User login
Cutis is a peer-reviewed clinical journal for the dermatologist, allergist, and general practitioner published monthly since 1965. Concise clinical articles present the practical side of dermatology, helping physicians to improve patient care. Cutis is referenced in Index Medicus/MEDLINE and is written and edited by industry leaders.
ass lick
assault rifle
balls
ballsac
black jack
bleach
Boko Haram
bondage
causas
cheap
child abuse
cocaine
compulsive behaviors
cost of miracles
cunt
Daech
display network stats
drug paraphernalia
explosion
fart
fda and death
fda AND warn
fda AND warning
fda AND warns
feom
fuck
gambling
gfc
gun
human trafficking
humira AND expensive
illegal
ISIL
ISIS
Islamic caliphate
Islamic state
madvocate
masturbation
mixed martial arts
MMA
molestation
national rifle association
NRA
nsfw
nuccitelli
pedophile
pedophilia
poker
porn
porn
pornography
psychedelic drug
recreational drug
sex slave rings
shit
slot machine
snort
substance abuse
terrorism
terrorist
texarkana
Texas hold 'em
UFC
section[contains(@class, 'nav-hidden')]
section[contains(@class, 'nav-hidden active')
A peer-reviewed, indexed journal for dermatologists with original research, image quizzes, cases and reviews, and columns.
Rupioid Psoriasis and Psoriatic Arthritis in a Patient With Skin of Color
To the Editor:
A 49-year-old black woman presented with multiple hyperkeratotic papules that progressed over the last 2 months to circular plaques with central thick black crust resembling eschar. She first noticed these lesions as firm, small, black papules on the legs and continued to develop new lesions that eventually evolved into large, coin-shaped, hyperkeratotic plaques. Her medical history was notable for stage III non-Hodgkin follicular lymphoma in remission after treatment with rituximab, cyclophosphamide, doxorubicin hydrochloride, vincristine sulfate, and prednisone 7 months earlier, and chronic hepatitis B infection being treated with entecavir. Her family history was not remarkable for psoriasis or inflammatory arthritis.
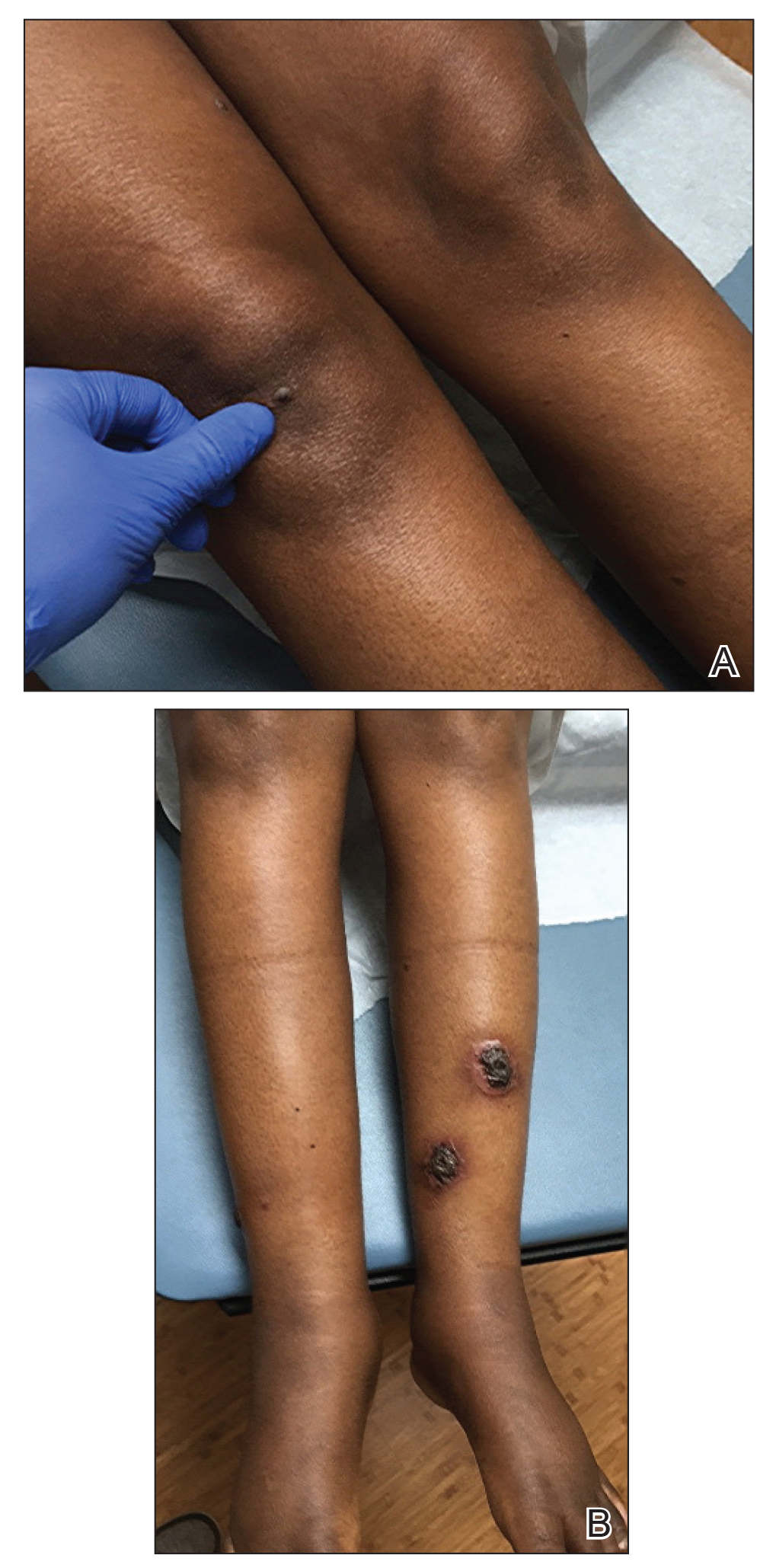
She initially was seen by internal medicine and was started on topical triamcinolone with no improvement of the lesions. At presentation to dermatology, physical examination revealed firm, small, black, hyperkeratotic papules (Figure 1A) and circular plaques with a rim of erythema and central thick, smooth, black crust resembling eschar (Figure 1B). No other skin changes were noted at the time. The bilateral metacarpophalangeal, bilateral proximal interphalangeal, left wrist, and bilateral ankle joints were remarkable for tenderness, swelling, and reduced range of motion. She noted concomitant arthralgia and stiffness but denied fever. She had no other systemic symptoms including night sweats, weight loss, fatigue, malaise, sun sensitivity, oral ulcers, or hair loss. A radiograph of the hand was negative for erosive changes but showed mild periarticular osteopenia and fusiform soft tissue swelling of the third digit. Given the central appearance of eschar in the larger lesions, the initial differential diagnosis included Sweet syndrome, invasive fungal infection, vasculitis, and recurrent lymphoma.
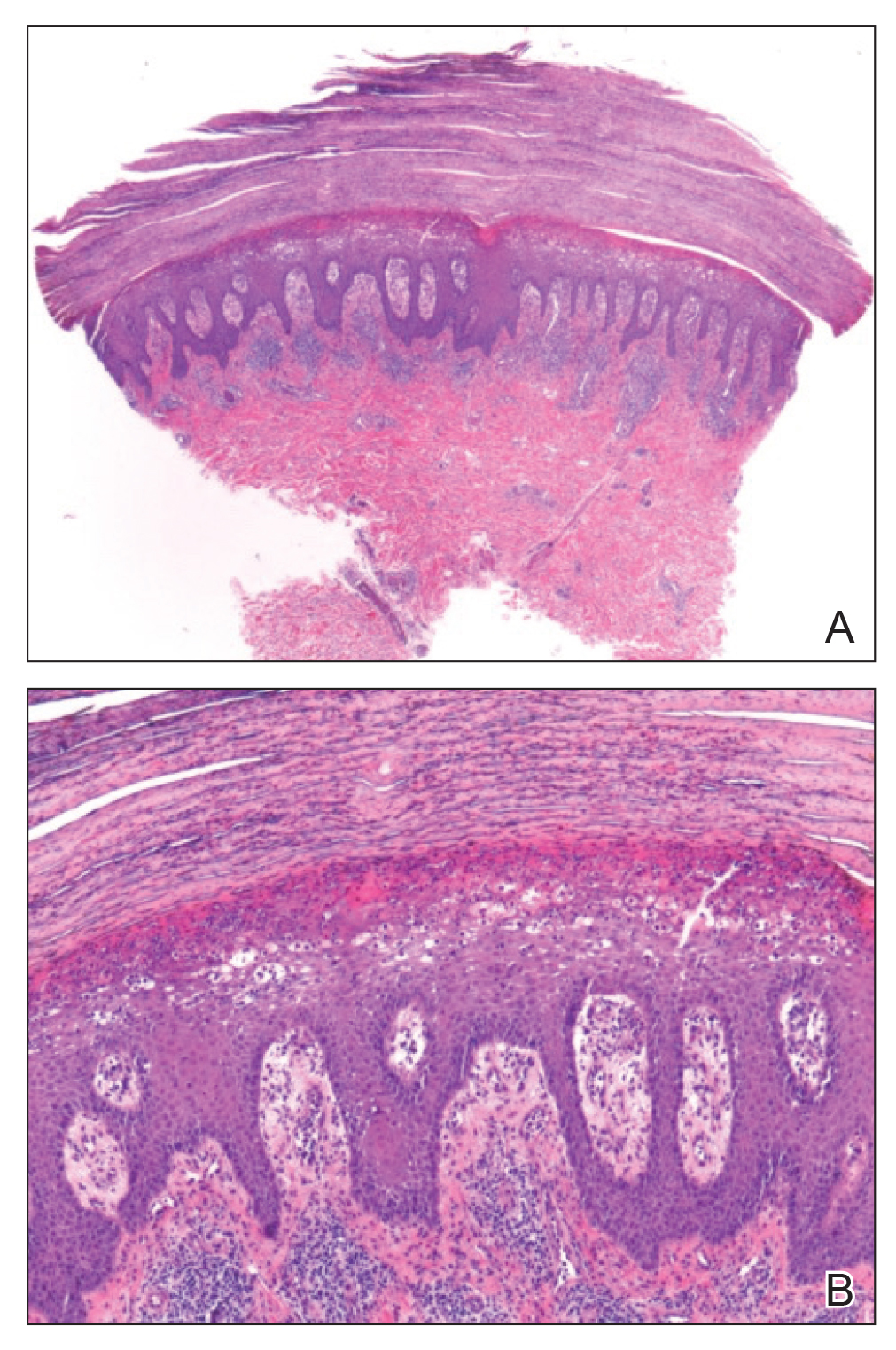
A 4-mm punch biopsy specimen of a representative lesion on the right leg revealed psoriasiform epidermal hyperplasia, parakeratosis, neutrophils in the stratum corneum and spinosum, elongation of the rete ridges, and superficial vascular ectasia, which favored a diagnosis of psoriasis (Figure 2). A periodic acid-Schiff stain was negative for fungal hyphae. Fungal culture, bacterial tissue culture, and acid-fast bacilli smear were negative. Absence of deep dermal inflammation precluded a diagnosis of Sweet syndrome. Further notable laboratory studies included negative human immunodeficiency virus (HIV) antibody, rapid plasma reagin, hepatitis C antibody, and rheumatoid factor.
At follow-up 2 weeks later, the initial lesions were still present, and she had developed new widespread, well-demarcated, erythematous plaques with silver scale along the scalp, back, chest, and abdomen that were more typical of psoriasis. Oil spots were noted on several fingernails and toenails. Based on the clinicopathologic findings, nail changes, and asymmetric inflammatory arthritis, a diagnosis of rupioid psoriasis with psoriatic arthritis (PsA) was established. Treatment with clobetasol ointment 0.05% twice daily to active lesions was started. Initiation of systemic therapy with a steroid-sparing agent was deferred in anticipation of care coordination with rheumatology, hepatology, and hematology/oncology due to the patient's history of follicular lymphoma and chronic hepatitis B. Although attempts were made to avoid systemic corticosteroids due to the risk for a psoriasis flare upon discontinuation, because of the severity of arthralgia she was started on oral prednisone 20 mg daily by rheumatology with plans for a slow taper once an alternative systemic agent was started.1
At 10-week follow-up, the patient had marked improvement of psoriatic plaques with no active lesions while only on prednisone 20 mg daily. In consultation with her care team, she subsequently was started on methotrexate 10 mg weekly for 2 weeks followed by titration to 15 mg weekly. Plans were to start a prednisone taper after a month of methotrexate to allow her new treatment time for therapeutic effect. Notably, the patient chose to discontinue prednisone 2 weeks into methotrexate therapy after only two 10-mg doses of methotrexate weekly and well before therapeutic levels were achieved. Despite stopping prednisone early and without a taper, she did not experience a relapse in psoriatic skin lesions. Three months following initiation of methotrexate, she sustained resolution of the cutaneous lesions with only residual postinflammatory hyperpigmentation.
Psoriasis is a common chronic inflammatory skin disorder with multiple clinical presentations. There are several variants of psoriasis that are classified by their morphologic appearance including chronic plaque, guttate, erythrodermic, and pustular, with more than 90% of cases representing the plaque variant. Less common clinical presentations of psoriasis include rupioid, ostraceous, inverse, elephantine, and HIV associated.2 Rupioid psoriasis is a rare variant that presents with cone-shaped, limpetlike lesions.3,4 Similar to the limited epidemiological and clinical data pertaining to psoriasis in nonwhite racial groups, there also is a paucity of documented reports of rupioid psoriasis in skin of color.
Rupioid comes from the Greek word rhupos, meaning dirt or filth, and is used to describe well-demarcated lesions with thick, yellow, dirty-appearing, adherent crusts resembling oyster shells with a surrounding rim of erythema.5 Rupioid psoriasis initially was reported in 1948 and remains an uncommon and infrequently reported variant.6 The majority of reported cases have been associated with arthropathy, similar to our patient.3,4 Rupioid lesions also have been observed in an array of other diseases, such as secondary syphilis, crusted scabies, disseminated histoplasmosis, HIV, reactive arthritis, and aminoaciduria.7-11
Diagnosis of rupioid psoriasis can be confirmed with a skin biopsy, which demonstrates characteristic histopathologic findings of psoriasis.3 Laboratory analysis should be performed to rule out other causes of rupioid lesions, and PsA should be differentiated from rheumatoid arthritis if arthropathy is present. In our case, serum rapid plasma reagin, anti-HIV antibody, rheumatoid factor, and fungal cultures were negative. Usin0)g clinical findings, histopathology, laboratory analyses, and radiograph findings, the diagnosis of rupioid psoriasis with PsA was confirmed in our patient.
Psoriasis was not originally suspected in our patient due to the noncharacteristic lesions with smooth black crust--similar appearing to eschar--and the patient's complicated medical history. Variations in the presentation of psoriasis among white individuals and those with skin of color have been reported in the literature.12,13 Psoriatic lesions in darker skin tones may appear more violaceous or hyperpigmented with more conspicuous erythema and thicker plaques. Our patient lacked the classic rupioid appearance of concentric circular layers of dirty, yellow, oysterlike scale, and instead had thick, lamellate, black crust. A PubMed search of articles indexed for MEDLINE using the terms rupioid, coral reef psoriasis, rupioides, and rhupus revealed no other cases of rupioid psoriasis reported in black patients and no cases detailing the variations of rupioid lesions in skin of color. A case of rupioid psoriasis has been reported in a Hispanic patient, but the described psoriatic lesions were more characteristic of the dirty-appearing, conic plaques previously reported.14 Our case highlights a unique example of the variable presentations of cutaneous disorders in skin of color and black patients.
Our patient's case of rupioid psoriasis with PsA presented unique challenges for systemic treatment due to her multiple comorbidities. Rupioid psoriasis most often is treated with combination topical and systemic therapy, with agents such as methotrexate and cyclosporine having prior success.3,4 This variant of psoriasis is highly responsive to treatment, and marked improvement of lesions has been achieved with topical steroids alone with proper adherence.15 Our patient was started on clobetasol ointment 0.05% while a systemic agent was debated for her PsA. Although she did not have improvement with topical therapy alone, she experienced rapid resolution of the skin lesions after initiation of low-dose prednisone 20 mg daily. Interestingly, our patient did not experience a flare of the skin lesions upon discontinuation of systemic steroids despite the lack of an appropriate taper and methotrexate not having reached therapeutic levels.
The clinical nuances of rupioid psoriasis in skin of color have not yet been described and remain an important diagnostic consideration. Our patient achieved remission of skin lesions with sequential treatment of topical clobetasol, a low-dose systemic steroid, and methotrexate. Based on available reports, rupioid psoriasis may represent a variant of psoriasis that is highly responsive to treatment.
- Mrowietz U, Domm S. Systemic steroids in the treatment of psoriasis: what is fact, what is fiction? J Eur Acad Dermatol Venereol. 2013;27:1022-1025.
- Goldsmith LA, Katz SI, Gilchrest BA, eds. Fitzpatrick's Dermatology in General Medicine. 8th ed. New York, NY: The McGraw-Hill Companies; 2012.
- Wang JL, Yang JH. Rupioid psoriasis associated with arthropathy. J Dermatol. 1997;24:46-49.
- Murakami T, Ohtsuki M, Nakagawa H. Rupioid psoriasis with arthropathy. Clin Exp Dermatol. 2000;25:409-412.
- Chung HJ, Marley-Kemp D, Keller M. Rupioid psoriasis and other skin diseases with rupioid manifestations. Cutis. 2014;94:119-121.
- Salamon M, Omulecki A, Sysa-Jedrzejowska A, et al. Psoriasisrupioides: a rare variant of a common disease. Cutis. 2011;88:135-137.
- Krase IZ, Cavanaugh K, Curiel-Lewandrowski C. A case of rupioid syphilis. JAAD Case Rep. 2016;2:141-143.
- Garofalo V, Saraceno R, Milana M, et al. Crusted scabies in a liver transplant patient mimicking rupioid psoriasis. Eur J Dermatol. 2016;26:495-496.
- Corti M, Villafane MF, Palmieri O, et al. Rupioid histoplasmosis: first case reported in an AIDS patient in Argentina. Rev Inst Med Trop Sao Paulo. 2010;52:279-280.
- Sehgal VN, Koranne RV, Shyam Prasad AL. Unusual manifestations of Reiter's disease in a child. Dermatologica. 1985;170:77-79.
- Haim S, Gilhar A, Cohen A. Cutaneous manifestations associated with aminoaciduria. report of two cases. Dermatologica. 1978;156:244-250.
- McMichael AJ, Vachiramon V, Guzman-Sanchez DA, et al. Psoriasis in African-Americans: a caregivers' survey. J Drugs Dermatol. 2012;11:478-482.
- Alexis AF, Blackcloud P. Psoriasis in skin of color: epidemiology, genetics, clinical presentation, and treatment nuances. J Clin Aesthet Dermatol. 2014;7:16-24.
- Posligua A, Maldonado C, Gonzalez MG. Rupioid psoriasis preceded by varicella presenting as Koebner phenomenon. J Am Acad Dermatol. 2016;74(5 suppl 1):AB268.
- Feldman SR, Feldman S, Brown K, et al. "Coral reef" psoriasis: a marker of resistance to topical treatment. J Dermatolog Treat. 2008;19:257-258.
To the Editor:
A 49-year-old black woman presented with multiple hyperkeratotic papules that progressed over the last 2 months to circular plaques with central thick black crust resembling eschar. She first noticed these lesions as firm, small, black papules on the legs and continued to develop new lesions that eventually evolved into large, coin-shaped, hyperkeratotic plaques. Her medical history was notable for stage III non-Hodgkin follicular lymphoma in remission after treatment with rituximab, cyclophosphamide, doxorubicin hydrochloride, vincristine sulfate, and prednisone 7 months earlier, and chronic hepatitis B infection being treated with entecavir. Her family history was not remarkable for psoriasis or inflammatory arthritis.
She initially was seen by internal medicine and was started on topical triamcinolone with no improvement of the lesions. At presentation to dermatology, physical examination revealed firm, small, black, hyperkeratotic papules (Figure 1A) and circular plaques with a rim of erythema and central thick, smooth, black crust resembling eschar (Figure 1B). No other skin changes were noted at the time. The bilateral metacarpophalangeal, bilateral proximal interphalangeal, left wrist, and bilateral ankle joints were remarkable for tenderness, swelling, and reduced range of motion. She noted concomitant arthralgia and stiffness but denied fever. She had no other systemic symptoms including night sweats, weight loss, fatigue, malaise, sun sensitivity, oral ulcers, or hair loss. A radiograph of the hand was negative for erosive changes but showed mild periarticular osteopenia and fusiform soft tissue swelling of the third digit. Given the central appearance of eschar in the larger lesions, the initial differential diagnosis included Sweet syndrome, invasive fungal infection, vasculitis, and recurrent lymphoma.
A 4-mm punch biopsy specimen of a representative lesion on the right leg revealed psoriasiform epidermal hyperplasia, parakeratosis, neutrophils in the stratum corneum and spinosum, elongation of the rete ridges, and superficial vascular ectasia, which favored a diagnosis of psoriasis (Figure 2). A periodic acid-Schiff stain was negative for fungal hyphae. Fungal culture, bacterial tissue culture, and acid-fast bacilli smear were negative. Absence of deep dermal inflammation precluded a diagnosis of Sweet syndrome. Further notable laboratory studies included negative human immunodeficiency virus (HIV) antibody, rapid plasma reagin, hepatitis C antibody, and rheumatoid factor.
At follow-up 2 weeks later, the initial lesions were still present, and she had developed new widespread, well-demarcated, erythematous plaques with silver scale along the scalp, back, chest, and abdomen that were more typical of psoriasis. Oil spots were noted on several fingernails and toenails. Based on the clinicopathologic findings, nail changes, and asymmetric inflammatory arthritis, a diagnosis of rupioid psoriasis with psoriatic arthritis (PsA) was established. Treatment with clobetasol ointment 0.05% twice daily to active lesions was started. Initiation of systemic therapy with a steroid-sparing agent was deferred in anticipation of care coordination with rheumatology, hepatology, and hematology/oncology due to the patient's history of follicular lymphoma and chronic hepatitis B. Although attempts were made to avoid systemic corticosteroids due to the risk for a psoriasis flare upon discontinuation, because of the severity of arthralgia she was started on oral prednisone 20 mg daily by rheumatology with plans for a slow taper once an alternative systemic agent was started.1
At 10-week follow-up, the patient had marked improvement of psoriatic plaques with no active lesions while only on prednisone 20 mg daily. In consultation with her care team, she subsequently was started on methotrexate 10 mg weekly for 2 weeks followed by titration to 15 mg weekly. Plans were to start a prednisone taper after a month of methotrexate to allow her new treatment time for therapeutic effect. Notably, the patient chose to discontinue prednisone 2 weeks into methotrexate therapy after only two 10-mg doses of methotrexate weekly and well before therapeutic levels were achieved. Despite stopping prednisone early and without a taper, she did not experience a relapse in psoriatic skin lesions. Three months following initiation of methotrexate, she sustained resolution of the cutaneous lesions with only residual postinflammatory hyperpigmentation.
Psoriasis is a common chronic inflammatory skin disorder with multiple clinical presentations. There are several variants of psoriasis that are classified by their morphologic appearance including chronic plaque, guttate, erythrodermic, and pustular, with more than 90% of cases representing the plaque variant. Less common clinical presentations of psoriasis include rupioid, ostraceous, inverse, elephantine, and HIV associated.2 Rupioid psoriasis is a rare variant that presents with cone-shaped, limpetlike lesions.3,4 Similar to the limited epidemiological and clinical data pertaining to psoriasis in nonwhite racial groups, there also is a paucity of documented reports of rupioid psoriasis in skin of color.
Rupioid comes from the Greek word rhupos, meaning dirt or filth, and is used to describe well-demarcated lesions with thick, yellow, dirty-appearing, adherent crusts resembling oyster shells with a surrounding rim of erythema.5 Rupioid psoriasis initially was reported in 1948 and remains an uncommon and infrequently reported variant.6 The majority of reported cases have been associated with arthropathy, similar to our patient.3,4 Rupioid lesions also have been observed in an array of other diseases, such as secondary syphilis, crusted scabies, disseminated histoplasmosis, HIV, reactive arthritis, and aminoaciduria.7-11
Diagnosis of rupioid psoriasis can be confirmed with a skin biopsy, which demonstrates characteristic histopathologic findings of psoriasis.3 Laboratory analysis should be performed to rule out other causes of rupioid lesions, and PsA should be differentiated from rheumatoid arthritis if arthropathy is present. In our case, serum rapid plasma reagin, anti-HIV antibody, rheumatoid factor, and fungal cultures were negative. Usin0)g clinical findings, histopathology, laboratory analyses, and radiograph findings, the diagnosis of rupioid psoriasis with PsA was confirmed in our patient.
Psoriasis was not originally suspected in our patient due to the noncharacteristic lesions with smooth black crust--similar appearing to eschar--and the patient's complicated medical history. Variations in the presentation of psoriasis among white individuals and those with skin of color have been reported in the literature.12,13 Psoriatic lesions in darker skin tones may appear more violaceous or hyperpigmented with more conspicuous erythema and thicker plaques. Our patient lacked the classic rupioid appearance of concentric circular layers of dirty, yellow, oysterlike scale, and instead had thick, lamellate, black crust. A PubMed search of articles indexed for MEDLINE using the terms rupioid, coral reef psoriasis, rupioides, and rhupus revealed no other cases of rupioid psoriasis reported in black patients and no cases detailing the variations of rupioid lesions in skin of color. A case of rupioid psoriasis has been reported in a Hispanic patient, but the described psoriatic lesions were more characteristic of the dirty-appearing, conic plaques previously reported.14 Our case highlights a unique example of the variable presentations of cutaneous disorders in skin of color and black patients.
Our patient's case of rupioid psoriasis with PsA presented unique challenges for systemic treatment due to her multiple comorbidities. Rupioid psoriasis most often is treated with combination topical and systemic therapy, with agents such as methotrexate and cyclosporine having prior success.3,4 This variant of psoriasis is highly responsive to treatment, and marked improvement of lesions has been achieved with topical steroids alone with proper adherence.15 Our patient was started on clobetasol ointment 0.05% while a systemic agent was debated for her PsA. Although she did not have improvement with topical therapy alone, she experienced rapid resolution of the skin lesions after initiation of low-dose prednisone 20 mg daily. Interestingly, our patient did not experience a flare of the skin lesions upon discontinuation of systemic steroids despite the lack of an appropriate taper and methotrexate not having reached therapeutic levels.
The clinical nuances of rupioid psoriasis in skin of color have not yet been described and remain an important diagnostic consideration. Our patient achieved remission of skin lesions with sequential treatment of topical clobetasol, a low-dose systemic steroid, and methotrexate. Based on available reports, rupioid psoriasis may represent a variant of psoriasis that is highly responsive to treatment.
To the Editor:
A 49-year-old black woman presented with multiple hyperkeratotic papules that progressed over the last 2 months to circular plaques with central thick black crust resembling eschar. She first noticed these lesions as firm, small, black papules on the legs and continued to develop new lesions that eventually evolved into large, coin-shaped, hyperkeratotic plaques. Her medical history was notable for stage III non-Hodgkin follicular lymphoma in remission after treatment with rituximab, cyclophosphamide, doxorubicin hydrochloride, vincristine sulfate, and prednisone 7 months earlier, and chronic hepatitis B infection being treated with entecavir. Her family history was not remarkable for psoriasis or inflammatory arthritis.
She initially was seen by internal medicine and was started on topical triamcinolone with no improvement of the lesions. At presentation to dermatology, physical examination revealed firm, small, black, hyperkeratotic papules (Figure 1A) and circular plaques with a rim of erythema and central thick, smooth, black crust resembling eschar (Figure 1B). No other skin changes were noted at the time. The bilateral metacarpophalangeal, bilateral proximal interphalangeal, left wrist, and bilateral ankle joints were remarkable for tenderness, swelling, and reduced range of motion. She noted concomitant arthralgia and stiffness but denied fever. She had no other systemic symptoms including night sweats, weight loss, fatigue, malaise, sun sensitivity, oral ulcers, or hair loss. A radiograph of the hand was negative for erosive changes but showed mild periarticular osteopenia and fusiform soft tissue swelling of the third digit. Given the central appearance of eschar in the larger lesions, the initial differential diagnosis included Sweet syndrome, invasive fungal infection, vasculitis, and recurrent lymphoma.
A 4-mm punch biopsy specimen of a representative lesion on the right leg revealed psoriasiform epidermal hyperplasia, parakeratosis, neutrophils in the stratum corneum and spinosum, elongation of the rete ridges, and superficial vascular ectasia, which favored a diagnosis of psoriasis (Figure 2). A periodic acid-Schiff stain was negative for fungal hyphae. Fungal culture, bacterial tissue culture, and acid-fast bacilli smear were negative. Absence of deep dermal inflammation precluded a diagnosis of Sweet syndrome. Further notable laboratory studies included negative human immunodeficiency virus (HIV) antibody, rapid plasma reagin, hepatitis C antibody, and rheumatoid factor.
At follow-up 2 weeks later, the initial lesions were still present, and she had developed new widespread, well-demarcated, erythematous plaques with silver scale along the scalp, back, chest, and abdomen that were more typical of psoriasis. Oil spots were noted on several fingernails and toenails. Based on the clinicopathologic findings, nail changes, and asymmetric inflammatory arthritis, a diagnosis of rupioid psoriasis with psoriatic arthritis (PsA) was established. Treatment with clobetasol ointment 0.05% twice daily to active lesions was started. Initiation of systemic therapy with a steroid-sparing agent was deferred in anticipation of care coordination with rheumatology, hepatology, and hematology/oncology due to the patient's history of follicular lymphoma and chronic hepatitis B. Although attempts were made to avoid systemic corticosteroids due to the risk for a psoriasis flare upon discontinuation, because of the severity of arthralgia she was started on oral prednisone 20 mg daily by rheumatology with plans for a slow taper once an alternative systemic agent was started.1
At 10-week follow-up, the patient had marked improvement of psoriatic plaques with no active lesions while only on prednisone 20 mg daily. In consultation with her care team, she subsequently was started on methotrexate 10 mg weekly for 2 weeks followed by titration to 15 mg weekly. Plans were to start a prednisone taper after a month of methotrexate to allow her new treatment time for therapeutic effect. Notably, the patient chose to discontinue prednisone 2 weeks into methotrexate therapy after only two 10-mg doses of methotrexate weekly and well before therapeutic levels were achieved. Despite stopping prednisone early and without a taper, she did not experience a relapse in psoriatic skin lesions. Three months following initiation of methotrexate, she sustained resolution of the cutaneous lesions with only residual postinflammatory hyperpigmentation.
Psoriasis is a common chronic inflammatory skin disorder with multiple clinical presentations. There are several variants of psoriasis that are classified by their morphologic appearance including chronic plaque, guttate, erythrodermic, and pustular, with more than 90% of cases representing the plaque variant. Less common clinical presentations of psoriasis include rupioid, ostraceous, inverse, elephantine, and HIV associated.2 Rupioid psoriasis is a rare variant that presents with cone-shaped, limpetlike lesions.3,4 Similar to the limited epidemiological and clinical data pertaining to psoriasis in nonwhite racial groups, there also is a paucity of documented reports of rupioid psoriasis in skin of color.
Rupioid comes from the Greek word rhupos, meaning dirt or filth, and is used to describe well-demarcated lesions with thick, yellow, dirty-appearing, adherent crusts resembling oyster shells with a surrounding rim of erythema.5 Rupioid psoriasis initially was reported in 1948 and remains an uncommon and infrequently reported variant.6 The majority of reported cases have been associated with arthropathy, similar to our patient.3,4 Rupioid lesions also have been observed in an array of other diseases, such as secondary syphilis, crusted scabies, disseminated histoplasmosis, HIV, reactive arthritis, and aminoaciduria.7-11
Diagnosis of rupioid psoriasis can be confirmed with a skin biopsy, which demonstrates characteristic histopathologic findings of psoriasis.3 Laboratory analysis should be performed to rule out other causes of rupioid lesions, and PsA should be differentiated from rheumatoid arthritis if arthropathy is present. In our case, serum rapid plasma reagin, anti-HIV antibody, rheumatoid factor, and fungal cultures were negative. Usin0)g clinical findings, histopathology, laboratory analyses, and radiograph findings, the diagnosis of rupioid psoriasis with PsA was confirmed in our patient.
Psoriasis was not originally suspected in our patient due to the noncharacteristic lesions with smooth black crust--similar appearing to eschar--and the patient's complicated medical history. Variations in the presentation of psoriasis among white individuals and those with skin of color have been reported in the literature.12,13 Psoriatic lesions in darker skin tones may appear more violaceous or hyperpigmented with more conspicuous erythema and thicker plaques. Our patient lacked the classic rupioid appearance of concentric circular layers of dirty, yellow, oysterlike scale, and instead had thick, lamellate, black crust. A PubMed search of articles indexed for MEDLINE using the terms rupioid, coral reef psoriasis, rupioides, and rhupus revealed no other cases of rupioid psoriasis reported in black patients and no cases detailing the variations of rupioid lesions in skin of color. A case of rupioid psoriasis has been reported in a Hispanic patient, but the described psoriatic lesions were more characteristic of the dirty-appearing, conic plaques previously reported.14 Our case highlights a unique example of the variable presentations of cutaneous disorders in skin of color and black patients.
Our patient's case of rupioid psoriasis with PsA presented unique challenges for systemic treatment due to her multiple comorbidities. Rupioid psoriasis most often is treated with combination topical and systemic therapy, with agents such as methotrexate and cyclosporine having prior success.3,4 This variant of psoriasis is highly responsive to treatment, and marked improvement of lesions has been achieved with topical steroids alone with proper adherence.15 Our patient was started on clobetasol ointment 0.05% while a systemic agent was debated for her PsA. Although she did not have improvement with topical therapy alone, she experienced rapid resolution of the skin lesions after initiation of low-dose prednisone 20 mg daily. Interestingly, our patient did not experience a flare of the skin lesions upon discontinuation of systemic steroids despite the lack of an appropriate taper and methotrexate not having reached therapeutic levels.
The clinical nuances of rupioid psoriasis in skin of color have not yet been described and remain an important diagnostic consideration. Our patient achieved remission of skin lesions with sequential treatment of topical clobetasol, a low-dose systemic steroid, and methotrexate. Based on available reports, rupioid psoriasis may represent a variant of psoriasis that is highly responsive to treatment.
- Mrowietz U, Domm S. Systemic steroids in the treatment of psoriasis: what is fact, what is fiction? J Eur Acad Dermatol Venereol. 2013;27:1022-1025.
- Goldsmith LA, Katz SI, Gilchrest BA, eds. Fitzpatrick's Dermatology in General Medicine. 8th ed. New York, NY: The McGraw-Hill Companies; 2012.
- Wang JL, Yang JH. Rupioid psoriasis associated with arthropathy. J Dermatol. 1997;24:46-49.
- Murakami T, Ohtsuki M, Nakagawa H. Rupioid psoriasis with arthropathy. Clin Exp Dermatol. 2000;25:409-412.
- Chung HJ, Marley-Kemp D, Keller M. Rupioid psoriasis and other skin diseases with rupioid manifestations. Cutis. 2014;94:119-121.
- Salamon M, Omulecki A, Sysa-Jedrzejowska A, et al. Psoriasisrupioides: a rare variant of a common disease. Cutis. 2011;88:135-137.
- Krase IZ, Cavanaugh K, Curiel-Lewandrowski C. A case of rupioid syphilis. JAAD Case Rep. 2016;2:141-143.
- Garofalo V, Saraceno R, Milana M, et al. Crusted scabies in a liver transplant patient mimicking rupioid psoriasis. Eur J Dermatol. 2016;26:495-496.
- Corti M, Villafane MF, Palmieri O, et al. Rupioid histoplasmosis: first case reported in an AIDS patient in Argentina. Rev Inst Med Trop Sao Paulo. 2010;52:279-280.
- Sehgal VN, Koranne RV, Shyam Prasad AL. Unusual manifestations of Reiter's disease in a child. Dermatologica. 1985;170:77-79.
- Haim S, Gilhar A, Cohen A. Cutaneous manifestations associated with aminoaciduria. report of two cases. Dermatologica. 1978;156:244-250.
- McMichael AJ, Vachiramon V, Guzman-Sanchez DA, et al. Psoriasis in African-Americans: a caregivers' survey. J Drugs Dermatol. 2012;11:478-482.
