User login
NIH Issues Final Rule for Registering Clinical Trials and Reporting Results
The National Institutes of Health (NIH) has published a new policy to assure that results of clinical trials are widely shared. NIH Director Francis Collins, MD, said the final rule and NIH policy that have been issued “will help maximize the value of clinical trials, whether publicly or privately supported, and help us honor our commitments to trial participants, who do so much to help society advance knowledge and improve health.”
The National Institutes of Health (NIH) has published a new policy to assure that results of clinical trials are widely shared. NIH Director Francis Collins, MD, said the final rule and NIH policy that have been issued “will help maximize the value of clinical trials, whether publicly or privately supported, and help us honor our commitments to trial participants, who do so much to help society advance knowledge and improve health.”
The National Institutes of Health (NIH) has published a new policy to assure that results of clinical trials are widely shared. NIH Director Francis Collins, MD, said the final rule and NIH policy that have been issued “will help maximize the value of clinical trials, whether publicly or privately supported, and help us honor our commitments to trial participants, who do so much to help society advance knowledge and improve health.”
Acute HIV Causes Transient Neurologic Findings
Clinical Question: How common are neurologic findings in acute HIV infection?
Background: The incidence of neurologic findings with acute HIV is unknown.
Study Design: Cohort study.
Setting: Bangkok, Thailand.
Synopsis: In this study, 134 patients were identified after presenting for voluntary HIV testing. Five others were enrolled through an ongoing local study. All 139 participants underwent structured neurologic evaluations at enrollment (median of 19 days after presumed exposure), then at four and 12 weeks. Combination antiretroviral therapy (cART) was initiated immediately after initial evaluation.
The cohort was 93% male. Mean age was younger than 30 years. Fifty-three percent of participants experienced some neurologic finding within 12 weeks of diagnosis. One-third (33%) were cognitive symptoms, predominantly problems of concentration (24% of patients) and memory (16% of patients). One-third (34%) were motor findings, and 11% were neuropathy. Forty-nine percent of the neurologic issues were present at diagnosis. Symptoms were mostly mild, although one patient developed fulminant Guillain-Barré syndrome. Patients with neurologic findings had higher viral loads at diagnosis (mean plasma log10 HIV RNA 5.9 versus 5.4; P = 0.006). Participants with and without neurologic findings had similar cerebral spinal fluid viral loads (mean log10 HIV RNA 3.7 versus 3.1, P = 0.14) and serum CD4 counts (339 versus 381 cells/mm3; P = 0.46). Neurologic findings resolved within one month of cART treatment in 90% of patients. Study limitations include lack of a control cohort and potential confounding from illicit drug use among participants.
Bottom Line: Acute HIV infection commonly causes mild neurologic problems, which remit with treatment.
Citation: Hellmuth J, Fletcher JL, Valcour V, et al. Neurologic signs and symptoms frequently manifest in acute HIV infection. Neurology. 2016;87(2):148-154.
Clinical Question: How common are neurologic findings in acute HIV infection?
Background: The incidence of neurologic findings with acute HIV is unknown.
Study Design: Cohort study.
Setting: Bangkok, Thailand.
Synopsis: In this study, 134 patients were identified after presenting for voluntary HIV testing. Five others were enrolled through an ongoing local study. All 139 participants underwent structured neurologic evaluations at enrollment (median of 19 days after presumed exposure), then at four and 12 weeks. Combination antiretroviral therapy (cART) was initiated immediately after initial evaluation.
The cohort was 93% male. Mean age was younger than 30 years. Fifty-three percent of participants experienced some neurologic finding within 12 weeks of diagnosis. One-third (33%) were cognitive symptoms, predominantly problems of concentration (24% of patients) and memory (16% of patients). One-third (34%) were motor findings, and 11% were neuropathy. Forty-nine percent of the neurologic issues were present at diagnosis. Symptoms were mostly mild, although one patient developed fulminant Guillain-Barré syndrome. Patients with neurologic findings had higher viral loads at diagnosis (mean plasma log10 HIV RNA 5.9 versus 5.4; P = 0.006). Participants with and without neurologic findings had similar cerebral spinal fluid viral loads (mean log10 HIV RNA 3.7 versus 3.1, P = 0.14) and serum CD4 counts (339 versus 381 cells/mm3; P = 0.46). Neurologic findings resolved within one month of cART treatment in 90% of patients. Study limitations include lack of a control cohort and potential confounding from illicit drug use among participants.
Bottom Line: Acute HIV infection commonly causes mild neurologic problems, which remit with treatment.
Citation: Hellmuth J, Fletcher JL, Valcour V, et al. Neurologic signs and symptoms frequently manifest in acute HIV infection. Neurology. 2016;87(2):148-154.
Clinical Question: How common are neurologic findings in acute HIV infection?
Background: The incidence of neurologic findings with acute HIV is unknown.
Study Design: Cohort study.
Setting: Bangkok, Thailand.
Synopsis: In this study, 134 patients were identified after presenting for voluntary HIV testing. Five others were enrolled through an ongoing local study. All 139 participants underwent structured neurologic evaluations at enrollment (median of 19 days after presumed exposure), then at four and 12 weeks. Combination antiretroviral therapy (cART) was initiated immediately after initial evaluation.
The cohort was 93% male. Mean age was younger than 30 years. Fifty-three percent of participants experienced some neurologic finding within 12 weeks of diagnosis. One-third (33%) were cognitive symptoms, predominantly problems of concentration (24% of patients) and memory (16% of patients). One-third (34%) were motor findings, and 11% were neuropathy. Forty-nine percent of the neurologic issues were present at diagnosis. Symptoms were mostly mild, although one patient developed fulminant Guillain-Barré syndrome. Patients with neurologic findings had higher viral loads at diagnosis (mean plasma log10 HIV RNA 5.9 versus 5.4; P = 0.006). Participants with and without neurologic findings had similar cerebral spinal fluid viral loads (mean log10 HIV RNA 3.7 versus 3.1, P = 0.14) and serum CD4 counts (339 versus 381 cells/mm3; P = 0.46). Neurologic findings resolved within one month of cART treatment in 90% of patients. Study limitations include lack of a control cohort and potential confounding from illicit drug use among participants.
Bottom Line: Acute HIV infection commonly causes mild neurologic problems, which remit with treatment.
Citation: Hellmuth J, Fletcher JL, Valcour V, et al. Neurologic signs and symptoms frequently manifest in acute HIV infection. Neurology. 2016;87(2):148-154.

Two-Minute Screen Effective for Post-Op Delirium
Clinical Question: Is the 10-point cognitive screener (10-CS) effective in screening for delirium in older adults with hip fracture?
Background: Delirium in elderly hip fracture patients has been established as a significant comorbidity. There is, however, no agreement on the most appropriate and practical screening tool. Commonly used screening methods, which focus on the detection of cognitive impairment as a surrogate, are time-consuming, insensitive for mild impairment, and limited in their application to patients with impaired dexterity and poor education.
Study Design: Prospective cohort study.
Setting: Tertiary referral hospital in São Paulo, Brazil.
Synopsis: In the study, 147 consecutive hip fracture patients over age 60 were screened using the 10-CS. This test stratifies patients into three categories: normal, possible, and probable cognitive impairment. Development of in-hospital delirium was evaluated by daily Confusion Assessment Method testing administered by a geriatrician. Patients categorized as probable cognitive impairment were more likely to develop delirium (hazard ratio, 7.48; 95% CI, 2.2–25.4).
Hospitalists involved in perioperative care should consider using this simple screening tool. With an area under ROC curve of 0.83 (95% CI, 0.76–0.89), it effectively detects delirium in this high-risk population. Independently, patients who developed delirium had a longer length of stay (median 11.0 versus 7.0; P < 0.001). This serves as a reminder of the importance of screening and preventing delirium in this population.
Bottom Line: The 10-CS tool is practical in its application and effective in identifying elderly hip fracture patients at risk for delirium.
Citation: Fortes-Filho SQ, Apolinario D, Melo JA, Suzuki I, Sitta MD, Garcez-Leme LE. Predicting delirium after hip fracture with a 2-min cognitive screen: prospective cohort study [published online ahead of print May 17, 2016]. Age Ageing. pii:afw084.
Clinical Question: Is the 10-point cognitive screener (10-CS) effective in screening for delirium in older adults with hip fracture?
Background: Delirium in elderly hip fracture patients has been established as a significant comorbidity. There is, however, no agreement on the most appropriate and practical screening tool. Commonly used screening methods, which focus on the detection of cognitive impairment as a surrogate, are time-consuming, insensitive for mild impairment, and limited in their application to patients with impaired dexterity and poor education.
Study Design: Prospective cohort study.
Setting: Tertiary referral hospital in São Paulo, Brazil.
Synopsis: In the study, 147 consecutive hip fracture patients over age 60 were screened using the 10-CS. This test stratifies patients into three categories: normal, possible, and probable cognitive impairment. Development of in-hospital delirium was evaluated by daily Confusion Assessment Method testing administered by a geriatrician. Patients categorized as probable cognitive impairment were more likely to develop delirium (hazard ratio, 7.48; 95% CI, 2.2–25.4).
Hospitalists involved in perioperative care should consider using this simple screening tool. With an area under ROC curve of 0.83 (95% CI, 0.76–0.89), it effectively detects delirium in this high-risk population. Independently, patients who developed delirium had a longer length of stay (median 11.0 versus 7.0; P < 0.001). This serves as a reminder of the importance of screening and preventing delirium in this population.
Bottom Line: The 10-CS tool is practical in its application and effective in identifying elderly hip fracture patients at risk for delirium.
Citation: Fortes-Filho SQ, Apolinario D, Melo JA, Suzuki I, Sitta MD, Garcez-Leme LE. Predicting delirium after hip fracture with a 2-min cognitive screen: prospective cohort study [published online ahead of print May 17, 2016]. Age Ageing. pii:afw084.
Clinical Question: Is the 10-point cognitive screener (10-CS) effective in screening for delirium in older adults with hip fracture?
Background: Delirium in elderly hip fracture patients has been established as a significant comorbidity. There is, however, no agreement on the most appropriate and practical screening tool. Commonly used screening methods, which focus on the detection of cognitive impairment as a surrogate, are time-consuming, insensitive for mild impairment, and limited in their application to patients with impaired dexterity and poor education.
Study Design: Prospective cohort study.
Setting: Tertiary referral hospital in São Paulo, Brazil.
Synopsis: In the study, 147 consecutive hip fracture patients over age 60 were screened using the 10-CS. This test stratifies patients into three categories: normal, possible, and probable cognitive impairment. Development of in-hospital delirium was evaluated by daily Confusion Assessment Method testing administered by a geriatrician. Patients categorized as probable cognitive impairment were more likely to develop delirium (hazard ratio, 7.48; 95% CI, 2.2–25.4).
Hospitalists involved in perioperative care should consider using this simple screening tool. With an area under ROC curve of 0.83 (95% CI, 0.76–0.89), it effectively detects delirium in this high-risk population. Independently, patients who developed delirium had a longer length of stay (median 11.0 versus 7.0; P < 0.001). This serves as a reminder of the importance of screening and preventing delirium in this population.
Bottom Line: The 10-CS tool is practical in its application and effective in identifying elderly hip fracture patients at risk for delirium.
Citation: Fortes-Filho SQ, Apolinario D, Melo JA, Suzuki I, Sitta MD, Garcez-Leme LE. Predicting delirium after hip fracture with a 2-min cognitive screen: prospective cohort study [published online ahead of print May 17, 2016]. Age Ageing. pii:afw084.
Analysis yields ‘strong evidence’ for benefit of physical activity in NAFLD
Regular physical exercise significantly improved measures of nonalcoholic fatty liver disease independently of dietary changes, according to a meta-analysis of randomized controlled* trials published in the October issue of Clinical Gastroenterology and Hepatology.
“On the basis of the current findings, physical activity should be recommended not only in combination with dietary changes but also independently as an effective approach to manage NAFLD,” wrote Lorenzo Orci, MD, and his associates at the University of Geneva. “We propose that the level of evidence surrounding the specific role of physical activity in the management of NAFLD is now sufficient to be awarded a grade of Ia.”
Nonalcoholic fatty liver disease, “the hepatic manifestation of metabolic syndrome,” affects at least one in four U.S. adults and 15%-35% of individuals in Europe, the Middle East, China, and Japan, the researchers noted. Dietary changes are the cornerstone of NAFLD management, and there is less evidence for how physical exercise affects liver fat content. Therefore, the researchers searched MEDLINE, Embase, and the Cochrane databases from inception through October 2015 to find randomized trials of the impact of physical activity on markers of liver steatosis and liver inflammation in patients diagnosed with NAFLD, obesity, type 2 diabetes, or metabolic syndrome. This approach yielded 28 trials with data from more than 1,600 patients. Only two trials were multicenter, 13 required participants to have an NAFLD diagnosis, four focused on type 2 diabetes, and most of the rest included sedentary obese patients without requiring a diagnosis of NAFLD, the researchers said (Clin Gastroenterol Hepatol. 2016 May 4. doi: 10.1016/j.cgh.2016.04.036).
After researchers accounted for dietary changes, physical activity led to a significant drop in intrahepatic lipid content with a standardized mean difference of –0.69 compared with controls (95% confidence interval, –0.90 to –0.48; P less than .0001). “Because effect sizes such as standard mean difference [SMD] are difficult to interpret, the translation of such a statistical measure into a clinically relevant notion has been the focus of research for more than a decade,” the investigators added. “A commonly used interpretation was proposed by Cohen, who suggested that SMDs of 0.2, 0.5, and 0.8 correspond to small, moderate, and large effect sizes, respectively. By using this rule of thumb, our results indicate that physical activity exerts a moderate-to-large impact on the reduction of intrahepatic lipid content.”
Exercise reduced liver fat content even more in pediatric patients (SMD, –0.75; 95% CI, –0.1 to –0.5; P less than .0001) and in patients who had been specifically diagnosed with NAFLD (SMD, –0.86; 95% CI, –1.26 to –0.46; P less than .0001). Patients with the highest baseline body mass index also seemed to benefit more than patients with lower baseline BMI (P = .04). Indeed, exercise reduced BMI itself by a weighted mean difference of 0.8 (95% CI, –1.22 to 0.38; P less than .001), the researchers noted. Exercise intensity did not seem to affect the likelihood of benefit. There was a trend toward a greater effect of aerobic over resistance training (P = .06), and few studies examined the effects of combining both types of exercise.
The multivariable analysis also linked physical activity to an average 3.30 IU/L drop in alanine aminotransferase levels (95% CI, –5.57 to –1.04) and to a 4.9 IU/L decrease in aspartate aminotransferase levels (95% CI, –8.68 to –1.02). The investigators were unable to assess the long-term effects of physical exercise, nor its effects on hepatic fibrosis or inflammation, they noted. Nonetheless, the moderate to large effect size “provides strong evidence for the recommendation of physical activity as an effective intervention in the treatment of NAFLD,” they concluded. “Physical activity is also associated with an improvement in blood levels of aminotransferases and is particularly beneficial in patients presenting with severe obesity at baseline.”
The work was funded by the Ligue Genevoise contre le Cancer and the Dr Henri Dubois-Ferrière/Dinu Lipatti Foundation and by the Swiss National Science Foundation. The investigators had no disclosures.
*Content was updated on 10/25/2016
There has been tremendous interest in developing pharmacologic treatments for nonalcoholic steatohepatitis, especially in the Western world. There has not been significant enthusiasm for investigating exercise-based lifestyle modification as a primary treatment for NASH. Although the meta-analysis by Orci et al. included 28 studies, there are only 2 studies (combined, fewer than 100 patients) that examined the effect of exercise on liver histology in NASH and they both suggest that lifestyle modification consisting of exercise in addition to dietary modification improves liver histology in NASH. A seminal study was published by Vilar-Gomez et al. (Gastroenterology. 2015;149:367-78) that showed that a lifestyle modification consisting of reduction in caloric intake by 750 kcal/d along with low-intensity exercise (200 minutes of walking each week) led to significant improvement in liver histology, especially in those who lost at least 5% of their body weight.

Naga Chalasani, MD, AGAF, FACG, FAASLD, is the David W. Crabb Professor and director of the division of gastroenterology and hepatology, Indiana University, Purdue. He had no relevant conflicts.
There has been tremendous interest in developing pharmacologic treatments for nonalcoholic steatohepatitis, especially in the Western world. There has not been significant enthusiasm for investigating exercise-based lifestyle modification as a primary treatment for NASH. Although the meta-analysis by Orci et al. included 28 studies, there are only 2 studies (combined, fewer than 100 patients) that examined the effect of exercise on liver histology in NASH and they both suggest that lifestyle modification consisting of exercise in addition to dietary modification improves liver histology in NASH. A seminal study was published by Vilar-Gomez et al. (Gastroenterology. 2015;149:367-78) that showed that a lifestyle modification consisting of reduction in caloric intake by 750 kcal/d along with low-intensity exercise (200 minutes of walking each week) led to significant improvement in liver histology, especially in those who lost at least 5% of their body weight.
Naga Chalasani, MD, AGAF, FACG, FAASLD, is the David W. Crabb Professor and director of the division of gastroenterology and hepatology, Indiana University, Purdue. He had no relevant conflicts.
There has been tremendous interest in developing pharmacologic treatments for nonalcoholic steatohepatitis, especially in the Western world. There has not been significant enthusiasm for investigating exercise-based lifestyle modification as a primary treatment for NASH. Although the meta-analysis by Orci et al. included 28 studies, there are only 2 studies (combined, fewer than 100 patients) that examined the effect of exercise on liver histology in NASH and they both suggest that lifestyle modification consisting of exercise in addition to dietary modification improves liver histology in NASH. A seminal study was published by Vilar-Gomez et al. (Gastroenterology. 2015;149:367-78) that showed that a lifestyle modification consisting of reduction in caloric intake by 750 kcal/d along with low-intensity exercise (200 minutes of walking each week) led to significant improvement in liver histology, especially in those who lost at least 5% of their body weight.
Naga Chalasani, MD, AGAF, FACG, FAASLD, is the David W. Crabb Professor and director of the division of gastroenterology and hepatology, Indiana University, Purdue. He had no relevant conflicts.
Regular physical exercise significantly improved measures of nonalcoholic fatty liver disease independently of dietary changes, according to a meta-analysis of randomized controlled* trials published in the October issue of Clinical Gastroenterology and Hepatology.
“On the basis of the current findings, physical activity should be recommended not only in combination with dietary changes but also independently as an effective approach to manage NAFLD,” wrote Lorenzo Orci, MD, and his associates at the University of Geneva. “We propose that the level of evidence surrounding the specific role of physical activity in the management of NAFLD is now sufficient to be awarded a grade of Ia.”
Nonalcoholic fatty liver disease, “the hepatic manifestation of metabolic syndrome,” affects at least one in four U.S. adults and 15%-35% of individuals in Europe, the Middle East, China, and Japan, the researchers noted. Dietary changes are the cornerstone of NAFLD management, and there is less evidence for how physical exercise affects liver fat content. Therefore, the researchers searched MEDLINE, Embase, and the Cochrane databases from inception through October 2015 to find randomized trials of the impact of physical activity on markers of liver steatosis and liver inflammation in patients diagnosed with NAFLD, obesity, type 2 diabetes, or metabolic syndrome. This approach yielded 28 trials with data from more than 1,600 patients. Only two trials were multicenter, 13 required participants to have an NAFLD diagnosis, four focused on type 2 diabetes, and most of the rest included sedentary obese patients without requiring a diagnosis of NAFLD, the researchers said (Clin Gastroenterol Hepatol. 2016 May 4. doi: 10.1016/j.cgh.2016.04.036).
After researchers accounted for dietary changes, physical activity led to a significant drop in intrahepatic lipid content with a standardized mean difference of –0.69 compared with controls (95% confidence interval, –0.90 to –0.48; P less than .0001). “Because effect sizes such as standard mean difference [SMD] are difficult to interpret, the translation of such a statistical measure into a clinically relevant notion has been the focus of research for more than a decade,” the investigators added. “A commonly used interpretation was proposed by Cohen, who suggested that SMDs of 0.2, 0.5, and 0.8 correspond to small, moderate, and large effect sizes, respectively. By using this rule of thumb, our results indicate that physical activity exerts a moderate-to-large impact on the reduction of intrahepatic lipid content.”
Exercise reduced liver fat content even more in pediatric patients (SMD, –0.75; 95% CI, –0.1 to –0.5; P less than .0001) and in patients who had been specifically diagnosed with NAFLD (SMD, –0.86; 95% CI, –1.26 to –0.46; P less than .0001). Patients with the highest baseline body mass index also seemed to benefit more than patients with lower baseline BMI (P = .04). Indeed, exercise reduced BMI itself by a weighted mean difference of 0.8 (95% CI, –1.22 to 0.38; P less than .001), the researchers noted. Exercise intensity did not seem to affect the likelihood of benefit. There was a trend toward a greater effect of aerobic over resistance training (P = .06), and few studies examined the effects of combining both types of exercise.
The multivariable analysis also linked physical activity to an average 3.30 IU/L drop in alanine aminotransferase levels (95% CI, –5.57 to –1.04) and to a 4.9 IU/L decrease in aspartate aminotransferase levels (95% CI, –8.68 to –1.02). The investigators were unable to assess the long-term effects of physical exercise, nor its effects on hepatic fibrosis or inflammation, they noted. Nonetheless, the moderate to large effect size “provides strong evidence for the recommendation of physical activity as an effective intervention in the treatment of NAFLD,” they concluded. “Physical activity is also associated with an improvement in blood levels of aminotransferases and is particularly beneficial in patients presenting with severe obesity at baseline.”
The work was funded by the Ligue Genevoise contre le Cancer and the Dr Henri Dubois-Ferrière/Dinu Lipatti Foundation and by the Swiss National Science Foundation. The investigators had no disclosures.
*Content was updated on 10/25/2016
Regular physical exercise significantly improved measures of nonalcoholic fatty liver disease independently of dietary changes, according to a meta-analysis of randomized controlled* trials published in the October issue of Clinical Gastroenterology and Hepatology.
“On the basis of the current findings, physical activity should be recommended not only in combination with dietary changes but also independently as an effective approach to manage NAFLD,” wrote Lorenzo Orci, MD, and his associates at the University of Geneva. “We propose that the level of evidence surrounding the specific role of physical activity in the management of NAFLD is now sufficient to be awarded a grade of Ia.”
Nonalcoholic fatty liver disease, “the hepatic manifestation of metabolic syndrome,” affects at least one in four U.S. adults and 15%-35% of individuals in Europe, the Middle East, China, and Japan, the researchers noted. Dietary changes are the cornerstone of NAFLD management, and there is less evidence for how physical exercise affects liver fat content. Therefore, the researchers searched MEDLINE, Embase, and the Cochrane databases from inception through October 2015 to find randomized trials of the impact of physical activity on markers of liver steatosis and liver inflammation in patients diagnosed with NAFLD, obesity, type 2 diabetes, or metabolic syndrome. This approach yielded 28 trials with data from more than 1,600 patients. Only two trials were multicenter, 13 required participants to have an NAFLD diagnosis, four focused on type 2 diabetes, and most of the rest included sedentary obese patients without requiring a diagnosis of NAFLD, the researchers said (Clin Gastroenterol Hepatol. 2016 May 4. doi: 10.1016/j.cgh.2016.04.036).
After researchers accounted for dietary changes, physical activity led to a significant drop in intrahepatic lipid content with a standardized mean difference of –0.69 compared with controls (95% confidence interval, –0.90 to –0.48; P less than .0001). “Because effect sizes such as standard mean difference [SMD] are difficult to interpret, the translation of such a statistical measure into a clinically relevant notion has been the focus of research for more than a decade,” the investigators added. “A commonly used interpretation was proposed by Cohen, who suggested that SMDs of 0.2, 0.5, and 0.8 correspond to small, moderate, and large effect sizes, respectively. By using this rule of thumb, our results indicate that physical activity exerts a moderate-to-large impact on the reduction of intrahepatic lipid content.”
Exercise reduced liver fat content even more in pediatric patients (SMD, –0.75; 95% CI, –0.1 to –0.5; P less than .0001) and in patients who had been specifically diagnosed with NAFLD (SMD, –0.86; 95% CI, –1.26 to –0.46; P less than .0001). Patients with the highest baseline body mass index also seemed to benefit more than patients with lower baseline BMI (P = .04). Indeed, exercise reduced BMI itself by a weighted mean difference of 0.8 (95% CI, –1.22 to 0.38; P less than .001), the researchers noted. Exercise intensity did not seem to affect the likelihood of benefit. There was a trend toward a greater effect of aerobic over resistance training (P = .06), and few studies examined the effects of combining both types of exercise.
The multivariable analysis also linked physical activity to an average 3.30 IU/L drop in alanine aminotransferase levels (95% CI, –5.57 to –1.04) and to a 4.9 IU/L decrease in aspartate aminotransferase levels (95% CI, –8.68 to –1.02). The investigators were unable to assess the long-term effects of physical exercise, nor its effects on hepatic fibrosis or inflammation, they noted. Nonetheless, the moderate to large effect size “provides strong evidence for the recommendation of physical activity as an effective intervention in the treatment of NAFLD,” they concluded. “Physical activity is also associated with an improvement in blood levels of aminotransferases and is particularly beneficial in patients presenting with severe obesity at baseline.”
The work was funded by the Ligue Genevoise contre le Cancer and the Dr Henri Dubois-Ferrière/Dinu Lipatti Foundation and by the Swiss National Science Foundation. The investigators had no disclosures.
*Content was updated on 10/25/2016
Key clinical point: Physical activity benefits measures of nonalcoholic fatty liver disease independently of diet.
Major finding: After researchers accounted for dietary changes, physical activity led to a significant drop in intrahepatic lipid content with a standardized mean difference of –0.69 compared with controls (95% confidence interval, –0.90 to –0.48; P less than .0001).
Data source: A systematic review and meta-analysis of 28 randomized controlled trials comprising more than 16,000 patients.
Disclosures: The work was funded by the Ligue Genevoise contre le Cancer and the Dr Henri Dubois-Ferrière/Dinu Lipatti Foundation and by the Swiss National Science Foundation. The researchers had no disclosures.
Investigational HCV drug combo yields high SVR12 rates in compensated cirrhosis
A once-daily regimen of two investigational, direct-acting anti-HCV agents, ABT-493 and ABT-530, was well tolerated and achieved sustained viral response at 12 weeks (SVR12) for nearly all patients with compensated cirrhosis and chronic genotype (GT) 1 or 3 hepatitis C virus infection, according to open-label phase II studies.
“The unique potency of these agents against all genotypes, even in the presence of common NS3 and/or NS5A baseline substitutions that confer resistance to most contemporary NS3/4A protease inhibitors and NS5A inhibitors, offers the potential for pangenotypic [HCV] therapy without ribavirin,” Edward J. Gane, MD, of the University of Auckland, New Zealand, and his associates wrote in the October issue of Gastroenterology. Phase III trials are now testing this hypothesis by focusing on cohorts of treatment-experienced, genotype 3–infected patients, on patients with renal impairment, and on patients who failed earlier-generation direct-acting antiviral regimens, they said.

The prevalence of HCV-related cirrhosis has yet to peak, and gold standard therapies for GT3 and GT1a infections can take weeks of treatment and the use of ribavirin, which causes undesirable side effects, the investigators noted. Attempts to surmount these residual barriers led to the development of ABT-493, an HCV nonstructural (NS) protein 3/4A protease inhibitor, and ABT-530, an HCV NS5A inhibitor. During in vitro studies, both agents showed “potent” activity against all major HCV genotypes, including variants with mutations that confer resistance to earlier, direct-acting antivirals, the researchers said (Gastroenterology. 2016 Jul 22. doi: 10.1053/j.gastro.2016.07.020). Their two open-label phase II studies enrolled adults with compensated cirrhosis and chronic GT3 (55 patients) or GT1 (27 patients) infection. Among GT1 patients, 41% had baseline NS3 substitutions conferring resistance to earlier-generation drugs, 19% had NS5A substitutions, and 11% had both mutations. The GT1-infected patients received 200 mg ABT-493 and 120 mg of ABT-530. The GT3-infected patients received 300 mg ABT-493 and 120 mg ABT-530, and half (27 patients) also received ribavirin. Most patients were treatment-naive, male, and white, with Child-Pugh scores of 5 and HCV RNA levels averaging about 6.2-6.6 log10 IU/mL.
In all, 26 patients with GT1 infection (96%) achieved SVR12 (95% confidence interval, 82% to 99%). The remaining patient relapsed after completing treatment. All treatment-naive GT3 patients achieved SVR12 whether or not they received ribavirin. However, one treatment-experienced GT3 patient who did not receive ribavirin relapsed after 16 weeks of treatment. Thus, rates of SVR12 were 96% (95% confidence interval, 82%-99%) for GT3 patients who did not receive ribavirin and 100% (95% CI, 88%-100%) for those who did. Notably, 94% of patients with baseline substitutions in NS3 and NS5A achieved SVR12, and there was no apparent link between treatment failure and any demographic or clinical characteristics, the investigators wrote.
Adverse events affected about 74% of patients and were usually mild or moderate in severity. Patients who did not receive ribavirin were most likely to report headache (15%), diarrhea (13%), and fatigue (11%). Only 4% of GT1 patients and 7% of the GT3 cohorts developed serious adverse events, and the only serious adverse event considered possibly treatment related involved a delusional disorder in a 57-year-old male who was receiving ribavirin and admitted amphetamine and alcohol use on the day it occurred. Treatment-related laboratory abnormalities were uncommon, no patients stopped treatment because of adverse events, and there were no deaths. “The rates of some adverse events were numerically higher with the higher ABT-493 dose, though the sample sizes are small and this was a cross-study comparison,” the investigators added. “Though not included in this study, patients with severe or end-stage kidney disease are predicted to be able to be treated with ABT-493 and ABT-530 because both agents have negligible renal excretion. These drugs were well tolerated in HCV-uninfected patients with renal impairment and can be administered without dose adjustment.”AbbVie funded the study and makes ABT-493 and ABT-530. Dr. Gane disclosed ties to AbbVie, Achillion Pharmaceuticals, Alnylam, Janssen, Merck, Novartis, and Novira.
