User login
Update on viral hepatitis in pregnancy
Viral hepatitis affects mother and child, and pregnancy can exacerbate the disease. Vertical transmission contributes significantly to the high prevalence of viral hepatitis and compromises the well-being and the prognosis in the newborn. The indications for therapy in pregnant women may differ from those in the general population, and new therapies are available.
HEPATITIS A
Hepatitis A virus (HAV) infection is associated with significant morbidity and death around the world, as 1.4 million cases are reported every year worldwide.1 However, in the United States, the prevalence has declined by 95% since HAV vaccination was introduced in 1995, and in 2013, the prevalence was 0.6 per 100,000 population.2 Acute HAV infection during pregnancy is rare. As a result, the incidence during pregnancy is difficult to ascertain.3
HAV is transmitted by the fecal-oral route from person to person contact and from contamination of food and water. Vertical transmission from pregnant mother to fetus has not been reported.4
Clinical outcomes of HAV in pregnancy
Acute HAV infection during pregnancy is rare, and teratogenicity associated with HAV has not been reported.3 The course of the disease during pregnancy is generally similar to that in nonpregnant patients, with excellent maternal and fetal outcomes in developed nations. There have been reports in developing nations of premature contractions and labor, placental separation, premature rupture of the membranes, and vaginal bleeding.5,6
Diagnosis
Routine screening for HAV is not recommended, but serologic testing by detection of anti-HAV immunoglobulin M (IgM) antibodies is done in high-risk patients suspected of having acute HAV infection.
Prevention
Prevention includes adherence to sanitary practices and active and passive immunoprophylaxis.3 Universal vaccination for pregnant mothers is not recommended,1,2,6 but vaccination is recommended for high-risk patients and mothers—those with chronic liver disease, those receiving clotting factors, those who use illegal drugs, and travelers to areas where HAV is endemic. Immune globulin is also available for postexposure prophylaxis. HAV vaccines and immune globulin are safe in pregnancy.3,6,7
Treatment
Treatment of acute HAV in pregnancy is supportive because of its benign nature; few patients require hospitalization.3
Pregnant patients with HAV can deliver vaginally, and breastfeeding is not contraindicated.8
HEPATITIS B
Hepatitis B virus (HBV) infection is a major global health problem. About 240 million people worldwide have chronic HBV infection, and more than 780,000 die every year from acute and chronic consequences.9
Vertical transmission is responsible for about half of chronic HBV infections worldwide. Thus, interruption of mother-to-child transmission is important. Universal maternal screening and passive-active immunoprophylaxis of newborns have lowered the transmission rates to between 5% and 10%. The 10% failure rate is unacceptably high and has been attributed to seropositivity for hepatitis B e antigen and a high viral load in the mother (ie, HBV DNA > 106 copies/mL). High viral load is an independent risk factor for failure of immunoprophylaxis.10 Therefore, antiviral therapy is suggested in pregnant women who have a high HBV viral load to further decrease the chance of mother-to-child transmission and to prevent failure of immunoprophylaxis.11
Clinical outcomes of HBV in pregnancy
Acute HBV infection during pregnancy is usually benign and is not associated with increased risk of death or teratogenicity.12 Symptomatic disease in the mother with acute hepatitis B includes nausea, vomiting, abdominal pain, fatigue, and jaundice.3 For the newborn, there is increased risk of low birth weight and prematurity.13
When acute HBV infection occurs early in pregnancy, the rate of perinatal transmission is about 10%, increasing to 60% if it occurs near delivery.12,13
Chronic HBV infection does not usually affect the outcome of pregnancy, but it may if the woman has cirrhosis or advanced liver disease14; however, pregnancy is very rare in women with HBV cirrhosis due to anovulation, and acute HBV flares have been described during pregnancy and postpartum.15
Pregnant patients with cirrhosis and portal hypertension are at risk of hepatic decompensation, variceal bleeding, and death.16 Risk is high with a score of 10 or more on the Model for End-stage Liver Disease scale, and is low with a score of 6 or less.17 Like nonpregnant patients, pregnant patients with cirrhosis should be monitored, and upper endoscopy should be performed in the 2nd trimester to assess for varices. A beta-blocker should be given or banding of varices should be done to avoid rupture. Rates of fetal demise, premature labor, spontaneous abortion, and stillbirth are high with portal hypertension.16
Risk of mother-to-child HBV transmission
Vertical HBV transmission can occur during the antepartum, intrapartum, and postpartum periods,18,19 but it most often occurs during the intrapartum period at the time of delivery. Without immunoprophylaxis of the newborn, the risk of mother-to-child transmission can be as high as 90% if the mother is hepatitis B e antigen-positive and has a viral load greater than 106 copies/mL. With active and passive immunoprophylaxis, the risk decreases substantially.
Screening and diagnosis
All pregnant women should be tested for hepatitis B surface antigen during the 1st trimester,20 or any time thereafter if early testing was not done, even if they were vaccinated before becoming pregnant.21
Prevention
HBV infection is best prevented before pregnancy by vaccinating the mother or, after delivery, by vaccinating the newborn. Universal vaccination of newborns has been the standard of care since the 1990s. Pregnant women should be tested early in the pregnancy; unvaccinated, uninfected women at high risk of acquiring HBV (eg, because of sexual contacts or intravenous drug use) should be vaccinated.2,3
HBV vaccine and immune globulin are both approved by the US Food and Drug Administration (FDA) for prevention of HBV infection and can be given during pregnancy and breastfeeding.3 All infants should be vaccinated for HBV at birth, and all infants born to mothers who test positive for hepatitis B s antigen should receive the HBV vaccine and the immune globulin within 12 to 24 hours after delivery. The vaccine series should be completed within 6 months.20,21 This will decrease the rate of neonatal infection.
Treatment of HBV infection in pregnancy
The main objectives of treating chronic HBV infection in pregnancy are to maintain stable liver function in the mother and to prevent neonatal infection, which may cause cirrhosis and hepatocellular carcinoma and contribute to the global burden of the disease.22 Therefore, maternal HBV DNA and liver aminotransferase levels should be tested regularly during gestation.
The current guidelines of the American Association for the Study of Liver Diseases suggest antiviral therapy to reduce the risk of perinatal transmission of HBV in pregnant women with an HBV DNA level greater than 200,000 IU/mL or greater than 106 copies/mL.23,24
In a meta-analysis,25 antiviral therapy with lamivudine, telbivudine, or tenofovir showed no apparent teratogenicity or safety concerns for maternal and fetal outcomes26 and significantly reduced the rate of mother-to-child transmission. Of these 3 drugs, telbivudine was associated with a higher rate of normalization of liver enzymes, HBV suppression, and e-antigen seroconversion.25 Lamivudine has proven the test of time in mothers co-infected with HBV and human immunodeficiency virus (HIV). However, tenofovir is considered the preferred treatment in pregnancy, owing to concerns about drug resistance to telbivudine and lamivudine and a high genetic barrier to resistance with tenofovir.26 In mothers with HBV and HIV treated with tenofovir, treatment was associated with lower bone mineral density in the newborns, with a propensity for renal injury in the mothers. No safety concerns for maternal or fetal outcomes were identified in pregnant women infected only with HBV.25
Many pregnant mothers choose to stop therapy around the time of conception because of safety concerns for the baby. In such situations, close monitoring is necessary to detect flares of HBV infection.
When the decision to treat is made, treatment should begin at 28 to 30 weeks of gestation, when organogenesis is complete and to allow enough time for HBV DNA levels to decline.
Breastfeeding is not contraindicated because antiviral drugs are minimally excreted in breast milk and are unlikely to cause toxicity. However, data are insufficient as to the long-term safety for the newborn when the mother took these drugs during pregancy and while breastfeeding.23,27
Alanine aminotransferase and HBV DNA levels should be monitored postpartum because of the possibility of a hepatitis flare. In this setting, any of the three drugs can be used.28 For mothers on therapy because of cirrhosis or an advanced histologic feature, antiviral therapy should be continued throughout pregnancy to prevent disease progression and decompensation.19,22,27
No drug therapy is necessary for pregnant carriers of HBV.
Delivery and breastfeeding
The mode of delivery does not appear to have a significant effect on the interruption of vertical transmission of HBV.29 Cesarean delivery is not recommended by the US Centers for Disease Control and Prevention (CDC)2 or the American College of Obstetricians and Gynecologists.6 Breastfeeding is encouraged if the infant has received appropriate immunoprophylaxis.6
Coinfection with hepatitis D
Coinfection with hepatitis D virus (HDV) and HBV is associated with severe acute hepatitis30,31 and increases the risk of death by a factor of 10. The World Health Organization recommends testing for HDV in pregnant women who are HBV-positive.8
Prevention of HDV infection requires prevention of HBV. The treatment of HDV in pregnancy is supportive. Pegylated interferon is successful outside pregnancy but is contraindicated during pregnancy.32 In patients with fulminant hepatic failure and end-stage liver disease, liver transplant can be lifesaving.
Take-home points
- HBV infection during pregnancy is usually benign and not severe but can be associated with an increased risk of mother-to-child transmission and progression of liver disease in the pregnant mother.
- Prevention of vertical transmission of HBV is important to reduce the burden of chronic HBV infection. Universal maternal screening early in pregnancy and passive-active immunoprophylaxis of newborns are usually sufficient to prevent vertical transmission of HBV, but antiviral therapy is needed for highly viremic mothers to further reduce the risk.
- Antiviral therapy is also indicated for pregnant women with moderate to severe hepatitis or cirrhosis to prevent disease progression and liver failure.
- Telbivudine, tenofovir, or lamivudine can be used during pregnancy, but more data are needed on the long-term safety of fetal exposure to these agents.
HEPATITIS C
The global prevalence of hepatitis C virus (HCV) infection is 2% to 3%, with 130 to 170 million HCV-positive people, most of whom are chronically infected.33 The incidence of HCV during pregnancy is 1% to 2.4%, but 3% to 5% of infected mothers transmit HCV to their child at the time of birth.6,34 Women coinfected with HIV and HCV have twice the risk of perinatal HCV transmission compared with women who have HCV infection alone.6,34
HCV infection is usually asymptomatic and is discovered either by screening high-risk patients or during evaluation of persistently elevated aminotransferase levels. Acute HCV infection during pregnancy has been reported only rarely, and most pregnant women who are infected have chronic disease with no effect on the pregnancy or the infant.6,34
Treatment
The CDC recommends that all adults (including pregnant women) born between 1945 and 1965 undergo 1-time testing for HCV without prior ascertainment of HCV risk (strong recommendation, with moderate quality of evidence).35 The most important risk factor for HCV infection is past or current injection drug use.33 Additional risk factors are similar to those for nonpregnant patients.
Because of the benign effect of HCV on the pregnancy, treatment is not recommended. To decrease the risk of maternal-child transmission, it is prudent to avoid amniocentesis, scalp instrumentation, and prolonged rupture of membranes.6
There is no vaccine or immune globulin for prevention. HCV infection should not influence the mode of delivery, and it is not a contraindication to breastfeeding.34,36,37
HEPATITIS E
Every year, 20 million cases of hepatitis E virus (HEV) infection are recorded worldwide. These numbers include 3.3 million symptomatic cases and 56,600 deaths.38 HEV infection is most common in developing countries, and pregnant women traveling to these areas are at high risk of acquiring this infection, of developing fulminant hepatitis, and of death.39 Sporadic cases not associated with travel are increasingly reported in developed countries and are attributed to immunocompromised status (due to HIV or solid-organ transplant).38,40
Modes of transmission of HEV are mainly via fecal-oral contamination and by vertical transmission.41
Diagnosis
HEV infection can be diagnosed either by detecting IgM antibody with an enzyme-linked immunosorbent assay or by detecting HEV RNA in the blood using reverse transcription polymerase chain reaction testing.42
Treatment and prevention
Hospitalization should be considered for pregnant women. Ribavirin or pegylated interferon alpha or both are effective but are contraindicated in pregnancy because of the risk of teratogenicity.41,42 Urgent liver transplant can be a successful option in acute liver failure.
Prevention relies primarily on good sanitation, clean drinking water, and avoiding raw pork and venison. Boiling and chlorination of water inactivate HEV.39,40 Pregnant women should be advised to avoid travel to highly endemic areas.
- World Health Organization (WHO).Hepatitis A fact sheet. www.who.int/mediacentre/factsheets/fs328/en/. Accessed December 7, 2016.
- US Centers for Disease Control and Prevention (CDC). Viral hepatitis—statistics & surveillance. www.cdc.gov/hepatitis/statistics/2013surveillance/commentary.htm#hepatitis A. Accessed December 7, 2016.
- Rac MW, Sheffield JS. Prevention and management of viral hepatitis in pregnancy. Obstet Gynecol Clin North Am 2014; 41:573–592.
- Workowski KA, Bolan GA; Centers for Disease Control and Prevention (CDC). Sexually transmitted diseases treatment guidelines, 2015. MMWR Recomm Rep 2015; 64:1–137.
- Elinav E, Ben-Dov IZ, Shapira Y, et al. Acute hepatitis A infection in pregnancy is associated with high rates of gestational complications and preterm labor. Gastroenterology 2006; 130:1129–1134.
- American College of Obstetricians and Gynecologists. ACOG Practice Bulletin No. 86: Viral hepatitis in pregnancy. Obstet Gynecol 2007; 110:941–956.
- Advisory Committee on Immunization Practices. Guidelines for Vaccinating Pregnant Women. www.cdc.gov/vaccines/pregnancy/hcp/guidelines.html. Accessed December 7, 2016.
- Daudi N, Shouval D, Stein-Zamir C, Ackerman Z. Breastmilk hepatitis A virus RNA in nursing mothers with acute hepatitis A virus infection. Breastfeed Med 2012; 7:313–315.
- World Health Organization (WHO). Hepatitis B fact sheet. www.who.int/mediacentre/factsheets/fs204/en/. Accessed December 7, 2016.
- Zou H, Chen Y, Duan Z, Zhang H, Pan C. Virologic factors associated with failure to passive-active immunoprophylaxis in infants born to HBsAg-positive mothers. J Viral Hepat 2012; 19:e18–e25.
- Pan CQ, Lee HM. Antiviral therapy for chronic hepatitis B in pregnancy. Semin Liver Dis 2013; 33:138–146.
- Sookoian S. Liver disease during pregnancy: acute viral hepatitis. Ann Hepatol 2006; 5:231–236.
- Jonas MM. Hepatitis B and pregnancy: an underestimated issue. Liver Int 2009; 29(suppl 1):133–139.
- Wong S, Chan LY, Yu V, Ho L. Hepatitis B carrier and perinatal outcome in singleton pregnancy. Am J Perinatol 1999; 16:485–488.
- Rawal BK, Parida S, Watkins RP, Ghosh P, Smith H. Symptomatic reactivation of hepatitis B in pregnancy. Lancet 1991; 337:364.
- Aggarwal N, Negi N, Aggarwal A, Bodh V, Dhiman RK. Pregnancy with portal hypertension. J Clin Exp Hepatol 2014; 4:163–171.
- Westbrook RH, Yeoman AD, O'Grady JG, Harrison PM, Devlin J, Heneghan MA. Model for end-stage liver disease score predicts outcome in cirrhotic patients during pregnancy. Clin Gastroenterol Hepatol 2011; 9:694–699.
- Cheung KW, Seto MT, Wong SF. Towards complete eradication of hepatitis B infection from perinatal transmission: review of the mechanisms of in utero infection and the use of antiviral treatment during pregnancy. Eur J Obstet Gynecol Reproduct Biol 2013; 169:17–23.
- Pan CQ, Duan AP, Bhamidimarri KR, et al. An algorithm for risk assessment and intervention of mother to child transmission of hepatitis B virus. Clin Gastroenterol Hepatol 2012; 10:452–459.
- Mast EE, Margolis HS, Fiore AE, et al; Advisory Committee on Immunization Practices (ACIP). A comprehensive immunization strategy to eliminate transmission of hepatitis B virus infection in the United States: recommendations of the Advisory Committee on Immunization Practices (ACIP) part 1: immunization of infants, children, and adolescents. MMWR Recomm Rep 2005; 54:1–31.
- US Centers for Disease Control (CDC). Prevention of perinatal transmission of hepatitis B virus: prenatal screening of all pregnant women for hepatitis B surface antigen. MMWR Morb Mortal Wkly Rep 1988; 37:341–346, 351.
- Han GR, Xu CL, Zhao W, Yang YF. Management of chronic hepatitis B in pregnancy. World J Gastroenterol 2012; 18:4517–4521.
- Terrault NA, Bzowej NH, Chang KM, Hwang JP, Jonas MM, Murad MH; American Association for the Study of Liver Diseases. AASLD guidelines for treatment of chronic hepatitis B. Hepatology 2016; 63:261–283.
- Tran TT, Ahn J, Reau N. ACG clinical guideline: liver disease and pregnancy. Am J Gastroenterol 2016: 111:176–194.
- Brown RS Jr, McMahon BJ, Lok AS, et al. Antiviral therapy in chronic hepatitis B viral infection during pregnancy: a systematic review and meta-analysis. Hepatol 2016; 63:319–333.
- Brown RS Jr, Verna EC, Pereira MR, et al. Hepatitis B virus and human immunodeficiency virus drugs in pregnancy: findings from the antiretroviral pregnancy registry. J Hepatol 2012; 57:953–959.
- Lamberth JR, Reddy SC, Pan JJ, Dasher KJ. Chronic hepatitis B infection in pregnancy. World J Hepatol 2015; 7:1233–1237.
- Potthoff A, Rifai K, Wedemeyer H, Deterding K, Manns M, Strassburg C. Successful treatment of fulminant hepatitis B during pregnancy. Z Gastroenterol 2009; 47:667–670.
- Yang J, Zeng XM, Men YL, Zhao LS. Elective caesarean section versus vaginal delivery for preventing mother to child transmission of hepatitis B virus—a systematic review. Virol J 2008; 5:100.
- Price J. An update on hepatitis B, D, and E viruses. Top Antivir Med 2014; 21:157–163.
- World Health Organization (WHO). Global alert and response. Hepatitis Delta. www.who.int/csr/resources/publications/hepatitis/who_cds_csr_ncs_2001_1/en/. Accessed December 7, 2016.
- Abbas Z, Memon MS, Mithani H, Jafri W, Hamid S. Treatment of chronic hepatitis D patients with pegylated interferon: a real-world experience. Antivir Ther 2014; 19:463–468.
- Baldo V, Baldovin T, Trivello R, Floreani A. Epidemiology of HCV infection. Curr Pharm Des 2008; 14:1646–1654.
- Floreani A. Hepatitis C and pregnancy. World J Gastroenterol 2013; 19:6714–6720.
- US Centers for Disease Control and Prevention. Viral hepatitis—CDC recommendations for specific populations and settings. www.cdc.gov/hepatitis/populations/1945-1965.htm. Accessed December 7, 2016.
- World Health Organization (WHO). Hepatitis C fact sheet. www.who.int/mediacentre/factsheets/fs164/en/. Accessed December 7, 2016.
- Moyer VA; US Preventive Services Task Force. Screening for hepatitis C virus infection in adults: US Preventive Services Task Force Recommendation Statement. Ann Intern Med 2013; 159:349–357.
- World Health Organization (WHO). Hepatitis E fact sheet. www.who.int/mediacentre/factsheets/fs280/en/. Accessed December 7, 2016.
- Velosa M, Figueiredo A, Gloria H, et al. Fulminant hepatitis E in a pregnant woman. GE Port J Gastroenterol 2013; 20:210–214.
- US Centers for Disease Control and Prevention (CDC). Viral hepatitis—hepatitis E information. Hepatitis E FAQs for health professionals. www.cdc.gov/hepatitis/hev/hevfaq.htm. Accessed December 7, 2016.
- Peters van Ton AM, Gevers TJ, Drenth JP. Antiviral therapy in chronic hepatitis E: a systematic review. J Viral Hepat 2015; 22:965–973.
- Kamar N, Bendall R, Legrand-Abravanel F, et al. Hepatitis E. Lancet 2012: 379:2477–2488.
Viral hepatitis affects mother and child, and pregnancy can exacerbate the disease. Vertical transmission contributes significantly to the high prevalence of viral hepatitis and compromises the well-being and the prognosis in the newborn. The indications for therapy in pregnant women may differ from those in the general population, and new therapies are available.
HEPATITIS A
Hepatitis A virus (HAV) infection is associated with significant morbidity and death around the world, as 1.4 million cases are reported every year worldwide.1 However, in the United States, the prevalence has declined by 95% since HAV vaccination was introduced in 1995, and in 2013, the prevalence was 0.6 per 100,000 population.2 Acute HAV infection during pregnancy is rare. As a result, the incidence during pregnancy is difficult to ascertain.3
HAV is transmitted by the fecal-oral route from person to person contact and from contamination of food and water. Vertical transmission from pregnant mother to fetus has not been reported.4
Clinical outcomes of HAV in pregnancy
Acute HAV infection during pregnancy is rare, and teratogenicity associated with HAV has not been reported.3 The course of the disease during pregnancy is generally similar to that in nonpregnant patients, with excellent maternal and fetal outcomes in developed nations. There have been reports in developing nations of premature contractions and labor, placental separation, premature rupture of the membranes, and vaginal bleeding.5,6
Diagnosis
Routine screening for HAV is not recommended, but serologic testing by detection of anti-HAV immunoglobulin M (IgM) antibodies is done in high-risk patients suspected of having acute HAV infection.
Prevention
Prevention includes adherence to sanitary practices and active and passive immunoprophylaxis.3 Universal vaccination for pregnant mothers is not recommended,1,2,6 but vaccination is recommended for high-risk patients and mothers—those with chronic liver disease, those receiving clotting factors, those who use illegal drugs, and travelers to areas where HAV is endemic. Immune globulin is also available for postexposure prophylaxis. HAV vaccines and immune globulin are safe in pregnancy.3,6,7
Treatment
Treatment of acute HAV in pregnancy is supportive because of its benign nature; few patients require hospitalization.3
Pregnant patients with HAV can deliver vaginally, and breastfeeding is not contraindicated.8
HEPATITIS B
Hepatitis B virus (HBV) infection is a major global health problem. About 240 million people worldwide have chronic HBV infection, and more than 780,000 die every year from acute and chronic consequences.9
Vertical transmission is responsible for about half of chronic HBV infections worldwide. Thus, interruption of mother-to-child transmission is important. Universal maternal screening and passive-active immunoprophylaxis of newborns have lowered the transmission rates to between 5% and 10%. The 10% failure rate is unacceptably high and has been attributed to seropositivity for hepatitis B e antigen and a high viral load in the mother (ie, HBV DNA > 106 copies/mL). High viral load is an independent risk factor for failure of immunoprophylaxis.10 Therefore, antiviral therapy is suggested in pregnant women who have a high HBV viral load to further decrease the chance of mother-to-child transmission and to prevent failure of immunoprophylaxis.11
Clinical outcomes of HBV in pregnancy
Acute HBV infection during pregnancy is usually benign and is not associated with increased risk of death or teratogenicity.12 Symptomatic disease in the mother with acute hepatitis B includes nausea, vomiting, abdominal pain, fatigue, and jaundice.3 For the newborn, there is increased risk of low birth weight and prematurity.13
When acute HBV infection occurs early in pregnancy, the rate of perinatal transmission is about 10%, increasing to 60% if it occurs near delivery.12,13
Chronic HBV infection does not usually affect the outcome of pregnancy, but it may if the woman has cirrhosis or advanced liver disease14; however, pregnancy is very rare in women with HBV cirrhosis due to anovulation, and acute HBV flares have been described during pregnancy and postpartum.15
Pregnant patients with cirrhosis and portal hypertension are at risk of hepatic decompensation, variceal bleeding, and death.16 Risk is high with a score of 10 or more on the Model for End-stage Liver Disease scale, and is low with a score of 6 or less.17 Like nonpregnant patients, pregnant patients with cirrhosis should be monitored, and upper endoscopy should be performed in the 2nd trimester to assess for varices. A beta-blocker should be given or banding of varices should be done to avoid rupture. Rates of fetal demise, premature labor, spontaneous abortion, and stillbirth are high with portal hypertension.16
Risk of mother-to-child HBV transmission
Vertical HBV transmission can occur during the antepartum, intrapartum, and postpartum periods,18,19 but it most often occurs during the intrapartum period at the time of delivery. Without immunoprophylaxis of the newborn, the risk of mother-to-child transmission can be as high as 90% if the mother is hepatitis B e antigen-positive and has a viral load greater than 106 copies/mL. With active and passive immunoprophylaxis, the risk decreases substantially.
Screening and diagnosis
All pregnant women should be tested for hepatitis B surface antigen during the 1st trimester,20 or any time thereafter if early testing was not done, even if they were vaccinated before becoming pregnant.21
Prevention
HBV infection is best prevented before pregnancy by vaccinating the mother or, after delivery, by vaccinating the newborn. Universal vaccination of newborns has been the standard of care since the 1990s. Pregnant women should be tested early in the pregnancy; unvaccinated, uninfected women at high risk of acquiring HBV (eg, because of sexual contacts or intravenous drug use) should be vaccinated.2,3
HBV vaccine and immune globulin are both approved by the US Food and Drug Administration (FDA) for prevention of HBV infection and can be given during pregnancy and breastfeeding.3 All infants should be vaccinated for HBV at birth, and all infants born to mothers who test positive for hepatitis B s antigen should receive the HBV vaccine and the immune globulin within 12 to 24 hours after delivery. The vaccine series should be completed within 6 months.20,21 This will decrease the rate of neonatal infection.
Treatment of HBV infection in pregnancy
The main objectives of treating chronic HBV infection in pregnancy are to maintain stable liver function in the mother and to prevent neonatal infection, which may cause cirrhosis and hepatocellular carcinoma and contribute to the global burden of the disease.22 Therefore, maternal HBV DNA and liver aminotransferase levels should be tested regularly during gestation.
The current guidelines of the American Association for the Study of Liver Diseases suggest antiviral therapy to reduce the risk of perinatal transmission of HBV in pregnant women with an HBV DNA level greater than 200,000 IU/mL or greater than 106 copies/mL.23,24
In a meta-analysis,25 antiviral therapy with lamivudine, telbivudine, or tenofovir showed no apparent teratogenicity or safety concerns for maternal and fetal outcomes26 and significantly reduced the rate of mother-to-child transmission. Of these 3 drugs, telbivudine was associated with a higher rate of normalization of liver enzymes, HBV suppression, and e-antigen seroconversion.25 Lamivudine has proven the test of time in mothers co-infected with HBV and human immunodeficiency virus (HIV). However, tenofovir is considered the preferred treatment in pregnancy, owing to concerns about drug resistance to telbivudine and lamivudine and a high genetic barrier to resistance with tenofovir.26 In mothers with HBV and HIV treated with tenofovir, treatment was associated with lower bone mineral density in the newborns, with a propensity for renal injury in the mothers. No safety concerns for maternal or fetal outcomes were identified in pregnant women infected only with HBV.25
Many pregnant mothers choose to stop therapy around the time of conception because of safety concerns for the baby. In such situations, close monitoring is necessary to detect flares of HBV infection.
When the decision to treat is made, treatment should begin at 28 to 30 weeks of gestation, when organogenesis is complete and to allow enough time for HBV DNA levels to decline.
Breastfeeding is not contraindicated because antiviral drugs are minimally excreted in breast milk and are unlikely to cause toxicity. However, data are insufficient as to the long-term safety for the newborn when the mother took these drugs during pregancy and while breastfeeding.23,27
Alanine aminotransferase and HBV DNA levels should be monitored postpartum because of the possibility of a hepatitis flare. In this setting, any of the three drugs can be used.28 For mothers on therapy because of cirrhosis or an advanced histologic feature, antiviral therapy should be continued throughout pregnancy to prevent disease progression and decompensation.19,22,27
No drug therapy is necessary for pregnant carriers of HBV.
Delivery and breastfeeding
The mode of delivery does not appear to have a significant effect on the interruption of vertical transmission of HBV.29 Cesarean delivery is not recommended by the US Centers for Disease Control and Prevention (CDC)2 or the American College of Obstetricians and Gynecologists.6 Breastfeeding is encouraged if the infant has received appropriate immunoprophylaxis.6
Coinfection with hepatitis D
Coinfection with hepatitis D virus (HDV) and HBV is associated with severe acute hepatitis30,31 and increases the risk of death by a factor of 10. The World Health Organization recommends testing for HDV in pregnant women who are HBV-positive.8
Prevention of HDV infection requires prevention of HBV. The treatment of HDV in pregnancy is supportive. Pegylated interferon is successful outside pregnancy but is contraindicated during pregnancy.32 In patients with fulminant hepatic failure and end-stage liver disease, liver transplant can be lifesaving.
Take-home points
- HBV infection during pregnancy is usually benign and not severe but can be associated with an increased risk of mother-to-child transmission and progression of liver disease in the pregnant mother.
- Prevention of vertical transmission of HBV is important to reduce the burden of chronic HBV infection. Universal maternal screening early in pregnancy and passive-active immunoprophylaxis of newborns are usually sufficient to prevent vertical transmission of HBV, but antiviral therapy is needed for highly viremic mothers to further reduce the risk.
- Antiviral therapy is also indicated for pregnant women with moderate to severe hepatitis or cirrhosis to prevent disease progression and liver failure.
- Telbivudine, tenofovir, or lamivudine can be used during pregnancy, but more data are needed on the long-term safety of fetal exposure to these agents.
HEPATITIS C
The global prevalence of hepatitis C virus (HCV) infection is 2% to 3%, with 130 to 170 million HCV-positive people, most of whom are chronically infected.33 The incidence of HCV during pregnancy is 1% to 2.4%, but 3% to 5% of infected mothers transmit HCV to their child at the time of birth.6,34 Women coinfected with HIV and HCV have twice the risk of perinatal HCV transmission compared with women who have HCV infection alone.6,34
HCV infection is usually asymptomatic and is discovered either by screening high-risk patients or during evaluation of persistently elevated aminotransferase levels. Acute HCV infection during pregnancy has been reported only rarely, and most pregnant women who are infected have chronic disease with no effect on the pregnancy or the infant.6,34
Treatment
The CDC recommends that all adults (including pregnant women) born between 1945 and 1965 undergo 1-time testing for HCV without prior ascertainment of HCV risk (strong recommendation, with moderate quality of evidence).35 The most important risk factor for HCV infection is past or current injection drug use.33 Additional risk factors are similar to those for nonpregnant patients.
Because of the benign effect of HCV on the pregnancy, treatment is not recommended. To decrease the risk of maternal-child transmission, it is prudent to avoid amniocentesis, scalp instrumentation, and prolonged rupture of membranes.6
There is no vaccine or immune globulin for prevention. HCV infection should not influence the mode of delivery, and it is not a contraindication to breastfeeding.34,36,37
HEPATITIS E
Every year, 20 million cases of hepatitis E virus (HEV) infection are recorded worldwide. These numbers include 3.3 million symptomatic cases and 56,600 deaths.38 HEV infection is most common in developing countries, and pregnant women traveling to these areas are at high risk of acquiring this infection, of developing fulminant hepatitis, and of death.39 Sporadic cases not associated with travel are increasingly reported in developed countries and are attributed to immunocompromised status (due to HIV or solid-organ transplant).38,40
Modes of transmission of HEV are mainly via fecal-oral contamination and by vertical transmission.41
Diagnosis
HEV infection can be diagnosed either by detecting IgM antibody with an enzyme-linked immunosorbent assay or by detecting HEV RNA in the blood using reverse transcription polymerase chain reaction testing.42
Treatment and prevention
Hospitalization should be considered for pregnant women. Ribavirin or pegylated interferon alpha or both are effective but are contraindicated in pregnancy because of the risk of teratogenicity.41,42 Urgent liver transplant can be a successful option in acute liver failure.
Prevention relies primarily on good sanitation, clean drinking water, and avoiding raw pork and venison. Boiling and chlorination of water inactivate HEV.39,40 Pregnant women should be advised to avoid travel to highly endemic areas.
Viral hepatitis affects mother and child, and pregnancy can exacerbate the disease. Vertical transmission contributes significantly to the high prevalence of viral hepatitis and compromises the well-being and the prognosis in the newborn. The indications for therapy in pregnant women may differ from those in the general population, and new therapies are available.
HEPATITIS A
Hepatitis A virus (HAV) infection is associated with significant morbidity and death around the world, as 1.4 million cases are reported every year worldwide.1 However, in the United States, the prevalence has declined by 95% since HAV vaccination was introduced in 1995, and in 2013, the prevalence was 0.6 per 100,000 population.2 Acute HAV infection during pregnancy is rare. As a result, the incidence during pregnancy is difficult to ascertain.3
HAV is transmitted by the fecal-oral route from person to person contact and from contamination of food and water. Vertical transmission from pregnant mother to fetus has not been reported.4
Clinical outcomes of HAV in pregnancy
Acute HAV infection during pregnancy is rare, and teratogenicity associated with HAV has not been reported.3 The course of the disease during pregnancy is generally similar to that in nonpregnant patients, with excellent maternal and fetal outcomes in developed nations. There have been reports in developing nations of premature contractions and labor, placental separation, premature rupture of the membranes, and vaginal bleeding.5,6
Diagnosis
Routine screening for HAV is not recommended, but serologic testing by detection of anti-HAV immunoglobulin M (IgM) antibodies is done in high-risk patients suspected of having acute HAV infection.
Prevention
Prevention includes adherence to sanitary practices and active and passive immunoprophylaxis.3 Universal vaccination for pregnant mothers is not recommended,1,2,6 but vaccination is recommended for high-risk patients and mothers—those with chronic liver disease, those receiving clotting factors, those who use illegal drugs, and travelers to areas where HAV is endemic. Immune globulin is also available for postexposure prophylaxis. HAV vaccines and immune globulin are safe in pregnancy.3,6,7
Treatment
Treatment of acute HAV in pregnancy is supportive because of its benign nature; few patients require hospitalization.3
Pregnant patients with HAV can deliver vaginally, and breastfeeding is not contraindicated.8
HEPATITIS B
Hepatitis B virus (HBV) infection is a major global health problem. About 240 million people worldwide have chronic HBV infection, and more than 780,000 die every year from acute and chronic consequences.9
Vertical transmission is responsible for about half of chronic HBV infections worldwide. Thus, interruption of mother-to-child transmission is important. Universal maternal screening and passive-active immunoprophylaxis of newborns have lowered the transmission rates to between 5% and 10%. The 10% failure rate is unacceptably high and has been attributed to seropositivity for hepatitis B e antigen and a high viral load in the mother (ie, HBV DNA > 106 copies/mL). High viral load is an independent risk factor for failure of immunoprophylaxis.10 Therefore, antiviral therapy is suggested in pregnant women who have a high HBV viral load to further decrease the chance of mother-to-child transmission and to prevent failure of immunoprophylaxis.11
Clinical outcomes of HBV in pregnancy
Acute HBV infection during pregnancy is usually benign and is not associated with increased risk of death or teratogenicity.12 Symptomatic disease in the mother with acute hepatitis B includes nausea, vomiting, abdominal pain, fatigue, and jaundice.3 For the newborn, there is increased risk of low birth weight and prematurity.13
When acute HBV infection occurs early in pregnancy, the rate of perinatal transmission is about 10%, increasing to 60% if it occurs near delivery.12,13
Chronic HBV infection does not usually affect the outcome of pregnancy, but it may if the woman has cirrhosis or advanced liver disease14; however, pregnancy is very rare in women with HBV cirrhosis due to anovulation, and acute HBV flares have been described during pregnancy and postpartum.15
Pregnant patients with cirrhosis and portal hypertension are at risk of hepatic decompensation, variceal bleeding, and death.16 Risk is high with a score of 10 or more on the Model for End-stage Liver Disease scale, and is low with a score of 6 or less.17 Like nonpregnant patients, pregnant patients with cirrhosis should be monitored, and upper endoscopy should be performed in the 2nd trimester to assess for varices. A beta-blocker should be given or banding of varices should be done to avoid rupture. Rates of fetal demise, premature labor, spontaneous abortion, and stillbirth are high with portal hypertension.16
Risk of mother-to-child HBV transmission
Vertical HBV transmission can occur during the antepartum, intrapartum, and postpartum periods,18,19 but it most often occurs during the intrapartum period at the time of delivery. Without immunoprophylaxis of the newborn, the risk of mother-to-child transmission can be as high as 90% if the mother is hepatitis B e antigen-positive and has a viral load greater than 106 copies/mL. With active and passive immunoprophylaxis, the risk decreases substantially.
Screening and diagnosis
All pregnant women should be tested for hepatitis B surface antigen during the 1st trimester,20 or any time thereafter if early testing was not done, even if they were vaccinated before becoming pregnant.21
Prevention
HBV infection is best prevented before pregnancy by vaccinating the mother or, after delivery, by vaccinating the newborn. Universal vaccination of newborns has been the standard of care since the 1990s. Pregnant women should be tested early in the pregnancy; unvaccinated, uninfected women at high risk of acquiring HBV (eg, because of sexual contacts or intravenous drug use) should be vaccinated.2,3
HBV vaccine and immune globulin are both approved by the US Food and Drug Administration (FDA) for prevention of HBV infection and can be given during pregnancy and breastfeeding.3 All infants should be vaccinated for HBV at birth, and all infants born to mothers who test positive for hepatitis B s antigen should receive the HBV vaccine and the immune globulin within 12 to 24 hours after delivery. The vaccine series should be completed within 6 months.20,21 This will decrease the rate of neonatal infection.
Treatment of HBV infection in pregnancy
The main objectives of treating chronic HBV infection in pregnancy are to maintain stable liver function in the mother and to prevent neonatal infection, which may cause cirrhosis and hepatocellular carcinoma and contribute to the global burden of the disease.22 Therefore, maternal HBV DNA and liver aminotransferase levels should be tested regularly during gestation.
The current guidelines of the American Association for the Study of Liver Diseases suggest antiviral therapy to reduce the risk of perinatal transmission of HBV in pregnant women with an HBV DNA level greater than 200,000 IU/mL or greater than 106 copies/mL.23,24
In a meta-analysis,25 antiviral therapy with lamivudine, telbivudine, or tenofovir showed no apparent teratogenicity or safety concerns for maternal and fetal outcomes26 and significantly reduced the rate of mother-to-child transmission. Of these 3 drugs, telbivudine was associated with a higher rate of normalization of liver enzymes, HBV suppression, and e-antigen seroconversion.25 Lamivudine has proven the test of time in mothers co-infected with HBV and human immunodeficiency virus (HIV). However, tenofovir is considered the preferred treatment in pregnancy, owing to concerns about drug resistance to telbivudine and lamivudine and a high genetic barrier to resistance with tenofovir.26 In mothers with HBV and HIV treated with tenofovir, treatment was associated with lower bone mineral density in the newborns, with a propensity for renal injury in the mothers. No safety concerns for maternal or fetal outcomes were identified in pregnant women infected only with HBV.25
Many pregnant mothers choose to stop therapy around the time of conception because of safety concerns for the baby. In such situations, close monitoring is necessary to detect flares of HBV infection.
When the decision to treat is made, treatment should begin at 28 to 30 weeks of gestation, when organogenesis is complete and to allow enough time for HBV DNA levels to decline.
Breastfeeding is not contraindicated because antiviral drugs are minimally excreted in breast milk and are unlikely to cause toxicity. However, data are insufficient as to the long-term safety for the newborn when the mother took these drugs during pregancy and while breastfeeding.23,27
Alanine aminotransferase and HBV DNA levels should be monitored postpartum because of the possibility of a hepatitis flare. In this setting, any of the three drugs can be used.28 For mothers on therapy because of cirrhosis or an advanced histologic feature, antiviral therapy should be continued throughout pregnancy to prevent disease progression and decompensation.19,22,27
No drug therapy is necessary for pregnant carriers of HBV.
Delivery and breastfeeding
The mode of delivery does not appear to have a significant effect on the interruption of vertical transmission of HBV.29 Cesarean delivery is not recommended by the US Centers for Disease Control and Prevention (CDC)2 or the American College of Obstetricians and Gynecologists.6 Breastfeeding is encouraged if the infant has received appropriate immunoprophylaxis.6
Coinfection with hepatitis D
Coinfection with hepatitis D virus (HDV) and HBV is associated with severe acute hepatitis30,31 and increases the risk of death by a factor of 10. The World Health Organization recommends testing for HDV in pregnant women who are HBV-positive.8
Prevention of HDV infection requires prevention of HBV. The treatment of HDV in pregnancy is supportive. Pegylated interferon is successful outside pregnancy but is contraindicated during pregnancy.32 In patients with fulminant hepatic failure and end-stage liver disease, liver transplant can be lifesaving.
Take-home points
- HBV infection during pregnancy is usually benign and not severe but can be associated with an increased risk of mother-to-child transmission and progression of liver disease in the pregnant mother.
- Prevention of vertical transmission of HBV is important to reduce the burden of chronic HBV infection. Universal maternal screening early in pregnancy and passive-active immunoprophylaxis of newborns are usually sufficient to prevent vertical transmission of HBV, but antiviral therapy is needed for highly viremic mothers to further reduce the risk.
- Antiviral therapy is also indicated for pregnant women with moderate to severe hepatitis or cirrhosis to prevent disease progression and liver failure.
- Telbivudine, tenofovir, or lamivudine can be used during pregnancy, but more data are needed on the long-term safety of fetal exposure to these agents.
HEPATITIS C
The global prevalence of hepatitis C virus (HCV) infection is 2% to 3%, with 130 to 170 million HCV-positive people, most of whom are chronically infected.33 The incidence of HCV during pregnancy is 1% to 2.4%, but 3% to 5% of infected mothers transmit HCV to their child at the time of birth.6,34 Women coinfected with HIV and HCV have twice the risk of perinatal HCV transmission compared with women who have HCV infection alone.6,34
HCV infection is usually asymptomatic and is discovered either by screening high-risk patients or during evaluation of persistently elevated aminotransferase levels. Acute HCV infection during pregnancy has been reported only rarely, and most pregnant women who are infected have chronic disease with no effect on the pregnancy or the infant.6,34
Treatment
The CDC recommends that all adults (including pregnant women) born between 1945 and 1965 undergo 1-time testing for HCV without prior ascertainment of HCV risk (strong recommendation, with moderate quality of evidence).35 The most important risk factor for HCV infection is past or current injection drug use.33 Additional risk factors are similar to those for nonpregnant patients.
Because of the benign effect of HCV on the pregnancy, treatment is not recommended. To decrease the risk of maternal-child transmission, it is prudent to avoid amniocentesis, scalp instrumentation, and prolonged rupture of membranes.6
There is no vaccine or immune globulin for prevention. HCV infection should not influence the mode of delivery, and it is not a contraindication to breastfeeding.34,36,37
HEPATITIS E
Every year, 20 million cases of hepatitis E virus (HEV) infection are recorded worldwide. These numbers include 3.3 million symptomatic cases and 56,600 deaths.38 HEV infection is most common in developing countries, and pregnant women traveling to these areas are at high risk of acquiring this infection, of developing fulminant hepatitis, and of death.39 Sporadic cases not associated with travel are increasingly reported in developed countries and are attributed to immunocompromised status (due to HIV or solid-organ transplant).38,40
Modes of transmission of HEV are mainly via fecal-oral contamination and by vertical transmission.41
Diagnosis
HEV infection can be diagnosed either by detecting IgM antibody with an enzyme-linked immunosorbent assay or by detecting HEV RNA in the blood using reverse transcription polymerase chain reaction testing.42
Treatment and prevention
Hospitalization should be considered for pregnant women. Ribavirin or pegylated interferon alpha or both are effective but are contraindicated in pregnancy because of the risk of teratogenicity.41,42 Urgent liver transplant can be a successful option in acute liver failure.
Prevention relies primarily on good sanitation, clean drinking water, and avoiding raw pork and venison. Boiling and chlorination of water inactivate HEV.39,40 Pregnant women should be advised to avoid travel to highly endemic areas.
- World Health Organization (WHO).Hepatitis A fact sheet. www.who.int/mediacentre/factsheets/fs328/en/. Accessed December 7, 2016.
- US Centers for Disease Control and Prevention (CDC). Viral hepatitis—statistics & surveillance. www.cdc.gov/hepatitis/statistics/2013surveillance/commentary.htm#hepatitis A. Accessed December 7, 2016.
- Rac MW, Sheffield JS. Prevention and management of viral hepatitis in pregnancy. Obstet Gynecol Clin North Am 2014; 41:573–592.
- Workowski KA, Bolan GA; Centers for Disease Control and Prevention (CDC). Sexually transmitted diseases treatment guidelines, 2015. MMWR Recomm Rep 2015; 64:1–137.
- Elinav E, Ben-Dov IZ, Shapira Y, et al. Acute hepatitis A infection in pregnancy is associated with high rates of gestational complications and preterm labor. Gastroenterology 2006; 130:1129–1134.
- American College of Obstetricians and Gynecologists. ACOG Practice Bulletin No. 86: Viral hepatitis in pregnancy. Obstet Gynecol 2007; 110:941–956.
- Advisory Committee on Immunization Practices. Guidelines for Vaccinating Pregnant Women. www.cdc.gov/vaccines/pregnancy/hcp/guidelines.html. Accessed December 7, 2016.
- Daudi N, Shouval D, Stein-Zamir C, Ackerman Z. Breastmilk hepatitis A virus RNA in nursing mothers with acute hepatitis A virus infection. Breastfeed Med 2012; 7:313–315.
- World Health Organization (WHO). Hepatitis B fact sheet. www.who.int/mediacentre/factsheets/fs204/en/. Accessed December 7, 2016.
- Zou H, Chen Y, Duan Z, Zhang H, Pan C. Virologic factors associated with failure to passive-active immunoprophylaxis in infants born to HBsAg-positive mothers. J Viral Hepat 2012; 19:e18–e25.
- Pan CQ, Lee HM. Antiviral therapy for chronic hepatitis B in pregnancy. Semin Liver Dis 2013; 33:138–146.
- Sookoian S. Liver disease during pregnancy: acute viral hepatitis. Ann Hepatol 2006; 5:231–236.
- Jonas MM. Hepatitis B and pregnancy: an underestimated issue. Liver Int 2009; 29(suppl 1):133–139.
- Wong S, Chan LY, Yu V, Ho L. Hepatitis B carrier and perinatal outcome in singleton pregnancy. Am J Perinatol 1999; 16:485–488.
- Rawal BK, Parida S, Watkins RP, Ghosh P, Smith H. Symptomatic reactivation of hepatitis B in pregnancy. Lancet 1991; 337:364.
- Aggarwal N, Negi N, Aggarwal A, Bodh V, Dhiman RK. Pregnancy with portal hypertension. J Clin Exp Hepatol 2014; 4:163–171.
- Westbrook RH, Yeoman AD, O'Grady JG, Harrison PM, Devlin J, Heneghan MA. Model for end-stage liver disease score predicts outcome in cirrhotic patients during pregnancy. Clin Gastroenterol Hepatol 2011; 9:694–699.
- Cheung KW, Seto MT, Wong SF. Towards complete eradication of hepatitis B infection from perinatal transmission: review of the mechanisms of in utero infection and the use of antiviral treatment during pregnancy. Eur J Obstet Gynecol Reproduct Biol 2013; 169:17–23.
- Pan CQ, Duan AP, Bhamidimarri KR, et al. An algorithm for risk assessment and intervention of mother to child transmission of hepatitis B virus. Clin Gastroenterol Hepatol 2012; 10:452–459.
- Mast EE, Margolis HS, Fiore AE, et al; Advisory Committee on Immunization Practices (ACIP). A comprehensive immunization strategy to eliminate transmission of hepatitis B virus infection in the United States: recommendations of the Advisory Committee on Immunization Practices (ACIP) part 1: immunization of infants, children, and adolescents. MMWR Recomm Rep 2005; 54:1–31.
- US Centers for Disease Control (CDC). Prevention of perinatal transmission of hepatitis B virus: prenatal screening of all pregnant women for hepatitis B surface antigen. MMWR Morb Mortal Wkly Rep 1988; 37:341–346, 351.
- Han GR, Xu CL, Zhao W, Yang YF. Management of chronic hepatitis B in pregnancy. World J Gastroenterol 2012; 18:4517–4521.
- Terrault NA, Bzowej NH, Chang KM, Hwang JP, Jonas MM, Murad MH; American Association for the Study of Liver Diseases. AASLD guidelines for treatment of chronic hepatitis B. Hepatology 2016; 63:261–283.
- Tran TT, Ahn J, Reau N. ACG clinical guideline: liver disease and pregnancy. Am J Gastroenterol 2016: 111:176–194.
- Brown RS Jr, McMahon BJ, Lok AS, et al. Antiviral therapy in chronic hepatitis B viral infection during pregnancy: a systematic review and meta-analysis. Hepatol 2016; 63:319–333.
- Brown RS Jr, Verna EC, Pereira MR, et al. Hepatitis B virus and human immunodeficiency virus drugs in pregnancy: findings from the antiretroviral pregnancy registry. J Hepatol 2012; 57:953–959.
- Lamberth JR, Reddy SC, Pan JJ, Dasher KJ. Chronic hepatitis B infection in pregnancy. World J Hepatol 2015; 7:1233–1237.
- Potthoff A, Rifai K, Wedemeyer H, Deterding K, Manns M, Strassburg C. Successful treatment of fulminant hepatitis B during pregnancy. Z Gastroenterol 2009; 47:667–670.
- Yang J, Zeng XM, Men YL, Zhao LS. Elective caesarean section versus vaginal delivery for preventing mother to child transmission of hepatitis B virus—a systematic review. Virol J 2008; 5:100.
- Price J. An update on hepatitis B, D, and E viruses. Top Antivir Med 2014; 21:157–163.
- World Health Organization (WHO). Global alert and response. Hepatitis Delta. www.who.int/csr/resources/publications/hepatitis/who_cds_csr_ncs_2001_1/en/. Accessed December 7, 2016.
- Abbas Z, Memon MS, Mithani H, Jafri W, Hamid S. Treatment of chronic hepatitis D patients with pegylated interferon: a real-world experience. Antivir Ther 2014; 19:463–468.
- Baldo V, Baldovin T, Trivello R, Floreani A. Epidemiology of HCV infection. Curr Pharm Des 2008; 14:1646–1654.
- Floreani A. Hepatitis C and pregnancy. World J Gastroenterol 2013; 19:6714–6720.
- US Centers for Disease Control and Prevention. Viral hepatitis—CDC recommendations for specific populations and settings. www.cdc.gov/hepatitis/populations/1945-1965.htm. Accessed December 7, 2016.
- World Health Organization (WHO). Hepatitis C fact sheet. www.who.int/mediacentre/factsheets/fs164/en/. Accessed December 7, 2016.
- Moyer VA; US Preventive Services Task Force. Screening for hepatitis C virus infection in adults: US Preventive Services Task Force Recommendation Statement. Ann Intern Med 2013; 159:349–357.
- World Health Organization (WHO). Hepatitis E fact sheet. www.who.int/mediacentre/factsheets/fs280/en/. Accessed December 7, 2016.
- Velosa M, Figueiredo A, Gloria H, et al. Fulminant hepatitis E in a pregnant woman. GE Port J Gastroenterol 2013; 20:210–214.
- US Centers for Disease Control and Prevention (CDC). Viral hepatitis—hepatitis E information. Hepatitis E FAQs for health professionals. www.cdc.gov/hepatitis/hev/hevfaq.htm. Accessed December 7, 2016.
- Peters van Ton AM, Gevers TJ, Drenth JP. Antiviral therapy in chronic hepatitis E: a systematic review. J Viral Hepat 2015; 22:965–973.
- Kamar N, Bendall R, Legrand-Abravanel F, et al. Hepatitis E. Lancet 2012: 379:2477–2488.
- World Health Organization (WHO).Hepatitis A fact sheet. www.who.int/mediacentre/factsheets/fs328/en/. Accessed December 7, 2016.
- US Centers for Disease Control and Prevention (CDC). Viral hepatitis—statistics & surveillance. www.cdc.gov/hepatitis/statistics/2013surveillance/commentary.htm#hepatitis A. Accessed December 7, 2016.
- Rac MW, Sheffield JS. Prevention and management of viral hepatitis in pregnancy. Obstet Gynecol Clin North Am 2014; 41:573–592.
- Workowski KA, Bolan GA; Centers for Disease Control and Prevention (CDC). Sexually transmitted diseases treatment guidelines, 2015. MMWR Recomm Rep 2015; 64:1–137.
- Elinav E, Ben-Dov IZ, Shapira Y, et al. Acute hepatitis A infection in pregnancy is associated with high rates of gestational complications and preterm labor. Gastroenterology 2006; 130:1129–1134.
- American College of Obstetricians and Gynecologists. ACOG Practice Bulletin No. 86: Viral hepatitis in pregnancy. Obstet Gynecol 2007; 110:941–956.
- Advisory Committee on Immunization Practices. Guidelines for Vaccinating Pregnant Women. www.cdc.gov/vaccines/pregnancy/hcp/guidelines.html. Accessed December 7, 2016.
- Daudi N, Shouval D, Stein-Zamir C, Ackerman Z. Breastmilk hepatitis A virus RNA in nursing mothers with acute hepatitis A virus infection. Breastfeed Med 2012; 7:313–315.
- World Health Organization (WHO). Hepatitis B fact sheet. www.who.int/mediacentre/factsheets/fs204/en/. Accessed December 7, 2016.
- Zou H, Chen Y, Duan Z, Zhang H, Pan C. Virologic factors associated with failure to passive-active immunoprophylaxis in infants born to HBsAg-positive mothers. J Viral Hepat 2012; 19:e18–e25.
- Pan CQ, Lee HM. Antiviral therapy for chronic hepatitis B in pregnancy. Semin Liver Dis 2013; 33:138–146.
- Sookoian S. Liver disease during pregnancy: acute viral hepatitis. Ann Hepatol 2006; 5:231–236.
- Jonas MM. Hepatitis B and pregnancy: an underestimated issue. Liver Int 2009; 29(suppl 1):133–139.
- Wong S, Chan LY, Yu V, Ho L. Hepatitis B carrier and perinatal outcome in singleton pregnancy. Am J Perinatol 1999; 16:485–488.
- Rawal BK, Parida S, Watkins RP, Ghosh P, Smith H. Symptomatic reactivation of hepatitis B in pregnancy. Lancet 1991; 337:364.
- Aggarwal N, Negi N, Aggarwal A, Bodh V, Dhiman RK. Pregnancy with portal hypertension. J Clin Exp Hepatol 2014; 4:163–171.
- Westbrook RH, Yeoman AD, O'Grady JG, Harrison PM, Devlin J, Heneghan MA. Model for end-stage liver disease score predicts outcome in cirrhotic patients during pregnancy. Clin Gastroenterol Hepatol 2011; 9:694–699.
- Cheung KW, Seto MT, Wong SF. Towards complete eradication of hepatitis B infection from perinatal transmission: review of the mechanisms of in utero infection and the use of antiviral treatment during pregnancy. Eur J Obstet Gynecol Reproduct Biol 2013; 169:17–23.
- Pan CQ, Duan AP, Bhamidimarri KR, et al. An algorithm for risk assessment and intervention of mother to child transmission of hepatitis B virus. Clin Gastroenterol Hepatol 2012; 10:452–459.
- Mast EE, Margolis HS, Fiore AE, et al; Advisory Committee on Immunization Practices (ACIP). A comprehensive immunization strategy to eliminate transmission of hepatitis B virus infection in the United States: recommendations of the Advisory Committee on Immunization Practices (ACIP) part 1: immunization of infants, children, and adolescents. MMWR Recomm Rep 2005; 54:1–31.
- US Centers for Disease Control (CDC). Prevention of perinatal transmission of hepatitis B virus: prenatal screening of all pregnant women for hepatitis B surface antigen. MMWR Morb Mortal Wkly Rep 1988; 37:341–346, 351.
- Han GR, Xu CL, Zhao W, Yang YF. Management of chronic hepatitis B in pregnancy. World J Gastroenterol 2012; 18:4517–4521.
- Terrault NA, Bzowej NH, Chang KM, Hwang JP, Jonas MM, Murad MH; American Association for the Study of Liver Diseases. AASLD guidelines for treatment of chronic hepatitis B. Hepatology 2016; 63:261–283.
- Tran TT, Ahn J, Reau N. ACG clinical guideline: liver disease and pregnancy. Am J Gastroenterol 2016: 111:176–194.
- Brown RS Jr, McMahon BJ, Lok AS, et al. Antiviral therapy in chronic hepatitis B viral infection during pregnancy: a systematic review and meta-analysis. Hepatol 2016; 63:319–333.
- Brown RS Jr, Verna EC, Pereira MR, et al. Hepatitis B virus and human immunodeficiency virus drugs in pregnancy: findings from the antiretroviral pregnancy registry. J Hepatol 2012; 57:953–959.
- Lamberth JR, Reddy SC, Pan JJ, Dasher KJ. Chronic hepatitis B infection in pregnancy. World J Hepatol 2015; 7:1233–1237.
- Potthoff A, Rifai K, Wedemeyer H, Deterding K, Manns M, Strassburg C. Successful treatment of fulminant hepatitis B during pregnancy. Z Gastroenterol 2009; 47:667–670.
- Yang J, Zeng XM, Men YL, Zhao LS. Elective caesarean section versus vaginal delivery for preventing mother to child transmission of hepatitis B virus—a systematic review. Virol J 2008; 5:100.
- Price J. An update on hepatitis B, D, and E viruses. Top Antivir Med 2014; 21:157–163.
- World Health Organization (WHO). Global alert and response. Hepatitis Delta. www.who.int/csr/resources/publications/hepatitis/who_cds_csr_ncs_2001_1/en/. Accessed December 7, 2016.
- Abbas Z, Memon MS, Mithani H, Jafri W, Hamid S. Treatment of chronic hepatitis D patients with pegylated interferon: a real-world experience. Antivir Ther 2014; 19:463–468.
- Baldo V, Baldovin T, Trivello R, Floreani A. Epidemiology of HCV infection. Curr Pharm Des 2008; 14:1646–1654.
- Floreani A. Hepatitis C and pregnancy. World J Gastroenterol 2013; 19:6714–6720.
- US Centers for Disease Control and Prevention. Viral hepatitis—CDC recommendations for specific populations and settings. www.cdc.gov/hepatitis/populations/1945-1965.htm. Accessed December 7, 2016.
- World Health Organization (WHO). Hepatitis C fact sheet. www.who.int/mediacentre/factsheets/fs164/en/. Accessed December 7, 2016.
- Moyer VA; US Preventive Services Task Force. Screening for hepatitis C virus infection in adults: US Preventive Services Task Force Recommendation Statement. Ann Intern Med 2013; 159:349–357.
- World Health Organization (WHO). Hepatitis E fact sheet. www.who.int/mediacentre/factsheets/fs280/en/. Accessed December 7, 2016.
- Velosa M, Figueiredo A, Gloria H, et al. Fulminant hepatitis E in a pregnant woman. GE Port J Gastroenterol 2013; 20:210–214.
- US Centers for Disease Control and Prevention (CDC). Viral hepatitis—hepatitis E information. Hepatitis E FAQs for health professionals. www.cdc.gov/hepatitis/hev/hevfaq.htm. Accessed December 7, 2016.
- Peters van Ton AM, Gevers TJ, Drenth JP. Antiviral therapy in chronic hepatitis E: a systematic review. J Viral Hepat 2015; 22:965–973.
- Kamar N, Bendall R, Legrand-Abravanel F, et al. Hepatitis E. Lancet 2012: 379:2477–2488.
KEY POINTS
- Preventing vertical transmission of HBV infection in pregnancy is key to decreasing the global burden of this infection. Universal maternal screening and passive-active immunoprophylaxis of newborns have reduced transmission of HBV, but the addition of antiviral therapy is necessary to further decrease immunoprophylaxis failure.
- Tenofovir, telbivudine, and lamivudine can be used safely in pregnancy without apparent teratogenicity or other harmful effects on mother or baby. But optimal outcome requires discussion of safety and the plan of care with the patient, obstetrician, and hepatologist.
- Most pregnant women with hepatitis C virus (HCV) infection have chronic disease, with no effects on the pregnancy or baby, but 3% to 5% transmit HCV to their child at the time of birth. All pregnant women at risk should be screened at the first prenatal visit. The safety and efficacy of treating pregnant women to prevent transmission to the fetus are not established; thus, treatment is not recommended for pregnant women.
Porcelain heart in a uremic patient
A 58-year-old man with end-stage renal disease due to diabetic nephropathy was admitted with aggravated exertional dyspnea and intermittent chest pain for 1 week. He had been on hemodialysis for 15 years.
His blood pressure was 124/69 mm Hg, pulse 96 beats per minute, and temperature 35.8°C. On physical examination, he had bilateral diffuse crackles, elevated jugular venous pressure (9.5 cm H2O) with positive hepatojugular reflux, and apparent dependent pedal edema. The Kussmaul sign was not observed.
Cardiac enzymes were in the normal range (creatine kinase 73 U/L, troponin I 0.032 ng/mL), but the brain-natriuretic peptide level was elevated at 340 pg/mL. Other laboratory findings included calcium 9 mg/dL (reference range 8.4–10.2 mg/dL), inorganic phosphate 5 mg/dL (2.5–4.5 mg/dL), and intact parathyroid hormone 1,457 pg/mL (10–69 pg/mL).

