User login
Pharmacist-Led Deprescribing of Aspirin for Primary Prevention of Cardiovascular Disease Among Geriatric Veterans
Pharmacist-Led Deprescribing of Aspirin for Primary Prevention of Cardiovascular Disease Among Geriatric Veterans
Low-dose aspirin commonly is used for the prevention of cardiovascular disease (CVD) but is associated with an increased risk of major bleeding.1 The use of aspirin for primary prevention is largely extrapolated from clinical trials showing benefit in the secondary prevention of myocardial infarction and ischemic stroke. However, results from the Aspirin in Reducing Events in the Elderly (ASPREE) trial challenged this practice.2 The ASPREE trial, conducted in the United States and Australia from 2010 to 2014, sought to determine whether daily 100 mg aspirin, was superior to placebo in promoting disability-free survival among older adults. Participants were aged ≥ 70 years (≥ 65 years for Hispanic and Black US participants), living in the community, and were free from preexisting CVD, cerebrovascular disease, or any chronic condition likely to limit survival to < 5 years. The study found no significant difference in the primary endpoints of death, dementia, or persistent physical disability, but there was a significantly higher risk of major hemorrhage in the aspirin group (3.8% vs 2.8%; hazard ratio, 1.38; 95% CI, 1.18-1.62; P < .001).
Several medical societies have updated their guideline recommendations for aspirin for primary prevention of CVD. The 2022 United States Public Service Task Force (USPSTF) provides a grade C recommendation (at least moderate certainty that the net benefit is small) to consider low-dose aspirin for the primary prevention of CVD on an individual patient basis for adults aged 40 to 59 years who have a ≥ 10% 10-year CVD risk. For adults aged ≥ 60 years, the USPSTF recommendation is grade D (moderate or high certainty that the practice has no net benefit or that harms outweigh the benefits) for low-dose aspirin use.1,3 The American College of Cardiology and American Heart Association (ACC/AHA) recommend considering low-dose aspirin for primary prevention of atherosclerotic cardiovascular disease (ASCVD) among select adults aged 40 to 70 years at higher CVD risk but not at increased risk of bleeding.4 The American Diabetes Association (ADA) recommends low-dose aspirin for primary prevention of CVD in patients with diabetes and additional risk factors such as family history of premature ASCVD, hypertension, dyslipidemia, smoking, or chronic kidney disease, and who are not at higher risk of bleeding.5 The ADA standards also caution against the use of aspirin as primary prevention in patients aged > 70 years. Low-dose aspirin use is not recommended for the primary prevention of CVD in older adults or adults of any age who are at increased risk of bleeding.
Recent literature using the US Department of Veterans Affairs (VA) Corporate Data Warehouse database confirms 86,555 of 1.8 million veterans aged > 70 years (5%) were taking low-dose aspirin for primary prevention of ASCVD despite guideline recommendations.6 Higher risk of gastrointestinal and other major bleeding from low-dose aspirin has been reported in the literature.1 Major bleeds represent a significant burden to the health care system with an estimated mean $13,093 cost for gastrointestinal bleed hospitalization.7
Considering the large scale aspirin use without appropriate indication within the veteran population, the risk of adverse effects, and the significant cost to patients and the health care system, it is imperative to determine the best approach to efficiently deprescribe aspirin for primary prevention among geriatric patients. Deprescribing refers to the systematic and supervised process of dose reduction or drug discontinuation with the goal of improving health and/or reducing the risk of adverse effects.8 During patient visits, primary care practitioners (PCPs) have opportunities to discontinue aspirin, but these encounters are time-limited and deprescribing might be secondary to more acute primary care needs. The shortage of PCPs is expected to worsen in coming years, which could further reduce their availability to assess inappropriate aspirin use.9
VA clinical pharmacist practitioners (CPPs) serve as medication experts and work autonomously under a broad scope of practice as part of the patient aligned care team.10-12 CPPs can free up time for PCPs and facilitate deprescribing efforts, especially for older adults. One retrospective cohort study conducted at a VA medical center found that CPPs deprescribed more potentially inappropriate medications among individuals aged ≥ 80 years compared with usual care with PCPs (26.8% vs 16.1%; P < .001).12,13 An aspirin deprescribing protocol conducted in 2022 resulted in nearly half of veterans aged ≥ 70 years contacted by phone agreeing to stop aspirin. Although this study supports the role pharmacists can play in reducing aspirin use in accordance with guidelines, the authors acknowledge that their interventions had a mean time of 12 minutes per patient and would require workflow changes.14 The purpose of this study is to evaluate the efficiency of aspirin deprescribing through 2 approaches: direct deprescribing by pharmacists using populationlevel review compared with clinicians following a pharmacist-led education.
Methods
This was a single-center quality improvement cohort study at the Durham VA Health Care System (DVAHCS) in North Carolina. Patients included were aged ≥ 70 years without known ASCVD who received care at any of 3 DVAHCS community-based outpatient clinics and prescribed aspirin. Patient data was obtained using the VIONE (Deprescribing Dashboard called Vital, Important, Optional, Not indicated, and Every medication has a specific indication or diagnosis) dashboard.15 VIONE was developed to identify potentially inappropriate medications (PIMs) that are eligible to deprescribe based on Beers Criteria, Screening Tool of Older Personsf Prescriptions criteria, and common clinical scenarios when clinicians determine the risk outweighs the benefit to continue a specific medication. 16,17 VIONE is used to reduce polypharmacy and improve patient safety, comfort, and medication adherence. Aspirin for patients aged ≥ 70 years without a history of ASCVD is a PIM identified by VIONE. Patients aged ≥ 70 years were chosen as an inclusion criteria in this study to match the ASPREE trial inclusion criteria and age inclusion criteria in the VIONE dashboard for aspirin deprescribing.2 Patient lists were generated for these potentially inappropriate aspirin prescriptions for 3 months before clinician staff education presentations, the day of the presentations, and 3 months after.
The primary endpoint was the number of veterans with aspirin deprescribed directly by 2 pharmacists over 12 weeks, divided by total patient care time spent, compared with the change in number of veterans with aspirin deprescribed by any DVAHCS physician, nurse practitioner, physician assistant, or CPP over 12 weeks, divided by the total pharmacist time spent on PCP education. Secondary endpoints were the number of aspirin orders discontinued by pharmacists and CPPs, the number of aspirin orders discontinued 12 weeks before pharmacist-led education compared with the number of aspirin orders discontinued 12 weeks after CPP-led education, average and median pharmacist time spent per patient encounter, and time of direct patient encounters vs time spent on PCP education.
Pharmacists reviewed each patient who met the inclusion criteria from the list generated by VIONE on December 1, 2022, for aspirin appropriateness according to the ACC/AHA and USPSTF guidelines, with the goal to discontinue aspirin for primary prevention of ASCVD and no other indications.1,4 Pharmacists documented their visits using VIONE methodology in the Computerized Patient Record System (CPRS) using a polypharmacy review note. CPPs contacted patients who were taking aspirin for primary prevention by unscheduled telephone call to assess for aspirin adherence, undocumented history of ASCVD, cardiovascular risk factors, and history of bleeding. Aspirin was discontinued if patients met guideline criteria recommendations and agreed to discontinuation. Risk-benefit discussions were completed when patients without known ASCVD were considered high risk because the ACC/AHA guidelines mention there is insufficient evidence of safety and efficacy of aspirin for primary prevention for patients with other known ASCVD risk factors (eg, strong family history of premature myocardial infarction, inability to achieve lipid, blood pressure, or glucose targets, or significant elevation in coronary artery calcium score).
High risk was defined as family history of premature ASCVD (in a male first-degree relative aged < 55 years or a female first-degree relative aged < 65 years), most recent blood pressure or 2 blood pressure results in the last 12 months > 160/100 mm Hg, recent hemoglobin A1c > 9%, and/or low-density lipoprotein > 190 mg/dL or not prescribed an indicated statin.3 Aspirin was continued or discontinued according to patient preference after the personalized risk-benefit discussion.
For patients with a clinical indication for aspirin use other than ASCVD (eg, atrial fibrillation not on anticoagulation, venous thromboembolism prophylaxis, carotid artery disease), CPPs documented their assessment and when appropriate deferred to the PCP for consideration of stopping aspirin. For patients with undocumented ASCVD, CPPs added their ASCVD history to their problem list and aspirin was continued. PCPs were notified by alert when aspirin was discontinued and when patients could not be reached by telephone.
presented a review of recent guideline updates and supporting literature at 2 online staff meetings. The education sessions lasted about 10 minutes and were presented to PCPs across 3 community-based outpatient clinics. An estimated 40 minutes were spent creating the PowerPoint education materials, seeking feedback, making edits, and answering questions or emails from PCPs after the presentation. During the presentation, pharmacists encouraged PCPs to discontinue aspirin (active VA prescriptions and reported over-the-counter use) for primary prevention of ASCVD in patients aged ≥ 70 years during their upcoming appointments and consider risk factors recommended by the ACC/AHA guidelines when applicable. PCPs were notified that CPPs planned to start a population review for discontinuing active VA aspirin prescriptions on December 1, 2022. The primary endpoint and secondary endpoints were analyzed using descriptive statistics. All data were analyzed using Microsoft Excel.
Results
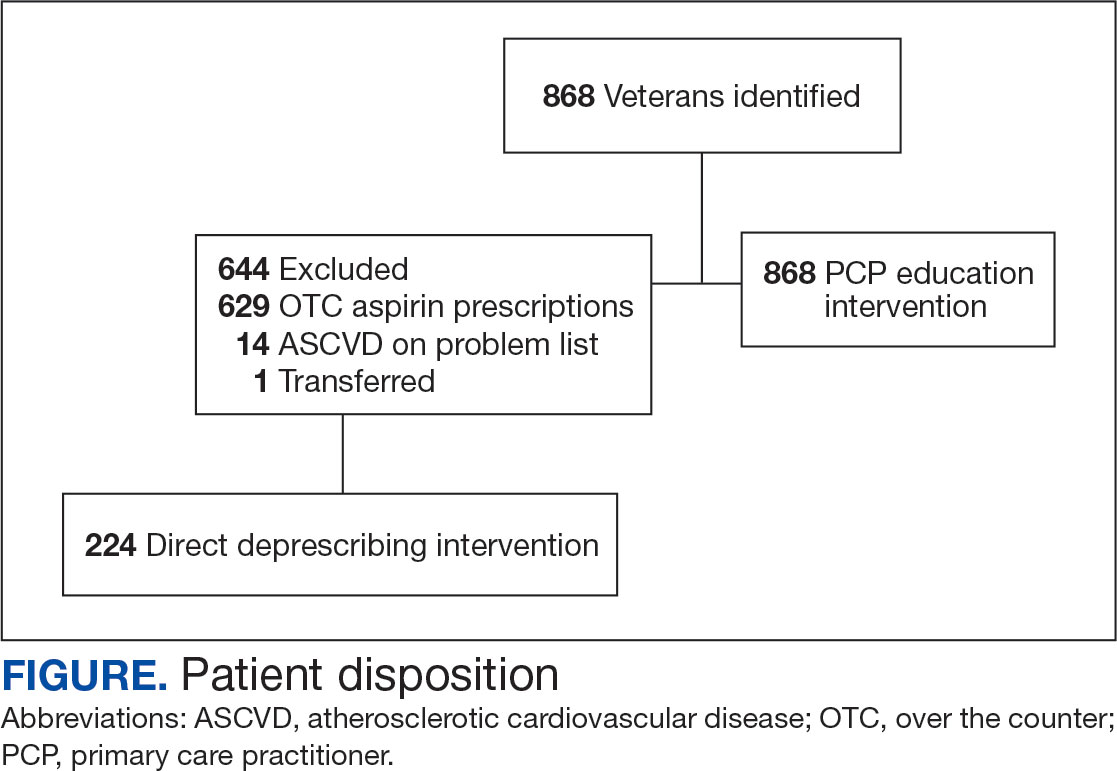
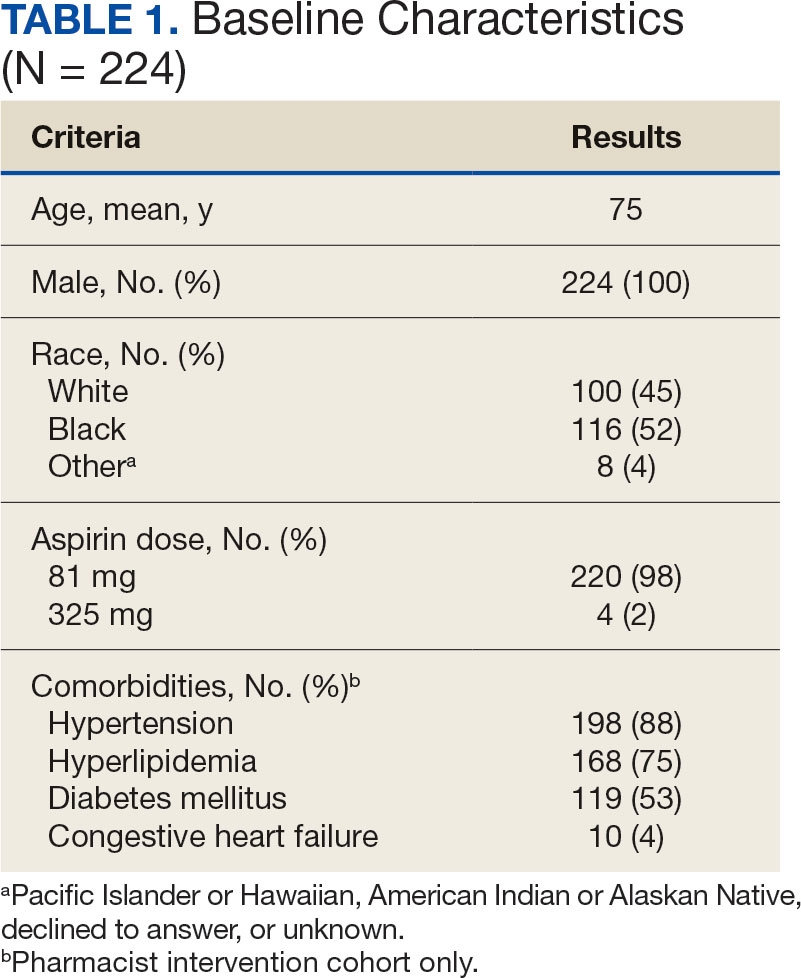
A total of 868 patients aged ≥ 70 years with active prescriptions for aspirin were identified on December 1, 2022. After applying inclusion and exclusion criteria for the pharmacist population review, 224 patients were included for cohort final analysis (Figure). All 868 patients were eligible for the CPP intervention. Primary reasons for exclusion from the CPP population included over-thecounter aspirin and a history of ASCVD in the patient’s problem list. All patients were male, with a mean (SD) age of 75 (4.4) years (Table 1). Most patients were prescribed aspirin, 81 mg daily (n = 220; 98%).
The direct CPP deprescribing intervention resulted in 2 aspirin prescriptions discontinued per hour of pharmacist time and 67 aspirin prescriptions discontinued per hour of pharmacist time via the PCP education intervention. CPPs discontinued 66 aspirin orders in the 12 weeks before the PCP education sessions. A total of 230 aspirin prescriptions were discontinued in the 12 weeks following the PCP education sessions, with 97 discontinued directly by CPPs and 133 discontinued by PCPs. The PCP education session yielded an additional 67 discontinued aspirin orders compared with the 12 weeks before the education sessions (Table 2).

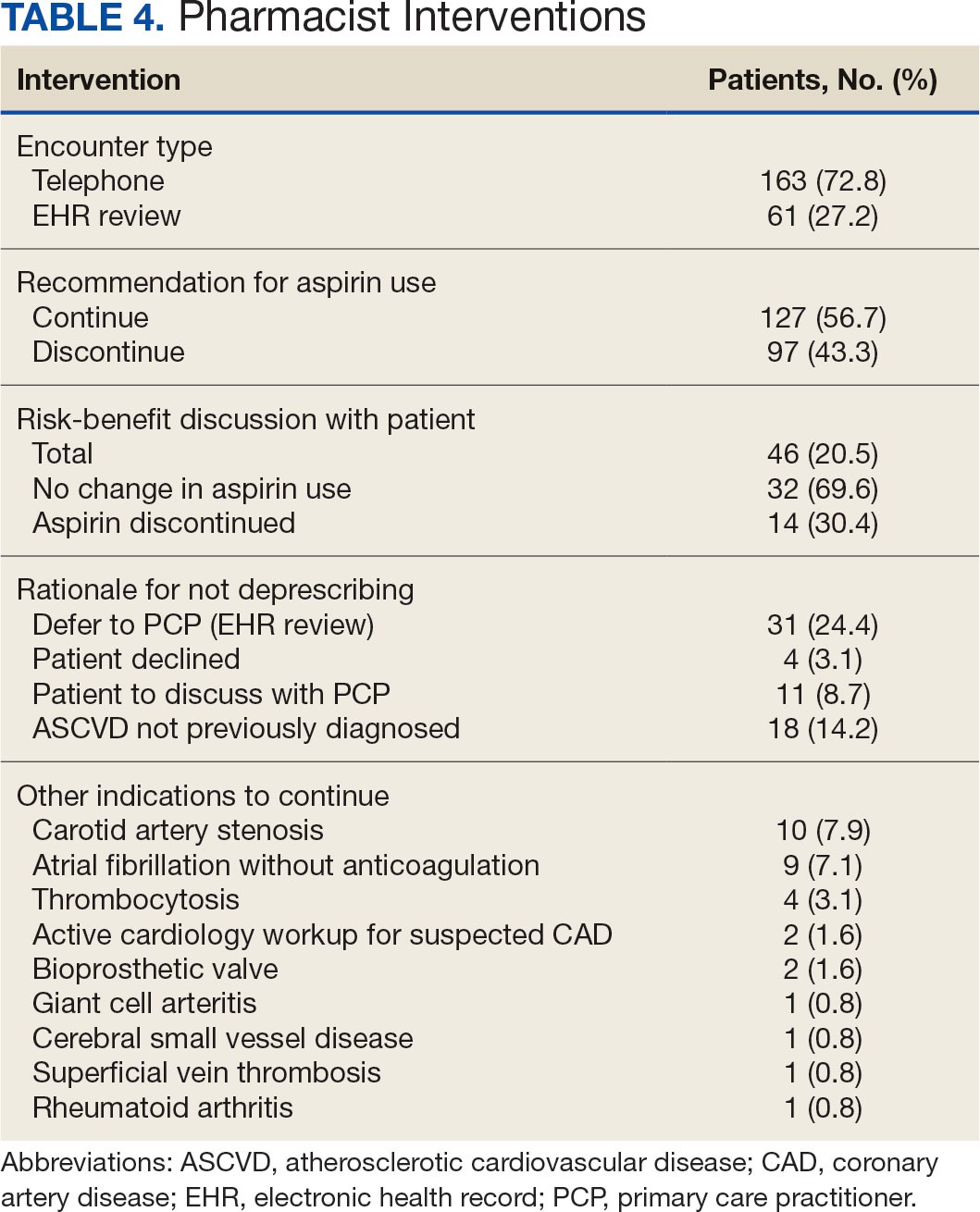
The CPP direct deprescribing intervention took about 48.3 hours, accounting for health record review and time interacting with patients. The PCP education intervention took about 60 minutes, which included time for preparing and delivering education materials (Table 3). CPP deprescribing encounter types, interventions, and related subcategories, and other identified indications to continue aspirin are listed in Table 4.

Discussion
Compared with direct deprescribing by pharmacists, the PCP education intervention was more efficient based on number of aspirin orders discontinued by pharmacist time. PCPs discontinued twice as many aspirin prescriptions in the 12 weeks after pharmacist-led education compared with the 12 weeks before.
Patients were primarily contacted by telephone (73%) for deprescribing. Among the 163 patients reached by phone and encouraged to discontinue aspirin, 97 patients (60%) accepted the recommendation, which was similar to the acceptance rates found in the literature (48% to 55%).14,18 Although many veterans continued taking aspirin (78%), most had indications for its continued use, such as a history of ASCVD, atrial fibrillation without anticoagulation, and carotid artery stenosis, and complex comorbidities that required further discussion with their PCP. Less common uses for aspirin were identified through CPRS review or patient reports included cerebral small vessel disease without history of ASCVD, subclavian artery stenosis, thrombocytosis, bioprosthetic valve replacement, giant cell arteritis, rheumatoid arthritis, and prevention of second eye involvement of ischemic optic neuropathy.
to describe the benefit of clinical pharmacy services for deprescribing aspirin for primary prevention of ASCVD through PCP education. Previously published literature has assessed alternative ways to identify or discontinue PIMs—including aspirin—among geriatric patients. One study evaluated the use of marking inappropriate aspirin prescriptions in the electronic health database, leading to a significant reduction in incidence of inappropriate aspirin prescribing; however, it did not assess changes in discontinuation rates of existing aspirin prescriptions.19 The previous VA pharmacist aspirin deprescribing protocol demonstrated pharmacists’ aptitude at discontinuing aspirin for primary prevention but only used direct patient contact and did not compare efficiency with other methods, including PCP education.14
This quality improvement project contributes new data to the existing literature to support the use of clinical pharmacists to discontinue aspirin for primary prevention and suggests a strong role for pharmacists as educators on clinical guidelines, in addition to their roles directly deprescribing PIMs in clinical practice. This study is further strengthened by its use of VIONE, which previously has demonstrated effectiveness in deprescribing a variety of PIMs in primary care settings.20
Despite using VIONE for generating a list of patients eligible for deprescription, our CPRS review found that this list was frequently inaccurate. For example, a small portion of patients were on the VIONE generated list indicating they had no ASCVD history, but had transient ischemic attack listed in their problem lists. Patient problem lists often were missing documented ASCVD history that was revealed by patient interview or CPRS review. It is possible that patients interviewed might have omitted relevant ASCVD history because of low health literacy, conditions affecting memory, or use of health care services outside the VA system.
There were several instances of aspirin used for other non-ASCVD indications, such as primary stroke prevention in atrial fibrillation. The ACC/AHA atrial fibrillation guidelines previously provided a Class IIb recommendation (benefit is greater than risk but additional studies are needed) for considering no antithrombic therapy or treatment with oral anticoagulant or aspirin for nonvalvular atrial fibrillation with CHA2DS2-VASc (Congestive heart failure, Hypertension, Age [> 65 y, 1 point; > 75 y, 2 points], Diabetes, previous Stroke/transient ischemic attack [2 points]) score of 1.21 The ACC/ AHA guidelines were updated in 2023 to recommend against antiplatelet therapy as an alternative to anticoagulation for reducing cardioembolic stroke risk among patients with atrial fibrillation with no indication for antiplatelet therapy because of risk of harm.22 If a patient has no risk factors for stroke, aspirin is not recommended to prevent thromboembolic events because of a lack of benefit. Interventions from this quality improvement study were completed before the 2023 atrial fibrillation guideline was published and therefore in this study aspirin was not discontinued when used for atrial fibrillation. Aspirin use for atrial fibrillation might benefit from similar discontinuation efforts analyzed within this study. Beyond atrial fibrillation, major guidelines do not comment on the use of aspirin for any other indications in the absence of clinical ASCVD.
Limitations
This study is limited by the lack of clinical consensus for complex patients and demonstrates the importance of individualized patient assessment when considering discontinuing aspirin. Because of the project’s relatively short intervention period, aspirin deprescribing rates could decrease over time and repeated education efforts might be necessary to see lasting impact. Health care professionals from services outside of primary care also might have discontinued aspirin during the study period unrelated to the education and these discontinued aspirin prescriptions could contribute to the higher rate observed among PCPs. This study included a specific population cohort of male, US veterans and might not reflect other populations where these interventions could be implemented.
The measurement of time spent by pharmacists and PCPs is an additional limitation. Although it is expected that PCPs attempt to discontinue aspirin during their existing patient care appointments, the time spent during visits was not measured or documented. Direct deprescribing by pharmacist CPRS review required a significant amount of time and could be a barrier to successful intervention by CPPs in patient aligned care teams.
To reduce the time pharmacists spent completing CPRS reviews, an aspirin deprescribing clinical reminder tool could be used to assess use and appropriate indication quickly during any primary care visit led by a PCP or CPP. In addition, it is recommended that pharmacists regularly educate health care professionals on guideline recommendations for aspirin use among geriatric patients. Future studies of the incidence of major cardiovascular events after aspirin deprescribing among geriatric patients and a longitudinal cost/benefit analysis could support these initiatives.
Conclusions
In this study, pharmacists successfully deprescribed inappropriate medications, such as aspirin. However, pharmacist-led PCP education is more efficient compared with direct deprescribing using a population-level review. PCP education requires less time and could allow ambulatory care pharmacists to spend more time on other direct patient care interventions to improve quality and access to care in primary care clinics. This study’s results further support the role of pharmacists in deprescribing PIMs for older adults and the use of a deprescribing tool, such as VIONE, in a primary care setting.
