User login
Cutaneous Angiosarcoma of the Lower Leg
Angiosarcoma is a rare and aggressive vascular malignant neoplasm derived from endothelial cells. In general, sarcomas account for approximately 1% of all malignancies, with approximately 2% being angiosarcomas.1 The risk of recurrence at 5 years is estimated to be 84%, and 5-year survival is estimated at 15% to 30%. Poor prognostic factors for angiosarcoma include large tumor size, depth of invasion greater than 3 mm, high mitotic rate, positive surgical margins, and metastasis.2 Approximately 20% to 40% of patients who are diagnosed with angiosarcoma already have distant metastasis, contributing to the aggressive nature of this neoplasm.3
Angiosarcoma can affect various anatomic locations, including the skin, soft tissue, breasts, and liver. Cutaneous angiosarcoma is the most common clinical manifestation, accounting for approximately 50% to 60% of all cases, and typically is known to occur in 3 distinct settings.2 Primary or idiopathic cutaneous angiosarcoma is most commonly seen in elderly individuals, with a peak incidence in the seventh to eighth decades of life, and presents as a bruiselike lesion predominantly on the head and neck. Angiosarcoma also is seen clinically in patients exposed to radiation treatment, with a median onset of symptoms occurring 5 to 10 years posttreatment, and in patients with chronic lymphedema, usually on the arm following radical mastectomy, which also is known as Stewart-Treves syndrome.2
With any sarcoma, treatment typically first involves surgical excision; however, there is no direct approach for treatment of cutaneous angiosarcoma, as an individual plan typically is needed for each patient. Treatment options include surgical excision, radiation, chemotherapy, or a combination of these therapies.2,4
We present a rare case of cutaneous angiosarcoma of the left leg in the setting of chronic venous insufficiency with some degree of lymphedema and a nonhealing ulcer. This case is unique in that it does not fit the classic presentation of cutaneous angiosarcoma previously described.
Case Report
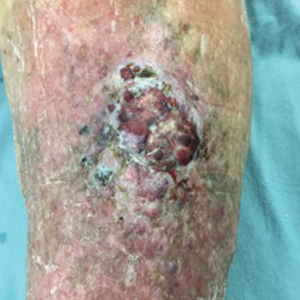
An 83-year-old woman with a medical history of advanced dementia, congestive heart failure, chronic obstructive pulmonary disease, chronic kidney disease, type 2 diabetes mellitus, hypertension, and chronic venous insufficiency with stasis dermatitis presented to the emergency department following a mechanical fall. Most of her medical history was obtained from the patient’s family. She had a history of multiple falls originally thought to be related to a chronic leg ulcer that had been managed with wound care. Recently, however, the lesion was noted to have increasing erythema surrounding the wound margins. An 8×8-cm erythematous plaque on the anterior lateral left leg with a firm central nodule with hemorrhagic crust that measured approximately 4 cm in diameter was noted by the emergency department physicians (Figure 1). In the emergency department, vitals and other laboratory values were within reference range, and a radiograph of the left tibia/fibula was unremarkable. Cellulitis initially was considered in the emergency department and cephalexin was started; however, since the patient was afebrile and had no leukocytosis, plastic surgery also was consulted. Biopsies were obtained from the superior and inferior parts of the lesion. Histologic analysis revealed a poorly differentiated vascular neoplasm of epithelioid endothelial cells with considerable cell atypia that extended through the entirety of the dermis (Figure 2). The tumor cells stained positive with vimentin and CD34. Pathology noted no immunohistochemistry stains to synaptophysin, S-100, human melanoma black 45, MART-1, CK20, CK7, CK8/18, CK5/6, and p63. The pathologic diagnosis was consistent with cutaneous angiosarcoma. Computed tomography of the chest, abdomen, and pelvis revealed no local or distant metastases.

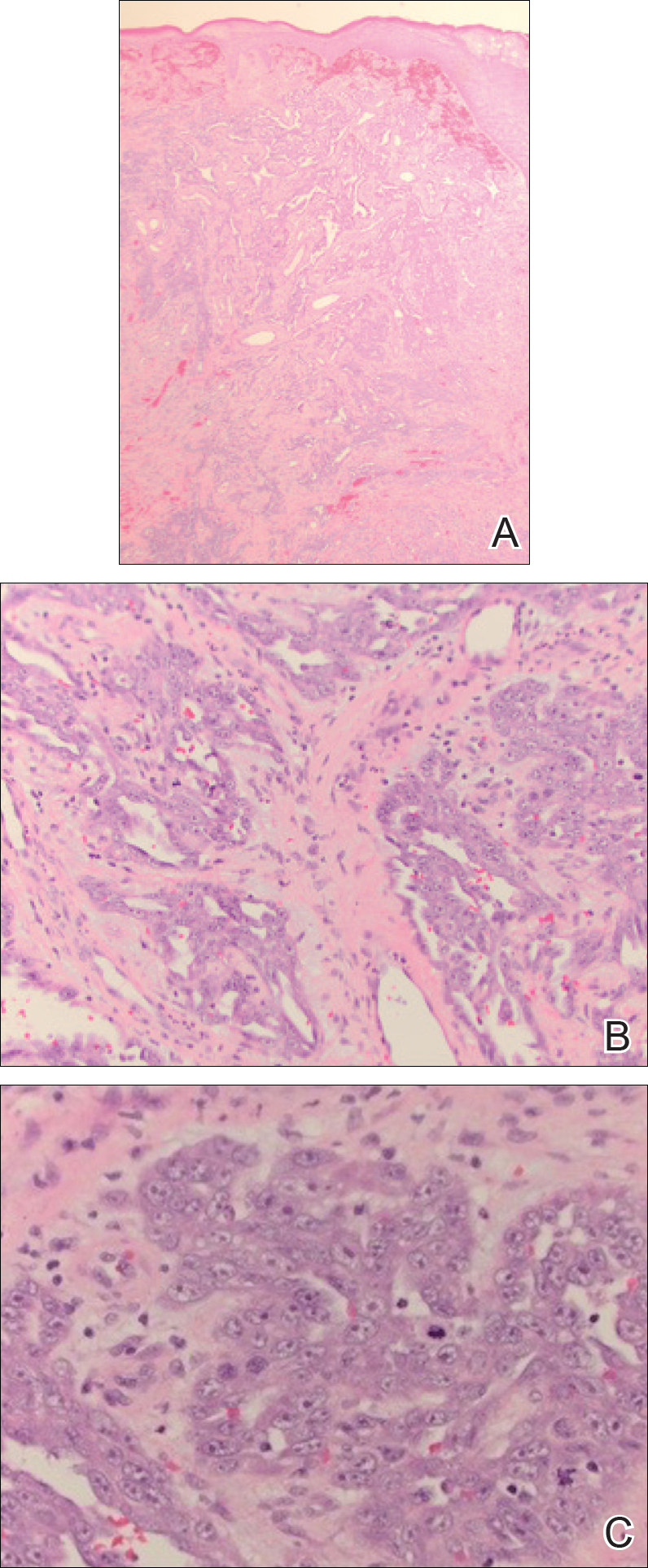
A wide excision of the cutaneous angiosarcoma was performed. The initial frozen section analysis revealed positive margins. Three additional excisions still showed positive margins, and further excision was held after obtaining family consent due to the extensive nature of the neoplasm and lengthy operating room time. The final defect after excision measured 15×10×2.5 cm (Figure 3A), and subsequent application of a split-thickness graft was performed. Additional treatment options were discussed with the family, including radiation therapy, amputation of the left lower leg, or no treatment. The family opted not to proceed with further treatment. The graft healed without signs of reoccurrence approximately 3 months later (Figure 3B), and the patient received physical therapy, which allowed her to gain strength and some independence.

Comment
Clinical Manifestation
Cutaneous angiosarcoma is a rare malignant vascular neoplasm that when clinically diagnosed is typically seen in 3 settings: (1) idiopathic (commonly on the face and neck), (2) following radiation treatment, and (3) classically following mastectomy with subsequent chronic lymphedema. Our patient did not classically fit these settings of cutaneous angiosarcoma due to the location of the lesion on the lower leg as well as its occurrence in the setting of a chronic nonhealing ulcer and lymphedema.
Chronic lymphedema is a common clinical manifestation that is likely secondary to other medical conditions, such as in our patient. As a result, these patients are at increased risk for developing chronic ulcers due to poor wound healing; however, as seen in our patient, chronic nonhealing ulcers require a broad differential because they may clinically mimic many processes. Patient history and visual presentation were crucial in this case because a biopsy was obtained that ultimately led to the patient’s diagnosis.
Differential Diagnosis
Initially, a venous ulcer secondary to chronic venous insufficiency was considered in the differential for our patient. She had a history of congestive heart failure, kidney disease, and type 2 diabetes mellitus, all of which contribute to lymphedema and/or poor wound healing. However, venous ulcers usually are located on the medial ankles and are irregularly shaped with an erythematous border and fibrinous exudate with central depression, making it a less likely diagnosis in our patient. Additionally, an infectious process was considered, but the patient was afebrile and laboratory values demonstrated no leukocytosis.
Marjolin ulcer was highly suspected because the clinical presentation revealed a nodule with hemorrhagic crust and induration in the setting of a chronic nonhealing ulcer. The pathogenesis of malignancy in chronic ulcers is thought to be due to continuous mitotic activity from regeneration and repair of the wound, especially in the setting of repeated trauma to the area.5 In our patient, the history of multiple falls with possible multitrauma injury to the chronic ulcer further increased suspicion of malignancy. The most common and frequently seen malignancy that develops in chronic ulcers is squamous cell carcinoma (SCC) followed by basal cell carcinoma. Plastic surgery suspected an SCC for the working diagnosis, which prompted a punch biopsy; however, the histologic analysis was not consistent with SCC or basal cell carcinoma. Marjolin ulcer also may demonstrate a periosteal reaction,5 which was not the case with our patient after a radiograph of the left tibia/fibula was unremarkable.
Another potential malignancy to consider is melanoma. There are few case reports of biopsy-proven melanoma from an enlarging chronic ulcer.6,7 Additionally, poorly differentiated angiosarcoma can mimic melanoma2; however, immunohistochemistry stain was negative for S-100, human melanoma black 45, and MART-1, making melanoma unlikely.
Kaposi sarcoma (KS) and angiosarcoma are both malignant vascular tumors that similarly present with red to purple patches, plaques, or nodules, making it difficult to distinguish between the two conditions. It is important to note that KS usually is lower grade, and the pathogenesis is linked to human herpesvirus 8, which can be identified on immunohistochemistry staining. There have been cases of KS reported in patients who have no history of human immunodeficiency virus/AIDS, thus the classic subtype of KS may have been considered in this patient.8 The histologic appearance of KS may vary from dilated irregular endothelial cells lining the vascular space to mild endothelial cell atypia. Histology also shows hemosiderin-laden macrophages, extravasated red blood cells, and an inflammatory infiltrate. An additional malignant vascular neoplasm that needs to be differentiated is epithelioid hemangioendothelioma. Cutaneous presentation of an epithelioid hemangioendothelioma may be similar to what was seen in our patient but histologically will usually show neoplastic cells with pale eosinophilic cytoplasm and vesicular nuclei of plump, oval, polygonal cells in cords or aggregates surrounding vascular channels. These neoplasms also tend to occur around medium- to large-sized veins.1,9 With our patient, even though human herpesvirus 8 was not tested with immunohistochemistry, gold standard immunohistochemistry confirmation with CD34 and vimentin staining combined with poorly differentiated endothelial atypia with mitotic figures on histologic analysis favored angiosarcoma versus KS or epithelioid hemangioendothelioma.10,11
Management
Cutaneous angiosarcoma is a rare and aggressive vascular neoplasm accounting for approximately 2% of all combined sarcomas, with an estimated 20% to 40% having distant metastasis at diagnosis.1,3 For this reason, computed tomography was performed in our patient and revealed no local or distant metastasis. Therefore, chemotherapy was not an appropriate adjuvant treatment option.12 With no evidence of metastasis, initial treatment began with surgical removal but proved to be difficult in our patient. Although the implications of positive surgical margins remain unclear with regard to overall patient survival, surgical resection followed by radiation therapy has been shown to be optimal, as it reduces the risk of local reoccurrence.3 There have been reported cases of cutaneous angiosarcoma of the leg that were treated with amputation without signs of reoccurrence or metastasis.10,13,14 Given the results from these cases and considering that our patient had no metastasis, amputation seemed to be a good prognostic option; however, considering other factors regarding the patient’s comorbidities and quality of life, her family decided not to pursue any further treatment with amputation or radiation therapy.
Conclusion
There should be low threshold for biopsy in patients who present with nonhealing wounds that do not progress in the normal phase of wound healing with suspicion for malignancy. As seen with our patient, cutaneous angiosarcoma can clinically mimic many disease processes, and although rare in nature, it should always be considered when a patient presents with a rapidly growing lesion in the setting of chronic lymphedema or venous ulcer.
- Kumar V, Abbas A, Aster J. Robbins Basic Pathology. 9th ed. Philadelphia, PA: Elsevier Saunders; 2013.
- Donghi D, Kerl K, Dummer R, et al. Cutaneous angiosarcoma: own experience over 13 years. clinical features, disease course and immunohistochemical profile. J Eur Acad Dermatol Venereol. 2010;24:1230-1234.
- Dossett LA, Harrington M, Cruse CW, et al. Cutaneous angiosarcoma. Curr Probl Cancer. 2015;39:258-263.
- Morgan MB, Swann M, Somach S, et al. Cutaneous angiosarcoma: a case series with prognostic correlation. J Am Acad Dermatol. 2004;50:867-874.
- Pekarek B, Buck S, Osher L. A comprehensive review on Marjolin’s ulcers: diagnosis and treatment. J Am Col Certif Wound Spec. 2011;3:60-64.
- Gerslova A, Pokorna A, Stukavcova A, et al. Rare cause of non-healing foot wound—acral lentiginous melanoma. Neuro Endocrinol Lett. 2012;37:12-17.
- Turk BG, Bozkurt A, Yaman B, et al. Melanoma arising in chronic ulceration associated with lymphoedema. J Wound Care. 2013;22:74-75.
- Phavixay L, Raynolds D, Simman R. Non AIDS Kaposi’s sarcoma leading to lower extremities wounds, case presentations and discussion.J Am Coll Clin Wound Spec. 2012;4:13-15.
- Requena L, Kutzner H. Hemangioendothelioma. Semin Diagn Pathol. 2013;30:29-44.
- Harrison WD, Chandrasekar CR. Stewart-Treves syndrome following idiopathic leg lymphoedema: remember sarcoma. J Wound Care. 2015;24(6 suppl):S5-S7.
- Kak I, Salama S, Gohla G, et al. A case of patch stage of Kaposi’s sarcoma and discussion of the differential diagnosis. Rare Tumors. 2016;8:6123.
- Agulnik M, Yarber JL, Okuno SH, et al. An open-label, multicenter, phase II study of bevacizumab for the treatment of angiosarcoma and epithelioid hemangioendotheliomas. Ann Oncol. 2013;24:257-263.
- Linda DD, Harish S, Alowami S, et al. Radiology-pathology conference: cutaneous angiosarcoma of the leg. Clin Imaging. 2013;37:602-607.
- Roy P, Clark MA, Thomas JM. Stewart-Treves syndrome—treatment and outcome in six patients from a single centre. Eur J Surg Oncol. 2004;30:982-986.
Angiosarcoma is a rare and aggressive vascular malignant neoplasm derived from endothelial cells. In general, sarcomas account for approximately 1% of all malignancies, with approximately 2% being angiosarcomas.1 The risk of recurrence at 5 years is estimated to be 84%, and 5-year survival is estimated at 15% to 30%. Poor prognostic factors for angiosarcoma include large tumor size, depth of invasion greater than 3 mm, high mitotic rate, positive surgical margins, and metastasis.2 Approximately 20% to 40% of patients who are diagnosed with angiosarcoma already have distant metastasis, contributing to the aggressive nature of this neoplasm.3
Angiosarcoma can affect various anatomic locations, including the skin, soft tissue, breasts, and liver. Cutaneous angiosarcoma is the most common clinical manifestation, accounting for approximately 50% to 60% of all cases, and typically is known to occur in 3 distinct settings.2 Primary or idiopathic cutaneous angiosarcoma is most commonly seen in elderly individuals, with a peak incidence in the seventh to eighth decades of life, and presents as a bruiselike lesion predominantly on the head and neck. Angiosarcoma also is seen clinically in patients exposed to radiation treatment, with a median onset of symptoms occurring 5 to 10 years posttreatment, and in patients with chronic lymphedema, usually on the arm following radical mastectomy, which also is known as Stewart-Treves syndrome.2
With any sarcoma, treatment typically first involves surgical excision; however, there is no direct approach for treatment of cutaneous angiosarcoma, as an individual plan typically is needed for each patient. Treatment options include surgical excision, radiation, chemotherapy, or a combination of these therapies.2,4
We present a rare case of cutaneous angiosarcoma of the left leg in the setting of chronic venous insufficiency with some degree of lymphedema and a nonhealing ulcer. This case is unique in that it does not fit the classic presentation of cutaneous angiosarcoma previously described.
Case Report
An 83-year-old woman with a medical history of advanced dementia, congestive heart failure, chronic obstructive pulmonary disease, chronic kidney disease, type 2 diabetes mellitus, hypertension, and chronic venous insufficiency with stasis dermatitis presented to the emergency department following a mechanical fall. Most of her medical history was obtained from the patient’s family. She had a history of multiple falls originally thought to be related to a chronic leg ulcer that had been managed with wound care. Recently, however, the lesion was noted to have increasing erythema surrounding the wound margins. An 8×8-cm erythematous plaque on the anterior lateral left leg with a firm central nodule with hemorrhagic crust that measured approximately 4 cm in diameter was noted by the emergency department physicians (Figure 1). In the emergency department, vitals and other laboratory values were within reference range, and a radiograph of the left tibia/fibula was unremarkable. Cellulitis initially was considered in the emergency department and cephalexin was started; however, since the patient was afebrile and had no leukocytosis, plastic surgery also was consulted. Biopsies were obtained from the superior and inferior parts of the lesion. Histologic analysis revealed a poorly differentiated vascular neoplasm of epithelioid endothelial cells with considerable cell atypia that extended through the entirety of the dermis (Figure 2). The tumor cells stained positive with vimentin and CD34. Pathology noted no immunohistochemistry stains to synaptophysin, S-100, human melanoma black 45, MART-1, CK20, CK7, CK8/18, CK5/6, and p63. The pathologic diagnosis was consistent with cutaneous angiosarcoma. Computed tomography of the chest, abdomen, and pelvis revealed no local or distant metastases.
A wide excision of the cutaneous angiosarcoma was performed. The initial frozen section analysis revealed positive margins. Three additional excisions still showed positive margins, and further excision was held after obtaining family consent due to the extensive nature of the neoplasm and lengthy operating room time. The final defect after excision measured 15×10×2.5 cm (Figure 3A), and subsequent application of a split-thickness graft was performed. Additional treatment options were discussed with the family, including radiation therapy, amputation of the left lower leg, or no treatment. The family opted not to proceed with further treatment. The graft healed without signs of reoccurrence approximately 3 months later (Figure 3B), and the patient received physical therapy, which allowed her to gain strength and some independence.
Comment
Clinical Manifestation
Cutaneous angiosarcoma is a rare malignant vascular neoplasm that when clinically diagnosed is typically seen in 3 settings: (1) idiopathic (commonly on the face and neck), (2) following radiation treatment, and (3) classically following mastectomy with subsequent chronic lymphedema. Our patient did not classically fit these settings of cutaneous angiosarcoma due to the location of the lesion on the lower leg as well as its occurrence in the setting of a chronic nonhealing ulcer and lymphedema.
Chronic lymphedema is a common clinical manifestation that is likely secondary to other medical conditions, such as in our patient. As a result, these patients are at increased risk for developing chronic ulcers due to poor wound healing; however, as seen in our patient, chronic nonhealing ulcers require a broad differential because they may clinically mimic many processes. Patient history and visual presentation were crucial in this case because a biopsy was obtained that ultimately led to the patient’s diagnosis.
Differential Diagnosis
Initially, a venous ulcer secondary to chronic venous insufficiency was considered in the differential for our patient. She had a history of congestive heart failure, kidney disease, and type 2 diabetes mellitus, all of which contribute to lymphedema and/or poor wound healing. However, venous ulcers usually are located on the medial ankles and are irregularly shaped with an erythematous border and fibrinous exudate with central depression, making it a less likely diagnosis in our patient. Additionally, an infectious process was considered, but the patient was afebrile and laboratory values demonstrated no leukocytosis.
Marjolin ulcer was highly suspected because the clinical presentation revealed a nodule with hemorrhagic crust and induration in the setting of a chronic nonhealing ulcer. The pathogenesis of malignancy in chronic ulcers is thought to be due to continuous mitotic activity from regeneration and repair of the wound, especially in the setting of repeated trauma to the area.5 In our patient, the history of multiple falls with possible multitrauma injury to the chronic ulcer further increased suspicion of malignancy. The most common and frequently seen malignancy that develops in chronic ulcers is squamous cell carcinoma (SCC) followed by basal cell carcinoma. Plastic surgery suspected an SCC for the working diagnosis, which prompted a punch biopsy; however, the histologic analysis was not consistent with SCC or basal cell carcinoma. Marjolin ulcer also may demonstrate a periosteal reaction,5 which was not the case with our patient after a radiograph of the left tibia/fibula was unremarkable.
Another potential malignancy to consider is melanoma. There are few case reports of biopsy-proven melanoma from an enlarging chronic ulcer.6,7 Additionally, poorly differentiated angiosarcoma can mimic melanoma2; however, immunohistochemistry stain was negative for S-100, human melanoma black 45, and MART-1, making melanoma unlikely.
Kaposi sarcoma (KS) and angiosarcoma are both malignant vascular tumors that similarly present with red to purple patches, plaques, or nodules, making it difficult to distinguish between the two conditions. It is important to note that KS usually is lower grade, and the pathogenesis is linked to human herpesvirus 8, which can be identified on immunohistochemistry staining. There have been cases of KS reported in patients who have no history of human immunodeficiency virus/AIDS, thus the classic subtype of KS may have been considered in this patient.8 The histologic appearance of KS may vary from dilated irregular endothelial cells lining the vascular space to mild endothelial cell atypia. Histology also shows hemosiderin-laden macrophages, extravasated red blood cells, and an inflammatory infiltrate. An additional malignant vascular neoplasm that needs to be differentiated is epithelioid hemangioendothelioma. Cutaneous presentation of an epithelioid hemangioendothelioma may be similar to what was seen in our patient but histologically will usually show neoplastic cells with pale eosinophilic cytoplasm and vesicular nuclei of plump, oval, polygonal cells in cords or aggregates surrounding vascular channels. These neoplasms also tend to occur around medium- to large-sized veins.1,9 With our patient, even though human herpesvirus 8 was not tested with immunohistochemistry, gold standard immunohistochemistry confirmation with CD34 and vimentin staining combined with poorly differentiated endothelial atypia with mitotic figures on histologic analysis favored angiosarcoma versus KS or epithelioid hemangioendothelioma.10,11
Management
Cutaneous angiosarcoma is a rare and aggressive vascular neoplasm accounting for approximately 2% of all combined sarcomas, with an estimated 20% to 40% having distant metastasis at diagnosis.1,3 For this reason, computed tomography was performed in our patient and revealed no local or distant metastasis. Therefore, chemotherapy was not an appropriate adjuvant treatment option.12 With no evidence of metastasis, initial treatment began with surgical removal but proved to be difficult in our patient. Although the implications of positive surgical margins remain unclear with regard to overall patient survival, surgical resection followed by radiation therapy has been shown to be optimal, as it reduces the risk of local reoccurrence.3 There have been reported cases of cutaneous angiosarcoma of the leg that were treated with amputation without signs of reoccurrence or metastasis.10,13,14 Given the results from these cases and considering that our patient had no metastasis, amputation seemed to be a good prognostic option; however, considering other factors regarding the patient’s comorbidities and quality of life, her family decided not to pursue any further treatment with amputation or radiation therapy.
Conclusion
There should be low threshold for biopsy in patients who present with nonhealing wounds that do not progress in the normal phase of wound healing with suspicion for malignancy. As seen with our patient, cutaneous angiosarcoma can clinically mimic many disease processes, and although rare in nature, it should always be considered when a patient presents with a rapidly growing lesion in the setting of chronic lymphedema or venous ulcer.
Angiosarcoma is a rare and aggressive vascular malignant neoplasm derived from endothelial cells. In general, sarcomas account for approximately 1% of all malignancies, with approximately 2% being angiosarcomas.1 The risk of recurrence at 5 years is estimated to be 84%, and 5-year survival is estimated at 15% to 30%. Poor prognostic factors for angiosarcoma include large tumor size, depth of invasion greater than 3 mm, high mitotic rate, positive surgical margins, and metastasis.2 Approximately 20% to 40% of patients who are diagnosed with angiosarcoma already have distant metastasis, contributing to the aggressive nature of this neoplasm.3
Angiosarcoma can affect various anatomic locations, including the skin, soft tissue, breasts, and liver. Cutaneous angiosarcoma is the most common clinical manifestation, accounting for approximately 50% to 60% of all cases, and typically is known to occur in 3 distinct settings.2 Primary or idiopathic cutaneous angiosarcoma is most commonly seen in elderly individuals, with a peak incidence in the seventh to eighth decades of life, and presents as a bruiselike lesion predominantly on the head and neck. Angiosarcoma also is seen clinically in patients exposed to radiation treatment, with a median onset of symptoms occurring 5 to 10 years posttreatment, and in patients with chronic lymphedema, usually on the arm following radical mastectomy, which also is known as Stewart-Treves syndrome.2
With any sarcoma, treatment typically first involves surgical excision; however, there is no direct approach for treatment of cutaneous angiosarcoma, as an individual plan typically is needed for each patient. Treatment options include surgical excision, radiation, chemotherapy, or a combination of these therapies.2,4
We present a rare case of cutaneous angiosarcoma of the left leg in the setting of chronic venous insufficiency with some degree of lymphedema and a nonhealing ulcer. This case is unique in that it does not fit the classic presentation of cutaneous angiosarcoma previously described.
Case Report
An 83-year-old woman with a medical history of advanced dementia, congestive heart failure, chronic obstructive pulmonary disease, chronic kidney disease, type 2 diabetes mellitus, hypertension, and chronic venous insufficiency with stasis dermatitis presented to the emergency department following a mechanical fall. Most of her medical history was obtained from the patient’s family. She had a history of multiple falls originally thought to be related to a chronic leg ulcer that had been managed with wound care. Recently, however, the lesion was noted to have increasing erythema surrounding the wound margins. An 8×8-cm erythematous plaque on the anterior lateral left leg with a firm central nodule with hemorrhagic crust that measured approximately 4 cm in diameter was noted by the emergency department physicians (Figure 1). In the emergency department, vitals and other laboratory values were within reference range, and a radiograph of the left tibia/fibula was unremarkable. Cellulitis initially was considered in the emergency department and cephalexin was started; however, since the patient was afebrile and had no leukocytosis, plastic surgery also was consulted. Biopsies were obtained from the superior and inferior parts of the lesion. Histologic analysis revealed a poorly differentiated vascular neoplasm of epithelioid endothelial cells with considerable cell atypia that extended through the entirety of the dermis (Figure 2). The tumor cells stained positive with vimentin and CD34. Pathology noted no immunohistochemistry stains to synaptophysin, S-100, human melanoma black 45, MART-1, CK20, CK7, CK8/18, CK5/6, and p63. The pathologic diagnosis was consistent with cutaneous angiosarcoma. Computed tomography of the chest, abdomen, and pelvis revealed no local or distant metastases.
A wide excision of the cutaneous angiosarcoma was performed. The initial frozen section analysis revealed positive margins. Three additional excisions still showed positive margins, and further excision was held after obtaining family consent due to the extensive nature of the neoplasm and lengthy operating room time. The final defect after excision measured 15×10×2.5 cm (Figure 3A), and subsequent application of a split-thickness graft was performed. Additional treatment options were discussed with the family, including radiation therapy, amputation of the left lower leg, or no treatment. The family opted not to proceed with further treatment. The graft healed without signs of reoccurrence approximately 3 months later (Figure 3B), and the patient received physical therapy, which allowed her to gain strength and some independence.
Comment
Clinical Manifestation
Cutaneous angiosarcoma is a rare malignant vascular neoplasm that when clinically diagnosed is typically seen in 3 settings: (1) idiopathic (commonly on the face and neck), (2) following radiation treatment, and (3) classically following mastectomy with subsequent chronic lymphedema. Our patient did not classically fit these settings of cutaneous angiosarcoma due to the location of the lesion on the lower leg as well as its occurrence in the setting of a chronic nonhealing ulcer and lymphedema.
Chronic lymphedema is a common clinical manifestation that is likely secondary to other medical conditions, such as in our patient. As a result, these patients are at increased risk for developing chronic ulcers due to poor wound healing; however, as seen in our patient, chronic nonhealing ulcers require a broad differential because they may clinically mimic many processes. Patient history and visual presentation were crucial in this case because a biopsy was obtained that ultimately led to the patient’s diagnosis.
Differential Diagnosis
Initially, a venous ulcer secondary to chronic venous insufficiency was considered in the differential for our patient. She had a history of congestive heart failure, kidney disease, and type 2 diabetes mellitus, all of which contribute to lymphedema and/or poor wound healing. However, venous ulcers usually are located on the medial ankles and are irregularly shaped with an erythematous border and fibrinous exudate with central depression, making it a less likely diagnosis in our patient. Additionally, an infectious process was considered, but the patient was afebrile and laboratory values demonstrated no leukocytosis.
Marjolin ulcer was highly suspected because the clinical presentation revealed a nodule with hemorrhagic crust and induration in the setting of a chronic nonhealing ulcer. The pathogenesis of malignancy in chronic ulcers is thought to be due to continuous mitotic activity from regeneration and repair of the wound, especially in the setting of repeated trauma to the area.5 In our patient, the history of multiple falls with possible multitrauma injury to the chronic ulcer further increased suspicion of malignancy. The most common and frequently seen malignancy that develops in chronic ulcers is squamous cell carcinoma (SCC) followed by basal cell carcinoma. Plastic surgery suspected an SCC for the working diagnosis, which prompted a punch biopsy; however, the histologic analysis was not consistent with SCC or basal cell carcinoma. Marjolin ulcer also may demonstrate a periosteal reaction,5 which was not the case with our patient after a radiograph of the left tibia/fibula was unremarkable.
Another potential malignancy to consider is melanoma. There are few case reports of biopsy-proven melanoma from an enlarging chronic ulcer.6,7 Additionally, poorly differentiated angiosarcoma can mimic melanoma2; however, immunohistochemistry stain was negative for S-100, human melanoma black 45, and MART-1, making melanoma unlikely.
Kaposi sarcoma (KS) and angiosarcoma are both malignant vascular tumors that similarly present with red to purple patches, plaques, or nodules, making it difficult to distinguish between the two conditions. It is important to note that KS usually is lower grade, and the pathogenesis is linked to human herpesvirus 8, which can be identified on immunohistochemistry staining. There have been cases of KS reported in patients who have no history of human immunodeficiency virus/AIDS, thus the classic subtype of KS may have been considered in this patient.8 The histologic appearance of KS may vary from dilated irregular endothelial cells lining the vascular space to mild endothelial cell atypia. Histology also shows hemosiderin-laden macrophages, extravasated red blood cells, and an inflammatory infiltrate. An additional malignant vascular neoplasm that needs to be differentiated is epithelioid hemangioendothelioma. Cutaneous presentation of an epithelioid hemangioendothelioma may be similar to what was seen in our patient but histologically will usually show neoplastic cells with pale eosinophilic cytoplasm and vesicular nuclei of plump, oval, polygonal cells in cords or aggregates surrounding vascular channels. These neoplasms also tend to occur around medium- to large-sized veins.1,9 With our patient, even though human herpesvirus 8 was not tested with immunohistochemistry, gold standard immunohistochemistry confirmation with CD34 and vimentin staining combined with poorly differentiated endothelial atypia with mitotic figures on histologic analysis favored angiosarcoma versus KS or epithelioid hemangioendothelioma.10,11
Management
Cutaneous angiosarcoma is a rare and aggressive vascular neoplasm accounting for approximately 2% of all combined sarcomas, with an estimated 20% to 40% having distant metastasis at diagnosis.1,3 For this reason, computed tomography was performed in our patient and revealed no local or distant metastasis. Therefore, chemotherapy was not an appropriate adjuvant treatment option.12 With no evidence of metastasis, initial treatment began with surgical removal but proved to be difficult in our patient. Although the implications of positive surgical margins remain unclear with regard to overall patient survival, surgical resection followed by radiation therapy has been shown to be optimal, as it reduces the risk of local reoccurrence.3 There have been reported cases of cutaneous angiosarcoma of the leg that were treated with amputation without signs of reoccurrence or metastasis.10,13,14 Given the results from these cases and considering that our patient had no metastasis, amputation seemed to be a good prognostic option; however, considering other factors regarding the patient’s comorbidities and quality of life, her family decided not to pursue any further treatment with amputation or radiation therapy.
Conclusion
There should be low threshold for biopsy in patients who present with nonhealing wounds that do not progress in the normal phase of wound healing with suspicion for malignancy. As seen with our patient, cutaneous angiosarcoma can clinically mimic many disease processes, and although rare in nature, it should always be considered when a patient presents with a rapidly growing lesion in the setting of chronic lymphedema or venous ulcer.
- Kumar V, Abbas A, Aster J. Robbins Basic Pathology. 9th ed. Philadelphia, PA: Elsevier Saunders; 2013.
- Donghi D, Kerl K, Dummer R, et al. Cutaneous angiosarcoma: own experience over 13 years. clinical features, disease course and immunohistochemical profile. J Eur Acad Dermatol Venereol. 2010;24:1230-1234.
- Dossett LA, Harrington M, Cruse CW, et al. Cutaneous angiosarcoma. Curr Probl Cancer. 2015;39:258-263.
- Morgan MB, Swann M, Somach S, et al. Cutaneous angiosarcoma: a case series with prognostic correlation. J Am Acad Dermatol. 2004;50:867-874.
- Pekarek B, Buck S, Osher L. A comprehensive review on Marjolin’s ulcers: diagnosis and treatment. J Am Col Certif Wound Spec. 2011;3:60-64.
- Gerslova A, Pokorna A, Stukavcova A, et al. Rare cause of non-healing foot wound—acral lentiginous melanoma. Neuro Endocrinol Lett. 2012;37:12-17.
- Turk BG, Bozkurt A, Yaman B, et al. Melanoma arising in chronic ulceration associated with lymphoedema. J Wound Care. 2013;22:74-75.
- Phavixay L, Raynolds D, Simman R. Non AIDS Kaposi’s sarcoma leading to lower extremities wounds, case presentations and discussion.J Am Coll Clin Wound Spec. 2012;4:13-15.
- Requena L, Kutzner H. Hemangioendothelioma. Semin Diagn Pathol. 2013;30:29-44.
- Harrison WD, Chandrasekar CR. Stewart-Treves syndrome following idiopathic leg lymphoedema: remember sarcoma. J Wound Care. 2015;24(6 suppl):S5-S7.
- Kak I, Salama S, Gohla G, et al. A case of patch stage of Kaposi’s sarcoma and discussion of the differential diagnosis. Rare Tumors. 2016;8:6123.
- Agulnik M, Yarber JL, Okuno SH, et al. An open-label, multicenter, phase II study of bevacizumab for the treatment of angiosarcoma and epithelioid hemangioendotheliomas. Ann Oncol. 2013;24:257-263.
- Linda DD, Harish S, Alowami S, et al. Radiology-pathology conference: cutaneous angiosarcoma of the leg. Clin Imaging. 2013;37:602-607.
- Roy P, Clark MA, Thomas JM. Stewart-Treves syndrome—treatment and outcome in six patients from a single centre. Eur J Surg Oncol. 2004;30:982-986.
- Kumar V, Abbas A, Aster J. Robbins Basic Pathology. 9th ed. Philadelphia, PA: Elsevier Saunders; 2013.
- Donghi D, Kerl K, Dummer R, et al. Cutaneous angiosarcoma: own experience over 13 years. clinical features, disease course and immunohistochemical profile. J Eur Acad Dermatol Venereol. 2010;24:1230-1234.
- Dossett LA, Harrington M, Cruse CW, et al. Cutaneous angiosarcoma. Curr Probl Cancer. 2015;39:258-263.
- Morgan MB, Swann M, Somach S, et al. Cutaneous angiosarcoma: a case series with prognostic correlation. J Am Acad Dermatol. 2004;50:867-874.
- Pekarek B, Buck S, Osher L. A comprehensive review on Marjolin’s ulcers: diagnosis and treatment. J Am Col Certif Wound Spec. 2011;3:60-64.
- Gerslova A, Pokorna A, Stukavcova A, et al. Rare cause of non-healing foot wound—acral lentiginous melanoma. Neuro Endocrinol Lett. 2012;37:12-17.
- Turk BG, Bozkurt A, Yaman B, et al. Melanoma arising in chronic ulceration associated with lymphoedema. J Wound Care. 2013;22:74-75.
- Phavixay L, Raynolds D, Simman R. Non AIDS Kaposi’s sarcoma leading to lower extremities wounds, case presentations and discussion.J Am Coll Clin Wound Spec. 2012;4:13-15.
- Requena L, Kutzner H. Hemangioendothelioma. Semin Diagn Pathol. 2013;30:29-44.
