User login
Dupilumab for Off-Label Treatment of Moderate to Severe Childhood Atopic Dermatitis
Case Report
A 7-year-old boy with a history of shellfish anaphylaxis, pollen allergy, asthma, rhinoconjunctivitis, frequent headaches and ear infections, sinusitis, bronchitis, vitiligo, warts, and cold sores presented to our dermatology clinic for evaluation of a widespread crusting, cracking, red rash that had been present since 6 months of age. The patient’s mother reported that he had many sleepless nights from uncontrolled itching. His medications included albuterol solution for nebulization, loratadine, and montelukast. Prior to the current presentation he had been treated with triamcinolone and betamethasone creams by the pediatrician. Despite compliance with topical therapy, his mother stated the itching persisted and lesions lingered with minimal improvement. He also was treated with oral corticosteroids for episodic sinusitis and bronchitis, which was beneficial to the skin lesions for only a short duration. The patient was adopted and therefore his family history was unavailable.
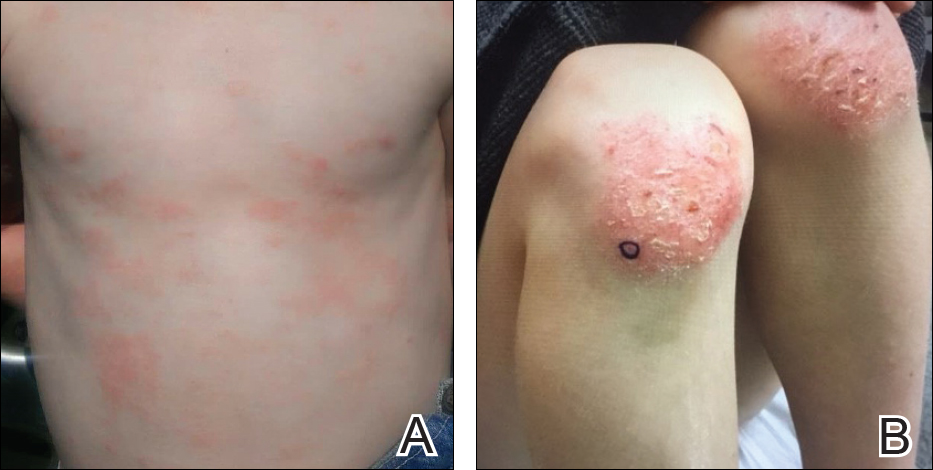
During physical examination, the patient was in the fetal position on the examination table and appeared uncomfortable, scratching himself. The patient admitted to severe widespread itching and burning. On skin examination, multiple thick, lichenified, highly pruritic plaques coalesced on the knees, ankles, arms, and wrists, and very discreet scaly patches were present on the scalp. Annular patches covered 50% of the patient’s body, with highly inflamed lesions concentrated in skin folds (Figure 1), leading to diagnosis of atopic dermatitis (AD).
Over the course of several months, a number of topical therapies were prescribed. The calcineurin inhibitor pimecrolimus cream 1% proffered minimal relief, and the patient experienced burning with crisaborole despite attempts to combine it with emollients and topical corticosteroids. The patient and his mother favored intermittent use of topical corticosteroids alone; however, he experienced frequent disease flares. Stabilized hypochlorous acid spray and mupirocin 2% antibiotic ointment were included in the treatment regimen as adjunctive topical therapies. Additionally, the patient underwent bleach and vinegar bath therapy without success.
Although UVA and UVB phototherapy has shown to be safe and effective in children, our patient had limited treatment options due to insurance restrictions. The patient had been taking oral corticosteroids on and off for years prior to presentation to our dermatology clinic.
Our patient weighed approximately 40 lb and was prescribed methotrexate 5 mg once weekly for 2 weeks along with oral folic acid 1 mg once daily, except when taking the methotrexate. Laboratory workup was ordered at 2- and then 4-week intervals. After 2 weeks of treatment, methotrexate was increased to 10 mg once weekly. His asthma was carefully monitored by the allergist, and his mother was instructed to stop the medication if he had worsening shortness of breath or exacerbation of asthma symptoms. He tolerated methotrexate at 10 mg once weekly well without clinical side effects for 6 months. His mother observed less frequent ear and sinus infections during methotrexate therapy; however, he developed anemia over time and the methotrexate was discontinued. Understanding the nature of off-label use in administering dupilumab, the patient’s mother consented to a scheduled dosage of 300 mg subcutaneous (SQ) injection every month in the absence of a loading dose with the assumption of future modifications pending his response to therapy.
Five days after treatment with a 300-mg SQ dupilumab injection, the patient returned to clinic for evaluation of a vesicular rash with subsequent peeling confined to the shoulders (Figure 2). He and his mother denied any UV exposure, citing he had been completely out of the sun. He denied constitutional symptoms including fever, malaise, swelling, joint pain, headache, muscle pain, nausea, vomiting, diarrhea, enlarged lymph glands, difficulty urinating, breathing, or neurological disturbance. Upon physical examination, the rash was not considered to be a drug eruption. Had a mild drug reaction been suspected, a careful rechallenge, weighing the risks and benefits, would have been considered and was discussed with the mother and patient. New-onset or worsening eye symptoms should be reported; therefore, a referral to ophthalmology was prompted due to our patient’s history of rhinoconjunctivitis and persistent conjunctival injection observed early after initiating dupilumab therapy. Nothing remarkable was found.
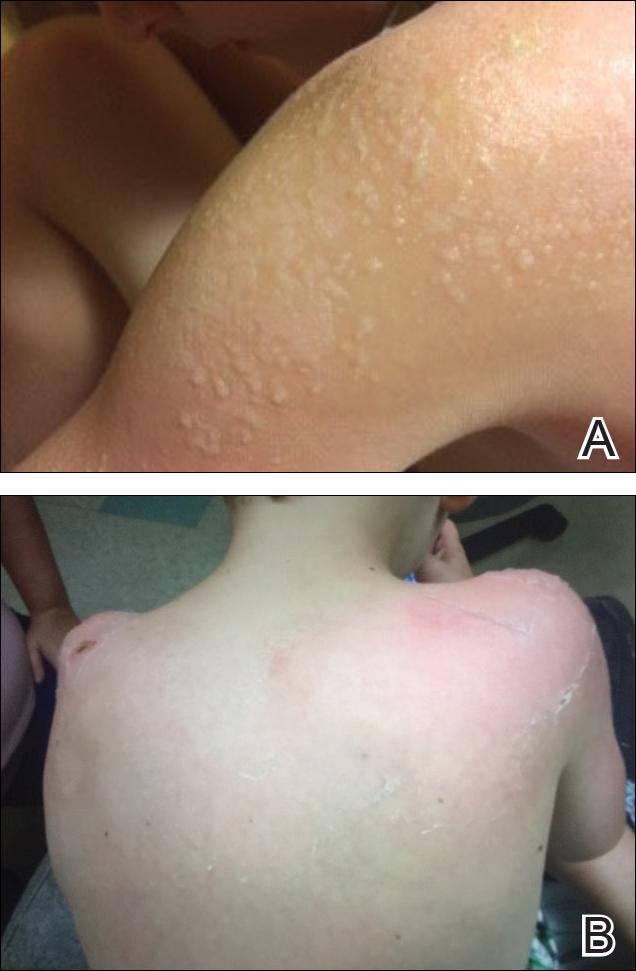
The patient was eager to continue dupilumab therapy due to considerable reduction of itching and elimination of lesions. His mother reported that the greatest benefit 1 month after starting dupilumab was almost no itching (Figure 3A). Additionally, he denied headache or nasopharyngitis at his 1-month office visit. After 2 months of dupilumab therapy, the patient reported persistent lesions on the feet and ankles despite concomitant treatment with topical corticosteroids. The decision to increase the dupilumab dose to 300-mg SQ injection once every 3 weeks for a total of 3 doses was made, which resulted in resolution of all lesions (Figure 3B).
Comment
Prevalence and Pathogenesis
Atopic dermatitis affects 31.6 million individuals in the United States, with 17.8 million experiencing moderate to severe lesions.1 The current prevalence of AD in the pediatric population ranges from 10% to 30% compared to 2% to 10% in adults. Fortunately, up to 70% of young children enter remission or improve by 12 years of age. Atopic diatheses may simultaneously occur, which includes asthma and rhinoconjunctivitis.2
Complications from AD include bacterial and viral infections and ocular disease. Furthermore, impaired growth in stature has been correlated with individuals who have extensive disease.2 Of interest, our 7-year-old patient gained 7 lb and grew almost 3 in within 6 months of being on immunosuppressant therapy. Children with AD have poorer sleep efficiency in contrast to children without AD.3 Eczema is associated with more frequent headaches in childhood, especially in those with sleep disturbances,4 as our patient had experienced prior to systemic therapy.
The pathogenesis of AD is complex, and one must take into consideration the multiple cellular activities including inflammatory mechanics in the absence of IgE-mediated sensitization, epidermal barrier changes, epicutaneous sensitization, dendritic cell roles, T-cell responses and cytokine orchestrations, actions of microbial colonization, and involvement of autoimmunity.5 Select patients with AD have IgE antibodies focused against self-proteins. Disease severity correlates with ubiquity of these antibodies. Moreover, certain autoallergens induce helper T cell (TH1) responses.5 Circulating TH2 cytokines and chemokines IL-4, IL4ra, and IL-13 also have been linked to AD pathogenesis. Additionally, nonlesional skin abnormalities have been observed.6 Most recently, researchers have identified a caspase recruitment domain family member 11 (CARD11) gene mutation possibly leading to AD.7 Clinically, our patient responded favorably to dupilumab, which inhibits TH2 cytokines IL-4 and IL13. He experienced a considerable decrease in itching and inflammation and reduced lesion count after 1 month of treatment with dupilumab. No skin lesions were identified on visual examination at week 17 and inevitably the patient discontinued messy topicals.
Treatment Options
Because AD is characterized by episodes of remission and relapse, management generally is comprised of trigger avoidance, including known allergens and irritants; a skin care regimen that promotes healthy epidermal barrier function; anti-inflammatory therapies to control both flares and subclinical inflammation; and adjunctive therapy for additional symptomatic control (eg, phototherapy, stabilized hypochlorous acid, topical antibiotic treatment) when needed. Avoidance of excessive washing or irritants, food provocation, and emotional stress, as well as toleration of body temperature fluctuations and humidity, is recommended to amend exacerbations.5
Current topical therapies include emollients; corticosteroids; calcineurin inhibitors; and crisaborole, a newer phosphodiesterase 4 inhibitor. There are a number of emollients and moisturizers available, and one over-the-counter preparation showed tolerability and improved skin hydration in AD patients and demonstrated less transepidermal water loss than the control group.8 Ointments such as petrolatum usually do not include ingredients such as preservatives, gelling agents, or humectants that can promote stinging or burning.9 Topical corticosteroids, which ameliorate inflammation by subduing proinflammatory cytokine expression, have been the mainstay of treatment for more than 60 years; however, caution should be used due to the potential for side effects, mainly but not limited to systemic absorption in children, development of striae, and skin atrophy. Calcineurin inhibitors prohibit T-cell activity, modify mast cell response, and decrease dendritic cells in the epidermis. Since 2000, calcineurin inhibitors have been utilized as steroid-sparing agents10; however, prior authorization is still necessary with some insurance providers. Crisaborole ointment 2%, the newest topical agent for AD treatment in the market, has shown improvement of erythema, exudation, and pruritus. Approved for patients aged 2 years and older, twice-daily application of topical crisaborole as a steroid-sparing agent has rendered AD symptom relief.11 It has been reported that 4% of patients encounter stinging or burning with topical crisaborole application, whereas up to 50% of calcineurin inhibitors induce these adverse effects.12 Stabilized hypochlorous acid spray or gel acts as an antipruritic and antimicrobial agent, relieving pain associated with skin irritations. Topical antimicrobial preparations such as mupirocin 2% antibiotic ointment can reduce Staphylococcus colonization when applied in the nasal passage as well as to affected skin lesions.2
In children, UVA and UVB phototherapy has proven safe and effective and can be utilized in AD when suitable.13 When patients inadequately respond to topical therapies and phototherapy, systemic immunomodulatory agents have been recommended as treatment options.A child’s developing immune system indeed may be sensitive to systemic therapies as the innate immune system fully matures in adolescence and his/her adaptive immune system is undergoing vigorous definition.14 Systemic immunomodulatory agents such as cyclosporine, azathioprine, mycophenolate mofetil, and methotrexate have been used off label for years and pose certain challenges in being identified as durable therapies due to potential side effects. Cyclosporine is effective for the treatment of AD; however, long-term administration should be dosed up to a 12-month period and then stopped to decrease cumulative exposure to the drug. Therefore, further treatment options must be considered. For children, cyclosporine should be administered in a dose of 3 to 6 mg/kg daily. Fluctuations in blood pressure and renal function should be monitored. The recommended pediatric dose for azathioprine is 1 to 4 mg/kg daily with laboratory monitoring, particularly of liver enzymes and complete blood cell count. Obtaining the patient’s thiopurine methyltransferase level may aid in dosing. Gastrointestinal tract symptoms such as nausea, vomiting, and diarrhea are common. Phototherapy is not advised in conjunction with azathioprine due to an increased risk of photocarcinogenicity.13 The literature supporting mycophenolate mofetil in children with AD is limited.
Biologic therapies targeting IgE, B-lymphocyte antigen CD20, IL-5, thymic stromal lymphopoietin, TH17 cells, IL-12, IL-23, interferon gamma, IL-6 receptors, tumor necrosis factor, phosphodiesterase 4, Janus kinase, chymase, and nuclear receptors expressed on adipocytes and immune cells have undergone investigation for treatment of AD.17 Additionally, biologic agents targeting IL-31, IL-13, and IL-22 also have been evaluated.1 Currently, there are no US Food and Drug Administration–approved biologic agents for moderate to severe childhood AD.
Dupilumab, an IL-4Rα and IL-13Rα antagonist, recently has been approved for treatment of moderate to severe AD in adults but not yet for children. Potential side effects include nasopharyngitis, headache, hypersensitivity reactions, and ocular symptoms,11 namely keratitis and conjunctivitis.18 Less than 1% of patients experienced keratitis in clinical trials, while conjunctivitis was reported in 4% of patients taking dupilumab with topical corticosteroids at 52 weeks.18 However, possible ocular findings on slit-lamp examination in AD patients include atopic keratoconjunctivitis, blepharitis, palpebral conjunctival scarring, papillary conjunctival reaction, Horner-Trantas dots, keratoconus, and atopic cataracts. Spontaneous retinal detachment is seen more commonly in individuals with AD than in the general population.19
In clinical trials, hypersensitivity reactions included urticaria and serum sickness or serum sickness–like reactions in less than 1% of patients taking dupilumab.18
Conclusion
Childhood AD can be debilitating, and affected individuals often lead a poorer quality of life if left untreated. Embarrassment and isolation are commonly experienced. Increased responsibility and work in tending for a child with eczema may result in parental exhaustion.21 As with psoriasis, AD can impair activity and productivity.22 Currently, dupilumab has proven to positively impact health-related quality of life for adults.23 Pending the outcome of ongoing pediatric clinical trials, dupilumab may become a benchmark therapy for children younger than 18 years.
- Samalonis L. What’s new in eczema and atopic dermatitis research. The Dermatologist. November 19, 2015. http://www.the-dermatologist.com/content/whats-new-eczema-and-atopic-dermatitis-research. Accessed July 19, 2018.
- Habif T. Atopic dermatitis. In: Bonnet C, Pinczewski A, Cook L, eds. Clinical Dermatology. 5th ed. Edinburgh, Scotland: Mosby Elsevier; 2010:160-180.
- Fishbein AB, Mueller K, Kruse L, et al. Sleep disturbance in children with moderate/severe atopic dermatitis: a case control study [published online October 28, 2017]. J Am Acad Dermatol. 2018;78:336-341.
- Silverberg J. Association between childhood eczema and headaches: an analysis of 19 US population-based studies [published online August 29, 2015]. J Allergy Clin Immunol. 2016;137:492-499.e5.
- Bieber T, Bussmann C. Atopic dermatitis. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. China: Saunders Elsevier; 2012:203-216.
- Suarez-Farinas M, Tintle S, Shemer A, et al. Non-lesional atopic dermatitis (AD) skin is characterized by broad terminal differentiation defects and variable immune abnormalities. J Allergy Clin Immunol. 2011;127:954-964.
- Hilton L. AD gene mutation identified: discovery may lead to new therapeutic option for patients. Dermatol Times. 2017;38:30.
- Zeichner JA, Dryer L. Effect of CeraVe Healing Ointment on skin hydration and barrier function on normal and barrier-impaired skin. Poster presented at: Orlando Dermatology Aesthetic & Clinical Conference; January 15-16, 2016; Orlando, FL.
- Garg T, Rath G, Goyal AK. Comprehensive review on additives of topical dosage forms for drug delivery. Drug Delivery. 2015;22:969-987.
- Eichenfield LF, Tom WL, Berger TG, et al. Guidelines of care for the management of atopic dermatitis section 2. management and treatment of atopic dermatitis with topical therapies. J Am Acad Dermatol. 2014;71:116-132.
- Koutnik-Fotopoulous E. Update on the latest eczema treatments. The Dermatologist. February 17, 2016. http://www.the-dermatologist.com/content/update-latest-eczema-treatments. Accessed August 16, 2018.
- Paller AS, Tom WL, Lebwohl MG, et al. Efficacy and safety of crisaborole ointment, a novel phosphodiesterase 4 inhibitor for the topical treatment of AD in children and adults [published online July 11, 2016]. J Am Acad Dermatol. 2016;75:494-503.
- Sidbury R, Davis D, Cohen D, et al. Guidelines of care for the management of atopic dermatitis: section 3. management and treatment with phototherapy and systemic agents . J Am Acad Dermatol. 2014;71:327-349.
- van der Merwe R, Gianella-Borradori A. Industry perspective on the clinical development of systemic products for the treatment of atopic dermatitis in pediatric patients with inadequate response to topical prescription therapy. Presented at: FDA Dermatologic and Ophthalmic Drugs Advisory Committee Meeting; March 9, 2015; Silver Spring, MD.
- Heller M, Shin HT, Orlow SJ, et al. Mycophenolate mofetil for severe childhood atopic dermatitis: experience in 14 patients. Br J Dermatol. 2007;157:127-132.
- Callen JP, Kulp-Shorten CL. Methotrexate. In: Wolverton SE, ed. Comprehensive Dermatologic Drug Therapy. 3rd ed. China: Saunders Elsevier; 2013:169-181.
- Guttman-Yassky E, Dhingra N, Leung DY. New era of biological therapeutics in atopic dermatitis [published online January 16, 2013]. Expert Opin Biol Ther. 2013;13:549-561.
- Dupixent [package insert]. Tarrytown, NY: Regeneron Pharmaceuticals, Inc; 2017.
- Lowery RS. Ophthalmologic manifestations of atopic dermatitis clinical presentation. Medscape website. emedicine.medscape.com/article/1197636-clinical#b4. Updated September 7, 2016. Accessed July 19, 2018.
- Lenz HJ. Management and preparedness for infusion and hypersensitivity reactions. Oncologist. 2007;12:601-609.
- Lewis-Jones S. Quality of life and childhood atopic dermatitis: the misery of living with childhood eczema. Int J Clin Pract. 2006;60:984-992.
- Eckert L, Gupta S, Amand C, et al. Impact of atopic dermatitis on health-related quality of life and productivity in adults in the Unites States: an analysis using the National Health and Wellness Survey. J Am Acad Dermatol, 2017;77:274-279.
- Tsianakas A, Luger TA, Radin A. Dupilumab treatment improves quality of life in adult patients with moderate-to-severe atopic dermatitis: results from a randomized, placebo-controlled clinical trial [published online January 11, 2018]. Br J Dermatol. 2018;178:406-414.
Case Report
A 7-year-old boy with a history of shellfish anaphylaxis, pollen allergy, asthma, rhinoconjunctivitis, frequent headaches and ear infections, sinusitis, bronchitis, vitiligo, warts, and cold sores presented to our dermatology clinic for evaluation of a widespread crusting, cracking, red rash that had been present since 6 months of age. The patient’s mother reported that he had many sleepless nights from uncontrolled itching. His medications included albuterol solution for nebulization, loratadine, and montelukast. Prior to the current presentation he had been treated with triamcinolone and betamethasone creams by the pediatrician. Despite compliance with topical therapy, his mother stated the itching persisted and lesions lingered with minimal improvement. He also was treated with oral corticosteroids for episodic sinusitis and bronchitis, which was beneficial to the skin lesions for only a short duration. The patient was adopted and therefore his family history was unavailable.
During physical examination, the patient was in the fetal position on the examination table and appeared uncomfortable, scratching himself. The patient admitted to severe widespread itching and burning. On skin examination, multiple thick, lichenified, highly pruritic plaques coalesced on the knees, ankles, arms, and wrists, and very discreet scaly patches were present on the scalp. Annular patches covered 50% of the patient’s body, with highly inflamed lesions concentrated in skin folds (Figure 1), leading to diagnosis of atopic dermatitis (AD).
Over the course of several months, a number of topical therapies were prescribed. The calcineurin inhibitor pimecrolimus cream 1% proffered minimal relief, and the patient experienced burning with crisaborole despite attempts to combine it with emollients and topical corticosteroids. The patient and his mother favored intermittent use of topical corticosteroids alone; however, he experienced frequent disease flares. Stabilized hypochlorous acid spray and mupirocin 2% antibiotic ointment were included in the treatment regimen as adjunctive topical therapies. Additionally, the patient underwent bleach and vinegar bath therapy without success.
Although UVA and UVB phototherapy has shown to be safe and effective in children, our patient had limited treatment options due to insurance restrictions. The patient had been taking oral corticosteroids on and off for years prior to presentation to our dermatology clinic.
Our patient weighed approximately 40 lb and was prescribed methotrexate 5 mg once weekly for 2 weeks along with oral folic acid 1 mg once daily, except when taking the methotrexate. Laboratory workup was ordered at 2- and then 4-week intervals. After 2 weeks of treatment, methotrexate was increased to 10 mg once weekly. His asthma was carefully monitored by the allergist, and his mother was instructed to stop the medication if he had worsening shortness of breath or exacerbation of asthma symptoms. He tolerated methotrexate at 10 mg once weekly well without clinical side effects for 6 months. His mother observed less frequent ear and sinus infections during methotrexate therapy; however, he developed anemia over time and the methotrexate was discontinued. Understanding the nature of off-label use in administering dupilumab, the patient’s mother consented to a scheduled dosage of 300 mg subcutaneous (SQ) injection every month in the absence of a loading dose with the assumption of future modifications pending his response to therapy.
Five days after treatment with a 300-mg SQ dupilumab injection, the patient returned to clinic for evaluation of a vesicular rash with subsequent peeling confined to the shoulders (Figure 2). He and his mother denied any UV exposure, citing he had been completely out of the sun. He denied constitutional symptoms including fever, malaise, swelling, joint pain, headache, muscle pain, nausea, vomiting, diarrhea, enlarged lymph glands, difficulty urinating, breathing, or neurological disturbance. Upon physical examination, the rash was not considered to be a drug eruption. Had a mild drug reaction been suspected, a careful rechallenge, weighing the risks and benefits, would have been considered and was discussed with the mother and patient. New-onset or worsening eye symptoms should be reported; therefore, a referral to ophthalmology was prompted due to our patient’s history of rhinoconjunctivitis and persistent conjunctival injection observed early after initiating dupilumab therapy. Nothing remarkable was found.
The patient was eager to continue dupilumab therapy due to considerable reduction of itching and elimination of lesions. His mother reported that the greatest benefit 1 month after starting dupilumab was almost no itching (Figure 3A). Additionally, he denied headache or nasopharyngitis at his 1-month office visit. After 2 months of dupilumab therapy, the patient reported persistent lesions on the feet and ankles despite concomitant treatment with topical corticosteroids. The decision to increase the dupilumab dose to 300-mg SQ injection once every 3 weeks for a total of 3 doses was made, which resulted in resolution of all lesions (Figure 3B).
Comment
Prevalence and Pathogenesis
Atopic dermatitis affects 31.6 million individuals in the United States, with 17.8 million experiencing moderate to severe lesions.1 The current prevalence of AD in the pediatric population ranges from 10% to 30% compared to 2% to 10% in adults. Fortunately, up to 70% of young children enter remission or improve by 12 years of age. Atopic diatheses may simultaneously occur, which includes asthma and rhinoconjunctivitis.2
Complications from AD include bacterial and viral infections and ocular disease. Furthermore, impaired growth in stature has been correlated with individuals who have extensive disease.2 Of interest, our 7-year-old patient gained 7 lb and grew almost 3 in within 6 months of being on immunosuppressant therapy. Children with AD have poorer sleep efficiency in contrast to children without AD.3 Eczema is associated with more frequent headaches in childhood, especially in those with sleep disturbances,4 as our patient had experienced prior to systemic therapy.
The pathogenesis of AD is complex, and one must take into consideration the multiple cellular activities including inflammatory mechanics in the absence of IgE-mediated sensitization, epidermal barrier changes, epicutaneous sensitization, dendritic cell roles, T-cell responses and cytokine orchestrations, actions of microbial colonization, and involvement of autoimmunity.5 Select patients with AD have IgE antibodies focused against self-proteins. Disease severity correlates with ubiquity of these antibodies. Moreover, certain autoallergens induce helper T cell (TH1) responses.5 Circulating TH2 cytokines and chemokines IL-4, IL4ra, and IL-13 also have been linked to AD pathogenesis. Additionally, nonlesional skin abnormalities have been observed.6 Most recently, researchers have identified a caspase recruitment domain family member 11 (CARD11) gene mutation possibly leading to AD.7 Clinically, our patient responded favorably to dupilumab, which inhibits TH2 cytokines IL-4 and IL13. He experienced a considerable decrease in itching and inflammation and reduced lesion count after 1 month of treatment with dupilumab. No skin lesions were identified on visual examination at week 17 and inevitably the patient discontinued messy topicals.
Treatment Options
Because AD is characterized by episodes of remission and relapse, management generally is comprised of trigger avoidance, including known allergens and irritants; a skin care regimen that promotes healthy epidermal barrier function; anti-inflammatory therapies to control both flares and subclinical inflammation; and adjunctive therapy for additional symptomatic control (eg, phototherapy, stabilized hypochlorous acid, topical antibiotic treatment) when needed. Avoidance of excessive washing or irritants, food provocation, and emotional stress, as well as toleration of body temperature fluctuations and humidity, is recommended to amend exacerbations.5
Current topical therapies include emollients; corticosteroids; calcineurin inhibitors; and crisaborole, a newer phosphodiesterase 4 inhibitor. There are a number of emollients and moisturizers available, and one over-the-counter preparation showed tolerability and improved skin hydration in AD patients and demonstrated less transepidermal water loss than the control group.8 Ointments such as petrolatum usually do not include ingredients such as preservatives, gelling agents, or humectants that can promote stinging or burning.9 Topical corticosteroids, which ameliorate inflammation by subduing proinflammatory cytokine expression, have been the mainstay of treatment for more than 60 years; however, caution should be used due to the potential for side effects, mainly but not limited to systemic absorption in children, development of striae, and skin atrophy. Calcineurin inhibitors prohibit T-cell activity, modify mast cell response, and decrease dendritic cells in the epidermis. Since 2000, calcineurin inhibitors have been utilized as steroid-sparing agents10; however, prior authorization is still necessary with some insurance providers. Crisaborole ointment 2%, the newest topical agent for AD treatment in the market, has shown improvement of erythema, exudation, and pruritus. Approved for patients aged 2 years and older, twice-daily application of topical crisaborole as a steroid-sparing agent has rendered AD symptom relief.11 It has been reported that 4% of patients encounter stinging or burning with topical crisaborole application, whereas up to 50% of calcineurin inhibitors induce these adverse effects.12 Stabilized hypochlorous acid spray or gel acts as an antipruritic and antimicrobial agent, relieving pain associated with skin irritations. Topical antimicrobial preparations such as mupirocin 2% antibiotic ointment can reduce Staphylococcus colonization when applied in the nasal passage as well as to affected skin lesions.2
In children, UVA and UVB phototherapy has proven safe and effective and can be utilized in AD when suitable.13 When patients inadequately respond to topical therapies and phototherapy, systemic immunomodulatory agents have been recommended as treatment options.A child’s developing immune system indeed may be sensitive to systemic therapies as the innate immune system fully matures in adolescence and his/her adaptive immune system is undergoing vigorous definition.14 Systemic immunomodulatory agents such as cyclosporine, azathioprine, mycophenolate mofetil, and methotrexate have been used off label for years and pose certain challenges in being identified as durable therapies due to potential side effects. Cyclosporine is effective for the treatment of AD; however, long-term administration should be dosed up to a 12-month period and then stopped to decrease cumulative exposure to the drug. Therefore, further treatment options must be considered. For children, cyclosporine should be administered in a dose of 3 to 6 mg/kg daily. Fluctuations in blood pressure and renal function should be monitored. The recommended pediatric dose for azathioprine is 1 to 4 mg/kg daily with laboratory monitoring, particularly of liver enzymes and complete blood cell count. Obtaining the patient’s thiopurine methyltransferase level may aid in dosing. Gastrointestinal tract symptoms such as nausea, vomiting, and diarrhea are common. Phototherapy is not advised in conjunction with azathioprine due to an increased risk of photocarcinogenicity.13 The literature supporting mycophenolate mofetil in children with AD is limited.
Biologic therapies targeting IgE, B-lymphocyte antigen CD20, IL-5, thymic stromal lymphopoietin, TH17 cells, IL-12, IL-23, interferon gamma, IL-6 receptors, tumor necrosis factor, phosphodiesterase 4, Janus kinase, chymase, and nuclear receptors expressed on adipocytes and immune cells have undergone investigation for treatment of AD.17 Additionally, biologic agents targeting IL-31, IL-13, and IL-22 also have been evaluated.1 Currently, there are no US Food and Drug Administration–approved biologic agents for moderate to severe childhood AD.
Dupilumab, an IL-4Rα and IL-13Rα antagonist, recently has been approved for treatment of moderate to severe AD in adults but not yet for children. Potential side effects include nasopharyngitis, headache, hypersensitivity reactions, and ocular symptoms,11 namely keratitis and conjunctivitis.18 Less than 1% of patients experienced keratitis in clinical trials, while conjunctivitis was reported in 4% of patients taking dupilumab with topical corticosteroids at 52 weeks.18 However, possible ocular findings on slit-lamp examination in AD patients include atopic keratoconjunctivitis, blepharitis, palpebral conjunctival scarring, papillary conjunctival reaction, Horner-Trantas dots, keratoconus, and atopic cataracts. Spontaneous retinal detachment is seen more commonly in individuals with AD than in the general population.19
In clinical trials, hypersensitivity reactions included urticaria and serum sickness or serum sickness–like reactions in less than 1% of patients taking dupilumab.18
Conclusion
Childhood AD can be debilitating, and affected individuals often lead a poorer quality of life if left untreated. Embarrassment and isolation are commonly experienced. Increased responsibility and work in tending for a child with eczema may result in parental exhaustion.21 As with psoriasis, AD can impair activity and productivity.22 Currently, dupilumab has proven to positively impact health-related quality of life for adults.23 Pending the outcome of ongoing pediatric clinical trials, dupilumab may become a benchmark therapy for children younger than 18 years.
Case Report
A 7-year-old boy with a history of shellfish anaphylaxis, pollen allergy, asthma, rhinoconjunctivitis, frequent headaches and ear infections, sinusitis, bronchitis, vitiligo, warts, and cold sores presented to our dermatology clinic for evaluation of a widespread crusting, cracking, red rash that had been present since 6 months of age. The patient’s mother reported that he had many sleepless nights from uncontrolled itching. His medications included albuterol solution for nebulization, loratadine, and montelukast. Prior to the current presentation he had been treated with triamcinolone and betamethasone creams by the pediatrician. Despite compliance with topical therapy, his mother stated the itching persisted and lesions lingered with minimal improvement. He also was treated with oral corticosteroids for episodic sinusitis and bronchitis, which was beneficial to the skin lesions for only a short duration. The patient was adopted and therefore his family history was unavailable.
During physical examination, the patient was in the fetal position on the examination table and appeared uncomfortable, scratching himself. The patient admitted to severe widespread itching and burning. On skin examination, multiple thick, lichenified, highly pruritic plaques coalesced on the knees, ankles, arms, and wrists, and very discreet scaly patches were present on the scalp. Annular patches covered 50% of the patient’s body, with highly inflamed lesions concentrated in skin folds (Figure 1), leading to diagnosis of atopic dermatitis (AD).
Over the course of several months, a number of topical therapies were prescribed. The calcineurin inhibitor pimecrolimus cream 1% proffered minimal relief, and the patient experienced burning with crisaborole despite attempts to combine it with emollients and topical corticosteroids. The patient and his mother favored intermittent use of topical corticosteroids alone; however, he experienced frequent disease flares. Stabilized hypochlorous acid spray and mupirocin 2% antibiotic ointment were included in the treatment regimen as adjunctive topical therapies. Additionally, the patient underwent bleach and vinegar bath therapy without success.
Although UVA and UVB phototherapy has shown to be safe and effective in children, our patient had limited treatment options due to insurance restrictions. The patient had been taking oral corticosteroids on and off for years prior to presentation to our dermatology clinic.
Our patient weighed approximately 40 lb and was prescribed methotrexate 5 mg once weekly for 2 weeks along with oral folic acid 1 mg once daily, except when taking the methotrexate. Laboratory workup was ordered at 2- and then 4-week intervals. After 2 weeks of treatment, methotrexate was increased to 10 mg once weekly. His asthma was carefully monitored by the allergist, and his mother was instructed to stop the medication if he had worsening shortness of breath or exacerbation of asthma symptoms. He tolerated methotrexate at 10 mg once weekly well without clinical side effects for 6 months. His mother observed less frequent ear and sinus infections during methotrexate therapy; however, he developed anemia over time and the methotrexate was discontinued. Understanding the nature of off-label use in administering dupilumab, the patient’s mother consented to a scheduled dosage of 300 mg subcutaneous (SQ) injection every month in the absence of a loading dose with the assumption of future modifications pending his response to therapy.
Five days after treatment with a 300-mg SQ dupilumab injection, the patient returned to clinic for evaluation of a vesicular rash with subsequent peeling confined to the shoulders (Figure 2). He and his mother denied any UV exposure, citing he had been completely out of the sun. He denied constitutional symptoms including fever, malaise, swelling, joint pain, headache, muscle pain, nausea, vomiting, diarrhea, enlarged lymph glands, difficulty urinating, breathing, or neurological disturbance. Upon physical examination, the rash was not considered to be a drug eruption. Had a mild drug reaction been suspected, a careful rechallenge, weighing the risks and benefits, would have been considered and was discussed with the mother and patient. New-onset or worsening eye symptoms should be reported; therefore, a referral to ophthalmology was prompted due to our patient’s history of rhinoconjunctivitis and persistent conjunctival injection observed early after initiating dupilumab therapy. Nothing remarkable was found.
The patient was eager to continue dupilumab therapy due to considerable reduction of itching and elimination of lesions. His mother reported that the greatest benefit 1 month after starting dupilumab was almost no itching (Figure 3A). Additionally, he denied headache or nasopharyngitis at his 1-month office visit. After 2 months of dupilumab therapy, the patient reported persistent lesions on the feet and ankles despite concomitant treatment with topical corticosteroids. The decision to increase the dupilumab dose to 300-mg SQ injection once every 3 weeks for a total of 3 doses was made, which resulted in resolution of all lesions (Figure 3B).
Comment
Prevalence and Pathogenesis
Atopic dermatitis affects 31.6 million individuals in the United States, with 17.8 million experiencing moderate to severe lesions.1 The current prevalence of AD in the pediatric population ranges from 10% to 30% compared to 2% to 10% in adults. Fortunately, up to 70% of young children enter remission or improve by 12 years of age. Atopic diatheses may simultaneously occur, which includes asthma and rhinoconjunctivitis.2
Complications from AD include bacterial and viral infections and ocular disease. Furthermore, impaired growth in stature has been correlated with individuals who have extensive disease.2 Of interest, our 7-year-old patient gained 7 lb and grew almost 3 in within 6 months of being on immunosuppressant therapy. Children with AD have poorer sleep efficiency in contrast to children without AD.3 Eczema is associated with more frequent headaches in childhood, especially in those with sleep disturbances,4 as our patient had experienced prior to systemic therapy.
The pathogenesis of AD is complex, and one must take into consideration the multiple cellular activities including inflammatory mechanics in the absence of IgE-mediated sensitization, epidermal barrier changes, epicutaneous sensitization, dendritic cell roles, T-cell responses and cytokine orchestrations, actions of microbial colonization, and involvement of autoimmunity.5 Select patients with AD have IgE antibodies focused against self-proteins. Disease severity correlates with ubiquity of these antibodies. Moreover, certain autoallergens induce helper T cell (TH1) responses.5 Circulating TH2 cytokines and chemokines IL-4, IL4ra, and IL-13 also have been linked to AD pathogenesis. Additionally, nonlesional skin abnormalities have been observed.6 Most recently, researchers have identified a caspase recruitment domain family member 11 (CARD11) gene mutation possibly leading to AD.7 Clinically, our patient responded favorably to dupilumab, which inhibits TH2 cytokines IL-4 and IL13. He experienced a considerable decrease in itching and inflammation and reduced lesion count after 1 month of treatment with dupilumab. No skin lesions were identified on visual examination at week 17 and inevitably the patient discontinued messy topicals.
Treatment Options
Because AD is characterized by episodes of remission and relapse, management generally is comprised of trigger avoidance, including known allergens and irritants; a skin care regimen that promotes healthy epidermal barrier function; anti-inflammatory therapies to control both flares and subclinical inflammation; and adjunctive therapy for additional symptomatic control (eg, phototherapy, stabilized hypochlorous acid, topical antibiotic treatment) when needed. Avoidance of excessive washing or irritants, food provocation, and emotional stress, as well as toleration of body temperature fluctuations and humidity, is recommended to amend exacerbations.5
Current topical therapies include emollients; corticosteroids; calcineurin inhibitors; and crisaborole, a newer phosphodiesterase 4 inhibitor. There are a number of emollients and moisturizers available, and one over-the-counter preparation showed tolerability and improved skin hydration in AD patients and demonstrated less transepidermal water loss than the control group.8 Ointments such as petrolatum usually do not include ingredients such as preservatives, gelling agents, or humectants that can promote stinging or burning.9 Topical corticosteroids, which ameliorate inflammation by subduing proinflammatory cytokine expression, have been the mainstay of treatment for more than 60 years; however, caution should be used due to the potential for side effects, mainly but not limited to systemic absorption in children, development of striae, and skin atrophy. Calcineurin inhibitors prohibit T-cell activity, modify mast cell response, and decrease dendritic cells in the epidermis. Since 2000, calcineurin inhibitors have been utilized as steroid-sparing agents10; however, prior authorization is still necessary with some insurance providers. Crisaborole ointment 2%, the newest topical agent for AD treatment in the market, has shown improvement of erythema, exudation, and pruritus. Approved for patients aged 2 years and older, twice-daily application of topical crisaborole as a steroid-sparing agent has rendered AD symptom relief.11 It has been reported that 4% of patients encounter stinging or burning with topical crisaborole application, whereas up to 50% of calcineurin inhibitors induce these adverse effects.12 Stabilized hypochlorous acid spray or gel acts as an antipruritic and antimicrobial agent, relieving pain associated with skin irritations. Topical antimicrobial preparations such as mupirocin 2% antibiotic ointment can reduce Staphylococcus colonization when applied in the nasal passage as well as to affected skin lesions.2
In children, UVA and UVB phototherapy has proven safe and effective and can be utilized in AD when suitable.13 When patients inadequately respond to topical therapies and phototherapy, systemic immunomodulatory agents have been recommended as treatment options.A child’s developing immune system indeed may be sensitive to systemic therapies as the innate immune system fully matures in adolescence and his/her adaptive immune system is undergoing vigorous definition.14 Systemic immunomodulatory agents such as cyclosporine, azathioprine, mycophenolate mofetil, and methotrexate have been used off label for years and pose certain challenges in being identified as durable therapies due to potential side effects. Cyclosporine is effective for the treatment of AD; however, long-term administration should be dosed up to a 12-month period and then stopped to decrease cumulative exposure to the drug. Therefore, further treatment options must be considered. For children, cyclosporine should be administered in a dose of 3 to 6 mg/kg daily. Fluctuations in blood pressure and renal function should be monitored. The recommended pediatric dose for azathioprine is 1 to 4 mg/kg daily with laboratory monitoring, particularly of liver enzymes and complete blood cell count. Obtaining the patient’s thiopurine methyltransferase level may aid in dosing. Gastrointestinal tract symptoms such as nausea, vomiting, and diarrhea are common. Phototherapy is not advised in conjunction with azathioprine due to an increased risk of photocarcinogenicity.13 The literature supporting mycophenolate mofetil in children with AD is limited.
