User login
PICCs linked to lower- and upper-extremity DVT

A study of more than 70,000 patients suggests the use of peripherally inserted central catheters (PICCs) is not only associated with upper-extremity deep vein thrombosis (DVT).
It’s linked to lower-extremity DVT as well. However, PICC use was not associated with pulmonary embolism (PE).
Vineet Chopra, MD, of the University of Michigan in Ann Arbor, and his colleagues reported these findings in The American Journal of Medicine.
“Our study confirmed that PICCs are strongly associated with DVT in upper limbs,” Dr Chopra said. “However, what is novel and noteworthy in this study is that the presence of a PICC was also associated with an increased risk of lower-extremity DVT. Prior studies had not assessed whether PICCs are independently associated with an increase in the risk of subsequent lower-extremity DVT.”
Dr Chopra and his colleagues analyzed data from 76,242 patients hospitalized at 48 Michigan hospitals.
The team reviewed PICC placement, existing medical conditions, risk factors for venous thromboembolism (VTE), and thrombotic events occurring within 90 days of hospital admission.
A total of 3790 patients received a PICC during hospitalization. And there were 876 VTEs, including 208 upper-extremity DVTs, 372 lower-extremity DVTs, and 296 PEs.
After adjusting for risk factors other than PICC use, the researchers found that PICCs were independently associated with all-cause VTE, upper-extremity DVT, and lower-extremity DVT, but not PE. The hazard ratios were 3.16, 10.49, 1.48, and 1.34, respectively.
The team also found that VTE prophylaxis did not reduce the risk of DVT.
“Taken together, these findings suggest that the thrombotic burden associated with peripherally inserted central catheters may not be restricted to the extremity where the device resides or easily attenuated after insertion,” Dr Chopra said.
He and his colleagues therefore concluded that clinicians should weigh the risks and benefits of PICC use and consider using alternative devices in patients at high risk of DVT. And clinicians should not focus only on the extremity where a PICC resides but the composite risk of VTE among patients who receive a PICC. ![]()
A study of more than 70,000 patients suggests the use of peripherally inserted central catheters (PICCs) is not only associated with upper-extremity deep vein thrombosis (DVT).
It’s linked to lower-extremity DVT as well. However, PICC use was not associated with pulmonary embolism (PE).
Vineet Chopra, MD, of the University of Michigan in Ann Arbor, and his colleagues reported these findings in The American Journal of Medicine.
“Our study confirmed that PICCs are strongly associated with DVT in upper limbs,” Dr Chopra said. “However, what is novel and noteworthy in this study is that the presence of a PICC was also associated with an increased risk of lower-extremity DVT. Prior studies had not assessed whether PICCs are independently associated with an increase in the risk of subsequent lower-extremity DVT.”
Dr Chopra and his colleagues analyzed data from 76,242 patients hospitalized at 48 Michigan hospitals.
The team reviewed PICC placement, existing medical conditions, risk factors for venous thromboembolism (VTE), and thrombotic events occurring within 90 days of hospital admission.
A total of 3790 patients received a PICC during hospitalization. And there were 876 VTEs, including 208 upper-extremity DVTs, 372 lower-extremity DVTs, and 296 PEs.
After adjusting for risk factors other than PICC use, the researchers found that PICCs were independently associated with all-cause VTE, upper-extremity DVT, and lower-extremity DVT, but not PE. The hazard ratios were 3.16, 10.49, 1.48, and 1.34, respectively.
The team also found that VTE prophylaxis did not reduce the risk of DVT.
“Taken together, these findings suggest that the thrombotic burden associated with peripherally inserted central catheters may not be restricted to the extremity where the device resides or easily attenuated after insertion,” Dr Chopra said.
He and his colleagues therefore concluded that clinicians should weigh the risks and benefits of PICC use and consider using alternative devices in patients at high risk of DVT. And clinicians should not focus only on the extremity where a PICC resides but the composite risk of VTE among patients who receive a PICC. ![]()
A study of more than 70,000 patients suggests the use of peripherally inserted central catheters (PICCs) is not only associated with upper-extremity deep vein thrombosis (DVT).
It’s linked to lower-extremity DVT as well. However, PICC use was not associated with pulmonary embolism (PE).
Vineet Chopra, MD, of the University of Michigan in Ann Arbor, and his colleagues reported these findings in The American Journal of Medicine.
“Our study confirmed that PICCs are strongly associated with DVT in upper limbs,” Dr Chopra said. “However, what is novel and noteworthy in this study is that the presence of a PICC was also associated with an increased risk of lower-extremity DVT. Prior studies had not assessed whether PICCs are independently associated with an increase in the risk of subsequent lower-extremity DVT.”
Dr Chopra and his colleagues analyzed data from 76,242 patients hospitalized at 48 Michigan hospitals.
The team reviewed PICC placement, existing medical conditions, risk factors for venous thromboembolism (VTE), and thrombotic events occurring within 90 days of hospital admission.
A total of 3790 patients received a PICC during hospitalization. And there were 876 VTEs, including 208 upper-extremity DVTs, 372 lower-extremity DVTs, and 296 PEs.
After adjusting for risk factors other than PICC use, the researchers found that PICCs were independently associated with all-cause VTE, upper-extremity DVT, and lower-extremity DVT, but not PE. The hazard ratios were 3.16, 10.49, 1.48, and 1.34, respectively.
The team also found that VTE prophylaxis did not reduce the risk of DVT.
“Taken together, these findings suggest that the thrombotic burden associated with peripherally inserted central catheters may not be restricted to the extremity where the device resides or easily attenuated after insertion,” Dr Chopra said.
He and his colleagues therefore concluded that clinicians should weigh the risks and benefits of PICC use and consider using alternative devices in patients at high risk of DVT. And clinicians should not focus only on the extremity where a PICC resides but the composite risk of VTE among patients who receive a PICC. ![]()
FDA approves new formulation of pain patch for cancer patients

receiving treatment
Photo by Rhoda Baer
The US Food and Drug Administration (FDA) has approved a new formulation of fentanyl buccal soluble film CII (Onsolis), a patch used to manage breakthrough pain in adult cancer patients who are opioid-tolerant.
This decision allows BioDelivery Sciences International, Inc. (BDSI), the company developing Onsolis, to bring the product back to the US marketplace.
However, the company said this will not happen before 2016.
Onsolis is an opioid agonist indicated for the management of breakthrough pain in cancer patients 18 years of age and older who are already receiving and are tolerant to opioid therapy for their underlying persistent cancer pain.
Onsolis utilizes BioErodible MucoAdhesive drug delivery technology, which consists of a small, bioerodible polymer film that is applied to the inner lining of the cheek. Onsolis is the only differentiated fentanyl-containing product for this indication that provides buccal administration.
Onsolis off the US market
Onsolis was originally approved by the FDA in July 2009, but BDSI stopped manufacturing the product in March 2012, after the FDA uncovered 2 issues with Onsolis.
The FDA found that, during Onsolis’s 24-month shelf-life, microscopic crystals formed on the product, and the color faded slightly. BDSI said these changes did not affect the product’s underlying integrity or safety, but the FDA thought the fading color in particular might cause patients to question the product’s efficacy.
So the FDA required that Onsolis be modified before additional product could be manufactured and distributed. Supplies of Onsolis that were already on the market remained on the market.
An analysis by BDSI showed that the changes in Onsolis were related to an excipient used in the manufacturing process that could be removed to resolve the problem.
The excipient was specific to the manufacture of Onsolis in the US. Therefore, it did not impact the launch of Breakyl, which is the brand name for Onsolis in the European Union.
Return to market
After BDSI reformulated Onsolis to prevent the aforementioned changes in the product’s appearance, the FDA approved the product’s return to market.
“We are pleased to have obtained FDA approval of our [supplemental new drug application] and to now be in a position to move toward returning Onsolis to the US marketplace,” said Mark A. Sirgo, PharmD, President and Chief Executive Officer of BDSI.
“Although we have options for Onsolis, including commercializing it on our own, our current plan is to determine the value we can secure in a partnership with a company that has access to the target physician audience. We have been engaged with a number of potential partners, and, with this approval, we can now proceed forward with those discussions in earnest. We will provide more definitive timing in the near future about the reintroduction, but this would not be prior to 2016.”
Once Onsolis does return to the market, it will only be available via the Transmucosal Immediate Release Fentanyl (TIRF) Risk Evaluation and Mitigation Strategy (REMS) program. This is an FDA-required program designed to mitigate the risk of misuse, abuse, addiction, overdose, and serious complications due to medication errors with the use of TIRF medicines.
Outpatients, healthcare professionals who prescribe to outpatients, pharmacies, and distributors must enroll in the program to receive Onsolis. Further information is available at www.TIRFREMSAccess.com. ![]()
receiving treatment
Photo by Rhoda Baer
The US Food and Drug Administration (FDA) has approved a new formulation of fentanyl buccal soluble film CII (Onsolis), a patch used to manage breakthrough pain in adult cancer patients who are opioid-tolerant.
This decision allows BioDelivery Sciences International, Inc. (BDSI), the company developing Onsolis, to bring the product back to the US marketplace.
However, the company said this will not happen before 2016.
Onsolis is an opioid agonist indicated for the management of breakthrough pain in cancer patients 18 years of age and older who are already receiving and are tolerant to opioid therapy for their underlying persistent cancer pain.
Onsolis utilizes BioErodible MucoAdhesive drug delivery technology, which consists of a small, bioerodible polymer film that is applied to the inner lining of the cheek. Onsolis is the only differentiated fentanyl-containing product for this indication that provides buccal administration.
Onsolis off the US market
Onsolis was originally approved by the FDA in July 2009, but BDSI stopped manufacturing the product in March 2012, after the FDA uncovered 2 issues with Onsolis.
The FDA found that, during Onsolis’s 24-month shelf-life, microscopic crystals formed on the product, and the color faded slightly. BDSI said these changes did not affect the product’s underlying integrity or safety, but the FDA thought the fading color in particular might cause patients to question the product’s efficacy.
So the FDA required that Onsolis be modified before additional product could be manufactured and distributed. Supplies of Onsolis that were already on the market remained on the market.
An analysis by BDSI showed that the changes in Onsolis were related to an excipient used in the manufacturing process that could be removed to resolve the problem.
The excipient was specific to the manufacture of Onsolis in the US. Therefore, it did not impact the launch of Breakyl, which is the brand name for Onsolis in the European Union.
Return to market
After BDSI reformulated Onsolis to prevent the aforementioned changes in the product’s appearance, the FDA approved the product’s return to market.
“We are pleased to have obtained FDA approval of our [supplemental new drug application] and to now be in a position to move toward returning Onsolis to the US marketplace,” said Mark A. Sirgo, PharmD, President and Chief Executive Officer of BDSI.
“Although we have options for Onsolis, including commercializing it on our own, our current plan is to determine the value we can secure in a partnership with a company that has access to the target physician audience. We have been engaged with a number of potential partners, and, with this approval, we can now proceed forward with those discussions in earnest. We will provide more definitive timing in the near future about the reintroduction, but this would not be prior to 2016.”
Once Onsolis does return to the market, it will only be available via the Transmucosal Immediate Release Fentanyl (TIRF) Risk Evaluation and Mitigation Strategy (REMS) program. This is an FDA-required program designed to mitigate the risk of misuse, abuse, addiction, overdose, and serious complications due to medication errors with the use of TIRF medicines.
Outpatients, healthcare professionals who prescribe to outpatients, pharmacies, and distributors must enroll in the program to receive Onsolis. Further information is available at www.TIRFREMSAccess.com. ![]()
receiving treatment
Photo by Rhoda Baer
The US Food and Drug Administration (FDA) has approved a new formulation of fentanyl buccal soluble film CII (Onsolis), a patch used to manage breakthrough pain in adult cancer patients who are opioid-tolerant.
This decision allows BioDelivery Sciences International, Inc. (BDSI), the company developing Onsolis, to bring the product back to the US marketplace.
However, the company said this will not happen before 2016.
Onsolis is an opioid agonist indicated for the management of breakthrough pain in cancer patients 18 years of age and older who are already receiving and are tolerant to opioid therapy for their underlying persistent cancer pain.
Onsolis utilizes BioErodible MucoAdhesive drug delivery technology, which consists of a small, bioerodible polymer film that is applied to the inner lining of the cheek. Onsolis is the only differentiated fentanyl-containing product for this indication that provides buccal administration.
Onsolis off the US market
Onsolis was originally approved by the FDA in July 2009, but BDSI stopped manufacturing the product in March 2012, after the FDA uncovered 2 issues with Onsolis.
The FDA found that, during Onsolis’s 24-month shelf-life, microscopic crystals formed on the product, and the color faded slightly. BDSI said these changes did not affect the product’s underlying integrity or safety, but the FDA thought the fading color in particular might cause patients to question the product’s efficacy.
So the FDA required that Onsolis be modified before additional product could be manufactured and distributed. Supplies of Onsolis that were already on the market remained on the market.
An analysis by BDSI showed that the changes in Onsolis were related to an excipient used in the manufacturing process that could be removed to resolve the problem.
The excipient was specific to the manufacture of Onsolis in the US. Therefore, it did not impact the launch of Breakyl, which is the brand name for Onsolis in the European Union.
Return to market
After BDSI reformulated Onsolis to prevent the aforementioned changes in the product’s appearance, the FDA approved the product’s return to market.
“We are pleased to have obtained FDA approval of our [supplemental new drug application] and to now be in a position to move toward returning Onsolis to the US marketplace,” said Mark A. Sirgo, PharmD, President and Chief Executive Officer of BDSI.
“Although we have options for Onsolis, including commercializing it on our own, our current plan is to determine the value we can secure in a partnership with a company that has access to the target physician audience. We have been engaged with a number of potential partners, and, with this approval, we can now proceed forward with those discussions in earnest. We will provide more definitive timing in the near future about the reintroduction, but this would not be prior to 2016.”
Once Onsolis does return to the market, it will only be available via the Transmucosal Immediate Release Fentanyl (TIRF) Risk Evaluation and Mitigation Strategy (REMS) program. This is an FDA-required program designed to mitigate the risk of misuse, abuse, addiction, overdose, and serious complications due to medication errors with the use of TIRF medicines.
Outpatients, healthcare professionals who prescribe to outpatients, pharmacies, and distributors must enroll in the program to receive Onsolis. Further information is available at www.TIRFREMSAccess.com. ![]()
The Use of Sodium Sulfacetamide in Dermatology
Sodium sulfacetamide has various uses in the field of dermatology due to its anti-inflammatory and antibacterial properties. It has been shown to be effective in the management of a variety of inflammatory facial dermatoses, including papulopustular rosacea, acne vulgaris, seborrheic dermatitis, and perioral dermatitis. We review the mechanism of action, pharmacology and formulations, clinical uses, and adverse effects of sodium sulfacetamide as a dermatologic treatment.
Mechanism of Action
Sodium sulfacetamide is a sulfonamide-type antibacterial agent. Its mechanism of action is the inhibition of bacterial dihydropteroate synthetase, which prevents the conversion of p-aminobenzoic acid to folic acid. This process causes a bacteriostatic effect on the growth of several gram-negative and gram-positive organisms, including Propionibacterium acnes.1,2
The effectiveness of sodium sulfacetamide is increased when used in combination with sulfur, which has keratolytic, antibacterial, antifungal, and antiparasitic effects. The addition of hydrocortisone has been reported to increase the effectiveness of both agents.3
Pharmacology
Sodium sulfacetamide is highly soluble at the physiologic pH of 7.4, which contributes to its high level of penetration and absorption.4 An in vitro study showed percutaneous absorption of sodium sulfacetamide to be around 4%.5 Sulfonamides are metabolized mainly by the liver and are excreted by the kidneys.
Formulations
The most common concentrations of sodium sulfacetamide and sulfur are 10% and 5%, respectively. A wide variety of sulfacetamide-containing products are available, many of which are marketed to treat specific conditions depending on additional ingredients or the type of delivery system.
Clinical Uses
Topical formulations of sodium sulfacetamide and sulfur have proven to be efficacious in the management of rosacea, with a typical regimen consisting of twice-daily application for 8 weeks.6 The sulfur in the formulation has the additional benefit of targeting Demodex mites, which are implicated as a contributing factor in some cases of rosacea.7 Sodium sulfacetamide 10%–sulfur 5% lotion was more effective in improving the erythema, papulopustules, and overall severity of rosacea as compared to metronidazole gel 0.75%.8 Other studies have reported increased efficacy when sodium sulfacetamide and topical sulfur are used along with metronidazole.9,10
Sodium sulfacetamide also has shown efficacy against acne. Its antibacterial and drying properties have been shown to decrease the number of inflammatory lesions and comedones, and in the treatment of acne vulgaris, no sensitivity reactions have been observed.2 Also, unlike topical antibiotics, cases of P acnes resistance to topical sulfur products have not been widely reported. Studies have demonstrated that twice-daily use of sodium sulfacetamide 10%–sulfur 5% for 12 weeks decreases inflammatory acne lesions by 80.4% to 83%.11,12
Seborrheic dermatitis is a common chronic infection of the skin caused by Malassezia species. One study investigated the use of sodium sulfacetamide ointment and soap to treat seborrheic dermatitis and found that the condition was either improved or completely controlled in 93% (71/76) of cases.4 Sodium sulfacetamide lotion was an effective treatment of seborrheic dermatitis in 89% (54/61) of patients with scalp involvement and 68% (30/44) of patients with glabrous skin involvement.13
Perioral dermatitis is characterized by groups of erythematous papules and pustules localized around the mouth. The use of topical sodium sulfacetamide along with oral tetracyclines has been demonstrated to consistently clear lesions in most patients with perioral dermatitis.14 Sodium sulfacetamide is unique in that it is not associated with the excessive erythema and irritation often found with retinoic acid and benzoyl peroxide.15 Unfortunately, however, there have been no well-controlled trials to compare the efficacy of sodium sulfacetamide to other topical therapies for this condition.
