postthrombotic syndrome had occurred in 41% of the catheter-directed thrombolysis group compared with 56% of the conventional treatment group (P = .047). At 5 years, the difference widened to 43% vs 71% (P < .01, number needed to treat = 4).3 Despite the superiority of lytic therapy, the incidence of postthrombotic syndrome remained high in patients who received this treatment.
The ATTRACT trial (Acute Venous Thrombosis: Thrombus Removal With Adjunctive Catheter-Directed Thrombolysis),4 a US multicenter, open-label, assessor-blind study, randomized 698 patients with femoral or more-proximal deep vein thrombosis to either standard care (anticoagulant therapy and graduated elastic compression stockings) or standard care plus catheter-directed thrombolysis. In preliminary results presented at the Society of Interventional Radiology meeting in March 2017, although no difference was found in the primary outcome (postthrombotic syndrome at 24 months), catheter-directed thrombolysis for iliofemoral deep vein thrombosis led to a 25% reduction in moderate to severe postthrombotic syndrome.
Although it is too early to draw conclusions before publication of the ATTRACT study, the preliminary results highlight the need to individualize treatment and to be selective about using catheter-directed thrombolysis. The trials provide reassurance that catheter-directed lysis is a reasonable and safe intervention when performed by physicians experienced in the procedure. The risk of major bleeding appears to be low (about 2%) and that for intracranial hemorrhage even lower (< 0.5%).
Catheter-directed thrombolysis is appropriate in some cases
The 2016 ACCP guidelines1 recommend anticoagulant therapy alone over catheter-directed thrombolysis for patients with acute proximal deep vein thrombosis of the leg. However, it is a grade 2C (weak) recommendation.
They provide no specific recommendation as to the clinical indications for catheter-directed thrombolysis, but identify patients who would be most likely to benefit, ie, those who have:
Iliofemoral deep vein thrombosis
Symptoms for less than 14 days
Good functional status
Life expectancy of more than 1 year
Low risk of bleeding.
Our patient satisfies these criteria, suggesting that catheter-directed thrombolysis is a reasonable option for him.
Timing is important. Catheter-directed lysis is more likely to be beneficial if used before fibrin deposits form and stiffen the venous valves, causing irreversible damage that leads to postthrombotic syndrome.
Role of direct oral anticoagulants
The availability of direct oral anticoagulants has generated interest in defining their therapeutic role in patients with venous thromboembolism.
In a meta-analysis5 of major trials comparing direct oral anticoagulants and vitamin K antagonists such as warfarin, no significant difference was found for the risk of recurrent venous thromboembolism or venous thromboembolism-related deaths. However, fewer patients experienced major bleeding with direct oral anticoagulants (relative risk 0.61, P = .002). Although significant, the absolute risk reduction was small; the incidence of major bleeding was 1.1% with direct oral anticoagulants vs 1.8% with vitamin K antagonists.
The main advantage of direct oral anticoagulants is greater convenience for the patient.
The 2016 ACCP guidelines1 on the treatment of venous thrombosis and pulmonary embolism are summarized in Table 1. They suggest using direct oral anticoagulants rather than vitamin K antagonists to manage venous thromboembolism, but this is a weak (ie, grade 2B) recommendation, likely because the net clinical benefit of direct oral anticoagulants over vitamin K antagonists is modest.
WHICH PATIENTS ON WARFARIN NEED BRIDGING PREOPERATIVELY?
Many patients still take warfarin, particularly those with atrial fibrillation, a mechanical heart valve, or venous thromboembolism. In many countries, warfarin remains the dominant anticoagulant for stroke prevention. Whether these patients need heparin during the period of perioperative warfarin interruption is a frequently encountered scenario that, until recently, was controversial. Recent studies have helped to inform the need for heparin bridging in many of these patients.
Case 2: An elderly woman on warfarin facing cancer surgery
A 75-year-old woman weighing 65 kg is scheduled for elective colon resection for incidentally found colon cancer. She is taking warfarin for atrial fibrillation. She also has hypertension and diabetes and had a transient ischemic attack 10 years ago.
One doctor told her she needs to be assessed for heparin bridging, but another told her she does not need bridging.
The default management should be not to bridge patients who have atrial fibrillation, but to consider bridging in selected patients, such as those with recent stroke or transient ischemic attack or a prior thromboembolic event during warfarin interruption. However, decisions about bridging should not be made on the basis of the CHADS2 score alone. For the patient described here, I would recommend not bridging.
Complex factors contribute to stroke risk
Stroke risk for patients with atrial fibrillation can be quickly estimated with the CHADS2 score, based on:
Congestive heart failure (1 point)
Hypertension (1 point)
Age at least 75 (1 point)
Diabetes (1 point)
Stroke or transient ischemic attack (2 points).
Our patient has a score of 5, corresponding to an annual adjusted stroke risk of 12.5%. Whether her transient ischemic attack of 10 years ago is comparable in significance to a recent stroke is debatable and highlights a weakness of clinical prediction rules. Moreover, such prediction scores were developed to estimate the long-term risk of stroke if anticoagulants are not given, and they have not been assessed in a perioperative setting where there is short-term interruption of anticoagulants. Also, the perioperative milieu is associated with additional factors not captured in these clinical prediction rules that may affect the risk of stroke.
Thus, the risk of perioperative stroke likely involves the interplay of multiple factors, including the type of surgery the patient is undergoing. Some factors may be mitigated:
Rebound hypercoagulability after stopping an oral anticoagulant can be prevented by intraoperative blood pressure and volume control
Elevated biochemical factors (eg, D-dimer, B-type natriuretic peptide, troponin) may be lowered with perioperative aspirin therapy
Lipid and genetic factors may be mitigated with perioperative statin use.
Can heparin bridging also mitigate the risk?
Bridging in patients with atrial fibrillation
Most patients who are taking warfarin are doing so because of atrial fibrillation, so most evidence about perioperative bridging was developed in such patients.
The BRIDGE trial (Bridging Anticoagulation in Patients Who Require Temporary Interruption of Warfarin Therapy for an Elective Invasive Procedure or Surgery)6 was the first randomized controlled trial to compare a bridging and no-bridging strategy for patients with atrial fibrillation who required warfarin interruption for elective surgery. Nearly 2,000 patients were given either low-molecular-weight heparin or placebo starting 3 days before until 24 hours before a procedure, and then for 5 to 10 days afterwards. For all patients, warfarin was stopped 5 days before the procedure and was resumed within 24 hours afterwards.
A no-bridging strategy was noninferior to bridging: the risk of perioperative arterial thromboembolism was 0.4% without bridging vs 0.3% with bridging (P = .01 for noninferiority). In addition, a no-bridging strategy conferred a lower risk of major bleeding than bridging: 1.3% vs 3.2% (relative risk 0.41, P = .005 for superiority).
Although the difference in absolute bleeding risk was small, bleeding rates were lower than those seen outside of clinical trials, as the bridging protocol used in BRIDGE was designed to minimize the risk of bleeding. Also, although only 5% of patients had a CHADS2 score of 5 or 6, such patients are infrequent in clinical practice, and BRIDGE did include a considerable proportion (17%) of patients with a prior stroke or transient ischemic attack who would be considered at high risk.
Other evidence about heparin bridging is derived from observational studies, more than 10 of which have been conducted. In general, they have found that not bridging is associated with low rates of arterial thromboembolism (< 0.5%) and that bridging is associated with high rates of major bleeding (4%–7%).7–12
Bridging in patients with a mechanical heart valve
Warfarin is the only anticoagulant option for patients who have a mechanical heart valve. No randomized controlled trials have evaluated the benefits of perioperative bridging vs no bridging in this setting.
Observational (cohort) studies suggest that the risk of perioperative arterial thromboembolism is similar with or without bridging anticoagulation, although most patients studied were bridged and those not bridged were considered at low risk (eg, with a bileaflet aortic valve and no additional risk factors).13 However, without stronger evidence from randomized controlled trials, bridging should be the default management for patients with a mechanical heart valve. In our practice, we bridge most patients who have a mechanical heart valve unless they are considered to be at low risk, such as those who have a bileaflet aortic valve.
Bridging in patients with prior venous thromboembolism
Even less evidence is available for periprocedural management of patients who have a history of venous thromboembolism. No randomized controlled trials exist evaluating bridging vs no bridging. In 1 cohort study in which more than 90% of patients had had thromboembolism more than 3 months before the procedure, the rate of recurrent venous thromboembolism without bridging was less than 0.5%.14
It is reasonable to bridge patients who need anticoagulant interruption within 3 months of diagnosis of a deep vein thrombosis or pulmonary embolism, and to consider using a temporary inferior vena cava filter for patients who have had a clot who need treatment interruption during the initial 3 to 4 weeks after diagnosis.
Practice guidelines: Perioperative anticoagulation
The ACCP,15 the American College of Cardiology,16 and the American Heart Association17 have published guidelines for perioperative management of antithrombotic therapy. Despite a paucity of evidence from randomized trials, there are sufficient data to inform clinical management. Some guidelines are complex. A simplified algorithm has been proposed that considers the type of procedure, the CHADS2 score, whether the patient has a mechanical heart valve, and whether there has been a recent venous thromboembolic event.18
Guidance for preoperative and postoperative bridging for patients taking warfarin is summarized in Table 2.
CARDIAC PROCEDURES
For patients facing a procedure to implant an implantable cardioverter-defibrillator (ICD) or pacemaker, a procedure-specific concern is the avoidance of pocket hematoma.
Patients on warfarin: Do not bridge
The BRUISE CONTROL-1 trial (Bridge or Continue Coumadin for Device Surgery Randomized Controlled Trial)19 randomized patients undergoing pacemaker or ICD implantation to either continued anticoagulation therapy and not bridging (ie, continued warfarin so long as the international normalized ratio was < 3) vs conventional bridging treatment (ie, stopping warfarin and bridging with low-molecular-weight heparin). A clinically significant device-pocket hematoma occurred in 3.5% of the continued-warfarin group vs 16.0% in the heparin-bridging group (P < .001). Thromboembolic complications were rare, and rates did not differ between the 2 groups.
Results of the BRUISE CONTROL-1 trial serve as a caution to at least not be too aggressive with bridging. The study design involved resuming heparin 24 hours after surgery, which is perhaps more aggressive than standard practice. In our practice, we wait at least 24 hours to reinstate heparin after minor surgery, and 48 to 72 hours after surgery with higher bleeding risk.
These results are perhaps not surprising if one considers how carefully surgeons try to control bleeding during surgery for patients taking anticoagulants. For patients who are not on an anticoagulant, small bleeding may be less of a concern during a procedure. When high doses of heparin are introduced soon after surgery, small concerns during surgery may become big problems afterward.
Based on these results, it is reasonable to undertake device implantation without interruption of a vitamin K antagonist such as warfarin.
Patients on direct oral anticoagulants: The jury is still out
The similar BRUISE CONTROL-2 trial is currently under way, comparing interruption vs continuation of dabigatran for patients undergoing cardiac device surgery.
In Europe, surgeons are less concerned than those in the United States about operating while a patient is on anticoagulant therapy. But the safety of this practice is not backed by strong evidence.
Direct oral anticoagulants: Consider pharmacokinetics
Direct oral anticoagulants are potent and fast-acting, with a peak effect 1 to 3 hours after intake. This rapid anticoagulant action is similar to that of bridging with low-molecular-weight heparin, and caution is needed when administering direct oral anticoagulants, especially after major surgery or surgery with a high bleeding risk.
Frost et al20 compared the pharmacokinetics of apixaban (with twice-daily dosing) and rivaroxaban (once-daily dosing) and found that peak anticoagulant activity is faster and higher with rivaroxaban. This is important, because many patients will take their anticoagulant first thing in the morning. Consequently, if patients require any kind of procedure (including dental), they should skip the morning dose of the direct oral anticoagulant to avoid having the procedure done during the peak anticoagulant effect, and they should either not take that day’s dose or defer the dose until the evening after the procedure.
MANAGING SURGERY FOR PATIENTS ON A DIRECT ORAL ANTICOAGULANT
Case 3: An elderly woman on apixaban facing surgery
Let us imagine that our previous patient takes apixaban instead of warfarin. She is 75 years old, has atrial fibrillation, and is about to undergo elective colon resection for cancer. One doctor advises her to simply stop apixaban for 2 days, while another says she should go off apixaban for 5 days and will need bridging. Which plan is best?
In the perioperative setting, our goal is to interrupt patients’ anticoagulant therapy for the shortest time that results in no residual anticoagulant effect at the time of the procedure.
The European Society of Regional Anaesthesia and Pain Therapy and the American Society of Regional Anesthesia and Pain Medicine21 recommend an extended period of interruption of direct oral anticoagulants (Table 3)
They further recommend that if the risk of venous thromboembolism is high, low-molecular-weight heparin bridging should be done while stopping the direct oral anticoagulant, with the heparin discontinued 24 hours before the procedure. This recommendation seems counterintuitive, as it is advising replacing a short-acting anticoagulant with low-molecular-weight heparin, another short-acting anticoagulant.
The guidelines committee was unable to provide strength and grading of their recommendations, as too few well-designed studies are available to support them. The doctor in case 3 who advised stopping apixaban for 5 days and bridging is following the guidelines, but without much evidence to support this strategy.
Is bridging needed during interruption of a direct oral anticoagulant?
There are no randomized, controlled trials of bridging vs no bridging in patients taking direct oral anticoagulants. Substudies exist of patients taking these drugs for atrial fibrillation who had treatment interrupted for procedures, but the studies did not randomize bridging vs no bridging, nor were bridging regimens standardized. Three of the four atrial fibrillation trials had a blinded design (warfarin vs direct oral anticoagulants), making perioperative management difficult, as physicians did not know the pharmacokinetics of the drugs their patients were taking.22–24
We used the database from the Randomized Evaluation of Long-Term Anticoagulation Therapy (RE-LY) trial22 to evaluate bridging in patients taking either warfarin or dabigatran. With an open-label study design (the blinding was only for the 110 mg and 150 mg dabigatran doses), clinicians were aware of whether patients were receiving warfarin or dabigatran, thereby facilitating perioperative management. Among dabigatran-treated patients, those who were bridged had significantly more major bleeding than those not bridged (6.5% vs 1.8%, P < .001), with no difference between the groups for stroke or systemic embolism. Although it is not a randomized controlled trial, it does provide evidence that bridging may not be advisable for patients taking a direct oral anticoagulant.
The 2017 American College of Cardiology guidelines25 conclude that parenteral bridging is not indicated for direct oral anticoagulants. Although this is not based on strong evidence, the guidance appears reasonable according to the evidence at hand.
The 2017 American Heart Association Guidelines16 recommend a somewhat complex approach based on periprocedural bleeding risk and thromboembolic risk.
How long to interrupt direct oral anticoagulants?
Table 4 shows a simplified approach to interrupting direct oral anticoagulants that we use in Canada. The approach takes into account the type of surgery and kidney function for patients taking dabigatran, a drug that depends more on renal clearance than the other direct oral anticoagulants do.26
Evidence for this approach comes from a prospective cohort study27 of 541 patients being treated with dabigatran who were having an elective surgery or invasive procedure. Patients received standard perioperative management, with the timing of the last dabigatran dose before the procedure (24 hours, 48 hours, or 96 hours) based on the bleeding risk of surgery and the patient’s creatinine clearance. Dabigatran was resumed 24 to 72 hours after the procedure. No heparin bridging was done. Patients were followed for up to 30 days postoperatively. The results were favorable with few complications: one transient ischemic attack (0.2%), 10 major bleeding episodes (1.8%), and 28 minor bleeding episodes (5.2%).
A subgroup of 181 patients in this study28 had a plasma sample drawn just before surgery, allowing the investigators to assess the level of coagulation factors after dabigatran interruption. Results were as follows:
93% had a normal prothrombin time
80% had a normal activated partial thromboplastin time
33% had a normal thrombin time
81% had a normal dilute thrombin time.
The dilute thrombin time is considered the most reliable test of the anticoagulant effect of dabigatran but is not widely available. The activated partial thromboplastin time can provide a more widely used coagulation test to assess (in a less precise manner) whether there is an anticoagulant effect of dabigatran present, and more sensitive activated partial thromboplastin time assays can be used to better detect any residual dabigatran effect.
Dabigatran levels were also measured. Although 66% of patients had low drug levels just before surgery, the others still had substantial dabigatran on board. The fact that bleeding event rates were so low in this study despite the presence of dabigatran in many patients raises the question of whether having some drug on board is a good predictor of bleeding risk.
An interruption protocol with a longer interruption interval—12 to 14 hours longer than in the previous study (3 days for high-bleed risk procedures, 2 days for low-bleed risk procedures)—brought the activated partial thromboplastin time and dilute thrombin time to normal levels for 100% of patients with the protocol for high-bleeding-risk surgery. This study was based on small numbers and its interruption strategy needs further investigation.29
Case 3 continued
Based on the current empiric evidence, we recommend interrupting direct oral anticoagulants for 2 days (or approximately a 60-hour interval between the last dose and surgery) for this 75-year-old woman who is taking apixaban (Table 5). This interruption interval corresponds to 5 elimination half-lives for apixaban, which should result in little to no residual anticoagulant and will facilitate major surgery and, if indicated, neuraxial anesthesia.
The PAUSE study (NCT02228798), a multicenter, prospective cohort study, is designed to establish a safe, standardized protocol for the perioperative management of patients with atrial fibrillation taking dabigatran, rivaroxaban, or apixaban and will include 3,300 patients.
PATIENTS WITH A CORONARY STENT WHO NEED SURGERY
Case 4: A woman with a stent facing surgery
A 70-year-old woman needs breast cancer resection. She has coronary artery disease and had a drug-eluting stent placed 5 months ago after elective cardiac catheterization. She also has hypertension, obesity, and type 2 diabetes. Her medications include an angiotensin II receptor blocker, hydrochlorothiazide, insulin, and an oral hypoglycemic. She is also taking aspirin 81 mg daily and ticagrelor (a P2Y12 receptor antagonist) 90 mg twice daily.
Her cardiologist is concerned that stopping antiplatelet therapy could trigger acute stent thrombosis, which has a 50% or higher mortality rate.
Should she stop taking aspirin before surgery? What about the ticagrelor?
Is aspirin safe during surgery?
Evidence concerning aspirin during surgery comes from Perioperative Ischemic Evaluation 2 (POISE-2), a double-blind, randomized controlled trial.30 Patients who had known cardiovascular disease or risk factors for cardiovascular disease and were about to undergo noncardiac surgery were stratified according to whether they had been taking aspirin before the study (patients taking aspirin within 72 hours of the surgery were excluded from randomization). Participants in each group were randomized to take either aspirin or placebo just before surgery. The primary outcome was the combined rate of death or nonfatal myocardial infarction 30 days after randomization.
The study found no differences in the primary end point between the two groups. However, major bleeding occurred significantly more often in the aspirin group (4.6% vs 3.8%, hazard ratio 1.2, 95% confidence interval 1.0–1.5).
Moreover, only 4% of the patients in this trial had a cardiac stent. The trial excluded patients who had had a bare-metal stent placed within 6 weeks or a drug-eluting stent placed within 1 year, so it does not help us answer whether aspirin should be stopped for our current patient.
Is surgery safe for patients with stents?
The safety of undergoing surgery with a stent was investigated in a large US Veterans Administration retrospective cohort study.31 More than 20,000 patients with stents who underwent noncardiac surgery within 2 years of stent placement were compared with a control group of more than 41,000 patients with stents who did not undergo surgery. Patients were matched by stent type and cardiac risk factors at the time of stent placement.
The risk of an adverse cardiac event in both the surgical and nonsurgical cohorts was highest in the initial 6 weeks after stent placement and plateaued 6 months after stent placement, when the risk difference between the surgical and nonsurgical groups leveled off to 1%.
The risk of a major adverse cardiac event postoperatively was much more dependent on the timing of stent placement in complex and inpatient surgeries. For outpatient surgeries, the risk of a major cardiac event was very low and the timing of stent placement did not matter.
A Danish observational study32 compared more than 4,000 patients with drug-eluting stents having surgery to more than 20,000 matched controls without coronary heart disease having similar surgery. The risk of myocardial infarction or cardiac death was much higher for patients undergoing surgery within 1 month after drug-eluting stent placement compared with controls without heart disease and patients with stent placement longer than 1 month before surgery.
Our practice is to continue aspirin for surgery in patients with coronary stents regardless of the timing of placement. Although there is a small increased risk of bleeding, this must be balanced against thrombotic risk. We typically stop clopidogrel 5 to 7 days before surgery and ticagrelor 3 to 5 days before surgery. We may decide to give platelets before very-high-risk surgery (eg, intracranial, spinal) if there is a decision to continue both antiplatelet drugs—for example, in a patient who recently received a drug-eluting stent (ie, within 3 months). It is essential to involve the cardiologist and surgeon in these decisions.
BOTTOM LINE
Navigating the anticoagulant landscape in 2017 is complex. Doctors should review professional society guidelines while considering the strength of evidence on which they are based and tailor management to individual patient characteristics. Table 6 summarizes the management recommendations reviewed in this article.
References
Kearon C, Aki EA, Ornelas J, et al. Antithrombotic therapy for VTE disease: CHEST guideline and expert panel report. Chest 2016; 149:315–352.
Enden T, Haig Y, Klow NE, et al; CaVenT Study Group. Long-term outcome after additional catheter-directed thrombolysis versus standard treatment for acute iliofemoral deep vein thrombosis (the CaVenT study): a randomised controlled trial. Lancet 2012; 379:31–38.
Haig Y, Enden T, Grotta O, et al; CaVenT Study Group. Post-thrombotic syndrome after catheter-directed thrombolysis for deep vein thrombosis (CaVenT): 5-year follow-up results of an open-label, randomized controlled trial. Lancet Haematol 2016; 3:e64–e71.
Vedantham S, Goldhaber SZ, Kahn SR, et al. Rationale and design of the ATTRACT Study: a multicenter randomized trial to evaluate pharmacomechanical catheter-directed thrombolysis for the prevention of postthrombotic syndrome in patients with proximal deep vein thrombosis. Am Heart J 2013; 165:523–530.
Van Es N, Coppens M, Schulman S, Middeldorp S, Buller HR. Direct oral anticoagulants compared with vitamin K antagonists for acute venous thromboembolism: evidence from phase 3 trials. Blood 2014; 124:1968–1975.
Douketis JD, Spyropoulos AC, Kaatz S, et al; BRIDGE Investigators. Perioperative bridging anticoagulation in patients with atrial fibrillation. N Engl J Med 2015; 373:823–833.
Douketis J, Johnson JA, Turpie AG. Low-molecular-weight heparin as bridging anticoagulation during interruption of warfarin: assessment of a standardized periprocedural anticoagulation regimen. Arch Intern Med 2004; 164:1319–1326.
Dunn AS, Spyropoulos AC, Turpie AG. Bridging therapy in patients on long-term oral anticoagulants who require surgery: the Prospective Peri-operative Enoxaparin Cohort Trial (PROSPECT). J Thromb Haemost 2007; 5:2211–2218.
Kovacs MJ, Kearon C, Rodger M, et al. Single-arm study of bridging therapy with low-molecular-weight heparin for patients at risk of arterial embolism who require temporary interruption of warfarin. Circulation 2004; 110:1658–1663.
Spyropoulos AC, Turpie AG, Dunn AS, et al; REGIMEN Investigators. Clinical outcomes with unfractionated heparin or low-molecular-weight heparin as bridging therapy in patients on long-term oral anticoagulants: the REGIMEN registry. J Thromb Haemost 2006; 4:1246–1252.
Douketis JD, Woods K, Foster GA, Crowther MA. Bridging anticoagulation with low-molecular-weight heparin after interruption of warfarin therapy is associated with a residual anticoagulant effect prior to surgery. Thromb Haemost 2005; 94:528–531.
Schulman S, Hwang HG, Eikelboom JW, Kearon C, Pai M, Delaney J. Loading dose vs. maintenance dose of warfarin for reinitiation after invasive procedures: a randomized trial. J Thromb Haemost 2014; 12:1254-1259.
Siegal D, Yudin J, Kaatz S, Douketis JD, Lim W, Spyropoulos AC. Periprocedural heparin bridging in patients receiving vitamin K antagonists: systematic review and meta-analysis of bleeding and thromboembolic rates. Circulation 2012; 126:1630–1639.
Skeith L, Taylor J, Lazo-Langner A, Kovacs MJ. Conservative perioperative anticoagulation management in patients with chronic venous thromboembolic disease: a cohort study. J Thromb Haemost 2012; 10:2298–2304.
Douketis JD, Spyropoulos AC, Spencer FA, et al. Perioperative management of antithrombotic therapy: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest 2012; 141(2 suppl):e326S–e350S.
Doherty JU, Gluckman TJ, Hucker WJ, et al. 2017 ACC expert consensus decision pathway for periprocedural management of anticoagulation in patients with nonvalvular atrial fibrillation: a report of the American College of Cardiology Clinical Expert Consensus Document Task Force. J Am Coll Cardiol 2017; 69:871–898.
Raval AN, Cigarroa JE, Chung MK, et al; American Heart Association Clinical Pharmacology Subcommittee of the Acute Cardiac Care and General Cardiology Committee of the Council on Clinical Cardiology; Council on Cardiovascular Disease in the Young; and Council on Quality of Care and Outcomes Research. Management of patients on non-vitamin K antagonist oral anticoagulants in the acute care and periprocedural setting: a scientific statement from the American Heart Association. Circulation 2017; 135:e604–e633.
Tafur A, Douketis J. Perioperative anticoagulant management in patients with atrial fibrillation: practical implications of recent clinical trials. Pol Arch Med Wewn 2015; 125:666–671.
Birnie DH, Healey JS, Wells GA, et al: BRUISE CONTROL Investigators. Pacemaker or defibrillator surgery without interruption of anticoagulation. N Engl J Med 2013; 368:2084–2093.
Frost C, Song Y, Barrett YC, et al. A randomized direct comparison of the pharmacokinetics and pharmacodynamics of apixaban and rivaroxaban. Clin Pharmacol 2014; 6:179–187.
Narouze S, Benzon HT, Provenzano DA, et al. Interventional spine and pain procedures in patients on antiplatelet and anticoagulant medications: guidelines from the American Society of Regional Anesthesia and Pain Medicine, the European Society of Regional Anesthesia and Pain Therapy, the American Academy of Pain Medicine, the International Neuromodulation Society, the North American Neuromodulation Society, and the World institute of Pain. Reg Anesth Pain Med 2015; 40:182–212.
Douketis JD, Healey JS, Brueckmann M, et al. Perioperative bridging anticoagulation during dabigatran or warfarin interruption among patients who had an elective surgery or procedure. Substudy of the RE-LY trial. Thromb Haemost 2015; 113:625–632.
Steinberg BA, Peterson ED, Kim S, et al; Outcomes Registry for Better Informed Treatment of Atrial Fibrillation Investigators and Patients. Use and outcomes associated with bridging during anticoagulation interruptions in patients with atrial fibrillation: findings from the Outcomes Registry for Better Informed Treatment of Atrial Fibrillation (ORBIT-AF). Circulation 2015; 131:488–494.
Garcia D, Alexander JH, Wallentin L, et al. Management and clinical outcomes in patients treated with apixaban vs warfarin undergoing procedures. Blood 2014; 124:3692–3698.
Doherty JU, Gluckman TJ, Hucker WJ, et al. 2017 ACC expert consensus decision pathway for periprocedural management of anticoagulation in patients with nonvalvular atrial fibrillation: a report of the American College of Cardiology Clinical Expert Consensus Document Task Force. J Am Coll Cardiol 2017; 69:871–898.
Schulman S, Carrier M, Lee AY, et al; Periop Dabigatran Study Group. Perioperative management of dabigatran: a prospective cohort study. Circulation 2015; 132:167–173.
Douketis JD, Wang G, Chan N, et al. Effect of standardized perioperative dabigatran interruption on the residual anticoagulation effect at the time of surgery or procedure. J Thromb Haemost 2016; 14:89–97.
Douketis JD, Syed S, Schulman S. Periprocedural management of direct oral anticoagulants: comment on the 2015 American Society of Regional Anesthesia and Pain Medicine guidelines. Reg Anesth Pain Med 2016; 41:127–129.
Devereaux PJ, Mrkobrada M, Sessler DI, et al; POISE-2 Investigators. Aspirin in patients undergoing noncardiac surgery. N Engl J Med 2014; 370:1494–1503.
Holcomb CN, Graham LA, Richman JS, et al. The incremental risk of noncardiac surgery on adverse cardiac events following coronary stenting. J Am Coll Cardiol 2014; 64:2730–2739.
Egholm G, Kristensen SD, Thim T, et al. Risk associated with surgery within 12 months after coronary drug-eluting stent implantation. J Am Coll Cardiol 2016; 68:2622–2632.
References
Kearon C, Aki EA, Ornelas J, et al. Antithrombotic therapy for VTE disease: CHEST guideline and expert panel report. Chest 2016; 149:315–352.
Enden T, Haig Y, Klow NE, et al; CaVenT Study Group. Long-term outcome after additional catheter-directed thrombolysis versus standard treatment for acute iliofemoral deep vein thrombosis (the CaVenT study): a randomised controlled trial. Lancet 2012; 379:31–38.
Haig Y, Enden T, Grotta O, et al; CaVenT Study Group. Post-thrombotic syndrome after catheter-directed thrombolysis for deep vein thrombosis (CaVenT): 5-year follow-up results of an open-label, randomized controlled trial. Lancet Haematol 2016; 3:e64–e71.
Vedantham S, Goldhaber SZ, Kahn SR, et al. Rationale and design of the ATTRACT Study: a multicenter randomized trial to evaluate pharmacomechanical catheter-directed thrombolysis for the prevention of postthrombotic syndrome in patients with proximal deep vein thrombosis. Am Heart J 2013; 165:523–530.
Van Es N, Coppens M, Schulman S, Middeldorp S, Buller HR. Direct oral anticoagulants compared with vitamin K antagonists for acute venous thromboembolism: evidence from phase 3 trials. Blood 2014; 124:1968–1975.
Douketis JD, Spyropoulos AC, Kaatz S, et al; BRIDGE Investigators. Perioperative bridging anticoagulation in patients with atrial fibrillation. N Engl J Med 2015; 373:823–833.
Douketis J, Johnson JA, Turpie AG. Low-molecular-weight heparin as bridging anticoagulation during interruption of warfarin: assessment of a standardized periprocedural anticoagulation regimen. Arch Intern Med 2004; 164:1319–1326.
Dunn AS, Spyropoulos AC, Turpie AG. Bridging therapy in patients on long-term oral anticoagulants who require surgery: the Prospective Peri-operative Enoxaparin Cohort Trial (PROSPECT). J Thromb Haemost 2007; 5:2211–2218.
Kovacs MJ, Kearon C, Rodger M, et al. Single-arm study of bridging therapy with low-molecular-weight heparin for patients at risk of arterial embolism who require temporary interruption of warfarin. Circulation 2004; 110:1658–1663.
Spyropoulos AC, Turpie AG, Dunn AS, et al; REGIMEN Investigators. Clinical outcomes with unfractionated heparin or low-molecular-weight heparin as bridging therapy in patients on long-term oral anticoagulants: the REGIMEN registry. J Thromb Haemost 2006; 4:1246–1252.
Douketis JD, Woods K, Foster GA, Crowther MA. Bridging anticoagulation with low-molecular-weight heparin after interruption of warfarin therapy is associated with a residual anticoagulant effect prior to surgery. Thromb Haemost 2005; 94:528–531.
Schulman S, Hwang HG, Eikelboom JW, Kearon C, Pai M, Delaney J. Loading dose vs. maintenance dose of warfarin for reinitiation after invasive procedures: a randomized trial. J Thromb Haemost 2014; 12:1254-1259.
Siegal D, Yudin J, Kaatz S, Douketis JD, Lim W, Spyropoulos AC. Periprocedural heparin bridging in patients receiving vitamin K antagonists: systematic review and meta-analysis of bleeding and thromboembolic rates. Circulation 2012; 126:1630–1639.
Skeith L, Taylor J, Lazo-Langner A, Kovacs MJ. Conservative perioperative anticoagulation management in patients with chronic venous thromboembolic disease: a cohort study. J Thromb Haemost 2012; 10:2298–2304.
Douketis JD, Spyropoulos AC, Spencer FA, et al. Perioperative management of antithrombotic therapy: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest 2012; 141(2 suppl):e326S–e350S.
Doherty JU, Gluckman TJ, Hucker WJ, et al. 2017 ACC expert consensus decision pathway for periprocedural management of anticoagulation in patients with nonvalvular atrial fibrillation: a report of the American College of Cardiology Clinical Expert Consensus Document Task Force. J Am Coll Cardiol 2017; 69:871–898.
Raval AN, Cigarroa JE, Chung MK, et al; American Heart Association Clinical Pharmacology Subcommittee of the Acute Cardiac Care and General Cardiology Committee of the Council on Clinical Cardiology; Council on Cardiovascular Disease in the Young; and Council on Quality of Care and Outcomes Research. Management of patients on non-vitamin K antagonist oral anticoagulants in the acute care and periprocedural setting: a scientific statement from the American Heart Association. Circulation 2017; 135:e604–e633.
Tafur A, Douketis J. Perioperative anticoagulant management in patients with atrial fibrillation: practical implications of recent clinical trials. Pol Arch Med Wewn 2015; 125:666–671.
Birnie DH, Healey JS, Wells GA, et al: BRUISE CONTROL Investigators. Pacemaker or defibrillator surgery without interruption of anticoagulation. N Engl J Med 2013; 368:2084–2093.
Frost C, Song Y, Barrett YC, et al. A randomized direct comparison of the pharmacokinetics and pharmacodynamics of apixaban and rivaroxaban. Clin Pharmacol 2014; 6:179–187.
Narouze S, Benzon HT, Provenzano DA, et al. Interventional spine and pain procedures in patients on antiplatelet and anticoagulant medications: guidelines from the American Society of Regional Anesthesia and Pain Medicine, the European Society of Regional Anesthesia and Pain Therapy, the American Academy of Pain Medicine, the International Neuromodulation Society, the North American Neuromodulation Society, and the World institute of Pain. Reg Anesth Pain Med 2015; 40:182–212.
Douketis JD, Healey JS, Brueckmann M, et al. Perioperative bridging anticoagulation during dabigatran or warfarin interruption among patients who had an elective surgery or procedure. Substudy of the RE-LY trial. Thromb Haemost 2015; 113:625–632.
Steinberg BA, Peterson ED, Kim S, et al; Outcomes Registry for Better Informed Treatment of Atrial Fibrillation Investigators and Patients. Use and outcomes associated with bridging during anticoagulation interruptions in patients with atrial fibrillation: findings from the Outcomes Registry for Better Informed Treatment of Atrial Fibrillation (ORBIT-AF). Circulation 2015; 131:488–494.
Garcia D, Alexander JH, Wallentin L, et al. Management and clinical outcomes in patients treated with apixaban vs warfarin undergoing procedures. Blood 2014; 124:3692–3698.
Doherty JU, Gluckman TJ, Hucker WJ, et al. 2017 ACC expert consensus decision pathway for periprocedural management of anticoagulation in patients with nonvalvular atrial fibrillation: a report of the American College of Cardiology Clinical Expert Consensus Document Task Force. J Am Coll Cardiol 2017; 69:871–898.
Schulman S, Carrier M, Lee AY, et al; Periop Dabigatran Study Group. Perioperative management of dabigatran: a prospective cohort study. Circulation 2015; 132:167–173.
Douketis JD, Wang G, Chan N, et al. Effect of standardized perioperative dabigatran interruption on the residual anticoagulation effect at the time of surgery or procedure. J Thromb Haemost 2016; 14:89–97.
Douketis JD, Syed S, Schulman S. Periprocedural management of direct oral anticoagulants: comment on the 2015 American Society of Regional Anesthesia and Pain Medicine guidelines. Reg Anesth Pain Med 2016; 41:127–129.
Devereaux PJ, Mrkobrada M, Sessler DI, et al; POISE-2 Investigators. Aspirin in patients undergoing noncardiac surgery. N Engl J Med 2014; 370:1494–1503.
Holcomb CN, Graham LA, Richman JS, et al. The incremental risk of noncardiac surgery on adverse cardiac events following coronary stenting. J Am Coll Cardiol 2014; 64:2730–2739.
Egholm G, Kristensen SD, Thim T, et al. Risk associated with surgery within 12 months after coronary drug-eluting stent implantation. J Am Coll Cardiol 2016; 68:2622–2632.
Venous thromboembolism has a myriad of clinical presentations, warranting a holistic management approach that incorporates multiple antithrombotic management strategies.
A direct oral anticoagulant is an acceptable treatment option in patients with submassive venous thromboembolism, whereas catheter-directed thrombolysis should be considered in patients with iliofemoral deep vein thrombosis, and low-molecular-weight heparin in patients with cancer-associated thrombosis.
Perioperative management of direct oral anticoagulants should be based on the pharmacokinetic properties of the drug, the patient’s renal function, and the risk of bleeding posed by the surgery or procedure.
Perioperative heparin bridging can be avoided in most patients who have atrial fibrillation or venous thromboembolism, but should be considered in most patients with a mechanical heart valve.
Disallow All Ads
Content Gating
No Gating (article Unlocked/Free)
Alternative CME
Disqus Comments
Default
Consolidated Pubs: Do Not Show Source Publication Logo
Since 2008, the US Food and Drug Administration (FDA) has required new diabetes drugs to demonstrate cardiovascular safety, resulting in large and lengthy clinical trials. Under the new regulations, several dipeptidyl peptidase-4 (DPP-4) inhibitors, sodium-glucose cotransporter-2 (SGLT-2) inhibitors, and glucagon-like peptide-1 (GLP-1) receptor agonists have demonstrated cardiovascular safety, with some demonstrating superior cardiovascular efficacy. In 2016, the SGLT-2 inhibitor empagliflozin became the first (and as of this writing, the only) diabetes drug approved by the FDA for a clinical outcome indication, ie, to reduce the risk of cardiovascular death.
DIABETES DRUG DEVELOPMENT
Changing priorities
The International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use (ICH) was formed in 1990 as a collaborative effort across global regulatory agencies and coordinated by the World Health Organization to universalize criteria for drug development. The ICH standards for type 2 diabetes drug development included the following requirements for patient exposure to investigational products to satisfy new drug application requirements:
1,500 individuals total (including single-dose exposure)
300–600 patients for 6 months
100 patients for 1 year.
Thus, just 250 patient-years of exposure were needed for approval of a drug that patients might take for decades. These standards were unlikely to reveal rare, serious complications and had no ability to assess clinical outcomes efficacy for either microvascular or macrovascular disease complications.
When the ICH regulatory standards were set in the early 1990s, only insulin and sulfonylureas were available in the United States. (Metformin had been available outside the United States since the 1950s.) Since 1990, the prevalence of type 2 diabetes in the United States has increased from around 2% to now over 10% of the US adult population. This increase, along with the known increased risk of atherosclerotic cardiovascular disease and heart failure associated with diabetes, created a sense of urgency for developing new therapies. With a burgeoning population with or at risk of diabetes, new drugs were needed and were rapidly developed.
Since 1995, when metformin was approved in the United States, a new class of antihyperglycemic medication has been approved about once every 2 years, so that by 2008, 12 classes of medications had become available for the treatment of type 2 diabetes. This extraordinary rate of drug development has now yielded more classes of medications to treat type 2 diabetes than we presently have for the treatment of hypertension.
This proliferation of new treatments resolved much of the pressure of the unmet medical need, over a period of increasing awareness of the cardiovascular complications of type 2 diabetes, along with numerous examples of adverse cardiovascular effects observed with some of the drugs. In this context, the FDA (and in parallel the European Medicines Agency) made paradigm-shifting changes in the requirements for the development of new type 2 diabetes drugs, requiring large-scale randomized clinical outcome data to assess cardiovascular safety of the new drugs. In December 2008, the FDA published a Guidance for Industry,1 recommending that sponsors of new drugs for type 2 diabetes demonstrate that therapy would not only improve glucose control, but also that it would, at a minimum, not result in an unacceptable increase in cardiovascular risk.1 To better assess new diabetes drugs, the requirement for patient-years of exposure to the studied drug was increased by over 60-fold from 250 patient-years to more than 15,000.
INCRETIN MODULATORS
The incretin system, a regulator of postprandial glucose metabolism, is an attractive target for glycemic control, as it promotes early satiety and lowers blood glucose.
After a meal, endocrine cells in the distal small intestine secrete the incretin hormones GLP-1 and gastric inhibitory polypeptide (GIP), among others, which reduce gastric motility, stimulate the pancreas to augment glucose-appropriate insulin secretion, and decrease postprandial glucagon release. GLP-1 also interacts with the satiety center of the hypothalamus, suppressing appetite. GLP-1 and GIP are rapidly inactivated by the circulating protease DPP-4. Injectable formulations of GLP-1 receptor agonists that are resistant to DPP-4 degradation have been developed.
Ten incretin modulators are now available in the United States. The 4 available DPP-4 inhibitors are all once-daily oral medications, and the 6 GLP-1 receptor agonists are all injectable (Table 1).
Small studies in humans and animals suggest that DPP-4 inhibitors and GLP-1-receptor agonists may have multiple favorable effects on the cardiovascular system independent of their glycemic effects. These include reducing myocardial infarct size,2–5 improving endothelial function,6 reducing inflammation and oxidative stress,7 reducing atherosclerotic plaque volume,8 improving left ventricular function, 9,10 and lowering triglyceride levels.11 However, large clinical trials are needed to determine clinical effectiveness.
DPP-4 INHIBITORS: NOT INFERIOR TO PLACEBO
Saxagliptin
Saxagliptin, a DPP-4 inhibitor, was found in a meta-analysis of phase 2B and early phase 3 trial data involving almost 5,000 patients to be associated with a dramatic 56% relative risk reduction in cardiovascular death, heart attack, and stroke. However, this analysis was limited by the extremely low number of events to analyze, with only 41 total patients with cardiovascular events in that dataset.12
The SAVOR-TIMI 53 trial13 subsequently compared saxagliptin and placebo in a randomized, double-blind trial conducted in 26 countries with nearly 16,500 patients with type 2 diabetes. All patients continued their conventional diabetes treatment at the discretion of their physicians.
During an average follow-up of 2 years, 1,222 events of cardiovascular death, myocardial infarction, or stroke occurred. No significant difference in event rates was found between the saxagliptin and placebo groups. This did not demonstrate the expected cardiovascular benefit based on prior meta-analysis of phase 2B and phase 3 data presented above, but saxagliptin did not increase cardiovascular risk and was the first diabetes drug to earn this distinction of robustly statistically proven cardiovascular safety.
Further analysis of the SAVOR-TIMI 53 trial data revealed a 27% increased relative risk of heart failure hospitalization with saxagliptin compared with placebo.14 Although the risk was statistically significant, the absolute difference in heart failure incidence between the drug and placebo groups was only 0.7% (3.5% vs 2.8%, respectively). As the average follow-up in the trial was 2 years, the absolute incremental risk of heart failure seen with saxagliptin is 0.35% annually—almost identical in magnitude to the increased heart failure risk with pioglitazone. The increased risk of heart failure was seen within the first 6 months of the trial and persisted throughout the trial, indicating an increased up-front risk of heart failure.
Alogliptin
The EXAMINE trial15 compared the DPP-4 inhibitor alogliptin and placebo in 5,380 patients with type 2 diabetes who had had a recent acute coronary event.15 Over the 30 months of the trial, more than 600 primary outcome events of cardiovascular death, myocardial infarction, or stroke occurred, with no significant difference between drug and placebo groups with established nominal statistical noninferiority. A numerically higher incidence of heart failure was noted in patients who received alogliptin than with placebo, but the difference was not statistically significant.16 However, this study was not powered to detect such an increased risk. In patients entering the trial with no history of heart failure, the risk of hospitalization for heart failure was 76% higher in the alogliptin group than in the placebo group, with a nominally significant P value less than .05 in this subgroup.
These analyses led the FDA in 2016 to mandate label warnings for saxagliptin and alogliptin regarding the increased risk of heart failure.17
Sitagliptin
The TECOS trial18 tested the DPP-4 inhibitor sitagliptin and, unlike the SAVOR or EXAMINE trials, included hospitalization for unstable angina in the composite end point. Nearly 15,000 patients with type 2 diabetes and established cardiovascular disease were enrolled, and almost 2,500 events occurred. No significant difference was found between the 2 groups.
In a series of analyses prospectively planned, sitagliptin was not associated with an increased risk of hospitalization for heart failure.19 But despite these robust analyses demonstrating no incremental heart failure risk with sitagliptin, in August 2017, the US product label for sitagliptin was modified to include a warning that other DPP-4 inhibitors have been associated with heart failure and to suggest caution. The label for linagliptin had the same FDA-required changes, with no data yet available from outcomes trials with linagliptin.
GLP-1 RECEPTOR AGONISTS
Lixisenatide: Noninferior to placebo
The ELIXA trial20 assessed the cardiovascular safety of the GLP-1 receptor agonist lixisenatide in patients with type 2 diabetes who recently had an acute coronary event. The study enrolled 6,068 patients from 49 countries, and nearly 1,000 events (cardiovascular death, myocardial infarction, stroke, or unstable angina) occurred during the median 25 months of the study. Results showed lixisenatide did not increase or decrease cardiovascular events or adverse events when compared with placebo.
Liraglutide: Evidence of benefit
The LEADER trial21 randomized 9,340 patients with or at increased risk for cardiovascular disease to receive the injectable GLP-1 receptor agonist liraglutide or placebo. After a median of 3.8 years of follow-up, liraglutide use was associated with a statistically significant 13% relative reduction in major adverse cardiovascular events, mostly driven by a 22% reduction in cardiovascular death.
Semaglutide: Evidence of benefit
The SUSTAIN-6 trial22 found a statistically significant 26% relative risk reduction in cardiovascular outcomes comparing once-weekly semaglutide (an injectable GLP-1 receptor agonist) and placebo in 3,297 patients with type 2 diabetes and established cardiovascular disease, chronic kidney disease, or risk factors for cardiovascular disease. The significant reduction in the incidence of nonfatal stroke with semaglutide was the main driver of the observed benefit.
Taspoglutide: Development halted
Taspoglutide was a candidate GLP-1 receptor agonist that underwent clinical trials for cardiovascular outcomes planned to involve about 8,000 patients. The trials were stopped early and drug development was halted after about 600 patient-years of exposure because of antibody formation in about half of patients exposed to taspoglutide, with anaphylactoid reactions and anaphylaxis reported.23
SGLT-2 INHIBITORS
The renal glomeruli filter about 180 g of glucose every day in normal adults; nearly all of it is reabsorbed by SGLT-2 in the proximal tubules, so that very little glucose is excreted in the urine.24–26 The benign condition hereditary glucosuria occurs due to loss-of-function mutations in the gene for SGLT-2. Individuals with this condition rarely if ever develop type 2 diabetes or obesity, and this observation led pharmaceutical researchers to probe SGLT-2 as a therapeutic target.
Inhibitors of SGLT-2 block glucose reabsorption in the renal proximal tubules and lead to glucosuria. Patients treated with an SGLT-2 inhibitor have lower serum glucose levels and lose weight. Inhibitors also reduce sodium reabsorption via SGLT-2 and lead to increased sodium excretion and decreased blood pressure.27
Three SGLT-2 antagonists are available in the United States: canagliflozin, dapagliflozin, and empagliflozin (Table 1). Ertugliflozin is currently in a phase 3B trial, and cardiovascular outcomes trials are in the planning phase for sotagliflozin, a dual SGLT-1/SGLT-2 inhibitor with SGLT-1 localized to the gastrointestinal tract.28
Empaglifozin: Evidence of benefit
The EMPA-REG OUTCOME trial29 randomized more than 7,200 patients with type 2 diabetes and atherosclerotic vascular disease to receive the SGLT-2 inhibitor empagliflozin or placebo as once-daily tablets, with both groups receiving off-study treatment for glycemic control at the discretion of their own care providers. Two doses of empagliflozin were evaluated in the trial (10 and 25 mg per day), with the 2 dosing groups pooled for all analyses as prospectively planned.
Patients taking empagliflozin had a 14% relative risk reduction of the composite outcome (cardiovascular death, myocardial infarction, and stroke) vs placebo, with no difference in effect between the 2 randomized doses. The improvement in the composite outcome was seen early in the empagliflozin group and persisted for the 4 years of the study.
This was the first trial of newly developed diabetes drugs that showed a statistically significant reduction in cardiovascular risk. The study revealed a 38% relative risk reduction in cardiovascular death in the treatment group. The risk reduction occurred early in the trial and improved throughout the duration of the study. This is a dramatic finding, unequaled even in trials of drugs that specifically target cardiovascular disease. Both doses of empagliflozin studied provided similar benefit over placebo, reinforcing the validity of the findings. Interestingly, in the empagliflozin group, there was a 35% relative risk reduction in heart failure hospitalizations.