Biologic therapies targeting IgE, B-lymphocyte antigen CD20, IL-5, thymic stromal lymphopoietin, TH17 cells, IL-12, IL-23, interferon gamma, IL-6 receptors, tumor necrosis factor, phosphodiesterase 4, Janus kinase, chymase, and nuclear receptors expressed on adipocytes and immune cells have undergone investigation for treatment of AD.17 Additionally, biologic agents targeting IL-31, IL-13, and IL-22 also have been evaluated.1 Currently, there are no US Food and Drug Administration–approved biologic agents for moderate to severe childhood AD.
Dupilumab, an IL-4Rα and IL-13Rα antagonist, recently has been approved for treatment of moderate to severe AD in adults but not yet for children. Potential side effects include nasopharyngitis, headache, hypersensitivity reactions, and ocular symptoms,11 namely keratitis and conjunctivitis.18 Less than 1% of patients experienced keratitis in clinical trials, while conjunctivitis was reported in 4% of patients taking dupilumab with topical corticosteroids at 52 weeks.18 However, possible ocular findings on slit-lamp examination in AD patients include atopic keratoconjunctivitis, blepharitis, palpebral conjunctival scarring, papillary conjunctival reaction, Horner-Trantas dots, keratoconus, and atopic cataracts. Spontaneous retinal detachment is seen more commonly in individuals with AD than in the general population.19
In clinical trials, hypersensitivity reactions included urticaria and serum sickness or serum sickness–like reactions in less than 1% of patients taking dupilumab.18
Conclusion
Childhood AD can be debilitating, and affected individuals often lead a poorer quality of life if left untreated. Embarrassment and isolation are commonly experienced. Increased responsibility and work in tending for a child with eczema may result in parental exhaustion.21 As with psoriasis, AD can impair activity and productivity.22 Currently, dupilumab has proven to positively impact health-related quality of life for adults.23 Pending the outcome of ongoing pediatric clinical trials, dupilumab may become a benchmark therapy for children younger than 18 years.
- Samalonis L. What’s new in eczema and atopic dermatitis research. The Dermatologist. November 19, 2015. http://www.the-dermatologist.com/content/whats-new-eczema-and-atopic-dermatitis-research. Accessed July 19, 2018.
- Habif T. Atopic dermatitis. In: Bonnet C, Pinczewski A, Cook L, eds. Clinical Dermatology. 5th ed. Edinburgh, Scotland: Mosby Elsevier; 2010:160-180.
- Fishbein AB, Mueller K, Kruse L, et al. Sleep disturbance in children with moderate/severe atopic dermatitis: a case control study [published online October 28, 2017]. J Am Acad Dermatol. 2018;78:336-341.
- Silverberg J. Association between childhood eczema and headaches: an analysis of 19 US population-based studies [published online August 29, 2015]. J Allergy Clin Immunol. 2016;137:492-499.e5.
- Bieber T, Bussmann C. Atopic dermatitis. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. China: Saunders Elsevier; 2012:203-216.
- Suarez-Farinas M, Tintle S, Shemer A, et al. Non-lesional atopic dermatitis (AD) skin is characterized by broad terminal differentiation defects and variable immune abnormalities. J Allergy Clin Immunol. 2011;127:954-964.
- Hilton L. AD gene mutation identified: discovery may lead to new therapeutic option for patients. Dermatol Times. 2017;38:30.
- Zeichner JA, Dryer L. Effect of CeraVe Healing Ointment on skin hydration and barrier function on normal and barrier-impaired skin. Poster presented at: Orlando Dermatology Aesthetic & Clinical Conference; January 15-16, 2016; Orlando, FL.
- Garg T, Rath G, Goyal AK. Comprehensive review on additives of topical dosage forms for drug delivery. Drug Delivery. 2015;22:969-987.
- Eichenfield LF, Tom WL, Berger TG, et al. Guidelines of care for the management of atopic dermatitis section 2. management and treatment of atopic dermatitis with topical therapies. J Am Acad Dermatol. 2014;71:116-132.
- Koutnik-Fotopoulous E. Update on the latest eczema treatments. The Dermatologist. February 17, 2016. http://www.the-dermatologist.com/content/update-latest-eczema-treatments. Accessed August 16, 2018.
- Paller AS, Tom WL, Lebwohl MG, et al. Efficacy and safety of crisaborole ointment, a novel phosphodiesterase 4 inhibitor for the topical treatment of AD in children and adults [published online July 11, 2016]. J Am Acad Dermatol. 2016;75:494-503.
- Sidbury R, Davis D, Cohen D, et al. Guidelines of care for the management of atopic dermatitis: section 3. management and treatment with phototherapy and systemic agents . J Am Acad Dermatol. 2014;71:327-349.
- van der Merwe R, Gianella-Borradori A. Industry perspective on the clinical development of systemic products for the treatment of atopic dermatitis in pediatric patients with inadequate response to topical prescription therapy. Presented at: FDA Dermatologic and Ophthalmic Drugs Advisory Committee Meeting; March 9, 2015; Silver Spring, MD.
- Heller M, Shin HT, Orlow SJ, et al. Mycophenolate mofetil for severe childhood atopic dermatitis: experience in 14 patients. Br J Dermatol. 2007;157:127-132.
- Callen JP, Kulp-Shorten CL. Methotrexate. In: Wolverton SE, ed. Comprehensive Dermatologic Drug Therapy. 3rd ed. China: Saunders Elsevier; 2013:169-181.
- Guttman-Yassky E, Dhingra N, Leung DY. New era of biological therapeutics in atopic dermatitis [published online January 16, 2013]. Expert Opin Biol Ther. 2013;13:549-561.
- Dupixent [package insert]. Tarrytown, NY: Regeneron Pharmaceuticals, Inc; 2017.
- Lowery RS. Ophthalmologic manifestations of atopic dermatitis clinical presentation. Medscape website. emedicine.medscape.com/article/1197636-clinical#b4. Updated September 7, 2016. Accessed July 19, 2018.
- Lenz HJ. Management and preparedness for infusion and hypersensitivity reactions. Oncologist. 2007;12:601-609.
- Lewis-Jones S. Quality of life and childhood atopic dermatitis: the misery of living with childhood eczema. Int J Clin Pract. 2006;60:984-992.
- Eckert L, Gupta S, Amand C, et al. Impact of atopic dermatitis on health-related quality of life and productivity in adults in the Unites States: an analysis using the National Health and Wellness Survey. J Am Acad Dermatol, 2017;77:274-279.
- Tsianakas A, Luger TA, Radin A. Dupilumab treatment improves quality of life in adult patients with moderate-to-severe atopic dermatitis: results from a randomized, placebo-controlled clinical trial [published online January 11, 2018]. Br J Dermatol. 2018;178:406-414.
- Samalonis L. What’s new in eczema and atopic dermatitis research. The Dermatologist. November 19, 2015. http://www.the-dermatologist.com/content/whats-new-eczema-and-atopic-dermatitis-research. Accessed July 19, 2018.
- Habif T. Atopic dermatitis. In: Bonnet C, Pinczewski A, Cook L, eds. Clinical Dermatology. 5th ed. Edinburgh, Scotland: Mosby Elsevier; 2010:160-180.
- Fishbein AB, Mueller K, Kruse L, et al. Sleep disturbance in children with moderate/severe atopic dermatitis: a case control study [published online October 28, 2017]. J Am Acad Dermatol. 2018;78:336-341.
- Silverberg J. Association between childhood eczema and headaches: an analysis of 19 US population-based studies [published online August 29, 2015]. J Allergy Clin Immunol. 2016;137:492-499.e5.
- Bieber T, Bussmann C. Atopic dermatitis. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. China: Saunders Elsevier; 2012:203-216.
- Suarez-Farinas M, Tintle S, Shemer A, et al. Non-lesional atopic dermatitis (AD) skin is characterized by broad terminal differentiation defects and variable immune abnormalities. J Allergy Clin Immunol. 2011;127:954-964.
- Hilton L. AD gene mutation identified: discovery may lead to new therapeutic option for patients. Dermatol Times. 2017;38:30.
- Zeichner JA, Dryer L. Effect of CeraVe Healing Ointment on skin hydration and barrier function on normal and barrier-impaired skin. Poster presented at: Orlando Dermatology Aesthetic & Clinical Conference; January 15-16, 2016; Orlando, FL.
- Garg T, Rath G, Goyal AK. Comprehensive review on additives of topical dosage forms for drug delivery. Drug Delivery. 2015;22:969-987.
- Eichenfield LF, Tom WL, Berger TG, et al. Guidelines of care for the management of atopic dermatitis section 2. management and treatment of atopic dermatitis with topical therapies. J Am Acad Dermatol. 2014;71:116-132.
- Koutnik-Fotopoulous E. Update on the latest eczema treatments. The Dermatologist. February 17, 2016. http://www.the-dermatologist.com/content/update-latest-eczema-treatments. Accessed August 16, 2018.
- Paller AS, Tom WL, Lebwohl MG, et al. Efficacy and safety of crisaborole ointment, a novel phosphodiesterase 4 inhibitor for the topical treatment of AD in children and adults [published online July 11, 2016]. J Am Acad Dermatol. 2016;75:494-503.
- Sidbury R, Davis D, Cohen D, et al. Guidelines of care for the management of atopic dermatitis: section 3. management and treatment with phototherapy and systemic agents . J Am Acad Dermatol. 2014;71:327-349.
- van der Merwe R, Gianella-Borradori A. Industry perspective on the clinical development of systemic products for the treatment of atopic dermatitis in pediatric patients with inadequate response to topical prescription therapy. Presented at: FDA Dermatologic and Ophthalmic Drugs Advisory Committee Meeting; March 9, 2015; Silver Spring, MD.
- Heller M, Shin HT, Orlow SJ, et al. Mycophenolate mofetil for severe childhood atopic dermatitis: experience in 14 patients. Br J Dermatol. 2007;157:127-132.
- Callen JP, Kulp-Shorten CL. Methotrexate. In: Wolverton SE, ed. Comprehensive Dermatologic Drug Therapy. 3rd ed. China: Saunders Elsevier; 2013:169-181.
- Guttman-Yassky E, Dhingra N, Leung DY. New era of biological therapeutics in atopic dermatitis [published online January 16, 2013]. Expert Opin Biol Ther. 2013;13:549-561.
- Dupixent [package insert]. Tarrytown, NY: Regeneron Pharmaceuticals, Inc; 2017.
- Lowery RS. Ophthalmologic manifestations of atopic dermatitis clinical presentation. Medscape website. emedicine.medscape.com/article/1197636-clinical#b4. Updated September 7, 2016. Accessed July 19, 2018.
- Lenz HJ. Management and preparedness for infusion and hypersensitivity reactions. Oncologist. 2007;12:601-609.
- Lewis-Jones S. Quality of life and childhood atopic dermatitis: the misery of living with childhood eczema. Int J Clin Pract. 2006;60:984-992.
- Eckert L, Gupta S, Amand C, et al. Impact of atopic dermatitis on health-related quality of life and productivity in adults in the Unites States: an analysis using the National Health and Wellness Survey. J Am Acad Dermatol, 2017;77:274-279.
- Tsianakas A, Luger TA, Radin A. Dupilumab treatment improves quality of life in adult patients with moderate-to-severe atopic dermatitis: results from a randomized, placebo-controlled clinical trial [published online January 11, 2018]. Br J Dermatol. 2018;178:406-414.
Practice Points
- Childhood atopic dermatitis can be debilitating, and affected individuals often experience poorer quality of life if left untreated.
- Dupilumab may become a benchmark therapy for children younger than 18 years.
Concurrent Notalgia Paresthetica and Brachioradial Pruritus Associated With Cervical Degenerative Disc Disease
Case Report
A 74-year-old man presented with persistent episodes of severe pruritus with exacerbations on the bilateral forearms, arms, and left side of the mid back of 4 years’ duration. He had a refractory and debilitating disease that had failed extensive therapies including topical antipruritics, antihistamines, oral hydroxyzine, capsaicin, potent topical steroids (ie, clobetasol, fluocinonide, triamcinolone), phototherapy with narrowband UVB, and various dietary modifications including a gluten-free trial. The patient reported he had exhausted all medical evaluation through care with more than 7 physicians and multiple dermatologists, including a university-based dermatology department for repeated consultations; he was seen by our dermatology center for an eighth opinion.
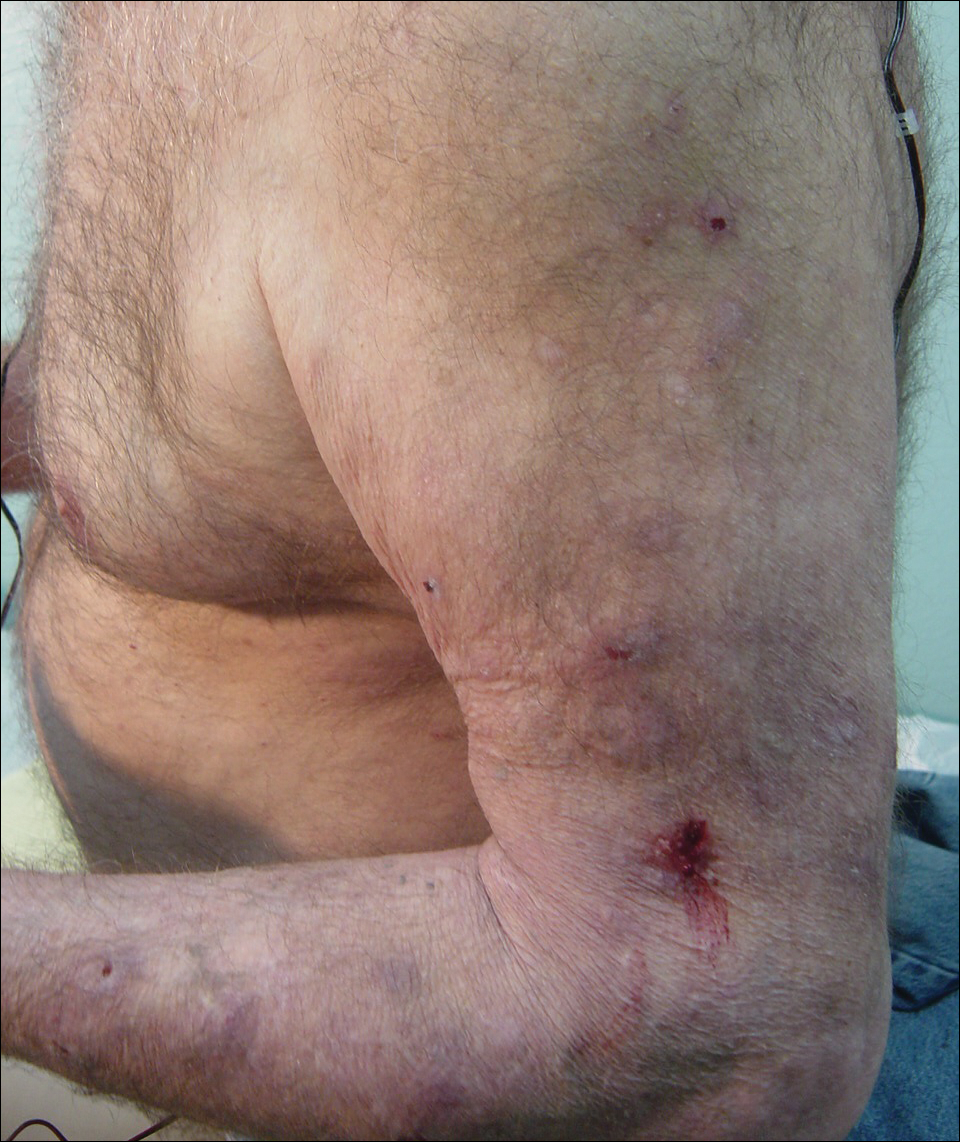
Initial dermatologic examination revealed multiple secondarily excoriated, hemorrhagic, hyperpigmented plaques and nodules on the right side of the mid upper back indicative of notalgia paresthetica (NP) with secondary chronic skin changes (Figure 1). Additional examination of the left arm and forearm revealed several open erosions, raised nodules, and lichenified skin plaques indicative of brachioradial pruritus (BP) with secondary skin changes (Figure 2). In addition, multiple lichenified plaques of the left side of the mid back were associated with decreased sensory alternations to light touch and pin prick. Of note, the localized pruritus pattern, particularly of the unilateral infrascapular back region, heralded the possibility of a neuropathic pruritus condition originating from the cervical spine. Examination confirmed decreased range of motion in the neck with associated marked palpable bilateral cervical muscle spasm and tenderness. Laboratory testing confirmed Staphylococcus aureus secondary skin infection that was treated empirically with chlorhexidine wash. General pruritus serology and imaging workup was ordered with contributory results. The patient’s medical history was notable for noninsulin-dependent diabetes mellitus, obesity, deep venous thrombosis, asthma, vein surgery, cardiovascular disease, atrial fibrillation, atopy, allergies, asthma, and keratosis pilaris, as well as drug intolerances of warfarin sodium, sitagliptin, and clopidogrel. His medications on presentation included glyburide, digoxin, prednisone, aspirin, cetirizine, cimetidine, and hydroxyzine. Based on the relatively classic localized pruritus symptoms and the anatomical distribution of skin findings, a clinical diagnosis of concurrent NP and BRP was made, and radiologic studies of the cervical spine were ordered.

Magnetic resonance imaging (MRI) of the cervical spine showed severe central canal stenosis at C3-C4 secondary to disc disease slight asymmetric toward the right side, severe central canal stenosis at C4-C5 slightly more prominent in the midline, severe central stenosis at C5-C6 more prominent in the midline, and mild changes at other levels as described. Laboratory workup revealed an abnormal complete blood cell count with mildly elevated white blood cell count (11,800/µL [reference range, 4000–10,500/µL]), elevated neutrophils (8600/µL [reference range, 1800–7800/µL]), elevated eosinophils (600/µL [reference range, 0–450/µL ]), and elevated IgE (160 IU/mL [reference range, 0–100 IU/mL]). Further testing revealed negative results for Helicobacter pylori IgG and IgM, human immunodeficiency virus, and hepatitis B and C screening panels; antinuclear antibody negative; normal thyroid-stimulating hormone; and normal thyroid peroxidase antibody. Chest radiograph and computed tomography of the chest, abdomen, and pelvis were negative.
We referred the patient for a neurosurgical consultation that uncovered newly diagnosed severe cervical stenosis with mild to moderate canal compromise at C3, C4, C5, and C6. His motor examination revealed full strength in the upper extremities (5/5). Sensory examination showed patchy sensory alteration on the mid back. He declined oral antibiotics as advised for the skin staphylococcal infection and neurosurgical treatment for the cervical disease.
During the 4 years prior to presentation at our center, the patient reported failure to improve with a dermatologically prescribed gluten-free diet as well as all topical and oral steroid treatments. He was presented at a university grand rounds where a suggestion for UVB light treatment was made; the patient reported possible worsening of symptoms with narrowband UVB phototherapy.
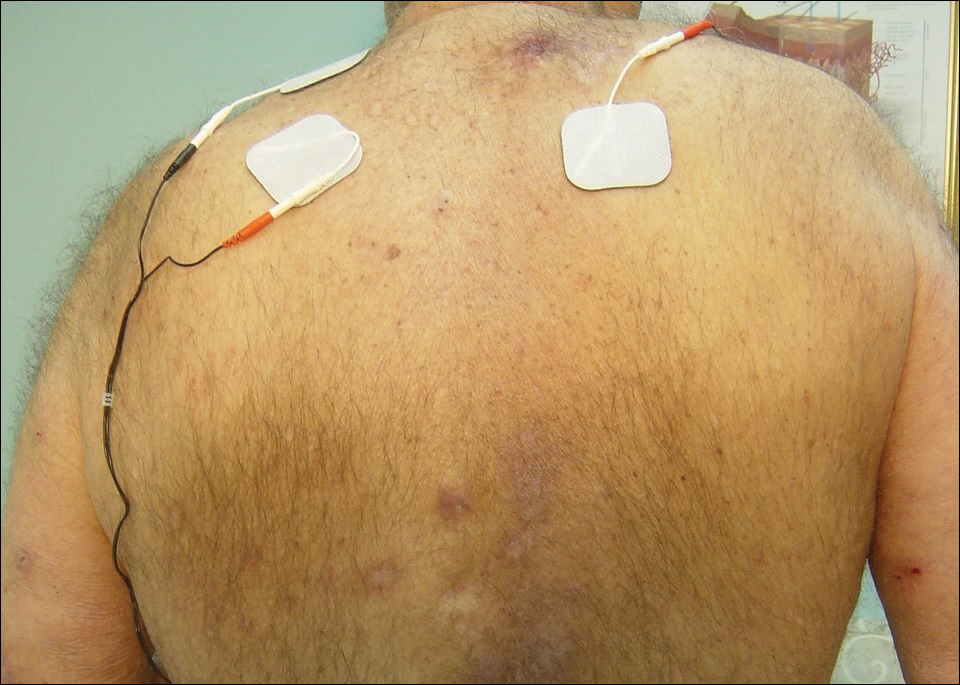
At the patient’s first visit at our center, for immediate symptom relief he underwent therapy with transcutaneous electronic nerve stimulation (TENS) with acupuncture of the cervicothoracic spine (Figure 3). He agreed to discontinue oral prednisone and begin chlorhexidine cleansing body wash, low-dose hydroxyzine 10-mg tablets up to 60 mg every 6 hours as required for pruritus, and mupirocin intranasal ointment. At 1-week follow-up, he reported at least 50% improvement in his symptoms with decreased pruritus, improved sleep, and enhanced quality of life. Within 2 weeks of initial assessment, there was a notable 70% clinical improvement of both the NP and BRP, with a notable decrease in cutaneous erosions and flattening of the pruritic skin nodules. He reported adequate control of symptoms with continued TENS for at-home use 3 times daily for 5- to 10-minute intervals.
Comment
NP Presentation
Notalgia paresthetica is a common, albeit heavily unrecognized and underdiagnosed, sensory neuropathic syndrome of the back, classically of the unilateral infrascapular region. Notalg
The dermatologic condition may consist of other symptoms that include but are not limited to localized burning, pain, tenderness, hyperalgesia, or dysesthesia. Notalgia paresthetica typically is associated with a poorly confined tan or hyperpigmented patch in the symptomatic area, though the skin may have no visible findings in many early cases. Notalgia paresthetica tends to be a chronic condition with periodic remissions and exacerbations. It is generally not associated with other comorbidities and is not life threatening; however, it does frequently decrease quality of life, causing much discomfort and annoyance to the affected patients.
Treatment
Topical therapies for NP have generally failed and are considered difficult because of the out-of-reach affected location. There is no uniformly effective treatment of NP.
Pathogenesis
The etiologies for NP and BRP are evolving and remain to be fully elucidated. Although the exact etiology remains uncertain, there are several possible mechanisms that have been proposed for NP: (1) neuropathy from degenerative cervicothoracic disc disease or direct nerve impingement,1 and (2) localized increased sensory innervations of the affected skin areas.2
Differential and Workup
The differential diagnosis in NP may include allergic or irritant contact dermatitis, fixed drug eruption, dermatophytosis, neoplasm, lichen amyloidosis, arthropod reaction, lichenified skin reactions including lichen simplex chronicus, neurodermatitis, infection, and other hypersensitivity reaction.
It is important during the initial assessment of patients with NP and/or BRP to obtain a thorough history of osteoarthritis, neck trauma, motor vehicle accident(s), vertebral fracture, cervical neoplasm or malignancy, family history of NP or BRP, or cervical disc disease. Radiographs or MRIs of the cervical spine may aid in diagnosis and treatment, and perhaps more so if there is an absence of contributory medical history. Radiographic imaging also may be indicated if there is a positive family history of osteoarthritis or vertebral disc disease.
A full laboratory workup including complete blood cell count, chemistry panel including renal and liver functions, and other laboratory tests (eg, IgE levels) may be warranted if pruritus is generalized and persistent to exclude other causes. Proper management of NP and BRP may involve a multispecialty effort of dermatology with radiology; orthopedic surgery; neurosurgery; neurology; and adjunctive fields including acupuncture, massage, chiropractic, and physical therapy.
NP and BRB Overlap
Because NP and BRP often originate from varying degrees of cervical disease, particularly at the C5-C6 level, traditional topical therapies aimed at treating the affected skin of the mid back and forearms may be ineffectual or partially effective as basic emollients. Due to NP and BRP’s periodic spontaneous remissions and exacerbations, it may be reasonably difficult to accurately measure direct response to various therapies. Some topical therapies aimed at the mid back skin or forearms may appear partially effective from a placebo-type perspective.
Currently, uniformly effective treatment of NP and BRP include the following: (1) therapies aimed at the cervical spine at C5-C6, including TENS, cervical massage, physical therapy, and acupuncture; and (2) therapies targeting the underlying lowered pruritus threshold such as oral antihistamines and narrowband UVB. Although uniformly effective treatments in this space had been previously lacking, traditional therapeutic options for NP and BRP included capsaicin cream, eutectic mixture of local anesthetic cream, topical steroids, pramoxine cream, topical cooling, oral steroids, menthol creams, various commercially available topical mixtures of menthol and methyl salicylate, cordran tape, intralesional corticosteroid injections, botulinum toxin injections,3 oral antihistamines, hydroxyzine, doxepin, topiramate, carbamazepine, antidepressants, gabapentin, oxcarbazepine, topiramate, thalidomide,4 paravertebral local anesthetic block, cervical epidural injection, surgical resection of the rib, and many others. Some of the tried systemic therapies exert their effect through the spinal nerves and central nervous system, thereby supporting the neuropathic etiology of NP.
Cervical Disc Disease
Alai et al1 reported a 37-year-old with documented NP on the right side of the back with MRI findings of disc disease at C5-C6 and mild nerve impingement that strongly suggest the association of cervical degenerative disc disease and NP.
Savk and Savk5 reported that 7 of 10 patients with NP demonstrated normal neurological examination and standard electrodiagnostic results. All had skin histopathology compatible with postinflammatory hyperpigmentation. There were no amyloid deposits or other described pathology on pathologic examination of the skin. Seven of 10 cases confirmed radiographic changes in the vertebra corresponding to the dermatome of the cutaneous lesion.5
An earlier study by Springall et al6 evaluating the mechanism of NP studied whether the cutaneous symptoms were caused by alternations on the cutaneous innervation of the involved infrascapular area. They p
Histologic studies have shown cutaneous changes in a few cases including lichen amyloidosis that may be secondary to the localized chronic scratching and rubbing.7 Clinical observations in orthopedics have established a clear relationship between the upper thoracic and interscapular region and the lower cervical spine. Frequently, cervical disc disease presents as referred pain in the upper thoracic and interscapular area. Similarly, some tumors of the cervical medulla also have presented as interscapular pain.8
Some have speculated direct involvement and actual entrapment of the posterior rami of T2-T6 spinal nerves.9 However, there are referred symptoms from the cervical area directly to the interscapular back. Degenerative vertebral and disc changes corresponding to the affected dermatome may be observed in some cases. Radiographic imaging of cervical and thoracic spine will help to exclude disc disease and possible nerve compromise.8
With advances in radiography and availability of MRI, earlier detection and intervention of cervical disc disease is possible. Early recognition may promote timely intervention and treatment to prevent cervical spine disease progression. In addition to degenerative cervical discs, osteoarthritis, and cervical spine strain and muscle spasm, there may be neoplasms or other pathology of the cervical spine contributing to NP and BRP.
There is some thought that there may be a relationship between NP and BRP. The described association of many cases of BRP and cervical spine disease6 and description of these diseases as likely neuropathic/neurogenic pruritic conditions also support a probable association of these two conditions. In contrast, NP has classically been described as unilateral in distribution, while BRP may involve unilateral or bilateral dorsolateral forearms. Most recently, as seen in our case, there are increasing incidences of nonclassic presentations of both of these diseases that may involve additional skin areas and be a basis for diagnostic challenges to the clinician.
First-line therapies for NP and BRP with associated cervical spinal disease are currently evolving and may include nondermatologic and noninvasive treatments such as spinal manipulation, physical therapy, acupuncture, cervical soft collars, massage, cervical traction, cervical muscle strengthening and increased range on motion, oral nonsteroidal anti-inflammatory medications (eg, ibuprofen, celecoxib, ketorolac), and oral muscle relaxants (eg, carisoprodol, cyclobenzaprine, methocarbamol, metaxalone). Curren
Conclusion
Notalgia paresthetica and BRP may not be solely skin diseases but rather cutaneous signs of an underlying cervical spine disease. The striking association of NP with BRP we present as well as the degenerative and/or traumatic cervicothoracic spine disease suggests that early spinal nerve impingement or cervical muscle spasm may contribute to the pathogenesis of these skin symptoms. Additional studies are needed to further assess the relationship of NP and BRP as well as the association of each disease entity independently with cervical spine disease, as it is unknown if these are causal or coincidental findings. Although topical therapies may seemingly help decrease the localized symptoms in NP and BRP in some cases, systemic or broader-scope cervical spinal evaluation may be warranted to fully evaluate refractory cases. Cervical spinal imaging and treatment, particularly at C5-C6 levels, may be appropriate as primary or first-line therapy in many cases of NP and BRP. The paradigm shift in thinking will more likely than not be to treat the cervical spine and the skin will follow.
- Alai NN, Skinner HB, Nabili S, et al. Notalgia paresthetica associated with cervical spinal stenosis and cervicothoracic disk disease at C4 through C7. Cutis. 2010;85:77-81.
- Savk E, Savk O, Bolukbasi O, et al. Notalgia paresthetica: a study on pathogenesis. Int J Dermatol. 2000;39:754-759.
- Tait CP, Grigg E, Quirk CJ. Brachioradial pruritus and cervical spine manipulation. Australas J Dermatol. 1998;39:168-170.
- Goodless DR, Eaglstein WH. Brachioradial pruritus treatment with topical capsaicin. J Am Acad Dermatol. 1993;29(5, pt 1):783-784.
- Savk O, Savk E. Investigation of spinal pathology in notalgia paresthetica. J Am Acad Dermatol. 2005;52:1085-1087.
- Springall DR, Karanth SS, Kirkham N, et al. Symptoms of notalgia paresthetica may be explained by increased dermal innervation. J Invest Dermatol. 1991;97:555-561.
- Weinfeld PK. Successful treatment of notalgia paresthetica with botulinum toxin type A. Arch Dermatol. 2007;143:980-982.
- Misery L. What is notalgia paresthetica? Dermatology. 2002;204:86-87.
- Pleet AB, Massey EW. Notalgia paresthetica. Neurology. 1978;28:1310-1312.
- Findlay C, Ayis S, Demetriades AK. Total disc replacement versus anterior cervical discectomy and fusion. Bone Joint J. 2018;100-B:991-1001.
Case Report
A 74-year-old man presented with persistent episodes of severe pruritus with exacerbations on the bilateral forearms, arms, and left side of the mid back of 4 years’ duration. He had a refractory and debilitating disease that had failed extensive therapies including topical antipruritics, antihistamines, oral hydroxyzine, capsaicin, potent topical steroids (ie, clobetasol, fluocinonide, triamcinolone), phototherapy with narrowband UVB, and various dietary modifications including a gluten-free trial. The patient reported he had exhausted all medical evaluation through care with more than 7 physicians and multiple dermatologists, including a university-based dermatology department for repeated consultations; he was seen by our dermatology center for an eighth opinion.
Initial dermatologic examination revealed multiple secondarily excoriated, hemorrhagic, hyperpigmented plaques and nodules on the right side of the mid upper back indicative of notalgia paresthetica (NP) with secondary chronic skin changes (Figure 1). Additional examination of the left arm and forearm revealed several open erosions, raised nodules, and lichenified skin plaques indicative of brachioradial pruritus (BP) with secondary skin changes (Figure 2). In addition, multiple lichenified plaques of the left side of the mid back were associated with decreased sensory alternations to light touch and pin prick. Of note, the localized pruritus pattern, particularly of the unilateral infrascapular back region, heralded the possibility of a neuropathic pruritus condition originating from the cervical spine. Examination confirmed decreased range of motion in the neck with associated marked palpable bilateral cervical muscle spasm and tenderness. Laboratory testing confirmed Staphylococcus aureus secondary skin infection that was treated empirically with chlorhexidine wash. General pruritus serology and imaging workup was ordered with contributory results. The patient’s medical history was notable for noninsulin-dependent diabetes mellitus, obesity, deep venous thrombosis, asthma, vein surgery, cardiovascular disease, atrial fibrillation, atopy, allergies, asthma, and keratosis pilaris, as well as drug intolerances of warfarin sodium, sitagliptin, and clopidogrel. His medications on presentation included glyburide, digoxin, prednisone, aspirin, cetirizine, cimetidine, and hydroxyzine. Based on the relatively classic localized pruritus symptoms and the anatomical distribution of skin findings, a clinical diagnosis of concurrent NP and BRP was made, and radiologic studies of the cervical spine were ordered.
Magnetic resonance imaging (MRI) of the cervical spine showed severe central canal stenosis at C3-C4 secondary to disc disease slight asymmetric toward the right side, severe central canal stenosis at C4-C5 slightly more prominent in the midline, severe central stenosis at C5-C6 more prominent in the midline, and mild changes at other levels as described. Laboratory workup revealed an abnormal complete blood cell count with mildly elevated white blood cell count (11,800/µL [reference range, 4000–10,500/µL]), elevated neutrophils (8600/µL [reference range, 1800–7800/µL]), elevated eosinophils (600/µL [reference range, 0–450/µL ]), and elevated IgE (160 IU/mL [reference range, 0–100 IU/mL]). Further testing revealed negative results for Helicobacter pylori IgG and IgM, human immunodeficiency virus, and hepatitis B and C screening panels; antinuclear antibody negative; normal thyroid-stimulating hormone; and normal thyroid peroxidase antibody. Chest radiograph and computed tomography of the chest, abdomen, and pelvis were negative.
We referred the patient for a neurosurgical consultation that uncovered newly diagnosed severe cervical stenosis with mild to moderate canal compromise at C3, C4, C5, and C6. His motor examination revealed full strength in the upper extremities (5/5). Sensory examination showed patchy sensory alteration on the mid back. He declined oral antibiotics as advised for the skin staphylococcal infection and neurosurgical treatment for the cervical disease.
During the 4 years prior to presentation at our center, the patient reported failure to improve with a dermatologically prescribed gluten-free diet as well as all topical and oral steroid treatments. He was presented at a university grand rounds where a suggestion for UVB light treatment was made; the patient reported possible worsening of symptoms with narrowband UVB phototherapy.
At the patient’s first visit at our center, for immediate symptom relief he underwent therapy with transcutaneous electronic nerve stimulation (TENS) with acupuncture of the cervicothoracic spine (Figure 3). He agreed to discontinue oral prednisone and begin chlorhexidine cleansing body wash, low-dose hydroxyzine 10-mg tablets up to 60 mg every 6 hours as required for pruritus, and mupirocin intranasal ointment. At 1-week follow-up, he reported at least 50% improvement in his symptoms with decreased pruritus, improved sleep, and enhanced quality of life. Within 2 weeks of initial assessment, there was a notable 70% clinical improvement of both the NP and BRP, with a notable decrease in cutaneous erosions and flattening of the pruritic skin nodules. He reported adequate control of symptoms with continued TENS for at-home use 3 times daily for 5- to 10-minute intervals.
Comment
NP Presentation
Notalgia paresthetica is a common, albeit heavily unrecognized and underdiagnosed, sensory neuropathic syndrome of the back, classically of the unilateral infrascapular region. Notalg
The dermatologic condition may consist of other symptoms that include but are not limited to localized burning, pain, tenderness, hyperalgesia, or dysesthesia. Notalgia paresthetica typically is associated with a poorly confined tan or hyperpigmented patch in the symptomatic area, though the skin may have no visible findings in many early cases. Notalgia paresthetica tends to be a chronic condition with periodic remissions and exacerbations. It is generally not associated with other comorbidities and is not life threatening; however, it does frequently decrease quality of life, causing much discomfort and annoyance to the affected patients.
Treatment
Topical therapies for NP have generally failed and are considered difficult because of the out-of-reach affected location. There is no uniformly effective treatment of NP.
Pathogenesis
The etiologies for NP and BRP are evolving and remain to be fully elucidated. Although the exact etiology remains uncertain, there are several possible mechanisms that have been proposed for NP: (1) neuropathy from degenerative cervicothoracic disc disease or direct nerve impingement,1 and (2) localized increased sensory innervations of the affected skin areas.2
Differential and Workup
The differential diagnosis in NP may include allergic or irritant contact dermatitis, fixed drug eruption, dermatophytosis, neoplasm, lichen amyloidosis, arthropod reaction, lichenified skin reactions including lichen simplex chronicus, neurodermatitis, infection, and other hypersensitivity reaction.
It is important during the initial assessment of patients with NP and/or BRP to obtain a thorough history of osteoarthritis, neck trauma, motor vehicle accident(s), vertebral fracture, cervical neoplasm or malignancy, family history of NP or BRP, or cervical disc disease. Radiographs or MRIs of the cervical spine may aid in diagnosis and treatment, and perhaps more so if there is an absence of contributory medical history. Radiographic imaging also may be indicated if there is a positive family history of osteoarthritis or vertebral disc disease.
A full laboratory workup including complete blood cell count, chemistry panel including renal and liver functions, and other laboratory tests (eg, IgE levels) may be warranted if pruritus is generalized and persistent to exclude other causes. Proper management of NP and BRP may involve a multispecialty effort of dermatology with radiology; orthopedic surgery; neurosurgery; neurology; and adjunctive fields including acupuncture, massage, chiropractic, and physical therapy.
NP and BRB Overlap
Because NP and BRP often originate from varying degrees of cervical disease, particularly at the C5-C6 level, traditional topical therapies aimed at treating the affected skin of the mid back and forearms may be ineffectual or partially effective as basic emollients. Due to NP and BRP’s periodic spontaneous remissions and exacerbations, it may be reasonably difficult to accurately measure direct response to various therapies. Some topical therapies aimed at the mid back skin or forearms may appear partially effective from a placebo-type perspective.