- US Preventive Services Task Force; Davidson KW, Barry MJ, et al. Aspirin use to prevent cardiovascular disease: US Preventive Services Task Force recommendation statement. JAMA. 2022;327(16):1577-1584. doi:10.1001/jama.2022.4983
- McNeil JJ, Nelson MR, Woods RL, et al. Effect of aspirin on all-cause mortality in the healthy elderly. N Engl J Med. 2018;379(16):1519-1528. doi:10.1056/NEJMoa1803955
- Barry MJ, Wolff TA, Pbert L, et al. Putting evidence into practice: an update on the US Preventive Services Task Force methods for developing recommendations for preventive services. Ann Fam Med. 2023;21(2):165-171. doi:10.1370/afm.2946
- Arnett DK, Blumenthal RS, Albert MA, et al. 2019 ACC/ AHA Guideline on the Primary Prevention of Cardiovascular Disease: A report of the American College of Cardiology/ American Heart Association Task Force on Clinical Practice Guidelines. Circulation. 2019;140(11):e596-e646. doi:10.1161/CIR.0000000000000678
- American Diabetes Association Professional Practice Committee. 10. Cardiovascular disease and risk management: Standards of care in diabetes-2024. Diabetes Care. 2024;47(Suppl 1):S179-S218. doi:10.2337/dc24-S010
- Ong SY, Chui P, Bhargava A, Justice A, Hauser RG. Estimating aspirin overuse for primary prevention of atherosclerotic cardiovascular disease (from a nationwide healthcare system). Am J Cardiol. 2020;137:25-30. doi:10.1016/j.amjcard.2020.09.042
- Weiss AJ, Jiang HJ. Overview of clinical conditions with frequent and costly hospital readmissions by payer, 2018. In: Healthcare Cost and Utilization Project (HCUP) Statistical Briefs. Agency for Healthcare Research and Quality (US); July 20, 2021.
- Krishnaswami A, Steinman MA, Goyal P, et al. Deprescribing in older adults with cardiovascular disease. J Am Coll Cardiol. 2019;73(20):2584-2595. doi:10.1016/j.jacc.2019.03.467
- Association of American Medical Colleges. The complexities of physician supply and demand: projections from 2019 to 2034. Accessed March 17, 2024. https://www.aamc.org/media/54681/download
- US Department of Veterans Affairs, Veterans Health Administration. VHA Directive 1108.07(1): General pharmacy service requirements. November 28, 2022. Accessed March 17, 2024. https://www.va.gov/vhapublications/ViewPublication.asp?pub_ID=10045
- US Department of Veterans Affairs, Veterans Health Administration. VHA Handbook 1108.11(3): Clinical pharmacy services. July 1, 2015. Accessed March 17, 2024. https://www.va.gov/vhapublications/ViewPublication.asp?pub_ID=3120
- US Department of Veterans Affairs. Clinical pharmacist practitioner (CPP) to improve access to and quality of care August 2021. August 2021. Accessed May 19, 2023. https://www.pbm.va.gov/PBM/CPPO/Documents/ExternalFactSheet_OptimizingtheCPPToImproveAccess_508.pdf
- Ammerman CA, Simpkins BA, Warman N, Downs TN. Potentially inappropriate medications in older adults: Deprescribing with a clinical pharmacist. J Am Geriatr Soc. 2019;67(1):115-118. doi:10.1111/jgs.15623
- Rothbauer K, Siodlak M, Dreischmeier E, Ranola TS, Welch L. Evaluation of a pharmacist-driven ambulatory aspirin deprescribing protocol. Fed Pract. 2022;39(suppl 5):S37- S41a. doi:10.12788/fp.0294
- US Department of Veterans Affairs. VIONE changes the way VA handles prescriptions. January 25, 2020. Accessed May 21, 2023. https://news.va.gov/70709/vione-changes-way-va-handles-prescriptions/
- 2023 American Geriatrics Society Beers Criteria Update Expert Panel. American Geriatrics Society 2023 updated AGS Beers Criteria for potentially inappropriate medication use in older adults. J Am Geriatr Soc. 2023;71(7):2052- 2081. doi:10.1111/jgs.18372
- O’Mahony D, Cherubini A, Guiteras AR, et al. STOPP/ START criteria for potentially inappropriate prescribing in older people: version 3. Eur Geriatr Med. 2023;14(4):625- 632. doi:10.1007/s41999-023-00777-y
- Draeger C, Lodhi F, Geissinger N, Larson T, Griesbach S. Interdisciplinary deprescribing of aspirin through prescriber education and provision of patient-specific recommendations. WMJ. 2022;121(3):220-225
- de Lusignan S, Hinton W, Seidu S, et al. Dashboards to reduce inappropriate prescribing of metformin and aspirin: A quality assurance programme in a primary care sentinel network. Prim Care Diabetes. 2021;15(6):1075-1079. doi:10.1016/j.pcd.2021.06.003
- Nelson MW, Downs TN, Puglisi GM, Simpkins BA, Collier AS. Use of a deprescribing tool in an interdisciplinary primary-care patient-aligned care team. Sr Care Pharm. 2022;37(1):34-43. doi:10.4140/TCP.n.2022.34
- January CT, Wann LS, Alpert JS, et al. 2014 AHA/ ACC/HRS guideline for the management of patients with atrial fibrillation: a report of the American College of Cardiology/American Heart Association Task Force on practice guidelines and the Heart Rhythm Society. Circulation. 2014;130(23):e199-e267. doi:10.1161/CIR.0000000000000041
- Joglar JA, Chung MK, Armbruster AL, et al. 2023 ACC/ AHA/ACCP/HRS Guideline for the Diagnosis and Management of Atrial Fibrillation: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines Circulation. 2024;149(1):e1- e156. doi:10.1161/CIR.0000000000001193
Low-dose aspirin commonly is used for the prevention of cardiovascular disease (CVD) but is associated with an increased risk of major bleeding.1 The use of aspirin for primary prevention is largely extrapolated from clinical trials showing benefit in the secondary prevention of myocardial infarction and ischemic stroke. However, results from the Aspirin in Reducing Events in the Elderly (ASPREE) trial challenged this practice.2 The ASPREE trial, conducted in the United States and Australia from 2010 to 2014, sought to determine whether daily 100 mg aspirin, was superior to placebo in promoting disability-free survival among older adults. Participants were aged ≥ 70 years (≥ 65 years for Hispanic and Black US participants), living in the community, and were free from preexisting CVD, cerebrovascular disease, or any chronic condition likely to limit survival to < 5 years. The study found no significant difference in the primary endpoints of death, dementia, or persistent physical disability, but there was a significantly higher risk of major hemorrhage in the aspirin group (3.8% vs 2.8%; hazard ratio, 1.38; 95% CI, 1.18-1.62; P < .001).
Several medical societies have updated their guideline recommendations for aspirin for primary prevention of CVD. The 2022 United States Public Service Task Force (USPSTF) provides a grade C recommendation (at least moderate certainty that the net benefit is small) to consider low-dose aspirin for the primary prevention of CVD on an individual patient basis for adults aged 40 to 59 years who have a ≥ 10% 10-year CVD risk. For adults aged ≥ 60 years, the USPSTF recommendation is grade D (moderate or high certainty that the practice has no net benefit or that harms outweigh the benefits) for low-dose aspirin use.1,3 The American College of Cardiology and American Heart Association (ACC/AHA) recommend considering low-dose aspirin for primary prevention of atherosclerotic cardiovascular disease (ASCVD) among select adults aged 40 to 70 years at higher CVD risk but not at increased risk of bleeding.4 The American Diabetes Association (ADA) recommends low-dose aspirin for primary prevention of CVD in patients with diabetes and additional risk factors such as family history of premature ASCVD, hypertension, dyslipidemia, smoking, or chronic kidney disease, and who are not at higher risk of bleeding.5 The ADA standards also caution against the use of aspirin as primary prevention in patients aged > 70 years. Low-dose aspirin use is not recommended for the primary prevention of CVD in older adults or adults of any age who are at increased risk of bleeding.
Recent literature using the US Department of Veterans Affairs (VA) Corporate Data Warehouse database confirms 86,555 of 1.8 million veterans aged > 70 years (5%) were taking low-dose aspirin for primary prevention of ASCVD despite guideline recommendations.6 Higher risk of gastrointestinal and other major bleeding from low-dose aspirin has been reported in the literature.1 Major bleeds represent a significant burden to the health care system with an estimated mean $13,093 cost for gastrointestinal bleed hospitalization.7
Considering the large scale aspirin use without appropriate indication within the veteran population, the risk of adverse effects, and the significant cost to patients and the health care system, it is imperative to determine the best approach to efficiently deprescribe aspirin for primary prevention among geriatric patients. Deprescribing refers to the systematic and supervised process of dose reduction or drug discontinuation with the goal of improving health and/or reducing the risk of adverse effects.8 During patient visits, primary care practitioners (PCPs) have opportunities to discontinue aspirin, but these encounters are time-limited and deprescribing might be secondary to more acute primary care needs. The shortage of PCPs is expected to worsen in coming years, which could further reduce their availability to assess inappropriate aspirin use.9
VA clinical pharmacist practitioners (CPPs) serve as medication experts and work autonomously under a broad scope of practice as part of the patient aligned care team.10-12 CPPs can free up time for PCPs and facilitate deprescribing efforts, especially for older adults. One retrospective cohort study conducted at a VA medical center found that CPPs deprescribed more potentially inappropriate medications among individuals aged ≥ 80 years compared with usual care with PCPs (26.8% vs 16.1%; P < .001).12,13 An aspirin deprescribing protocol conducted in 2022 resulted in nearly half of veterans aged ≥ 70 years contacted by phone agreeing to stop aspirin. Although this study supports the role pharmacists can play in reducing aspirin use in accordance with guidelines, the authors acknowledge that their interventions had a mean time of 12 minutes per patient and would require workflow changes.14 The purpose of this study is to evaluate the efficiency of aspirin deprescribing through 2 approaches: direct deprescribing by pharmacists using populationlevel review compared with clinicians following a pharmacist-led education.
Methods
This was a single-center quality improvement cohort study at the Durham VA Health Care System (DVAHCS) in North Carolina. Patients included were aged ≥ 70 years without known ASCVD who received care at any of 3 DVAHCS community-based outpatient clinics and prescribed aspirin. Patient data was obtained using the VIONE (Deprescribing Dashboard called Vital, Important, Optional, Not indicated, and Every medication has a specific indication or diagnosis) dashboard.15 VIONE was developed to identify potentially inappropriate medications (PIMs) that are eligible to deprescribe based on Beers Criteria, Screening Tool of Older Personsf Prescriptions criteria, and common clinical scenarios when clinicians determine the risk outweighs the benefit to continue a specific medication. 16,17 VIONE is used to reduce polypharmacy and improve patient safety, comfort, and medication adherence. Aspirin for patients aged ≥ 70 years without a history of ASCVD is a PIM identified by VIONE. Patients aged ≥ 70 years were chosen as an inclusion criteria in this study to match the ASPREE trial inclusion criteria and age inclusion criteria in the VIONE dashboard for aspirin deprescribing.2 Patient lists were generated for these potentially inappropriate aspirin prescriptions for 3 months before clinician staff education presentations, the day of the presentations, and 3 months after.
The primary endpoint was the number of veterans with aspirin deprescribed directly by 2 pharmacists over 12 weeks, divided by total patient care time spent, compared with the change in number of veterans with aspirin deprescribed by any DVAHCS physician, nurse practitioner, physician assistant, or CPP over 12 weeks, divided by the total pharmacist time spent on PCP education. Secondary endpoints were the number of aspirin orders discontinued by pharmacists and CPPs, the number of aspirin orders discontinued 12 weeks before pharmacist-led education compared with the number of aspirin orders discontinued 12 weeks after CPP-led education, average and median pharmacist time spent per patient encounter, and time of direct patient encounters vs time spent on PCP education.
Pharmacists reviewed each patient who met the inclusion criteria from the list generated by VIONE on December 1, 2022, for aspirin appropriateness according to the ACC/AHA and USPSTF guidelines, with the goal to discontinue aspirin for primary prevention of ASCVD and no other indications.1,4 Pharmacists documented their visits using VIONE methodology in the Computerized Patient Record System (CPRS) using a polypharmacy review note. CPPs contacted patients who were taking aspirin for primary prevention by unscheduled telephone call to assess for aspirin adherence, undocumented history of ASCVD, cardiovascular risk factors, and history of bleeding. Aspirin was discontinued if patients met guideline criteria recommendations and agreed to discontinuation. Risk-benefit discussions were completed when patients without known ASCVD were considered high risk because the ACC/AHA guidelines mention there is insufficient evidence of safety and efficacy of aspirin for primary prevention for patients with other known ASCVD risk factors (eg, strong family history of premature myocardial infarction, inability to achieve lipid, blood pressure, or glucose targets, or significant elevation in coronary artery calcium score).
High risk was defined as family history of premature ASCVD (in a male first-degree relative aged < 55 years or a female first-degree relative aged < 65 years), most recent blood pressure or 2 blood pressure results in the last 12 months > 160/100 mm Hg, recent hemoglobin A1c > 9%, and/or low-density lipoprotein > 190 mg/dL or not prescribed an indicated statin.3 Aspirin was continued or discontinued according to patient preference after the personalized risk-benefit discussion.
For patients with a clinical indication for aspirin use other than ASCVD (eg, atrial fibrillation not on anticoagulation, venous thromboembolism prophylaxis, carotid artery disease), CPPs documented their assessment and when appropriate deferred to the PCP for consideration of stopping aspirin. For patients with undocumented ASCVD, CPPs added their ASCVD history to their problem list and aspirin was continued. PCPs were notified by alert when aspirin was discontinued and when patients could not be reached by telephone.
presented a review of recent guideline updates and supporting literature at 2 online staff meetings. The education sessions lasted about 10 minutes and were presented to PCPs across 3 community-based outpatient clinics. An estimated 40 minutes were spent creating the PowerPoint education materials, seeking feedback, making edits, and answering questions or emails from PCPs after the presentation. During the presentation, pharmacists encouraged PCPs to discontinue aspirin (active VA prescriptions and reported over-the-counter use) for primary prevention of ASCVD in patients aged ≥ 70 years during their upcoming appointments and consider risk factors recommended by the ACC/AHA guidelines when applicable. PCPs were notified that CPPs planned to start a population review for discontinuing active VA aspirin prescriptions on December 1, 2022. The primary endpoint and secondary endpoints were analyzed using descriptive statistics. All data were analyzed using Microsoft Excel.
Results
A total of 868 patients aged ≥ 70 years with active prescriptions for aspirin were identified on December 1, 2022. After applying inclusion and exclusion criteria for the pharmacist population review, 224 patients were included for cohort final analysis (Figure). All 868 patients were eligible for the CPP intervention. Primary reasons for exclusion from the CPP population included over-thecounter aspirin and a history of ASCVD in the patient’s problem list. All patients were male, with a mean (SD) age of 75 (4.4) years (Table 1). Most patients were prescribed aspirin, 81 mg daily (n = 220; 98%).
The direct CPP deprescribing intervention resulted in 2 aspirin prescriptions discontinued per hour of pharmacist time and 67 aspirin prescriptions discontinued per hour of pharmacist time via the PCP education intervention. CPPs discontinued 66 aspirin orders in the 12 weeks before the PCP education sessions. A total of 230 aspirin prescriptions were discontinued in the 12 weeks following the PCP education sessions, with 97 discontinued directly by CPPs and 133 discontinued by PCPs. The PCP education session yielded an additional 67 discontinued aspirin orders compared with the 12 weeks before the education sessions (Table 2).
The CPP direct deprescribing intervention took about 48.3 hours, accounting for health record review and time interacting with patients. The PCP education intervention took about 60 minutes, which included time for preparing and delivering education materials (Table 3). CPP deprescribing encounter types, interventions, and related subcategories, and other identified indications to continue aspirin are listed in Table 4.
Discussion
Compared with direct deprescribing by pharmacists, the PCP education intervention was more efficient based on number of aspirin orders discontinued by pharmacist time. PCPs discontinued twice as many aspirin prescriptions in the 12 weeks after pharmacist-led education compared with the 12 weeks before.
Patients were primarily contacted by telephone (73%) for deprescribing. Among the 163 patients reached by phone and encouraged to discontinue aspirin, 97 patients (60%) accepted the recommendation, which was similar to the acceptance rates found in the literature (48% to 55%).14,18 Although many veterans continued taking aspirin (78%), most had indications for its continued use, such as a history of ASCVD, atrial fibrillation without anticoagulation, and carotid artery stenosis, and complex comorbidities that required further discussion with their PCP. Less common uses for aspirin were identified through CPRS review or patient reports included cerebral small vessel disease without history of ASCVD, subclavian artery stenosis, thrombocytosis, bioprosthetic valve replacement, giant cell arteritis, rheumatoid arthritis, and prevention of second eye involvement of ischemic optic neuropathy.
to describe the benefit of clinical pharmacy services for deprescribing aspirin for primary prevention of ASCVD through PCP education. Previously published literature has assessed alternative ways to identify or discontinue PIMs—including aspirin—among geriatric patients. One study evaluated the use of marking inappropriate aspirin prescriptions in the electronic health database, leading to a significant reduction in incidence of inappropriate aspirin prescribing; however, it did not assess changes in discontinuation rates of existing aspirin prescriptions.19 The previous VA pharmacist aspirin deprescribing protocol demonstrated pharmacists’ aptitude at discontinuing aspirin for primary prevention but only used direct patient contact and did not compare efficiency with other methods, including PCP education.14
This quality improvement project contributes new data to the existing literature to support the use of clinical pharmacists to discontinue aspirin for primary prevention and suggests a strong role for pharmacists as educators on clinical guidelines, in addition to their roles directly deprescribing PIMs in clinical practice. This study is further strengthened by its use of VIONE, which previously has demonstrated effectiveness in deprescribing a variety of PIMs in primary care settings.20
Despite using VIONE for generating a list of patients eligible for deprescription, our CPRS review found that this list was frequently inaccurate. For example, a small portion of patients were on the VIONE generated list indicating they had no ASCVD history, but had transient ischemic attack listed in their problem lists. Patient problem lists often were missing documented ASCVD history that was revealed by patient interview or CPRS review. It is possible that patients interviewed might have omitted relevant ASCVD history because of low health literacy, conditions affecting memory, or use of health care services outside the VA system.
There were several instances of aspirin used for other non-ASCVD indications, such as primary stroke prevention in atrial fibrillation. The ACC/AHA atrial fibrillation guidelines previously provided a Class IIb recommendation (benefit is greater than risk but additional studies are needed) for considering no antithrombic therapy or treatment with oral anticoagulant or aspirin for nonvalvular atrial fibrillation with CHA2DS2-VASc (Congestive heart failure, Hypertension, Age [> 65 y, 1 point; > 75 y, 2 points], Diabetes, previous Stroke/transient ischemic attack [2 points]) score of 1.21 The ACC/ AHA guidelines were updated in 2023 to recommend against antiplatelet therapy as an alternative to anticoagulation for reducing cardioembolic stroke risk among patients with atrial fibrillation with no indication for antiplatelet therapy because of risk of harm.22 If a patient has no risk factors for stroke, aspirin is not recommended to prevent thromboembolic events because of a lack of benefit. Interventions from this quality improvement study were completed before the 2023 atrial fibrillation guideline was published and therefore in this study aspirin was not discontinued when used for atrial fibrillation. Aspirin use for atrial fibrillation might benefit from similar discontinuation efforts analyzed within this study. Beyond atrial fibrillation, major guidelines do not comment on the use of aspirin for any other indications in the absence of clinical ASCVD.
Limitations
This study is limited by the lack of clinical consensus for complex patients and demonstrates the importance of individualized patient assessment when considering discontinuing aspirin. Because of the project’s relatively short intervention period, aspirin deprescribing rates could decrease over time and repeated education efforts might be necessary to see lasting impact. Health care professionals from services outside of primary care also might have discontinued aspirin during the study period unrelated to the education and these discontinued aspirin prescriptions could contribute to the higher rate observed among PCPs. This study included a specific population cohort of male, US veterans and might not reflect other populations where these interventions could be implemented.
The measurement of time spent by pharmacists and PCPs is an additional limitation. Although it is expected that PCPs attempt to discontinue aspirin during their existing patient care appointments, the time spent during visits was not measured or documented. Direct deprescribing by pharmacist CPRS review required a significant amount of time and could be a barrier to successful intervention by CPPs in patient aligned care teams.
To reduce the time pharmacists spent completing CPRS reviews, an aspirin deprescribing clinical reminder tool could be used to assess use and appropriate indication quickly during any primary care visit led by a PCP or CPP. In addition, it is recommended that pharmacists regularly educate health care professionals on guideline recommendations for aspirin use among geriatric patients. Future studies of the incidence of major cardiovascular events after aspirin deprescribing among geriatric patients and a longitudinal cost/benefit analysis could support these initiatives.
Conclusions
In this study, pharmacists successfully deprescribed inappropriate medications, such as aspirin. However, pharmacist-led PCP education is more efficient compared with direct deprescribing using a population-level review. PCP education requires less time and could allow ambulatory care pharmacists to spend more time on other direct patient care interventions to improve quality and access to care in primary care clinics. This study’s results further support the role of pharmacists in deprescribing PIMs for older adults and the use of a deprescribing tool, such as VIONE, in a primary care setting.
Low-dose aspirin commonly is used for the prevention of cardiovascular disease (CVD) but is associated with an increased risk of major bleeding.1 The use of aspirin for primary prevention is largely extrapolated from clinical trials showing benefit in the secondary prevention of myocardial infarction and ischemic stroke. However, results from the Aspirin in Reducing Events in the Elderly (ASPREE) trial challenged this practice.2 The ASPREE trial, conducted in the United States and Australia from 2010 to 2014, sought to determine whether daily 100 mg aspirin, was superior to placebo in promoting disability-free survival among older adults. Participants were aged ≥ 70 years (≥ 65 years for Hispanic and Black US participants), living in the community, and were free from preexisting CVD, cerebrovascular disease, or any chronic condition likely to limit survival to < 5 years. The study found no significant difference in the primary endpoints of death, dementia, or persistent physical disability, but there was a significantly higher risk of major hemorrhage in the aspirin group (3.8% vs 2.8%; hazard ratio, 1.38; 95% CI, 1.18-1.62; P < .001).
Several medical societies have updated their guideline recommendations for aspirin for primary prevention of CVD. The 2022 United States Public Service Task Force (USPSTF) provides a grade C recommendation (at least moderate certainty that the net benefit is small) to consider low-dose aspirin for the primary prevention of CVD on an individual patient basis for adults aged 40 to 59 years who have a ≥ 10% 10-year CVD risk. For adults aged ≥ 60 years, the USPSTF recommendation is grade D (moderate or high certainty that the practice has no net benefit or that harms outweigh the benefits) for low-dose aspirin use.1,3 The American College of Cardiology and American Heart Association (ACC/AHA) recommend considering low-dose aspirin for primary prevention of atherosclerotic cardiovascular disease (ASCVD) among select adults aged 40 to 70 years at higher CVD risk but not at increased risk of bleeding.4 The American Diabetes Association (ADA) recommends low-dose aspirin for primary prevention of CVD in patients with diabetes and additional risk factors such as family history of premature ASCVD, hypertension, dyslipidemia, smoking, or chronic kidney disease, and who are not at higher risk of bleeding.5 The ADA standards also caution against the use of aspirin as primary prevention in patients aged > 70 years. Low-dose aspirin use is not recommended for the primary prevention of CVD in older adults or adults of any age who are at increased risk of bleeding.