In phase II and III clinical trials of direct-acting antivirals (DAAs), sustained viral response (SVR) rates over 90% were achieved in most patient groups and the combinations were well tolerated, results confirmed in real-world studies. However, a number of patients remain “difficult to cure.” Among them, patients infected with genotype 3, especially those with advanced liver disease, do not respond as well as patients infected with other genotypes and often need ribavirin.
In this study, a combination of two “next- generation” drugs with potent pangenotypic antiviral activity and a high barrier to resistance was administered to patients infected with HCV genotype 1 or 3 with compensated cirrhosis. Overall, 96% of patients infected with genotype 1 and 98% of patients infected with genotype 3 achieved SVR, with no apparent effect of ribavirin. The combination was well tolerated. Pending confirmation in phase III trials, these results suggest that pangenotypic combination regimens will be available in the very near future (approval expected in 2017) and that genotype 3 will become as easy to cure as other genotypes, while less ribavirin will be used. Unfortunately, patients with decompensated cirrhosis will not benefit from these advances, as protease inhibitors such as ABT-493 cannot be used in this population. This pangenotypic regimen may also prove particularly useful in patients with severe or end-stage kidney disease who should not receive the nucleotide analogue sofosbuvir. High SVR rates appear to be achievable when retreating patients who failed a prior DAA-based treatment with this combination, but relapses may still occur with highly resistant viruses. This next generation of HCV drugs will be the last generation. With this armamentarium, it will be technically possible to cure the vast majority of HCV-infected patients. Thus, screening and diagnosing HCV-infected patients are now mandatory in order to provide them with efficient care and make the world almost free of hepatitis C by 2030.
Jean-Michel Pawlotsky, MD, PhD, director of the National Reference Center for Viral Hepatitis B, C, and D, and professor of medicine in the department of virology, Hôpital Henri Mondor, Université Paris-Est, Créteil, France. He has received research grants from Gilead and Abbvie and has served as an adviser for Abbvie, Bristol-Myers Squibb, Gilead, Janssen, and Merck.
In phase II and III clinical trials of direct-acting antivirals (DAAs), sustained viral response (SVR) rates over 90% were achieved in most patient groups and the combinations were well tolerated, results confirmed in real-world studies. However, a number of patients remain “difficult to cure.” Among them, patients infected with genotype 3, especially those with advanced liver disease, do not respond as well as patients infected with other genotypes and often need ribavirin.
In this study, a combination of two “next- generation” drugs with potent pangenotypic antiviral activity and a high barrier to resistance was administered to patients infected with HCV genotype 1 or 3 with compensated cirrhosis. Overall, 96% of patients infected with genotype 1 and 98% of patients infected with genotype 3 achieved SVR, with no apparent effect of ribavirin. The combination was well tolerated. Pending confirmation in phase III trials, these results suggest that pangenotypic combination regimens will be available in the very near future (approval expected in 2017) and that genotype 3 will become as easy to cure as other genotypes, while less ribavirin will be used. Unfortunately, patients with decompensated cirrhosis will not benefit from these advances, as protease inhibitors such as ABT-493 cannot be used in this population. This pangenotypic regimen may also prove particularly useful in patients with severe or end-stage kidney disease who should not receive the nucleotide analogue sofosbuvir. High SVR rates appear to be achievable when retreating patients who failed a prior DAA-based treatment with this combination, but relapses may still occur with highly resistant viruses. This next generation of HCV drugs will be the last generation. With this armamentarium, it will be technically possible to cure the vast majority of HCV-infected patients. Thus, screening and diagnosing HCV-infected patients are now mandatory in order to provide them with efficient care and make the world almost free of hepatitis C by 2030.
Jean-Michel Pawlotsky, MD, PhD, director of the National Reference Center for Viral Hepatitis B, C, and D, and professor of medicine in the department of virology, Hôpital Henri Mondor, Université Paris-Est, Créteil, France. He has received research grants from Gilead and Abbvie and has served as an adviser for Abbvie, Bristol-Myers Squibb, Gilead, Janssen, and Merck.
In phase II and III clinical trials of direct-acting antivirals (DAAs), sustained viral response (SVR) rates over 90% were achieved in most patient groups and the combinations were well tolerated, results confirmed in real-world studies. However, a number of patients remain “difficult to cure.” Among them, patients infected with genotype 3, especially those with advanced liver disease, do not respond as well as patients infected with other genotypes and often need ribavirin.
In this study, a combination of two “next- generation” drugs with potent pangenotypic antiviral activity and a high barrier to resistance was administered to patients infected with HCV genotype 1 or 3 with compensated cirrhosis. Overall, 96% of patients infected with genotype 1 and 98% of patients infected with genotype 3 achieved SVR, with no apparent effect of ribavirin. The combination was well tolerated. Pending confirmation in phase III trials, these results suggest that pangenotypic combination regimens will be available in the very near future (approval expected in 2017) and that genotype 3 will become as easy to cure as other genotypes, while less ribavirin will be used. Unfortunately, patients with decompensated cirrhosis will not benefit from these advances, as protease inhibitors such as ABT-493 cannot be used in this population. This pangenotypic regimen may also prove particularly useful in patients with severe or end-stage kidney disease who should not receive the nucleotide analogue sofosbuvir. High SVR rates appear to be achievable when retreating patients who failed a prior DAA-based treatment with this combination, but relapses may still occur with highly resistant viruses. This next generation of HCV drugs will be the last generation. With this armamentarium, it will be technically possible to cure the vast majority of HCV-infected patients. Thus, screening and diagnosing HCV-infected patients are now mandatory in order to provide them with efficient care and make the world almost free of hepatitis C by 2030.
Jean-Michel Pawlotsky, MD, PhD, director of the National Reference Center for Viral Hepatitis B, C, and D, and professor of medicine in the department of virology, Hôpital Henri Mondor, Université Paris-Est, Créteil, France. He has received research grants from Gilead and Abbvie and has served as an adviser for Abbvie, Bristol-Myers Squibb, Gilead, Janssen, and Merck.
A once-daily regimen of two investigational, direct-acting anti-HCV agents, ABT-493 and ABT-530, was well tolerated and achieved sustained viral response at 12 weeks (SVR12) for nearly all patients with compensated cirrhosis and chronic genotype (GT) 1 or 3 hepatitis C virus infection, according to open-label phase II studies.
“The unique potency of these agents against all genotypes, even in the presence of common NS3 and/or NS5A baseline substitutions that confer resistance to most contemporary NS3/4A protease inhibitors and NS5A inhibitors, offers the potential for pangenotypic [HCV] therapy without ribavirin,” Edward J. Gane, MD, of the University of Auckland, New Zealand, and his associates wrote in the October issue of Gastroenterology. Phase III trials are now testing this hypothesis by focusing on cohorts of treatment-experienced, genotype 3–infected patients, on patients with renal impairment, and on patients who failed earlier-generation direct-acting antiviral regimens, they said.
The prevalence of HCV-related cirrhosis has yet to peak, and gold standard therapies for GT3 and GT1a infections can take weeks of treatment and the use of ribavirin, which causes undesirable side effects, the investigators noted. Attempts to surmount these residual barriers led to the development of ABT-493, an HCV nonstructural (NS) protein 3/4A protease inhibitor, and ABT-530, an HCV NS5A inhibitor. During in vitro studies, both agents showed “potent” activity against all major HCV genotypes, including variants with mutations that confer resistance to earlier, direct-acting antivirals, the researchers said (Gastroenterology. 2016 Jul 22. doi: 10.1053/j.gastro.2016.07.020). Their two open-label phase II studies enrolled adults with compensated cirrhosis and chronic GT3 (55 patients) or GT1 (27 patients) infection. Among GT1 patients, 41% had baseline NS3 substitutions conferring resistance to earlier-generation drugs, 19% had NS5A substitutions, and 11% had both mutations. The GT1-infected patients received 200 mg ABT-493 and 120 mg of ABT-530. The GT3-infected patients received 300 mg ABT-493 and 120 mg ABT-530, and half (27 patients) also received ribavirin. Most patients were treatment-naive, male, and white, with Child-Pugh scores of 5 and HCV RNA levels averaging about 6.2-6.6 log10 IU/mL.
In all, 26 patients with GT1 infection (96%) achieved SVR12 (95% confidence interval, 82% to 99%). The remaining patient relapsed after completing treatment. All treatment-naive GT3 patients achieved SVR12 whether or not they received ribavirin. However, one treatment-experienced GT3 patient who did not receive ribavirin relapsed after 16 weeks of treatment. Thus, rates of SVR12 were 96% (95% confidence interval, 82%-99%) for GT3 patients who did not receive ribavirin and 100% (95% CI, 88%-100%) for those who did. Notably, 94% of patients with baseline substitutions in NS3 and NS5A achieved SVR12, and there was no apparent link between treatment failure and any demographic or clinical characteristics, the investigators wrote.
Adverse events affected about 74% of patients and were usually mild or moderate in severity. Patients who did not receive ribavirin were most likely to report headache (15%), diarrhea (13%), and fatigue (11%). Only 4% of GT1 patients and 7% of the GT3 cohorts developed serious adverse events, and the only serious adverse event considered possibly treatment related involved a delusional disorder in a 57-year-old male who was receiving ribavirin and admitted amphetamine and alcohol use on the day it occurred. Treatment-related laboratory abnormalities were uncommon, no patients stopped treatment because of adverse events, and there were no deaths. “The rates of some adverse events were numerically higher with the higher ABT-493 dose, though the sample sizes are small and this was a cross-study comparison,” the investigators added. “Though not included in this study, patients with severe or end-stage kidney disease are predicted to be able to be treated with ABT-493 and ABT-530 because both agents have negligible renal excretion. These drugs were well tolerated in HCV-uninfected patients with renal impairment and can be administered without dose adjustment.”AbbVie funded the study and makes ABT-493 and ABT-530. Dr. Gane disclosed ties to AbbVie, Achillion Pharmaceuticals, Alnylam, Janssen, Merck, Novartis, and Novira.
A once-daily regimen of two investigational, direct-acting anti-HCV agents, ABT-493 and ABT-530, was well tolerated and achieved sustained viral response at 12 weeks (SVR12) for nearly all patients with compensated cirrhosis and chronic genotype (GT) 1 or 3 hepatitis C virus infection, according to open-label phase II studies.
“The unique potency of these agents against all genotypes, even in the presence of common NS3 and/or NS5A baseline substitutions that confer resistance to most contemporary NS3/4A protease inhibitors and NS5A inhibitors, offers the potential for pangenotypic [HCV] therapy without ribavirin,” Edward J. Gane, MD, of the University of Auckland, New Zealand, and his associates wrote in the October issue of Gastroenterology. Phase III trials are now testing this hypothesis by focusing on cohorts of treatment-experienced, genotype 3–infected patients, on patients with renal impairment, and on patients who failed earlier-generation direct-acting antiviral regimens, they said.
The prevalence of HCV-related cirrhosis has yet to peak, and gold standard therapies for GT3 and GT1a infections can take weeks of treatment and the use of ribavirin, which causes undesirable side effects, the investigators noted. Attempts to surmount these residual barriers led to the development of ABT-493, an HCV nonstructural (NS) protein 3/4A protease inhibitor, and ABT-530, an HCV NS5A inhibitor. During in vitro studies, both agents showed “potent” activity against all major HCV genotypes, including variants with mutations that confer resistance to earlier, direct-acting antivirals, the researchers said (Gastroenterology. 2016 Jul 22. doi: 10.1053/j.gastro.2016.07.020). Their two open-label phase II studies enrolled adults with compensated cirrhosis and chronic GT3 (55 patients) or GT1 (27 patients) infection. Among GT1 patients, 41% had baseline NS3 substitutions conferring resistance to earlier-generation drugs, 19% had NS5A substitutions, and 11% had both mutations. The GT1-infected patients received 200 mg ABT-493 and 120 mg of ABT-530. The GT3-infected patients received 300 mg ABT-493 and 120 mg ABT-530, and half (27 patients) also received ribavirin. Most patients were treatment-naive, male, and white, with Child-Pugh scores of 5 and HCV RNA levels averaging about 6.2-6.6 log10 IU/mL.
In all, 26 patients with GT1 infection (96%) achieved SVR12 (95% confidence interval, 82% to 99%). The remaining patient relapsed after completing treatment. All treatment-naive GT3 patients achieved SVR12 whether or not they received ribavirin. However, one treatment-experienced GT3 patient who did not receive ribavirin relapsed after 16 weeks of treatment. Thus, rates of SVR12 were 96% (95% confidence interval, 82%-99%) for GT3 patients who did not receive ribavirin and 100% (95% CI, 88%-100%) for those who did. Notably, 94% of patients with baseline substitutions in NS3 and NS5A achieved SVR12, and there was no apparent link between treatment failure and any demographic or clinical characteristics, the investigators wrote.
Adverse events affected about 74% of patients and were usually mild or moderate in severity. Patients who did not receive ribavirin were most likely to report headache (15%), diarrhea (13%), and fatigue (11%). Only 4% of GT1 patients and 7% of the GT3 cohorts developed serious adverse events, and the only serious adverse event considered possibly treatment related involved a delusional disorder in a 57-year-old male who was receiving ribavirin and admitted amphetamine and alcohol use on the day it occurred. Treatment-related laboratory abnormalities were uncommon, no patients stopped treatment because of adverse events, and there were no deaths. “The rates of some adverse events were numerically higher with the higher ABT-493 dose, though the sample sizes are small and this was a cross-study comparison,” the investigators added. “Though not included in this study, patients with severe or end-stage kidney disease are predicted to be able to be treated with ABT-493 and ABT-530 because both agents have negligible renal excretion. These drugs were well tolerated in HCV-uninfected patients with renal impairment and can be administered without dose adjustment.”AbbVie funded the study and makes ABT-493 and ABT-530. Dr. Gane disclosed ties to AbbVie, Achillion Pharmaceuticals, Alnylam, Janssen, Merck, Novartis, and Novira.
FROM GASTROENTEROLOGY
Key Clinical Point: The ABT-493/ABT-530 investigational direct-acting antiviral combination cured nearly all patients with compensated cirrhosis and genotype 1 or 3 hepatitis C virus infection.
Major finding: Rates of sustained viral response at 12 weeks (SVR12) were 96% for genotype 1–infected patients; 96% for genotype 3, ribavirin-free patients; and 100% for genotype 3 patients who received ribavirin.
Data source: Two open-label phase II trials of 27 GT1 patients and 55 GT3 patients in compensated cirrhosis.
Disclosures: AbbVie makes these agents and funded the study. Dr. Gane disclosed ties to AbbVie, Achillion Pharmaceuticals, Alnylam, Janssen, Merck, Novartis, and Novira.

How you can aid your patient’s claim for long-term disability
Neuropsychiatric disorders are associated with high rates of impaired work capacity despite the best efforts of treating clinicians to help their patients stay employed or resume working after symptoms improve.1
In the past, a note from the psychiatrist stating that the patient was unable to work because of a neuropsychiatric condition often was sufficient to approve a disability claim. This is no longer the case in today’s more restrictive climate, and what constitutes prima facie evidence of a patient’s inability to sustain competitive employment secondary to neuropsychiatric illness has significantly changed.
The following practices can help facilitate approval of your patient’s disability claim.
Document as you go. Progress notes should include the type, frequency, context, duration, and severity of symptoms supporting ≥1 psychiatric diagnoses which prevent your patient from holding a job. It also is important to document the parameters of treatment and the patient’s response, including compliance with treatment recommendations. Preferably, progress notes should include quantitative ratings over time that pertain to everyday functioning, highlighting how your patient is coping with the psychosocial, cognitive, and executive functioning demands of his (her) job.
When documented over time, ratings based on the Global Assessment of Functioning scale or a comparable scale are useful in quantifying the nature and degree of impaired functioning related to work capacity. Consider administering rating scales at periodic intervals to show changes over time. When feasible, scales should be based on a patient’s and informant’s report of symptomatic status and everyday functioning, and could include use of instruments such as the World Health Organization’s Disability Assessment Schedule.2,3
Include documentation specific to work capacity. Disability claims often are denied, in part, because the treating psychiatrist’s judgment regarding work capacity seems to “come out of the blue,” appears premature, or lacks discussion of the functional implications of the patient’s clinical status in regards to recent or current job expectations. Therefore, progress notes should include reference to long-standing, emerging, or worsening behaviors or symptoms that have clear implications for your patient’s ability to work.
Outline the functional implications of the patient’s preserved and impaired abilities and skills as they relate to work capacity, vocational history, and recent or current job situation. For example, work requirements that are highly dependent on interaction with the public, supervisors, or coworkers would be significantly affected by recurrent or persistent psychosis, even if the patient adheres to treatment and symptoms are relatively mild. Problems with working memory or anterograde memory could impair work that routinely involves learning and retention of new instructions and procedures.
Provide psychoeducation and support. Educate your patient and their family about the disability claims process, including the high rate that claims are initially denied. Consider retaining an advocate—clinical case manager, family member, or non-family third party—to assist your patient in navigating the disability application process, such as help completing paperwork, setting up appointments, and providing transportation.
Remain responsive to inquiries from disability examiners. Return forms and phone calls from disability examiners, psychiatrists, and other health care professionals reviewing your patient’s claim for long-term disability in a timely manner. Failure to do so can be used to support denial of the claim.
Consider referral for consultations and diagnostics to support the claim of impaired work capacity. Depending on the nature of the case, this could involve additional medical workup (including neuroimaging), a consultation from a vocational rehabilitation specialist, or referral for psychological or neuropsychological testing.
Psychometric assessment is becoming the preferred method for garnering support for impaired work capacity caused by neuropsychiatric factors. Findings from psychometric assessment hold up to scrutiny better if the evaluation includes symptom validity testing to rule out factitious disorder, malingering, or somatization, and results from self-report and informant-based measures of adaptive behavior and functioning.4
1. Gold LH, Shuman DW. Evaluating mental health disability in the workplace: models, process and analysis. New York, NY: Springer; 2009.
2. Traxler J. Mental health disability: a resident’s perspective of problems and solutions. Psychiatric Times. http://www.psychiatrictimes.com/residents-corner/mental-health-disability-residents-perspective-problems-and-solutions. Published November 26, 2014. Accessed August 31, 2016.
3. Zimmerman M. The importance of measuring outcomes in clinical practice. Psychiatric Times. http://www.psychiatrictimes.com/uspc2014/importance-measuring-outcomes-clinical-practice. Published October 1, 2014. Accessed August 31, 2016.
4. Schwarz L, Roskos PT, Grossberg GT. Answers to 7 questions about using neuropsychological testing in your practice. Current Psychiatry. 2014;13(3):34-39.
Neuropsychiatric disorders are associated with high rates of impaired work capacity despite the best efforts of treating clinicians to help their patients stay employed or resume working after symptoms improve.1
In the past, a note from the psychiatrist stating that the patient was unable to work because of a neuropsychiatric condition often was sufficient to approve a disability claim. This is no longer the case in today’s more restrictive climate, and what constitutes prima facie evidence of a patient’s inability to sustain competitive employment secondary to neuropsychiatric illness has significantly changed.
The following practices can help facilitate approval of your patient’s disability claim.
Document as you go. Progress notes should include the type, frequency, context, duration, and severity of symptoms supporting ≥1 psychiatric diagnoses which prevent your patient from holding a job. It also is important to document the parameters of treatment and the patient’s response, including compliance with treatment recommendations. Preferably, progress notes should include quantitative ratings over time that pertain to everyday functioning, highlighting how your patient is coping with the psychosocial, cognitive, and executive functioning demands of his (her) job.
When documented over time, ratings based on the Global Assessment of Functioning scale or a comparable scale are useful in quantifying the nature and degree of impaired functioning related to work capacity. Consider administering rating scales at periodic intervals to show changes over time. When feasible, scales should be based on a patient’s and informant’s report of symptomatic status and everyday functioning, and could include use of instruments such as the World Health Organization’s Disability Assessment Schedule.2,3
Include documentation specific to work capacity. Disability claims often are denied, in part, because the treating psychiatrist’s judgment regarding work capacity seems to “come out of the blue,” appears premature, or lacks discussion of the functional implications of the patient’s clinical status in regards to recent or current job expectations. Therefore, progress notes should include reference to long-standing, emerging, or worsening behaviors or symptoms that have clear implications for your patient’s ability to work.
Outline the functional implications of the patient’s preserved and impaired abilities and skills as they relate to work capacity, vocational history, and recent or current job situation. For example, work requirements that are highly dependent on interaction with the public, supervisors, or coworkers would be significantly affected by recurrent or persistent psychosis, even if the patient adheres to treatment and symptoms are relatively mild. Problems with working memory or anterograde memory could impair work that routinely involves learning and retention of new instructions and procedures.
Provide psychoeducation and support. Educate your patient and their family about the disability claims process, including the high rate that claims are initially denied. Consider retaining an advocate—clinical case manager, family member, or non-family third party—to assist your patient in navigating the disability application process, such as help completing paperwork, setting up appointments, and providing transportation.
Remain responsive to inquiries from disability examiners. Return forms and phone calls from disability examiners, psychiatrists, and other health care professionals reviewing your patient’s claim for long-term disability in a timely manner. Failure to do so can be used to support denial of the claim.
Consider referral for consultations and diagnostics to support the claim of impaired work capacity. Depending on the nature of the case, this could involve additional medical workup (including neuroimaging), a consultation from a vocational rehabilitation specialist, or referral for psychological or neuropsychological testing.
Psychometric assessment is becoming the preferred method for garnering support for impaired work capacity caused by neuropsychiatric factors. Findings from psychometric assessment hold up to scrutiny better if the evaluation includes symptom validity testing to rule out factitious disorder, malingering, or somatization, and results from self-report and informant-based measures of adaptive behavior and functioning.4
Neuropsychiatric disorders are associated with high rates of impaired work capacity despite the best efforts of treating clinicians to help their patients stay employed or resume working after symptoms improve.1
In the past, a note from the psychiatrist stating that the patient was unable to work because of a neuropsychiatric condition often was sufficient to approve a disability claim. This is no longer the case in today’s more restrictive climate, and what constitutes prima facie evidence of a patient’s inability to sustain competitive employment secondary to neuropsychiatric illness has significantly changed.
The following practices can help facilitate approval of your patient’s disability claim.
Document as you go. Progress notes should include the type, frequency, context, duration, and severity of symptoms supporting ≥1 psychiatric diagnoses which prevent your patient from holding a job. It also is important to document the parameters of treatment and the patient’s response, including compliance with treatment recommendations. Preferably, progress notes should include quantitative ratings over time that pertain to everyday functioning, highlighting how your patient is coping with the psychosocial, cognitive, and executive functioning demands of his (her) job.
When documented over time, ratings based on the Global Assessment of Functioning scale or a comparable scale are useful in quantifying the nature and degree of impaired functioning related to work capacity. Consider administering rating scales at periodic intervals to show changes over time. When feasible, scales should be based on a patient’s and informant’s report of symptomatic status and everyday functioning, and could include use of instruments such as the World Health Organization’s Disability Assessment Schedule.2,3
Include documentation specific to work capacity. Disability claims often are denied, in part, because the treating psychiatrist’s judgment regarding work capacity seems to “come out of the blue,” appears premature, or lacks discussion of the functional implications of the patient’s clinical status in regards to recent or current job expectations. Therefore, progress notes should include reference to long-standing, emerging, or worsening behaviors or symptoms that have clear implications for your patient’s ability to work.
Outline the functional implications of the patient’s preserved and impaired abilities and skills as they relate to work capacity, vocational history, and recent or current job situation. For example, work requirements that are highly dependent on interaction with the public, supervisors, or coworkers would be significantly affected by recurrent or persistent psychosis, even if the patient adheres to treatment and symptoms are relatively mild. Problems with working memory or anterograde memory could impair work that routinely involves learning and retention of new instructions and procedures.
Provide psychoeducation and support. Educate your patient and their family about the disability claims process, including the high rate that claims are initially denied. Consider retaining an advocate—clinical case manager, family member, or non-family third party—to assist your patient in navigating the disability application process, such as help completing paperwork, setting up appointments, and providing transportation.
Remain responsive to inquiries from disability examiners. Return forms and phone calls from disability examiners, psychiatrists, and other health care professionals reviewing your patient’s claim for long-term disability in a timely manner. Failure to do so can be used to support denial of the claim.
Consider referral for consultations and diagnostics to support the claim of impaired work capacity. Depending on the nature of the case, this could involve additional medical workup (including neuroimaging), a consultation from a vocational rehabilitation specialist, or referral for psychological or neuropsychological testing.
Psychometric assessment is becoming the preferred method for garnering support for impaired work capacity caused by neuropsychiatric factors. Findings from psychometric assessment hold up to scrutiny better if the evaluation includes symptom validity testing to rule out factitious disorder, malingering, or somatization, and results from self-report and informant-based measures of adaptive behavior and functioning.4
1. Gold LH, Shuman DW. Evaluating mental health disability in the workplace: models, process and analysis. New York, NY: Springer; 2009.
2. Traxler J. Mental health disability: a resident’s perspective of problems and solutions. Psychiatric Times. http://www.psychiatrictimes.com/residents-corner/mental-health-disability-residents-perspective-problems-and-solutions. Published November 26, 2014. Accessed August 31, 2016.
3. Zimmerman M. The importance of measuring outcomes in clinical practice. Psychiatric Times. http://www.psychiatrictimes.com/uspc2014/importance-measuring-outcomes-clinical-practice. Published October 1, 2014. Accessed August 31, 2016.
4. Schwarz L, Roskos PT, Grossberg GT. Answers to 7 questions about using neuropsychological testing in your practice. Current Psychiatry. 2014;13(3):34-39.
1. Gold LH, Shuman DW. Evaluating mental health disability in the workplace: models, process and analysis. New York, NY: Springer; 2009.
2. Traxler J. Mental health disability: a resident’s perspective of problems and solutions. Psychiatric Times. http://www.psychiatrictimes.com/residents-corner/mental-health-disability-residents-perspective-problems-and-solutions. Published November 26, 2014. Accessed August 31, 2016.
3. Zimmerman M. The importance of measuring outcomes in clinical practice. Psychiatric Times. http://www.psychiatrictimes.com/uspc2014/importance-measuring-outcomes-clinical-practice. Published October 1, 2014. Accessed August 31, 2016.
4. Schwarz L, Roskos PT, Grossberg GT. Answers to 7 questions about using neuropsychological testing in your practice. Current Psychiatry. 2014;13(3):34-39.
Stabilized schizoaffective disorder; later confusion and depression appears
CASE
Disoriented and confused
Mr. D, age 42, presents to our emergency department (ED) accompanied by his family with recent onset of disorientation, confusion, depressive mood with labile affect, sleep disturbances, purposeless movements, and grossly reduced kinetics/verbal output. He has a history of schizoaffective disorder, bipolar type, and recurrent admissions for psychotic mood instability.
A few months earlier, Mr. D was treated at our facility for acute exacerbation of his schizoaffective disorder. He was stabilized and discharged with aripiprazole, 30 mg/d, and mirtazapine, 15 mg/d—he had been taking both medications for some time—and newly started extended-release divalproex, 500 mg in the morning/1000 mg nightly (13.2 mg/kg). His trough valproic acid serum level was 70 µg/mL at discharge. He continued on this medication regimen until he returns to our ED with his family.
Mr. D has several medical problems, such as type 2 diabetes mellitus and hypertension, for which he has been receiving metformin, 1,000 mg/d, lisinopril, 10 mg/d, and simvastatin, 20 mg/d. He has no history of alcohol or substance abuse and does not smoke.
Serum and urine analyses are unremarkable and include finger-stick blood glucose, complete blood count, urinalysis, urine drug screen, comprehensive metabolic panel, magnesium, γ-glutamyl transpeptidase (GGTP), amylase, thyroid-stimulating hormone, and blood alcohol level. Random valproic acid serum level taken in the ED is 64 µg/mL. Non-contrast head CT is interpreted as non-acute. There are no documented abnormal findings during the physical exam.
What could be causing Mr. D’s altered mental status?
a) symptoms of a medical illness
b) medication, undetected substance intoxication, or withdrawal-related symptoms
c) acute exacerbation of schizoaffective disorder
d) delirium
e) catatonia of undetected and/or multiple causes
The authors’ observations
The differential diagnosis was broad at the time of Mr. D’s presentation to the ED because his symptoms overlapped across clinical considerations. The initial medical evaluation was negative, which suggested an active primary mental illness. However, Mr. D’s presenting symptoms warranted continued vigilance for concurrent or emergent delirium or catatonia, especially because of the potential morbidity if these conditions are not detected and managed.
EVALUATION
Fluctuating status
Mr. D is admitted to the mental health unit for treatment of presumptive bipolar depression with catatonic features. The initial admitting team continues aripiprazole, increased divalproex extended release to 1,000 mg in the morning/1,500 mg at night, held mirtazapine, and started lorazepam, 2 mg, 3 times daily, for catatonia. Metformin, lisinopril, and simvastatin are continued. Mr. D’s mental status and behavior fluctuates over the next 48 hours prompting the treatment team to consider an emergent delirious process.
On day 3, the primary team assumes care and observes fluctuations in level of arousal with disorientation, inattention, labile affect, disorganized speech and behavior, and responsiveness to internal (visual) stimuli. Finger-stick blood glucose level remains stable. Review of physical symptoms is notable for nausea and examination reveals unsteady gait and asterixis. His family denies that Mr. D used alcohol or drugs before admission. Collateral information from the family and review of Mr. D’s outpatient records is consistent with an acutely fluctuating confusional state that began 10 days before admission.