- Alexis AF, Blackcloud P. Psoriasis in skin of color: epidemiology, genetics, clinical presentation, and treatment nuances. J Clin Aesthet Dermatol. 2014;7:16-24.
- Posligua A, Maldonado C, Gonzalez MG. Rupioid psoriasis preceded by varicella presenting as Koebner phenomenon. J Am Acad Dermatol. 2016;74(5 suppl 1):AB268.
- Feldman SR, Feldman S, Brown K, et al. "Coral reef" psoriasis: a marker of resistance to topical treatment. J Dermatolog Treat. 2008;19:257-258.
- Mrowietz U, Domm S. Systemic steroids in the treatment of psoriasis: what is fact, what is fiction? J Eur Acad Dermatol Venereol. 2013;27:1022-1025.
- Goldsmith LA, Katz SI, Gilchrest BA, eds. Fitzpatrick's Dermatology in General Medicine. 8th ed. New York, NY: The McGraw-Hill Companies; 2012.
- Wang JL, Yang JH. Rupioid psoriasis associated with arthropathy. J Dermatol. 1997;24:46-49.
- Murakami T, Ohtsuki M, Nakagawa H. Rupioid psoriasis with arthropathy. Clin Exp Dermatol. 2000;25:409-412.
- Chung HJ, Marley-Kemp D, Keller M. Rupioid psoriasis and other skin diseases with rupioid manifestations. Cutis. 2014;94:119-121.
- Salamon M, Omulecki A, Sysa-Jedrzejowska A, et al. Psoriasisrupioides: a rare variant of a common disease. Cutis. 2011;88:135-137.
- Krase IZ, Cavanaugh K, Curiel-Lewandrowski C. A case of rupioid syphilis. JAAD Case Rep. 2016;2:141-143.
- Garofalo V, Saraceno R, Milana M, et al. Crusted scabies in a liver transplant patient mimicking rupioid psoriasis. Eur J Dermatol. 2016;26:495-496.
- Corti M, Villafane MF, Palmieri O, et al. Rupioid histoplasmosis: first case reported in an AIDS patient in Argentina. Rev Inst Med Trop Sao Paulo. 2010;52:279-280.
- Sehgal VN, Koranne RV, Shyam Prasad AL. Unusual manifestations of Reiter's disease in a child. Dermatologica. 1985;170:77-79.
- Haim S, Gilhar A, Cohen A. Cutaneous manifestations associated with aminoaciduria. report of two cases. Dermatologica. 1978;156:244-250.
- McMichael AJ, Vachiramon V, Guzman-Sanchez DA, et al. Psoriasis in African-Americans: a caregivers' survey. J Drugs Dermatol. 2012;11:478-482.
- Alexis AF, Blackcloud P. Psoriasis in skin of color: epidemiology, genetics, clinical presentation, and treatment nuances. J Clin Aesthet Dermatol. 2014;7:16-24.
- Posligua A, Maldonado C, Gonzalez MG. Rupioid psoriasis preceded by varicella presenting as Koebner phenomenon. J Am Acad Dermatol. 2016;74(5 suppl 1):AB268.
- Feldman SR, Feldman S, Brown K, et al. "Coral reef" psoriasis: a marker of resistance to topical treatment. J Dermatolog Treat. 2008;19:257-258.
Practice Points
- Rupioid psoriasis in skin of color may present a diagnostic challenge for health care providers.
- Rupioid psoriasis may represent a psoriasis variant that is highly responsive to treatment.

PD-1 Signaling in Extramammary Paget Disease
Primary extramammary Paget disease (EMPD) is an adnexal carcinoma of the apocrine gland ducts that presents as an erythematous patch on cutaneous sites rich with apocrine glands.1 Primary EMPD can be in situ or invasive with the potential to become metastatic.2 Treatment of primary EMPD is challenging due to the difficulty of achieving clear surgical margins, as the tumor has microscopic spread throughout the epidermis in a skipping fashion.3 Mohs micrographic surgery is the treatment of choice; however, there is a clinical need to identify additional treatment modalities, especially for patients with unresectable, invasive, or metastatic primary EMPD,4 which partly is due to lack of data to understand the pathogenesis of primary EMPD. Recently, there have been studies investigating the genetic characteristics of EMPD tumors. The interaction between the programmed cell death receptor 1 (PD-1) and its ligand (PD-L1) is one of the pathways recently studied and has been reported to be a potential target in EMPD.5-7 Programmed cell death receptor 1 signaling constitutes an immune checkpoint pathway that regulates the activation of tumor-specific T cells.8 In several malignancies, cancer cells express PD-L1 on their surface to activate PD-1 signaling in T cells as a mechanism to dampen the tumor-specific immune response and evade antitumor immunity.9 Thus, blocking PD-1 signaling widely is used to activate tumor-specific T cells and decrease tumor burden.10 Given the advances of immunotherapy in many neoplasms and the paucity of effective agents to treat EMPD, this article serves to shed light on recent data studying PD-1 signaling in EMPD and highlights the potential clinical use of immunotherapy for EMPD.
EMPD and Its Subtypes
Extramammary Paget disease is a rare adenocarcinoma typically affecting older patients (age >60 years) in cutaneous sites with abundant apocrine glands such as the genital and perianal skin.3 Extramammary Paget disease presents as an erythematous patch and frequently is treated initially as a skin dermatosis, resulting in a delay in diagnosis. Histologically, EMPD is characterized by the presence of single cells or a nest of cells having abundant pale cytoplasm and large vesicular nuclei distributed in the epidermis in a pagetoid fashion.11
Extramammary Paget disease can be primary or secondary; the 2 subtypes behave differently both clinically and prognostically. Although primary EMPD is considered to be an adnexal carcinoma of the apocrine gland ducts, secondary EMPD is considered to be an intraepithelial extension of malignant cells from an underlying internal neoplasm.12 The underlying malignancies usually are located within dermal adnexal glands or organs in the vicinity of the cutaneous lesion, such as the colon in the case of perianal EMPD. Histologically, primary and secondary EMPD can be differentiated based on their immunophenotypic staining profiles. Although all cases of EMPD show positive immunohistochemistry staining for cytokeratin 7, carcinoembryonic antigen, and epithelial membrane antigen, only primary EMPD will additionally stain for GCDFP-15 (gross cystic disease fluid protein 15) and GATA.11 Regardless of the immunohistochemistry stains, every patient newly diagnosed with EMPD deserves a full workup for malignancy screening, including a colonoscopy, cystoscopy, mammography and Papanicolaou test in women, pelvic ultrasound, and computed tomography of the abdomen and pelvis.13
The first-line treatment of EMPD is surgery; however, obtaining clear surgical margins can be a challenge, with high recurrence rates due to the microscopic spread of the disease throughout the epidermis.4 In addition, anatomic location affects the surgical approach and patient survival. Recent studies on EMPD mortality outcomes in women show that mortality is higher in patients with vaginal EMPD than in those with vulvar/labial EMPD, partly due to the sensitive location that makes it difficult to perform wide local excisions.13,14 Assessing the entire margins with tissue preservation using Mohs micrographic surgery has been shown to be successful in decreasing the recurrence rate, especially when coupled with the use of cytokeratin 7 immunohistochemistry.4 Other treatment modalities include radiation, topical imiquimod, and photodynamic therapy.15,16 Regardless of treatment modality, EMPD requires long‐term follow-up to monitor for disease recurrence, regional lymphadenopathy, distant metastasis, or development of an internal malignancy.
The pathogenesis of primary EMPD remains unclear. The tumor is thought to be derived from Toker cells, which are pluripotent adnexal stem cells located in the epidermis that normally give rise to apocrine glands.17 There have been few studies investigating the genetic characteristics of EMPD lesions in an attempt to understand pathogenesis as well as to find druggable targets. Current data for targeted therapy have focused on HER2 (human epidermal growth factor receptor 2) hormone receptor expression,18 ERBB (erythroblastic oncogene B) amplification,19 CDK4 (cyclin-dependent kinase 4)–cyclin D1 signaling,20 and most recently PD-1/PD-L1 pathway.5-7
PD-1 Expression in EMPD: Implication for Immunotherapy
Most tumors display novel antigens that are recognized by the host immune system and thus stimulate cell-mediated and humoral pathways. The immune system naturally provides regulatory immune checkpoints to T cell–mediated immune responses. One of these checkpoints involves the interaction between PD-1 on T cells and its ligand PD-L1 on tumor cells.21 When PD-1 binds to PD-L1 on tumor cells, there is inhibition of T-cell proliferation, a decrease in cytokine production, and induction of T-cell cytolysis.22 The Figure summarizes the dynamics for T-cell regulation.

Naturally, tumor-infiltrating T cells trigger their own inhibition by binding to PD-L1. However, certain tumor cells constitutively upregulate the expression of PD-L1. With that, the tumor cells gain the ability to suppress T cells and avoid T cell–mediated cytotoxicity,23 which is known as the adoptive immune resistance mechanism. There have been several studies in the literature investigating the PD-1 signaling pathway in EMPD as a way to determine if EMPD would be susceptible to immune checkpoint blockade. The success of checkpoint inhibitor immunotherapy generally correlates with increased PD-L1 expression by tumor cells.
One study evaluated the expression of PD-L1 in tumor cells and tumor-infiltrating T cells in 18 cases of EMPD.6 The authors identified that even though tumor cell PD-L1 expression was detected in only 3 (17%) cases, tumor-infiltrating lymphocytes expressed PD-L1 in the majority of the cases analyzed and in all of the cases positive for tumor cell PD-L1.6
Another study evaluated PD-1 and PD-L1 expression in EMPD tumor cells and tumor-associated immune infiltrate.5 They found that PD-1 was expressed heavily by the tumor-associated immune infiltrate in all EMPD cases analyzed. Similar to the previously mentioned study,6 PD-L1 was expressed by tumor cells in a few cases only. Interestingly, they found that the density of CD3 in the tumor-associated immune infiltrate was significantly (P=.049) higher in patients who were alive than in those who died, suggesting the importance of an exuberant T-cell response for survival in EMPD.5
A third study investigated protein expression of the B7 family members as well as PD-1 and PD-L1/2 in 55 EMPD samples. In this study the authors also found that tumor cell PD-L1 was minimal. Interestingly, they also found that tumor cells expressed B7 proteins in the majority of the cases.7
Finally, another study examined activity levels of T cells in EMPD by measuring the number and expression levels of cytotoxic T-cell cytokines.24 The authors first found that EMPD tumors had a significantly higher number of CD8+ tumor-infiltrating lymphocytes compared to peripheral blood (P<.01). These CD8+ tumor-infiltrating lymphocytes also had a significantly higher expression of PD-1 (P<.01). They also found that tumor cells produced an immunosuppressive molecule called indoleamine 2,3-dyoxygenae that functions by suppressing T-cell activity levels. They concluded that in EMPD, tumor-specific T lymphocytes have an exhausted phenotype due to PD-1 activation as well as indoleamine 2,3-dyoxygenase release to the tumor microenvironment.24
These studies highlight that restoring the effector functions of tumor-specific T lymphocytes could be an effective treatment strategy for EMPD. In fact, immunotherapy has been used with success for EMPD in the form of topical immunomodulators such as imiquimod.16,25 More than 40 cases of EMPD treated with imiquimod 5% have been published; of these, only 6 were considered nonresponders,5 which suggests that EMPD may respond to other immunotherapies such as checkpoint inhibitors. It is an exciting time for immunotherapy as more checkpoint inhibitors are being developed. Among the newer agents is cemiplimab, which is a PD-1 inhibitor now US Food and Drug Administration approved for the treatment of locally advanced or metastatic cutaneous squamous cell carcinoma in patients who are not candidates for curative surgery or curative radiation.26 Programmed cell death receptor 1 signaling can serve as a potential target in EMPD, and further studies need to be performed to test the clinical efficacy, especially in unresectable or invasive/metastatic EMPD. As the PD-1 pathway is more studied in EMPD, and as more PD-1 inhibitors get developed, it would be a clinical need to establish clinical studies for PD-1 inhibitors in EMPD.
- Ito T, Kaku-Ito Y, Furue M. The diagnosis and management of extramammary Paget’s disease. Expert Rev Anticancer Ther. 2018;18:543-553.
- van der Zwan JM, Siesling S, Blokx WAM, et al. Invasive extramammary Paget’s disease and the risk for secondary tumours in Europe. Eur J Surg Oncol. 2012;38:214-221.
- Simonds RM, Segal RJ, Sharma A. Extramammary Paget’s disease: a review of the literature. Int J Dermatol. 2019;58:871-879.
- Wollina U, Goldman A, Bieneck A, et al. Surgical treatment for extramammary Paget’s disease. Curr Treat Options Oncol. 2018;19:27.
- Mauzo SH, Tetzlaff MT, Milton DR, et al. Expression of PD-1 and PD-L1 in extramammary Paget disease: implications for immune-targeted therapy. Cancers (Basel). 2019;11:754.
- Fowler MR, Flanigan KL, Googe PB. PD-L1 expression in extramammary Paget disease [published online March 6, 2020]. Am J Dermatopathol. doi:10.1097/dad.0000000000001622.
- Pourmaleki M, Young JH, Socci ND, et al. Extramammary Paget disease shows differential expression of B7 family members B7-H3, B7-H4, PD-L1, PD-L2 and cancer/testis antigens NY-ESO-1 and MAGE-A. Oncotarget. 2019;10:6152-6167.
- Mahoney KM, Freeman GJ, McDermott DF. The next immune-checkpoint inhibitors: PD-1/PD-L1 blockade in melanoma. Clin Ther. 2015;37:764-782.
- Dany M, Nganga R, Chidiac A, et al. Advances in immunotherapy for melanoma management. Hum Vaccines Immunother. 2016;12:2501-2511.
- Richter MD, Hughes GC, Chung SH, et al. Immunologic adverse events from immune checkpoint therapy [published online April 13, 2020]. Best Pract Res Clin Rheumatol. doi:10.1016/j.berh.2020.101511.
- Kang Z, Zhang Q, Zhang Q, et al. Clinical and pathological characteristics of extramammary Paget’s disease: report of 246 Chinese male patients. Int J Clin Exp Pathol. 2015;8:13233-13240.
- Ohara K, Fujisawa Y, Yoshino K, et al. A proposal for a TNM staging system for extramammary Paget disease: retrospective analysis of 301 patients with invasive primary tumors. J Dermatol Sci. 2016;83:234-239.
- Hatta N. Prognostic factors of extramammary Paget’s disease. Curr Treat Options Oncol. 2018;19:47.
- Yao H, Xie M, Fu S, et al. Survival analysis of patients with invasive extramammary Paget disease: implications of anatomic sites. BMC Cancer. 2018;18:403.
- Herrel LA, Weiss AD, Goodman M, et al. Extramammary Paget’s disease in males: survival outcomes in 495 patients. Ann Surg Oncol. 2015;22:1625-1630.
- Sanderson P, Innamaa A, Palmer J, et al. Imiquimod therapy for extramammary Paget’s disease of the vulva: a viable non-surgical alternative. J Obstet Gynaecol. 2013;33:479-483.
- Smith AA. Pre-Paget cells: evidence of keratinocyte origin of extramammary Paget’s disease. Intractable Rare Dis Res. 2019;8:203-205.
- Garganese G, Inzani F, Mantovani G, et al. The vulvar immunohistochemical panel (VIP) project: molecular profiles of vulvar Paget’s disease. J Cancer Res Clin Oncol. 2019;145:2211-2225.
- Dias-Santagata D, Lam Q, Bergethon K, et al. A potential role for targeted therapy in a subset of metastasizing adnexal carcinomas. Mod Pathol. 2011;24:974-982.
- Cohen JM, Granter SR, Werchniak AE. Risk stratification in extramammary Paget disease. Clin Exp Dermatol. 2015;40:473-478.
- Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018;8:1069-1086.
- Shi Y. Regulatory mechanisms of PD-L1 expression in cancer cells. Cancer Immunol Immunother. 2018;67:1481-1489.
- Cui C, Yu B, Jiang Q, et al. The roles of PD-1/PD-L1 and its signalling pathway in gastrointestinal tract cancers. Clin Exp Pharmacol Physiol. 2019;46:3-10.
- Iga N, Otsuka A, Yamamoto Y, et al. Accumulation of exhausted CD8+ T cells in extramammary Paget’s disease. PLoS One. 2019;14:E0211135.
- Frances L, Pascual JC, Leiva-Salinas M, et al. Extramammary Paget disease successfully treated with topical imiquimod 5% and tazarotene. Dermatol Ther. 2014;27:19-20.
- Lee A, Duggan S, Deeks ED. Cemiplimab: a review in advanced cutaneous squamous cell carcinoma. Drugs. 2020;80:813-819.
Primary extramammary Paget disease (EMPD) is an adnexal carcinoma of the apocrine gland ducts that presents as an erythematous patch on cutaneous sites rich with apocrine glands.1 Primary EMPD can be in situ or invasive with the potential to become metastatic.2 Treatment of primary EMPD is challenging due to the difficulty of achieving clear surgical margins, as the tumor has microscopic spread throughout the epidermis in a skipping fashion.3 Mohs micrographic surgery is the treatment of choice; however, there is a clinical need to identify additional treatment modalities, especially for patients with unresectable, invasive, or metastatic primary EMPD,4 which partly is due to lack of data to understand the pathogenesis of primary EMPD. Recently, there have been studies investigating the genetic characteristics of EMPD tumors. The interaction between the programmed cell death receptor 1 (PD-1) and its ligand (PD-L1) is one of the pathways recently studied and has been reported to be a potential target in EMPD.5-7 Programmed cell death receptor 1 signaling constitutes an immune checkpoint pathway that regulates the activation of tumor-specific T cells.8 In several malignancies, cancer cells express PD-L1 on their surface to activate PD-1 signaling in T cells as a mechanism to dampen the tumor-specific immune response and evade antitumor immunity.9 Thus, blocking PD-1 signaling widely is used to activate tumor-specific T cells and decrease tumor burden.10 Given the advances of immunotherapy in many neoplasms and the paucity of effective agents to treat EMPD, this article serves to shed light on recent data studying PD-1 signaling in EMPD and highlights the potential clinical use of immunotherapy for EMPD.
EMPD and Its Subtypes
Extramammary Paget disease is a rare adenocarcinoma typically affecting older patients (age >60 years) in cutaneous sites with abundant apocrine glands such as the genital and perianal skin.3 Extramammary Paget disease presents as an erythematous patch and frequently is treated initially as a skin dermatosis, resulting in a delay in diagnosis. Histologically, EMPD is characterized by the presence of single cells or a nest of cells having abundant pale cytoplasm and large vesicular nuclei distributed in the epidermis in a pagetoid fashion.11
Extramammary Paget disease can be primary or secondary; the 2 subtypes behave differently both clinically and prognostically. Although primary EMPD is considered to be an adnexal carcinoma of the apocrine gland ducts, secondary EMPD is considered to be an intraepithelial extension of malignant cells from an underlying internal neoplasm.12 The underlying malignancies usually are located within dermal adnexal glands or organs in the vicinity of the cutaneous lesion, such as the colon in the case of perianal EMPD. Histologically, primary and secondary EMPD can be differentiated based on their immunophenotypic staining profiles. Although all cases of EMPD show positive immunohistochemistry staining for cytokeratin 7, carcinoembryonic antigen, and epithelial membrane antigen, only primary EMPD will additionally stain for GCDFP-15 (gross cystic disease fluid protein 15) and GATA.11 Regardless of the immunohistochemistry stains, every patient newly diagnosed with EMPD deserves a full workup for malignancy screening, including a colonoscopy, cystoscopy, mammography and Papanicolaou test in women, pelvic ultrasound, and computed tomography of the abdomen and pelvis.13
The first-line treatment of EMPD is surgery; however, obtaining clear surgical margins can be a challenge, with high recurrence rates due to the microscopic spread of the disease throughout the epidermis.4 In addition, anatomic location affects the surgical approach and patient survival. Recent studies on EMPD mortality outcomes in women show that mortality is higher in patients with vaginal EMPD than in those with vulvar/labial EMPD, partly due to the sensitive location that makes it difficult to perform wide local excisions.13,14 Assessing the entire margins with tissue preservation using Mohs micrographic surgery has been shown to be successful in decreasing the recurrence rate, especially when coupled with the use of cytokeratin 7 immunohistochemistry.4 Other treatment modalities include radiation, topical imiquimod, and photodynamic therapy.15,16 Regardless of treatment modality, EMPD requires long‐term follow-up to monitor for disease recurrence, regional lymphadenopathy, distant metastasis, or development of an internal malignancy.
The pathogenesis of primary EMPD remains unclear. The tumor is thought to be derived from Toker cells, which are pluripotent adnexal stem cells located in the epidermis that normally give rise to apocrine glands.17 There have been few studies investigating the genetic characteristics of EMPD lesions in an attempt to understand pathogenesis as well as to find druggable targets. Current data for targeted therapy have focused on HER2 (human epidermal growth factor receptor 2) hormone receptor expression,18 ERBB (erythroblastic oncogene B) amplification,19 CDK4 (cyclin-dependent kinase 4)–cyclin D1 signaling,20 and most recently PD-1/PD-L1 pathway.5-7
PD-1 Expression in EMPD: Implication for Immunotherapy
Most tumors display novel antigens that are recognized by the host immune system and thus stimulate cell-mediated and humoral pathways. The immune system naturally provides regulatory immune checkpoints to T cell–mediated immune responses. One of these checkpoints involves the interaction between PD-1 on T cells and its ligand PD-L1 on tumor cells.21 When PD-1 binds to PD-L1 on tumor cells, there is inhibition of T-cell proliferation, a decrease in cytokine production, and induction of T-cell cytolysis.22 The Figure summarizes the dynamics for T-cell regulation.
Naturally, tumor-infiltrating T cells trigger their own inhibition by binding to PD-L1. However, certain tumor cells constitutively upregulate the expression of PD-L1. With that, the tumor cells gain the ability to suppress T cells and avoid T cell–mediated cytotoxicity,23 which is known as the adoptive immune resistance mechanism. There have been several studies in the literature investigating the PD-1 signaling pathway in EMPD as a way to determine if EMPD would be susceptible to immune checkpoint blockade. The success of checkpoint inhibitor immunotherapy generally correlates with increased PD-L1 expression by tumor cells.
One study evaluated the expression of PD-L1 in tumor cells and tumor-infiltrating T cells in 18 cases of EMPD.6 The authors identified that even though tumor cell PD-L1 expression was detected in only 3 (17%) cases, tumor-infiltrating lymphocytes expressed PD-L1 in the majority of the cases analyzed and in all of the cases positive for tumor cell PD-L1.6
Another study evaluated PD-1 and PD-L1 expression in EMPD tumor cells and tumor-associated immune infiltrate.5 They found that PD-1 was expressed heavily by the tumor-associated immune infiltrate in all EMPD cases analyzed. Similar to the previously mentioned study,6 PD-L1 was expressed by tumor cells in a few cases only. Interestingly, they found that the density of CD3 in the tumor-associated immune infiltrate was significantly (P=.049) higher in patients who were alive than in those who died, suggesting the importance of an exuberant T-cell response for survival in EMPD.5
A third study investigated protein expression of the B7 family members as well as PD-1 and PD-L1/2 in 55 EMPD samples. In this study the authors also found that tumor cell PD-L1 was minimal. Interestingly, they also found that tumor cells expressed B7 proteins in the majority of the cases.7
Finally, another study examined activity levels of T cells in EMPD by measuring the number and expression levels of cytotoxic T-cell cytokines.24 The authors first found that EMPD tumors had a significantly higher number of CD8+ tumor-infiltrating lymphocytes compared to peripheral blood (P<.01). These CD8+ tumor-infiltrating lymphocytes also had a significantly higher expression of PD-1 (P<.01). They also found that tumor cells produced an immunosuppressive molecule called indoleamine 2,3-dyoxygenae that functions by suppressing T-cell activity levels. They concluded that in EMPD, tumor-specific T lymphocytes have an exhausted phenotype due to PD-1 activation as well as indoleamine 2,3-dyoxygenase release to the tumor microenvironment.24
These studies highlight that restoring the effector functions of tumor-specific T lymphocytes could be an effective treatment strategy for EMPD. In fact, immunotherapy has been used with success for EMPD in the form of topical immunomodulators such as imiquimod.16,25 More than 40 cases of EMPD treated with imiquimod 5% have been published; of these, only 6 were considered nonresponders,5 which suggests that EMPD may respond to other immunotherapies such as checkpoint inhibitors. It is an exciting time for immunotherapy as more checkpoint inhibitors are being developed. Among the newer agents is cemiplimab, which is a PD-1 inhibitor now US Food and Drug Administration approved for the treatment of locally advanced or metastatic cutaneous squamous cell carcinoma in patients who are not candidates for curative surgery or curative radiation.26 Programmed cell death receptor 1 signaling can serve as a potential target in EMPD, and further studies need to be performed to test the clinical efficacy, especially in unresectable or invasive/metastatic EMPD. As the PD-1 pathway is more studied in EMPD, and as more PD-1 inhibitors get developed, it would be a clinical need to establish clinical studies for PD-1 inhibitors in EMPD.
Primary extramammary Paget disease (EMPD) is an adnexal carcinoma of the apocrine gland ducts that presents as an erythematous patch on cutaneous sites rich with apocrine glands.1 Primary EMPD can be in situ or invasive with the potential to become metastatic.2 Treatment of primary EMPD is challenging due to the difficulty of achieving clear surgical margins, as the tumor has microscopic spread throughout the epidermis in a skipping fashion.3 Mohs micrographic surgery is the treatment of choice; however, there is a clinical need to identify additional treatment modalities, especially for patients with unresectable, invasive, or metastatic primary EMPD,4 which partly is due to lack of data to understand the pathogenesis of primary EMPD. Recently, there have been studies investigating the genetic characteristics of EMPD tumors. The interaction between the programmed cell death receptor 1 (PD-1) and its ligand (PD-L1) is one of the pathways recently studied and has been reported to be a potential target in EMPD.5-7 Programmed cell death receptor 1 signaling constitutes an immune checkpoint pathway that regulates the activation of tumor-specific T cells.8 In several malignancies, cancer cells express PD-L1 on their surface to activate PD-1 signaling in T cells as a mechanism to dampen the tumor-specific immune response and evade antitumor immunity.9 Thus, blocking PD-1 signaling widely is used to activate tumor-specific T cells and decrease tumor burden.10 Given the advances of immunotherapy in many neoplasms and the paucity of effective agents to treat EMPD, this article serves to shed light on recent data studying PD-1 signaling in EMPD and highlights the potential clinical use of immunotherapy for EMPD.
EMPD and Its Subtypes
Extramammary Paget disease is a rare adenocarcinoma typically affecting older patients (age >60 years) in cutaneous sites with abundant apocrine glands such as the genital and perianal skin.3 Extramammary Paget disease presents as an erythematous patch and frequently is treated initially as a skin dermatosis, resulting in a delay in diagnosis. Histologically, EMPD is characterized by the presence of single cells or a nest of cells having abundant pale cytoplasm and large vesicular nuclei distributed in the epidermis in a pagetoid fashion.11
Extramammary Paget disease can be primary or secondary; the 2 subtypes behave differently both clinically and prognostically. Although primary EMPD is considered to be an adnexal carcinoma of the apocrine gland ducts, secondary EMPD is considered to be an intraepithelial extension of malignant cells from an underlying internal neoplasm.12 The underlying malignancies usually are located within dermal adnexal glands or organs in the vicinity of the cutaneous lesion, such as the colon in the case of perianal EMPD. Histologically, primary and secondary EMPD can be differentiated based on their immunophenotypic staining profiles. Although all cases of EMPD show positive immunohistochemistry staining for cytokeratin 7, carcinoembryonic antigen, and epithelial membrane antigen, only primary EMPD will additionally stain for GCDFP-15 (gross cystic disease fluid protein 15) and GATA.11 Regardless of the immunohistochemistry stains, every patient newly diagnosed with EMPD deserves a full workup for malignancy screening, including a colonoscopy, cystoscopy, mammography and Papanicolaou test in women, pelvic ultrasound, and computed tomography of the abdomen and pelvis.13
The first-line treatment of EMPD is surgery; however, obtaining clear surgical margins can be a challenge, with high recurrence rates due to the microscopic spread of the disease throughout the epidermis.4 In addition, anatomic location affects the surgical approach and patient survival. Recent studies on EMPD mortality outcomes in women show that mortality is higher in patients with vaginal EMPD than in those with vulvar/labial EMPD, partly due to the sensitive location that makes it difficult to perform wide local excisions.13,14 Assessing the entire margins with tissue preservation using Mohs micrographic surgery has been shown to be successful in decreasing the recurrence rate, especially when coupled with the use of cytokeratin 7 immunohistochemistry.4 Other treatment modalities include radiation, topical imiquimod, and photodynamic therapy.15,16 Regardless of treatment modality, EMPD requires long‐term follow-up to monitor for disease recurrence, regional lymphadenopathy, distant metastasis, or development of an internal malignancy.
The pathogenesis of primary EMPD remains unclear. The tumor is thought to be derived from Toker cells, which are pluripotent adnexal stem cells located in the epidermis that normally give rise to apocrine glands.17 There have been few studies investigating the genetic characteristics of EMPD lesions in an attempt to understand pathogenesis as well as to find druggable targets. Current data for targeted therapy have focused on HER2 (human epidermal growth factor receptor 2) hormone receptor expression,18 ERBB (erythroblastic oncogene B) amplification,19 CDK4 (cyclin-dependent kinase 4)–cyclin D1 signaling,20 and most recently PD-1/PD-L1 pathway.5-7
PD-1 Expression in EMPD: Implication for Immunotherapy
Most tumors display novel antigens that are recognized by the host immune system and thus stimulate cell-mediated and humoral pathways. The immune system naturally provides regulatory immune checkpoints to T cell–mediated immune responses. One of these checkpoints involves the interaction between PD-1 on T cells and its ligand PD-L1 on tumor cells.21 When PD-1 binds to PD-L1 on tumor cells, there is inhibition of T-cell proliferation, a decrease in cytokine production, and induction of T-cell cytolysis.22 The Figure summarizes the dynamics for T-cell regulation.
Naturally, tumor-infiltrating T cells trigger their own inhibition by binding to PD-L1. However, certain tumor cells constitutively upregulate the expression of PD-L1. With that, the tumor cells gain the ability to suppress T cells and avoid T cell–mediated cytotoxicity,23 which is known as the adoptive immune resistance mechanism. There have been several studies in the literature investigating the PD-1 signaling pathway in EMPD as a way to determine if EMPD would be susceptible to immune checkpoint blockade. The success of checkpoint inhibitor immunotherapy generally correlates with increased PD-L1 expression by tumor cells.
One study evaluated the expression of PD-L1 in tumor cells and tumor-infiltrating T cells in 18 cases of EMPD.6 The authors identified that even though tumor cell PD-L1 expression was detected in only 3 (17%) cases, tumor-infiltrating lymphocytes expressed PD-L1 in the majority of the cases analyzed and in all of the cases positive for tumor cell PD-L1.6
Another study evaluated PD-1 and PD-L1 expression in EMPD tumor cells and tumor-associated immune infiltrate.5 They found that PD-1 was expressed heavily by the tumor-associated immune infiltrate in all EMPD cases analyzed. Similar to the previously mentioned study,6 PD-L1 was expressed by tumor cells in a few cases only. Interestingly, they found that the density of CD3 in the tumor-associated immune infiltrate was significantly (P=.049) higher in patients who were alive than in those who died, suggesting the importance of an exuberant T-cell response for survival in EMPD.5
A third study investigated protein expression of the B7 family members as well as PD-1 and PD-L1/2 in 55 EMPD samples. In this study the authors also found that tumor cell PD-L1 was minimal. Interestingly, they also found that tumor cells expressed B7 proteins in the majority of the cases.7
Finally, another study examined activity levels of T cells in EMPD by measuring the number and expression levels of cytotoxic T-cell cytokines.24 The authors first found that EMPD tumors had a significantly higher number of CD8+ tumor-infiltrating lymphocytes compared to peripheral blood (P<.01). These CD8+ tumor-infiltrating lymphocytes also had a significantly higher expression of PD-1 (P<.01). They also found that tumor cells produced an immunosuppressive molecule called indoleamine 2,3-dyoxygenae that functions by suppressing T-cell activity levels. They concluded that in EMPD, tumor-specific T lymphocytes have an exhausted phenotype due to PD-1 activation as well as indoleamine 2,3-dyoxygenase release to the tumor microenvironment.24
These studies highlight that restoring the effector functions of tumor-specific T lymphocytes could be an effective treatment strategy for EMPD. In fact, immunotherapy has been used with success for EMPD in the form of topical immunomodulators such as imiquimod.16,25 More than 40 cases of EMPD treated with imiquimod 5% have been published; of these, only 6 were considered nonresponders,5 which suggests that EMPD may respond to other immunotherapies such as checkpoint inhibitors. It is an exciting time for immunotherapy as more checkpoint inhibitors are being developed. Among the newer agents is cemiplimab, which is a PD-1 inhibitor now US Food and Drug Administration approved for the treatment of locally advanced or metastatic cutaneous squamous cell carcinoma in patients who are not candidates for curative surgery or curative radiation.26 Programmed cell death receptor 1 signaling can serve as a potential target in EMPD, and further studies need to be performed to test the clinical efficacy, especially in unresectable or invasive/metastatic EMPD. As the PD-1 pathway is more studied in EMPD, and as more PD-1 inhibitors get developed, it would be a clinical need to establish clinical studies for PD-1 inhibitors in EMPD.
- Ito T, Kaku-Ito Y, Furue M. The diagnosis and management of extramammary Paget’s disease. Expert Rev Anticancer Ther. 2018;18:543-553.
- van der Zwan JM, Siesling S, Blokx WAM, et al. Invasive extramammary Paget’s disease and the risk for secondary tumours in Europe. Eur J Surg Oncol. 2012;38:214-221.
- Simonds RM, Segal RJ, Sharma A. Extramammary Paget’s disease: a review of the literature. Int J Dermatol. 2019;58:871-879.
- Wollina U, Goldman A, Bieneck A, et al. Surgical treatment for extramammary Paget’s disease. Curr Treat Options Oncol. 2018;19:27.
- Mauzo SH, Tetzlaff MT, Milton DR, et al. Expression of PD-1 and PD-L1 in extramammary Paget disease: implications for immune-targeted therapy. Cancers (Basel). 2019;11:754.
- Fowler MR, Flanigan KL, Googe PB. PD-L1 expression in extramammary Paget disease [published online March 6, 2020]. Am J Dermatopathol. doi:10.1097/dad.0000000000001622.
- Pourmaleki M, Young JH, Socci ND, et al. Extramammary Paget disease shows differential expression of B7 family members B7-H3, B7-H4, PD-L1, PD-L2 and cancer/testis antigens NY-ESO-1 and MAGE-A. Oncotarget. 2019;10:6152-6167.
- Mahoney KM, Freeman GJ, McDermott DF. The next immune-checkpoint inhibitors: PD-1/PD-L1 blockade in melanoma. Clin Ther. 2015;37:764-782.
- Dany M, Nganga R, Chidiac A, et al. Advances in immunotherapy for melanoma management. Hum Vaccines Immunother. 2016;12:2501-2511.
- Richter MD, Hughes GC, Chung SH, et al. Immunologic adverse events from immune checkpoint therapy [published online April 13, 2020]. Best Pract Res Clin Rheumatol. doi:10.1016/j.berh.2020.101511.
- Kang Z, Zhang Q, Zhang Q, et al. Clinical and pathological characteristics of extramammary Paget’s disease: report of 246 Chinese male patients. Int J Clin Exp Pathol. 2015;8:13233-13240.
- Ohara K, Fujisawa Y, Yoshino K, et al. A proposal for a TNM staging system for extramammary Paget disease: retrospective analysis of 301 patients with invasive primary tumors. J Dermatol Sci. 2016;83:234-239.
- Hatta N. Prognostic factors of extramammary Paget’s disease. Curr Treat Options Oncol. 2018;19:47.
- Yao H, Xie M, Fu S, et al. Survival analysis of patients with invasive extramammary Paget disease: implications of anatomic sites. BMC Cancer. 2018;18:403.
- Herrel LA, Weiss AD, Goodman M, et al. Extramammary Paget’s disease in males: survival outcomes in 495 patients. Ann Surg Oncol. 2015;22:1625-1630.
- Sanderson P, Innamaa A, Palmer J, et al. Imiquimod therapy for extramammary Paget’s disease of the vulva: a viable non-surgical alternative. J Obstet Gynaecol. 2013;33:479-483.
- Smith AA. Pre-Paget cells: evidence of keratinocyte origin of extramammary Paget’s disease. Intractable Rare Dis Res. 2019;8:203-205.