Recent literature using the US Department of Veterans Affairs (VA) Corporate Data Warehouse database confirms 86,555 of 1.8 million veterans aged > 70 years (5%) were taking low-dose aspirin for primary prevention of ASCVD despite guideline recommendations.6 Higher risk of gastrointestinal and other major bleeding from low-dose aspirin has been reported in the literature.1 Major bleeds represent a significant burden to the health care system with an estimated mean $13,093 cost for gastrointestinal bleed hospitalization.7
Considering the large scale aspirin use without appropriate indication within the veteran population, the risk of adverse effects, and the significant cost to patients and the health care system, it is imperative to determine the best approach to efficiently deprescribe aspirin for primary prevention among geriatric patients. Deprescribing refers to the systematic and supervised process of dose reduction or drug discontinuation with the goal of improving health and/or reducing the risk of adverse effects.8 During patient visits, primary care practitioners (PCPs) have opportunities to discontinue aspirin, but these encounters are time-limited and deprescribing might be secondary to more acute primary care needs. The shortage of PCPs is expected to worsen in coming years, which could further reduce their availability to assess inappropriate aspirin use.9
VA clinical pharmacist practitioners (CPPs) serve as medication experts and work autonomously under a broad scope of practice as part of the patient aligned care team.10-12 CPPs can free up time for PCPs and facilitate deprescribing efforts, especially for older adults. One retrospective cohort study conducted at a VA medical center found that CPPs deprescribed more potentially inappropriate medications among individuals aged ≥ 80 years compared with usual care with PCPs (26.8% vs 16.1%; P < .001).12,13 An aspirin deprescribing protocol conducted in 2022 resulted in nearly half of veterans aged ≥ 70 years contacted by phone agreeing to stop aspirin. Although this study supports the role pharmacists can play in reducing aspirin use in accordance with guidelines, the authors acknowledge that their interventions had a mean time of 12 minutes per patient and would require workflow changes.14 The purpose of this study is to evaluate the efficiency of aspirin deprescribing through 2 approaches: direct deprescribing by pharmacists using populationlevel review compared with clinicians following a pharmacist-led education.
Methods
This was a single-center quality improvement cohort study at the Durham VA Health Care System (DVAHCS) in North Carolina. Patients included were aged ≥ 70 years without known ASCVD who received care at any of 3 DVAHCS community-based outpatient clinics and prescribed aspirin. Patient data was obtained using the VIONE (Deprescribing Dashboard called Vital, Important, Optional, Not indicated, and Every medication has a specific indication or diagnosis) dashboard.15 VIONE was developed to identify potentially inappropriate medications (PIMs) that are eligible to deprescribe based on Beers Criteria, Screening Tool of Older Personsf Prescriptions criteria, and common clinical scenarios when clinicians determine the risk outweighs the benefit to continue a specific medication. 16,17 VIONE is used to reduce polypharmacy and improve patient safety, comfort, and medication adherence. Aspirin for patients aged ≥ 70 years without a history of ASCVD is a PIM identified by VIONE. Patients aged ≥ 70 years were chosen as an inclusion criteria in this study to match the ASPREE trial inclusion criteria and age inclusion criteria in the VIONE dashboard for aspirin deprescribing.2 Patient lists were generated for these potentially inappropriate aspirin prescriptions for 3 months before clinician staff education presentations, the day of the presentations, and 3 months after.
The primary endpoint was the number of veterans with aspirin deprescribed directly by 2 pharmacists over 12 weeks, divided by total patient care time spent, compared with the change in number of veterans with aspirin deprescribed by any DVAHCS physician, nurse practitioner, physician assistant, or CPP over 12 weeks, divided by the total pharmacist time spent on PCP education. Secondary endpoints were the number of aspirin orders discontinued by pharmacists and CPPs, the number of aspirin orders discontinued 12 weeks before pharmacist-led education compared with the number of aspirin orders discontinued 12 weeks after CPP-led education, average and median pharmacist time spent per patient encounter, and time of direct patient encounters vs time spent on PCP education.
Pharmacists reviewed each patient who met the inclusion criteria from the list generated by VIONE on December 1, 2022, for aspirin appropriateness according to the ACC/AHA and USPSTF guidelines, with the goal to discontinue aspirin for primary prevention of ASCVD and no other indications.1,4 Pharmacists documented their visits using VIONE methodology in the Computerized Patient Record System (CPRS) using a polypharmacy review note. CPPs contacted patients who were taking aspirin for primary prevention by unscheduled telephone call to assess for aspirin adherence, undocumented history of ASCVD, cardiovascular risk factors, and history of bleeding. Aspirin was discontinued if patients met guideline criteria recommendations and agreed to discontinuation. Risk-benefit discussions were completed when patients without known ASCVD were considered high risk because the ACC/AHA guidelines mention there is insufficient evidence of safety and efficacy of aspirin for primary prevention for patients with other known ASCVD risk factors (eg, strong family history of premature myocardial infarction, inability to achieve lipid, blood pressure, or glucose targets, or significant elevation in coronary artery calcium score).
High risk was defined as family history of premature ASCVD (in a male first-degree relative aged < 55 years or a female first-degree relative aged < 65 years), most recent blood pressure or 2 blood pressure results in the last 12 months > 160/100 mm Hg, recent hemoglobin A1c > 9%, and/or low-density lipoprotein > 190 mg/dL or not prescribed an indicated statin.3 Aspirin was continued or discontinued according to patient preference after the personalized risk-benefit discussion.
For patients with a clinical indication for aspirin use other than ASCVD (eg, atrial fibrillation not on anticoagulation, venous thromboembolism prophylaxis, carotid artery disease), CPPs documented their assessment and when appropriate deferred to the PCP for consideration of stopping aspirin. For patients with undocumented ASCVD, CPPs added their ASCVD history to their problem list and aspirin was continued. PCPs were notified by alert when aspirin was discontinued and when patients could not be reached by telephone.
presented a review of recent guideline updates and supporting literature at 2 online staff meetings. The education sessions lasted about 10 minutes and were presented to PCPs across 3 community-based outpatient clinics. An estimated 40 minutes were spent creating the PowerPoint education materials, seeking feedback, making edits, and answering questions or emails from PCPs after the presentation. During the presentation, pharmacists encouraged PCPs to discontinue aspirin (active VA prescriptions and reported over-the-counter use) for primary prevention of ASCVD in patients aged ≥ 70 years during their upcoming appointments and consider risk factors recommended by the ACC/AHA guidelines when applicable. PCPs were notified that CPPs planned to start a population review for discontinuing active VA aspirin prescriptions on December 1, 2022. The primary endpoint and secondary endpoints were analyzed using descriptive statistics. All data were analyzed using Microsoft Excel.
Results
A total of 868 patients aged ≥ 70 years with active prescriptions for aspirin were identified on December 1, 2022. After applying inclusion and exclusion criteria for the pharmacist population review, 224 patients were included for cohort final analysis (Figure). All 868 patients were eligible for the CPP intervention. Primary reasons for exclusion from the CPP population included over-thecounter aspirin and a history of ASCVD in the patient’s problem list. All patients were male, with a mean (SD) age of 75 (4.4) years (Table 1). Most patients were prescribed aspirin, 81 mg daily (n = 220; 98%).
The direct CPP deprescribing intervention resulted in 2 aspirin prescriptions discontinued per hour of pharmacist time and 67 aspirin prescriptions discontinued per hour of pharmacist time via the PCP education intervention. CPPs discontinued 66 aspirin orders in the 12 weeks before the PCP education sessions. A total of 230 aspirin prescriptions were discontinued in the 12 weeks following the PCP education sessions, with 97 discontinued directly by CPPs and 133 discontinued by PCPs. The PCP education session yielded an additional 67 discontinued aspirin orders compared with the 12 weeks before the education sessions (Table 2).
The CPP direct deprescribing intervention took about 48.3 hours, accounting for health record review and time interacting with patients. The PCP education intervention took about 60 minutes, which included time for preparing and delivering education materials (Table 3). CPP deprescribing encounter types, interventions, and related subcategories, and other identified indications to continue aspirin are listed in Table 4.
Discussion
Compared with direct deprescribing by pharmacists, the PCP education intervention was more efficient based on number of aspirin orders discontinued by pharmacist time. PCPs discontinued twice as many aspirin prescriptions in the 12 weeks after pharmacist-led education compared with the 12 weeks before.
Patients were primarily contacted by telephone (73%) for deprescribing. Among the 163 patients reached by phone and encouraged to discontinue aspirin, 97 patients (60%) accepted the recommendation, which was similar to the acceptance rates found in the literature (48% to 55%).14,18 Although many veterans continued taking aspirin (78%), most had indications for its continued use, such as a history of ASCVD, atrial fibrillation without anticoagulation, and carotid artery stenosis, and complex comorbidities that required further discussion with their PCP. Less common uses for aspirin were identified through CPRS review or patient reports included cerebral small vessel disease without history of ASCVD, subclavian artery stenosis, thrombocytosis, bioprosthetic valve replacement, giant cell arteritis, rheumatoid arthritis, and prevention of second eye involvement of ischemic optic neuropathy.
to describe the benefit of clinical pharmacy services for deprescribing aspirin for primary prevention of ASCVD through PCP education. Previously published literature has assessed alternative ways to identify or discontinue PIMs—including aspirin—among geriatric patients. One study evaluated the use of marking inappropriate aspirin prescriptions in the electronic health database, leading to a significant reduction in incidence of inappropriate aspirin prescribing; however, it did not assess changes in discontinuation rates of existing aspirin prescriptions.19 The previous VA pharmacist aspirin deprescribing protocol demonstrated pharmacists’ aptitude at discontinuing aspirin for primary prevention but only used direct patient contact and did not compare efficiency with other methods, including PCP education.14
This quality improvement project contributes new data to the existing literature to support the use of clinical pharmacists to discontinue aspirin for primary prevention and suggests a strong role for pharmacists as educators on clinical guidelines, in addition to their roles directly deprescribing PIMs in clinical practice. This study is further strengthened by its use of VIONE, which previously has demonstrated effectiveness in deprescribing a variety of PIMs in primary care settings.20
Despite using VIONE for generating a list of patients eligible for deprescription, our CPRS review found that this list was frequently inaccurate. For example, a small portion of patients were on the VIONE generated list indicating they had no ASCVD history, but had transient ischemic attack listed in their problem lists. Patient problem lists often were missing documented ASCVD history that was revealed by patient interview or CPRS review. It is possible that patients interviewed might have omitted relevant ASCVD history because of low health literacy, conditions affecting memory, or use of health care services outside the VA system.
There were several instances of aspirin used for other non-ASCVD indications, such as primary stroke prevention in atrial fibrillation. The ACC/AHA atrial fibrillation guidelines previously provided a Class IIb recommendation (benefit is greater than risk but additional studies are needed) for considering no antithrombic therapy or treatment with oral anticoagulant or aspirin for nonvalvular atrial fibrillation with CHA2DS2-VASc (Congestive heart failure, Hypertension, Age [> 65 y, 1 point; > 75 y, 2 points], Diabetes, previous Stroke/transient ischemic attack [2 points]) score of 1.21 The ACC/ AHA guidelines were updated in 2023 to recommend against antiplatelet therapy as an alternative to anticoagulation for reducing cardioembolic stroke risk among patients with atrial fibrillation with no indication for antiplatelet therapy because of risk of harm.22 If a patient has no risk factors for stroke, aspirin is not recommended to prevent thromboembolic events because of a lack of benefit. Interventions from this quality improvement study were completed before the 2023 atrial fibrillation guideline was published and therefore in this study aspirin was not discontinued when used for atrial fibrillation. Aspirin use for atrial fibrillation might benefit from similar discontinuation efforts analyzed within this study. Beyond atrial fibrillation, major guidelines do not comment on the use of aspirin for any other indications in the absence of clinical ASCVD.
Limitations
This study is limited by the lack of clinical consensus for complex patients and demonstrates the importance of individualized patient assessment when considering discontinuing aspirin. Because of the project’s relatively short intervention period, aspirin deprescribing rates could decrease over time and repeated education efforts might be necessary to see lasting impact. Health care professionals from services outside of primary care also might have discontinued aspirin during the study period unrelated to the education and these discontinued aspirin prescriptions could contribute to the higher rate observed among PCPs. This study included a specific population cohort of male, US veterans and might not reflect other populations where these interventions could be implemented.
The measurement of time spent by pharmacists and PCPs is an additional limitation. Although it is expected that PCPs attempt to discontinue aspirin during their existing patient care appointments, the time spent during visits was not measured or documented. Direct deprescribing by pharmacist CPRS review required a significant amount of time and could be a barrier to successful intervention by CPPs in patient aligned care teams.
To reduce the time pharmacists spent completing CPRS reviews, an aspirin deprescribing clinical reminder tool could be used to assess use and appropriate indication quickly during any primary care visit led by a PCP or CPP. In addition, it is recommended that pharmacists regularly educate health care professionals on guideline recommendations for aspirin use among geriatric patients. Future studies of the incidence of major cardiovascular events after aspirin deprescribing among geriatric patients and a longitudinal cost/benefit analysis could support these initiatives.
Conclusions
In this study, pharmacists successfully deprescribed inappropriate medications, such as aspirin. However, pharmacist-led PCP education is more efficient compared with direct deprescribing using a population-level review. PCP education requires less time and could allow ambulatory care pharmacists to spend more time on other direct patient care interventions to improve quality and access to care in primary care clinics. This study’s results further support the role of pharmacists in deprescribing PIMs for older adults and the use of a deprescribing tool, such as VIONE, in a primary care setting.
- US Preventive Services Task Force; Davidson KW, Barry MJ, et al. Aspirin use to prevent cardiovascular disease: US Preventive Services Task Force recommendation statement. JAMA. 2022;327(16):1577-1584. doi:10.1001/jama.2022.4983
- McNeil JJ, Nelson MR, Woods RL, et al. Effect of aspirin on all-cause mortality in the healthy elderly. N Engl J Med. 2018;379(16):1519-1528. doi:10.1056/NEJMoa1803955
- Barry MJ, Wolff TA, Pbert L, et al. Putting evidence into practice: an update on the US Preventive Services Task Force methods for developing recommendations for preventive services. Ann Fam Med. 2023;21(2):165-171. doi:10.1370/afm.2946
- Arnett DK, Blumenthal RS, Albert MA, et al. 2019 ACC/ AHA Guideline on the Primary Prevention of Cardiovascular Disease: A report of the American College of Cardiology/ American Heart Association Task Force on Clinical Practice Guidelines. Circulation. 2019;140(11):e596-e646. doi:10.1161/CIR.0000000000000678
- American Diabetes Association Professional Practice Committee. 10. Cardiovascular disease and risk management: Standards of care in diabetes-2024. Diabetes Care. 2024;47(Suppl 1):S179-S218. doi:10.2337/dc24-S010
- Ong SY, Chui P, Bhargava A, Justice A, Hauser RG. Estimating aspirin overuse for primary prevention of atherosclerotic cardiovascular disease (from a nationwide healthcare system). Am J Cardiol. 2020;137:25-30. doi:10.1016/j.amjcard.2020.09.042
- Weiss AJ, Jiang HJ. Overview of clinical conditions with frequent and costly hospital readmissions by payer, 2018. In: Healthcare Cost and Utilization Project (HCUP) Statistical Briefs. Agency for Healthcare Research and Quality (US); July 20, 2021.
- Krishnaswami A, Steinman MA, Goyal P, et al. Deprescribing in older adults with cardiovascular disease. J Am Coll Cardiol. 2019;73(20):2584-2595. doi:10.1016/j.jacc.2019.03.467
- Association of American Medical Colleges. The complexities of physician supply and demand: projections from 2019 to 2034. Accessed March 17, 2024. https://www.aamc.org/media/54681/download
- US Department of Veterans Affairs, Veterans Health Administration. VHA Directive 1108.07(1): General pharmacy service requirements. November 28, 2022. Accessed March 17, 2024. https://www.va.gov/vhapublications/ViewPublication.asp?pub_ID=10045
- US Department of Veterans Affairs, Veterans Health Administration. VHA Handbook 1108.11(3): Clinical pharmacy services. July 1, 2015. Accessed March 17, 2024. https://www.va.gov/vhapublications/ViewPublication.asp?pub_ID=3120
- US Department of Veterans Affairs. Clinical pharmacist practitioner (CPP) to improve access to and quality of care August 2021. August 2021. Accessed May 19, 2023. https://www.pbm.va.gov/PBM/CPPO/Documents/ExternalFactSheet_OptimizingtheCPPToImproveAccess_508.pdf
- Ammerman CA, Simpkins BA, Warman N, Downs TN. Potentially inappropriate medications in older adults: Deprescribing with a clinical pharmacist. J Am Geriatr Soc. 2019;67(1):115-118. doi:10.1111/jgs.15623
- Rothbauer K, Siodlak M, Dreischmeier E, Ranola TS, Welch L. Evaluation of a pharmacist-driven ambulatory aspirin deprescribing protocol. Fed Pract. 2022;39(suppl 5):S37- S41a. doi:10.12788/fp.0294
- US Department of Veterans Affairs. VIONE changes the way VA handles prescriptions. January 25, 2020. Accessed May 21, 2023. https://news.va.gov/70709/vione-changes-way-va-handles-prescriptions/
- 2023 American Geriatrics Society Beers Criteria Update Expert Panel. American Geriatrics Society 2023 updated AGS Beers Criteria for potentially inappropriate medication use in older adults. J Am Geriatr Soc. 2023;71(7):2052- 2081. doi:10.1111/jgs.18372
- O’Mahony D, Cherubini A, Guiteras AR, et al. STOPP/ START criteria for potentially inappropriate prescribing in older people: version 3. Eur Geriatr Med. 2023;14(4):625- 632. doi:10.1007/s41999-023-00777-y
- Draeger C, Lodhi F, Geissinger N, Larson T, Griesbach S. Interdisciplinary deprescribing of aspirin through prescriber education and provision of patient-specific recommendations. WMJ. 2022;121(3):220-225
- de Lusignan S, Hinton W, Seidu S, et al. Dashboards to reduce inappropriate prescribing of metformin and aspirin: A quality assurance programme in a primary care sentinel network. Prim Care Diabetes. 2021;15(6):1075-1079. doi:10.1016/j.pcd.2021.06.003
- Nelson MW, Downs TN, Puglisi GM, Simpkins BA, Collier AS. Use of a deprescribing tool in an interdisciplinary primary-care patient-aligned care team. Sr Care Pharm. 2022;37(1):34-43. doi:10.4140/TCP.n.2022.34
- January CT, Wann LS, Alpert JS, et al. 2014 AHA/ ACC/HRS guideline for the management of patients with atrial fibrillation: a report of the American College of Cardiology/American Heart Association Task Force on practice guidelines and the Heart Rhythm Society. Circulation. 2014;130(23):e199-e267. doi:10.1161/CIR.0000000000000041
- Joglar JA, Chung MK, Armbruster AL, et al. 2023 ACC/ AHA/ACCP/HRS Guideline for the Diagnosis and Management of Atrial Fibrillation: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines Circulation. 2024;149(1):e1- e156. doi:10.1161/CIR.0000000000001193
- US Preventive Services Task Force; Davidson KW, Barry MJ, et al. Aspirin use to prevent cardiovascular disease: US Preventive Services Task Force recommendation statement. JAMA. 2022;327(16):1577-1584. doi:10.1001/jama.2022.4983
- McNeil JJ, Nelson MR, Woods RL, et al. Effect of aspirin on all-cause mortality in the healthy elderly. N Engl J Med. 2018;379(16):1519-1528. doi:10.1056/NEJMoa1803955
- Barry MJ, Wolff TA, Pbert L, et al. Putting evidence into practice: an update on the US Preventive Services Task Force methods for developing recommendations for preventive services. Ann Fam Med. 2023;21(2):165-171. doi:10.1370/afm.2946
- Arnett DK, Blumenthal RS, Albert MA, et al. 2019 ACC/ AHA Guideline on the Primary Prevention of Cardiovascular Disease: A report of the American College of Cardiology/ American Heart Association Task Force on Clinical Practice Guidelines. Circulation. 2019;140(11):e596-e646. doi:10.1161/CIR.0000000000000678
- American Diabetes Association Professional Practice Committee. 10. Cardiovascular disease and risk management: Standards of care in diabetes-2024. Diabetes Care. 2024;47(Suppl 1):S179-S218. doi:10.2337/dc24-S010
- Ong SY, Chui P, Bhargava A, Justice A, Hauser RG. Estimating aspirin overuse for primary prevention of atherosclerotic cardiovascular disease (from a nationwide healthcare system). Am J Cardiol. 2020;137:25-30. doi:10.1016/j.amjcard.2020.09.042
- Weiss AJ, Jiang HJ. Overview of clinical conditions with frequent and costly hospital readmissions by payer, 2018. In: Healthcare Cost and Utilization Project (HCUP) Statistical Briefs. Agency for Healthcare Research and Quality (US); July 20, 2021.
- Krishnaswami A, Steinman MA, Goyal P, et al. Deprescribing in older adults with cardiovascular disease. J Am Coll Cardiol. 2019;73(20):2584-2595. doi:10.1016/j.jacc.2019.03.467
- Association of American Medical Colleges. The complexities of physician supply and demand: projections from 2019 to 2034. Accessed March 17, 2024. https://www.aamc.org/media/54681/download
- US Department of Veterans Affairs, Veterans Health Administration. VHA Directive 1108.07(1): General pharmacy service requirements. November 28, 2022. Accessed March 17, 2024. https://www.va.gov/vhapublications/ViewPublication.asp?pub_ID=10045
- US Department of Veterans Affairs, Veterans Health Administration. VHA Handbook 1108.11(3): Clinical pharmacy services. July 1, 2015. Accessed March 17, 2024. https://www.va.gov/vhapublications/ViewPublication.asp?pub_ID=3120
- US Department of Veterans Affairs. Clinical pharmacist practitioner (CPP) to improve access to and quality of care August 2021. August 2021. Accessed May 19, 2023. https://www.pbm.va.gov/PBM/CPPO/Documents/ExternalFactSheet_OptimizingtheCPPToImproveAccess_508.pdf
- Ammerman CA, Simpkins BA, Warman N, Downs TN. Potentially inappropriate medications in older adults: Deprescribing with a clinical pharmacist. J Am Geriatr Soc. 2019;67(1):115-118. doi:10.1111/jgs.15623
- Rothbauer K, Siodlak M, Dreischmeier E, Ranola TS, Welch L. Evaluation of a pharmacist-driven ambulatory aspirin deprescribing protocol. Fed Pract. 2022;39(suppl 5):S37- S41a. doi:10.12788/fp.0294
- US Department of Veterans Affairs. VIONE changes the way VA handles prescriptions. January 25, 2020. Accessed May 21, 2023. https://news.va.gov/70709/vione-changes-way-va-handles-prescriptions/
- 2023 American Geriatrics Society Beers Criteria Update Expert Panel. American Geriatrics Society 2023 updated AGS Beers Criteria for potentially inappropriate medication use in older adults. J Am Geriatr Soc. 2023;71(7):2052- 2081. doi:10.1111/jgs.18372
- O’Mahony D, Cherubini A, Guiteras AR, et al. STOPP/ START criteria for potentially inappropriate prescribing in older people: version 3. Eur Geriatr Med. 2023;14(4):625- 632. doi:10.1007/s41999-023-00777-y
- Draeger C, Lodhi F, Geissinger N, Larson T, Griesbach S. Interdisciplinary deprescribing of aspirin through prescriber education and provision of patient-specific recommendations. WMJ. 2022;121(3):220-225
- de Lusignan S, Hinton W, Seidu S, et al. Dashboards to reduce inappropriate prescribing of metformin and aspirin: A quality assurance programme in a primary care sentinel network. Prim Care Diabetes. 2021;15(6):1075-1079. doi:10.1016/j.pcd.2021.06.003
- Nelson MW, Downs TN, Puglisi GM, Simpkins BA, Collier AS. Use of a deprescribing tool in an interdisciplinary primary-care patient-aligned care team. Sr Care Pharm. 2022;37(1):34-43. doi:10.4140/TCP.n.2022.34
- January CT, Wann LS, Alpert JS, et al. 2014 AHA/ ACC/HRS guideline for the management of patients with atrial fibrillation: a report of the American College of Cardiology/American Heart Association Task Force on practice guidelines and the Heart Rhythm Society. Circulation. 2014;130(23):e199-e267. doi:10.1161/CIR.0000000000000041
- Joglar JA, Chung MK, Armbruster AL, et al. 2023 ACC/ AHA/ACCP/HRS Guideline for the Diagnosis and Management of Atrial Fibrillation: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines Circulation. 2024;149(1):e1- e156. doi:10.1161/CIR.0000000000001193
Pharmacist-Led Deprescribing of Aspirin for Primary Prevention of Cardiovascular Disease Among Geriatric Veterans
Pharmacist-Led Deprescribing of Aspirin for Primary Prevention of Cardiovascular Disease Among Geriatric Veterans
Hemophilia A: Bleeds Plummet After Experimental Gene Therapy
SAN DIEGO — Promising early results from an ongoing randomized, open-label, single-arm, phase 3 study could pave the way for a Pfizer product to become the second US Food and Drug Administration (FDA)–approved gene therapy for hemophilia A.
In an efficacy population of 50 patients with hemophilia A, the AFFINE trial found that their mean annualized bleeding rate (ABR) fell from 4.73 pre-infusion with giroctocogene fitelparvovec to 1.24 post-infusion (week 12 to 15 or more months, −3.49, −6.06 to −0.91; P = .004), researchers reported earlier this month at American Society of Hematology (ASH) 2024 Annual Meeting. Sixty-four percent had no bleeding events over a median follow-up of 33.6 months (14.5-44.4).