Adverse Effects
Adverse effects from sodium sulfacetamide are rare and generally are limited to cutaneous reactions including dryness, erythema, pruritus, and discomfort.1 Periocular use of sodium sulfacetamide can cause conjunctival irritation. One study reported that 19% (6/31) of patients experienced local reactions but most were considered mild.9 Rare but serious reactions including erythema multiforme and Stevens-Johnson syndrome have been reported from ophthalmic use.16,17
A common limiting factor to sodium sulfacetamide preparations that include elemental sulfur is the offensive smell, which has hindered patient compliance in the past; however, pharmaceutical companies have attempted to create more tolerable products without the odor.10 One study found that the tolerability of a sodium sulfacetamide 10%–sulfur 5% foam using a rinse-off method of application was excellent, with only 33% (8/24) of participants commenting on the smell.18 Another limiting factor of sodium sulfacetamide preparations containing sulfur is orange-brown discoloration when combined with benzoyl peroxide, which does not affect the skin but may stain clothing.19
Sodium sulfacetamide is rendered less effective when combined with silver-containing products.20 No other notable drug interactions are known; however, oral sulfonamides are known to interact with several drugs, including cyclosporine and phenytoin.21,22
Contraindications
Sodium sulfacetamide is contraindicated in patients with known hypersensitivity to sulfonamides, sulfur, or any other component of the preparation. It is a pregnancy category C drug, and pregnant women should only use sodium sulfacetamide if it is the only modality to treat the condition or the benefits outweigh the risks. Although there are no known reports of problems related to topical sodium sulfacetamide during pregnancy, the use of oral sulfonamides during pregnancy can increase the risk for neonatal jaundice.23 Likewise, caution should be exercised in prescribing this product to nursing women, as systemic sulfonamide antibacterials are well known to cause kernicterus in nursing neonates.1
Conclusion
The efficacy and safety of sodium sulfacetamide, used alone or in combination with sulfur, has been demonstrated in the treatment of rosacea, acne, seborrheic dermatitis, and perioral dermatitis. Advances in formulation technology to decrease odor and irritation have allowed for more use of this product. Further studies will help elucidate the role that sodium sulfacetamide should play in the treatment of inflammatory dermatoses in comparison to other available products.
1. Akhavan A, Bershad S. Topical acne drugs: review of clinical properties, systemic exposure, and safety. Am J Clin Dermatol. 2003;4:473-492.
2. Gupta AK, Nicol K. The use of sulfur in dermatology. J Drugs Dermatol. 2004;3:427-431.
3. Motaparthi K, Hsu S. Topical antibacterial agents. In: Wolverton SE, ed. Comprehensive Dermatologic Drug Therapy. 3rd ed. Philadelphia, PA: Saunders; 2012:445-459.
4. Duemling WM. Sodium sulfacetamide in topical therapy. AMA Arch Derm Syphilol. 1954;69:75-82.
5. Sodium sulfacetamide. Drugs.com Web site. http://drugs.com/pro/sodium-sulfacetamide.html. Revised December 2012. Accessed June 16, 2015.
6. Sauder DN, Miller R, Gratton D, et al. The treatment of rosacea: the safety and efficacy of sodium sulfacetamide 10% and sulfur 5% lotion (Novacet) is demonstrated in a double-blind study. J Dermatol Treat. 1997;8:79-85.
7. Trumbore MW, Goldstein JA, Gurge RM. Treatment of papulopustular rosacea with sodium sulfacetamide 10%/sulfur 5% emollient foam. J Drugs Dermatol. 2009;8:299-304.
8. Lebwohl MG, Medansky RS, Russo CL, et al. The comparative efficacy of sodium sulfacetamide 10%/sulfur 5% lotion and metronidazole 0.75% gel in the treatment of rosacea. J Geriatr Dermatol. 1995;3:183-185.
9. Nally JB, Berson DS. Topical therapies for rosacea. J Drugs Dermatol. 2006;5:23-26.
10. Pelle MT, Crawford GH, James WD. Rosacea II: therapy. J Am Acad Dermatol. 2004;51:499-512.
11. Tarimci N, Sener S, Kilinç T. Topical sodium sulfacetamide/sulfur lotion. J Clin Pharm Ther. 1997;22:301.
12. Breneman DL, Ariano MC. Successful treatment of acne vulgaris in women with a new topical sodium sulfacetamide/sulfur lotion. Int J Dermatol. 1993;32:365-367.
13. Whelan ST. Sodium sulfacetamide for seborrheic dermatitis. AMA Arch Derm. 1955;71:724.
14. Bendl BJ. Perioral dermatitis: etiology and treatment. Cutis. 1976;17:903-908.
15. Olansky S. Old drug—in a new system—revisited. Cutis. 1977;19:852-854.
16. Genvert GI, Cohen EJ, Donnenfeld ED, et al. Erythema multiforme after use of topical sulfacetamide. Am J Ophthalmol. 1985;99:465-468.
17. Rubin Z. Ophthalmic sulfonamide-induced Stevens-Johnson syndrome. Arch Dermatol. 1977;113:235-236.
18. Draelos ZD. The multifunctionality of 10% sodium sulfacetamide, 5% sulfur emollient foam in the treatment of inflammatory facial dermatoses. J Drugs Dermatol. 2010;9:234-246.
19. Dubina MI, Fleischer AB. Interaction of topical sulfacetamide and topical dapsone with benzoyl peroxide. Arch Dermatol. 2009;145:1027-1029.
20. Sodium sulfacetamide – sulfacetamide sodium liquid. DailyMed Web site. http://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=0d92c55b-5b54-4f5d-8921-24e4e877ae50. Accessed June 17, 2015.
21. Spes CH, Angermann CE, Stempfle HU, et al. Sulfadiazine therapy for toxoplasmosis in heart transplant recipients decreases cyclosporine concentration. Clin Investig. 1992;70:752-754.
22. Hansen JM, Kampmann JP, Siersbaek-Nielsen K, et al. The effect of different sulfonamides on phenytoin metabolism in man. Acta Med Scand Suppl. 1979;624:106-110.
23. Bradley JS, Sauberan JB. Antimicrobial agents. In: Long SS, Pickering LK, Prober CG. Principles and Practices of Pediatric Infectious Diseases. 4th ed. Philadelphia, PA: Elsevier Saunders; 2012:1453-1483.
Sodium sulfacetamide has various uses in the field of dermatology due to its anti-inflammatory and antibacterial properties. It has been shown to be effective in the management of a variety of inflammatory facial dermatoses, including papulopustular rosacea, acne vulgaris, seborrheic dermatitis, and perioral dermatitis. We review the mechanism of action, pharmacology and formulations, clinical uses, and adverse effects of sodium sulfacetamide as a dermatologic treatment.
Mechanism of Action
Sodium sulfacetamide is a sulfonamide-type antibacterial agent. Its mechanism of action is the inhibition of bacterial dihydropteroate synthetase, which prevents the conversion of p-aminobenzoic acid to folic acid. This process causes a bacteriostatic effect on the growth of several gram-negative and gram-positive organisms, including Propionibacterium acnes.1,2
The effectiveness of sodium sulfacetamide is increased when used in combination with sulfur, which has keratolytic, antibacterial, antifungal, and antiparasitic effects. The addition of hydrocortisone has been reported to increase the effectiveness of both agents.3
Pharmacology
Sodium sulfacetamide is highly soluble at the physiologic pH of 7.4, which contributes to its high level of penetration and absorption.4 An in vitro study showed percutaneous absorption of sodium sulfacetamide to be around 4%.5 Sulfonamides are metabolized mainly by the liver and are excreted by the kidneys.
Formulations
The most common concentrations of sodium sulfacetamide and sulfur are 10% and 5%, respectively. A wide variety of sulfacetamide-containing products are available, many of which are marketed to treat specific conditions depending on additional ingredients or the type of delivery system.
Clinical Uses
Topical formulations of sodium sulfacetamide and sulfur have proven to be efficacious in the management of rosacea, with a typical regimen consisting of twice-daily application for 8 weeks.6 The sulfur in the formulation has the additional benefit of targeting Demodex mites, which are implicated as a contributing factor in some cases of rosacea.7 Sodium sulfacetamide 10%–sulfur 5% lotion was more effective in improving the erythema, papulopustules, and overall severity of rosacea as compared to metronidazole gel 0.75%.8 Other studies have reported increased efficacy when sodium sulfacetamide and topical sulfur are used along with metronidazole.9,10
Sodium sulfacetamide also has shown efficacy against acne. Its antibacterial and drying properties have been shown to decrease the number of inflammatory lesions and comedones, and in the treatment of acne vulgaris, no sensitivity reactions have been observed.2 Also, unlike topical antibiotics, cases of P acnes resistance to topical sulfur products have not been widely reported. Studies have demonstrated that twice-daily use of sodium sulfacetamide 10%–sulfur 5% for 12 weeks decreases inflammatory acne lesions by 80.4% to 83%.11,12
Seborrheic dermatitis is a common chronic infection of the skin caused by Malassezia species. One study investigated the use of sodium sulfacetamide ointment and soap to treat seborrheic dermatitis and found that the condition was either improved or completely controlled in 93% (71/76) of cases.4 Sodium sulfacetamide lotion was an effective treatment of seborrheic dermatitis in 89% (54/61) of patients with scalp involvement and 68% (30/44) of patients with glabrous skin involvement.13
Perioral dermatitis is characterized by groups of erythematous papules and pustules localized around the mouth. The use of topical sodium sulfacetamide along with oral tetracyclines has been demonstrated to consistently clear lesions in most patients with perioral dermatitis.14 Sodium sulfacetamide is unique in that it is not associated with the excessive erythema and irritation often found with retinoic acid and benzoyl peroxide.15 Unfortunately, however, there have been no well-controlled trials to compare the efficacy of sodium sulfacetamide to other topical therapies for this condition.
Adverse Effects
Adverse effects from sodium sulfacetamide are rare and generally are limited to cutaneous reactions including dryness, erythema, pruritus, and discomfort.1 Periocular use of sodium sulfacetamide can cause conjunctival irritation. One study reported that 19% (6/31) of patients experienced local reactions but most were considered mild.9 Rare but serious reactions including erythema multiforme and Stevens-Johnson syndrome have been reported from ophthalmic use.16,17
A common limiting factor to sodium sulfacetamide preparations that include elemental sulfur is the offensive smell, which has hindered patient compliance in the past; however, pharmaceutical companies have attempted to create more tolerable products without the odor.10 One study found that the tolerability of a sodium sulfacetamide 10%–sulfur 5% foam using a rinse-off method of application was excellent, with only 33% (8/24) of participants commenting on the smell.18 Another limiting factor of sodium sulfacetamide preparations containing sulfur is orange-brown discoloration when combined with benzoyl peroxide, which does not affect the skin but may stain clothing.19
Sodium sulfacetamide is rendered less effective when combined with silver-containing products.20 No other notable drug interactions are known; however, oral sulfonamides are known to interact with several drugs, including cyclosporine and phenytoin.21,22
Contraindications
Sodium sulfacetamide is contraindicated in patients with known hypersensitivity to sulfonamides, sulfur, or any other component of the preparation. It is a pregnancy category C drug, and pregnant women should only use sodium sulfacetamide if it is the only modality to treat the condition or the benefits outweigh the risks. Although there are no known reports of problems related to topical sodium sulfacetamide during pregnancy, the use of oral sulfonamides during pregnancy can increase the risk for neonatal jaundice.23 Likewise, caution should be exercised in prescribing this product to nursing women, as systemic sulfonamide antibacterials are well known to cause kernicterus in nursing neonates.1
Conclusion
The efficacy and safety of sodium sulfacetamide, used alone or in combination with sulfur, has been demonstrated in the treatment of rosacea, acne, seborrheic dermatitis, and perioral dermatitis. Advances in formulation technology to decrease odor and irritation have allowed for more use of this product. Further studies will help elucidate the role that sodium sulfacetamide should play in the treatment of inflammatory dermatoses in comparison to other available products.
Sodium sulfacetamide has various uses in the field of dermatology due to its anti-inflammatory and antibacterial properties. It has been shown to be effective in the management of a variety of inflammatory facial dermatoses, including papulopustular rosacea, acne vulgaris, seborrheic dermatitis, and perioral dermatitis. We review the mechanism of action, pharmacology and formulations, clinical uses, and adverse effects of sodium sulfacetamide as a dermatologic treatment.
Mechanism of Action
Sodium sulfacetamide is a sulfonamide-type antibacterial agent. Its mechanism of action is the inhibition of bacterial dihydropteroate synthetase, which prevents the conversion of p-aminobenzoic acid to folic acid. This process causes a bacteriostatic effect on the growth of several gram-negative and gram-positive organisms, including Propionibacterium acnes.1,2
The effectiveness of sodium sulfacetamide is increased when used in combination with sulfur, which has keratolytic, antibacterial, antifungal, and antiparasitic effects. The addition of hydrocortisone has been reported to increase the effectiveness of both agents.3
Pharmacology
Sodium sulfacetamide is highly soluble at the physiologic pH of 7.4, which contributes to its high level of penetration and absorption.4 An in vitro study showed percutaneous absorption of sodium sulfacetamide to be around 4%.5 Sulfonamides are metabolized mainly by the liver and are excreted by the kidneys.
Formulations
The most common concentrations of sodium sulfacetamide and sulfur are 10% and 5%, respectively. A wide variety of sulfacetamide-containing products are available, many of which are marketed to treat specific conditions depending on additional ingredients or the type of delivery system.
Clinical Uses
Topical formulations of sodium sulfacetamide and sulfur have proven to be efficacious in the management of rosacea, with a typical regimen consisting of twice-daily application for 8 weeks.6 The sulfur in the formulation has the additional benefit of targeting Demodex mites, which are implicated as a contributing factor in some cases of rosacea.7 Sodium sulfacetamide 10%–sulfur 5% lotion was more effective in improving the erythema, papulopustules, and overall severity of rosacea as compared to metronidazole gel 0.75%.8 Other studies have reported increased efficacy when sodium sulfacetamide and topical sulfur are used along with metronidazole.9,10
Sodium sulfacetamide also has shown efficacy against acne. Its antibacterial and drying properties have been shown to decrease the number of inflammatory lesions and comedones, and in the treatment of acne vulgaris, no sensitivity reactions have been observed.2 Also, unlike topical antibiotics, cases of P acnes resistance to topical sulfur products have not been widely reported. Studies have demonstrated that twice-daily use of sodium sulfacetamide 10%–sulfur 5% for 12 weeks decreases inflammatory acne lesions by 80.4% to 83%.11,12
Seborrheic dermatitis is a common chronic infection of the skin caused by Malassezia species. One study investigated the use of sodium sulfacetamide ointment and soap to treat seborrheic dermatitis and found that the condition was either improved or completely controlled in 93% (71/76) of cases.4 Sodium sulfacetamide lotion was an effective treatment of seborrheic dermatitis in 89% (54/61) of patients with scalp involvement and 68% (30/44) of patients with glabrous skin involvement.13
Perioral dermatitis is characterized by groups of erythematous papules and pustules localized around the mouth. The use of topical sodium sulfacetamide along with oral tetracyclines has been demonstrated to consistently clear lesions in most patients with perioral dermatitis.14 Sodium sulfacetamide is unique in that it is not associated with the excessive erythema and irritation often found with retinoic acid and benzoyl peroxide.15 Unfortunately, however, there have been no well-controlled trials to compare the efficacy of sodium sulfacetamide to other topical therapies for this condition.
Adverse Effects
Adverse effects from sodium sulfacetamide are rare and generally are limited to cutaneous reactions including dryness, erythema, pruritus, and discomfort.1 Periocular use of sodium sulfacetamide can cause conjunctival irritation. One study reported that 19% (6/31) of patients experienced local reactions but most were considered mild.9 Rare but serious reactions including erythema multiforme and Stevens-Johnson syndrome have been reported from ophthalmic use.16,17
A common limiting factor to sodium sulfacetamide preparations that include elemental sulfur is the offensive smell, which has hindered patient compliance in the past; however, pharmaceutical companies have attempted to create more tolerable products without the odor.10 One study found that the tolerability of a sodium sulfacetamide 10%–sulfur 5% foam using a rinse-off method of application was excellent, with only 33% (8/24) of participants commenting on the smell.18 Another limiting factor of sodium sulfacetamide preparations containing sulfur is orange-brown discoloration when combined with benzoyl peroxide, which does not affect the skin but may stain clothing.19
Sodium sulfacetamide is rendered less effective when combined with silver-containing products.20 No other notable drug interactions are known; however, oral sulfonamides are known to interact with several drugs, including cyclosporine and phenytoin.21,22
Contraindications
Sodium sulfacetamide is contraindicated in patients with known hypersensitivity to sulfonamides, sulfur, or any other component of the preparation. It is a pregnancy category C drug, and pregnant women should only use sodium sulfacetamide if it is the only modality to treat the condition or the benefits outweigh the risks. Although there are no known reports of problems related to topical sodium sulfacetamide during pregnancy, the use of oral sulfonamides during pregnancy can increase the risk for neonatal jaundice.23 Likewise, caution should be exercised in prescribing this product to nursing women, as systemic sulfonamide antibacterials are well known to cause kernicterus in nursing neonates.1
Conclusion
The efficacy and safety of sodium sulfacetamide, used alone or in combination with sulfur, has been demonstrated in the treatment of rosacea, acne, seborrheic dermatitis, and perioral dermatitis. Advances in formulation technology to decrease odor and irritation have allowed for more use of this product. Further studies will help elucidate the role that sodium sulfacetamide should play in the treatment of inflammatory dermatoses in comparison to other available products.
