User login
Scalp Psoriasis With Increased Hair Density
Case Report
A 19-year-old man first presented to our outpatient dermatology clinic for evaluation of a rash on the elbows and knees of 2 to 3 months’ duration. The lesions were asymptomatic. A review of symptoms including joint pain was largely negative. His medical history was remarkable for terminal ileitis, Crohn disease, anal fissure, rhabdomyolysis, and viral gastroenteritis. Physical examination revealed a well-nourished man with red, scaly, indurated papules and plaques involving approximately 0.5% of the body surface area. A diagnosis of plaque psoriasis was made, and he was treated with topical corticosteroids for 2 weeks and as needed thereafter.
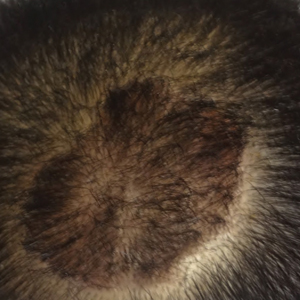
The patient remained stable for 5 years before presenting again to the dermatology clinic for psoriasis that had now spread to the scalp. Clinical examination revealed a very thin, faintly erythematous, scaly patch associated with increased hair density of the right frontal and parietal scalp (Figure). The patient denied any trauma or injury to the area or application of hair dye. We prescribed clobetasol solution 0.05% twice daily to the affected area of the scalp for 2 weeks, which resulted in minimal resolution of the psoriatic scalp lesion.

Comment
The scalp is a site of predilection in psoriasis, as approximately 80% of psoriasis patients report involvement of the scalp.1 Scalp involvement can dramatically affect a patient’s quality of life and often poses considerable therapeutic challenges for dermatologists.1 Alopecia in the setting of scalp psoriasis is common but is not well understood.2 First described by Shuster3 in 1972, psoriatic alopecia is associated with diminished hair density, follicular miniaturization, sebaceous gland atrophy, and an increased number of dystrophic bulbs in psoriatic plaques.4 It clinically presents as pink scaly plaques consistent with psoriasis with overlying alopecia. There are few instances of psoriatic alopecia reported as cicatricial hair loss and generalized telogen effluvium.2 It is known that a higher proportion of telogen and catagen hairs exist in patients with psoriatic alopecia.5 Additionally, psoriasis patients have more dystrophic hairs in affected and unaffected skin despite no differences in skin when compared to unaffected patients. Many patients achieve hair regrowth following treatment of psoriasis.2
We described a patient with scalp psoriasis who had increased and preserved hair density. Our case suggests that while most patients with scalp psoriasis experience psoriatic alopecia of the lesional skin, some may unconventionally experience increased hair density, which is contradictory to propositions that the friction associated with the application of topical treatments results in breakage of telogen hairs.2 Additionally, the presence of increased hair density in scalp psoriasis can further complicate antipsoriatic treatment by making the scalp inaccessible and topical therapies even more difficult to apply.
- Krueger G, Koo J, Lebwohl M, et al. The impact of psoriasis on quality of life: results of a 1998 National Psoriasis Foundation patient-membership survey. Arch Dermatol. 2001;137:280-284.
- George SM, Taylor MR, Farrant PB. Psoriatic alopecia. Clin Exp Dermatol. 2015;40:717-721.
- Shuster S. Psoriatic alopecia. Br J Dermatol. 1972;87:73-77.
- Wyatt E, Bottoms E, Comaish S. Abnormal hair shafts in psoriasis on scanning electron microscopy. Br J Dermatol. 1972;87:368-373.
- Schoorl WJ, van Baar HJ, van de Kerkhof PC. The hair root pattern in psoriasis of the scalp. Acta Derm Venereol. 1992;72:141-142.
Case Report
A 19-year-old man first presented to our outpatient dermatology clinic for evaluation of a rash on the elbows and knees of 2 to 3 months’ duration. The lesions were asymptomatic. A review of symptoms including joint pain was largely negative. His medical history was remarkable for terminal ileitis, Crohn disease, anal fissure, rhabdomyolysis, and viral gastroenteritis. Physical examination revealed a well-nourished man with red, scaly, indurated papules and plaques involving approximately 0.5% of the body surface area. A diagnosis of plaque psoriasis was made, and he was treated with topical corticosteroids for 2 weeks and as needed thereafter.
The patient remained stable for 5 years before presenting again to the dermatology clinic for psoriasis that had now spread to the scalp. Clinical examination revealed a very thin, faintly erythematous, scaly patch associated with increased hair density of the right frontal and parietal scalp (Figure). The patient denied any trauma or injury to the area or application of hair dye. We prescribed clobetasol solution 0.05% twice daily to the affected area of the scalp for 2 weeks, which resulted in minimal resolution of the psoriatic scalp lesion.
Comment
The scalp is a site of predilection in psoriasis, as approximately 80% of psoriasis patients report involvement of the scalp.1 Scalp involvement can dramatically affect a patient’s quality of life and often poses considerable therapeutic challenges for dermatologists.1 Alopecia in the setting of scalp psoriasis is common but is not well understood.2 First described by Shuster3 in 1972, psoriatic alopecia is associated with diminished hair density, follicular miniaturization, sebaceous gland atrophy, and an increased number of dystrophic bulbs in psoriatic plaques.4 It clinically presents as pink scaly plaques consistent with psoriasis with overlying alopecia. There are few instances of psoriatic alopecia reported as cicatricial hair loss and generalized telogen effluvium.2 It is known that a higher proportion of telogen and catagen hairs exist in patients with psoriatic alopecia.5 Additionally, psoriasis patients have more dystrophic hairs in affected and unaffected skin despite no differences in skin when compared to unaffected patients. Many patients achieve hair regrowth following treatment of psoriasis.2
We described a patient with scalp psoriasis who had increased and preserved hair density. Our case suggests that while most patients with scalp psoriasis experience psoriatic alopecia of the lesional skin, some may unconventionally experience increased hair density, which is contradictory to propositions that the friction associated with the application of topical treatments results in breakage of telogen hairs.2 Additionally, the presence of increased hair density in scalp psoriasis can further complicate antipsoriatic treatment by making the scalp inaccessible and topical therapies even more difficult to apply.
Case Report
A 19-year-old man first presented to our outpatient dermatology clinic for evaluation of a rash on the elbows and knees of 2 to 3 months’ duration. The lesions were asymptomatic. A review of symptoms including joint pain was largely negative. His medical history was remarkable for terminal ileitis, Crohn disease, anal fissure, rhabdomyolysis, and viral gastroenteritis. Physical examination revealed a well-nourished man with red, scaly, indurated papules and plaques involving approximately 0.5% of the body surface area. A diagnosis of plaque psoriasis was made, and he was treated with topical corticosteroids for 2 weeks and as needed thereafter.
The patient remained stable for 5 years before presenting again to the dermatology clinic for psoriasis that had now spread to the scalp. Clinical examination revealed a very thin, faintly erythematous, scaly patch associated with increased hair density of the right frontal and parietal scalp (Figure). The patient denied any trauma or injury to the area or application of hair dye. We prescribed clobetasol solution 0.05% twice daily to the affected area of the scalp for 2 weeks, which resulted in minimal resolution of the psoriatic scalp lesion.
Comment
The scalp is a site of predilection in psoriasis, as approximately 80% of psoriasis patients report involvement of the scalp.1 Scalp involvement can dramatically affect a patient’s quality of life and often poses considerable therapeutic challenges for dermatologists.1 Alopecia in the setting of scalp psoriasis is common but is not well understood.2 First described by Shuster3 in 1972, psoriatic alopecia is associated with diminished hair density, follicular miniaturization, sebaceous gland atrophy, and an increased number of dystrophic bulbs in psoriatic plaques.4 It clinically presents as pink scaly plaques consistent with psoriasis with overlying alopecia. There are few instances of psoriatic alopecia reported as cicatricial hair loss and generalized telogen effluvium.2 It is known that a higher proportion of telogen and catagen hairs exist in patients with psoriatic alopecia.5 Additionally, psoriasis patients have more dystrophic hairs in affected and unaffected skin despite no differences in skin when compared to unaffected patients. Many patients achieve hair regrowth following treatment of psoriasis.2
We described a patient with scalp psoriasis who had increased and preserved hair density. Our case suggests that while most patients with scalp psoriasis experience psoriatic alopecia of the lesional skin, some may unconventionally experience increased hair density, which is contradictory to propositions that the friction associated with the application of topical treatments results in breakage of telogen hairs.2 Additionally, the presence of increased hair density in scalp psoriasis can further complicate antipsoriatic treatment by making the scalp inaccessible and topical therapies even more difficult to apply.
- Krueger G, Koo J, Lebwohl M, et al. The impact of psoriasis on quality of life: results of a 1998 National Psoriasis Foundation patient-membership survey. Arch Dermatol. 2001;137:280-284.
- George SM, Taylor MR, Farrant PB. Psoriatic alopecia. Clin Exp Dermatol. 2015;40:717-721.
- Shuster S. Psoriatic alopecia. Br J Dermatol. 1972;87:73-77.
- Wyatt E, Bottoms E, Comaish S. Abnormal hair shafts in psoriasis on scanning electron microscopy. Br J Dermatol. 1972;87:368-373.
- Schoorl WJ, van Baar HJ, van de Kerkhof PC. The hair root pattern in psoriasis of the scalp. Acta Derm Venereol. 1992;72:141-142.
- Krueger G, Koo J, Lebwohl M, et al. The impact of psoriasis on quality of life: results of a 1998 National Psoriasis Foundation patient-membership survey. Arch Dermatol. 2001;137:280-284.
- George SM, Taylor MR, Farrant PB. Psoriatic alopecia. Clin Exp Dermatol. 2015;40:717-721.
- Shuster S. Psoriatic alopecia. Br J Dermatol. 1972;87:73-77.
- Wyatt E, Bottoms E, Comaish S. Abnormal hair shafts in psoriasis on scanning electron microscopy. Br J Dermatol. 1972;87:368-373.
- Schoorl WJ, van Baar HJ, van de Kerkhof PC. The hair root pattern in psoriasis of the scalp. Acta Derm Venereol. 1992;72:141-142.
Practice Points
- Scalp psoriasis may present with hair loss or increased hair density.
- Psoriasis with increased hair density may make topical medications more difficult to apply.
Reflectance Confocal Microscopy as a First-Line Diagnostic Technique for Mycosis Fungoides
Case Report
A 60-year-old man with a history of Hodgkin lymphoma that had been treated with chemotherapy 6 years prior presented to our dermatology clinic with a persistent pruritic rash on the back, abdomen, and bilateral arms and legs. The eruption initially began as localized discrete lesions on the lower back 1 year prior to the current presentation; at that time a diagnosis of psoriasis was made at an outside dermatology clinic, and treatment with mometasone furoate cream was initiated. Despite the patient’s compliance with this treatment, the lesions did not resolve and began spreading to the arms, legs, chest, and abdomen. His current medications included lisinopril, escitalopram, aspirin, and omeprazole.
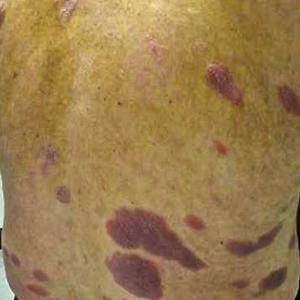
On presentation to our clinic, physical examination revealed round, scaly, pink plaques and tumors of variable sizes (3–10 cm) distributed asymmetrically on the chest, back, abdomen, arms, and legs (Figure 1). The lesions were grouped in well-defined areas encompassing approximately 30% of the body surface area. No lymphadenopathy was appreciated. In vivo reflectance confocal microscopy (RCM) performed on one of the lesions revealed disarray of the epidermis with small, weakly refractile, round to oval cells scattered within the spinous layer and dermoepidermal junction (Figure 2). Additionally, these weakly refractile, round to oval cells also were seen in vesiclelike dark spaces, and hyporefractile basal cells were appreciated surrounding the dermal papillae. Mycosis fungoides (MF) was diagnosed following correlation of the RCM findings with the clinical picture.
.")
 scattered among keratinocytes in vesiclelike dark spaces (A)...")
A biopsy was performed, with pathologic examination confirming the diagnosis of tumor-stage MF. Parakeratosis with epidermotropism of lymphocytes was noted along the basal layer and into the spinous layer of the epidermis (Figure 3). Underlying the epidermis there was a dense mononuclear infiltrate and conspicuous eosinophils extending to the deeper reticular dermis. The infiltrating cells had cerebriform nuclei and large pale cytoplasm. On immunostaining, the lymphocytes were positive for CD3 and CD4, and negative for CD5, CD7, and CD8. The patient was referred to the oncology department for disease management. Staging workup including computed tomography, flow cytometry, and T-cell receptor gene rearrangement were consistent with tumor-stage MF (T3N0M0B0).
 as well as a dense infiltrate in the dermis (A)(H&E, original magnifications ×10 and ×50 [inset])...")
Comment
Clinical Presentation of MF
Mycosis fungoides, a non-Hodgkin lymphoma of T-cell origin, is the most commonly diagnosed cutaneous lymphoma worldwide.1 It has an annual incidence of approximately 0.36 per 100,000 persons, and this number continues to rise.2,3 The median age of diagnosis is 55 to 60 years, and MF occurs twice as often in men versus women.4
The clinical presentation of MF varies and is classified by stages including patches, plaques, tumors, and erythroderma.5 Classically, MF is slowly progressive and begins as pruritic erythematous patches that have a predilection for non–sun-exposed areas of the skin. Over time, these patches may evolve into plaques and tumors. Early or patch-stage MF often presents as well-demarcated lesions of various sizes and shapes that tend to enlarge.6 These lesions may resemble eczema or psoriasis if there is scaling, such as in our patient. At the tumor stage, flat or dome-shaped nodules that may vary in color and are deeper than plaques begin to appear. Ulcerations, which were absent in our case, may often be seen.
Because of the diverse clinical manifestations of MF, which can mimic other common dermatoses, diagnosis often is challenging for clinicians. Furthermore, histology can yield nonspecific diagnostic results and may even resemble chronic inflammatory dermatoses.7 As a result, patients frequently are subjected to multiple skin biopsies to establish the diagnosis,8 and diagnosis may be delayed, with the median time from onset of skin symptoms to diagnosis being approximately 6 years.9
Reflectance Confocal Microscopy
In vivo RCM is a noninvasive technique that allows visualization of the skin at a cellular level and recently has been evaluated as a diagnostic tool for many skin conditions.10,11 Reflectance confocal microscopy findings have been well established for many cutaneous malignancies as well as inflammatory conditions such as psoriasis and atopic dermatitis.12,13 Specifically, 2 preliminary descriptive studies utilized RCM to visualize the characteristic features of MF in vivo.14,15 These studies reported the histopathologic correlation of RCM findings in biopsy-proven MF lesions. Consistent in all stages of MF is the presence of small, weakly refractile, round to oval cells within the spinous layer that correlate with atypical lymphocytes, in addition to hyporefractile basal cells surrounding the dermal papillae. Patch-stage MF lesions have more subtle epidermal findings compared to plaque-stage lesions, which tend to have more prominent vesiclelike dark spaces filled with collections of monomorphous, weakly refractile, round to oval cells corresponding with Pautrier microabscesses and evidence of spongiosis.14,15 The first descriptive study of RCM in the diagnosis of MF failed to identify features of tumor-stage MF that would distinguish it from patch- or plaque-stage disease. The investigators also stated that deep nodular collections of atypical lymphocytes seen on histopathology in tumor-stage MF were missed on RCM evaluation.14 Furthermore, the second descriptive study of RCM and MF, which included 2 patients with tumor-stage disease, also failed to differentiate tumor-stage MF from the patch or plaque stages.15
Because of these 2 descriptive studies, a pilot study was conducted to determine the applicability and reproducibility of RCM findings for MF diagnosis.16 Two blinded confocalists were asked to diagnose RCM images as MF when compared to either normal skin or a variety of lymphoproliferative disorders. Of 15 patients, the confocalists correctly diagnosed MF in 84% and 90% of cases, respectively. Additionally, they reported the specificity and sensitivity of the following RCM features in the diagnosis of MF: spongiosis, 88.9% and 94.7%; loss of demarcation, 88.9% and 94.7%; disarray of the epidermis, 77.8% and 89.5%; hyporefractile rings, 88.9% and 78.9%; junctional atypical lymphocytes, 100% and 73.7%; and vesiclelike structures (Pautrier microabscesses), 100% and 73.7%. Importantly, this study did not evaluate the specificity and sensitivity of MF diagnosis compared to other eczematous or inflammatory conditions that may share similar RCM findings; therefore, these results are not generalizable, and many of the RCM findings characteristically seen in MF are not specific to its diagnosis.16
One study assessed the diagnostic accuracy of RCM in evaluating erythematosquamous diseases including MF, psoriasis, contact dermatitis, discoid lupus, and subacute cutaneous lupus.17 In this study, 3 blinded confocalists achieved a 95.41% and 92.89% specificity and 89.13% and 63.33% sensitivity for psoriasis and MF, respectively. Typical features of psoriasis on RCM included parakeratosis, reduction or absence of the granular layer, papillomatosis, acanthosis with normal honeycomb pattern of the epidermis, and dilated vessels in the upper dermis. Features that were more specific to MF included epidermotropic atypical lymphocytes, interface dermatitis, pleomorphic tumor cells, and dendritic cells.17 However, atypical lymphocytes and interface dermatitis also may be seen in cutaneous lupus; therefore, additional studies are still needed to validate RCM’s utility in differentiating between erythematosquamous skin diseases, including psoriasis, cutaneous lupus, and MF. Currently, RCM findings must be interpreted in conjunction with the clinical and histologic picture.
Importantly, RCM also is limited when evaluating MF due to its limited depth of visualization, as it allows imaging only to the superficial papillary dermis. Furthermore, any infiltrative process such as epidermal hyperplasia, spongiosis, or scaling, which can be seen in MF, may further impair the imaging quality of the deeper dermis.
Conclusion
Despite its limitations, RCM has the potential to be advantageous in evaluating skin lesions suspicious for MF in real time and is a promising technology for a quick noninvasive bedside adjunct tool. Its utility in selecting the optimal site for biopsy for better yield of histopathologic results in suspected MF cases has been demonstrated.16 However, large-scale studies still are needed to evaluate RCM in the diagnosis of the wide diversity of MF lesions as well as its efficacy in selecting optimal biopsy sites.
- Lutzner M, Edelson R, Schein P, et al. Cutaneous T-cell lymphomas: the Sézary syndrome, mycosis fungoides, and related disorders. Ann Intern Med. 1975;83:534-552.
- Akinbami AA, Osikomaiya BI, John-Olabode SO, et al. Mycosis fungoides: case report and literature review. Clin Med Insights Case Rep. 2014;7:95-98.
- Criscione VD, Weinstock MA. Incidence of cutaneous T-cell lymphoma in the United States, 1973-2002. Arch Dermatol. 2007;143:854-959.
- Bradford PT, Devesa SS, Anderson WF, et al. Cutaneous lymphoma incidence patterns in the United States: a population-based study of 3884 cases. Blood. 2009;113:5064-5073.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785.
- Nashan D, Faulhaber D, Stander S. Mycosis fungoides: a dermatological masquerader. Br J Dermatol. 2007;157:1-10.
- Santucci M, Biggeri A, Feller AC, et al. Efficacy of histologic criteria for diagnosing early mycosis fungoides: an EORTC cutaneous lymphoma study group investigation. European Organization for Research and Treatment of Cancer. Am J Surg Pathol. 2000;24:40-50.
- Glass LF, Keller KL, Messina JL, et al. Cutaneous T-cell lymphoma. Cancer Control. 1998;5:11-18.
- Hoppe RT, Wood GS, Abel EA. Mycosis fungoides and the Sézary syndrome: pathology, staging, and treatment. Curr Probl Cancer. 1990;14:293-371.
- Tannous ZS, Mihm MC, Flotte TJ, et al. In vivo examination of lentigo maligna and malignant melanoma in situ, lentigo maligna type by near-infrared reflectance confocal microscopy: comparison of in vivo confocal images with histologic sections. J Am Acad Dermatol. 2002;46:260-263.
- Gerger A, Koller S, Weger W, et al. Sensitivity and specificity of confocal laser-scanning microscopy for in vivo diagnosis of malignant skin tumors. Cancer. 2006;107:193-200.
- Branzan AL, Landthaler M, Szeimies RM. In vivo confocal scanning laser microscopy in dermatology [published online November 18, 2006]. Lasers Med Sci. 2007;22:73-82.
- González S. Confocal reflectance microscopy in dermatology: promise and reality of non-invasive diagnosis and monitoring. Actas Dermosifiliogr. 2009;100(suppl 2):59-69.
- Agero AL, Gill M, Ardigo M, et al. In vivo reflectance confocal microscopy of mycosis fungoides: a preliminary study [published online April 16, 2007]. J Am Acad Dermatol. 2007;57:435-441.
- Wi L, Dai H, Li Z, et al. Reflectance confocal microscopy for the characteristics of mycosis fungoides and correlation with histology: a pilot study [published online April 18, 2013]. Skin Res Technol. 2013;19:352-355.
- Lange-Asschenfeldt S, Babilli J, Beyer M, et al. Consistency and distribution of reflectance confocal microscopy features for diagnosis of cutaneous T cell lymphoma. J Biomed Opt. 2012;17:016001.
- Koller S, Gerger A, Ahlgrimm-Siess V. In vivo reflectance confocal microscopy of erythematosquamous skin diseases [published online March 6, 2009]. Exp Dermatol. 2009;18:536-540.
Case Report
A 60-year-old man with a history of Hodgkin lymphoma that had been treated with chemotherapy 6 years prior presented to our dermatology clinic with a persistent pruritic rash on the back, abdomen, and bilateral arms and legs. The eruption initially began as localized discrete lesions on the lower back 1 year prior to the current presentation; at that time a diagnosis of psoriasis was made at an outside dermatology clinic, and treatment with mometasone furoate cream was initiated. Despite the patient’s compliance with this treatment, the lesions did not resolve and began spreading to the arms, legs, chest, and abdomen. His current medications included lisinopril, escitalopram, aspirin, and omeprazole.
On presentation to our clinic, physical examination revealed round, scaly, pink plaques and tumors of variable sizes (3–10 cm) distributed asymmetrically on the chest, back, abdomen, arms, and legs (Figure 1). The lesions were grouped in well-defined areas encompassing approximately 30% of the body surface area. No lymphadenopathy was appreciated. In vivo reflectance confocal microscopy (RCM) performed on one of the lesions revealed disarray of the epidermis with small, weakly refractile, round to oval cells scattered within the spinous layer and dermoepidermal junction (Figure 2). Additionally, these weakly refractile, round to oval cells also were seen in vesiclelike dark spaces, and hyporefractile basal cells were appreciated surrounding the dermal papillae. Mycosis fungoides (MF) was diagnosed following correlation of the RCM findings with the clinical picture.
A biopsy was performed, with pathologic examination confirming the diagnosis of tumor-stage MF. Parakeratosis with epidermotropism of lymphocytes was noted along the basal layer and into the spinous layer of the epidermis (Figure 3). Underlying the epidermis there was a dense mononuclear infiltrate and conspicuous eosinophils extending to the deeper reticular dermis. The infiltrating cells had cerebriform nuclei and large pale cytoplasm. On immunostaining, the lymphocytes were positive for CD3 and CD4, and negative for CD5, CD7, and CD8. The patient was referred to the oncology department for disease management. Staging workup including computed tomography, flow cytometry, and T-cell receptor gene rearrangement were consistent with tumor-stage MF (T3N0M0B0).
Comment
Clinical Presentation of MF
Mycosis fungoides, a non-Hodgkin lymphoma of T-cell origin, is the most commonly diagnosed cutaneous lymphoma worldwide.1 It has an annual incidence of approximately 0.36 per 100,000 persons, and this number continues to rise.2,3 The median age of diagnosis is 55 to 60 years, and MF occurs twice as often in men versus women.4
The clinical presentation of MF varies and is classified by stages including patches, plaques, tumors, and erythroderma.5 Classically, MF is slowly progressive and begins as pruritic erythematous patches that have a predilection for non–sun-exposed areas of the skin. Over time, these patches may evolve into plaques and tumors. Early or patch-stage MF often presents as well-demarcated lesions of various sizes and shapes that tend to enlarge.6 These lesions may resemble eczema or psoriasis if there is scaling, such as in our patient. At the tumor stage, flat or dome-shaped nodules that may vary in color and are deeper than plaques begin to appear. Ulcerations, which were absent in our case, may often be seen.
Because of the diverse clinical manifestations of MF, which can mimic other common dermatoses, diagnosis often is challenging for clinicians. Furthermore, histology can yield nonspecific diagnostic results and may even resemble chronic inflammatory dermatoses.7 As a result, patients frequently are subjected to multiple skin biopsies to establish the diagnosis,8 and diagnosis may be delayed, with the median time from onset of skin symptoms to diagnosis being approximately 6 years.9
Reflectance Confocal Microscopy
In vivo RCM is a noninvasive technique that allows visualization of the skin at a cellular level and recently has been evaluated as a diagnostic tool for many skin conditions.10,11 Reflectance confocal microscopy findings have been well established for many cutaneous malignancies as well as inflammatory conditions such as psoriasis and atopic dermatitis.12,13 Specifically, 2 preliminary descriptive studies utilized RCM to visualize the characteristic features of MF in vivo.14,15 These studies reported the histopathologic correlation of RCM findings in biopsy-proven MF lesions. Consistent in all stages of MF is the presence of small, weakly refractile, round to oval cells within the spinous layer that correlate with atypical lymphocytes, in addition to hyporefractile basal cells surrounding the dermal papillae. Patch-stage MF lesions have more subtle epidermal findings compared to plaque-stage lesions, which tend to have more prominent vesiclelike dark spaces filled with collections of monomorphous, weakly refractile, round to oval cells corresponding with Pautrier microabscesses and evidence of spongiosis.14,15 The first descriptive study of RCM in the diagnosis of MF failed to identify features of tumor-stage MF that would distinguish it from patch- or plaque-stage disease. The investigators also stated that deep nodular collections of atypical lymphocytes seen on histopathology in tumor-stage MF were missed on RCM evaluation.14 Furthermore, the second descriptive study of RCM and MF, which included 2 patients with tumor-stage disease, also failed to differentiate tumor-stage MF from the patch or plaque stages.15
Because of these 2 descriptive studies, a pilot study was conducted to determine the applicability and reproducibility of RCM findings for MF diagnosis.16 Two blinded confocalists were asked to diagnose RCM images as MF when compared to either normal skin or a variety of lymphoproliferative disorders. Of 15 patients, the confocalists correctly diagnosed MF in 84% and 90% of cases, respectively. Additionally, they reported the specificity and sensitivity of the following RCM features in the diagnosis of MF: spongiosis, 88.9% and 94.7%; loss of demarcation, 88.9% and 94.7%; disarray of the epidermis, 77.8% and 89.5%; hyporefractile rings, 88.9% and 78.9%; junctional atypical lymphocytes, 100% and 73.7%; and vesiclelike structures (Pautrier microabscesses), 100% and 73.7%. Importantly, this study did not evaluate the specificity and sensitivity of MF diagnosis compared to other eczematous or inflammatory conditions that may share similar RCM findings; therefore, these results are not generalizable, and many of the RCM findings characteristically seen in MF are not specific to its diagnosis.16
One study assessed the diagnostic accuracy of RCM in evaluating erythematosquamous diseases including MF, psoriasis, contact dermatitis, discoid lupus, and subacute cutaneous lupus.17 In this study, 3 blinded confocalists achieved a 95.41% and 92.89% specificity and 89.13% and 63.33% sensitivity for psoriasis and MF, respectively. Typical features of psoriasis on RCM included parakeratosis, reduction or absence of the granular layer, papillomatosis, acanthosis with normal honeycomb pattern of the epidermis, and dilated vessels in the upper dermis. Features that were more specific to MF included epidermotropic atypical lymphocytes, interface dermatitis, pleomorphic tumor cells, and dendritic cells.17 However, atypical lymphocytes and interface dermatitis also may be seen in cutaneous lupus; therefore, additional studies are still needed to validate RCM’s utility in differentiating between erythematosquamous skin diseases, including psoriasis, cutaneous lupus, and MF. Currently, RCM findings must be interpreted in conjunction with the clinical and histologic picture.
Importantly, RCM also is limited when evaluating MF due to its limited depth of visualization, as it allows imaging only to the superficial papillary dermis. Furthermore, any infiltrative process such as epidermal hyperplasia, spongiosis, or scaling, which can be seen in MF, may further impair the imaging quality of the deeper dermis.
Conclusion
Despite its limitations, RCM has the potential to be advantageous in evaluating skin lesions suspicious for MF in real time and is a promising technology for a quick noninvasive bedside adjunct tool. Its utility in selecting the optimal site for biopsy for better yield of histopathologic results in suspected MF cases has been demonstrated.16 However, large-scale studies still are needed to evaluate RCM in the diagnosis of the wide diversity of MF lesions as well as its efficacy in selecting optimal biopsy sites.
Case Report
A 60-year-old man with a history of Hodgkin lymphoma that had been treated with chemotherapy 6 years prior presented to our dermatology clinic with a persistent pruritic rash on the back, abdomen, and bilateral arms and legs. The eruption initially began as localized discrete lesions on the lower back 1 year prior to the current presentation; at that time a diagnosis of psoriasis was made at an outside dermatology clinic, and treatment with mometasone furoate cream was initiated. Despite the patient’s compliance with this treatment, the lesions did not resolve and began spreading to the arms, legs, chest, and abdomen. His current medications included lisinopril, escitalopram, aspirin, and omeprazole.
On presentation to our clinic, physical examination revealed round, scaly, pink plaques and tumors of variable sizes (3–10 cm) distributed asymmetrically on the chest, back, abdomen, arms, and legs (Figure 1). The lesions were grouped in well-defined areas encompassing approximately 30% of the body surface area. No lymphadenopathy was appreciated. In vivo reflectance confocal microscopy (RCM) performed on one of the lesions revealed disarray of the epidermis with small, weakly refractile, round to oval cells scattered within the spinous layer and dermoepidermal junction (Figure 2). Additionally, these weakly refractile, round to oval cells also were seen in vesiclelike dark spaces, and hyporefractile basal cells were appreciated surrounding the dermal papillae. Mycosis fungoides (MF) was diagnosed following correlation of the RCM findings with the clinical picture.
A biopsy was performed, with pathologic examination confirming the diagnosis of tumor-stage MF. Parakeratosis with epidermotropism of lymphocytes was noted along the basal layer and into the spinous layer of the epidermis (Figure 3). Underlying the epidermis there was a dense mononuclear infiltrate and conspicuous eosinophils extending to the deeper reticular dermis. The infiltrating cells had cerebriform nuclei and large pale cytoplasm. On immunostaining, the lymphocytes were positive for CD3 and CD4, and negative for CD5, CD7, and CD8. The patient was referred to the oncology department for disease management. Staging workup including computed tomography, flow cytometry, and T-cell receptor gene rearrangement were consistent with tumor-stage MF (T3N0M0B0).
Comment
Clinical Presentation of MF
Mycosis fungoides, a non-Hodgkin lymphoma of T-cell origin, is the most commonly diagnosed cutaneous lymphoma worldwide.1 It has an annual incidence of approximately 0.36 per 100,000 persons, and this number continues to rise.2,3 The median age of diagnosis is 55 to 60 years, and MF occurs twice as often in men versus women.4
The clinical presentation of MF varies and is classified by stages including patches, plaques, tumors, and erythroderma.5 Classically, MF is slowly progressive and begins as pruritic erythematous patches that have a predilection for non–sun-exposed areas of the skin. Over time, these patches may evolve into plaques and tumors. Early or patch-stage MF often presents as well-demarcated lesions of various sizes and shapes that tend to enlarge.6 These lesions may resemble eczema or psoriasis if there is scaling, such as in our patient. At the tumor stage, flat or dome-shaped nodules that may vary in color and are deeper than plaques begin to appear. Ulcerations, which were absent in our case, may often be seen.