Currently, uniformly effective treatment of NP and BRP include the following: (1) therapies aimed at the cervical spine at C5-C6, including TENS, cervical massage, physical therapy, and acupuncture; and (2) therapies targeting the underlying lowered pruritus threshold such as oral antihistamines and narrowband UVB. Although uniformly effective treatments in this space had been previously lacking, traditional therapeutic options for NP and BRP included capsaicin cream, eutectic mixture of local anesthetic cream, topical steroids, pramoxine cream, topical cooling, oral steroids, menthol creams, various commercially available topical mixtures of menthol and methyl salicylate, cordran tape, intralesional corticosteroid injections, botulinum toxin injections,3 oral antihistamines, hydroxyzine, doxepin, topiramate, carbamazepine, antidepressants, gabapentin, oxcarbazepine, topiramate, thalidomide,4 paravertebral local anesthetic block, cervical epidural injection, surgical resection of the rib, and many others. Some of the tried systemic therapies exert their effect through the spinal nerves and central nervous system, thereby supporting the neuropathic etiology of NP.
Cervical Disc Disease
Alai et al1 reported a 37-year-old with documented NP on the right side of the back with MRI findings of disc disease at C5-C6 and mild nerve impingement that strongly suggest the association of cervical degenerative disc disease and NP.
Savk and Savk5 reported that 7 of 10 patients with NP demonstrated normal neurological examination and standard electrodiagnostic results. All had skin histopathology compatible with postinflammatory hyperpigmentation. There were no amyloid deposits or other described pathology on pathologic examination of the skin. Seven of 10 cases confirmed radiographic changes in the vertebra corresponding to the dermatome of the cutaneous lesion.5
An earlier study by Springall et al6 evaluating the mechanism of NP studied whether the cutaneous symptoms were caused by alternations on the cutaneous innervation of the involved infrascapular area. They p
Histologic studies have shown cutaneous changes in a few cases including lichen amyloidosis that may be secondary to the localized chronic scratching and rubbing.7 Clinical observations in orthopedics have established a clear relationship between the upper thoracic and interscapular region and the lower cervical spine. Frequently, cervical disc disease presents as referred pain in the upper thoracic and interscapular area. Similarly, some tumors of the cervical medulla also have presented as interscapular pain.8
Some have speculated direct involvement and actual entrapment of the posterior rami of T2-T6 spinal nerves.9 However, there are referred symptoms from the cervical area directly to the interscapular back. Degenerative vertebral and disc changes corresponding to the affected dermatome may be observed in some cases. Radiographic imaging of cervical and thoracic spine will help to exclude disc disease and possible nerve compromise.8
With advances in radiography and availability of MRI, earlier detection and intervention of cervical disc disease is possible. Early recognition may promote timely intervention and treatment to prevent cervical spine disease progression. In addition to degenerative cervical discs, osteoarthritis, and cervical spine strain and muscle spasm, there may be neoplasms or other pathology of the cervical spine contributing to NP and BRP.
There is some thought that there may be a relationship between NP and BRP. The described association of many cases of BRP and cervical spine disease6 and description of these diseases as likely neuropathic/neurogenic pruritic conditions also support a probable association of these two conditions. In contrast, NP has classically been described as unilateral in distribution, while BRP may involve unilateral or bilateral dorsolateral forearms. Most recently, as seen in our case, there are increasing incidences of nonclassic presentations of both of these diseases that may involve additional skin areas and be a basis for diagnostic challenges to the clinician.
First-line therapies for NP and BRP with associated cervical spinal disease are currently evolving and may include nondermatologic and noninvasive treatments such as spinal manipulation, physical therapy, acupuncture, cervical soft collars, massage, cervical traction, cervical muscle strengthening and increased range on motion, oral nonsteroidal anti-inflammatory medications (eg, ibuprofen, celecoxib, ketorolac), and oral muscle relaxants (eg, carisoprodol, cyclobenzaprine, methocarbamol, metaxalone). Curren
Conclusion
Notalgia paresthetica and BRP may not be solely skin diseases but rather cutaneous signs of an underlying cervical spine disease. The striking association of NP with BRP we present as well as the degenerative and/or traumatic cervicothoracic spine disease suggests that early spinal nerve impingement or cervical muscle spasm may contribute to the pathogenesis of these skin symptoms. Additional studies are needed to further assess the relationship of NP and BRP as well as the association of each disease entity independently with cervical spine disease, as it is unknown if these are causal or coincidental findings. Although topical therapies may seemingly help decrease the localized symptoms in NP and BRP in some cases, systemic or broader-scope cervical spinal evaluation may be warranted to fully evaluate refractory cases. Cervical spinal imaging and treatment, particularly at C5-C6 levels, may be appropriate as primary or first-line therapy in many cases of NP and BRP. The paradigm shift in thinking will more likely than not be to treat the cervical spine and the skin will follow.
Case Report
A 74-year-old man presented with persistent episodes of severe pruritus with exacerbations on the bilateral forearms, arms, and left side of the mid back of 4 years’ duration. He had a refractory and debilitating disease that had failed extensive therapies including topical antipruritics, antihistamines, oral hydroxyzine, capsaicin, potent topical steroids (ie, clobetasol, fluocinonide, triamcinolone), phototherapy with narrowband UVB, and various dietary modifications including a gluten-free trial. The patient reported he had exhausted all medical evaluation through care with more than 7 physicians and multiple dermatologists, including a university-based dermatology department for repeated consultations; he was seen by our dermatology center for an eighth opinion.
Initial dermatologic examination revealed multiple secondarily excoriated, hemorrhagic, hyperpigmented plaques and nodules on the right side of the mid upper back indicative of notalgia paresthetica (NP) with secondary chronic skin changes (Figure 1). Additional examination of the left arm and forearm revealed several open erosions, raised nodules, and lichenified skin plaques indicative of brachioradial pruritus (BP) with secondary skin changes (Figure 2). In addition, multiple lichenified plaques of the left side of the mid back were associated with decreased sensory alternations to light touch and pin prick. Of note, the localized pruritus pattern, particularly of the unilateral infrascapular back region, heralded the possibility of a neuropathic pruritus condition originating from the cervical spine. Examination confirmed decreased range of motion in the neck with associated marked palpable bilateral cervical muscle spasm and tenderness. Laboratory testing confirmed Staphylococcus aureus secondary skin infection that was treated empirically with chlorhexidine wash. General pruritus serology and imaging workup was ordered with contributory results. The patient’s medical history was notable for noninsulin-dependent diabetes mellitus, obesity, deep venous thrombosis, asthma, vein surgery, cardiovascular disease, atrial fibrillation, atopy, allergies, asthma, and keratosis pilaris, as well as drug intolerances of warfarin sodium, sitagliptin, and clopidogrel. His medications on presentation included glyburide, digoxin, prednisone, aspirin, cetirizine, cimetidine, and hydroxyzine. Based on the relatively classic localized pruritus symptoms and the anatomical distribution of skin findings, a clinical diagnosis of concurrent NP and BRP was made, and radiologic studies of the cervical spine were ordered.
Magnetic resonance imaging (MRI) of the cervical spine showed severe central canal stenosis at C3-C4 secondary to disc disease slight asymmetric toward the right side, severe central canal stenosis at C4-C5 slightly more prominent in the midline, severe central stenosis at C5-C6 more prominent in the midline, and mild changes at other levels as described. Laboratory workup revealed an abnormal complete blood cell count with mildly elevated white blood cell count (11,800/µL [reference range, 4000–10,500/µL]), elevated neutrophils (8600/µL [reference range, 1800–7800/µL]), elevated eosinophils (600/µL [reference range, 0–450/µL ]), and elevated IgE (160 IU/mL [reference range, 0–100 IU/mL]). Further testing revealed negative results for Helicobacter pylori IgG and IgM, human immunodeficiency virus, and hepatitis B and C screening panels; antinuclear antibody negative; normal thyroid-stimulating hormone; and normal thyroid peroxidase antibody. Chest radiograph and computed tomography of the chest, abdomen, and pelvis were negative.
We referred the patient for a neurosurgical consultation that uncovered newly diagnosed severe cervical stenosis with mild to moderate canal compromise at C3, C4, C5, and C6. His motor examination revealed full strength in the upper extremities (5/5). Sensory examination showed patchy sensory alteration on the mid back. He declined oral antibiotics as advised for the skin staphylococcal infection and neurosurgical treatment for the cervical disease.
During the 4 years prior to presentation at our center, the patient reported failure to improve with a dermatologically prescribed gluten-free diet as well as all topical and oral steroid treatments. He was presented at a university grand rounds where a suggestion for UVB light treatment was made; the patient reported possible worsening of symptoms with narrowband UVB phototherapy.
At the patient’s first visit at our center, for immediate symptom relief he underwent therapy with transcutaneous electronic nerve stimulation (TENS) with acupuncture of the cervicothoracic spine (Figure 3). He agreed to discontinue oral prednisone and begin chlorhexidine cleansing body wash, low-dose hydroxyzine 10-mg tablets up to 60 mg every 6 hours as required for pruritus, and mupirocin intranasal ointment. At 1-week follow-up, he reported at least 50% improvement in his symptoms with decreased pruritus, improved sleep, and enhanced quality of life. Within 2 weeks of initial assessment, there was a notable 70% clinical improvement of both the NP and BRP, with a notable decrease in cutaneous erosions and flattening of the pruritic skin nodules. He reported adequate control of symptoms with continued TENS for at-home use 3 times daily for 5- to 10-minute intervals.
Comment
NP Presentation
Notalgia paresthetica is a common, albeit heavily unrecognized and underdiagnosed, sensory neuropathic syndrome of the back, classically of the unilateral infrascapular region. Notalg
The dermatologic condition may consist of other symptoms that include but are not limited to localized burning, pain, tenderness, hyperalgesia, or dysesthesia. Notalgia paresthetica typically is associated with a poorly confined tan or hyperpigmented patch in the symptomatic area, though the skin may have no visible findings in many early cases. Notalgia paresthetica tends to be a chronic condition with periodic remissions and exacerbations. It is generally not associated with other comorbidities and is not life threatening; however, it does frequently decrease quality of life, causing much discomfort and annoyance to the affected patients.
Treatment
Topical therapies for NP have generally failed and are considered difficult because of the out-of-reach affected location. There is no uniformly effective treatment of NP.
Pathogenesis
The etiologies for NP and BRP are evolving and remain to be fully elucidated. Although the exact etiology remains uncertain, there are several possible mechanisms that have been proposed for NP: (1) neuropathy from degenerative cervicothoracic disc disease or direct nerve impingement,1 and (2) localized increased sensory innervations of the affected skin areas.2
Differential and Workup
The differential diagnosis in NP may include allergic or irritant contact dermatitis, fixed drug eruption, dermatophytosis, neoplasm, lichen amyloidosis, arthropod reaction, lichenified skin reactions including lichen simplex chronicus, neurodermatitis, infection, and other hypersensitivity reaction.
It is important during the initial assessment of patients with NP and/or BRP to obtain a thorough history of osteoarthritis, neck trauma, motor vehicle accident(s), vertebral fracture, cervical neoplasm or malignancy, family history of NP or BRP, or cervical disc disease. Radiographs or MRIs of the cervical spine may aid in diagnosis and treatment, and perhaps more so if there is an absence of contributory medical history. Radiographic imaging also may be indicated if there is a positive family history of osteoarthritis or vertebral disc disease.
A full laboratory workup including complete blood cell count, chemistry panel including renal and liver functions, and other laboratory tests (eg, IgE levels) may be warranted if pruritus is generalized and persistent to exclude other causes. Proper management of NP and BRP may involve a multispecialty effort of dermatology with radiology; orthopedic surgery; neurosurgery; neurology; and adjunctive fields including acupuncture, massage, chiropractic, and physical therapy.
NP and BRB Overlap
Because NP and BRP often originate from varying degrees of cervical disease, particularly at the C5-C6 level, traditional topical therapies aimed at treating the affected skin of the mid back and forearms may be ineffectual or partially effective as basic emollients. Due to NP and BRP’s periodic spontaneous remissions and exacerbations, it may be reasonably difficult to accurately measure direct response to various therapies. Some topical therapies aimed at the mid back skin or forearms may appear partially effective from a placebo-type perspective.
Currently, uniformly effective treatment of NP and BRP include the following: (1) therapies aimed at the cervical spine at C5-C6, including TENS, cervical massage, physical therapy, and acupuncture; and (2) therapies targeting the underlying lowered pruritus threshold such as oral antihistamines and narrowband UVB. Although uniformly effective treatments in this space had been previously lacking, traditional therapeutic options for NP and BRP included capsaicin cream, eutectic mixture of local anesthetic cream, topical steroids, pramoxine cream, topical cooling, oral steroids, menthol creams, various commercially available topical mixtures of menthol and methyl salicylate, cordran tape, intralesional corticosteroid injections, botulinum toxin injections,3 oral antihistamines, hydroxyzine, doxepin, topiramate, carbamazepine, antidepressants, gabapentin, oxcarbazepine, topiramate, thalidomide,4 paravertebral local anesthetic block, cervical epidural injection, surgical resection of the rib, and many others. Some of the tried systemic therapies exert their effect through the spinal nerves and central nervous system, thereby supporting the neuropathic etiology of NP.
Cervical Disc Disease
Alai et al1 reported a 37-year-old with documented NP on the right side of the back with MRI findings of disc disease at C5-C6 and mild nerve impingement that strongly suggest the association of cervical degenerative disc disease and NP.
Savk and Savk5 reported that 7 of 10 patients with NP demonstrated normal neurological examination and standard electrodiagnostic results. All had skin histopathology compatible with postinflammatory hyperpigmentation. There were no amyloid deposits or other described pathology on pathologic examination of the skin. Seven of 10 cases confirmed radiographic changes in the vertebra corresponding to the dermatome of the cutaneous lesion.5
An earlier study by Springall et al6 evaluating the mechanism of NP studied whether the cutaneous symptoms were caused by alternations on the cutaneous innervation of the involved infrascapular area. They p
Histologic studies have shown cutaneous changes in a few cases including lichen amyloidosis that may be secondary to the localized chronic scratching and rubbing.7 Clinical observations in orthopedics have established a clear relationship between the upper thoracic and interscapular region and the lower cervical spine. Frequently, cervical disc disease presents as referred pain in the upper thoracic and interscapular area. Similarly, some tumors of the cervical medulla also have presented as interscapular pain.8
Some have speculated direct involvement and actual entrapment of the posterior rami of T2-T6 spinal nerves.9 However, there are referred symptoms from the cervical area directly to the interscapular back. Degenerative vertebral and disc changes corresponding to the affected dermatome may be observed in some cases. Radiographic imaging of cervical and thoracic spine will help to exclude disc disease and possible nerve compromise.8
With advances in radiography and availability of MRI, earlier detection and intervention of cervical disc disease is possible. Early recognition may promote timely intervention and treatment to prevent cervical spine disease progression. In addition to degenerative cervical discs, osteoarthritis, and cervical spine strain and muscle spasm, there may be neoplasms or other pathology of the cervical spine contributing to NP and BRP.
There is some thought that there may be a relationship between NP and BRP. The described association of many cases of BRP and cervical spine disease6 and description of these diseases as likely neuropathic/neurogenic pruritic conditions also support a probable association of these two conditions. In contrast, NP has classically been described as unilateral in distribution, while BRP may involve unilateral or bilateral dorsolateral forearms. Most recently, as seen in our case, there are increasing incidences of nonclassic presentations of both of these diseases that may involve additional skin areas and be a basis for diagnostic challenges to the clinician.
First-line therapies for NP and BRP with associated cervical spinal disease are currently evolving and may include nondermatologic and noninvasive treatments such as spinal manipulation, physical therapy, acupuncture, cervical soft collars, massage, cervical traction, cervical muscle strengthening and increased range on motion, oral nonsteroidal anti-inflammatory medications (eg, ibuprofen, celecoxib, ketorolac), and oral muscle relaxants (eg, carisoprodol, cyclobenzaprine, methocarbamol, metaxalone). Curren
Conclusion
Notalgia paresthetica and BRP may not be solely skin diseases but rather cutaneous signs of an underlying cervical spine disease. The striking association of NP with BRP we present as well as the degenerative and/or traumatic cervicothoracic spine disease suggests that early spinal nerve impingement or cervical muscle spasm may contribute to the pathogenesis of these skin symptoms. Additional studies are needed to further assess the relationship of NP and BRP as well as the association of each disease entity independently with cervical spine disease, as it is unknown if these are causal or coincidental findings. Although topical therapies may seemingly help decrease the localized symptoms in NP and BRP in some cases, systemic or broader-scope cervical spinal evaluation may be warranted to fully evaluate refractory cases. Cervical spinal imaging and treatment, particularly at C5-C6 levels, may be appropriate as primary or first-line therapy in many cases of NP and BRP. The paradigm shift in thinking will more likely than not be to treat the cervical spine and the skin will follow.
- Alai NN, Skinner HB, Nabili S, et al. Notalgia paresthetica associated with cervical spinal stenosis and cervicothoracic disk disease at C4 through C7. Cutis. 2010;85:77-81.
- Savk E, Savk O, Bolukbasi O, et al. Notalgia paresthetica: a study on pathogenesis. Int J Dermatol. 2000;39:754-759.
- Tait CP, Grigg E, Quirk CJ. Brachioradial pruritus and cervical spine manipulation. Australas J Dermatol. 1998;39:168-170.
- Goodless DR, Eaglstein WH. Brachioradial pruritus treatment with topical capsaicin. J Am Acad Dermatol. 1993;29(5, pt 1):783-784.
- Savk O, Savk E. Investigation of spinal pathology in notalgia paresthetica. J Am Acad Dermatol. 2005;52:1085-1087.
- Springall DR, Karanth SS, Kirkham N, et al. Symptoms of notalgia paresthetica may be explained by increased dermal innervation. J Invest Dermatol. 1991;97:555-561.
- Weinfeld PK. Successful treatment of notalgia paresthetica with botulinum toxin type A. Arch Dermatol. 2007;143:980-982.
- Misery L. What is notalgia paresthetica? Dermatology. 2002;204:86-87.
- Pleet AB, Massey EW. Notalgia paresthetica. Neurology. 1978;28:1310-1312.
- Findlay C, Ayis S, Demetriades AK. Total disc replacement versus anterior cervical discectomy and fusion. Bone Joint J. 2018;100-B:991-1001.
- Alai NN, Skinner HB, Nabili S, et al. Notalgia paresthetica associated with cervical spinal stenosis and cervicothoracic disk disease at C4 through C7. Cutis. 2010;85:77-81.
- Savk E, Savk O, Bolukbasi O, et al. Notalgia paresthetica: a study on pathogenesis. Int J Dermatol. 2000;39:754-759.
- Tait CP, Grigg E, Quirk CJ. Brachioradial pruritus and cervical spine manipulation. Australas J Dermatol. 1998;39:168-170.
- Goodless DR, Eaglstein WH. Brachioradial pruritus treatment with topical capsaicin. J Am Acad Dermatol. 1993;29(5, pt 1):783-784.
- Savk O, Savk E. Investigation of spinal pathology in notalgia paresthetica. J Am Acad Dermatol. 2005;52:1085-1087.
- Springall DR, Karanth SS, Kirkham N, et al. Symptoms of notalgia paresthetica may be explained by increased dermal innervation. J Invest Dermatol. 1991;97:555-561.
- Weinfeld PK. Successful treatment of notalgia paresthetica with botulinum toxin type A. Arch Dermatol. 2007;143:980-982.
- Misery L. What is notalgia paresthetica? Dermatology. 2002;204:86-87.
- Pleet AB, Massey EW. Notalgia paresthetica. Neurology. 1978;28:1310-1312.
- Findlay C, Ayis S, Demetriades AK. Total disc replacement versus anterior cervical discectomy and fusion. Bone Joint J. 2018;100-B:991-1001.
Practice Points
- Assess the spine in patients with notalgia paresthetica (NP) and brachioradial pruritus (BRP). Cervical spinal disease, especially at the C5-C6 level, may be the root cause of BRP and/or NP.
- Consider a multimodality approach to treatment, including physical therapy, acupuncture, massage, and transcutaneous electronic nerve stimulation.
- Treat the neck (underlying cause) and expect the skin (cutaneous symptoms) to follow.
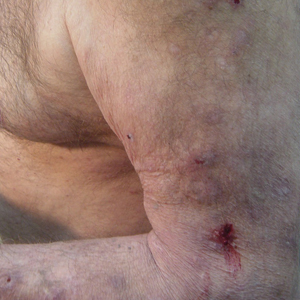
Angioimmunoblastic T-Cell Lymphoma Mimicking Diffuse Large B-Cell Lymphoma
Angioimmunoblastic T-cell lymphoma (AITL) is a rare, often aggressive type of peripheral T-cell lymphoma. It comprises 18% of peripheral T-cell lymphomas and 1% to 2% of all non-Hodgkin lymphomas.1 The incidence of AITL in the United States is estimated to be 0.05 cases per 100,000 person-years,2 and there is a slight male predominance.1,3,4 It typically presents in the seventh decade of life; however, cases have been reported in adults ranging from 20 to 91 years of age.3
Angioimmunoblastic T-cell lymphoma presents with lymphadenopathy, hepatosplenomegaly, and systemic B symptoms (eg, fever, night sweats, weight loss, generalized pruritus).4-6 There are cutaneous manifestations in up to 50% of cases4,5,7 and frequently signs of autoimmune disorder.4,5 The diagnosis often is made by excisional lymph node biopsy. Lymph node specimens characteristically have a mixed inflammatory infiltrate that includes numerous B cells often infected with Epstein-Barr virus (EBV) and a relatively small population of atypical T lymphocytes.8 Identification of this neoplastic population of CD4+CD8− T lymphocytes expressing normal follicular helper T-cell markers CD10, chemokine CXCL13, programmed cell death protein 1 (PD-1), and B-cell lymphoma 6 (BCL-6) confirms the diagnosis of AITL.9,10 These malignant cells can be identified in skin specimens in cases of cutaneous metastatic disease.11,12 We present a case originally misdiagnosed as diffuse large B-cell lymphoma that was later identified as AITL on skin biopsy.
Case Report
A 72-year-old woman presented with a pruritic erythematous eruption around the neck of 3 weeks’ duration (Figure 1). Her medical history was notable for diffuse large B-cell non-Hodgkin lymphoma diagnosed 3 months prior based on results from a right cervical lymph node biopsy. She was treated with bendamustine and rituximab. On physical examination there were erythematous edematous papules coalescing into indurated plaques around the neck. The differential diagnosis included drug hypersensitivity reaction, herpes zoster, urticaria, and cutaneous metastasis. Two punch biopsies were taken for hematoxylin and eosin and tissue culture.
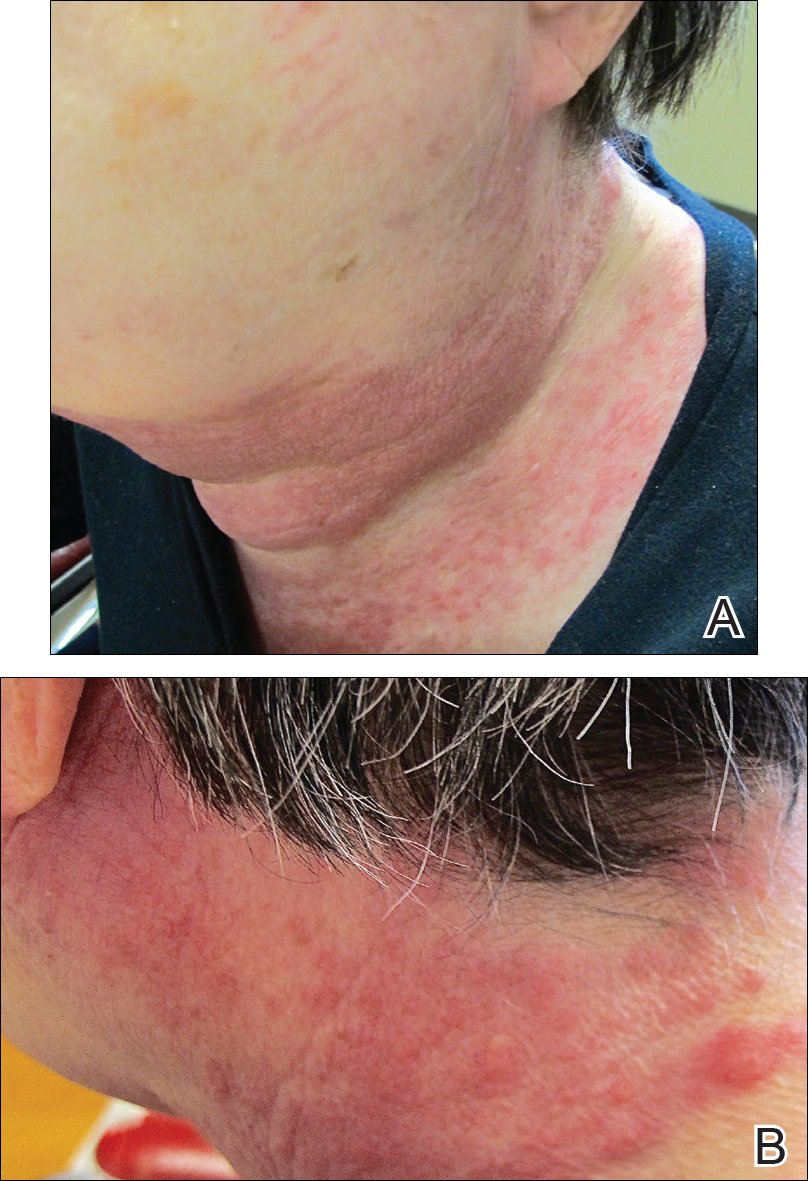
Tissue cultures and viral polymerase chain reaction were negative. Histopathologic examination revealed a scant atypical lymphoid infiltrate focally involving the deep dermis. The cells were medium to large in size and contained hyperchromatic pleomorphic nuclei (Figure 2). They were positive for CD3 and CD4, which was concerning for T-cell lymphoma. The histologic report of the excisional lymph node biopsy done 3 months prior described an atypical lymphoid neoplasm with extensive necrosis and extranodal spread that stained positively for CD20 (Figure 3).
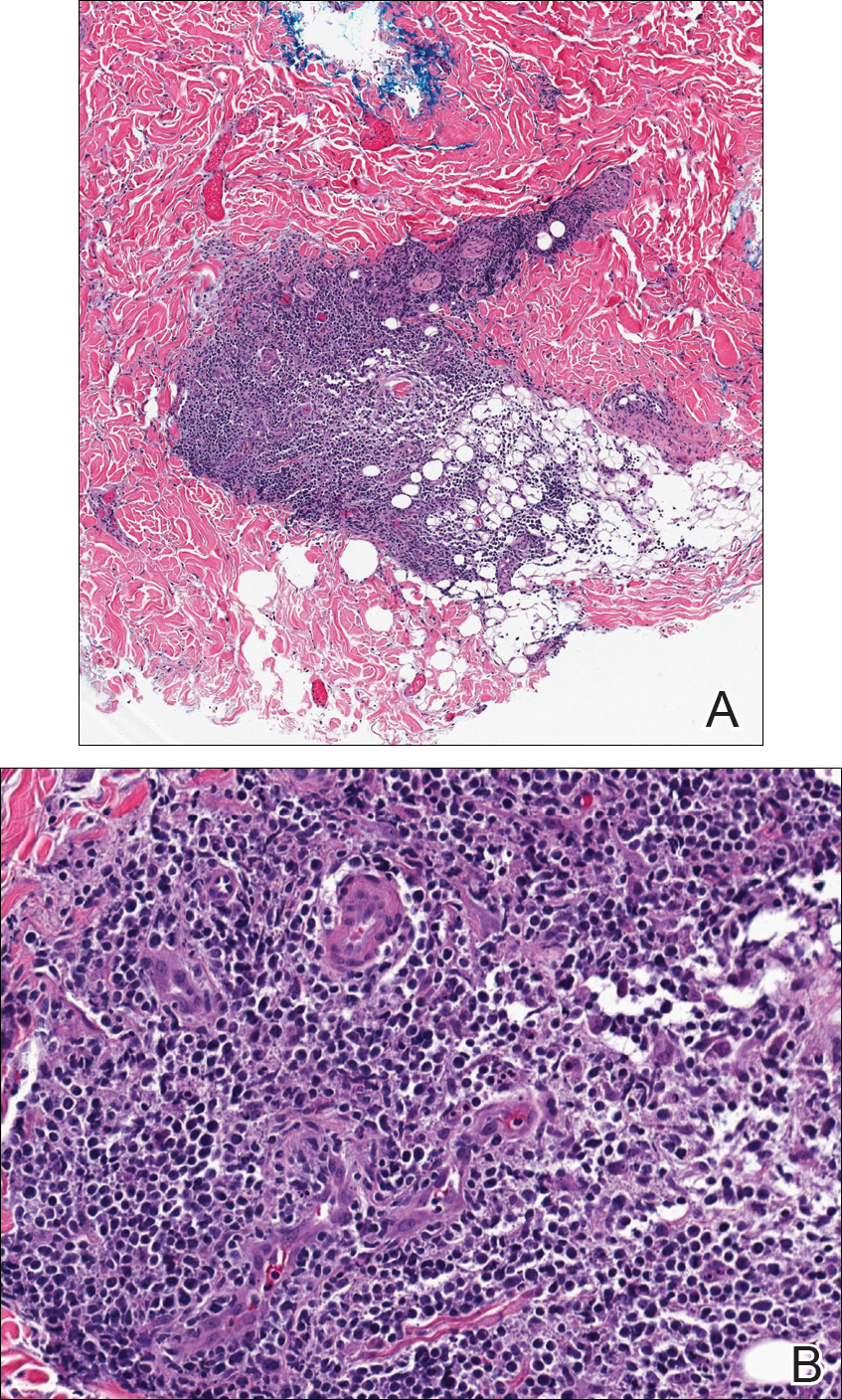
Further staining of this cervical lymph node specimen revealed large atypical lymphoid cells positive for CD3, CD10, B-cell lymphoma 2 (BCL-2), BCL-6, and PD-1. There were intermixed mature B lymphocytes positive for CD20 and BCL-2. Chromogenic in situ hybridization with probes for EBV showed numerous positive cells throughout the infiltrate. Polymerase chain reaction demonstrated a T-cell population with clonally rearranged T-cell receptor genes. Primers for immunoglobulin heavy and light chains showed no evidence of a clonal B-cell population.
Additional staining of the atypical cutaneous lymphocytes revealed positivity for CD3, CD10, and PD-1. The morphologic and immunophenotypic findings of both specimens supported the diagnosis of AITL.
The patient declined further treatment and chose hospice care.
Comment
Etiology
Angioimmunoblastic T-cell lymphoma was originally named angioimmunoblastic lymphadenopathy with dysproteinemia. It was initially thought to be a benign hyperreactive immune process driven by B cells, and patients often died of infectious complications not long after the diagnosis was made.13 As more cases were reported with clonal rearrangements and signs of progressive lymphoma, AITL was recognized as a malignancy.
Presentation
Patients with AITL often present with advanced stage III or IV disease with extranodal and bone marrow involvement.3-6 Cutaneous disease occurs in up to half of patients and portends a poor prognosis.7 The rash often is a nonspecific erythematous macular and papular eruption mimicking a morbilliform viral exanthem or drug reaction. Urticarial, nodular, petechial, purpuric, eczematous, erythrodermic, and vesiculobullous presentations have been described.4,11,12 In up to one-third of cases, the eruption occurs in association with a new medication, often leading to an initial misdiagnosis of drug hypersensitivity reaction.4,11 In a review conducted by Balaraman et al,14 84% of patients with AITL reported having pruritus.
There is an association of autoimmune phenomena in patients with AITL, which is likely a result of immune dysregulation associated with poorly functioning follicular helper T cells. Patients may present with arthralgia, hemolytic anemia, or thrombocytopenic purpura. Hypergammaglobulinemia has been reported in 30% to 50% of AITL patients.4,6 Other pertinent immunologic findings include positive Coombs test, cold agglutinins, cryoglobulinemia, hypocomplementemia, and positive antinuclear antibodies.4-7
Gene Analysis
Affected lymph nodes have a characteristically effaced architecture with proliferative arborizing venules; a hyperplastic population of follicular dendritic cells; and a mixed inflammatory infiltrate that is comprised of atypical lymphocytes and variable numbers of reactive lymphocytes, histiocytes, eosinophils, and plasma cells. The malignant T lymphocytes often account for only a small portion of the infiltrate.8 T-cell gene rearrangement studies identify clonal cells with β and γ rearrangements in the majority of cases.4 These cells are predominantly CD4+CD8− and express normal follicular helper T-cell markers CD10, CXCL13, BCL-6,5,9 and PD-1.10 Numerous B cells are seen intermixed with follicular dendritic cells. They are frequently infected with EBV and can have an atypical Reed-Sternberg cell–like appearance.4,5,15 In the evaluation of AITL, polymerase chain reaction studies with primers for immunoglobulin heavy and light chain should be performed to look for clonal B-cell populations and rule out a possible secondary B-cell lymphoma.
Histology
Five histologic patterns have been described with cutaneous AITL: (1) superficial perivascular infiltrate of eosinophils and lymphocytes that lack atypia, (2) sparse perivascular infiltrate with atypical lymphocytes, (3) dense dermal infiltrate of pleomorphic lymphocytes, (4) leukocytoclastic vasculitis without atypical lymphocytes,11 and (5) necrotizing vasculitis.12 The finding of vascular hyperplasia, perivascular infiltrate, or vasculitis has been reported in 91% of cases in the literature. Although these findings are nonspecific, an analysis of cutaneous cases reported in the literature found that 87% demonstrated T-cell receptor gene rearrangements.14 Lymphoid cells are positive for CD10 and PD-1, as was demonstrated in our case, and are CXCL13 positive in the majority of cases.12 Atypical and EBV-infected B cells also can be found in the skin.11,12
Differential Diagnosis
Angioimmunoblastic T-cell lymphoma can mimic infectious, autoimmune, or allergic etiologies, and misdiagnosis of another type of lymphoma is not uncommon, as occurred in our case. Patients who have a delay in the correct diagnosis have similar outcomes to those correctly diagnosed at first presentation.4
Treatment
There are no effective therapies for AITL. Poor prognostic factors include age (>60 years), stages III to IV disease, male gender, elevated serum lactate dehydrogenase level,3,5,10 and cutaneous involvement.7 Corticosteroids, anthracycline-based chemotherapy, and autologous stem cell transplant are currently the mainstays of therapy. Initial response to chemotherapy is promising, but duration of response is poor overall and there is no increased survival.5,15 A large population-based study of 1207 cases by Xu and Liu3 showed the overall survival rate at 2 and 10 years was 46.8% and 21.9%, respectively. Ten-year disease-specific survival was 35.9%, and there was no demonstrable improvement in survival over the last 2 decades.3 Case reports have demonstrated that thalidomide,16 lenalidomide,17 and cyclosporine plus dexamethasone18 have been successfully used to achieve remission for up to 3 years.
Conclusion
Angioimmunoblastic T-cell lymphoma is difficult to diagnose due to nonspecific clinical and histologic findings. Cutaneous manifestations are seen in AITL in up to half of cases that may occur early or in advanced disease. Similar to all cutaneous metastases, the appearance of the lesions can greatly vary. Our case demonstrates that dermatologists and dermatopathologists can make this diagnosis in the appropriate clinicopathologic context utilizing appropriate immunohistochemical staining and gene rearrangement studies.
- Rudiger T, Weisenburger DD, Anderson JR, et al. Peripheral T-cell lymphoma (excluding anaplastic large-cell lymphoma): results from the Non-Hodgkins Lymphoma Classification Project. Ann Oncol. 2002;13:140-149.
- Morton LM, Wang SS, Devesa SS, et al. Lymphoma incidence patterns by WHO subtype in the United States, 1992-2001. Blood. 2006;107:265-276.
- Xu B, Liu P. No survival improvement for patients with angioimmunoblastic T-cell lymphoma over the past two decades: a population-based study of 1207 cases. PLoS One. 2014;9:e92585.
- Lachenal F, Berger F, Ghesquieres H, et al. Angioimmunoblastic T-cell lymphoma: clinical and laboratory features at diagnosis in 77 patients. Medicine (Baltimore). 2007;86:282-292.
- Mourad N, Mounier N, Briére J, et al. Clinical, biologic, and pathologic features in 157 patients with angioimmunoblastic T-cell lymphoma treated within the Groupe d’Etude des Lymphomes de l’Adulte (GELA) trials. Blood. 2008;111:4463-4470.
- Frederico M, Rudiger T, Bellei M, et al. Clinicopathologic characteristics of angioimmunoblastic T-cell lymphoma: analysis of the International Peripheral T-cell Lymphoma Project. J Clin Oncol. 2013;31:240-246.
- Siegert W, Nerl C, Agthe A, et al. Angioimmunoblastic lym-phadenopathy (AILD)-type T-cell lymphoma: prognostic impact of clinical observations and laboratory findings at presentation. The Kiel Lymphoma Study Group. Ann Oncol. 1995;6:659-664.
- Attygalle AD, Chuang SS, Diss TC, et al. Distinguishing angioimmunoblastic T-cell lymphoma from peripheral T-cell lymphoma, unspecified, using morphology, immunophenotype, and molecular genetics. Histopathology. 2007;50:498-508.
- Dupuis J, Boye K, Martin N, et al. Expression of CXCL13 by neoplastic cells in angioimmunoblastic T-cell lymphoma (AITL): a new diagnostic marker providing evidence that AITL derives from follicular helper cells. Am J Surg Pathol. 2006;30:490-494.
- Odejide O, Weigert O, Lane AA, et al. A targeted mutational landscape of angioimmunoblastic T-cell lymphoma. Blood. 2014;123:1293-1296.
- Martel P, Laroche L, Courville P, et al. Cutaneous involvementin patients with angioimmunoblastic lymphadenopathy with dysproteinemia: a clinical, immunohistological, and molecular analysis. Arch Dermatol. 2000;136:881-886.
- Ortonne N, Dupuis J, Plonquet A, et al. Characterization of CXCL13+ neoplastic t cells in cutaneous lesions of angioimmunoblastic T-cell lymphoma (AITL). Am J Surg Pathol. 2007;31:1068-1076.
- Frizzera G, Moran E, Rappaport H. Angioimmunoblastic lymphadenopathy with dysproteinemia. Lancet. 1974;1:1070-1073.
- Balaraman B, Conley JA, Sheinbein DM. Evaluation of cutaneous angioimmunoblastic T-cell lymphoma [published online May 6, 2011]. J Am Acad Dermatol. 2011;65:855-862.
- Tokunaga T, Shimada K, Yamamoto K, et al. Retrospective analysis of prognostic factors for angioimmunoblastic T-cell lymphoma: a multicenter cooperative study in Japan. Blood. 2012;119:2837-2843.
- Dogan A, Ngu LSP, Ng SH, et al. Pathology and clinical features of angioimmunoblastic T-cell lymphoma after successful treatment with thalidomide. Leukemia. 2005;19:873-875.
- Fabbri A, Cencini E, Pietrini A, et al. Impressive activity of lenalidomide monotherapy in refractory angioimmunoblastic T-cell lymphoma: report of a case with long-term follow-up. Hematol Oncol. 2013;31:213-217.