1. Akhavan A, Bershad S. Topical acne drugs: review of clinical properties, systemic exposure, and safety. Am J Clin Dermatol. 2003;4:473-492.
2. Gupta AK, Nicol K. The use of sulfur in dermatology. J Drugs Dermatol. 2004;3:427-431.
3. Motaparthi K, Hsu S. Topical antibacterial agents. In: Wolverton SE, ed. Comprehensive Dermatologic Drug Therapy. 3rd ed. Philadelphia, PA: Saunders; 2012:445-459.
4. Duemling WM. Sodium sulfacetamide in topical therapy. AMA Arch Derm Syphilol. 1954;69:75-82.
5. Sodium sulfacetamide. Drugs.com Web site. http://drugs.com/pro/sodium-sulfacetamide.html. Revised December 2012. Accessed June 16, 2015.
6. Sauder DN, Miller R, Gratton D, et al. The treatment of rosacea: the safety and efficacy of sodium sulfacetamide 10% and sulfur 5% lotion (Novacet) is demonstrated in a double-blind study. J Dermatol Treat. 1997;8:79-85.
7. Trumbore MW, Goldstein JA, Gurge RM. Treatment of papulopustular rosacea with sodium sulfacetamide 10%/sulfur 5% emollient foam. J Drugs Dermatol. 2009;8:299-304.
8. Lebwohl MG, Medansky RS, Russo CL, et al. The comparative efficacy of sodium sulfacetamide 10%/sulfur 5% lotion and metronidazole 0.75% gel in the treatment of rosacea. J Geriatr Dermatol. 1995;3:183-185.
9. Nally JB, Berson DS. Topical therapies for rosacea. J Drugs Dermatol. 2006;5:23-26.
10. Pelle MT, Crawford GH, James WD. Rosacea II: therapy. J Am Acad Dermatol. 2004;51:499-512.
11. Tarimci N, Sener S, Kilinç T. Topical sodium sulfacetamide/sulfur lotion. J Clin Pharm Ther. 1997;22:301.
12. Breneman DL, Ariano MC. Successful treatment of acne vulgaris in women with a new topical sodium sulfacetamide/sulfur lotion. Int J Dermatol. 1993;32:365-367.
13. Whelan ST. Sodium sulfacetamide for seborrheic dermatitis. AMA Arch Derm. 1955;71:724.
14. Bendl BJ. Perioral dermatitis: etiology and treatment. Cutis. 1976;17:903-908.
15. Olansky S. Old drug—in a new system—revisited. Cutis. 1977;19:852-854.
16. Genvert GI, Cohen EJ, Donnenfeld ED, et al. Erythema multiforme after use of topical sulfacetamide. Am J Ophthalmol. 1985;99:465-468.
17. Rubin Z. Ophthalmic sulfonamide-induced Stevens-Johnson syndrome. Arch Dermatol. 1977;113:235-236.
18. Draelos ZD. The multifunctionality of 10% sodium sulfacetamide, 5% sulfur emollient foam in the treatment of inflammatory facial dermatoses. J Drugs Dermatol. 2010;9:234-246.
19. Dubina MI, Fleischer AB. Interaction of topical sulfacetamide and topical dapsone with benzoyl peroxide. Arch Dermatol. 2009;145:1027-1029.
20. Sodium sulfacetamide – sulfacetamide sodium liquid. DailyMed Web site. http://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=0d92c55b-5b54-4f5d-8921-24e4e877ae50. Accessed June 17, 2015.
21. Spes CH, Angermann CE, Stempfle HU, et al. Sulfadiazine therapy for toxoplasmosis in heart transplant recipients decreases cyclosporine concentration. Clin Investig. 1992;70:752-754.
22. Hansen JM, Kampmann JP, Siersbaek-Nielsen K, et al. The effect of different sulfonamides on phenytoin metabolism in man. Acta Med Scand Suppl. 1979;624:106-110.
23. Bradley JS, Sauberan JB. Antimicrobial agents. In: Long SS, Pickering LK, Prober CG. Principles and Practices of Pediatric Infectious Diseases. 4th ed. Philadelphia, PA: Elsevier Saunders; 2012:1453-1483.
1. Akhavan A, Bershad S. Topical acne drugs: review of clinical properties, systemic exposure, and safety. Am J Clin Dermatol. 2003;4:473-492.
2. Gupta AK, Nicol K. The use of sulfur in dermatology. J Drugs Dermatol. 2004;3:427-431.
3. Motaparthi K, Hsu S. Topical antibacterial agents. In: Wolverton SE, ed. Comprehensive Dermatologic Drug Therapy. 3rd ed. Philadelphia, PA: Saunders; 2012:445-459.
4. Duemling WM. Sodium sulfacetamide in topical therapy. AMA Arch Derm Syphilol. 1954;69:75-82.
5. Sodium sulfacetamide. Drugs.com Web site. http://drugs.com/pro/sodium-sulfacetamide.html. Revised December 2012. Accessed June 16, 2015.
6. Sauder DN, Miller R, Gratton D, et al. The treatment of rosacea: the safety and efficacy of sodium sulfacetamide 10% and sulfur 5% lotion (Novacet) is demonstrated in a double-blind study. J Dermatol Treat. 1997;8:79-85.
7. Trumbore MW, Goldstein JA, Gurge RM. Treatment of papulopustular rosacea with sodium sulfacetamide 10%/sulfur 5% emollient foam. J Drugs Dermatol. 2009;8:299-304.
8. Lebwohl MG, Medansky RS, Russo CL, et al. The comparative efficacy of sodium sulfacetamide 10%/sulfur 5% lotion and metronidazole 0.75% gel in the treatment of rosacea. J Geriatr Dermatol. 1995;3:183-185.
9. Nally JB, Berson DS. Topical therapies for rosacea. J Drugs Dermatol. 2006;5:23-26.
10. Pelle MT, Crawford GH, James WD. Rosacea II: therapy. J Am Acad Dermatol. 2004;51:499-512.
11. Tarimci N, Sener S, Kilinç T. Topical sodium sulfacetamide/sulfur lotion. J Clin Pharm Ther. 1997;22:301.
12. Breneman DL, Ariano MC. Successful treatment of acne vulgaris in women with a new topical sodium sulfacetamide/sulfur lotion. Int J Dermatol. 1993;32:365-367.
13. Whelan ST. Sodium sulfacetamide for seborrheic dermatitis. AMA Arch Derm. 1955;71:724.
14. Bendl BJ. Perioral dermatitis: etiology and treatment. Cutis. 1976;17:903-908.
15. Olansky S. Old drug—in a new system—revisited. Cutis. 1977;19:852-854.
16. Genvert GI, Cohen EJ, Donnenfeld ED, et al. Erythema multiforme after use of topical sulfacetamide. Am J Ophthalmol. 1985;99:465-468.
17. Rubin Z. Ophthalmic sulfonamide-induced Stevens-Johnson syndrome. Arch Dermatol. 1977;113:235-236.
18. Draelos ZD. The multifunctionality of 10% sodium sulfacetamide, 5% sulfur emollient foam in the treatment of inflammatory facial dermatoses. J Drugs Dermatol. 2010;9:234-246.
19. Dubina MI, Fleischer AB. Interaction of topical sulfacetamide and topical dapsone with benzoyl peroxide. Arch Dermatol. 2009;145:1027-1029.
20. Sodium sulfacetamide – sulfacetamide sodium liquid. DailyMed Web site. http://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=0d92c55b-5b54-4f5d-8921-24e4e877ae50. Accessed June 17, 2015.
21. Spes CH, Angermann CE, Stempfle HU, et al. Sulfadiazine therapy for toxoplasmosis in heart transplant recipients decreases cyclosporine concentration. Clin Investig. 1992;70:752-754.
22. Hansen JM, Kampmann JP, Siersbaek-Nielsen K, et al. The effect of different sulfonamides on phenytoin metabolism in man. Acta Med Scand Suppl. 1979;624:106-110.
23. Bradley JS, Sauberan JB. Antimicrobial agents. In: Long SS, Pickering LK, Prober CG. Principles and Practices of Pediatric Infectious Diseases. 4th ed. Philadelphia, PA: Elsevier Saunders; 2012:1453-1483.
Practice Points
- Sodium sulfacetamide is a useful agent in the management of papulopustular rosacea, acne vulgaris, seborrheic dermatitis, and perioral dermatitis.
- Adverse effects are rare and generally are limited to dryness, erythema, pruritus, and discomfort.

Onychomatricoma: An Often Misdiagnosed Tumor of the Nails
Changes in the appearance of the nail apparatus can be produced by a variety of conditions. Onychomatricoma is an unusual benign tumor with specific clinical characteristics that was first described more than 2 decades ago.1 It is often and easily misdiagnosed because the condition rarely has been described. We report a case of onychomatricoma in a 54-year-old Colombian man who presented with a deformity of the nail plate on the right index finger that corresponded with the classic description of onychomatricoma. We emphasize the importance of reporting such lesions to prevent misdiagnosis and delay of proper treatment.
Case Report
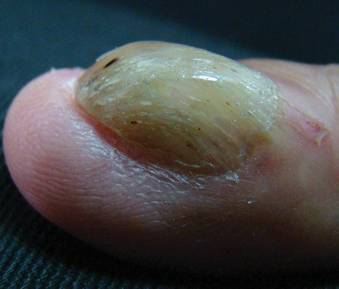
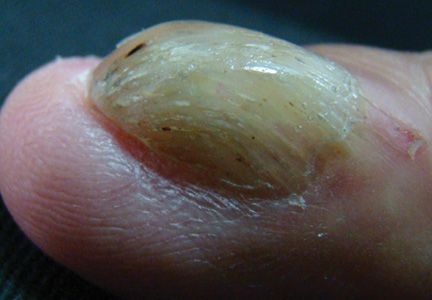
A 54-year-old Colombian man presented with nail dystrophy involving the right index finger of 2 years’ duration. He did not recall any trauma prior to the onset of the nail abnormalities. Several topical treatments had previously been ineffective. Physical examination revealed a longitudinally banded thickening of the lateral half of the nail plate on the right index finger with yellowish brown discoloration, transverse overcurvature of the nail, longitudinal white lines, and splinter hemorrhages (Figure 1). Direct microscopy and fungal culture were performed to diagnose or rule out onychomycosis.
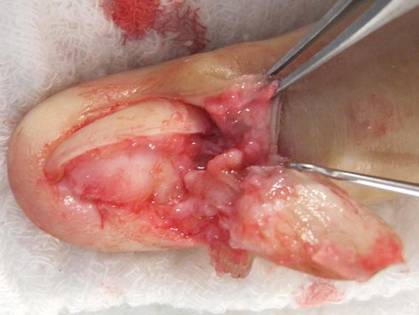
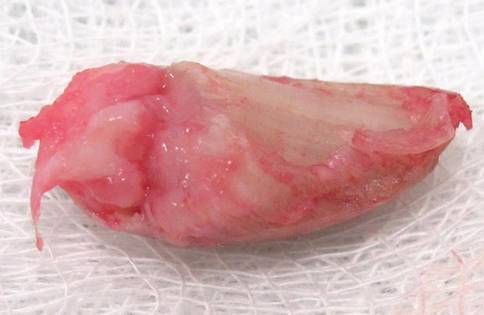
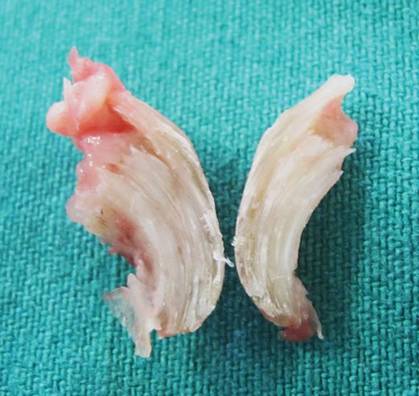
A clinical diagnosis of onychomatricoma was made, and the lesion was surgically removed and sent for histopathologic study (Figure 2). The radial half of the nail plate was avulsed, and the proximal part of the removed nail plate contained a large, firmly attached, filamentous tumor arising from the nail matrix (Figure 3) with multiple fine filiform projections (Figure 4). The nail bed was cleaned with a curette to remove any debris, the ulnar half of the nail plate and nail bed was left in place, and the radial border was reconstructed. Histology confirmed the clinical diagnosis (Figure 5). No recurrences of the tumor were seen 36 months following surgery.
|  |
Comment
Since the original report of this tumor,1 fewer than 10 cases of onychomatricoma have been reported in Latin America,2-5 with no more than 80 cases reported worldwide.6 Clinicians and academicians are becoming interested in the topic, which will result in better recognition and more reports in the literature.
The clinical differential diagnosis of onycho-matricoma is extensive,7,8 but onychomatricoma has characteristic clinical and histopathologic features that allow its separation from other nail disorders and subungual tumors (Table).9 There are 4 cardinal clinical signs that suggest a diagnosis of onychomatricoma: (1) banded or diffuse thickening of the nail plate of variable widths; (2) yellowish discoloration of the involved nail plate, often showing fine splinter hemorrhages in the proximal nail portion; (3) transverse overcurvature of the nail; and (4) exposure of a filamentous tufted tumor emerging from the matrix in a funnel-shaped nail by avulsion.1
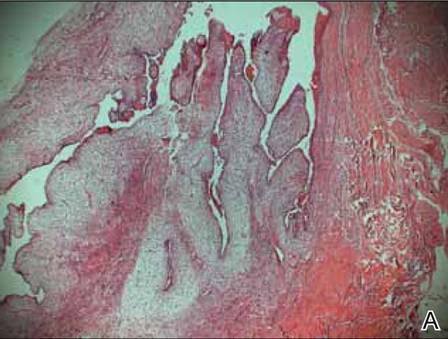 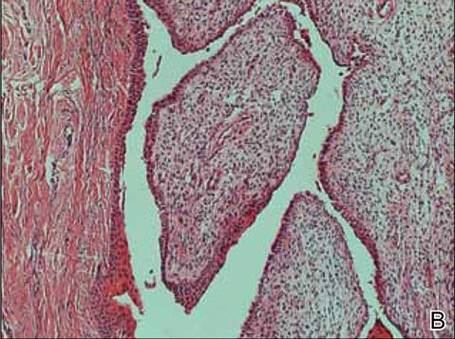 |
Histologic findings of onychomatricoma typically demonstrate a fibroepithelial tumor with a biphasic growth pattern mimicking normal nail matrix histology, including a proximal zone, which corresponds to the base of the fibroepithelial tumor, and a distal zone, which is composed of multiple epithelial digitations that extend into the small cavities present in the attached nail.10-12 Nevertheless, the anatomic tumor location, the often fragmented aspect of the tissue specimen, and the choice of the section planes may change the typical histologic features seen in onychomatricoma.13 Stromal prominence, cellularity, and atypia may vary in individual cases.10-12
The etiology of onychomatricoma is not yet known. Although it has been suggested that onychomatricoma could be an epithelial and connective tissue hamartoma simulating the nail matrix structure,1,10 the more recent concept of an epithelial onychogenic tumor with onychogenic mesenchyme will help to clarify its etiology because new histopathologic and immunohistochemical features suggest a neoplastic nature.14 On the other hand, predisposing factors such as trauma to the nail plate and onychomycosis may play a role,7 as the thumbs, index fingers, and great toes are more susceptible to onychomycosis and accidental trauma.
Conclusion
Our patient fulfilled the criteria of onychomatricoma.1 Onychomatricoma should be kept in mind in the differential diagnosis of subungual or periungual tumors to avoid misdiagnosis and erroneous treatments.
1. Baran R, Kint A. Onychomatrixoma: filamentous tufted tumor in the matrix of a funnel-shaped nail: a new entity (report of three cases). Br J Dermatol. 1992;126:510-515.
2. Estrada-Chavez G, Vega-Memije ME, Toussaint-Caire S, et al. Giant onychomatricoma: report of two cases with rare clinical presentation. Int J Dermatol. 2007;46: 634-636.
3. Soto R, Wortsman X, Corredoira Y. Onychomatricoma: clinical and sonographic findings. Arch Dermatol. 2009;145:1461-1462.
4. Tavares GT, Chiacchio NG, Chiacchio ND, et al. Onychomatricoma: a tumor unknown to dermatologists. An Bras Dermatol. 2015;90:265-267.
5. Fernández-Sánchez M, Saeb-Lima M, Charli-Joseph Y, et al. Onychomatricoma: an infrequent nail tumor. Indian J Dermatol Venereol Leprol. 2012;78:382-383.
6. Tavares G, Di-Chiacchio N, Di-Santis E, et al. Onycho-matricoma: epidemiological and clinical findings in a large series of 30 cases [published online ahead of print May 12, 2015]. Br J Dermatol. doi:10.1111/bjd.13900.
7. Rashid RM, Swan J. Onychomatricoma: benign sporadic nail lesion or much more? Dermatol Online J. 2006;12:4.
8. Goutos I, Furniss D, Smith GD. Onychomatricoma: an unusual case of ungual pathology. case report and review of the literature. J Plast Reconstr Aesthet Surg. 2010;63:54-57.
