User login
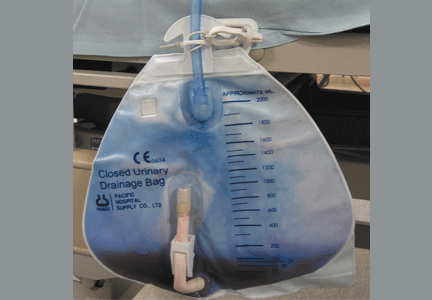
Purple urine in a woman with chronic kidney disease
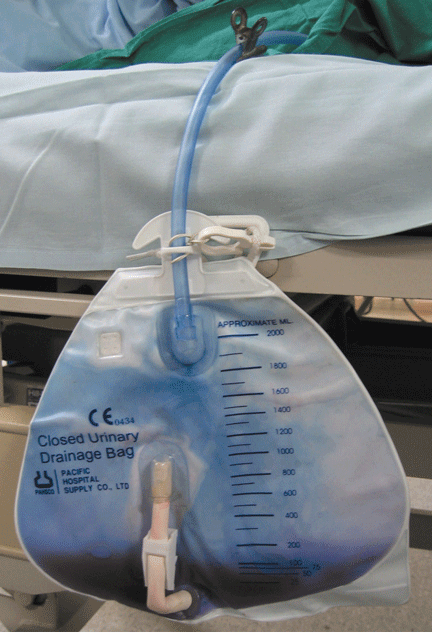
On examination, her blood pressure is 138/70 mm Hg, respiratory rate 18 breaths/minute, and heart rate 80 beats/minute. Her abdomen is soft. She has never undergone abdominal surgery. She is not taking any medications that may have caused urine discoloration.
Plain radiography of the abdomen reveals no abnormal gas. Laboratory test results are as follows:
- White blood cell count 15.1 × 109/L (reference range 4–11), with 85% neutrophils (reference range 39.5%–74%)
- C-reactive protein 6.27 mg/dL (reference range 0.0–1.0)
- Blood urea nitrogen 54 mg/dL (reference range 8–25)
- Serum creatinine 2.5 mg/dL (0.70–1.40)
- Estimated glomerular filtration rate 20 mL/min/1.73 m2 (< 60 is sufficient for the diagnosis of chronic kidney disease)
- Liver function tests are normal
- Urine pH 8.0 (4.80–7.80); urine is positive for nitrates and for marked pyuria and bacteriuria.
Urine culture yields more than 100,000 colony-forming units of Pseudomonas aeruginosa, Morganella morganii, and Proteus vulgaris. These results and the patient’s presentation point to a diagnosis of purple urine bag syndrome. After placement of a new urinary catheter and 7 days of intravenous ciprofloxacin (Cipro) 250 mg every 12 hours, the color of her urine returns to normal.
PURPLE URINE BAG SYNDROME
Purple urine bag syndrome is rare, and catheter-associated urinary tract infection is the main cause.1 However, it has also been associated with intestinal intussusception.2 In our patient, the examination and radiography ruled out intussusception.
Factors reported to be involved in the development of this syndrome include older age, female sex, chronic constipation, chronic urinary catheterization, alkaline (common) or acidic (uncommon) urine, and a higher bacterial load in the urine.3,4
The pathogenesis of purple-colored urine4,5 is thought to start with the metabolism of dietary tryptophan by intestinal bacteria to indole. Indole is then absorbed into the portal circulation and is converted to indoxyl sulfate, which is excreted into the urine. In vitro experiments4,5 have shown that certain bacteria in the urine produce indoxyl sulfatase and indoxyl phosphatase, which break down indoxyl sulfate to indoxyl. Indoxyl can then be converted to indigo or indirubin in alkaline5 or acidic4 urine. When blue indigo and red indirubin mix together, the result is purple.4,5
Bacteria that possess indoxyl sulfatase or indoxyl phosphatase include P aeruginosa, M morganii, P vulgaris, Escherichia coli, and Providencia species.5,6 However, not all bacteria of the same species produce the enzymes required for the formation of purple urine.5 This may explain the rarity of this syndrome despite the common occurrence of urinary tract infection in patients with risk factors for purple urine bag syndrome.
CHRONIC KIDNEY DISEASE: A POTENTIAL RISK FACTOR
Chronic kidney disease was shown to be a risk factor for purple urine bag syndrome in a small cohort study of Taiwanese patients.7 The serum and urine levels of indoxyl sulfate increased markedly in patients who had chronic kidney disease or who were undergoing dialysis because of impaired renal clearance.6 Furthermore, indoxyl sulfate, which plays an important role in this syndrome, is also cytotoxic and may increase the rate of renal failure in uremic rats.4
Although purple urine itself is usually considered benign,3 it should prompt an evaluation for urinary tract infection, especially in patients with kidney disease. Failure to treat the underlying infection can lead to septicemia or Fourier gangrene.1,3
- Tasi YM, Huang MS, Yang CJ, Yeh SM, Liu CC. Purple urine bag syndrome, not always a benign process. Am J Emerg Med 2009; 27:895–897.
- Pillai RN, Clavijo J, Narayanan M, Zaman K. An association of purple urine bag syndrome with intussusception. Urology 2007; 70:812.e1–812.e2.
- Pillai BP, Chong VH, Yong AM. Purple urine bag syndrome. Singapore Med J 2009; 50:e193–e194.
- Bar-Or D, Rael LT, Bar-Or R, Craun ML, Statz J, Garrett RE. Mass spectrometry analysis of urine and catheter of a patient with purple urinary bag syndrome. Clin Chim Acta 2007; 378:216–218.
- Dealler SF, Hawkey PM, Millar MR. Enzymatic degradation of urinary indoxyl sulfate by Providencia stuartii and Klebsiella pneumoniae causes the purple urine bag syndrome. J Clin Microbiol 1988; 26:2152–2156.
- Wang IK, Ho DR, Chang HY, Lin CL, Chuang FR. Purple urine bag syndrome in a hemodialysis patient. Intern Med 2005; 44:859–861.
- Yang CJ, Lu PL, Chen TC, et al. Chronic kidney disease is a potential risk factor for the development of purple urine bag syndrome. J Am Geriatr Soc 2009; 57:1937–1938.
On examination, her blood pressure is 138/70 mm Hg, respiratory rate 18 breaths/minute, and heart rate 80 beats/minute. Her abdomen is soft. She has never undergone abdominal surgery. She is not taking any medications that may have caused urine discoloration.
Plain radiography of the abdomen reveals no abnormal gas. Laboratory test results are as follows:
- White blood cell count 15.1 × 109/L (reference range 4–11), with 85% neutrophils (reference range 39.5%–74%)
- C-reactive protein 6.27 mg/dL (reference range 0.0–1.0)
- Blood urea nitrogen 54 mg/dL (reference range 8–25)
- Serum creatinine 2.5 mg/dL (0.70–1.40)
- Estimated glomerular filtration rate 20 mL/min/1.73 m2 (< 60 is sufficient for the diagnosis of chronic kidney disease)
- Liver function tests are normal
- Urine pH 8.0 (4.80–7.80); urine is positive for nitrates and for marked pyuria and bacteriuria.
Urine culture yields more than 100,000 colony-forming units of Pseudomonas aeruginosa, Morganella morganii, and Proteus vulgaris. These results and the patient’s presentation point to a diagnosis of purple urine bag syndrome. After placement of a new urinary catheter and 7 days of intravenous ciprofloxacin (Cipro) 250 mg every 12 hours, the color of her urine returns to normal.
PURPLE URINE BAG SYNDROME
Purple urine bag syndrome is rare, and catheter-associated urinary tract infection is the main cause.1 However, it has also been associated with intestinal intussusception.2 In our patient, the examination and radiography ruled out intussusception.
Factors reported to be involved in the development of this syndrome include older age, female sex, chronic constipation, chronic urinary catheterization, alkaline (common) or acidic (uncommon) urine, and a higher bacterial load in the urine.3,4
The pathogenesis of purple-colored urine4,5 is thought to start with the metabolism of dietary tryptophan by intestinal bacteria to indole. Indole is then absorbed into the portal circulation and is converted to indoxyl sulfate, which is excreted into the urine. In vitro experiments4,5 have shown that certain bacteria in the urine produce indoxyl sulfatase and indoxyl phosphatase, which break down indoxyl sulfate to indoxyl. Indoxyl can then be converted to indigo or indirubin in alkaline5 or acidic4 urine. When blue indigo and red indirubin mix together, the result is purple.4,5
Bacteria that possess indoxyl sulfatase or indoxyl phosphatase include P aeruginosa, M morganii, P vulgaris, Escherichia coli, and Providencia species.5,6 However, not all bacteria of the same species produce the enzymes required for the formation of purple urine.5 This may explain the rarity of this syndrome despite the common occurrence of urinary tract infection in patients with risk factors for purple urine bag syndrome.
CHRONIC KIDNEY DISEASE: A POTENTIAL RISK FACTOR
Chronic kidney disease was shown to be a risk factor for purple urine bag syndrome in a small cohort study of Taiwanese patients.7 The serum and urine levels of indoxyl sulfate increased markedly in patients who had chronic kidney disease or who were undergoing dialysis because of impaired renal clearance.6 Furthermore, indoxyl sulfate, which plays an important role in this syndrome, is also cytotoxic and may increase the rate of renal failure in uremic rats.4
Although purple urine itself is usually considered benign,3 it should prompt an evaluation for urinary tract infection, especially in patients with kidney disease. Failure to treat the underlying infection can lead to septicemia or Fourier gangrene.1,3
On examination, her blood pressure is 138/70 mm Hg, respiratory rate 18 breaths/minute, and heart rate 80 beats/minute. Her abdomen is soft. She has never undergone abdominal surgery. She is not taking any medications that may have caused urine discoloration.
Plain radiography of the abdomen reveals no abnormal gas. Laboratory test results are as follows:
- White blood cell count 15.1 × 109/L (reference range 4–11), with 85% neutrophils (reference range 39.5%–74%)
- C-reactive protein 6.27 mg/dL (reference range 0.0–1.0)
- Blood urea nitrogen 54 mg/dL (reference range 8–25)
- Serum creatinine 2.5 mg/dL (0.70–1.40)
- Estimated glomerular filtration rate 20 mL/min/1.73 m2 (< 60 is sufficient for the diagnosis of chronic kidney disease)
- Liver function tests are normal
- Urine pH 8.0 (4.80–7.80); urine is positive for nitrates and for marked pyuria and bacteriuria.
Urine culture yields more than 100,000 colony-forming units of Pseudomonas aeruginosa, Morganella morganii, and Proteus vulgaris. These results and the patient’s presentation point to a diagnosis of purple urine bag syndrome. After placement of a new urinary catheter and 7 days of intravenous ciprofloxacin (Cipro) 250 mg every 12 hours, the color of her urine returns to normal.
PURPLE URINE BAG SYNDROME
Purple urine bag syndrome is rare, and catheter-associated urinary tract infection is the main cause.1 However, it has also been associated with intestinal intussusception.2 In our patient, the examination and radiography ruled out intussusception.
Factors reported to be involved in the development of this syndrome include older age, female sex, chronic constipation, chronic urinary catheterization, alkaline (common) or acidic (uncommon) urine, and a higher bacterial load in the urine.3,4
The pathogenesis of purple-colored urine4,5 is thought to start with the metabolism of dietary tryptophan by intestinal bacteria to indole. Indole is then absorbed into the portal circulation and is converted to indoxyl sulfate, which is excreted into the urine. In vitro experiments4,5 have shown that certain bacteria in the urine produce indoxyl sulfatase and indoxyl phosphatase, which break down indoxyl sulfate to indoxyl. Indoxyl can then be converted to indigo or indirubin in alkaline5 or acidic4 urine. When blue indigo and red indirubin mix together, the result is purple.4,5
Bacteria that possess indoxyl sulfatase or indoxyl phosphatase include P aeruginosa, M morganii, P vulgaris, Escherichia coli, and Providencia species.5,6 However, not all bacteria of the same species produce the enzymes required for the formation of purple urine.5 This may explain the rarity of this syndrome despite the common occurrence of urinary tract infection in patients with risk factors for purple urine bag syndrome.
CHRONIC KIDNEY DISEASE: A POTENTIAL RISK FACTOR
Chronic kidney disease was shown to be a risk factor for purple urine bag syndrome in a small cohort study of Taiwanese patients.7 The serum and urine levels of indoxyl sulfate increased markedly in patients who had chronic kidney disease or who were undergoing dialysis because of impaired renal clearance.6 Furthermore, indoxyl sulfate, which plays an important role in this syndrome, is also cytotoxic and may increase the rate of renal failure in uremic rats.4
Although purple urine itself is usually considered benign,3 it should prompt an evaluation for urinary tract infection, especially in patients with kidney disease. Failure to treat the underlying infection can lead to septicemia or Fourier gangrene.1,3
- Tasi YM, Huang MS, Yang CJ, Yeh SM, Liu CC. Purple urine bag syndrome, not always a benign process. Am J Emerg Med 2009; 27:895–897.
- Pillai RN, Clavijo J, Narayanan M, Zaman K. An association of purple urine bag syndrome with intussusception. Urology 2007; 70:812.e1–812.e2.
- Pillai BP, Chong VH, Yong AM. Purple urine bag syndrome. Singapore Med J 2009; 50:e193–e194.