- Kobayashi T, Kuroda J, Uchiyama H, et al. Successful treatment of chemotherapy-refractory angioimmunoblastic T cell lymphoma with cyclosporin A. Acta Haematol. 2012;127:10-15.
Angioimmunoblastic T-cell lymphoma (AITL) is a rare, often aggressive type of peripheral T-cell lymphoma. It comprises 18% of peripheral T-cell lymphomas and 1% to 2% of all non-Hodgkin lymphomas.1 The incidence of AITL in the United States is estimated to be 0.05 cases per 100,000 person-years,2 and there is a slight male predominance.1,3,4 It typically presents in the seventh decade of life; however, cases have been reported in adults ranging from 20 to 91 years of age.3
Angioimmunoblastic T-cell lymphoma presents with lymphadenopathy, hepatosplenomegaly, and systemic B symptoms (eg, fever, night sweats, weight loss, generalized pruritus).4-6 There are cutaneous manifestations in up to 50% of cases4,5,7 and frequently signs of autoimmune disorder.4,5 The diagnosis often is made by excisional lymph node biopsy. Lymph node specimens characteristically have a mixed inflammatory infiltrate that includes numerous B cells often infected with Epstein-Barr virus (EBV) and a relatively small population of atypical T lymphocytes.8 Identification of this neoplastic population of CD4+CD8− T lymphocytes expressing normal follicular helper T-cell markers CD10, chemokine CXCL13, programmed cell death protein 1 (PD-1), and B-cell lymphoma 6 (BCL-6) confirms the diagnosis of AITL.9,10 These malignant cells can be identified in skin specimens in cases of cutaneous metastatic disease.11,12 We present a case originally misdiagnosed as diffuse large B-cell lymphoma that was later identified as AITL on skin biopsy.
Case Report
A 72-year-old woman presented with a pruritic erythematous eruption around the neck of 3 weeks’ duration (Figure 1). Her medical history was notable for diffuse large B-cell non-Hodgkin lymphoma diagnosed 3 months prior based on results from a right cervical lymph node biopsy. She was treated with bendamustine and rituximab. On physical examination there were erythematous edematous papules coalescing into indurated plaques around the neck. The differential diagnosis included drug hypersensitivity reaction, herpes zoster, urticaria, and cutaneous metastasis. Two punch biopsies were taken for hematoxylin and eosin and tissue culture.
Tissue cultures and viral polymerase chain reaction were negative. Histopathologic examination revealed a scant atypical lymphoid infiltrate focally involving the deep dermis. The cells were medium to large in size and contained hyperchromatic pleomorphic nuclei (Figure 2). They were positive for CD3 and CD4, which was concerning for T-cell lymphoma. The histologic report of the excisional lymph node biopsy done 3 months prior described an atypical lymphoid neoplasm with extensive necrosis and extranodal spread that stained positively for CD20 (Figure 3).
Further staining of this cervical lymph node specimen revealed large atypical lymphoid cells positive for CD3, CD10, B-cell lymphoma 2 (BCL-2), BCL-6, and PD-1. There were intermixed mature B lymphocytes positive for CD20 and BCL-2. Chromogenic in situ hybridization with probes for EBV showed numerous positive cells throughout the infiltrate. Polymerase chain reaction demonstrated a T-cell population with clonally rearranged T-cell receptor genes. Primers for immunoglobulin heavy and light chains showed no evidence of a clonal B-cell population.
Additional staining of the atypical cutaneous lymphocytes revealed positivity for CD3, CD10, and PD-1. The morphologic and immunophenotypic findings of both specimens supported the diagnosis of AITL.
The patient declined further treatment and chose hospice care.
Comment
Etiology
Angioimmunoblastic T-cell lymphoma was originally named angioimmunoblastic lymphadenopathy with dysproteinemia. It was initially thought to be a benign hyperreactive immune process driven by B cells, and patients often died of infectious complications not long after the diagnosis was made.13 As more cases were reported with clonal rearrangements and signs of progressive lymphoma, AITL was recognized as a malignancy.
Presentation
Patients with AITL often present with advanced stage III or IV disease with extranodal and bone marrow involvement.3-6 Cutaneous disease occurs in up to half of patients and portends a poor prognosis.7 The rash often is a nonspecific erythematous macular and papular eruption mimicking a morbilliform viral exanthem or drug reaction. Urticarial, nodular, petechial, purpuric, eczematous, erythrodermic, and vesiculobullous presentations have been described.4,11,12 In up to one-third of cases, the eruption occurs in association with a new medication, often leading to an initial misdiagnosis of drug hypersensitivity reaction.4,11 In a review conducted by Balaraman et al,14 84% of patients with AITL reported having pruritus.
There is an association of autoimmune phenomena in patients with AITL, which is likely a result of immune dysregulation associated with poorly functioning follicular helper T cells. Patients may present with arthralgia, hemolytic anemia, or thrombocytopenic purpura. Hypergammaglobulinemia has been reported in 30% to 50% of AITL patients.4,6 Other pertinent immunologic findings include positive Coombs test, cold agglutinins, cryoglobulinemia, hypocomplementemia, and positive antinuclear antibodies.4-7
Gene Analysis
Affected lymph nodes have a characteristically effaced architecture with proliferative arborizing venules; a hyperplastic population of follicular dendritic cells; and a mixed inflammatory infiltrate that is comprised of atypical lymphocytes and variable numbers of reactive lymphocytes, histiocytes, eosinophils, and plasma cells. The malignant T lymphocytes often account for only a small portion of the infiltrate.8 T-cell gene rearrangement studies identify clonal cells with β and γ rearrangements in the majority of cases.4 These cells are predominantly CD4+CD8− and express normal follicular helper T-cell markers CD10, CXCL13, BCL-6,5,9 and PD-1.10 Numerous B cells are seen intermixed with follicular dendritic cells. They are frequently infected with EBV and can have an atypical Reed-Sternberg cell–like appearance.4,5,15 In the evaluation of AITL, polymerase chain reaction studies with primers for immunoglobulin heavy and light chain should be performed to look for clonal B-cell populations and rule out a possible secondary B-cell lymphoma.
Histology
Five histologic patterns have been described with cutaneous AITL: (1) superficial perivascular infiltrate of eosinophils and lymphocytes that lack atypia, (2) sparse perivascular infiltrate with atypical lymphocytes, (3) dense dermal infiltrate of pleomorphic lymphocytes, (4) leukocytoclastic vasculitis without atypical lymphocytes,11 and (5) necrotizing vasculitis.12 The finding of vascular hyperplasia, perivascular infiltrate, or vasculitis has been reported in 91% of cases in the literature. Although these findings are nonspecific, an analysis of cutaneous cases reported in the literature found that 87% demonstrated T-cell receptor gene rearrangements.14 Lymphoid cells are positive for CD10 and PD-1, as was demonstrated in our case, and are CXCL13 positive in the majority of cases.12 Atypical and EBV-infected B cells also can be found in the skin.11,12
Differential Diagnosis
Angioimmunoblastic T-cell lymphoma can mimic infectious, autoimmune, or allergic etiologies, and misdiagnosis of another type of lymphoma is not uncommon, as occurred in our case. Patients who have a delay in the correct diagnosis have similar outcomes to those correctly diagnosed at first presentation.4
Treatment
There are no effective therapies for AITL. Poor prognostic factors include age (>60 years), stages III to IV disease, male gender, elevated serum lactate dehydrogenase level,3,5,10 and cutaneous involvement.7 Corticosteroids, anthracycline-based chemotherapy, and autologous stem cell transplant are currently the mainstays of therapy. Initial response to chemotherapy is promising, but duration of response is poor overall and there is no increased survival.5,15 A large population-based study of 1207 cases by Xu and Liu3 showed the overall survival rate at 2 and 10 years was 46.8% and 21.9%, respectively. Ten-year disease-specific survival was 35.9%, and there was no demonstrable improvement in survival over the last 2 decades.3 Case reports have demonstrated that thalidomide,16 lenalidomide,17 and cyclosporine plus dexamethasone18 have been successfully used to achieve remission for up to 3 years.
Conclusion
Angioimmunoblastic T-cell lymphoma is difficult to diagnose due to nonspecific clinical and histologic findings. Cutaneous manifestations are seen in AITL in up to half of cases that may occur early or in advanced disease. Similar to all cutaneous metastases, the appearance of the lesions can greatly vary. Our case demonstrates that dermatologists and dermatopathologists can make this diagnosis in the appropriate clinicopathologic context utilizing appropriate immunohistochemical staining and gene rearrangement studies.
Angioimmunoblastic T-cell lymphoma (AITL) is a rare, often aggressive type of peripheral T-cell lymphoma. It comprises 18% of peripheral T-cell lymphomas and 1% to 2% of all non-Hodgkin lymphomas.1 The incidence of AITL in the United States is estimated to be 0.05 cases per 100,000 person-years,2 and there is a slight male predominance.1,3,4 It typically presents in the seventh decade of life; however, cases have been reported in adults ranging from 20 to 91 years of age.3
Angioimmunoblastic T-cell lymphoma presents with lymphadenopathy, hepatosplenomegaly, and systemic B symptoms (eg, fever, night sweats, weight loss, generalized pruritus).4-6 There are cutaneous manifestations in up to 50% of cases4,5,7 and frequently signs of autoimmune disorder.4,5 The diagnosis often is made by excisional lymph node biopsy. Lymph node specimens characteristically have a mixed inflammatory infiltrate that includes numerous B cells often infected with Epstein-Barr virus (EBV) and a relatively small population of atypical T lymphocytes.8 Identification of this neoplastic population of CD4+CD8− T lymphocytes expressing normal follicular helper T-cell markers CD10, chemokine CXCL13, programmed cell death protein 1 (PD-1), and B-cell lymphoma 6 (BCL-6) confirms the diagnosis of AITL.9,10 These malignant cells can be identified in skin specimens in cases of cutaneous metastatic disease.11,12 We present a case originally misdiagnosed as diffuse large B-cell lymphoma that was later identified as AITL on skin biopsy.
Case Report
A 72-year-old woman presented with a pruritic erythematous eruption around the neck of 3 weeks’ duration (Figure 1). Her medical history was notable for diffuse large B-cell non-Hodgkin lymphoma diagnosed 3 months prior based on results from a right cervical lymph node biopsy. She was treated with bendamustine and rituximab. On physical examination there were erythematous edematous papules coalescing into indurated plaques around the neck. The differential diagnosis included drug hypersensitivity reaction, herpes zoster, urticaria, and cutaneous metastasis. Two punch biopsies were taken for hematoxylin and eosin and tissue culture.
Tissue cultures and viral polymerase chain reaction were negative. Histopathologic examination revealed a scant atypical lymphoid infiltrate focally involving the deep dermis. The cells were medium to large in size and contained hyperchromatic pleomorphic nuclei (Figure 2). They were positive for CD3 and CD4, which was concerning for T-cell lymphoma. The histologic report of the excisional lymph node biopsy done 3 months prior described an atypical lymphoid neoplasm with extensive necrosis and extranodal spread that stained positively for CD20 (Figure 3).
Further staining of this cervical lymph node specimen revealed large atypical lymphoid cells positive for CD3, CD10, B-cell lymphoma 2 (BCL-2), BCL-6, and PD-1. There were intermixed mature B lymphocytes positive for CD20 and BCL-2. Chromogenic in situ hybridization with probes for EBV showed numerous positive cells throughout the infiltrate. Polymerase chain reaction demonstrated a T-cell population with clonally rearranged T-cell receptor genes. Primers for immunoglobulin heavy and light chains showed no evidence of a clonal B-cell population.
Additional staining of the atypical cutaneous lymphocytes revealed positivity for CD3, CD10, and PD-1. The morphologic and immunophenotypic findings of both specimens supported the diagnosis of AITL.
The patient declined further treatment and chose hospice care.
Comment
Etiology
Angioimmunoblastic T-cell lymphoma was originally named angioimmunoblastic lymphadenopathy with dysproteinemia. It was initially thought to be a benign hyperreactive immune process driven by B cells, and patients often died of infectious complications not long after the diagnosis was made.13 As more cases were reported with clonal rearrangements and signs of progressive lymphoma, AITL was recognized as a malignancy.
Presentation
Patients with AITL often present with advanced stage III or IV disease with extranodal and bone marrow involvement.3-6 Cutaneous disease occurs in up to half of patients and portends a poor prognosis.7 The rash often is a nonspecific erythematous macular and papular eruption mimicking a morbilliform viral exanthem or drug reaction. Urticarial, nodular, petechial, purpuric, eczematous, erythrodermic, and vesiculobullous presentations have been described.4,11,12 In up to one-third of cases, the eruption occurs in association with a new medication, often leading to an initial misdiagnosis of drug hypersensitivity reaction.4,11 In a review conducted by Balaraman et al,14 84% of patients with AITL reported having pruritus.
There is an association of autoimmune phenomena in patients with AITL, which is likely a result of immune dysregulation associated with poorly functioning follicular helper T cells. Patients may present with arthralgia, hemolytic anemia, or thrombocytopenic purpura. Hypergammaglobulinemia has been reported in 30% to 50% of AITL patients.4,6 Other pertinent immunologic findings include positive Coombs test, cold agglutinins, cryoglobulinemia, hypocomplementemia, and positive antinuclear antibodies.4-7
Gene Analysis
Affected lymph nodes have a characteristically effaced architecture with proliferative arborizing venules; a hyperplastic population of follicular dendritic cells; and a mixed inflammatory infiltrate that is comprised of atypical lymphocytes and variable numbers of reactive lymphocytes, histiocytes, eosinophils, and plasma cells. The malignant T lymphocytes often account for only a small portion of the infiltrate.8 T-cell gene rearrangement studies identify clonal cells with β and γ rearrangements in the majority of cases.4 These cells are predominantly CD4+CD8− and express normal follicular helper T-cell markers CD10, CXCL13, BCL-6,5,9 and PD-1.10 Numerous B cells are seen intermixed with follicular dendritic cells. They are frequently infected with EBV and can have an atypical Reed-Sternberg cell–like appearance.4,5,15 In the evaluation of AITL, polymerase chain reaction studies with primers for immunoglobulin heavy and light chain should be performed to look for clonal B-cell populations and rule out a possible secondary B-cell lymphoma.
Histology
Five histologic patterns have been described with cutaneous AITL: (1) superficial perivascular infiltrate of eosinophils and lymphocytes that lack atypia, (2) sparse perivascular infiltrate with atypical lymphocytes, (3) dense dermal infiltrate of pleomorphic lymphocytes, (4) leukocytoclastic vasculitis without atypical lymphocytes,11 and (5) necrotizing vasculitis.12 The finding of vascular hyperplasia, perivascular infiltrate, or vasculitis has been reported in 91% of cases in the literature. Although these findings are nonspecific, an analysis of cutaneous cases reported in the literature found that 87% demonstrated T-cell receptor gene rearrangements.14 Lymphoid cells are positive for CD10 and PD-1, as was demonstrated in our case, and are CXCL13 positive in the majority of cases.12 Atypical and EBV-infected B cells also can be found in the skin.11,12
Differential Diagnosis
Angioimmunoblastic T-cell lymphoma can mimic infectious, autoimmune, or allergic etiologies, and misdiagnosis of another type of lymphoma is not uncommon, as occurred in our case. Patients who have a delay in the correct diagnosis have similar outcomes to those correctly diagnosed at first presentation.4
Treatment
There are no effective therapies for AITL. Poor prognostic factors include age (>60 years), stages III to IV disease, male gender, elevated serum lactate dehydrogenase level,3,5,10 and cutaneous involvement.7 Corticosteroids, anthracycline-based chemotherapy, and autologous stem cell transplant are currently the mainstays of therapy. Initial response to chemotherapy is promising, but duration of response is poor overall and there is no increased survival.5,15 A large population-based study of 1207 cases by Xu and Liu3 showed the overall survival rate at 2 and 10 years was 46.8% and 21.9%, respectively. Ten-year disease-specific survival was 35.9%, and there was no demonstrable improvement in survival over the last 2 decades.3 Case reports have demonstrated that thalidomide,16 lenalidomide,17 and cyclosporine plus dexamethasone18 have been successfully used to achieve remission for up to 3 years.
Conclusion
Angioimmunoblastic T-cell lymphoma is difficult to diagnose due to nonspecific clinical and histologic findings. Cutaneous manifestations are seen in AITL in up to half of cases that may occur early or in advanced disease. Similar to all cutaneous metastases, the appearance of the lesions can greatly vary. Our case demonstrates that dermatologists and dermatopathologists can make this diagnosis in the appropriate clinicopathologic context utilizing appropriate immunohistochemical staining and gene rearrangement studies.
- Rudiger T, Weisenburger DD, Anderson JR, et al. Peripheral T-cell lymphoma (excluding anaplastic large-cell lymphoma): results from the Non-Hodgkins Lymphoma Classification Project. Ann Oncol. 2002;13:140-149.
- Morton LM, Wang SS, Devesa SS, et al. Lymphoma incidence patterns by WHO subtype in the United States, 1992-2001. Blood. 2006;107:265-276.
- Xu B, Liu P. No survival improvement for patients with angioimmunoblastic T-cell lymphoma over the past two decades: a population-based study of 1207 cases. PLoS One. 2014;9:e92585.
- Lachenal F, Berger F, Ghesquieres H, et al. Angioimmunoblastic T-cell lymphoma: clinical and laboratory features at diagnosis in 77 patients. Medicine (Baltimore). 2007;86:282-292.
- Mourad N, Mounier N, Briére J, et al. Clinical, biologic, and pathologic features in 157 patients with angioimmunoblastic T-cell lymphoma treated within the Groupe d’Etude des Lymphomes de l’Adulte (GELA) trials. Blood. 2008;111:4463-4470.
- Frederico M, Rudiger T, Bellei M, et al. Clinicopathologic characteristics of angioimmunoblastic T-cell lymphoma: analysis of the International Peripheral T-cell Lymphoma Project. J Clin Oncol. 2013;31:240-246.
- Siegert W, Nerl C, Agthe A, et al. Angioimmunoblastic lym-phadenopathy (AILD)-type T-cell lymphoma: prognostic impact of clinical observations and laboratory findings at presentation. The Kiel Lymphoma Study Group. Ann Oncol. 1995;6:659-664.
- Attygalle AD, Chuang SS, Diss TC, et al. Distinguishing angioimmunoblastic T-cell lymphoma from peripheral T-cell lymphoma, unspecified, using morphology, immunophenotype, and molecular genetics. Histopathology. 2007;50:498-508.
- Dupuis J, Boye K, Martin N, et al. Expression of CXCL13 by neoplastic cells in angioimmunoblastic T-cell lymphoma (AITL): a new diagnostic marker providing evidence that AITL derives from follicular helper cells. Am J Surg Pathol. 2006;30:490-494.
- Odejide O, Weigert O, Lane AA, et al. A targeted mutational landscape of angioimmunoblastic T-cell lymphoma. Blood. 2014;123:1293-1296.
- Martel P, Laroche L, Courville P, et al. Cutaneous involvementin patients with angioimmunoblastic lymphadenopathy with dysproteinemia: a clinical, immunohistological, and molecular analysis. Arch Dermatol. 2000;136:881-886.
- Ortonne N, Dupuis J, Plonquet A, et al. Characterization of CXCL13+ neoplastic t cells in cutaneous lesions of angioimmunoblastic T-cell lymphoma (AITL). Am J Surg Pathol. 2007;31:1068-1076.
- Frizzera G, Moran E, Rappaport H. Angioimmunoblastic lymphadenopathy with dysproteinemia. Lancet. 1974;1:1070-1073.
- Balaraman B, Conley JA, Sheinbein DM. Evaluation of cutaneous angioimmunoblastic T-cell lymphoma [published online May 6, 2011]. J Am Acad Dermatol. 2011;65:855-862.
- Tokunaga T, Shimada K, Yamamoto K, et al. Retrospective analysis of prognostic factors for angioimmunoblastic T-cell lymphoma: a multicenter cooperative study in Japan. Blood. 2012;119:2837-2843.
- Dogan A, Ngu LSP, Ng SH, et al. Pathology and clinical features of angioimmunoblastic T-cell lymphoma after successful treatment with thalidomide. Leukemia. 2005;19:873-875.
- Fabbri A, Cencini E, Pietrini A, et al. Impressive activity of lenalidomide monotherapy in refractory angioimmunoblastic T-cell lymphoma: report of a case with long-term follow-up. Hematol Oncol. 2013;31:213-217.
- Kobayashi T, Kuroda J, Uchiyama H, et al. Successful treatment of chemotherapy-refractory angioimmunoblastic T cell lymphoma with cyclosporin A. Acta Haematol. 2012;127:10-15.
- Rudiger T, Weisenburger DD, Anderson JR, et al. Peripheral T-cell lymphoma (excluding anaplastic large-cell lymphoma): results from the Non-Hodgkins Lymphoma Classification Project. Ann Oncol. 2002;13:140-149.
- Morton LM, Wang SS, Devesa SS, et al. Lymphoma incidence patterns by WHO subtype in the United States, 1992-2001. Blood. 2006;107:265-276.
- Xu B, Liu P. No survival improvement for patients with angioimmunoblastic T-cell lymphoma over the past two decades: a population-based study of 1207 cases. PLoS One. 2014;9:e92585.
- Lachenal F, Berger F, Ghesquieres H, et al. Angioimmunoblastic T-cell lymphoma: clinical and laboratory features at diagnosis in 77 patients. Medicine (Baltimore). 2007;86:282-292.
- Mourad N, Mounier N, Briére J, et al. Clinical, biologic, and pathologic features in 157 patients with angioimmunoblastic T-cell lymphoma treated within the Groupe d’Etude des Lymphomes de l’Adulte (GELA) trials. Blood. 2008;111:4463-4470.
- Frederico M, Rudiger T, Bellei M, et al. Clinicopathologic characteristics of angioimmunoblastic T-cell lymphoma: analysis of the International Peripheral T-cell Lymphoma Project. J Clin Oncol. 2013;31:240-246.
- Siegert W, Nerl C, Agthe A, et al. Angioimmunoblastic lym-phadenopathy (AILD)-type T-cell lymphoma: prognostic impact of clinical observations and laboratory findings at presentation. The Kiel Lymphoma Study Group. Ann Oncol. 1995;6:659-664.
- Attygalle AD, Chuang SS, Diss TC, et al. Distinguishing angioimmunoblastic T-cell lymphoma from peripheral T-cell lymphoma, unspecified, using morphology, immunophenotype, and molecular genetics. Histopathology. 2007;50:498-508.
- Dupuis J, Boye K, Martin N, et al. Expression of CXCL13 by neoplastic cells in angioimmunoblastic T-cell lymphoma (AITL): a new diagnostic marker providing evidence that AITL derives from follicular helper cells. Am J Surg Pathol. 2006;30:490-494.
- Odejide O, Weigert O, Lane AA, et al. A targeted mutational landscape of angioimmunoblastic T-cell lymphoma. Blood. 2014;123:1293-1296.
- Martel P, Laroche L, Courville P, et al. Cutaneous involvementin patients with angioimmunoblastic lymphadenopathy with dysproteinemia: a clinical, immunohistological, and molecular analysis. Arch Dermatol. 2000;136:881-886.
- Ortonne N, Dupuis J, Plonquet A, et al. Characterization of CXCL13+ neoplastic t cells in cutaneous lesions of angioimmunoblastic T-cell lymphoma (AITL). Am J Surg Pathol. 2007;31:1068-1076.
- Frizzera G, Moran E, Rappaport H. Angioimmunoblastic lymphadenopathy with dysproteinemia. Lancet. 1974;1:1070-1073.
- Balaraman B, Conley JA, Sheinbein DM. Evaluation of cutaneous angioimmunoblastic T-cell lymphoma [published online May 6, 2011]. J Am Acad Dermatol. 2011;65:855-862.
- Tokunaga T, Shimada K, Yamamoto K, et al. Retrospective analysis of prognostic factors for angioimmunoblastic T-cell lymphoma: a multicenter cooperative study in Japan. Blood. 2012;119:2837-2843.
- Dogan A, Ngu LSP, Ng SH, et al. Pathology and clinical features of angioimmunoblastic T-cell lymphoma after successful treatment with thalidomide. Leukemia. 2005;19:873-875.
- Fabbri A, Cencini E, Pietrini A, et al. Impressive activity of lenalidomide monotherapy in refractory angioimmunoblastic T-cell lymphoma: report of a case with long-term follow-up. Hematol Oncol. 2013;31:213-217.
- Kobayashi T, Kuroda J, Uchiyama H, et al. Successful treatment of chemotherapy-refractory angioimmunoblastic T cell lymphoma with cyclosporin A. Acta Haematol. 2012;127:10-15.
Practice Points
- Angioimmunoblastic T-cell lymphoma (AITL) is a rare, often aggressive type of peripheral T-cell lymphoma.
- Cutaneous manifestations have been seen in up to 50% of cases.
- Immunohistochemical markers for normal follicular helper T cells—CD-10, chemokine CXCL-13, and programmed cell death protein 1 (PD-1)—can be used to differentiate AITL from other types of lymphoma.
- The prognosis of AITL is poor.
Levamisole-Induced Vasculopathy With Gastric Involvement in a Cocaine User
In 2010, two separate reports of cutaneous vasculitic/vasculopathic eruptions in patients with recent exposure to levamisole-contaminated cocaine (LCC) were published in the literature.1,2 Since then, additional reports have been published.3-6 Retiform purpura associated with cocaine use appears to be a similar condition, perhaps lying at one end of the spectrum of LCC-induced cutaneous vascular disease.7,8 Although some patients have been described as having nausea and vomiting,8,9 including one with a sudden drop in hemoglobin to 5.8 g/dL (reference range, 14.0–17.5 g/dL),10 there are no known reported cases of LCC and levamisole-induced vasculopathy in organ systems other than the skin. Herein, we report the case of a patient with levamisole-induced vasculopathy (LIV) demonstrating endoscopic evidence of gastric hemorrhage with features similar to those involving the skin.
Case Report
A 35-year-old woman with a history of hepatitis C, intravenous drug abuse, and bipolar disorder presented to the emergency department with painful necrotic lesions on the head, neck, arms, and legs of several days’ duration. Approximately 1 year prior she had been admitted to the hospital with similar lesions, with eventual partial necrosis of the left earlobe. The patient reported she had last used crack cocaine 3 days prior to the development of the lesions. A urine drug screen was positive for lorazepam, alprazolam, buprenorphine, methadone, tetrahydrocannabinol, and cocaine. She also reported abdominal pain and gastric reflux of recent onset but denied any history of gastrointestinal tract disease. During the previous admission, the patient demonstrated antinuclear antibodies at a titer of greater than 1:160 (normal, <1:40) in a smooth pattern as well as positive perinuclear antineutrophil cytoplasmic antibodies (p-ANCA) and cytoplasmic antineutrophil cytoplasmic antibodies (c-ANCA) and positive cryoglobulins. Physical examination yielded purpuric and hemorrhagic patches and plaques on the nose, bilateral ears (Figure 1A), face (Figure 1B), arms, and legs. Older lesions exhibited evidence of evolving erosion and ulceration. A biopsy of a lesion on the right arm was obtained, demonstrating extensive epidermal necrosis, hemorrhage, fibrin thrombi within dermal blood vessels, fibrinoid mural necrosis, perivascular neutrophils, and leukocytoclasis (Figure 2). These findings were consistent with LIV caused by exposure to LCC. A complete blood cell count was unremarkable. She was started on pain management and was given prednisone to treat the cutaneous eruption. Because of continued reports of epigastric pain and discomfort on swallowing, an upper gastrointestinal endoscopy was performed. Numerous esophageal erosions and gastric submucosal hemorrhages similar to those on the skin were noted (Figure 3). Pathology taken at the time of the endoscopy demonstrated mucosal erosions, but an evaluation for vascular insult was not possible, as submucosal tissue was not obtained. As the skin lesions began to heal, the gastric symptoms gradually subsided, and the patient was released from the hospital after 7 days.
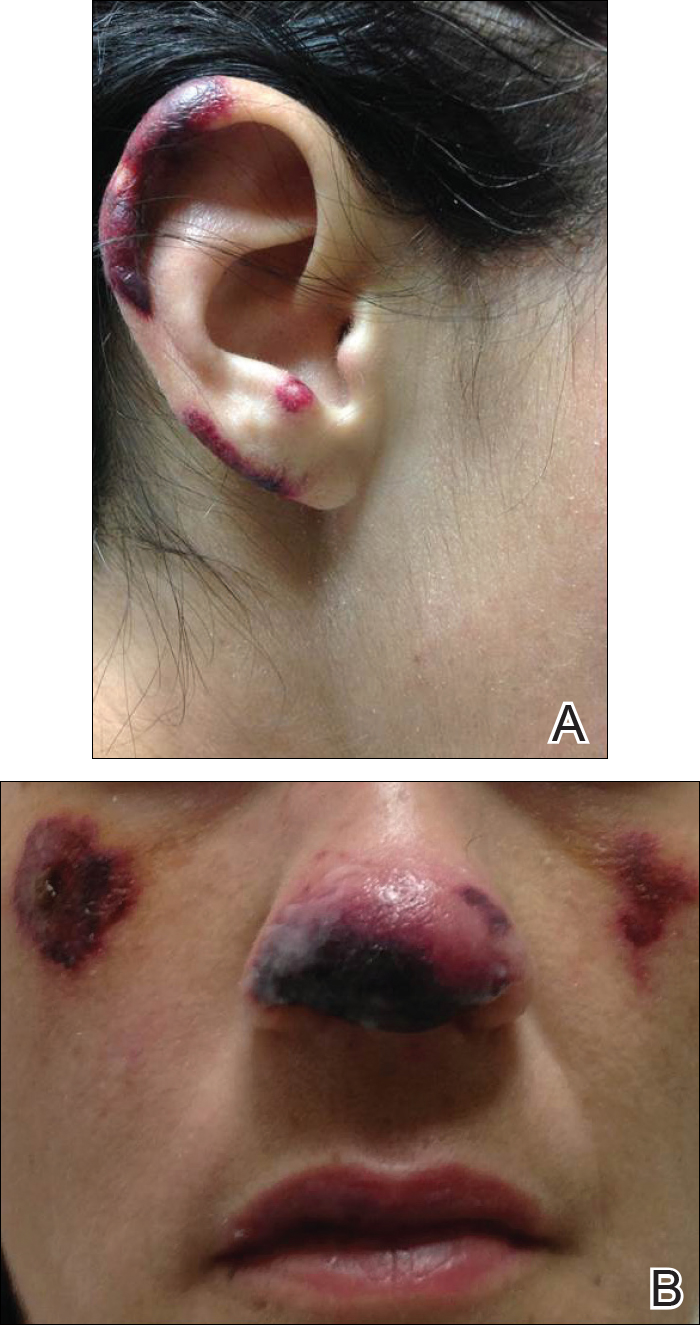
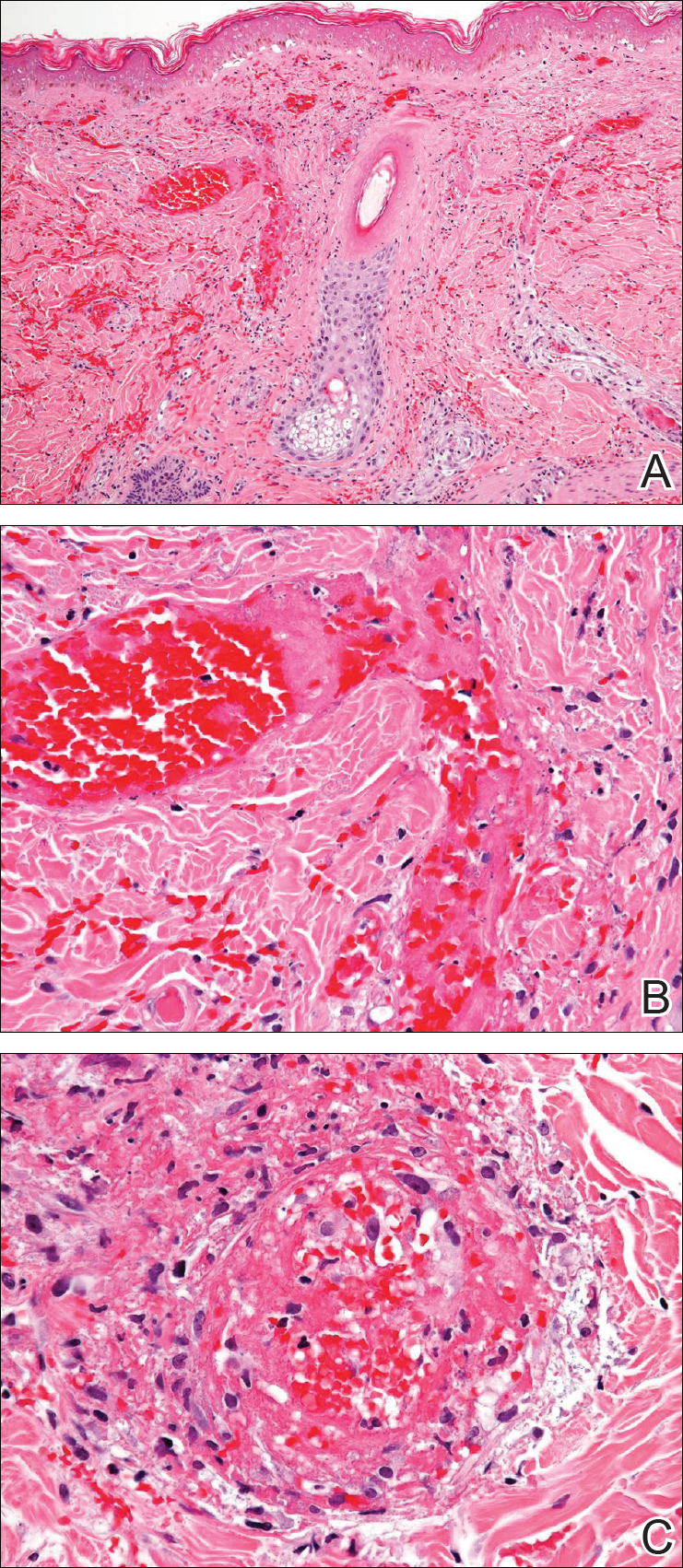
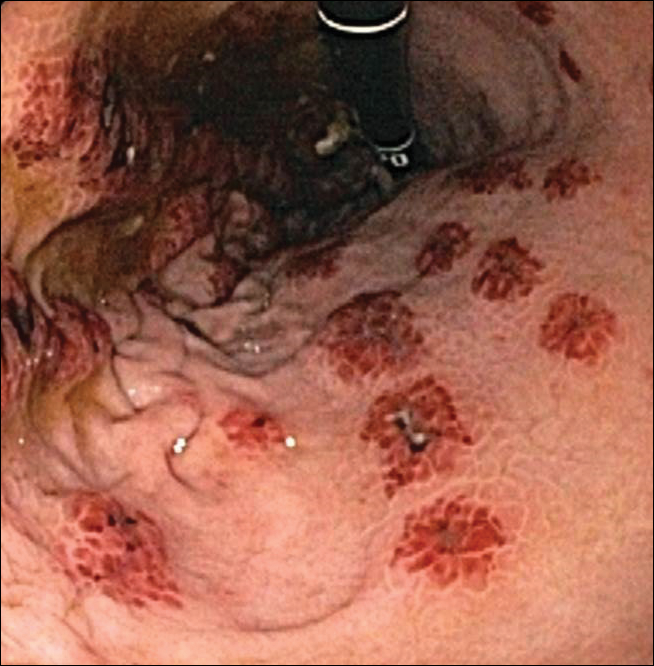
Comment
Levamisole-Contaminated Cocaine
Cocaine is a crystalline alkaloid obtained from the leaves of the coca plant.7 Fifty percent of globally produced cocaine is consumed in the United States.10 There are 2 to 5 million cocaine users in the United States; in 2009, a reported 1.6 million US adults admitted to having used cocaine in the previous month.4,11,12 Cocaine has been known to be cut with similar-appearing substances including lactose and mannitol, though caffeine, acetaminophen, methylphenidate, and other ingredients have been utilized.7
Levamisole is a synthetic imidazothiazole derivative initially developed for use as an immunomodulatory agent in patients with rheumatoid arthritis.4 It was later paired with 5-fluorouracil for administration in patients with carcinomas of the colon and breasts.4,13 In 2000, the drug was withdrawn from the US market for use in humans after an association between levamisole and agranulocytosis was noted in 2.5% to 13% of patients taking the drug for rheumatoid arthritis or as an adjuvant therapy for breast carcinoma.9,12 It still is available for veterinary use as an anthelmintic and is administered to humans in other countries. Levamisole acts as an immunomodulator by enhancing macrophage chemotaxis and upregulating T-cell functions as well as stimulating neutrophil chemotaxis and dendritic cell maturation.4 It also is known to generate autoantibodies including lupus anticoagulant, p-ANCA, c-ANCA, and antinuclear antibodies.7,14 Levamisole is known to exhibit cutaneous reactions. In 1999, Rongioletti et al14 reported 5 children with purpura of the ears who had been given levamisole for pediatric nephrotic syndrome. Involvement of other body areas was noted. Three patients developed lupus anticoagulant antibodies, 3 exhibited p-ANCA antibodies, and 1 was positive for c-ANCA antibodies. The investigators noted an exceptionally long latency period of 12 to 44 months after starting the drug. Histologically a vasculopathic/vasculitic process was noted.14 Direct immunofluorescence studies of affected skin in LIV have demonstrated IgM, IgA, IgG, C3, and fibrin staining of blood vessels.4,15 Anti–human elastase antibodies are considered both sensitive and specific for LIV and serve to differentiate it from cocaine-induced pseudovasculitis.4,7
In April 2008, the New Mexico Department of Health began evaluating several unexplained cases of agranulocytosis and noted that 11 of 21 cases were associated with cocaine use.9 Later that year, public health workers in Alberta and British Columbia, Canada, reported finding traces of levamisole in clinical specimens and drug paraphernalia of cocaine users with agranulocytosis. Officials from the New Mexico Department of Health learned of these findings and investigated the cases, finding 7 of 9 patients with idiopathic agranulocytosis had recent exposure to cocaine. None of the 21 total patients experienced any skin findings. Nausea and vomiting were common symptoms, but abdominal pain was described in only 2 patients from an additional investigation in Washington. Both of these patients used crack cocaine, and one had a positive urine test for levamisole.9
The presence of levamisole initially was detected by the US Drug Enforcement Administration in 2003. By July 2009, 69% of cocaine and 3% of heroin seized by this agency was noted to contain levamisole.16 From 2003 to 2009, the concentration of levamisole contamination rose to 10%.4 A 2011 study found levamisole in 194 of 249 cocaine-positive urine samples.16
It is unclear why cocaine producers add levamisoleto their product. Possibilities include increasing the drug’s bulk or enhancing its stimulatory effects.12 Chang et al17 posited that levamisole increases the stimulatory and euphoric effects of cocaine by increasing dopamine levels in the brain. Additionally, levamisole is metabolized to aminorex, an amphetaminelike hallucinogen that suppresses appetite, in patients with LCC.13 Vagi et al12 interviewed 10 patients who had been hospitalized for agranulocytosis secondary to use of LCC. None were aware of the presence of this additive, suggesting it was not used as a marketing tool.