9. Fraga GR, Patterson JW, McHargue CA. Onychomatricoma: report of a case and its comparison with fibrokeratoma of the nailbed. Am J Dermatopathol. 2001;23:36-40.
10. Perrin C, Goettmann S, Baran R. Onychomatricoma: clinical and histopathologic findings in 12 cases. J Am Acad Dermatol. 1998;39:560-564.
11. Gaertner EM, Gordon M, Reed T. Onychomatricoma: case report of an unusual subungual tumor with literature review. J Cutan Pathol. 2009;36(suppl 1):S66-S69.
12. Perrin C, Baran R, Pisani A, et al. The onychomatricoma: additional histologic criteria and immunohistochemical study. Am J Dermatopathol. 2002;24:199-203.
13. Perrin C, Baran R, Balaguer T, et al. Onychomatricoma: new clinical and histological features. a review of 19 tumors. Am J Dermatopathol. 2010;32:1-8.
14. Perrin C, Langbein L, Schweizer J, et al. Onychomatricoma in the light of the microanatomy of the normal nail unit. Am J Dermatopathol. 2011;33:131-139.
Changes in the appearance of the nail apparatus can be produced by a variety of conditions. Onychomatricoma is an unusual benign tumor with specific clinical characteristics that was first described more than 2 decades ago.1 It is often and easily misdiagnosed because the condition rarely has been described. We report a case of onychomatricoma in a 54-year-old Colombian man who presented with a deformity of the nail plate on the right index finger that corresponded with the classic description of onychomatricoma. We emphasize the importance of reporting such lesions to prevent misdiagnosis and delay of proper treatment.
Case Report
A 54-year-old Colombian man presented with nail dystrophy involving the right index finger of 2 years’ duration. He did not recall any trauma prior to the onset of the nail abnormalities. Several topical treatments had previously been ineffective. Physical examination revealed a longitudinally banded thickening of the lateral half of the nail plate on the right index finger with yellowish brown discoloration, transverse overcurvature of the nail, longitudinal white lines, and splinter hemorrhages (Figure 1). Direct microscopy and fungal culture were performed to diagnose or rule out onychomycosis.
A clinical diagnosis of onychomatricoma was made, and the lesion was surgically removed and sent for histopathologic study (Figure 2). The radial half of the nail plate was avulsed, and the proximal part of the removed nail plate contained a large, firmly attached, filamentous tumor arising from the nail matrix (Figure 3) with multiple fine filiform projections (Figure 4). The nail bed was cleaned with a curette to remove any debris, the ulnar half of the nail plate and nail bed was left in place, and the radial border was reconstructed. Histology confirmed the clinical diagnosis (Figure 5). No recurrences of the tumor were seen 36 months following surgery.
| |
Comment
Since the original report of this tumor,1 fewer than 10 cases of onychomatricoma have been reported in Latin America,2-5 with no more than 80 cases reported worldwide.6 Clinicians and academicians are becoming interested in the topic, which will result in better recognition and more reports in the literature.
The clinical differential diagnosis of onycho-matricoma is extensive,7,8 but onychomatricoma has characteristic clinical and histopathologic features that allow its separation from other nail disorders and subungual tumors (Table).9 There are 4 cardinal clinical signs that suggest a diagnosis of onychomatricoma: (1) banded or diffuse thickening of the nail plate of variable widths; (2) yellowish discoloration of the involved nail plate, often showing fine splinter hemorrhages in the proximal nail portion; (3) transverse overcurvature of the nail; and (4) exposure of a filamentous tufted tumor emerging from the matrix in a funnel-shaped nail by avulsion.1
|
Histologic findings of onychomatricoma typically demonstrate a fibroepithelial tumor with a biphasic growth pattern mimicking normal nail matrix histology, including a proximal zone, which corresponds to the base of the fibroepithelial tumor, and a distal zone, which is composed of multiple epithelial digitations that extend into the small cavities present in the attached nail.10-12 Nevertheless, the anatomic tumor location, the often fragmented aspect of the tissue specimen, and the choice of the section planes may change the typical histologic features seen in onychomatricoma.13 Stromal prominence, cellularity, and atypia may vary in individual cases.10-12
The etiology of onychomatricoma is not yet known. Although it has been suggested that onychomatricoma could be an epithelial and connective tissue hamartoma simulating the nail matrix structure,1,10 the more recent concept of an epithelial onychogenic tumor with onychogenic mesenchyme will help to clarify its etiology because new histopathologic and immunohistochemical features suggest a neoplastic nature.14 On the other hand, predisposing factors such as trauma to the nail plate and onychomycosis may play a role,7 as the thumbs, index fingers, and great toes are more susceptible to onychomycosis and accidental trauma.
Conclusion
Our patient fulfilled the criteria of onychomatricoma.1 Onychomatricoma should be kept in mind in the differential diagnosis of subungual or periungual tumors to avoid misdiagnosis and erroneous treatments.
Changes in the appearance of the nail apparatus can be produced by a variety of conditions. Onychomatricoma is an unusual benign tumor with specific clinical characteristics that was first described more than 2 decades ago.1 It is often and easily misdiagnosed because the condition rarely has been described. We report a case of onychomatricoma in a 54-year-old Colombian man who presented with a deformity of the nail plate on the right index finger that corresponded with the classic description of onychomatricoma. We emphasize the importance of reporting such lesions to prevent misdiagnosis and delay of proper treatment.
Case Report
A 54-year-old Colombian man presented with nail dystrophy involving the right index finger of 2 years’ duration. He did not recall any trauma prior to the onset of the nail abnormalities. Several topical treatments had previously been ineffective. Physical examination revealed a longitudinally banded thickening of the lateral half of the nail plate on the right index finger with yellowish brown discoloration, transverse overcurvature of the nail, longitudinal white lines, and splinter hemorrhages (Figure 1). Direct microscopy and fungal culture were performed to diagnose or rule out onychomycosis.
A clinical diagnosis of onychomatricoma was made, and the lesion was surgically removed and sent for histopathologic study (Figure 2). The radial half of the nail plate was avulsed, and the proximal part of the removed nail plate contained a large, firmly attached, filamentous tumor arising from the nail matrix (Figure 3) with multiple fine filiform projections (Figure 4). The nail bed was cleaned with a curette to remove any debris, the ulnar half of the nail plate and nail bed was left in place, and the radial border was reconstructed. Histology confirmed the clinical diagnosis (Figure 5). No recurrences of the tumor were seen 36 months following surgery.
| |
Comment
Since the original report of this tumor,1 fewer than 10 cases of onychomatricoma have been reported in Latin America,2-5 with no more than 80 cases reported worldwide.6 Clinicians and academicians are becoming interested in the topic, which will result in better recognition and more reports in the literature.
The clinical differential diagnosis of onycho-matricoma is extensive,7,8 but onychomatricoma has characteristic clinical and histopathologic features that allow its separation from other nail disorders and subungual tumors (Table).9 There are 4 cardinal clinical signs that suggest a diagnosis of onychomatricoma: (1) banded or diffuse thickening of the nail plate of variable widths; (2) yellowish discoloration of the involved nail plate, often showing fine splinter hemorrhages in the proximal nail portion; (3) transverse overcurvature of the nail; and (4) exposure of a filamentous tufted tumor emerging from the matrix in a funnel-shaped nail by avulsion.1
|
Histologic findings of onychomatricoma typically demonstrate a fibroepithelial tumor with a biphasic growth pattern mimicking normal nail matrix histology, including a proximal zone, which corresponds to the base of the fibroepithelial tumor, and a distal zone, which is composed of multiple epithelial digitations that extend into the small cavities present in the attached nail.10-12 Nevertheless, the anatomic tumor location, the often fragmented aspect of the tissue specimen, and the choice of the section planes may change the typical histologic features seen in onychomatricoma.13 Stromal prominence, cellularity, and atypia may vary in individual cases.10-12
The etiology of onychomatricoma is not yet known. Although it has been suggested that onychomatricoma could be an epithelial and connective tissue hamartoma simulating the nail matrix structure,1,10 the more recent concept of an epithelial onychogenic tumor with onychogenic mesenchyme will help to clarify its etiology because new histopathologic and immunohistochemical features suggest a neoplastic nature.14 On the other hand, predisposing factors such as trauma to the nail plate and onychomycosis may play a role,7 as the thumbs, index fingers, and great toes are more susceptible to onychomycosis and accidental trauma.
Conclusion
Our patient fulfilled the criteria of onychomatricoma.1 Onychomatricoma should be kept in mind in the differential diagnosis of subungual or periungual tumors to avoid misdiagnosis and erroneous treatments.
1. Baran R, Kint A. Onychomatrixoma: filamentous tufted tumor in the matrix of a funnel-shaped nail: a new entity (report of three cases). Br J Dermatol. 1992;126:510-515.
2. Estrada-Chavez G, Vega-Memije ME, Toussaint-Caire S, et al. Giant onychomatricoma: report of two cases with rare clinical presentation. Int J Dermatol. 2007;46: 634-636.
3. Soto R, Wortsman X, Corredoira Y. Onychomatricoma: clinical and sonographic findings. Arch Dermatol. 2009;145:1461-1462.
4. Tavares GT, Chiacchio NG, Chiacchio ND, et al. Onychomatricoma: a tumor unknown to dermatologists. An Bras Dermatol. 2015;90:265-267.
5. Fernández-Sánchez M, Saeb-Lima M, Charli-Joseph Y, et al. Onychomatricoma: an infrequent nail tumor. Indian J Dermatol Venereol Leprol. 2012;78:382-383.
6. Tavares G, Di-Chiacchio N, Di-Santis E, et al. Onycho-matricoma: epidemiological and clinical findings in a large series of 30 cases [published online ahead of print May 12, 2015]. Br J Dermatol. doi:10.1111/bjd.13900.
7. Rashid RM, Swan J. Onychomatricoma: benign sporadic nail lesion or much more? Dermatol Online J. 2006;12:4.
8. Goutos I, Furniss D, Smith GD. Onychomatricoma: an unusual case of ungual pathology. case report and review of the literature. J Plast Reconstr Aesthet Surg. 2010;63:54-57.
9. Fraga GR, Patterson JW, McHargue CA. Onychomatricoma: report of a case and its comparison with fibrokeratoma of the nailbed. Am J Dermatopathol. 2001;23:36-40.
10. Perrin C, Goettmann S, Baran R. Onychomatricoma: clinical and histopathologic findings in 12 cases. J Am Acad Dermatol. 1998;39:560-564.
11. Gaertner EM, Gordon M, Reed T. Onychomatricoma: case report of an unusual subungual tumor with literature review. J Cutan Pathol. 2009;36(suppl 1):S66-S69.
12. Perrin C, Baran R, Pisani A, et al. The onychomatricoma: additional histologic criteria and immunohistochemical study. Am J Dermatopathol. 2002;24:199-203.
13. Perrin C, Baran R, Balaguer T, et al. Onychomatricoma: new clinical and histological features. a review of 19 tumors. Am J Dermatopathol. 2010;32:1-8.
14. Perrin C, Langbein L, Schweizer J, et al. Onychomatricoma in the light of the microanatomy of the normal nail unit. Am J Dermatopathol. 2011;33:131-139.
1. Baran R, Kint A. Onychomatrixoma: filamentous tufted tumor in the matrix of a funnel-shaped nail: a new entity (report of three cases). Br J Dermatol. 1992;126:510-515.
2. Estrada-Chavez G, Vega-Memije ME, Toussaint-Caire S, et al. Giant onychomatricoma: report of two cases with rare clinical presentation. Int J Dermatol. 2007;46: 634-636.
3. Soto R, Wortsman X, Corredoira Y. Onychomatricoma: clinical and sonographic findings. Arch Dermatol. 2009;145:1461-1462.
4. Tavares GT, Chiacchio NG, Chiacchio ND, et al. Onychomatricoma: a tumor unknown to dermatologists. An Bras Dermatol. 2015;90:265-267.
5. Fernández-Sánchez M, Saeb-Lima M, Charli-Joseph Y, et al. Onychomatricoma: an infrequent nail tumor. Indian J Dermatol Venereol Leprol. 2012;78:382-383.
6. Tavares G, Di-Chiacchio N, Di-Santis E, et al. Onycho-matricoma: epidemiological and clinical findings in a large series of 30 cases [published online ahead of print May 12, 2015]. Br J Dermatol. doi:10.1111/bjd.13900.
7. Rashid RM, Swan J. Onychomatricoma: benign sporadic nail lesion or much more? Dermatol Online J. 2006;12:4.
8. Goutos I, Furniss D, Smith GD. Onychomatricoma: an unusual case of ungual pathology. case report and review of the literature. J Plast Reconstr Aesthet Surg. 2010;63:54-57.
9. Fraga GR, Patterson JW, McHargue CA. Onychomatricoma: report of a case and its comparison with fibrokeratoma of the nailbed. Am J Dermatopathol. 2001;23:36-40.
10. Perrin C, Goettmann S, Baran R. Onychomatricoma: clinical and histopathologic findings in 12 cases. J Am Acad Dermatol. 1998;39:560-564.
11. Gaertner EM, Gordon M, Reed T. Onychomatricoma: case report of an unusual subungual tumor with literature review. J Cutan Pathol. 2009;36(suppl 1):S66-S69.
12. Perrin C, Baran R, Pisani A, et al. The onychomatricoma: additional histologic criteria and immunohistochemical study. Am J Dermatopathol. 2002;24:199-203.
13. Perrin C, Baran R, Balaguer T, et al. Onychomatricoma: new clinical and histological features. a review of 19 tumors. Am J Dermatopathol. 2010;32:1-8.
14. Perrin C, Langbein L, Schweizer J, et al. Onychomatricoma in the light of the microanatomy of the normal nail unit. Am J Dermatopathol. 2011;33:131-139.
Practice Points
- Onychomatricoma has been described mostly in white individuals, but it can occur in all races and ethnic groups.
- Onychomatricoma should be kept in mind in the differential diagnosis of subungual or periungual tumors.
- Treatment of onychomatricoma is complete surgical excision.
How to Teach the Potassium Hydroxide Preparation: A Disappearing Clinical Art Form
Potassium hydroxide (KOH) preparations remain an important bedside test for prompt and accurate diagnosis of superficial fungal infections known as dermatophytoses. This tool has been used for at least 100 years, with early terminology referring to it as potash; for the last century, it has largely been a technique passed down as a skill from master technician to learning apprentice. The original pioneer of the KOH preparation remains a mystery.1
Variations on techniques for performing the KOH preparation exist, and tips and tricks on the use of this test are a hot topic among dermatologists.2 Although primary care and dermatology-specific publications espouse the importance of the KOH preparation,3,4 it has unfortunately been identified and labeled as one of the forgotten diagnostic tools.5
It is incumbent on dermatologists to educate medical students and residents using a simple and specific method to ensure that this simple and effective technique, with sensitivity reported between 87% and 91% depending on the expertise of the examiner,6 remains part of the clinical armamentarium. One concern in the instruction of large groups of students and clinicians is the ready accessibility or availability of viable skin samples. This article describes a method of collecting and storing skin samples that will allow educators to train large groups of students on performing KOH preparations without having to repeatedly seek skin samples or patients with superficial skin infections. A detailed description of the pedagogy used to teach the preparation and interpretation of KOH slides to a large group of students also is reviewed.
Specimen Collection
The first step in teaching the KOH preparation to a large group is the collection of a suitable number of skin scrapings from patients with a superficial fungal skin infection (eg, tinea corporis, tinea versicolor). A common technique for obtaining skin samples is to use a no. 15 scalpel blade (Figure 1) to scrape the scale of the lesion at its scaly border once the area is moistened with an alcohol pad or soap and water.7 The moisture from the alcohol pad allows the scale to stick to the no. 15 blade, facilitating collection. Once a suitable amount of scale is collected, it is placed on a glass microscope slide by smearing the scale from the blade onto the slide. This process has been modified to facilitate a larger quantity of specimen as follows: dermatophyte-infected plaques with scale are rubbed with the no. 15 blade and the free scale drops into a standard urine specimen cup. This process is repeated multiple times from different sites to capture the displaced scale with the dermatophyte. We have found that as long as the specimen cups are sealed tightly and stored in a relatively dry and cool environment (room temperature), the samples can be used to construct KOH teaching slides for at least 3 years. We have not used them beyond 3 years but suspect that they would continue to be viable after this time.

Preparation of Slides
Given that time for teaching often is limited, it is beneficial to fix many skin scrapings on a large number of glass slides prior to the session, which enables students to simply add KOH to the slides on the teaching day. To prepare the slides in advance, it is necessary to gather the following materials: a specimen cup with skin samples, glass slides, pickups or tweezers, a small pipette, a cup of water, protective gloves, and a pencil. After donning protective gloves, the pickups or tweezers are used to retrieve a few flakes of scale from the specimen cup and place them on the center of a glass slide. Using the pipette, 1 or 2 drops of water are added to the scale, and the slide is then allowed to dry. The slides are marked with the pencil to indicate the “up” side to prevent the students from applying KOH solution to the wrong side of the slide. The skin scale is fixed in place on the slide as the water evaporates and may be stored until needed for use in a standard slide box or folder.