The ABR for treated bleeds fell from 4.08 to 0.07 (−4.01, −5.57 to −2.45; P < .0001), and 88% had no treated bleeds over the same follow-up period.
“The primary endpoint for this trial was a reduction in total bleeds in patients, and that was achieved,” hematologist and first author Andrew D. Leavitt, MD, of the University of California at San Francisco, said in an interview. “More impressive was the reduction in treated bleeds, a kind of a surrogate marker for bleeds of clinical significance to the individual. And as one would expect or hope to see in a gene therapy trial, there was a significant and marked reduction in the use of factor.”
Moving forward, he said, the message to clinicians is that “there’s every reason to believe that they will have yet another option for their patients.”
Gene Therapy on the Rise in Hemophilia
Gene therapy has arisen as an approved therapy for hemophilia over just the last few years. Two gene therapies for hemophilia B have been approved by the FDA since 2022, and one was approved for hemophilia A, the more common type, in 2023.
As Leavitt noted, one-time treatment with gene therapy offers an alternative to treatment with blood factor, long the mainstay of hemophilia therapy.
“One of the real pluses of gene therapy is the potential to remove the burden of hemophilia, which is large and, I suspect, underappreciated even by providers,” he said. “You have to sit down with your patients and really get a real good sense of just how difficult it is for them to manage with many products on the market over the last few decades.”
Why is there a need for multiple gene therapy products? “A patient may have neutralizing antibodies against the proteins on the surface of gene therapy product A that prevents its use, but not on gene therapy B, which allows use of product B,” Leavitt said. “We need a few flavors so that we can offer gene therapy to the maximum number of interested patients.”
High Efficacy and an ‘Acceptable’ Safety Profile
For the study, researchers dosed 75 patients (mean age, 32.3 [19-59]; 100% men, 74.7% White and 18.7% Asian,) with hemophilia A with giroctocogene fitelparvovec, a hepatocyte-directed recombinant adeno-associated virus serotype 6 vector encoding a B-domain–deleted variant of human factor VIII. The efficacy population is 50 patients with at least 6 months of follow-up in the lead-in study.
The annualized infusion rate of exogenous FVIII was 124.39 mean annualized infusion rate prior to the treatment infusion vs 0.21 post-infusion, week 12 through at least 15 months (−124.18, −139.47 to −108.89; P < .0001).
Leavitt said the results are similar to other gene therapies for hemophilia in that “it is difficult to predict how high your factor level will become. There’s a broad range of outcomes for individuals, and the duration of expression remains an unknown.”
The study authors described the treatment as “generally well tolerated” with “an acceptable and manageable safety profile.”
Of the 75 subjects, 98.7% had adverse effects (AEs, 740 events) and 90.7% had treatment-related AEs. Common treatment-related AEs included hepatotoxicity (62.7%) and infusion-related reactions (73.3%). No subjects discontinued therapy due to AEs.
Nearly two thirds — 62.7% — of subjects used corticosteroids for a mean 114.6 days (11-296).
Study Findings ‘Look Really Good’
In an interview, Guy Young, MD, director of the Hemostasis and Thrombosis Program at Children’s Hospital Los Angeles and professor of pediatrics, Keck School of Medicine of University of Southern California, Los Angeles, said that “generally speaking, the new data looks really good.” Young, who didn’t take part in the study, noted that factor levels following treatment were high, and one subject actually had a thrombotic event and needed to be treated with an anticoagulant.
The high factor levels could actually be a sign of lasting benefit vs valoctocogene roxaparvovec (Roctavian), the sole FDA-approved gene therapy for hemophilia A, which is linked to significant drops in factor level after 6 months, he said. “Wouldn’t it be better to start really high?”
Gene therapy for hemophilia is highly expensive, although proponents noted that insurers may save money over the long run if patients don’t require prophylactic treatment or therapy for bleeds.
A Pfizer spokesman declined to comment on the new therapy’s potential cost. In regard to when the therapy may receive FDA approval, he said “Pfizer is discussing this data with regulatory authorities.”
Pfizer funded this study. Leavitt disclosed ties with HEMA, Merck, Catalyst, Genentech, Pfizer, BioMarin, and Sangamo. Other study authors reported relationships with Pfizer. Young disclosed ties with Pfizer and BioMarin.
A version of this article appeared on Medscape.com.
SAN DIEGO — Promising early results from an ongoing randomized, open-label, single-arm, phase 3 study could pave the way for a Pfizer product to become the second US Food and Drug Administration (FDA)–approved gene therapy for hemophilia A.
In an efficacy population of 50 patients with hemophilia A, the AFFINE trial found that their mean annualized bleeding rate (ABR) fell from 4.73 pre-infusion with giroctocogene fitelparvovec to 1.24 post-infusion (week 12 to 15 or more months, −3.49, −6.06 to −0.91; P = .004), researchers reported earlier this month at American Society of Hematology (ASH) 2024 Annual Meeting. Sixty-four percent had no bleeding events over a median follow-up of 33.6 months (14.5-44.4).
The ABR for treated bleeds fell from 4.08 to 0.07 (−4.01, −5.57 to −2.45; P < .0001), and 88% had no treated bleeds over the same follow-up period.
“The primary endpoint for this trial was a reduction in total bleeds in patients, and that was achieved,” hematologist and first author Andrew D. Leavitt, MD, of the University of California at San Francisco, said in an interview. “More impressive was the reduction in treated bleeds, a kind of a surrogate marker for bleeds of clinical significance to the individual. And as one would expect or hope to see in a gene therapy trial, there was a significant and marked reduction in the use of factor.”
Moving forward, he said, the message to clinicians is that “there’s every reason to believe that they will have yet another option for their patients.”
Gene Therapy on the Rise in Hemophilia
Gene therapy has arisen as an approved therapy for hemophilia over just the last few years. Two gene therapies for hemophilia B have been approved by the FDA since 2022, and one was approved for hemophilia A, the more common type, in 2023.
As Leavitt noted, one-time treatment with gene therapy offers an alternative to treatment with blood factor, long the mainstay of hemophilia therapy.
“One of the real pluses of gene therapy is the potential to remove the burden of hemophilia, which is large and, I suspect, underappreciated even by providers,” he said. “You have to sit down with your patients and really get a real good sense of just how difficult it is for them to manage with many products on the market over the last few decades.”
Why is there a need for multiple gene therapy products? “A patient may have neutralizing antibodies against the proteins on the surface of gene therapy product A that prevents its use, but not on gene therapy B, which allows use of product B,” Leavitt said. “We need a few flavors so that we can offer gene therapy to the maximum number of interested patients.”
High Efficacy and an ‘Acceptable’ Safety Profile
For the study, researchers dosed 75 patients (mean age, 32.3 [19-59]; 100% men, 74.7% White and 18.7% Asian,) with hemophilia A with giroctocogene fitelparvovec, a hepatocyte-directed recombinant adeno-associated virus serotype 6 vector encoding a B-domain–deleted variant of human factor VIII. The efficacy population is 50 patients with at least 6 months of follow-up in the lead-in study.
The annualized infusion rate of exogenous FVIII was 124.39 mean annualized infusion rate prior to the treatment infusion vs 0.21 post-infusion, week 12 through at least 15 months (−124.18, −139.47 to −108.89; P < .0001).
Leavitt said the results are similar to other gene therapies for hemophilia in that “it is difficult to predict how high your factor level will become. There’s a broad range of outcomes for individuals, and the duration of expression remains an unknown.”
The study authors described the treatment as “generally well tolerated” with “an acceptable and manageable safety profile.”
Of the 75 subjects, 98.7% had adverse effects (AEs, 740 events) and 90.7% had treatment-related AEs. Common treatment-related AEs included hepatotoxicity (62.7%) and infusion-related reactions (73.3%). No subjects discontinued therapy due to AEs.
Nearly two thirds — 62.7% — of subjects used corticosteroids for a mean 114.6 days (11-296).
Study Findings ‘Look Really Good’
In an interview, Guy Young, MD, director of the Hemostasis and Thrombosis Program at Children’s Hospital Los Angeles and professor of pediatrics, Keck School of Medicine of University of Southern California, Los Angeles, said that “generally speaking, the new data looks really good.” Young, who didn’t take part in the study, noted that factor levels following treatment were high, and one subject actually had a thrombotic event and needed to be treated with an anticoagulant.
The high factor levels could actually be a sign of lasting benefit vs valoctocogene roxaparvovec (Roctavian), the sole FDA-approved gene therapy for hemophilia A, which is linked to significant drops in factor level after 6 months, he said. “Wouldn’t it be better to start really high?”
Gene therapy for hemophilia is highly expensive, although proponents noted that insurers may save money over the long run if patients don’t require prophylactic treatment or therapy for bleeds.
A Pfizer spokesman declined to comment on the new therapy’s potential cost. In regard to when the therapy may receive FDA approval, he said “Pfizer is discussing this data with regulatory authorities.”
Pfizer funded this study. Leavitt disclosed ties with HEMA, Merck, Catalyst, Genentech, Pfizer, BioMarin, and Sangamo. Other study authors reported relationships with Pfizer. Young disclosed ties with Pfizer and BioMarin.
A version of this article appeared on Medscape.com.
SAN DIEGO — Promising early results from an ongoing randomized, open-label, single-arm, phase 3 study could pave the way for a Pfizer product to become the second US Food and Drug Administration (FDA)–approved gene therapy for hemophilia A.
In an efficacy population of 50 patients with hemophilia A, the AFFINE trial found that their mean annualized bleeding rate (ABR) fell from 4.73 pre-infusion with giroctocogene fitelparvovec to 1.24 post-infusion (week 12 to 15 or more months, −3.49, −6.06 to −0.91; P = .004), researchers reported earlier this month at American Society of Hematology (ASH) 2024 Annual Meeting. Sixty-four percent had no bleeding events over a median follow-up of 33.6 months (14.5-44.4).
The ABR for treated bleeds fell from 4.08 to 0.07 (−4.01, −5.57 to −2.45; P < .0001), and 88% had no treated bleeds over the same follow-up period.
“The primary endpoint for this trial was a reduction in total bleeds in patients, and that was achieved,” hematologist and first author Andrew D. Leavitt, MD, of the University of California at San Francisco, said in an interview. “More impressive was the reduction in treated bleeds, a kind of a surrogate marker for bleeds of clinical significance to the individual. And as one would expect or hope to see in a gene therapy trial, there was a significant and marked reduction in the use of factor.”
Moving forward, he said, the message to clinicians is that “there’s every reason to believe that they will have yet another option for their patients.”
Gene Therapy on the Rise in Hemophilia
Gene therapy has arisen as an approved therapy for hemophilia over just the last few years. Two gene therapies for hemophilia B have been approved by the FDA since 2022, and one was approved for hemophilia A, the more common type, in 2023.
As Leavitt noted, one-time treatment with gene therapy offers an alternative to treatment with blood factor, long the mainstay of hemophilia therapy.
“One of the real pluses of gene therapy is the potential to remove the burden of hemophilia, which is large and, I suspect, underappreciated even by providers,” he said. “You have to sit down with your patients and really get a real good sense of just how difficult it is for them to manage with many products on the market over the last few decades.”
Why is there a need for multiple gene therapy products? “A patient may have neutralizing antibodies against the proteins on the surface of gene therapy product A that prevents its use, but not on gene therapy B, which allows use of product B,” Leavitt said. “We need a few flavors so that we can offer gene therapy to the maximum number of interested patients.”
High Efficacy and an ‘Acceptable’ Safety Profile
For the study, researchers dosed 75 patients (mean age, 32.3 [19-59]; 100% men, 74.7% White and 18.7% Asian,) with hemophilia A with giroctocogene fitelparvovec, a hepatocyte-directed recombinant adeno-associated virus serotype 6 vector encoding a B-domain–deleted variant of human factor VIII. The efficacy population is 50 patients with at least 6 months of follow-up in the lead-in study.
The annualized infusion rate of exogenous FVIII was 124.39 mean annualized infusion rate prior to the treatment infusion vs 0.21 post-infusion, week 12 through at least 15 months (−124.18, −139.47 to −108.89; P < .0001).
Leavitt said the results are similar to other gene therapies for hemophilia in that “it is difficult to predict how high your factor level will become. There’s a broad range of outcomes for individuals, and the duration of expression remains an unknown.”
The study authors described the treatment as “generally well tolerated” with “an acceptable and manageable safety profile.”
Of the 75 subjects, 98.7% had adverse effects (AEs, 740 events) and 90.7% had treatment-related AEs. Common treatment-related AEs included hepatotoxicity (62.7%) and infusion-related reactions (73.3%). No subjects discontinued therapy due to AEs.
Nearly two thirds — 62.7% — of subjects used corticosteroids for a mean 114.6 days (11-296).
Study Findings ‘Look Really Good’
In an interview, Guy Young, MD, director of the Hemostasis and Thrombosis Program at Children’s Hospital Los Angeles and professor of pediatrics, Keck School of Medicine of University of Southern California, Los Angeles, said that “generally speaking, the new data looks really good.” Young, who didn’t take part in the study, noted that factor levels following treatment were high, and one subject actually had a thrombotic event and needed to be treated with an anticoagulant.
The high factor levels could actually be a sign of lasting benefit vs valoctocogene roxaparvovec (Roctavian), the sole FDA-approved gene therapy for hemophilia A, which is linked to significant drops in factor level after 6 months, he said. “Wouldn’t it be better to start really high?”
Gene therapy for hemophilia is highly expensive, although proponents noted that insurers may save money over the long run if patients don’t require prophylactic treatment or therapy for bleeds.
A Pfizer spokesman declined to comment on the new therapy’s potential cost. In regard to when the therapy may receive FDA approval, he said “Pfizer is discussing this data with regulatory authorities.”
Pfizer funded this study. Leavitt disclosed ties with HEMA, Merck, Catalyst, Genentech, Pfizer, BioMarin, and Sangamo. Other study authors reported relationships with Pfizer. Young disclosed ties with Pfizer and BioMarin.
A version of this article appeared on Medscape.com.
FROM ASH 2024
Daratumumab Confirmed as SOC for AL Amyloidosis
Adding DARA to VCd (D-VCd; Darzalex Faspro; Janssen Biotech) provided deeper and more rapid hematologic response and clinically meaningful and statistically significant improvement in overall survival (OS) and major organ deterioration progression-free survival (MOD-PFS), combined with 40.7% cardiac complete response (CR), first author Efstathios Kastritis, MD, said during presentation of an oral abstract at the American Society of Hematology (ASH) 2024 Annual Meeting.
“The Andromeda study is the first comparing two contemporary regimens that shows a significant survival improvement for patients with AL amyloidosis,” said Kastritis, an associate professor at the National and Kapodistrian University of Athens in Greece. “These findings reaffirm frontline D-VCd as the standard of care in this difficult-to-treat disease.”
The regimen was approved for this indication in 2021 based on prior earlier findings from the Andromeda trial. The current results are from a preplanned analysis for MOD-PFS and OS.
At a median follow-up of 61.4 months, the overall hematologic CR rates were 59.5% and 19.2% among 388 patients randomized to receive D-VCd or VCd, respectively (odds ratio, 6.03), which showed continued improvement with additional DARA vs the 53.3% and 18.1% rates observed at the primary analysis, Kastritis reported.
Time to hematologic CR was 67.5 days and 85.0 days in the treatment groups, respectively, and median duration of hematologic CR was not reached in either group.
A significant 56% improvement was also observed in MOD-PFS (hazard ratio [HR], 0.44). Median MOD-PFS was not reached in the D-VCd group and was 30.3 months in the VCd group.
A significant 38% improvement was observed in OS (HR, 0.62), 5-year OS was 76.1% vs 64.7% in the D-VCd and VCd groups, respectively, he said.
“[The OS] benefit occurred even though more than 70% of the patients in the VCd arm who received a subsequent therapy were treated with a DARA-based regimen,” he stressed. “This further emphasizes the importance of using DSTS in the frontline setting.”
Trial participants had newly diagnosed AL amyloidosis with measurable hematologic disease, one or more involved organs, cardiac stage I-IIIA, estimated glomerular filtration rate of at least 20 mL/min, and absence of symptomatic multiple myeloma. They were randomized 1:1 to the two treatment groups. All patients received 1.3 mg/m2 of bortezomib by weekly injection, 300 mg/m2 of cyclophosphamide either by weekly oral or intravenous administration, and 20-40 mg of dexamethasone by weekly oral or intravenous administration for six 28-day cycles.
Those in the D-VCd group also received 1800 mg of DARA coformulated with rHuPH20 as a weekly injection in cycles 1-2, as a biweekly injection in cycles 3-6, and by injection every 4 weeks thereafter for up to 24 28-day cycles.
The median duration of treatment was 21.3 months for D-VCd and 5.3 months for VCd, and of 122 patients who received subsequent therapy, 82 (67%) received subsequent DARA.
Patients who achieved hematologic CR had better MOD-PFS and OS (HR, 0.30 and 0.41, respectively), regardless of the treatment received, he noted, adding that “this further supports that complete hematologic response is a valid early endpoint for the evaluation of anti-monoclonal therapies in AL amyloidosis.”
“I think this is very important for the further development of new treatments in this disease,” he said.
Of note, cardiac and renal response rates in the D-VCd group were about two to three times greater than those in the VCd group at 6, 12, 24, 36, and 48 months, Kastritis said.
Among 235 patients with an evaluable cardiac response, 113 achieved a very good partial response or better, including 76 of 118 (64.4%) in the D-VCd group and 37 of 117 (31.6%) in the VCd group. Of these, 48 (40.7%) and 16 (13.7%) achieved a cardiac CR.
Grade 3 or 4 treatment-emergent adverse events (TEAEs) occurring in at least 5% of patients in the D-VCd and VCd groups, respectively, were lymphopenia (13% and 10%), pneumonia (8% and 4%), hypokalemia (2% and 5%), and peripheral edema (3% and 6%), he noted, adding that systemic administration-related reactions occurred in 14 (7%) of patients receiving D-VCd; all were grade 1 or 2 and most (86%) occurred after the first injection. TEAEs led to treatment discontinuation in 5% and 4% of patients in the groups, respectively.
No new safety signals were observed during the extended follow-up, he said.
Kastritis reported relationships with Pfizer, Genesis Pharma, Sanofi, AbbVie, GSK, Prothena, Janssen, and Amgen.
A version of this article first appeared on Medscape.com.
Adding DARA to VCd (D-VCd; Darzalex Faspro; Janssen Biotech) provided deeper and more rapid hematologic response and clinically meaningful and statistically significant improvement in overall survival (OS) and major organ deterioration progression-free survival (MOD-PFS), combined with 40.7% cardiac complete response (CR), first author Efstathios Kastritis, MD, said during presentation of an oral abstract at the American Society of Hematology (ASH) 2024 Annual Meeting.
“The Andromeda study is the first comparing two contemporary regimens that shows a significant survival improvement for patients with AL amyloidosis,” said Kastritis, an associate professor at the National and Kapodistrian University of Athens in Greece. “These findings reaffirm frontline D-VCd as the standard of care in this difficult-to-treat disease.”
The regimen was approved for this indication in 2021 based on prior earlier findings from the Andromeda trial. The current results are from a preplanned analysis for MOD-PFS and OS.
At a median follow-up of 61.4 months, the overall hematologic CR rates were 59.5% and 19.2% among 388 patients randomized to receive D-VCd or VCd, respectively (odds ratio, 6.03), which showed continued improvement with additional DARA vs the 53.3% and 18.1% rates observed at the primary analysis, Kastritis reported.
Time to hematologic CR was 67.5 days and 85.0 days in the treatment groups, respectively, and median duration of hematologic CR was not reached in either group.
A significant 56% improvement was also observed in MOD-PFS (hazard ratio [HR], 0.44). Median MOD-PFS was not reached in the D-VCd group and was 30.3 months in the VCd group.
A significant 38% improvement was observed in OS (HR, 0.62), 5-year OS was 76.1% vs 64.7% in the D-VCd and VCd groups, respectively, he said.
“[The OS] benefit occurred even though more than 70% of the patients in the VCd arm who received a subsequent therapy were treated with a DARA-based regimen,” he stressed. “This further emphasizes the importance of using DSTS in the frontline setting.”
Trial participants had newly diagnosed AL amyloidosis with measurable hematologic disease, one or more involved organs, cardiac stage I-IIIA, estimated glomerular filtration rate of at least 20 mL/min, and absence of symptomatic multiple myeloma. They were randomized 1:1 to the two treatment groups. All patients received 1.3 mg/m2 of bortezomib by weekly injection, 300 mg/m2 of cyclophosphamide either by weekly oral or intravenous administration, and 20-40 mg of dexamethasone by weekly oral or intravenous administration for six 28-day cycles.
Those in the D-VCd group also received 1800 mg of DARA coformulated with rHuPH20 as a weekly injection in cycles 1-2, as a biweekly injection in cycles 3-6, and by injection every 4 weeks thereafter for up to 24 28-day cycles.
The median duration of treatment was 21.3 months for D-VCd and 5.3 months for VCd, and of 122 patients who received subsequent therapy, 82 (67%) received subsequent DARA.
Patients who achieved hematologic CR had better MOD-PFS and OS (HR, 0.30 and 0.41, respectively), regardless of the treatment received, he noted, adding that “this further supports that complete hematologic response is a valid early endpoint for the evaluation of anti-monoclonal therapies in AL amyloidosis.”
“I think this is very important for the further development of new treatments in this disease,” he said.
Of note, cardiac and renal response rates in the D-VCd group were about two to three times greater than those in the VCd group at 6, 12, 24, 36, and 48 months, Kastritis said.
Among 235 patients with an evaluable cardiac response, 113 achieved a very good partial response or better, including 76 of 118 (64.4%) in the D-VCd group and 37 of 117 (31.6%) in the VCd group. Of these, 48 (40.7%) and 16 (13.7%) achieved a cardiac CR.
Grade 3 or 4 treatment-emergent adverse events (TEAEs) occurring in at least 5% of patients in the D-VCd and VCd groups, respectively, were lymphopenia (13% and 10%), pneumonia (8% and 4%), hypokalemia (2% and 5%), and peripheral edema (3% and 6%), he noted, adding that systemic administration-related reactions occurred in 14 (7%) of patients receiving D-VCd; all were grade 1 or 2 and most (86%) occurred after the first injection. TEAEs led to treatment discontinuation in 5% and 4% of patients in the groups, respectively.
No new safety signals were observed during the extended follow-up, he said.
Kastritis reported relationships with Pfizer, Genesis Pharma, Sanofi, AbbVie, GSK, Prothena, Janssen, and Amgen.
A version of this article first appeared on Medscape.com.
Adding DARA to VCd (D-VCd; Darzalex Faspro; Janssen Biotech) provided deeper and more rapid hematologic response and clinically meaningful and statistically significant improvement in overall survival (OS) and major organ deterioration progression-free survival (MOD-PFS), combined with 40.7% cardiac complete response (CR), first author Efstathios Kastritis, MD, said during presentation of an oral abstract at the American Society of Hematology (ASH) 2024 Annual Meeting.
“The Andromeda study is the first comparing two contemporary regimens that shows a significant survival improvement for patients with AL amyloidosis,” said Kastritis, an associate professor at the National and Kapodistrian University of Athens in Greece. “These findings reaffirm frontline D-VCd as the standard of care in this difficult-to-treat disease.”
The regimen was approved for this indication in 2021 based on prior earlier findings from the Andromeda trial. The current results are from a preplanned analysis for MOD-PFS and OS.
At a median follow-up of 61.4 months, the overall hematologic CR rates were 59.5% and 19.2% among 388 patients randomized to receive D-VCd or VCd, respectively (odds ratio, 6.03), which showed continued improvement with additional DARA vs the 53.3% and 18.1% rates observed at the primary analysis, Kastritis reported.
Time to hematologic CR was 67.5 days and 85.0 days in the treatment groups, respectively, and median duration of hematologic CR was not reached in either group.
A significant 56% improvement was also observed in MOD-PFS (hazard ratio [HR], 0.44). Median MOD-PFS was not reached in the D-VCd group and was 30.3 months in the VCd group.
A significant 38% improvement was observed in OS (HR, 0.62), 5-year OS was 76.1% vs 64.7% in the D-VCd and VCd groups, respectively, he said.
“[The OS] benefit occurred even though more than 70% of the patients in the VCd arm who received a subsequent therapy were treated with a DARA-based regimen,” he stressed. “This further emphasizes the importance of using DSTS in the frontline setting.”