- Garganese G, Inzani F, Mantovani G, et al. The vulvar immunohistochemical panel (VIP) project: molecular profiles of vulvar Paget’s disease. J Cancer Res Clin Oncol. 2019;145:2211-2225.
- Dias-Santagata D, Lam Q, Bergethon K, et al. A potential role for targeted therapy in a subset of metastasizing adnexal carcinomas. Mod Pathol. 2011;24:974-982.
- Cohen JM, Granter SR, Werchniak AE. Risk stratification in extramammary Paget disease. Clin Exp Dermatol. 2015;40:473-478.
- Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018;8:1069-1086.
- Shi Y. Regulatory mechanisms of PD-L1 expression in cancer cells. Cancer Immunol Immunother. 2018;67:1481-1489.
- Cui C, Yu B, Jiang Q, et al. The roles of PD-1/PD-L1 and its signalling pathway in gastrointestinal tract cancers. Clin Exp Pharmacol Physiol. 2019;46:3-10.
- Iga N, Otsuka A, Yamamoto Y, et al. Accumulation of exhausted CD8+ T cells in extramammary Paget’s disease. PLoS One. 2019;14:E0211135.
- Frances L, Pascual JC, Leiva-Salinas M, et al. Extramammary Paget disease successfully treated with topical imiquimod 5% and tazarotene. Dermatol Ther. 2014;27:19-20.
- Lee A, Duggan S, Deeks ED. Cemiplimab: a review in advanced cutaneous squamous cell carcinoma. Drugs. 2020;80:813-819.
- Ito T, Kaku-Ito Y, Furue M. The diagnosis and management of extramammary Paget’s disease. Expert Rev Anticancer Ther. 2018;18:543-553.
- van der Zwan JM, Siesling S, Blokx WAM, et al. Invasive extramammary Paget’s disease and the risk for secondary tumours in Europe. Eur J Surg Oncol. 2012;38:214-221.
- Simonds RM, Segal RJ, Sharma A. Extramammary Paget’s disease: a review of the literature. Int J Dermatol. 2019;58:871-879.
- Wollina U, Goldman A, Bieneck A, et al. Surgical treatment for extramammary Paget’s disease. Curr Treat Options Oncol. 2018;19:27.
- Mauzo SH, Tetzlaff MT, Milton DR, et al. Expression of PD-1 and PD-L1 in extramammary Paget disease: implications for immune-targeted therapy. Cancers (Basel). 2019;11:754.
- Fowler MR, Flanigan KL, Googe PB. PD-L1 expression in extramammary Paget disease [published online March 6, 2020]. Am J Dermatopathol. doi:10.1097/dad.0000000000001622.
- Pourmaleki M, Young JH, Socci ND, et al. Extramammary Paget disease shows differential expression of B7 family members B7-H3, B7-H4, PD-L1, PD-L2 and cancer/testis antigens NY-ESO-1 and MAGE-A. Oncotarget. 2019;10:6152-6167.
- Mahoney KM, Freeman GJ, McDermott DF. The next immune-checkpoint inhibitors: PD-1/PD-L1 blockade in melanoma. Clin Ther. 2015;37:764-782.
- Dany M, Nganga R, Chidiac A, et al. Advances in immunotherapy for melanoma management. Hum Vaccines Immunother. 2016;12:2501-2511.
- Richter MD, Hughes GC, Chung SH, et al. Immunologic adverse events from immune checkpoint therapy [published online April 13, 2020]. Best Pract Res Clin Rheumatol. doi:10.1016/j.berh.2020.101511.
- Kang Z, Zhang Q, Zhang Q, et al. Clinical and pathological characteristics of extramammary Paget’s disease: report of 246 Chinese male patients. Int J Clin Exp Pathol. 2015;8:13233-13240.
- Ohara K, Fujisawa Y, Yoshino K, et al. A proposal for a TNM staging system for extramammary Paget disease: retrospective analysis of 301 patients with invasive primary tumors. J Dermatol Sci. 2016;83:234-239.
- Hatta N. Prognostic factors of extramammary Paget’s disease. Curr Treat Options Oncol. 2018;19:47.
- Yao H, Xie M, Fu S, et al. Survival analysis of patients with invasive extramammary Paget disease: implications of anatomic sites. BMC Cancer. 2018;18:403.
- Herrel LA, Weiss AD, Goodman M, et al. Extramammary Paget’s disease in males: survival outcomes in 495 patients. Ann Surg Oncol. 2015;22:1625-1630.
- Sanderson P, Innamaa A, Palmer J, et al. Imiquimod therapy for extramammary Paget’s disease of the vulva: a viable non-surgical alternative. J Obstet Gynaecol. 2013;33:479-483.
- Smith AA. Pre-Paget cells: evidence of keratinocyte origin of extramammary Paget’s disease. Intractable Rare Dis Res. 2019;8:203-205.
- Garganese G, Inzani F, Mantovani G, et al. The vulvar immunohistochemical panel (VIP) project: molecular profiles of vulvar Paget’s disease. J Cancer Res Clin Oncol. 2019;145:2211-2225.
- Dias-Santagata D, Lam Q, Bergethon K, et al. A potential role for targeted therapy in a subset of metastasizing adnexal carcinomas. Mod Pathol. 2011;24:974-982.
- Cohen JM, Granter SR, Werchniak AE. Risk stratification in extramammary Paget disease. Clin Exp Dermatol. 2015;40:473-478.
- Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018;8:1069-1086.
- Shi Y. Regulatory mechanisms of PD-L1 expression in cancer cells. Cancer Immunol Immunother. 2018;67:1481-1489.
- Cui C, Yu B, Jiang Q, et al. The roles of PD-1/PD-L1 and its signalling pathway in gastrointestinal tract cancers. Clin Exp Pharmacol Physiol. 2019;46:3-10.
- Iga N, Otsuka A, Yamamoto Y, et al. Accumulation of exhausted CD8+ T cells in extramammary Paget’s disease. PLoS One. 2019;14:E0211135.
- Frances L, Pascual JC, Leiva-Salinas M, et al. Extramammary Paget disease successfully treated with topical imiquimod 5% and tazarotene. Dermatol Ther. 2014;27:19-20.
- Lee A, Duggan S, Deeks ED. Cemiplimab: a review in advanced cutaneous squamous cell carcinoma. Drugs. 2020;80:813-819.
Resident Pearls
- Primary extramammary Paget disease (EMPD) is an adnexal carcinoma of the apocrine gland ducts, while secondary EMPD is an extension of malignant cells from an underlying internal neoplasm.
- Surgical margin clearance in EMPD often is problematic, with high recurrence rates indicating the need for additional treatment modalities.
- Programmed cell death receptor 1 (PD-1) signaling can serve as a potential target in EMPD. Further studies and clinical trials are needed to test the efficacy of PD-1 inhibitors in unresectable or invasive/metastatic EMPD.

Erythematous Plaque on the Back of a Newborn
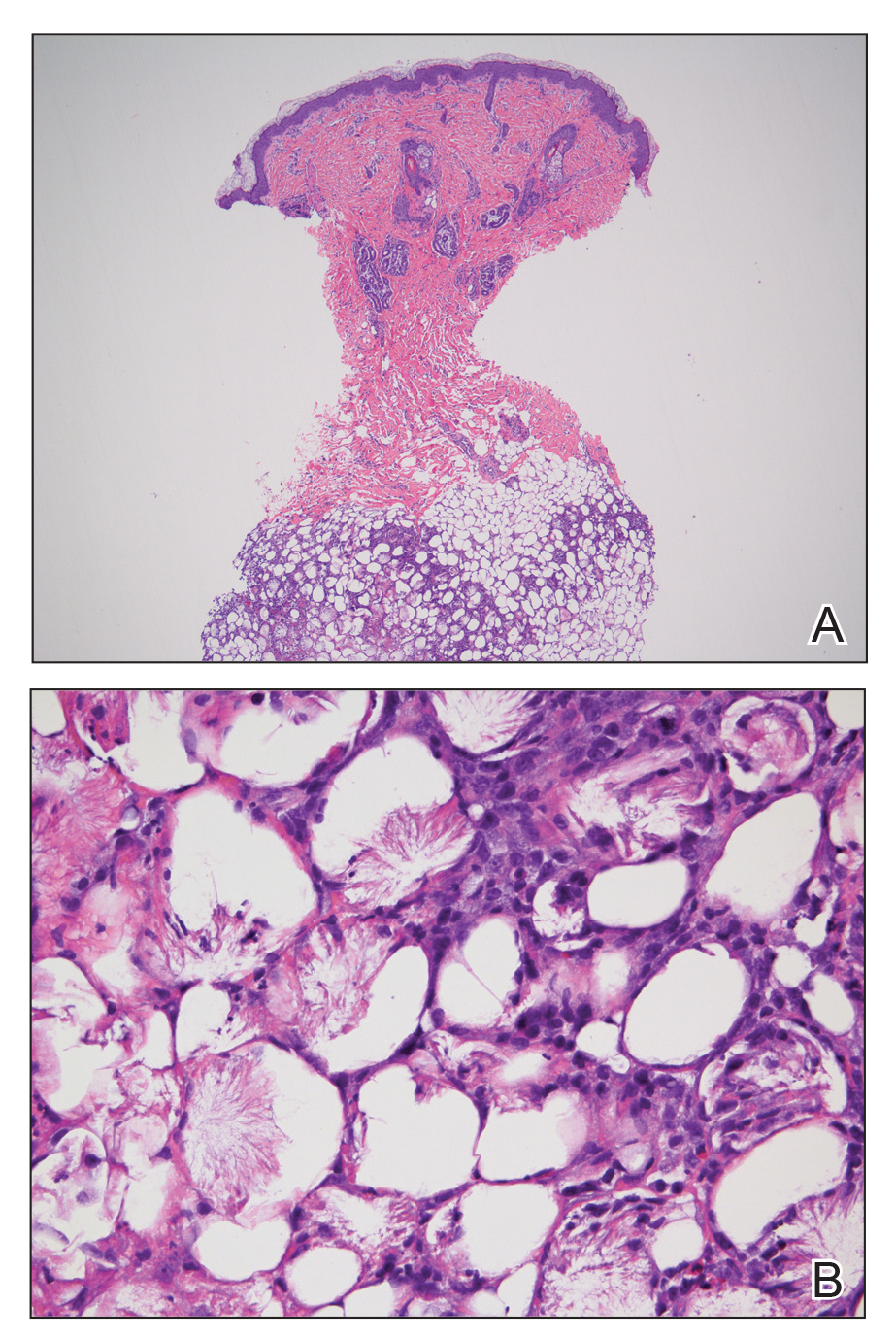
Subcutaneous fat necrosis of the newborn is a benign and self-limited condition that commonly occurs in term to postterm infants.1 However, it is an important diagnosis to recognize, as the potential exists for co-occurring metabolic derangements, most commonly hypercalcemia.1-4 Subcutaneous fat necrosis of the newborn is characterized by a panniculitis, most often on the back, shoulders, face, and buttocks. Lesions commonly present as erythematous nodules and plaques with overlying induration and can appear from birth to up to the first 6 weeks of life; calcification can be present in long-standing cases.2 Biopsy is diagnostic, showing a normal epidermis and dermis with a diffuse lobular panniculitis (Figure, A). Fat degeneration, radial crystal formation, and interstitial histiocytes also can be seen (Figure, B).
Patients with suspected subcutaneous fat necrosis should have their calcium levels checked, as up to 25% of patients may have coexisting hypercalcemia, which can contribute to morbidity and mortality.2 The hypercalcemia can occur with the onset of the lesions; however, it may be seen after they resolve completely.3 Thus, it is recommended that calcium levels be monitored for at least 1 month after lesions resolve. The exact etiology of subcutaneous fat necrosis is unknown, but it has been associated with perinatal stress and neonatal and maternal risk factors such as umbilical cord prolapse, meconium aspiration, neonatal sepsis, preeclampsia, and Rh incompatibility.1 The prognosis generally is excellent, with no treatment necessary for the skin lesions, as they resolve within a few months without subsequent sequelae or scarring.1,2 Patients with hypercalcemia should be treated appropriately with measures such as hydration and restriction of vitamin D; severe cases can be treated with bisphosphonates or loop diuretics.4
Cutis marmorata presents symmetrically on the trunk and may affect the upper and lower extremities as a reticulated erythema, often in response to cold temperature. Lesions are transient and resolve with warming. The isolated location of the skin lesions on the back, consistent course, and induration is unlikely to be seen in cutis marmorata. Infantile hemangiomas present several weeks to months after birth, and they undergo a rapid growth phase and subsequent slower involution phase. Furthermore, infantile hemangiomas have a rubbery feel and typically are not hard plaques, as seen in our patient.5 Patients with bacterial cellulitis often have systemic symptoms such as fever or chills, and the lesion generally is an ill-defined area of erythema and edema that can enlarge and become fluctuant.6 Sclerema neonatorum is a rare condition characterized by diffuse thickening of the skin that occurs in premature infants.7 These patients often are severely ill, as opposed to our asymptomatic full-term patient.
- Rubin G, Spagnut G, Morandi F, et al. Subcutaneous fat necrosis of the newborn. Clin Case Rep. 2015;3:1017-1020.
- de Campos Luciano Gomes MP, Porro AM, Simões da Silva Enokihara MM, et al. Subcutaneous fat necrosis of the newborn: clinical manifestations in two cases. An Bras Dermatol. 2013;88(6 suppl 1):154-157.
- Karochristou K, Siahanidou T, Kakourou-Tsivitanidou T, et al. Subcutaneous fat necrosis associated with severe hypocalcemia in a neonate. J Perinatol. 2005;26:64-66.
- Salas IV, Miralbell AR, Peinado CM, et al. Subcutaneous fat necrosis of the newborn and hypercalcemia: a case report. J Am Acad Dermatol. 2014;70:AB149.
- Darrow DH, Greene AK, Mancini AJ, et al. Diagnosis and management of infantile hemangioma. Pediatrics. 2015;136:E1060-E1104.
- Linder KA, Malani PN. Cellulitis. JAMA. 2017;317:2142.
- Jardine D, Atherton DJ, Trompeter RS. Sclerema neonaturm and subcutaneous fat necrosis of the newborn in the same infant. Eur J Pediatr. 1990;150:125-126.
Subcutaneous fat necrosis of the newborn is a benign and self-limited condition that commonly occurs in term to postterm infants.1 However, it is an important diagnosis to recognize, as the potential exists for co-occurring metabolic derangements, most commonly hypercalcemia.1-4 Subcutaneous fat necrosis of the newborn is characterized by a panniculitis, most often on the back, shoulders, face, and buttocks. Lesions commonly present as erythematous nodules and plaques with overlying induration and can appear from birth to up to the first 6 weeks of life; calcification can be present in long-standing cases.2 Biopsy is diagnostic, showing a normal epidermis and dermis with a diffuse lobular panniculitis (Figure, A). Fat degeneration, radial crystal formation, and interstitial histiocytes also can be seen (Figure, B).
Patients with suspected subcutaneous fat necrosis should have their calcium levels checked, as up to 25% of patients may have coexisting hypercalcemia, which can contribute to morbidity and mortality.2 The hypercalcemia can occur with the onset of the lesions; however, it may be seen after they resolve completely.3 Thus, it is recommended that calcium levels be monitored for at least 1 month after lesions resolve. The exact etiology of subcutaneous fat necrosis is unknown, but it has been associated with perinatal stress and neonatal and maternal risk factors such as umbilical cord prolapse, meconium aspiration, neonatal sepsis, preeclampsia, and Rh incompatibility.1 The prognosis generally is excellent, with no treatment necessary for the skin lesions, as they resolve within a few months without subsequent sequelae or scarring.1,2 Patients with hypercalcemia should be treated appropriately with measures such as hydration and restriction of vitamin D; severe cases can be treated with bisphosphonates or loop diuretics.4
Cutis marmorata presents symmetrically on the trunk and may affect the upper and lower extremities as a reticulated erythema, often in response to cold temperature. Lesions are transient and resolve with warming. The isolated location of the skin lesions on the back, consistent course, and induration is unlikely to be seen in cutis marmorata. Infantile hemangiomas present several weeks to months after birth, and they undergo a rapid growth phase and subsequent slower involution phase. Furthermore, infantile hemangiomas have a rubbery feel and typically are not hard plaques, as seen in our patient.5 Patients with bacterial cellulitis often have systemic symptoms such as fever or chills, and the lesion generally is an ill-defined area of erythema and edema that can enlarge and become fluctuant.6 Sclerema neonatorum is a rare condition characterized by diffuse thickening of the skin that occurs in premature infants.7 These patients often are severely ill, as opposed to our asymptomatic full-term patient.
Subcutaneous fat necrosis of the newborn is a benign and self-limited condition that commonly occurs in term to postterm infants.1 However, it is an important diagnosis to recognize, as the potential exists for co-occurring metabolic derangements, most commonly hypercalcemia.1-4 Subcutaneous fat necrosis of the newborn is characterized by a panniculitis, most often on the back, shoulders, face, and buttocks. Lesions commonly present as erythematous nodules and plaques with overlying induration and can appear from birth to up to the first 6 weeks of life; calcification can be present in long-standing cases.2 Biopsy is diagnostic, showing a normal epidermis and dermis with a diffuse lobular panniculitis (Figure, A). Fat degeneration, radial crystal formation, and interstitial histiocytes also can be seen (Figure, B).
Patients with suspected subcutaneous fat necrosis should have their calcium levels checked, as up to 25% of patients may have coexisting hypercalcemia, which can contribute to morbidity and mortality.2 The hypercalcemia can occur with the onset of the lesions; however, it may be seen after they resolve completely.3 Thus, it is recommended that calcium levels be monitored for at least 1 month after lesions resolve. The exact etiology of subcutaneous fat necrosis is unknown, but it has been associated with perinatal stress and neonatal and maternal risk factors such as umbilical cord prolapse, meconium aspiration, neonatal sepsis, preeclampsia, and Rh incompatibility.1 The prognosis generally is excellent, with no treatment necessary for the skin lesions, as they resolve within a few months without subsequent sequelae or scarring.1,2 Patients with hypercalcemia should be treated appropriately with measures such as hydration and restriction of vitamin D; severe cases can be treated with bisphosphonates or loop diuretics.4
Cutis marmorata presents symmetrically on the trunk and may affect the upper and lower extremities as a reticulated erythema, often in response to cold temperature. Lesions are transient and resolve with warming. The isolated location of the skin lesions on the back, consistent course, and induration is unlikely to be seen in cutis marmorata. Infantile hemangiomas present several weeks to months after birth, and they undergo a rapid growth phase and subsequent slower involution phase. Furthermore, infantile hemangiomas have a rubbery feel and typically are not hard plaques, as seen in our patient.5 Patients with bacterial cellulitis often have systemic symptoms such as fever or chills, and the lesion generally is an ill-defined area of erythema and edema that can enlarge and become fluctuant.6 Sclerema neonatorum is a rare condition characterized by diffuse thickening of the skin that occurs in premature infants.7 These patients often are severely ill, as opposed to our asymptomatic full-term patient.
- Rubin G, Spagnut G, Morandi F, et al. Subcutaneous fat necrosis of the newborn. Clin Case Rep. 2015;3:1017-1020.
- de Campos Luciano Gomes MP, Porro AM, Simões da Silva Enokihara MM, et al. Subcutaneous fat necrosis of the newborn: clinical manifestations in two cases. An Bras Dermatol. 2013;88(6 suppl 1):154-157.
- Karochristou K, Siahanidou T, Kakourou-Tsivitanidou T, et al. Subcutaneous fat necrosis associated with severe hypocalcemia in a neonate. J Perinatol. 2005;26:64-66.
- Salas IV, Miralbell AR, Peinado CM, et al. Subcutaneous fat necrosis of the newborn and hypercalcemia: a case report. J Am Acad Dermatol. 2014;70:AB149.
- Darrow DH, Greene AK, Mancini AJ, et al. Diagnosis and management of infantile hemangioma. Pediatrics. 2015;136:E1060-E1104.
- Linder KA, Malani PN. Cellulitis. JAMA. 2017;317:2142.
- Jardine D, Atherton DJ, Trompeter RS. Sclerema neonaturm and subcutaneous fat necrosis of the newborn in the same infant. Eur J Pediatr. 1990;150:125-126.
- Rubin G, Spagnut G, Morandi F, et al. Subcutaneous fat necrosis of the newborn. Clin Case Rep. 2015;3:1017-1020.
- de Campos Luciano Gomes MP, Porro AM, Simões da Silva Enokihara MM, et al. Subcutaneous fat necrosis of the newborn: clinical manifestations in two cases. An Bras Dermatol. 2013;88(6 suppl 1):154-157.
- Karochristou K, Siahanidou T, Kakourou-Tsivitanidou T, et al. Subcutaneous fat necrosis associated with severe hypocalcemia in a neonate. J Perinatol. 2005;26:64-66.
- Salas IV, Miralbell AR, Peinado CM, et al. Subcutaneous fat necrosis of the newborn and hypercalcemia: a case report. J Am Acad Dermatol. 2014;70:AB149.
- Darrow DH, Greene AK, Mancini AJ, et al. Diagnosis and management of infantile hemangioma. Pediatrics. 2015;136:E1060-E1104.
- Linder KA, Malani PN. Cellulitis. JAMA. 2017;317:2142.
- Jardine D, Atherton DJ, Trompeter RS. Sclerema neonaturm and subcutaneous fat necrosis of the newborn in the same infant. Eur J Pediatr. 1990;150:125-126.
An 8-day-old female infant presented with a mass on the lower back that had been present since birth. The patient was well appearing, alert, and active. Physical examination revealed a 6×5-cm, erythematous, ill-defined, indurated plaque on the lower thoracic back. There was no associated family history of similar findings. According to the mother, the patient was feeding well with no recent fever, irritability, or lethargy. The patient was born via elective induction of labor at term due to maternal intrauterine infection from chorioamnionitis. The birth was complicated by shoulder dystocia with an Erb palsy, and she was hospitalized for 5 days after delivery for management of hypotension and ABO isoimmunization and to rule out sepsis; blood cultures were negative for neonatal infection.
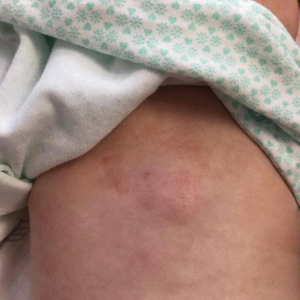
Melanoma in Hispanics: We May Have It All Wrong
To the Editor:
We read with interest the commentary by Srivastava et al,1 "The Dayanara Effect: Increasing Skin Cancer Awareness in the Hispanic Community," concerning former Miss Universe Dayanara Torres and her diagnosis of metastatic melanoma; however, we believe it misses the mark. A quick Google search shows that Ms. Torres has fair skin and blue eyes. She has lived most of her life in Puerto Rico, the Philippines, and California--places where sun exposure is high and may have contributed to her diagnosis. Factors that have been linked to an increased risk for melanoma are fair skin, red or blonde hair, blue or green eyes, intense intermittent sun exposure and sunburns, a weakened immune system, and a family history of skin cancer.2 Although we do not know her complete medical history, Ms. Torres' skin phenotype and likely chronic UV exposure made her a candidate for skin cancer. Although Srivastava et al1 acknowledged that the Hispanic community encompasses a wide variety of individuals with varying levels of skin pigmentation and sun sensitivity, they overlooked Ms. Torres' risk for skin cancer because of her ethnic background. This form of generalization may negatively affect patient care and safety. By 2060, Hispanics are projected to account for almost 30% of the US population,3 and we must acknowledge the flaws that exist in our overall methodology for assessing skin cancer risk among this population to provide patients with unbiased care.
In the early 1970s, the United States adopted the ethnonym Hispanic as a way of conglomerating Spanish-speaking individuals from Spain, the Caribbean, and Central and South America.4 The goal was to implement a common identifier that enabled the US Government to study the economic and social development of these groups. Nevertheless, considerable differences exist among distinct Hispanic communities, and variations in skin pigmentation and sun sensitivity are no exception. Although Hispanic countries are an amalgam of diverse races due to colonization, some have stronger European, African, or Amerindian influences, limiting the use of ethnicity during melanoma risk assessment. Another misconception reflected in the commentary by Srivastava et al1 is the belief that the terms white and Hispanic are mutually exclusive. A study examining melanoma rates in the Chilean population supports this claim.5 The genetic composition of the Chilean high socioeconomic strata is 5% Amerindian and 95% white, while the low socioeconomic strata is approximately 40% Amerindian and 60% white. Patients from the low socioeconomic strata had higher rates of acral malignant melanoma, which typically is seen in patients with skin of color. On the other hand, males from the high socioeconomic strata had higher rates of truncal melanoma, which is more common among the white population.5 These results suggest that while both groups are considered Hispanic, it is ancestral origin that contributes to the differential rates and types of malignant melanoma.
When analyzing data regarding melanoma rates in Hispanics, particularly data collected in the United States, we must question if the results are representative of the entire population. One point worth emphasizing is that melanoma data in the Hispanic community often is flawed. The North American Association of Central Cancer Registries considers Europeans such as Spaniards, as well as citizens of Andorra, the Canary Islands, and the Balearic Islands as Hispanic.6 Additionally, the Florida Cancer Data System uses data such as country of birth, ethnicity, and surname or maiden name recorded by the hospital tumor registry to identify Hispanic patients with melanoma.7 In 2006, Hu et al7 used the Florida Cancer Data System to analyze melanoma data in Miami-Dade County in South Florida, which has the second largest Hispanic community in the United States. One limitation to such data is that ethnicity often is self-reported by patients or assigned by a health care provider. In addition, women whose maiden names are not available may be misclassified through marriage depending on whether their husbands have Spanish or non-Spanish last names.7 Finally, with societal norms evolving, Americans are now more accepting of interracial marriages. In 2017, the Pew Research Center reported that 17% of all newlyweds in the United States were intermarried and 42% of these marriages were between a white individual and a Hispanic individual, comprising the most prevalent form of intermarriage reported.8 In 2015, 27% of newlywed Hispanics were intermarried. This percentage varied depending on whether they were born in the United States or abroad. Although 15% of Hispanic immigrants married a spouse from another race, 39% of Hispanics born in the United States married a non-Hispanic (eg, white, black, Asian, or Native American who is not Hispanic).8 This type of marriage and subsequent offspring might lead to an increase in the white genetic pool. As a result, the risk for melanoma development may be increased or misrepresented. Remaining aware of these changes in the population is crucial, as it exemplifies why the current methodology for gathering and reporting melanoma data is unreliable.
Labeling Ms. Torres as Hispanic due to her Puerto Rican nationality did not tell us anything about her risk for developing melanoma. To correctly assess the risk for melanoma among Hispanics, it is imperative that we re-evaluate our approach. We agree with He et al9 that our efforts should be dedicated to better understanding the impact of pigmentation, race, genetics, and sunburn on the risk for melanoma. Until we know more about this possible correlation, we should reconsider how we study melanoma using Hispanics as an ethnicity. We may have it all wrong.
- Srivastava R, Wassef C, Rao BK. The Dayanara effect: increasing skin cancer awareness in the Hispanic community. Cutis. 2019;103:257-258.
- Curiel-Lewandrowski C. Risk factors for the development of melanoma. UpToDate. https://www.uptodate.com/contents/risk-factors-for-the-development-of-melanoma. Updated February 27, 2020. Accessed April 16, 2020.
- Colby SL, Ortman JM. Projections of the size and composition of the U.S. population: 2014 to 2060. United States Census Bureau website. https://www.census.gov/library/publications/2015/demo/p25-1143.html. Published March 3, 2015. Accessed April 16, 2020.
- National Research Council (US) Panel on Hispanics in the United States; Tienda M, Mitchell F, editors. Multiple Origins, Uncertain Destinies: Hispanics and the American Future. Washington (DC): National Academies Press (US); 2006. 3, Defining Hispanicity: E Pluribus Unum or E Pluribus Plures? Available from: https://www.ncbi.nlm.nih.gov/books/NBK19811/
- Zemelman VB, Valenzuela CY, Sazunic I, et al. Malignant melanoma in Chile: different site distribution between private and state patients. Biol Res. 2014;47:34.
- NAACCR Race and Ethnicity Work Group. NAACCR guideline for enhancing Hispanic-Latino identification: revised NAACCR Hispanic/Latino identification algorithm [NHIA v2.2.1]. NAACCR website. https://www.naaccr.org/wp-content/uploads/2016/11/NHIA_v2_2_1_09122011.pdf. Revised September 12, 2011. Accessed April 15, 2020.
- Hu S, Soza-Vento RM, Parker DF, et al. Comparison of stage at diagnosis of melanoma among Hispanic, black, and white patients in Miami-Dade County, Florida. Arch Dermatol. 2006;142:704-708.
- Livingston G, Brown A. Intermarriage in the U.S. 50 years after Loving v. Virginia. Pew Research Center website. https://www.pewsocialtrends.org/2017/05/18/intermarriage-in-the-u-s-50-years-after-loving-v-virginia/. Published May 18, 2017. Accessed April 15, 2020.
- He SY, McCulloch CE, Boscardin WJ, et al. Self-reported pigmentary phenotypes and race are significant but incomplete predictors of Fitzpatrick skin phototype in an ethnically diverse population. J Am Acad Dermatol. 2014;71:731-737.
Authors' Response
While Ms. Cruzval-O'Reilly and Dr. Lugo-Somolinos highlight many important points on conducting meaningful research for the Hispanic community, they seem to have misunderstood the overall purpose of our commentary,1 which was to highlight the increased skin cancer awareness that a notable and vocal member of the Hispanic community brought to our academic dermatology clinic, rather than to discuss skin types within the Hispanic community. As the authors mentioned, the term Hispanic is a descriptor of ethnicity rather than race, and Hispanic patients may have varying levels of skin pigmentation and sun sensitivity. While Dayanara Torres may have risk factors for developing melanoma, minimizing her connection with the Hispanic community because of her fair skin and light eyes would be a mistake. It not only isolates members of the Hispanic community that are of skin types I and II, but it also discounts the power of her story and language in raising awareness. We observed an increase in Hispanic patients presenting to our clinic who were concerned about skin cancer after Ms. Torres shared her diagnosis of metastatic melanoma through social media, followed by Spanish language educational videos on melanoma.
Several studies have described disparities among Hispanic patients diagnosed with melanoma as compared to their non-Hispanic white counterparts, including younger age at diagnosis, later stage of presentation, increased presence of regional involvement, and worse mortality.2-6 Furthermore, a small study of high school students by Ma et al7 showed disparities in skin cancer knowledge, perceived risk, and sun-protective behaviors among Hispanic whites and non-Hispanic whites, which remained significant (P<.05) after controlling for skin pigmentation and sun sensitivity. We agree with the authors that further analysis of skin type, race, genetics, and other risk factors may help refine the research on skin cancer disparities within the Hispanic community. We suspect that disparities may persist even when examining these factors. There have been several studies showing that knowledge-based interventions, especially when delivered in Spanish, improve understanding of skin cancer, personal risk, and self-examinations, and we support Ms. Torres' efforts in utilizing her platform to provide information about melanoma in Spanish.8-12
Radhika Srivastava, MD; Cindy Wassef, MD; Babar K. Rao, MD
From the Department of Dermatology, Rutgers Robert Wood Johnson Medical School, Somerset, New Jersey.
The authors report no conflict of interest.
Correspondence: Radhika Srivastava, MD, 1 World's Fair Dr, Ste 2400, Somerset, NJ 08873 (rsrivastavamd@gmail.com).
References
- Srivastava R, Wassef C, Rao BK. The Dayanara effect: increasing skin cancer awareness in the Hispanic community. Cutis. 2019;103:257-258.
- Perez MI. Skin cancer in Hispanics in the United States. J Drugs Dermatol. 2019;18:s117-s120.
- Higgins S, Nazemi A, Feinstein S, et al. Clinical presentations of melanoma in African Americans, Hispanics, and Asians. Dermatol Surg. 2019;45:791-801.
- Harvey VM, Oldfield CW, Chen JT, et al. Melanoma disparities among US Hispanics: use of the social ecological model to contextualize reasons for inequitable outcomes and frame a research agenda [published online August 29, 2016]. J Skin Cancer. doi:10.1155/2016/4635740
- Garnett E, Townsend J, Steele B, et al. Characteristics, rates, and trends of melanoma incidence among Hispanics in the USA. Cancer Causes Control. 2016;27:647-659.
- Rouhani P, Hu S, Kirsner RS. Melanoma in Hispanic and black Americans. Cancer Control. 2008;15:248-253.
- Ma F, Collado-Mesa F, Hu S, et al. Skin cancer awareness and sun protection behaviors in white Hispanic and white non-Hispanic high school students in Miami, Florida. Arch Dermatol. 2007;143:983-988.
- Kundu RV, Kamaria M, Ortiz S, et al. Effectiveness of a knowledge-based intervention for melanoma among those with ethnic skin. J Am Acad Dermatol. 2010;62:777-784.
- Kailas A, Botwin AL, Pritchett EN, et al. Assessing the effectiveness of knowledge-based interventions in increasing skin cancer awareness, knowledge, and protective behaviors in skin of color populations. Cutis. 2017;100:235-240.
- Roman CJ, Guan X, Barnholtz-Sloan J, et al. A trial online educational melanoma program aimed at the Hispanic population improves knowledge and behaviors. Dermatol Surg. 2016;42:672-676.
- Hernandez C, Kim H, Mauleon G, et al. A pilot program in collaboration with community centers to increase awareness and participation in skin cancer screening among Latinos in Chicago. J Cancer Educ. 2013;28:342-345.
- Chung GY, Brown G, Gibson D. Increasing melanoma screening among Hispanic/Latino Americans: a community-based educational intervention. Health Educ Behav. 2015;42:627-632.
To the Editor:
We read with interest the commentary by Srivastava et al,1 "The Dayanara Effect: Increasing Skin Cancer Awareness in the Hispanic Community," concerning former Miss Universe Dayanara Torres and her diagnosis of metastatic melanoma; however, we believe it misses the mark. A quick Google search shows that Ms. Torres has fair skin and blue eyes. She has lived most of her life in Puerto Rico, the Philippines, and California--places where sun exposure is high and may have contributed to her diagnosis. Factors that have been linked to an increased risk for melanoma are fair skin, red or blonde hair, blue or green eyes, intense intermittent sun exposure and sunburns, a weakened immune system, and a family history of skin cancer.2 Although we do not know her complete medical history, Ms. Torres' skin phenotype and likely chronic UV exposure made her a candidate for skin cancer. Although Srivastava et al1 acknowledged that the Hispanic community encompasses a wide variety of individuals with varying levels of skin pigmentation and sun sensitivity, they overlooked Ms. Torres' risk for skin cancer because of her ethnic background. This form of generalization may negatively affect patient care and safety. By 2060, Hispanics are projected to account for almost 30% of the US population,3 and we must acknowledge the flaws that exist in our overall methodology for assessing skin cancer risk among this population to provide patients with unbiased care.
In the early 1970s, the United States adopted the ethnonym Hispanic as a way of conglomerating Spanish-speaking individuals from Spain, the Caribbean, and Central and South America.4 The goal was to implement a common identifier that enabled the US Government to study the economic and social development of these groups. Nevertheless, considerable differences exist among distinct Hispanic communities, and variations in skin pigmentation and sun sensitivity are no exception. Although Hispanic countries are an amalgam of diverse races due to colonization, some have stronger European, African, or Amerindian influences, limiting the use of ethnicity during melanoma risk assessment. Another misconception reflected in the commentary by Srivastava et al1 is the belief that the terms white and Hispanic are mutually exclusive. A study examining melanoma rates in the Chilean population supports this claim.5 The genetic composition of the Chilean high socioeconomic strata is 5% Amerindian and 95% white, while the low socioeconomic strata is approximately 40% Amerindian and 60% white. Patients from the low socioeconomic strata had higher rates of acral malignant melanoma, which typically is seen in patients with skin of color. On the other hand, males from the high socioeconomic strata had higher rates of truncal melanoma, which is more common among the white population.5 These results suggest that while both groups are considered Hispanic, it is ancestral origin that contributes to the differential rates and types of malignant melanoma.
