User login
Novel triple therapy in ITP provides enduring responses
For patients with chronic primary immune thrombocytopenia, a three-drug regimen was associated with a high response rate and relapse-free survival, according to the results of a single-arm, phase IIb trial published online in Blood.
Twenty patients with primary immune thrombocytopenia (ITP) received an investigative triple therapy of oral dexamethasone 40 mg (days 1-4), oral cyclosporine 2.5-3.0 mg/kg daily (days 1-28), and intravenous rituximab 100 mg (day 7, 14, 21, and 28), and of this group, 12 patients responded. The median time to response was 7.4 days, and all patients maintained their response for at least 7 months, Dr. Philip Young-Ill Choi, of St. George Clinical School, University of New South Wales, Kogarah, Australia, and his colleagues reported (Blood 2015;126[4]:500-3).
Complete response was 30% at 6 months, and only two patients relapsed during a median follow-up period of 17.5 months (range, 7-47 months). Among patients who responded, relapse-free survival at 12 and 24 months was 92% and 76%, respectively (95% confidence intervals, 53%-98% and 30%-93%, respectively).
Peripheral T cells declined for all patients, irrespective of response, but responders had lower CD4+ T cells than did nonresponders for 6 months after treatment (median, 0.62 vs. 0.91 x 109/L; P less than .0001). Peripheral CD19+ B cells became undetectable for all patients by day 28, but recovery was earlier for patients younger than 50 years (median, 6.5 months vs. not reached; P = .0105).
The regimen was generally well tolerated, without any deaths, treatment-related serious adverse events, serum sickness, interruptions, or delays caused by toxicity.
A major advantage of this regimen is its short duration of therapy, and yet 12 of 20 patients enjoyed a prolonged remission of 7 months or longer without needing further treatment. However, interpretation of the data was limited by the small sample size, the investigators noted.
“Although our study shows encouraging results, the incremental benefit of cyclosporine to rituximab and dexamethasone remains unresolved, and randomized controlled trials are required,” they wrote.
No funding source for the study was given. One author reported receiving speaker’s fees from Roche, and another is on the speakers bureau and receives research funding from GlaxoSmithKline and Amgen. The remaining authors declared no competing financial interests.
For patients with chronic primary immune thrombocytopenia, a three-drug regimen was associated with a high response rate and relapse-free survival, according to the results of a single-arm, phase IIb trial published online in Blood.
Twenty patients with primary immune thrombocytopenia (ITP) received an investigative triple therapy of oral dexamethasone 40 mg (days 1-4), oral cyclosporine 2.5-3.0 mg/kg daily (days 1-28), and intravenous rituximab 100 mg (day 7, 14, 21, and 28), and of this group, 12 patients responded. The median time to response was 7.4 days, and all patients maintained their response for at least 7 months, Dr. Philip Young-Ill Choi, of St. George Clinical School, University of New South Wales, Kogarah, Australia, and his colleagues reported (Blood 2015;126[4]:500-3).
Complete response was 30% at 6 months, and only two patients relapsed during a median follow-up period of 17.5 months (range, 7-47 months). Among patients who responded, relapse-free survival at 12 and 24 months was 92% and 76%, respectively (95% confidence intervals, 53%-98% and 30%-93%, respectively).
Peripheral T cells declined for all patients, irrespective of response, but responders had lower CD4+ T cells than did nonresponders for 6 months after treatment (median, 0.62 vs. 0.91 x 109/L; P less than .0001). Peripheral CD19+ B cells became undetectable for all patients by day 28, but recovery was earlier for patients younger than 50 years (median, 6.5 months vs. not reached; P = .0105).
The regimen was generally well tolerated, without any deaths, treatment-related serious adverse events, serum sickness, interruptions, or delays caused by toxicity.
A major advantage of this regimen is its short duration of therapy, and yet 12 of 20 patients enjoyed a prolonged remission of 7 months or longer without needing further treatment. However, interpretation of the data was limited by the small sample size, the investigators noted.
“Although our study shows encouraging results, the incremental benefit of cyclosporine to rituximab and dexamethasone remains unresolved, and randomized controlled trials are required,” they wrote.
No funding source for the study was given. One author reported receiving speaker’s fees from Roche, and another is on the speakers bureau and receives research funding from GlaxoSmithKline and Amgen. The remaining authors declared no competing financial interests.
For patients with chronic primary immune thrombocytopenia, a three-drug regimen was associated with a high response rate and relapse-free survival, according to the results of a single-arm, phase IIb trial published online in Blood.
Twenty patients with primary immune thrombocytopenia (ITP) received an investigative triple therapy of oral dexamethasone 40 mg (days 1-4), oral cyclosporine 2.5-3.0 mg/kg daily (days 1-28), and intravenous rituximab 100 mg (day 7, 14, 21, and 28), and of this group, 12 patients responded. The median time to response was 7.4 days, and all patients maintained their response for at least 7 months, Dr. Philip Young-Ill Choi, of St. George Clinical School, University of New South Wales, Kogarah, Australia, and his colleagues reported (Blood 2015;126[4]:500-3).
Complete response was 30% at 6 months, and only two patients relapsed during a median follow-up period of 17.5 months (range, 7-47 months). Among patients who responded, relapse-free survival at 12 and 24 months was 92% and 76%, respectively (95% confidence intervals, 53%-98% and 30%-93%, respectively).
Peripheral T cells declined for all patients, irrespective of response, but responders had lower CD4+ T cells than did nonresponders for 6 months after treatment (median, 0.62 vs. 0.91 x 109/L; P less than .0001). Peripheral CD19+ B cells became undetectable for all patients by day 28, but recovery was earlier for patients younger than 50 years (median, 6.5 months vs. not reached; P = .0105).
The regimen was generally well tolerated, without any deaths, treatment-related serious adverse events, serum sickness, interruptions, or delays caused by toxicity.
A major advantage of this regimen is its short duration of therapy, and yet 12 of 20 patients enjoyed a prolonged remission of 7 months or longer without needing further treatment. However, interpretation of the data was limited by the small sample size, the investigators noted.
“Although our study shows encouraging results, the incremental benefit of cyclosporine to rituximab and dexamethasone remains unresolved, and randomized controlled trials are required,” they wrote.
No funding source for the study was given. One author reported receiving speaker’s fees from Roche, and another is on the speakers bureau and receives research funding from GlaxoSmithKline and Amgen. The remaining authors declared no competing financial interests.
FROM BLOOD
Key clinical point: Patients with primary immune thrombocytopenia can achieve an enduring response with a novel triple drug regimen.
Major finding: Relapse-free survival was 92% at 12 months for responders and 76% at 24 months
Data source: Prospective, single-arm, phase IIb study involving 20 patients.
Disclosures: No funding source for the study was given. One author reported receiving speaker’s fees from Roche, and another is on the speakers bureau and receives research funding from GlaxoSmithKline and Amgen. The remaining authors declared no competing financial interests.
ACO Insider: Avoid the ‘default future’
As readers of this column know, the move to value-based payment for population health management can lead to a golden era for proactive primary care physicians. This conclusion is only strengthened by recent legislation mandating value incentives and penalties: the Medicare Access and CHIP Reauthorization Act of 2015 (MACRA), sometimes called the “SGR fix.”
This radical change, tellingly supported by both parties and both houses of Congress, would have been unthinkable just a few years ago. Under MACRA’s new Merit-Based Incentive Payment System (MIPS), you are looking at fee increases or reductions ranging from an upside of 4%-9% over time and an equal potential for reduction.
But, if you participate in a Medicare ACO or similar entity under the new alternative payment model, you get a 5% bump and are excluded from any MIPS and meaningful use requirements or penalties.
This merely adds to the growing list of incentives for primary care physician–led coordinated care. There is an extra compensation for wellness exams and chronic care management amounting to potentially more than $100,000 per primary care physician per year. Do not forget the $840 million the Centers for Medicare & Medicaid Services is designating to the Transforming Clinical Practice Initiative limited to training clinicians, and the $800 million for rural accountable care organizations (ACO) operations costs limited to physicians, critical access hospitals, and small hospitals.
Oh, by the way, all of the high-value opportunities for ACOs are in the primary care physician’s wheelhouse. Success stories of primary care–led ACOs are impressive.
A no-brainer, right? Well, apparently not for most primary care physicians. Why? This all will require change. It can be a very beneficial change of your status – measured by professional and financial reward – but it is big-time change.
As Mark Twain is quoted as saying, “I’m all for progress; it’s change I object to.”
You have not been in such a position of influence before, you don’t have teams of advisors like others in health care, and you don’t have the experience for this. You do not have spare intellectual bandwidth to deal with this and everything else. You are accustomed to things being run by the big health systems and managed care companies.
It is human nature to deal with stress with the survivalist instincts of fight, flight, or freeze. You may be feeling an almost irresistible urge to hunker down and do nothing. It’s natural. It is your “default future.”
But being unprepared is not an option. This shift is coming inexorably and rapidly. You can either stay sitting on the tracks or drive the train. It’s up to you.
Your default future is one controlled by others. It is one of the missed opportunity of a lifetime for primary care. The government is paying you for training, ACO start-up and operations, and incentivizing your leadership through both coding- and value-based financial inducements.
The bottom line is that America is asking you to run the new health care system and wants to pay you to do it, on top of your fee-for-service payments.
Think of the impact on your patients. Isn’t this why you went to medical school? Failure to do anything means you actually have made a bigger choice for your default future – guaranteeing even greater change being imposed on you by others. Control your agenda; do not wait to become part of someone else’s.
In closing, a recent email comment by one of your fellow readers sums it up best: “The default future (or the ostrich option) is a destiny of marginalization and consumption by the beast, an outcome not in our patients’ best interest.”
Mr. Bobbitt is a senior partner and head of the health law group at the Smith Anderson law firm in Raleigh, N.C. He has many years’ experience assisting physicians form integrated delivery systems. He has spoken and written nationally to primary care physicians on the strategies and practicalities of forming or joining ACOs. This article is meant to be educational and does not constitute legal advice. For additional information, readers may contact the author at bbobbitt@smithlaw.com or 919-821-6612.
As readers of this column know, the move to value-based payment for population health management can lead to a golden era for proactive primary care physicians. This conclusion is only strengthened by recent legislation mandating value incentives and penalties: the Medicare Access and CHIP Reauthorization Act of 2015 (MACRA), sometimes called the “SGR fix.”
This radical change, tellingly supported by both parties and both houses of Congress, would have been unthinkable just a few years ago. Under MACRA’s new Merit-Based Incentive Payment System (MIPS), you are looking at fee increases or reductions ranging from an upside of 4%-9% over time and an equal potential for reduction.
But, if you participate in a Medicare ACO or similar entity under the new alternative payment model, you get a 5% bump and are excluded from any MIPS and meaningful use requirements or penalties.
This merely adds to the growing list of incentives for primary care physician–led coordinated care. There is an extra compensation for wellness exams and chronic care management amounting to potentially more than $100,000 per primary care physician per year. Do not forget the $840 million the Centers for Medicare & Medicaid Services is designating to the Transforming Clinical Practice Initiative limited to training clinicians, and the $800 million for rural accountable care organizations (ACO) operations costs limited to physicians, critical access hospitals, and small hospitals.
Oh, by the way, all of the high-value opportunities for ACOs are in the primary care physician’s wheelhouse. Success stories of primary care–led ACOs are impressive.
A no-brainer, right? Well, apparently not for most primary care physicians. Why? This all will require change. It can be a very beneficial change of your status – measured by professional and financial reward – but it is big-time change.
As Mark Twain is quoted as saying, “I’m all for progress; it’s change I object to.”