Canaglifozin: Evidence of benefit
The CANVAS Program consisted of two sister trials, CANVAS and CANVAS-R, and examined the safety and efficacy of canagliflozin.30 More than 10,000 participants with type 2 diabetes and atherosclerotic disease or at increased risk of cardiovascular disease were randomized to receive canagliflozin or placebo. Canagliflozin led to a 14% relative risk reduction in the composite outcome of cardiovascular death, nonfatal myocardial infarction, or nonfatal stroke, but there was a statistically significant doubling in the incidence of amputations. Unlike empagliflozin, canagliflozin did not demonstrate a significant reduction in death from cardiovascular causes, suggesting that this may not be a class effect of SGLT-2 inhibitors. As with empagliflozin, canagliflozin led to a 33% relative risk reduction in heart failure hospitalizations.
Cardiovascular benefits independent of glucose-lowering
The cardiovascular benefits of empagliflozin in EMPA-REG OUTCOME and canagliflozin in CANVAS were observed early, suggesting that the mechanism may be due to the direct effects on the cardiovascular system rather than glycemic modification.
Improved glycemic control with the SGLT-2 inhibitor was seen early in both studies, but with the trials designed for glycemic equipoise encouraging open-label therapy targeting hemoglobin A1c to standard-of-care targets in both groups, the contrast in hemoglobin A1c between groups diminished throughout the trial after its first assessment. Although hemoglobin A1c levels in the SGLT-2 inhibitor groups decreased in the first 12 weeks, they increased over time nearly to the level seen in the placebo group. The adjusted mean hemoglobin A1c level in the placebo groups remained near 8.0% throughout the studies, a target consistent with guidelines from the American Diabetes Association and the European Association for the Study of Diabetes31 for the high-risk populations recruited and enrolled.
Blood pressure reduction and weight loss do not explain cardiovascular benefits
SGLT-2 inhibitors lower blood pressure independent of their diuretic effects. In the EMPA-REG OUTCOME trial, the adjusted mean systolic blood pressure was 3 to 4 mm Hg lower in the treatment groups than in the placebo group throughout the trial.29 This level of blood pressure lowering translates to an estimated 10% to 12% relative risk reduction for major adverse cardiovascular events, including heart failure. Although the risk reduction from blood pressure lowering is not insignificant, it does not explain the 38% reduction in cardiovascular deaths seen in the trial. Canagliflozin led to a similar 4-mm Hg reduction in systolic pressure compared with the placebo group.30
Weight loss was seen with both empagliflozin and canagliflozin but was not dramatic and is unlikely to account for the described cardiovascular benefits.
Theories of cardiovascular benefit
Several mechanisms have been proposed to help explain the observed cardiovascular benefits of SGLT-2 inhibitors.32
Ketone-body elevation. Ferrannini et al33 found that the blood concentration of the ketone-body beta-hydroxybutyrate is about twice as high in patients with type 2 diabetes in the fasting state who are chronically taking empagliflozin as in patients not receiving the drug. Beta-hydroxybutyrate levels peak after a meal and then return to baseline over several hours before rising again during the fasting period. Although the ketone elevation is not nearly as extreme as in diabetic ketoacidosis (about a 1,000-fold increase), the observed increase may reduce myocardial oxygen demand, as beta-hydroxybutyrate is among the most efficient metabolic substrates for the myocardium.
Red blood cell expansion. Perhaps a more likely explanation of the cardiovascular benefit seen with SGLT-2 inhibitor therapy is the increase in hemoglobin and hematocrit levels. At first attributed to hemoconcentration secondary to diuresis, this has been disproven by a number of studies. The EMPA-REG OUTCOME trial29 found that within 12 weeks of exposure to empagliflozin, hematocrit levels rose nearly 4% absolutely compared with the levels in the placebo group. This increase is equivalent to transfusing a unit of red blood cells, favorably affecting myocardial oxygen supply.
Reduction in glomerular hypertension. The kidneys regulate glomerular filtration in a process involving the macula densa, an area of specialized cells in the juxtaglomerular apparatus in the loop of Henle that responds to sodium concentration in the urine. Normally, SGLT-2 receptors upstream from the loop of Henle reabsorb sodium and glucose into the bloodstream, reducing sodium delivery to the macula densa, which senses this as a low-volume state. The macula densa cells respond by releasing factors that dilate afferent arterioles and increase glomerular filtration. People with diabetes have more glucose to reabsorb and therefore also reabsorb more sodium, leading to glomerular hypertension.
SGLT-2 inhibitors block both glucose and sodium reuptake at SGLT-2 receptors, normalizing the response at the macula densa, restoring a normal glomerular filtration rate, and alleviating glomerular hypertension. As the kidney perceives a more normal volume status, renin-angiotensin-aldosterone stimulation is attenuated and sympathetic nervous system activity improves.27,34 If this model of SGLT-2 inhibitor effects on the kidney is correct, these drugs have similar effects as angiotensin-converting enzyme (ACE) inhibitors, angiotensin II receptor blockers (ARBs), mineralocorticoid antagonists, and beta-blockers combined.
Kidney benefits
Empagliflozin35 and canagliflozin30 both reduced the rate of progression of kidney dysfunction and led to fewer clinically relevant renal events compared with placebo. Treatment and placebo groups also received standard care, so many patients were treated with renin-angiotensin-aldosterone system inhibitors and with good blood pressure control, making the finding that SGLT-2 inhibitors had a significant beneficial effect even more dramatic. Beneficial effects on markers of kidney function were seen early on, suggesting a more favorable hemodynamic effect on the kidney rather than improved glycemic control attenuating microvascular disease.
Empagliflozin approved to reduce clinical events
In December 2016, the FDA approved the indication for empagliflozin to reduce the risk of cardiovascular death in patients with type 2 diabetes,36 the first-ever clinical outcome indication for a type 2 diabetes medication. The European Society of Cardiology guidelines now include empagliflozin as preferred therapy for type 2 diabetes, recommending it to prevent the onset of heart failure and prolong life.37 This recommendation goes beyond the evidence from the EMPA-REG OUTCOME trial on which it is based, as the trial only studied patients with known atherosclerotic vascular disease.
The 2016 European Guidelines on cardiovascular disease prevention also recommend that an SGLT-2 inhibitor be considered early for patients with type 2 diabetes and cardiovascular disease to reduce cardiovascular and total mortality.38 The American Diabetes Association in their 2017 guidelines also endorse empagliflozin for treating patients with type 2 diabetes and cardiovascular disease.39 The fact that the American Diabetes Association recommendation is not based on glycemic control, in line with the product-labeled indication, is a major shift in the association’s guidance.
Cautions with SGLT-2 inhibitors
Use SGLT-2 inhibitors in patients with low blood pressure with caution, and with increased blood pressure monitoring just following initiation.
Consider modifying antihypertensive drugs in patients with labile blood pressure.
Consider stopping or reducing background diuretics when starting an SGLT-2 inhibitor, and reassess volume status after 1 to 2 weeks.
For patients on insulin, sulfonylureas, or both, consider decreasing dosages when starting an SGLT-2 inhibitor, and reassess glycemic control periodically.
Counsel patients about urinary hygiene. Although bacterial urinary tract infections have not emerged as a problem, fungal genital infections have, particularly in women and uncircumcised men.
Consider SGLT-2 inhibitors to be “sick-day” medications. Patients with diabetes must adjust their diabetes medications if their oral intake is reduced for a day or more, such as while sick or fasting. SGLT-2 inhibitors should not be taken on these days. Cases of diabetic ketoacidosis have arisen in patients who reduced oral intake while continuing their SGLT-2 inhibitor.
OTHER DRUGS WITH DEVELOPMENT HALTED
Aleglitazar, a peroxisome proliferator-activated receptor agonist taken orally once daily, raised high expectations when it was found in early studies to lower serum triglycerides and raise high-density lipoprotein cholesterol levels in addition to lowering blood glucose. However, a phase 3 trial in more than 7,000 patients was terminated after a median follow up of 2 years because of increased rates of heart failure, worsened kidney function, bone fractures, and gastrointestinal bleeding.40 Development of this drug was stopped.
Fasiglifam, a G-protein-coupled receptor 40 agonist, was tested in a cardiovascular clinical outcomes trial. Compared with placebo, fasiglifam reduced hemoglobin A1c levels with low risk of hypoglycemia.41 However, safety concerns about increased liver enzyme levels led to the cessation of the drug’s development.42
HOW WILL THIS AFFECT DIABETES MANAGEMENT?
Metformin is still the most commonly prescribed drug for type 2 diabetes but has only marginal evidence for its cardiovascular benefits and may not be the first-line therapy for the management of diabetes in the future. In the EMPA REG OUTCOME, LEADER, and SUSTAIN-6 trials, the novel diabetes medications were given to patients who were already treated with available therapies, often including metformin. Treatment with empagliflozin, liraglutide, and semaglutide may be indicated for patients with diabetes and atherosclerotic vascular disease as first-line therapies in the future.
SGLT-2 inhibitor therapy can cost about $500 per month, and GLP-1 inhibitors are only slightly less expensive. The cost may be prohibitive for many patients. As more evidence, guidelines, and FDA criteria support the use of these novel diabetes drugs, third-party payers and pharmaceutical companies may be motivated to lower costs to help reach more patients who can benefit from these therapies.
References
US Food and Drug Administration. Guidance for industry. Diabetes mellitus—evaluating cardiovascular risk in new antidiabetic therapies to treat type 2 diabetes. www.fda.gov/downloads/Drugs/.../Guidances/ucm071627.pdf. Accessed September 1, 2017.
Ye Y, Keyes KT, Zhang C, Perez-Polo JR, Lin Y, Birnbaum Y. The myocardial infarct size-limiting effect of sitagliptin is PKA-dependent, whereas the protective effect of pioglitazone is partially dependent on PKA. Am J Physiol Heart Circ Physiol 2010; 298:H1454–H1465.
Hocher B, Sharkovska Y, Mark M, Klein T, Pfab T. The novel DPP-4 inhibitors linagliptin and BI 14361 reduce infarct size after myocardial ischemia/reperfusion in rats. Int J Cardiol 2013; 167:87–93.
Woo JS, Kim W, Ha SJ, et al. Cardioprotective effects of exenatide in patients with ST-segment-elevation myocardial infarction undergoing primary percutaneous coronary intervention: results of exenatide myocardial protection in revascularization study. Arterioscler Thromb Vasc Biol 2013; 33:2252–2260.
Lønborg J, Vejlstrup N, Kelbæk H, et al. Exenatide reduces reperfusion injury in patients with ST-segment elevation myocardial infarction. Eur Heart J 2012; 33:1491–1499.
van Poppel PC, Netea MG, Smits P, Tack CJ. Vildagliptin improves endothelium-dependent vasodilatation in type 2 diabetes. Diabetes Care 2011; 34:2072–2077.
Kröller-Schön S, Knorr M, Hausding M, et al. Glucose-independent improvement of vascular dysfunction in experimental sepsis by dipeptidyl-peptidase 4 inhibition. Cardiovasc Res 2012; 96:140–149.
Ta NN, Schuyler CA, Li Y, Lopes-Virella MF, Huang Y. DPP-4 (CD26) inhibitor alogliptin inhibits atherosclerosis in diabetic apolipoprotein E-deficient mice. J Cardiovasc Pharmacol 2011; 58:157–166.
Sauvé M, Ban K, Momen MA, et al. Genetic deletion or pharmacological inhibition of dipeptidyl peptidase-4 improves cardiovascular outcomes after myocardial infarction in mice. Diabetes 2010; 59:1063–1073.
Read PA, Khan FZ, Heck PM, Hoole SP, Dutka DP. DPP-4 inhibition by sitagliptin improves the myocardial response to dobutamine stress and mitigates stunning in a pilot study of patients with coronary artery disease. Circ Cardiovasc Imaging 2010; 3:195–201.
Matikainen N, Mänttäri S, Schweizer A, et al. Vildagliptin therapy reduces postprandial intestinal triglyceride-rich lipoprotein particles in patients with type 2 diabetes. Diabetologia 2006; 49:2049–2057.
Frederich R, Alexander JH, Fiedorek FT, et al. A systematic assessment of cardiovascular outcomes in the saxagliptin drug development program for type 2 diabetes. Postgrad Med 2010; 122:16–27.
Scirica BM, Bhatt DL, Braunwald E, et al; SAVOR-TIMI 53 Steering Committee and Investigators. Saxagliptin and cardiovascular outcomes in patients with type 2 diabetes mellitus. N Engl J Med 2013; 369:1317–1326.
Scirica BM, Braunwald E, Raz I, et al; SAVOR-TIMI 53 Steering Committee and Investigators. Heart failure, saxagliptin, and diabetes mellitus: observations from the SAVOR-TIMI 53 randomized trial. Circulation 2014; 130:1579–1588.
White WB, Cannon CP, Heller SR, et al; EXAMINE Investigators. Alogliptin after acute coronary syndrome in patients with type 2 diabetes. N Engl J Med 2013; 369:1327–1335.
Zannad F, Cannon CP, Cushman WC, et al; EXAMINE Investigators. Heart failure and mortality outcomes in patients with type 2 diabetes taking alogliptin versus placebo in EXAMINE: a multicentre, randomised, double-blind trial. Lancet 2015; 385:2067–2076.
Green JB, Bethel MA, Armstrong PW, et al; TECOS Study Group. Effect of sitagliptin on cardiovascular outcomes in type 2 diabetes. N Engl J Med 2015; 373:232–242.
McGuire DK, Van de Werf F, Armstrong PW, et al; Trial Evaluating Cardiovascular Outcomes With Sitagliptin (TECOS) Study Group. Association between sitagliptin use and heart failure hospitalization and related outcomes in type 2 diabetes mellitus: secondary analysis of a randomized clinical trial. JAMA Cardiol 2016; 1:126–135.
Pfeffer MA, Claggett B, Diaz R, et al; ELIXA Investigators. Lixisenatide in patients with type 2 diabetes and acute coronary syndrome. N Engl J Med 2015; 373:2247–2257.
Marso SP, Daniels GH, Brown-Frandsen K, et al; LEADER Steering Committee; LEADER Trial Investigators. Liraglutide and cardiovascular outcomes in type 2 diabetes. N Engl J Med 2016; 375:311–322.
Marso SP, Bain SC, Consoli A, et al; SUSTAIN-6 Investigators. Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N Engl J Med 2016; 375:1834–1844.
Rosenstock J, Balas B, Charbonnel B, et al; T-EMERGE 2 Study Group. The fate of taspoglutide, a weekly GLP-1 receptor agonist, versus twice-daily exenatide for type 2 diabetes: the T-EMERGE 2 trial. Diabetes Care 2013; 36:498–504.
Wright EM. Renal Na(+)-glucose cotransporters. Am J Physiol 2001; 280:F10–F18.
Lee YJ, Lee YJ, Han HJ. Regulatory mechanisms of Na(+)/glucose cotransporters in renal proximal tubule cells. Kidney Int 2007; 72(suppl 106):S27–S35.
Hummel CS, Lu C, Loo DD, Hirayama BA, Voss AA, Wright EM. Glucose transport by human renal Na+/D-glucose cotransporters SGLT1 and SGLT2. Am J Physiol Cell Physiol 2011; 300:C14–C21.
Heerspink HJ, Perkins BA, Fitchett DH, Husain M, Cherney DZ. Sodium glucose cotransporter 2 inhibitors in the treatment of diabetes mellitus: cardiovascular and kidney effects, potential mechanisms, and clinical applications. Circulation 2016; 134:752–772.
Lapuerta P, Zambrowicz, Strumph P, Sands A. Development of sotagliflozin, a dual sodium-dependent glucose transporter 1/2 inhibitor. Diabetes Vasc Dis Res 2015; 12:101–110.
Zinman B, Wanner C, Lachin JM, et al, for the EMPA-REG OUTCOME Investigators. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med 2015; 373:2117–2128.
Neal B, Vlado-Perkovic V, Mahaffey KW, et al, for the CANVAS Program Collaborative Group. Canagloflozin and cardiovascular and renal events in type 2 diabetes. N Engl J Med 2017; 377:644–657.
Inzucchi SE, Bergenstal RM, Buse JB, et al. Management of hyperglycemia in type 2 diabetes, 2015: a patient-centered approach: update to a position statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care 2015; 38:140–149.
Ferrannini E, Mark M, Mayoux E. CV protection in the EMPA-REG OUTCOME trial: a “thrifty substrate” hypothesis. Diabetes Care 2016; 39:1108–1114.
Cherney DZ, Perkins BA, Soleymanlou N, et al. Renal hemodynamic effect of sodium-glucose cotransporter 2 inhibition in patients with type 1 diabetes mellitus. Circulation 2014; 129:587–597.
Wanner C, Inzucchi SE, Lachin JM, et al, for the EMPA-REG OUTCOME Investigators. Empagliflozin and progression of kidney disease in type 2 diabetes. N Engl J Med 2016; 375:323–334.
Ponikowski P, Voors AA, Anker SD, et al; Authors/Task Force Members; Document Reviewers. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: the Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur J Heart Fail 2016; 18:891–975.
Piepoli MF, Hoes AW, Agewall S, et al; Authors/Task Force Members. 2016 European guidelines on cardiovascular disease prevention in clinical practice. The Sixth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (constituted by representatives of 10 societies and by invited experts). Developed with the special contribution of the European Association for Cardiovascular Prevention & Rehabilitation. Eur Heart J 2016; 37:2315–2381.
American Diabetes Association. American Diabetes Association standards of medical care in diabetes. Diabetes Care 2017; 40(suppl 1):S1–S135.
Lincoff AM, Tardif JC, Schwartz GG, et al; AleCardio Investigators. Effect of aleglitazar on cardiovascular outcomes after acute coronary syndrome in patients with type 2 diabetes mellitus: the AleCardio randomized clinical trial. JAMA 2014; 311:1515–1525.
Kaku K, Enya K, Nakaya R, Ohira T, Matsuno R. Efficacy and safety of fasiglifam (TAK0*&%), a G protein-coupled receptor 40 agonist, in Japanese patients with type 2 diabetes inadequately controlled by diet and exercise: a randomized, double-blind, placebocontrolled, phase III trial. Diabetes Obes Metab 2015; 17: 675–681.
Kershaw V. Patel, MD Department of Internal Medicine, Division of Cardiology, University of Texas Southwestern Medical Center, Dallas
Natalia de Albuquerque Rocha, MD Department of Internal Medicine, Division of Cardiology, University of Texas Southwestern Medical Center, Dallas
Darren K. McGuire, MD, MHSc Department of Internal Medicine, Division of Cardiology, University of Texas Southwestern Medical Center, Dallas
Address: Darren K. McGuire, MD, UT Southwestern Medical Center, 5323 Harry Hines Boulevard, E5.726, Dallas, TX 75390-8830; darren.mcguire@utsouthwestern.edu
Darren K. McGuire has disclosed clinical trial leadership for AstraZeneca, Boehringer Ingelheim, Eisai, Eli Lilly, GlaxoSmithKline, Janssen, Lexicon Genetics, Merck, Novo Nordisk, and Sanofi Aventis; and consultancy for Boehringer Ingelheim, Merck, Novo Nordisk, and Sanofi Aventis.
Medical Grand Rounds articles are based on edited transcripts from Medicine Grand Rounds presentations at Cleveland Clinic. They are approved by the authors but are not peer-reviewed.
Kershaw V. Patel, MD Department of Internal Medicine, Division of Cardiology, University of Texas Southwestern Medical Center, Dallas
Natalia de Albuquerque Rocha, MD Department of Internal Medicine, Division of Cardiology, University of Texas Southwestern Medical Center, Dallas
Darren K. McGuire, MD, MHSc Department of Internal Medicine, Division of Cardiology, University of Texas Southwestern Medical Center, Dallas
Address: Darren K. McGuire, MD, UT Southwestern Medical Center, 5323 Harry Hines Boulevard, E5.726, Dallas, TX 75390-8830; darren.mcguire@utsouthwestern.edu
Darren K. McGuire has disclosed clinical trial leadership for AstraZeneca, Boehringer Ingelheim, Eisai, Eli Lilly, GlaxoSmithKline, Janssen, Lexicon Genetics, Merck, Novo Nordisk, and Sanofi Aventis; and consultancy for Boehringer Ingelheim, Merck, Novo Nordisk, and Sanofi Aventis.
Medical Grand Rounds articles are based on edited transcripts from Medicine Grand Rounds presentations at Cleveland Clinic. They are approved by the authors but are not peer-reviewed.
Author and Disclosure Information
Kershaw V. Patel, MD Department of Internal Medicine, Division of Cardiology, University of Texas Southwestern Medical Center, Dallas
Natalia de Albuquerque Rocha, MD Department of Internal Medicine, Division of Cardiology, University of Texas Southwestern Medical Center, Dallas
Darren K. McGuire, MD, MHSc Department of Internal Medicine, Division of Cardiology, University of Texas Southwestern Medical Center, Dallas
Address: Darren K. McGuire, MD, UT Southwestern Medical Center, 5323 Harry Hines Boulevard, E5.726, Dallas, TX 75390-8830; darren.mcguire@utsouthwestern.edu
Darren K. McGuire has disclosed clinical trial leadership for AstraZeneca, Boehringer Ingelheim, Eisai, Eli Lilly, GlaxoSmithKline, Janssen, Lexicon Genetics, Merck, Novo Nordisk, and Sanofi Aventis; and consultancy for Boehringer Ingelheim, Merck, Novo Nordisk, and Sanofi Aventis.
Medical Grand Rounds articles are based on edited transcripts from Medicine Grand Rounds presentations at Cleveland Clinic. They are approved by the authors but are not peer-reviewed.
Since 2008, the US Food and Drug Administration (FDA) has required new diabetes drugs to demonstrate cardiovascular safety, resulting in large and lengthy clinical trials. Under the new regulations, several dipeptidyl peptidase-4 (DPP-4) inhibitors, sodium-glucose cotransporter-2 (SGLT-2) inhibitors, and glucagon-like peptide-1 (GLP-1) receptor agonists have demonstrated cardiovascular safety, with some demonstrating superior cardiovascular efficacy. In 2016, the SGLT-2 inhibitor empagliflozin became the first (and as of this writing, the only) diabetes drug approved by the FDA for a clinical outcome indication, ie, to reduce the risk of cardiovascular death.
DIABETES DRUG DEVELOPMENT
Changing priorities
The International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use (ICH) was formed in 1990 as a collaborative effort across global regulatory agencies and coordinated by the World Health Organization to universalize criteria for drug development. The ICH standards for type 2 diabetes drug development included the following requirements for patient exposure to investigational products to satisfy new drug application requirements:
1,500 individuals total (including single-dose exposure)
300–600 patients for 6 months
100 patients for 1 year.
Thus, just 250 patient-years of exposure were needed for approval of a drug that patients might take for decades. These standards were unlikely to reveal rare, serious complications and had no ability to assess clinical outcomes efficacy for either microvascular or macrovascular disease complications.
When the ICH regulatory standards were set in the early 1990s, only insulin and sulfonylureas were available in the United States. (Metformin had been available outside the United States since the 1950s.) Since 1990, the prevalence of type 2 diabetes in the United States has increased from around 2% to now over 10% of the US adult population. This increase, along with the known increased risk of atherosclerotic cardiovascular disease and heart failure associated with diabetes, created a sense of urgency for developing new therapies. With a burgeoning population with or at risk of diabetes, new drugs were needed and were rapidly developed.
Since 1995, when metformin was approved in the United States, a new class of antihyperglycemic medication has been approved about once every 2 years, so that by 2008, 12 classes of medications had become available for the treatment of type 2 diabetes. This extraordinary rate of drug development has now yielded more classes of medications to treat type 2 diabetes than we presently have for the treatment of hypertension.
This proliferation of new treatments resolved much of the pressure of the unmet medical need, over a period of increasing awareness of the cardiovascular complications of type 2 diabetes, along with numerous examples of adverse cardiovascular effects observed with some of the drugs. In this context, the FDA (and in parallel the European Medicines Agency) made paradigm-shifting changes in the requirements for the development of new type 2 diabetes drugs, requiring large-scale randomized clinical outcome data to assess cardiovascular safety of the new drugs. In December 2008, the FDA published a Guidance for Industry,1 recommending that sponsors of new drugs for type 2 diabetes demonstrate that therapy would not only improve glucose control, but also that it would, at a minimum, not result in an unacceptable increase in cardiovascular risk.1 To better assess new diabetes drugs, the requirement for patient-years of exposure to the studied drug was increased by over 60-fold from 250 patient-years to more than 15,000.
INCRETIN MODULATORS
The incretin system, a regulator of postprandial glucose metabolism, is an attractive target for glycemic control, as it promotes early satiety and lowers blood glucose.
After a meal, endocrine cells in the distal small intestine secrete the incretin hormones GLP-1 and gastric inhibitory polypeptide (GIP), among others, which reduce gastric motility, stimulate the pancreas to augment glucose-appropriate insulin secretion, and decrease postprandial glucagon release. GLP-1 also interacts with the satiety center of the hypothalamus, suppressing appetite. GLP-1 and GIP are rapidly inactivated by the circulating protease DPP-4. Injectable formulations of GLP-1 receptor agonists that are resistant to DPP-4 degradation have been developed.
Ten incretin modulators are now available in the United States. The 4 available DPP-4 inhibitors are all once-daily oral medications, and the 6 GLP-1 receptor agonists are all injectable (Table 1).
Small studies in humans and animals suggest that DPP-4 inhibitors and GLP-1-receptor agonists may have multiple favorable effects on the cardiovascular system independent of their glycemic effects. These include reducing myocardial infarct size,2–5 improving endothelial function,6 reducing inflammation and oxidative stress,7 reducing atherosclerotic plaque volume,8 improving left ventricular function, 9,10 and lowering triglyceride levels.11 However, large clinical trials are needed to determine clinical effectiveness.
DPP-4 INHIBITORS: NOT INFERIOR TO PLACEBO
Saxagliptin
Saxagliptin, a DPP-4 inhibitor, was found in a meta-analysis of phase 2B and early phase 3 trial data involving almost 5,000 patients to be associated with a dramatic 56% relative risk reduction in cardiovascular death, heart attack, and stroke. However, this analysis was limited by the extremely low number of events to analyze, with only 41 total patients with cardiovascular events in that dataset.12
The SAVOR-TIMI 53 trial13 subsequently compared saxagliptin and placebo in a randomized, double-blind trial conducted in 26 countries with nearly 16,500 patients with type 2 diabetes. All patients continued their conventional diabetes treatment at the discretion of their physicians.
During an average follow-up of 2 years, 1,222 events of cardiovascular death, myocardial infarction, or stroke occurred. No significant difference in event rates was found between the saxagliptin and placebo groups. This did not demonstrate the expected cardiovascular benefit based on prior meta-analysis of phase 2B and phase 3 data presented above, but saxagliptin did not increase cardiovascular risk and was the first diabetes drug to earn this distinction of robustly statistically proven cardiovascular safety.
Further analysis of the SAVOR-TIMI 53 trial data revealed a 27% increased relative risk of heart failure hospitalization with saxagliptin compared with placebo.14 Although the risk was statistically significant, the absolute difference in heart failure incidence between the drug and placebo groups was only 0.7% (3.5% vs 2.8%, respectively). As the average follow-up in the trial was 2 years, the absolute incremental risk of heart failure seen with saxagliptin is 0.35% annually—almost identical in magnitude to the increased heart failure risk with pioglitazone. The increased risk of heart failure was seen within the first 6 months of the trial and persisted throughout the trial, indicating an increased up-front risk of heart failure.
Alogliptin
The EXAMINE trial15 compared the DPP-4 inhibitor alogliptin and placebo in 5,380 patients with type 2 diabetes who had had a recent acute coronary event.15 Over the 30 months of the trial, more than 600 primary outcome events of cardiovascular death, myocardial infarction, or stroke occurred, with no significant difference between drug and placebo groups with established nominal statistical noninferiority. A numerically higher incidence of heart failure was noted in patients who received alogliptin than with placebo, but the difference was not statistically significant.16 However, this study was not powered to detect such an increased risk. In patients entering the trial with no history of heart failure, the risk of hospitalization for heart failure was 76% higher in the alogliptin group than in the placebo group, with a nominally significant P value less than .05 in this subgroup.
These analyses led the FDA in 2016 to mandate label warnings for saxagliptin and alogliptin regarding the increased risk of heart failure.17
Sitagliptin
The TECOS trial18 tested the DPP-4 inhibitor sitagliptin and, unlike the SAVOR or EXAMINE trials, included hospitalization for unstable angina in the composite end point. Nearly 15,000 patients with type 2 diabetes and established cardiovascular disease were enrolled, and almost 2,500 events occurred. No significant difference was found between the 2 groups.
In a series of analyses prospectively planned, sitagliptin was not associated with an increased risk of hospitalization for heart failure.19 But despite these robust analyses demonstrating no incremental heart failure risk with sitagliptin, in August 2017, the US product label for sitagliptin was modified to include a warning that other DPP-4 inhibitors have been associated with heart failure and to suggest caution. The label for linagliptin had the same FDA-required changes, with no data yet available from outcomes trials with linagliptin.
GLP-1 RECEPTOR AGONISTS
Lixisenatide: Noninferior to placebo
The ELIXA trial20 assessed the cardiovascular safety of the GLP-1 receptor agonist lixisenatide in patients with type 2 diabetes who recently had an acute coronary event. The study enrolled 6,068 patients from 49 countries, and nearly 1,000 events (cardiovascular death, myocardial infarction, stroke, or unstable angina) occurred during the median 25 months of the study. Results showed lixisenatide did not increase or decrease cardiovascular events or adverse events when compared with placebo.
Liraglutide: Evidence of benefit
The LEADER trial21 randomized 9,340 patients with or at increased risk for cardiovascular disease to receive the injectable GLP-1 receptor agonist liraglutide or placebo. After a median of 3.8 years of follow-up, liraglutide use was associated with a statistically significant 13% relative reduction in major adverse cardiovascular events, mostly driven by a 22% reduction in cardiovascular death.
Semaglutide: Evidence of benefit
The SUSTAIN-6 trial22 found a statistically significant 26% relative risk reduction in cardiovascular outcomes comparing once-weekly semaglutide (an injectable GLP-1 receptor agonist) and placebo in 3,297 patients with type 2 diabetes and established cardiovascular disease, chronic kidney disease, or risk factors for cardiovascular disease. The significant reduction in the incidence of nonfatal stroke with semaglutide was the main driver of the observed benefit.
Taspoglutide: Development halted
Taspoglutide was a candidate GLP-1 receptor agonist that underwent clinical trials for cardiovascular outcomes planned to involve about 8,000 patients. The trials were stopped early and drug development was halted after about 600 patient-years of exposure because of antibody formation in about half of patients exposed to taspoglutide, with anaphylactoid reactions and anaphylaxis reported.23
SGLT-2 INHIBITORS
The renal glomeruli filter about 180 g of glucose every day in normal adults; nearly all of it is reabsorbed by SGLT-2 in the proximal tubules, so that very little glucose is excreted in the urine.24–26 The benign condition hereditary glucosuria occurs due to loss-of-function mutations in the gene for SGLT-2. Individuals with this condition rarely if ever develop type 2 diabetes or obesity, and this observation led pharmaceutical researchers to probe SGLT-2 as a therapeutic target.
Inhibitors of SGLT-2 block glucose reabsorption in the renal proximal tubules and lead to glucosuria. Patients treated with an SGLT-2 inhibitor have lower serum glucose levels and lose weight. Inhibitors also reduce sodium reabsorption via SGLT-2 and lead to increased sodium excretion and decreased blood pressure.27
Three SGLT-2 antagonists are available in the United States: canagliflozin, dapagliflozin, and empagliflozin (Table 1). Ertugliflozin is currently in a phase 3B trial, and cardiovascular outcomes trials are in the planning phase for sotagliflozin, a dual SGLT-1/SGLT-2 inhibitor with SGLT-1 localized to the gastrointestinal tract.28
Empaglifozin: Evidence of benefit
The EMPA-REG OUTCOME trial29 randomized more than 7,200 patients with type 2 diabetes and atherosclerotic vascular disease to receive the SGLT-2 inhibitor empagliflozin or placebo as once-daily tablets, with both groups receiving off-study treatment for glycemic control at the discretion of their own care providers. Two doses of empagliflozin were evaluated in the trial (10 and 25 mg per day), with the 2 dosing groups pooled for all analyses as prospectively planned.
Patients taking empagliflozin had a 14% relative risk reduction of the composite outcome (cardiovascular death, myocardial infarction, and stroke) vs placebo, with no difference in effect between the 2 randomized doses. The improvement in the composite outcome was seen early in the empagliflozin group and persisted for the 4 years of the study.
This was the first trial of newly developed diabetes drugs that showed a statistically significant reduction in cardiovascular risk. The study revealed a 38% relative risk reduction in cardiovascular death in the treatment group. The risk reduction occurred early in the trial and improved throughout the duration of the study. This is a dramatic finding, unequaled even in trials of drugs that specifically target cardiovascular disease. Both doses of empagliflozin studied provided similar benefit over placebo, reinforcing the validity of the findings. Interestingly, in the empagliflozin group, there was a 35% relative risk reduction in heart failure hospitalizations.
Canaglifozin: Evidence of benefit
The CANVAS Program consisted of two sister trials, CANVAS and CANVAS-R, and examined the safety and efficacy of canagliflozin.30 More than 10,000 participants with type 2 diabetes and atherosclerotic disease or at increased risk of cardiovascular disease were randomized to receive canagliflozin or placebo. Canagliflozin led to a 14% relative risk reduction in the composite outcome of cardiovascular death, nonfatal myocardial infarction, or nonfatal stroke, but there was a statistically significant doubling in the incidence of amputations. Unlike empagliflozin, canagliflozin did not demonstrate a significant reduction in death from cardiovascular causes, suggesting that this may not be a class effect of SGLT-2 inhibitors. As with empagliflozin, canagliflozin led to a 33% relative risk reduction in heart failure hospitalizations.
Cardiovascular benefits independent of glucose-lowering
The cardiovascular benefits of empagliflozin in EMPA-REG OUTCOME and canagliflozin in CANVAS were observed early, suggesting that the mechanism may be due to the direct effects on the cardiovascular system rather than glycemic modification.
Improved glycemic control with the SGLT-2 inhibitor was seen early in both studies, but with the trials designed for glycemic equipoise encouraging open-label therapy targeting hemoglobin A1c to standard-of-care targets in both groups, the contrast in hemoglobin A1c between groups diminished throughout the trial after its first assessment. Although hemoglobin A1c levels in the SGLT-2 inhibitor groups decreased in the first 12 weeks, they increased over time nearly to the level seen in the placebo group. The adjusted mean hemoglobin A1c level in the placebo groups remained near 8.0% throughout the studies, a target consistent with guidelines from the American Diabetes Association and the European Association for the Study of Diabetes31 for the high-risk populations recruited and enrolled.
Blood pressure reduction and weight loss do not explain cardiovascular benefits
SGLT-2 inhibitors lower blood pressure independent of their diuretic effects. In the EMPA-REG OUTCOME trial, the adjusted mean systolic blood pressure was 3 to 4 mm Hg lower in the treatment groups than in the placebo group throughout the trial.29 This level of blood pressure lowering translates to an estimated 10% to 12% relative risk reduction for major adverse cardiovascular events, including heart failure. Although the risk reduction from blood pressure lowering is not insignificant, it does not explain the 38% reduction in cardiovascular deaths seen in the trial. Canagliflozin led to a similar 4-mm Hg reduction in systolic pressure compared with the placebo group.30
Weight loss was seen with both empagliflozin and canagliflozin but was not dramatic and is unlikely to account for the described cardiovascular benefits.
Theories of cardiovascular benefit
Several mechanisms have been proposed to help explain the observed cardiovascular benefits of SGLT-2 inhibitors.32
Ketone-body elevation. Ferrannini et al33 found that the blood concentration of the ketone-body beta-hydroxybutyrate is about twice as high in patients with type 2 diabetes in the fasting state who are chronically taking empagliflozin as in patients not receiving the drug. Beta-hydroxybutyrate levels peak after a meal and then return to baseline over several hours before rising again during the fasting period. Although the ketone elevation is not nearly as extreme as in diabetic ketoacidosis (about a 1,000-fold increase), the observed increase may reduce myocardial oxygen demand, as beta-hydroxybutyrate is among the most efficient metabolic substrates for the myocardium.
Red blood cell expansion. Perhaps a more likely explanation of the cardiovascular benefit seen with SGLT-2 inhibitor therapy is the increase in hemoglobin and hematocrit levels. At first attributed to hemoconcentration secondary to diuresis, this has been disproven by a number of studies. The EMPA-REG OUTCOME trial29 found that within 12 weeks of exposure to empagliflozin, hematocrit levels rose nearly 4% absolutely compared with the levels in the placebo group. This increase is equivalent to transfusing a unit of red blood cells, favorably affecting myocardial oxygen supply.
Reduction in glomerular hypertension. The kidneys regulate glomerular filtration in a process involving the macula densa, an area of specialized cells in the juxtaglomerular apparatus in the loop of Henle that responds to sodium concentration in the urine. Normally, SGLT-2 receptors upstream from the loop of Henle reabsorb sodium and glucose into the bloodstream, reducing sodium delivery to the macula densa, which senses this as a low-volume state. The macula densa cells respond by releasing factors that dilate afferent arterioles and increase glomerular filtration. People with diabetes have more glucose to reabsorb and therefore also reabsorb more sodium, leading to glomerular hypertension.
SGLT-2 inhibitors block both glucose and sodium reuptake at SGLT-2 receptors, normalizing the response at the macula densa, restoring a normal glomerular filtration rate, and alleviating glomerular hypertension. As the kidney perceives a more normal volume status, renin-angiotensin-aldosterone stimulation is attenuated and sympathetic nervous system activity improves.27,34 If this model of SGLT-2 inhibitor effects on the kidney is correct, these drugs have similar effects as angiotensin-converting enzyme (ACE) inhibitors, angiotensin II receptor blockers (ARBs), mineralocorticoid antagonists, and beta-blockers combined.
Kidney benefits
Empagliflozin35 and canagliflozin30 both reduced the rate of progression of kidney dysfunction and led to fewer clinically relevant renal events compared with placebo. Treatment and placebo groups also received standard care, so many patients were treated with renin-angiotensin-aldosterone system inhibitors and with good blood pressure control, making the finding that SGLT-2 inhibitors had a significant beneficial effect even more dramatic. Beneficial effects on markers of kidney function were seen early on, suggesting a more favorable hemodynamic effect on the kidney rather than improved glycemic control attenuating microvascular disease.
Empagliflozin approved to reduce clinical events
In December 2016, the FDA approved the indication for empagliflozin to reduce the risk of cardiovascular death in patients with type 2 diabetes,36 the first-ever clinical outcome indication for a type 2 diabetes medication. The European Society of Cardiology guidelines now include empagliflozin as preferred therapy for type 2 diabetes, recommending it to prevent the onset of heart failure and prolong life.37 This recommendation goes beyond the evidence from the EMPA-REG OUTCOME trial on which it is based, as the trial only studied patients with known atherosclerotic vascular disease.
The 2016 European Guidelines on cardiovascular disease prevention also recommend that an SGLT-2 inhibitor be considered early for patients with type 2 diabetes and cardiovascular disease to reduce cardiovascular and total mortality.38 The American Diabetes Association in their 2017 guidelines also endorse empagliflozin for treating patients with type 2 diabetes and cardiovascular disease.39 The fact that the American Diabetes Association recommendation is not based on glycemic control, in line with the product-labeled indication, is a major shift in the association’s guidance.
Cautions with SGLT-2 inhibitors
Use SGLT-2 inhibitors in patients with low blood pressure with caution, and with increased blood pressure monitoring just following initiation.
Consider modifying antihypertensive drugs in patients with labile blood pressure.
Consider stopping or reducing background diuretics when starting an SGLT-2 inhibitor, and reassess volume status after 1 to 2 weeks.
For patients on insulin, sulfonylureas, or both, consider decreasing dosages when starting an SGLT-2 inhibitor, and reassess glycemic control periodically.
Counsel patients about urinary hygiene. Although bacterial urinary tract infections have not emerged as a problem, fungal genital infections have, particularly in women and uncircumcised men.
Consider SGLT-2 inhibitors to be “sick-day” medications. Patients with diabetes must adjust their diabetes medications if their oral intake is reduced for a day or more, such as while sick or fasting. SGLT-2 inhibitors should not be taken on these days. Cases of diabetic ketoacidosis have arisen in patients who reduced oral intake while continuing their SGLT-2 inhibitor.
OTHER DRUGS WITH DEVELOPMENT HALTED
Aleglitazar, a peroxisome proliferator-activated receptor agonist taken orally once daily, raised high expectations when it was found in early studies to lower serum triglycerides and raise high-density lipoprotein cholesterol levels in addition to lowering blood glucose. However, a phase 3 trial in more than 7,000 patients was terminated after a median follow up of 2 years because of increased rates of heart failure, worsened kidney function, bone fractures, and gastrointestinal bleeding.40 Development of this drug was stopped.
Fasiglifam, a G-protein-coupled receptor 40 agonist, was tested in a cardiovascular clinical outcomes trial. Compared with placebo, fasiglifam reduced hemoglobin A1c levels with low risk of hypoglycemia.41 However, safety concerns about increased liver enzyme levels led to the cessation of the drug’s development.42
HOW WILL THIS AFFECT DIABETES MANAGEMENT?
Metformin is still the most commonly prescribed drug for type 2 diabetes but has only marginal evidence for its cardiovascular benefits and may not be the first-line therapy for the management of diabetes in the future. In the EMPA REG OUTCOME, LEADER, and SUSTAIN-6 trials, the novel diabetes medications were given to patients who were already treated with available therapies, often including metformin. Treatment with empagliflozin, liraglutide, and semaglutide may be indicated for patients with diabetes and atherosclerotic vascular disease as first-line therapies in the future.
SGLT-2 inhibitor therapy can cost about $500 per month, and GLP-1 inhibitors are only slightly less expensive. The cost may be prohibitive for many patients. As more evidence, guidelines, and FDA criteria support the use of these novel diabetes drugs, third-party payers and pharmaceutical companies may be motivated to lower costs to help reach more patients who can benefit from these therapies.
Since 2008, the US Food and Drug Administration (FDA) has required new diabetes drugs to demonstrate cardiovascular safety, resulting in large and lengthy clinical trials. Under the new regulations, several dipeptidyl peptidase-4 (DPP-4) inhibitors, sodium-glucose cotransporter-2 (SGLT-2) inhibitors, and glucagon-like peptide-1 (GLP-1) receptor agonists have demonstrated cardiovascular safety, with some demonstrating superior cardiovascular efficacy. In 2016, the SGLT-2 inhibitor empagliflozin became the first (and as of this writing, the only) diabetes drug approved by the FDA for a clinical outcome indication, ie, to reduce the risk of cardiovascular death.
DIABETES DRUG DEVELOPMENT
Changing priorities
The International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use (ICH) was formed in 1990 as a collaborative effort across global regulatory agencies and coordinated by the World Health Organization to universalize criteria for drug development. The ICH standards for type 2 diabetes drug development included the following requirements for patient exposure to investigational products to satisfy new drug application requirements:
1,500 individuals total (including single-dose exposure)
300–600 patients for 6 months
100 patients for 1 year.
Thus, just 250 patient-years of exposure were needed for approval of a drug that patients might take for decades. These standards were unlikely to reveal rare, serious complications and had no ability to assess clinical outcomes efficacy for either microvascular or macrovascular disease complications.
When the ICH regulatory standards were set in the early 1990s, only insulin and sulfonylureas were available in the United States. (Metformin had been available outside the United States since the 1950s.) Since 1990, the prevalence of type 2 diabetes in the United States has increased from around 2% to now over 10% of the US adult population. This increase, along with the known increased risk of atherosclerotic cardiovascular disease and heart failure associated with diabetes, created a sense of urgency for developing new therapies. With a burgeoning population with or at risk of diabetes, new drugs were needed and were rapidly developed.
Since 1995, when metformin was approved in the United States, a new class of antihyperglycemic medication has been approved about once every 2 years, so that by 2008, 12 classes of medications had become available for the treatment of type 2 diabetes. This extraordinary rate of drug development has now yielded more classes of medications to treat type 2 diabetes than we presently have for the treatment of hypertension.
This proliferation of new treatments resolved much of the pressure of the unmet medical need, over a period of increasing awareness of the cardiovascular complications of type 2 diabetes, along with numerous examples of adverse cardiovascular effects observed with some of the drugs. In this context, the FDA (and in parallel the European Medicines Agency) made paradigm-shifting changes in the requirements for the development of new type 2 diabetes drugs, requiring large-scale randomized clinical outcome data to assess cardiovascular safety of the new drugs. In December 2008, the FDA published a Guidance for Industry,1 recommending that sponsors of new drugs for type 2 diabetes demonstrate that therapy would not only improve glucose control, but also that it would, at a minimum, not result in an unacceptable increase in cardiovascular risk.1 To better assess new diabetes drugs, the requirement for patient-years of exposure to the studied drug was increased by over 60-fold from 250 patient-years to more than 15,000.
INCRETIN MODULATORS
The incretin system, a regulator of postprandial glucose metabolism, is an attractive target for glycemic control, as it promotes early satiety and lowers blood glucose.
After a meal, endocrine cells in the distal small intestine secrete the incretin hormones GLP-1 and gastric inhibitory polypeptide (GIP), among others, which reduce gastric motility, stimulate the pancreas to augment glucose-appropriate insulin secretion, and decrease postprandial glucagon release. GLP-1 also interacts with the satiety center of the hypothalamus, suppressing appetite. GLP-1 and GIP are rapidly inactivated by the circulating protease DPP-4. Injectable formulations of GLP-1 receptor agonists that are resistant to DPP-4 degradation have been developed.
Ten incretin modulators are now available in the United States. The 4 available DPP-4 inhibitors are all once-daily oral medications, and the 6 GLP-1 receptor agonists are all injectable (Table 1).
Small studies in humans and animals suggest that DPP-4 inhibitors and GLP-1-receptor agonists may have multiple favorable effects on the cardiovascular system independent of their glycemic effects. These include reducing myocardial infarct size,2–5 improving endothelial function,6 reducing inflammation and oxidative stress,7 reducing atherosclerotic plaque volume,8 improving left ventricular function, 9,10 and lowering triglyceride levels.11 However, large clinical trials are needed to determine clinical effectiveness.
DPP-4 INHIBITORS: NOT INFERIOR TO PLACEBO
Saxagliptin
Saxagliptin, a DPP-4 inhibitor, was found in a meta-analysis of phase 2B and early phase 3 trial data involving almost 5,000 patients to be associated with a dramatic 56% relative risk reduction in cardiovascular death, heart attack, and stroke. However, this analysis was limited by the extremely low number of events to analyze, with only 41 total patients with cardiovascular events in that dataset.12
The SAVOR-TIMI 53 trial13 subsequently compared saxagliptin and placebo in a randomized, double-blind trial conducted in 26 countries with nearly 16,500 patients with type 2 diabetes. All patients continued their conventional diabetes treatment at the discretion of their physicians.
During an average follow-up of 2 years, 1,222 events of cardiovascular death, myocardial infarction, or stroke occurred. No significant difference in event rates was found between the saxagliptin and placebo groups. This did not demonstrate the expected cardiovascular benefit based on prior meta-analysis of phase 2B and phase 3 data presented above, but saxagliptin did not increase cardiovascular risk and was the first diabetes drug to earn this distinction of robustly statistically proven cardiovascular safety.
Further analysis of the SAVOR-TIMI 53 trial data revealed a 27% increased relative risk of heart failure hospitalization with saxagliptin compared with placebo.14 Although the risk was statistically significant, the absolute difference in heart failure incidence between the drug and placebo groups was only 0.7% (3.5% vs 2.8%, respectively). As the average follow-up in the trial was 2 years, the absolute incremental risk of heart failure seen with saxagliptin is 0.35% annually—almost identical in magnitude to the increased heart failure risk with pioglitazone. The increased risk of heart failure was seen within the first 6 months of the trial and persisted throughout the trial, indicating an increased up-front risk of heart failure.
Alogliptin
The EXAMINE trial15 compared the DPP-4 inhibitor alogliptin and placebo in 5,380 patients with type 2 diabetes who had had a recent acute coronary event.15 Over the 30 months of the trial, more than 600 primary outcome events of cardiovascular death, myocardial infarction, or stroke occurred, with no significant difference between drug and placebo groups with established nominal statistical noninferiority. A numerically higher incidence of heart failure was noted in patients who received alogliptin than with placebo, but the difference was not statistically significant.16 However, this study was not powered to detect such an increased risk. In patients entering the trial with no history of heart failure, the risk of hospitalization for heart failure was 76% higher in the alogliptin group than in the placebo group, with a nominally significant P value less than .05 in this subgroup.