Because of the diverse clinical manifestations of MF, which can mimic other common dermatoses, diagnosis often is challenging for clinicians. Furthermore, histology can yield nonspecific diagnostic results and may even resemble chronic inflammatory dermatoses.7 As a result, patients frequently are subjected to multiple skin biopsies to establish the diagnosis,8 and diagnosis may be delayed, with the median time from onset of skin symptoms to diagnosis being approximately 6 years.9
Reflectance Confocal Microscopy
In vivo RCM is a noninvasive technique that allows visualization of the skin at a cellular level and recently has been evaluated as a diagnostic tool for many skin conditions.10,11 Reflectance confocal microscopy findings have been well established for many cutaneous malignancies as well as inflammatory conditions such as psoriasis and atopic dermatitis.12,13 Specifically, 2 preliminary descriptive studies utilized RCM to visualize the characteristic features of MF in vivo.14,15 These studies reported the histopathologic correlation of RCM findings in biopsy-proven MF lesions. Consistent in all stages of MF is the presence of small, weakly refractile, round to oval cells within the spinous layer that correlate with atypical lymphocytes, in addition to hyporefractile basal cells surrounding the dermal papillae. Patch-stage MF lesions have more subtle epidermal findings compared to plaque-stage lesions, which tend to have more prominent vesiclelike dark spaces filled with collections of monomorphous, weakly refractile, round to oval cells corresponding with Pautrier microabscesses and evidence of spongiosis.14,15 The first descriptive study of RCM in the diagnosis of MF failed to identify features of tumor-stage MF that would distinguish it from patch- or plaque-stage disease. The investigators also stated that deep nodular collections of atypical lymphocytes seen on histopathology in tumor-stage MF were missed on RCM evaluation.14 Furthermore, the second descriptive study of RCM and MF, which included 2 patients with tumor-stage disease, also failed to differentiate tumor-stage MF from the patch or plaque stages.15
Because of these 2 descriptive studies, a pilot study was conducted to determine the applicability and reproducibility of RCM findings for MF diagnosis.16 Two blinded confocalists were asked to diagnose RCM images as MF when compared to either normal skin or a variety of lymphoproliferative disorders. Of 15 patients, the confocalists correctly diagnosed MF in 84% and 90% of cases, respectively. Additionally, they reported the specificity and sensitivity of the following RCM features in the diagnosis of MF: spongiosis, 88.9% and 94.7%; loss of demarcation, 88.9% and 94.7%; disarray of the epidermis, 77.8% and 89.5%; hyporefractile rings, 88.9% and 78.9%; junctional atypical lymphocytes, 100% and 73.7%; and vesiclelike structures (Pautrier microabscesses), 100% and 73.7%. Importantly, this study did not evaluate the specificity and sensitivity of MF diagnosis compared to other eczematous or inflammatory conditions that may share similar RCM findings; therefore, these results are not generalizable, and many of the RCM findings characteristically seen in MF are not specific to its diagnosis.16
One study assessed the diagnostic accuracy of RCM in evaluating erythematosquamous diseases including MF, psoriasis, contact dermatitis, discoid lupus, and subacute cutaneous lupus.17 In this study, 3 blinded confocalists achieved a 95.41% and 92.89% specificity and 89.13% and 63.33% sensitivity for psoriasis and MF, respectively. Typical features of psoriasis on RCM included parakeratosis, reduction or absence of the granular layer, papillomatosis, acanthosis with normal honeycomb pattern of the epidermis, and dilated vessels in the upper dermis. Features that were more specific to MF included epidermotropic atypical lymphocytes, interface dermatitis, pleomorphic tumor cells, and dendritic cells.17 However, atypical lymphocytes and interface dermatitis also may be seen in cutaneous lupus; therefore, additional studies are still needed to validate RCM’s utility in differentiating between erythematosquamous skin diseases, including psoriasis, cutaneous lupus, and MF. Currently, RCM findings must be interpreted in conjunction with the clinical and histologic picture.
Importantly, RCM also is limited when evaluating MF due to its limited depth of visualization, as it allows imaging only to the superficial papillary dermis. Furthermore, any infiltrative process such as epidermal hyperplasia, spongiosis, or scaling, which can be seen in MF, may further impair the imaging quality of the deeper dermis.
Conclusion
Despite its limitations, RCM has the potential to be advantageous in evaluating skin lesions suspicious for MF in real time and is a promising technology for a quick noninvasive bedside adjunct tool. Its utility in selecting the optimal site for biopsy for better yield of histopathologic results in suspected MF cases has been demonstrated.16 However, large-scale studies still are needed to evaluate RCM in the diagnosis of the wide diversity of MF lesions as well as its efficacy in selecting optimal biopsy sites.
- Lutzner M, Edelson R, Schein P, et al. Cutaneous T-cell lymphomas: the Sézary syndrome, mycosis fungoides, and related disorders. Ann Intern Med. 1975;83:534-552.
- Akinbami AA, Osikomaiya BI, John-Olabode SO, et al. Mycosis fungoides: case report and literature review. Clin Med Insights Case Rep. 2014;7:95-98.
- Criscione VD, Weinstock MA. Incidence of cutaneous T-cell lymphoma in the United States, 1973-2002. Arch Dermatol. 2007;143:854-959.
- Bradford PT, Devesa SS, Anderson WF, et al. Cutaneous lymphoma incidence patterns in the United States: a population-based study of 3884 cases. Blood. 2009;113:5064-5073.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785.
- Nashan D, Faulhaber D, Stander S. Mycosis fungoides: a dermatological masquerader. Br J Dermatol. 2007;157:1-10.
- Santucci M, Biggeri A, Feller AC, et al. Efficacy of histologic criteria for diagnosing early mycosis fungoides: an EORTC cutaneous lymphoma study group investigation. European Organization for Research and Treatment of Cancer. Am J Surg Pathol. 2000;24:40-50.
- Glass LF, Keller KL, Messina JL, et al. Cutaneous T-cell lymphoma. Cancer Control. 1998;5:11-18.
- Hoppe RT, Wood GS, Abel EA. Mycosis fungoides and the Sézary syndrome: pathology, staging, and treatment. Curr Probl Cancer. 1990;14:293-371.
- Tannous ZS, Mihm MC, Flotte TJ, et al. In vivo examination of lentigo maligna and malignant melanoma in situ, lentigo maligna type by near-infrared reflectance confocal microscopy: comparison of in vivo confocal images with histologic sections. J Am Acad Dermatol. 2002;46:260-263.
- Gerger A, Koller S, Weger W, et al. Sensitivity and specificity of confocal laser-scanning microscopy for in vivo diagnosis of malignant skin tumors. Cancer. 2006;107:193-200.
- Branzan AL, Landthaler M, Szeimies RM. In vivo confocal scanning laser microscopy in dermatology [published online November 18, 2006]. Lasers Med Sci. 2007;22:73-82.
- González S. Confocal reflectance microscopy in dermatology: promise and reality of non-invasive diagnosis and monitoring. Actas Dermosifiliogr. 2009;100(suppl 2):59-69.
- Agero AL, Gill M, Ardigo M, et al. In vivo reflectance confocal microscopy of mycosis fungoides: a preliminary study [published online April 16, 2007]. J Am Acad Dermatol. 2007;57:435-441.
- Wi L, Dai H, Li Z, et al. Reflectance confocal microscopy for the characteristics of mycosis fungoides and correlation with histology: a pilot study [published online April 18, 2013]. Skin Res Technol. 2013;19:352-355.
- Lange-Asschenfeldt S, Babilli J, Beyer M, et al. Consistency and distribution of reflectance confocal microscopy features for diagnosis of cutaneous T cell lymphoma. J Biomed Opt. 2012;17:016001.
- Koller S, Gerger A, Ahlgrimm-Siess V. In vivo reflectance confocal microscopy of erythematosquamous skin diseases [published online March 6, 2009]. Exp Dermatol. 2009;18:536-540.
- Lutzner M, Edelson R, Schein P, et al. Cutaneous T-cell lymphomas: the Sézary syndrome, mycosis fungoides, and related disorders. Ann Intern Med. 1975;83:534-552.
- Akinbami AA, Osikomaiya BI, John-Olabode SO, et al. Mycosis fungoides: case report and literature review. Clin Med Insights Case Rep. 2014;7:95-98.
- Criscione VD, Weinstock MA. Incidence of cutaneous T-cell lymphoma in the United States, 1973-2002. Arch Dermatol. 2007;143:854-959.
- Bradford PT, Devesa SS, Anderson WF, et al. Cutaneous lymphoma incidence patterns in the United States: a population-based study of 3884 cases. Blood. 2009;113:5064-5073.
- Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005;105:3768-3785.
- Nashan D, Faulhaber D, Stander S. Mycosis fungoides: a dermatological masquerader. Br J Dermatol. 2007;157:1-10.
- Santucci M, Biggeri A, Feller AC, et al. Efficacy of histologic criteria for diagnosing early mycosis fungoides: an EORTC cutaneous lymphoma study group investigation. European Organization for Research and Treatment of Cancer. Am J Surg Pathol. 2000;24:40-50.
- Glass LF, Keller KL, Messina JL, et al. Cutaneous T-cell lymphoma. Cancer Control. 1998;5:11-18.
- Hoppe RT, Wood GS, Abel EA. Mycosis fungoides and the Sézary syndrome: pathology, staging, and treatment. Curr Probl Cancer. 1990;14:293-371.
- Tannous ZS, Mihm MC, Flotte TJ, et al. In vivo examination of lentigo maligna and malignant melanoma in situ, lentigo maligna type by near-infrared reflectance confocal microscopy: comparison of in vivo confocal images with histologic sections. J Am Acad Dermatol. 2002;46:260-263.
- Gerger A, Koller S, Weger W, et al. Sensitivity and specificity of confocal laser-scanning microscopy for in vivo diagnosis of malignant skin tumors. Cancer. 2006;107:193-200.
- Branzan AL, Landthaler M, Szeimies RM. In vivo confocal scanning laser microscopy in dermatology [published online November 18, 2006]. Lasers Med Sci. 2007;22:73-82.
- González S. Confocal reflectance microscopy in dermatology: promise and reality of non-invasive diagnosis and monitoring. Actas Dermosifiliogr. 2009;100(suppl 2):59-69.
- Agero AL, Gill M, Ardigo M, et al. In vivo reflectance confocal microscopy of mycosis fungoides: a preliminary study [published online April 16, 2007]. J Am Acad Dermatol. 2007;57:435-441.
- Wi L, Dai H, Li Z, et al. Reflectance confocal microscopy for the characteristics of mycosis fungoides and correlation with histology: a pilot study [published online April 18, 2013]. Skin Res Technol. 2013;19:352-355.
- Lange-Asschenfeldt S, Babilli J, Beyer M, et al. Consistency and distribution of reflectance confocal microscopy features for diagnosis of cutaneous T cell lymphoma. J Biomed Opt. 2012;17:016001.
- Koller S, Gerger A, Ahlgrimm-Siess V. In vivo reflectance confocal microscopy of erythematosquamous skin diseases [published online March 6, 2009]. Exp Dermatol. 2009;18:536-540.
Practice Points
- Mycosis fungoides (MF) can be a challenging diagnosis to establish and often requires multiple biopsies.
- Reflectance confocal microscopy (RCM) may be helpful as a bedside noninvasive diagnostic technique.
- In suspected MF cases, RCM may assist in selecting the optimal biopsy site for better yield of histopathologic results.
Nonscarring Alopecia Associated With Vitamin D Deficiency
Vitamin D receptors are found in every cell of the body and have been shown to play a role in bone, neural, and cardiovascular health; immune regulation; and possibly cancer prevention via the regulation of cell differentiation, proliferation, and apoptosis.1 Although it is controversial, vitamin D deficiency has been associated with various forms of nonscarring hair loss,2-4 including telogen effluvium, androgenetic alopecia, and alopecia areata. We describe a notable case of nonscarring alopecia associated with vitamin D deficiency in which vitamin D replacement therapy promoted hair regrowth.
Case Report
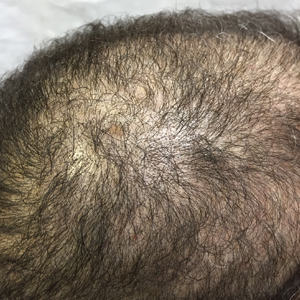
An otherwise healthy 34-year-old black woman presented to the Hair and Nail Clinic at the University of Pittsburgh Medical Center (Pittsburgh, Pennsylvania) for evaluation of progressive hair loss of 4 years’ duration that began shortly after her fourth child was born. Although she denied any history of excessive shedding, she stated that she used to have shoulder-length hair and somehow it had become extremely short without shaving or cutting the hair (Figure 1). Her current medications included a progestin intrauterine device and biotin 10 mg once daily, the latter of which she had taken for several months for the hair loss without any improvement.
.")
On physical examination, the patient was noted to have diffusely thinning, short, brittle hair. Trichoscopy was notable for hairs of varying diameters, with some fractured at the level of the follicular ostia but no yellow dots at the follicular openings or exclamation point hairs. No scarring or erythema was seen on the scalp. The patient refused several of our team’s recommendations for scalp biopsy due to needle phobia. A hair growth window was made that showed good regrowth at 2 weeks after the initial presentation. Initial blood work revealed a total serum 25-hydroxyvitamin D level of 12 ng/mL (optimal, >30 ng/mL). Complete blood cell count, hormonal panel, zinc level, iron level, and thyroid studies were all normal.
The patient was started on vitamin D3 replacement therapy 50,000 IU once weekly for 4 weeks followed by 1000 IU once daily for 6 months. No other topical or systemic treatments were administered for the nonscarring alopecia. At a follow-up visit 6 months later, the patient’s vitamin D level was 36 ng/mL, and she had noticeable hair regrowth (Figure 2). At this time, the diagnosis of nonscarring alopecia associated with vitamin D deficiency was made.
.")
Comment
Vitamin D is a fat-soluble vitamin that can be obtained via sun exposure, food sources (eg, fish, vitamin D–fortified foods), and direct supplementation.5 It has been estimated that nearly 1 billion individuals worldwide6 and approximately 41.6% of US adults are vitamin D deficient.7 Certainly not all of these individuals will present with alopecia, but in patients with hair loss, we suggest that vitamin D deficiency is an important factor to consider. Risk factors for vitamin D deficiency include older age, obesity, darker skin types, residence in northern latitudes, and malabsorption syndromes.7
Pathogenesis
Vitamin D is thought to play a role in the normal initiation and completion of the hair cycle as well as the differentiation of the follicular and interfollicular epidermis. The vitamin D receptor (VDR) is thought to induce the development of mature anagen hairs via the canonical WNT-β-catenin and hedgehog signaling pathways.8 In the absence of VDRs, the stem cells in the bulge of the hair follicle have an impaired ability to replicate, and as a result, VDR-deficient mice have shown near-total hair loss.9-12 We propose that vitamin D deficiency can not only be a trigger for hair loss but also can perpetuate hair loss and poor regrowth.
Diagnosis and Prevention of Vitamin D Deficiency
In the skin, 7-dehydrocholesterol is converted to previtamin D3 via UVB light, followed by subsequent conversion to vitamin D3. Dietary sources are in the form of either vitamin D2 or D3, both of which are converted in the liver to 25-hydroxyvitamin D, the major circulating metabolite. In the kidneys, 25-hydroxyvitamin D is then converted to 1,25-dihydroxyvitamin D, the biologically active form. Paradoxically, serum levels of 1,25-dihydroxyvitamin D can be normal or high in the setting of vitamin D deficiency; therefore, serum total 25-hydroxyvitamin D is the best way to assess a patient’s vitamin D status.5,13
The optimal serum 25-hydroxyvitamin D level is controversial. Recommendations range between 20 to 40 ng/mL14 and 30 to 50 ng/mL.13,15,16 Vitamin D levels higher than 50 ng/mL have been correlated with an increased risk of bone fractures and certain cancers.16-18 Vitamin D toxicity usually is noted in serum levels greater than 88 ng/mL; symptoms of toxicity include hypercalcemia, nausea, vomiting, and muscle weakness. For nondeficient patients, the National Academy of Medicine (formerly the Institute of Medicine) recommended an upper limit of 4000 IU daily.14 The optimal dose in preventing vitamin D deficiency ranges from 600 to 1000 IU daily.13-15
Treatment of Vitamin D Deficiency
In the setting of vitamin D deficiency, the amount required for repletion often is dependent on each individual’s ability to absorb and convert to 25-hydroxyvitamin D. Typically every 100 IU of vitamin D correlates with a 0.7 to 1.0 ng/mL increase in serum 25-hydroxyvitamin D levels.19 There are multiple dosing regimens used to achieve the desired serum 25-hydroxyvitamin D levels in deficient patients. One recommendation from the Endocrine Society is 50,000 IU once weekly for 6 to 8 weeks (single doses >50,000 IU typically are not recommended due to increased risk for toxicity), followed by 600 to 1000 IU once daily in children and 1500 to 2000 IU once daily in adults thereafter.13 In patients with vitamin D deficiency, reassessment of serum 25-hydroxyvitamin D levels is recommended after 3 to 4 months of treatment, and adjustments to the repletion regimen should be made as needed.15,16 Generally, vitamin D3 is recommended over vitamin D2 due to enhanced efficacy in raising serum 25-hydroxyvitamin D levels.20
Vitamin D Deficiency in Alopecia
Although most recommendations are given in the interest of optimizing bone health, in the setting of alopecia, we set a similar serum 25-hydroxyvitamin D goal of greater than 30 ng/mL. We recommend treatment with vitamin D3 and practice the following repletion protocol: 50,000 IU once weekly for 4 weeks, followed by 1000 IU once daily for at least 8 weeks for serum 25-hydroxyvitamin D levels less than 20 ng/mL. For serum hydroxyvitamin D levels between 20 and 29 ng/mL, we recommend 1000 IU once daily for at least 12 weeks. We recheck blood levels again in 3 months. If levels fail to normalize, we will refer the patient to endocrinology. If levels return to normal, we transition to a daily multivitamin with vitamin D (400–800 IU) once daily and refer the patient back to the primary care physician for long-term monitoring.
- Nagpal S, Na S, Rathnachalam R. Noncalcemic actions of vitamin D receptor ligands. Endocr Rev. 2005;26:662-687.
- Cheung EJ, Sink JR, English III JC. Vitamin and mineral deficiencies in patients with telogen effluvium: a retrospective cross-sectional study. J Drugs Dermatol. 2016;15:1235-1237.
- Rasheed H, Mahgoub D, Hegazy R, et al. Serum ferritin and vitamin D in female hair loss: do they play a role? Skin Pharmacol Physiol. 2013;26:101-107.
- Aksu Cerman A, Sarikaya Solak S, Kivanc Altunay I. Vitamin D deficiency in alopecia areata. Br J Dermatol. 2014;170:1299-1304.
- Holick MF. Vitamin D deficiency. N Engl J Med. 2007;357:266-281.
- Holick MF. High prevalence of vitamin D inadequacy and implications for health. Mayo Clin Proc. 2006;81:353-373.
- Yetley EA. Assessing the vitamin D status of the US population. Am J Clin Nutr. 2008;88:558S-564S.
- Lisse TS, Saini V, Zhao H, et al. The vitamin D receptor is required for activation of cWnt and hedgehog signaling in keratinocytes. Mol Endocrinol. 2014;28:1698-1706.
- Cianferotti L, Cox M, Skorjia K, et al. Vitamin D receptor is essential for normal keratinocyte stem cell function [published online May 17, 2007]. Porc Natl Acad Sci U S A. 2007;104:9428-9433.
- Xie Z, Komuves L, Yu QC, et al. Lack of the vitamin D receptor is associated with reduced epidermal differentiation and hair follicle growth. J Invest Dermatol. 2002;118:11-16.
- Kong J, Li XJ, Gavin D, et al. Targeted expression of human vitamin D receptor in the skin promotes the initiation of the postnatal hair follicle cycle and rescues the alopecia in vitamin D receptor null mice. J Invest Dermatol. 2002;118:631-638.
- Bikle DD, Elalieh H, Chang S, et al. Development and progression of alopecia in the vitamin D receptor null mouse. J Cell Physiol. 2006;207:340-353.
- Holick MF, Binkley NC, Bischoff-Ferrari HA, et al; Endocrine Society. Evaluation, treatment, and prevention of vitamin D deficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2011;96:1911-1930.
- Ross AC, Manson JE, Abrams SA, et al. The 2011 report on dietary reference intakes for calcium and vitamin D from the Institute of Medicine: what clinicians need to know. J Clin Endocrinol Metab. 2011;96:53-58.
- Dawson-Hughes B, Mithal A, Bonjour JP, et al. IOF position statement: vitamin D recommendations for older adults. Osteoporos Int. 2010;21:1151-1154.
- Judge J, Birge S, Gloth F 3rd; American Geriatrics Society Workgroup on Vitamin D Supplementation for Older Adults. Recommendations abstracted from the American Geriatrics Society Consensus Statement on vitamin D for prevention of falls and their consequences. J Am Geriatr Soc. 2014;62:147-152.
- Ahn J, Peters U, Albanes D, et al; Prostate, Lung, Colorectal, and Ovarian Cancer Screening Trial Project Team. Serum vitamin D concentration and prostate cancer risk: a nested case-control study. J Natl Cancer Inst. 2008;4:100:796-804.
- Stolzenberg-Solomon RZ, Jacobs EJ, Arslan AA, et al. Circulating 25-hydroxyvitamin D and risk of pancreatic cancer: Cohort Consortium Vitamin D Pooling Project of Rarer Cancers [published online June 18, 2010]. Am J Epidemiol. 2010;172:81-93.
- Heaney RP, Davies KM, Chen TC, et al. Human serum 25-hydroxycholecalciferol response to extended oral dosing with cholecalciferol. Am J Clin Nutr. 2003;77:204-210. Erratum in: 2003;78:1047.
- Tripkovic L, Lambert H, Hart K, et al. Comparison of vitamin D2 and vitamin D3 supplementation in raising serum 25-hydroxyvitamin D status: a systematic review and meta-analysis. Am J Clin Nutr. 2012;95:1357-1364.
Vitamin D receptors are found in every cell of the body and have been shown to play a role in bone, neural, and cardiovascular health; immune regulation; and possibly cancer prevention via the regulation of cell differentiation, proliferation, and apoptosis.1 Although it is controversial, vitamin D deficiency has been associated with various forms of nonscarring hair loss,2-4 including telogen effluvium, androgenetic alopecia, and alopecia areata. We describe a notable case of nonscarring alopecia associated with vitamin D deficiency in which vitamin D replacement therapy promoted hair regrowth.
Case Report
An otherwise healthy 34-year-old black woman presented to the Hair and Nail Clinic at the University of Pittsburgh Medical Center (Pittsburgh, Pennsylvania) for evaluation of progressive hair loss of 4 years’ duration that began shortly after her fourth child was born. Although she denied any history of excessive shedding, she stated that she used to have shoulder-length hair and somehow it had become extremely short without shaving or cutting the hair (Figure 1). Her current medications included a progestin intrauterine device and biotin 10 mg once daily, the latter of which she had taken for several months for the hair loss without any improvement.
On physical examination, the patient was noted to have diffusely thinning, short, brittle hair. Trichoscopy was notable for hairs of varying diameters, with some fractured at the level of the follicular ostia but no yellow dots at the follicular openings or exclamation point hairs. No scarring or erythema was seen on the scalp. The patient refused several of our team’s recommendations for scalp biopsy due to needle phobia. A hair growth window was made that showed good regrowth at 2 weeks after the initial presentation. Initial blood work revealed a total serum 25-hydroxyvitamin D level of 12 ng/mL (optimal, >30 ng/mL). Complete blood cell count, hormonal panel, zinc level, iron level, and thyroid studies were all normal.
The patient was started on vitamin D3 replacement therapy 50,000 IU once weekly for 4 weeks followed by 1000 IU once daily for 6 months. No other topical or systemic treatments were administered for the nonscarring alopecia. At a follow-up visit 6 months later, the patient’s vitamin D level was 36 ng/mL, and she had noticeable hair regrowth (Figure 2). At this time, the diagnosis of nonscarring alopecia associated with vitamin D deficiency was made.
Comment
Vitamin D is a fat-soluble vitamin that can be obtained via sun exposure, food sources (eg, fish, vitamin D–fortified foods), and direct supplementation.5 It has been estimated that nearly 1 billion individuals worldwide6 and approximately 41.6% of US adults are vitamin D deficient.7 Certainly not all of these individuals will present with alopecia, but in patients with hair loss, we suggest that vitamin D deficiency is an important factor to consider. Risk factors for vitamin D deficiency include older age, obesity, darker skin types, residence in northern latitudes, and malabsorption syndromes.7
Pathogenesis
Vitamin D is thought to play a role in the normal initiation and completion of the hair cycle as well as the differentiation of the follicular and interfollicular epidermis. The vitamin D receptor (VDR) is thought to induce the development of mature anagen hairs via the canonical WNT-β-catenin and hedgehog signaling pathways.8 In the absence of VDRs, the stem cells in the bulge of the hair follicle have an impaired ability to replicate, and as a result, VDR-deficient mice have shown near-total hair loss.9-12 We propose that vitamin D deficiency can not only be a trigger for hair loss but also can perpetuate hair loss and poor regrowth.
Diagnosis and Prevention of Vitamin D Deficiency
In the skin, 7-dehydrocholesterol is converted to previtamin D3 via UVB light, followed by subsequent conversion to vitamin D3. Dietary sources are in the form of either vitamin D2 or D3, both of which are converted in the liver to 25-hydroxyvitamin D, the major circulating metabolite. In the kidneys, 25-hydroxyvitamin D is then converted to 1,25-dihydroxyvitamin D, the biologically active form. Paradoxically, serum levels of 1,25-dihydroxyvitamin D can be normal or high in the setting of vitamin D deficiency; therefore, serum total 25-hydroxyvitamin D is the best way to assess a patient’s vitamin D status.5,13
The optimal serum 25-hydroxyvitamin D level is controversial. Recommendations range between 20 to 40 ng/mL14 and 30 to 50 ng/mL.13,15,16 Vitamin D levels higher than 50 ng/mL have been correlated with an increased risk of bone fractures and certain cancers.16-18 Vitamin D toxicity usually is noted in serum levels greater than 88 ng/mL; symptoms of toxicity include hypercalcemia, nausea, vomiting, and muscle weakness. For nondeficient patients, the National Academy of Medicine (formerly the Institute of Medicine) recommended an upper limit of 4000 IU daily.14 The optimal dose in preventing vitamin D deficiency ranges from 600 to 1000 IU daily.13-15
Treatment of Vitamin D Deficiency
In the setting of vitamin D deficiency, the amount required for repletion often is dependent on each individual’s ability to absorb and convert to 25-hydroxyvitamin D. Typically every 100 IU of vitamin D correlates with a 0.7 to 1.0 ng/mL increase in serum 25-hydroxyvitamin D levels.19 There are multiple dosing regimens used to achieve the desired serum 25-hydroxyvitamin D levels in deficient patients. One recommendation from the Endocrine Society is 50,000 IU once weekly for 6 to 8 weeks (single doses >50,000 IU typically are not recommended due to increased risk for toxicity), followed by 600 to 1000 IU once daily in children and 1500 to 2000 IU once daily in adults thereafter.13 In patients with vitamin D deficiency, reassessment of serum 25-hydroxyvitamin D levels is recommended after 3 to 4 months of treatment, and adjustments to the repletion regimen should be made as needed.15,16 Generally, vitamin D3 is recommended over vitamin D2 due to enhanced efficacy in raising serum 25-hydroxyvitamin D levels.20
Vitamin D Deficiency in Alopecia
Although most recommendations are given in the interest of optimizing bone health, in the setting of alopecia, we set a similar serum 25-hydroxyvitamin D goal of greater than 30 ng/mL. We recommend treatment with vitamin D3 and practice the following repletion protocol: 50,000 IU once weekly for 4 weeks, followed by 1000 IU once daily for at least 8 weeks for serum 25-hydroxyvitamin D levels less than 20 ng/mL. For serum hydroxyvitamin D levels between 20 and 29 ng/mL, we recommend 1000 IU once daily for at least 12 weeks. We recheck blood levels again in 3 months. If levels fail to normalize, we will refer the patient to endocrinology. If levels return to normal, we transition to a daily multivitamin with vitamin D (400–800 IU) once daily and refer the patient back to the primary care physician for long-term monitoring.
Vitamin D receptors are found in every cell of the body and have been shown to play a role in bone, neural, and cardiovascular health; immune regulation; and possibly cancer prevention via the regulation of cell differentiation, proliferation, and apoptosis.1 Although it is controversial, vitamin D deficiency has been associated with various forms of nonscarring hair loss,2-4 including telogen effluvium, androgenetic alopecia, and alopecia areata. We describe a notable case of nonscarring alopecia associated with vitamin D deficiency in which vitamin D replacement therapy promoted hair regrowth.
Case Report
An otherwise healthy 34-year-old black woman presented to the Hair and Nail Clinic at the University of Pittsburgh Medical Center (Pittsburgh, Pennsylvania) for evaluation of progressive hair loss of 4 years’ duration that began shortly after her fourth child was born. Although she denied any history of excessive shedding, she stated that she used to have shoulder-length hair and somehow it had become extremely short without shaving or cutting the hair (Figure 1). Her current medications included a progestin intrauterine device and biotin 10 mg once daily, the latter of which she had taken for several months for the hair loss without any improvement.
On physical examination, the patient was noted to have diffusely thinning, short, brittle hair. Trichoscopy was notable for hairs of varying diameters, with some fractured at the level of the follicular ostia but no yellow dots at the follicular openings or exclamation point hairs. No scarring or erythema was seen on the scalp. The patient refused several of our team’s recommendations for scalp biopsy due to needle phobia. A hair growth window was made that showed good regrowth at 2 weeks after the initial presentation. Initial blood work revealed a total serum 25-hydroxyvitamin D level of 12 ng/mL (optimal, >30 ng/mL). Complete blood cell count, hormonal panel, zinc level, iron level, and thyroid studies were all normal.
The patient was started on vitamin D3 replacement therapy 50,000 IU once weekly for 4 weeks followed by 1000 IU once daily for 6 months. No other topical or systemic treatments were administered for the nonscarring alopecia. At a follow-up visit 6 months later, the patient’s vitamin D level was 36 ng/mL, and she had noticeable hair regrowth (Figure 2). At this time, the diagnosis of nonscarring alopecia associated with vitamin D deficiency was made.
Comment
Vitamin D is a fat-soluble vitamin that can be obtained via sun exposure, food sources (eg, fish, vitamin D–fortified foods), and direct supplementation.5 It has been estimated that nearly 1 billion individuals worldwide6 and approximately 41.6% of US adults are vitamin D deficient.7 Certainly not all of these individuals will present with alopecia, but in patients with hair loss, we suggest that vitamin D deficiency is an important factor to consider. Risk factors for vitamin D deficiency include older age, obesity, darker skin types, residence in northern latitudes, and malabsorption syndromes.7
Pathogenesis
Vitamin D is thought to play a role in the normal initiation and completion of the hair cycle as well as the differentiation of the follicular and interfollicular epidermis. The vitamin D receptor (VDR) is thought to induce the development of mature anagen hairs via the canonical WNT-β-catenin and hedgehog signaling pathways.8 In the absence of VDRs, the stem cells in the bulge of the hair follicle have an impaired ability to replicate, and as a result, VDR-deficient mice have shown near-total hair loss.9-12 We propose that vitamin D deficiency can not only be a trigger for hair loss but also can perpetuate hair loss and poor regrowth.