Trial participants had newly diagnosed AL amyloidosis with measurable hematologic disease, one or more involved organs, cardiac stage I-IIIA, estimated glomerular filtration rate of at least 20 mL/min, and absence of symptomatic multiple myeloma. They were randomized 1:1 to the two treatment groups. All patients received 1.3 mg/m2 of bortezomib by weekly injection, 300 mg/m2 of cyclophosphamide either by weekly oral or intravenous administration, and 20-40 mg of dexamethasone by weekly oral or intravenous administration for six 28-day cycles.
Those in the D-VCd group also received 1800 mg of DARA coformulated with rHuPH20 as a weekly injection in cycles 1-2, as a biweekly injection in cycles 3-6, and by injection every 4 weeks thereafter for up to 24 28-day cycles.
The median duration of treatment was 21.3 months for D-VCd and 5.3 months for VCd, and of 122 patients who received subsequent therapy, 82 (67%) received subsequent DARA.
Patients who achieved hematologic CR had better MOD-PFS and OS (HR, 0.30 and 0.41, respectively), regardless of the treatment received, he noted, adding that “this further supports that complete hematologic response is a valid early endpoint for the evaluation of anti-monoclonal therapies in AL amyloidosis.”
“I think this is very important for the further development of new treatments in this disease,” he said.
Of note, cardiac and renal response rates in the D-VCd group were about two to three times greater than those in the VCd group at 6, 12, 24, 36, and 48 months, Kastritis said.
Among 235 patients with an evaluable cardiac response, 113 achieved a very good partial response or better, including 76 of 118 (64.4%) in the D-VCd group and 37 of 117 (31.6%) in the VCd group. Of these, 48 (40.7%) and 16 (13.7%) achieved a cardiac CR.
Grade 3 or 4 treatment-emergent adverse events (TEAEs) occurring in at least 5% of patients in the D-VCd and VCd groups, respectively, were lymphopenia (13% and 10%), pneumonia (8% and 4%), hypokalemia (2% and 5%), and peripheral edema (3% and 6%), he noted, adding that systemic administration-related reactions occurred in 14 (7%) of patients receiving D-VCd; all were grade 1 or 2 and most (86%) occurred after the first injection. TEAEs led to treatment discontinuation in 5% and 4% of patients in the groups, respectively.
No new safety signals were observed during the extended follow-up, he said.
Kastritis reported relationships with Pfizer, Genesis Pharma, Sanofi, AbbVie, GSK, Prothena, Janssen, and Amgen.
A version of this article first appeared on Medscape.com.
FROM ASH 2024
Upfront Therapy for ITP in Children: New Drug a Game-Changer?
“This is the first time in 30 years that a new drug is being tested for newly diagnosed pediatric ITP,” said the study’s lead author, Kristin A. Shimano, MD, professor of pediatrics at the Benioff Children’s Hospital, University of California San Francisco, in a press statement for the study, presented at the American Society of Hematology (ASH) 2024 Annual Meeting earlier this month.
“We really think that this has the potential to transform the approach to the management of ITP in the newly diagnosed phase with the use of a therapy that can provide sustained hemostatic platelet counts to bridge the time that patients are at risk of bleeding events with the goal to wean off the medication for patients who have a natural resolution of their disease,” Shimano said in her talk.
While children with ITP, a rare autoimmune blood disorder, very often improve without the need for any treatment, some do require intervention, and the condition can become chronic. First-line therapies for those patients commonly include corticosteroids, intravenous immunoglobulin (IVIg), and anti-D globulin; however, side effects can be undesirable, and with their efficacy often temporary, patients can require monitoring and juggling of treatments.
Eltrombopag, an oral, daily thrombopoietin receptor agonist, was approved by the US Food and Drug Administration for children and adults with chronic ITP in 2015; however, research has been lacking on the benefits of the therapy for newly diagnosed pediatric patients.
To investigate the drug’s efficacy at that stage, Shimano and colleagues with the ITP Consortium of North America launched the prospective, open-label Pediatric ITP Newly diagnosed pts Epag vs Standard therapy (PINES) trial, enrolling 118 patients at 23 institutions between May 2019 and January 2024.
All enrollees had been diagnosed with ITP within 3 months and had been determined by their treating hematologist to require pharmacologic treatment.
Of the patients, about 40% were untreated, and 60% had been treated with at least one medication prior to the trial but did not have a lasting response.
The patients were stratified by age and prior treatment and randomized 2:1 to receive either eltrombopag (n = 78) or the investigator’s choice of one of three standard first-line therapies, including prednisone, IVIg, or anti-D globulin at specified doses (n = 40). Overall, 29 in the standard-of-care arm received prednisone and 11 received IVIg. The patients had a median age of 8 years.
For the study’s primary endpoint, patients in the eltrombopag group had a significantly greater sustained response at 12 weeks, defined as having at least three of four platelet counts > 50 × 109/L during weeks 6-12 without the need for rescue treatment, with a rate of 63% vs 35% in the standard-of-care group (P = .0054).
There were no significant differences between the two groups in terms of the proportion of patients with a high bleeding score at weeks 1-4 and week 12.
However, those in the eltrombopag arm had a significantly lower rate of receiving rescue therapy (18% vs 38% with the standard of care; P = .02).
Both groups showed clinically meaningful improvements from baseline in terms of health-related quality of life, as assessed by parent proxy-reported KIT overall scores.
Twenty adverse events that were grade 3 or higher, including six serious adverse events, occurred in each of the study’s arms, with the most common events including headache and epistaxis.
Treatment-related serious adverse events occurred among six patients in the eltrombopag group and one in the control group, but importantly, no thromboembolic events were reported.
One intracranial hemorrhage occurred in the eltrombopag arm.
With eltrombopag having a slower effect than some other treatments, Shimano cautioned that the therapy is not recommended for patients with severe bleeding.
“Patients with grade 4 or 5 bleeding at the time of screening were specifically excluded from the study, so for patients who have very severe bleeding who need to get their platelets up very quickly, this would not be the ideal therapy for them,” she noted.
On the basis of results, the trial was recommended to close early due to efficacy; however, the participants are being followed for a total of 12 months to determine the durability of the responses, including in terms of bleeding events, quality of life, or the development of chronic ITP.
“We have shown that in pediatric patients with newly diagnosed ITP requiring pharmacologic treatment, eltrombopag resulted in a significant, clinically relevant higher rate of a durable platelet response in the absence of rescue treatment as compared with standard first-line therapies,” Shimano said.
“Eltrombopag could certainly be added to the medication choices hematologists consider as they are making treatment decisions with families, and it is an option that could potentially raise platelets for a more sustained period in children with ITP in the newly diagnosed period, which is one of the most difficult times for patients with regard to the impact of the disease on bleeding symptoms and quality of life,” she added.
Commenting on the study, James B. Bussel, MD, emeritus professor of pediatrics, medicine and obstetrics and gynecology at Weill Cornell Medicine in New York City, commented that “generally, a short-term increase in platelets is the biggest challenge, which is getting the patient to the point of not requiring future treatment to get better.”
“If more children can be shown to be going into remission earlier, that would be great,” he said.
While eltrombopag is known to be effective in chronic ITP, a key caveat of its use in newly diagnosed patients is the question of whether patients will get better on their own and feasibly be able to be spared the cost and burden of treatment in the first place.
However, identifying which patients will fit that profile isn’t always easy.
“Exactly which child needs treatment can be hard to determine, and there is some debate about that,” Bussel noted.
“The theoretic standard is that the platelet count doesn’t matter — only whether the patient is bleeding a lot, and then there is debate over treatment based on bleeding scores,” he said.
Quality-of-life issues, such as patients’ ability to take part in activities, are also a key consideration.
“It would be great if eltrombopag can support children who really need it and provide clear unequivocal benefit beyond just increasing the platelet count, but also leading to better quality of life,” Bussel said.
The new findings are “a very encouraging start, but I’d really like to see what the story is at 1 year.”
The study was funded by Novartis, maker of eltrombopag, and sponsored by the ITP Consortium of North America. Shimano disclosed ties with Sanofi, Sobi, Daiichi Sankyo, Novartis, and Pfizer. Bussel reported a relationship with Novartis that ended more than 2 years ago.
A version of this article first appeared on Medscape.com.
“This is the first time in 30 years that a new drug is being tested for newly diagnosed pediatric ITP,” said the study’s lead author, Kristin A. Shimano, MD, professor of pediatrics at the Benioff Children’s Hospital, University of California San Francisco, in a press statement for the study, presented at the American Society of Hematology (ASH) 2024 Annual Meeting earlier this month.
“We really think that this has the potential to transform the approach to the management of ITP in the newly diagnosed phase with the use of a therapy that can provide sustained hemostatic platelet counts to bridge the time that patients are at risk of bleeding events with the goal to wean off the medication for patients who have a natural resolution of their disease,” Shimano said in her talk.
While children with ITP, a rare autoimmune blood disorder, very often improve without the need for any treatment, some do require intervention, and the condition can become chronic. First-line therapies for those patients commonly include corticosteroids, intravenous immunoglobulin (IVIg), and anti-D globulin; however, side effects can be undesirable, and with their efficacy often temporary, patients can require monitoring and juggling of treatments.
Eltrombopag, an oral, daily thrombopoietin receptor agonist, was approved by the US Food and Drug Administration for children and adults with chronic ITP in 2015; however, research has been lacking on the benefits of the therapy for newly diagnosed pediatric patients.
To investigate the drug’s efficacy at that stage, Shimano and colleagues with the ITP Consortium of North America launched the prospective, open-label Pediatric ITP Newly diagnosed pts Epag vs Standard therapy (PINES) trial, enrolling 118 patients at 23 institutions between May 2019 and January 2024.
All enrollees had been diagnosed with ITP within 3 months and had been determined by their treating hematologist to require pharmacologic treatment.
Of the patients, about 40% were untreated, and 60% had been treated with at least one medication prior to the trial but did not have a lasting response.
The patients were stratified by age and prior treatment and randomized 2:1 to receive either eltrombopag (n = 78) or the investigator’s choice of one of three standard first-line therapies, including prednisone, IVIg, or anti-D globulin at specified doses (n = 40). Overall, 29 in the standard-of-care arm received prednisone and 11 received IVIg. The patients had a median age of 8 years.
For the study’s primary endpoint, patients in the eltrombopag group had a significantly greater sustained response at 12 weeks, defined as having at least three of four platelet counts > 50 × 109/L during weeks 6-12 without the need for rescue treatment, with a rate of 63% vs 35% in the standard-of-care group (P = .0054).
There were no significant differences between the two groups in terms of the proportion of patients with a high bleeding score at weeks 1-4 and week 12.
However, those in the eltrombopag arm had a significantly lower rate of receiving rescue therapy (18% vs 38% with the standard of care; P = .02).
Both groups showed clinically meaningful improvements from baseline in terms of health-related quality of life, as assessed by parent proxy-reported KIT overall scores.
Twenty adverse events that were grade 3 or higher, including six serious adverse events, occurred in each of the study’s arms, with the most common events including headache and epistaxis.
Treatment-related serious adverse events occurred among six patients in the eltrombopag group and one in the control group, but importantly, no thromboembolic events were reported.
One intracranial hemorrhage occurred in the eltrombopag arm.
With eltrombopag having a slower effect than some other treatments, Shimano cautioned that the therapy is not recommended for patients with severe bleeding.
“Patients with grade 4 or 5 bleeding at the time of screening were specifically excluded from the study, so for patients who have very severe bleeding who need to get their platelets up very quickly, this would not be the ideal therapy for them,” she noted.
On the basis of results, the trial was recommended to close early due to efficacy; however, the participants are being followed for a total of 12 months to determine the durability of the responses, including in terms of bleeding events, quality of life, or the development of chronic ITP.
“We have shown that in pediatric patients with newly diagnosed ITP requiring pharmacologic treatment, eltrombopag resulted in a significant, clinically relevant higher rate of a durable platelet response in the absence of rescue treatment as compared with standard first-line therapies,” Shimano said.
“Eltrombopag could certainly be added to the medication choices hematologists consider as they are making treatment decisions with families, and it is an option that could potentially raise platelets for a more sustained period in children with ITP in the newly diagnosed period, which is one of the most difficult times for patients with regard to the impact of the disease on bleeding symptoms and quality of life,” she added.
Commenting on the study, James B. Bussel, MD, emeritus professor of pediatrics, medicine and obstetrics and gynecology at Weill Cornell Medicine in New York City, commented that “generally, a short-term increase in platelets is the biggest challenge, which is getting the patient to the point of not requiring future treatment to get better.”
“If more children can be shown to be going into remission earlier, that would be great,” he said.
While eltrombopag is known to be effective in chronic ITP, a key caveat of its use in newly diagnosed patients is the question of whether patients will get better on their own and feasibly be able to be spared the cost and burden of treatment in the first place.
However, identifying which patients will fit that profile isn’t always easy.
“Exactly which child needs treatment can be hard to determine, and there is some debate about that,” Bussel noted.
“The theoretic standard is that the platelet count doesn’t matter — only whether the patient is bleeding a lot, and then there is debate over treatment based on bleeding scores,” he said.
Quality-of-life issues, such as patients’ ability to take part in activities, are also a key consideration.
“It would be great if eltrombopag can support children who really need it and provide clear unequivocal benefit beyond just increasing the platelet count, but also leading to better quality of life,” Bussel said.
The new findings are “a very encouraging start, but I’d really like to see what the story is at 1 year.”
The study was funded by Novartis, maker of eltrombopag, and sponsored by the ITP Consortium of North America. Shimano disclosed ties with Sanofi, Sobi, Daiichi Sankyo, Novartis, and Pfizer. Bussel reported a relationship with Novartis that ended more than 2 years ago.
A version of this article first appeared on Medscape.com.
“This is the first time in 30 years that a new drug is being tested for newly diagnosed pediatric ITP,” said the study’s lead author, Kristin A. Shimano, MD, professor of pediatrics at the Benioff Children’s Hospital, University of California San Francisco, in a press statement for the study, presented at the American Society of Hematology (ASH) 2024 Annual Meeting earlier this month.
“We really think that this has the potential to transform the approach to the management of ITP in the newly diagnosed phase with the use of a therapy that can provide sustained hemostatic platelet counts to bridge the time that patients are at risk of bleeding events with the goal to wean off the medication for patients who have a natural resolution of their disease,” Shimano said in her talk.
While children with ITP, a rare autoimmune blood disorder, very often improve without the need for any treatment, some do require intervention, and the condition can become chronic. First-line therapies for those patients commonly include corticosteroids, intravenous immunoglobulin (IVIg), and anti-D globulin; however, side effects can be undesirable, and with their efficacy often temporary, patients can require monitoring and juggling of treatments.
Eltrombopag, an oral, daily thrombopoietin receptor agonist, was approved by the US Food and Drug Administration for children and adults with chronic ITP in 2015; however, research has been lacking on the benefits of the therapy for newly diagnosed pediatric patients.
To investigate the drug’s efficacy at that stage, Shimano and colleagues with the ITP Consortium of North America launched the prospective, open-label Pediatric ITP Newly diagnosed pts Epag vs Standard therapy (PINES) trial, enrolling 118 patients at 23 institutions between May 2019 and January 2024.
All enrollees had been diagnosed with ITP within 3 months and had been determined by their treating hematologist to require pharmacologic treatment.
Of the patients, about 40% were untreated, and 60% had been treated with at least one medication prior to the trial but did not have a lasting response.
The patients were stratified by age and prior treatment and randomized 2:1 to receive either eltrombopag (n = 78) or the investigator’s choice of one of three standard first-line therapies, including prednisone, IVIg, or anti-D globulin at specified doses (n = 40). Overall, 29 in the standard-of-care arm received prednisone and 11 received IVIg. The patients had a median age of 8 years.
For the study’s primary endpoint, patients in the eltrombopag group had a significantly greater sustained response at 12 weeks, defined as having at least three of four platelet counts > 50 × 109/L during weeks 6-12 without the need for rescue treatment, with a rate of 63% vs 35% in the standard-of-care group (P = .0054).
There were no significant differences between the two groups in terms of the proportion of patients with a high bleeding score at weeks 1-4 and week 12.
However, those in the eltrombopag arm had a significantly lower rate of receiving rescue therapy (18% vs 38% with the standard of care; P = .02).
Both groups showed clinically meaningful improvements from baseline in terms of health-related quality of life, as assessed by parent proxy-reported KIT overall scores.
Twenty adverse events that were grade 3 or higher, including six serious adverse events, occurred in each of the study’s arms, with the most common events including headache and epistaxis.
Treatment-related serious adverse events occurred among six patients in the eltrombopag group and one in the control group, but importantly, no thromboembolic events were reported.
One intracranial hemorrhage occurred in the eltrombopag arm.
With eltrombopag having a slower effect than some other treatments, Shimano cautioned that the therapy is not recommended for patients with severe bleeding.
“Patients with grade 4 or 5 bleeding at the time of screening were specifically excluded from the study, so for patients who have very severe bleeding who need to get their platelets up very quickly, this would not be the ideal therapy for them,” she noted.
On the basis of results, the trial was recommended to close early due to efficacy; however, the participants are being followed for a total of 12 months to determine the durability of the responses, including in terms of bleeding events, quality of life, or the development of chronic ITP.
“We have shown that in pediatric patients with newly diagnosed ITP requiring pharmacologic treatment, eltrombopag resulted in a significant, clinically relevant higher rate of a durable platelet response in the absence of rescue treatment as compared with standard first-line therapies,” Shimano said.
“Eltrombopag could certainly be added to the medication choices hematologists consider as they are making treatment decisions with families, and it is an option that could potentially raise platelets for a more sustained period in children with ITP in the newly diagnosed period, which is one of the most difficult times for patients with regard to the impact of the disease on bleeding symptoms and quality of life,” she added.
Commenting on the study, James B. Bussel, MD, emeritus professor of pediatrics, medicine and obstetrics and gynecology at Weill Cornell Medicine in New York City, commented that “generally, a short-term increase in platelets is the biggest challenge, which is getting the patient to the point of not requiring future treatment to get better.”
“If more children can be shown to be going into remission earlier, that would be great,” he said.
While eltrombopag is known to be effective in chronic ITP, a key caveat of its use in newly diagnosed patients is the question of whether patients will get better on their own and feasibly be able to be spared the cost and burden of treatment in the first place.
However, identifying which patients will fit that profile isn’t always easy.
“Exactly which child needs treatment can be hard to determine, and there is some debate about that,” Bussel noted.
“The theoretic standard is that the platelet count doesn’t matter — only whether the patient is bleeding a lot, and then there is debate over treatment based on bleeding scores,” he said.
Quality-of-life issues, such as patients’ ability to take part in activities, are also a key consideration.
“It would be great if eltrombopag can support children who really need it and provide clear unequivocal benefit beyond just increasing the platelet count, but also leading to better quality of life,” Bussel said.
The new findings are “a very encouraging start, but I’d really like to see what the story is at 1 year.”
The study was funded by Novartis, maker of eltrombopag, and sponsored by the ITP Consortium of North America. Shimano disclosed ties with Sanofi, Sobi, Daiichi Sankyo, Novartis, and Pfizer. Bussel reported a relationship with Novartis that ended more than 2 years ago.
A version of this article first appeared on Medscape.com.
FROM ASH 2024
Fertility Preservation in SCD: Women Have More Complications
Of 46 patients with SCD, complications occurred in 25 of 55 controlled ovarian hyperstimulation cycles, including 29 vaso-occlusive episodes (VOEs), researchers reported at the American Society of Hematology (ASH) 2024 Annual Meeting.
Of 21 post-retrieval VOEs, 19 required emergency department care or hospitalization.
“Baseline sickle cell disease severity is most likely associated with a patient’s risk of complications from an egg retrieval cycle,” study co-author Sarah Cromack, MD, a reproductive endocrinology and infertility fellow at Northwestern University, Chicago, said in an interview.
“Both hematologists and reproductive endocrinologists can use this information to plan ahead and anticipate possible issues, check blood counts prior to and after egg retrieval to see if transfusion is needed, and plan close follow-up during stimulation and immediately after egg retrieval to evaluate and treat pain.”
SCD Accelerates Decline in Ovarian Reserve
Pediatric hematologist Lydia H. Pecker, MD, MS, of Johns Hopkins University School of Medicine, Baltimore, the study’s corresponding author, said in an interview that SCD is “a disease of accelerated aging” that leads to accelerated decline in ovarian reserve. “The common indication for fertility preservation in SCD is before bone marrow transplant or gene therapy,” she said, although FP can also be offered to other patients with SCD.
According to Cromack, researchers launched the study to expand information about SCD and FP in light of sparse data about outcomes.
All the 46 patients had hemoglobin SS (HbSS, 93%) and HbSβ0-thalassemia (7%) and a median age of 23.7 (18-28) years. Almost all (44 patients) underwent FP prior to curative treatments, and all had at least one SCD-related complication, mainly cerebrovascular disease (16), acute chest syndrome (23), and more than two VOEs per year (31).
Median anti-Mullerian hormone (AMH) level (2.1 ng/mL), a measurement of ovarian reserve, was lower than the expected level of 2.8-3.4 ng/mL among women in the age range of the patients, the researchers reported. “This is consistent with previous studies showing lower AMH for age in women with sickle cell disease,” Pecker said.
Complications in 45% of Retrieval Cycles
“In terms of success of oocyte cryopreservation, the median number of mature eggs frozen was 11,” said co-author and reproductive endocrinologist Jessica Walter, MD, of Northwestern University, in an interview. “Given the average age of 24 years in the cohort, this would give each patient about a 70% estimated probability of at least one live birth from their cohort of frozen eggs. Thus, patients hoping for more than one child may want to consider more than one cycle of egg freezing.”
The rate of complications was “fairly high” at 45% of all cycles, Walter said. “These were mostly complications from underlying sickle cell disease, including unplanned transfusions and admissions for vaso-occlusive crises. Surprisingly, there were very few cases of ovarian hyperstimulation syndrome in this young patient group, which may be due to a combination of underlying vascular disease, lower peak estradiol levels, and slightly less eggs retrieved then would be expected compared to an age-matched healthy controls.”
Any FP complication was associated with more than three VOEs in the year before controlled ovarian hyperstimulation (mean of three VOEs per patient without complications vs six per patient with complications; P = .036).
Higher Than Normal Need for Multiple Cycles
Reproductive endocrinologist H. Irene Su, MD, professor and co-director of the Center for OB/GYN Research Innovations at Moores Cancer Center, University of California San Diego, praised the study as “an important report” in an interview.
Su, who wasn’t involved in the research, said the percentage of patients requiring more than one cycle due to cancellation or low oocyte yield — 13% — is “higher than expected, given the young age of this cohort.”
This could reflect the hypothesis that “sickle cell crises and hypoxia adversely affect the finite number of oocytes in the ovary,” she said.
As for the study findings regarding complications, she said the rate “is very high compared to the general infertility or fertility preservation population. It would be good to learn predictors of these outcomes so that fertility and hematology clinicians can work together to stratify risk and supportive services around FP cycles. It would also be good to know if the post-retrieval VOE were unexpected given the patient’s disease activity prior to FP.”
Message: FP in SCD Is Feasible, Acceptable
A.D. Mishkin, MD, MPH, associate professor of psychiatry and liaison to the Blood and Marrow Transplantation Program at NewYork–Presbyterian/Columbia University Irving Medical Center, New York City, said in an interview that the study “establishes the feasibility and acceptability of oocyte harvest and preservation in a population of patients with active ongoing symptoms from SCD. It also indicates their interest in pursuing fertility preservation in the setting of frequent crises and the potential for management of ensuing complications.”
Mishkin, who didn’t take part in the research, highlighted the finding that half the patients got access to FP via public insurance or research funding. “Even in this population where most women had multiple complications in the year prior to FP, and even among patients who needed multiple retrievals, these patients wanted to go through that risk to preserve their fertility,” Mishkin said. “This is an important finding given the very limited access many individuals have to FP due to its high cost and limited insurance coverage, which is also largely state-dependent.”
There’s another factor to consider regarding SCD and FP: The potential danger of pregnancy.
Corresponding author Pecker noted that “pregnancy is high risk for people with sickle cell disease. There are very high rates of severe maternal mortality and morbidity even in high-income countries. However, some of this is modifiable with routine use of chronic transfusions during pregnancy and with high-quality and integrated expert SCD and expert maternal fetal medicine care during pregnancy.”
The National Institutes of Health supported the research. Pecker reported receiving research funding from Alexion, Novartis, and Aummune and consulting for Novo Nordisk. Other authors reported no disclosures. Su and Mishkin reported no disclosures.
A version of this article appeared on Medscape.com.
Of 46 patients with SCD, complications occurred in 25 of 55 controlled ovarian hyperstimulation cycles, including 29 vaso-occlusive episodes (VOEs), researchers reported at the American Society of Hematology (ASH) 2024 Annual Meeting.
Of 21 post-retrieval VOEs, 19 required emergency department care or hospitalization.