Performing the KOH Preparation
On the day of teaching, it is helpful to engage the entire group of students with an introductory lecture on the purpose and use of the KOH preparation. Upon completion, students move to a workstation with all of the materials needed to prepare the slide. Additional items needed at this time are 10% KOH solution, coverslips, and a heating device (eg, lighter, Bunsen burner, match)(optional). Students are instructed to place 1 or 2 skin scales onto a glass slide or retrieve a slide with skin scales already fixed, and then add 1 drop of 10% KOH solution directly to the sample (Figure 2). Next, they should place a slide coverslip onto the KOH drop and skin sample using a side-to-side technique that will move the scale into a thin layer within the KOH solution and push away any excess solution to the periphery (Figure 3). Large amounts of excess KOH solution should be cleared away with a paper towel, lens paper, or tissue. The heat source can be used to gently heat the underside of the glass slide (Figure 4), but it often is sufficient to simply wait 3 to 5 minutes for the KOH solution to take effect. The heat accelerates the maceration of the scale and makes it easier to see the hyphae among the keratinocytes. Some physicians advocate the use of dimethyl sulfoxide in lieu of heating,8 but this solution may not be available in all primary care settings.
 |  |

Microscopic Examination
Prior to examining the slides under the microscope, students may complete a self-guided tutorial (eg, digital or paper slide show) on the various features seen through the microscope that are indicative of dermatophytes, including branching hyphae and yeast buds. They also should be educated about the common appearance of artifacts that may resemble hyphae. Once the students have completed the tutorial, they may proceed to microscopic examination.
While the students are viewing their slides under the microscope, we find it helpful to have at least 1 experienced faculty member for every group of 10 students. This instructor should encourage the students to lower the microscope condenser all the way to facilitate better observation. Students should start with low power (×4 or red band) and scan for areas that are rich in skin scale. Once a collection of scale is found, the student can switch to higher power (×10 or yellow band) and start scanning for hyphae. Students should be reminded to search for filamentous and branching tubes that are refractile. The term refractile may be confusing to some students, so we explain that shifting the focus up or down will show the hyphae to change in brightness and may reveal a greenish tint. Another helpful indicator to point out is the feature that hyphae will cross the border of epidermal skin cells, whereas artifacts will not (Figure 5). Once the students have identified evidence of a dermatophyte infection, they must call the instructor to their station to verify the presence of hyphae or yeast buds, which helps confirm their understanding of the procedure. Once the student accurately identifies these items, the session is complete.
Comment
The use of a KOH preparation is a fast, simple, accurate, and cost-effective way to diagnose superficial fungal infections; however, because of insufficient familiarity with this tool, the technique often is replaced by initiation of empiric antifungal therapy in patients with suspected dermatophytosis. This empiric treatment has the potential to delay appropriate diagnosis and treatment (eg, in a patient with nummular dermatitis, which can clinically mimic tinea corporis). One way to encourage the use of the KOH preparation in the primary care and dermatologic setting is to educate large groups of next-generation physicians while in medical training. This article describes a teaching technique that allows for long-term storage of positive skin samples and a detailed description of the pedagogy used to train and educate a large group of students in a relatively short period of time.
All KOH preparations fall under the US federal government’s Clinical Laboratory Improvement Amendments and require proficiency testing.9 Although the teaching method presented here is designed for teaching medical students, it may be utilized to educate or refamiliarize experienced physicians with the procedure in an effort to improve proficiency in point-of-care testing programs used in many health care systems to comply with the Clinical Laboratories Improvement Amendments. Future analyses could assess whether the method described here improves provider performance on such proficiency measures and whether it ultimately helps ensure quality patient care.
1. Dasgupta T, Sahu J. Origins of the KOH technique. Clin Dermatol. 2012;2:238-242.
2. Stone S. Editor’s commentary. Clin Dermatol. 2012;2:241-242.
3. Monroe JR. The diagnostic value of a KOH. JAAPA. 2001;4:50-51.
4. Hainer BL. Dermatophyte infections. Am Fam Physician. 2003;1:101-109.
5. Ponka D, Baddar F. Microscopic potassium hydroxide preparation. Can Fam Physician. 2014;60:57.
6. Lilly KK, Koshnick RL, Grill JP, et al. Cost-effectiveness of diagnostic tests for toenail onychomycosis: a repeated-measure, single-blinded, cross-sectional evaluation of 7 diagnostic tests. J Am Acad Dermatol. 2006;4:620-626.
7. Bolognia JL, Jorizzo JL, Schaffer JV. Dermatology. 3rd ed. New York, NY: Elsevier Saunders; 2012.
8. James WD, Berger T, Elston D. Andrew’s Diseases of the Skin: Clinical Dermatology. 11th ed. New York, NY: Elsevier Saunders; 2011.
9. Clinical Laboratory Improvement Amendments (CLIA). Centers for Medicare & Medicaid Services Web site. https://www.cms.gov/Regulations-and-Guidance/Legislation/CLIA/index.html?redirect=/clia/. Updated June 6, 2015. Accessed July 21, 2015.
Potassium hydroxide (KOH) preparations remain an important bedside test for prompt and accurate diagnosis of superficial fungal infections known as dermatophytoses. This tool has been used for at least 100 years, with early terminology referring to it as potash; for the last century, it has largely been a technique passed down as a skill from master technician to learning apprentice. The original pioneer of the KOH preparation remains a mystery.1
Variations on techniques for performing the KOH preparation exist, and tips and tricks on the use of this test are a hot topic among dermatologists.2 Although primary care and dermatology-specific publications espouse the importance of the KOH preparation,3,4 it has unfortunately been identified and labeled as one of the forgotten diagnostic tools.5
It is incumbent on dermatologists to educate medical students and residents using a simple and specific method to ensure that this simple and effective technique, with sensitivity reported between 87% and 91% depending on the expertise of the examiner,6 remains part of the clinical armamentarium. One concern in the instruction of large groups of students and clinicians is the ready accessibility or availability of viable skin samples. This article describes a method of collecting and storing skin samples that will allow educators to train large groups of students on performing KOH preparations without having to repeatedly seek skin samples or patients with superficial skin infections. A detailed description of the pedagogy used to teach the preparation and interpretation of KOH slides to a large group of students also is reviewed.
Specimen Collection
The first step in teaching the KOH preparation to a large group is the collection of a suitable number of skin scrapings from patients with a superficial fungal skin infection (eg, tinea corporis, tinea versicolor). A common technique for obtaining skin samples is to use a no. 15 scalpel blade (Figure 1) to scrape the scale of the lesion at its scaly border once the area is moistened with an alcohol pad or soap and water.7 The moisture from the alcohol pad allows the scale to stick to the no. 15 blade, facilitating collection. Once a suitable amount of scale is collected, it is placed on a glass microscope slide by smearing the scale from the blade onto the slide. This process has been modified to facilitate a larger quantity of specimen as follows: dermatophyte-infected plaques with scale are rubbed with the no. 15 blade and the free scale drops into a standard urine specimen cup. This process is repeated multiple times from different sites to capture the displaced scale with the dermatophyte. We have found that as long as the specimen cups are sealed tightly and stored in a relatively dry and cool environment (room temperature), the samples can be used to construct KOH teaching slides for at least 3 years. We have not used them beyond 3 years but suspect that they would continue to be viable after this time.
Preparation of Slides
Given that time for teaching often is limited, it is beneficial to fix many skin scrapings on a large number of glass slides prior to the session, which enables students to simply add KOH to the slides on the teaching day. To prepare the slides in advance, it is necessary to gather the following materials: a specimen cup with skin samples, glass slides, pickups or tweezers, a small pipette, a cup of water, protective gloves, and a pencil. After donning protective gloves, the pickups or tweezers are used to retrieve a few flakes of scale from the specimen cup and place them on the center of a glass slide. Using the pipette, 1 or 2 drops of water are added to the scale, and the slide is then allowed to dry. The slides are marked with the pencil to indicate the “up” side to prevent the students from applying KOH solution to the wrong side of the slide. The skin scale is fixed in place on the slide as the water evaporates and may be stored until needed for use in a standard slide box or folder.
Performing the KOH Preparation
On the day of teaching, it is helpful to engage the entire group of students with an introductory lecture on the purpose and use of the KOH preparation. Upon completion, students move to a workstation with all of the materials needed to prepare the slide. Additional items needed at this time are 10% KOH solution, coverslips, and a heating device (eg, lighter, Bunsen burner, match)(optional). Students are instructed to place 1 or 2 skin scales onto a glass slide or retrieve a slide with skin scales already fixed, and then add 1 drop of 10% KOH solution directly to the sample (Figure 2). Next, they should place a slide coverslip onto the KOH drop and skin sample using a side-to-side technique that will move the scale into a thin layer within the KOH solution and push away any excess solution to the periphery (Figure 3). Large amounts of excess KOH solution should be cleared away with a paper towel, lens paper, or tissue. The heat source can be used to gently heat the underside of the glass slide (Figure 4), but it often is sufficient to simply wait 3 to 5 minutes for the KOH solution to take effect. The heat accelerates the maceration of the scale and makes it easier to see the hyphae among the keratinocytes. Some physicians advocate the use of dimethyl sulfoxide in lieu of heating,8 but this solution may not be available in all primary care settings.
| |
Microscopic Examination
Prior to examining the slides under the microscope, students may complete a self-guided tutorial (eg, digital or paper slide show) on the various features seen through the microscope that are indicative of dermatophytes, including branching hyphae and yeast buds. They also should be educated about the common appearance of artifacts that may resemble hyphae. Once the students have completed the tutorial, they may proceed to microscopic examination.
While the students are viewing their slides under the microscope, we find it helpful to have at least 1 experienced faculty member for every group of 10 students. This instructor should encourage the students to lower the microscope condenser all the way to facilitate better observation. Students should start with low power (×4 or red band) and scan for areas that are rich in skin scale. Once a collection of scale is found, the student can switch to higher power (×10 or yellow band) and start scanning for hyphae. Students should be reminded to search for filamentous and branching tubes that are refractile. The term refractile may be confusing to some students, so we explain that shifting the focus up or down will show the hyphae to change in brightness and may reveal a greenish tint. Another helpful indicator to point out is the feature that hyphae will cross the border of epidermal skin cells, whereas artifacts will not (Figure 5). Once the students have identified evidence of a dermatophyte infection, they must call the instructor to their station to verify the presence of hyphae or yeast buds, which helps confirm their understanding of the procedure. Once the student accurately identifies these items, the session is complete.
Comment
The use of a KOH preparation is a fast, simple, accurate, and cost-effective way to diagnose superficial fungal infections; however, because of insufficient familiarity with this tool, the technique often is replaced by initiation of empiric antifungal therapy in patients with suspected dermatophytosis. This empiric treatment has the potential to delay appropriate diagnosis and treatment (eg, in a patient with nummular dermatitis, which can clinically mimic tinea corporis). One way to encourage the use of the KOH preparation in the primary care and dermatologic setting is to educate large groups of next-generation physicians while in medical training. This article describes a teaching technique that allows for long-term storage of positive skin samples and a detailed description of the pedagogy used to train and educate a large group of students in a relatively short period of time.
All KOH preparations fall under the US federal government’s Clinical Laboratory Improvement Amendments and require proficiency testing.9 Although the teaching method presented here is designed for teaching medical students, it may be utilized to educate or refamiliarize experienced physicians with the procedure in an effort to improve proficiency in point-of-care testing programs used in many health care systems to comply with the Clinical Laboratories Improvement Amendments. Future analyses could assess whether the method described here improves provider performance on such proficiency measures and whether it ultimately helps ensure quality patient care.
Potassium hydroxide (KOH) preparations remain an important bedside test for prompt and accurate diagnosis of superficial fungal infections known as dermatophytoses. This tool has been used for at least 100 years, with early terminology referring to it as potash; for the last century, it has largely been a technique passed down as a skill from master technician to learning apprentice. The original pioneer of the KOH preparation remains a mystery.1
Variations on techniques for performing the KOH preparation exist, and tips and tricks on the use of this test are a hot topic among dermatologists.2 Although primary care and dermatology-specific publications espouse the importance of the KOH preparation,3,4 it has unfortunately been identified and labeled as one of the forgotten diagnostic tools.5
It is incumbent on dermatologists to educate medical students and residents using a simple and specific method to ensure that this simple and effective technique, with sensitivity reported between 87% and 91% depending on the expertise of the examiner,6 remains part of the clinical armamentarium. One concern in the instruction of large groups of students and clinicians is the ready accessibility or availability of viable skin samples. This article describes a method of collecting and storing skin samples that will allow educators to train large groups of students on performing KOH preparations without having to repeatedly seek skin samples or patients with superficial skin infections. A detailed description of the pedagogy used to teach the preparation and interpretation of KOH slides to a large group of students also is reviewed.
Specimen Collection
The first step in teaching the KOH preparation to a large group is the collection of a suitable number of skin scrapings from patients with a superficial fungal skin infection (eg, tinea corporis, tinea versicolor). A common technique for obtaining skin samples is to use a no. 15 scalpel blade (Figure 1) to scrape the scale of the lesion at its scaly border once the area is moistened with an alcohol pad or soap and water.7 The moisture from the alcohol pad allows the scale to stick to the no. 15 blade, facilitating collection. Once a suitable amount of scale is collected, it is placed on a glass microscope slide by smearing the scale from the blade onto the slide. This process has been modified to facilitate a larger quantity of specimen as follows: dermatophyte-infected plaques with scale are rubbed with the no. 15 blade and the free scale drops into a standard urine specimen cup. This process is repeated multiple times from different sites to capture the displaced scale with the dermatophyte. We have found that as long as the specimen cups are sealed tightly and stored in a relatively dry and cool environment (room temperature), the samples can be used to construct KOH teaching slides for at least 3 years. We have not used them beyond 3 years but suspect that they would continue to be viable after this time.
Preparation of Slides
Given that time for teaching often is limited, it is beneficial to fix many skin scrapings on a large number of glass slides prior to the session, which enables students to simply add KOH to the slides on the teaching day. To prepare the slides in advance, it is necessary to gather the following materials: a specimen cup with skin samples, glass slides, pickups or tweezers, a small pipette, a cup of water, protective gloves, and a pencil. After donning protective gloves, the pickups or tweezers are used to retrieve a few flakes of scale from the specimen cup and place them on the center of a glass slide. Using the pipette, 1 or 2 drops of water are added to the scale, and the slide is then allowed to dry. The slides are marked with the pencil to indicate the “up” side to prevent the students from applying KOH solution to the wrong side of the slide. The skin scale is fixed in place on the slide as the water evaporates and may be stored until needed for use in a standard slide box or folder.
Performing the KOH Preparation
On the day of teaching, it is helpful to engage the entire group of students with an introductory lecture on the purpose and use of the KOH preparation. Upon completion, students move to a workstation with all of the materials needed to prepare the slide. Additional items needed at this time are 10% KOH solution, coverslips, and a heating device (eg, lighter, Bunsen burner, match)(optional). Students are instructed to place 1 or 2 skin scales onto a glass slide or retrieve a slide with skin scales already fixed, and then add 1 drop of 10% KOH solution directly to the sample (Figure 2). Next, they should place a slide coverslip onto the KOH drop and skin sample using a side-to-side technique that will move the scale into a thin layer within the KOH solution and push away any excess solution to the periphery (Figure 3). Large amounts of excess KOH solution should be cleared away with a paper towel, lens paper, or tissue. The heat source can be used to gently heat the underside of the glass slide (Figure 4), but it often is sufficient to simply wait 3 to 5 minutes for the KOH solution to take effect. The heat accelerates the maceration of the scale and makes it easier to see the hyphae among the keratinocytes. Some physicians advocate the use of dimethyl sulfoxide in lieu of heating,8 but this solution may not be available in all primary care settings.
| |
Microscopic Examination
Prior to examining the slides under the microscope, students may complete a self-guided tutorial (eg, digital or paper slide show) on the various features seen through the microscope that are indicative of dermatophytes, including branching hyphae and yeast buds. They also should be educated about the common appearance of artifacts that may resemble hyphae. Once the students have completed the tutorial, they may proceed to microscopic examination.
While the students are viewing their slides under the microscope, we find it helpful to have at least 1 experienced faculty member for every group of 10 students. This instructor should encourage the students to lower the microscope condenser all the way to facilitate better observation. Students should start with low power (×4 or red band) and scan for areas that are rich in skin scale. Once a collection of scale is found, the student can switch to higher power (×10 or yellow band) and start scanning for hyphae. Students should be reminded to search for filamentous and branching tubes that are refractile. The term refractile may be confusing to some students, so we explain that shifting the focus up or down will show the hyphae to change in brightness and may reveal a greenish tint. Another helpful indicator to point out is the feature that hyphae will cross the border of epidermal skin cells, whereas artifacts will not (Figure 5). Once the students have identified evidence of a dermatophyte infection, they must call the instructor to their station to verify the presence of hyphae or yeast buds, which helps confirm their understanding of the procedure. Once the student accurately identifies these items, the session is complete.
Comment
The use of a KOH preparation is a fast, simple, accurate, and cost-effective way to diagnose superficial fungal infections; however, because of insufficient familiarity with this tool, the technique often is replaced by initiation of empiric antifungal therapy in patients with suspected dermatophytosis. This empiric treatment has the potential to delay appropriate diagnosis and treatment (eg, in a patient with nummular dermatitis, which can clinically mimic tinea corporis). One way to encourage the use of the KOH preparation in the primary care and dermatologic setting is to educate large groups of next-generation physicians while in medical training. This article describes a teaching technique that allows for long-term storage of positive skin samples and a detailed description of the pedagogy used to train and educate a large group of students in a relatively short period of time.
All KOH preparations fall under the US federal government’s Clinical Laboratory Improvement Amendments and require proficiency testing.9 Although the teaching method presented here is designed for teaching medical students, it may be utilized to educate or refamiliarize experienced physicians with the procedure in an effort to improve proficiency in point-of-care testing programs used in many health care systems to comply with the Clinical Laboratories Improvement Amendments. Future analyses could assess whether the method described here improves provider performance on such proficiency measures and whether it ultimately helps ensure quality patient care.