Diagnosis and Prevention of Vitamin D Deficiency
In the skin, 7-dehydrocholesterol is converted to previtamin D3 via UVB light, followed by subsequent conversion to vitamin D3. Dietary sources are in the form of either vitamin D2 or D3, both of which are converted in the liver to 25-hydroxyvitamin D, the major circulating metabolite. In the kidneys, 25-hydroxyvitamin D is then converted to 1,25-dihydroxyvitamin D, the biologically active form. Paradoxically, serum levels of 1,25-dihydroxyvitamin D can be normal or high in the setting of vitamin D deficiency; therefore, serum total 25-hydroxyvitamin D is the best way to assess a patient’s vitamin D status.5,13
The optimal serum 25-hydroxyvitamin D level is controversial. Recommendations range between 20 to 40 ng/mL14 and 30 to 50 ng/mL.13,15,16 Vitamin D levels higher than 50 ng/mL have been correlated with an increased risk of bone fractures and certain cancers.16-18 Vitamin D toxicity usually is noted in serum levels greater than 88 ng/mL; symptoms of toxicity include hypercalcemia, nausea, vomiting, and muscle weakness. For nondeficient patients, the National Academy of Medicine (formerly the Institute of Medicine) recommended an upper limit of 4000 IU daily.14 The optimal dose in preventing vitamin D deficiency ranges from 600 to 1000 IU daily.13-15
Treatment of Vitamin D Deficiency
In the setting of vitamin D deficiency, the amount required for repletion often is dependent on each individual’s ability to absorb and convert to 25-hydroxyvitamin D. Typically every 100 IU of vitamin D correlates with a 0.7 to 1.0 ng/mL increase in serum 25-hydroxyvitamin D levels.19 There are multiple dosing regimens used to achieve the desired serum 25-hydroxyvitamin D levels in deficient patients. One recommendation from the Endocrine Society is 50,000 IU once weekly for 6 to 8 weeks (single doses >50,000 IU typically are not recommended due to increased risk for toxicity), followed by 600 to 1000 IU once daily in children and 1500 to 2000 IU once daily in adults thereafter.13 In patients with vitamin D deficiency, reassessment of serum 25-hydroxyvitamin D levels is recommended after 3 to 4 months of treatment, and adjustments to the repletion regimen should be made as needed.15,16 Generally, vitamin D3 is recommended over vitamin D2 due to enhanced efficacy in raising serum 25-hydroxyvitamin D levels.20
Vitamin D Deficiency in Alopecia
Although most recommendations are given in the interest of optimizing bone health, in the setting of alopecia, we set a similar serum 25-hydroxyvitamin D goal of greater than 30 ng/mL. We recommend treatment with vitamin D3 and practice the following repletion protocol: 50,000 IU once weekly for 4 weeks, followed by 1000 IU once daily for at least 8 weeks for serum 25-hydroxyvitamin D levels less than 20 ng/mL. For serum hydroxyvitamin D levels between 20 and 29 ng/mL, we recommend 1000 IU once daily for at least 12 weeks. We recheck blood levels again in 3 months. If levels fail to normalize, we will refer the patient to endocrinology. If levels return to normal, we transition to a daily multivitamin with vitamin D (400–800 IU) once daily and refer the patient back to the primary care physician for long-term monitoring.
- Nagpal S, Na S, Rathnachalam R. Noncalcemic actions of vitamin D receptor ligands. Endocr Rev. 2005;26:662-687.
- Cheung EJ, Sink JR, English III JC. Vitamin and mineral deficiencies in patients with telogen effluvium: a retrospective cross-sectional study. J Drugs Dermatol. 2016;15:1235-1237.
- Rasheed H, Mahgoub D, Hegazy R, et al. Serum ferritin and vitamin D in female hair loss: do they play a role? Skin Pharmacol Physiol. 2013;26:101-107.
- Aksu Cerman A, Sarikaya Solak S, Kivanc Altunay I. Vitamin D deficiency in alopecia areata. Br J Dermatol. 2014;170:1299-1304.
- Holick MF. Vitamin D deficiency. N Engl J Med. 2007;357:266-281.
- Holick MF. High prevalence of vitamin D inadequacy and implications for health. Mayo Clin Proc. 2006;81:353-373.
- Yetley EA. Assessing the vitamin D status of the US population. Am J Clin Nutr. 2008;88:558S-564S.
- Lisse TS, Saini V, Zhao H, et al. The vitamin D receptor is required for activation of cWnt and hedgehog signaling in keratinocytes. Mol Endocrinol. 2014;28:1698-1706.
- Cianferotti L, Cox M, Skorjia K, et al. Vitamin D receptor is essential for normal keratinocyte stem cell function [published online May 17, 2007]. Porc Natl Acad Sci U S A. 2007;104:9428-9433.
- Xie Z, Komuves L, Yu QC, et al. Lack of the vitamin D receptor is associated with reduced epidermal differentiation and hair follicle growth. J Invest Dermatol. 2002;118:11-16.
- Kong J, Li XJ, Gavin D, et al. Targeted expression of human vitamin D receptor in the skin promotes the initiation of the postnatal hair follicle cycle and rescues the alopecia in vitamin D receptor null mice. J Invest Dermatol. 2002;118:631-638.
- Bikle DD, Elalieh H, Chang S, et al. Development and progression of alopecia in the vitamin D receptor null mouse. J Cell Physiol. 2006;207:340-353.
- Holick MF, Binkley NC, Bischoff-Ferrari HA, et al; Endocrine Society. Evaluation, treatment, and prevention of vitamin D deficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2011;96:1911-1930.
- Ross AC, Manson JE, Abrams SA, et al. The 2011 report on dietary reference intakes for calcium and vitamin D from the Institute of Medicine: what clinicians need to know. J Clin Endocrinol Metab. 2011;96:53-58.
- Dawson-Hughes B, Mithal A, Bonjour JP, et al. IOF position statement: vitamin D recommendations for older adults. Osteoporos Int. 2010;21:1151-1154.
- Judge J, Birge S, Gloth F 3rd; American Geriatrics Society Workgroup on Vitamin D Supplementation for Older Adults. Recommendations abstracted from the American Geriatrics Society Consensus Statement on vitamin D for prevention of falls and their consequences. J Am Geriatr Soc. 2014;62:147-152.
- Ahn J, Peters U, Albanes D, et al; Prostate, Lung, Colorectal, and Ovarian Cancer Screening Trial Project Team. Serum vitamin D concentration and prostate cancer risk: a nested case-control study. J Natl Cancer Inst. 2008;4:100:796-804.
- Stolzenberg-Solomon RZ, Jacobs EJ, Arslan AA, et al. Circulating 25-hydroxyvitamin D and risk of pancreatic cancer: Cohort Consortium Vitamin D Pooling Project of Rarer Cancers [published online June 18, 2010]. Am J Epidemiol. 2010;172:81-93.
- Heaney RP, Davies KM, Chen TC, et al. Human serum 25-hydroxycholecalciferol response to extended oral dosing with cholecalciferol. Am J Clin Nutr. 2003;77:204-210. Erratum in: 2003;78:1047.
- Tripkovic L, Lambert H, Hart K, et al. Comparison of vitamin D2 and vitamin D3 supplementation in raising serum 25-hydroxyvitamin D status: a systematic review and meta-analysis. Am J Clin Nutr. 2012;95:1357-1364.
- Nagpal S, Na S, Rathnachalam R. Noncalcemic actions of vitamin D receptor ligands. Endocr Rev. 2005;26:662-687.
- Cheung EJ, Sink JR, English III JC. Vitamin and mineral deficiencies in patients with telogen effluvium: a retrospective cross-sectional study. J Drugs Dermatol. 2016;15:1235-1237.
- Rasheed H, Mahgoub D, Hegazy R, et al. Serum ferritin and vitamin D in female hair loss: do they play a role? Skin Pharmacol Physiol. 2013;26:101-107.
- Aksu Cerman A, Sarikaya Solak S, Kivanc Altunay I. Vitamin D deficiency in alopecia areata. Br J Dermatol. 2014;170:1299-1304.
- Holick MF. Vitamin D deficiency. N Engl J Med. 2007;357:266-281.
- Holick MF. High prevalence of vitamin D inadequacy and implications for health. Mayo Clin Proc. 2006;81:353-373.
- Yetley EA. Assessing the vitamin D status of the US population. Am J Clin Nutr. 2008;88:558S-564S.
- Lisse TS, Saini V, Zhao H, et al. The vitamin D receptor is required for activation of cWnt and hedgehog signaling in keratinocytes. Mol Endocrinol. 2014;28:1698-1706.
- Cianferotti L, Cox M, Skorjia K, et al. Vitamin D receptor is essential for normal keratinocyte stem cell function [published online May 17, 2007]. Porc Natl Acad Sci U S A. 2007;104:9428-9433.
- Xie Z, Komuves L, Yu QC, et al. Lack of the vitamin D receptor is associated with reduced epidermal differentiation and hair follicle growth. J Invest Dermatol. 2002;118:11-16.
- Kong J, Li XJ, Gavin D, et al. Targeted expression of human vitamin D receptor in the skin promotes the initiation of the postnatal hair follicle cycle and rescues the alopecia in vitamin D receptor null mice. J Invest Dermatol. 2002;118:631-638.
- Bikle DD, Elalieh H, Chang S, et al. Development and progression of alopecia in the vitamin D receptor null mouse. J Cell Physiol. 2006;207:340-353.
- Holick MF, Binkley NC, Bischoff-Ferrari HA, et al; Endocrine Society. Evaluation, treatment, and prevention of vitamin D deficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2011;96:1911-1930.
- Ross AC, Manson JE, Abrams SA, et al. The 2011 report on dietary reference intakes for calcium and vitamin D from the Institute of Medicine: what clinicians need to know. J Clin Endocrinol Metab. 2011;96:53-58.
- Dawson-Hughes B, Mithal A, Bonjour JP, et al. IOF position statement: vitamin D recommendations for older adults. Osteoporos Int. 2010;21:1151-1154.
- Judge J, Birge S, Gloth F 3rd; American Geriatrics Society Workgroup on Vitamin D Supplementation for Older Adults. Recommendations abstracted from the American Geriatrics Society Consensus Statement on vitamin D for prevention of falls and their consequences. J Am Geriatr Soc. 2014;62:147-152.
- Ahn J, Peters U, Albanes D, et al; Prostate, Lung, Colorectal, and Ovarian Cancer Screening Trial Project Team. Serum vitamin D concentration and prostate cancer risk: a nested case-control study. J Natl Cancer Inst. 2008;4:100:796-804.
- Stolzenberg-Solomon RZ, Jacobs EJ, Arslan AA, et al. Circulating 25-hydroxyvitamin D and risk of pancreatic cancer: Cohort Consortium Vitamin D Pooling Project of Rarer Cancers [published online June 18, 2010]. Am J Epidemiol. 2010;172:81-93.
- Heaney RP, Davies KM, Chen TC, et al. Human serum 25-hydroxycholecalciferol response to extended oral dosing with cholecalciferol. Am J Clin Nutr. 2003;77:204-210. Erratum in: 2003;78:1047.
- Tripkovic L, Lambert H, Hart K, et al. Comparison of vitamin D2 and vitamin D3 supplementation in raising serum 25-hydroxyvitamin D status: a systematic review and meta-analysis. Am J Clin Nutr. 2012;95:1357-1364.
Practice Points
- The evaluation of vitamin D levels is important in the management of nonscarring alopecia.
- Vitamin D deficiency can present as nonscarring alopecia not associated with alopecia areata, androgenetic alopecia, or telogen effluvium.
Vertebral Artery Dissection in Active-Duty Soldier Due to Mixed Martial Arts Choke Hold
Knowledge of the potential dangers of mixed martial arts is valuable for Department of Defense (DoD) health care providers as the military continues to implement combatives training into regular military instruction. This case study presents an active-duty service member who developed a spontaneous vertebral artery dissection (sVAD) during mixed martial arts training, which led to a cerebellar stroke.
To the authors’ knowledge this is the first documented case of a sVAD with associated stroke related to a mixed martial arts choke hold. Understanding the diagnosis, management, and prognosis of this condition will remain important as hand-to-hand combat instruction continues to be a part of regular military training.
Case Presentation
A 39-year-old active-duty male without significant past medical history presented to the emergency department (ED) at the San Antonio Military Medical Center in Texas for evaluation of severe vertigo with associated nausea and vomiting. He had participated in a Jiu-Jitsu match the evening prior to his presentation and reported that he was placed in a choke hold within the last 12 seconds of the match. He denied losing consciousness during this hold.
Once released, he attempted to stand and developed sudden onset vertigo with severe nausea, leading to multiple bouts of emesis. He additionally developed a throbbing, left-sided headache radiating down the left side of his neck. While the vertigo resolved within an hour, he continued to experience bouts of nausea and emesis, prompting him to present to the ED for further evaluation. The patient’s past medical history was remarkable only for multiple prior concussions, and his only medication was occasional ibuprofen. He denied the usage of recreational drugs.
Upon presentation to the ED, the patient’s vital signs were 139/93 mm Hg blood pressure, 73 beats per minute heart rate, 16 breaths per minute respiration, 100% oxygen saturation on room air, and 97.7° F temperature. 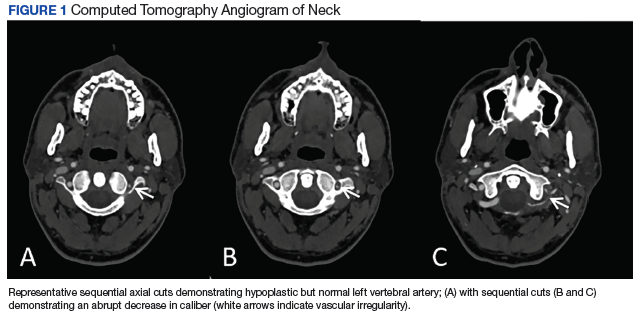
The patient demonstrated normal balance and exhibited no nystagmus or limb/truncal ataxia as evaluated with finger-to-nose/heel-to-shin testing and gait exam. Complete blood count, comprehensive metabolic panel, and coagulation panel all demonstrated no abnormalities. 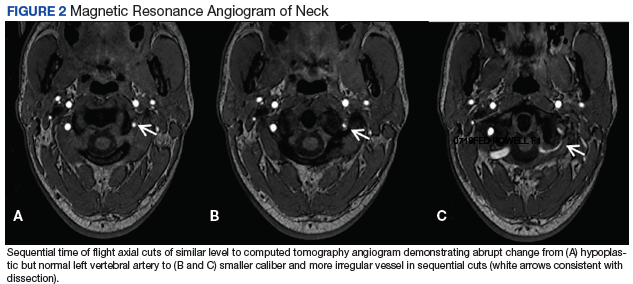
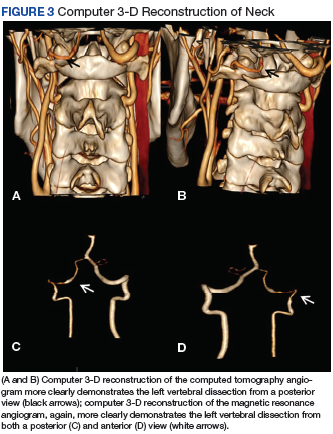
The patient was admitted to the hospital for symptom control and further monitoring. His headache and nausea were managed with medications, and he began antiplatelet therapy with aspirin 325 mg daily. Given the size of his cerebellar infarction, it was decided that he would be monitored in the hospital for 72 hours for the development of significant cerebellar edema. He remained stable throughout his hospitalization and had only a mild headache at the time of discharge.
The patient was last seen 3 months postinjury with no subjective complaints and a completely normal neurologic exam. The treatment plan for the patient is to continue aspirin for 6 months postinjury at which time a repeat CT will be performed to ensure resolution. He has been counseled to avoid heavy lifting and any activity with potential for sudden movement/force of the neck (grappling/wrestling, chiropractic manipulation, roller coasters, or sit-ups) until the repeat CT has been completed.
Discussion
Spontaneous vertebral artery and carotid artery dissections are collectively referred to as sCADs. Spontaneous cervical artery dissections are a rare condition with a higher incidence of internal carotid dissections than are VADs (1.72 vs 0.97 per 100,000 people).1 In contrast to the general stroke population, patients with sCADs are typically younger (mean age 45.3 years); and more than half of the patients are male.1,2
Spontaneous cervical artery dissections are typically characterized by subintimal tears of the vertebral artery leading to the accumulation of an intramural hematoma and creation of a “false lumen” in the arterial wall.3 A sVAD is more often found in the pars transversaria (V2; 35%) or atlas loop (V3; 34%) segments of the vertebral artery than in the prevertebral (V1; 20%) or intracranial (V4; 11%) segments.3-5 The etiology of these injuries is thought to be minor trauma to the neck in the context of a likely underlying connective tissue disease, though no direct association with a particular disease has been shown.
Biopsy evaluation of the superficial temporal arteries of patients with sCADs have revealed pathologic changes of the media and adventitial layers, including vacuolar degeneration and capillary neoangiogenesis, which are not found in the arteries of control patients.5 Although definitive association with a known connective tissue disease is rare, angiographic evidence of fibromuscular dysplasia, a nonspecific marker of connective tissue disease, is noted in as many as 15% to 20% of patients.6 Consequently, routine connective tissue disease screening is not recommended in these patients. One study found that about 40% of sCAD patients can recall minor cervical trauma in the preceding month in comparison to only 10% of other patients with stroke, leading to the moniker of “bottoms-up” or “beauty-parlor strokes” for these injuries. The most common mechanisms of minor neck trauma causing sCADs include tennis and golf swings, yoga, and roller-coaster rides.7,8
Usually symptomatic at presentation, the most frequently encountered sCAD symptoms are head or neck pain (80%), brain ischemia (56%), and Horner syndrome (25%).1 A study of 161 consecutive patients with internal carotid (n = 135) or vertebral artery (n = 26) dissections revealed that headache was reported by 69% of those with sVADs, and when present, was the initial manifestation in 33%. Headaches typically were ipsilateral to the dissection, located posteriorly in 83% of patients, and lasted an average duration of 72 hours. Neck pain, which was noted in 46% of sVAD patients, was predominantly posterior and ipsilateral in location as well.9 Ischemic symptoms of sVAD may include posterior circulation symptoms, such as vertigo, ataxia, diplopia, and leg weakness as well as lateral medullary (Wallenberg) syndrome characterized by dizziness, postural instability, limb hypotonia/ataxia, blurred vision, and nystagmus.
In a study of 169 patients with sCAD, brain ischemia occurred in 77% (131 patients) including 67% (n = 114) with ischemic stroke and 10% (n = 17) with transient ischemic attack. Head and/or neck pain was noted in 88% of those with brain ischemia.4 Etiologies for infarction included thromboembolic (85%), hemodynamic (12%), and mixed (3%).10 Isolated local symptoms are rare with one study of 245 patients with sCAD revealing only 20 (8%) presenting with pain only. Of those with pain only, 6 presented with headache, 2 with neck pain, and 12 with both.11
Diagnosis of sVAD requires a high index of suspicion and is confirmed by diagnostic testing. Previously, invasive angiography was the diagnostic gold standard, but with the improvement in quality of CT and MR angiography, these noninvasive modalities have become the tests of choice. There have been no studies to date revealing a definitive benefit of one modality over the other. A meta-analysis of 25 articles that compared the use of CT and MR angiography for the diagnosis of carotid and VAD revealed similar sensitivity and specificity.12 In contrast, a study involving 10 patients with confirmed sVAD who had both CT and MR angiographies during evaluation showed more total findings consistent with dissection on CT than with MR angiography when graded by 2 neuroradiologists. Additionally, the neuroradiologists subjectively rated CT angiography as preferential to MR in showing the imaging findings of dissection in 8 of 10 cases of vertebral dissection.13
Treatment for sCAD remains heavily debated. The use of IV thrombolysis within the standard time window for acute ischemic stroke is advocated for these patients. A meta-analysis of patients with sCAD vs matched patients with stroke from other causes treated with IV thrombolysis showed no difference in mortality at 3 months (9.0% vs 8.8%) or symptomatic intracranial hemorrhage (3.3% vs 3.0%). Additionally, similar percentages of patients had excellent (30.9% vs 37.4%) and favorable (58.2% vs 52.2%) 3-month functional statuses as expressed by the Modified Rankin Score (mRS).14,15
Debate remains regarding subacute therapy for sCAD with either antiplatelet or anticoagulant therapy. A randomized study of 250 patients with cervical artery dissection (118 carotid, 132 vertebral) in which 126 patients were assigned to antiplatelet therapy and 124 patients were assigned to anticoagulant therapy showed an overall low rate of recurrent stroke (2%). There was no significant difference in efficacy between the therapy groups with stroke or death occurring in 3 antiplatelet patients and 1 anticoagulated patient. Adverse effects were very low in both groups with no deaths and only 1 major bleed in the anticoagulation group. Of note, stroke rates were lower in this study than prior observational studies.16
A nonrandomized study of 88 patients with extracranial sCAD showed overall low rates of recurrent ischemic stroke at 3 months with 1/59 (1.7%) in the antiplatelet group and 1/28 (3.6%) in the anticoagulation group (P 17 Given this low overall rate of recurrent stroke in prior studies, a guideline recommendation for antiplatelet or anticoagulant therapy cannot be made at this time.
The overall prognosis for this condition is fair. Functional status and recurrence risk are favorable, with one study finding a mRS score of 1 Additionally, a historic cohort study of 432 patients with first event of sCAD revealed that after a mean follow-up of 31 months, only 4 (0.9%) patients had a recurrent ischemic stroke either due to incomplete recanalization of the artery (n = 2) or recurrent sCAD (n = 2), and only 4 (0.9%) total recurrences of sCAD were report (2 without associated ischemic strokes).18 Further, a prospective study of 61 patients with confirmed sVAD revealed complete recanalization of 45.9% at 3 months, 62.3% at 6 months, and 63.9% at 12 months, suggesting that recanalization occurs mostly during the initial 6 months. There was no identified association between outcome and complete recanalization with favorable outcomes observed in 55 (90.2%) of patients and no further ischemic symptoms during follow-up.19
Neck maneuvers have been cited as a more common cause of sCAD in several previous studies. One retrospective study found chiropractic neck manipulation to be the etiology in 12 of 141 patients with CT- or MR- confirmed sCAD.20 As noted previously, to the authors’ knowledge this is the first reported case of a sVAD occurring after a mixed martial arts choke hold. While sports-related strokes are rare, one evaluation of 70 published cases found that 80% were due to sCAD. Commonly associated sports in this study included football, yoga, wrestling, tennis, golf, and swimming.21 Grappling-related neck manipulation has been noted as an etiology in a few case reports.
Hyperextension of the neck was deemed to be the etiology in boys aged 11 years and 17 years who developed a sCAD while participating in Judo and backyard wrestling, respectively.22,23 In the martial arts realm, there is a case report of a 26-year-old male who developed a sVAD after rapid head turning during a solo Kung Fu maneuver as well as a report of a 41-year-old male experiencing a right VAD complicated by a posterior infarction several days after straining his neck during a mixed martial arts competition.24,25 The patient denied any choke hold or direct blow to the neck.
The present case is different in that it is the first reported case of a sVAD occurring after a submission maneuver. Prior grappling-related sVADs were associated with hyperextension or rapid acceleration/deceleration forces on the neck. Isometric force to the neck is a rarely described mechanism for development of this injury. Although there are isolated and infrequent forensic case reports of carotid dissection with strangulation injuries, the authors believe this is the first documented case of a sVAD attributed to a combatives submission.
In the context of the military health system, it is important to be aware of this potential complication of combatives as instruction in close-quarters combat continues to be an important part of military training.
1. Lee VH, Brown RD Jr, Mandrekar J, Mokri B. Incidence and outcome of cervical artery dissection: a population-based study. Neurology. 2006;67(10):1809-1812.
2. Arnold M, Kappeler L, Georgiadis D, et al. Gender differences in spontaneous cervical artery dissection. Neurology. 2006;67(6):1050-1052.
3. Schvienk W. Spontaneous dissection of the carotid and vertebral arteries. N Engl J Med. 2001;344(12):898-906.
4. Arnold M, Bousser MG, Fahrni G, et al. Vertebral artery dissection: presenting findings and predictors of outcome. Stroke. 2006;37(10):2499-2503.
5. Völker W, Dittrich R, Grewe S, et al. The outer arterial wall layers are primarily affected in spontaneous cervical artery dissection. Neurology. 2011;76(17):1463-1471.
6. Debette S, Markus HS. The genetics of cervical artery dissection: a systematic review. Stroke. 2009;40(6):459-466.
7. DeBehnke D, Brady W. Vertebral artery dissection due to minor neck trauma. J Emerg Med. 1994;12(1):27-31.
8. Engelter ST, Grond-Ginsbach C, Metso TM, et al; Cervical Artery Dissection and Ischemic Stroke Patients Study Group. Cervical artery dissection: trauma and other potential mechanical trigger events. Neurology. 2013;80(21):1950-1957.
9. Silbert PL, Mokri B, Schievink WI. Headache and neck pain in spontaneous internal carotid and vertebral artery dissections. Neurology. 1995;45(8):1517-1522.
10. Morel A, Naggara O, Touzé E, et al. Mechanism of ischemic infarct in spontaneous cervical artery dissection. Stroke. 2012;43(5):1354-1361.
11. Arnold M, Cumurciuc R, Stapf C, Favrole P, Berthet K, Bousser MG. Pain as the only symptom of cervical artery dissection. J Neurol Neurosurg Psychiatry. 2006;77(9):1021-1024.
12. Provenzale J, Sarikaya B. Comparison of test performance characteristics of MRI, MR angiography, and CT angiography in the diagnosis of carotid and vertebral artery dissection: a review of the medical literature. AJR Am J Roentgenol. 2009;1939(4):1167-1174.
13. Vertinsky AT, Schwartz NE, Fishbein NJ, Rosenberg J, Albers GW, Zaharchuk G. Comparison of multidetector CT angiography and MR imaging of cervical artery dissection. AJNR Am J Neuroradiol. 2008;29(9):1753-1760.
14. Zinkstok SM, Vergouwen MD, Engelter ST, et al. Safety and functional outcome of thrombolysis in dissection-related ischemic stroke: a meta-analysis of individual patient data. Stroke. 2011;42(9):2515-2520.
15. Engelter S, Rutgers M, Hatz F, et al. Intravenous thrombolysis in stroke attributable to cervical artery dissection. Stroke. 2009;40(12):3772-3776.
16. CADISS trial investigators, Markus HS, Hayter E, et al. Antiplatelet treatment compared with anticoagulation treatment for cervical artery dissection (CADISS): a randomized trial. Lancet Neurol. 2015;14(4):361-367.
17. Kennedy F, Lanfranconi S, Hicks C, et al; CADISS Investigators. Antiplatelets vs. anticoagulation for dissection: CADISS nonrandomized arm and meta-analysis. Neurology. 2012;79(7):686-689.
18. Touze E, Gauvrit JY, Moulin T, Meder JF, Bracard S, Mas JL; Multicenter Survey on Natural History of Cervical Artery Dissection. Risk of stroke and recurrent dissection after a cervical artery dissection: a multicenter study. Neurology. 2003;61(10):1347-1351.
19. Arauz A, Marquez J, Artigas C, Balderrama J, Orrego H. Recanalization of vertebral artery dissection. Stroke. 2010;41(4):717-721.
20. Kennell KA, Daghfal MM, Patel SG, et DeSanto JR, Waterman GS, Bertino RE. Cervical artery dissection related to chiropractic manipulation: one institution’s experience. J Fam Pract. 2017;66(9):556-562.
21. McCrory P. Vertebral artery dissection causing stroke in sport. J Clin Neurosci. 2000;7(4):298-300.
22. Lannuzel A, Moulin T, Amsallem D, Galmiche J, Rumbach L. Vertebral artery dissection following a judo session: a case report. Neuropediatrics. 1994;25(2):106-108.
23. Gupta V, Dhawan N, Bahl J. Minor trauma causing stroke in a young athlete. Case Rep Neurol Med. 2015;2015: 182875.
24. Pacei F, Valvasorri L, Bet L. Vertebral artery dissection during Kung-Fu training. Neurol Sci. 2014;35(2):331-332.
25. Slowey M, Maw G, Furyk J. Case report on vertebral artery dissection in mixed martial arts. Emerg Med Australas. 2012;24(2):203-206.
Knowledge of the potential dangers of mixed martial arts is valuable for Department of Defense (DoD) health care providers as the military continues to implement combatives training into regular military instruction. This case study presents an active-duty service member who developed a spontaneous vertebral artery dissection (sVAD) during mixed martial arts training, which led to a cerebellar stroke.
To the authors’ knowledge this is the first documented case of a sVAD with associated stroke related to a mixed martial arts choke hold. Understanding the diagnosis, management, and prognosis of this condition will remain important as hand-to-hand combat instruction continues to be a part of regular military training.
Case Presentation
A 39-year-old active-duty male without significant past medical history presented to the emergency department (ED) at the San Antonio Military Medical Center in Texas for evaluation of severe vertigo with associated nausea and vomiting. He had participated in a Jiu-Jitsu match the evening prior to his presentation and reported that he was placed in a choke hold within the last 12 seconds of the match. He denied losing consciousness during this hold.
Once released, he attempted to stand and developed sudden onset vertigo with severe nausea, leading to multiple bouts of emesis. He additionally developed a throbbing, left-sided headache radiating down the left side of his neck. While the vertigo resolved within an hour, he continued to experience bouts of nausea and emesis, prompting him to present to the ED for further evaluation. The patient’s past medical history was remarkable only for multiple prior concussions, and his only medication was occasional ibuprofen. He denied the usage of recreational drugs.
Upon presentation to the ED, the patient’s vital signs were 139/93 mm Hg blood pressure, 73 beats per minute heart rate, 16 breaths per minute respiration, 100% oxygen saturation on room air, and 97.7° F temperature.
The patient demonstrated normal balance and exhibited no nystagmus or limb/truncal ataxia as evaluated with finger-to-nose/heel-to-shin testing and gait exam. Complete blood count, comprehensive metabolic panel, and coagulation panel all demonstrated no abnormalities.
The patient was admitted to the hospital for symptom control and further monitoring. His headache and nausea were managed with medications, and he began antiplatelet therapy with aspirin 325 mg daily. Given the size of his cerebellar infarction, it was decided that he would be monitored in the hospital for 72 hours for the development of significant cerebellar edema. He remained stable throughout his hospitalization and had only a mild headache at the time of discharge.
The patient was last seen 3 months postinjury with no subjective complaints and a completely normal neurologic exam. The treatment plan for the patient is to continue aspirin for 6 months postinjury at which time a repeat CT will be performed to ensure resolution. He has been counseled to avoid heavy lifting and any activity with potential for sudden movement/force of the neck (grappling/wrestling, chiropractic manipulation, roller coasters, or sit-ups) until the repeat CT has been completed.
Discussion
Spontaneous vertebral artery and carotid artery dissections are collectively referred to as sCADs. Spontaneous cervical artery dissections are a rare condition with a higher incidence of internal carotid dissections than are VADs (1.72 vs 0.97 per 100,000 people).1 In contrast to the general stroke population, patients with sCADs are typically younger (mean age 45.3 years); and more than half of the patients are male.1,2
Spontaneous cervical artery dissections are typically characterized by subintimal tears of the vertebral artery leading to the accumulation of an intramural hematoma and creation of a “false lumen” in the arterial wall.3 A sVAD is more often found in the pars transversaria (V2; 35%) or atlas loop (V3; 34%) segments of the vertebral artery than in the prevertebral (V1; 20%) or intracranial (V4; 11%) segments.3-5 The etiology of these injuries is thought to be minor trauma to the neck in the context of a likely underlying connective tissue disease, though no direct association with a particular disease has been shown.
Biopsy evaluation of the superficial temporal arteries of patients with sCADs have revealed pathologic changes of the media and adventitial layers, including vacuolar degeneration and capillary neoangiogenesis, which are not found in the arteries of control patients.5 Although definitive association with a known connective tissue disease is rare, angiographic evidence of fibromuscular dysplasia, a nonspecific marker of connective tissue disease, is noted in as many as 15% to 20% of patients.6 Consequently, routine connective tissue disease screening is not recommended in these patients. One study found that about 40% of sCAD patients can recall minor cervical trauma in the preceding month in comparison to only 10% of other patients with stroke, leading to the moniker of “bottoms-up” or “beauty-parlor strokes” for these injuries. The most common mechanisms of minor neck trauma causing sCADs include tennis and golf swings, yoga, and roller-coaster rides.7,8
Usually symptomatic at presentation, the most frequently encountered sCAD symptoms are head or neck pain (80%), brain ischemia (56%), and Horner syndrome (25%).1 A study of 161 consecutive patients with internal carotid (n = 135) or vertebral artery (n = 26) dissections revealed that headache was reported by 69% of those with sVADs, and when present, was the initial manifestation in 33%. Headaches typically were ipsilateral to the dissection, located posteriorly in 83% of patients, and lasted an average duration of 72 hours. Neck pain, which was noted in 46% of sVAD patients, was predominantly posterior and ipsilateral in location as well.9 Ischemic symptoms of sVAD may include posterior circulation symptoms, such as vertigo, ataxia, diplopia, and leg weakness as well as lateral medullary (Wallenberg) syndrome characterized by dizziness, postural instability, limb hypotonia/ataxia, blurred vision, and nystagmus.