- Harrison WD, Chandrasekar CR. Stewart-Treves syndrome following idiopathic leg lymphoedema: remember sarcoma. J Wound Care. 2015;24(6 suppl):S5-S7.
- Kak I, Salama S, Gohla G, et al. A case of patch stage of Kaposi’s sarcoma and discussion of the differential diagnosis. Rare Tumors. 2016;8:6123.
- Agulnik M, Yarber JL, Okuno SH, et al. An open-label, multicenter, phase II study of bevacizumab for the treatment of angiosarcoma and epithelioid hemangioendotheliomas. Ann Oncol. 2013;24:257-263.
- Linda DD, Harish S, Alowami S, et al. Radiology-pathology conference: cutaneous angiosarcoma of the leg. Clin Imaging. 2013;37:602-607.
- Roy P, Clark MA, Thomas JM. Stewart-Treves syndrome—treatment and outcome in six patients from a single centre. Eur J Surg Oncol. 2004;30:982-986.
Practice Points
- Cutaneous angiosarcoma is a rare malignant vascular neoplasm typically seen in 3 settings: (1) idiopathic (commonly on the face and neck), (2) following radiation treatment, and (3) classically in the setting of chronic lymphedema following mastectomy (Stewart-Treves syndrome).
- There should be a low threshold for biopsy in patients who present with nonhealing wounds that do not progress in the normal phase of wound healing with suspicion for malignancy.
- Histologic analysis of angiosarcoma shows positive staining for CD34 and vimentin with poorly differentiated endothelial atypia with mitotic figures.
Acute Superior Mesenteric Venous Thrombosis in a Young Patient Without Risk Factors
In this case report, the authors address the diagnostic challenges of a young, healthy patient who presented to the ED with unrelenting abdominal pain.
Acute mesenteric ischemia (AMI) results when oxygen delivery to the mesenteric artery is compromised, and is a serious diagnosis that should be considered in patients of all ages to avoid significant morbidity and mortality. The majority of cases are due to arterial embolism, arterial thrombus, or intestinal hypoperfusion (non-occlusive). Acute mesenteric venous thrombosis (MVT) accounts for only 2% to 10% of AMI cases, and only 0.01% of emergency surgery admissions.1 A large systematic review showed a 44% mortality rate for MVT, in contrast to 66% to 89% for all other forms of AMI.2 The typical age range for MVT is reported between 45 and 60 years, with a slight male predominance.3 Dull, central abdominal pain is the most frequently reported symptom of MVT, although it is generally less impressive than the pain described in other forms of AMI.3Along with the hallmark of abdominal pain out of proportion to the examination, other gastrointestinal symptoms include weight loss and non-specific altered bowel function (constipation, diarrhea, abdominal distention, and bloating), which are present in half of all patients with MVT.1 Peritoneal signs and bloody stools portend poor outcomes, as they often occur with disease progression.4
Case
A 26-year-old man presented to the ED with periumbilical and lower abdominal pain for 1 week. The pain was described as constant and dull, worsened by movement and oral intake, and improved with lying flat. He described bloating and decreased volume of bowel movements. He denied nausea, vomiting, fever, colicky pain, blood in stool, testicular pain, urinary complaints, trauma, or any similar episodes in the past. The patient had no known medical conditions or surgical history, except for a remote history of alcohol dependence (in remission) and tobacco use. There was no personal or family history of coagulopathy. Of note, he was seen by his primary care physician a few days prior to his ED presentation and had been instructed to take acetaminophen, which did not provide relief.
The patient’s vital signs at presentation were: blood pressure, 122/70 mm Hg; heart rate, 93 beats/min; respiratory rate, 18 breaths/min; and temperature, 37.5°C (99.5°F). Oxygen saturation was 99% on room air. The physical examination was remarkable only for mild abdominal tenderness diffusely, greater in the lower and central abdomen than in the upper abdomen. The remainder of the physical examination was unremarkable.
Laboratory studies ordered included a complete blood count, comprehensive metabolic profile, lipase, and urinalysis. The patient did have a mild transaminitis (aspartate aminotransferase, 48 U/L; alanine aminotransferase, 84 U/L); the remainder of the studies were normal. A serum lactate, drawn after the 1 L of normal saline was administered intravenously (IV), was within normal limits (0.7 mmol/L). No prior laboratory studies were available for comparison.
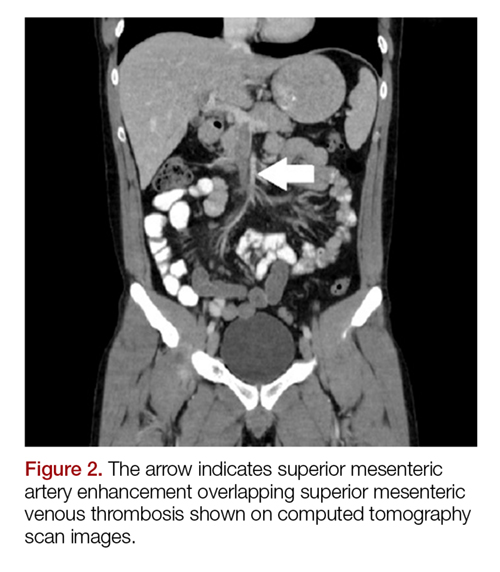
The patient’s continued abdominal pain and transaminitis prompted an ED bedside right upper quadrant ultrasound, which showed a small gallbladder polyp; no signs of gallbladder disease were present. The patient required three doses of morphine 4 mg IV without complete pain relief. Given the concern for pain out of proportion to physical examination, a computed tomography (CT) scan of the abdomen/pelvis with IV and oral contrast was ordered. The radiologist interpreted the scan as showing a superior mesenteric vein (SMV) thrombus extending into the splenic/portal vein confluence and the intrahepatic portal veins (Figures 1 and 2). 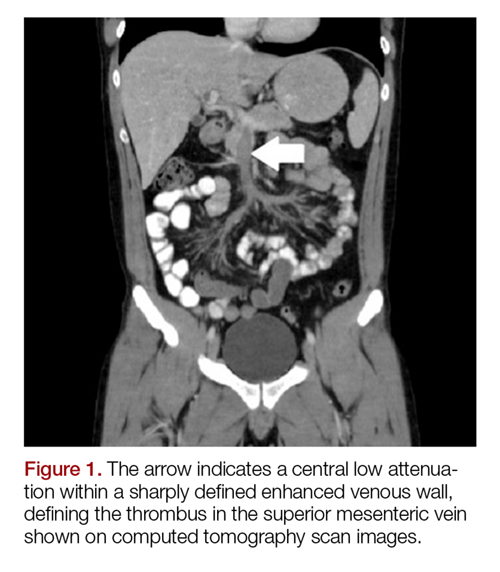
Ciprofloxacin and metronidazole were administered IV for antibiotic prophylaxis, and the patient was placed on bowel rest with advancement to regular diet as tolerated. Propranolol was given for variceal prophylaxis. The patient was discharged home the following day in stable condition. Although he still had mild abdominal tenderness, the vital signs and physical examination were within normal limits. The patient was placed on a 6-month course of rivaroxaban therapy. Coagulopathy testing was scheduled at a later date, since ongoing anticoagulation treatment could interfere with test results. Unfortunately, the patient did not attend follow-up appointments to obtain testing.
Discussion
Mesenteric venous thrombosis is seen predominantly in middle-aged patients presenting with vague symptoms, which makes this a challenging diagnosis to make in the acute care setting. Risk factors for MVT include recent injury (causing trauma to the vasculature), recent surgery (causing stagnant blood flow), inflammatory conditions, and hypercoagulable states.1 In this patient’s case, no risk factors were identified; although the majority of cases of MVT will have an identifiable risk factor.2 Still, 21% to 49% of cases of MVT are considered idiopathic.1,3It is possible that our patient had a prior undiagnosed pancreatitis associated with his history of alcoholism that contributed to his thrombosis. Pancreatitis and other inflammatory conditions, including diverticulitis or inflammatory bowel disease, are more commonly associated with thrombus formation in the large veins, as opposed to an undiagnosed hypercoagulable state, which would more likely affect distal venuoles, vasa recta, or venous arcades.1,5 The patient’s mild transaminitis was likely secondary to hepatic congestion from the venous thrombus extending to the splenic-portal vein confluence and intrahepatic portal vein. One study looked at patients with pancreatitis and found that 16.7% of their study population had an SMV thrombus, while 4.1% had a SMV thrombus with a concomitant portal vein thrombus.6
Although there are no pathognomonic laboratory findings of MVT, elevated lactate, leukocytosis, and elevated D-dimer levels may be helpful in supporting the diagnosis.7,8 A recent study found that elevated D-dimer levels may be a specific marker in the early recognition of acute SMV thrombosis, as well as predicting risk, outcomes, and treatment options.8 However, emergency physicians should maintain a high index of suspicion in patients with concerning features of the disease, since normal laboratory values, including lactate, do not reliably exclude the diagnosis.
Computed tomography scanning and CT angiography (CTA) are quite helpful in diagnosing MVT. Ultrasound of the upper abdomen may also play a role, noting dilated or thickened bowel wall with intraluminal air or echogenic material in the superior mesenteric vein or portal vein.9 Although magnetic resonance venography most reliably demonstrates thrombi, its lack of widespread availability makes CT with IV contrast the preferred initial study.3Computed tomography not only has high sensitivity, but also offers alternative diagnoses in the undifferentiated presentation.1One study found CT to be 100% sensitive in detecting any abnormality associated with MVT or bowel ischemia.10 Common CT findings of MVT include dilated and thickened bowel loops, mesenteric fat standing, ascites, a halo or target appearance of bowel, vessel filling defects from a thrombus, and pneumatosis intestinalis.11 The latter usually indicates transmural infarction, and can extend as portomesenteric vein gas.11 Of note, if the initial CT scan is non-diagnostic and a high clinical suspicion for mesenteric ischemia remains with no alternative diagnosis, CTA is the gold standard.3,7Expeditious diagnosis of MVT is imperative, given the potential complications of intestinal infarction, submucosal hemorrhage secondary to edema, and third spacing of the venous outflow into the bowel wall due to collateral vessels being unable to redirect blood flow in conjunction with complete venous occlusion.12Not all MVTs progress to infarction, given the extensive collateral circulation. Early diagnosis, however, is crucial for conservative management to be effective.9Acute MVT without signs of infarction necessitates anticoagulation therapy to decrease clot propagation and recurrence.1 In addition, prophylactic antibiotics to limit bacterial translocation, and bowel rest are advised.13,14 If the patient is unresponsive to anticoagulation, thrombolytic and endovascular therapies may be of benefit in select patients.15 Once intestinal ischemia or infarction develops, the prognosis is poor: mortality approaches 75% with infarction.1 If signs of bowel infarction are present, a laparotomy must be performed promptly, although in most cases, delayed patient presentation makes small bowel resection unavoidable.9 Further testing for hypercoagulability is recommended, particularly in isolated thrombosis, since long-term anticoagulation therapy may be necessary if a coagulopathy is discovered.1
Conclusion
Mesenteric venous thrombosis is atypical in a young, healthy patient. However, due to high mortality rates with disease progression, it is important to consider in any patient with unrelenting abdominal pain and vague gastrointestinal symptoms of uncertain cause, even in those without risk factors. Early detection and management of MVT before progression to mesenteric ischemia and infarction considerably lowers the mortality rate. Emergency physicians must be vigilant when treating a patient with abdominal pain out of proportion to physical examination, unrelenting pain despite analgesic medications, or repeat ED visits for the same abdominal complaints.
1. Harnik IG, Brandt LJ. Mesenteric venous thrombosis. Vasc Med. 2010;15(5):407-418. doi:10.1177/1358863x10379673.
2. Tilsed JV, Casamassima A, Kurihara H, et al. ESTES guidelines: acute mesenteric ischaemia. Eur J Trauma Emerg Surg. 2016;42(2):253-270. doi:10.1007/s00068-016-0634-0.
3. Tendler DA, Lamont JT, Grubel P. Mesenteric venous thrombosis in adults. UpToDate Web site. https://www.uptodate.com/contents/mesenteric-venous-thrombosis-in-adults. Accessed November 16, 2017.
4. Al-Zahrani HA, Lindsay T. Mesenteric ischemia. In: Hall JB, Schmidt GA, Kress JP, eds. Principles of Critical Care. 4th ed. New York, NY: McGraw Hill; 2015:1036-1044.
5. Kumar S, Sarr MG, Kamath PS. Mesenteric venous thrombosis. N Engl J Med. 2001;345(23):1683-1688. doi:10.1056/nejmra010076.
6. Al-Khazraji A, Hasan AQ, Patel I, Alkhawam H, Ghrair F, Lieber J. The role of abdominal computed tomography scan in acute pancreatitis. Pancreas. 2017;46(6):e52-e54. doi:10.1097/mpa.0000000000000837.
7. Bradbury MS, Kavanagh PV, Bechtold RE, et al. Mesenteric venous thrombosis: diagnosis and noninvasive imaging. Radiographics. 2002;22(3):527-541.
8. Yang S, Fan X, Ding W, et al. D-dimer as an early marker of severity in patients with acute superior mesenteric venous thrombosis. Medicine (Baltimore). 2014;93(29):e270. doi:10.1097/md.0000000000000270.
9. Matos C, Van Gansbeke D, Zalcman M, et al. Mesenteric vein thrombosis: early CT and US diagnosis and conservative management. Gastrointest Radiol. 1986;11(4):322-325.
10. Rhee RY, Gloviczki P, Mendonca CT, et al. Mesenteric venous thrombosis: still a lethal disease in the 1990s. J Vasc Surg. 1994;20(5):688-697.
11. Furukawa A, Kanasaki S, Kono N, et al. CT diagnosis of acute mesenteric ischemia from various causes. AJR Am J Roentgenol. 2009;192(2):408-416. doi:10.2214/ajr.08.1138.
12. Johnson CC, Baggenstoss AH. Mesenteric vascular occlusion; study of 99 cases of occlusion of veins. Proc Staff Meet Mayo Clin. 1949;24(25):628-636.13. Hmoud B, Singal AK, Kamath PS. Mesenteric venous thrombosis. J Clin Exp Hepatol. 2014;4(3):257-263. doi:10.1016/j.jceh.2014.03.052.
14. Schoots IG, Koffeman GI, Legemate DA, Levi M, van Gulik TM. Systematic review of survival after acute mesenteric ischaemia according to disease aetiology. Br J Surg. 2004;91(1):17-27.
15. Yang S, Fan X, Ding W, et al. Multidisciplinary stepwise management strategy for acute superior mesenteric venous thrombosis: an intestinal stroke center experience. Thromb Res. 2015;135(1):36-45. doi:10.1016/j.thromres.2014.10.018.
In this case report, the authors address the diagnostic challenges of a young, healthy patient who presented to the ED with unrelenting abdominal pain.
In this case report, the authors address the diagnostic challenges of a young, healthy patient who presented to the ED with unrelenting abdominal pain.
Acute mesenteric ischemia (AMI) results when oxygen delivery to the mesenteric artery is compromised, and is a serious diagnosis that should be considered in patients of all ages to avoid significant morbidity and mortality. The majority of cases are due to arterial embolism, arterial thrombus, or intestinal hypoperfusion (non-occlusive). Acute mesenteric venous thrombosis (MVT) accounts for only 2% to 10% of AMI cases, and only 0.01% of emergency surgery admissions.1 A large systematic review showed a 44% mortality rate for MVT, in contrast to 66% to 89% for all other forms of AMI.2 The typical age range for MVT is reported between 45 and 60 years, with a slight male predominance.3 Dull, central abdominal pain is the most frequently reported symptom of MVT, although it is generally less impressive than the pain described in other forms of AMI.3Along with the hallmark of abdominal pain out of proportion to the examination, other gastrointestinal symptoms include weight loss and non-specific altered bowel function (constipation, diarrhea, abdominal distention, and bloating), which are present in half of all patients with MVT.1 Peritoneal signs and bloody stools portend poor outcomes, as they often occur with disease progression.4
Case
A 26-year-old man presented to the ED with periumbilical and lower abdominal pain for 1 week. The pain was described as constant and dull, worsened by movement and oral intake, and improved with lying flat. He described bloating and decreased volume of bowel movements. He denied nausea, vomiting, fever, colicky pain, blood in stool, testicular pain, urinary complaints, trauma, or any similar episodes in the past. The patient had no known medical conditions or surgical history, except for a remote history of alcohol dependence (in remission) and tobacco use. There was no personal or family history of coagulopathy. Of note, he was seen by his primary care physician a few days prior to his ED presentation and had been instructed to take acetaminophen, which did not provide relief.
The patient’s vital signs at presentation were: blood pressure, 122/70 mm Hg; heart rate, 93 beats/min; respiratory rate, 18 breaths/min; and temperature, 37.5°C (99.5°F). Oxygen saturation was 99% on room air. The physical examination was remarkable only for mild abdominal tenderness diffusely, greater in the lower and central abdomen than in the upper abdomen. The remainder of the physical examination was unremarkable.
Laboratory studies ordered included a complete blood count, comprehensive metabolic profile, lipase, and urinalysis. The patient did have a mild transaminitis (aspartate aminotransferase, 48 U/L; alanine aminotransferase, 84 U/L); the remainder of the studies were normal. A serum lactate, drawn after the 1 L of normal saline was administered intravenously (IV), was within normal limits (0.7 mmol/L). No prior laboratory studies were available for comparison.
The patient’s continued abdominal pain and transaminitis prompted an ED bedside right upper quadrant ultrasound, which showed a small gallbladder polyp; no signs of gallbladder disease were present. The patient required three doses of morphine 4 mg IV without complete pain relief. Given the concern for pain out of proportion to physical examination, a computed tomography (CT) scan of the abdomen/pelvis with IV and oral contrast was ordered. The radiologist interpreted the scan as showing a superior mesenteric vein (SMV) thrombus extending into the splenic/portal vein confluence and the intrahepatic portal veins (Figures 1 and 2).
Ciprofloxacin and metronidazole were administered IV for antibiotic prophylaxis, and the patient was placed on bowel rest with advancement to regular diet as tolerated. Propranolol was given for variceal prophylaxis. The patient was discharged home the following day in stable condition. Although he still had mild abdominal tenderness, the vital signs and physical examination were within normal limits. The patient was placed on a 6-month course of rivaroxaban therapy. Coagulopathy testing was scheduled at a later date, since ongoing anticoagulation treatment could interfere with test results. Unfortunately, the patient did not attend follow-up appointments to obtain testing.
Discussion
Mesenteric venous thrombosis is seen predominantly in middle-aged patients presenting with vague symptoms, which makes this a challenging diagnosis to make in the acute care setting. Risk factors for MVT include recent injury (causing trauma to the vasculature), recent surgery (causing stagnant blood flow), inflammatory conditions, and hypercoagulable states.1 In this patient’s case, no risk factors were identified; although the majority of cases of MVT will have an identifiable risk factor.2 Still, 21% to 49% of cases of MVT are considered idiopathic.1,3It is possible that our patient had a prior undiagnosed pancreatitis associated with his history of alcoholism that contributed to his thrombosis. Pancreatitis and other inflammatory conditions, including diverticulitis or inflammatory bowel disease, are more commonly associated with thrombus formation in the large veins, as opposed to an undiagnosed hypercoagulable state, which would more likely affect distal venuoles, vasa recta, or venous arcades.1,5 The patient’s mild transaminitis was likely secondary to hepatic congestion from the venous thrombus extending to the splenic-portal vein confluence and intrahepatic portal vein. One study looked at patients with pancreatitis and found that 16.7% of their study population had an SMV thrombus, while 4.1% had a SMV thrombus with a concomitant portal vein thrombus.6
Although there are no pathognomonic laboratory findings of MVT, elevated lactate, leukocytosis, and elevated D-dimer levels may be helpful in supporting the diagnosis.7,8 A recent study found that elevated D-dimer levels may be a specific marker in the early recognition of acute SMV thrombosis, as well as predicting risk, outcomes, and treatment options.8 However, emergency physicians should maintain a high index of suspicion in patients with concerning features of the disease, since normal laboratory values, including lactate, do not reliably exclude the diagnosis.
Computed tomography scanning and CT angiography (CTA) are quite helpful in diagnosing MVT. Ultrasound of the upper abdomen may also play a role, noting dilated or thickened bowel wall with intraluminal air or echogenic material in the superior mesenteric vein or portal vein.9 Although magnetic resonance venography most reliably demonstrates thrombi, its lack of widespread availability makes CT with IV contrast the preferred initial study.3Computed tomography not only has high sensitivity, but also offers alternative diagnoses in the undifferentiated presentation.1One study found CT to be 100% sensitive in detecting any abnormality associated with MVT or bowel ischemia.10 Common CT findings of MVT include dilated and thickened bowel loops, mesenteric fat standing, ascites, a halo or target appearance of bowel, vessel filling defects from a thrombus, and pneumatosis intestinalis.11 The latter usually indicates transmural infarction, and can extend as portomesenteric vein gas.11 Of note, if the initial CT scan is non-diagnostic and a high clinical suspicion for mesenteric ischemia remains with no alternative diagnosis, CTA is the gold standard.3,7Expeditious diagnosis of MVT is imperative, given the potential complications of intestinal infarction, submucosal hemorrhage secondary to edema, and third spacing of the venous outflow into the bowel wall due to collateral vessels being unable to redirect blood flow in conjunction with complete venous occlusion.12Not all MVTs progress to infarction, given the extensive collateral circulation. Early diagnosis, however, is crucial for conservative management to be effective.9Acute MVT without signs of infarction necessitates anticoagulation therapy to decrease clot propagation and recurrence.1 In addition, prophylactic antibiotics to limit bacterial translocation, and bowel rest are advised.13,14 If the patient is unresponsive to anticoagulation, thrombolytic and endovascular therapies may be of benefit in select patients.15 Once intestinal ischemia or infarction develops, the prognosis is poor: mortality approaches 75% with infarction.1 If signs of bowel infarction are present, a laparotomy must be performed promptly, although in most cases, delayed patient presentation makes small bowel resection unavoidable.9 Further testing for hypercoagulability is recommended, particularly in isolated thrombosis, since long-term anticoagulation therapy may be necessary if a coagulopathy is discovered.1
Conclusion
Mesenteric venous thrombosis is atypical in a young, healthy patient. However, due to high mortality rates with disease progression, it is important to consider in any patient with unrelenting abdominal pain and vague gastrointestinal symptoms of uncertain cause, even in those without risk factors. Early detection and management of MVT before progression to mesenteric ischemia and infarction considerably lowers the mortality rate. Emergency physicians must be vigilant when treating a patient with abdominal pain out of proportion to physical examination, unrelenting pain despite analgesic medications, or repeat ED visits for the same abdominal complaints.
Acute mesenteric ischemia (AMI) results when oxygen delivery to the mesenteric artery is compromised, and is a serious diagnosis that should be considered in patients of all ages to avoid significant morbidity and mortality. The majority of cases are due to arterial embolism, arterial thrombus, or intestinal hypoperfusion (non-occlusive). Acute mesenteric venous thrombosis (MVT) accounts for only 2% to 10% of AMI cases, and only 0.01% of emergency surgery admissions.1 A large systematic review showed a 44% mortality rate for MVT, in contrast to 66% to 89% for all other forms of AMI.2 The typical age range for MVT is reported between 45 and 60 years, with a slight male predominance.3 Dull, central abdominal pain is the most frequently reported symptom of MVT, although it is generally less impressive than the pain described in other forms of AMI.3Along with the hallmark of abdominal pain out of proportion to the examination, other gastrointestinal symptoms include weight loss and non-specific altered bowel function (constipation, diarrhea, abdominal distention, and bloating), which are present in half of all patients with MVT.1 Peritoneal signs and bloody stools portend poor outcomes, as they often occur with disease progression.4
Case
A 26-year-old man presented to the ED with periumbilical and lower abdominal pain for 1 week. The pain was described as constant and dull, worsened by movement and oral intake, and improved with lying flat. He described bloating and decreased volume of bowel movements. He denied nausea, vomiting, fever, colicky pain, blood in stool, testicular pain, urinary complaints, trauma, or any similar episodes in the past. The patient had no known medical conditions or surgical history, except for a remote history of alcohol dependence (in remission) and tobacco use. There was no personal or family history of coagulopathy. Of note, he was seen by his primary care physician a few days prior to his ED presentation and had been instructed to take acetaminophen, which did not provide relief.
The patient’s vital signs at presentation were: blood pressure, 122/70 mm Hg; heart rate, 93 beats/min; respiratory rate, 18 breaths/min; and temperature, 37.5°C (99.5°F). Oxygen saturation was 99% on room air. The physical examination was remarkable only for mild abdominal tenderness diffusely, greater in the lower and central abdomen than in the upper abdomen. The remainder of the physical examination was unremarkable.
Laboratory studies ordered included a complete blood count, comprehensive metabolic profile, lipase, and urinalysis. The patient did have a mild transaminitis (aspartate aminotransferase, 48 U/L; alanine aminotransferase, 84 U/L); the remainder of the studies were normal. A serum lactate, drawn after the 1 L of normal saline was administered intravenously (IV), was within normal limits (0.7 mmol/L). No prior laboratory studies were available for comparison.
The patient’s continued abdominal pain and transaminitis prompted an ED bedside right upper quadrant ultrasound, which showed a small gallbladder polyp; no signs of gallbladder disease were present. The patient required three doses of morphine 4 mg IV without complete pain relief. Given the concern for pain out of proportion to physical examination, a computed tomography (CT) scan of the abdomen/pelvis with IV and oral contrast was ordered. The radiologist interpreted the scan as showing a superior mesenteric vein (SMV) thrombus extending into the splenic/portal vein confluence and the intrahepatic portal veins (Figures 1 and 2).
Ciprofloxacin and metronidazole were administered IV for antibiotic prophylaxis, and the patient was placed on bowel rest with advancement to regular diet as tolerated. Propranolol was given for variceal prophylaxis. The patient was discharged home the following day in stable condition. Although he still had mild abdominal tenderness, the vital signs and physical examination were within normal limits. The patient was placed on a 6-month course of rivaroxaban therapy. Coagulopathy testing was scheduled at a later date, since ongoing anticoagulation treatment could interfere with test results. Unfortunately, the patient did not attend follow-up appointments to obtain testing.
Discussion
Mesenteric venous thrombosis is seen predominantly in middle-aged patients presenting with vague symptoms, which makes this a challenging diagnosis to make in the acute care setting. Risk factors for MVT include recent injury (causing trauma to the vasculature), recent surgery (causing stagnant blood flow), inflammatory conditions, and hypercoagulable states.1 In this patient’s case, no risk factors were identified; although the majority of cases of MVT will have an identifiable risk factor.2 Still, 21% to 49% of cases of MVT are considered idiopathic.1,3It is possible that our patient had a prior undiagnosed pancreatitis associated with his history of alcoholism that contributed to his thrombosis. Pancreatitis and other inflammatory conditions, including diverticulitis or inflammatory bowel disease, are more commonly associated with thrombus formation in the large veins, as opposed to an undiagnosed hypercoagulable state, which would more likely affect distal venuoles, vasa recta, or venous arcades.1,5 The patient’s mild transaminitis was likely secondary to hepatic congestion from the venous thrombus extending to the splenic-portal vein confluence and intrahepatic portal vein. One study looked at patients with pancreatitis and found that 16.7% of their study population had an SMV thrombus, while 4.1% had a SMV thrombus with a concomitant portal vein thrombus.6
Although there are no pathognomonic laboratory findings of MVT, elevated lactate, leukocytosis, and elevated D-dimer levels may be helpful in supporting the diagnosis.7,8 A recent study found that elevated D-dimer levels may be a specific marker in the early recognition of acute SMV thrombosis, as well as predicting risk, outcomes, and treatment options.8 However, emergency physicians should maintain a high index of suspicion in patients with concerning features of the disease, since normal laboratory values, including lactate, do not reliably exclude the diagnosis.
Computed tomography scanning and CT angiography (CTA) are quite helpful in diagnosing MVT. Ultrasound of the upper abdomen may also play a role, noting dilated or thickened bowel wall with intraluminal air or echogenic material in the superior mesenteric vein or portal vein.9 Although magnetic resonance venography most reliably demonstrates thrombi, its lack of widespread availability makes CT with IV contrast the preferred initial study.3Computed tomography not only has high sensitivity, but also offers alternative diagnoses in the undifferentiated presentation.1One study found CT to be 100% sensitive in detecting any abnormality associated with MVT or bowel ischemia.10 Common CT findings of MVT include dilated and thickened bowel loops, mesenteric fat standing, ascites, a halo or target appearance of bowel, vessel filling defects from a thrombus, and pneumatosis intestinalis.11 The latter usually indicates transmural infarction, and can extend as portomesenteric vein gas.11 Of note, if the initial CT scan is non-diagnostic and a high clinical suspicion for mesenteric ischemia remains with no alternative diagnosis, CTA is the gold standard.3,7Expeditious diagnosis of MVT is imperative, given the potential complications of intestinal infarction, submucosal hemorrhage secondary to edema, and third spacing of the venous outflow into the bowel wall due to collateral vessels being unable to redirect blood flow in conjunction with complete venous occlusion.12Not all MVTs progress to infarction, given the extensive collateral circulation. Early diagnosis, however, is crucial for conservative management to be effective.9Acute MVT without signs of infarction necessitates anticoagulation therapy to decrease clot propagation and recurrence.1 In addition, prophylactic antibiotics to limit bacterial translocation, and bowel rest are advised.13,14 If the patient is unresponsive to anticoagulation, thrombolytic and endovascular therapies may be of benefit in select patients.15 Once intestinal ischemia or infarction develops, the prognosis is poor: mortality approaches 75% with infarction.1 If signs of bowel infarction are present, a laparotomy must be performed promptly, although in most cases, delayed patient presentation makes small bowel resection unavoidable.9 Further testing for hypercoagulability is recommended, particularly in isolated thrombosis, since long-term anticoagulation therapy may be necessary if a coagulopathy is discovered.1
Conclusion
Mesenteric venous thrombosis is atypical in a young, healthy patient. However, due to high mortality rates with disease progression, it is important to consider in any patient with unrelenting abdominal pain and vague gastrointestinal symptoms of uncertain cause, even in those without risk factors. Early detection and management of MVT before progression to mesenteric ischemia and infarction considerably lowers the mortality rate. Emergency physicians must be vigilant when treating a patient with abdominal pain out of proportion to physical examination, unrelenting pain despite analgesic medications, or repeat ED visits for the same abdominal complaints.
1. Harnik IG, Brandt LJ. Mesenteric venous thrombosis. Vasc Med. 2010;15(5):407-418. doi:10.1177/1358863x10379673.
2. Tilsed JV, Casamassima A, Kurihara H, et al. ESTES guidelines: acute mesenteric ischaemia. Eur J Trauma Emerg Surg. 2016;42(2):253-270. doi:10.1007/s00068-016-0634-0.
3. Tendler DA, Lamont JT, Grubel P. Mesenteric venous thrombosis in adults. UpToDate Web site. https://www.uptodate.com/contents/mesenteric-venous-thrombosis-in-adults. Accessed November 16, 2017.
4. Al-Zahrani HA, Lindsay T. Mesenteric ischemia. In: Hall JB, Schmidt GA, Kress JP, eds. Principles of Critical Care. 4th ed. New York, NY: McGraw Hill; 2015:1036-1044.
5. Kumar S, Sarr MG, Kamath PS. Mesenteric venous thrombosis. N Engl J Med. 2001;345(23):1683-1688. doi:10.1056/nejmra010076.
6. Al-Khazraji A, Hasan AQ, Patel I, Alkhawam H, Ghrair F, Lieber J. The role of abdominal computed tomography scan in acute pancreatitis. Pancreas. 2017;46(6):e52-e54. doi:10.1097/mpa.0000000000000837.
7. Bradbury MS, Kavanagh PV, Bechtold RE, et al. Mesenteric venous thrombosis: diagnosis and noninvasive imaging. Radiographics. 2002;22(3):527-541.
8. Yang S, Fan X, Ding W, et al. D-dimer as an early marker of severity in patients with acute superior mesenteric venous thrombosis. Medicine (Baltimore). 2014;93(29):e270. doi:10.1097/md.0000000000000270.
9. Matos C, Van Gansbeke D, Zalcman M, et al. Mesenteric vein thrombosis: early CT and US diagnosis and conservative management. Gastrointest Radiol. 1986;11(4):322-325.
10. Rhee RY, Gloviczki P, Mendonca CT, et al. Mesenteric venous thrombosis: still a lethal disease in the 1990s. J Vasc Surg. 1994;20(5):688-697.
11. Furukawa A, Kanasaki S, Kono N, et al. CT diagnosis of acute mesenteric ischemia from various causes. AJR Am J Roentgenol. 2009;192(2):408-416. doi:10.2214/ajr.08.1138.
12. Johnson CC, Baggenstoss AH. Mesenteric vascular occlusion; study of 99 cases of occlusion of veins. Proc Staff Meet Mayo Clin. 1949;24(25):628-636.13. Hmoud B, Singal AK, Kamath PS. Mesenteric venous thrombosis. J Clin Exp Hepatol. 2014;4(3):257-263. doi:10.1016/j.jceh.2014.03.052.
14. Schoots IG, Koffeman GI, Legemate DA, Levi M, van Gulik TM. Systematic review of survival after acute mesenteric ischaemia according to disease aetiology. Br J Surg. 2004;91(1):17-27.
15. Yang S, Fan X, Ding W, et al. Multidisciplinary stepwise management strategy for acute superior mesenteric venous thrombosis: an intestinal stroke center experience. Thromb Res. 2015;135(1):36-45. doi:10.1016/j.thromres.2014.10.018.
1. Harnik IG, Brandt LJ. Mesenteric venous thrombosis. Vasc Med. 2010;15(5):407-418. doi:10.1177/1358863x10379673.
2. Tilsed JV, Casamassima A, Kurihara H, et al. ESTES guidelines: acute mesenteric ischaemia. Eur J Trauma Emerg Surg. 2016;42(2):253-270. doi:10.1007/s00068-016-0634-0.
3. Tendler DA, Lamont JT, Grubel P. Mesenteric venous thrombosis in adults. UpToDate Web site. https://www.uptodate.com/contents/mesenteric-venous-thrombosis-in-adults. Accessed November 16, 2017.
4. Al-Zahrani HA, Lindsay T. Mesenteric ischemia. In: Hall JB, Schmidt GA, Kress JP, eds. Principles of Critical Care. 4th ed. New York, NY: McGraw Hill; 2015:1036-1044.
5. Kumar S, Sarr MG, Kamath PS. Mesenteric venous thrombosis. N Engl J Med. 2001;345(23):1683-1688. doi:10.1056/nejmra010076.
6. Al-Khazraji A, Hasan AQ, Patel I, Alkhawam H, Ghrair F, Lieber J. The role of abdominal computed tomography scan in acute pancreatitis. Pancreas. 2017;46(6):e52-e54. doi:10.1097/mpa.0000000000000837.
7. Bradbury MS, Kavanagh PV, Bechtold RE, et al. Mesenteric venous thrombosis: diagnosis and noninvasive imaging. Radiographics. 2002;22(3):527-541.
8. Yang S, Fan X, Ding W, et al. D-dimer as an early marker of severity in patients with acute superior mesenteric venous thrombosis. Medicine (Baltimore). 2014;93(29):e270. doi:10.1097/md.0000000000000270.
9. Matos C, Van Gansbeke D, Zalcman M, et al. Mesenteric vein thrombosis: early CT and US diagnosis and conservative management. Gastrointest Radiol. 1986;11(4):322-325.
10. Rhee RY, Gloviczki P, Mendonca CT, et al. Mesenteric venous thrombosis: still a lethal disease in the 1990s. J Vasc Surg. 1994;20(5):688-697.
11. Furukawa A, Kanasaki S, Kono N, et al. CT diagnosis of acute mesenteric ischemia from various causes. AJR Am J Roentgenol. 2009;192(2):408-416. doi:10.2214/ajr.08.1138.
12. Johnson CC, Baggenstoss AH. Mesenteric vascular occlusion; study of 99 cases of occlusion of veins. Proc Staff Meet Mayo Clin. 1949;24(25):628-636.13. Hmoud B, Singal AK, Kamath PS. Mesenteric venous thrombosis. J Clin Exp Hepatol. 2014;4(3):257-263. doi:10.1016/j.jceh.2014.03.052.
14. Schoots IG, Koffeman GI, Legemate DA, Levi M, van Gulik TM. Systematic review of survival after acute mesenteric ischaemia according to disease aetiology. Br J Surg. 2004;91(1):17-27.
15. Yang S, Fan X, Ding W, et al. Multidisciplinary stepwise management strategy for acute superior mesenteric venous thrombosis: an intestinal stroke center experience. Thromb Res. 2015;135(1):36-45. doi:10.1016/j.thromres.2014.10.018.

A Ticking Noise From the Chest: Recognition of the Hamman Sign
The authors describe a case of a 21-year-old woman who presented with shortness of breath and exhibited a Hamman sign, an uncommon clinical finding.
Traditionally, a physician develops a differential diagnosis based primarily (>70%) on the history and the physical examination of a patient.1 While modern medicine has developed with new technological devices and a growing number of diagnostic tests, one must not forget the value of a thorough physical examination.
Case
A 21-year-old woman, in previous good health, presented to the ED with the chief complaint of shortness of breath. She stated that she woke up with acute dyspnea and a stabbing pain on the left side of her thorax, related to her breathing. The patient looked distressed upon presentation.
