User login
Relatively Asymptomatic, but Still Problematic
ANSWER
The correct answer is seborrheic dermatitis (choice “d”), a common cause of penile rashes that typically manifests initially as chronic dandruff or in some other form on the head or neck.
Herpes simplex (choice “a”) is certainly common, but it likely would have presented with grouped vesicles on an erythematous base. Furthermore, each episode would have been limited to about two weeks, and the eruption would have produced noticeable symptoms and responded to the valacyclovir.
Yeast infection (choice “b”), while often diagnosed, is in reality unusual, especially in the circumcised and otherwise healthy male. Nystatin, although far from the ideal treatment, should have had some effect.
Fixed drug eruption (FDE; choice “c”) could have been a suspect, had there been a drug to blame. FDE usually presents as a brownish red, shiny round macule that appears and reappears in the same area with repeated exposure to the same drug. The penile shaft is a favorite area for it. Drugs known to trigger FDE include NSAIDs, sulfa, tetracycline, penicillin, pseudoephedrine, and aspirin.
DISCUSSION
Seborrheic dermatitis (SD), also known as seborrhea, is an extremely common chronic papulosquamous disorder patterned on the sebum-rich areas of the scalp, face, and trunk. Although not directly caused by the highly lipophilic commensal yeast Malassezia furfur, it does appear to be related to increases in the number of those organisms, as well as to immunologic abnormalities and increased production of sebum. It can range from a mild scaly rash to whole-body erythroderma and can affect an astonishing range of areas, including the genitals.
SD almost always manifests with dandruff (or “cradle cap” in the infant), followed by faint scaling in and around the ears or on the face (eg, nasolabial folds, brows, and glabella), mid chest, axillae, periumbilical region, and genitals. Below the head and neck, SD often mystifies the nondermatology provider, who tends to call it “fungal infection” or, when it’s seen in moist intertriginous skin, “yeast infection.”
SD, especially in this case, represents the perfect example of the need to “look elsewhere” for clues when confronted with a mysterious rash. Patients can certainly have more than one dermatologic diagnosis at a time, but a single explanation is considerably more likely and should therefore be sought. In this case, corroboration for the diagnosis of SD was readily found by looking for it in its known locations.
SD can take on different looks, including a distinctly annular morphology, especially in patients with darker skin. It can occasionally be severe in patients with Parkinson’s disease, multiple sclerosis, or a history of stroke. This case mirrors my experience in that I see increased stress as a major precipitating factor in the worsening of pre-existing SD.
In addition to the items already mentioned, the differential for penile rashes includes lichen planus. However, the lesions of lichen planus tend to have a distinctly purple appearance and well-defined margins, and on the penis, they tend to spill over onto the penile corona and glans.
TREATMENT/PROGNOSIS
In this case, treatment comprised a combination of oxiconazole lotion and 2.5% hydrocortisone cream. Many other combinations have been used successfully, including pimecrolimus or tacrolimus combined with ketoconazole cream.
Whatever is used, a cure will not be forthcoming, since the condition is almost always chronic. The main value of an accurate diagnosis in such a case lies in easing the patient’s mind regarding the terrible diseases he doesn’t have.
ANSWER
The correct answer is seborrheic dermatitis (choice “d”), a common cause of penile rashes that typically manifests initially as chronic dandruff or in some other form on the head or neck.
Herpes simplex (choice “a”) is certainly common, but it likely would have presented with grouped vesicles on an erythematous base. Furthermore, each episode would have been limited to about two weeks, and the eruption would have produced noticeable symptoms and responded to the valacyclovir.
Yeast infection (choice “b”), while often diagnosed, is in reality unusual, especially in the circumcised and otherwise healthy male. Nystatin, although far from the ideal treatment, should have had some effect.
Fixed drug eruption (FDE; choice “c”) could have been a suspect, had there been a drug to blame. FDE usually presents as a brownish red, shiny round macule that appears and reappears in the same area with repeated exposure to the same drug. The penile shaft is a favorite area for it. Drugs known to trigger FDE include NSAIDs, sulfa, tetracycline, penicillin, pseudoephedrine, and aspirin.
DISCUSSION
Seborrheic dermatitis (SD), also known as seborrhea, is an extremely common chronic papulosquamous disorder patterned on the sebum-rich areas of the scalp, face, and trunk. Although not directly caused by the highly lipophilic commensal yeast Malassezia furfur, it does appear to be related to increases in the number of those organisms, as well as to immunologic abnormalities and increased production of sebum. It can range from a mild scaly rash to whole-body erythroderma and can affect an astonishing range of areas, including the genitals.
SD almost always manifests with dandruff (or “cradle cap” in the infant), followed by faint scaling in and around the ears or on the face (eg, nasolabial folds, brows, and glabella), mid chest, axillae, periumbilical region, and genitals. Below the head and neck, SD often mystifies the nondermatology provider, who tends to call it “fungal infection” or, when it’s seen in moist intertriginous skin, “yeast infection.”
SD, especially in this case, represents the perfect example of the need to “look elsewhere” for clues when confronted with a mysterious rash. Patients can certainly have more than one dermatologic diagnosis at a time, but a single explanation is considerably more likely and should therefore be sought. In this case, corroboration for the diagnosis of SD was readily found by looking for it in its known locations.
SD can take on different looks, including a distinctly annular morphology, especially in patients with darker skin. It can occasionally be severe in patients with Parkinson’s disease, multiple sclerosis, or a history of stroke. This case mirrors my experience in that I see increased stress as a major precipitating factor in the worsening of pre-existing SD.
In addition to the items already mentioned, the differential for penile rashes includes lichen planus. However, the lesions of lichen planus tend to have a distinctly purple appearance and well-defined margins, and on the penis, they tend to spill over onto the penile corona and glans.
TREATMENT/PROGNOSIS
In this case, treatment comprised a combination of oxiconazole lotion and 2.5% hydrocortisone cream. Many other combinations have been used successfully, including pimecrolimus or tacrolimus combined with ketoconazole cream.
Whatever is used, a cure will not be forthcoming, since the condition is almost always chronic. The main value of an accurate diagnosis in such a case lies in easing the patient’s mind regarding the terrible diseases he doesn’t have.
ANSWER
The correct answer is seborrheic dermatitis (choice “d”), a common cause of penile rashes that typically manifests initially as chronic dandruff or in some other form on the head or neck.
Herpes simplex (choice “a”) is certainly common, but it likely would have presented with grouped vesicles on an erythematous base. Furthermore, each episode would have been limited to about two weeks, and the eruption would have produced noticeable symptoms and responded to the valacyclovir.
Yeast infection (choice “b”), while often diagnosed, is in reality unusual, especially in the circumcised and otherwise healthy male. Nystatin, although far from the ideal treatment, should have had some effect.
Fixed drug eruption (FDE; choice “c”) could have been a suspect, had there been a drug to blame. FDE usually presents as a brownish red, shiny round macule that appears and reappears in the same area with repeated exposure to the same drug. The penile shaft is a favorite area for it. Drugs known to trigger FDE include NSAIDs, sulfa, tetracycline, penicillin, pseudoephedrine, and aspirin.
DISCUSSION
Seborrheic dermatitis (SD), also known as seborrhea, is an extremely common chronic papulosquamous disorder patterned on the sebum-rich areas of the scalp, face, and trunk. Although not directly caused by the highly lipophilic commensal yeast Malassezia furfur, it does appear to be related to increases in the number of those organisms, as well as to immunologic abnormalities and increased production of sebum. It can range from a mild scaly rash to whole-body erythroderma and can affect an astonishing range of areas, including the genitals.
SD almost always manifests with dandruff (or “cradle cap” in the infant), followed by faint scaling in and around the ears or on the face (eg, nasolabial folds, brows, and glabella), mid chest, axillae, periumbilical region, and genitals. Below the head and neck, SD often mystifies the nondermatology provider, who tends to call it “fungal infection” or, when it’s seen in moist intertriginous skin, “yeast infection.”
SD, especially in this case, represents the perfect example of the need to “look elsewhere” for clues when confronted with a mysterious rash. Patients can certainly have more than one dermatologic diagnosis at a time, but a single explanation is considerably more likely and should therefore be sought. In this case, corroboration for the diagnosis of SD was readily found by looking for it in its known locations.
SD can take on different looks, including a distinctly annular morphology, especially in patients with darker skin. It can occasionally be severe in patients with Parkinson’s disease, multiple sclerosis, or a history of stroke. This case mirrors my experience in that I see increased stress as a major precipitating factor in the worsening of pre-existing SD.
In addition to the items already mentioned, the differential for penile rashes includes lichen planus. However, the lesions of lichen planus tend to have a distinctly purple appearance and well-defined margins, and on the penis, they tend to spill over onto the penile corona and glans.
TREATMENT/PROGNOSIS
In this case, treatment comprised a combination of oxiconazole lotion and 2.5% hydrocortisone cream. Many other combinations have been used successfully, including pimecrolimus or tacrolimus combined with ketoconazole cream.
Whatever is used, a cure will not be forthcoming, since the condition is almost always chronic. The main value of an accurate diagnosis in such a case lies in easing the patient’s mind regarding the terrible diseases he doesn’t have.

A 31-year-old man is referred to dermatology for evaluation of a penile rash that has repeatedly manifested and resolved over a period of months. Relatively asymptomatic, the eruption has persisted despite a two-week course of valacyclovir 500 mg bid, followed by a month-long course of topical nystatin cream tid. The patient says he has been in otherwise good health. However, he reports being under a great deal of stress, as his job and his marriage ended within the space of a few weeks. He denies any sexual exposure outside his marriage. Other than those already mentioned, the patient has taken no medications, prescription or OTC. The problem area is obvious: a bright pink papulosquamous patch on the distal right shaft of his circumcised penis. This round lesion, which measures more than 3 cm in diameter, has a shiny appearance and slightly irregular margins. No other areas of involvement are noted in the genital area. However, there is a similar scaly pink rash behind both of the patient’s ears, as well as patches of dandruff in the scalp, especially over and behind the ears. A similar rash is seen in the patient’s umbilicus and surrounding area.

Thickened Velvety Plaques in a 75-Year-Old Woman
The Diagnosis: Tripe Palms and Acanthosis Nigricans
A shave biopsy specimen from the left palm showed slight epidermal hyperplasia with substantial papillomatosis and compact orthokeratosis. The complete blood cell count, thyrotropin level, uric acid level, liver function tests, mammogram, Papanicolaou test, and chest radiograph were unremarkable. A basic metabolic panel showed mildly elevated blood glucose at 111 mg/dL (reference range, 70-99 mg/dL) and hemoglobin A1c at 6.3% (reference range, <6.0%). Full-body computed tomography, endoscopy, and colonoscopy initially were normal. One year later after presenting with tripe palms, the patient had a bowel obstruction secondary to omental carcinomatosis from a primary ovarian tumor.
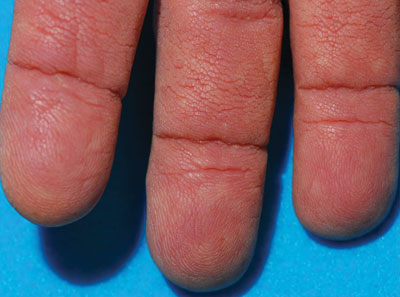
The term tripe in tripe palms refers to the resemblance to the edible lining of the bovine foregut. It originated in 1963 from a patient's own description of the rugose velvety texture of the palms.1 In the literature, tripe palms also is called acanthosis palmaris, acanthosis nigricans of the palms, palmar hyperkeratosis, palmar keratoderma, and pachydermatoglyphy. It is a rare cutaneous finding. Tripe palms is associated with other cutaneous paraneoplastic syndromes such as malignant acanthosis nigricans (72% of cases) and Leser-Trélat sign (10% of cases). It affects more men than women (63% vs 37%) and is almost exclusively seen in adults (median age, 62 years).1
The clinical appearance of tripe palms includes hypertrophy of the palms and often the soles with papillations producing a velvety or honeycomb appearance. In addition, the dermatoglyphics are pronounced. The histologic findings typically show hyperkeratosis and acanthosis. Other features that can be seen include dermal mucinosis and increased mast cells in the dermis. To differentiate tripe palms from other keratodermas, substantial papillations can be seen with less diffuse hyperkeratosis.1
Tripe palms has been associated with an underlying malignancy in more than 90% of published cases. In two-thirds of cases, tripe palms either appears before or concurrent with the diagnosis of cancer.2 It is rarely reported as an idiopathic finding or associated with nonneoplastic disorders. Malignancies most commonly associated are adenocarcinomas, especially of the stomach (27%) and lungs (22%). Other neoplasms, such as in our patient, include those of the genitourinary tract and breast. In a patient with tripe palms in the absence of acanthosis nigricans, the most common neoplasm is of the lung, especially when clubbing of the nails also is present.2 Thus, after a diagnosis of tripe palms is established, a thorough investigation for an underlying malignancy is the next most important step to direct specific therapy.