Electrocardiography showed sinus tachycardia with low voltage in diffuse leads and generalized flattening of the T wave. Chest radiography showed a bilateral reticulonodular pattern, mild costophrenic angle obliteration, and notable calcifications along the cardiac contour. Thoracic computed tomography showed a porcelain-like encasement of the heart (Figure 1). Transthoracic echocardiography showed thickened pericardium, pericardial calcification, and mild interventricular septal bounce in diastole, with no dyskinesia of ventricular wall motion. We decided not to perform an invasive hemodynamic assessment.
CAUSES OF PERICARDIAL CALCIFICATION
Pericardial calcification, abnormal calcium deposits in response to inflammation,1 has become more widely reported as the use of chest computed tomography has become more widespread. The common identifiable causes of pericardial calcification include recurrent or chronic pericarditis, radiation therapy for Hodgkin lymphoma or breast cancer, tuberculosis, and end-stage kidney disease.2,3 Other possible causes are retention of uremic metabolites, metastatic calcification induced by secondary hyperparathyroidism, and calcium-phosphate deposition induced by hyperphosphatemia.4
In chronic kidney disease, the amount of pericardial fluid and fibrinous pericardial deposition is thought to contribute to increased pericardial thickness and constriction. In some patients, pericardial calcification and thickening would lead to constrictive pericarditis, which could be confirmed by echocardiography and cardiac catheterization. About 25% to 50% of cases of pericardial calcification are complicated by constrictive pericarditis.5,6 Constrictive pericarditis occurs in up to 4% of patients with end-stage renal disease, even with successful dialysis.7
Partial clinical improvement may be obtained with intensive hemodialysis, strict volume control, and decreased catabolism in patients with multiple comorbidities.8 However, the definite treatment is total pericardiectomy, which reduces symptoms substantially and offers a favorable long-term outcome.7
SECONDARY HYPERPARATHYROIDISM
Secondary hyperparathyroidism is a common complication in patients with end-stage renal disease and is characterized by derangements in the homeostasis of calcium, phosphorus, and vitamin D.9
Because renal function is decreased, phosphate is retained and calcitriol synthesis is reduced, resulting in hypocalcemia, which induces parathyroid gland hyperplasia and parathyroid hormone secretion.10 Moreover, some patents with long-standing secondary hyperparathyroidism may develop tertiary hyperparathyroidism associated with autonomous parathyroid hormone secretion, hypercalcemia, and hyperphosphatemia.11
The Kidney Disease: Improving Global Outcomes (KDIGO) Work Group recommends screening for and managing secondary hyperparathyroidism in all patients with stage 3 chronic kidney disease (estimated glomerular filtration rate < 60 mL/min). In patients with stage 5 chronic kidney disease or on dialysis, the serum calcium and phosphorus levels should be monitored every 1 to 3 months and the parathyroid hormone levels every 3 to 6 months.12
According to KDIGO guidelines, the target level of calcium is less than 10.2 mg/dL, and the target phosphorus level is less than 4.6 mg/dL. The level of parathyroid hormone should be maintained at 2 to 9 times the upper limit of normal for the assay.
The management of secondary hyperparathyroidism includes a low-phosphorus diet, calcium-containing or calcium-free phosphate binders, a calcitriol supplement, and calcimimetics. If medical treatment fails and manifestations are significant, parathyroidectomy may be indicated.13
- Alpert MA, Ravenscraft MD. Pericardial involvement in end-stage renal disease. Am J Med Sci 2003; 325:228–236.
- Gowda RM, Boxt LM. Calcifications of the heart. Radiol Clin North Am 2004; 42:603–617.
- Kleynberg RL, Kleynberg VM, Kleynberg LM, Farahmandian D. Chronic constrictive pericarditis in association with end-stage renal disease. Int J Nephrol 2011; 2011:469602.
- Rao N, Crail S. Metastatic calcification and long-term hemodialysis. N Engl J Med 2013; 368:2415.
- Ling LH, Oh JK, Schaff HV, et al. Constrictive pericarditis in the modern era: evolving clinical spectrum and impact on outcome after pericardiectomy. Circulation 1999; 100:1380–1386.
- Bergman M, Vitrai J, Salman H. Constrictive pericarditis: a reminder of a not so rare disease. Eur J Intern Med 2006; 17:457–464.
- Szabó G, Schmack B, Bulut C, et al. Constrictive pericarditis: risks, aetiologies and outcomes after total pericardiectomy: 24 years of experience. Eur J Cardiothorac Surg 2013; 44:1023–1028.
- Feldman V, Dovrish Z, Weisenberg N, Neuman Y, Amital H. Uremic pericarditis. Isr Med Assoc J 2011; 13:256–257.
- Levin A, Bakris GL, Molitch M, et al. Prevalence of abnormal serum vitamin D, PTH, calcium, and phosphorus in patients with chronic kidney disease: results of the study to evaluate early kidney disease. Kidney Int 2007; 71:31–38.
- Martin KJ, Gonzalez EA. Metabolic bone disease in chronic kidney disease. J Am Soc Nephrol 2007; 18:875–885.
- Kerby J, Rue LW, Blair H, Hudson S, Sellers MT, Diethelm AG. Operative treatment of tertiary hyperparathyroidism: a single-center experience. Ann Surg 1998; 227:878–886.
- Kidney Disease: Improving Global Outcomes (KDIGO) CKDMBD Work Group. KDIGO clinical practice guideline for the diagnosis, evaluation, prevention, and treatment of chronic kidney disease—mineral and bone disorder (CKD-MBD). Kidney Int Suppl 2009; 76:S1–130.
- National Kidney Foundation. K/DOQI clinical practice guidelines for bone metabolism and disease in chronic kidney disease. Am J Kidney Dis 2003; 42(4 suppl 3):S1–201.
A 58-year-old man with end-stage renal disease due to diabetic nephropathy was admitted with aggravated exertional dyspnea and intermittent chest pain for 1 week. He had been on hemodialysis for 15 years.
His blood pressure was 124/69 mm Hg, pulse 96 beats per minute, and temperature 35.8°C. On physical examination, he had bilateral diffuse crackles, elevated jugular venous pressure (9.5 cm H2O) with positive hepatojugular reflux, and apparent dependent pedal edema. The Kussmaul sign was not observed.
Cardiac enzymes were in the normal range (creatine kinase 73 U/L, troponin I 0.032 ng/mL), but the brain-natriuretic peptide level was elevated at 340 pg/mL. Other laboratory findings included calcium 9 mg/dL (reference range 8.4–10.2 mg/dL), inorganic phosphate 5 mg/dL (2.5–4.5 mg/dL), and intact parathyroid hormone 1,457 pg/mL (10–69 pg/mL).
Electrocardiography showed sinus tachycardia with low voltage in diffuse leads and generalized flattening of the T wave. Chest radiography showed a bilateral reticulonodular pattern, mild costophrenic angle obliteration, and notable calcifications along the cardiac contour. Thoracic computed tomography showed a porcelain-like encasement of the heart (Figure 1). Transthoracic echocardiography showed thickened pericardium, pericardial calcification, and mild interventricular septal bounce in diastole, with no dyskinesia of ventricular wall motion. We decided not to perform an invasive hemodynamic assessment.
CAUSES OF PERICARDIAL CALCIFICATION
Pericardial calcification, abnormal calcium deposits in response to inflammation,1 has become more widely reported as the use of chest computed tomography has become more widespread. The common identifiable causes of pericardial calcification include recurrent or chronic pericarditis, radiation therapy for Hodgkin lymphoma or breast cancer, tuberculosis, and end-stage kidney disease.2,3 Other possible causes are retention of uremic metabolites, metastatic calcification induced by secondary hyperparathyroidism, and calcium-phosphate deposition induced by hyperphosphatemia.4
In chronic kidney disease, the amount of pericardial fluid and fibrinous pericardial deposition is thought to contribute to increased pericardial thickness and constriction. In some patients, pericardial calcification and thickening would lead to constrictive pericarditis, which could be confirmed by echocardiography and cardiac catheterization. About 25% to 50% of cases of pericardial calcification are complicated by constrictive pericarditis.5,6 Constrictive pericarditis occurs in up to 4% of patients with end-stage renal disease, even with successful dialysis.7
Partial clinical improvement may be obtained with intensive hemodialysis, strict volume control, and decreased catabolism in patients with multiple comorbidities.8 However, the definite treatment is total pericardiectomy, which reduces symptoms substantially and offers a favorable long-term outcome.7
SECONDARY HYPERPARATHYROIDISM
Secondary hyperparathyroidism is a common complication in patients with end-stage renal disease and is characterized by derangements in the homeostasis of calcium, phosphorus, and vitamin D.9
Because renal function is decreased, phosphate is retained and calcitriol synthesis is reduced, resulting in hypocalcemia, which induces parathyroid gland hyperplasia and parathyroid hormone secretion.10 Moreover, some patents with long-standing secondary hyperparathyroidism may develop tertiary hyperparathyroidism associated with autonomous parathyroid hormone secretion, hypercalcemia, and hyperphosphatemia.11
The Kidney Disease: Improving Global Outcomes (KDIGO) Work Group recommends screening for and managing secondary hyperparathyroidism in all patients with stage 3 chronic kidney disease (estimated glomerular filtration rate < 60 mL/min). In patients with stage 5 chronic kidney disease or on dialysis, the serum calcium and phosphorus levels should be monitored every 1 to 3 months and the parathyroid hormone levels every 3 to 6 months.12
According to KDIGO guidelines, the target level of calcium is less than 10.2 mg/dL, and the target phosphorus level is less than 4.6 mg/dL. The level of parathyroid hormone should be maintained at 2 to 9 times the upper limit of normal for the assay.
The management of secondary hyperparathyroidism includes a low-phosphorus diet, calcium-containing or calcium-free phosphate binders, a calcitriol supplement, and calcimimetics. If medical treatment fails and manifestations are significant, parathyroidectomy may be indicated.13
A 58-year-old man with end-stage renal disease due to diabetic nephropathy was admitted with aggravated exertional dyspnea and intermittent chest pain for 1 week. He had been on hemodialysis for 15 years.
His blood pressure was 124/69 mm Hg, pulse 96 beats per minute, and temperature 35.8°C. On physical examination, he had bilateral diffuse crackles, elevated jugular venous pressure (9.5 cm H2O) with positive hepatojugular reflux, and apparent dependent pedal edema. The Kussmaul sign was not observed.
Cardiac enzymes were in the normal range (creatine kinase 73 U/L, troponin I 0.032 ng/mL), but the brain-natriuretic peptide level was elevated at 340 pg/mL. Other laboratory findings included calcium 9 mg/dL (reference range 8.4–10.2 mg/dL), inorganic phosphate 5 mg/dL (2.5–4.5 mg/dL), and intact parathyroid hormone 1,457 pg/mL (10–69 pg/mL).
Electrocardiography showed sinus tachycardia with low voltage in diffuse leads and generalized flattening of the T wave. Chest radiography showed a bilateral reticulonodular pattern, mild costophrenic angle obliteration, and notable calcifications along the cardiac contour. Thoracic computed tomography showed a porcelain-like encasement of the heart (Figure 1). Transthoracic echocardiography showed thickened pericardium, pericardial calcification, and mild interventricular septal bounce in diastole, with no dyskinesia of ventricular wall motion. We decided not to perform an invasive hemodynamic assessment.
CAUSES OF PERICARDIAL CALCIFICATION
Pericardial calcification, abnormal calcium deposits in response to inflammation,1 has become more widely reported as the use of chest computed tomography has become more widespread. The common identifiable causes of pericardial calcification include recurrent or chronic pericarditis, radiation therapy for Hodgkin lymphoma or breast cancer, tuberculosis, and end-stage kidney disease.2,3 Other possible causes are retention of uremic metabolites, metastatic calcification induced by secondary hyperparathyroidism, and calcium-phosphate deposition induced by hyperphosphatemia.4
In chronic kidney disease, the amount of pericardial fluid and fibrinous pericardial deposition is thought to contribute to increased pericardial thickness and constriction. In some patients, pericardial calcification and thickening would lead to constrictive pericarditis, which could be confirmed by echocardiography and cardiac catheterization. About 25% to 50% of cases of pericardial calcification are complicated by constrictive pericarditis.5,6 Constrictive pericarditis occurs in up to 4% of patients with end-stage renal disease, even with successful dialysis.7
Partial clinical improvement may be obtained with intensive hemodialysis, strict volume control, and decreased catabolism in patients with multiple comorbidities.8 However, the definite treatment is total pericardiectomy, which reduces symptoms substantially and offers a favorable long-term outcome.7
SECONDARY HYPERPARATHYROIDISM
Secondary hyperparathyroidism is a common complication in patients with end-stage renal disease and is characterized by derangements in the homeostasis of calcium, phosphorus, and vitamin D.9
Because renal function is decreased, phosphate is retained and calcitriol synthesis is reduced, resulting in hypocalcemia, which induces parathyroid gland hyperplasia and parathyroid hormone secretion.10 Moreover, some patents with long-standing secondary hyperparathyroidism may develop tertiary hyperparathyroidism associated with autonomous parathyroid hormone secretion, hypercalcemia, and hyperphosphatemia.11
The Kidney Disease: Improving Global Outcomes (KDIGO) Work Group recommends screening for and managing secondary hyperparathyroidism in all patients with stage 3 chronic kidney disease (estimated glomerular filtration rate < 60 mL/min). In patients with stage 5 chronic kidney disease or on dialysis, the serum calcium and phosphorus levels should be monitored every 1 to 3 months and the parathyroid hormone levels every 3 to 6 months.12
According to KDIGO guidelines, the target level of calcium is less than 10.2 mg/dL, and the target phosphorus level is less than 4.6 mg/dL. The level of parathyroid hormone should be maintained at 2 to 9 times the upper limit of normal for the assay.
The management of secondary hyperparathyroidism includes a low-phosphorus diet, calcium-containing or calcium-free phosphate binders, a calcitriol supplement, and calcimimetics. If medical treatment fails and manifestations are significant, parathyroidectomy may be indicated.13
- Alpert MA, Ravenscraft MD. Pericardial involvement in end-stage renal disease. Am J Med Sci 2003; 325:228–236.
- Gowda RM, Boxt LM. Calcifications of the heart. Radiol Clin North Am 2004; 42:603–617.
- Kleynberg RL, Kleynberg VM, Kleynberg LM, Farahmandian D. Chronic constrictive pericarditis in association with end-stage renal disease. Int J Nephrol 2011; 2011:469602.
- Rao N, Crail S. Metastatic calcification and long-term hemodialysis. N Engl J Med 2013; 368:2415.
- Ling LH, Oh JK, Schaff HV, et al. Constrictive pericarditis in the modern era: evolving clinical spectrum and impact on outcome after pericardiectomy. Circulation 1999; 100:1380–1386.
- Bergman M, Vitrai J, Salman H. Constrictive pericarditis: a reminder of a not so rare disease. Eur J Intern Med 2006; 17:457–464.
- Szabó G, Schmack B, Bulut C, et al. Constrictive pericarditis: risks, aetiologies and outcomes after total pericardiectomy: 24 years of experience. Eur J Cardiothorac Surg 2013; 44:1023–1028.
- Feldman V, Dovrish Z, Weisenberg N, Neuman Y, Amital H. Uremic pericarditis. Isr Med Assoc J 2011; 13:256–257.
- Levin A, Bakris GL, Molitch M, et al. Prevalence of abnormal serum vitamin D, PTH, calcium, and phosphorus in patients with chronic kidney disease: results of the study to evaluate early kidney disease. Kidney Int 2007; 71:31–38.
- Martin KJ, Gonzalez EA. Metabolic bone disease in chronic kidney disease. J Am Soc Nephrol 2007; 18:875–885.
- Kerby J, Rue LW, Blair H, Hudson S, Sellers MT, Diethelm AG. Operative treatment of tertiary hyperparathyroidism: a single-center experience. Ann Surg 1998; 227:878–886.
- Kidney Disease: Improving Global Outcomes (KDIGO) CKDMBD Work Group. KDIGO clinical practice guideline for the diagnosis, evaluation, prevention, and treatment of chronic kidney disease—mineral and bone disorder (CKD-MBD). Kidney Int Suppl 2009; 76:S1–130.
- National Kidney Foundation. K/DOQI clinical practice guidelines for bone metabolism and disease in chronic kidney disease. Am J Kidney Dis 2003; 42(4 suppl 3):S1–201.
- Alpert MA, Ravenscraft MD. Pericardial involvement in end-stage renal disease. Am J Med Sci 2003; 325:228–236.
- Gowda RM, Boxt LM. Calcifications of the heart. Radiol Clin North Am 2004; 42:603–617.
- Kleynberg RL, Kleynberg VM, Kleynberg LM, Farahmandian D. Chronic constrictive pericarditis in association with end-stage renal disease. Int J Nephrol 2011; 2011:469602.
- Rao N, Crail S. Metastatic calcification and long-term hemodialysis. N Engl J Med 2013; 368:2415.
- Ling LH, Oh JK, Schaff HV, et al. Constrictive pericarditis in the modern era: evolving clinical spectrum and impact on outcome after pericardiectomy. Circulation 1999; 100:1380–1386.
- Bergman M, Vitrai J, Salman H. Constrictive pericarditis: a reminder of a not so rare disease. Eur J Intern Med 2006; 17:457–464.
- Szabó G, Schmack B, Bulut C, et al. Constrictive pericarditis: risks, aetiologies and outcomes after total pericardiectomy: 24 years of experience. Eur J Cardiothorac Surg 2013; 44:1023–1028.
- Feldman V, Dovrish Z, Weisenberg N, Neuman Y, Amital H. Uremic pericarditis. Isr Med Assoc J 2011; 13:256–257.
- Levin A, Bakris GL, Molitch M, et al. Prevalence of abnormal serum vitamin D, PTH, calcium, and phosphorus in patients with chronic kidney disease: results of the study to evaluate early kidney disease. Kidney Int 2007; 71:31–38.
- Martin KJ, Gonzalez EA. Metabolic bone disease in chronic kidney disease. J Am Soc Nephrol 2007; 18:875–885.
- Kerby J, Rue LW, Blair H, Hudson S, Sellers MT, Diethelm AG. Operative treatment of tertiary hyperparathyroidism: a single-center experience. Ann Surg 1998; 227:878–886.
- Kidney Disease: Improving Global Outcomes (KDIGO) CKDMBD Work Group. KDIGO clinical practice guideline for the diagnosis, evaluation, prevention, and treatment of chronic kidney disease—mineral and bone disorder (CKD-MBD). Kidney Int Suppl 2009; 76:S1–130.
- National Kidney Foundation. K/DOQI clinical practice guidelines for bone metabolism and disease in chronic kidney disease. Am J Kidney Dis 2003; 42(4 suppl 3):S1–201.
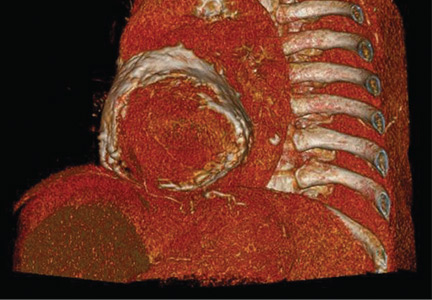
Disseminated molluscum contagiosum lesions in an HIV patient