These analyses led the FDA in 2016 to mandate label warnings for saxagliptin and alogliptin regarding the increased risk of heart failure.17
Sitagliptin
The TECOS trial18 tested the DPP-4 inhibitor sitagliptin and, unlike the SAVOR or EXAMINE trials, included hospitalization for unstable angina in the composite end point. Nearly 15,000 patients with type 2 diabetes and established cardiovascular disease were enrolled, and almost 2,500 events occurred. No significant difference was found between the 2 groups.
In a series of analyses prospectively planned, sitagliptin was not associated with an increased risk of hospitalization for heart failure.19 But despite these robust analyses demonstrating no incremental heart failure risk with sitagliptin, in August 2017, the US product label for sitagliptin was modified to include a warning that other DPP-4 inhibitors have been associated with heart failure and to suggest caution. The label for linagliptin had the same FDA-required changes, with no data yet available from outcomes trials with linagliptin.
GLP-1 RECEPTOR AGONISTS
Lixisenatide: Noninferior to placebo
The ELIXA trial20 assessed the cardiovascular safety of the GLP-1 receptor agonist lixisenatide in patients with type 2 diabetes who recently had an acute coronary event. The study enrolled 6,068 patients from 49 countries, and nearly 1,000 events (cardiovascular death, myocardial infarction, stroke, or unstable angina) occurred during the median 25 months of the study. Results showed lixisenatide did not increase or decrease cardiovascular events or adverse events when compared with placebo.
Liraglutide: Evidence of benefit
The LEADER trial21 randomized 9,340 patients with or at increased risk for cardiovascular disease to receive the injectable GLP-1 receptor agonist liraglutide or placebo. After a median of 3.8 years of follow-up, liraglutide use was associated with a statistically significant 13% relative reduction in major adverse cardiovascular events, mostly driven by a 22% reduction in cardiovascular death.
Semaglutide: Evidence of benefit
The SUSTAIN-6 trial22 found a statistically significant 26% relative risk reduction in cardiovascular outcomes comparing once-weekly semaglutide (an injectable GLP-1 receptor agonist) and placebo in 3,297 patients with type 2 diabetes and established cardiovascular disease, chronic kidney disease, or risk factors for cardiovascular disease. The significant reduction in the incidence of nonfatal stroke with semaglutide was the main driver of the observed benefit.
Taspoglutide: Development halted
Taspoglutide was a candidate GLP-1 receptor agonist that underwent clinical trials for cardiovascular outcomes planned to involve about 8,000 patients. The trials were stopped early and drug development was halted after about 600 patient-years of exposure because of antibody formation in about half of patients exposed to taspoglutide, with anaphylactoid reactions and anaphylaxis reported.23
SGLT-2 INHIBITORS
The renal glomeruli filter about 180 g of glucose every day in normal adults; nearly all of it is reabsorbed by SGLT-2 in the proximal tubules, so that very little glucose is excreted in the urine.24–26 The benign condition hereditary glucosuria occurs due to loss-of-function mutations in the gene for SGLT-2. Individuals with this condition rarely if ever develop type 2 diabetes or obesity, and this observation led pharmaceutical researchers to probe SGLT-2 as a therapeutic target.
Inhibitors of SGLT-2 block glucose reabsorption in the renal proximal tubules and lead to glucosuria. Patients treated with an SGLT-2 inhibitor have lower serum glucose levels and lose weight. Inhibitors also reduce sodium reabsorption via SGLT-2 and lead to increased sodium excretion and decreased blood pressure.27
Three SGLT-2 antagonists are available in the United States: canagliflozin, dapagliflozin, and empagliflozin (Table 1). Ertugliflozin is currently in a phase 3B trial, and cardiovascular outcomes trials are in the planning phase for sotagliflozin, a dual SGLT-1/SGLT-2 inhibitor with SGLT-1 localized to the gastrointestinal tract.28
Empaglifozin: Evidence of benefit
The EMPA-REG OUTCOME trial29 randomized more than 7,200 patients with type 2 diabetes and atherosclerotic vascular disease to receive the SGLT-2 inhibitor empagliflozin or placebo as once-daily tablets, with both groups receiving off-study treatment for glycemic control at the discretion of their own care providers. Two doses of empagliflozin were evaluated in the trial (10 and 25 mg per day), with the 2 dosing groups pooled for all analyses as prospectively planned.
Patients taking empagliflozin had a 14% relative risk reduction of the composite outcome (cardiovascular death, myocardial infarction, and stroke) vs placebo, with no difference in effect between the 2 randomized doses. The improvement in the composite outcome was seen early in the empagliflozin group and persisted for the 4 years of the study.
This was the first trial of newly developed diabetes drugs that showed a statistically significant reduction in cardiovascular risk. The study revealed a 38% relative risk reduction in cardiovascular death in the treatment group. The risk reduction occurred early in the trial and improved throughout the duration of the study. This is a dramatic finding, unequaled even in trials of drugs that specifically target cardiovascular disease. Both doses of empagliflozin studied provided similar benefit over placebo, reinforcing the validity of the findings. Interestingly, in the empagliflozin group, there was a 35% relative risk reduction in heart failure hospitalizations.
Canaglifozin: Evidence of benefit
The CANVAS Program consisted of two sister trials, CANVAS and CANVAS-R, and examined the safety and efficacy of canagliflozin.30 More than 10,000 participants with type 2 diabetes and atherosclerotic disease or at increased risk of cardiovascular disease were randomized to receive canagliflozin or placebo. Canagliflozin led to a 14% relative risk reduction in the composite outcome of cardiovascular death, nonfatal myocardial infarction, or nonfatal stroke, but there was a statistically significant doubling in the incidence of amputations. Unlike empagliflozin, canagliflozin did not demonstrate a significant reduction in death from cardiovascular causes, suggesting that this may not be a class effect of SGLT-2 inhibitors. As with empagliflozin, canagliflozin led to a 33% relative risk reduction in heart failure hospitalizations.
Cardiovascular benefits independent of glucose-lowering
The cardiovascular benefits of empagliflozin in EMPA-REG OUTCOME and canagliflozin in CANVAS were observed early, suggesting that the mechanism may be due to the direct effects on the cardiovascular system rather than glycemic modification.
Improved glycemic control with the SGLT-2 inhibitor was seen early in both studies, but with the trials designed for glycemic equipoise encouraging open-label therapy targeting hemoglobin A1c to standard-of-care targets in both groups, the contrast in hemoglobin A1c between groups diminished throughout the trial after its first assessment. Although hemoglobin A1c levels in the SGLT-2 inhibitor groups decreased in the first 12 weeks, they increased over time nearly to the level seen in the placebo group. The adjusted mean hemoglobin A1c level in the placebo groups remained near 8.0% throughout the studies, a target consistent with guidelines from the American Diabetes Association and the European Association for the Study of Diabetes31 for the high-risk populations recruited and enrolled.
Blood pressure reduction and weight loss do not explain cardiovascular benefits
SGLT-2 inhibitors lower blood pressure independent of their diuretic effects. In the EMPA-REG OUTCOME trial, the adjusted mean systolic blood pressure was 3 to 4 mm Hg lower in the treatment groups than in the placebo group throughout the trial.29 This level of blood pressure lowering translates to an estimated 10% to 12% relative risk reduction for major adverse cardiovascular events, including heart failure. Although the risk reduction from blood pressure lowering is not insignificant, it does not explain the 38% reduction in cardiovascular deaths seen in the trial. Canagliflozin led to a similar 4-mm Hg reduction in systolic pressure compared with the placebo group.30
Weight loss was seen with both empagliflozin and canagliflozin but was not dramatic and is unlikely to account for the described cardiovascular benefits.
Theories of cardiovascular benefit
Several mechanisms have been proposed to help explain the observed cardiovascular benefits of SGLT-2 inhibitors.32
Ketone-body elevation. Ferrannini et al33 found that the blood concentration of the ketone-body beta-hydroxybutyrate is about twice as high in patients with type 2 diabetes in the fasting state who are chronically taking empagliflozin as in patients not receiving the drug. Beta-hydroxybutyrate levels peak after a meal and then return to baseline over several hours before rising again during the fasting period. Although the ketone elevation is not nearly as extreme as in diabetic ketoacidosis (about a 1,000-fold increase), the observed increase may reduce myocardial oxygen demand, as beta-hydroxybutyrate is among the most efficient metabolic substrates for the myocardium.
Red blood cell expansion. Perhaps a more likely explanation of the cardiovascular benefit seen with SGLT-2 inhibitor therapy is the increase in hemoglobin and hematocrit levels. At first attributed to hemoconcentration secondary to diuresis, this has been disproven by a number of studies. The EMPA-REG OUTCOME trial29 found that within 12 weeks of exposure to empagliflozin, hematocrit levels rose nearly 4% absolutely compared with the levels in the placebo group. This increase is equivalent to transfusing a unit of red blood cells, favorably affecting myocardial oxygen supply.
Reduction in glomerular hypertension. The kidneys regulate glomerular filtration in a process involving the macula densa, an area of specialized cells in the juxtaglomerular apparatus in the loop of Henle that responds to sodium concentration in the urine. Normally, SGLT-2 receptors upstream from the loop of Henle reabsorb sodium and glucose into the bloodstream, reducing sodium delivery to the macula densa, which senses this as a low-volume state. The macula densa cells respond by releasing factors that dilate afferent arterioles and increase glomerular filtration. People with diabetes have more glucose to reabsorb and therefore also reabsorb more sodium, leading to glomerular hypertension.
SGLT-2 inhibitors block both glucose and sodium reuptake at SGLT-2 receptors, normalizing the response at the macula densa, restoring a normal glomerular filtration rate, and alleviating glomerular hypertension. As the kidney perceives a more normal volume status, renin-angiotensin-aldosterone stimulation is attenuated and sympathetic nervous system activity improves.27,34 If this model of SGLT-2 inhibitor effects on the kidney is correct, these drugs have similar effects as angiotensin-converting enzyme (ACE) inhibitors, angiotensin II receptor blockers (ARBs), mineralocorticoid antagonists, and beta-blockers combined.
Kidney benefits
Empagliflozin35 and canagliflozin30 both reduced the rate of progression of kidney dysfunction and led to fewer clinically relevant renal events compared with placebo. Treatment and placebo groups also received standard care, so many patients were treated with renin-angiotensin-aldosterone system inhibitors and with good blood pressure control, making the finding that SGLT-2 inhibitors had a significant beneficial effect even more dramatic. Beneficial effects on markers of kidney function were seen early on, suggesting a more favorable hemodynamic effect on the kidney rather than improved glycemic control attenuating microvascular disease.
Empagliflozin approved to reduce clinical events
In December 2016, the FDA approved the indication for empagliflozin to reduce the risk of cardiovascular death in patients with type 2 diabetes,36 the first-ever clinical outcome indication for a type 2 diabetes medication. The European Society of Cardiology guidelines now include empagliflozin as preferred therapy for type 2 diabetes, recommending it to prevent the onset of heart failure and prolong life.37 This recommendation goes beyond the evidence from the EMPA-REG OUTCOME trial on which it is based, as the trial only studied patients with known atherosclerotic vascular disease.
The 2016 European Guidelines on cardiovascular disease prevention also recommend that an SGLT-2 inhibitor be considered early for patients with type 2 diabetes and cardiovascular disease to reduce cardiovascular and total mortality.38 The American Diabetes Association in their 2017 guidelines also endorse empagliflozin for treating patients with type 2 diabetes and cardiovascular disease.39 The fact that the American Diabetes Association recommendation is not based on glycemic control, in line with the product-labeled indication, is a major shift in the association’s guidance.
Cautions with SGLT-2 inhibitors
Use SGLT-2 inhibitors in patients with low blood pressure with caution, and with increased blood pressure monitoring just following initiation.
Consider modifying antihypertensive drugs in patients with labile blood pressure.
Consider stopping or reducing background diuretics when starting an SGLT-2 inhibitor, and reassess volume status after 1 to 2 weeks.
For patients on insulin, sulfonylureas, or both, consider decreasing dosages when starting an SGLT-2 inhibitor, and reassess glycemic control periodically.
Counsel patients about urinary hygiene. Although bacterial urinary tract infections have not emerged as a problem, fungal genital infections have, particularly in women and uncircumcised men.
Consider SGLT-2 inhibitors to be “sick-day” medications. Patients with diabetes must adjust their diabetes medications if their oral intake is reduced for a day or more, such as while sick or fasting. SGLT-2 inhibitors should not be taken on these days. Cases of diabetic ketoacidosis have arisen in patients who reduced oral intake while continuing their SGLT-2 inhibitor.
OTHER DRUGS WITH DEVELOPMENT HALTED
Aleglitazar, a peroxisome proliferator-activated receptor agonist taken orally once daily, raised high expectations when it was found in early studies to lower serum triglycerides and raise high-density lipoprotein cholesterol levels in addition to lowering blood glucose. However, a phase 3 trial in more than 7,000 patients was terminated after a median follow up of 2 years because of increased rates of heart failure, worsened kidney function, bone fractures, and gastrointestinal bleeding.40 Development of this drug was stopped.
Fasiglifam, a G-protein-coupled receptor 40 agonist, was tested in a cardiovascular clinical outcomes trial. Compared with placebo, fasiglifam reduced hemoglobin A1c levels with low risk of hypoglycemia.41 However, safety concerns about increased liver enzyme levels led to the cessation of the drug’s development.42
HOW WILL THIS AFFECT DIABETES MANAGEMENT?
Metformin is still the most commonly prescribed drug for type 2 diabetes but has only marginal evidence for its cardiovascular benefits and may not be the first-line therapy for the management of diabetes in the future. In the EMPA REG OUTCOME, LEADER, and SUSTAIN-6 trials, the novel diabetes medications were given to patients who were already treated with available therapies, often including metformin. Treatment with empagliflozin, liraglutide, and semaglutide may be indicated for patients with diabetes and atherosclerotic vascular disease as first-line therapies in the future.
SGLT-2 inhibitor therapy can cost about $500 per month, and GLP-1 inhibitors are only slightly less expensive. The cost may be prohibitive for many patients. As more evidence, guidelines, and FDA criteria support the use of these novel diabetes drugs, third-party payers and pharmaceutical companies may be motivated to lower costs to help reach more patients who can benefit from these therapies.
References
US Food and Drug Administration. Guidance for industry. Diabetes mellitus—evaluating cardiovascular risk in new antidiabetic therapies to treat type 2 diabetes. www.fda.gov/downloads/Drugs/.../Guidances/ucm071627.pdf. Accessed September 1, 2017.
Ye Y, Keyes KT, Zhang C, Perez-Polo JR, Lin Y, Birnbaum Y. The myocardial infarct size-limiting effect of sitagliptin is PKA-dependent, whereas the protective effect of pioglitazone is partially dependent on PKA. Am J Physiol Heart Circ Physiol 2010; 298:H1454–H1465.
Hocher B, Sharkovska Y, Mark M, Klein T, Pfab T. The novel DPP-4 inhibitors linagliptin and BI 14361 reduce infarct size after myocardial ischemia/reperfusion in rats. Int J Cardiol 2013; 167:87–93.
Woo JS, Kim W, Ha SJ, et al. Cardioprotective effects of exenatide in patients with ST-segment-elevation myocardial infarction undergoing primary percutaneous coronary intervention: results of exenatide myocardial protection in revascularization study. Arterioscler Thromb Vasc Biol 2013; 33:2252–2260.
Lønborg J, Vejlstrup N, Kelbæk H, et al. Exenatide reduces reperfusion injury in patients with ST-segment elevation myocardial infarction. Eur Heart J 2012; 33:1491–1499.
van Poppel PC, Netea MG, Smits P, Tack CJ. Vildagliptin improves endothelium-dependent vasodilatation in type 2 diabetes. Diabetes Care 2011; 34:2072–2077.
Kröller-Schön S, Knorr M, Hausding M, et al. Glucose-independent improvement of vascular dysfunction in experimental sepsis by dipeptidyl-peptidase 4 inhibition. Cardiovasc Res 2012; 96:140–149.
Ta NN, Schuyler CA, Li Y, Lopes-Virella MF, Huang Y. DPP-4 (CD26) inhibitor alogliptin inhibits atherosclerosis in diabetic apolipoprotein E-deficient mice. J Cardiovasc Pharmacol 2011; 58:157–166.
Sauvé M, Ban K, Momen MA, et al. Genetic deletion or pharmacological inhibition of dipeptidyl peptidase-4 improves cardiovascular outcomes after myocardial infarction in mice. Diabetes 2010; 59:1063–1073.
Read PA, Khan FZ, Heck PM, Hoole SP, Dutka DP. DPP-4 inhibition by sitagliptin improves the myocardial response to dobutamine stress and mitigates stunning in a pilot study of patients with coronary artery disease. Circ Cardiovasc Imaging 2010; 3:195–201.
Matikainen N, Mänttäri S, Schweizer A, et al. Vildagliptin therapy reduces postprandial intestinal triglyceride-rich lipoprotein particles in patients with type 2 diabetes. Diabetologia 2006; 49:2049–2057.
Frederich R, Alexander JH, Fiedorek FT, et al. A systematic assessment of cardiovascular outcomes in the saxagliptin drug development program for type 2 diabetes. Postgrad Med 2010; 122:16–27.
Scirica BM, Bhatt DL, Braunwald E, et al; SAVOR-TIMI 53 Steering Committee and Investigators. Saxagliptin and cardiovascular outcomes in patients with type 2 diabetes mellitus. N Engl J Med 2013; 369:1317–1326.
Scirica BM, Braunwald E, Raz I, et al; SAVOR-TIMI 53 Steering Committee and Investigators. Heart failure, saxagliptin, and diabetes mellitus: observations from the SAVOR-TIMI 53 randomized trial. Circulation 2014; 130:1579–1588.
White WB, Cannon CP, Heller SR, et al; EXAMINE Investigators. Alogliptin after acute coronary syndrome in patients with type 2 diabetes. N Engl J Med 2013; 369:1327–1335.
Zannad F, Cannon CP, Cushman WC, et al; EXAMINE Investigators. Heart failure and mortality outcomes in patients with type 2 diabetes taking alogliptin versus placebo in EXAMINE: a multicentre, randomised, double-blind trial. Lancet 2015; 385:2067–2076.
Green JB, Bethel MA, Armstrong PW, et al; TECOS Study Group. Effect of sitagliptin on cardiovascular outcomes in type 2 diabetes. N Engl J Med 2015; 373:232–242.
McGuire DK, Van de Werf F, Armstrong PW, et al; Trial Evaluating Cardiovascular Outcomes With Sitagliptin (TECOS) Study Group. Association between sitagliptin use and heart failure hospitalization and related outcomes in type 2 diabetes mellitus: secondary analysis of a randomized clinical trial. JAMA Cardiol 2016; 1:126–135.
Pfeffer MA, Claggett B, Diaz R, et al; ELIXA Investigators. Lixisenatide in patients with type 2 diabetes and acute coronary syndrome. N Engl J Med 2015; 373:2247–2257.
Marso SP, Daniels GH, Brown-Frandsen K, et al; LEADER Steering Committee; LEADER Trial Investigators. Liraglutide and cardiovascular outcomes in type 2 diabetes. N Engl J Med 2016; 375:311–322.
Marso SP, Bain SC, Consoli A, et al; SUSTAIN-6 Investigators. Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N Engl J Med 2016; 375:1834–1844.
Rosenstock J, Balas B, Charbonnel B, et al; T-EMERGE 2 Study Group. The fate of taspoglutide, a weekly GLP-1 receptor agonist, versus twice-daily exenatide for type 2 diabetes: the T-EMERGE 2 trial. Diabetes Care 2013; 36:498–504.
Wright EM. Renal Na(+)-glucose cotransporters. Am J Physiol 2001; 280:F10–F18.
Lee YJ, Lee YJ, Han HJ. Regulatory mechanisms of Na(+)/glucose cotransporters in renal proximal tubule cells. Kidney Int 2007; 72(suppl 106):S27–S35.
Hummel CS, Lu C, Loo DD, Hirayama BA, Voss AA, Wright EM. Glucose transport by human renal Na+/D-glucose cotransporters SGLT1 and SGLT2. Am J Physiol Cell Physiol 2011; 300:C14–C21.
Heerspink HJ, Perkins BA, Fitchett DH, Husain M, Cherney DZ. Sodium glucose cotransporter 2 inhibitors in the treatment of diabetes mellitus: cardiovascular and kidney effects, potential mechanisms, and clinical applications. Circulation 2016; 134:752–772.
Lapuerta P, Zambrowicz, Strumph P, Sands A. Development of sotagliflozin, a dual sodium-dependent glucose transporter 1/2 inhibitor. Diabetes Vasc Dis Res 2015; 12:101–110.
Zinman B, Wanner C, Lachin JM, et al, for the EMPA-REG OUTCOME Investigators. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med 2015; 373:2117–2128.
Neal B, Vlado-Perkovic V, Mahaffey KW, et al, for the CANVAS Program Collaborative Group. Canagloflozin and cardiovascular and renal events in type 2 diabetes. N Engl J Med 2017; 377:644–657.
Inzucchi SE, Bergenstal RM, Buse JB, et al. Management of hyperglycemia in type 2 diabetes, 2015: a patient-centered approach: update to a position statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care 2015; 38:140–149.
Ferrannini E, Mark M, Mayoux E. CV protection in the EMPA-REG OUTCOME trial: a “thrifty substrate” hypothesis. Diabetes Care 2016; 39:1108–1114.
Cherney DZ, Perkins BA, Soleymanlou N, et al. Renal hemodynamic effect of sodium-glucose cotransporter 2 inhibition in patients with type 1 diabetes mellitus. Circulation 2014; 129:587–597.
Wanner C, Inzucchi SE, Lachin JM, et al, for the EMPA-REG OUTCOME Investigators. Empagliflozin and progression of kidney disease in type 2 diabetes. N Engl J Med 2016; 375:323–334.
Ponikowski P, Voors AA, Anker SD, et al; Authors/Task Force Members; Document Reviewers. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: the Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur J Heart Fail 2016; 18:891–975.
Piepoli MF, Hoes AW, Agewall S, et al; Authors/Task Force Members. 2016 European guidelines on cardiovascular disease prevention in clinical practice. The Sixth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (constituted by representatives of 10 societies and by invited experts). Developed with the special contribution of the European Association for Cardiovascular Prevention & Rehabilitation. Eur Heart J 2016; 37:2315–2381.
American Diabetes Association. American Diabetes Association standards of medical care in diabetes. Diabetes Care 2017; 40(suppl 1):S1–S135.
Lincoff AM, Tardif JC, Schwartz GG, et al; AleCardio Investigators. Effect of aleglitazar on cardiovascular outcomes after acute coronary syndrome in patients with type 2 diabetes mellitus: the AleCardio randomized clinical trial. JAMA 2014; 311:1515–1525.
Kaku K, Enya K, Nakaya R, Ohira T, Matsuno R. Efficacy and safety of fasiglifam (TAK0*&%), a G protein-coupled receptor 40 agonist, in Japanese patients with type 2 diabetes inadequately controlled by diet and exercise: a randomized, double-blind, placebocontrolled, phase III trial. Diabetes Obes Metab 2015; 17: 675–681.
US Food and Drug Administration. Guidance for industry. Diabetes mellitus—evaluating cardiovascular risk in new antidiabetic therapies to treat type 2 diabetes. www.fda.gov/downloads/Drugs/.../Guidances/ucm071627.pdf. Accessed September 1, 2017.
Ye Y, Keyes KT, Zhang C, Perez-Polo JR, Lin Y, Birnbaum Y. The myocardial infarct size-limiting effect of sitagliptin is PKA-dependent, whereas the protective effect of pioglitazone is partially dependent on PKA. Am J Physiol Heart Circ Physiol 2010; 298:H1454–H1465.
Hocher B, Sharkovska Y, Mark M, Klein T, Pfab T. The novel DPP-4 inhibitors linagliptin and BI 14361 reduce infarct size after myocardial ischemia/reperfusion in rats. Int J Cardiol 2013; 167:87–93.
Woo JS, Kim W, Ha SJ, et al. Cardioprotective effects of exenatide in patients with ST-segment-elevation myocardial infarction undergoing primary percutaneous coronary intervention: results of exenatide myocardial protection in revascularization study. Arterioscler Thromb Vasc Biol 2013; 33:2252–2260.
Lønborg J, Vejlstrup N, Kelbæk H, et al. Exenatide reduces reperfusion injury in patients with ST-segment elevation myocardial infarction. Eur Heart J 2012; 33:1491–1499.
van Poppel PC, Netea MG, Smits P, Tack CJ. Vildagliptin improves endothelium-dependent vasodilatation in type 2 diabetes. Diabetes Care 2011; 34:2072–2077.
Kröller-Schön S, Knorr M, Hausding M, et al. Glucose-independent improvement of vascular dysfunction in experimental sepsis by dipeptidyl-peptidase 4 inhibition. Cardiovasc Res 2012; 96:140–149.
Ta NN, Schuyler CA, Li Y, Lopes-Virella MF, Huang Y. DPP-4 (CD26) inhibitor alogliptin inhibits atherosclerosis in diabetic apolipoprotein E-deficient mice. J Cardiovasc Pharmacol 2011; 58:157–166.
Sauvé M, Ban K, Momen MA, et al. Genetic deletion or pharmacological inhibition of dipeptidyl peptidase-4 improves cardiovascular outcomes after myocardial infarction in mice. Diabetes 2010; 59:1063–1073.
Read PA, Khan FZ, Heck PM, Hoole SP, Dutka DP. DPP-4 inhibition by sitagliptin improves the myocardial response to dobutamine stress and mitigates stunning in a pilot study of patients with coronary artery disease. Circ Cardiovasc Imaging 2010; 3:195–201.
Matikainen N, Mänttäri S, Schweizer A, et al. Vildagliptin therapy reduces postprandial intestinal triglyceride-rich lipoprotein particles in patients with type 2 diabetes. Diabetologia 2006; 49:2049–2057.
Frederich R, Alexander JH, Fiedorek FT, et al. A systematic assessment of cardiovascular outcomes in the saxagliptin drug development program for type 2 diabetes. Postgrad Med 2010; 122:16–27.
Scirica BM, Bhatt DL, Braunwald E, et al; SAVOR-TIMI 53 Steering Committee and Investigators. Saxagliptin and cardiovascular outcomes in patients with type 2 diabetes mellitus. N Engl J Med 2013; 369:1317–1326.
Scirica BM, Braunwald E, Raz I, et al; SAVOR-TIMI 53 Steering Committee and Investigators. Heart failure, saxagliptin, and diabetes mellitus: observations from the SAVOR-TIMI 53 randomized trial. Circulation 2014; 130:1579–1588.
White WB, Cannon CP, Heller SR, et al; EXAMINE Investigators. Alogliptin after acute coronary syndrome in patients with type 2 diabetes. N Engl J Med 2013; 369:1327–1335.
Zannad F, Cannon CP, Cushman WC, et al; EXAMINE Investigators. Heart failure and mortality outcomes in patients with type 2 diabetes taking alogliptin versus placebo in EXAMINE: a multicentre, randomised, double-blind trial. Lancet 2015; 385:2067–2076.
Green JB, Bethel MA, Armstrong PW, et al; TECOS Study Group. Effect of sitagliptin on cardiovascular outcomes in type 2 diabetes. N Engl J Med 2015; 373:232–242.
McGuire DK, Van de Werf F, Armstrong PW, et al; Trial Evaluating Cardiovascular Outcomes With Sitagliptin (TECOS) Study Group. Association between sitagliptin use and heart failure hospitalization and related outcomes in type 2 diabetes mellitus: secondary analysis of a randomized clinical trial. JAMA Cardiol 2016; 1:126–135.
Pfeffer MA, Claggett B, Diaz R, et al; ELIXA Investigators. Lixisenatide in patients with type 2 diabetes and acute coronary syndrome. N Engl J Med 2015; 373:2247–2257.
Marso SP, Daniels GH, Brown-Frandsen K, et al; LEADER Steering Committee; LEADER Trial Investigators. Liraglutide and cardiovascular outcomes in type 2 diabetes. N Engl J Med 2016; 375:311–322.
Marso SP, Bain SC, Consoli A, et al; SUSTAIN-6 Investigators. Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N Engl J Med 2016; 375:1834–1844.
Rosenstock J, Balas B, Charbonnel B, et al; T-EMERGE 2 Study Group. The fate of taspoglutide, a weekly GLP-1 receptor agonist, versus twice-daily exenatide for type 2 diabetes: the T-EMERGE 2 trial. Diabetes Care 2013; 36:498–504.
Wright EM. Renal Na(+)-glucose cotransporters. Am J Physiol 2001; 280:F10–F18.
Lee YJ, Lee YJ, Han HJ. Regulatory mechanisms of Na(+)/glucose cotransporters in renal proximal tubule cells. Kidney Int 2007; 72(suppl 106):S27–S35.
Hummel CS, Lu C, Loo DD, Hirayama BA, Voss AA, Wright EM. Glucose transport by human renal Na+/D-glucose cotransporters SGLT1 and SGLT2. Am J Physiol Cell Physiol 2011; 300:C14–C21.
Heerspink HJ, Perkins BA, Fitchett DH, Husain M, Cherney DZ. Sodium glucose cotransporter 2 inhibitors in the treatment of diabetes mellitus: cardiovascular and kidney effects, potential mechanisms, and clinical applications. Circulation 2016; 134:752–772.
Lapuerta P, Zambrowicz, Strumph P, Sands A. Development of sotagliflozin, a dual sodium-dependent glucose transporter 1/2 inhibitor. Diabetes Vasc Dis Res 2015; 12:101–110.
Zinman B, Wanner C, Lachin JM, et al, for the EMPA-REG OUTCOME Investigators. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med 2015; 373:2117–2128.
Neal B, Vlado-Perkovic V, Mahaffey KW, et al, for the CANVAS Program Collaborative Group. Canagloflozin and cardiovascular and renal events in type 2 diabetes. N Engl J Med 2017; 377:644–657.
Inzucchi SE, Bergenstal RM, Buse JB, et al. Management of hyperglycemia in type 2 diabetes, 2015: a patient-centered approach: update to a position statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care 2015; 38:140–149.
Ferrannini E, Mark M, Mayoux E. CV protection in the EMPA-REG OUTCOME trial: a “thrifty substrate” hypothesis. Diabetes Care 2016; 39:1108–1114.
Cherney DZ, Perkins BA, Soleymanlou N, et al. Renal hemodynamic effect of sodium-glucose cotransporter 2 inhibition in patients with type 1 diabetes mellitus. Circulation 2014; 129:587–597.
Wanner C, Inzucchi SE, Lachin JM, et al, for the EMPA-REG OUTCOME Investigators. Empagliflozin and progression of kidney disease in type 2 diabetes. N Engl J Med 2016; 375:323–334.
Ponikowski P, Voors AA, Anker SD, et al; Authors/Task Force Members; Document Reviewers. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: the Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur J Heart Fail 2016; 18:891–975.
Piepoli MF, Hoes AW, Agewall S, et al; Authors/Task Force Members. 2016 European guidelines on cardiovascular disease prevention in clinical practice. The Sixth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (constituted by representatives of 10 societies and by invited experts). Developed with the special contribution of the European Association for Cardiovascular Prevention & Rehabilitation. Eur Heart J 2016; 37:2315–2381.
American Diabetes Association. American Diabetes Association standards of medical care in diabetes. Diabetes Care 2017; 40(suppl 1):S1–S135.
Lincoff AM, Tardif JC, Schwartz GG, et al; AleCardio Investigators. Effect of aleglitazar on cardiovascular outcomes after acute coronary syndrome in patients with type 2 diabetes mellitus: the AleCardio randomized clinical trial. JAMA 2014; 311:1515–1525.
Kaku K, Enya K, Nakaya R, Ohira T, Matsuno R. Efficacy and safety of fasiglifam (TAK0*&%), a G protein-coupled receptor 40 agonist, in Japanese patients with type 2 diabetes inadequately controlled by diet and exercise: a randomized, double-blind, placebocontrolled, phase III trial. Diabetes Obes Metab 2015; 17: 675–681.
Saxagliptin, alogliptin, and sitagliptin confer neither benefit nor harm for the composite outcome of cardiovascular death, myocardial infarction, or stroke. Saxagliptin and alogliptin carry warnings of increased risk of heart failure; sitagliptin was shown to not affect heart failure risk.
Liraglutide and semaglutide showed evidence of cardiovascular benefit; lixisenatide was noninferior to placebo.
Empagliflozin is now approved to reduce risk of cardiovascular death in patients with type 2 diabetes and atherosclerotic cardiovascular disease.
Canagliflozin decreased the composite outcome of cardiovascular death, nonfatal myocardial infarction, or nonfatal stroke in patients with type 2 diabetes with or at risk of cardiovascular disease, but also increased the risk of amputation and did not significantly reduce the individual outcome of cardiovascular death.
Since the first successful pregnancy in a kidney transplant recipient in 1958,1 hundreds of kidney recipients have had successful pregnancies. Chronic kidney disease disrupts the hypothalamic-pituitary-gonadal axis, leading to anovulation and infertility. However, within 6 months of kidney transplant, the hypothalamic-pituitary-gonadal axis and sex hormone levels return to normal,2 and the renal allograft is able to adapt to the various physiologic changes of pregnancy.3
Successful pregnancy after kidney transplant requires a team approach to care that includes the primary care physician, a transplant nephrologist, and an obstetrician with expertise in high-risk pregnancies. But equally important is educating and counseling the patient about the risks and challenges. This should begin at the first pretransplant visit.4
Below are answers to questions often asked by renal transplant recipients who wish to become pregnant.
WHAT IS THE IDEAL TIME TO BECOME PREGNANT AFTER KIDNEY TRANSPLANT?
According to American Society of Transplantation and European best-practice guidelines, as outlined in Table 1, the ideal time to conceive is 1 to 2 years after renal transplant if graft function is stable, proteinuria is minimal, there are no recent episodes of acute rejection, and the patient is not taking teratogenic medications. Because transplant recipients take teratogenic immunosuppressive drugs such as mycophenolate mofetil, women should be counseled to start contraception as soon as possible after kidney transplant.5,6
Mycophenolate mofetil and sirolimus are contraindicated in pregnancy and should be stopped at least 6 weeks before conception. Mycophenolate mofetil increases the risk of congenital malformations and spontaneous abortion. Data on sirolimus from clinical studies are limited, but in animal studies it is associated with delay in ossification of skeletal structure and with an increase in fetal mortality.7
WHAT INCREASES THE RISK OF A POOR PREGNANCY OUTCOME AFTER RENAL TRANSPLANT?
Risk factors for poor maternal and fetal outcomes include an elevated prepregnancy serum creatinine level (≥ 1.4 mg/dL), hypertension, and proteinuria (≥ 500 mg/24 hours). Younger age at transplant and at conception is associated with better pregnancy outcome.5,8
WHAT ARE THE POSSIBLE MATERNAL COMPLICATIONS?
Kidney transplant recipients who become pregnant have a risk of developing preeclampsia 6 times higher than normal, and the incidence rate ranges between 24% and 38%.9,10 The risk of cesarean delivery is 5 times higher than in the general population, and the incidence rate is 43% to 64%.10,11
Low-dose aspirin reduces the risk of preeclampsia and should be prescribed to all pregnant women who are kidney transplant recipients. Angiotensin-converting enzyme inhibitors are contraindicated due to the risk of teratogenic effects, ie, pulmonary hypoplasia and oligohydramnios.4
WHAT ARE THE POSSIBLE FETAL COMPLICATIONS?
Women who become pregnant after kidney transplant are at greater risk of preterm delivery (40% to 60% higher risk), having a baby with low birth weight (42% to 46% higher risk), and intrauterine growth restriction (30% to 50% higher risk). But the risk of perinatal mortality is not increased in the absence of the above-mentioned risk factors.10,11
DOES PREGNANCY INCREASE THE RISK OF GRAFT FAILURE?
Pregnancy does not increase the risk of allograft loss as long as the patient has a prepregnancy serum creatinine below 1.4 mg/dL, no hypertension, and urine protein excretion less than 500 mg/24 hours.12
WHAT CHANGES TO IMMUNE SUPPRESSION ARE REQUIRED BEFORE AND DURING PREGNANCY?
Careful management of immunosuppression is critical in renal transplant recipients before and during pregnancy because of the risks of teratogenicity and other adverse effects.
As stated above, mycophenolate mofetil and sirolimus are teratogenic and should be stopped 6 weeks before conception. The recommended maintenance immunosuppression during pregnancy includes calcineurin inhibitors (tacrolimus and cyclosporine), azathioprine, and low-dose prednisone.
A 20% to 25% increase in the dose of calcineurin inhibitor is required during pregnancy due to an increase in metabolic activity of cytochrome P450 and an increase in the volume of distribution.5,6,13 However, this dosing increase requires more frequent monitoring throughout the pregnancy to ensure the safest possible therapeutic levels.
DOES PREGNANCY INCREASE THE RISK OF INFECTION?
Because of their immunosuppressed state, renal transplant recipients are prone to infection; the incidence rate of urinary tract infection is as high as 40% due to mild reflux and pregnancy-related dilation of ureters and collecting ducts.6 Women should be screened for urinary tract infection at every visit with urine dipstick testing and with urine culture every 4 weeks. Antibiotics such as nitrofurantoin, amoxicillin, and cephalexin are safe to treat urinary tract infection during pregnancy.6
IS BREAST-FEEDING SAFE IN RENAL TRANSPLANT RECIPIENTS?
Breast-feeding is considered safe for women with renal transplant who are on prednisone, azathioprine, cyclosporine, and tacrolimus. Women should avoid breast-feeding if they are taking mycophenolate mofetil, sirolimus, everolimus, or belatacept, as clinical data on safety are not adequate.14
References
Murray JE, Reid DE, Harrison JH, Merrill JP. Successful pregnancies after human renal transplantation. N Engl J Med 1963; 269:341–343.
Saha MT, Saha HH, Niskanen LK, Salmela KT, Pasternack AI. Time course of serum prolactin and sex hormones following successful renal transplantation. Nephron 2002; 92:735–737.
Davison JM. The effect of pregnancy on kidney function in renal allograft recipients. Kidney Int 1985; 27:74–79.
Shah S, Verma P. Overview of pregnancy in renal transplant patients. Int J Nephrol 2016; 2016:4539342.
McKay DB, Josephson MA, Armenti VT, et al; Women’s Health Committee of the American Society of Transplantation. Reproduction and transplantation: report on the AST Consensus Conference on Reproductive Issues and Transplantation. Am J Transplant 2005; 5:1592–1599.
EBPG Expert Group on Renal Transplantation. European best practice guidelines for renal transplantation. Section IV: long-term management of the transplant recipient. IV.10. Pregnancy in renal transplant recipients. Nephrol Dial Transplant 2002; 17(suppl 4):50–55.
Armenti VT, Moitz MJ, Cardonick EH, Davison JM. Immunosuppression in pregnancy: choices for infant and maternal health. Drugs 2002; 62:2361–2375.
Bramham K, Chusney G, Lee J, Lightstone L, Nelson-Piercy C. Breastfeeding and tacrolimus: serial monitoring in breast-fed and bottle-fed infants. Clin J Am Soc Nephrol 2013; 8:563–567.
Deshpande NA, James NT, Kucirka LM, et al. Pregnancy outcomes in kidney transplant recipients: a systematic review and meta-analysis. Am J Transplant 2011; 11:2388–2404.
Bramham K, Nelson-Piercy C, Gao H, et al. Pregnancy in renal transplant recipients: a UK national cohort study. Clin J Am Soc Nephrol 2013; 8:290–298.
Coscia LA, Constantinescu S, Moritz MJ, et al. Report from the National Transplantation Pregnancy Registry (NTPR): outcomes of pregnancy after transplantation. Clin Transpl 2010: 65–85.
Sibanda N, Briggs JD, Davison JM, Johnson RJ, Rudge CJ. Pregnancy after organ transplantation: a report from the UK transplant pregnancy registry. Transplantation 2007; 83:1301–1307.
Kim H, Jeong JC, Yang J, et al. The optimal therapy of calcineurin inhibitors for pregnancy in kidney transplantation. Clin Transplant 2015; 29:142–148.
Constantinescu S, Pai A, Coscia LA, Davison JM, Moritz MJ, Armenti VT. Breast-feeding after transplantation. Best Pract Res Clin Obstet Gynaecol 2014; 28:1163–1173.
Silvi Shah, MD Division of Nephrology, University of Cincinnati, Cincinnati, OH
Address: Silvi Shah, MD, Division of Nephrology, University of Cincinnati, Department of Internal Medicine, 231 Albert Sabin Way, Medical Sciences Building Room 6065, PO Box 670557, Cincinnati, OH 45267-0557; silvishah2108@gmail.com
Silvi Shah, MD Division of Nephrology, University of Cincinnati, Cincinnati, OH
Address: Silvi Shah, MD, Division of Nephrology, University of Cincinnati, Department of Internal Medicine, 231 Albert Sabin Way, Medical Sciences Building Room 6065, PO Box 670557, Cincinnati, OH 45267-0557; silvishah2108@gmail.com
Author and Disclosure Information
Silvi Shah, MD Division of Nephrology, University of Cincinnati, Cincinnati, OH
Address: Silvi Shah, MD, Division of Nephrology, University of Cincinnati, Department of Internal Medicine, 231 Albert Sabin Way, Medical Sciences Building Room 6065, PO Box 670557, Cincinnati, OH 45267-0557; silvishah2108@gmail.com
Since the first successful pregnancy in a kidney transplant recipient in 1958,1 hundreds of kidney recipients have had successful pregnancies. Chronic kidney disease disrupts the hypothalamic-pituitary-gonadal axis, leading to anovulation and infertility. However, within 6 months of kidney transplant, the hypothalamic-pituitary-gonadal axis and sex hormone levels return to normal,2 and the renal allograft is able to adapt to the various physiologic changes of pregnancy.3
Successful pregnancy after kidney transplant requires a team approach to care that includes the primary care physician, a transplant nephrologist, and an obstetrician with expertise in high-risk pregnancies. But equally important is educating and counseling the patient about the risks and challenges. This should begin at the first pretransplant visit.4
Below are answers to questions often asked by renal transplant recipients who wish to become pregnant.
WHAT IS THE IDEAL TIME TO BECOME PREGNANT AFTER KIDNEY TRANSPLANT?
According to American Society of Transplantation and European best-practice guidelines, as outlined in Table 1, the ideal time to conceive is 1 to 2 years after renal transplant if graft function is stable, proteinuria is minimal, there are no recent episodes of acute rejection, and the patient is not taking teratogenic medications. Because transplant recipients take teratogenic immunosuppressive drugs such as mycophenolate mofetil, women should be counseled to start contraception as soon as possible after kidney transplant.5,6
Mycophenolate mofetil and sirolimus are contraindicated in pregnancy and should be stopped at least 6 weeks before conception. Mycophenolate mofetil increases the risk of congenital malformations and spontaneous abortion. Data on sirolimus from clinical studies are limited, but in animal studies it is associated with delay in ossification of skeletal structure and with an increase in fetal mortality.7
WHAT INCREASES THE RISK OF A POOR PREGNANCY OUTCOME AFTER RENAL TRANSPLANT?
Risk factors for poor maternal and fetal outcomes include an elevated prepregnancy serum creatinine level (≥ 1.4 mg/dL), hypertension, and proteinuria (≥ 500 mg/24 hours). Younger age at transplant and at conception is associated with better pregnancy outcome.5,8
WHAT ARE THE POSSIBLE MATERNAL COMPLICATIONS?
Kidney transplant recipients who become pregnant have a risk of developing preeclampsia 6 times higher than normal, and the incidence rate ranges between 24% and 38%.9,10 The risk of cesarean delivery is 5 times higher than in the general population, and the incidence rate is 43% to 64%.10,11
Low-dose aspirin reduces the risk of preeclampsia and should be prescribed to all pregnant women who are kidney transplant recipients. Angiotensin-converting enzyme inhibitors are contraindicated due to the risk of teratogenic effects, ie, pulmonary hypoplasia and oligohydramnios.4
WHAT ARE THE POSSIBLE FETAL COMPLICATIONS?
Women who become pregnant after kidney transplant are at greater risk of preterm delivery (40% to 60% higher risk), having a baby with low birth weight (42% to 46% higher risk), and intrauterine growth restriction (30% to 50% higher risk). But the risk of perinatal mortality is not increased in the absence of the above-mentioned risk factors.10,11
DOES PREGNANCY INCREASE THE RISK OF GRAFT FAILURE?
Pregnancy does not increase the risk of allograft loss as long as the patient has a prepregnancy serum creatinine below 1.4 mg/dL, no hypertension, and urine protein excretion less than 500 mg/24 hours.12
WHAT CHANGES TO IMMUNE SUPPRESSION ARE REQUIRED BEFORE AND DURING PREGNANCY?
Careful management of immunosuppression is critical in renal transplant recipients before and during pregnancy because of the risks of teratogenicity and other adverse effects.
As stated above, mycophenolate mofetil and sirolimus are teratogenic and should be stopped 6 weeks before conception. The recommended maintenance immunosuppression during pregnancy includes calcineurin inhibitors (tacrolimus and cyclosporine), azathioprine, and low-dose prednisone.
A 20% to 25% increase in the dose of calcineurin inhibitor is required during pregnancy due to an increase in metabolic activity of cytochrome P450 and an increase in the volume of distribution.5,6,13 However, this dosing increase requires more frequent monitoring throughout the pregnancy to ensure the safest possible therapeutic levels.
DOES PREGNANCY INCREASE THE RISK OF INFECTION?
Because of their immunosuppressed state, renal transplant recipients are prone to infection; the incidence rate of urinary tract infection is as high as 40% due to mild reflux and pregnancy-related dilation of ureters and collecting ducts.6 Women should be screened for urinary tract infection at every visit with urine dipstick testing and with urine culture every 4 weeks. Antibiotics such as nitrofurantoin, amoxicillin, and cephalexin are safe to treat urinary tract infection during pregnancy.6
IS BREAST-FEEDING SAFE IN RENAL TRANSPLANT RECIPIENTS?
Breast-feeding is considered safe for women with renal transplant who are on prednisone, azathioprine, cyclosporine, and tacrolimus. Women should avoid breast-feeding if they are taking mycophenolate mofetil, sirolimus, everolimus, or belatacept, as clinical data on safety are not adequate.14
Since the first successful pregnancy in a kidney transplant recipient in 1958,1 hundreds of kidney recipients have had successful pregnancies. Chronic kidney disease disrupts the hypothalamic-pituitary-gonadal axis, leading to anovulation and infertility. However, within 6 months of kidney transplant, the hypothalamic-pituitary-gonadal axis and sex hormone levels return to normal,2 and the renal allograft is able to adapt to the various physiologic changes of pregnancy.3
Successful pregnancy after kidney transplant requires a team approach to care that includes the primary care physician, a transplant nephrologist, and an obstetrician with expertise in high-risk pregnancies. But equally important is educating and counseling the patient about the risks and challenges. This should begin at the first pretransplant visit.4
Below are answers to questions often asked by renal transplant recipients who wish to become pregnant.
WHAT IS THE IDEAL TIME TO BECOME PREGNANT AFTER KIDNEY TRANSPLANT?
According to American Society of Transplantation and European best-practice guidelines, as outlined in Table 1, the ideal time to conceive is 1 to 2 years after renal transplant if graft function is stable, proteinuria is minimal, there are no recent episodes of acute rejection, and the patient is not taking teratogenic medications. Because transplant recipients take teratogenic immunosuppressive drugs such as mycophenolate mofetil, women should be counseled to start contraception as soon as possible after kidney transplant.5,6
Mycophenolate mofetil and sirolimus are contraindicated in pregnancy and should be stopped at least 6 weeks before conception. Mycophenolate mofetil increases the risk of congenital malformations and spontaneous abortion. Data on sirolimus from clinical studies are limited, but in animal studies it is associated with delay in ossification of skeletal structure and with an increase in fetal mortality.7
WHAT INCREASES THE RISK OF A POOR PREGNANCY OUTCOME AFTER RENAL TRANSPLANT?
Risk factors for poor maternal and fetal outcomes include an elevated prepregnancy serum creatinine level (≥ 1.4 mg/dL), hypertension, and proteinuria (≥ 500 mg/24 hours). Younger age at transplant and at conception is associated with better pregnancy outcome.5,8
WHAT ARE THE POSSIBLE MATERNAL COMPLICATIONS?
Kidney transplant recipients who become pregnant have a risk of developing preeclampsia 6 times higher than normal, and the incidence rate ranges between 24% and 38%.9,10 The risk of cesarean delivery is 5 times higher than in the general population, and the incidence rate is 43% to 64%.10,11
Low-dose aspirin reduces the risk of preeclampsia and should be prescribed to all pregnant women who are kidney transplant recipients. Angiotensin-converting enzyme inhibitors are contraindicated due to the risk of teratogenic effects, ie, pulmonary hypoplasia and oligohydramnios.4
WHAT ARE THE POSSIBLE FETAL COMPLICATIONS?
Women who become pregnant after kidney transplant are at greater risk of preterm delivery (40% to 60% higher risk), having a baby with low birth weight (42% to 46% higher risk), and intrauterine growth restriction (30% to 50% higher risk). But the risk of perinatal mortality is not increased in the absence of the above-mentioned risk factors.10,11
DOES PREGNANCY INCREASE THE RISK OF GRAFT FAILURE?