1. Cohen PR, Grossman ME, Silvers DN, et al. Tripe palms and cancer. Clin Dermatol. 1993;11:165-173.
2. Moore RL, Devere TS. Epidermal manifestations of internal malignancy. Dermatol Clin. 2008;26:17-29.
The Diagnosis: Tripe Palms and Acanthosis Nigricans
A shave biopsy specimen from the left palm showed slight epidermal hyperplasia with substantial papillomatosis and compact orthokeratosis. The complete blood cell count, thyrotropin level, uric acid level, liver function tests, mammogram, Papanicolaou test, and chest radiograph were unremarkable. A basic metabolic panel showed mildly elevated blood glucose at 111 mg/dL (reference range, 70-99 mg/dL) and hemoglobin A1c at 6.3% (reference range, <6.0%). Full-body computed tomography, endoscopy, and colonoscopy initially were normal. One year later after presenting with tripe palms, the patient had a bowel obstruction secondary to omental carcinomatosis from a primary ovarian tumor.
The term tripe in tripe palms refers to the resemblance to the edible lining of the bovine foregut. It originated in 1963 from a patient's own description of the rugose velvety texture of the palms.1 In the literature, tripe palms also is called acanthosis palmaris, acanthosis nigricans of the palms, palmar hyperkeratosis, palmar keratoderma, and pachydermatoglyphy. It is a rare cutaneous finding. Tripe palms is associated with other cutaneous paraneoplastic syndromes such as malignant acanthosis nigricans (72% of cases) and Leser-Trélat sign (10% of cases). It affects more men than women (63% vs 37%) and is almost exclusively seen in adults (median age, 62 years).1
The clinical appearance of tripe palms includes hypertrophy of the palms and often the soles with papillations producing a velvety or honeycomb appearance. In addition, the dermatoglyphics are pronounced. The histologic findings typically show hyperkeratosis and acanthosis. Other features that can be seen include dermal mucinosis and increased mast cells in the dermis. To differentiate tripe palms from other keratodermas, substantial papillations can be seen with less diffuse hyperkeratosis.1
Tripe palms has been associated with an underlying malignancy in more than 90% of published cases. In two-thirds of cases, tripe palms either appears before or concurrent with the diagnosis of cancer.2 It is rarely reported as an idiopathic finding or associated with nonneoplastic disorders. Malignancies most commonly associated are adenocarcinomas, especially of the stomach (27%) and lungs (22%). Other neoplasms, such as in our patient, include those of the genitourinary tract and breast. In a patient with tripe palms in the absence of acanthosis nigricans, the most common neoplasm is of the lung, especially when clubbing of the nails also is present.2 Thus, after a diagnosis of tripe palms is established, a thorough investigation for an underlying malignancy is the next most important step to direct specific therapy.
The Diagnosis: Tripe Palms and Acanthosis Nigricans
A shave biopsy specimen from the left palm showed slight epidermal hyperplasia with substantial papillomatosis and compact orthokeratosis. The complete blood cell count, thyrotropin level, uric acid level, liver function tests, mammogram, Papanicolaou test, and chest radiograph were unremarkable. A basic metabolic panel showed mildly elevated blood glucose at 111 mg/dL (reference range, 70-99 mg/dL) and hemoglobin A1c at 6.3% (reference range, <6.0%). Full-body computed tomography, endoscopy, and colonoscopy initially were normal. One year later after presenting with tripe palms, the patient had a bowel obstruction secondary to omental carcinomatosis from a primary ovarian tumor.
The term tripe in tripe palms refers to the resemblance to the edible lining of the bovine foregut. It originated in 1963 from a patient's own description of the rugose velvety texture of the palms.1 In the literature, tripe palms also is called acanthosis palmaris, acanthosis nigricans of the palms, palmar hyperkeratosis, palmar keratoderma, and pachydermatoglyphy. It is a rare cutaneous finding. Tripe palms is associated with other cutaneous paraneoplastic syndromes such as malignant acanthosis nigricans (72% of cases) and Leser-Trélat sign (10% of cases). It affects more men than women (63% vs 37%) and is almost exclusively seen in adults (median age, 62 years).1
The clinical appearance of tripe palms includes hypertrophy of the palms and often the soles with papillations producing a velvety or honeycomb appearance. In addition, the dermatoglyphics are pronounced. The histologic findings typically show hyperkeratosis and acanthosis. Other features that can be seen include dermal mucinosis and increased mast cells in the dermis. To differentiate tripe palms from other keratodermas, substantial papillations can be seen with less diffuse hyperkeratosis.1
Tripe palms has been associated with an underlying malignancy in more than 90% of published cases. In two-thirds of cases, tripe palms either appears before or concurrent with the diagnosis of cancer.2 It is rarely reported as an idiopathic finding or associated with nonneoplastic disorders. Malignancies most commonly associated are adenocarcinomas, especially of the stomach (27%) and lungs (22%). Other neoplasms, such as in our patient, include those of the genitourinary tract and breast. In a patient with tripe palms in the absence of acanthosis nigricans, the most common neoplasm is of the lung, especially when clubbing of the nails also is present.2 Thus, after a diagnosis of tripe palms is established, a thorough investigation for an underlying malignancy is the next most important step to direct specific therapy.
1. Cohen PR, Grossman ME, Silvers DN, et al. Tripe palms and cancer. Clin Dermatol. 1993;11:165-173.
2. Moore RL, Devere TS. Epidermal manifestations of internal malignancy. Dermatol Clin. 2008;26:17-29.
1. Cohen PR, Grossman ME, Silvers DN, et al. Tripe palms and cancer. Clin Dermatol. 1993;11:165-173.
2. Moore RL, Devere TS. Epidermal manifestations of internal malignancy. Dermatol Clin. 2008;26:17-29.
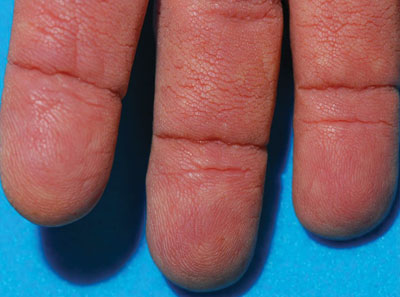
A 75-year-old woman presented with progressive, velvety, thick skin involving the bilateral axillae, inner thighs, palms, and buccal mucosa. She also reported weight loss of approximately 25 pounds over the last 12 months. Her medical history was notable for metabolic syndrome, allergic rhinitis, and colon polyps. She denied a family history of malignancy. On physical examination, she was a healthy-appearing overweight woman. The palmar surface of the bilateral hands was thickened and velvety with exaggerated dermatoglyphics. She had similarly thickened, velvety, gray-brown plaques on the bilateral axillae and proximal aspects of the inner thighs. The buccal mucosa had a thickened rugose texture.
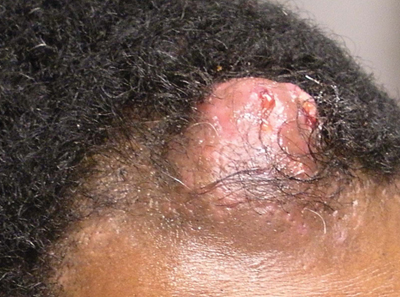
Solitary Nodular Lesion on the Scalp
The Diagnosis: Pilomatricoma
Pilomatricoma, first described by Malherbe and Chenantais1 in 1880, is a benign appendageal tumor derived from hair follicle matrix cells. It classically manifests as a solitary, asymptomatic, firm dermal nodule with a normal overlying epidermis. Less common morphologic variants include perforating, lymphagiectatic, keratoacanthomalike, pigmented, and anetodermalike surface changes.2 Inflammation and erosion through the skin surface are observed in the rare perforating variant, as seen in our patient. The average size is 1 cm, and it rarely exceeds 3 cm in diameter.3 The tumors predominantly occur on the head, neck, and upper extremities, with only 9.5% on the scalp.2 It may occur at any age, though it has a bimodal distribution with peaks in childhood and in adults older than 60 years. A slight preponderance in females has been observed with a female to male ratio of 1.5 to 1.2 Although our patient is black, most reported cases have occurred in individuals of European descent. Because cases of pilomatricoma are not systematically reported, it is uncertain if this finding represents a publication bias or if race is an actual risk factor. Multiple pilomatricomas and familial cases have been described in association with myotonic dystrophy, Turner syndrome, Gardner syndrome, Rubinstein-Taybi syndrome, polyfactorial coagulopathy, trisomy 9, xeroderma pigmentosum, and basal cell nevus syndrome.2,4
It has been shown that the proliferating cells of pilomatricomas stain with antibodies directed against Lef1 (lymphoid enhancer binding factor 1), a marker from hair matrix cells, providing biochemical evidence for the morphologic appearance of these neoplasms.5 Pilomatricomas have been associated with B-cell/chronic lymphocytic leukemia lymphoma 2 gene, BCL2, expression, a proto-oncogene that suppresses apoptosis in benign and malignant neoplasms, which may contribute to the pathogenesis of these tumors.6 Pilomatricomas also have been associated with β-catenin mutation, expression of Bmp2 (bone morphogenetic protein 2), and human hair keratin basic 1.7-9
Definitive diagnosis is obtained through biopsy, looking for characteristic histopathologic findings. The lesion usually is found in the lower dermis and subcutaneous fat. However, in the perforating variant, the lesion is more superficial, located in the papillary and mid dermis, as seen in our patient.10
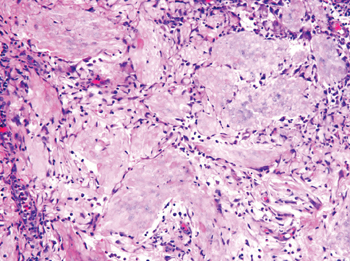
Pilomatricomas are sharply demarcated, often surrounded by a connective-tissue capsule. Histopathologic analysis reveals islands of epithelial cells comprised of 3 subtypes: basophilic cells with scant cytoplasm, shadow cells with a central pallor (Figure), and transitional cells between the former 2 cellular types.11 The number of basophilic and transitional cells is inversely related to the number of shadow cells. In older lesions, the shadow cells predominate, while the basophilic cells are few in number or absent. Calcium deposits are seen in 80% of lesions with von Kossa staining.12
Transformation into malignancy, known as pilomatrical carcinoma, is rare. These malignant neoplasms are characterized by aggressive biologic behavior such as recurrence, diffuse spread, or metastasis, or by cytologic abnormalities such as poor cellular organization, squamous differentiation, and conspicuous mitotic activity.13 The recent growth of the long-standing lesion in our patient might be interpreted as a sign of malignant transformation. However, this observation may be related to the intense inflammatory reaction supported by the histopathology.
Pilomatricomas are not associated with mortality. Pilomatrical carcinomas are uncommon but are locally invasive and can cause visceral metastases and death. Spontaneous regression has never been observed and medical treatment is ineffective. The treatment of choice is incision and curettage or surgical excision.14 Although recurrence has only been reported in 2.6% of cases from a large case series (N=228), patients should be monitored after surgical excision.12
1. Malherbe A, Chenantais J. Note sur l'epithelioma calcifie des glandes sebacees. Prog Med. 1880;8:826-828.
2. Julian CG, Bowers PW. A clinical review of 209 pilomatricomas. J Am Acad Dermatol. 1998;39(2, pt 1):191-195.
3. Lozzi GP, Soyer HP, Fruehauf J, et al. Giant pilomatricoma. Am J Dermatopathol. 2007;29:286-289.
4. Hubbard VG, Whittaker SJ. Multiple familial pilomatricomas: an unusual case. J Cutan Pathol. 2004;31:281-283.
5. Kizawa K, Toyoda M, Ito M, et al. Aberrantly differentiated cells in benign pilomatrixoma reflect the normal hair follicle: immunohistochemical analysis of Ca-binding S100A2, S100A3 and S100A6 proteins. Br J Dermatol. 2005;152:314-320.