A 37-year-old woman with a 3-month history of disorientation and depression was admitted to the infectious disease unit. In addition, she had had multiple painless exophytic lesions on the face, abdomen, and genital area for the past 3 years.
Physical examination revealed multiple waxy lesions, which were skin-colored dome-shaped papules with an umbilicated top, with diameters of 2 to 10 mm (Figure 1).
")
Skin biopsy study (Figure 2) showed lobulated endophytic hyperplasia with an intradermal pseudotumor (Henderson-Paterson bodies). Dimorphic fungal infection with Cryptococcus species was excluded. A final diagnosis of molluscum contagiosum was made based on the clinical appearance of the lesions and the histologic findings.
The patient was known to be positive for human immunodeficiency virus (HIV) and to have discontinued medications and follow-up visits in 2011. She was severely immunodepressed, at stage C3 (the worst stage) in the US Centers for Disease Control and Prevention classification. Her CD4 cell count was 26 × 106/L (reference range 533–1,674) and 11% (34%–61%); her viral load was 252,085 copies/mm3.
Subsequently, she was diagnosed with HIV-related encephalopathy and disseminated Mycobacterium tuberculosis infection. Highly active antiretoroviral therapy (HAART) and tuberculosis treatment were started.
LINKED TO IMMUNOCOMPROMISE
Molluscum contagiosum virus is an important human skin pathogen. Transmitted through direct skin-to-skin contact, it can cause disfigurement and suffering in affected patients. It often affects children, but abundant or atypical lesions in an adult usually indicate underlying immunodeficiency1 and are usually related to impaired cell-mediated immunity.2
In the mid-1980s, atypical molluscum contagiosum was recognized as a feature of HIV infection,3 but with widespread use of HAART, lesions are now less frequently observed in Western countries. Cases of molluscum contagiosum have also been reported in patients receiving immunosuppressive drugs such as methrotrexate and tumor necrosis factor alpha inhibitors.4 A high burden of lesions such as our patient had is uncommon.
Optimal treatment in HIV patients is restoration of immunologic competence with HAART. Adjunctive treatment with surgical excision, curettage, cryotherapy, and various chemical removal methods can also be applied.4,5 Severe infection secondary to iatrogenic immunosuppression may be resistant to standard therapy, and when the condition does not respond to combination treatment, withdrawal of immunosuppressive therapies may be necessary.4
The bottom line. Molluscum contagiosum is less frequently seen in the HAART era; however, when present it usually indicates a high level of immunosuppression. Clinicians need to keep the relation in mind.
Acknowledgment: We thank Dr. Isabel Faro Viana for the histologic image.
- Chen X, Anstey AV, Bugert JJ. Molluscum contagiosum virus infection. Lancet Infect Dis 2013; 13:877–888.
- Jung AC, Paauw DS. Diagnosing HIV-related disease: using the CD4 count as a guide. J Gen Intern Med 1998; 13:131–136.
- Beutler BD, Cohen PR. Molluscum contagiosum of the eyelid: case report in a man receiving methotrexate and literature review of molluscum contagiosum in patients who are immunosuppressed secondary to methotrexate or HIV infection. Dermatol Online J 2016; 22:pii:13030/qt8vz669cj.
- Gur I. The epidemiology of Molluscum contagiosum in HIV-seropositive patients: a unique entity or insignificant finding? Int J STD AIDS 2008; 19:503–506.
- Filo-Rogulska M, Pindycka-Piaszczynska M, Januszewski K, Jarzab J. Disseminated atypical molluscum contagiosum as a presenting symptom of HIV infection. Postepy Dermatol Alergol 2013; 30:56–58.
A 37-year-old woman with a 3-month history of disorientation and depression was admitted to the infectious disease unit. In addition, she had had multiple painless exophytic lesions on the face, abdomen, and genital area for the past 3 years.
Physical examination revealed multiple waxy lesions, which were skin-colored dome-shaped papules with an umbilicated top, with diameters of 2 to 10 mm (Figure 1).
Skin biopsy study (Figure 2) showed lobulated endophytic hyperplasia with an intradermal pseudotumor (Henderson-Paterson bodies). Dimorphic fungal infection with Cryptococcus species was excluded. A final diagnosis of molluscum contagiosum was made based on the clinical appearance of the lesions and the histologic findings.
The patient was known to be positive for human immunodeficiency virus (HIV) and to have discontinued medications and follow-up visits in 2011. She was severely immunodepressed, at stage C3 (the worst stage) in the US Centers for Disease Control and Prevention classification. Her CD4 cell count was 26 × 106/L (reference range 533–1,674) and 11% (34%–61%); her viral load was 252,085 copies/mm3.
Subsequently, she was diagnosed with HIV-related encephalopathy and disseminated Mycobacterium tuberculosis infection. Highly active antiretoroviral therapy (HAART) and tuberculosis treatment were started.
LINKED TO IMMUNOCOMPROMISE
Molluscum contagiosum virus is an important human skin pathogen. Transmitted through direct skin-to-skin contact, it can cause disfigurement and suffering in affected patients. It often affects children, but abundant or atypical lesions in an adult usually indicate underlying immunodeficiency1 and are usually related to impaired cell-mediated immunity.2
In the mid-1980s, atypical molluscum contagiosum was recognized as a feature of HIV infection,3 but with widespread use of HAART, lesions are now less frequently observed in Western countries. Cases of molluscum contagiosum have also been reported in patients receiving immunosuppressive drugs such as methrotrexate and tumor necrosis factor alpha inhibitors.4 A high burden of lesions such as our patient had is uncommon.
Optimal treatment in HIV patients is restoration of immunologic competence with HAART. Adjunctive treatment with surgical excision, curettage, cryotherapy, and various chemical removal methods can also be applied.4,5 Severe infection secondary to iatrogenic immunosuppression may be resistant to standard therapy, and when the condition does not respond to combination treatment, withdrawal of immunosuppressive therapies may be necessary.4
The bottom line. Molluscum contagiosum is less frequently seen in the HAART era; however, when present it usually indicates a high level of immunosuppression. Clinicians need to keep the relation in mind.
Acknowledgment: We thank Dr. Isabel Faro Viana for the histologic image.
A 37-year-old woman with a 3-month history of disorientation and depression was admitted to the infectious disease unit. In addition, she had had multiple painless exophytic lesions on the face, abdomen, and genital area for the past 3 years.
Physical examination revealed multiple waxy lesions, which were skin-colored dome-shaped papules with an umbilicated top, with diameters of 2 to 10 mm (Figure 1).
Skin biopsy study (Figure 2) showed lobulated endophytic hyperplasia with an intradermal pseudotumor (Henderson-Paterson bodies). Dimorphic fungal infection with Cryptococcus species was excluded. A final diagnosis of molluscum contagiosum was made based on the clinical appearance of the lesions and the histologic findings.
The patient was known to be positive for human immunodeficiency virus (HIV) and to have discontinued medications and follow-up visits in 2011. She was severely immunodepressed, at stage C3 (the worst stage) in the US Centers for Disease Control and Prevention classification. Her CD4 cell count was 26 × 106/L (reference range 533–1,674) and 11% (34%–61%); her viral load was 252,085 copies/mm3.
Subsequently, she was diagnosed with HIV-related encephalopathy and disseminated Mycobacterium tuberculosis infection. Highly active antiretoroviral therapy (HAART) and tuberculosis treatment were started.
LINKED TO IMMUNOCOMPROMISE
Molluscum contagiosum virus is an important human skin pathogen. Transmitted through direct skin-to-skin contact, it can cause disfigurement and suffering in affected patients. It often affects children, but abundant or atypical lesions in an adult usually indicate underlying immunodeficiency1 and are usually related to impaired cell-mediated immunity.2
In the mid-1980s, atypical molluscum contagiosum was recognized as a feature of HIV infection,3 but with widespread use of HAART, lesions are now less frequently observed in Western countries. Cases of molluscum contagiosum have also been reported in patients receiving immunosuppressive drugs such as methrotrexate and tumor necrosis factor alpha inhibitors.4 A high burden of lesions such as our patient had is uncommon.
Optimal treatment in HIV patients is restoration of immunologic competence with HAART. Adjunctive treatment with surgical excision, curettage, cryotherapy, and various chemical removal methods can also be applied.4,5 Severe infection secondary to iatrogenic immunosuppression may be resistant to standard therapy, and when the condition does not respond to combination treatment, withdrawal of immunosuppressive therapies may be necessary.4
The bottom line. Molluscum contagiosum is less frequently seen in the HAART era; however, when present it usually indicates a high level of immunosuppression. Clinicians need to keep the relation in mind.
Acknowledgment: We thank Dr. Isabel Faro Viana for the histologic image.
- Chen X, Anstey AV, Bugert JJ. Molluscum contagiosum virus infection. Lancet Infect Dis 2013; 13:877–888.
- Jung AC, Paauw DS. Diagnosing HIV-related disease: using the CD4 count as a guide. J Gen Intern Med 1998; 13:131–136.
- Beutler BD, Cohen PR. Molluscum contagiosum of the eyelid: case report in a man receiving methotrexate and literature review of molluscum contagiosum in patients who are immunosuppressed secondary to methotrexate or HIV infection. Dermatol Online J 2016; 22:pii:13030/qt8vz669cj.
- Gur I. The epidemiology of Molluscum contagiosum in HIV-seropositive patients: a unique entity or insignificant finding? Int J STD AIDS 2008; 19:503–506.
- Filo-Rogulska M, Pindycka-Piaszczynska M, Januszewski K, Jarzab J. Disseminated atypical molluscum contagiosum as a presenting symptom of HIV infection. Postepy Dermatol Alergol 2013; 30:56–58.
- Chen X, Anstey AV, Bugert JJ. Molluscum contagiosum virus infection. Lancet Infect Dis 2013; 13:877–888.
- Jung AC, Paauw DS. Diagnosing HIV-related disease: using the CD4 count as a guide. J Gen Intern Med 1998; 13:131–136.
- Beutler BD, Cohen PR. Molluscum contagiosum of the eyelid: case report in a man receiving methotrexate and literature review of molluscum contagiosum in patients who are immunosuppressed secondary to methotrexate or HIV infection. Dermatol Online J 2016; 22:pii:13030/qt8vz669cj.
- Gur I. The epidemiology of Molluscum contagiosum in HIV-seropositive patients: a unique entity or insignificant finding? Int J STD AIDS 2008; 19:503–506.
- Filo-Rogulska M, Pindycka-Piaszczynska M, Januszewski K, Jarzab J. Disseminated atypical molluscum contagiosum as a presenting symptom of HIV infection. Postepy Dermatol Alergol 2013; 30:56–58.

Worsening migraine due to neurocysticercosis
A 35-year-old woman with a history of migraine presented with a headache that had worsened over the past 2 weeks. The headache was occipital and was associated with blurred vision, photophobia, tingling of the hands, episodes of flashing lights and images, and difficulty concentrating. The headache was similar to her typical migraines, but with the addition of flashing lights and images.
Her medical history included a cystic mass in the right occipital lobe that had been found incidentally on magnetic resonance imaging (MRI) during a workup for pituitary adenoma. The mass was thought to be a congenital lesion or arachnoid cyst, and intermittent screening had been recommended.
The patient had grown up in Honduras and had lived in the jungle until age 12, when she moved to the United States.
EVALUATION AND MANAGEMENT
Physical examination was remarkable for partial visual field loss in the periphery of the left temporal quadrant in both eyes (partial homonymous hemianopia). Repeat MRI showed a cystic lesion with scolex (the anterior end of a tapeworm) in the right occipital lobe, with surrounding edema (Figure 1).
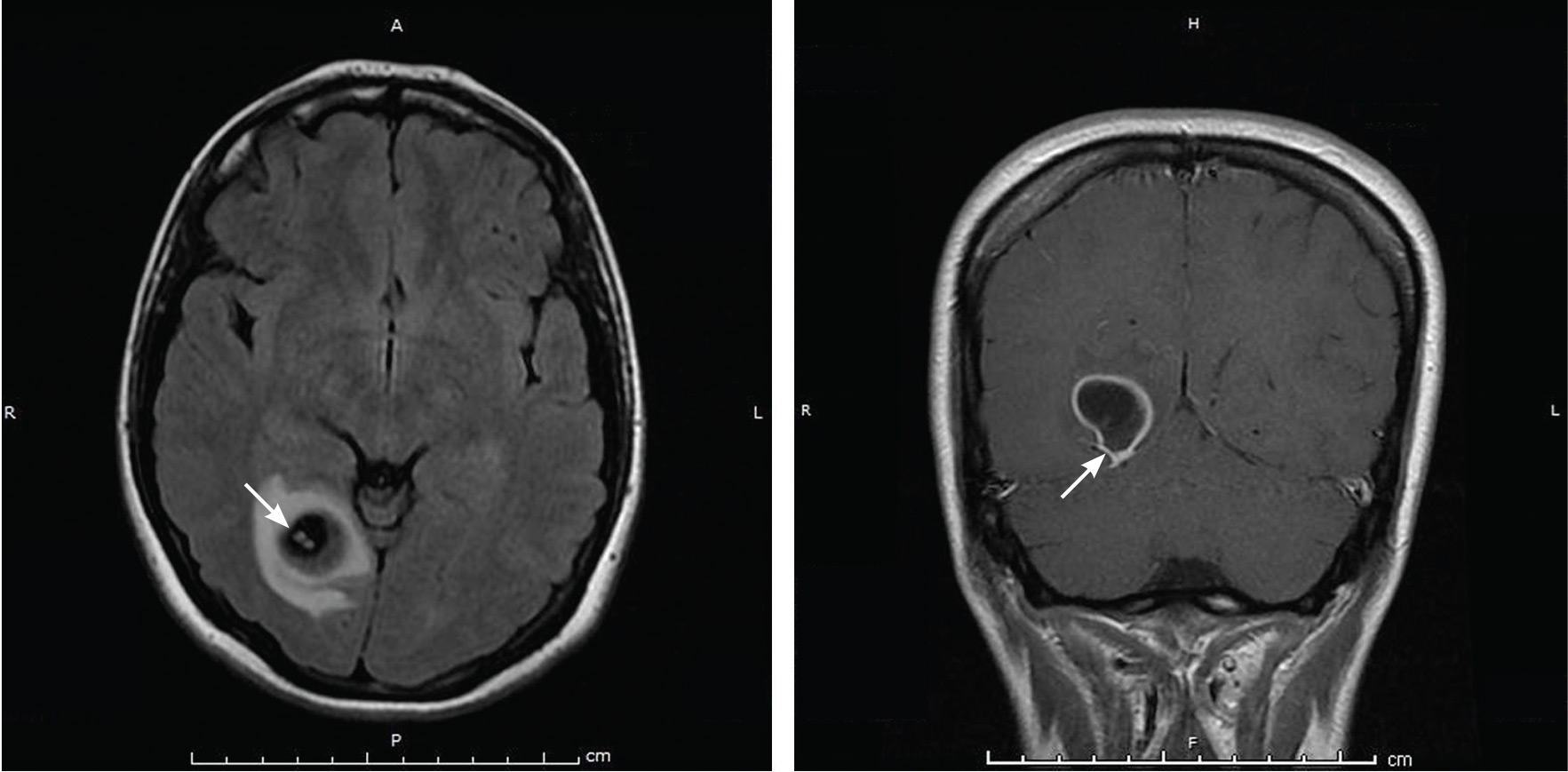
Cystic brain lesions are associated with arachnoid cyst, glioma, and malignancy, but the presence of the scolex placed neurocysticercosis as the leading diagnosis. Testing for cysticercus antibody was negative. This test was done in the hope of confirming our high suspicion; while a negative test result does not exclude this diagnosis, a positive test would have been helpful to corroborate what we suspected. However, her imaging and clinical features were sufficient to warrant treating her for neurocysticercosis
She was treated with albendazole 400 mg twice a day for 10 days, and prednisone 1 mg/kg/day for 10 days followed by a taper. Because of the frequency with which neurocysticercosis causes seizures, an antiepileptic drug is also recommended, at least until active lesions have subsided.1 In this patient, levetiracetam 1,000 mg twice a day was prescribed for 6 months for seizure prophylaxis.
Repeat MRI 2 months later showed improvement (Figure 2). Her acute neurologic signs and symptoms had resolved, but she continued to be followed for chronic migraines (Figure 3). She has had no seizures despite weaning from levetiracetam.


TAPEWORM AND MIGRAINE
Neurocysticercosis is caused by the cestode Taenia solium, acquired by eating undercooked pork contaminated with the cysts or eggs.1 The oncospheres released by the eggs migrate through the host body and encyst in end organs.
Neuroimaging can show 4 stages of the cysts—vesicular with living larva, colloidal with larva degeneration, granulonodular with thickening of the cyst, and calcification.1
For patients who have lived in or visited high-risk areas of the world such as Central America, South America, sub-Saharan Africa, India, and Asia, it is important to include neurocysticercosis in the differential diagnosis of migraine with focal deficits or migraine with an evolving quality. Encysted larvae can remain asymptomatic for years but can cause brain edema, often leading to seizures.
Serum testing for cysticercus antibody can indicate acute infection, chronic infection, and possibly the immune response to treatment; however, serum testing has limited sensitivity in patients who have single or calcified lesions.2 A negative test result does not exclude infection and is more likely to be a false negative in patients with a single or calcified lesion.
Current treatment guidelines recommend albendazole 400 mg twice daily along with dexamethasone or prednisolone to decrease the number of cysts and the development of lesional epilepsy.1 Albendazole in combination with praziquantel 50 mg/kg/day kills more cysts than albendazole alone and should be considered in patients with more than 2 cysts.3
- Baird RA, Wiebe S, Zunt JR, Halperin JJ, Gronseth G, Roos KL. Evidence-based guideline: treatment of parenchymal neurocysticercosis: report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology 2013; 80:1424–1429.
- Garcia HH, Wittner M, Coyle CM, Tanowitz HB, White AC Jr. Cysticercosis. In: Guerrant RL, Walker DH, Weller PF, editors. Tropical Infectious Diseases: Principles, Pathogens, and Practice. Philadelphia, PA: Elsevier Churchill Livingstone; 2006:1289–1303.
- Garcia HH, Gonzales I, Lescano AG, et al; Cysticercosis Working Group in Peru. Efficacy of combined antiparasitic therapy with praziquantel and albendazole for neurocysticercosis: a double blind, randomized controlled trial. Lancet Infect Dis 2014; 14:687–695.
A 35-year-old woman with a history of migraine presented with a headache that had worsened over the past 2 weeks. The headache was occipital and was associated with blurred vision, photophobia, tingling of the hands, episodes of flashing lights and images, and difficulty concentrating. The headache was similar to her typical migraines, but with the addition of flashing lights and images.
Her medical history included a cystic mass in the right occipital lobe that had been found incidentally on magnetic resonance imaging (MRI) during a workup for pituitary adenoma. The mass was thought to be a congenital lesion or arachnoid cyst, and intermittent screening had been recommended.
The patient had grown up in Honduras and had lived in the jungle until age 12, when she moved to the United States.
EVALUATION AND MANAGEMENT
Physical examination was remarkable for partial visual field loss in the periphery of the left temporal quadrant in both eyes (partial homonymous hemianopia). Repeat MRI showed a cystic lesion with scolex (the anterior end of a tapeworm) in the right occipital lobe, with surrounding edema (Figure 1).
Cystic brain lesions are associated with arachnoid cyst, glioma, and malignancy, but the presence of the scolex placed neurocysticercosis as the leading diagnosis. Testing for cysticercus antibody was negative. This test was done in the hope of confirming our high suspicion; while a negative test result does not exclude this diagnosis, a positive test would have been helpful to corroborate what we suspected. However, her imaging and clinical features were sufficient to warrant treating her for neurocysticercosis
She was treated with albendazole 400 mg twice a day for 10 days, and prednisone 1 mg/kg/day for 10 days followed by a taper. Because of the frequency with which neurocysticercosis causes seizures, an antiepileptic drug is also recommended, at least until active lesions have subsided.1 In this patient, levetiracetam 1,000 mg twice a day was prescribed for 6 months for seizure prophylaxis.
Repeat MRI 2 months later showed improvement (Figure 2). Her acute neurologic signs and symptoms had resolved, but she continued to be followed for chronic migraines (Figure 3). She has had no seizures despite weaning from levetiracetam.
TAPEWORM AND MIGRAINE
Neurocysticercosis is caused by the cestode Taenia solium, acquired by eating undercooked pork contaminated with the cysts or eggs.1 The oncospheres released by the eggs migrate through the host body and encyst in end organs.
Neuroimaging can show 4 stages of the cysts—vesicular with living larva, colloidal with larva degeneration, granulonodular with thickening of the cyst, and calcification.1
For patients who have lived in or visited high-risk areas of the world such as Central America, South America, sub-Saharan Africa, India, and Asia, it is important to include neurocysticercosis in the differential diagnosis of migraine with focal deficits or migraine with an evolving quality. Encysted larvae can remain asymptomatic for years but can cause brain edema, often leading to seizures.
Serum testing for cysticercus antibody can indicate acute infection, chronic infection, and possibly the immune response to treatment; however, serum testing has limited sensitivity in patients who have single or calcified lesions.2 A negative test result does not exclude infection and is more likely to be a false negative in patients with a single or calcified lesion.
Current treatment guidelines recommend albendazole 400 mg twice daily along with dexamethasone or prednisolone to decrease the number of cysts and the development of lesional epilepsy.1 Albendazole in combination with praziquantel 50 mg/kg/day kills more cysts than albendazole alone and should be considered in patients with more than 2 cysts.3
A 35-year-old woman with a history of migraine presented with a headache that had worsened over the past 2 weeks. The headache was occipital and was associated with blurred vision, photophobia, tingling of the hands, episodes of flashing lights and images, and difficulty concentrating. The headache was similar to her typical migraines, but with the addition of flashing lights and images.
Her medical history included a cystic mass in the right occipital lobe that had been found incidentally on magnetic resonance imaging (MRI) during a workup for pituitary adenoma. The mass was thought to be a congenital lesion or arachnoid cyst, and intermittent screening had been recommended.
The patient had grown up in Honduras and had lived in the jungle until age 12, when she moved to the United States.
EVALUATION AND MANAGEMENT
Physical examination was remarkable for partial visual field loss in the periphery of the left temporal quadrant in both eyes (partial homonymous hemianopia). Repeat MRI showed a cystic lesion with scolex (the anterior end of a tapeworm) in the right occipital lobe, with surrounding edema (Figure 1).
Cystic brain lesions are associated with arachnoid cyst, glioma, and malignancy, but the presence of the scolex placed neurocysticercosis as the leading diagnosis. Testing for cysticercus antibody was negative. This test was done in the hope of confirming our high suspicion; while a negative test result does not exclude this diagnosis, a positive test would have been helpful to corroborate what we suspected. However, her imaging and clinical features were sufficient to warrant treating her for neurocysticercosis
She was treated with albendazole 400 mg twice a day for 10 days, and prednisone 1 mg/kg/day for 10 days followed by a taper. Because of the frequency with which neurocysticercosis causes seizures, an antiepileptic drug is also recommended, at least until active lesions have subsided.1 In this patient, levetiracetam 1,000 mg twice a day was prescribed for 6 months for seizure prophylaxis.
Repeat MRI 2 months later showed improvement (Figure 2). Her acute neurologic signs and symptoms had resolved, but she continued to be followed for chronic migraines (Figure 3). She has had no seizures despite weaning from levetiracetam.
TAPEWORM AND MIGRAINE
Neurocysticercosis is caused by the cestode Taenia solium, acquired by eating undercooked pork contaminated with the cysts or eggs.1 The oncospheres released by the eggs migrate through the host body and encyst in end organs.
Neuroimaging can show 4 stages of the cysts—vesicular with living larva, colloidal with larva degeneration, granulonodular with thickening of the cyst, and calcification.1
For patients who have lived in or visited high-risk areas of the world such as Central America, South America, sub-Saharan Africa, India, and Asia, it is important to include neurocysticercosis in the differential diagnosis of migraine with focal deficits or migraine with an evolving quality. Encysted larvae can remain asymptomatic for years but can cause brain edema, often leading to seizures.
Serum testing for cysticercus antibody can indicate acute infection, chronic infection, and possibly the immune response to treatment; however, serum testing has limited sensitivity in patients who have single or calcified lesions.2 A negative test result does not exclude infection and is more likely to be a false negative in patients with a single or calcified lesion.
Current treatment guidelines recommend albendazole 400 mg twice daily along with dexamethasone or prednisolone to decrease the number of cysts and the development of lesional epilepsy.1 Albendazole in combination with praziquantel 50 mg/kg/day kills more cysts than albendazole alone and should be considered in patients with more than 2 cysts.3
- Baird RA, Wiebe S, Zunt JR, Halperin JJ, Gronseth G, Roos KL. Evidence-based guideline: treatment of parenchymal neurocysticercosis: report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology 2013; 80:1424–1429.
- Garcia HH, Wittner M, Coyle CM, Tanowitz HB, White AC Jr. Cysticercosis. In: Guerrant RL, Walker DH, Weller PF, editors. Tropical Infectious Diseases: Principles, Pathogens, and Practice. Philadelphia, PA: Elsevier Churchill Livingstone; 2006:1289–1303.
- Garcia HH, Gonzales I, Lescano AG, et al; Cysticercosis Working Group in Peru. Efficacy of combined antiparasitic therapy with praziquantel and albendazole for neurocysticercosis: a double blind, randomized controlled trial. Lancet Infect Dis 2014; 14:687–695.
- Baird RA, Wiebe S, Zunt JR, Halperin JJ, Gronseth G, Roos KL. Evidence-based guideline: treatment of parenchymal neurocysticercosis: report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology 2013; 80:1424–1429.
- Garcia HH, Wittner M, Coyle CM, Tanowitz HB, White AC Jr. Cysticercosis. In: Guerrant RL, Walker DH, Weller PF, editors. Tropical Infectious Diseases: Principles, Pathogens, and Practice. Philadelphia, PA: Elsevier Churchill Livingstone; 2006:1289–1303.
- Garcia HH, Gonzales I, Lescano AG, et al; Cysticercosis Working Group in Peru. Efficacy of combined antiparasitic therapy with praziquantel and albendazole for neurocysticercosis: a double blind, randomized controlled trial. Lancet Infect Dis 2014; 14:687–695.
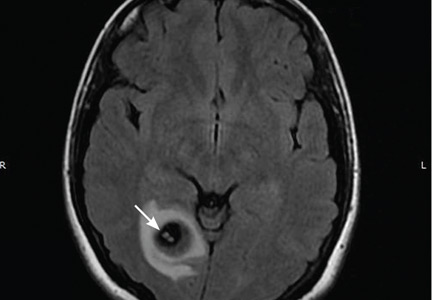
Bleeding esophageal varices: Who should receive a shunt?
A transjugular intrahepatic portosystemic shunt (TIPS) has been shown in randomized controlled trials to be effective for:
- Secondary prevention of variceal bleeding
- Controlling refractory ascites in patients with liver cirrhosis.
In addition, findings from retrospective case series have suggested that it helps in cases of:
- Acute variceal bleeding refractory to endoscopic therapy
- Gastropathy due to portal hypertension
- Bleeding gastric varices
- Refractory hepatic hydrothorax
- Hepatorenal syndrome
- Budd-Chiari syndrome
- Veno-occlusive disease
- Hepatopulmonary syndrome.
Here, we discuss the indications for a TIPS in cirrhotic patients with esophageal variceal bleeding.
CIRRHOSIS CAN LEAD TO PORTAL HYPERTENSION, BLEEDING
Cirrhosis of the liver alters the hepatic architecture. Development of regenerating nodules and deposition of connective tissue between these nodules increase the resistance to portal blood flow, which can lead to portal hypertension.1
Esophageal variceal bleeding is a complication of portal hypertension and a major cause of death in patients with liver cirrhosis. Combined treatment with vasoactive drugs, prophylactic antibiotics, and endoscopic band ligation is the standard of care for patients with acute bleeding. However, this treatment fails in about 10% to 15% of these patients. A TIPS creates a connection between the portal and hepatic veins, resulting in portal decompression and homeostasis.2
PRE-TIPS EVALUATION
Patients being considered for a TIPS should be medically assessed before the procedure. The workup should include the following:
- Routine blood tests, including blood type and screen (indirect Coombs test), complete blood cell count, basic metabolic panel, liver function tests, prothrombin time, and partial thromboplastin time
- Doppler ultrasonography of the liver to ensure that the portal and hepatic veins are patent
- Echocardiography to assess pulmonary arterial pressure and right-side heart function
- The hepatic venous pressure gradient, which is measured at the time of TIPS placement, reflects the degree of portal hypertension. A hepatic vein is catheterized, and the right atrial pressure or the free hepatic venous pressure is subtracted from the wedged hepatic venous pressure. The gradient is normally 1 to 5 mm Hg. A gradient greater than 5 mm Hg indicates portal hypertension, and esophageal varices may start to bleed when the gradient is greater than 12 mm Hg. The goal of TIPS placement is to reduce the gradient to less than 12 mm Hg, or at least by 50%.
Heart failure is a contraindication
Pulmonary hypertension may follow TIPS placement because the shunt increases venous return to the heart. Additionally, systemic vascular resistance decreases in patients who have a shunt. This further worsens the hyperdynamic circulatory state already present in patients with cirrhosis. Cardiac output increases in response to these changes. When the heart’s ability to handle this “volume overload” is exceeded, pulmonary venous pressures rise, with increasing ventilation-perfusion mismatch, hypoxia, and pulmonary vasoconstriction; pulmonary edema may ensue.
Congestive heart failure, severe tricuspid regurgitation, and severe pulmonary hypertension (mean pulmonary pressures > 45 mm Hg) are therefore considered absolute contraindications to TIPS placement.3,4 This is why echocardiography is recommended to assess pulmonary pressure along with the size and function of the right side of the heart before proceeding with TIPS insertion.
Other considerations
TIPS insertion is not recommended in patients with active hepatic encephalopathy, which should be adequately controlled before insertion of a TIPS. This can be achieved with lactulose and rifaximin. Lactulose is a laxative; the recommended target is 3 to 4 bowel movements daily. Rifaximin is a poorly absorbed antibiotic that has a wide spectrum of coverage, affecting gram-negative and gram-positive aerobes and anaerobes. It wipes out the gut bacteria and so decreases the production of ammonia by the gut.
Paracentesis is recommended before TIPS placement if a large volume of ascites is present. Draining the fluid allows the liver to drop down and makes it easier to access the portal vein from the hepatic vein.
WHEN TO CONSIDER A TIPS IN ESOPHAGEAL VARICEAL BLEEDING
Acute bleeding refractory to endoscopic therapy