Her vital signs at presentation were: blood pressure, 150/85 mm Hg; heart rate, 120 beats/min; respiratory
During physical examination, a loud ticking noise was heard originating from the thorax, even without a stethoscope (an example of the sound can be heard at https://www.youtube.com/watch?v=mXJHtJeL1mM). During auscultation, the ticking noise was prominent in early systole and audible over all parts of the thorax. The sound was only heard when the patient was in the supine position and disappeared when she sat up. It persisted when the patient was holding her breath. Breath sounds were equal and clear bilaterally. There was no subcutaneous emphysema palpable over the thorax or neck region.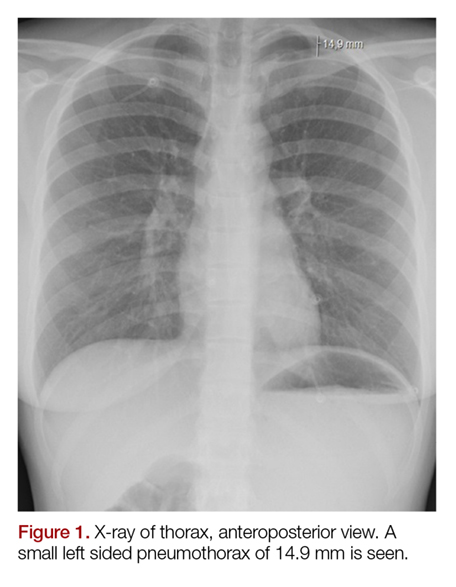
The electrocardiogram and blood results, including D-dimer, were normal. The chest X-ray showed an apical pneumothorax of 1.5 cm on the left side (Figures 1 and 2). There was no evidence of pneumomediastinum or pneumopericardium. The patient received acetaminophen and ibuprofen tablets for pain, and she was discharged home. At the follow-up 2 weeks later, she had no remaining symptoms and the ticking sound had disappeared.
Discussion
These loud intermittent noises originating from the thorax were described for the first time at the beginning of the 19th century.2,3 However, it was Louis Virgil Hamman whose name would be linked to this physical examination finding. In 1937 he described typical clicking, crackling, and popping sounds over the precordium, synchronized with the heartbeat. This was usually in combination with subcutaneous emphysema in the neck region. Hamman presumed that the symptoms were due to mediastinal air caused by rupture alveoli or bronchioles, resulting in interstitial emphysema of the lung parenchyma. In addition, air could leak into the pleural space, causing a pneumothorax. He concluded that the clinical findings were pathognomonic for spontaneous mediastinal emphysema, and this physical examination finding became known as the “Hamman sign”.4-11
However, in the following years it was demonstrated that the appearance of loud, systolic clicking noises over the precordium could also be present in patients with a small spontaneous left-sided pneumothorax.5-9 It was assumed the pneumothorax caused a small amount of air to accumulate in the pleural space in the major fissure inferiorly, which shifted with the cardiac contraction. This results in the noise being present while in the supine position. In the sitting position, the air moves cranially above the heart, meaning it is not influenced by the cardiac contractions and the noise disappears.5-8 The Hamman sign is absent in right-sided pneumothorax, presumably because of the smaller contact surface between the lung pleura and the mediastinal pleura overlying the heart in comparison to the left side. Also, the contractions of the right side of the heart are much weaker and generate less pressure in comparison to the left atrium and ventricle.8,11Only a small amount of air, approximately 25 mL, is enough to produce the typical sound. In larger pneumothorax’s, with more than 125 mL of intrapleural air, these sounds are absent, because the contractions of the heart cannot create enough pressure to cause the accumulated air to shift in the pleural space.5,8-9Sound analysis in left-sided pneumothorax by Roelandt et al7 showed multiple murmurs which can be present in both systole and diastole. In contrast to pulmonic noises, the Hamman sign persists when the patient is holding their breath and disappears with sitting or standing.3,6-10 Furthermore, it must not be confused with extra heart sounds, which present as a “gallop rhythm”, with a strong resemblance in quality to the first normal heart sound (S1). In addition, extra heart sounds are uncommon in healthy patients and do not appear suddenly or temporarily.5,8
The Hamman sign is a rare physical examination finding, only identified in less than 1% of all patients with a pneumothorax.9 However, its presence is so specific that it is strong evidence for an underlying pneumothorax or pneumomediastinum, even if radiographic imaging is normal.10 As previously stated, since the Hamman sign is mostly commonly associated with a pneumothorax consisting of less than 125 mL of air, these can usually be treated conservatively, without the necessity of placing a chest tube or aspiration. However, when a patient experiences significant shortness of breath, the emergency physician should consider ordering additional imaging, in the form of an ultrasound or a computed tomography scan to identify the underlying cause of the Hamman sign and place a chest tube when clinically indicated.
Conclusion
The Hamman sign is a rare clinical examination finding in left-sided pneumothorax or pneumomediastinum, in which a ticking or crackling noise is heard over the thorax. This is mostly synchronous with the heartbeat and not related to respiration. It is caused by a small amount of accumulated air in the pleural space, which is being displaced by cardiac contractions during the cardiac cycle. Although typically small, pneumothoraces have a good prognosis. Recognition of the Hamman sign is important, and physicians must realize that even a normal chest X-ray does not rule out the diagnosis.
1. Sandler G. Costs of unnecessary tests. Br Med J. 1979;2(6181):21-24.
2. Laennec RTH, Andral G, Laennec M. Traité De L’auscultation Médiate, Et Des Maladies Des Poumons Et Du Coeur. Paris, France: Paris, J.S.Chaudé, 1826; 1837.
3. Lister WA, Camb MB, Lond MRCP. A case of pericardial knock associated with spontaneous pneumothorax. Lancet. 1928;211(5468):1225-1226.
4. Hamman L. Spontaneous interstitial emphysema of the lungs. Tr A Am Physicians. 1937;52:311-319.
5. Scadding JG, Lond MRCP, Wood P. Systolic clicks due to left-sided pneumothorax. Lancet. 1939;234(6067):1208-1211.
6. Scott JT. Mediastinal emphysema and left pneumothorax. Dist Chest. 1957;32(4):421-434.
7. Roelandt J, Willems J, van der Hauwaert LG, de Geest H. Clicks and sounds (whoops) in left-sided pneumothorax. Clinical and phonocardiographic study. Dis Chest. 1969;56(1):31-36.
8. Smit FW, van Embden Andresen GH, Ubbens R. “Hammans’s sign”, pneumomediastinum and pneumothorax. Ned Tijdschr Geneeskd. 1974;118(22):828-833.
9. Baumann MH, Sahn SA. Hamman’s sign revisited. Pneumothorax or pneumomediastinum? Chest. 1992;102(4):1281-1282.
10. Remmelts HH, Banga JD. Popping pneumothorax. Neth J Med. 2010;68(4):187.
11. Jaiganesh T, Wright K, Sadana A. Mobile diagnosis: Hamman’s crunch in a primary spontaneous pneumothorax. Emerg Med J. 2010;27(6):482-483. doi:10.1136/emj.2009.079681.
The authors describe a case of a 21-year-old woman who presented with shortness of breath and exhibited a Hamman sign, an uncommon clinical finding.
The authors describe a case of a 21-year-old woman who presented with shortness of breath and exhibited a Hamman sign, an uncommon clinical finding.
Traditionally, a physician develops a differential diagnosis based primarily (>70%) on the history and the physical examination of a patient.1 While modern medicine has developed with new technological devices and a growing number of diagnostic tests, one must not forget the value of a thorough physical examination.
Case
A 21-year-old woman, in previous good health, presented to the ED with the chief complaint of shortness of breath. She stated that she woke up with acute dyspnea and a stabbing pain on the left side of her thorax, related to her breathing. The patient looked distressed upon presentation.
Her vital signs at presentation were: blood pressure, 150/85 mm Hg; heart rate, 120 beats/min; respiratory
During physical examination, a loud ticking noise was heard originating from the thorax, even without a stethoscope (an example of the sound can be heard at https://www.youtube.com/watch?v=mXJHtJeL1mM). During auscultation, the ticking noise was prominent in early systole and audible over all parts of the thorax. The sound was only heard when the patient was in the supine position and disappeared when she sat up. It persisted when the patient was holding her breath. Breath sounds were equal and clear bilaterally. There was no subcutaneous emphysema palpable over the thorax or neck region.
The electrocardiogram and blood results, including D-dimer, were normal. The chest X-ray showed an apical pneumothorax of 1.5 cm on the left side (Figures 1 and 2). There was no evidence of pneumomediastinum or pneumopericardium. The patient received acetaminophen and ibuprofen tablets for pain, and she was discharged home. At the follow-up 2 weeks later, she had no remaining symptoms and the ticking sound had disappeared.
Discussion
These loud intermittent noises originating from the thorax were described for the first time at the beginning of the 19th century.2,3 However, it was Louis Virgil Hamman whose name would be linked to this physical examination finding. In 1937 he described typical clicking, crackling, and popping sounds over the precordium, synchronized with the heartbeat. This was usually in combination with subcutaneous emphysema in the neck region. Hamman presumed that the symptoms were due to mediastinal air caused by rupture alveoli or bronchioles, resulting in interstitial emphysema of the lung parenchyma. In addition, air could leak into the pleural space, causing a pneumothorax. He concluded that the clinical findings were pathognomonic for spontaneous mediastinal emphysema, and this physical examination finding became known as the “Hamman sign”.4-11
However, in the following years it was demonstrated that the appearance of loud, systolic clicking noises over the precordium could also be present in patients with a small spontaneous left-sided pneumothorax.5-9 It was assumed the pneumothorax caused a small amount of air to accumulate in the pleural space in the major fissure inferiorly, which shifted with the cardiac contraction. This results in the noise being present while in the supine position. In the sitting position, the air moves cranially above the heart, meaning it is not influenced by the cardiac contractions and the noise disappears.5-8 The Hamman sign is absent in right-sided pneumothorax, presumably because of the smaller contact surface between the lung pleura and the mediastinal pleura overlying the heart in comparison to the left side. Also, the contractions of the right side of the heart are much weaker and generate less pressure in comparison to the left atrium and ventricle.8,11Only a small amount of air, approximately 25 mL, is enough to produce the typical sound. In larger pneumothorax’s, with more than 125 mL of intrapleural air, these sounds are absent, because the contractions of the heart cannot create enough pressure to cause the accumulated air to shift in the pleural space.5,8-9Sound analysis in left-sided pneumothorax by Roelandt et al7 showed multiple murmurs which can be present in both systole and diastole. In contrast to pulmonic noises, the Hamman sign persists when the patient is holding their breath and disappears with sitting or standing.3,6-10 Furthermore, it must not be confused with extra heart sounds, which present as a “gallop rhythm”, with a strong resemblance in quality to the first normal heart sound (S1). In addition, extra heart sounds are uncommon in healthy patients and do not appear suddenly or temporarily.5,8
The Hamman sign is a rare physical examination finding, only identified in less than 1% of all patients with a pneumothorax.9 However, its presence is so specific that it is strong evidence for an underlying pneumothorax or pneumomediastinum, even if radiographic imaging is normal.10 As previously stated, since the Hamman sign is mostly commonly associated with a pneumothorax consisting of less than 125 mL of air, these can usually be treated conservatively, without the necessity of placing a chest tube or aspiration. However, when a patient experiences significant shortness of breath, the emergency physician should consider ordering additional imaging, in the form of an ultrasound or a computed tomography scan to identify the underlying cause of the Hamman sign and place a chest tube when clinically indicated.
Conclusion
The Hamman sign is a rare clinical examination finding in left-sided pneumothorax or pneumomediastinum, in which a ticking or crackling noise is heard over the thorax. This is mostly synchronous with the heartbeat and not related to respiration. It is caused by a small amount of accumulated air in the pleural space, which is being displaced by cardiac contractions during the cardiac cycle. Although typically small, pneumothoraces have a good prognosis. Recognition of the Hamman sign is important, and physicians must realize that even a normal chest X-ray does not rule out the diagnosis.
Traditionally, a physician develops a differential diagnosis based primarily (>70%) on the history and the physical examination of a patient.1 While modern medicine has developed with new technological devices and a growing number of diagnostic tests, one must not forget the value of a thorough physical examination.
Case
A 21-year-old woman, in previous good health, presented to the ED with the chief complaint of shortness of breath. She stated that she woke up with acute dyspnea and a stabbing pain on the left side of her thorax, related to her breathing. The patient looked distressed upon presentation.
Her vital signs at presentation were: blood pressure, 150/85 mm Hg; heart rate, 120 beats/min; respiratory
During physical examination, a loud ticking noise was heard originating from the thorax, even without a stethoscope (an example of the sound can be heard at https://www.youtube.com/watch?v=mXJHtJeL1mM). During auscultation, the ticking noise was prominent in early systole and audible over all parts of the thorax. The sound was only heard when the patient was in the supine position and disappeared when she sat up. It persisted when the patient was holding her breath. Breath sounds were equal and clear bilaterally. There was no subcutaneous emphysema palpable over the thorax or neck region.
The electrocardiogram and blood results, including D-dimer, were normal. The chest X-ray showed an apical pneumothorax of 1.5 cm on the left side (Figures 1 and 2). There was no evidence of pneumomediastinum or pneumopericardium. The patient received acetaminophen and ibuprofen tablets for pain, and she was discharged home. At the follow-up 2 weeks later, she had no remaining symptoms and the ticking sound had disappeared.
Discussion
These loud intermittent noises originating from the thorax were described for the first time at the beginning of the 19th century.2,3 However, it was Louis Virgil Hamman whose name would be linked to this physical examination finding. In 1937 he described typical clicking, crackling, and popping sounds over the precordium, synchronized with the heartbeat. This was usually in combination with subcutaneous emphysema in the neck region. Hamman presumed that the symptoms were due to mediastinal air caused by rupture alveoli or bronchioles, resulting in interstitial emphysema of the lung parenchyma. In addition, air could leak into the pleural space, causing a pneumothorax. He concluded that the clinical findings were pathognomonic for spontaneous mediastinal emphysema, and this physical examination finding became known as the “Hamman sign”.4-11
However, in the following years it was demonstrated that the appearance of loud, systolic clicking noises over the precordium could also be present in patients with a small spontaneous left-sided pneumothorax.5-9 It was assumed the pneumothorax caused a small amount of air to accumulate in the pleural space in the major fissure inferiorly, which shifted with the cardiac contraction. This results in the noise being present while in the supine position. In the sitting position, the air moves cranially above the heart, meaning it is not influenced by the cardiac contractions and the noise disappears.5-8 The Hamman sign is absent in right-sided pneumothorax, presumably because of the smaller contact surface between the lung pleura and the mediastinal pleura overlying the heart in comparison to the left side. Also, the contractions of the right side of the heart are much weaker and generate less pressure in comparison to the left atrium and ventricle.8,11Only a small amount of air, approximately 25 mL, is enough to produce the typical sound. In larger pneumothorax’s, with more than 125 mL of intrapleural air, these sounds are absent, because the contractions of the heart cannot create enough pressure to cause the accumulated air to shift in the pleural space.5,8-9Sound analysis in left-sided pneumothorax by Roelandt et al7 showed multiple murmurs which can be present in both systole and diastole. In contrast to pulmonic noises, the Hamman sign persists when the patient is holding their breath and disappears with sitting or standing.3,6-10 Furthermore, it must not be confused with extra heart sounds, which present as a “gallop rhythm”, with a strong resemblance in quality to the first normal heart sound (S1). In addition, extra heart sounds are uncommon in healthy patients and do not appear suddenly or temporarily.5,8
The Hamman sign is a rare physical examination finding, only identified in less than 1% of all patients with a pneumothorax.9 However, its presence is so specific that it is strong evidence for an underlying pneumothorax or pneumomediastinum, even if radiographic imaging is normal.10 As previously stated, since the Hamman sign is mostly commonly associated with a pneumothorax consisting of less than 125 mL of air, these can usually be treated conservatively, without the necessity of placing a chest tube or aspiration. However, when a patient experiences significant shortness of breath, the emergency physician should consider ordering additional imaging, in the form of an ultrasound or a computed tomography scan to identify the underlying cause of the Hamman sign and place a chest tube when clinically indicated.
Conclusion
The Hamman sign is a rare clinical examination finding in left-sided pneumothorax or pneumomediastinum, in which a ticking or crackling noise is heard over the thorax. This is mostly synchronous with the heartbeat and not related to respiration. It is caused by a small amount of accumulated air in the pleural space, which is being displaced by cardiac contractions during the cardiac cycle. Although typically small, pneumothoraces have a good prognosis. Recognition of the Hamman sign is important, and physicians must realize that even a normal chest X-ray does not rule out the diagnosis.
1. Sandler G. Costs of unnecessary tests. Br Med J. 1979;2(6181):21-24.
2. Laennec RTH, Andral G, Laennec M. Traité De L’auscultation Médiate, Et Des Maladies Des Poumons Et Du Coeur. Paris, France: Paris, J.S.Chaudé, 1826; 1837.
3. Lister WA, Camb MB, Lond MRCP. A case of pericardial knock associated with spontaneous pneumothorax. Lancet. 1928;211(5468):1225-1226.
4. Hamman L. Spontaneous interstitial emphysema of the lungs. Tr A Am Physicians. 1937;52:311-319.
5. Scadding JG, Lond MRCP, Wood P. Systolic clicks due to left-sided pneumothorax. Lancet. 1939;234(6067):1208-1211.
6. Scott JT. Mediastinal emphysema and left pneumothorax. Dist Chest. 1957;32(4):421-434.
7. Roelandt J, Willems J, van der Hauwaert LG, de Geest H. Clicks and sounds (whoops) in left-sided pneumothorax. Clinical and phonocardiographic study. Dis Chest. 1969;56(1):31-36.
8. Smit FW, van Embden Andresen GH, Ubbens R. “Hammans’s sign”, pneumomediastinum and pneumothorax. Ned Tijdschr Geneeskd. 1974;118(22):828-833.
9. Baumann MH, Sahn SA. Hamman’s sign revisited. Pneumothorax or pneumomediastinum? Chest. 1992;102(4):1281-1282.
10. Remmelts HH, Banga JD. Popping pneumothorax. Neth J Med. 2010;68(4):187.
11. Jaiganesh T, Wright K, Sadana A. Mobile diagnosis: Hamman’s crunch in a primary spontaneous pneumothorax. Emerg Med J. 2010;27(6):482-483. doi:10.1136/emj.2009.079681.
1. Sandler G. Costs of unnecessary tests. Br Med J. 1979;2(6181):21-24.
2. Laennec RTH, Andral G, Laennec M. Traité De L’auscultation Médiate, Et Des Maladies Des Poumons Et Du Coeur. Paris, France: Paris, J.S.Chaudé, 1826; 1837.
3. Lister WA, Camb MB, Lond MRCP. A case of pericardial knock associated with spontaneous pneumothorax. Lancet. 1928;211(5468):1225-1226.
4. Hamman L. Spontaneous interstitial emphysema of the lungs. Tr A Am Physicians. 1937;52:311-319.
5. Scadding JG, Lond MRCP, Wood P. Systolic clicks due to left-sided pneumothorax. Lancet. 1939;234(6067):1208-1211.
6. Scott JT. Mediastinal emphysema and left pneumothorax. Dist Chest. 1957;32(4):421-434.
7. Roelandt J, Willems J, van der Hauwaert LG, de Geest H. Clicks and sounds (whoops) in left-sided pneumothorax. Clinical and phonocardiographic study. Dis Chest. 1969;56(1):31-36.
8. Smit FW, van Embden Andresen GH, Ubbens R. “Hammans’s sign”, pneumomediastinum and pneumothorax. Ned Tijdschr Geneeskd. 1974;118(22):828-833.
9. Baumann MH, Sahn SA. Hamman’s sign revisited. Pneumothorax or pneumomediastinum? Chest. 1992;102(4):1281-1282.
10. Remmelts HH, Banga JD. Popping pneumothorax. Neth J Med. 2010;68(4):187.
11. Jaiganesh T, Wright K, Sadana A. Mobile diagnosis: Hamman’s crunch in a primary spontaneous pneumothorax. Emerg Med J. 2010;27(6):482-483. doi:10.1136/emj.2009.079681.
Primary Cutaneous Apocrine Carcinoma Arising Within a Nevus Sebaceus
Nevus sebaceus (NS) is a benign hair follicle neoplasm present in approximately 1.3% of the population, typically involving the scalp, neck, or face.1 These lesions usually are present at birth or identified soon after, during the first year. They present as a yellowish hairless patch or plaque but can develop a more papillomatous appearance, especially after puberty. Historically, the concern with NS was its tendency to transform into basal cell carcinoma (BCC), which prompted surgical excision of the lesion during childhood. This theory has been discounted more recently, as further research has suggested that what was once thought to be BCC may have been confused with the similarly appearing trichoblastoma; however, malignant transformation of NS does still occur, with BCC still being the most common.2 We present the case of a long-standing NS with rare transformation to apocrine carcinoma.
Case Report
A 76-year-old woman presented with several new lesions within a previously diagnosed NS. She reported having the large plaque for as long as she could recall but reported that several new growths developed within the plaque over the last 2 months, slowly increasing in size. She reported a prior biopsy within the growth several years prior, which she described as an irritated seborrheic keratosis.
Physical examination demonstrated 4 distinct lesions within the flesh-colored, verrucous plaque located on the left side of the temporal scalp (Figure 1). The first lesion was a 2.5-cm pearly, pink, exophytic tumor (labeled as A in Figure 1). The next 2 lesions were brown, pedunculated, verrucous papules (labeled as B and C in Figure 1). The last lesion was a purple papule (labeled as D in Figure 1). Four shave biopsies were performed for histologic analysis of the lesions. Lesions B, C, and D were consistent with trichoblastomas, as pathology showed basaloid epithelial tumors that displayed primitive follicular structures, areas of stromal induction, and some pigmentation. Lesion A, originally thought to be suspicious for a BCC, was determined to be a primary cutaneous apocrine adenocarcinoma upon pathologic review. The pathology showed a dermal tumor displaying solid and tubular areas with decapitation secretion. Nuclear pleomorphism and mitoses were present (Figure 2), and staining for carcinoembryonic antigen was positive (Figure 3). Immunoreactivity with epithelial membrane antigen and cytokeratin 7 was noted as well as focal positivity for mammaglobin. Primary apocrine carcinoma was favored over metastatic carcinoma due to the location of the lesion within an NS along with a negative history of internal malignancy. Dermatopathology recommended complete removal of all lesions within the NS.
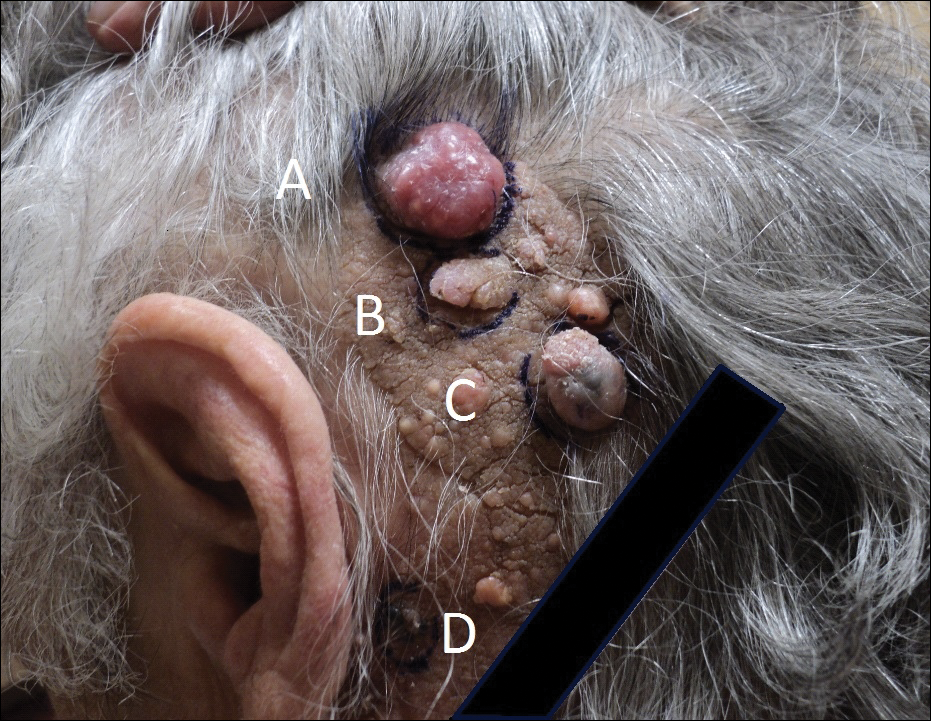

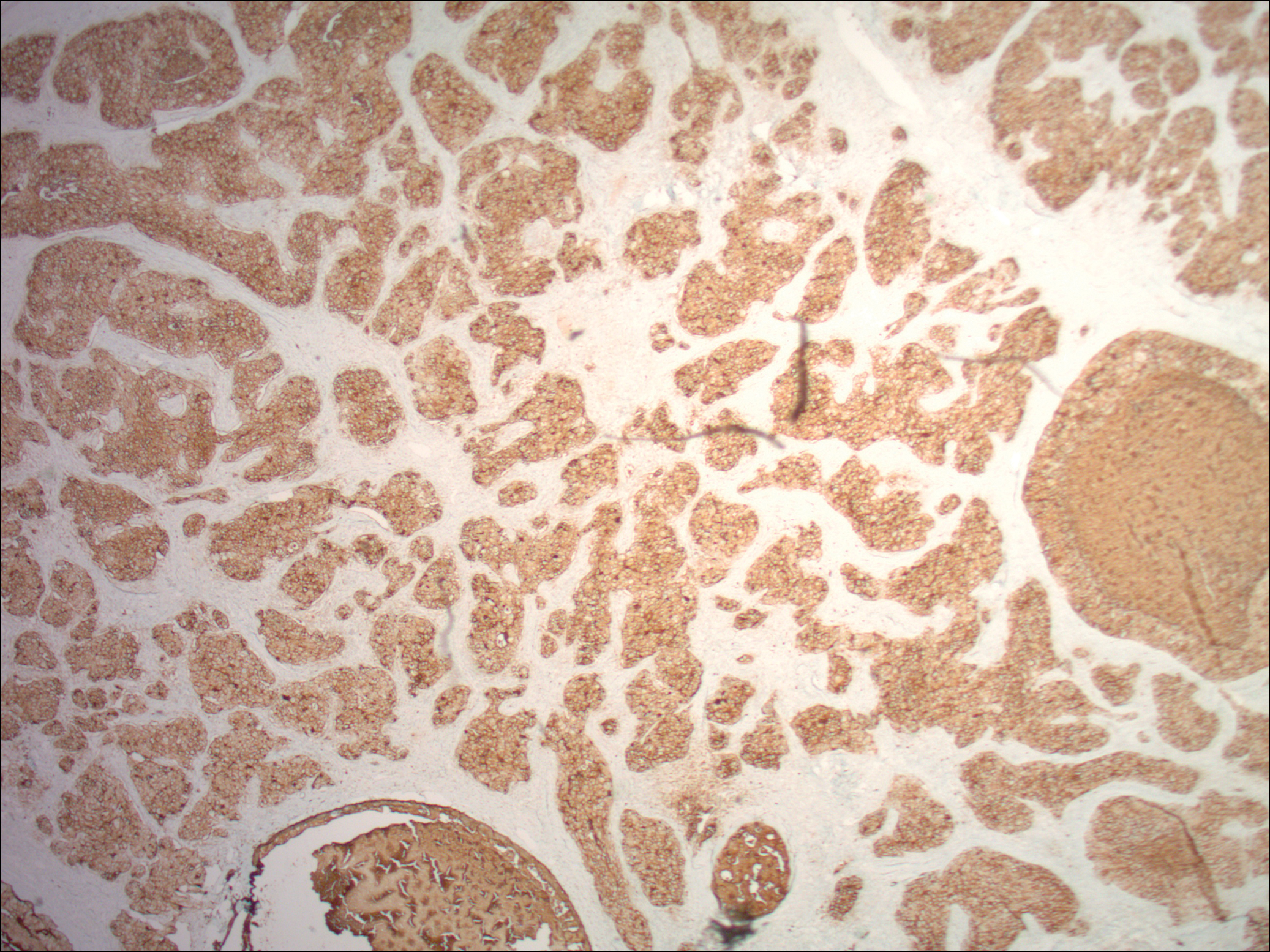
Upon discussing biopsy results and recommendations with our patient, she agreed to undergo excision with intraoperative pathology by a plastic surgeon within our practice to ensure clear margins. The surgical defect following excision was sizeable and closed utilizing a rhomboid flap, full-thickness skin graft, and a split-thickness skin graft. At surgical follow-up, she was doing well and there have been no signs of local recurrence for 10 months since excision.
Comment
Presentation
Nevus sebaceus is the most common adnexal tumor and is classified as a benign congenital hair follicle tumor that is located most commonly on the scalp but also occurs on the face and neck.1 The lesions usually are present at birth but also can develop during the first year of life.2 Diagnosis may be later, during adolescence, when patients seek medical attention during the lesion’s rapid growth phase.1 Nevus sebaceus also is known as an organoid nevus because it may contain all components of the skin. It was originally identified by Jadassohn in 1895.3 It presents as a yellowish, smooth, hairless patch or plaque in prepubertal patients. During adolescence, the lesion typically becomes more yellowish, as well as papillomatous, scaly, or warty. The reported incidence of NS is 0.05% to 1% in dermatology patients.2
Differential
Nevus sebaceus also is a component of several syndromes that should be kept in mind, including Schimmelpenning-Feuerstein-Mims syndrome, which presents with neurologic, skeletal, genitourinary, cardiovascular, and ophthalmic disorders, in addition to cutaneous features. Others include phacomatosis pigmentokeratotica, didmyosis aplasticosebacea, SCALP syndrome (sebaceus nevus, central nervous system malformations, aplasia cutis congenita, limbal dermoid, and pigmented nevus), and more.4,5
Etiology
The etiology of NS has not been completely determined. One study that evaluated 44 NS tissue samples suggested the presence of human papillomavirus (HPV) in NS formation, finding that 82% of NS lesions studied contained HPV DNA. From these results, Carlson et al6 suggested a possible maternal transmission of HPV and infection of ectodermal cells as a potential cause of NS; however, this hypothesis was soon challenged by a study that showed a complete absence of HPV in 16 samples via histological evaluation and polymerase chain reaction for a broad range of HPV types.7 There were investigations into a patched (PTCH) deletion as the cause of NS and thus explained the historically high rate of secondary BCC.8 Further studies showed no mutations at the PTCH locus in trichoblastomas or other tumors arising from NS.9,10
More recent studies have recognized HRAS and KRAS mutations as a causative factor in NS.11 Nevus sebaceus belongs to a group of syndromes resulting from lethal mutations that survive via mosaicism. Nevus sebaceus is caused by postzygotic HRAS or KRAS mutations and is known as a mosaic RASopathy.12 In fact, there is growing evidence to suggest that other nevoid proliferations including keratinocytic epidermal nevi and melanocytic nevi also fall into the spectrum of mosaic RASopathies.13
Staging
There are 3 clinical stages of NS, originally described by Mehregan and Pinkus.14 In stage I (historically known as the infantile stage), the lesion presents as a yellow to pink, smooth, hairless patch. Histologic features include immature hair follicles and hypoplastic sebaceous glands. In stage II (also known as the puberty stage), the lesion becomes more pronounced. Firmer plaques can develop with hyperkeratosis. Hormonal changes cause sebaceous glands to develop, accompanied by epidermal hyperplasia and maturation of apocrine glands. Stage III (the tumoral stage) is a period that various neoplasms have the highest likelihood of occurring. Nevus sebaceus in an adolescent or adult demonstrates mature adnexal structures and greater epidermal hyperplasia.2,4,15
Malignancy
By virtue of these stages of NS development, malignant transformation is expected most often during stage III. However, cases have been reported of malignant tumor development in NS in children before puberty. Two case reports described a 7-year-old boy and a 10-year-old boy diagnosed with a BCC arising from an NS.16,17 However, secondary BCC formation before 16 years of age is rare. Basal cell carcinoma arising from an NS has been commonly reported and is the most common malignant neoplasm in NS (1.1%).2,3 However, the most common neoplasm overall is trichoblastoma (7.4%). The second most common tumor was syringocystadenoma papilliferum, occurring in approximately 5.2% of NS cases. The neoplasm rate in NS was found to be proportional to the patient age.2,18 Multiple studies have shown the overall rate of secondary neoplasms in NS to be 13% to 21.4%, with malignant tumors composing 0.8% to 2.5%.2,15,19 Other neoplasms that have been reported include keratoacanthoma, trichilemmoma, sebaceoma, nevocellular nevus, squamous cell carcinoma, adnexal carcinoma, apocrine adenocarcinoma, and malignant melanoma.19-21
It is argued that the reported rate of BCC formation is overestimated, as prior studies incorrectly labeled trichoblastomas as BCCs. In fact, the largest studies of NS from the 1990s revealed lower rates of malignant secondary tumors than previously determined.4
The identification of apocrine adenocarcinoma tumors arising from NS is exceedingly rare. A study performed by Cribier et al19 in 2000 retrospect
Histopathology
Histopathologic examination reveals considerable variation in morphology, and an underlying pattern has been difficult to recognize. Unfortunately, some authors have concluded that the diagnosis of apocrine carcinoma is relatively subjective.26 Robson et al26 identified 3 general architectural patterns: tubular, tubulopapillary, and solid. Tubular structures consisted of glands and ducts lined by a single or multilayered epithelium. Tubulopapillary architecture was characterized by epithelium forming papillary folds without a fibrovascular core. The solid morphology showed sheets of cells with limited ductal or tubular formation.26 The most specific criteria of these apocrine carcinomas are identification of decapitation secretion, periodic acid–Schiff–positive diastase-resistant material present in the cells or lumen, and positive immunostaining for gross cystic disease fluid protein-15.27
Robson et al26 reported estrogen receptor positivity and androgen receptor positivity in 62% and 64% of 24 primary apocrine carcinoma cases, respectively. However, whether these markers are as common in NS-related apocrine carcinomas has yet to be noted in the literature. One study reports a case of apocrine carcinoma from NS with positive staining for human epidermal growth factor-2, a cell membrane receptor tyrosine kinase commonly investigated in breast cancers and extramammary Paget disease.22
These apocrine carcinomas do have the potential for lymphatic metastasis, as seen with multiple studies. Domingo and Helwig21 identified regional lymph node metastasis in 2 of its 4 apocrine carcinoma patients. Robson et al26 reported lymphovascular invasion in 4 cases and perineural invasion in 2 of 24 patients studied. However, even in the context of recurrence and regional metastasis, the prognosis was good and seldom fatal.26
Treatment
The most effective treatment of NS is excision of dermal and epidermal components. Excision should be completed with a minimum of 2- to 3-mm margins and full thickness down to the underlying supporting fat.28 Historically, the practice of prophylactic excision of NS was supported by the potential for malignant transformation; however, early excision of NS may be less reasonable in light of these more recent studies showing lower incidence of BCC (0.8%), replaced by benign trichoblastomas.19 In the case of apocrine carcinoma development, excision is undoubtedly recommended, with unclear recommendations regarding further evaluation for metastasis.
Excision also may be favored for cosmetic purposes, given the visible regions where NS tends to develop. Chepla and Gosain29 argued that surgical intervention should be based on other factors such as location on the scalp, alopecia, and other issues affecting appearance and monitoring rather than incidence of malignant transformation. Close monitoring and biopsy of suspicious areas is a more conservative option.
Other therapies include CO2 laser, as demonstrated by Kiedrowicz et al,30 on linear NS in a patient with Schimmelpenning-Feuerstein-Mims syndrome.31 However, this approach is palliative and not effective in removing the entire lesion. Electrodesiccation and curettage and dermabrasion also are not good options for the same reason.4
Occurrence in Children
Nevus sebaceus in children, accompanied by other findings suggestive of epidermal nevus syndromes, should prompt further investigation. Schimmelpenning-Feuerstein-Mims syndrome includes major neurological abnormalities including hemimegalencephaly and seizures.32
Conclusion
Apocrine carcinomas are malignant neoplasms that may rarely arise within an NS. Their clinical identification is difficult and requires histopathologic evaluation. Upon recognition, prompt excision with tumor-free margins is recommended. As a rare entity, little data is available regarding its metastatic potential or overall survival rates. Further investigation is clearly necessary as new cases arise.
- Kamyab-Hesari K, Balochi K, Afshar N, et al. Clinicopathological study of 1016 consecutive adnexal skin tumors. Acta Med Iran. 2013;51:879-885.
- Idriss MH, Elston DM. Secondary neoplasms associated with nevus sebaceus of Jadassohn: a study of 707 cases. J Am Acad Dermatol. 2014;70:332-337.
- Ball EA, Hussain M, Moss AL. Squamous cell carcinoma and basal cell carcinoma arising in a naevus sebaceous of Jadassohn: case report and literature review. Clin Exp Dermatol. 2005;30:259-260.
- Moody MN, Landau JM, Goldberg LH. Nevus sebaceous revisited. Pediatr Dermatol. 2012;29:15-23.
- Happle R. The group of epidermal nevus syndromes part I. well defined phenotypes. J Am Acad Dermatol. 2010;63:1-22; quiz 23-24.
- Carlson JA, Cribier B, Nuovo G, et al. Epidermodysplasia verruciformis-associated and genital-mucosal high-risk human papillomavirus DNA are prevalent in nevus sebaceus of Jadassohn. J Am Acad Dermatol. 2008;59:279-294.