6. Farrier S, Morgan M. bcl-2 expression in pilomatricoma. Am J Dermatopathol. 1997;19:254-257.
7. Park SW, Suh KS, Wang HY, et al. Beta-catenin expression in the transitional cell zone of pilomatricoma. Br J Dermatol. 2001;145:624-629.
8. Kurokawa I, Kusumoto K, Bessho K, et al. Immunohistochemical expression of bone morphogenetic protein-2 in pilomatricoma. Br J Dermatol. 2000;143:754-758.
9. Cribier B, Asch PH, Regnier C, et al. Expression of human hair keratin basic 1 in pilomatrixoma: a study of 128 cases. Br J Dermatol. 1999;140:600-604.
10. Bayle P, Bazex J, Lamant L, et al. Multiple perforating and non perforating pilomatricomas in a patient with Churg-Strauss syndrome and Rubinstein-Taybi syndrome. J Eur Acad Dermatol Venereol. 2004;18:607-610.
11. Elder D, Elenitsas R, Ragsdale BD. Pilomatricoma. In: Elder D, Elenitsas R, Jaworsky C, et al, eds. Histopathology of the Skin. 8th ed. Philadelphia, PA: Lippincott-Raven; 1997:757-759.
12. Forbis R Jr, Helwig EB. Pilomatrixoma (calcifying epithelioma). Arch Dermatol. 1961;83:606-618.
13. Wood MG, Parhizgar B, Beerman H. Malignant pilomatricoma. Arch Dermatol. 1984;120:770-773.
14. Thomas RW, Perkins JA, Ruegemer JL, et al. Surgical excision of pilomatrixoma of the head and neck: a retrospective review of 26 cases. Ear Nose Throat J. 1999;78:541, 544-546, 548.
The Diagnosis: Pilomatricoma
Pilomatricoma, first described by Malherbe and Chenantais1 in 1880, is a benign appendageal tumor derived from hair follicle matrix cells. It classically manifests as a solitary, asymptomatic, firm dermal nodule with a normal overlying epidermis. Less common morphologic variants include perforating, lymphagiectatic, keratoacanthomalike, pigmented, and anetodermalike surface changes.2 Inflammation and erosion through the skin surface are observed in the rare perforating variant, as seen in our patient. The average size is 1 cm, and it rarely exceeds 3 cm in diameter.3 The tumors predominantly occur on the head, neck, and upper extremities, with only 9.5% on the scalp.2 It may occur at any age, though it has a bimodal distribution with peaks in childhood and in adults older than 60 years. A slight preponderance in females has been observed with a female to male ratio of 1.5 to 1.2 Although our patient is black, most reported cases have occurred in individuals of European descent. Because cases of pilomatricoma are not systematically reported, it is uncertain if this finding represents a publication bias or if race is an actual risk factor. Multiple pilomatricomas and familial cases have been described in association with myotonic dystrophy, Turner syndrome, Gardner syndrome, Rubinstein-Taybi syndrome, polyfactorial coagulopathy, trisomy 9, xeroderma pigmentosum, and basal cell nevus syndrome.2,4
It has been shown that the proliferating cells of pilomatricomas stain with antibodies directed against Lef1 (lymphoid enhancer binding factor 1), a marker from hair matrix cells, providing biochemical evidence for the morphologic appearance of these neoplasms.5 Pilomatricomas have been associated with B-cell/chronic lymphocytic leukemia lymphoma 2 gene, BCL2, expression, a proto-oncogene that suppresses apoptosis in benign and malignant neoplasms, which may contribute to the pathogenesis of these tumors.6 Pilomatricomas also have been associated with β-catenin mutation, expression of Bmp2 (bone morphogenetic protein 2), and human hair keratin basic 1.7-9
Definitive diagnosis is obtained through biopsy, looking for characteristic histopathologic findings. The lesion usually is found in the lower dermis and subcutaneous fat. However, in the perforating variant, the lesion is more superficial, located in the papillary and mid dermis, as seen in our patient.10
Pilomatricomas are sharply demarcated, often surrounded by a connective-tissue capsule. Histopathologic analysis reveals islands of epithelial cells comprised of 3 subtypes: basophilic cells with scant cytoplasm, shadow cells with a central pallor (Figure), and transitional cells between the former 2 cellular types.11 The number of basophilic and transitional cells is inversely related to the number of shadow cells. In older lesions, the shadow cells predominate, while the basophilic cells are few in number or absent. Calcium deposits are seen in 80% of lesions with von Kossa staining.12
Transformation into malignancy, known as pilomatrical carcinoma, is rare. These malignant neoplasms are characterized by aggressive biologic behavior such as recurrence, diffuse spread, or metastasis, or by cytologic abnormalities such as poor cellular organization, squamous differentiation, and conspicuous mitotic activity.13 The recent growth of the long-standing lesion in our patient might be interpreted as a sign of malignant transformation. However, this observation may be related to the intense inflammatory reaction supported by the histopathology.
Pilomatricomas are not associated with mortality. Pilomatrical carcinomas are uncommon but are locally invasive and can cause visceral metastases and death. Spontaneous regression has never been observed and medical treatment is ineffective. The treatment of choice is incision and curettage or surgical excision.14 Although recurrence has only been reported in 2.6% of cases from a large case series (N=228), patients should be monitored after surgical excision.12
The Diagnosis: Pilomatricoma
Pilomatricoma, first described by Malherbe and Chenantais1 in 1880, is a benign appendageal tumor derived from hair follicle matrix cells. It classically manifests as a solitary, asymptomatic, firm dermal nodule with a normal overlying epidermis. Less common morphologic variants include perforating, lymphagiectatic, keratoacanthomalike, pigmented, and anetodermalike surface changes.2 Inflammation and erosion through the skin surface are observed in the rare perforating variant, as seen in our patient. The average size is 1 cm, and it rarely exceeds 3 cm in diameter.3 The tumors predominantly occur on the head, neck, and upper extremities, with only 9.5% on the scalp.2 It may occur at any age, though it has a bimodal distribution with peaks in childhood and in adults older than 60 years. A slight preponderance in females has been observed with a female to male ratio of 1.5 to 1.2 Although our patient is black, most reported cases have occurred in individuals of European descent. Because cases of pilomatricoma are not systematically reported, it is uncertain if this finding represents a publication bias or if race is an actual risk factor. Multiple pilomatricomas and familial cases have been described in association with myotonic dystrophy, Turner syndrome, Gardner syndrome, Rubinstein-Taybi syndrome, polyfactorial coagulopathy, trisomy 9, xeroderma pigmentosum, and basal cell nevus syndrome.2,4
It has been shown that the proliferating cells of pilomatricomas stain with antibodies directed against Lef1 (lymphoid enhancer binding factor 1), a marker from hair matrix cells, providing biochemical evidence for the morphologic appearance of these neoplasms.5 Pilomatricomas have been associated with B-cell/chronic lymphocytic leukemia lymphoma 2 gene, BCL2, expression, a proto-oncogene that suppresses apoptosis in benign and malignant neoplasms, which may contribute to the pathogenesis of these tumors.6 Pilomatricomas also have been associated with β-catenin mutation, expression of Bmp2 (bone morphogenetic protein 2), and human hair keratin basic 1.7-9
Definitive diagnosis is obtained through biopsy, looking for characteristic histopathologic findings. The lesion usually is found in the lower dermis and subcutaneous fat. However, in the perforating variant, the lesion is more superficial, located in the papillary and mid dermis, as seen in our patient.10
Pilomatricomas are sharply demarcated, often surrounded by a connective-tissue capsule. Histopathologic analysis reveals islands of epithelial cells comprised of 3 subtypes: basophilic cells with scant cytoplasm, shadow cells with a central pallor (Figure), and transitional cells between the former 2 cellular types.11 The number of basophilic and transitional cells is inversely related to the number of shadow cells. In older lesions, the shadow cells predominate, while the basophilic cells are few in number or absent. Calcium deposits are seen in 80% of lesions with von Kossa staining.12
Transformation into malignancy, known as pilomatrical carcinoma, is rare. These malignant neoplasms are characterized by aggressive biologic behavior such as recurrence, diffuse spread, or metastasis, or by cytologic abnormalities such as poor cellular organization, squamous differentiation, and conspicuous mitotic activity.13 The recent growth of the long-standing lesion in our patient might be interpreted as a sign of malignant transformation. However, this observation may be related to the intense inflammatory reaction supported by the histopathology.
Pilomatricomas are not associated with mortality. Pilomatrical carcinomas are uncommon but are locally invasive and can cause visceral metastases and death. Spontaneous regression has never been observed and medical treatment is ineffective. The treatment of choice is incision and curettage or surgical excision.14 Although recurrence has only been reported in 2.6% of cases from a large case series (N=228), patients should be monitored after surgical excision.12
1. Malherbe A, Chenantais J. Note sur l'epithelioma calcifie des glandes sebacees. Prog Med. 1880;8:826-828.
2. Julian CG, Bowers PW. A clinical review of 209 pilomatricomas. J Am Acad Dermatol. 1998;39(2, pt 1):191-195.
3. Lozzi GP, Soyer HP, Fruehauf J, et al. Giant pilomatricoma. Am J Dermatopathol. 2007;29:286-289.
4. Hubbard VG, Whittaker SJ. Multiple familial pilomatricomas: an unusual case. J Cutan Pathol. 2004;31:281-283.
5. Kizawa K, Toyoda M, Ito M, et al. Aberrantly differentiated cells in benign pilomatrixoma reflect the normal hair follicle: immunohistochemical analysis of Ca-binding S100A2, S100A3 and S100A6 proteins. Br J Dermatol. 2005;152:314-320.
6. Farrier S, Morgan M. bcl-2 expression in pilomatricoma. Am J Dermatopathol. 1997;19:254-257.
7. Park SW, Suh KS, Wang HY, et al. Beta-catenin expression in the transitional cell zone of pilomatricoma. Br J Dermatol. 2001;145:624-629.
8. Kurokawa I, Kusumoto K, Bessho K, et al. Immunohistochemical expression of bone morphogenetic protein-2 in pilomatricoma. Br J Dermatol. 2000;143:754-758.
9. Cribier B, Asch PH, Regnier C, et al. Expression of human hair keratin basic 1 in pilomatrixoma: a study of 128 cases. Br J Dermatol. 1999;140:600-604.
10. Bayle P, Bazex J, Lamant L, et al. Multiple perforating and non perforating pilomatricomas in a patient with Churg-Strauss syndrome and Rubinstein-Taybi syndrome. J Eur Acad Dermatol Venereol. 2004;18:607-610.
11. Elder D, Elenitsas R, Ragsdale BD. Pilomatricoma. In: Elder D, Elenitsas R, Jaworsky C, et al, eds. Histopathology of the Skin. 8th ed. Philadelphia, PA: Lippincott-Raven; 1997:757-759.
12. Forbis R Jr, Helwig EB. Pilomatrixoma (calcifying epithelioma). Arch Dermatol. 1961;83:606-618.
13. Wood MG, Parhizgar B, Beerman H. Malignant pilomatricoma. Arch Dermatol. 1984;120:770-773.
14. Thomas RW, Perkins JA, Ruegemer JL, et al. Surgical excision of pilomatrixoma of the head and neck: a retrospective review of 26 cases. Ear Nose Throat J. 1999;78:541, 544-546, 548.