At this point, what is your differential diagnosis for Mr. D’s altered mental status?
a) symptoms of a medical illness
b) medication, undetected substance intoxication, or withdrawal-related symptoms
c) acute exacerbation of schizoaffective disorder
d) delirium
e) catatonia of undetected or multiple causes
TREATMENT
Valproate stopped
Mr. D’s ammonia level is 119 µg/dL (reference range, 15 to 45 μg/dL) on hospital day 3. Divalproex and lorazepam are discontinued, and standing lactulose is started because it is evident that he has active valproate-related hyperammonemic encephalopathy (VHE), also known as delirium due to valproate-related hyperammonemia.
Awake and drowsy EEG within 24 hours reveals “diffuse irregular slow activity” without epileptogenic features. HIV, syphilis, and vitamin B12 and red blood cell folate screening are negative. We confirm that Mr. D is not a vegetarian (dietary carnitine deficiency is a risk factor for VHE). He is not screened for a urea cycle disorder.
The authors’ observations
Divalproex is a commonly used FDA-approved treatment for a variety of neurologic and psychiatric conditions including acute bipolar mania.1-3 It also is used for off-label control of various psychiatric symptoms. It is a stable coordination compound composed of sodium valproate and valproic acid that dissipates into the valproate ion in the gastrointestinal tract.1 (In this article, references to valproate [VPA] include valproic acid and divalproex.) The drug is relatively well-tolerated; however, use may carry teratogenic risk and can adversely impact a variety of body systems, especially hematopoietic, gastrointestinal, and neurologic systems.1-3 Adverse effects can be idiosyncratic or in part related to VPA serum levels.1,4 VPA toxicity increases the likelihood of some adverse health outcomes, such as nausea, diarrhea, and tremors.1
Identifying and treating VHE
Asymptomatic elevations in ammonia without evidence of hepatic injury are common, might be related to valproic serum levels, and may occur in up to one-half of psychiatric patients receiving VPA.2-4 In contrast, VHE is a rare and potentially lethal idiosyncratic event unrelated to duration of VPA treatment, dosage, or valproic serum level.2-4 In addition, prior safe use might not protect against future VHE.3,4
VHE presents as delirium with characteristic acute changes in mental status, including alterations in cognition or level of consciousness ranging from lethargy to coma, along with possible focal neurological findings or vomiting.1,3,4 Although more common among patients with a seizure disorder, VHE also might be associated with new seizure activity in patients who do not have a seizure disorder.5
Although symptomatically acute in onset, emergence is unpredictable and can occur within days or up to years of use with therapeutic VPA dosing and valproic serum levels.2,4 Complicating identification, laboratory transaminase or ammonia elevations may or may not be present2-4; however, VHE typically occurs in the setting of hyperammonemia and normal transaminase levels.2 Reversible EEG findings are nonspecific2 and could show generalized slowing with occasional bursts of frontal intermittent rhythmic delta activity and triphasic waves.2,4
Pathophysiological descriptions of emergent VHE have been hypothesized,2-4 but the definitive causal mechanism remains unclear.6 Published VHE risk factors2-6 include:
- polypharmacy (especially anti-convulsants)
- inherited or dietary-based carnitine deficiency
- urea cycle disorders
- mental retardation.

How would you treat VHE?
a) cholinesterase inhibitors
b) antipsychotic therapy
c) supportive care
d) ammonia-reducing agents such as lactulose, carnitine, and neomycin
e) discontinue valproate
Outcome Normalized ammonia
Four days after discontinuing divalproex and starting lactulose, Mr. D’s fluctuating level of arousal, orientation, attention, and perceptual disturbances resolve along with restoration of environmental relatedness in setting of normalized ammonia level to 39 µg/dL. He is euthymic, non-psychotic, and without cognitive impairment at time of discharge. An “allergy” to divalproex is entered in his electronic medical record in an effort to discourage future retrial.
The authors’ observations
Once identified, management of VHE invariably includes consideration for discontinuation of valproate1,2,4,19; other adjunctive, expediting, ammonia-reducing strategies, including lactulose and carnitine, have also been described.2,4,5,20 Although lactulose is more commonly used, carnitine supplementation might be associated with a preferable dosing schedule and drug interaction and side-effect profile.20 Rapidly deteriorating clinical status could indicate hemodialysis.4
Of critical importance, these management strategies rely on awareness of and prompt identification of the condition, which includes an ability to distinguish emergent VHE from the mental illness VPA is used to treat.
Stopping the offending agent generally results in complete recovery in VHE patients with psychiatric illness.4 Most (>90%, n = 31) psychiatric patients in our and prior5 case series reviews recovered within 2 weeks of intervention.5 Cautious resumption of divalproex could be considered if there is a compelling clinical indication and you suspect that a putative polypharmacy agent such as topiramate has been removed; otherwise future retrial of VPA should be avoided.14
Mr. D’s case was consistent with a valproate-related hyperammonemic delirious event. He had preadmission acute onset, intra-daily fluctuating confusion, and visual perceptual disturbances with nausea, asterixis, gait disturbance, elevated ammonia, and a supportive EEG months after starting divalproex. Similar to our case, some challenging aspects of identifying emergent VHE include:
- earlier safe use of divalproex over extended periods
- lack of elevated VPA serum level
- lack of transaminase elevation
- lack of apparent risk factors
- presence of background serious mental illness, which can distract from VHE detection via misattribution to uncontrolled primary mental illness.
This last point is critical because it can delay VHE identification and treatment or worse, result in misdiagnosis with accompanying continuation or escalation of VPA dosing as has initially occurred in Mr. D’s case. Similar concerns have been raised2,5 and occurred,5,19 which is not surprising given the frequency of VPA use for psychiatric conditions and symptoms.
Providers should have a low threshold for checking an ammonia level in clinical scenarios that involve any alteration in mental status that may resemble delirium in psychiatric patients treated with valproate. From a preventative perspective, it may be prudent to avoid valproate in psychiatric patients with known VHE risk factors. Either way, promotion of VHE awareness and detection across medical disciplines is paramount.
1. Depakote [package insert]. Chicago, IL: AbbVie; 2016.
2. Lewis C, Deshpande A, Tesar G, et al. Valproate-induced hyperammonemic encephalopathy: a brief review. Curr Med Res Opin. 2012;28(6):1039-1042.
3. Nanau RM, Neuman MG. Adverse drug reactions induced by valproic acid. Clin Biochem. 2013;46(15):1323-1338.
4. Chopra A, Kolla BP, Mansukhani MP, et al. Valproate-induced hyperammonemic encephalopathy: an update on risk factors, clinical correlates and management. Gen Hosp Psychiatry. 2012;34(3):290-298.
5. Carr RB, Shrewsbury K. Hyperammonemia due to valproic acid in the psychiatric setting. Am J Psychiatry. 2007;164(7):1020-1027.
6. Hung C, Li T, Wei I, et al. The real mechanism of VPA-induced hyperammonemia remains unknown. Gen Hosp Psychiatry. 2011;33(1):84.e3-84.e4.
7. Starer J, Chang G. Hyperammonemic encephalopathy, valproic acid, and benzodiazepine withdrawal: a case series. Am J Drug Alcohol Abuse. 2010;36(2):98-101.
8. Eubanks AL, Aguirre B, Bourgeois JA. Severe acute hyperammonemia after brief exposure to valproate. Psychosomatics. 2008;49(1):82-83.
9. Fan CC, Huang MC, Liu HC. Lamotrigine might potentiate valproic acid-induced hyperammonemic encephalopathy. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(7):1747-1748.
10. Deutsch SI, Burket JA, Rosse RB. Valproate-induced hyperammonemic encephalopathy and normal liver functions: possible synergism with topiramate. Clin Neuropharmacol. 2009;32(6):350-352.
11. Rodrigues-Silva N, Venâncio Ä, Bouça J. Risperidone, a risk for valproate induced encephalopathy? Gen Hosp Psychiatry. 2013;35(4):452.e5-452.e6.
12. Sunkavalli KK, Iqbal FM, Singh B, et al. Valproate-induced hyperammonemic encephalopathy: a case report and brief review of the literature. Am J Ther. 2013;20(5):569-571.
13. Abreu LN, Issler C, Lafer B. Valproate-induced reversible pseudoatrophy of the brain and hyperammonemic encephalopathy in a bipolar patient. Aust N Z J Psychiatry. 2009;43(5):484-485.
14. Hong L, Schutz J, Nance M. A case of valproate-induced encephalopathy. Aust N Z J Psychiatry. 2012;46(12):1200-1201.
15. Kimmel RJ, Irwin SA, Meyer JM. Valproic acid-associated hyperammonemic encephalopathy: a case report from the psychiatric setting. Int Clin Psychopharmacol. 2005;20(1):57-58.
16. Elgudin L, Hall Y, Schubert D. Ammonia induced encephalopathy from valproic acid in a bipolar patient: case report. Int J Psychiatry Med. 2003;33(1):91-96.
17. Stewart JT. A case of hyperammonemic encephalopathy after 11 years of valproate therapy. J Clin Psychopharmacol. 2008;28(3):361-362.
18. Wadzinski J, Franks R, Roane D, et al. Valproate-associated hyperammonemic encephalopathy. J Am Board Fam Med. 2007;20(5):499-502.
19. Chang M, Tang X, Wen S, et al. Valproate (VPA)-associated hyperammonemic encephalopathy independent of elevated serum VPA levels: 21 cases in China from May 2000 to May 2012. Compr Psychiatry. 2013;54(5):562-567.
20. Sonik P, Hilty DM, Rossaro L, et al. Carnitine supplementation for valproate-related hyperammonemia to maintain therapeutic valproate level. J Clin Psychopharmacol. 2011;31(5):680-682.
CASE
Disoriented and confused
Mr. D, age 42, presents to our emergency department (ED) accompanied by his family with recent onset of disorientation, confusion, depressive mood with labile affect, sleep disturbances, purposeless movements, and grossly reduced kinetics/verbal output. He has a history of schizoaffective disorder, bipolar type, and recurrent admissions for psychotic mood instability.
A few months earlier, Mr. D was treated at our facility for acute exacerbation of his schizoaffective disorder. He was stabilized and discharged with aripiprazole, 30 mg/d, and mirtazapine, 15 mg/d—he had been taking both medications for some time—and newly started extended-release divalproex, 500 mg in the morning/1000 mg nightly (13.2 mg/kg). His trough valproic acid serum level was 70 µg/mL at discharge. He continued on this medication regimen until he returns to our ED with his family.
Mr. D has several medical problems, such as type 2 diabetes mellitus and hypertension, for which he has been receiving metformin, 1,000 mg/d, lisinopril, 10 mg/d, and simvastatin, 20 mg/d. He has no history of alcohol or substance abuse and does not smoke.
Serum and urine analyses are unremarkable and include finger-stick blood glucose, complete blood count, urinalysis, urine drug screen, comprehensive metabolic panel, magnesium, γ-glutamyl transpeptidase (GGTP), amylase, thyroid-stimulating hormone, and blood alcohol level. Random valproic acid serum level taken in the ED is 64 µg/mL. Non-contrast head CT is interpreted as non-acute. There are no documented abnormal findings during the physical exam.
What could be causing Mr. D’s altered mental status?
a) symptoms of a medical illness
b) medication, undetected substance intoxication, or withdrawal-related symptoms
c) acute exacerbation of schizoaffective disorder
d) delirium
e) catatonia of undetected and/or multiple causes
The authors’ observations
The differential diagnosis was broad at the time of Mr. D’s presentation to the ED because his symptoms overlapped across clinical considerations. The initial medical evaluation was negative, which suggested an active primary mental illness. However, Mr. D’s presenting symptoms warranted continued vigilance for concurrent or emergent delirium or catatonia, especially because of the potential morbidity if these conditions are not detected and managed.
EVALUATION
Fluctuating status
Mr. D is admitted to the mental health unit for treatment of presumptive bipolar depression with catatonic features. The initial admitting team continues aripiprazole, increased divalproex extended release to 1,000 mg in the morning/1,500 mg at night, held mirtazapine, and started lorazepam, 2 mg, 3 times daily, for catatonia. Metformin, lisinopril, and simvastatin are continued. Mr. D’s mental status and behavior fluctuates over the next 48 hours prompting the treatment team to consider an emergent delirious process.
On day 3, the primary team assumes care and observes fluctuations in level of arousal with disorientation, inattention, labile affect, disorganized speech and behavior, and responsiveness to internal (visual) stimuli. Finger-stick blood glucose level remains stable. Review of physical symptoms is notable for nausea and examination reveals unsteady gait and asterixis. His family denies that Mr. D used alcohol or drugs before admission. Collateral information from the family and review of Mr. D’s outpatient records is consistent with an acutely fluctuating confusional state that began 10 days before admission.
At this point, what is your differential diagnosis for Mr. D’s altered mental status?
a) symptoms of a medical illness
b) medication, undetected substance intoxication, or withdrawal-related symptoms
c) acute exacerbation of schizoaffective disorder
d) delirium
e) catatonia of undetected or multiple causes
TREATMENT
Valproate stopped
Mr. D’s ammonia level is 119 µg/dL (reference range, 15 to 45 μg/dL) on hospital day 3. Divalproex and lorazepam are discontinued, and standing lactulose is started because it is evident that he has active valproate-related hyperammonemic encephalopathy (VHE), also known as delirium due to valproate-related hyperammonemia.
Awake and drowsy EEG within 24 hours reveals “diffuse irregular slow activity” without epileptogenic features. HIV, syphilis, and vitamin B12 and red blood cell folate screening are negative. We confirm that Mr. D is not a vegetarian (dietary carnitine deficiency is a risk factor for VHE). He is not screened for a urea cycle disorder.
The authors’ observations
Divalproex is a commonly used FDA-approved treatment for a variety of neurologic and psychiatric conditions including acute bipolar mania.1-3 It also is used for off-label control of various psychiatric symptoms. It is a stable coordination compound composed of sodium valproate and valproic acid that dissipates into the valproate ion in the gastrointestinal tract.1 (In this article, references to valproate [VPA] include valproic acid and divalproex.) The drug is relatively well-tolerated; however, use may carry teratogenic risk and can adversely impact a variety of body systems, especially hematopoietic, gastrointestinal, and neurologic systems.1-3 Adverse effects can be idiosyncratic or in part related to VPA serum levels.1,4 VPA toxicity increases the likelihood of some adverse health outcomes, such as nausea, diarrhea, and tremors.1
Identifying and treating VHE
Asymptomatic elevations in ammonia without evidence of hepatic injury are common, might be related to valproic serum levels, and may occur in up to one-half of psychiatric patients receiving VPA.2-4 In contrast, VHE is a rare and potentially lethal idiosyncratic event unrelated to duration of VPA treatment, dosage, or valproic serum level.2-4 In addition, prior safe use might not protect against future VHE.3,4
VHE presents as delirium with characteristic acute changes in mental status, including alterations in cognition or level of consciousness ranging from lethargy to coma, along with possible focal neurological findings or vomiting.1,3,4 Although more common among patients with a seizure disorder, VHE also might be associated with new seizure activity in patients who do not have a seizure disorder.5
Although symptomatically acute in onset, emergence is unpredictable and can occur within days or up to years of use with therapeutic VPA dosing and valproic serum levels.2,4 Complicating identification, laboratory transaminase or ammonia elevations may or may not be present2-4; however, VHE typically occurs in the setting of hyperammonemia and normal transaminase levels.2 Reversible EEG findings are nonspecific2 and could show generalized slowing with occasional bursts of frontal intermittent rhythmic delta activity and triphasic waves.2,4
Pathophysiological descriptions of emergent VHE have been hypothesized,2-4 but the definitive causal mechanism remains unclear.6 Published VHE risk factors2-6 include:
- polypharmacy (especially anti-convulsants)
- inherited or dietary-based carnitine deficiency
- urea cycle disorders
- mental retardation.
How would you treat VHE?
a) cholinesterase inhibitors
b) antipsychotic therapy
c) supportive care
d) ammonia-reducing agents such as lactulose, carnitine, and neomycin
e) discontinue valproate
Outcome Normalized ammonia
Four days after discontinuing divalproex and starting lactulose, Mr. D’s fluctuating level of arousal, orientation, attention, and perceptual disturbances resolve along with restoration of environmental relatedness in setting of normalized ammonia level to 39 µg/dL. He is euthymic, non-psychotic, and without cognitive impairment at time of discharge. An “allergy” to divalproex is entered in his electronic medical record in an effort to discourage future retrial.
The authors’ observations
Once identified, management of VHE invariably includes consideration for discontinuation of valproate1,2,4,19; other adjunctive, expediting, ammonia-reducing strategies, including lactulose and carnitine, have also been described.2,4,5,20 Although lactulose is more commonly used, carnitine supplementation might be associated with a preferable dosing schedule and drug interaction and side-effect profile.20 Rapidly deteriorating clinical status could indicate hemodialysis.4
Of critical importance, these management strategies rely on awareness of and prompt identification of the condition, which includes an ability to distinguish emergent VHE from the mental illness VPA is used to treat.
Stopping the offending agent generally results in complete recovery in VHE patients with psychiatric illness.4 Most (>90%, n = 31) psychiatric patients in our and prior5 case series reviews recovered within 2 weeks of intervention.5 Cautious resumption of divalproex could be considered if there is a compelling clinical indication and you suspect that a putative polypharmacy agent such as topiramate has been removed; otherwise future retrial of VPA should be avoided.14
Mr. D’s case was consistent with a valproate-related hyperammonemic delirious event. He had preadmission acute onset, intra-daily fluctuating confusion, and visual perceptual disturbances with nausea, asterixis, gait disturbance, elevated ammonia, and a supportive EEG months after starting divalproex. Similar to our case, some challenging aspects of identifying emergent VHE include:
- earlier safe use of divalproex over extended periods
- lack of elevated VPA serum level
- lack of transaminase elevation
- lack of apparent risk factors
- presence of background serious mental illness, which can distract from VHE detection via misattribution to uncontrolled primary mental illness.
This last point is critical because it can delay VHE identification and treatment or worse, result in misdiagnosis with accompanying continuation or escalation of VPA dosing as has initially occurred in Mr. D’s case. Similar concerns have been raised2,5 and occurred,5,19 which is not surprising given the frequency of VPA use for psychiatric conditions and symptoms.
Providers should have a low threshold for checking an ammonia level in clinical scenarios that involve any alteration in mental status that may resemble delirium in psychiatric patients treated with valproate. From a preventative perspective, it may be prudent to avoid valproate in psychiatric patients with known VHE risk factors. Either way, promotion of VHE awareness and detection across medical disciplines is paramount.
CASE
Disoriented and confused
Mr. D, age 42, presents to our emergency department (ED) accompanied by his family with recent onset of disorientation, confusion, depressive mood with labile affect, sleep disturbances, purposeless movements, and grossly reduced kinetics/verbal output. He has a history of schizoaffective disorder, bipolar type, and recurrent admissions for psychotic mood instability.
A few months earlier, Mr. D was treated at our facility for acute exacerbation of his schizoaffective disorder. He was stabilized and discharged with aripiprazole, 30 mg/d, and mirtazapine, 15 mg/d—he had been taking both medications for some time—and newly started extended-release divalproex, 500 mg in the morning/1000 mg nightly (13.2 mg/kg). His trough valproic acid serum level was 70 µg/mL at discharge. He continued on this medication regimen until he returns to our ED with his family.
Mr. D has several medical problems, such as type 2 diabetes mellitus and hypertension, for which he has been receiving metformin, 1,000 mg/d, lisinopril, 10 mg/d, and simvastatin, 20 mg/d. He has no history of alcohol or substance abuse and does not smoke.
Serum and urine analyses are unremarkable and include finger-stick blood glucose, complete blood count, urinalysis, urine drug screen, comprehensive metabolic panel, magnesium, γ-glutamyl transpeptidase (GGTP), amylase, thyroid-stimulating hormone, and blood alcohol level. Random valproic acid serum level taken in the ED is 64 µg/mL. Non-contrast head CT is interpreted as non-acute. There are no documented abnormal findings during the physical exam.
What could be causing Mr. D’s altered mental status?
a) symptoms of a medical illness
b) medication, undetected substance intoxication, or withdrawal-related symptoms
c) acute exacerbation of schizoaffective disorder
d) delirium
e) catatonia of undetected and/or multiple causes
The authors’ observations
The differential diagnosis was broad at the time of Mr. D’s presentation to the ED because his symptoms overlapped across clinical considerations. The initial medical evaluation was negative, which suggested an active primary mental illness. However, Mr. D’s presenting symptoms warranted continued vigilance for concurrent or emergent delirium or catatonia, especially because of the potential morbidity if these conditions are not detected and managed.
EVALUATION
Fluctuating status
Mr. D is admitted to the mental health unit for treatment of presumptive bipolar depression with catatonic features. The initial admitting team continues aripiprazole, increased divalproex extended release to 1,000 mg in the morning/1,500 mg at night, held mirtazapine, and started lorazepam, 2 mg, 3 times daily, for catatonia. Metformin, lisinopril, and simvastatin are continued. Mr. D’s mental status and behavior fluctuates over the next 48 hours prompting the treatment team to consider an emergent delirious process.
On day 3, the primary team assumes care and observes fluctuations in level of arousal with disorientation, inattention, labile affect, disorganized speech and behavior, and responsiveness to internal (visual) stimuli. Finger-stick blood glucose level remains stable. Review of physical symptoms is notable for nausea and examination reveals unsteady gait and asterixis. His family denies that Mr. D used alcohol or drugs before admission. Collateral information from the family and review of Mr. D’s outpatient records is consistent with an acutely fluctuating confusional state that began 10 days before admission.
At this point, what is your differential diagnosis for Mr. D’s altered mental status?
a) symptoms of a medical illness
b) medication, undetected substance intoxication, or withdrawal-related symptoms
c) acute exacerbation of schizoaffective disorder
d) delirium
e) catatonia of undetected or multiple causes
TREATMENT
Valproate stopped
Mr. D’s ammonia level is 119 µg/dL (reference range, 15 to 45 μg/dL) on hospital day 3. Divalproex and lorazepam are discontinued, and standing lactulose is started because it is evident that he has active valproate-related hyperammonemic encephalopathy (VHE), also known as delirium due to valproate-related hyperammonemia.
Awake and drowsy EEG within 24 hours reveals “diffuse irregular slow activity” without epileptogenic features. HIV, syphilis, and vitamin B12 and red blood cell folate screening are negative. We confirm that Mr. D is not a vegetarian (dietary carnitine deficiency is a risk factor for VHE). He is not screened for a urea cycle disorder.
The authors’ observations
Divalproex is a commonly used FDA-approved treatment for a variety of neurologic and psychiatric conditions including acute bipolar mania.1-3 It also is used for off-label control of various psychiatric symptoms. It is a stable coordination compound composed of sodium valproate and valproic acid that dissipates into the valproate ion in the gastrointestinal tract.1 (In this article, references to valproate [VPA] include valproic acid and divalproex.) The drug is relatively well-tolerated; however, use may carry teratogenic risk and can adversely impact a variety of body systems, especially hematopoietic, gastrointestinal, and neurologic systems.1-3 Adverse effects can be idiosyncratic or in part related to VPA serum levels.1,4 VPA toxicity increases the likelihood of some adverse health outcomes, such as nausea, diarrhea, and tremors.1
Identifying and treating VHE
Asymptomatic elevations in ammonia without evidence of hepatic injury are common, might be related to valproic serum levels, and may occur in up to one-half of psychiatric patients receiving VPA.2-4 In contrast, VHE is a rare and potentially lethal idiosyncratic event unrelated to duration of VPA treatment, dosage, or valproic serum level.2-4 In addition, prior safe use might not protect against future VHE.3,4
VHE presents as delirium with characteristic acute changes in mental status, including alterations in cognition or level of consciousness ranging from lethargy to coma, along with possible focal neurological findings or vomiting.1,3,4 Although more common among patients with a seizure disorder, VHE also might be associated with new seizure activity in patients who do not have a seizure disorder.5
Although symptomatically acute in onset, emergence is unpredictable and can occur within days or up to years of use with therapeutic VPA dosing and valproic serum levels.2,4 Complicating identification, laboratory transaminase or ammonia elevations may or may not be present2-4; however, VHE typically occurs in the setting of hyperammonemia and normal transaminase levels.2 Reversible EEG findings are nonspecific2 and could show generalized slowing with occasional bursts of frontal intermittent rhythmic delta activity and triphasic waves.2,4
Pathophysiological descriptions of emergent VHE have been hypothesized,2-4 but the definitive causal mechanism remains unclear.6 Published VHE risk factors2-6 include:
- polypharmacy (especially anti-convulsants)
- inherited or dietary-based carnitine deficiency
- urea cycle disorders
- mental retardation.
How would you treat VHE?
a) cholinesterase inhibitors
b) antipsychotic therapy
c) supportive care
d) ammonia-reducing agents such as lactulose, carnitine, and neomycin
e) discontinue valproate
Outcome Normalized ammonia
Four days after discontinuing divalproex and starting lactulose, Mr. D’s fluctuating level of arousal, orientation, attention, and perceptual disturbances resolve along with restoration of environmental relatedness in setting of normalized ammonia level to 39 µg/dL. He is euthymic, non-psychotic, and without cognitive impairment at time of discharge. An “allergy” to divalproex is entered in his electronic medical record in an effort to discourage future retrial.
The authors’ observations
Once identified, management of VHE invariably includes consideration for discontinuation of valproate1,2,4,19; other adjunctive, expediting, ammonia-reducing strategies, including lactulose and carnitine, have also been described.2,4,5,20 Although lactulose is more commonly used, carnitine supplementation might be associated with a preferable dosing schedule and drug interaction and side-effect profile.20 Rapidly deteriorating clinical status could indicate hemodialysis.4
Of critical importance, these management strategies rely on awareness of and prompt identification of the condition, which includes an ability to distinguish emergent VHE from the mental illness VPA is used to treat.
Stopping the offending agent generally results in complete recovery in VHE patients with psychiatric illness.4 Most (>90%, n = 31) psychiatric patients in our and prior5 case series reviews recovered within 2 weeks of intervention.5 Cautious resumption of divalproex could be considered if there is a compelling clinical indication and you suspect that a putative polypharmacy agent such as topiramate has been removed; otherwise future retrial of VPA should be avoided.14
Mr. D’s case was consistent with a valproate-related hyperammonemic delirious event. He had preadmission acute onset, intra-daily fluctuating confusion, and visual perceptual disturbances with nausea, asterixis, gait disturbance, elevated ammonia, and a supportive EEG months after starting divalproex. Similar to our case, some challenging aspects of identifying emergent VHE include:
- earlier safe use of divalproex over extended periods
- lack of elevated VPA serum level
- lack of transaminase elevation
- lack of apparent risk factors
- presence of background serious mental illness, which can distract from VHE detection via misattribution to uncontrolled primary mental illness.
This last point is critical because it can delay VHE identification and treatment or worse, result in misdiagnosis with accompanying continuation or escalation of VPA dosing as has initially occurred in Mr. D’s case. Similar concerns have been raised2,5 and occurred,5,19 which is not surprising given the frequency of VPA use for psychiatric conditions and symptoms.
Providers should have a low threshold for checking an ammonia level in clinical scenarios that involve any alteration in mental status that may resemble delirium in psychiatric patients treated with valproate. From a preventative perspective, it may be prudent to avoid valproate in psychiatric patients with known VHE risk factors. Either way, promotion of VHE awareness and detection across medical disciplines is paramount.
1. Depakote [package insert]. Chicago, IL: AbbVie; 2016.
2. Lewis C, Deshpande A, Tesar G, et al. Valproate-induced hyperammonemic encephalopathy: a brief review. Curr Med Res Opin. 2012;28(6):1039-1042.
3. Nanau RM, Neuman MG. Adverse drug reactions induced by valproic acid. Clin Biochem. 2013;46(15):1323-1338.
4. Chopra A, Kolla BP, Mansukhani MP, et al. Valproate-induced hyperammonemic encephalopathy: an update on risk factors, clinical correlates and management. Gen Hosp Psychiatry. 2012;34(3):290-298.
5. Carr RB, Shrewsbury K. Hyperammonemia due to valproic acid in the psychiatric setting. Am J Psychiatry. 2007;164(7):1020-1027.
6. Hung C, Li T, Wei I, et al. The real mechanism of VPA-induced hyperammonemia remains unknown. Gen Hosp Psychiatry. 2011;33(1):84.e3-84.e4.
7. Starer J, Chang G. Hyperammonemic encephalopathy, valproic acid, and benzodiazepine withdrawal: a case series. Am J Drug Alcohol Abuse. 2010;36(2):98-101.
8. Eubanks AL, Aguirre B, Bourgeois JA. Severe acute hyperammonemia after brief exposure to valproate. Psychosomatics. 2008;49(1):82-83.
9. Fan CC, Huang MC, Liu HC. Lamotrigine might potentiate valproic acid-induced hyperammonemic encephalopathy. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(7):1747-1748.
10. Deutsch SI, Burket JA, Rosse RB. Valproate-induced hyperammonemic encephalopathy and normal liver functions: possible synergism with topiramate. Clin Neuropharmacol. 2009;32(6):350-352.
11. Rodrigues-Silva N, Venâncio Ä, Bouça J. Risperidone, a risk for valproate induced encephalopathy? Gen Hosp Psychiatry. 2013;35(4):452.e5-452.e6.
12. Sunkavalli KK, Iqbal FM, Singh B, et al. Valproate-induced hyperammonemic encephalopathy: a case report and brief review of the literature. Am J Ther. 2013;20(5):569-571.