- Kim D, Benjamin LT, Sahoo MK, et al. Human papilloma virus is not prevalent in nevus sebaceus [published online November 14, 2013]. Pediatr Dermatol. 2014;31:326-330.
- Xin H, Matt D, Qin JZ, et al. The sebaceous nevus: a nevus with deletions of the PTCH gene. Cancer Res. 1999;59:1834-1836.
- Hafner C, Schmiemann V, Ruetten A, et al. PTCH mutations are not mainly involved in the pathogenesis of sporadic trichoblastomas. Hum Pathol. 2007;38:1496-1500.
- Takata M, Tojo M, Hatta N, et al. No evidence of deregulated patched-hedgehog signaling pathway in trichoblastomas and other tumors arising within nevus sebaceous. J Invest Dermatol. 2001;117:1666-1670.
- Levinsohn JL, Tian LC, Boyden LM, et al. Whole-exome sequencing reveals somatic mutations in HRAS and KRAS, which cause nevus sebaceus [published online October 25, 2012]. J Invest Dermatol. 2013;133:827-830.
- Happle R. Nevus sebaceus is a mosaic RASopathy. J Invest Dermatol. 2013;133:597-600.
- Luo S, Tsao H. Epidermal, sebaceous, and melanocytic nevoid proliferations are spectrums of mosaic RASopathies. J Invest Dermatol. 2014;134:2493-2496.
- Mehregan AH, Pinkus H. Life history of organoid nevi. special reference to nevus sebaceus of Jadassohn. Arch Dermatol. 1965;91:574-588.
- Muñoz-Pérez MA, García-Hernandez MJ, Ríos JJ, et al. Sebaceus naevi: a clinicopathologic study. J Eur Acad Dermatol Venereol. 2002;16:319-324.
- Altaykan A, Ersoy-Evans S, Erkin G, et al. Basal cell carcinoma arising in nevus sebaceous during childhood. Pediatr Dermatol. 2008;25:616-619.
- Turner CD, Shea CR, Rosoff PM. Basal cell carcinoma originating from a nevus sebaceus on the scalp of a 7-year-old boy. J Pediatr Hematol Oncol. 2001;23:247-249.
- Jaqueti G, Requena L, Sánchez Yus E. Trichoblastoma is the most common neoplasm developed in nevus sebaceus of Jadassohn: a clinicopathologic study of a series of 155 cases. Am J Dermatopathol. 2000;22:108-118.
- Cribier B, Scrivener Y, Grosshans E. Tumors arising in nevus sebaceus: a study of 596 cases. J Am Acad Dermatol. 2000;42(2, pt 1):263-268.
- Paudel U, Jha A, Pokhrel DB, et al. Apocrine carcinoma developing in a naevus sebaceous of scalp. Kathmandu Univ Med J (KUMJ). 2012;10:103-105.
- Domingo J, Helwig EB. Malignant neoplasms associated with nevus sebaceus of Jadassohn. J Am Acad Dermatol. 1979;1:545-556.
- Tanese K, Wakabayashi A, Suzuki T, et al. Immunoexpression of human epidermal growth factor receptor-2 in apocrine carcinoma arising in naevus sebaceous, case report [published online August 23, 2009]. J Eur Acad Dermatol Venereol. 2010;24:360-362.
- Dalle S, Skowron F, Balme B, et al. Apocrine carcinoma developed in nevus sebaceus of Jadassohn. Eur J Dermatol. 2003;13:487-489.
- Jacyk WK, Requena L, Sánchez Yus E, et al. Tubular apocrine carcinoma arising in a nevus sebaceus of Jadassohn. Am J Dermatopathol. 1998;20:389-392.
- Ansai S, Koseki S, Hashimoto H, et al. A case of ductal sweat gland carcinoma connected to syringocystadenoma papilliferum arising in nevus sebaceus. J Cutan Pathol. 1994;21:557-563.
- Robson A, Lazar AJ, Ben Nagi J, et al. Primary cutaneous apocrine carcinoma: a clinico-pathologic analysis of 24 cases. Am J Surg Pathol. 2008;32:682-690.
- Paties C, Taccagni GL, Papotti M, et al. Apocrine carcinoma of the skin. a clinicopathologic, immunocytochemical, and ultrastructural study. Cancer. 1993;71:375-381.
- Davison SP, Khachemoune A, Yu D, et al. Nevus sebaceus of Jadassohn revisited with reconstruction options. Int J Dermatol. 2005;44:145-150.
- Chepla KJ, Gosain AK. Giant nevus sebaceus: definition, surgical techniques, and rationale for treatment. Plast Reconstr Surg. 2012;130:296E-304E.
- Kiedrowicz M, Kacalak-Rzepka A, Królicki A et al. Therapeutic effects of CO2 laser therapy of linear nevus sebaceous in the course of the Schimmelpenning-Feuerstein-Mims syndrome. Postepy Dermatol Allergol. 2013;30:320-323.
- Ashinoff R. Linear nevus sebaceus of Jadassohn treated with the carbon dioxide laser. Pediatr Dermatol. 1993;10:189-191.
- van de Warrenburg BP, van Gulik S, Renier WO, et al. The linear naevus sebaceus syndrome. Clin Neurol Neurosurg. 1998;100:126-132.
Nevus sebaceus (NS) is a benign hair follicle neoplasm present in approximately 1.3% of the population, typically involving the scalp, neck, or face.1 These lesions usually are present at birth or identified soon after, during the first year. They present as a yellowish hairless patch or plaque but can develop a more papillomatous appearance, especially after puberty. Historically, the concern with NS was its tendency to transform into basal cell carcinoma (BCC), which prompted surgical excision of the lesion during childhood. This theory has been discounted more recently, as further research has suggested that what was once thought to be BCC may have been confused with the similarly appearing trichoblastoma; however, malignant transformation of NS does still occur, with BCC still being the most common.2 We present the case of a long-standing NS with rare transformation to apocrine carcinoma.
Case Report
A 76-year-old woman presented with several new lesions within a previously diagnosed NS. She reported having the large plaque for as long as she could recall but reported that several new growths developed within the plaque over the last 2 months, slowly increasing in size. She reported a prior biopsy within the growth several years prior, which she described as an irritated seborrheic keratosis.
Physical examination demonstrated 4 distinct lesions within the flesh-colored, verrucous plaque located on the left side of the temporal scalp (Figure 1). The first lesion was a 2.5-cm pearly, pink, exophytic tumor (labeled as A in Figure 1). The next 2 lesions were brown, pedunculated, verrucous papules (labeled as B and C in Figure 1). The last lesion was a purple papule (labeled as D in Figure 1). Four shave biopsies were performed for histologic analysis of the lesions. Lesions B, C, and D were consistent with trichoblastomas, as pathology showed basaloid epithelial tumors that displayed primitive follicular structures, areas of stromal induction, and some pigmentation. Lesion A, originally thought to be suspicious for a BCC, was determined to be a primary cutaneous apocrine adenocarcinoma upon pathologic review. The pathology showed a dermal tumor displaying solid and tubular areas with decapitation secretion. Nuclear pleomorphism and mitoses were present (Figure 2), and staining for carcinoembryonic antigen was positive (Figure 3). Immunoreactivity with epithelial membrane antigen and cytokeratin 7 was noted as well as focal positivity for mammaglobin. Primary apocrine carcinoma was favored over metastatic carcinoma due to the location of the lesion within an NS along with a negative history of internal malignancy. Dermatopathology recommended complete removal of all lesions within the NS.
Upon discussing biopsy results and recommendations with our patient, she agreed to undergo excision with intraoperative pathology by a plastic surgeon within our practice to ensure clear margins. The surgical defect following excision was sizeable and closed utilizing a rhomboid flap, full-thickness skin graft, and a split-thickness skin graft. At surgical follow-up, she was doing well and there have been no signs of local recurrence for 10 months since excision.
Comment
Presentation
Nevus sebaceus is the most common adnexal tumor and is classified as a benign congenital hair follicle tumor that is located most commonly on the scalp but also occurs on the face and neck.1 The lesions usually are present at birth but also can develop during the first year of life.2 Diagnosis may be later, during adolescence, when patients seek medical attention during the lesion’s rapid growth phase.1 Nevus sebaceus also is known as an organoid nevus because it may contain all components of the skin. It was originally identified by Jadassohn in 1895.3 It presents as a yellowish, smooth, hairless patch or plaque in prepubertal patients. During adolescence, the lesion typically becomes more yellowish, as well as papillomatous, scaly, or warty. The reported incidence of NS is 0.05% to 1% in dermatology patients.2
Differential
Nevus sebaceus also is a component of several syndromes that should be kept in mind, including Schimmelpenning-Feuerstein-Mims syndrome, which presents with neurologic, skeletal, genitourinary, cardiovascular, and ophthalmic disorders, in addition to cutaneous features. Others include phacomatosis pigmentokeratotica, didmyosis aplasticosebacea, SCALP syndrome (sebaceus nevus, central nervous system malformations, aplasia cutis congenita, limbal dermoid, and pigmented nevus), and more.4,5
Etiology
The etiology of NS has not been completely determined. One study that evaluated 44 NS tissue samples suggested the presence of human papillomavirus (HPV) in NS formation, finding that 82% of NS lesions studied contained HPV DNA. From these results, Carlson et al6 suggested a possible maternal transmission of HPV and infection of ectodermal cells as a potential cause of NS; however, this hypothesis was soon challenged by a study that showed a complete absence of HPV in 16 samples via histological evaluation and polymerase chain reaction for a broad range of HPV types.7 There were investigations into a patched (PTCH) deletion as the cause of NS and thus explained the historically high rate of secondary BCC.8 Further studies showed no mutations at the PTCH locus in trichoblastomas or other tumors arising from NS.9,10
More recent studies have recognized HRAS and KRAS mutations as a causative factor in NS.11 Nevus sebaceus belongs to a group of syndromes resulting from lethal mutations that survive via mosaicism. Nevus sebaceus is caused by postzygotic HRAS or KRAS mutations and is known as a mosaic RASopathy.12 In fact, there is growing evidence to suggest that other nevoid proliferations including keratinocytic epidermal nevi and melanocytic nevi also fall into the spectrum of mosaic RASopathies.13
Staging
There are 3 clinical stages of NS, originally described by Mehregan and Pinkus.14 In stage I (historically known as the infantile stage), the lesion presents as a yellow to pink, smooth, hairless patch. Histologic features include immature hair follicles and hypoplastic sebaceous glands. In stage II (also known as the puberty stage), the lesion becomes more pronounced. Firmer plaques can develop with hyperkeratosis. Hormonal changes cause sebaceous glands to develop, accompanied by epidermal hyperplasia and maturation of apocrine glands. Stage III (the tumoral stage) is a period that various neoplasms have the highest likelihood of occurring. Nevus sebaceus in an adolescent or adult demonstrates mature adnexal structures and greater epidermal hyperplasia.2,4,15
Malignancy
By virtue of these stages of NS development, malignant transformation is expected most often during stage III. However, cases have been reported of malignant tumor development in NS in children before puberty. Two case reports described a 7-year-old boy and a 10-year-old boy diagnosed with a BCC arising from an NS.16,17 However, secondary BCC formation before 16 years of age is rare. Basal cell carcinoma arising from an NS has been commonly reported and is the most common malignant neoplasm in NS (1.1%).2,3 However, the most common neoplasm overall is trichoblastoma (7.4%). The second most common tumor was syringocystadenoma papilliferum, occurring in approximately 5.2% of NS cases. The neoplasm rate in NS was found to be proportional to the patient age.2,18 Multiple studies have shown the overall rate of secondary neoplasms in NS to be 13% to 21.4%, with malignant tumors composing 0.8% to 2.5%.2,15,19 Other neoplasms that have been reported include keratoacanthoma, trichilemmoma, sebaceoma, nevocellular nevus, squamous cell carcinoma, adnexal carcinoma, apocrine adenocarcinoma, and malignant melanoma.19-21
It is argued that the reported rate of BCC formation is overestimated, as prior studies incorrectly labeled trichoblastomas as BCCs. In fact, the largest studies of NS from the 1990s revealed lower rates of malignant secondary tumors than previously determined.4
The identification of apocrine adenocarcinoma tumors arising from NS is exceedingly rare. A study performed by Cribier et al19 in 2000 retrospect
Histopathology
Histopathologic examination reveals considerable variation in morphology, and an underlying pattern has been difficult to recognize. Unfortunately, some authors have concluded that the diagnosis of apocrine carcinoma is relatively subjective.26 Robson et al26 identified 3 general architectural patterns: tubular, tubulopapillary, and solid. Tubular structures consisted of glands and ducts lined by a single or multilayered epithelium. Tubulopapillary architecture was characterized by epithelium forming papillary folds without a fibrovascular core. The solid morphology showed sheets of cells with limited ductal or tubular formation.26 The most specific criteria of these apocrine carcinomas are identification of decapitation secretion, periodic acid–Schiff–positive diastase-resistant material present in the cells or lumen, and positive immunostaining for gross cystic disease fluid protein-15.27
Robson et al26 reported estrogen receptor positivity and androgen receptor positivity in 62% and 64% of 24 primary apocrine carcinoma cases, respectively. However, whether these markers are as common in NS-related apocrine carcinomas has yet to be noted in the literature. One study reports a case of apocrine carcinoma from NS with positive staining for human epidermal growth factor-2, a cell membrane receptor tyrosine kinase commonly investigated in breast cancers and extramammary Paget disease.22
These apocrine carcinomas do have the potential for lymphatic metastasis, as seen with multiple studies. Domingo and Helwig21 identified regional lymph node metastasis in 2 of its 4 apocrine carcinoma patients. Robson et al26 reported lymphovascular invasion in 4 cases and perineural invasion in 2 of 24 patients studied. However, even in the context of recurrence and regional metastasis, the prognosis was good and seldom fatal.26
Treatment
The most effective treatment of NS is excision of dermal and epidermal components. Excision should be completed with a minimum of 2- to 3-mm margins and full thickness down to the underlying supporting fat.28 Historically, the practice of prophylactic excision of NS was supported by the potential for malignant transformation; however, early excision of NS may be less reasonable in light of these more recent studies showing lower incidence of BCC (0.8%), replaced by benign trichoblastomas.19 In the case of apocrine carcinoma development, excision is undoubtedly recommended, with unclear recommendations regarding further evaluation for metastasis.
Excision also may be favored for cosmetic purposes, given the visible regions where NS tends to develop. Chepla and Gosain29 argued that surgical intervention should be based on other factors such as location on the scalp, alopecia, and other issues affecting appearance and monitoring rather than incidence of malignant transformation. Close monitoring and biopsy of suspicious areas is a more conservative option.
Other therapies include CO2 laser, as demonstrated by Kiedrowicz et al,30 on linear NS in a patient with Schimmelpenning-Feuerstein-Mims syndrome.31 However, this approach is palliative and not effective in removing the entire lesion. Electrodesiccation and curettage and dermabrasion also are not good options for the same reason.4
Occurrence in Children
Nevus sebaceus in children, accompanied by other findings suggestive of epidermal nevus syndromes, should prompt further investigation. Schimmelpenning-Feuerstein-Mims syndrome includes major neurological abnormalities including hemimegalencephaly and seizures.32
Conclusion
Apocrine carcinomas are malignant neoplasms that may rarely arise within an NS. Their clinical identification is difficult and requires histopathologic evaluation. Upon recognition, prompt excision with tumor-free margins is recommended. As a rare entity, little data is available regarding its metastatic potential or overall survival rates. Further investigation is clearly necessary as new cases arise.
Nevus sebaceus (NS) is a benign hair follicle neoplasm present in approximately 1.3% of the population, typically involving the scalp, neck, or face.1 These lesions usually are present at birth or identified soon after, during the first year. They present as a yellowish hairless patch or plaque but can develop a more papillomatous appearance, especially after puberty. Historically, the concern with NS was its tendency to transform into basal cell carcinoma (BCC), which prompted surgical excision of the lesion during childhood. This theory has been discounted more recently, as further research has suggested that what was once thought to be BCC may have been confused with the similarly appearing trichoblastoma; however, malignant transformation of NS does still occur, with BCC still being the most common.2 We present the case of a long-standing NS with rare transformation to apocrine carcinoma.
Case Report
A 76-year-old woman presented with several new lesions within a previously diagnosed NS. She reported having the large plaque for as long as she could recall but reported that several new growths developed within the plaque over the last 2 months, slowly increasing in size. She reported a prior biopsy within the growth several years prior, which she described as an irritated seborrheic keratosis.
Physical examination demonstrated 4 distinct lesions within the flesh-colored, verrucous plaque located on the left side of the temporal scalp (Figure 1). The first lesion was a 2.5-cm pearly, pink, exophytic tumor (labeled as A in Figure 1). The next 2 lesions were brown, pedunculated, verrucous papules (labeled as B and C in Figure 1). The last lesion was a purple papule (labeled as D in Figure 1). Four shave biopsies were performed for histologic analysis of the lesions. Lesions B, C, and D were consistent with trichoblastomas, as pathology showed basaloid epithelial tumors that displayed primitive follicular structures, areas of stromal induction, and some pigmentation. Lesion A, originally thought to be suspicious for a BCC, was determined to be a primary cutaneous apocrine adenocarcinoma upon pathologic review. The pathology showed a dermal tumor displaying solid and tubular areas with decapitation secretion. Nuclear pleomorphism and mitoses were present (Figure 2), and staining for carcinoembryonic antigen was positive (Figure 3). Immunoreactivity with epithelial membrane antigen and cytokeratin 7 was noted as well as focal positivity for mammaglobin. Primary apocrine carcinoma was favored over metastatic carcinoma due to the location of the lesion within an NS along with a negative history of internal malignancy. Dermatopathology recommended complete removal of all lesions within the NS.
Upon discussing biopsy results and recommendations with our patient, she agreed to undergo excision with intraoperative pathology by a plastic surgeon within our practice to ensure clear margins. The surgical defect following excision was sizeable and closed utilizing a rhomboid flap, full-thickness skin graft, and a split-thickness skin graft. At surgical follow-up, she was doing well and there have been no signs of local recurrence for 10 months since excision.
Comment
Presentation
Nevus sebaceus is the most common adnexal tumor and is classified as a benign congenital hair follicle tumor that is located most commonly on the scalp but also occurs on the face and neck.1 The lesions usually are present at birth but also can develop during the first year of life.2 Diagnosis may be later, during adolescence, when patients seek medical attention during the lesion’s rapid growth phase.1 Nevus sebaceus also is known as an organoid nevus because it may contain all components of the skin. It was originally identified by Jadassohn in 1895.3 It presents as a yellowish, smooth, hairless patch or plaque in prepubertal patients. During adolescence, the lesion typically becomes more yellowish, as well as papillomatous, scaly, or warty. The reported incidence of NS is 0.05% to 1% in dermatology patients.2
Differential
Nevus sebaceus also is a component of several syndromes that should be kept in mind, including Schimmelpenning-Feuerstein-Mims syndrome, which presents with neurologic, skeletal, genitourinary, cardiovascular, and ophthalmic disorders, in addition to cutaneous features. Others include phacomatosis pigmentokeratotica, didmyosis aplasticosebacea, SCALP syndrome (sebaceus nevus, central nervous system malformations, aplasia cutis congenita, limbal dermoid, and pigmented nevus), and more.4,5
Etiology
The etiology of NS has not been completely determined. One study that evaluated 44 NS tissue samples suggested the presence of human papillomavirus (HPV) in NS formation, finding that 82% of NS lesions studied contained HPV DNA. From these results, Carlson et al6 suggested a possible maternal transmission of HPV and infection of ectodermal cells as a potential cause of NS; however, this hypothesis was soon challenged by a study that showed a complete absence of HPV in 16 samples via histological evaluation and polymerase chain reaction for a broad range of HPV types.7 There were investigations into a patched (PTCH) deletion as the cause of NS and thus explained the historically high rate of secondary BCC.8 Further studies showed no mutations at the PTCH locus in trichoblastomas or other tumors arising from NS.9,10
More recent studies have recognized HRAS and KRAS mutations as a causative factor in NS.11 Nevus sebaceus belongs to a group of syndromes resulting from lethal mutations that survive via mosaicism. Nevus sebaceus is caused by postzygotic HRAS or KRAS mutations and is known as a mosaic RASopathy.12 In fact, there is growing evidence to suggest that other nevoid proliferations including keratinocytic epidermal nevi and melanocytic nevi also fall into the spectrum of mosaic RASopathies.13
Staging
There are 3 clinical stages of NS, originally described by Mehregan and Pinkus.14 In stage I (historically known as the infantile stage), the lesion presents as a yellow to pink, smooth, hairless patch. Histologic features include immature hair follicles and hypoplastic sebaceous glands. In stage II (also known as the puberty stage), the lesion becomes more pronounced. Firmer plaques can develop with hyperkeratosis. Hormonal changes cause sebaceous glands to develop, accompanied by epidermal hyperplasia and maturation of apocrine glands. Stage III (the tumoral stage) is a period that various neoplasms have the highest likelihood of occurring. Nevus sebaceus in an adolescent or adult demonstrates mature adnexal structures and greater epidermal hyperplasia.2,4,15
Malignancy
By virtue of these stages of NS development, malignant transformation is expected most often during stage III. However, cases have been reported of malignant tumor development in NS in children before puberty. Two case reports described a 7-year-old boy and a 10-year-old boy diagnosed with a BCC arising from an NS.16,17 However, secondary BCC formation before 16 years of age is rare. Basal cell carcinoma arising from an NS has been commonly reported and is the most common malignant neoplasm in NS (1.1%).2,3 However, the most common neoplasm overall is trichoblastoma (7.4%). The second most common tumor was syringocystadenoma papilliferum, occurring in approximately 5.2% of NS cases. The neoplasm rate in NS was found to be proportional to the patient age.2,18 Multiple studies have shown the overall rate of secondary neoplasms in NS to be 13% to 21.4%, with malignant tumors composing 0.8% to 2.5%.2,15,19 Other neoplasms that have been reported include keratoacanthoma, trichilemmoma, sebaceoma, nevocellular nevus, squamous cell carcinoma, adnexal carcinoma, apocrine adenocarcinoma, and malignant melanoma.19-21
It is argued that the reported rate of BCC formation is overestimated, as prior studies incorrectly labeled trichoblastomas as BCCs. In fact, the largest studies of NS from the 1990s revealed lower rates of malignant secondary tumors than previously determined.4
The identification of apocrine adenocarcinoma tumors arising from NS is exceedingly rare. A study performed by Cribier et al19 in 2000 retrospect
Histopathology
Histopathologic examination reveals considerable variation in morphology, and an underlying pattern has been difficult to recognize. Unfortunately, some authors have concluded that the diagnosis of apocrine carcinoma is relatively subjective.26 Robson et al26 identified 3 general architectural patterns: tubular, tubulopapillary, and solid. Tubular structures consisted of glands and ducts lined by a single or multilayered epithelium. Tubulopapillary architecture was characterized by epithelium forming papillary folds without a fibrovascular core. The solid morphology showed sheets of cells with limited ductal or tubular formation.26 The most specific criteria of these apocrine carcinomas are identification of decapitation secretion, periodic acid–Schiff–positive diastase-resistant material present in the cells or lumen, and positive immunostaining for gross cystic disease fluid protein-15.27
Robson et al26 reported estrogen receptor positivity and androgen receptor positivity in 62% and 64% of 24 primary apocrine carcinoma cases, respectively. However, whether these markers are as common in NS-related apocrine carcinomas has yet to be noted in the literature. One study reports a case of apocrine carcinoma from NS with positive staining for human epidermal growth factor-2, a cell membrane receptor tyrosine kinase commonly investigated in breast cancers and extramammary Paget disease.22
These apocrine carcinomas do have the potential for lymphatic metastasis, as seen with multiple studies. Domingo and Helwig21 identified regional lymph node metastasis in 2 of its 4 apocrine carcinoma patients. Robson et al26 reported lymphovascular invasion in 4 cases and perineural invasion in 2 of 24 patients studied. However, even in the context of recurrence and regional metastasis, the prognosis was good and seldom fatal.26
Treatment
The most effective treatment of NS is excision of dermal and epidermal components. Excision should be completed with a minimum of 2- to 3-mm margins and full thickness down to the underlying supporting fat.28 Historically, the practice of prophylactic excision of NS was supported by the potential for malignant transformation; however, early excision of NS may be less reasonable in light of these more recent studies showing lower incidence of BCC (0.8%), replaced by benign trichoblastomas.19 In the case of apocrine carcinoma development, excision is undoubtedly recommended, with unclear recommendations regarding further evaluation for metastasis.
Excision also may be favored for cosmetic purposes, given the visible regions where NS tends to develop. Chepla and Gosain29 argued that surgical intervention should be based on other factors such as location on the scalp, alopecia, and other issues affecting appearance and monitoring rather than incidence of malignant transformation. Close monitoring and biopsy of suspicious areas is a more conservative option.
Other therapies include CO2 laser, as demonstrated by Kiedrowicz et al,30 on linear NS in a patient with Schimmelpenning-Feuerstein-Mims syndrome.31 However, this approach is palliative and not effective in removing the entire lesion. Electrodesiccation and curettage and dermabrasion also are not good options for the same reason.4
Occurrence in Children
Nevus sebaceus in children, accompanied by other findings suggestive of epidermal nevus syndromes, should prompt further investigation. Schimmelpenning-Feuerstein-Mims syndrome includes major neurological abnormalities including hemimegalencephaly and seizures.32
Conclusion
Apocrine carcinomas are malignant neoplasms that may rarely arise within an NS. Their clinical identification is difficult and requires histopathologic evaluation. Upon recognition, prompt excision with tumor-free margins is recommended. As a rare entity, little data is available regarding its metastatic potential or overall survival rates. Further investigation is clearly necessary as new cases arise.
- Kamyab-Hesari K, Balochi K, Afshar N, et al. Clinicopathological study of 1016 consecutive adnexal skin tumors. Acta Med Iran. 2013;51:879-885.
- Idriss MH, Elston DM. Secondary neoplasms associated with nevus sebaceus of Jadassohn: a study of 707 cases. J Am Acad Dermatol. 2014;70:332-337.
- Ball EA, Hussain M, Moss AL. Squamous cell carcinoma and basal cell carcinoma arising in a naevus sebaceous of Jadassohn: case report and literature review. Clin Exp Dermatol. 2005;30:259-260.
- Moody MN, Landau JM, Goldberg LH. Nevus sebaceous revisited. Pediatr Dermatol. 2012;29:15-23.
- Happle R. The group of epidermal nevus syndromes part I. well defined phenotypes. J Am Acad Dermatol. 2010;63:1-22; quiz 23-24.
- Carlson JA, Cribier B, Nuovo G, et al. Epidermodysplasia verruciformis-associated and genital-mucosal high-risk human papillomavirus DNA are prevalent in nevus sebaceus of Jadassohn. J Am Acad Dermatol. 2008;59:279-294.
- Kim D, Benjamin LT, Sahoo MK, et al. Human papilloma virus is not prevalent in nevus sebaceus [published online November 14, 2013]. Pediatr Dermatol. 2014;31:326-330.
- Xin H, Matt D, Qin JZ, et al. The sebaceous nevus: a nevus with deletions of the PTCH gene. Cancer Res. 1999;59:1834-1836.
- Hafner C, Schmiemann V, Ruetten A, et al. PTCH mutations are not mainly involved in the pathogenesis of sporadic trichoblastomas. Hum Pathol. 2007;38:1496-1500.
- Takata M, Tojo M, Hatta N, et al. No evidence of deregulated patched-hedgehog signaling pathway in trichoblastomas and other tumors arising within nevus sebaceous. J Invest Dermatol. 2001;117:1666-1670.
- Levinsohn JL, Tian LC, Boyden LM, et al. Whole-exome sequencing reveals somatic mutations in HRAS and KRAS, which cause nevus sebaceus [published online October 25, 2012]. J Invest Dermatol. 2013;133:827-830.
- Happle R. Nevus sebaceus is a mosaic RASopathy. J Invest Dermatol. 2013;133:597-600.
- Luo S, Tsao H. Epidermal, sebaceous, and melanocytic nevoid proliferations are spectrums of mosaic RASopathies. J Invest Dermatol. 2014;134:2493-2496.
- Mehregan AH, Pinkus H. Life history of organoid nevi. special reference to nevus sebaceus of Jadassohn. Arch Dermatol. 1965;91:574-588.
- Muñoz-Pérez MA, García-Hernandez MJ, Ríos JJ, et al. Sebaceus naevi: a clinicopathologic study. J Eur Acad Dermatol Venereol. 2002;16:319-324.
- Altaykan A, Ersoy-Evans S, Erkin G, et al. Basal cell carcinoma arising in nevus sebaceous during childhood. Pediatr Dermatol. 2008;25:616-619.
- Turner CD, Shea CR, Rosoff PM. Basal cell carcinoma originating from a nevus sebaceus on the scalp of a 7-year-old boy. J Pediatr Hematol Oncol. 2001;23:247-249.
- Jaqueti G, Requena L, Sánchez Yus E. Trichoblastoma is the most common neoplasm developed in nevus sebaceus of Jadassohn: a clinicopathologic study of a series of 155 cases. Am J Dermatopathol. 2000;22:108-118.
- Cribier B, Scrivener Y, Grosshans E. Tumors arising in nevus sebaceus: a study of 596 cases. J Am Acad Dermatol. 2000;42(2, pt 1):263-268.
- Paudel U, Jha A, Pokhrel DB, et al. Apocrine carcinoma developing in a naevus sebaceous of scalp. Kathmandu Univ Med J (KUMJ). 2012;10:103-105.
- Domingo J, Helwig EB. Malignant neoplasms associated with nevus sebaceus of Jadassohn. J Am Acad Dermatol. 1979;1:545-556.
- Tanese K, Wakabayashi A, Suzuki T, et al. Immunoexpression of human epidermal growth factor receptor-2 in apocrine carcinoma arising in naevus sebaceous, case report [published online August 23, 2009]. J Eur Acad Dermatol Venereol. 2010;24:360-362.
- Dalle S, Skowron F, Balme B, et al. Apocrine carcinoma developed in nevus sebaceus of Jadassohn. Eur J Dermatol. 2003;13:487-489.
- Jacyk WK, Requena L, Sánchez Yus E, et al. Tubular apocrine carcinoma arising in a nevus sebaceus of Jadassohn. Am J Dermatopathol. 1998;20:389-392.
- Ansai S, Koseki S, Hashimoto H, et al. A case of ductal sweat gland carcinoma connected to syringocystadenoma papilliferum arising in nevus sebaceus. J Cutan Pathol. 1994;21:557-563.
- Robson A, Lazar AJ, Ben Nagi J, et al. Primary cutaneous apocrine carcinoma: a clinico-pathologic analysis of 24 cases. Am J Surg Pathol. 2008;32:682-690.
- Paties C, Taccagni GL, Papotti M, et al. Apocrine carcinoma of the skin. a clinicopathologic, immunocytochemical, and ultrastructural study. Cancer. 1993;71:375-381.
- Davison SP, Khachemoune A, Yu D, et al. Nevus sebaceus of Jadassohn revisited with reconstruction options. Int J Dermatol. 2005;44:145-150.
- Chepla KJ, Gosain AK. Giant nevus sebaceus: definition, surgical techniques, and rationale for treatment. Plast Reconstr Surg. 2012;130:296E-304E.
- Kiedrowicz M, Kacalak-Rzepka A, Królicki A et al. Therapeutic effects of CO2 laser therapy of linear nevus sebaceous in the course of the Schimmelpenning-Feuerstein-Mims syndrome. Postepy Dermatol Allergol. 2013;30:320-323.
- Ashinoff R. Linear nevus sebaceus of Jadassohn treated with the carbon dioxide laser. Pediatr Dermatol. 1993;10:189-191.
- van de Warrenburg BP, van Gulik S, Renier WO, et al. The linear naevus sebaceus syndrome. Clin Neurol Neurosurg. 1998;100:126-132.
- Kamyab-Hesari K, Balochi K, Afshar N, et al. Clinicopathological study of 1016 consecutive adnexal skin tumors. Acta Med Iran. 2013;51:879-885.
- Idriss MH, Elston DM. Secondary neoplasms associated with nevus sebaceus of Jadassohn: a study of 707 cases. J Am Acad Dermatol. 2014;70:332-337.
- Ball EA, Hussain M, Moss AL. Squamous cell carcinoma and basal cell carcinoma arising in a naevus sebaceous of Jadassohn: case report and literature review. Clin Exp Dermatol. 2005;30:259-260.
- Moody MN, Landau JM, Goldberg LH. Nevus sebaceous revisited. Pediatr Dermatol. 2012;29:15-23.
- Happle R. The group of epidermal nevus syndromes part I. well defined phenotypes. J Am Acad Dermatol. 2010;63:1-22; quiz 23-24.
- Carlson JA, Cribier B, Nuovo G, et al. Epidermodysplasia verruciformis-associated and genital-mucosal high-risk human papillomavirus DNA are prevalent in nevus sebaceus of Jadassohn. J Am Acad Dermatol. 2008;59:279-294.
- Kim D, Benjamin LT, Sahoo MK, et al. Human papilloma virus is not prevalent in nevus sebaceus [published online November 14, 2013]. Pediatr Dermatol. 2014;31:326-330.
- Xin H, Matt D, Qin JZ, et al. The sebaceous nevus: a nevus with deletions of the PTCH gene. Cancer Res. 1999;59:1834-1836.
- Hafner C, Schmiemann V, Ruetten A, et al. PTCH mutations are not mainly involved in the pathogenesis of sporadic trichoblastomas. Hum Pathol. 2007;38:1496-1500.
- Takata M, Tojo M, Hatta N, et al. No evidence of deregulated patched-hedgehog signaling pathway in trichoblastomas and other tumors arising within nevus sebaceous. J Invest Dermatol. 2001;117:1666-1670.
- Levinsohn JL, Tian LC, Boyden LM, et al. Whole-exome sequencing reveals somatic mutations in HRAS and KRAS, which cause nevus sebaceus [published online October 25, 2012]. J Invest Dermatol. 2013;133:827-830.
- Happle R. Nevus sebaceus is a mosaic RASopathy. J Invest Dermatol. 2013;133:597-600.
- Luo S, Tsao H. Epidermal, sebaceous, and melanocytic nevoid proliferations are spectrums of mosaic RASopathies. J Invest Dermatol. 2014;134:2493-2496.
- Mehregan AH, Pinkus H. Life history of organoid nevi. special reference to nevus sebaceus of Jadassohn. Arch Dermatol. 1965;91:574-588.
- Muñoz-Pérez MA, García-Hernandez MJ, Ríos JJ, et al. Sebaceus naevi: a clinicopathologic study. J Eur Acad Dermatol Venereol. 2002;16:319-324.
- Altaykan A, Ersoy-Evans S, Erkin G, et al. Basal cell carcinoma arising in nevus sebaceous during childhood. Pediatr Dermatol. 2008;25:616-619.
- Turner CD, Shea CR, Rosoff PM. Basal cell carcinoma originating from a nevus sebaceus on the scalp of a 7-year-old boy. J Pediatr Hematol Oncol. 2001;23:247-249.
- Jaqueti G, Requena L, Sánchez Yus E. Trichoblastoma is the most common neoplasm developed in nevus sebaceus of Jadassohn: a clinicopathologic study of a series of 155 cases. Am J Dermatopathol. 2000;22:108-118.
- Cribier B, Scrivener Y, Grosshans E. Tumors arising in nevus sebaceus: a study of 596 cases. J Am Acad Dermatol. 2000;42(2, pt 1):263-268.
- Paudel U, Jha A, Pokhrel DB, et al. Apocrine carcinoma developing in a naevus sebaceous of scalp. Kathmandu Univ Med J (KUMJ). 2012;10:103-105.
- Domingo J, Helwig EB. Malignant neoplasms associated with nevus sebaceus of Jadassohn. J Am Acad Dermatol. 1979;1:545-556.
- Tanese K, Wakabayashi A, Suzuki T, et al. Immunoexpression of human epidermal growth factor receptor-2 in apocrine carcinoma arising in naevus sebaceous, case report [published online August 23, 2009]. J Eur Acad Dermatol Venereol. 2010;24:360-362.
- Dalle S, Skowron F, Balme B, et al. Apocrine carcinoma developed in nevus sebaceus of Jadassohn. Eur J Dermatol. 2003;13:487-489.
- Jacyk WK, Requena L, Sánchez Yus E, et al. Tubular apocrine carcinoma arising in a nevus sebaceus of Jadassohn. Am J Dermatopathol. 1998;20:389-392.
- Ansai S, Koseki S, Hashimoto H, et al. A case of ductal sweat gland carcinoma connected to syringocystadenoma papilliferum arising in nevus sebaceus. J Cutan Pathol. 1994;21:557-563.
- Robson A, Lazar AJ, Ben Nagi J, et al. Primary cutaneous apocrine carcinoma: a clinico-pathologic analysis of 24 cases. Am J Surg Pathol. 2008;32:682-690.
- Paties C, Taccagni GL, Papotti M, et al. Apocrine carcinoma of the skin. a clinicopathologic, immunocytochemical, and ultrastructural study. Cancer. 1993;71:375-381.
- Davison SP, Khachemoune A, Yu D, et al. Nevus sebaceus of Jadassohn revisited with reconstruction options. Int J Dermatol. 2005;44:145-150.
- Chepla KJ, Gosain AK. Giant nevus sebaceus: definition, surgical techniques, and rationale for treatment. Plast Reconstr Surg. 2012;130:296E-304E.