1. Malherbe A, Chenantais J. Note sur l'epithelioma calcifie des glandes sebacees. Prog Med. 1880;8:826-828.
2. Julian CG, Bowers PW. A clinical review of 209 pilomatricomas. J Am Acad Dermatol. 1998;39(2, pt 1):191-195.
3. Lozzi GP, Soyer HP, Fruehauf J, et al. Giant pilomatricoma. Am J Dermatopathol. 2007;29:286-289.
4. Hubbard VG, Whittaker SJ. Multiple familial pilomatricomas: an unusual case. J Cutan Pathol. 2004;31:281-283.
5. Kizawa K, Toyoda M, Ito M, et al. Aberrantly differentiated cells in benign pilomatrixoma reflect the normal hair follicle: immunohistochemical analysis of Ca-binding S100A2, S100A3 and S100A6 proteins. Br J Dermatol. 2005;152:314-320.
6. Farrier S, Morgan M. bcl-2 expression in pilomatricoma. Am J Dermatopathol. 1997;19:254-257.
7. Park SW, Suh KS, Wang HY, et al. Beta-catenin expression in the transitional cell zone of pilomatricoma. Br J Dermatol. 2001;145:624-629.
8. Kurokawa I, Kusumoto K, Bessho K, et al. Immunohistochemical expression of bone morphogenetic protein-2 in pilomatricoma. Br J Dermatol. 2000;143:754-758.
9. Cribier B, Asch PH, Regnier C, et al. Expression of human hair keratin basic 1 in pilomatrixoma: a study of 128 cases. Br J Dermatol. 1999;140:600-604.
10. Bayle P, Bazex J, Lamant L, et al. Multiple perforating and non perforating pilomatricomas in a patient with Churg-Strauss syndrome and Rubinstein-Taybi syndrome. J Eur Acad Dermatol Venereol. 2004;18:607-610.
11. Elder D, Elenitsas R, Ragsdale BD. Pilomatricoma. In: Elder D, Elenitsas R, Jaworsky C, et al, eds. Histopathology of the Skin. 8th ed. Philadelphia, PA: Lippincott-Raven; 1997:757-759.
12. Forbis R Jr, Helwig EB. Pilomatrixoma (calcifying epithelioma). Arch Dermatol. 1961;83:606-618.
13. Wood MG, Parhizgar B, Beerman H. Malignant pilomatricoma. Arch Dermatol. 1984;120:770-773.
14. Thomas RW, Perkins JA, Ruegemer JL, et al. Surgical excision of pilomatrixoma of the head and neck: a retrospective review of 26 cases. Ear Nose Throat J. 1999;78:541, 544-546, 548.
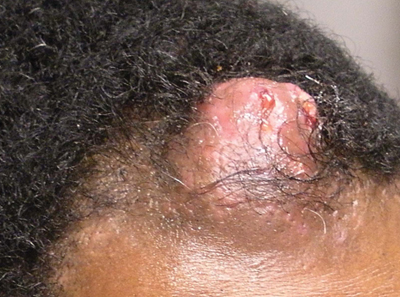
An otherwise healthy 40-year-old man presented for examination of a solitary nodular lesion on the frontal aspect of the scalp of 1 year’s duration. The lesion had rapidly increased in size in the 2 weeks prior to presentation. He presented to the emergency department after he noted pain and drainage from the lesion. Biopsy of the lesion revealed islands of pale eosinophilic shadow cells with an intense dermal infiltrate consisting of lymphocytes, histiocytes, plasma cells, and neutrophils.
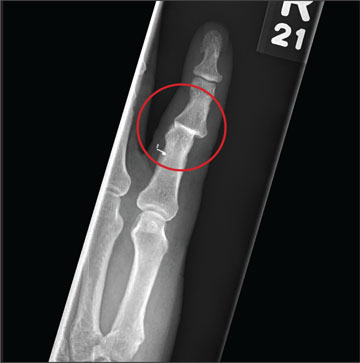
Hand Pain Following an Altercation
ANSWER
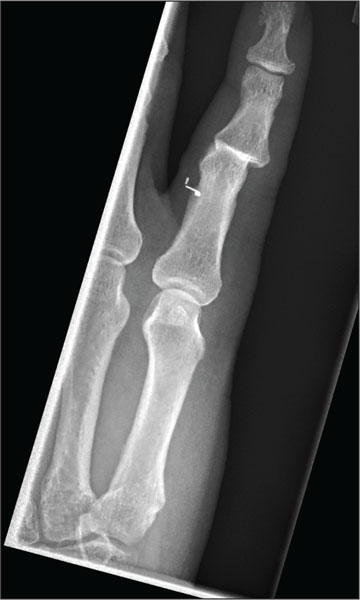
The radiograph shows moderate soft-tissue swelling with dislocation of the proximal interphalangeal joint. No definite fracture is seen. In addition, there are some metallic-appearing foreign bodies.
The patient was treated with closed reduction and splinting. He also received a referral to outpatient orthopedics for follow-up.
ANSWER
The radiograph shows moderate soft-tissue swelling with dislocation of the proximal interphalangeal joint. No definite fracture is seen. In addition, there are some metallic-appearing foreign bodies.
The patient was treated with closed reduction and splinting. He also received a referral to outpatient orthopedics for follow-up.
ANSWER
The radiograph shows moderate soft-tissue swelling with dislocation of the proximal interphalangeal joint. No definite fracture is seen. In addition, there are some metallic-appearing foreign bodies.
The patient was treated with closed reduction and splinting. He also received a referral to outpatient orthopedics for follow-up.
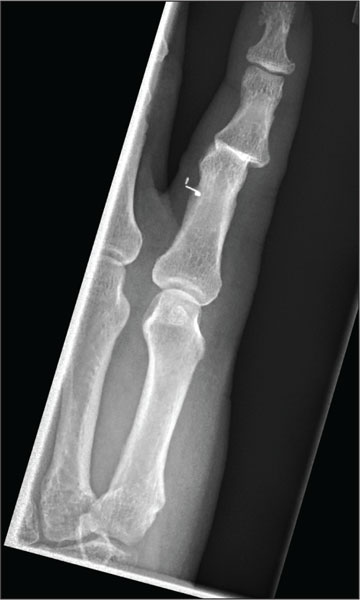
A 60-year-old man presents with a complaint of pain in his right fifth finger following an altercation. He is not sure exactly how the injury occurred, but he does recall that at one point his hand was twisted awkwardly. He denies any significant medical history. His vital signs are normal. Primary survey appears normal as well. On examination, you notice moderate swelling around the fifth finger of his right hand, which does appear to be slightly deformed. There are no obvious wounds or lacerations. He has moderate tenderness at the base of his finger. Range of motion is limited due to the swelling. Good capillary refill time is noted. The triage nurse already sent the patient for a radiograph of his finger (shown). What is your impression?
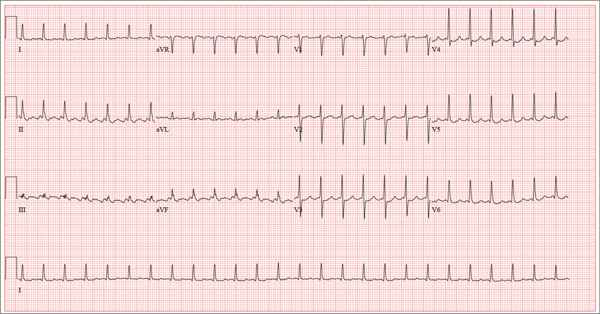
For Lethargic Patient, Trouble Is Brewing
ANSWER
The correct interpretation is an atrial tachycardia with 2:1 ventricular conduction. The ventricular rate is 87 beats/min (690 ms), and the atrial rate is 174 beats/min (345 ms). Two P waves are present for each QRS, which excludes a first-degree atrioventricular block. The less obvious P wave is found in the terminal portion of the QRS complex. (You may convince yourself of this by using calipers to measure the R-R interval, dividing that measurement in half, and then applying it to the ECG. You will see the P waves march through without changing the ventricular response.)
A nonspecific intraventricular conduction delay is also present. The QRS duration is > 100 ms; however, the criteria for right or left bundle branch block are absent.
A thorough investigation revealed that the clerk formulating the herbs for the tea was using, among other things, dried foxglove. Foxglove has been used as a remedy for lethargy in the elderly, presumably because it inadvertently treats symptoms of congestive heart failure. It was the tea consumption that accounted for the presence of digoxin in the patient’s blood. (Recall that there is a substantial overlap between therapeutic and toxic serum concentrations of digoxin.) When the patient stopped consuming the tea, his atrial tachycardia resolved, as did his symptoms.
ANSWER
The correct interpretation is an atrial tachycardia with 2:1 ventricular conduction. The ventricular rate is 87 beats/min (690 ms), and the atrial rate is 174 beats/min (345 ms). Two P waves are present for each QRS, which excludes a first-degree atrioventricular block. The less obvious P wave is found in the terminal portion of the QRS complex. (You may convince yourself of this by using calipers to measure the R-R interval, dividing that measurement in half, and then applying it to the ECG. You will see the P waves march through without changing the ventricular response.)
A nonspecific intraventricular conduction delay is also present. The QRS duration is > 100 ms; however, the criteria for right or left bundle branch block are absent.
A thorough investigation revealed that the clerk formulating the herbs for the tea was using, among other things, dried foxglove. Foxglove has been used as a remedy for lethargy in the elderly, presumably because it inadvertently treats symptoms of congestive heart failure. It was the tea consumption that accounted for the presence of digoxin in the patient’s blood. (Recall that there is a substantial overlap between therapeutic and toxic serum concentrations of digoxin.) When the patient stopped consuming the tea, his atrial tachycardia resolved, as did his symptoms.
ANSWER
The correct interpretation is an atrial tachycardia with 2:1 ventricular conduction. The ventricular rate is 87 beats/min (690 ms), and the atrial rate is 174 beats/min (345 ms). Two P waves are present for each QRS, which excludes a first-degree atrioventricular block. The less obvious P wave is found in the terminal portion of the QRS complex. (You may convince yourself of this by using calipers to measure the R-R interval, dividing that measurement in half, and then applying it to the ECG. You will see the P waves march through without changing the ventricular response.)
A nonspecific intraventricular conduction delay is also present. The QRS duration is > 100 ms; however, the criteria for right or left bundle branch block are absent.
A thorough investigation revealed that the clerk formulating the herbs for the tea was using, among other things, dried foxglove. Foxglove has been used as a remedy for lethargy in the elderly, presumably because it inadvertently treats symptoms of congestive heart failure. It was the tea consumption that accounted for the presence of digoxin in the patient’s blood. (Recall that there is a substantial overlap between therapeutic and toxic serum concentrations of digoxin.) When the patient stopped consuming the tea, his atrial tachycardia resolved, as did his symptoms.