- Bar-Or D, Rael LT, Bar-Or R, Craun ML, Statz J, Garrett RE. Mass spectrometry analysis of urine and catheter of a patient with purple urinary bag syndrome. Clin Chim Acta 2007; 378:216–218.
- Dealler SF, Hawkey PM, Millar MR. Enzymatic degradation of urinary indoxyl sulfate by Providencia stuartii and Klebsiella pneumoniae causes the purple urine bag syndrome. J Clin Microbiol 1988; 26:2152–2156.
- Wang IK, Ho DR, Chang HY, Lin CL, Chuang FR. Purple urine bag syndrome in a hemodialysis patient. Intern Med 2005; 44:859–861.
- Yang CJ, Lu PL, Chen TC, et al. Chronic kidney disease is a potential risk factor for the development of purple urine bag syndrome. J Am Geriatr Soc 2009; 57:1937–1938.
- Tasi YM, Huang MS, Yang CJ, Yeh SM, Liu CC. Purple urine bag syndrome, not always a benign process. Am J Emerg Med 2009; 27:895–897.
- Pillai RN, Clavijo J, Narayanan M, Zaman K. An association of purple urine bag syndrome with intussusception. Urology 2007; 70:812.e1–812.e2.
- Pillai BP, Chong VH, Yong AM. Purple urine bag syndrome. Singapore Med J 2009; 50:e193–e194.
- Bar-Or D, Rael LT, Bar-Or R, Craun ML, Statz J, Garrett RE. Mass spectrometry analysis of urine and catheter of a patient with purple urinary bag syndrome. Clin Chim Acta 2007; 378:216–218.
- Dealler SF, Hawkey PM, Millar MR. Enzymatic degradation of urinary indoxyl sulfate by Providencia stuartii and Klebsiella pneumoniae causes the purple urine bag syndrome. J Clin Microbiol 1988; 26:2152–2156.
- Wang IK, Ho DR, Chang HY, Lin CL, Chuang FR. Purple urine bag syndrome in a hemodialysis patient. Intern Med 2005; 44:859–861.
- Yang CJ, Lu PL, Chen TC, et al. Chronic kidney disease is a potential risk factor for the development of purple urine bag syndrome. J Am Geriatr Soc 2009; 57:1937–1938.
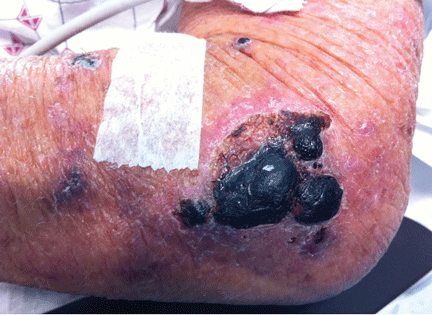
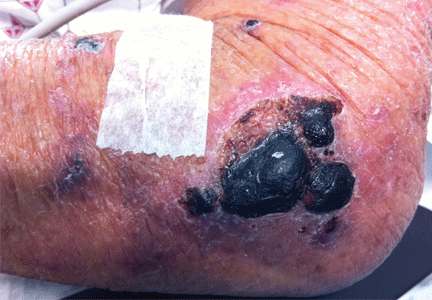
Purpuric lesion on the elbow
A 75-year-old man was admitted to the hospital with new-onset atrial fibrillation. He underwent rate control, and a heparin infusion was started. Warfarin (Coumadin) 10 mg was added on the second hospital day. Two days later, the heparin infusion was discontinued when the international normalized ratio (INR) was in the therapeutic range.
Q: Which is the most likely diagnosis?
- Pyoderma gangrenosum
- Cutaneous vasculitis
- Warfarin-induced skin necrosis
- Ecthyma gangrenosum
- Dermatitis herpetiformis
A: The most likely diagnosis is warfarin-induced skin necrosis, a rare paradoxical complication that occurs in 0.01% to 0.1% of patients receiving this drug.1 Microthrombosis leads to necrosis of the skin and subcutaneous tissues, arising within 2 to 10 days after the start of anticoagulation therapy, although in rare cases it can occur months to years later.2,3
The most common risk factors include the unopposed use of warfarin (ie, unopposed by heparin at the start of therapy), using higher doses of warfarin during the initiation of anticoagulation, and inadequate overlap with an effective parenteral anticoagulant. Patients with protein C or S deficiency, heparin-induced thrombocytopenia,4 resistance to activated protein C, antithrombin deficiency, and lupus anticoagulant have also been reported to be at risk.
The most common sites affected are areas with high subcutaneous fat content, such as the abdomen, thighs, breasts, and buttocks. Skin presentations can vary from dermal plaques to petechial lesions, which rapidly progress to well-demarcated, bluish-black, painful lesions and eventually to hemorrhagic bullae and necrosis.1
At the start of warfarin therapy, the levels of protein C and factor VII (with half-lives of 5 to 8 hours) fall faster than those of other vitamin-K-dependent factors (ie, factors II, IX, and X). This causes a transient imbalance in procoagulant and anticoagulant pathways favoring thrombosis of the microvasculature, with resulting necrosis. Patients with hereditary protein C deficiency are at higher risk.5
Histologic review of lesions often shows venous thrombosis and diffuse necrosis of the dermis and subcutaneous tissue.2
Promptly stopping the warfarin and choosing alternative anticoagulation may help prevent further progression of this condition. Wound care, debridement, and sometimes skin grafting may be necessary, depending on the extent of the lesions. A rechallenge with warfarin is often difficult, but cases have been reported in which treatment was resumed without adverse consequences.6 Avoiding large loading doses of warfarin, gradually increasing doses over an extended period (about 10 days),3 and starting warfarin with a heparin bridge for at least 5 days (which was not done in this patient) would prevent the condition.
Early recognition, differentiation, and diagnosis are essential to minimize morbidity and to prevent death.
CASE CONTINUED
Warfarin was discontinued once the patient developed the skin lesions. He received vitamin K and fresh frozen plasma to normalize his INR, and he was started on a heparin infusion, after which the lesions began to heal. The patient refused a skin biopsy. Platelet counts remained stable during his hospital course. Protein C levels were not checked, given his recent use of warfarin. He was started on dabigatran (Pradaxa) and was discharged a week later.
THE OTHER DIAGNOSTIC CHOICES
Pyoderma gangrenosum is an uncommon ulcerative skin condition often associated with autoimmune disease. It usually starts at the site of a minor injury, more commonly on the legs, and gradually progresses to a painful ulcer.
Cutaneous vasculitis is an inflammation of small blood vessels characterized by palpable purpura. The lesions can resemble urticaria, petechia, or erythema multiforme. It is commonly associated with infection, drug therapy, inflammatory disease, and malignancy.
Ecthyma gangrenosum is an infection of skin caused by Pseudomonas aeruginosa. Usually, it presents as hemorrhagic pustules or infarct-like areas with surrounding erythema that evolve into necrotic ulcers surrounded by erythema.
Dermatitis herpetiformis is a chronic skin condition, presenting with fluid-filled blisters and commonly involving the neck, back, scalp, and elbows. This condition is associated with celiac disease, and the lesions are extremely pruritic.
- Nazarian RM, Van Cott EM, Zembowicz A, Duncan LM. Warfarin-induced skin necrosis. J Am Acad Dermatol 2009; 61:325–332.
- Ward CT, Chavalitanonda N. Atypical warfarin-induced skin necrosis. Pharmacotherapy 2006; 26:1175–1179.
- Chan YC, Valenti D, Mansfield AO, Stansby G. Warfarin induced skin necrosis. Br J Surg 2000; 87:266–272.
- Warkentin TE, Sikov WM, Lillicrap DP. Multicentric warfarin-induced skin necrosis complicating heparin-induced thrombocytopenia. Am J Hematol 1999; 62:44–48.
- Ad-El DD, Meirovitz A, Weinberg A, et al. Warfarin skin necrosis: local and systemic factors. Br J Plast Surg 2000; 53:624–626.
- Jillella AP, Lutcher CL. Reinstituting warfarin in patients who develop warfarin skin necrosis. Am J Hematol 1996; 52:117–119.
A 75-year-old man was admitted to the hospital with new-onset atrial fibrillation. He underwent rate control, and a heparin infusion was started. Warfarin (Coumadin) 10 mg was added on the second hospital day. Two days later, the heparin infusion was discontinued when the international normalized ratio (INR) was in the therapeutic range.
Q: Which is the most likely diagnosis?
- Pyoderma gangrenosum
- Cutaneous vasculitis
- Warfarin-induced skin necrosis
- Ecthyma gangrenosum
- Dermatitis herpetiformis
A: The most likely diagnosis is warfarin-induced skin necrosis, a rare paradoxical complication that occurs in 0.01% to 0.1% of patients receiving this drug.1 Microthrombosis leads to necrosis of the skin and subcutaneous tissues, arising within 2 to 10 days after the start of anticoagulation therapy, although in rare cases it can occur months to years later.2,3
The most common risk factors include the unopposed use of warfarin (ie, unopposed by heparin at the start of therapy), using higher doses of warfarin during the initiation of anticoagulation, and inadequate overlap with an effective parenteral anticoagulant. Patients with protein C or S deficiency, heparin-induced thrombocytopenia,4 resistance to activated protein C, antithrombin deficiency, and lupus anticoagulant have also been reported to be at risk.
The most common sites affected are areas with high subcutaneous fat content, such as the abdomen, thighs, breasts, and buttocks. Skin presentations can vary from dermal plaques to petechial lesions, which rapidly progress to well-demarcated, bluish-black, painful lesions and eventually to hemorrhagic bullae and necrosis.1
At the start of warfarin therapy, the levels of protein C and factor VII (with half-lives of 5 to 8 hours) fall faster than those of other vitamin-K-dependent factors (ie, factors II, IX, and X). This causes a transient imbalance in procoagulant and anticoagulant pathways favoring thrombosis of the microvasculature, with resulting necrosis. Patients with hereditary protein C deficiency are at higher risk.5
Histologic review of lesions often shows venous thrombosis and diffuse necrosis of the dermis and subcutaneous tissue.2
Promptly stopping the warfarin and choosing alternative anticoagulation may help prevent further progression of this condition. Wound care, debridement, and sometimes skin grafting may be necessary, depending on the extent of the lesions. A rechallenge with warfarin is often difficult, but cases have been reported in which treatment was resumed without adverse consequences.6 Avoiding large loading doses of warfarin, gradually increasing doses over an extended period (about 10 days),3 and starting warfarin with a heparin bridge for at least 5 days (which was not done in this patient) would prevent the condition.
Early recognition, differentiation, and diagnosis are essential to minimize morbidity and to prevent death.
CASE CONTINUED
Warfarin was discontinued once the patient developed the skin lesions. He received vitamin K and fresh frozen plasma to normalize his INR, and he was started on a heparin infusion, after which the lesions began to heal. The patient refused a skin biopsy. Platelet counts remained stable during his hospital course. Protein C levels were not checked, given his recent use of warfarin. He was started on dabigatran (Pradaxa) and was discharged a week later.
THE OTHER DIAGNOSTIC CHOICES
Pyoderma gangrenosum is an uncommon ulcerative skin condition often associated with autoimmune disease. It usually starts at the site of a minor injury, more commonly on the legs, and gradually progresses to a painful ulcer.
Cutaneous vasculitis is an inflammation of small blood vessels characterized by palpable purpura. The lesions can resemble urticaria, petechia, or erythema multiforme. It is commonly associated with infection, drug therapy, inflammatory disease, and malignancy.
Ecthyma gangrenosum is an infection of skin caused by Pseudomonas aeruginosa. Usually, it presents as hemorrhagic pustules or infarct-like areas with surrounding erythema that evolve into necrotic ulcers surrounded by erythema.
Dermatitis herpetiformis is a chronic skin condition, presenting with fluid-filled blisters and commonly involving the neck, back, scalp, and elbows. This condition is associated with celiac disease, and the lesions are extremely pruritic.
A 75-year-old man was admitted to the hospital with new-onset atrial fibrillation. He underwent rate control, and a heparin infusion was started. Warfarin (Coumadin) 10 mg was added on the second hospital day. Two days later, the heparin infusion was discontinued when the international normalized ratio (INR) was in the therapeutic range.
Q: Which is the most likely diagnosis?
- Pyoderma gangrenosum
- Cutaneous vasculitis
- Warfarin-induced skin necrosis
- Ecthyma gangrenosum
- Dermatitis herpetiformis
A: The most likely diagnosis is warfarin-induced skin necrosis, a rare paradoxical complication that occurs in 0.01% to 0.1% of patients receiving this drug.1 Microthrombosis leads to necrosis of the skin and subcutaneous tissues, arising within 2 to 10 days after the start of anticoagulation therapy, although in rare cases it can occur months to years later.2,3
The most common risk factors include the unopposed use of warfarin (ie, unopposed by heparin at the start of therapy), using higher doses of warfarin during the initiation of anticoagulation, and inadequate overlap with an effective parenteral anticoagulant. Patients with protein C or S deficiency, heparin-induced thrombocytopenia,4 resistance to activated protein C, antithrombin deficiency, and lupus anticoagulant have also been reported to be at risk.