1. Dasgupta T, Sahu J. Origins of the KOH technique. Clin Dermatol. 2012;2:238-242.
2. Stone S. Editor’s commentary. Clin Dermatol. 2012;2:241-242.
3. Monroe JR. The diagnostic value of a KOH. JAAPA. 2001;4:50-51.
4. Hainer BL. Dermatophyte infections. Am Fam Physician. 2003;1:101-109.
5. Ponka D, Baddar F. Microscopic potassium hydroxide preparation. Can Fam Physician. 2014;60:57.
6. Lilly KK, Koshnick RL, Grill JP, et al. Cost-effectiveness of diagnostic tests for toenail onychomycosis: a repeated-measure, single-blinded, cross-sectional evaluation of 7 diagnostic tests. J Am Acad Dermatol. 2006;4:620-626.
7. Bolognia JL, Jorizzo JL, Schaffer JV. Dermatology. 3rd ed. New York, NY: Elsevier Saunders; 2012.
8. James WD, Berger T, Elston D. Andrew’s Diseases of the Skin: Clinical Dermatology. 11th ed. New York, NY: Elsevier Saunders; 2011.
9. Clinical Laboratory Improvement Amendments (CLIA). Centers for Medicare & Medicaid Services Web site. https://www.cms.gov/Regulations-and-Guidance/Legislation/CLIA/index.html?redirect=/clia/. Updated June 6, 2015. Accessed July 21, 2015.
1. Dasgupta T, Sahu J. Origins of the KOH technique. Clin Dermatol. 2012;2:238-242.
2. Stone S. Editor’s commentary. Clin Dermatol. 2012;2:241-242.
3. Monroe JR. The diagnostic value of a KOH. JAAPA. 2001;4:50-51.
4. Hainer BL. Dermatophyte infections. Am Fam Physician. 2003;1:101-109.
5. Ponka D, Baddar F. Microscopic potassium hydroxide preparation. Can Fam Physician. 2014;60:57.
6. Lilly KK, Koshnick RL, Grill JP, et al. Cost-effectiveness of diagnostic tests for toenail onychomycosis: a repeated-measure, single-blinded, cross-sectional evaluation of 7 diagnostic tests. J Am Acad Dermatol. 2006;4:620-626.
7. Bolognia JL, Jorizzo JL, Schaffer JV. Dermatology. 3rd ed. New York, NY: Elsevier Saunders; 2012.
8. James WD, Berger T, Elston D. Andrew’s Diseases of the Skin: Clinical Dermatology. 11th ed. New York, NY: Elsevier Saunders; 2011.
9. Clinical Laboratory Improvement Amendments (CLIA). Centers for Medicare & Medicaid Services Web site. https://www.cms.gov/Regulations-and-Guidance/Legislation/CLIA/index.html?redirect=/clia/. Updated June 6, 2015. Accessed July 21, 2015.
Practice Points
- Potassium hydroxide (KOH) preparations can lead to diagnostic confidence and direct appropriate therapy.
- Refreshing the basics of this simple technique can lead to better patient outcomes in the primary care setting and in the dermatology specialty clinic.
- Teaching the KOH preparation to the next generation of physicians will ensure its longevity and assure future benefit to patients.
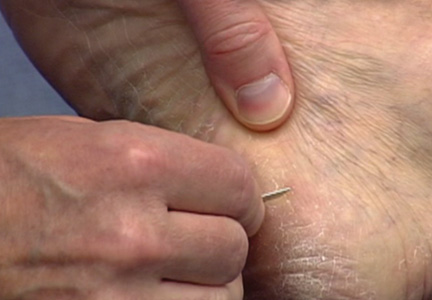
Diffuse Crusting of the Skin
The Diagnosis: Crusted Scabies
Approximately 1 year prior to presentation the patient completed a course of therapy comprised of repeat doses of ivermectin and permethrin for crusted scabies. He stated that the crusts had recurred shortly after treatment (Figure 1). The initial differential diagnosis included crusted scabies, pityriasis rubra pilaris, and psoriasis. Scraping from the interdigital web space (Figure 2) revealed multiple scabies mites, scabella, and ova (Figure 3). The patient was diagnosed with crusted scabies. Laboratory test results revealed a normal white blood cell count of 7300/μL, with an elevated eosinophil count of 1900/μL (reference range, <500/μL). Human T-lymphotropic virus 1 (HTLV-1) was found to be positive. Serum protein electrophoresis showed a moderate polyclonal increase in gamma globulins. Treatment was promptly initiated with permethrin cream 5% applied to the whole body for a total of 2 treatments that were administered 7 days apart. Ivermectin also was started at 200 μg/kg and was given on days 1, 7, 14, and 29. Salicylic acid cream 25% was applied daily to help hasten resolution of the crusts.
 |  |
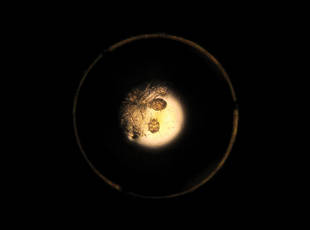
Human T-lymphotropic virus 1 is endemic to Japan, Central and South America, the Middle East, and regions of Africa and the Caribbean.1 Multiple cases of HTLV-1 associated with crusted scabies have been reported in endemic areas.2-6 In the present case, we encountered a patient with crusted scabies and HTLV-1 in a nonendemic area.
Cases of HTLV-1 and crusted scabies have been reported in Brazil, Peru, and Central Australia.3,4,7 A retrospective study in Brazil showed a notable association between HTLV-1 infection and crusted scabies; of 23 patients with crusted scabies, 21 were HTLV-1 positive.3 In another small study from Peru, 23 patients with crusted scabies were tested for HTLV-1 and 16 (70%) were seropositive.4 Neither study had a control group for comparison; however, interestingly, the estimated seroprevalence in the general population (based on blood donors) is only 0.04% to 1% and 1.2% to 1.7% in Brazil and Peru, respectively, thus reinforcing the association between HTLV-1 and crusted scabies.1 Crusted scabies and HTLV-1 seropositivity acting as a predictor for adult T-cell leukemia/lymphoma (ATL) has been reported8 but not consistently.3 Our patient had abnormal serum protein electrophoresis findings. Although the results were not diagnostic of ATL, patients with HTLV-1 are at increased risk for ATL and the pres-ence of this virus is important to document when there is a high index of suspicion for ATL, as with crusted scabies.
Our patient resided in the northeastern United States and there was no evidence in support of underlying immunosuppression. This case of crusted scabies is unique because the patient was HTLV-1 positive without other underlying immunocompromise and he resided in a region where HTLV-1 is nonendemic. In light of these findings, the present case serves as a reminder to check for HTLV-1 infection in those patients with severe or crusted scabies and without an identifiable reason for immunosuppression, even if the patient resides in an area that is nonendemic for HTLV-1.
- Gessain A, Cassar O. Epidemiological aspects and world distribution of HTLV-1 infection. Front Microbiol. 2012;3:388.
- Nobre V, Guedes AC, Martins ML, et al. Dermatological findings in 3 generations of a family with a high prevalence of human T cell lymphotropic virus type 1 infection in Brazil. Clin Infect Dis. 2006;43:1257-1263.
- Brites C, Weyll M, Pedroso C, et al. Severe and Norwegian scabies are strongly associated with retroviral (HIV-1/HTLV-1) infection in Bahia, Brazil. AIDS. 2002;16:1292-1293.
- Blas M, Bravo F, Castillo W, et al. Norwegian scabies in Peru: the impact of human T cell lymphotropic virus type I infection. Am J Trop Med Hyg. 2005;72:855-857.
- Bergman JN, Dodd WA, Trotter MJ, et al. Crusted scabies in association with human T-cell lymphotropic virus 1. J Cutan Med Surg. 1999;3:148-152.
- Daisley H, Charles W, Suite M. Crusted (Norwegian) scabies as a pre-diagnostic indicator for HTLV-1 infection. Trans R Soc Trop Med Hyg. 1993;87:295.
- Mollison LC. HTLV-I and clinical disease correlates in central Australian aborigines. Med J Aust. 1994;160:238.
- del Giudice P, Sainte Marie D, Gérard Y, et al. Is crusted (Norwegian) scabies a marker of adult T cell leukemia/lymphoma in human T lymphotropic virus type I–seropositive patients? J Infect Dis. 1997;176:1090-1092.
The Diagnosis: Crusted Scabies
Approximately 1 year prior to presentation the patient completed a course of therapy comprised of repeat doses of ivermectin and permethrin for crusted scabies. He stated that the crusts had recurred shortly after treatment (Figure 1). The initial differential diagnosis included crusted scabies, pityriasis rubra pilaris, and psoriasis. Scraping from the interdigital web space (Figure 2) revealed multiple scabies mites, scabella, and ova (Figure 3). The patient was diagnosed with crusted scabies. Laboratory test results revealed a normal white blood cell count of 7300/μL, with an elevated eosinophil count of 1900/μL (reference range, <500/μL). Human T-lymphotropic virus 1 (HTLV-1) was found to be positive. Serum protein electrophoresis showed a moderate polyclonal increase in gamma globulins. Treatment was promptly initiated with permethrin cream 5% applied to the whole body for a total of 2 treatments that were administered 7 days apart. Ivermectin also was started at 200 μg/kg and was given on days 1, 7, 14, and 29. Salicylic acid cream 25% was applied daily to help hasten resolution of the crusts.
| |
Human T-lymphotropic virus 1 is endemic to Japan, Central and South America, the Middle East, and regions of Africa and the Caribbean.1 Multiple cases of HTLV-1 associated with crusted scabies have been reported in endemic areas.2-6 In the present case, we encountered a patient with crusted scabies and HTLV-1 in a nonendemic area.
Cases of HTLV-1 and crusted scabies have been reported in Brazil, Peru, and Central Australia.3,4,7 A retrospective study in Brazil showed a notable association between HTLV-1 infection and crusted scabies; of 23 patients with crusted scabies, 21 were HTLV-1 positive.3 In another small study from Peru, 23 patients with crusted scabies were tested for HTLV-1 and 16 (70%) were seropositive.4 Neither study had a control group for comparison; however, interestingly, the estimated seroprevalence in the general population (based on blood donors) is only 0.04% to 1% and 1.2% to 1.7% in Brazil and Peru, respectively, thus reinforcing the association between HTLV-1 and crusted scabies.1 Crusted scabies and HTLV-1 seropositivity acting as a predictor for adult T-cell leukemia/lymphoma (ATL) has been reported8 but not consistently.3 Our patient had abnormal serum protein electrophoresis findings. Although the results were not diagnostic of ATL, patients with HTLV-1 are at increased risk for ATL and the pres-ence of this virus is important to document when there is a high index of suspicion for ATL, as with crusted scabies.
Our patient resided in the northeastern United States and there was no evidence in support of underlying immunosuppression. This case of crusted scabies is unique because the patient was HTLV-1 positive without other underlying immunocompromise and he resided in a region where HTLV-1 is nonendemic. In light of these findings, the present case serves as a reminder to check for HTLV-1 infection in those patients with severe or crusted scabies and without an identifiable reason for immunosuppression, even if the patient resides in an area that is nonendemic for HTLV-1.
The Diagnosis: Crusted Scabies
Approximately 1 year prior to presentation the patient completed a course of therapy comprised of repeat doses of ivermectin and permethrin for crusted scabies. He stated that the crusts had recurred shortly after treatment (Figure 1). The initial differential diagnosis included crusted scabies, pityriasis rubra pilaris, and psoriasis. Scraping from the interdigital web space (Figure 2) revealed multiple scabies mites, scabella, and ova (Figure 3). The patient was diagnosed with crusted scabies. Laboratory test results revealed a normal white blood cell count of 7300/μL, with an elevated eosinophil count of 1900/μL (reference range, <500/μL). Human T-lymphotropic virus 1 (HTLV-1) was found to be positive. Serum protein electrophoresis showed a moderate polyclonal increase in gamma globulins. Treatment was promptly initiated with permethrin cream 5% applied to the whole body for a total of 2 treatments that were administered 7 days apart. Ivermectin also was started at 200 μg/kg and was given on days 1, 7, 14, and 29. Salicylic acid cream 25% was applied daily to help hasten resolution of the crusts.
| |
Human T-lymphotropic virus 1 is endemic to Japan, Central and South America, the Middle East, and regions of Africa and the Caribbean.1 Multiple cases of HTLV-1 associated with crusted scabies have been reported in endemic areas.2-6 In the present case, we encountered a patient with crusted scabies and HTLV-1 in a nonendemic area.
Cases of HTLV-1 and crusted scabies have been reported in Brazil, Peru, and Central Australia.3,4,7 A retrospective study in Brazil showed a notable association between HTLV-1 infection and crusted scabies; of 23 patients with crusted scabies, 21 were HTLV-1 positive.3 In another small study from Peru, 23 patients with crusted scabies were tested for HTLV-1 and 16 (70%) were seropositive.4 Neither study had a control group for comparison; however, interestingly, the estimated seroprevalence in the general population (based on blood donors) is only 0.04% to 1% and 1.2% to 1.7% in Brazil and Peru, respectively, thus reinforcing the association between HTLV-1 and crusted scabies.1 Crusted scabies and HTLV-1 seropositivity acting as a predictor for adult T-cell leukemia/lymphoma (ATL) has been reported8 but not consistently.3 Our patient had abnormal serum protein electrophoresis findings. Although the results were not diagnostic of ATL, patients with HTLV-1 are at increased risk for ATL and the pres-ence of this virus is important to document when there is a high index of suspicion for ATL, as with crusted scabies.
Our patient resided in the northeastern United States and there was no evidence in support of underlying immunosuppression. This case of crusted scabies is unique because the patient was HTLV-1 positive without other underlying immunocompromise and he resided in a region where HTLV-1 is nonendemic. In light of these findings, the present case serves as a reminder to check for HTLV-1 infection in those patients with severe or crusted scabies and without an identifiable reason for immunosuppression, even if the patient resides in an area that is nonendemic for HTLV-1.
- Gessain A, Cassar O. Epidemiological aspects and world distribution of HTLV-1 infection. Front Microbiol. 2012;3:388.
- Nobre V, Guedes AC, Martins ML, et al. Dermatological findings in 3 generations of a family with a high prevalence of human T cell lymphotropic virus type 1 infection in Brazil. Clin Infect Dis. 2006;43:1257-1263.
- Brites C, Weyll M, Pedroso C, et al. Severe and Norwegian scabies are strongly associated with retroviral (HIV-1/HTLV-1) infection in Bahia, Brazil. AIDS. 2002;16:1292-1293.
- Blas M, Bravo F, Castillo W, et al. Norwegian scabies in Peru: the impact of human T cell lymphotropic virus type I infection. Am J Trop Med Hyg. 2005;72:855-857.
- Bergman JN, Dodd WA, Trotter MJ, et al. Crusted scabies in association with human T-cell lymphotropic virus 1. J Cutan Med Surg. 1999;3:148-152.
- Daisley H, Charles W, Suite M. Crusted (Norwegian) scabies as a pre-diagnostic indicator for HTLV-1 infection. Trans R Soc Trop Med Hyg. 1993;87:295.
- Mollison LC. HTLV-I and clinical disease correlates in central Australian aborigines. Med J Aust. 1994;160:238.
- del Giudice P, Sainte Marie D, Gérard Y, et al. Is crusted (Norwegian) scabies a marker of adult T cell leukemia/lymphoma in human T lymphotropic virus type I–seropositive patients? J Infect Dis. 1997;176:1090-1092.
- Gessain A, Cassar O. Epidemiological aspects and world distribution of HTLV-1 infection. Front Microbiol. 2012;3:388.
- Nobre V, Guedes AC, Martins ML, et al. Dermatological findings in 3 generations of a family with a high prevalence of human T cell lymphotropic virus type 1 infection in Brazil. Clin Infect Dis. 2006;43:1257-1263.
- Brites C, Weyll M, Pedroso C, et al. Severe and Norwegian scabies are strongly associated with retroviral (HIV-1/HTLV-1) infection in Bahia, Brazil. AIDS. 2002;16:1292-1293.
- Blas M, Bravo F, Castillo W, et al. Norwegian scabies in Peru: the impact of human T cell lymphotropic virus type I infection. Am J Trop Med Hyg. 2005;72:855-857.
- Bergman JN, Dodd WA, Trotter MJ, et al. Crusted scabies in association with human T-cell lymphotropic virus 1. J Cutan Med Surg. 1999;3:148-152.
- Daisley H, Charles W, Suite M. Crusted (Norwegian) scabies as a pre-diagnostic indicator for HTLV-1 infection. Trans R Soc Trop Med Hyg. 1993;87:295.
- Mollison LC. HTLV-I and clinical disease correlates in central Australian aborigines. Med J Aust. 1994;160:238.
- del Giudice P, Sainte Marie D, Gérard Y, et al. Is crusted (Norwegian) scabies a marker of adult T cell leukemia/lymphoma in human T lymphotropic virus type I–seropositive patients? J Infect Dis. 1997;176:1090-1092.
An 82-year-old white man who resided in the northeastern United States was admitted to the hospital for weakness. He was evaluated for diffuse crusting of the skin including the face. The patient reported a history of minimally itchy, generalized crusts of 10 years’ duration. He lived alone with one cat. His medications included fluticasone furoate, terazosin, and levothyroxine sodium. Human immunodeficiency virus screening was negative. A scraping was taken from one of the thick crusts within an interdigital web space.