You have not been in such a position of influence before, you don’t have teams of advisors like others in health care, and you don’t have the experience for this. You do not have spare intellectual bandwidth to deal with this and everything else. You are accustomed to things being run by the big health systems and managed care companies.
It is human nature to deal with stress with the survivalist instincts of fight, flight, or freeze. You may be feeling an almost irresistible urge to hunker down and do nothing. It’s natural. It is your “default future.”
But being unprepared is not an option. This shift is coming inexorably and rapidly. You can either stay sitting on the tracks or drive the train. It’s up to you.
Your default future is one controlled by others. It is one of the missed opportunity of a lifetime for primary care. The government is paying you for training, ACO start-up and operations, and incentivizing your leadership through both coding- and value-based financial inducements.
The bottom line is that America is asking you to run the new health care system and wants to pay you to do it, on top of your fee-for-service payments.
Think of the impact on your patients. Isn’t this why you went to medical school? Failure to do anything means you actually have made a bigger choice for your default future – guaranteeing even greater change being imposed on you by others. Control your agenda; do not wait to become part of someone else’s.
In closing, a recent email comment by one of your fellow readers sums it up best: “The default future (or the ostrich option) is a destiny of marginalization and consumption by the beast, an outcome not in our patients’ best interest.”
Mr. Bobbitt is a senior partner and head of the health law group at the Smith Anderson law firm in Raleigh, N.C. He has many years’ experience assisting physicians form integrated delivery systems. He has spoken and written nationally to primary care physicians on the strategies and practicalities of forming or joining ACOs. This article is meant to be educational and does not constitute legal advice. For additional information, readers may contact the author at bbobbitt@smithlaw.com or 919-821-6612.
As readers of this column know, the move to value-based payment for population health management can lead to a golden era for proactive primary care physicians. This conclusion is only strengthened by recent legislation mandating value incentives and penalties: the Medicare Access and CHIP Reauthorization Act of 2015 (MACRA), sometimes called the “SGR fix.”
This radical change, tellingly supported by both parties and both houses of Congress, would have been unthinkable just a few years ago. Under MACRA’s new Merit-Based Incentive Payment System (MIPS), you are looking at fee increases or reductions ranging from an upside of 4%-9% over time and an equal potential for reduction.
But, if you participate in a Medicare ACO or similar entity under the new alternative payment model, you get a 5% bump and are excluded from any MIPS and meaningful use requirements or penalties.
This merely adds to the growing list of incentives for primary care physician–led coordinated care. There is an extra compensation for wellness exams and chronic care management amounting to potentially more than $100,000 per primary care physician per year. Do not forget the $840 million the Centers for Medicare & Medicaid Services is designating to the Transforming Clinical Practice Initiative limited to training clinicians, and the $800 million for rural accountable care organizations (ACO) operations costs limited to physicians, critical access hospitals, and small hospitals.
Oh, by the way, all of the high-value opportunities for ACOs are in the primary care physician’s wheelhouse. Success stories of primary care–led ACOs are impressive.
A no-brainer, right? Well, apparently not for most primary care physicians. Why? This all will require change. It can be a very beneficial change of your status – measured by professional and financial reward – but it is big-time change.
As Mark Twain is quoted as saying, “I’m all for progress; it’s change I object to.”
You have not been in such a position of influence before, you don’t have teams of advisors like others in health care, and you don’t have the experience for this. You do not have spare intellectual bandwidth to deal with this and everything else. You are accustomed to things being run by the big health systems and managed care companies.
It is human nature to deal with stress with the survivalist instincts of fight, flight, or freeze. You may be feeling an almost irresistible urge to hunker down and do nothing. It’s natural. It is your “default future.”
But being unprepared is not an option. This shift is coming inexorably and rapidly. You can either stay sitting on the tracks or drive the train. It’s up to you.
Your default future is one controlled by others. It is one of the missed opportunity of a lifetime for primary care. The government is paying you for training, ACO start-up and operations, and incentivizing your leadership through both coding- and value-based financial inducements.
The bottom line is that America is asking you to run the new health care system and wants to pay you to do it, on top of your fee-for-service payments.
Think of the impact on your patients. Isn’t this why you went to medical school? Failure to do anything means you actually have made a bigger choice for your default future – guaranteeing even greater change being imposed on you by others. Control your agenda; do not wait to become part of someone else’s.
In closing, a recent email comment by one of your fellow readers sums it up best: “The default future (or the ostrich option) is a destiny of marginalization and consumption by the beast, an outcome not in our patients’ best interest.”
Mr. Bobbitt is a senior partner and head of the health law group at the Smith Anderson law firm in Raleigh, N.C. He has many years’ experience assisting physicians form integrated delivery systems. He has spoken and written nationally to primary care physicians on the strategies and practicalities of forming or joining ACOs. This article is meant to be educational and does not constitute legal advice. For additional information, readers may contact the author at bbobbitt@smithlaw.com or 919-821-6612.

What Matters: Sleep restriction
At one time or another, insomnia afflicts nearly one-half of U.S. adults, half of whom have a clinically diagnosable disorder. This presents perpetual challenges in the face of patient populations that have been told to “ask your doctor about” sleeping medications or have received them already.
We know that the Z-drugs (zolpidem, zaleplon, and eszopiclone), some of the most widely used pharmacologics for insomnia, are benzodiazepine receptor agonists. As such, tolerance develops, and this tolerance leads to escalating doses, increased side effects, and sleepier patients.
Cognitive-behavioral therapy has been shown to be effective for insomnia, but this clinical service is not widely available. For busy clinicians trying to help these patients, we need a simple tool that can be easily explained to patients, giving them a project on which to work.
This tool is sleep restriction. The goal of sleep restriction is to consolidate fragmented sleep to increase the intrinsic sleep drive.
You might have heard your patients describe their bedroom as a “torture chamber.” Some of this torture relates to sleepless staring at the ceiling for hours on end. Sleep restriction gets them out of the chamber.
Karen Falloon, Ph.D., of the University of Auckland (New Zealand), and her colleagues conducted a randomized trial in New Zealand investigating the impact of simplified sleep restriction (SSR) for patients with primary insomnia (Br J Gen Pract. 2015 Aug;65(637):e508-15).
A total of 97 patients were randomized. All patients received sleep hygiene advice, including avoiding caffeine and developing a consistent bedtime routine. Patients in the SSR arm received a verbal and written sleep prescription establishing bedtime and wake-up times informed by a baseline 2-week daily sleep diary.
The sleep prescription was average total sleep duration plus 50% of the total time spent awake in bed. The minimum time in bed was 5 hours. If participants were sleeping less than 85% of the time in bed, the time allowed in bed was reduced to total sleep time plus 30 minutes. Sleepy patients could spend 30 more minutes in bed. All changes were made at bedtime, with wake-up time held constant.
At 6 months, the SSR group had improved perceived sleep quality and fatigue, and improved sleep efficiency as measured by actigraphy. A total of 67% of patients responded to SSR, compared with 41% of controls (number needed to treat = 4).
The efficacy of this intervention is extremely impressive. Importantly, it was delivered by a general practitioner without specialized training during two “slightly extended” visits.
Potential participants were excluded if they were on a sleeping medication, which does not imply that this would not work in a population already on Z-drugs. Consideration should to be given to possible risks when implementing sleep restriction with patients taking Z-drugs with longer half-lives (for example, eszopiclone is 6 hours, zolpidem is 3 hours, and zaleplon is 1 hour), because of higher serum concentrations upon waking.
But when these medications fail, or you have Z-drug–naive patients with insomnia, have this intervention ready.
Dr. Ebbert is professor of medicine, a general internist at the Mayo Clinic in Rochester, Minn., and a diplomate of the American Board of Addiction Medicine. The opinions expressed are those of the author and do not necessarily represent the views and opinions of the Mayo Clinic. The opinions expressed in this article should not be used to diagnose or treat any medical condition, nor should they be used as a substitute for medical advice from a qualified, board-certified practicing clinician. Dr. Ebbert has no relevant financial disclosures about this article. Follow him on Twitter @jonebbert.
At one time or another, insomnia afflicts nearly one-half of U.S. adults, half of whom have a clinically diagnosable disorder. This presents perpetual challenges in the face of patient populations that have been told to “ask your doctor about” sleeping medications or have received them already.
We know that the Z-drugs (zolpidem, zaleplon, and eszopiclone), some of the most widely used pharmacologics for insomnia, are benzodiazepine receptor agonists. As such, tolerance develops, and this tolerance leads to escalating doses, increased side effects, and sleepier patients.
Cognitive-behavioral therapy has been shown to be effective for insomnia, but this clinical service is not widely available. For busy clinicians trying to help these patients, we need a simple tool that can be easily explained to patients, giving them a project on which to work.
This tool is sleep restriction. The goal of sleep restriction is to consolidate fragmented sleep to increase the intrinsic sleep drive.
You might have heard your patients describe their bedroom as a “torture chamber.” Some of this torture relates to sleepless staring at the ceiling for hours on end. Sleep restriction gets them out of the chamber.
Karen Falloon, Ph.D., of the University of Auckland (New Zealand), and her colleagues conducted a randomized trial in New Zealand investigating the impact of simplified sleep restriction (SSR) for patients with primary insomnia (Br J Gen Pract. 2015 Aug;65(637):e508-15).
A total of 97 patients were randomized. All patients received sleep hygiene advice, including avoiding caffeine and developing a consistent bedtime routine. Patients in the SSR arm received a verbal and written sleep prescription establishing bedtime and wake-up times informed by a baseline 2-week daily sleep diary.
The sleep prescription was average total sleep duration plus 50% of the total time spent awake in bed. The minimum time in bed was 5 hours. If participants were sleeping less than 85% of the time in bed, the time allowed in bed was reduced to total sleep time plus 30 minutes. Sleepy patients could spend 30 more minutes in bed. All changes were made at bedtime, with wake-up time held constant.
At 6 months, the SSR group had improved perceived sleep quality and fatigue, and improved sleep efficiency as measured by actigraphy. A total of 67% of patients responded to SSR, compared with 41% of controls (number needed to treat = 4).
The efficacy of this intervention is extremely impressive. Importantly, it was delivered by a general practitioner without specialized training during two “slightly extended” visits.
Potential participants were excluded if they were on a sleeping medication, which does not imply that this would not work in a population already on Z-drugs. Consideration should to be given to possible risks when implementing sleep restriction with patients taking Z-drugs with longer half-lives (for example, eszopiclone is 6 hours, zolpidem is 3 hours, and zaleplon is 1 hour), because of higher serum concentrations upon waking.
But when these medications fail, or you have Z-drug–naive patients with insomnia, have this intervention ready.
Dr. Ebbert is professor of medicine, a general internist at the Mayo Clinic in Rochester, Minn., and a diplomate of the American Board of Addiction Medicine. The opinions expressed are those of the author and do not necessarily represent the views and opinions of the Mayo Clinic. The opinions expressed in this article should not be used to diagnose or treat any medical condition, nor should they be used as a substitute for medical advice from a qualified, board-certified practicing clinician. Dr. Ebbert has no relevant financial disclosures about this article. Follow him on Twitter @jonebbert.
At one time or another, insomnia afflicts nearly one-half of U.S. adults, half of whom have a clinically diagnosable disorder. This presents perpetual challenges in the face of patient populations that have been told to “ask your doctor about” sleeping medications or have received them already.
We know that the Z-drugs (zolpidem, zaleplon, and eszopiclone), some of the most widely used pharmacologics for insomnia, are benzodiazepine receptor agonists. As such, tolerance develops, and this tolerance leads to escalating doses, increased side effects, and sleepier patients.
Cognitive-behavioral therapy has been shown to be effective for insomnia, but this clinical service is not widely available. For busy clinicians trying to help these patients, we need a simple tool that can be easily explained to patients, giving them a project on which to work.