The most common sites affected are areas with high subcutaneous fat content, such as the abdomen, thighs, breasts, and buttocks. Skin presentations can vary from dermal plaques to petechial lesions, which rapidly progress to well-demarcated, bluish-black, painful lesions and eventually to hemorrhagic bullae and necrosis.1
At the start of warfarin therapy, the levels of protein C and factor VII (with half-lives of 5 to 8 hours) fall faster than those of other vitamin-K-dependent factors (ie, factors II, IX, and X). This causes a transient imbalance in procoagulant and anticoagulant pathways favoring thrombosis of the microvasculature, with resulting necrosis. Patients with hereditary protein C deficiency are at higher risk.5
Histologic review of lesions often shows venous thrombosis and diffuse necrosis of the dermis and subcutaneous tissue.2
Promptly stopping the warfarin and choosing alternative anticoagulation may help prevent further progression of this condition. Wound care, debridement, and sometimes skin grafting may be necessary, depending on the extent of the lesions. A rechallenge with warfarin is often difficult, but cases have been reported in which treatment was resumed without adverse consequences.6 Avoiding large loading doses of warfarin, gradually increasing doses over an extended period (about 10 days),3 and starting warfarin with a heparin bridge for at least 5 days (which was not done in this patient) would prevent the condition.
Early recognition, differentiation, and diagnosis are essential to minimize morbidity and to prevent death.
CASE CONTINUED
Warfarin was discontinued once the patient developed the skin lesions. He received vitamin K and fresh frozen plasma to normalize his INR, and he was started on a heparin infusion, after which the lesions began to heal. The patient refused a skin biopsy. Platelet counts remained stable during his hospital course. Protein C levels were not checked, given his recent use of warfarin. He was started on dabigatran (Pradaxa) and was discharged a week later.
THE OTHER DIAGNOSTIC CHOICES
Pyoderma gangrenosum is an uncommon ulcerative skin condition often associated with autoimmune disease. It usually starts at the site of a minor injury, more commonly on the legs, and gradually progresses to a painful ulcer.
Cutaneous vasculitis is an inflammation of small blood vessels characterized by palpable purpura. The lesions can resemble urticaria, petechia, or erythema multiforme. It is commonly associated with infection, drug therapy, inflammatory disease, and malignancy.
Ecthyma gangrenosum is an infection of skin caused by Pseudomonas aeruginosa. Usually, it presents as hemorrhagic pustules or infarct-like areas with surrounding erythema that evolve into necrotic ulcers surrounded by erythema.
Dermatitis herpetiformis is a chronic skin condition, presenting with fluid-filled blisters and commonly involving the neck, back, scalp, and elbows. This condition is associated with celiac disease, and the lesions are extremely pruritic.
- Nazarian RM, Van Cott EM, Zembowicz A, Duncan LM. Warfarin-induced skin necrosis. J Am Acad Dermatol 2009; 61:325–332.
- Ward CT, Chavalitanonda N. Atypical warfarin-induced skin necrosis. Pharmacotherapy 2006; 26:1175–1179.
- Chan YC, Valenti D, Mansfield AO, Stansby G. Warfarin induced skin necrosis. Br J Surg 2000; 87:266–272.
- Warkentin TE, Sikov WM, Lillicrap DP. Multicentric warfarin-induced skin necrosis complicating heparin-induced thrombocytopenia. Am J Hematol 1999; 62:44–48.
- Ad-El DD, Meirovitz A, Weinberg A, et al. Warfarin skin necrosis: local and systemic factors. Br J Plast Surg 2000; 53:624–626.
- Jillella AP, Lutcher CL. Reinstituting warfarin in patients who develop warfarin skin necrosis. Am J Hematol 1996; 52:117–119.
- Nazarian RM, Van Cott EM, Zembowicz A, Duncan LM. Warfarin-induced skin necrosis. J Am Acad Dermatol 2009; 61:325–332.
- Ward CT, Chavalitanonda N. Atypical warfarin-induced skin necrosis. Pharmacotherapy 2006; 26:1175–1179.
- Chan YC, Valenti D, Mansfield AO, Stansby G. Warfarin induced skin necrosis. Br J Surg 2000; 87:266–272.
- Warkentin TE, Sikov WM, Lillicrap DP. Multicentric warfarin-induced skin necrosis complicating heparin-induced thrombocytopenia. Am J Hematol 1999; 62:44–48.
- Ad-El DD, Meirovitz A, Weinberg A, et al. Warfarin skin necrosis: local and systemic factors. Br J Plast Surg 2000; 53:624–626.
- Jillella AP, Lutcher CL. Reinstituting warfarin in patients who develop warfarin skin necrosis. Am J Hematol 1996; 52:117–119.
Examine before ordering: An algorithm unchanged by new tests

We rheumatologists may have inadvertently encouraged this practice. We teach about the prevalence of specific autoantibodies in patients with specific, accurately diagnosed autoimmune disorders as opposed to that in the general population (ie, the test’s sensitivity and specificity). But that is different than using a test to diagnose a specific disease in an ill patient with a heretofore undiagnosed condition (ie, the test’s predictive value). When I ask trainees or nonrheumatologists, “Why order all those tests?” the response I often get is that they thought the rheumatologist would want them when he or she was consulted. The fact that I also see our rheumatology fellows requesting the same tests before fully evaluating the patient clinically suggests that we have not done a great job at explaining the clinical utility and limitations of these tests. A serologic test should be used to strengthen or refute the clinician’s preliminary diagnosis, depending on the test’s specificity and sensitivity. It should not be used to generate a diagnosis.
So with these concerns, why would we invite a paper encouraging the use of the relatively new anti-cyclic citrullinated peptide (anti-CCP) test to evaluate patients with possible rheumatoid arthritis (Bose and Calabrese)?
As discussed in that paper, this test has characteristics that are useful when evaluating patients with polyarthritis compatible with the diagnosis of rheumatoid arthritis. Specifically, this test, unlike the traditional test for rheumatoid factor, can help discern whether the arthritis is a reaction to an infection like hepatitis C or endocarditis. Like rheumatoid factor, anti-CCP may precede the appearance of clinically meaningful arthritis and helps to predict prognosis in established rheumatoid arthritis. But, like other serologic tests, the anti-CCP test cannot supplant the listening ears and examining fingers of the clinician in establishing the pretest likelihood of the diagnosis. Clinical evaluation must precede laboratory testing.
We rheumatologists may have inadvertently encouraged this practice. We teach about the prevalence of specific autoantibodies in patients with specific, accurately diagnosed autoimmune disorders as opposed to that in the general population (ie, the test’s sensitivity and specificity). But that is different than using a test to diagnose a specific disease in an ill patient with a heretofore undiagnosed condition (ie, the test’s predictive value). When I ask trainees or nonrheumatologists, “Why order all those tests?” the response I often get is that they thought the rheumatologist would want them when he or she was consulted. The fact that I also see our rheumatology fellows requesting the same tests before fully evaluating the patient clinically suggests that we have not done a great job at explaining the clinical utility and limitations of these tests. A serologic test should be used to strengthen or refute the clinician’s preliminary diagnosis, depending on the test’s specificity and sensitivity. It should not be used to generate a diagnosis.
So with these concerns, why would we invite a paper encouraging the use of the relatively new anti-cyclic citrullinated peptide (anti-CCP) test to evaluate patients with possible rheumatoid arthritis (Bose and Calabrese)?
As discussed in that paper, this test has characteristics that are useful when evaluating patients with polyarthritis compatible with the diagnosis of rheumatoid arthritis. Specifically, this test, unlike the traditional test for rheumatoid factor, can help discern whether the arthritis is a reaction to an infection like hepatitis C or endocarditis. Like rheumatoid factor, anti-CCP may precede the appearance of clinically meaningful arthritis and helps to predict prognosis in established rheumatoid arthritis. But, like other serologic tests, the anti-CCP test cannot supplant the listening ears and examining fingers of the clinician in establishing the pretest likelihood of the diagnosis. Clinical evaluation must precede laboratory testing.
We rheumatologists may have inadvertently encouraged this practice. We teach about the prevalence of specific autoantibodies in patients with specific, accurately diagnosed autoimmune disorders as opposed to that in the general population (ie, the test’s sensitivity and specificity). But that is different than using a test to diagnose a specific disease in an ill patient with a heretofore undiagnosed condition (ie, the test’s predictive value). When I ask trainees or nonrheumatologists, “Why order all those tests?” the response I often get is that they thought the rheumatologist would want them when he or she was consulted. The fact that I also see our rheumatology fellows requesting the same tests before fully evaluating the patient clinically suggests that we have not done a great job at explaining the clinical utility and limitations of these tests. A serologic test should be used to strengthen or refute the clinician’s preliminary diagnosis, depending on the test’s specificity and sensitivity. It should not be used to generate a diagnosis.
So with these concerns, why would we invite a paper encouraging the use of the relatively new anti-cyclic citrullinated peptide (anti-CCP) test to evaluate patients with possible rheumatoid arthritis (Bose and Calabrese)?
As discussed in that paper, this test has characteristics that are useful when evaluating patients with polyarthritis compatible with the diagnosis of rheumatoid arthritis. Specifically, this test, unlike the traditional test for rheumatoid factor, can help discern whether the arthritis is a reaction to an infection like hepatitis C or endocarditis. Like rheumatoid factor, anti-CCP may precede the appearance of clinically meaningful arthritis and helps to predict prognosis in established rheumatoid arthritis. But, like other serologic tests, the anti-CCP test cannot supplant the listening ears and examining fingers of the clinician in establishing the pretest likelihood of the diagnosis. Clinical evaluation must precede laboratory testing.
Should I order an anti-CCP antibody test to diagnose rheumatoid arthritis?
Yes. Testing for anti-cyclic citrullinated peptide (anti-CCP) antibody can help diagnose rheumatoid arthritis (RA) because it is a highly specific test.
For many years, the diagnosis of RA has been based on the presentation of symmetrical small- and large-joint polyarthritis that spares the lower spine, further supported by the presence of characteristic joint damage on radiography and an elevated rheumatoid factor while also excluding clinical mimics. However, rheumatoid factor is often not detected early in RA, and detection of rheumatoid factor is not specific for RA. Testing for anti-CCP antibody can provide additional information and, in some cases, enable earlier and more specific diagnosis.
An important advance in our understanding of the pathogenesis of RA and in improving our ability to diagnose it early is the recognition that RA patients often produce autoantibodies directed against proteins and peptides containing the amino acid citrulline. Citrulline is generated in an inflammatory environment by the modification of the amino acid arginine by the enzyme peptidylarginine deiminase. Antibodies against cyclic citrulline are generated by patients with a certain genetic makeup, although citrulline can be detected in inflammatory tissues in conditions other than RA (without the antibody).
Anti-CCP antibody has been found in sera up to 10 years before the onset of joint symptoms in patients who later develop RA and may appear somewhat earlier than rheumatoid factor.1 From 10% to 15% of RA patients remain seronegative for rheumatoid factor throughout the disease course.
INFORMAL GUIDELINES FOR ANTI-CCP ANTIBODY TESTING
The role of anti-CCP antibody testing in the management of RA is still being defined, but we suggest several informal guidelines.
Anti-CCP antibody testing can help interpret the significance of an inexplicably high rheumatoid factor titer in the absence of classic RA. In such situations, a negative anti-CCP antibody test suggests a nonrheumatic disorder such as hepatitis C virus infection or endocarditis, whereas a positive anti-CCP antibody test is more consistent with early or even preclinical RA since this test, unlike rheumatoid factor testing, is generally negative in the setting of infection.

However, in a patient who has documented RA and who is seropositive for rheumatoid factor, anti-CCP antibody testing has limited value, as the information it provides may be redundant. In a patient with a low to intermediate probability for RA and with a negative or low level of rheumatoid factor, a positive anti-CCP antibody test helps confirm the diagnosis. Rheumatoid factor positivity and anti-CCP antibody positivity are each associated with more severe RA. Neither test varies with the activity of RA.
Finally, in smokers with a particular genotype, the presence of anti-CCP antibody predicts a particularly worse course for RA.
THE ROLE OF RHEUMATOID FACTOR TESTING
Rheumatoid factor, first described in 1940,4 is an antibody against the Fc portion of immunoglobulin G. The cutoff value for positivity varies by laboratory but is usually greater than 45 IU/mL by enzyme-linked immunosorbent assay or laser nephelometry, or greater than 1:80 by latex fixation. However, serum titers or serum levels expressed as “IU/mL” cannot accurately be compared between laboratories; instead, when using tests for rheumatoid factor, physicians should refer to specificity and sensitivity measurements for each analyzing laboratory.
Around 50% of patients with RA become positive for rheumatoid factor in the first 6 months, and 85% become positive over the first 2 years. Also, rheumatoid factor testing suffers from low specificity, since it can be detected (although sometimes in low levels) in a variety of infectious and inflammatory conditions, such as bacterial endocarditis, malaria, tuberculosis, osteomyelitis, hepatitis C (with or without cryoglobulinemia), Sjögren syndrome, systemic lupus erythematosus, primary biliary cirrhosis, postvaccination arthropathy, and aging.
Current detection methods cannot differentiate between naturally occurring, transiently induced, and RA-associated rheumatoid factor. The levels are generally higher in RA than in many non-RA disorders, but significant overlap occurs. Rheumatoid factor positivity serves as a marker of poor prognosis, predicting generally more aggressive, erosive disease, and it is correlated with extra-articular manifestations such as rheumatoid nodules and lung involvement.
The classification criteria for RA published in 2010 by the American College of Rheumatology and the European League Against Rheumatism provide references for the measurement of rheumatoid factor: “low-level positive” refers to values less than or equal to three times the upper limit of normal for a particular laboratory; “high-level positive” refers to values more than three times the upper limit of normal.5 This is an attempt to provide a clinically useful benchmark for the measurement of rheumatoid factor, the values of which may vary between laboratories.