“Baseline sickle cell disease severity is most likely associated with a patient’s risk of complications from an egg retrieval cycle,” study co-author Sarah Cromack, MD, a reproductive endocrinology and infertility fellow at Northwestern University, Chicago, said in an interview.
“Both hematologists and reproductive endocrinologists can use this information to plan ahead and anticipate possible issues, check blood counts prior to and after egg retrieval to see if transfusion is needed, and plan close follow-up during stimulation and immediately after egg retrieval to evaluate and treat pain.”
SCD Accelerates Decline in Ovarian Reserve
Pediatric hematologist Lydia H. Pecker, MD, MS, of Johns Hopkins University School of Medicine, Baltimore, the study’s corresponding author, said in an interview that SCD is “a disease of accelerated aging” that leads to accelerated decline in ovarian reserve. “The common indication for fertility preservation in SCD is before bone marrow transplant or gene therapy,” she said, although FP can also be offered to other patients with SCD.
According to Cromack, researchers launched the study to expand information about SCD and FP in light of sparse data about outcomes.
All the 46 patients had hemoglobin SS (HbSS, 93%) and HbSβ0-thalassemia (7%) and a median age of 23.7 (18-28) years. Almost all (44 patients) underwent FP prior to curative treatments, and all had at least one SCD-related complication, mainly cerebrovascular disease (16), acute chest syndrome (23), and more than two VOEs per year (31).
Median anti-Mullerian hormone (AMH) level (2.1 ng/mL), a measurement of ovarian reserve, was lower than the expected level of 2.8-3.4 ng/mL among women in the age range of the patients, the researchers reported. “This is consistent with previous studies showing lower AMH for age in women with sickle cell disease,” Pecker said.
Complications in 45% of Retrieval Cycles
“In terms of success of oocyte cryopreservation, the median number of mature eggs frozen was 11,” said co-author and reproductive endocrinologist Jessica Walter, MD, of Northwestern University, in an interview. “Given the average age of 24 years in the cohort, this would give each patient about a 70% estimated probability of at least one live birth from their cohort of frozen eggs. Thus, patients hoping for more than one child may want to consider more than one cycle of egg freezing.”
The rate of complications was “fairly high” at 45% of all cycles, Walter said. “These were mostly complications from underlying sickle cell disease, including unplanned transfusions and admissions for vaso-occlusive crises. Surprisingly, there were very few cases of ovarian hyperstimulation syndrome in this young patient group, which may be due to a combination of underlying vascular disease, lower peak estradiol levels, and slightly less eggs retrieved then would be expected compared to an age-matched healthy controls.”
Any FP complication was associated with more than three VOEs in the year before controlled ovarian hyperstimulation (mean of three VOEs per patient without complications vs six per patient with complications; P = .036).
Higher Than Normal Need for Multiple Cycles
Reproductive endocrinologist H. Irene Su, MD, professor and co-director of the Center for OB/GYN Research Innovations at Moores Cancer Center, University of California San Diego, praised the study as “an important report” in an interview.
Su, who wasn’t involved in the research, said the percentage of patients requiring more than one cycle due to cancellation or low oocyte yield — 13% — is “higher than expected, given the young age of this cohort.”
This could reflect the hypothesis that “sickle cell crises and hypoxia adversely affect the finite number of oocytes in the ovary,” she said.
As for the study findings regarding complications, she said the rate “is very high compared to the general infertility or fertility preservation population. It would be good to learn predictors of these outcomes so that fertility and hematology clinicians can work together to stratify risk and supportive services around FP cycles. It would also be good to know if the post-retrieval VOE were unexpected given the patient’s disease activity prior to FP.”
Message: FP in SCD Is Feasible, Acceptable
A.D. Mishkin, MD, MPH, associate professor of psychiatry and liaison to the Blood and Marrow Transplantation Program at NewYork–Presbyterian/Columbia University Irving Medical Center, New York City, said in an interview that the study “establishes the feasibility and acceptability of oocyte harvest and preservation in a population of patients with active ongoing symptoms from SCD. It also indicates their interest in pursuing fertility preservation in the setting of frequent crises and the potential for management of ensuing complications.”
Mishkin, who didn’t take part in the research, highlighted the finding that half the patients got access to FP via public insurance or research funding. “Even in this population where most women had multiple complications in the year prior to FP, and even among patients who needed multiple retrievals, these patients wanted to go through that risk to preserve their fertility,” Mishkin said. “This is an important finding given the very limited access many individuals have to FP due to its high cost and limited insurance coverage, which is also largely state-dependent.”
There’s another factor to consider regarding SCD and FP: The potential danger of pregnancy.
Corresponding author Pecker noted that “pregnancy is high risk for people with sickle cell disease. There are very high rates of severe maternal mortality and morbidity even in high-income countries. However, some of this is modifiable with routine use of chronic transfusions during pregnancy and with high-quality and integrated expert SCD and expert maternal fetal medicine care during pregnancy.”
The National Institutes of Health supported the research. Pecker reported receiving research funding from Alexion, Novartis, and Aummune and consulting for Novo Nordisk. Other authors reported no disclosures. Su and Mishkin reported no disclosures.
A version of this article appeared on Medscape.com.
Of 46 patients with SCD, complications occurred in 25 of 55 controlled ovarian hyperstimulation cycles, including 29 vaso-occlusive episodes (VOEs), researchers reported at the American Society of Hematology (ASH) 2024 Annual Meeting.
Of 21 post-retrieval VOEs, 19 required emergency department care or hospitalization.
“Baseline sickle cell disease severity is most likely associated with a patient’s risk of complications from an egg retrieval cycle,” study co-author Sarah Cromack, MD, a reproductive endocrinology and infertility fellow at Northwestern University, Chicago, said in an interview.
“Both hematologists and reproductive endocrinologists can use this information to plan ahead and anticipate possible issues, check blood counts prior to and after egg retrieval to see if transfusion is needed, and plan close follow-up during stimulation and immediately after egg retrieval to evaluate and treat pain.”
SCD Accelerates Decline in Ovarian Reserve
Pediatric hematologist Lydia H. Pecker, MD, MS, of Johns Hopkins University School of Medicine, Baltimore, the study’s corresponding author, said in an interview that SCD is “a disease of accelerated aging” that leads to accelerated decline in ovarian reserve. “The common indication for fertility preservation in SCD is before bone marrow transplant or gene therapy,” she said, although FP can also be offered to other patients with SCD.
According to Cromack, researchers launched the study to expand information about SCD and FP in light of sparse data about outcomes.
All the 46 patients had hemoglobin SS (HbSS, 93%) and HbSβ0-thalassemia (7%) and a median age of 23.7 (18-28) years. Almost all (44 patients) underwent FP prior to curative treatments, and all had at least one SCD-related complication, mainly cerebrovascular disease (16), acute chest syndrome (23), and more than two VOEs per year (31).
Median anti-Mullerian hormone (AMH) level (2.1 ng/mL), a measurement of ovarian reserve, was lower than the expected level of 2.8-3.4 ng/mL among women in the age range of the patients, the researchers reported. “This is consistent with previous studies showing lower AMH for age in women with sickle cell disease,” Pecker said.
Complications in 45% of Retrieval Cycles
“In terms of success of oocyte cryopreservation, the median number of mature eggs frozen was 11,” said co-author and reproductive endocrinologist Jessica Walter, MD, of Northwestern University, in an interview. “Given the average age of 24 years in the cohort, this would give each patient about a 70% estimated probability of at least one live birth from their cohort of frozen eggs. Thus, patients hoping for more than one child may want to consider more than one cycle of egg freezing.”
The rate of complications was “fairly high” at 45% of all cycles, Walter said. “These were mostly complications from underlying sickle cell disease, including unplanned transfusions and admissions for vaso-occlusive crises. Surprisingly, there were very few cases of ovarian hyperstimulation syndrome in this young patient group, which may be due to a combination of underlying vascular disease, lower peak estradiol levels, and slightly less eggs retrieved then would be expected compared to an age-matched healthy controls.”
Any FP complication was associated with more than three VOEs in the year before controlled ovarian hyperstimulation (mean of three VOEs per patient without complications vs six per patient with complications; P = .036).
Higher Than Normal Need for Multiple Cycles
Reproductive endocrinologist H. Irene Su, MD, professor and co-director of the Center for OB/GYN Research Innovations at Moores Cancer Center, University of California San Diego, praised the study as “an important report” in an interview.
Su, who wasn’t involved in the research, said the percentage of patients requiring more than one cycle due to cancellation or low oocyte yield — 13% — is “higher than expected, given the young age of this cohort.”
This could reflect the hypothesis that “sickle cell crises and hypoxia adversely affect the finite number of oocytes in the ovary,” she said.
As for the study findings regarding complications, she said the rate “is very high compared to the general infertility or fertility preservation population. It would be good to learn predictors of these outcomes so that fertility and hematology clinicians can work together to stratify risk and supportive services around FP cycles. It would also be good to know if the post-retrieval VOE were unexpected given the patient’s disease activity prior to FP.”
Message: FP in SCD Is Feasible, Acceptable
A.D. Mishkin, MD, MPH, associate professor of psychiatry and liaison to the Blood and Marrow Transplantation Program at NewYork–Presbyterian/Columbia University Irving Medical Center, New York City, said in an interview that the study “establishes the feasibility and acceptability of oocyte harvest and preservation in a population of patients with active ongoing symptoms from SCD. It also indicates their interest in pursuing fertility preservation in the setting of frequent crises and the potential for management of ensuing complications.”
Mishkin, who didn’t take part in the research, highlighted the finding that half the patients got access to FP via public insurance or research funding. “Even in this population where most women had multiple complications in the year prior to FP, and even among patients who needed multiple retrievals, these patients wanted to go through that risk to preserve their fertility,” Mishkin said. “This is an important finding given the very limited access many individuals have to FP due to its high cost and limited insurance coverage, which is also largely state-dependent.”
There’s another factor to consider regarding SCD and FP: The potential danger of pregnancy.
Corresponding author Pecker noted that “pregnancy is high risk for people with sickle cell disease. There are very high rates of severe maternal mortality and morbidity even in high-income countries. However, some of this is modifiable with routine use of chronic transfusions during pregnancy and with high-quality and integrated expert SCD and expert maternal fetal medicine care during pregnancy.”
The National Institutes of Health supported the research. Pecker reported receiving research funding from Alexion, Novartis, and Aummune and consulting for Novo Nordisk. Other authors reported no disclosures. Su and Mishkin reported no disclosures.
A version of this article appeared on Medscape.com.
FROM ASH 2024
Rilzabrutinib Shines in Phase 3 Trial of Tough-to-Treat Immune Thrombocytopenia
In the LUNA 3 trial, treatment with rilzabrutinib (Sanofi) led to rapid and durable platelet responses, reduced bleeding and need for rescue therapy, and improved health-related quality of life in patients with persistent or chronic immune thrombocytopenia.
Notably, rilzabrutinib also “significantly improved fatigue, even among patients who did not have a significant platelet count rise,” said David J. Kuter, MD, DPhil, director of clinical hematology, Massachusetts General Hospital, Boston, who reported the findings during a press briefing at the American Society of Hematology (ASH) 2024 Annual Meeting.
Briefing moderator Charles Abrams, MD, University of Pennsylvania, Philadelphia, noted that LUNA 3 enrolled a “remarkably tough group of patients, really the hardest of the hard” and showed that rilzabrutinib was “well-tolerated and caused an increase in platelet counts.”
The study, Abrams added, demonstrates “significant progress” in treatment of a disease that has historically been viewed as “benign,” which is “good for our patients.”
Immune thrombocytopenia is a relatively rare autoimmune disease that affects 10 to 23 patients per 100,000 in the United States. For those with the condition, the body’s immune system attacks platelets, causing platelet counts to drop below 100,000/μL of blood. The disease leads to increased bleeding risk and thrombosis, impaired clotting and health-related quality of life, as well as greater fatigue.
“People living with immune thrombocytopenia who cannot tolerate or do not respond to medications aimed at raising platelet counts are at risk of uncontrolled bleeding and often endure side effects from steroids and other available therapies,” Kuter noted in a Sanofi news release.
Rilzabrutinib, which received fast-track designation in November 2020 from the US Food and Drug Administration to treat immune thrombocytopenia, is currently under regulatory review and has a target action date of August 29, 2025.
In the LUNA 3 study, adults with persistent or chronic immune thrombocytopenia and severely low platelet counts (median, 15,000/μL) received oral rilzabrutinib 400 mg twice a day (133 patients) or placebo (69 patients) for up to 24 weeks during a blinded treatment period, followed by a 28-week open-label period.
Platelet response — defined as counts at or above 50,000/μL or counts between 30,000/μL and 50,000/μL but doubled from baseline — was achieved in nearly two thirds of patients taking rilzabrutinib compared with almost one third of patients taking placebo at week 13.
The primary endpoint was durable platelet response, defined as the proportion of patients able to achieve platelet counts at or above 50,000/μL for at least eight out of the last 12 weeks of the 24-week blinded period, without the need for rescue therapy.
No patient taking placebo met this endpoint, compared with 23% of patients taking rilzabrutinib (P < .0001).
For the combined double-blind and open-label periods, a durable response was achieved in 29% of the 133 patients randomized to rilzabrutinib and 25% of the 193 patients receiving the drug in the open-label period at the data cutoff.
Rilzabrutinib also led to significant improvements in bleeding (based on the Immune Thrombocytopenic Purpura Bleeding Score), with a mean change from baseline at week 25 of –0.04 with rilzabrutinib versus 0.05 with placebo (P = .0006).
Patients on rilzabrutinib were three times more likely to achieve a platelet response than their peers on placebo (hazard ratio, 3.1; P < .0001), with a median time to first platelet response of 36 days (vs median not achieved by patients on placebo). Among patients randomized to rilzabrutinib who achieved a response, the median time to response was 15 days.
Compared with placebo, rilzabrutinib significantly reduced the need for rescue therapy by 52% (P = .0007).
Rilzabrutinib was also associated with significant and sustained improvement in physical fatigue (based on the Immune Thrombocytopenic Purpura Patient Assessment Questionnaire [ITP-PAQ] Item 10 score).
“To our surprise, those patients who got active therapy but did not have a durable response still had an improvement in their fatigue levels and that suggests rilzabrutinib may affect fatigue or have anti-inflammatory properties since BTK inhibition has many different elements to it,” Kuter said during the briefing.
The most common treatment-related adverse events with rilzabrutinib versus placebo were mild to moderate (grade 1/2) diarrhea (23% vs 4%), nausea (17% vs 6%), headache (8% vs 1%), and abdominal pain (6% vs 1%). Rates of grade 2 or higher gastrointestinal adverse events were comparable between groups: 6% with rilzabrutinib versus 4% with placebo. In the rilzabrutinib group, one patient who had numerous risk factors had a treatment-related grade 3 peripheral embolism and one patient died due to pneumonia unrelated to treatment.
“I’m encouraged by the robust therapeutic effects I’ve seen in patients of the LUNA 3 study across all aspects of the disease, including clinically meaningful and sustained improvements in platelet count, quality of life metrics, reduction in bleeding, and a favorable safety profile,” Kuter said in the Sanofi news release.
The LUNA 3 study was funded by Sanofi. Kuter has disclosed various relationships with Sanofi and other pharmaceutical companies.
A version of this article first appeared on Medscape.com.
In the LUNA 3 trial, treatment with rilzabrutinib (Sanofi) led to rapid and durable platelet responses, reduced bleeding and need for rescue therapy, and improved health-related quality of life in patients with persistent or chronic immune thrombocytopenia.
Notably, rilzabrutinib also “significantly improved fatigue, even among patients who did not have a significant platelet count rise,” said David J. Kuter, MD, DPhil, director of clinical hematology, Massachusetts General Hospital, Boston, who reported the findings during a press briefing at the American Society of Hematology (ASH) 2024 Annual Meeting.
Briefing moderator Charles Abrams, MD, University of Pennsylvania, Philadelphia, noted that LUNA 3 enrolled a “remarkably tough group of patients, really the hardest of the hard” and showed that rilzabrutinib was “well-tolerated and caused an increase in platelet counts.”
The study, Abrams added, demonstrates “significant progress” in treatment of a disease that has historically been viewed as “benign,” which is “good for our patients.”
Immune thrombocytopenia is a relatively rare autoimmune disease that affects 10 to 23 patients per 100,000 in the United States. For those with the condition, the body’s immune system attacks platelets, causing platelet counts to drop below 100,000/μL of blood. The disease leads to increased bleeding risk and thrombosis, impaired clotting and health-related quality of life, as well as greater fatigue.
“People living with immune thrombocytopenia who cannot tolerate or do not respond to medications aimed at raising platelet counts are at risk of uncontrolled bleeding and often endure side effects from steroids and other available therapies,” Kuter noted in a Sanofi news release.
Rilzabrutinib, which received fast-track designation in November 2020 from the US Food and Drug Administration to treat immune thrombocytopenia, is currently under regulatory review and has a target action date of August 29, 2025.
In the LUNA 3 study, adults with persistent or chronic immune thrombocytopenia and severely low platelet counts (median, 15,000/μL) received oral rilzabrutinib 400 mg twice a day (133 patients) or placebo (69 patients) for up to 24 weeks during a blinded treatment period, followed by a 28-week open-label period.
Platelet response — defined as counts at or above 50,000/μL or counts between 30,000/μL and 50,000/μL but doubled from baseline — was achieved in nearly two thirds of patients taking rilzabrutinib compared with almost one third of patients taking placebo at week 13.
The primary endpoint was durable platelet response, defined as the proportion of patients able to achieve platelet counts at or above 50,000/μL for at least eight out of the last 12 weeks of the 24-week blinded period, without the need for rescue therapy.
No patient taking placebo met this endpoint, compared with 23% of patients taking rilzabrutinib (P < .0001).
For the combined double-blind and open-label periods, a durable response was achieved in 29% of the 133 patients randomized to rilzabrutinib and 25% of the 193 patients receiving the drug in the open-label period at the data cutoff.
Rilzabrutinib also led to significant improvements in bleeding (based on the Immune Thrombocytopenic Purpura Bleeding Score), with a mean change from baseline at week 25 of –0.04 with rilzabrutinib versus 0.05 with placebo (P = .0006).
Patients on rilzabrutinib were three times more likely to achieve a platelet response than their peers on placebo (hazard ratio, 3.1; P < .0001), with a median time to first platelet response of 36 days (vs median not achieved by patients on placebo). Among patients randomized to rilzabrutinib who achieved a response, the median time to response was 15 days.
Compared with placebo, rilzabrutinib significantly reduced the need for rescue therapy by 52% (P = .0007).
Rilzabrutinib was also associated with significant and sustained improvement in physical fatigue (based on the Immune Thrombocytopenic Purpura Patient Assessment Questionnaire [ITP-PAQ] Item 10 score).
“To our surprise, those patients who got active therapy but did not have a durable response still had an improvement in their fatigue levels and that suggests rilzabrutinib may affect fatigue or have anti-inflammatory properties since BTK inhibition has many different elements to it,” Kuter said during the briefing.
The most common treatment-related adverse events with rilzabrutinib versus placebo were mild to moderate (grade 1/2) diarrhea (23% vs 4%), nausea (17% vs 6%), headache (8% vs 1%), and abdominal pain (6% vs 1%). Rates of grade 2 or higher gastrointestinal adverse events were comparable between groups: 6% with rilzabrutinib versus 4% with placebo. In the rilzabrutinib group, one patient who had numerous risk factors had a treatment-related grade 3 peripheral embolism and one patient died due to pneumonia unrelated to treatment.
“I’m encouraged by the robust therapeutic effects I’ve seen in patients of the LUNA 3 study across all aspects of the disease, including clinically meaningful and sustained improvements in platelet count, quality of life metrics, reduction in bleeding, and a favorable safety profile,” Kuter said in the Sanofi news release.
The LUNA 3 study was funded by Sanofi. Kuter has disclosed various relationships with Sanofi and other pharmaceutical companies.
A version of this article first appeared on Medscape.com.
In the LUNA 3 trial, treatment with rilzabrutinib (Sanofi) led to rapid and durable platelet responses, reduced bleeding and need for rescue therapy, and improved health-related quality of life in patients with persistent or chronic immune thrombocytopenia.
Notably, rilzabrutinib also “significantly improved fatigue, even among patients who did not have a significant platelet count rise,” said David J. Kuter, MD, DPhil, director of clinical hematology, Massachusetts General Hospital, Boston, who reported the findings during a press briefing at the American Society of Hematology (ASH) 2024 Annual Meeting.
Briefing moderator Charles Abrams, MD, University of Pennsylvania, Philadelphia, noted that LUNA 3 enrolled a “remarkably tough group of patients, really the hardest of the hard” and showed that rilzabrutinib was “well-tolerated and caused an increase in platelet counts.”
The study, Abrams added, demonstrates “significant progress” in treatment of a disease that has historically been viewed as “benign,” which is “good for our patients.”
Immune thrombocytopenia is a relatively rare autoimmune disease that affects 10 to 23 patients per 100,000 in the United States. For those with the condition, the body’s immune system attacks platelets, causing platelet counts to drop below 100,000/μL of blood. The disease leads to increased bleeding risk and thrombosis, impaired clotting and health-related quality of life, as well as greater fatigue.
“People living with immune thrombocytopenia who cannot tolerate or do not respond to medications aimed at raising platelet counts are at risk of uncontrolled bleeding and often endure side effects from steroids and other available therapies,” Kuter noted in a Sanofi news release.
Rilzabrutinib, which received fast-track designation in November 2020 from the US Food and Drug Administration to treat immune thrombocytopenia, is currently under regulatory review and has a target action date of August 29, 2025.
In the LUNA 3 study, adults with persistent or chronic immune thrombocytopenia and severely low platelet counts (median, 15,000/μL) received oral rilzabrutinib 400 mg twice a day (133 patients) or placebo (69 patients) for up to 24 weeks during a blinded treatment period, followed by a 28-week open-label period.
Platelet response — defined as counts at or above 50,000/μL or counts between 30,000/μL and 50,000/μL but doubled from baseline — was achieved in nearly two thirds of patients taking rilzabrutinib compared with almost one third of patients taking placebo at week 13.
The primary endpoint was durable platelet response, defined as the proportion of patients able to achieve platelet counts at or above 50,000/μL for at least eight out of the last 12 weeks of the 24-week blinded period, without the need for rescue therapy.
No patient taking placebo met this endpoint, compared with 23% of patients taking rilzabrutinib (P < .0001).
For the combined double-blind and open-label periods, a durable response was achieved in 29% of the 133 patients randomized to rilzabrutinib and 25% of the 193 patients receiving the drug in the open-label period at the data cutoff.
Rilzabrutinib also led to significant improvements in bleeding (based on the Immune Thrombocytopenic Purpura Bleeding Score), with a mean change from baseline at week 25 of –0.04 with rilzabrutinib versus 0.05 with placebo (P = .0006).
Patients on rilzabrutinib were three times more likely to achieve a platelet response than their peers on placebo (hazard ratio, 3.1; P < .0001), with a median time to first platelet response of 36 days (vs median not achieved by patients on placebo). Among patients randomized to rilzabrutinib who achieved a response, the median time to response was 15 days.
Compared with placebo, rilzabrutinib significantly reduced the need for rescue therapy by 52% (P = .0007).
Rilzabrutinib was also associated with significant and sustained improvement in physical fatigue (based on the Immune Thrombocytopenic Purpura Patient Assessment Questionnaire [ITP-PAQ] Item 10 score).
“To our surprise, those patients who got active therapy but did not have a durable response still had an improvement in their fatigue levels and that suggests rilzabrutinib may affect fatigue or have anti-inflammatory properties since BTK inhibition has many different elements to it,” Kuter said during the briefing.
The most common treatment-related adverse events with rilzabrutinib versus placebo were mild to moderate (grade 1/2) diarrhea (23% vs 4%), nausea (17% vs 6%), headache (8% vs 1%), and abdominal pain (6% vs 1%). Rates of grade 2 or higher gastrointestinal adverse events were comparable between groups: 6% with rilzabrutinib versus 4% with placebo. In the rilzabrutinib group, one patient who had numerous risk factors had a treatment-related grade 3 peripheral embolism and one patient died due to pneumonia unrelated to treatment.
“I’m encouraged by the robust therapeutic effects I’ve seen in patients of the LUNA 3 study across all aspects of the disease, including clinically meaningful and sustained improvements in platelet count, quality of life metrics, reduction in bleeding, and a favorable safety profile,” Kuter said in the Sanofi news release.
The LUNA 3 study was funded by Sanofi. Kuter has disclosed various relationships with Sanofi and other pharmaceutical companies.
A version of this article first appeared on Medscape.com.