This tool is sleep restriction. The goal of sleep restriction is to consolidate fragmented sleep to increase the intrinsic sleep drive.
You might have heard your patients describe their bedroom as a “torture chamber.” Some of this torture relates to sleepless staring at the ceiling for hours on end. Sleep restriction gets them out of the chamber.
Karen Falloon, Ph.D., of the University of Auckland (New Zealand), and her colleagues conducted a randomized trial in New Zealand investigating the impact of simplified sleep restriction (SSR) for patients with primary insomnia (Br J Gen Pract. 2015 Aug;65(637):e508-15).
A total of 97 patients were randomized. All patients received sleep hygiene advice, including avoiding caffeine and developing a consistent bedtime routine. Patients in the SSR arm received a verbal and written sleep prescription establishing bedtime and wake-up times informed by a baseline 2-week daily sleep diary.
The sleep prescription was average total sleep duration plus 50% of the total time spent awake in bed. The minimum time in bed was 5 hours. If participants were sleeping less than 85% of the time in bed, the time allowed in bed was reduced to total sleep time plus 30 minutes. Sleepy patients could spend 30 more minutes in bed. All changes were made at bedtime, with wake-up time held constant.
At 6 months, the SSR group had improved perceived sleep quality and fatigue, and improved sleep efficiency as measured by actigraphy. A total of 67% of patients responded to SSR, compared with 41% of controls (number needed to treat = 4).
The efficacy of this intervention is extremely impressive. Importantly, it was delivered by a general practitioner without specialized training during two “slightly extended” visits.
Potential participants were excluded if they were on a sleeping medication, which does not imply that this would not work in a population already on Z-drugs. Consideration should to be given to possible risks when implementing sleep restriction with patients taking Z-drugs with longer half-lives (for example, eszopiclone is 6 hours, zolpidem is 3 hours, and zaleplon is 1 hour), because of higher serum concentrations upon waking.
But when these medications fail, or you have Z-drug–naive patients with insomnia, have this intervention ready.
Dr. Ebbert is professor of medicine, a general internist at the Mayo Clinic in Rochester, Minn., and a diplomate of the American Board of Addiction Medicine. The opinions expressed are those of the author and do not necessarily represent the views and opinions of the Mayo Clinic. The opinions expressed in this article should not be used to diagnose or treat any medical condition, nor should they be used as a substitute for medical advice from a qualified, board-certified practicing clinician. Dr. Ebbert has no relevant financial disclosures about this article. Follow him on Twitter @jonebbert.

Hemophilia A drug appears safe, effective long-term
Interim results of the ASPIRE trial suggest extended treatment or prophylaxis with a recombinant factor VIII Fc fusion protein (rFVIIIFc/efmoroctocog alfa, Eloctate/Elocta) can be safe and effective for hemophilia A patients of all ages.
Patients could enroll in the phase 3 ASPIRE trial after completing the A-LONG and Kids A-LONG studies.
At the time of the interim analysis, most patients had at least 100 days of cumulative exposure to rFVIIIFc.
None of the patients developed inhibitors, and the investigators said adverse events (AEs) were generally consistent with those expected in the general hemophilia A population.
Furthermore, the median annualized bleeding rates (ABRs) were low among patients receiving prophylaxis, and most patients had no change in prophylactic infusion frequency or total weekly prophylactic dose.
These results appear in Haemophilia. The trial is sponsored by Biogen, the company developing rFVIIIFc.
Trial design
ASPIRE has enrolled 211 males with hemophilia A, including 150 (98%) of those who completed A-LONG and 61 (91%) of those who completed Kids A-LONG.
ASPIRE has an on-demand treatment group and 3 prophylactic treatment groups: individualized, weekly, and modified prophylaxis. In the on-demand group, dosing is based on the type and severity of bleeding episodes.
Subjects in the individualized prophylaxis group receive rFVIIIFc at 25-65 IU kg−1 every 3 to 5 days or twice-weekly rFVIIIFc at 20–65 IU kg−1 on day 1 and 40–65 IU kg−1 on day 4. In subjects younger than 12, the investigators can make dose adjustments.
The weekly prophylaxis group receives rFVIIIFc at 65 IU kg−1 every 7 days. Patients who cannot receive optimal treatment in either the individualized or weekly prophylaxis groups can be placed in the modified prophylaxis group.
Patients can change their treatment group at any time during the study. However, subjects younger than 12 can only participate in the individualized and modified prophylaxis groups.
“The design of the ASPIRE study provides physicians a high degree of dosing flexibility, with the goal of reflecting their real-world treatment practices,” said Guy Young, MD, of Children’s Hospital of Los Angeles in California.
Treatment
As of the interim analysis, the median time in the ASPIRE study was 80.9 weeks for adults and adolescents completing the A-LONG study and 23.9 weeks for children completing the Kids A-LONG study. The median cumulative duration of treatment was 117.7 weeks for adults and adolescents and 51.5 weeks for children.
Nearly all Kids A-LONG subjects (96.7%) continued on individualized prophylaxis. Two subjects switched to the modified prophylaxis group upon enrollment in ASPIRE, but none of the subjects changed their treatment group during ASPIRE.
Of the A-LONG subjects, 16.7% changed treatment groups at enrollment in ASPIRE, and 11.3% made a change to their treatment group during ASPIRE. None of the subjects changed treatment groups more than once.
Most patients who were previously on a prophylactic regimen in A-LONG had either no change to their infusion interval (71.9%) or had a longer infusion interval (21.9%) during ASPIRE. Most patients on Kids A-LONG (95.1%) had no change to their prophylactic infusion interval on ASPIRE.
Safety
Overall, 65.4% of subjects had at least 1 AE, and 10.9% had at least 1 serious AE. All of the serious AEs were considered unrelated to rFVIIIFc, and all had resolved by the time of the interim data cut. There were no serious allergic reactions, serious vascular thrombotic events, or deaths.
The most common AEs (incidence of 5% or greater) were nasopharyngitis (12.8%), upper respiratory infection (7.6%), and arthralgia (5.2%). Three adults (1.4%) experienced 4 mild AEs that were thought to be related to rFVIIIFc—chromaturia, elevated blood creatinine, and headache/hot flashes.
Efficacy
For adults and adolescents, the overall ABR was 0.66 in the individualized prophylaxis arm, 2.03 in the weekly prophylaxis arm, 1.97 in the modified prophylaxis arm, and 18.36 in the on-demand treatment arm.
For children in the individualized prophylaxis arm, the overall ABR was 0.00 in children younger than 6 and 1.56 in those ages 6 to 11. For children in the modified prophylaxis arm, the overall ABR was 6.55 in those younger than 6 and 0.00 for children 6 to 11.
A total of 566 bleeding episodes occurred in adults and adolescents who were treated prophylactically. Patients in the on-demand treatment arm experienced 262 bleeding episodes.
Overall, 90.8% of these bleeding episodes were controlled with a single infusion of rFVIIIFc, and 96.9% of the episodes were controlled with 1 or 2 infusions.
Children had a total of 51 bleeding episodes—23 among children younger than 6 and 28 among children 6 to 11.
Most of these episodes were controlled with a single infusion of rFVIIIFc—82.6% among children younger than 6 and 82.1% among children 6 to 11. And 95.7% and 89.3%, respectively, were controlled with 1 to 2 infusions.
“The results suggest prophylaxis with Eloctate shows efficacy and safety for the long-term treatment of hemophilia A,” Dr Young concluded. ![]()
Interim results of the ASPIRE trial suggest extended treatment or prophylaxis with a recombinant factor VIII Fc fusion protein (rFVIIIFc/efmoroctocog alfa, Eloctate/Elocta) can be safe and effective for hemophilia A patients of all ages.
Patients could enroll in the phase 3 ASPIRE trial after completing the A-LONG and Kids A-LONG studies.
At the time of the interim analysis, most patients had at least 100 days of cumulative exposure to rFVIIIFc.
None of the patients developed inhibitors, and the investigators said adverse events (AEs) were generally consistent with those expected in the general hemophilia A population.
Furthermore, the median annualized bleeding rates (ABRs) were low among patients receiving prophylaxis, and most patients had no change in prophylactic infusion frequency or total weekly prophylactic dose.
These results appear in Haemophilia. The trial is sponsored by Biogen, the company developing rFVIIIFc.
Trial design
ASPIRE has enrolled 211 males with hemophilia A, including 150 (98%) of those who completed A-LONG and 61 (91%) of those who completed Kids A-LONG.
ASPIRE has an on-demand treatment group and 3 prophylactic treatment groups: individualized, weekly, and modified prophylaxis. In the on-demand group, dosing is based on the type and severity of bleeding episodes.
Subjects in the individualized prophylaxis group receive rFVIIIFc at 25-65 IU kg−1 every 3 to 5 days or twice-weekly rFVIIIFc at 20–65 IU kg−1 on day 1 and 40–65 IU kg−1 on day 4. In subjects younger than 12, the investigators can make dose adjustments.
The weekly prophylaxis group receives rFVIIIFc at 65 IU kg−1 every 7 days. Patients who cannot receive optimal treatment in either the individualized or weekly prophylaxis groups can be placed in the modified prophylaxis group.
Patients can change their treatment group at any time during the study. However, subjects younger than 12 can only participate in the individualized and modified prophylaxis groups.
“The design of the ASPIRE study provides physicians a high degree of dosing flexibility, with the goal of reflecting their real-world treatment practices,” said Guy Young, MD, of Children’s Hospital of Los Angeles in California.
Treatment
As of the interim analysis, the median time in the ASPIRE study was 80.9 weeks for adults and adolescents completing the A-LONG study and 23.9 weeks for children completing the Kids A-LONG study. The median cumulative duration of treatment was 117.7 weeks for adults and adolescents and 51.5 weeks for children.
Nearly all Kids A-LONG subjects (96.7%) continued on individualized prophylaxis. Two subjects switched to the modified prophylaxis group upon enrollment in ASPIRE, but none of the subjects changed their treatment group during ASPIRE.
Of the A-LONG subjects, 16.7% changed treatment groups at enrollment in ASPIRE, and 11.3% made a change to their treatment group during ASPIRE. None of the subjects changed treatment groups more than once.
Most patients who were previously on a prophylactic regimen in A-LONG had either no change to their infusion interval (71.9%) or had a longer infusion interval (21.9%) during ASPIRE. Most patients on Kids A-LONG (95.1%) had no change to their prophylactic infusion interval on ASPIRE.
Safety
Overall, 65.4% of subjects had at least 1 AE, and 10.9% had at least 1 serious AE. All of the serious AEs were considered unrelated to rFVIIIFc, and all had resolved by the time of the interim data cut. There were no serious allergic reactions, serious vascular thrombotic events, or deaths.
The most common AEs (incidence of 5% or greater) were nasopharyngitis (12.8%), upper respiratory infection (7.6%), and arthralgia (5.2%). Three adults (1.4%) experienced 4 mild AEs that were thought to be related to rFVIIIFc—chromaturia, elevated blood creatinine, and headache/hot flashes.
Efficacy
For adults and adolescents, the overall ABR was 0.66 in the individualized prophylaxis arm, 2.03 in the weekly prophylaxis arm, 1.97 in the modified prophylaxis arm, and 18.36 in the on-demand treatment arm.
For children in the individualized prophylaxis arm, the overall ABR was 0.00 in children younger than 6 and 1.56 in those ages 6 to 11. For children in the modified prophylaxis arm, the overall ABR was 6.55 in those younger than 6 and 0.00 for children 6 to 11.
A total of 566 bleeding episodes occurred in adults and adolescents who were treated prophylactically. Patients in the on-demand treatment arm experienced 262 bleeding episodes.