STUDIES COMPARING THE TWO TESTS
Several studies have evaluated the utility and validity of anti-CCP antibody testing vs rheumatoid factor testing.
In a study of 826 US veterans with RA,6 75% tested positive for anti-CCP antibody and 80% were positive for rheumatoid factor. It was found that a higher anti-CCP antibody titer was associated with increased disease activity and inversely correlated with remission, especially in those also positive for rheumatoid factor.6
In another study,1 in which blood samples from 79 patients with RA who had been blood donors were analyzed, 39 patients (49.4%) were positive for either rheumatoid factor or anti-CCP antibody, or both, a median of 4.5 years (range 0.1 to 13.8 years) before the onset of RA symptoms; 32 patients (40.5%) became positive for anti-CCP antibody before symptom onset.
Whiting et al,7 in a systematic review of 151 studies, showed that anti-CCP antibody testing had greater specificity than rheumatoid factor testing (96% vs 86%), with similar sensitivity (56% vs 58%)—most notably in eight cohort studies of patients with early RA.7 In the 15 cohort studies analyzed, the test was found to have a positive likelihood ratio of 12.7 and a negative likelihood ratio of 0.45, supporting this as a test of high positive predictive value for RA.
In view of the evidence from these studies, it is not surprising that the 2010 collaborative classification of RA of the American College of Rheumatology and the European League Against Rheumatism places equal weight on anti-CCP antibody testing and rheumatoid factor testing in the early diagnosis of RA.5
GENETICS AND THE PROGNOSIS OF RHEUMATOID ARTHRITIS
In recent years, there has been a growing recognition that the pathogenesis of RA in patients who are seropositive for rheumatoid factor or anti-CCP antibody is different from the pathogenesis of RA in patients who are seronegative for rheumatoid factor and anti-CCP antibody. This may help us guide therapy.
Patients positive for rheumatoid factor or anti-CCP antibody who have a specific allelic subset of a region of the immune-response gene DRB1*04 appear to be highly vulnerable to smoking as an environmental trigger or to worsening RA.8
Patients positive for anti-CCP antibody tend also to have severe joint destruction and, hence, have a worse prognosis. Kaltenhäuser et al9 found that determining the presence of the shared epitope (an RA-specific genetic marker) and positivity for anti-CCP antibody facilitates prediction of the disease course and prognosis.9
Studies have shown that patients with confirmed RA who test positive for anti-CCP antibody may also have more-severe extraarticular manifestations. Recent studies have found anti-CCP antibody positivity in 15.7% to 17.5% of patients with psoriatic arthritis and in 85% of patients with RA. Patients with psoriatic arthritis who were positive for anti-CCP antibody had more joints that were tender and swollen, erosive arthritis, deformities, and functional impairment of peripheral joints.10,11
THE COST DIFFERENCE IS TRIVIAL IN THE LONG RUN
Cost is the major differentiating factor between rheumatoid factor testing and anti-CCP antibody testing. Rheumatoid factor testing costs around $43, and anti-CCP antibody testing costs $102 in the reference laboratory at Cleveland Clinic. However, the difference in cost is trivial, since this is only a one-time cost, whereas the information anti-CCP antibody testing provides can have a major impact on predicting the prognosis and determining the choice of therapy for a disease associated with high direct and indirect costs over a lifetime. Also, Medicare and other insurers would likely reimburse for anti-CCP antibody testing as long as it was associated with a related diagnosis such as arthralgia or arthritis.
Given that there will be a small number of patients with confirmed RA who will be negative for rheumatoid factor yet positive for anti-CCP antibody, one can support ordering both tests in tandem in a patient whom you strongly suspect of having RA. Or, at $100, one could make the argument that it would be cost-effective to order anti-CCP antibody testing only if rheumatoid factor testing is negative.
Testing for rheumatoid factor and anti-CCP antibody should not be done serially to assess treatment response or disease activity in these patients: these markers do not vary with inflammatory activity or disappear with clinical “remission.”
- Nielen MM, van Schaardenburg D, Reesink HW, et al. Specific autoantibodies precede the symptoms of rheumatoid arthritis: a study of serial measurements in blood donors. Arthritis Rheum 2004; 50:380–386.
- Egerer K, Feist E, Burmester GR. The serological diagnosis of rheumatoid arthritis: antibodies to citrullinated antigens. Dtsch Arztebl Int 2009; 106:159–163.
- Conrad K, Roggenbuck D, Reinhold D, Dörner T. Profiling of rheumatoid arthritis associated autoantibodies. Autoimmun Rev 2010; 9:431–435.
- Waaler E. On the occurrence of a factor in human serum activating the specific agglutintion of sheep blood corpuscles. 1939. APMIS 2007; 115:422–438.
- Aletaha D, Neogi T, Silman AJ, et al. 2010 Rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum 2010; 62:2569–2581.
- Miriovsky BJ, Michaud K, Thiele GM, et al. Anti-CCP antibody and rheumatoid factor concentrations predict greater disease activity in men with rheumatoid arthritis. Ann Rheum Dis 2010; 69:1292–1297.
- Whiting PF, Smidt N, Sterne JA, et al. Systematic review: accuracy of anti-citrullinated peptide antibodies for diagnosing rheumatoid arthritis. Ann Intern Med 2010; 152:456–464;W155–W166.
- van Venrooij WJ, van Beers JJ, Pruijn GJ. Anti-CCP antibody, a marker for the early detection of rheumatoid arthritis. Ann N Y Acad Sci 2008; 1143:268–285.
- Kaltenhäuser S, Pierer M, Arnold S, et al. Antibodies against cyclic citrullinated peptide are associated with the DRB1 shared epitope and predict joint erosion in rheumatoid arthritis. Rheumatology (Oxford) 2007; 46:100–104.
- Bogliolo L, Alpini C, Caporali R, Scirè CA, Moratti R, Montecucco C. Antibodies to cyclic citrullinated peptides in psoriatic arthritis. J Rheumatol 2005; 32:511–515.
- Abdel Fattah NS, Hassan HE, Galal ZA, El Okda el SE. Assessment of anti-cyclic citrullinated peptide in psoriatic arthritis. BMC Res Notes 2009; 2:44.
Yes. Testing for anti-cyclic citrullinated peptide (anti-CCP) antibody can help diagnose rheumatoid arthritis (RA) because it is a highly specific test.
For many years, the diagnosis of RA has been based on the presentation of symmetrical small- and large-joint polyarthritis that spares the lower spine, further supported by the presence of characteristic joint damage on radiography and an elevated rheumatoid factor while also excluding clinical mimics. However, rheumatoid factor is often not detected early in RA, and detection of rheumatoid factor is not specific for RA. Testing for anti-CCP antibody can provide additional information and, in some cases, enable earlier and more specific diagnosis.
An important advance in our understanding of the pathogenesis of RA and in improving our ability to diagnose it early is the recognition that RA patients often produce autoantibodies directed against proteins and peptides containing the amino acid citrulline. Citrulline is generated in an inflammatory environment by the modification of the amino acid arginine by the enzyme peptidylarginine deiminase. Antibodies against cyclic citrulline are generated by patients with a certain genetic makeup, although citrulline can be detected in inflammatory tissues in conditions other than RA (without the antibody).
Anti-CCP antibody has been found in sera up to 10 years before the onset of joint symptoms in patients who later develop RA and may appear somewhat earlier than rheumatoid factor.1 From 10% to 15% of RA patients remain seronegative for rheumatoid factor throughout the disease course.
INFORMAL GUIDELINES FOR ANTI-CCP ANTIBODY TESTING
The role of anti-CCP antibody testing in the management of RA is still being defined, but we suggest several informal guidelines.
Anti-CCP antibody testing can help interpret the significance of an inexplicably high rheumatoid factor titer in the absence of classic RA. In such situations, a negative anti-CCP antibody test suggests a nonrheumatic disorder such as hepatitis C virus infection or endocarditis, whereas a positive anti-CCP antibody test is more consistent with early or even preclinical RA since this test, unlike rheumatoid factor testing, is generally negative in the setting of infection.
However, in a patient who has documented RA and who is seropositive for rheumatoid factor, anti-CCP antibody testing has limited value, as the information it provides may be redundant. In a patient with a low to intermediate probability for RA and with a negative or low level of rheumatoid factor, a positive anti-CCP antibody test helps confirm the diagnosis. Rheumatoid factor positivity and anti-CCP antibody positivity are each associated with more severe RA. Neither test varies with the activity of RA.
Finally, in smokers with a particular genotype, the presence of anti-CCP antibody predicts a particularly worse course for RA.
THE ROLE OF RHEUMATOID FACTOR TESTING
Rheumatoid factor, first described in 1940,4 is an antibody against the Fc portion of immunoglobulin G. The cutoff value for positivity varies by laboratory but is usually greater than 45 IU/mL by enzyme-linked immunosorbent assay or laser nephelometry, or greater than 1:80 by latex fixation. However, serum titers or serum levels expressed as “IU/mL” cannot accurately be compared between laboratories; instead, when using tests for rheumatoid factor, physicians should refer to specificity and sensitivity measurements for each analyzing laboratory.
Around 50% of patients with RA become positive for rheumatoid factor in the first 6 months, and 85% become positive over the first 2 years. Also, rheumatoid factor testing suffers from low specificity, since it can be detected (although sometimes in low levels) in a variety of infectious and inflammatory conditions, such as bacterial endocarditis, malaria, tuberculosis, osteomyelitis, hepatitis C (with or without cryoglobulinemia), Sjögren syndrome, systemic lupus erythematosus, primary biliary cirrhosis, postvaccination arthropathy, and aging.
Current detection methods cannot differentiate between naturally occurring, transiently induced, and RA-associated rheumatoid factor. The levels are generally higher in RA than in many non-RA disorders, but significant overlap occurs. Rheumatoid factor positivity serves as a marker of poor prognosis, predicting generally more aggressive, erosive disease, and it is correlated with extra-articular manifestations such as rheumatoid nodules and lung involvement.
The classification criteria for RA published in 2010 by the American College of Rheumatology and the European League Against Rheumatism provide references for the measurement of rheumatoid factor: “low-level positive” refers to values less than or equal to three times the upper limit of normal for a particular laboratory; “high-level positive” refers to values more than three times the upper limit of normal.5 This is an attempt to provide a clinically useful benchmark for the measurement of rheumatoid factor, the values of which may vary between laboratories.
STUDIES COMPARING THE TWO TESTS
Several studies have evaluated the utility and validity of anti-CCP antibody testing vs rheumatoid factor testing.
In a study of 826 US veterans with RA,6 75% tested positive for anti-CCP antibody and 80% were positive for rheumatoid factor. It was found that a higher anti-CCP antibody titer was associated with increased disease activity and inversely correlated with remission, especially in those also positive for rheumatoid factor.6
In another study,1 in which blood samples from 79 patients with RA who had been blood donors were analyzed, 39 patients (49.4%) were positive for either rheumatoid factor or anti-CCP antibody, or both, a median of 4.5 years (range 0.1 to 13.8 years) before the onset of RA symptoms; 32 patients (40.5%) became positive for anti-CCP antibody before symptom onset.
Whiting et al,7 in a systematic review of 151 studies, showed that anti-CCP antibody testing had greater specificity than rheumatoid factor testing (96% vs 86%), with similar sensitivity (56% vs 58%)—most notably in eight cohort studies of patients with early RA.7 In the 15 cohort studies analyzed, the test was found to have a positive likelihood ratio of 12.7 and a negative likelihood ratio of 0.45, supporting this as a test of high positive predictive value for RA.
In view of the evidence from these studies, it is not surprising that the 2010 collaborative classification of RA of the American College of Rheumatology and the European League Against Rheumatism places equal weight on anti-CCP antibody testing and rheumatoid factor testing in the early diagnosis of RA.5
GENETICS AND THE PROGNOSIS OF RHEUMATOID ARTHRITIS
In recent years, there has been a growing recognition that the pathogenesis of RA in patients who are seropositive for rheumatoid factor or anti-CCP antibody is different from the pathogenesis of RA in patients who are seronegative for rheumatoid factor and anti-CCP antibody. This may help us guide therapy.
Patients positive for rheumatoid factor or anti-CCP antibody who have a specific allelic subset of a region of the immune-response gene DRB1*04 appear to be highly vulnerable to smoking as an environmental trigger or to worsening RA.8
Patients positive for anti-CCP antibody tend also to have severe joint destruction and, hence, have a worse prognosis. Kaltenhäuser et al9 found that determining the presence of the shared epitope (an RA-specific genetic marker) and positivity for anti-CCP antibody facilitates prediction of the disease course and prognosis.9
Studies have shown that patients with confirmed RA who test positive for anti-CCP antibody may also have more-severe extraarticular manifestations. Recent studies have found anti-CCP antibody positivity in 15.7% to 17.5% of patients with psoriatic arthritis and in 85% of patients with RA. Patients with psoriatic arthritis who were positive for anti-CCP antibody had more joints that were tender and swollen, erosive arthritis, deformities, and functional impairment of peripheral joints.10,11
THE COST DIFFERENCE IS TRIVIAL IN THE LONG RUN
Cost is the major differentiating factor between rheumatoid factor testing and anti-CCP antibody testing. Rheumatoid factor testing costs around $43, and anti-CCP antibody testing costs $102 in the reference laboratory at Cleveland Clinic. However, the difference in cost is trivial, since this is only a one-time cost, whereas the information anti-CCP antibody testing provides can have a major impact on predicting the prognosis and determining the choice of therapy for a disease associated with high direct and indirect costs over a lifetime. Also, Medicare and other insurers would likely reimburse for anti-CCP antibody testing as long as it was associated with a related diagnosis such as arthralgia or arthritis.