- Kiedrowicz M, Kacalak-Rzepka A, Królicki A et al. Therapeutic effects of CO2 laser therapy of linear nevus sebaceous in the course of the Schimmelpenning-Feuerstein-Mims syndrome. Postepy Dermatol Allergol. 2013;30:320-323.
- Ashinoff R. Linear nevus sebaceus of Jadassohn treated with the carbon dioxide laser. Pediatr Dermatol. 1993;10:189-191.
- van de Warrenburg BP, van Gulik S, Renier WO, et al. The linear naevus sebaceus syndrome. Clin Neurol Neurosurg. 1998;100:126-132.
Practice Points
- Nevus sebaceus (NS) in the centrofacial region has been correlated with a higher risk for neurological abnormalities, including intellectual disability and seizures.
- Historically, basal cell carcinomas (BCCs) were considered a common occurrence arising from an NS, prompting prophylactic surgical excision of such lesions.
- More recently, it has been recognized that the most common tumor to arise from NS is trichoblastoma rather than BCC; in fact, BCC and other malignancies have been found to be relatively rare compared to their benign counterparts.
- In light of this discovery, observation of NS may be a more prudent course of treatment versus prophylactic surgical excision.
Xanthogranulomatous Reaction to Trametinib for Metastatic Malignant Melanoma
A decade ago, the few agents approved by the US Food and Drug Administration for treatment of metastatic melanoma demonstrated low therapeutic success rates (ie, <15%–20%).1 Since then, advances in molecular biology have identified oncogenes that contribute to melanoma progression.2 Inhibition of the mitogen-activated protein kinase (MAPK) pathway by targeting mutant BRAF and mitogen-activated extracellular signal-regulated kinase (MEK) has created promising pharmacologic treatment opportunities.3 Due to the recent US Food and Drug Administration approval of these therapies for treatment of melanoma, it is important to better characterize these adverse events (AEs) so that we can manage them. We present the development of an unusual cutaneous reaction to trametinib, a MEK inhibitor, in a man with stage IV M1b malignant melanoma.
Case Report
A 66-year-old man with stage IV M1b malignant melanoma with metastases to the brain and lungs presented with recurring pruritic erythematous papules on the face and bilateral forearms that began shortly after initiating therapy with trametinib. The cutaneous eruption had initially presented on the face, forearms, and dorsal hands when trametinib was used in combination with vemurafenib, a BRAF inhibitor, and ipilimumab, a human cytotoxic T-lymphocyte antigen 4–blocking antibody; however, lesions initially were minimal and self-resolving. When trametinib was reintroduced as monotherapy due to fever attributed to the combination treatment regimen, the cutaneous eruption recurred more severely. Physical examination revealed erythematous scaly papules limited to the face and bilateral upper extremities, including the flexural surfaces.
A biopsy from the flexural surface of the right forearm revealed a dense perivascular lymphoid and xanthomatous infiltrate in the dermis (Figure 1). Poorly formed granulomas within the mid reticular dermis demonstrated focal palisading of histiocytes with prominent giant cells at the periphery. Histiocytes and giant cells showed foamy or xanthomatous cytoplasm. Within the reaction, degenerative and swollen collagen fibers were noted with no mucin deposition, which was confirmed with negative colloidal iron staining.
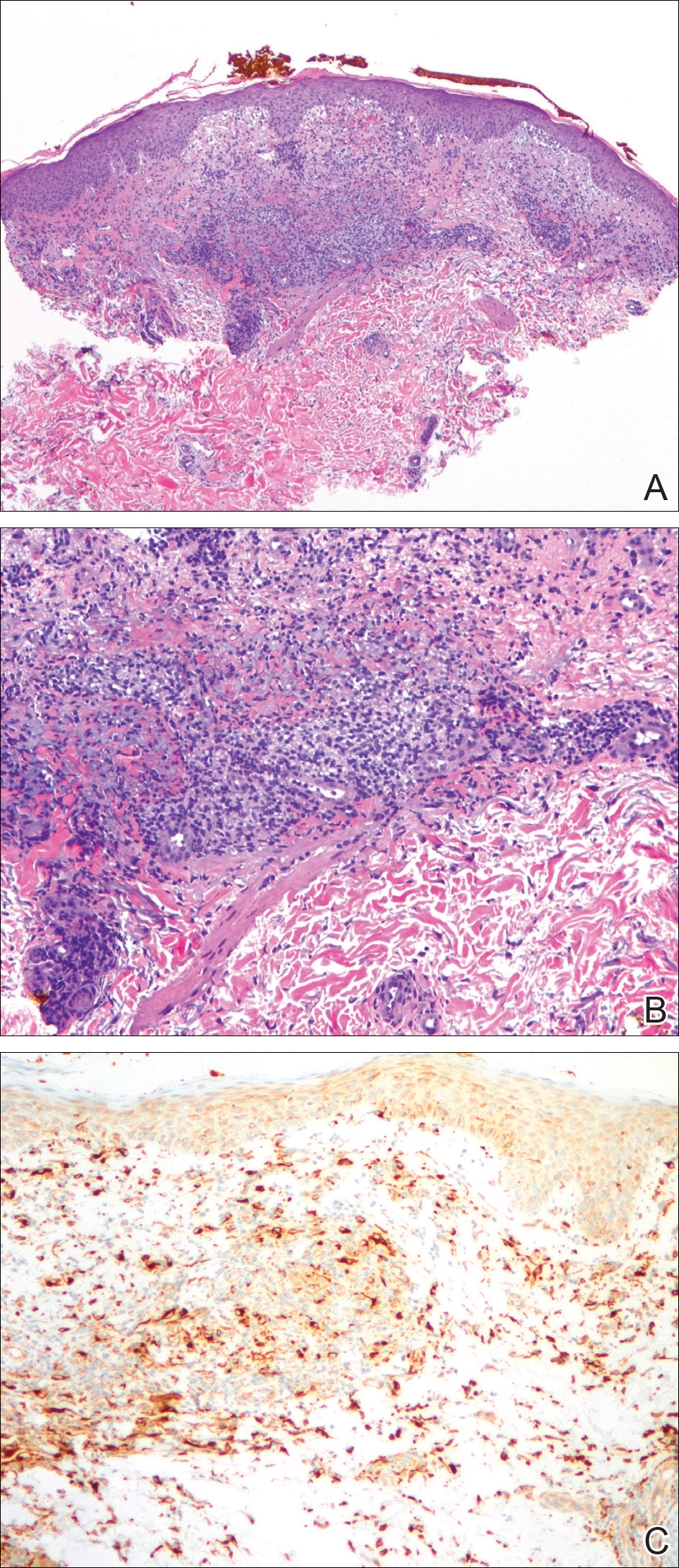
Brief cessation of trametinib along with application of clobetasol propionate ointment 0.05% resulted in resolution of the cutaneous eruption. Later, trametinib was reintroduced in combination with vemurafenib, though therapy was intermittently discontinued due to various side effects. Skin lesions continued to recur (Figure 2) while the patient was on trametinib but remained minimal and continued to respond to topical clobetasol propionate. One year later, the patient continues to tolerate combination therapy with trametinib and vemurafenib.
Comment
BRAF Inhibitors
Normally, activated BRAF phosphorylates and stimulates MEK proteins, ultimately influencing cell proliferation, survival, and differentiation.3-5 BRAF mutations that constitutively activate this pathway have been detected in several malignancies, including papillary thyroid cancer, colorectal cancer, and brain tumors, but they are particularly prevalent in melanoma.4,6 The majority of BRAF-positive malignant melanomas are associated with V600E, in which valine is substituted for glutamic acid at codon 600. The next most common BRAF mutation is V600K, in which valine is substituted for lysine.2,7 Together these constitute approximately 95% of BRAF mutations in melanoma patients.5
MEK Inhibitors
Initially, BRAF inhibitors (BRAFi) were introduced to the market for treating melanoma with great success; however, resistance to BRAFi therapy quickly was identified within months of initiating therapy, leading to investigations for combination therapy with MEK inhibitors (MEKi).2,5 MEK inhibition decreases cellular proliferation and also leads to apoptosis of melanoma cells in patients with BRAF V600E or V600K mutations.2,8 Trametinib, in particular, is a reversible, highly selective allosteric inhibitor of both MEK1 and MEK2. While on trametinib, patients with metastatic melanoma have experienced 3 times as long progression-free survival as well as 81% overall survival compared to 67% overall survival at 6 months in patients on chemotherapy, dacarbazine, or paclitaxel.5 However, AEs are quite common with trametinib, with cutaneous AEs being a leading side effect. Several large trials have reported that 57% to 92% of patients on trametinib report cutaneous AEs, with the majority of cases being described as papulopustular or acneform (Table).5,9

Combination Therapy
Fortunately, combination treatment with a BRAFi may alleviate MEKi-induced cutaneous drug reactions. In one study, acneform eruptions were identified in only 10% of those on combination therapy—trametinib with the BRAFi dabrafenib—compared to 77% of patients on trametinib monotherapy.10 Strikingly, cutaneous AEs occurred in 100% of trametinib-treated mice compared to 30% of combination-treated mice in another study, while the benefits of MEKi remained similar in both groups.11 Because BRAFi and MEKi combination therapy improves progression-free survival while minimizing AEs, we support the use of combination therapy instead of BRAFi or MEKi monotherapy.5
Histologic Evidence of AEs
Histology of trametinib-associated cutaneous reactions is not well characterized, which is in contrast to our understanding of cutaneous AEs associated with BRAFi in which transient acantholytic dermatosis (seen in 45% of patients) and verrucal keratosis (seen in 18% of patients) have been well characterized on histology.12 Interestingly, cutaneous granulomatous eruptions have been attributed to BRAFi therapy in 4 patients.13,14 One patient was on monotherapy with vemurafenib and granulomatous dermatitis with focal necrosis was seen on histology.13 The other 3 patients were on combination therapy with trametinib; 2 had histology-proven sarcoidal granulomatous inflammation, and 1 demonstrated perifollicular granulomatous inflammation and granulomatous inflammation surrounding a focus of melanoma cells.13,14 Although these granulomatous reactions were attributed to BRAFi or combination therapy, the association with trametinib remains unclear. On the other hand, our patient’s granulomatous reaction was exacerbated on trametinib monotherapy, suggesting a relationship to trametinib itself rather than BRAFi.
Conclusion
With the discovery of molecular targeting in melanoma, BRAFi and MEKi therapies provide major milestones in metastatic melanoma management. As more patients are treated with these agents, it is important that we better characterize their associated side effects. Our case of an unusual xanthogranulomatous reaction to trametinib adds to the knowledge base of possible cutaneous reactions caused by this drug. We hope that prospective studies will further investigate and differentiate the cutaneous AEs described so that we can better manage these patients.
- Eggermont AM, Schadendorf D. Melanoma and immunotherapy. Hematol Oncol Clin North Am. 2009;23:547-564.
- Chung C, Reilly S. Trametinib: a novel signal transduction inhibitors for the treatment of metastatic cutaneous melanoma. Am J Health Syst Pharm. 2015;72:101-110.
- Montagut C, Settleman J. Targeting the RAF-MEK-ERK pathway in cancer therapy [published online February 12, 2009]. Cancer Lett. 2009;283:125-134.
- Hertzman Johansson C, Egyhazi Brage S. BRAF inhibitors in cancer therapy [published online December 8, 2013]. Pharmacol Ther. 2014;142:176-182.
- Flaherty KT, Robert C, Hersey P, et al; METRIC Study Group. Improved survival with MEK inhibition in BRAF-mutated melanoma [published online June 4, 2012]. N Engl J Med. 2012;367:107-114.
- Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer [published online June 9, 2002]. Nature. 2002;417:949-954.
- Houben R, Becker JC, Kappel A, et al. Constitutive activation of the Ras-Raf signaling pathway in metastatic melanoma is associated with poor prognosis. J Carcinog. 2004;3:6.
- Roberts PF, Der CJ. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene. 2007;26:3291-3310.
- Falchook GS, Lewis KD, Infante JR, et al. Activity of the oral MEK inhibitor trametinib in patients with advanced melanoma: a phase 2 dose-escalation trial [published online July 16, 2012]. Lancet Oncol. 2012;13:782-789.
- Anforth R, Liu M, Nguyen B, et al. Acneiform eruptions: a common cutaneous toxicity of the MEK inhibitor trametinib [published online December 9, 2013]. Australas J Dermatol. 2014;55:250-254.
- Gadiot J, Hooijkaas AI, Deken MA, et al. Synchronous BRAF(V600E) and MEK inhibition leads to superior control of murine melanoma by limiting MEK inhibitor induced skin toxicity. Onco Targets Ther. 2013;6:1649-1658.
- Anforth R, Carlos G, Clements A, et al. Cutaneous adverse events in patients treated with BRAF inhibitor-based therapies for metastatic melanoma for longer than 52 weeks [published online November 21, 2014]. Br J Dermatol. 2015;172:239-243.
- Park JJ, Hawryluk EB, Tahan SR, et al. Cutaneous granulomatous eruption and successful response to potent topical steroids in patients undergoing targeted BRAF inhibitor treatment for metastatic melanoma. JAMA Dermatol. 2014;150:307-311.
- Green JS, Norris DA, Wisell K. Novel cutaneous effects of combination chemotherapy with BRAF and MEK inhibitors: a report of two cases. Br J Dermatol. 2013;169:172-176.
A decade ago, the few agents approved by the US Food and Drug Administration for treatment of metastatic melanoma demonstrated low therapeutic success rates (ie, <15%–20%).1 Since then, advances in molecular biology have identified oncogenes that contribute to melanoma progression.2 Inhibition of the mitogen-activated protein kinase (MAPK) pathway by targeting mutant BRAF and mitogen-activated extracellular signal-regulated kinase (MEK) has created promising pharmacologic treatment opportunities.3 Due to the recent US Food and Drug Administration approval of these therapies for treatment of melanoma, it is important to better characterize these adverse events (AEs) so that we can manage them. We present the development of an unusual cutaneous reaction to trametinib, a MEK inhibitor, in a man with stage IV M1b malignant melanoma.
Case Report
A 66-year-old man with stage IV M1b malignant melanoma with metastases to the brain and lungs presented with recurring pruritic erythematous papules on the face and bilateral forearms that began shortly after initiating therapy with trametinib. The cutaneous eruption had initially presented on the face, forearms, and dorsal hands when trametinib was used in combination with vemurafenib, a BRAF inhibitor, and ipilimumab, a human cytotoxic T-lymphocyte antigen 4–blocking antibody; however, lesions initially were minimal and self-resolving. When trametinib was reintroduced as monotherapy due to fever attributed to the combination treatment regimen, the cutaneous eruption recurred more severely. Physical examination revealed erythematous scaly papules limited to the face and bilateral upper extremities, including the flexural surfaces.
A biopsy from the flexural surface of the right forearm revealed a dense perivascular lymphoid and xanthomatous infiltrate in the dermis (Figure 1). Poorly formed granulomas within the mid reticular dermis demonstrated focal palisading of histiocytes with prominent giant cells at the periphery. Histiocytes and giant cells showed foamy or xanthomatous cytoplasm. Within the reaction, degenerative and swollen collagen fibers were noted with no mucin deposition, which was confirmed with negative colloidal iron staining.
Brief cessation of trametinib along with application of clobetasol propionate ointment 0.05% resulted in resolution of the cutaneous eruption. Later, trametinib was reintroduced in combination with vemurafenib, though therapy was intermittently discontinued due to various side effects. Skin lesions continued to recur (Figure 2) while the patient was on trametinib but remained minimal and continued to respond to topical clobetasol propionate. One year later, the patient continues to tolerate combination therapy with trametinib and vemurafenib.
Comment
BRAF Inhibitors
Normally, activated BRAF phosphorylates and stimulates MEK proteins, ultimately influencing cell proliferation, survival, and differentiation.3-5 BRAF mutations that constitutively activate this pathway have been detected in several malignancies, including papillary thyroid cancer, colorectal cancer, and brain tumors, but they are particularly prevalent in melanoma.4,6 The majority of BRAF-positive malignant melanomas are associated with V600E, in which valine is substituted for glutamic acid at codon 600. The next most common BRAF mutation is V600K, in which valine is substituted for lysine.2,7 Together these constitute approximately 95% of BRAF mutations in melanoma patients.5
MEK Inhibitors
Initially, BRAF inhibitors (BRAFi) were introduced to the market for treating melanoma with great success; however, resistance to BRAFi therapy quickly was identified within months of initiating therapy, leading to investigations for combination therapy with MEK inhibitors (MEKi).2,5 MEK inhibition decreases cellular proliferation and also leads to apoptosis of melanoma cells in patients with BRAF V600E or V600K mutations.2,8 Trametinib, in particular, is a reversible, highly selective allosteric inhibitor of both MEK1 and MEK2. While on trametinib, patients with metastatic melanoma have experienced 3 times as long progression-free survival as well as 81% overall survival compared to 67% overall survival at 6 months in patients on chemotherapy, dacarbazine, or paclitaxel.5 However, AEs are quite common with trametinib, with cutaneous AEs being a leading side effect. Several large trials have reported that 57% to 92% of patients on trametinib report cutaneous AEs, with the majority of cases being described as papulopustular or acneform (Table).5,9
Combination Therapy
Fortunately, combination treatment with a BRAFi may alleviate MEKi-induced cutaneous drug reactions. In one study, acneform eruptions were identified in only 10% of those on combination therapy—trametinib with the BRAFi dabrafenib—compared to 77% of patients on trametinib monotherapy.10 Strikingly, cutaneous AEs occurred in 100% of trametinib-treated mice compared to 30% of combination-treated mice in another study, while the benefits of MEKi remained similar in both groups.11 Because BRAFi and MEKi combination therapy improves progression-free survival while minimizing AEs, we support the use of combination therapy instead of BRAFi or MEKi monotherapy.5
Histologic Evidence of AEs
Histology of trametinib-associated cutaneous reactions is not well characterized, which is in contrast to our understanding of cutaneous AEs associated with BRAFi in which transient acantholytic dermatosis (seen in 45% of patients) and verrucal keratosis (seen in 18% of patients) have been well characterized on histology.12 Interestingly, cutaneous granulomatous eruptions have been attributed to BRAFi therapy in 4 patients.13,14 One patient was on monotherapy with vemurafenib and granulomatous dermatitis with focal necrosis was seen on histology.13 The other 3 patients were on combination therapy with trametinib; 2 had histology-proven sarcoidal granulomatous inflammation, and 1 demonstrated perifollicular granulomatous inflammation and granulomatous inflammation surrounding a focus of melanoma cells.13,14 Although these granulomatous reactions were attributed to BRAFi or combination therapy, the association with trametinib remains unclear. On the other hand, our patient’s granulomatous reaction was exacerbated on trametinib monotherapy, suggesting a relationship to trametinib itself rather than BRAFi.
Conclusion
With the discovery of molecular targeting in melanoma, BRAFi and MEKi therapies provide major milestones in metastatic melanoma management. As more patients are treated with these agents, it is important that we better characterize their associated side effects. Our case of an unusual xanthogranulomatous reaction to trametinib adds to the knowledge base of possible cutaneous reactions caused by this drug. We hope that prospective studies will further investigate and differentiate the cutaneous AEs described so that we can better manage these patients.
A decade ago, the few agents approved by the US Food and Drug Administration for treatment of metastatic melanoma demonstrated low therapeutic success rates (ie, <15%–20%).1 Since then, advances in molecular biology have identified oncogenes that contribute to melanoma progression.2 Inhibition of the mitogen-activated protein kinase (MAPK) pathway by targeting mutant BRAF and mitogen-activated extracellular signal-regulated kinase (MEK) has created promising pharmacologic treatment opportunities.3 Due to the recent US Food and Drug Administration approval of these therapies for treatment of melanoma, it is important to better characterize these adverse events (AEs) so that we can manage them. We present the development of an unusual cutaneous reaction to trametinib, a MEK inhibitor, in a man with stage IV M1b malignant melanoma.
Case Report
A 66-year-old man with stage IV M1b malignant melanoma with metastases to the brain and lungs presented with recurring pruritic erythematous papules on the face and bilateral forearms that began shortly after initiating therapy with trametinib. The cutaneous eruption had initially presented on the face, forearms, and dorsal hands when trametinib was used in combination with vemurafenib, a BRAF inhibitor, and ipilimumab, a human cytotoxic T-lymphocyte antigen 4–blocking antibody; however, lesions initially were minimal and self-resolving. When trametinib was reintroduced as monotherapy due to fever attributed to the combination treatment regimen, the cutaneous eruption recurred more severely. Physical examination revealed erythematous scaly papules limited to the face and bilateral upper extremities, including the flexural surfaces.
A biopsy from the flexural surface of the right forearm revealed a dense perivascular lymphoid and xanthomatous infiltrate in the dermis (Figure 1). Poorly formed granulomas within the mid reticular dermis demonstrated focal palisading of histiocytes with prominent giant cells at the periphery. Histiocytes and giant cells showed foamy or xanthomatous cytoplasm. Within the reaction, degenerative and swollen collagen fibers were noted with no mucin deposition, which was confirmed with negative colloidal iron staining.
Brief cessation of trametinib along with application of clobetasol propionate ointment 0.05% resulted in resolution of the cutaneous eruption. Later, trametinib was reintroduced in combination with vemurafenib, though therapy was intermittently discontinued due to various side effects. Skin lesions continued to recur (Figure 2) while the patient was on trametinib but remained minimal and continued to respond to topical clobetasol propionate. One year later, the patient continues to tolerate combination therapy with trametinib and vemurafenib.
Comment
BRAF Inhibitors
Normally, activated BRAF phosphorylates and stimulates MEK proteins, ultimately influencing cell proliferation, survival, and differentiation.3-5 BRAF mutations that constitutively activate this pathway have been detected in several malignancies, including papillary thyroid cancer, colorectal cancer, and brain tumors, but they are particularly prevalent in melanoma.4,6 The majority of BRAF-positive malignant melanomas are associated with V600E, in which valine is substituted for glutamic acid at codon 600. The next most common BRAF mutation is V600K, in which valine is substituted for lysine.2,7 Together these constitute approximately 95% of BRAF mutations in melanoma patients.5
MEK Inhibitors
Initially, BRAF inhibitors (BRAFi) were introduced to the market for treating melanoma with great success; however, resistance to BRAFi therapy quickly was identified within months of initiating therapy, leading to investigations for combination therapy with MEK inhibitors (MEKi).2,5 MEK inhibition decreases cellular proliferation and also leads to apoptosis of melanoma cells in patients with BRAF V600E or V600K mutations.2,8 Trametinib, in particular, is a reversible, highly selective allosteric inhibitor of both MEK1 and MEK2. While on trametinib, patients with metastatic melanoma have experienced 3 times as long progression-free survival as well as 81% overall survival compared to 67% overall survival at 6 months in patients on chemotherapy, dacarbazine, or paclitaxel.5 However, AEs are quite common with trametinib, with cutaneous AEs being a leading side effect. Several large trials have reported that 57% to 92% of patients on trametinib report cutaneous AEs, with the majority of cases being described as papulopustular or acneform (Table).5,9
Combination Therapy
Fortunately, combination treatment with a BRAFi may alleviate MEKi-induced cutaneous drug reactions. In one study, acneform eruptions were identified in only 10% of those on combination therapy—trametinib with the BRAFi dabrafenib—compared to 77% of patients on trametinib monotherapy.10 Strikingly, cutaneous AEs occurred in 100% of trametinib-treated mice compared to 30% of combination-treated mice in another study, while the benefits of MEKi remained similar in both groups.11 Because BRAFi and MEKi combination therapy improves progression-free survival while minimizing AEs, we support the use of combination therapy instead of BRAFi or MEKi monotherapy.5
Histologic Evidence of AEs
Histology of trametinib-associated cutaneous reactions is not well characterized, which is in contrast to our understanding of cutaneous AEs associated with BRAFi in which transient acantholytic dermatosis (seen in 45% of patients) and verrucal keratosis (seen in 18% of patients) have been well characterized on histology.12 Interestingly, cutaneous granulomatous eruptions have been attributed to BRAFi therapy in 4 patients.13,14 One patient was on monotherapy with vemurafenib and granulomatous dermatitis with focal necrosis was seen on histology.13 The other 3 patients were on combination therapy with trametinib; 2 had histology-proven sarcoidal granulomatous inflammation, and 1 demonstrated perifollicular granulomatous inflammation and granulomatous inflammation surrounding a focus of melanoma cells.13,14 Although these granulomatous reactions were attributed to BRAFi or combination therapy, the association with trametinib remains unclear. On the other hand, our patient’s granulomatous reaction was exacerbated on trametinib monotherapy, suggesting a relationship to trametinib itself rather than BRAFi.
Conclusion
With the discovery of molecular targeting in melanoma, BRAFi and MEKi therapies provide major milestones in metastatic melanoma management. As more patients are treated with these agents, it is important that we better characterize their associated side effects. Our case of an unusual xanthogranulomatous reaction to trametinib adds to the knowledge base of possible cutaneous reactions caused by this drug. We hope that prospective studies will further investigate and differentiate the cutaneous AEs described so that we can better manage these patients.
- Eggermont AM, Schadendorf D. Melanoma and immunotherapy. Hematol Oncol Clin North Am. 2009;23:547-564.
- Chung C, Reilly S. Trametinib: a novel signal transduction inhibitors for the treatment of metastatic cutaneous melanoma. Am J Health Syst Pharm. 2015;72:101-110.
- Montagut C, Settleman J. Targeting the RAF-MEK-ERK pathway in cancer therapy [published online February 12, 2009]. Cancer Lett. 2009;283:125-134.
- Hertzman Johansson C, Egyhazi Brage S. BRAF inhibitors in cancer therapy [published online December 8, 2013]. Pharmacol Ther. 2014;142:176-182.
- Flaherty KT, Robert C, Hersey P, et al; METRIC Study Group. Improved survival with MEK inhibition in BRAF-mutated melanoma [published online June 4, 2012]. N Engl J Med. 2012;367:107-114.
- Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer [published online June 9, 2002]. Nature. 2002;417:949-954.
- Houben R, Becker JC, Kappel A, et al. Constitutive activation of the Ras-Raf signaling pathway in metastatic melanoma is associated with poor prognosis. J Carcinog. 2004;3:6.
- Roberts PF, Der CJ. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene. 2007;26:3291-3310.
- Falchook GS, Lewis KD, Infante JR, et al. Activity of the oral MEK inhibitor trametinib in patients with advanced melanoma: a phase 2 dose-escalation trial [published online July 16, 2012]. Lancet Oncol. 2012;13:782-789.
- Anforth R, Liu M, Nguyen B, et al. Acneiform eruptions: a common cutaneous toxicity of the MEK inhibitor trametinib [published online December 9, 2013]. Australas J Dermatol. 2014;55:250-254.
- Gadiot J, Hooijkaas AI, Deken MA, et al. Synchronous BRAF(V600E) and MEK inhibition leads to superior control of murine melanoma by limiting MEK inhibitor induced skin toxicity. Onco Targets Ther. 2013;6:1649-1658.
- Anforth R, Carlos G, Clements A, et al. Cutaneous adverse events in patients treated with BRAF inhibitor-based therapies for metastatic melanoma for longer than 52 weeks [published online November 21, 2014]. Br J Dermatol. 2015;172:239-243.
- Park JJ, Hawryluk EB, Tahan SR, et al. Cutaneous granulomatous eruption and successful response to potent topical steroids in patients undergoing targeted BRAF inhibitor treatment for metastatic melanoma. JAMA Dermatol. 2014;150:307-311.
- Green JS, Norris DA, Wisell K. Novel cutaneous effects of combination chemotherapy with BRAF and MEK inhibitors: a report of two cases. Br J Dermatol. 2013;169:172-176.
- Eggermont AM, Schadendorf D. Melanoma and immunotherapy. Hematol Oncol Clin North Am. 2009;23:547-564.
- Chung C, Reilly S. Trametinib: a novel signal transduction inhibitors for the treatment of metastatic cutaneous melanoma. Am J Health Syst Pharm. 2015;72:101-110.
- Montagut C, Settleman J. Targeting the RAF-MEK-ERK pathway in cancer therapy [published online February 12, 2009]. Cancer Lett. 2009;283:125-134.
- Hertzman Johansson C, Egyhazi Brage S. BRAF inhibitors in cancer therapy [published online December 8, 2013]. Pharmacol Ther. 2014;142:176-182.
- Flaherty KT, Robert C, Hersey P, et al; METRIC Study Group. Improved survival with MEK inhibition in BRAF-mutated melanoma [published online June 4, 2012]. N Engl J Med. 2012;367:107-114.
- Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer [published online June 9, 2002]. Nature. 2002;417:949-954.
- Houben R, Becker JC, Kappel A, et al. Constitutive activation of the Ras-Raf signaling pathway in metastatic melanoma is associated with poor prognosis. J Carcinog. 2004;3:6.
- Roberts PF, Der CJ. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene. 2007;26:3291-3310.
- Falchook GS, Lewis KD, Infante JR, et al. Activity of the oral MEK inhibitor trametinib in patients with advanced melanoma: a phase 2 dose-escalation trial [published online July 16, 2012]. Lancet Oncol. 2012;13:782-789.
- Anforth R, Liu M, Nguyen B, et al. Acneiform eruptions: a common cutaneous toxicity of the MEK inhibitor trametinib [published online December 9, 2013]. Australas J Dermatol. 2014;55:250-254.
- Gadiot J, Hooijkaas AI, Deken MA, et al. Synchronous BRAF(V600E) and MEK inhibition leads to superior control of murine melanoma by limiting MEK inhibitor induced skin toxicity. Onco Targets Ther. 2013;6:1649-1658.
- Anforth R, Carlos G, Clements A, et al. Cutaneous adverse events in patients treated with BRAF inhibitor-based therapies for metastatic melanoma for longer than 52 weeks [published online November 21, 2014]. Br J Dermatol. 2015;172:239-243.
- Park JJ, Hawryluk EB, Tahan SR, et al. Cutaneous granulomatous eruption and successful response to potent topical steroids in patients undergoing targeted BRAF inhibitor treatment for metastatic melanoma. JAMA Dermatol. 2014;150:307-311.
- Green JS, Norris DA, Wisell K. Novel cutaneous effects of combination chemotherapy with BRAF and MEK inhibitors: a report of two cases. Br J Dermatol. 2013;169:172-176.
Practice Points
- With the discovery of molecular targeting in melanoma, BRAF and MEK inhibitors have been increasingly utilized as therapies in metastatic melanoma management.
- Trametinib, a MEK inhibitor, is commonly associated with cutaneous adverse reactions, particularly acneform eruptions.
- We report a patient on trametinib who developed an eruption with an unusual xanthogranulomatous reaction pattern noted on histology.
Acquired Perforating Dermatosis in a Skin Graft
Case Report
A 57-year-old black woman with a history of dialysis-dependent end-stage renal disease, diabetes mellitus (DM), hypertension, diastolic congestive heart failure, and chronic bronchitis was admitted to Howard University Hospital (Washington, DC) for acute chest pain and shortness of breath. During her hospital stay the dermatology team was consulted for evaluation of two 1.6-cm teardrop-shaped, yellow-white-chalky plaques noted in the center of an atrophic, hyperpigmented, shiny, contracted split-thickness skin graft (STSG) on the right posterior forearm (Figure 1). Twenty years prior, the patient received STSGs on the right and left forearm secondary to caustic burns. Two months before the current admission she noticed 2 adjacent teardrop-shaped white plaques within the center of the STSG on the right forearm. At a 3-month follow-up, she had developed more lesions within both graft sites of the bilateral forearm. There was no notable pruritus associated with the lesions.
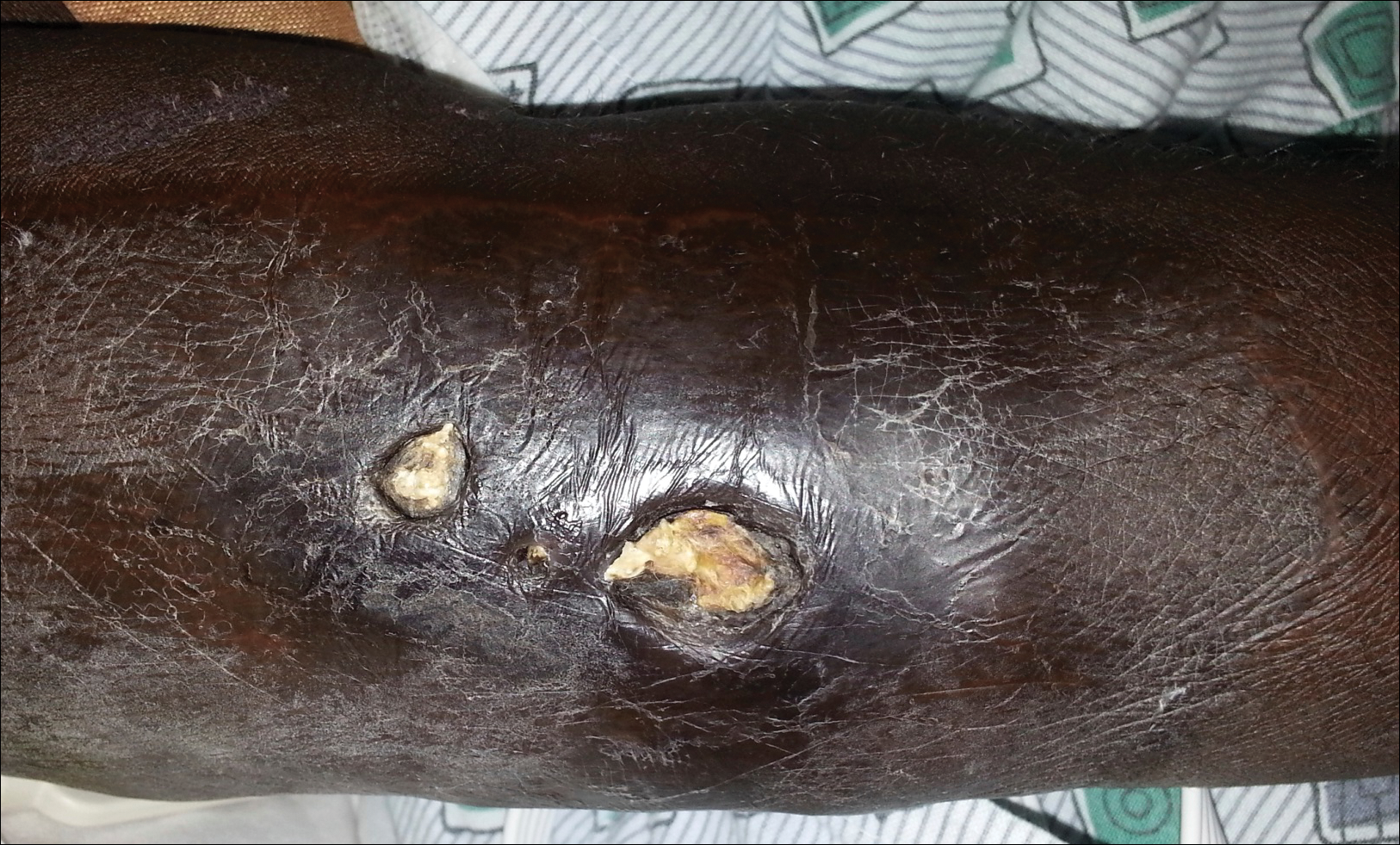
A 4-mm punch biopsy showed an orthokeratotic plug with basophilic inflammatory debris adjacent to acanthotic epidermis, necrotic basophilic debris at the superficial dermis with epidermal canals extending from the base of the lesion superiorly, and transepidermal elimination of elastic fibers (Figure 2A). A Verhoeff-van Gieson stain revealed the necrotic basophilic debris located in the superficial dermis admixed with a cluster of black wavy elastic fibers establishing the identity of the perforating substance (Figure 2B). Masson trichrome stain revealed loss of collagen structure within the aggregate of elastic fibers adjacent to the epidermis and no collagen within epidermal canals (Figure 2C). These histopathologic findings together with the clinical presentation were consistent with a diagnosis of acquired perforating dermatosis (APD).