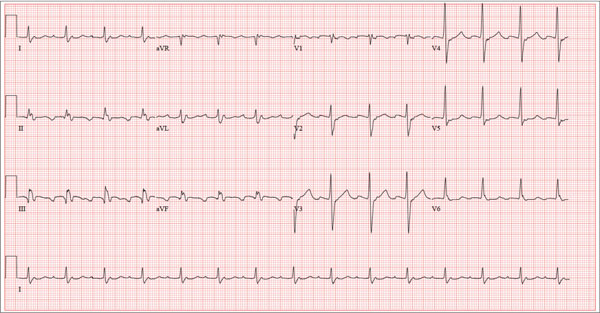
A 72-year-old man presents with a primary complaint of lethargy. He emigrated from Southeast Asia to the United States about a year ago and neither speaks nor understands English. His grandson, who is fluent, accompanies him to his appointment. Through his grandson, the patient explains that he has become increasingly tired in the past four months—to the extent that exercise and activities of daily living have become difficult. The patient’s libido also has been affected. In an effort to correct this, he visited a local Asian goods store, where he was given a mixture of herbs from which to brew tea to treat his symptoms. For three weeks, he consumed the tea twice daily. Initially, his energy, stamina, and libido improved. However, his symptoms eventually returned, so he doubled his tea consumption with the idea that this would improve his condition. Unfortunately, in addition to his lethargy, he is now experiencing palpitations, a fluttering sensation in his chest, and occasional dizziness. He denies chest pain, shortness of breath, nocturnal dyspnea, syncope, or near syncope. Medical history is difficult to elicit. He denies prior history of hypertension, myocardial infarction, congestive heart failure, or diabetes. Neither he nor his grandson understands the concept of arrhythmias (eg, atrial fibrillation). He was treated for tuberculosis as a child and has had no recurrence. He has had no surgeries. The patient takes no prescribed medications. He does, however, use herbal products including ginseng, horny goat weed, and fenugreek (in addition to his herbal tea). He has no known drug allergies. Social history reveals that the patient lives with his son’s family, having moved to the US from Thailand after his wife died of old age. He worked as a farmer his entire life. He drinks one ounce of whiskey daily and smokes 1 to 1.5 packs of cigarettes a day. The review of systems is noncontributory. His grandson is reluctant to ask the patient many questions regarding his health, once he notices his grandfather’s agitation at answering questions. The physical exam reveals a thin, elderly male with weathered skin who is in no acute distress. Vital signs include a blood pressure of 118/62 mm Hg; pulse, 80 beats/min and regular; respiratory rate, 16 breaths/min; and temperature, 97.8°F. His height is 62 in and his weight, 117 lb. The HEENT exam is remarkable for arcus senilis and multiple missing teeth. There is no jugular distention, and the thyroid is not enlarged. The lungs reveal coarse breath sounds that clear with coughing in all lung fields. (The patient has an occasional harsh cough.) The cardiac exam is positive for a grade II/VI systolic murmur best heard at the left upper sternal border, which radiates to the carotid arteries. The rhythm is regular at a rate of 80 beats/min, and there are no clicks or rubs. The abdomen is scaphoid, soft, and nontender, with no palpable masses. The peripheral pulses are strong and equal bilaterally. Extremities demonstrate full range of motion, and the neurologic exam is grossly intact. Routine laboratory tests including a complete blood count and electrolyte panel are obtained. Because you are unsure of his medication regimen, you order a toxicology screen. You are surprised to see a serum digoxin level of 0.7 ng/mL. Finally, given the patient’s symptoms of palpitations and dizziness, you order an ECG. It shows the following: a ventricular rate of 87 beats/min; PR interval, 218 ms; QRS duration, 130 ms; QT/QTc interval, 416/500 ms; P axis, 24°; R axis, 49°; and T axis, 45°. What is your interpretation of this ECG?
These Old Lesions? She’s Had Them for Years …
ANSWER
The correct answer is disseminated superficial actinic porokeratosis (DSAP; choice “a”). This condition, caused by an inherited defect of the SART3 gene, is seen mostly on the sun-exposed skin of middle-aged women.
Stasis dermatitis (choice “b”) can cause a number of skin changes, but not the discrete annular lesions seen with DSAP.
Seborrheic keratoses (choice “c”) are common on the legs. However, they don’t display this same morphology.
Nummular eczema (choice “d”) presents with annular papulosquamous lesions (as opposed to the fixed lesions seen with DSAP), often on the legs and lower trunk, but without the thready circumferential scaly border.
Continue reading for Joe Monroe's discussion...
DISCUSSION
Leg skin is prey to an astonishing array of problems; many have to do with increased hydrostatic pressure (eg, venous stasis disease), with the almost complete lack of sebaceous glands (eg, nummular eczema), or with the simple fact of being “in harm’s way.” And there is no law that says a given patient can’t have more than one problem at a time, co-existing and serving to confuse the examiner. Such is the case with this patient.
Her concern about possible blood clots is misplaced but understandable. Deep vein thromboses would not present in multiples, would not be on the surface or scaly, and would almost certainly be painful.
The fixed nature of this patient’s scaly lesions is extremely significant—but only if you know about DSAP, which typically manifests in the third decade of life and slowly worsens. The lesions’ highly palpable and unique scaly border makes them hard to leave alone. This might not be a problem except for the warfarin, which makes otherwise minor trauma visible as purpuric macules. Chronic sun damage tends to accentuate them as well. The positive family history is nicely corroborative and quite common.
The brown macules on the patient’s legs are solar lentigines (sun-caused freckles), which many patients (and even younger providers) erroneously call “age spots.” When these individuals become “aged,” they’ll understand that there is no such thing as an age spot.
This patient could easily have had nummular eczema, but not for 30 years! Those lesions, treated or not, will come and go. But not DSAP, about which many questions remain: If they’re caused by sun exposure, why don’t we see them more often on the face and arms? And why don’t we see them on the sun-damaged skin of older men?
If needed, a biopsy could have been performed. It would have been confirmatory of the diagnosis and effectively would have ruled out the other items in the differential, including wart, squamous cell carcinoma, and actinic or seborrheic keratosis.
ANSWER
The correct answer is disseminated superficial actinic porokeratosis (DSAP; choice “a”). This condition, caused by an inherited defect of the SART3 gene, is seen mostly on the sun-exposed skin of middle-aged women.
Stasis dermatitis (choice “b”) can cause a number of skin changes, but not the discrete annular lesions seen with DSAP.
Seborrheic keratoses (choice “c”) are common on the legs. However, they don’t display this same morphology.
Nummular eczema (choice “d”) presents with annular papulosquamous lesions (as opposed to the fixed lesions seen with DSAP), often on the legs and lower trunk, but without the thready circumferential scaly border.
Continue reading for Joe Monroe's discussion...
DISCUSSION
Leg skin is prey to an astonishing array of problems; many have to do with increased hydrostatic pressure (eg, venous stasis disease), with the almost complete lack of sebaceous glands (eg, nummular eczema), or with the simple fact of being “in harm’s way.” And there is no law that says a given patient can’t have more than one problem at a time, co-existing and serving to confuse the examiner. Such is the case with this patient.
Her concern about possible blood clots is misplaced but understandable. Deep vein thromboses would not present in multiples, would not be on the surface or scaly, and would almost certainly be painful.
The fixed nature of this patient’s scaly lesions is extremely significant—but only if you know about DSAP, which typically manifests in the third decade of life and slowly worsens. The lesions’ highly palpable and unique scaly border makes them hard to leave alone. This might not be a problem except for the warfarin, which makes otherwise minor trauma visible as purpuric macules. Chronic sun damage tends to accentuate them as well. The positive family history is nicely corroborative and quite common.
The brown macules on the patient’s legs are solar lentigines (sun-caused freckles), which many patients (and even younger providers) erroneously call “age spots.” When these individuals become “aged,” they’ll understand that there is no such thing as an age spot.
This patient could easily have had nummular eczema, but not for 30 years! Those lesions, treated or not, will come and go. But not DSAP, about which many questions remain: If they’re caused by sun exposure, why don’t we see them more often on the face and arms? And why don’t we see them on the sun-damaged skin of older men?
If needed, a biopsy could have been performed. It would have been confirmatory of the diagnosis and effectively would have ruled out the other items in the differential, including wart, squamous cell carcinoma, and actinic or seborrheic keratosis.
ANSWER
The correct answer is disseminated superficial actinic porokeratosis (DSAP; choice “a”). This condition, caused by an inherited defect of the SART3 gene, is seen mostly on the sun-exposed skin of middle-aged women.
Stasis dermatitis (choice “b”) can cause a number of skin changes, but not the discrete annular lesions seen with DSAP.
Seborrheic keratoses (choice “c”) are common on the legs. However, they don’t display this same morphology.
Nummular eczema (choice “d”) presents with annular papulosquamous lesions (as opposed to the fixed lesions seen with DSAP), often on the legs and lower trunk, but without the thready circumferential scaly border.
Continue reading for Joe Monroe's discussion...
DISCUSSION
Leg skin is prey to an astonishing array of problems; many have to do with increased hydrostatic pressure (eg, venous stasis disease), with the almost complete lack of sebaceous glands (eg, nummular eczema), or with the simple fact of being “in harm’s way.” And there is no law that says a given patient can’t have more than one problem at a time, co-existing and serving to confuse the examiner. Such is the case with this patient.
Her concern about possible blood clots is misplaced but understandable. Deep vein thromboses would not present in multiples, would not be on the surface or scaly, and would almost certainly be painful.
The fixed nature of this patient’s scaly lesions is extremely significant—but only if you know about DSAP, which typically manifests in the third decade of life and slowly worsens. The lesions’ highly palpable and unique scaly border makes them hard to leave alone. This might not be a problem except for the warfarin, which makes otherwise minor trauma visible as purpuric macules. Chronic sun damage tends to accentuate them as well. The positive family history is nicely corroborative and quite common.
The brown macules on the patient’s legs are solar lentigines (sun-caused freckles), which many patients (and even younger providers) erroneously call “age spots.” When these individuals become “aged,” they’ll understand that there is no such thing as an age spot.
This patient could easily have had nummular eczema, but not for 30 years! Those lesions, treated or not, will come and go. But not DSAP, about which many questions remain: If they’re caused by sun exposure, why don’t we see them more often on the face and arms? And why don’t we see them on the sun-damaged skin of older men?
If needed, a biopsy could have been performed. It would have been confirmatory of the diagnosis and effectively would have ruled out the other items in the differential, including wart, squamous cell carcinoma, and actinic or seborrheic keratosis.
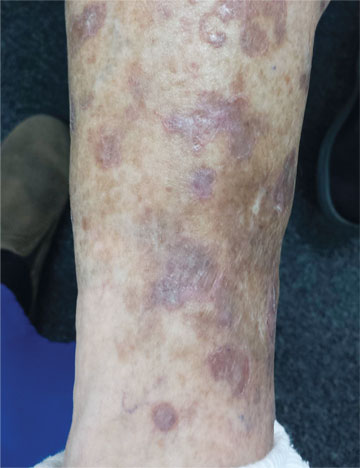
A 65-year-old woman is referred to dermatology with discoloration of her legs that started several weeks ago. Her family suggested it might be “blood clots,” although she has been taking warfarin since she was diagnosed with atrial fibrillation several months ago. Her dermatologic condition is basically asymptomatic, but the patient admits to scratching her legs, saying it’s “hard to leave them alone.” On further questioning, she reveals that she has had “rough places” on her legs for at least 20 years and volunteers that her sister had the same problem, which was diagnosed years ago as “fungal infection.” Both she and her sister spent a great deal of time in the sun as children, long before sunscreen was invented. The patient is otherwise fairly healthy. She takes medication for her lipids, as well as daily vitamins. Her atrial fibrillation is under control and requires no medications other than the warfarin. A great deal of focal discoloration is seen on both legs, circumferentially distributed from well below the knees to just above the ankles. Many of the lesions are brown macules, but more are purplish-red, annular, and scaly. On closer examination, these lesions—the ones the patient says she has had for decades—have a very fine, thready, scaly border that palpation reveals to be tough and adherent. They average about 2 cm in diameter. There are no such lesions noted elsewhere on the patient’s skin. There is, however, abundant evidence of excessive sun exposure, characterized by a multitude of solar lentigines, many fine wrinkles, and extremely thin arm skin.

Verrucous Nodule on the Upper Lip
The Diagnosis: Disseminated Coccidioidomycosis
Fungi of the genus Coccidioides cause coccidioidomycosis and live in the soil of endemic areas including the southwestern United States (eg, Arizona, New Mexico, California) and Mexico. Coccidioides is a dimorphic fungus with parasitic and infectious saprophytic phases. Each year there are approximately 150,000 new infections of coccidioidomycosis in the United States, almost exclusively in the southwest.1 Coccidioidomycosis typically is an asymptomatic or mild infection in an immunocompetent patient. Although the lungs are nearly always the primary sites of infection, common sites of dissemination include the skin, meninges, bones, and joints. The skin is the most common site of disseminated, or secondary, coccidioidomycosis.2 Less commonly and usually caused by traumatic implantation, the skin is the site of primary infection.