In a study of 169 patients with sCAD, brain ischemia occurred in 77% (131 patients) including 67% (n = 114) with ischemic stroke and 10% (n = 17) with transient ischemic attack. Head and/or neck pain was noted in 88% of those with brain ischemia.4 Etiologies for infarction included thromboembolic (85%), hemodynamic (12%), and mixed (3%).10 Isolated local symptoms are rare with one study of 245 patients with sCAD revealing only 20 (8%) presenting with pain only. Of those with pain only, 6 presented with headache, 2 with neck pain, and 12 with both.11
Diagnosis of sVAD requires a high index of suspicion and is confirmed by diagnostic testing. Previously, invasive angiography was the diagnostic gold standard, but with the improvement in quality of CT and MR angiography, these noninvasive modalities have become the tests of choice. There have been no studies to date revealing a definitive benefit of one modality over the other. A meta-analysis of 25 articles that compared the use of CT and MR angiography for the diagnosis of carotid and VAD revealed similar sensitivity and specificity.12 In contrast, a study involving 10 patients with confirmed sVAD who had both CT and MR angiographies during evaluation showed more total findings consistent with dissection on CT than with MR angiography when graded by 2 neuroradiologists. Additionally, the neuroradiologists subjectively rated CT angiography as preferential to MR in showing the imaging findings of dissection in 8 of 10 cases of vertebral dissection.13
Treatment for sCAD remains heavily debated. The use of IV thrombolysis within the standard time window for acute ischemic stroke is advocated for these patients. A meta-analysis of patients with sCAD vs matched patients with stroke from other causes treated with IV thrombolysis showed no difference in mortality at 3 months (9.0% vs 8.8%) or symptomatic intracranial hemorrhage (3.3% vs 3.0%). Additionally, similar percentages of patients had excellent (30.9% vs 37.4%) and favorable (58.2% vs 52.2%) 3-month functional statuses as expressed by the Modified Rankin Score (mRS).14,15
Debate remains regarding subacute therapy for sCAD with either antiplatelet or anticoagulant therapy. A randomized study of 250 patients with cervical artery dissection (118 carotid, 132 vertebral) in which 126 patients were assigned to antiplatelet therapy and 124 patients were assigned to anticoagulant therapy showed an overall low rate of recurrent stroke (2%). There was no significant difference in efficacy between the therapy groups with stroke or death occurring in 3 antiplatelet patients and 1 anticoagulated patient. Adverse effects were very low in both groups with no deaths and only 1 major bleed in the anticoagulation group. Of note, stroke rates were lower in this study than prior observational studies.16
A nonrandomized study of 88 patients with extracranial sCAD showed overall low rates of recurrent ischemic stroke at 3 months with 1/59 (1.7%) in the antiplatelet group and 1/28 (3.6%) in the anticoagulation group (P 17 Given this low overall rate of recurrent stroke in prior studies, a guideline recommendation for antiplatelet or anticoagulant therapy cannot be made at this time.
The overall prognosis for this condition is fair. Functional status and recurrence risk are favorable, with one study finding a mRS score of 1 Additionally, a historic cohort study of 432 patients with first event of sCAD revealed that after a mean follow-up of 31 months, only 4 (0.9%) patients had a recurrent ischemic stroke either due to incomplete recanalization of the artery (n = 2) or recurrent sCAD (n = 2), and only 4 (0.9%) total recurrences of sCAD were report (2 without associated ischemic strokes).18 Further, a prospective study of 61 patients with confirmed sVAD revealed complete recanalization of 45.9% at 3 months, 62.3% at 6 months, and 63.9% at 12 months, suggesting that recanalization occurs mostly during the initial 6 months. There was no identified association between outcome and complete recanalization with favorable outcomes observed in 55 (90.2%) of patients and no further ischemic symptoms during follow-up.19
Neck maneuvers have been cited as a more common cause of sCAD in several previous studies. One retrospective study found chiropractic neck manipulation to be the etiology in 12 of 141 patients with CT- or MR- confirmed sCAD.20 As noted previously, to the authors’ knowledge this is the first reported case of a sVAD occurring after a mixed martial arts choke hold. While sports-related strokes are rare, one evaluation of 70 published cases found that 80% were due to sCAD. Commonly associated sports in this study included football, yoga, wrestling, tennis, golf, and swimming.21 Grappling-related neck manipulation has been noted as an etiology in a few case reports.
Hyperextension of the neck was deemed to be the etiology in boys aged 11 years and 17 years who developed a sCAD while participating in Judo and backyard wrestling, respectively.22,23 In the martial arts realm, there is a case report of a 26-year-old male who developed a sVAD after rapid head turning during a solo Kung Fu maneuver as well as a report of a 41-year-old male experiencing a right VAD complicated by a posterior infarction several days after straining his neck during a mixed martial arts competition.24,25 The patient denied any choke hold or direct blow to the neck.
The present case is different in that it is the first reported case of a sVAD occurring after a submission maneuver. Prior grappling-related sVADs were associated with hyperextension or rapid acceleration/deceleration forces on the neck. Isometric force to the neck is a rarely described mechanism for development of this injury. Although there are isolated and infrequent forensic case reports of carotid dissection with strangulation injuries, the authors believe this is the first documented case of a sVAD attributed to a combatives submission.
In the context of the military health system, it is important to be aware of this potential complication of combatives as instruction in close-quarters combat continues to be an important part of military training.
Knowledge of the potential dangers of mixed martial arts is valuable for Department of Defense (DoD) health care providers as the military continues to implement combatives training into regular military instruction. This case study presents an active-duty service member who developed a spontaneous vertebral artery dissection (sVAD) during mixed martial arts training, which led to a cerebellar stroke.
To the authors’ knowledge this is the first documented case of a sVAD with associated stroke related to a mixed martial arts choke hold. Understanding the diagnosis, management, and prognosis of this condition will remain important as hand-to-hand combat instruction continues to be a part of regular military training.
Case Presentation
A 39-year-old active-duty male without significant past medical history presented to the emergency department (ED) at the San Antonio Military Medical Center in Texas for evaluation of severe vertigo with associated nausea and vomiting. He had participated in a Jiu-Jitsu match the evening prior to his presentation and reported that he was placed in a choke hold within the last 12 seconds of the match. He denied losing consciousness during this hold.
Once released, he attempted to stand and developed sudden onset vertigo with severe nausea, leading to multiple bouts of emesis. He additionally developed a throbbing, left-sided headache radiating down the left side of his neck. While the vertigo resolved within an hour, he continued to experience bouts of nausea and emesis, prompting him to present to the ED for further evaluation. The patient’s past medical history was remarkable only for multiple prior concussions, and his only medication was occasional ibuprofen. He denied the usage of recreational drugs.
Upon presentation to the ED, the patient’s vital signs were 139/93 mm Hg blood pressure, 73 beats per minute heart rate, 16 breaths per minute respiration, 100% oxygen saturation on room air, and 97.7° F temperature.
The patient demonstrated normal balance and exhibited no nystagmus or limb/truncal ataxia as evaluated with finger-to-nose/heel-to-shin testing and gait exam. Complete blood count, comprehensive metabolic panel, and coagulation panel all demonstrated no abnormalities.
The patient was admitted to the hospital for symptom control and further monitoring. His headache and nausea were managed with medications, and he began antiplatelet therapy with aspirin 325 mg daily. Given the size of his cerebellar infarction, it was decided that he would be monitored in the hospital for 72 hours for the development of significant cerebellar edema. He remained stable throughout his hospitalization and had only a mild headache at the time of discharge.
The patient was last seen 3 months postinjury with no subjective complaints and a completely normal neurologic exam. The treatment plan for the patient is to continue aspirin for 6 months postinjury at which time a repeat CT will be performed to ensure resolution. He has been counseled to avoid heavy lifting and any activity with potential for sudden movement/force of the neck (grappling/wrestling, chiropractic manipulation, roller coasters, or sit-ups) until the repeat CT has been completed.
Discussion
Spontaneous vertebral artery and carotid artery dissections are collectively referred to as sCADs. Spontaneous cervical artery dissections are a rare condition with a higher incidence of internal carotid dissections than are VADs (1.72 vs 0.97 per 100,000 people).1 In contrast to the general stroke population, patients with sCADs are typically younger (mean age 45.3 years); and more than half of the patients are male.1,2
Spontaneous cervical artery dissections are typically characterized by subintimal tears of the vertebral artery leading to the accumulation of an intramural hematoma and creation of a “false lumen” in the arterial wall.3 A sVAD is more often found in the pars transversaria (V2; 35%) or atlas loop (V3; 34%) segments of the vertebral artery than in the prevertebral (V1; 20%) or intracranial (V4; 11%) segments.3-5 The etiology of these injuries is thought to be minor trauma to the neck in the context of a likely underlying connective tissue disease, though no direct association with a particular disease has been shown.
Biopsy evaluation of the superficial temporal arteries of patients with sCADs have revealed pathologic changes of the media and adventitial layers, including vacuolar degeneration and capillary neoangiogenesis, which are not found in the arteries of control patients.5 Although definitive association with a known connective tissue disease is rare, angiographic evidence of fibromuscular dysplasia, a nonspecific marker of connective tissue disease, is noted in as many as 15% to 20% of patients.6 Consequently, routine connective tissue disease screening is not recommended in these patients. One study found that about 40% of sCAD patients can recall minor cervical trauma in the preceding month in comparison to only 10% of other patients with stroke, leading to the moniker of “bottoms-up” or “beauty-parlor strokes” for these injuries. The most common mechanisms of minor neck trauma causing sCADs include tennis and golf swings, yoga, and roller-coaster rides.7,8
Usually symptomatic at presentation, the most frequently encountered sCAD symptoms are head or neck pain (80%), brain ischemia (56%), and Horner syndrome (25%).1 A study of 161 consecutive patients with internal carotid (n = 135) or vertebral artery (n = 26) dissections revealed that headache was reported by 69% of those with sVADs, and when present, was the initial manifestation in 33%. Headaches typically were ipsilateral to the dissection, located posteriorly in 83% of patients, and lasted an average duration of 72 hours. Neck pain, which was noted in 46% of sVAD patients, was predominantly posterior and ipsilateral in location as well.9 Ischemic symptoms of sVAD may include posterior circulation symptoms, such as vertigo, ataxia, diplopia, and leg weakness as well as lateral medullary (Wallenberg) syndrome characterized by dizziness, postural instability, limb hypotonia/ataxia, blurred vision, and nystagmus.
In a study of 169 patients with sCAD, brain ischemia occurred in 77% (131 patients) including 67% (n = 114) with ischemic stroke and 10% (n = 17) with transient ischemic attack. Head and/or neck pain was noted in 88% of those with brain ischemia.4 Etiologies for infarction included thromboembolic (85%), hemodynamic (12%), and mixed (3%).10 Isolated local symptoms are rare with one study of 245 patients with sCAD revealing only 20 (8%) presenting with pain only. Of those with pain only, 6 presented with headache, 2 with neck pain, and 12 with both.11
Diagnosis of sVAD requires a high index of suspicion and is confirmed by diagnostic testing. Previously, invasive angiography was the diagnostic gold standard, but with the improvement in quality of CT and MR angiography, these noninvasive modalities have become the tests of choice. There have been no studies to date revealing a definitive benefit of one modality over the other. A meta-analysis of 25 articles that compared the use of CT and MR angiography for the diagnosis of carotid and VAD revealed similar sensitivity and specificity.12 In contrast, a study involving 10 patients with confirmed sVAD who had both CT and MR angiographies during evaluation showed more total findings consistent with dissection on CT than with MR angiography when graded by 2 neuroradiologists. Additionally, the neuroradiologists subjectively rated CT angiography as preferential to MR in showing the imaging findings of dissection in 8 of 10 cases of vertebral dissection.13
Treatment for sCAD remains heavily debated. The use of IV thrombolysis within the standard time window for acute ischemic stroke is advocated for these patients. A meta-analysis of patients with sCAD vs matched patients with stroke from other causes treated with IV thrombolysis showed no difference in mortality at 3 months (9.0% vs 8.8%) or symptomatic intracranial hemorrhage (3.3% vs 3.0%). Additionally, similar percentages of patients had excellent (30.9% vs 37.4%) and favorable (58.2% vs 52.2%) 3-month functional statuses as expressed by the Modified Rankin Score (mRS).14,15
Debate remains regarding subacute therapy for sCAD with either antiplatelet or anticoagulant therapy. A randomized study of 250 patients with cervical artery dissection (118 carotid, 132 vertebral) in which 126 patients were assigned to antiplatelet therapy and 124 patients were assigned to anticoagulant therapy showed an overall low rate of recurrent stroke (2%). There was no significant difference in efficacy between the therapy groups with stroke or death occurring in 3 antiplatelet patients and 1 anticoagulated patient. Adverse effects were very low in both groups with no deaths and only 1 major bleed in the anticoagulation group. Of note, stroke rates were lower in this study than prior observational studies.16
A nonrandomized study of 88 patients with extracranial sCAD showed overall low rates of recurrent ischemic stroke at 3 months with 1/59 (1.7%) in the antiplatelet group and 1/28 (3.6%) in the anticoagulation group (P 17 Given this low overall rate of recurrent stroke in prior studies, a guideline recommendation for antiplatelet or anticoagulant therapy cannot be made at this time.
The overall prognosis for this condition is fair. Functional status and recurrence risk are favorable, with one study finding a mRS score of 1 Additionally, a historic cohort study of 432 patients with first event of sCAD revealed that after a mean follow-up of 31 months, only 4 (0.9%) patients had a recurrent ischemic stroke either due to incomplete recanalization of the artery (n = 2) or recurrent sCAD (n = 2), and only 4 (0.9%) total recurrences of sCAD were report (2 without associated ischemic strokes).18 Further, a prospective study of 61 patients with confirmed sVAD revealed complete recanalization of 45.9% at 3 months, 62.3% at 6 months, and 63.9% at 12 months, suggesting that recanalization occurs mostly during the initial 6 months. There was no identified association between outcome and complete recanalization with favorable outcomes observed in 55 (90.2%) of patients and no further ischemic symptoms during follow-up.19
Neck maneuvers have been cited as a more common cause of sCAD in several previous studies. One retrospective study found chiropractic neck manipulation to be the etiology in 12 of 141 patients with CT- or MR- confirmed sCAD.20 As noted previously, to the authors’ knowledge this is the first reported case of a sVAD occurring after a mixed martial arts choke hold. While sports-related strokes are rare, one evaluation of 70 published cases found that 80% were due to sCAD. Commonly associated sports in this study included football, yoga, wrestling, tennis, golf, and swimming.21 Grappling-related neck manipulation has been noted as an etiology in a few case reports.
Hyperextension of the neck was deemed to be the etiology in boys aged 11 years and 17 years who developed a sCAD while participating in Judo and backyard wrestling, respectively.22,23 In the martial arts realm, there is a case report of a 26-year-old male who developed a sVAD after rapid head turning during a solo Kung Fu maneuver as well as a report of a 41-year-old male experiencing a right VAD complicated by a posterior infarction several days after straining his neck during a mixed martial arts competition.24,25 The patient denied any choke hold or direct blow to the neck.
The present case is different in that it is the first reported case of a sVAD occurring after a submission maneuver. Prior grappling-related sVADs were associated with hyperextension or rapid acceleration/deceleration forces on the neck. Isometric force to the neck is a rarely described mechanism for development of this injury. Although there are isolated and infrequent forensic case reports of carotid dissection with strangulation injuries, the authors believe this is the first documented case of a sVAD attributed to a combatives submission.
In the context of the military health system, it is important to be aware of this potential complication of combatives as instruction in close-quarters combat continues to be an important part of military training.
1. Lee VH, Brown RD Jr, Mandrekar J, Mokri B. Incidence and outcome of cervical artery dissection: a population-based study. Neurology. 2006;67(10):1809-1812.
2. Arnold M, Kappeler L, Georgiadis D, et al. Gender differences in spontaneous cervical artery dissection. Neurology. 2006;67(6):1050-1052.
3. Schvienk W. Spontaneous dissection of the carotid and vertebral arteries. N Engl J Med. 2001;344(12):898-906.
4. Arnold M, Bousser MG, Fahrni G, et al. Vertebral artery dissection: presenting findings and predictors of outcome. Stroke. 2006;37(10):2499-2503.
5. Völker W, Dittrich R, Grewe S, et al. The outer arterial wall layers are primarily affected in spontaneous cervical artery dissection. Neurology. 2011;76(17):1463-1471.
6. Debette S, Markus HS. The genetics of cervical artery dissection: a systematic review. Stroke. 2009;40(6):459-466.
7. DeBehnke D, Brady W. Vertebral artery dissection due to minor neck trauma. J Emerg Med. 1994;12(1):27-31.
8. Engelter ST, Grond-Ginsbach C, Metso TM, et al; Cervical Artery Dissection and Ischemic Stroke Patients Study Group. Cervical artery dissection: trauma and other potential mechanical trigger events. Neurology. 2013;80(21):1950-1957.
9. Silbert PL, Mokri B, Schievink WI. Headache and neck pain in spontaneous internal carotid and vertebral artery dissections. Neurology. 1995;45(8):1517-1522.
10. Morel A, Naggara O, Touzé E, et al. Mechanism of ischemic infarct in spontaneous cervical artery dissection. Stroke. 2012;43(5):1354-1361.
11. Arnold M, Cumurciuc R, Stapf C, Favrole P, Berthet K, Bousser MG. Pain as the only symptom of cervical artery dissection. J Neurol Neurosurg Psychiatry. 2006;77(9):1021-1024.
12. Provenzale J, Sarikaya B. Comparison of test performance characteristics of MRI, MR angiography, and CT angiography in the diagnosis of carotid and vertebral artery dissection: a review of the medical literature. AJR Am J Roentgenol. 2009;1939(4):1167-1174.
13. Vertinsky AT, Schwartz NE, Fishbein NJ, Rosenberg J, Albers GW, Zaharchuk G. Comparison of multidetector CT angiography and MR imaging of cervical artery dissection. AJNR Am J Neuroradiol. 2008;29(9):1753-1760.
14. Zinkstok SM, Vergouwen MD, Engelter ST, et al. Safety and functional outcome of thrombolysis in dissection-related ischemic stroke: a meta-analysis of individual patient data. Stroke. 2011;42(9):2515-2520.
15. Engelter S, Rutgers M, Hatz F, et al. Intravenous thrombolysis in stroke attributable to cervical artery dissection. Stroke. 2009;40(12):3772-3776.
16. CADISS trial investigators, Markus HS, Hayter E, et al. Antiplatelet treatment compared with anticoagulation treatment for cervical artery dissection (CADISS): a randomized trial. Lancet Neurol. 2015;14(4):361-367.
17. Kennedy F, Lanfranconi S, Hicks C, et al; CADISS Investigators. Antiplatelets vs. anticoagulation for dissection: CADISS nonrandomized arm and meta-analysis. Neurology. 2012;79(7):686-689.
18. Touze E, Gauvrit JY, Moulin T, Meder JF, Bracard S, Mas JL; Multicenter Survey on Natural History of Cervical Artery Dissection. Risk of stroke and recurrent dissection after a cervical artery dissection: a multicenter study. Neurology. 2003;61(10):1347-1351.
19. Arauz A, Marquez J, Artigas C, Balderrama J, Orrego H. Recanalization of vertebral artery dissection. Stroke. 2010;41(4):717-721.
20. Kennell KA, Daghfal MM, Patel SG, et DeSanto JR, Waterman GS, Bertino RE. Cervical artery dissection related to chiropractic manipulation: one institution’s experience. J Fam Pract. 2017;66(9):556-562.
21. McCrory P. Vertebral artery dissection causing stroke in sport. J Clin Neurosci. 2000;7(4):298-300.
22. Lannuzel A, Moulin T, Amsallem D, Galmiche J, Rumbach L. Vertebral artery dissection following a judo session: a case report. Neuropediatrics. 1994;25(2):106-108.
23. Gupta V, Dhawan N, Bahl J. Minor trauma causing stroke in a young athlete. Case Rep Neurol Med. 2015;2015: 182875.
24. Pacei F, Valvasorri L, Bet L. Vertebral artery dissection during Kung-Fu training. Neurol Sci. 2014;35(2):331-332.
25. Slowey M, Maw G, Furyk J. Case report on vertebral artery dissection in mixed martial arts. Emerg Med Australas. 2012;24(2):203-206.
1. Lee VH, Brown RD Jr, Mandrekar J, Mokri B. Incidence and outcome of cervical artery dissection: a population-based study. Neurology. 2006;67(10):1809-1812.
2. Arnold M, Kappeler L, Georgiadis D, et al. Gender differences in spontaneous cervical artery dissection. Neurology. 2006;67(6):1050-1052.
3. Schvienk W. Spontaneous dissection of the carotid and vertebral arteries. N Engl J Med. 2001;344(12):898-906.
4. Arnold M, Bousser MG, Fahrni G, et al. Vertebral artery dissection: presenting findings and predictors of outcome. Stroke. 2006;37(10):2499-2503.
5. Völker W, Dittrich R, Grewe S, et al. The outer arterial wall layers are primarily affected in spontaneous cervical artery dissection. Neurology. 2011;76(17):1463-1471.
6. Debette S, Markus HS. The genetics of cervical artery dissection: a systematic review. Stroke. 2009;40(6):459-466.
7. DeBehnke D, Brady W. Vertebral artery dissection due to minor neck trauma. J Emerg Med. 1994;12(1):27-31.
8. Engelter ST, Grond-Ginsbach C, Metso TM, et al; Cervical Artery Dissection and Ischemic Stroke Patients Study Group. Cervical artery dissection: trauma and other potential mechanical trigger events. Neurology. 2013;80(21):1950-1957.
9. Silbert PL, Mokri B, Schievink WI. Headache and neck pain in spontaneous internal carotid and vertebral artery dissections. Neurology. 1995;45(8):1517-1522.
10. Morel A, Naggara O, Touzé E, et al. Mechanism of ischemic infarct in spontaneous cervical artery dissection. Stroke. 2012;43(5):1354-1361.
11. Arnold M, Cumurciuc R, Stapf C, Favrole P, Berthet K, Bousser MG. Pain as the only symptom of cervical artery dissection. J Neurol Neurosurg Psychiatry. 2006;77(9):1021-1024.
12. Provenzale J, Sarikaya B. Comparison of test performance characteristics of MRI, MR angiography, and CT angiography in the diagnosis of carotid and vertebral artery dissection: a review of the medical literature. AJR Am J Roentgenol. 2009;1939(4):1167-1174.
13. Vertinsky AT, Schwartz NE, Fishbein NJ, Rosenberg J, Albers GW, Zaharchuk G. Comparison of multidetector CT angiography and MR imaging of cervical artery dissection. AJNR Am J Neuroradiol. 2008;29(9):1753-1760.
14. Zinkstok SM, Vergouwen MD, Engelter ST, et al. Safety and functional outcome of thrombolysis in dissection-related ischemic stroke: a meta-analysis of individual patient data. Stroke. 2011;42(9):2515-2520.
15. Engelter S, Rutgers M, Hatz F, et al. Intravenous thrombolysis in stroke attributable to cervical artery dissection. Stroke. 2009;40(12):3772-3776.
16. CADISS trial investigators, Markus HS, Hayter E, et al. Antiplatelet treatment compared with anticoagulation treatment for cervical artery dissection (CADISS): a randomized trial. Lancet Neurol. 2015;14(4):361-367.
17. Kennedy F, Lanfranconi S, Hicks C, et al; CADISS Investigators. Antiplatelets vs. anticoagulation for dissection: CADISS nonrandomized arm and meta-analysis. Neurology. 2012;79(7):686-689.
18. Touze E, Gauvrit JY, Moulin T, Meder JF, Bracard S, Mas JL; Multicenter Survey on Natural History of Cervical Artery Dissection. Risk of stroke and recurrent dissection after a cervical artery dissection: a multicenter study. Neurology. 2003;61(10):1347-1351.
19. Arauz A, Marquez J, Artigas C, Balderrama J, Orrego H. Recanalization of vertebral artery dissection. Stroke. 2010;41(4):717-721.
20. Kennell KA, Daghfal MM, Patel SG, et DeSanto JR, Waterman GS, Bertino RE. Cervical artery dissection related to chiropractic manipulation: one institution’s experience. J Fam Pract. 2017;66(9):556-562.
21. McCrory P. Vertebral artery dissection causing stroke in sport. J Clin Neurosci. 2000;7(4):298-300.
22. Lannuzel A, Moulin T, Amsallem D, Galmiche J, Rumbach L. Vertebral artery dissection following a judo session: a case report. Neuropediatrics. 1994;25(2):106-108.
23. Gupta V, Dhawan N, Bahl J. Minor trauma causing stroke in a young athlete. Case Rep Neurol Med. 2015;2015: 182875.
24. Pacei F, Valvasorri L, Bet L. Vertebral artery dissection during Kung-Fu training. Neurol Sci. 2014;35(2):331-332.
25. Slowey M, Maw G, Furyk J. Case report on vertebral artery dissection in mixed martial arts. Emerg Med Australas. 2012;24(2):203-206.
Body-wide, pruritic, papular rash • scalp lesion • excoriation • Dx?
THE CASE
A 7-year-old boy presented with a one-week history of a pruritic rash, which first appeared on his back and continued to spread across his entire body. The patient’s medical history was significant for a scalp lesion (FIGURE 1) that was being treated with oral griseofulvin (started 3 days earlier). He had no history of seasonal allergies, asthma, recent illness, or recent immunizations.

The physical exam was significant for a body-wide, nonerythematous, papular rash (FIGURE 2). There was evidence of excoriation due to itching. No mucosal involvement was appreciated. The remainder of the examination was unremarkable.
QUESTION
Based on the patient’s history and physical exam, which of the following is the most likely diagnosis?
A. Gianotti-Crosti syndrome
B. Atopic dermatitis
C. Dermatophytid reaction
D. Morbilliform drug eruption.
Continue to: THE DIAGNOSIS
THE DIAGNOSIS
The answer is C, dermatophytid reaction.
DISCUSSION
A dermatophytid reaction is a type of id reaction, or autoeczematization. An id reaction is when a localized dermatitis becomes a generalized pruritic eruption.1 In this case, the patient’s dermatitis was the result of a dermatophyte infection (tinea capitis), but an id reaction can also occur in response to noninfectious dermatitides and may be of an atopic, contact, or seborrheic nature.1
Dermatophytid reactions occur in up to 5% of all dermatophyte infections (most commonly tinea pedis) and are proposed to be type IV hypersensitivity reactions to the release of fungal antigens.1 These reactions can occur either before or after the initiation of antifungal treatment. They manifest as symmetric, pruritic, papulovesicular eruptions with fine scaling and commonly affect the face, trunk, extremities, palms, and interdigital spaces.1
What about other possible diagnoses?
Gianotti-Crosti syndrome is an asymptomatic, symmetric, papulovesicular dermatosis that involves the face, limbs, and buttocks of children 2 to 6 years of age.2 The lesions develop in response to a respiratory or gastrointestinal illness.2 They are typically associated with Epstein-Barr virus, hepatitis B, cytomegalovirus, respiratory syncytial virus, and coxsackievirus, but can occur with bacterial infections or following administration of routine immunizations.2
The lesions are self-limited and resolve within 2 months.2 Symptomatic lesions may be treated with oral antihistamines or steroids (topical or systemic).2
Continue to: Atopic dermatitis
Atopic dermatitis is characterized by symmetric involvement of the flexural surfaces of the body with a pruritic, erythematous rash that may have a fine scale.3 It usually manifests prior to 2 years of age, is recurrent, and is commonly associated with allergic rhinitis and asthma.3 Treatment involves trigger avoidance, topical emollients, topical corticosteroids, dilute bleach baths, and topical calcineurin inhibitors.3,4 For patients with significant nocturnal symptoms and sleep loss, oral antihistamines may be helpful.4
Morbilliform drug eruptions are the most common type of dermatologic drug reaction.5 These rashes occur approximately one to 2 weeks after exposure to a causative drug; they consist of pruritic, erythematous papules or macules that start centrally and may spread to the proximal extremities.5 Treatment involves discontinuation of the offending agent. Symptomatic relief may be achieved with oral antihistamines or topical or systemic corticosteroids.5
Treatment of dermatophytid reactions
While the initial impulse in the treatment of a dermatophytid reaction may be to discontinue oral antifungals, these treatments actually help resolve the underlying dermatophyte infection and should be continued. For children with tinea capitis, at least 6 weeks of treatment with an oral antifungal agent is warranted. Medications approved by the US Food and Drug Administration include terbinafine (for patients >4 years of age) and griseofulvin (for patients >2 years of age). Dosages are weight-based. (Fluconazole and itraconazole are not approved for this indication.) Lubricants, topical corticosteroids, and oral antihistamines can be used for acute management of pruritus.1
Our patient was treated successfully with griseofulvin and an oral antihistamine. However, he experienced headaches attributed to griseofulvin and was switched to terbinafine 5 mg/kg/d for 4 weeks. His tinea capitis was resolved at 8 weeks.
CORRESPONDENCE
Richard Temple, MD, CAPT, MC, USN. Department of Family Medicine, Naval Medical Center Camp Lejeune, 100 Brewster Blvd, Camp Lejeune, NC 28547; Richard.w.temple2.mil@mail.mil.
1. Cheng N, Rucker Wright D, Cohen BA. Dermatophytid in tinea capitis: rarely reported common phenomenon with clinical implications. Pediatrics. 2011;128:e453-e457.
2. Brandt O, Abeck D, Gianotti R, et al. Gianotti-Crosti syndrome. J Am Acad Dermatol. 2006;54:136-145.
3. Berke R, Singh A, Guralnick M. Atopic dermatitis: an overview. Am Fam Physician. 2012;86:35-42.
4. Eichenfield LF, Boguniewicz M, Simpson EL, et al. Translating atopic dermatitis management guidelines into practice for primary care providers. Pediatrics. 2015;136:554-565.
5. Riedl MA, Casillas AM. Adverse drug reactions: types and treatment options. Am Fam Physician. 2003;68:1781-1790.
THE CASE
A 7-year-old boy presented with a one-week history of a pruritic rash, which first appeared on his back and continued to spread across his entire body. The patient’s medical history was significant for a scalp lesion (FIGURE 1) that was being treated with oral griseofulvin (started 3 days earlier). He had no history of seasonal allergies, asthma, recent illness, or recent immunizations.
The physical exam was significant for a body-wide, nonerythematous, papular rash (FIGURE 2). There was evidence of excoriation due to itching. No mucosal involvement was appreciated. The remainder of the examination was unremarkable.
QUESTION
Based on the patient’s history and physical exam, which of the following is the most likely diagnosis?
A. Gianotti-Crosti syndrome
B. Atopic dermatitis
C. Dermatophytid reaction
D. Morbilliform drug eruption.
Continue to: THE DIAGNOSIS
THE DIAGNOSIS
The answer is C, dermatophytid reaction.
DISCUSSION
A dermatophytid reaction is a type of id reaction, or autoeczematization. An id reaction is when a localized dermatitis becomes a generalized pruritic eruption.1 In this case, the patient’s dermatitis was the result of a dermatophyte infection (tinea capitis), but an id reaction can also occur in response to noninfectious dermatitides and may be of an atopic, contact, or seborrheic nature.1
Dermatophytid reactions occur in up to 5% of all dermatophyte infections (most commonly tinea pedis) and are proposed to be type IV hypersensitivity reactions to the release of fungal antigens.1 These reactions can occur either before or after the initiation of antifungal treatment. They manifest as symmetric, pruritic, papulovesicular eruptions with fine scaling and commonly affect the face, trunk, extremities, palms, and interdigital spaces.1
What about other possible diagnoses?