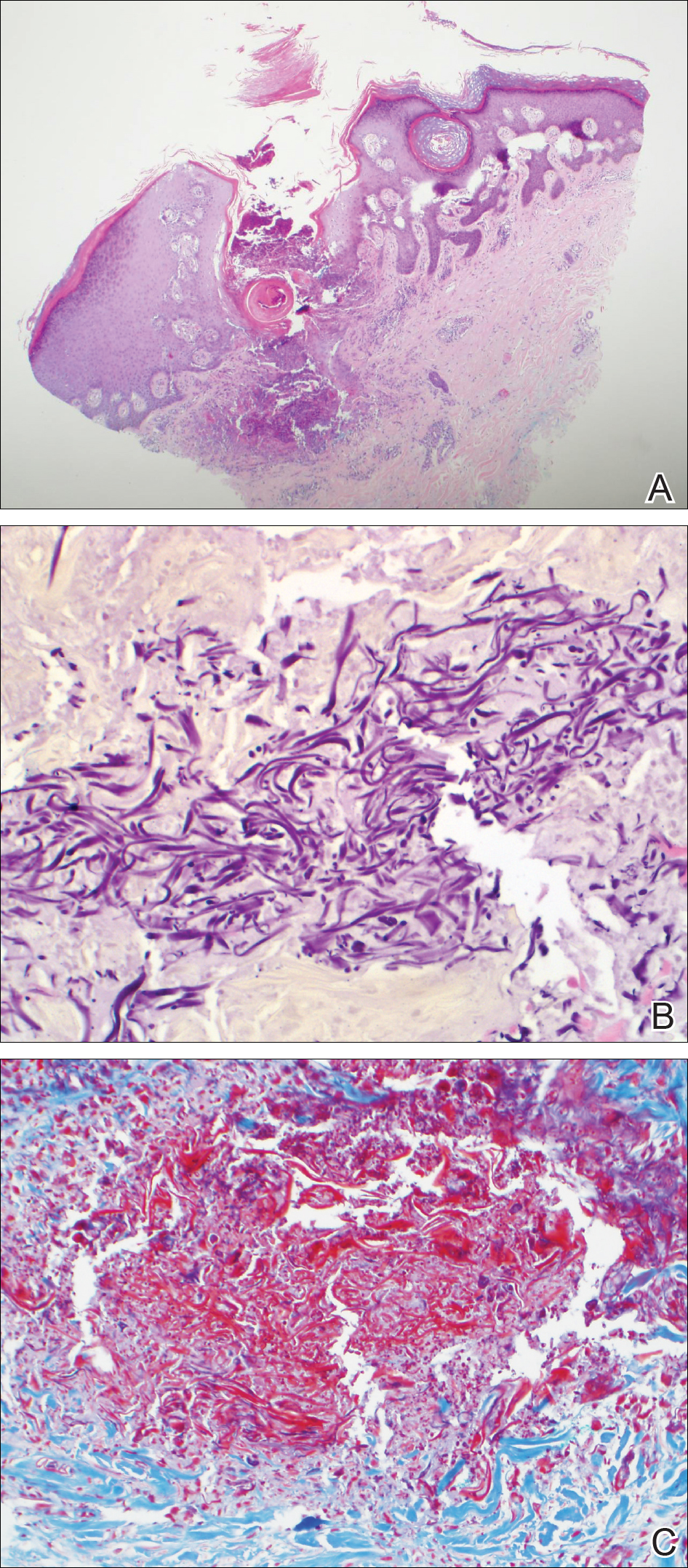
Comment
Presentation
Acquired perforating dermatosis is a dermatologic condition characterized by multiple pruritic, dome-shaped papules and plaques with central keratotic plugs giving a craterlike appearance.1-4 A green-brown or black crust with an erythematous border typically surrounds the primary lesions.4 Acquired perforating dermatosis favors a distribution over the trunk, gluteal region, and the extensor surfaces of the upper and lower extremities. Palmoplantar, intertriginous, and mucous membrane regions typically are spared.4 Occasionally, APD may present as generalized nodules and papules. Our case consisting of lesions that were localized to STSGs on the forearms supports the typical distribution; however, the presentation of APD occurring within a skin graft is unique.
From an epidemiologic standpoint, APD is more likely to affect men than women (1.5:1 ratio). Additionally, APD’s affected age range is 29 to 96 years (mean, 56.8 years),5 which is consistent with our patient’s age. Acquired perforating dermatosis has no racial predilection, though there is a predominance among black patients with concomitant chronic renal failure, as seen in our patient.3
Pathogenesis
The etiology of APD remains unknown.6 Some believe that the uremic or calcium deposits on the skin of patients with chronic kidney disease may trigger chronic pruritus, leading to epithelial hyperplasia and the development of perforating lesions.1,3 A prominent theory in the literature is that superficial trauma, such as scratching, induces necrosis of tissue, facilitating transepidermal elimination of connective tissue components.7 The Köbner phenomenon, which can easily be induced by scratching the skin, supports this idea.8 Fujimoto et al9 suggested that scratching exposes keratinocytes to advanced glycation end product–modified extracellular matrix proteins, particularly types I and III collagen. This exposure leads to the terminal differentiation of keratinocytes with the advanced glycation end receptor (CD36) followed by the upward movement of keratinocytes with glycated collagen. Others postulate fibronectin, involved in epidermal cell signaling, locomotion, and differentiation, is an antigenic trigger because patients with DM and uremia have increased levels of fibronectin in the serum and at sites of perforating skin lesions.10
Diseases Associated With APD
Acquired perforating dermatosis is an umbrella term for perforating disease found in adults. It is associated with systemic diseases, such as DM and pruritus of renal failure.11 Our patient had both dialysis-dependent end-stage renal disease and DM. Acquired perforating dermatosis is observed in 4.5% to 11% of patients on hemodialysis12,13; however, APD may occur prior to or in the absence of dialysis.3 Other examples of systemic conditions associated with APD include obstructive uropathy, chronic nephritis, anuria, and hypertensive nephrosclerosis. Koebnerization also may trigger lesions to manifest in a linear pattern after localized trauma to the skin.7 Acquired perforating dermatosis is associated with other types of trauma, such as healing herpes zoster, or following exposure to drugs, such as tumor necrosis factor α inhibitors, bevacizumab, telaprevir, sorafenib, sirolimus, and indinavir.14-16 Rarely, there have been associations with a history of insect bites, scabies, lymphoma, and hepatobiliary disease.1-3
Histopathology
Acquired perforating dermatosis is classified as a perforating disease, along with reactive perforating collagenosis, elastosis perforans serpiginosa (EPS), perforating folliculitis, and perforating calcific elastosis. Perforating diseases are histologically characterized by the transepidermal penetration and elimination of altered connective tissue and inflammatory cells.5 Each disease differs based on their clinical and histological characteristics.
Histologic sections of APD show a plug of crusting or hyperkeratosis with variable parakeratosis, acanthosis, and occasional dyskeratotic keratinocytes. In the dermis, aggregates of neutrophils, lymphocytes, macrophages, or multinucleated giant cells may be found.17 The histologic findings vary depending on the stage of evolution of the individual lesion. Early lesions show a concave depression with acanthosis, vacuolation of basal keratinocytes, and dermal inflammation.4 Additionally, transepidermal channels filled with keratin, pyknotic nuclear debris, inflammatory cells, elastin, or collagen can be noted.3 Over time, the elastic fibers, as detected by the Verhoeff-van Gieson stain, dissipate and the collagen acquires a basophilic staining. Adjacent to the channels, the basement membrane remains intact in early lesions but later shows discontinuities and electron-dense fibrinlike material.3 Occasionally, amorphous degenerated material within the perforations is the major histologic finding.11 Usually, the material cannot be clearly identified as collagen or elastin, but sometimes both are present.
In our case, we identified elastin as the perforating substance, which is less common than collagen, the typical perforating substance in APD. Elastin has occasionally been seen to serve as the only perforating substance from APD lesions among patients. Abe et al18 reported that the biopsy of a Japanese patient with keratotic follicular papules and serpiginous-arranged papules demonstrated elimination of atypical elastin fibers from the transepidermal channels. This patient was diagnosed with APD as well as EPS and perforating folliculitis based on the clinical presentation.18 Kim et al19 studied 30 Korean patients with APD. One had serpiginous hyperkeratotic plaques along the upper extremity and trunk that revealed transepidermal channels containing coarse elastic fibers and basophilic debris; however, due to the serpiginous morphology of lesions, both Abe et al18 and Kim et al19 favored a diagnosis of acquired EPS. Saray et al20 conducted a retrospective study of 22 Turkish patients with APD; 1 patient had a painful hyperkeratotic papule on the auricle that on histopathology showed degenerated elastin perforating through the keratotic plug, features similar to our case.
Differential Diagnosis
The differential diagnoses include perforating diseases14,19 as well as other disorders that exhibit the Köbner phenomenon, such as psoriasis, lichen planus, and verruca vulgaris.21,22 Also, it is not uncommon for patients with APD to have coexisting folliculitis or prurigo nodularis.22
Treatment
Management is focused on treating the symptoms. For pruritus, sedating antihistamines and other antipruritic agents are efficacious.23 Topical, intra-lesional, or systemic corticosteroids and topical retinoids have shown variable resolution in APD lesions.24 Some case reports describe topical menthol, salicylic acid, sulfur, benzoyl peroxide, systemic antibiotics (eg, clindamycin, doxycycline), and allopurinol for elevated uric acid levels as effective treatment methods.6 Narrowband UVB phototherapy is beneficial for APD and renal disease.25,26 Renal transplantation has been curative for some patients with APD.27 Given that our patient’s lesions were asymptomatic, no treatment was offered at the time.
Conclusion
Our patient presented with APD localized exclusively to the site of a skin graft, and histologic examination identified elastin as the primary perforating substance. A medical history of DM and chronic kidney disease predisposes patients to APD. This case suggests that skin graft sites may be predisposed to the development of APD.
- Rodney IJ, Taylor CS, Cohen G. Derm Dx: what are these pruritic nodules? The Dermatologist. October 15, 2009. http://www.the-dermatologist.com/content/derm-dx-what-are-these-pruritic-nodules. Accessed September 18, 2018.
- Gagnon, AL, Desai T. Dermatological diseases in patients with chronic kidney disease. J Nephropathol. 2013;2:104-109.
- Kurban MS, Boueiz A, Kibbi AG. Cutaneous manifestations of chronic kidney disease. Clin Dermatol. 2008;26:255-264.
- Wagner G, Sachse MM. Acquired reactive perforating dermatosis [published online May 29, 2013]. J Dtsch Dermatol Ges. 2013;11:723-729; 723-730.
- Karpouzis A, Giatromanolaki A, Sivridis E, et al. Acquired reactive perforating collagenosis: current status. J Dermatol. 2010;37:585-592.
- Healy R, Cerio R, Hollingsworth A, et al. Acquired perforating dermatosis associated with pregnancy. Clin Exp Dermatol. 2010;35:621-623.
- Cordova KB, Oberg TJ, Malik M, et al. Dermatologic conditions seen in end-stage renal disease. Semin Dial. 2009;22:45-55.
- Satchell AC, Crotty K, Lee S. Reactive perforating collagenosis: a condition that may be underdiagnosed. Australas J Dermatol. 2001;42:284-287.
- Fujimoto E, Kobayashi T, Fujimoto N, et al. AGE-modified collagens I and III induce keratinocyte terminal differentiation through AGE receptor CD36: epidermal-dermal interaction in acquired perforating dermatosis. J Invest Dermatol. 2010;130:405-414.
- Bilezikci B, Sechkin D, Demirhan B. Acquired perforating dermatosis in patients with chronic renal failure: a possible role for fibronectin. J Eur Acad Dermatol Venereol. 2003;17:230-232.
- Rapini RP, Herbert AA, Drucker CR. Acquired perforating dermatosis. evidence for combined transepidermal elimination of both collagen and elastic fibers. Arch Dermatol. 1989;125:1074-1078.
- Hurwitz RM, Melton ME, Creech FT, et al. Perforating folliculitis in association with hemodialysis. Am J Dermatopathol. 1982;4:101-108.
- Morton CA, Henderson IS, Jones MC, et al. Acquired perforating dermatosis in a British dialysis population. Br J Dermatol. 1996;135:671-677.
- Lübbe J, Sorg O, Malé PJ, et al. Sirolimus-induced inflammatory papules with acquired reactive perforating collagenosis [published online January 9, 2008]. Dermatology. 2008;216:239-242.
- Pernet C, Pageaux GP, Guillot B, et al. Telaprevir-induced acquired perforating dermatosis. JAMA Dermatol. 2014;150:1371-1372.
- Severino-Freire M, Sibaud V, Tournier E, et al. Acquired perforating dermatosis associated with sorafenib therapy [published online September 11, 2014]. J Eur Acad Dermatol Venereol. 2016;30:328-330.
- Zelger B, Hintner H, Auböck J, et al. Acquired perforating dermatosis. transepidermal elimination of DNA material and possible role of leukocytes in pathogenesis. Arch Dermatol. 1991;127:695-700.
- Abe R, Murase S, Nomura Y, et al. Acquired perforating dermatosis appearing as elastosis perforans serpiginosa and perforating folliculitis. Clin Exp Dermatol. 2008;33:653-654.
- Kim SW, Kim MS, Lee JH, et al. A clinicopathologic study of thirty cases of acquired perforating dermatosis in Korea. Ann Dermatol. 2014;26:162-171.
- Saray Y, Seçkin D, Bilezikçi B. Acquired perforating dermatosis: clinicopathological features in twenty-two cases. J Eur Acad Dermatol Venereol. 2006;20:679-688.
- Carter VH, Constantine VS. Kyrle’s disease. I. clinical findings in five cases and review of literature. Arch Dermatol. 1968;97:624-632.
- Robinson-Bostom L, Digiovanna JJ. Cutaneous manifestations of end-stage renal disease. J Am Acad Dermatol. 2000;43:975-986.
- Hong SB, Park JH, Ihm CG, et al. Acquired perforating dermatosis in patients with chronic renal failure and diabetes mellitus. J Korean Med Sci. 2004;19:283-288.
- Morton CA, Henderson IS, Jones MC, et al. Acquired perforating dermatosis in a British dialysis population. Br J Dermatol. 1996;135:671-677.
- Ohe S, Danno K, Sasaki H, et al. Treatment of acquired perforating dermatosis with narrowband ultraviolet B. J Am Acad Dermatol. 2004;50:892-894.
- Sezer E, Erkek E. Acquired perforating dermatosis successfully treated with photodynamic therapy. Photodermatol Photoimmunol Photomed. 2012;28:50-52.
- Saldanha LF, Gonick HC, Rodriguez HJ, et al. Silicon-related syndrome in dialysis patients. Nephron. 1997;77:48-56.
Case Report
A 57-year-old black woman with a history of dialysis-dependent end-stage renal disease, diabetes mellitus (DM), hypertension, diastolic congestive heart failure, and chronic bronchitis was admitted to Howard University Hospital (Washington, DC) for acute chest pain and shortness of breath. During her hospital stay the dermatology team was consulted for evaluation of two 1.6-cm teardrop-shaped, yellow-white-chalky plaques noted in the center of an atrophic, hyperpigmented, shiny, contracted split-thickness skin graft (STSG) on the right posterior forearm (Figure 1). Twenty years prior, the patient received STSGs on the right and left forearm secondary to caustic burns. Two months before the current admission she noticed 2 adjacent teardrop-shaped white plaques within the center of the STSG on the right forearm. At a 3-month follow-up, she had developed more lesions within both graft sites of the bilateral forearm. There was no notable pruritus associated with the lesions.
A 4-mm punch biopsy showed an orthokeratotic plug with basophilic inflammatory debris adjacent to acanthotic epidermis, necrotic basophilic debris at the superficial dermis with epidermal canals extending from the base of the lesion superiorly, and transepidermal elimination of elastic fibers (Figure 2A). A Verhoeff-van Gieson stain revealed the necrotic basophilic debris located in the superficial dermis admixed with a cluster of black wavy elastic fibers establishing the identity of the perforating substance (Figure 2B). Masson trichrome stain revealed loss of collagen structure within the aggregate of elastic fibers adjacent to the epidermis and no collagen within epidermal canals (Figure 2C). These histopathologic findings together with the clinical presentation were consistent with a diagnosis of acquired perforating dermatosis (APD).
Comment
Presentation
Acquired perforating dermatosis is a dermatologic condition characterized by multiple pruritic, dome-shaped papules and plaques with central keratotic plugs giving a craterlike appearance.1-4 A green-brown or black crust with an erythematous border typically surrounds the primary lesions.4 Acquired perforating dermatosis favors a distribution over the trunk, gluteal region, and the extensor surfaces of the upper and lower extremities. Palmoplantar, intertriginous, and mucous membrane regions typically are spared.4 Occasionally, APD may present as generalized nodules and papules. Our case consisting of lesions that were localized to STSGs on the forearms supports the typical distribution; however, the presentation of APD occurring within a skin graft is unique.
From an epidemiologic standpoint, APD is more likely to affect men than women (1.5:1 ratio). Additionally, APD’s affected age range is 29 to 96 years (mean, 56.8 years),5 which is consistent with our patient’s age. Acquired perforating dermatosis has no racial predilection, though there is a predominance among black patients with concomitant chronic renal failure, as seen in our patient.3
Pathogenesis
The etiology of APD remains unknown.6 Some believe that the uremic or calcium deposits on the skin of patients with chronic kidney disease may trigger chronic pruritus, leading to epithelial hyperplasia and the development of perforating lesions.1,3 A prominent theory in the literature is that superficial trauma, such as scratching, induces necrosis of tissue, facilitating transepidermal elimination of connective tissue components.7 The Köbner phenomenon, which can easily be induced by scratching the skin, supports this idea.8 Fujimoto et al9 suggested that scratching exposes keratinocytes to advanced glycation end product–modified extracellular matrix proteins, particularly types I and III collagen. This exposure leads to the terminal differentiation of keratinocytes with the advanced glycation end receptor (CD36) followed by the upward movement of keratinocytes with glycated collagen. Others postulate fibronectin, involved in epidermal cell signaling, locomotion, and differentiation, is an antigenic trigger because patients with DM and uremia have increased levels of fibronectin in the serum and at sites of perforating skin lesions.10
Diseases Associated With APD
Acquired perforating dermatosis is an umbrella term for perforating disease found in adults. It is associated with systemic diseases, such as DM and pruritus of renal failure.11 Our patient had both dialysis-dependent end-stage renal disease and DM. Acquired perforating dermatosis is observed in 4.5% to 11% of patients on hemodialysis12,13; however, APD may occur prior to or in the absence of dialysis.3 Other examples of systemic conditions associated with APD include obstructive uropathy, chronic nephritis, anuria, and hypertensive nephrosclerosis. Koebnerization also may trigger lesions to manifest in a linear pattern after localized trauma to the skin.7 Acquired perforating dermatosis is associated with other types of trauma, such as healing herpes zoster, or following exposure to drugs, such as tumor necrosis factor α inhibitors, bevacizumab, telaprevir, sorafenib, sirolimus, and indinavir.14-16 Rarely, there have been associations with a history of insect bites, scabies, lymphoma, and hepatobiliary disease.1-3
Histopathology
Acquired perforating dermatosis is classified as a perforating disease, along with reactive perforating collagenosis, elastosis perforans serpiginosa (EPS), perforating folliculitis, and perforating calcific elastosis. Perforating diseases are histologically characterized by the transepidermal penetration and elimination of altered connective tissue and inflammatory cells.5 Each disease differs based on their clinical and histological characteristics.
Histologic sections of APD show a plug of crusting or hyperkeratosis with variable parakeratosis, acanthosis, and occasional dyskeratotic keratinocytes. In the dermis, aggregates of neutrophils, lymphocytes, macrophages, or multinucleated giant cells may be found.17 The histologic findings vary depending on the stage of evolution of the individual lesion. Early lesions show a concave depression with acanthosis, vacuolation of basal keratinocytes, and dermal inflammation.4 Additionally, transepidermal channels filled with keratin, pyknotic nuclear debris, inflammatory cells, elastin, or collagen can be noted.3 Over time, the elastic fibers, as detected by the Verhoeff-van Gieson stain, dissipate and the collagen acquires a basophilic staining. Adjacent to the channels, the basement membrane remains intact in early lesions but later shows discontinuities and electron-dense fibrinlike material.3 Occasionally, amorphous degenerated material within the perforations is the major histologic finding.11 Usually, the material cannot be clearly identified as collagen or elastin, but sometimes both are present.
In our case, we identified elastin as the perforating substance, which is less common than collagen, the typical perforating substance in APD. Elastin has occasionally been seen to serve as the only perforating substance from APD lesions among patients. Abe et al18 reported that the biopsy of a Japanese patient with keratotic follicular papules and serpiginous-arranged papules demonstrated elimination of atypical elastin fibers from the transepidermal channels. This patient was diagnosed with APD as well as EPS and perforating folliculitis based on the clinical presentation.18 Kim et al19 studied 30 Korean patients with APD. One had serpiginous hyperkeratotic plaques along the upper extremity and trunk that revealed transepidermal channels containing coarse elastic fibers and basophilic debris; however, due to the serpiginous morphology of lesions, both Abe et al18 and Kim et al19 favored a diagnosis of acquired EPS. Saray et al20 conducted a retrospective study of 22 Turkish patients with APD; 1 patient had a painful hyperkeratotic papule on the auricle that on histopathology showed degenerated elastin perforating through the keratotic plug, features similar to our case.
Differential Diagnosis
The differential diagnoses include perforating diseases14,19 as well as other disorders that exhibit the Köbner phenomenon, such as psoriasis, lichen planus, and verruca vulgaris.21,22 Also, it is not uncommon for patients with APD to have coexisting folliculitis or prurigo nodularis.22
Treatment
Management is focused on treating the symptoms. For pruritus, sedating antihistamines and other antipruritic agents are efficacious.23 Topical, intra-lesional, or systemic corticosteroids and topical retinoids have shown variable resolution in APD lesions.24 Some case reports describe topical menthol, salicylic acid, sulfur, benzoyl peroxide, systemic antibiotics (eg, clindamycin, doxycycline), and allopurinol for elevated uric acid levels as effective treatment methods.6 Narrowband UVB phototherapy is beneficial for APD and renal disease.25,26 Renal transplantation has been curative for some patients with APD.27 Given that our patient’s lesions were asymptomatic, no treatment was offered at the time.
Conclusion
Our patient presented with APD localized exclusively to the site of a skin graft, and histologic examination identified elastin as the primary perforating substance. A medical history of DM and chronic kidney disease predisposes patients to APD. This case suggests that skin graft sites may be predisposed to the development of APD.
Case Report
A 57-year-old black woman with a history of dialysis-dependent end-stage renal disease, diabetes mellitus (DM), hypertension, diastolic congestive heart failure, and chronic bronchitis was admitted to Howard University Hospital (Washington, DC) for acute chest pain and shortness of breath. During her hospital stay the dermatology team was consulted for evaluation of two 1.6-cm teardrop-shaped, yellow-white-chalky plaques noted in the center of an atrophic, hyperpigmented, shiny, contracted split-thickness skin graft (STSG) on the right posterior forearm (Figure 1). Twenty years prior, the patient received STSGs on the right and left forearm secondary to caustic burns. Two months before the current admission she noticed 2 adjacent teardrop-shaped white plaques within the center of the STSG on the right forearm. At a 3-month follow-up, she had developed more lesions within both graft sites of the bilateral forearm. There was no notable pruritus associated with the lesions.
A 4-mm punch biopsy showed an orthokeratotic plug with basophilic inflammatory debris adjacent to acanthotic epidermis, necrotic basophilic debris at the superficial dermis with epidermal canals extending from the base of the lesion superiorly, and transepidermal elimination of elastic fibers (Figure 2A). A Verhoeff-van Gieson stain revealed the necrotic basophilic debris located in the superficial dermis admixed with a cluster of black wavy elastic fibers establishing the identity of the perforating substance (Figure 2B). Masson trichrome stain revealed loss of collagen structure within the aggregate of elastic fibers adjacent to the epidermis and no collagen within epidermal canals (Figure 2C). These histopathologic findings together with the clinical presentation were consistent with a diagnosis of acquired perforating dermatosis (APD).
Comment
Presentation
Acquired perforating dermatosis is a dermatologic condition characterized by multiple pruritic, dome-shaped papules and plaques with central keratotic plugs giving a craterlike appearance.1-4 A green-brown or black crust with an erythematous border typically surrounds the primary lesions.4 Acquired perforating dermatosis favors a distribution over the trunk, gluteal region, and the extensor surfaces of the upper and lower extremities. Palmoplantar, intertriginous, and mucous membrane regions typically are spared.4 Occasionally, APD may present as generalized nodules and papules. Our case consisting of lesions that were localized to STSGs on the forearms supports the typical distribution; however, the presentation of APD occurring within a skin graft is unique.
From an epidemiologic standpoint, APD is more likely to affect men than women (1.5:1 ratio). Additionally, APD’s affected age range is 29 to 96 years (mean, 56.8 years),5 which is consistent with our patient’s age. Acquired perforating dermatosis has no racial predilection, though there is a predominance among black patients with concomitant chronic renal failure, as seen in our patient.3
Pathogenesis
The etiology of APD remains unknown.6 Some believe that the uremic or calcium deposits on the skin of patients with chronic kidney disease may trigger chronic pruritus, leading to epithelial hyperplasia and the development of perforating lesions.1,3 A prominent theory in the literature is that superficial trauma, such as scratching, induces necrosis of tissue, facilitating transepidermal elimination of connective tissue components.7 The Köbner phenomenon, which can easily be induced by scratching the skin, supports this idea.8 Fujimoto et al9 suggested that scratching exposes keratinocytes to advanced glycation end product–modified extracellular matrix proteins, particularly types I and III collagen. This exposure leads to the terminal differentiation of keratinocytes with the advanced glycation end receptor (CD36) followed by the upward movement of keratinocytes with glycated collagen. Others postulate fibronectin, involved in epidermal cell signaling, locomotion, and differentiation, is an antigenic trigger because patients with DM and uremia have increased levels of fibronectin in the serum and at sites of perforating skin lesions.10
Diseases Associated With APD
Acquired perforating dermatosis is an umbrella term for perforating disease found in adults. It is associated with systemic diseases, such as DM and pruritus of renal failure.11 Our patient had both dialysis-dependent end-stage renal disease and DM. Acquired perforating dermatosis is observed in 4.5% to 11% of patients on hemodialysis12,13; however, APD may occur prior to or in the absence of dialysis.3 Other examples of systemic conditions associated with APD include obstructive uropathy, chronic nephritis, anuria, and hypertensive nephrosclerosis. Koebnerization also may trigger lesions to manifest in a linear pattern after localized trauma to the skin.7 Acquired perforating dermatosis is associated with other types of trauma, such as healing herpes zoster, or following exposure to drugs, such as tumor necrosis factor α inhibitors, bevacizumab, telaprevir, sorafenib, sirolimus, and indinavir.14-16 Rarely, there have been associations with a history of insect bites, scabies, lymphoma, and hepatobiliary disease.1-3
Histopathology
Acquired perforating dermatosis is classified as a perforating disease, along with reactive perforating collagenosis, elastosis perforans serpiginosa (EPS), perforating folliculitis, and perforating calcific elastosis. Perforating diseases are histologically characterized by the transepidermal penetration and elimination of altered connective tissue and inflammatory cells.5 Each disease differs based on their clinical and histological characteristics.
Histologic sections of APD show a plug of crusting or hyperkeratosis with variable parakeratosis, acanthosis, and occasional dyskeratotic keratinocytes. In the dermis, aggregates of neutrophils, lymphocytes, macrophages, or multinucleated giant cells may be found.17 The histologic findings vary depending on the stage of evolution of the individual lesion. Early lesions show a concave depression with acanthosis, vacuolation of basal keratinocytes, and dermal inflammation.4 Additionally, transepidermal channels filled with keratin, pyknotic nuclear debris, inflammatory cells, elastin, or collagen can be noted.3 Over time, the elastic fibers, as detected by the Verhoeff-van Gieson stain, dissipate and the collagen acquires a basophilic staining. Adjacent to the channels, the basement membrane remains intact in early lesions but later shows discontinuities and electron-dense fibrinlike material.3 Occasionally, amorphous degenerated material within the perforations is the major histologic finding.11 Usually, the material cannot be clearly identified as collagen or elastin, but sometimes both are present.
In our case, we identified elastin as the perforating substance, which is less common than collagen, the typical perforating substance in APD. Elastin has occasionally been seen to serve as the only perforating substance from APD lesions among patients. Abe et al18 reported that the biopsy of a Japanese patient with keratotic follicular papules and serpiginous-arranged papules demonstrated elimination of atypical elastin fibers from the transepidermal channels. This patient was diagnosed with APD as well as EPS and perforating folliculitis based on the clinical presentation.18 Kim et al19 studied 30 Korean patients with APD. One had serpiginous hyperkeratotic plaques along the upper extremity and trunk that revealed transepidermal channels containing coarse elastic fibers and basophilic debris; however, due to the serpiginous morphology of lesions, both Abe et al18 and Kim et al19 favored a diagnosis of acquired EPS. Saray et al20 conducted a retrospective study of 22 Turkish patients with APD; 1 patient had a painful hyperkeratotic papule on the auricle that on histopathology showed degenerated elastin perforating through the keratotic plug, features similar to our case.
Differential Diagnosis
The differential diagnoses include perforating diseases14,19 as well as other disorders that exhibit the Köbner phenomenon, such as psoriasis, lichen planus, and verruca vulgaris.21,22 Also, it is not uncommon for patients with APD to have coexisting folliculitis or prurigo nodularis.22
Treatment
Management is focused on treating the symptoms. For pruritus, sedating antihistamines and other antipruritic agents are efficacious.23 Topical, intra-lesional, or systemic corticosteroids and topical retinoids have shown variable resolution in APD lesions.24 Some case reports describe topical menthol, salicylic acid, sulfur, benzoyl peroxide, systemic antibiotics (eg, clindamycin, doxycycline), and allopurinol for elevated uric acid levels as effective treatment methods.6 Narrowband UVB phototherapy is beneficial for APD and renal disease.25,26 Renal transplantation has been curative for some patients with APD.27 Given that our patient’s lesions were asymptomatic, no treatment was offered at the time.
Conclusion
Our patient presented with APD localized exclusively to the site of a skin graft, and histologic examination identified elastin as the primary perforating substance. A medical history of DM and chronic kidney disease predisposes patients to APD. This case suggests that skin graft sites may be predisposed to the development of APD.
- Rodney IJ, Taylor CS, Cohen G. Derm Dx: what are these pruritic nodules? The Dermatologist. October 15, 2009. http://www.the-dermatologist.com/content/derm-dx-what-are-these-pruritic-nodules. Accessed September 18, 2018.
- Gagnon, AL, Desai T. Dermatological diseases in patients with chronic kidney disease. J Nephropathol. 2013;2:104-109.
- Kurban MS, Boueiz A, Kibbi AG. Cutaneous manifestations of chronic kidney disease. Clin Dermatol. 2008;26:255-264.
- Wagner G, Sachse MM. Acquired reactive perforating dermatosis [published online May 29, 2013]. J Dtsch Dermatol Ges. 2013;11:723-729; 723-730.
- Karpouzis A, Giatromanolaki A, Sivridis E, et al. Acquired reactive perforating collagenosis: current status. J Dermatol. 2010;37:585-592.
- Healy R, Cerio R, Hollingsworth A, et al. Acquired perforating dermatosis associated with pregnancy. Clin Exp Dermatol. 2010;35:621-623.
- Cordova KB, Oberg TJ, Malik M, et al. Dermatologic conditions seen in end-stage renal disease. Semin Dial. 2009;22:45-55.
- Satchell AC, Crotty K, Lee S. Reactive perforating collagenosis: a condition that may be underdiagnosed. Australas J Dermatol. 2001;42:284-287.
- Fujimoto E, Kobayashi T, Fujimoto N, et al. AGE-modified collagens I and III induce keratinocyte terminal differentiation through AGE receptor CD36: epidermal-dermal interaction in acquired perforating dermatosis. J Invest Dermatol. 2010;130:405-414.
- Bilezikci B, Sechkin D, Demirhan B. Acquired perforating dermatosis in patients with chronic renal failure: a possible role for fibronectin. J Eur Acad Dermatol Venereol. 2003;17:230-232.
- Rapini RP, Herbert AA, Drucker CR. Acquired perforating dermatosis. evidence for combined transepidermal elimination of both collagen and elastic fibers. Arch Dermatol. 1989;125:1074-1078.
- Hurwitz RM, Melton ME, Creech FT, et al. Perforating folliculitis in association with hemodialysis. Am J Dermatopathol. 1982;4:101-108.
- Morton CA, Henderson IS, Jones MC, et al. Acquired perforating dermatosis in a British dialysis population. Br J Dermatol. 1996;135:671-677.
- Lübbe J, Sorg O, Malé PJ, et al. Sirolimus-induced inflammatory papules with acquired reactive perforating collagenosis [published online January 9, 2008]. Dermatology. 2008;216:239-242.
- Pernet C, Pageaux GP, Guillot B, et al. Telaprevir-induced acquired perforating dermatosis. JAMA Dermatol. 2014;150:1371-1372.
- Severino-Freire M, Sibaud V, Tournier E, et al. Acquired perforating dermatosis associated with sorafenib therapy [published online September 11, 2014]. J Eur Acad Dermatol Venereol. 2016;30:328-330.
- Zelger B, Hintner H, Auböck J, et al. Acquired perforating dermatosis. transepidermal elimination of DNA material and possible role of leukocytes in pathogenesis. Arch Dermatol. 1991;127:695-700.
- Abe R, Murase S, Nomura Y, et al. Acquired perforating dermatosis appearing as elastosis perforans serpiginosa and perforating folliculitis. Clin Exp Dermatol. 2008;33:653-654.
- Kim SW, Kim MS, Lee JH, et al. A clinicopathologic study of thirty cases of acquired perforating dermatosis in Korea. Ann Dermatol. 2014;26:162-171.
- Saray Y, Seçkin D, Bilezikçi B. Acquired perforating dermatosis: clinicopathological features in twenty-two cases. J Eur Acad Dermatol Venereol. 2006;20:679-688.
- Carter VH, Constantine VS. Kyrle’s disease. I. clinical findings in five cases and review of literature. Arch Dermatol. 1968;97:624-632.
- Robinson-Bostom L, Digiovanna JJ. Cutaneous manifestations of end-stage renal disease. J Am Acad Dermatol. 2000;43:975-986.
- Hong SB, Park JH, Ihm CG, et al. Acquired perforating dermatosis in patients with chronic renal failure and diabetes mellitus. J Korean Med Sci. 2004;19:283-288.
- Morton CA, Henderson IS, Jones MC, et al. Acquired perforating dermatosis in a British dialysis population. Br J Dermatol. 1996;135:671-677.
- Ohe S, Danno K, Sasaki H, et al. Treatment of acquired perforating dermatosis with narrowband ultraviolet B. J Am Acad Dermatol. 2004;50:892-894.
- Sezer E, Erkek E. Acquired perforating dermatosis successfully treated with photodynamic therapy. Photodermatol Photoimmunol Photomed. 2012;28:50-52.
- Saldanha LF, Gonick HC, Rodriguez HJ, et al. Silicon-related syndrome in dialysis patients. Nephron. 1997;77:48-56.
- Rodney IJ, Taylor CS, Cohen G. Derm Dx: what are these pruritic nodules? The Dermatologist. October 15, 2009. http://www.the-dermatologist.com/content/derm-dx-what-are-these-pruritic-nodules. Accessed September 18, 2018.
- Gagnon, AL, Desai T. Dermatological diseases in patients with chronic kidney disease. J Nephropathol. 2013;2:104-109.
- Kurban MS, Boueiz A, Kibbi AG. Cutaneous manifestations of chronic kidney disease. Clin Dermatol. 2008;26:255-264.
- Wagner G, Sachse MM. Acquired reactive perforating dermatosis [published online May 29, 2013]. J Dtsch Dermatol Ges. 2013;11:723-729; 723-730.
- Karpouzis A, Giatromanolaki A, Sivridis E, et al. Acquired reactive perforating collagenosis: current status. J Dermatol. 2010;37:585-592.
- Healy R, Cerio R, Hollingsworth A, et al. Acquired perforating dermatosis associated with pregnancy. Clin Exp Dermatol. 2010;35:621-623.
- Cordova KB, Oberg TJ, Malik M, et al. Dermatologic conditions seen in end-stage renal disease. Semin Dial. 2009;22:45-55.
- Satchell AC, Crotty K, Lee S. Reactive perforating collagenosis: a condition that may be underdiagnosed. Australas J Dermatol. 2001;42:284-287.
- Fujimoto E, Kobayashi T, Fujimoto N, et al. AGE-modified collagens I and III induce keratinocyte terminal differentiation through AGE receptor CD36: epidermal-dermal interaction in acquired perforating dermatosis. J Invest Dermatol. 2010;130:405-414.
- Bilezikci B, Sechkin D, Demirhan B. Acquired perforating dermatosis in patients with chronic renal failure: a possible role for fibronectin. J Eur Acad Dermatol Venereol. 2003;17:230-232.
- Rapini RP, Herbert AA, Drucker CR. Acquired perforating dermatosis. evidence for combined transepidermal elimination of both collagen and elastic fibers. Arch Dermatol. 1989;125:1074-1078.
- Hurwitz RM, Melton ME, Creech FT, et al. Perforating folliculitis in association with hemodialysis. Am J Dermatopathol. 1982;4:101-108.
- Morton CA, Henderson IS, Jones MC, et al. Acquired perforating dermatosis in a British dialysis population. Br J Dermatol. 1996;135:671-677.
- Lübbe J, Sorg O, Malé PJ, et al. Sirolimus-induced inflammatory papules with acquired reactive perforating collagenosis [published online January 9, 2008]. Dermatology. 2008;216:239-242.
- Pernet C, Pageaux GP, Guillot B, et al. Telaprevir-induced acquired perforating dermatosis. JAMA Dermatol. 2014;150:1371-1372.
- Severino-Freire M, Sibaud V, Tournier E, et al. Acquired perforating dermatosis associated with sorafenib therapy [published online September 11, 2014]. J Eur Acad Dermatol Venereol. 2016;30:328-330.
- Zelger B, Hintner H, Auböck J, et al. Acquired perforating dermatosis. transepidermal elimination of DNA material and possible role of leukocytes in pathogenesis. Arch Dermatol. 1991;127:695-700.
- Abe R, Murase S, Nomura Y, et al. Acquired perforating dermatosis appearing as elastosis perforans serpiginosa and perforating folliculitis. Clin Exp Dermatol. 2008;33:653-654.
- Kim SW, Kim MS, Lee JH, et al. A clinicopathologic study of thirty cases of acquired perforating dermatosis in Korea. Ann Dermatol. 2014;26:162-171.
- Saray Y, Seçkin D, Bilezikçi B. Acquired perforating dermatosis: clinicopathological features in twenty-two cases. J Eur Acad Dermatol Venereol. 2006;20:679-688.