Pregnancy does not increase the risk of allograft loss as long as the patient has a prepregnancy serum creatinine below 1.4 mg/dL, no hypertension, and urine protein excretion less than 500 mg/24 hours.12
WHAT CHANGES TO IMMUNE SUPPRESSION ARE REQUIRED BEFORE AND DURING PREGNANCY?
Careful management of immunosuppression is critical in renal transplant recipients before and during pregnancy because of the risks of teratogenicity and other adverse effects.
As stated above, mycophenolate mofetil and sirolimus are teratogenic and should be stopped 6 weeks before conception. The recommended maintenance immunosuppression during pregnancy includes calcineurin inhibitors (tacrolimus and cyclosporine), azathioprine, and low-dose prednisone.
A 20% to 25% increase in the dose of calcineurin inhibitor is required during pregnancy due to an increase in metabolic activity of cytochrome P450 and an increase in the volume of distribution.5,6,13 However, this dosing increase requires more frequent monitoring throughout the pregnancy to ensure the safest possible therapeutic levels.
DOES PREGNANCY INCREASE THE RISK OF INFECTION?
Because of their immunosuppressed state, renal transplant recipients are prone to infection; the incidence rate of urinary tract infection is as high as 40% due to mild reflux and pregnancy-related dilation of ureters and collecting ducts.6 Women should be screened for urinary tract infection at every visit with urine dipstick testing and with urine culture every 4 weeks. Antibiotics such as nitrofurantoin, amoxicillin, and cephalexin are safe to treat urinary tract infection during pregnancy.6
IS BREAST-FEEDING SAFE IN RENAL TRANSPLANT RECIPIENTS?
Breast-feeding is considered safe for women with renal transplant who are on prednisone, azathioprine, cyclosporine, and tacrolimus. Women should avoid breast-feeding if they are taking mycophenolate mofetil, sirolimus, everolimus, or belatacept, as clinical data on safety are not adequate.14
References
Murray JE, Reid DE, Harrison JH, Merrill JP. Successful pregnancies after human renal transplantation. N Engl J Med 1963; 269:341–343.
Saha MT, Saha HH, Niskanen LK, Salmela KT, Pasternack AI. Time course of serum prolactin and sex hormones following successful renal transplantation. Nephron 2002; 92:735–737.
Davison JM. The effect of pregnancy on kidney function in renal allograft recipients. Kidney Int 1985; 27:74–79.
Shah S, Verma P. Overview of pregnancy in renal transplant patients. Int J Nephrol 2016; 2016:4539342.
McKay DB, Josephson MA, Armenti VT, et al; Women’s Health Committee of the American Society of Transplantation. Reproduction and transplantation: report on the AST Consensus Conference on Reproductive Issues and Transplantation. Am J Transplant 2005; 5:1592–1599.
EBPG Expert Group on Renal Transplantation. European best practice guidelines for renal transplantation. Section IV: long-term management of the transplant recipient. IV.10. Pregnancy in renal transplant recipients. Nephrol Dial Transplant 2002; 17(suppl 4):50–55.
Armenti VT, Moitz MJ, Cardonick EH, Davison JM. Immunosuppression in pregnancy: choices for infant and maternal health. Drugs 2002; 62:2361–2375.
Bramham K, Chusney G, Lee J, Lightstone L, Nelson-Piercy C. Breastfeeding and tacrolimus: serial monitoring in breast-fed and bottle-fed infants. Clin J Am Soc Nephrol 2013; 8:563–567.
Deshpande NA, James NT, Kucirka LM, et al. Pregnancy outcomes in kidney transplant recipients: a systematic review and meta-analysis. Am J Transplant 2011; 11:2388–2404.
Bramham K, Nelson-Piercy C, Gao H, et al. Pregnancy in renal transplant recipients: a UK national cohort study. Clin J Am Soc Nephrol 2013; 8:290–298.
Coscia LA, Constantinescu S, Moritz MJ, et al. Report from the National Transplantation Pregnancy Registry (NTPR): outcomes of pregnancy after transplantation. Clin Transpl 2010: 65–85.
Sibanda N, Briggs JD, Davison JM, Johnson RJ, Rudge CJ. Pregnancy after organ transplantation: a report from the UK transplant pregnancy registry. Transplantation 2007; 83:1301–1307.
Kim H, Jeong JC, Yang J, et al. The optimal therapy of calcineurin inhibitors for pregnancy in kidney transplantation. Clin Transplant 2015; 29:142–148.
Constantinescu S, Pai A, Coscia LA, Davison JM, Moritz MJ, Armenti VT. Breast-feeding after transplantation. Best Pract Res Clin Obstet Gynaecol 2014; 28:1163–1173.
References
Murray JE, Reid DE, Harrison JH, Merrill JP. Successful pregnancies after human renal transplantation. N Engl J Med 1963; 269:341–343.
Saha MT, Saha HH, Niskanen LK, Salmela KT, Pasternack AI. Time course of serum prolactin and sex hormones following successful renal transplantation. Nephron 2002; 92:735–737.
Davison JM. The effect of pregnancy on kidney function in renal allograft recipients. Kidney Int 1985; 27:74–79.
Shah S, Verma P. Overview of pregnancy in renal transplant patients. Int J Nephrol 2016; 2016:4539342.
McKay DB, Josephson MA, Armenti VT, et al; Women’s Health Committee of the American Society of Transplantation. Reproduction and transplantation: report on the AST Consensus Conference on Reproductive Issues and Transplantation. Am J Transplant 2005; 5:1592–1599.
EBPG Expert Group on Renal Transplantation. European best practice guidelines for renal transplantation. Section IV: long-term management of the transplant recipient. IV.10. Pregnancy in renal transplant recipients. Nephrol Dial Transplant 2002; 17(suppl 4):50–55.
Armenti VT, Moitz MJ, Cardonick EH, Davison JM. Immunosuppression in pregnancy: choices for infant and maternal health. Drugs 2002; 62:2361–2375.
Bramham K, Chusney G, Lee J, Lightstone L, Nelson-Piercy C. Breastfeeding and tacrolimus: serial monitoring in breast-fed and bottle-fed infants. Clin J Am Soc Nephrol 2013; 8:563–567.
Deshpande NA, James NT, Kucirka LM, et al. Pregnancy outcomes in kidney transplant recipients: a systematic review and meta-analysis. Am J Transplant 2011; 11:2388–2404.
Bramham K, Nelson-Piercy C, Gao H, et al. Pregnancy in renal transplant recipients: a UK national cohort study. Clin J Am Soc Nephrol 2013; 8:290–298.
Coscia LA, Constantinescu S, Moritz MJ, et al. Report from the National Transplantation Pregnancy Registry (NTPR): outcomes of pregnancy after transplantation. Clin Transpl 2010: 65–85.
Sibanda N, Briggs JD, Davison JM, Johnson RJ, Rudge CJ. Pregnancy after organ transplantation: a report from the UK transplant pregnancy registry. Transplantation 2007; 83:1301–1307.
Kim H, Jeong JC, Yang J, et al. The optimal therapy of calcineurin inhibitors for pregnancy in kidney transplantation. Clin Transplant 2015; 29:142–148.
Constantinescu S, Pai A, Coscia LA, Davison JM, Moritz MJ, Armenti VT. Breast-feeding after transplantation. Best Pract Res Clin Obstet Gynaecol 2014; 28:1163–1173.
Azithromycin is prescribed for a variety of acute respiratory and nonrespiratory infections. However, it is also used in several chronic respiratory diseases.
MECHANISM OF ACTION
Macrolide antibiotics like azithromycin inhibit bacterial growth and replication by interrupting protein synthesis. But azithromycin also has immunomodulatory properties.1
In the acute phase of inflammation, azithromycin exerts an initial neutrophil degranulation effect and enhances the oxidative response that is primed by particulate stimulus, which could facilitate its antibacterial effects. In the late phase, it down-regulates the oxidative burst and increases apoptosis of neutrophils to promote healing without compromising immunity. Azithromycin also attenuates airway mucus hypersecretion, improves ciliary function, and promotes pulmonary epithelial cell healing.2,3
Collectively, these effects make the drug effective in many chronic inflammatory lung conditions (Table 1).
CYSTIC FIBROSIS
Cystic fibrosis is a genetic disease affecting many organs, but its effect on the upper and lower airways has the greatest impact on quality of life and survival. Impaired mucociliary clearance and repeated respiratory infections contribute to chronic inflammation and a progressive decline in lung function.4,5
A 2012 Cochrane review of 5 studies in 549 patients found that, compared with those taking placebo, patients taking azithromycin 250–500 mg 3 times a week had improvement in forced expiratory volume in 1 second (FEV1). The mean difference at 6 months was 3.97% (95% cofidence interval [CI] 1.74– 6.19). Patients on azithromycin were free from pulmonary exacerbation approximately twice as long as patients on placebo (odds ratio 1.96, 95% CI 1.15–3.33).6,7
The Cystic Fibrosis Foundation recommends long-term azithromycin therapy to improve lung function and reduce exacerbations in patients age 6 or older who have persistent Pseudomonas aeruginosa airway cultures (level of evidence: fair).8
DIFFUSE PANBRONCHIOLITIS
Diffuse panbronchiolitis, or diffuse chronic inflammatory bronchiolitis and sinusitis, is seen mainly in patients of Asian descent.9 In the past, the mortality rate was greater than 90%, but between 1970 and 1979 the 10-year survival rate increased by more than 40% with chronic macrolide therapy, ie, with erythromycin.10,11
Later retrospective studies of azithromycin 500 mg 3 times a week showed results comparable to those with erythromycin, with improvement in symptoms, lung function, arterial partial pressure of oxygen, and radiologic findings, as well as fewer adverse effects.12 These benefits justify the current recommendation for azithromycin as the mainstay of therapy in diffuse panbronchiolitis.
BRONCHIOLITIS OBLITERANS SYNDROME
Bronchiolitis obliterans syndrome is an airflow limitation that arises without infection or imaging evidence of bronchiolitis in patients who received allogeneic hematopoietic stem cell or lung transplant. It occurs in 50% of lung transplant recipients as a form of chronic graft rejection and in 6% to 20% of allogeneic stem cell transplant recipients as a manifestation of chronic graft-vs-host disease.13,14
Azithromycin has been used in its management. A meta-analysis of lung transplant recipients found a significant improvement in the survival rate and overall lung function after an average of 7 months of treatment with azithromycin, with a mean increase in FEV1 of 8.8% (95% CI 5.1–12.47, P < .001).14 The evidence currently supports long-term azithromycin 250 mg 3 times a week after lung transplant to reduce any decline in lung function and to lower the mortality rate.14,15
In allogeneic stem cell transplant recipients, the evidence for long-term azithromycin treatment is sparse. A recent prospective multicenter study evaluated the effect of an azithromycin-based regimen (fluticasone, azithromycin, and montelukast, plus a steroid pulse) in stem cell recipients with bronchiolitis obliterans syndrome during the first 3 months after diagnosis. In the treated group, 6% had a drop in FEV1 of more than 10% at 3-month follow-up compared with 40% of historical controls (95% CI 1%–19%, P < .001). Also, treatment resulted in a 50% reduction in the dose of systemic steroids and a substantial improvement in functional status.16
Given the limited options in the management of these patients and until further studies are available, azithromycin 3 times weekly is suggested.
NON-CYSTIC FIBROSIS BRONCHIECTASIS
Non-cystic fibrosis bronchiectasis is a chronic inflammatory lung condition characterized by irreversible dilation of the bronchi and bronchioles due to a variety of causes including recurrent or old infection, immunodeficiency, autoimmune conditions, and connective tissue disease; it can also be idiopathic.17
Altenburg et al,18 in a randomized, double-blind, placebo-controlled trial, found that azithromycin 250 mg 3 times a week for 12 months reduced the number of exacerbations from a median number of 2 per patient with placebo to 0 per patient with azithromycin (P < .001). At 3 months, the FEV1 as a percent of predicted had increased by 1.03% in the azithromycin group and decreased by 0.10% in the placebo group (P = .047). The number needed to treat with azithromycin to maintain clinical stability was 3.0.
Wong et al19 randomized patients to receive azithromycin 500 mg 3 times a week or placebo for 6 months. The rate of exacerbations was 0.59 per patient in the azithromycin group and 1.57 per patient in the placebo group (P < .0001). The FEV1 remained unchanged from baseline in the azithromycin group while decreasing in the placebo group, but the difference was not significant.
EXACERBATIONS OF CHRONIC OBSTRUCTIVE PULMONARY DISEASE
Acute exacerbations of chronic obstructive pulmonary disease (COPD) are a major cause of death, poor quality of life, and healthcare expenditures.20 Prevention is therefore of the utmost importance.
Several studies have shown that azithromycin prophylaxis can reduce acute exacerbations of COPD. A recent meta-analysis showed that long-term macrolide prophylaxis significantly reduced exacerbations compared with rates in controls (risk ratio = 0.70, 95% CI 0.56–0.87, P < .01) and increased the median time to first COPD exacerbation by more than 90 days (P < .01).21 Long-term azithromycin therapy may be considered in selected patients who have frequent exacerbations despite optimal maintenance inhaler therapy.
PROPHYLAXIS IN IMMUNODEFICIENCY
Disseminated Mycobacterium avium complex (MAC) is an opportunistic infection most commonly occurring in patients with acquired immunodeficiency syndrome with CD4 counts below 50 cells/µL.22,23
In a double-blinded, randomized trial, patients who received azithromycin had a 47% reduction in the incidence of MAC infection.
Given the long half-life of azithromycin, it is effective with once-weekly dosing of 1,200 mg.23 Ideally, patients are placed on a prophylactic agent for disseminated MAC infection until the CD4 count reaches 100 cells/µL and remains at or above this level for 3 consecutive months.24
ADVERSE EFFECTS AND PRECAUTIONS
Long-term azithromycin therapy may produce bacterial resistance; the risk has been estimated at 2.7 times greater in patients who are on long-term azithromycin treatment.25 Also, patients at risk for MAC infection, such as those with cystic fibrosis, should be screened for it before starting treatment in order to prevent resistance to azithromycin.
The US Food and Drug Administration warns that azithromycin can lead to a prolonged corrected QT interval and potential fatal arrhythmias such as torsades de pointes. Major reviews have largely agreed that arrhythmias are more pronounced in patients with a coexisting cardiac risk factor such as existing QT-interval prolongation, low blood levels of potassium or magnesium, a slower than normal heart rate, or arrhythmias, or who are on class IA and III antiarrhythmic drugs.26–28
Other potential adverse effects of long-term azithromycin treatment are gastrointestinal symptoms and hearing impairment.29,30 A review of potential drug interactions is advised when patients are placed on long-term azithromycin therapy.
Although azithromycin is generally well tolerated, long-term treatment should be individualized and the benefits weighed against the risks. Patients should be monitored during treatment for any of the above adverse effects.
References
Bailly S, Pocidalo JJ, Fay M, Gougerot-Pocidalo MA. Differential modulation of cytokine production by macrolides: interleukin-6 production is increased by spiramycin and erythromycin. Antimicrob Agents Chemother 1991; 35:2016–2019.
Kanoh S, Rubin BK. Mechanisms of action and clinical application of macrolides as immunomodulatory medications. Clin Microbiol Rev 2010; 23:590–615.
Culić O, Eraković V, Cepelak I, et al. Azithromycin modulates neutrophil function and circulating inflammatory mediators in healthy human subjects. Eur J Pharmacol 2002; 450:277–289.
Cohen-Cymberknoh M, Kerem E, Ferkol T, Elizur A. Airway inflammation in cystic fibrosis: molecular mechanisms and clinical implications. Thorax 2013; 68:1157–1162.
Sagel SD, Wagner BD, Anthony MM, Emmett P, Zemanick ET. Sputum biomarkers of inflammation and lung function decline in children with cystic fibrosis. Am J Respir Crit Care Med 2012; 186:857–865.
Saiman L, Anstead M, Mayer-Hamblett N, et al; AZ0004 Azithromycin Study Group. Effect of azithromycin on pulmonary function in patients with cystic fibrosis uninfected with Pseudomonas aeruginosa: a randomized controlled trial. JAMA 2010; 303:1707–1715.
Southern KW, Barker PM, Solis-Moya A, Patel L. Macrolide antibiotics for cystic fibrosis. Cochrane Database Syst Rev 2012;11:CD002203.
Flume PA, O’Sullivan BP, Robinson KA, et al; Cystic Fibrosis Foundation, Pulmonary Therapies Committee. Cystic fibrosis pulmonary guidelines: chronic medications for maintenance of lung health. Am J Respir Crit Care Med 2007; 176:957–969.
Yanagihara K, Kadoto J, Kohno S. Diffuse panbronchiolitis—pathophysiology and treatment mechanisms. Int J Antimicrob Agents 2001; 18(suppl 1):S83–S87.
Kudoh S, Azuma A, Yamamoto M, Izumi T, Ando M. Improvement of survival in patients with diffuse panbronchiolitis treated with low-dose erythromycin. Am J Respir Crit Care Med 1998; 157:1829–1832.
Schultz MJ. Macrolide activities beyond their antimicrobial effects: macrolides in diffuse panbronchiolitis and cystic fibrosis. J Antimicrob Chemother 2004; 54:21–28.
Hui D, Yan F, Chen RH. The effects of azithromycin on patients with diffuse panbronchiolitis: a retrospective study of 29 cases. J Thorac Dis 2013; 5:613–617.
Khalid M, Al Saghir A, Saleemi S, et al. Azithromycin in bronchiolitis obliterans complicating bone marrow transplantation: a preliminary study. Eur Respir J 2005; 25:490–493.
Kingah PL, Muma G, Soubani A. Azithromycin improves lung function in patients with post-lung transplant bronchiolitis obliterans syndrome: a meta-analysis. Clin Transplant 2014; 28:906–910.
Corris PA, Ryan VA, Small T, et al. A randomised controlled trial of azithromycin therapy in bronchiolitis obliterans syndrome (BOS) post lung transplantation. Thorax 2015; 70:442–450.
Williams KM, Cheng GS, Pusic I, et al. Fluticasone, azithromycin, and montelukast treatment for new-onset bronchiolitis obliterans syndrome after hematopoietic cell transplantation. Biol Blood Marrow Transplant 2016; 22:710–716.
Haworth CS, Bilton D, Elborn JS. Long-term macrolide maintenance therapy in non-CF bronchiectasis: evidence and questions. Respir Med 2014; 108:1397–1408.
Altenburg J, de Graaff CS, Stienstra Y, et al. Effect of azithromycin maintenance treatment on infectious exacerbations among patients with non-cystic fibrosis bronchiectasis: the BAT randomized controlled trial. JAMA 2013; 309:1251–1259.
Wong C, Jayaram L, Karalus N, et al. Azithromycin for prevention of exacerbations in non-cystic fibrosis bronchiectasis (EMBRACE): a randomised, double-blind, placebo-controlled trial. Lancet 2012; 380:660–667.
Suissa S, Dell’Aniello S, Ernst P. Long-term natural history of chronic obstructive pulmonary disease: severe exacerbations and mortality. Thorax 2012; 67:957–963.
Ni W, Shao X, Cai X, et al. Prophylactic use of macrolide antibiotics for the prevention of chronic obstructive pulmonary disease exacerbation: a meta-analysis. PLoS One 2015; 10:e0121257.
Griffith DE, Aksamit T, Brown-Elliott BA, et al; ATS Mycobacterial Diseases Subcommittee; American Thoracic Society; Infectious Disease Society of America. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med 2007; 175:367–416.
Havlir DV, Dubé MP, Sattler FR, et al. Prophylaxis against disseminated Mycobacterium avium complex with weekly azithromycin, daily rifabutin, or both. California Collaborative Treatment Group. N Engl J Med 1996; 335:392–398.
Uthman MM, Uthman OA, Yahaya I. Interventions for the prevention of Mycobacterium avium complex in adults and children with HIV. Cochrane Database Syst Rev 2013; 4:CD007191.
Li H, Liu DH, Chen LL, et al. Meta-analysis of the adverse effects of long-term azithromycin use in patients with chronic lung diseases. Antimicrob Agents Chemother 2014; 58:511–517.
Svanström H, Pasternak B, Hviid A. Use of azithromycin and death from cardiovascular causes. N Engl J Med 2013; 368:1704–1712.
Albert RK, Schuller JL; COPD Clinical Research Network. Macrolide antibiotics and the risk of cardiac arrhythmias. Am J Respir Crit Care Med 2014; 189:1173–1180.
Ray WA, Murray KT, Hall K, Arbogast PG, Stein CM. Azithromycin and the risk of cardiovascular death. N Engl J Med 2012; 366:1881–1890.
Albert RK, Connett J, Bailey WC, et al; COPD Clinical Research Network. Azithromycin for prevention of exacerbations of COPD. N Engl J Med 2011; 365:689–698.
Broad J, Sanger GJ. The antibiotic azithromycin is a motilin receptor agonist in human stomach: comparison with erythromycin. Br J Pharmacol 2013; 168:1859–1867.
Abdulrazak Alchakaki, MD Division of Pulmonary, Critical Care, and Sleep Medicine, Wayne State University School of Medicine, Detroit, MI
Cassondra Cramer Division of Pulmonary, Critical Care and Sleep Medicine, Wayne State University School of Medicine, Detroit, MI
Allie Patterson Division of Pulmonary, Critical Care, and Sleep Medicine, Wayne State University School of Medicine, Detroit, MI
Ayman O. Soubani Division of Pulmonary, Critical Care, and Sleep Medicine, Wayne State University School of Medicine, Detroit, MI
Address: Abdulrazak Alchakaki, MD, Division of Pulmonary, Critical Care, and Sleep Medicine, Wayne State University School of Medicine, 3990 John R, 3 Hudson, Detroit, MI 48201; aalchaka@med.wayne.edu
Abdulrazak Alchakaki, MD Division of Pulmonary, Critical Care, and Sleep Medicine, Wayne State University School of Medicine, Detroit, MI
Cassondra Cramer Division of Pulmonary, Critical Care and Sleep Medicine, Wayne State University School of Medicine, Detroit, MI
Allie Patterson Division of Pulmonary, Critical Care, and Sleep Medicine, Wayne State University School of Medicine, Detroit, MI
Ayman O. Soubani Division of Pulmonary, Critical Care, and Sleep Medicine, Wayne State University School of Medicine, Detroit, MI
Address: Abdulrazak Alchakaki, MD, Division of Pulmonary, Critical Care, and Sleep Medicine, Wayne State University School of Medicine, 3990 John R, 3 Hudson, Detroit, MI 48201; aalchaka@med.wayne.edu
Author and Disclosure Information
Abdulrazak Alchakaki, MD Division of Pulmonary, Critical Care, and Sleep Medicine, Wayne State University School of Medicine, Detroit, MI
Cassondra Cramer Division of Pulmonary, Critical Care and Sleep Medicine, Wayne State University School of Medicine, Detroit, MI
Allie Patterson Division of Pulmonary, Critical Care, and Sleep Medicine, Wayne State University School of Medicine, Detroit, MI
Ayman O. Soubani Division of Pulmonary, Critical Care, and Sleep Medicine, Wayne State University School of Medicine, Detroit, MI
Address: Abdulrazak Alchakaki, MD, Division of Pulmonary, Critical Care, and Sleep Medicine, Wayne State University School of Medicine, 3990 John R, 3 Hudson, Detroit, MI 48201; aalchaka@med.wayne.edu
Azithromycin is prescribed for a variety of acute respiratory and nonrespiratory infections. However, it is also used in several chronic respiratory diseases.
MECHANISM OF ACTION
Macrolide antibiotics like azithromycin inhibit bacterial growth and replication by interrupting protein synthesis. But azithromycin also has immunomodulatory properties.1
In the acute phase of inflammation, azithromycin exerts an initial neutrophil degranulation effect and enhances the oxidative response that is primed by particulate stimulus, which could facilitate its antibacterial effects. In the late phase, it down-regulates the oxidative burst and increases apoptosis of neutrophils to promote healing without compromising immunity. Azithromycin also attenuates airway mucus hypersecretion, improves ciliary function, and promotes pulmonary epithelial cell healing.2,3
Collectively, these effects make the drug effective in many chronic inflammatory lung conditions (Table 1).
CYSTIC FIBROSIS
Cystic fibrosis is a genetic disease affecting many organs, but its effect on the upper and lower airways has the greatest impact on quality of life and survival. Impaired mucociliary clearance and repeated respiratory infections contribute to chronic inflammation and a progressive decline in lung function.4,5
A 2012 Cochrane review of 5 studies in 549 patients found that, compared with those taking placebo, patients taking azithromycin 250–500 mg 3 times a week had improvement in forced expiratory volume in 1 second (FEV1). The mean difference at 6 months was 3.97% (95% cofidence interval [CI] 1.74– 6.19). Patients on azithromycin were free from pulmonary exacerbation approximately twice as long as patients on placebo (odds ratio 1.96, 95% CI 1.15–3.33).6,7
The Cystic Fibrosis Foundation recommends long-term azithromycin therapy to improve lung function and reduce exacerbations in patients age 6 or older who have persistent Pseudomonas aeruginosa airway cultures (level of evidence: fair).8
DIFFUSE PANBRONCHIOLITIS
Diffuse panbronchiolitis, or diffuse chronic inflammatory bronchiolitis and sinusitis, is seen mainly in patients of Asian descent.9 In the past, the mortality rate was greater than 90%, but between 1970 and 1979 the 10-year survival rate increased by more than 40% with chronic macrolide therapy, ie, with erythromycin.10,11
Later retrospective studies of azithromycin 500 mg 3 times a week showed results comparable to those with erythromycin, with improvement in symptoms, lung function, arterial partial pressure of oxygen, and radiologic findings, as well as fewer adverse effects.12 These benefits justify the current recommendation for azithromycin as the mainstay of therapy in diffuse panbronchiolitis.
BRONCHIOLITIS OBLITERANS SYNDROME
Bronchiolitis obliterans syndrome is an airflow limitation that arises without infection or imaging evidence of bronchiolitis in patients who received allogeneic hematopoietic stem cell or lung transplant. It occurs in 50% of lung transplant recipients as a form of chronic graft rejection and in 6% to 20% of allogeneic stem cell transplant recipients as a manifestation of chronic graft-vs-host disease.13,14
Azithromycin has been used in its management. A meta-analysis of lung transplant recipients found a significant improvement in the survival rate and overall lung function after an average of 7 months of treatment with azithromycin, with a mean increase in FEV1 of 8.8% (95% CI 5.1–12.47, P < .001).14 The evidence currently supports long-term azithromycin 250 mg 3 times a week after lung transplant to reduce any decline in lung function and to lower the mortality rate.14,15
In allogeneic stem cell transplant recipients, the evidence for long-term azithromycin treatment is sparse. A recent prospective multicenter study evaluated the effect of an azithromycin-based regimen (fluticasone, azithromycin, and montelukast, plus a steroid pulse) in stem cell recipients with bronchiolitis obliterans syndrome during the first 3 months after diagnosis. In the treated group, 6% had a drop in FEV1 of more than 10% at 3-month follow-up compared with 40% of historical controls (95% CI 1%–19%, P < .001). Also, treatment resulted in a 50% reduction in the dose of systemic steroids and a substantial improvement in functional status.16
Given the limited options in the management of these patients and until further studies are available, azithromycin 3 times weekly is suggested.
NON-CYSTIC FIBROSIS BRONCHIECTASIS
Non-cystic fibrosis bronchiectasis is a chronic inflammatory lung condition characterized by irreversible dilation of the bronchi and bronchioles due to a variety of causes including recurrent or old infection, immunodeficiency, autoimmune conditions, and connective tissue disease; it can also be idiopathic.17
Altenburg et al,18 in a randomized, double-blind, placebo-controlled trial, found that azithromycin 250 mg 3 times a week for 12 months reduced the number of exacerbations from a median number of 2 per patient with placebo to 0 per patient with azithromycin (P < .001). At 3 months, the FEV1 as a percent of predicted had increased by 1.03% in the azithromycin group and decreased by 0.10% in the placebo group (P = .047). The number needed to treat with azithromycin to maintain clinical stability was 3.0.
Wong et al19 randomized patients to receive azithromycin 500 mg 3 times a week or placebo for 6 months. The rate of exacerbations was 0.59 per patient in the azithromycin group and 1.57 per patient in the placebo group (P < .0001). The FEV1 remained unchanged from baseline in the azithromycin group while decreasing in the placebo group, but the difference was not significant.
EXACERBATIONS OF CHRONIC OBSTRUCTIVE PULMONARY DISEASE
Acute exacerbations of chronic obstructive pulmonary disease (COPD) are a major cause of death, poor quality of life, and healthcare expenditures.20 Prevention is therefore of the utmost importance.
Several studies have shown that azithromycin prophylaxis can reduce acute exacerbations of COPD. A recent meta-analysis showed that long-term macrolide prophylaxis significantly reduced exacerbations compared with rates in controls (risk ratio = 0.70, 95% CI 0.56–0.87, P < .01) and increased the median time to first COPD exacerbation by more than 90 days (P < .01).21 Long-term azithromycin therapy may be considered in selected patients who have frequent exacerbations despite optimal maintenance inhaler therapy.
PROPHYLAXIS IN IMMUNODEFICIENCY
Disseminated Mycobacterium avium complex (MAC) is an opportunistic infection most commonly occurring in patients with acquired immunodeficiency syndrome with CD4 counts below 50 cells/µL.22,23
In a double-blinded, randomized trial, patients who received azithromycin had a 47% reduction in the incidence of MAC infection.
Given the long half-life of azithromycin, it is effective with once-weekly dosing of 1,200 mg.23 Ideally, patients are placed on a prophylactic agent for disseminated MAC infection until the CD4 count reaches 100 cells/µL and remains at or above this level for 3 consecutive months.24
ADVERSE EFFECTS AND PRECAUTIONS
Long-term azithromycin therapy may produce bacterial resistance; the risk has been estimated at 2.7 times greater in patients who are on long-term azithromycin treatment.25 Also, patients at risk for MAC infection, such as those with cystic fibrosis, should be screened for it before starting treatment in order to prevent resistance to azithromycin.
The US Food and Drug Administration warns that azithromycin can lead to a prolonged corrected QT interval and potential fatal arrhythmias such as torsades de pointes. Major reviews have largely agreed that arrhythmias are more pronounced in patients with a coexisting cardiac risk factor such as existing QT-interval prolongation, low blood levels of potassium or magnesium, a slower than normal heart rate, or arrhythmias, or who are on class IA and III antiarrhythmic drugs.26–28
Other potential adverse effects of long-term azithromycin treatment are gastrointestinal symptoms and hearing impairment.29,30 A review of potential drug interactions is advised when patients are placed on long-term azithromycin therapy.
Although azithromycin is generally well tolerated, long-term treatment should be individualized and the benefits weighed against the risks. Patients should be monitored during treatment for any of the above adverse effects.
Azithromycin is prescribed for a variety of acute respiratory and nonrespiratory infections. However, it is also used in several chronic respiratory diseases.
MECHANISM OF ACTION
Macrolide antibiotics like azithromycin inhibit bacterial growth and replication by interrupting protein synthesis. But azithromycin also has immunomodulatory properties.1
In the acute phase of inflammation, azithromycin exerts an initial neutrophil degranulation effect and enhances the oxidative response that is primed by particulate stimulus, which could facilitate its antibacterial effects. In the late phase, it down-regulates the oxidative burst and increases apoptosis of neutrophils to promote healing without compromising immunity. Azithromycin also attenuates airway mucus hypersecretion, improves ciliary function, and promotes pulmonary epithelial cell healing.2,3
Collectively, these effects make the drug effective in many chronic inflammatory lung conditions (Table 1).
CYSTIC FIBROSIS
Cystic fibrosis is a genetic disease affecting many organs, but its effect on the upper and lower airways has the greatest impact on quality of life and survival. Impaired mucociliary clearance and repeated respiratory infections contribute to chronic inflammation and a progressive decline in lung function.4,5
A 2012 Cochrane review of 5 studies in 549 patients found that, compared with those taking placebo, patients taking azithromycin 250–500 mg 3 times a week had improvement in forced expiratory volume in 1 second (FEV1). The mean difference at 6 months was 3.97% (95% cofidence interval [CI] 1.74– 6.19). Patients on azithromycin were free from pulmonary exacerbation approximately twice as long as patients on placebo (odds ratio 1.96, 95% CI 1.15–3.33).6,7
The Cystic Fibrosis Foundation recommends long-term azithromycin therapy to improve lung function and reduce exacerbations in patients age 6 or older who have persistent Pseudomonas aeruginosa airway cultures (level of evidence: fair).8
DIFFUSE PANBRONCHIOLITIS
Diffuse panbronchiolitis, or diffuse chronic inflammatory bronchiolitis and sinusitis, is seen mainly in patients of Asian descent.9 In the past, the mortality rate was greater than 90%, but between 1970 and 1979 the 10-year survival rate increased by more than 40% with chronic macrolide therapy, ie, with erythromycin.10,11
Later retrospective studies of azithromycin 500 mg 3 times a week showed results comparable to those with erythromycin, with improvement in symptoms, lung function, arterial partial pressure of oxygen, and radiologic findings, as well as fewer adverse effects.12 These benefits justify the current recommendation for azithromycin as the mainstay of therapy in diffuse panbronchiolitis.
BRONCHIOLITIS OBLITERANS SYNDROME
Bronchiolitis obliterans syndrome is an airflow limitation that arises without infection or imaging evidence of bronchiolitis in patients who received allogeneic hematopoietic stem cell or lung transplant. It occurs in 50% of lung transplant recipients as a form of chronic graft rejection and in 6% to 20% of allogeneic stem cell transplant recipients as a manifestation of chronic graft-vs-host disease.13,14
Azithromycin has been used in its management. A meta-analysis of lung transplant recipients found a significant improvement in the survival rate and overall lung function after an average of 7 months of treatment with azithromycin, with a mean increase in FEV1 of 8.8% (95% CI 5.1–12.47, P < .001).14 The evidence currently supports long-term azithromycin 250 mg 3 times a week after lung transplant to reduce any decline in lung function and to lower the mortality rate.14,15
In allogeneic stem cell transplant recipients, the evidence for long-term azithromycin treatment is sparse. A recent prospective multicenter study evaluated the effect of an azithromycin-based regimen (fluticasone, azithromycin, and montelukast, plus a steroid pulse) in stem cell recipients with bronchiolitis obliterans syndrome during the first 3 months after diagnosis. In the treated group, 6% had a drop in FEV1 of more than 10% at 3-month follow-up compared with 40% of historical controls (95% CI 1%–19%, P < .001). Also, treatment resulted in a 50% reduction in the dose of systemic steroids and a substantial improvement in functional status.16
Given the limited options in the management of these patients and until further studies are available, azithromycin 3 times weekly is suggested.
NON-CYSTIC FIBROSIS BRONCHIECTASIS
Non-cystic fibrosis bronchiectasis is a chronic inflammatory lung condition characterized by irreversible dilation of the bronchi and bronchioles due to a variety of causes including recurrent or old infection, immunodeficiency, autoimmune conditions, and connective tissue disease; it can also be idiopathic.17
Altenburg et al,18 in a randomized, double-blind, placebo-controlled trial, found that azithromycin 250 mg 3 times a week for 12 months reduced the number of exacerbations from a median number of 2 per patient with placebo to 0 per patient with azithromycin (P < .001). At 3 months, the FEV1 as a percent of predicted had increased by 1.03% in the azithromycin group and decreased by 0.10% in the placebo group (P = .047). The number needed to treat with azithromycin to maintain clinical stability was 3.0.
Wong et al19 randomized patients to receive azithromycin 500 mg 3 times a week or placebo for 6 months. The rate of exacerbations was 0.59 per patient in the azithromycin group and 1.57 per patient in the placebo group (P < .0001). The FEV1 remained unchanged from baseline in the azithromycin group while decreasing in the placebo group, but the difference was not significant.
EXACERBATIONS OF CHRONIC OBSTRUCTIVE PULMONARY DISEASE
Acute exacerbations of chronic obstructive pulmonary disease (COPD) are a major cause of death, poor quality of life, and healthcare expenditures.20 Prevention is therefore of the utmost importance.
Several studies have shown that azithromycin prophylaxis can reduce acute exacerbations of COPD. A recent meta-analysis showed that long-term macrolide prophylaxis significantly reduced exacerbations compared with rates in controls (risk ratio = 0.70, 95% CI 0.56–0.87, P < .01) and increased the median time to first COPD exacerbation by more than 90 days (P < .01).21 Long-term azithromycin therapy may be considered in selected patients who have frequent exacerbations despite optimal maintenance inhaler therapy.
PROPHYLAXIS IN IMMUNODEFICIENCY
Disseminated Mycobacterium avium complex (MAC) is an opportunistic infection most commonly occurring in patients with acquired immunodeficiency syndrome with CD4 counts below 50 cells/µL.22,23
In a double-blinded, randomized trial, patients who received azithromycin had a 47% reduction in the incidence of MAC infection.
Given the long half-life of azithromycin, it is effective with once-weekly dosing of 1,200 mg.23 Ideally, patients are placed on a prophylactic agent for disseminated MAC infection until the CD4 count reaches 100 cells/µL and remains at or above this level for 3 consecutive months.24
ADVERSE EFFECTS AND PRECAUTIONS
Long-term azithromycin therapy may produce bacterial resistance; the risk has been estimated at 2.7 times greater in patients who are on long-term azithromycin treatment.25 Also, patients at risk for MAC infection, such as those with cystic fibrosis, should be screened for it before starting treatment in order to prevent resistance to azithromycin.
The US Food and Drug Administration warns that azithromycin can lead to a prolonged corrected QT interval and potential fatal arrhythmias such as torsades de pointes. Major reviews have largely agreed that arrhythmias are more pronounced in patients with a coexisting cardiac risk factor such as existing QT-interval prolongation, low blood levels of potassium or magnesium, a slower than normal heart rate, or arrhythmias, or who are on class IA and III antiarrhythmic drugs.26–28
Other potential adverse effects of long-term azithromycin treatment are gastrointestinal symptoms and hearing impairment.29,30 A review of potential drug interactions is advised when patients are placed on long-term azithromycin therapy.
Although azithromycin is generally well tolerated, long-term treatment should be individualized and the benefits weighed against the risks. Patients should be monitored during treatment for any of the above adverse effects.
References
Bailly S, Pocidalo JJ, Fay M, Gougerot-Pocidalo MA. Differential modulation of cytokine production by macrolides: interleukin-6 production is increased by spiramycin and erythromycin. Antimicrob Agents Chemother 1991; 35:2016–2019.
Kanoh S, Rubin BK. Mechanisms of action and clinical application of macrolides as immunomodulatory medications. Clin Microbiol Rev 2010; 23:590–615.
Culić O, Eraković V, Cepelak I, et al. Azithromycin modulates neutrophil function and circulating inflammatory mediators in healthy human subjects. Eur J Pharmacol 2002; 450:277–289.
Cohen-Cymberknoh M, Kerem E, Ferkol T, Elizur A. Airway inflammation in cystic fibrosis: molecular mechanisms and clinical implications. Thorax 2013; 68:1157–1162.
Sagel SD, Wagner BD, Anthony MM, Emmett P, Zemanick ET. Sputum biomarkers of inflammation and lung function decline in children with cystic fibrosis. Am J Respir Crit Care Med 2012; 186:857–865.
Saiman L, Anstead M, Mayer-Hamblett N, et al; AZ0004 Azithromycin Study Group. Effect of azithromycin on pulmonary function in patients with cystic fibrosis uninfected with Pseudomonas aeruginosa: a randomized controlled trial. JAMA 2010; 303:1707–1715.
Southern KW, Barker PM, Solis-Moya A, Patel L. Macrolide antibiotics for cystic fibrosis. Cochrane Database Syst Rev 2012;11:CD002203.
Flume PA, O’Sullivan BP, Robinson KA, et al; Cystic Fibrosis Foundation, Pulmonary Therapies Committee. Cystic fibrosis pulmonary guidelines: chronic medications for maintenance of lung health. Am J Respir Crit Care Med 2007; 176:957–969.
Yanagihara K, Kadoto J, Kohno S. Diffuse panbronchiolitis—pathophysiology and treatment mechanisms. Int J Antimicrob Agents 2001; 18(suppl 1):S83–S87.
Kudoh S, Azuma A, Yamamoto M, Izumi T, Ando M. Improvement of survival in patients with diffuse panbronchiolitis treated with low-dose erythromycin. Am J Respir Crit Care Med 1998; 157:1829–1832.
Schultz MJ. Macrolide activities beyond their antimicrobial effects: macrolides in diffuse panbronchiolitis and cystic fibrosis. J Antimicrob Chemother 2004; 54:21–28.
Hui D, Yan F, Chen RH. The effects of azithromycin on patients with diffuse panbronchiolitis: a retrospective study of 29 cases. J Thorac Dis 2013; 5:613–617.
Khalid M, Al Saghir A, Saleemi S, et al. Azithromycin in bronchiolitis obliterans complicating bone marrow transplantation: a preliminary study. Eur Respir J 2005; 25:490–493.
Kingah PL, Muma G, Soubani A. Azithromycin improves lung function in patients with post-lung transplant bronchiolitis obliterans syndrome: a meta-analysis. Clin Transplant 2014; 28:906–910.
Corris PA, Ryan VA, Small T, et al. A randomised controlled trial of azithromycin therapy in bronchiolitis obliterans syndrome (BOS) post lung transplantation. Thorax 2015; 70:442–450.
Williams KM, Cheng GS, Pusic I, et al. Fluticasone, azithromycin, and montelukast treatment for new-onset bronchiolitis obliterans syndrome after hematopoietic cell transplantation. Biol Blood Marrow Transplant 2016; 22:710–716.
Haworth CS, Bilton D, Elborn JS. Long-term macrolide maintenance therapy in non-CF bronchiectasis: evidence and questions. Respir Med 2014; 108:1397–1408.
Altenburg J, de Graaff CS, Stienstra Y, et al. Effect of azithromycin maintenance treatment on infectious exacerbations among patients with non-cystic fibrosis bronchiectasis: the BAT randomized controlled trial. JAMA 2013; 309:1251–1259.
Wong C, Jayaram L, Karalus N, et al. Azithromycin for prevention of exacerbations in non-cystic fibrosis bronchiectasis (EMBRACE): a randomised, double-blind, placebo-controlled trial. Lancet 2012; 380:660–667.
Suissa S, Dell’Aniello S, Ernst P. Long-term natural history of chronic obstructive pulmonary disease: severe exacerbations and mortality. Thorax 2012; 67:957–963.
Ni W, Shao X, Cai X, et al. Prophylactic use of macrolide antibiotics for the prevention of chronic obstructive pulmonary disease exacerbation: a meta-analysis. PLoS One 2015; 10:e0121257.
Griffith DE, Aksamit T, Brown-Elliott BA, et al; ATS Mycobacterial Diseases Subcommittee; American Thoracic Society; Infectious Disease Society of America. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med 2007; 175:367–416.
Havlir DV, Dubé MP, Sattler FR, et al. Prophylaxis against disseminated Mycobacterium avium complex with weekly azithromycin, daily rifabutin, or both. California Collaborative Treatment Group. N Engl J Med 1996; 335:392–398.
Uthman MM, Uthman OA, Yahaya I. Interventions for the prevention of Mycobacterium avium complex in adults and children with HIV. Cochrane Database Syst Rev 2013; 4:CD007191.
Li H, Liu DH, Chen LL, et al. Meta-analysis of the adverse effects of long-term azithromycin use in patients with chronic lung diseases. Antimicrob Agents Chemother 2014; 58:511–517.
Svanström H, Pasternak B, Hviid A. Use of azithromycin and death from cardiovascular causes. N Engl J Med 2013; 368:1704–1712.
Albert RK, Schuller JL; COPD Clinical Research Network. Macrolide antibiotics and the risk of cardiac arrhythmias. Am J Respir Crit Care Med 2014; 189:1173–1180.
Ray WA, Murray KT, Hall K, Arbogast PG, Stein CM. Azithromycin and the risk of cardiovascular death. N Engl J Med 2012; 366:1881–1890.
Albert RK, Connett J, Bailey WC, et al; COPD Clinical Research Network. Azithromycin for prevention of exacerbations of COPD. N Engl J Med 2011; 365:689–698.
Broad J, Sanger GJ. The antibiotic azithromycin is a motilin receptor agonist in human stomach: comparison with erythromycin. Br J Pharmacol 2013; 168:1859–1867.
References
Bailly S, Pocidalo JJ, Fay M, Gougerot-Pocidalo MA. Differential modulation of cytokine production by macrolides: interleukin-6 production is increased by spiramycin and erythromycin. Antimicrob Agents Chemother 1991; 35:2016–2019.
Kanoh S, Rubin BK. Mechanisms of action and clinical application of macrolides as immunomodulatory medications. Clin Microbiol Rev 2010; 23:590–615.
Culić O, Eraković V, Cepelak I, et al. Azithromycin modulates neutrophil function and circulating inflammatory mediators in healthy human subjects. Eur J Pharmacol 2002; 450:277–289.
Cohen-Cymberknoh M, Kerem E, Ferkol T, Elizur A. Airway inflammation in cystic fibrosis: molecular mechanisms and clinical implications. Thorax 2013; 68:1157–1162.
Sagel SD, Wagner BD, Anthony MM, Emmett P, Zemanick ET. Sputum biomarkers of inflammation and lung function decline in children with cystic fibrosis. Am J Respir Crit Care Med 2012; 186:857–865.
Saiman L, Anstead M, Mayer-Hamblett N, et al; AZ0004 Azithromycin Study Group. Effect of azithromycin on pulmonary function in patients with cystic fibrosis uninfected with Pseudomonas aeruginosa: a randomized controlled trial. JAMA 2010; 303:1707–1715.
Southern KW, Barker PM, Solis-Moya A, Patel L. Macrolide antibiotics for cystic fibrosis. Cochrane Database Syst Rev 2012;11:CD002203.
Flume PA, O’Sullivan BP, Robinson KA, et al; Cystic Fibrosis Foundation, Pulmonary Therapies Committee. Cystic fibrosis pulmonary guidelines: chronic medications for maintenance of lung health. Am J Respir Crit Care Med 2007; 176:957–969.
Yanagihara K, Kadoto J, Kohno S. Diffuse panbronchiolitis—pathophysiology and treatment mechanisms. Int J Antimicrob Agents 2001; 18(suppl 1):S83–S87.
Kudoh S, Azuma A, Yamamoto M, Izumi T, Ando M. Improvement of survival in patients with diffuse panbronchiolitis treated with low-dose erythromycin. Am J Respir Crit Care Med 1998; 157:1829–1832.
Schultz MJ. Macrolide activities beyond their antimicrobial effects: macrolides in diffuse panbronchiolitis and cystic fibrosis. J Antimicrob Chemother 2004; 54:21–28.
Hui D, Yan F, Chen RH. The effects of azithromycin on patients with diffuse panbronchiolitis: a retrospective study of 29 cases. J Thorac Dis 2013; 5:613–617.
Khalid M, Al Saghir A, Saleemi S, et al. Azithromycin in bronchiolitis obliterans complicating bone marrow transplantation: a preliminary study. Eur Respir J 2005; 25:490–493.
Kingah PL, Muma G, Soubani A. Azithromycin improves lung function in patients with post-lung transplant bronchiolitis obliterans syndrome: a meta-analysis. Clin Transplant 2014; 28:906–910.
Corris PA, Ryan VA, Small T, et al. A randomised controlled trial of azithromycin therapy in bronchiolitis obliterans syndrome (BOS) post lung transplantation. Thorax 2015; 70:442–450.
Williams KM, Cheng GS, Pusic I, et al. Fluticasone, azithromycin, and montelukast treatment for new-onset bronchiolitis obliterans syndrome after hematopoietic cell transplantation. Biol Blood Marrow Transplant 2016; 22:710–716.
Haworth CS, Bilton D, Elborn JS. Long-term macrolide maintenance therapy in non-CF bronchiectasis: evidence and questions. Respir Med 2014; 108:1397–1408.
Altenburg J, de Graaff CS, Stienstra Y, et al. Effect of azithromycin maintenance treatment on infectious exacerbations among patients with non-cystic fibrosis bronchiectasis: the BAT randomized controlled trial. JAMA 2013; 309:1251–1259.
Wong C, Jayaram L, Karalus N, et al. Azithromycin for prevention of exacerbations in non-cystic fibrosis bronchiectasis (EMBRACE): a randomised, double-blind, placebo-controlled trial. Lancet 2012; 380:660–667.
Suissa S, Dell’Aniello S, Ernst P. Long-term natural history of chronic obstructive pulmonary disease: severe exacerbations and mortality. Thorax 2012; 67:957–963.
Ni W, Shao X, Cai X, et al. Prophylactic use of macrolide antibiotics for the prevention of chronic obstructive pulmonary disease exacerbation: a meta-analysis. PLoS One 2015; 10:e0121257.