Disseminated coccidioidomycosis occurs in approximately 1 in 200 infected individuals.2,3 Certain populations of patients are more likely to be affected by disseminated coccidioidomycosis, including specific ethnic groups such as black individuals, Filipinos, and Mexicans4,5; pregnant women6; and immunosuppressed patients such as those with human immunodeficiency virus, hematogenous malignancy, or organ transplantation.7-9 When skin lesions are present, they usually develop after the initial lung manifestation. The location of the lesion in cutaneous disseminated disease can be highly variable, but the face and head are the most common locations (30%).10
Cutaneous manifestations of coccidioidomycosis may be classified as being caused by the presence of the organism in the skin (organism specific) or a reactive process. Organism-specific cutaneous lesions are commonly due to systemic disease with secondary skin involvement, but they also may be due to a primary infection. These organism-specific lesions can present as papules, nodules, macules, verrucous plaques, abscesses, or pustules. Reactive cutaneous manifestations are only associated with disseminated disease; do not contain any organisms; and include manifestations such as erythema nodosum, acute exanthem, erythema multiforme, and possibly Sweet syndrome.11
The clinical differential diagnosis of cutaneous coccidioidomycosis includes other
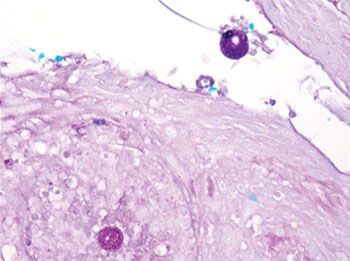
mycoses such as histoplasmosis and blastomycosis, as well as tuberculosis, sarcoidosis, basal cell and squamous cell carcinoma, and verruca vulgaris. The diagnosis of cutaneous coccidioidomycosis can be made with skin biopsy, culture, and serologic tests. The characteristic spherules can be visualized on routine hematoxylin and eosin stain or more readily with fungal stains (Figure). Spherules are thick walled and distinguishable from other fungi because of the characteristic endospores inside as well as their larger size. Organisms also may be detected via culture within 2 to 5 days.
Distinguishing primary cutaneous from disseminated skin lesions can be challenging but can have notable treatment implications. Although histology typically cannot distinguish primary from disseminated cutaneous infections, clinical history and serologic studies have been found to be useful. In disseminated disease, IgG antibodies are elevated, while in primary cutaneous disease, IgM antibodies are elevated but typically not IgG.12 Therefore, tube precipitin and latex particle agglutination tests that detect IgM antibodies should be positive in primary infections.13 Primary lesions can spontaneously resolve within months to years and may not require treatment if symptomatic, while secondary lesions must be therapeutically addressed.12 Despite the lack of treatment needed, primary cutaneous infections often are treated with azoles.14 In contrast, disseminated cutaneous infection requires systemic therapy. Treatment of disseminated infection with cutaneous coccidioidomycosis typically includes amphotericin B until a clinical response is achieved and antibody titers decline. Amphotericin B can then be replaced with an oral azole such as itraconozole or fluconazole.15
The patient discussed in this case demonstrates the typical clinical presentation of disseminated coccidioidomycosis with classical and diagnostic pathology. This case also highlights the importance of a detailed travel history; in the era of globalization, patients often present with diseases in nonendemic areas. Clinicians must consider the diagnosis of coccidioidomycosis, even in immunocompetent patients in nonendemic areas when their history and presentation are appropriate. The diagnosis should be confirmed with biopsy, culture, and/or serology.
1. Galgiani JN, Ampel NM, Blair JE, et al. Coccidioidomycosis [published online ahead of print September 20, 2005]. Clin Infect Dis. 2005;41:1217-1223.
2. Chiller TM, Galgiani JN, Stevens DA. Coccidioidomycosis. Infect Dis Clin North Am. 2003;17:41-57, vii.
3. Rance BR, Elston DM. Disseminated coccidioidomyclosis discovered during routine skin cancer screening. Cutis. 2002;70:70-72.
4. Einstein HE, Johnson RH. Coccidioidomycosis: new aspects of epidemiology and therapy [comment in Clin Infect Dis. 1994;18:470]. Clin Infect Dis. 1993;16:349- 354.
5. Crum NF, Ballon-Landa G. Coccidioidomycosis in pregnancy: case report and review of the literature. Am J Med. 2006;119:e11-e17.
6. Caldwell JW, Arsura EL, Kilgore WB, et al. Coccidioidomycosis in pregnancy during an epidemic in California. Obstet Gynecol. 2000;95:236-239.
7. Singh VR, Smith DK, Lawerence J, et al. Coccidioidomy-cosis in patients infected with human immunodeficiency virus: review of 91 cases at a single institution. Clin Infect Dis. 1996;23:563-568.
8. Blair JE, Logan JL. Coccidioidomycosis in solid organ transplantation [published online ahead of print October 4, 2001]. Clin Infect Dis. 2001;33:1536-1544.
9. Riley DK, Galgiani JN, O’Donnell MR, et al. Coccidioidomycosis in bone marrow transplant recipients. Transplantation. 1993;56:1531-1533.
10. Carpenter JB, Feldman JS, Leyva WH, et al. Clinical and pathologic characteristics of disseminated cutaneous coccidioidomycosis. J Am Acad Dermatol. 2010;62:831-837.
11. DiCaudo DJ. Coccidioidomycosis: a review and update. J Am Acad Dermatol. 2006;55:929-942; quiz 943-945.
12. Chang A, Tung RC, McGillis TS, et al. Primary cutaneous coccidioidomycosis. J Am Acad Dermatol. 2003;49:944-949.
13. Wilson JW, Smith CE, Plunkett OA. Primary cutaneous coccidioidomycosis: the criteria for diagnosis and report of a case. Calif Med. 1953;79:233-239.
14. Blair JE. State-of-the-art treatment of coccidioidomycosis: skin and soft-tissue infections [published online ahead of print March 1, 2007]. Ann N Y Acad Sci. 2007;1111:411-421.
15. Galgiani JN, Ampel NM, Catanzaro A, et al. Practice guidelines for the treatment of coccidioidomycosis. Infectious Diseases Society of America [published online ahead of print April 20, 2000]. Clin Infect Dis. 2000;30:658-661.
The Diagnosis: Disseminated Coccidioidomycosis
Fungi of the genus Coccidioides cause coccidioidomycosis and live in the soil of endemic areas including the southwestern United States (eg, Arizona, New Mexico, California) and Mexico. Coccidioides is a dimorphic fungus with parasitic and infectious saprophytic phases. Each year there are approximately 150,000 new infections of coccidioidomycosis in the United States, almost exclusively in the southwest.1 Coccidioidomycosis typically is an asymptomatic or mild infection in an immunocompetent patient. Although the lungs are nearly always the primary sites of infection, common sites of dissemination include the skin, meninges, bones, and joints. The skin is the most common site of disseminated, or secondary, coccidioidomycosis.2 Less commonly and usually caused by traumatic implantation, the skin is the site of primary infection.
Disseminated coccidioidomycosis occurs in approximately 1 in 200 infected individuals.2,3 Certain populations of patients are more likely to be affected by disseminated coccidioidomycosis, including specific ethnic groups such as black individuals, Filipinos, and Mexicans4,5; pregnant women6; and immunosuppressed patients such as those with human immunodeficiency virus, hematogenous malignancy, or organ transplantation.7-9 When skin lesions are present, they usually develop after the initial lung manifestation. The location of the lesion in cutaneous disseminated disease can be highly variable, but the face and head are the most common locations (30%).10
Cutaneous manifestations of coccidioidomycosis may be classified as being caused by the presence of the organism in the skin (organism specific) or a reactive process. Organism-specific cutaneous lesions are commonly due to systemic disease with secondary skin involvement, but they also may be due to a primary infection. These organism-specific lesions can present as papules, nodules, macules, verrucous plaques, abscesses, or pustules. Reactive cutaneous manifestations are only associated with disseminated disease; do not contain any organisms; and include manifestations such as erythema nodosum, acute exanthem, erythema multiforme, and possibly Sweet syndrome.11
The clinical differential diagnosis of cutaneous coccidioidomycosis includes other
mycoses such as histoplasmosis and blastomycosis, as well as tuberculosis, sarcoidosis, basal cell and squamous cell carcinoma, and verruca vulgaris. The diagnosis of cutaneous coccidioidomycosis can be made with skin biopsy, culture, and serologic tests. The characteristic spherules can be visualized on routine hematoxylin and eosin stain or more readily with fungal stains (Figure). Spherules are thick walled and distinguishable from other fungi because of the characteristic endospores inside as well as their larger size. Organisms also may be detected via culture within 2 to 5 days.
Distinguishing primary cutaneous from disseminated skin lesions can be challenging but can have notable treatment implications. Although histology typically cannot distinguish primary from disseminated cutaneous infections, clinical history and serologic studies have been found to be useful. In disseminated disease, IgG antibodies are elevated, while in primary cutaneous disease, IgM antibodies are elevated but typically not IgG.12 Therefore, tube precipitin and latex particle agglutination tests that detect IgM antibodies should be positive in primary infections.13 Primary lesions can spontaneously resolve within months to years and may not require treatment if symptomatic, while secondary lesions must be therapeutically addressed.12 Despite the lack of treatment needed, primary cutaneous infections often are treated with azoles.14 In contrast, disseminated cutaneous infection requires systemic therapy. Treatment of disseminated infection with cutaneous coccidioidomycosis typically includes amphotericin B until a clinical response is achieved and antibody titers decline. Amphotericin B can then be replaced with an oral azole such as itraconozole or fluconazole.15
The patient discussed in this case demonstrates the typical clinical presentation of disseminated coccidioidomycosis with classical and diagnostic pathology. This case also highlights the importance of a detailed travel history; in the era of globalization, patients often present with diseases in nonendemic areas. Clinicians must consider the diagnosis of coccidioidomycosis, even in immunocompetent patients in nonendemic areas when their history and presentation are appropriate. The diagnosis should be confirmed with biopsy, culture, and/or serology.
The Diagnosis: Disseminated Coccidioidomycosis
Fungi of the genus Coccidioides cause coccidioidomycosis and live in the soil of endemic areas including the southwestern United States (eg, Arizona, New Mexico, California) and Mexico. Coccidioides is a dimorphic fungus with parasitic and infectious saprophytic phases. Each year there are approximately 150,000 new infections of coccidioidomycosis in the United States, almost exclusively in the southwest.1 Coccidioidomycosis typically is an asymptomatic or mild infection in an immunocompetent patient. Although the lungs are nearly always the primary sites of infection, common sites of dissemination include the skin, meninges, bones, and joints. The skin is the most common site of disseminated, or secondary, coccidioidomycosis.2 Less commonly and usually caused by traumatic implantation, the skin is the site of primary infection.
Disseminated coccidioidomycosis occurs in approximately 1 in 200 infected individuals.2,3 Certain populations of patients are more likely to be affected by disseminated coccidioidomycosis, including specific ethnic groups such as black individuals, Filipinos, and Mexicans4,5; pregnant women6; and immunosuppressed patients such as those with human immunodeficiency virus, hematogenous malignancy, or organ transplantation.7-9 When skin lesions are present, they usually develop after the initial lung manifestation. The location of the lesion in cutaneous disseminated disease can be highly variable, but the face and head are the most common locations (30%).10
Cutaneous manifestations of coccidioidomycosis may be classified as being caused by the presence of the organism in the skin (organism specific) or a reactive process. Organism-specific cutaneous lesions are commonly due to systemic disease with secondary skin involvement, but they also may be due to a primary infection. These organism-specific lesions can present as papules, nodules, macules, verrucous plaques, abscesses, or pustules. Reactive cutaneous manifestations are only associated with disseminated disease; do not contain any organisms; and include manifestations such as erythema nodosum, acute exanthem, erythema multiforme, and possibly Sweet syndrome.11
The clinical differential diagnosis of cutaneous coccidioidomycosis includes other
mycoses such as histoplasmosis and blastomycosis, as well as tuberculosis, sarcoidosis, basal cell and squamous cell carcinoma, and verruca vulgaris. The diagnosis of cutaneous coccidioidomycosis can be made with skin biopsy, culture, and serologic tests. The characteristic spherules can be visualized on routine hematoxylin and eosin stain or more readily with fungal stains (Figure). Spherules are thick walled and distinguishable from other fungi because of the characteristic endospores inside as well as their larger size. Organisms also may be detected via culture within 2 to 5 days.