Given that there will be a small number of patients with confirmed RA who will be negative for rheumatoid factor yet positive for anti-CCP antibody, one can support ordering both tests in tandem in a patient whom you strongly suspect of having RA. Or, at $100, one could make the argument that it would be cost-effective to order anti-CCP antibody testing only if rheumatoid factor testing is negative.
Testing for rheumatoid factor and anti-CCP antibody should not be done serially to assess treatment response or disease activity in these patients: these markers do not vary with inflammatory activity or disappear with clinical “remission.”
Yes. Testing for anti-cyclic citrullinated peptide (anti-CCP) antibody can help diagnose rheumatoid arthritis (RA) because it is a highly specific test.
For many years, the diagnosis of RA has been based on the presentation of symmetrical small- and large-joint polyarthritis that spares the lower spine, further supported by the presence of characteristic joint damage on radiography and an elevated rheumatoid factor while also excluding clinical mimics. However, rheumatoid factor is often not detected early in RA, and detection of rheumatoid factor is not specific for RA. Testing for anti-CCP antibody can provide additional information and, in some cases, enable earlier and more specific diagnosis.
An important advance in our understanding of the pathogenesis of RA and in improving our ability to diagnose it early is the recognition that RA patients often produce autoantibodies directed against proteins and peptides containing the amino acid citrulline. Citrulline is generated in an inflammatory environment by the modification of the amino acid arginine by the enzyme peptidylarginine deiminase. Antibodies against cyclic citrulline are generated by patients with a certain genetic makeup, although citrulline can be detected in inflammatory tissues in conditions other than RA (without the antibody).
Anti-CCP antibody has been found in sera up to 10 years before the onset of joint symptoms in patients who later develop RA and may appear somewhat earlier than rheumatoid factor.1 From 10% to 15% of RA patients remain seronegative for rheumatoid factor throughout the disease course.
INFORMAL GUIDELINES FOR ANTI-CCP ANTIBODY TESTING
The role of anti-CCP antibody testing in the management of RA is still being defined, but we suggest several informal guidelines.
Anti-CCP antibody testing can help interpret the significance of an inexplicably high rheumatoid factor titer in the absence of classic RA. In such situations, a negative anti-CCP antibody test suggests a nonrheumatic disorder such as hepatitis C virus infection or endocarditis, whereas a positive anti-CCP antibody test is more consistent with early or even preclinical RA since this test, unlike rheumatoid factor testing, is generally negative in the setting of infection.
However, in a patient who has documented RA and who is seropositive for rheumatoid factor, anti-CCP antibody testing has limited value, as the information it provides may be redundant. In a patient with a low to intermediate probability for RA and with a negative or low level of rheumatoid factor, a positive anti-CCP antibody test helps confirm the diagnosis. Rheumatoid factor positivity and anti-CCP antibody positivity are each associated with more severe RA. Neither test varies with the activity of RA.
Finally, in smokers with a particular genotype, the presence of anti-CCP antibody predicts a particularly worse course for RA.
THE ROLE OF RHEUMATOID FACTOR TESTING
Rheumatoid factor, first described in 1940,4 is an antibody against the Fc portion of immunoglobulin G. The cutoff value for positivity varies by laboratory but is usually greater than 45 IU/mL by enzyme-linked immunosorbent assay or laser nephelometry, or greater than 1:80 by latex fixation. However, serum titers or serum levels expressed as “IU/mL” cannot accurately be compared between laboratories; instead, when using tests for rheumatoid factor, physicians should refer to specificity and sensitivity measurements for each analyzing laboratory.
Around 50% of patients with RA become positive for rheumatoid factor in the first 6 months, and 85% become positive over the first 2 years. Also, rheumatoid factor testing suffers from low specificity, since it can be detected (although sometimes in low levels) in a variety of infectious and inflammatory conditions, such as bacterial endocarditis, malaria, tuberculosis, osteomyelitis, hepatitis C (with or without cryoglobulinemia), Sjögren syndrome, systemic lupus erythematosus, primary biliary cirrhosis, postvaccination arthropathy, and aging.
Current detection methods cannot differentiate between naturally occurring, transiently induced, and RA-associated rheumatoid factor. The levels are generally higher in RA than in many non-RA disorders, but significant overlap occurs. Rheumatoid factor positivity serves as a marker of poor prognosis, predicting generally more aggressive, erosive disease, and it is correlated with extra-articular manifestations such as rheumatoid nodules and lung involvement.
The classification criteria for RA published in 2010 by the American College of Rheumatology and the European League Against Rheumatism provide references for the measurement of rheumatoid factor: “low-level positive” refers to values less than or equal to three times the upper limit of normal for a particular laboratory; “high-level positive” refers to values more than three times the upper limit of normal.5 This is an attempt to provide a clinically useful benchmark for the measurement of rheumatoid factor, the values of which may vary between laboratories.
STUDIES COMPARING THE TWO TESTS
Several studies have evaluated the utility and validity of anti-CCP antibody testing vs rheumatoid factor testing.
In a study of 826 US veterans with RA,6 75% tested positive for anti-CCP antibody and 80% were positive for rheumatoid factor. It was found that a higher anti-CCP antibody titer was associated with increased disease activity and inversely correlated with remission, especially in those also positive for rheumatoid factor.6
In another study,1 in which blood samples from 79 patients with RA who had been blood donors were analyzed, 39 patients (49.4%) were positive for either rheumatoid factor or anti-CCP antibody, or both, a median of 4.5 years (range 0.1 to 13.8 years) before the onset of RA symptoms; 32 patients (40.5%) became positive for anti-CCP antibody before symptom onset.
Whiting et al,7 in a systematic review of 151 studies, showed that anti-CCP antibody testing had greater specificity than rheumatoid factor testing (96% vs 86%), with similar sensitivity (56% vs 58%)—most notably in eight cohort studies of patients with early RA.7 In the 15 cohort studies analyzed, the test was found to have a positive likelihood ratio of 12.7 and a negative likelihood ratio of 0.45, supporting this as a test of high positive predictive value for RA.
In view of the evidence from these studies, it is not surprising that the 2010 collaborative classification of RA of the American College of Rheumatology and the European League Against Rheumatism places equal weight on anti-CCP antibody testing and rheumatoid factor testing in the early diagnosis of RA.5
GENETICS AND THE PROGNOSIS OF RHEUMATOID ARTHRITIS
In recent years, there has been a growing recognition that the pathogenesis of RA in patients who are seropositive for rheumatoid factor or anti-CCP antibody is different from the pathogenesis of RA in patients who are seronegative for rheumatoid factor and anti-CCP antibody. This may help us guide therapy.
Patients positive for rheumatoid factor or anti-CCP antibody who have a specific allelic subset of a region of the immune-response gene DRB1*04 appear to be highly vulnerable to smoking as an environmental trigger or to worsening RA.8
Patients positive for anti-CCP antibody tend also to have severe joint destruction and, hence, have a worse prognosis. Kaltenhäuser et al9 found that determining the presence of the shared epitope (an RA-specific genetic marker) and positivity for anti-CCP antibody facilitates prediction of the disease course and prognosis.9
Studies have shown that patients with confirmed RA who test positive for anti-CCP antibody may also have more-severe extraarticular manifestations. Recent studies have found anti-CCP antibody positivity in 15.7% to 17.5% of patients with psoriatic arthritis and in 85% of patients with RA. Patients with psoriatic arthritis who were positive for anti-CCP antibody had more joints that were tender and swollen, erosive arthritis, deformities, and functional impairment of peripheral joints.10,11
THE COST DIFFERENCE IS TRIVIAL IN THE LONG RUN
Cost is the major differentiating factor between rheumatoid factor testing and anti-CCP antibody testing. Rheumatoid factor testing costs around $43, and anti-CCP antibody testing costs $102 in the reference laboratory at Cleveland Clinic. However, the difference in cost is trivial, since this is only a one-time cost, whereas the information anti-CCP antibody testing provides can have a major impact on predicting the prognosis and determining the choice of therapy for a disease associated with high direct and indirect costs over a lifetime. Also, Medicare and other insurers would likely reimburse for anti-CCP antibody testing as long as it was associated with a related diagnosis such as arthralgia or arthritis.
Given that there will be a small number of patients with confirmed RA who will be negative for rheumatoid factor yet positive for anti-CCP antibody, one can support ordering both tests in tandem in a patient whom you strongly suspect of having RA. Or, at $100, one could make the argument that it would be cost-effective to order anti-CCP antibody testing only if rheumatoid factor testing is negative.
Testing for rheumatoid factor and anti-CCP antibody should not be done serially to assess treatment response or disease activity in these patients: these markers do not vary with inflammatory activity or disappear with clinical “remission.”
- Nielen MM, van Schaardenburg D, Reesink HW, et al. Specific autoantibodies precede the symptoms of rheumatoid arthritis: a study of serial measurements in blood donors. Arthritis Rheum 2004; 50:380–386.
- Egerer K, Feist E, Burmester GR. The serological diagnosis of rheumatoid arthritis: antibodies to citrullinated antigens. Dtsch Arztebl Int 2009; 106:159–163.
- Conrad K, Roggenbuck D, Reinhold D, Dörner T. Profiling of rheumatoid arthritis associated autoantibodies. Autoimmun Rev 2010; 9:431–435.
- Waaler E. On the occurrence of a factor in human serum activating the specific agglutintion of sheep blood corpuscles. 1939. APMIS 2007; 115:422–438.
- Aletaha D, Neogi T, Silman AJ, et al. 2010 Rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum 2010; 62:2569–2581.
- Miriovsky BJ, Michaud K, Thiele GM, et al. Anti-CCP antibody and rheumatoid factor concentrations predict greater disease activity in men with rheumatoid arthritis. Ann Rheum Dis 2010; 69:1292–1297.
- Whiting PF, Smidt N, Sterne JA, et al. Systematic review: accuracy of anti-citrullinated peptide antibodies for diagnosing rheumatoid arthritis. Ann Intern Med 2010; 152:456–464;W155–W166.
- van Venrooij WJ, van Beers JJ, Pruijn GJ. Anti-CCP antibody, a marker for the early detection of rheumatoid arthritis. Ann N Y Acad Sci 2008; 1143:268–285.
- Kaltenhäuser S, Pierer M, Arnold S, et al. Antibodies against cyclic citrullinated peptide are associated with the DRB1 shared epitope and predict joint erosion in rheumatoid arthritis. Rheumatology (Oxford) 2007; 46:100–104.
- Bogliolo L, Alpini C, Caporali R, Scirè CA, Moratti R, Montecucco C. Antibodies to cyclic citrullinated peptides in psoriatic arthritis. J Rheumatol 2005; 32:511–515.
- Abdel Fattah NS, Hassan HE, Galal ZA, El Okda el SE. Assessment of anti-cyclic citrullinated peptide in psoriatic arthritis. BMC Res Notes 2009; 2:44.
- Nielen MM, van Schaardenburg D, Reesink HW, et al. Specific autoantibodies precede the symptoms of rheumatoid arthritis: a study of serial measurements in blood donors. Arthritis Rheum 2004; 50:380–386.
- Egerer K, Feist E, Burmester GR. The serological diagnosis of rheumatoid arthritis: antibodies to citrullinated antigens. Dtsch Arztebl Int 2009; 106:159–163.
- Conrad K, Roggenbuck D, Reinhold D, Dörner T. Profiling of rheumatoid arthritis associated autoantibodies. Autoimmun Rev 2010; 9:431–435.
- Waaler E. On the occurrence of a factor in human serum activating the specific agglutintion of sheep blood corpuscles. 1939. APMIS 2007; 115:422–438.
- Aletaha D, Neogi T, Silman AJ, et al. 2010 Rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum 2010; 62:2569–2581.
- Miriovsky BJ, Michaud K, Thiele GM, et al. Anti-CCP antibody and rheumatoid factor concentrations predict greater disease activity in men with rheumatoid arthritis. Ann Rheum Dis 2010; 69:1292–1297.
- Whiting PF, Smidt N, Sterne JA, et al. Systematic review: accuracy of anti-citrullinated peptide antibodies for diagnosing rheumatoid arthritis. Ann Intern Med 2010; 152:456–464;W155–W166.
- van Venrooij WJ, van Beers JJ, Pruijn GJ. Anti-CCP antibody, a marker for the early detection of rheumatoid arthritis. Ann N Y Acad Sci 2008; 1143:268–285.
- Kaltenhäuser S, Pierer M, Arnold S, et al. Antibodies against cyclic citrullinated peptide are associated with the DRB1 shared epitope and predict joint erosion in rheumatoid arthritis. Rheumatology (Oxford) 2007; 46:100–104.
- Bogliolo L, Alpini C, Caporali R, Scirè CA, Moratti R, Montecucco C. Antibodies to cyclic citrullinated peptides in psoriatic arthritis. J Rheumatol 2005; 32:511–515.