Overall, 90.8% of these bleeding episodes were controlled with a single infusion of rFVIIIFc, and 96.9% of the episodes were controlled with 1 or 2 infusions.
Children had a total of 51 bleeding episodes—23 among children younger than 6 and 28 among children 6 to 11.
Most of these episodes were controlled with a single infusion of rFVIIIFc—82.6% among children younger than 6 and 82.1% among children 6 to 11. And 95.7% and 89.3%, respectively, were controlled with 1 to 2 infusions.
“The results suggest prophylaxis with Eloctate shows efficacy and safety for the long-term treatment of hemophilia A,” Dr Young concluded. ![]()
Interim results of the ASPIRE trial suggest extended treatment or prophylaxis with a recombinant factor VIII Fc fusion protein (rFVIIIFc/efmoroctocog alfa, Eloctate/Elocta) can be safe and effective for hemophilia A patients of all ages.
Patients could enroll in the phase 3 ASPIRE trial after completing the A-LONG and Kids A-LONG studies.
At the time of the interim analysis, most patients had at least 100 days of cumulative exposure to rFVIIIFc.
None of the patients developed inhibitors, and the investigators said adverse events (AEs) were generally consistent with those expected in the general hemophilia A population.
Furthermore, the median annualized bleeding rates (ABRs) were low among patients receiving prophylaxis, and most patients had no change in prophylactic infusion frequency or total weekly prophylactic dose.
These results appear in Haemophilia. The trial is sponsored by Biogen, the company developing rFVIIIFc.
Trial design
ASPIRE has enrolled 211 males with hemophilia A, including 150 (98%) of those who completed A-LONG and 61 (91%) of those who completed Kids A-LONG.
ASPIRE has an on-demand treatment group and 3 prophylactic treatment groups: individualized, weekly, and modified prophylaxis. In the on-demand group, dosing is based on the type and severity of bleeding episodes.
Subjects in the individualized prophylaxis group receive rFVIIIFc at 25-65 IU kg−1 every 3 to 5 days or twice-weekly rFVIIIFc at 20–65 IU kg−1 on day 1 and 40–65 IU kg−1 on day 4. In subjects younger than 12, the investigators can make dose adjustments.
The weekly prophylaxis group receives rFVIIIFc at 65 IU kg−1 every 7 days. Patients who cannot receive optimal treatment in either the individualized or weekly prophylaxis groups can be placed in the modified prophylaxis group.
Patients can change their treatment group at any time during the study. However, subjects younger than 12 can only participate in the individualized and modified prophylaxis groups.
“The design of the ASPIRE study provides physicians a high degree of dosing flexibility, with the goal of reflecting their real-world treatment practices,” said Guy Young, MD, of Children’s Hospital of Los Angeles in California.
Treatment
As of the interim analysis, the median time in the ASPIRE study was 80.9 weeks for adults and adolescents completing the A-LONG study and 23.9 weeks for children completing the Kids A-LONG study. The median cumulative duration of treatment was 117.7 weeks for adults and adolescents and 51.5 weeks for children.
Nearly all Kids A-LONG subjects (96.7%) continued on individualized prophylaxis. Two subjects switched to the modified prophylaxis group upon enrollment in ASPIRE, but none of the subjects changed their treatment group during ASPIRE.
Of the A-LONG subjects, 16.7% changed treatment groups at enrollment in ASPIRE, and 11.3% made a change to their treatment group during ASPIRE. None of the subjects changed treatment groups more than once.
Most patients who were previously on a prophylactic regimen in A-LONG had either no change to their infusion interval (71.9%) or had a longer infusion interval (21.9%) during ASPIRE. Most patients on Kids A-LONG (95.1%) had no change to their prophylactic infusion interval on ASPIRE.
Safety
Overall, 65.4% of subjects had at least 1 AE, and 10.9% had at least 1 serious AE. All of the serious AEs were considered unrelated to rFVIIIFc, and all had resolved by the time of the interim data cut. There were no serious allergic reactions, serious vascular thrombotic events, or deaths.
The most common AEs (incidence of 5% or greater) were nasopharyngitis (12.8%), upper respiratory infection (7.6%), and arthralgia (5.2%). Three adults (1.4%) experienced 4 mild AEs that were thought to be related to rFVIIIFc—chromaturia, elevated blood creatinine, and headache/hot flashes.
Efficacy
For adults and adolescents, the overall ABR was 0.66 in the individualized prophylaxis arm, 2.03 in the weekly prophylaxis arm, 1.97 in the modified prophylaxis arm, and 18.36 in the on-demand treatment arm.
For children in the individualized prophylaxis arm, the overall ABR was 0.00 in children younger than 6 and 1.56 in those ages 6 to 11. For children in the modified prophylaxis arm, the overall ABR was 6.55 in those younger than 6 and 0.00 for children 6 to 11.
A total of 566 bleeding episodes occurred in adults and adolescents who were treated prophylactically. Patients in the on-demand treatment arm experienced 262 bleeding episodes.
Overall, 90.8% of these bleeding episodes were controlled with a single infusion of rFVIIIFc, and 96.9% of the episodes were controlled with 1 or 2 infusions.
Children had a total of 51 bleeding episodes—23 among children younger than 6 and 28 among children 6 to 11.
Most of these episodes were controlled with a single infusion of rFVIIIFc—82.6% among children younger than 6 and 82.1% among children 6 to 11. And 95.7% and 89.3%, respectively, were controlled with 1 to 2 infusions.
“The results suggest prophylaxis with Eloctate shows efficacy and safety for the long-term treatment of hemophilia A,” Dr Young concluded. ![]()
Groups draft guidelines for acute leukemia

Photo courtesy of the CDC
The American Society of Hematology (ASH) and the College of American Pathologists (CAP) have opened a public comment period for a draft guideline that addresses the initial work-up of acute leukemia.
The guideline details the information required for the diagnosis of acute leukemias, as well as recommended testing and how test results and diagnosis should be correlated.
The document will be available for comment through August 31.
The guideline authors examined evidence from more than 170 articles to devise the draft guidelines. The resulting document answers the following questions:
- What clinical and laboratory information should be available during the initial diagnostic evaluation of a patient with acute leukemia?
- What specimens and sample types should be evaluated during the initial work-up of a patient with acute leukemia?
- At the time of diagnosis, what tests are required for all patients for the initial evaluation of an acute leukemia?
- What tests should be performed only on a subset of patients, including in response to results of initial tests and morphology?
- Where should testing be performed?
- How should test results and the diagnosis be correlated and reported?
“Evidence-based guidelines like these are increasingly vital to the continued improvement and continuity of patient care,” said ASH guideline co-chair James W. Vardiman, MD, of the University of Chicago in Illinois.
He and Daniel A. Arber, MD, of Stanford School of Medicine in California, (the CAP representative co-chair) are leading an interdisciplinary team of 8 physicians representing sub-specialties that include hematopathology and oncology.
“Our work on these guidelines aims at integrating the very best practices to improve outcomes for [acute leukemia] patients and their families,” Dr Arber said.
At the close of the comment period, the CAP/ASH team will review any comments and make final recommendations, which are targeted for publication in the first quarter of 2016. ![]()
Photo courtesy of the CDC
The American Society of Hematology (ASH) and the College of American Pathologists (CAP) have opened a public comment period for a draft guideline that addresses the initial work-up of acute leukemia.
The guideline details the information required for the diagnosis of acute leukemias, as well as recommended testing and how test results and diagnosis should be correlated.
The document will be available for comment through August 31.
The guideline authors examined evidence from more than 170 articles to devise the draft guidelines. The resulting document answers the following questions:
- What clinical and laboratory information should be available during the initial diagnostic evaluation of a patient with acute leukemia?
- What specimens and sample types should be evaluated during the initial work-up of a patient with acute leukemia?
- At the time of diagnosis, what tests are required for all patients for the initial evaluation of an acute leukemia?
- What tests should be performed only on a subset of patients, including in response to results of initial tests and morphology?
- Where should testing be performed?
- How should test results and the diagnosis be correlated and reported?
“Evidence-based guidelines like these are increasingly vital to the continued improvement and continuity of patient care,” said ASH guideline co-chair James W. Vardiman, MD, of the University of Chicago in Illinois.
He and Daniel A. Arber, MD, of Stanford School of Medicine in California, (the CAP representative co-chair) are leading an interdisciplinary team of 8 physicians representing sub-specialties that include hematopathology and oncology.
“Our work on these guidelines aims at integrating the very best practices to improve outcomes for [acute leukemia] patients and their families,” Dr Arber said.
At the close of the comment period, the CAP/ASH team will review any comments and make final recommendations, which are targeted for publication in the first quarter of 2016. ![]()
Photo courtesy of the CDC
The American Society of Hematology (ASH) and the College of American Pathologists (CAP) have opened a public comment period for a draft guideline that addresses the initial work-up of acute leukemia.
The guideline details the information required for the diagnosis of acute leukemias, as well as recommended testing and how test results and diagnosis should be correlated.
The document will be available for comment through August 31.
The guideline authors examined evidence from more than 170 articles to devise the draft guidelines. The resulting document answers the following questions:
- What clinical and laboratory information should be available during the initial diagnostic evaluation of a patient with acute leukemia?
- What specimens and sample types should be evaluated during the initial work-up of a patient with acute leukemia?
- At the time of diagnosis, what tests are required for all patients for the initial evaluation of an acute leukemia?
- What tests should be performed only on a subset of patients, including in response to results of initial tests and morphology?
- Where should testing be performed?
- How should test results and the diagnosis be correlated and reported?
“Evidence-based guidelines like these are increasingly vital to the continued improvement and continuity of patient care,” said ASH guideline co-chair James W. Vardiman, MD, of the University of Chicago in Illinois.
He and Daniel A. Arber, MD, of Stanford School of Medicine in California, (the CAP representative co-chair) are leading an interdisciplinary team of 8 physicians representing sub-specialties that include hematopathology and oncology.
“Our work on these guidelines aims at integrating the very best practices to improve outcomes for [acute leukemia] patients and their families,” Dr Arber said.
At the close of the comment period, the CAP/ASH team will review any comments and make final recommendations, which are targeted for publication in the first quarter of 2016. ![]()
Mutations may contribute to CTCL

mycosis fungoides
Researchers have identified 15 mutations that may drive cutaneous T-cell lymphoma (CTCL).
The team sequenced normal and cancerous samples from 73 patients with mycosis fungoides or Sézary syndrome.
This revealed recurrent alterations in the TNFR2 pathway, as well as mutations in phosphoinositide 3-kinase (PI3K)-related genes, NF-κB pathway genes, and other genes that regulate T-cell survival and proliferation.
Specifically, the researchers identified TNFRSF1B point mutations, TNFRSF1B gains, CTLA4-CD28 fusions, a TRAF3 deletion, and mutations in NFAT5, TEC, PIK3CD, PIK3R6, PIK3CG, PIK3R5, PIK3R4, VAV1, MALT1, CD28, and ITK.
Paul Khavari, MD, PhD, of Stanford University in California, and his colleagues conducted this research and described their findings in a letter to Nature Genetics.
TNFR2 mutations
The researchers noted that the most frequent recurrent point mutation they identified occurred at codon 377 of TNFRSF1B (5%; 4/73), resulting in a recurrent TNFR2 Thr377Ile mutant.
TNFR2 is a receptor that regulates T-cell signaling pathways, and the mutation locked the receptor into an always-on state, preventing the T-cell-survival pathway from shutting down.
Previous studies showed that patients with increased TNFR2 in their bloodstream had more aggressive forms of CTCL that were more likely to return quickly after treatment.
