User login
Cutis is a peer-reviewed clinical journal for the dermatologist, allergist, and general practitioner published monthly since 1965. Concise clinical articles present the practical side of dermatology, helping physicians to improve patient care. Cutis is referenced in Index Medicus/MEDLINE and is written and edited by industry leaders.
ass lick
assault rifle
balls
ballsac
black jack
bleach
Boko Haram
bondage
causas
cheap
child abuse
cocaine
compulsive behaviors
cost of miracles
cunt
Daech
display network stats
drug paraphernalia
explosion
fart
fda and death
fda AND warn
fda AND warning
fda AND warns
feom
fuck
gambling
gfc
gun
human trafficking
humira AND expensive
illegal
ISIL
ISIS
Islamic caliphate
Islamic state
madvocate
masturbation
mixed martial arts
MMA
molestation
national rifle association
NRA
nsfw
nuccitelli
pedophile
pedophilia
poker
porn
porn
pornography
psychedelic drug
recreational drug
sex slave rings
shit
slot machine
snort
substance abuse
terrorism
terrorist
texarkana
Texas hold 'em
UFC
section[contains(@class, 'nav-hidden')]
section[contains(@class, 'nav-hidden active')
A peer-reviewed, indexed journal for dermatologists with original research, image quizzes, cases and reviews, and columns.
Don't Let the Bedbugs Bite: An Unusual Presentation of Bedbug Infestation Resulting in Life-Threatening Anemia
To the Editor:
A 61-year-old man presented to the emergency department with a rash on the right leg, generalized pruritus, and chest pain. The patient described intermittent exertional pressure-like chest pain over the last few days but had no known prior cardiac history. He also noted worsening edema of the right leg with erythema. Three months prior he had been hospitalized for a similar presentation and was diagnosed with cellulitis of the right leg. The patient was treated with a course of trimethoprim-sulfamethoxazole and permethrin cream for presumed scabies and followed up with dermatology for the persistent generalized pruritic rash and cellulitis. At that time, he was diagnosed with stasis dermatitis with dermatitis neglecta and excoriations. He was educated on general hygiene and treated with triamcinolone, hydrophilic ointment, and pramoxine lotion for pruritus. He also was empirically treated again for scabies.