A TIPS remains the only choice to control acute variceal bleeding refractory to medical and endoscopic therapy (Figure 1), with a success rate of 90% to 100%.5 The urgency of TIPS placement is an independent predictor of early mortality.
Esophageal variceal rebleeding
Once varices bleed, the risk of rebleeding is higher than 50%, and rebleeding is associated with a high mortality rate. TIPS should be considered if nonselective beta-blockers and surveillance with upper endoscopy and banding fail to prevent rebleeding, with many studies showing a TIPS to be superior to pharmacologic and endoscopic therapies.6
A meta-analysis in 1999 by Papatheodoridis et al6 found that variceal rebleeding was significantly more frequent with endoscopic therapies, at 47% vs 19% with a TIPS, but the incidence of hepatic encephalopathy was higher with TIPS (34% vs 19%; P < .001), and there was no difference in mortality rates.
Hepatic encephalopathy occurs in 15% to 25% of patients after TIPS procedures. Risk factors include advanced age, poor renal function, and a history of hepatic encephalopathy. Hepatic encephalopathy can be managed with lactulose or rifaximin, or both (see above). Narcotics, antihistamines, and benzodiazepines should be avoided. In rare cases (5%) when hepatic encephalopathy is refractory to medical therapy, liver transplant should be considered.
A surgical distal splenorenal shunt is another option for patients with refractory or recurrent variceal bleeding. In a large randomized controlled trial,7 140 cirrhotic patients with recurrent variceal bleeding were randomized to receive either a distal splenorenal shunt or a TIPS. At a mean follow-up of 48 months, there was no difference in the rates of rebleeding between the two groups (5.5% with a surgical shunt vs 10.5% with a TIPS, P = .29) or in hepatic encephalopathy (50% in both groups). Survival rates were comparable between the two groups at 2 years (81% with a surgical shunt vs 88% with a TIPS) and 5 years (62% vs 61%).
Early use of TIPS after first variceal bleeding
In a 2010 randomized controlled trial,8 63 patients with cirrhosis (Child-Pugh class B or C) and acute variceal bleeding who had received standard medical and endoscopic therapy were randomized to receive either a TIPS within 72 hours of admission or long-term conservative treatment with nonselective beta-blockers and endoscopic band ligation. The 1-year actuarial probability of remaining free of rebleeding or failure to control bleeding was 50% in the conservative treatment group vs 97% in the early-TIPS group (P < .001). The 1-year actuarial survival rate was 61% in the conservative treatment group vs 86% in the early-TIPS group (P < .001).
The authors8 concluded that early use of TIPS in patients with cirrhosis and Child-Pugh scores of 7 to 13 who were hospitalized for acute variceal bleeding was associated with significant reductions in rates of treatment failure and mortality.
- Brenner D, Rippe RA. Pathogenesis of hepatic fibrosis. In: Yamada T, Alpers DH, Laine L, Kaplowitz N, Owyang C, Powell DW, editors. Textbook of Gastroenterology. 4th edition. Philadelphia, PA: Lippincott Williams & Wilkins; 2003.
- Bhogal HK, Sanyal AJ. Using transjugular intrahepatic portosystemic shunts for complications of cirrhosis. Clin Gastroenterol Hepatol 2011; 9:936–946.
- Garcia-Tsao G, Sanyal AJ, Grace ND, Carey WD; Practice Guidelines Committee of American Association for Study of Liver Diseases; Practice Parameters Committee of American College of Gastroenterology. Prevention and management of gastroesophageal varices and variceal hemorrhage in cirrhosis. Am J Gastroenterol 2007; 102:2086–2102.
- Azoulay D, Castaing D, Dennison A, Martino W, Eyraud D, Bismuth H. Transjugular intrahepatic portosystemic shunt worsens the hyperdynamic circulatory state of the cirrhotic patient: preliminary report of a prospective study. Hepatology 1994; 19:129–132.
- Rodríguez-Laiz JM, Bañares R, Echenagusia A, et al. Effects of transjugular intrahepatic portasystemic shunt (TIPS) on splanchnic and systemic hemodynamics, and hepatic function in patients with portal hypertension. Preliminary results. Dig Dis Sci 1995; 40:2121–2127.
- Papatheodoridis GV, Goulis J, Leandro G, Patch D, Burroughs AK. Transjugular intrahepatic portosystemic shunt compared with endoscopic treatment for prevention of variceal rebleeding: a meta-analysis. Hepatology 1999; 30:612–622.
- Henderson JM, Boyer TD, Kutner MH, et al; DIVERT Study Group. Distal splenorenal shunt versus transjugular intrahepatic portal systemic shunt for variceal bleeding: a randomized trial. Gastroenterology 2006; 130:1643–1651.
- García-Pagán JC, Caca K, Bureau C, et al; Early TIPS (Transjugular Intrahepatic Portosystemic Shunt) Cooperative Study Group. Early use of TIPS in patients with cirrhosis and variceal bleeding. N Engl J Med 2010; 362:2370–2379.
A transjugular intrahepatic portosystemic shunt (TIPS) has been shown in randomized controlled trials to be effective for:
- Secondary prevention of variceal bleeding
- Controlling refractory ascites in patients with liver cirrhosis.
In addition, findings from retrospective case series have suggested that it helps in cases of:
- Acute variceal bleeding refractory to endoscopic therapy
- Gastropathy due to portal hypertension
- Bleeding gastric varices
- Refractory hepatic hydrothorax
- Hepatorenal syndrome
- Budd-Chiari syndrome
- Veno-occlusive disease
- Hepatopulmonary syndrome.
Here, we discuss the indications for a TIPS in cirrhotic patients with esophageal variceal bleeding.
CIRRHOSIS CAN LEAD TO PORTAL HYPERTENSION, BLEEDING
Cirrhosis of the liver alters the hepatic architecture. Development of regenerating nodules and deposition of connective tissue between these nodules increase the resistance to portal blood flow, which can lead to portal hypertension.1
Esophageal variceal bleeding is a complication of portal hypertension and a major cause of death in patients with liver cirrhosis. Combined treatment with vasoactive drugs, prophylactic antibiotics, and endoscopic band ligation is the standard of care for patients with acute bleeding. However, this treatment fails in about 10% to 15% of these patients. A TIPS creates a connection between the portal and hepatic veins, resulting in portal decompression and homeostasis.2
PRE-TIPS EVALUATION
Patients being considered for a TIPS should be medically assessed before the procedure. The workup should include the following:
- Routine blood tests, including blood type and screen (indirect Coombs test), complete blood cell count, basic metabolic panel, liver function tests, prothrombin time, and partial thromboplastin time
- Doppler ultrasonography of the liver to ensure that the portal and hepatic veins are patent
- Echocardiography to assess pulmonary arterial pressure and right-side heart function
- The hepatic venous pressure gradient, which is measured at the time of TIPS placement, reflects the degree of portal hypertension. A hepatic vein is catheterized, and the right atrial pressure or the free hepatic venous pressure is subtracted from the wedged hepatic venous pressure. The gradient is normally 1 to 5 mm Hg. A gradient greater than 5 mm Hg indicates portal hypertension, and esophageal varices may start to bleed when the gradient is greater than 12 mm Hg. The goal of TIPS placement is to reduce the gradient to less than 12 mm Hg, or at least by 50%.
Heart failure is a contraindication
Pulmonary hypertension may follow TIPS placement because the shunt increases venous return to the heart. Additionally, systemic vascular resistance decreases in patients who have a shunt. This further worsens the hyperdynamic circulatory state already present in patients with cirrhosis. Cardiac output increases in response to these changes. When the heart’s ability to handle this “volume overload” is exceeded, pulmonary venous pressures rise, with increasing ventilation-perfusion mismatch, hypoxia, and pulmonary vasoconstriction; pulmonary edema may ensue.
Congestive heart failure, severe tricuspid regurgitation, and severe pulmonary hypertension (mean pulmonary pressures > 45 mm Hg) are therefore considered absolute contraindications to TIPS placement.3,4 This is why echocardiography is recommended to assess pulmonary pressure along with the size and function of the right side of the heart before proceeding with TIPS insertion.
Other considerations
TIPS insertion is not recommended in patients with active hepatic encephalopathy, which should be adequately controlled before insertion of a TIPS. This can be achieved with lactulose and rifaximin. Lactulose is a laxative; the recommended target is 3 to 4 bowel movements daily. Rifaximin is a poorly absorbed antibiotic that has a wide spectrum of coverage, affecting gram-negative and gram-positive aerobes and anaerobes. It wipes out the gut bacteria and so decreases the production of ammonia by the gut.
Paracentesis is recommended before TIPS placement if a large volume of ascites is present. Draining the fluid allows the liver to drop down and makes it easier to access the portal vein from the hepatic vein.
WHEN TO CONSIDER A TIPS IN ESOPHAGEAL VARICEAL BLEEDING
Acute bleeding refractory to endoscopic therapy
A TIPS remains the only choice to control acute variceal bleeding refractory to medical and endoscopic therapy (Figure 1), with a success rate of 90% to 100%.5 The urgency of TIPS placement is an independent predictor of early mortality.
Esophageal variceal rebleeding
Once varices bleed, the risk of rebleeding is higher than 50%, and rebleeding is associated with a high mortality rate. TIPS should be considered if nonselective beta-blockers and surveillance with upper endoscopy and banding fail to prevent rebleeding, with many studies showing a TIPS to be superior to pharmacologic and endoscopic therapies.6
A meta-analysis in 1999 by Papatheodoridis et al6 found that variceal rebleeding was significantly more frequent with endoscopic therapies, at 47% vs 19% with a TIPS, but the incidence of hepatic encephalopathy was higher with TIPS (34% vs 19%; P < .001), and there was no difference in mortality rates.
Hepatic encephalopathy occurs in 15% to 25% of patients after TIPS procedures. Risk factors include advanced age, poor renal function, and a history of hepatic encephalopathy. Hepatic encephalopathy can be managed with lactulose or rifaximin, or both (see above). Narcotics, antihistamines, and benzodiazepines should be avoided. In rare cases (5%) when hepatic encephalopathy is refractory to medical therapy, liver transplant should be considered.
A surgical distal splenorenal shunt is another option for patients with refractory or recurrent variceal bleeding. In a large randomized controlled trial,7 140 cirrhotic patients with recurrent variceal bleeding were randomized to receive either a distal splenorenal shunt or a TIPS. At a mean follow-up of 48 months, there was no difference in the rates of rebleeding between the two groups (5.5% with a surgical shunt vs 10.5% with a TIPS, P = .29) or in hepatic encephalopathy (50% in both groups). Survival rates were comparable between the two groups at 2 years (81% with a surgical shunt vs 88% with a TIPS) and 5 years (62% vs 61%).
Early use of TIPS after first variceal bleeding
In a 2010 randomized controlled trial,8 63 patients with cirrhosis (Child-Pugh class B or C) and acute variceal bleeding who had received standard medical and endoscopic therapy were randomized to receive either a TIPS within 72 hours of admission or long-term conservative treatment with nonselective beta-blockers and endoscopic band ligation. The 1-year actuarial probability of remaining free of rebleeding or failure to control bleeding was 50% in the conservative treatment group vs 97% in the early-TIPS group (P < .001). The 1-year actuarial survival rate was 61% in the conservative treatment group vs 86% in the early-TIPS group (P < .001).
The authors8 concluded that early use of TIPS in patients with cirrhosis and Child-Pugh scores of 7 to 13 who were hospitalized for acute variceal bleeding was associated with significant reductions in rates of treatment failure and mortality.
A transjugular intrahepatic portosystemic shunt (TIPS) has been shown in randomized controlled trials to be effective for:
- Secondary prevention of variceal bleeding
- Controlling refractory ascites in patients with liver cirrhosis.
In addition, findings from retrospective case series have suggested that it helps in cases of:
- Acute variceal bleeding refractory to endoscopic therapy
- Gastropathy due to portal hypertension
- Bleeding gastric varices
- Refractory hepatic hydrothorax
- Hepatorenal syndrome
- Budd-Chiari syndrome
- Veno-occlusive disease
- Hepatopulmonary syndrome.
Here, we discuss the indications for a TIPS in cirrhotic patients with esophageal variceal bleeding.
CIRRHOSIS CAN LEAD TO PORTAL HYPERTENSION, BLEEDING
Cirrhosis of the liver alters the hepatic architecture. Development of regenerating nodules and deposition of connective tissue between these nodules increase the resistance to portal blood flow, which can lead to portal hypertension.1
Esophageal variceal bleeding is a complication of portal hypertension and a major cause of death in patients with liver cirrhosis. Combined treatment with vasoactive drugs, prophylactic antibiotics, and endoscopic band ligation is the standard of care for patients with acute bleeding. However, this treatment fails in about 10% to 15% of these patients. A TIPS creates a connection between the portal and hepatic veins, resulting in portal decompression and homeostasis.2
PRE-TIPS EVALUATION
Patients being considered for a TIPS should be medically assessed before the procedure. The workup should include the following:
- Routine blood tests, including blood type and screen (indirect Coombs test), complete blood cell count, basic metabolic panel, liver function tests, prothrombin time, and partial thromboplastin time
- Doppler ultrasonography of the liver to ensure that the portal and hepatic veins are patent
- Echocardiography to assess pulmonary arterial pressure and right-side heart function
- The hepatic venous pressure gradient, which is measured at the time of TIPS placement, reflects the degree of portal hypertension. A hepatic vein is catheterized, and the right atrial pressure or the free hepatic venous pressure is subtracted from the wedged hepatic venous pressure. The gradient is normally 1 to 5 mm Hg. A gradient greater than 5 mm Hg indicates portal hypertension, and esophageal varices may start to bleed when the gradient is greater than 12 mm Hg. The goal of TIPS placement is to reduce the gradient to less than 12 mm Hg, or at least by 50%.
Heart failure is a contraindication
Pulmonary hypertension may follow TIPS placement because the shunt increases venous return to the heart. Additionally, systemic vascular resistance decreases in patients who have a shunt. This further worsens the hyperdynamic circulatory state already present in patients with cirrhosis. Cardiac output increases in response to these changes. When the heart’s ability to handle this “volume overload” is exceeded, pulmonary venous pressures rise, with increasing ventilation-perfusion mismatch, hypoxia, and pulmonary vasoconstriction; pulmonary edema may ensue.
Congestive heart failure, severe tricuspid regurgitation, and severe pulmonary hypertension (mean pulmonary pressures > 45 mm Hg) are therefore considered absolute contraindications to TIPS placement.3,4 This is why echocardiography is recommended to assess pulmonary pressure along with the size and function of the right side of the heart before proceeding with TIPS insertion.
Other considerations
TIPS insertion is not recommended in patients with active hepatic encephalopathy, which should be adequately controlled before insertion of a TIPS. This can be achieved with lactulose and rifaximin. Lactulose is a laxative; the recommended target is 3 to 4 bowel movements daily. Rifaximin is a poorly absorbed antibiotic that has a wide spectrum of coverage, affecting gram-negative and gram-positive aerobes and anaerobes. It wipes out the gut bacteria and so decreases the production of ammonia by the gut.
Paracentesis is recommended before TIPS placement if a large volume of ascites is present. Draining the fluid allows the liver to drop down and makes it easier to access the portal vein from the hepatic vein.
WHEN TO CONSIDER A TIPS IN ESOPHAGEAL VARICEAL BLEEDING
Acute bleeding refractory to endoscopic therapy
A TIPS remains the only choice to control acute variceal bleeding refractory to medical and endoscopic therapy (Figure 1), with a success rate of 90% to 100%.5 The urgency of TIPS placement is an independent predictor of early mortality.
Esophageal variceal rebleeding
Once varices bleed, the risk of rebleeding is higher than 50%, and rebleeding is associated with a high mortality rate. TIPS should be considered if nonselective beta-blockers and surveillance with upper endoscopy and banding fail to prevent rebleeding, with many studies showing a TIPS to be superior to pharmacologic and endoscopic therapies.6
A meta-analysis in 1999 by Papatheodoridis et al6 found that variceal rebleeding was significantly more frequent with endoscopic therapies, at 47% vs 19% with a TIPS, but the incidence of hepatic encephalopathy was higher with TIPS (34% vs 19%; P < .001), and there was no difference in mortality rates.
Hepatic encephalopathy occurs in 15% to 25% of patients after TIPS procedures. Risk factors include advanced age, poor renal function, and a history of hepatic encephalopathy. Hepatic encephalopathy can be managed with lactulose or rifaximin, or both (see above). Narcotics, antihistamines, and benzodiazepines should be avoided. In rare cases (5%) when hepatic encephalopathy is refractory to medical therapy, liver transplant should be considered.
A surgical distal splenorenal shunt is another option for patients with refractory or recurrent variceal bleeding. In a large randomized controlled trial,7 140 cirrhotic patients with recurrent variceal bleeding were randomized to receive either a distal splenorenal shunt or a TIPS. At a mean follow-up of 48 months, there was no difference in the rates of rebleeding between the two groups (5.5% with a surgical shunt vs 10.5% with a TIPS, P = .29) or in hepatic encephalopathy (50% in both groups). Survival rates were comparable between the two groups at 2 years (81% with a surgical shunt vs 88% with a TIPS) and 5 years (62% vs 61%).
Early use of TIPS after first variceal bleeding
In a 2010 randomized controlled trial,8 63 patients with cirrhosis (Child-Pugh class B or C) and acute variceal bleeding who had received standard medical and endoscopic therapy were randomized to receive either a TIPS within 72 hours of admission or long-term conservative treatment with nonselective beta-blockers and endoscopic band ligation. The 1-year actuarial probability of remaining free of rebleeding or failure to control bleeding was 50% in the conservative treatment group vs 97% in the early-TIPS group (P < .001). The 1-year actuarial survival rate was 61% in the conservative treatment group vs 86% in the early-TIPS group (P < .001).
The authors8 concluded that early use of TIPS in patients with cirrhosis and Child-Pugh scores of 7 to 13 who were hospitalized for acute variceal bleeding was associated with significant reductions in rates of treatment failure and mortality.
- Brenner D, Rippe RA. Pathogenesis of hepatic fibrosis. In: Yamada T, Alpers DH, Laine L, Kaplowitz N, Owyang C, Powell DW, editors. Textbook of Gastroenterology. 4th edition. Philadelphia, PA: Lippincott Williams & Wilkins; 2003.
- Bhogal HK, Sanyal AJ. Using transjugular intrahepatic portosystemic shunts for complications of cirrhosis. Clin Gastroenterol Hepatol 2011; 9:936–946.
- Garcia-Tsao G, Sanyal AJ, Grace ND, Carey WD; Practice Guidelines Committee of American Association for Study of Liver Diseases; Practice Parameters Committee of American College of Gastroenterology. Prevention and management of gastroesophageal varices and variceal hemorrhage in cirrhosis. Am J Gastroenterol 2007; 102:2086–2102.
- Azoulay D, Castaing D, Dennison A, Martino W, Eyraud D, Bismuth H. Transjugular intrahepatic portosystemic shunt worsens the hyperdynamic circulatory state of the cirrhotic patient: preliminary report of a prospective study. Hepatology 1994; 19:129–132.
- Rodríguez-Laiz JM, Bañares R, Echenagusia A, et al. Effects of transjugular intrahepatic portasystemic shunt (TIPS) on splanchnic and systemic hemodynamics, and hepatic function in patients with portal hypertension. Preliminary results. Dig Dis Sci 1995; 40:2121–2127.
- Papatheodoridis GV, Goulis J, Leandro G, Patch D, Burroughs AK. Transjugular intrahepatic portosystemic shunt compared with endoscopic treatment for prevention of variceal rebleeding: a meta-analysis. Hepatology 1999; 30:612–622.
- Henderson JM, Boyer TD, Kutner MH, et al; DIVERT Study Group. Distal splenorenal shunt versus transjugular intrahepatic portal systemic shunt for variceal bleeding: a randomized trial. Gastroenterology 2006; 130:1643–1651.
- García-Pagán JC, Caca K, Bureau C, et al; Early TIPS (Transjugular Intrahepatic Portosystemic Shunt) Cooperative Study Group. Early use of TIPS in patients with cirrhosis and variceal bleeding. N Engl J Med 2010; 362:2370–2379.
- Brenner D, Rippe RA. Pathogenesis of hepatic fibrosis. In: Yamada T, Alpers DH, Laine L, Kaplowitz N, Owyang C, Powell DW, editors. Textbook of Gastroenterology. 4th edition. Philadelphia, PA: Lippincott Williams & Wilkins; 2003.
- Bhogal HK, Sanyal AJ. Using transjugular intrahepatic portosystemic shunts for complications of cirrhosis. Clin Gastroenterol Hepatol 2011; 9:936–946.
- Garcia-Tsao G, Sanyal AJ, Grace ND, Carey WD; Practice Guidelines Committee of American Association for Study of Liver Diseases; Practice Parameters Committee of American College of Gastroenterology. Prevention and management of gastroesophageal varices and variceal hemorrhage in cirrhosis. Am J Gastroenterol 2007; 102:2086–2102.
- Azoulay D, Castaing D, Dennison A, Martino W, Eyraud D, Bismuth H. Transjugular intrahepatic portosystemic shunt worsens the hyperdynamic circulatory state of the cirrhotic patient: preliminary report of a prospective study. Hepatology 1994; 19:129–132.
- Rodríguez-Laiz JM, Bañares R, Echenagusia A, et al. Effects of transjugular intrahepatic portasystemic shunt (TIPS) on splanchnic and systemic hemodynamics, and hepatic function in patients with portal hypertension. Preliminary results. Dig Dis Sci 1995; 40:2121–2127.
- Papatheodoridis GV, Goulis J, Leandro G, Patch D, Burroughs AK. Transjugular intrahepatic portosystemic shunt compared with endoscopic treatment for prevention of variceal rebleeding: a meta-analysis. Hepatology 1999; 30:612–622.
- Henderson JM, Boyer TD, Kutner MH, et al; DIVERT Study Group. Distal splenorenal shunt versus transjugular intrahepatic portal systemic shunt for variceal bleeding: a randomized trial. Gastroenterology 2006; 130:1643–1651.
- García-Pagán JC, Caca K, Bureau C, et al; Early TIPS (Transjugular Intrahepatic Portosystemic Shunt) Cooperative Study Group. Early use of TIPS in patients with cirrhosis and variceal bleeding. N Engl J Med 2010; 362:2370–2379.

Minimizing use of antipsychotics

Iodine deficiency: Clinical implications
A 65-year-old woman is found to have a goiter. She is clinically euthyroid. She is a strict vegan and only uses noniodized Himalayan salt for cooking. Her thyroid gland is diffusely enlarged with no nodules. The estimated weight of the thyroid gland is 50 g (normal 10–20 g) based on ultrasonography. Her thyroid-stimulating hormone (TSH) level is 2.95 mU/L (reference range 0.5–5 mU/L), and her free thyroxine level is 0.8 ng/dL (0.7–1.8 ng/dL). Testing for TSH receptor antibody is negative. Her 24-hour urine iodine is undetectable (urine iodine concentration < 10 μg/L with urine volume 3,175 mL). What may be the cause of her goiter?
Iodine is an essential element needed for the production of thyroid hormone, which controls metabolism and plays a major role in fetal neurodevelopment. Its ionized form is called iodide. Iodine deficiency results in impairment of thyroid hormone synthesis and may lead to several undesirable consequences. Physicians should be aware of the risks iodine deficiency poses, especially during pregnancy, and should be familiar with approaches to testing and current indications for iodine supplementation.
SOURCES OF IODINE AND SALT IODIZATION
The major environmental source of iodine is the ocean. Elemental iodine in the ocean volatilizes into the atmosphere and returns to the soil by rain. The effects of glaciation, flooding, and leaching into soil have resulted in the variable geographic distribution of iodine. Mountainous areas (eg, the Alps, Andes, Himalayas) and areas with frequent flooding typically have iodine-deficient soil due to slow iodine cycling.1 Seafood is a good source of iodine because marine plants and animals concentrate iodine from seawater. The iodine content of other foods varies widely, depending on the source and any additives.
In the United States, the major sources of dietary iodine are dairy products (due to livestock iodine supplements and use of iodophors for cleaning milk udders) and iodized salt.1,2 Seafood contains a higher amount of iodine by weight than dairy products but is consumed far less than dairy.3,4 Further, the iodine content of milk can range from 88 to 168 μg per 250 mL (about 1 cup), depending on the product manufacturer. Also, iodine content is often omitted from the food label. Even if it is reported, the package labeling may not accurately predict the iodine content.5
Less common sources of iodine are radiographic contrast, bread with iodate dough conditioners, red food coloring (erythrosine), and drugs such as amiodarone.1
Using iodized salt is an effective and stable way to ensure adequate iodine intake. In the United States, only table salt is iodized, and the salt typically used in processed food has only minimal iodine content.6 Nearly 70% of the salt we ingest is from processed food. Table salt provides only 15% of dietary salt intake, and only 70% of consumers choose iodized salt for home cooking.7
IODINE REQUIREMENTS
Daily requirements of iodine suggested by the World Health Organization (WHO) and by the US Institute of Medicine are in the range of 90 to 150 µg/day.8,9 The iodine requirement is higher in pregnancy (220–250 µg/day) because of increased maternal thyroid hormone production required to maintain euthyroidism and increased renal iodine clearance, and it is even higher in lactating women (250–290 µg/day).
IODINE STATUS IN POPULATIONS
Since the establishment of universal salt iodization programs under the influence of the WHO and the International Council for Control of Iodine Deficiency Disorders (ICCIDD) in 1990, global iodine status has continued to improve. Yet only 70% of households worldwide currently have access to adequately iodized salt, because many countries lack a national program for iodine supplementation. The population of the United States was historically iodine-deficient, but since the introduction of salt iodization in the 1920s, the iodine status in the United States has been considered adequate.1
The WHO defines iodine status for a population by the median spot urinary iodine concentration. Because a urinary iodine concentration of 100 μg/L represents an iodine intake of about 150 μg/day, the WHO uses a median urinary iodine concentration of 100 to 199 μg/L to define adequate iodine intake for a nonpregnant population.9
The National Health and Nutrition Examination Survey (NHANES) found that the median urinary iodine concentration decreased by more than 50% from the 1970s to the 1990s, indicating declining iodine status in the US population.2 Of particular concern, the percentage of women of childbearing age with moderate iodine deficiency increased from 4% to 15% over this period.2 Still, the NHANES survey in 2009–2010 indicated that the overall US population is still iodine-sufficient (median urinary iodine concentration 144 μg/L).10 The decline in the US iodine status may be due to reduction of iodine content in dairy products, increased use of noniodized salt by the food industry, and recommendations to avoid salt for blood pressure control.
Although US iodine status has been considered generally adequate, iodine intake varies greatly across the population. Vegans tend to have iodine-deficient diets, while kelp consumers may have excessive iodine intake.11 Individuals with lactose intolerance are at risk of iodine deficiency, given that dairy products are a major source of iodine in the United States. Physicians should be aware of these risk factors for iodine deficiency.
PREGNANCY AND LACTATION
It is crucial to maintain euthyroidism during pregnancy. In early gestation, maternal thyroid hormone production increases 50% due to an increase in thyroid-binding globulin and stimulation by human chorionic gonadotropin. The glomerular filtration rate increases by 30% to 50% during pregnancy, thus increasing renal iodine clearance. Fetal thyroid hormone production increases during the second half of pregnancy, further contributing to increased maternal iodine requirements because iodine readily crosses the placenta.12
Women with sufficient iodine intake before and during pregnancy generally have adequate intrathyroidal iodine storage and can adapt to the increased demand for thyroid hormone throughout gestation. But in the setting of even mild iodine deficiency, total body iodine stores decline gradually from the first to third trimester of pregnancy.13
The fetal thyroid gland does not begin to concentrate iodine until 10 to 12 weeks of gestation and is not controlled by TSH until the full development of the pituitary-portal vascular system at 20 weeks of gestation.12 Therefore, the fetus relies on maternal thyroid hormone during this critical stage of neurodevelopment. Thyroid hormone is essential for oligodendrocyte differentiation and myelin distribution14 as well as fetal neuronal proliferation and migration in the first and second trimesters. Iodine deficiency leading to maternal hypothyroidism can result in irreversible fetal brain damage.
Because of the greater requirement during pregnancy, the WHO recommends using a median urinary iodine concentration of 150 to 249 μg/L to define a population that has no iodine deficiency.9 The NHANES data from 2007 to 2010 showed that pregnant US women were mildly iodine-deficient (median urinary iodine concentration 135 μg/L),10 and the National Children’s Study of 501 pregnant US women during the third trimester in 2009 to 2010 showed they had adequate iodine intake (median urinary iodine concentration 167 μg/L). Interestingly, pregnant non-Hispanic blacks were the only ethnic group with a median urinary iodine concentration less than 150 μg/L, suggesting that race or ethnicity is a predictor of iodine status in pregnant women.10
Iodine requirements during lactation
During lactation, thyroid hormone production and renal iodine clearance return to the prepregnancy state. However, a significant amount of iodine is excreted into breast milk at a concentration 20 to 50 times greater than that in plasma.15 It is recommended that lactating women continue high iodine intake to ensure sufficient iodine in breast milk to build reserves in the newborn’s thyroid gland.
The iodine requirement during lactation is 225 to 350 μg/day.16 Breast milk containing 100 to 200 μg/L of iodine appears to provide adequate iodine to meet Institute of Medicine recommendations for infants.17 The amount of iodine excreted into breast milk depends on maternal iodine intake. In the setting of iodine sufficiency, the iodine content of breast milk is 150 to 180 μg/L, but it is much lower (9–32 μg/L) in women from iodine-deficient areas, eg, the “goiter belt,” which included the Great Lakes, the Appalachians, and northwestern states. While iodized salt has virtually eliminated the goiter belt, the risk of iodine deficiency remains for people who avoid iodized salt and dairy.15
To ensure adequate iodine intake, the American Thyroid Association recommends that women receive iodine supplementation daily during pregnancy and lactation.11 However, the iodine content of prenatal multivitamins is currently not mandated in the United States. Only half of marketed prenatal vitamins in the United States contain iodine, in the form of either potassium iodide or kelp. Though most iodine-containing products claim to contain at least 150 μg of iodine per daily dose, when measured, the actual iodine content varied between 33 and 610 μg.18
CONSEQUENCES OF IODINE DEFICIENCY
Goiter
Goiter in iodine-deficient areas is considered to be an adaptation to chronic iodine deficiency. Low iodine intake leads to reduced thyroid hormone production, which in turn stimulates TSH secretion from the pituitary. TSH increases iodine uptake by the thyroid, stimulates thyroid growth, and leads to goiter development.
Initially, goiter is characterized by diffuse thyroid enlargement, but over time it may become nodular from progressive accumulation of new thyroid follicles. Goiter in children from iodine-deficient areas is diffusely enlarged, whereas in older adults it tends to be multinodular.
Iodine deficiency and chronic TSH stimulation may play a role in TSH receptor-activating mutations of thyroid follicles. These “gain-of-function” mutations are more common in the glands of patients with goiter in areas of iodine deficiency but are relatively rare in areas of iodine sufficiency.19 Toxic multinodular goiter may eventually develop, and hyperthyroidism may occur if iodine deficiency is not severe.
Goiter generally does not cause obstructive symptoms, since the thyroid usually grows outward. However, a very large goiter may descend to the thoracic inlet and compress the trachea and esophagus. The obstructive effect of a large goiter can be demonstrated by having a patient raise the arms adjacent to the face (the Pemberton maneuver). Signs suggesting obstruction are engorged neck veins, facial plethora, increased dyspnea, and stridor during the maneuver. Computed tomography of the neck and upper thorax may provide information on the degree of tracheal compression.20
Hypothyroidism
A normal or low triiodothyronine (T3), a low serum thyroxine (T4), and a variably elevated TSH are features of thyroid function tests in iodine deficiency.11,21,22 As long as daily iodine intake exceeds 50 μg/day, the absolute uptake of iodine by the thyroid gland usually remains adequate to maintain euthyroidism. Below 50 μg/day, iodine storage in the thyroid becomes depleted, leading to hypothyroidism.1
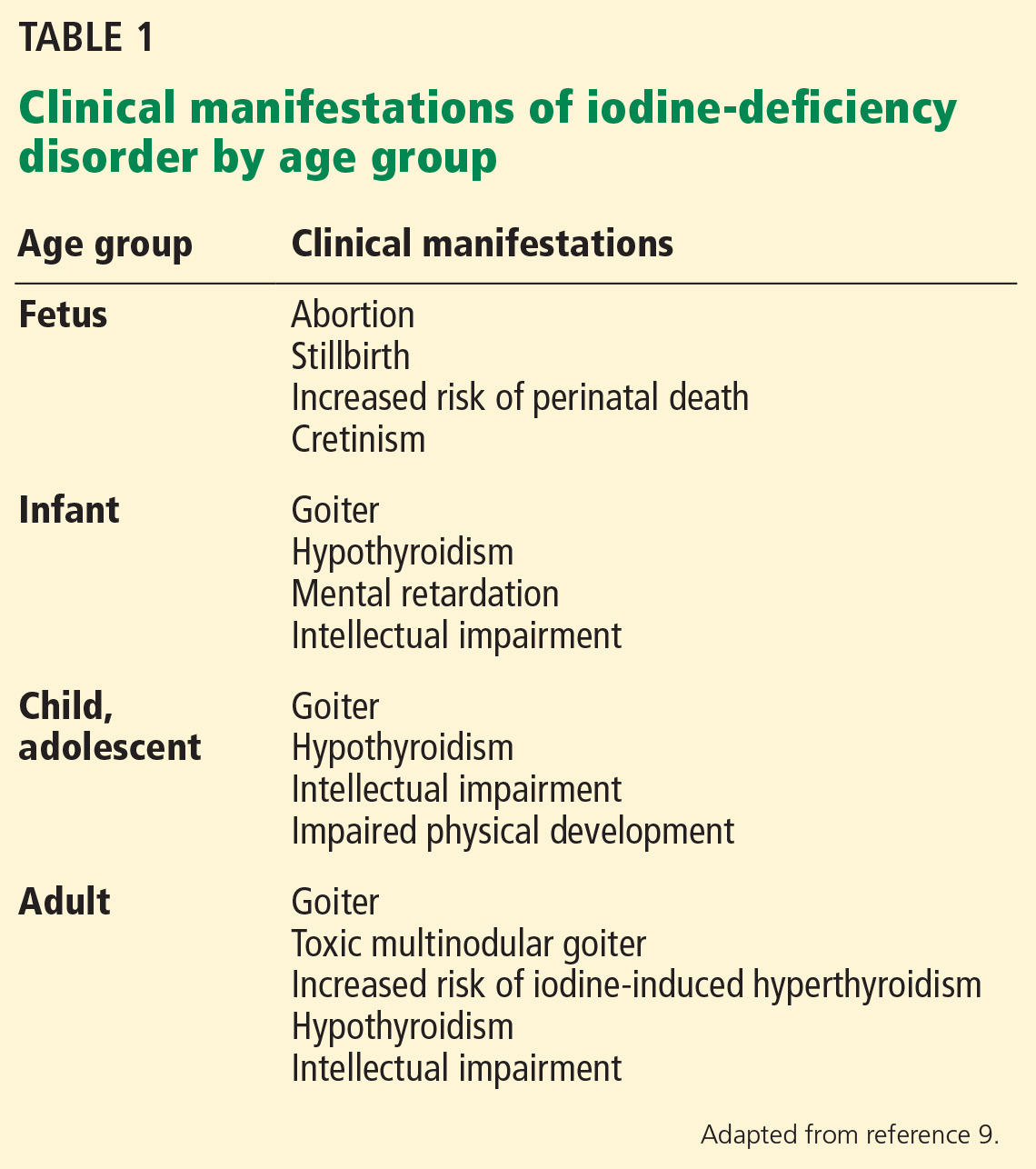
The clinical manifestations of hypothyroidism from iodine deficiency are similar to those of hypothyroidism from other causes. Because of thyroid hormone’s role in neural and somatic development, the manifestations of hypothyroidism differ among age groups (Table 1).
Cretinism
Before the development of fetal thyroid tissue in the 10th to 12th week of gestation, the fetus is dependent on maternal thyroid hormone, which crosses the placenta to support general and neural development. Iodine deficiency leading to maternal hypothyroidism (in early gestation) or inadequate fetal thyroid hormone production (in late gestation) may result in various degrees of mental retardation or lower than expected IQ.
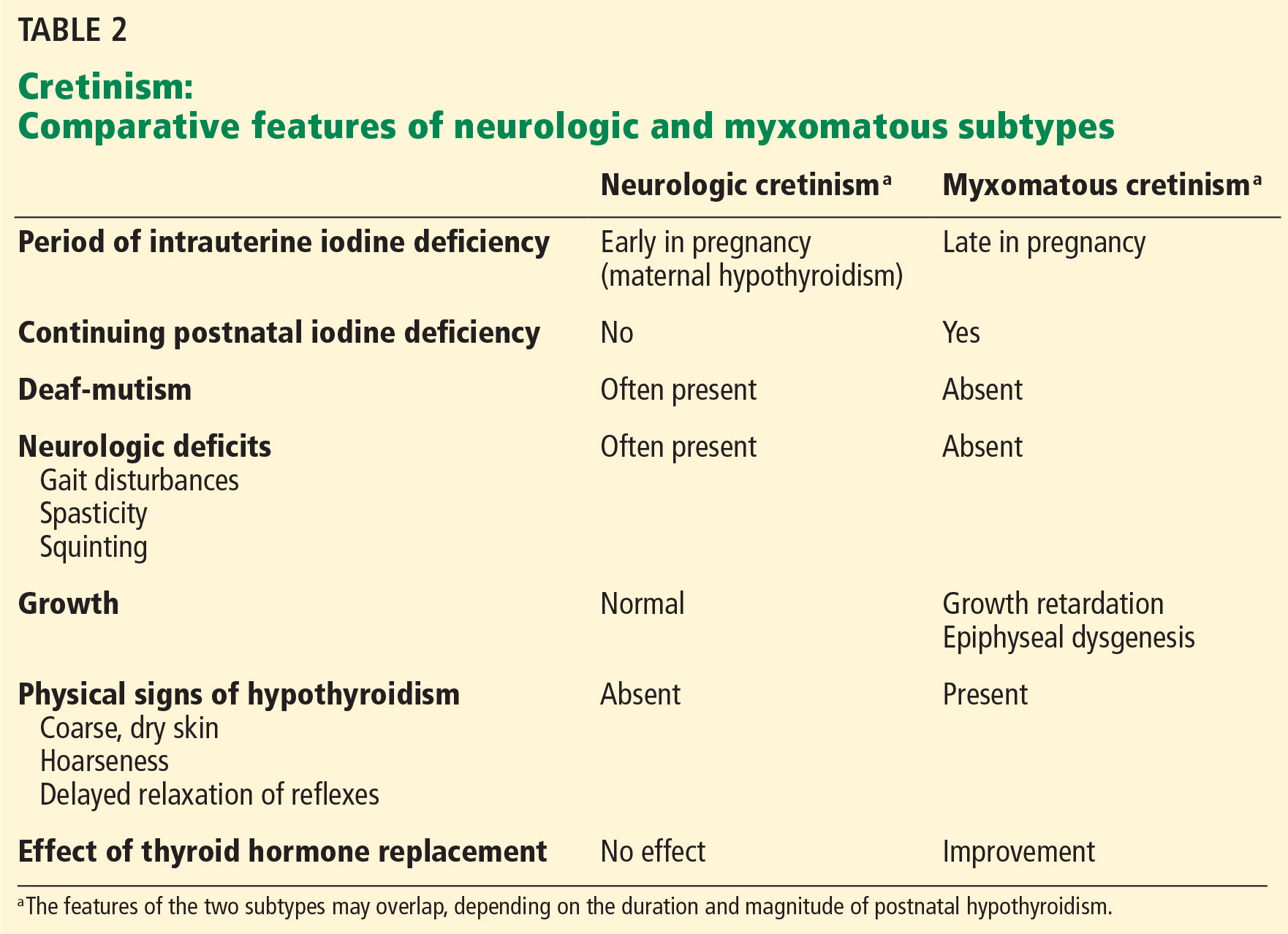
Severe iodine deficiency during gestation typically results in cretinism, characterized by severe mental retardation accompanied by other neurologic or physical defects. Cretinism is divided into two subtypes according to clinical manifestations (neurologic and myxomatous cretinism; Table 2), which may reflect the different timing of intrauterine insult to the developing fetal nervous system and whether the iodine deficiency continues into the postnatal period. Both types can be prevented by adequate maternal iodine intake before and during pregnancy.23,24
Although mild gestational iodine deficiency does not result in cretinism, it nevertheless has an adverse impact on fetal neurodevelopment and subsequent functioning. Children of mothers with mild gestational iodine deficiency were found to have reductions in spelling, grammar, and English literacy performance despite growing up in iodine-replete environments.25
Impaired cognitive development
Reduction in IQ has been noted in affected youth from regions of severe and mild iodine deficiency. A meta-analysis of studies relating iodine deficiency to cognitive development suggested that chronic moderate to severe iodine deficiency reduced expected average IQ by about 13.5 points.26
The effects of mild iodine deficiency during childhood are more difficult to quantify. The results of one study suggested that mild iodine deficiency was associated with subtle neurodevelopmental deficits and that iodine supplementation might improve cognitive function in mildly iodine-deficient children.27
In a 2009 randomized, placebo-controlled study in New Zealand, 184 children ages 10 to 13 with mild iodine deficiency (median urinary iodine concentration of 63 μg/L) received iodine supplementation (150 μg/day) or placebo for 28 weeks. Iodine supplementation increased the median urinary iodine concentration to 145 μg/L and significantly improved perceptual reasoning measures and overall cognitive score compared with placebo.28
These findings suggest that correcting mild iodine deficiency in children could improve certain components of cognition. More research is needed to understand the effects of mild iodine deficiency and iodine supplementation on cognitive function.
ASSESSING IODINE STATUS
The diagnosis of iodine deficiency is based on clinical and laboratory assessments. Clinical manifestations compatible with iodine deficiency and careful history-taking focused on the patient’s dietary iodine intake and geographic data are keys to the diagnosis.
Four main methods are used to assess iodine status at a population level: urinary iodine, serum thyroglobulin, serum TSH, and thyroid size. Urinary iodine is a sensitive marker for recent iodine intake (within days); thyroglobulin represents iodine nutrition over a period of months and thyroid size over a period of years.1
Urinary iodine
Most dietary iodine is excreted into the urine within 24 hours of ingestion, and the 24-hour urinary iodine is considered a reference standard for the measurement of individual daily iodine intake. However, the process of collection is cumbersome, and the 24-hour urinary iodine can vary from day to day in the same person, depending on the amount of iodine ingested.
A study in healthy women from an iodine-sufficient area suggested that 10 repeated 24-hour urine collections estimated the person’s iodine status at a precision of 20% because of variable daily iodine intake.29 Therefore, when necessary, several 24-hour urine iodine determinations should be performed.
A single, random, spot urinary iodine is expressed as the urinary iodine concentration and is affected by the amount of iodine and fluids the individual ingests in a day, thus resulting in high variation both within an individual person and between individuals. Expressing the urinary iodine concentration as the ratio of urine iodine to creatinine is useful in correcting for the influence of fluid intake. The ratio of urine iodine to creatinine can be used to estimate 24-hour urine iodine with the following formula: urine iodine (μg/L)/creatinine (g/L)× age- and sex-specific estimated 24-hour creatinine excretion (g/day). Another clinical use of the spot urine iodine is to screen for exposure to a large amount of iodine from a source such as radiographic contrast.30
Although individual urine iodine excretion and urine volume can vary from day to day, this variation tends to even out in a large number of samples. In study populations of at least 500, the median value of the spot urinary iodine concentration is considered a reliable measure of iodine intake in that population.30 The spot urine iodine test is convenient, making it the test of choice to study iodine status in a large cohort. The WHO recommends using the median value of the spot urine iodine to evaluate the iodine status of a population.9
Thyroglobulin
Thyroglobulin is a thyroid-specific protein involved in the synthesis of thyroid hormone. Small amounts can be detected in the blood of healthy people. In the absence of thyroid damage, the amount of serum thyroglobulin depends on thyroid cell mass and TSH stimulation. The serum level is elevated in iodine deficiency as a result of chronic TSH stimulation and thyroid hyperplasia. Thus, thyroglobulin can serve as a marker of iodine deficiency.
Serum thyroglobulin assays have been adapted for use on dried whole-blood spots, which require only a few drops of whole blood collected on filter paper and left to air-dry. The results of the dried whole-blood assay correlate closely with those of the serum assay.31 An established international dried whole-blood thyroglobulin reference range for iodine-sufficient school-age children is 4 to 40 μg/L.32 A median level of less than 13 μg/L in school-age children indicates iodine sufficiency in the population.33
Thyroid-stimulating hormone
Iodine deficiency lowers serum T4, which in turn leads to increased serum TSH. Therefore, iodine-deficient populations generally have higher TSH than iodine-sufficient groups. However, the TSH values in older children and adults with iodine deficiency are not significantly different from values of those with adequate iodine intake. Therefore, TSH is not a practical marker of iodine deficiency in the general population.
In contrast, TSH in newborns is a reasonable indicator of population iodine status. The newborn thyroid has limited iodine stores compared with that of an adult and hence a much higher iodine turnover rate. TSH from the cord blood is markedly elevated in newborns of mothers with moderate to severe iodine deficiency.34 A high prevalence of newborns with elevated TSH should therefore reflect iodine deficiency in the area where the mothers of the newborns live.
TSH is now routinely checked in newborns to screen for congenital hypothyroidism. TSH is typically checked 2 to 5 days after delivery to avoid confusion with transient physiologic TSH elevation, which occurs within a few hours after birth and decreases rapidly in 24 hours. The WHO has proposed that a more than 3% prevalence of newborns with TSH values higher than 5 mU/L from blood samples collected 3 to 4 days after birth indicates iodine deficiency in a population.1 This threshold appears to correlate well with the iodine status of the population defined by the WHO’s median urinary iodine concentration.35,36
But several other factors can influence the measurement of newborn TSH, such as prematurity, time of blood collection, maternal or newborn exposure to iodine-containing antiseptics, and the TSH assay methodology. These potential confounding factors limit the role of neonatal TSH as a reliable monitoring tool for iodine deficiency.35,37
Thyroid size