13. Abreu LN, Issler C, Lafer B. Valproate-induced reversible pseudoatrophy of the brain and hyperammonemic encephalopathy in a bipolar patient. Aust N Z J Psychiatry. 2009;43(5):484-485.
14. Hong L, Schutz J, Nance M. A case of valproate-induced encephalopathy. Aust N Z J Psychiatry. 2012;46(12):1200-1201.
15. Kimmel RJ, Irwin SA, Meyer JM. Valproic acid-associated hyperammonemic encephalopathy: a case report from the psychiatric setting. Int Clin Psychopharmacol. 2005;20(1):57-58.
16. Elgudin L, Hall Y, Schubert D. Ammonia induced encephalopathy from valproic acid in a bipolar patient: case report. Int J Psychiatry Med. 2003;33(1):91-96.
17. Stewart JT. A case of hyperammonemic encephalopathy after 11 years of valproate therapy. J Clin Psychopharmacol. 2008;28(3):361-362.
18. Wadzinski J, Franks R, Roane D, et al. Valproate-associated hyperammonemic encephalopathy. J Am Board Fam Med. 2007;20(5):499-502.
19. Chang M, Tang X, Wen S, et al. Valproate (VPA)-associated hyperammonemic encephalopathy independent of elevated serum VPA levels: 21 cases in China from May 2000 to May 2012. Compr Psychiatry. 2013;54(5):562-567.
20. Sonik P, Hilty DM, Rossaro L, et al. Carnitine supplementation for valproate-related hyperammonemia to maintain therapeutic valproate level. J Clin Psychopharmacol. 2011;31(5):680-682.
1. Depakote [package insert]. Chicago, IL: AbbVie; 2016.
2. Lewis C, Deshpande A, Tesar G, et al. Valproate-induced hyperammonemic encephalopathy: a brief review. Curr Med Res Opin. 2012;28(6):1039-1042.
3. Nanau RM, Neuman MG. Adverse drug reactions induced by valproic acid. Clin Biochem. 2013;46(15):1323-1338.
4. Chopra A, Kolla BP, Mansukhani MP, et al. Valproate-induced hyperammonemic encephalopathy: an update on risk factors, clinical correlates and management. Gen Hosp Psychiatry. 2012;34(3):290-298.
5. Carr RB, Shrewsbury K. Hyperammonemia due to valproic acid in the psychiatric setting. Am J Psychiatry. 2007;164(7):1020-1027.
6. Hung C, Li T, Wei I, et al. The real mechanism of VPA-induced hyperammonemia remains unknown. Gen Hosp Psychiatry. 2011;33(1):84.e3-84.e4.
7. Starer J, Chang G. Hyperammonemic encephalopathy, valproic acid, and benzodiazepine withdrawal: a case series. Am J Drug Alcohol Abuse. 2010;36(2):98-101.
8. Eubanks AL, Aguirre B, Bourgeois JA. Severe acute hyperammonemia after brief exposure to valproate. Psychosomatics. 2008;49(1):82-83.
9. Fan CC, Huang MC, Liu HC. Lamotrigine might potentiate valproic acid-induced hyperammonemic encephalopathy. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(7):1747-1748.
10. Deutsch SI, Burket JA, Rosse RB. Valproate-induced hyperammonemic encephalopathy and normal liver functions: possible synergism with topiramate. Clin Neuropharmacol. 2009;32(6):350-352.
11. Rodrigues-Silva N, Venâncio Ä, Bouça J. Risperidone, a risk for valproate induced encephalopathy? Gen Hosp Psychiatry. 2013;35(4):452.e5-452.e6.
12. Sunkavalli KK, Iqbal FM, Singh B, et al. Valproate-induced hyperammonemic encephalopathy: a case report and brief review of the literature. Am J Ther. 2013;20(5):569-571.
13. Abreu LN, Issler C, Lafer B. Valproate-induced reversible pseudoatrophy of the brain and hyperammonemic encephalopathy in a bipolar patient. Aust N Z J Psychiatry. 2009;43(5):484-485.
14. Hong L, Schutz J, Nance M. A case of valproate-induced encephalopathy. Aust N Z J Psychiatry. 2012;46(12):1200-1201.
15. Kimmel RJ, Irwin SA, Meyer JM. Valproic acid-associated hyperammonemic encephalopathy: a case report from the psychiatric setting. Int Clin Psychopharmacol. 2005;20(1):57-58.
16. Elgudin L, Hall Y, Schubert D. Ammonia induced encephalopathy from valproic acid in a bipolar patient: case report. Int J Psychiatry Med. 2003;33(1):91-96.
17. Stewart JT. A case of hyperammonemic encephalopathy after 11 years of valproate therapy. J Clin Psychopharmacol. 2008;28(3):361-362.
18. Wadzinski J, Franks R, Roane D, et al. Valproate-associated hyperammonemic encephalopathy. J Am Board Fam Med. 2007;20(5):499-502.
19. Chang M, Tang X, Wen S, et al. Valproate (VPA)-associated hyperammonemic encephalopathy independent of elevated serum VPA levels: 21 cases in China from May 2000 to May 2012. Compr Psychiatry. 2013;54(5):562-567.
20. Sonik P, Hilty DM, Rossaro L, et al. Carnitine supplementation for valproate-related hyperammonemia to maintain therapeutic valproate level. J Clin Psychopharmacol. 2011;31(5):680-682.
Rediscovering clozapine: Clinically relevant off-label uses
Clozapine has been available for decades, but relatively little has been published regarding its off-label uses. This data shortage likely is due in part to clozapine’s strict monitoring requirements, and we suspect off-label use is more commonplace than the literature reflects.
Refractory schizophrenia and reduction in suicidal behavior in schizophrenia or schizoaffective disorder are clozapine’s 2 FDA-approved indications. Clozapine also may be prescribed for other indications, and off-label uses have varying degrees of scientific support.
Our goal in “Rediscovering clozapine” has been to deepen clinicians’ appreciation for this unique medication and provide practical clinical guidance for its safe and effective use.1,2 This final segment reviews representative literature regarding clozapine’s off-label use for bipolar disorder and other indications (Table).

At this point, clozapine still is generally most appropriate for use in refractory cases, regardless of the primary condition being treated. We suggest, however, that physicians should at least consider, “Why is clozapine NOT appropriate for this refractory patient?”
7 Steps define off-label use

Seven steps are useful to consider when prescribing a medication off-label (Figure).3 Off-label prescribing is common in medicine and remains an important component of clinical practice. Sixty percent of antipsychotic prescriptions are written off-label,4 and physicians can prescribe any available medication to any patient for any purpose.
The FDA endorses off-label prescribing: “Good medical practice and the best interests of the patient require that physicians use legally available drugs, biologics and devices according to their best knowledge and judgment.”5 Published case reports and case series provide guidance about the scientific support behind specific off-label indications.
Prescribing off-label based on clinical experience alone is legal, and 1 study reported that 73% of off-label prescriptions written by office-based physicians had little or no scientific support.6 From a medicolegal perspective, prescribing off-label with scientific support is preferred.
Bipolar disorder
Clozapine clearly is established as the most effective antipsychotic for treating refractory schizophrenia. A growing body of evidence supports the off-label use of clozapine for patients with bipolar disorder as well. This literature includes:
- a randomized, open-label trial of maintenance treatment of refractory bipolar disorder7
- 2 studies of treatment of acute mania8,9
- a case series of 3 patients with refractory bipolar disorder and psychotic features who were effectively treated during acute manic episodes with ultra-rapid dose titrations of clozapine.10
In China, clozapine commonly is used to treat bipolar disorder. Results have been positive, and some clinicians there consider clozapine a first-line treatment for this indication.11
In the largest published study of clozapine’s benefits for bipolar disorder, a Danish group presented a retrospective analysis of 326 patients with bipolar disorder (and no history of a schizophrenia-spectrum disorder) treated with clozapine between 1996 and 2007. The study group displayed a significant and clinically relevant reduction in psychiatric hospitalizations, polypharmacy, and self-harm. The authors concluded that clozapine appeared to be an appropriate choice for refractory bipolar disorder and encouraged future investigators to consider randomized controlled studies.12
Major depressive disorder
Published evidence supporting clozapine’s use for refractory unipolar depression is less robust than the evidence for refractory bipolar disorder. One retrospective analysis comparing clozapine treatment for bipolar disorder and unipolar depression concluded that patients with bipolar disorder responded better overall.13
Most case reports involve psychotic depression. One case series discussed clozapine treatment of 3 patients with psychotic depression and reported significant improvement in both depressive and psychotic symptoms.14 Other case reports also described patients with refractory psychotic depression.15,16
We located only 1 case report about using clozapine for depressive symptoms absent psychosis. This case involved a patient who developed recurrent depression, hypersomnia, and behavioral disturbances at age 13 after a viral febrile infection. At age 27, she was hospitalized during an episode and started on low-dose clozapine. After discharge, she remained symptom-free for 30 months on clozapine, 50 to 100 mg/d. Although her symptoms included recurrent depression, her overall clinical picture seemed most consistent with Kleine-Levin syndrome (also known as “Sleeping Beauty” syndrome) rather than a primary mood disorder.17
Borderline personality disorder
Psychotherapy is the mainstay for treating borderline personality disorder (BPD), with pharmacotherapy often added to target symptoms such as anger and impulsivity.18 Some small studies and case series have examined clozapine use for BPD.
An open-label study of 15 inpatients with BPD and psychotic disorder not otherwise specified showed improvement on multiple rating scales with clozapine dosages averaging 250 mg/d.19 In a case series of 22 female inpatients with a primary diagnosis of BPD, clozapine showed beneficial effects in several clinical domains, including symptom severity and frequency of aggressive incidents. The greatest improvement occurred within the first 6 months of treatment.20
Eight patients who continued clozapine after hospital discharge had fewer and shorter subsequent hospitalizations than others with BPD who were not prescribed clozapine at discharge.21 Individual case reports have discussed benefits of clozapine in challenging BPD cases.22-24
Substance use treatment
A growing body of literature suggests that clozapine may reduce cravings for alcohol and illicit drugs because of its unique receptor profile. Much of the data has been collected in dual diagnosis patients taking clozapine primarily to treat schizophrenia or schizoaffective disorder. Patients in 1 study showed a comparable response to clozapine therapy whether they had a history of substance abuse or not. The authors opined that their results demonstrated a more generalizable decrease in cravings and recommended further study.25
In a naturalistic study of 151 dual diagnosis patients with schizophrenia, alcohol use rates decreased significantly among those who received clozapine for psychiatric symptoms. After 3 years, 79% of patients treated with clozapine were in remission from alcohol use, compared with 33.7% of patients treated with other antipsychotics.26
Other studies have reported decreased alcohol and illicit drug use in patients with schizophrenia and concomitant substance use.27,28 Animal studies have displayed similar results, showing decreased alcohol intake with clozapine.29,30
Compelling results have been shown in patients with schizophrenia and Cannabis use disorder. A small randomized trial compared clozapine with other antipsychotics in individuals with schizophrenia and Cannabis use disorder. Clozapine was associated with significantly decreased Cannabis use, independent of overall symptom response or level of functioning.31 An animal study demonstrated an attenuated development of conditioned place preference (classical conditioning) to cocaine. The authors suggested that clozapine should be considered as a future pharmacotherapy to treat cocaine use.32
The literature does not support prescribing clozapine solely for alcohol or illicit drug use, but clozapine merits consideration in patients with schizophrenia and comorbid substance use. This approach may be most beneficial in controlled environments, such as inpatient or residential facilities.
Suicidality
The 2-year International Suicide Prevention Trial (InterSePT) was the first to support clozapine’s efficacy in reducing the risk of recurrent suicidal behavior in schizophrenia or schizoaffective disorder.33 InterSePT data were in line with earlier observations, including improvement in reported depression and hopelessness in patients with primary psychotic disorders.34,35 Clozapine’s action at serotonin receptors (in addition to dopamine receptors) may explain the benefits, based on the suspected link between suicide risk and serotonin.34,36
Most published reports regarding clozapine for suicidality involve patients with schizophrenia or schizoaffective disorder. We found only 1 published case report describing clozapine’s use for recurrent suicidality in a patient with bipolar disorder. The authors described a dramatic reduction in suicidal ideation, suicide attempts, and hospitalizations after other attempted interventions—including electroconvulsive therapy—had been ineffective.37
Aggression
In the absence of FDA-approved treatments for long-term management of aggression, many clinicians prescribe atypical antipsychotics. With the exception of clozapine, the demonstrated benefits of these medications for reducing aggression are equivocal. Clozapine is thought to be superior among atypical antipsychotics for addressing aggression because of its unique and broad combination of dopaminergic and serotonergic activity. Its effects on the D1-dopamine receptor likely target aggression, and its effects on the serotonin 2A receptor (5-HT2A) likely target the impulsivity commonly associated with aggression.38,39
Clozapine has been shown to reduce long-term aggression in patients with psychotic disorders.40-44 Most reports involve individuals with schizophrenia or schizoaffective disorder because this population is most commonly treated with clozapine. However, clozapine’s anti-aggressive benefits appear not to be solely related to sedation or improvement in psychosis.42,45
What is known about clozapine’s mechanism suggests that its anti-aggressive benefits would extend beyond patients with schizophrenia and schizoaffective disorder. In a case series of 7 nonpsychotic patients with antisocial personality disorder and psychopathic traits, all displayed benefits with clozapine—particularly in domains of impulsive behavioral dyscontrol and anger.46
Self-injurious behaviors (SIB) and aggression in 2 patients with profound mental retardation were reduced significantly after treatment was switched from risperidone to clozapine.47 In a similar case, SIB and aggression improved in a man with cognitive impairment.48 The case of Mr. C recounts our experience with using clozapine in a patient with cognitive impairment.
CASE REPORT
Daily assaults keep patient hospitalized
Mr. C, age 19 at the end of treatment, had moderate intellectual disability and an extensive history of violence. He grew up in group homes and long-term psychiatric facilities. Immediately after turning 18, he was transferred from an adolescent facility to an adult psychiatric hospital.
Our treatment team tried various combinations of benzodiazepines, mood stabilizers, and antipsychotics, but Mr. C consistently assaulted 1 or 2 peers daily without clear provocation. Eventually we started him on clozapine, which we titrated to an effective dose (based on a therapeutic serum level). We also added a therapeutic dosage of lithium to address his residual aggression. With the regimen of clozapine and lithium, Mr. C’s assaultive behavior improved dramatically. After going more than 1 year without assaulting a peer, he was placed in the community.
Movement disorders
Parkinson’s disease. The most extensive evidence for treating movement disorders with clozapine involves patients with Parkinson’s disease (PD). Geriatric psychiatrists commonly use clozapine, particularly at low doses, to treat psychotic symptoms in patients with PD. Because of a relatively low likelihood of extrapyramidal side effects, clozapine and quetiapine are the 2 antipsychotics most often used to treat dopamimetic psychosis in PD.49 In a randomized, placebo-controlled study, low-dose clozapine showed benefits in treating dopamimetic psychosis in PD, without worsening overall motor function.50 (The recent approval of pimavanserin for PD psychosis likely will impact off-label use of clozapine for this condition.)
A retrospective review of patients with PD and Lewy body dementia described benefits of treating psychosis with clozapine.51 Benefits also have been reported in using clozapine to address levodopa-induced dyskinesia (LID) absent psychotic symptoms. In an evidence-based review, the Movement Disorder Society described clozapine for LID as “efficacious and possibly useful.”52
Tardive syndromes. In a retrospective review of clozapine use for tardive dyskinesia, 43% of the 30 patients showed improvement, particularly those with concomitant dystonia.53 Another retrospective analysis reported similar outcomes for 48 patients with tardive dyskinesia treated with clozapine.54 Case series and case reports show support for clozapine as monotherapy for tardive dystonia.55
Huntington’s disease. A randomized, double-blind study found little benefit in using clozapine for patients with Huntington’s disease. The authors concluded that, although individual patients may be able to tolerate sufficiently high dosages to improve chorea, clinicians should use restraint when considering clozapine for this population.56
Precautions in older patients. Caution is advised when using clozapine for movement disorders in older individuals, particularly those with concurrent dementia. All antipsychotics, including clozapine,57 carry a “black-box” warning of increased mortality in older adults with dementia.
We hope that this series, “Rediscovering clozapine,” has helped you get reacquainted with this effective medication, employ appropriate caution, and explore off-label uses.
1. Newman WJ, Newman BM. Rediscovering clozapine: after a turbulent history, current guidance on initiating and monitoring. Current Psychiatry. 2016;15(7):42-46,48-49.
2. Newman BM, Newman WJ. Rediscovering clozapine: adverse effects develop—what should you do now? Current Psychiatry. 2016;15(8):40-46,48-49.
3. Newman WJ, Xiong GL, Barnhorst AV. Beta-blockers: off-label use in psychiatric disorders. Psychopharm Review. 2013;48(10):73-80.
4. Stafford RS. Regulating off-label drug use—rethinking the role of the FDA. N Engl J Med. 2008;358(14):1427-1429.
5. U.S. Food and Drug Administration. “Off-label” and investigational use of marketed drugs, biologics, and medical devices—information sheet. http://www.fda.gov/RegulatoryInformation/Guidances/ucm126486.htm. Updated January 25, 2016. Accessed November 24, 2015.
6. Radley DC, Finkelstein SN, Stafford RS. Off-label prescribing among office-based physicians. Arch Intern Med. 2006;166(9):1021-1026.
7. Suppes T, Webb A, Paul B, et al. Clinical outcome in a randomized 1-year trial of clozapine versus treatment as usual for patients with treatment-resistant illness and a history of mania. Am J Psychiatry. 1999;156(8):1164-1169.
8. Barbini B, Scherillo P, Benedetti F, et al. Response to clozapine in acute mania is more rapid than that of chlorpromazine. Int Clin Psychopharmacol. 1997;12(2):109-112.
9. Green AI, Tohen M, Patel JK, et al. Clozapine in the treatment of refractory psychotic mania. Am J Psychiatry. 2000;157(6):982-986.
10. Aksoy-Poyraz C, Turan ¸S, Demirel ÖF, et al. Effectiveness of ultra-rapid dose titration of clozapine for treatment-resistant bipolar mania: case series. Ther Adv Psychopharmacol. 2015;5(4):237-242.
11. Li XB, Tang YL, Wang CY, et al. Clozapine for treatment-resistant bipolar disorder: a systematic review. Bipolar Disord. 2015;17(3):235-247.
12. Nielsen J, Kane JM, Correll CU. Real-world effectiveness of clozapine in patients with bipolar disorder: results from a 2-year mirror-image study. Bipolar Disord. 2012;14(8):863-869.
13. Banov MD, Zarate CA Jr, Tohen M, et al. Clozapine therapy in refractory affective disorders: polarity predicts response in long-term follow-up. J Clin Psychiatry. 1994;55(7):295-300.
14. Ranjan R, Meltzer HY. Acute and long-term effectiveness of clozapine in treatment-resistant psychotic depression. Biol Psychiatry. 1996;40(4):253-258.
15. Dassa D, Kaladjian A, Azorin JM, et al. Clozapine in the treatment of psychotic refractory depression. Br J Psychiatry. 1993;163:822-824.
16. Jeyapaul P, Vieweg R. A case study evaluating the use of clozapine in depression with psychotic features. Ann Gen Psychiatry. 2006;5:20.
17. Havaki-Kontaxaki BJ, Ferentinos PP, Kontaxakis VP, et al. Low-dose clozapine monotherapy for recurring episodes of depression, hypersomnia and behavioural disturbances: a case report. Acta Neuropsychiatr. 2011;23(4):191-193.
18. Stoffers J, Völlm BA, Rücker G, et al. Pharmacological interventions for borderline personality disorder. Cochrane Database Syst Rev. 2010;(6):CD005653. doi: 10.1002/14651858.CD005653.pub2.
19. Frankenburg FR, Zanarini MC. Clozapine treatment of borderline patients: a preliminary study. Compr Psychiatry. 1993;34(6):402-405.
20. Frogley C, Anagnostakis K, Mitchell S, et al. A case series of clozapine for borderline personality disorder. Ann Clin Psychiatry. 2013;25(2):125-134.
21. Parker GF. Clozapine and borderline personality disorder. Psychiatr Serv. 2002;53(3):348-349.
22. Chengappa KNR, Baker RW, Sirri C. The successful use of clozapine in ameliorating severe self mutilation in a patient with borderline personality disorder. J Pers Disord. 1995;9(1):76-82.
23. Rutledge E, O’Regan M, Mohan D. Borderline personality disorder and clozapine. Ir J Psychol Med. 2007;24(1):40-41.
24. Vohra AK. Treatment of severe borderline personality disorder with clozapine. Indian J Psychiatry. 2010;52(3):267-269.
25. Buckley P, Thompson P, Way L, et al. Substance abuse among patients with treatment-resistant schizophrenia: characteristics and implications for clozapine therapy. Am J Psychiatry. 1994;151(3):385-389.
26. Drake RE, Xie H, McHugo GJ, et al. The effects of clozapine on alcohol and drug use disorders among patients with schizophrenia. Schizophr Bull. 2000;26(2):441-449.
27. Zimmet SV, Strous RD, Burgess ES, et al. Effects of clozapine on substance use in patients with schizophrenia and schizoaffective disorder: a retrospective survey. J Clin Psychopharmacol. 2000;20(1):94-98.
28. Green AI, Noordsy DL, Brunette MF, et al. Substance abuse and schizophrenia: pharmacotherapeutic intervention. J Subst Abuse Treat. 2008;34(1):61-71.
29. Green AI, Chau DT, Keung WM, et al. Clozapine reduces alcohol drinking in Syrian golden hamsters. Psychiatry Res. 2004;128(1):9-20.
30. Chau DT, Gulick D, Xie H, et al. Clozapine chronically suppresses alcohol drinking in Syrian golden hamsters. Neuropharmacology. 2010;58(2):351-356.
31. Brunette MF, Dawson R, O’Keefe CD, et al. A randomized trial of clozapine vs. other antipsychotics for cannabis use disorder in patients with schizophrenia. J Dual Diagn. 2011;7(1-2):50-63.
32. Kosten TA, Nestler EJ. Clozapine attenuates cocaine conditioned place preference. Life Sci. 1994;55(1):9-14.
33. Meltzer HY, Alphs L, Green AI, et al; International Suicide Prevention Trial Study Group. Clozapine treatment for suicidality in schizophrenia: International Suicide Prevention Trial (InterSePT) [Erratum in: Arch Gen Psychiatry. 2003;60(7):735]. Arch Gen Psychiatry. 2003;60(1):82-91.
34. Meltzer HY, Okayli G. Reduction of suicidality during clozapine treatment of neuroleptic-resistant schizophrenia: impact on risk-benefit assessment. Am J Psychiatry. 1995;152(2):183-190.
35. Sernyak MJ, Desai R, Stolar M, et al. Impact of clozapine on completed suicide. Am J Psychiatry. 2001;158(6):931-937.
36. Nordström P, Asberg M. Suicide risk and serotonin. Int Clin Psychopharmacol. 1992;6(suppl 6):12-21.
37. Vangala VR, Brown ES, Suppes T. Clozapine associated with decreased suicidality in bipolar disorder: a case report. Bipolar Disord. 1999;1(2):123-124.
38. Meltzer HY. The mechanism of action of novel antipsychotic drugs. Schizophr Bull. 1991;17(2):263-287.
39. Meltzer HY. An overview of the mechanism of action of clozapine. J Clin Psychiatry. 1994;55(suppl B):47-52.
40. Rabinowitz J, Avnon M, Rosenberg V. Effect of clozapine on physical and verbal aggression. Schizophr Res. 1996;22(3):249-255.
41. Spivak B, Roitman S, Vered Y, et al. Diminished suicidal and aggressive behavior, high plasma norepinephrine levels, and serum triglyceride levels in chronic neuroleptic-resistant schizophrenic patients maintained on clozapine. Clin Neuropharmacol. 1998;21(4):245-250.
42. Citrome L, Volavka J, Czobor P, et al. Effects of clozapine, olanzapine, risperidone, and haloperidol on hostility among patients with schizophrenia. Psychiatr Serv. 2001;52(11):1510-1514.
43. Volavka J, Czobor P, Nolan K, et al. Overt aggression and psychotic symptoms in patients with schizophrenia treated with clozapine, olanzapine, risperidone, or haloperidol. J Clin Psychopharmacol. 2004;24(2):225-228.
44. Krakowski MI, Czobar P, Citrome L, et al. Atypical antipsychotic agents in the treatment of violent patients with schizophrenia and schizoaffective disorder. Arch Gen Psychiatry. 2006;63(6):622-629.
45. Chiles JA, Davidson P, McBride D. Effects of clozapine on use of seclusion and restraint at a state hospital. Hosp Community Psychiatry. 1994;45(3):269-271.
46. Brown D, Larkin F, Sengupta S, et al. Clozapine: an effective treatment for seriously violent and psychopathic men with antisocial personality disorder in a UK high-security hospital. CNS Spectr. 2014;19(5):391-402.
47. Hammock R, Levine WR, Schroeder SR. Brief report: effects of clozapine on self-injurious behavior of two risperidone nonresponders with mental retardation. J Autism Dev Disord. 2001;31(1):109-113.
48. Hammock RG, Schroeder SR, Levine WR. The effect of clozapine on self-injurious behavior. J Autism Dev Disord. 1995;25(6):611-626.
49. Morgante L, Epifanio A, Spina E, et al. Quetiapine and clozapine in parkinsonian patients with dopaminergic psychosis [Erratum in: Clin Neuropharmacol. 2004;27(5):256]. Clin Neuropharmacol. 2004;27(4):153-156.
50. Pollak P, Tison F, Rascol O. Clozapine in drug induced psychosis in Parkinson’s disease: a randomised, placebo controlled study with open follow up. J Neurol Neurosurg Psychiatry. 2004;75(5):689-695.
51. Lutz UC, Sirfy A, Wiatr G, et al. Clozapine serum concentrations in dopamimetic psychosis in Parkinson’s disease and related disorders. Eur J Clin Pharmacol. 2014;70(12):1471-1476.
52. Fox SH, Katzenschlager R, Lim SY, et al. The Movement Disorder Society Evidence-Based Medicine Review Update: treatment for the motor symptoms of Parkinson’s disease. Mov Disord. 2011;26(suppl 3):S2-S41.
53. Lieberman JA, Saltz BL, Johns CA, et al. The effects of clozapine on tardive dyskinesia. Br J Psychiatry. 1991;158:503-510.
54. Naber D, Leppig M, Grohmann R, et al. Efficacy and adverse effects of clozapine in the treatment of schizophrenia and tardive dyskinesia—a retrospective study. Psychopharmacology (Berl). 1989;99(suppl):S73-S76.
55. Pinninti NR, Faden J, Adityanjee A. Are second-generation antipsychotics useful in tardive dystonia? Clin Neuropharmacol. 2015;38(5):183-197.
56. van Vugt JP, Siesling S, Vergeer M, et al. Clozapine versus placebo in Huntington’s disease: a double blind randomised comparative study. J Neurol Neurosurg Psychiatry. 1997;63(1):35-39.
57. Novartis Pharmaceuticals Corporation. Clozaril (clozapine). Prescribing information. http://clozaril.com/wp-content/themes/eyesite/pi/Clozaril-2015A507-10022015-Approved.pdf. Accessed September 2, 2016.
Clozapine has been available for decades, but relatively little has been published regarding its off-label uses. This data shortage likely is due in part to clozapine’s strict monitoring requirements, and we suspect off-label use is more commonplace than the literature reflects.
Refractory schizophrenia and reduction in suicidal behavior in schizophrenia or schizoaffective disorder are clozapine’s 2 FDA-approved indications. Clozapine also may be prescribed for other indications, and off-label uses have varying degrees of scientific support.
Our goal in “Rediscovering clozapine” has been to deepen clinicians’ appreciation for this unique medication and provide practical clinical guidance for its safe and effective use.1,2 This final segment reviews representative literature regarding clozapine’s off-label use for bipolar disorder and other indications (Table).
At this point, clozapine still is generally most appropriate for use in refractory cases, regardless of the primary condition being treated. We suggest, however, that physicians should at least consider, “Why is clozapine NOT appropriate for this refractory patient?”
7 Steps define off-label use
Seven steps are useful to consider when prescribing a medication off-label (Figure).3 Off-label prescribing is common in medicine and remains an important component of clinical practice. Sixty percent of antipsychotic prescriptions are written off-label,4 and physicians can prescribe any available medication to any patient for any purpose.
The FDA endorses off-label prescribing: “Good medical practice and the best interests of the patient require that physicians use legally available drugs, biologics and devices according to their best knowledge and judgment.”5 Published case reports and case series provide guidance about the scientific support behind specific off-label indications.
Prescribing off-label based on clinical experience alone is legal, and 1 study reported that 73% of off-label prescriptions written by office-based physicians had little or no scientific support.6 From a medicolegal perspective, prescribing off-label with scientific support is preferred.