At the current presentation, preliminary investigation showed profound anemia with a hemoglobin level of 6.2 g/dL (baseline hemoglobin level 3 months prior, 13.1 g/dL). He was subsequently admitted to the general medicine ward for further investigation of severe symptomatic anemia. A medical history revealed moderate chronic obstructive pulmonary disease, hypertension, gastroesophageal reflux disease, xerosis, and fracture of the right ankle following open reduction internal fixation 6 years prior to admission. There was no history of blood loss, antiplatelet agents, or anticoagulants. He was on disability and lived in a single-room occupancy hotel. He did not report any high-risk sexual behaviors or abuse of alcohol or drugs. He actively smoked 1.5 packs of cigarettes per day for the last 30 years. He denied any allergies.
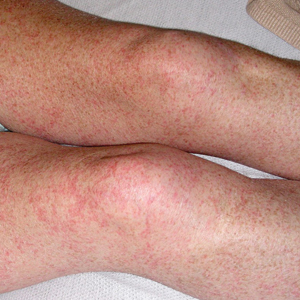
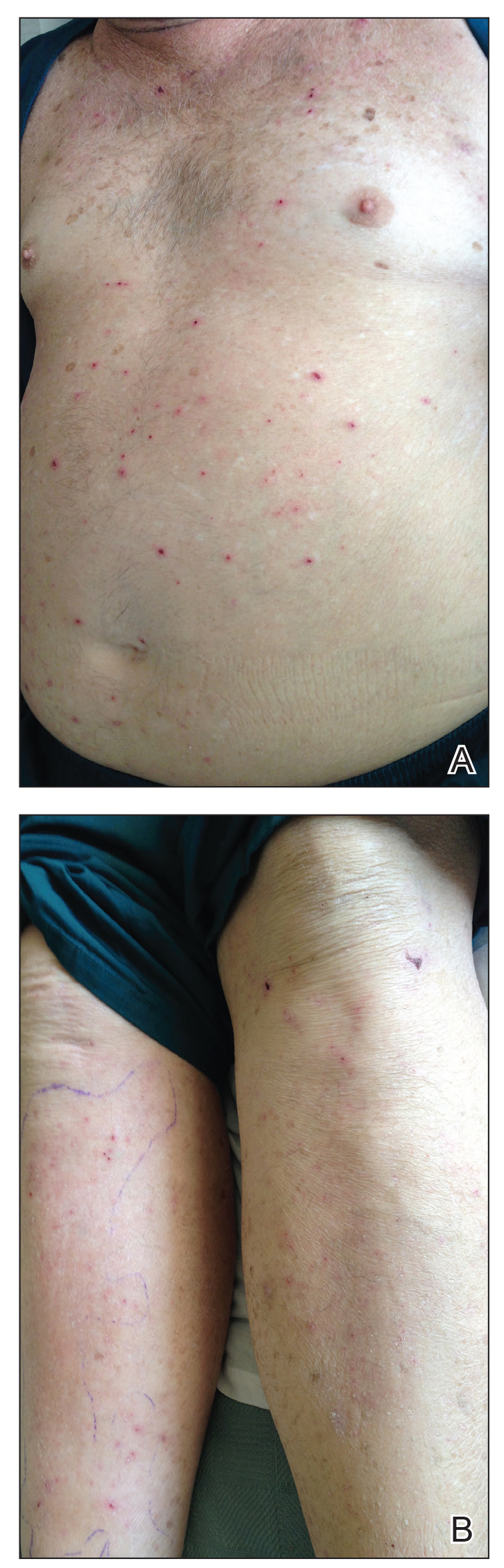
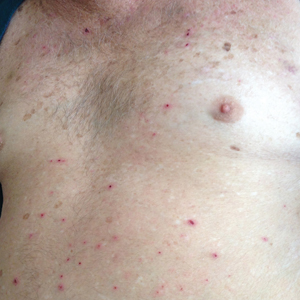
Physical examination revealed the patient was afebrile, nontoxic, disheveled, and in no acute distress. He had anicteric sclera and pale conjunctiva. The right leg appeared more erythematous and edematous compared to the left leg but without warmth or tenderness to palpation. He had innumerable 4- to 5-mm, erythematous, excoriated papules on the skin (Figure). His bed sheets were noted to have multiple rusty-black specks thought to be related to the crusted lesions. Physical examination was otherwise unremarkable.
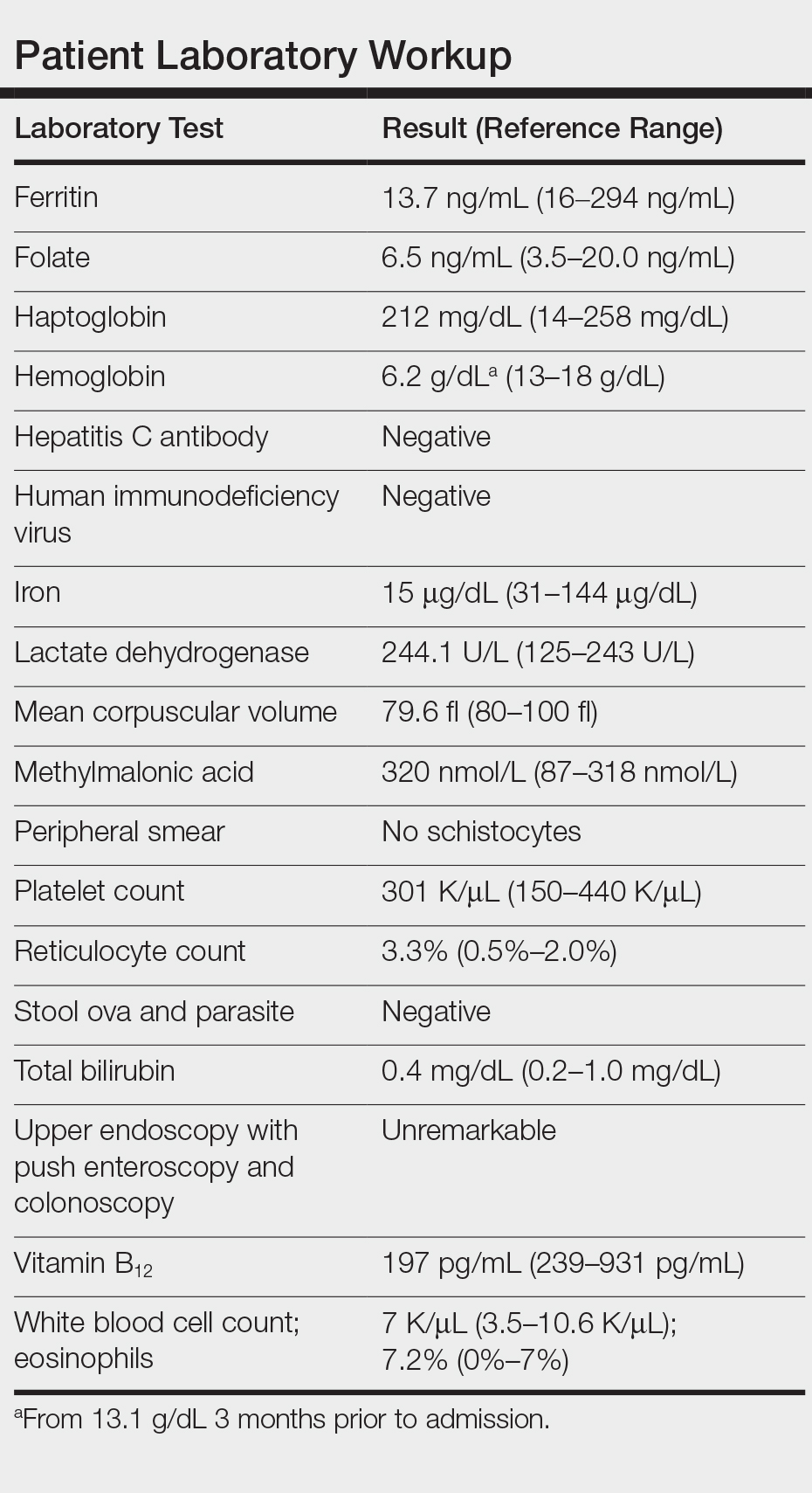
Laboratory workup revealed severe iron-deficiency anemia without any evidence of hemolysis, marrow suppression, infection, or immune compromise (Table). He had a vitamin B12 deficiency (197 pg/mL [reference range, 239-931 pg/mL]), but we felt it was very unlikely to be responsible for his profound, sudden-onset microcytic anemia. Further evaluation for occult bleeding revealed an unremarkable upper endoscopy with push enteroscopy and colonoscopy. An alternate etiology of the anemia could not be identified.
Subsequently, he reported multiple pruritic bug bites sustained at the hotel room where he resided and continued to note pruritus while hospitalized. Pest control inspected the hospital room and identified bedbugs, Cimex lectularius, among his belongings. Upon further review, his clothes and walker were found to be completely infested with these organisms in different stages of development. Treatment included blood transfusions, iron supplementation, and environmental control of the infested living space both in the hospital and at his residence, with subsequent resolution of symptoms and anemia. Two weeks following discharge, the patient no longer reported pruritus, and his hemoglobin level had returned to baseline.