Gianotti-Crosti syndrome is an asymptomatic, symmetric, papulovesicular dermatosis that involves the face, limbs, and buttocks of children 2 to 6 years of age.2 The lesions develop in response to a respiratory or gastrointestinal illness.2 They are typically associated with Epstein-Barr virus, hepatitis B, cytomegalovirus, respiratory syncytial virus, and coxsackievirus, but can occur with bacterial infections or following administration of routine immunizations.2
The lesions are self-limited and resolve within 2 months.2 Symptomatic lesions may be treated with oral antihistamines or steroids (topical or systemic).2
Continue to: Atopic dermatitis
Atopic dermatitis is characterized by symmetric involvement of the flexural surfaces of the body with a pruritic, erythematous rash that may have a fine scale.3 It usually manifests prior to 2 years of age, is recurrent, and is commonly associated with allergic rhinitis and asthma.3 Treatment involves trigger avoidance, topical emollients, topical corticosteroids, dilute bleach baths, and topical calcineurin inhibitors.3,4 For patients with significant nocturnal symptoms and sleep loss, oral antihistamines may be helpful.4
Morbilliform drug eruptions are the most common type of dermatologic drug reaction.5 These rashes occur approximately one to 2 weeks after exposure to a causative drug; they consist of pruritic, erythematous papules or macules that start centrally and may spread to the proximal extremities.5 Treatment involves discontinuation of the offending agent. Symptomatic relief may be achieved with oral antihistamines or topical or systemic corticosteroids.5
Treatment of dermatophytid reactions
While the initial impulse in the treatment of a dermatophytid reaction may be to discontinue oral antifungals, these treatments actually help resolve the underlying dermatophyte infection and should be continued. For children with tinea capitis, at least 6 weeks of treatment with an oral antifungal agent is warranted. Medications approved by the US Food and Drug Administration include terbinafine (for patients >4 years of age) and griseofulvin (for patients >2 years of age). Dosages are weight-based. (Fluconazole and itraconazole are not approved for this indication.) Lubricants, topical corticosteroids, and oral antihistamines can be used for acute management of pruritus.1
Our patient was treated successfully with griseofulvin and an oral antihistamine. However, he experienced headaches attributed to griseofulvin and was switched to terbinafine 5 mg/kg/d for 4 weeks. His tinea capitis was resolved at 8 weeks.
CORRESPONDENCE
Richard Temple, MD, CAPT, MC, USN. Department of Family Medicine, Naval Medical Center Camp Lejeune, 100 Brewster Blvd, Camp Lejeune, NC 28547; Richard.w.temple2.mil@mail.mil.
THE CASE
A 7-year-old boy presented with a one-week history of a pruritic rash, which first appeared on his back and continued to spread across his entire body. The patient’s medical history was significant for a scalp lesion (FIGURE 1) that was being treated with oral griseofulvin (started 3 days earlier). He had no history of seasonal allergies, asthma, recent illness, or recent immunizations.
The physical exam was significant for a body-wide, nonerythematous, papular rash (FIGURE 2). There was evidence of excoriation due to itching. No mucosal involvement was appreciated. The remainder of the examination was unremarkable.
QUESTION
Based on the patient’s history and physical exam, which of the following is the most likely diagnosis?
A. Gianotti-Crosti syndrome
B. Atopic dermatitis
C. Dermatophytid reaction
D. Morbilliform drug eruption.
Continue to: THE DIAGNOSIS
THE DIAGNOSIS
The answer is C, dermatophytid reaction.
DISCUSSION
A dermatophytid reaction is a type of id reaction, or autoeczematization. An id reaction is when a localized dermatitis becomes a generalized pruritic eruption.1 In this case, the patient’s dermatitis was the result of a dermatophyte infection (tinea capitis), but an id reaction can also occur in response to noninfectious dermatitides and may be of an atopic, contact, or seborrheic nature.1
Dermatophytid reactions occur in up to 5% of all dermatophyte infections (most commonly tinea pedis) and are proposed to be type IV hypersensitivity reactions to the release of fungal antigens.1 These reactions can occur either before or after the initiation of antifungal treatment. They manifest as symmetric, pruritic, papulovesicular eruptions with fine scaling and commonly affect the face, trunk, extremities, palms, and interdigital spaces.1
What about other possible diagnoses?
Gianotti-Crosti syndrome is an asymptomatic, symmetric, papulovesicular dermatosis that involves the face, limbs, and buttocks of children 2 to 6 years of age.2 The lesions develop in response to a respiratory or gastrointestinal illness.2 They are typically associated with Epstein-Barr virus, hepatitis B, cytomegalovirus, respiratory syncytial virus, and coxsackievirus, but can occur with bacterial infections or following administration of routine immunizations.2
The lesions are self-limited and resolve within 2 months.2 Symptomatic lesions may be treated with oral antihistamines or steroids (topical or systemic).2
Continue to: Atopic dermatitis
Atopic dermatitis is characterized by symmetric involvement of the flexural surfaces of the body with a pruritic, erythematous rash that may have a fine scale.3 It usually manifests prior to 2 years of age, is recurrent, and is commonly associated with allergic rhinitis and asthma.3 Treatment involves trigger avoidance, topical emollients, topical corticosteroids, dilute bleach baths, and topical calcineurin inhibitors.3,4 For patients with significant nocturnal symptoms and sleep loss, oral antihistamines may be helpful.4
Morbilliform drug eruptions are the most common type of dermatologic drug reaction.5 These rashes occur approximately one to 2 weeks after exposure to a causative drug; they consist of pruritic, erythematous papules or macules that start centrally and may spread to the proximal extremities.5 Treatment involves discontinuation of the offending agent. Symptomatic relief may be achieved with oral antihistamines or topical or systemic corticosteroids.5
Treatment of dermatophytid reactions
While the initial impulse in the treatment of a dermatophytid reaction may be to discontinue oral antifungals, these treatments actually help resolve the underlying dermatophyte infection and should be continued. For children with tinea capitis, at least 6 weeks of treatment with an oral antifungal agent is warranted. Medications approved by the US Food and Drug Administration include terbinafine (for patients >4 years of age) and griseofulvin (for patients >2 years of age). Dosages are weight-based. (Fluconazole and itraconazole are not approved for this indication.) Lubricants, topical corticosteroids, and oral antihistamines can be used for acute management of pruritus.1
Our patient was treated successfully with griseofulvin and an oral antihistamine. However, he experienced headaches attributed to griseofulvin and was switched to terbinafine 5 mg/kg/d for 4 weeks. His tinea capitis was resolved at 8 weeks.
CORRESPONDENCE
Richard Temple, MD, CAPT, MC, USN. Department of Family Medicine, Naval Medical Center Camp Lejeune, 100 Brewster Blvd, Camp Lejeune, NC 28547; Richard.w.temple2.mil@mail.mil.
1. Cheng N, Rucker Wright D, Cohen BA. Dermatophytid in tinea capitis: rarely reported common phenomenon with clinical implications. Pediatrics. 2011;128:e453-e457.
2. Brandt O, Abeck D, Gianotti R, et al. Gianotti-Crosti syndrome. J Am Acad Dermatol. 2006;54:136-145.
3. Berke R, Singh A, Guralnick M. Atopic dermatitis: an overview. Am Fam Physician. 2012;86:35-42.
4. Eichenfield LF, Boguniewicz M, Simpson EL, et al. Translating atopic dermatitis management guidelines into practice for primary care providers. Pediatrics. 2015;136:554-565.
5. Riedl MA, Casillas AM. Adverse drug reactions: types and treatment options. Am Fam Physician. 2003;68:1781-1790.
1. Cheng N, Rucker Wright D, Cohen BA. Dermatophytid in tinea capitis: rarely reported common phenomenon with clinical implications. Pediatrics. 2011;128:e453-e457.
2. Brandt O, Abeck D, Gianotti R, et al. Gianotti-Crosti syndrome. J Am Acad Dermatol. 2006;54:136-145.
3. Berke R, Singh A, Guralnick M. Atopic dermatitis: an overview. Am Fam Physician. 2012;86:35-42.
4. Eichenfield LF, Boguniewicz M, Simpson EL, et al. Translating atopic dermatitis management guidelines into practice for primary care providers. Pediatrics. 2015;136:554-565.
5. Riedl MA, Casillas AM. Adverse drug reactions: types and treatment options. Am Fam Physician. 2003;68:1781-1790.

Clostridium difficile Colitis in a Patient With Abdominal Distention, Pain, and Severe Constipation
A 66-year-old man with steroid-dependent asthma, well-controlled diabetes mellitus (DM), and chronic pain on hospice presented to Georg
On presentation, the patient reported taking the following medications: daily oxycodone 20 to 30 mg, tramadol 200 mg, gabapentin 1,200 mg, and frequent doses of morphine concentrate. Due to episodes of constipation and diarrhea, the veteran had recently self-discontinued taking stool softener (Senna plus). One month prior to this admission, the patient was enrolled in hospice service by his primary physician for severe COPD due to chronic hypoxic respiratory failure and worsening frailty. His baseline oxygen requirement was 4 to 5 L of supplemental oxygen with continued dyspnea upon any ambulation. The patient reported frequent falls prior to admission. Despite chronic steroid use, the patient’s DM was well controlled with metformin His hemoglobin A1c ranged from 6.0 to 7.8.
The patient was supine and appeared to be uncomfortable but not in acute distress on exam. His body habitus was Cushingoid, and he appeared much older than his stated age. His vitals were as follows: temperature 100.2°F, heart rate of 104 beats per minute, blood pressure of 98/56 mm Hg, and 95% oxygen on 4L nasal cannula (baseline 4-5L). A respiratory exam revealed distant breath sounds without wheeze, rhonchi, or rales, and a cardiac exam revealed no murmurs. He was in sinus rhythm with tachycardia. The abdomen was obese with purple straie and markedly distended. On percussion, his abdomen was tympanic with tinkling bowel sounds. He had no rebound tenderness, peritoneal signs, or fluid wave.
Laboratory results revealed a white blood cell (WBC) count of 13,790 cells/μL with a neutrophilic shift of 82.0, and an elevated creatinine of 2.16 mg/dL up from a baseline of 1.12 mg/dL. The chemistry panel was abnormal with a 125 mmol/L sodium (reference range 137-145 mmol/L). 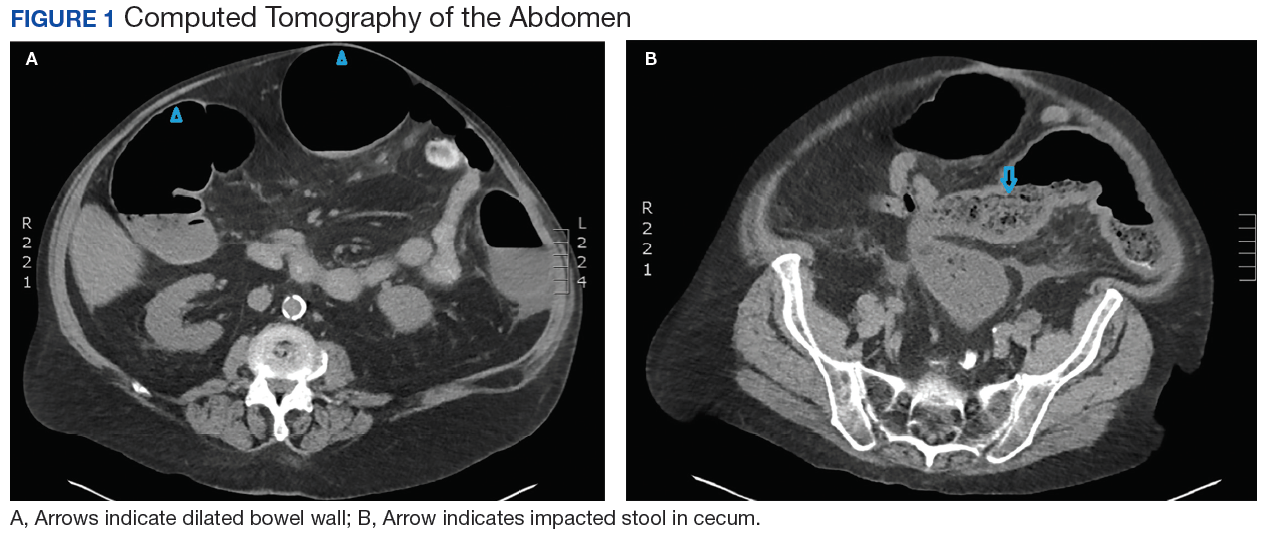
Diagnosis
On admission, the authors’ differential diagnosis included fecal impaction with large bowel obstruction, colitis, narcotic induced ileus, dehydration leading to severe constipation, and delayed gastric emptying secondary to long-standing DM. Ciprofloxacin and metronidazole antibiotics were initiated out of concern for possible colitis and potential bacterial translocation. Intravenous fluids were initiated, and the patient was instructed to have nothing by mouth (NPO) aside from the antibiotics. All opioids, including tramadol, were held. Out of concern for narcotic-induced constipation, a dose of methylnaltrexone to induce stooling was administered but had no effect on the constipation.
The gastroenterology department was consulted for a possible endoscopy to aid in decompression of the sigmoid. However, given the amount of distention and concern for perforation with endoscopy, the patient did not undergo endoscopy on admission. The patient remained afebrile on hospital day 3, and all antibiotics were discontinued. His WBC count normalized with complete resolution of the kidney injury. Antibiotic stewardship and infectious disease consults at George E. Wahlen VAMC reviewed the case and supported the decision to stop all antibiotics since it was not clear whether or not the patient was infected. Despite aggressive bowel care that included a nasogastric tube for large-volume polyethylene glycol and lactulose, various enemas and suppositories, the patient remained constipated.
On hospital day 5, still NPO, the patient had several bilious liquid stools that appeared to have a sediment quality to them. His abdomen remained distended, tympanic, and uncomfortable to palpation., He was examined frequently due to concern for possible perforation. On hospital day 8, gastroenterology reevaluated the need for endoscopy and proceeded with a flexible sigmoidoscopy. 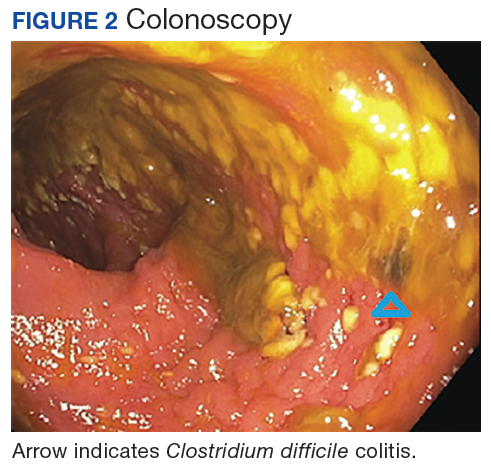
Polymerase chain reaction analysis of the colonoscopy stool samples were positive for Clostridium difficile (C difficile). The patient was started on IV metronidazole and oral vancomycin. His diet advanced and over the next few days he began stooling. He was subsequently discharged back to an extended care facility for rehabilitation. During this hospitalization, he made it clear he wished to be discharged from hospice services. He wanted to regain his strength through aggressive physical and occupational therapies.
Conclusion
Typical clinical manifestations of fulminant colitis include fever, diarrhea, abdominal pain, distention, and frequently WBC counts > 20,000 cells/μL. However, C difficile colitis, also known as pseudomembranous colitis, occasionally can present as an acute ileus, with little or no diarrhea.1 This veteran had several risk factors for C difficile infection, which included long-term residence in an extended care facility, frequent asthma exacerbations that required antibiotics, severe chronic disease, aged > 65 years,and ciprofloxacin given the first 3 days of this hospitalization.2 Until the endoscopy results were presented, no one on the patient’s care team, including gastroenterology and infectious disease, had included an infectious etiology in the differential diagnosis. This case reinforces the need to broaden differential diagnoses and look beyond assumptions that opioids without an adequate bowel regime were the cause. Avoiding anchoring heuristics can be a challenge as this case demonstrates.
1. Kawsar HI, Gopal KV, Shahnewaz J, Daw HA. Constipation in Clostridium difficile infection. BMJ Case Rep. 2012;2012: pii: bcr0220125938.
2. Leffler D, Lamont T. Clostridium difficile infection. N Engl J Med. 2015;372(16)1539-1548.
A 66-year-old man with steroid-dependent asthma, well-controlled diabetes mellitus (DM), and chronic pain on hospice presented to Georg
On presentation, the patient reported taking the following medications: daily oxycodone 20 to 30 mg, tramadol 200 mg, gabapentin 1,200 mg, and frequent doses of morphine concentrate. Due to episodes of constipation and diarrhea, the veteran had recently self-discontinued taking stool softener (Senna plus). One month prior to this admission, the patient was enrolled in hospice service by his primary physician for severe COPD due to chronic hypoxic respiratory failure and worsening frailty. His baseline oxygen requirement was 4 to 5 L of supplemental oxygen with continued dyspnea upon any ambulation. The patient reported frequent falls prior to admission. Despite chronic steroid use, the patient’s DM was well controlled with metformin His hemoglobin A1c ranged from 6.0 to 7.8.
The patient was supine and appeared to be uncomfortable but not in acute distress on exam. His body habitus was Cushingoid, and he appeared much older than his stated age. His vitals were as follows: temperature 100.2°F, heart rate of 104 beats per minute, blood pressure of 98/56 mm Hg, and 95% oxygen on 4L nasal cannula (baseline 4-5L). A respiratory exam revealed distant breath sounds without wheeze, rhonchi, or rales, and a cardiac exam revealed no murmurs. He was in sinus rhythm with tachycardia. The abdomen was obese with purple straie and markedly distended. On percussion, his abdomen was tympanic with tinkling bowel sounds. He had no rebound tenderness, peritoneal signs, or fluid wave.
Laboratory results revealed a white blood cell (WBC) count of 13,790 cells/μL with a neutrophilic shift of 82.0, and an elevated creatinine of 2.16 mg/dL up from a baseline of 1.12 mg/dL. The chemistry panel was abnormal with a 125 mmol/L sodium (reference range 137-145 mmol/L).
Diagnosis
On admission, the authors’ differential diagnosis included fecal impaction with large bowel obstruction, colitis, narcotic induced ileus, dehydration leading to severe constipation, and delayed gastric emptying secondary to long-standing DM. Ciprofloxacin and metronidazole antibiotics were initiated out of concern for possible colitis and potential bacterial translocation. Intravenous fluids were initiated, and the patient was instructed to have nothing by mouth (NPO) aside from the antibiotics. All opioids, including tramadol, were held. Out of concern for narcotic-induced constipation, a dose of methylnaltrexone to induce stooling was administered but had no effect on the constipation.
The gastroenterology department was consulted for a possible endoscopy to aid in decompression of the sigmoid. However, given the amount of distention and concern for perforation with endoscopy, the patient did not undergo endoscopy on admission. The patient remained afebrile on hospital day 3, and all antibiotics were discontinued. His WBC count normalized with complete resolution of the kidney injury. Antibiotic stewardship and infectious disease consults at George E. Wahlen VAMC reviewed the case and supported the decision to stop all antibiotics since it was not clear whether or not the patient was infected. Despite aggressive bowel care that included a nasogastric tube for large-volume polyethylene glycol and lactulose, various enemas and suppositories, the patient remained constipated.
On hospital day 5, still NPO, the patient had several bilious liquid stools that appeared to have a sediment quality to them. His abdomen remained distended, tympanic, and uncomfortable to palpation., He was examined frequently due to concern for possible perforation. On hospital day 8, gastroenterology reevaluated the need for endoscopy and proceeded with a flexible sigmoidoscopy.
Polymerase chain reaction analysis of the colonoscopy stool samples were positive for Clostridium difficile (C difficile). The patient was started on IV metronidazole and oral vancomycin. His diet advanced and over the next few days he began stooling. He was subsequently discharged back to an extended care facility for rehabilitation. During this hospitalization, he made it clear he wished to be discharged from hospice services. He wanted to regain his strength through aggressive physical and occupational therapies.
Conclusion
Typical clinical manifestations of fulminant colitis include fever, diarrhea, abdominal pain, distention, and frequently WBC counts > 20,000 cells/μL. However, C difficile colitis, also known as pseudomembranous colitis, occasionally can present as an acute ileus, with little or no diarrhea.1 This veteran had several risk factors for C difficile infection, which included long-term residence in an extended care facility, frequent asthma exacerbations that required antibiotics, severe chronic disease, aged > 65 years,and ciprofloxacin given the first 3 days of this hospitalization.2 Until the endoscopy results were presented, no one on the patient’s care team, including gastroenterology and infectious disease, had included an infectious etiology in the differential diagnosis. This case reinforces the need to broaden differential diagnoses and look beyond assumptions that opioids without an adequate bowel regime were the cause. Avoiding anchoring heuristics can be a challenge as this case demonstrates.
A 66-year-old man with steroid-dependent asthma, well-controlled diabetes mellitus (DM), and chronic pain on hospice presented to Georg
On presentation, the patient reported taking the following medications: daily oxycodone 20 to 30 mg, tramadol 200 mg, gabapentin 1,200 mg, and frequent doses of morphine concentrate. Due to episodes of constipation and diarrhea, the veteran had recently self-discontinued taking stool softener (Senna plus). One month prior to this admission, the patient was enrolled in hospice service by his primary physician for severe COPD due to chronic hypoxic respiratory failure and worsening frailty. His baseline oxygen requirement was 4 to 5 L of supplemental oxygen with continued dyspnea upon any ambulation. The patient reported frequent falls prior to admission. Despite chronic steroid use, the patient’s DM was well controlled with metformin His hemoglobin A1c ranged from 6.0 to 7.8.
The patient was supine and appeared to be uncomfortable but not in acute distress on exam. His body habitus was Cushingoid, and he appeared much older than his stated age. His vitals were as follows: temperature 100.2°F, heart rate of 104 beats per minute, blood pressure of 98/56 mm Hg, and 95% oxygen on 4L nasal cannula (baseline 4-5L). A respiratory exam revealed distant breath sounds without wheeze, rhonchi, or rales, and a cardiac exam revealed no murmurs. He was in sinus rhythm with tachycardia. The abdomen was obese with purple straie and markedly distended. On percussion, his abdomen was tympanic with tinkling bowel sounds. He had no rebound tenderness, peritoneal signs, or fluid wave.
Laboratory results revealed a white blood cell (WBC) count of 13,790 cells/μL with a neutrophilic shift of 82.0, and an elevated creatinine of 2.16 mg/dL up from a baseline of 1.12 mg/dL. The chemistry panel was abnormal with a 125 mmol/L sodium (reference range 137-145 mmol/L).
Diagnosis
On admission, the authors’ differential diagnosis included fecal impaction with large bowel obstruction, colitis, narcotic induced ileus, dehydration leading to severe constipation, and delayed gastric emptying secondary to long-standing DM. Ciprofloxacin and metronidazole antibiotics were initiated out of concern for possible colitis and potential bacterial translocation. Intravenous fluids were initiated, and the patient was instructed to have nothing by mouth (NPO) aside from the antibiotics. All opioids, including tramadol, were held. Out of concern for narcotic-induced constipation, a dose of methylnaltrexone to induce stooling was administered but had no effect on the constipation.
The gastroenterology department was consulted for a possible endoscopy to aid in decompression of the sigmoid. However, given the amount of distention and concern for perforation with endoscopy, the patient did not undergo endoscopy on admission. The patient remained afebrile on hospital day 3, and all antibiotics were discontinued. His WBC count normalized with complete resolution of the kidney injury. Antibiotic stewardship and infectious disease consults at George E. Wahlen VAMC reviewed the case and supported the decision to stop all antibiotics since it was not clear whether or not the patient was infected. Despite aggressive bowel care that included a nasogastric tube for large-volume polyethylene glycol and lactulose, various enemas and suppositories, the patient remained constipated.
On hospital day 5, still NPO, the patient had several bilious liquid stools that appeared to have a sediment quality to them. His abdomen remained distended, tympanic, and uncomfortable to palpation., He was examined frequently due to concern for possible perforation. On hospital day 8, gastroenterology reevaluated the need for endoscopy and proceeded with a flexible sigmoidoscopy.
Polymerase chain reaction analysis of the colonoscopy stool samples were positive for Clostridium difficile (C difficile). The patient was started on IV metronidazole and oral vancomycin. His diet advanced and over the next few days he began stooling. He was subsequently discharged back to an extended care facility for rehabilitation. During this hospitalization, he made it clear he wished to be discharged from hospice services. He wanted to regain his strength through aggressive physical and occupational therapies.
Conclusion
Typical clinical manifestations of fulminant colitis include fever, diarrhea, abdominal pain, distention, and frequently WBC counts > 20,000 cells/μL. However, C difficile colitis, also known as pseudomembranous colitis, occasionally can present as an acute ileus, with little or no diarrhea.1 This veteran had several risk factors for C difficile infection, which included long-term residence in an extended care facility, frequent asthma exacerbations that required antibiotics, severe chronic disease, aged > 65 years,and ciprofloxacin given the first 3 days of this hospitalization.2 Until the endoscopy results were presented, no one on the patient’s care team, including gastroenterology and infectious disease, had included an infectious etiology in the differential diagnosis. This case reinforces the need to broaden differential diagnoses and look beyond assumptions that opioids without an adequate bowel regime were the cause. Avoiding anchoring heuristics can be a challenge as this case demonstrates.
1. Kawsar HI, Gopal KV, Shahnewaz J, Daw HA. Constipation in Clostridium difficile infection. BMJ Case Rep. 2012;2012: pii: bcr0220125938.
2. Leffler D, Lamont T. Clostridium difficile infection. N Engl J Med. 2015;372(16)1539-1548.
1. Kawsar HI, Gopal KV, Shahnewaz J, Daw HA. Constipation in Clostridium difficile infection. BMJ Case Rep. 2012;2012: pii: bcr0220125938.
2. Leffler D, Lamont T. Clostridium difficile infection. N Engl J Med. 2015;372(16)1539-1548.
Nephrogenic Systemic Fibrosis in a Patient With Multiple Inflammatory Disorders
First described in 2000 in a case series of 15 patients, nephrogenic systemic fibrosis (NSF) is a rare scleroderma-like fibrosing skin condition associated with gadolinium exposure in end stage renal disease (ESRD).1 Patients with advanced chronic kidney disease (CKD) or ESRD are at the highest risk for this condition when exposed to gadolinium-based contrast dyes.
Nephrogenic systemic fibrosis is a devastating and rapidly progressive condition, making its prevention in at-risk populations of utmost importance. In this article, the authors describe a case of a patient who developed NSF in the setting of gadolinium exposure and multiple inflammatory dermatologic conditions. This case illustrates the possible role of a pro-inflammatory state in predisposing to NSF, which may help further elucidate its mechanism of action.
Case Presentation
A 61-year-old Hispanic male with a history of IV heroin use with ESRD secondary to membranous glomerulonephritis on hemodialysis and chronic hepatitis C infection presented to the West Los Angeles VAMC with fevers and night sweats that had persisted for 2 weeks. His physical examination was notable for diffuse tender palpable purpura and petechiae (including his palms and soles), altered mental status, and diffuse myoclonic jerks, which necessitated endotracheal intubation and mechanical ventilation for airway protection. Blood cultures were positive for methicillin-sensitive Staphylococcus aureus (MSSA). Laboratory results were notable for an elevated sedimentation rate of 53 mm/h (0-10 mm/h), C-reactive protein of 19.8 mg/L (< 0.744 mg/dL), and albumin of 1.2 g/dL (3.2-4.8 g/dL). An extensive rheumatologic workup was unrevealing, and a lumbar puncture was unremarkable. A biopsy of his skin lesions was consistent with leukocytoclastic vasculitis.
The patient’s prior hemodialysis access, a tunneled dialysis catheter in the right subclavian vein, was removed given concern for line infection and replaced with an internal jugular temporary hemodialysis line. Given his altered mental status and myoclonic jerks, the decision was made to pursue a magnetic resonance imaging (MRI) scan of the brain and spine with gadolinium contrast to evaluate for cerebral vasculitis and/or septic emboli to the brain.
The patient received 15 mL of gadoversetamide contrast in accordance with hospital imaging protocol. The MRI revealed only chronic ischemic changes. The patient underwent hemodialysis about 18 hours later. The patient was treated with a 6-week course of IV penicillin G. His altered mental status and myoclonic jerks resolved without intervention, and he was then discharged to an acute rehabilitation unit.
Eight weeks after his initial presentation the patient developed a purulent wound on his right forearm (Figure 1) 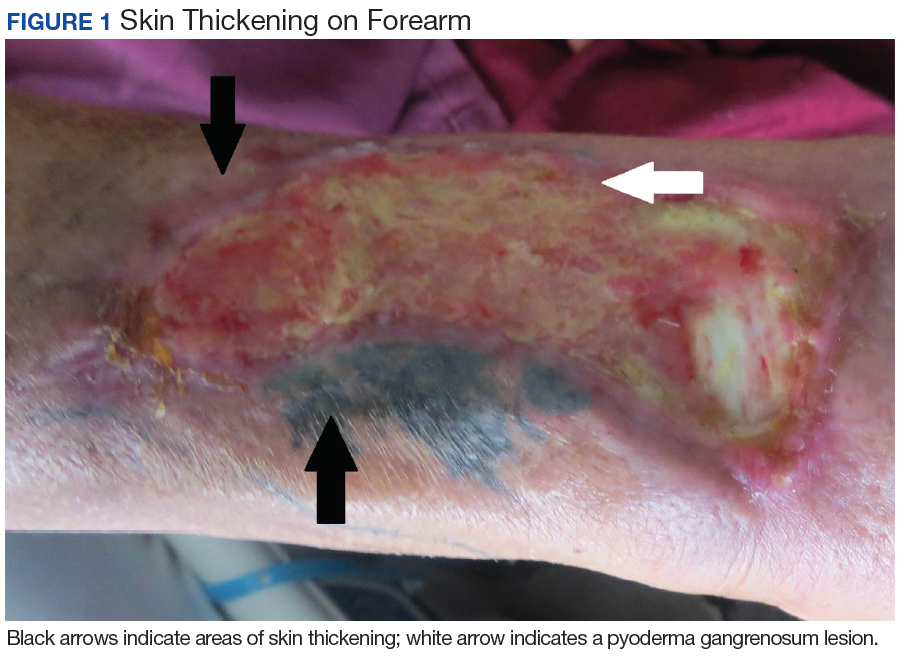
The patient was discharged to continue physical and occupational therapy to preserve his functional mobility, as no other treatment options were available. 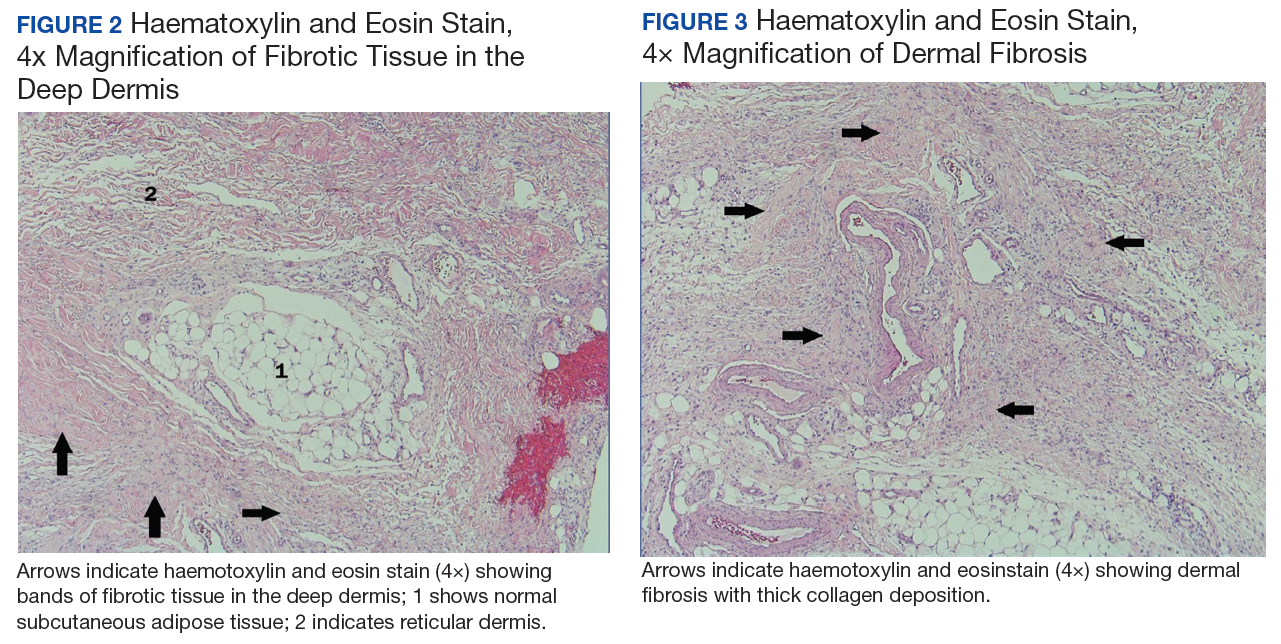
Discussion
Nephrogenic systemic fibrosis is a poorly understood inflammatory condition that produces diffuse fibrosis of the skin. Typically, the disease begins with progressive skin induration of the extremities. Systemic involvement may occur, leading to fibrosis of skeletal muscle, fascia, and multiple organs. Flexion contractures may develop that limit physical function. Fibrosis can become apparent within days to months after exposure to gadolinium contrast.