Inflammation Contributes to Effect of Diabetes on Brain
NEW YORK - Inflammation may contribute to impaired cerebral vasoregulation in type 2 diabetes, research suggests.
In a two-year study, participants with type 2 diabetes experienced diminished regional and global vasoreactivity in the brain, as well as a decline in cognitive function and the ability to perform daily tasks.
Higher blood levels of inflammatory markers were correlated with decreases in cerebral vasoreactivity and vasodilation in the diabetic subjects, but not in controls.
"Normal blood flow regulation allows the brain to redistribute blood to areas of the brain that have increased activity while performing certain tasks," senior author Dr. Vera Novak, of Beth Israel Deaconess Medical Center in Boston, said in a news release. "People with type 2 diabetes have impaired blood flow regulation. Our results suggest that diabetes and high blood sugar impose a chronic negative effect on cognitive and decision-making skills."
The study's final analysis involved 40 people, average age 69, including 19 with diabetes and 21 controls. The diabetes patients had been treated for the disease an average of 13 years. Smokers were excluded.
The researchers administered a number of cognition and memory tests, magnetic resonance imaging (MRI) scans and blood tests at the beginning of the study and at 24 months.
At the two-year visit, the diabetics had lower global gray matter volume, lower composite scores on learning and memory, lower regional and global cerebral vasoreactivity, and worse glycemic control, compared to baseline.
Among the diabetics, impaired cerebral vasoreactivity at baseline correlated with worse performance of daily activities. In addition, worsening vasodilation correlated with greater decreases in executive function, independent of age, education, and other factors.
"Higher serum soluble intercellular and vascular adhesion molecules, higher cortisol, and higher C-reactive protein levels at baseline were associated with greater decreases in cerebral vasoreactivity and vasodilation only in the (diabetes) group, independent of diabetes control and 24-hour blood pressure," the researchers wrote online July 8 in Neurology.
"Inflammation may further impair cerebral vasoregulation, which consequently accelerates decline in executive function and daily activities performance in older people with (diabetes)," they said.
"Early detection and monitoring of blood flow regulation may be an important predictor of accelerated changes in cognitive and decision-making skills," Dr. Novak said in the news release. She called for additional studies in a greater number of people and for a longer duration.
"We are currently starting a Phase 2-3 clinical trial to see if intranasal insulin could prevent/slow down cognitive decline," she told Reuters Health by email.
She also noted that while no specific treatment exists to prevent cognitive decline, healthy life styles help people to have less decline.
Large clinical trials have shown that even strict control of blood sugar does not prevent cognitive decline. The high fluctuation in blood glucose that occurs with diabetes damages the nerves of the brain, she said.
The study was funded by the National Institute on Aging, American Diabetes Association, Harvard Clinical and Translational Science Center and National Center for Research Resources.
NEW YORK - Inflammation may contribute to impaired cerebral vasoregulation in type 2 diabetes, research suggests.
In a two-year study, participants with type 2 diabetes experienced diminished regional and global vasoreactivity in the brain, as well as a decline in cognitive function and the ability to perform daily tasks.
Higher blood levels of inflammatory markers were correlated with decreases in cerebral vasoreactivity and vasodilation in the diabetic subjects, but not in controls.
"Normal blood flow regulation allows the brain to redistribute blood to areas of the brain that have increased activity while performing certain tasks," senior author Dr. Vera Novak, of Beth Israel Deaconess Medical Center in Boston, said in a news release. "People with type 2 diabetes have impaired blood flow regulation. Our results suggest that diabetes and high blood sugar impose a chronic negative effect on cognitive and decision-making skills."
The study's final analysis involved 40 people, average age 69, including 19 with diabetes and 21 controls. The diabetes patients had been treated for the disease an average of 13 years. Smokers were excluded.
The researchers administered a number of cognition and memory tests, magnetic resonance imaging (MRI) scans and blood tests at the beginning of the study and at 24 months.
At the two-year visit, the diabetics had lower global gray matter volume, lower composite scores on learning and memory, lower regional and global cerebral vasoreactivity, and worse glycemic control, compared to baseline.
Among the diabetics, impaired cerebral vasoreactivity at baseline correlated with worse performance of daily activities. In addition, worsening vasodilation correlated with greater decreases in executive function, independent of age, education, and other factors.
"Higher serum soluble intercellular and vascular adhesion molecules, higher cortisol, and higher C-reactive protein levels at baseline were associated with greater decreases in cerebral vasoreactivity and vasodilation only in the (diabetes) group, independent of diabetes control and 24-hour blood pressure," the researchers wrote online July 8 in Neurology.
"Inflammation may further impair cerebral vasoregulation, which consequently accelerates decline in executive function and daily activities performance in older people with (diabetes)," they said.
"Early detection and monitoring of blood flow regulation may be an important predictor of accelerated changes in cognitive and decision-making skills," Dr. Novak said in the news release. She called for additional studies in a greater number of people and for a longer duration.
"We are currently starting a Phase 2-3 clinical trial to see if intranasal insulin could prevent/slow down cognitive decline," she told Reuters Health by email.
She also noted that while no specific treatment exists to prevent cognitive decline, healthy life styles help people to have less decline.
Large clinical trials have shown that even strict control of blood sugar does not prevent cognitive decline. The high fluctuation in blood glucose that occurs with diabetes damages the nerves of the brain, she said.
The study was funded by the National Institute on Aging, American Diabetes Association, Harvard Clinical and Translational Science Center and National Center for Research Resources.
NEW YORK - Inflammation may contribute to impaired cerebral vasoregulation in type 2 diabetes, research suggests.
In a two-year study, participants with type 2 diabetes experienced diminished regional and global vasoreactivity in the brain, as well as a decline in cognitive function and the ability to perform daily tasks.
Higher blood levels of inflammatory markers were correlated with decreases in cerebral vasoreactivity and vasodilation in the diabetic subjects, but not in controls.
"Normal blood flow regulation allows the brain to redistribute blood to areas of the brain that have increased activity while performing certain tasks," senior author Dr. Vera Novak, of Beth Israel Deaconess Medical Center in Boston, said in a news release. "People with type 2 diabetes have impaired blood flow regulation. Our results suggest that diabetes and high blood sugar impose a chronic negative effect on cognitive and decision-making skills."
The study's final analysis involved 40 people, average age 69, including 19 with diabetes and 21 controls. The diabetes patients had been treated for the disease an average of 13 years. Smokers were excluded.
The researchers administered a number of cognition and memory tests, magnetic resonance imaging (MRI) scans and blood tests at the beginning of the study and at 24 months.
At the two-year visit, the diabetics had lower global gray matter volume, lower composite scores on learning and memory, lower regional and global cerebral vasoreactivity, and worse glycemic control, compared to baseline.
Among the diabetics, impaired cerebral vasoreactivity at baseline correlated with worse performance of daily activities. In addition, worsening vasodilation correlated with greater decreases in executive function, independent of age, education, and other factors.
"Higher serum soluble intercellular and vascular adhesion molecules, higher cortisol, and higher C-reactive protein levels at baseline were associated with greater decreases in cerebral vasoreactivity and vasodilation only in the (diabetes) group, independent of diabetes control and 24-hour blood pressure," the researchers wrote online July 8 in Neurology.
"Inflammation may further impair cerebral vasoregulation, which consequently accelerates decline in executive function and daily activities performance in older people with (diabetes)," they said.
"Early detection and monitoring of blood flow regulation may be an important predictor of accelerated changes in cognitive and decision-making skills," Dr. Novak said in the news release. She called for additional studies in a greater number of people and for a longer duration.
"We are currently starting a Phase 2-3 clinical trial to see if intranasal insulin could prevent/slow down cognitive decline," she told Reuters Health by email.
She also noted that while no specific treatment exists to prevent cognitive decline, healthy life styles help people to have less decline.
Large clinical trials have shown that even strict control of blood sugar does not prevent cognitive decline. The high fluctuation in blood glucose that occurs with diabetes damages the nerves of the brain, she said.
The study was funded by the National Institute on Aging, American Diabetes Association, Harvard Clinical and Translational Science Center and National Center for Research Resources.
Hemophilia A product performs well in 3 trials
DALLAS—Results of 3 studies suggest the full-length recombinant factor VIII product BAY 81-8973 can be safe and effective in children, adolescents, and adults with severe hemophilia A.
Patients who received BAY 81-8973 as routine prophylaxis had low annualized bleeding rates (ABRs)—significantly lower than patients who received on-demand treatment.
None of the patients developed inhibitors, and no adverse event occurred in more than 10% of patients.
These results were presented in posters at the National Hemophilia Foundation’s 67th Annual Meeting. All 3 trials were sponsored by Bayer Healthcare AG, the company developing BAY 81-8973.
LEOPOLD trials
The 3 trials are part of the LEOPOLD Clinical Development Program, which was designed to evaluate BAY 81-8973 in patients with severe hemophilia A.
The first trial, LEOPOLD I, included male patients ages 12 to 65 who had at least 150 previous exposure days with a factor VIII product. For this study, they received BAY 81-8973 at 20-50 IU/kg 2 or 3 times a week (according to investigator decision) for 12 months.
The second trial, LEOPOLD II, also enrolled previously treated (≥ 150 previous exposure days) male subjects ages 12 to 65. The subjects were randomized to receive BAY 81-8973 as a low-dose prophylaxis regimen (20-30 IU/kg) twice a week, high-dose prophylaxis (30-40 IU/kg) 3 times a week, or on-demand.
In the third trial, LEOPOLD Kids, researchers evaluated BAY 81-8973 in previously treated children ages 12 and younger. Patients received BAY 81-8973 at 20-50 IU/kg twice a week, 3 times a week, or every other day (according to investigator decision) for at least 50 exposure days.
Another part of LEOPOLD KIDS, in which researchers are evaluating BAY 81-8973 in previously untreated children, is ongoing.
Efficacy results
The ABR was the primary efficacy endpoint in LEOPOLD I and II. In LEOPOLD KIDS, the primary efficacy endpoint was the ABR for total bleeds occurring within 48 hours of previous prophylaxis treatment, but the researchers also analyzed the ABR independent of the time of injection.
For all 3 trials, 193 patients were evaluable for efficacy. One hundred and forty patients received BAY 81-8973 for 12 months or more. The median exposure days were 155 for LEOPOLD I and 153 for LEOPOLD II.
In LEOPOLD I (n=62), the median ABR was 1.0 for all patients who received prophylaxis, 1.0 for patients who received twice-weekly prophylaxis, and 2.0 for patients who received thrice-weekly prophylaxis.
Sixteen patients did not experience any bleeds while on study—6 patients on the twice-weekly prophylaxis regimen and 10 on the thrice-weekly prophylaxis regimen.
LEOPOLD II included 28 patients who received twice-weekly prophylaxis, 31 who received thrice-weekly prophylaxis, and 21 who received on-demand treatment.
The median ABR was significantly lower in patients who received either prophylactic regimen than those who received on-demand treatment—2.0 and 60.0, respectively (P<0.0001). The median ABR was 4.0 for patients who received twice-weekly prophylaxis and 2.0 for patients who received thrice-weekly prophylaxis.
Again, 16 patients who received prophylaxis did not have any bleeds during the study period. This included 8 patients on the twice-weekly prophylaxis regimen and 8 on the thrice-weekly prophylaxis regimen.
In LEOPOLD KIDS (n=51), the median ABR within 48 hours of prophylactic injection was 0, and the median ABR independent of the time of injection was 1.9.
Twenty-three patients did not have any bleeds during the 6-month treatment period—10 children younger than 6 and 13 children ages 6 to 12.
Safety results
For all 3 trials, 193 patients were evaluable for safety. Adverse reactions were defined as treatment-emergent adverse events with
at least a reasonable suspected causal relationship to BAY 81-8973.
The researchers said the frequency, type, and severity of adverse reactions in children were similar to those observed in adults and adolescents.
The adverse reactions included headache (7.3%), pyrexia (4.1%), pruritus (3.1%), injection site reactions (2.6%), insomnia (2.6%), rash (2.6%), abdominal pain (2.1%), dyspepsia (2.1%), abdominal discomfort (1.6%), lymphadenopathy (1%), dizziness (1%), allergic dermatitis (1%), heart palpitations (1%), sinus tachycardia (1%), chest discomfort (1%), hypersensitivity (0.5%), dysgeusia (0.5%), urticaria (0.5%), and flushing (0.5%).
None of the patients developed factor VIII inhibitors. ![]()
DALLAS—Results of 3 studies suggest the full-length recombinant factor VIII product BAY 81-8973 can be safe and effective in children, adolescents, and adults with severe hemophilia A.
Patients who received BAY 81-8973 as routine prophylaxis had low annualized bleeding rates (ABRs)—significantly lower than patients who received on-demand treatment.
None of the patients developed inhibitors, and no adverse event occurred in more than 10% of patients.
These results were presented in posters at the National Hemophilia Foundation’s 67th Annual Meeting. All 3 trials were sponsored by Bayer Healthcare AG, the company developing BAY 81-8973.
LEOPOLD trials
The 3 trials are part of the LEOPOLD Clinical Development Program, which was designed to evaluate BAY 81-8973 in patients with severe hemophilia A.
The first trial, LEOPOLD I, included male patients ages 12 to 65 who had at least 150 previous exposure days with a factor VIII product. For this study, they received BAY 81-8973 at 20-50 IU/kg 2 or 3 times a week (according to investigator decision) for 12 months.
The second trial, LEOPOLD II, also enrolled previously treated (≥ 150 previous exposure days) male subjects ages 12 to 65. The subjects were randomized to receive BAY 81-8973 as a low-dose prophylaxis regimen (20-30 IU/kg) twice a week, high-dose prophylaxis (30-40 IU/kg) 3 times a week, or on-demand.
In the third trial, LEOPOLD Kids, researchers evaluated BAY 81-8973 in previously treated children ages 12 and younger. Patients received BAY 81-8973 at 20-50 IU/kg twice a week, 3 times a week, or every other day (according to investigator decision) for at least 50 exposure days.
Another part of LEOPOLD KIDS, in which researchers are evaluating BAY 81-8973 in previously untreated children, is ongoing.
Efficacy results
The ABR was the primary efficacy endpoint in LEOPOLD I and II. In LEOPOLD KIDS, the primary efficacy endpoint was the ABR for total bleeds occurring within 48 hours of previous prophylaxis treatment, but the researchers also analyzed the ABR independent of the time of injection.
For all 3 trials, 193 patients were evaluable for efficacy. One hundred and forty patients received BAY 81-8973 for 12 months or more. The median exposure days were 155 for LEOPOLD I and 153 for LEOPOLD II.
In LEOPOLD I (n=62), the median ABR was 1.0 for all patients who received prophylaxis, 1.0 for patients who received twice-weekly prophylaxis, and 2.0 for patients who received thrice-weekly prophylaxis.
Sixteen patients did not experience any bleeds while on study—6 patients on the twice-weekly prophylaxis regimen and 10 on the thrice-weekly prophylaxis regimen.
LEOPOLD II included 28 patients who received twice-weekly prophylaxis, 31 who received thrice-weekly prophylaxis, and 21 who received on-demand treatment.
The median ABR was significantly lower in patients who received either prophylactic regimen than those who received on-demand treatment—2.0 and 60.0, respectively (P<0.0001). The median ABR was 4.0 for patients who received twice-weekly prophylaxis and 2.0 for patients who received thrice-weekly prophylaxis.
Again, 16 patients who received prophylaxis did not have any bleeds during the study period. This included 8 patients on the twice-weekly prophylaxis regimen and 8 on the thrice-weekly prophylaxis regimen.
In LEOPOLD KIDS (n=51), the median ABR within 48 hours of prophylactic injection was 0, and the median ABR independent of the time of injection was 1.9.
Twenty-three patients did not have any bleeds during the 6-month treatment period—10 children younger than 6 and 13 children ages 6 to 12.
Safety results
For all 3 trials, 193 patients were evaluable for safety. Adverse reactions were defined as treatment-emergent adverse events with
at least a reasonable suspected causal relationship to BAY 81-8973.
The researchers said the frequency, type, and severity of adverse reactions in children were similar to those observed in adults and adolescents.
The adverse reactions included headache (7.3%), pyrexia (4.1%), pruritus (3.1%), injection site reactions (2.6%), insomnia (2.6%), rash (2.6%), abdominal pain (2.1%), dyspepsia (2.1%), abdominal discomfort (1.6%), lymphadenopathy (1%), dizziness (1%), allergic dermatitis (1%), heart palpitations (1%), sinus tachycardia (1%), chest discomfort (1%), hypersensitivity (0.5%), dysgeusia (0.5%), urticaria (0.5%), and flushing (0.5%).
None of the patients developed factor VIII inhibitors. ![]()
DALLAS—Results of 3 studies suggest the full-length recombinant factor VIII product BAY 81-8973 can be safe and effective in children, adolescents, and adults with severe hemophilia A.
Patients who received BAY 81-8973 as routine prophylaxis had low annualized bleeding rates (ABRs)—significantly lower than patients who received on-demand treatment.
None of the patients developed inhibitors, and no adverse event occurred in more than 10% of patients.
These results were presented in posters at the National Hemophilia Foundation’s 67th Annual Meeting. All 3 trials were sponsored by Bayer Healthcare AG, the company developing BAY 81-8973.
LEOPOLD trials
The 3 trials are part of the LEOPOLD Clinical Development Program, which was designed to evaluate BAY 81-8973 in patients with severe hemophilia A.