Griffith DE, Aksamit T, Brown-Elliott BA, et al; ATS Mycobacterial Diseases Subcommittee; American Thoracic Society; Infectious Disease Society of America. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med 2007; 175:367–416.
Havlir DV, Dubé MP, Sattler FR, et al. Prophylaxis against disseminated Mycobacterium avium complex with weekly azithromycin, daily rifabutin, or both. California Collaborative Treatment Group. N Engl J Med 1996; 335:392–398.
Uthman MM, Uthman OA, Yahaya I. Interventions for the prevention of Mycobacterium avium complex in adults and children with HIV. Cochrane Database Syst Rev 2013; 4:CD007191.
Li H, Liu DH, Chen LL, et al. Meta-analysis of the adverse effects of long-term azithromycin use in patients with chronic lung diseases. Antimicrob Agents Chemother 2014; 58:511–517.
Svanström H, Pasternak B, Hviid A. Use of azithromycin and death from cardiovascular causes. N Engl J Med 2013; 368:1704–1712.
Albert RK, Schuller JL; COPD Clinical Research Network. Macrolide antibiotics and the risk of cardiac arrhythmias. Am J Respir Crit Care Med 2014; 189:1173–1180.
Ray WA, Murray KT, Hall K, Arbogast PG, Stein CM. Azithromycin and the risk of cardiovascular death. N Engl J Med 2012; 366:1881–1890.
Albert RK, Connett J, Bailey WC, et al; COPD Clinical Research Network. Azithromycin for prevention of exacerbations of COPD. N Engl J Med 2011; 365:689–698.
Broad J, Sanger GJ. The antibiotic azithromycin is a motilin receptor agonist in human stomach: comparison with erythromycin. Br J Pharmacol 2013; 168:1859–1867.
African American, Hispanic, American Indian, and Alaskan Native women continue to be disproportionately affected by cervical cancer compared with white women. From 2006 to 2010, the incidence of cervical cancer in African American women was 10.3 per 100,000; in white women it was 7.2.1 The mortality rate from cervical cancer in African American women is twice that in white women.1 Although cervical cancer rates have decreased nationwide, significant racial health disparities persist.
As the first-line healthcare providers for many women, the primary care physician and the general obstetrician-gynecologist are optimally positioned to reduce these disparities.
Cervical cancer is the third most common gynecologic cancer, after uterine and ovarian cancer. Nearly 13,000 new cases are diagnosed each year in the United States, and more than 4,000 women die of it.2 Fortunately, cervical cancer can be significantly prevented with adequate screening and vaccination against human papillomavirus (HPV).
WHY ARE BLACK WOMEN MORE LIKELY TO DIE OF CERVICAL CANCER?
Later stage at diagnosis. African American women are more likely to present with advanced cervical cancer than non-Hispanic white women.3–6
Less-aggressive treatment. African American women are more likely to receive no treatment after a cancer diagnosis.6 Differences in treatment may be attributed to comorbid conditions, stage at cancer diagnosis, and patient refusal.5,7
Less access to care. A study from the Surveillance, Epidemiology, and End Results program of the National Cancer Institute looked at 7,267 women (4,431 non-Hispanic white women, 1,830 Hispanic white women, and 1,006 non-Hispanic African American women) who were diagnosed with primary invasive cervical cancer from 1992 to 1996 and followed through 2000. African American women had a 19% higher mortality rate compared with non-Hispanic white women during follow-up despite adjusting for age, stage, histology, and time of first treatment.8
However, a later study from the same program found no such difference after 1995, when the data were adjusted for marital status, disease stage, age, treatment, grade, and histology.6
Equal access to healthcare may eliminate most of the disparity.7 A study in women with cervical cancer who sought treatment within the United States military healthcare system found no difference in treatment or 5- and 10-year survival rates between African American and white women.5 Equal access to comprehensive healthcare eliminated any disparity once cervical cancer was diagnosed.
CERVICAL CANCER SCREENING
The value of cervical cancer screening and prevention is well established. In 1941, Papanicolau reported that cervical cancer could be detected from vaginal smears.9 Since the development and widespread implementation of the “Pap” smear, cervical cancer rates have decreased dramatically in the United States.
Another major advance was the discovery that persistent infection with HPV is necessary for the development of cervical cancer, precancerous lesions, and genital warts.10
With advancing research, guidelines for cervical cancer screening have changed considerably over the years. Today, combined cervical cytologic and HPV testing is the mainstay. (Isolated HPV testing is generally not available outside clinical trials.)
Who should be screened?
Previous recommendations called for women to undergo Pap testing when they first became sexually active and then every year. However, cervical lesions are likely to regress in young women.11 One study found that 28% of cervical intimal neoplasia (CIN) grade 2 and 3 lesions spontaneously regressed by 15 weeks, although lesions associated with HPV 16 infection were less likely to regress than with other HPV types.12 A study of college women found that HPV infection persisted in only 9% of women after 24 months.13
To minimize unnecessary treatment of young women with dysplasia, the American Society for Colposcopy and Cervical Pathology in 2012 recommended cytologic screening for all women 21 years or older, regardless of age at first sexual encounter.14 Screening intervals were changed from every year to every 3 years until age 30, at which time cotesting with cytology and HPV testing is performed every 5 years. Routine cotesting is not recommended for women younger than 30, who have a high likelihood of HPV infection and spontaneous regression.
In 2014, the US Food and Drug Administration approved primary HPV screening (ie, testing for HPV first, and then performing cytology in samples that test positive) for women age 25 and older.15
Patients who need further evaluation and testing should be referred for colposcopy. The current guidelines for patients who have abnormal results on cervical cancer screening16 can be reviewed at www.asccp.org/asccp-guidelines.
As screening guidelines continue to evolve, primary care physicians will need to stay current and also help educate their patients. For example, many of our patients have undergone annual Pap screening for most of their lives and may not yet know about the new testing intervals.
Are there disparities in screening and follow-up?
Disparities in screening and follow-up may exist, but the evidence is not clear-cut.
In a 2013 National Health Interview Survey report, the rates of cervical cancer screening with Pap tests did not differ between African American and white women.17 However, the information on Pap testing was based on a single question asking participants if they had had a Pap test in the last 3 years. In our experience, patients may confuse Pap tests with speculum examinations.
Once women are screened, adequate and timely follow-up of abnormal results is key.
In a study from the National Breast and Cervical Cancer Early Detection Program,18 women who had cytology findings of atypical squamous cells of undetermined significance or low-grade squamous intraepithelial lesions were to undergo repeat Pap testing every 4 to 6 months for 2 years. African American women were the least likely to have a follow-up Pap smear compared with other racial groups.
On the other hand, there was no difference related to race in follow-up rates of abnormal Pap tests in women ages 47 to 64 in the South Carolina Breast and Cervical Cancer Early Detection Program.19
In a study in an urban population (predominantly African African), the overall follow-up rate was only 26% at 12 months from an initial abnormal Pap smear. This study did not find any differences in follow-up according to race or ethnicity; however, it had insufficient power to detect a difference because only 15% of the study participants were white.20
What is in a genotype?
HPV is implicated in progression to both squamous cell carcinoma and adenocarcinoma of the cervix. Worldwide, HPV genotypes 16 and 18 are associated with 73% of cases of invasive cervical cancer; most of the remainder are associated with, in order of decreasing prevalence, genotypes 58, 33, 45, 31, 52, 35, 59, 39, 51, and 56.21
High-grade cervical lesions in African American women may less often be positive for HPV 16 and 18 than in white women.22,23 On the other hand, the proportion of non-Hispanic black women infected with HPV 35 and 58 was significantly higher than in non-Hispanic white women.22 Regardless, HPV screening is recommended for women of all races and ethnicities.
The 2-valent and 4-valent HPV vaccines do not cover HPV 35 or 58. The newer 9-valent vaccine covers HPV 58 (but not 35) and so may in theory decrease any potential disparity related to infection with a specific oncogenic subtype.
THE ROLE OF PREVENTION
HPV vaccination
Currently, 3 vaccines against HPV are available in the United States, a 2-valent, a 4-valent, and since 2015, a 9-valent preparation (Table 1).
The Females United to Unilaterally Reduce Endo/Ectocervical Disease study demonstrated that the 4-valent vaccine was highly effective against cervical intraepithelial neoplasia due to HPV 16 and 18.24 In another study, the 2-valent vaccine reduced the incidence of CIN 3 or higher by 87% in women who received all 3 doses and who had no evidence of HPV infection at baseline.25
HPV vaccination is expensive. Each shot costs about $130, plus the cost of administering it. Although the Vaccines for Children program covers the HPV vaccine for uninsured and underinsured children and adolescents under age 19, Medicaid coverage varies from state to state for adults over age 21.
The Advisory Committee on Immunization Practices (ACIP)26 recommends routine vaccination for:
Males 11 or 12 years old
Females ages 9 to 26.
In October 2016, the ACIP approved a 2-dose series given 6 to 12 months apart for patients starting vaccination at ages 9 through 14 years who are not immunocompromised. Others should receive a 3-dose series, with the second dose given 1 to 2 months after the first dose and the third dose given 6 months after the first dose.27 Previously, 3 doses were recommended for everyone.
Disparities in HPV vaccination rates
HPV vaccination rates among adolescents in the United States increased from 33.6% in 2013 to 41.7% in 2014.28 However, HPV vaccination rates continue to lag behind those of other routine vaccines, such as Tdap and meningococcal conjugate.
Reagan-Steiner et al28 reported that more black than white girls age 13 through 17 received at least 1 dose of a 3-dose HPV vaccination series, but more white girls received all 3 doses (70.6% vs 61.6%). In contrast, a meta-analysis by Fisher et al29 found African American and uninsured women generally less likely to initiate the HPV vaccination series. Kessels et al30 reported similar findings.
Barriers to HPV vaccination
Barriers to HPV vaccination can be provider-dependent, parental, or institutional.
Malo et al31 surveyed Florida Medicaid providers and found that those who participated in the Vaccines for Children program were less likely to cite lack of reimbursement as a barrier to vaccination.
Meites et al32 surveyed sexually transmitted disease clinics and found that common reasons for not offering HPV vaccine were cost, staff time, and difficulty coordinating follow-up visits to complete the series.
Providers report lack of urgency or lack of perception of cervical cancer as a true public health threat, safety concerns regarding the vaccine, and the inability to coadminister vaccines as barriers.33
Studies have shown that relatively few parents (up to 18%) of parents are concerned about the effect of the vaccine on sexual activity.34 Rather, they are most likely to cite lack of information regarding the vaccine, lack of physician recommendation, and not knowing where to receive the vaccine as barriers.35,36
Guerry et al37 determined that the single most important factor in vaccine initiation was physician recommendation, a finding reiterated in other studies.35,38 A study in North Carolina identified failure of physician recommendation as one of the missed opportunities for vaccination of young women.39
Therefore, the primary care physician, as the initial contact with the child or young adult, holds a responsibility to narrow this gap. In simply discussing and recommending the vaccine, physicians could increase vaccination rates.
REPRODUCTIVE HEALTH
Although 80% of women will be infected with HPV in their lifetime, only a small proportion will develop cervical cancer, suggesting there are other cofactors in the progression to cervical cancer.40
Given the infectious etiology of cervical cancer, other contributing reproductive health factors have been described. As expected, the number of sexual partners correlates with HPV infection.41,42 Younger age at first intercourse has been linked to development of cervical neoplasia, consistent with persistent infection leading to neoplasia.41,42
Primary care physicians should provide timely and comprehensive sexual education, including information on safe sexual practices and pregnancy prevention.
Human immunodeficiency virus
In 2010, the estimated rate of new human immunodeficiency virus (HIV) infections in African American women was nearly 20 times greater than in white women.43 Previous studies have shown a clear relationship between HIV and HPV-associated cancers, including cervical neoplasia and invasive cervical cancer.44,45
Women with HIV should receive screening for cervical cancer at the time of diagnosis, 6 months after the initial diagnosis, and annually thereafter.46
Conflicting evidence exists regarding the effect of highly active antiretroviral therapy on the incidence of HPV-related disease, so aggressive screening and management of cervical neoplasia is recommended for women with HIV, regardless of CD4+ levels or viral load.47–49
Additional infectious culprits
Coinfection with other sexually transmitted infections, specifically Chlamydia, herpes, and HIV, has been associated with cervical neoplasia and invasive cervical cancer. A positive linear association exists between the number of sexually transmitted infections and cervical neoplasia.50
C trachomatis is the most common sexually transmitted infection in the United States, with a 6-times higher rate in African American women.51 Women who are seropositive for C trachomatis are at twofold higher risk of developing squamous cell cervical cancer.52,53 Women who are seropositive for Chlamydia infection, herpes virus 2, or HPV are at markedly increased risk of invasive cervical cancer.50
Tobacco use
The negative impact of smoking on numerous other cancers resulted in investigation of its role in cervical cancer.
Early case-control studies found an association between cervical cancer and smoking,54 but because these studies did not account for HPV infection status, they could not establish causality. Subsequently, several studies did control for HPV infection; the risk of squamous cervical cancer was twice as high in women who had ever smoked.55 Furthermore, the more cigarettes smoked per day, the higher the risk of cervical neoplasia.41,56
According to the US Centers for Disease Control and Prevention in 2014, the highest prevalence of smoking was among American Indian and Alaskan Native women, 32.5% of whom said they smoked every day, compared with 17.2% of white women and 13.7% of African American women.57
HOW CAN PRIMARY CARE PHYSICIANS CLOSE THE GAP?
Primary care physicians are the first point of contact for patients of all ages and so can help minimize such disparities. They can tackle 2 important cervical cancer prevention interventions first-hand: vaccination and screening (Table 2), including follow-up of abnormal screening results.
By promoting HPV vaccination to children and young adults, primary care physicians can help prevent cervical cancer. Moreover, primary care physicians will see most adolescents for a nonpreventive health visit, an optimal opportunity to discuss sexual activity practices and HPV vaccination.58 Including the HPV vaccine as routine with other vaccinations can close the gap.38
Screening and treatment of sexually transmitted infection during these visits can affect the risk that future HPV infection will progress to neoplasia or cancer. Persistent lifestyle modification counseling, especially smoking cessation through motivational interviewing, can lessen the risk of cervical cancer neoplasia progression.
Additionally, in light of recent changes in cervical cancer screening guidelines, the primary care physician’s role as educator is of utmost importance. In one study, although 99% of women had received a Pap test, 87% could not identify the purpose of the Pap test.59 The primary care physician’s role is perhaps the most influential in preventing disease and, as such, has the greatest impact on a patient’s disease process.
References
Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin 2014; 64:9–29.
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin 2015; 65:5–29.
Koh WJ, Greer BE, Abu-Rustum NR, et al. Cervical cancer, version 2.2015. J Natl Compr Canc Netw 2015; 13:395-404.
Farley J, Risinger JI, Rose GS, Maxwell GL. Racial disparities in blacks with gynecologic cancers. Cancer 2007; 110:234–243.
Farley JH, Hines JF, Taylor RR, et al. Equal care ensures equal survival for African-American women with cervical carcinoma. Cancer 2001; 91:869–873.
Rauh-Hain JA, Clemmer JT, Bradford LS, et al. Racial disparities in cervical cancer survival over time. Cancer 2013; 119:3644–3652.
Collins Y, Holcomb K, Chapman-Davis E, Khabele D, Farley JH. Gynecologic cancer disparities: a report from the Health Disparities Taskforce of the Society of Gynecologic Oncology. Gynecol Oncol 2014; 133:353–361.
Patel DA, Barnholtz-Sloan JS, Patel MK, Malone JM Jr, Chuba PJ, Schwartz K. A population-based study of racial and ethnic differences in survival among women with invasive cervical cancer: analysis of surveillance, epidemiology, and end results data. Gynecol Oncol 2005; 97:550–558.
Papanicolaou GN, Traut HF. The diagnostic value of vaginal smears in carcinoma of the uterus. 1941. Arch Pathol Lab Med 1997; 121:211–224.
Walboomers JM, Jacobs M V, Manos MM, et al. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J Pathol 1999; 189:12–19.
Moscicki AB, Shiboski S, Hills NK, et al. Regression of low-grade squamous intra-epithelial lesions in young women. Lancet 2004; 364:1678–1683.
Trimble CL, Piantadosi S, Gravitt P, et al. Spontaneous regression of high-grade cervical dysplasia: effects of human papillomavirus type and HLA phenotype. Clin Cancer Res 2005; 11:4717–4723.
Ho GY, Bierman R, Beardsley L, Chang CJ, Burk RD. Natural history of cervicovaginal papillomavirus infection in young women. N Engl J Med 1998; 338:423-428.
Saslow D, Solomon D, Lawson HW, et al; ACS-ASCCP-ASCP Cervical Cancer Guideline Committee. American Cancer Society, American Society for Colposcopy and Cervical Pathology, and American Society for Clinical Pathology screening guidelines for the prevention and early detection of cervical cancer. CA Cancer J Clin 2012; 62:147–172.
Huh WK, Ault KA, Chelmow D, et al. Use of primary high-risk human papillomavirus testing for cervical cancer screening: interim clinical guidance. Obstet Gynecol 2015; 125:330–337.
Massad LS, Einstein MH, Huh WK, et al; 2012 ASCCP Consensus Guidelines Conference. 2012 updated consensus guidelines for the management of abnormal cervical cancer screening tests and cancer precursors. J Low Genit Tract Dis 2013; 17(suppl 1):S1–S27.
Sabatino SA, White MC, Thompson TD, Klabunde CN. Cancer screening test use—United States, 2013. MMWR 2015; 64:464–468.
Benard VB, Lawson HW, Eheman CR, Anderson C, Helsel W. Adherence to guidelines for follow-up of low-grade cytologic abnormalities among medically underserved women. Obstet Gynecol 2005; 105:1323–1328.
Eggleston KS, Coker AL, Luchok KJ, Meyer TE. Adherence to recommendations for follow-up to abnormal Pap tests. Obstet Gynecol 2007; 109:1332–1341.
Peterson NB, Han J, Freund KM. Inadequate follow-up for abnormal Pap smears in an urban population. J Natl Med Assoc 2003; 95:825–832.
Li N, Franceschi S, Howell-Jones R, Snijders PJ, Clifford GM. Human papillomavirus type distribution in 30,848 invasive cervical cancers worldwide: variation by geographical region, histological type and year of publication. Int J Cancer 2011; 128:927–935.
Hariri S, Unger ER, Powell SE, et al; HPV-IMPACT Working Group. Human papillomavirus genotypes in high-grade cervical lesions in the United States. J Infect Dis 2012; 206:1878–1886.
Niccolai LM, Russ C, Julian PJ, et al. Individual and geographic disparities in human papillomavirus types 16/18 in high-grade cervical lesions: associations with race, ethnicity, and poverty. Cancer 2013; 119:3052–3058.
FUTURE II Study Group. Quadrivalent vaccine against human papillomavirus to prevent high-grade cervical lesions. N Engl J Med 2007; 356:1915–1927.
Paavonen J, Naud P, Salmerón J, et al; HPV PATRICIA Study Group. Efficacy of human papillomavirus (HPV)-16/18 AS04-adjuvanted vaccine against cervical infection and precancer caused by oncogenic HPV types (PATRICIA): final analysis of a double-blind, randomised study in young women. Lancet 2009; 374:301–314.
Centers for Disease Control and Prevention (CDC). Recommendations on the use of quadrivalent human papillomavirus vaccine in males—Advisory Committee on Immunization Practices (ACIP), 2011. MMWR Morb Mortal Wkly Rep 2011; 60:1705–1708.
Meites E, Kempe A, Markowitz LE. Use of a 2-dose schedule for human papillomavirus vaccination—updated recommendations of the Advisory Committee on Immunization Practices. MMWR Morb Mortal Wkly Rep 2016; 65:1405–1408.
Reagan-Steiner S, Yankey D, Jeyarajah J, et al. National, regional, state, and selected local area vaccination coverage among adolescents aged 13–17 years—United States, 2014. MMWR Morb Mortal Wkly Rep 2015; 64:784–792.
Fisher H, Trotter CL, Audrey S, MacDonald-Wallis K, Hickman M. Inequalities in the uptake of human papillomavirus vaccination: a systematic review and meta-analysis. Int J Epidemiol 2013; 42:896–908.
Kessels SJ, Marshall HS, Watson M, Braunack-Mayer AJ, Reuzel R, Tooher RL. Factors associated with HPV vaccine uptake in teenage girls: a systematic review. Vaccine 2012; 30:3546–3556.
Malo TL, Hassani D, Staras SA, Shenkman EA, Giuliano AR, Vadaparampil ST. Do Florida Medicaid providers’ barriers to HPV vaccination vary based on VFC program participation? Matern Child Health J 2013; 17:609–615.
Meites E, Llata E, Hariri S, et al. HPV vaccine implementation in STD clinics—STD Surveillance Network. Sex Transm Dis 2012; 39:32–34.
Perkins RB, Clark JA. What affects human papillomavirus vaccination rates? A qualitative analysis of providers’ perceptions. Womens Health Issues 2012; 22:e379–e386.
Holman DM, Benard V, Roland KB, Watson M, Liddon N, Stokley S. Barriers to human papillomavirus vaccination among US adolescents: a systematic review of the literature. JAMA Pediatr 2014; 168:76–82.
Dorell CG, Yankey D, Santibanez TA, Markowitz LE. Human papillomavirus vaccination series initiation and completion, 2008–2009. Pediatrics 2011; 128:830–839.
Bastani R, Glenn BA, Tsui J, et al. Understanding suboptimal human papillomavirus vaccine uptake among ethnic minority girls. Cancer Epidemiol Biomarkers Prev 2011; 20:1463–1472.
Guerry SL, De Rosa CJ, Markowitz LE, et al. Human papillomavirus vaccine initiation among adolescent girls in high-risk communities. Vaccine 2011; 29:2235–2241.
Hull PC, Williams EA, Khabele D, Dean C, Bond B, Sanderson M. HPV vaccine use among African American girls: qualitative formative research using a participatory social marketing approach. Gynecol Oncol 2014; 132(suppl 1):S13–S20.
Brewer NT, Gottlieb SL, Reiter PL, et al. Longitudinal predictors of human papillomavirus vaccine initiation among adolescent girls in a high-risk geographic area. Sex Transm Dis 2011; 38:197–204.
Wang SS, Zuna RE, Wentzensen N, et al. Human papillomavirus cofactors by disease progression and human papillomavirus types in the study to understand cervical cancer early endpoints and determinants. Cancer Epidemiol Biomarkers Prev 2009; 18:113–120.
Deacon JM, Evans CD, Yule R, et al. Sexual behaviour and smoking as determinants of cervical HPV infection and of CIN3 among those infected: a case-control study nested within the Manchester cohort. Br J Cancer 2000; 83:1565–1572.
International Collaboration of Epidemiological Studies of Cervical Cancer. Cervical carcinoma and sexual behavior: collaborative reanalysis of individual data on 15,461 women with cervical carcinoma and 29,164 women without cervical carcinoma from 21 epidemiological studies. Cancer Epidemiol Biomarkers Prev 2009; 18:1060–1069.
Centers for Disease Control and Prevention (CDC). Estimated HIV incidence in the United States, 2007–2010. HIV Surveillance Supplemental Report 2012; 17(No. 4). https://www.cdc.gov/hiv/pdf/statistics_hssr_vol_17_no_4.pdf. Accessed September 12, 2017.
Frisch M, Biggar RJ, Goedert JJ. Human papillomavirus-associated cancers in patients with human immunodeficiency virus infection and acquired immunodeficiency syndrome. J Natl Cancer Inst 2000; 92:1500–1510.
Schäfer A, Friedmann W, Mielke M, Schwartländer B, Koch MA. The increased frequency of cervical dysplasia-neoplasia in women infected with the human immunodeficiency virus is related to the degree of immunosuppression. Am J Obstet Gynecol 1991; 164:593–599.
De Vuyst H, Lillo F, Broutet N, Smith JS. HIV, human papillomavirus, and cervical neoplasia and cancer in the era of highly active antiretroviral therapy. Eur J Cancer Prev 2008; 17:545–554.
Palefsky JM. Cervical human papillomavirus infection and cervical intraepithelial neoplasia in women positive for human immunodeficiency virus in the era of highly active antiretroviral therapy. Curr Opin Oncol 2003; 15:382–388.
Adler DH. The impact of HAART on HPV-related cervical disease. Curr HIV Res 2010; 8:493–497.
Castellsagué X, Pawlita M, Roura E, et al. Prospective seroepidemiologic study on the role of human papillomavirus and other infections in cervical carcinogenesis: evidence from the EPIC cohort. Int J Cancer 2014; 135:440–452.
Centers for Disease Control and Prevention (CDC). 2013 sexually transmitted disease surveillance. www.cdc.gov/std/stats13/exordium.htm. Accessed September 12, 2017.
Smith JS, Bosetti C, Muñoz N, et al; IARC multicentric case-control study. Chlamydia trachomatis and invasive cervical cancer: a pooled analysis of the IARC multicentric case-control study. Int J Cancer 2004; 111:431–439.
Koskela P, Anttila T, Bjørge T, et al. Chlamydia trachomatis infection as a risk factor for invasive cervical cancer. Int J Cancer 2000; 85:35–39.
Office on Smoking and Health (US). Women and smoking: a report of the Surgeon General: Chapter 3. Health consequences of tobacco use among women. http://www.ncbi.nlm.nih.gov/books/NBK44312. Accessed September 12, 2017.
Plummer M, Herrero R, Franceschi S, et al; IARC Multi-centre Cervical Cancer Study Group. Smoking and cervical cancer: pooled analysis of the IARC multi-centric case—control study. Cancer Causes Control 2003; 14:805–814.
Ho GY, Kadish AS, Burk RD, et al. HPV 16 and cigarette smoking as risk factors for high-grade cervical intra-epithelial neoplasia. Int J Cancer 1998; 78:281–285.
Jamal A, Homa DM, O’Connor E, et al. Current cigarette smoking among adults - United States, 2005-2014. MMWR Morb Mortal Wkly Rep 2015; 64:1233–1240.
Nordin JD, Solberg LI, Parker ED. Adolescent primary care visit patterns. Ann Fam Med 2010; 8:511–516.
Lindau ST, Tomori C, Lyons T, Langseth L, Bennett CL, Garcia P. The association of health literacy with cervical cancer prevention knowledge and health behaviors in a multiethnic cohort of women. Am J Obstet Gynecol 2002; 186:938–943.
Cynthia Arvizo, MD Department of Obstetrics and Gynecology, Vanderbilt University Medical Center, Nashville, TN
Haider Mahdi, MD Department of Obstetrics and Gynecology, Women’s Health Institute, Cleveland Clinic; Assistant Professor, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, Cleveland, OH
Address: Haider Mahdi, MD, Women’s Health Institute, A81, Cleveland Clinic, 9500 Euclid Avenue, Cleveland, OH 44195; mahdih@ccf.org
Cynthia Arvizo, MD Department of Obstetrics and Gynecology, Vanderbilt University Medical Center, Nashville, TN
Haider Mahdi, MD Department of Obstetrics and Gynecology, Women’s Health Institute, Cleveland Clinic; Assistant Professor, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, Cleveland, OH
Address: Haider Mahdi, MD, Women’s Health Institute, A81, Cleveland Clinic, 9500 Euclid Avenue, Cleveland, OH 44195; mahdih@ccf.org
Author and Disclosure Information
Cynthia Arvizo, MD Department of Obstetrics and Gynecology, Vanderbilt University Medical Center, Nashville, TN
Haider Mahdi, MD Department of Obstetrics and Gynecology, Women’s Health Institute, Cleveland Clinic; Assistant Professor, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, Cleveland, OH
Address: Haider Mahdi, MD, Women’s Health Institute, A81, Cleveland Clinic, 9500 Euclid Avenue, Cleveland, OH 44195; mahdih@ccf.org
African American, Hispanic, American Indian, and Alaskan Native women continue to be disproportionately affected by cervical cancer compared with white women. From 2006 to 2010, the incidence of cervical cancer in African American women was 10.3 per 100,000; in white women it was 7.2.1 The mortality rate from cervical cancer in African American women is twice that in white women.1 Although cervical cancer rates have decreased nationwide, significant racial health disparities persist.
As the first-line healthcare providers for many women, the primary care physician and the general obstetrician-gynecologist are optimally positioned to reduce these disparities.
Cervical cancer is the third most common gynecologic cancer, after uterine and ovarian cancer. Nearly 13,000 new cases are diagnosed each year in the United States, and more than 4,000 women die of it.2 Fortunately, cervical cancer can be significantly prevented with adequate screening and vaccination against human papillomavirus (HPV).
WHY ARE BLACK WOMEN MORE LIKELY TO DIE OF CERVICAL CANCER?
Later stage at diagnosis. African American women are more likely to present with advanced cervical cancer than non-Hispanic white women.3–6
Less-aggressive treatment. African American women are more likely to receive no treatment after a cancer diagnosis.6 Differences in treatment may be attributed to comorbid conditions, stage at cancer diagnosis, and patient refusal.5,7
Less access to care. A study from the Surveillance, Epidemiology, and End Results program of the National Cancer Institute looked at 7,267 women (4,431 non-Hispanic white women, 1,830 Hispanic white women, and 1,006 non-Hispanic African American women) who were diagnosed with primary invasive cervical cancer from 1992 to 1996 and followed through 2000. African American women had a 19% higher mortality rate compared with non-Hispanic white women during follow-up despite adjusting for age, stage, histology, and time of first treatment.8
However, a later study from the same program found no such difference after 1995, when the data were adjusted for marital status, disease stage, age, treatment, grade, and histology.6
Equal access to healthcare may eliminate most of the disparity.7 A study in women with cervical cancer who sought treatment within the United States military healthcare system found no difference in treatment or 5- and 10-year survival rates between African American and white women.5 Equal access to comprehensive healthcare eliminated any disparity once cervical cancer was diagnosed.
CERVICAL CANCER SCREENING
The value of cervical cancer screening and prevention is well established. In 1941, Papanicolau reported that cervical cancer could be detected from vaginal smears.9 Since the development and widespread implementation of the “Pap” smear, cervical cancer rates have decreased dramatically in the United States.
Another major advance was the discovery that persistent infection with HPV is necessary for the development of cervical cancer, precancerous lesions, and genital warts.10
With advancing research, guidelines for cervical cancer screening have changed considerably over the years. Today, combined cervical cytologic and HPV testing is the mainstay. (Isolated HPV testing is generally not available outside clinical trials.)
Who should be screened?
Previous recommendations called for women to undergo Pap testing when they first became sexually active and then every year. However, cervical lesions are likely to regress in young women.11 One study found that 28% of cervical intimal neoplasia (CIN) grade 2 and 3 lesions spontaneously regressed by 15 weeks, although lesions associated with HPV 16 infection were less likely to regress than with other HPV types.12 A study of college women found that HPV infection persisted in only 9% of women after 24 months.13
To minimize unnecessary treatment of young women with dysplasia, the American Society for Colposcopy and Cervical Pathology in 2012 recommended cytologic screening for all women 21 years or older, regardless of age at first sexual encounter.14 Screening intervals were changed from every year to every 3 years until age 30, at which time cotesting with cytology and HPV testing is performed every 5 years. Routine cotesting is not recommended for women younger than 30, who have a high likelihood of HPV infection and spontaneous regression.
In 2014, the US Food and Drug Administration approved primary HPV screening (ie, testing for HPV first, and then performing cytology in samples that test positive) for women age 25 and older.15
Patients who need further evaluation and testing should be referred for colposcopy. The current guidelines for patients who have abnormal results on cervical cancer screening16 can be reviewed at www.asccp.org/asccp-guidelines.
As screening guidelines continue to evolve, primary care physicians will need to stay current and also help educate their patients. For example, many of our patients have undergone annual Pap screening for most of their lives and may not yet know about the new testing intervals.
Are there disparities in screening and follow-up?
Disparities in screening and follow-up may exist, but the evidence is not clear-cut.
In a 2013 National Health Interview Survey report, the rates of cervical cancer screening with Pap tests did not differ between African American and white women.17 However, the information on Pap testing was based on a single question asking participants if they had had a Pap test in the last 3 years. In our experience, patients may confuse Pap tests with speculum examinations.
Once women are screened, adequate and timely follow-up of abnormal results is key.
In a study from the National Breast and Cervical Cancer Early Detection Program,18 women who had cytology findings of atypical squamous cells of undetermined significance or low-grade squamous intraepithelial lesions were to undergo repeat Pap testing every 4 to 6 months for 2 years. African American women were the least likely to have a follow-up Pap smear compared with other racial groups.
On the other hand, there was no difference related to race in follow-up rates of abnormal Pap tests in women ages 47 to 64 in the South Carolina Breast and Cervical Cancer Early Detection Program.19
In a study in an urban population (predominantly African African), the overall follow-up rate was only 26% at 12 months from an initial abnormal Pap smear. This study did not find any differences in follow-up according to race or ethnicity; however, it had insufficient power to detect a difference because only 15% of the study participants were white.20
What is in a genotype?
HPV is implicated in progression to both squamous cell carcinoma and adenocarcinoma of the cervix. Worldwide, HPV genotypes 16 and 18 are associated with 73% of cases of invasive cervical cancer; most of the remainder are associated with, in order of decreasing prevalence, genotypes 58, 33, 45, 31, 52, 35, 59, 39, 51, and 56.21
High-grade cervical lesions in African American women may less often be positive for HPV 16 and 18 than in white women.22,23 On the other hand, the proportion of non-Hispanic black women infected with HPV 35 and 58 was significantly higher than in non-Hispanic white women.22 Regardless, HPV screening is recommended for women of all races and ethnicities.
The 2-valent and 4-valent HPV vaccines do not cover HPV 35 or 58. The newer 9-valent vaccine covers HPV 58 (but not 35) and so may in theory decrease any potential disparity related to infection with a specific oncogenic subtype.
THE ROLE OF PREVENTION
HPV vaccination
Currently, 3 vaccines against HPV are available in the United States, a 2-valent, a 4-valent, and since 2015, a 9-valent preparation (Table 1).
The Females United to Unilaterally Reduce Endo/Ectocervical Disease study demonstrated that the 4-valent vaccine was highly effective against cervical intraepithelial neoplasia due to HPV 16 and 18.24 In another study, the 2-valent vaccine reduced the incidence of CIN 3 or higher by 87% in women who received all 3 doses and who had no evidence of HPV infection at baseline.25
HPV vaccination is expensive. Each shot costs about $130, plus the cost of administering it. Although the Vaccines for Children program covers the HPV vaccine for uninsured and underinsured children and adolescents under age 19, Medicaid coverage varies from state to state for adults over age 21.
The Advisory Committee on Immunization Practices (ACIP)26 recommends routine vaccination for:
Males 11 or 12 years old
Females ages 9 to 26.
In October 2016, the ACIP approved a 2-dose series given 6 to 12 months apart for patients starting vaccination at ages 9 through 14 years who are not immunocompromised. Others should receive a 3-dose series, with the second dose given 1 to 2 months after the first dose and the third dose given 6 months after the first dose.27 Previously, 3 doses were recommended for everyone.
Disparities in HPV vaccination rates
HPV vaccination rates among adolescents in the United States increased from 33.6% in 2013 to 41.7% in 2014.28 However, HPV vaccination rates continue to lag behind those of other routine vaccines, such as Tdap and meningococcal conjugate.
Reagan-Steiner et al28 reported that more black than white girls age 13 through 17 received at least 1 dose of a 3-dose HPV vaccination series, but more white girls received all 3 doses (70.6% vs 61.6%). In contrast, a meta-analysis by Fisher et al29 found African American and uninsured women generally less likely to initiate the HPV vaccination series. Kessels et al30 reported similar findings.
Barriers to HPV vaccination
Barriers to HPV vaccination can be provider-dependent, parental, or institutional.
Malo et al31 surveyed Florida Medicaid providers and found that those who participated in the Vaccines for Children program were less likely to cite lack of reimbursement as a barrier to vaccination.
Meites et al32 surveyed sexually transmitted disease clinics and found that common reasons for not offering HPV vaccine were cost, staff time, and difficulty coordinating follow-up visits to complete the series.
Providers report lack of urgency or lack of perception of cervical cancer as a true public health threat, safety concerns regarding the vaccine, and the inability to coadminister vaccines as barriers.33
Studies have shown that relatively few parents (up to 18%) of parents are concerned about the effect of the vaccine on sexual activity.34 Rather, they are most likely to cite lack of information regarding the vaccine, lack of physician recommendation, and not knowing where to receive the vaccine as barriers.35,36
Guerry et al37 determined that the single most important factor in vaccine initiation was physician recommendation, a finding reiterated in other studies.35,38 A study in North Carolina identified failure of physician recommendation as one of the missed opportunities for vaccination of young women.39
Therefore, the primary care physician, as the initial contact with the child or young adult, holds a responsibility to narrow this gap. In simply discussing and recommending the vaccine, physicians could increase vaccination rates.
REPRODUCTIVE HEALTH
Although 80% of women will be infected with HPV in their lifetime, only a small proportion will develop cervical cancer, suggesting there are other cofactors in the progression to cervical cancer.40
Given the infectious etiology of cervical cancer, other contributing reproductive health factors have been described. As expected, the number of sexual partners correlates with HPV infection.41,42 Younger age at first intercourse has been linked to development of cervical neoplasia, consistent with persistent infection leading to neoplasia.41,42
Primary care physicians should provide timely and comprehensive sexual education, including information on safe sexual practices and pregnancy prevention.
Human immunodeficiency virus
In 2010, the estimated rate of new human immunodeficiency virus (HIV) infections in African American women was nearly 20 times greater than in white women.43 Previous studies have shown a clear relationship between HIV and HPV-associated cancers, including cervical neoplasia and invasive cervical cancer.44,45
Women with HIV should receive screening for cervical cancer at the time of diagnosis, 6 months after the initial diagnosis, and annually thereafter.46
Conflicting evidence exists regarding the effect of highly active antiretroviral therapy on the incidence of HPV-related disease, so aggressive screening and management of cervical neoplasia is recommended for women with HIV, regardless of CD4+ levels or viral load.47–49
Additional infectious culprits
Coinfection with other sexually transmitted infections, specifically Chlamydia, herpes, and HIV, has been associated with cervical neoplasia and invasive cervical cancer. A positive linear association exists between the number of sexually transmitted infections and cervical neoplasia.50
C trachomatis is the most common sexually transmitted infection in the United States, with a 6-times higher rate in African American women.51 Women who are seropositive for C trachomatis are at twofold higher risk of developing squamous cell cervical cancer.52,53 Women who are seropositive for Chlamydia infection, herpes virus 2, or HPV are at markedly increased risk of invasive cervical cancer.50
Tobacco use
The negative impact of smoking on numerous other cancers resulted in investigation of its role in cervical cancer.
Early case-control studies found an association between cervical cancer and smoking,54 but because these studies did not account for HPV infection status, they could not establish causality. Subsequently, several studies did control for HPV infection; the risk of squamous cervical cancer was twice as high in women who had ever smoked.55 Furthermore, the more cigarettes smoked per day, the higher the risk of cervical neoplasia.41,56
According to the US Centers for Disease Control and Prevention in 2014, the highest prevalence of smoking was among American Indian and Alaskan Native women, 32.5% of whom said they smoked every day, compared with 17.2% of white women and 13.7% of African American women.57
HOW CAN PRIMARY CARE PHYSICIANS CLOSE THE GAP?
Primary care physicians are the first point of contact for patients of all ages and so can help minimize such disparities. They can tackle 2 important cervical cancer prevention interventions first-hand: vaccination and screening (Table 2), including follow-up of abnormal screening results.
By promoting HPV vaccination to children and young adults, primary care physicians can help prevent cervical cancer. Moreover, primary care physicians will see most adolescents for a nonpreventive health visit, an optimal opportunity to discuss sexual activity practices and HPV vaccination.58 Including the HPV vaccine as routine with other vaccinations can close the gap.38
Screening and treatment of sexually transmitted infection during these visits can affect the risk that future HPV infection will progress to neoplasia or cancer. Persistent lifestyle modification counseling, especially smoking cessation through motivational interviewing, can lessen the risk of cervical cancer neoplasia progression.
Additionally, in light of recent changes in cervical cancer screening guidelines, the primary care physician’s role as educator is of utmost importance. In one study, although 99% of women had received a Pap test, 87% could not identify the purpose of the Pap test.59 The primary care physician’s role is perhaps the most influential in preventing disease and, as such, has the greatest impact on a patient’s disease process.
African American, Hispanic, American Indian, and Alaskan Native women continue to be disproportionately affected by cervical cancer compared with white women. From 2006 to 2010, the incidence of cervical cancer in African American women was 10.3 per 100,000; in white women it was 7.2.1 The mortality rate from cervical cancer in African American women is twice that in white women.1 Although cervical cancer rates have decreased nationwide, significant racial health disparities persist.
As the first-line healthcare providers for many women, the primary care physician and the general obstetrician-gynecologist are optimally positioned to reduce these disparities.
Cervical cancer is the third most common gynecologic cancer, after uterine and ovarian cancer. Nearly 13,000 new cases are diagnosed each year in the United States, and more than 4,000 women die of it.2 Fortunately, cervical cancer can be significantly prevented with adequate screening and vaccination against human papillomavirus (HPV).
WHY ARE BLACK WOMEN MORE LIKELY TO DIE OF CERVICAL CANCER?
Later stage at diagnosis. African American women are more likely to present with advanced cervical cancer than non-Hispanic white women.3–6
Less-aggressive treatment. African American women are more likely to receive no treatment after a cancer diagnosis.6 Differences in treatment may be attributed to comorbid conditions, stage at cancer diagnosis, and patient refusal.5,7
Less access to care. A study from the Surveillance, Epidemiology, and End Results program of the National Cancer Institute looked at 7,267 women (4,431 non-Hispanic white women, 1,830 Hispanic white women, and 1,006 non-Hispanic African American women) who were diagnosed with primary invasive cervical cancer from 1992 to 1996 and followed through 2000. African American women had a 19% higher mortality rate compared with non-Hispanic white women during follow-up despite adjusting for age, stage, histology, and time of first treatment.8
However, a later study from the same program found no such difference after 1995, when the data were adjusted for marital status, disease stage, age, treatment, grade, and histology.6
Equal access to healthcare may eliminate most of the disparity.7 A study in women with cervical cancer who sought treatment within the United States military healthcare system found no difference in treatment or 5- and 10-year survival rates between African American and white women.5 Equal access to comprehensive healthcare eliminated any disparity once cervical cancer was diagnosed.
CERVICAL CANCER SCREENING
The value of cervical cancer screening and prevention is well established. In 1941, Papanicolau reported that cervical cancer could be detected from vaginal smears.9 Since the development and widespread implementation of the “Pap” smear, cervical cancer rates have decreased dramatically in the United States.
Another major advance was the discovery that persistent infection with HPV is necessary for the development of cervical cancer, precancerous lesions, and genital warts.10
With advancing research, guidelines for cervical cancer screening have changed considerably over the years. Today, combined cervical cytologic and HPV testing is the mainstay. (Isolated HPV testing is generally not available outside clinical trials.)
Who should be screened?
Previous recommendations called for women to undergo Pap testing when they first became sexually active and then every year. However, cervical lesions are likely to regress in young women.11 One study found that 28% of cervical intimal neoplasia (CIN) grade 2 and 3 lesions spontaneously regressed by 15 weeks, although lesions associated with HPV 16 infection were less likely to regress than with other HPV types.12 A study of college women found that HPV infection persisted in only 9% of women after 24 months.13
To minimize unnecessary treatment of young women with dysplasia, the American Society for Colposcopy and Cervical Pathology in 2012 recommended cytologic screening for all women 21 years or older, regardless of age at first sexual encounter.14 Screening intervals were changed from every year to every 3 years until age 30, at which time cotesting with cytology and HPV testing is performed every 5 years. Routine cotesting is not recommended for women younger than 30, who have a high likelihood of HPV infection and spontaneous regression.
In 2014, the US Food and Drug Administration approved primary HPV screening (ie, testing for HPV first, and then performing cytology in samples that test positive) for women age 25 and older.15
Patients who need further evaluation and testing should be referred for colposcopy. The current guidelines for patients who have abnormal results on cervical cancer screening16 can be reviewed at www.asccp.org/asccp-guidelines.
As screening guidelines continue to evolve, primary care physicians will need to stay current and also help educate their patients. For example, many of our patients have undergone annual Pap screening for most of their lives and may not yet know about the new testing intervals.
Are there disparities in screening and follow-up?
Disparities in screening and follow-up may exist, but the evidence is not clear-cut.
In a 2013 National Health Interview Survey report, the rates of cervical cancer screening with Pap tests did not differ between African American and white women.17 However, the information on Pap testing was based on a single question asking participants if they had had a Pap test in the last 3 years. In our experience, patients may confuse Pap tests with speculum examinations.
Once women are screened, adequate and timely follow-up of abnormal results is key.
In a study from the National Breast and Cervical Cancer Early Detection Program,18 women who had cytology findings of atypical squamous cells of undetermined significance or low-grade squamous intraepithelial lesions were to undergo repeat Pap testing every 4 to 6 months for 2 years. African American women were the least likely to have a follow-up Pap smear compared with other racial groups.
On the other hand, there was no difference related to race in follow-up rates of abnormal Pap tests in women ages 47 to 64 in the South Carolina Breast and Cervical Cancer Early Detection Program.19
In a study in an urban population (predominantly African African), the overall follow-up rate was only 26% at 12 months from an initial abnormal Pap smear. This study did not find any differences in follow-up according to race or ethnicity; however, it had insufficient power to detect a difference because only 15% of the study participants were white.20
What is in a genotype?
HPV is implicated in progression to both squamous cell carcinoma and adenocarcinoma of the cervix. Worldwide, HPV genotypes 16 and 18 are associated with 73% of cases of invasive cervical cancer; most of the remainder are associated with, in order of decreasing prevalence, genotypes 58, 33, 45, 31, 52, 35, 59, 39, 51, and 56.21
High-grade cervical lesions in African American women may less often be positive for HPV 16 and 18 than in white women.22,23 On the other hand, the proportion of non-Hispanic black women infected with HPV 35 and 58 was significantly higher than in non-Hispanic white women.22 Regardless, HPV screening is recommended for women of all races and ethnicities.
The 2-valent and 4-valent HPV vaccines do not cover HPV 35 or 58. The newer 9-valent vaccine covers HPV 58 (but not 35) and so may in theory decrease any potential disparity related to infection with a specific oncogenic subtype.
THE ROLE OF PREVENTION
HPV vaccination
Currently, 3 vaccines against HPV are available in the United States, a 2-valent, a 4-valent, and since 2015, a 9-valent preparation (Table 1).
The Females United to Unilaterally Reduce Endo/Ectocervical Disease study demonstrated that the 4-valent vaccine was highly effective against cervical intraepithelial neoplasia due to HPV 16 and 18.24 In another study, the 2-valent vaccine reduced the incidence of CIN 3 or higher by 87% in women who received all 3 doses and who had no evidence of HPV infection at baseline.25
HPV vaccination is expensive. Each shot costs about $130, plus the cost of administering it. Although the Vaccines for Children program covers the HPV vaccine for uninsured and underinsured children and adolescents under age 19, Medicaid coverage varies from state to state for adults over age 21.
The Advisory Committee on Immunization Practices (ACIP)26 recommends routine vaccination for:
Males 11 or 12 years old
Females ages 9 to 26.
In October 2016, the ACIP approved a 2-dose series given 6 to 12 months apart for patients starting vaccination at ages 9 through 14 years who are not immunocompromised. Others should receive a 3-dose series, with the second dose given 1 to 2 months after the first dose and the third dose given 6 months after the first dose.27 Previously, 3 doses were recommended for everyone.
Disparities in HPV vaccination rates
HPV vaccination rates among adolescents in the United States increased from 33.6% in 2013 to 41.7% in 2014.28 However, HPV vaccination rates continue to lag behind those of other routine vaccines, such as Tdap and meningococcal conjugate.
Reagan-Steiner et al28 reported that more black than white girls age 13 through 17 received at least 1 dose of a 3-dose HPV vaccination series, but more white girls received all 3 doses (70.6% vs 61.6%). In contrast, a meta-analysis by Fisher et al29 found African American and uninsured women generally less likely to initiate the HPV vaccination series. Kessels et al30 reported similar findings.
Barriers to HPV vaccination
Barriers to HPV vaccination can be provider-dependent, parental, or institutional.
Malo et al31 surveyed Florida Medicaid providers and found that those who participated in the Vaccines for Children program were less likely to cite lack of reimbursement as a barrier to vaccination.
Meites et al32 surveyed sexually transmitted disease clinics and found that common reasons for not offering HPV vaccine were cost, staff time, and difficulty coordinating follow-up visits to complete the series.