Beyond renal insufficiency, it is unclear what other risk factors predispose patients to developing this condition. Only a minority of patients with CKD stages 1 through 4 will develop NSF on exposure to gadolinium contrast. However, the incidence of NSF among patients with CKD stage 5 who are exposed to gadolinium has been estimated to be about 13.4% in a prospective study involving 18 patients.2
In a 2015 meta-analysis by Zhang and colleagues, the only clear risk factor identified for the development of NSF, aside from gadolinium exposure, was severe renal insufficiency with a glomerular filtration rate of < 30 mL/min/1.75m2.3 Due to the limited number of patients identified with this disease, it is difficult to identify other risk factors associated with the development of NSF. Based on in vitro studies, it has been postulated that a pro-inflammatory state predisposes patients to develop NSF.4,5 The proposed mechanism for NSF involves extravasation of gadolinium in the setting of vascular endothelial permeability.5,6 Gadolinium then interacts with tissue macrophages, which induce the release of inflammatory cytokines and the secretion of smooth muscle actin by dermal fibroblasts.6,7
Treatment of NSF has been largely unsuccessful. Multiple modalities of treatment that included topical and oral steroids, immunosuppression, plasmapheresis, and ultraviolent therapy have been attempted, none of which have been proven to consistently limit progression of the disease.8 The most effective intervention is early physical therapy to preserve functionality and prevent contracture formation. For patients who are eligible, early renal transplantation may offer the best chance of improved mobility. In a case series review by Cuffy and colleagues, 5 of 6 patients who underwent renal transplantation after the development of NSF experienced softening of the involved skin, and 2 patients had improved mobility of joints.9
Conclusion
The case presented here illustrates a possible association between a pro-inflammatory state and the development of NSF. This patient had multiple inflammatory conditions, including MSSA bacteremia, leukocytoclastic vasculitis, and pyoderma gangrenosum (the latter 2 conditions were thought to be associated with his underlying chronic hepatitis C infection), which the authors believe predisposed him to endothelial permeability and risk for developing NSF. The risk of developing NSF in at-risk patients with each episode of gadolinium exposure is estimated around 2.4%, or an incidence of 4.3 cases per 1,000 patient-years, leading the American College of Radiologists to recommend against the administration of gadolinium-based contrast except in cases in which benefits clearly outweigh risks.10 However, an MRI with gadolinium contrast can offer high diagnostic yield in cases such as the one presented here in which a diagnosis remains elusive. Moreover, the use of linear gadolinium-based contrast agents such as gadoversetamide, as in this case, has been reported to be associated with higher incidence of NSF.5 Since this case, the West Los Angeles VAMC has switched to gadobutrol contrast for its MRI protocol, which has been purported to be a lower risk agent compared with that of linear gadolinium-based contrast agents (although several cases of NSF have been reported with gadobutrol in the literature).11
Providers weighing the decision to administer gadolinium contrast to patients with ESRD should discuss the risks and benefits thoroughly, especially in patients with preexisting inflammatory conditions. In addition, although it has not been shown to effectively reduce the risk of NSF after administration of gadolinium, hemodialysis is recommended 2 hours after contrast administration for individuals at risk (the study patient received hemodialysis approximately 18 hours after).12 Given the lack of effective treatment options for NSF, prevention is key. A deeper understanding of the pathophysiology of NSF and identification of its risk factors is paramount to the prevention of this devastating disease.
1. Cowper SE, Robin HS, Steinberg SM, Su LD, Gupta S, LeBoit PE. Scleromyxoedema-like cutaneous diseases in renal-dialysis patients. Lancet. 2000;356(9234):1000-1001.
2. Todd DJ, Kagan A, Chibnik LB, Kay J. Cutaneous changes of nephrogenic systemic fibrosis. Arthritis Rheum. 2007;56(10):3433-3441.
3. Zhang B, Liang L, Chen W, Liang C, Zhang S. An updated study to determine association between gadolinium-based contrast agents and nephrogenic systemic fibrosis. PLoS One. 2015;10(6):e0129720.
4. Wermuth PJ, Del Galdo F, Jiménez SA. Induction of the expression of profibrotic cytokines and growth factors in normal human peripheral blood monocytes by gadolinium contrast agents. Arthritis Rheum. 2009;60(5):1508-1518.
5. Daftari Besheli L, Aran S, Shaqdan K, Kay J, Abujudeh H. Current status of nephrogenic systemic fibrosis. Clin Radiol. 2014;69(7):661-668.
6. Wagner B, Drel V, Gorin Y. Pathophysiology of gadolinium-associated systemic fibrosis. Am J Physiol Renal Physiol. 2016;31(1):F1-F11.
7. Idée JM, Fretellier N, Robic C, Corot C. The role of gadolinium chelates in the mechanism of nephrogenic systemic fibrosis: a critical update. Crit Rev Toxicol. 2014;44(10):895-913.
8. Mendoza FA, Artlett CM, Sandorfi N, Latinis K, Piera-Velazquez S, Jimenez SA. Description of 12 cases of nephrogenic fibrosing dermopathy and review of the literature. Semin Arthritis Rheum. 2006;35(4):238-249.
9. Cuffy MC, Singh M, Formica R, et al. Renal transplantation for nephrogenic systemic fibrosis: a case report and review of the literature. Nephrol Dial Transplant. 2011;26(3):1099-1109.
10. Deo A, Fogel M, Cowper SE. Nephrogenic systemic fibrosis: a population study examining the relationship of disease development of gadolinium exposure. Clin J Am Soc Nephrol. 2007;2(2):264-267
11. Elmholdt TR, Jørgensen B, Ramsing M, Pedersen M, Olesen AB. Two cases of nephrogenic systemic fibrosis after exposure to the macrocyclic compound gadobutrol. NDT Plus. 2010;3(3):285-287.
12. Abu-Alfa AK. Nephrogenic systemic fibrosis and gadolinium-based contrast agents. Adv Chronic Kidney Dis. 2011;18(3);188-198.
First described in 2000 in a case series of 15 patients, nephrogenic systemic fibrosis (NSF) is a rare scleroderma-like fibrosing skin condition associated with gadolinium exposure in end stage renal disease (ESRD).1 Patients with advanced chronic kidney disease (CKD) or ESRD are at the highest risk for this condition when exposed to gadolinium-based contrast dyes.
Nephrogenic systemic fibrosis is a devastating and rapidly progressive condition, making its prevention in at-risk populations of utmost importance. In this article, the authors describe a case of a patient who developed NSF in the setting of gadolinium exposure and multiple inflammatory dermatologic conditions. This case illustrates the possible role of a pro-inflammatory state in predisposing to NSF, which may help further elucidate its mechanism of action.
Case Presentation
A 61-year-old Hispanic male with a history of IV heroin use with ESRD secondary to membranous glomerulonephritis on hemodialysis and chronic hepatitis C infection presented to the West Los Angeles VAMC with fevers and night sweats that had persisted for 2 weeks. His physical examination was notable for diffuse tender palpable purpura and petechiae (including his palms and soles), altered mental status, and diffuse myoclonic jerks, which necessitated endotracheal intubation and mechanical ventilation for airway protection. Blood cultures were positive for methicillin-sensitive Staphylococcus aureus (MSSA). Laboratory results were notable for an elevated sedimentation rate of 53 mm/h (0-10 mm/h), C-reactive protein of 19.8 mg/L (< 0.744 mg/dL), and albumin of 1.2 g/dL (3.2-4.8 g/dL). An extensive rheumatologic workup was unrevealing, and a lumbar puncture was unremarkable. A biopsy of his skin lesions was consistent with leukocytoclastic vasculitis.
The patient’s prior hemodialysis access, a tunneled dialysis catheter in the right subclavian vein, was removed given concern for line infection and replaced with an internal jugular temporary hemodialysis line. Given his altered mental status and myoclonic jerks, the decision was made to pursue a magnetic resonance imaging (MRI) scan of the brain and spine with gadolinium contrast to evaluate for cerebral vasculitis and/or septic emboli to the brain.
The patient received 15 mL of gadoversetamide contrast in accordance with hospital imaging protocol. The MRI revealed only chronic ischemic changes. The patient underwent hemodialysis about 18 hours later. The patient was treated with a 6-week course of IV penicillin G. His altered mental status and myoclonic jerks resolved without intervention, and he was then discharged to an acute rehabilitation unit.
Eight weeks after his initial presentation the patient developed a purulent wound on his right forearm (Figure 1)
The patient was discharged to continue physical and occupational therapy to preserve his functional mobility, as no other treatment options were available.
Discussion
Nephrogenic systemic fibrosis is a poorly understood inflammatory condition that produces diffuse fibrosis of the skin. Typically, the disease begins with progressive skin induration of the extremities. Systemic involvement may occur, leading to fibrosis of skeletal muscle, fascia, and multiple organs. Flexion contractures may develop that limit physical function. Fibrosis can become apparent within days to months after exposure to gadolinium contrast.
Beyond renal insufficiency, it is unclear what other risk factors predispose patients to developing this condition. Only a minority of patients with CKD stages 1 through 4 will develop NSF on exposure to gadolinium contrast. However, the incidence of NSF among patients with CKD stage 5 who are exposed to gadolinium has been estimated to be about 13.4% in a prospective study involving 18 patients.2
In a 2015 meta-analysis by Zhang and colleagues, the only clear risk factor identified for the development of NSF, aside from gadolinium exposure, was severe renal insufficiency with a glomerular filtration rate of < 30 mL/min/1.75m2.3 Due to the limited number of patients identified with this disease, it is difficult to identify other risk factors associated with the development of NSF. Based on in vitro studies, it has been postulated that a pro-inflammatory state predisposes patients to develop NSF.4,5 The proposed mechanism for NSF involves extravasation of gadolinium in the setting of vascular endothelial permeability.5,6 Gadolinium then interacts with tissue macrophages, which induce the release of inflammatory cytokines and the secretion of smooth muscle actin by dermal fibroblasts.6,7
Treatment of NSF has been largely unsuccessful. Multiple modalities of treatment that included topical and oral steroids, immunosuppression, plasmapheresis, and ultraviolent therapy have been attempted, none of which have been proven to consistently limit progression of the disease.8 The most effective intervention is early physical therapy to preserve functionality and prevent contracture formation. For patients who are eligible, early renal transplantation may offer the best chance of improved mobility. In a case series review by Cuffy and colleagues, 5 of 6 patients who underwent renal transplantation after the development of NSF experienced softening of the involved skin, and 2 patients had improved mobility of joints.9
Conclusion
The case presented here illustrates a possible association between a pro-inflammatory state and the development of NSF. This patient had multiple inflammatory conditions, including MSSA bacteremia, leukocytoclastic vasculitis, and pyoderma gangrenosum (the latter 2 conditions were thought to be associated with his underlying chronic hepatitis C infection), which the authors believe predisposed him to endothelial permeability and risk for developing NSF. The risk of developing NSF in at-risk patients with each episode of gadolinium exposure is estimated around 2.4%, or an incidence of 4.3 cases per 1,000 patient-years, leading the American College of Radiologists to recommend against the administration of gadolinium-based contrast except in cases in which benefits clearly outweigh risks.10 However, an MRI with gadolinium contrast can offer high diagnostic yield in cases such as the one presented here in which a diagnosis remains elusive. Moreover, the use of linear gadolinium-based contrast agents such as gadoversetamide, as in this case, has been reported to be associated with higher incidence of NSF.5 Since this case, the West Los Angeles VAMC has switched to gadobutrol contrast for its MRI protocol, which has been purported to be a lower risk agent compared with that of linear gadolinium-based contrast agents (although several cases of NSF have been reported with gadobutrol in the literature).11
Providers weighing the decision to administer gadolinium contrast to patients with ESRD should discuss the risks and benefits thoroughly, especially in patients with preexisting inflammatory conditions. In addition, although it has not been shown to effectively reduce the risk of NSF after administration of gadolinium, hemodialysis is recommended 2 hours after contrast administration for individuals at risk (the study patient received hemodialysis approximately 18 hours after).12 Given the lack of effective treatment options for NSF, prevention is key. A deeper understanding of the pathophysiology of NSF and identification of its risk factors is paramount to the prevention of this devastating disease.
First described in 2000 in a case series of 15 patients, nephrogenic systemic fibrosis (NSF) is a rare scleroderma-like fibrosing skin condition associated with gadolinium exposure in end stage renal disease (ESRD).1 Patients with advanced chronic kidney disease (CKD) or ESRD are at the highest risk for this condition when exposed to gadolinium-based contrast dyes.
Nephrogenic systemic fibrosis is a devastating and rapidly progressive condition, making its prevention in at-risk populations of utmost importance. In this article, the authors describe a case of a patient who developed NSF in the setting of gadolinium exposure and multiple inflammatory dermatologic conditions. This case illustrates the possible role of a pro-inflammatory state in predisposing to NSF, which may help further elucidate its mechanism of action.
Case Presentation
A 61-year-old Hispanic male with a history of IV heroin use with ESRD secondary to membranous glomerulonephritis on hemodialysis and chronic hepatitis C infection presented to the West Los Angeles VAMC with fevers and night sweats that had persisted for 2 weeks. His physical examination was notable for diffuse tender palpable purpura and petechiae (including his palms and soles), altered mental status, and diffuse myoclonic jerks, which necessitated endotracheal intubation and mechanical ventilation for airway protection. Blood cultures were positive for methicillin-sensitive Staphylococcus aureus (MSSA). Laboratory results were notable for an elevated sedimentation rate of 53 mm/h (0-10 mm/h), C-reactive protein of 19.8 mg/L (< 0.744 mg/dL), and albumin of 1.2 g/dL (3.2-4.8 g/dL). An extensive rheumatologic workup was unrevealing, and a lumbar puncture was unremarkable. A biopsy of his skin lesions was consistent with leukocytoclastic vasculitis.
The patient’s prior hemodialysis access, a tunneled dialysis catheter in the right subclavian vein, was removed given concern for line infection and replaced with an internal jugular temporary hemodialysis line. Given his altered mental status and myoclonic jerks, the decision was made to pursue a magnetic resonance imaging (MRI) scan of the brain and spine with gadolinium contrast to evaluate for cerebral vasculitis and/or septic emboli to the brain.
The patient received 15 mL of gadoversetamide contrast in accordance with hospital imaging protocol. The MRI revealed only chronic ischemic changes. The patient underwent hemodialysis about 18 hours later. The patient was treated with a 6-week course of IV penicillin G. His altered mental status and myoclonic jerks resolved without intervention, and he was then discharged to an acute rehabilitation unit.
Eight weeks after his initial presentation the patient developed a purulent wound on his right forearm (Figure 1)
The patient was discharged to continue physical and occupational therapy to preserve his functional mobility, as no other treatment options were available.
Discussion
Nephrogenic systemic fibrosis is a poorly understood inflammatory condition that produces diffuse fibrosis of the skin. Typically, the disease begins with progressive skin induration of the extremities. Systemic involvement may occur, leading to fibrosis of skeletal muscle, fascia, and multiple organs. Flexion contractures may develop that limit physical function. Fibrosis can become apparent within days to months after exposure to gadolinium contrast.
Beyond renal insufficiency, it is unclear what other risk factors predispose patients to developing this condition. Only a minority of patients with CKD stages 1 through 4 will develop NSF on exposure to gadolinium contrast. However, the incidence of NSF among patients with CKD stage 5 who are exposed to gadolinium has been estimated to be about 13.4% in a prospective study involving 18 patients.2
In a 2015 meta-analysis by Zhang and colleagues, the only clear risk factor identified for the development of NSF, aside from gadolinium exposure, was severe renal insufficiency with a glomerular filtration rate of < 30 mL/min/1.75m2.3 Due to the limited number of patients identified with this disease, it is difficult to identify other risk factors associated with the development of NSF. Based on in vitro studies, it has been postulated that a pro-inflammatory state predisposes patients to develop NSF.4,5 The proposed mechanism for NSF involves extravasation of gadolinium in the setting of vascular endothelial permeability.5,6 Gadolinium then interacts with tissue macrophages, which induce the release of inflammatory cytokines and the secretion of smooth muscle actin by dermal fibroblasts.6,7
Treatment of NSF has been largely unsuccessful. Multiple modalities of treatment that included topical and oral steroids, immunosuppression, plasmapheresis, and ultraviolent therapy have been attempted, none of which have been proven to consistently limit progression of the disease.8 The most effective intervention is early physical therapy to preserve functionality and prevent contracture formation. For patients who are eligible, early renal transplantation may offer the best chance of improved mobility. In a case series review by Cuffy and colleagues, 5 of 6 patients who underwent renal transplantation after the development of NSF experienced softening of the involved skin, and 2 patients had improved mobility of joints.9
Conclusion
The case presented here illustrates a possible association between a pro-inflammatory state and the development of NSF. This patient had multiple inflammatory conditions, including MSSA bacteremia, leukocytoclastic vasculitis, and pyoderma gangrenosum (the latter 2 conditions were thought to be associated with his underlying chronic hepatitis C infection), which the authors believe predisposed him to endothelial permeability and risk for developing NSF. The risk of developing NSF in at-risk patients with each episode of gadolinium exposure is estimated around 2.4%, or an incidence of 4.3 cases per 1,000 patient-years, leading the American College of Radiologists to recommend against the administration of gadolinium-based contrast except in cases in which benefits clearly outweigh risks.10 However, an MRI with gadolinium contrast can offer high diagnostic yield in cases such as the one presented here in which a diagnosis remains elusive. Moreover, the use of linear gadolinium-based contrast agents such as gadoversetamide, as in this case, has been reported to be associated with higher incidence of NSF.5 Since this case, the West Los Angeles VAMC has switched to gadobutrol contrast for its MRI protocol, which has been purported to be a lower risk agent compared with that of linear gadolinium-based contrast agents (although several cases of NSF have been reported with gadobutrol in the literature).11
Providers weighing the decision to administer gadolinium contrast to patients with ESRD should discuss the risks and benefits thoroughly, especially in patients with preexisting inflammatory conditions. In addition, although it has not been shown to effectively reduce the risk of NSF after administration of gadolinium, hemodialysis is recommended 2 hours after contrast administration for individuals at risk (the study patient received hemodialysis approximately 18 hours after).12 Given the lack of effective treatment options for NSF, prevention is key. A deeper understanding of the pathophysiology of NSF and identification of its risk factors is paramount to the prevention of this devastating disease.
1. Cowper SE, Robin HS, Steinberg SM, Su LD, Gupta S, LeBoit PE. Scleromyxoedema-like cutaneous diseases in renal-dialysis patients. Lancet. 2000;356(9234):1000-1001.
2. Todd DJ, Kagan A, Chibnik LB, Kay J. Cutaneous changes of nephrogenic systemic fibrosis. Arthritis Rheum. 2007;56(10):3433-3441.
3. Zhang B, Liang L, Chen W, Liang C, Zhang S. An updated study to determine association between gadolinium-based contrast agents and nephrogenic systemic fibrosis. PLoS One. 2015;10(6):e0129720.
4. Wermuth PJ, Del Galdo F, Jiménez SA. Induction of the expression of profibrotic cytokines and growth factors in normal human peripheral blood monocytes by gadolinium contrast agents. Arthritis Rheum. 2009;60(5):1508-1518.
5. Daftari Besheli L, Aran S, Shaqdan K, Kay J, Abujudeh H. Current status of nephrogenic systemic fibrosis. Clin Radiol. 2014;69(7):661-668.
6. Wagner B, Drel V, Gorin Y. Pathophysiology of gadolinium-associated systemic fibrosis. Am J Physiol Renal Physiol. 2016;31(1):F1-F11.
7. Idée JM, Fretellier N, Robic C, Corot C. The role of gadolinium chelates in the mechanism of nephrogenic systemic fibrosis: a critical update. Crit Rev Toxicol. 2014;44(10):895-913.
8. Mendoza FA, Artlett CM, Sandorfi N, Latinis K, Piera-Velazquez S, Jimenez SA. Description of 12 cases of nephrogenic fibrosing dermopathy and review of the literature. Semin Arthritis Rheum. 2006;35(4):238-249.
9. Cuffy MC, Singh M, Formica R, et al. Renal transplantation for nephrogenic systemic fibrosis: a case report and review of the literature. Nephrol Dial Transplant. 2011;26(3):1099-1109.
10. Deo A, Fogel M, Cowper SE. Nephrogenic systemic fibrosis: a population study examining the relationship of disease development of gadolinium exposure. Clin J Am Soc Nephrol. 2007;2(2):264-267
11. Elmholdt TR, Jørgensen B, Ramsing M, Pedersen M, Olesen AB. Two cases of nephrogenic systemic fibrosis after exposure to the macrocyclic compound gadobutrol. NDT Plus. 2010;3(3):285-287.
12. Abu-Alfa AK. Nephrogenic systemic fibrosis and gadolinium-based contrast agents. Adv Chronic Kidney Dis. 2011;18(3);188-198.
1. Cowper SE, Robin HS, Steinberg SM, Su LD, Gupta S, LeBoit PE. Scleromyxoedema-like cutaneous diseases in renal-dialysis patients. Lancet. 2000;356(9234):1000-1001.
2. Todd DJ, Kagan A, Chibnik LB, Kay J. Cutaneous changes of nephrogenic systemic fibrosis. Arthritis Rheum. 2007;56(10):3433-3441.
3. Zhang B, Liang L, Chen W, Liang C, Zhang S. An updated study to determine association between gadolinium-based contrast agents and nephrogenic systemic fibrosis. PLoS One. 2015;10(6):e0129720.
4. Wermuth PJ, Del Galdo F, Jiménez SA. Induction of the expression of profibrotic cytokines and growth factors in normal human peripheral blood monocytes by gadolinium contrast agents. Arthritis Rheum. 2009;60(5):1508-1518.
5. Daftari Besheli L, Aran S, Shaqdan K, Kay J, Abujudeh H. Current status of nephrogenic systemic fibrosis. Clin Radiol. 2014;69(7):661-668.
6. Wagner B, Drel V, Gorin Y. Pathophysiology of gadolinium-associated systemic fibrosis. Am J Physiol Renal Physiol. 2016;31(1):F1-F11.
7. Idée JM, Fretellier N, Robic C, Corot C. The role of gadolinium chelates in the mechanism of nephrogenic systemic fibrosis: a critical update. Crit Rev Toxicol. 2014;44(10):895-913.
8. Mendoza FA, Artlett CM, Sandorfi N, Latinis K, Piera-Velazquez S, Jimenez SA. Description of 12 cases of nephrogenic fibrosing dermopathy and review of the literature. Semin Arthritis Rheum. 2006;35(4):238-249.
9. Cuffy MC, Singh M, Formica R, et al. Renal transplantation for nephrogenic systemic fibrosis: a case report and review of the literature. Nephrol Dial Transplant. 2011;26(3):1099-1109.
10. Deo A, Fogel M, Cowper SE. Nephrogenic systemic fibrosis: a population study examining the relationship of disease development of gadolinium exposure. Clin J Am Soc Nephrol. 2007;2(2):264-267
11. Elmholdt TR, Jørgensen B, Ramsing M, Pedersen M, Olesen AB. Two cases of nephrogenic systemic fibrosis after exposure to the macrocyclic compound gadobutrol. NDT Plus. 2010;3(3):285-287.
12. Abu-Alfa AK. Nephrogenic systemic fibrosis and gadolinium-based contrast agents. Adv Chronic Kidney Dis. 2011;18(3);188-198.
Recurrence of a small gastric gastrointestinal stromal tumor with high mitotic index
Gastrointestinal stromal tumor (GIST) is the most common soft tissue sarcoma of the gastrointestinal tract, usually arising from the interstitial cells of Cajal or similar cells in the outer wall of the gastrointestinal tract.1,2 Most GISTs have an activating mutation in KIT or platelet-derived growth factor receptor alpha (PDGFRA). Tumor size, mitotic rate, and anatomic site are the most common pathological features used to risk stratify GIST tumors.3-10 It is important to note when using such risk calculators that preoperative imatinib before determining tumor characteristics (such as mitoses per 50 high-power fields [hpf]) often changes the relevant parameters so that the same risk calculations may not apply. Tumors with a mitotic rate ≤5 mitoses per 50 hpf and a size ≤5 cm in greatest dimension have a lower recurrence rate after resection than tumors with a mitotic rate >5 mitoses per 50 hpf and a size >10 cm, and larger tumors can have a recurrence rate of up to 86%.11,12 Findings from a large observational study have suggested that the prognosis of gastric GIST in Korea and Japan may be more favorable compared with that in Western countries.13
The primary treatment of a localized primary GIST is surgical excision, but a cure is limited by recurrence.14,15 Imatinib is useful in the treatment of metastatic or recurrent GIST, and adjuvant treatment with imatinib after surgery has been shown to improve progression-free and overall survival in some cases.3,16-18 Responses to adjuvant imatinib depend on tumor sensitivity to the drug and the risk of recurrence. Drug sensitivity is largely dependent on the presence of mutations in KIT or PDGFRA.3,18 Recurrence risk is highly dependent on tumor size, tumor site, tumor rupture, and mitotic index.1,3,5,6,8,9,18,19 Findings on the use of gene expression patterns to predict recurrence risk have also been reported.20-27 However, recurrence risk is poorly understood for categories in which there are few cases with known outcomes, such as very small gastric GIST with a high mitotic index. For example, few cases of gastric GIST have been reported with a tumor size ≤2 cm, a mitotic rate >5 mitoses per 50 hpf, and adequate clinical follow-up. In such cases, it is difficult to assess the risk of recurrence.6 We report here the long-term outcome of a patient with a 1.8 cm gastric GIST with a mitotic index of 36 mitoses per 50 hpf and a KIT exon 11 mutation.
Case presentation and summary
A 69-year-old man presented with periumbilical and epigastric pain of 6-month duration. His medical history was notable for hyperlipidemia, hypertension, coronary angioplasty, and spinal surgery. He had a 40 pack-year smoking history and consumed 2 to 4 alcoholic drinks per day. The results of a physical examination were unremarkable. A computedtomographic (CT) scan showed no abnormalities. An esophagoduodenoscopy (EGD) revealed gastric ulcers. He was treated successfully with omeprazole 20 mg by mouth daily.
A month later, a follow-up EGD revealed a 1.8 × 1.5 cm submucosal mass 3 cm from the gastroesophageal junction. The patient underwent a fundus wedge resection, and a submucosal mass 1.8 cm in greatest dimension was removed. Pathologic examination revealed a GIST, spindle cell type, with a mitotic rate of 36 mitoses per 50 hpf with negative margins. Immunohistochemistry was positive for CD117. An exon 11 deletion (KVV558-560NV) was present in KIT. The patient’s risk of recurrence was unclear, and his follow-up included CT scans of the abdomen and pelvis every 3 to 4 months for the first 2 years, then every 6 months for the next 2.5 years.
A CT scan about 3.5 years after primary resection revealed small nonspecific liver hypodensities that became more prominent during the next year. About 5 years after primary resection, magnetic resonance imaging (MRI) revealed several liver lesions, the largest of which measured at 1.3 cm in greatest dimension. The patient’s liver metastases were readily identified by MRI (Figure 1) and CT imaging (Figure 2A).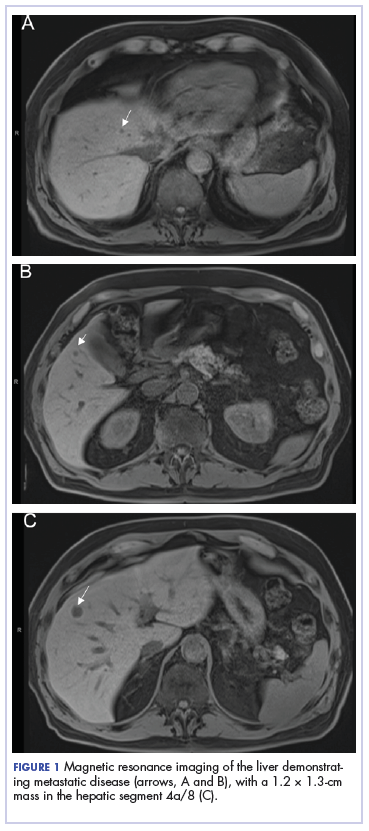
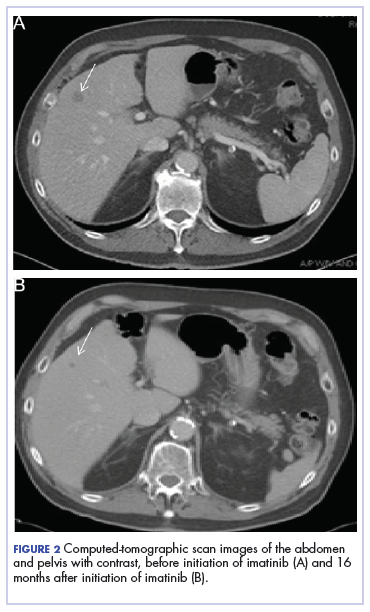
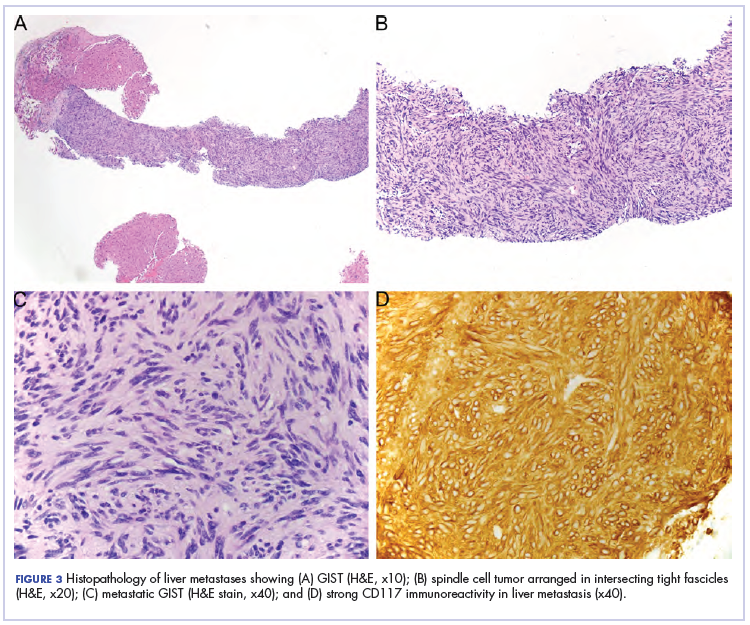
Discussion
Small gastric GISTs are sometimes found by endoscopy performed for unrelated reasons. Recent data suggest that the incidence of gastric GIST may be higher than previously thought. In a Japanese study of patients with gastric cancer in which 100 stomachs were systematically examined pathologically, 50 microscopic GISTs were found in 35 patients.28 Most small gastric GISTs have a low mitotic index. Few cases have been described with a high mitotic index. In a study of 1765 cases of GIST of the stomach, 8 patients had a tumor size less than 2 cm and a mitotic index greater than 5. Of those, only 6 patients had long-term follow-up, and 3 were alive without disease at 2, 17, and 20 years of follow-up.7 These limited data make it impossible to predict outcomes in patients with small gastric GIST with a high mitotic index.