When analyzing data regarding melanoma rates in Hispanics, particularly data collected in the United States, we must question if the results are representative of the entire population. One point worth emphasizing is that melanoma data in the Hispanic community often is flawed. The North American Association of Central Cancer Registries considers Europeans such as Spaniards, as well as citizens of Andorra, the Canary Islands, and the Balearic Islands as Hispanic.6 Additionally, the Florida Cancer Data System uses data such as country of birth, ethnicity, and surname or maiden name recorded by the hospital tumor registry to identify Hispanic patients with melanoma.7 In 2006, Hu et al7 used the Florida Cancer Data System to analyze melanoma data in Miami-Dade County in South Florida, which has the second largest Hispanic community in the United States. One limitation to such data is that ethnicity often is self-reported by patients or assigned by a health care provider. In addition, women whose maiden names are not available may be misclassified through marriage depending on whether their husbands have Spanish or non-Spanish last names.7 Finally, with societal norms evolving, Americans are now more accepting of interracial marriages. In 2017, the Pew Research Center reported that 17% of all newlyweds in the United States were intermarried and 42% of these marriages were between a white individual and a Hispanic individual, comprising the most prevalent form of intermarriage reported.8 In 2015, 27% of newlywed Hispanics were intermarried. This percentage varied depending on whether they were born in the United States or abroad. Although 15% of Hispanic immigrants married a spouse from another race, 39% of Hispanics born in the United States married a non-Hispanic (eg, white, black, Asian, or Native American who is not Hispanic).8 This type of marriage and subsequent offspring might lead to an increase in the white genetic pool. As a result, the risk for melanoma development may be increased or misrepresented. Remaining aware of these changes in the population is crucial, as it exemplifies why the current methodology for gathering and reporting melanoma data is unreliable.
Labeling Ms. Torres as Hispanic due to her Puerto Rican nationality did not tell us anything about her risk for developing melanoma. To correctly assess the risk for melanoma among Hispanics, it is imperative that we re-evaluate our approach. We agree with He et al9 that our efforts should be dedicated to better understanding the impact of pigmentation, race, genetics, and sunburn on the risk for melanoma. Until we know more about this possible correlation, we should reconsider how we study melanoma using Hispanics as an ethnicity. We may have it all wrong.
- Srivastava R, Wassef C, Rao BK. The Dayanara effect: increasing skin cancer awareness in the Hispanic community. Cutis. 2019;103:257-258.
- Curiel-Lewandrowski C. Risk factors for the development of melanoma. UpToDate. https://www.uptodate.com/contents/risk-factors-for-the-development-of-melanoma. Updated February 27, 2020. Accessed April 16, 2020.
- Colby SL, Ortman JM. Projections of the size and composition of the U.S. population: 2014 to 2060. United States Census Bureau website. https://www.census.gov/library/publications/2015/demo/p25-1143.html. Published March 3, 2015. Accessed April 16, 2020.
- National Research Council (US) Panel on Hispanics in the United States; Tienda M, Mitchell F, editors. Multiple Origins, Uncertain Destinies: Hispanics and the American Future. Washington (DC): National Academies Press (US); 2006. 3, Defining Hispanicity: E Pluribus Unum or E Pluribus Plures? Available from: https://www.ncbi.nlm.nih.gov/books/NBK19811/
- Zemelman VB, Valenzuela CY, Sazunic I, et al. Malignant melanoma in Chile: different site distribution between private and state patients. Biol Res. 2014;47:34.
- NAACCR Race and Ethnicity Work Group. NAACCR guideline for enhancing Hispanic-Latino identification: revised NAACCR Hispanic/Latino identification algorithm [NHIA v2.2.1]. NAACCR website. https://www.naaccr.org/wp-content/uploads/2016/11/NHIA_v2_2_1_09122011.pdf. Revised September 12, 2011. Accessed April 15, 2020.
- Hu S, Soza-Vento RM, Parker DF, et al. Comparison of stage at diagnosis of melanoma among Hispanic, black, and white patients in Miami-Dade County, Florida. Arch Dermatol. 2006;142:704-708.
- Livingston G, Brown A. Intermarriage in the U.S. 50 years after Loving v. Virginia. Pew Research Center website. https://www.pewsocialtrends.org/2017/05/18/intermarriage-in-the-u-s-50-years-after-loving-v-virginia/. Published May 18, 2017. Accessed April 15, 2020.
- He SY, McCulloch CE, Boscardin WJ, et al. Self-reported pigmentary phenotypes and race are significant but incomplete predictors of Fitzpatrick skin phototype in an ethnically diverse population. J Am Acad Dermatol. 2014;71:731-737.
Authors' Response
While Ms. Cruzval-O'Reilly and Dr. Lugo-Somolinos highlight many important points on conducting meaningful research for the Hispanic community, they seem to have misunderstood the overall purpose of our commentary,1 which was to highlight the increased skin cancer awareness that a notable and vocal member of the Hispanic community brought to our academic dermatology clinic, rather than to discuss skin types within the Hispanic community. As the authors mentioned, the term Hispanic is a descriptor of ethnicity rather than race, and Hispanic patients may have varying levels of skin pigmentation and sun sensitivity. While Dayanara Torres may have risk factors for developing melanoma, minimizing her connection with the Hispanic community because of her fair skin and light eyes would be a mistake. It not only isolates members of the Hispanic community that are of skin types I and II, but it also discounts the power of her story and language in raising awareness. We observed an increase in Hispanic patients presenting to our clinic who were concerned about skin cancer after Ms. Torres shared her diagnosis of metastatic melanoma through social media, followed by Spanish language educational videos on melanoma.
Several studies have described disparities among Hispanic patients diagnosed with melanoma as compared to their non-Hispanic white counterparts, including younger age at diagnosis, later stage of presentation, increased presence of regional involvement, and worse mortality.2-6 Furthermore, a small study of high school students by Ma et al7 showed disparities in skin cancer knowledge, perceived risk, and sun-protective behaviors among Hispanic whites and non-Hispanic whites, which remained significant (P<.05) after controlling for skin pigmentation and sun sensitivity. We agree with the authors that further analysis of skin type, race, genetics, and other risk factors may help refine the research on skin cancer disparities within the Hispanic community. We suspect that disparities may persist even when examining these factors. There have been several studies showing that knowledge-based interventions, especially when delivered in Spanish, improve understanding of skin cancer, personal risk, and self-examinations, and we support Ms. Torres' efforts in utilizing her platform to provide information about melanoma in Spanish.8-12
Radhika Srivastava, MD; Cindy Wassef, MD; Babar K. Rao, MD
From the Department of Dermatology, Rutgers Robert Wood Johnson Medical School, Somerset, New Jersey.
The authors report no conflict of interest.
Correspondence: Radhika Srivastava, MD, 1 World's Fair Dr, Ste 2400, Somerset, NJ 08873 (rsrivastavamd@gmail.com).
References
- Srivastava R, Wassef C, Rao BK. The Dayanara effect: increasing skin cancer awareness in the Hispanic community. Cutis. 2019;103:257-258.
- Perez MI. Skin cancer in Hispanics in the United States. J Drugs Dermatol. 2019;18:s117-s120.
- Higgins S, Nazemi A, Feinstein S, et al. Clinical presentations of melanoma in African Americans, Hispanics, and Asians. Dermatol Surg. 2019;45:791-801.
- Harvey VM, Oldfield CW, Chen JT, et al. Melanoma disparities among US Hispanics: use of the social ecological model to contextualize reasons for inequitable outcomes and frame a research agenda [published online August 29, 2016]. J Skin Cancer. doi:10.1155/2016/4635740
- Garnett E, Townsend J, Steele B, et al. Characteristics, rates, and trends of melanoma incidence among Hispanics in the USA. Cancer Causes Control. 2016;27:647-659.
- Rouhani P, Hu S, Kirsner RS. Melanoma in Hispanic and black Americans. Cancer Control. 2008;15:248-253.
- Ma F, Collado-Mesa F, Hu S, et al. Skin cancer awareness and sun protection behaviors in white Hispanic and white non-Hispanic high school students in Miami, Florida. Arch Dermatol. 2007;143:983-988.
- Kundu RV, Kamaria M, Ortiz S, et al. Effectiveness of a knowledge-based intervention for melanoma among those with ethnic skin. J Am Acad Dermatol. 2010;62:777-784.
- Kailas A, Botwin AL, Pritchett EN, et al. Assessing the effectiveness of knowledge-based interventions in increasing skin cancer awareness, knowledge, and protective behaviors in skin of color populations. Cutis. 2017;100:235-240.
- Roman CJ, Guan X, Barnholtz-Sloan J, et al. A trial online educational melanoma program aimed at the Hispanic population improves knowledge and behaviors. Dermatol Surg. 2016;42:672-676.
- Hernandez C, Kim H, Mauleon G, et al. A pilot program in collaboration with community centers to increase awareness and participation in skin cancer screening among Latinos in Chicago. J Cancer Educ. 2013;28:342-345.
- Chung GY, Brown G, Gibson D. Increasing melanoma screening among Hispanic/Latino Americans: a community-based educational intervention. Health Educ Behav. 2015;42:627-632.
To the Editor:
We read with interest the commentary by Srivastava et al,1 "The Dayanara Effect: Increasing Skin Cancer Awareness in the Hispanic Community," concerning former Miss Universe Dayanara Torres and her diagnosis of metastatic melanoma; however, we believe it misses the mark. A quick Google search shows that Ms. Torres has fair skin and blue eyes. She has lived most of her life in Puerto Rico, the Philippines, and California--places where sun exposure is high and may have contributed to her diagnosis. Factors that have been linked to an increased risk for melanoma are fair skin, red or blonde hair, blue or green eyes, intense intermittent sun exposure and sunburns, a weakened immune system, and a family history of skin cancer.2 Although we do not know her complete medical history, Ms. Torres' skin phenotype and likely chronic UV exposure made her a candidate for skin cancer. Although Srivastava et al1 acknowledged that the Hispanic community encompasses a wide variety of individuals with varying levels of skin pigmentation and sun sensitivity, they overlooked Ms. Torres' risk for skin cancer because of her ethnic background. This form of generalization may negatively affect patient care and safety. By 2060, Hispanics are projected to account for almost 30% of the US population,3 and we must acknowledge the flaws that exist in our overall methodology for assessing skin cancer risk among this population to provide patients with unbiased care.
In the early 1970s, the United States adopted the ethnonym Hispanic as a way of conglomerating Spanish-speaking individuals from Spain, the Caribbean, and Central and South America.4 The goal was to implement a common identifier that enabled the US Government to study the economic and social development of these groups. Nevertheless, considerable differences exist among distinct Hispanic communities, and variations in skin pigmentation and sun sensitivity are no exception. Although Hispanic countries are an amalgam of diverse races due to colonization, some have stronger European, African, or Amerindian influences, limiting the use of ethnicity during melanoma risk assessment. Another misconception reflected in the commentary by Srivastava et al1 is the belief that the terms white and Hispanic are mutually exclusive. A study examining melanoma rates in the Chilean population supports this claim.5 The genetic composition of the Chilean high socioeconomic strata is 5% Amerindian and 95% white, while the low socioeconomic strata is approximately 40% Amerindian and 60% white. Patients from the low socioeconomic strata had higher rates of acral malignant melanoma, which typically is seen in patients with skin of color. On the other hand, males from the high socioeconomic strata had higher rates of truncal melanoma, which is more common among the white population.5 These results suggest that while both groups are considered Hispanic, it is ancestral origin that contributes to the differential rates and types of malignant melanoma.
When analyzing data regarding melanoma rates in Hispanics, particularly data collected in the United States, we must question if the results are representative of the entire population. One point worth emphasizing is that melanoma data in the Hispanic community often is flawed. The North American Association of Central Cancer Registries considers Europeans such as Spaniards, as well as citizens of Andorra, the Canary Islands, and the Balearic Islands as Hispanic.6 Additionally, the Florida Cancer Data System uses data such as country of birth, ethnicity, and surname or maiden name recorded by the hospital tumor registry to identify Hispanic patients with melanoma.7 In 2006, Hu et al7 used the Florida Cancer Data System to analyze melanoma data in Miami-Dade County in South Florida, which has the second largest Hispanic community in the United States. One limitation to such data is that ethnicity often is self-reported by patients or assigned by a health care provider. In addition, women whose maiden names are not available may be misclassified through marriage depending on whether their husbands have Spanish or non-Spanish last names.7 Finally, with societal norms evolving, Americans are now more accepting of interracial marriages. In 2017, the Pew Research Center reported that 17% of all newlyweds in the United States were intermarried and 42% of these marriages were between a white individual and a Hispanic individual, comprising the most prevalent form of intermarriage reported.8 In 2015, 27% of newlywed Hispanics were intermarried. This percentage varied depending on whether they were born in the United States or abroad. Although 15% of Hispanic immigrants married a spouse from another race, 39% of Hispanics born in the United States married a non-Hispanic (eg, white, black, Asian, or Native American who is not Hispanic).8 This type of marriage and subsequent offspring might lead to an increase in the white genetic pool. As a result, the risk for melanoma development may be increased or misrepresented. Remaining aware of these changes in the population is crucial, as it exemplifies why the current methodology for gathering and reporting melanoma data is unreliable.
Labeling Ms. Torres as Hispanic due to her Puerto Rican nationality did not tell us anything about her risk for developing melanoma. To correctly assess the risk for melanoma among Hispanics, it is imperative that we re-evaluate our approach. We agree with He et al9 that our efforts should be dedicated to better understanding the impact of pigmentation, race, genetics, and sunburn on the risk for melanoma. Until we know more about this possible correlation, we should reconsider how we study melanoma using Hispanics as an ethnicity. We may have it all wrong.
- Srivastava R, Wassef C, Rao BK. The Dayanara effect: increasing skin cancer awareness in the Hispanic community. Cutis. 2019;103:257-258.
- Curiel-Lewandrowski C. Risk factors for the development of melanoma. UpToDate. https://www.uptodate.com/contents/risk-factors-for-the-development-of-melanoma. Updated February 27, 2020. Accessed April 16, 2020.
- Colby SL, Ortman JM. Projections of the size and composition of the U.S. population: 2014 to 2060. United States Census Bureau website. https://www.census.gov/library/publications/2015/demo/p25-1143.html. Published March 3, 2015. Accessed April 16, 2020.
- National Research Council (US) Panel on Hispanics in the United States; Tienda M, Mitchell F, editors. Multiple Origins, Uncertain Destinies: Hispanics and the American Future. Washington (DC): National Academies Press (US); 2006. 3, Defining Hispanicity: E Pluribus Unum or E Pluribus Plures? Available from: https://www.ncbi.nlm.nih.gov/books/NBK19811/
- Zemelman VB, Valenzuela CY, Sazunic I, et al. Malignant melanoma in Chile: different site distribution between private and state patients. Biol Res. 2014;47:34.
- NAACCR Race and Ethnicity Work Group. NAACCR guideline for enhancing Hispanic-Latino identification: revised NAACCR Hispanic/Latino identification algorithm [NHIA v2.2.1]. NAACCR website. https://www.naaccr.org/wp-content/uploads/2016/11/NHIA_v2_2_1_09122011.pdf. Revised September 12, 2011. Accessed April 15, 2020.
- Hu S, Soza-Vento RM, Parker DF, et al. Comparison of stage at diagnosis of melanoma among Hispanic, black, and white patients in Miami-Dade County, Florida. Arch Dermatol. 2006;142:704-708.
- Livingston G, Brown A. Intermarriage in the U.S. 50 years after Loving v. Virginia. Pew Research Center website. https://www.pewsocialtrends.org/2017/05/18/intermarriage-in-the-u-s-50-years-after-loving-v-virginia/. Published May 18, 2017. Accessed April 15, 2020.
- He SY, McCulloch CE, Boscardin WJ, et al. Self-reported pigmentary phenotypes and race are significant but incomplete predictors of Fitzpatrick skin phototype in an ethnically diverse population. J Am Acad Dermatol. 2014;71:731-737.
Authors' Response
While Ms. Cruzval-O'Reilly and Dr. Lugo-Somolinos highlight many important points on conducting meaningful research for the Hispanic community, they seem to have misunderstood the overall purpose of our commentary,1 which was to highlight the increased skin cancer awareness that a notable and vocal member of the Hispanic community brought to our academic dermatology clinic, rather than to discuss skin types within the Hispanic community. As the authors mentioned, the term Hispanic is a descriptor of ethnicity rather than race, and Hispanic patients may have varying levels of skin pigmentation and sun sensitivity. While Dayanara Torres may have risk factors for developing melanoma, minimizing her connection with the Hispanic community because of her fair skin and light eyes would be a mistake. It not only isolates members of the Hispanic community that are of skin types I and II, but it also discounts the power of her story and language in raising awareness. We observed an increase in Hispanic patients presenting to our clinic who were concerned about skin cancer after Ms. Torres shared her diagnosis of metastatic melanoma through social media, followed by Spanish language educational videos on melanoma.
Several studies have described disparities among Hispanic patients diagnosed with melanoma as compared to their non-Hispanic white counterparts, including younger age at diagnosis, later stage of presentation, increased presence of regional involvement, and worse mortality.2-6 Furthermore, a small study of high school students by Ma et al7 showed disparities in skin cancer knowledge, perceived risk, and sun-protective behaviors among Hispanic whites and non-Hispanic whites, which remained significant (P<.05) after controlling for skin pigmentation and sun sensitivity. We agree with the authors that further analysis of skin type, race, genetics, and other risk factors may help refine the research on skin cancer disparities within the Hispanic community. We suspect that disparities may persist even when examining these factors. There have been several studies showing that knowledge-based interventions, especially when delivered in Spanish, improve understanding of skin cancer, personal risk, and self-examinations, and we support Ms. Torres' efforts in utilizing her platform to provide information about melanoma in Spanish.8-12
Radhika Srivastava, MD; Cindy Wassef, MD; Babar K. Rao, MD
From the Department of Dermatology, Rutgers Robert Wood Johnson Medical School, Somerset, New Jersey.
The authors report no conflict of interest.
Correspondence: Radhika Srivastava, MD, 1 World's Fair Dr, Ste 2400, Somerset, NJ 08873 (rsrivastavamd@gmail.com).
References
- Srivastava R, Wassef C, Rao BK. The Dayanara effect: increasing skin cancer awareness in the Hispanic community. Cutis. 2019;103:257-258.
- Perez MI. Skin cancer in Hispanics in the United States. J Drugs Dermatol. 2019;18:s117-s120.
- Higgins S, Nazemi A, Feinstein S, et al. Clinical presentations of melanoma in African Americans, Hispanics, and Asians. Dermatol Surg. 2019;45:791-801.
- Harvey VM, Oldfield CW, Chen JT, et al. Melanoma disparities among US Hispanics: use of the social ecological model to contextualize reasons for inequitable outcomes and frame a research agenda [published online August 29, 2016]. J Skin Cancer. doi:10.1155/2016/4635740
- Garnett E, Townsend J, Steele B, et al. Characteristics, rates, and trends of melanoma incidence among Hispanics in the USA. Cancer Causes Control. 2016;27:647-659.
- Rouhani P, Hu S, Kirsner RS. Melanoma in Hispanic and black Americans. Cancer Control. 2008;15:248-253.
- Ma F, Collado-Mesa F, Hu S, et al. Skin cancer awareness and sun protection behaviors in white Hispanic and white non-Hispanic high school students in Miami, Florida. Arch Dermatol. 2007;143:983-988.
- Kundu RV, Kamaria M, Ortiz S, et al. Effectiveness of a knowledge-based intervention for melanoma among those with ethnic skin. J Am Acad Dermatol. 2010;62:777-784.
- Kailas A, Botwin AL, Pritchett EN, et al. Assessing the effectiveness of knowledge-based interventions in increasing skin cancer awareness, knowledge, and protective behaviors in skin of color populations. Cutis. 2017;100:235-240.
- Roman CJ, Guan X, Barnholtz-Sloan J, et al. A trial online educational melanoma program aimed at the Hispanic population improves knowledge and behaviors. Dermatol Surg. 2016;42:672-676.
- Hernandez C, Kim H, Mauleon G, et al. A pilot program in collaboration with community centers to increase awareness and participation in skin cancer screening among Latinos in Chicago. J Cancer Educ. 2013;28:342-345.
- Chung GY, Brown G, Gibson D. Increasing melanoma screening among Hispanic/Latino Americans: a community-based educational intervention. Health Educ Behav. 2015;42:627-632.
Cutaneous Metastases Masquerading as Solitary or Multiple Keratoacanthomas
To the Editor:
We read with interest the excellent Cutis articles on cutaneous metastases by Tarantino et al1 and Agnetta et al.2 Tarantino et al1 reported a 59-year-old man who developed cutaneous metastases on the scalp from an esophageal adenocarcinoma. Agnetta et al2 described a 76-year-old woman with metastatic melanoma mimicking eruptive keratoacanthomas.
Cutaneous metastases are not common. They may herald the unsuspected diagnosis of a solid tumor recurrence or progression of systemic disease in an oncology patient. Occasionally, they are the primary manifestation of a visceral tumor in a previously cancer-free patient. Less often, skin lesions are the manifestation of a new or recurrent hematologic malignancy.3,4
The morphology of cutaneous metastases is variable. Most commonly they appear as papules and nodules. However, they can mimic bacterial (eg, erysipelas) and viral (eg, herpes zoster) infections or present as scalp alopecia.5-7
Cutaneous metastases also can mimic benign (eg, epidermoid cysts) or malignant (eg, keratoacanthoma) neoplasms. Keratoacanthomalike cutaneous metastases are rare.8 They can present as single or multiple tumors.9,10
In the case reported by Tarantino et al,1 the patient had a history of metastatic adenocarcinoma of the esophagus. His unsuspected recurrence presented not only with a single keratoacanthomalike cutaneous metastasis on the scalp but also with another metastasis-related scalp lesion that appeared as a smooth pearly papule. We also observed a 53-year-old man whose metastatic esophageal adenocarcinoma presented with a keratoacanthomalike nodule on the right upper lip; additionally, the patient had other cutaneous metastases that appeared as an erythematous papule on the forehead and a cystic nodule on the scalp.8 Other investigators also observed a single keratoacanthomalike lesion on the left cheek of a 49-year-old man with metastatic esophageal adenocarcinoma.11
Agnetta et al2 described a patient with a history of malignant melanoma on the left upper back that had been excised 2 years prior. She presented with the eruptive onset of multiple keratoacanthomalike cutaneous metastases on the chest, back, and right arm.2 The important observation of metastatic malignant melanoma presenting as multiple keratoacanthomalike cutaneous metastases pointed out by Agnetta et al2 confirms a similar occurrence reported by Reed et al12 in a patient with metastatic malignant melanoma.
We also previously reported the case of a 68-year-old man with metastatic laryngeal squamous cell carcinoma (SCC) who developed more than 10 keratoacanthomalike nodules within a radiation port that extended from the face to the mid chest.10 In addition, other researchers have noted a similar phenomenon of keratoacanthomalike cutaneous metastases mimicking eruptive keratoacanthomas.13 Gil et al14 described a 40-year-old woman whose metastatic epithelioid trophoblastic tumor initially presented as 11 keratoacanthomalike scalp nodules; interestingly, the first nodule spontaneously regressed. Araghi et al15 reported a 58-year-old woman--with a stable SCC of the larynx that had been diagnosed 2 years prior and treated with chemoradiotherapy--in whom cancer progression presented as multiple keratoacanthomalike lesions in an area of prior radiotherapy.
In conclusion, cutaneous metastases presenting as new-onset solitary or multiple keratoacanthomalike nodules in either a cancer-free individual or a patient with a prior history of a visceral malignancy is uncommon. Although the clinical features mimic those of a single or eruptive keratoacanthomas, a biopsy will readily establish the diagnosis of cutaneous metastatic cancer. Metastatic esophageal carcinoma--either adenocarcinoma or SCC--can present, albeit rarely, with cutaneous lesions that can have various morphologies.8 Whether there is an increased predilection for patients with metastatic esophageal adenocarcinoma to present with single keratoacanthomalike cutaneous metastases with or without concurrent additional skin lesions of cutaneous metastases of other morphologies remains to be determined.
- Tarantino IS, Tennill T, Fraga G, et al. Cutaneous metastases from esophageal adenocarcinoma on the scalp. Cutis. 2020;105:E3-E5.
- Agnetta V, Hamstra A, Hirokane J, et al. Metastatic melanoma mimicking eruptive keratoacanthomas. Cutis. 2020;105:E29-E31.
- Cohen PR. Skin clues to primary and metastatic malignancy. Am Fam Physician. 1995;51:1199-1204.
- Cohen PR. Leukemia cutis-associated leonine facies and eyebrow loss. Cutis. 2019;103:212.
- Cohen PR, Prieto VG, Piha-Paul SA, et al. The "shield sign" in two men with metastatic salivary duct carcinoma to the skin: cutaneous metastases presenting as carcinoma hemorrhagiectoides. J Clin Aesthet Dermatol. 2012;5:27-36.
- Manteaux A, Cohen PR, Rapini RP. Zosteriform and epidermotropic metastasis. report of two cases. J Dermatol Surg Oncol. 1992;18:97-100.
- Conner KB, Cohen PR. Cutaneous metastases of breast carcinoma presenting as alopecia neoplastica. South Med J. 2009;102:385-389.
- Riahi RR, Cohen PR. Clinical manifestations of cutaneous metastases: a review with special emphasis on cutaneous metastases mimicking keratoacanthoma. Am J Clin Dermatol. 2012;13:103-112.
- Riahi RR, Cohen PR. Malignancies with skin lesions mimicking keratoacanthoma. Dermatol Online J. 2013;19:20397.
- Ellis DL, Riahi RR, Murina AT, et al. Metastatic laryngeal carcinoma mimicking eruptive keratoacanthomas: report of keratoacanthoma-like cutaneous metastases in a radiation port. Dermatol Online J. 2014;20. pii:13030/qt3s43b81f.
- Hani AC, Nuñez E, Cuellar I, et al. Cutaneous metastases as a manifestation of esophageal adenocarcinoma recurrence: a case report [published online September 5, 2019]. Rev Gastroenterol Mex. doi:10.1016/j.rgmx.2019.06.002.
- Reed KB, Cook-Norris RH, Brewer JD. The cutaneous manifestations of metastatic malignant melanoma. Int J Dermatol. 2012;51:243-249.
- Cohen PR, Riahi RR. Cutaneous metastases mimicking keratoacanthoma. Int J Dermatol. 2014;53:E320-E322.
- Gil F, Elvas L, Raposo S, et al. Keratoacanthoma-like nodules as first manifestation of metastatic epithelioid trophoblastic tumor. Dermatol Online J. 2019;25. pii:13030/qt9xx6p2tt.
- Araghi F, Fatemi A, Rakhshan A, et al. Skin metastasis of laryngeal carcinoma presenting as multiple eruptive nodules [published online February 10, 2020]. Head Neck Pathol. doi:10.1007/s12105-020-01143-1.
To the Editor:
We read with interest the excellent Cutis articles on cutaneous metastases by Tarantino et al1 and Agnetta et al.2 Tarantino et al1 reported a 59-year-old man who developed cutaneous metastases on the scalp from an esophageal adenocarcinoma. Agnetta et al2 described a 76-year-old woman with metastatic melanoma mimicking eruptive keratoacanthomas.
Cutaneous metastases are not common. They may herald the unsuspected diagnosis of a solid tumor recurrence or progression of systemic disease in an oncology patient. Occasionally, they are the primary manifestation of a visceral tumor in a previously cancer-free patient. Less often, skin lesions are the manifestation of a new or recurrent hematologic malignancy.3,4
The morphology of cutaneous metastases is variable. Most commonly they appear as papules and nodules. However, they can mimic bacterial (eg, erysipelas) and viral (eg, herpes zoster) infections or present as scalp alopecia.5-7
Cutaneous metastases also can mimic benign (eg, epidermoid cysts) or malignant (eg, keratoacanthoma) neoplasms. Keratoacanthomalike cutaneous metastases are rare.8 They can present as single or multiple tumors.9,10
In the case reported by Tarantino et al,1 the patient had a history of metastatic adenocarcinoma of the esophagus. His unsuspected recurrence presented not only with a single keratoacanthomalike cutaneous metastasis on the scalp but also with another metastasis-related scalp lesion that appeared as a smooth pearly papule. We also observed a 53-year-old man whose metastatic esophageal adenocarcinoma presented with a keratoacanthomalike nodule on the right upper lip; additionally, the patient had other cutaneous metastases that appeared as an erythematous papule on the forehead and a cystic nodule on the scalp.8 Other investigators also observed a single keratoacanthomalike lesion on the left cheek of a 49-year-old man with metastatic esophageal adenocarcinoma.11
Agnetta et al2 described a patient with a history of malignant melanoma on the left upper back that had been excised 2 years prior. She presented with the eruptive onset of multiple keratoacanthomalike cutaneous metastases on the chest, back, and right arm.2 The important observation of metastatic malignant melanoma presenting as multiple keratoacanthomalike cutaneous metastases pointed out by Agnetta et al2 confirms a similar occurrence reported by Reed et al12 in a patient with metastatic malignant melanoma.
We also previously reported the case of a 68-year-old man with metastatic laryngeal squamous cell carcinoma (SCC) who developed more than 10 keratoacanthomalike nodules within a radiation port that extended from the face to the mid chest.10 In addition, other researchers have noted a similar phenomenon of keratoacanthomalike cutaneous metastases mimicking eruptive keratoacanthomas.13 Gil et al14 described a 40-year-old woman whose metastatic epithelioid trophoblastic tumor initially presented as 11 keratoacanthomalike scalp nodules; interestingly, the first nodule spontaneously regressed. Araghi et al15 reported a 58-year-old woman--with a stable SCC of the larynx that had been diagnosed 2 years prior and treated with chemoradiotherapy--in whom cancer progression presented as multiple keratoacanthomalike lesions in an area of prior radiotherapy.
In conclusion, cutaneous metastases presenting as new-onset solitary or multiple keratoacanthomalike nodules in either a cancer-free individual or a patient with a prior history of a visceral malignancy is uncommon. Although the clinical features mimic those of a single or eruptive keratoacanthomas, a biopsy will readily establish the diagnosis of cutaneous metastatic cancer. Metastatic esophageal carcinoma--either adenocarcinoma or SCC--can present, albeit rarely, with cutaneous lesions that can have various morphologies.8 Whether there is an increased predilection for patients with metastatic esophageal adenocarcinoma to present with single keratoacanthomalike cutaneous metastases with or without concurrent additional skin lesions of cutaneous metastases of other morphologies remains to be determined.
To the Editor:
We read with interest the excellent Cutis articles on cutaneous metastases by Tarantino et al1 and Agnetta et al.2 Tarantino et al1 reported a 59-year-old man who developed cutaneous metastases on the scalp from an esophageal adenocarcinoma. Agnetta et al2 described a 76-year-old woman with metastatic melanoma mimicking eruptive keratoacanthomas.
Cutaneous metastases are not common. They may herald the unsuspected diagnosis of a solid tumor recurrence or progression of systemic disease in an oncology patient. Occasionally, they are the primary manifestation of a visceral tumor in a previously cancer-free patient. Less often, skin lesions are the manifestation of a new or recurrent hematologic malignancy.3,4
The morphology of cutaneous metastases is variable. Most commonly they appear as papules and nodules. However, they can mimic bacterial (eg, erysipelas) and viral (eg, herpes zoster) infections or present as scalp alopecia.5-7
Cutaneous metastases also can mimic benign (eg, epidermoid cysts) or malignant (eg, keratoacanthoma) neoplasms. Keratoacanthomalike cutaneous metastases are rare.8 They can present as single or multiple tumors.9,10
In the case reported by Tarantino et al,1 the patient had a history of metastatic adenocarcinoma of the esophagus. His unsuspected recurrence presented not only with a single keratoacanthomalike cutaneous metastasis on the scalp but also with another metastasis-related scalp lesion that appeared as a smooth pearly papule. We also observed a 53-year-old man whose metastatic esophageal adenocarcinoma presented with a keratoacanthomalike nodule on the right upper lip; additionally, the patient had other cutaneous metastases that appeared as an erythematous papule on the forehead and a cystic nodule on the scalp.8 Other investigators also observed a single keratoacanthomalike lesion on the left cheek of a 49-year-old man with metastatic esophageal adenocarcinoma.11
Agnetta et al2 described a patient with a history of malignant melanoma on the left upper back that had been excised 2 years prior. She presented with the eruptive onset of multiple keratoacanthomalike cutaneous metastases on the chest, back, and right arm.2 The important observation of metastatic malignant melanoma presenting as multiple keratoacanthomalike cutaneous metastases pointed out by Agnetta et al2 confirms a similar occurrence reported by Reed et al12 in a patient with metastatic malignant melanoma.
We also previously reported the case of a 68-year-old man with metastatic laryngeal squamous cell carcinoma (SCC) who developed more than 10 keratoacanthomalike nodules within a radiation port that extended from the face to the mid chest.10 In addition, other researchers have noted a similar phenomenon of keratoacanthomalike cutaneous metastases mimicking eruptive keratoacanthomas.13 Gil et al14 described a 40-year-old woman whose metastatic epithelioid trophoblastic tumor initially presented as 11 keratoacanthomalike scalp nodules; interestingly, the first nodule spontaneously regressed. Araghi et al15 reported a 58-year-old woman--with a stable SCC of the larynx that had been diagnosed 2 years prior and treated with chemoradiotherapy--in whom cancer progression presented as multiple keratoacanthomalike lesions in an area of prior radiotherapy.
In conclusion, cutaneous metastases presenting as new-onset solitary or multiple keratoacanthomalike nodules in either a cancer-free individual or a patient with a prior history of a visceral malignancy is uncommon. Although the clinical features mimic those of a single or eruptive keratoacanthomas, a biopsy will readily establish the diagnosis of cutaneous metastatic cancer. Metastatic esophageal carcinoma--either adenocarcinoma or SCC--can present, albeit rarely, with cutaneous lesions that can have various morphologies.8 Whether there is an increased predilection for patients with metastatic esophageal adenocarcinoma to present with single keratoacanthomalike cutaneous metastases with or without concurrent additional skin lesions of cutaneous metastases of other morphologies remains to be determined.
- Tarantino IS, Tennill T, Fraga G, et al. Cutaneous metastases from esophageal adenocarcinoma on the scalp. Cutis. 2020;105:E3-E5.
- Agnetta V, Hamstra A, Hirokane J, et al. Metastatic melanoma mimicking eruptive keratoacanthomas. Cutis. 2020;105:E29-E31.
- Cohen PR. Skin clues to primary and metastatic malignancy. Am Fam Physician. 1995;51:1199-1204.
- Cohen PR. Leukemia cutis-associated leonine facies and eyebrow loss. Cutis. 2019;103:212.
- Cohen PR, Prieto VG, Piha-Paul SA, et al. The "shield sign" in two men with metastatic salivary duct carcinoma to the skin: cutaneous metastases presenting as carcinoma hemorrhagiectoides. J Clin Aesthet Dermatol. 2012;5:27-36.
- Manteaux A, Cohen PR, Rapini RP. Zosteriform and epidermotropic metastasis. report of two cases. J Dermatol Surg Oncol. 1992;18:97-100.
- Conner KB, Cohen PR. Cutaneous metastases of breast carcinoma presenting as alopecia neoplastica. South Med J. 2009;102:385-389.
- Riahi RR, Cohen PR. Clinical manifestations of cutaneous metastases: a review with special emphasis on cutaneous metastases mimicking keratoacanthoma. Am J Clin Dermatol. 2012;13:103-112.
- Riahi RR, Cohen PR. Malignancies with skin lesions mimicking keratoacanthoma. Dermatol Online J. 2013;19:20397.
- Ellis DL, Riahi RR, Murina AT, et al. Metastatic laryngeal carcinoma mimicking eruptive keratoacanthomas: report of keratoacanthoma-like cutaneous metastases in a radiation port. Dermatol Online J. 2014;20. pii:13030/qt3s43b81f.
- Hani AC, Nuñez E, Cuellar I, et al. Cutaneous metastases as a manifestation of esophageal adenocarcinoma recurrence: a case report [published online September 5, 2019]. Rev Gastroenterol Mex. doi:10.1016/j.rgmx.2019.06.002.
- Reed KB, Cook-Norris RH, Brewer JD. The cutaneous manifestations of metastatic malignant melanoma. Int J Dermatol. 2012;51:243-249.
- Cohen PR, Riahi RR. Cutaneous metastases mimicking keratoacanthoma. Int J Dermatol. 2014;53:E320-E322.
- Gil F, Elvas L, Raposo S, et al. Keratoacanthoma-like nodules as first manifestation of metastatic epithelioid trophoblastic tumor. Dermatol Online J. 2019;25. pii:13030/qt9xx6p2tt.
- Araghi F, Fatemi A, Rakhshan A, et al. Skin metastasis of laryngeal carcinoma presenting as multiple eruptive nodules [published online February 10, 2020]. Head Neck Pathol. doi:10.1007/s12105-020-01143-1.