Cutaneous Vasculopathy
Levamisole-induced vasculopathy (also called levamisole-induced cutaneous vasculopathy11) initially was reported by 2 separate groups in 2010.1,2 Patients typically present with tender purpuric to hemorrhagic papules, plaques, and bullae with an affinity to affect the ears, nose, and face, though other areas of the body can be affected. A pattern of retiform purpura may precede these findings in some patients. Women are disproportionately affected.11 Crack cocaine use is overrepresented in LIV compared to insufflation or snorting of the drug. Affected patients may exhibit systemic symptoms including myalgia, arthralgia, and frank arthritis.10 Additionally, 15% to 80% of patients exhibit positive antinuclear antibodies, anticardiolipin antibodies, lupus anticoagulant antibodies, p-ANCA antibodies, and c-ANCA antibodies. Magro and Wang8 hypothesized that levamisole acting in conjunction with cocaine rather than the effects of levamisole alone is responsible for some of these findings.
Histologically, the features of a vasculopathic process are noted in some patients with the presence of frank vasculitis.1 The vasculopathic component demonstrates vessel dilatation with thrombosis, eosinophilic deposits, and erythrocyte extravasation. Patients with frank vasculitis exhibit fibrinoid vessel wall necrosis and fibrin deposition, extravasated erythrocytes, endothelial cell atypia, and leukocytoclasia.3 Jacob et al3 noted interstitial and perivascular neovascularization in affected tissue, believed to represent one stage in the evolution of medium vessel vasculitis. Intercellular adhesion molecule 1 has been reported in affected vessel walls with endothelial caspase 3 expression and C5b-9 deposition.8 Magro and Wang8 believe the retiform purpura seen in the early stages of some of these patients with LIV represents a thrombotic dynamic with C5b-9 deposition and enhanced apoptosis. Overt vasculitis follows later, subsequent to the effect of ANCA antibodies and upregulated intercellular adhesion molecule 1 expression on vessel walls.
The clinical course of LIV typically is 2 to 3 weeks for lesion resolution; however, normalization of serologies may require 2 to 14 months. Observation and pain control with or without administration of systemic steroids is sufficient for most patients, but skin grafting, wound debridement, cyclosporine, mycophenolate mofetil, and plasmapheresis also have been employed.4,5 Morbidity may be substantive. One report noted LCC to be responsible for 3 cases of pulmonary hemorrhage and acute progression to chronic renal failure in another 2 patients.15 Ching and Smith18 described a patient with 52% total body surface area involvement who required skin grafting, nasal amputation, patellectomy, central upper lip excision, and amputation of the leg above the knee.
Gastrointestinal Presentation
Patients with LIV have been reported to exhibit abdominal pain, but our patient exhibited a rare presentation of visualized gastrointestinal purpura. Although support for a vasculitic/vasculopathic process requires a tissue diagnosis, the endoscopic appearance of gastric vasculitis is similar to that of cutaneous vasculitis.19 Clinicians caring for patients exposed to LCC should bear in mind that the vascular insults associated with LIV are not restricted solely to the skin.
- Waller JM, Feramisco JD, Alberta-Wszolek L, et al. Cocaine-associated retiform purpura and neutropenia: is levamisole the culprit? J Am Acad Dermatol. 2010;63:530-535.
- Bradford M, Rosenberg B, Moreno J, et al. Bilateral necrosis of earlobes and cheeks: another complication of cocaine contaminated with levamisole. Ann Int Med. 2010;152:758-759.
- Jacob RS, Silva CY, Powers JG, et al. Levamisole-induced vasculopathy: a report of 2 cases and a novel histopathologic finding. Am J Dermatopathol. 2012;34:208-213.
- Lee KC, Ladizinski B, Federman DG. Complications associated with use of levamisole-contaminated cocaine: an emerging public health challenge. Mayo Clin Proc. 2012;87:581-586.
- Pavenski K, Vandenberghe H, Jakubovic H, et al. Plasmapheresis and steroid treatment of levamisole-induced vasculopathy and associated skin necrosis in crack/cocaine users. J Cutan Med Surg. 2013;17:123-126.
- Mandrell J, Kranc CL. Prednisone and vardenafil hydrochloride refractory levamisole-induced vasculitis. Cutis. 2016;98:E15-E19.
- Walsh NM, Green PJ, Burlingame RW, et al. Cocaine-related retiform purpura: evidence to incriminate the adulterant, levamisole [published online August 25, 2010]. J Cutan Pathol. 2010;37:1212-1219.
- Magro CM, Wang X. Cocaine-associated retiform purpura: a C5b-9 mediated microangiopathy syndrome associated with enhanced apoptosis and high levels of intercellular adhesion molecule-1 expression. Am J Dermatopathol 2013;35:722-730.
- Centers for Disease Control and Prevention (CDC). Agranulocytosis associated with cocaine use—four states, March 2008-November 2009. MMWR Morb Mortal Wkly Rep. 2009;58:1381-1385.
- Espinoza LR, Alamino RP. Cocaine-induced vasculitis: clinical and immunological spectrum. Curr Rhematol Rep. 2012;14:532-538.
- Arora NP. Cutaneous vasculopathy and neutropenia associated with levamisole-adulterated cocaine. Am J Med Sci. 2013;345:45-51.
- Vagi SJ, Sheikh S, Brackney M, et al. Passive multistate surveillance for neutropenia after of cocaine or heroin possibly contaminated with levamisole. Ann Emerg Med. 2013;61:468-474.
- Lee KC, Ladizinski, Nutan FN. Systemic complications of levamisole toxicity. J Am Acad Dermatol. 2012;67:791-792.
- Rongioletti E, Ghio L, Ginervri E, et al. Purpura of the ears: a distinctive vasculopathy with circulating autoantibodies complicating longer-term treatment with levamisole in children. Br J Dermatol. 1999;140:948-951.
- McGrath MM, Isakova T, Rennke HG, et al. Contaminated cocaine and antineutrophil cytoplasmic antibody-associated diseases. Clin J Am Soc Nephrol. 2011;6:2799-2805.
- Buchanan JA, Heard K, Burbach C, et al. Prevalence of levamisole in urine toxicology screens positive for cocaine in an inner-city hospital. JAMA. 2011;305:1657-1658.
- Chang A, Osterloh J, Thomas J. Levamisole: a dangerous new cocaine adulterant. Clin Pharmacol Ther. 2010;88:408-411.
- Ching JA, Smith DJ. Levamisole-induced necrosis of skin, soft-tissue and bone: case report and review of literature. J Burn Care Res. 2012;33:E1-E5.
- Naruse G, Shimata K. Cutaneous and gastrointestinal purpura. N Engl J Med. 2013;369:1843.
In 2010, two separate reports of cutaneous vasculitic/vasculopathic eruptions in patients with recent exposure to levamisole-contaminated cocaine (LCC) were published in the literature.1,2 Since then, additional reports have been published.3-6 Retiform purpura associated with cocaine use appears to be a similar condition, perhaps lying at one end of the spectrum of LCC-induced cutaneous vascular disease.7,8 Although some patients have been described as having nausea and vomiting,8,9 including one with a sudden drop in hemoglobin to 5.8 g/dL (reference range, 14.0–17.5 g/dL),10 there are no known reported cases of LCC and levamisole-induced vasculopathy in organ systems other than the skin. Herein, we report the case of a patient with levamisole-induced vasculopathy (LIV) demonstrating endoscopic evidence of gastric hemorrhage with features similar to those involving the skin.
Case Report
A 35-year-old woman with a history of hepatitis C, intravenous drug abuse, and bipolar disorder presented to the emergency department with painful necrotic lesions on the head, neck, arms, and legs of several days’ duration. Approximately 1 year prior she had been admitted to the hospital with similar lesions, with eventual partial necrosis of the left earlobe. The patient reported she had last used crack cocaine 3 days prior to the development of the lesions. A urine drug screen was positive for lorazepam, alprazolam, buprenorphine, methadone, tetrahydrocannabinol, and cocaine. She also reported abdominal pain and gastric reflux of recent onset but denied any history of gastrointestinal tract disease. During the previous admission, the patient demonstrated antinuclear antibodies at a titer of greater than 1:160 (normal, <1:40) in a smooth pattern as well as positive perinuclear antineutrophil cytoplasmic antibodies (p-ANCA) and cytoplasmic antineutrophil cytoplasmic antibodies (c-ANCA) and positive cryoglobulins. Physical examination yielded purpuric and hemorrhagic patches and plaques on the nose, bilateral ears (Figure 1A), face (Figure 1B), arms, and legs. Older lesions exhibited evidence of evolving erosion and ulceration. A biopsy of a lesion on the right arm was obtained, demonstrating extensive epidermal necrosis, hemorrhage, fibrin thrombi within dermal blood vessels, fibrinoid mural necrosis, perivascular neutrophils, and leukocytoclasis (Figure 2). These findings were consistent with LIV caused by exposure to LCC. A complete blood cell count was unremarkable. She was started on pain management and was given prednisone to treat the cutaneous eruption. Because of continued reports of epigastric pain and discomfort on swallowing, an upper gastrointestinal endoscopy was performed. Numerous esophageal erosions and gastric submucosal hemorrhages similar to those on the skin were noted (Figure 3). Pathology taken at the time of the endoscopy demonstrated mucosal erosions, but an evaluation for vascular insult was not possible, as submucosal tissue was not obtained. As the skin lesions began to heal, the gastric symptoms gradually subsided, and the patient was released from the hospital after 7 days.
Comment
Levamisole-Contaminated Cocaine
Cocaine is a crystalline alkaloid obtained from the leaves of the coca plant.7 Fifty percent of globally produced cocaine is consumed in the United States.10 There are 2 to 5 million cocaine users in the United States; in 2009, a reported 1.6 million US adults admitted to having used cocaine in the previous month.4,11,12 Cocaine has been known to be cut with similar-appearing substances including lactose and mannitol, though caffeine, acetaminophen, methylphenidate, and other ingredients have been utilized.7
Levamisole is a synthetic imidazothiazole derivative initially developed for use as an immunomodulatory agent in patients with rheumatoid arthritis.4 It was later paired with 5-fluorouracil for administration in patients with carcinomas of the colon and breasts.4,13 In 2000, the drug was withdrawn from the US market for use in humans after an association between levamisole and agranulocytosis was noted in 2.5% to 13% of patients taking the drug for rheumatoid arthritis or as an adjuvant therapy for breast carcinoma.9,12 It still is available for veterinary use as an anthelmintic and is administered to humans in other countries. Levamisole acts as an immunomodulator by enhancing macrophage chemotaxis and upregulating T-cell functions as well as stimulating neutrophil chemotaxis and dendritic cell maturation.4 It also is known to generate autoantibodies including lupus anticoagulant, p-ANCA, c-ANCA, and antinuclear antibodies.7,14 Levamisole is known to exhibit cutaneous reactions. In 1999, Rongioletti et al14 reported 5 children with purpura of the ears who had been given levamisole for pediatric nephrotic syndrome. Involvement of other body areas was noted. Three patients developed lupus anticoagulant antibodies, 3 exhibited p-ANCA antibodies, and 1 was positive for c-ANCA antibodies. The investigators noted an exceptionally long latency period of 12 to 44 months after starting the drug. Histologically a vasculopathic/vasculitic process was noted.14 Direct immunofluorescence studies of affected skin in LIV have demonstrated IgM, IgA, IgG, C3, and fibrin staining of blood vessels.4,15 Anti–human elastase antibodies are considered both sensitive and specific for LIV and serve to differentiate it from cocaine-induced pseudovasculitis.4,7
In April 2008, the New Mexico Department of Health began evaluating several unexplained cases of agranulocytosis and noted that 11 of 21 cases were associated with cocaine use.9 Later that year, public health workers in Alberta and British Columbia, Canada, reported finding traces of levamisole in clinical specimens and drug paraphernalia of cocaine users with agranulocytosis. Officials from the New Mexico Department of Health learned of these findings and investigated the cases, finding 7 of 9 patients with idiopathic agranulocytosis had recent exposure to cocaine. None of the 21 total patients experienced any skin findings. Nausea and vomiting were common symptoms, but abdominal pain was described in only 2 patients from an additional investigation in Washington. Both of these patients used crack cocaine, and one had a positive urine test for levamisole.9
The presence of levamisole initially was detected by the US Drug Enforcement Administration in 2003. By July 2009, 69% of cocaine and 3% of heroin seized by this agency was noted to contain levamisole.16 From 2003 to 2009, the concentration of levamisole contamination rose to 10%.4 A 2011 study found levamisole in 194 of 249 cocaine-positive urine samples.16
It is unclear why cocaine producers add levamisoleto their product. Possibilities include increasing the drug’s bulk or enhancing its stimulatory effects.12 Chang et al17 posited that levamisole increases the stimulatory and euphoric effects of cocaine by increasing dopamine levels in the brain. Additionally, levamisole is metabolized to aminorex, an amphetaminelike hallucinogen that suppresses appetite, in patients with LCC.13 Vagi et al12 interviewed 10 patients who had been hospitalized for agranulocytosis secondary to use of LCC. None were aware of the presence of this additive, suggesting it was not used as a marketing tool.
Cutaneous Vasculopathy
Levamisole-induced vasculopathy (also called levamisole-induced cutaneous vasculopathy11) initially was reported by 2 separate groups in 2010.1,2 Patients typically present with tender purpuric to hemorrhagic papules, plaques, and bullae with an affinity to affect the ears, nose, and face, though other areas of the body can be affected. A pattern of retiform purpura may precede these findings in some patients. Women are disproportionately affected.11 Crack cocaine use is overrepresented in LIV compared to insufflation or snorting of the drug. Affected patients may exhibit systemic symptoms including myalgia, arthralgia, and frank arthritis.10 Additionally, 15% to 80% of patients exhibit positive antinuclear antibodies, anticardiolipin antibodies, lupus anticoagulant antibodies, p-ANCA antibodies, and c-ANCA antibodies. Magro and Wang8 hypothesized that levamisole acting in conjunction with cocaine rather than the effects of levamisole alone is responsible for some of these findings.
Histologically, the features of a vasculopathic process are noted in some patients with the presence of frank vasculitis.1 The vasculopathic component demonstrates vessel dilatation with thrombosis, eosinophilic deposits, and erythrocyte extravasation. Patients with frank vasculitis exhibit fibrinoid vessel wall necrosis and fibrin deposition, extravasated erythrocytes, endothelial cell atypia, and leukocytoclasia.3 Jacob et al3 noted interstitial and perivascular neovascularization in affected tissue, believed to represent one stage in the evolution of medium vessel vasculitis. Intercellular adhesion molecule 1 has been reported in affected vessel walls with endothelial caspase 3 expression and C5b-9 deposition.8 Magro and Wang8 believe the retiform purpura seen in the early stages of some of these patients with LIV represents a thrombotic dynamic with C5b-9 deposition and enhanced apoptosis. Overt vasculitis follows later, subsequent to the effect of ANCA antibodies and upregulated intercellular adhesion molecule 1 expression on vessel walls.
The clinical course of LIV typically is 2 to 3 weeks for lesion resolution; however, normalization of serologies may require 2 to 14 months. Observation and pain control with or without administration of systemic steroids is sufficient for most patients, but skin grafting, wound debridement, cyclosporine, mycophenolate mofetil, and plasmapheresis also have been employed.4,5 Morbidity may be substantive. One report noted LCC to be responsible for 3 cases of pulmonary hemorrhage and acute progression to chronic renal failure in another 2 patients.15 Ching and Smith18 described a patient with 52% total body surface area involvement who required skin grafting, nasal amputation, patellectomy, central upper lip excision, and amputation of the leg above the knee.
Gastrointestinal Presentation
Patients with LIV have been reported to exhibit abdominal pain, but our patient exhibited a rare presentation of visualized gastrointestinal purpura. Although support for a vasculitic/vasculopathic process requires a tissue diagnosis, the endoscopic appearance of gastric vasculitis is similar to that of cutaneous vasculitis.19 Clinicians caring for patients exposed to LCC should bear in mind that the vascular insults associated with LIV are not restricted solely to the skin.
In 2010, two separate reports of cutaneous vasculitic/vasculopathic eruptions in patients with recent exposure to levamisole-contaminated cocaine (LCC) were published in the literature.1,2 Since then, additional reports have been published.3-6 Retiform purpura associated with cocaine use appears to be a similar condition, perhaps lying at one end of the spectrum of LCC-induced cutaneous vascular disease.7,8 Although some patients have been described as having nausea and vomiting,8,9 including one with a sudden drop in hemoglobin to 5.8 g/dL (reference range, 14.0–17.5 g/dL),10 there are no known reported cases of LCC and levamisole-induced vasculopathy in organ systems other than the skin. Herein, we report the case of a patient with levamisole-induced vasculopathy (LIV) demonstrating endoscopic evidence of gastric hemorrhage with features similar to those involving the skin.
Case Report
A 35-year-old woman with a history of hepatitis C, intravenous drug abuse, and bipolar disorder presented to the emergency department with painful necrotic lesions on the head, neck, arms, and legs of several days’ duration. Approximately 1 year prior she had been admitted to the hospital with similar lesions, with eventual partial necrosis of the left earlobe. The patient reported she had last used crack cocaine 3 days prior to the development of the lesions. A urine drug screen was positive for lorazepam, alprazolam, buprenorphine, methadone, tetrahydrocannabinol, and cocaine. She also reported abdominal pain and gastric reflux of recent onset but denied any history of gastrointestinal tract disease. During the previous admission, the patient demonstrated antinuclear antibodies at a titer of greater than 1:160 (normal, <1:40) in a smooth pattern as well as positive perinuclear antineutrophil cytoplasmic antibodies (p-ANCA) and cytoplasmic antineutrophil cytoplasmic antibodies (c-ANCA) and positive cryoglobulins. Physical examination yielded purpuric and hemorrhagic patches and plaques on the nose, bilateral ears (Figure 1A), face (Figure 1B), arms, and legs. Older lesions exhibited evidence of evolving erosion and ulceration. A biopsy of a lesion on the right arm was obtained, demonstrating extensive epidermal necrosis, hemorrhage, fibrin thrombi within dermal blood vessels, fibrinoid mural necrosis, perivascular neutrophils, and leukocytoclasis (Figure 2). These findings were consistent with LIV caused by exposure to LCC. A complete blood cell count was unremarkable. She was started on pain management and was given prednisone to treat the cutaneous eruption. Because of continued reports of epigastric pain and discomfort on swallowing, an upper gastrointestinal endoscopy was performed. Numerous esophageal erosions and gastric submucosal hemorrhages similar to those on the skin were noted (Figure 3). Pathology taken at the time of the endoscopy demonstrated mucosal erosions, but an evaluation for vascular insult was not possible, as submucosal tissue was not obtained. As the skin lesions began to heal, the gastric symptoms gradually subsided, and the patient was released from the hospital after 7 days.
Comment
Levamisole-Contaminated Cocaine
Cocaine is a crystalline alkaloid obtained from the leaves of the coca plant.7 Fifty percent of globally produced cocaine is consumed in the United States.10 There are 2 to 5 million cocaine users in the United States; in 2009, a reported 1.6 million US adults admitted to having used cocaine in the previous month.4,11,12 Cocaine has been known to be cut with similar-appearing substances including lactose and mannitol, though caffeine, acetaminophen, methylphenidate, and other ingredients have been utilized.7
Levamisole is a synthetic imidazothiazole derivative initially developed for use as an immunomodulatory agent in patients with rheumatoid arthritis.4 It was later paired with 5-fluorouracil for administration in patients with carcinomas of the colon and breasts.4,13 In 2000, the drug was withdrawn from the US market for use in humans after an association between levamisole and agranulocytosis was noted in 2.5% to 13% of patients taking the drug for rheumatoid arthritis or as an adjuvant therapy for breast carcinoma.9,12 It still is available for veterinary use as an anthelmintic and is administered to humans in other countries. Levamisole acts as an immunomodulator by enhancing macrophage chemotaxis and upregulating T-cell functions as well as stimulating neutrophil chemotaxis and dendritic cell maturation.4 It also is known to generate autoantibodies including lupus anticoagulant, p-ANCA, c-ANCA, and antinuclear antibodies.7,14 Levamisole is known to exhibit cutaneous reactions. In 1999, Rongioletti et al14 reported 5 children with purpura of the ears who had been given levamisole for pediatric nephrotic syndrome. Involvement of other body areas was noted. Three patients developed lupus anticoagulant antibodies, 3 exhibited p-ANCA antibodies, and 1 was positive for c-ANCA antibodies. The investigators noted an exceptionally long latency period of 12 to 44 months after starting the drug. Histologically a vasculopathic/vasculitic process was noted.14 Direct immunofluorescence studies of affected skin in LIV have demonstrated IgM, IgA, IgG, C3, and fibrin staining of blood vessels.4,15 Anti–human elastase antibodies are considered both sensitive and specific for LIV and serve to differentiate it from cocaine-induced pseudovasculitis.4,7
In April 2008, the New Mexico Department of Health began evaluating several unexplained cases of agranulocytosis and noted that 11 of 21 cases were associated with cocaine use.9 Later that year, public health workers in Alberta and British Columbia, Canada, reported finding traces of levamisole in clinical specimens and drug paraphernalia of cocaine users with agranulocytosis. Officials from the New Mexico Department of Health learned of these findings and investigated the cases, finding 7 of 9 patients with idiopathic agranulocytosis had recent exposure to cocaine. None of the 21 total patients experienced any skin findings. Nausea and vomiting were common symptoms, but abdominal pain was described in only 2 patients from an additional investigation in Washington. Both of these patients used crack cocaine, and one had a positive urine test for levamisole.9
The presence of levamisole initially was detected by the US Drug Enforcement Administration in 2003. By July 2009, 69% of cocaine and 3% of heroin seized by this agency was noted to contain levamisole.16 From 2003 to 2009, the concentration of levamisole contamination rose to 10%.4 A 2011 study found levamisole in 194 of 249 cocaine-positive urine samples.16
It is unclear why cocaine producers add levamisoleto their product. Possibilities include increasing the drug’s bulk or enhancing its stimulatory effects.12 Chang et al17 posited that levamisole increases the stimulatory and euphoric effects of cocaine by increasing dopamine levels in the brain. Additionally, levamisole is metabolized to aminorex, an amphetaminelike hallucinogen that suppresses appetite, in patients with LCC.13 Vagi et al12 interviewed 10 patients who had been hospitalized for agranulocytosis secondary to use of LCC. None were aware of the presence of this additive, suggesting it was not used as a marketing tool.
Cutaneous Vasculopathy
Levamisole-induced vasculopathy (also called levamisole-induced cutaneous vasculopathy11) initially was reported by 2 separate groups in 2010.1,2 Patients typically present with tender purpuric to hemorrhagic papules, plaques, and bullae with an affinity to affect the ears, nose, and face, though other areas of the body can be affected. A pattern of retiform purpura may precede these findings in some patients. Women are disproportionately affected.11 Crack cocaine use is overrepresented in LIV compared to insufflation or snorting of the drug. Affected patients may exhibit systemic symptoms including myalgia, arthralgia, and frank arthritis.10 Additionally, 15% to 80% of patients exhibit positive antinuclear antibodies, anticardiolipin antibodies, lupus anticoagulant antibodies, p-ANCA antibodies, and c-ANCA antibodies. Magro and Wang8 hypothesized that levamisole acting in conjunction with cocaine rather than the effects of levamisole alone is responsible for some of these findings.
Histologically, the features of a vasculopathic process are noted in some patients with the presence of frank vasculitis.1 The vasculopathic component demonstrates vessel dilatation with thrombosis, eosinophilic deposits, and erythrocyte extravasation. Patients with frank vasculitis exhibit fibrinoid vessel wall necrosis and fibrin deposition, extravasated erythrocytes, endothelial cell atypia, and leukocytoclasia.3 Jacob et al3 noted interstitial and perivascular neovascularization in affected tissue, believed to represent one stage in the evolution of medium vessel vasculitis. Intercellular adhesion molecule 1 has been reported in affected vessel walls with endothelial caspase 3 expression and C5b-9 deposition.8 Magro and Wang8 believe the retiform purpura seen in the early stages of some of these patients with LIV represents a thrombotic dynamic with C5b-9 deposition and enhanced apoptosis. Overt vasculitis follows later, subsequent to the effect of ANCA antibodies and upregulated intercellular adhesion molecule 1 expression on vessel walls.
The clinical course of LIV typically is 2 to 3 weeks for lesion resolution; however, normalization of serologies may require 2 to 14 months. Observation and pain control with or without administration of systemic steroids is sufficient for most patients, but skin grafting, wound debridement, cyclosporine, mycophenolate mofetil, and plasmapheresis also have been employed.4,5 Morbidity may be substantive. One report noted LCC to be responsible for 3 cases of pulmonary hemorrhage and acute progression to chronic renal failure in another 2 patients.15 Ching and Smith18 described a patient with 52% total body surface area involvement who required skin grafting, nasal amputation, patellectomy, central upper lip excision, and amputation of the leg above the knee.
Gastrointestinal Presentation
Patients with LIV have been reported to exhibit abdominal pain, but our patient exhibited a rare presentation of visualized gastrointestinal purpura. Although support for a vasculitic/vasculopathic process requires a tissue diagnosis, the endoscopic appearance of gastric vasculitis is similar to that of cutaneous vasculitis.19 Clinicians caring for patients exposed to LCC should bear in mind that the vascular insults associated with LIV are not restricted solely to the skin.
- Waller JM, Feramisco JD, Alberta-Wszolek L, et al. Cocaine-associated retiform purpura and neutropenia: is levamisole the culprit? J Am Acad Dermatol. 2010;63:530-535.
- Bradford M, Rosenberg B, Moreno J, et al. Bilateral necrosis of earlobes and cheeks: another complication of cocaine contaminated with levamisole. Ann Int Med. 2010;152:758-759.
- Jacob RS, Silva CY, Powers JG, et al. Levamisole-induced vasculopathy: a report of 2 cases and a novel histopathologic finding. Am J Dermatopathol. 2012;34:208-213.
- Lee KC, Ladizinski B, Federman DG. Complications associated with use of levamisole-contaminated cocaine: an emerging public health challenge. Mayo Clin Proc. 2012;87:581-586.
- Pavenski K, Vandenberghe H, Jakubovic H, et al. Plasmapheresis and steroid treatment of levamisole-induced vasculopathy and associated skin necrosis in crack/cocaine users. J Cutan Med Surg. 2013;17:123-126.
- Mandrell J, Kranc CL. Prednisone and vardenafil hydrochloride refractory levamisole-induced vasculitis. Cutis. 2016;98:E15-E19.
- Walsh NM, Green PJ, Burlingame RW, et al. Cocaine-related retiform purpura: evidence to incriminate the adulterant, levamisole [published online August 25, 2010]. J Cutan Pathol. 2010;37:1212-1219.
- Magro CM, Wang X. Cocaine-associated retiform purpura: a C5b-9 mediated microangiopathy syndrome associated with enhanced apoptosis and high levels of intercellular adhesion molecule-1 expression. Am J Dermatopathol 2013;35:722-730.
- Centers for Disease Control and Prevention (CDC). Agranulocytosis associated with cocaine use—four states, March 2008-November 2009. MMWR Morb Mortal Wkly Rep. 2009;58:1381-1385.
- Espinoza LR, Alamino RP. Cocaine-induced vasculitis: clinical and immunological spectrum. Curr Rhematol Rep. 2012;14:532-538.
- Arora NP. Cutaneous vasculopathy and neutropenia associated with levamisole-adulterated cocaine. Am J Med Sci. 2013;345:45-51.
- Vagi SJ, Sheikh S, Brackney M, et al. Passive multistate surveillance for neutropenia after of cocaine or heroin possibly contaminated with levamisole. Ann Emerg Med. 2013;61:468-474.
- Lee KC, Ladizinski, Nutan FN. Systemic complications of levamisole toxicity. J Am Acad Dermatol. 2012;67:791-792.
- Rongioletti E, Ghio L, Ginervri E, et al. Purpura of the ears: a distinctive vasculopathy with circulating autoantibodies complicating longer-term treatment with levamisole in children. Br J Dermatol. 1999;140:948-951.
- McGrath MM, Isakova T, Rennke HG, et al. Contaminated cocaine and antineutrophil cytoplasmic antibody-associated diseases. Clin J Am Soc Nephrol. 2011;6:2799-2805.
- Buchanan JA, Heard K, Burbach C, et al. Prevalence of levamisole in urine toxicology screens positive for cocaine in an inner-city hospital. JAMA. 2011;305:1657-1658.
- Chang A, Osterloh J, Thomas J. Levamisole: a dangerous new cocaine adulterant. Clin Pharmacol Ther. 2010;88:408-411.
- Ching JA, Smith DJ. Levamisole-induced necrosis of skin, soft-tissue and bone: case report and review of literature. J Burn Care Res. 2012;33:E1-E5.
- Naruse G, Shimata K. Cutaneous and gastrointestinal purpura. N Engl J Med. 2013;369:1843.
- Waller JM, Feramisco JD, Alberta-Wszolek L, et al. Cocaine-associated retiform purpura and neutropenia: is levamisole the culprit? J Am Acad Dermatol. 2010;63:530-535.
- Bradford M, Rosenberg B, Moreno J, et al. Bilateral necrosis of earlobes and cheeks: another complication of cocaine contaminated with levamisole. Ann Int Med. 2010;152:758-759.
- Jacob RS, Silva CY, Powers JG, et al. Levamisole-induced vasculopathy: a report of 2 cases and a novel histopathologic finding. Am J Dermatopathol. 2012;34:208-213.
- Lee KC, Ladizinski B, Federman DG. Complications associated with use of levamisole-contaminated cocaine: an emerging public health challenge. Mayo Clin Proc. 2012;87:581-586.
- Pavenski K, Vandenberghe H, Jakubovic H, et al. Plasmapheresis and steroid treatment of levamisole-induced vasculopathy and associated skin necrosis in crack/cocaine users. J Cutan Med Surg. 2013;17:123-126.
- Mandrell J, Kranc CL. Prednisone and vardenafil hydrochloride refractory levamisole-induced vasculitis. Cutis. 2016;98:E15-E19.
- Walsh NM, Green PJ, Burlingame RW, et al. Cocaine-related retiform purpura: evidence to incriminate the adulterant, levamisole [published online August 25, 2010]. J Cutan Pathol. 2010;37:1212-1219.
- Magro CM, Wang X. Cocaine-associated retiform purpura: a C5b-9 mediated microangiopathy syndrome associated with enhanced apoptosis and high levels of intercellular adhesion molecule-1 expression. Am J Dermatopathol 2013;35:722-730.
- Centers for Disease Control and Prevention (CDC). Agranulocytosis associated with cocaine use—four states, March 2008-November 2009. MMWR Morb Mortal Wkly Rep. 2009;58:1381-1385.
- Espinoza LR, Alamino RP. Cocaine-induced vasculitis: clinical and immunological spectrum. Curr Rhematol Rep. 2012;14:532-538.
- Arora NP. Cutaneous vasculopathy and neutropenia associated with levamisole-adulterated cocaine. Am J Med Sci. 2013;345:45-51.
- Vagi SJ, Sheikh S, Brackney M, et al. Passive multistate surveillance for neutropenia after of cocaine or heroin possibly contaminated with levamisole. Ann Emerg Med. 2013;61:468-474.
- Lee KC, Ladizinski, Nutan FN. Systemic complications of levamisole toxicity. J Am Acad Dermatol. 2012;67:791-792.
- Rongioletti E, Ghio L, Ginervri E, et al. Purpura of the ears: a distinctive vasculopathy with circulating autoantibodies complicating longer-term treatment with levamisole in children. Br J Dermatol. 1999;140:948-951.
- McGrath MM, Isakova T, Rennke HG, et al. Contaminated cocaine and antineutrophil cytoplasmic antibody-associated diseases. Clin J Am Soc Nephrol. 2011;6:2799-2805.
- Buchanan JA, Heard K, Burbach C, et al. Prevalence of levamisole in urine toxicology screens positive for cocaine in an inner-city hospital. JAMA. 2011;305:1657-1658.
- Chang A, Osterloh J, Thomas J. Levamisole: a dangerous new cocaine adulterant. Clin Pharmacol Ther. 2010;88:408-411.
- Ching JA, Smith DJ. Levamisole-induced necrosis of skin, soft-tissue and bone: case report and review of literature. J Burn Care Res. 2012;33:E1-E5.
- Naruse G, Shimata K. Cutaneous and gastrointestinal purpura. N Engl J Med. 2013;369:1843.
Practice Points
- More than half of the cocaine illicitly consumed in the United States is contaminated with levamisole, a veterinary drug that can incite a vasculitic/vasculopathic response in the skin as well as in other organ systems.
- Because dermatologists often are the specialists to make the diagnosis of levamisole-induced vasculopathy, clinicians should be made aware that consumption of levamisole-contaminated cocaine may affect more than the skin alone.
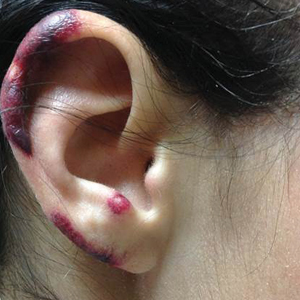
Extramedullary plasmacytoma of the thyroid, refractory to radiation therapy and treated with bortezomib
Plasma cell neoplasms involving tissues other than the bone marrow are known as extramedullary plasmacytoma (EMP).1 EMPs mostly involve the head and neck region.2 Solitary EMP involving only the thyroid gland is very rare.3,4 Because of the limited knowledge about this condition and its rarity, its management can be challenging and is often extrapolated from plasma cell myeloma.5,6 In general, surgery or radiation are considered as front-line therapy.3,5 EMPs usually respond well to radiotherapy with almost complete remission. No definite guidelines outlining the treatment of radio-resistant EMP of the thyroid have yet been published. Data supporting the use of chemotherapy is particularly limited.4,7,8
Here, we describe the case of a 53-year-old woman with a long history of thyroiditis who presented with rapidly worsening symptomatic thyroid enlargement. She was diagnosed with EMP of the thyroid gland that was not amenable to surgery and was refractory to radiotherapy but responded to adjuvant chemotherapy with bortezomib. This report highlights 2 unique aspects of this condition: it focuses on a rare case of EMP and, as far as we know, it reports for the first time on EMP that was resistant to radiotherapy. It also highlights the need for guidelines for the treatment of EMPs.
Case presentation and summary
A 53-year-old woman presented to the emergency department with complaints of difficulty swallowing, hoarseness, and neck pain during the previous 1 month. She had a known history of Hashimoto’s thyroiditis, and an ultrasound scan of her neck 6 years previously had demonstrated diffuse thyromegaly without discrete nodules. On presentation, the patient’s vitals were stable, and a neck examination revealed a firm and enlarged thyroid without any cervical adenopathy. Laboratory investigations revealed a normal complete blood count and comprehensive metabolic panel. She had an elevated thyroid-stimulating hormone level of 13.40 mIU/L (reference range, 0.47-4.68 mIU/L) and normal thyroxine level of 4.5 pmol/L (reference range, 4.5-12.0 pmol/L). A computerized tomography (CT) scan of the neck revealed an enlarged thyroid gland (right lobe length, 10.3 cm; isthmus, 2 cm; left lobe, 8 cm) with a focal area of increased echogenicity in the midpole of the left lobe measuring 9.5 mm × 5.5 mm. The patient was discharged to home with pain medications, and urgent follow-up with an otolaryngologist was arranged. A flexible laryngoscopy was done in the otolaryngology clinic, which revealed retropharyngeal bulging that correlated with the thyromegaly evident on the CT scan.
Because of the patient’s significant symptoms, we decided to proceed with surgery with a clinical diagnosis of likely thyroiditis. A left subtotal thyroidectomy with extension to the superior mediastinum was performed, but a right thyroidectomy could not be done safely. On gross examination, a well-capsulated left lobe with a tan-white, lobulated, soft cut surface was seen. Microscopic examination revealed replacement of thyroid parenchyma with sheets of mature-appearing plasma cells with eccentric round nuclei, abundant eosinophilic cytoplasm without atypia, and few scattered thyroid follicles with lymphoepithelial lesions (Figure 1A). Immunohistochemistry confirmed plasma cells with expression of CD138 (Figure 1B).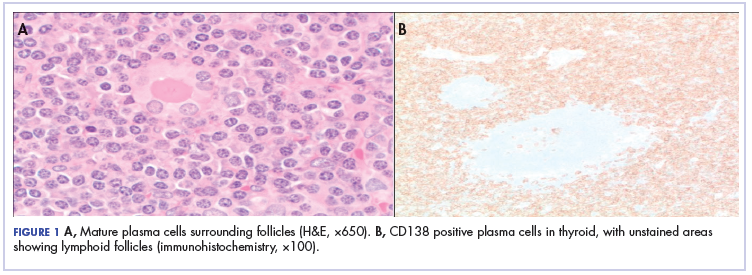
Fluorescence in situ hybridization (FISH) showed that the neoplastic plasma cells contained monotypic kappa immunoglobulin light chain messenger RNA. Clonal immunoglobulin gene rearrangement was detected on polymerase chain reaction. A diagnosis of plasmacytoma of the thyroid gland in a background of thyroiditis was made on the basis of the aforementioned observations.
After that diagnosis, we performed an extensive work-up for plasma cell myeloma. Bone marrow biopsy showed normal maturing trilineage hematopoiesis with scattered mature-appearing plasma cells Figure 2A. Flow cytometry showed a rare (0.2%) population of polytypic plasma cells and was confirmed by CD138 immunohistochemistry. FISH showed proportionate distribution (2-5:1) of kappa and lambda light chains in plasma cells (Figure 2B).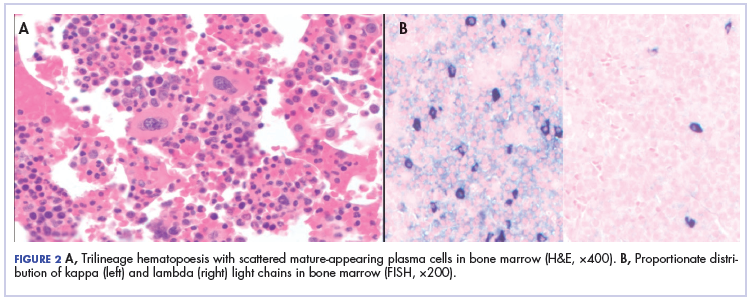
Serum protein electrophoresis showed normal levels of serum proteins with no M spike. Serum total protein was 7.9 g/dL, albumin 5.0 g/dL, α1-globulin 0.3 g/dL, α2-globulin 0.8 g/dL, β-globulin 0.7 g/dL, and γ-globulin 1.6 g/dL, with an albumin–globulin ratio of 1.47. Calcium and β2-microglobulin were also in the normal ranges. Serum-free kappa light chain was found to be elevated (20.9 mg/L; reference range, 3.3-19.4 mg/L). The immunoglobulin G level was also elevated at 3,104 mg/dL (reference range, 700-1,600 mg/dL).