For patients who are at high risk of recurrence after surgery, 3 years of adjuvant imatinib treatment compared with 1 year has been shown to improve overall survival and is the current standard of care.10,17 A study comparing 5 and 3 years of imatinib is ongoing to establish whether a longer period of adjuvant treatment is warranted. In patients with metastatic GIST, lifelong imatinib until lack of benefit is considered optimal treatment.10 All patients should undergo KIT mutation analysis. Those with the PDGFRA D842V mutation, SDH (succinate dehydrogenase) deficiency, or neurofibromatosis-related GIST should not receive adjuvant imatinib.
This case has several unusual features. The small tumor size with a very high mitotic rate is rare. Such cases have not been reported in large numbers and have therefore not been reliably incorporated into risk prediction algorithms. In addition, despite a high mitotic index, the tumor was not FDG avid on PET imaging. The diagnosis of GIST is strongly supported by the KIT mutation and response to imatinib. This particular KIT mutation in larger GISTs is associated with aggressive disease. The present case adds to the data on the biology of small gastric GISTs with a high mitotic index and suggests the mitotic index in these tumors may be a more important predictor than size.
Acknowledgment
The authors thank Michael Franklin, MS, for editorial assistance, and Sabrina Porter for media edits.
1. Corless CL, Barnett CM, Heinrich MC. Gastrointestinal stromal tumours: origin and molecular oncology. Nat Rev Cancer. 2011;11(12):865-878.
2. Hirota S, Isozaki K, Moriyama Y, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998;279(5350):577-580.
3. Corless CL, Ballman KV, Antonescu CR, et al. Pathologic and molecular features correlate with long-term outcome after adjuvant therapy of resected primary GI stromal tumor: the ACOSOG Z9001 trial. J Clin Oncol. 2014;32(15):1563-1570.
4. Huang J, Zheng DL, Qin FS, et al. Genetic and epigenetic silencing of SCARA5 may contribute to human hepatocellular carcinoma by activating FAK signaling. J Clin Invest. 2010;120(1):223-241.
5. Joensuu H, Vehtari A, Riihimaki J, et al. Risk of recurrence of gastrointestinal stromal tumour after surgery: an analysis of pooled population-based cohorts. Lancet Oncol. 2012;13(3):265-274.
6. Miettinen M, Lasota J. Gastrointestinal stromal tumors: review on morphology, molecular pathology, prognosis, and differential diagnosis. Arch Pathol Lab Med. 2006;130(10):1466-1478.
7. Miettinen M, Sobin LH, Lasota J. Gastrointestinal stromal tumors of the stomach: a clinicopathologic, immunohistochemical, and molecular genetic study of 1765 cases with long-term follow-up. Am J Surg Pathol. 2005;29(1):52-68.
8. Patel S. Navigating risk stratification systems for the management of patients with GIST. Ann Surg Oncol. 2011;18(6):1698-1704.
9. Rossi S, Miceli R, Messerini L, et al. Natural history of imatinib-naive GISTs: a retrospective analysis of 929 cases with long-term follow-up and development of a survival nomogram based on mitotic index and size as continuous variables. Am J Surg Pathol. 2011;35(11):1646-1656.
10. National Comprehensive Cancer Network. Sarcoma. https://www.nccn.org. Accessed March 27, 2018.
11. Fletcher CD, Berman JJ, Corless C, et al. Diagnosis of gastrointestinal stromal tumors: a consensus approach. Int J Surg Pathol. 2002;10(2):81-89.
12. Huang HY, Li CF, Huang WW, et al. A modification of NIH consensus criteria to better distinguish the highly lethal subset of primary localized gastrointestinal stromal tumors: a subdivision of the original high-risk group on the basis of outcome. Surgery. 2007;141(6):748-756.
13. Kim MC, Yook JH, Yang HK, et al. Long-term surgical outcome of 1057 gastric GISTs according to 7th UICC/AJCC TNM system: multicenter observational study From Korea and Japan. Medicine (Baltimore). 2015;94(41):e1526.
14. Casali PG, Blay JY; ESMO/CONTICANET/EUROBONET Consensus Panel of experts. Soft tissue sarcomas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2010;21(Suppl 5):v198-v203.
15. Joensuu H, DeMatteo RP. The management of gastrointestinal stromal tumors: a model for targeted and multidisciplinary therapy of malignancy. Annu Rev Med. 2012;63:247-258.
16. Dematteo RP, Ballman KV, Antonescu CR, et al. Adjuvant imatinib mesylate after resection of localised, primary gastrointestinal stromal tumour: a randomised, double-blind, placebo-controlled trial. Lancet. 2009;373(9669):1097-1104.
17. Joensuu H, Eriksson M, Sundby Hall K, et al. One vs three years of adjuvant imatinib for operable gastrointestinal stromal tumor: a randomized trial. JAMA. 2012;307(12):1265-1272.
18. Joensuu H, Rutkowski P, Nishida T, et al. KIT and PDGFRA mutations and the risk of GI stromal tumor recurrence. J Clin Oncol. 2015;33(6):634-642.
19. Fletcher CD, Berman JJ, Corless C, et al. Diagnosis of gastrointestinal stromal tumors: A consensus approach. Hum Pathol. 2002;33(5):459-465.
20. Antonescu CR, Viale A, Sarran L, et al. Gene expression in gastrointestinal stromal tumors is distinguished by KIT genotype and anatomic site. Clin Cancer Res. 2004;10(10):3282-3290.
21. Arne G, Kristiansson E, Nerman O, et al. Expression profiling of GIST: CD133 is associated with KIT exon 11 mutations, gastric location and poor prognosis. Int J Cancer. 2011;129(5):1149-1161.
22. Bertucci F, Finetti P, Ostrowski J, et al. Genomic Grade Index predicts postoperative clinical outcome of GIST. Br J Cancer. 2012;107(8):1433-1441.
23. Koon N, Schneider-Stock R, Sarlomo-Rikala M, et al. Molecular targets for tumour progression in gastrointestinal stromal tumours. Gut. 2004;53(2):235-240.
24. Lagarde P, Perot G, Kauffmann A, et al. Mitotic checkpoints and chromosome instability are strong predictors of clinical outcome in gastrointestinal stromal tumors. Clin Cancer Res. 2012;18(3):826-838.
25. Skubitz KM, Geschwind K, Xu WW, Koopmeiners JS, Skubitz AP. Gene expression identifies heterogeneity of metastatic behavior among gastrointestinal stromal tumors. J Transl Med. 2016;14:51.
26. Yamaguchi U, Nakayama R, Honda K, et al. Distinct gene expression-defined classes of gastrointestinal stromal tumor. J Clin Oncol. 2008;26(25):4100-4108.
27. Ylipaa A, Hunt KK, Yang J, et al. Integrative genomic characterization and a genomic staging system for gastrointestinal stromal tumors. Cancer. 2011;117(2):380-389.
28. Kawanowa K, Sakuma Y, Sakurai S, et al. High incidence of microscopic gastrointestinal stromal tumors in the stomach. Hum Pathol. 2006;37(12):1527-1535.
Gastrointestinal stromal tumor (GIST) is the most common soft tissue sarcoma of the gastrointestinal tract, usually arising from the interstitial cells of Cajal or similar cells in the outer wall of the gastrointestinal tract.1,2 Most GISTs have an activating mutation in KIT or platelet-derived growth factor receptor alpha (PDGFRA). Tumor size, mitotic rate, and anatomic site are the most common pathological features used to risk stratify GIST tumors.3-10 It is important to note when using such risk calculators that preoperative imatinib before determining tumor characteristics (such as mitoses per 50 high-power fields [hpf]) often changes the relevant parameters so that the same risk calculations may not apply. Tumors with a mitotic rate ≤5 mitoses per 50 hpf and a size ≤5 cm in greatest dimension have a lower recurrence rate after resection than tumors with a mitotic rate >5 mitoses per 50 hpf and a size >10 cm, and larger tumors can have a recurrence rate of up to 86%.11,12 Findings from a large observational study have suggested that the prognosis of gastric GIST in Korea and Japan may be more favorable compared with that in Western countries.13
The primary treatment of a localized primary GIST is surgical excision, but a cure is limited by recurrence.14,15 Imatinib is useful in the treatment of metastatic or recurrent GIST, and adjuvant treatment with imatinib after surgery has been shown to improve progression-free and overall survival in some cases.3,16-18 Responses to adjuvant imatinib depend on tumor sensitivity to the drug and the risk of recurrence. Drug sensitivity is largely dependent on the presence of mutations in KIT or PDGFRA.3,18 Recurrence risk is highly dependent on tumor size, tumor site, tumor rupture, and mitotic index.1,3,5,6,8,9,18,19 Findings on the use of gene expression patterns to predict recurrence risk have also been reported.20-27 However, recurrence risk is poorly understood for categories in which there are few cases with known outcomes, such as very small gastric GIST with a high mitotic index. For example, few cases of gastric GIST have been reported with a tumor size ≤2 cm, a mitotic rate >5 mitoses per 50 hpf, and adequate clinical follow-up. In such cases, it is difficult to assess the risk of recurrence.6 We report here the long-term outcome of a patient with a 1.8 cm gastric GIST with a mitotic index of 36 mitoses per 50 hpf and a KIT exon 11 mutation.
Case presentation and summary
A 69-year-old man presented with periumbilical and epigastric pain of 6-month duration. His medical history was notable for hyperlipidemia, hypertension, coronary angioplasty, and spinal surgery. He had a 40 pack-year smoking history and consumed 2 to 4 alcoholic drinks per day. The results of a physical examination were unremarkable. A computedtomographic (CT) scan showed no abnormalities. An esophagoduodenoscopy (EGD) revealed gastric ulcers. He was treated successfully with omeprazole 20 mg by mouth daily.
A month later, a follow-up EGD revealed a 1.8 × 1.5 cm submucosal mass 3 cm from the gastroesophageal junction. The patient underwent a fundus wedge resection, and a submucosal mass 1.8 cm in greatest dimension was removed. Pathologic examination revealed a GIST, spindle cell type, with a mitotic rate of 36 mitoses per 50 hpf with negative margins. Immunohistochemistry was positive for CD117. An exon 11 deletion (KVV558-560NV) was present in KIT. The patient’s risk of recurrence was unclear, and his follow-up included CT scans of the abdomen and pelvis every 3 to 4 months for the first 2 years, then every 6 months for the next 2.5 years.
A CT scan about 3.5 years after primary resection revealed small nonspecific liver hypodensities that became more prominent during the next year. About 5 years after primary resection, magnetic resonance imaging (MRI) revealed several liver lesions, the largest of which measured at 1.3 cm in greatest dimension. The patient’s liver metastases were readily identified by MRI (Figure 1) and CT imaging (Figure 2A).
Discussion
Small gastric GISTs are sometimes found by endoscopy performed for unrelated reasons. Recent data suggest that the incidence of gastric GIST may be higher than previously thought. In a Japanese study of patients with gastric cancer in which 100 stomachs were systematically examined pathologically, 50 microscopic GISTs were found in 35 patients.28 Most small gastric GISTs have a low mitotic index. Few cases have been described with a high mitotic index. In a study of 1765 cases of GIST of the stomach, 8 patients had a tumor size less than 2 cm and a mitotic index greater than 5. Of those, only 6 patients had long-term follow-up, and 3 were alive without disease at 2, 17, and 20 years of follow-up.7 These limited data make it impossible to predict outcomes in patients with small gastric GIST with a high mitotic index.
For patients who are at high risk of recurrence after surgery, 3 years of adjuvant imatinib treatment compared with 1 year has been shown to improve overall survival and is the current standard of care.10,17 A study comparing 5 and 3 years of imatinib is ongoing to establish whether a longer period of adjuvant treatment is warranted. In patients with metastatic GIST, lifelong imatinib until lack of benefit is considered optimal treatment.10 All patients should undergo KIT mutation analysis. Those with the PDGFRA D842V mutation, SDH (succinate dehydrogenase) deficiency, or neurofibromatosis-related GIST should not receive adjuvant imatinib.
This case has several unusual features. The small tumor size with a very high mitotic rate is rare. Such cases have not been reported in large numbers and have therefore not been reliably incorporated into risk prediction algorithms. In addition, despite a high mitotic index, the tumor was not FDG avid on PET imaging. The diagnosis of GIST is strongly supported by the KIT mutation and response to imatinib. This particular KIT mutation in larger GISTs is associated with aggressive disease. The present case adds to the data on the biology of small gastric GISTs with a high mitotic index and suggests the mitotic index in these tumors may be a more important predictor than size.
Acknowledgment
The authors thank Michael Franklin, MS, for editorial assistance, and Sabrina Porter for media edits.
Gastrointestinal stromal tumor (GIST) is the most common soft tissue sarcoma of the gastrointestinal tract, usually arising from the interstitial cells of Cajal or similar cells in the outer wall of the gastrointestinal tract.1,2 Most GISTs have an activating mutation in KIT or platelet-derived growth factor receptor alpha (PDGFRA). Tumor size, mitotic rate, and anatomic site are the most common pathological features used to risk stratify GIST tumors.3-10 It is important to note when using such risk calculators that preoperative imatinib before determining tumor characteristics (such as mitoses per 50 high-power fields [hpf]) often changes the relevant parameters so that the same risk calculations may not apply. Tumors with a mitotic rate ≤5 mitoses per 50 hpf and a size ≤5 cm in greatest dimension have a lower recurrence rate after resection than tumors with a mitotic rate >5 mitoses per 50 hpf and a size >10 cm, and larger tumors can have a recurrence rate of up to 86%.11,12 Findings from a large observational study have suggested that the prognosis of gastric GIST in Korea and Japan may be more favorable compared with that in Western countries.13
The primary treatment of a localized primary GIST is surgical excision, but a cure is limited by recurrence.14,15 Imatinib is useful in the treatment of metastatic or recurrent GIST, and adjuvant treatment with imatinib after surgery has been shown to improve progression-free and overall survival in some cases.3,16-18 Responses to adjuvant imatinib depend on tumor sensitivity to the drug and the risk of recurrence. Drug sensitivity is largely dependent on the presence of mutations in KIT or PDGFRA.3,18 Recurrence risk is highly dependent on tumor size, tumor site, tumor rupture, and mitotic index.1,3,5,6,8,9,18,19 Findings on the use of gene expression patterns to predict recurrence risk have also been reported.20-27 However, recurrence risk is poorly understood for categories in which there are few cases with known outcomes, such as very small gastric GIST with a high mitotic index. For example, few cases of gastric GIST have been reported with a tumor size ≤2 cm, a mitotic rate >5 mitoses per 50 hpf, and adequate clinical follow-up. In such cases, it is difficult to assess the risk of recurrence.6 We report here the long-term outcome of a patient with a 1.8 cm gastric GIST with a mitotic index of 36 mitoses per 50 hpf and a KIT exon 11 mutation.
Case presentation and summary
A 69-year-old man presented with periumbilical and epigastric pain of 6-month duration. His medical history was notable for hyperlipidemia, hypertension, coronary angioplasty, and spinal surgery. He had a 40 pack-year smoking history and consumed 2 to 4 alcoholic drinks per day. The results of a physical examination were unremarkable. A computedtomographic (CT) scan showed no abnormalities. An esophagoduodenoscopy (EGD) revealed gastric ulcers. He was treated successfully with omeprazole 20 mg by mouth daily.
A month later, a follow-up EGD revealed a 1.8 × 1.5 cm submucosal mass 3 cm from the gastroesophageal junction. The patient underwent a fundus wedge resection, and a submucosal mass 1.8 cm in greatest dimension was removed. Pathologic examination revealed a GIST, spindle cell type, with a mitotic rate of 36 mitoses per 50 hpf with negative margins. Immunohistochemistry was positive for CD117. An exon 11 deletion (KVV558-560NV) was present in KIT. The patient’s risk of recurrence was unclear, and his follow-up included CT scans of the abdomen and pelvis every 3 to 4 months for the first 2 years, then every 6 months for the next 2.5 years.
A CT scan about 3.5 years after primary resection revealed small nonspecific liver hypodensities that became more prominent during the next year. About 5 years after primary resection, magnetic resonance imaging (MRI) revealed several liver lesions, the largest of which measured at 1.3 cm in greatest dimension. The patient’s liver metastases were readily identified by MRI (Figure 1) and CT imaging (Figure 2A).
Discussion
Small gastric GISTs are sometimes found by endoscopy performed for unrelated reasons. Recent data suggest that the incidence of gastric GIST may be higher than previously thought. In a Japanese study of patients with gastric cancer in which 100 stomachs were systematically examined pathologically, 50 microscopic GISTs were found in 35 patients.28 Most small gastric GISTs have a low mitotic index. Few cases have been described with a high mitotic index. In a study of 1765 cases of GIST of the stomach, 8 patients had a tumor size less than 2 cm and a mitotic index greater than 5. Of those, only 6 patients had long-term follow-up, and 3 were alive without disease at 2, 17, and 20 years of follow-up.7 These limited data make it impossible to predict outcomes in patients with small gastric GIST with a high mitotic index.
For patients who are at high risk of recurrence after surgery, 3 years of adjuvant imatinib treatment compared with 1 year has been shown to improve overall survival and is the current standard of care.10,17 A study comparing 5 and 3 years of imatinib is ongoing to establish whether a longer period of adjuvant treatment is warranted. In patients with metastatic GIST, lifelong imatinib until lack of benefit is considered optimal treatment.10 All patients should undergo KIT mutation analysis. Those with the PDGFRA D842V mutation, SDH (succinate dehydrogenase) deficiency, or neurofibromatosis-related GIST should not receive adjuvant imatinib.
This case has several unusual features. The small tumor size with a very high mitotic rate is rare. Such cases have not been reported in large numbers and have therefore not been reliably incorporated into risk prediction algorithms. In addition, despite a high mitotic index, the tumor was not FDG avid on PET imaging. The diagnosis of GIST is strongly supported by the KIT mutation and response to imatinib. This particular KIT mutation in larger GISTs is associated with aggressive disease. The present case adds to the data on the biology of small gastric GISTs with a high mitotic index and suggests the mitotic index in these tumors may be a more important predictor than size.
Acknowledgment
The authors thank Michael Franklin, MS, for editorial assistance, and Sabrina Porter for media edits.
1. Corless CL, Barnett CM, Heinrich MC. Gastrointestinal stromal tumours: origin and molecular oncology. Nat Rev Cancer. 2011;11(12):865-878.
2. Hirota S, Isozaki K, Moriyama Y, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998;279(5350):577-580.
3. Corless CL, Ballman KV, Antonescu CR, et al. Pathologic and molecular features correlate with long-term outcome after adjuvant therapy of resected primary GI stromal tumor: the ACOSOG Z9001 trial. J Clin Oncol. 2014;32(15):1563-1570.
4. Huang J, Zheng DL, Qin FS, et al. Genetic and epigenetic silencing of SCARA5 may contribute to human hepatocellular carcinoma by activating FAK signaling. J Clin Invest. 2010;120(1):223-241.
5. Joensuu H, Vehtari A, Riihimaki J, et al. Risk of recurrence of gastrointestinal stromal tumour after surgery: an analysis of pooled population-based cohorts. Lancet Oncol. 2012;13(3):265-274.
6. Miettinen M, Lasota J. Gastrointestinal stromal tumors: review on morphology, molecular pathology, prognosis, and differential diagnosis. Arch Pathol Lab Med. 2006;130(10):1466-1478.
7. Miettinen M, Sobin LH, Lasota J. Gastrointestinal stromal tumors of the stomach: a clinicopathologic, immunohistochemical, and molecular genetic study of 1765 cases with long-term follow-up. Am J Surg Pathol. 2005;29(1):52-68.
8. Patel S. Navigating risk stratification systems for the management of patients with GIST. Ann Surg Oncol. 2011;18(6):1698-1704.
9. Rossi S, Miceli R, Messerini L, et al. Natural history of imatinib-naive GISTs: a retrospective analysis of 929 cases with long-term follow-up and development of a survival nomogram based on mitotic index and size as continuous variables. Am J Surg Pathol. 2011;35(11):1646-1656.
10. National Comprehensive Cancer Network. Sarcoma. https://www.nccn.org. Accessed March 27, 2018.
11. Fletcher CD, Berman JJ, Corless C, et al. Diagnosis of gastrointestinal stromal tumors: a consensus approach. Int J Surg Pathol. 2002;10(2):81-89.
12. Huang HY, Li CF, Huang WW, et al. A modification of NIH consensus criteria to better distinguish the highly lethal subset of primary localized gastrointestinal stromal tumors: a subdivision of the original high-risk group on the basis of outcome. Surgery. 2007;141(6):748-756.
13. Kim MC, Yook JH, Yang HK, et al. Long-term surgical outcome of 1057 gastric GISTs according to 7th UICC/AJCC TNM system: multicenter observational study From Korea and Japan. Medicine (Baltimore). 2015;94(41):e1526.
14. Casali PG, Blay JY; ESMO/CONTICANET/EUROBONET Consensus Panel of experts. Soft tissue sarcomas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2010;21(Suppl 5):v198-v203.
15. Joensuu H, DeMatteo RP. The management of gastrointestinal stromal tumors: a model for targeted and multidisciplinary therapy of malignancy. Annu Rev Med. 2012;63:247-258.
16. Dematteo RP, Ballman KV, Antonescu CR, et al. Adjuvant imatinib mesylate after resection of localised, primary gastrointestinal stromal tumour: a randomised, double-blind, placebo-controlled trial. Lancet. 2009;373(9669):1097-1104.
17. Joensuu H, Eriksson M, Sundby Hall K, et al. One vs three years of adjuvant imatinib for operable gastrointestinal stromal tumor: a randomized trial. JAMA. 2012;307(12):1265-1272.
18. Joensuu H, Rutkowski P, Nishida T, et al. KIT and PDGFRA mutations and the risk of GI stromal tumor recurrence. J Clin Oncol. 2015;33(6):634-642.
19. Fletcher CD, Berman JJ, Corless C, et al. Diagnosis of gastrointestinal stromal tumors: A consensus approach. Hum Pathol. 2002;33(5):459-465.
20. Antonescu CR, Viale A, Sarran L, et al. Gene expression in gastrointestinal stromal tumors is distinguished by KIT genotype and anatomic site. Clin Cancer Res. 2004;10(10):3282-3290.
21. Arne G, Kristiansson E, Nerman O, et al. Expression profiling of GIST: CD133 is associated with KIT exon 11 mutations, gastric location and poor prognosis. Int J Cancer. 2011;129(5):1149-1161.
22. Bertucci F, Finetti P, Ostrowski J, et al. Genomic Grade Index predicts postoperative clinical outcome of GIST. Br J Cancer. 2012;107(8):1433-1441.
23. Koon N, Schneider-Stock R, Sarlomo-Rikala M, et al. Molecular targets for tumour progression in gastrointestinal stromal tumours. Gut. 2004;53(2):235-240.
24. Lagarde P, Perot G, Kauffmann A, et al. Mitotic checkpoints and chromosome instability are strong predictors of clinical outcome in gastrointestinal stromal tumors. Clin Cancer Res. 2012;18(3):826-838.
25. Skubitz KM, Geschwind K, Xu WW, Koopmeiners JS, Skubitz AP. Gene expression identifies heterogeneity of metastatic behavior among gastrointestinal stromal tumors. J Transl Med. 2016;14:51.
26. Yamaguchi U, Nakayama R, Honda K, et al. Distinct gene expression-defined classes of gastrointestinal stromal tumor. J Clin Oncol. 2008;26(25):4100-4108.
27. Ylipaa A, Hunt KK, Yang J, et al. Integrative genomic characterization and a genomic staging system for gastrointestinal stromal tumors. Cancer. 2011;117(2):380-389.
28. Kawanowa K, Sakuma Y, Sakurai S, et al. High incidence of microscopic gastrointestinal stromal tumors in the stomach. Hum Pathol. 2006;37(12):1527-1535.
1. Corless CL, Barnett CM, Heinrich MC. Gastrointestinal stromal tumours: origin and molecular oncology. Nat Rev Cancer. 2011;11(12):865-878.
2. Hirota S, Isozaki K, Moriyama Y, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998;279(5350):577-580.
3. Corless CL, Ballman KV, Antonescu CR, et al. Pathologic and molecular features correlate with long-term outcome after adjuvant therapy of resected primary GI stromal tumor: the ACOSOG Z9001 trial. J Clin Oncol. 2014;32(15):1563-1570.
4. Huang J, Zheng DL, Qin FS, et al. Genetic and epigenetic silencing of SCARA5 may contribute to human hepatocellular carcinoma by activating FAK signaling. J Clin Invest. 2010;120(1):223-241.
5. Joensuu H, Vehtari A, Riihimaki J, et al. Risk of recurrence of gastrointestinal stromal tumour after surgery: an analysis of pooled population-based cohorts. Lancet Oncol. 2012;13(3):265-274.
6. Miettinen M, Lasota J. Gastrointestinal stromal tumors: review on morphology, molecular pathology, prognosis, and differential diagnosis. Arch Pathol Lab Med. 2006;130(10):1466-1478.
7. Miettinen M, Sobin LH, Lasota J. Gastrointestinal stromal tumors of the stomach: a clinicopathologic, immunohistochemical, and molecular genetic study of 1765 cases with long-term follow-up. Am J Surg Pathol. 2005;29(1):52-68.
8. Patel S. Navigating risk stratification systems for the management of patients with GIST. Ann Surg Oncol. 2011;18(6):1698-1704.
9. Rossi S, Miceli R, Messerini L, et al. Natural history of imatinib-naive GISTs: a retrospective analysis of 929 cases with long-term follow-up and development of a survival nomogram based on mitotic index and size as continuous variables. Am J Surg Pathol. 2011;35(11):1646-1656.
10. National Comprehensive Cancer Network. Sarcoma. https://www.nccn.org. Accessed March 27, 2018.
11. Fletcher CD, Berman JJ, Corless C, et al. Diagnosis of gastrointestinal stromal tumors: a consensus approach. Int J Surg Pathol. 2002;10(2):81-89.
12. Huang HY, Li CF, Huang WW, et al. A modification of NIH consensus criteria to better distinguish the highly lethal subset of primary localized gastrointestinal stromal tumors: a subdivision of the original high-risk group on the basis of outcome. Surgery. 2007;141(6):748-756.
13. Kim MC, Yook JH, Yang HK, et al. Long-term surgical outcome of 1057 gastric GISTs according to 7th UICC/AJCC TNM system: multicenter observational study From Korea and Japan. Medicine (Baltimore). 2015;94(41):e1526.
14. Casali PG, Blay JY; ESMO/CONTICANET/EUROBONET Consensus Panel of experts. Soft tissue sarcomas: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2010;21(Suppl 5):v198-v203.
15. Joensuu H, DeMatteo RP. The management of gastrointestinal stromal tumors: a model for targeted and multidisciplinary therapy of malignancy. Annu Rev Med. 2012;63:247-258.
16. Dematteo RP, Ballman KV, Antonescu CR, et al. Adjuvant imatinib mesylate after resection of localised, primary gastrointestinal stromal tumour: a randomised, double-blind, placebo-controlled trial. Lancet. 2009;373(9669):1097-1104.
17. Joensuu H, Eriksson M, Sundby Hall K, et al. One vs three years of adjuvant imatinib for operable gastrointestinal stromal tumor: a randomized trial. JAMA. 2012;307(12):1265-1272.
18. Joensuu H, Rutkowski P, Nishida T, et al. KIT and PDGFRA mutations and the risk of GI stromal tumor recurrence. J Clin Oncol. 2015;33(6):634-642.
19. Fletcher CD, Berman JJ, Corless C, et al. Diagnosis of gastrointestinal stromal tumors: A consensus approach. Hum Pathol. 2002;33(5):459-465.
20. Antonescu CR, Viale A, Sarran L, et al. Gene expression in gastrointestinal stromal tumors is distinguished by KIT genotype and anatomic site. Clin Cancer Res. 2004;10(10):3282-3290.
21. Arne G, Kristiansson E, Nerman O, et al. Expression profiling of GIST: CD133 is associated with KIT exon 11 mutations, gastric location and poor prognosis. Int J Cancer. 2011;129(5):1149-1161.
22. Bertucci F, Finetti P, Ostrowski J, et al. Genomic Grade Index predicts postoperative clinical outcome of GIST. Br J Cancer. 2012;107(8):1433-1441.
23. Koon N, Schneider-Stock R, Sarlomo-Rikala M, et al. Molecular targets for tumour progression in gastrointestinal stromal tumours. Gut. 2004;53(2):235-240.
24. Lagarde P, Perot G, Kauffmann A, et al. Mitotic checkpoints and chromosome instability are strong predictors of clinical outcome in gastrointestinal stromal tumors. Clin Cancer Res. 2012;18(3):826-838.
25. Skubitz KM, Geschwind K, Xu WW, Koopmeiners JS, Skubitz AP. Gene expression identifies heterogeneity of metastatic behavior among gastrointestinal stromal tumors. J Transl Med. 2016;14:51.
26. Yamaguchi U, Nakayama R, Honda K, et al. Distinct gene expression-defined classes of gastrointestinal stromal tumor. J Clin Oncol. 2008;26(25):4100-4108.
27. Ylipaa A, Hunt KK, Yang J, et al. Integrative genomic characterization and a genomic staging system for gastrointestinal stromal tumors. Cancer. 2011;117(2):380-389.
28. Kawanowa K, Sakuma Y, Sakurai S, et al. High incidence of microscopic gastrointestinal stromal tumors in the stomach. Hum Pathol. 2006;37(12):1527-1535.
Striking rash in a patient with lung cancer on a checkpoint inhibitor
Lung cancer remains the most common cause of cancer death in the United States and worldwide.1 Despite advances in the treatment of the disease and development of targeted therapy, the 5-year overall survival in stage IV non–small-cell lung cancer remains poor, ranging from 6% to 10%.2 More recently, checkpoint inhibitors have had a major impact on the treatment of lung cancer. Nivolumab was the first program cell death protein-1 (PD-1) inhibitor approved for malignant melanoma.3 In July 2015, it was approved as a second-line treatment of squamous cell carcinoma of the lung.4 Since then, the use of nivolumab has extended to other malignancies such as head and neck cancer, renal cell carcinoma, and the list continues to expand. In lung cancer, it demonstrated superior overall survival of 9 months, compared with 6 months with docetaxel.4 Other checkpoint inhibitors such as pembrolizumab5 and atezolizumab6 were subsequently developed, and are also used in the treatment of lung cancer.
Serious potential autoimmune complications arise in up to 30% of patients treated with PD-1 inhibitors. Dermatologic toxicity is the most common immune-related adverse event in these patients. In addition to vitiligo, most common is a reticular maculopapular rash on the trunk and extremities. Other adverse events, such as photosensitivity, alopecia, xerosis, and hair color changes, are reported less frequently.7 We report here a case of rash at an unusual location (auricular and periauricular) with skin exfoliation mimicking other common skin conditions such as eczema and psoriasis.
Case presentation and summary
A 57-year-old woman with a history of cerebrovascular accident with residual left lower-leg paresis presented for acute onset expressive aphasia in the absence of other constitutional or neurological findings. Magnetic resonance imaging of the brain showed a posterior, left parietal lobe lesion of 1.6 cm with intralesional hemorrhage and surrounding edema suggestive of brain metastasis. The patient had a 35 pack-year history of smoking. A staging work-up with computed-tomographic (CT) scans showed a spiculated enhancing nodule in the superior segment of the right lower lobe plus mediastinal adenopathy.