- Tarantino IS, Tennill T, Fraga G, et al. Cutaneous metastases from esophageal adenocarcinoma on the scalp. Cutis. 2020;105:E3-E5.
- Agnetta V, Hamstra A, Hirokane J, et al. Metastatic melanoma mimicking eruptive keratoacanthomas. Cutis. 2020;105:E29-E31.
- Cohen PR. Skin clues to primary and metastatic malignancy. Am Fam Physician. 1995;51:1199-1204.
- Cohen PR. Leukemia cutis-associated leonine facies and eyebrow loss. Cutis. 2019;103:212.
- Cohen PR, Prieto VG, Piha-Paul SA, et al. The "shield sign" in two men with metastatic salivary duct carcinoma to the skin: cutaneous metastases presenting as carcinoma hemorrhagiectoides. J Clin Aesthet Dermatol. 2012;5:27-36.
- Manteaux A, Cohen PR, Rapini RP. Zosteriform and epidermotropic metastasis. report of two cases. J Dermatol Surg Oncol. 1992;18:97-100.
- Conner KB, Cohen PR. Cutaneous metastases of breast carcinoma presenting as alopecia neoplastica. South Med J. 2009;102:385-389.
- Riahi RR, Cohen PR. Clinical manifestations of cutaneous metastases: a review with special emphasis on cutaneous metastases mimicking keratoacanthoma. Am J Clin Dermatol. 2012;13:103-112.
- Riahi RR, Cohen PR. Malignancies with skin lesions mimicking keratoacanthoma. Dermatol Online J. 2013;19:20397.
- Ellis DL, Riahi RR, Murina AT, et al. Metastatic laryngeal carcinoma mimicking eruptive keratoacanthomas: report of keratoacanthoma-like cutaneous metastases in a radiation port. Dermatol Online J. 2014;20. pii:13030/qt3s43b81f.
- Hani AC, Nuñez E, Cuellar I, et al. Cutaneous metastases as a manifestation of esophageal adenocarcinoma recurrence: a case report [published online September 5, 2019]. Rev Gastroenterol Mex. doi:10.1016/j.rgmx.2019.06.002.
- Reed KB, Cook-Norris RH, Brewer JD. The cutaneous manifestations of metastatic malignant melanoma. Int J Dermatol. 2012;51:243-249.
- Cohen PR, Riahi RR. Cutaneous metastases mimicking keratoacanthoma. Int J Dermatol. 2014;53:E320-E322.
- Gil F, Elvas L, Raposo S, et al. Keratoacanthoma-like nodules as first manifestation of metastatic epithelioid trophoblastic tumor. Dermatol Online J. 2019;25. pii:13030/qt9xx6p2tt.
- Araghi F, Fatemi A, Rakhshan A, et al. Skin metastasis of laryngeal carcinoma presenting as multiple eruptive nodules [published online February 10, 2020]. Head Neck Pathol. doi:10.1007/s12105-020-01143-1.
Woody Erythematous Induration on the Posterior Neck
The Diagnosis: Scleredema Diabeticorum
Histologically, scleredema is characterized by mucin deposition between collagen bundles in the deep dermis. Clinically, it is characterized by a progressive indurated plaque with associated stiffness of the involved area. It most commonly presents on the posterior aspect of the neck, though it can extend to involve the shoulders and upper torso.1 Scleredema is divided into 3 subtypes based on clinical associations. Type 1 often is preceded by an infection, most commonly group A Streptococcus. This type occurs acutely and often resolves completely over a few months.2 Type 2, which has progressive onset, is associated with monoclonal gammopathy.3 Type 3 is the most common type and is associated with diabetes mellitus. A study of 484 patients with type 2 diabetes mellitus demonstrated a prevalence of 2.5%.4 Although the exact pathogenesis has not been defined, it is hypothesized that irreversible glycosylation of collagen and alterations in collagenase activity may lead to accumulation of collagen and mucin in the dermis.5 Similar to type 2, type 3 scleredema appears subtly, progresses slowly, and tends to be chronic.1,6 Scleredema is characterized by marked dermal thickening and enlarged collagen bundles separated by mucin deposition (Figure 1). Fibroblast proliferation is characteristically absent.1
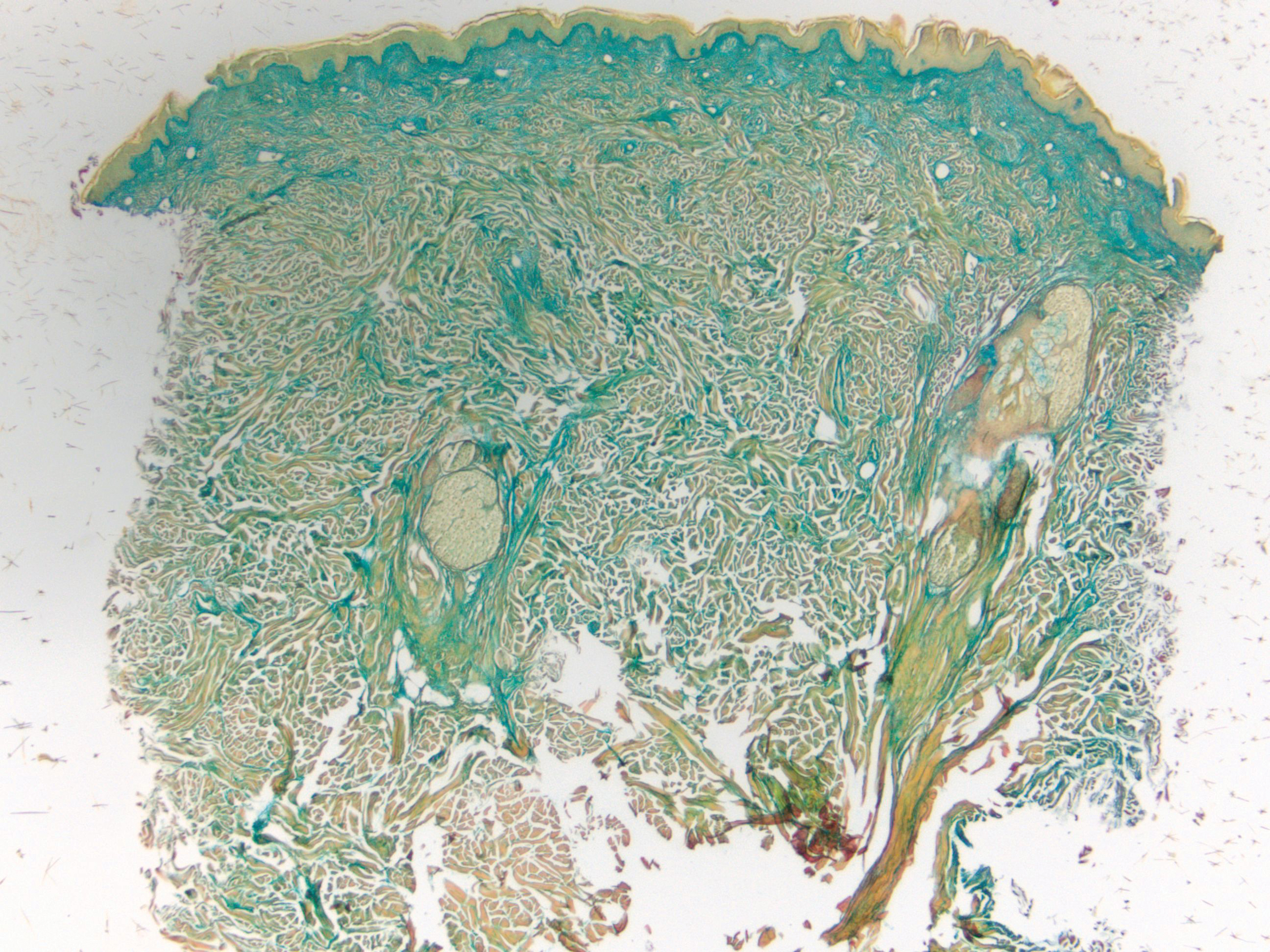
Clinically, tumid lupus erythematosus presents with erythematous edematous plaques on sun-exposed areas.7 Pretibial myxedema (PM) classically is associated with Graves disease; however, it can present in association with other types of thyroid dysfunction. Classically, PM presents on the pretibial regions as well-demarcated erythematous or hyperpigmented plaques.8 Similar to scleredema, histologic examination of tumid lupus erythematosus and PM reveals mucin deposition. Tumid lupus erythematosus also may demonstrate periadnexal and perivascular lymphocytic inflammation (Figure 2).7 The collagen bundles present in PM often are thin in comparison to scleredema (Figure 3).8

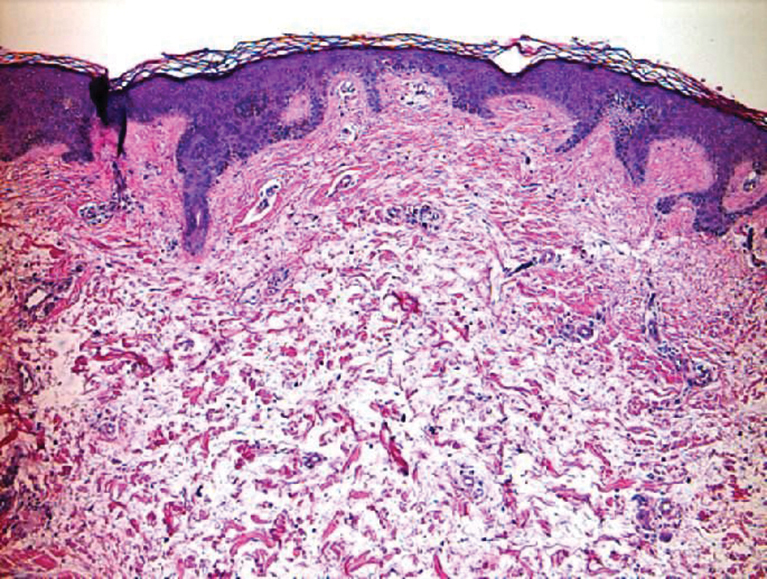
Scleroderma also presents with skin induration, erythema, and stiffening. However, unlike scleredema, scleroderma commonly involves the fingers, toes, and face. It presents with symptoms of Raynaud phenomenon, painful digital nonpitting edema, perioral skin tightening, mucocutaneous telangiectasia, and calcinosis cutis. Scleroderma also can involve organs such as the lungs, heart, kidneys, and gastrointestinal tract.9 Histologically, scleroderma is characterized by a compact dermis with closely packed collagen bundles. Other features of scleroderma can include perivascular mononuclear inflammatory cell infiltration, progressive atrophy of intradermal and perieccrine fat, and fibrosis (Figure 4).10
Scleromyxedema, also called papular mucinosis, is primary dermal mucinosis that often presents with waxy, dome-shaped papules that may coalesce into plaques. Similar to scleredema, scleromyxedema shows increased mucin deposition. However, scleromyxedema commonly is associated with fibroblast proliferation, which is characteristically absent in scleredema (Figure 5).11
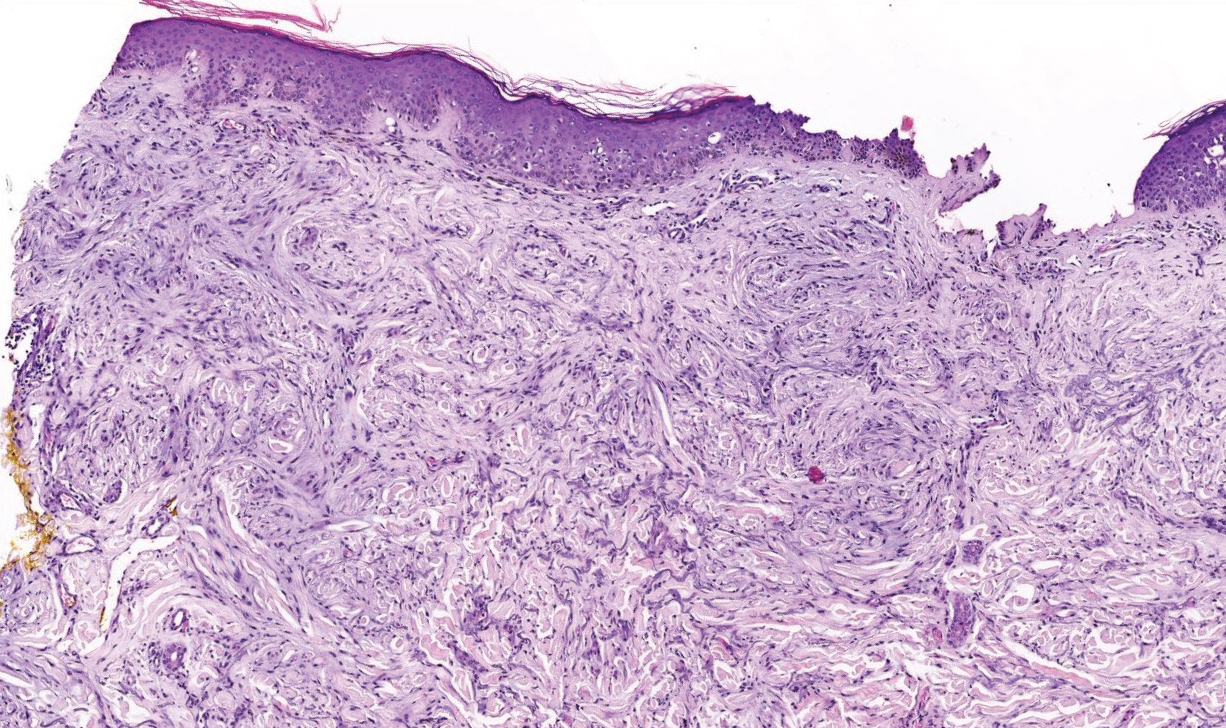
- Beers WH, Ince A, Moore TL. Scleredema adultorum of Buschke: a case report and review of the literature. Semin Arthritis Rheum. 2006;35:355-359.
- Cron RQ, Swetter SM. Scleredema revisited. a poststreptococcal complication. Clin Pediatr (Phila). 1994;33:606-610.
- Kövary PM, Vakilzadeh F, Macher E, et al. Monoclonal gammopathy in scleredema. observations in three cases. Arch Dermatol. 1981;117:536-539.
- Cole GW, Headley J, Skowsky R. Scleredema diabeticorum: a common and distinct cutaneous manifestation of diabetes mellitus. Diabetes Care. 1983;6:189-192.
- Namas R, Ashraf A. Scleredema of Buschke. Eur J Rheumatol. 2016;3:191-192.
- Knobler R, Moinzadeh P, Hunzelmann N, et al. European Dermatology Forum S1-guideline on the diagnosis and treatment of sclerosing diseases of the skin, part 2: scleromyxedema, scleredema and nephrogenic systemic fibrosis. J Eur Acad Dermatol Venereol. 2017;31:1581-1594.
- Kuhn A, Richter-Hintz D, Oslislo C, et al. Lupus erythematosus tumidus--a neglected subset of cutaneous lupus erythematosus: report of 40 cases. Arch Dermatol. 2000;136:1033-1041.
- Fatourechi V. Pretibial myxedema: pathophysiology and treatment options. Am J Clin Dermatol. 2005;6:295-309.
- van den Hoogen F, Khanna D, Fransen J, et al. 2013 Classification Criteria for Systemic Sclerosis: An American College of Rheumatology/European League Against Rheumatism Collaborative Initiative. 2013;65:2737-2747.
- Ferreli C, Gasparini G, Parodi A, et al. Cutaneous manifestations of scleroderma and scleroderma-like disorders: a comprehensive review. Clin Rev Allergy Immunol. 2017;53:306-336.
- Rongioletti F, Merlo G, Cinotti E, et al. Scleromyxedema: a multicenter study of characteristics, comorbidities, course, and therapy in 30 patients. J Am Acad Dermatol. 2013;69:66-72.
The Diagnosis: Scleredema Diabeticorum
Histologically, scleredema is characterized by mucin deposition between collagen bundles in the deep dermis. Clinically, it is characterized by a progressive indurated plaque with associated stiffness of the involved area. It most commonly presents on the posterior aspect of the neck, though it can extend to involve the shoulders and upper torso.1 Scleredema is divided into 3 subtypes based on clinical associations. Type 1 often is preceded by an infection, most commonly group A Streptococcus. This type occurs acutely and often resolves completely over a few months.2 Type 2, which has progressive onset, is associated with monoclonal gammopathy.3 Type 3 is the most common type and is associated with diabetes mellitus. A study of 484 patients with type 2 diabetes mellitus demonstrated a prevalence of 2.5%.4 Although the exact pathogenesis has not been defined, it is hypothesized that irreversible glycosylation of collagen and alterations in collagenase activity may lead to accumulation of collagen and mucin in the dermis.5 Similar to type 2, type 3 scleredema appears subtly, progresses slowly, and tends to be chronic.1,6 Scleredema is characterized by marked dermal thickening and enlarged collagen bundles separated by mucin deposition (Figure 1). Fibroblast proliferation is characteristically absent.1
Clinically, tumid lupus erythematosus presents with erythematous edematous plaques on sun-exposed areas.7 Pretibial myxedema (PM) classically is associated with Graves disease; however, it can present in association with other types of thyroid dysfunction. Classically, PM presents on the pretibial regions as well-demarcated erythematous or hyperpigmented plaques.8 Similar to scleredema, histologic examination of tumid lupus erythematosus and PM reveals mucin deposition. Tumid lupus erythematosus also may demonstrate periadnexal and perivascular lymphocytic inflammation (Figure 2).7 The collagen bundles present in PM often are thin in comparison to scleredema (Figure 3).8
Scleroderma also presents with skin induration, erythema, and stiffening. However, unlike scleredema, scleroderma commonly involves the fingers, toes, and face. It presents with symptoms of Raynaud phenomenon, painful digital nonpitting edema, perioral skin tightening, mucocutaneous telangiectasia, and calcinosis cutis. Scleroderma also can involve organs such as the lungs, heart, kidneys, and gastrointestinal tract.9 Histologically, scleroderma is characterized by a compact dermis with closely packed collagen bundles. Other features of scleroderma can include perivascular mononuclear inflammatory cell infiltration, progressive atrophy of intradermal and perieccrine fat, and fibrosis (Figure 4).10
Scleromyxedema, also called papular mucinosis, is primary dermal mucinosis that often presents with waxy, dome-shaped papules that may coalesce into plaques. Similar to scleredema, scleromyxedema shows increased mucin deposition. However, scleromyxedema commonly is associated with fibroblast proliferation, which is characteristically absent in scleredema (Figure 5).11
The Diagnosis: Scleredema Diabeticorum
Histologically, scleredema is characterized by mucin deposition between collagen bundles in the deep dermis. Clinically, it is characterized by a progressive indurated plaque with associated stiffness of the involved area. It most commonly presents on the posterior aspect of the neck, though it can extend to involve the shoulders and upper torso.1 Scleredema is divided into 3 subtypes based on clinical associations. Type 1 often is preceded by an infection, most commonly group A Streptococcus. This type occurs acutely and often resolves completely over a few months.2 Type 2, which has progressive onset, is associated with monoclonal gammopathy.3 Type 3 is the most common type and is associated with diabetes mellitus. A study of 484 patients with type 2 diabetes mellitus demonstrated a prevalence of 2.5%.4 Although the exact pathogenesis has not been defined, it is hypothesized that irreversible glycosylation of collagen and alterations in collagenase activity may lead to accumulation of collagen and mucin in the dermis.5 Similar to type 2, type 3 scleredema appears subtly, progresses slowly, and tends to be chronic.1,6 Scleredema is characterized by marked dermal thickening and enlarged collagen bundles separated by mucin deposition (Figure 1). Fibroblast proliferation is characteristically absent.1
Clinically, tumid lupus erythematosus presents with erythematous edematous plaques on sun-exposed areas.7 Pretibial myxedema (PM) classically is associated with Graves disease; however, it can present in association with other types of thyroid dysfunction. Classically, PM presents on the pretibial regions as well-demarcated erythematous or hyperpigmented plaques.8 Similar to scleredema, histologic examination of tumid lupus erythematosus and PM reveals mucin deposition. Tumid lupus erythematosus also may demonstrate periadnexal and perivascular lymphocytic inflammation (Figure 2).7 The collagen bundles present in PM often are thin in comparison to scleredema (Figure 3).8
Scleroderma also presents with skin induration, erythema, and stiffening. However, unlike scleredema, scleroderma commonly involves the fingers, toes, and face. It presents with symptoms of Raynaud phenomenon, painful digital nonpitting edema, perioral skin tightening, mucocutaneous telangiectasia, and calcinosis cutis. Scleroderma also can involve organs such as the lungs, heart, kidneys, and gastrointestinal tract.9 Histologically, scleroderma is characterized by a compact dermis with closely packed collagen bundles. Other features of scleroderma can include perivascular mononuclear inflammatory cell infiltration, progressive atrophy of intradermal and perieccrine fat, and fibrosis (Figure 4).10
Scleromyxedema, also called papular mucinosis, is primary dermal mucinosis that often presents with waxy, dome-shaped papules that may coalesce into plaques. Similar to scleredema, scleromyxedema shows increased mucin deposition. However, scleromyxedema commonly is associated with fibroblast proliferation, which is characteristically absent in scleredema (Figure 5).11
- Beers WH, Ince A, Moore TL. Scleredema adultorum of Buschke: a case report and review of the literature. Semin Arthritis Rheum. 2006;35:355-359.
- Cron RQ, Swetter SM. Scleredema revisited. a poststreptococcal complication. Clin Pediatr (Phila). 1994;33:606-610.
- Kövary PM, Vakilzadeh F, Macher E, et al. Monoclonal gammopathy in scleredema. observations in three cases. Arch Dermatol. 1981;117:536-539.
- Cole GW, Headley J, Skowsky R. Scleredema diabeticorum: a common and distinct cutaneous manifestation of diabetes mellitus. Diabetes Care. 1983;6:189-192.
- Namas R, Ashraf A. Scleredema of Buschke. Eur J Rheumatol. 2016;3:191-192.
- Knobler R, Moinzadeh P, Hunzelmann N, et al. European Dermatology Forum S1-guideline on the diagnosis and treatment of sclerosing diseases of the skin, part 2: scleromyxedema, scleredema and nephrogenic systemic fibrosis. J Eur Acad Dermatol Venereol. 2017;31:1581-1594.
- Kuhn A, Richter-Hintz D, Oslislo C, et al. Lupus erythematosus tumidus--a neglected subset of cutaneous lupus erythematosus: report of 40 cases. Arch Dermatol. 2000;136:1033-1041.
- Fatourechi V. Pretibial myxedema: pathophysiology and treatment options. Am J Clin Dermatol. 2005;6:295-309.
- van den Hoogen F, Khanna D, Fransen J, et al. 2013 Classification Criteria for Systemic Sclerosis: An American College of Rheumatology/European League Against Rheumatism Collaborative Initiative. 2013;65:2737-2747.
- Ferreli C, Gasparini G, Parodi A, et al. Cutaneous manifestations of scleroderma and scleroderma-like disorders: a comprehensive review. Clin Rev Allergy Immunol. 2017;53:306-336.
- Rongioletti F, Merlo G, Cinotti E, et al. Scleromyxedema: a multicenter study of characteristics, comorbidities, course, and therapy in 30 patients. J Am Acad Dermatol. 2013;69:66-72.
- Beers WH, Ince A, Moore TL. Scleredema adultorum of Buschke: a case report and review of the literature. Semin Arthritis Rheum. 2006;35:355-359.
- Cron RQ, Swetter SM. Scleredema revisited. a poststreptococcal complication. Clin Pediatr (Phila). 1994;33:606-610.
- Kövary PM, Vakilzadeh F, Macher E, et al. Monoclonal gammopathy in scleredema. observations in three cases. Arch Dermatol. 1981;117:536-539.
- Cole GW, Headley J, Skowsky R. Scleredema diabeticorum: a common and distinct cutaneous manifestation of diabetes mellitus. Diabetes Care. 1983;6:189-192.
- Namas R, Ashraf A. Scleredema of Buschke. Eur J Rheumatol. 2016;3:191-192.
- Knobler R, Moinzadeh P, Hunzelmann N, et al. European Dermatology Forum S1-guideline on the diagnosis and treatment of sclerosing diseases of the skin, part 2: scleromyxedema, scleredema and nephrogenic systemic fibrosis. J Eur Acad Dermatol Venereol. 2017;31:1581-1594.
- Kuhn A, Richter-Hintz D, Oslislo C, et al. Lupus erythematosus tumidus--a neglected subset of cutaneous lupus erythematosus: report of 40 cases. Arch Dermatol. 2000;136:1033-1041.
- Fatourechi V. Pretibial myxedema: pathophysiology and treatment options. Am J Clin Dermatol. 2005;6:295-309.
- van den Hoogen F, Khanna D, Fransen J, et al. 2013 Classification Criteria for Systemic Sclerosis: An American College of Rheumatology/European League Against Rheumatism Collaborative Initiative. 2013;65:2737-2747.
- Ferreli C, Gasparini G, Parodi A, et al. Cutaneous manifestations of scleroderma and scleroderma-like disorders: a comprehensive review. Clin Rev Allergy Immunol. 2017;53:306-336.
- Rongioletti F, Merlo G, Cinotti E, et al. Scleromyxedema: a multicenter study of characteristics, comorbidities, course, and therapy in 30 patients. J Am Acad Dermatol. 2013;69:66-72.
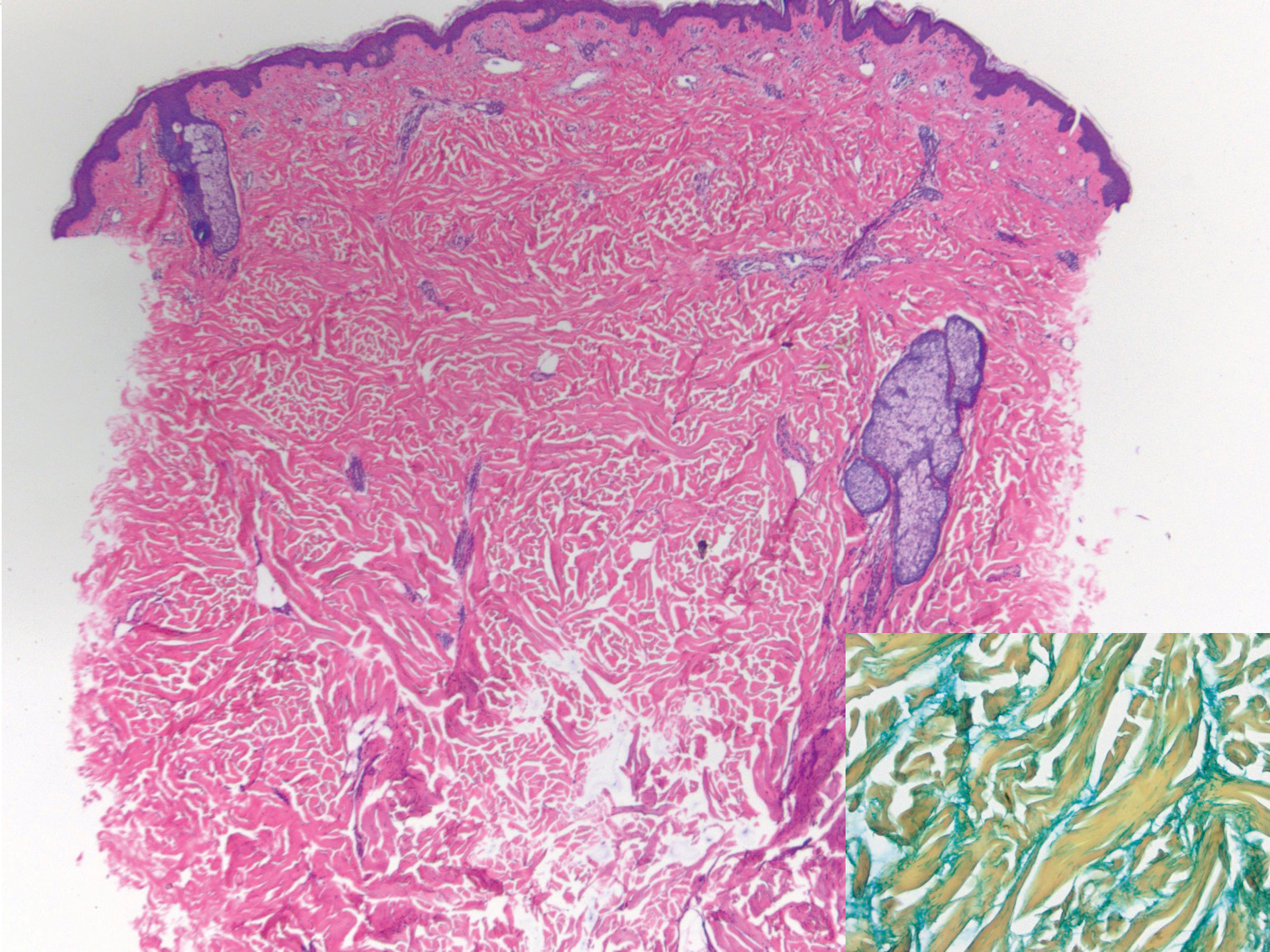
A 39-year-old white woman with a medical history of type 1 diabetes mellitus and rheumatoid arthritis presented to the dermatology clinic with pain and thickened skin on the posterior neck of 4 weeks’ duration. The patient noted stiffness in the neck and shoulders but denied any pain, pruritus, fever, chills, night sweats, fatigue, cough, dyspnea, dysphagia, weight loss, or change in appetite. Physical examination revealed a woody indurated plaque with slight erythema that was present diffusely on the posterior neck and upper back. The patient reported that a recent complete blood cell count and complete metabolic panel performed by her primary care physician were within reference range. Hemoglobin A1C was 8.6% of total hemoglobin (reference range, 4%–7%). A punch biopsy was performed.
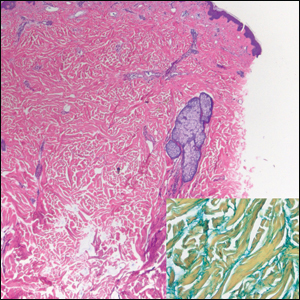
Granular Parakeratosis
To the Editor:
A 46-year-old overweight woman presented with a rash in the axillae of 2 months’ duration. She did not report any additional symptoms such as pruritus or pain. She reported changing her deodorant recently from Secret Original to Secret Clinical Strength (both Procter & Gamble). Her medical history was remarkable for asthma and gastroesophageal reflux disease. Clinical examination revealed erythematous-brown, stuccolike, hyperkeratotic papules coalescing into plaques in recently shaved axillae, affecting the left axilla more than the right axilla (Figure 1). The clinical differential diagnosis included granular parakeratosis, intertrigo, Hailey-Hailey disease, Darier disease, pemphigus vegetans, confluent and reticulated papillomatosis, acanthosis nigricans, seborrheic keratoses, and irritant or allergic contact dermatitis. A punch biopsy revealed a marked compact parakeratotic horn with retention of keratohyalin granules (Figure 2). The subjacent epidermis showed some acanthosis and spongiosis with mild chronic inflammation of the dermal rim. Based on histopathology, granular parakeratosis was diagnosed.
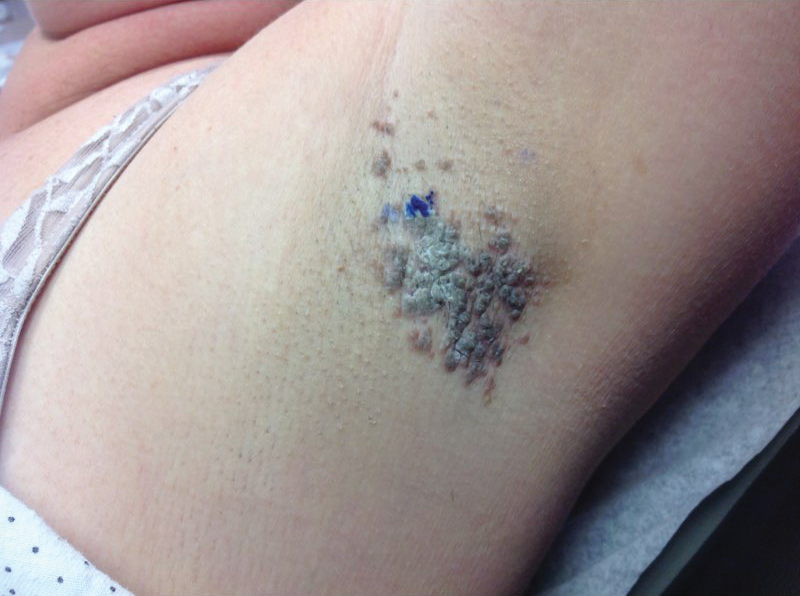
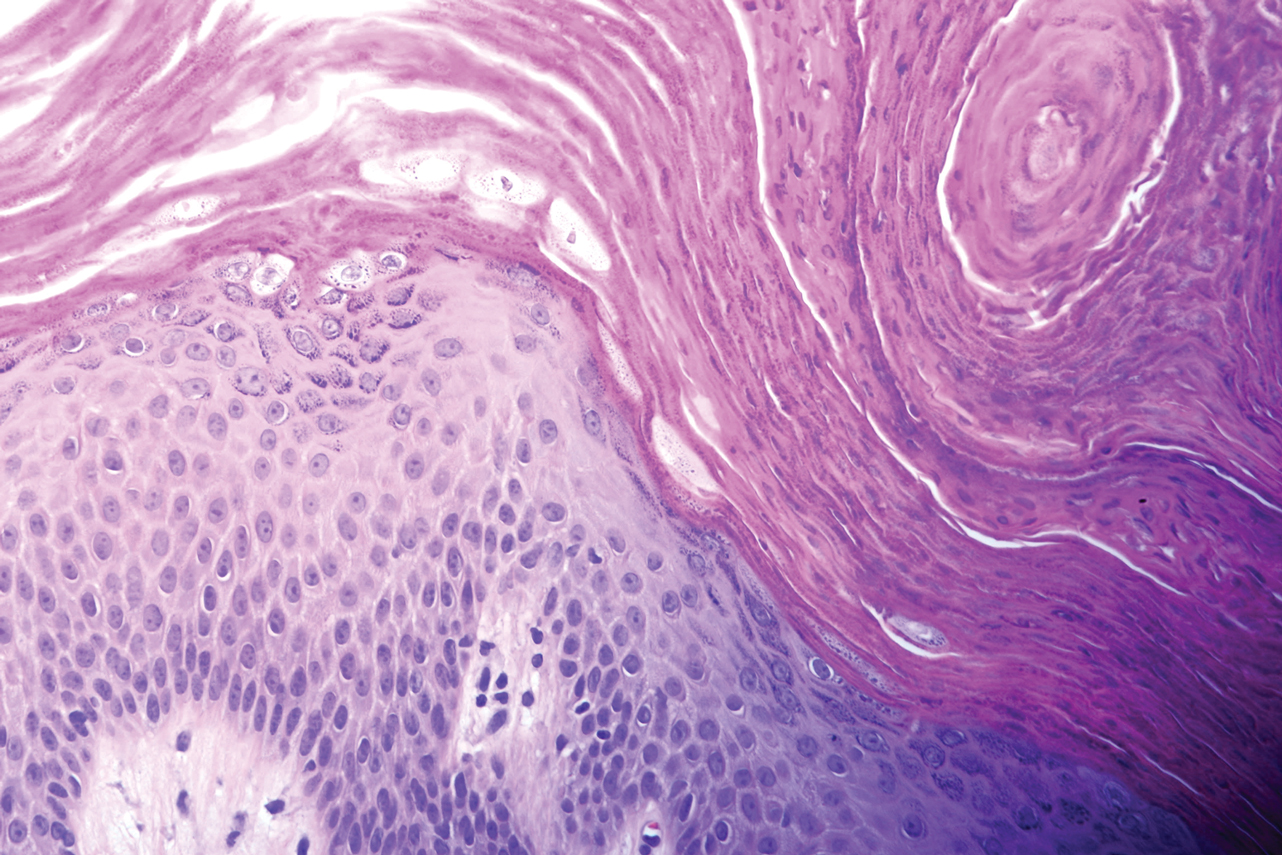
At a subsequent visit 2 weeks later, we prescribed glycolic acid lotion 10% applied to the axillae twice daily, plus tretinoin gel 0.05% applied to the axillae each evening. She reported clearing after 1 week of therapy. She also had changed her deodorant from Secret Clinical Strength back to the usual Secret Original. The patient discontinued topical treatment after clearing of the lesions. Three weeks later, clinical examination revealed postinflammatory hyperpigmentation in the axillae, and the prior lesions had resolved (Figure 3).