Providers report lack of urgency or lack of perception of cervical cancer as a true public health threat, safety concerns regarding the vaccine, and the inability to coadminister vaccines as barriers.33
Studies have shown that relatively few parents (up to 18%) of parents are concerned about the effect of the vaccine on sexual activity.34 Rather, they are most likely to cite lack of information regarding the vaccine, lack of physician recommendation, and not knowing where to receive the vaccine as barriers.35,36
Guerry et al37 determined that the single most important factor in vaccine initiation was physician recommendation, a finding reiterated in other studies.35,38 A study in North Carolina identified failure of physician recommendation as one of the missed opportunities for vaccination of young women.39
Therefore, the primary care physician, as the initial contact with the child or young adult, holds a responsibility to narrow this gap. In simply discussing and recommending the vaccine, physicians could increase vaccination rates.
REPRODUCTIVE HEALTH
Although 80% of women will be infected with HPV in their lifetime, only a small proportion will develop cervical cancer, suggesting there are other cofactors in the progression to cervical cancer.40
Given the infectious etiology of cervical cancer, other contributing reproductive health factors have been described. As expected, the number of sexual partners correlates with HPV infection.41,42 Younger age at first intercourse has been linked to development of cervical neoplasia, consistent with persistent infection leading to neoplasia.41,42
Primary care physicians should provide timely and comprehensive sexual education, including information on safe sexual practices and pregnancy prevention.
Human immunodeficiency virus
In 2010, the estimated rate of new human immunodeficiency virus (HIV) infections in African American women was nearly 20 times greater than in white women.43 Previous studies have shown a clear relationship between HIV and HPV-associated cancers, including cervical neoplasia and invasive cervical cancer.44,45
Women with HIV should receive screening for cervical cancer at the time of diagnosis, 6 months after the initial diagnosis, and annually thereafter.46
Conflicting evidence exists regarding the effect of highly active antiretroviral therapy on the incidence of HPV-related disease, so aggressive screening and management of cervical neoplasia is recommended for women with HIV, regardless of CD4+ levels or viral load.47–49
Additional infectious culprits
Coinfection with other sexually transmitted infections, specifically Chlamydia, herpes, and HIV, has been associated with cervical neoplasia and invasive cervical cancer. A positive linear association exists between the number of sexually transmitted infections and cervical neoplasia.50
C trachomatis is the most common sexually transmitted infection in the United States, with a 6-times higher rate in African American women.51 Women who are seropositive for C trachomatis are at twofold higher risk of developing squamous cell cervical cancer.52,53 Women who are seropositive for Chlamydia infection, herpes virus 2, or HPV are at markedly increased risk of invasive cervical cancer.50
Tobacco use
The negative impact of smoking on numerous other cancers resulted in investigation of its role in cervical cancer.
Early case-control studies found an association between cervical cancer and smoking,54 but because these studies did not account for HPV infection status, they could not establish causality. Subsequently, several studies did control for HPV infection; the risk of squamous cervical cancer was twice as high in women who had ever smoked.55 Furthermore, the more cigarettes smoked per day, the higher the risk of cervical neoplasia.41,56
According to the US Centers for Disease Control and Prevention in 2014, the highest prevalence of smoking was among American Indian and Alaskan Native women, 32.5% of whom said they smoked every day, compared with 17.2% of white women and 13.7% of African American women.57
HOW CAN PRIMARY CARE PHYSICIANS CLOSE THE GAP?
Primary care physicians are the first point of contact for patients of all ages and so can help minimize such disparities. They can tackle 2 important cervical cancer prevention interventions first-hand: vaccination and screening (Table 2), including follow-up of abnormal screening results.
By promoting HPV vaccination to children and young adults, primary care physicians can help prevent cervical cancer. Moreover, primary care physicians will see most adolescents for a nonpreventive health visit, an optimal opportunity to discuss sexual activity practices and HPV vaccination.58 Including the HPV vaccine as routine with other vaccinations can close the gap.38
Screening and treatment of sexually transmitted infection during these visits can affect the risk that future HPV infection will progress to neoplasia or cancer. Persistent lifestyle modification counseling, especially smoking cessation through motivational interviewing, can lessen the risk of cervical cancer neoplasia progression.
Additionally, in light of recent changes in cervical cancer screening guidelines, the primary care physician’s role as educator is of utmost importance. In one study, although 99% of women had received a Pap test, 87% could not identify the purpose of the Pap test.59 The primary care physician’s role is perhaps the most influential in preventing disease and, as such, has the greatest impact on a patient’s disease process.
References
Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin 2014; 64:9–29.
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin 2015; 65:5–29.
Koh WJ, Greer BE, Abu-Rustum NR, et al. Cervical cancer, version 2.2015. J Natl Compr Canc Netw 2015; 13:395-404.
Farley J, Risinger JI, Rose GS, Maxwell GL. Racial disparities in blacks with gynecologic cancers. Cancer 2007; 110:234–243.
Farley JH, Hines JF, Taylor RR, et al. Equal care ensures equal survival for African-American women with cervical carcinoma. Cancer 2001; 91:869–873.
Rauh-Hain JA, Clemmer JT, Bradford LS, et al. Racial disparities in cervical cancer survival over time. Cancer 2013; 119:3644–3652.
Collins Y, Holcomb K, Chapman-Davis E, Khabele D, Farley JH. Gynecologic cancer disparities: a report from the Health Disparities Taskforce of the Society of Gynecologic Oncology. Gynecol Oncol 2014; 133:353–361.
Patel DA, Barnholtz-Sloan JS, Patel MK, Malone JM Jr, Chuba PJ, Schwartz K. A population-based study of racial and ethnic differences in survival among women with invasive cervical cancer: analysis of surveillance, epidemiology, and end results data. Gynecol Oncol 2005; 97:550–558.
Papanicolaou GN, Traut HF. The diagnostic value of vaginal smears in carcinoma of the uterus. 1941. Arch Pathol Lab Med 1997; 121:211–224.
Walboomers JM, Jacobs M V, Manos MM, et al. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J Pathol 1999; 189:12–19.
Moscicki AB, Shiboski S, Hills NK, et al. Regression of low-grade squamous intra-epithelial lesions in young women. Lancet 2004; 364:1678–1683.
Trimble CL, Piantadosi S, Gravitt P, et al. Spontaneous regression of high-grade cervical dysplasia: effects of human papillomavirus type and HLA phenotype. Clin Cancer Res 2005; 11:4717–4723.
Ho GY, Bierman R, Beardsley L, Chang CJ, Burk RD. Natural history of cervicovaginal papillomavirus infection in young women. N Engl J Med 1998; 338:423-428.
Saslow D, Solomon D, Lawson HW, et al; ACS-ASCCP-ASCP Cervical Cancer Guideline Committee. American Cancer Society, American Society for Colposcopy and Cervical Pathology, and American Society for Clinical Pathology screening guidelines for the prevention and early detection of cervical cancer. CA Cancer J Clin 2012; 62:147–172.
Huh WK, Ault KA, Chelmow D, et al. Use of primary high-risk human papillomavirus testing for cervical cancer screening: interim clinical guidance. Obstet Gynecol 2015; 125:330–337.
Massad LS, Einstein MH, Huh WK, et al; 2012 ASCCP Consensus Guidelines Conference. 2012 updated consensus guidelines for the management of abnormal cervical cancer screening tests and cancer precursors. J Low Genit Tract Dis 2013; 17(suppl 1):S1–S27.
Sabatino SA, White MC, Thompson TD, Klabunde CN. Cancer screening test use—United States, 2013. MMWR 2015; 64:464–468.
Benard VB, Lawson HW, Eheman CR, Anderson C, Helsel W. Adherence to guidelines for follow-up of low-grade cytologic abnormalities among medically underserved women. Obstet Gynecol 2005; 105:1323–1328.
Eggleston KS, Coker AL, Luchok KJ, Meyer TE. Adherence to recommendations for follow-up to abnormal Pap tests. Obstet Gynecol 2007; 109:1332–1341.
Peterson NB, Han J, Freund KM. Inadequate follow-up for abnormal Pap smears in an urban population. J Natl Med Assoc 2003; 95:825–832.
Li N, Franceschi S, Howell-Jones R, Snijders PJ, Clifford GM. Human papillomavirus type distribution in 30,848 invasive cervical cancers worldwide: variation by geographical region, histological type and year of publication. Int J Cancer 2011; 128:927–935.
Hariri S, Unger ER, Powell SE, et al; HPV-IMPACT Working Group. Human papillomavirus genotypes in high-grade cervical lesions in the United States. J Infect Dis 2012; 206:1878–1886.
Niccolai LM, Russ C, Julian PJ, et al. Individual and geographic disparities in human papillomavirus types 16/18 in high-grade cervical lesions: associations with race, ethnicity, and poverty. Cancer 2013; 119:3052–3058.
FUTURE II Study Group. Quadrivalent vaccine against human papillomavirus to prevent high-grade cervical lesions. N Engl J Med 2007; 356:1915–1927.
Paavonen J, Naud P, Salmerón J, et al; HPV PATRICIA Study Group. Efficacy of human papillomavirus (HPV)-16/18 AS04-adjuvanted vaccine against cervical infection and precancer caused by oncogenic HPV types (PATRICIA): final analysis of a double-blind, randomised study in young women. Lancet 2009; 374:301–314.
Centers for Disease Control and Prevention (CDC). Recommendations on the use of quadrivalent human papillomavirus vaccine in males—Advisory Committee on Immunization Practices (ACIP), 2011. MMWR Morb Mortal Wkly Rep 2011; 60:1705–1708.
Meites E, Kempe A, Markowitz LE. Use of a 2-dose schedule for human papillomavirus vaccination—updated recommendations of the Advisory Committee on Immunization Practices. MMWR Morb Mortal Wkly Rep 2016; 65:1405–1408.
Reagan-Steiner S, Yankey D, Jeyarajah J, et al. National, regional, state, and selected local area vaccination coverage among adolescents aged 13–17 years—United States, 2014. MMWR Morb Mortal Wkly Rep 2015; 64:784–792.
Fisher H, Trotter CL, Audrey S, MacDonald-Wallis K, Hickman M. Inequalities in the uptake of human papillomavirus vaccination: a systematic review and meta-analysis. Int J Epidemiol 2013; 42:896–908.
Kessels SJ, Marshall HS, Watson M, Braunack-Mayer AJ, Reuzel R, Tooher RL. Factors associated with HPV vaccine uptake in teenage girls: a systematic review. Vaccine 2012; 30:3546–3556.
Malo TL, Hassani D, Staras SA, Shenkman EA, Giuliano AR, Vadaparampil ST. Do Florida Medicaid providers’ barriers to HPV vaccination vary based on VFC program participation? Matern Child Health J 2013; 17:609–615.
Meites E, Llata E, Hariri S, et al. HPV vaccine implementation in STD clinics—STD Surveillance Network. Sex Transm Dis 2012; 39:32–34.
Perkins RB, Clark JA. What affects human papillomavirus vaccination rates? A qualitative analysis of providers’ perceptions. Womens Health Issues 2012; 22:e379–e386.
Holman DM, Benard V, Roland KB, Watson M, Liddon N, Stokley S. Barriers to human papillomavirus vaccination among US adolescents: a systematic review of the literature. JAMA Pediatr 2014; 168:76–82.
Dorell CG, Yankey D, Santibanez TA, Markowitz LE. Human papillomavirus vaccination series initiation and completion, 2008–2009. Pediatrics 2011; 128:830–839.
Bastani R, Glenn BA, Tsui J, et al. Understanding suboptimal human papillomavirus vaccine uptake among ethnic minority girls. Cancer Epidemiol Biomarkers Prev 2011; 20:1463–1472.
Guerry SL, De Rosa CJ, Markowitz LE, et al. Human papillomavirus vaccine initiation among adolescent girls in high-risk communities. Vaccine 2011; 29:2235–2241.
Hull PC, Williams EA, Khabele D, Dean C, Bond B, Sanderson M. HPV vaccine use among African American girls: qualitative formative research using a participatory social marketing approach. Gynecol Oncol 2014; 132(suppl 1):S13–S20.
Brewer NT, Gottlieb SL, Reiter PL, et al. Longitudinal predictors of human papillomavirus vaccine initiation among adolescent girls in a high-risk geographic area. Sex Transm Dis 2011; 38:197–204.
Wang SS, Zuna RE, Wentzensen N, et al. Human papillomavirus cofactors by disease progression and human papillomavirus types in the study to understand cervical cancer early endpoints and determinants. Cancer Epidemiol Biomarkers Prev 2009; 18:113–120.
Deacon JM, Evans CD, Yule R, et al. Sexual behaviour and smoking as determinants of cervical HPV infection and of CIN3 among those infected: a case-control study nested within the Manchester cohort. Br J Cancer 2000; 83:1565–1572.
International Collaboration of Epidemiological Studies of Cervical Cancer. Cervical carcinoma and sexual behavior: collaborative reanalysis of individual data on 15,461 women with cervical carcinoma and 29,164 women without cervical carcinoma from 21 epidemiological studies. Cancer Epidemiol Biomarkers Prev 2009; 18:1060–1069.
Centers for Disease Control and Prevention (CDC). Estimated HIV incidence in the United States, 2007–2010. HIV Surveillance Supplemental Report 2012; 17(No. 4). https://www.cdc.gov/hiv/pdf/statistics_hssr_vol_17_no_4.pdf. Accessed September 12, 2017.
Frisch M, Biggar RJ, Goedert JJ. Human papillomavirus-associated cancers in patients with human immunodeficiency virus infection and acquired immunodeficiency syndrome. J Natl Cancer Inst 2000; 92:1500–1510.
Schäfer A, Friedmann W, Mielke M, Schwartländer B, Koch MA. The increased frequency of cervical dysplasia-neoplasia in women infected with the human immunodeficiency virus is related to the degree of immunosuppression. Am J Obstet Gynecol 1991; 164:593–599.
De Vuyst H, Lillo F, Broutet N, Smith JS. HIV, human papillomavirus, and cervical neoplasia and cancer in the era of highly active antiretroviral therapy. Eur J Cancer Prev 2008; 17:545–554.
Palefsky JM. Cervical human papillomavirus infection and cervical intraepithelial neoplasia in women positive for human immunodeficiency virus in the era of highly active antiretroviral therapy. Curr Opin Oncol 2003; 15:382–388.
Adler DH. The impact of HAART on HPV-related cervical disease. Curr HIV Res 2010; 8:493–497.
Castellsagué X, Pawlita M, Roura E, et al. Prospective seroepidemiologic study on the role of human papillomavirus and other infections in cervical carcinogenesis: evidence from the EPIC cohort. Int J Cancer 2014; 135:440–452.
Centers for Disease Control and Prevention (CDC). 2013 sexually transmitted disease surveillance. www.cdc.gov/std/stats13/exordium.htm. Accessed September 12, 2017.
Smith JS, Bosetti C, Muñoz N, et al; IARC multicentric case-control study. Chlamydia trachomatis and invasive cervical cancer: a pooled analysis of the IARC multicentric case-control study. Int J Cancer 2004; 111:431–439.
Koskela P, Anttila T, Bjørge T, et al. Chlamydia trachomatis infection as a risk factor for invasive cervical cancer. Int J Cancer 2000; 85:35–39.
Office on Smoking and Health (US). Women and smoking: a report of the Surgeon General: Chapter 3. Health consequences of tobacco use among women. http://www.ncbi.nlm.nih.gov/books/NBK44312. Accessed September 12, 2017.
Plummer M, Herrero R, Franceschi S, et al; IARC Multi-centre Cervical Cancer Study Group. Smoking and cervical cancer: pooled analysis of the IARC multi-centric case—control study. Cancer Causes Control 2003; 14:805–814.
Ho GY, Kadish AS, Burk RD, et al. HPV 16 and cigarette smoking as risk factors for high-grade cervical intra-epithelial neoplasia. Int J Cancer 1998; 78:281–285.
Jamal A, Homa DM, O’Connor E, et al. Current cigarette smoking among adults - United States, 2005-2014. MMWR Morb Mortal Wkly Rep 2015; 64:1233–1240.
Nordin JD, Solberg LI, Parker ED. Adolescent primary care visit patterns. Ann Fam Med 2010; 8:511–516.
Lindau ST, Tomori C, Lyons T, Langseth L, Bennett CL, Garcia P. The association of health literacy with cervical cancer prevention knowledge and health behaviors in a multiethnic cohort of women. Am J Obstet Gynecol 2002; 186:938–943.
References
Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin 2014; 64:9–29.
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin 2015; 65:5–29.
Koh WJ, Greer BE, Abu-Rustum NR, et al. Cervical cancer, version 2.2015. J Natl Compr Canc Netw 2015; 13:395-404.
Farley J, Risinger JI, Rose GS, Maxwell GL. Racial disparities in blacks with gynecologic cancers. Cancer 2007; 110:234–243.
Farley JH, Hines JF, Taylor RR, et al. Equal care ensures equal survival for African-American women with cervical carcinoma. Cancer 2001; 91:869–873.
Rauh-Hain JA, Clemmer JT, Bradford LS, et al. Racial disparities in cervical cancer survival over time. Cancer 2013; 119:3644–3652.
Collins Y, Holcomb K, Chapman-Davis E, Khabele D, Farley JH. Gynecologic cancer disparities: a report from the Health Disparities Taskforce of the Society of Gynecologic Oncology. Gynecol Oncol 2014; 133:353–361.
Patel DA, Barnholtz-Sloan JS, Patel MK, Malone JM Jr, Chuba PJ, Schwartz K. A population-based study of racial and ethnic differences in survival among women with invasive cervical cancer: analysis of surveillance, epidemiology, and end results data. Gynecol Oncol 2005; 97:550–558.
Papanicolaou GN, Traut HF. The diagnostic value of vaginal smears in carcinoma of the uterus. 1941. Arch Pathol Lab Med 1997; 121:211–224.
Walboomers JM, Jacobs M V, Manos MM, et al. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J Pathol 1999; 189:12–19.
Moscicki AB, Shiboski S, Hills NK, et al. Regression of low-grade squamous intra-epithelial lesions in young women. Lancet 2004; 364:1678–1683.
Trimble CL, Piantadosi S, Gravitt P, et al. Spontaneous regression of high-grade cervical dysplasia: effects of human papillomavirus type and HLA phenotype. Clin Cancer Res 2005; 11:4717–4723.
Ho GY, Bierman R, Beardsley L, Chang CJ, Burk RD. Natural history of cervicovaginal papillomavirus infection in young women. N Engl J Med 1998; 338:423-428.
Saslow D, Solomon D, Lawson HW, et al; ACS-ASCCP-ASCP Cervical Cancer Guideline Committee. American Cancer Society, American Society for Colposcopy and Cervical Pathology, and American Society for Clinical Pathology screening guidelines for the prevention and early detection of cervical cancer. CA Cancer J Clin 2012; 62:147–172.
Huh WK, Ault KA, Chelmow D, et al. Use of primary high-risk human papillomavirus testing for cervical cancer screening: interim clinical guidance. Obstet Gynecol 2015; 125:330–337.
Massad LS, Einstein MH, Huh WK, et al; 2012 ASCCP Consensus Guidelines Conference. 2012 updated consensus guidelines for the management of abnormal cervical cancer screening tests and cancer precursors. J Low Genit Tract Dis 2013; 17(suppl 1):S1–S27.
Sabatino SA, White MC, Thompson TD, Klabunde CN. Cancer screening test use—United States, 2013. MMWR 2015; 64:464–468.
Benard VB, Lawson HW, Eheman CR, Anderson C, Helsel W. Adherence to guidelines for follow-up of low-grade cytologic abnormalities among medically underserved women. Obstet Gynecol 2005; 105:1323–1328.
Eggleston KS, Coker AL, Luchok KJ, Meyer TE. Adherence to recommendations for follow-up to abnormal Pap tests. Obstet Gynecol 2007; 109:1332–1341.
Peterson NB, Han J, Freund KM. Inadequate follow-up for abnormal Pap smears in an urban population. J Natl Med Assoc 2003; 95:825–832.
Li N, Franceschi S, Howell-Jones R, Snijders PJ, Clifford GM. Human papillomavirus type distribution in 30,848 invasive cervical cancers worldwide: variation by geographical region, histological type and year of publication. Int J Cancer 2011; 128:927–935.
Hariri S, Unger ER, Powell SE, et al; HPV-IMPACT Working Group. Human papillomavirus genotypes in high-grade cervical lesions in the United States. J Infect Dis 2012; 206:1878–1886.
Niccolai LM, Russ C, Julian PJ, et al. Individual and geographic disparities in human papillomavirus types 16/18 in high-grade cervical lesions: associations with race, ethnicity, and poverty. Cancer 2013; 119:3052–3058.
FUTURE II Study Group. Quadrivalent vaccine against human papillomavirus to prevent high-grade cervical lesions. N Engl J Med 2007; 356:1915–1927.
Paavonen J, Naud P, Salmerón J, et al; HPV PATRICIA Study Group. Efficacy of human papillomavirus (HPV)-16/18 AS04-adjuvanted vaccine against cervical infection and precancer caused by oncogenic HPV types (PATRICIA): final analysis of a double-blind, randomised study in young women. Lancet 2009; 374:301–314.
Centers for Disease Control and Prevention (CDC). Recommendations on the use of quadrivalent human papillomavirus vaccine in males—Advisory Committee on Immunization Practices (ACIP), 2011. MMWR Morb Mortal Wkly Rep 2011; 60:1705–1708.
Meites E, Kempe A, Markowitz LE. Use of a 2-dose schedule for human papillomavirus vaccination—updated recommendations of the Advisory Committee on Immunization Practices. MMWR Morb Mortal Wkly Rep 2016; 65:1405–1408.
Reagan-Steiner S, Yankey D, Jeyarajah J, et al. National, regional, state, and selected local area vaccination coverage among adolescents aged 13–17 years—United States, 2014. MMWR Morb Mortal Wkly Rep 2015; 64:784–792.
Fisher H, Trotter CL, Audrey S, MacDonald-Wallis K, Hickman M. Inequalities in the uptake of human papillomavirus vaccination: a systematic review and meta-analysis. Int J Epidemiol 2013; 42:896–908.
Kessels SJ, Marshall HS, Watson M, Braunack-Mayer AJ, Reuzel R, Tooher RL. Factors associated with HPV vaccine uptake in teenage girls: a systematic review. Vaccine 2012; 30:3546–3556.
Malo TL, Hassani D, Staras SA, Shenkman EA, Giuliano AR, Vadaparampil ST. Do Florida Medicaid providers’ barriers to HPV vaccination vary based on VFC program participation? Matern Child Health J 2013; 17:609–615.
Meites E, Llata E, Hariri S, et al. HPV vaccine implementation in STD clinics—STD Surveillance Network. Sex Transm Dis 2012; 39:32–34.
Perkins RB, Clark JA. What affects human papillomavirus vaccination rates? A qualitative analysis of providers’ perceptions. Womens Health Issues 2012; 22:e379–e386.
Holman DM, Benard V, Roland KB, Watson M, Liddon N, Stokley S. Barriers to human papillomavirus vaccination among US adolescents: a systematic review of the literature. JAMA Pediatr 2014; 168:76–82.
Dorell CG, Yankey D, Santibanez TA, Markowitz LE. Human papillomavirus vaccination series initiation and completion, 2008–2009. Pediatrics 2011; 128:830–839.
Bastani R, Glenn BA, Tsui J, et al. Understanding suboptimal human papillomavirus vaccine uptake among ethnic minority girls. Cancer Epidemiol Biomarkers Prev 2011; 20:1463–1472.
Guerry SL, De Rosa CJ, Markowitz LE, et al. Human papillomavirus vaccine initiation among adolescent girls in high-risk communities. Vaccine 2011; 29:2235–2241.
Hull PC, Williams EA, Khabele D, Dean C, Bond B, Sanderson M. HPV vaccine use among African American girls: qualitative formative research using a participatory social marketing approach. Gynecol Oncol 2014; 132(suppl 1):S13–S20.
Brewer NT, Gottlieb SL, Reiter PL, et al. Longitudinal predictors of human papillomavirus vaccine initiation among adolescent girls in a high-risk geographic area. Sex Transm Dis 2011; 38:197–204.
Wang SS, Zuna RE, Wentzensen N, et al. Human papillomavirus cofactors by disease progression and human papillomavirus types in the study to understand cervical cancer early endpoints and determinants. Cancer Epidemiol Biomarkers Prev 2009; 18:113–120.
Deacon JM, Evans CD, Yule R, et al. Sexual behaviour and smoking as determinants of cervical HPV infection and of CIN3 among those infected: a case-control study nested within the Manchester cohort. Br J Cancer 2000; 83:1565–1572.
International Collaboration of Epidemiological Studies of Cervical Cancer. Cervical carcinoma and sexual behavior: collaborative reanalysis of individual data on 15,461 women with cervical carcinoma and 29,164 women without cervical carcinoma from 21 epidemiological studies. Cancer Epidemiol Biomarkers Prev 2009; 18:1060–1069.
Centers for Disease Control and Prevention (CDC). Estimated HIV incidence in the United States, 2007–2010. HIV Surveillance Supplemental Report 2012; 17(No. 4). https://www.cdc.gov/hiv/pdf/statistics_hssr_vol_17_no_4.pdf. Accessed September 12, 2017.
Frisch M, Biggar RJ, Goedert JJ. Human papillomavirus-associated cancers in patients with human immunodeficiency virus infection and acquired immunodeficiency syndrome. J Natl Cancer Inst 2000; 92:1500–1510.
Schäfer A, Friedmann W, Mielke M, Schwartländer B, Koch MA. The increased frequency of cervical dysplasia-neoplasia in women infected with the human immunodeficiency virus is related to the degree of immunosuppression. Am J Obstet Gynecol 1991; 164:593–599.
De Vuyst H, Lillo F, Broutet N, Smith JS. HIV, human papillomavirus, and cervical neoplasia and cancer in the era of highly active antiretroviral therapy. Eur J Cancer Prev 2008; 17:545–554.
Palefsky JM. Cervical human papillomavirus infection and cervical intraepithelial neoplasia in women positive for human immunodeficiency virus in the era of highly active antiretroviral therapy. Curr Opin Oncol 2003; 15:382–388.
Adler DH. The impact of HAART on HPV-related cervical disease. Curr HIV Res 2010; 8:493–497.
Castellsagué X, Pawlita M, Roura E, et al. Prospective seroepidemiologic study on the role of human papillomavirus and other infections in cervical carcinogenesis: evidence from the EPIC cohort. Int J Cancer 2014; 135:440–452.
Centers for Disease Control and Prevention (CDC). 2013 sexually transmitted disease surveillance. www.cdc.gov/std/stats13/exordium.htm. Accessed September 12, 2017.
Smith JS, Bosetti C, Muñoz N, et al; IARC multicentric case-control study. Chlamydia trachomatis and invasive cervical cancer: a pooled analysis of the IARC multicentric case-control study. Int J Cancer 2004; 111:431–439.
Koskela P, Anttila T, Bjørge T, et al. Chlamydia trachomatis infection as a risk factor for invasive cervical cancer. Int J Cancer 2000; 85:35–39.
Office on Smoking and Health (US). Women and smoking: a report of the Surgeon General: Chapter 3. Health consequences of tobacco use among women. http://www.ncbi.nlm.nih.gov/books/NBK44312. Accessed September 12, 2017.
Plummer M, Herrero R, Franceschi S, et al; IARC Multi-centre Cervical Cancer Study Group. Smoking and cervical cancer: pooled analysis of the IARC multi-centric case—control study. Cancer Causes Control 2003; 14:805–814.
Ho GY, Kadish AS, Burk RD, et al. HPV 16 and cigarette smoking as risk factors for high-grade cervical intra-epithelial neoplasia. Int J Cancer 1998; 78:281–285.
Jamal A, Homa DM, O’Connor E, et al. Current cigarette smoking among adults - United States, 2005-2014. MMWR Morb Mortal Wkly Rep 2015; 64:1233–1240.
Nordin JD, Solberg LI, Parker ED. Adolescent primary care visit patterns. Ann Fam Med 2010; 8:511–516.
Lindau ST, Tomori C, Lyons T, Langseth L, Bennett CL, Garcia P. The association of health literacy with cervical cancer prevention knowledge and health behaviors in a multiethnic cohort of women. Am J Obstet Gynecol 2002; 186:938–943.
Primary care providers play a crucial role in cancer control, including screening and follow-up.1,2 In particular, they are often responsible for performing the initial screening and, when necessary, discussing appropriate treatment options. However, cancer screening practices in primary care can vary significantly, leading to disparities in access to these services.3
Arvizo and Mahdi,4 in this issue of the Journal, discuss disparities in cervical cancer screening, noting that African American women have a higher risk of developing and dying of cervical cancer than white women, possibly because they are diagnosed at a later stage and have lower stage-specific survival rates. The authors state that equal access to healthcare may help mitigate these factors, and they also discuss how primary care providers can reduce these disparities.
PRIORITIZING CERVICAL CANCER SCREENING
Even in patients who have access to regular primary care, other barriers to cancer screening may exist. A 2014 study used self-reported data from the Behavioral Risk Factor Surveillance System survey to assess barriers to cervical cancer screening in older women (ages 40 to 65) who reported having health insurance and a personal healthcare provider.5 Those who were never or rarely screened for cervical cancer were more likely than those who were regularly screened to have a chronic condition, such as heart disease, chronic obstructive pulmonary disease, arthritis, depression, kidney disease, or diabetes.
This finding suggests that cancer screening may be a low priority during an adult primary care visit in which multiple chronic diseases must be addressed. To reduce disparities in cancer screening, primary care systems need to be designed to optimize delivery of preventive care and disease management using a team approach.
SYSTEMATIC FOLLOW-UP
Arvizo and Mahdi also discuss the follow-up of abnormal screening Papanicolaou (Pap) smears. While appropriate follow-up is a key factor in the management of cervical dysplasia, follow-up rates vary among African American women. System-level interventions such as the use of an electronic medical record-based tracking system in primary care settings6 with established protocols for follow-up may be effective.
But even with such systems in place, patients may face psychosocial barriers (eg, lack of health literacy, distress after receiving an abnormal cervical cytology test result7) that prevent them from seeking additional care. To improve follow-up rates, providers must be aware of these barriers and know how to address them through effective communication.
VACCINATION FOR HPV
Finally, the association between human papilloma virus (HPV) infection and cervical cancer makes HPV vaccination a crucial step in cervical cancer prevention. Continued provider education regarding HPV vaccination can improve knowledge about the HPV vaccine,8 as well as improve vaccination rates.9 The recent approval of a 2-dose vaccine schedule for younger girls10 may also help improve vaccine series completion rates.
The authors also suggest that primary care providers counsel all patients about risk factors for cervical cancer, including unsafe sex practices and tobacco use.
OPTIMIZING SCREENING AND PREVENTION
I commend the authors for their discussion of cervical cancer disparities and for raising awareness of the important role primary care providers play in reducing these disparities. Improving cervical cancer screening rates and follow-up will require providers and patients to be aware of cervical cancer risk factors. Further, system-level practice interventions will optimize primary care providers’ ability to engage patients in cancer screening conversations and ensure timely follow-up of screening tests.
References
Emery JD, Shaw K, Williams B, et al. The role of primary care in early detection and follow-up of cancer. Nat Rev Clin Oncol 2014; 11:38–48.
Rubin G, Berendsen A, Crawford SM, et al. The expanding role of primary care in cancer control. Lancet Oncol 2015; 16:1231–1272.
Martires KJ, Kurlander DE, Minwell GJ, Dahms EB, Bordeaux JS. Patterns of cancer screening in primary care from 2005 to 2010. Cancer 2014; 120:253–261.
Arvizo C, Mahdi H. Disparities in cervical cancer in African-American women: what primary care physicians can do. Cleve Clin J Med 2017; 84:788–794.
Crawford A, Benard V, King J, Thomas CC. Understanding barriers to cervical cancer screening in women with access to care, behavioral risk factor surveillance system, 2014. Prev Chronic Dis 2016; 13:E154.
Dupuis EA, White HF, Newman D, Sobieraj JE, Gokhale M, Freund KM. Tracking abnormal cervical cancer screening: evaluation of an EMR-based intervention. J Gen Intern Med 2010; 25:575–580.
Hui SK, Miller SM, Wen KY, et al. Psychosocial barriers to follow-up adherence after an abnormal cervical cytology test result among low-income, inner-city women. J Prim Care Community Health 2014; 5:234–241.
Berenson AB, Rahman M, Hirth JM, Rupp RE, Sarpong KO. A brief educational intervention increases providers’ human papillomavirus vaccine knowledge. Hum Vaccin Immunother 2015; 11:1331–1336.
Perkins RB, Zisblatt L, Legler A, Trucks E, Hanchate A, Gorin SS. Effectiveness of a provider-focused intervention to improve HPV vaccination rates in boys and girls. Vaccine 2015; 33:1223–1229.
Meites E, Kempe A, Markowitz LE. Use of a 2-dose schedule for human papillomavirus vaccination—updated recommendations of the Advisory Committee on Immunization Practices. MMWR Morb Mortal Wkly Rep 2016; 65:1405–1408.
Anita D. Misra-Hebert MD, MPH Department of Internal Medicine, Center for Value-Based Care Research, Medicine Institute, and Department of Quantitative Health Sciences, Lerner Research Institute, Cleveland Clinic; Assistant Professor, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, Cleveland, OH
Address: Anita D. Misra-Hebert MD, MPH, Department of Internal Medicine, Center for Value-Based Care Research, Medicine Institute, G10, Cleveland Clinic, 9500 Euclid Avenue, Cleveland, OH 44195; misraa@ccf.org
Dr. Misra-Hebert is supported by an Agency for Healthcare Research and Quality grant K08HS024128.
Anita D. Misra-Hebert MD, MPH Department of Internal Medicine, Center for Value-Based Care Research, Medicine Institute, and Department of Quantitative Health Sciences, Lerner Research Institute, Cleveland Clinic; Assistant Professor, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, Cleveland, OH
Address: Anita D. Misra-Hebert MD, MPH, Department of Internal Medicine, Center for Value-Based Care Research, Medicine Institute, G10, Cleveland Clinic, 9500 Euclid Avenue, Cleveland, OH 44195; misraa@ccf.org
Dr. Misra-Hebert is supported by an Agency for Healthcare Research and Quality grant K08HS024128.
Author and Disclosure Information
Anita D. Misra-Hebert MD, MPH Department of Internal Medicine, Center for Value-Based Care Research, Medicine Institute, and Department of Quantitative Health Sciences, Lerner Research Institute, Cleveland Clinic; Assistant Professor, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, Cleveland, OH
Address: Anita D. Misra-Hebert MD, MPH, Department of Internal Medicine, Center for Value-Based Care Research, Medicine Institute, G10, Cleveland Clinic, 9500 Euclid Avenue, Cleveland, OH 44195; misraa@ccf.org
Dr. Misra-Hebert is supported by an Agency for Healthcare Research and Quality grant K08HS024128.
Primary care providers play a crucial role in cancer control, including screening and follow-up.1,2 In particular, they are often responsible for performing the initial screening and, when necessary, discussing appropriate treatment options. However, cancer screening practices in primary care can vary significantly, leading to disparities in access to these services.3
Arvizo and Mahdi,4 in this issue of the Journal, discuss disparities in cervical cancer screening, noting that African American women have a higher risk of developing and dying of cervical cancer than white women, possibly because they are diagnosed at a later stage and have lower stage-specific survival rates. The authors state that equal access to healthcare may help mitigate these factors, and they also discuss how primary care providers can reduce these disparities.
PRIORITIZING CERVICAL CANCER SCREENING
Even in patients who have access to regular primary care, other barriers to cancer screening may exist. A 2014 study used self-reported data from the Behavioral Risk Factor Surveillance System survey to assess barriers to cervical cancer screening in older women (ages 40 to 65) who reported having health insurance and a personal healthcare provider.5 Those who were never or rarely screened for cervical cancer were more likely than those who were regularly screened to have a chronic condition, such as heart disease, chronic obstructive pulmonary disease, arthritis, depression, kidney disease, or diabetes.
This finding suggests that cancer screening may be a low priority during an adult primary care visit in which multiple chronic diseases must be addressed. To reduce disparities in cancer screening, primary care systems need to be designed to optimize delivery of preventive care and disease management using a team approach.
SYSTEMATIC FOLLOW-UP
Arvizo and Mahdi also discuss the follow-up of abnormal screening Papanicolaou (Pap) smears. While appropriate follow-up is a key factor in the management of cervical dysplasia, follow-up rates vary among African American women. System-level interventions such as the use of an electronic medical record-based tracking system in primary care settings6 with established protocols for follow-up may be effective.
But even with such systems in place, patients may face psychosocial barriers (eg, lack of health literacy, distress after receiving an abnormal cervical cytology test result7) that prevent them from seeking additional care. To improve follow-up rates, providers must be aware of these barriers and know how to address them through effective communication.
VACCINATION FOR HPV
Finally, the association between human papilloma virus (HPV) infection and cervical cancer makes HPV vaccination a crucial step in cervical cancer prevention. Continued provider education regarding HPV vaccination can improve knowledge about the HPV vaccine,8 as well as improve vaccination rates.9 The recent approval of a 2-dose vaccine schedule for younger girls10 may also help improve vaccine series completion rates.
The authors also suggest that primary care providers counsel all patients about risk factors for cervical cancer, including unsafe sex practices and tobacco use.
OPTIMIZING SCREENING AND PREVENTION
I commend the authors for their discussion of cervical cancer disparities and for raising awareness of the important role primary care providers play in reducing these disparities. Improving cervical cancer screening rates and follow-up will require providers and patients to be aware of cervical cancer risk factors. Further, system-level practice interventions will optimize primary care providers’ ability to engage patients in cancer screening conversations and ensure timely follow-up of screening tests.
Primary care providers play a crucial role in cancer control, including screening and follow-up.1,2 In particular, they are often responsible for performing the initial screening and, when necessary, discussing appropriate treatment options. However, cancer screening practices in primary care can vary significantly, leading to disparities in access to these services.3
Arvizo and Mahdi,4 in this issue of the Journal, discuss disparities in cervical cancer screening, noting that African American women have a higher risk of developing and dying of cervical cancer than white women, possibly because they are diagnosed at a later stage and have lower stage-specific survival rates. The authors state that equal access to healthcare may help mitigate these factors, and they also discuss how primary care providers can reduce these disparities.
PRIORITIZING CERVICAL CANCER SCREENING
Even in patients who have access to regular primary care, other barriers to cancer screening may exist. A 2014 study used self-reported data from the Behavioral Risk Factor Surveillance System survey to assess barriers to cervical cancer screening in older women (ages 40 to 65) who reported having health insurance and a personal healthcare provider.5 Those who were never or rarely screened for cervical cancer were more likely than those who were regularly screened to have a chronic condition, such as heart disease, chronic obstructive pulmonary disease, arthritis, depression, kidney disease, or diabetes.
This finding suggests that cancer screening may be a low priority during an adult primary care visit in which multiple chronic diseases must be addressed. To reduce disparities in cancer screening, primary care systems need to be designed to optimize delivery of preventive care and disease management using a team approach.
SYSTEMATIC FOLLOW-UP
Arvizo and Mahdi also discuss the follow-up of abnormal screening Papanicolaou (Pap) smears. While appropriate follow-up is a key factor in the management of cervical dysplasia, follow-up rates vary among African American women. System-level interventions such as the use of an electronic medical record-based tracking system in primary care settings6 with established protocols for follow-up may be effective.
But even with such systems in place, patients may face psychosocial barriers (eg, lack of health literacy, distress after receiving an abnormal cervical cytology test result7) that prevent them from seeking additional care. To improve follow-up rates, providers must be aware of these barriers and know how to address them through effective communication.
VACCINATION FOR HPV
Finally, the association between human papilloma virus (HPV) infection and cervical cancer makes HPV vaccination a crucial step in cervical cancer prevention. Continued provider education regarding HPV vaccination can improve knowledge about the HPV vaccine,8 as well as improve vaccination rates.9 The recent approval of a 2-dose vaccine schedule for younger girls10 may also help improve vaccine series completion rates.
The authors also suggest that primary care providers counsel all patients about risk factors for cervical cancer, including unsafe sex practices and tobacco use.
OPTIMIZING SCREENING AND PREVENTION
I commend the authors for their discussion of cervical cancer disparities and for raising awareness of the important role primary care providers play in reducing these disparities. Improving cervical cancer screening rates and follow-up will require providers and patients to be aware of cervical cancer risk factors. Further, system-level practice interventions will optimize primary care providers’ ability to engage patients in cancer screening conversations and ensure timely follow-up of screening tests.
References
Emery JD, Shaw K, Williams B, et al. The role of primary care in early detection and follow-up of cancer. Nat Rev Clin Oncol 2014; 11:38–48.
Rubin G, Berendsen A, Crawford SM, et al. The expanding role of primary care in cancer control. Lancet Oncol 2015; 16:1231–1272.
Martires KJ, Kurlander DE, Minwell GJ, Dahms EB, Bordeaux JS. Patterns of cancer screening in primary care from 2005 to 2010. Cancer 2014; 120:253–261.
Arvizo C, Mahdi H. Disparities in cervical cancer in African-American women: what primary care physicians can do. Cleve Clin J Med 2017; 84:788–794.
Crawford A, Benard V, King J, Thomas CC. Understanding barriers to cervical cancer screening in women with access to care, behavioral risk factor surveillance system, 2014. Prev Chronic Dis 2016; 13:E154.
Dupuis EA, White HF, Newman D, Sobieraj JE, Gokhale M, Freund KM. Tracking abnormal cervical cancer screening: evaluation of an EMR-based intervention. J Gen Intern Med 2010; 25:575–580.
Hui SK, Miller SM, Wen KY, et al. Psychosocial barriers to follow-up adherence after an abnormal cervical cytology test result among low-income, inner-city women. J Prim Care Community Health 2014; 5:234–241.
Berenson AB, Rahman M, Hirth JM, Rupp RE, Sarpong KO. A brief educational intervention increases providers’ human papillomavirus vaccine knowledge. Hum Vaccin Immunother 2015; 11:1331–1336.
Perkins RB, Zisblatt L, Legler A, Trucks E, Hanchate A, Gorin SS. Effectiveness of a provider-focused intervention to improve HPV vaccination rates in boys and girls. Vaccine 2015; 33:1223–1229.
Meites E, Kempe A, Markowitz LE. Use of a 2-dose schedule for human papillomavirus vaccination—updated recommendations of the Advisory Committee on Immunization Practices. MMWR Morb Mortal Wkly Rep 2016; 65:1405–1408.
References
Emery JD, Shaw K, Williams B, et al. The role of primary care in early detection and follow-up of cancer. Nat Rev Clin Oncol 2014; 11:38–48.
Rubin G, Berendsen A, Crawford SM, et al. The expanding role of primary care in cancer control. Lancet Oncol 2015; 16:1231–1272.
Martires KJ, Kurlander DE, Minwell GJ, Dahms EB, Bordeaux JS. Patterns of cancer screening in primary care from 2005 to 2010. Cancer 2014; 120:253–261.
Arvizo C, Mahdi H. Disparities in cervical cancer in African-American women: what primary care physicians can do. Cleve Clin J Med 2017; 84:788–794.
Crawford A, Benard V, King J, Thomas CC. Understanding barriers to cervical cancer screening in women with access to care, behavioral risk factor surveillance system, 2014. Prev Chronic Dis 2016; 13:E154.
Dupuis EA, White HF, Newman D, Sobieraj JE, Gokhale M, Freund KM. Tracking abnormal cervical cancer screening: evaluation of an EMR-based intervention. J Gen Intern Med 2010; 25:575–580.
Hui SK, Miller SM, Wen KY, et al. Psychosocial barriers to follow-up adherence after an abnormal cervical cytology test result among low-income, inner-city women. J Prim Care Community Health 2014; 5:234–241.
Berenson AB, Rahman M, Hirth JM, Rupp RE, Sarpong KO. A brief educational intervention increases providers’ human papillomavirus vaccine knowledge. Hum Vaccin Immunother 2015; 11:1331–1336.
Perkins RB, Zisblatt L, Legler A, Trucks E, Hanchate A, Gorin SS. Effectiveness of a provider-focused intervention to improve HPV vaccination rates in boys and girls. Vaccine 2015; 33:1223–1229.
Meites E, Kempe A, Markowitz LE. Use of a 2-dose schedule for human papillomavirus vaccination—updated recommendations of the Advisory Committee on Immunization Practices. MMWR Morb Mortal Wkly Rep 2016; 65:1405–1408.
A 40-year-old man was brought to the emergency department with altered mental status. His roommate had found him lying unconscious in snow on the lawn outside his residence. When the emergency medical services team arrived, they recorded a core body temperature of 28.3°C (82.9°F) and instituted advanced cardiac life support.
During transit to the hospital, the patient’s heart rhythm changed from asystole to ventricular fibrillation, and defibrillation was performed twice.
Upon his arrival at the emergency room, advanced life support was continued, resulting in return of spontaneous circulation, with slow, wide-complex QRS rhythm noted on electrocardiography (ECG).
On examination, the patient’s pupils were fixed and dilated. The extremities were cold to palpation. The core body temperature dropped to 27.7°C (81.9°F).
Laboratory test results showed severe acidemia (arterial pH 6.8), elevated aspartate aminotransferase and alanine aminotransferase levels, and elevated creatinine and troponin. The troponin was measured 3 times and rose from 0.8 ng/mL to 0.9 ng/mL. A urine toxicology screen was positive for cannabinoids and cocaine.
Figure 1. On 12-lead electrocardiography, elevation of the J point (Osborn wave; arrows) is typically seen in precordial leads V3 to V6 and is a marker for hypothermia. The amplitude of Osborn waves is directly proportional to the degree of hypothermia.
ECG revealed J-point elevation (Osborn waves) in the precordial leads (Figure 1). A baseline electrocardiogram in the medical record from a previous admission had been normal.
An aggressive hypothermia protocol was initiated, but the patient died despite resuscitation efforts.
HYPOTHERMIA AND HEART RHYTHMS
Hypothermia—a core body temperature below 35°C (95°F)—causes generalized slowing of impulse conduction through cardiac tissues, shown on ECG as a prolongation of the PR, RR, QRS, and QT intervals.1
A characteristic feature is elevation of the J point, also called the J wave or Osborn wave, most prominent in precordial leads V2 to V5 and caused by abnormal membrane repolarization in the early phase. The degree of hypothermia correlates linearly with the amplitude of the Osborn wave.2,3
Laboratory tests can identify complications such as rhabdomyolysis, spontaneous bleeding, and lactic acidosis. Moderate to severe hypothermia may cause prolongation of all ECG intervals. Management requires resuscitation and rewarming.
Conditions to consider in the differential diagnosis are Brugada syndrome, hypercalcemia, and early repolarization syndrome.
References
Doshi HH, Giudici MC. The EKG in hypothermia and hyperthermia. J Electrocardiol 2015; 48:203–208.
Alsafwah S. Electrocardiographic changes in hypothermia. Heart Lung 2001; 30:161–163.
Graham CA, McNaughton GW, Wyatt JP. The electrocardiogram in hypothermia. Wilderness Environ Med 2001; 12:232–235.
A 40-year-old man was brought to the emergency department with altered mental status. His roommate had found him lying unconscious in snow on the lawn outside his residence. When the emergency medical services team arrived, they recorded a core body temperature of 28.3°C (82.9°F) and instituted advanced cardiac life support.
During transit to the hospital, the patient’s heart rhythm changed from asystole to ventricular fibrillation, and defibrillation was performed twice.
Upon his arrival at the emergency room, advanced life support was continued, resulting in return of spontaneous circulation, with slow, wide-complex QRS rhythm noted on electrocardiography (ECG).
On examination, the patient’s pupils were fixed and dilated. The extremities were cold to palpation. The core body temperature dropped to 27.7°C (81.9°F).
Laboratory test results showed severe acidemia (arterial pH 6.8), elevated aspartate aminotransferase and alanine aminotransferase levels, and elevated creatinine and troponin. The troponin was measured 3 times and rose from 0.8 ng/mL to 0.9 ng/mL. A urine toxicology screen was positive for cannabinoids and cocaine.
Figure 1. On 12-lead electrocardiography, elevation of the J point (Osborn wave; arrows) is typically seen in precordial leads V3 to V6 and is a marker for hypothermia. The amplitude of Osborn waves is directly proportional to the degree of hypothermia.
ECG revealed J-point elevation (Osborn waves) in the precordial leads (Figure 1). A baseline electrocardiogram in the medical record from a previous admission had been normal.
An aggressive hypothermia protocol was initiated, but the patient died despite resuscitation efforts.
HYPOTHERMIA AND HEART RHYTHMS
Hypothermia—a core body temperature below 35°C (95°F)—causes generalized slowing of impulse conduction through cardiac tissues, shown on ECG as a prolongation of the PR, RR, QRS, and QT intervals.1
A characteristic feature is elevation of the J point, also called the J wave or Osborn wave, most prominent in precordial leads V2 to V5 and caused by abnormal membrane repolarization in the early phase. The degree of hypothermia correlates linearly with the amplitude of the Osborn wave.2,3
Laboratory tests can identify complications such as rhabdomyolysis, spontaneous bleeding, and lactic acidosis. Moderate to severe hypothermia may cause prolongation of all ECG intervals. Management requires resuscitation and rewarming.