The first trial, LEOPOLD I, included male patients ages 12 to 65 who had at least 150 previous exposure days with a factor VIII product. For this study, they received BAY 81-8973 at 20-50 IU/kg 2 or 3 times a week (according to investigator decision) for 12 months.
The second trial, LEOPOLD II, also enrolled previously treated (≥ 150 previous exposure days) male subjects ages 12 to 65. The subjects were randomized to receive BAY 81-8973 as a low-dose prophylaxis regimen (20-30 IU/kg) twice a week, high-dose prophylaxis (30-40 IU/kg) 3 times a week, or on-demand.
In the third trial, LEOPOLD Kids, researchers evaluated BAY 81-8973 in previously treated children ages 12 and younger. Patients received BAY 81-8973 at 20-50 IU/kg twice a week, 3 times a week, or every other day (according to investigator decision) for at least 50 exposure days.
Another part of LEOPOLD KIDS, in which researchers are evaluating BAY 81-8973 in previously untreated children, is ongoing.
Efficacy results
The ABR was the primary efficacy endpoint in LEOPOLD I and II. In LEOPOLD KIDS, the primary efficacy endpoint was the ABR for total bleeds occurring within 48 hours of previous prophylaxis treatment, but the researchers also analyzed the ABR independent of the time of injection.
For all 3 trials, 193 patients were evaluable for efficacy. One hundred and forty patients received BAY 81-8973 for 12 months or more. The median exposure days were 155 for LEOPOLD I and 153 for LEOPOLD II.
In LEOPOLD I (n=62), the median ABR was 1.0 for all patients who received prophylaxis, 1.0 for patients who received twice-weekly prophylaxis, and 2.0 for patients who received thrice-weekly prophylaxis.
Sixteen patients did not experience any bleeds while on study—6 patients on the twice-weekly prophylaxis regimen and 10 on the thrice-weekly prophylaxis regimen.
LEOPOLD II included 28 patients who received twice-weekly prophylaxis, 31 who received thrice-weekly prophylaxis, and 21 who received on-demand treatment.
The median ABR was significantly lower in patients who received either prophylactic regimen than those who received on-demand treatment—2.0 and 60.0, respectively (P<0.0001). The median ABR was 4.0 for patients who received twice-weekly prophylaxis and 2.0 for patients who received thrice-weekly prophylaxis.
Again, 16 patients who received prophylaxis did not have any bleeds during the study period. This included 8 patients on the twice-weekly prophylaxis regimen and 8 on the thrice-weekly prophylaxis regimen.
In LEOPOLD KIDS (n=51), the median ABR within 48 hours of prophylactic injection was 0, and the median ABR independent of the time of injection was 1.9.
Twenty-three patients did not have any bleeds during the 6-month treatment period—10 children younger than 6 and 13 children ages 6 to 12.
Safety results
For all 3 trials, 193 patients were evaluable for safety. Adverse reactions were defined as treatment-emergent adverse events with
at least a reasonable suspected causal relationship to BAY 81-8973.
The researchers said the frequency, type, and severity of adverse reactions in children were similar to those observed in adults and adolescents.
The adverse reactions included headache (7.3%), pyrexia (4.1%), pruritus (3.1%), injection site reactions (2.6%), insomnia (2.6%), rash (2.6%), abdominal pain (2.1%), dyspepsia (2.1%), abdominal discomfort (1.6%), lymphadenopathy (1%), dizziness (1%), allergic dermatitis (1%), heart palpitations (1%), sinus tachycardia (1%), chest discomfort (1%), hypersensitivity (0.5%), dysgeusia (0.5%), urticaria (0.5%), and flushing (0.5%).
None of the patients developed factor VIII inhibitors. ![]()
FDA expands indication for VWD drug
Photo by Piotr Bodzek
The US Food and Drug Administration (FDA) has expanded the approved indication for a human von Willebrand factor/coagulation factor VIII complex (Wilate) to include prevention of excessive bleeding during and after minor and major surgery in adult and pediatric patients with von Willebrand disease (VWD).
The product was previously approved for the treatment of spontaneous and trauma-induced bleeding episodes in patients with severe VWD, as well as patients with mild or moderate VWD in whom the use of desmopressin is known or suspected to be ineffective or contraindicated.
About Wilate
Wilate is a plasma-derived, highly purified concentrate of freeze-dried human von Willebrand factor and coagulation factor VIII. Two virus-inactivation steps are incorporated into the manufacturing process of the product: a solvent/detergent and terminal dry-heat treatment.
Investigators evaluated Wilate’s safety and efficacy in surgical procedures in a single-arm, phase 3 study known as WONDERS. The investigators enrolled 41 patients, 30 of whom completed the trial.
The patients had type 1, type 2 or type 3 VWD. They had a median age of 39.7, and most were female. All patients underwent surgery—21 major and 9 minor surgeries.
The hemostatic efficacy of Wilate was assessed intra-operatively by the surgeon and post-operatively by investigators. The overall hemostatic efficacy—success or failure—of Wilate was based on both assessments, using a 4-point ordinal efficacy scale.
In the 29 evaluable surgeries, the success rate was 96.7%. Wilate was successful in 100% of minor surgeries (n=9) and 95.2% of major surgeries (n=20).
There were 2 serious adverse events—erosive gastritis and vaginal hemorrhage. Nonserious events included nausea (7 cases in 6 patients), vomiting (n=6), pain (n=4), pyrexia (n=4), procedural pain (10 cases in 8 patients), decrease in hemoglobin (6 cases in 4 patients), and hypertension (n=4).
Wilate is under development by Octapharma. For more information on the product, visit www.WILATEusa.com. ![]()
Photo by Piotr Bodzek
The US Food and Drug Administration (FDA) has expanded the approved indication for a human von Willebrand factor/coagulation factor VIII complex (Wilate) to include prevention of excessive bleeding during and after minor and major surgery in adult and pediatric patients with von Willebrand disease (VWD).
The product was previously approved for the treatment of spontaneous and trauma-induced bleeding episodes in patients with severe VWD, as well as patients with mild or moderate VWD in whom the use of desmopressin is known or suspected to be ineffective or contraindicated.
About Wilate
Wilate is a plasma-derived, highly purified concentrate of freeze-dried human von Willebrand factor and coagulation factor VIII. Two virus-inactivation steps are incorporated into the manufacturing process of the product: a solvent/detergent and terminal dry-heat treatment.
Investigators evaluated Wilate’s safety and efficacy in surgical procedures in a single-arm, phase 3 study known as WONDERS. The investigators enrolled 41 patients, 30 of whom completed the trial.
The patients had type 1, type 2 or type 3 VWD. They had a median age of 39.7, and most were female. All patients underwent surgery—21 major and 9 minor surgeries.
The hemostatic efficacy of Wilate was assessed intra-operatively by the surgeon and post-operatively by investigators. The overall hemostatic efficacy—success or failure—of Wilate was based on both assessments, using a 4-point ordinal efficacy scale.
In the 29 evaluable surgeries, the success rate was 96.7%. Wilate was successful in 100% of minor surgeries (n=9) and 95.2% of major surgeries (n=20).
There were 2 serious adverse events—erosive gastritis and vaginal hemorrhage. Nonserious events included nausea (7 cases in 6 patients), vomiting (n=6), pain (n=4), pyrexia (n=4), procedural pain (10 cases in 8 patients), decrease in hemoglobin (6 cases in 4 patients), and hypertension (n=4).
Wilate is under development by Octapharma. For more information on the product, visit www.WILATEusa.com. ![]()
Photo by Piotr Bodzek
The US Food and Drug Administration (FDA) has expanded the approved indication for a human von Willebrand factor/coagulation factor VIII complex (Wilate) to include prevention of excessive bleeding during and after minor and major surgery in adult and pediatric patients with von Willebrand disease (VWD).
The product was previously approved for the treatment of spontaneous and trauma-induced bleeding episodes in patients with severe VWD, as well as patients with mild or moderate VWD in whom the use of desmopressin is known or suspected to be ineffective or contraindicated.
About Wilate
Wilate is a plasma-derived, highly purified concentrate of freeze-dried human von Willebrand factor and coagulation factor VIII. Two virus-inactivation steps are incorporated into the manufacturing process of the product: a solvent/detergent and terminal dry-heat treatment.
Investigators evaluated Wilate’s safety and efficacy in surgical procedures in a single-arm, phase 3 study known as WONDERS. The investigators enrolled 41 patients, 30 of whom completed the trial.
The patients had type 1, type 2 or type 3 VWD. They had a median age of 39.7, and most were female. All patients underwent surgery—21 major and 9 minor surgeries.
The hemostatic efficacy of Wilate was assessed intra-operatively by the surgeon and post-operatively by investigators. The overall hemostatic efficacy—success or failure—of Wilate was based on both assessments, using a 4-point ordinal efficacy scale.
In the 29 evaluable surgeries, the success rate was 96.7%. Wilate was successful in 100% of minor surgeries (n=9) and 95.2% of major surgeries (n=20).
There were 2 serious adverse events—erosive gastritis and vaginal hemorrhage. Nonserious events included nausea (7 cases in 6 patients), vomiting (n=6), pain (n=4), pyrexia (n=4), procedural pain (10 cases in 8 patients), decrease in hemoglobin (6 cases in 4 patients), and hypertension (n=4).
Wilate is under development by Octapharma. For more information on the product, visit www.WILATEusa.com. ![]()
Warfarin associated with real-world benefits in AF

Routine use of warfarin is associated with clinical benefits in ischemic stroke patients with atrial fibrillation (AF), according to research published in BMJ.
When compared to patients who did not receive anticoagulant therapy, patients who received warfarin at hospital discharge had a lower incidence of major adverse cardiovascular events (MACE), less time spent in an institutional care facility, and a lower risk of all-cause mortality.
Warfarin seemed particularly beneficial for patients older than 80 years of age, women, and patients with more severe strokes.
Ying Xian, MD, PhD, of the Duke Clinical Research Institute in Durham, North Carolina, and his colleagues conducted this research, analyzing the association between warfarin treatment and longitudinal outcomes after ischemic stroke among AF patients.
The researchers evaluated 12,552 warfarin-naive AF patients who were admitted to 1487 hospitals across the US and were discharged between 2009 and 2011. Each patient had at least 1 year of follow-up after discharge.
In all, 11,039 (87.9%) were treated with warfarin at discharge. These patients were slightly younger and less likely to have a history of previous stroke or coronary artery disease than patients who did not receive warfarin.
Unadjusted results
At 2 years of follow-up, the incidence of MACE was significantly lower in patients treated with warfarin than in untreated patients—54.7% and 66.8%, respectively (P<0.001).
In addition, on average, patients who received warfarin had 86 more days alive and out of institutional care in the 2-year follow-up period than patients who did not receive warfarin (P<0.001).
The incidence of readmission due to ischemic stroke was significantly lower among warfarin-treated patients than untreated patients—7.9% and 11.8%, respectively (P<0.001)—but there was no significant difference in readmission due to hemorrhagic stroke—1.4% and 1.1%, respectively (P=0.50).
The incidence of all-cause mortality was significantly lower among warfarin-treated patients—32.4% and 50%, respectively (P<0.001).
Adjusted results
In adjusted analysis (weighting by the inverse probability of treatment and control for other discharge drugs), patients treated with warfarin at discharge had a significantly lower risk of MACE over 2 years. The adjusted hazard ratio (aHR) was 0.87.
Warfarin-treated patients were also more likely to spend more days alive and out of institutional care. The adjusted home-time difference was 47.6 days.
Patients on warfarin had a lower risk of all-cause mortality (aHR=0.72) and ischemic stroke readmission (aHR=0.63), but there was no significant difference between treated and untreated patients with regard to hemorrhagic stroke readmission (aHR=1.37).
The researchers said the benefits associated with warfarin were consistent across clinically relevant groups by age, sex, stroke severity, and history of stroke and coronary artery disease.
Patients aged 80 and older, women, and those with more severe stroke seemed to enjoy greater benefits from warfarin treatment, even though these groups were less likely to receive warfarin.
The researchers speculated that this might result from clinicians’ misperception of warfarin’s risks and benefits for these patient groups. ![]()
Routine use of warfarin is associated with clinical benefits in ischemic stroke patients with atrial fibrillation (AF), according to research published in BMJ.
When compared to patients who did not receive anticoagulant therapy, patients who received warfarin at hospital discharge had a lower incidence of major adverse cardiovascular events (MACE), less time spent in an institutional care facility, and a lower risk of all-cause mortality.
Warfarin seemed particularly beneficial for patients older than 80 years of age, women, and patients with more severe strokes.
Ying Xian, MD, PhD, of the Duke Clinical Research Institute in Durham, North Carolina, and his colleagues conducted this research, analyzing the association between warfarin treatment and longitudinal outcomes after ischemic stroke among AF patients.
The researchers evaluated 12,552 warfarin-naive AF patients who were admitted to 1487 hospitals across the US and were discharged between 2009 and 2011. Each patient had at least 1 year of follow-up after discharge.
In all, 11,039 (87.9%) were treated with warfarin at discharge. These patients were slightly younger and less likely to have a history of previous stroke or coronary artery disease than patients who did not receive warfarin.
Unadjusted results
At 2 years of follow-up, the incidence of MACE was significantly lower in patients treated with warfarin than in untreated patients—54.7% and 66.8%, respectively (P<0.001).
In addition, on average, patients who received warfarin had 86 more days alive and out of institutional care in the 2-year follow-up period than patients who did not receive warfarin (P<0.001).
The incidence of readmission due to ischemic stroke was significantly lower among warfarin-treated patients than untreated patients—7.9% and 11.8%, respectively (P<0.001)—but there was no significant difference in readmission due to hemorrhagic stroke—1.4% and 1.1%, respectively (P=0.50).
The incidence of all-cause mortality was significantly lower among warfarin-treated patients—32.4% and 50%, respectively (P<0.001).
Adjusted results
In adjusted analysis (weighting by the inverse probability of treatment and control for other discharge drugs), patients treated with warfarin at discharge had a significantly lower risk of MACE over 2 years. The adjusted hazard ratio (aHR) was 0.87.
Warfarin-treated patients were also more likely to spend more days alive and out of institutional care. The adjusted home-time difference was 47.6 days.
Patients on warfarin had a lower risk of all-cause mortality (aHR=0.72) and ischemic stroke readmission (aHR=0.63), but there was no significant difference between treated and untreated patients with regard to hemorrhagic stroke readmission (aHR=1.37).
The researchers said the benefits associated with warfarin were consistent across clinically relevant groups by age, sex, stroke severity, and history of stroke and coronary artery disease.
Patients aged 80 and older, women, and those with more severe stroke seemed to enjoy greater benefits from warfarin treatment, even though these groups were less likely to receive warfarin.
The researchers speculated that this might result from clinicians’ misperception of warfarin’s risks and benefits for these patient groups. ![]()
Routine use of warfarin is associated with clinical benefits in ischemic stroke patients with atrial fibrillation (AF), according to research published in BMJ.
When compared to patients who did not receive anticoagulant therapy, patients who received warfarin at hospital discharge had a lower incidence of major adverse cardiovascular events (MACE), less time spent in an institutional care facility, and a lower risk of all-cause mortality.
Warfarin seemed particularly beneficial for patients older than 80 years of age, women, and patients with more severe strokes.
Ying Xian, MD, PhD, of the Duke Clinical Research Institute in Durham, North Carolina, and his colleagues conducted this research, analyzing the association between warfarin treatment and longitudinal outcomes after ischemic stroke among AF patients.
The researchers evaluated 12,552 warfarin-naive AF patients who were admitted to 1487 hospitals across the US and were discharged between 2009 and 2011. Each patient had at least 1 year of follow-up after discharge.
In all, 11,039 (87.9%) were treated with warfarin at discharge. These patients were slightly younger and less likely to have a history of previous stroke or coronary artery disease than patients who did not receive warfarin.
Unadjusted results
At 2 years of follow-up, the incidence of MACE was significantly lower in patients treated with warfarin than in untreated patients—54.7% and 66.8%, respectively (P<0.001).
In addition, on average, patients who received warfarin had 86 more days alive and out of institutional care in the 2-year follow-up period than patients who did not receive warfarin (P<0.001).
The incidence of readmission due to ischemic stroke was significantly lower among warfarin-treated patients than untreated patients—7.9% and 11.8%, respectively (P<0.001)—but there was no significant difference in readmission due to hemorrhagic stroke—1.4% and 1.1%, respectively (P=0.50).
The incidence of all-cause mortality was significantly lower among warfarin-treated patients—32.4% and 50%, respectively (P<0.001).
Adjusted results
In adjusted analysis (weighting by the inverse probability of treatment and control for other discharge drugs), patients treated with warfarin at discharge had a significantly lower risk of MACE over 2 years. The adjusted hazard ratio (aHR) was 0.87.
Warfarin-treated patients were also more likely to spend more days alive and out of institutional care. The adjusted home-time difference was 47.6 days.
Patients on warfarin had a lower risk of all-cause mortality (aHR=0.72) and ischemic stroke readmission (aHR=0.63), but there was no significant difference between treated and untreated patients with regard to hemorrhagic stroke readmission (aHR=1.37).
The researchers said the benefits associated with warfarin were consistent across clinically relevant groups by age, sex, stroke severity, and history of stroke and coronary artery disease.
Patients aged 80 and older, women, and those with more severe stroke seemed to enjoy greater benefits from warfarin treatment, even though these groups were less likely to receive warfarin.
The researchers speculated that this might result from clinicians’ misperception of warfarin’s risks and benefits for these patient groups. ![]()