- Abdel Fattah NS, Hassan HE, Galal ZA, El Okda el SE. Assessment of anti-cyclic citrullinated peptide in psoriatic arthritis. BMC Res Notes 2009; 2:44.
In reply: Parkinson disease
In Reply: I thank Dr. Keller for his thoughtful comments. They are most appreciated.
It is true that with availability of generic ropinirole and pramipexole, there are now cheaper alternatives to levodopa. Nonetheless, levodopa remains the cheapest and most efficacious medication for Parkinson disease to date. Whenever levodopa is compared head-to-head with any dopamine agonist, the general results remain consistent: levodopa affords better motor improvement with lesser side effects, but is more likely to lead to motor fluctuations, specifically dyskinesias. Therefore, in general, levodopa is the first choice in elderly patients where tolerability may be an issue, whereas a dopamine agonist may be the initial treatment of choice in younger Parkinson patients, who are able to tolerate the drug better and have a higher likelihood of developing dyskinesias.
It is a tougher task to determine which among the dopamine agonists is superior. The newer dopamine agonists have not been compared head-to-head. Therefore, it is practically a “coin toss” when selecting which dopamine agonist to try. Their mechanism of action (D2 and D3 receptor agonist activity) and frequency of intake (three times per day for generics; once daily for long-acting formulations), cost, and side effect profile are nearly identical, despite minor differences in their half-lives.
Regarding putative neuroprotective agents in Parkinson disease, indeed, isradipine is one of the medications currently undergoing investigation for its potential neuroprotective effect. While I personally have no objection to using it for a Parkinson disease patient who also happens to need an antihypertensive agent, I am more cautious about endorsing it as a neuroprotective agent until results of clinical trials have been released. Similarly, while a large epidemiologic study has shown that people who take ibuprofen are less likely to develop Parkinson disease, there has been no robust human trial that has shown the drug to slow the progression of Parkinson disease among patients who are already suffering from the disorder. Therefore, the current use of ibuprofen in Parkinson disease should be based more on its anti-inflammatory indications rather than its possible neuroprotective effect. Finally, we have shown, in a large, multicenter, global randomized controlled trial with a delayed-start design, that pramipexole is unlikely to possess any meaningful neuroprotective effect. Therefore, I am personally not that optimistic that dexpramipexole would demonstrate such an effect.
While in theory combining the use of catechol-O-methyltransferase (COMT) inhibitors and monoamine oxidase (MAO) type B inhibitors can synergistically work to inhibit the breakdown of other catecholamines and lead to adrenergic crisis when taken concomitantly, this has not been our experience. Perhaps it is because at recommended doses, the MAO inhibition is selective to type B (where receptors are more confined to the brain) and not type A (where receptors are more distributed throughout blood vessels, thereby having a higher likelihood of causing a hypertensive crisis as is seen in the use of nonselective MAO inhibitors). Therefore, at our center, we routinely use the two classes of agents concomitantly with minimal safety concerns.
In Reply: I thank Dr. Keller for his thoughtful comments. They are most appreciated.
It is true that with availability of generic ropinirole and pramipexole, there are now cheaper alternatives to levodopa. Nonetheless, levodopa remains the cheapest and most efficacious medication for Parkinson disease to date. Whenever levodopa is compared head-to-head with any dopamine agonist, the general results remain consistent: levodopa affords better motor improvement with lesser side effects, but is more likely to lead to motor fluctuations, specifically dyskinesias. Therefore, in general, levodopa is the first choice in elderly patients where tolerability may be an issue, whereas a dopamine agonist may be the initial treatment of choice in younger Parkinson patients, who are able to tolerate the drug better and have a higher likelihood of developing dyskinesias.
It is a tougher task to determine which among the dopamine agonists is superior. The newer dopamine agonists have not been compared head-to-head. Therefore, it is practically a “coin toss” when selecting which dopamine agonist to try. Their mechanism of action (D2 and D3 receptor agonist activity) and frequency of intake (three times per day for generics; once daily for long-acting formulations), cost, and side effect profile are nearly identical, despite minor differences in their half-lives.
Regarding putative neuroprotective agents in Parkinson disease, indeed, isradipine is one of the medications currently undergoing investigation for its potential neuroprotective effect. While I personally have no objection to using it for a Parkinson disease patient who also happens to need an antihypertensive agent, I am more cautious about endorsing it as a neuroprotective agent until results of clinical trials have been released. Similarly, while a large epidemiologic study has shown that people who take ibuprofen are less likely to develop Parkinson disease, there has been no robust human trial that has shown the drug to slow the progression of Parkinson disease among patients who are already suffering from the disorder. Therefore, the current use of ibuprofen in Parkinson disease should be based more on its anti-inflammatory indications rather than its possible neuroprotective effect. Finally, we have shown, in a large, multicenter, global randomized controlled trial with a delayed-start design, that pramipexole is unlikely to possess any meaningful neuroprotective effect. Therefore, I am personally not that optimistic that dexpramipexole would demonstrate such an effect.
While in theory combining the use of catechol-O-methyltransferase (COMT) inhibitors and monoamine oxidase (MAO) type B inhibitors can synergistically work to inhibit the breakdown of other catecholamines and lead to adrenergic crisis when taken concomitantly, this has not been our experience. Perhaps it is because at recommended doses, the MAO inhibition is selective to type B (where receptors are more confined to the brain) and not type A (where receptors are more distributed throughout blood vessels, thereby having a higher likelihood of causing a hypertensive crisis as is seen in the use of nonselective MAO inhibitors). Therefore, at our center, we routinely use the two classes of agents concomitantly with minimal safety concerns.
In Reply: I thank Dr. Keller for his thoughtful comments. They are most appreciated.
It is true that with availability of generic ropinirole and pramipexole, there are now cheaper alternatives to levodopa. Nonetheless, levodopa remains the cheapest and most efficacious medication for Parkinson disease to date. Whenever levodopa is compared head-to-head with any dopamine agonist, the general results remain consistent: levodopa affords better motor improvement with lesser side effects, but is more likely to lead to motor fluctuations, specifically dyskinesias. Therefore, in general, levodopa is the first choice in elderly patients where tolerability may be an issue, whereas a dopamine agonist may be the initial treatment of choice in younger Parkinson patients, who are able to tolerate the drug better and have a higher likelihood of developing dyskinesias.
It is a tougher task to determine which among the dopamine agonists is superior. The newer dopamine agonists have not been compared head-to-head. Therefore, it is practically a “coin toss” when selecting which dopamine agonist to try. Their mechanism of action (D2 and D3 receptor agonist activity) and frequency of intake (three times per day for generics; once daily for long-acting formulations), cost, and side effect profile are nearly identical, despite minor differences in their half-lives.
Regarding putative neuroprotective agents in Parkinson disease, indeed, isradipine is one of the medications currently undergoing investigation for its potential neuroprotective effect. While I personally have no objection to using it for a Parkinson disease patient who also happens to need an antihypertensive agent, I am more cautious about endorsing it as a neuroprotective agent until results of clinical trials have been released. Similarly, while a large epidemiologic study has shown that people who take ibuprofen are less likely to develop Parkinson disease, there has been no robust human trial that has shown the drug to slow the progression of Parkinson disease among patients who are already suffering from the disorder. Therefore, the current use of ibuprofen in Parkinson disease should be based more on its anti-inflammatory indications rather than its possible neuroprotective effect. Finally, we have shown, in a large, multicenter, global randomized controlled trial with a delayed-start design, that pramipexole is unlikely to possess any meaningful neuroprotective effect. Therefore, I am personally not that optimistic that dexpramipexole would demonstrate such an effect.
While in theory combining the use of catechol-O-methyltransferase (COMT) inhibitors and monoamine oxidase (MAO) type B inhibitors can synergistically work to inhibit the breakdown of other catecholamines and lead to adrenergic crisis when taken concomitantly, this has not been our experience. Perhaps it is because at recommended doses, the MAO inhibition is selective to type B (where receptors are more confined to the brain) and not type A (where receptors are more distributed throughout blood vessels, thereby having a higher likelihood of causing a hypertensive crisis as is seen in the use of nonselective MAO inhibitors). Therefore, at our center, we routinely use the two classes of agents concomitantly with minimal safety concerns.
Parkinson disease
To the Editor: I have the following comments and questions regarding the excellent Medical Grand Rounds article on Parkinson disease by Dr. Fernandez in your January 2012 issue.1
The author mentions that when “cost may be of concern, levodopa is the preferred starting drug.”1 Generic versions of pramipexole and ropinirole are now available and have made these medications more affordable. For example, the cash price of generic ropinirole 5 mg was recently $66 for 100 tablets, comparable with generic carbidopa/levodopa (25/100 mg priced at $46 for 100 tablets.2 And even though the price of generic pramipexole was $240 for 90 tablets, seniors with Medicare Part D drug coverage can usually get any generic medication for a low copay.
When choosing a dopamine agonist, how does Dr. Fernandez decide between ropinirole and pramipexole (aside from the price difference noted above)? Pramipexole has a longer elimination half-life (8 to 12 hours) compared with ropinirole (6 hours).3 Does this imply a significantly longer effective dosing interval for pramipexole? Are there other significant clinical differences between these agents?
Isradipine (DynaCirc CR), a dihydropyridine calcium channel blocker, has shown promise as a neuroprotective agent for slowing the progression of Parkinson disease in epidemiologic and laboratory studies, as noted by the author. In addition, immediate-release isradipine, with its relatively short elimination half-life of 8 hours,3 may be well suited for treating Parkinson patients whose essential hypertension is complicated by episodes of orthostatic hypotension. It should be noted that dihydropyridines that do not cross the blood-brain barrier (such as amlodipine [Norvasc]) have shown no evidence of neuroprotection.
Ibuprofen is another drug that has fairly strong epidemiologic and laboratory evidence that it might be neuroprotective,4 although the other nonsteroidal anti-inflammatory drugs (NSAIDs) have proven disappointing as a class.5 Lacking any prospective randomized trials, the evidence is not strong enough to recommend ibuprofen solely for neuroprotection. Does Dr. Fernandez, however, consider it reasonable to suggest ibuprofen to Parkinson patients who need to take an NSAID for an approved indication (such as pain)?
Dexpramipexole has recently demonstrated great promise in a phase 3 clinical trial as a neuroprotective agent in amyotrophic lateral sclerosis.6 How does this compound relate to pramipexole, and does the author believe it may offer neuroprotection in other neurodegenerative diseases like Parkinson disease?
The author discusses the use of catechol-O-methyltransferase (COMT) inhibitors (such as Comtan and Tasmar) and the monoamine oxidase (MAO) type-B inhibitors rasagiline (Azilect) and selegiline (Eldepryl, Zelapar) for prolonging the effects of levodopa by slowing the breakdown of dopamine. However, it is important to note that it is contraindicated to prescribe both a COMT inhibitor and an MAO-B inhibitor, because these agents also inhibit the breakdown of other catecholamines and can lead to adrenergic crisis when taken concomitantly.
- Fernandez HH. Updates in the medical management of Parkinson disease. Cleve Clin J Med 2012; 79:28–35.
- Drugstore.com. www.Drugstore.com. Accessed February 5, 2012.
- PDR.net. www.PDR.net. Accessed February 25, 2012.
- Gao X, Chen H, Schwarzschild MA, Ascherio A. Use of ibuprofen and risk of Parkinson disease. Neurology 2011; 76:863–869.
- Driver JA, Logroscino G, Lu L, Gaziano JM, Kurth T. Use of non-steroidal anti-inflammatory drugs and risk of Parkinson’s disease: nested case-control study. BMJ 2011; 342:d198.
- Cudkowicz M, Bozik ME, Ingersoll EW, et al. The effects of dexpramipexole (KNS-760704) in individuals with amyotrophic lateral sclerosis. Nat Med 2011; 17:1652–1656.
To the Editor: I have the following comments and questions regarding the excellent Medical Grand Rounds article on Parkinson disease by Dr. Fernandez in your January 2012 issue.1
The author mentions that when “cost may be of concern, levodopa is the preferred starting drug.”1 Generic versions of pramipexole and ropinirole are now available and have made these medications more affordable. For example, the cash price of generic ropinirole 5 mg was recently $66 for 100 tablets, comparable with generic carbidopa/levodopa (25/100 mg priced at $46 for 100 tablets.2 And even though the price of generic pramipexole was $240 for 90 tablets, seniors with Medicare Part D drug coverage can usually get any generic medication for a low copay.
When choosing a dopamine agonist, how does Dr. Fernandez decide between ropinirole and pramipexole (aside from the price difference noted above)? Pramipexole has a longer elimination half-life (8 to 12 hours) compared with ropinirole (6 hours).3 Does this imply a significantly longer effective dosing interval for pramipexole? Are there other significant clinical differences between these agents?
Isradipine (DynaCirc CR), a dihydropyridine calcium channel blocker, has shown promise as a neuroprotective agent for slowing the progression of Parkinson disease in epidemiologic and laboratory studies, as noted by the author. In addition, immediate-release isradipine, with its relatively short elimination half-life of 8 hours,3 may be well suited for treating Parkinson patients whose essential hypertension is complicated by episodes of orthostatic hypotension. It should be noted that dihydropyridines that do not cross the blood-brain barrier (such as amlodipine [Norvasc]) have shown no evidence of neuroprotection.