A positron-emission tomographic (PET) scan done 1 month after the surgery showed no other sites of disease except the thyroid. No lytic bone lesions were present. The patient was treated with 50.4 Gy of radiation by external beam radiotherapy to the thyroid in 28 fractions as definitive therapy. Despite treatment with surgery and radiation, she continued to have pain around the neck, and a repeat PET scan 3 months after completion of radiation showed persistent uptake in the thyroid. Because of her refractoriness to radiotherapy, she was started on systemic therapy with a weekly regimen of bortezomib and dexamethasone for 9 cycles. Her symptoms began to resolve, and a repeat PET scan done after completion of chemotherapy showed no evidence of uptake, suggesting adequate response to chemotherapy, and chemotherapy was therefore stopped. The patient was scheduled a regular follow-up in 3 months. Because of continued local symptoms in this follow-up period, the decision was made to perform surgical gland removal, and she underwent completion of
Discussion
Plasma cells are well-differentiated B-lymphocytes that secrete antibodies and provide protective immunity to the human body.9 Plasma cell neoplasms are clonal proliferation of plasma cells, producing monoclonal immunoglobulins. They are of the following different types: plasma cell myeloma, monoclonal gammopathy of unknown significance, immunoglobulin deposition disease, POEMS (polyneuropathy, organomegaly, endocrinopathy, monoclonal protein, skin changes) syndrome, and plasmacytomas, which are divided into 2 types – solitary plasmacytoma of the bone, and extramedullary plasmacytoma (EMP).10 EMP is a rare condition and encompasses 3% to 5% of all plasma cell neoplasms, depending on the study.1,2,5 It is more common in men than in women (2.6:1, respectively), with equal incidence among black and white patients. Median age at diagnosis is 62 years, and it is more common among those aged 40 to 70 years.2,11 The most common sites of occurrence are the respiratory tract, the mouth, and the pharynx, but other sites such as the eyes, brain, skin, and lymph nodes may also be involved.2
EMP involving the thyroid gland is a very rare occurrence, but plasma cell myeloma has been shown to secondarily involve the thyroid.4 Similar to our report, EMP of the thyroid in the setting of thyroiditis has been reported by other authors.3,4 The incidence of EMP occurring in the thyroid varies according to different authors. Wiltshaw found 7 cases involving the thyroid out of 272 cases of EMP.1 Galieni and colleagues reported only 1 case that involved the thyroid out of 46 described cases of EMP.12
El- Siemińska and colleagues showed that levels of interleukin (IL)-6 are elevated in thyroiditis.13 IL-6 promotes monoclonal as well as polyclonal proliferation of plasma cells. Kovalchuk and colleagues showed an increase in EMP in IL-6 transgenic mice, suggesting a pathophysiologic explanation.14
The diagnostic requirements of EMP include the following: histology showing monoclonal plasma cell infiltration in tissue; bone marrow biopsy with normal plasma cell aspirate and biopsy (plasma cells, <5%); no lytic lesions on skeletal survey; no anemia, renal impairment, or hypercalcemia; and absent or low serum M protein.12
Our case fulfilled those criteria.
The treatment options for EMP include surgery, radiotherapy, or a combined approach including both. Usually, EMPs are very sensitive to radiotherapy, and complete remission can be achieved by radiotherapy alone in 80% to 100% of cases.6,11,15 Surgery is considered if the tumor is diffuse or is causing symptoms secondary to pressure on surrounding structures. A combined approach is recommended in cases with incomplete surgical margin or lymph node involvement.5,6
There is limited evidence about and experience with the use of chemotherapy in the treatment of EMP. It has been recommended that chemotherapy be considered in patients with refractory or relapsed disease using the same regimen used in plasma cell myeloma.5 Katodritou and colleagues have reported using bortezomib and dexamethasone without surgery in a solitary gastric plasmacytoma to avoid the toxicity of gastrointestinal irradiation.7 Wei and colleagues treated a patient with EMP in the pancreas with bortezomib and achieved a near-complete remission.8 To our knowledge, there is no documented literature about the treatment of EMP of the thyroid with chemotherapy. In our patient, despite the treatment with surgery and radiation, there was persistent uptake on the PET scan, so we treated her with bortezomib and dexamethasone. Because there is an 11% to 30% risk of progression to multiple myeloma, long-term follow-up is recommended in EMP.11
Conclusions
Solitary EMP of the thyroid gland is a rare condition. Plasma cell myeloma must be ruled out to make a diagnosis. Data on the incidence of EMP and its clinicopathological features are sparse, and literature describing proper guidelines on treatment is limited. It can be treated with radiotherapy, surgery, or a combined approach. There is limited data on the role of chemotherapy; our case adds to the available literature on using myeloma-based therapy in refractory disease and, to our knowledge, is the only case report using this in the literature on cases of EMP of the thyroid. Regular follow-up, even after the disease is in remission, is necessary because of the high risk of progression to plasma cell myeloma.
1. Wiltshaw E. The natural history of extramedullary plasmacytoma and its relation to solitary myeloma of bone and myelomatosis. Medicine (Baltimore). 1976;55(3):217-238.
2. Dores GM, Landgren O, McGlynn KA, Curtis RE, Linet MS, Devesa SS. Plasmacytoma of bone, extramedullary plasmacytoma, and multiple myeloma: incidence and survival in the United States, 1992-2004. Br J Haematol. 2009;144(1):86-94.
3. Kovacs CS, Mant MJ, Nguyen GK, Ginsberg J. Plasma cell lesions of the thyroid: report of a case of solitary plasmacytoma and a review of the literature. Thyroid. 1994;4(1):65-71.
4. Avila A, Villalpando A, Montoya G, Luna MA. Clinical features and differential diagnoses of solitary extramedullary plasmacytoma of the thyroid: a case report. Ann Diagn Pathol. 2009;13(2):119-123.
5. Hughes M, Soutar R, Lucraft H, Owen R, Bird J. Guidelines on the diagnosis and management of solitary plasmacytoma of bone, extramedullary plasmacytoma and multiple solitary plasmacytomas: 2009 update. London, United Kingdom: British Committee for Standards in Haematology; 2009.
6. Weber DM. Solitary bone and extramedullary plasmacytoma. Hematology Am Soc Hematol Educ Program. 2005;373-376.
7. Katodritou E, Kartsios C, Gastari V, et al. Successful treatment of extramedullary gastric plasmacytoma with the combination of bortezomib and dexamethasone: first reported case. Leuk Res. 2008;32(2):339-341.
8. Wei JY, Tong HY, Zhu WF, et al. Bortezomib in treatment of extramedullary plasmacytoma of the pancreas. Hepatobiliary Pancreat Dis Int. 2009;8(3):329-331.
9. Roth K, Oehme L, Zehentmeier S, Zhang Y, Niesner R, Hauser AE. Tracking plasma cell differentiation and survival. Cytometry A. 2014;85(1):15-24.
10. Swerdlow SH, Campo E, Harris NL, et al. WHO classification of tumours of haematopoietic and lymphoid tissues. 4th ed. Lyon, France: International Agency for Research on Cancer; 2008.
11. Alexiou C, Kau RJ, Dietzfelbinger H, et al. Extramedullary plasmacytoma: tumor occurrence and therapeutic concepts. Cancer. 1999;85(11):2305-2314.
12. Galieni P, Cavo M, Pulsoni A, et al. Clinical outcome of extramedullary plasmacytoma. Haematologica. 2000;85(1):47-51.
13. Siemińska L, Wojciechowska C, Kos-Kudła B, et al. Serum concentrations of leptin, adiponectin, and interleukin-6 in postmenopausal women with Hashimoto’s thyroiditis. Endokrynol Pol. 2010;61(1):112-116.
14. Kovalchuk AL, Kim JS, Park SS, et al. IL-6 transgenic mouse model for extraosseous plasmacytoma. Proc Natl Acad Sci US
15. Chao MW, Gibbs P, Wirth A, Quong G, Guiney MJ, Liew KH. Radiotherapy in the management of solitary extramedullary plasmacytoma. Intern Med J. 2005;35(4):211-215.
Plasma cell neoplasms involving tissues other than the bone marrow are known as extramedullary plasmacytoma (EMP).1 EMPs mostly involve the head and neck region.2 Solitary EMP involving only the thyroid gland is very rare.3,4 Because of the limited knowledge about this condition and its rarity, its management can be challenging and is often extrapolated from plasma cell myeloma.5,6 In general, surgery or radiation are considered as front-line therapy.3,5 EMPs usually respond well to radiotherapy with almost complete remission. No definite guidelines outlining the treatment of radio-resistant EMP of the thyroid have yet been published. Data supporting the use of chemotherapy is particularly limited.4,7,8
Here, we describe the case of a 53-year-old woman with a long history of thyroiditis who presented with rapidly worsening symptomatic thyroid enlargement. She was diagnosed with EMP of the thyroid gland that was not amenable to surgery and was refractory to radiotherapy but responded to adjuvant chemotherapy with bortezomib. This report highlights 2 unique aspects of this condition: it focuses on a rare case of EMP and, as far as we know, it reports for the first time on EMP that was resistant to radiotherapy. It also highlights the need for guidelines for the treatment of EMPs.
Case presentation and summary
A 53-year-old woman presented to the emergency department with complaints of difficulty swallowing, hoarseness, and neck pain during the previous 1 month. She had a known history of Hashimoto’s thyroiditis, and an ultrasound scan of her neck 6 years previously had demonstrated diffuse thyromegaly without discrete nodules. On presentation, the patient’s vitals were stable, and a neck examination revealed a firm and enlarged thyroid without any cervical adenopathy. Laboratory investigations revealed a normal complete blood count and comprehensive metabolic panel. She had an elevated thyroid-stimulating hormone level of 13.40 mIU/L (reference range, 0.47-4.68 mIU/L) and normal thyroxine level of 4.5 pmol/L (reference range, 4.5-12.0 pmol/L). A computerized tomography (CT) scan of the neck revealed an enlarged thyroid gland (right lobe length, 10.3 cm; isthmus, 2 cm; left lobe, 8 cm) with a focal area of increased echogenicity in the midpole of the left lobe measuring 9.5 mm × 5.5 mm. The patient was discharged to home with pain medications, and urgent follow-up with an otolaryngologist was arranged. A flexible laryngoscopy was done in the otolaryngology clinic, which revealed retropharyngeal bulging that correlated with the thyromegaly evident on the CT scan.
Because of the patient’s significant symptoms, we decided to proceed with surgery with a clinical diagnosis of likely thyroiditis. A left subtotal thyroidectomy with extension to the superior mediastinum was performed, but a right thyroidectomy could not be done safely. On gross examination, a well-capsulated left lobe with a tan-white, lobulated, soft cut surface was seen. Microscopic examination revealed replacement of thyroid parenchyma with sheets of mature-appearing plasma cells with eccentric round nuclei, abundant eosinophilic cytoplasm without atypia, and few scattered thyroid follicles with lymphoepithelial lesions (Figure 1A). Immunohistochemistry confirmed plasma cells with expression of CD138 (Figure 1B).
Fluorescence in situ hybridization (FISH) showed that the neoplastic plasma cells contained monotypic kappa immunoglobulin light chain messenger RNA. Clonal immunoglobulin gene rearrangement was detected on polymerase chain reaction. A diagnosis of plasmacytoma of the thyroid gland in a background of thyroiditis was made on the basis of the aforementioned observations.
After that diagnosis, we performed an extensive work-up for plasma cell myeloma. Bone marrow biopsy showed normal maturing trilineage hematopoiesis with scattered mature-appearing plasma cells Figure 2A. Flow cytometry showed a rare (0.2%) population of polytypic plasma cells and was confirmed by CD138 immunohistochemistry. FISH showed proportionate distribution (2-5:1) of kappa and lambda light chains in plasma cells (Figure 2B).
Serum protein electrophoresis showed normal levels of serum proteins with no M spike. Serum total protein was 7.9 g/dL, albumin 5.0 g/dL, α1-globulin 0.3 g/dL, α2-globulin 0.8 g/dL, β-globulin 0.7 g/dL, and γ-globulin 1.6 g/dL, with an albumin–globulin ratio of 1.47. Calcium and β2-microglobulin were also in the normal ranges. Serum-free kappa light chain was found to be elevated (20.9 mg/L; reference range, 3.3-19.4 mg/L). The immunoglobulin G level was also elevated at 3,104 mg/dL (reference range, 700-1,600 mg/dL).
A positron-emission tomographic (PET) scan done 1 month after the surgery showed no other sites of disease except the thyroid. No lytic bone lesions were present. The patient was treated with 50.4 Gy of radiation by external beam radiotherapy to the thyroid in 28 fractions as definitive therapy. Despite treatment with surgery and radiation, she continued to have pain around the neck, and a repeat PET scan 3 months after completion of radiation showed persistent uptake in the thyroid. Because of her refractoriness to radiotherapy, she was started on systemic therapy with a weekly regimen of bortezomib and dexamethasone for 9 cycles. Her symptoms began to resolve, and a repeat PET scan done after completion of chemotherapy showed no evidence of uptake, suggesting adequate response to chemotherapy, and chemotherapy was therefore stopped. The patient was scheduled a regular follow-up in 3 months. Because of continued local symptoms in this follow-up period, the decision was made to perform surgical gland removal, and she underwent completion of
Discussion
Plasma cells are well-differentiated B-lymphocytes that secrete antibodies and provide protective immunity to the human body.9 Plasma cell neoplasms are clonal proliferation of plasma cells, producing monoclonal immunoglobulins. They are of the following different types: plasma cell myeloma, monoclonal gammopathy of unknown significance, immunoglobulin deposition disease, POEMS (polyneuropathy, organomegaly, endocrinopathy, monoclonal protein, skin changes) syndrome, and plasmacytomas, which are divided into 2 types – solitary plasmacytoma of the bone, and extramedullary plasmacytoma (EMP).10 EMP is a rare condition and encompasses 3% to 5% of all plasma cell neoplasms, depending on the study.1,2,5 It is more common in men than in women (2.6:1, respectively), with equal incidence among black and white patients. Median age at diagnosis is 62 years, and it is more common among those aged 40 to 70 years.2,11 The most common sites of occurrence are the respiratory tract, the mouth, and the pharynx, but other sites such as the eyes, brain, skin, and lymph nodes may also be involved.2
EMP involving the thyroid gland is a very rare occurrence, but plasma cell myeloma has been shown to secondarily involve the thyroid.4 Similar to our report, EMP of the thyroid in the setting of thyroiditis has been reported by other authors.3,4 The incidence of EMP occurring in the thyroid varies according to different authors. Wiltshaw found 7 cases involving the thyroid out of 272 cases of EMP.1 Galieni and colleagues reported only 1 case that involved the thyroid out of 46 described cases of EMP.12
El- Siemińska and colleagues showed that levels of interleukin (IL)-6 are elevated in thyroiditis.13 IL-6 promotes monoclonal as well as polyclonal proliferation of plasma cells. Kovalchuk and colleagues showed an increase in EMP in IL-6 transgenic mice, suggesting a pathophysiologic explanation.14
The diagnostic requirements of EMP include the following: histology showing monoclonal plasma cell infiltration in tissue; bone marrow biopsy with normal plasma cell aspirate and biopsy (plasma cells, <5%); no lytic lesions on skeletal survey; no anemia, renal impairment, or hypercalcemia; and absent or low serum M protein.12
Our case fulfilled those criteria.
The treatment options for EMP include surgery, radiotherapy, or a combined approach including both. Usually, EMPs are very sensitive to radiotherapy, and complete remission can be achieved by radiotherapy alone in 80% to 100% of cases.6,11,15 Surgery is considered if the tumor is diffuse or is causing symptoms secondary to pressure on surrounding structures. A combined approach is recommended in cases with incomplete surgical margin or lymph node involvement.5,6
There is limited evidence about and experience with the use of chemotherapy in the treatment of EMP. It has been recommended that chemotherapy be considered in patients with refractory or relapsed disease using the same regimen used in plasma cell myeloma.5 Katodritou and colleagues have reported using bortezomib and dexamethasone without surgery in a solitary gastric plasmacytoma to avoid the toxicity of gastrointestinal irradiation.7 Wei and colleagues treated a patient with EMP in the pancreas with bortezomib and achieved a near-complete remission.8 To our knowledge, there is no documented literature about the treatment of EMP of the thyroid with chemotherapy. In our patient, despite the treatment with surgery and radiation, there was persistent uptake on the PET scan, so we treated her with bortezomib and dexamethasone. Because there is an 11% to 30% risk of progression to multiple myeloma, long-term follow-up is recommended in EMP.11
Conclusions
Solitary EMP of the thyroid gland is a rare condition. Plasma cell myeloma must be ruled out to make a diagnosis. Data on the incidence of EMP and its clinicopathological features are sparse, and literature describing proper guidelines on treatment is limited. It can be treated with radiotherapy, surgery, or a combined approach. There is limited data on the role of chemotherapy; our case adds to the available literature on using myeloma-based therapy in refractory disease and, to our knowledge, is the only case report using this in the literature on cases of EMP of the thyroid. Regular follow-up, even after the disease is in remission, is necessary because of the high risk of progression to plasma cell myeloma.
Plasma cell neoplasms involving tissues other than the bone marrow are known as extramedullary plasmacytoma (EMP).1 EMPs mostly involve the head and neck region.2 Solitary EMP involving only the thyroid gland is very rare.3,4 Because of the limited knowledge about this condition and its rarity, its management can be challenging and is often extrapolated from plasma cell myeloma.5,6 In general, surgery or radiation are considered as front-line therapy.3,5 EMPs usually respond well to radiotherapy with almost complete remission. No definite guidelines outlining the treatment of radio-resistant EMP of the thyroid have yet been published. Data supporting the use of chemotherapy is particularly limited.4,7,8
Here, we describe the case of a 53-year-old woman with a long history of thyroiditis who presented with rapidly worsening symptomatic thyroid enlargement. She was diagnosed with EMP of the thyroid gland that was not amenable to surgery and was refractory to radiotherapy but responded to adjuvant chemotherapy with bortezomib. This report highlights 2 unique aspects of this condition: it focuses on a rare case of EMP and, as far as we know, it reports for the first time on EMP that was resistant to radiotherapy. It also highlights the need for guidelines for the treatment of EMPs.
Case presentation and summary
A 53-year-old woman presented to the emergency department with complaints of difficulty swallowing, hoarseness, and neck pain during the previous 1 month. She had a known history of Hashimoto’s thyroiditis, and an ultrasound scan of her neck 6 years previously had demonstrated diffuse thyromegaly without discrete nodules. On presentation, the patient’s vitals were stable, and a neck examination revealed a firm and enlarged thyroid without any cervical adenopathy. Laboratory investigations revealed a normal complete blood count and comprehensive metabolic panel. She had an elevated thyroid-stimulating hormone level of 13.40 mIU/L (reference range, 0.47-4.68 mIU/L) and normal thyroxine level of 4.5 pmol/L (reference range, 4.5-12.0 pmol/L). A computerized tomography (CT) scan of the neck revealed an enlarged thyroid gland (right lobe length, 10.3 cm; isthmus, 2 cm; left lobe, 8 cm) with a focal area of increased echogenicity in the midpole of the left lobe measuring 9.5 mm × 5.5 mm. The patient was discharged to home with pain medications, and urgent follow-up with an otolaryngologist was arranged. A flexible laryngoscopy was done in the otolaryngology clinic, which revealed retropharyngeal bulging that correlated with the thyromegaly evident on the CT scan.
Because of the patient’s significant symptoms, we decided to proceed with surgery with a clinical diagnosis of likely thyroiditis. A left subtotal thyroidectomy with extension to the superior mediastinum was performed, but a right thyroidectomy could not be done safely. On gross examination, a well-capsulated left lobe with a tan-white, lobulated, soft cut surface was seen. Microscopic examination revealed replacement of thyroid parenchyma with sheets of mature-appearing plasma cells with eccentric round nuclei, abundant eosinophilic cytoplasm without atypia, and few scattered thyroid follicles with lymphoepithelial lesions (Figure 1A). Immunohistochemistry confirmed plasma cells with expression of CD138 (Figure 1B).
Fluorescence in situ hybridization (FISH) showed that the neoplastic plasma cells contained monotypic kappa immunoglobulin light chain messenger RNA. Clonal immunoglobulin gene rearrangement was detected on polymerase chain reaction. A diagnosis of plasmacytoma of the thyroid gland in a background of thyroiditis was made on the basis of the aforementioned observations.
After that diagnosis, we performed an extensive work-up for plasma cell myeloma. Bone marrow biopsy showed normal maturing trilineage hematopoiesis with scattered mature-appearing plasma cells Figure 2A. Flow cytometry showed a rare (0.2%) population of polytypic plasma cells and was confirmed by CD138 immunohistochemistry. FISH showed proportionate distribution (2-5:1) of kappa and lambda light chains in plasma cells (Figure 2B).
Serum protein electrophoresis showed normal levels of serum proteins with no M spike. Serum total protein was 7.9 g/dL, albumin 5.0 g/dL, α1-globulin 0.3 g/dL, α2-globulin 0.8 g/dL, β-globulin 0.7 g/dL, and γ-globulin 1.6 g/dL, with an albumin–globulin ratio of 1.47. Calcium and β2-microglobulin were also in the normal ranges. Serum-free kappa light chain was found to be elevated (20.9 mg/L; reference range, 3.3-19.4 mg/L). The immunoglobulin G level was also elevated at 3,104 mg/dL (reference range, 700-1,600 mg/dL).
A positron-emission tomographic (PET) scan done 1 month after the surgery showed no other sites of disease except the thyroid. No lytic bone lesions were present. The patient was treated with 50.4 Gy of radiation by external beam radiotherapy to the thyroid in 28 fractions as definitive therapy. Despite treatment with surgery and radiation, she continued to have pain around the neck, and a repeat PET scan 3 months after completion of radiation showed persistent uptake in the thyroid. Because of her refractoriness to radiotherapy, she was started on systemic therapy with a weekly regimen of bortezomib and dexamethasone for 9 cycles. Her symptoms began to resolve, and a repeat PET scan done after completion of chemotherapy showed no evidence of uptake, suggesting adequate response to chemotherapy, and chemotherapy was therefore stopped. The patient was scheduled a regular follow-up in 3 months. Because of continued local symptoms in this follow-up period, the decision was made to perform surgical gland removal, and she underwent completion of
Discussion
Plasma cells are well-differentiated B-lymphocytes that secrete antibodies and provide protective immunity to the human body.9 Plasma cell neoplasms are clonal proliferation of plasma cells, producing monoclonal immunoglobulins. They are of the following different types: plasma cell myeloma, monoclonal gammopathy of unknown significance, immunoglobulin deposition disease, POEMS (polyneuropathy, organomegaly, endocrinopathy, monoclonal protein, skin changes) syndrome, and plasmacytomas, which are divided into 2 types – solitary plasmacytoma of the bone, and extramedullary plasmacytoma (EMP).10 EMP is a rare condition and encompasses 3% to 5% of all plasma cell neoplasms, depending on the study.1,2,5 It is more common in men than in women (2.6:1, respectively), with equal incidence among black and white patients. Median age at diagnosis is 62 years, and it is more common among those aged 40 to 70 years.2,11 The most common sites of occurrence are the respiratory tract, the mouth, and the pharynx, but other sites such as the eyes, brain, skin, and lymph nodes may also be involved.2
EMP involving the thyroid gland is a very rare occurrence, but plasma cell myeloma has been shown to secondarily involve the thyroid.4 Similar to our report, EMP of the thyroid in the setting of thyroiditis has been reported by other authors.3,4 The incidence of EMP occurring in the thyroid varies according to different authors. Wiltshaw found 7 cases involving the thyroid out of 272 cases of EMP.1 Galieni and colleagues reported only 1 case that involved the thyroid out of 46 described cases of EMP.12
El- Siemińska and colleagues showed that levels of interleukin (IL)-6 are elevated in thyroiditis.13 IL-6 promotes monoclonal as well as polyclonal proliferation of plasma cells. Kovalchuk and colleagues showed an increase in EMP in IL-6 transgenic mice, suggesting a pathophysiologic explanation.14
The diagnostic requirements of EMP include the following: histology showing monoclonal plasma cell infiltration in tissue; bone marrow biopsy with normal plasma cell aspirate and biopsy (plasma cells, <5%); no lytic lesions on skeletal survey; no anemia, renal impairment, or hypercalcemia; and absent or low serum M protein.12
Our case fulfilled those criteria.
The treatment options for EMP include surgery, radiotherapy, or a combined approach including both. Usually, EMPs are very sensitive to radiotherapy, and complete remission can be achieved by radiotherapy alone in 80% to 100% of cases.6,11,15 Surgery is considered if the tumor is diffuse or is causing symptoms secondary to pressure on surrounding structures. A combined approach is recommended in cases with incomplete surgical margin or lymph node involvement.5,6
There is limited evidence about and experience with the use of chemotherapy in the treatment of EMP. It has been recommended that chemotherapy be considered in patients with refractory or relapsed disease using the same regimen used in plasma cell myeloma.5 Katodritou and colleagues have reported using bortezomib and dexamethasone without surgery in a solitary gastric plasmacytoma to avoid the toxicity of gastrointestinal irradiation.7 Wei and colleagues treated a patient with EMP in the pancreas with bortezomib and achieved a near-complete remission.8 To our knowledge, there is no documented literature about the treatment of EMP of the thyroid with chemotherapy. In our patient, despite the treatment with surgery and radiation, there was persistent uptake on the PET scan, so we treated her with bortezomib and dexamethasone. Because there is an 11% to 30% risk of progression to multiple myeloma, long-term follow-up is recommended in EMP.11
Conclusions
Solitary EMP of the thyroid gland is a rare condition. Plasma cell myeloma must be ruled out to make a diagnosis. Data on the incidence of EMP and its clinicopathological features are sparse, and literature describing proper guidelines on treatment is limited. It can be treated with radiotherapy, surgery, or a combined approach. There is limited data on the role of chemotherapy; our case adds to the available literature on using myeloma-based therapy in refractory disease and, to our knowledge, is the only case report using this in the literature on cases of EMP of the thyroid. Regular follow-up, even after the disease is in remission, is necessary because of the high risk of progression to plasma cell myeloma.
1. Wiltshaw E. The natural history of extramedullary plasmacytoma and its relation to solitary myeloma of bone and myelomatosis. Medicine (Baltimore). 1976;55(3):217-238.
2. Dores GM, Landgren O, McGlynn KA, Curtis RE, Linet MS, Devesa SS. Plasmacytoma of bone, extramedullary plasmacytoma, and multiple myeloma: incidence and survival in the United States, 1992-2004. Br J Haematol. 2009;144(1):86-94.
3. Kovacs CS, Mant MJ, Nguyen GK, Ginsberg J. Plasma cell lesions of the thyroid: report of a case of solitary plasmacytoma and a review of the literature. Thyroid. 1994;4(1):65-71.
4. Avila A, Villalpando A, Montoya G, Luna MA. Clinical features and differential diagnoses of solitary extramedullary plasmacytoma of the thyroid: a case report. Ann Diagn Pathol. 2009;13(2):119-123.
5. Hughes M, Soutar R, Lucraft H, Owen R, Bird J. Guidelines on the diagnosis and management of solitary plasmacytoma of bone, extramedullary plasmacytoma and multiple solitary plasmacytomas: 2009 update. London, United Kingdom: British Committee for Standards in Haematology; 2009.
6. Weber DM. Solitary bone and extramedullary plasmacytoma. Hematology Am Soc Hematol Educ Program. 2005;373-376.
7. Katodritou E, Kartsios C, Gastari V, et al. Successful treatment of extramedullary gastric plasmacytoma with the combination of bortezomib and dexamethasone: first reported case. Leuk Res. 2008;32(2):339-341.
8. Wei JY, Tong HY, Zhu WF, et al. Bortezomib in treatment of extramedullary plasmacytoma of the pancreas. Hepatobiliary Pancreat Dis Int. 2009;8(3):329-331.
9. Roth K, Oehme L, Zehentmeier S, Zhang Y, Niesner R, Hauser AE. Tracking plasma cell differentiation and survival. Cytometry A. 2014;85(1):15-24.
10. Swerdlow SH, Campo E, Harris NL, et al. WHO classification of tumours of haematopoietic and lymphoid tissues. 4th ed. Lyon, France: International Agency for Research on Cancer; 2008.
11. Alexiou C, Kau RJ, Dietzfelbinger H, et al. Extramedullary plasmacytoma: tumor occurrence and therapeutic concepts. Cancer. 1999;85(11):2305-2314.
12. Galieni P, Cavo M, Pulsoni A, et al. Clinical outcome of extramedullary plasmacytoma. Haematologica. 2000;85(1):47-51.
13. Siemińska L, Wojciechowska C, Kos-Kudła B, et al. Serum concentrations of leptin, adiponectin, and interleukin-6 in postmenopausal women with Hashimoto’s thyroiditis. Endokrynol Pol. 2010;61(1):112-116.
14. Kovalchuk AL, Kim JS, Park SS, et al. IL-6 transgenic mouse model for extraosseous plasmacytoma. Proc Natl Acad Sci US
15. Chao MW, Gibbs P, Wirth A, Quong G, Guiney MJ, Liew KH. Radiotherapy in the management of solitary extramedullary plasmacytoma. Intern Med J. 2005;35(4):211-215.
1. Wiltshaw E. The natural history of extramedullary plasmacytoma and its relation to solitary myeloma of bone and myelomatosis. Medicine (Baltimore). 1976;55(3):217-238.
2. Dores GM, Landgren O, McGlynn KA, Curtis RE, Linet MS, Devesa SS. Plasmacytoma of bone, extramedullary plasmacytoma, and multiple myeloma: incidence and survival in the United States, 1992-2004. Br J Haematol. 2009;144(1):86-94.
3. Kovacs CS, Mant MJ, Nguyen GK, Ginsberg J. Plasma cell lesions of the thyroid: report of a case of solitary plasmacytoma and a review of the literature. Thyroid. 1994;4(1):65-71.
4. Avila A, Villalpando A, Montoya G, Luna MA. Clinical features and differential diagnoses of solitary extramedullary plasmacytoma of the thyroid: a case report. Ann Diagn Pathol. 2009;13(2):119-123.
5. Hughes M, Soutar R, Lucraft H, Owen R, Bird J. Guidelines on the diagnosis and management of solitary plasmacytoma of bone, extramedullary plasmacytoma and multiple solitary plasmacytomas: 2009 update. London, United Kingdom: British Committee for Standards in Haematology; 2009.
6. Weber DM. Solitary bone and extramedullary plasmacytoma. Hematology Am Soc Hematol Educ Program. 2005;373-376.
7. Katodritou E, Kartsios C, Gastari V, et al. Successful treatment of extramedullary gastric plasmacytoma with the combination of bortezomib and dexamethasone: first reported case. Leuk Res. 2008;32(2):339-341.
8. Wei JY, Tong HY, Zhu WF, et al. Bortezomib in treatment of extramedullary plasmacytoma of the pancreas. Hepatobiliary Pancreat Dis Int. 2009;8(3):329-331.
9. Roth K, Oehme L, Zehentmeier S, Zhang Y, Niesner R, Hauser AE. Tracking plasma cell differentiation and survival. Cytometry A. 2014;85(1):15-24.
10. Swerdlow SH, Campo E, Harris NL, et al. WHO classification of tumours of haematopoietic and lymphoid tissues. 4th ed. Lyon, France: International Agency for Research on Cancer; 2008.
11. Alexiou C, Kau RJ, Dietzfelbinger H, et al. Extramedullary plasmacytoma: tumor occurrence and therapeutic concepts. Cancer. 1999;85(11):2305-2314.
12. Galieni P, Cavo M, Pulsoni A, et al. Clinical outcome of extramedullary plasmacytoma. Haematologica. 2000;85(1):47-51.
13. Siemińska L, Wojciechowska C, Kos-Kudła B, et al. Serum concentrations of leptin, adiponectin, and interleukin-6 in postmenopausal women with Hashimoto’s thyroiditis. Endokrynol Pol. 2010;61(1):112-116.
14. Kovalchuk AL, Kim JS, Park SS, et al. IL-6 transgenic mouse model for extraosseous plasmacytoma. Proc Natl Acad Sci US
15. Chao MW, Gibbs P, Wirth A, Quong G, Guiney MJ, Liew KH. Radiotherapy in the management of solitary extramedullary plasmacytoma. Intern Med J. 2005;35(4):211-215.
Salivary ductal adenocarcinoma with complete response to androgen blockade
Salivary ductal adenocarcinomas make up about 9% of malignant salivary gland tumors and occur mostly in men older than 50 years, with a peak incidence in the sixth and seventh decades. It is the most aggressive of salivary gland tumors and is histologically similar to high-grade, invasive ductal carcinoma of the breast. In all, 65% of patients will die of the disease, and most will experience skin ulceration and nerve palsy.1 With such an aggressive clinical picture, the temptation for many oncologists and patients is to use aggressive cytotoxic chemotherapies. Considering the lack of large trials exploring treatment options in this less-common subtype of salivary gland carcinoma, practice guidelines also recommend the use of aggressive chemotherapies. Unlike other types of malignant cancers of the salivary glands, 70% to 90% of ductal adenocarcinomas express the androgen receptor (AR) by immunohistochemistry.2 There are reported cases of androgen deprivation therapy (ADT) as a successful treatment for salivary ductal adenocarcinomas that express the AR (Table).In 2003, Locati and colleagues reported the case of a man with salivary ductal adenocarcinomas who had a complete response with ADT.3 In 2016, the same group of authors published a retrospective analysis of 17 patients with recurrent or metastatic AR-positive salivary gland cancers who were treated with ADT and reported a 64.7% overall response rate among the patients.4 A 10-patient case series in the Netherlands demonstrated a 50% response rate to ADT plus bicalutamide, including a palliative effect in the form of pain relief.5 A retrospective analysis by Price and colleagues of 5 patients with AR-positive metastatic salivary duct adenocarcinoma showed a 60% response rate to a combination of leuprolide and bicalutamide.6
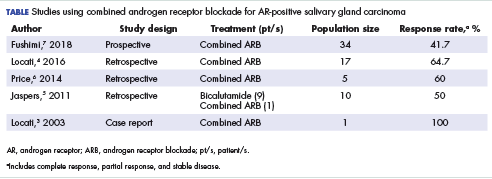
Case presentation and summary
A 91-year-old man was diagnosed with salivary ductal adenocarcinoma of the left parotid gland in September 2013 and underwent left parotidectomy and lymph node dissection, which revealed AJCC stage IVA (pT2 pN3 M0) disease. The following year, in December 2014, he had an enlarging left neck mass that was pathologically confirmed to be recurrent disease, and he underwent left level V neck dissection in February 2015. Five months after surgery, in July 2015, he presented with left neck fullness and new skin nodules, and the results of a biopsy confirmed recurrent disease. Given his relatively asymptomatic state and advanced age, the oncology care team decided to follow the patient without any pharmacologic therapy.
The patient felt relatively well for 11 months but slowly developed increasing pain in the left neck in June 2016. The skin nodules also began to spread inferiorly from his left neck to his upper chest with the development of open sores that wept serous fluid with scab formation (Figure 1). He and his wife lived independently and managed all their own instrumental activities of daily living (IADL). Eventually, the pain in his neck became so severe that it began to interfere with his ability to drive. He declined radiation therapy because of side effects and transportation issues, but he desired something to alleviate the burden of the disease. During a multidisciplinary cancer conference, the staff pathologist and oncologist discussed AR immunohistochemistry to assist with management. In June 2016, the patient’s tumor was found to have AR immunostaining (nuclear pattern) in 100% of cells, and he was treated with combined androgen blockade, consisting of monthly 3.6 mg goserelin injections and daily bicalutamide 50 mg orally.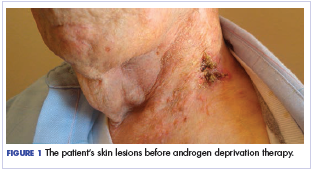
Within a week, the patient noticed that the skin lesions stopped weeping fluid. Within 2 weeks, the pain had begun to resolve. At his formal follow-up visit 11 weeks after starting treatment, he was not taking any pain medications and reported no pain. In addition, his visually apparent disease had almost completely resolved (Figure 2). He was fully able to manage his own IADL and reported a marked increase in satisfaction with the quality of his life.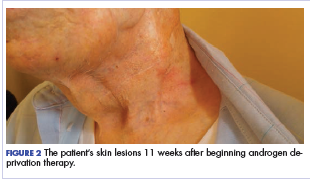
Discussion
The oncology care team clearly defined the goal of care for this patient as palliative and conveyed as such to the patient. The team considered the risks and side effects of cytotoxic chemotherapy agents to be contrary to the patient’s stated primary goal of independence. We selected the combined androgen blockade because it has a low toxicity rate and thus met the primary goals of therapy.
The European Organization for Research and Treatment of Cancer is presently conducting a trial in which cytotoxic chemotherapy is being compared with ADT in AR-positive salivary duct tumors. Findings from a recent prospective, phase-2 trial conducted in Japan suggested that combined AR blockade has similar efficacy and less toxicity than conventional cytotoxic chemotherapy for recurrent and/or metastatic and unresectable locally advanced AR-positive salivary gland carcinoma.7 As more data become available from other studies, it is possible that practice guidelines will be revised to recommend this treatment approach for these cancers.
1. Eveson JW, Thompson LDR. Malignant neoplasms of the salivary glands. In: Thompson LDR, ed. Head and neck pathology. 2nd ed. Philadelphia, PA: Elsevier Inc; 2013:304-305.
2. Luk PP, Weston JD, Yu B, et al. Salivary duct carcinoma: clinicopathologic features, morphologic spectrum, and somatic mutations. Head Neck. 2016;38(suppl 1):E1838-E1847.
3. Locati LD, Quattrone P, Bossi P, Marchianò AV, Cantù G, Licitra L. A complete remission with androgen-deprivation therapy in a recurrent androgen receptor-expressing adenocarcinoma of the parotid gland. Ann Oncol. 2003;14(8):1327-1328.
4. Locati LD, Perrone F, Cortelazzi B, et al. Clinical activity of androgen deprivation therapy in patients with metastatic/relapsed androgen receptor-positive salivary gland cancers. Head Neck. 2016;38(5):724-731.
5. Jaspers HC, Verbist BM, Schoffelen R, et al. Androgen receptor-positive salivary duct carcinoma: a disease entity with promising new treatment options. J Clin Oncol. 2011;29(16):e473-e476.
6. Price KAR, Okuno SH, Molina JR, Garcia JJ. Treatment of metastatic salivary duct carcinoma with combined androgen blockade (CAB) with leuprolide acetate and bicalutamide. Int J Radiat Oncol Biol Phys. 2014;88(2):521-522.
7. Fushimi C, Tada Y, Takahashi H, et al. A prospective phase II study of combined androgen blockade in patients with androgen receptor-positive metastatic or locally advanced unresectable salivary gland carcinoma. Ann Oncol. 2018;29(4):979-984.
Salivary ductal adenocarcinomas make up about 9% of malignant salivary gland tumors and occur mostly in men older than 50 years, with a peak incidence in the sixth and seventh decades. It is the most aggressive of salivary gland tumors and is histologically similar to high-grade, invasive ductal carcinoma of the breast. In all, 65% of patients will die of the disease, and most will experience skin ulceration and nerve palsy.1 With such an aggressive clinical picture, the temptation for many oncologists and patients is to use aggressive cytotoxic chemotherapies. Considering the lack of large trials exploring treatment options in this less-common subtype of salivary gland carcinoma, practice guidelines also recommend the use of aggressive chemotherapies. Unlike other types of malignant cancers of the salivary glands, 70% to 90% of ductal adenocarcinomas express the androgen receptor (AR) by immunohistochemistry.2 There are reported cases of androgen deprivation therapy (ADT) as a successful treatment for salivary ductal adenocarcinomas that express the AR (Table).In 2003, Locati and colleagues reported the case of a man with salivary ductal adenocarcinomas who had a complete response with ADT.3 In 2016, the same group of authors published a retrospective analysis of 17 patients with recurrent or metastatic AR-positive salivary gland cancers who were treated with ADT and reported a 64.7% overall response rate among the patients.4 A 10-patient case series in the Netherlands demonstrated a 50% response rate to ADT plus bicalutamide, including a palliative effect in the form of pain relief.5 A retrospective analysis by Price and colleagues of 5 patients with AR-positive metastatic salivary duct adenocarcinoma showed a 60% response rate to a combination of leuprolide and bicalutamide.6
Case presentation and summary
A 91-year-old man was diagnosed with salivary ductal adenocarcinoma of the left parotid gland in September 2013 and underwent left parotidectomy and lymph node dissection, which revealed AJCC stage IVA (pT2 pN3 M0) disease. The following year, in December 2014, he had an enlarging left neck mass that was pathologically confirmed to be recurrent disease, and he underwent left level V neck dissection in February 2015. Five months after surgery, in July 2015, he presented with left neck fullness and new skin nodules, and the results of a biopsy confirmed recurrent disease. Given his relatively asymptomatic state and advanced age, the oncology care team decided to follow the patient without any pharmacologic therapy.