This led Dr Khavari and his colleagues to look at the other patients’ DNA to see if duplications could account for both the elevated levels in the blood and increased signaling to activate the T-cell-survival pathway. The team found that 10 of the patients had TNFRSF1B gains.
In total, TNFRSF1B was altered in 18% of patients (13/73), by point mutation or gain (both in 1 patient). The researchers said this suggests a potential role of oncogenic TNFR2 signaling in the development of CTCL.
The team uncovered evidence to support this role by growing cells in the lab with either the point mutation or the gain. Their experiment showed the T-cell-survival pathway was more active in these cells than in normal cells.
Now, the researchers are working to incorporate the mutations they identified into the DNA of mice to study the mutated genes’ effects and the actions of drugs on those genes. ![]()
mycosis fungoides
Researchers have identified 15 mutations that may drive cutaneous T-cell lymphoma (CTCL).
The team sequenced normal and cancerous samples from 73 patients with mycosis fungoides or Sézary syndrome.
This revealed recurrent alterations in the TNFR2 pathway, as well as mutations in phosphoinositide 3-kinase (PI3K)-related genes, NF-κB pathway genes, and other genes that regulate T-cell survival and proliferation.
Specifically, the researchers identified TNFRSF1B point mutations, TNFRSF1B gains, CTLA4-CD28 fusions, a TRAF3 deletion, and mutations in NFAT5, TEC, PIK3CD, PIK3R6, PIK3CG, PIK3R5, PIK3R4, VAV1, MALT1, CD28, and ITK.
Paul Khavari, MD, PhD, of Stanford University in California, and his colleagues conducted this research and described their findings in a letter to Nature Genetics.
TNFR2 mutations
The researchers noted that the most frequent recurrent point mutation they identified occurred at codon 377 of TNFRSF1B (5%; 4/73), resulting in a recurrent TNFR2 Thr377Ile mutant.
TNFR2 is a receptor that regulates T-cell signaling pathways, and the mutation locked the receptor into an always-on state, preventing the T-cell-survival pathway from shutting down.
Previous studies showed that patients with increased TNFR2 in their bloodstream had more aggressive forms of CTCL that were more likely to return quickly after treatment.
This led Dr Khavari and his colleagues to look at the other patients’ DNA to see if duplications could account for both the elevated levels in the blood and increased signaling to activate the T-cell-survival pathway. The team found that 10 of the patients had TNFRSF1B gains.
In total, TNFRSF1B was altered in 18% of patients (13/73), by point mutation or gain (both in 1 patient). The researchers said this suggests a potential role of oncogenic TNFR2 signaling in the development of CTCL.
The team uncovered evidence to support this role by growing cells in the lab with either the point mutation or the gain. Their experiment showed the T-cell-survival pathway was more active in these cells than in normal cells.
Now, the researchers are working to incorporate the mutations they identified into the DNA of mice to study the mutated genes’ effects and the actions of drugs on those genes. ![]()
mycosis fungoides
Researchers have identified 15 mutations that may drive cutaneous T-cell lymphoma (CTCL).
The team sequenced normal and cancerous samples from 73 patients with mycosis fungoides or Sézary syndrome.
This revealed recurrent alterations in the TNFR2 pathway, as well as mutations in phosphoinositide 3-kinase (PI3K)-related genes, NF-κB pathway genes, and other genes that regulate T-cell survival and proliferation.
Specifically, the researchers identified TNFRSF1B point mutations, TNFRSF1B gains, CTLA4-CD28 fusions, a TRAF3 deletion, and mutations in NFAT5, TEC, PIK3CD, PIK3R6, PIK3CG, PIK3R5, PIK3R4, VAV1, MALT1, CD28, and ITK.
Paul Khavari, MD, PhD, of Stanford University in California, and his colleagues conducted this research and described their findings in a letter to Nature Genetics.
TNFR2 mutations
The researchers noted that the most frequent recurrent point mutation they identified occurred at codon 377 of TNFRSF1B (5%; 4/73), resulting in a recurrent TNFR2 Thr377Ile mutant.
TNFR2 is a receptor that regulates T-cell signaling pathways, and the mutation locked the receptor into an always-on state, preventing the T-cell-survival pathway from shutting down.
Previous studies showed that patients with increased TNFR2 in their bloodstream had more aggressive forms of CTCL that were more likely to return quickly after treatment.
This led Dr Khavari and his colleagues to look at the other patients’ DNA to see if duplications could account for both the elevated levels in the blood and increased signaling to activate the T-cell-survival pathway. The team found that 10 of the patients had TNFRSF1B gains.
In total, TNFRSF1B was altered in 18% of patients (13/73), by point mutation or gain (both in 1 patient). The researchers said this suggests a potential role of oncogenic TNFR2 signaling in the development of CTCL.
The team uncovered evidence to support this role by growing cells in the lab with either the point mutation or the gain. Their experiment showed the T-cell-survival pathway was more active in these cells than in normal cells.
Now, the researchers are working to incorporate the mutations they identified into the DNA of mice to study the mutated genes’ effects and the actions of drugs on those genes. ![]()
Selinexor dose lowered due to sepsis in AML patients

Photo by Esther Dyson
An excess of sepsis cases has prompted dose reductions in a phase 2 trial of selinexor in older patients with relapsed/refractory acute myeloid leukemia (AML).
For this study, known as SOPRA, researchers are comparing selinexor to physician’s choice of treatment in AML patients age 60 and older.
There have only been 8 cases of sepsis in the selinexor arm thus far, but this is more than observed in the physician’s
choice arm.
So the company developing selinexor, Karyopharm Therapeutics Inc., said it has reduced the selinexor dose used in this study.
The company decided to reduce the dose from 55 mg/m2 to a fixed dose of 60 mg, which corresponds to approximately 35 mg/m2. Dosing will remain twice weekly.
The change was implemented based on ongoing safety and tolerability evaluations in the SOPRA study, as well as maturing data from AML patients in the phase 1, first-in-human trial of selinexor.
The SOPRA study uses a 2 to 1 randomization of AML patients to selinexor or physician’s choice and, therefore, approximately twice as many cases of sepsis would be expected on the selinexor arm compared with the physician’s choice arm.
As of the end of July 2015, there have been 8 reports of sepsis in 7 patients receiving selinexor at 55 mg/m2, compared with 2 reports of sepsis in 2 patients receiving physician’s choice.
Karyopharm pointed out that these numbers are small, and sepsis is often observed in patients with AML, but the incidence of sepsis appears to be higher in the patients receiving selinexor.
In addition, the company noted an apparent increase in the incidence of sepsis in patients with relapsed or refractory AML receiving high doses of selinexor twice weekly in the phase 1 trial of patients with hematologic malignancies.
Karyopharm said selinexor doses of 60 mg twice weekly do not appear to be associated with any increase in sepsis or other infection-related events in patients with hematologic malignancies or solid tumors.
In addition, most patients with AML in the phase 1 study who showed a response to selinexor, including complete responses, received the drug at doses of approximately 60 mg or lower.
As a result of the change in dose, the SOPRA study will now have an interim assessment in mid-2016.
Karyopharm is actively enrolling patients in the SOPRA study, as well as a study of selinexor in patients with relapsed/refractory diffuse large B-cell lymphoma (SADAL trial), and a study of the drug in patients with Richter’s transformation (SIRRT trial).
Preliminary top-line data from all 3 studies are anticipated in the fourth quarter of 2016.
The company has also initiated a single-arm trial of selinexor plus dexamethasone in patients with multiple myeloma (STORM trial), which will initially include 80 patients. Preliminary top-line data from this study are anticipated in mid-2016. ![]()
Photo by Esther Dyson
An excess of sepsis cases has prompted dose reductions in a phase 2 trial of selinexor in older patients with relapsed/refractory acute myeloid leukemia (AML).
For this study, known as SOPRA, researchers are comparing selinexor to physician’s choice of treatment in AML patients age 60 and older.
There have only been 8 cases of sepsis in the selinexor arm thus far, but this is more than observed in the physician’s
choice arm.
So the company developing selinexor, Karyopharm Therapeutics Inc., said it has reduced the selinexor dose used in this study.
The company decided to reduce the dose from 55 mg/m2 to a fixed dose of 60 mg, which corresponds to approximately 35 mg/m2. Dosing will remain twice weekly.
The change was implemented based on ongoing safety and tolerability evaluations in the SOPRA study, as well as maturing data from AML patients in the phase 1, first-in-human trial of selinexor.
The SOPRA study uses a 2 to 1 randomization of AML patients to selinexor or physician’s choice and, therefore, approximately twice as many cases of sepsis would be expected on the selinexor arm compared with the physician’s choice arm.
As of the end of July 2015, there have been 8 reports of sepsis in 7 patients receiving selinexor at 55 mg/m2, compared with 2 reports of sepsis in 2 patients receiving physician’s choice.
Karyopharm pointed out that these numbers are small, and sepsis is often observed in patients with AML, but the incidence of sepsis appears to be higher in the patients receiving selinexor.
In addition, the company noted an apparent increase in the incidence of sepsis in patients with relapsed or refractory AML receiving high doses of selinexor twice weekly in the phase 1 trial of patients with hematologic malignancies.
Karyopharm said selinexor doses of 60 mg twice weekly do not appear to be associated with any increase in sepsis or other infection-related events in patients with hematologic malignancies or solid tumors.
In addition, most patients with AML in the phase 1 study who showed a response to selinexor, including complete responses, received the drug at doses of approximately 60 mg or lower.
As a result of the change in dose, the SOPRA study will now have an interim assessment in mid-2016.
Karyopharm is actively enrolling patients in the SOPRA study, as well as a study of selinexor in patients with relapsed/refractory diffuse large B-cell lymphoma (SADAL trial), and a study of the drug in patients with Richter’s transformation (SIRRT trial).
Preliminary top-line data from all 3 studies are anticipated in the fourth quarter of 2016.
The company has also initiated a single-arm trial of selinexor plus dexamethasone in patients with multiple myeloma (STORM trial), which will initially include 80 patients. Preliminary top-line data from this study are anticipated in mid-2016. ![]()
Photo by Esther Dyson
An excess of sepsis cases has prompted dose reductions in a phase 2 trial of selinexor in older patients with relapsed/refractory acute myeloid leukemia (AML).
For this study, known as SOPRA, researchers are comparing selinexor to physician’s choice of treatment in AML patients age 60 and older.
There have only been 8 cases of sepsis in the selinexor arm thus far, but this is more than observed in the physician’s
choice arm.
So the company developing selinexor, Karyopharm Therapeutics Inc., said it has reduced the selinexor dose used in this study.
The company decided to reduce the dose from 55 mg/m2 to a fixed dose of 60 mg, which corresponds to approximately 35 mg/m2. Dosing will remain twice weekly.
The change was implemented based on ongoing safety and tolerability evaluations in the SOPRA study, as well as maturing data from AML patients in the phase 1, first-in-human trial of selinexor.
The SOPRA study uses a 2 to 1 randomization of AML patients to selinexor or physician’s choice and, therefore, approximately twice as many cases of sepsis would be expected on the selinexor arm compared with the physician’s choice arm.
As of the end of July 2015, there have been 8 reports of sepsis in 7 patients receiving selinexor at 55 mg/m2, compared with 2 reports of sepsis in 2 patients receiving physician’s choice.
Karyopharm pointed out that these numbers are small, and sepsis is often observed in patients with AML, but the incidence of sepsis appears to be higher in the patients receiving selinexor.
In addition, the company noted an apparent increase in the incidence of sepsis in patients with relapsed or refractory AML receiving high doses of selinexor twice weekly in the phase 1 trial of patients with hematologic malignancies.