Bipolar disorder
Clozapine clearly is established as the most effective antipsychotic for treating refractory schizophrenia. A growing body of evidence supports the off-label use of clozapine for patients with bipolar disorder as well. This literature includes:
- a randomized, open-label trial of maintenance treatment of refractory bipolar disorder7
- 2 studies of treatment of acute mania8,9
- a case series of 3 patients with refractory bipolar disorder and psychotic features who were effectively treated during acute manic episodes with ultra-rapid dose titrations of clozapine.10
In China, clozapine commonly is used to treat bipolar disorder. Results have been positive, and some clinicians there consider clozapine a first-line treatment for this indication.11
In the largest published study of clozapine’s benefits for bipolar disorder, a Danish group presented a retrospective analysis of 326 patients with bipolar disorder (and no history of a schizophrenia-spectrum disorder) treated with clozapine between 1996 and 2007. The study group displayed a significant and clinically relevant reduction in psychiatric hospitalizations, polypharmacy, and self-harm. The authors concluded that clozapine appeared to be an appropriate choice for refractory bipolar disorder and encouraged future investigators to consider randomized controlled studies.12
Major depressive disorder
Published evidence supporting clozapine’s use for refractory unipolar depression is less robust than the evidence for refractory bipolar disorder. One retrospective analysis comparing clozapine treatment for bipolar disorder and unipolar depression concluded that patients with bipolar disorder responded better overall.13
Most case reports involve psychotic depression. One case series discussed clozapine treatment of 3 patients with psychotic depression and reported significant improvement in both depressive and psychotic symptoms.14 Other case reports also described patients with refractory psychotic depression.15,16
We located only 1 case report about using clozapine for depressive symptoms absent psychosis. This case involved a patient who developed recurrent depression, hypersomnia, and behavioral disturbances at age 13 after a viral febrile infection. At age 27, she was hospitalized during an episode and started on low-dose clozapine. After discharge, she remained symptom-free for 30 months on clozapine, 50 to 100 mg/d. Although her symptoms included recurrent depression, her overall clinical picture seemed most consistent with Kleine-Levin syndrome (also known as “Sleeping Beauty” syndrome) rather than a primary mood disorder.17
Borderline personality disorder
Psychotherapy is the mainstay for treating borderline personality disorder (BPD), with pharmacotherapy often added to target symptoms such as anger and impulsivity.18 Some small studies and case series have examined clozapine use for BPD.
An open-label study of 15 inpatients with BPD and psychotic disorder not otherwise specified showed improvement on multiple rating scales with clozapine dosages averaging 250 mg/d.19 In a case series of 22 female inpatients with a primary diagnosis of BPD, clozapine showed beneficial effects in several clinical domains, including symptom severity and frequency of aggressive incidents. The greatest improvement occurred within the first 6 months of treatment.20
Eight patients who continued clozapine after hospital discharge had fewer and shorter subsequent hospitalizations than others with BPD who were not prescribed clozapine at discharge.21 Individual case reports have discussed benefits of clozapine in challenging BPD cases.22-24
Substance use treatment
A growing body of literature suggests that clozapine may reduce cravings for alcohol and illicit drugs because of its unique receptor profile. Much of the data has been collected in dual diagnosis patients taking clozapine primarily to treat schizophrenia or schizoaffective disorder. Patients in 1 study showed a comparable response to clozapine therapy whether they had a history of substance abuse or not. The authors opined that their results demonstrated a more generalizable decrease in cravings and recommended further study.25
In a naturalistic study of 151 dual diagnosis patients with schizophrenia, alcohol use rates decreased significantly among those who received clozapine for psychiatric symptoms. After 3 years, 79% of patients treated with clozapine were in remission from alcohol use, compared with 33.7% of patients treated with other antipsychotics.26
Other studies have reported decreased alcohol and illicit drug use in patients with schizophrenia and concomitant substance use.27,28 Animal studies have displayed similar results, showing decreased alcohol intake with clozapine.29,30
Compelling results have been shown in patients with schizophrenia and Cannabis use disorder. A small randomized trial compared clozapine with other antipsychotics in individuals with schizophrenia and Cannabis use disorder. Clozapine was associated with significantly decreased Cannabis use, independent of overall symptom response or level of functioning.31 An animal study demonstrated an attenuated development of conditioned place preference (classical conditioning) to cocaine. The authors suggested that clozapine should be considered as a future pharmacotherapy to treat cocaine use.32
The literature does not support prescribing clozapine solely for alcohol or illicit drug use, but clozapine merits consideration in patients with schizophrenia and comorbid substance use. This approach may be most beneficial in controlled environments, such as inpatient or residential facilities.
Suicidality
The 2-year International Suicide Prevention Trial (InterSePT) was the first to support clozapine’s efficacy in reducing the risk of recurrent suicidal behavior in schizophrenia or schizoaffective disorder.33 InterSePT data were in line with earlier observations, including improvement in reported depression and hopelessness in patients with primary psychotic disorders.34,35 Clozapine’s action at serotonin receptors (in addition to dopamine receptors) may explain the benefits, based on the suspected link between suicide risk and serotonin.34,36
Most published reports regarding clozapine for suicidality involve patients with schizophrenia or schizoaffective disorder. We found only 1 published case report describing clozapine’s use for recurrent suicidality in a patient with bipolar disorder. The authors described a dramatic reduction in suicidal ideation, suicide attempts, and hospitalizations after other attempted interventions—including electroconvulsive therapy—had been ineffective.37
Aggression
In the absence of FDA-approved treatments for long-term management of aggression, many clinicians prescribe atypical antipsychotics. With the exception of clozapine, the demonstrated benefits of these medications for reducing aggression are equivocal. Clozapine is thought to be superior among atypical antipsychotics for addressing aggression because of its unique and broad combination of dopaminergic and serotonergic activity. Its effects on the D1-dopamine receptor likely target aggression, and its effects on the serotonin 2A receptor (5-HT2A) likely target the impulsivity commonly associated with aggression.38,39
Clozapine has been shown to reduce long-term aggression in patients with psychotic disorders.40-44 Most reports involve individuals with schizophrenia or schizoaffective disorder because this population is most commonly treated with clozapine. However, clozapine’s anti-aggressive benefits appear not to be solely related to sedation or improvement in psychosis.42,45
What is known about clozapine’s mechanism suggests that its anti-aggressive benefits would extend beyond patients with schizophrenia and schizoaffective disorder. In a case series of 7 nonpsychotic patients with antisocial personality disorder and psychopathic traits, all displayed benefits with clozapine—particularly in domains of impulsive behavioral dyscontrol and anger.46
Self-injurious behaviors (SIB) and aggression in 2 patients with profound mental retardation were reduced significantly after treatment was switched from risperidone to clozapine.47 In a similar case, SIB and aggression improved in a man with cognitive impairment.48 The case of Mr. C recounts our experience with using clozapine in a patient with cognitive impairment.
CASE REPORT
Daily assaults keep patient hospitalized
Mr. C, age 19 at the end of treatment, had moderate intellectual disability and an extensive history of violence. He grew up in group homes and long-term psychiatric facilities. Immediately after turning 18, he was transferred from an adolescent facility to an adult psychiatric hospital.
Our treatment team tried various combinations of benzodiazepines, mood stabilizers, and antipsychotics, but Mr. C consistently assaulted 1 or 2 peers daily without clear provocation. Eventually we started him on clozapine, which we titrated to an effective dose (based on a therapeutic serum level). We also added a therapeutic dosage of lithium to address his residual aggression. With the regimen of clozapine and lithium, Mr. C’s assaultive behavior improved dramatically. After going more than 1 year without assaulting a peer, he was placed in the community.
Movement disorders
Parkinson’s disease. The most extensive evidence for treating movement disorders with clozapine involves patients with Parkinson’s disease (PD). Geriatric psychiatrists commonly use clozapine, particularly at low doses, to treat psychotic symptoms in patients with PD. Because of a relatively low likelihood of extrapyramidal side effects, clozapine and quetiapine are the 2 antipsychotics most often used to treat dopamimetic psychosis in PD.49 In a randomized, placebo-controlled study, low-dose clozapine showed benefits in treating dopamimetic psychosis in PD, without worsening overall motor function.50 (The recent approval of pimavanserin for PD psychosis likely will impact off-label use of clozapine for this condition.)
A retrospective review of patients with PD and Lewy body dementia described benefits of treating psychosis with clozapine.51 Benefits also have been reported in using clozapine to address levodopa-induced dyskinesia (LID) absent psychotic symptoms. In an evidence-based review, the Movement Disorder Society described clozapine for LID as “efficacious and possibly useful.”52
Tardive syndromes. In a retrospective review of clozapine use for tardive dyskinesia, 43% of the 30 patients showed improvement, particularly those with concomitant dystonia.53 Another retrospective analysis reported similar outcomes for 48 patients with tardive dyskinesia treated with clozapine.54 Case series and case reports show support for clozapine as monotherapy for tardive dystonia.55
Huntington’s disease. A randomized, double-blind study found little benefit in using clozapine for patients with Huntington’s disease. The authors concluded that, although individual patients may be able to tolerate sufficiently high dosages to improve chorea, clinicians should use restraint when considering clozapine for this population.56
Precautions in older patients. Caution is advised when using clozapine for movement disorders in older individuals, particularly those with concurrent dementia. All antipsychotics, including clozapine,57 carry a “black-box” warning of increased mortality in older adults with dementia.
We hope that this series, “Rediscovering clozapine,” has helped you get reacquainted with this effective medication, employ appropriate caution, and explore off-label uses.
Clozapine has been available for decades, but relatively little has been published regarding its off-label uses. This data shortage likely is due in part to clozapine’s strict monitoring requirements, and we suspect off-label use is more commonplace than the literature reflects.
Refractory schizophrenia and reduction in suicidal behavior in schizophrenia or schizoaffective disorder are clozapine’s 2 FDA-approved indications. Clozapine also may be prescribed for other indications, and off-label uses have varying degrees of scientific support.
Our goal in “Rediscovering clozapine” has been to deepen clinicians’ appreciation for this unique medication and provide practical clinical guidance for its safe and effective use.1,2 This final segment reviews representative literature regarding clozapine’s off-label use for bipolar disorder and other indications (Table).
At this point, clozapine still is generally most appropriate for use in refractory cases, regardless of the primary condition being treated. We suggest, however, that physicians should at least consider, “Why is clozapine NOT appropriate for this refractory patient?”
7 Steps define off-label use
Seven steps are useful to consider when prescribing a medication off-label (Figure).3 Off-label prescribing is common in medicine and remains an important component of clinical practice. Sixty percent of antipsychotic prescriptions are written off-label,4 and physicians can prescribe any available medication to any patient for any purpose.
The FDA endorses off-label prescribing: “Good medical practice and the best interests of the patient require that physicians use legally available drugs, biologics and devices according to their best knowledge and judgment.”5 Published case reports and case series provide guidance about the scientific support behind specific off-label indications.
Prescribing off-label based on clinical experience alone is legal, and 1 study reported that 73% of off-label prescriptions written by office-based physicians had little or no scientific support.6 From a medicolegal perspective, prescribing off-label with scientific support is preferred.
Bipolar disorder
Clozapine clearly is established as the most effective antipsychotic for treating refractory schizophrenia. A growing body of evidence supports the off-label use of clozapine for patients with bipolar disorder as well. This literature includes:
- a randomized, open-label trial of maintenance treatment of refractory bipolar disorder7
- 2 studies of treatment of acute mania8,9
- a case series of 3 patients with refractory bipolar disorder and psychotic features who were effectively treated during acute manic episodes with ultra-rapid dose titrations of clozapine.10
In China, clozapine commonly is used to treat bipolar disorder. Results have been positive, and some clinicians there consider clozapine a first-line treatment for this indication.11
In the largest published study of clozapine’s benefits for bipolar disorder, a Danish group presented a retrospective analysis of 326 patients with bipolar disorder (and no history of a schizophrenia-spectrum disorder) treated with clozapine between 1996 and 2007. The study group displayed a significant and clinically relevant reduction in psychiatric hospitalizations, polypharmacy, and self-harm. The authors concluded that clozapine appeared to be an appropriate choice for refractory bipolar disorder and encouraged future investigators to consider randomized controlled studies.12
Major depressive disorder
Published evidence supporting clozapine’s use for refractory unipolar depression is less robust than the evidence for refractory bipolar disorder. One retrospective analysis comparing clozapine treatment for bipolar disorder and unipolar depression concluded that patients with bipolar disorder responded better overall.13
Most case reports involve psychotic depression. One case series discussed clozapine treatment of 3 patients with psychotic depression and reported significant improvement in both depressive and psychotic symptoms.14 Other case reports also described patients with refractory psychotic depression.15,16
We located only 1 case report about using clozapine for depressive symptoms absent psychosis. This case involved a patient who developed recurrent depression, hypersomnia, and behavioral disturbances at age 13 after a viral febrile infection. At age 27, she was hospitalized during an episode and started on low-dose clozapine. After discharge, she remained symptom-free for 30 months on clozapine, 50 to 100 mg/d. Although her symptoms included recurrent depression, her overall clinical picture seemed most consistent with Kleine-Levin syndrome (also known as “Sleeping Beauty” syndrome) rather than a primary mood disorder.17
Borderline personality disorder
Psychotherapy is the mainstay for treating borderline personality disorder (BPD), with pharmacotherapy often added to target symptoms such as anger and impulsivity.18 Some small studies and case series have examined clozapine use for BPD.
An open-label study of 15 inpatients with BPD and psychotic disorder not otherwise specified showed improvement on multiple rating scales with clozapine dosages averaging 250 mg/d.19 In a case series of 22 female inpatients with a primary diagnosis of BPD, clozapine showed beneficial effects in several clinical domains, including symptom severity and frequency of aggressive incidents. The greatest improvement occurred within the first 6 months of treatment.20
Eight patients who continued clozapine after hospital discharge had fewer and shorter subsequent hospitalizations than others with BPD who were not prescribed clozapine at discharge.21 Individual case reports have discussed benefits of clozapine in challenging BPD cases.22-24
Substance use treatment
A growing body of literature suggests that clozapine may reduce cravings for alcohol and illicit drugs because of its unique receptor profile. Much of the data has been collected in dual diagnosis patients taking clozapine primarily to treat schizophrenia or schizoaffective disorder. Patients in 1 study showed a comparable response to clozapine therapy whether they had a history of substance abuse or not. The authors opined that their results demonstrated a more generalizable decrease in cravings and recommended further study.25
In a naturalistic study of 151 dual diagnosis patients with schizophrenia, alcohol use rates decreased significantly among those who received clozapine for psychiatric symptoms. After 3 years, 79% of patients treated with clozapine were in remission from alcohol use, compared with 33.7% of patients treated with other antipsychotics.26
Other studies have reported decreased alcohol and illicit drug use in patients with schizophrenia and concomitant substance use.27,28 Animal studies have displayed similar results, showing decreased alcohol intake with clozapine.29,30
Compelling results have been shown in patients with schizophrenia and Cannabis use disorder. A small randomized trial compared clozapine with other antipsychotics in individuals with schizophrenia and Cannabis use disorder. Clozapine was associated with significantly decreased Cannabis use, independent of overall symptom response or level of functioning.31 An animal study demonstrated an attenuated development of conditioned place preference (classical conditioning) to cocaine. The authors suggested that clozapine should be considered as a future pharmacotherapy to treat cocaine use.32
The literature does not support prescribing clozapine solely for alcohol or illicit drug use, but clozapine merits consideration in patients with schizophrenia and comorbid substance use. This approach may be most beneficial in controlled environments, such as inpatient or residential facilities.
Suicidality
The 2-year International Suicide Prevention Trial (InterSePT) was the first to support clozapine’s efficacy in reducing the risk of recurrent suicidal behavior in schizophrenia or schizoaffective disorder.33 InterSePT data were in line with earlier observations, including improvement in reported depression and hopelessness in patients with primary psychotic disorders.34,35 Clozapine’s action at serotonin receptors (in addition to dopamine receptors) may explain the benefits, based on the suspected link between suicide risk and serotonin.34,36
Most published reports regarding clozapine for suicidality involve patients with schizophrenia or schizoaffective disorder. We found only 1 published case report describing clozapine’s use for recurrent suicidality in a patient with bipolar disorder. The authors described a dramatic reduction in suicidal ideation, suicide attempts, and hospitalizations after other attempted interventions—including electroconvulsive therapy—had been ineffective.37
Aggression
In the absence of FDA-approved treatments for long-term management of aggression, many clinicians prescribe atypical antipsychotics. With the exception of clozapine, the demonstrated benefits of these medications for reducing aggression are equivocal. Clozapine is thought to be superior among atypical antipsychotics for addressing aggression because of its unique and broad combination of dopaminergic and serotonergic activity. Its effects on the D1-dopamine receptor likely target aggression, and its effects on the serotonin 2A receptor (5-HT2A) likely target the impulsivity commonly associated with aggression.38,39
Clozapine has been shown to reduce long-term aggression in patients with psychotic disorders.40-44 Most reports involve individuals with schizophrenia or schizoaffective disorder because this population is most commonly treated with clozapine. However, clozapine’s anti-aggressive benefits appear not to be solely related to sedation or improvement in psychosis.42,45
What is known about clozapine’s mechanism suggests that its anti-aggressive benefits would extend beyond patients with schizophrenia and schizoaffective disorder. In a case series of 7 nonpsychotic patients with antisocial personality disorder and psychopathic traits, all displayed benefits with clozapine—particularly in domains of impulsive behavioral dyscontrol and anger.46
Self-injurious behaviors (SIB) and aggression in 2 patients with profound mental retardation were reduced significantly after treatment was switched from risperidone to clozapine.47 In a similar case, SIB and aggression improved in a man with cognitive impairment.48 The case of Mr. C recounts our experience with using clozapine in a patient with cognitive impairment.
CASE REPORT
Daily assaults keep patient hospitalized
Mr. C, age 19 at the end of treatment, had moderate intellectual disability and an extensive history of violence. He grew up in group homes and long-term psychiatric facilities. Immediately after turning 18, he was transferred from an adolescent facility to an adult psychiatric hospital.
Our treatment team tried various combinations of benzodiazepines, mood stabilizers, and antipsychotics, but Mr. C consistently assaulted 1 or 2 peers daily without clear provocation. Eventually we started him on clozapine, which we titrated to an effective dose (based on a therapeutic serum level). We also added a therapeutic dosage of lithium to address his residual aggression. With the regimen of clozapine and lithium, Mr. C’s assaultive behavior improved dramatically. After going more than 1 year without assaulting a peer, he was placed in the community.
Movement disorders
Parkinson’s disease. The most extensive evidence for treating movement disorders with clozapine involves patients with Parkinson’s disease (PD). Geriatric psychiatrists commonly use clozapine, particularly at low doses, to treat psychotic symptoms in patients with PD. Because of a relatively low likelihood of extrapyramidal side effects, clozapine and quetiapine are the 2 antipsychotics most often used to treat dopamimetic psychosis in PD.49 In a randomized, placebo-controlled study, low-dose clozapine showed benefits in treating dopamimetic psychosis in PD, without worsening overall motor function.50 (The recent approval of pimavanserin for PD psychosis likely will impact off-label use of clozapine for this condition.)
A retrospective review of patients with PD and Lewy body dementia described benefits of treating psychosis with clozapine.51 Benefits also have been reported in using clozapine to address levodopa-induced dyskinesia (LID) absent psychotic symptoms. In an evidence-based review, the Movement Disorder Society described clozapine for LID as “efficacious and possibly useful.”52
Tardive syndromes. In a retrospective review of clozapine use for tardive dyskinesia, 43% of the 30 patients showed improvement, particularly those with concomitant dystonia.53 Another retrospective analysis reported similar outcomes for 48 patients with tardive dyskinesia treated with clozapine.54 Case series and case reports show support for clozapine as monotherapy for tardive dystonia.55
Huntington’s disease. A randomized, double-blind study found little benefit in using clozapine for patients with Huntington’s disease. The authors concluded that, although individual patients may be able to tolerate sufficiently high dosages to improve chorea, clinicians should use restraint when considering clozapine for this population.56
Precautions in older patients. Caution is advised when using clozapine for movement disorders in older individuals, particularly those with concurrent dementia. All antipsychotics, including clozapine,57 carry a “black-box” warning of increased mortality in older adults with dementia.
We hope that this series, “Rediscovering clozapine,” has helped you get reacquainted with this effective medication, employ appropriate caution, and explore off-label uses.
1. Newman WJ, Newman BM. Rediscovering clozapine: after a turbulent history, current guidance on initiating and monitoring. Current Psychiatry. 2016;15(7):42-46,48-49.
2. Newman BM, Newman WJ. Rediscovering clozapine: adverse effects develop—what should you do now? Current Psychiatry. 2016;15(8):40-46,48-49.
3. Newman WJ, Xiong GL, Barnhorst AV. Beta-blockers: off-label use in psychiatric disorders. Psychopharm Review. 2013;48(10):73-80.
4. Stafford RS. Regulating off-label drug use—rethinking the role of the FDA. N Engl J Med. 2008;358(14):1427-1429.
5. U.S. Food and Drug Administration. “Off-label” and investigational use of marketed drugs, biologics, and medical devices—information sheet. http://www.fda.gov/RegulatoryInformation/Guidances/ucm126486.htm. Updated January 25, 2016. Accessed November 24, 2015.
6. Radley DC, Finkelstein SN, Stafford RS. Off-label prescribing among office-based physicians. Arch Intern Med. 2006;166(9):1021-1026.
7. Suppes T, Webb A, Paul B, et al. Clinical outcome in a randomized 1-year trial of clozapine versus treatment as usual for patients with treatment-resistant illness and a history of mania. Am J Psychiatry. 1999;156(8):1164-1169.
8. Barbini B, Scherillo P, Benedetti F, et al. Response to clozapine in acute mania is more rapid than that of chlorpromazine. Int Clin Psychopharmacol. 1997;12(2):109-112.
9. Green AI, Tohen M, Patel JK, et al. Clozapine in the treatment of refractory psychotic mania. Am J Psychiatry. 2000;157(6):982-986.
10. Aksoy-Poyraz C, Turan ¸S, Demirel ÖF, et al. Effectiveness of ultra-rapid dose titration of clozapine for treatment-resistant bipolar mania: case series. Ther Adv Psychopharmacol. 2015;5(4):237-242.
11. Li XB, Tang YL, Wang CY, et al. Clozapine for treatment-resistant bipolar disorder: a systematic review. Bipolar Disord. 2015;17(3):235-247.
12. Nielsen J, Kane JM, Correll CU. Real-world effectiveness of clozapine in patients with bipolar disorder: results from a 2-year mirror-image study. Bipolar Disord. 2012;14(8):863-869.
13. Banov MD, Zarate CA Jr, Tohen M, et al. Clozapine therapy in refractory affective disorders: polarity predicts response in long-term follow-up. J Clin Psychiatry. 1994;55(7):295-300.
14. Ranjan R, Meltzer HY. Acute and long-term effectiveness of clozapine in treatment-resistant psychotic depression. Biol Psychiatry. 1996;40(4):253-258.
15. Dassa D, Kaladjian A, Azorin JM, et al. Clozapine in the treatment of psychotic refractory depression. Br J Psychiatry. 1993;163:822-824.
16. Jeyapaul P, Vieweg R. A case study evaluating the use of clozapine in depression with psychotic features. Ann Gen Psychiatry. 2006;5:20.
17. Havaki-Kontaxaki BJ, Ferentinos PP, Kontaxakis VP, et al. Low-dose clozapine monotherapy for recurring episodes of depression, hypersomnia and behavioural disturbances: a case report. Acta Neuropsychiatr. 2011;23(4):191-193.
18. Stoffers J, Völlm BA, Rücker G, et al. Pharmacological interventions for borderline personality disorder. Cochrane Database Syst Rev. 2010;(6):CD005653. doi: 10.1002/14651858.CD005653.pub2.
19. Frankenburg FR, Zanarini MC. Clozapine treatment of borderline patients: a preliminary study. Compr Psychiatry. 1993;34(6):402-405.
20. Frogley C, Anagnostakis K, Mitchell S, et al. A case series of clozapine for borderline personality disorder. Ann Clin Psychiatry. 2013;25(2):125-134.
21. Parker GF. Clozapine and borderline personality disorder. Psychiatr Serv. 2002;53(3):348-349.
22. Chengappa KNR, Baker RW, Sirri C. The successful use of clozapine in ameliorating severe self mutilation in a patient with borderline personality disorder. J Pers Disord. 1995;9(1):76-82.
23. Rutledge E, O’Regan M, Mohan D. Borderline personality disorder and clozapine. Ir J Psychol Med. 2007;24(1):40-41.
24. Vohra AK. Treatment of severe borderline personality disorder with clozapine. Indian J Psychiatry. 2010;52(3):267-269.
25. Buckley P, Thompson P, Way L, et al. Substance abuse among patients with treatment-resistant schizophrenia: characteristics and implications for clozapine therapy. Am J Psychiatry. 1994;151(3):385-389.
26. Drake RE, Xie H, McHugo GJ, et al. The effects of clozapine on alcohol and drug use disorders among patients with schizophrenia. Schizophr Bull. 2000;26(2):441-449.
27. Zimmet SV, Strous RD, Burgess ES, et al. Effects of clozapine on substance use in patients with schizophrenia and schizoaffective disorder: a retrospective survey. J Clin Psychopharmacol. 2000;20(1):94-98.
28. Green AI, Noordsy DL, Brunette MF, et al. Substance abuse and schizophrenia: pharmacotherapeutic intervention. J Subst Abuse Treat. 2008;34(1):61-71.
29. Green AI, Chau DT, Keung WM, et al. Clozapine reduces alcohol drinking in Syrian golden hamsters. Psychiatry Res. 2004;128(1):9-20.
30. Chau DT, Gulick D, Xie H, et al. Clozapine chronically suppresses alcohol drinking in Syrian golden hamsters. Neuropharmacology. 2010;58(2):351-356.
31. Brunette MF, Dawson R, O’Keefe CD, et al. A randomized trial of clozapine vs. other antipsychotics for cannabis use disorder in patients with schizophrenia. J Dual Diagn. 2011;7(1-2):50-63.
32. Kosten TA, Nestler EJ. Clozapine attenuates cocaine conditioned place preference. Life Sci. 1994;55(1):9-14.
33. Meltzer HY, Alphs L, Green AI, et al; International Suicide Prevention Trial Study Group. Clozapine treatment for suicidality in schizophrenia: International Suicide Prevention Trial (InterSePT) [Erratum in: Arch Gen Psychiatry. 2003;60(7):735]. Arch Gen Psychiatry. 2003;60(1):82-91.
34. Meltzer HY, Okayli G. Reduction of suicidality during clozapine treatment of neuroleptic-resistant schizophrenia: impact on risk-benefit assessment. Am J Psychiatry. 1995;152(2):183-190.
35. Sernyak MJ, Desai R, Stolar M, et al. Impact of clozapine on completed suicide. Am J Psychiatry. 2001;158(6):931-937.
36. Nordström P, Asberg M. Suicide risk and serotonin. Int Clin Psychopharmacol. 1992;6(suppl 6):12-21.
37. Vangala VR, Brown ES, Suppes T. Clozapine associated with decreased suicidality in bipolar disorder: a case report. Bipolar Disord. 1999;1(2):123-124.
38. Meltzer HY. The mechanism of action of novel antipsychotic drugs. Schizophr Bull. 1991;17(2):263-287.
39. Meltzer HY. An overview of the mechanism of action of clozapine. J Clin Psychiatry. 1994;55(suppl B):47-52.
40. Rabinowitz J, Avnon M, Rosenberg V. Effect of clozapine on physical and verbal aggression. Schizophr Res. 1996;22(3):249-255.
41. Spivak B, Roitman S, Vered Y, et al. Diminished suicidal and aggressive behavior, high plasma norepinephrine levels, and serum triglyceride levels in chronic neuroleptic-resistant schizophrenic patients maintained on clozapine. Clin Neuropharmacol. 1998;21(4):245-250.
42. Citrome L, Volavka J, Czobor P, et al. Effects of clozapine, olanzapine, risperidone, and haloperidol on hostility among patients with schizophrenia. Psychiatr Serv. 2001;52(11):1510-1514.
43. Volavka J, Czobor P, Nolan K, et al. Overt aggression and psychotic symptoms in patients with schizophrenia treated with clozapine, olanzapine, risperidone, or haloperidol. J Clin Psychopharmacol. 2004;24(2):225-228.
44. Krakowski MI, Czobar P, Citrome L, et al. Atypical antipsychotic agents in the treatment of violent patients with schizophrenia and schizoaffective disorder. Arch Gen Psychiatry. 2006;63(6):622-629.
45. Chiles JA, Davidson P, McBride D. Effects of clozapine on use of seclusion and restraint at a state hospital. Hosp Community Psychiatry. 1994;45(3):269-271.
46. Brown D, Larkin F, Sengupta S, et al. Clozapine: an effective treatment for seriously violent and psychopathic men with antisocial personality disorder in a UK high-security hospital. CNS Spectr. 2014;19(5):391-402.
47. Hammock R, Levine WR, Schroeder SR. Brief report: effects of clozapine on self-injurious behavior of two risperidone nonresponders with mental retardation. J Autism Dev Disord. 2001;31(1):109-113.
48. Hammock RG, Schroeder SR, Levine WR. The effect of clozapine on self-injurious behavior. J Autism Dev Disord. 1995;25(6):611-626.
49. Morgante L, Epifanio A, Spina E, et al. Quetiapine and clozapine in parkinsonian patients with dopaminergic psychosis [Erratum in: Clin Neuropharmacol. 2004;27(5):256]. Clin Neuropharmacol. 2004;27(4):153-156.
50. Pollak P, Tison F, Rascol O. Clozapine in drug induced psychosis in Parkinson’s disease: a randomised, placebo controlled study with open follow up. J Neurol Neurosurg Psychiatry. 2004;75(5):689-695.
51. Lutz UC, Sirfy A, Wiatr G, et al. Clozapine serum concentrations in dopamimetic psychosis in Parkinson’s disease and related disorders. Eur J Clin Pharmacol. 2014;70(12):1471-1476.
52. Fox SH, Katzenschlager R, Lim SY, et al. The Movement Disorder Society Evidence-Based Medicine Review Update: treatment for the motor symptoms of Parkinson’s disease. Mov Disord. 2011;26(suppl 3):S2-S41.
53. Lieberman JA, Saltz BL, Johns CA, et al. The effects of clozapine on tardive dyskinesia. Br J Psychiatry. 1991;158:503-510.
54. Naber D, Leppig M, Grohmann R, et al. Efficacy and adverse effects of clozapine in the treatment of schizophrenia and tardive dyskinesia—a retrospective study. Psychopharmacology (Berl). 1989;99(suppl):S73-S76.