The size of the thyroid gland varies inversely with iodine intake. Thyroid size can be assessed by either palpation or ultrasonography, with the latter being more sensitive. The goiter rate in school-age children can be used to determine the severity of iodine deficiency in the population (Table 3). A goiter rate of 5% or more in school-age children suggests the presence of iodine deficiency in the community.
Although thyroid size is easy to estimate by palpation, it has low sensitivity and specificity to detect iodine deficiency and high interobserver variation. Thyroid ultrasonography provides a more precise measurement of thyroid gland volume. Zimmermann et al38 provided reference data on thyroid volume stratified by age, sex, and body surface area of school-age children in iodine-sufficient areas.38 Results of ultrasonography in a population is then compared with these reference data. The higher the percentage of the population with thyroid volume exceeding the 97th percentile of the reference range, the more severe the iodine deficiency. However, the WHO does not specify how to grade the degree of iodine deficiency based on the thyroid volume obtained with ultrasonography. Follow-up studies showed no significant correlation between urinary iodine concentration and thyroid size.39,40
Thyroid size decreases slowly after iodine repletion. Therefore, the goiter rate may remain high for several years after iodine supplementation begins.1,9
TREATMENT AND PREVENTION

Treatment of iodine deficiency should be instituted at the levels recommended by the Institute of Medicine and the WHO. The tolerable upper intake levels for iodine are outlined in Table 4. In a nonpregnant adult, 150 μg/day is sufficient for normal thyroid function. Iodine intake should be higher for pregnant and lactating women (250 μg/day according to the WHO recommendation).
Iodine supplementation is easily achieved by using iodized salt or an iodine-containing daily multivitamin.
In patients with overt hypothyroidism from iodine deficiency, we recommend initiating levothyroxine treatment along with iodine supplementation to restore euthyroidism, with consideration of possible interruption in 6 to 12 months when the urine iodine has normalized and goiter size has decreased. Thyroid function should be reassessed 4 to 6 weeks after discontinuation of levothyroxine.
At the population level, iodine deficiency can usually be prevented by iodization of food products or the water supply. In developing countries where salt iodization is not practical, iodine deficiency has been eradicated by adding iodine drops to well water or by injecting people with iodized oil.
TAKE-HOME POINTS
Iodine is essential for thyroid hormone synthesis. It can be obtained by eating iodine-containing foods or by using iodized salt. The WHO classifies iodine deficiency based on the median urinary iodine concentration. Iodine nutrition at the community level is best assessed by measurements of urinary iodine, thyroglobulin, serum TSH, and thyroid size.
Adequate iodine intake during pregnancy is important for fetal development. Iodine deficiency is associated with goiter and hypothyroidism. Severe iodine deficiency during pregnancy is associated with cretinism.
There is evidence that mild to moderate iodine deficiency can cause impaired cognitive development and that correcting the iodine deficiency can significantly improve cognitive function.
CASE FOLLOW-UP
This patient had been following a strict vegan diet with very little intake of iodized salt. Her dietary history and the presence of goiter suggested iodine deficiency. She was instructed to take an iodine supplement 150 μg/day to meet her daily requirement. After 2 months of iodine supplementation, her urine iodine concentration had increased to 58 μg/L. She remained biochemically euthyroid.
- Zimmermann MB. Iodine deficiency. Endocr Rev 2009; 30:376–408.
- Pearce EN, Andersson M, Zimmermann MB. Global iodine nutrition: where do we stand in 2013? Thyroid 2013; 23:523–528.
- Huang SW. Seafood and iodine: an analysis of a medical myth. Allergy Asthma Proc 2005; 26:468–469.
- US Census Bureau. The 2012 Statistical abstract. Health and nutrition. www.census.gov/prod/2011pubs/12statab/health.pdf. Accessed December 1, 2016.
- Pearce EN, Pino S, He X, Bazrafshan HR, Lee SL, Braverman LE. Sources of dietary iodine: bread, cows’ milk, and infant formula in the Boston area. J Clin Endocrinol Metab 2004; 89:3421–3424.
- Salt Institute. Production and Industry. www.saltinstitute.org/salt-101/production-industry. Accessed September 20, 2016.
- Dunn JT. Guarding our nation's thyroid health. J Clin Endocrinol Metab 2002; 87:486–488.
- Institute of Medicine (US) Panel on Micronutrients. Dietary reference intakes for vitamin A, vitamin K, arsenic, boron, chromium, copper, iodine, iron, manganese, molybdenum, nickel, silicon, vanadium, and zinc. Washington, DC: National Academy Press; 2001.
- World Health Organization (WHO). Assessment of iodine deficiency disorders and monitoring their elimination: a guide for programme managers. www.who.int/nutrition/publications/micronutrients/iodine_deficiency/9789241595827/en. Accessed December 1, 2016.
- Caldwell KL, Pan Y, Mortensen ME, Makhmudov A, Merrill L, Moye J. Iodine status in pregnant women in the National Children's Study and in US women (15–44 years), National Health and Nutrition Examination Survey 2005–2010. Thyroid 2013; 23:927–937.
- Public Health Committee of the American Thyroid Association; Becker DV, Braverman LE, Delange F, et al. Iodine supplementation for pregnancy and lactation—United States and Canada: recommendations of the American Thyroid Association. Thyroid 2006; 16:949–951.
- Leung AM, Pearce EN, Braverman LE. Iodine nutrition in pregnancy and lactation. Endocrinol Metab Clin North Am 2011; 40:765–777.
- Pearce EN. Iodine in pregnancy: is salt iodization enough? J Clin Endocrinol Metab 2008; 93:2466–2468.
- Younes-Rapozo V, Berendonk J, Savignon T, Manhaes AC, Barradas PC. Thyroid hormone deficiency changes the distribution of oligodendrocyte/myelin markers during oligodendroglial differentiation in vitro. Int J Dev Neurosci 2006; 24:445–453.
- Azizi F, Smyth P. Breastfeeding and maternal and infant iodine nutrition. Clin Endocrinol (Oxf) 2009; 70:803–809.
- Delange F. Iodine requirements during pregnancy, lactation and the neonatal period and indicators of optimal iodine nutrition. Public Health Nutr 2007; 10:1571–1583.
- Semba RD, Delange F. Iodine in human milk: perspectives for infant health. Nutr Rev 2001; 59:269–278.
- Leung AM, Pearce EN, Braverman LE. Iodine content of prenatal multivitamins in the United States. N Engl J Med 2009; 360:939–940.
- Tonacchera M, Agretti P, Chiovato L, et al. Activating thyrotropin receptor mutations are present in nonadenomatous hyperfunctioning nodules of toxic or autonomous multinodular goiter. J Clin Endocrinol Metab 2000; 85:2270–2274.
- Medeiros-Neto G, Camargo RY, Tomimori EK. Approach to and treatment of goiters. Med Clin North Am 2012; 96:351–368.
- Heidemann P, Stubbe P. Serum 3,5,3'-triiodothyronine, thyroxine, and thyrotropin in hypothyroid infants with congenital goiter and the response to iodine. J Clin Endocrinol Metab 1978; 47:189–192.
- Patel YC, Pharoah PO, Hornabrook RW, Hetzel BS. Serum triiodothyronine, thyroxine and thyroid-stimulating hormone in endemic goiter: a comparison of goitrous and nongoitrous subjects in New Guinea. J Clin Endocrinol Metab 1973; 37:783–789.
- Boyages SC, Halpern JP. Endemic cretinism: toward a unifying hypothesis. Thyroid 1993; 3:59–69.
- Chen ZP, Hetzel BS. Cretinism revisited. Best Pract Res Clin Endocrinol Metab 2010; 24:39–50.
- Hynes KL, Otahal P, Hay I, Burgess JR. Mild iodine deficiency during pregnancy is associated with reduced educational outcomes in the offspring: 9-year follow-up of the gestational iodine cohort. J Clin Endocrinol Metab 2013; 98:1954–1962.
- Bleichrodt N, Born MP. A metaanalysis of research on iodine and its relationship to cognitive development. In: Stanbury JB, ed. The Damaged Brain of Iodine Deficiency. 1st ed. New York, NY: Cognizant Communication; 1994:195–200.
- Melse-Boonstra A, Jaiswal N. Iodine deficiency in pregnancy, infancy and childhood and its consequences for brain development. Best Pract Res Clin Endocrinol Metab 2010; 24:29–38.
- Gordon RC, Rose MC, Skeaff SA, Gray AR, Morgan KM, Ruffman T. Iodine supplementation improves cognition in mildly iodine-deficient children. Am J Clin Nutr 2009; 90:1264–1271.
- Konig F, Andersson M, Hotz K, Aeberli I, Zimmermann MB. Ten repeat collections for urinary iodine from spot samples or 24-hour samples are needed to reliably estimate individual iodine status in women. J Nutr 2011; 141:2049–2054.
- Vejbjerg P, Knudsen N, Perrild H, et al. Estimation of iodine intake from various urinary iodine measurements in population studies. Thyroid 2009; 19:1281–1286.
- Zimmermann MB, Hess SY, Adou P, Toresanni T, Wegmuller R, Hurrell RF. Thyroid size and goiter prevalence after introduction of iodized salt: a 5-y prospective study in schoolchildren in Cote d’Ivoire. Am J Clin Nutr 2003; 77:663–667.
- Zimmermann MB, de Benoist B, Corigliano S, et al. Assessment of iodine status using dried blood spot thyroglobulin: development of reference material and establishment of an international reference range in iodine-sufficient children. J Clin Endocrinol Metab 2006; 91:4881–4887.
- Zimmermann MB, Aeberli I, Andersson M, et al. Thyroglobulin is a sensitive measure of both deficient and excess iodine intakes in children and indicates no adverse effects on thyroid function in the UIC range of 100-299 mug/L: a UNICEF/ICCIDD study group report. J Clin Endocrinol Metab 2013; 98:1271–1280.
- Thilly CH, Delange F, Lagasse R, et al. Fetal hypothyroidism and maternal thyroid status in severe endemic goiter. J Clin Endocrinol Metab 1978; 47:354–360.
- Sullivan KM, May W, Nordenberg D, Houston R, Maberly GF. Use of thyroid stimulating hormone testing in newborns to identify iodine deficiency. J Nutr 1997; 127:55–58.
- Delange F. Neonatal thyroid screening as a monitoring tool for the control of iodine deficiency. Acta Paediatr Suppl 1999; 88:21–24.
- Li M, Eastman CJ. Neonatal TSH screening: is it a sensitive and reliable tool for monitoring iodine status in populations? Best Pract Res Clin Endocrinol Metab 2010; 24:63–75.
- Zimmermann MB, Moretti D, Chaouki N, Torresani T. Development of a dried whole-blood spot thyroglobulin assay and its evaluation as an indicator of thyroid status in goitrous children receiving iodized salt. Am J Clin Nutr 2003; 77:1453–1458.
- Brahmbhatt S, Brahmbhatt RM, Boyages SC. Thyroid ultrasound is the best prevalence indicator for assessment of iodine deficiency disorders: a study in rural/tribal schoolchildren from Gujarat (Western India). Eur J Endocrinol 2000; 143:37–46.
- Moradi M, Hashemipour M, Akbari S, Kor Z, Mirbod SA, Kooshanmehr MR. Ultrasonographic evaluation of the thyroid gland volume among 8-15-year-old children in Isfahan, Iran. Adv Biomed Res 2014; 3:9.
A 65-year-old woman is found to have a goiter. She is clinically euthyroid. She is a strict vegan and only uses noniodized Himalayan salt for cooking. Her thyroid gland is diffusely enlarged with no nodules. The estimated weight of the thyroid gland is 50 g (normal 10–20 g) based on ultrasonography. Her thyroid-stimulating hormone (TSH) level is 2.95 mU/L (reference range 0.5–5 mU/L), and her free thyroxine level is 0.8 ng/dL (0.7–1.8 ng/dL). Testing for TSH receptor antibody is negative. Her 24-hour urine iodine is undetectable (urine iodine concentration < 10 μg/L with urine volume 3,175 mL). What may be the cause of her goiter?
Iodine is an essential element needed for the production of thyroid hormone, which controls metabolism and plays a major role in fetal neurodevelopment. Its ionized form is called iodide. Iodine deficiency results in impairment of thyroid hormone synthesis and may lead to several undesirable consequences. Physicians should be aware of the risks iodine deficiency poses, especially during pregnancy, and should be familiar with approaches to testing and current indications for iodine supplementation.
SOURCES OF IODINE AND SALT IODIZATION
The major environmental source of iodine is the ocean. Elemental iodine in the ocean volatilizes into the atmosphere and returns to the soil by rain. The effects of glaciation, flooding, and leaching into soil have resulted in the variable geographic distribution of iodine. Mountainous areas (eg, the Alps, Andes, Himalayas) and areas with frequent flooding typically have iodine-deficient soil due to slow iodine cycling.1 Seafood is a good source of iodine because marine plants and animals concentrate iodine from seawater. The iodine content of other foods varies widely, depending on the source and any additives.
In the United States, the major sources of dietary iodine are dairy products (due to livestock iodine supplements and use of iodophors for cleaning milk udders) and iodized salt.1,2 Seafood contains a higher amount of iodine by weight than dairy products but is consumed far less than dairy.3,4 Further, the iodine content of milk can range from 88 to 168 μg per 250 mL (about 1 cup), depending on the product manufacturer. Also, iodine content is often omitted from the food label. Even if it is reported, the package labeling may not accurately predict the iodine content.5
Less common sources of iodine are radiographic contrast, bread with iodate dough conditioners, red food coloring (erythrosine), and drugs such as amiodarone.1
Using iodized salt is an effective and stable way to ensure adequate iodine intake. In the United States, only table salt is iodized, and the salt typically used in processed food has only minimal iodine content.6 Nearly 70% of the salt we ingest is from processed food. Table salt provides only 15% of dietary salt intake, and only 70% of consumers choose iodized salt for home cooking.7
IODINE REQUIREMENTS
Daily requirements of iodine suggested by the World Health Organization (WHO) and by the US Institute of Medicine are in the range of 90 to 150 µg/day.8,9 The iodine requirement is higher in pregnancy (220–250 µg/day) because of increased maternal thyroid hormone production required to maintain euthyroidism and increased renal iodine clearance, and it is even higher in lactating women (250–290 µg/day).
IODINE STATUS IN POPULATIONS
Since the establishment of universal salt iodization programs under the influence of the WHO and the International Council for Control of Iodine Deficiency Disorders (ICCIDD) in 1990, global iodine status has continued to improve. Yet only 70% of households worldwide currently have access to adequately iodized salt, because many countries lack a national program for iodine supplementation. The population of the United States was historically iodine-deficient, but since the introduction of salt iodization in the 1920s, the iodine status in the United States has been considered adequate.1
The WHO defines iodine status for a population by the median spot urinary iodine concentration. Because a urinary iodine concentration of 100 μg/L represents an iodine intake of about 150 μg/day, the WHO uses a median urinary iodine concentration of 100 to 199 μg/L to define adequate iodine intake for a nonpregnant population.9
The National Health and Nutrition Examination Survey (NHANES) found that the median urinary iodine concentration decreased by more than 50% from the 1970s to the 1990s, indicating declining iodine status in the US population.2 Of particular concern, the percentage of women of childbearing age with moderate iodine deficiency increased from 4% to 15% over this period.2 Still, the NHANES survey in 2009–2010 indicated that the overall US population is still iodine-sufficient (median urinary iodine concentration 144 μg/L).10 The decline in the US iodine status may be due to reduction of iodine content in dairy products, increased use of noniodized salt by the food industry, and recommendations to avoid salt for blood pressure control.
Although US iodine status has been considered generally adequate, iodine intake varies greatly across the population. Vegans tend to have iodine-deficient diets, while kelp consumers may have excessive iodine intake.11 Individuals with lactose intolerance are at risk of iodine deficiency, given that dairy products are a major source of iodine in the United States. Physicians should be aware of these risk factors for iodine deficiency.
PREGNANCY AND LACTATION
It is crucial to maintain euthyroidism during pregnancy. In early gestation, maternal thyroid hormone production increases 50% due to an increase in thyroid-binding globulin and stimulation by human chorionic gonadotropin. The glomerular filtration rate increases by 30% to 50% during pregnancy, thus increasing renal iodine clearance. Fetal thyroid hormone production increases during the second half of pregnancy, further contributing to increased maternal iodine requirements because iodine readily crosses the placenta.12
Women with sufficient iodine intake before and during pregnancy generally have adequate intrathyroidal iodine storage and can adapt to the increased demand for thyroid hormone throughout gestation. But in the setting of even mild iodine deficiency, total body iodine stores decline gradually from the first to third trimester of pregnancy.13
The fetal thyroid gland does not begin to concentrate iodine until 10 to 12 weeks of gestation and is not controlled by TSH until the full development of the pituitary-portal vascular system at 20 weeks of gestation.12 Therefore, the fetus relies on maternal thyroid hormone during this critical stage of neurodevelopment. Thyroid hormone is essential for oligodendrocyte differentiation and myelin distribution14 as well as fetal neuronal proliferation and migration in the first and second trimesters. Iodine deficiency leading to maternal hypothyroidism can result in irreversible fetal brain damage.
Because of the greater requirement during pregnancy, the WHO recommends using a median urinary iodine concentration of 150 to 249 μg/L to define a population that has no iodine deficiency.9 The NHANES data from 2007 to 2010 showed that pregnant US women were mildly iodine-deficient (median urinary iodine concentration 135 μg/L),10 and the National Children’s Study of 501 pregnant US women during the third trimester in 2009 to 2010 showed they had adequate iodine intake (median urinary iodine concentration 167 μg/L). Interestingly, pregnant non-Hispanic blacks were the only ethnic group with a median urinary iodine concentration less than 150 μg/L, suggesting that race or ethnicity is a predictor of iodine status in pregnant women.10
Iodine requirements during lactation
During lactation, thyroid hormone production and renal iodine clearance return to the prepregnancy state. However, a significant amount of iodine is excreted into breast milk at a concentration 20 to 50 times greater than that in plasma.15 It is recommended that lactating women continue high iodine intake to ensure sufficient iodine in breast milk to build reserves in the newborn’s thyroid gland.
The iodine requirement during lactation is 225 to 350 μg/day.16 Breast milk containing 100 to 200 μg/L of iodine appears to provide adequate iodine to meet Institute of Medicine recommendations for infants.17 The amount of iodine excreted into breast milk depends on maternal iodine intake. In the setting of iodine sufficiency, the iodine content of breast milk is 150 to 180 μg/L, but it is much lower (9–32 μg/L) in women from iodine-deficient areas, eg, the “goiter belt,” which included the Great Lakes, the Appalachians, and northwestern states. While iodized salt has virtually eliminated the goiter belt, the risk of iodine deficiency remains for people who avoid iodized salt and dairy.15
To ensure adequate iodine intake, the American Thyroid Association recommends that women receive iodine supplementation daily during pregnancy and lactation.11 However, the iodine content of prenatal multivitamins is currently not mandated in the United States. Only half of marketed prenatal vitamins in the United States contain iodine, in the form of either potassium iodide or kelp. Though most iodine-containing products claim to contain at least 150 μg of iodine per daily dose, when measured, the actual iodine content varied between 33 and 610 μg.18
CONSEQUENCES OF IODINE DEFICIENCY
Goiter
Goiter in iodine-deficient areas is considered to be an adaptation to chronic iodine deficiency. Low iodine intake leads to reduced thyroid hormone production, which in turn stimulates TSH secretion from the pituitary. TSH increases iodine uptake by the thyroid, stimulates thyroid growth, and leads to goiter development.
Initially, goiter is characterized by diffuse thyroid enlargement, but over time it may become nodular from progressive accumulation of new thyroid follicles. Goiter in children from iodine-deficient areas is diffusely enlarged, whereas in older adults it tends to be multinodular.
Iodine deficiency and chronic TSH stimulation may play a role in TSH receptor-activating mutations of thyroid follicles. These “gain-of-function” mutations are more common in the glands of patients with goiter in areas of iodine deficiency but are relatively rare in areas of iodine sufficiency.19 Toxic multinodular goiter may eventually develop, and hyperthyroidism may occur if iodine deficiency is not severe.
Goiter generally does not cause obstructive symptoms, since the thyroid usually grows outward. However, a very large goiter may descend to the thoracic inlet and compress the trachea and esophagus. The obstructive effect of a large goiter can be demonstrated by having a patient raise the arms adjacent to the face (the Pemberton maneuver). Signs suggesting obstruction are engorged neck veins, facial plethora, increased dyspnea, and stridor during the maneuver. Computed tomography of the neck and upper thorax may provide information on the degree of tracheal compression.20
Hypothyroidism
A normal or low triiodothyronine (T3), a low serum thyroxine (T4), and a variably elevated TSH are features of thyroid function tests in iodine deficiency.11,21,22 As long as daily iodine intake exceeds 50 μg/day, the absolute uptake of iodine by the thyroid gland usually remains adequate to maintain euthyroidism. Below 50 μg/day, iodine storage in the thyroid becomes depleted, leading to hypothyroidism.1
The clinical manifestations of hypothyroidism from iodine deficiency are similar to those of hypothyroidism from other causes. Because of thyroid hormone’s role in neural and somatic development, the manifestations of hypothyroidism differ among age groups (Table 1).
Cretinism
Before the development of fetal thyroid tissue in the 10th to 12th week of gestation, the fetus is dependent on maternal thyroid hormone, which crosses the placenta to support general and neural development. Iodine deficiency leading to maternal hypothyroidism (in early gestation) or inadequate fetal thyroid hormone production (in late gestation) may result in various degrees of mental retardation or lower than expected IQ.
Severe iodine deficiency during gestation typically results in cretinism, characterized by severe mental retardation accompanied by other neurologic or physical defects. Cretinism is divided into two subtypes according to clinical manifestations (neurologic and myxomatous cretinism; Table 2), which may reflect the different timing of intrauterine insult to the developing fetal nervous system and whether the iodine deficiency continues into the postnatal period. Both types can be prevented by adequate maternal iodine intake before and during pregnancy.23,24
Although mild gestational iodine deficiency does not result in cretinism, it nevertheless has an adverse impact on fetal neurodevelopment and subsequent functioning. Children of mothers with mild gestational iodine deficiency were found to have reductions in spelling, grammar, and English literacy performance despite growing up in iodine-replete environments.25
Impaired cognitive development
Reduction in IQ has been noted in affected youth from regions of severe and mild iodine deficiency. A meta-analysis of studies relating iodine deficiency to cognitive development suggested that chronic moderate to severe iodine deficiency reduced expected average IQ by about 13.5 points.26
The effects of mild iodine deficiency during childhood are more difficult to quantify. The results of one study suggested that mild iodine deficiency was associated with subtle neurodevelopmental deficits and that iodine supplementation might improve cognitive function in mildly iodine-deficient children.27
In a 2009 randomized, placebo-controlled study in New Zealand, 184 children ages 10 to 13 with mild iodine deficiency (median urinary iodine concentration of 63 μg/L) received iodine supplementation (150 μg/day) or placebo for 28 weeks. Iodine supplementation increased the median urinary iodine concentration to 145 μg/L and significantly improved perceptual reasoning measures and overall cognitive score compared with placebo.28
These findings suggest that correcting mild iodine deficiency in children could improve certain components of cognition. More research is needed to understand the effects of mild iodine deficiency and iodine supplementation on cognitive function.
ASSESSING IODINE STATUS
The diagnosis of iodine deficiency is based on clinical and laboratory assessments. Clinical manifestations compatible with iodine deficiency and careful history-taking focused on the patient’s dietary iodine intake and geographic data are keys to the diagnosis.
Four main methods are used to assess iodine status at a population level: urinary iodine, serum thyroglobulin, serum TSH, and thyroid size. Urinary iodine is a sensitive marker for recent iodine intake (within days); thyroglobulin represents iodine nutrition over a period of months and thyroid size over a period of years.1
Urinary iodine
Most dietary iodine is excreted into the urine within 24 hours of ingestion, and the 24-hour urinary iodine is considered a reference standard for the measurement of individual daily iodine intake. However, the process of collection is cumbersome, and the 24-hour urinary iodine can vary from day to day in the same person, depending on the amount of iodine ingested.
A study in healthy women from an iodine-sufficient area suggested that 10 repeated 24-hour urine collections estimated the person’s iodine status at a precision of 20% because of variable daily iodine intake.29 Therefore, when necessary, several 24-hour urine iodine determinations should be performed.
A single, random, spot urinary iodine is expressed as the urinary iodine concentration and is affected by the amount of iodine and fluids the individual ingests in a day, thus resulting in high variation both within an individual person and between individuals. Expressing the urinary iodine concentration as the ratio of urine iodine to creatinine is useful in correcting for the influence of fluid intake. The ratio of urine iodine to creatinine can be used to estimate 24-hour urine iodine with the following formula: urine iodine (μg/L)/creatinine (g/L)× age- and sex-specific estimated 24-hour creatinine excretion (g/day). Another clinical use of the spot urine iodine is to screen for exposure to a large amount of iodine from a source such as radiographic contrast.30
Although individual urine iodine excretion and urine volume can vary from day to day, this variation tends to even out in a large number of samples. In study populations of at least 500, the median value of the spot urinary iodine concentration is considered a reliable measure of iodine intake in that population.30 The spot urine iodine test is convenient, making it the test of choice to study iodine status in a large cohort. The WHO recommends using the median value of the spot urine iodine to evaluate the iodine status of a population.9
Thyroglobulin
Thyroglobulin is a thyroid-specific protein involved in the synthesis of thyroid hormone. Small amounts can be detected in the blood of healthy people. In the absence of thyroid damage, the amount of serum thyroglobulin depends on thyroid cell mass and TSH stimulation. The serum level is elevated in iodine deficiency as a result of chronic TSH stimulation and thyroid hyperplasia. Thus, thyroglobulin can serve as a marker of iodine deficiency.
Serum thyroglobulin assays have been adapted for use on dried whole-blood spots, which require only a few drops of whole blood collected on filter paper and left to air-dry. The results of the dried whole-blood assay correlate closely with those of the serum assay.31 An established international dried whole-blood thyroglobulin reference range for iodine-sufficient school-age children is 4 to 40 μg/L.32 A median level of less than 13 μg/L in school-age children indicates iodine sufficiency in the population.33
Thyroid-stimulating hormone
Iodine deficiency lowers serum T4, which in turn leads to increased serum TSH. Therefore, iodine-deficient populations generally have higher TSH than iodine-sufficient groups. However, the TSH values in older children and adults with iodine deficiency are not significantly different from values of those with adequate iodine intake. Therefore, TSH is not a practical marker of iodine deficiency in the general population.
In contrast, TSH in newborns is a reasonable indicator of population iodine status. The newborn thyroid has limited iodine stores compared with that of an adult and hence a much higher iodine turnover rate. TSH from the cord blood is markedly elevated in newborns of mothers with moderate to severe iodine deficiency.34 A high prevalence of newborns with elevated TSH should therefore reflect iodine deficiency in the area where the mothers of the newborns live.
TSH is now routinely checked in newborns to screen for congenital hypothyroidism. TSH is typically checked 2 to 5 days after delivery to avoid confusion with transient physiologic TSH elevation, which occurs within a few hours after birth and decreases rapidly in 24 hours. The WHO has proposed that a more than 3% prevalence of newborns with TSH values higher than 5 mU/L from blood samples collected 3 to 4 days after birth indicates iodine deficiency in a population.1 This threshold appears to correlate well with the iodine status of the population defined by the WHO’s median urinary iodine concentration.35,36
But several other factors can influence the measurement of newborn TSH, such as prematurity, time of blood collection, maternal or newborn exposure to iodine-containing antiseptics, and the TSH assay methodology. These potential confounding factors limit the role of neonatal TSH as a reliable monitoring tool for iodine deficiency.35,37
Thyroid size
The size of the thyroid gland varies inversely with iodine intake. Thyroid size can be assessed by either palpation or ultrasonography, with the latter being more sensitive. The goiter rate in school-age children can be used to determine the severity of iodine deficiency in the population (Table 3). A goiter rate of 5% or more in school-age children suggests the presence of iodine deficiency in the community.
Although thyroid size is easy to estimate by palpation, it has low sensitivity and specificity to detect iodine deficiency and high interobserver variation. Thyroid ultrasonography provides a more precise measurement of thyroid gland volume. Zimmermann et al38 provided reference data on thyroid volume stratified by age, sex, and body surface area of school-age children in iodine-sufficient areas.38 Results of ultrasonography in a population is then compared with these reference data. The higher the percentage of the population with thyroid volume exceeding the 97th percentile of the reference range, the more severe the iodine deficiency. However, the WHO does not specify how to grade the degree of iodine deficiency based on the thyroid volume obtained with ultrasonography. Follow-up studies showed no significant correlation between urinary iodine concentration and thyroid size.39,40
Thyroid size decreases slowly after iodine repletion. Therefore, the goiter rate may remain high for several years after iodine supplementation begins.1,9
TREATMENT AND PREVENTION
Treatment of iodine deficiency should be instituted at the levels recommended by the Institute of Medicine and the WHO. The tolerable upper intake levels for iodine are outlined in Table 4. In a nonpregnant adult, 150 μg/day is sufficient for normal thyroid function. Iodine intake should be higher for pregnant and lactating women (250 μg/day according to the WHO recommendation).
Iodine supplementation is easily achieved by using iodized salt or an iodine-containing daily multivitamin.
In patients with overt hypothyroidism from iodine deficiency, we recommend initiating levothyroxine treatment along with iodine supplementation to restore euthyroidism, with consideration of possible interruption in 6 to 12 months when the urine iodine has normalized and goiter size has decreased. Thyroid function should be reassessed 4 to 6 weeks after discontinuation of levothyroxine.
At the population level, iodine deficiency can usually be prevented by iodization of food products or the water supply. In developing countries where salt iodization is not practical, iodine deficiency has been eradicated by adding iodine drops to well water or by injecting people with iodized oil.
TAKE-HOME POINTS
Iodine is essential for thyroid hormone synthesis. It can be obtained by eating iodine-containing foods or by using iodized salt. The WHO classifies iodine deficiency based on the median urinary iodine concentration. Iodine nutrition at the community level is best assessed by measurements of urinary iodine, thyroglobulin, serum TSH, and thyroid size.
Adequate iodine intake during pregnancy is important for fetal development. Iodine deficiency is associated with goiter and hypothyroidism. Severe iodine deficiency during pregnancy is associated with cretinism.
There is evidence that mild to moderate iodine deficiency can cause impaired cognitive development and that correcting the iodine deficiency can significantly improve cognitive function.
CASE FOLLOW-UP
This patient had been following a strict vegan diet with very little intake of iodized salt. Her dietary history and the presence of goiter suggested iodine deficiency. She was instructed to take an iodine supplement 150 μg/day to meet her daily requirement. After 2 months of iodine supplementation, her urine iodine concentration had increased to 58 μg/L. She remained biochemically euthyroid.
A 65-year-old woman is found to have a goiter. She is clinically euthyroid. She is a strict vegan and only uses noniodized Himalayan salt for cooking. Her thyroid gland is diffusely enlarged with no nodules. The estimated weight of the thyroid gland is 50 g (normal 10–20 g) based on ultrasonography. Her thyroid-stimulating hormone (TSH) level is 2.95 mU/L (reference range 0.5–5 mU/L), and her free thyroxine level is 0.8 ng/dL (0.7–1.8 ng/dL). Testing for TSH receptor antibody is negative. Her 24-hour urine iodine is undetectable (urine iodine concentration < 10 μg/L with urine volume 3,175 mL). What may be the cause of her goiter?
Iodine is an essential element needed for the production of thyroid hormone, which controls metabolism and plays a major role in fetal neurodevelopment. Its ionized form is called iodide. Iodine deficiency results in impairment of thyroid hormone synthesis and may lead to several undesirable consequences. Physicians should be aware of the risks iodine deficiency poses, especially during pregnancy, and should be familiar with approaches to testing and current indications for iodine supplementation.
SOURCES OF IODINE AND SALT IODIZATION
The major environmental source of iodine is the ocean. Elemental iodine in the ocean volatilizes into the atmosphere and returns to the soil by rain. The effects of glaciation, flooding, and leaching into soil have resulted in the variable geographic distribution of iodine. Mountainous areas (eg, the Alps, Andes, Himalayas) and areas with frequent flooding typically have iodine-deficient soil due to slow iodine cycling.1 Seafood is a good source of iodine because marine plants and animals concentrate iodine from seawater. The iodine content of other foods varies widely, depending on the source and any additives.
In the United States, the major sources of dietary iodine are dairy products (due to livestock iodine supplements and use of iodophors for cleaning milk udders) and iodized salt.1,2 Seafood contains a higher amount of iodine by weight than dairy products but is consumed far less than dairy.3,4 Further, the iodine content of milk can range from 88 to 168 μg per 250 mL (about 1 cup), depending on the product manufacturer. Also, iodine content is often omitted from the food label. Even if it is reported, the package labeling may not accurately predict the iodine content.5
Less common sources of iodine are radiographic contrast, bread with iodate dough conditioners, red food coloring (erythrosine), and drugs such as amiodarone.1
Using iodized salt is an effective and stable way to ensure adequate iodine intake. In the United States, only table salt is iodized, and the salt typically used in processed food has only minimal iodine content.6 Nearly 70% of the salt we ingest is from processed food. Table salt provides only 15% of dietary salt intake, and only 70% of consumers choose iodized salt for home cooking.7
IODINE REQUIREMENTS
Daily requirements of iodine suggested by the World Health Organization (WHO) and by the US Institute of Medicine are in the range of 90 to 150 µg/day.8,9 The iodine requirement is higher in pregnancy (220–250 µg/day) because of increased maternal thyroid hormone production required to maintain euthyroidism and increased renal iodine clearance, and it is even higher in lactating women (250–290 µg/day).
IODINE STATUS IN POPULATIONS
Since the establishment of universal salt iodization programs under the influence of the WHO and the International Council for Control of Iodine Deficiency Disorders (ICCIDD) in 1990, global iodine status has continued to improve. Yet only 70% of households worldwide currently have access to adequately iodized salt, because many countries lack a national program for iodine supplementation. The population of the United States was historically iodine-deficient, but since the introduction of salt iodization in the 1920s, the iodine status in the United States has been considered adequate.1
The WHO defines iodine status for a population by the median spot urinary iodine concentration. Because a urinary iodine concentration of 100 μg/L represents an iodine intake of about 150 μg/day, the WHO uses a median urinary iodine concentration of 100 to 199 μg/L to define adequate iodine intake for a nonpregnant population.9
The National Health and Nutrition Examination Survey (NHANES) found that the median urinary iodine concentration decreased by more than 50% from the 1970s to the 1990s, indicating declining iodine status in the US population.2 Of particular concern, the percentage of women of childbearing age with moderate iodine deficiency increased from 4% to 15% over this period.2 Still, the NHANES survey in 2009–2010 indicated that the overall US population is still iodine-sufficient (median urinary iodine concentration 144 μg/L).10 The decline in the US iodine status may be due to reduction of iodine content in dairy products, increased use of noniodized salt by the food industry, and recommendations to avoid salt for blood pressure control.
Although US iodine status has been considered generally adequate, iodine intake varies greatly across the population. Vegans tend to have iodine-deficient diets, while kelp consumers may have excessive iodine intake.11 Individuals with lactose intolerance are at risk of iodine deficiency, given that dairy products are a major source of iodine in the United States. Physicians should be aware of these risk factors for iodine deficiency.
PREGNANCY AND LACTATION
It is crucial to maintain euthyroidism during pregnancy. In early gestation, maternal thyroid hormone production increases 50% due to an increase in thyroid-binding globulin and stimulation by human chorionic gonadotropin. The glomerular filtration rate increases by 30% to 50% during pregnancy, thus increasing renal iodine clearance. Fetal thyroid hormone production increases during the second half of pregnancy, further contributing to increased maternal iodine requirements because iodine readily crosses the placenta.12
Women with sufficient iodine intake before and during pregnancy generally have adequate intrathyroidal iodine storage and can adapt to the increased demand for thyroid hormone throughout gestation. But in the setting of even mild iodine deficiency, total body iodine stores decline gradually from the first to third trimester of pregnancy.13
The fetal thyroid gland does not begin to concentrate iodine until 10 to 12 weeks of gestation and is not controlled by TSH until the full development of the pituitary-portal vascular system at 20 weeks of gestation.12 Therefore, the fetus relies on maternal thyroid hormone during this critical stage of neurodevelopment. Thyroid hormone is essential for oligodendrocyte differentiation and myelin distribution14 as well as fetal neuronal proliferation and migration in the first and second trimesters. Iodine deficiency leading to maternal hypothyroidism can result in irreversible fetal brain damage.
Because of the greater requirement during pregnancy, the WHO recommends using a median urinary iodine concentration of 150 to 249 μg/L to define a population that has no iodine deficiency.9 The NHANES data from 2007 to 2010 showed that pregnant US women were mildly iodine-deficient (median urinary iodine concentration 135 μg/L),10 and the National Children’s Study of 501 pregnant US women during the third trimester in 2009 to 2010 showed they had adequate iodine intake (median urinary iodine concentration 167 μg/L). Interestingly, pregnant non-Hispanic blacks were the only ethnic group with a median urinary iodine concentration less than 150 μg/L, suggesting that race or ethnicity is a predictor of iodine status in pregnant women.10
Iodine requirements during lactation
During lactation, thyroid hormone production and renal iodine clearance return to the prepregnancy state. However, a significant amount of iodine is excreted into breast milk at a concentration 20 to 50 times greater than that in plasma.15 It is recommended that lactating women continue high iodine intake to ensure sufficient iodine in breast milk to build reserves in the newborn’s thyroid gland.
The iodine requirement during lactation is 225 to 350 μg/day.16 Breast milk containing 100 to 200 μg/L of iodine appears to provide adequate iodine to meet Institute of Medicine recommendations for infants.17 The amount of iodine excreted into breast milk depends on maternal iodine intake. In the setting of iodine sufficiency, the iodine content of breast milk is 150 to 180 μg/L, but it is much lower (9–32 μg/L) in women from iodine-deficient areas, eg, the “goiter belt,” which included the Great Lakes, the Appalachians, and northwestern states. While iodized salt has virtually eliminated the goiter belt, the risk of iodine deficiency remains for people who avoid iodized salt and dairy.15
To ensure adequate iodine intake, the American Thyroid Association recommends that women receive iodine supplementation daily during pregnancy and lactation.11 However, the iodine content of prenatal multivitamins is currently not mandated in the United States. Only half of marketed prenatal vitamins in the United States contain iodine, in the form of either potassium iodide or kelp. Though most iodine-containing products claim to contain at least 150 μg of iodine per daily dose, when measured, the actual iodine content varied between 33 and 610 μg.18
CONSEQUENCES OF IODINE DEFICIENCY
Goiter
Goiter in iodine-deficient areas is considered to be an adaptation to chronic iodine deficiency. Low iodine intake leads to reduced thyroid hormone production, which in turn stimulates TSH secretion from the pituitary. TSH increases iodine uptake by the thyroid, stimulates thyroid growth, and leads to goiter development.
Initially, goiter is characterized by diffuse thyroid enlargement, but over time it may become nodular from progressive accumulation of new thyroid follicles. Goiter in children from iodine-deficient areas is diffusely enlarged, whereas in older adults it tends to be multinodular.
Iodine deficiency and chronic TSH stimulation may play a role in TSH receptor-activating mutations of thyroid follicles. These “gain-of-function” mutations are more common in the glands of patients with goiter in areas of iodine deficiency but are relatively rare in areas of iodine sufficiency.19 Toxic multinodular goiter may eventually develop, and hyperthyroidism may occur if iodine deficiency is not severe.
Goiter generally does not cause obstructive symptoms, since the thyroid usually grows outward. However, a very large goiter may descend to the thoracic inlet and compress the trachea and esophagus. The obstructive effect of a large goiter can be demonstrated by having a patient raise the arms adjacent to the face (the Pemberton maneuver). Signs suggesting obstruction are engorged neck veins, facial plethora, increased dyspnea, and stridor during the maneuver. Computed tomography of the neck and upper thorax may provide information on the degree of tracheal compression.20
Hypothyroidism
A normal or low triiodothyronine (T3), a low serum thyroxine (T4), and a variably elevated TSH are features of thyroid function tests in iodine deficiency.11,21,22 As long as daily iodine intake exceeds 50 μg/day, the absolute uptake of iodine by the thyroid gland usually remains adequate to maintain euthyroidism. Below 50 μg/day, iodine storage in the thyroid becomes depleted, leading to hypothyroidism.1
The clinical manifestations of hypothyroidism from iodine deficiency are similar to those of hypothyroidism from other causes. Because of thyroid hormone’s role in neural and somatic development, the manifestations of hypothyroidism differ among age groups (Table 1).
Cretinism
Before the development of fetal thyroid tissue in the 10th to 12th week of gestation, the fetus is dependent on maternal thyroid hormone, which crosses the placenta to support general and neural development. Iodine deficiency leading to maternal hypothyroidism (in early gestation) or inadequate fetal thyroid hormone production (in late gestation) may result in various degrees of mental retardation or lower than expected IQ.
Severe iodine deficiency during gestation typically results in cretinism, characterized by severe mental retardation accompanied by other neurologic or physical defects. Cretinism is divided into two subtypes according to clinical manifestations (neurologic and myxomatous cretinism; Table 2), which may reflect the different timing of intrauterine insult to the developing fetal nervous system and whether the iodine deficiency continues into the postnatal period. Both types can be prevented by adequate maternal iodine intake before and during pregnancy.23,24
Although mild gestational iodine deficiency does not result in cretinism, it nevertheless has an adverse impact on fetal neurodevelopment and subsequent functioning. Children of mothers with mild gestational iodine deficiency were found to have reductions in spelling, grammar, and English literacy performance despite growing up in iodine-replete environments.25
Impaired cognitive development
Reduction in IQ has been noted in affected youth from regions of severe and mild iodine deficiency. A meta-analysis of studies relating iodine deficiency to cognitive development suggested that chronic moderate to severe iodine deficiency reduced expected average IQ by about 13.5 points.26
The effects of mild iodine deficiency during childhood are more difficult to quantify. The results of one study suggested that mild iodine deficiency was associated with subtle neurodevelopmental deficits and that iodine supplementation might improve cognitive function in mildly iodine-deficient children.27
In a 2009 randomized, placebo-controlled study in New Zealand, 184 children ages 10 to 13 with mild iodine deficiency (median urinary iodine concentration of 63 μg/L) received iodine supplementation (150 μg/day) or placebo for 28 weeks. Iodine supplementation increased the median urinary iodine concentration to 145 μg/L and significantly improved perceptual reasoning measures and overall cognitive score compared with placebo.28
These findings suggest that correcting mild iodine deficiency in children could improve certain components of cognition. More research is needed to understand the effects of mild iodine deficiency and iodine supplementation on cognitive function.
ASSESSING IODINE STATUS
The diagnosis of iodine deficiency is based on clinical and laboratory assessments. Clinical manifestations compatible with iodine deficiency and careful history-taking focused on the patient’s dietary iodine intake and geographic data are keys to the diagnosis.
Four main methods are used to assess iodine status at a population level: urinary iodine, serum thyroglobulin, serum TSH, and thyroid size. Urinary iodine is a sensitive marker for recent iodine intake (within days); thyroglobulin represents iodine nutrition over a period of months and thyroid size over a period of years.1
Urinary iodine
Most dietary iodine is excreted into the urine within 24 hours of ingestion, and the 24-hour urinary iodine is considered a reference standard for the measurement of individual daily iodine intake. However, the process of collection is cumbersome, and the 24-hour urinary iodine can vary from day to day in the same person, depending on the amount of iodine ingested.
A study in healthy women from an iodine-sufficient area suggested that 10 repeated 24-hour urine collections estimated the person’s iodine status at a precision of 20% because of variable daily iodine intake.29 Therefore, when necessary, several 24-hour urine iodine determinations should be performed.
A single, random, spot urinary iodine is expressed as the urinary iodine concentration and is affected by the amount of iodine and fluids the individual ingests in a day, thus resulting in high variation both within an individual person and between individuals. Expressing the urinary iodine concentration as the ratio of urine iodine to creatinine is useful in correcting for the influence of fluid intake. The ratio of urine iodine to creatinine can be used to estimate 24-hour urine iodine with the following formula: urine iodine (μg/L)/creatinine (g/L)× age- and sex-specific estimated 24-hour creatinine excretion (g/day). Another clinical use of the spot urine iodine is to screen for exposure to a large amount of iodine from a source such as radiographic contrast.30
Although individual urine iodine excretion and urine volume can vary from day to day, this variation tends to even out in a large number of samples. In study populations of at least 500, the median value of the spot urinary iodine concentration is considered a reliable measure of iodine intake in that population.30 The spot urine iodine test is convenient, making it the test of choice to study iodine status in a large cohort. The WHO recommends using the median value of the spot urine iodine to evaluate the iodine status of a population.9
Thyroglobulin
Thyroglobulin is a thyroid-specific protein involved in the synthesis of thyroid hormone. Small amounts can be detected in the blood of healthy people. In the absence of thyroid damage, the amount of serum thyroglobulin depends on thyroid cell mass and TSH stimulation. The serum level is elevated in iodine deficiency as a result of chronic TSH stimulation and thyroid hyperplasia. Thus, thyroglobulin can serve as a marker of iodine deficiency.
Serum thyroglobulin assays have been adapted for use on dried whole-blood spots, which require only a few drops of whole blood collected on filter paper and left to air-dry. The results of the dried whole-blood assay correlate closely with those of the serum assay.31 An established international dried whole-blood thyroglobulin reference range for iodine-sufficient school-age children is 4 to 40 μg/L.32 A median level of less than 13 μg/L in school-age children indicates iodine sufficiency in the population.33
Thyroid-stimulating hormone
Iodine deficiency lowers serum T4, which in turn leads to increased serum TSH. Therefore, iodine-deficient populations generally have higher TSH than iodine-sufficient groups. However, the TSH values in older children and adults with iodine deficiency are not significantly different from values of those with adequate iodine intake. Therefore, TSH is not a practical marker of iodine deficiency in the general population.
In contrast, TSH in newborns is a reasonable indicator of population iodine status. The newborn thyroid has limited iodine stores compared with that of an adult and hence a much higher iodine turnover rate. TSH from the cord blood is markedly elevated in newborns of mothers with moderate to severe iodine deficiency.34 A high prevalence of newborns with elevated TSH should therefore reflect iodine deficiency in the area where the mothers of the newborns live.
TSH is now routinely checked in newborns to screen for congenital hypothyroidism. TSH is typically checked 2 to 5 days after delivery to avoid confusion with transient physiologic TSH elevation, which occurs within a few hours after birth and decreases rapidly in 24 hours. The WHO has proposed that a more than 3% prevalence of newborns with TSH values higher than 5 mU/L from blood samples collected 3 to 4 days after birth indicates iodine deficiency in a population.1 This threshold appears to correlate well with the iodine status of the population defined by the WHO’s median urinary iodine concentration.35,36
But several other factors can influence the measurement of newborn TSH, such as prematurity, time of blood collection, maternal or newborn exposure to iodine-containing antiseptics, and the TSH assay methodology. These potential confounding factors limit the role of neonatal TSH as a reliable monitoring tool for iodine deficiency.35,37
Thyroid size
The size of the thyroid gland varies inversely with iodine intake. Thyroid size can be assessed by either palpation or ultrasonography, with the latter being more sensitive. The goiter rate in school-age children can be used to determine the severity of iodine deficiency in the population (Table 3). A goiter rate of 5% or more in school-age children suggests the presence of iodine deficiency in the community.
Although thyroid size is easy to estimate by palpation, it has low sensitivity and specificity to detect iodine deficiency and high interobserver variation. Thyroid ultrasonography provides a more precise measurement of thyroid gland volume. Zimmermann et al38 provided reference data on thyroid volume stratified by age, sex, and body surface area of school-age children in iodine-sufficient areas.38 Results of ultrasonography in a population is then compared with these reference data. The higher the percentage of the population with thyroid volume exceeding the 97th percentile of the reference range, the more severe the iodine deficiency. However, the WHO does not specify how to grade the degree of iodine deficiency based on the thyroid volume obtained with ultrasonography. Follow-up studies showed no significant correlation between urinary iodine concentration and thyroid size.39,40
Thyroid size decreases slowly after iodine repletion. Therefore, the goiter rate may remain high for several years after iodine supplementation begins.1,9
TREATMENT AND PREVENTION
Treatment of iodine deficiency should be instituted at the levels recommended by the Institute of Medicine and the WHO. The tolerable upper intake levels for iodine are outlined in Table 4. In a nonpregnant adult, 150 μg/day is sufficient for normal thyroid function. Iodine intake should be higher for pregnant and lactating women (250 μg/day according to the WHO recommendation).
Iodine supplementation is easily achieved by using iodized salt or an iodine-containing daily multivitamin.
In patients with overt hypothyroidism from iodine deficiency, we recommend initiating levothyroxine treatment along with iodine supplementation to restore euthyroidism, with consideration of possible interruption in 6 to 12 months when the urine iodine has normalized and goiter size has decreased. Thyroid function should be reassessed 4 to 6 weeks after discontinuation of levothyroxine.
At the population level, iodine deficiency can usually be prevented by iodization of food products or the water supply. In developing countries where salt iodization is not practical, iodine deficiency has been eradicated by adding iodine drops to well water or by injecting people with iodized oil.
TAKE-HOME POINTS
Iodine is essential for thyroid hormone synthesis. It can be obtained by eating iodine-containing foods or by using iodized salt. The WHO classifies iodine deficiency based on the median urinary iodine concentration. Iodine nutrition at the community level is best assessed by measurements of urinary iodine, thyroglobulin, serum TSH, and thyroid size.
Adequate iodine intake during pregnancy is important for fetal development. Iodine deficiency is associated with goiter and hypothyroidism. Severe iodine deficiency during pregnancy is associated with cretinism.
There is evidence that mild to moderate iodine deficiency can cause impaired cognitive development and that correcting the iodine deficiency can significantly improve cognitive function.
CASE FOLLOW-UP
This patient had been following a strict vegan diet with very little intake of iodized salt. Her dietary history and the presence of goiter suggested iodine deficiency. She was instructed to take an iodine supplement 150 μg/day to meet her daily requirement. After 2 months of iodine supplementation, her urine iodine concentration had increased to 58 μg/L. She remained biochemically euthyroid.
- Zimmermann MB. Iodine deficiency. Endocr Rev 2009; 30:376–408.
- Pearce EN, Andersson M, Zimmermann MB. Global iodine nutrition: where do we stand in 2013? Thyroid 2013; 23:523–528.
- Huang SW. Seafood and iodine: an analysis of a medical myth. Allergy Asthma Proc 2005; 26:468–469.
- US Census Bureau. The 2012 Statistical abstract. Health and nutrition. www.census.gov/prod/2011pubs/12statab/health.pdf. Accessed December 1, 2016.
- Pearce EN, Pino S, He X, Bazrafshan HR, Lee SL, Braverman LE. Sources of dietary iodine: bread, cows’ milk, and infant formula in the Boston area. J Clin Endocrinol Metab 2004; 89:3421–3424.
- Salt Institute. Production and Industry. www.saltinstitute.org/salt-101/production-industry. Accessed September 20, 2016.
- Dunn JT. Guarding our nation's thyroid health. J Clin Endocrinol Metab 2002; 87:486–488.
- Institute of Medicine (US) Panel on Micronutrients. Dietary reference intakes for vitamin A, vitamin K, arsenic, boron, chromium, copper, iodine, iron, manganese, molybdenum, nickel, silicon, vanadium, and zinc. Washington, DC: National Academy Press; 2001.
- World Health Organization (WHO). Assessment of iodine deficiency disorders and monitoring their elimination: a guide for programme managers. www.who.int/nutrition/publications/micronutrients/iodine_deficiency/9789241595827/en. Accessed December 1, 2016.
- Caldwell KL, Pan Y, Mortensen ME, Makhmudov A, Merrill L, Moye J. Iodine status in pregnant women in the National Children's Study and in US women (15–44 years), National Health and Nutrition Examination Survey 2005–2010. Thyroid 2013; 23:927–937.
- Public Health Committee of the American Thyroid Association; Becker DV, Braverman LE, Delange F, et al. Iodine supplementation for pregnancy and lactation—United States and Canada: recommendations of the American Thyroid Association. Thyroid 2006; 16:949–951.
- Leung AM, Pearce EN, Braverman LE. Iodine nutrition in pregnancy and lactation. Endocrinol Metab Clin North Am 2011; 40:765–777.
- Pearce EN. Iodine in pregnancy: is salt iodization enough? J Clin Endocrinol Metab 2008; 93:2466–2468.
- Younes-Rapozo V, Berendonk J, Savignon T, Manhaes AC, Barradas PC. Thyroid hormone deficiency changes the distribution of oligodendrocyte/myelin markers during oligodendroglial differentiation in vitro. Int J Dev Neurosci 2006; 24:445–453.
- Azizi F, Smyth P. Breastfeeding and maternal and infant iodine nutrition. Clin Endocrinol (Oxf) 2009; 70:803–809.
- Delange F. Iodine requirements during pregnancy, lactation and the neonatal period and indicators of optimal iodine nutrition. Public Health Nutr 2007; 10:1571–1583.
- Semba RD, Delange F. Iodine in human milk: perspectives for infant health. Nutr Rev 2001; 59:269–278.
- Leung AM, Pearce EN, Braverman LE. Iodine content of prenatal multivitamins in the United States. N Engl J Med 2009; 360:939–940.
- Tonacchera M, Agretti P, Chiovato L, et al. Activating thyrotropin receptor mutations are present in nonadenomatous hyperfunctioning nodules of toxic or autonomous multinodular goiter. J Clin Endocrinol Metab 2000; 85:2270–2274.
- Medeiros-Neto G, Camargo RY, Tomimori EK. Approach to and treatment of goiters. Med Clin North Am 2012; 96:351–368.
- Heidemann P, Stubbe P. Serum 3,5,3'-triiodothyronine, thyroxine, and thyrotropin in hypothyroid infants with congenital goiter and the response to iodine. J Clin Endocrinol Metab 1978; 47:189–192.
- Patel YC, Pharoah PO, Hornabrook RW, Hetzel BS. Serum triiodothyronine, thyroxine and thyroid-stimulating hormone in endemic goiter: a comparison of goitrous and nongoitrous subjects in New Guinea. J Clin Endocrinol Metab 1973; 37:783–789.
- Boyages SC, Halpern JP. Endemic cretinism: toward a unifying hypothesis. Thyroid 1993; 3:59–69.
- Chen ZP, Hetzel BS. Cretinism revisited. Best Pract Res Clin Endocrinol Metab 2010; 24:39–50.
- Hynes KL, Otahal P, Hay I, Burgess JR. Mild iodine deficiency during pregnancy is associated with reduced educational outcomes in the offspring: 9-year follow-up of the gestational iodine cohort. J Clin Endocrinol Metab 2013; 98:1954–1962.
- Bleichrodt N, Born MP. A metaanalysis of research on iodine and its relationship to cognitive development. In: Stanbury JB, ed. The Damaged Brain of Iodine Deficiency. 1st ed. New York, NY: Cognizant Communication; 1994:195–200.
- Melse-Boonstra A, Jaiswal N. Iodine deficiency in pregnancy, infancy and childhood and its consequences for brain development. Best Pract Res Clin Endocrinol Metab 2010; 24:29–38.
- Gordon RC, Rose MC, Skeaff SA, Gray AR, Morgan KM, Ruffman T. Iodine supplementation improves cognition in mildly iodine-deficient children. Am J Clin Nutr 2009; 90:1264–1271.
- Konig F, Andersson M, Hotz K, Aeberli I, Zimmermann MB. Ten repeat collections for urinary iodine from spot samples or 24-hour samples are needed to reliably estimate individual iodine status in women. J Nutr 2011; 141:2049–2054.
- Vejbjerg P, Knudsen N, Perrild H, et al. Estimation of iodine intake from various urinary iodine measurements in population studies. Thyroid 2009; 19:1281–1286.
- Zimmermann MB, Hess SY, Adou P, Toresanni T, Wegmuller R, Hurrell RF. Thyroid size and goiter prevalence after introduction of iodized salt: a 5-y prospective study in schoolchildren in Cote d’Ivoire. Am J Clin Nutr 2003; 77:663–667.
- Zimmermann MB, de Benoist B, Corigliano S, et al. Assessment of iodine status using dried blood spot thyroglobulin: development of reference material and establishment of an international reference range in iodine-sufficient children. J Clin Endocrinol Metab 2006; 91:4881–4887.
- Zimmermann MB, Aeberli I, Andersson M, et al. Thyroglobulin is a sensitive measure of both deficient and excess iodine intakes in children and indicates no adverse effects on thyroid function in the UIC range of 100-299 mug/L: a UNICEF/ICCIDD study group report. J Clin Endocrinol Metab 2013; 98:1271–1280.
- Thilly CH, Delange F, Lagasse R, et al. Fetal hypothyroidism and maternal thyroid status in severe endemic goiter. J Clin Endocrinol Metab 1978; 47:354–360.
- Sullivan KM, May W, Nordenberg D, Houston R, Maberly GF. Use of thyroid stimulating hormone testing in newborns to identify iodine deficiency. J Nutr 1997; 127:55–58.
- Delange F. Neonatal thyroid screening as a monitoring tool for the control of iodine deficiency. Acta Paediatr Suppl 1999; 88:21–24.
- Li M, Eastman CJ. Neonatal TSH screening: is it a sensitive and reliable tool for monitoring iodine status in populations? Best Pract Res Clin Endocrinol Metab 2010; 24:63–75.
- Zimmermann MB, Moretti D, Chaouki N, Torresani T. Development of a dried whole-blood spot thyroglobulin assay and its evaluation as an indicator of thyroid status in goitrous children receiving iodized salt. Am J Clin Nutr 2003; 77:1453–1458.
- Brahmbhatt S, Brahmbhatt RM, Boyages SC. Thyroid ultrasound is the best prevalence indicator for assessment of iodine deficiency disorders: a study in rural/tribal schoolchildren from Gujarat (Western India). Eur J Endocrinol 2000; 143:37–46.
- Moradi M, Hashemipour M, Akbari S, Kor Z, Mirbod SA, Kooshanmehr MR. Ultrasonographic evaluation of the thyroid gland volume among 8-15-year-old children in Isfahan, Iran. Adv Biomed Res 2014; 3:9.
- Zimmermann MB. Iodine deficiency. Endocr Rev 2009; 30:376–408.
- Pearce EN, Andersson M, Zimmermann MB. Global iodine nutrition: where do we stand in 2013? Thyroid 2013; 23:523–528.
- Huang SW. Seafood and iodine: an analysis of a medical myth. Allergy Asthma Proc 2005; 26:468–469.
- US Census Bureau. The 2012 Statistical abstract. Health and nutrition. www.census.gov/prod/2011pubs/12statab/health.pdf. Accessed December 1, 2016.
- Pearce EN, Pino S, He X, Bazrafshan HR, Lee SL, Braverman LE. Sources of dietary iodine: bread, cows’ milk, and infant formula in the Boston area. J Clin Endocrinol Metab 2004; 89:3421–3424.
- Salt Institute. Production and Industry. www.saltinstitute.org/salt-101/production-industry. Accessed September 20, 2016.
- Dunn JT. Guarding our nation's thyroid health. J Clin Endocrinol Metab 2002; 87:486–488.
- Institute of Medicine (US) Panel on Micronutrients. Dietary reference intakes for vitamin A, vitamin K, arsenic, boron, chromium, copper, iodine, iron, manganese, molybdenum, nickel, silicon, vanadium, and zinc. Washington, DC: National Academy Press; 2001.
- World Health Organization (WHO). Assessment of iodine deficiency disorders and monitoring their elimination: a guide for programme managers. www.who.int/nutrition/publications/micronutrients/iodine_deficiency/9789241595827/en. Accessed December 1, 2016.
- Caldwell KL, Pan Y, Mortensen ME, Makhmudov A, Merrill L, Moye J. Iodine status in pregnant women in the National Children's Study and in US women (15–44 years), National Health and Nutrition Examination Survey 2005–2010. Thyroid 2013; 23:927–937.
- Public Health Committee of the American Thyroid Association; Becker DV, Braverman LE, Delange F, et al. Iodine supplementation for pregnancy and lactation—United States and Canada: recommendations of the American Thyroid Association. Thyroid 2006; 16:949–951.
- Leung AM, Pearce EN, Braverman LE. Iodine nutrition in pregnancy and lactation. Endocrinol Metab Clin North Am 2011; 40:765–777.
- Pearce EN. Iodine in pregnancy: is salt iodization enough? J Clin Endocrinol Metab 2008; 93:2466–2468.
- Younes-Rapozo V, Berendonk J, Savignon T, Manhaes AC, Barradas PC. Thyroid hormone deficiency changes the distribution of oligodendrocyte/myelin markers during oligodendroglial differentiation in vitro. Int J Dev Neurosci 2006; 24:445–453.
- Azizi F, Smyth P. Breastfeeding and maternal and infant iodine nutrition. Clin Endocrinol (Oxf) 2009; 70:803–809.
- Delange F. Iodine requirements during pregnancy, lactation and the neonatal period and indicators of optimal iodine nutrition. Public Health Nutr 2007; 10:1571–1583.
- Semba RD, Delange F. Iodine in human milk: perspectives for infant health. Nutr Rev 2001; 59:269–278.
- Leung AM, Pearce EN, Braverman LE. Iodine content of prenatal multivitamins in the United States. N Engl J Med 2009; 360:939–940.
- Tonacchera M, Agretti P, Chiovato L, et al. Activating thyrotropin receptor mutations are present in nonadenomatous hyperfunctioning nodules of toxic or autonomous multinodular goiter. J Clin Endocrinol Metab 2000; 85:2270–2274.
- Medeiros-Neto G, Camargo RY, Tomimori EK. Approach to and treatment of goiters. Med Clin North Am 2012; 96:351–368.
- Heidemann P, Stubbe P. Serum 3,5,3'-triiodothyronine, thyroxine, and thyrotropin in hypothyroid infants with congenital goiter and the response to iodine. J Clin Endocrinol Metab 1978; 47:189–192.
- Patel YC, Pharoah PO, Hornabrook RW, Hetzel BS. Serum triiodothyronine, thyroxine and thyroid-stimulating hormone in endemic goiter: a comparison of goitrous and nongoitrous subjects in New Guinea. J Clin Endocrinol Metab 1973; 37:783–789.
- Boyages SC, Halpern JP. Endemic cretinism: toward a unifying hypothesis. Thyroid 1993; 3:59–69.
- Chen ZP, Hetzel BS. Cretinism revisited. Best Pract Res Clin Endocrinol Metab 2010; 24:39–50.
- Hynes KL, Otahal P, Hay I, Burgess JR. Mild iodine deficiency during pregnancy is associated with reduced educational outcomes in the offspring: 9-year follow-up of the gestational iodine cohort. J Clin Endocrinol Metab 2013; 98:1954–1962.
- Bleichrodt N, Born MP. A metaanalysis of research on iodine and its relationship to cognitive development. In: Stanbury JB, ed. The Damaged Brain of Iodine Deficiency. 1st ed. New York, NY: Cognizant Communication; 1994:195–200.
- Melse-Boonstra A, Jaiswal N. Iodine deficiency in pregnancy, infancy and childhood and its consequences for brain development. Best Pract Res Clin Endocrinol Metab 2010; 24:29–38.
- Gordon RC, Rose MC, Skeaff SA, Gray AR, Morgan KM, Ruffman T. Iodine supplementation improves cognition in mildly iodine-deficient children. Am J Clin Nutr 2009; 90:1264–1271.
- Konig F, Andersson M, Hotz K, Aeberli I, Zimmermann MB. Ten repeat collections for urinary iodine from spot samples or 24-hour samples are needed to reliably estimate individual iodine status in women. J Nutr 2011; 141:2049–2054.
- Vejbjerg P, Knudsen N, Perrild H, et al. Estimation of iodine intake from various urinary iodine measurements in population studies. Thyroid 2009; 19:1281–1286.
- Zimmermann MB, Hess SY, Adou P, Toresanni T, Wegmuller R, Hurrell RF. Thyroid size and goiter prevalence after introduction of iodized salt: a 5-y prospective study in schoolchildren in Cote d’Ivoire. Am J Clin Nutr 2003; 77:663–667.
- Zimmermann MB, de Benoist B, Corigliano S, et al. Assessment of iodine status using dried blood spot thyroglobulin: development of reference material and establishment of an international reference range in iodine-sufficient children. J Clin Endocrinol Metab 2006; 91:4881–4887.
- Zimmermann MB, Aeberli I, Andersson M, et al. Thyroglobulin is a sensitive measure of both deficient and excess iodine intakes in children and indicates no adverse effects on thyroid function in the UIC range of 100-299 mug/L: a UNICEF/ICCIDD study group report. J Clin Endocrinol Metab 2013; 98:1271–1280.
- Thilly CH, Delange F, Lagasse R, et al. Fetal hypothyroidism and maternal thyroid status in severe endemic goiter. J Clin Endocrinol Metab 1978; 47:354–360.
- Sullivan KM, May W, Nordenberg D, Houston R, Maberly GF. Use of thyroid stimulating hormone testing in newborns to identify iodine deficiency. J Nutr 1997; 127:55–58.
- Delange F. Neonatal thyroid screening as a monitoring tool for the control of iodine deficiency. Acta Paediatr Suppl 1999; 88:21–24.
- Li M, Eastman CJ. Neonatal TSH screening: is it a sensitive and reliable tool for monitoring iodine status in populations? Best Pract Res Clin Endocrinol Metab 2010; 24:63–75.
- Zimmermann MB, Moretti D, Chaouki N, Torresani T. Development of a dried whole-blood spot thyroglobulin assay and its evaluation as an indicator of thyroid status in goitrous children receiving iodized salt. Am J Clin Nutr 2003; 77:1453–1458.
- Brahmbhatt S, Brahmbhatt RM, Boyages SC. Thyroid ultrasound is the best prevalence indicator for assessment of iodine deficiency disorders: a study in rural/tribal schoolchildren from Gujarat (Western India). Eur J Endocrinol 2000; 143:37–46.
- Moradi M, Hashemipour M, Akbari S, Kor Z, Mirbod SA, Kooshanmehr MR. Ultrasonographic evaluation of the thyroid gland volume among 8-15-year-old children in Isfahan, Iran. Adv Biomed Res 2014; 3:9.
KEY POINTS
- Adequate iodine intake during pregnancy is critical for normal fetal development.
- Major sources of dietary iodine in the United States are dairy products and iodized salt.
- The daily iodine requirement for nonpregnant adults is 150 µg, and for pregnant women it is 220 to 250 μg. Pregnant and lactating women should take a daily iodine supplement to ensure adequate iodine intake.
- Assessing the risk of iodine deficiency from clinical signs and from the history is key to diagnosing iodine deficiency. Individual urine iodine concentrations may vary from day to day. Repeated samples can be used to confirm iodine deficiency.