- Carter VH, Constantine VS. Kyrle’s disease. I. clinical findings in five cases and review of literature. Arch Dermatol. 1968;97:624-632.
- Robinson-Bostom L, Digiovanna JJ. Cutaneous manifestations of end-stage renal disease. J Am Acad Dermatol. 2000;43:975-986.
- Hong SB, Park JH, Ihm CG, et al. Acquired perforating dermatosis in patients with chronic renal failure and diabetes mellitus. J Korean Med Sci. 2004;19:283-288.
- Morton CA, Henderson IS, Jones MC, et al. Acquired perforating dermatosis in a British dialysis population. Br J Dermatol. 1996;135:671-677.
- Ohe S, Danno K, Sasaki H, et al. Treatment of acquired perforating dermatosis with narrowband ultraviolet B. J Am Acad Dermatol. 2004;50:892-894.
- Sezer E, Erkek E. Acquired perforating dermatosis successfully treated with photodynamic therapy. Photodermatol Photoimmunol Photomed. 2012;28:50-52.
- Saldanha LF, Gonick HC, Rodriguez HJ, et al. Silicon-related syndrome in dialysis patients. Nephron. 1997;77:48-56.
Practice Points
- Acquired perforating dermatosis (APD) presents as pruritic crateriform papules and plaques with central keratotic plugs.
- A medical history of diabetes mellitus and chronic kidney disease predisposes patients to APD. This case suggests that skin graft sites may be predisposed to the development of APD.
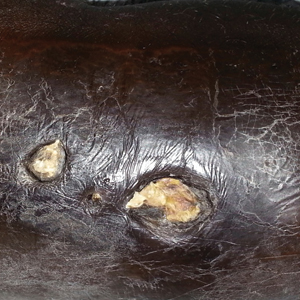
Leukemia Cutis in Acute Myeloid Leukemia Signifies a Poor Prognosis
Case Report
A 66-year-old man with a history of type 2 diabetes mellitus presented with considerable muscle weakness and infiltrative, flesh-colored plaques on the face, trunk, and arms of 3 months’ duration. The patient required the use of a wheelchair due to muscle weakness. On physical examination he had diffuse, infiltrative, flesh-colored plaques on the entire face (Figure 1A), trunk, and arms. The eyelids and lips were swollen, and the nose was distorted due to the infiltrative plaques (Figure 1B). Additionally, there were hypopigmented macules and patches scattered among the infiltrative plaques on the face, trunk, and arms (Figure 1C).
Punch biopsy specimens were obtained from the left cheek and left upper arm and were submitted for histologic examination with routine hematoxylin and eosin staining (Figure 2). Histopathology showed infiltrating and diffuse monomorphic cells in the dermis with large and hyperchromatic nuclei. Some nuclei were cleaved or folded in configuration. The cells displayed ample surrounding cytoplasm, which was finely granular or vacuolated. The infiltrate was accentuated in the perifollicular adventitial dermis. Immunohistochemistry was positive for CD33 and negative for CD3, CD20, and myeloperoxidase. Additionally, periodic acid–Schiff and Fite stains were negative for microorganisms. These morphologic and immunohistochemical findings were consistent with acute myeloid leukemia (AML). Further testing with complete blood cell count, peripheral blood smear, and bone marrow biopsy confirmed the diagnosis of AML. The patient subsequently died 5 weeks later.
Comment
Presentation of LC
Thirty percent to 40% of leukemia patients present with a variety of nonspecific cutaneous signs, including those related to hemorrhage, infection, and drug eruptions, as well as paraneoplastic lesions.1 Cutaneous signs of leukemia are less commonly due to leukemia cutis (LC), defined as the neoplastic infiltration of the skin or subcutaneous tissue by leukemic cells. The clinical presentation of LC varies, making it difficult to diagnose without immunohistochemistry. It can pre-sent as single or multiple erythematous papules and/or nodules, infiltrated plaques, macules, palpable purpura, ulcers, ecchymoses, and/or vesicles.2 Leukemia cutis most often presents on the head, neck, trunk, and sites of current or prior trauma. Gingival hyperplasia is another associated finding in the acute monocytic and myelomonocytic types of AML.3 Additionally, chloromas or granulocytic sarcomas are dermal nodules that can pre-sent in myelogenous leukemia.4
LC and AML
Leukemia cutis most commonly is observed in AML compared to the other types of leukemia. The myelomonocytic and monocytic subtypes of AML are most often implicated.5,6 The majority of patients with LC present with a pre-established (55%–77%) or simultaneous diagnosis of systemic leukemia (23%–38%).
Histopathology
In LC, histology typically reveals a normal epidermis and nodular or diffuse infiltrating cells in the dermis. The cells can appear monomorphic, atypical, or immature, and there is occasional single-filing between collagen bundles. Causative types of neoplasms can be distinguished based on their morphologic, immunophenotypic, and cytogenetic properties.8-10
Incidence
Of the acute leukemias, AML accounts for the highest prevalence in adults,11 with an annual incidence of 14,590 cases in the United States.12 The incidence of AML increases with age; the mean age of patients diagnosed with AML is 67 years.12 Risk is increased with a history of exposure to radiotherapy, chemotherapy, or cigarette smoke; preexisting myeloproliferative or myelodysplastic syndromes and mutations in DNA repair (eg, Fanconi anemia); neutropenia (eg, from elastase mutations); and Down syndrome.13
Diagnosis
More than 20% blasts in the bone marrow is required for a diagnosis of AML.14 Specific to AML is the presence of large immature precursor cells with a granular cytoplasm and, when present, highly diagnostic Auer rods.12Acute myeloid leukemia can be distinguished by staining for myeloperoxidase; Sudan Black B; or the antigens CD13, CD33, or c-kit.15
In our case, CD33 was positive, which is a characteristic finding in AML. Myeloperoxidase also can be positive in AML; however, in our case it was negative, and it can be an insensitive marker in the context of LC. Although most cases of LC present concurrently with bone marrow infiltration, some cases present before systemic involvement; for example, granulocytic sarcomas can occur months earlier than the development of systemic leukemia. Thus, early detection by a dermatologist is essential. Depending on the lesion’s appearance, the differential diagnoses can include lymphoma, drug eruptions, infectious etiologies, sarcoidosis, metastases from other malignancies, and blistering dermatoses.
Management
Systemic therapy should be the cornerstone of therapy. Induction therapy includes the combined use of cytarabine (except in acute promyelocytic leukemia [M3], for which all-trans retinoic acid is indicated) and anthracycline derivatives in a “7+3” regimen to achieve complete remission. Specifically, cytarabine (100–200 mg/m2) typically is continuously administered intravenously for 7 days combined with intravenous administration of either daunorubicin (60–90 mg/m2) or idarubicin (12 mg/m2) on days 1, 2, and 3. Postremission therapy is highly individualized depending on patients’ prognostic factors and is indicated to reduce the likelihood of relapse and to improve patient mortality. High doses of cytarabine and hematopoietic stem cell transplantation commonly are utilized.12 Resolution of hematologic atypia may result in complete or partial resolution of LC.10
Conclusion
We diagnosed AML with systemic involvement in our patient based on the cutaneous manifestation of LC. Diagnosis of LC relies on immunohistochemistry and strong clinical suspicion, as cutaneous findings are diverse and nonspecific. Early recognition is essential, as LC in the context of systemic involvement portends a poor prognosis. Our patient died 5 weeks following initial presentation.
- Rao AG, Danturty I. Leukemia cutis. Indian J Dermatol. 2012;57:504.
- Su WPD, Buechner SA, Chin-Yang L. Clinicopathologic correlations in leukemia cutis. J Am Acad Dermatol. 1984;11:121-128.
- Kumar M, Nair V, Mishra L, et al. Gingival hyperplasia—a clue to the diagnosis of acute leukemia? Arch Oral Sci Res. 2012;2:165-168.
- Winfield H, Smoller B. Other lymphoproliferative and myeloproliferative diseases. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Philadelphia, PA: Mosby/Elsevier; 2012:2037-2048.
- Babina T, Miller L, Thomas B. Leukemia cutis. J Drugs Dermatol. 2012;11:416-417.
- Tziotzios C, Makrygeorgou A. Leukemia cutis. Cleve Clin J Med. 2011;78:226-227.
- Ratnam KV, Khor CJL, Su WPD. Leukemia cutis. Dermatol Clin 1994;12:419-431.
- Cho-Vega JH, Medeiros LJ, Prieto VG, et al. Leukemia cutis. Am J Clin Pathol. 2008;129:130-142.
- Buechner SA, Li CY, Su WP. Leukemia cutis. a histopathologic study of 42 cases. Am J Dermatopathol. 1985;7:109-119.
- Wagner G, Fenchel K, Back W, et al. Leukemia cutis—epidemiology, clinical presentation, and differential diagnoses. J Dtsch Dermatol Ges. 2012;10:27-36.
- O’Donnell MR, Abboud CN, Altman J, et al. NCCN Clinical Practice Guidelines acute myeloid leukemia. J Natl Compr Canc Netw. 2012;10:984-1021.
- Marcucci G, Bloomfield CD. Acute myeloid leukemia. In: Kasper DL, Fauci AS, Hauser SL, et al, eds. Harrison’s Principles of Internal Medicine. 19th ed. New York, NY: McGraw-Hill; 2015:678-686.
- Aster JC, DeAngelo DJ. Acute leukemias. In: Bunn HF, Aster JC, eds. Pathophysiology of Blood Disorders. New York, NY: McGraw-Hill; 2010:244-259.
- Damon LE, Andreadis C. Blood disorders. In: Papadakis MA, McPhee SJ, Rabow MW, eds. Current Medical Diagnosis & Treatment 2016. New York, NY: McGraw-Hill; 2016:495-541.
- Parikh SA, Jabbour E, Koller CA. Adult acute myeloid leukemia. In: Kantarjian HM, Wolff RA, eds. The MD Anderson Manual of Medical Oncology. 2nd ed. New York, NY: McGraw-Hill; 2011:15-32.
Case Report
A 66-year-old man with a history of type 2 diabetes mellitus presented with considerable muscle weakness and infiltrative, flesh-colored plaques on the face, trunk, and arms of 3 months’ duration. The patient required the use of a wheelchair due to muscle weakness. On physical examination he had diffuse, infiltrative, flesh-colored plaques on the entire face (Figure 1A), trunk, and arms. The eyelids and lips were swollen, and the nose was distorted due to the infiltrative plaques (Figure 1B). Additionally, there were hypopigmented macules and patches scattered among the infiltrative plaques on the face, trunk, and arms (Figure 1C).
Punch biopsy specimens were obtained from the left cheek and left upper arm and were submitted for histologic examination with routine hematoxylin and eosin staining (Figure 2). Histopathology showed infiltrating and diffuse monomorphic cells in the dermis with large and hyperchromatic nuclei. Some nuclei were cleaved or folded in configuration. The cells displayed ample surrounding cytoplasm, which was finely granular or vacuolated. The infiltrate was accentuated in the perifollicular adventitial dermis. Immunohistochemistry was positive for CD33 and negative for CD3, CD20, and myeloperoxidase. Additionally, periodic acid–Schiff and Fite stains were negative for microorganisms. These morphologic and immunohistochemical findings were consistent with acute myeloid leukemia (AML). Further testing with complete blood cell count, peripheral blood smear, and bone marrow biopsy confirmed the diagnosis of AML. The patient subsequently died 5 weeks later.
Comment
Presentation of LC
Thirty percent to 40% of leukemia patients present with a variety of nonspecific cutaneous signs, including those related to hemorrhage, infection, and drug eruptions, as well as paraneoplastic lesions.1 Cutaneous signs of leukemia are less commonly due to leukemia cutis (LC), defined as the neoplastic infiltration of the skin or subcutaneous tissue by leukemic cells. The clinical presentation of LC varies, making it difficult to diagnose without immunohistochemistry. It can pre-sent as single or multiple erythematous papules and/or nodules, infiltrated plaques, macules, palpable purpura, ulcers, ecchymoses, and/or vesicles.2 Leukemia cutis most often presents on the head, neck, trunk, and sites of current or prior trauma. Gingival hyperplasia is another associated finding in the acute monocytic and myelomonocytic types of AML.3 Additionally, chloromas or granulocytic sarcomas are dermal nodules that can pre-sent in myelogenous leukemia.4
LC and AML
Leukemia cutis most commonly is observed in AML compared to the other types of leukemia. The myelomonocytic and monocytic subtypes of AML are most often implicated.5,6 The majority of patients with LC present with a pre-established (55%–77%) or simultaneous diagnosis of systemic leukemia (23%–38%).
Histopathology
In LC, histology typically reveals a normal epidermis and nodular or diffuse infiltrating cells in the dermis. The cells can appear monomorphic, atypical, or immature, and there is occasional single-filing between collagen bundles. Causative types of neoplasms can be distinguished based on their morphologic, immunophenotypic, and cytogenetic properties.8-10
Incidence
Of the acute leukemias, AML accounts for the highest prevalence in adults,11 with an annual incidence of 14,590 cases in the United States.12 The incidence of AML increases with age; the mean age of patients diagnosed with AML is 67 years.12 Risk is increased with a history of exposure to radiotherapy, chemotherapy, or cigarette smoke; preexisting myeloproliferative or myelodysplastic syndromes and mutations in DNA repair (eg, Fanconi anemia); neutropenia (eg, from elastase mutations); and Down syndrome.13
Diagnosis
More than 20% blasts in the bone marrow is required for a diagnosis of AML.14 Specific to AML is the presence of large immature precursor cells with a granular cytoplasm and, when present, highly diagnostic Auer rods.12Acute myeloid leukemia can be distinguished by staining for myeloperoxidase; Sudan Black B; or the antigens CD13, CD33, or c-kit.15
In our case, CD33 was positive, which is a characteristic finding in AML. Myeloperoxidase also can be positive in AML; however, in our case it was negative, and it can be an insensitive marker in the context of LC. Although most cases of LC present concurrently with bone marrow infiltration, some cases present before systemic involvement; for example, granulocytic sarcomas can occur months earlier than the development of systemic leukemia. Thus, early detection by a dermatologist is essential. Depending on the lesion’s appearance, the differential diagnoses can include lymphoma, drug eruptions, infectious etiologies, sarcoidosis, metastases from other malignancies, and blistering dermatoses.
Management
Systemic therapy should be the cornerstone of therapy. Induction therapy includes the combined use of cytarabine (except in acute promyelocytic leukemia [M3], for which all-trans retinoic acid is indicated) and anthracycline derivatives in a “7+3” regimen to achieve complete remission. Specifically, cytarabine (100–200 mg/m2) typically is continuously administered intravenously for 7 days combined with intravenous administration of either daunorubicin (60–90 mg/m2) or idarubicin (12 mg/m2) on days 1, 2, and 3. Postremission therapy is highly individualized depending on patients’ prognostic factors and is indicated to reduce the likelihood of relapse and to improve patient mortality. High doses of cytarabine and hematopoietic stem cell transplantation commonly are utilized.12 Resolution of hematologic atypia may result in complete or partial resolution of LC.10
Conclusion
We diagnosed AML with systemic involvement in our patient based on the cutaneous manifestation of LC. Diagnosis of LC relies on immunohistochemistry and strong clinical suspicion, as cutaneous findings are diverse and nonspecific. Early recognition is essential, as LC in the context of systemic involvement portends a poor prognosis. Our patient died 5 weeks following initial presentation.
Case Report
A 66-year-old man with a history of type 2 diabetes mellitus presented with considerable muscle weakness and infiltrative, flesh-colored plaques on the face, trunk, and arms of 3 months’ duration. The patient required the use of a wheelchair due to muscle weakness. On physical examination he had diffuse, infiltrative, flesh-colored plaques on the entire face (Figure 1A), trunk, and arms. The eyelids and lips were swollen, and the nose was distorted due to the infiltrative plaques (Figure 1B). Additionally, there were hypopigmented macules and patches scattered among the infiltrative plaques on the face, trunk, and arms (Figure 1C).
Punch biopsy specimens were obtained from the left cheek and left upper arm and were submitted for histologic examination with routine hematoxylin and eosin staining (Figure 2). Histopathology showed infiltrating and diffuse monomorphic cells in the dermis with large and hyperchromatic nuclei. Some nuclei were cleaved or folded in configuration. The cells displayed ample surrounding cytoplasm, which was finely granular or vacuolated. The infiltrate was accentuated in the perifollicular adventitial dermis. Immunohistochemistry was positive for CD33 and negative for CD3, CD20, and myeloperoxidase. Additionally, periodic acid–Schiff and Fite stains were negative for microorganisms. These morphologic and immunohistochemical findings were consistent with acute myeloid leukemia (AML). Further testing with complete blood cell count, peripheral blood smear, and bone marrow biopsy confirmed the diagnosis of AML. The patient subsequently died 5 weeks later.
Comment
Presentation of LC
Thirty percent to 40% of leukemia patients present with a variety of nonspecific cutaneous signs, including those related to hemorrhage, infection, and drug eruptions, as well as paraneoplastic lesions.1 Cutaneous signs of leukemia are less commonly due to leukemia cutis (LC), defined as the neoplastic infiltration of the skin or subcutaneous tissue by leukemic cells. The clinical presentation of LC varies, making it difficult to diagnose without immunohistochemistry. It can pre-sent as single or multiple erythematous papules and/or nodules, infiltrated plaques, macules, palpable purpura, ulcers, ecchymoses, and/or vesicles.2 Leukemia cutis most often presents on the head, neck, trunk, and sites of current or prior trauma. Gingival hyperplasia is another associated finding in the acute monocytic and myelomonocytic types of AML.3 Additionally, chloromas or granulocytic sarcomas are dermal nodules that can pre-sent in myelogenous leukemia.4
LC and AML
Leukemia cutis most commonly is observed in AML compared to the other types of leukemia. The myelomonocytic and monocytic subtypes of AML are most often implicated.5,6 The majority of patients with LC present with a pre-established (55%–77%) or simultaneous diagnosis of systemic leukemia (23%–38%).
Histopathology
In LC, histology typically reveals a normal epidermis and nodular or diffuse infiltrating cells in the dermis. The cells can appear monomorphic, atypical, or immature, and there is occasional single-filing between collagen bundles. Causative types of neoplasms can be distinguished based on their morphologic, immunophenotypic, and cytogenetic properties.8-10
Incidence
Of the acute leukemias, AML accounts for the highest prevalence in adults,11 with an annual incidence of 14,590 cases in the United States.12 The incidence of AML increases with age; the mean age of patients diagnosed with AML is 67 years.12 Risk is increased with a history of exposure to radiotherapy, chemotherapy, or cigarette smoke; preexisting myeloproliferative or myelodysplastic syndromes and mutations in DNA repair (eg, Fanconi anemia); neutropenia (eg, from elastase mutations); and Down syndrome.13
Diagnosis
More than 20% blasts in the bone marrow is required for a diagnosis of AML.14 Specific to AML is the presence of large immature precursor cells with a granular cytoplasm and, when present, highly diagnostic Auer rods.12Acute myeloid leukemia can be distinguished by staining for myeloperoxidase; Sudan Black B; or the antigens CD13, CD33, or c-kit.15
In our case, CD33 was positive, which is a characteristic finding in AML. Myeloperoxidase also can be positive in AML; however, in our case it was negative, and it can be an insensitive marker in the context of LC. Although most cases of LC present concurrently with bone marrow infiltration, some cases present before systemic involvement; for example, granulocytic sarcomas can occur months earlier than the development of systemic leukemia. Thus, early detection by a dermatologist is essential. Depending on the lesion’s appearance, the differential diagnoses can include lymphoma, drug eruptions, infectious etiologies, sarcoidosis, metastases from other malignancies, and blistering dermatoses.
Management
Systemic therapy should be the cornerstone of therapy. Induction therapy includes the combined use of cytarabine (except in acute promyelocytic leukemia [M3], for which all-trans retinoic acid is indicated) and anthracycline derivatives in a “7+3” regimen to achieve complete remission. Specifically, cytarabine (100–200 mg/m2) typically is continuously administered intravenously for 7 days combined with intravenous administration of either daunorubicin (60–90 mg/m2) or idarubicin (12 mg/m2) on days 1, 2, and 3. Postremission therapy is highly individualized depending on patients’ prognostic factors and is indicated to reduce the likelihood of relapse and to improve patient mortality. High doses of cytarabine and hematopoietic stem cell transplantation commonly are utilized.12 Resolution of hematologic atypia may result in complete or partial resolution of LC.10
Conclusion
We diagnosed AML with systemic involvement in our patient based on the cutaneous manifestation of LC. Diagnosis of LC relies on immunohistochemistry and strong clinical suspicion, as cutaneous findings are diverse and nonspecific. Early recognition is essential, as LC in the context of systemic involvement portends a poor prognosis. Our patient died 5 weeks following initial presentation.
- Rao AG, Danturty I. Leukemia cutis. Indian J Dermatol. 2012;57:504.
- Su WPD, Buechner SA, Chin-Yang L. Clinicopathologic correlations in leukemia cutis. J Am Acad Dermatol. 1984;11:121-128.
- Kumar M, Nair V, Mishra L, et al. Gingival hyperplasia—a clue to the diagnosis of acute leukemia? Arch Oral Sci Res. 2012;2:165-168.
- Winfield H, Smoller B. Other lymphoproliferative and myeloproliferative diseases. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Philadelphia, PA: Mosby/Elsevier; 2012:2037-2048.
- Babina T, Miller L, Thomas B. Leukemia cutis. J Drugs Dermatol. 2012;11:416-417.
- Tziotzios C, Makrygeorgou A. Leukemia cutis. Cleve Clin J Med. 2011;78:226-227.
- Ratnam KV, Khor CJL, Su WPD. Leukemia cutis. Dermatol Clin 1994;12:419-431.
- Cho-Vega JH, Medeiros LJ, Prieto VG, et al. Leukemia cutis. Am J Clin Pathol. 2008;129:130-142.
- Buechner SA, Li CY, Su WP. Leukemia cutis. a histopathologic study of 42 cases. Am J Dermatopathol. 1985;7:109-119.
- Wagner G, Fenchel K, Back W, et al. Leukemia cutis—epidemiology, clinical presentation, and differential diagnoses. J Dtsch Dermatol Ges. 2012;10:27-36.
- O’Donnell MR, Abboud CN, Altman J, et al. NCCN Clinical Practice Guidelines acute myeloid leukemia. J Natl Compr Canc Netw. 2012;10:984-1021.
- Marcucci G, Bloomfield CD. Acute myeloid leukemia. In: Kasper DL, Fauci AS, Hauser SL, et al, eds. Harrison’s Principles of Internal Medicine. 19th ed. New York, NY: McGraw-Hill; 2015:678-686.
- Aster JC, DeAngelo DJ. Acute leukemias. In: Bunn HF, Aster JC, eds. Pathophysiology of Blood Disorders. New York, NY: McGraw-Hill; 2010:244-259.
- Damon LE, Andreadis C. Blood disorders. In: Papadakis MA, McPhee SJ, Rabow MW, eds. Current Medical Diagnosis & Treatment 2016. New York, NY: McGraw-Hill; 2016:495-541.
- Parikh SA, Jabbour E, Koller CA. Adult acute myeloid leukemia. In: Kantarjian HM, Wolff RA, eds. The MD Anderson Manual of Medical Oncology. 2nd ed. New York, NY: McGraw-Hill; 2011:15-32.
- Rao AG, Danturty I. Leukemia cutis. Indian J Dermatol. 2012;57:504.
- Su WPD, Buechner SA, Chin-Yang L. Clinicopathologic correlations in leukemia cutis. J Am Acad Dermatol. 1984;11:121-128.
- Kumar M, Nair V, Mishra L, et al. Gingival hyperplasia—a clue to the diagnosis of acute leukemia? Arch Oral Sci Res. 2012;2:165-168.
- Winfield H, Smoller B. Other lymphoproliferative and myeloproliferative diseases. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Philadelphia, PA: Mosby/Elsevier; 2012:2037-2048.
- Babina T, Miller L, Thomas B. Leukemia cutis. J Drugs Dermatol. 2012;11:416-417.
- Tziotzios C, Makrygeorgou A. Leukemia cutis. Cleve Clin J Med. 2011;78:226-227.
- Ratnam KV, Khor CJL, Su WPD. Leukemia cutis. Dermatol Clin 1994;12:419-431.
- Cho-Vega JH, Medeiros LJ, Prieto VG, et al. Leukemia cutis. Am J Clin Pathol. 2008;129:130-142.
- Buechner SA, Li CY, Su WP. Leukemia cutis. a histopathologic study of 42 cases. Am J Dermatopathol. 1985;7:109-119.
- Wagner G, Fenchel K, Back W, et al. Leukemia cutis—epidemiology, clinical presentation, and differential diagnoses. J Dtsch Dermatol Ges. 2012;10:27-36.
- O’Donnell MR, Abboud CN, Altman J, et al. NCCN Clinical Practice Guidelines acute myeloid leukemia. J Natl Compr Canc Netw. 2012;10:984-1021.
- Marcucci G, Bloomfield CD. Acute myeloid leukemia. In: Kasper DL, Fauci AS, Hauser SL, et al, eds. Harrison’s Principles of Internal Medicine. 19th ed. New York, NY: McGraw-Hill; 2015:678-686.
- Aster JC, DeAngelo DJ. Acute leukemias. In: Bunn HF, Aster JC, eds. Pathophysiology of Blood Disorders. New York, NY: McGraw-Hill; 2010:244-259.
- Damon LE, Andreadis C. Blood disorders. In: Papadakis MA, McPhee SJ, Rabow MW, eds. Current Medical Diagnosis & Treatment 2016. New York, NY: McGraw-Hill; 2016:495-541.
- Parikh SA, Jabbour E, Koller CA. Adult acute myeloid leukemia. In: Kantarjian HM, Wolff RA, eds. The MD Anderson Manual of Medical Oncology. 2nd ed. New York, NY: McGraw-Hill; 2011:15-32.
Practice Points
- Leukemia cutis (LC) describes cutaneous and/or subcutaneous infiltration by leukemic cells and most commonly occurs in patients with acute myeloid leukemia.
- The vast majority of patients presenting with LC already have systemic involvement.
- Cutaneous presentation of LC is diverse, thus diagnosis often is dependent on immunohisto-chemical findings.
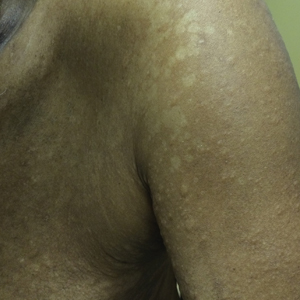
Trichodysplasia Spinulosa in the Setting of Colon Cancer
Case Report
An 82-year-old woman presented to the clinic with a rash on the face that had been present for a few months. She denied any treatment or prior occurrence. Her medical history was remarkable for non-Hodgkin lymphoma that had been successfully treated with chemotherapy 4 years prior. Additionally, she recently had been diagnosed with stage IV colon cancer. She reported that surgery had been scheduled and she would start adjuvant chemotherapy soon after.
On physical examination she exhibited perioral and perinasal erythematous papules with sparing of the vermilion border. A diagnosis of perioral dermatitis was made, and she was started on topical metronidazole. At 1-month follow-up, her condition had slightly worsened and she was subsequently started on doxycycline. When she returned to the clinic again the following month, physical examination revealed agminated folliculocentric papules with central spicules on the face, nose, ears, upper extremities (Figure 1), and trunk. The differential diagnosis included multiple minute digitate hyperkeratosis, spiculosis of multiple myeloma, and trichodysplasia spinulosa (TS).
A punch biopsy of 2 separate papules on the face and upper extremity revealed dilated follicles with enlarged trichohyalin granules and dyskeratosis (Figure 2), consistent with TS. Additional testing such as electron microscopy or polymerase chain reaction was not performed to keep the patient’s medical costs down; also, the strong clinical and histopathologic evidence did not warrant further testing.
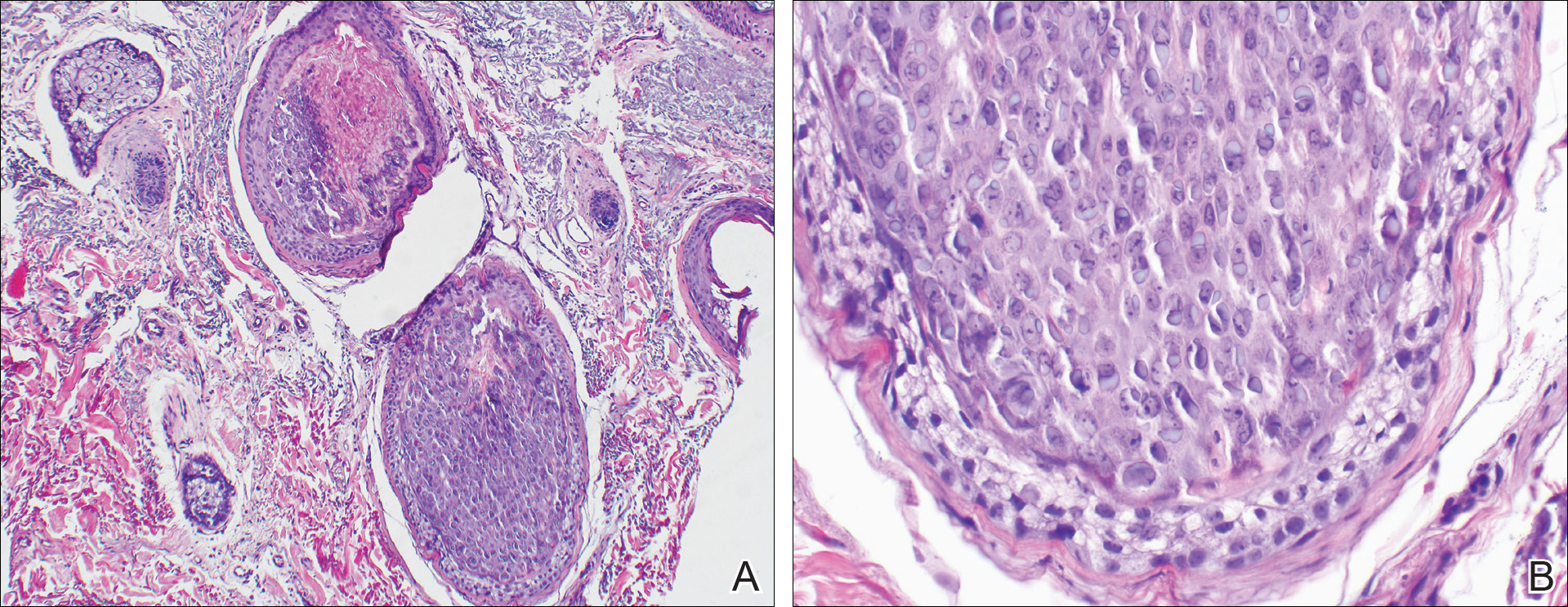
The plan was to start split-face treatment with topical acyclovir and a topical retinoid to see which agent was more effective, but the patient declined until her chemotherapy regimen had concluded. Unfortunately, the patient died 3 months later due to colon cancer.
Comment
History and Presentation
Trichodysplasia spinulosa was first recognized as hairlike hyperkeratosis.1 The name by which it is currently known was later championed by Haycox et al.2 They reported a case of a 44-year-old man who underwent a combined renal-pancreas transplant and while taking immunosuppressive medication developed erythematous papules with follicular spinous processes and progressive alopecia.2 Other synonymous terms used for this condition include pilomatrix dysplasia, cyclosporine-induced folliculodystrophy, virus-associated trichodysplasia,3 and follicular dystrophy of immunosuppression.4 Trichodysplasia spinulosa can affect both adult and pediatric immunocompromised patients, including organ transplant recipients on immunosuppressants and cancer patients on chemotherapy.3 The condition also has been reported to precede the recurrence of lymphoma.5
Etiology
The connection of TS with a viral etiology was first demonstrated in 1999, and subsequently it was confirmed to be a polyomavirus.2 The family name of Polyomaviridae possesses a Greek derivation with poly- meaning many and -oma meaning cancer.3 This name was given after the polyomavirus induced multiple tumors in mice.3,6 This viral family consists of multiple naked viruses with a surrounding icosahedral capsid containing 3 structural proteins known as VP1, VP2, and VP3. Their life cycle is characterized by early and late phases with respective early and late protein formation.3
Polyomavirus infections maintain an asymptomatic and latent course in immunocompetent patients.7 The prevalence and manifestation of these viruses change when the host’s immune system is altered. The first identified JC virus and BK virus of the same family have been found at increased frequencies in blood and lymphoid tissue during host immunosuppression.6 Moreover, the Merkel cell polyomavirus detected in Merkel cell carcinoma is well documented in the dermatologic literature.6,8
A specific polyomavirus has been implicated in the majority of TS cases and has subsequently received the name of TS polyomavirus.9 As a polyomavirus, it similarly produces capsid antigens and large/small T antigens. Among the viral protein antigens produced, the large tumor or LT antigen represents one of the most potent viral proteins. It has been postulated to inhibit the retinoblastoma family of proteins, leading to increased inner root sheath cells that allow for further viral replication.9,10
The disease presents with folliculocentric papules localized mainly on the central face and ears, which grow central keratin spines or spicules that can become 1 to 3 mm in length. Coinciding alopecia and madarosis also may be present.9
Diagnosis
Histologic examination reveals abnormal follicular maturation and distension. Additionally, increased proliferation and amount of trichohyalin is seen within the inner root sheath cells. Further testing via viral culture, polymerase chain reaction, electron microscopy, or immunohistochemical stains can confirm the diagnosis. Such testing may not be warranted in all cases given that classic clinical findings coupled with routine histopathology staining can provide enough evidence.10,11
Management
Currently, a universal successful treatment for TS does not exist. There have been anecdotal successes reported with topical medications such as cidofovir ointment 1%, acyclovir combined with 2-deoxy-D-glucose and epigallocatechin, corticosteroids, topical tacrolimus, topical retinoids, and imiquimod. Additionally, success has been seen with oral minocycline, oral retinoids, valacyclovir, and valganciclovir, with the latter showing the best results. Patients also have shown improvement after modifying their immunosuppressive treatment regimen.10,12
Conclusion
Given the previously published case of TS preceding the recurrence of lymphoma,5 we notified our patient’s oncologist of this potential risk. Her history of lymphoma and immunosuppressive treatment 4 years prior may represent the etiology of the cutaneous presentation; however, the TS with concurrent colon cancer presented prior to starting immunosuppressive therapy, suggesting that it also may have been a paraneoplastic process and not just a sign of immunosuppression. Therefore, we recommend that patients who present with TS should be evaluated for underlying malignancy if not already diagnosed.
- Linke M, Geraud C, Sauer C, et al. Follicular erythematous papules with keratotic spicules. Acta Derm Venereol . 2014;94:493-494.
- Haycox CL, Kim S, Fleckman P, et al. Trichodysplasia spinulosa—a newly described folliculocentric viral infection in an immunocompromised host. J Investig Dermatol Symp Proc. 1999;4:268-271.
- Moens U, Ludvigsen M, Van Ghelue M. Human polyomaviruses in skin diseases [published online September 12, 2011]. Patholog Res Int. 2011;2011:123491.
- Matthews MR, Wang RC, Reddick RL, et al. Viral-associated trichodysplasia spinulosa: a case with electron microscopic and molecular detection of the trichodysplasia spinulosa–associated human polyomavirus. J Cutan Pathol. 2011;38:420-431.
- Osswald SS, Kulick KB, Tomaszewski MM, et al. Viral-associated trichodysplasia in a patient with lymphoma: a case report and review. J Cutan Pathol. 2007;34:721-725.
- Dalianis T, Hirsch HH. Human polyomavirus in disease and cancer. Virology. 2013;437:63-72.
- Tsuzuki S, Fukumoto H, Mine S, et al. Detection of trichodysplasia spinulosa–associated polyomavirus in a fatal case of myocarditis in a seven-month-old girl. Int J Clin Exp Pathol. 2014;7:5308-5312.
- Sadeghi M, Aronen M, Chen T, et al. Merkel cell polyomavirus and trichodysplasia spinulosa–associated polyomavirus DNAs and antibodies in blood among the elderly. BMC Infect Dis. 2012;12:383.
- Van der Meijden E, Kazem S, Burgers MM, et al. Seroprevalence of trichodysplasia spinulosa-associated polyomavirus. Emerg Infect Dis. 2011;17:1355-1363.
- Krichhof MG, Shojania K, Hull MW, et al. Trichodysplasia spinulosa: rare presentation of polyomavirus infection in immunocompromised patients. J Cutan Med Surg. 2014;18:430-435.
- Rianthavorn P, Posuwan N, Payungporn S, et al. Polyomavirus reactivation in pediatric patients with systemic lupus erythematosus. Tohoku J Exp Med. 2012;228:197-204.
- Wanat KA, Holler PD, Dentchev T, et al. Viral-associated trichodysplasia: characterization of a novel polyomavirus infection with therapeutic insights. Arch Dermatol. 2012;148:219-223.
Case Report
An 82-year-old woman presented to the clinic with a rash on the face that had been present for a few months. She denied any treatment or prior occurrence. Her medical history was remarkable for non-Hodgkin lymphoma that had been successfully treated with chemotherapy 4 years prior. Additionally, she recently had been diagnosed with stage IV colon cancer. She reported that surgery had been scheduled and she would start adjuvant chemotherapy soon after.