55. Pinninti NR, Faden J, Adityanjee A. Are second-generation antipsychotics useful in tardive dystonia? Clin Neuropharmacol. 2015;38(5):183-197.
56. van Vugt JP, Siesling S, Vergeer M, et al. Clozapine versus placebo in Huntington’s disease: a double blind randomised comparative study. J Neurol Neurosurg Psychiatry. 1997;63(1):35-39.
57. Novartis Pharmaceuticals Corporation. Clozaril (clozapine). Prescribing information. http://clozaril.com/wp-content/themes/eyesite/pi/Clozaril-2015A507-10022015-Approved.pdf. Accessed September 2, 2016.
1. Newman WJ, Newman BM. Rediscovering clozapine: after a turbulent history, current guidance on initiating and monitoring. Current Psychiatry. 2016;15(7):42-46,48-49.
2. Newman BM, Newman WJ. Rediscovering clozapine: adverse effects develop—what should you do now? Current Psychiatry. 2016;15(8):40-46,48-49.
3. Newman WJ, Xiong GL, Barnhorst AV. Beta-blockers: off-label use in psychiatric disorders. Psychopharm Review. 2013;48(10):73-80.
4. Stafford RS. Regulating off-label drug use—rethinking the role of the FDA. N Engl J Med. 2008;358(14):1427-1429.
5. U.S. Food and Drug Administration. “Off-label” and investigational use of marketed drugs, biologics, and medical devices—information sheet. http://www.fda.gov/RegulatoryInformation/Guidances/ucm126486.htm. Updated January 25, 2016. Accessed November 24, 2015.
6. Radley DC, Finkelstein SN, Stafford RS. Off-label prescribing among office-based physicians. Arch Intern Med. 2006;166(9):1021-1026.
7. Suppes T, Webb A, Paul B, et al. Clinical outcome in a randomized 1-year trial of clozapine versus treatment as usual for patients with treatment-resistant illness and a history of mania. Am J Psychiatry. 1999;156(8):1164-1169.
8. Barbini B, Scherillo P, Benedetti F, et al. Response to clozapine in acute mania is more rapid than that of chlorpromazine. Int Clin Psychopharmacol. 1997;12(2):109-112.
9. Green AI, Tohen M, Patel JK, et al. Clozapine in the treatment of refractory psychotic mania. Am J Psychiatry. 2000;157(6):982-986.
10. Aksoy-Poyraz C, Turan ¸S, Demirel ÖF, et al. Effectiveness of ultra-rapid dose titration of clozapine for treatment-resistant bipolar mania: case series. Ther Adv Psychopharmacol. 2015;5(4):237-242.
11. Li XB, Tang YL, Wang CY, et al. Clozapine for treatment-resistant bipolar disorder: a systematic review. Bipolar Disord. 2015;17(3):235-247.
12. Nielsen J, Kane JM, Correll CU. Real-world effectiveness of clozapine in patients with bipolar disorder: results from a 2-year mirror-image study. Bipolar Disord. 2012;14(8):863-869.
13. Banov MD, Zarate CA Jr, Tohen M, et al. Clozapine therapy in refractory affective disorders: polarity predicts response in long-term follow-up. J Clin Psychiatry. 1994;55(7):295-300.
14. Ranjan R, Meltzer HY. Acute and long-term effectiveness of clozapine in treatment-resistant psychotic depression. Biol Psychiatry. 1996;40(4):253-258.
15. Dassa D, Kaladjian A, Azorin JM, et al. Clozapine in the treatment of psychotic refractory depression. Br J Psychiatry. 1993;163:822-824.
16. Jeyapaul P, Vieweg R. A case study evaluating the use of clozapine in depression with psychotic features. Ann Gen Psychiatry. 2006;5:20.
17. Havaki-Kontaxaki BJ, Ferentinos PP, Kontaxakis VP, et al. Low-dose clozapine monotherapy for recurring episodes of depression, hypersomnia and behavioural disturbances: a case report. Acta Neuropsychiatr. 2011;23(4):191-193.
18. Stoffers J, Völlm BA, Rücker G, et al. Pharmacological interventions for borderline personality disorder. Cochrane Database Syst Rev. 2010;(6):CD005653. doi: 10.1002/14651858.CD005653.pub2.
19. Frankenburg FR, Zanarini MC. Clozapine treatment of borderline patients: a preliminary study. Compr Psychiatry. 1993;34(6):402-405.
20. Frogley C, Anagnostakis K, Mitchell S, et al. A case series of clozapine for borderline personality disorder. Ann Clin Psychiatry. 2013;25(2):125-134.
21. Parker GF. Clozapine and borderline personality disorder. Psychiatr Serv. 2002;53(3):348-349.
22. Chengappa KNR, Baker RW, Sirri C. The successful use of clozapine in ameliorating severe self mutilation in a patient with borderline personality disorder. J Pers Disord. 1995;9(1):76-82.
23. Rutledge E, O’Regan M, Mohan D. Borderline personality disorder and clozapine. Ir J Psychol Med. 2007;24(1):40-41.
24. Vohra AK. Treatment of severe borderline personality disorder with clozapine. Indian J Psychiatry. 2010;52(3):267-269.
25. Buckley P, Thompson P, Way L, et al. Substance abuse among patients with treatment-resistant schizophrenia: characteristics and implications for clozapine therapy. Am J Psychiatry. 1994;151(3):385-389.
26. Drake RE, Xie H, McHugo GJ, et al. The effects of clozapine on alcohol and drug use disorders among patients with schizophrenia. Schizophr Bull. 2000;26(2):441-449.
27. Zimmet SV, Strous RD, Burgess ES, et al. Effects of clozapine on substance use in patients with schizophrenia and schizoaffective disorder: a retrospective survey. J Clin Psychopharmacol. 2000;20(1):94-98.
28. Green AI, Noordsy DL, Brunette MF, et al. Substance abuse and schizophrenia: pharmacotherapeutic intervention. J Subst Abuse Treat. 2008;34(1):61-71.
29. Green AI, Chau DT, Keung WM, et al. Clozapine reduces alcohol drinking in Syrian golden hamsters. Psychiatry Res. 2004;128(1):9-20.
30. Chau DT, Gulick D, Xie H, et al. Clozapine chronically suppresses alcohol drinking in Syrian golden hamsters. Neuropharmacology. 2010;58(2):351-356.
31. Brunette MF, Dawson R, O’Keefe CD, et al. A randomized trial of clozapine vs. other antipsychotics for cannabis use disorder in patients with schizophrenia. J Dual Diagn. 2011;7(1-2):50-63.
32. Kosten TA, Nestler EJ. Clozapine attenuates cocaine conditioned place preference. Life Sci. 1994;55(1):9-14.
33. Meltzer HY, Alphs L, Green AI, et al; International Suicide Prevention Trial Study Group. Clozapine treatment for suicidality in schizophrenia: International Suicide Prevention Trial (InterSePT) [Erratum in: Arch Gen Psychiatry. 2003;60(7):735]. Arch Gen Psychiatry. 2003;60(1):82-91.
34. Meltzer HY, Okayli G. Reduction of suicidality during clozapine treatment of neuroleptic-resistant schizophrenia: impact on risk-benefit assessment. Am J Psychiatry. 1995;152(2):183-190.
35. Sernyak MJ, Desai R, Stolar M, et al. Impact of clozapine on completed suicide. Am J Psychiatry. 2001;158(6):931-937.
36. Nordström P, Asberg M. Suicide risk and serotonin. Int Clin Psychopharmacol. 1992;6(suppl 6):12-21.
37. Vangala VR, Brown ES, Suppes T. Clozapine associated with decreased suicidality in bipolar disorder: a case report. Bipolar Disord. 1999;1(2):123-124.
38. Meltzer HY. The mechanism of action of novel antipsychotic drugs. Schizophr Bull. 1991;17(2):263-287.
39. Meltzer HY. An overview of the mechanism of action of clozapine. J Clin Psychiatry. 1994;55(suppl B):47-52.
40. Rabinowitz J, Avnon M, Rosenberg V. Effect of clozapine on physical and verbal aggression. Schizophr Res. 1996;22(3):249-255.
41. Spivak B, Roitman S, Vered Y, et al. Diminished suicidal and aggressive behavior, high plasma norepinephrine levels, and serum triglyceride levels in chronic neuroleptic-resistant schizophrenic patients maintained on clozapine. Clin Neuropharmacol. 1998;21(4):245-250.
42. Citrome L, Volavka J, Czobor P, et al. Effects of clozapine, olanzapine, risperidone, and haloperidol on hostility among patients with schizophrenia. Psychiatr Serv. 2001;52(11):1510-1514.
43. Volavka J, Czobor P, Nolan K, et al. Overt aggression and psychotic symptoms in patients with schizophrenia treated with clozapine, olanzapine, risperidone, or haloperidol. J Clin Psychopharmacol. 2004;24(2):225-228.
44. Krakowski MI, Czobar P, Citrome L, et al. Atypical antipsychotic agents in the treatment of violent patients with schizophrenia and schizoaffective disorder. Arch Gen Psychiatry. 2006;63(6):622-629.
45. Chiles JA, Davidson P, McBride D. Effects of clozapine on use of seclusion and restraint at a state hospital. Hosp Community Psychiatry. 1994;45(3):269-271.
46. Brown D, Larkin F, Sengupta S, et al. Clozapine: an effective treatment for seriously violent and psychopathic men with antisocial personality disorder in a UK high-security hospital. CNS Spectr. 2014;19(5):391-402.
47. Hammock R, Levine WR, Schroeder SR. Brief report: effects of clozapine on self-injurious behavior of two risperidone nonresponders with mental retardation. J Autism Dev Disord. 2001;31(1):109-113.
48. Hammock RG, Schroeder SR, Levine WR. The effect of clozapine on self-injurious behavior. J Autism Dev Disord. 1995;25(6):611-626.
49. Morgante L, Epifanio A, Spina E, et al. Quetiapine and clozapine in parkinsonian patients with dopaminergic psychosis [Erratum in: Clin Neuropharmacol. 2004;27(5):256]. Clin Neuropharmacol. 2004;27(4):153-156.
50. Pollak P, Tison F, Rascol O. Clozapine in drug induced psychosis in Parkinson’s disease: a randomised, placebo controlled study with open follow up. J Neurol Neurosurg Psychiatry. 2004;75(5):689-695.
51. Lutz UC, Sirfy A, Wiatr G, et al. Clozapine serum concentrations in dopamimetic psychosis in Parkinson’s disease and related disorders. Eur J Clin Pharmacol. 2014;70(12):1471-1476.
52. Fox SH, Katzenschlager R, Lim SY, et al. The Movement Disorder Society Evidence-Based Medicine Review Update: treatment for the motor symptoms of Parkinson’s disease. Mov Disord. 2011;26(suppl 3):S2-S41.
53. Lieberman JA, Saltz BL, Johns CA, et al. The effects of clozapine on tardive dyskinesia. Br J Psychiatry. 1991;158:503-510.
54. Naber D, Leppig M, Grohmann R, et al. Efficacy and adverse effects of clozapine in the treatment of schizophrenia and tardive dyskinesia—a retrospective study. Psychopharmacology (Berl). 1989;99(suppl):S73-S76.
55. Pinninti NR, Faden J, Adityanjee A. Are second-generation antipsychotics useful in tardive dystonia? Clin Neuropharmacol. 2015;38(5):183-197.
56. van Vugt JP, Siesling S, Vergeer M, et al. Clozapine versus placebo in Huntington’s disease: a double blind randomised comparative study. J Neurol Neurosurg Psychiatry. 1997;63(1):35-39.
57. Novartis Pharmaceuticals Corporation. Clozaril (clozapine). Prescribing information. http://clozaril.com/wp-content/themes/eyesite/pi/Clozaril-2015A507-10022015-Approved.pdf. Accessed September 2, 2016.

ADHD symptoms are stable, then a sudden relapse
CASE
Sudden deterioration
R, age 11, has attention-deficit/hyperactivity disorder (ADHD), combined type, and oppositional defiant disorder, which has been stable for more than a year on extended-release (ER) methylphenidate (brand name: Concerta), 54 mg/d (1.2 mg/kg). With combined pharmacotherapy and behavioral management, his symptoms of hyperactivity, inattention, and impulsivity improved at school and at home. He shows some academic gains as evidenced by improved achievement at school.
Over 2 months, R experiences a substantial deterioration in behavioral and academic performance. Along with core symptoms of ADHD, he begins to exhibit physical and verbal aggression. A report from school states that R has been using obscene language and destroying property, and has had episodes of provoked aggression toward his peers. His grades drop and he receives 2 school suspensions because of aggressive behavior.
What could be causing R’s ADHD symptoms to reemerge?
a) nonadherence to treatment
b) substance abuse
c) medication change
d) all of the above
The authors’ observations


Worsening of psychiatric symptoms in a stable patient is relatively common. Many factors can contribute to patient destabilization. Treatment nonadherence is a leading cause, along with psychosocial stressors and substance use (Table).
EVALUATION
Adherence confirmed
R is hyperactive and distracted during his visit, a clear deterioration from his baseline status. R is oppositional and defiant toward his mother during the session, but shows good social skills when communicating with the physician.
R’s mother reports that her son seldom forgets to take his medication, and she ensures that he is swallowing the pill, rather than chewing it. Data from the prescription drug-monitoring program show that the family is filling the prescriptions regularly. The ER methylphenidate dosage is raised to 72 mg/d. The clinicians provide psychoeducation about adherence to a medication regimen to R and his family. Also, his parents and teachers receive Vanderbilt Assessment Scales for ADHD to assess the symptoms in different settings.
At a follow-up visit a week later, R’s mother reports that her son continues to have problems in school and at home. The Vanderbilt scales reveal that R is having clinically significant problems with attention, hyperactivity, impulse control, and oppositional behavior.
A urine drug screen is ordered to rule out the possibility of a sudden deterioration of ADHD symptoms secondary to substance use disorder. To ensure compliance, we recommend that R take his medication at the school nurse’s office in the morning.
A week later
Although R takes his medication at school, he continues to show core symptoms of ADHD without improvement. The urine drug screen is negative. A physical examination does not reveal any medical illness. The treatment team calls the pharmacist to obtain a complete list of medications R is taking, who confirms that he is only receiving ER methylphenidate, 72 mg/d. The pharmacist also notes that R’s medication was switched from the brand-name drug to a generic 3 months ago because of a change in insurance coverage. This change coincided with the reemergence of his ADHD symptoms.
R’s mother reports that the new pills do not look like the old ones even before the dosage was raised. A new brand-necessary prescription is sent to the pharmacy. With the brand-name medication, R’s symptoms quickly improve, and remain improved when the dosage is decreased to the previous dosage of 54 mg/d.
With osmotic-controlled release oral delivery system (OROS) and outer coating of ER methylphenidate, how much drug is released immediately vs slow release?
a) 22% immediate release and 78% slow release
b) 78% immediate release and 22% slow release
c) 50% immediate release and 50% slow release
The authors’ observations
Generic substitution of a brand medication can result in worsening of symptoms and increased adverse effects. Possible bioequivalence issues can lead to failure of drug therapy.1
In 2013, the FDA determined that 2 specific generic formulations of ER methylphenidate do not have therapeutic equivalency to the brand-name medication, Concerta. The FDA stated, “Based on an analysis of data, FDA has concerns about whether or not two approved generic versions of Concerta tablets (methylphenidate hydrochloride extended-release tablets), used to treat attention-deficit hyperactivity disorder in adults and children, are therapeutically equiv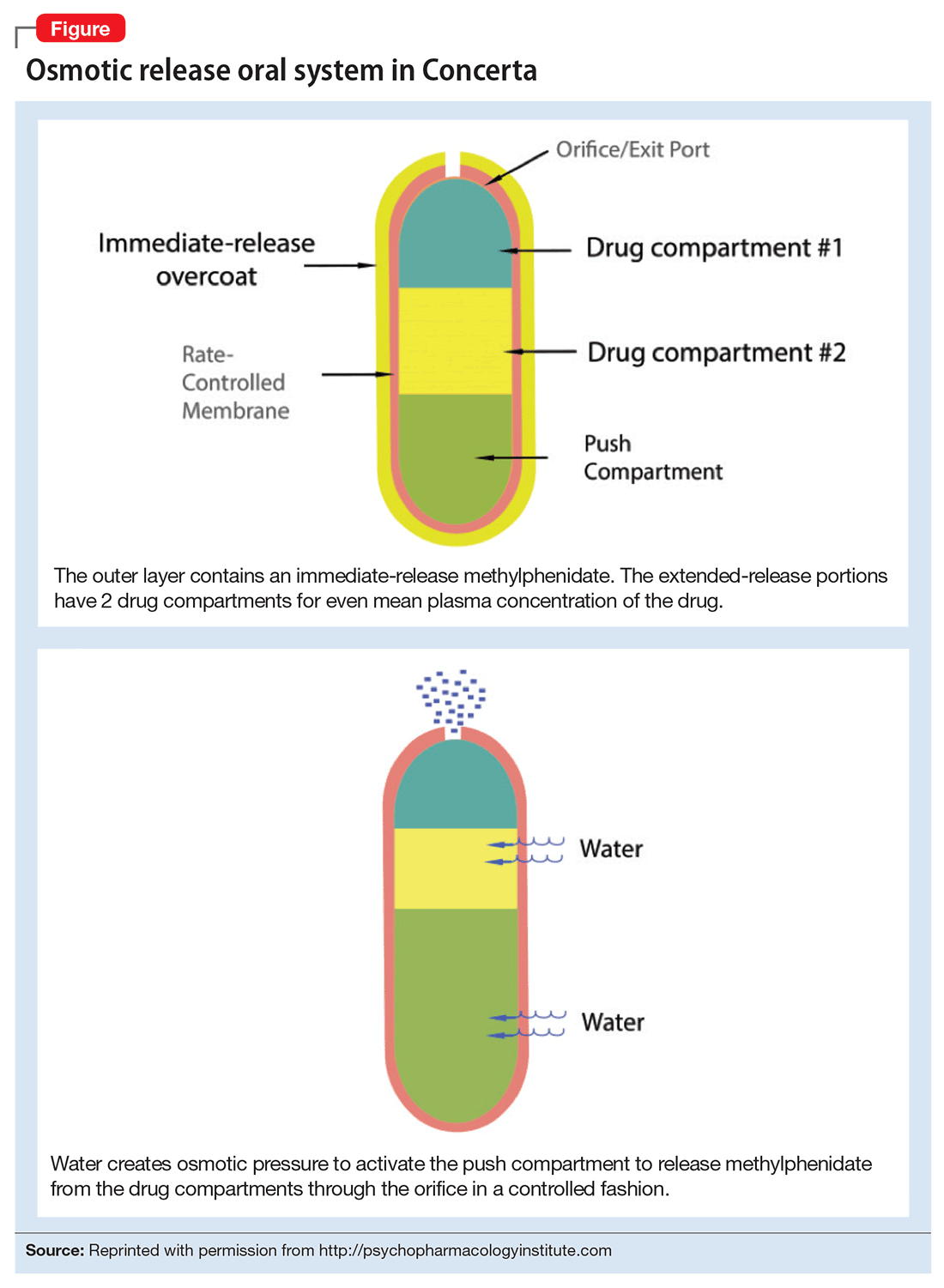
In an apparent confirmation of the FDA’s concerns, a case series of children and adolescents with ADHD observed that almost all of the patients showed symptom improvement when they switched from a non-OROS formulation to an OROS preparation at the same dosage.3
The OROS preparation is thought to provide more predictable medication delivery over an extended period of time (Figure). A patient taking an ER formulation without OROS might lose this benefit, which could lead to symptom destabilization, even if the patient is taking the medication as instructed.
Brand vs generic
Under FDA regulations, companies seeking approval for generic formulations of approved drugs must demonstrate that their products are the same as the brand-name drug in terms of:
- active ingredients
- strength
- dosage form
- route of administration
- packaging label.
In addition, the pharmaceutical company must demonstrate that the generic form is absorbed and distributed to the part of the body at which it has its effect at acceptably similar levels to the brand-name drug. All medications—new or generic, in clinical trials or approved, prescription or over-the-counter—must be manufactured under controlled conditions that assure product quality.
However, some studies have disputed this equivalency. In 1 study, patients with schizophrenia receiving generic olanzapine had lower serum concentration than patients with schizophrenia taking equivalent dosages of brand-name olanzapine.4 Similarly, studies comparing generic and brand-name venlafaxine showed significant differences in peak plasma concentration (Cmax)between generic and brand-name compounds.5
The FDA has considered upgrading the manufacturers’ warnings about the risk of generic medications, but has delayed the decision to 2017.6
FDA’s approval process for generic drugs
To receive approval of a generic formulation in the United States, the FDA requires that the generic drug should be compared with the corresponding brand-name drug in small crossover trials involving at least 24 to 36 healthy volunteers.
Bioequivalence is then established based on assessments of the rate of absorption (Cmax and area under the plasma concentration-time curve [AUC]). The FDA’s criteria are designed to achieve 90% confidence that the ratios of the test-to-reference log-transformed mean values for AUC and Cmax are within the interval of 80% to 125%. The FDA accepts −20% to 25% variation in Cmax and AUC in products that are considered bioequivalent. This is much less stringent than its −5% to 5% standard used for brand-name products. The FDA publishes a list of generic drugs that have been certified as bioequivalent, known as the “Orange Book.”5
Considerations when substituting generic medication
Because of the growing number of generic formulations of the same medication, generic–generic switches are becoming more commonplace. Theoretically, any 2 generic versions of the same medication can have a variation of up to 40% in AUC and Cmax. Generic medications are tested in healthy human controls through single-dose studies, which raises concerns about their applicability to the entire patient population.
Bioequivalence. It is a matter of debate whether bioequivalence translates to therapeutic equivalency. For medications with a narrow therapeutic index, the FDA has accepted that these 2 phenomena are not necessarily linked. With the exception of a few medications, including lithium and some anticonvulsants such as divalproex sodium and carbamazepine, serum level of the medications usually does not predict clinical response.
Inert ingredients. Generic medications can include inert ingredients (excipients) that are different from those in their branded counterparts. Some of these inactive ingredients can cause adverse effects. A study comparing paroxetine mesylate and paroxetine hydrochloride showed differences in bioequivalence and clinical efficacy.7
In some cases, brand-to-generic substitution can thwart clinical progress in a stable patient. This small change in the medication could destabilize the patient’s condition, which, in turn, may lead to unnecessary and significant social and financial burdens on the patient’s family, school, community, and the health care system.
Recommendations
In the event of a change in clinical response, clinicians first should evaluate adherence and explore other factors, such as biological, psychological, medical, and social issues. Adherence can be adversely affected by a change in the physical characteristics of the pill. Prescribers should remain cognizant of brand–generic and generic–generic switches. It may be reasonable to adjust the dosage of the new generic medication to address changes in clinical effectiveness.
If these strategies are ineffective, consider switching to a brand-name medication. Write “Dispense As Written” on the prescription to ensure delivery of the branded medication or a specific generic version of the medication.
An insurance company might require prior authorization to approve payment for the brand medication. To save time, use electronic forms or fax for communicating with the insurance company. Adding references to FDA statements and research papers, along with the patient’s history and presentations, would be prudent to demonstrate doubts about efficacy of the generic medication.
1. Atif M, Azeem M, Sarwar MR. Potential problems and recommendations regarding substitution of generic antiepileptic drugs: a systematic review of literature. Springerplus. 2016;5:182. doi: 10.1186/s40064-016-1824-2.
2. U.S. Food and Drug Administration. Methylphenidate hydrochloride extended release tablets (generic Concerta) made by Mallinckrodt and Kudco. http://www.fda.gov/Drugs/DrugSafety/ucm422568.htm. Updated November 13, 2014. Accessed August 29, 2016.
3. Lally MD, Kral MC, Boan AD. Not all generic Concerta is created equal: comparison of OROS versus non-OROS for the treatment of ADHD [published online October 14, 2015]. Clin Pediatr (Phila). doi:10.1177/0009922815611647.
4. Italiano DD, Bruno A, Santoro V, et al. Generic olanzapine substitution in patients with schizophrenia: assessment of serum concentrations and therapeutic response after switching. Ther Drug Monit. 2015;37(6):827-830.
5. Borgheini GG. The bioequivalence and therapeutic efficacy of generic versus brand-name psychoactive drugs. Clin Ther. 2003;25(6):1578-1592.
6. Thomas K. F.D.A. delays rule on generic drug labels. http://www.nytimes.com/2016/05/20/business/fda-delays-rule-on-generic-drug-labels.html. Published May 19, 2016. Accessed August 29, 2016.
7. Pae CU, Misra A, Ham BJ, et al. Paroxetine mesylate: comparable to paroxetine hydrochloride? Expert Opin Pharmacother. 2010;11(2):185-193
CASE
Sudden deterioration
R, age 11, has attention-deficit/hyperactivity disorder (ADHD), combined type, and oppositional defiant disorder, which has been stable for more than a year on extended-release (ER) methylphenidate (brand name: Concerta), 54 mg/d (1.2 mg/kg). With combined pharmacotherapy and behavioral management, his symptoms of hyperactivity, inattention, and impulsivity improved at school and at home. He shows some academic gains as evidenced by improved achievement at school.
Over 2 months, R experiences a substantial deterioration in behavioral and academic performance. Along with core symptoms of ADHD, he begins to exhibit physical and verbal aggression. A report from school states that R has been using obscene language and destroying property, and has had episodes of provoked aggression toward his peers. His grades drop and he receives 2 school suspensions because of aggressive behavior.
What could be causing R’s ADHD symptoms to reemerge?
a) nonadherence to treatment
b) substance abuse
c) medication change
d) all of the above
The authors’ observations
Worsening of psychiatric symptoms in a stable patient is relatively common. Many factors can contribute to patient destabilization. Treatment nonadherence is a leading cause, along with psychosocial stressors and substance use (Table).
EVALUATION
Adherence confirmed
R is hyperactive and distracted during his visit, a clear deterioration from his baseline status. R is oppositional and defiant toward his mother during the session, but shows good social skills when communicating with the physician.
R’s mother reports that her son seldom forgets to take his medication, and she ensures that he is swallowing the pill, rather than chewing it. Data from the prescription drug-monitoring program show that the family is filling the prescriptions regularly. The ER methylphenidate dosage is raised to 72 mg/d. The clinicians provide psychoeducation about adherence to a medication regimen to R and his family. Also, his parents and teachers receive Vanderbilt Assessment Scales for ADHD to assess the symptoms in different settings.
At a follow-up visit a week later, R’s mother reports that her son continues to have problems in school and at home. The Vanderbilt scales reveal that R is having clinically significant problems with attention, hyperactivity, impulse control, and oppositional behavior.
A urine drug screen is ordered to rule out the possibility of a sudden deterioration of ADHD symptoms secondary to substance use disorder. To ensure compliance, we recommend that R take his medication at the school nurse’s office in the morning.
A week later
Although R takes his medication at school, he continues to show core symptoms of ADHD without improvement. The urine drug screen is negative. A physical examination does not reveal any medical illness. The treatment team calls the pharmacist to obtain a complete list of medications R is taking, who confirms that he is only receiving ER methylphenidate, 72 mg/d. The pharmacist also notes that R’s medication was switched from the brand-name drug to a generic 3 months ago because of a change in insurance coverage. This change coincided with the reemergence of his ADHD symptoms.
R’s mother reports that the new pills do not look like the old ones even before the dosage was raised. A new brand-necessary prescription is sent to the pharmacy. With the brand-name medication, R’s symptoms quickly improve, and remain improved when the dosage is decreased to the previous dosage of 54 mg/d.
With osmotic-controlled release oral delivery system (OROS) and outer coating of ER methylphenidate, how much drug is released immediately vs slow release?
a) 22% immediate release and 78% slow release
b) 78% immediate release and 22% slow release
c) 50% immediate release and 50% slow release
The authors’ observations
Generic substitution of a brand medication can result in worsening of symptoms and increased adverse effects. Possible bioequivalence issues can lead to failure of drug therapy.1
In 2013, the FDA determined that 2 specific generic formulations of ER methylphenidate do not have therapeutic equivalency to the brand-name medication, Concerta. The FDA stated, “Based on an analysis of data, FDA has concerns about whether or not two approved generic versions of Concerta tablets (methylphenidate hydrochloride extended-release tablets), used to treat attention-deficit hyperactivity disorder in adults and children, are therapeutically equiv
In an apparent confirmation of the FDA’s concerns, a case series of children and adolescents with ADHD observed that almost all of the patients showed symptom improvement when they switched from a non-OROS formulation to an OROS preparation at the same dosage.3
The OROS preparation is thought to provide more predictable medication delivery over an extended period of time (Figure). A patient taking an ER formulation without OROS might lose this benefit, which could lead to symptom destabilization, even if the patient is taking the medication as instructed.
Brand vs generic
Under FDA regulations, companies seeking approval for generic formulations of approved drugs must demonstrate that their products are the same as the brand-name drug in terms of:
- active ingredients
- strength
- dosage form
- route of administration
- packaging label.
In addition, the pharmaceutical company must demonstrate that the generic form is absorbed and distributed to the part of the body at which it has its effect at acceptably similar levels to the brand-name drug. All medications—new or generic, in clinical trials or approved, prescription or over-the-counter—must be manufactured under controlled conditions that assure product quality.
However, some studies have disputed this equivalency. In 1 study, patients with schizophrenia receiving generic olanzapine had lower serum concentration than patients with schizophrenia taking equivalent dosages of brand-name olanzapine.4 Similarly, studies comparing generic and brand-name venlafaxine showed significant differences in peak plasma concentration (Cmax)between generic and brand-name compounds.5
The FDA has considered upgrading the manufacturers’ warnings about the risk of generic medications, but has delayed the decision to 2017.6
FDA’s approval process for generic drugs
To receive approval of a generic formulation in the United States, the FDA requires that the generic drug should be compared with the corresponding brand-name drug in small crossover trials involving at least 24 to 36 healthy volunteers.