Conditions to consider in the differential diagnosis are Brugada syndrome, hypercalcemia, and early repolarization syndrome.
A 40-year-old man was brought to the emergency department with altered mental status. His roommate had found him lying unconscious in snow on the lawn outside his residence. When the emergency medical services team arrived, they recorded a core body temperature of 28.3°C (82.9°F) and instituted advanced cardiac life support.
During transit to the hospital, the patient’s heart rhythm changed from asystole to ventricular fibrillation, and defibrillation was performed twice.
Upon his arrival at the emergency room, advanced life support was continued, resulting in return of spontaneous circulation, with slow, wide-complex QRS rhythm noted on electrocardiography (ECG).
On examination, the patient’s pupils were fixed and dilated. The extremities were cold to palpation. The core body temperature dropped to 27.7°C (81.9°F).
Laboratory test results showed severe acidemia (arterial pH 6.8), elevated aspartate aminotransferase and alanine aminotransferase levels, and elevated creatinine and troponin. The troponin was measured 3 times and rose from 0.8 ng/mL to 0.9 ng/mL. A urine toxicology screen was positive for cannabinoids and cocaine.
Figure 1. On 12-lead electrocardiography, elevation of the J point (Osborn wave; arrows) is typically seen in precordial leads V3 to V6 and is a marker for hypothermia. The amplitude of Osborn waves is directly proportional to the degree of hypothermia.
ECG revealed J-point elevation (Osborn waves) in the precordial leads (Figure 1). A baseline electrocardiogram in the medical record from a previous admission had been normal.
An aggressive hypothermia protocol was initiated, but the patient died despite resuscitation efforts.
HYPOTHERMIA AND HEART RHYTHMS
Hypothermia—a core body temperature below 35°C (95°F)—causes generalized slowing of impulse conduction through cardiac tissues, shown on ECG as a prolongation of the PR, RR, QRS, and QT intervals.1
A characteristic feature is elevation of the J point, also called the J wave or Osborn wave, most prominent in precordial leads V2 to V5 and caused by abnormal membrane repolarization in the early phase. The degree of hypothermia correlates linearly with the amplitude of the Osborn wave.2,3
Laboratory tests can identify complications such as rhabdomyolysis, spontaneous bleeding, and lactic acidosis. Moderate to severe hypothermia may cause prolongation of all ECG intervals. Management requires resuscitation and rewarming.
Conditions to consider in the differential diagnosis are Brugada syndrome, hypercalcemia, and early repolarization syndrome.
References
Doshi HH, Giudici MC. The EKG in hypothermia and hyperthermia. J Electrocardiol 2015; 48:203–208.
Alsafwah S. Electrocardiographic changes in hypothermia. Heart Lung 2001; 30:161–163.
Graham CA, McNaughton GW, Wyatt JP. The electrocardiogram in hypothermia. Wilderness Environ Med 2001; 12:232–235.
References
Doshi HH, Giudici MC. The EKG in hypothermia and hyperthermia. J Electrocardiol 2015; 48:203–208.
Alsafwah S. Electrocardiographic changes in hypothermia. Heart Lung 2001; 30:161–163.
Graham CA, McNaughton GW, Wyatt JP. The electrocardiogram in hypothermia. Wilderness Environ Med 2001; 12:232–235.
A 41-year-old woman presented with coughing, wheezing, and painful subcutaneous nodules on her legs. She had presented 6 years ago with similar nodules and enlarged retroauricular, occipital, and maxillary lymph nodes. At that time, biopsy study of submaxillary lymph nodes and skin showed nonnecrotizing granulomas, with negative microbiology studies. Chest radiography and spirometry were normal. A diagnosis of sarcoidosis was made. Treatment was offered but refused.
Figure 1. Magnetic resonance imaging revealed low-intensity lesions (arrows) on T1-weighted sequences (upper panel) and high-intensity lesions on short tau inversion recovery sequences (lower panel). The lesions have defined borders, no cortical disruption, and no mass effect, features suggesting that the lesions are benign. Lack of signal homogeneity suggests chronic lesions.
Based on this history, computed tomography of the thorax and abdomen was performed and showed mediastinal and hilar lymphadenopathy, small bilateral lung nodules, and osseous cystic areas in both iliac blades. Magnetic resonance imaging (MRI) showed numerous discrete lesions in both iliac bones (Figure 1). Biopsy study of iliac bone revealed preserved architecture with no evidence of malignancy or granulomas.
Radiography of the hands showed an osseous lytic lesion in the third proximal phalanx of the right hand. No other radiographic abnormalities were noted.
The clinical and radiographic features and the patient’s clinical course were consistent with osseous sarcoidosis. She was started on methotrexate and a low-dose corticosteroid and was symptom-free at 12-month follow-up. Follow-up MRI showed reduction in the lymphadenopathies and stabilization of the bone lesions.
SARCOIDOSIS AND BONE
Sarcoidosis is a systemic granulomatous disease that involves the lung in more than 90% of cases. Skeletal involvement has been reported in 1% to 14% of patients.1,2 Typical osseous involvement is cystic osteitis of the phalangeal bones of the hands and feet, but any part of the skeleton may be involved.3
Bone sarcoidosis is usually asymptomatic and is discovered incidentally. The diagnosis of sarcoidosis has usually been established clinically before bone lesions are detected on MRI. However, sarcoidosis-related bone lesions resembling bone metastases on MRI may be the initial presentation. The presence of intralesional fat has been described as a feature that excludes malignancy.
No treatment has been shown to be of benefit.4 Sarcoidosis is a diagnosis of exclusion and radiographic lytic bone features are not specific, so a neoplastic cause (such as primary osteoblastoma, metastasis, or multiple myeloma) must always be ruled out, as well as other bone conditions such as osteomyelitis or bone cyst.
References
Valeyre D, Prasse A, Nunes H, Uzunhan Y, Brillet PY, Müller-Quernheim J. Sarcoidosis. Lancet 2014; 383:1155–1167.
James DG, Neville E, Carstairs LS. Bone and joint sarcoidosis. Semin Arthritis Rheum 1976; 6:53–81.
Moore SL, Kransdorf MJ, Schweitzer ME, Murphey MD, Babb JS. Can sarcoidosis and metastatic bone lesions be reliably differentiated on routine MRI? AJR Am J Roentgenol 2012; 198:1387–1393.
Hamoud S, Srour S, Fruchter O, Vlodavsky E, Hayek T. Lytic bone lesion: presenting finding of sarcoidosis. Isr Med Assoc J 2010; 12:59–60.
Anahy M. Brandy-García Servicio de Reumatología, Hospital Universitario Central de Asturias Oviedo, Spain
Ivan Cabezas-Rodriguez Servicio de Reumatología, Hospital Universitario Central de Asturias, Oviedo, Spain
Luis Caminal-Montero Consulta-Unidad Enfermedades Autoinmunes Sistémicas, Unidad Clínica de Medicina Interna Hospital Universitario Central de Asturias, Oviedo, Spain
Carlos Suarez-Cuervo Department of General Medicine, Borders General Hospital, Melrose, Scotland, UK
Pilar Redondo-Buil Servicio de Radiología, Hospital Universitario Central de Asturias, Oviedo, Spain
Address: Anahy M. Brandy-García, Servicio de Reumatología, Hospital Universitario Central de Asturias, Av Roma s/n, 33011 Asturias, Spain; anahymbg@gmail.com
sarcoidosis, lung nodules, bone metastases, lytic osseous metastases, Anahy Brandy-Garcia, Ivan Cabezas-Rodriguez, Luis Caminal-Montero, Carlos Suarez-Cuervo, Pilar Redondo-Buil
Anahy M. Brandy-García Servicio de Reumatología, Hospital Universitario Central de Asturias Oviedo, Spain
Ivan Cabezas-Rodriguez Servicio de Reumatología, Hospital Universitario Central de Asturias, Oviedo, Spain
Luis Caminal-Montero Consulta-Unidad Enfermedades Autoinmunes Sistémicas, Unidad Clínica de Medicina Interna Hospital Universitario Central de Asturias, Oviedo, Spain
Carlos Suarez-Cuervo Department of General Medicine, Borders General Hospital, Melrose, Scotland, UK
Pilar Redondo-Buil Servicio de Radiología, Hospital Universitario Central de Asturias, Oviedo, Spain
Address: Anahy M. Brandy-García, Servicio de Reumatología, Hospital Universitario Central de Asturias, Av Roma s/n, 33011 Asturias, Spain; anahymbg@gmail.com
Author and Disclosure Information
Anahy M. Brandy-García Servicio de Reumatología, Hospital Universitario Central de Asturias Oviedo, Spain
Ivan Cabezas-Rodriguez Servicio de Reumatología, Hospital Universitario Central de Asturias, Oviedo, Spain
Luis Caminal-Montero Consulta-Unidad Enfermedades Autoinmunes Sistémicas, Unidad Clínica de Medicina Interna Hospital Universitario Central de Asturias, Oviedo, Spain
Carlos Suarez-Cuervo Department of General Medicine, Borders General Hospital, Melrose, Scotland, UK
Pilar Redondo-Buil Servicio de Radiología, Hospital Universitario Central de Asturias, Oviedo, Spain
Address: Anahy M. Brandy-García, Servicio de Reumatología, Hospital Universitario Central de Asturias, Av Roma s/n, 33011 Asturias, Spain; anahymbg@gmail.com
A 41-year-old woman presented with coughing, wheezing, and painful subcutaneous nodules on her legs. She had presented 6 years ago with similar nodules and enlarged retroauricular, occipital, and maxillary lymph nodes. At that time, biopsy study of submaxillary lymph nodes and skin showed nonnecrotizing granulomas, with negative microbiology studies. Chest radiography and spirometry were normal. A diagnosis of sarcoidosis was made. Treatment was offered but refused.
Figure 1. Magnetic resonance imaging revealed low-intensity lesions (arrows) on T1-weighted sequences (upper panel) and high-intensity lesions on short tau inversion recovery sequences (lower panel). The lesions have defined borders, no cortical disruption, and no mass effect, features suggesting that the lesions are benign. Lack of signal homogeneity suggests chronic lesions.
Based on this history, computed tomography of the thorax and abdomen was performed and showed mediastinal and hilar lymphadenopathy, small bilateral lung nodules, and osseous cystic areas in both iliac blades. Magnetic resonance imaging (MRI) showed numerous discrete lesions in both iliac bones (Figure 1). Biopsy study of iliac bone revealed preserved architecture with no evidence of malignancy or granulomas.
Radiography of the hands showed an osseous lytic lesion in the third proximal phalanx of the right hand. No other radiographic abnormalities were noted.
The clinical and radiographic features and the patient’s clinical course were consistent with osseous sarcoidosis. She was started on methotrexate and a low-dose corticosteroid and was symptom-free at 12-month follow-up. Follow-up MRI showed reduction in the lymphadenopathies and stabilization of the bone lesions.
SARCOIDOSIS AND BONE
Sarcoidosis is a systemic granulomatous disease that involves the lung in more than 90% of cases. Skeletal involvement has been reported in 1% to 14% of patients.1,2 Typical osseous involvement is cystic osteitis of the phalangeal bones of the hands and feet, but any part of the skeleton may be involved.3
Bone sarcoidosis is usually asymptomatic and is discovered incidentally. The diagnosis of sarcoidosis has usually been established clinically before bone lesions are detected on MRI. However, sarcoidosis-related bone lesions resembling bone metastases on MRI may be the initial presentation. The presence of intralesional fat has been described as a feature that excludes malignancy.
No treatment has been shown to be of benefit.4 Sarcoidosis is a diagnosis of exclusion and radiographic lytic bone features are not specific, so a neoplastic cause (such as primary osteoblastoma, metastasis, or multiple myeloma) must always be ruled out, as well as other bone conditions such as osteomyelitis or bone cyst.
A 41-year-old woman presented with coughing, wheezing, and painful subcutaneous nodules on her legs. She had presented 6 years ago with similar nodules and enlarged retroauricular, occipital, and maxillary lymph nodes. At that time, biopsy study of submaxillary lymph nodes and skin showed nonnecrotizing granulomas, with negative microbiology studies. Chest radiography and spirometry were normal. A diagnosis of sarcoidosis was made. Treatment was offered but refused.
Figure 1. Magnetic resonance imaging revealed low-intensity lesions (arrows) on T1-weighted sequences (upper panel) and high-intensity lesions on short tau inversion recovery sequences (lower panel). The lesions have defined borders, no cortical disruption, and no mass effect, features suggesting that the lesions are benign. Lack of signal homogeneity suggests chronic lesions.
Based on this history, computed tomography of the thorax and abdomen was performed and showed mediastinal and hilar lymphadenopathy, small bilateral lung nodules, and osseous cystic areas in both iliac blades. Magnetic resonance imaging (MRI) showed numerous discrete lesions in both iliac bones (Figure 1). Biopsy study of iliac bone revealed preserved architecture with no evidence of malignancy or granulomas.
Radiography of the hands showed an osseous lytic lesion in the third proximal phalanx of the right hand. No other radiographic abnormalities were noted.
The clinical and radiographic features and the patient’s clinical course were consistent with osseous sarcoidosis. She was started on methotrexate and a low-dose corticosteroid and was symptom-free at 12-month follow-up. Follow-up MRI showed reduction in the lymphadenopathies and stabilization of the bone lesions.
SARCOIDOSIS AND BONE
Sarcoidosis is a systemic granulomatous disease that involves the lung in more than 90% of cases. Skeletal involvement has been reported in 1% to 14% of patients.1,2 Typical osseous involvement is cystic osteitis of the phalangeal bones of the hands and feet, but any part of the skeleton may be involved.3
Bone sarcoidosis is usually asymptomatic and is discovered incidentally. The diagnosis of sarcoidosis has usually been established clinically before bone lesions are detected on MRI. However, sarcoidosis-related bone lesions resembling bone metastases on MRI may be the initial presentation. The presence of intralesional fat has been described as a feature that excludes malignancy.
No treatment has been shown to be of benefit.4 Sarcoidosis is a diagnosis of exclusion and radiographic lytic bone features are not specific, so a neoplastic cause (such as primary osteoblastoma, metastasis, or multiple myeloma) must always be ruled out, as well as other bone conditions such as osteomyelitis or bone cyst.
References
Valeyre D, Prasse A, Nunes H, Uzunhan Y, Brillet PY, Müller-Quernheim J. Sarcoidosis. Lancet 2014; 383:1155–1167.
James DG, Neville E, Carstairs LS. Bone and joint sarcoidosis. Semin Arthritis Rheum 1976; 6:53–81.
Moore SL, Kransdorf MJ, Schweitzer ME, Murphey MD, Babb JS. Can sarcoidosis and metastatic bone lesions be reliably differentiated on routine MRI? AJR Am J Roentgenol 2012; 198:1387–1393.
Hamoud S, Srour S, Fruchter O, Vlodavsky E, Hayek T. Lytic bone lesion: presenting finding of sarcoidosis. Isr Med Assoc J 2010; 12:59–60.
References
Valeyre D, Prasse A, Nunes H, Uzunhan Y, Brillet PY, Müller-Quernheim J. Sarcoidosis. Lancet 2014; 383:1155–1167.
James DG, Neville E, Carstairs LS. Bone and joint sarcoidosis. Semin Arthritis Rheum 1976; 6:53–81.
Moore SL, Kransdorf MJ, Schweitzer ME, Murphey MD, Babb JS. Can sarcoidosis and metastatic bone lesions be reliably differentiated on routine MRI? AJR Am J Roentgenol 2012; 198:1387–1393.
Hamoud S, Srour S, Fruchter O, Vlodavsky E, Hayek T. Lytic bone lesion: presenting finding of sarcoidosis. Isr Med Assoc J 2010; 12:59–60.
sarcoidosis, lung nodules, bone metastases, lytic osseous metastases, Anahy Brandy-Garcia, Ivan Cabezas-Rodriguez, Luis Caminal-Montero, Carlos Suarez-Cuervo, Pilar Redondo-Buil
Legacy Keywords
sarcoidosis, lung nodules, bone metastases, lytic osseous metastases, Anahy Brandy-Garcia, Ivan Cabezas-Rodriguez, Luis Caminal-Montero, Carlos Suarez-Cuervo, Pilar Redondo-Buil
Raynaud phenomenon is an overactive vascular response to cold and emotional stress that results in cutaneous color changes and sensory symptoms of the digits (Figure 1). It can occur in isolation as primary Raynaud phenomenon or secondary to another disease process. It is thought to be triggered by a heightened sympathetic vasoconstrictive response of small arteriovenous anastomoses in the fingers, toes, ears, and tip of the nose. These structures play a key role in maintaining a stable core body temperature by cutaneous thermoregulation.1
Figure 1. (A) White digits with intense vasoconstriction in Raynaud phenomenon; (B) blue digits with
hypoxemic venous stasis; (C) red digits with hyperemic reperfusion.
Secondary Raynaud phenomenon can be seen with a wide array of systemic conditions as well as environmental and drug exposures. It is a frequent feature of autoimmune rheumatic conditions such as systemic sclerosis, mixed connective tissue disease, systemic lupus erythematosus, and dermatomyositis. Less commonly, cryoproteinemias, paraneoplastic syndromes, hypothyroidism, and carpal tunnel syndrome can be associated with or cause Raynaud phenomenon. Vibratory trauma (eg, from using a jackhammer) and drugs (eg, vasopressors, stimulants, ergots, chemotherapeutic agents) can also cause Raynaud phenomenon.1
A variety of disorders that cause vasospasm or vascular occlusion of the peripheral circulation can mimic typical Raynaud phenomenon, including peripheral nerve injury,2 complex regional pain syndrome,3 occlusive vascular disease, vasculitis, acrocyanosis,4 and thoracic outlet syndrome.
The prevalence of Raynaud phenomenon is not exactly known, in part due to geographic differences in climate and variation in methods of assessment. However, a 2015 systematic review and meta-analysis of primary Raynaud phenomenon determined a pooled prevalence of 4.85% (95% confidence interval [CI] 2.08%–8.71%) in the general population.5 Accordingly, accurate identification and management of this condition is a useful skill for the internist.
COLD SENSITIVITY AND COLOR CHANGES
Because there are no confirmatory diagnostic tests for this condition, there are no formal diagnostic criteria. However, many experts agree that Raynaud phenomenon can be diagnosed clinically when patients report:
Unusual sensitivity of the fingers to cold, manifesting as pain or paresthesia (eg, tingling, pricking, numbness), and
Color changes of the fingers when exposed to cold, specifically pale white or blue-black, or both.6
Provocative testing such as submerging patients’ hands in cold water is not recommended, as it is distressing to the patient and inconsistent in triggering an event.
Pain is a symptom of critical digital ischemia.
The skin color changes are due to rapid alterations in blood flow in digital skin. The pale white is due to markedly reduced or absent flow secondary to intense vasoconstriction, the blue-black is due to hypoxemic venous stasis, and the red blush is due to hyperemic reperfusion (Figure 1). However, not all patients have all 3 phases of the classic triphasic color changes, and color changes may not follow a set sequence.
Raynaud phenomenon can also occur in other areas of the body that have thermoregulatory vessels, such as the toes, ears, nipples, tongue, and nose. While some patients with Raynaud phenomenon have a finger that is more sensitive than the others, repeated isolated single-digit or asymmetric events without typical progression to all fingers suggest a secondary local structural disease requiring further investigation (see below).
Symptoms related to Raynaud often mimic sensory changes including paresthesias, numbness, aching, and clumsiness of the hand. Abnormal vascular reactivity has been implicated as a causative factor in several disorders, such as migraine headache, preeclampsia, and variant angina. While case reports, case series, and some controlled studies have linked Raynaud phenomenon and these conditions, there is no solid evidence of a systemic vasospastic disorder in patients with primary Raynaud phenomenon.
Raynaud phenomenon is triggered by more than just a cold ambient temperature. Provocation can occur during movement from warmer to relatively cooler temperatures, as well as during episodes of elevated sympathetic activity (eg, emotional distress or fear). In fact, maintaining full body warmth as well as emotional equilibrium are the most important strategies to reduce the frequency of attacks.
PRIMARY VS SECONDARY RAYNAUD PHENOMENON
To distinguish between primary and secondary Raynaud phenomenon, a careful history and physical examination are paramount.
Primary Raynaud phenomenon
In uncomplicated primary Raynaud phenomenon, the episodes typically last 15 to 20 minutes after rewarming and usually start in a single finger and spread to other digits symmetrically and bilaterally.7 The thumb is often spared, and ischemic digital ulcers do not occur. Vasoconstrictive episodes are mild.
Females under age 20 are most commonly affected. In our experience, a young woman with the above clinical picture, no signs or symptoms suggestive of connective tissue disease (see below), and normal nailfold capillaries can be diagnosed as having primary Raynaud phenomenon without any further workup.
Careful clinical follow-up is recommended, because if an occult secondary process is indeed present, most patients will begin to show additional symptoms or signs of it within 2 years of the onset of Raynaud phenomenon.
Should a clinician be unfamiliar with nailfold capillary examination, or if symptoms (eg, fatigue or arthralgia) or signs (eg, rash, arthritis) suggestive of connective tissue disease are present, referral to a rheumatologist for further evaluation is appropriate. Results of further diagnostic testing dictated by the history and physical such as a screening antinuclear antibody test can be sent before referral.
Secondary Raynaud phenomenon
Several clinical features suggest secondary Raynaud phenomenon and warrant referral to a rheumatologist:
Age 20 or older at onset
Frequent severe vasoconstrictive episodes
Male sex
Thumb involvement
Figure 2. (A) Dilated nailfold capillaries in a patient with scleroderma (blue arrow); (B) dilation and dropout of nailfold capillaries (white arrow) viewed with a magnifier.Signs of an autoimmune rheumatic disease, eg, sclerodactyly, cutaneous or mucosal matted telangiectasia, inflammatory arthritis, an abnormal lung examination, severe digital ischemia with ulceration or gangrene, or nailfold capillary dilation or dropout (Figure 2)8
Isolated single-limb or 1-finger ischemic events, seen in macrovascular occlusive disease or inflammatory disease mimicking Raynaud phenomenon (eg, atherosclerosis, vasculitis); when isolated acute ischemic events occur in the upper or lower extremity, a further workup is necessary.
Figure 3 shows our approach to evaluation.
NONPHARMACOLOGIC THERAPY
Figure 3. Our approach to diagnosis of Raynaud phenomenon and differentiating primary from secondary Raynaud phenomenon.
Cold avoidance and stress management are first-line therapies for preventing Raynaud attacks and must be part of any treatment strategy. Digital arteries and thermoregulatory vessels of the skin are predominantly under sympathetic adrenergic control, so temperature changes and emotional stressors trigger vasoconstriction. Patients should be counseled to:
Keep the whole body warm. Patients should wear multiple layers of clothing, a hat, warm gloves, and warm socks. Commercially available hand-warmers can help, especially for patients who live in cold climates.
Learn to avoid or manage stress. Good communication, attention to the patient’s needs, and regular follow-up for reassurance are paramount. For some patients, psychotropic medications to manage mood may help. Behavioral approaches have been suggested for acute stress management. One approach, autogenic training, is a form of relaxation with temperature biofeedback in which finger temperature data are provided to patients to help them learn to relax by monitoring their internal states and changes in temperature. However, there are no strong data to support the routine use of this technique or the use of one behavioral approach over another. Trials have generally been of low quality and limited by small sample size.9
Stop smoking!10
Stop a Raynaud attack should one occur, eg, place the hands under warm water or in a warm part of the body, such as under legs when sitting. This can help speed recovery.
In addition, the physician should:
Eliminate vasoconstricting agents such as nonselective beta-blockers, ergots, triptans, and amphetamines.
PHARMACOLOGIC THERAPY
For many patients, nonpharmacologic interventions are enough to decrease the severity and frequency of attacks. However, if Raynaud phenomenon continues to negatively affect quality of life, drug therapy can be added (Table 1).
Calcium channel blockers
Calcium channel blockers are first-line agents for both primary and secondary Raynaud phenomenon that does not adequately respond to nonpharmacologic interventions. These agents are effective, available, and reasonably inexpensive.
Dihydropyridine calcium channel blockers such as nifedipine and amlodipine are commonly used. Both drugs are acceptable options, though some patients may respond better to one than the other in terms of symptoms and side effects. Nondihydropyridines such as diltiazem can also be used, but they have less potent vasodilatory effects because they are less selective for vascular smooth muscle.
These medications should be started at the lowest dose and titrated up over several weeks as tolerated to achieve their maximal effect. Intermittent therapy (eg, during the winter months only) is reasonable for primary Raynaud without risk of digital ulceration, as relief of symptoms and improvement in quality of life are the main indications for therapy in this circumstance.
A 2016 Cochrane review and meta-analysis of the use of calcium channel blockers to treat primary Raynaud phenomenon included 7 randomized controlled trials with 296 patients treated with either nifedipine or nicardipine.11 There was moderate-quality evidence that these drugs minimally decreased the frequency of attacks (standardized mean difference of 0.23; 95% CI 0.08–0.38, P = .003). This translated to 1.72 fewer attacks per week with treatment than with no pharmacologic therapy (95% CI 0.60–2.84). When analyzed individually, only nifedipine was effective; nicardipine did not decrease the frequency of attacks.
Unfortunately, calcium channel blockers failed to decrease the severity of attacks (according to unvalidated severity scoring systems) or make any differences in physiologic measurement outcomes. Attacks were not completely eliminated, just less frequent than before treatment.11
Most commonly reported side effects included headache, flushing, hypotension, edema, and, rarely, gastrointestinal reflux. Use of these medications may be limited by hypotension.
The review was limited by the small sample size, short duration of treatment, and relatively low doses of calcium channel blockers used in the available studies.11
A 2005 meta-analysis also indicated a statistically significant decrease of 2.8 to 5 attacks per week with nifedipine treatment, though this study also included some patients with secondary Raynaud phenomenon.12
Phosphodiesterase type 5 inhibitors
When calcium channel blockers do not adequately control symptoms, phosphodiesterase type 5 (PDE5) inhibitors can be added or substituted. These medications work by preventing breakdown of cyclic guanosine monophosphate, which induces relaxation in vascular smooth muscle and vasodilation.
Sildenafil can be started at a low dose (20 mg daily) and up-titrated to the maximum dose (20 mg 3 times daily) as tolerated.
A 2014 meta-analysis of 6 randomized controlled trials included 244 patients with secondary Raynaud phenomenon treated with sildenafil, tadalafil, or vardenafil.13 These drugs decreased the daily frequency of attacks by about 0.5 per day vs placebo (–0.49, 95% CI –0.71 to –0.28, P < .0001). PDE5 inhibitors also decreased the severity of attacks (based on the Raynaud’s Condition Score, a popular scoring system) and the duration of attacks by a statistically significant amount.
Almost all patients in these 6 trials were on PDE5 monotherapy. Data on the cumulative benefit of calcium channel blocker and PDE5 inhibitor combination therapy are not yet available. Not all patients tolerate combination therapy, as it can cause symptomatic hypotension, but it can be a successful option in some.
There are also no data showing that either calcium channel blockers or PDE5 inhibitors are superior, though the former are less expensive. A small double-blind, randomized, crossover study of udenafil vs amlodipine in the treatment of secondary Raynaud phenomenon showed that both medications significantly decreased the frequency of attacks and had comparable efficacy.14
Cost and insurance coverage. We have generally been successful in obtaining coverage for this off-label use of PDE5 inhibitors, though additional effort may be required. No drug (not even a calcium channel blocker) is approved by the US Food and Drug Administration for use in Raynaud phenomenon. In our experience, a letter of appeal outlining the rationale for use and citing supporting publications can lead to successful coverage of a medication. If the drug is still not approved, the patient either pays for it out of pocket or another agent is selected. In certain circumstances, pharmaceutical companies may provide prescription assistance for compassionate use of these drugs in Raynaud phenomenon, although this also takes letter-writing, phone calls, or both on the part of the physician.
Topical nitrates
Patients who have an unsatisfactory response to calcium channel blockers with or without PDE5 inhibitors can try topical nitrates, available as sustained-release transdermal patches, tapes, creams, gels, and ointments.
Small trials have noted slight improvement in the Raynaud Condition Score15 and finger temperature16 with these therapies. Another trial noted decreased frequency of attacks and symptoms with the use of sustained-release glyceryl trinitrate patches, but use was limited by intolerable headache.17
In our experience, topical nitrates are most helpful for patients who have 1 or a few digits that are more severely affected than the others, and we reserve these drugs for this indication. Localized vasodilation can provide targeted rapid relief of more ischemic areas.
Topical nitroglycerin can be applied to the base of the ischemic digit for 6 to 12 hours. Preparations vary, and patients should be closely monitored for dose response and tolerance.
Combining a topical nitrate with a calcium channel blocker is safe, but the use of a nitrate with a PDE5 inhibitor is contraindicated due to the risk of hypotension. The use of topical nitrates may be limited by systemic side effects such as headache and flushing and a lack of benefit over time.
Other therapies
If the aforementioned agents are not tolerated or not effective, there is limited evidence that other therapies reduce the frequency and sometimes the severity of attacks. These are not first-line agents but may be tried when other options have been exhausted and symptoms persist. There are no data to support combining these therapies, but in our experience doing so may help some patients in whom drug-drug interactions are not prohibitive.
Prazosin, an alpha-1-adrenergic receptor antagonist, was reported to improve Raynaud phenomenon in 2 small studies in the 1980s, but we do not use it since better options are available. In addition, the vasoactive blood vessels involved do not have alpha-1 receptors, so there is no theoretical basis for using prazosin.18,19
Fluoxetine, a selective serotonin reuptake inhibitor, reduced the frequency and severity of attacks in a 6-week crossover study with nifedipine.20
Losartan, an angiotensin II receptor blocker, also reduced the severity and frequency of attacks when compared with nifedipine.21
Pentoxifylline, a nonselective phosphodiesterase inhibitor, showed some benefit in a trial in 11 patients with primary Raynaud.22
Atorvastatin, a lipid-lowering drug, reduced the number of digital ulcers in patients with secondary Raynaud already on first-line vasodilatory therapy, and might be added in this situation.23
Botulinum toxin A injections have some data to support their use, but evidence is based on uncontrolled case series.24 A controlled trial in scleroderma patients with severe Raynaud phenomenon found botulinum toxin to be no better than placebo.25
Prostacyclin preparations are available. Intermittent intravenous doses of prostacyclin analogues over several days can be used in resistant cases. Oral prostacyclin agents have not shown consistent benefit. New prostacyclin receptor agonists are under investigation.
Overall, we move to other options only in patients with persistent symptoms that impair quality of life, or in patients with recurrent digital ischemic lesions that have not responded to calcium channel blockers and PDE5 inhibitors or nitrates, either alone or in combination.
DIGITAL ULCERATION AND ACUTE DIGITAL ISCHEMIC CRISIS
Patients with secondary Raynaud phenomenon may be at risk of recurrent digital ulceration and acute digital ischemia with gangrene. These patients should be comanaged with a rheumatologist so that the underlying disease process is fully addressed. Digital ulcers should be inspected closely for signs of infection, which may require treatment with antibiotics.
Acute digital ischemia is a medical emergency and should prompt inpatient admission with warming, emotional regulation, and pain control (often with narcotics) to decrease sympathetic vasoconstriction. These patients require aggressive vasodilatory therapy to reverse the ischemic event.
A short-acting calcium channel blocker or combination therapy with a calcium channel blocker and a PDE5 inhibitor or topical nitrate should be started. If there is no benefit, then transient intravenous vasodilatory therapy with a prostacyclin (epoprostenol) or localized digital sympathectomy is used to prevent digital loss.
The endothelin receptor inhibitor bosentan has been shown to decrease recurrent digital ulcers in patients with scleroderma, and while bosentan does not decrease the frequency of Raynaud attacks, it can be used in this select group to prevent new digital ulcers.
Treatment options may be limited by insurance coverage or access to intravenous infusions.
TAKE-HOME RECOMMENDATIONS
For many patients with primary or secondary Raynaud phenomenon, nonpharmacologic interventions are all that are required to decrease the frequency of attacks and improve quality of life. The goal should not be to eliminate attacks completely, as aggressive drug treatment may cause more harm than benefit. From our perspective, the goals of treatment should be to improve quality of life and prevent ischemic complications.
Pharmacologic therapies should be added only if attacks remain poorly controlled with incapacitating symptoms, or if the patient has digital ischemic ulcers. Calcium channel blockers are first-line therapy, given proven efficacy and low cost, and should be titrated to the maximum tolerated dose before adding or substituting other agents.
References
Wigley FM, Flavahan NA. Raynaud’s phenomenon. N Engl J Med 2016; 375:556–565.
Irwin MS, Gilbert SE, Terenghi G, Smith RW, Green CJ. Cold intolerance following peripheral nerve injury. Natural history and factors predicting severity of symptoms. J Hand Surg Br 1997; 22:308–316.
Wasner G. Vasomotor disturbances in complex regional pain syndrome—a review. Pain Med 2010; 11:1267–1273.
Kurklinsky AK, Miller VM, Rooke TW. Acrocyanosis: the Flying Dutchman. Vasc Med 2011; 16:288–301.
Garner R, Kumari R, Lanyon P, Doherty M, Zhang W. Prevalence, risk factors and associations of primary Raynaud’s phenomenon: systematic review and meta-analysis of observational studies. BMJ Open 2015; 5:e006389.
Wigley FM. Clinical practice. Raynaud’s phenomenon. N Engl J Med 2002; 347:1001–1008.
Chikura B, Moore TL, Manning JB, Vail A, Herrick AL. Sparing of the thumb in Raynaud’s phenomenon. Rheumatology (Oxford) 2008; 47:219–221.
Kallenerg CG. Early detection of connective tissue disease in patients with Raynaud’s phenomenon. Rheum Dis Clin North Am 1990; 16:11–30.
Kwakkenbos L, Thombs BD. Non-drug approaches to treating Raynaud’s phenomenon. In: Wigley FM, Herrick AL, Flavahan NA, editors. Raynaud’s Phenomenon. A Guide to Pathogenesis and Treatment. New York: Springer Science+Business Media, 2015:299–313.
Goodfield MJ, Hume A, Rowell NR. The acute effects of cigarette smoking on cutaneous blood flow in smoking and non-smoking subjects with and without Raynaud’s phenomenon. Br J Rheumatol 1990; 29:89–91.
Ennis H, Hughes M, Anderson ME, Wilkinson J, Herrick AL. Calcium channel blockers for primary Raynaud’s phenomenon. Cochrane Database Sys Review 2016; 2:CD002069.
Thompson AE, Pope JE. Calcium channel blockers for primary Raynaud’s phenomenon: a meta-analysis. Rheumatology (Oxford) 2005; 44:145–150.
Roustit M, Blaise S, Allanore Y, Carpentier P, Caglayan E, Cracowski J. Phosphodiesterase-5 inhibitors for the treatment of secondary Raynaud’s phenomenon: systematic review and meta-analysis of randomized trials. Ann Rheum Dis 2013; 72:1696–1699.
Lee EY, Park JK, Lee W, et al. Head-to-head comparison of udenafil vs amlodipine in the treatment of secondary Raynaud's phenomenon: a double-blind, randomized, cross-over study. Rheumatology (Oxford) 2014; 53:658–664.
Chung L, Shapiro L, Fiorentino D, et al. MQX-503, a novel formulation of nitroglycerin, improves the severity of Raynaud’s phenomenon: a randomized, controlled trial. Arthritis Rheum 2009; 60:870–877.
Kan C, Akimoto S, Abe M, Okada K, Ishikawa O. Preliminary thermographic evaluation of a new nitroglycerine tape on the peripheral circulatory disturbance in systemic sclerosis. Ann Rheum Dis 2002; 61:177–179.
Teh LS, Manning J, Moore T, Tully MP, O’Reilly D, Jayson MI. Sustained-release transdermal glyceryl trinitrate patches as a treatment for primary and secondary Raynaud’s phenomenon. Br J Rheumatol 1995; 34:636–641.
Russell IJ, Lessard JA. Prazosin treatment of Raynaud’s phenomenon: a double blind single crossover study. J Rheumatol 1985; 12:94–98.
Wollersheim H, Thien T, Fennis J, van Elteren P, van ‘t Laar A. Double-blind, placebo-controlled study of prazosin in Raynaud’s phenomenon. Clin Pharmacol Ther 1986; 40:219–225.
Coleiro B, Marshall SE, Denton CP, et al. Treatment of Raynaud’s phenomenon with the selective serotonin reuptake inhibitor fluoxetine. Rheumatology (Oxford) 2001; 40:1038–1043.
Didazio M, Denton CP, Smith R, et al. Losartan therapy for Raynaud’s phenomenon and scleroderma: clinical and biochemical findings in a fifteen-week randomized, parallel-group, controlled trial. Arthritis Rheum 1999; 42:2646–2655.
Neirotti M, Longo F, Molaschi M, Macchione C, Pernigotti L. Functional vascular disorders: treatment with pentoxifylline. Angiology 1987; 38:575–580.
Abou-Raya A, Abou-Raya S, Helmii M. Statins: potentially useful in therapy of systemic sclerosis-related Raynaud’s phenomenon and digital ulcers. J Rheumatol 2008; 35:1801–1808.
Iorio ML, Masden DL, Higgins JP. Botulinum toxin A treatment of Raynaud’s phenomenon: a review. Semin Arthritis Rheum 2012; 41: 599–603.
Bello RJ, Cooney CM, Melamed E, et al. The therapeutic efficacy of botulinum toxin in treating scleroderma-associated Raynaud’s phenomenon: a randomized, double-blind, placebo-controlled clinical trial. Arthritis Rheumatol 2017. Epub ahead of print.
Samantha C. Shapiro, MD Department of Medicine, Division of Rheumatology, Johns Hopkins University School of Medicine; Postdoctoral Fellow, Johns Hopkins Division of Rheumatology, Baltimore, MD
Fredrick M. Wigley, MD Department of Medicine, Division of Rheumatology, Johns Hopkins University School of Medicine; Martha McCrory Professor of Medicine, Johns Hopkins Division of Rheumatology, Baltimore, MD
Address: Samantha C. Shapiro, MD, Department of Medicine, Division of Rheumatology, Johns Hopkins University School of Medicine; 5200 Eastern Avenue, Suite 4100, Mason F. Lord Building, Center Tower, Baltimore, MD 21224; sshapi28@jhmi.edu
Samantha C. Shapiro, MD Department of Medicine, Division of Rheumatology, Johns Hopkins University School of Medicine; Postdoctoral Fellow, Johns Hopkins Division of Rheumatology, Baltimore, MD
Fredrick M. Wigley, MD Department of Medicine, Division of Rheumatology, Johns Hopkins University School of Medicine; Martha McCrory Professor of Medicine, Johns Hopkins Division of Rheumatology, Baltimore, MD
Address: Samantha C. Shapiro, MD, Department of Medicine, Division of Rheumatology, Johns Hopkins University School of Medicine; 5200 Eastern Avenue, Suite 4100, Mason F. Lord Building, Center Tower, Baltimore, MD 21224; sshapi28@jhmi.edu
Author and Disclosure Information
Samantha C. Shapiro, MD Department of Medicine, Division of Rheumatology, Johns Hopkins University School of Medicine; Postdoctoral Fellow, Johns Hopkins Division of Rheumatology, Baltimore, MD
Fredrick M. Wigley, MD Department of Medicine, Division of Rheumatology, Johns Hopkins University School of Medicine; Martha McCrory Professor of Medicine, Johns Hopkins Division of Rheumatology, Baltimore, MD
Address: Samantha C. Shapiro, MD, Department of Medicine, Division of Rheumatology, Johns Hopkins University School of Medicine; 5200 Eastern Avenue, Suite 4100, Mason F. Lord Building, Center Tower, Baltimore, MD 21224; sshapi28@jhmi.edu
Raynaud phenomenon is an overactive vascular response to cold and emotional stress that results in cutaneous color changes and sensory symptoms of the digits (Figure 1). It can occur in isolation as primary Raynaud phenomenon or secondary to another disease process. It is thought to be triggered by a heightened sympathetic vasoconstrictive response of small arteriovenous anastomoses in the fingers, toes, ears, and tip of the nose. These structures play a key role in maintaining a stable core body temperature by cutaneous thermoregulation.1
Figure 1. (A) White digits with intense vasoconstriction in Raynaud phenomenon; (B) blue digits with
hypoxemic venous stasis; (C) red digits with hyperemic reperfusion.
Secondary Raynaud phenomenon can be seen with a wide array of systemic conditions as well as environmental and drug exposures. It is a frequent feature of autoimmune rheumatic conditions such as systemic sclerosis, mixed connective tissue disease, systemic lupus erythematosus, and dermatomyositis. Less commonly, cryoproteinemias, paraneoplastic syndromes, hypothyroidism, and carpal tunnel syndrome can be associated with or cause Raynaud phenomenon. Vibratory trauma (eg, from using a jackhammer) and drugs (eg, vasopressors, stimulants, ergots, chemotherapeutic agents) can also cause Raynaud phenomenon.1
A variety of disorders that cause vasospasm or vascular occlusion of the peripheral circulation can mimic typical Raynaud phenomenon, including peripheral nerve injury,2 complex regional pain syndrome,3 occlusive vascular disease, vasculitis, acrocyanosis,4 and thoracic outlet syndrome.
The prevalence of Raynaud phenomenon is not exactly known, in part due to geographic differences in climate and variation in methods of assessment. However, a 2015 systematic review and meta-analysis of primary Raynaud phenomenon determined a pooled prevalence of 4.85% (95% confidence interval [CI] 2.08%–8.71%) in the general population.5 Accordingly, accurate identification and management of this condition is a useful skill for the internist.
COLD SENSITIVITY AND COLOR CHANGES
Because there are no confirmatory diagnostic tests for this condition, there are no formal diagnostic criteria. However, many experts agree that Raynaud phenomenon can be diagnosed clinically when patients report:
Unusual sensitivity of the fingers to cold, manifesting as pain or paresthesia (eg, tingling, pricking, numbness), and
Color changes of the fingers when exposed to cold, specifically pale white or blue-black, or both.6
Provocative testing such as submerging patients’ hands in cold water is not recommended, as it is distressing to the patient and inconsistent in triggering an event.
Pain is a symptom of critical digital ischemia.
The skin color changes are due to rapid alterations in blood flow in digital skin. The pale white is due to markedly reduced or absent flow secondary to intense vasoconstriction, the blue-black is due to hypoxemic venous stasis, and the red blush is due to hyperemic reperfusion (Figure 1). However, not all patients have all 3 phases of the classic triphasic color changes, and color changes may not follow a set sequence.
Raynaud phenomenon can also occur in other areas of the body that have thermoregulatory vessels, such as the toes, ears, nipples, tongue, and nose. While some patients with Raynaud phenomenon have a finger that is more sensitive than the others, repeated isolated single-digit or asymmetric events without typical progression to all fingers suggest a secondary local structural disease requiring further investigation (see below).
Symptoms related to Raynaud often mimic sensory changes including paresthesias, numbness, aching, and clumsiness of the hand. Abnormal vascular reactivity has been implicated as a causative factor in several disorders, such as migraine headache, preeclampsia, and variant angina. While case reports, case series, and some controlled studies have linked Raynaud phenomenon and these conditions, there is no solid evidence of a systemic vasospastic disorder in patients with primary Raynaud phenomenon.
Raynaud phenomenon is triggered by more than just a cold ambient temperature. Provocation can occur during movement from warmer to relatively cooler temperatures, as well as during episodes of elevated sympathetic activity (eg, emotional distress or fear). In fact, maintaining full body warmth as well as emotional equilibrium are the most important strategies to reduce the frequency of attacks.
PRIMARY VS SECONDARY RAYNAUD PHENOMENON
To distinguish between primary and secondary Raynaud phenomenon, a careful history and physical examination are paramount.
Primary Raynaud phenomenon
In uncomplicated primary Raynaud phenomenon, the episodes typically last 15 to 20 minutes after rewarming and usually start in a single finger and spread to other digits symmetrically and bilaterally.7 The thumb is often spared, and ischemic digital ulcers do not occur. Vasoconstrictive episodes are mild.
Females under age 20 are most commonly affected. In our experience, a young woman with the above clinical picture, no signs or symptoms suggestive of connective tissue disease (see below), and normal nailfold capillaries can be diagnosed as having primary Raynaud phenomenon without any further workup.
Careful clinical follow-up is recommended, because if an occult secondary process is indeed present, most patients will begin to show additional symptoms or signs of it within 2 years of the onset of Raynaud phenomenon.
Should a clinician be unfamiliar with nailfold capillary examination, or if symptoms (eg, fatigue or arthralgia) or signs (eg, rash, arthritis) suggestive of connective tissue disease are present, referral to a rheumatologist for further evaluation is appropriate. Results of further diagnostic testing dictated by the history and physical such as a screening antinuclear antibody test can be sent before referral.
Secondary Raynaud phenomenon
Several clinical features suggest secondary Raynaud phenomenon and warrant referral to a rheumatologist:
Age 20 or older at onset
Frequent severe vasoconstrictive episodes
Male sex
Thumb involvement
Figure 2. (A) Dilated nailfold capillaries in a patient with scleroderma (blue arrow); (B) dilation and dropout of nailfold capillaries (white arrow) viewed with a magnifier.Signs of an autoimmune rheumatic disease, eg, sclerodactyly, cutaneous or mucosal matted telangiectasia, inflammatory arthritis, an abnormal lung examination, severe digital ischemia with ulceration or gangrene, or nailfold capillary dilation or dropout (Figure 2)8
Isolated single-limb or 1-finger ischemic events, seen in macrovascular occlusive disease or inflammatory disease mimicking Raynaud phenomenon (eg, atherosclerosis, vasculitis); when isolated acute ischemic events occur in the upper or lower extremity, a further workup is necessary.
Figure 3 shows our approach to evaluation.
NONPHARMACOLOGIC THERAPY
Figure 3. Our approach to diagnosis of Raynaud phenomenon and differentiating primary from secondary Raynaud phenomenon.
Cold avoidance and stress management are first-line therapies for preventing Raynaud attacks and must be part of any treatment strategy. Digital arteries and thermoregulatory vessels of the skin are predominantly under sympathetic adrenergic control, so temperature changes and emotional stressors trigger vasoconstriction. Patients should be counseled to:
Keep the whole body warm. Patients should wear multiple layers of clothing, a hat, warm gloves, and warm socks. Commercially available hand-warmers can help, especially for patients who live in cold climates.
Learn to avoid or manage stress. Good communication, attention to the patient’s needs, and regular follow-up for reassurance are paramount. For some patients, psychotropic medications to manage mood may help. Behavioral approaches have been suggested for acute stress management. One approach, autogenic training, is a form of relaxation with temperature biofeedback in which finger temperature data are provided to patients to help them learn to relax by monitoring their internal states and changes in temperature. However, there are no strong data to support the routine use of this technique or the use of one behavioral approach over another. Trials have generally been of low quality and limited by small sample size.9
Stop smoking!10
Stop a Raynaud attack should one occur, eg, place the hands under warm water or in a warm part of the body, such as under legs when sitting. This can help speed recovery.
In addition, the physician should:
Eliminate vasoconstricting agents such as nonselective beta-blockers, ergots, triptans, and amphetamines.
PHARMACOLOGIC THERAPY
For many patients, nonpharmacologic interventions are enough to decrease the severity and frequency of attacks. However, if Raynaud phenomenon continues to negatively affect quality of life, drug therapy can be added (Table 1).
Calcium channel blockers
Calcium channel blockers are first-line agents for both primary and secondary Raynaud phenomenon that does not adequately respond to nonpharmacologic interventions. These agents are effective, available, and reasonably inexpensive.
Dihydropyridine calcium channel blockers such as nifedipine and amlodipine are commonly used. Both drugs are acceptable options, though some patients may respond better to one than the other in terms of symptoms and side effects. Nondihydropyridines such as diltiazem can also be used, but they have less potent vasodilatory effects because they are less selective for vascular smooth muscle.
These medications should be started at the lowest dose and titrated up over several weeks as tolerated to achieve their maximal effect. Intermittent therapy (eg, during the winter months only) is reasonable for primary Raynaud without risk of digital ulceration, as relief of symptoms and improvement in quality of life are the main indications for therapy in this circumstance.
A 2016 Cochrane review and meta-analysis of the use of calcium channel blockers to treat primary Raynaud phenomenon included 7 randomized controlled trials with 296 patients treated with either nifedipine or nicardipine.11 There was moderate-quality evidence that these drugs minimally decreased the frequency of attacks (standardized mean difference of 0.23; 95% CI 0.08–0.38, P = .003). This translated to 1.72 fewer attacks per week with treatment than with no pharmacologic therapy (95% CI 0.60–2.84). When analyzed individually, only nifedipine was effective; nicardipine did not decrease the frequency of attacks.