The patient felt relatively well for 11 months but slowly developed increasing pain in the left neck in June 2016. The skin nodules also began to spread inferiorly from his left neck to his upper chest with the development of open sores that wept serous fluid with scab formation (Figure 1). He and his wife lived independently and managed all their own instrumental activities of daily living (IADL). Eventually, the pain in his neck became so severe that it began to interfere with his ability to drive. He declined radiation therapy because of side effects and transportation issues, but he desired something to alleviate the burden of the disease. During a multidisciplinary cancer conference, the staff pathologist and oncologist discussed AR immunohistochemistry to assist with management. In June 2016, the patient’s tumor was found to have AR immunostaining (nuclear pattern) in 100% of cells, and he was treated with combined androgen blockade, consisting of monthly 3.6 mg goserelin injections and daily bicalutamide 50 mg orally.
Within a week, the patient noticed that the skin lesions stopped weeping fluid. Within 2 weeks, the pain had begun to resolve. At his formal follow-up visit 11 weeks after starting treatment, he was not taking any pain medications and reported no pain. In addition, his visually apparent disease had almost completely resolved (Figure 2). He was fully able to manage his own IADL and reported a marked increase in satisfaction with the quality of his life.
Discussion
The oncology care team clearly defined the goal of care for this patient as palliative and conveyed as such to the patient. The team considered the risks and side effects of cytotoxic chemotherapy agents to be contrary to the patient’s stated primary goal of independence. We selected the combined androgen blockade because it has a low toxicity rate and thus met the primary goals of therapy.
The European Organization for Research and Treatment of Cancer is presently conducting a trial in which cytotoxic chemotherapy is being compared with ADT in AR-positive salivary duct tumors. Findings from a recent prospective, phase-2 trial conducted in Japan suggested that combined AR blockade has similar efficacy and less toxicity than conventional cytotoxic chemotherapy for recurrent and/or metastatic and unresectable locally advanced AR-positive salivary gland carcinoma.7 As more data become available from other studies, it is possible that practice guidelines will be revised to recommend this treatment approach for these cancers.
Salivary ductal adenocarcinomas make up about 9% of malignant salivary gland tumors and occur mostly in men older than 50 years, with a peak incidence in the sixth and seventh decades. It is the most aggressive of salivary gland tumors and is histologically similar to high-grade, invasive ductal carcinoma of the breast. In all, 65% of patients will die of the disease, and most will experience skin ulceration and nerve palsy.1 With such an aggressive clinical picture, the temptation for many oncologists and patients is to use aggressive cytotoxic chemotherapies. Considering the lack of large trials exploring treatment options in this less-common subtype of salivary gland carcinoma, practice guidelines also recommend the use of aggressive chemotherapies. Unlike other types of malignant cancers of the salivary glands, 70% to 90% of ductal adenocarcinomas express the androgen receptor (AR) by immunohistochemistry.2 There are reported cases of androgen deprivation therapy (ADT) as a successful treatment for salivary ductal adenocarcinomas that express the AR (Table).In 2003, Locati and colleagues reported the case of a man with salivary ductal adenocarcinomas who had a complete response with ADT.3 In 2016, the same group of authors published a retrospective analysis of 17 patients with recurrent or metastatic AR-positive salivary gland cancers who were treated with ADT and reported a 64.7% overall response rate among the patients.4 A 10-patient case series in the Netherlands demonstrated a 50% response rate to ADT plus bicalutamide, including a palliative effect in the form of pain relief.5 A retrospective analysis by Price and colleagues of 5 patients with AR-positive metastatic salivary duct adenocarcinoma showed a 60% response rate to a combination of leuprolide and bicalutamide.6
Case presentation and summary
A 91-year-old man was diagnosed with salivary ductal adenocarcinoma of the left parotid gland in September 2013 and underwent left parotidectomy and lymph node dissection, which revealed AJCC stage IVA (pT2 pN3 M0) disease. The following year, in December 2014, he had an enlarging left neck mass that was pathologically confirmed to be recurrent disease, and he underwent left level V neck dissection in February 2015. Five months after surgery, in July 2015, he presented with left neck fullness and new skin nodules, and the results of a biopsy confirmed recurrent disease. Given his relatively asymptomatic state and advanced age, the oncology care team decided to follow the patient without any pharmacologic therapy.
The patient felt relatively well for 11 months but slowly developed increasing pain in the left neck in June 2016. The skin nodules also began to spread inferiorly from his left neck to his upper chest with the development of open sores that wept serous fluid with scab formation (Figure 1). He and his wife lived independently and managed all their own instrumental activities of daily living (IADL). Eventually, the pain in his neck became so severe that it began to interfere with his ability to drive. He declined radiation therapy because of side effects and transportation issues, but he desired something to alleviate the burden of the disease. During a multidisciplinary cancer conference, the staff pathologist and oncologist discussed AR immunohistochemistry to assist with management. In June 2016, the patient’s tumor was found to have AR immunostaining (nuclear pattern) in 100% of cells, and he was treated with combined androgen blockade, consisting of monthly 3.6 mg goserelin injections and daily bicalutamide 50 mg orally.
Within a week, the patient noticed that the skin lesions stopped weeping fluid. Within 2 weeks, the pain had begun to resolve. At his formal follow-up visit 11 weeks after starting treatment, he was not taking any pain medications and reported no pain. In addition, his visually apparent disease had almost completely resolved (Figure 2). He was fully able to manage his own IADL and reported a marked increase in satisfaction with the quality of his life.
Discussion
The oncology care team clearly defined the goal of care for this patient as palliative and conveyed as such to the patient. The team considered the risks and side effects of cytotoxic chemotherapy agents to be contrary to the patient’s stated primary goal of independence. We selected the combined androgen blockade because it has a low toxicity rate and thus met the primary goals of therapy.
The European Organization for Research and Treatment of Cancer is presently conducting a trial in which cytotoxic chemotherapy is being compared with ADT in AR-positive salivary duct tumors. Findings from a recent prospective, phase-2 trial conducted in Japan suggested that combined AR blockade has similar efficacy and less toxicity than conventional cytotoxic chemotherapy for recurrent and/or metastatic and unresectable locally advanced AR-positive salivary gland carcinoma.7 As more data become available from other studies, it is possible that practice guidelines will be revised to recommend this treatment approach for these cancers.
1. Eveson JW, Thompson LDR. Malignant neoplasms of the salivary glands. In: Thompson LDR, ed. Head and neck pathology. 2nd ed. Philadelphia, PA: Elsevier Inc; 2013:304-305.
2. Luk PP, Weston JD, Yu B, et al. Salivary duct carcinoma: clinicopathologic features, morphologic spectrum, and somatic mutations. Head Neck. 2016;38(suppl 1):E1838-E1847.
3. Locati LD, Quattrone P, Bossi P, Marchianò AV, Cantù G, Licitra L. A complete remission with androgen-deprivation therapy in a recurrent androgen receptor-expressing adenocarcinoma of the parotid gland. Ann Oncol. 2003;14(8):1327-1328.
4. Locati LD, Perrone F, Cortelazzi B, et al. Clinical activity of androgen deprivation therapy in patients with metastatic/relapsed androgen receptor-positive salivary gland cancers. Head Neck. 2016;38(5):724-731.
5. Jaspers HC, Verbist BM, Schoffelen R, et al. Androgen receptor-positive salivary duct carcinoma: a disease entity with promising new treatment options. J Clin Oncol. 2011;29(16):e473-e476.
6. Price KAR, Okuno SH, Molina JR, Garcia JJ. Treatment of metastatic salivary duct carcinoma with combined androgen blockade (CAB) with leuprolide acetate and bicalutamide. Int J Radiat Oncol Biol Phys. 2014;88(2):521-522.
7. Fushimi C, Tada Y, Takahashi H, et al. A prospective phase II study of combined androgen blockade in patients with androgen receptor-positive metastatic or locally advanced unresectable salivary gland carcinoma. Ann Oncol. 2018;29(4):979-984.
1. Eveson JW, Thompson LDR. Malignant neoplasms of the salivary glands. In: Thompson LDR, ed. Head and neck pathology. 2nd ed. Philadelphia, PA: Elsevier Inc; 2013:304-305.
2. Luk PP, Weston JD, Yu B, et al. Salivary duct carcinoma: clinicopathologic features, morphologic spectrum, and somatic mutations. Head Neck. 2016;38(suppl 1):E1838-E1847.
3. Locati LD, Quattrone P, Bossi P, Marchianò AV, Cantù G, Licitra L. A complete remission with androgen-deprivation therapy in a recurrent androgen receptor-expressing adenocarcinoma of the parotid gland. Ann Oncol. 2003;14(8):1327-1328.
4. Locati LD, Perrone F, Cortelazzi B, et al. Clinical activity of androgen deprivation therapy in patients with metastatic/relapsed androgen receptor-positive salivary gland cancers. Head Neck. 2016;38(5):724-731.
5. Jaspers HC, Verbist BM, Schoffelen R, et al. Androgen receptor-positive salivary duct carcinoma: a disease entity with promising new treatment options. J Clin Oncol. 2011;29(16):e473-e476.
6. Price KAR, Okuno SH, Molina JR, Garcia JJ. Treatment of metastatic salivary duct carcinoma with combined androgen blockade (CAB) with leuprolide acetate and bicalutamide. Int J Radiat Oncol Biol Phys. 2014;88(2):521-522.
7. Fushimi C, Tada Y, Takahashi H, et al. A prospective phase II study of combined androgen blockade in patients with androgen receptor-positive metastatic or locally advanced unresectable salivary gland carcinoma. Ann Oncol. 2018;29(4):979-984.
Metastatic Vulvovaginal Crohn Disease in the Setting of Well-Controlled Intestinal Disease
The cutaneous manifestations of Crohn disease (CD) are varied, including pyoderma gangrenosum, erythema nodosum, and metastatic CD (MCD). First described by Parks et al,1 MCD is defined as the occurrence of granulomatous lesions at a skin site distant from the gastrointestinal tract.1-20 Metastatic CD presents a diagnostic challenge because it is a rare component in the spectrum of inflammatory bowel disease complications, and many physicians are unaware of its existence. It may precede, coincide with, or develop after the diagnosis of intestinal disease.2-5 Vulvoperineal involvement is particularly problematic because a multitude of other, more likely disease processes are considered first. Typically it is initially diagnosed as a presumed infection prompting reflexive treatment with antivirals, antifungals, and antibiotics. Patients may experience symptoms for years prior to correct diagnosis and institution of proper therapy. A variety of clinical presentations have been described, including nonspecific pain and swelling, erythematous papules and plaques, and nonhealing ulcers. Skin biopsy characteristically confirms the diagnosis and reveals dermal noncaseating granulomas. Multiple oral and parenteral therapies are available, with surgical intervention reserved for resistant cases. We present a case of vulvovaginal MCD in the setting of well-controlled intestinal disease. We also provide a review of the literature regarding genital CD and emphasize the need to keep MCD in the differential of vulvoperineal pathology.
Case Report
A 29-year-old woman was referred to the dermatology clinic with vulvar pain, swelling, and pruritus of 14 months’ duration. Her medical history was remarkable for CD following a colectomy with colostomy. Prior therapies included methotrexate with infliximab for 5 years followed by a 2-year regimen with adalimumab, which induced remission of the intestinal disease.
The patient previously had taken a variety of topical and oral antimicrobials based on treatment from a primary care physician because fungal, bacterial, and viral infections initially were suspected; however, the vulvar disease persisted, and drug-induced immunosuppression was considered to be an underlying factor. Thus, adalimumab was discontinued. Despite elimination of the biologic, the vulvar disease progressed, which prompted referral to the dermatology clinic.
Physical examination revealed diffuse vulvar edema with overlying erythema and scale (Figure 1A). Upon closer inspection, scattered violaceous papules atop a backdrop of lichenification were evident, imparting a cobblestone appearance (Figure 1B). Additionally, a fissure was present on the gluteal cleft. Biopsy from the left labia majora demonstrated well-formed granulomas within a fibrotic reticular dermis (Figures 2A and 2B). The granulomas consisted of both mononucleated and multinucleated histiocytes, rimmed peripherally by lymphocytes and plasma cells (Figure 2C). Periodic acid–Schiff–diastase and acid-fast bacilli stains as well as polarizing microscopy were negative.
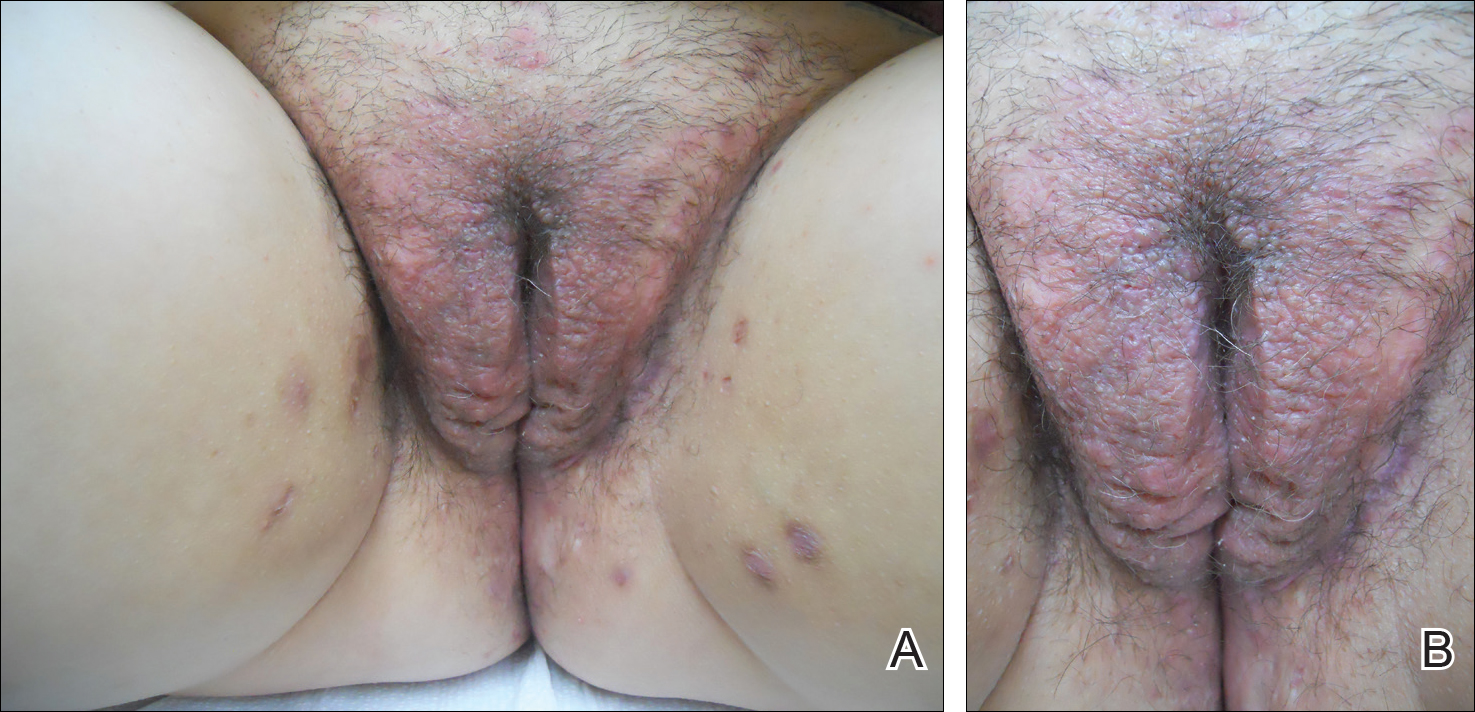
Given the patient’s history, a diagnosis of vulvoperineal MCD was rendered. The patient was started on oral metronidazole 250 mg 3 times daily with topical fluocinonide and tacrolimus. She responded well to this treatment regimen and was referred back to the gastroenterologist for management of the intestinal disease.
Comment
Crohn disease is an idiopathic chronic inflammatory condition that primarily affects the gastrointestinal tract, anywhere from the mouth to the anus. It is characterized by transmural inflammation and fissures that can extend beyond the muscularis propria.4,6 Extraintestinal manifestations are common.3
Cutaneous CD often presents as perianal, perifistular, or peristomal inflammation or ulceration.7 Other skin manifestations include pyoderma gangrenosum, erythema nodosum, erythema multiforme, epidermolysis bullosa acquisita, and palmar erythema.7 Metastatic CD involves skin noncontiguous with the gastrointestinal tract1-20 and may involve any portion of the cutis. Although rare, MCD is the typical etiology underlying vulvar CD.8
Approximately 20% of MCD patients have cutaneous lesions without a history of gastrointestinal disease. More than half of cases in adults and approximately two-thirds in children involve the genitalia. Although more common in adults, vulvar involvement has been reported in children as young as 6 years of age.2 Diagnosis is especially challenging when bowel symptoms are absent; those patients should be evaluated and followed for subsequent intestinal involvement.6
Clinically, symptoms may include general discomfort, pain, pruritus, and dyspareunia. Psychosocial and sexual dysfunction are prevalent and also should be addressed.9 Depending on the stage of the disease, physical examination may reveal erythema, edema, papules, pustules, nodules, condylomatous lesions, abscesses, fissures, fistulas, ulceration, acrochordons, and scarring.2-6,10,11
A host of infections (ie, mycobacterial, actinomycosis, deep fungal, sexually transmitted, schistosomiasis), inflammatory conditions (ie, sarcoid, hidradenitis suppurativa), foreign body reactions, Melkersson-Rosenthal syndrome, and sexual abuse should be included in the differential diagnosis.2,6,10-12 Once infection, sarcoid, and foreign body reaction have been ruled out, noncaseating granulomas in skin are highly suggestive of CD.7
Histopathologic findings of MCD reveal myriad morphological reaction patterns,5,13 including high-grade dysplasia and carcinoma of the vulva; therefore, it may be imprudent to withhold diagnosis based on the absence of the historically pathognomonic noncaseating granulomas.5
The etiopathogenesis of MCD remains an enigma. Dermatopathologic examinations consistently reveal a vascular injury syndrome,13 implicating a possible circulatory system contribution via deposition of immune complexes or antigens in skin.7 Bacterial infection has been implicated in the intestinal manifestations of CD; however, failure to detect microbial ribosomal RNA in MCD biopsies refutes theories of hematogenous spread of microbes.13 Another plausible explanation is that antibodies are formed to conserved microbial epitopes following loss of tolerance to gut flora, which results in an excessive immunologic response at distinct sites in susceptible individuals.13 A T-lymphocyte–mediated type IV hypersensitivity reaction also has been proposed via cross-reactivity of lymphocytes, with skin antigens precipitating extraintestinal granuloma formation and vascular injury.3 Clearly, further investigation is needed.
Magnetic resonanance imaging can identify the extent and anatomy of intestinal and pelvic disease and can assist in the diagnosis of vulvar CD.10,11,14 For these reasons, some experts propose that imaging should be instituted prior to therapy,12,15,16 especially when direct extension is suspected.17
Treatment is challenging and often involves collaboration among several specialties.12 Many treatment options exist because therapeutic responses vary and genital MCD is frequently recalcitrant to therapy.4 Medical therapy includes antibiotics such as metronidazole, corticosteroids (ie, topical, intralesional, systemic), and immune modulators (eg, azathioprine, 6-mercaptopurine, cyclosporine, methotrexate, mycophenolate mofetil, tumor necrosis factor α inhibitors).2,3,6,10,16,18 Thalidomide has been used for refractory cases.19 These treatments can be used alone or in combination. Patients should be monitored for side effects and informed that many treatment regimens may be required before a sustained response is achieved.4,16,18 Surgery is reserved for the most resistant cases. Extensive radical excision of the involved area is the best approach, as limited local excision often is followed by recurrence.20
Conclusion
Our case highlights that vulvar CD can develop in the setting of well-controlled intestinal disease. Vulvoperineal CD should be considered in the differential diagnosis of chronic vulvar pain, swelling, and pruritus, especially in cases resistant to standard therapies and regardless of whether or not gastrointestinal tract symptoms are present. Physicians must be cognizant that vulvar signs and symptoms may precede, coincide with, or follow the diagnosis of intestinal CD. Increased awareness of this entity may facilitate its early recognition and prompt more timely treatment among women with vulvar disease caused by MCD.
- Parks AG, Morson BC, Pegum JS. Crohn’s disease with cutaneous involvement. Proc R Soc Med. 1965;58:241-242.
- Ploysangam T, Heubi JE, Eisen D, et al. Cutaneous Crohn’s disease in children. J Am Acad Dermatol. 1997;36:697-704.
- Palamaras I, El-Jabbour J, Pietropaolo N, et al. Metastatic Crohn’s disease: a review. J Eur Acad Dermatol Venereol. 2008;22:1033-1043.
- Leu S, Sun PK, Collyer J, et al. Clinical spectrum of vulvar metastatic Crohn’s disease. Dig Dis Sci. 2009;54:1565-1571.
- Foo WC, Papalas JA, Robboy SJ, et al. Vulvar manifestations of Crohn’s disease. Am J Dermatopathol. 2001;33:588-593.
- Urbanek M, Neill SM, McKee PH. Vulval Crohn’s disease: difficulties in diagnosis. Clin Exp Dermatol. 1996;21:211-214.
- Burgdorf W. Cutaneous manifestations of Crohn’s disease. J Am Acad Dermatol. 1981;5:689-695.
- Andreani SM, Ratnasingham K, Dang HH, et al. Crohn’s disease of the vulva. Int J Surg. 2010;8:2-5.
- Feller E, Ribaudo S, Jackson N. Gynecologic aspects of Crohn’s disease. Am Fam Physician. 2001;64:1725-1728.
- Corbett SL, Walsh CM, Spitzer RF, et al. Vulvar inflammation as the only clinical manifestation of Crohn disease in an 8-year-old girl [published online May 10, 2010]. Pediatrics. 2010;125:E1518-E1522.
- Tonolini M, Villa C, Campari A, et al. Common and unusual urogenital Crohn’s disease complications: spectrum of cross-sectional imaging findings. Abdom Imaging. 2013;38:32-41.
- Bhaduri S, Jenkinson S, Lewis F. Vulval Crohn’s disease—a multi-specialty approach. Int J STD AIDS. 2005;16:512-514.
- Crowson AN, Nuovo GJ, Mihm MC Jr, et al. Cutaneous manifestations of Crohn’s disease, its spectrum, and its pathogenesis: intracellular consensus bacterial 16S rRNA is associated with the gastrointestinal but not the cutaneous manifestations of Crohn’s disease. Hum Pathol. 2003;34:1185-1192.
- Pai D, Dillman JR, Mahani MG, et al. MRI of vulvar Crohn disease. Pediatr Radiol. 2011;41:537-541.
- Madnani NA, Desai D, Gandhi N, et al. Isolated Crohn’s disease of the vulva. Indian J Dermatol Venereol Leprol. 2011;77:342-344.
- Makhija S, Trotter M, Wagner E, et al. Refractory Crohn’s disease of the vulva treated with infliximab: a case report. Can J Gastroenterol. 2007;21:835-837.
- Fahmy N, Kalidindi M, Khan R. Direct colo-labial Crohn’s abscess mimicking bartholinitis. Am J Obstret Gynecol. 2010;30:741-742.
- Preston PW, Hudson N, Lewis FM. Treatment of vulval Crohn’s disease with infliximab. Clin Exp Derm. 2006;31:378-380.
- Kolivras A, De Maubeuge J, André J, et al. Thalidomide in refractory vulvar ulcerations associated with Crohn’s disease. Dermatology. 2003;206:381-383.
- Kao MS, Paulson JD, Askin FB. Crohn’s disease of the vulva. Obstet Gynecol. 1975;46:329-333.
The cutaneous manifestations of Crohn disease (CD) are varied, including pyoderma gangrenosum, erythema nodosum, and metastatic CD (MCD). First described by Parks et al,1 MCD is defined as the occurrence of granulomatous lesions at a skin site distant from the gastrointestinal tract.1-20 Metastatic CD presents a diagnostic challenge because it is a rare component in the spectrum of inflammatory bowel disease complications, and many physicians are unaware of its existence. It may precede, coincide with, or develop after the diagnosis of intestinal disease.2-5 Vulvoperineal involvement is particularly problematic because a multitude of other, more likely disease processes are considered first. Typically it is initially diagnosed as a presumed infection prompting reflexive treatment with antivirals, antifungals, and antibiotics. Patients may experience symptoms for years prior to correct diagnosis and institution of proper therapy. A variety of clinical presentations have been described, including nonspecific pain and swelling, erythematous papules and plaques, and nonhealing ulcers. Skin biopsy characteristically confirms the diagnosis and reveals dermal noncaseating granulomas. Multiple oral and parenteral therapies are available, with surgical intervention reserved for resistant cases. We present a case of vulvovaginal MCD in the setting of well-controlled intestinal disease. We also provide a review of the literature regarding genital CD and emphasize the need to keep MCD in the differential of vulvoperineal pathology.
Case Report
A 29-year-old woman was referred to the dermatology clinic with vulvar pain, swelling, and pruritus of 14 months’ duration. Her medical history was remarkable for CD following a colectomy with colostomy. Prior therapies included methotrexate with infliximab for 5 years followed by a 2-year regimen with adalimumab, which induced remission of the intestinal disease.
The patient previously had taken a variety of topical and oral antimicrobials based on treatment from a primary care physician because fungal, bacterial, and viral infections initially were suspected; however, the vulvar disease persisted, and drug-induced immunosuppression was considered to be an underlying factor. Thus, adalimumab was discontinued. Despite elimination of the biologic, the vulvar disease progressed, which prompted referral to the dermatology clinic.
Physical examination revealed diffuse vulvar edema with overlying erythema and scale (Figure 1A). Upon closer inspection, scattered violaceous papules atop a backdrop of lichenification were evident, imparting a cobblestone appearance (Figure 1B). Additionally, a fissure was present on the gluteal cleft. Biopsy from the left labia majora demonstrated well-formed granulomas within a fibrotic reticular dermis (Figures 2A and 2B). The granulomas consisted of both mononucleated and multinucleated histiocytes, rimmed peripherally by lymphocytes and plasma cells (Figure 2C). Periodic acid–Schiff–diastase and acid-fast bacilli stains as well as polarizing microscopy were negative.
Given the patient’s history, a diagnosis of vulvoperineal MCD was rendered. The patient was started on oral metronidazole 250 mg 3 times daily with topical fluocinonide and tacrolimus. She responded well to this treatment regimen and was referred back to the gastroenterologist for management of the intestinal disease.
Comment
Crohn disease is an idiopathic chronic inflammatory condition that primarily affects the gastrointestinal tract, anywhere from the mouth to the anus. It is characterized by transmural inflammation and fissures that can extend beyond the muscularis propria.4,6 Extraintestinal manifestations are common.3
Cutaneous CD often presents as perianal, perifistular, or peristomal inflammation or ulceration.7 Other skin manifestations include pyoderma gangrenosum, erythema nodosum, erythema multiforme, epidermolysis bullosa acquisita, and palmar erythema.7 Metastatic CD involves skin noncontiguous with the gastrointestinal tract1-20 and may involve any portion of the cutis. Although rare, MCD is the typical etiology underlying vulvar CD.8
Approximately 20% of MCD patients have cutaneous lesions without a history of gastrointestinal disease. More than half of cases in adults and approximately two-thirds in children involve the genitalia. Although more common in adults, vulvar involvement has been reported in children as young as 6 years of age.2 Diagnosis is especially challenging when bowel symptoms are absent; those patients should be evaluated and followed for subsequent intestinal involvement.6
Clinically, symptoms may include general discomfort, pain, pruritus, and dyspareunia. Psychosocial and sexual dysfunction are prevalent and also should be addressed.9 Depending on the stage of the disease, physical examination may reveal erythema, edema, papules, pustules, nodules, condylomatous lesions, abscesses, fissures, fistulas, ulceration, acrochordons, and scarring.2-6,10,11
A host of infections (ie, mycobacterial, actinomycosis, deep fungal, sexually transmitted, schistosomiasis), inflammatory conditions (ie, sarcoid, hidradenitis suppurativa), foreign body reactions, Melkersson-Rosenthal syndrome, and sexual abuse should be included in the differential diagnosis.2,6,10-12 Once infection, sarcoid, and foreign body reaction have been ruled out, noncaseating granulomas in skin are highly suggestive of CD.7
Histopathologic findings of MCD reveal myriad morphological reaction patterns,5,13 including high-grade dysplasia and carcinoma of the vulva; therefore, it may be imprudent to withhold diagnosis based on the absence of the historically pathognomonic noncaseating granulomas.5
The etiopathogenesis of MCD remains an enigma. Dermatopathologic examinations consistently reveal a vascular injury syndrome,13 implicating a possible circulatory system contribution via deposition of immune complexes or antigens in skin.7 Bacterial infection has been implicated in the intestinal manifestations of CD; however, failure to detect microbial ribosomal RNA in MCD biopsies refutes theories of hematogenous spread of microbes.13 Another plausible explanation is that antibodies are formed to conserved microbial epitopes following loss of tolerance to gut flora, which results in an excessive immunologic response at distinct sites in susceptible individuals.13 A T-lymphocyte–mediated type IV hypersensitivity reaction also has been proposed via cross-reactivity of lymphocytes, with skin antigens precipitating extraintestinal granuloma formation and vascular injury.3 Clearly, further investigation is needed.
Magnetic resonanance imaging can identify the extent and anatomy of intestinal and pelvic disease and can assist in the diagnosis of vulvar CD.10,11,14 For these reasons, some experts propose that imaging should be instituted prior to therapy,12,15,16 especially when direct extension is suspected.17
Treatment is challenging and often involves collaboration among several specialties.12 Many treatment options exist because therapeutic responses vary and genital MCD is frequently recalcitrant to therapy.4 Medical therapy includes antibiotics such as metronidazole, corticosteroids (ie, topical, intralesional, systemic), and immune modulators (eg, azathioprine, 6-mercaptopurine, cyclosporine, methotrexate, mycophenolate mofetil, tumor necrosis factor α inhibitors).2,3,6,10,16,18 Thalidomide has been used for refractory cases.19 These treatments can be used alone or in combination. Patients should be monitored for side effects and informed that many treatment regimens may be required before a sustained response is achieved.4,16,18 Surgery is reserved for the most resistant cases. Extensive radical excision of the involved area is the best approach, as limited local excision often is followed by recurrence.20
Conclusion
Our case highlights that vulvar CD can develop in the setting of well-controlled intestinal disease. Vulvoperineal CD should be considered in the differential diagnosis of chronic vulvar pain, swelling, and pruritus, especially in cases resistant to standard therapies and regardless of whether or not gastrointestinal tract symptoms are present. Physicians must be cognizant that vulvar signs and symptoms may precede, coincide with, or follow the diagnosis of intestinal CD. Increased awareness of this entity may facilitate its early recognition and prompt more timely treatment among women with vulvar disease caused by MCD.
The cutaneous manifestations of Crohn disease (CD) are varied, including pyoderma gangrenosum, erythema nodosum, and metastatic CD (MCD). First described by Parks et al,1 MCD is defined as the occurrence of granulomatous lesions at a skin site distant from the gastrointestinal tract.1-20 Metastatic CD presents a diagnostic challenge because it is a rare component in the spectrum of inflammatory bowel disease complications, and many physicians are unaware of its existence. It may precede, coincide with, or develop after the diagnosis of intestinal disease.2-5 Vulvoperineal involvement is particularly problematic because a multitude of other, more likely disease processes are considered first. Typically it is initially diagnosed as a presumed infection prompting reflexive treatment with antivirals, antifungals, and antibiotics. Patients may experience symptoms for years prior to correct diagnosis and institution of proper therapy. A variety of clinical presentations have been described, including nonspecific pain and swelling, erythematous papules and plaques, and nonhealing ulcers. Skin biopsy characteristically confirms the diagnosis and reveals dermal noncaseating granulomas. Multiple oral and parenteral therapies are available, with surgical intervention reserved for resistant cases. We present a case of vulvovaginal MCD in the setting of well-controlled intestinal disease. We also provide a review of the literature regarding genital CD and emphasize the need to keep MCD in the differential of vulvoperineal pathology.
Case Report
A 29-year-old woman was referred to the dermatology clinic with vulvar pain, swelling, and pruritus of 14 months’ duration. Her medical history was remarkable for CD following a colectomy with colostomy. Prior therapies included methotrexate with infliximab for 5 years followed by a 2-year regimen with adalimumab, which induced remission of the intestinal disease.
The patient previously had taken a variety of topical and oral antimicrobials based on treatment from a primary care physician because fungal, bacterial, and viral infections initially were suspected; however, the vulvar disease persisted, and drug-induced immunosuppression was considered to be an underlying factor. Thus, adalimumab was discontinued. Despite elimination of the biologic, the vulvar disease progressed, which prompted referral to the dermatology clinic.
Physical examination revealed diffuse vulvar edema with overlying erythema and scale (Figure 1A). Upon closer inspection, scattered violaceous papules atop a backdrop of lichenification were evident, imparting a cobblestone appearance (Figure 1B). Additionally, a fissure was present on the gluteal cleft. Biopsy from the left labia majora demonstrated well-formed granulomas within a fibrotic reticular dermis (Figures 2A and 2B). The granulomas consisted of both mononucleated and multinucleated histiocytes, rimmed peripherally by lymphocytes and plasma cells (Figure 2C). Periodic acid–Schiff–diastase and acid-fast bacilli stains as well as polarizing microscopy were negative.
Given the patient’s history, a diagnosis of vulvoperineal MCD was rendered. The patient was started on oral metronidazole 250 mg 3 times daily with topical fluocinonide and tacrolimus. She responded well to this treatment regimen and was referred back to the gastroenterologist for management of the intestinal disease.
Comment
Crohn disease is an idiopathic chronic inflammatory condition that primarily affects the gastrointestinal tract, anywhere from the mouth to the anus. It is characterized by transmural inflammation and fissures that can extend beyond the muscularis propria.4,6 Extraintestinal manifestations are common.3
Cutaneous CD often presents as perianal, perifistular, or peristomal inflammation or ulceration.7 Other skin manifestations include pyoderma gangrenosum, erythema nodosum, erythema multiforme, epidermolysis bullosa acquisita, and palmar erythema.7 Metastatic CD involves skin noncontiguous with the gastrointestinal tract1-20 and may involve any portion of the cutis. Although rare, MCD is the typical etiology underlying vulvar CD.8
Approximately 20% of MCD patients have cutaneous lesions without a history of gastrointestinal disease. More than half of cases in adults and approximately two-thirds in children involve the genitalia. Although more common in adults, vulvar involvement has been reported in children as young as 6 years of age.2 Diagnosis is especially challenging when bowel symptoms are absent; those patients should be evaluated and followed for subsequent intestinal involvement.6
Clinically, symptoms may include general discomfort, pain, pruritus, and dyspareunia. Psychosocial and sexual dysfunction are prevalent and also should be addressed.9 Depending on the stage of the disease, physical examination may reveal erythema, edema, papules, pustules, nodules, condylomatous lesions, abscesses, fissures, fistulas, ulceration, acrochordons, and scarring.2-6,10,11
A host of infections (ie, mycobacterial, actinomycosis, deep fungal, sexually transmitted, schistosomiasis), inflammatory conditions (ie, sarcoid, hidradenitis suppurativa), foreign body reactions, Melkersson-Rosenthal syndrome, and sexual abuse should be included in the differential diagnosis.2,6,10-12 Once infection, sarcoid, and foreign body reaction have been ruled out, noncaseating granulomas in skin are highly suggestive of CD.7
Histopathologic findings of MCD reveal myriad morphological reaction patterns,5,13 including high-grade dysplasia and carcinoma of the vulva; therefore, it may be imprudent to withhold diagnosis based on the absence of the historically pathognomonic noncaseating granulomas.5
The etiopathogenesis of MCD remains an enigma. Dermatopathologic examinations consistently reveal a vascular injury syndrome,13 implicating a possible circulatory system contribution via deposition of immune complexes or antigens in skin.7 Bacterial infection has been implicated in the intestinal manifestations of CD; however, failure to detect microbial ribosomal RNA in MCD biopsies refutes theories of hematogenous spread of microbes.13 Another plausible explanation is that antibodies are formed to conserved microbial epitopes following loss of tolerance to gut flora, which results in an excessive immunologic response at distinct sites in susceptible individuals.13 A T-lymphocyte–mediated type IV hypersensitivity reaction also has been proposed via cross-reactivity of lymphocytes, with skin antigens precipitating extraintestinal granuloma formation and vascular injury.3 Clearly, further investigation is needed.
Magnetic resonanance imaging can identify the extent and anatomy of intestinal and pelvic disease and can assist in the diagnosis of vulvar CD.10,11,14 For these reasons, some experts propose that imaging should be instituted prior to therapy,12,15,16 especially when direct extension is suspected.17
Treatment is challenging and often involves collaboration among several specialties.12 Many treatment options exist because therapeutic responses vary and genital MCD is frequently recalcitrant to therapy.4 Medical therapy includes antibiotics such as metronidazole, corticosteroids (ie, topical, intralesional, systemic), and immune modulators (eg, azathioprine, 6-mercaptopurine, cyclosporine, methotrexate, mycophenolate mofetil, tumor necrosis factor α inhibitors).2,3,6,10,16,18 Thalidomide has been used for refractory cases.19 These treatments can be used alone or in combination. Patients should be monitored for side effects and informed that many treatment regimens may be required before a sustained response is achieved.4,16,18 Surgery is reserved for the most resistant cases. Extensive radical excision of the involved area is the best approach, as limited local excision often is followed by recurrence.20
Conclusion
Our case highlights that vulvar CD can develop in the setting of well-controlled intestinal disease. Vulvoperineal CD should be considered in the differential diagnosis of chronic vulvar pain, swelling, and pruritus, especially in cases resistant to standard therapies and regardless of whether or not gastrointestinal tract symptoms are present. Physicians must be cognizant that vulvar signs and symptoms may precede, coincide with, or follow the diagnosis of intestinal CD. Increased awareness of this entity may facilitate its early recognition and prompt more timely treatment among women with vulvar disease caused by MCD.
- Parks AG, Morson BC, Pegum JS. Crohn’s disease with cutaneous involvement. Proc R Soc Med. 1965;58:241-242.
- Ploysangam T, Heubi JE, Eisen D, et al. Cutaneous Crohn’s disease in children. J Am Acad Dermatol. 1997;36:697-704.
- Palamaras I, El-Jabbour J, Pietropaolo N, et al. Metastatic Crohn’s disease: a review. J Eur Acad Dermatol Venereol. 2008;22:1033-1043.