Bioequivalence is then established based on assessments of the rate of absorption (Cmax and area under the plasma concentration-time curve [AUC]). The FDA’s criteria are designed to achieve 90% confidence that the ratios of the test-to-reference log-transformed mean values for AUC and Cmax are within the interval of 80% to 125%. The FDA accepts −20% to 25% variation in Cmax and AUC in products that are considered bioequivalent. This is much less stringent than its −5% to 5% standard used for brand-name products. The FDA publishes a list of generic drugs that have been certified as bioequivalent, known as the “Orange Book.”5
Considerations when substituting generic medication
Because of the growing number of generic formulations of the same medication, generic–generic switches are becoming more commonplace. Theoretically, any 2 generic versions of the same medication can have a variation of up to 40% in AUC and Cmax. Generic medications are tested in healthy human controls through single-dose studies, which raises concerns about their applicability to the entire patient population.
Bioequivalence. It is a matter of debate whether bioequivalence translates to therapeutic equivalency. For medications with a narrow therapeutic index, the FDA has accepted that these 2 phenomena are not necessarily linked. With the exception of a few medications, including lithium and some anticonvulsants such as divalproex sodium and carbamazepine, serum level of the medications usually does not predict clinical response.
Inert ingredients. Generic medications can include inert ingredients (excipients) that are different from those in their branded counterparts. Some of these inactive ingredients can cause adverse effects. A study comparing paroxetine mesylate and paroxetine hydrochloride showed differences in bioequivalence and clinical efficacy.7
In some cases, brand-to-generic substitution can thwart clinical progress in a stable patient. This small change in the medication could destabilize the patient’s condition, which, in turn, may lead to unnecessary and significant social and financial burdens on the patient’s family, school, community, and the health care system.
Recommendations
In the event of a change in clinical response, clinicians first should evaluate adherence and explore other factors, such as biological, psychological, medical, and social issues. Adherence can be adversely affected by a change in the physical characteristics of the pill. Prescribers should remain cognizant of brand–generic and generic–generic switches. It may be reasonable to adjust the dosage of the new generic medication to address changes in clinical effectiveness.
If these strategies are ineffective, consider switching to a brand-name medication. Write “Dispense As Written” on the prescription to ensure delivery of the branded medication or a specific generic version of the medication.
An insurance company might require prior authorization to approve payment for the brand medication. To save time, use electronic forms or fax for communicating with the insurance company. Adding references to FDA statements and research papers, along with the patient’s history and presentations, would be prudent to demonstrate doubts about efficacy of the generic medication.
CASE
Sudden deterioration
R, age 11, has attention-deficit/hyperactivity disorder (ADHD), combined type, and oppositional defiant disorder, which has been stable for more than a year on extended-release (ER) methylphenidate (brand name: Concerta), 54 mg/d (1.2 mg/kg). With combined pharmacotherapy and behavioral management, his symptoms of hyperactivity, inattention, and impulsivity improved at school and at home. He shows some academic gains as evidenced by improved achievement at school.
Over 2 months, R experiences a substantial deterioration in behavioral and academic performance. Along with core symptoms of ADHD, he begins to exhibit physical and verbal aggression. A report from school states that R has been using obscene language and destroying property, and has had episodes of provoked aggression toward his peers. His grades drop and he receives 2 school suspensions because of aggressive behavior.
What could be causing R’s ADHD symptoms to reemerge?
a) nonadherence to treatment
b) substance abuse
c) medication change
d) all of the above
The authors’ observations
Worsening of psychiatric symptoms in a stable patient is relatively common. Many factors can contribute to patient destabilization. Treatment nonadherence is a leading cause, along with psychosocial stressors and substance use (Table).
EVALUATION
Adherence confirmed
R is hyperactive and distracted during his visit, a clear deterioration from his baseline status. R is oppositional and defiant toward his mother during the session, but shows good social skills when communicating with the physician.
R’s mother reports that her son seldom forgets to take his medication, and she ensures that he is swallowing the pill, rather than chewing it. Data from the prescription drug-monitoring program show that the family is filling the prescriptions regularly. The ER methylphenidate dosage is raised to 72 mg/d. The clinicians provide psychoeducation about adherence to a medication regimen to R and his family. Also, his parents and teachers receive Vanderbilt Assessment Scales for ADHD to assess the symptoms in different settings.
At a follow-up visit a week later, R’s mother reports that her son continues to have problems in school and at home. The Vanderbilt scales reveal that R is having clinically significant problems with attention, hyperactivity, impulse control, and oppositional behavior.
A urine drug screen is ordered to rule out the possibility of a sudden deterioration of ADHD symptoms secondary to substance use disorder. To ensure compliance, we recommend that R take his medication at the school nurse’s office in the morning.
A week later
Although R takes his medication at school, he continues to show core symptoms of ADHD without improvement. The urine drug screen is negative. A physical examination does not reveal any medical illness. The treatment team calls the pharmacist to obtain a complete list of medications R is taking, who confirms that he is only receiving ER methylphenidate, 72 mg/d. The pharmacist also notes that R’s medication was switched from the brand-name drug to a generic 3 months ago because of a change in insurance coverage. This change coincided with the reemergence of his ADHD symptoms.
R’s mother reports that the new pills do not look like the old ones even before the dosage was raised. A new brand-necessary prescription is sent to the pharmacy. With the brand-name medication, R’s symptoms quickly improve, and remain improved when the dosage is decreased to the previous dosage of 54 mg/d.
With osmotic-controlled release oral delivery system (OROS) and outer coating of ER methylphenidate, how much drug is released immediately vs slow release?
a) 22% immediate release and 78% slow release
b) 78% immediate release and 22% slow release
c) 50% immediate release and 50% slow release
The authors’ observations
Generic substitution of a brand medication can result in worsening of symptoms and increased adverse effects. Possible bioequivalence issues can lead to failure of drug therapy.1
In 2013, the FDA determined that 2 specific generic formulations of ER methylphenidate do not have therapeutic equivalency to the brand-name medication, Concerta. The FDA stated, “Based on an analysis of data, FDA has concerns about whether or not two approved generic versions of Concerta tablets (methylphenidate hydrochloride extended-release tablets), used to treat attention-deficit hyperactivity disorder in adults and children, are therapeutically equiv
In an apparent confirmation of the FDA’s concerns, a case series of children and adolescents with ADHD observed that almost all of the patients showed symptom improvement when they switched from a non-OROS formulation to an OROS preparation at the same dosage.3
The OROS preparation is thought to provide more predictable medication delivery over an extended period of time (Figure). A patient taking an ER formulation without OROS might lose this benefit, which could lead to symptom destabilization, even if the patient is taking the medication as instructed.
Brand vs generic
Under FDA regulations, companies seeking approval for generic formulations of approved drugs must demonstrate that their products are the same as the brand-name drug in terms of:
- active ingredients
- strength
- dosage form
- route of administration
- packaging label.
In addition, the pharmaceutical company must demonstrate that the generic form is absorbed and distributed to the part of the body at which it has its effect at acceptably similar levels to the brand-name drug. All medications—new or generic, in clinical trials or approved, prescription or over-the-counter—must be manufactured under controlled conditions that assure product quality.
However, some studies have disputed this equivalency. In 1 study, patients with schizophrenia receiving generic olanzapine had lower serum concentration than patients with schizophrenia taking equivalent dosages of brand-name olanzapine.4 Similarly, studies comparing generic and brand-name venlafaxine showed significant differences in peak plasma concentration (Cmax)between generic and brand-name compounds.5
The FDA has considered upgrading the manufacturers’ warnings about the risk of generic medications, but has delayed the decision to 2017.6
FDA’s approval process for generic drugs
To receive approval of a generic formulation in the United States, the FDA requires that the generic drug should be compared with the corresponding brand-name drug in small crossover trials involving at least 24 to 36 healthy volunteers.
Bioequivalence is then established based on assessments of the rate of absorption (Cmax and area under the plasma concentration-time curve [AUC]). The FDA’s criteria are designed to achieve 90% confidence that the ratios of the test-to-reference log-transformed mean values for AUC and Cmax are within the interval of 80% to 125%. The FDA accepts −20% to 25% variation in Cmax and AUC in products that are considered bioequivalent. This is much less stringent than its −5% to 5% standard used for brand-name products. The FDA publishes a list of generic drugs that have been certified as bioequivalent, known as the “Orange Book.”5
Considerations when substituting generic medication
Because of the growing number of generic formulations of the same medication, generic–generic switches are becoming more commonplace. Theoretically, any 2 generic versions of the same medication can have a variation of up to 40% in AUC and Cmax. Generic medications are tested in healthy human controls through single-dose studies, which raises concerns about their applicability to the entire patient population.
Bioequivalence. It is a matter of debate whether bioequivalence translates to therapeutic equivalency. For medications with a narrow therapeutic index, the FDA has accepted that these 2 phenomena are not necessarily linked. With the exception of a few medications, including lithium and some anticonvulsants such as divalproex sodium and carbamazepine, serum level of the medications usually does not predict clinical response.
Inert ingredients. Generic medications can include inert ingredients (excipients) that are different from those in their branded counterparts. Some of these inactive ingredients can cause adverse effects. A study comparing paroxetine mesylate and paroxetine hydrochloride showed differences in bioequivalence and clinical efficacy.7
In some cases, brand-to-generic substitution can thwart clinical progress in a stable patient. This small change in the medication could destabilize the patient’s condition, which, in turn, may lead to unnecessary and significant social and financial burdens on the patient’s family, school, community, and the health care system.
Recommendations
In the event of a change in clinical response, clinicians first should evaluate adherence and explore other factors, such as biological, psychological, medical, and social issues. Adherence can be adversely affected by a change in the physical characteristics of the pill. Prescribers should remain cognizant of brand–generic and generic–generic switches. It may be reasonable to adjust the dosage of the new generic medication to address changes in clinical effectiveness.
If these strategies are ineffective, consider switching to a brand-name medication. Write “Dispense As Written” on the prescription to ensure delivery of the branded medication or a specific generic version of the medication.
An insurance company might require prior authorization to approve payment for the brand medication. To save time, use electronic forms or fax for communicating with the insurance company. Adding references to FDA statements and research papers, along with the patient’s history and presentations, would be prudent to demonstrate doubts about efficacy of the generic medication.
1. Atif M, Azeem M, Sarwar MR. Potential problems and recommendations regarding substitution of generic antiepileptic drugs: a systematic review of literature. Springerplus. 2016;5:182. doi: 10.1186/s40064-016-1824-2.
2. U.S. Food and Drug Administration. Methylphenidate hydrochloride extended release tablets (generic Concerta) made by Mallinckrodt and Kudco. http://www.fda.gov/Drugs/DrugSafety/ucm422568.htm. Updated November 13, 2014. Accessed August 29, 2016.
3. Lally MD, Kral MC, Boan AD. Not all generic Concerta is created equal: comparison of OROS versus non-OROS for the treatment of ADHD [published online October 14, 2015]. Clin Pediatr (Phila). doi:10.1177/0009922815611647.
4. Italiano DD, Bruno A, Santoro V, et al. Generic olanzapine substitution in patients with schizophrenia: assessment of serum concentrations and therapeutic response after switching. Ther Drug Monit. 2015;37(6):827-830.
5. Borgheini GG. The bioequivalence and therapeutic efficacy of generic versus brand-name psychoactive drugs. Clin Ther. 2003;25(6):1578-1592.
6. Thomas K. F.D.A. delays rule on generic drug labels. http://www.nytimes.com/2016/05/20/business/fda-delays-rule-on-generic-drug-labels.html. Published May 19, 2016. Accessed August 29, 2016.
7. Pae CU, Misra A, Ham BJ, et al. Paroxetine mesylate: comparable to paroxetine hydrochloride? Expert Opin Pharmacother. 2010;11(2):185-193
1. Atif M, Azeem M, Sarwar MR. Potential problems and recommendations regarding substitution of generic antiepileptic drugs: a systematic review of literature. Springerplus. 2016;5:182. doi: 10.1186/s40064-016-1824-2.
2. U.S. Food and Drug Administration. Methylphenidate hydrochloride extended release tablets (generic Concerta) made by Mallinckrodt and Kudco. http://www.fda.gov/Drugs/DrugSafety/ucm422568.htm. Updated November 13, 2014. Accessed August 29, 2016.
3. Lally MD, Kral MC, Boan AD. Not all generic Concerta is created equal: comparison of OROS versus non-OROS for the treatment of ADHD [published online October 14, 2015]. Clin Pediatr (Phila). doi:10.1177/0009922815611647.
4. Italiano DD, Bruno A, Santoro V, et al. Generic olanzapine substitution in patients with schizophrenia: assessment of serum concentrations and therapeutic response after switching. Ther Drug Monit. 2015;37(6):827-830.
5. Borgheini GG. The bioequivalence and therapeutic efficacy of generic versus brand-name psychoactive drugs. Clin Ther. 2003;25(6):1578-1592.
6. Thomas K. F.D.A. delays rule on generic drug labels. http://www.nytimes.com/2016/05/20/business/fda-delays-rule-on-generic-drug-labels.html. Published May 19, 2016. Accessed August 29, 2016.
7. Pae CU, Misra A, Ham BJ, et al. Paroxetine mesylate: comparable to paroxetine hydrochloride? Expert Opin Pharmacother. 2010;11(2):185-193
High-value intervention: Providing colorectal cancer screening
Cancer screening is an important example of secondary prevention—the aim being to detect disease at an early stage, when treatment can prevent symptomatic disease. Over the years, screening tests for breast cancer, colorectal cancer (CRC), cervical cancer, and, most recently, lung cancer have been developed and recommended by the U.S. Preventive Services Task Force (USPSTF). Among breast cancer, cervical cancer, and CRC, the screening rate for CRC remains lowest, at 58.6%.1
The importance of screening for CRC is highlighted by the facts that:
- CRC is the third most commonly diagnosed form of cancer in the United States among both men and women
- CRC is the second leading cause of cancer-related death.2
The overall decrease in the incidence of CRC in the United States has been credited to improvements in screening and removal of potentially precancerous lesions.3
Harmful disparity puts the mentally ill at exceptional risk
Screening patterns for CRC among patients with mental illness are poorly characterized, but it is known that the overall cancer screening rate among patients with severe psychiatric illness lags significantly behind the rate in the general population.4,5 In addition, studies have shown that mortality among patients with CRC who have a mental disorder is elevated, compared with CRC patients who do not have a psychiatric diagnosis.6
Why this disparity? It might be that CRC is more likely to be diagnosed at an advanced stage among these patients, or that they are less likely to receive cancer treatment after diagnosis, or are more likely to have a longer delay between diagnosis and initial treatment than patients who do not have a psychiatric diagnosis.7
Regardless, psychiatric practitioners can make a significant impact on reducing this health disparity by leveraging their unique therapeutic relationship to educate patients about screening options and dispel myths about cancer screening. In this article, we outline practical strategies for CRC screening and weigh the advantages and disadvantages for the use of several tools and guidelines in psychiatric patients.
What is the pathogenesis of colorectal cancer?
Most cases of CRC evolve from polyps, abnormal growths on the lining of the colon or rectum. Constituting an estimated 96% of all polyps, adenomas are by far the most common form in the colon and rectum.
Adenomas also are most likely to transform over time to dysplasia, and then to progress to cancer.8 Although all adenomas have malignant potential, <10% evolve to adenocarcinoma. This proposed adenoma➝carcinoma sequence is not well understood; however, it is known that CRC usually develops slowly—over 10 to 15 years.9 Detection and removal of adenomas and treatable, localized carcinomas form the basis of screening for CRC.
Risk factors for colorectal cancer
A number of risk factors for CRC have been identified.
Specific heritable conditions, such as Lynch syndrome and familial adenomatous polyposis, pose the greatest risk of CRC, particularly at younger ages and compared with people without such a history.10
Family history. One of the strongest risk factors for CRC remains a family history of the disease. People who have a first-degree relative with a diagnosis of CRC are at 2 to 3 times the risk of CRC, compared with people without a family history of the disease. This risk increases further if multiple family members are affected or if the diagnosis was made in a relative at a young age.11,12
Other non-modifiable risk factors include a personal history of inflammatory bowel disease, type 2 diabetes mellitus, male sex, African American heritage, and increasing age.13-15
Common modifiable risk factors include obesity, smoking, and alcohol consumption.16-18
What is the role of screening?
CRC screening is only appropriate for patients who are asymptomatic. CRC generally is asymptomatic in early stages. Prognosis also is most favorable when CRC is detected in the asymptomatic stage.
As lesions of CRC grow, the presentation might include hematochezia, melena, abdominal pain, weight loss, occult anemia, constipation or diarrhea, and changes in stool caliber.19 These signs and symptoms are not highly specific for CRC, however, and might be indicative of other gastrointestinal pathology, including inflammatory bowel disease, diverticulitis, irritable bowel syndrome, infectious colitis, hemorrhoids, and mesenteric ischemia.
Symptomatic patients should be referred directly for diagnostic evaluation. Colonoscopy with biopsy is the standard for diagnosing CRC. Once a diagnosis of CRC is made, patients should be referred to a specialist to discuss treatment; options largely depend on the stage of the cancer at diagnosis.
What screening tests are available?
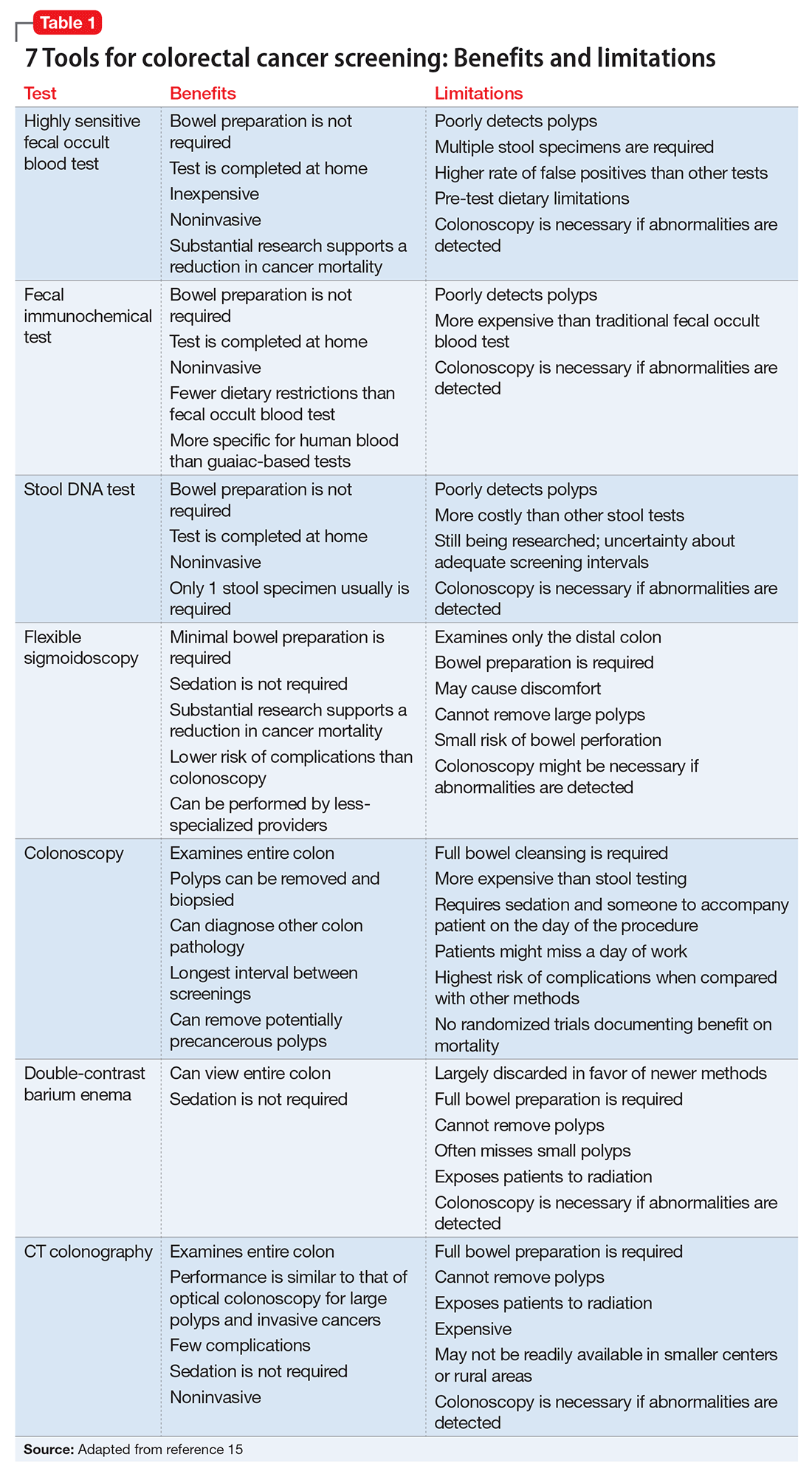
Unlike screening for other cancers, there are a number of reasonable options for CRC screening; Table 115 compares their relative pros and cons. Each test has its benefits and drawbacks, allowing the screening strategy to be customized based on patient preference and characteristics, but this variability also can lead to confusion by patient and provider about those options.
Stool-based tests detect trace amounts of blood from early-stage treatable cancers. Highly sensitive fecal occult blood testing (FOBT) has been shown specifically to decrease mortality from CRC.20 Stool-based tests are inexpensive and noninvasive, but require:
- more frequent testing
- that the patient collect the stool specimen
- follow-up colonoscopy when test results are positive.
Endoscopic and imaging tests detect polyps and early-stage treatable cancers; all require some degree of bowel preparation, and some require sedation. Testing intervals vary but, as a group, are longer than the interval between stool-based tests because polyps grow slowly. Because colonoscopy with biopsy is the preferred screening method for diagnosing CRC, it is the only screening option that also is a diagnostic procedure.
Where can screening guidelines be found?
Several professional organizations have developed guidelines for CRC screening. The 2 major 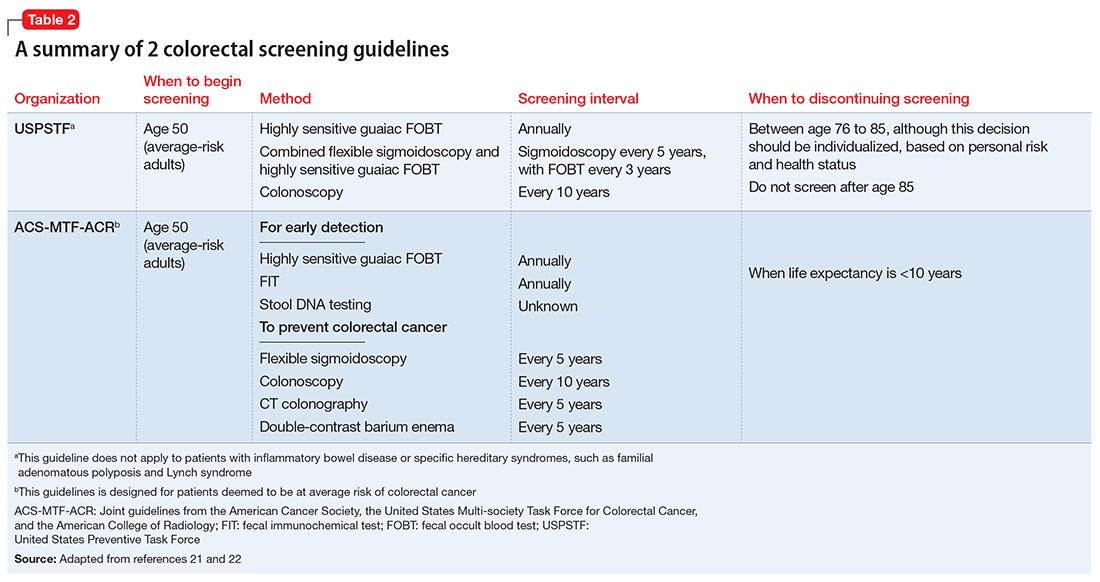
An update to both guidelines was released in 2008. Table 221,22 summarizes their recommendations.
Both guidelines recommend that screening begin at age 50 (Box). The primary differences between the 2 guidelines lie in the scope of recommended options for screening and the time frame for discontinuing screening:
- USPSTF requires a higher level of evidence for screening options and limits recommended options to FOBT, sigmoidoscopy combined with FOBT, and colonoscopy.
- ACS-MSTF-ACR emphasizes options that detect premalignant polyps, and generally is more inclusive of testing options; it also delineates tests as useful for either (1) early detection of cancer (stool-based studies) or (2) cancer prevention (endoscopic and imaging tests).
On the question of when to stop screening, ACS-MSTF-ACR bases its recommendations on life expectancy; USPSTF sets a specific age for ending screening.21,22
Recommendations of a third entity, the American College of Gastroenterology (ACG), are similar to those of ACS-MSTF-ACR; however, ACG (1) recommends beginning screening African American patients at age 45 because of their increased risk of CRC and (2) gives preference to colonoscopy as the preferred screening modality.23
Guidelines vary for high-risk patients (those with a history of familial adenomatous polyposis or another inherited syndrome associated with CRC; those with a family history of CRC in the young; those with a history of radiation exposure, history of CRC, or inflammatory bowel disease; and those with several first-degree relatives with CRC). Patients who fall into any of these categories should be referred for specialty care to establish the time of initial screening and the interval of subsequent screening.
CRC screening in the presence of psychiatric illness
Psychiatrists have an opportunity to support their patients when considering potentially confusing CRC screening recommendations. This opportunity might occur during a discussion about general preventive care, or a patient might come to an appointment after visiting a primary care provider, and ask for advice about screening options.
The potential benefits of CRC screening are negated if a patient is unable or unwilling to complete the test or undergo timely follow-up of positive results. It is important, therefore, to individualize screening recommendations—keeping in mind the degree of impairment from mental illness and the patient’s preferences and reliability to engage in follow-up. To date, there are no agreed-on screening guidelines specifically for patients with comorbid mental illness.
Adapting USPSTF guidelines for CRC screening of average-risk patients with mental illness, we offer the following recommendations:
Recommend screening. Begin routine screening at age 50. Patients with well-controlled or mild symptoms should be screened with a stool study with or without flexible sigmoidoscopy. Stool studies are safe, noninvasive, and require no bowel preparation; when used alone, however, they need to be performed yearly.
Screening accuracy is increased when a stool-based test is combined with flexible sigmoidoscopy; screening then can be performed less often. Unlike colonoscopy, flexible sigmoidoscopy does not involve sedation; a high-functioning patient might find this appealing and tolerate the greater frequency of screening. On the other hand, some patients might not accept the inconvenience of collecting the stool sample with the kit provided and returning it to the lab for processing.
Manage psychiatric illness optimally. For a patient with moderate or severe psychiatric symptoms, first attempt to optimize treatment of the underlying psychiatric condition before establishing a CRC screening program. If control of symptoms is likely to improve over the next 1 or 2 visits, it might be reasonable to defer screening until symptoms are better controlled and then reassess the patient before making specific screening recommendations. Screening should not be delayed, however, if significant improvement in symptoms is not expected in the near future. Lengthy delay might lead to failure in initiating screening at all.
We recommend that patients with persistent moderate or severe symptoms be screened with traditional colonoscopy. The sedation associated with colonoscopy (1) may be preferable to some patients with more severe illness and (2) allows for screening and diagnostic biopsy if needed during the same procedure. Screening with colonoscopy also:
- avoids the yearly adherence to a screening program that is needed with stool cards alone
- does not rely on patients collecting and returning stool kits for processing.
A potential challenge for patients with limited social support is the requirement to have someone accompany the patient on the day of colonoscopy.
Take steps to improve the screening rate. In addition to specific recommendations based on symptom severity, there are systems-level interventions that should be considered to improve the screening rate. These include:
- addressing transportation issues that are a barrier to screening
- considering the use of health navigators or peer advocates to help guide patients through the sometimes complex systems of care.
A more comprehensive systems-level intervention for mental health clinics that work primarily with persistent and severe mentally ill populations might include employing a care coordinator to organize referrals to primary care or even exploring reverse integration. In reverse integration, primary care providers co-locate within the mental health clinic, (1) allowing for “one-stop shopping” of mental health and primary care needs and (2) facilitating collaboration and shared treatment planning between primary care and mental health for complex patients.
2. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63(1):11-30.
3. Edwards BK, Ward E, Kohler BA, et al. Annual report to the nation on the status of cancer, 1975-2006, featuring colorectal cancer trends and impact of interventions (risk factors, screening, and treatment) to reduce future rates. Cancer. 2010;116(3):544-573.
4. Miller E, Lasser KE, Becker AE. Breast and cervical cancer screening for women with mental illness: patient and provider perspectives on improving linkages between primary care and mental health. Arch Womens Ment Health. 2007;10(5):189-197.
5. Howard LM, Barley EA, Davies E, et al. Cancer diagnosis in people with severe mental illness: practical and ethical issues. Lancet Oncol. 2010;11(8):797-804.
6. Baillargeon J, Kuo YF, Lin YL, et al. Effect of mental disorders on diagnosis, treatment, and survival of older adults with colon cancer. J Am Geriatr Soc. 2011;59(7):1268-1273.
7. Robertson R, Campbell NC, Smith S, et al. Factors influencing time from presentation to treatment of colorectal and breast cancer in urban and rural areas. Br J Cancer. 2004;90(8):1479-1485.
8. Stewart SL, Wike JM, Kato I, et al. A population-based study of colorectal cancer histology in the United States, 1998-2001. Cancer. 2006;107(suppl 5):1128-1141.