Distinguishing primary cutaneous from disseminated skin lesions can be challenging but can have notable treatment implications. Although histology typically cannot distinguish primary from disseminated cutaneous infections, clinical history and serologic studies have been found to be useful. In disseminated disease, IgG antibodies are elevated, while in primary cutaneous disease, IgM antibodies are elevated but typically not IgG.12 Therefore, tube precipitin and latex particle agglutination tests that detect IgM antibodies should be positive in primary infections.13 Primary lesions can spontaneously resolve within months to years and may not require treatment if symptomatic, while secondary lesions must be therapeutically addressed.12 Despite the lack of treatment needed, primary cutaneous infections often are treated with azoles.14 In contrast, disseminated cutaneous infection requires systemic therapy. Treatment of disseminated infection with cutaneous coccidioidomycosis typically includes amphotericin B until a clinical response is achieved and antibody titers decline. Amphotericin B can then be replaced with an oral azole such as itraconozole or fluconazole.15
The patient discussed in this case demonstrates the typical clinical presentation of disseminated coccidioidomycosis with classical and diagnostic pathology. This case also highlights the importance of a detailed travel history; in the era of globalization, patients often present with diseases in nonendemic areas. Clinicians must consider the diagnosis of coccidioidomycosis, even in immunocompetent patients in nonendemic areas when their history and presentation are appropriate. The diagnosis should be confirmed with biopsy, culture, and/or serology.
1. Galgiani JN, Ampel NM, Blair JE, et al. Coccidioidomycosis [published online ahead of print September 20, 2005]. Clin Infect Dis. 2005;41:1217-1223.
2. Chiller TM, Galgiani JN, Stevens DA. Coccidioidomycosis. Infect Dis Clin North Am. 2003;17:41-57, vii.
3. Rance BR, Elston DM. Disseminated coccidioidomyclosis discovered during routine skin cancer screening. Cutis. 2002;70:70-72.
4. Einstein HE, Johnson RH. Coccidioidomycosis: new aspects of epidemiology and therapy [comment in Clin Infect Dis. 1994;18:470]. Clin Infect Dis. 1993;16:349- 354.
5. Crum NF, Ballon-Landa G. Coccidioidomycosis in pregnancy: case report and review of the literature. Am J Med. 2006;119:e11-e17.
6. Caldwell JW, Arsura EL, Kilgore WB, et al. Coccidioidomycosis in pregnancy during an epidemic in California. Obstet Gynecol. 2000;95:236-239.
7. Singh VR, Smith DK, Lawerence J, et al. Coccidioidomy-cosis in patients infected with human immunodeficiency virus: review of 91 cases at a single institution. Clin Infect Dis. 1996;23:563-568.
8. Blair JE, Logan JL. Coccidioidomycosis in solid organ transplantation [published online ahead of print October 4, 2001]. Clin Infect Dis. 2001;33:1536-1544.
9. Riley DK, Galgiani JN, O’Donnell MR, et al. Coccidioidomycosis in bone marrow transplant recipients. Transplantation. 1993;56:1531-1533.
10. Carpenter JB, Feldman JS, Leyva WH, et al. Clinical and pathologic characteristics of disseminated cutaneous coccidioidomycosis. J Am Acad Dermatol. 2010;62:831-837.
11. DiCaudo DJ. Coccidioidomycosis: a review and update. J Am Acad Dermatol. 2006;55:929-942; quiz 943-945.
12. Chang A, Tung RC, McGillis TS, et al. Primary cutaneous coccidioidomycosis. J Am Acad Dermatol. 2003;49:944-949.
13. Wilson JW, Smith CE, Plunkett OA. Primary cutaneous coccidioidomycosis: the criteria for diagnosis and report of a case. Calif Med. 1953;79:233-239.
14. Blair JE. State-of-the-art treatment of coccidioidomycosis: skin and soft-tissue infections [published online ahead of print March 1, 2007]. Ann N Y Acad Sci. 2007;1111:411-421.
15. Galgiani JN, Ampel NM, Catanzaro A, et al. Practice guidelines for the treatment of coccidioidomycosis. Infectious Diseases Society of America [published online ahead of print April 20, 2000]. Clin Infect Dis. 2000;30:658-661.
1. Galgiani JN, Ampel NM, Blair JE, et al. Coccidioidomycosis [published online ahead of print September 20, 2005]. Clin Infect Dis. 2005;41:1217-1223.
2. Chiller TM, Galgiani JN, Stevens DA. Coccidioidomycosis. Infect Dis Clin North Am. 2003;17:41-57, vii.
3. Rance BR, Elston DM. Disseminated coccidioidomyclosis discovered during routine skin cancer screening. Cutis. 2002;70:70-72.
4. Einstein HE, Johnson RH. Coccidioidomycosis: new aspects of epidemiology and therapy [comment in Clin Infect Dis. 1994;18:470]. Clin Infect Dis. 1993;16:349- 354.
5. Crum NF, Ballon-Landa G. Coccidioidomycosis in pregnancy: case report and review of the literature. Am J Med. 2006;119:e11-e17.
6. Caldwell JW, Arsura EL, Kilgore WB, et al. Coccidioidomycosis in pregnancy during an epidemic in California. Obstet Gynecol. 2000;95:236-239.
7. Singh VR, Smith DK, Lawerence J, et al. Coccidioidomy-cosis in patients infected with human immunodeficiency virus: review of 91 cases at a single institution. Clin Infect Dis. 1996;23:563-568.
8. Blair JE, Logan JL. Coccidioidomycosis in solid organ transplantation [published online ahead of print October 4, 2001]. Clin Infect Dis. 2001;33:1536-1544.
9. Riley DK, Galgiani JN, O’Donnell MR, et al. Coccidioidomycosis in bone marrow transplant recipients. Transplantation. 1993;56:1531-1533.
10. Carpenter JB, Feldman JS, Leyva WH, et al. Clinical and pathologic characteristics of disseminated cutaneous coccidioidomycosis. J Am Acad Dermatol. 2010;62:831-837.
11. DiCaudo DJ. Coccidioidomycosis: a review and update. J Am Acad Dermatol. 2006;55:929-942; quiz 943-945.
12. Chang A, Tung RC, McGillis TS, et al. Primary cutaneous coccidioidomycosis. J Am Acad Dermatol. 2003;49:944-949.
13. Wilson JW, Smith CE, Plunkett OA. Primary cutaneous coccidioidomycosis: the criteria for diagnosis and report of a case. Calif Med. 1953;79:233-239.
14. Blair JE. State-of-the-art treatment of coccidioidomycosis: skin and soft-tissue infections [published online ahead of print March 1, 2007]. Ann N Y Acad Sci. 2007;1111:411-421.
15. Galgiani JN, Ampel NM, Catanzaro A, et al. Practice guidelines for the treatment of coccidioidomycosis. Infectious Diseases Society of America [published online ahead of print April 20, 2000]. Clin Infect Dis. 2000;30:658-661.
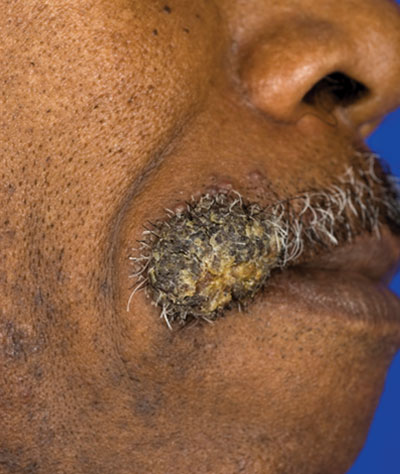
A 62-year-old black man presented to a dermatologist in the northeastern United States with a verrucous nontender nodule on the upper lip after traveling to southern California 1 month prior. The patient was not immunocompromised but reported a recent febrile upper respiratory illness.
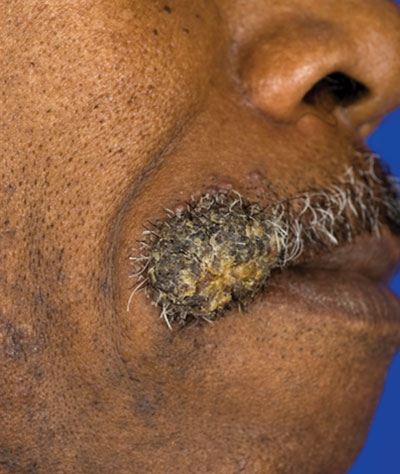
Thick Plaques on the Foot
The Diagnosis: Prayer Callus
After our patient demonstrated his routine position for prayer (Figure), the
diagnosis of prayer callus was confirmed. We suggested he use a foam pad under his foot during prayer. The patient did not return for follow-up.
Prayer calluses have been documented in the literature as prayer nodules1-3 or marks.4 Callus is a preferred term, as it implies the act of repeated pressure or friction to an area of the skin. It is more descriptive than marks and avoids the inclusion of nodules caused by different prayer or religious activities such as ritual scarring.5
Abanmi et al4 studied a large group (N=349) of Muslim patients with regular praying habits and found a high prevalence of what they referred to as prayer marks, defined by lichenification and/or hyperpigmentation, on the knees, forehead, ankles, and dorsal aspects of the feet. They studied the histopathology of 33 marks. No statistical analysis was performed. They reported common histologic findings of compact orthokeratosis, hypergranulosis, dermal papillary fibrosis, and dermal vascularization. In contrast to lichen simplex chronicus, the authors found that the dermal fibrosis did not exhibit collagen bundles perpendicular to the epidermis.4
This study found that a higher prevalence of lichenification was observed on the left foot (males, 57%; females, 39%) than the right foot (males, 14%; females, 19%), which was attributed to a more typical prayer position that placed pressure on the left foot.4 Our patient only had calluses on his left foot, which was consistent with the prayer position.
Treatment options for prayer calluses include debridement, either mechanical or with topical keratolytic preparations. There is a high likelihood of recurrence if prayer practices are not changed. Optimally, more definitive treatment, which can be combined with initial debridement, would be position adjustment to lessen pressure or friction to the area or protection over the area with a cushion, as we attempted with our patient in the form of a foam pad.
1. Kahana M, Cohen M, Ronnen M, et al. Prayer nodules in Moslem men. Cutis. 1986;38:281-282.
2. Vollum DI, Azadeh B. Prayer nodules. Clin Exp Dermatol. 1979;4:39-47.
3. Kumar PV, Hambarsoomian B. Prayer nodules fine needle aspiration cytologic findings. Acta Cytol. 1988;32:83-85.
4. Abanmi AA, Al Zouman AY, Al Hussaini H, et al. Prayer marks. Int J Dermatol. 2002;41:411-414.
5. Bourrel P. Problems related to African customs and ritual mutilations [in French]. Contracept Fertil Sex (Paris). 1983;11:1351-1358.
The Diagnosis: Prayer Callus
After our patient demonstrated his routine position for prayer (Figure), the
diagnosis of prayer callus was confirmed. We suggested he use a foam pad under his foot during prayer. The patient did not return for follow-up.
Prayer calluses have been documented in the literature as prayer nodules1-3 or marks.4 Callus is a preferred term, as it implies the act of repeated pressure or friction to an area of the skin. It is more descriptive than marks and avoids the inclusion of nodules caused by different prayer or religious activities such as ritual scarring.5
Abanmi et al4 studied a large group (N=349) of Muslim patients with regular praying habits and found a high prevalence of what they referred to as prayer marks, defined by lichenification and/or hyperpigmentation, on the knees, forehead, ankles, and dorsal aspects of the feet. They studied the histopathology of 33 marks. No statistical analysis was performed. They reported common histologic findings of compact orthokeratosis, hypergranulosis, dermal papillary fibrosis, and dermal vascularization. In contrast to lichen simplex chronicus, the authors found that the dermal fibrosis did not exhibit collagen bundles perpendicular to the epidermis.4
This study found that a higher prevalence of lichenification was observed on the left foot (males, 57%; females, 39%) than the right foot (males, 14%; females, 19%), which was attributed to a more typical prayer position that placed pressure on the left foot.4 Our patient only had calluses on his left foot, which was consistent with the prayer position.
Treatment options for prayer calluses include debridement, either mechanical or with topical keratolytic preparations. There is a high likelihood of recurrence if prayer practices are not changed. Optimally, more definitive treatment, which can be combined with initial debridement, would be position adjustment to lessen pressure or friction to the area or protection over the area with a cushion, as we attempted with our patient in the form of a foam pad.
The Diagnosis: Prayer Callus
After our patient demonstrated his routine position for prayer (Figure), the
diagnosis of prayer callus was confirmed. We suggested he use a foam pad under his foot during prayer. The patient did not return for follow-up.