Granular parakeratosis is an unusual condition most commonly presenting in middle-aged women in the axillae, with a clinical presentation of erythematous to brownish hyperkeratotic papules coalescing into plaques. Although few cases have been reported, granular parakeratosis likely is more common than has been reported. There have been reports involving the scalp, cheeks, abdomen, thighs, and other intertriginous areas including inguinal folds and the submammary region.1-4 There also is an infantile form related to diapers and zinc oxide paste.5 Although uncommon, granular parakeratosis can occur as a single papule or plaque and is termed granular parakeratotic acanthoma.6 Lesions may persist for months, spontaneously resolve and recur, and occasionally evolve into fissures and erosions due to irritation. Pruritus is a common concern. Histology of granular parakeratosis reveals hyperkeratosis with eosinophilic staining, compact parakeratosis with retention of basophilic keratohyalin granules, and vascular proliferation and ectasia.5
The cause is unknown but possibly related to irritation from rubbing, occlusion, sweating, or deodorants.5,7 Cases indicate a link to obesity. Hypotheses as to the etiology include the disruption of cornification. Normally, filaggrin maintains the keratohyaline granules in the stratum corneum during cornification. Therefore, the retention of keratohyaline granules in granular parakeratosis may be due to a defect in processing profilaggrin to filaggrin, which has been proposed based on ultrastructural and immunohistochemical studies.8
The differential diagnosis includes granular parakeratosis, intertrigo (caused by seborrheic dermatitis, candidiasis, inverse psoriasis, or erythrasma), Hailey-Hailey disease, Darier disease, pemphigus vegetans, confluent and reticulated papillomatosis, and irritant or allergic contact dermatitis. The papules may resemble seborrheic keratoses, while the plaques can be mistaken for acanthosis nigricans.
Therapeutic success has been reported with topical corticosteroids, vitamin D analogues, topical or oral retinoids, ammonium lactate, calcineurin inhibitors, topical or oral antifungals, cryotherapy, and botulinum toxin injections.3,9-11 In addition, parakeratosis has decreased in biopsies from psoriatic patients after acitretin, methotrexate, and phototherapy, which may be alternative treatments for unusually difficult or recalcitrant cases of granular parakeratosis. To minimize side effects and resolve the papules quickly, we combined 2 synergistic agents—glycolic acid and tretinoin—each with different mechanisms of action, and we observed excellent clinical response.
Granular parakeratosis is possibly related to a combination of topical products that potentiate irritation, rubbing, and occlusion of sweat. Multiple treatment modalities likely contribute to clearing, the most important being removal of any triggering topical products. Our patient’s change in deodorant may have been the inciting factor for the disease. Withdrawal of the Secret Clinical Strength deodorant prompted clearing, though topical retinoid and glycolic acid acted as facilitating therapies for timely results. A thorough history, as highlighted by this case, may help pinpoint etiologic factors. By identifying a seemingly innocuous change in hygienic routine, we were able to minimize the need for ongoing therapy.
- Graham R. Intertriginous granular parakeratosis: a case report and review of the literature. J Am Acad Dermatol. 2011;64:AB45-AB45.
- Compton AK, Jackson JM. Isotretinoin as a treatment for axillary granular parakeratosis. Cutis. 2007;80:55-56.
- Channual J, Fife DJ, Wu JJ. Axillary granular parakeratosis. Cutis. 2013;92;61, 65-66.
- Streams S, Gottwald L, Zaher A, et al. Granular parakeratosis of the scalp: a case report. J Am Acad Dermatol. 2007;56:AB81-AB81.
- James WD, Berger T, Elston D. Andrews’ Diseases of the Skin. 12th ed. Philadelphia, PA: Elsevier, Inc; 2015.
- Resnik KS, Kantor GR, DiLeonardo M. Granular parakeratotic acanthoma. Am J Dermatopathol. 2005;27:393-396.
- Naylor E, Wartman D, Telang G, et al. Granular parakeratosis secondary to postsurgical occlusion. J Am Acad Dermatol. 2008;58:AB126.
- Bolognia JL, Jorizzo JL, Schaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier, Inc; 2012.
- Baum B, Skopit S. Granular parakeratosis treatment with tacrolimus 0.1% ointment: a case presentation and discussion. J Am Osteo Coll Dermatol. 2013;26:40-41.
- Brown SK, Heilman ER. Granular parakeratosis: resolution with topical tretinoin. J Am Acad Dermatol. 2002;47:S279-S280.
- Webster CG, Resnik KS, Webster GF. Axillary granular parakeratosis: response to isotretinoin. J Am Acad Dermatol. 1997;37:789790.
To the Editor:
A 46-year-old overweight woman presented with a rash in the axillae of 2 months’ duration. She did not report any additional symptoms such as pruritus or pain. She reported changing her deodorant recently from Secret Original to Secret Clinical Strength (both Procter & Gamble). Her medical history was remarkable for asthma and gastroesophageal reflux disease. Clinical examination revealed erythematous-brown, stuccolike, hyperkeratotic papules coalescing into plaques in recently shaved axillae, affecting the left axilla more than the right axilla (Figure 1). The clinical differential diagnosis included granular parakeratosis, intertrigo, Hailey-Hailey disease, Darier disease, pemphigus vegetans, confluent and reticulated papillomatosis, acanthosis nigricans, seborrheic keratoses, and irritant or allergic contact dermatitis. A punch biopsy revealed a marked compact parakeratotic horn with retention of keratohyalin granules (Figure 2). The subjacent epidermis showed some acanthosis and spongiosis with mild chronic inflammation of the dermal rim. Based on histopathology, granular parakeratosis was diagnosed.
At a subsequent visit 2 weeks later, we prescribed glycolic acid lotion 10% applied to the axillae twice daily, plus tretinoin gel 0.05% applied to the axillae each evening. She reported clearing after 1 week of therapy. She also had changed her deodorant from Secret Clinical Strength back to the usual Secret Original. The patient discontinued topical treatment after clearing of the lesions. Three weeks later, clinical examination revealed postinflammatory hyperpigmentation in the axillae, and the prior lesions had resolved (Figure 3).
Granular parakeratosis is an unusual condition most commonly presenting in middle-aged women in the axillae, with a clinical presentation of erythematous to brownish hyperkeratotic papules coalescing into plaques. Although few cases have been reported, granular parakeratosis likely is more common than has been reported. There have been reports involving the scalp, cheeks, abdomen, thighs, and other intertriginous areas including inguinal folds and the submammary region.1-4 There also is an infantile form related to diapers and zinc oxide paste.5 Although uncommon, granular parakeratosis can occur as a single papule or plaque and is termed granular parakeratotic acanthoma.6 Lesions may persist for months, spontaneously resolve and recur, and occasionally evolve into fissures and erosions due to irritation. Pruritus is a common concern. Histology of granular parakeratosis reveals hyperkeratosis with eosinophilic staining, compact parakeratosis with retention of basophilic keratohyalin granules, and vascular proliferation and ectasia.5
The cause is unknown but possibly related to irritation from rubbing, occlusion, sweating, or deodorants.5,7 Cases indicate a link to obesity. Hypotheses as to the etiology include the disruption of cornification. Normally, filaggrin maintains the keratohyaline granules in the stratum corneum during cornification. Therefore, the retention of keratohyaline granules in granular parakeratosis may be due to a defect in processing profilaggrin to filaggrin, which has been proposed based on ultrastructural and immunohistochemical studies.8
The differential diagnosis includes granular parakeratosis, intertrigo (caused by seborrheic dermatitis, candidiasis, inverse psoriasis, or erythrasma), Hailey-Hailey disease, Darier disease, pemphigus vegetans, confluent and reticulated papillomatosis, and irritant or allergic contact dermatitis. The papules may resemble seborrheic keratoses, while the plaques can be mistaken for acanthosis nigricans.
Therapeutic success has been reported with topical corticosteroids, vitamin D analogues, topical or oral retinoids, ammonium lactate, calcineurin inhibitors, topical or oral antifungals, cryotherapy, and botulinum toxin injections.3,9-11 In addition, parakeratosis has decreased in biopsies from psoriatic patients after acitretin, methotrexate, and phototherapy, which may be alternative treatments for unusually difficult or recalcitrant cases of granular parakeratosis. To minimize side effects and resolve the papules quickly, we combined 2 synergistic agents—glycolic acid and tretinoin—each with different mechanisms of action, and we observed excellent clinical response.
Granular parakeratosis is possibly related to a combination of topical products that potentiate irritation, rubbing, and occlusion of sweat. Multiple treatment modalities likely contribute to clearing, the most important being removal of any triggering topical products. Our patient’s change in deodorant may have been the inciting factor for the disease. Withdrawal of the Secret Clinical Strength deodorant prompted clearing, though topical retinoid and glycolic acid acted as facilitating therapies for timely results. A thorough history, as highlighted by this case, may help pinpoint etiologic factors. By identifying a seemingly innocuous change in hygienic routine, we were able to minimize the need for ongoing therapy.
To the Editor:
A 46-year-old overweight woman presented with a rash in the axillae of 2 months’ duration. She did not report any additional symptoms such as pruritus or pain. She reported changing her deodorant recently from Secret Original to Secret Clinical Strength (both Procter & Gamble). Her medical history was remarkable for asthma and gastroesophageal reflux disease. Clinical examination revealed erythematous-brown, stuccolike, hyperkeratotic papules coalescing into plaques in recently shaved axillae, affecting the left axilla more than the right axilla (Figure 1). The clinical differential diagnosis included granular parakeratosis, intertrigo, Hailey-Hailey disease, Darier disease, pemphigus vegetans, confluent and reticulated papillomatosis, acanthosis nigricans, seborrheic keratoses, and irritant or allergic contact dermatitis. A punch biopsy revealed a marked compact parakeratotic horn with retention of keratohyalin granules (Figure 2). The subjacent epidermis showed some acanthosis and spongiosis with mild chronic inflammation of the dermal rim. Based on histopathology, granular parakeratosis was diagnosed.
At a subsequent visit 2 weeks later, we prescribed glycolic acid lotion 10% applied to the axillae twice daily, plus tretinoin gel 0.05% applied to the axillae each evening. She reported clearing after 1 week of therapy. She also had changed her deodorant from Secret Clinical Strength back to the usual Secret Original. The patient discontinued topical treatment after clearing of the lesions. Three weeks later, clinical examination revealed postinflammatory hyperpigmentation in the axillae, and the prior lesions had resolved (Figure 3).
Granular parakeratosis is an unusual condition most commonly presenting in middle-aged women in the axillae, with a clinical presentation of erythematous to brownish hyperkeratotic papules coalescing into plaques. Although few cases have been reported, granular parakeratosis likely is more common than has been reported. There have been reports involving the scalp, cheeks, abdomen, thighs, and other intertriginous areas including inguinal folds and the submammary region.1-4 There also is an infantile form related to diapers and zinc oxide paste.5 Although uncommon, granular parakeratosis can occur as a single papule or plaque and is termed granular parakeratotic acanthoma.6 Lesions may persist for months, spontaneously resolve and recur, and occasionally evolve into fissures and erosions due to irritation. Pruritus is a common concern. Histology of granular parakeratosis reveals hyperkeratosis with eosinophilic staining, compact parakeratosis with retention of basophilic keratohyalin granules, and vascular proliferation and ectasia.5
The cause is unknown but possibly related to irritation from rubbing, occlusion, sweating, or deodorants.5,7 Cases indicate a link to obesity. Hypotheses as to the etiology include the disruption of cornification. Normally, filaggrin maintains the keratohyaline granules in the stratum corneum during cornification. Therefore, the retention of keratohyaline granules in granular parakeratosis may be due to a defect in processing profilaggrin to filaggrin, which has been proposed based on ultrastructural and immunohistochemical studies.8
The differential diagnosis includes granular parakeratosis, intertrigo (caused by seborrheic dermatitis, candidiasis, inverse psoriasis, or erythrasma), Hailey-Hailey disease, Darier disease, pemphigus vegetans, confluent and reticulated papillomatosis, and irritant or allergic contact dermatitis. The papules may resemble seborrheic keratoses, while the plaques can be mistaken for acanthosis nigricans.
Therapeutic success has been reported with topical corticosteroids, vitamin D analogues, topical or oral retinoids, ammonium lactate, calcineurin inhibitors, topical or oral antifungals, cryotherapy, and botulinum toxin injections.3,9-11 In addition, parakeratosis has decreased in biopsies from psoriatic patients after acitretin, methotrexate, and phototherapy, which may be alternative treatments for unusually difficult or recalcitrant cases of granular parakeratosis. To minimize side effects and resolve the papules quickly, we combined 2 synergistic agents—glycolic acid and tretinoin—each with different mechanisms of action, and we observed excellent clinical response.
Granular parakeratosis is possibly related to a combination of topical products that potentiate irritation, rubbing, and occlusion of sweat. Multiple treatment modalities likely contribute to clearing, the most important being removal of any triggering topical products. Our patient’s change in deodorant may have been the inciting factor for the disease. Withdrawal of the Secret Clinical Strength deodorant prompted clearing, though topical retinoid and glycolic acid acted as facilitating therapies for timely results. A thorough history, as highlighted by this case, may help pinpoint etiologic factors. By identifying a seemingly innocuous change in hygienic routine, we were able to minimize the need for ongoing therapy.
- Graham R. Intertriginous granular parakeratosis: a case report and review of the literature. J Am Acad Dermatol. 2011;64:AB45-AB45.
- Compton AK, Jackson JM. Isotretinoin as a treatment for axillary granular parakeratosis. Cutis. 2007;80:55-56.
- Channual J, Fife DJ, Wu JJ. Axillary granular parakeratosis. Cutis. 2013;92;61, 65-66.
- Streams S, Gottwald L, Zaher A, et al. Granular parakeratosis of the scalp: a case report. J Am Acad Dermatol. 2007;56:AB81-AB81.
- James WD, Berger T, Elston D. Andrews’ Diseases of the Skin. 12th ed. Philadelphia, PA: Elsevier, Inc; 2015.
- Resnik KS, Kantor GR, DiLeonardo M. Granular parakeratotic acanthoma. Am J Dermatopathol. 2005;27:393-396.
- Naylor E, Wartman D, Telang G, et al. Granular parakeratosis secondary to postsurgical occlusion. J Am Acad Dermatol. 2008;58:AB126.
- Bolognia JL, Jorizzo JL, Schaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier, Inc; 2012.
- Baum B, Skopit S. Granular parakeratosis treatment with tacrolimus 0.1% ointment: a case presentation and discussion. J Am Osteo Coll Dermatol. 2013;26:40-41.
- Brown SK, Heilman ER. Granular parakeratosis: resolution with topical tretinoin. J Am Acad Dermatol. 2002;47:S279-S280.
- Webster CG, Resnik KS, Webster GF. Axillary granular parakeratosis: response to isotretinoin. J Am Acad Dermatol. 1997;37:789790.
- Graham R. Intertriginous granular parakeratosis: a case report and review of the literature. J Am Acad Dermatol. 2011;64:AB45-AB45.
- Compton AK, Jackson JM. Isotretinoin as a treatment for axillary granular parakeratosis. Cutis. 2007;80:55-56.
- Channual J, Fife DJ, Wu JJ. Axillary granular parakeratosis. Cutis. 2013;92;61, 65-66.
- Streams S, Gottwald L, Zaher A, et al. Granular parakeratosis of the scalp: a case report. J Am Acad Dermatol. 2007;56:AB81-AB81.
- James WD, Berger T, Elston D. Andrews’ Diseases of the Skin. 12th ed. Philadelphia, PA: Elsevier, Inc; 2015.
- Resnik KS, Kantor GR, DiLeonardo M. Granular parakeratotic acanthoma. Am J Dermatopathol. 2005;27:393-396.
- Naylor E, Wartman D, Telang G, et al. Granular parakeratosis secondary to postsurgical occlusion. J Am Acad Dermatol. 2008;58:AB126.
- Bolognia JL, Jorizzo JL, Schaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier, Inc; 2012.
- Baum B, Skopit S. Granular parakeratosis treatment with tacrolimus 0.1% ointment: a case presentation and discussion. J Am Osteo Coll Dermatol. 2013;26:40-41.
- Brown SK, Heilman ER. Granular parakeratosis: resolution with topical tretinoin. J Am Acad Dermatol. 2002;47:S279-S280.
- Webster CG, Resnik KS, Webster GF. Axillary granular parakeratosis: response to isotretinoin. J Am Acad Dermatol. 1997;37:789790.
Practice Points
- Granular parakeratosis most commonly presents in middle-aged women in the axillae.
- The cause is unknown but possibly related to irritation from rubbing, occlusion, sweating, or deodorants.
- Multiple treatment modalities likely contribute to clearing, the most important being removal of any triggering topical products.
Eczema Herpeticum in a Patient With Hailey-Hailey Disease Confounded by Coexistent Psoriasis
To the Editor:
Hailey-Hailey disease (HHD), also known as benign familial pemphigus, is an uncommon autosomal-dominant skin disease.1 Defects in the ATPase type 2C member 1 gene, ATP2C1, result in abnormal intracellular epidermal adherence, and patients experience recurring blisters in skin folds. Longitudinal white streaks of the fingernails also may be present.1 The illness does not appear until puberty and is heightened by the second or third decade of life. Family history often suggests the presence of disease.2 Misdiagnosis of HHD occurs because of a wide spectrum of presentations. The presence of superimposed infections and carcinomas may both obscure and exacerbate this disease.2
Herpes simplex viruse types 1 and 2 (HSV-1 and HSV-2) are DNA viruses that cause common recurrent diseases. Usually, HSV-1 is associated with infection of the mouth and HSV-2 is associated with infection of the genitalia.3 Longitudinal cutaneous lesions manifest as grouped vesicles on an erythematous base. Tzanck smear of herpetic vesicles will reveal the presence of multinucleated giant cells. A direct fluorescent antibody technique also may be used to confirm the diagnosis.3
Erythrodermic HHD disease is a rare condition; moreover, there are only a few reported cases with coexistence of HHD and HSV in the literature.3-6 We report a rare presentation of erythrodermic HHD and coexistent psoriasis with HSV superinfection.
A 69-year-old man presented to an outpatient dermatology clinic for evaluation and treatment of a rash on the scalp, face, back, and lower legs. The patient confirmed a dandruff diagnosis on the scalp and face as well as psoriasis on the trunk and extremities for the last 45 years. He described a history of successful treatment with topical agents and UV light therapy. A family history revealed that the patient’s father and 1 of 2 siblings had a similar rash and “skin problems.” The patient had a medical history of thyroid cancer treated with radiation treatment and a partial thyroidectomy 35 years prior to the current presentation as well as incompletely treated chronic hepatitis C.
A search of medical records revealed a punch biopsy from the posterior neck that demonstrated an acantholytic dyskeratosis with suprabasal acantholysis. Clinicians were unable to differentiate if it was Darier disease (DAR) or HHD. Treatment of the patient’s seborrheic dermatitis and acantholytic disorder was successful at that time with ketoconazole shampoo, ketoconazole cream, desonide cream, and triamcinolone cream. The patient remained stable for 5 years before presenting again to the dermatology clinic for worsening rash despite topical therapies.
At the current presentation, physical examination at the outpatient dermatology clinic revealed few scaly, erythematous, eroded papules distributed on the mid-back; erythematous greasy scaling on the scalp, face, and chest; and pink scaly plaques with white-silvery scale on the anterior lower legs. Histopathology of a specimen from the right mid-back demonstrated acantholysis with suprabasal clefting, hyperkeratosis, and parakeratosis with no dyskeratotic cells identified. The pathologic differential diagnosis included primary acantholytic processes including Grover disease, DAR, HHD, and pemphigus. Pathology from the right shin demonstrated acanthosis, confluent parakeratosis with associated decreased granular cell layer and collections of neutrophils within the stratum corneum, spongiosis, and superficial dermal perivascular chronic inflammation with focal exocytosis and dilated blood vessels in the papillary dermis. The clinical and pathological diagnosis on the lower legs was consistent with psoriasis. Diagnoses of seborrheic dermatitis, psoriasis on the lower legs, and HHD vs DAR on the back and chest were made. The patient was instructed to continue ketoconazole shampoo, ketoconazole cream, and desonide for seborrheic dermatitis; fluocinonide ointment 0.05% to the lower legs for psoriasis; and triamcinolone cream and a bland moisturizer to the back and chest for HHD.
Over the ensuing months, the rash worsened with erythema and scaling affecting more than half of the body surface area. Topical corticosteroids and bland emollients resulted in minimal success. Biologics and acitretin were considered for the psoriasiform dermatitis but avoided due to the patient’s medical history of thyroid cancer and chronic hepatitis C infection. Because the patient described prior success with UV light therapy for psoriasis, he requested light therapy. A subsequent trial of narrowband UVB light therapy initially improved some of the psoriasiform dermatitis on the trunk and extremities; however, after 4 weeks of treatment, the patient described pain in some of the skin and felt he was burned by minimal exposure to light therapy on one particular visit, which caused him to stop light therapy.
Approximately 2 weeks later, the patient presented to the emergency department stating his psoriasis was infected; he was diagnosed with psoriasis with secondary cellulitis and received intravenous vancomycin and piperacillin-tazobactam, with bacterial cultures demonstrating Corynebacterium and methicillin-resistant Staphylococcus aureus. Some improvement was noted in the patient’s skin after antibiotics were initiated, but he continued to describe worsening “burning and pain” throughout the psoriasis lesions. The patient’s care was transferred to the Veterans Affairs hospital where a dermatology inpatient consultation was placed.
Our initial dermatologic examination revealed generalized scaly erythroderma on the neck, trunk, and extremities, sparing the face, palms, and soles (Figure 1). Multiple crusted and intact vesicles also were present overlying the erythematous plaques on the chest, back, and proximal extremities, most grouped in clusters. The patient endorsed new symptoms of pain and burning. Tzanck smear from the abdomen along with shave biopsies from the left flank and right abdomen were performed, and intravenous acyclovir was initiated immediately after these procedures.
Viral cultures were taken but were incorrectly processed by the laboratory. Tzanck smear showed severe acute inflammation with numerous neutrophils, multinucleated giant cells with viral nuclear changes, and positive immunostaining for HSV and negative immunostaining for herpes zoster. Both pathology specimens revealed an intense acute mixed, mainly neutrophilic, inflammatory infiltrate extending into the deeper dermis as well as distorted and necrotic hair follicles, some of which displayed multinucleated epithelial cells with margination of chromatin that were positive for both HSV-1 and HSV-2 and negative for herpes zoster (Figure 2). The positivity of both HSV strains might represent co-infection or could be a cross-reaction of antibodies used in immunohistochemistry to the HSV antigens. There was acantholysis surrounding the ulceration and extending through the full thickness of the epidermis with a dilapidated brick wall pattern (Figure 3) as well as negative immunohistochemical staining for HSV-1 and HSV-2 antigens. The clinical and histological picture together, along with prior clinical and pathological reports, confirmed the diagnoses of acute erythrodermic HHD with HSV superinfection.
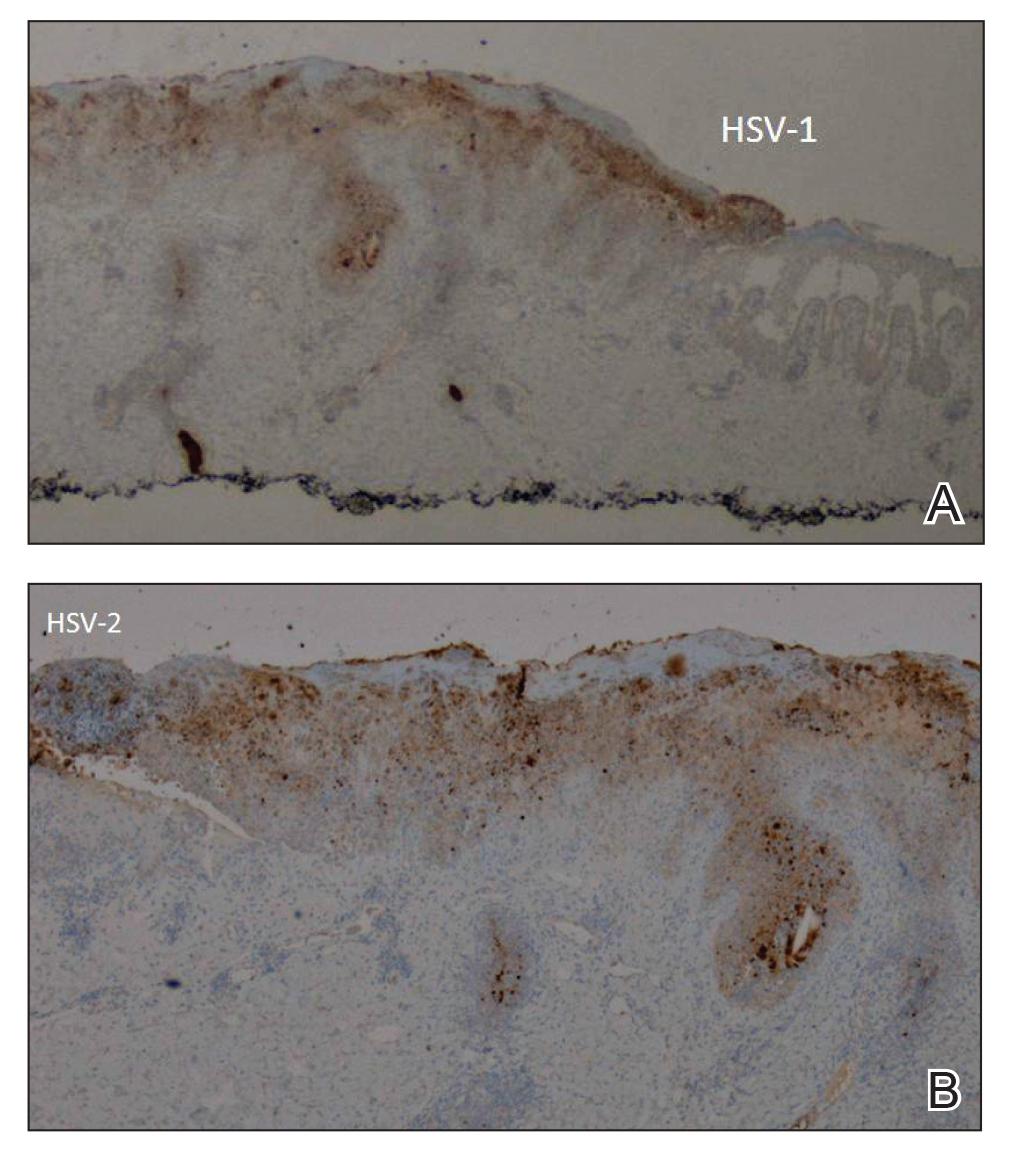
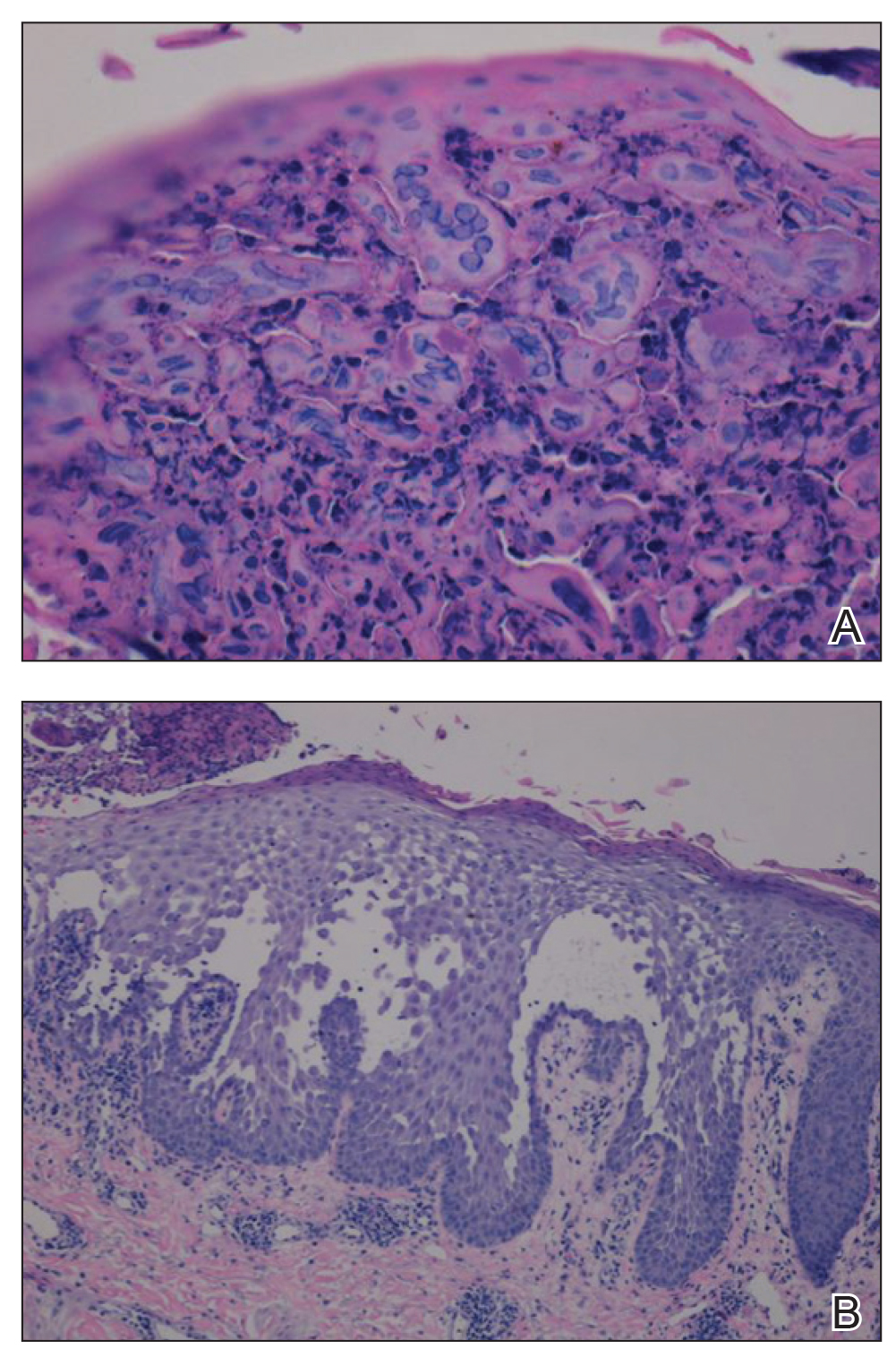
The patient’s condition and pain improved within 24 hours on intravenous acyclovir. On the third day, his lesions were resolving and symptoms improved, so he was transitioned to oral acyclovir and discharged from the hospital. Follow-up in the dermatology outpatient clinic 1 week later revealed that all vesicles and papules had cleared, but the patient was still erythrodermic. Because HHD cannot always be distinguished histologically from other forms of pemphigus but yields a negative immunofluorescence, direct immunofluorescence and indirect immunofluorescence were obtained upon patient follow-up in the clinic and were both negative. Hepatitis C viral loads were undetectable. Consultations to gastroenterology and oncology teams were placed for consideration of systemic agents, and the patient was initiated on oral acitretin 25 mg daily, along with clobetasol as adjuvant therapy for any residual skin plaques. The laboratory results were closely monitored. Within 4 weeks after starting acitretin, the patient’s erythroderma had completely resolved. The patient has remained stable since then, except for one episode of secondary Staphylococcus infection that cleared on oral antibiotics. The patient remains stable and clear on oral acitretin 25 mg daily, with concomitant desonide cream and fluocinonide ointment as needed.
Hailey-Hailey disease is characterized by recurrent episodes of erythema, blisters, and plaques localized to intertriginous and perianal areas.1,2 Patients display a spectrum of lesions that vary in severity.8 Typical histologic examination reveals a dilapidated brick wall appearance. Pathology of well-developed lesions will show suprabasal acantholysis with minimal dyskeratosis.2
The generalized form of HHD is an extremely rare variant of the disease.10 Generalized HHD may resemble acute hypersensitivity reaction, erythema multiforme, and toxic epidermal necrolysis.1 Chronic diseases, such as psoriasis (as in this patient), also may contribute to a clinically confusing picture.8 Hailey-Hailey disease and psoriasis are thought to occasionally koebnerize (isomorphic response) to areas of trauma.16 Our patient experienced widespread erythematous papules and plaques not restricted to skin folds. His skin lesions continued to worsen over several months progressing to erythroderma. The presence of suprabasal acantholysis in a dilapidated brick wall pattern, along with the patient’s history, prior pathology reports, clinical picture, and negative direct immunofluorescence and indirect immunofluorescence studies helped to confirm the diagnosis of erythrodermic HHD.
Hailey-Hailey disease is caused by heterozygous mutations in the ATP2C1 gene on chromosome 3q21-24 coding for a Golgi ATPase called SPCA1 (secretory pathway calcium/manganese-ATPase).9 Subsequent disturbances in cytosolic-Golgi calcium concentrations interfere with epidermal keratinocyte adherence resulting in acantholytic disease. Studies of interfamilial and intrafamilial mutations fail to pinpoint a common mutation pattern among patients with generalized phenotypes,9 which further supports theories that intrinsic or extrinsic factors such as friction, heat, radiation, contact allergens, and infection affect the severity of HHD disease and not the type of mutation.3,9
Generalization of HHD is likely caused by nonspecific triggers in an already genetically disturbed epidermis.10 Interrupted epithelial function exposes skin to infections that exacerbate the underlying disease. Superimposing bacterial infections are commonly reported in HHD. Staphylococcus, Streptococcus, and Candida species colonize the skin and aggravate the disease.11 Much less commonly, HSV superinfection can complicate HHD.3-7 No data are currently available about the frequency or incidence of Herpesviridae in HHD.7 Some studies suggest that UVB light therapy can be an exacerbating factor in DAR and some but not all HHD patients,12,13 while other case reports14,15 document clinically improved responses using phototherapy for patients with HHD. Clinicians should remain suspicious and evaluate for HSV infection in refractory or sudden exacerbation of HHD.7 Furthermore, coexistent psoriasis and HHD also is a rare entity but has been described,8 which illustrates the importance of not attributing all skin manifestations to a previously diagnosed disorder but instead keeping an open mind in case new dermatologic conditions present themselves at a later time.
We present a rare case of erythrodermic HHD and coexistent psoriasis with HSV superinfection. We hope to draw awareness to this association of generalized HHD with both HSV and psoriasis to help clinicians make the correct diagnosis promptly in similar cases in the future.
- Chave TA, Milligan A. Acute generalized Hailey-Hailey disease. Clin Exp Dermatol. 2002;27:290-292.
- Mohr MR, Erdag G, Shada Al, et al. Two patients with Hailey-Hailey disease, multiple primary melanomas, and other cancers. Arch Dermatol. 2011;147:211-215.
- Lee GM, Kim YM, Lee SY, et al. A case of eczema herpeticum with Hailey-Hailey Disease. Ann Dermatol. 2009;21:311-314.
- Zaim MT, Bickers DR. Herpes simplex associated with Hailey-Hailey disease. J Am Acad Dermatol. 1987;17:701-702.
- Peppiatt T, Keefe M, White JE. Hailey-Hailey disease-exacerbation by herpes simplex virus and patch tests. Clin Exp Dermatol. 2006;17:201-202.
- Almeida L, Grossman ME. Benign familial pemphigus complicated by herpes simplex virus. Cutis. 1989;44:261-262.
- Nikkels AF, Delvenne P, Herfs M, et al. Occult herpes simplex virus colonization of bullous dermatitides. Am J Clin Dermatol. 2008;9:163-168.
- Chao SC, Lee JY, Wu MC, et al. A novel splice mutation in the ATP2C1 gene in a woman with concomitant psoriasis vulgaris and disseminated Hailey-Hailey disease. Int J Dermatol. 2012;51:947-951.
- Ikeda S, Shigihara T, Mayuzumi N, et al. Mutations of ATP2C1 in Japanese patients with Hailey-Hailey disease: intrafamilial and interfamilial phenotype variations and lack of correlation with mutation patterns. J Invest Dermatol. 2001;117:1654-1656.
- Marsch W, Stuttgen G. Generalized Hailey-Hailey disease. Br J Dermatol. 1978;99:553-559.
- Friedman-Birnbaum R, Haim S, Marcus S. Generalized familial benign chronic pemphigus. Dermatologica. 1980;161:112-115.
- Richard G, Linse R, Harth W. Hailey-Hailey disease. early detection of heterozygotes by an ultraviolet provocation tests—clinical relevance of the method. Hautarzt. 1993;44:376-379.