Vulvovaginitis: Find the cause to treat it
Although vulvovaginitis has several possible causes, the typical presenting symptoms are similar regardless of the cause: itching, burning, and vaginal discharge. Physical examination often reveals atrophy, redness, excoriations, and fissures in the vulvovaginal and perianal areas. Determining the cause is key to successful treatment.
This article reviews the diagnosis and treatment of many common and less common infectious and noninfectious causes of vulvovaginitis, the use of special tests, and the management of persistent cases.
DIAGNOSIS CAN BE CHALLENGING

Vulvar and vaginal symptoms are most commonly caused by local infections, but other causes must be also be considered, including several noninfectious ones (Table 1). Challenges in diagnosing vulvovaginitis are many and include distinguishing contact from allergic dermatitis, recognizing vaginal atrophy, and recognizing a parasitic infection. Determining whether a patient has an infectious process is important so that antibiotics can be used only when truly needed.
Foreign bodies in the vagina should also be considered, especially in children,1 as should sexual abuse. A 15-year retrospective review of prepubertal girls presenting with recurrent vaginal discharge found that sexual abuse might have been involved in about 5% of cases.2
Systemic diseases, such as eczema and psoriasis, may also present with gynecologic symptoms.
Heavy vaginal discharge may also be normal. This situation is a diagnosis of exclusion but is important to recognize in order to allay the patient’s anxiety and avoid unnecessary treatment.
SIMPLE OFFICE-BASED ASSESSMENT
A thorough history and physical examination are always warranted.

Simple tests of vaginal secretions can often determine the diagnosis (Table 2). Vaginal secretions should be analyzed in the following order:
Testing the pH. The pH can help determine likely diagnoses and streamline further testing (Figure 1).

Saline microscopy. Some of the vaginal discharge sample should be diluted with 1 or 2 drops of normal saline and examined under a microscope, first at × 10 magnification, then at × 40. The sample should be searched for epithelial cells, blood cells, “clue” cells (ie, epithelial cells with borders studded or obscured by bacteria), and motile trichomonads.
10% KOH whiff test and microscopy. To a second vaginal sample, a small amount of 10% potassium hydroxide should be added, and the examiner should sniff it. An amine or fishy odor is a sign of bacterial vaginosis.

If pH paper, KOH, and a microscope are unavailable, other point-of-care tests can be used for specific conditions as discussed below.
INFECTIOUS CAUSES
Infectious causes of vulvovaginitis include bacterial vaginosis, candidiasis, trichomoniasis, and herpes simplex virus (HSV) infection.
BACTERIAL VAGINOSIS
Bacterial vaginosis is the most common vaginal disorder worldwide. It has been linked to preterm delivery, intra-amniotic infection, endometritis, postabortion infection, and vaginal cuff cellulitis after hysterectomy.3 It may also be a risk factor for human immunodeficiency virus (HIV) infection.4
The condition reflects a microbial imbalance in the vaginal ecosystem, characterized by depletion of the dominant hydrogen peroxide-producing lactobacilli and overgrowth of anaerobic and facultative aerobic organisms such as Gardnerella vaginalis, Mycoplasma hominis, Atopobium vaginae, and Prevotella and Mobiluncus species.
Diagnosis of bacterial vaginosis
The Amsel criteria consist of the following:
- pH greater than 4.5
- Positive whiff test
- Homogeneous discharge
- Clue cells.
Three of the four criteria must be present for a diagnosis of bacterial vaginosis. This method is inexpensive and provides immediate results in the clinic.
The Nugent score, based on seeing certain bacteria from a vaginal swab on Gram stain microscopy, is the diagnostic standard for research.5
DNA tests. Affirm VPIII (BD Diagnostics, Sparks, MD) is a nonamplified nucleic acid probe hybridization test that detects Trichomonas vaginalis, Candida albicans, and G vaginalis. Although it is more expensive than testing for the Amsel criteria, it is commonly used in private offices because it is simple to use, gives rapid results, and does not require a microscope.6 Insurance pays for it when the test is indicated, but we know of a patient who received a bill for approximately $500 when the insurance company thought the test was not indicated.
In a study of 109 patients with symptoms of vulvovaginitis, the Affirm VPIII was found comparable to saline microscopy when tested on residual vaginal samples. Compared with Gram stain using Nugent scoring, the test has a sensitivity of 87.7% to 95.2% and a specificity of 81% to 99.1% for bacterial vaginosis.7
In 323 symptomatic women, a polymerase chain reaction (PCR) assay for bacterial vaginosis was 96.9% sensitive and 92.6% specific for bacterial vaginosis, and Affirm VPIII was 90.1% sensitive and 67.6% specific, compared with a reference standard incorporating Nugent Gram-stain scores and Amsel criteria.8 The test is commercially available.
Management of bacterial vaginosis
Initial treatment. Bacterial vaginosis can be treated with oral or topical metronidazole, oral tinidazole, or oral or topical clindamycin.9 All options offer equivalent efficacy as initial treatments, so the choice may be based on cost and preferred route of administration.
Treatment for recurrent disease. Women who have 3 or more episodes in 12 months should receive initial treatment each time as described above and should then be offered additional suppressive therapy with 0.75% metronidazole intravaginal gel 2 times a week for 4 months. A side effect of therapy is vulvovaginal candidiasis, which should be treated as needed.
In a multicenter study, Sobel et al10 randomized patients who had recurrent bacterial vaginosis to twice-weekly metronidazole gel or placebo for 16 weeks after their initial treatment. During the 28 weeks of follow-up, recurrences occurred in 51% of treated women vs 75% of those on placebo.
Another option for chronic therapy is oral metronidazole and boric acid vaginal suppositories.
Reichman et al11 treated women with oral metronidazole or tinidazole 500 mg twice a day for 7 days, followed by vaginal boric acid 600 mg daily for 21 days. This was followed by twice-weekly vaginal metronidazole gel for 16 weeks. At follow-up, the cure rate was 92% at 7 weeks, dropping to 88% at 12 weeks and 50% at 36 weeks.
Patients with recurrent bacterial vaginosis despite therapy should be referred to a vulvovaginal or infectious disease specialist.
VULVOVAGINAL CANDIDIASIS
Vulvovaginal candidiasis is the second most common cause of vaginitis.
Diagnosis can be clinical