FROM ASH 2024
ASH 2024: New Leukemia Txs, Fewer Blood Clots With GLP-1 Rxs
Children’s Disorders: Major Progress in B-Cell Acute Lymphoblastic Leukemia (B-ALL), Immune Thrombocytopenic Purpura (ITP)
While B-ALL is the most common childhood cancer and one of the most treatable, some patients face grim outcomes after they relapse following chemotherapy, said Cynthia E. Dunbar, MD, chief of the Translational Stem Cell Biology Branch at the National Heart, Lung, and Blood Institute.
A new study reports that adding the targeted cancer drug blinatumomab (Blincyto) to chemotherapy boosted disease-free survival in standard-risk pediatric patients. “They definitively demonstrate a benefit with the addition of this immunotherapeutic drug, achieving 97% disease-free survival at 3 years on the blinatumomab plus chemotherapy arm compared to 90% for the control arm with standard therapies alone,” Dunbar said. “This trial will establish the addition of blinatumomab for childhood B-ALL as standard of care.”
A reporter asked Dunbar about the risk for severe immune activation syndrome. “These immune cell engagers can result in cytokine release syndrome and other severe immune activation consequences,” she said. “However, it appears that children seem to be less susceptible to those, at least in terms of severity, than adults. In this study, the complications that occurred didn’t result in mortality and were easily treatable. So that was not a major drawback to the addition of this drug.”
The blinatumomab study is sponsored by Children’s Oncology Group.
In ITP, thrombopoietin (TPO) agonists such as eltrombopag (Promacta) are a mainstay of second- or third-line treatment in children and adults with severe cases, Dunbar said. “However, TPO agonists are generally only given after months to years of failures of corticosteroids, IVIG [intravenous immunoglobulin], or splenectomy.”
In the phase 3, randomized, controlled PINES trial, researchers explored whether the drug could improve outcomes in children with untreated or very recent-onset severe ITP vs standard of care.
“The children treated with eltrombopag had double the response rate with a much lower need for rescue therapies,” Dunbar said. The percentage of patients who received rescue therapy was 19% in the eltrombopag arm (15/78) vs 46% in the control arm (18/39, P = .002).
“Given the potential short- and long-term consequences of corticosteroids and other standard treatments in children, this study is encouraging and will likely result in a change in the standard of care for pediatric ITP,” Dunbar said.
The eltrombopag study is sponsored by the ITP Consortium of North America and funded by Novartis.
Fewer Blood Clots: Another Big Benefit for Weight Loss Drugs?
Mikkael A. Sekeres, MD, MS, of the Sylvester Comprehensive Cancer Center at the University of Miami in Florida, highlighted an analysis of whether GLP-1 receptor agonists, initially approved as type 2 diabetes treatments, affect the risk for VTE.
Researchers tracked patients with type 2 diabetes — 366,369 who received the drugs and 290,219 who took dipeptidyl peptidase 4 inhibitors. The patients who took GLP-1 agonists “had lower rates of venous thromboembolic events after 1 year,” Sekeres said. “The risk reduction was actually pretty substantial.”
In these patients, the risk for VTE was 18% lower (hazard ratio [HR], 0.82; 95% CI, 0.77-0.88), and there were 22% and 15% reductions in pulmonary embolisms and deep venous thrombosis, respectively (HR, 0.78; 95% CI, 0.71-0.86 and HR, 0.85; 95% CI, 0.79-0.92).
Drug Regimen Improves Outcomes in Chronic Lymphocytic Leukemia (CLL)
An interim analysis of an open-label, randomized, phase 3 trial of patients with untreated CLL “demonstrated superior progression-free survival with acalabrutinib and venetoclax versus what we consider more classic chemotherapy of fludarabine, Cytoxan [cyclophosphamide], and rituximab or bendamustine and rituximab,” Sekeres said. “Similar findings were seen with acalabrutinib, venetoclax, and obinutuzumab vs that classic chemotherapy.”
Overall response rates were 93% for both the acalabrutinib/venetoclax regimens vs 75% for bendamustine/rituximab, Sekeres noted, and overall survival was higher for acalabrutinib/venetoclax vs the two classic chemotherapy regimens (HR, 0.33; P < .0001).
However, Sekeres questioned the value of comparing acalabrutinib/venetoclax with classical chemotherapy regimens. “A lot of times we have a lot of new, really good, really effective therapy to offer to patients that isn’t as toxic as previous chemotherapy.”
In contrast, fludarabine, cyclophosphamide, and rituximab are “your grandmother’s or your grandfather’s chemotherapy. It’s pretty toxic stuff,” he said.
Sekeres said it would have been better to compare acalabrutinib/venetoclax with a Bruton tyrosine kinase inhibitor–based regimen.
The German CLL Study Group is listed as the trial’s sponsor, and AstraZeneca is a collaborator. Dunbar disclosed research funding from Novartis. Sekeres had no relevant disclosures.
A version of this article appeared on Medscape.com.
Children’s Disorders: Major Progress in B-Cell Acute Lymphoblastic Leukemia (B-ALL), Immune Thrombocytopenic Purpura (ITP)
While B-ALL is the most common childhood cancer and one of the most treatable, some patients face grim outcomes after they relapse following chemotherapy, said Cynthia E. Dunbar, MD, chief of the Translational Stem Cell Biology Branch at the National Heart, Lung, and Blood Institute.
A new study reports that adding the targeted cancer drug blinatumomab (Blincyto) to chemotherapy boosted disease-free survival in standard-risk pediatric patients. “They definitively demonstrate a benefit with the addition of this immunotherapeutic drug, achieving 97% disease-free survival at 3 years on the blinatumomab plus chemotherapy arm compared to 90% for the control arm with standard therapies alone,” Dunbar said. “This trial will establish the addition of blinatumomab for childhood B-ALL as standard of care.”
A reporter asked Dunbar about the risk for severe immune activation syndrome. “These immune cell engagers can result in cytokine release syndrome and other severe immune activation consequences,” she said. “However, it appears that children seem to be less susceptible to those, at least in terms of severity, than adults. In this study, the complications that occurred didn’t result in mortality and were easily treatable. So that was not a major drawback to the addition of this drug.”
The blinatumomab study is sponsored by Children’s Oncology Group.
In ITP, thrombopoietin (TPO) agonists such as eltrombopag (Promacta) are a mainstay of second- or third-line treatment in children and adults with severe cases, Dunbar said. “However, TPO agonists are generally only given after months to years of failures of corticosteroids, IVIG [intravenous immunoglobulin], or splenectomy.”
In the phase 3, randomized, controlled PINES trial, researchers explored whether the drug could improve outcomes in children with untreated or very recent-onset severe ITP vs standard of care.
“The children treated with eltrombopag had double the response rate with a much lower need for rescue therapies,” Dunbar said. The percentage of patients who received rescue therapy was 19% in the eltrombopag arm (15/78) vs 46% in the control arm (18/39, P = .002).
“Given the potential short- and long-term consequences of corticosteroids and other standard treatments in children, this study is encouraging and will likely result in a change in the standard of care for pediatric ITP,” Dunbar said.
The eltrombopag study is sponsored by the ITP Consortium of North America and funded by Novartis.
Fewer Blood Clots: Another Big Benefit for Weight Loss Drugs?
Mikkael A. Sekeres, MD, MS, of the Sylvester Comprehensive Cancer Center at the University of Miami in Florida, highlighted an analysis of whether GLP-1 receptor agonists, initially approved as type 2 diabetes treatments, affect the risk for VTE.
Researchers tracked patients with type 2 diabetes — 366,369 who received the drugs and 290,219 who took dipeptidyl peptidase 4 inhibitors. The patients who took GLP-1 agonists “had lower rates of venous thromboembolic events after 1 year,” Sekeres said. “The risk reduction was actually pretty substantial.”
In these patients, the risk for VTE was 18% lower (hazard ratio [HR], 0.82; 95% CI, 0.77-0.88), and there were 22% and 15% reductions in pulmonary embolisms and deep venous thrombosis, respectively (HR, 0.78; 95% CI, 0.71-0.86 and HR, 0.85; 95% CI, 0.79-0.92).
Drug Regimen Improves Outcomes in Chronic Lymphocytic Leukemia (CLL)
An interim analysis of an open-label, randomized, phase 3 trial of patients with untreated CLL “demonstrated superior progression-free survival with acalabrutinib and venetoclax versus what we consider more classic chemotherapy of fludarabine, Cytoxan [cyclophosphamide], and rituximab or bendamustine and rituximab,” Sekeres said. “Similar findings were seen with acalabrutinib, venetoclax, and obinutuzumab vs that classic chemotherapy.”
Overall response rates were 93% for both the acalabrutinib/venetoclax regimens vs 75% for bendamustine/rituximab, Sekeres noted, and overall survival was higher for acalabrutinib/venetoclax vs the two classic chemotherapy regimens (HR, 0.33; P < .0001).
However, Sekeres questioned the value of comparing acalabrutinib/venetoclax with classical chemotherapy regimens. “A lot of times we have a lot of new, really good, really effective therapy to offer to patients that isn’t as toxic as previous chemotherapy.”
In contrast, fludarabine, cyclophosphamide, and rituximab are “your grandmother’s or your grandfather’s chemotherapy. It’s pretty toxic stuff,” he said.
Sekeres said it would have been better to compare acalabrutinib/venetoclax with a Bruton tyrosine kinase inhibitor–based regimen.
The German CLL Study Group is listed as the trial’s sponsor, and AstraZeneca is a collaborator. Dunbar disclosed research funding from Novartis. Sekeres had no relevant disclosures.
A version of this article appeared on Medscape.com.
Children’s Disorders: Major Progress in B-Cell Acute Lymphoblastic Leukemia (B-ALL), Immune Thrombocytopenic Purpura (ITP)
While B-ALL is the most common childhood cancer and one of the most treatable, some patients face grim outcomes after they relapse following chemotherapy, said Cynthia E. Dunbar, MD, chief of the Translational Stem Cell Biology Branch at the National Heart, Lung, and Blood Institute.
A new study reports that adding the targeted cancer drug blinatumomab (Blincyto) to chemotherapy boosted disease-free survival in standard-risk pediatric patients. “They definitively demonstrate a benefit with the addition of this immunotherapeutic drug, achieving 97% disease-free survival at 3 years on the blinatumomab plus chemotherapy arm compared to 90% for the control arm with standard therapies alone,” Dunbar said. “This trial will establish the addition of blinatumomab for childhood B-ALL as standard of care.”
A reporter asked Dunbar about the risk for severe immune activation syndrome. “These immune cell engagers can result in cytokine release syndrome and other severe immune activation consequences,” she said. “However, it appears that children seem to be less susceptible to those, at least in terms of severity, than adults. In this study, the complications that occurred didn’t result in mortality and were easily treatable. So that was not a major drawback to the addition of this drug.”
The blinatumomab study is sponsored by Children’s Oncology Group.
In ITP, thrombopoietin (TPO) agonists such as eltrombopag (Promacta) are a mainstay of second- or third-line treatment in children and adults with severe cases, Dunbar said. “However, TPO agonists are generally only given after months to years of failures of corticosteroids, IVIG [intravenous immunoglobulin], or splenectomy.”
In the phase 3, randomized, controlled PINES trial, researchers explored whether the drug could improve outcomes in children with untreated or very recent-onset severe ITP vs standard of care.
“The children treated with eltrombopag had double the response rate with a much lower need for rescue therapies,” Dunbar said. The percentage of patients who received rescue therapy was 19% in the eltrombopag arm (15/78) vs 46% in the control arm (18/39, P = .002).
“Given the potential short- and long-term consequences of corticosteroids and other standard treatments in children, this study is encouraging and will likely result in a change in the standard of care for pediatric ITP,” Dunbar said.
The eltrombopag study is sponsored by the ITP Consortium of North America and funded by Novartis.
Fewer Blood Clots: Another Big Benefit for Weight Loss Drugs?
Mikkael A. Sekeres, MD, MS, of the Sylvester Comprehensive Cancer Center at the University of Miami in Florida, highlighted an analysis of whether GLP-1 receptor agonists, initially approved as type 2 diabetes treatments, affect the risk for VTE.
Researchers tracked patients with type 2 diabetes — 366,369 who received the drugs and 290,219 who took dipeptidyl peptidase 4 inhibitors. The patients who took GLP-1 agonists “had lower rates of venous thromboembolic events after 1 year,” Sekeres said. “The risk reduction was actually pretty substantial.”
In these patients, the risk for VTE was 18% lower (hazard ratio [HR], 0.82; 95% CI, 0.77-0.88), and there were 22% and 15% reductions in pulmonary embolisms and deep venous thrombosis, respectively (HR, 0.78; 95% CI, 0.71-0.86 and HR, 0.85; 95% CI, 0.79-0.92).
Drug Regimen Improves Outcomes in Chronic Lymphocytic Leukemia (CLL)
An interim analysis of an open-label, randomized, phase 3 trial of patients with untreated CLL “demonstrated superior progression-free survival with acalabrutinib and venetoclax versus what we consider more classic chemotherapy of fludarabine, Cytoxan [cyclophosphamide], and rituximab or bendamustine and rituximab,” Sekeres said. “Similar findings were seen with acalabrutinib, venetoclax, and obinutuzumab vs that classic chemotherapy.”
Overall response rates were 93% for both the acalabrutinib/venetoclax regimens vs 75% for bendamustine/rituximab, Sekeres noted, and overall survival was higher for acalabrutinib/venetoclax vs the two classic chemotherapy regimens (HR, 0.33; P < .0001).
However, Sekeres questioned the value of comparing acalabrutinib/venetoclax with classical chemotherapy regimens. “A lot of times we have a lot of new, really good, really effective therapy to offer to patients that isn’t as toxic as previous chemotherapy.”
In contrast, fludarabine, cyclophosphamide, and rituximab are “your grandmother’s or your grandfather’s chemotherapy. It’s pretty toxic stuff,” he said.
Sekeres said it would have been better to compare acalabrutinib/venetoclax with a Bruton tyrosine kinase inhibitor–based regimen.
The German CLL Study Group is listed as the trial’s sponsor, and AstraZeneca is a collaborator. Dunbar disclosed research funding from Novartis. Sekeres had no relevant disclosures.
A version of this article appeared on Medscape.com.
FROM ASH 2024
Blood Buddies: Can Mentorship Revive Classical Hematology?
But when it comes to turning people on to the idea of a career in classical hematology (CH), there may be no more powerful influence than a mentor who loves their job. That’s why the field is focusing so much on supporting mentors and mentees amid a stark shortage of classical hematologists.
“Mentorship is key for maintaining trainee interest in the field and for providing role models for career growth,” said Rakhi P. Naik, MD, MHS, associate professor of medicine and director of the Hematology Fellowship Track at Johns Hopkins University, Baltimore, Maryland, in an interview. “This collaboration is especially critical because there are so few trainees and so few mentors currently in the field.”
Now there’s new research backing up the power of mentorship, even when it’s only provided virtually, and a brand-new program aims to unite more mentors and mentees.
Here’s a closer look at mentor-focused efforts to attract medical students to CH.
How Severe Is the Shortage in CH?
Patients with conditions treated by classical hematologists are waiting months for appointments at many outpatient centers, with some being forced to wait 6 months or more, said Srikanth Nagalla, MD, chief of benign hematology at Miami Cancer Institute, Florida, in an interview.
The shortage is creating dire problems in the inpatient setting too, Nagalla said. “Serious blood disorders like heparin-induced thrombocytopenia, acute chest syndrome [a complication of sickle cell disease], and thrombotic thrombocytopenic purpura have to be diagnosed and treated in a timely manner. If not, the morbidity and mortality are really high.”
If classical hematologists aren’t available, he said, oncologists and others not trained in hematology will need to cover these patients.
Hematologist Ariela Marshall, MD, associate professor of medicine at the University of Minnesota in Minneapolis, noted in an interview that the CH shortage comes at a time when medical advances and an aging population are boosting the number of patients with noncancerous blood disorders. Older people are at greater risk for blood clots, she said. And lifespans for patients with bleeding and clotting disorders are rising thanks to effective new treatments.
“Because of our larger patient population in CH, we are going to need more classical hematologists to follow them for longer and longer periods of time,” she said.
There’s no sign yet that newly minted physicians will take up the slack in CH. A 2019 study found that just 4.6% of 626 of hematology/oncology fellows said they planned to go into CH, also known as benign hematology, vs 67.1% who expected to treat patients with solid tumors, blood cancer, or both. The rest, 24.6%, planned to work in CH plus the two oncology fields.
Why Does a Shortage Exist?
“The reasons are complex, but one of the most important factors was the combining of the adult hematology and medical oncology training programs by the Accreditation Council for Graduate Medical Education in 1995,” Naik said. “After that time, the majority of fellowship training programs went from having separate programs for hematology and medical oncology to combining the training for the two specialties into one. Because most of these combined training programs resided within Cancer Centers, classical hematology training slowly became de-emphasized.”
As a result, fewer fellows ended up specializing in CH, she said.
The field of CH also appears to suffer from a less than enticing reputation. According to a 2019 study coauthored by Marshall, surveys of thousands of hematology/oncology fellows found that “hematology, particularly benign hematology, was viewed as having poorer income potential, research funding, job availability, and job security than oncology.”
Regarding pay, Marshall said the good news is that many classical hematologists work in academia, where it’s common for pay to be “equitable across hematology/oncology divisions and based more on academic rank and other factors rather than subspecialty within hematology oncology.”
However, she noted, “this may differ at institutions where hematology and oncology are different departments. For example, centers where oncology is its own department, and hematology is part of the department of medicine.”
As for job availability, Naik said that there’s plenty of demand. “In academics, it is clear that there are jobs available everywhere, but trainees are often worried about job prospects in private practice. While classical hematology jobs in private practice are not widely advertised, I can attest that there is no shortage of need,” she said. “Many private practices do not specifically advertise for classical hematologists because they assume that classical hematology experts are not available. But I assure you that every private practice my trainees have ever approached is always ecstatic to hire a classical hematologist.”
Why Are Mentors Important?
Mentorship is crucial to promoting the value of CH as a great career choice in a competitive environment, classical hematologists say. “We can motivate trainees by showing how the disease states themselves are so fascinating and how the treatments are showing great outcomes,” Nagalla said. “We can show positive results, how patient lives can be changed, and how well-respected across the system [we] are.”
As a selling point, classical hematologists like to emphasize that their field requires intensive detective work. “Let’s say a patient comes with anemia, which might have 15 different causes. You get some labs, and then you systemically rule in or rule out most of these on the differential diagnosis,” Nagalla said. “Then once you narrow it down, you get more labs. You keep going to the next step and next step, and so finally you come to a conclusion.”
As for therapy, Marshall said that “while for many cancers there are specific treatment recommendations for patients with a specific cancer type at a specific stage, there is not always a specific treatment recommendation (or a ‘right answer’) for our CH patients. Treatment planning depends strongly on a patient’s preferences, other medical conditions, and a discussion about risks [and] benefits of different treatment options such that two patients with the same condition may choose two different treatment options.”
Marshall also emphasizes to trainees that “CH is a broad field. Physicians and trainees are able to interact and collaborate with physicians in other specialties such as gastroenterology, cardiology, ob/gyn, and surgical specialties.”
Does Research Support Mentorship in CH?
The 2019 study that revealed just 4.6% of fellows planned to go into CH found that “fellows who planned to enter hematology-only careers were significantly more likely to report having clinical training and mentorship experiences in hematology throughout their training relative to fellows with oncology-only or combined hematology/oncology career plans.”
Now there are more data to support mentorships. For a study published in Blood Advances in September 2024, Zoya Qureshy, MD, an internal medicine chief resident at the University of California at San Diego, and colleagues evaluated a year-long external membership program implemented by the American Society of Hematology (ASH) Medical Educators Institute.
The program linked 35 US hematology/oncology fellows (80% female, 46% White, 35% Asian) who were interested in CH to 34 North American faculty members. The pairs were told to meet virtually once a month.
Of 30 mentees and 23 mentors surveyed, 94% and 85%, respectively, said their pairings were good matches. Two thirds of the mentees accepted faculty positions in CH after their mentorships.
“Our study showed that external mentorship in a virtual format is feasible,” Qureshy said in an interview. “Additionally, external mentorship provided benefits such as different perspectives and the opportunity for mentorship for those who may not have it in their field of interest at their home institution.”
Qureshy added that “one strength of our mentorship program was that mentoring pairs were meticulously assigned based on shared interests and background. Many participants cited this common ground as a reason why they thought their mentoring pair was a good match.”
There’s an important caveat: Most of the mentees weren’t new to CH. About 70% had previously worked with a mentor in the CH field, and 86% had previously conducted research in the field.
What’s Next for Mentorship in CH?
The ASH Hematology-Focused Fellowship Training Program Consortium aims to mint 50 new academic hematologists by 2030 through programs at 12 institutions. “Mentorship is an exciting aspect of the program since it allows classical hematology trainees to form a network of peers nationally and also provides access to mentors across institutions,” Naik said. “And as the workforce grows, there will be more and more role models for future trainees to look up to.”
Moving forward, she said, “we hope to inspire even more institutions to adopt hematology training tracks throughout the country.”
Meanwhile, ASH’s new Classical Hematology Advancement Mentorship is taking applications for its debut 2025 program through January 9, 2025. Trainees will meet monthly with mentors both virtually and in person. Applicants must have been in their first or second year of hematology/oncology fellowship training at accredited programs in the United States as of July 15, 2024.
Naik, Marshall, Nagalla, and Qureshy have no relevant disclosures.
A version of this article appeared on Medscape.com.
But when it comes to turning people on to the idea of a career in classical hematology (CH), there may be no more powerful influence than a mentor who loves their job. That’s why the field is focusing so much on supporting mentors and mentees amid a stark shortage of classical hematologists.
“Mentorship is key for maintaining trainee interest in the field and for providing role models for career growth,” said Rakhi P. Naik, MD, MHS, associate professor of medicine and director of the Hematology Fellowship Track at Johns Hopkins University, Baltimore, Maryland, in an interview. “This collaboration is especially critical because there are so few trainees and so few mentors currently in the field.”
Now there’s new research backing up the power of mentorship, even when it’s only provided virtually, and a brand-new program aims to unite more mentors and mentees.
Here’s a closer look at mentor-focused efforts to attract medical students to CH.
How Severe Is the Shortage in CH?
Patients with conditions treated by classical hematologists are waiting months for appointments at many outpatient centers, with some being forced to wait 6 months or more, said Srikanth Nagalla, MD, chief of benign hematology at Miami Cancer Institute, Florida, in an interview.
The shortage is creating dire problems in the inpatient setting too, Nagalla said. “Serious blood disorders like heparin-induced thrombocytopenia, acute chest syndrome [a complication of sickle cell disease], and thrombotic thrombocytopenic purpura have to be diagnosed and treated in a timely manner. If not, the morbidity and mortality are really high.”
If classical hematologists aren’t available, he said, oncologists and others not trained in hematology will need to cover these patients.
Hematologist Ariela Marshall, MD, associate professor of medicine at the University of Minnesota in Minneapolis, noted in an interview that the CH shortage comes at a time when medical advances and an aging population are boosting the number of patients with noncancerous blood disorders. Older people are at greater risk for blood clots, she said. And lifespans for patients with bleeding and clotting disorders are rising thanks to effective new treatments.
“Because of our larger patient population in CH, we are going to need more classical hematologists to follow them for longer and longer periods of time,” she said.
There’s no sign yet that newly minted physicians will take up the slack in CH. A 2019 study found that just 4.6% of 626 of hematology/oncology fellows said they planned to go into CH, also known as benign hematology, vs 67.1% who expected to treat patients with solid tumors, blood cancer, or both. The rest, 24.6%, planned to work in CH plus the two oncology fields.
Why Does a Shortage Exist?
“The reasons are complex, but one of the most important factors was the combining of the adult hematology and medical oncology training programs by the Accreditation Council for Graduate Medical Education in 1995,” Naik said. “After that time, the majority of fellowship training programs went from having separate programs for hematology and medical oncology to combining the training for the two specialties into one. Because most of these combined training programs resided within Cancer Centers, classical hematology training slowly became de-emphasized.”
As a result, fewer fellows ended up specializing in CH, she said.
The field of CH also appears to suffer from a less than enticing reputation. According to a 2019 study coauthored by Marshall, surveys of thousands of hematology/oncology fellows found that “hematology, particularly benign hematology, was viewed as having poorer income potential, research funding, job availability, and job security than oncology.”
Regarding pay, Marshall said the good news is that many classical hematologists work in academia, where it’s common for pay to be “equitable across hematology/oncology divisions and based more on academic rank and other factors rather than subspecialty within hematology oncology.”
However, she noted, “this may differ at institutions where hematology and oncology are different departments. For example, centers where oncology is its own department, and hematology is part of the department of medicine.”