9. Levine JS, Ahnen DJ. Clinical practice. Adenomatous polyps of the colon. N Engl J Med. 2006;355(24):2551-2557.
10. Lynch HT, de la Chapelle A. Hereditary colorectal cancer. N Engl J Med. 2003;348(10):919-932.
11. Butterworth AS, Higgins JP, Pharoah P. Relative and absolute risk of colorectal cancer for individuals with a family history: a meta-analysis. Eur J Cancer. 2006;42(2):216-227.
12. Johns LE, Houlston RS. A systematic review and meta-analysis of familial colorectal cancer risk. Am J Gastroenterol. 2001;96(10):2992-3003.
13. Ekbom A, Helmick C, Zack M, et al. Ulcerative colitis and colorectal cancer. A population-based study. N Engl J Med. 1990;323(18):1228-1233.
14. Yang YX, Hennessy S, Lewis JD. Type 2 diabetes mellitus and the risk of colorectal cancer. Clin Gastroenterol Hepatol. 2005;3(6):587-594.
15. American Cancer Society. Colorectal cancer facts & figures 2011-2013. http://www.cancer.org/acs/groups/content/@epidemiologysurveilance/documents/document/acspc-028323.pdf. Published 2011. Accessed July 5, 2016.
16. Botteri E, Iodice S, Bagnardi V, et al. Smoking and colorectal cancer: a meta-analysis. JAMA. 2008;300(23):2765-2778.
17. Cho E, Smith-Warner SA, Ritz J, et al. Alcohol intake and colorectal cancer: a pooled analysis of 8 cohort studies. Ann Intern Med. 2004;140(8):603-613.
18. Larsson SC, Wolk A. Obesity and colon and rectal cancer risk: a meta-analysis of prospective studies. Am J Clin Nutr. 2007;86(3):556-565.
19. Speights VO, Johnston MW, Stoltenberg PH, et al. Colorectal cancer: current trends in initial clinical manifestations. South Med J. 1991;84(5):575-578.
20. Shaukat A, Mongin SJ, Geisser MS, et al. Long-term mortality after screening for colorectal cancer. N Engl J Med. 2013;369(12):1106-1114.
21. U.S. Preventive Services Task Force. Screening for colorectal cancer: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med. 2008;149(9):627-637.
22. Levin B, Lieberman DA, McFarland B, et al; American Cancer Society Colorectal Cancer Advisory Group; US Multi-Society Task Force; American College of Radiology Colon Cancer Committee. Screening and surveillance for the early detection of colorectal cancer and adenomatous polyps, 2008: a joint guideline from the American Cancer Society, the US Multi-Society Task Force on Colorectal Cancer, and the American College of Radiology. CA Cancer J Clin. 2008;58(3):130-160.
23. Rex DK, Johnson DA, Anderson JC, et al; American College of Gastroenterology. American College of Gastroenterology Guidelines for Colorectal Cancer Screening 2009 [corrected] [Erratum in: Am J Gastroenetrol. 2009;104(6):1613]. Am J Gastroenterol. 2009;104(3):739-750.
Cancer screening is an important example of secondary prevention—the aim being to detect disease at an early stage, when treatment can prevent symptomatic disease. Over the years, screening tests for breast cancer, colorectal cancer (CRC), cervical cancer, and, most recently, lung cancer have been developed and recommended by the U.S. Preventive Services Task Force (USPSTF). Among breast cancer, cervical cancer, and CRC, the screening rate for CRC remains lowest, at 58.6%.1
The importance of screening for CRC is highlighted by the facts that:
- CRC is the third most commonly diagnosed form of cancer in the United States among both men and women
- CRC is the second leading cause of cancer-related death.2
The overall decrease in the incidence of CRC in the United States has been credited to improvements in screening and removal of potentially precancerous lesions.3
Harmful disparity puts the mentally ill at exceptional risk
Screening patterns for CRC among patients with mental illness are poorly characterized, but it is known that the overall cancer screening rate among patients with severe psychiatric illness lags significantly behind the rate in the general population.4,5 In addition, studies have shown that mortality among patients with CRC who have a mental disorder is elevated, compared with CRC patients who do not have a psychiatric diagnosis.6
Why this disparity? It might be that CRC is more likely to be diagnosed at an advanced stage among these patients, or that they are less likely to receive cancer treatment after diagnosis, or are more likely to have a longer delay between diagnosis and initial treatment than patients who do not have a psychiatric diagnosis.7
Regardless, psychiatric practitioners can make a significant impact on reducing this health disparity by leveraging their unique therapeutic relationship to educate patients about screening options and dispel myths about cancer screening. In this article, we outline practical strategies for CRC screening and weigh the advantages and disadvantages for the use of several tools and guidelines in psychiatric patients.
What is the pathogenesis of colorectal cancer?
Most cases of CRC evolve from polyps, abnormal growths on the lining of the colon or rectum. Constituting an estimated 96% of all polyps, adenomas are by far the most common form in the colon and rectum.
Adenomas also are most likely to transform over time to dysplasia, and then to progress to cancer.8 Although all adenomas have malignant potential, <10% evolve to adenocarcinoma. This proposed adenoma➝carcinoma sequence is not well understood; however, it is known that CRC usually develops slowly—over 10 to 15 years.9 Detection and removal of adenomas and treatable, localized carcinomas form the basis of screening for CRC.
Risk factors for colorectal cancer
A number of risk factors for CRC have been identified.
Specific heritable conditions, such as Lynch syndrome and familial adenomatous polyposis, pose the greatest risk of CRC, particularly at younger ages and compared with people without such a history.10
Family history. One of the strongest risk factors for CRC remains a family history of the disease. People who have a first-degree relative with a diagnosis of CRC are at 2 to 3 times the risk of CRC, compared with people without a family history of the disease. This risk increases further if multiple family members are affected or if the diagnosis was made in a relative at a young age.11,12
Other non-modifiable risk factors include a personal history of inflammatory bowel disease, type 2 diabetes mellitus, male sex, African American heritage, and increasing age.13-15
Common modifiable risk factors include obesity, smoking, and alcohol consumption.16-18
What is the role of screening?
CRC screening is only appropriate for patients who are asymptomatic. CRC generally is asymptomatic in early stages. Prognosis also is most favorable when CRC is detected in the asymptomatic stage.
As lesions of CRC grow, the presentation might include hematochezia, melena, abdominal pain, weight loss, occult anemia, constipation or diarrhea, and changes in stool caliber.19 These signs and symptoms are not highly specific for CRC, however, and might be indicative of other gastrointestinal pathology, including inflammatory bowel disease, diverticulitis, irritable bowel syndrome, infectious colitis, hemorrhoids, and mesenteric ischemia.
Symptomatic patients should be referred directly for diagnostic evaluation. Colonoscopy with biopsy is the standard for diagnosing CRC. Once a diagnosis of CRC is made, patients should be referred to a specialist to discuss treatment; options largely depend on the stage of the cancer at diagnosis.
What screening tests are available?
Unlike screening for other cancers, there are a number of reasonable options for CRC screening; Table 115 compares their relative pros and cons. Each test has its benefits and drawbacks, allowing the screening strategy to be customized based on patient preference and characteristics, but this variability also can lead to confusion by patient and provider about those options.
Stool-based tests detect trace amounts of blood from early-stage treatable cancers. Highly sensitive fecal occult blood testing (FOBT) has been shown specifically to decrease mortality from CRC.20 Stool-based tests are inexpensive and noninvasive, but require:
- more frequent testing
- that the patient collect the stool specimen
- follow-up colonoscopy when test results are positive.
Endoscopic and imaging tests detect polyps and early-stage treatable cancers; all require some degree of bowel preparation, and some require sedation. Testing intervals vary but, as a group, are longer than the interval between stool-based tests because polyps grow slowly. Because colonoscopy with biopsy is the preferred screening method for diagnosing CRC, it is the only screening option that also is a diagnostic procedure.
Where can screening guidelines be found?
Several professional organizations have developed guidelines for CRC screening. The 2 major
An update to both guidelines was released in 2008. Table 221,22 summarizes their recommendations.
Both guidelines recommend that screening begin at age 50 (Box). The primary differences between the 2 guidelines lie in the scope of recommended options for screening and the time frame for discontinuing screening:
- USPSTF requires a higher level of evidence for screening options and limits recommended options to FOBT, sigmoidoscopy combined with FOBT, and colonoscopy.
- ACS-MSTF-ACR emphasizes options that detect premalignant polyps, and generally is more inclusive of testing options; it also delineates tests as useful for either (1) early detection of cancer (stool-based studies) or (2) cancer prevention (endoscopic and imaging tests).
On the question of when to stop screening, ACS-MSTF-ACR bases its recommendations on life expectancy; USPSTF sets a specific age for ending screening.21,22
Recommendations of a third entity, the American College of Gastroenterology (ACG), are similar to those of ACS-MSTF-ACR; however, ACG (1) recommends beginning screening African American patients at age 45 because of their increased risk of CRC and (2) gives preference to colonoscopy as the preferred screening modality.23
Guidelines vary for high-risk patients (those with a history of familial adenomatous polyposis or another inherited syndrome associated with CRC; those with a family history of CRC in the young; those with a history of radiation exposure, history of CRC, or inflammatory bowel disease; and those with several first-degree relatives with CRC). Patients who fall into any of these categories should be referred for specialty care to establish the time of initial screening and the interval of subsequent screening.
CRC screening in the presence of psychiatric illness
Psychiatrists have an opportunity to support their patients when considering potentially confusing CRC screening recommendations. This opportunity might occur during a discussion about general preventive care, or a patient might come to an appointment after visiting a primary care provider, and ask for advice about screening options.
The potential benefits of CRC screening are negated if a patient is unable or unwilling to complete the test or undergo timely follow-up of positive results. It is important, therefore, to individualize screening recommendations—keeping in mind the degree of impairment from mental illness and the patient’s preferences and reliability to engage in follow-up. To date, there are no agreed-on screening guidelines specifically for patients with comorbid mental illness.
Adapting USPSTF guidelines for CRC screening of average-risk patients with mental illness, we offer the following recommendations:
Recommend screening. Begin routine screening at age 50. Patients with well-controlled or mild symptoms should be screened with a stool study with or without flexible sigmoidoscopy. Stool studies are safe, noninvasive, and require no bowel preparation; when used alone, however, they need to be performed yearly.
Screening accuracy is increased when a stool-based test is combined with flexible sigmoidoscopy; screening then can be performed less often. Unlike colonoscopy, flexible sigmoidoscopy does not involve sedation; a high-functioning patient might find this appealing and tolerate the greater frequency of screening. On the other hand, some patients might not accept the inconvenience of collecting the stool sample with the kit provided and returning it to the lab for processing.
Manage psychiatric illness optimally. For a patient with moderate or severe psychiatric symptoms, first attempt to optimize treatment of the underlying psychiatric condition before establishing a CRC screening program. If control of symptoms is likely to improve over the next 1 or 2 visits, it might be reasonable to defer screening until symptoms are better controlled and then reassess the patient before making specific screening recommendations. Screening should not be delayed, however, if significant improvement in symptoms is not expected in the near future. Lengthy delay might lead to failure in initiating screening at all.
We recommend that patients with persistent moderate or severe symptoms be screened with traditional colonoscopy. The sedation associated with colonoscopy (1) may be preferable to some patients with more severe illness and (2) allows for screening and diagnostic biopsy if needed during the same procedure. Screening with colonoscopy also:
- avoids the yearly adherence to a screening program that is needed with stool cards alone
- does not rely on patients collecting and returning stool kits for processing.
A potential challenge for patients with limited social support is the requirement to have someone accompany the patient on the day of colonoscopy.
Take steps to improve the screening rate. In addition to specific recommendations based on symptom severity, there are systems-level interventions that should be considered to improve the screening rate. These include:
- addressing transportation issues that are a barrier to screening
- considering the use of health navigators or peer advocates to help guide patients through the sometimes complex systems of care.
A more comprehensive systems-level intervention for mental health clinics that work primarily with persistent and severe mentally ill populations might include employing a care coordinator to organize referrals to primary care or even exploring reverse integration. In reverse integration, primary care providers co-locate within the mental health clinic, (1) allowing for “one-stop shopping” of mental health and primary care needs and (2) facilitating collaboration and shared treatment planning between primary care and mental health for complex patients.
Cancer screening is an important example of secondary prevention—the aim being to detect disease at an early stage, when treatment can prevent symptomatic disease. Over the years, screening tests for breast cancer, colorectal cancer (CRC), cervical cancer, and, most recently, lung cancer have been developed and recommended by the U.S. Preventive Services Task Force (USPSTF). Among breast cancer, cervical cancer, and CRC, the screening rate for CRC remains lowest, at 58.6%.1
The importance of screening for CRC is highlighted by the facts that:
- CRC is the third most commonly diagnosed form of cancer in the United States among both men and women
- CRC is the second leading cause of cancer-related death.2
The overall decrease in the incidence of CRC in the United States has been credited to improvements in screening and removal of potentially precancerous lesions.3
Harmful disparity puts the mentally ill at exceptional risk
Screening patterns for CRC among patients with mental illness are poorly characterized, but it is known that the overall cancer screening rate among patients with severe psychiatric illness lags significantly behind the rate in the general population.4,5 In addition, studies have shown that mortality among patients with CRC who have a mental disorder is elevated, compared with CRC patients who do not have a psychiatric diagnosis.6
Why this disparity? It might be that CRC is more likely to be diagnosed at an advanced stage among these patients, or that they are less likely to receive cancer treatment after diagnosis, or are more likely to have a longer delay between diagnosis and initial treatment than patients who do not have a psychiatric diagnosis.7
Regardless, psychiatric practitioners can make a significant impact on reducing this health disparity by leveraging their unique therapeutic relationship to educate patients about screening options and dispel myths about cancer screening. In this article, we outline practical strategies for CRC screening and weigh the advantages and disadvantages for the use of several tools and guidelines in psychiatric patients.
What is the pathogenesis of colorectal cancer?
Most cases of CRC evolve from polyps, abnormal growths on the lining of the colon or rectum. Constituting an estimated 96% of all polyps, adenomas are by far the most common form in the colon and rectum.
Adenomas also are most likely to transform over time to dysplasia, and then to progress to cancer.8 Although all adenomas have malignant potential, <10% evolve to adenocarcinoma. This proposed adenoma➝carcinoma sequence is not well understood; however, it is known that CRC usually develops slowly—over 10 to 15 years.9 Detection and removal of adenomas and treatable, localized carcinomas form the basis of screening for CRC.
Risk factors for colorectal cancer
A number of risk factors for CRC have been identified.
Specific heritable conditions, such as Lynch syndrome and familial adenomatous polyposis, pose the greatest risk of CRC, particularly at younger ages and compared with people without such a history.10
Family history. One of the strongest risk factors for CRC remains a family history of the disease. People who have a first-degree relative with a diagnosis of CRC are at 2 to 3 times the risk of CRC, compared with people without a family history of the disease. This risk increases further if multiple family members are affected or if the diagnosis was made in a relative at a young age.11,12
Other non-modifiable risk factors include a personal history of inflammatory bowel disease, type 2 diabetes mellitus, male sex, African American heritage, and increasing age.13-15
Common modifiable risk factors include obesity, smoking, and alcohol consumption.16-18
What is the role of screening?
CRC screening is only appropriate for patients who are asymptomatic. CRC generally is asymptomatic in early stages. Prognosis also is most favorable when CRC is detected in the asymptomatic stage.
As lesions of CRC grow, the presentation might include hematochezia, melena, abdominal pain, weight loss, occult anemia, constipation or diarrhea, and changes in stool caliber.19 These signs and symptoms are not highly specific for CRC, however, and might be indicative of other gastrointestinal pathology, including inflammatory bowel disease, diverticulitis, irritable bowel syndrome, infectious colitis, hemorrhoids, and mesenteric ischemia.
Symptomatic patients should be referred directly for diagnostic evaluation. Colonoscopy with biopsy is the standard for diagnosing CRC. Once a diagnosis of CRC is made, patients should be referred to a specialist to discuss treatment; options largely depend on the stage of the cancer at diagnosis.
What screening tests are available?
Unlike screening for other cancers, there are a number of reasonable options for CRC screening; Table 115 compares their relative pros and cons. Each test has its benefits and drawbacks, allowing the screening strategy to be customized based on patient preference and characteristics, but this variability also can lead to confusion by patient and provider about those options.
Stool-based tests detect trace amounts of blood from early-stage treatable cancers. Highly sensitive fecal occult blood testing (FOBT) has been shown specifically to decrease mortality from CRC.20 Stool-based tests are inexpensive and noninvasive, but require:
- more frequent testing
- that the patient collect the stool specimen
- follow-up colonoscopy when test results are positive.
Endoscopic and imaging tests detect polyps and early-stage treatable cancers; all require some degree of bowel preparation, and some require sedation. Testing intervals vary but, as a group, are longer than the interval between stool-based tests because polyps grow slowly. Because colonoscopy with biopsy is the preferred screening method for diagnosing CRC, it is the only screening option that also is a diagnostic procedure.
Where can screening guidelines be found?
Several professional organizations have developed guidelines for CRC screening. The 2 major
An update to both guidelines was released in 2008. Table 221,22 summarizes their recommendations.
Both guidelines recommend that screening begin at age 50 (Box). The primary differences between the 2 guidelines lie in the scope of recommended options for screening and the time frame for discontinuing screening:
- USPSTF requires a higher level of evidence for screening options and limits recommended options to FOBT, sigmoidoscopy combined with FOBT, and colonoscopy.
- ACS-MSTF-ACR emphasizes options that detect premalignant polyps, and generally is more inclusive of testing options; it also delineates tests as useful for either (1) early detection of cancer (stool-based studies) or (2) cancer prevention (endoscopic and imaging tests).
On the question of when to stop screening, ACS-MSTF-ACR bases its recommendations on life expectancy; USPSTF sets a specific age for ending screening.21,22
Recommendations of a third entity, the American College of Gastroenterology (ACG), are similar to those of ACS-MSTF-ACR; however, ACG (1) recommends beginning screening African American patients at age 45 because of their increased risk of CRC and (2) gives preference to colonoscopy as the preferred screening modality.23
Guidelines vary for high-risk patients (those with a history of familial adenomatous polyposis or another inherited syndrome associated with CRC; those with a family history of CRC in the young; those with a history of radiation exposure, history of CRC, or inflammatory bowel disease; and those with several first-degree relatives with CRC). Patients who fall into any of these categories should be referred for specialty care to establish the time of initial screening and the interval of subsequent screening.
CRC screening in the presence of psychiatric illness
Psychiatrists have an opportunity to support their patients when considering potentially confusing CRC screening recommendations. This opportunity might occur during a discussion about general preventive care, or a patient might come to an appointment after visiting a primary care provider, and ask for advice about screening options.
The potential benefits of CRC screening are negated if a patient is unable or unwilling to complete the test or undergo timely follow-up of positive results. It is important, therefore, to individualize screening recommendations—keeping in mind the degree of impairment from mental illness and the patient’s preferences and reliability to engage in follow-up. To date, there are no agreed-on screening guidelines specifically for patients with comorbid mental illness.
Adapting USPSTF guidelines for CRC screening of average-risk patients with mental illness, we offer the following recommendations:
Recommend screening. Begin routine screening at age 50. Patients with well-controlled or mild symptoms should be screened with a stool study with or without flexible sigmoidoscopy. Stool studies are safe, noninvasive, and require no bowel preparation; when used alone, however, they need to be performed yearly.
Screening accuracy is increased when a stool-based test is combined with flexible sigmoidoscopy; screening then can be performed less often. Unlike colonoscopy, flexible sigmoidoscopy does not involve sedation; a high-functioning patient might find this appealing and tolerate the greater frequency of screening. On the other hand, some patients might not accept the inconvenience of collecting the stool sample with the kit provided and returning it to the lab for processing.
Manage psychiatric illness optimally. For a patient with moderate or severe psychiatric symptoms, first attempt to optimize treatment of the underlying psychiatric condition before establishing a CRC screening program. If control of symptoms is likely to improve over the next 1 or 2 visits, it might be reasonable to defer screening until symptoms are better controlled and then reassess the patient before making specific screening recommendations. Screening should not be delayed, however, if significant improvement in symptoms is not expected in the near future. Lengthy delay might lead to failure in initiating screening at all.
We recommend that patients with persistent moderate or severe symptoms be screened with traditional colonoscopy. The sedation associated with colonoscopy (1) may be preferable to some patients with more severe illness and (2) allows for screening and diagnostic biopsy if needed during the same procedure. Screening with colonoscopy also:
- avoids the yearly adherence to a screening program that is needed with stool cards alone
- does not rely on patients collecting and returning stool kits for processing.
A potential challenge for patients with limited social support is the requirement to have someone accompany the patient on the day of colonoscopy.
Take steps to improve the screening rate. In addition to specific recommendations based on symptom severity, there are systems-level interventions that should be considered to improve the screening rate. These include:
- addressing transportation issues that are a barrier to screening
- considering the use of health navigators or peer advocates to help guide patients through the sometimes complex systems of care.
A more comprehensive systems-level intervention for mental health clinics that work primarily with persistent and severe mentally ill populations might include employing a care coordinator to organize referrals to primary care or even exploring reverse integration. In reverse integration, primary care providers co-locate within the mental health clinic, (1) allowing for “one-stop shopping” of mental health and primary care needs and (2) facilitating collaboration and shared treatment planning between primary care and mental health for complex patients.
2. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63(1):11-30.
3. Edwards BK, Ward E, Kohler BA, et al. Annual report to the nation on the status of cancer, 1975-2006, featuring colorectal cancer trends and impact of interventions (risk factors, screening, and treatment) to reduce future rates. Cancer. 2010;116(3):544-573.
4. Miller E, Lasser KE, Becker AE. Breast and cervical cancer screening for women with mental illness: patient and provider perspectives on improving linkages between primary care and mental health. Arch Womens Ment Health. 2007;10(5):189-197.
5. Howard LM, Barley EA, Davies E, et al. Cancer diagnosis in people with severe mental illness: practical and ethical issues. Lancet Oncol. 2010;11(8):797-804.
6. Baillargeon J, Kuo YF, Lin YL, et al. Effect of mental disorders on diagnosis, treatment, and survival of older adults with colon cancer. J Am Geriatr Soc. 2011;59(7):1268-1273.
7. Robertson R, Campbell NC, Smith S, et al. Factors influencing time from presentation to treatment of colorectal and breast cancer in urban and rural areas. Br J Cancer. 2004;90(8):1479-1485.
8. Stewart SL, Wike JM, Kato I, et al. A population-based study of colorectal cancer histology in the United States, 1998-2001. Cancer. 2006;107(suppl 5):1128-1141.
9. Levine JS, Ahnen DJ. Clinical practice. Adenomatous polyps of the colon. N Engl J Med. 2006;355(24):2551-2557.
10. Lynch HT, de la Chapelle A. Hereditary colorectal cancer. N Engl J Med. 2003;348(10):919-932.
11. Butterworth AS, Higgins JP, Pharoah P. Relative and absolute risk of colorectal cancer for individuals with a family history: a meta-analysis. Eur J Cancer. 2006;42(2):216-227.
12. Johns LE, Houlston RS. A systematic review and meta-analysis of familial colorectal cancer risk. Am J Gastroenterol. 2001;96(10):2992-3003.
13. Ekbom A, Helmick C, Zack M, et al. Ulcerative colitis and colorectal cancer. A population-based study. N Engl J Med. 1990;323(18):1228-1233.
14. Yang YX, Hennessy S, Lewis JD. Type 2 diabetes mellitus and the risk of colorectal cancer. Clin Gastroenterol Hepatol. 2005;3(6):587-594.
15. American Cancer Society. Colorectal cancer facts & figures 2011-2013. http://www.cancer.org/acs/groups/content/@epidemiologysurveilance/documents/document/acspc-028323.pdf. Published 2011. Accessed July 5, 2016.
16. Botteri E, Iodice S, Bagnardi V, et al. Smoking and colorectal cancer: a meta-analysis. JAMA. 2008;300(23):2765-2778.
17. Cho E, Smith-Warner SA, Ritz J, et al. Alcohol intake and colorectal cancer: a pooled analysis of 8 cohort studies. Ann Intern Med. 2004;140(8):603-613.
18. Larsson SC, Wolk A. Obesity and colon and rectal cancer risk: a meta-analysis of prospective studies. Am J Clin Nutr. 2007;86(3):556-565.
19. Speights VO, Johnston MW, Stoltenberg PH, et al. Colorectal cancer: current trends in initial clinical manifestations. South Med J. 1991;84(5):575-578.
20. Shaukat A, Mongin SJ, Geisser MS, et al. Long-term mortality after screening for colorectal cancer. N Engl J Med. 2013;369(12):1106-1114.
21. U.S. Preventive Services Task Force. Screening for colorectal cancer: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med. 2008;149(9):627-637.
22. Levin B, Lieberman DA, McFarland B, et al; American Cancer Society Colorectal Cancer Advisory Group; US Multi-Society Task Force; American College of Radiology Colon Cancer Committee. Screening and surveillance for the early detection of colorectal cancer and adenomatous polyps, 2008: a joint guideline from the American Cancer Society, the US Multi-Society Task Force on Colorectal Cancer, and the American College of Radiology. CA Cancer J Clin. 2008;58(3):130-160.
23. Rex DK, Johnson DA, Anderson JC, et al; American College of Gastroenterology. American College of Gastroenterology Guidelines for Colorectal Cancer Screening 2009 [corrected] [Erratum in: Am J Gastroenetrol. 2009;104(6):1613]. Am J Gastroenterol. 2009;104(3):739-750.
2. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63(1):11-30.
3. Edwards BK, Ward E, Kohler BA, et al. Annual report to the nation on the status of cancer, 1975-2006, featuring colorectal cancer trends and impact of interventions (risk factors, screening, and treatment) to reduce future rates. Cancer. 2010;116(3):544-573.
4. Miller E, Lasser KE, Becker AE. Breast and cervical cancer screening for women with mental illness: patient and provider perspectives on improving linkages between primary care and mental health. Arch Womens Ment Health. 2007;10(5):189-197.
5. Howard LM, Barley EA, Davies E, et al. Cancer diagnosis in people with severe mental illness: practical and ethical issues. Lancet Oncol. 2010;11(8):797-804.
6. Baillargeon J, Kuo YF, Lin YL, et al. Effect of mental disorders on diagnosis, treatment, and survival of older adults with colon cancer. J Am Geriatr Soc. 2011;59(7):1268-1273.
7. Robertson R, Campbell NC, Smith S, et al. Factors influencing time from presentation to treatment of colorectal and breast cancer in urban and rural areas. Br J Cancer. 2004;90(8):1479-1485.
8. Stewart SL, Wike JM, Kato I, et al. A population-based study of colorectal cancer histology in the United States, 1998-2001. Cancer. 2006;107(suppl 5):1128-1141.
9. Levine JS, Ahnen DJ. Clinical practice. Adenomatous polyps of the colon. N Engl J Med. 2006;355(24):2551-2557.
10. Lynch HT, de la Chapelle A. Hereditary colorectal cancer. N Engl J Med. 2003;348(10):919-932.
11. Butterworth AS, Higgins JP, Pharoah P. Relative and absolute risk of colorectal cancer for individuals with a family history: a meta-analysis. Eur J Cancer. 2006;42(2):216-227.
12. Johns LE, Houlston RS. A systematic review and meta-analysis of familial colorectal cancer risk. Am J Gastroenterol. 2001;96(10):2992-3003.
13. Ekbom A, Helmick C, Zack M, et al. Ulcerative colitis and colorectal cancer. A population-based study. N Engl J Med. 1990;323(18):1228-1233.
14. Yang YX, Hennessy S, Lewis JD. Type 2 diabetes mellitus and the risk of colorectal cancer. Clin Gastroenterol Hepatol. 2005;3(6):587-594.
15. American Cancer Society. Colorectal cancer facts & figures 2011-2013. http://www.cancer.org/acs/groups/content/@epidemiologysurveilance/documents/document/acspc-028323.pdf. Published 2011. Accessed July 5, 2016.
16. Botteri E, Iodice S, Bagnardi V, et al. Smoking and colorectal cancer: a meta-analysis. JAMA. 2008;300(23):2765-2778.
17. Cho E, Smith-Warner SA, Ritz J, et al. Alcohol intake and colorectal cancer: a pooled analysis of 8 cohort studies. Ann Intern Med. 2004;140(8):603-613.
18. Larsson SC, Wolk A. Obesity and colon and rectal cancer risk: a meta-analysis of prospective studies. Am J Clin Nutr. 2007;86(3):556-565.
19. Speights VO, Johnston MW, Stoltenberg PH, et al. Colorectal cancer: current trends in initial clinical manifestations. South Med J. 1991;84(5):575-578.
20. Shaukat A, Mongin SJ, Geisser MS, et al. Long-term mortality after screening for colorectal cancer. N Engl J Med. 2013;369(12):1106-1114.
21. U.S. Preventive Services Task Force. Screening for colorectal cancer: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med. 2008;149(9):627-637.
22. Levin B, Lieberman DA, McFarland B, et al; American Cancer Society Colorectal Cancer Advisory Group; US Multi-Society Task Force; American College of Radiology Colon Cancer Committee. Screening and surveillance for the early detection of colorectal cancer and adenomatous polyps, 2008: a joint guideline from the American Cancer Society, the US Multi-Society Task Force on Colorectal Cancer, and the American College of Radiology. CA Cancer J Clin. 2008;58(3):130-160.
23. Rex DK, Johnson DA, Anderson JC, et al; American College of Gastroenterology. American College of Gastroenterology Guidelines for Colorectal Cancer Screening 2009 [corrected] [Erratum in: Am J Gastroenetrol. 2009;104(6):1613]. Am J Gastroenterol. 2009;104(3):739-750.