On physical examination she exhibited perioral and perinasal erythematous papules with sparing of the vermilion border. A diagnosis of perioral dermatitis was made, and she was started on topical metronidazole. At 1-month follow-up, her condition had slightly worsened and she was subsequently started on doxycycline. When she returned to the clinic again the following month, physical examination revealed agminated folliculocentric papules with central spicules on the face, nose, ears, upper extremities (Figure 1), and trunk. The differential diagnosis included multiple minute digitate hyperkeratosis, spiculosis of multiple myeloma, and trichodysplasia spinulosa (TS).
A punch biopsy of 2 separate papules on the face and upper extremity revealed dilated follicles with enlarged trichohyalin granules and dyskeratosis (Figure 2), consistent with TS. Additional testing such as electron microscopy or polymerase chain reaction was not performed to keep the patient’s medical costs down; also, the strong clinical and histopathologic evidence did not warrant further testing.
The plan was to start split-face treatment with topical acyclovir and a topical retinoid to see which agent was more effective, but the patient declined until her chemotherapy regimen had concluded. Unfortunately, the patient died 3 months later due to colon cancer.
Comment
History and Presentation
Trichodysplasia spinulosa was first recognized as hairlike hyperkeratosis.1 The name by which it is currently known was later championed by Haycox et al.2 They reported a case of a 44-year-old man who underwent a combined renal-pancreas transplant and while taking immunosuppressive medication developed erythematous papules with follicular spinous processes and progressive alopecia.2 Other synonymous terms used for this condition include pilomatrix dysplasia, cyclosporine-induced folliculodystrophy, virus-associated trichodysplasia,3 and follicular dystrophy of immunosuppression.4 Trichodysplasia spinulosa can affect both adult and pediatric immunocompromised patients, including organ transplant recipients on immunosuppressants and cancer patients on chemotherapy.3 The condition also has been reported to precede the recurrence of lymphoma.5
Etiology
The connection of TS with a viral etiology was first demonstrated in 1999, and subsequently it was confirmed to be a polyomavirus.2 The family name of Polyomaviridae possesses a Greek derivation with poly- meaning many and -oma meaning cancer.3 This name was given after the polyomavirus induced multiple tumors in mice.3,6 This viral family consists of multiple naked viruses with a surrounding icosahedral capsid containing 3 structural proteins known as VP1, VP2, and VP3. Their life cycle is characterized by early and late phases with respective early and late protein formation.3
Polyomavirus infections maintain an asymptomatic and latent course in immunocompetent patients.7 The prevalence and manifestation of these viruses change when the host’s immune system is altered. The first identified JC virus and BK virus of the same family have been found at increased frequencies in blood and lymphoid tissue during host immunosuppression.6 Moreover, the Merkel cell polyomavirus detected in Merkel cell carcinoma is well documented in the dermatologic literature.6,8
A specific polyomavirus has been implicated in the majority of TS cases and has subsequently received the name of TS polyomavirus.9 As a polyomavirus, it similarly produces capsid antigens and large/small T antigens. Among the viral protein antigens produced, the large tumor or LT antigen represents one of the most potent viral proteins. It has been postulated to inhibit the retinoblastoma family of proteins, leading to increased inner root sheath cells that allow for further viral replication.9,10
The disease presents with folliculocentric papules localized mainly on the central face and ears, which grow central keratin spines or spicules that can become 1 to 3 mm in length. Coinciding alopecia and madarosis also may be present.9
Diagnosis
Histologic examination reveals abnormal follicular maturation and distension. Additionally, increased proliferation and amount of trichohyalin is seen within the inner root sheath cells. Further testing via viral culture, polymerase chain reaction, electron microscopy, or immunohistochemical stains can confirm the diagnosis. Such testing may not be warranted in all cases given that classic clinical findings coupled with routine histopathology staining can provide enough evidence.10,11
Management
Currently, a universal successful treatment for TS does not exist. There have been anecdotal successes reported with topical medications such as cidofovir ointment 1%, acyclovir combined with 2-deoxy-D-glucose and epigallocatechin, corticosteroids, topical tacrolimus, topical retinoids, and imiquimod. Additionally, success has been seen with oral minocycline, oral retinoids, valacyclovir, and valganciclovir, with the latter showing the best results. Patients also have shown improvement after modifying their immunosuppressive treatment regimen.10,12
Conclusion
Given the previously published case of TS preceding the recurrence of lymphoma,5 we notified our patient’s oncologist of this potential risk. Her history of lymphoma and immunosuppressive treatment 4 years prior may represent the etiology of the cutaneous presentation; however, the TS with concurrent colon cancer presented prior to starting immunosuppressive therapy, suggesting that it also may have been a paraneoplastic process and not just a sign of immunosuppression. Therefore, we recommend that patients who present with TS should be evaluated for underlying malignancy if not already diagnosed.
Case Report
An 82-year-old woman presented to the clinic with a rash on the face that had been present for a few months. She denied any treatment or prior occurrence. Her medical history was remarkable for non-Hodgkin lymphoma that had been successfully treated with chemotherapy 4 years prior. Additionally, she recently had been diagnosed with stage IV colon cancer. She reported that surgery had been scheduled and she would start adjuvant chemotherapy soon after.
On physical examination she exhibited perioral and perinasal erythematous papules with sparing of the vermilion border. A diagnosis of perioral dermatitis was made, and she was started on topical metronidazole. At 1-month follow-up, her condition had slightly worsened and she was subsequently started on doxycycline. When she returned to the clinic again the following month, physical examination revealed agminated folliculocentric papules with central spicules on the face, nose, ears, upper extremities (Figure 1), and trunk. The differential diagnosis included multiple minute digitate hyperkeratosis, spiculosis of multiple myeloma, and trichodysplasia spinulosa (TS).
A punch biopsy of 2 separate papules on the face and upper extremity revealed dilated follicles with enlarged trichohyalin granules and dyskeratosis (Figure 2), consistent with TS. Additional testing such as electron microscopy or polymerase chain reaction was not performed to keep the patient’s medical costs down; also, the strong clinical and histopathologic evidence did not warrant further testing.
The plan was to start split-face treatment with topical acyclovir and a topical retinoid to see which agent was more effective, but the patient declined until her chemotherapy regimen had concluded. Unfortunately, the patient died 3 months later due to colon cancer.
Comment
History and Presentation
Trichodysplasia spinulosa was first recognized as hairlike hyperkeratosis.1 The name by which it is currently known was later championed by Haycox et al.2 They reported a case of a 44-year-old man who underwent a combined renal-pancreas transplant and while taking immunosuppressive medication developed erythematous papules with follicular spinous processes and progressive alopecia.2 Other synonymous terms used for this condition include pilomatrix dysplasia, cyclosporine-induced folliculodystrophy, virus-associated trichodysplasia,3 and follicular dystrophy of immunosuppression.4 Trichodysplasia spinulosa can affect both adult and pediatric immunocompromised patients, including organ transplant recipients on immunosuppressants and cancer patients on chemotherapy.3 The condition also has been reported to precede the recurrence of lymphoma.5
Etiology
The connection of TS with a viral etiology was first demonstrated in 1999, and subsequently it was confirmed to be a polyomavirus.2 The family name of Polyomaviridae possesses a Greek derivation with poly- meaning many and -oma meaning cancer.3 This name was given after the polyomavirus induced multiple tumors in mice.3,6 This viral family consists of multiple naked viruses with a surrounding icosahedral capsid containing 3 structural proteins known as VP1, VP2, and VP3. Their life cycle is characterized by early and late phases with respective early and late protein formation.3
Polyomavirus infections maintain an asymptomatic and latent course in immunocompetent patients.7 The prevalence and manifestation of these viruses change when the host’s immune system is altered. The first identified JC virus and BK virus of the same family have been found at increased frequencies in blood and lymphoid tissue during host immunosuppression.6 Moreover, the Merkel cell polyomavirus detected in Merkel cell carcinoma is well documented in the dermatologic literature.6,8
A specific polyomavirus has been implicated in the majority of TS cases and has subsequently received the name of TS polyomavirus.9 As a polyomavirus, it similarly produces capsid antigens and large/small T antigens. Among the viral protein antigens produced, the large tumor or LT antigen represents one of the most potent viral proteins. It has been postulated to inhibit the retinoblastoma family of proteins, leading to increased inner root sheath cells that allow for further viral replication.9,10
The disease presents with folliculocentric papules localized mainly on the central face and ears, which grow central keratin spines or spicules that can become 1 to 3 mm in length. Coinciding alopecia and madarosis also may be present.9
Diagnosis
Histologic examination reveals abnormal follicular maturation and distension. Additionally, increased proliferation and amount of trichohyalin is seen within the inner root sheath cells. Further testing via viral culture, polymerase chain reaction, electron microscopy, or immunohistochemical stains can confirm the diagnosis. Such testing may not be warranted in all cases given that classic clinical findings coupled with routine histopathology staining can provide enough evidence.10,11
Management
Currently, a universal successful treatment for TS does not exist. There have been anecdotal successes reported with topical medications such as cidofovir ointment 1%, acyclovir combined with 2-deoxy-D-glucose and epigallocatechin, corticosteroids, topical tacrolimus, topical retinoids, and imiquimod. Additionally, success has been seen with oral minocycline, oral retinoids, valacyclovir, and valganciclovir, with the latter showing the best results. Patients also have shown improvement after modifying their immunosuppressive treatment regimen.10,12
Conclusion
Given the previously published case of TS preceding the recurrence of lymphoma,5 we notified our patient’s oncologist of this potential risk. Her history of lymphoma and immunosuppressive treatment 4 years prior may represent the etiology of the cutaneous presentation; however, the TS with concurrent colon cancer presented prior to starting immunosuppressive therapy, suggesting that it also may have been a paraneoplastic process and not just a sign of immunosuppression. Therefore, we recommend that patients who present with TS should be evaluated for underlying malignancy if not already diagnosed.
- Linke M, Geraud C, Sauer C, et al. Follicular erythematous papules with keratotic spicules. Acta Derm Venereol . 2014;94:493-494.
- Haycox CL, Kim S, Fleckman P, et al. Trichodysplasia spinulosa—a newly described folliculocentric viral infection in an immunocompromised host. J Investig Dermatol Symp Proc. 1999;4:268-271.
- Moens U, Ludvigsen M, Van Ghelue M. Human polyomaviruses in skin diseases [published online September 12, 2011]. Patholog Res Int. 2011;2011:123491.
- Matthews MR, Wang RC, Reddick RL, et al. Viral-associated trichodysplasia spinulosa: a case with electron microscopic and molecular detection of the trichodysplasia spinulosa–associated human polyomavirus. J Cutan Pathol. 2011;38:420-431.
- Osswald SS, Kulick KB, Tomaszewski MM, et al. Viral-associated trichodysplasia in a patient with lymphoma: a case report and review. J Cutan Pathol. 2007;34:721-725.
- Dalianis T, Hirsch HH. Human polyomavirus in disease and cancer. Virology. 2013;437:63-72.
- Tsuzuki S, Fukumoto H, Mine S, et al. Detection of trichodysplasia spinulosa–associated polyomavirus in a fatal case of myocarditis in a seven-month-old girl. Int J Clin Exp Pathol. 2014;7:5308-5312.
- Sadeghi M, Aronen M, Chen T, et al. Merkel cell polyomavirus and trichodysplasia spinulosa–associated polyomavirus DNAs and antibodies in blood among the elderly. BMC Infect Dis. 2012;12:383.
- Van der Meijden E, Kazem S, Burgers MM, et al. Seroprevalence of trichodysplasia spinulosa-associated polyomavirus. Emerg Infect Dis. 2011;17:1355-1363.
- Krichhof MG, Shojania K, Hull MW, et al. Trichodysplasia spinulosa: rare presentation of polyomavirus infection in immunocompromised patients. J Cutan Med Surg. 2014;18:430-435.
- Rianthavorn P, Posuwan N, Payungporn S, et al. Polyomavirus reactivation in pediatric patients with systemic lupus erythematosus. Tohoku J Exp Med. 2012;228:197-204.
- Wanat KA, Holler PD, Dentchev T, et al. Viral-associated trichodysplasia: characterization of a novel polyomavirus infection with therapeutic insights. Arch Dermatol. 2012;148:219-223.
- Linke M, Geraud C, Sauer C, et al. Follicular erythematous papules with keratotic spicules. Acta Derm Venereol . 2014;94:493-494.
- Haycox CL, Kim S, Fleckman P, et al. Trichodysplasia spinulosa—a newly described folliculocentric viral infection in an immunocompromised host. J Investig Dermatol Symp Proc. 1999;4:268-271.
- Moens U, Ludvigsen M, Van Ghelue M. Human polyomaviruses in skin diseases [published online September 12, 2011]. Patholog Res Int. 2011;2011:123491.
- Matthews MR, Wang RC, Reddick RL, et al. Viral-associated trichodysplasia spinulosa: a case with electron microscopic and molecular detection of the trichodysplasia spinulosa–associated human polyomavirus. J Cutan Pathol. 2011;38:420-431.
- Osswald SS, Kulick KB, Tomaszewski MM, et al. Viral-associated trichodysplasia in a patient with lymphoma: a case report and review. J Cutan Pathol. 2007;34:721-725.
- Dalianis T, Hirsch HH. Human polyomavirus in disease and cancer. Virology. 2013;437:63-72.
- Tsuzuki S, Fukumoto H, Mine S, et al. Detection of trichodysplasia spinulosa–associated polyomavirus in a fatal case of myocarditis in a seven-month-old girl. Int J Clin Exp Pathol. 2014;7:5308-5312.
- Sadeghi M, Aronen M, Chen T, et al. Merkel cell polyomavirus and trichodysplasia spinulosa–associated polyomavirus DNAs and antibodies in blood among the elderly. BMC Infect Dis. 2012;12:383.
- Van der Meijden E, Kazem S, Burgers MM, et al. Seroprevalence of trichodysplasia spinulosa-associated polyomavirus. Emerg Infect Dis. 2011;17:1355-1363.
- Krichhof MG, Shojania K, Hull MW, et al. Trichodysplasia spinulosa: rare presentation of polyomavirus infection in immunocompromised patients. J Cutan Med Surg. 2014;18:430-435.
- Rianthavorn P, Posuwan N, Payungporn S, et al. Polyomavirus reactivation in pediatric patients with systemic lupus erythematosus. Tohoku J Exp Med. 2012;228:197-204.
- Wanat KA, Holler PD, Dentchev T, et al. Viral-associated trichodysplasia: characterization of a novel polyomavirus infection with therapeutic insights. Arch Dermatol. 2012;148:219-223.
Practice Points
- Rashes have a life span and can evolve with time.
- If apparent straightforward conditions do not appear to respond to standard therapy, start to think outside the box for underlying potential causes.

Nausea and vomiting • sensitivity to smell • history of hypertension and alcohol abuse • Dx?
THE CASE
A 57-year-old woman presented to a family physician with acute encephalopathy and complaints of recent gastritis. She reported a 2-month history of nausea, vomiting, decreased oral intake, and extreme sensitivity to smell. The patient had a history of hypertension, and a family member privately disclosed to the FP that she also had a history of alcohol abuse. The patient was taking lorazepam daily, as needed, for anxiety.
On initial assessment, the patient was alert, but not oriented to time or situation. She was ataxic and agitated but did not exhibit pupillary constriction or tremor. The FP sent her to the emergency department (ED).
After being assessed in the ED, the patient was admitted. Over the course of several days, she showed worsening mentation; she persistently believed she was in Chicago, her childhood home. On memory testing, she was unable to recall any of 3 objects after 5 minutes. She exhibited horizontal nystagmus and dysmetria bilaterally and continued to be ataxic, requiring 2-point assistance. Her agitation was managed nonpharmacologically.
A work-up was performed, which included laboratory testing, a urinalysis, and computed tomography (CT) of the head. A comprehensive metabolic panel, complete blood count, and thyroid stimulating hormone test were unremarkable except for electrolyte disturbances, with a sodium level of 158 mEq/L and a potassium level of 2.6 mEq/L (reference ranges: 135-145 mEq/L and 3.5-5 mEq/L, respectively).
Her blood alcohol level was zero, and not surprisingly given her use of lorazepam, a urine drug screen was positive for benzodiazepines. The urinalysis results were consistent with a urinary tract infection (UTI), for which she was treated with an antibiotic. A carbohydrate-deficient transferrin test may have been useful to establish chronic alcohol abuse, but was not ordered. The head CT was negative.
After a few days with fluids and electrolyte replacement, the patient’s electrolytes normalized.
THE DIAGNOSIS
The differential diagnosis included sepsis, metabolic encephalopathy, and alcoholic encephalopathy. Given that the patient’s urine drug screen was positive, benzodiazepine withdrawal was also considered a plausible explanation for her continued cognitive disturbances. (It was surmised that she had likely taken her last lorazepam several days prior.) However, the lack of other signs of withdrawal prompted further investigation.
Continue to: Since her encephalopathy...
Since her encephalopathy, ataxia, and nystagmus persisted, magnetic resonance imaging (MRI) of the brain was performed on Day 3 of hospitalization (FIGURE). A lumbar puncture and an electroencephalogram were also considered but were not performed because the MRI results revealed bilateral enhancement of the m

DISCUSSION
WKS is the concurrence of Wernicke’s encephalopathy (an acute, life-threatening condition marked by ataxia, confusion, and ocular signs) and Korsakoff’s psychosis (a long-term, debilitating amnestic syndrome). WKS is a neuropsychiatric disorder in which patients experience profound short-term amnesia; it is precipitated by thiamine deficiency (defined as a whole blood thiamine level <0.7 ng/ml1).The link to thiamine was confirmed during World War II, when thiamine treatment resolved symptoms in starving prisoners. If recognized early, treatment of thiamine deficiency can prevent long-term morbidity from WKS.
Etiology of thiamine deficiency
Our patient’s alcohol abuse placed her at risk for WKS, and her olfactory aversion to certain foods was a diagnostic clue. In this case, we inadvertently administered dextrose with antibiotics for the UTI prior to administering thiamine; this exacerbated the thiamine deficiency because glucose and thiamine compete for the same substrate.
Is alcohol abuse always to blame for WKS?
The quantity and type of alcohol that results in the development of WKS has not been well studied, but the Caine diagnostic criteria defines chronic alcoholism as the consumption of 80 g/d of ethanol (8 drinks/d).2 While WKS is commonly associated with alcoholism, other causative conditions may be overlooked. Other associated illnesses include acquired immune deficiency syndrome (AIDS), cancer, hyperemesis gravidarum, prolonged total parenteral nutrition, and psychiatric illnesses such as eating disorders and schizophrenia. Procedures such as gastric bypass and dialysis can also precipitate WKS.3
Men and women are both at risk of developing WKS. A lack of consumption of thiamine-rich sources such as cereals, rice, and legumes puts patients at risk for WKS. The recommended dietary allowance of thiamine increases with age and may be higher for obese patients.4
Continue to: Suspect thiamine deficiency and obstain a thorough history
Suspect thiamine deficiency and obtain a thorough history
A high index of suspicion for thiamine deficiency is essential for diagnosis of WKS. History of alcohol use should be obtained, including quantity, frequency, pattern, duration, and time of last use. Physicians should assess nutrition and ask about vomiting and diarrhea. It is important to collaborate with the patient’s family and friends and inquire into other substance misuse.5
Since WKS targets the dorsomedial thalamus, which is responsible for olfactory processing, patients may complain of a distorted perception of smell.6 On physical examination, look for signs of protein-calorie malnutrition, including cheilitis, glossitis, and bleeding gums; signs of alcohol abuse, such as hepatomegaly; and evidence of injuries or poor self-care.5
Varied presentation leads to under- and misdiagnosis
Diagnosis of WKS can be difficult due to the varied presentation; there is a broad spectrum of clinical features. The clinical triad of mental status change, ophthalmoplegia, and gait ataxia is present in as few as 10% of cases.3 Mental status changes may include a global confusional state ranging from disorientation, apathy, anxiety, fear, and mild memory impairment to pronounced amnesia. Ophthalmoplegia can include nystagmus, ocular palsies, retinal hemorrhages, scotoma, or photophobia; and ataxia can range from a mild gait abnormality to an inability to stand.7 This varied presentation ultimately leads to underdiagnosis and misdiagnosis.
MRI findings are also varied in WKS. However, the mammillary bodies are involved in many cases, where atrophy of these structures have high specificity. The dorsomedial thalamus is associated with the reported impairment in memory and can be identified antemortem on MRI.3 There is no quantifiable evidence of how much thiamine should be used to prevent WKS. However, thiamine should be given before the administration of glucose whenever WKS is considered.
Our patient. Despite the administration of thiamine (100 mg parenterally for 5 d, followed by oral thiamine 300 mg/d indefinitely), our patient’s memory and cognition remained unchanged. She underwent intensive inpatient rehabilitation for 2 months and was eventually placed in long-term nursing care.
Continue to: THE TAKEAWAY
THE TAKEAWAY
A high index of suspicion is crucial to prevent possible long-term neurologic sequelae in WKS. Appropriate care starts at the beginning, with the patient’s story.
CORRESPONDENCE
Romith Naug, MD, 15 St. Andrew Street, Unit 601, Brockville, ON Canada K6V0B8; Romith.naug@gmail.com.
1. Doshi S, Velpandian T, Seth S, et al. Prevalence of thiamine deficiency in heart failure patients on long-term diuretic therapy. J Prac Cardiovasc Sci. 2015;1:25-29.
2. Caine D, Halliday GM, Kril JJ, et al. Operational criteria for the classification of chronic alcoholics: identification of Wernicke’s encephalopathy. J Neurol Neurosurg Psychiatry. 1997;62:51-60.
3. Donnino MW, Vega J, Miller J, et al. Myths and misconceptions of Wernicke’s encephalopathy: what every emergency physician should know. Ann Emerg Med. 2007;50:715-721.
4. Kerns J, Arundel C, Chawla LS. Thiamin deficiency in people with obesity. Adv Nutr. 2015;6:147-153.
5. Latt N, Dore G. Thiamine in the treatment of Wernicke encephalopathy in patients with alcohol use disorders. Intern Med J. 2014;44:911-915.
6. Wilson DA, Xu W, Sadrian B, et al. Cortical odor processing in health and disease. Prog Brain Res. 2014;208:275-305.
7. Isenberg-Grzeda E, Kutner HE, Nicolson SE. Wernicke-Korsakoff syndrome: under-recognized and under-treated. Psychosomatics. 2012;53:507-516.
THE CASE
A 57-year-old woman presented to a family physician with acute encephalopathy and complaints of recent gastritis. She reported a 2-month history of nausea, vomiting, decreased oral intake, and extreme sensitivity to smell. The patient had a history of hypertension, and a family member privately disclosed to the FP that she also had a history of alcohol abuse. The patient was taking lorazepam daily, as needed, for anxiety.
On initial assessment, the patient was alert, but not oriented to time or situation. She was ataxic and agitated but did not exhibit pupillary constriction or tremor. The FP sent her to the emergency department (ED).
After being assessed in the ED, the patient was admitted. Over the course of several days, she showed worsening mentation; she persistently believed she was in Chicago, her childhood home. On memory testing, she was unable to recall any of 3 objects after 5 minutes. She exhibited horizontal nystagmus and dysmetria bilaterally and continued to be ataxic, requiring 2-point assistance. Her agitation was managed nonpharmacologically.
A work-up was performed, which included laboratory testing, a urinalysis, and computed tomography (CT) of the head. A comprehensive metabolic panel, complete blood count, and thyroid stimulating hormone test were unremarkable except for electrolyte disturbances, with a sodium level of 158 mEq/L and a potassium level of 2.6 mEq/L (reference ranges: 135-145 mEq/L and 3.5-5 mEq/L, respectively).
Her blood alcohol level was zero, and not surprisingly given her use of lorazepam, a urine drug screen was positive for benzodiazepines. The urinalysis results were consistent with a urinary tract infection (UTI), for which she was treated with an antibiotic. A carbohydrate-deficient transferrin test may have been useful to establish chronic alcohol abuse, but was not ordered. The head CT was negative.
After a few days with fluids and electrolyte replacement, the patient’s electrolytes normalized.
THE DIAGNOSIS
The differential diagnosis included sepsis, metabolic encephalopathy, and alcoholic encephalopathy. Given that the patient’s urine drug screen was positive, benzodiazepine withdrawal was also considered a plausible explanation for her continued cognitive disturbances. (It was surmised that she had likely taken her last lorazepam several days prior.) However, the lack of other signs of withdrawal prompted further investigation.
Continue to: Since her encephalopathy...
Since her encephalopathy, ataxia, and nystagmus persisted, magnetic resonance imaging (MRI) of the brain was performed on Day 3 of hospitalization (FIGURE). A lumbar puncture and an electroencephalogram were also considered but were not performed because the MRI results revealed bilateral enhancement of the m
DISCUSSION
WKS is the concurrence of Wernicke’s encephalopathy (an acute, life-threatening condition marked by ataxia, confusion, and ocular signs) and Korsakoff’s psychosis (a long-term, debilitating amnestic syndrome). WKS is a neuropsychiatric disorder in which patients experience profound short-term amnesia; it is precipitated by thiamine deficiency (defined as a whole blood thiamine level <0.7 ng/ml1).The link to thiamine was confirmed during World War II, when thiamine treatment resolved symptoms in starving prisoners. If recognized early, treatment of thiamine deficiency can prevent long-term morbidity from WKS.
Etiology of thiamine deficiency
Our patient’s alcohol abuse placed her at risk for WKS, and her olfactory aversion to certain foods was a diagnostic clue. In this case, we inadvertently administered dextrose with antibiotics for the UTI prior to administering thiamine; this exacerbated the thiamine deficiency because glucose and thiamine compete for the same substrate.
Is alcohol abuse always to blame for WKS?
The quantity and type of alcohol that results in the development of WKS has not been well studied, but the Caine diagnostic criteria defines chronic alcoholism as the consumption of 80 g/d of ethanol (8 drinks/d).2 While WKS is commonly associated with alcoholism, other causative conditions may be overlooked. Other associated illnesses include acquired immune deficiency syndrome (AIDS), cancer, hyperemesis gravidarum, prolonged total parenteral nutrition, and psychiatric illnesses such as eating disorders and schizophrenia. Procedures such as gastric bypass and dialysis can also precipitate WKS.3
Men and women are both at risk of developing WKS. A lack of consumption of thiamine-rich sources such as cereals, rice, and legumes puts patients at risk for WKS. The recommended dietary allowance of thiamine increases with age and may be higher for obese patients.4
Continue to: Suspect thiamine deficiency and obstain a thorough history
Suspect thiamine deficiency and obtain a thorough history
A high index of suspicion for thiamine deficiency is essential for diagnosis of WKS. History of alcohol use should be obtained, including quantity, frequency, pattern, duration, and time of last use. Physicians should assess nutrition and ask about vomiting and diarrhea. It is important to collaborate with the patient’s family and friends and inquire into other substance misuse.5
Since WKS targets the dorsomedial thalamus, which is responsible for olfactory processing, patients may complain of a distorted perception of smell.6 On physical examination, look for signs of protein-calorie malnutrition, including cheilitis, glossitis, and bleeding gums; signs of alcohol abuse, such as hepatomegaly; and evidence of injuries or poor self-care.5
Varied presentation leads to under- and misdiagnosis
Diagnosis of WKS can be difficult due to the varied presentation; there is a broad spectrum of clinical features. The clinical triad of mental status change, ophthalmoplegia, and gait ataxia is present in as few as 10% of cases.3 Mental status changes may include a global confusional state ranging from disorientation, apathy, anxiety, fear, and mild memory impairment to pronounced amnesia. Ophthalmoplegia can include nystagmus, ocular palsies, retinal hemorrhages, scotoma, or photophobia; and ataxia can range from a mild gait abnormality to an inability to stand.7 This varied presentation ultimately leads to underdiagnosis and misdiagnosis.
MRI findings are also varied in WKS. However, the mammillary bodies are involved in many cases, where atrophy of these structures have high specificity. The dorsomedial thalamus is associated with the reported impairment in memory and can be identified antemortem on MRI.3 There is no quantifiable evidence of how much thiamine should be used to prevent WKS. However, thiamine should be given before the administration of glucose whenever WKS is considered.
Our patient. Despite the administration of thiamine (100 mg parenterally for 5 d, followed by oral thiamine 300 mg/d indefinitely), our patient’s memory and cognition remained unchanged. She underwent intensive inpatient rehabilitation for 2 months and was eventually placed in long-term nursing care.
Continue to: THE TAKEAWAY
THE TAKEAWAY
A high index of suspicion is crucial to prevent possible long-term neurologic sequelae in WKS. Appropriate care starts at the beginning, with the patient’s story.
CORRESPONDENCE
Romith Naug, MD, 15 St. Andrew Street, Unit 601, Brockville, ON Canada K6V0B8; Romith.naug@gmail.com.
THE CASE
A 57-year-old woman presented to a family physician with acute encephalopathy and complaints of recent gastritis. She reported a 2-month history of nausea, vomiting, decreased oral intake, and extreme sensitivity to smell. The patient had a history of hypertension, and a family member privately disclosed to the FP that she also had a history of alcohol abuse. The patient was taking lorazepam daily, as needed, for anxiety.
On initial assessment, the patient was alert, but not oriented to time or situation. She was ataxic and agitated but did not exhibit pupillary constriction or tremor. The FP sent her to the emergency department (ED).
After being assessed in the ED, the patient was admitted. Over the course of several days, she showed worsening mentation; she persistently believed she was in Chicago, her childhood home. On memory testing, she was unable to recall any of 3 objects after 5 minutes. She exhibited horizontal nystagmus and dysmetria bilaterally and continued to be ataxic, requiring 2-point assistance. Her agitation was managed nonpharmacologically.
A work-up was performed, which included laboratory testing, a urinalysis, and computed tomography (CT) of the head. A comprehensive metabolic panel, complete blood count, and thyroid stimulating hormone test were unremarkable except for electrolyte disturbances, with a sodium level of 158 mEq/L and a potassium level of 2.6 mEq/L (reference ranges: 135-145 mEq/L and 3.5-5 mEq/L, respectively).
Her blood alcohol level was zero, and not surprisingly given her use of lorazepam, a urine drug screen was positive for benzodiazepines. The urinalysis results were consistent with a urinary tract infection (UTI), for which she was treated with an antibiotic. A carbohydrate-deficient transferrin test may have been useful to establish chronic alcohol abuse, but was not ordered. The head CT was negative.
After a few days with fluids and electrolyte replacement, the patient’s electrolytes normalized.
THE DIAGNOSIS
The differential diagnosis included sepsis, metabolic encephalopathy, and alcoholic encephalopathy. Given that the patient’s urine drug screen was positive, benzodiazepine withdrawal was also considered a plausible explanation for her continued cognitive disturbances. (It was surmised that she had likely taken her last lorazepam several days prior.) However, the lack of other signs of withdrawal prompted further investigation.
Continue to: Since her encephalopathy...
Since her encephalopathy, ataxia, and nystagmus persisted, magnetic resonance imaging (MRI) of the brain was performed on Day 3 of hospitalization (FIGURE). A lumbar puncture and an electroencephalogram were also considered but were not performed because the MRI results revealed bilateral enhancement of the m
DISCUSSION
WKS is the concurrence of Wernicke’s encephalopathy (an acute, life-threatening condition marked by ataxia, confusion, and ocular signs) and Korsakoff’s psychosis (a long-term, debilitating amnestic syndrome). WKS is a neuropsychiatric disorder in which patients experience profound short-term amnesia; it is precipitated by thiamine deficiency (defined as a whole blood thiamine level <0.7 ng/ml1).The link to thiamine was confirmed during World War II, when thiamine treatment resolved symptoms in starving prisoners. If recognized early, treatment of thiamine deficiency can prevent long-term morbidity from WKS.
Etiology of thiamine deficiency
Our patient’s alcohol abuse placed her at risk for WKS, and her olfactory aversion to certain foods was a diagnostic clue. In this case, we inadvertently administered dextrose with antibiotics for the UTI prior to administering thiamine; this exacerbated the thiamine deficiency because glucose and thiamine compete for the same substrate.
Is alcohol abuse always to blame for WKS?
The quantity and type of alcohol that results in the development of WKS has not been well studied, but the Caine diagnostic criteria defines chronic alcoholism as the consumption of 80 g/d of ethanol (8 drinks/d).2 While WKS is commonly associated with alcoholism, other causative conditions may be overlooked. Other associated illnesses include acquired immune deficiency syndrome (AIDS), cancer, hyperemesis gravidarum, prolonged total parenteral nutrition, and psychiatric illnesses such as eating disorders and schizophrenia. Procedures such as gastric bypass and dialysis can also precipitate WKS.3
Men and women are both at risk of developing WKS. A lack of consumption of thiamine-rich sources such as cereals, rice, and legumes puts patients at risk for WKS. The recommended dietary allowance of thiamine increases with age and may be higher for obese patients.4
Continue to: Suspect thiamine deficiency and obstain a thorough history
Suspect thiamine deficiency and obtain a thorough history
A high index of suspicion for thiamine deficiency is essential for diagnosis of WKS. History of alcohol use should be obtained, including quantity, frequency, pattern, duration, and time of last use. Physicians should assess nutrition and ask about vomiting and diarrhea. It is important to collaborate with the patient’s family and friends and inquire into other substance misuse.5
Since WKS targets the dorsomedial thalamus, which is responsible for olfactory processing, patients may complain of a distorted perception of smell.6 On physical examination, look for signs of protein-calorie malnutrition, including cheilitis, glossitis, and bleeding gums; signs of alcohol abuse, such as hepatomegaly; and evidence of injuries or poor self-care.5
Varied presentation leads to under- and misdiagnosis
Diagnosis of WKS can be difficult due to the varied presentation; there is a broad spectrum of clinical features. The clinical triad of mental status change, ophthalmoplegia, and gait ataxia is present in as few as 10% of cases.3 Mental status changes may include a global confusional state ranging from disorientation, apathy, anxiety, fear, and mild memory impairment to pronounced amnesia. Ophthalmoplegia can include nystagmus, ocular palsies, retinal hemorrhages, scotoma, or photophobia; and ataxia can range from a mild gait abnormality to an inability to stand.7 This varied presentation ultimately leads to underdiagnosis and misdiagnosis.
MRI findings are also varied in WKS. However, the mammillary bodies are involved in many cases, where atrophy of these structures have high specificity. The dorsomedial thalamus is associated with the reported impairment in memory and can be identified antemortem on MRI.3 There is no quantifiable evidence of how much thiamine should be used to prevent WKS. However, thiamine should be given before the administration of glucose whenever WKS is considered.
Our patient. Despite the administration of thiamine (100 mg parenterally for 5 d, followed by oral thiamine 300 mg/d indefinitely), our patient’s memory and cognition remained unchanged. She underwent intensive inpatient rehabilitation for 2 months and was eventually placed in long-term nursing care.
Continue to: THE TAKEAWAY
THE TAKEAWAY
A high index of suspicion is crucial to prevent possible long-term neurologic sequelae in WKS. Appropriate care starts at the beginning, with the patient’s story.
CORRESPONDENCE
Romith Naug, MD, 15 St. Andrew Street, Unit 601, Brockville, ON Canada K6V0B8; Romith.naug@gmail.com.
1. Doshi S, Velpandian T, Seth S, et al. Prevalence of thiamine deficiency in heart failure patients on long-term diuretic therapy. J Prac Cardiovasc Sci. 2015;1:25-29.
2. Caine D, Halliday GM, Kril JJ, et al. Operational criteria for the classification of chronic alcoholics: identification of Wernicke’s encephalopathy. J Neurol Neurosurg Psychiatry. 1997;62:51-60.
3. Donnino MW, Vega J, Miller J, et al. Myths and misconceptions of Wernicke’s encephalopathy: what every emergency physician should know. Ann Emerg Med. 2007;50:715-721.
4. Kerns J, Arundel C, Chawla LS. Thiamin deficiency in people with obesity. Adv Nutr. 2015;6:147-153.
5. Latt N, Dore G. Thiamine in the treatment of Wernicke encephalopathy in patients with alcohol use disorders. Intern Med J. 2014;44:911-915.
6. Wilson DA, Xu W, Sadrian B, et al. Cortical odor processing in health and disease. Prog Brain Res. 2014;208:275-305.
7. Isenberg-Grzeda E, Kutner HE, Nicolson SE. Wernicke-Korsakoff syndrome: under-recognized and under-treated. Psychosomatics. 2012;53:507-516.
1. Doshi S, Velpandian T, Seth S, et al. Prevalence of thiamine deficiency in heart failure patients on long-term diuretic therapy. J Prac Cardiovasc Sci. 2015;1:25-29.
2. Caine D, Halliday GM, Kril JJ, et al. Operational criteria for the classification of chronic alcoholics: identification of Wernicke’s encephalopathy. J Neurol Neurosurg Psychiatry. 1997;62:51-60.
3. Donnino MW, Vega J, Miller J, et al. Myths and misconceptions of Wernicke’s encephalopathy: what every emergency physician should know. Ann Emerg Med. 2007;50:715-721.
4. Kerns J, Arundel C, Chawla LS. Thiamin deficiency in people with obesity. Adv Nutr. 2015;6:147-153.
5. Latt N, Dore G. Thiamine in the treatment of Wernicke encephalopathy in patients with alcohol use disorders. Intern Med J. 2014;44:911-915.
6. Wilson DA, Xu W, Sadrian B, et al. Cortical odor processing in health and disease. Prog Brain Res. 2014;208:275-305.
7. Isenberg-Grzeda E, Kutner HE, Nicolson SE. Wernicke-Korsakoff syndrome: under-recognized and under-treated. Psychosomatics. 2012;53:507-516.