Vulvovaginal candidiasis can be clinically diagnosed on the basis of cottage cheese-like clumpy discharge; external dysuria (a burning sensation when urine comes in contact with the vulva); and vulvar itching, pain, swelling, and redness. Edema, fissures, and excoriations may be seen on examination of the vulva. (Figure 3).
Saline microscopy (Figure 2) with the addition of 10% KOH may reveal the characteristic fungal elements, but its sensitivity is only 50%.
Fungal culture remains the gold standard for diagnosis and is needed to determine the sensitivity of specific strains of Candida to therapy.12
DNA tests can also be helpful. In a study of patients with symptomatic vaginitis, Affirm VPIII detected Candida in 11% of samples, whereas microscopy detected it in only 7%.13 Another study7 found that Affirm VPIII produced comparable results whether the sample was collected from residual vaginal discharge found on the speculum or was collected in the traditional way (by swabbing).
Cartwright et al8 compared the performance of a multiplexed, real-time PCR assay and Affirm VPIII in 102 patients. PCR was much more sensitive (97.7% vs 58.1%) but less specific (93.2% vs 100%), with culture serving as the gold standard.
Management of candidiasis
Uncomplicated cases can be managed with prescription or over-the-counter topical or oral antifungal medications for 1 to 7 days, depending on the medication.9 However, most of the common antifungals may not be effective against non-albicans Candida.
In immunosuppressed patients and diabetic patients, if symptoms do not improve with regular treatment, a vaginal sample should be cultured for C albicans. If the culture is positive, the patient should be treated with fluconazole 150 mg orally every 3 days for 3 doses.14
Patients with recurrent episodes (3 or more in 12 months) should follow initial treatment with maintenance therapy of weekly fluconazole 150 mg orally for 6 months.15
Non-albicans Candida may be azole-resistant, and fungal culture and sensitivity should be obtained. Sobel et al13 documented successful treatment of non-albicans Candida using boric acid and flucytosine. Phillips16 documented successful use of compounded amphotericin B in a 50-mg vaginal suppository for 14 days. Therefore, in patients who have Candida species other than C albicans, treatment should be one of the following:
- Vaginal boric acid 600 mg daily for 14 to 21 days
- Flucytosine in 15.5% vaginal cream, intravaginally administered as 5 g for 14 days
- Amphotericin B 50 mg vaginal suppositories for 14 days.
Boric acid is readily available, but flucytosine vaginal cream and amphotericin B vaginal suppositories must usually be compounded by a pharmacist.
Of note: All that itches is not yeast. Patients with persistent itching despite treatment should be referred to a specialist to search for another cause.
TRICHOMONIASIS
The incidence of T vaginalis infection is higher than that of Neisseria gonorrhoeae and Chlamydia trachomatis combined, with an estimated 7.4 million new cases occurring in the year 2000 in the United States.17 Infection increases the sexual transmission of HIV.18–20 It is often asymptomatic and so is likely underdiagnosed.
Diagnosis of trichomoniasis
Vaginal pH may be normal or elevated (> 4.5).
Direct microscopy. Observation by saline microscopy of motile trichomonads with their characteristic jerky movements is 100% specific but only 50% sensitive. Sensitivity is reduced by delaying microscopy on the sample by as little as 10 minutes.21
The incidental finding of T vaginalis on a conventional Papanicolaou (Pap) smear has poor sensitivity and specificity, and patients diagnosed with T vaginalis by conventional Pap smear should have a second test performed. The liquid-based Pap test is more accurate for microscopic diagnosis, and its results can be used to determine if treatment is needed (sensitivity 60%–90%; specificity 98%–100%).22,23
Culture. Amplification of T vaginalis in liquid culture usually provides results within 3 days.24 It is more sensitive than microscopy but less sensitive than a nucleic acid amplification test: compared with a nucleic acid amplification test, culture is 44% to 75% sensitive for detecting T vaginalis and 100% specific.19 Culture is the preferred test for resistant strains.
Non–culture-based or nucleic acid tests do not require viable organisms, so they allow for a wider range of specimen storage temperatures and time intervals between collection and processing. This quality limits them for testing treatment success; if performed too early, they may detect nonviable organisms. A 2-week interval is recommended between the end of treatment and retesting.25
Nonamplified tests such as Affirm VPIII and the Osom Trichomonas Test (Sekisui Diagnostics, Lexington, MA) are 40% to 95% sensitive, depending on the test and reference standard used, and 92% to 100% specific.26,27
Nucleic acid amplification tests are usually not performed as point-of-care tests. They are more expensive and require special equipment with trained personnel. Sensitivities range from 76% to 100%, making these tests more suitable for screening and testing of asymptomatic women, in whom the concentration of organisms may be lower.
Treatment of trichomoniasis
Treatment is a single 2-g oral dose of metronidazole or tinidazole.9
If initial treatment is ineffective, an additional regimen can be either of the following:
- Oral metronidazole 500 mg twice a day for 7 days
- Oral metronidazole or tinidazole, 2 g daily for 5 days.
Patients allergic to nitroimidazoles should be referred for desensitization.
If these treatments are unsuccessful, the patient should be referred to an infectious disease specialist or gynecologist who specializes in vulvovaginal disorders. Treatment failure is uncommon and is usually related to noncompliance, reinfection, or metronidazole resistance.28 The US Centers for Disease Control and Prevention offers testing for resistance by request.
Reportedly successful regimens for refractory trichomoniasis include 14 days of either:
- Oral tinidazole 500 mg 4 times daily plus vaginal tinidazole 500 mg twice daily29
- Oral tinidazole 1 g 3 times daily plus compounded 5% intravaginal paromomycin 5 g nightly.30
HERPES SIMPLEX VIRUS INFECTION
HSV (HSV-1 and HSV-2) causes lifelong infection. About 50 million people in the United States are infected with HSV-2, the most common cause of recurrent infections.31 Owing to changes in sexual practices, an increasing number of young people are acquiring anogenital HSV-1 infection.32,33
Diagnosis of herpes

Diagnosis may be difficult because the painful vesicular or ulcerative lesions (Figure 4) may not be visible at the time of presentation. Diagnosis is based on specific virologic and serologic tests. Nonspecific tests (eg, Tzanck smear, direct immunofluorescence) are neither sensitive nor specific and should not be relied on for diagnosis.34 HSV culture or HSV-PCR testing of a lesion is preferred. The sensitivity of viral culture can be low and is dependent on the stage of healing of a lesion and obtaining an adequate sample.
Accurate type-specific HSV serologic assays are based on HSV-specific glycoprotein G1 (HSV-1) and glycoprotein G2 (HSV-2). Unless a patient’s serologic status has already been determined, serologic testing should be done concurrently with HSV culture or PCR testing. Serologic testing enables classification of an infection as primary, nonprimary, or recurrent. For example, a patient with a positive HSV culture and negative serology most likely has primary HSV infection, and serologic study should be repeated after 6 to 8 weeks to assess for seroconversion.
Immunoglobulin M (IgM) testing for HSV-1 or HSV-2 is not diagnostic or type-specific and may be positive during recurrent genital or oral episodes of herpes.35
Treatment of herpes
In general, antiviral medications (eg, acyclovir, valacyclovir, famciclovir) are effective for managing HSV.12 Episodic or continuous suppression therapy may be needed for patients experiencing more than four outbreaks in 12 months. Patients who do not respond to treatment should be referred to an infectious disease specialist and undergo a viral culture with sensitivities.
NONINFECTIOUS CAUSES
Desquamative inflammatory vaginitis
Desquamative inflammatory vaginitis is a chronic vaginal disorder of unknown cause. It is a diagnosis of exclusion, and some patients may have a superimposed bacterial infection. It occurs mostly in perimenopausal woman and is often associated with low estrogen levels.
Diagnosis. Patients may report copious green-yellow mucoid discharge, vulvar or vaginal pain, and dyspareunia. On examination, the vulva may be erythematous, friable, and tender to the touch. The vagina may have ecchymoses, be diffusely erythematous, and have linear lesions. Mucoid or purulent discharge may be seen.
.")
The vaginal pH is greater than 4.5.
Saline microscopy shows increased parabasal cells and leukorrhea (Figure 5).
Diagnosis is based on all of the following:
- At least 1 symptom (ie, vaginal discharge, dyspareunia, pruritus, pain, irritation, or burning)
- Vaginal inflammation on examination
- pH higher than 4.5
- Presence of parabasal cells and leukorrhea on microscopy (a ratio of leukocytes to vaginal epithelial cells > 1:1).36
Treatment involves use of 2% intravaginal clindamycin or 10% intravaginal compounded hydrocortisone cream for 4 to 6 weeks. Patients who are not cured with single-agent therapy may benefit from compounded clindamycin and hydrocortisone, with estrogen added to the formulation for hypoestrogenic patients.
Atrophic vaginitis
Atrophic vaginitis is often seen in menopausal or hypoestrogenic women. Presenting symptoms include vulvar or vaginal pain and dyspareunia.
Diagnosis. On physical examination, the vulva appears pale and atrophic, with narrowing of the introitus. Vaginal examination may reveal a pale mucosa that lacks elasticity and rugation. The examination should be performed with caution, as the vagina may bleed easily.
The vaginal pH is usually elevated.

Saline microscopy may show parabasal cells and a paucity of epithelial cells. (Figure 6).
The Vaginal Maturation Index is an indicator of the maturity of the epithelial cell types being exfoliated; these normally include parabasal (immature) cells, intermediate, and superficial (mature) cells. A predominance of immature cells indicates a hypoestrogenic state.
Infection should be considered and treated as needed.
Treatment. Patients with no contraindication may benefit from systemic hormone therapy or topical estrogen, or both.
Contact dermatitis
Contact dermatitis is classified into two types:
Irritant dermatitis, caused by the destructive action of contactants, eg, urine, feces, topical agents, feminine wipes
Allergic dermatitis, also contactant-induced, but immunologically mediated.
If a diagnosis cannot be made from the patient history and physical examination, biopsy should be performed.
Treatment of contact dermatitis involves removing the irritant, hydrating the skin with sitz baths, and using an emollient (eg, petroleum jelly) and midpotent topical steroids until resolution. Some patients benefit from topical immunosuppressive agents (eg, tacrolimus). Patients with severe symptoms may be treated with a tapering course of oral steroids for 5 to 7 days. Recalcitrant cases should be referred to a specialist.
Lichen planus


Lichen sclerosus is a benign, chronic, progressive dermatologic condition characterized by marked inflammation, epithelial thinning, and distinctive dermal changes accompanied by pruritus and pain (Figures 7 and 8).
Treatment. High-potency topical steroids are the mainstay of therapy for lichen disease. Although these are not infectious processes, superimposed infections (mostly bacterial and fungal) may also be present and should be treated.
- Van Eyk N, Allen L, Giesbrecht E, et al. Pediatric vulvovaginal disorders: a diagnostic approach and review of the literature. J Obstet Gynaecol Can 2009; 31:850–862.
- McGreal S, Wood P. Recurrent vaginal discharge in children. J Pediatr Adolesc Gynecol 2013; 26:205–208.
- Livengood CH. Bacterial vaginosis: an overview for 2009. Rev Obstet Gynecol 2009; 2:28–37.
- Atashili J, Poole C, Ndumbe PM, Adimora AA, Smith JS. Bacterial vaginosis and HIV acquisition: a meta-analysis of published studies. AIDS 2008; 22:1493–1501.
- Money D. The laboratory diagnosis of bacterial vaginosis. Can J Infect Dis Med Microbiol 2005; 16:77–79.
- Lowe NK, Neal JL, Ryan-Wenger NA, et al. Accuracy of the clinical diagnosis of vaginitis compared with a DNA probe laboratory standard. Obstet Gynecol 2009; 113:89–95.
- Mulhem E, Boyanton BL Jr, Robinson-Dunn B, Ebert C, Dzebo R. Performance of the Affirm VP-III using residual vaginal discharge collected from the speculum to characterize vaginitis in symptomatic women. J Low Genit Tract Dis 2014; 18:344–346.
- Cartwright CP, Lembke BD, Ramachandran K, et al. Comparison of nucleic acid amplification assays with BD Affirm VPIII for diagnosis of vaginitis in symptomatic women. J Clin Microbiol 2013; 51:3694–3699.
- Workowski KA, Bolan GA; Centers for Disease Control and Prevention (CDC). Sexually transmitted diseases treatment guidelines, 2015. MMWR Recomm Rep 2015; 64:1–137.
- Sobel JD, Ferris D, Schwebke J, et al. Suppressive antibacterial therapy with 0.75% metronidazole vaginal gel to prevent recurrent bacterial vaginosis. Am J Obstet Gynecol 2006; 194:1283–1289.
- Reichman O, Akins R, Sobel JD. Boric acid addition to suppressive antimicrobial therapy for recurrent bacterial vaginosis. Sex Transm Dis 2009; 36:732–734.
- Carr PL, Felsenstein D, Friedman RH. Evaluation and management of vaginitis. J Gen Intern Med 1998; 13:335–346.
- Sobel JD, Chaim W, Nagappan V, Leaman D. Treatment of vaginitis caused by Candida glabrata: use of topical boric acid and flucytosine. Am J Obstet Gynecol 2003; 189:1297–1300.
- Sobel JD, Kapernick PS, Zervos M, et al. Treatment of complicated Candida vaginitis: comparison of single and sequential doses of fluconazole. Am J Obstet Gynecol 2001; 185:363–369.
- Sobel JD, Wiesenfeld HC, Martens M, et al. Maintenance fluconazole therapy for recurrent vulvovaginal candidiasis. N Engl J Med 2004; 351:876–883.
- Phillips AJ. Treatment of non-albicans Candida vaginitis with amphotericin B vaginal suppositories. Am J Obstet Gynecol 2005; 192:2009–2013.
- Weinstock H, Berman S, Cates W Jr. Sexually transmitted diseases among American youth: incidence and prevalence estimates, 2000. Perspect Sex Reprod Health 2004; 36:6–10.
- Gatski M, Martin DH, Clark RA, Harville E, Schmidt N, Kissinger P. Co-occurrence of Trichomonas vaginalis and bacterial vaginosis among HIV-positive women. Sex Transm Dis 2011; 38:163–166.
- Hobbs MM, Seña AC. Modern diagnosis of Trichomonas vaginalis infection. Sex Transm Infect 2013; 89:434–438.
- McClelland RS, Sangare L, Hassan WM, et al. Infection with Trichomonas vaginalis increases the risk of HIV-1 acquisition. J Infect Dis 2007; 195:698–702.
- Kingston MA, Bansal D, Carlin EM. ‘Shelf life’ of Trichomonas vaginalis. Int J STD AIDS 2003; 14:28–29.
- Aslan DL, Gulbahce HE, Stelow EB, et al. The diagnosis of Trichomonas vaginalis in liquid-based Pap tests: correlation with PCR. Diagn Cytopathol 2005; 32:341–344.
- Lara-Torre E, Pinkerton JS. Accuracy of detection of Trichomonas vaginalis organisms on a liquid-based papanicolaou smear. Am J Obstet Gynecol 2003; 188:354–356.
- Hobbs MM, Lapple DM, Lawing LF, et al. Methods for detection of Trichomonas vaginalis in the male partners of infected women: implications for control of trichomoniasis. J Clin Microbiol 2006; 44:3994–3999.
- Van Der Pol B, Williams JA, Orr DP, Batteiger BE, Fortenberry JD. Prevalence, incidence, natural history, and response to treatment of Trichomonas vaginalis infection among adolescent women. J Infect Dis 2005; 192:2039–2044.
- Campbell L, Woods V, Lloyd T, Elsayed S, Church DL. Evaluation of the OSOM Trichomonas rapid test versus wet preparation examination for detection of Trichomonas vaginalis vaginitis in specimens from women with a low prevalence of infection. J Clin Microbiol 2008; 46:3467–3469.
- Chapin K, Andrea S. APTIMA Trichomonas vaginalis, a transcription-mediated amplification assay for detection of Trichomonas vaginalis in urogenital specimens. Expert Rev Mol Diagn 2011; 11:679–688.
- Krashin JW, Koumans EH, Bradshaw-Sydnor AC, et al. Trichomonas vaginalis prevalence, incidence, risk factors and antibiotic-resistance in an adolescent population. Sex Transm Dis 2010; 37:440–444.
- Sobel JD, Nyirjesy P, Brown W. Tinidazole therapy for metronidazole-resistant vaginal trichomoniasis. Clin Infect Dis 2001; 33:1341–1346.
- Nyirjesy P, Gilbert J, Mulcahy LJ. Resistant trichomoniasis: successful treatment with combination therapy. Sex Transm Dis 2011; 38:962–963.
- Bradley H, Markowitz LE, Gibson T, McQuillan GM. Seroprevalence of herpes simplex virus types 1 and 2—United States, 1999–2010. J Infect Dis 2014; 209:325–333.
- Roberts CM, Pfister JR, Spear SJ. Increasing proportion of herpes simplex virus type 1 as a cause of genital herpes infection in college students. Sex Transm Dis 2003; 30:797–800.
- Ryder N, Jin F, McNulty AM, Grulich AE, Donovan B. Increasing role of herpes simplex virus type 1 in first-episode anogenital herpes in heterosexual women and younger men who have sex with men, 1992-2006. Sex Transm Infect 2009; 85:416–419.
- Caviness AC, Oelze LL, Saz UE, Greer JM, Demmler-Harrison GJ. Direct immunofluorescence assay compared to cell culture for the diagnosis of mucocutaneous herpes simplex virus infections in children. J Clin Virol 2010; 49:58–60.
- Morrow R, Friedrich D. Performance of a novel test for IgM and IgG antibodies in subjects with culture-documented genital herpes simplex virus-1 or -2 infection. Clin Microbiol Infect 2006; 12:463–469.
- Bradford J, Fischer G. Desquamative inflammatory vaginitis: differential diagnosis and alternate diagnostic criteria. J Low Genit Tract Dis 2010; 14:306–310.
Although vulvovaginitis has several possible causes, the typical presenting symptoms are similar regardless of the cause: itching, burning, and vaginal discharge. Physical examination often reveals atrophy, redness, excoriations, and fissures in the vulvovaginal and perianal areas. Determining the cause is key to successful treatment.
This article reviews the diagnosis and treatment of many common and less common infectious and noninfectious causes of vulvovaginitis, the use of special tests, and the management of persistent cases.
DIAGNOSIS CAN BE CHALLENGING
Vulvar and vaginal symptoms are most commonly caused by local infections, but other causes must be also be considered, including several noninfectious ones (Table 1). Challenges in diagnosing vulvovaginitis are many and include distinguishing contact from allergic dermatitis, recognizing vaginal atrophy, and recognizing a parasitic infection. Determining whether a patient has an infectious process is important so that antibiotics can be used only when truly needed.
Foreign bodies in the vagina should also be considered, especially in children,1 as should sexual abuse. A 15-year retrospective review of prepubertal girls presenting with recurrent vaginal discharge found that sexual abuse might have been involved in about 5% of cases.2
Systemic diseases, such as eczema and psoriasis, may also present with gynecologic symptoms.
Heavy vaginal discharge may also be normal. This situation is a diagnosis of exclusion but is important to recognize in order to allay the patient’s anxiety and avoid unnecessary treatment.
SIMPLE OFFICE-BASED ASSESSMENT
A thorough history and physical examination are always warranted.
Simple tests of vaginal secretions can often determine the diagnosis (Table 2). Vaginal secretions should be analyzed in the following order:
Testing the pH. The pH can help determine likely diagnoses and streamline further testing (Figure 1).
Saline microscopy. Some of the vaginal discharge sample should be diluted with 1 or 2 drops of normal saline and examined under a microscope, first at × 10 magnification, then at × 40. The sample should be searched for epithelial cells, blood cells, “clue” cells (ie, epithelial cells with borders studded or obscured by bacteria), and motile trichomonads.
10% KOH whiff test and microscopy. To a second vaginal sample, a small amount of 10% potassium hydroxide should be added, and the examiner should sniff it. An amine or fishy odor is a sign of bacterial vaginosis.
If pH paper, KOH, and a microscope are unavailable, other point-of-care tests can be used for specific conditions as discussed below.
INFECTIOUS CAUSES
Infectious causes of vulvovaginitis include bacterial vaginosis, candidiasis, trichomoniasis, and herpes simplex virus (HSV) infection.
BACTERIAL VAGINOSIS
Bacterial vaginosis is the most common vaginal disorder worldwide. It has been linked to preterm delivery, intra-amniotic infection, endometritis, postabortion infection, and vaginal cuff cellulitis after hysterectomy.3 It may also be a risk factor for human immunodeficiency virus (HIV) infection.4
The condition reflects a microbial imbalance in the vaginal ecosystem, characterized by depletion of the dominant hydrogen peroxide-producing lactobacilli and overgrowth of anaerobic and facultative aerobic organisms such as Gardnerella vaginalis, Mycoplasma hominis, Atopobium vaginae, and Prevotella and Mobiluncus species.
Diagnosis of bacterial vaginosis
The Amsel criteria consist of the following:
- pH greater than 4.5
- Positive whiff test
- Homogeneous discharge
- Clue cells.
Three of the four criteria must be present for a diagnosis of bacterial vaginosis. This method is inexpensive and provides immediate results in the clinic.
The Nugent score, based on seeing certain bacteria from a vaginal swab on Gram stain microscopy, is the diagnostic standard for research.5
DNA tests. Affirm VPIII (BD Diagnostics, Sparks, MD) is a nonamplified nucleic acid probe hybridization test that detects Trichomonas vaginalis, Candida albicans, and G vaginalis. Although it is more expensive than testing for the Amsel criteria, it is commonly used in private offices because it is simple to use, gives rapid results, and does not require a microscope.6 Insurance pays for it when the test is indicated, but we know of a patient who received a bill for approximately $500 when the insurance company thought the test was not indicated.
In a study of 109 patients with symptoms of vulvovaginitis, the Affirm VPIII was found comparable to saline microscopy when tested on residual vaginal samples. Compared with Gram stain using Nugent scoring, the test has a sensitivity of 87.7% to 95.2% and a specificity of 81% to 99.1% for bacterial vaginosis.7
In 323 symptomatic women, a polymerase chain reaction (PCR) assay for bacterial vaginosis was 96.9% sensitive and 92.6% specific for bacterial vaginosis, and Affirm VPIII was 90.1% sensitive and 67.6% specific, compared with a reference standard incorporating Nugent Gram-stain scores and Amsel criteria.8 The test is commercially available.
Management of bacterial vaginosis
Initial treatment. Bacterial vaginosis can be treated with oral or topical metronidazole, oral tinidazole, or oral or topical clindamycin.9 All options offer equivalent efficacy as initial treatments, so the choice may be based on cost and preferred route of administration.
Treatment for recurrent disease. Women who have 3 or more episodes in 12 months should receive initial treatment each time as described above and should then be offered additional suppressive therapy with 0.75% metronidazole intravaginal gel 2 times a week for 4 months. A side effect of therapy is vulvovaginal candidiasis, which should be treated as needed.
In a multicenter study, Sobel et al10 randomized patients who had recurrent bacterial vaginosis to twice-weekly metronidazole gel or placebo for 16 weeks after their initial treatment. During the 28 weeks of follow-up, recurrences occurred in 51% of treated women vs 75% of those on placebo.
Another option for chronic therapy is oral metronidazole and boric acid vaginal suppositories.
Reichman et al11 treated women with oral metronidazole or tinidazole 500 mg twice a day for 7 days, followed by vaginal boric acid 600 mg daily for 21 days. This was followed by twice-weekly vaginal metronidazole gel for 16 weeks. At follow-up, the cure rate was 92% at 7 weeks, dropping to 88% at 12 weeks and 50% at 36 weeks.
Patients with recurrent bacterial vaginosis despite therapy should be referred to a vulvovaginal or infectious disease specialist.
VULVOVAGINAL CANDIDIASIS
Vulvovaginal candidiasis is the second most common cause of vaginitis.
Diagnosis can be clinical
Vulvovaginal candidiasis can be clinically diagnosed on the basis of cottage cheese-like clumpy discharge; external dysuria (a burning sensation when urine comes in contact with the vulva); and vulvar itching, pain, swelling, and redness. Edema, fissures, and excoriations may be seen on examination of the vulva. (Figure 3).
Saline microscopy (Figure 2) with the addition of 10% KOH may reveal the characteristic fungal elements, but its sensitivity is only 50%.
Fungal culture remains the gold standard for diagnosis and is needed to determine the sensitivity of specific strains of Candida to therapy.12
DNA tests can also be helpful. In a study of patients with symptomatic vaginitis, Affirm VPIII detected Candida in 11% of samples, whereas microscopy detected it in only 7%.13 Another study7 found that Affirm VPIII produced comparable results whether the sample was collected from residual vaginal discharge found on the speculum or was collected in the traditional way (by swabbing).
Cartwright et al8 compared the performance of a multiplexed, real-time PCR assay and Affirm VPIII in 102 patients. PCR was much more sensitive (97.7% vs 58.1%) but less specific (93.2% vs 100%), with culture serving as the gold standard.
Management of candidiasis
Uncomplicated cases can be managed with prescription or over-the-counter topical or oral antifungal medications for 1 to 7 days, depending on the medication.9 However, most of the common antifungals may not be effective against non-albicans Candida.
In immunosuppressed patients and diabetic patients, if symptoms do not improve with regular treatment, a vaginal sample should be cultured for C albicans. If the culture is positive, the patient should be treated with fluconazole 150 mg orally every 3 days for 3 doses.14
Patients with recurrent episodes (3 or more in 12 months) should follow initial treatment with maintenance therapy of weekly fluconazole 150 mg orally for 6 months.15
Non-albicans Candida may be azole-resistant, and fungal culture and sensitivity should be obtained. Sobel et al13 documented successful treatment of non-albicans Candida using boric acid and flucytosine. Phillips16 documented successful use of compounded amphotericin B in a 50-mg vaginal suppository for 14 days. Therefore, in patients who have Candida species other than C albicans, treatment should be one of the following:
- Vaginal boric acid 600 mg daily for 14 to 21 days
- Flucytosine in 15.5% vaginal cream, intravaginally administered as 5 g for 14 days
- Amphotericin B 50 mg vaginal suppositories for 14 days.
Boric acid is readily available, but flucytosine vaginal cream and amphotericin B vaginal suppositories must usually be compounded by a pharmacist.
Of note: All that itches is not yeast. Patients with persistent itching despite treatment should be referred to a specialist to search for another cause.
TRICHOMONIASIS
The incidence of T vaginalis infection is higher than that of Neisseria gonorrhoeae and Chlamydia trachomatis combined, with an estimated 7.4 million new cases occurring in the year 2000 in the United States.17 Infection increases the sexual transmission of HIV.18–20 It is often asymptomatic and so is likely underdiagnosed.
Diagnosis of trichomoniasis
Vaginal pH may be normal or elevated (> 4.5).
Direct microscopy. Observation by saline microscopy of motile trichomonads with their characteristic jerky movements is 100% specific but only 50% sensitive. Sensitivity is reduced by delaying microscopy on the sample by as little as 10 minutes.21
The incidental finding of T vaginalis on a conventional Papanicolaou (Pap) smear has poor sensitivity and specificity, and patients diagnosed with T vaginalis by conventional Pap smear should have a second test performed. The liquid-based Pap test is more accurate for microscopic diagnosis, and its results can be used to determine if treatment is needed (sensitivity 60%–90%; specificity 98%–100%).22,23
Culture. Amplification of T vaginalis in liquid culture usually provides results within 3 days.24 It is more sensitive than microscopy but less sensitive than a nucleic acid amplification test: compared with a nucleic acid amplification test, culture is 44% to 75% sensitive for detecting T vaginalis and 100% specific.19 Culture is the preferred test for resistant strains.
Non–culture-based or nucleic acid tests do not require viable organisms, so they allow for a wider range of specimen storage temperatures and time intervals between collection and processing. This quality limits them for testing treatment success; if performed too early, they may detect nonviable organisms. A 2-week interval is recommended between the end of treatment and retesting.25
Nonamplified tests such as Affirm VPIII and the Osom Trichomonas Test (Sekisui Diagnostics, Lexington, MA) are 40% to 95% sensitive, depending on the test and reference standard used, and 92% to 100% specific.26,27
Nucleic acid amplification tests are usually not performed as point-of-care tests. They are more expensive and require special equipment with trained personnel. Sensitivities range from 76% to 100%, making these tests more suitable for screening and testing of asymptomatic women, in whom the concentration of organisms may be lower.
Treatment of trichomoniasis
Treatment is a single 2-g oral dose of metronidazole or tinidazole.9
If initial treatment is ineffective, an additional regimen can be either of the following:
- Oral metronidazole 500 mg twice a day for 7 days
- Oral metronidazole or tinidazole, 2 g daily for 5 days.
Patients allergic to nitroimidazoles should be referred for desensitization.
If these treatments are unsuccessful, the patient should be referred to an infectious disease specialist or gynecologist who specializes in vulvovaginal disorders. Treatment failure is uncommon and is usually related to noncompliance, reinfection, or metronidazole resistance.28 The US Centers for Disease Control and Prevention offers testing for resistance by request.
Reportedly successful regimens for refractory trichomoniasis include 14 days of either:
- Oral tinidazole 500 mg 4 times daily plus vaginal tinidazole 500 mg twice daily29
- Oral tinidazole 1 g 3 times daily plus compounded 5% intravaginal paromomycin 5 g nightly.30
HERPES SIMPLEX VIRUS INFECTION
HSV (HSV-1 and HSV-2) causes lifelong infection. About 50 million people in the United States are infected with HSV-2, the most common cause of recurrent infections.31 Owing to changes in sexual practices, an increasing number of young people are acquiring anogenital HSV-1 infection.32,33
Diagnosis of herpes
Diagnosis may be difficult because the painful vesicular or ulcerative lesions (Figure 4) may not be visible at the time of presentation. Diagnosis is based on specific virologic and serologic tests. Nonspecific tests (eg, Tzanck smear, direct immunofluorescence) are neither sensitive nor specific and should not be relied on for diagnosis.34 HSV culture or HSV-PCR testing of a lesion is preferred. The sensitivity of viral culture can be low and is dependent on the stage of healing of a lesion and obtaining an adequate sample.
Accurate type-specific HSV serologic assays are based on HSV-specific glycoprotein G1 (HSV-1) and glycoprotein G2 (HSV-2). Unless a patient’s serologic status has already been determined, serologic testing should be done concurrently with HSV culture or PCR testing. Serologic testing enables classification of an infection as primary, nonprimary, or recurrent. For example, a patient with a positive HSV culture and negative serology most likely has primary HSV infection, and serologic study should be repeated after 6 to 8 weeks to assess for seroconversion.
Immunoglobulin M (IgM) testing for HSV-1 or HSV-2 is not diagnostic or type-specific and may be positive during recurrent genital or oral episodes of herpes.35
Treatment of herpes
In general, antiviral medications (eg, acyclovir, valacyclovir, famciclovir) are effective for managing HSV.12 Episodic or continuous suppression therapy may be needed for patients experiencing more than four outbreaks in 12 months. Patients who do not respond to treatment should be referred to an infectious disease specialist and undergo a viral culture with sensitivities.
NONINFECTIOUS CAUSES
Desquamative inflammatory vaginitis
Desquamative inflammatory vaginitis is a chronic vaginal disorder of unknown cause. It is a diagnosis of exclusion, and some patients may have a superimposed bacterial infection. It occurs mostly in perimenopausal woman and is often associated with low estrogen levels.
Diagnosis. Patients may report copious green-yellow mucoid discharge, vulvar or vaginal pain, and dyspareunia. On examination, the vulva may be erythematous, friable, and tender to the touch. The vagina may have ecchymoses, be diffusely erythematous, and have linear lesions. Mucoid or purulent discharge may be seen.
The vaginal pH is greater than 4.5.
Saline microscopy shows increased parabasal cells and leukorrhea (Figure 5).
Diagnosis is based on all of the following:
- At least 1 symptom (ie, vaginal discharge, dyspareunia, pruritus, pain, irritation, or burning)
- Vaginal inflammation on examination
- pH higher than 4.5
- Presence of parabasal cells and leukorrhea on microscopy (a ratio of leukocytes to vaginal epithelial cells > 1:1).36
Treatment involves use of 2% intravaginal clindamycin or 10% intravaginal compounded hydrocortisone cream for 4 to 6 weeks. Patients who are not cured with single-agent therapy may benefit from compounded clindamycin and hydrocortisone, with estrogen added to the formulation for hypoestrogenic patients.
Atrophic vaginitis
Atrophic vaginitis is often seen in menopausal or hypoestrogenic women. Presenting symptoms include vulvar or vaginal pain and dyspareunia.
Diagnosis. On physical examination, the vulva appears pale and atrophic, with narrowing of the introitus. Vaginal examination may reveal a pale mucosa that lacks elasticity and rugation. The examination should be performed with caution, as the vagina may bleed easily.
The vaginal pH is usually elevated.
Saline microscopy may show parabasal cells and a paucity of epithelial cells. (Figure 6).
The Vaginal Maturation Index is an indicator of the maturity of the epithelial cell types being exfoliated; these normally include parabasal (immature) cells, intermediate, and superficial (mature) cells. A predominance of immature cells indicates a hypoestrogenic state.
Infection should be considered and treated as needed.
Treatment. Patients with no contraindication may benefit from systemic hormone therapy or topical estrogen, or both.
Contact dermatitis
Contact dermatitis is classified into two types:
Irritant dermatitis, caused by the destructive action of contactants, eg, urine, feces, topical agents, feminine wipes
Allergic dermatitis, also contactant-induced, but immunologically mediated.
If a diagnosis cannot be made from the patient history and physical examination, biopsy should be performed.
Treatment of contact dermatitis involves removing the irritant, hydrating the skin with sitz baths, and using an emollient (eg, petroleum jelly) and midpotent topical steroids until resolution. Some patients benefit from topical immunosuppressive agents (eg, tacrolimus). Patients with severe symptoms may be treated with a tapering course of oral steroids for 5 to 7 days. Recalcitrant cases should be referred to a specialist.
Lichen planus
Lichen sclerosus is a benign, chronic, progressive dermatologic condition characterized by marked inflammation, epithelial thinning, and distinctive dermal changes accompanied by pruritus and pain (Figures 7 and 8).
Treatment. High-potency topical steroids are the mainstay of therapy for lichen disease. Although these are not infectious processes, superimposed infections (mostly bacterial and fungal) may also be present and should be treated.
Although vulvovaginitis has several possible causes, the typical presenting symptoms are similar regardless of the cause: itching, burning, and vaginal discharge. Physical examination often reveals atrophy, redness, excoriations, and fissures in the vulvovaginal and perianal areas. Determining the cause is key to successful treatment.
This article reviews the diagnosis and treatment of many common and less common infectious and noninfectious causes of vulvovaginitis, the use of special tests, and the management of persistent cases.
DIAGNOSIS CAN BE CHALLENGING
Vulvar and vaginal symptoms are most commonly caused by local infections, but other causes must be also be considered, including several noninfectious ones (Table 1). Challenges in diagnosing vulvovaginitis are many and include distinguishing contact from allergic dermatitis, recognizing vaginal atrophy, and recognizing a parasitic infection. Determining whether a patient has an infectious process is important so that antibiotics can be used only when truly needed.
Foreign bodies in the vagina should also be considered, especially in children,1 as should sexual abuse. A 15-year retrospective review of prepubertal girls presenting with recurrent vaginal discharge found that sexual abuse might have been involved in about 5% of cases.2
Systemic diseases, such as eczema and psoriasis, may also present with gynecologic symptoms.
Heavy vaginal discharge may also be normal. This situation is a diagnosis of exclusion but is important to recognize in order to allay the patient’s anxiety and avoid unnecessary treatment.
SIMPLE OFFICE-BASED ASSESSMENT
A thorough history and physical examination are always warranted.
Simple tests of vaginal secretions can often determine the diagnosis (Table 2). Vaginal secretions should be analyzed in the following order:
Testing the pH. The pH can help determine likely diagnoses and streamline further testing (Figure 1).
Saline microscopy. Some of the vaginal discharge sample should be diluted with 1 or 2 drops of normal saline and examined under a microscope, first at × 10 magnification, then at × 40. The sample should be searched for epithelial cells, blood cells, “clue” cells (ie, epithelial cells with borders studded or obscured by bacteria), and motile trichomonads.
10% KOH whiff test and microscopy. To a second vaginal sample, a small amount of 10% potassium hydroxide should be added, and the examiner should sniff it. An amine or fishy odor is a sign of bacterial vaginosis.
If pH paper, KOH, and a microscope are unavailable, other point-of-care tests can be used for specific conditions as discussed below.
INFECTIOUS CAUSES
Infectious causes of vulvovaginitis include bacterial vaginosis, candidiasis, trichomoniasis, and herpes simplex virus (HSV) infection.
BACTERIAL VAGINOSIS
Bacterial vaginosis is the most common vaginal disorder worldwide. It has been linked to preterm delivery, intra-amniotic infection, endometritis, postabortion infection, and vaginal cuff cellulitis after hysterectomy.3 It may also be a risk factor for human immunodeficiency virus (HIV) infection.4
The condition reflects a microbial imbalance in the vaginal ecosystem, characterized by depletion of the dominant hydrogen peroxide-producing lactobacilli and overgrowth of anaerobic and facultative aerobic organisms such as Gardnerella vaginalis, Mycoplasma hominis, Atopobium vaginae, and Prevotella and Mobiluncus species.
Diagnosis of bacterial vaginosis
The Amsel criteria consist of the following:
- pH greater than 4.5
- Positive whiff test
- Homogeneous discharge
- Clue cells.
Three of the four criteria must be present for a diagnosis of bacterial vaginosis. This method is inexpensive and provides immediate results in the clinic.
The Nugent score, based on seeing certain bacteria from a vaginal swab on Gram stain microscopy, is the diagnostic standard for research.5
DNA tests. Affirm VPIII (BD Diagnostics, Sparks, MD) is a nonamplified nucleic acid probe hybridization test that detects Trichomonas vaginalis, Candida albicans, and G vaginalis. Although it is more expensive than testing for the Amsel criteria, it is commonly used in private offices because it is simple to use, gives rapid results, and does not require a microscope.6 Insurance pays for it when the test is indicated, but we know of a patient who received a bill for approximately $500 when the insurance company thought the test was not indicated.
In a study of 109 patients with symptoms of vulvovaginitis, the Affirm VPIII was found comparable to saline microscopy when tested on residual vaginal samples. Compared with Gram stain using Nugent scoring, the test has a sensitivity of 87.7% to 95.2% and a specificity of 81% to 99.1% for bacterial vaginosis.7
In 323 symptomatic women, a polymerase chain reaction (PCR) assay for bacterial vaginosis was 96.9% sensitive and 92.6% specific for bacterial vaginosis, and Affirm VPIII was 90.1% sensitive and 67.6% specific, compared with a reference standard incorporating Nugent Gram-stain scores and Amsel criteria.8 The test is commercially available.
Management of bacterial vaginosis
Initial treatment. Bacterial vaginosis can be treated with oral or topical metronidazole, oral tinidazole, or oral or topical clindamycin.9 All options offer equivalent efficacy as initial treatments, so the choice may be based on cost and preferred route of administration.
Treatment for recurrent disease. Women who have 3 or more episodes in 12 months should receive initial treatment each time as described above and should then be offered additional suppressive therapy with 0.75% metronidazole intravaginal gel 2 times a week for 4 months. A side effect of therapy is vulvovaginal candidiasis, which should be treated as needed.
In a multicenter study, Sobel et al10 randomized patients who had recurrent bacterial vaginosis to twice-weekly metronidazole gel or placebo for 16 weeks after their initial treatment. During the 28 weeks of follow-up, recurrences occurred in 51% of treated women vs 75% of those on placebo.
Another option for chronic therapy is oral metronidazole and boric acid vaginal suppositories.
Reichman et al11 treated women with oral metronidazole or tinidazole 500 mg twice a day for 7 days, followed by vaginal boric acid 600 mg daily for 21 days. This was followed by twice-weekly vaginal metronidazole gel for 16 weeks. At follow-up, the cure rate was 92% at 7 weeks, dropping to 88% at 12 weeks and 50% at 36 weeks.
Patients with recurrent bacterial vaginosis despite therapy should be referred to a vulvovaginal or infectious disease specialist.
VULVOVAGINAL CANDIDIASIS
Vulvovaginal candidiasis is the second most common cause of vaginitis.
Diagnosis can be clinical
Vulvovaginal candidiasis can be clinically diagnosed on the basis of cottage cheese-like clumpy discharge; external dysuria (a burning sensation when urine comes in contact with the vulva); and vulvar itching, pain, swelling, and redness. Edema, fissures, and excoriations may be seen on examination of the vulva. (Figure 3).
Saline microscopy (Figure 2) with the addition of 10% KOH may reveal the characteristic fungal elements, but its sensitivity is only 50%.
Fungal culture remains the gold standard for diagnosis and is needed to determine the sensitivity of specific strains of Candida to therapy.12
DNA tests can also be helpful. In a study of patients with symptomatic vaginitis, Affirm VPIII detected Candida in 11% of samples, whereas microscopy detected it in only 7%.13 Another study7 found that Affirm VPIII produced comparable results whether the sample was collected from residual vaginal discharge found on the speculum or was collected in the traditional way (by swabbing).
Cartwright et al8 compared the performance of a multiplexed, real-time PCR assay and Affirm VPIII in 102 patients. PCR was much more sensitive (97.7% vs 58.1%) but less specific (93.2% vs 100%), with culture serving as the gold standard.
Management of candidiasis
Uncomplicated cases can be managed with prescription or over-the-counter topical or oral antifungal medications for 1 to 7 days, depending on the medication.9 However, most of the common antifungals may not be effective against non-albicans Candida.
In immunosuppressed patients and diabetic patients, if symptoms do not improve with regular treatment, a vaginal sample should be cultured for C albicans. If the culture is positive, the patient should be treated with fluconazole 150 mg orally every 3 days for 3 doses.14
Patients with recurrent episodes (3 or more in 12 months) should follow initial treatment with maintenance therapy of weekly fluconazole 150 mg orally for 6 months.15
Non-albicans Candida may be azole-resistant, and fungal culture and sensitivity should be obtained. Sobel et al13 documented successful treatment of non-albicans Candida using boric acid and flucytosine. Phillips16 documented successful use of compounded amphotericin B in a 50-mg vaginal suppository for 14 days. Therefore, in patients who have Candida species other than C albicans, treatment should be one of the following:
- Vaginal boric acid 600 mg daily for 14 to 21 days
- Flucytosine in 15.5% vaginal cream, intravaginally administered as 5 g for 14 days
- Amphotericin B 50 mg vaginal suppositories for 14 days.
Boric acid is readily available, but flucytosine vaginal cream and amphotericin B vaginal suppositories must usually be compounded by a pharmacist.
Of note: All that itches is not yeast. Patients with persistent itching despite treatment should be referred to a specialist to search for another cause.
TRICHOMONIASIS
The incidence of T vaginalis infection is higher than that of Neisseria gonorrhoeae and Chlamydia trachomatis combined, with an estimated 7.4 million new cases occurring in the year 2000 in the United States.17 Infection increases the sexual transmission of HIV.18–20 It is often asymptomatic and so is likely underdiagnosed.
Diagnosis of trichomoniasis
Vaginal pH may be normal or elevated (> 4.5).
Direct microscopy. Observation by saline microscopy of motile trichomonads with their characteristic jerky movements is 100% specific but only 50% sensitive. Sensitivity is reduced by delaying microscopy on the sample by as little as 10 minutes.21
The incidental finding of T vaginalis on a conventional Papanicolaou (Pap) smear has poor sensitivity and specificity, and patients diagnosed with T vaginalis by conventional Pap smear should have a second test performed. The liquid-based Pap test is more accurate for microscopic diagnosis, and its results can be used to determine if treatment is needed (sensitivity 60%–90%; specificity 98%–100%).22,23
Culture. Amplification of T vaginalis in liquid culture usually provides results within 3 days.24 It is more sensitive than microscopy but less sensitive than a nucleic acid amplification test: compared with a nucleic acid amplification test, culture is 44% to 75% sensitive for detecting T vaginalis and 100% specific.19 Culture is the preferred test for resistant strains.
Non–culture-based or nucleic acid tests do not require viable organisms, so they allow for a wider range of specimen storage temperatures and time intervals between collection and processing. This quality limits them for testing treatment success; if performed too early, they may detect nonviable organisms. A 2-week interval is recommended between the end of treatment and retesting.25
Nonamplified tests such as Affirm VPIII and the Osom Trichomonas Test (Sekisui Diagnostics, Lexington, MA) are 40% to 95% sensitive, depending on the test and reference standard used, and 92% to 100% specific.26,27
Nucleic acid amplification tests are usually not performed as point-of-care tests. They are more expensive and require special equipment with trained personnel. Sensitivities range from 76% to 100%, making these tests more suitable for screening and testing of asymptomatic women, in whom the concentration of organisms may be lower.
Treatment of trichomoniasis
Treatment is a single 2-g oral dose of metronidazole or tinidazole.9
If initial treatment is ineffective, an additional regimen can be either of the following:
- Oral metronidazole 500 mg twice a day for 7 days
- Oral metronidazole or tinidazole, 2 g daily for 5 days.
Patients allergic to nitroimidazoles should be referred for desensitization.
If these treatments are unsuccessful, the patient should be referred to an infectious disease specialist or gynecologist who specializes in vulvovaginal disorders. Treatment failure is uncommon and is usually related to noncompliance, reinfection, or metronidazole resistance.28 The US Centers for Disease Control and Prevention offers testing for resistance by request.
Reportedly successful regimens for refractory trichomoniasis include 14 days of either:
- Oral tinidazole 500 mg 4 times daily plus vaginal tinidazole 500 mg twice daily29
- Oral tinidazole 1 g 3 times daily plus compounded 5% intravaginal paromomycin 5 g nightly.30
HERPES SIMPLEX VIRUS INFECTION
HSV (HSV-1 and HSV-2) causes lifelong infection. About 50 million people in the United States are infected with HSV-2, the most common cause of recurrent infections.31 Owing to changes in sexual practices, an increasing number of young people are acquiring anogenital HSV-1 infection.32,33
Diagnosis of herpes
Diagnosis may be difficult because the painful vesicular or ulcerative lesions (Figure 4) may not be visible at the time of presentation. Diagnosis is based on specific virologic and serologic tests. Nonspecific tests (eg, Tzanck smear, direct immunofluorescence) are neither sensitive nor specific and should not be relied on for diagnosis.34 HSV culture or HSV-PCR testing of a lesion is preferred. The sensitivity of viral culture can be low and is dependent on the stage of healing of a lesion and obtaining an adequate sample.
Accurate type-specific HSV serologic assays are based on HSV-specific glycoprotein G1 (HSV-1) and glycoprotein G2 (HSV-2). Unless a patient’s serologic status has already been determined, serologic testing should be done concurrently with HSV culture or PCR testing. Serologic testing enables classification of an infection as primary, nonprimary, or recurrent. For example, a patient with a positive HSV culture and negative serology most likely has primary HSV infection, and serologic study should be repeated after 6 to 8 weeks to assess for seroconversion.
Immunoglobulin M (IgM) testing for HSV-1 or HSV-2 is not diagnostic or type-specific and may be positive during recurrent genital or oral episodes of herpes.35
Treatment of herpes
In general, antiviral medications (eg, acyclovir, valacyclovir, famciclovir) are effective for managing HSV.12 Episodic or continuous suppression therapy may be needed for patients experiencing more than four outbreaks in 12 months. Patients who do not respond to treatment should be referred to an infectious disease specialist and undergo a viral culture with sensitivities.
NONINFECTIOUS CAUSES
Desquamative inflammatory vaginitis
Desquamative inflammatory vaginitis is a chronic vaginal disorder of unknown cause. It is a diagnosis of exclusion, and some patients may have a superimposed bacterial infection. It occurs mostly in perimenopausal woman and is often associated with low estrogen levels.
Diagnosis. Patients may report copious green-yellow mucoid discharge, vulvar or vaginal pain, and dyspareunia. On examination, the vulva may be erythematous, friable, and tender to the touch. The vagina may have ecchymoses, be diffusely erythematous, and have linear lesions. Mucoid or purulent discharge may be seen.
The vaginal pH is greater than 4.5.
Saline microscopy shows increased parabasal cells and leukorrhea (Figure 5).
Diagnosis is based on all of the following:
- At least 1 symptom (ie, vaginal discharge, dyspareunia, pruritus, pain, irritation, or burning)
- Vaginal inflammation on examination
- pH higher than 4.5
- Presence of parabasal cells and leukorrhea on microscopy (a ratio of leukocytes to vaginal epithelial cells > 1:1).36
Treatment involves use of 2% intravaginal clindamycin or 10% intravaginal compounded hydrocortisone cream for 4 to 6 weeks. Patients who are not cured with single-agent therapy may benefit from compounded clindamycin and hydrocortisone, with estrogen added to the formulation for hypoestrogenic patients.
Atrophic vaginitis
Atrophic vaginitis is often seen in menopausal or hypoestrogenic women. Presenting symptoms include vulvar or vaginal pain and dyspareunia.
Diagnosis. On physical examination, the vulva appears pale and atrophic, with narrowing of the introitus. Vaginal examination may reveal a pale mucosa that lacks elasticity and rugation. The examination should be performed with caution, as the vagina may bleed easily.
The vaginal pH is usually elevated.
Saline microscopy may show parabasal cells and a paucity of epithelial cells. (Figure 6).
The Vaginal Maturation Index is an indicator of the maturity of the epithelial cell types being exfoliated; these normally include parabasal (immature) cells, intermediate, and superficial (mature) cells. A predominance of immature cells indicates a hypoestrogenic state.
Infection should be considered and treated as needed.
Treatment. Patients with no contraindication may benefit from systemic hormone therapy or topical estrogen, or both.
Contact dermatitis
Contact dermatitis is classified into two types:
Irritant dermatitis, caused by the destructive action of contactants, eg, urine, feces, topical agents, feminine wipes
Allergic dermatitis, also contactant-induced, but immunologically mediated.
If a diagnosis cannot be made from the patient history and physical examination, biopsy should be performed.
Treatment of contact dermatitis involves removing the irritant, hydrating the skin with sitz baths, and using an emollient (eg, petroleum jelly) and midpotent topical steroids until resolution. Some patients benefit from topical immunosuppressive agents (eg, tacrolimus). Patients with severe symptoms may be treated with a tapering course of oral steroids for 5 to 7 days. Recalcitrant cases should be referred to a specialist.
Lichen planus
Lichen sclerosus is a benign, chronic, progressive dermatologic condition characterized by marked inflammation, epithelial thinning, and distinctive dermal changes accompanied by pruritus and pain (Figures 7 and 8).
Treatment. High-potency topical steroids are the mainstay of therapy for lichen disease. Although these are not infectious processes, superimposed infections (mostly bacterial and fungal) may also be present and should be treated.
- Van Eyk N, Allen L, Giesbrecht E, et al. Pediatric vulvovaginal disorders: a diagnostic approach and review of the literature. J Obstet Gynaecol Can 2009; 31:850–862.
- McGreal S, Wood P. Recurrent vaginal discharge in children. J Pediatr Adolesc Gynecol 2013; 26:205–208.
- Livengood CH. Bacterial vaginosis: an overview for 2009. Rev Obstet Gynecol 2009; 2:28–37.
- Atashili J, Poole C, Ndumbe PM, Adimora AA, Smith JS. Bacterial vaginosis and HIV acquisition: a meta-analysis of published studies. AIDS 2008; 22:1493–1501.
- Money D. The laboratory diagnosis of bacterial vaginosis. Can J Infect Dis Med Microbiol 2005; 16:77–79.
- Lowe NK, Neal JL, Ryan-Wenger NA, et al. Accuracy of the clinical diagnosis of vaginitis compared with a DNA probe laboratory standard. Obstet Gynecol 2009; 113:89–95.
- Mulhem E, Boyanton BL Jr, Robinson-Dunn B, Ebert C, Dzebo R. Performance of the Affirm VP-III using residual vaginal discharge collected from the speculum to characterize vaginitis in symptomatic women. J Low Genit Tract Dis 2014; 18:344–346.
- Cartwright CP, Lembke BD, Ramachandran K, et al. Comparison of nucleic acid amplification assays with BD Affirm VPIII for diagnosis of vaginitis in symptomatic women. J Clin Microbiol 2013; 51:3694–3699.
- Workowski KA, Bolan GA; Centers for Disease Control and Prevention (CDC). Sexually transmitted diseases treatment guidelines, 2015. MMWR Recomm Rep 2015; 64:1–137.
- Sobel JD, Ferris D, Schwebke J, et al. Suppressive antibacterial therapy with 0.75% metronidazole vaginal gel to prevent recurrent bacterial vaginosis. Am J Obstet Gynecol 2006; 194:1283–1289.
- Reichman O, Akins R, Sobel JD. Boric acid addition to suppressive antimicrobial therapy for recurrent bacterial vaginosis. Sex Transm Dis 2009; 36:732–734.
- Carr PL, Felsenstein D, Friedman RH. Evaluation and management of vaginitis. J Gen Intern Med 1998; 13:335–346.
- Sobel JD, Chaim W, Nagappan V, Leaman D. Treatment of vaginitis caused by Candida glabrata: use of topical boric acid and flucytosine. Am J Obstet Gynecol 2003; 189:1297–1300.
- Sobel JD, Kapernick PS, Zervos M, et al. Treatment of complicated Candida vaginitis: comparison of single and sequential doses of fluconazole. Am J Obstet Gynecol 2001; 185:363–369.
- Sobel JD, Wiesenfeld HC, Martens M, et al. Maintenance fluconazole therapy for recurrent vulvovaginal candidiasis. N Engl J Med 2004; 351:876–883.
- Phillips AJ. Treatment of non-albicans Candida vaginitis with amphotericin B vaginal suppositories. Am J Obstet Gynecol 2005; 192:2009–2013.
- Weinstock H, Berman S, Cates W Jr. Sexually transmitted diseases among American youth: incidence and prevalence estimates, 2000. Perspect Sex Reprod Health 2004; 36:6–10.
- Gatski M, Martin DH, Clark RA, Harville E, Schmidt N, Kissinger P. Co-occurrence of Trichomonas vaginalis and bacterial vaginosis among HIV-positive women. Sex Transm Dis 2011; 38:163–166.
- Hobbs MM, Seña AC. Modern diagnosis of Trichomonas vaginalis infection. Sex Transm Infect 2013; 89:434–438.
- McClelland RS, Sangare L, Hassan WM, et al. Infection with Trichomonas vaginalis increases the risk of HIV-1 acquisition. J Infect Dis 2007; 195:698–702.
- Kingston MA, Bansal D, Carlin EM. ‘Shelf life’ of Trichomonas vaginalis. Int J STD AIDS 2003; 14:28–29.
- Aslan DL, Gulbahce HE, Stelow EB, et al. The diagnosis of Trichomonas vaginalis in liquid-based Pap tests: correlation with PCR. Diagn Cytopathol 2005; 32:341–344.
- Lara-Torre E, Pinkerton JS. Accuracy of detection of Trichomonas vaginalis organisms on a liquid-based papanicolaou smear. Am J Obstet Gynecol 2003; 188:354–356.
- Hobbs MM, Lapple DM, Lawing LF, et al. Methods for detection of Trichomonas vaginalis in the male partners of infected women: implications for control of trichomoniasis. J Clin Microbiol 2006; 44:3994–3999.
- Van Der Pol B, Williams JA, Orr DP, Batteiger BE, Fortenberry JD. Prevalence, incidence, natural history, and response to treatment of Trichomonas vaginalis infection among adolescent women. J Infect Dis 2005; 192:2039–2044.
- Campbell L, Woods V, Lloyd T, Elsayed S, Church DL. Evaluation of the OSOM Trichomonas rapid test versus wet preparation examination for detection of Trichomonas vaginalis vaginitis in specimens from women with a low prevalence of infection. J Clin Microbiol 2008; 46:3467–3469.
- Chapin K, Andrea S. APTIMA Trichomonas vaginalis, a transcription-mediated amplification assay for detection of Trichomonas vaginalis in urogenital specimens. Expert Rev Mol Diagn 2011; 11:679–688.
- Krashin JW, Koumans EH, Bradshaw-Sydnor AC, et al. Trichomonas vaginalis prevalence, incidence, risk factors and antibiotic-resistance in an adolescent population. Sex Transm Dis 2010; 37:440–444.
- Sobel JD, Nyirjesy P, Brown W. Tinidazole therapy for metronidazole-resistant vaginal trichomoniasis. Clin Infect Dis 2001; 33:1341–1346.
- Nyirjesy P, Gilbert J, Mulcahy LJ. Resistant trichomoniasis: successful treatment with combination therapy. Sex Transm Dis 2011; 38:962–963.
- Bradley H, Markowitz LE, Gibson T, McQuillan GM. Seroprevalence of herpes simplex virus types 1 and 2—United States, 1999–2010. J Infect Dis 2014; 209:325–333.
- Roberts CM, Pfister JR, Spear SJ. Increasing proportion of herpes simplex virus type 1 as a cause of genital herpes infection in college students. Sex Transm Dis 2003; 30:797–800.
- Ryder N, Jin F, McNulty AM, Grulich AE, Donovan B. Increasing role of herpes simplex virus type 1 in first-episode anogenital herpes in heterosexual women and younger men who have sex with men, 1992-2006. Sex Transm Infect 2009; 85:416–419.
- Caviness AC, Oelze LL, Saz UE, Greer JM, Demmler-Harrison GJ. Direct immunofluorescence assay compared to cell culture for the diagnosis of mucocutaneous herpes simplex virus infections in children. J Clin Virol 2010; 49:58–60.
- Morrow R, Friedrich D. Performance of a novel test for IgM and IgG antibodies in subjects with culture-documented genital herpes simplex virus-1 or -2 infection. Clin Microbiol Infect 2006; 12:463–469.
- Bradford J, Fischer G. Desquamative inflammatory vaginitis: differential diagnosis and alternate diagnostic criteria. J Low Genit Tract Dis 2010; 14:306–310.
- Van Eyk N, Allen L, Giesbrecht E, et al. Pediatric vulvovaginal disorders: a diagnostic approach and review of the literature. J Obstet Gynaecol Can 2009; 31:850–862.
- McGreal S, Wood P. Recurrent vaginal discharge in children. J Pediatr Adolesc Gynecol 2013; 26:205–208.
- Livengood CH. Bacterial vaginosis: an overview for 2009. Rev Obstet Gynecol 2009; 2:28–37.
- Atashili J, Poole C, Ndumbe PM, Adimora AA, Smith JS. Bacterial vaginosis and HIV acquisition: a meta-analysis of published studies. AIDS 2008; 22:1493–1501.
- Money D. The laboratory diagnosis of bacterial vaginosis. Can J Infect Dis Med Microbiol 2005; 16:77–79.
- Lowe NK, Neal JL, Ryan-Wenger NA, et al. Accuracy of the clinical diagnosis of vaginitis compared with a DNA probe laboratory standard. Obstet Gynecol 2009; 113:89–95.
- Mulhem E, Boyanton BL Jr, Robinson-Dunn B, Ebert C, Dzebo R. Performance of the Affirm VP-III using residual vaginal discharge collected from the speculum to characterize vaginitis in symptomatic women. J Low Genit Tract Dis 2014; 18:344–346.
- Cartwright CP, Lembke BD, Ramachandran K, et al. Comparison of nucleic acid amplification assays with BD Affirm VPIII for diagnosis of vaginitis in symptomatic women. J Clin Microbiol 2013; 51:3694–3699.
- Workowski KA, Bolan GA; Centers for Disease Control and Prevention (CDC). Sexually transmitted diseases treatment guidelines, 2015. MMWR Recomm Rep 2015; 64:1–137.
- Sobel JD, Ferris D, Schwebke J, et al. Suppressive antibacterial therapy with 0.75% metronidazole vaginal gel to prevent recurrent bacterial vaginosis. Am J Obstet Gynecol 2006; 194:1283–1289.
- Reichman O, Akins R, Sobel JD. Boric acid addition to suppressive antimicrobial therapy for recurrent bacterial vaginosis. Sex Transm Dis 2009; 36:732–734.
- Carr PL, Felsenstein D, Friedman RH. Evaluation and management of vaginitis. J Gen Intern Med 1998; 13:335–346.
- Sobel JD, Chaim W, Nagappan V, Leaman D. Treatment of vaginitis caused by Candida glabrata: use of topical boric acid and flucytosine. Am J Obstet Gynecol 2003; 189:1297–1300.
- Sobel JD, Kapernick PS, Zervos M, et al. Treatment of complicated Candida vaginitis: comparison of single and sequential doses of fluconazole. Am J Obstet Gynecol 2001; 185:363–369.
- Sobel JD, Wiesenfeld HC, Martens M, et al. Maintenance fluconazole therapy for recurrent vulvovaginal candidiasis. N Engl J Med 2004; 351:876–883.
- Phillips AJ. Treatment of non-albicans Candida vaginitis with amphotericin B vaginal suppositories. Am J Obstet Gynecol 2005; 192:2009–2013.
- Weinstock H, Berman S, Cates W Jr. Sexually transmitted diseases among American youth: incidence and prevalence estimates, 2000. Perspect Sex Reprod Health 2004; 36:6–10.
- Gatski M, Martin DH, Clark RA, Harville E, Schmidt N, Kissinger P. Co-occurrence of Trichomonas vaginalis and bacterial vaginosis among HIV-positive women. Sex Transm Dis 2011; 38:163–166.
- Hobbs MM, Seña AC. Modern diagnosis of Trichomonas vaginalis infection. Sex Transm Infect 2013; 89:434–438.
- McClelland RS, Sangare L, Hassan WM, et al. Infection with Trichomonas vaginalis increases the risk of HIV-1 acquisition. J Infect Dis 2007; 195:698–702.
- Kingston MA, Bansal D, Carlin EM. ‘Shelf life’ of Trichomonas vaginalis. Int J STD AIDS 2003; 14:28–29.
- Aslan DL, Gulbahce HE, Stelow EB, et al. The diagnosis of Trichomonas vaginalis in liquid-based Pap tests: correlation with PCR. Diagn Cytopathol 2005; 32:341–344.
- Lara-Torre E, Pinkerton JS. Accuracy of detection of Trichomonas vaginalis organisms on a liquid-based papanicolaou smear. Am J Obstet Gynecol 2003; 188:354–356.
- Hobbs MM, Lapple DM, Lawing LF, et al. Methods for detection of Trichomonas vaginalis in the male partners of infected women: implications for control of trichomoniasis. J Clin Microbiol 2006; 44:3994–3999.
- Van Der Pol B, Williams JA, Orr DP, Batteiger BE, Fortenberry JD. Prevalence, incidence, natural history, and response to treatment of Trichomonas vaginalis infection among adolescent women. J Infect Dis 2005; 192:2039–2044.
- Campbell L, Woods V, Lloyd T, Elsayed S, Church DL. Evaluation of the OSOM Trichomonas rapid test versus wet preparation examination for detection of Trichomonas vaginalis vaginitis in specimens from women with a low prevalence of infection. J Clin Microbiol 2008; 46:3467–3469.
- Chapin K, Andrea S. APTIMA Trichomonas vaginalis, a transcription-mediated amplification assay for detection of Trichomonas vaginalis in urogenital specimens. Expert Rev Mol Diagn 2011; 11:679–688.
- Krashin JW, Koumans EH, Bradshaw-Sydnor AC, et al. Trichomonas vaginalis prevalence, incidence, risk factors and antibiotic-resistance in an adolescent population. Sex Transm Dis 2010; 37:440–444.
- Sobel JD, Nyirjesy P, Brown W. Tinidazole therapy for metronidazole-resistant vaginal trichomoniasis. Clin Infect Dis 2001; 33:1341–1346.
- Nyirjesy P, Gilbert J, Mulcahy LJ. Resistant trichomoniasis: successful treatment with combination therapy. Sex Transm Dis 2011; 38:962–963.
- Bradley H, Markowitz LE, Gibson T, McQuillan GM. Seroprevalence of herpes simplex virus types 1 and 2—United States, 1999–2010. J Infect Dis 2014; 209:325–333.
- Roberts CM, Pfister JR, Spear SJ. Increasing proportion of herpes simplex virus type 1 as a cause of genital herpes infection in college students. Sex Transm Dis 2003; 30:797–800.
- Ryder N, Jin F, McNulty AM, Grulich AE, Donovan B. Increasing role of herpes simplex virus type 1 in first-episode anogenital herpes in heterosexual women and younger men who have sex with men, 1992-2006. Sex Transm Infect 2009; 85:416–419.
- Caviness AC, Oelze LL, Saz UE, Greer JM, Demmler-Harrison GJ. Direct immunofluorescence assay compared to cell culture for the diagnosis of mucocutaneous herpes simplex virus infections in children. J Clin Virol 2010; 49:58–60.
- Morrow R, Friedrich D. Performance of a novel test for IgM and IgG antibodies in subjects with culture-documented genital herpes simplex virus-1 or -2 infection. Clin Microbiol Infect 2006; 12:463–469.
- Bradford J, Fischer G. Desquamative inflammatory vaginitis: differential diagnosis and alternate diagnostic criteria. J Low Genit Tract Dis 2010; 14:306–310.
KEY POINTS
- Typical presenting symptoms of vulvovaginitis are itching, burning, and abnormal discharge.
- Evaluating vaginal secretions with simple office-based tools is often sufficient for diagnosis, although DNA testing is also available.
- Depending on the cause, vulvovaginitis is generally treated with a course of oral or topical antibiotics, antiviral or antifungal drugs, anti-inflammatory agents, or hormonal therapy.
- Cases that do not resolve may require maintenance therapy. Patients who have persistent or unusual symptoms should be referred to a specialist.
Channeling the flow of medical information