- Mayuzumi N, Ikeda S, Kawada H, et al. Effects of ultraviolet B irradiation, proinflammatory cytokines and raised extracellular calcium concentration on the expression of ATP2A2 and ATP2C1. Br J Dermatol. 2005;152:697-701.
- Vanderbeck KA, Giroux L, Murugan NJ, et al. Combined therapeutic use of oral alitretinoin and narrowband ultraviolet-B therapy in the treatment of Hailey-Hailey disease. Dermatol Rep. 2014;6:5604.
- Mizuno K, Hamada T, Hasimoto T, et al. Successful treatment with narrow-band UVB therapy for a case of generalized Hailey-Hailey disease with a novel splice-site mutation in ATP2C1 gene. Dermatol Ther. 2014;27:233-235.
- Thappa DM. The isomorphic phenomenon of Koebner. Indian J Dermatol Venereol Leprol. 2004;70:187-189.
To the Editor:
Hailey-Hailey disease (HHD), also known as benign familial pemphigus, is an uncommon autosomal-dominant skin disease.1 Defects in the ATPase type 2C member 1 gene, ATP2C1, result in abnormal intracellular epidermal adherence, and patients experience recurring blisters in skin folds. Longitudinal white streaks of the fingernails also may be present.1 The illness does not appear until puberty and is heightened by the second or third decade of life. Family history often suggests the presence of disease.2 Misdiagnosis of HHD occurs because of a wide spectrum of presentations. The presence of superimposed infections and carcinomas may both obscure and exacerbate this disease.2
Herpes simplex viruse types 1 and 2 (HSV-1 and HSV-2) are DNA viruses that cause common recurrent diseases. Usually, HSV-1 is associated with infection of the mouth and HSV-2 is associated with infection of the genitalia.3 Longitudinal cutaneous lesions manifest as grouped vesicles on an erythematous base. Tzanck smear of herpetic vesicles will reveal the presence of multinucleated giant cells. A direct fluorescent antibody technique also may be used to confirm the diagnosis.3
Erythrodermic HHD disease is a rare condition; moreover, there are only a few reported cases with coexistence of HHD and HSV in the literature.3-6 We report a rare presentation of erythrodermic HHD and coexistent psoriasis with HSV superinfection.
A 69-year-old man presented to an outpatient dermatology clinic for evaluation and treatment of a rash on the scalp, face, back, and lower legs. The patient confirmed a dandruff diagnosis on the scalp and face as well as psoriasis on the trunk and extremities for the last 45 years. He described a history of successful treatment with topical agents and UV light therapy. A family history revealed that the patient’s father and 1 of 2 siblings had a similar rash and “skin problems.” The patient had a medical history of thyroid cancer treated with radiation treatment and a partial thyroidectomy 35 years prior to the current presentation as well as incompletely treated chronic hepatitis C.
A search of medical records revealed a punch biopsy from the posterior neck that demonstrated an acantholytic dyskeratosis with suprabasal acantholysis. Clinicians were unable to differentiate if it was Darier disease (DAR) or HHD. Treatment of the patient’s seborrheic dermatitis and acantholytic disorder was successful at that time with ketoconazole shampoo, ketoconazole cream, desonide cream, and triamcinolone cream. The patient remained stable for 5 years before presenting again to the dermatology clinic for worsening rash despite topical therapies.
At the current presentation, physical examination at the outpatient dermatology clinic revealed few scaly, erythematous, eroded papules distributed on the mid-back; erythematous greasy scaling on the scalp, face, and chest; and pink scaly plaques with white-silvery scale on the anterior lower legs. Histopathology of a specimen from the right mid-back demonstrated acantholysis with suprabasal clefting, hyperkeratosis, and parakeratosis with no dyskeratotic cells identified. The pathologic differential diagnosis included primary acantholytic processes including Grover disease, DAR, HHD, and pemphigus. Pathology from the right shin demonstrated acanthosis, confluent parakeratosis with associated decreased granular cell layer and collections of neutrophils within the stratum corneum, spongiosis, and superficial dermal perivascular chronic inflammation with focal exocytosis and dilated blood vessels in the papillary dermis. The clinical and pathological diagnosis on the lower legs was consistent with psoriasis. Diagnoses of seborrheic dermatitis, psoriasis on the lower legs, and HHD vs DAR on the back and chest were made. The patient was instructed to continue ketoconazole shampoo, ketoconazole cream, and desonide for seborrheic dermatitis; fluocinonide ointment 0.05% to the lower legs for psoriasis; and triamcinolone cream and a bland moisturizer to the back and chest for HHD.
Over the ensuing months, the rash worsened with erythema and scaling affecting more than half of the body surface area. Topical corticosteroids and bland emollients resulted in minimal success. Biologics and acitretin were considered for the psoriasiform dermatitis but avoided due to the patient’s medical history of thyroid cancer and chronic hepatitis C infection. Because the patient described prior success with UV light therapy for psoriasis, he requested light therapy. A subsequent trial of narrowband UVB light therapy initially improved some of the psoriasiform dermatitis on the trunk and extremities; however, after 4 weeks of treatment, the patient described pain in some of the skin and felt he was burned by minimal exposure to light therapy on one particular visit, which caused him to stop light therapy.
Approximately 2 weeks later, the patient presented to the emergency department stating his psoriasis was infected; he was diagnosed with psoriasis with secondary cellulitis and received intravenous vancomycin and piperacillin-tazobactam, with bacterial cultures demonstrating Corynebacterium and methicillin-resistant Staphylococcus aureus. Some improvement was noted in the patient’s skin after antibiotics were initiated, but he continued to describe worsening “burning and pain” throughout the psoriasis lesions. The patient’s care was transferred to the Veterans Affairs hospital where a dermatology inpatient consultation was placed.
Our initial dermatologic examination revealed generalized scaly erythroderma on the neck, trunk, and extremities, sparing the face, palms, and soles (Figure 1). Multiple crusted and intact vesicles also were present overlying the erythematous plaques on the chest, back, and proximal extremities, most grouped in clusters. The patient endorsed new symptoms of pain and burning. Tzanck smear from the abdomen along with shave biopsies from the left flank and right abdomen were performed, and intravenous acyclovir was initiated immediately after these procedures.
Viral cultures were taken but were incorrectly processed by the laboratory. Tzanck smear showed severe acute inflammation with numerous neutrophils, multinucleated giant cells with viral nuclear changes, and positive immunostaining for HSV and negative immunostaining for herpes zoster. Both pathology specimens revealed an intense acute mixed, mainly neutrophilic, inflammatory infiltrate extending into the deeper dermis as well as distorted and necrotic hair follicles, some of which displayed multinucleated epithelial cells with margination of chromatin that were positive for both HSV-1 and HSV-2 and negative for herpes zoster (Figure 2). The positivity of both HSV strains might represent co-infection or could be a cross-reaction of antibodies used in immunohistochemistry to the HSV antigens. There was acantholysis surrounding the ulceration and extending through the full thickness of the epidermis with a dilapidated brick wall pattern (Figure 3) as well as negative immunohistochemical staining for HSV-1 and HSV-2 antigens. The clinical and histological picture together, along with prior clinical and pathological reports, confirmed the diagnoses of acute erythrodermic HHD with HSV superinfection.
The patient’s condition and pain improved within 24 hours on intravenous acyclovir. On the third day, his lesions were resolving and symptoms improved, so he was transitioned to oral acyclovir and discharged from the hospital. Follow-up in the dermatology outpatient clinic 1 week later revealed that all vesicles and papules had cleared, but the patient was still erythrodermic. Because HHD cannot always be distinguished histologically from other forms of pemphigus but yields a negative immunofluorescence, direct immunofluorescence and indirect immunofluorescence were obtained upon patient follow-up in the clinic and were both negative. Hepatitis C viral loads were undetectable. Consultations to gastroenterology and oncology teams were placed for consideration of systemic agents, and the patient was initiated on oral acitretin 25 mg daily, along with clobetasol as adjuvant therapy for any residual skin plaques. The laboratory results were closely monitored. Within 4 weeks after starting acitretin, the patient’s erythroderma had completely resolved. The patient has remained stable since then, except for one episode of secondary Staphylococcus infection that cleared on oral antibiotics. The patient remains stable and clear on oral acitretin 25 mg daily, with concomitant desonide cream and fluocinonide ointment as needed.
Hailey-Hailey disease is characterized by recurrent episodes of erythema, blisters, and plaques localized to intertriginous and perianal areas.1,2 Patients display a spectrum of lesions that vary in severity.8 Typical histologic examination reveals a dilapidated brick wall appearance. Pathology of well-developed lesions will show suprabasal acantholysis with minimal dyskeratosis.2
The generalized form of HHD is an extremely rare variant of the disease.10 Generalized HHD may resemble acute hypersensitivity reaction, erythema multiforme, and toxic epidermal necrolysis.1 Chronic diseases, such as psoriasis (as in this patient), also may contribute to a clinically confusing picture.8 Hailey-Hailey disease and psoriasis are thought to occasionally koebnerize (isomorphic response) to areas of trauma.16 Our patient experienced widespread erythematous papules and plaques not restricted to skin folds. His skin lesions continued to worsen over several months progressing to erythroderma. The presence of suprabasal acantholysis in a dilapidated brick wall pattern, along with the patient’s history, prior pathology reports, clinical picture, and negative direct immunofluorescence and indirect immunofluorescence studies helped to confirm the diagnosis of erythrodermic HHD.
Hailey-Hailey disease is caused by heterozygous mutations in the ATP2C1 gene on chromosome 3q21-24 coding for a Golgi ATPase called SPCA1 (secretory pathway calcium/manganese-ATPase).9 Subsequent disturbances in cytosolic-Golgi calcium concentrations interfere with epidermal keratinocyte adherence resulting in acantholytic disease. Studies of interfamilial and intrafamilial mutations fail to pinpoint a common mutation pattern among patients with generalized phenotypes,9 which further supports theories that intrinsic or extrinsic factors such as friction, heat, radiation, contact allergens, and infection affect the severity of HHD disease and not the type of mutation.3,9
Generalization of HHD is likely caused by nonspecific triggers in an already genetically disturbed epidermis.10 Interrupted epithelial function exposes skin to infections that exacerbate the underlying disease. Superimposing bacterial infections are commonly reported in HHD. Staphylococcus, Streptococcus, and Candida species colonize the skin and aggravate the disease.11 Much less commonly, HSV superinfection can complicate HHD.3-7 No data are currently available about the frequency or incidence of Herpesviridae in HHD.7 Some studies suggest that UVB light therapy can be an exacerbating factor in DAR and some but not all HHD patients,12,13 while other case reports14,15 document clinically improved responses using phototherapy for patients with HHD. Clinicians should remain suspicious and evaluate for HSV infection in refractory or sudden exacerbation of HHD.7 Furthermore, coexistent psoriasis and HHD also is a rare entity but has been described,8 which illustrates the importance of not attributing all skin manifestations to a previously diagnosed disorder but instead keeping an open mind in case new dermatologic conditions present themselves at a later time.
We present a rare case of erythrodermic HHD and coexistent psoriasis with HSV superinfection. We hope to draw awareness to this association of generalized HHD with both HSV and psoriasis to help clinicians make the correct diagnosis promptly in similar cases in the future.
To the Editor:
Hailey-Hailey disease (HHD), also known as benign familial pemphigus, is an uncommon autosomal-dominant skin disease.1 Defects in the ATPase type 2C member 1 gene, ATP2C1, result in abnormal intracellular epidermal adherence, and patients experience recurring blisters in skin folds. Longitudinal white streaks of the fingernails also may be present.1 The illness does not appear until puberty and is heightened by the second or third decade of life. Family history often suggests the presence of disease.2 Misdiagnosis of HHD occurs because of a wide spectrum of presentations. The presence of superimposed infections and carcinomas may both obscure and exacerbate this disease.2
Herpes simplex viruse types 1 and 2 (HSV-1 and HSV-2) are DNA viruses that cause common recurrent diseases. Usually, HSV-1 is associated with infection of the mouth and HSV-2 is associated with infection of the genitalia.3 Longitudinal cutaneous lesions manifest as grouped vesicles on an erythematous base. Tzanck smear of herpetic vesicles will reveal the presence of multinucleated giant cells. A direct fluorescent antibody technique also may be used to confirm the diagnosis.3
Erythrodermic HHD disease is a rare condition; moreover, there are only a few reported cases with coexistence of HHD and HSV in the literature.3-6 We report a rare presentation of erythrodermic HHD and coexistent psoriasis with HSV superinfection.
A 69-year-old man presented to an outpatient dermatology clinic for evaluation and treatment of a rash on the scalp, face, back, and lower legs. The patient confirmed a dandruff diagnosis on the scalp and face as well as psoriasis on the trunk and extremities for the last 45 years. He described a history of successful treatment with topical agents and UV light therapy. A family history revealed that the patient’s father and 1 of 2 siblings had a similar rash and “skin problems.” The patient had a medical history of thyroid cancer treated with radiation treatment and a partial thyroidectomy 35 years prior to the current presentation as well as incompletely treated chronic hepatitis C.
A search of medical records revealed a punch biopsy from the posterior neck that demonstrated an acantholytic dyskeratosis with suprabasal acantholysis. Clinicians were unable to differentiate if it was Darier disease (DAR) or HHD. Treatment of the patient’s seborrheic dermatitis and acantholytic disorder was successful at that time with ketoconazole shampoo, ketoconazole cream, desonide cream, and triamcinolone cream. The patient remained stable for 5 years before presenting again to the dermatology clinic for worsening rash despite topical therapies.
At the current presentation, physical examination at the outpatient dermatology clinic revealed few scaly, erythematous, eroded papules distributed on the mid-back; erythematous greasy scaling on the scalp, face, and chest; and pink scaly plaques with white-silvery scale on the anterior lower legs. Histopathology of a specimen from the right mid-back demonstrated acantholysis with suprabasal clefting, hyperkeratosis, and parakeratosis with no dyskeratotic cells identified. The pathologic differential diagnosis included primary acantholytic processes including Grover disease, DAR, HHD, and pemphigus. Pathology from the right shin demonstrated acanthosis, confluent parakeratosis with associated decreased granular cell layer and collections of neutrophils within the stratum corneum, spongiosis, and superficial dermal perivascular chronic inflammation with focal exocytosis and dilated blood vessels in the papillary dermis. The clinical and pathological diagnosis on the lower legs was consistent with psoriasis. Diagnoses of seborrheic dermatitis, psoriasis on the lower legs, and HHD vs DAR on the back and chest were made. The patient was instructed to continue ketoconazole shampoo, ketoconazole cream, and desonide for seborrheic dermatitis; fluocinonide ointment 0.05% to the lower legs for psoriasis; and triamcinolone cream and a bland moisturizer to the back and chest for HHD.
Over the ensuing months, the rash worsened with erythema and scaling affecting more than half of the body surface area. Topical corticosteroids and bland emollients resulted in minimal success. Biologics and acitretin were considered for the psoriasiform dermatitis but avoided due to the patient’s medical history of thyroid cancer and chronic hepatitis C infection. Because the patient described prior success with UV light therapy for psoriasis, he requested light therapy. A subsequent trial of narrowband UVB light therapy initially improved some of the psoriasiform dermatitis on the trunk and extremities; however, after 4 weeks of treatment, the patient described pain in some of the skin and felt he was burned by minimal exposure to light therapy on one particular visit, which caused him to stop light therapy.
Approximately 2 weeks later, the patient presented to the emergency department stating his psoriasis was infected; he was diagnosed with psoriasis with secondary cellulitis and received intravenous vancomycin and piperacillin-tazobactam, with bacterial cultures demonstrating Corynebacterium and methicillin-resistant Staphylococcus aureus. Some improvement was noted in the patient’s skin after antibiotics were initiated, but he continued to describe worsening “burning and pain” throughout the psoriasis lesions. The patient’s care was transferred to the Veterans Affairs hospital where a dermatology inpatient consultation was placed.
Our initial dermatologic examination revealed generalized scaly erythroderma on the neck, trunk, and extremities, sparing the face, palms, and soles (Figure 1). Multiple crusted and intact vesicles also were present overlying the erythematous plaques on the chest, back, and proximal extremities, most grouped in clusters. The patient endorsed new symptoms of pain and burning. Tzanck smear from the abdomen along with shave biopsies from the left flank and right abdomen were performed, and intravenous acyclovir was initiated immediately after these procedures.
Viral cultures were taken but were incorrectly processed by the laboratory. Tzanck smear showed severe acute inflammation with numerous neutrophils, multinucleated giant cells with viral nuclear changes, and positive immunostaining for HSV and negative immunostaining for herpes zoster. Both pathology specimens revealed an intense acute mixed, mainly neutrophilic, inflammatory infiltrate extending into the deeper dermis as well as distorted and necrotic hair follicles, some of which displayed multinucleated epithelial cells with margination of chromatin that were positive for both HSV-1 and HSV-2 and negative for herpes zoster (Figure 2). The positivity of both HSV strains might represent co-infection or could be a cross-reaction of antibodies used in immunohistochemistry to the HSV antigens. There was acantholysis surrounding the ulceration and extending through the full thickness of the epidermis with a dilapidated brick wall pattern (Figure 3) as well as negative immunohistochemical staining for HSV-1 and HSV-2 antigens. The clinical and histological picture together, along with prior clinical and pathological reports, confirmed the diagnoses of acute erythrodermic HHD with HSV superinfection.
The patient’s condition and pain improved within 24 hours on intravenous acyclovir. On the third day, his lesions were resolving and symptoms improved, so he was transitioned to oral acyclovir and discharged from the hospital. Follow-up in the dermatology outpatient clinic 1 week later revealed that all vesicles and papules had cleared, but the patient was still erythrodermic. Because HHD cannot always be distinguished histologically from other forms of pemphigus but yields a negative immunofluorescence, direct immunofluorescence and indirect immunofluorescence were obtained upon patient follow-up in the clinic and were both negative. Hepatitis C viral loads were undetectable. Consultations to gastroenterology and oncology teams were placed for consideration of systemic agents, and the patient was initiated on oral acitretin 25 mg daily, along with clobetasol as adjuvant therapy for any residual skin plaques. The laboratory results were closely monitored. Within 4 weeks after starting acitretin, the patient’s erythroderma had completely resolved. The patient has remained stable since then, except for one episode of secondary Staphylococcus infection that cleared on oral antibiotics. The patient remains stable and clear on oral acitretin 25 mg daily, with concomitant desonide cream and fluocinonide ointment as needed.
Hailey-Hailey disease is characterized by recurrent episodes of erythema, blisters, and plaques localized to intertriginous and perianal areas.1,2 Patients display a spectrum of lesions that vary in severity.8 Typical histologic examination reveals a dilapidated brick wall appearance. Pathology of well-developed lesions will show suprabasal acantholysis with minimal dyskeratosis.2
The generalized form of HHD is an extremely rare variant of the disease.10 Generalized HHD may resemble acute hypersensitivity reaction, erythema multiforme, and toxic epidermal necrolysis.1 Chronic diseases, such as psoriasis (as in this patient), also may contribute to a clinically confusing picture.8 Hailey-Hailey disease and psoriasis are thought to occasionally koebnerize (isomorphic response) to areas of trauma.16 Our patient experienced widespread erythematous papules and plaques not restricted to skin folds. His skin lesions continued to worsen over several months progressing to erythroderma. The presence of suprabasal acantholysis in a dilapidated brick wall pattern, along with the patient’s history, prior pathology reports, clinical picture, and negative direct immunofluorescence and indirect immunofluorescence studies helped to confirm the diagnosis of erythrodermic HHD.
Hailey-Hailey disease is caused by heterozygous mutations in the ATP2C1 gene on chromosome 3q21-24 coding for a Golgi ATPase called SPCA1 (secretory pathway calcium/manganese-ATPase).9 Subsequent disturbances in cytosolic-Golgi calcium concentrations interfere with epidermal keratinocyte adherence resulting in acantholytic disease. Studies of interfamilial and intrafamilial mutations fail to pinpoint a common mutation pattern among patients with generalized phenotypes,9 which further supports theories that intrinsic or extrinsic factors such as friction, heat, radiation, contact allergens, and infection affect the severity of HHD disease and not the type of mutation.3,9
Generalization of HHD is likely caused by nonspecific triggers in an already genetically disturbed epidermis.10 Interrupted epithelial function exposes skin to infections that exacerbate the underlying disease. Superimposing bacterial infections are commonly reported in HHD. Staphylococcus, Streptococcus, and Candida species colonize the skin and aggravate the disease.11 Much less commonly, HSV superinfection can complicate HHD.3-7 No data are currently available about the frequency or incidence of Herpesviridae in HHD.7 Some studies suggest that UVB light therapy can be an exacerbating factor in DAR and some but not all HHD patients,12,13 while other case reports14,15 document clinically improved responses using phototherapy for patients with HHD. Clinicians should remain suspicious and evaluate for HSV infection in refractory or sudden exacerbation of HHD.7 Furthermore, coexistent psoriasis and HHD also is a rare entity but has been described,8 which illustrates the importance of not attributing all skin manifestations to a previously diagnosed disorder but instead keeping an open mind in case new dermatologic conditions present themselves at a later time.
We present a rare case of erythrodermic HHD and coexistent psoriasis with HSV superinfection. We hope to draw awareness to this association of generalized HHD with both HSV and psoriasis to help clinicians make the correct diagnosis promptly in similar cases in the future.
- Chave TA, Milligan A. Acute generalized Hailey-Hailey disease. Clin Exp Dermatol. 2002;27:290-292.
- Mohr MR, Erdag G, Shada Al, et al. Two patients with Hailey-Hailey disease, multiple primary melanomas, and other cancers. Arch Dermatol. 2011;147:211-215.
- Lee GM, Kim YM, Lee SY, et al. A case of eczema herpeticum with Hailey-Hailey Disease. Ann Dermatol. 2009;21:311-314.
- Zaim MT, Bickers DR. Herpes simplex associated with Hailey-Hailey disease. J Am Acad Dermatol. 1987;17:701-702.
- Peppiatt T, Keefe M, White JE. Hailey-Hailey disease-exacerbation by herpes simplex virus and patch tests. Clin Exp Dermatol. 2006;17:201-202.
- Almeida L, Grossman ME. Benign familial pemphigus complicated by herpes simplex virus. Cutis. 1989;44:261-262.
- Nikkels AF, Delvenne P, Herfs M, et al. Occult herpes simplex virus colonization of bullous dermatitides. Am J Clin Dermatol. 2008;9:163-168.
- Chao SC, Lee JY, Wu MC, et al. A novel splice mutation in the ATP2C1 gene in a woman with concomitant psoriasis vulgaris and disseminated Hailey-Hailey disease. Int J Dermatol. 2012;51:947-951.
- Ikeda S, Shigihara T, Mayuzumi N, et al. Mutations of ATP2C1 in Japanese patients with Hailey-Hailey disease: intrafamilial and interfamilial phenotype variations and lack of correlation with mutation patterns. J Invest Dermatol. 2001;117:1654-1656.
- Marsch W, Stuttgen G. Generalized Hailey-Hailey disease. Br J Dermatol. 1978;99:553-559.
- Friedman-Birnbaum R, Haim S, Marcus S. Generalized familial benign chronic pemphigus. Dermatologica. 1980;161:112-115.
- Richard G, Linse R, Harth W. Hailey-Hailey disease. early detection of heterozygotes by an ultraviolet provocation tests—clinical relevance of the method. Hautarzt. 1993;44:376-379.
- Mayuzumi N, Ikeda S, Kawada H, et al. Effects of ultraviolet B irradiation, proinflammatory cytokines and raised extracellular calcium concentration on the expression of ATP2A2 and ATP2C1. Br J Dermatol. 2005;152:697-701.
- Vanderbeck KA, Giroux L, Murugan NJ, et al. Combined therapeutic use of oral alitretinoin and narrowband ultraviolet-B therapy in the treatment of Hailey-Hailey disease. Dermatol Rep. 2014;6:5604.
- Mizuno K, Hamada T, Hasimoto T, et al. Successful treatment with narrow-band UVB therapy for a case of generalized Hailey-Hailey disease with a novel splice-site mutation in ATP2C1 gene. Dermatol Ther. 2014;27:233-235.
- Thappa DM. The isomorphic phenomenon of Koebner. Indian J Dermatol Venereol Leprol. 2004;70:187-189.
- Chave TA, Milligan A. Acute generalized Hailey-Hailey disease. Clin Exp Dermatol. 2002;27:290-292.
- Mohr MR, Erdag G, Shada Al, et al. Two patients with Hailey-Hailey disease, multiple primary melanomas, and other cancers. Arch Dermatol. 2011;147:211-215.
- Lee GM, Kim YM, Lee SY, et al. A case of eczema herpeticum with Hailey-Hailey Disease. Ann Dermatol. 2009;21:311-314.
- Zaim MT, Bickers DR. Herpes simplex associated with Hailey-Hailey disease. J Am Acad Dermatol. 1987;17:701-702.
- Peppiatt T, Keefe M, White JE. Hailey-Hailey disease-exacerbation by herpes simplex virus and patch tests. Clin Exp Dermatol. 2006;17:201-202.
- Almeida L, Grossman ME. Benign familial pemphigus complicated by herpes simplex virus. Cutis. 1989;44:261-262.
- Nikkels AF, Delvenne P, Herfs M, et al. Occult herpes simplex virus colonization of bullous dermatitides. Am J Clin Dermatol. 2008;9:163-168.
- Chao SC, Lee JY, Wu MC, et al. A novel splice mutation in the ATP2C1 gene in a woman with concomitant psoriasis vulgaris and disseminated Hailey-Hailey disease. Int J Dermatol. 2012;51:947-951.
- Ikeda S, Shigihara T, Mayuzumi N, et al. Mutations of ATP2C1 in Japanese patients with Hailey-Hailey disease: intrafamilial and interfamilial phenotype variations and lack of correlation with mutation patterns. J Invest Dermatol. 2001;117:1654-1656.
- Marsch W, Stuttgen G. Generalized Hailey-Hailey disease. Br J Dermatol. 1978;99:553-559.
- Friedman-Birnbaum R, Haim S, Marcus S. Generalized familial benign chronic pemphigus. Dermatologica. 1980;161:112-115.
- Richard G, Linse R, Harth W. Hailey-Hailey disease. early detection of heterozygotes by an ultraviolet provocation tests—clinical relevance of the method. Hautarzt. 1993;44:376-379.
- Mayuzumi N, Ikeda S, Kawada H, et al. Effects of ultraviolet B irradiation, proinflammatory cytokines and raised extracellular calcium concentration on the expression of ATP2A2 and ATP2C1. Br J Dermatol. 2005;152:697-701.
- Vanderbeck KA, Giroux L, Murugan NJ, et al. Combined therapeutic use of oral alitretinoin and narrowband ultraviolet-B therapy in the treatment of Hailey-Hailey disease. Dermatol Rep. 2014;6:5604.
- Mizuno K, Hamada T, Hasimoto T, et al. Successful treatment with narrow-band UVB therapy for a case of generalized Hailey-Hailey disease with a novel splice-site mutation in ATP2C1 gene. Dermatol Ther. 2014;27:233-235.
- Thappa DM. The isomorphic phenomenon of Koebner. Indian J Dermatol Venereol Leprol. 2004;70:187-189.
Practice Points
- Misdiagnosis of Hailey-Hailey disease (HHD) occurs because of a wide spectrum of presentations.
- Hailey-Hailey disease and psoriasis are thought to occasionally koebnerize (isomorphic response) to areas of trauma.
- Clinicians should remain suspicious and evaluate for herpes simplex virus infection in refractory or sudden exacerbation of HHD.
Variations in Preference for Topical Vehicles Among Demographic Groups
Topical medication is a mainstay in the treatment of dermatologic conditions. Adherence to medication regimens can be challenging in patients requiring long-term topical treatment, and nonadherence is multifactorial. A major modifiable contributing factor is patient dissatisfaction with the vehicle used. Medications often have options for different topical preparations. Therefore, it is important to consider patient preference when prescribing topical treatments to maximize adherence, ensure patient satisfaction, and optimize outcomes.
We hypothesized that notable differences exist among demographic groups regarding preference for topical vehicles. Little research has been conducted to delineate trends. This study aimed to identify variations in preference for creams, lotions, and ointments by age, gender, and ethnicity.
Methods
Data were collected through surveys distributed to all patients seen at the Truman Medical Center University Health Dermatology Clinic in Kansas City, Missouri, between September 2018 and June 2019. The study was approved by the University of Missouri Kansas City institutional review board. An estimated response rate of 95% was achieved. Each patient was informed that the survey was voluntary and anonymous, and declining to complete the survey had no effect on the care provided. Each patient completed only 1 survey and returned it to a collection box before departing from clinic.
In the survey, patients provided demographic information, including age, gender, and ethnicity. Age groups included patients younger than 40 years, 40 to 60 years, and older than 60 years. Gender groups included male and female. Ethnicity included white, black, Hispanic/Latino, and Asian/Pacific Islander or other. Patients then chose 1 of 3 options for topical vehicle preference: cream, lotion, or ointment. Each of these options was accompanied by a brief description of the vehicle, a photograph, and examples of common commercial products to aid in decision-making. The expected values were calculated based on a probability distribution under the assumption that variables have no association. Therefore, the discrepancy between the expected value and the observed value was used to describe the significance of the association between variables.
Data were analyzed using χ2 tests with the aid of a statistician. P<.05 was considered statistically significant.
Results
A total of 404 surveys were collected and recorded. Data showed statistically significant trends in each demographic parameter.
Age
First, we analyzed differences in preference based on age (Table 1). Of 404 patients, 163 were younger than 40 years, 171 were aged 40 to 60 years, and 70 were older than 60 years. Patients younger than 40 years preferred lotion (68 vs 46.0 expected). Patients aged 40 to 60 years showed preference for cream (83 vs 76.6 expected) and ointment (56 vs 46.1 expected). Patients older than 60 years preferred cream (41 vs 31.4 expected). These findings were statistically significant (P<.0001).
Gender
Next, we evaluated variations based on gender (Table 2). Of 404 patients, 254 were female and 150 were male. Females preferred cream (127 vs 113.8 expected). Males exhibited preference for lotion (50 vs 42.3 expected) and ointment (46 vs 40.5 expected). Differences between genders were statistically significant (P=.023).

Ethnicity
We then analyzed preferences based on ethnicity (Table 3). Of 404 patients, 30 were Hispanic/Latino, 26 were Asian/Pacific Islander or other, 227 were white, and 121 were black. Hispanic/Latino patients showed equivocal findings, aligning with expected counts. Asian/Pacific Islander or other patients exhibited slight preferences for cream (14 vs 11.6 expected) and lotion (10 vs 7.3 expected). White patients preferred cream (119 vs 101.7 expected) and lotion (82 vs 64.1 expected). Black patients showed strong preference for ointment (72 vs 32.6 expected). Differences in preferences based on ethnicity were statistically significant (P<.0001).

Comment
Topical medication is a mainstay of dermatologic therapy. Many topical preparations (or vehicles) exist, including ointments, creams, lotions, gels, solutions, and foams. Vehicle type not only influences bioavailability of the prepared medication but also has a notable impact on adherence and subsequent efficacy of the topical therapy.
Medication adherence is especially challenging in dermatology, as topical medications play a central role in treatment. Compliance with the medication regimen is paramount in treatment efficacy.1 In dermatology, adherence with oral medications is higher than it is for topical medications2; various factors contribute to this difference. Compliance may decline with topical treatment due to time-consuming application, misunderstanding about the disease or the treatment regimen, frequency of administration, dissatisfaction with efficacy or appearance, and other variables.3
Other factors have been found to be important to topical medication adherence; younger age, female gender, marriage, employment, nonsmoking, nondrinking, and higher cognitive ability were associated with higher topical medication adherence.4 Our study focused on one factor: identification of demographic-specific preferences that might have implications on adherence within the studied demographic groups.
It is known that individual preferences exist when patients are choosing a topical preparation. However, a PubMed search of articles indexed for MEDLINE using the terms topical, vehicle, preparation, adherence, and preference revealed few studies that examined the preference for topical vehicle by age, gender, or ethnicity.
Existing studies have examined preferences for topical preparations based on specific disease states; this literature, albeit limited, demonstrates that preferences for topical product formulations vary among acne, atopic dermatitis, and plaque psoriasis patients.5 Other studies focus on specific patient populations or medications. For example, one study found that preference for corticosteroid vehicles among psoriasis patients was highly variable and choice of vehicle was critical to adherence.6 Another study highlighted differences in vehicle choice between younger and older age groups with psoriasis.7
Given the limited data overall, it was our goal to determine if any patterns of preference existed by age, gender, or ethnicity, regardless of disease state or indication for topical product. Importantly, over-the-counter products—cosmetic or otherwise—were not differentiated from prescribed topical medications. Our survey elucidated significant differences in preference by age, gender, and ethnicity.
Notable Findings
Regarding age, patients younger than 40 years preferred lotion, patients aged 40 to 60 years preferred cream, and patients older than 60 years preferred cream. Analysis based on gender showed that females preferred cream, and males preferred lotion and ointment. Analysis based on ethnicity most notably demonstrated a strong preference for ointment in black patients while showing preference for cream in white patients.
Potential Biases and Pitfalls
Limitations of this study included the small Hispanic/Latino and Asian/Pacific Islander populations surveyed, possible misunderstanding of the survey by respondents, and the potential for surveys being filled out twice by the same patient. Future surveys could be conducted over a longer period to increase the total sample size and to better characterize less-represented populations, such as Hispanic and Asian patients. To avoid repeat participation, the first question of the survey asked patients to indicate if they had previously completed the survey and instructed patients who had to return the repeat survey to the front desk.
To limit other errors, our survey included concise accessible descriptions of each preparation along with clear representative photographs and examples of common brands. Still, it is possible that some mistakes could have been made while patients filled out the survey based on comprehension deficits, oversight, or other reasons. It also is possible that preference might vary individually depending on the indication of the topical product—cosmetic or therapeutic—or even by anatomic site of application. Neither of these considerations was assessed specifically in our survey.
Conclusion
Our hope is that this study helps practitioners better anticipate topical preferences among patients with the ultimate goal of increasing medication adherence and patient outcomes. Nevertheless, although these general trends can provide helpful guidance, we acknowledge that individual preferences vary, and care should always be patient centered.
Acknowledgment
We thank An-Lin Cheng, PhD (Kansas City, Missouri), for assistance with the statistical analysis.
- Kircik LH. Vehicles always matter. J Drugs Dermatol. 2019;18:s99.
- Furue M, Onozuka D, Takeuchi S, et al. Poor adherence to oral andtopical medication in 3096 dermatological patients as assessed by the Morisky Medication Adherence Scale-8. Br J Dermatol. 2015;172:272-275.
- Tan X, Feldman SR, Chang, J, et al. Topical drug delivery systems in dermatology: a review of patient adherence issues. Expert Opin Drug Deliv. 2012;9:1263-1271.
- Ahn CS, Culp L, Huang WW, et al. Adherence in dermatology. J Dermatolog Treat. 2017;28:94-103.
- Eastman WJ, Malahias S, Delconte J, et al. Assessing attributes of topical vehicles for the treatment of acne, atopic dermatitis, and plaque psoriasis. Cutis. 2014;94:46-53.
- Felix K, Unrue E, Inyang M, et al. Patients preferences for different corticosteroid vehicles are highly variable. J Dermatolog Treat. 2019;31:147-151.
- Hong C-H, Papp KA, Lophaven KW, et al. Patients with psoriasis have different preferences for topical therapy, highlighting the importance of individualized treatment approaches: randomized phase IIIb PSO-INSIGHTFUL study. J Eur Acad Dermatol Venereol. 2017;31:1876-1883.
Topical medication is a mainstay in the treatment of dermatologic conditions. Adherence to medication regimens can be challenging in patients requiring long-term topical treatment, and nonadherence is multifactorial. A major modifiable contributing factor is patient dissatisfaction with the vehicle used. Medications often have options for different topical preparations. Therefore, it is important to consider patient preference when prescribing topical treatments to maximize adherence, ensure patient satisfaction, and optimize outcomes.
We hypothesized that notable differences exist among demographic groups regarding preference for topical vehicles. Little research has been conducted to delineate trends. This study aimed to identify variations in preference for creams, lotions, and ointments by age, gender, and ethnicity.
Methods
Data were collected through surveys distributed to all patients seen at the Truman Medical Center University Health Dermatology Clinic in Kansas City, Missouri, between September 2018 and June 2019. The study was approved by the University of Missouri Kansas City institutional review board. An estimated response rate of 95% was achieved. Each patient was informed that the survey was voluntary and anonymous, and declining to complete the survey had no effect on the care provided. Each patient completed only 1 survey and returned it to a collection box before departing from clinic.