- Leu S, Sun PK, Collyer J, et al. Clinical spectrum of vulvar metastatic Crohn’s disease. Dig Dis Sci. 2009;54:1565-1571.
- Foo WC, Papalas JA, Robboy SJ, et al. Vulvar manifestations of Crohn’s disease. Am J Dermatopathol. 2001;33:588-593.
- Urbanek M, Neill SM, McKee PH. Vulval Crohn’s disease: difficulties in diagnosis. Clin Exp Dermatol. 1996;21:211-214.
- Burgdorf W. Cutaneous manifestations of Crohn’s disease. J Am Acad Dermatol. 1981;5:689-695.
- Andreani SM, Ratnasingham K, Dang HH, et al. Crohn’s disease of the vulva. Int J Surg. 2010;8:2-5.
- Feller E, Ribaudo S, Jackson N. Gynecologic aspects of Crohn’s disease. Am Fam Physician. 2001;64:1725-1728.
- Corbett SL, Walsh CM, Spitzer RF, et al. Vulvar inflammation as the only clinical manifestation of Crohn disease in an 8-year-old girl [published online May 10, 2010]. Pediatrics. 2010;125:E1518-E1522.
- Tonolini M, Villa C, Campari A, et al. Common and unusual urogenital Crohn’s disease complications: spectrum of cross-sectional imaging findings. Abdom Imaging. 2013;38:32-41.
- Bhaduri S, Jenkinson S, Lewis F. Vulval Crohn’s disease—a multi-specialty approach. Int J STD AIDS. 2005;16:512-514.
- Crowson AN, Nuovo GJ, Mihm MC Jr, et al. Cutaneous manifestations of Crohn’s disease, its spectrum, and its pathogenesis: intracellular consensus bacterial 16S rRNA is associated with the gastrointestinal but not the cutaneous manifestations of Crohn’s disease. Hum Pathol. 2003;34:1185-1192.
- Pai D, Dillman JR, Mahani MG, et al. MRI of vulvar Crohn disease. Pediatr Radiol. 2011;41:537-541.
- Madnani NA, Desai D, Gandhi N, et al. Isolated Crohn’s disease of the vulva. Indian J Dermatol Venereol Leprol. 2011;77:342-344.
- Makhija S, Trotter M, Wagner E, et al. Refractory Crohn’s disease of the vulva treated with infliximab: a case report. Can J Gastroenterol. 2007;21:835-837.
- Fahmy N, Kalidindi M, Khan R. Direct colo-labial Crohn’s abscess mimicking bartholinitis. Am J Obstret Gynecol. 2010;30:741-742.
- Preston PW, Hudson N, Lewis FM. Treatment of vulval Crohn’s disease with infliximab. Clin Exp Derm. 2006;31:378-380.
- Kolivras A, De Maubeuge J, André J, et al. Thalidomide in refractory vulvar ulcerations associated with Crohn’s disease. Dermatology. 2003;206:381-383.
- Kao MS, Paulson JD, Askin FB. Crohn’s disease of the vulva. Obstet Gynecol. 1975;46:329-333.
- Parks AG, Morson BC, Pegum JS. Crohn’s disease with cutaneous involvement. Proc R Soc Med. 1965;58:241-242.
- Ploysangam T, Heubi JE, Eisen D, et al. Cutaneous Crohn’s disease in children. J Am Acad Dermatol. 1997;36:697-704.
- Palamaras I, El-Jabbour J, Pietropaolo N, et al. Metastatic Crohn’s disease: a review. J Eur Acad Dermatol Venereol. 2008;22:1033-1043.
- Leu S, Sun PK, Collyer J, et al. Clinical spectrum of vulvar metastatic Crohn’s disease. Dig Dis Sci. 2009;54:1565-1571.
- Foo WC, Papalas JA, Robboy SJ, et al. Vulvar manifestations of Crohn’s disease. Am J Dermatopathol. 2001;33:588-593.
- Urbanek M, Neill SM, McKee PH. Vulval Crohn’s disease: difficulties in diagnosis. Clin Exp Dermatol. 1996;21:211-214.
- Burgdorf W. Cutaneous manifestations of Crohn’s disease. J Am Acad Dermatol. 1981;5:689-695.
- Andreani SM, Ratnasingham K, Dang HH, et al. Crohn’s disease of the vulva. Int J Surg. 2010;8:2-5.
- Feller E, Ribaudo S, Jackson N. Gynecologic aspects of Crohn’s disease. Am Fam Physician. 2001;64:1725-1728.
- Corbett SL, Walsh CM, Spitzer RF, et al. Vulvar inflammation as the only clinical manifestation of Crohn disease in an 8-year-old girl [published online May 10, 2010]. Pediatrics. 2010;125:E1518-E1522.
- Tonolini M, Villa C, Campari A, et al. Common and unusual urogenital Crohn’s disease complications: spectrum of cross-sectional imaging findings. Abdom Imaging. 2013;38:32-41.
- Bhaduri S, Jenkinson S, Lewis F. Vulval Crohn’s disease—a multi-specialty approach. Int J STD AIDS. 2005;16:512-514.
- Crowson AN, Nuovo GJ, Mihm MC Jr, et al. Cutaneous manifestations of Crohn’s disease, its spectrum, and its pathogenesis: intracellular consensus bacterial 16S rRNA is associated with the gastrointestinal but not the cutaneous manifestations of Crohn’s disease. Hum Pathol. 2003;34:1185-1192.
- Pai D, Dillman JR, Mahani MG, et al. MRI of vulvar Crohn disease. Pediatr Radiol. 2011;41:537-541.
- Madnani NA, Desai D, Gandhi N, et al. Isolated Crohn’s disease of the vulva. Indian J Dermatol Venereol Leprol. 2011;77:342-344.
- Makhija S, Trotter M, Wagner E, et al. Refractory Crohn’s disease of the vulva treated with infliximab: a case report. Can J Gastroenterol. 2007;21:835-837.
- Fahmy N, Kalidindi M, Khan R. Direct colo-labial Crohn’s abscess mimicking bartholinitis. Am J Obstret Gynecol. 2010;30:741-742.
- Preston PW, Hudson N, Lewis FM. Treatment of vulval Crohn’s disease with infliximab. Clin Exp Derm. 2006;31:378-380.
- Kolivras A, De Maubeuge J, André J, et al. Thalidomide in refractory vulvar ulcerations associated with Crohn’s disease. Dermatology. 2003;206:381-383.
- Kao MS, Paulson JD, Askin FB. Crohn’s disease of the vulva. Obstet Gynecol. 1975;46:329-333.

Carcinoma of the colon in a child
Colon cancer is not common in childhood even though cases have been reported in children and adolescents.1,2 Although it is sporadic, it can arise in the setting of predisposing illnesses such as familial polyposis syndrome or inflammatory bowel disease.2-5 Only 1 or 2 cases per million children are reported globally each year, but the incidence has been noted to be on the rise.2 The nonspecific gastrointestinal symptoms and anemia as features of the disease could also be seen in other common childhood ailments, such as helminthiasis in our region in West Africa. As a result, unless there is a high index of suspicion at the outset, there is a risk that colon cancer will be diagnosed at a late stage, especially in children with no apparent predisposing factor.
In this case, an 11-year-old girl presented to our institution with abdominal pain, melena, abdominal swelling, and iron deficiency anemia. A positive family history of colon cancer in the mother and a brain tumor in an elder sibling prompted a search for and subsequent diagnosis of colon cancer. Her case highlights the importance of a high index of suspicion in making an early diagnosis to achieve the best possible outcomes. This case is being reported in line with the SCARE guidelines.6
Case summary and presentation
An 11-year-old girl presented to our facilty with recurrent abdominal pain of 8 months duration, a 4-month history of progressive paleness of the palms, and a month-long fever. There was an associated change in bowel habit to about 2-3 times per day, weight loss despite a preserved appetite, and black, tarry stools. A month before she presented, she developed low-grade pyrexia, dysuria, and pica. She was treated for iron deficiency anemia at a peripheral hospital where she first sought for care with oral iron, folic acid, and vitamin C, but with no improvement in symptoms.
She was the youngest of 8 children born to parents who were first cousins. Her father had died in a car accident when she was a year old, and her mother had died 6 years later after being diagnosed with and treated for colon cancer. An elder sibling died of a brain tumor at the age of 9 years.
On admission to our institution, the girl looked acutely ill. She was severely pale, but afebrile and anicteric. She had no petechial or purpuric skin rashes, but had glossitis with areas of papules on the anterior two-thirds of the dorsum of the tongue. She had no gingival hypertrophy, but had significant peripheral lymphadenopathy and weighed 67% of the weight for her age. In addition, she had generalized abdominal pain and a soft, well-circumscribed tender mass located at the right iliac fossa was palpated and estimated to be 8 cm x 6 cm.
A full blood count showed severe hypochromic microcytic anemia, with a red blood cell count of 2.53 x 1012/L, packed cell volume of 9%, white blood cell count 9.4 x109/L, platelet cell count of 453 x 109/L, mean corpuscular volume of 48.6 fl, and a red cell distribution width of 23.7%. Iron studies could not be done because we lacked the facilities, but a bone marrow aspiration biopsy showed reduced bone marrow iron stores. A fecal occult blood test was positive for blood, but negative for culture, ova, or cysts. An abdominopelvic ultrasound showed the well-circumscribed mass at the right iliac fossa, and that was confirmed by a computed-tomographic scan (Figure 1).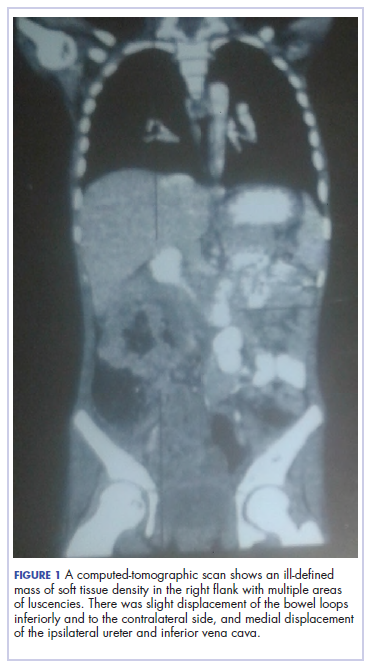
An upper endoscopy revealed fundal and prepyloric erosions and reflux eosophagitis. Although findings from a sigmoidoscopy were normal, a histology of biopsied tissues showed features of chronic inflammation.
There was a delay in arriving at the final diagnosis because the patient’s family faced financial difficulties and some of the imaging procedures were not available at our institution. Other diagnoses that were entertained and managed in this case were iron deficiency anemia from peptic ulcer disease. Six weeks after her initial presentation to our institution, the patient had an exploratory laparotomy. The findings intra-operatively were those of a huge tumor involving the ascending colon measuring 16 x14 cm and extending to involve the cecum and mesenteric lymph nodes (Figure 2).
Kidneys, liver and spleen were macroscopically normal. An assessment of Duke’s stage 3C colon cancer was made and she had an extended radical hemicolectomy with anastomosis.
A 44.5-cm long right hemicolectomy segment comprising a 17-cm ileal segment, a 6-cm cecum, 21.5-cm ascending colon, and an 8-cm appendix was removed. The tumor was located in the ascending colon at 7.5 cm from the distal resection margin and extending 1 cm into the cecum. It had a circumference of 27 cm with fibrinous exudates on its peritoneal surface. Dissection revealed uneven circumferential thickening of the bowel wall, luminal dilatation, marked mucosal ulcerations, and liquid content made up of fecal material and necrotic debris. The tumor cut surface was solid white. We also removed 4 lymph nodes. Other uninvolved areas showed focal mucosal hyperemia, but no polyps were observed. Histology showed moderately differentiated adenocarcinoma (pT4) with ¼ nodal involvement (Figure 3).
The patient’s postoperative course was uneventful, and she had adjuvant chemotherapy with oral capecitabine and intravenous oxaliplatin. She completed the 8-cycle protocol with excellent clinical response and minimal adverse events were recorded. A repeat abdominal CT scan showed no residual tumor (Figure 4), and her full blood count showed normal hematological profile with no evidence of iron deficiency.
She is presently on follow up 2 years after confirmation of the diagnosis. (Her histological diagnosis was made June 2016, and her last clinic follow-up was March 2018.
Discussion
Our patient presented with symptoms of abdominal pain, dysuria, melena, and pallor as in other case reports.7-10 A diagnosis of iron deficiency anemia was initially entertained in view of the hematologic profile, and for which management was instituted. The findings of gastric and duodenal erosions on endoscopy further supported the assumption for and treatment of peptic ulcer disease. Iron deficiency in this patient was owing to chronic blood loss from a tumour located at the upper parts of the. Vague and nonspecific symptoms are associated with delayed diagnosis and poor prognosis.1-5,11 Nonspecificity of symptoms is typical feature of colon cancer as reported in other studies.1,11-13 However, the strong family history of colon cancer heightened suspicion in this case, otherwise the diagnosis of an ascending colon tumor could have been delayed until much later and with graver consequences.
The diagnosis of colon cancer in this child was made about a year after her initial symptoms, and 3 months after her presentation to us. Ascending and transverse colon cancers are usually diagnosed late because the symptoms of intestinal obstruction – frank bleeding – will not present until the illness is substantially advanced. Ameh and Nmadu reported a case series of 8 patients from our facility with rectosigmoid tumor, of whom 6 had mucinous adenocarcinaoma and 5 of those 6 had stage 3C disease. Although the patient in the present case had an advanced disease at diagnosis, she had a moderately differentiated histology in contrast to the 6 previously reported cases, who had mucinous histology.14
Previous studies have shown that colorectal carcinoma is a rare disease worldwide, with an annual age-adjusted incidence of 0.38 people/million.1,2 When it occurs in the young, familial or hereditary predisposition should be highly suspected.1-3 To date, there is scant literature on children younger than 16 years in Nigeria.15 Various studies have found a relationship between patients with early-stage colon cancer and inherited genetic predisposition to the disease.2,5 Familial adenomatous polyposis syndrome is an autosomal dominant disorder characterized by the development of polyps during the first decade of life, extensive polyposis in the second decade, and transformation into frank carcinoma in early adulthood.1-5
Although our patient’s mother was diagnosed with and died of colon cancer, the type of which could not be ascertained because her records could not be traced. However, the operative and histological findings in this patient did not suggest the presence of polyposis. The clinical phenotype for the autosomal recessive mismatch repair deficiency includes susceptibity to glioma, leukemia, lymphoma, and colorectal carcinoma in children and young adults.1,5 Screening for genetic markers in the child in the present case might have identified the genetic abnormalities involved and would have been invaluable in the evaluation of her 6 surviving siblings and further management of this family. In conclusion. A high index of suspicion should prompt inclusion of colon cancer in the differential diagnosis of nonspecific gastrointestinal symptoms associated with colon cancer in children.
Acknowledg
The authors obtained written informed consent from the patient and her elder sibling before writing this report. In addition, the authors thank all the staff involved in the management of this child in the pediatric medical and surgical wards.
1. Sultan I, Rodriguez-Galindo C, El-Taani H, Pastore G, Casanova M, Gallino G, Ferrari A. Distinct features of colorectal cancer in children and adolescents. A population-based study of 159 cases. Cancer. 2010;1;116(3):758-65.
2. Ferrari A. Intestinal carcinomas. In: Schneider DT, Brecht IB, Olson TA, Ferrari A (eds). Rare tumors in children and adolescents. 1st ed. Copyright, Springer-Verlag Berlin Heidelberg; 2012; chap 32.
3. Hill DA, Furman WL, Bilups CA, Riedly SE, Cain AM, Rao BN. Colorectal carcinoma in childhood and adolescence: a clinicopathological review. J Clin Oncol. 2007;25(36):5808-5814.
4. Saab OKR, Furman WL. Epidemiology and management options for colorectal cancer in children. Paediatr Drugs. 2008;10(3):177-192.
5. Bertario L, Signoroni S. Gastrointestinal cancer predisposition syndromes. In: Schneider DT, Brecht IB, Olson TA, Ferrari A (eds). Rare tumors in children and adolescents. Copyright, Springer-Verlag Berlin Heidelberg; 2012; chap 30.
6. Agha RA, Fowler AJ, Saetta A, et al, for the SCARE Group. The SCARE Statement: consensus-based surgical case report guidelines. Int J Surg. 2016;34:180-186.
7. Tricoli JV, Seibel NL, Blair DG, Albritton K, Hayes-Lattin B. Unique characteristics of adolescent and young adult acute lymphoblastic leukemia, breast cancer, and colon cancer. J Natl Cancer Inst. 2011;103(8):628-635.
8. Begum M, Khan ZJ, Hassan K, Karim S. Carcinoma colon of a child presenting with abdominal pain. Bangaladesh J Child Health. 2014;38(1):44-47.
9. Woods R, Larkin JO, Muldoon C, Kennedy MJ, Mehigan B, McCormick P. Metastatic paediatric colorectal carcinoma. Ir Med J. 2012;105(3):88-89.
10. Bjoernsen LP, Lindsay MB. An unusual case of pediatric abdominal pain. CJEM. 2011;13(2):133-138.
11. Takalkar UV, Asegaonkar SB, Kulkarni U, Jadhav A, Advani S, Reddy DN. Carcinoma of colon in an adolescent: a case report with review of literature. Int J Sci Rep 2015;1(2):151-3.
12. Zamir N, Ahmad S, Akhtar J. Mucinous adenocarcinoma of colon. APSP J Case Rep. 2010;1(2):20.
13. Al-Tonbary Y, Darwish A, El-Hussein A, Fouda A. Adenocarcinoma of the colon in children: case series and mini-review of the literature. Hematol Oncol Stem Cell Ther. 2013;6(1):29-33.
14. Ameh EA, Nmadu PT. Colorectal adenocarcinoma in children and adolescents: a report of 8 patients from Zaria, Nigeria. West Afr J Med. 2000;19(4):273-276.
15. Ibrahim, AE, Afolayan KA, Adeniji OM, Buhari KB. Colorectal carcinoma in children and young adults in Ilorin, Nigeria. West Afr J Med. 2011;30(3):202-205.
Colon cancer is not common in childhood even though cases have been reported in children and adolescents.1,2 Although it is sporadic, it can arise in the setting of predisposing illnesses such as familial polyposis syndrome or inflammatory bowel disease.2-5 Only 1 or 2 cases per million children are reported globally each year, but the incidence has been noted to be on the rise.2 The nonspecific gastrointestinal symptoms and anemia as features of the disease could also be seen in other common childhood ailments, such as helminthiasis in our region in West Africa. As a result, unless there is a high index of suspicion at the outset, there is a risk that colon cancer will be diagnosed at a late stage, especially in children with no apparent predisposing factor.
In this case, an 11-year-old girl presented to our institution with abdominal pain, melena, abdominal swelling, and iron deficiency anemia. A positive family history of colon cancer in the mother and a brain tumor in an elder sibling prompted a search for and subsequent diagnosis of colon cancer. Her case highlights the importance of a high index of suspicion in making an early diagnosis to achieve the best possible outcomes. This case is being reported in line with the SCARE guidelines.6
Case summary and presentation
An 11-year-old girl presented to our facilty with recurrent abdominal pain of 8 months duration, a 4-month history of progressive paleness of the palms, and a month-long fever. There was an associated change in bowel habit to about 2-3 times per day, weight loss despite a preserved appetite, and black, tarry stools. A month before she presented, she developed low-grade pyrexia, dysuria, and pica. She was treated for iron deficiency anemia at a peripheral hospital where she first sought for care with oral iron, folic acid, and vitamin C, but with no improvement in symptoms.
She was the youngest of 8 children born to parents who were first cousins. Her father had died in a car accident when she was a year old, and her mother had died 6 years later after being diagnosed with and treated for colon cancer. An elder sibling died of a brain tumor at the age of 9 years.
On admission to our institution, the girl looked acutely ill. She was severely pale, but afebrile and anicteric. She had no petechial or purpuric skin rashes, but had glossitis with areas of papules on the anterior two-thirds of the dorsum of the tongue. She had no gingival hypertrophy, but had significant peripheral lymphadenopathy and weighed 67% of the weight for her age. In addition, she had generalized abdominal pain and a soft, well-circumscribed tender mass located at the right iliac fossa was palpated and estimated to be 8 cm x 6 cm.
A full blood count showed severe hypochromic microcytic anemia, with a red blood cell count of 2.53 x 1012/L, packed cell volume of 9%, white blood cell count 9.4 x109/L, platelet cell count of 453 x 109/L, mean corpuscular volume of 48.6 fl, and a red cell distribution width of 23.7%. Iron studies could not be done because we lacked the facilities, but a bone marrow aspiration biopsy showed reduced bone marrow iron stores. A fecal occult blood test was positive for blood, but negative for culture, ova, or cysts. An abdominopelvic ultrasound showed the well-circumscribed mass at the right iliac fossa, and that was confirmed by a computed-tomographic scan (Figure 1).
An upper endoscopy revealed fundal and prepyloric erosions and reflux eosophagitis. Although findings from a sigmoidoscopy were normal, a histology of biopsied tissues showed features of chronic inflammation.
There was a delay in arriving at the final diagnosis because the patient’s family faced financial difficulties and some of the imaging procedures were not available at our institution. Other diagnoses that were entertained and managed in this case were iron deficiency anemia from peptic ulcer disease. Six weeks after her initial presentation to our institution, the patient had an exploratory laparotomy. The findings intra-operatively were those of a huge tumor involving the ascending colon measuring 16 x14 cm and extending to involve the cecum and mesenteric lymph nodes (Figure 2).
Kidneys, liver and spleen were macroscopically normal. An assessment of Duke’s stage 3C colon cancer was made and she had an extended radical hemicolectomy with anastomosis.
A 44.5-cm long right hemicolectomy segment comprising a 17-cm ileal segment, a 6-cm cecum, 21.5-cm ascending colon, and an 8-cm appendix was removed. The tumor was located in the ascending colon at 7.5 cm from the distal resection margin and extending 1 cm into the cecum. It had a circumference of 27 cm with fibrinous exudates on its peritoneal surface. Dissection revealed uneven circumferential thickening of the bowel wall, luminal dilatation, marked mucosal ulcerations, and liquid content made up of fecal material and necrotic debris. The tumor cut surface was solid white. We also removed 4 lymph nodes. Other uninvolved areas showed focal mucosal hyperemia, but no polyps were observed. Histology showed moderately differentiated adenocarcinoma (pT4) with ¼ nodal involvement (Figure 3).
The patient’s postoperative course was uneventful, and she had adjuvant chemotherapy with oral capecitabine and intravenous oxaliplatin. She completed the 8-cycle protocol with excellent clinical response and minimal adverse events were recorded. A repeat abdominal CT scan showed no residual tumor (Figure 4), and her full blood count showed normal hematological profile with no evidence of iron deficiency.
She is presently on follow up 2 years after confirmation of the diagnosis. (Her histological diagnosis was made June 2016, and her last clinic follow-up was March 2018.
Discussion
Our patient presented with symptoms of abdominal pain, dysuria, melena, and pallor as in other case reports.7-10 A diagnosis of iron deficiency anemia was initially entertained in view of the hematologic profile, and for which management was instituted. The findings of gastric and duodenal erosions on endoscopy further supported the assumption for and treatment of peptic ulcer disease. Iron deficiency in this patient was owing to chronic blood loss from a tumour located at the upper parts of the. Vague and nonspecific symptoms are associated with delayed diagnosis and poor prognosis.1-5,11 Nonspecificity of symptoms is typical feature of colon cancer as reported in other studies.1,11-13 However, the strong family history of colon cancer heightened suspicion in this case, otherwise the diagnosis of an ascending colon tumor could have been delayed until much later and with graver consequences.
The diagnosis of colon cancer in this child was made about a year after her initial symptoms, and 3 months after her presentation to us. Ascending and transverse colon cancers are usually diagnosed late because the symptoms of intestinal obstruction – frank bleeding – will not present until the illness is substantially advanced. Ameh and Nmadu reported a case series of 8 patients from our facility with rectosigmoid tumor, of whom 6 had mucinous adenocarcinaoma and 5 of those 6 had stage 3C disease. Although the patient in the present case had an advanced disease at diagnosis, she had a moderately differentiated histology in contrast to the 6 previously reported cases, who had mucinous histology.14
Previous studies have shown that colorectal carcinoma is a rare disease worldwide, with an annual age-adjusted incidence of 0.38 people/million.1,2 When it occurs in the young, familial or hereditary predisposition should be highly suspected.1-3 To date, there is scant literature on children younger than 16 years in Nigeria.15 Various studies have found a relationship between patients with early-stage colon cancer and inherited genetic predisposition to the disease.2,5 Familial adenomatous polyposis syndrome is an autosomal dominant disorder characterized by the development of polyps during the first decade of life, extensive polyposis in the second decade, and transformation into frank carcinoma in early adulthood.1-5
Although our patient’s mother was diagnosed with and died of colon cancer, the type of which could not be ascertained because her records could not be traced. However, the operative and histological findings in this patient did not suggest the presence of polyposis. The clinical phenotype for the autosomal recessive mismatch repair deficiency includes susceptibity to glioma, leukemia, lymphoma, and colorectal carcinoma in children and young adults.1,5 Screening for genetic markers in the child in the present case might have identified the genetic abnormalities involved and would have been invaluable in the evaluation of her 6 surviving siblings and further management of this family. In conclusion. A high index of suspicion should prompt inclusion of colon cancer in the differential diagnosis of nonspecific gastrointestinal symptoms associated with colon cancer in children.
Acknowledg
The authors obtained written informed consent from the patient and her elder sibling before writing this report. In addition, the authors thank all the staff involved in the management of this child in the pediatric medical and surgical wards.
Colon cancer is not common in childhood even though cases have been reported in children and adolescents.1,2 Although it is sporadic, it can arise in the setting of predisposing illnesses such as familial polyposis syndrome or inflammatory bowel disease.2-5 Only 1 or 2 cases per million children are reported globally each year, but the incidence has been noted to be on the rise.2 The nonspecific gastrointestinal symptoms and anemia as features of the disease could also be seen in other common childhood ailments, such as helminthiasis in our region in West Africa. As a result, unless there is a high index of suspicion at the outset, there is a risk that colon cancer will be diagnosed at a late stage, especially in children with no apparent predisposing factor.
In this case, an 11-year-old girl presented to our institution with abdominal pain, melena, abdominal swelling, and iron deficiency anemia. A positive family history of colon cancer in the mother and a brain tumor in an elder sibling prompted a search for and subsequent diagnosis of colon cancer. Her case highlights the importance of a high index of suspicion in making an early diagnosis to achieve the best possible outcomes. This case is being reported in line with the SCARE guidelines.6
Case summary and presentation
An 11-year-old girl presented to our facilty with recurrent abdominal pain of 8 months duration, a 4-month history of progressive paleness of the palms, and a month-long fever. There was an associated change in bowel habit to about 2-3 times per day, weight loss despite a preserved appetite, and black, tarry stools. A month before she presented, she developed low-grade pyrexia, dysuria, and pica. She was treated for iron deficiency anemia at a peripheral hospital where she first sought for care with oral iron, folic acid, and vitamin C, but with no improvement in symptoms.
She was the youngest of 8 children born to parents who were first cousins. Her father had died in a car accident when she was a year old, and her mother had died 6 years later after being diagnosed with and treated for colon cancer. An elder sibling died of a brain tumor at the age of 9 years.
On admission to our institution, the girl looked acutely ill. She was severely pale, but afebrile and anicteric. She had no petechial or purpuric skin rashes, but had glossitis with areas of papules on the anterior two-thirds of the dorsum of the tongue. She had no gingival hypertrophy, but had significant peripheral lymphadenopathy and weighed 67% of the weight for her age. In addition, she had generalized abdominal pain and a soft, well-circumscribed tender mass located at the right iliac fossa was palpated and estimated to be 8 cm x 6 cm.
A full blood count showed severe hypochromic microcytic anemia, with a red blood cell count of 2.53 x 1012/L, packed cell volume of 9%, white blood cell count 9.4 x109/L, platelet cell count of 453 x 109/L, mean corpuscular volume of 48.6 fl, and a red cell distribution width of 23.7%. Iron studies could not be done because we lacked the facilities, but a bone marrow aspiration biopsy showed reduced bone marrow iron stores. A fecal occult blood test was positive for blood, but negative for culture, ova, or cysts. An abdominopelvic ultrasound showed the well-circumscribed mass at the right iliac fossa, and that was confirmed by a computed-tomographic scan (Figure 1).
An upper endoscopy revealed fundal and prepyloric erosions and reflux eosophagitis. Although findings from a sigmoidoscopy were normal, a histology of biopsied tissues showed features of chronic inflammation.
There was a delay in arriving at the final diagnosis because the patient’s family faced financial difficulties and some of the imaging procedures were not available at our institution. Other diagnoses that were entertained and managed in this case were iron deficiency anemia from peptic ulcer disease. Six weeks after her initial presentation to our institution, the patient had an exploratory laparotomy. The findings intra-operatively were those of a huge tumor involving the ascending colon measuring 16 x14 cm and extending to involve the cecum and mesenteric lymph nodes (Figure 2).
Kidneys, liver and spleen were macroscopically normal. An assessment of Duke’s stage 3C colon cancer was made and she had an extended radical hemicolectomy with anastomosis.
A 44.5-cm long right hemicolectomy segment comprising a 17-cm ileal segment, a 6-cm cecum, 21.5-cm ascending colon, and an 8-cm appendix was removed. The tumor was located in the ascending colon at 7.5 cm from the distal resection margin and extending 1 cm into the cecum. It had a circumference of 27 cm with fibrinous exudates on its peritoneal surface. Dissection revealed uneven circumferential thickening of the bowel wall, luminal dilatation, marked mucosal ulcerations, and liquid content made up of fecal material and necrotic debris. The tumor cut surface was solid white. We also removed 4 lymph nodes. Other uninvolved areas showed focal mucosal hyperemia, but no polyps were observed. Histology showed moderately differentiated adenocarcinoma (pT4) with ¼ nodal involvement (Figure 3).
The patient’s postoperative course was uneventful, and she had adjuvant chemotherapy with oral capecitabine and intravenous oxaliplatin. She completed the 8-cycle protocol with excellent clinical response and minimal adverse events were recorded. A repeat abdominal CT scan showed no residual tumor (Figure 4), and her full blood count showed normal hematological profile with no evidence of iron deficiency.
She is presently on follow up 2 years after confirmation of the diagnosis. (Her histological diagnosis was made June 2016, and her last clinic follow-up was March 2018.
Discussion
Our patient presented with symptoms of abdominal pain, dysuria, melena, and pallor as in other case reports.7-10 A diagnosis of iron deficiency anemia was initially entertained in view of the hematologic profile, and for which management was instituted. The findings of gastric and duodenal erosions on endoscopy further supported the assumption for and treatment of peptic ulcer disease. Iron deficiency in this patient was owing to chronic blood loss from a tumour located at the upper parts of the. Vague and nonspecific symptoms are associated with delayed diagnosis and poor prognosis.1-5,11 Nonspecificity of symptoms is typical feature of colon cancer as reported in other studies.1,11-13 However, the strong family history of colon cancer heightened suspicion in this case, otherwise the diagnosis of an ascending colon tumor could have been delayed until much later and with graver consequences.
The diagnosis of colon cancer in this child was made about a year after her initial symptoms, and 3 months after her presentation to us. Ascending and transverse colon cancers are usually diagnosed late because the symptoms of intestinal obstruction – frank bleeding – will not present until the illness is substantially advanced. Ameh and Nmadu reported a case series of 8 patients from our facility with rectosigmoid tumor, of whom 6 had mucinous adenocarcinaoma and 5 of those 6 had stage 3C disease. Although the patient in the present case had an advanced disease at diagnosis, she had a moderately differentiated histology in contrast to the 6 previously reported cases, who had mucinous histology.14
Previous studies have shown that colorectal carcinoma is a rare disease worldwide, with an annual age-adjusted incidence of 0.38 people/million.1,2 When it occurs in the young, familial or hereditary predisposition should be highly suspected.1-3 To date, there is scant literature on children younger than 16 years in Nigeria.15 Various studies have found a relationship between patients with early-stage colon cancer and inherited genetic predisposition to the disease.2,5 Familial adenomatous polyposis syndrome is an autosomal dominant disorder characterized by the development of polyps during the first decade of life, extensive polyposis in the second decade, and transformation into frank carcinoma in early adulthood.1-5
Although our patient’s mother was diagnosed with and died of colon cancer, the type of which could not be ascertained because her records could not be traced. However, the operative and histological findings in this patient did not suggest the presence of polyposis. The clinical phenotype for the autosomal recessive mismatch repair deficiency includes susceptibity to glioma, leukemia, lymphoma, and colorectal carcinoma in children and young adults.1,5 Screening for genetic markers in the child in the present case might have identified the genetic abnormalities involved and would have been invaluable in the evaluation of her 6 surviving siblings and further management of this family. In conclusion. A high index of suspicion should prompt inclusion of colon cancer in the differential diagnosis of nonspecific gastrointestinal symptoms associated with colon cancer in children.
Acknowledg
The authors obtained written informed consent from the patient and her elder sibling before writing this report. In addition, the authors thank all the staff involved in the management of this child in the pediatric medical and surgical wards.
1. Sultan I, Rodriguez-Galindo C, El-Taani H, Pastore G, Casanova M, Gallino G, Ferrari A. Distinct features of colorectal cancer in children and adolescents. A population-based study of 159 cases. Cancer. 2010;1;116(3):758-65.
2. Ferrari A. Intestinal carcinomas. In: Schneider DT, Brecht IB, Olson TA, Ferrari A (eds). Rare tumors in children and adolescents. 1st ed. Copyright, Springer-Verlag Berlin Heidelberg; 2012; chap 32.
3. Hill DA, Furman WL, Bilups CA, Riedly SE, Cain AM, Rao BN. Colorectal carcinoma in childhood and adolescence: a clinicopathological review. J Clin Oncol. 2007;25(36):5808-5814.
4. Saab OKR, Furman WL. Epidemiology and management options for colorectal cancer in children. Paediatr Drugs. 2008;10(3):177-192.
5. Bertario L, Signoroni S. Gastrointestinal cancer predisposition syndromes. In: Schneider DT, Brecht IB, Olson TA, Ferrari A (eds). Rare tumors in children and adolescents. Copyright, Springer-Verlag Berlin Heidelberg; 2012; chap 30.
6. Agha RA, Fowler AJ, Saetta A, et al, for the SCARE Group. The SCARE Statement: consensus-based surgical case report guidelines. Int J Surg. 2016;34:180-186.
7. Tricoli JV, Seibel NL, Blair DG, Albritton K, Hayes-Lattin B. Unique characteristics of adolescent and young adult acute lymphoblastic leukemia, breast cancer, and colon cancer. J Natl Cancer Inst. 2011;103(8):628-635.
8. Begum M, Khan ZJ, Hassan K, Karim S. Carcinoma colon of a child presenting with abdominal pain. Bangaladesh J Child Health. 2014;38(1):44-47.
9. Woods R, Larkin JO, Muldoon C, Kennedy MJ, Mehigan B, McCormick P. Metastatic paediatric colorectal carcinoma. Ir Med J. 2012;105(3):88-89.
10. Bjoernsen LP, Lindsay MB. An unusual case of pediatric abdominal pain. CJEM. 2011;13(2):133-138.
11. Takalkar UV, Asegaonkar SB, Kulkarni U, Jadhav A, Advani S, Reddy DN. Carcinoma of colon in an adolescent: a case report with review of literature. Int J Sci Rep 2015;1(2):151-3.
12. Zamir N, Ahmad S, Akhtar J. Mucinous adenocarcinoma of colon. APSP J Case Rep. 2010;1(2):20.
13. Al-Tonbary Y, Darwish A, El-Hussein A, Fouda A. Adenocarcinoma of the colon in children: case series and mini-review of the literature. Hematol Oncol Stem Cell Ther. 2013;6(1):29-33.
14. Ameh EA, Nmadu PT. Colorectal adenocarcinoma in children and adolescents: a report of 8 patients from Zaria, Nigeria. West Afr J Med. 2000;19(4):273-276.
15. Ibrahim, AE, Afolayan KA, Adeniji OM, Buhari KB. Colorectal carcinoma in children and young adults in Ilorin, Nigeria. West Afr J Med. 2011;30(3):202-205.
1. Sultan I, Rodriguez-Galindo C, El-Taani H, Pastore G, Casanova M, Gallino G, Ferrari A. Distinct features of colorectal cancer in children and adolescents. A population-based study of 159 cases. Cancer. 2010;1;116(3):758-65.
2. Ferrari A. Intestinal carcinomas. In: Schneider DT, Brecht IB, Olson TA, Ferrari A (eds). Rare tumors in children and adolescents. 1st ed. Copyright, Springer-Verlag Berlin Heidelberg; 2012; chap 32.
3. Hill DA, Furman WL, Bilups CA, Riedly SE, Cain AM, Rao BN. Colorectal carcinoma in childhood and adolescence: a clinicopathological review. J Clin Oncol. 2007;25(36):5808-5814.
4. Saab OKR, Furman WL. Epidemiology and management options for colorectal cancer in children. Paediatr Drugs. 2008;10(3):177-192.
5. Bertario L, Signoroni S. Gastrointestinal cancer predisposition syndromes. In: Schneider DT, Brecht IB, Olson TA, Ferrari A (eds). Rare tumors in children and adolescents. Copyright, Springer-Verlag Berlin Heidelberg; 2012; chap 30.
6. Agha RA, Fowler AJ, Saetta A, et al, for the SCARE Group. The SCARE Statement: consensus-based surgical case report guidelines. Int J Surg. 2016;34:180-186.
7. Tricoli JV, Seibel NL, Blair DG, Albritton K, Hayes-Lattin B. Unique characteristics of adolescent and young adult acute lymphoblastic leukemia, breast cancer, and colon cancer. J Natl Cancer Inst. 2011;103(8):628-635.
8. Begum M, Khan ZJ, Hassan K, Karim S. Carcinoma colon of a child presenting with abdominal pain. Bangaladesh J Child Health. 2014;38(1):44-47.
9. Woods R, Larkin JO, Muldoon C, Kennedy MJ, Mehigan B, McCormick P. Metastatic paediatric colorectal carcinoma. Ir Med J. 2012;105(3):88-89.
10. Bjoernsen LP, Lindsay MB. An unusual case of pediatric abdominal pain. CJEM. 2011;13(2):133-138.
11. Takalkar UV, Asegaonkar SB, Kulkarni U, Jadhav A, Advani S, Reddy DN. Carcinoma of colon in an adolescent: a case report with review of literature. Int J Sci Rep 2015;1(2):151-3.
12. Zamir N, Ahmad S, Akhtar J. Mucinous adenocarcinoma of colon. APSP J Case Rep. 2010;1(2):20.
13. Al-Tonbary Y, Darwish A, El-Hussein A, Fouda A. Adenocarcinoma of the colon in children: case series and mini-review of the literature. Hematol Oncol Stem Cell Ther. 2013;6(1):29-33.
14. Ameh EA, Nmadu PT. Colorectal adenocarcinoma in children and adolescents: a report of 8 patients from Zaria, Nigeria. West Afr J Med. 2000;19(4):273-276.
15. Ibrahim, AE, Afolayan KA, Adeniji OM, Buhari KB. Colorectal carcinoma in children and young adults in Ilorin, Nigeria. West Afr J Med. 2011;30(3):202-205.