The patient underwent a CT-guided core biopsy of the spiculated nodule, which was found to be consistent with adenocarcinoma of the lung. It was negative for EGFR mutation or ALK rearrangement. She received stereotactic radiosurgery to the left posterior parietal lesion, and after completion of radiation, was started on systemic chemotherapy with cisplatin plus pemetrexed for adenocarcinoma of the lung. She received 4 cycles of chemotherapy. Repeat imaging with a PET-CT showed interval increase of the mediastinal hypermetabolic lymphadenopathy with new hypermetabolic pretracheal lymph nodes and interval development of multiple liver metastases in the right and left lobes of the liver (Figure 1). She was started on second-line therapy with nivolumab at a dose of 240 mg every 2 weeks. The treatment was complicated initially by new onset grade 2 papular pruritic rash after cycle 2 of therapy. The rash involved the upper and lower extremities, sparing the palms, soles, trunk, abdomen, and the back. It resolved with treatment delay and topical steroids.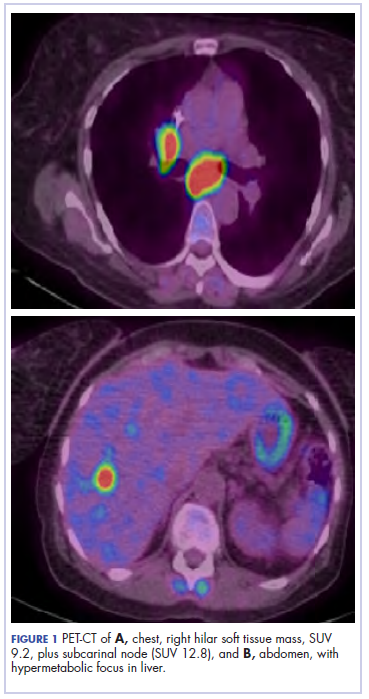
The patient resumed treatment with nivolumab after complete resolution of the rash. However, she developed grade 2 nephritis after cycle 5 with a creatinine level of 1.98 mg/dL (reference range, 0.6-1.2 mg/ dL). This was resolved after treatment with oral prednisone, at a starting dose of 1 mg/kg and tapered over 4 weeks. PET CT scans obtained after cycles 5 and 11 showed no metabolic activity in the mediastinum or the liver and markedly decreased uptake in the right lower lobe nodule, down to an SUV of 1.7 with no new nodules. An MRI of the brain was stable (Figure 2).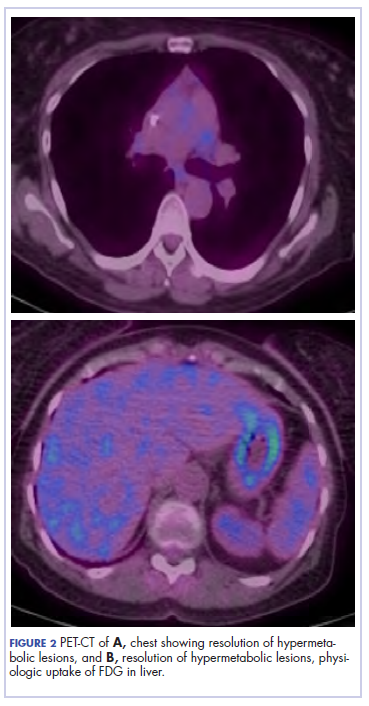
After cycle 16 of nivolumab, the patient developed a severe eczematous rash with excoriations at the base of both ears involving the periauricular and auricular areas bilaterally (Figure 3).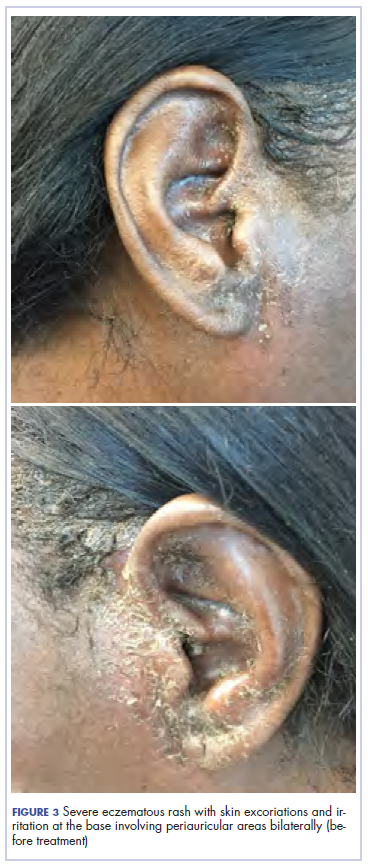
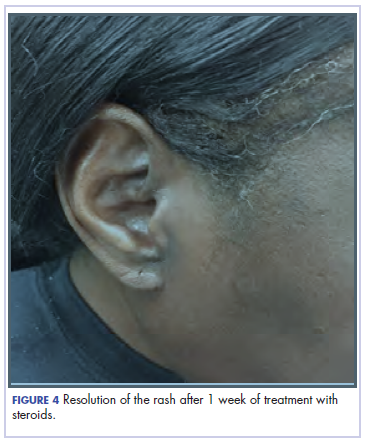
She completed 4 weeks of steroid therapy on a tapering schedule. Treatment with nivolumab was resumed afterward with no adverse autoimmune complications. At her last visit (25 months after initiating a PD-1 inhibitor), there was no clinical or radiologic evidence of lung cancer nor any of autoimmune adverse effects.
Discussion
Among multiple autoimmune complications, dermatologic toxicity is the most common immune-related adverse event, occuring in about 30% to 40% of patients7,8 and with an average onset of 3-4 weeks after initiating treatment with checkpoint inhibitors.9 In addition to vitiligo, the most common type of rash described is a reticular maculopapular rash on the trunk and extremities.10 Other findings, such as photosensitivity, alopecia, xerosis, and hair color changes, have been reported in smaller numbers. Skin exfoliation, as seen in the present case, has been reported in fewer than 1% of the cases.4 Perivascular lymphocytic infiltrates extending deep into the dermis are most likely to be seen if the lesions are biopsied. Both the location of the rash in our patient and its relapsing nature are rare and make it more interesting as it presents a diagnostic dilemma for treating physicians. Ear, nose, and throat surgeons are more likely to encounter such a complication with the expanded use of PD-1 and PD-ligand 1 inhibitors in advanced head and neck cancers. The differential diagnosis includes localized eczema, psoriatic rash, skin infection, or an autoimmune phenomenon.
The location of the rash was also of concern because there have been reports of autoimmune inner-ear disease related to immunotherapy.11 After the failure of treatment with empiric antibiotics and topical steroids, in addition to the development of a new rash on her abdomen, we concluded that this case might represent an unusual autoimmune skin complication. The resolution of the skin lesions in both locations (the ears and the abdomen) with the oral steroid therapy, supported our suspected diagnosis of autoimmune dermatitis.
It is essential that these complications are detected early and misdiagnosis is avoided because timely treatment with steroids will prevent progression to more severe problems such as Steven-Johnson syndrome, toxic epidermal necrolysis,12 or extension into the inner ear.11This case is part of a growing spectrum of other unusual cases seen with immunotherapy treatment, such as erythema nodosum-like reactions,13 bullous dermatitis,14 and psoriasiform eruptions.15 It highlights the need for an awareness of expanding dermatologic complications from immunotherapy beyond the reported common manifestations. Established guidelines and algorithms for the management of immune-related dermatologic toxicity are available to assist the physician in treatment (Table 1).16 Skin biopsy should be considered if the diagnosis remains uncertain, although starting empiric treatment with steroids is a widely acceptable approach. Reassessing the skin rash in 48 hours to 1 week after treatment initiation is crucial because steroid-refractory cases will need additional immunosuppression. Early termination of steroids is associated with higher recurrence rate, therefore tapering steroids over 4 weeks is highly recommended before resuming treatment with checkpoint inhibitors.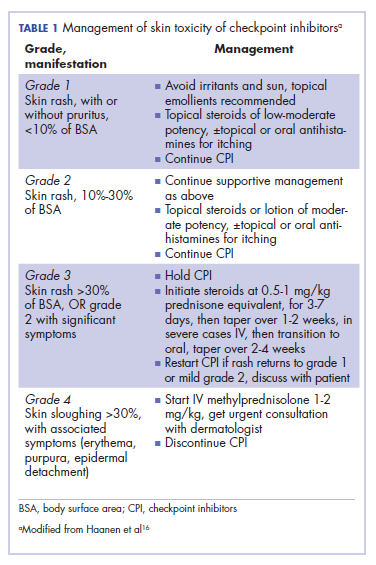
In summary, increased awareness among health care professionals of the common and unusual complications of immunotherapy agents is important and essential in patient care. In addition to oncologists, head and neck surgeons, pulmonologists, urologists, dermatologists, and general internists will encounter patients with immunotherapy-related complications. Patient education should be emphasized to ensure prompt investigation and treatment of complications. Finally, it is not yet clear whether the development of autoimmune reactions predicts disease response to treatment. In a series of 134 patients with lung cancer, the occurrence of autoimmune adverse events correlated with improved survival.17 More research is needed to identify prognostic and predictive biomarkers for response to immunotherapy.
Conclusion
This pattern of autoimmune dermatitis localizing to the ears is rare (<1% of cases of dermatitis). Nevertheless, it raises the awareness for dermatologic complications of immunotherapy beyond the classical reported manifestations. Prompt diagnosis and treatment is essential to avoid serious complications such as Steven-Johnson syndrome, toxic epidermal necrolysis, and potentially damage to the inner ear.
1.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87-108.
2. Goldstraw P, Chansky K, Crowley J, et al. The IASLC Lung Cancer Staging Project: proposals for revision of the TNM stage groupings in the forthcoming (Eighth) edition of the TNM classification for lung cancer. J Thorac Oncol. 2016;11:39-51.
3. Robert C, Long GV, Brady B, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372:320-330.
4. Brahmer J, Reckamp KL, Baas P, et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N Engl J Med. 2015;373:123-135.
5. Reck M, Rodriguez-Abreu D, Robinson AG, et al. Pembrolizumab versus chemo-therapy for PD- L1- positive non-small-cell lung cancer. N Engl J Med. 2016;375:1823- 1833.
6. Rittmeyer A, Barlesi F, Waterkamp D, et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): a phase 3, open-label, multicentre randomised controlled trial. Lancet. 2017;389:255-265.
7. Collins LK, Chapman MS, Carter JB, Samie FH. Cutaneous adverse events of the immune checkpoint inhibitors. Curr Prob Cancer. 2017;41:125-128.
8. Naidoo J, Page DB, Li BT, et al. Toxicities of the anti-PD-1 and anti-PD-L1 immune checkpoint antibodies. Ann Oncol. 2015;26(12):2375.
9. Weber JS, Kähler KC, Hauschild A. Management of immune-related adverse events and kinetics of response with ipilimumab. J Clin Oncol. 2012;30(21):2691-2697.
10. Belum VR, Benhuri B, Postow MA, et al. Characterisation and management of dermatologic adverse events to agents targeting the PD-1 receptor. Eur J Cancer. 2016;60:12-25.
11. Zibelman M, Pollak N, Olszanski AJ. Autoimmune inner ear disease in a melanoma patient treated with pembrolizumab. J Immunother Cancer. 2016;4:8.
12. Nayar N, Briscoe K, Penas PF. Toxic epidermal necrolysis-like reaction with severe satellite cell necrosis associated with nivolumab in a patient with ipilimumab refractory metastatic melanoma. J Immunother. 2016;39(3):149-152.
13. Tetzlaff MT, Jazaeri AA, Torres-Cabala CA, et al. Erythema nodosum-like panniculitis mimicking disease recurrence: a novel toxicity from immune checkpoint blockade therapy - report of 2 patients. J Cutan Pathol. 2017;44(12):1080-1086.
14. Naidoo J, Schindler K, Querfeld C, et al. Autoimmune bullous skin disorders with immune checkpoint inhibitors targeting PD-1 and PD-L1. Cancer Immunol Res. 2016;4(5):383-389.
15. Ohtsuka M, Miura T, Mori T, Ishikawa M, Yamamoto T. Occurrence of psoriasiform eruption during nivolumab therapy for primary oral mucosal melanoma. JAMA Dermatol. 2015;151(7):797-799.
16. Haanen JBAG, Carbonnel F, Robert C, et al; ESMO Guidelines Committee. Management of toxicities from immunotherapy: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2017;28(suppl 4):iv119-iv142.
17. Haratani K, Hayashi H, Chiba Y, et al. Association of immune-related adverse events with nivolumab efficacy in non-small-cell lung cancer. JAMA Oncol. 2018;4(3):374-378.
Lung cancer remains the most common cause of cancer death in the United States and worldwide.1 Despite advances in the treatment of the disease and development of targeted therapy, the 5-year overall survival in stage IV non–small-cell lung cancer remains poor, ranging from 6% to 10%.2 More recently, checkpoint inhibitors have had a major impact on the treatment of lung cancer. Nivolumab was the first program cell death protein-1 (PD-1) inhibitor approved for malignant melanoma.3 In July 2015, it was approved as a second-line treatment of squamous cell carcinoma of the lung.4 Since then, the use of nivolumab has extended to other malignancies such as head and neck cancer, renal cell carcinoma, and the list continues to expand. In lung cancer, it demonstrated superior overall survival of 9 months, compared with 6 months with docetaxel.4 Other checkpoint inhibitors such as pembrolizumab5 and atezolizumab6 were subsequently developed, and are also used in the treatment of lung cancer.
Serious potential autoimmune complications arise in up to 30% of patients treated with PD-1 inhibitors. Dermatologic toxicity is the most common immune-related adverse event in these patients. In addition to vitiligo, most common is a reticular maculopapular rash on the trunk and extremities. Other adverse events, such as photosensitivity, alopecia, xerosis, and hair color changes, are reported less frequently.7 We report here a case of rash at an unusual location (auricular and periauricular) with skin exfoliation mimicking other common skin conditions such as eczema and psoriasis.
Case presentation and summary
A 57-year-old woman with a history of cerebrovascular accident with residual left lower-leg paresis presented for acute onset expressive aphasia in the absence of other constitutional or neurological findings. Magnetic resonance imaging of the brain showed a posterior, left parietal lobe lesion of 1.6 cm with intralesional hemorrhage and surrounding edema suggestive of brain metastasis. The patient had a 35 pack-year history of smoking. A staging work-up with computed-tomographic (CT) scans showed a spiculated enhancing nodule in the superior segment of the right lower lobe plus mediastinal adenopathy.
The patient underwent a CT-guided core biopsy of the spiculated nodule, which was found to be consistent with adenocarcinoma of the lung. It was negative for EGFR mutation or ALK rearrangement. She received stereotactic radiosurgery to the left posterior parietal lesion, and after completion of radiation, was started on systemic chemotherapy with cisplatin plus pemetrexed for adenocarcinoma of the lung. She received 4 cycles of chemotherapy. Repeat imaging with a PET-CT showed interval increase of the mediastinal hypermetabolic lymphadenopathy with new hypermetabolic pretracheal lymph nodes and interval development of multiple liver metastases in the right and left lobes of the liver (Figure 1). She was started on second-line therapy with nivolumab at a dose of 240 mg every 2 weeks. The treatment was complicated initially by new onset grade 2 papular pruritic rash after cycle 2 of therapy. The rash involved the upper and lower extremities, sparing the palms, soles, trunk, abdomen, and the back. It resolved with treatment delay and topical steroids.
The patient resumed treatment with nivolumab after complete resolution of the rash. However, she developed grade 2 nephritis after cycle 5 with a creatinine level of 1.98 mg/dL (reference range, 0.6-1.2 mg/ dL). This was resolved after treatment with oral prednisone, at a starting dose of 1 mg/kg and tapered over 4 weeks. PET CT scans obtained after cycles 5 and 11 showed no metabolic activity in the mediastinum or the liver and markedly decreased uptake in the right lower lobe nodule, down to an SUV of 1.7 with no new nodules. An MRI of the brain was stable (Figure 2).
After cycle 16 of nivolumab, the patient developed a severe eczematous rash with excoriations at the base of both ears involving the periauricular and auricular areas bilaterally (Figure 3).
She completed 4 weeks of steroid therapy on a tapering schedule. Treatment with nivolumab was resumed afterward with no adverse autoimmune complications. At her last visit (25 months after initiating a PD-1 inhibitor), there was no clinical or radiologic evidence of lung cancer nor any of autoimmune adverse effects.
Discussion
Among multiple autoimmune complications, dermatologic toxicity is the most common immune-related adverse event, occuring in about 30% to 40% of patients7,8 and with an average onset of 3-4 weeks after initiating treatment with checkpoint inhibitors.9 In addition to vitiligo, the most common type of rash described is a reticular maculopapular rash on the trunk and extremities.10 Other findings, such as photosensitivity, alopecia, xerosis, and hair color changes, have been reported in smaller numbers. Skin exfoliation, as seen in the present case, has been reported in fewer than 1% of the cases.4 Perivascular lymphocytic infiltrates extending deep into the dermis are most likely to be seen if the lesions are biopsied. Both the location of the rash in our patient and its relapsing nature are rare and make it more interesting as it presents a diagnostic dilemma for treating physicians. Ear, nose, and throat surgeons are more likely to encounter such a complication with the expanded use of PD-1 and PD-ligand 1 inhibitors in advanced head and neck cancers. The differential diagnosis includes localized eczema, psoriatic rash, skin infection, or an autoimmune phenomenon.
The location of the rash was also of concern because there have been reports of autoimmune inner-ear disease related to immunotherapy.11 After the failure of treatment with empiric antibiotics and topical steroids, in addition to the development of a new rash on her abdomen, we concluded that this case might represent an unusual autoimmune skin complication. The resolution of the skin lesions in both locations (the ears and the abdomen) with the oral steroid therapy, supported our suspected diagnosis of autoimmune dermatitis.
It is essential that these complications are detected early and misdiagnosis is avoided because timely treatment with steroids will prevent progression to more severe problems such as Steven-Johnson syndrome, toxic epidermal necrolysis,12 or extension into the inner ear.11This case is part of a growing spectrum of other unusual cases seen with immunotherapy treatment, such as erythema nodosum-like reactions,13 bullous dermatitis,14 and psoriasiform eruptions.15 It highlights the need for an awareness of expanding dermatologic complications from immunotherapy beyond the reported common manifestations. Established guidelines and algorithms for the management of immune-related dermatologic toxicity are available to assist the physician in treatment (Table 1).16 Skin biopsy should be considered if the diagnosis remains uncertain, although starting empiric treatment with steroids is a widely acceptable approach. Reassessing the skin rash in 48 hours to 1 week after treatment initiation is crucial because steroid-refractory cases will need additional immunosuppression. Early termination of steroids is associated with higher recurrence rate, therefore tapering steroids over 4 weeks is highly recommended before resuming treatment with checkpoint inhibitors.
In summary, increased awareness among health care professionals of the common and unusual complications of immunotherapy agents is important and essential in patient care. In addition to oncologists, head and neck surgeons, pulmonologists, urologists, dermatologists, and general internists will encounter patients with immunotherapy-related complications. Patient education should be emphasized to ensure prompt investigation and treatment of complications. Finally, it is not yet clear whether the development of autoimmune reactions predicts disease response to treatment. In a series of 134 patients with lung cancer, the occurrence of autoimmune adverse events correlated with improved survival.17 More research is needed to identify prognostic and predictive biomarkers for response to immunotherapy.
Conclusion
This pattern of autoimmune dermatitis localizing to the ears is rare (<1% of cases of dermatitis). Nevertheless, it raises the awareness for dermatologic complications of immunotherapy beyond the classical reported manifestations. Prompt diagnosis and treatment is essential to avoid serious complications such as Steven-Johnson syndrome, toxic epidermal necrolysis, and potentially damage to the inner ear.
Lung cancer remains the most common cause of cancer death in the United States and worldwide.1 Despite advances in the treatment of the disease and development of targeted therapy, the 5-year overall survival in stage IV non–small-cell lung cancer remains poor, ranging from 6% to 10%.2 More recently, checkpoint inhibitors have had a major impact on the treatment of lung cancer. Nivolumab was the first program cell death protein-1 (PD-1) inhibitor approved for malignant melanoma.3 In July 2015, it was approved as a second-line treatment of squamous cell carcinoma of the lung.4 Since then, the use of nivolumab has extended to other malignancies such as head and neck cancer, renal cell carcinoma, and the list continues to expand. In lung cancer, it demonstrated superior overall survival of 9 months, compared with 6 months with docetaxel.4 Other checkpoint inhibitors such as pembrolizumab5 and atezolizumab6 were subsequently developed, and are also used in the treatment of lung cancer.
Serious potential autoimmune complications arise in up to 30% of patients treated with PD-1 inhibitors. Dermatologic toxicity is the most common immune-related adverse event in these patients. In addition to vitiligo, most common is a reticular maculopapular rash on the trunk and extremities. Other adverse events, such as photosensitivity, alopecia, xerosis, and hair color changes, are reported less frequently.7 We report here a case of rash at an unusual location (auricular and periauricular) with skin exfoliation mimicking other common skin conditions such as eczema and psoriasis.
Case presentation and summary
A 57-year-old woman with a history of cerebrovascular accident with residual left lower-leg paresis presented for acute onset expressive aphasia in the absence of other constitutional or neurological findings. Magnetic resonance imaging of the brain showed a posterior, left parietal lobe lesion of 1.6 cm with intralesional hemorrhage and surrounding edema suggestive of brain metastasis. The patient had a 35 pack-year history of smoking. A staging work-up with computed-tomographic (CT) scans showed a spiculated enhancing nodule in the superior segment of the right lower lobe plus mediastinal adenopathy.
The patient underwent a CT-guided core biopsy of the spiculated nodule, which was found to be consistent with adenocarcinoma of the lung. It was negative for EGFR mutation or ALK rearrangement. She received stereotactic radiosurgery to the left posterior parietal lesion, and after completion of radiation, was started on systemic chemotherapy with cisplatin plus pemetrexed for adenocarcinoma of the lung. She received 4 cycles of chemotherapy. Repeat imaging with a PET-CT showed interval increase of the mediastinal hypermetabolic lymphadenopathy with new hypermetabolic pretracheal lymph nodes and interval development of multiple liver metastases in the right and left lobes of the liver (Figure 1). She was started on second-line therapy with nivolumab at a dose of 240 mg every 2 weeks. The treatment was complicated initially by new onset grade 2 papular pruritic rash after cycle 2 of therapy. The rash involved the upper and lower extremities, sparing the palms, soles, trunk, abdomen, and the back. It resolved with treatment delay and topical steroids.
The patient resumed treatment with nivolumab after complete resolution of the rash. However, she developed grade 2 nephritis after cycle 5 with a creatinine level of 1.98 mg/dL (reference range, 0.6-1.2 mg/ dL). This was resolved after treatment with oral prednisone, at a starting dose of 1 mg/kg and tapered over 4 weeks. PET CT scans obtained after cycles 5 and 11 showed no metabolic activity in the mediastinum or the liver and markedly decreased uptake in the right lower lobe nodule, down to an SUV of 1.7 with no new nodules. An MRI of the brain was stable (Figure 2).
After cycle 16 of nivolumab, the patient developed a severe eczematous rash with excoriations at the base of both ears involving the periauricular and auricular areas bilaterally (Figure 3).
She completed 4 weeks of steroid therapy on a tapering schedule. Treatment with nivolumab was resumed afterward with no adverse autoimmune complications. At her last visit (25 months after initiating a PD-1 inhibitor), there was no clinical or radiologic evidence of lung cancer nor any of autoimmune adverse effects.
Discussion
Among multiple autoimmune complications, dermatologic toxicity is the most common immune-related adverse event, occuring in about 30% to 40% of patients7,8 and with an average onset of 3-4 weeks after initiating treatment with checkpoint inhibitors.9 In addition to vitiligo, the most common type of rash described is a reticular maculopapular rash on the trunk and extremities.10 Other findings, such as photosensitivity, alopecia, xerosis, and hair color changes, have been reported in smaller numbers. Skin exfoliation, as seen in the present case, has been reported in fewer than 1% of the cases.4 Perivascular lymphocytic infiltrates extending deep into the dermis are most likely to be seen if the lesions are biopsied. Both the location of the rash in our patient and its relapsing nature are rare and make it more interesting as it presents a diagnostic dilemma for treating physicians. Ear, nose, and throat surgeons are more likely to encounter such a complication with the expanded use of PD-1 and PD-ligand 1 inhibitors in advanced head and neck cancers. The differential diagnosis includes localized eczema, psoriatic rash, skin infection, or an autoimmune phenomenon.
The location of the rash was also of concern because there have been reports of autoimmune inner-ear disease related to immunotherapy.11 After the failure of treatment with empiric antibiotics and topical steroids, in addition to the development of a new rash on her abdomen, we concluded that this case might represent an unusual autoimmune skin complication. The resolution of the skin lesions in both locations (the ears and the abdomen) with the oral steroid therapy, supported our suspected diagnosis of autoimmune dermatitis.
It is essential that these complications are detected early and misdiagnosis is avoided because timely treatment with steroids will prevent progression to more severe problems such as Steven-Johnson syndrome, toxic epidermal necrolysis,12 or extension into the inner ear.11This case is part of a growing spectrum of other unusual cases seen with immunotherapy treatment, such as erythema nodosum-like reactions,13 bullous dermatitis,14 and psoriasiform eruptions.15 It highlights the need for an awareness of expanding dermatologic complications from immunotherapy beyond the reported common manifestations. Established guidelines and algorithms for the management of immune-related dermatologic toxicity are available to assist the physician in treatment (Table 1).16 Skin biopsy should be considered if the diagnosis remains uncertain, although starting empiric treatment with steroids is a widely acceptable approach. Reassessing the skin rash in 48 hours to 1 week after treatment initiation is crucial because steroid-refractory cases will need additional immunosuppression. Early termination of steroids is associated with higher recurrence rate, therefore tapering steroids over 4 weeks is highly recommended before resuming treatment with checkpoint inhibitors.
In summary, increased awareness among health care professionals of the common and unusual complications of immunotherapy agents is important and essential in patient care. In addition to oncologists, head and neck surgeons, pulmonologists, urologists, dermatologists, and general internists will encounter patients with immunotherapy-related complications. Patient education should be emphasized to ensure prompt investigation and treatment of complications. Finally, it is not yet clear whether the development of autoimmune reactions predicts disease response to treatment. In a series of 134 patients with lung cancer, the occurrence of autoimmune adverse events correlated with improved survival.17 More research is needed to identify prognostic and predictive biomarkers for response to immunotherapy.
Conclusion
This pattern of autoimmune dermatitis localizing to the ears is rare (<1% of cases of dermatitis). Nevertheless, it raises the awareness for dermatologic complications of immunotherapy beyond the classical reported manifestations. Prompt diagnosis and treatment is essential to avoid serious complications such as Steven-Johnson syndrome, toxic epidermal necrolysis, and potentially damage to the inner ear.
1.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87-108.
2. Goldstraw P, Chansky K, Crowley J, et al. The IASLC Lung Cancer Staging Project: proposals for revision of the TNM stage groupings in the forthcoming (Eighth) edition of the TNM classification for lung cancer. J Thorac Oncol. 2016;11:39-51.
3. Robert C, Long GV, Brady B, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372:320-330.
4. Brahmer J, Reckamp KL, Baas P, et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N Engl J Med. 2015;373:123-135.
5. Reck M, Rodriguez-Abreu D, Robinson AG, et al. Pembrolizumab versus chemo-therapy for PD- L1- positive non-small-cell lung cancer. N Engl J Med. 2016;375:1823- 1833.
6. Rittmeyer A, Barlesi F, Waterkamp D, et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): a phase 3, open-label, multicentre randomised controlled trial. Lancet. 2017;389:255-265.
7. Collins LK, Chapman MS, Carter JB, Samie FH. Cutaneous adverse events of the immune checkpoint inhibitors. Curr Prob Cancer. 2017;41:125-128.
8. Naidoo J, Page DB, Li BT, et al. Toxicities of the anti-PD-1 and anti-PD-L1 immune checkpoint antibodies. Ann Oncol. 2015;26(12):2375.
9. Weber JS, Kähler KC, Hauschild A. Management of immune-related adverse events and kinetics of response with ipilimumab. J Clin Oncol. 2012;30(21):2691-2697.
10. Belum VR, Benhuri B, Postow MA, et al. Characterisation and management of dermatologic adverse events to agents targeting the PD-1 receptor. Eur J Cancer. 2016;60:12-25.
11. Zibelman M, Pollak N, Olszanski AJ. Autoimmune inner ear disease in a melanoma patient treated with pembrolizumab. J Immunother Cancer. 2016;4:8.
12. Nayar N, Briscoe K, Penas PF. Toxic epidermal necrolysis-like reaction with severe satellite cell necrosis associated with nivolumab in a patient with ipilimumab refractory metastatic melanoma. J Immunother. 2016;39(3):149-152.
13. Tetzlaff MT, Jazaeri AA, Torres-Cabala CA, et al. Erythema nodosum-like panniculitis mimicking disease recurrence: a novel toxicity from immune checkpoint blockade therapy - report of 2 patients. J Cutan Pathol. 2017;44(12):1080-1086.
14. Naidoo J, Schindler K, Querfeld C, et al. Autoimmune bullous skin disorders with immune checkpoint inhibitors targeting PD-1 and PD-L1. Cancer Immunol Res. 2016;4(5):383-389.
15. Ohtsuka M, Miura T, Mori T, Ishikawa M, Yamamoto T. Occurrence of psoriasiform eruption during nivolumab therapy for primary oral mucosal melanoma. JAMA Dermatol. 2015;151(7):797-799.
16. Haanen JBAG, Carbonnel F, Robert C, et al; ESMO Guidelines Committee. Management of toxicities from immunotherapy: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2017;28(suppl 4):iv119-iv142.
17. Haratani K, Hayashi H, Chiba Y, et al. Association of immune-related adverse events with nivolumab efficacy in non-small-cell lung cancer. JAMA Oncol. 2018;4(3):374-378.
1.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87-108.
2. Goldstraw P, Chansky K, Crowley J, et al. The IASLC Lung Cancer Staging Project: proposals for revision of the TNM stage groupings in the forthcoming (Eighth) edition of the TNM classification for lung cancer. J Thorac Oncol. 2016;11:39-51.
3. Robert C, Long GV, Brady B, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372:320-330.
4. Brahmer J, Reckamp KL, Baas P, et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N Engl J Med. 2015;373:123-135.
5. Reck M, Rodriguez-Abreu D, Robinson AG, et al. Pembrolizumab versus chemo-therapy for PD- L1- positive non-small-cell lung cancer. N Engl J Med. 2016;375:1823- 1833.
6. Rittmeyer A, Barlesi F, Waterkamp D, et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): a phase 3, open-label, multicentre randomised controlled trial. Lancet. 2017;389:255-265.
7. Collins LK, Chapman MS, Carter JB, Samie FH. Cutaneous adverse events of the immune checkpoint inhibitors. Curr Prob Cancer. 2017;41:125-128.
8. Naidoo J, Page DB, Li BT, et al. Toxicities of the anti-PD-1 and anti-PD-L1 immune checkpoint antibodies. Ann Oncol. 2015;26(12):2375.
9. Weber JS, Kähler KC, Hauschild A. Management of immune-related adverse events and kinetics of response with ipilimumab. J Clin Oncol. 2012;30(21):2691-2697.
10. Belum VR, Benhuri B, Postow MA, et al. Characterisation and management of dermatologic adverse events to agents targeting the PD-1 receptor. Eur J Cancer. 2016;60:12-25.
11. Zibelman M, Pollak N, Olszanski AJ. Autoimmune inner ear disease in a melanoma patient treated with pembrolizumab. J Immunother Cancer. 2016;4:8.
12. Nayar N, Briscoe K, Penas PF. Toxic epidermal necrolysis-like reaction with severe satellite cell necrosis associated with nivolumab in a patient with ipilimumab refractory metastatic melanoma. J Immunother. 2016;39(3):149-152.
13. Tetzlaff MT, Jazaeri AA, Torres-Cabala CA, et al. Erythema nodosum-like panniculitis mimicking disease recurrence: a novel toxicity from immune checkpoint blockade therapy - report of 2 patients. J Cutan Pathol. 2017;44(12):1080-1086.
14. Naidoo J, Schindler K, Querfeld C, et al. Autoimmune bullous skin disorders with immune checkpoint inhibitors targeting PD-1 and PD-L1. Cancer Immunol Res. 2016;4(5):383-389.
15. Ohtsuka M, Miura T, Mori T, Ishikawa M, Yamamoto T. Occurrence of psoriasiform eruption during nivolumab therapy for primary oral mucosal melanoma. JAMA Dermatol. 2015;151(7):797-799.
16. Haanen JBAG, Carbonnel F, Robert C, et al; ESMO Guidelines Committee. Management of toxicities from immunotherapy: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2017;28(suppl 4):iv119-iv142.
17. Haratani K, Hayashi H, Chiba Y, et al. Association of immune-related adverse events with nivolumab efficacy in non-small-cell lung cancer. JAMA Oncol. 2018;4(3):374-378.