Ibuprofen is another drug that has fairly strong epidemiologic and laboratory evidence that it might be neuroprotective,4 although the other nonsteroidal anti-inflammatory drugs (NSAIDs) have proven disappointing as a class.5 Lacking any prospective randomized trials, the evidence is not strong enough to recommend ibuprofen solely for neuroprotection. Does Dr. Fernandez, however, consider it reasonable to suggest ibuprofen to Parkinson patients who need to take an NSAID for an approved indication (such as pain)?
Dexpramipexole has recently demonstrated great promise in a phase 3 clinical trial as a neuroprotective agent in amyotrophic lateral sclerosis.6 How does this compound relate to pramipexole, and does the author believe it may offer neuroprotection in other neurodegenerative diseases like Parkinson disease?
The author discusses the use of catechol-O-methyltransferase (COMT) inhibitors (such as Comtan and Tasmar) and the monoamine oxidase (MAO) type-B inhibitors rasagiline (Azilect) and selegiline (Eldepryl, Zelapar) for prolonging the effects of levodopa by slowing the breakdown of dopamine. However, it is important to note that it is contraindicated to prescribe both a COMT inhibitor and an MAO-B inhibitor, because these agents also inhibit the breakdown of other catecholamines and can lead to adrenergic crisis when taken concomitantly.
To the Editor: I have the following comments and questions regarding the excellent Medical Grand Rounds article on Parkinson disease by Dr. Fernandez in your January 2012 issue.1
The author mentions that when “cost may be of concern, levodopa is the preferred starting drug.”1 Generic versions of pramipexole and ropinirole are now available and have made these medications more affordable. For example, the cash price of generic ropinirole 5 mg was recently $66 for 100 tablets, comparable with generic carbidopa/levodopa (25/100 mg priced at $46 for 100 tablets.2 And even though the price of generic pramipexole was $240 for 90 tablets, seniors with Medicare Part D drug coverage can usually get any generic medication for a low copay.
When choosing a dopamine agonist, how does Dr. Fernandez decide between ropinirole and pramipexole (aside from the price difference noted above)? Pramipexole has a longer elimination half-life (8 to 12 hours) compared with ropinirole (6 hours).3 Does this imply a significantly longer effective dosing interval for pramipexole? Are there other significant clinical differences between these agents?
Isradipine (DynaCirc CR), a dihydropyridine calcium channel blocker, has shown promise as a neuroprotective agent for slowing the progression of Parkinson disease in epidemiologic and laboratory studies, as noted by the author. In addition, immediate-release isradipine, with its relatively short elimination half-life of 8 hours,3 may be well suited for treating Parkinson patients whose essential hypertension is complicated by episodes of orthostatic hypotension. It should be noted that dihydropyridines that do not cross the blood-brain barrier (such as amlodipine [Norvasc]) have shown no evidence of neuroprotection.
Ibuprofen is another drug that has fairly strong epidemiologic and laboratory evidence that it might be neuroprotective,4 although the other nonsteroidal anti-inflammatory drugs (NSAIDs) have proven disappointing as a class.5 Lacking any prospective randomized trials, the evidence is not strong enough to recommend ibuprofen solely for neuroprotection. Does Dr. Fernandez, however, consider it reasonable to suggest ibuprofen to Parkinson patients who need to take an NSAID for an approved indication (such as pain)?
Dexpramipexole has recently demonstrated great promise in a phase 3 clinical trial as a neuroprotective agent in amyotrophic lateral sclerosis.6 How does this compound relate to pramipexole, and does the author believe it may offer neuroprotection in other neurodegenerative diseases like Parkinson disease?
The author discusses the use of catechol-O-methyltransferase (COMT) inhibitors (such as Comtan and Tasmar) and the monoamine oxidase (MAO) type-B inhibitors rasagiline (Azilect) and selegiline (Eldepryl, Zelapar) for prolonging the effects of levodopa by slowing the breakdown of dopamine. However, it is important to note that it is contraindicated to prescribe both a COMT inhibitor and an MAO-B inhibitor, because these agents also inhibit the breakdown of other catecholamines and can lead to adrenergic crisis when taken concomitantly.
- Fernandez HH. Updates in the medical management of Parkinson disease. Cleve Clin J Med 2012; 79:28–35.
- Drugstore.com. www.Drugstore.com. Accessed February 5, 2012.
- PDR.net. www.PDR.net. Accessed February 25, 2012.
- Gao X, Chen H, Schwarzschild MA, Ascherio A. Use of ibuprofen and risk of Parkinson disease. Neurology 2011; 76:863–869.
- Driver JA, Logroscino G, Lu L, Gaziano JM, Kurth T. Use of non-steroidal anti-inflammatory drugs and risk of Parkinson’s disease: nested case-control study. BMJ 2011; 342:d198.
- Cudkowicz M, Bozik ME, Ingersoll EW, et al. The effects of dexpramipexole (KNS-760704) in individuals with amyotrophic lateral sclerosis. Nat Med 2011; 17:1652–1656.
- Fernandez HH. Updates in the medical management of Parkinson disease. Cleve Clin J Med 2012; 79:28–35.
- Drugstore.com. www.Drugstore.com. Accessed February 5, 2012.
- PDR.net. www.PDR.net. Accessed February 25, 2012.
- Gao X, Chen H, Schwarzschild MA, Ascherio A. Use of ibuprofen and risk of Parkinson disease. Neurology 2011; 76:863–869.
- Driver JA, Logroscino G, Lu L, Gaziano JM, Kurth T. Use of non-steroidal anti-inflammatory drugs and risk of Parkinson’s disease: nested case-control study. BMJ 2011; 342:d198.
- Cudkowicz M, Bozik ME, Ingersoll EW, et al. The effects of dexpramipexole (KNS-760704) in individuals with amyotrophic lateral sclerosis. Nat Med 2011; 17:1652–1656.
In reply: Essential tremor, beta-blockers, calcium channel blockers
In Reply: We agree and thank Dr. Keller for raising this valid point. The two classes of calcium channel blockers are distinct in their actions, and the warning about not combining a calcium channel blocker with a beta-blocker because of the increased risk of developing significant bradycardia applies only to the nondihydropyridine class.
In Reply: We agree and thank Dr. Keller for raising this valid point. The two classes of calcium channel blockers are distinct in their actions, and the warning about not combining a calcium channel blocker with a beta-blocker because of the increased risk of developing significant bradycardia applies only to the nondihydropyridine class.
In Reply: We agree and thank Dr. Keller for raising this valid point. The two classes of calcium channel blockers are distinct in their actions, and the warning about not combining a calcium channel blocker with a beta-blocker because of the increased risk of developing significant bradycardia applies only to the nondihydropyridine class.
Essential tremor, beta-blockers, and calcium channel blockers
To the Editor: In their thorough review of essential tremor,1Drs. Abboud, Ahmed, and Fernandez make a statement that needs clarification. In their list of absolute contraindications to propranolol (Inderal), the authors include “concurrent use of a calcium channel blocker.” This warning applies only to the nondihydropyridine calcium channel blockers, which are diltiazem (Cardizem) and verapamil (Calan). These two medications slow the heart rate and generally should not be combined with beta-blockers such as propranolol unless the patient requires this combination to control tachycardia. Most calcium channel blockers are dihydropyridines, which include amlodipine (Norvasc), nifedipine (Procardia), felodipine (Plendil), nisoldipine (Sular), isradipine (DynaCirc CR), and nicardipine (Cardene). These agents do not slow the heart rate significantly and therefore can be used freely in combination with propranolol. Of course, the dose of the calcium channel blocker may need to be decreased because of the antihypertensive effect of propranolol.
- Abboud H, Ahmed A, Fernandez HH. Essential tremor: choosing the right management plan for your patient. Cleve Clin J Med 2011; 78:821–828.
To the Editor: In their thorough review of essential tremor,1Drs. Abboud, Ahmed, and Fernandez make a statement that needs clarification. In their list of absolute contraindications to propranolol (Inderal), the authors include “concurrent use of a calcium channel blocker.” This warning applies only to the nondihydropyridine calcium channel blockers, which are diltiazem (Cardizem) and verapamil (Calan). These two medications slow the heart rate and generally should not be combined with beta-blockers such as propranolol unless the patient requires this combination to control tachycardia. Most calcium channel blockers are dihydropyridines, which include amlodipine (Norvasc), nifedipine (Procardia), felodipine (Plendil), nisoldipine (Sular), isradipine (DynaCirc CR), and nicardipine (Cardene). These agents do not slow the heart rate significantly and therefore can be used freely in combination with propranolol. Of course, the dose of the calcium channel blocker may need to be decreased because of the antihypertensive effect of propranolol.
To the Editor: In their thorough review of essential tremor,1Drs. Abboud, Ahmed, and Fernandez make a statement that needs clarification. In their list of absolute contraindications to propranolol (Inderal), the authors include “concurrent use of a calcium channel blocker.” This warning applies only to the nondihydropyridine calcium channel blockers, which are diltiazem (Cardizem) and verapamil (Calan). These two medications slow the heart rate and generally should not be combined with beta-blockers such as propranolol unless the patient requires this combination to control tachycardia. Most calcium channel blockers are dihydropyridines, which include amlodipine (Norvasc), nifedipine (Procardia), felodipine (Plendil), nisoldipine (Sular), isradipine (DynaCirc CR), and nicardipine (Cardene). These agents do not slow the heart rate significantly and therefore can be used freely in combination with propranolol. Of course, the dose of the calcium channel blocker may need to be decreased because of the antihypertensive effect of propranolol.
- Abboud H, Ahmed A, Fernandez HH. Essential tremor: choosing the right management plan for your patient. Cleve Clin J Med 2011; 78:821–828.
- Abboud H, Ahmed A, Fernandez HH. Essential tremor: choosing the right management plan for your patient. Cleve Clin J Med 2011; 78:821–828.
In reply: Glucocorticoid-induced diabetes and adrenal suppression
In Reply: We thank Drs. Rodríguez-Gutiérrez and Gonzálvez-Gonzálvez and Dr. Keller for their thoughtful comments.
In our paper, we did not elaborate on the low-dose cosyntropin stimulation test. The 1-μg test, in particular, has been shown to have similar or better sensitivity, with similar or lower specificity, compared with the 250-μg dose, depending on the study design. Unfortunately, the administration of the 1-μg dose presents more technical difficulty than the 250-μg dose, thus limiting its use. Cosyntropin (used in the United States) comes in a vial with 250 μg of powder. This must be reconstituted with 250 mL of normal saline, and only 1 mL is to be given. Adherence to the plastic tubing may occur, and more precise timing is needed as the cortisol levels may decrease.1–3
Responding to Dr. Keller, we were unable to find any systematic reviews comparing inhaled corticosteroids that have a “higher therapeutic index” as a class vs older inhaled corticosteroids. There are several studies, however, comparing individual inhaled corticosteroid preparations with each other in terms of adrenal effects, and we feel that it is beyond the scope of this response to perform a systematic analysis. In addition, the determination of adrenal function used in studies comparing one inhaled corticosteroid with another were varied, including cosyntropin stimulation tests and surrogates such as the urinary cortisolcreatinine ratio, a morning plasma cortisol level less than 5 μg/L, and serum cortisol concentration curves, preventing more definitive conclusions even if the data were to be pooled.4–6 A double-blind, randomized study comparing the adrenal effects of ciclesonide and fluticasone showed a smaller reduction in the peak serum cortisol level achieved with ciclesonide compared with fluticasone, in both low-dose and high-dose cosyntropin stimulation tests, with the results in the ciclesonide group being similar to placebo.7 However, the mean peak serum cortisol levels after exposure to these inhaled corticosteroids were not presented in table format, and the results have to be inferred from the figures and the narrative description of the baseline mean peak cortisol levels8 (ie, before exposure to these inhaled corticosteroids). Case reports have suggested that changing the inhaled corticostseroid formulation from fluticasone to ciclesonide allowed for improvement of adrenal function.8 The purported mechanism of decreased adrenal effects of ciclesonide is its greater deposition in the lungs and, hence, less entry into the systemic circulation and fewer systemic adverse effects.9
- Dorin RI, Qualls CR, Crapo LM. Diagnosis of adrenalin sufficiency. Ann Intern Med 2003; 139:194–204.
- Dickstein G. High-dose and low-dose cosyntropin stimulation tests for diagnosis of adrenal insufficiency. Ann Intern Med 2004; 140:312–314.
- Rose SR, Lustig RH, Burstein S, Pitukcheewanont P, Broome DC, Burthen GA. Diagnosis of ACTH deficiency. Comparison of overnight metyrapone test to either low-dose or high-dose ACTH test. Horm Res 1999; 52:73–79.
- Chrousos GP, Ghaly L, Shedden A, Iezzoni DG, Harris AG. Effects of mometasone furoate dry powder inhaler and beclomethasone dipropionate hydrofluoroalkane and chlorofluorocarbon on the hypothalamic-pituitary-adrenal axis in asthmatic subjects. Chest 2005; 128:70–77.
- White M, Crisalida T, Li H, Economides A, Kaliner M. Effects of long-term inhaled corticosteroids on adrenal function in asthmatics. Ann Allergy Asthma Immunol 2006; 96:437–444.
- Fardon TC, Lee DK, Haggart K, McFarlane LC, Lipworth BJ. Adrenal suppression with dry powder formulations of fluticasone propionate and mometasone furoate. Am J Respir Crit Care Med 2004; 170:960–966.