Karyopharm said selinexor doses of 60 mg twice weekly do not appear to be associated with any increase in sepsis or other infection-related events in patients with hematologic malignancies or solid tumors.
In addition, most patients with AML in the phase 1 study who showed a response to selinexor, including complete responses, received the drug at doses of approximately 60 mg or lower.
As a result of the change in dose, the SOPRA study will now have an interim assessment in mid-2016.
Karyopharm is actively enrolling patients in the SOPRA study, as well as a study of selinexor in patients with relapsed/refractory diffuse large B-cell lymphoma (SADAL trial), and a study of the drug in patients with Richter’s transformation (SIRRT trial).
Preliminary top-line data from all 3 studies are anticipated in the fourth quarter of 2016.
The company has also initiated a single-arm trial of selinexor plus dexamethasone in patients with multiple myeloma (STORM trial), which will initially include 80 patients. Preliminary top-line data from this study are anticipated in mid-2016. ![]()
Yet another insurer demand: patient care notifications
Some insurance companies recently started sending notifications about patient care. For example, one recently sent my boss a letter about one of his patients with rheumatoid arthritis, asking why the patient is not on a disease modifier. There is an invitation to explain why by ticking any one of several boxes. Is the patient perhaps not compliant? Did the patient discontinue the medication against the doctor’s advice? Has the patient passed away? Does the patient not have rheumatoid arthritis?
Oh, to be a fly on the wall when the insurance company decided that they would start doing this! This must impose a financial burden on the insurer, one that I cannot imagine they take on out of sheer altruism. What is the end game? What do they do with this information? Will they hold this information against the patient somehow, raise their premium in the next enrollment period? Or hold it against the physician, perhaps ding their reimbursement or use the information to include or exclude physicians from their panels?
When I decided to come to the United States, one of the biggest draws was the availability of health insurance. Most of my medical school education came from American textbooks, after all, so I thought it would be fabulous to be able to practice medicine the way it should be practiced because insurance will pay for it. (I know I sound like your elderly aunt that likes to repeat herself, but if you have not read any of my columns before, I come from the Philippines where health care is mostly paid for out of pocket, so how we treated patients was severely limited by how much the patient could afford.) I was wrong. I had no idea that part of my job description would include having to ask a corporate entity’s permission to administer treatments.
My boss replied to the letter. He said: “Patient is on dialysis and cannot be on methotrexate. He was prescribed a biologic, but your insurance does not cover its cost sufficiently to make it affordable. We had obtained the biologic through foundation support, but they ran out of money. So when you send out a letter like this blaming either the patient or MD for a compliance issue, I urge you to do some soul-searching.”
Dr. Chan practices rheumatology in Pawtucket, R.I.
Some insurance companies recently started sending notifications about patient care. For example, one recently sent my boss a letter about one of his patients with rheumatoid arthritis, asking why the patient is not on a disease modifier. There is an invitation to explain why by ticking any one of several boxes. Is the patient perhaps not compliant? Did the patient discontinue the medication against the doctor’s advice? Has the patient passed away? Does the patient not have rheumatoid arthritis?
Oh, to be a fly on the wall when the insurance company decided that they would start doing this! This must impose a financial burden on the insurer, one that I cannot imagine they take on out of sheer altruism. What is the end game? What do they do with this information? Will they hold this information against the patient somehow, raise their premium in the next enrollment period? Or hold it against the physician, perhaps ding their reimbursement or use the information to include or exclude physicians from their panels?
When I decided to come to the United States, one of the biggest draws was the availability of health insurance. Most of my medical school education came from American textbooks, after all, so I thought it would be fabulous to be able to practice medicine the way it should be practiced because insurance will pay for it. (I know I sound like your elderly aunt that likes to repeat herself, but if you have not read any of my columns before, I come from the Philippines where health care is mostly paid for out of pocket, so how we treated patients was severely limited by how much the patient could afford.) I was wrong. I had no idea that part of my job description would include having to ask a corporate entity’s permission to administer treatments.
My boss replied to the letter. He said: “Patient is on dialysis and cannot be on methotrexate. He was prescribed a biologic, but your insurance does not cover its cost sufficiently to make it affordable. We had obtained the biologic through foundation support, but they ran out of money. So when you send out a letter like this blaming either the patient or MD for a compliance issue, I urge you to do some soul-searching.”
Dr. Chan practices rheumatology in Pawtucket, R.I.
Some insurance companies recently started sending notifications about patient care. For example, one recently sent my boss a letter about one of his patients with rheumatoid arthritis, asking why the patient is not on a disease modifier. There is an invitation to explain why by ticking any one of several boxes. Is the patient perhaps not compliant? Did the patient discontinue the medication against the doctor’s advice? Has the patient passed away? Does the patient not have rheumatoid arthritis?
Oh, to be a fly on the wall when the insurance company decided that they would start doing this! This must impose a financial burden on the insurer, one that I cannot imagine they take on out of sheer altruism. What is the end game? What do they do with this information? Will they hold this information against the patient somehow, raise their premium in the next enrollment period? Or hold it against the physician, perhaps ding their reimbursement or use the information to include or exclude physicians from their panels?
When I decided to come to the United States, one of the biggest draws was the availability of health insurance. Most of my medical school education came from American textbooks, after all, so I thought it would be fabulous to be able to practice medicine the way it should be practiced because insurance will pay for it. (I know I sound like your elderly aunt that likes to repeat herself, but if you have not read any of my columns before, I come from the Philippines where health care is mostly paid for out of pocket, so how we treated patients was severely limited by how much the patient could afford.) I was wrong. I had no idea that part of my job description would include having to ask a corporate entity’s permission to administer treatments.
My boss replied to the letter. He said: “Patient is on dialysis and cannot be on methotrexate. He was prescribed a biologic, but your insurance does not cover its cost sufficiently to make it affordable. We had obtained the biologic through foundation support, but they ran out of money. So when you send out a letter like this blaming either the patient or MD for a compliance issue, I urge you to do some soul-searching.”
Dr. Chan practices rheumatology in Pawtucket, R.I.

Linear Scleroderma Associated With Neurofibromatosis Type I
To the Editor:
A 12-year-old girl presented with an asymptomatic hypopigmented area on the right cheek of 2 months’ duration. Two years prior to presentation she was diagnosed with neurofibromatosis type I (NF1) based on the findings of 13 café au lait spots on the trunk, axillary and groin freckling, bilateral Lisch nodules, and mild scoliosis. She was otherwise well and had no relevant medical history or family history of neurofibromatosis.
Physical examination revealed a 3×7-cm linear, shiny, sclerotic plaque extending from the right temple to the preauricular area and lower aspect of the right cheek (Figure 1) with associated facial asymmetry (Figure 2). A smaller similar plaque on the chin measured 0.5×0.5 cm. Examination of the oral cavity was unremarkable and there were no neurological signs. No other features were present to suggest a mixed connective tissue disease or lupus erythematosus, and nuclear antibodies were negative.
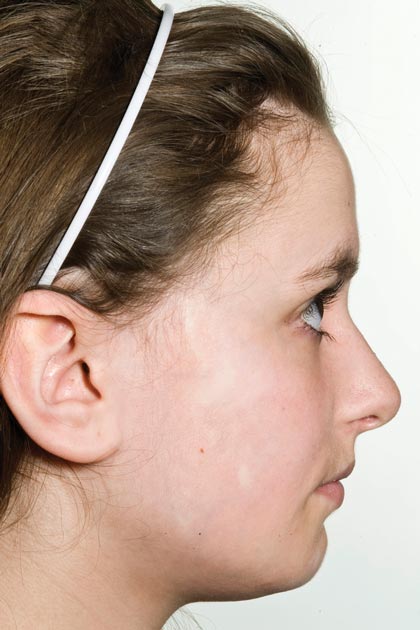 |  |
An incisional biopsy from the sclerotic plaque revealed swollen eosinophilic bundles in the reticular dermis with a moderate perivascular lymphohistiocytic infiltrate extending into the subcutaneous layer. These features were compatible with the diagnosis of linear scleroderma. Due to the progressive nature of the condition and its anatomical location, she was treated with pulsed intravenous methylprednisolone 1 g daily for 3 days and commenced on oral methotrexate 20 mg weekly as well as tacrolimus ointment 0.1% daily. The plaque gradually softened and faded over a period of 8 months. The patient continued on methotrexate for another 10 months. The facial asymmetry persisted with a discernible reduction in the volume of the right cheek. New onset of ipsilateral jaw locking and pain associated with spasms of the muscles of mastication suggested the diagnosis of Parry-Romberg syndrome.
Neurofibromatosis type I is a neuroectodermal abnormality first described by Friedrich von Recklinghausen in 1882 with an incidence of 1 in 3000 births. Neurofibromatosis type I gene mutations lead to increased Ras activity, which is implicated in many NF1-related conditions such as neurofibromas and schwannomas.1 Autoimmune conditions including systemic lupus erythematosus (SLE) and mixed connective tissue rarely have been reported in NF1, but the mechanism of their association is not clear.2 Based on a review of 5 reported cases of NF1 and SLE, most patients were female, and the predominant features of NF1 were café au lait macules and neurofibromas.3-6 One case documented a family history of NF1,6 suggesting predominance of sporadic mutations in these cases. Interestingly, in 3 cases the diagnosis of SLE preceded the diagnosis of NF1, prompting the authors to suggest a viral trigger for the development of NF1 lesions.3,4 Linear scleroderma is immunologically mediated and is characterized by the onset of smooth indurated cutaneous plaques. According to a PubMed search of articles indexed for MEDLINE using the search terms morphea and neurofibromatosis as well as linear scleroderma and neurofibromatosis, there have been no reports of linear scleroderma or morphea associated with NF1. Cichowski et al7 demonstrated enhanced activation of Ras and prolonged activities of both Ras and extracellular signal-regulated kinase (ERK) signaling pathways in NF1-deficient mice. Chen et al8 showed that heparin sulfate–dependent ERK activation contributes to the development of scleroderma by promoting the expression of profibrotic proteins in scleroderma fibroblasts. It was previously noted that increased Ras/ERK signaling activities were important in connective tissue growth factor expression in normal mesenchymal cells.9
Based on these findings, we speculated that hyperactivation of Ras/ERK signaling from NF1 mutations could lead to the promotion of fibrosis seen in scleroderma. The lack of similar reports, however, suggests that the presence of both conditions in this case is coincidental. However, the growing number of reports on autoimmune and connective tissue disorders in NF1 reflects the need for further research in this area.
1. Harrisingh MC, Lloyd AC. Ras/Raf/ERK signalling and NF1. Cell Cycle. 2004;3:1255-1258.
2. Migita K, Kawabe Y, Mori M, et al. Mixed connective tissue disease associated with von Recklinghausen’s neurofibromatosis. Intern Med. 2001;40:363-364.
3. Bitnun S, Bassan H. Letter: neurofibromatosis and SLE. N Engl J Med. 1975;292:429-430.
4. Riccardi VM. Neurofibromatosis in a patient with systemic lupus erythematosus. Arthritis Rheum. 1983;26:574.
5. Corominas H, Guardiola JM, Matas L, et al. Neurofibromatosis and systemic lupus erythematosus. A matter of coincidence? Clin Rheum. 2003;22:496-497.
6. Akyüz SG, Çatlik A, Bülbül M, et al. An unusual pediatric case with neurofibromatosis and systemic lupus erythematosus. Rheum Int. 2012;32:2345-2347.
7. Cichowski K, Santiago S, Jardim M, et al. Dynamic regulation of the Ras pathway via proteolysis of the NF1 tumour suppressor. Genes Dev. 2003;17:449-454.