Prayer calluses have been documented in the literature as prayer nodules1-3 or marks.4 Callus is a preferred term, as it implies the act of repeated pressure or friction to an area of the skin. It is more descriptive than marks and avoids the inclusion of nodules caused by different prayer or religious activities such as ritual scarring.5
Abanmi et al4 studied a large group (N=349) of Muslim patients with regular praying habits and found a high prevalence of what they referred to as prayer marks, defined by lichenification and/or hyperpigmentation, on the knees, forehead, ankles, and dorsal aspects of the feet. They studied the histopathology of 33 marks. No statistical analysis was performed. They reported common histologic findings of compact orthokeratosis, hypergranulosis, dermal papillary fibrosis, and dermal vascularization. In contrast to lichen simplex chronicus, the authors found that the dermal fibrosis did not exhibit collagen bundles perpendicular to the epidermis.4
This study found that a higher prevalence of lichenification was observed on the left foot (males, 57%; females, 39%) than the right foot (males, 14%; females, 19%), which was attributed to a more typical prayer position that placed pressure on the left foot.4 Our patient only had calluses on his left foot, which was consistent with the prayer position.
Treatment options for prayer calluses include debridement, either mechanical or with topical keratolytic preparations. There is a high likelihood of recurrence if prayer practices are not changed. Optimally, more definitive treatment, which can be combined with initial debridement, would be position adjustment to lessen pressure or friction to the area or protection over the area with a cushion, as we attempted with our patient in the form of a foam pad.
1. Kahana M, Cohen M, Ronnen M, et al. Prayer nodules in Moslem men. Cutis. 1986;38:281-282.
2. Vollum DI, Azadeh B. Prayer nodules. Clin Exp Dermatol. 1979;4:39-47.
3. Kumar PV, Hambarsoomian B. Prayer nodules fine needle aspiration cytologic findings. Acta Cytol. 1988;32:83-85.
4. Abanmi AA, Al Zouman AY, Al Hussaini H, et al. Prayer marks. Int J Dermatol. 2002;41:411-414.
5. Bourrel P. Problems related to African customs and ritual mutilations [in French]. Contracept Fertil Sex (Paris). 1983;11:1351-1358.
1. Kahana M, Cohen M, Ronnen M, et al. Prayer nodules in Moslem men. Cutis. 1986;38:281-282.
2. Vollum DI, Azadeh B. Prayer nodules. Clin Exp Dermatol. 1979;4:39-47.
3. Kumar PV, Hambarsoomian B. Prayer nodules fine needle aspiration cytologic findings. Acta Cytol. 1988;32:83-85.
4. Abanmi AA, Al Zouman AY, Al Hussaini H, et al. Prayer marks. Int J Dermatol. 2002;41:411-414.
5. Bourrel P. Problems related to African customs and ritual mutilations [in French]. Contracept Fertil Sex (Paris). 1983;11:1351-1358.
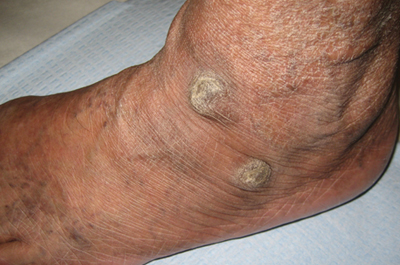
A 60-year-old man presented to the dermatology clinic with 2 thick plaques on the top of the left foot of at least 5 years’ duration with no recent changes. The bumps were not itchy or painful. The patient had a medical history of diabetes mellitus and hypertension. He did not report any recent travel. He denied cough, shortness of breath, weight loss, and fatigue. The patient was asked about any hobbies or activities that involved repeated pressure to the dorsal aspect of the foot. He revealed that his religious obligations required him to pray 5 times daily.
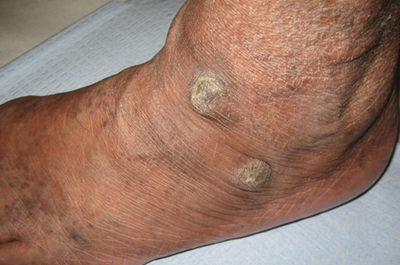
Is Headache a Sign of a Larger Problem?
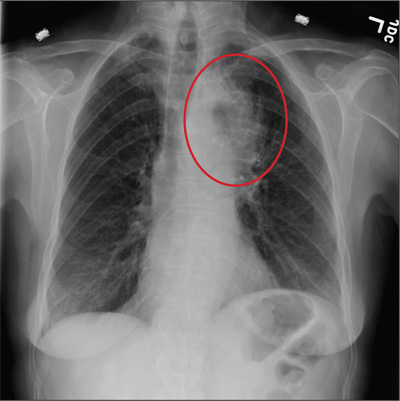
Answer
The radiograph shows a masslike density within the left suprahilar and mediastinal region, most likely consistent with a carcinoma. Given the entire clinical picture, this patient likely has a primary lung carcinoma that metastasized to the brain.
Answer
The radiograph shows a masslike density within the left suprahilar and mediastinal region, most likely consistent with a carcinoma. Given the entire clinical picture, this patient likely has a primary lung carcinoma that metastasized to the brain.
Answer
The radiograph shows a masslike density within the left suprahilar and mediastinal region, most likely consistent with a carcinoma. Given the entire clinical picture, this patient likely has a primary lung carcinoma that metastasized to the brain.
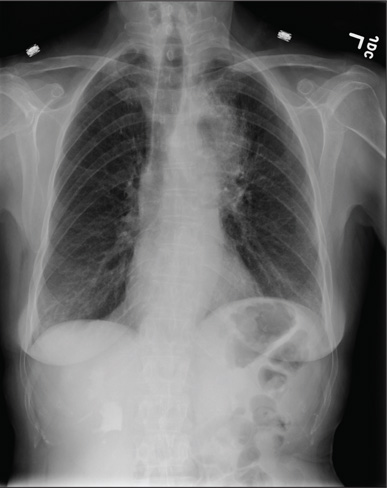
A 60-year-old woman presents with a complaint of severe headache, hoarseness, and weight loss, which have worsened in the past few days. Her headache is bifrontal, and at times she rates its severity as 10/10. She is not aware of any medical problems, but she admits she doesn’t have a primary care provider due to lack of insurance. She has a 30-year history of smoking one to one-and-a-half packs of cigarettes per day. Family history is positive for cancer. On examination, you note that she is uncomfortable but in no obvious distress. Her vital signs are normal. She is able to move all four extremities well and is neurovascularly intact. She has no other focal deficits. Noncontrast CT of the head is obtained. It shows a large right frontal lesion with surrounding vasogenic edema. You also order a chest radiograph (shown). What is your impression?
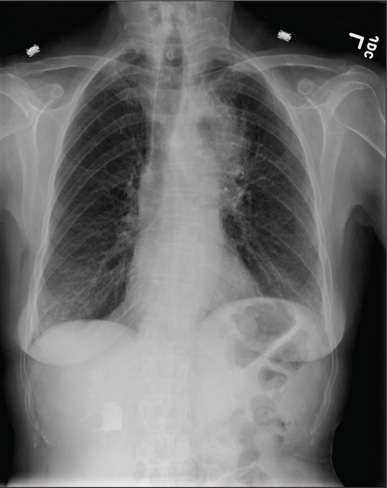
What Caused Patient’s Palpitations?
ANSWER
This ECG shows atrial flutter with 2:1 atrioventricular conduction. Additionally, ST depressions are seen in the anterior leads.
Typical sinus node P waves are absent, and atrial conduction at a rate of 310 beats/min is indicated by the sawtooth pattern in leads II and aVF. The ventricular rate is half that of the atrial rate (hence the 2:1 ratio). The ST depressions seen in the anterior leads, thought to be rate related, resolved upon cardioversion to terminate the atrial flutter.
Atrial flutter is uncommon in patients with structurally normal hearts and occurs far less frequently than atrial fibrillation. The etiology of this man’s arrhythmia may be due to pericarditis, based on his history and physical examination.
ANSWER
This ECG shows atrial flutter with 2:1 atrioventricular conduction. Additionally, ST depressions are seen in the anterior leads.
Typical sinus node P waves are absent, and atrial conduction at a rate of 310 beats/min is indicated by the sawtooth pattern in leads II and aVF. The ventricular rate is half that of the atrial rate (hence the 2:1 ratio). The ST depressions seen in the anterior leads, thought to be rate related, resolved upon cardioversion to terminate the atrial flutter.
Atrial flutter is uncommon in patients with structurally normal hearts and occurs far less frequently than atrial fibrillation. The etiology of this man’s arrhythmia may be due to pericarditis, based on his history and physical examination.
ANSWER
This ECG shows atrial flutter with 2:1 atrioventricular conduction. Additionally, ST depressions are seen in the anterior leads.
Typical sinus node P waves are absent, and atrial conduction at a rate of 310 beats/min is indicated by the sawtooth pattern in leads II and aVF. The ventricular rate is half that of the atrial rate (hence the 2:1 ratio). The ST depressions seen in the anterior leads, thought to be rate related, resolved upon cardioversion to terminate the atrial flutter.
Atrial flutter is uncommon in patients with structurally normal hearts and occurs far less frequently than atrial fibrillation. The etiology of this man’s arrhythmia may be due to pericarditis, based on his history and physical examination.
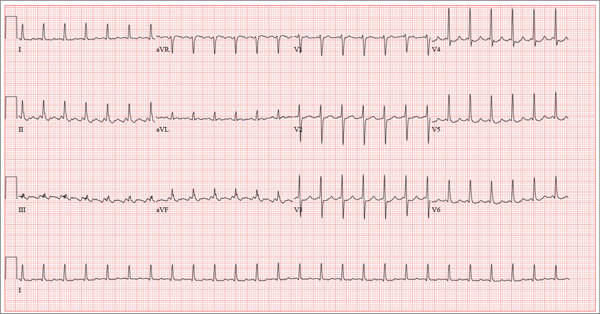
A 52-year-old man developed acute-onset palpitations, shortness of breath, and lightheadedness while sitting at his desk at work. He noticed his heart rate was rapid and asked a coworker to take his pulse for confirmation. He did not experience chest pain, syncope, or near syncope, but if he stood up and tried to walk, he very quickly became fatigued. His coworker tried to call 911; however, the patient asked to be driven to the urgent care center six blocks from their office instead. The patient’s heart rate and symptoms did not change en route. There is no previous history of heart disease. Although the patient works in an office, he is very active. He played hockey in high school and college and continues to play in an amateur league as well as coaching a youth group at the local ice rink. He is also an active member of a local bicycling club and recently completed a 150-mile recreational ride. He has no history of hypertension, diabetes, or pulmonary disease. Surgical history is remarkable for a medial meniscus repair of his right knee and a laparoscopic cholecystectomy, both performed more than 10 years ago. He works as a certified public accountant, does not smoke, and drinks one or two glasses of wine in the evening with meals. He is married and has two adult children. He denies using recreational drugs or herbal medicines. The only medication he uses is ibuprofen as needed for musculoskeletal aches and pains associated with his active lifestyle. He has no known drug allergies, and his immunizations are current. The review of systems is positive for a recent viral upper respiratory illness. He reports having vague, nonspecific substernal chest discomfort, but no pain, at the time of his illness. Symptoms have resolved. There are no other complaints. On arrival, the patient appears anxious and in mild distress, but without pain. Vital signs include a heart rate of 160 beats/min; blood pressure, 100/64 mm Hg; respiratory rate, 18 breaths/min-1; and temperature, 98.4°F. The HEENT exam is unremarkable except for corrective lenses. The chest is clear in all lung fields. There is no jugular venous distention, and carotid upstrokes are brisk. The cardiac exam reveals a regular rhythm at a rate of 150 beats/min with no murmurs or gallops; however, a rub is noted. The abdomen is soft and nontender with no organomegaly. Well-healed scars from his laparoscopic ports are present. The lower extremities show no evidence of edema. Peripheral pulses are strong and equal in both upper and lower extremities, and the neurologic exam is normal. Laboratory studies including a metabolic panel, complete blood count, and cardiac enzymes all yield normal results. An ECG reveals the following: a ventricular rate of 155 beats/min; PR interval, not measured; QRS duration, 78 ms; QT/QTc interval, 272/437 ms; P axis, unmeasurable; R axis, 34°; and T axis, –50°. What is your interpretation of this ECG?