- Lipworth BJ, Kaliner MA, LaForde CF, et al. Effects of ciclesonide and fluticasone on hypothalamic-pituitary-adrenal axis function in adults with mild-to-moderate persistent asthma. Ann Allergy Asthma Immunol 2005; 94:465–472.
- Heller MK, Laks J, Kovesi TA, Ahmet A. Reversal of adrenal suppression with ciclesonide. J Asthma 2010; 47:337–339.
- Kaliner MA. Pharmacologic characteristics and adrenal suppression with newer inhaled corticosteroids: a comparison of ciclesonide and fluticasone propionate. Clin Ther 2006; 28:319–3.
In Reply: We thank Drs. Rodríguez-Gutiérrez and Gonzálvez-Gonzálvez and Dr. Keller for their thoughtful comments.
In our paper, we did not elaborate on the low-dose cosyntropin stimulation test. The 1-μg test, in particular, has been shown to have similar or better sensitivity, with similar or lower specificity, compared with the 250-μg dose, depending on the study design. Unfortunately, the administration of the 1-μg dose presents more technical difficulty than the 250-μg dose, thus limiting its use. Cosyntropin (used in the United States) comes in a vial with 250 μg of powder. This must be reconstituted with 250 mL of normal saline, and only 1 mL is to be given. Adherence to the plastic tubing may occur, and more precise timing is needed as the cortisol levels may decrease.1–3
Responding to Dr. Keller, we were unable to find any systematic reviews comparing inhaled corticosteroids that have a “higher therapeutic index” as a class vs older inhaled corticosteroids. There are several studies, however, comparing individual inhaled corticosteroid preparations with each other in terms of adrenal effects, and we feel that it is beyond the scope of this response to perform a systematic analysis. In addition, the determination of adrenal function used in studies comparing one inhaled corticosteroid with another were varied, including cosyntropin stimulation tests and surrogates such as the urinary cortisolcreatinine ratio, a morning plasma cortisol level less than 5 μg/L, and serum cortisol concentration curves, preventing more definitive conclusions even if the data were to be pooled.4–6 A double-blind, randomized study comparing the adrenal effects of ciclesonide and fluticasone showed a smaller reduction in the peak serum cortisol level achieved with ciclesonide compared with fluticasone, in both low-dose and high-dose cosyntropin stimulation tests, with the results in the ciclesonide group being similar to placebo.7 However, the mean peak serum cortisol levels after exposure to these inhaled corticosteroids were not presented in table format, and the results have to be inferred from the figures and the narrative description of the baseline mean peak cortisol levels8 (ie, before exposure to these inhaled corticosteroids). Case reports have suggested that changing the inhaled corticostseroid formulation from fluticasone to ciclesonide allowed for improvement of adrenal function.8 The purported mechanism of decreased adrenal effects of ciclesonide is its greater deposition in the lungs and, hence, less entry into the systemic circulation and fewer systemic adverse effects.9
In Reply: We thank Drs. Rodríguez-Gutiérrez and Gonzálvez-Gonzálvez and Dr. Keller for their thoughtful comments.
In our paper, we did not elaborate on the low-dose cosyntropin stimulation test. The 1-μg test, in particular, has been shown to have similar or better sensitivity, with similar or lower specificity, compared with the 250-μg dose, depending on the study design. Unfortunately, the administration of the 1-μg dose presents more technical difficulty than the 250-μg dose, thus limiting its use. Cosyntropin (used in the United States) comes in a vial with 250 μg of powder. This must be reconstituted with 250 mL of normal saline, and only 1 mL is to be given. Adherence to the plastic tubing may occur, and more precise timing is needed as the cortisol levels may decrease.1–3
Responding to Dr. Keller, we were unable to find any systematic reviews comparing inhaled corticosteroids that have a “higher therapeutic index” as a class vs older inhaled corticosteroids. There are several studies, however, comparing individual inhaled corticosteroid preparations with each other in terms of adrenal effects, and we feel that it is beyond the scope of this response to perform a systematic analysis. In addition, the determination of adrenal function used in studies comparing one inhaled corticosteroid with another were varied, including cosyntropin stimulation tests and surrogates such as the urinary cortisolcreatinine ratio, a morning plasma cortisol level less than 5 μg/L, and serum cortisol concentration curves, preventing more definitive conclusions even if the data were to be pooled.4–6 A double-blind, randomized study comparing the adrenal effects of ciclesonide and fluticasone showed a smaller reduction in the peak serum cortisol level achieved with ciclesonide compared with fluticasone, in both low-dose and high-dose cosyntropin stimulation tests, with the results in the ciclesonide group being similar to placebo.7 However, the mean peak serum cortisol levels after exposure to these inhaled corticosteroids were not presented in table format, and the results have to be inferred from the figures and the narrative description of the baseline mean peak cortisol levels8 (ie, before exposure to these inhaled corticosteroids). Case reports have suggested that changing the inhaled corticostseroid formulation from fluticasone to ciclesonide allowed for improvement of adrenal function.8 The purported mechanism of decreased adrenal effects of ciclesonide is its greater deposition in the lungs and, hence, less entry into the systemic circulation and fewer systemic adverse effects.9
- Dorin RI, Qualls CR, Crapo LM. Diagnosis of adrenalin sufficiency. Ann Intern Med 2003; 139:194–204.
- Dickstein G. High-dose and low-dose cosyntropin stimulation tests for diagnosis of adrenal insufficiency. Ann Intern Med 2004; 140:312–314.
- Rose SR, Lustig RH, Burstein S, Pitukcheewanont P, Broome DC, Burthen GA. Diagnosis of ACTH deficiency. Comparison of overnight metyrapone test to either low-dose or high-dose ACTH test. Horm Res 1999; 52:73–79.
- Chrousos GP, Ghaly L, Shedden A, Iezzoni DG, Harris AG. Effects of mometasone furoate dry powder inhaler and beclomethasone dipropionate hydrofluoroalkane and chlorofluorocarbon on the hypothalamic-pituitary-adrenal axis in asthmatic subjects. Chest 2005; 128:70–77.
- White M, Crisalida T, Li H, Economides A, Kaliner M. Effects of long-term inhaled corticosteroids on adrenal function in asthmatics. Ann Allergy Asthma Immunol 2006; 96:437–444.
- Fardon TC, Lee DK, Haggart K, McFarlane LC, Lipworth BJ. Adrenal suppression with dry powder formulations of fluticasone propionate and mometasone furoate. Am J Respir Crit Care Med 2004; 170:960–966.
- Lipworth BJ, Kaliner MA, LaForde CF, et al. Effects of ciclesonide and fluticasone on hypothalamic-pituitary-adrenal axis function in adults with mild-to-moderate persistent asthma. Ann Allergy Asthma Immunol 2005; 94:465–472.
- Heller MK, Laks J, Kovesi TA, Ahmet A. Reversal of adrenal suppression with ciclesonide. J Asthma 2010; 47:337–339.
- Kaliner MA. Pharmacologic characteristics and adrenal suppression with newer inhaled corticosteroids: a comparison of ciclesonide and fluticasone propionate. Clin Ther 2006; 28:319–3.
- Dorin RI, Qualls CR, Crapo LM. Diagnosis of adrenalin sufficiency. Ann Intern Med 2003; 139:194–204.
- Dickstein G. High-dose and low-dose cosyntropin stimulation tests for diagnosis of adrenal insufficiency. Ann Intern Med 2004; 140:312–314.
- Rose SR, Lustig RH, Burstein S, Pitukcheewanont P, Broome DC, Burthen GA. Diagnosis of ACTH deficiency. Comparison of overnight metyrapone test to either low-dose or high-dose ACTH test. Horm Res 1999; 52:73–79.
- Chrousos GP, Ghaly L, Shedden A, Iezzoni DG, Harris AG. Effects of mometasone furoate dry powder inhaler and beclomethasone dipropionate hydrofluoroalkane and chlorofluorocarbon on the hypothalamic-pituitary-adrenal axis in asthmatic subjects. Chest 2005; 128:70–77.
- White M, Crisalida T, Li H, Economides A, Kaliner M. Effects of long-term inhaled corticosteroids on adrenal function in asthmatics. Ann Allergy Asthma Immunol 2006; 96:437–444.
- Fardon TC, Lee DK, Haggart K, McFarlane LC, Lipworth BJ. Adrenal suppression with dry powder formulations of fluticasone propionate and mometasone furoate. Am J Respir Crit Care Med 2004; 170:960–966.
- Lipworth BJ, Kaliner MA, LaForde CF, et al. Effects of ciclesonide and fluticasone on hypothalamic-pituitary-adrenal axis function in adults with mild-to-moderate persistent asthma. Ann Allergy Asthma Immunol 2005; 94:465–472.
- Heller MK, Laks J, Kovesi TA, Ahmet A. Reversal of adrenal suppression with ciclesonide. J Asthma 2010; 47:337–339.
- Kaliner MA. Pharmacologic characteristics and adrenal suppression with newer inhaled corticosteroids: a comparison of ciclesonide and fluticasone propionate. Clin Ther 2006; 28:319–3.
Glucocorticoid-induced diabetes and adrenal suppression
To the Editor: Drs. Lansang and Hustak1 provide a comprehensive and useful review of steroid-induced diabetes and adrenal suppression.
In their section on local steroids, they discuss the side effects of topical and inhaled glucocorticosteroids. Much has been made of the fact that certain steroids, such as mometasone (Elocon, Nasonex) and fluticasone (Flonase), have a higher “therapeutic index” or ratio of local anti-inflammatory effect to systemic side effects, due to extensive hepatic first-pass metabolism, than older agents such as beclomethasone (Qvar) and betamethasone (Diprosone).2 Ciclesonide (Alvesco, Omnaris), a newer inhaled steroid, is said to have an enhanced therapeutic index because it is a prodrug that is activated by metabolism in the lungs; it reportedly has an even less suppressive effect on hypothalamic-pituitaryadrenal axis function.3
Are the authors aware of any other evidence that clinical outcome, such as adrenal suppression or hyperglycemia, is improved by the use of steroids with a higher therapeutic index?
- Lansang MC, Hustak LK. Glucocorticoid-induced diabetes and adrenal suppression: how to detect and manage them. Cleve Clin J Med 2011; 78:748–756.
- Drug Bank. Mometasone. http://www.drugbank.ca/drugs/DB00764. Accessed February 17, 2012.
- Derom E, Louis R, Tiesler C, Engelsätter R, Kaufman JM, Joos GF. Effects of ciclesonide and fluticasone on cortisol secretion in patients with persistent asthma. Eur Respir J 2009; 33:1277–1286.
To the Editor: Drs. Lansang and Hustak1 provide a comprehensive and useful review of steroid-induced diabetes and adrenal suppression.
In their section on local steroids, they discuss the side effects of topical and inhaled glucocorticosteroids. Much has been made of the fact that certain steroids, such as mometasone (Elocon, Nasonex) and fluticasone (Flonase), have a higher “therapeutic index” or ratio of local anti-inflammatory effect to systemic side effects, due to extensive hepatic first-pass metabolism, than older agents such as beclomethasone (Qvar) and betamethasone (Diprosone).2 Ciclesonide (Alvesco, Omnaris), a newer inhaled steroid, is said to have an enhanced therapeutic index because it is a prodrug that is activated by metabolism in the lungs; it reportedly has an even less suppressive effect on hypothalamic-pituitaryadrenal axis function.3
Are the authors aware of any other evidence that clinical outcome, such as adrenal suppression or hyperglycemia, is improved by the use of steroids with a higher therapeutic index?
To the Editor: Drs. Lansang and Hustak1 provide a comprehensive and useful review of steroid-induced diabetes and adrenal suppression.
In their section on local steroids, they discuss the side effects of topical and inhaled glucocorticosteroids. Much has been made of the fact that certain steroids, such as mometasone (Elocon, Nasonex) and fluticasone (Flonase), have a higher “therapeutic index” or ratio of local anti-inflammatory effect to systemic side effects, due to extensive hepatic first-pass metabolism, than older agents such as beclomethasone (Qvar) and betamethasone (Diprosone).2 Ciclesonide (Alvesco, Omnaris), a newer inhaled steroid, is said to have an enhanced therapeutic index because it is a prodrug that is activated by metabolism in the lungs; it reportedly has an even less suppressive effect on hypothalamic-pituitaryadrenal axis function.3
Are the authors aware of any other evidence that clinical outcome, such as adrenal suppression or hyperglycemia, is improved by the use of steroids with a higher therapeutic index?
- Lansang MC, Hustak LK. Glucocorticoid-induced diabetes and adrenal suppression: how to detect and manage them. Cleve Clin J Med 2011; 78:748–756.
- Drug Bank. Mometasone. http://www.drugbank.ca/drugs/DB00764. Accessed February 17, 2012.
- Derom E, Louis R, Tiesler C, Engelsätter R, Kaufman JM, Joos GF. Effects of ciclesonide and fluticasone on cortisol secretion in patients with persistent asthma. Eur Respir J 2009; 33:1277–1286.
- Lansang MC, Hustak LK. Glucocorticoid-induced diabetes and adrenal suppression: how to detect and manage them. Cleve Clin J Med 2011; 78:748–756.
- Drug Bank. Mometasone. http://www.drugbank.ca/drugs/DB00764. Accessed February 17, 2012.
- Derom E, Louis R, Tiesler C, Engelsätter R, Kaufman JM, Joos GF. Effects of ciclesonide and fluticasone on cortisol secretion in patients with persistent asthma. Eur Respir J 2009; 33:1277–1286.