8. Chen Y, Leask A, Abraham DJ, et al. Heparan sulfate-dependent ERK activation contributes to the overexpression of fibrotic proteins and enhanced contraction by scleroderma fibroblasts. Arthritis Rheum. 2008;58:577-585.
9. Chen Y, Shi-wen X, van Beek J, et al. Matrix contraction by dermal fibroblasts requiring transforming growth factor-â/activin-linked kinase 5, heparin sulphate-containing proteoglycans and MEK/ERK: insights into pathological scarring in chronic fibrotic disease. Am J Pathol. 2005;167:1699-1711.
To the Editor:
A 12-year-old girl presented with an asymptomatic hypopigmented area on the right cheek of 2 months’ duration. Two years prior to presentation she was diagnosed with neurofibromatosis type I (NF1) based on the findings of 13 café au lait spots on the trunk, axillary and groin freckling, bilateral Lisch nodules, and mild scoliosis. She was otherwise well and had no relevant medical history or family history of neurofibromatosis.
Physical examination revealed a 3×7-cm linear, shiny, sclerotic plaque extending from the right temple to the preauricular area and lower aspect of the right cheek (Figure 1) with associated facial asymmetry (Figure 2). A smaller similar plaque on the chin measured 0.5×0.5 cm. Examination of the oral cavity was unremarkable and there were no neurological signs. No other features were present to suggest a mixed connective tissue disease or lupus erythematosus, and nuclear antibodies were negative.
| |
An incisional biopsy from the sclerotic plaque revealed swollen eosinophilic bundles in the reticular dermis with a moderate perivascular lymphohistiocytic infiltrate extending into the subcutaneous layer. These features were compatible with the diagnosis of linear scleroderma. Due to the progressive nature of the condition and its anatomical location, she was treated with pulsed intravenous methylprednisolone 1 g daily for 3 days and commenced on oral methotrexate 20 mg weekly as well as tacrolimus ointment 0.1% daily. The plaque gradually softened and faded over a period of 8 months. The patient continued on methotrexate for another 10 months. The facial asymmetry persisted with a discernible reduction in the volume of the right cheek. New onset of ipsilateral jaw locking and pain associated with spasms of the muscles of mastication suggested the diagnosis of Parry-Romberg syndrome.
Neurofibromatosis type I is a neuroectodermal abnormality first described by Friedrich von Recklinghausen in 1882 with an incidence of 1 in 3000 births. Neurofibromatosis type I gene mutations lead to increased Ras activity, which is implicated in many NF1-related conditions such as neurofibromas and schwannomas.1 Autoimmune conditions including systemic lupus erythematosus (SLE) and mixed connective tissue rarely have been reported in NF1, but the mechanism of their association is not clear.2 Based on a review of 5 reported cases of NF1 and SLE, most patients were female, and the predominant features of NF1 were café au lait macules and neurofibromas.3-6 One case documented a family history of NF1,6 suggesting predominance of sporadic mutations in these cases. Interestingly, in 3 cases the diagnosis of SLE preceded the diagnosis of NF1, prompting the authors to suggest a viral trigger for the development of NF1 lesions.3,4 Linear scleroderma is immunologically mediated and is characterized by the onset of smooth indurated cutaneous plaques. According to a PubMed search of articles indexed for MEDLINE using the search terms morphea and neurofibromatosis as well as linear scleroderma and neurofibromatosis, there have been no reports of linear scleroderma or morphea associated with NF1. Cichowski et al7 demonstrated enhanced activation of Ras and prolonged activities of both Ras and extracellular signal-regulated kinase (ERK) signaling pathways in NF1-deficient mice. Chen et al8 showed that heparin sulfate–dependent ERK activation contributes to the development of scleroderma by promoting the expression of profibrotic proteins in scleroderma fibroblasts. It was previously noted that increased Ras/ERK signaling activities were important in connective tissue growth factor expression in normal mesenchymal cells.9
Based on these findings, we speculated that hyperactivation of Ras/ERK signaling from NF1 mutations could lead to the promotion of fibrosis seen in scleroderma. The lack of similar reports, however, suggests that the presence of both conditions in this case is coincidental. However, the growing number of reports on autoimmune and connective tissue disorders in NF1 reflects the need for further research in this area.
To the Editor:
A 12-year-old girl presented with an asymptomatic hypopigmented area on the right cheek of 2 months’ duration. Two years prior to presentation she was diagnosed with neurofibromatosis type I (NF1) based on the findings of 13 café au lait spots on the trunk, axillary and groin freckling, bilateral Lisch nodules, and mild scoliosis. She was otherwise well and had no relevant medical history or family history of neurofibromatosis.
Physical examination revealed a 3×7-cm linear, shiny, sclerotic plaque extending from the right temple to the preauricular area and lower aspect of the right cheek (Figure 1) with associated facial asymmetry (Figure 2). A smaller similar plaque on the chin measured 0.5×0.5 cm. Examination of the oral cavity was unremarkable and there were no neurological signs. No other features were present to suggest a mixed connective tissue disease or lupus erythematosus, and nuclear antibodies were negative.
| |
An incisional biopsy from the sclerotic plaque revealed swollen eosinophilic bundles in the reticular dermis with a moderate perivascular lymphohistiocytic infiltrate extending into the subcutaneous layer. These features were compatible with the diagnosis of linear scleroderma. Due to the progressive nature of the condition and its anatomical location, she was treated with pulsed intravenous methylprednisolone 1 g daily for 3 days and commenced on oral methotrexate 20 mg weekly as well as tacrolimus ointment 0.1% daily. The plaque gradually softened and faded over a period of 8 months. The patient continued on methotrexate for another 10 months. The facial asymmetry persisted with a discernible reduction in the volume of the right cheek. New onset of ipsilateral jaw locking and pain associated with spasms of the muscles of mastication suggested the diagnosis of Parry-Romberg syndrome.
Neurofibromatosis type I is a neuroectodermal abnormality first described by Friedrich von Recklinghausen in 1882 with an incidence of 1 in 3000 births. Neurofibromatosis type I gene mutations lead to increased Ras activity, which is implicated in many NF1-related conditions such as neurofibromas and schwannomas.1 Autoimmune conditions including systemic lupus erythematosus (SLE) and mixed connective tissue rarely have been reported in NF1, but the mechanism of their association is not clear.2 Based on a review of 5 reported cases of NF1 and SLE, most patients were female, and the predominant features of NF1 were café au lait macules and neurofibromas.3-6 One case documented a family history of NF1,6 suggesting predominance of sporadic mutations in these cases. Interestingly, in 3 cases the diagnosis of SLE preceded the diagnosis of NF1, prompting the authors to suggest a viral trigger for the development of NF1 lesions.3,4 Linear scleroderma is immunologically mediated and is characterized by the onset of smooth indurated cutaneous plaques. According to a PubMed search of articles indexed for MEDLINE using the search terms morphea and neurofibromatosis as well as linear scleroderma and neurofibromatosis, there have been no reports of linear scleroderma or morphea associated with NF1. Cichowski et al7 demonstrated enhanced activation of Ras and prolonged activities of both Ras and extracellular signal-regulated kinase (ERK) signaling pathways in NF1-deficient mice. Chen et al8 showed that heparin sulfate–dependent ERK activation contributes to the development of scleroderma by promoting the expression of profibrotic proteins in scleroderma fibroblasts. It was previously noted that increased Ras/ERK signaling activities were important in connective tissue growth factor expression in normal mesenchymal cells.9
Based on these findings, we speculated that hyperactivation of Ras/ERK signaling from NF1 mutations could lead to the promotion of fibrosis seen in scleroderma. The lack of similar reports, however, suggests that the presence of both conditions in this case is coincidental. However, the growing number of reports on autoimmune and connective tissue disorders in NF1 reflects the need for further research in this area.
1. Harrisingh MC, Lloyd AC. Ras/Raf/ERK signalling and NF1. Cell Cycle. 2004;3:1255-1258.
2. Migita K, Kawabe Y, Mori M, et al. Mixed connective tissue disease associated with von Recklinghausen’s neurofibromatosis. Intern Med. 2001;40:363-364.
3. Bitnun S, Bassan H. Letter: neurofibromatosis and SLE. N Engl J Med. 1975;292:429-430.
4. Riccardi VM. Neurofibromatosis in a patient with systemic lupus erythematosus. Arthritis Rheum. 1983;26:574.
5. Corominas H, Guardiola JM, Matas L, et al. Neurofibromatosis and systemic lupus erythematosus. A matter of coincidence? Clin Rheum. 2003;22:496-497.
6. Akyüz SG, Çatlik A, Bülbül M, et al. An unusual pediatric case with neurofibromatosis and systemic lupus erythematosus. Rheum Int. 2012;32:2345-2347.
7. Cichowski K, Santiago S, Jardim M, et al. Dynamic regulation of the Ras pathway via proteolysis of the NF1 tumour suppressor. Genes Dev. 2003;17:449-454.
8. Chen Y, Leask A, Abraham DJ, et al. Heparan sulfate-dependent ERK activation contributes to the overexpression of fibrotic proteins and enhanced contraction by scleroderma fibroblasts. Arthritis Rheum. 2008;58:577-585.
9. Chen Y, Shi-wen X, van Beek J, et al. Matrix contraction by dermal fibroblasts requiring transforming growth factor-â/activin-linked kinase 5, heparin sulphate-containing proteoglycans and MEK/ERK: insights into pathological scarring in chronic fibrotic disease. Am J Pathol. 2005;167:1699-1711.
1. Harrisingh MC, Lloyd AC. Ras/Raf/ERK signalling and NF1. Cell Cycle. 2004;3:1255-1258.
2. Migita K, Kawabe Y, Mori M, et al. Mixed connective tissue disease associated with von Recklinghausen’s neurofibromatosis. Intern Med. 2001;40:363-364.
3. Bitnun S, Bassan H. Letter: neurofibromatosis and SLE. N Engl J Med. 1975;292:429-430.
4. Riccardi VM. Neurofibromatosis in a patient with systemic lupus erythematosus. Arthritis Rheum. 1983;26:574.
5. Corominas H, Guardiola JM, Matas L, et al. Neurofibromatosis and systemic lupus erythematosus. A matter of coincidence? Clin Rheum. 2003;22:496-497.
6. Akyüz SG, Çatlik A, Bülbül M, et al. An unusual pediatric case with neurofibromatosis and systemic lupus erythematosus. Rheum Int. 2012;32:2345-2347.
7. Cichowski K, Santiago S, Jardim M, et al. Dynamic regulation of the Ras pathway via proteolysis of the NF1 tumour suppressor. Genes Dev. 2003;17:449-454.
8. Chen Y, Leask A, Abraham DJ, et al. Heparan sulfate-dependent ERK activation contributes to the overexpression of fibrotic proteins and enhanced contraction by scleroderma fibroblasts. Arthritis Rheum. 2008;58:577-585.
9. Chen Y, Shi-wen X, van Beek J, et al. Matrix contraction by dermal fibroblasts requiring transforming growth factor-â/activin-linked kinase 5, heparin sulphate-containing proteoglycans and MEK/ERK: insights into pathological scarring in chronic fibrotic disease. Am J Pathol. 2005;167:1699-1711.
Stopping intergenerational cycles of trauma
Understanding the root of stress and trauma experienced by patients as individuals is a key charge of psychiatry. But patients also can experience psychological distress because of the traumatic experiences of previous generations. The mental health community must be prepared to treat patients who fall into the latter category.
My colleagues and I recently analyzed representative samples of First Nations adults and youth living on a reserve in Canada. We found that the number of previous generations in which families were forced to attend Indian residential schools was cumulatively linked with higher levels of psychological distress and suicidal ideation among those who did not attend themselves (Transcultural Psychiatry. 2014, 51:320-338).