Unfortunately, calcium channel blockers failed to decrease the severity of attacks (according to unvalidated severity scoring systems) or make any differences in physiologic measurement outcomes. Attacks were not completely eliminated, just less frequent than before treatment.11
Most commonly reported side effects included headache, flushing, hypotension, edema, and, rarely, gastrointestinal reflux. Use of these medications may be limited by hypotension.
The review was limited by the small sample size, short duration of treatment, and relatively low doses of calcium channel blockers used in the available studies.11
A 2005 meta-analysis also indicated a statistically significant decrease of 2.8 to 5 attacks per week with nifedipine treatment, though this study also included some patients with secondary Raynaud phenomenon.12
Phosphodiesterase type 5 inhibitors
When calcium channel blockers do not adequately control symptoms, phosphodiesterase type 5 (PDE5) inhibitors can be added or substituted. These medications work by preventing breakdown of cyclic guanosine monophosphate, which induces relaxation in vascular smooth muscle and vasodilation.
Sildenafil can be started at a low dose (20 mg daily) and up-titrated to the maximum dose (20 mg 3 times daily) as tolerated.
A 2014 meta-analysis of 6 randomized controlled trials included 244 patients with secondary Raynaud phenomenon treated with sildenafil, tadalafil, or vardenafil.13 These drugs decreased the daily frequency of attacks by about 0.5 per day vs placebo (–0.49, 95% CI –0.71 to –0.28, P < .0001). PDE5 inhibitors also decreased the severity of attacks (based on the Raynaud’s Condition Score, a popular scoring system) and the duration of attacks by a statistically significant amount.
Almost all patients in these 6 trials were on PDE5 monotherapy. Data on the cumulative benefit of calcium channel blocker and PDE5 inhibitor combination therapy are not yet available. Not all patients tolerate combination therapy, as it can cause symptomatic hypotension, but it can be a successful option in some.
There are also no data showing that either calcium channel blockers or PDE5 inhibitors are superior, though the former are less expensive. A small double-blind, randomized, crossover study of udenafil vs amlodipine in the treatment of secondary Raynaud phenomenon showed that both medications significantly decreased the frequency of attacks and had comparable efficacy.14
Cost and insurance coverage. We have generally been successful in obtaining coverage for this off-label use of PDE5 inhibitors, though additional effort may be required. No drug (not even a calcium channel blocker) is approved by the US Food and Drug Administration for use in Raynaud phenomenon. In our experience, a letter of appeal outlining the rationale for use and citing supporting publications can lead to successful coverage of a medication. If the drug is still not approved, the patient either pays for it out of pocket or another agent is selected. In certain circumstances, pharmaceutical companies may provide prescription assistance for compassionate use of these drugs in Raynaud phenomenon, although this also takes letter-writing, phone calls, or both on the part of the physician.
Topical nitrates
Patients who have an unsatisfactory response to calcium channel blockers with or without PDE5 inhibitors can try topical nitrates, available as sustained-release transdermal patches, tapes, creams, gels, and ointments.
Small trials have noted slight improvement in the Raynaud Condition Score15 and finger temperature16 with these therapies. Another trial noted decreased frequency of attacks and symptoms with the use of sustained-release glyceryl trinitrate patches, but use was limited by intolerable headache.17
In our experience, topical nitrates are most helpful for patients who have 1 or a few digits that are more severely affected than the others, and we reserve these drugs for this indication. Localized vasodilation can provide targeted rapid relief of more ischemic areas.
Topical nitroglycerin can be applied to the base of the ischemic digit for 6 to 12 hours. Preparations vary, and patients should be closely monitored for dose response and tolerance.
Combining a topical nitrate with a calcium channel blocker is safe, but the use of a nitrate with a PDE5 inhibitor is contraindicated due to the risk of hypotension. The use of topical nitrates may be limited by systemic side effects such as headache and flushing and a lack of benefit over time.
Other therapies
If the aforementioned agents are not tolerated or not effective, there is limited evidence that other therapies reduce the frequency and sometimes the severity of attacks. These are not first-line agents but may be tried when other options have been exhausted and symptoms persist. There are no data to support combining these therapies, but in our experience doing so may help some patients in whom drug-drug interactions are not prohibitive.
Prazosin, an alpha-1-adrenergic receptor antagonist, was reported to improve Raynaud phenomenon in 2 small studies in the 1980s, but we do not use it since better options are available. In addition, the vasoactive blood vessels involved do not have alpha-1 receptors, so there is no theoretical basis for using prazosin.18,19
Fluoxetine, a selective serotonin reuptake inhibitor, reduced the frequency and severity of attacks in a 6-week crossover study with nifedipine.20
Losartan, an angiotensin II receptor blocker, also reduced the severity and frequency of attacks when compared with nifedipine.21
Pentoxifylline, a nonselective phosphodiesterase inhibitor, showed some benefit in a trial in 11 patients with primary Raynaud.22
Atorvastatin, a lipid-lowering drug, reduced the number of digital ulcers in patients with secondary Raynaud already on first-line vasodilatory therapy, and might be added in this situation.23
Botulinum toxin A injections have some data to support their use, but evidence is based on uncontrolled case series.24 A controlled trial in scleroderma patients with severe Raynaud phenomenon found botulinum toxin to be no better than placebo.25
Prostacyclin preparations are available. Intermittent intravenous doses of prostacyclin analogues over several days can be used in resistant cases. Oral prostacyclin agents have not shown consistent benefit. New prostacyclin receptor agonists are under investigation.
Overall, we move to other options only in patients with persistent symptoms that impair quality of life, or in patients with recurrent digital ischemic lesions that have not responded to calcium channel blockers and PDE5 inhibitors or nitrates, either alone or in combination.
DIGITAL ULCERATION AND ACUTE DIGITAL ISCHEMIC CRISIS
Patients with secondary Raynaud phenomenon may be at risk of recurrent digital ulceration and acute digital ischemia with gangrene. These patients should be comanaged with a rheumatologist so that the underlying disease process is fully addressed. Digital ulcers should be inspected closely for signs of infection, which may require treatment with antibiotics.
Acute digital ischemia is a medical emergency and should prompt inpatient admission with warming, emotional regulation, and pain control (often with narcotics) to decrease sympathetic vasoconstriction. These patients require aggressive vasodilatory therapy to reverse the ischemic event.
A short-acting calcium channel blocker or combination therapy with a calcium channel blocker and a PDE5 inhibitor or topical nitrate should be started. If there is no benefit, then transient intravenous vasodilatory therapy with a prostacyclin (epoprostenol) or localized digital sympathectomy is used to prevent digital loss.
The endothelin receptor inhibitor bosentan has been shown to decrease recurrent digital ulcers in patients with scleroderma, and while bosentan does not decrease the frequency of Raynaud attacks, it can be used in this select group to prevent new digital ulcers.
Treatment options may be limited by insurance coverage or access to intravenous infusions.
TAKE-HOME RECOMMENDATIONS
For many patients with primary or secondary Raynaud phenomenon, nonpharmacologic interventions are all that are required to decrease the frequency of attacks and improve quality of life. The goal should not be to eliminate attacks completely, as aggressive drug treatment may cause more harm than benefit. From our perspective, the goals of treatment should be to improve quality of life and prevent ischemic complications.
Pharmacologic therapies should be added only if attacks remain poorly controlled with incapacitating symptoms, or if the patient has digital ischemic ulcers. Calcium channel blockers are first-line therapy, given proven efficacy and low cost, and should be titrated to the maximum tolerated dose before adding or substituting other agents.
Raynaud phenomenon is an overactive vascular response to cold and emotional stress that results in cutaneous color changes and sensory symptoms of the digits (Figure 1). It can occur in isolation as primary Raynaud phenomenon or secondary to another disease process. It is thought to be triggered by a heightened sympathetic vasoconstrictive response of small arteriovenous anastomoses in the fingers, toes, ears, and tip of the nose. These structures play a key role in maintaining a stable core body temperature by cutaneous thermoregulation.1
Figure 1. (A) White digits with intense vasoconstriction in Raynaud phenomenon; (B) blue digits with
hypoxemic venous stasis; (C) red digits with hyperemic reperfusion.
Secondary Raynaud phenomenon can be seen with a wide array of systemic conditions as well as environmental and drug exposures. It is a frequent feature of autoimmune rheumatic conditions such as systemic sclerosis, mixed connective tissue disease, systemic lupus erythematosus, and dermatomyositis. Less commonly, cryoproteinemias, paraneoplastic syndromes, hypothyroidism, and carpal tunnel syndrome can be associated with or cause Raynaud phenomenon. Vibratory trauma (eg, from using a jackhammer) and drugs (eg, vasopressors, stimulants, ergots, chemotherapeutic agents) can also cause Raynaud phenomenon.1
A variety of disorders that cause vasospasm or vascular occlusion of the peripheral circulation can mimic typical Raynaud phenomenon, including peripheral nerve injury,2 complex regional pain syndrome,3 occlusive vascular disease, vasculitis, acrocyanosis,4 and thoracic outlet syndrome.
The prevalence of Raynaud phenomenon is not exactly known, in part due to geographic differences in climate and variation in methods of assessment. However, a 2015 systematic review and meta-analysis of primary Raynaud phenomenon determined a pooled prevalence of 4.85% (95% confidence interval [CI] 2.08%–8.71%) in the general population.5 Accordingly, accurate identification and management of this condition is a useful skill for the internist.
COLD SENSITIVITY AND COLOR CHANGES
Because there are no confirmatory diagnostic tests for this condition, there are no formal diagnostic criteria. However, many experts agree that Raynaud phenomenon can be diagnosed clinically when patients report:
Unusual sensitivity of the fingers to cold, manifesting as pain or paresthesia (eg, tingling, pricking, numbness), and
Color changes of the fingers when exposed to cold, specifically pale white or blue-black, or both.6
Provocative testing such as submerging patients’ hands in cold water is not recommended, as it is distressing to the patient and inconsistent in triggering an event.
Pain is a symptom of critical digital ischemia.
The skin color changes are due to rapid alterations in blood flow in digital skin. The pale white is due to markedly reduced or absent flow secondary to intense vasoconstriction, the blue-black is due to hypoxemic venous stasis, and the red blush is due to hyperemic reperfusion (Figure 1). However, not all patients have all 3 phases of the classic triphasic color changes, and color changes may not follow a set sequence.
Raynaud phenomenon can also occur in other areas of the body that have thermoregulatory vessels, such as the toes, ears, nipples, tongue, and nose. While some patients with Raynaud phenomenon have a finger that is more sensitive than the others, repeated isolated single-digit or asymmetric events without typical progression to all fingers suggest a secondary local structural disease requiring further investigation (see below).
Symptoms related to Raynaud often mimic sensory changes including paresthesias, numbness, aching, and clumsiness of the hand. Abnormal vascular reactivity has been implicated as a causative factor in several disorders, such as migraine headache, preeclampsia, and variant angina. While case reports, case series, and some controlled studies have linked Raynaud phenomenon and these conditions, there is no solid evidence of a systemic vasospastic disorder in patients with primary Raynaud phenomenon.
Raynaud phenomenon is triggered by more than just a cold ambient temperature. Provocation can occur during movement from warmer to relatively cooler temperatures, as well as during episodes of elevated sympathetic activity (eg, emotional distress or fear). In fact, maintaining full body warmth as well as emotional equilibrium are the most important strategies to reduce the frequency of attacks.
PRIMARY VS SECONDARY RAYNAUD PHENOMENON
To distinguish between primary and secondary Raynaud phenomenon, a careful history and physical examination are paramount.
Primary Raynaud phenomenon
In uncomplicated primary Raynaud phenomenon, the episodes typically last 15 to 20 minutes after rewarming and usually start in a single finger and spread to other digits symmetrically and bilaterally.7 The thumb is often spared, and ischemic digital ulcers do not occur. Vasoconstrictive episodes are mild.
Females under age 20 are most commonly affected. In our experience, a young woman with the above clinical picture, no signs or symptoms suggestive of connective tissue disease (see below), and normal nailfold capillaries can be diagnosed as having primary Raynaud phenomenon without any further workup.
Careful clinical follow-up is recommended, because if an occult secondary process is indeed present, most patients will begin to show additional symptoms or signs of it within 2 years of the onset of Raynaud phenomenon.
Should a clinician be unfamiliar with nailfold capillary examination, or if symptoms (eg, fatigue or arthralgia) or signs (eg, rash, arthritis) suggestive of connective tissue disease are present, referral to a rheumatologist for further evaluation is appropriate. Results of further diagnostic testing dictated by the history and physical such as a screening antinuclear antibody test can be sent before referral.
Secondary Raynaud phenomenon
Several clinical features suggest secondary Raynaud phenomenon and warrant referral to a rheumatologist:
Age 20 or older at onset
Frequent severe vasoconstrictive episodes
Male sex
Thumb involvement
Figure 2. (A) Dilated nailfold capillaries in a patient with scleroderma (blue arrow); (B) dilation and dropout of nailfold capillaries (white arrow) viewed with a magnifier.Signs of an autoimmune rheumatic disease, eg, sclerodactyly, cutaneous or mucosal matted telangiectasia, inflammatory arthritis, an abnormal lung examination, severe digital ischemia with ulceration or gangrene, or nailfold capillary dilation or dropout (Figure 2)8
Isolated single-limb or 1-finger ischemic events, seen in macrovascular occlusive disease or inflammatory disease mimicking Raynaud phenomenon (eg, atherosclerosis, vasculitis); when isolated acute ischemic events occur in the upper or lower extremity, a further workup is necessary.
Figure 3 shows our approach to evaluation.
NONPHARMACOLOGIC THERAPY
Figure 3. Our approach to diagnosis of Raynaud phenomenon and differentiating primary from secondary Raynaud phenomenon.
Cold avoidance and stress management are first-line therapies for preventing Raynaud attacks and must be part of any treatment strategy. Digital arteries and thermoregulatory vessels of the skin are predominantly under sympathetic adrenergic control, so temperature changes and emotional stressors trigger vasoconstriction. Patients should be counseled to:
Keep the whole body warm. Patients should wear multiple layers of clothing, a hat, warm gloves, and warm socks. Commercially available hand-warmers can help, especially for patients who live in cold climates.
Learn to avoid or manage stress. Good communication, attention to the patient’s needs, and regular follow-up for reassurance are paramount. For some patients, psychotropic medications to manage mood may help. Behavioral approaches have been suggested for acute stress management. One approach, autogenic training, is a form of relaxation with temperature biofeedback in which finger temperature data are provided to patients to help them learn to relax by monitoring their internal states and changes in temperature. However, there are no strong data to support the routine use of this technique or the use of one behavioral approach over another. Trials have generally been of low quality and limited by small sample size.9
Stop smoking!10
Stop a Raynaud attack should one occur, eg, place the hands under warm water or in a warm part of the body, such as under legs when sitting. This can help speed recovery.
In addition, the physician should:
Eliminate vasoconstricting agents such as nonselective beta-blockers, ergots, triptans, and amphetamines.
PHARMACOLOGIC THERAPY
For many patients, nonpharmacologic interventions are enough to decrease the severity and frequency of attacks. However, if Raynaud phenomenon continues to negatively affect quality of life, drug therapy can be added (Table 1).
Calcium channel blockers
Calcium channel blockers are first-line agents for both primary and secondary Raynaud phenomenon that does not adequately respond to nonpharmacologic interventions. These agents are effective, available, and reasonably inexpensive.
Dihydropyridine calcium channel blockers such as nifedipine and amlodipine are commonly used. Both drugs are acceptable options, though some patients may respond better to one than the other in terms of symptoms and side effects. Nondihydropyridines such as diltiazem can also be used, but they have less potent vasodilatory effects because they are less selective for vascular smooth muscle.
These medications should be started at the lowest dose and titrated up over several weeks as tolerated to achieve their maximal effect. Intermittent therapy (eg, during the winter months only) is reasonable for primary Raynaud without risk of digital ulceration, as relief of symptoms and improvement in quality of life are the main indications for therapy in this circumstance.
A 2016 Cochrane review and meta-analysis of the use of calcium channel blockers to treat primary Raynaud phenomenon included 7 randomized controlled trials with 296 patients treated with either nifedipine or nicardipine.11 There was moderate-quality evidence that these drugs minimally decreased the frequency of attacks (standardized mean difference of 0.23; 95% CI 0.08–0.38, P = .003). This translated to 1.72 fewer attacks per week with treatment than with no pharmacologic therapy (95% CI 0.60–2.84). When analyzed individually, only nifedipine was effective; nicardipine did not decrease the frequency of attacks.
Unfortunately, calcium channel blockers failed to decrease the severity of attacks (according to unvalidated severity scoring systems) or make any differences in physiologic measurement outcomes. Attacks were not completely eliminated, just less frequent than before treatment.11
Most commonly reported side effects included headache, flushing, hypotension, edema, and, rarely, gastrointestinal reflux. Use of these medications may be limited by hypotension.
The review was limited by the small sample size, short duration of treatment, and relatively low doses of calcium channel blockers used in the available studies.11
A 2005 meta-analysis also indicated a statistically significant decrease of 2.8 to 5 attacks per week with nifedipine treatment, though this study also included some patients with secondary Raynaud phenomenon.12
Phosphodiesterase type 5 inhibitors
When calcium channel blockers do not adequately control symptoms, phosphodiesterase type 5 (PDE5) inhibitors can be added or substituted. These medications work by preventing breakdown of cyclic guanosine monophosphate, which induces relaxation in vascular smooth muscle and vasodilation.
Sildenafil can be started at a low dose (20 mg daily) and up-titrated to the maximum dose (20 mg 3 times daily) as tolerated.
A 2014 meta-analysis of 6 randomized controlled trials included 244 patients with secondary Raynaud phenomenon treated with sildenafil, tadalafil, or vardenafil.13 These drugs decreased the daily frequency of attacks by about 0.5 per day vs placebo (–0.49, 95% CI –0.71 to –0.28, P < .0001). PDE5 inhibitors also decreased the severity of attacks (based on the Raynaud’s Condition Score, a popular scoring system) and the duration of attacks by a statistically significant amount.
Almost all patients in these 6 trials were on PDE5 monotherapy. Data on the cumulative benefit of calcium channel blocker and PDE5 inhibitor combination therapy are not yet available. Not all patients tolerate combination therapy, as it can cause symptomatic hypotension, but it can be a successful option in some.
There are also no data showing that either calcium channel blockers or PDE5 inhibitors are superior, though the former are less expensive. A small double-blind, randomized, crossover study of udenafil vs amlodipine in the treatment of secondary Raynaud phenomenon showed that both medications significantly decreased the frequency of attacks and had comparable efficacy.14
Cost and insurance coverage. We have generally been successful in obtaining coverage for this off-label use of PDE5 inhibitors, though additional effort may be required. No drug (not even a calcium channel blocker) is approved by the US Food and Drug Administration for use in Raynaud phenomenon. In our experience, a letter of appeal outlining the rationale for use and citing supporting publications can lead to successful coverage of a medication. If the drug is still not approved, the patient either pays for it out of pocket or another agent is selected. In certain circumstances, pharmaceutical companies may provide prescription assistance for compassionate use of these drugs in Raynaud phenomenon, although this also takes letter-writing, phone calls, or both on the part of the physician.
Topical nitrates
Patients who have an unsatisfactory response to calcium channel blockers with or without PDE5 inhibitors can try topical nitrates, available as sustained-release transdermal patches, tapes, creams, gels, and ointments.
Small trials have noted slight improvement in the Raynaud Condition Score15 and finger temperature16 with these therapies. Another trial noted decreased frequency of attacks and symptoms with the use of sustained-release glyceryl trinitrate patches, but use was limited by intolerable headache.17
In our experience, topical nitrates are most helpful for patients who have 1 or a few digits that are more severely affected than the others, and we reserve these drugs for this indication. Localized vasodilation can provide targeted rapid relief of more ischemic areas.
Topical nitroglycerin can be applied to the base of the ischemic digit for 6 to 12 hours. Preparations vary, and patients should be closely monitored for dose response and tolerance.
Combining a topical nitrate with a calcium channel blocker is safe, but the use of a nitrate with a PDE5 inhibitor is contraindicated due to the risk of hypotension. The use of topical nitrates may be limited by systemic side effects such as headache and flushing and a lack of benefit over time.
Other therapies
If the aforementioned agents are not tolerated or not effective, there is limited evidence that other therapies reduce the frequency and sometimes the severity of attacks. These are not first-line agents but may be tried when other options have been exhausted and symptoms persist. There are no data to support combining these therapies, but in our experience doing so may help some patients in whom drug-drug interactions are not prohibitive.
Prazosin, an alpha-1-adrenergic receptor antagonist, was reported to improve Raynaud phenomenon in 2 small studies in the 1980s, but we do not use it since better options are available. In addition, the vasoactive blood vessels involved do not have alpha-1 receptors, so there is no theoretical basis for using prazosin.18,19
Fluoxetine, a selective serotonin reuptake inhibitor, reduced the frequency and severity of attacks in a 6-week crossover study with nifedipine.20
Losartan, an angiotensin II receptor blocker, also reduced the severity and frequency of attacks when compared with nifedipine.21
Pentoxifylline, a nonselective phosphodiesterase inhibitor, showed some benefit in a trial in 11 patients with primary Raynaud.22
Atorvastatin, a lipid-lowering drug, reduced the number of digital ulcers in patients with secondary Raynaud already on first-line vasodilatory therapy, and might be added in this situation.23
Botulinum toxin A injections have some data to support their use, but evidence is based on uncontrolled case series.24 A controlled trial in scleroderma patients with severe Raynaud phenomenon found botulinum toxin to be no better than placebo.25
Prostacyclin preparations are available. Intermittent intravenous doses of prostacyclin analogues over several days can be used in resistant cases. Oral prostacyclin agents have not shown consistent benefit. New prostacyclin receptor agonists are under investigation.
Overall, we move to other options only in patients with persistent symptoms that impair quality of life, or in patients with recurrent digital ischemic lesions that have not responded to calcium channel blockers and PDE5 inhibitors or nitrates, either alone or in combination.
DIGITAL ULCERATION AND ACUTE DIGITAL ISCHEMIC CRISIS
Patients with secondary Raynaud phenomenon may be at risk of recurrent digital ulceration and acute digital ischemia with gangrene. These patients should be comanaged with a rheumatologist so that the underlying disease process is fully addressed. Digital ulcers should be inspected closely for signs of infection, which may require treatment with antibiotics.
Acute digital ischemia is a medical emergency and should prompt inpatient admission with warming, emotional regulation, and pain control (often with narcotics) to decrease sympathetic vasoconstriction. These patients require aggressive vasodilatory therapy to reverse the ischemic event.
A short-acting calcium channel blocker or combination therapy with a calcium channel blocker and a PDE5 inhibitor or topical nitrate should be started. If there is no benefit, then transient intravenous vasodilatory therapy with a prostacyclin (epoprostenol) or localized digital sympathectomy is used to prevent digital loss.
The endothelin receptor inhibitor bosentan has been shown to decrease recurrent digital ulcers in patients with scleroderma, and while bosentan does not decrease the frequency of Raynaud attacks, it can be used in this select group to prevent new digital ulcers.
Treatment options may be limited by insurance coverage or access to intravenous infusions.
TAKE-HOME RECOMMENDATIONS
For many patients with primary or secondary Raynaud phenomenon, nonpharmacologic interventions are all that are required to decrease the frequency of attacks and improve quality of life. The goal should not be to eliminate attacks completely, as aggressive drug treatment may cause more harm than benefit. From our perspective, the goals of treatment should be to improve quality of life and prevent ischemic complications.
Pharmacologic therapies should be added only if attacks remain poorly controlled with incapacitating symptoms, or if the patient has digital ischemic ulcers. Calcium channel blockers are first-line therapy, given proven efficacy and low cost, and should be titrated to the maximum tolerated dose before adding or substituting other agents.
References
Wigley FM, Flavahan NA. Raynaud’s phenomenon. N Engl J Med 2016; 375:556–565.
Irwin MS, Gilbert SE, Terenghi G, Smith RW, Green CJ. Cold intolerance following peripheral nerve injury. Natural history and factors predicting severity of symptoms. J Hand Surg Br 1997; 22:308–316.
Wasner G. Vasomotor disturbances in complex regional pain syndrome—a review. Pain Med 2010; 11:1267–1273.
Kurklinsky AK, Miller VM, Rooke TW. Acrocyanosis: the Flying Dutchman. Vasc Med 2011; 16:288–301.
Garner R, Kumari R, Lanyon P, Doherty M, Zhang W. Prevalence, risk factors and associations of primary Raynaud’s phenomenon: systematic review and meta-analysis of observational studies. BMJ Open 2015; 5:e006389.
Wigley FM. Clinical practice. Raynaud’s phenomenon. N Engl J Med 2002; 347:1001–1008.
Chikura B, Moore TL, Manning JB, Vail A, Herrick AL. Sparing of the thumb in Raynaud’s phenomenon. Rheumatology (Oxford) 2008; 47:219–221.
Kallenerg CG. Early detection of connective tissue disease in patients with Raynaud’s phenomenon. Rheum Dis Clin North Am 1990; 16:11–30.
Kwakkenbos L, Thombs BD. Non-drug approaches to treating Raynaud’s phenomenon. In: Wigley FM, Herrick AL, Flavahan NA, editors. Raynaud’s Phenomenon. A Guide to Pathogenesis and Treatment. New York: Springer Science+Business Media, 2015:299–313.
Goodfield MJ, Hume A, Rowell NR. The acute effects of cigarette smoking on cutaneous blood flow in smoking and non-smoking subjects with and without Raynaud’s phenomenon. Br J Rheumatol 1990; 29:89–91.
Ennis H, Hughes M, Anderson ME, Wilkinson J, Herrick AL. Calcium channel blockers for primary Raynaud’s phenomenon. Cochrane Database Sys Review 2016; 2:CD002069.
Thompson AE, Pope JE. Calcium channel blockers for primary Raynaud’s phenomenon: a meta-analysis. Rheumatology (Oxford) 2005; 44:145–150.
Roustit M, Blaise S, Allanore Y, Carpentier P, Caglayan E, Cracowski J. Phosphodiesterase-5 inhibitors for the treatment of secondary Raynaud’s phenomenon: systematic review and meta-analysis of randomized trials. Ann Rheum Dis 2013; 72:1696–1699.
Lee EY, Park JK, Lee W, et al. Head-to-head comparison of udenafil vs amlodipine in the treatment of secondary Raynaud's phenomenon: a double-blind, randomized, cross-over study. Rheumatology (Oxford) 2014; 53:658–664.
Chung L, Shapiro L, Fiorentino D, et al. MQX-503, a novel formulation of nitroglycerin, improves the severity of Raynaud’s phenomenon: a randomized, controlled trial. Arthritis Rheum 2009; 60:870–877.
Kan C, Akimoto S, Abe M, Okada K, Ishikawa O. Preliminary thermographic evaluation of a new nitroglycerine tape on the peripheral circulatory disturbance in systemic sclerosis. Ann Rheum Dis 2002; 61:177–179.
Teh LS, Manning J, Moore T, Tully MP, O’Reilly D, Jayson MI. Sustained-release transdermal glyceryl trinitrate patches as a treatment for primary and secondary Raynaud’s phenomenon. Br J Rheumatol 1995; 34:636–641.
Russell IJ, Lessard JA. Prazosin treatment of Raynaud’s phenomenon: a double blind single crossover study. J Rheumatol 1985; 12:94–98.
Wollersheim H, Thien T, Fennis J, van Elteren P, van ‘t Laar A. Double-blind, placebo-controlled study of prazosin in Raynaud’s phenomenon. Clin Pharmacol Ther 1986; 40:219–225.
Coleiro B, Marshall SE, Denton CP, et al. Treatment of Raynaud’s phenomenon with the selective serotonin reuptake inhibitor fluoxetine. Rheumatology (Oxford) 2001; 40:1038–1043.
Didazio M, Denton CP, Smith R, et al. Losartan therapy for Raynaud’s phenomenon and scleroderma: clinical and biochemical findings in a fifteen-week randomized, parallel-group, controlled trial. Arthritis Rheum 1999; 42:2646–2655.
Neirotti M, Longo F, Molaschi M, Macchione C, Pernigotti L. Functional vascular disorders: treatment with pentoxifylline. Angiology 1987; 38:575–580.
Abou-Raya A, Abou-Raya S, Helmii M. Statins: potentially useful in therapy of systemic sclerosis-related Raynaud’s phenomenon and digital ulcers. J Rheumatol 2008; 35:1801–1808.
Iorio ML, Masden DL, Higgins JP. Botulinum toxin A treatment of Raynaud’s phenomenon: a review. Semin Arthritis Rheum 2012; 41: 599–603.
Bello RJ, Cooney CM, Melamed E, et al. The therapeutic efficacy of botulinum toxin in treating scleroderma-associated Raynaud’s phenomenon: a randomized, double-blind, placebo-controlled clinical trial. Arthritis Rheumatol 2017. Epub ahead of print.
References
Wigley FM, Flavahan NA. Raynaud’s phenomenon. N Engl J Med 2016; 375:556–565.
Irwin MS, Gilbert SE, Terenghi G, Smith RW, Green CJ. Cold intolerance following peripheral nerve injury. Natural history and factors predicting severity of symptoms. J Hand Surg Br 1997; 22:308–316.
Wasner G. Vasomotor disturbances in complex regional pain syndrome—a review. Pain Med 2010; 11:1267–1273.
Kurklinsky AK, Miller VM, Rooke TW. Acrocyanosis: the Flying Dutchman. Vasc Med 2011; 16:288–301.
Garner R, Kumari R, Lanyon P, Doherty M, Zhang W. Prevalence, risk factors and associations of primary Raynaud’s phenomenon: systematic review and meta-analysis of observational studies. BMJ Open 2015; 5:e006389.
Wigley FM. Clinical practice. Raynaud’s phenomenon. N Engl J Med 2002; 347:1001–1008.
Chikura B, Moore TL, Manning JB, Vail A, Herrick AL. Sparing of the thumb in Raynaud’s phenomenon. Rheumatology (Oxford) 2008; 47:219–221.
Kallenerg CG. Early detection of connective tissue disease in patients with Raynaud’s phenomenon. Rheum Dis Clin North Am 1990; 16:11–30.
Kwakkenbos L, Thombs BD. Non-drug approaches to treating Raynaud’s phenomenon. In: Wigley FM, Herrick AL, Flavahan NA, editors. Raynaud’s Phenomenon. A Guide to Pathogenesis and Treatment. New York: Springer Science+Business Media, 2015:299–313.
Goodfield MJ, Hume A, Rowell NR. The acute effects of cigarette smoking on cutaneous blood flow in smoking and non-smoking subjects with and without Raynaud’s phenomenon. Br J Rheumatol 1990; 29:89–91.
Ennis H, Hughes M, Anderson ME, Wilkinson J, Herrick AL. Calcium channel blockers for primary Raynaud’s phenomenon. Cochrane Database Sys Review 2016; 2:CD002069.
Thompson AE, Pope JE. Calcium channel blockers for primary Raynaud’s phenomenon: a meta-analysis. Rheumatology (Oxford) 2005; 44:145–150.
Roustit M, Blaise S, Allanore Y, Carpentier P, Caglayan E, Cracowski J. Phosphodiesterase-5 inhibitors for the treatment of secondary Raynaud’s phenomenon: systematic review and meta-analysis of randomized trials. Ann Rheum Dis 2013; 72:1696–1699.
Lee EY, Park JK, Lee W, et al. Head-to-head comparison of udenafil vs amlodipine in the treatment of secondary Raynaud's phenomenon: a double-blind, randomized, cross-over study. Rheumatology (Oxford) 2014; 53:658–664.
Chung L, Shapiro L, Fiorentino D, et al. MQX-503, a novel formulation of nitroglycerin, improves the severity of Raynaud’s phenomenon: a randomized, controlled trial. Arthritis Rheum 2009; 60:870–877.
Kan C, Akimoto S, Abe M, Okada K, Ishikawa O. Preliminary thermographic evaluation of a new nitroglycerine tape on the peripheral circulatory disturbance in systemic sclerosis. Ann Rheum Dis 2002; 61:177–179.
Teh LS, Manning J, Moore T, Tully MP, O’Reilly D, Jayson MI. Sustained-release transdermal glyceryl trinitrate patches as a treatment for primary and secondary Raynaud’s phenomenon. Br J Rheumatol 1995; 34:636–641.
Russell IJ, Lessard JA. Prazosin treatment of Raynaud’s phenomenon: a double blind single crossover study. J Rheumatol 1985; 12:94–98.
Wollersheim H, Thien T, Fennis J, van Elteren P, van ‘t Laar A. Double-blind, placebo-controlled study of prazosin in Raynaud’s phenomenon. Clin Pharmacol Ther 1986; 40:219–225.
Coleiro B, Marshall SE, Denton CP, et al. Treatment of Raynaud’s phenomenon with the selective serotonin reuptake inhibitor fluoxetine. Rheumatology (Oxford) 2001; 40:1038–1043.
Didazio M, Denton CP, Smith R, et al. Losartan therapy for Raynaud’s phenomenon and scleroderma: clinical and biochemical findings in a fifteen-week randomized, parallel-group, controlled trial. Arthritis Rheum 1999; 42:2646–2655.
Neirotti M, Longo F, Molaschi M, Macchione C, Pernigotti L. Functional vascular disorders: treatment with pentoxifylline. Angiology 1987; 38:575–580.
Abou-Raya A, Abou-Raya S, Helmii M. Statins: potentially useful in therapy of systemic sclerosis-related Raynaud’s phenomenon and digital ulcers. J Rheumatol 2008; 35:1801–1808.
Iorio ML, Masden DL, Higgins JP. Botulinum toxin A treatment of Raynaud’s phenomenon: a review. Semin Arthritis Rheum 2012; 41: 599–603.
Bello RJ, Cooney CM, Melamed E, et al. The therapeutic efficacy of botulinum toxin in treating scleroderma-associated Raynaud’s phenomenon: a randomized, double-blind, placebo-controlled clinical trial. Arthritis Rheumatol 2017. Epub ahead of print.
Primary Raynaud phenomenon occurs in the absence of any underlying disease process. Secondary Raynaud phenomenon occurs in concert with another disease, frequently rheumatic.
Young patients with mild Raynaud phenomenon, normal nailfold capillaries, and no additional symptoms or signs to suggest a rheumatic or other underlying disease can be followed carefully by the primary care doctor and do not require further serologic workup or referral to a specialist.
Nonpharmacologic interventions, ie, cold avoidance and stress management, are first-line for all patients.
Calcium channel blockers are first-line drugs and should be titrated to the maximum tolerated dose before adding or switching to other agents.
The goal of treatment should not be to eliminate Raynaud attacks completely but to improve quality of life and prevent ischemic complications.
Disallow All Ads
Content Gating
No Gating (article Unlocked/Free)
Alternative CME
Disqus Comments
Default
Consolidated Pubs: Do Not Show Source Publication Logo
For practitioners who see a lot of patients, particularly a lot of young women, patients describing cold-induced color changes of the fingers sometimes accompanied by tingling or burning are common. For most patients it is mild, but for some the discoloration or dysesthesia may be striking and disconcerting. For a minority, this reversible vasoconstrictive phenomenon (Raynaud “disease” if it occurs in isolation, without any associated underlying condition) may be the presenting sign of a systemic disorder.
For many patients, Raynaud symptoms are mild enough to not even mention to their primary care provider, and conversely, there is little reason for most clinicians to routinely inquire about such symptoms. So it may surprise some readers to read about the nuances of diagnosis and treatment discussed by Shapiro and Wigley in this issue of the Journal.
To a rheumatologist, Raynaud phenomenon, particularly of recent onset in an adult, raises the specter of an underlying systemic inflammatory disease. The phenomenon is not linked to a specific diagnosis; it is associated with lupus, rheumatoid arthritis, cryoglobulinemia, inflammatory myopathy, Sjögren syndrome, and, in its severe form, with the scleroderma syndromes. We focus on differentiating between these rheumatic disorders once we have discarded nonrheumatic causes such as atherosclerotic arterial disease, carcinoma, embolism, Buerger disease, medications, smoking, or thrombosis.
But rheumatologists are toward the bottom of the diagnostic funnel—we see these patients when an underlying disease is already suspected. The real challenge is for the primary care providers who first recognize the digital vasospasm on examination or are told of the symptoms by their patient. These clinicians need to know which initial reflexive actions are warranted and which can wait, for, as noted by Shapiro and Wigley, there are several options.
The first action is to try to determine the timeline, although Raynaud disease often has an insidious onset or the patient doesn’t recall the onset. New and sudden onset likely has a stronger association with an underlying disease. A focused physical examination should look for digital stigmata of ischemic damage; the presence of digital ulcers or healed digital pits indicates a possible vascular occlusive component in addition to the vascular spasm. This strongly suggests scleroderma or Buerger disease, as tissue damage doesn’t occur in (primary) Raynaud disease or generally even with Raynaud phenomenon associated with lupus or other rheumatic disorders. Sclerodactyly should be looked for: diffuse finger puffiness, skin-tightening, or early signs such as loss of the usual finger skin creases. Telangiectasia (not vascular spiders or cherry angiomata) should be searched for, particularly on the palms, face, and inner lips, as these vascular lesions are common in patients with limited scleroderma. Careful auscultation for basilar lung crackles should be done. Distal pulses should all be assessed, and bruits in the neck, abdomen and inguinal areas should be carefully sought.
Patients should be questioned about any symptom-associated reduction in exercise tolerance and particularly about trouble swallowing, “heartburn,” and symptoms of reflux. Although patients with Raynaud disease may have demonstrable esophageal dysmotility, the presence of significant, new, or worsened symptoms raises the concern of scleroderma. Patients should be asked about symptoms of malabsorption. Specific questioning should be directed at eliciting a history of joint stiffness and especially muscle weakness. The latter can be approached by inquiring about new or progressive difficulty in specific tasks such as walking up steps, brushing hair, and arising from low chairs or the toilet. Distinguishing muscle weakness from general fatigue is not always easy, but it is important.
Shapiro and Wigley discuss the extremely useful evaluation of nailfold capillaries, which can be done with a standard magnifier or ophthalmoscope. This is very valuable to help predict the development or current presence of a systemic rheumatic disease. But this is not a technique that most clinicians are familiar with. A potentially useful surrogate or adjunctive test, especially in the setting of new-onset Raynaud, is the antinuclear antibody (ANA) test; I prefer the immunofluorescent assay. While a positive test alone (with Raynaud) does not define the presence of any rheumatic disease, several older studies suggest that patients with a new onset of Raynaud phenomenon and a positive ANA test are more likely to develop a systemic autoimmune disorder than if the test is negative. Those who do so (and this is far from all) are most likely to have the disease manifest within a few years. Hence, if the ANA test is positive but the history, physical examination, and limited laboratory testing (complete blood cell count with differential, complete metabolic panel, creatine kinase, and urinalysis) are normal, it is reasonable to reexamine the patient in 3 months and then every 6 months for 2 to 3 years, repeating the focused history and physical examination. It is also reasonable at some point to refer these patients to a rheumatologist.
Since Raynaud phenomenon is common, and the associated severe rheumatic disorders associated with it are rare, it is easy to not recognize Raynaud phenomenon as a clue to the onset of a potentially severe systemic disease. Yet with a few simple questions, a focused examination, and minimal laboratory testing, patients who are more likely to harbor a systemic disease can usually be treated symptomatically if necessary, and appropriately triaged to observation or for subspecialty referral.
For practitioners who see a lot of patients, particularly a lot of young women, patients describing cold-induced color changes of the fingers sometimes accompanied by tingling or burning are common. For most patients it is mild, but for some the discoloration or dysesthesia may be striking and disconcerting. For a minority, this reversible vasoconstrictive phenomenon (Raynaud “disease” if it occurs in isolation, without any associated underlying condition) may be the presenting sign of a systemic disorder.
For many patients, Raynaud symptoms are mild enough to not even mention to their primary care provider, and conversely, there is little reason for most clinicians to routinely inquire about such symptoms. So it may surprise some readers to read about the nuances of diagnosis and treatment discussed by Shapiro and Wigley in this issue of the Journal.
To a rheumatologist, Raynaud phenomenon, particularly of recent onset in an adult, raises the specter of an underlying systemic inflammatory disease. The phenomenon is not linked to a specific diagnosis; it is associated with lupus, rheumatoid arthritis, cryoglobulinemia, inflammatory myopathy, Sjögren syndrome, and, in its severe form, with the scleroderma syndromes. We focus on differentiating between these rheumatic disorders once we have discarded nonrheumatic causes such as atherosclerotic arterial disease, carcinoma, embolism, Buerger disease, medications, smoking, or thrombosis.
But rheumatologists are toward the bottom of the diagnostic funnel—we see these patients when an underlying disease is already suspected. The real challenge is for the primary care providers who first recognize the digital vasospasm on examination or are told of the symptoms by their patient. These clinicians need to know which initial reflexive actions are warranted and which can wait, for, as noted by Shapiro and Wigley, there are several options.
The first action is to try to determine the timeline, although Raynaud disease often has an insidious onset or the patient doesn’t recall the onset. New and sudden onset likely has a stronger association with an underlying disease. A focused physical examination should look for digital stigmata of ischemic damage; the presence of digital ulcers or healed digital pits indicates a possible vascular occlusive component in addition to the vascular spasm. This strongly suggests scleroderma or Buerger disease, as tissue damage doesn’t occur in (primary) Raynaud disease or generally even with Raynaud phenomenon associated with lupus or other rheumatic disorders. Sclerodactyly should be looked for: diffuse finger puffiness, skin-tightening, or early signs such as loss of the usual finger skin creases. Telangiectasia (not vascular spiders or cherry angiomata) should be searched for, particularly on the palms, face, and inner lips, as these vascular lesions are common in patients with limited scleroderma. Careful auscultation for basilar lung crackles should be done. Distal pulses should all be assessed, and bruits in the neck, abdomen and inguinal areas should be carefully sought.
Patients should be questioned about any symptom-associated reduction in exercise tolerance and particularly about trouble swallowing, “heartburn,” and symptoms of reflux. Although patients with Raynaud disease may have demonstrable esophageal dysmotility, the presence of significant, new, or worsened symptoms raises the concern of scleroderma. Patients should be asked about symptoms of malabsorption. Specific questioning should be directed at eliciting a history of joint stiffness and especially muscle weakness. The latter can be approached by inquiring about new or progressive difficulty in specific tasks such as walking up steps, brushing hair, and arising from low chairs or the toilet. Distinguishing muscle weakness from general fatigue is not always easy, but it is important.
Shapiro and Wigley discuss the extremely useful evaluation of nailfold capillaries, which can be done with a standard magnifier or ophthalmoscope. This is very valuable to help predict the development or current presence of a systemic rheumatic disease. But this is not a technique that most clinicians are familiar with. A potentially useful surrogate or adjunctive test, especially in the setting of new-onset Raynaud, is the antinuclear antibody (ANA) test; I prefer the immunofluorescent assay. While a positive test alone (with Raynaud) does not define the presence of any rheumatic disease, several older studies suggest that patients with a new onset of Raynaud phenomenon and a positive ANA test are more likely to develop a systemic autoimmune disorder than if the test is negative. Those who do so (and this is far from all) are most likely to have the disease manifest within a few years. Hence, if the ANA test is positive but the history, physical examination, and limited laboratory testing (complete blood cell count with differential, complete metabolic panel, creatine kinase, and urinalysis) are normal, it is reasonable to reexamine the patient in 3 months and then every 6 months for 2 to 3 years, repeating the focused history and physical examination. It is also reasonable at some point to refer these patients to a rheumatologist.
Since Raynaud phenomenon is common, and the associated severe rheumatic disorders associated with it are rare, it is easy to not recognize Raynaud phenomenon as a clue to the onset of a potentially severe systemic disease. Yet with a few simple questions, a focused examination, and minimal laboratory testing, patients who are more likely to harbor a systemic disease can usually be treated symptomatically if necessary, and appropriately triaged to observation or for subspecialty referral.
For practitioners who see a lot of patients, particularly a lot of young women, patients describing cold-induced color changes of the fingers sometimes accompanied by tingling or burning are common. For most patients it is mild, but for some the discoloration or dysesthesia may be striking and disconcerting. For a minority, this reversible vasoconstrictive phenomenon (Raynaud “disease” if it occurs in isolation, without any associated underlying condition) may be the presenting sign of a systemic disorder.
For many patients, Raynaud symptoms are mild enough to not even mention to their primary care provider, and conversely, there is little reason for most clinicians to routinely inquire about such symptoms. So it may surprise some readers to read about the nuances of diagnosis and treatment discussed by Shapiro and Wigley in this issue of the Journal.
To a rheumatologist, Raynaud phenomenon, particularly of recent onset in an adult, raises the specter of an underlying systemic inflammatory disease. The phenomenon is not linked to a specific diagnosis; it is associated with lupus, rheumatoid arthritis, cryoglobulinemia, inflammatory myopathy, Sjögren syndrome, and, in its severe form, with the scleroderma syndromes. We focus on differentiating between these rheumatic disorders once we have discarded nonrheumatic causes such as atherosclerotic arterial disease, carcinoma, embolism, Buerger disease, medications, smoking, or thrombosis.
But rheumatologists are toward the bottom of the diagnostic funnel—we see these patients when an underlying disease is already suspected. The real challenge is for the primary care providers who first recognize the digital vasospasm on examination or are told of the symptoms by their patient. These clinicians need to know which initial reflexive actions are warranted and which can wait, for, as noted by Shapiro and Wigley, there are several options.
The first action is to try to determine the timeline, although Raynaud disease often has an insidious onset or the patient doesn’t recall the onset. New and sudden onset likely has a stronger association with an underlying disease. A focused physical examination should look for digital stigmata of ischemic damage; the presence of digital ulcers or healed digital pits indicates a possible vascular occlusive component in addition to the vascular spasm. This strongly suggests scleroderma or Buerger disease, as tissue damage doesn’t occur in (primary) Raynaud disease or generally even with Raynaud phenomenon associated with lupus or other rheumatic disorders. Sclerodactyly should be looked for: diffuse finger puffiness, skin-tightening, or early signs such as loss of the usual finger skin creases. Telangiectasia (not vascular spiders or cherry angiomata) should be searched for, particularly on the palms, face, and inner lips, as these vascular lesions are common in patients with limited scleroderma. Careful auscultation for basilar lung crackles should be done. Distal pulses should all be assessed, and bruits in the neck, abdomen and inguinal areas should be carefully sought.
Patients should be questioned about any symptom-associated reduction in exercise tolerance and particularly about trouble swallowing, “heartburn,” and symptoms of reflux. Although patients with Raynaud disease may have demonstrable esophageal dysmotility, the presence of significant, new, or worsened symptoms raises the concern of scleroderma. Patients should be asked about symptoms of malabsorption. Specific questioning should be directed at eliciting a history of joint stiffness and especially muscle weakness. The latter can be approached by inquiring about new or progressive difficulty in specific tasks such as walking up steps, brushing hair, and arising from low chairs or the toilet. Distinguishing muscle weakness from general fatigue is not always easy, but it is important.
Shapiro and Wigley discuss the extremely useful evaluation of nailfold capillaries, which can be done with a standard magnifier or ophthalmoscope. This is very valuable to help predict the development or current presence of a systemic rheumatic disease. But this is not a technique that most clinicians are familiar with. A potentially useful surrogate or adjunctive test, especially in the setting of new-onset Raynaud, is the antinuclear antibody (ANA) test; I prefer the immunofluorescent assay. While a positive test alone (with Raynaud) does not define the presence of any rheumatic disease, several older studies suggest that patients with a new onset of Raynaud phenomenon and a positive ANA test are more likely to develop a systemic autoimmune disorder than if the test is negative. Those who do so (and this is far from all) are most likely to have the disease manifest within a few years. Hence, if the ANA test is positive but the history, physical examination, and limited laboratory testing (complete blood cell count with differential, complete metabolic panel, creatine kinase, and urinalysis) are normal, it is reasonable to reexamine the patient in 3 months and then every 6 months for 2 to 3 years, repeating the focused history and physical examination. It is also reasonable at some point to refer these patients to a rheumatologist.
Since Raynaud phenomenon is common, and the associated severe rheumatic disorders associated with it are rare, it is easy to not recognize Raynaud phenomenon as a clue to the onset of a potentially severe systemic disease. Yet with a few simple questions, a focused examination, and minimal laboratory testing, patients who are more likely to harbor a systemic disease can usually be treated symptomatically if necessary, and appropriately triaged to observation or for subspecialty referral.















 is typically seen in precordial leads V3 to V6 and is a marker for hypothermia.")
 White digits with intense vasoconstriction in Raynaud phenomenon; (B) blue digits with hypoxemic venous stasis; (C) red digits with hyperemic reperfusion.")
 Dilated nailfold capillaries in a patient with scleroderma (blue arrow); (B) dilation and dropout of nailfold capillaries (white arrow) viewed with a magnifier.")
 Dilated nailfold capillaries in a patient with scleroderma (blue arrow); (B) dilation and dropout of nailfold capillaries (white arrow) viewed with a magnifier.")