In the survey, patients provided demographic information, including age, gender, and ethnicity. Age groups included patients younger than 40 years, 40 to 60 years, and older than 60 years. Gender groups included male and female. Ethnicity included white, black, Hispanic/Latino, and Asian/Pacific Islander or other. Patients then chose 1 of 3 options for topical vehicle preference: cream, lotion, or ointment. Each of these options was accompanied by a brief description of the vehicle, a photograph, and examples of common commercial products to aid in decision-making. The expected values were calculated based on a probability distribution under the assumption that variables have no association. Therefore, the discrepancy between the expected value and the observed value was used to describe the significance of the association between variables.
Data were analyzed using χ2 tests with the aid of a statistician. P<.05 was considered statistically significant.
Results
A total of 404 surveys were collected and recorded. Data showed statistically significant trends in each demographic parameter.
Age
First, we analyzed differences in preference based on age (Table 1). Of 404 patients, 163 were younger than 40 years, 171 were aged 40 to 60 years, and 70 were older than 60 years. Patients younger than 40 years preferred lotion (68 vs 46.0 expected). Patients aged 40 to 60 years showed preference for cream (83 vs 76.6 expected) and ointment (56 vs 46.1 expected). Patients older than 60 years preferred cream (41 vs 31.4 expected). These findings were statistically significant (P<.0001).
Gender
Next, we evaluated variations based on gender (Table 2). Of 404 patients, 254 were female and 150 were male. Females preferred cream (127 vs 113.8 expected). Males exhibited preference for lotion (50 vs 42.3 expected) and ointment (46 vs 40.5 expected). Differences between genders were statistically significant (P=.023).
Ethnicity
We then analyzed preferences based on ethnicity (Table 3). Of 404 patients, 30 were Hispanic/Latino, 26 were Asian/Pacific Islander or other, 227 were white, and 121 were black. Hispanic/Latino patients showed equivocal findings, aligning with expected counts. Asian/Pacific Islander or other patients exhibited slight preferences for cream (14 vs 11.6 expected) and lotion (10 vs 7.3 expected). White patients preferred cream (119 vs 101.7 expected) and lotion (82 vs 64.1 expected). Black patients showed strong preference for ointment (72 vs 32.6 expected). Differences in preferences based on ethnicity were statistically significant (P<.0001).
Comment
Topical medication is a mainstay of dermatologic therapy. Many topical preparations (or vehicles) exist, including ointments, creams, lotions, gels, solutions, and foams. Vehicle type not only influences bioavailability of the prepared medication but also has a notable impact on adherence and subsequent efficacy of the topical therapy.
Medication adherence is especially challenging in dermatology, as topical medications play a central role in treatment. Compliance with the medication regimen is paramount in treatment efficacy.1 In dermatology, adherence with oral medications is higher than it is for topical medications2; various factors contribute to this difference. Compliance may decline with topical treatment due to time-consuming application, misunderstanding about the disease or the treatment regimen, frequency of administration, dissatisfaction with efficacy or appearance, and other variables.3
Other factors have been found to be important to topical medication adherence; younger age, female gender, marriage, employment, nonsmoking, nondrinking, and higher cognitive ability were associated with higher topical medication adherence.4 Our study focused on one factor: identification of demographic-specific preferences that might have implications on adherence within the studied demographic groups.
It is known that individual preferences exist when patients are choosing a topical preparation. However, a PubMed search of articles indexed for MEDLINE using the terms topical, vehicle, preparation, adherence, and preference revealed few studies that examined the preference for topical vehicle by age, gender, or ethnicity.
Existing studies have examined preferences for topical preparations based on specific disease states; this literature, albeit limited, demonstrates that preferences for topical product formulations vary among acne, atopic dermatitis, and plaque psoriasis patients.5 Other studies focus on specific patient populations or medications. For example, one study found that preference for corticosteroid vehicles among psoriasis patients was highly variable and choice of vehicle was critical to adherence.6 Another study highlighted differences in vehicle choice between younger and older age groups with psoriasis.7
Given the limited data overall, it was our goal to determine if any patterns of preference existed by age, gender, or ethnicity, regardless of disease state or indication for topical product. Importantly, over-the-counter products—cosmetic or otherwise—were not differentiated from prescribed topical medications. Our survey elucidated significant differences in preference by age, gender, and ethnicity.
Notable Findings
Regarding age, patients younger than 40 years preferred lotion, patients aged 40 to 60 years preferred cream, and patients older than 60 years preferred cream. Analysis based on gender showed that females preferred cream, and males preferred lotion and ointment. Analysis based on ethnicity most notably demonstrated a strong preference for ointment in black patients while showing preference for cream in white patients.
Potential Biases and Pitfalls
Limitations of this study included the small Hispanic/Latino and Asian/Pacific Islander populations surveyed, possible misunderstanding of the survey by respondents, and the potential for surveys being filled out twice by the same patient. Future surveys could be conducted over a longer period to increase the total sample size and to better characterize less-represented populations, such as Hispanic and Asian patients. To avoid repeat participation, the first question of the survey asked patients to indicate if they had previously completed the survey and instructed patients who had to return the repeat survey to the front desk.
To limit other errors, our survey included concise accessible descriptions of each preparation along with clear representative photographs and examples of common brands. Still, it is possible that some mistakes could have been made while patients filled out the survey based on comprehension deficits, oversight, or other reasons. It also is possible that preference might vary individually depending on the indication of the topical product—cosmetic or therapeutic—or even by anatomic site of application. Neither of these considerations was assessed specifically in our survey.
Conclusion
Our hope is that this study helps practitioners better anticipate topical preferences among patients with the ultimate goal of increasing medication adherence and patient outcomes. Nevertheless, although these general trends can provide helpful guidance, we acknowledge that individual preferences vary, and care should always be patient centered.
Acknowledgment
We thank An-Lin Cheng, PhD (Kansas City, Missouri), for assistance with the statistical analysis.
Topical medication is a mainstay in the treatment of dermatologic conditions. Adherence to medication regimens can be challenging in patients requiring long-term topical treatment, and nonadherence is multifactorial. A major modifiable contributing factor is patient dissatisfaction with the vehicle used. Medications often have options for different topical preparations. Therefore, it is important to consider patient preference when prescribing topical treatments to maximize adherence, ensure patient satisfaction, and optimize outcomes.
We hypothesized that notable differences exist among demographic groups regarding preference for topical vehicles. Little research has been conducted to delineate trends. This study aimed to identify variations in preference for creams, lotions, and ointments by age, gender, and ethnicity.
Methods
Data were collected through surveys distributed to all patients seen at the Truman Medical Center University Health Dermatology Clinic in Kansas City, Missouri, between September 2018 and June 2019. The study was approved by the University of Missouri Kansas City institutional review board. An estimated response rate of 95% was achieved. Each patient was informed that the survey was voluntary and anonymous, and declining to complete the survey had no effect on the care provided. Each patient completed only 1 survey and returned it to a collection box before departing from clinic.
In the survey, patients provided demographic information, including age, gender, and ethnicity. Age groups included patients younger than 40 years, 40 to 60 years, and older than 60 years. Gender groups included male and female. Ethnicity included white, black, Hispanic/Latino, and Asian/Pacific Islander or other. Patients then chose 1 of 3 options for topical vehicle preference: cream, lotion, or ointment. Each of these options was accompanied by a brief description of the vehicle, a photograph, and examples of common commercial products to aid in decision-making. The expected values were calculated based on a probability distribution under the assumption that variables have no association. Therefore, the discrepancy between the expected value and the observed value was used to describe the significance of the association between variables.
Data were analyzed using χ2 tests with the aid of a statistician. P<.05 was considered statistically significant.
Results
A total of 404 surveys were collected and recorded. Data showed statistically significant trends in each demographic parameter.
Age
First, we analyzed differences in preference based on age (Table 1). Of 404 patients, 163 were younger than 40 years, 171 were aged 40 to 60 years, and 70 were older than 60 years. Patients younger than 40 years preferred lotion (68 vs 46.0 expected). Patients aged 40 to 60 years showed preference for cream (83 vs 76.6 expected) and ointment (56 vs 46.1 expected). Patients older than 60 years preferred cream (41 vs 31.4 expected). These findings were statistically significant (P<.0001).
Gender
Next, we evaluated variations based on gender (Table 2). Of 404 patients, 254 were female and 150 were male. Females preferred cream (127 vs 113.8 expected). Males exhibited preference for lotion (50 vs 42.3 expected) and ointment (46 vs 40.5 expected). Differences between genders were statistically significant (P=.023).
Ethnicity
We then analyzed preferences based on ethnicity (Table 3). Of 404 patients, 30 were Hispanic/Latino, 26 were Asian/Pacific Islander or other, 227 were white, and 121 were black. Hispanic/Latino patients showed equivocal findings, aligning with expected counts. Asian/Pacific Islander or other patients exhibited slight preferences for cream (14 vs 11.6 expected) and lotion (10 vs 7.3 expected). White patients preferred cream (119 vs 101.7 expected) and lotion (82 vs 64.1 expected). Black patients showed strong preference for ointment (72 vs 32.6 expected). Differences in preferences based on ethnicity were statistically significant (P<.0001).
Comment
Topical medication is a mainstay of dermatologic therapy. Many topical preparations (or vehicles) exist, including ointments, creams, lotions, gels, solutions, and foams. Vehicle type not only influences bioavailability of the prepared medication but also has a notable impact on adherence and subsequent efficacy of the topical therapy.
Medication adherence is especially challenging in dermatology, as topical medications play a central role in treatment. Compliance with the medication regimen is paramount in treatment efficacy.1 In dermatology, adherence with oral medications is higher than it is for topical medications2; various factors contribute to this difference. Compliance may decline with topical treatment due to time-consuming application, misunderstanding about the disease or the treatment regimen, frequency of administration, dissatisfaction with efficacy or appearance, and other variables.3
Other factors have been found to be important to topical medication adherence; younger age, female gender, marriage, employment, nonsmoking, nondrinking, and higher cognitive ability were associated with higher topical medication adherence.4 Our study focused on one factor: identification of demographic-specific preferences that might have implications on adherence within the studied demographic groups.
It is known that individual preferences exist when patients are choosing a topical preparation. However, a PubMed search of articles indexed for MEDLINE using the terms topical, vehicle, preparation, adherence, and preference revealed few studies that examined the preference for topical vehicle by age, gender, or ethnicity.
Existing studies have examined preferences for topical preparations based on specific disease states; this literature, albeit limited, demonstrates that preferences for topical product formulations vary among acne, atopic dermatitis, and plaque psoriasis patients.5 Other studies focus on specific patient populations or medications. For example, one study found that preference for corticosteroid vehicles among psoriasis patients was highly variable and choice of vehicle was critical to adherence.6 Another study highlighted differences in vehicle choice between younger and older age groups with psoriasis.7
Given the limited data overall, it was our goal to determine if any patterns of preference existed by age, gender, or ethnicity, regardless of disease state or indication for topical product. Importantly, over-the-counter products—cosmetic or otherwise—were not differentiated from prescribed topical medications. Our survey elucidated significant differences in preference by age, gender, and ethnicity.
Notable Findings
Regarding age, patients younger than 40 years preferred lotion, patients aged 40 to 60 years preferred cream, and patients older than 60 years preferred cream. Analysis based on gender showed that females preferred cream, and males preferred lotion and ointment. Analysis based on ethnicity most notably demonstrated a strong preference for ointment in black patients while showing preference for cream in white patients.
Potential Biases and Pitfalls
Limitations of this study included the small Hispanic/Latino and Asian/Pacific Islander populations surveyed, possible misunderstanding of the survey by respondents, and the potential for surveys being filled out twice by the same patient. Future surveys could be conducted over a longer period to increase the total sample size and to better characterize less-represented populations, such as Hispanic and Asian patients. To avoid repeat participation, the first question of the survey asked patients to indicate if they had previously completed the survey and instructed patients who had to return the repeat survey to the front desk.
To limit other errors, our survey included concise accessible descriptions of each preparation along with clear representative photographs and examples of common brands. Still, it is possible that some mistakes could have been made while patients filled out the survey based on comprehension deficits, oversight, or other reasons. It also is possible that preference might vary individually depending on the indication of the topical product—cosmetic or therapeutic—or even by anatomic site of application. Neither of these considerations was assessed specifically in our survey.
Conclusion
Our hope is that this study helps practitioners better anticipate topical preferences among patients with the ultimate goal of increasing medication adherence and patient outcomes. Nevertheless, although these general trends can provide helpful guidance, we acknowledge that individual preferences vary, and care should always be patient centered.
Acknowledgment
We thank An-Lin Cheng, PhD (Kansas City, Missouri), for assistance with the statistical analysis.
- Kircik LH. Vehicles always matter. J Drugs Dermatol. 2019;18:s99.
- Furue M, Onozuka D, Takeuchi S, et al. Poor adherence to oral andtopical medication in 3096 dermatological patients as assessed by the Morisky Medication Adherence Scale-8. Br J Dermatol. 2015;172:272-275.
- Tan X, Feldman SR, Chang, J, et al. Topical drug delivery systems in dermatology: a review of patient adherence issues. Expert Opin Drug Deliv. 2012;9:1263-1271.
- Ahn CS, Culp L, Huang WW, et al. Adherence in dermatology. J Dermatolog Treat. 2017;28:94-103.
- Eastman WJ, Malahias S, Delconte J, et al. Assessing attributes of topical vehicles for the treatment of acne, atopic dermatitis, and plaque psoriasis. Cutis. 2014;94:46-53.
- Felix K, Unrue E, Inyang M, et al. Patients preferences for different corticosteroid vehicles are highly variable. J Dermatolog Treat. 2019;31:147-151.
- Hong C-H, Papp KA, Lophaven KW, et al. Patients with psoriasis have different preferences for topical therapy, highlighting the importance of individualized treatment approaches: randomized phase IIIb PSO-INSIGHTFUL study. J Eur Acad Dermatol Venereol. 2017;31:1876-1883.
- Kircik LH. Vehicles always matter. J Drugs Dermatol. 2019;18:s99.
- Furue M, Onozuka D, Takeuchi S, et al. Poor adherence to oral andtopical medication in 3096 dermatological patients as assessed by the Morisky Medication Adherence Scale-8. Br J Dermatol. 2015;172:272-275.
- Tan X, Feldman SR, Chang, J, et al. Topical drug delivery systems in dermatology: a review of patient adherence issues. Expert Opin Drug Deliv. 2012;9:1263-1271.
- Ahn CS, Culp L, Huang WW, et al. Adherence in dermatology. J Dermatolog Treat. 2017;28:94-103.
- Eastman WJ, Malahias S, Delconte J, et al. Assessing attributes of topical vehicles for the treatment of acne, atopic dermatitis, and plaque psoriasis. Cutis. 2014;94:46-53.
- Felix K, Unrue E, Inyang M, et al. Patients preferences for different corticosteroid vehicles are highly variable. J Dermatolog Treat. 2019;31:147-151.
- Hong C-H, Papp KA, Lophaven KW, et al. Patients with psoriasis have different preferences for topical therapy, highlighting the importance of individualized treatment approaches: randomized phase IIIb PSO-INSIGHTFUL study. J Eur Acad Dermatol Venereol. 2017;31:1876-1883.
Practice Points
- Variations exist in preference for topical vehicles by age group, gender, and ethnicity.
- Identifying and utilizing preferred treatment options can help maximize patient outcomes.
How to Obtain a Dermatology Residency: A Guide Targeted to Underrepresented in Medicine Medical Students
There has been increasing attention focused on the lack of diversity within dermatology academic and residency programs.1-6 Several factors have been identified as contributing to this narrow pipeline of qualified applicants, including lack of mentorship, delayed exposure to the field, implicit bias, and lack of an overall holistic review of applications with overemphasis on board scores.1,5 In an effort to provide guidance to underrepresented in medicine (UIM) students who are interested in dermatology, the Skin of Color Society (SOCS) has created a detailed, step-by-step guide on how to obtain a position in a dermatology residency program,7 which was modeled after a similar resource created by the American Academy of Orthopaedic Surgeons.8 Here, we highlight the main SOCS recommendations to help guide medical students through a systematic approach to becoming successful applicants for dermatology residency.
Start Early
Competitive fields such as dermatology require intentional efforts starting at the beginning of medical school. Regardless of what specialty is right for you, begin by constructing a well-rounded application for residency immediately. Start by shadowing dermatologists and attending Grand Rounds held in your institution’s dermatology department to ensure that this field is right for you. Students are encouraged to meet with academic advisors and upperclassmen to seek guidance on gaining early exposure to dermatology at their home institutions (or nearby programs) during their first year. As a platform for learning about community-based dermatology activities, join your school’s Dermatology Interest Group, keeping in mind that an executive position in such a group can help foster relationships with faculty and residents of the dermatology department. A long-term commitment to community service also contributes to your depth as an applicant. Getting involved early helps students uncover health disparities in medicine and allows time to formulate ideas to implement change. Forming a well-rounded application mandates maintaining good academic standing, and students should prioritize mastering the curriculum, excelling in clinical rotations, and studying for the US Medical Licensing Examination (USMLE).
Choose a Mentor
The summer between your first and second years of medical school is an opportune time to explore research opportunities. Students successfully complete research by taking ownership of a project, efficiently meeting deadlines, maintaining contact with research mentors by quickly responding to emails, and producing quality work. Research outside of dermatology also is valued. Research mentors often provide future letters of recommendation, so commit to doing an outstanding job. For those finding it difficult to locate a mentor, consider searching the American Academy of Dermatology (AAD)(https://www.aad.org/mentorship/) or SOCS (https://skinofcolorsociety.org/) websites. The AAD has an established Diversity Mentorship Program (https://www.aad.org/member/career/volunteer/diversity-mentorship) that provides members with direct guidance from dermatologists for 4 weeks. Students use this time to conduct research, learn more about the specialty, and foster a relationship with their mentor. Students can apply any year of medical school; however, the typical awardee usually is a third-year or fourth-year student. The AAD may provide a stipend to help offset expenses.
Prepare for Boards
Second year is a continuation of the agenda set forth in first year, now with the focus shifting toward board preparation and excelling in clinical core didactics and rotations. According to data from the 2018 National Resident Matching Program,9 the mean USMLE Step 1 score for US allopathic senior medical students who matched into dermatology was 249 compared to 241 who did not match into dermatology. However, the mean score is just that—a mean—and people have matched with lower scores. Do not be intimidated by this number; instead, be driven to commit the time and resources to master the content and do your personal best on the USMLE Step examinations. Given the shift in some programs for earlier clinical exposure and postponement of boards until the third year, the recommendations in this timeline can be catered to fit a medical student’s specific situation.
Build Your Application
The third year of medical school is a busy year. Prepare for third-year clinical rotations by speaking with upperclassmen and clinical preceptors as you progress through your rotations. Evaluations and recommendations are weighed heavily by residency program directors, as this information is used to ascertain your clinical abilities. Seek feedback from your preceptors early and often with a sincere attempt to integrate suggested improvements. Schedule a dermatology rotation at your home institution after completing the core rotations. Although they are not required, applicants may complete away rotations early in their fourth year; the application period for visiting student learning opportunities typically opens April 1 of the third year, if not earlier. Free resources are available to help prepare for your dermatology rotations. Start by reviewing the Basic Dermatology Curriculum on the AAD website (https://www.aad.org/member/education/residents/bdc). Make contributions to
Interviewing for Residency
During your fourth year of medical school, you will be completing dermatology rotations, submitting your applications through the Electronic Residency Application Service, and interviewing with residency programs. When deciding which programs to apply to, consider referencing the American Medical Association Residency and Fellowship Database (https://freida.ama-assn.org/Freida/#/). Also keep in mind that, depending on your competitiveness, you should expect to receive 1 interview for every 10 programs you apply to, thus the application process can be quite costly. It is highly encouraged that you ask for letters of recommendation prior to August 15 and that you submit your applications by September 15. Complete mock interviews with a mentor and research commonly asked questions. Prior to your interview day, you want to spend time researching the program, browsing faculty publications, and reviewing your application. Dress in a comfortable suit, shoes, and minimal accessories; arrive early knowing that your interview begins even before you meet your interviewer, so treat everyone you meet with respect. Refrain from speaking to anyone in a casual way and have questions prepared to ask each interviewer. After your interviews, be sure to write thank you notes or emails if a program does not specifically discourage postinterview communication. Continuous efforts will improve your success in obtaining a dermatology residency position.
Final Thoughts
Recent articles have underscored and emphasized the importance of diversity in our field, with a call to action to find meaningful and overdue solutions.2,6 We acknowledge the important role that mentors play in providing timely, honest, and encouraging guidance to UIM students interested in careers in dermatology. We hope to provide readily available and detailed guidance to these students on how they can present themselves as excellent and qualified applicants through this summary and other platforms.
Acknowledgment
The authors would like to thank the members of the SOCS Diversity Task Force for their assistance in creating the original guide.
- Chen A, Shinkai K. Rethinking how we select dermatology applicants—turning the tide. JAMA Dermatol. 2017;153:259-260.
- Granstein RD, Cornelius L, Shinkai K. Diversity in dermatology—a call for action. JAMA Dermatol. 2017;153:499-500.
- Imadojemu S, James WD. Increasing African American representation in dermatology. JAMA Dermatol. 2016;152:15-16.
- Pandya AG, Alexis AF, Berger TG, et al. Increasing racial and ethnic diversity in dermatology: a call to action. J Am Acad Dermatol. 2016;74:584-587.
- Pritchett EN, Pandya AG, Ferguson NN, et al. Diversity in dermatology: roadmap for improvement. J Am Acad Dermatol. 2018;79:337-341.
- Taylor SC. Meeting the unique dermatologic needs of black patients [published online August 21, 2019]. JAMA Dermatol. doi:10.1001/jamadermatol.2019.1963.
- Skin of Color Society. How to obtain a position in a dermatology residency program. https://skinofcolorsociety.org/wp-content/uploads/2019/10/How-to-Obtain-a-Position-in-a-Dermatology-Residency-Program-10-08-2019.pdf. Accessed June 24, 2020.
- American Academy of Orthopaedic Surgeons. How to obtain an orthopedic residency by the American Academy of Orthopaedic Surgeons. https://www.aaos.org/globalassets/about/diversity/how-to-obtain-an-orthopaedic-residency.pdf. Accessed June 24, 2020.
- Results and Data—2018 Main Residency Match. Washington, DC: National Resident Matching Program; 2018. Published April 2018. Accessed June 24, 2020.
There has been increasing attention focused on the lack of diversity within dermatology academic and residency programs.1-6 Several factors have been identified as contributing to this narrow pipeline of qualified applicants, including lack of mentorship, delayed exposure to the field, implicit bias, and lack of an overall holistic review of applications with overemphasis on board scores.1,5 In an effort to provide guidance to underrepresented in medicine (UIM) students who are interested in dermatology, the Skin of Color Society (SOCS) has created a detailed, step-by-step guide on how to obtain a position in a dermatology residency program,7 which was modeled after a similar resource created by the American Academy of Orthopaedic Surgeons.8 Here, we highlight the main SOCS recommendations to help guide medical students through a systematic approach to becoming successful applicants for dermatology residency.
Start Early
Competitive fields such as dermatology require intentional efforts starting at the beginning of medical school. Regardless of what specialty is right for you, begin by constructing a well-rounded application for residency immediately. Start by shadowing dermatologists and attending Grand Rounds held in your institution’s dermatology department to ensure that this field is right for you. Students are encouraged to meet with academic advisors and upperclassmen to seek guidance on gaining early exposure to dermatology at their home institutions (or nearby programs) during their first year. As a platform for learning about community-based dermatology activities, join your school’s Dermatology Interest Group, keeping in mind that an executive position in such a group can help foster relationships with faculty and residents of the dermatology department. A long-term commitment to community service also contributes to your depth as an applicant. Getting involved early helps students uncover health disparities in medicine and allows time to formulate ideas to implement change. Forming a well-rounded application mandates maintaining good academic standing, and students should prioritize mastering the curriculum, excelling in clinical rotations, and studying for the US Medical Licensing Examination (USMLE).
Choose a Mentor
The summer between your first and second years of medical school is an opportune time to explore research opportunities. Students successfully complete research by taking ownership of a project, efficiently meeting deadlines, maintaining contact with research mentors by quickly responding to emails, and producing quality work. Research outside of dermatology also is valued. Research mentors often provide future letters of recommendation, so commit to doing an outstanding job. For those finding it difficult to locate a mentor, consider searching the American Academy of Dermatology (AAD)(https://www.aad.org/mentorship/) or SOCS (https://skinofcolorsociety.org/) websites. The AAD has an established Diversity Mentorship Program (https://www.aad.org/member/career/volunteer/diversity-mentorship) that provides members with direct guidance from dermatologists for 4 weeks. Students use this time to conduct research, learn more about the specialty, and foster a relationship with their mentor. Students can apply any year of medical school; however, the typical awardee usually is a third-year or fourth-year student. The AAD may provide a stipend to help offset expenses.
Prepare for Boards
Second year is a continuation of the agenda set forth in first year, now with the focus shifting toward board preparation and excelling in clinical core didactics and rotations. According to data from the 2018 National Resident Matching Program,9 the mean USMLE Step 1 score for US allopathic senior medical students who matched into dermatology was 249 compared to 241 who did not match into dermatology. However, the mean score is just that—a mean—and people have matched with lower scores. Do not be intimidated by this number; instead, be driven to commit the time and resources to master the content and do your personal best on the USMLE Step examinations. Given the shift in some programs for earlier clinical exposure and postponement of boards until the third year, the recommendations in this timeline can be catered to fit a medical student’s specific situation.
Build Your Application
The third year of medical school is a busy year. Prepare for third-year clinical rotations by speaking with upperclassmen and clinical preceptors as you progress through your rotations. Evaluations and recommendations are weighed heavily by residency program directors, as this information is used to ascertain your clinical abilities. Seek feedback from your preceptors early and often with a sincere attempt to integrate suggested improvements. Schedule a dermatology rotation at your home institution after completing the core rotations. Although they are not required, applicants may complete away rotations early in their fourth year; the application period for visiting student learning opportunities typically opens April 1 of the third year, if not earlier. Free resources are available to help prepare for your dermatology rotations. Start by reviewing the Basic Dermatology Curriculum on the AAD website (https://www.aad.org/member/education/residents/bdc). Make contributions to
Interviewing for Residency
During your fourth year of medical school, you will be completing dermatology rotations, submitting your applications through the Electronic Residency Application Service, and interviewing with residency programs. When deciding which programs to apply to, consider referencing the American Medical Association Residency and Fellowship Database (https://freida.ama-assn.org/Freida/#/). Also keep in mind that, depending on your competitiveness, you should expect to receive 1 interview for every 10 programs you apply to, thus the application process can be quite costly. It is highly encouraged that you ask for letters of recommendation prior to August 15 and that you submit your applications by September 15. Complete mock interviews with a mentor and research commonly asked questions. Prior to your interview day, you want to spend time researching the program, browsing faculty publications, and reviewing your application. Dress in a comfortable suit, shoes, and minimal accessories; arrive early knowing that your interview begins even before you meet your interviewer, so treat everyone you meet with respect. Refrain from speaking to anyone in a casual way and have questions prepared to ask each interviewer. After your interviews, be sure to write thank you notes or emails if a program does not specifically discourage postinterview communication. Continuous efforts will improve your success in obtaining a dermatology residency position.
Final Thoughts
Recent articles have underscored and emphasized the importance of diversity in our field, with a call to action to find meaningful and overdue solutions.2,6 We acknowledge the important role that mentors play in providing timely, honest, and encouraging guidance to UIM students interested in careers in dermatology. We hope to provide readily available and detailed guidance to these students on how they can present themselves as excellent and qualified applicants through this summary and other platforms.
Acknowledgment
The authors would like to thank the members of the SOCS Diversity Task Force for their assistance in creating the original guide.
There has been increasing attention focused on the lack of diversity within dermatology academic and residency programs.1-6 Several factors have been identified as contributing to this narrow pipeline of qualified applicants, including lack of mentorship, delayed exposure to the field, implicit bias, and lack of an overall holistic review of applications with overemphasis on board scores.1,5 In an effort to provide guidance to underrepresented in medicine (UIM) students who are interested in dermatology, the Skin of Color Society (SOCS) has created a detailed, step-by-step guide on how to obtain a position in a dermatology residency program,7 which was modeled after a similar resource created by the American Academy of Orthopaedic Surgeons.8 Here, we highlight the main SOCS recommendations to help guide medical students through a systematic approach to becoming successful applicants for dermatology residency.
Start Early
Competitive fields such as dermatology require intentional efforts starting at the beginning of medical school. Regardless of what specialty is right for you, begin by constructing a well-rounded application for residency immediately. Start by shadowing dermatologists and attending Grand Rounds held in your institution’s dermatology department to ensure that this field is right for you. Students are encouraged to meet with academic advisors and upperclassmen to seek guidance on gaining early exposure to dermatology at their home institutions (or nearby programs) during their first year. As a platform for learning about community-based dermatology activities, join your school’s Dermatology Interest Group, keeping in mind that an executive position in such a group can help foster relationships with faculty and residents of the dermatology department. A long-term commitment to community service also contributes to your depth as an applicant. Getting involved early helps students uncover health disparities in medicine and allows time to formulate ideas to implement change. Forming a well-rounded application mandates maintaining good academic standing, and students should prioritize mastering the curriculum, excelling in clinical rotations, and studying for the US Medical Licensing Examination (USMLE).
Choose a Mentor
The summer between your first and second years of medical school is an opportune time to explore research opportunities. Students successfully complete research by taking ownership of a project, efficiently meeting deadlines, maintaining contact with research mentors by quickly responding to emails, and producing quality work. Research outside of dermatology also is valued. Research mentors often provide future letters of recommendation, so commit to doing an outstanding job. For those finding it difficult to locate a mentor, consider searching the American Academy of Dermatology (AAD)(https://www.aad.org/mentorship/) or SOCS (https://skinofcolorsociety.org/) websites. The AAD has an established Diversity Mentorship Program (https://www.aad.org/member/career/volunteer/diversity-mentorship) that provides members with direct guidance from dermatologists for 4 weeks. Students use this time to conduct research, learn more about the specialty, and foster a relationship with their mentor. Students can apply any year of medical school; however, the typical awardee usually is a third-year or fourth-year student. The AAD may provide a stipend to help offset expenses.
Prepare for Boards
Second year is a continuation of the agenda set forth in first year, now with the focus shifting toward board preparation and excelling in clinical core didactics and rotations. According to data from the 2018 National Resident Matching Program,9 the mean USMLE Step 1 score for US allopathic senior medical students who matched into dermatology was 249 compared to 241 who did not match into dermatology. However, the mean score is just that—a mean—and people have matched with lower scores. Do not be intimidated by this number; instead, be driven to commit the time and resources to master the content and do your personal best on the USMLE Step examinations. Given the shift in some programs for earlier clinical exposure and postponement of boards until the third year, the recommendations in this timeline can be catered to fit a medical student’s specific situation.
Build Your Application
The third year of medical school is a busy year. Prepare for third-year clinical rotations by speaking with upperclassmen and clinical preceptors as you progress through your rotations. Evaluations and recommendations are weighed heavily by residency program directors, as this information is used to ascertain your clinical abilities. Seek feedback from your preceptors early and often with a sincere attempt to integrate suggested improvements. Schedule a dermatology rotation at your home institution after completing the core rotations. Although they are not required, applicants may complete away rotations early in their fourth year; the application period for visiting student learning opportunities typically opens April 1 of the third year, if not earlier. Free resources are available to help prepare for your dermatology rotations. Start by reviewing the Basic Dermatology Curriculum on the AAD website (https://www.aad.org/member/education/residents/bdc). Make contributions to
Interviewing for Residency
During your fourth year of medical school, you will be completing dermatology rotations, submitting your applications through the Electronic Residency Application Service, and interviewing with residency programs. When deciding which programs to apply to, consider referencing the American Medical Association Residency and Fellowship Database (https://freida.ama-assn.org/Freida/#/). Also keep in mind that, depending on your competitiveness, you should expect to receive 1 interview for every 10 programs you apply to, thus the application process can be quite costly. It is highly encouraged that you ask for letters of recommendation prior to August 15 and that you submit your applications by September 15. Complete mock interviews with a mentor and research commonly asked questions. Prior to your interview day, you want to spend time researching the program, browsing faculty publications, and reviewing your application. Dress in a comfortable suit, shoes, and minimal accessories; arrive early knowing that your interview begins even before you meet your interviewer, so treat everyone you meet with respect. Refrain from speaking to anyone in a casual way and have questions prepared to ask each interviewer. After your interviews, be sure to write thank you notes or emails if a program does not specifically discourage postinterview communication. Continuous efforts will improve your success in obtaining a dermatology residency position.
Final Thoughts
Recent articles have underscored and emphasized the importance of diversity in our field, with a call to action to find meaningful and overdue solutions.2,6 We acknowledge the important role that mentors play in providing timely, honest, and encouraging guidance to UIM students interested in careers in dermatology. We hope to provide readily available and detailed guidance to these students on how they can present themselves as excellent and qualified applicants through this summary and other platforms.
Acknowledgment
The authors would like to thank the members of the SOCS Diversity Task Force for their assistance in creating the original guide.
- Chen A, Shinkai K. Rethinking how we select dermatology applicants—turning the tide. JAMA Dermatol. 2017;153:259-260.
- Granstein RD, Cornelius L, Shinkai K. Diversity in dermatology—a call for action. JAMA Dermatol. 2017;153:499-500.
- Imadojemu S, James WD. Increasing African American representation in dermatology. JAMA Dermatol. 2016;152:15-16.
- Pandya AG, Alexis AF, Berger TG, et al. Increasing racial and ethnic diversity in dermatology: a call to action. J Am Acad Dermatol. 2016;74:584-587.
- Pritchett EN, Pandya AG, Ferguson NN, et al. Diversity in dermatology: roadmap for improvement. J Am Acad Dermatol. 2018;79:337-341.
- Taylor SC. Meeting the unique dermatologic needs of black patients [published online August 21, 2019]. JAMA Dermatol. doi:10.1001/jamadermatol.2019.1963.
- Skin of Color Society. How to obtain a position in a dermatology residency program. https://skinofcolorsociety.org/wp-content/uploads/2019/10/How-to-Obtain-a-Position-in-a-Dermatology-Residency-Program-10-08-2019.pdf. Accessed June 24, 2020.
- American Academy of Orthopaedic Surgeons. How to obtain an orthopedic residency by the American Academy of Orthopaedic Surgeons. https://www.aaos.org/globalassets/about/diversity/how-to-obtain-an-orthopaedic-residency.pdf. Accessed June 24, 2020.
- Results and Data—2018 Main Residency Match. Washington, DC: National Resident Matching Program; 2018. Published April 2018. Accessed June 24, 2020.
- Chen A, Shinkai K. Rethinking how we select dermatology applicants—turning the tide. JAMA Dermatol. 2017;153:259-260.
- Granstein RD, Cornelius L, Shinkai K. Diversity in dermatology—a call for action. JAMA Dermatol. 2017;153:499-500.
- Imadojemu S, James WD. Increasing African American representation in dermatology. JAMA Dermatol. 2016;152:15-16.
- Pandya AG, Alexis AF, Berger TG, et al. Increasing racial and ethnic diversity in dermatology: a call to action. J Am Acad Dermatol. 2016;74:584-587.
- Pritchett EN, Pandya AG, Ferguson NN, et al. Diversity in dermatology: roadmap for improvement. J Am Acad Dermatol. 2018;79:337-341.
- Taylor SC. Meeting the unique dermatologic needs of black patients [published online August 21, 2019]. JAMA Dermatol. doi:10.1001/jamadermatol.2019.1963.
- Skin of Color Society. How to obtain a position in a dermatology residency program. https://skinofcolorsociety.org/wp-content/uploads/2019/10/How-to-Obtain-a-Position-in-a-Dermatology-Residency-Program-10-08-2019.pdf. Accessed June 24, 2020.
- American Academy of Orthopaedic Surgeons. How to obtain an orthopedic residency by the American Academy of Orthopaedic Surgeons. https://www.aaos.org/globalassets/about/diversity/how-to-obtain-an-orthopaedic-residency.pdf. Accessed June 24, 2020.
- Results and Data—2018 Main Residency Match. Washington, DC: National Resident Matching Program; 2018. Published April 2018. Accessed June 24, 2020.
Practice Points
- Students interested in dermatology are encouraged to seek mentorship, strive for their academic best, and maintain their unique personal interests that make them a well-rounded applicant.
- Increasing diversity in dermatology requires initiative from students as well as dermatologists who are willing to mentor and sponsor.