As for job availability, Naik said that there’s plenty of demand. “In academics, it is clear that there are jobs available everywhere, but trainees are often worried about job prospects in private practice. While classical hematology jobs in private practice are not widely advertised, I can attest that there is no shortage of need,” she said. “Many private practices do not specifically advertise for classical hematologists because they assume that classical hematology experts are not available. But I assure you that every private practice my trainees have ever approached is always ecstatic to hire a classical hematologist.”
Why Are Mentors Important?
Mentorship is crucial to promoting the value of CH as a great career choice in a competitive environment, classical hematologists say. “We can motivate trainees by showing how the disease states themselves are so fascinating and how the treatments are showing great outcomes,” Nagalla said. “We can show positive results, how patient lives can be changed, and how well-respected across the system [we] are.”
As a selling point, classical hematologists like to emphasize that their field requires intensive detective work. “Let’s say a patient comes with anemia, which might have 15 different causes. You get some labs, and then you systemically rule in or rule out most of these on the differential diagnosis,” Nagalla said. “Then once you narrow it down, you get more labs. You keep going to the next step and next step, and so finally you come to a conclusion.”
As for therapy, Marshall said that “while for many cancers there are specific treatment recommendations for patients with a specific cancer type at a specific stage, there is not always a specific treatment recommendation (or a ‘right answer’) for our CH patients. Treatment planning depends strongly on a patient’s preferences, other medical conditions, and a discussion about risks [and] benefits of different treatment options such that two patients with the same condition may choose two different treatment options.”
Marshall also emphasizes to trainees that “CH is a broad field. Physicians and trainees are able to interact and collaborate with physicians in other specialties such as gastroenterology, cardiology, ob/gyn, and surgical specialties.”
Does Research Support Mentorship in CH?
The 2019 study that revealed just 4.6% of fellows planned to go into CH found that “fellows who planned to enter hematology-only careers were significantly more likely to report having clinical training and mentorship experiences in hematology throughout their training relative to fellows with oncology-only or combined hematology/oncology career plans.”
Now there are more data to support mentorships. For a study published in Blood Advances in September 2024, Zoya Qureshy, MD, an internal medicine chief resident at the University of California at San Diego, and colleagues evaluated a year-long external membership program implemented by the American Society of Hematology (ASH) Medical Educators Institute.
The program linked 35 US hematology/oncology fellows (80% female, 46% White, 35% Asian) who were interested in CH to 34 North American faculty members. The pairs were told to meet virtually once a month.
Of 30 mentees and 23 mentors surveyed, 94% and 85%, respectively, said their pairings were good matches. Two thirds of the mentees accepted faculty positions in CH after their mentorships.
“Our study showed that external mentorship in a virtual format is feasible,” Qureshy said in an interview. “Additionally, external mentorship provided benefits such as different perspectives and the opportunity for mentorship for those who may not have it in their field of interest at their home institution.”
Qureshy added that “one strength of our mentorship program was that mentoring pairs were meticulously assigned based on shared interests and background. Many participants cited this common ground as a reason why they thought their mentoring pair was a good match.”
There’s an important caveat: Most of the mentees weren’t new to CH. About 70% had previously worked with a mentor in the CH field, and 86% had previously conducted research in the field.
What’s Next for Mentorship in CH?
The ASH Hematology-Focused Fellowship Training Program Consortium aims to mint 50 new academic hematologists by 2030 through programs at 12 institutions. “Mentorship is an exciting aspect of the program since it allows classical hematology trainees to form a network of peers nationally and also provides access to mentors across institutions,” Naik said. “And as the workforce grows, there will be more and more role models for future trainees to look up to.”
Moving forward, she said, “we hope to inspire even more institutions to adopt hematology training tracks throughout the country.”
Meanwhile, ASH’s new Classical Hematology Advancement Mentorship is taking applications for its debut 2025 program through January 9, 2025. Trainees will meet monthly with mentors both virtually and in person. Applicants must have been in their first or second year of hematology/oncology fellowship training at accredited programs in the United States as of July 15, 2024.
Naik, Marshall, Nagalla, and Qureshy have no relevant disclosures.
A version of this article appeared on Medscape.com.
But when it comes to turning people on to the idea of a career in classical hematology (CH), there may be no more powerful influence than a mentor who loves their job. That’s why the field is focusing so much on supporting mentors and mentees amid a stark shortage of classical hematologists.
“Mentorship is key for maintaining trainee interest in the field and for providing role models for career growth,” said Rakhi P. Naik, MD, MHS, associate professor of medicine and director of the Hematology Fellowship Track at Johns Hopkins University, Baltimore, Maryland, in an interview. “This collaboration is especially critical because there are so few trainees and so few mentors currently in the field.”
Now there’s new research backing up the power of mentorship, even when it’s only provided virtually, and a brand-new program aims to unite more mentors and mentees.
Here’s a closer look at mentor-focused efforts to attract medical students to CH.
How Severe Is the Shortage in CH?
Patients with conditions treated by classical hematologists are waiting months for appointments at many outpatient centers, with some being forced to wait 6 months or more, said Srikanth Nagalla, MD, chief of benign hematology at Miami Cancer Institute, Florida, in an interview.
The shortage is creating dire problems in the inpatient setting too, Nagalla said. “Serious blood disorders like heparin-induced thrombocytopenia, acute chest syndrome [a complication of sickle cell disease], and thrombotic thrombocytopenic purpura have to be diagnosed and treated in a timely manner. If not, the morbidity and mortality are really high.”
If classical hematologists aren’t available, he said, oncologists and others not trained in hematology will need to cover these patients.
Hematologist Ariela Marshall, MD, associate professor of medicine at the University of Minnesota in Minneapolis, noted in an interview that the CH shortage comes at a time when medical advances and an aging population are boosting the number of patients with noncancerous blood disorders. Older people are at greater risk for blood clots, she said. And lifespans for patients with bleeding and clotting disorders are rising thanks to effective new treatments.
“Because of our larger patient population in CH, we are going to need more classical hematologists to follow them for longer and longer periods of time,” she said.
There’s no sign yet that newly minted physicians will take up the slack in CH. A 2019 study found that just 4.6% of 626 of hematology/oncology fellows said they planned to go into CH, also known as benign hematology, vs 67.1% who expected to treat patients with solid tumors, blood cancer, or both. The rest, 24.6%, planned to work in CH plus the two oncology fields.
Why Does a Shortage Exist?
“The reasons are complex, but one of the most important factors was the combining of the adult hematology and medical oncology training programs by the Accreditation Council for Graduate Medical Education in 1995,” Naik said. “After that time, the majority of fellowship training programs went from having separate programs for hematology and medical oncology to combining the training for the two specialties into one. Because most of these combined training programs resided within Cancer Centers, classical hematology training slowly became de-emphasized.”
As a result, fewer fellows ended up specializing in CH, she said.
The field of CH also appears to suffer from a less than enticing reputation. According to a 2019 study coauthored by Marshall, surveys of thousands of hematology/oncology fellows found that “hematology, particularly benign hematology, was viewed as having poorer income potential, research funding, job availability, and job security than oncology.”
Regarding pay, Marshall said the good news is that many classical hematologists work in academia, where it’s common for pay to be “equitable across hematology/oncology divisions and based more on academic rank and other factors rather than subspecialty within hematology oncology.”
However, she noted, “this may differ at institutions where hematology and oncology are different departments. For example, centers where oncology is its own department, and hematology is part of the department of medicine.”
As for job availability, Naik said that there’s plenty of demand. “In academics, it is clear that there are jobs available everywhere, but trainees are often worried about job prospects in private practice. While classical hematology jobs in private practice are not widely advertised, I can attest that there is no shortage of need,” she said. “Many private practices do not specifically advertise for classical hematologists because they assume that classical hematology experts are not available. But I assure you that every private practice my trainees have ever approached is always ecstatic to hire a classical hematologist.”
Why Are Mentors Important?
Mentorship is crucial to promoting the value of CH as a great career choice in a competitive environment, classical hematologists say. “We can motivate trainees by showing how the disease states themselves are so fascinating and how the treatments are showing great outcomes,” Nagalla said. “We can show positive results, how patient lives can be changed, and how well-respected across the system [we] are.”
As a selling point, classical hematologists like to emphasize that their field requires intensive detective work. “Let’s say a patient comes with anemia, which might have 15 different causes. You get some labs, and then you systemically rule in or rule out most of these on the differential diagnosis,” Nagalla said. “Then once you narrow it down, you get more labs. You keep going to the next step and next step, and so finally you come to a conclusion.”
As for therapy, Marshall said that “while for many cancers there are specific treatment recommendations for patients with a specific cancer type at a specific stage, there is not always a specific treatment recommendation (or a ‘right answer’) for our CH patients. Treatment planning depends strongly on a patient’s preferences, other medical conditions, and a discussion about risks [and] benefits of different treatment options such that two patients with the same condition may choose two different treatment options.”
Marshall also emphasizes to trainees that “CH is a broad field. Physicians and trainees are able to interact and collaborate with physicians in other specialties such as gastroenterology, cardiology, ob/gyn, and surgical specialties.”
Does Research Support Mentorship in CH?
The 2019 study that revealed just 4.6% of fellows planned to go into CH found that “fellows who planned to enter hematology-only careers were significantly more likely to report having clinical training and mentorship experiences in hematology throughout their training relative to fellows with oncology-only or combined hematology/oncology career plans.”
Now there are more data to support mentorships. For a study published in Blood Advances in September 2024, Zoya Qureshy, MD, an internal medicine chief resident at the University of California at San Diego, and colleagues evaluated a year-long external membership program implemented by the American Society of Hematology (ASH) Medical Educators Institute.
The program linked 35 US hematology/oncology fellows (80% female, 46% White, 35% Asian) who were interested in CH to 34 North American faculty members. The pairs were told to meet virtually once a month.
Of 30 mentees and 23 mentors surveyed, 94% and 85%, respectively, said their pairings were good matches. Two thirds of the mentees accepted faculty positions in CH after their mentorships.
“Our study showed that external mentorship in a virtual format is feasible,” Qureshy said in an interview. “Additionally, external mentorship provided benefits such as different perspectives and the opportunity for mentorship for those who may not have it in their field of interest at their home institution.”
Qureshy added that “one strength of our mentorship program was that mentoring pairs were meticulously assigned based on shared interests and background. Many participants cited this common ground as a reason why they thought their mentoring pair was a good match.”
There’s an important caveat: Most of the mentees weren’t new to CH. About 70% had previously worked with a mentor in the CH field, and 86% had previously conducted research in the field.
What’s Next for Mentorship in CH?
The ASH Hematology-Focused Fellowship Training Program Consortium aims to mint 50 new academic hematologists by 2030 through programs at 12 institutions. “Mentorship is an exciting aspect of the program since it allows classical hematology trainees to form a network of peers nationally and also provides access to mentors across institutions,” Naik said. “And as the workforce grows, there will be more and more role models for future trainees to look up to.”
Moving forward, she said, “we hope to inspire even more institutions to adopt hematology training tracks throughout the country.”
Meanwhile, ASH’s new Classical Hematology Advancement Mentorship is taking applications for its debut 2025 program through January 9, 2025. Trainees will meet monthly with mentors both virtually and in person. Applicants must have been in their first or second year of hematology/oncology fellowship training at accredited programs in the United States as of July 15, 2024.
Naik, Marshall, Nagalla, and Qureshy have no relevant disclosures.
A version of this article appeared on Medscape.com.
SCD: Can Atrial Arrhythmias Predict Strokes?
TOPLINE:
METHODOLOGY:
- A total of 130 adult patients with SCD were included in the DREPACOEUR prospective registry from November 2018 to November 2022.
- The patients underwent a comprehensive cardiac evaluation, including 24-hour electrocardiogram monitoring, echocardiography, and laboratory tests.
- The primary endpoint was the occurrence of atrial arrhythmias, defined by excessive supraventricular ectopic activity or any recent history of atrial fibrillation.
- Patients with a history of stroke or transient ischemic attack were also included in the PCDREP prospective registry for further assessment.
- Written informed consent was collected from all participating patients, and the study was approved by the ethics committee.
TAKEAWAY:
- Atrial arrhythmias were found in 26% of patients with SCD, with a significant association with stroke history (P = .001).
- Age and left atrial volume were independently associated with atrial arrhythmias, with optimal cutoffs of 47 years and 55 mL/m2, respectively.
- Patients with atrial arrhythmias had higher diastolic blood pressure, worse kidney function, and higher NT pro-BNP levels than those without arrhythmias.
- Atrial arrhythmias were associated with an increased risk for stroke unrelated to cerebral vasculopathy or other defined causes (odds ratio, 6.6; P = .009).
“Atrial arrhythmias were found in 26% of patients with sickle cell anemia, with a significant association with stroke history,” wrote the authors of the study. In a commentary published concurrently, Jonathan Uniat, MD, of Children’s Hospital Los Angeles in California, wrote, “Early detection and treatment of atrial arrhythmias may help prevent strokes in this population.”
SOURCE:
The study was led by Thomas d’Humières, Henri Mondor Hospital in Créteil, France. It was published online on November 12 in Blood Advances.
LIMITATIONS:
This study was a pilot prospective study and was underpowered with atrial arrhythmias occurring in only 34 patients. The population was relatively old for sickle cell anemia (45 years), and the study was biased because patients were selected based on clinical criteria indicative of underlying cardiovascular abnormalities. The population was heterogeneous in terms of antiarrhythmic therapy, and overall, at an advanced stage of the disease with frequent organ complications.
DISCLOSURES:
The study was supported by grants from FHU-SENEC. Pablo Bartolucci received grants from Addmedica, the Fabre Foundation, Novartis, and Bluebird in the past 36 months; received consulting fees from Addmedica, Novartis, Roche, GBT, Bluebird, Emmaus, Hemanext, and Agios; received honoraria for lectures from Novartis, Addmedica, and Jazz Pharmaceuticals; and reported being a member of the Novartis steering committee and cofounder of Innovhem. Additional disclosures are noted in the original article.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
TOPLINE:
METHODOLOGY:
- A total of 130 adult patients with SCD were included in the DREPACOEUR prospective registry from November 2018 to November 2022.
- The patients underwent a comprehensive cardiac evaluation, including 24-hour electrocardiogram monitoring, echocardiography, and laboratory tests.
- The primary endpoint was the occurrence of atrial arrhythmias, defined by excessive supraventricular ectopic activity or any recent history of atrial fibrillation.
- Patients with a history of stroke or transient ischemic attack were also included in the PCDREP prospective registry for further assessment.
- Written informed consent was collected from all participating patients, and the study was approved by the ethics committee.
TAKEAWAY:
- Atrial arrhythmias were found in 26% of patients with SCD, with a significant association with stroke history (P = .001).
- Age and left atrial volume were independently associated with atrial arrhythmias, with optimal cutoffs of 47 years and 55 mL/m2, respectively.
- Patients with atrial arrhythmias had higher diastolic blood pressure, worse kidney function, and higher NT pro-BNP levels than those without arrhythmias.
- Atrial arrhythmias were associated with an increased risk for stroke unrelated to cerebral vasculopathy or other defined causes (odds ratio, 6.6; P = .009).
“Atrial arrhythmias were found in 26% of patients with sickle cell anemia, with a significant association with stroke history,” wrote the authors of the study. In a commentary published concurrently, Jonathan Uniat, MD, of Children’s Hospital Los Angeles in California, wrote, “Early detection and treatment of atrial arrhythmias may help prevent strokes in this population.”
SOURCE:
The study was led by Thomas d’Humières, Henri Mondor Hospital in Créteil, France. It was published online on November 12 in Blood Advances.
LIMITATIONS:
This study was a pilot prospective study and was underpowered with atrial arrhythmias occurring in only 34 patients. The population was relatively old for sickle cell anemia (45 years), and the study was biased because patients were selected based on clinical criteria indicative of underlying cardiovascular abnormalities. The population was heterogeneous in terms of antiarrhythmic therapy, and overall, at an advanced stage of the disease with frequent organ complications.
DISCLOSURES:
The study was supported by grants from FHU-SENEC. Pablo Bartolucci received grants from Addmedica, the Fabre Foundation, Novartis, and Bluebird in the past 36 months; received consulting fees from Addmedica, Novartis, Roche, GBT, Bluebird, Emmaus, Hemanext, and Agios; received honoraria for lectures from Novartis, Addmedica, and Jazz Pharmaceuticals; and reported being a member of the Novartis steering committee and cofounder of Innovhem. Additional disclosures are noted in the original article.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
TOPLINE:
METHODOLOGY:
- A total of 130 adult patients with SCD were included in the DREPACOEUR prospective registry from November 2018 to November 2022.
- The patients underwent a comprehensive cardiac evaluation, including 24-hour electrocardiogram monitoring, echocardiography, and laboratory tests.
- The primary endpoint was the occurrence of atrial arrhythmias, defined by excessive supraventricular ectopic activity or any recent history of atrial fibrillation.
- Patients with a history of stroke or transient ischemic attack were also included in the PCDREP prospective registry for further assessment.
- Written informed consent was collected from all participating patients, and the study was approved by the ethics committee.
TAKEAWAY:
- Atrial arrhythmias were found in 26% of patients with SCD, with a significant association with stroke history (P = .001).
- Age and left atrial volume were independently associated with atrial arrhythmias, with optimal cutoffs of 47 years and 55 mL/m2, respectively.
- Patients with atrial arrhythmias had higher diastolic blood pressure, worse kidney function, and higher NT pro-BNP levels than those without arrhythmias.
- Atrial arrhythmias were associated with an increased risk for stroke unrelated to cerebral vasculopathy or other defined causes (odds ratio, 6.6; P = .009).
“Atrial arrhythmias were found in 26% of patients with sickle cell anemia, with a significant association with stroke history,” wrote the authors of the study. In a commentary published concurrently, Jonathan Uniat, MD, of Children’s Hospital Los Angeles in California, wrote, “Early detection and treatment of atrial arrhythmias may help prevent strokes in this population.”
SOURCE:
The study was led by Thomas d’Humières, Henri Mondor Hospital in Créteil, France. It was published online on November 12 in Blood Advances.
LIMITATIONS:
This study was a pilot prospective study and was underpowered with atrial arrhythmias occurring in only 34 patients. The population was relatively old for sickle cell anemia (45 years), and the study was biased because patients were selected based on clinical criteria indicative of underlying cardiovascular abnormalities. The population was heterogeneous in terms of antiarrhythmic therapy, and overall, at an advanced stage of the disease with frequent organ complications.
DISCLOSURES:
The study was supported by grants from FHU-SENEC. Pablo Bartolucci received grants from Addmedica, the Fabre Foundation, Novartis, and Bluebird in the past 36 months; received consulting fees from Addmedica, Novartis, Roche, GBT, Bluebird, Emmaus, Hemanext, and Agios; received honoraria for lectures from Novartis, Addmedica, and Jazz Pharmaceuticals; and reported being a member of the Novartis steering committee and cofounder of Innovhem. Additional disclosures are noted in the original article.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
FDA Approves Pfizer’s Hympavzi for Hemophilia A, B
The once-weekly subcutaneous injection targets an anticoagulation protein called tissue factor pathway inhibitor (TFPI). Reducing TFPI’s amount and activity in the blood subsequently increases the amount of thrombin, a pro-clotting enzyme, in circulation.
“Today’s approval of Hympavzi provides patients with hemophilia a new treatment option that is the first of its kind to work by targeting a protein in the blood clotting process,” Ann Farrell, MD, director of FDA’s Division of Non-Malignant Hematology, said in an agency press release.
Hympavzi is the first non-factor, once-weekly treatment for hemophilia B in the United States. The subcutaneous injection emicizumab (Hemlibra, Genentech), which works by a different mechanism, is already on the market for hemophilia A.
The current approval was based on the open-label BASIS trial in 116 men and boys with either severe hemophilia A or B without factor inhibitors.
During the trial’s first 6 months, patients received standard treatment with clotting factor replacement either on-demand (33 patients) or prophylactically (83 patients). Patients were then switched to Hympavzi prophylaxis for a year.
Among patients receiving on-demand standard treatment during the first 6 months, the annualized bleeding rate was 38 episodes. That rate fell to 3.2 episodes during treatment with Hympavzi.
Among patients receiving prophylactic standard treatment during the first 6 months, the estimated annualized bleeding rate was 7.85 episodes, which then fell to 5.08 during the year of Hympavzi prophylaxis, FDA said.
Injection-site reactions, headaches, and itching were the most common side effects with marstacimab, occurring in 3% or more of patients. Labeling warns of the potential for circulating blood clots, hypersensitivity, and embryofetal toxicity. Marstacimab is supplied in prefilled syringes.
Marstacimab is Pfizer’s second hemophilia approval in 2024. FDA approved the company’s hemophilia B gene therapy fidanacogene elaparvovec (Beqvez) in April.
Pfizer noted in a press release that results for another arm of the BASIS trial in patients with clotting factor inhibitors are expected in 2025.
A version of this article first appeared on Medscape.com.
The once-weekly subcutaneous injection targets an anticoagulation protein called tissue factor pathway inhibitor (TFPI). Reducing TFPI’s amount and activity in the blood subsequently increases the amount of thrombin, a pro-clotting enzyme, in circulation.
“Today’s approval of Hympavzi provides patients with hemophilia a new treatment option that is the first of its kind to work by targeting a protein in the blood clotting process,” Ann Farrell, MD, director of FDA’s Division of Non-Malignant Hematology, said in an agency press release.
Hympavzi is the first non-factor, once-weekly treatment for hemophilia B in the United States. The subcutaneous injection emicizumab (Hemlibra, Genentech), which works by a different mechanism, is already on the market for hemophilia A.
The current approval was based on the open-label BASIS trial in 116 men and boys with either severe hemophilia A or B without factor inhibitors.
During the trial’s first 6 months, patients received standard treatment with clotting factor replacement either on-demand (33 patients) or prophylactically (83 patients). Patients were then switched to Hympavzi prophylaxis for a year.
Among patients receiving on-demand standard treatment during the first 6 months, the annualized bleeding rate was 38 episodes. That rate fell to 3.2 episodes during treatment with Hympavzi.
Among patients receiving prophylactic standard treatment during the first 6 months, the estimated annualized bleeding rate was 7.85 episodes, which then fell to 5.08 during the year of Hympavzi prophylaxis, FDA said.
Injection-site reactions, headaches, and itching were the most common side effects with marstacimab, occurring in 3% or more of patients. Labeling warns of the potential for circulating blood clots, hypersensitivity, and embryofetal toxicity. Marstacimab is supplied in prefilled syringes.
Marstacimab is Pfizer’s second hemophilia approval in 2024. FDA approved the company’s hemophilia B gene therapy fidanacogene elaparvovec (Beqvez) in April.
Pfizer noted in a press release that results for another arm of the BASIS trial in patients with clotting factor inhibitors are expected in 2025.
A version of this article first appeared on Medscape.com.
The once-weekly subcutaneous injection targets an anticoagulation protein called tissue factor pathway inhibitor (TFPI). Reducing TFPI’s amount and activity in the blood subsequently increases the amount of thrombin, a pro-clotting enzyme, in circulation.
“Today’s approval of Hympavzi provides patients with hemophilia a new treatment option that is the first of its kind to work by targeting a protein in the blood clotting process,” Ann Farrell, MD, director of FDA’s Division of Non-Malignant Hematology, said in an agency press release.
Hympavzi is the first non-factor, once-weekly treatment for hemophilia B in the United States. The subcutaneous injection emicizumab (Hemlibra, Genentech), which works by a different mechanism, is already on the market for hemophilia A.
The current approval was based on the open-label BASIS trial in 116 men and boys with either severe hemophilia A or B without factor inhibitors.
During the trial’s first 6 months, patients received standard treatment with clotting factor replacement either on-demand (33 patients) or prophylactically (83 patients). Patients were then switched to Hympavzi prophylaxis for a year.
Among patients receiving on-demand standard treatment during the first 6 months, the annualized bleeding rate was 38 episodes. That rate fell to 3.2 episodes during treatment with Hympavzi.
Among patients receiving prophylactic standard treatment during the first 6 months, the estimated annualized bleeding rate was 7.85 episodes, which then fell to 5.08 during the year of Hympavzi prophylaxis, FDA said.
Injection-site reactions, headaches, and itching were the most common side effects with marstacimab, occurring in 3% or more of patients. Labeling warns of the potential for circulating blood clots, hypersensitivity, and embryofetal toxicity. Marstacimab is supplied in prefilled syringes.
Marstacimab is Pfizer’s second hemophilia approval in 2024. FDA approved the company’s hemophilia B gene therapy fidanacogene elaparvovec (Beqvez) in April.
Pfizer noted in a press release that results for another arm of the BASIS trial in patients with clotting factor inhibitors are expected in 2025.
A version of this article first appeared on Medscape.com.