New, changing technologies are being incorporated into every aspect of medical care and education, and the impact cannot be overstated. While the ultimate qualitative impact (beneficial, intrusive, efficient, obstructive, or neutral) of specific individual implementations remains to be seen, there is no doubt that the faces of patient care and the financial management of healthcare delivery have been forever altered.
The amount of information now literally at our fingertips is overwhelming. Some of my patients bring the latest from www.clinicaltrials.gov to their appointments. One patient recently brought downloaded online testimony from patients who were being given an oil supplement to treat disorders including gout, neuropathy, carpal tunnel, sinusitis, headache, and postop hip and knee pain and wanted to know why I hadn’t suggested it for her. Accessing the information pipeline is like drinking from a firehose, and there is no perfect valve that can adjust the flow to every thirst.
Lest we think that only patients use sites of variable reliability in getting their online medical information, in a recent survey by Kantar Media, when 508 physicians were asked which of 49 sites they looked at most often, Wikipedia came in at number 3 (UpToDate was number 1 and Cleveland Clinic Journal of Medicine was number 25). But when asked to rate the 49 sites for “quality clinical content,” responders listed Wikipedia as 47 (UpToDate was still number 1 and CCJM was number 6).
We healthcare providers can assess the accuracy and quality of clinical content. But it is not always easy, especially when trying to access, digest, and utilize information within the real-time constraints of an office visit or inpatient consultation. The now almost universal use of electronic medical records (EMRs) in major health systems provides access to true point-of-care information to assist in clinical decision-making, but how we filter and channel this information and apply it to the patient sitting in the exam room is not always straightforward. It is naïve to think that one source can fit all of our information needs.
A “smart” EMR can reflexively direct me to diagnosis-linked clinical guidelines or care paths. But without knowing the specifics of how the guidelines were written, I can’t know if they are ideally applicable to the patient in front of me. Guidelines based solely on clinical trial data may not be ideal for a given patient due to constraints of the clinical trial design and the tested clinical populations. There are times in areas outside my clinical field that I seek clinically nuanced expertise in interpreting clinical trials rather than the actual clinical trial data. Other times, when approaching problems within my own expertise, I want to see the raw data to reach a conclusion on its applicability and likely magnitude of effect in a specific patient. Using a trusted source of predigested, summarized data (as opposed to the raw data), knowing whether an author has a relationship with a specific company, and knowing that the clinical trials of a specific therapy are not intrinsically bad or good—all of these contribute to contextual decisions that lead to my clinical recommendations. I always want to know the nature of authors’ commercial relationships, as well as the track record of my information sources.
It is in this context that the paper by Andrews et al in this issue of the Journal addresses in a very practical way some of the nuances in using several popular point-of-care information resources. This is the second of 2 papers that these authors have contributed to the CCJM in an effort to inform and direct us how to quench our thirst for information without getting bloated.
New, changing technologies are being incorporated into every aspect of medical care and education, and the impact cannot be overstated. While the ultimate qualitative impact (beneficial, intrusive, efficient, obstructive, or neutral) of specific individual implementations remains to be seen, there is no doubt that the faces of patient care and the financial management of healthcare delivery have been forever altered.
The amount of information now literally at our fingertips is overwhelming. Some of my patients bring the latest from www.clinicaltrials.gov to their appointments. One patient recently brought downloaded online testimony from patients who were being given an oil supplement to treat disorders including gout, neuropathy, carpal tunnel, sinusitis, headache, and postop hip and knee pain and wanted to know why I hadn’t suggested it for her. Accessing the information pipeline is like drinking from a firehose, and there is no perfect valve that can adjust the flow to every thirst.
Lest we think that only patients use sites of variable reliability in getting their online medical information, in a recent survey by Kantar Media, when 508 physicians were asked which of 49 sites they looked at most often, Wikipedia came in at number 3 (UpToDate was number 1 and Cleveland Clinic Journal of Medicine was number 25). But when asked to rate the 49 sites for “quality clinical content,” responders listed Wikipedia as 47 (UpToDate was still number 1 and CCJM was number 6).
We healthcare providers can assess the accuracy and quality of clinical content. But it is not always easy, especially when trying to access, digest, and utilize information within the real-time constraints of an office visit or inpatient consultation. The now almost universal use of electronic medical records (EMRs) in major health systems provides access to true point-of-care information to assist in clinical decision-making, but how we filter and channel this information and apply it to the patient sitting in the exam room is not always straightforward. It is naïve to think that one source can fit all of our information needs.
A “smart” EMR can reflexively direct me to diagnosis-linked clinical guidelines or care paths. But without knowing the specifics of how the guidelines were written, I can’t know if they are ideally applicable to the patient in front of me. Guidelines based solely on clinical trial data may not be ideal for a given patient due to constraints of the clinical trial design and the tested clinical populations. There are times in areas outside my clinical field that I seek clinically nuanced expertise in interpreting clinical trials rather than the actual clinical trial data. Other times, when approaching problems within my own expertise, I want to see the raw data to reach a conclusion on its applicability and likely magnitude of effect in a specific patient. Using a trusted source of predigested, summarized data (as opposed to the raw data), knowing whether an author has a relationship with a specific company, and knowing that the clinical trials of a specific therapy are not intrinsically bad or good—all of these contribute to contextual decisions that lead to my clinical recommendations. I always want to know the nature of authors’ commercial relationships, as well as the track record of my information sources.
It is in this context that the paper by Andrews et al in this issue of the Journal addresses in a very practical way some of the nuances in using several popular point-of-care information resources. This is the second of 2 papers that these authors have contributed to the CCJM in an effort to inform and direct us how to quench our thirst for information without getting bloated.
New, changing technologies are being incorporated into every aspect of medical care and education, and the impact cannot be overstated. While the ultimate qualitative impact (beneficial, intrusive, efficient, obstructive, or neutral) of specific individual implementations remains to be seen, there is no doubt that the faces of patient care and the financial management of healthcare delivery have been forever altered.
The amount of information now literally at our fingertips is overwhelming. Some of my patients bring the latest from www.clinicaltrials.gov to their appointments. One patient recently brought downloaded online testimony from patients who were being given an oil supplement to treat disorders including gout, neuropathy, carpal tunnel, sinusitis, headache, and postop hip and knee pain and wanted to know why I hadn’t suggested it for her. Accessing the information pipeline is like drinking from a firehose, and there is no perfect valve that can adjust the flow to every thirst.
Lest we think that only patients use sites of variable reliability in getting their online medical information, in a recent survey by Kantar Media, when 508 physicians were asked which of 49 sites they looked at most often, Wikipedia came in at number 3 (UpToDate was number 1 and Cleveland Clinic Journal of Medicine was number 25). But when asked to rate the 49 sites for “quality clinical content,” responders listed Wikipedia as 47 (UpToDate was still number 1 and CCJM was number 6).
We healthcare providers can assess the accuracy and quality of clinical content. But it is not always easy, especially when trying to access, digest, and utilize information within the real-time constraints of an office visit or inpatient consultation. The now almost universal use of electronic medical records (EMRs) in major health systems provides access to true point-of-care information to assist in clinical decision-making, but how we filter and channel this information and apply it to the patient sitting in the exam room is not always straightforward. It is naïve to think that one source can fit all of our information needs.
A “smart” EMR can reflexively direct me to diagnosis-linked clinical guidelines or care paths. But without knowing the specifics of how the guidelines were written, I can’t know if they are ideally applicable to the patient in front of me. Guidelines based solely on clinical trial data may not be ideal for a given patient due to constraints of the clinical trial design and the tested clinical populations. There are times in areas outside my clinical field that I seek clinically nuanced expertise in interpreting clinical trials rather than the actual clinical trial data. Other times, when approaching problems within my own expertise, I want to see the raw data to reach a conclusion on its applicability and likely magnitude of effect in a specific patient. Using a trusted source of predigested, summarized data (as opposed to the raw data), knowing whether an author has a relationship with a specific company, and knowing that the clinical trials of a specific therapy are not intrinsically bad or good—all of these contribute to contextual decisions that lead to my clinical recommendations. I always want to know the nature of authors’ commercial relationships, as well as the track record of my information sources.
It is in this context that the paper by Andrews et al in this issue of the Journal addresses in a very practical way some of the nuances in using several popular point-of-care information resources. This is the second of 2 papers that these authors have contributed to the CCJM in an effort to inform and direct us how to quench our thirst for information without getting bloated.

Caring for international patients
To the Editor: We read with great interest the article by Drs. Cawcutt and Wilson on caring for international patients.1 They provide an overview of the challenges of delivering medical care for these patients (eg, cultural differences) and the likely benefits from such interactions (eg, gaining cultural knowledge). Having practiced medicine in 3 different continents and experienced working in various medical centers caring for international patients, we would like to offer a slightly different viewpoint.
First, gaining cultural knowledge should be regarded as a prerequisite for healthcare workers involved in the care of international patients, rather than the expected benefit and consequence of such encounters. Healthcare workers with some knowledge of an international patient’s culture are best able to serve that patient.2 Indeed, unless knowledge of cultural differences is obtained before such interactions, there is a significant risk of stereotyping by amplifying the sense of “otherness,” which is unfortunately too often mistaken for cultural sensitivity. The perception of the stereotypes and prejudices during the second stage of cultural adaptation (ie, irritation, hostility) often stems from the host’s lack of cultural knowledge. Table 1 of their article clearly reflects such risk: the authors have tried to exemplify the concepts they discussed through a number of real-life scenarios. But indeed some of those cases (eg, the man from Saudi Arabia) could be interpreted more as examples of stereotyping than cultural sensitivity.
Second, the authors do not mention requests by family members of international patients for nondisclosure of serious medical diagnoses, one we have frequently received from family members from different cultural backgrounds. These requests represent another challenge of caring for these patients as they counter our obligation for full disclosure and the patients’ right to know in order to be able to make informed decisions regarding their medical care.3
- Cawcutt KA, Wilson JW. Benefits and challenges of caring for international patients. Cleve Clin J Med 2016; 83:794–800.
- Martin DR. Challenges and opportunities in the care of international patients: clinical and health services issues for academic medical centers. Acad Med 2006;81:189–192.
- American Medical Association Code of Ethics. https://www.ama-assn.org/sites/default/files/media-browser/code-of-medical-ethics-chapter-2.pdf. Accessed November 28, 2016.
To the Editor: We read with great interest the article by Drs. Cawcutt and Wilson on caring for international patients.1 They provide an overview of the challenges of delivering medical care for these patients (eg, cultural differences) and the likely benefits from such interactions (eg, gaining cultural knowledge). Having practiced medicine in 3 different continents and experienced working in various medical centers caring for international patients, we would like to offer a slightly different viewpoint.
First, gaining cultural knowledge should be regarded as a prerequisite for healthcare workers involved in the care of international patients, rather than the expected benefit and consequence of such encounters. Healthcare workers with some knowledge of an international patient’s culture are best able to serve that patient.2 Indeed, unless knowledge of cultural differences is obtained before such interactions, there is a significant risk of stereotyping by amplifying the sense of “otherness,” which is unfortunately too often mistaken for cultural sensitivity. The perception of the stereotypes and prejudices during the second stage of cultural adaptation (ie, irritation, hostility) often stems from the host’s lack of cultural knowledge. Table 1 of their article clearly reflects such risk: the authors have tried to exemplify the concepts they discussed through a number of real-life scenarios. But indeed some of those cases (eg, the man from Saudi Arabia) could be interpreted more as examples of stereotyping than cultural sensitivity.
Second, the authors do not mention requests by family members of international patients for nondisclosure of serious medical diagnoses, one we have frequently received from family members from different cultural backgrounds. These requests represent another challenge of caring for these patients as they counter our obligation for full disclosure and the patients’ right to know in order to be able to make informed decisions regarding their medical care.3
To the Editor: We read with great interest the article by Drs. Cawcutt and Wilson on caring for international patients.1 They provide an overview of the challenges of delivering medical care for these patients (eg, cultural differences) and the likely benefits from such interactions (eg, gaining cultural knowledge). Having practiced medicine in 3 different continents and experienced working in various medical centers caring for international patients, we would like to offer a slightly different viewpoint.
First, gaining cultural knowledge should be regarded as a prerequisite for healthcare workers involved in the care of international patients, rather than the expected benefit and consequence of such encounters. Healthcare workers with some knowledge of an international patient’s culture are best able to serve that patient.2 Indeed, unless knowledge of cultural differences is obtained before such interactions, there is a significant risk of stereotyping by amplifying the sense of “otherness,” which is unfortunately too often mistaken for cultural sensitivity. The perception of the stereotypes and prejudices during the second stage of cultural adaptation (ie, irritation, hostility) often stems from the host’s lack of cultural knowledge. Table 1 of their article clearly reflects such risk: the authors have tried to exemplify the concepts they discussed through a number of real-life scenarios. But indeed some of those cases (eg, the man from Saudi Arabia) could be interpreted more as examples of stereotyping than cultural sensitivity.
Second, the authors do not mention requests by family members of international patients for nondisclosure of serious medical diagnoses, one we have frequently received from family members from different cultural backgrounds. These requests represent another challenge of caring for these patients as they counter our obligation for full disclosure and the patients’ right to know in order to be able to make informed decisions regarding their medical care.3
- Cawcutt KA, Wilson JW. Benefits and challenges of caring for international patients. Cleve Clin J Med 2016; 83:794–800.
- Martin DR. Challenges and opportunities in the care of international patients: clinical and health services issues for academic medical centers. Acad Med 2006;81:189–192.
- American Medical Association Code of Ethics. https://www.ama-assn.org/sites/default/files/media-browser/code-of-medical-ethics-chapter-2.pdf. Accessed November 28, 2016.
- Cawcutt KA, Wilson JW. Benefits and challenges of caring for international patients. Cleve Clin J Med 2016; 83:794–800.
- Martin DR. Challenges and opportunities in the care of international patients: clinical and health services issues for academic medical centers. Acad Med 2006;81:189–192.
- American Medical Association Code of Ethics. https://www.ama-assn.org/sites/default/files/media-browser/code-of-medical-ethics-chapter-2.pdf. Accessed November 28, 2016.