The forced removal of Indigenous children from their homes for the purposes of assimilation occurred over generations in many countries around the world. In Canada, these government-mandated church-run residential schools ran from the mid-1800s until 1996, and resulted in generations of indigenous children being exposed to chronic neglect, abuse, trauma, racism, and cultural shaming (“Honouring the Truth: Reconciling for the Future,” Truth and Reconciliation Commission of Canada, 2015).

Some of our more recent analyses revealed that having a family history of residential school attendance has been linked to a greater likelihood of early-onset mental health symptoms, which in turn, has been tied to an increased risk of suicidality and other unique outcomes that have implications for treatment and prevention. Furthermore, other negative outcomes experienced by indigenous peoples, such as low family income and limited educational opportunities, also appear to be involved in the intergenerational transmission of residential school trauma.
Against this backdrop in Canada, we have a prevailing sense of blaming the victim that is counterproductive to healing. According to a 2014 report by the Royal Canadian Mounted Police (RCMP), 1,017 indigenous women and girls were murdered between 1980 and 2012 across Canada. About one-third of the women died as a result of physical beating, and more than 90% of the victims had a “previous relationship” with the person who killed them, according to the RCMP report. Yet, despite evidence to the contrary, the prime minister of Canada shared his view last year that those staggering numbers should be viewed as a criminal issue and not a “sociological phenomenon” (CBC News, Aug. 21, 2014).
Members of other groups whose previous generations also were exposed indirectly to unspeakable trauma include Americans of African descent, Native Americans, and adult offspring of Holocaust survivors. Rachel Yehuda, Ph.D., has studied the latter group extensively.
In one study, Dr. Yehuda and her associates found significantly reduced cortisol excretion in Holocaust offspring, compared with controls (Psychoneuroendocrinology. 2014 Oct;48:1-10). In another study of the adult offspring of Holocaust survivors, Dr. Yehuda, professor of psychiatry at the Icahn School of Medicine at Mount Sinai in New York, found that “offspring with paternal [posttraumatic stress disorder] showed higher GR-1F promoter methylation, whereas offspring with both maternal and paternal PTSD showed lower methylation.” Furthermore, lower methylation was tied to greater suppression of cortisol (Am J Psychiatry. 2014 Aug;171(8)872-880).
An appreciation for the power of epigenetics, or “soul wounds,” can help us stop blaming indigenous and other traumatized populations, and start tending to their mental health needs. Furthermore, getting individuals to understand these concepts can be yet another step on the path toward healing.
A study of 19 staff members and clients in a Native American healing lodge who began a discourse on the legacy of historical trauma found that counselors understood that their clients carried pain “leading to adult dysfunction, including substance abuse” (J Consult Clin Psychol. 2009 Aug;77(4):751-762). Second, the counselors believed that the pain needed to be confessed. Third, the counselors thought that expressing the pain would help the clients become more introspective. Finally, the process included reclaiming “indigenous heritage, identity, and spirituality that program staff thought would neutralize the pathogenic effects of colonization.”
We do not know the extent to which epigenetics are involved in specific diseases. Epigenetic mechanisms have not yet been tested as pathways involved in the intergenerational transmission of trauma in indigenous peoples. But Dr. Yehuda’s work with the children of Holocaust survivors shows that these mechanisms do indeed exist.
In light of what we do understand and the DSM-5’s emphasis on cultural competency, mental health professionals have a role to play in reversing the negative intergenerational cycles experienced by people across the globe. Only when we start paying attention to the plight of indigenous peoples across the globe will true healing begin.
Dr. Bombay is assistant professor of psychiatry at Dalhousie University, Halifax, N.S.
Understanding the root of stress and trauma experienced by patients as individuals is a key charge of psychiatry. But patients also can experience psychological distress because of the traumatic experiences of previous generations. The mental health community must be prepared to treat patients who fall into the latter category.
My colleagues and I recently analyzed representative samples of First Nations adults and youth living on a reserve in Canada. We found that the number of previous generations in which families were forced to attend Indian residential schools was cumulatively linked with higher levels of psychological distress and suicidal ideation among those who did not attend themselves (Transcultural Psychiatry. 2014, 51:320-338).
The forced removal of Indigenous children from their homes for the purposes of assimilation occurred over generations in many countries around the world. In Canada, these government-mandated church-run residential schools ran from the mid-1800s until 1996, and resulted in generations of indigenous children being exposed to chronic neglect, abuse, trauma, racism, and cultural shaming (“Honouring the Truth: Reconciling for the Future,” Truth and Reconciliation Commission of Canada, 2015).
Some of our more recent analyses revealed that having a family history of residential school attendance has been linked to a greater likelihood of early-onset mental health symptoms, which in turn, has been tied to an increased risk of suicidality and other unique outcomes that have implications for treatment and prevention. Furthermore, other negative outcomes experienced by indigenous peoples, such as low family income and limited educational opportunities, also appear to be involved in the intergenerational transmission of residential school trauma.
Against this backdrop in Canada, we have a prevailing sense of blaming the victim that is counterproductive to healing. According to a 2014 report by the Royal Canadian Mounted Police (RCMP), 1,017 indigenous women and girls were murdered between 1980 and 2012 across Canada. About one-third of the women died as a result of physical beating, and more than 90% of the victims had a “previous relationship” with the person who killed them, according to the RCMP report. Yet, despite evidence to the contrary, the prime minister of Canada shared his view last year that those staggering numbers should be viewed as a criminal issue and not a “sociological phenomenon” (CBC News, Aug. 21, 2014).
Members of other groups whose previous generations also were exposed indirectly to unspeakable trauma include Americans of African descent, Native Americans, and adult offspring of Holocaust survivors. Rachel Yehuda, Ph.D., has studied the latter group extensively.
In one study, Dr. Yehuda and her associates found significantly reduced cortisol excretion in Holocaust offspring, compared with controls (Psychoneuroendocrinology. 2014 Oct;48:1-10). In another study of the adult offspring of Holocaust survivors, Dr. Yehuda, professor of psychiatry at the Icahn School of Medicine at Mount Sinai in New York, found that “offspring with paternal [posttraumatic stress disorder] showed higher GR-1F promoter methylation, whereas offspring with both maternal and paternal PTSD showed lower methylation.” Furthermore, lower methylation was tied to greater suppression of cortisol (Am J Psychiatry. 2014 Aug;171(8)872-880).
An appreciation for the power of epigenetics, or “soul wounds,” can help us stop blaming indigenous and other traumatized populations, and start tending to their mental health needs. Furthermore, getting individuals to understand these concepts can be yet another step on the path toward healing.
A study of 19 staff members and clients in a Native American healing lodge who began a discourse on the legacy of historical trauma found that counselors understood that their clients carried pain “leading to adult dysfunction, including substance abuse” (J Consult Clin Psychol. 2009 Aug;77(4):751-762). Second, the counselors believed that the pain needed to be confessed. Third, the counselors thought that expressing the pain would help the clients become more introspective. Finally, the process included reclaiming “indigenous heritage, identity, and spirituality that program staff thought would neutralize the pathogenic effects of colonization.”
We do not know the extent to which epigenetics are involved in specific diseases. Epigenetic mechanisms have not yet been tested as pathways involved in the intergenerational transmission of trauma in indigenous peoples. But Dr. Yehuda’s work with the children of Holocaust survivors shows that these mechanisms do indeed exist.
In light of what we do understand and the DSM-5’s emphasis on cultural competency, mental health professionals have a role to play in reversing the negative intergenerational cycles experienced by people across the globe. Only when we start paying attention to the plight of indigenous peoples across the globe will true healing begin.
Dr. Bombay is assistant professor of psychiatry at Dalhousie University, Halifax, N.S.
Understanding the root of stress and trauma experienced by patients as individuals is a key charge of psychiatry. But patients also can experience psychological distress because of the traumatic experiences of previous generations. The mental health community must be prepared to treat patients who fall into the latter category.
My colleagues and I recently analyzed representative samples of First Nations adults and youth living on a reserve in Canada. We found that the number of previous generations in which families were forced to attend Indian residential schools was cumulatively linked with higher levels of psychological distress and suicidal ideation among those who did not attend themselves (Transcultural Psychiatry. 2014, 51:320-338).
The forced removal of Indigenous children from their homes for the purposes of assimilation occurred over generations in many countries around the world. In Canada, these government-mandated church-run residential schools ran from the mid-1800s until 1996, and resulted in generations of indigenous children being exposed to chronic neglect, abuse, trauma, racism, and cultural shaming (“Honouring the Truth: Reconciling for the Future,” Truth and Reconciliation Commission of Canada, 2015).
Some of our more recent analyses revealed that having a family history of residential school attendance has been linked to a greater likelihood of early-onset mental health symptoms, which in turn, has been tied to an increased risk of suicidality and other unique outcomes that have implications for treatment and prevention. Furthermore, other negative outcomes experienced by indigenous peoples, such as low family income and limited educational opportunities, also appear to be involved in the intergenerational transmission of residential school trauma.
Against this backdrop in Canada, we have a prevailing sense of blaming the victim that is counterproductive to healing. According to a 2014 report by the Royal Canadian Mounted Police (RCMP), 1,017 indigenous women and girls were murdered between 1980 and 2012 across Canada. About one-third of the women died as a result of physical beating, and more than 90% of the victims had a “previous relationship” with the person who killed them, according to the RCMP report. Yet, despite evidence to the contrary, the prime minister of Canada shared his view last year that those staggering numbers should be viewed as a criminal issue and not a “sociological phenomenon” (CBC News, Aug. 21, 2014).
Members of other groups whose previous generations also were exposed indirectly to unspeakable trauma include Americans of African descent, Native Americans, and adult offspring of Holocaust survivors. Rachel Yehuda, Ph.D., has studied the latter group extensively.
In one study, Dr. Yehuda and her associates found significantly reduced cortisol excretion in Holocaust offspring, compared with controls (Psychoneuroendocrinology. 2014 Oct;48:1-10). In another study of the adult offspring of Holocaust survivors, Dr. Yehuda, professor of psychiatry at the Icahn School of Medicine at Mount Sinai in New York, found that “offspring with paternal [posttraumatic stress disorder] showed higher GR-1F promoter methylation, whereas offspring with both maternal and paternal PTSD showed lower methylation.” Furthermore, lower methylation was tied to greater suppression of cortisol (Am J Psychiatry. 2014 Aug;171(8)872-880).
An appreciation for the power of epigenetics, or “soul wounds,” can help us stop blaming indigenous and other traumatized populations, and start tending to their mental health needs. Furthermore, getting individuals to understand these concepts can be yet another step on the path toward healing.
A study of 19 staff members and clients in a Native American healing lodge who began a discourse on the legacy of historical trauma found that counselors understood that their clients carried pain “leading to adult dysfunction, including substance abuse” (J Consult Clin Psychol. 2009 Aug;77(4):751-762). Second, the counselors believed that the pain needed to be confessed. Third, the counselors thought that expressing the pain would help the clients become more introspective. Finally, the process included reclaiming “indigenous heritage, identity, and spirituality that program staff thought would neutralize the pathogenic effects of colonization.”
We do not know the extent to which epigenetics are involved in specific diseases. Epigenetic mechanisms have not yet been tested as pathways involved in the intergenerational transmission of trauma in indigenous peoples. But Dr. Yehuda’s work with the children of Holocaust survivors shows that these mechanisms do indeed exist.
In light of what we do understand and the DSM-5’s emphasis on cultural competency, mental health professionals have a role to play in reversing the negative intergenerational cycles experienced by people across the globe. Only when we start paying attention to the plight of indigenous peoples across the globe will true healing begin.
Dr. Bombay is assistant professor of psychiatry at Dalhousie University, Halifax, N.S.