Over the last decade there has been an exponential resurgence in C lectularius infestations in developed countries attributed to increasing global travel, growing pesticide resistance, lack of public awareness, and inadequate pest control programs. This re-emergence has resulted in a public health problem. Although bedbugs are not known to transmit infectious diseases, severe infestation can result in notable dermatitis, iron-deficiency anemia from chronic blood loss, superinfection, allergic reactions including anaphylaxis in rare cases, and psychologic distress.
Iron-deficiency anemia caused by excessive bedbug biting in infants and children has been documented as early as the 1960s.1 Our knowledge of severe anemia due to bedbug infestation is limited to only 4 cases in the literature, according to a PubMed search of articles indexed for MEDLINE using the terms bedbugs anemia and cimex anemia.1-4 All cases reported bedbug infestations involving personal clothing, belongings, and/or living spaces. Patient concerns at presentation ranged from lethargy and fatigue with pruritic rash to chest pain and syncope with findings of severe microcytic or normocytic anemia (hemoglobin level, 5-8 g/dL). All cases were treated supportively with blood transfusion and iron supplementation, with hemoglobin recovery after several weeks. Environmental extermination also was required to prevent recurrence.1-4 Given that each bedbug blood meal is on average 7 mm3, one would have to incur a minimum of 143,000 bites to experience a blood loss of 1 L.3
The differential diagnosis for a patient with generalized pruritus should be broad and includes dermatologic conditions (eg, xerosis, atopic dermatitis, contact dermatitis, urticaria, dermatophytosis, lichen simplex chronicus, psoriasis, scabies, pediculosis corporis and pubis, other arthropod bites, bullous pemphigoid), systemic disorders (eg, renal disease, diabetes mellitus, thyroid disease, cholestasis, human immunodeficiency virus), malignancy, connective tissue disease, medication side effects, and psychogenic and neuropathic itch.
The diagnosis of C lectularius infestation is confirmed by finding the wingless, reddish brown, flat and ovular arthropod, with adult lengths of 4 to 7 mm, approximately the size of an apple seed.5-11 Bedbugs typically are active at night and feed for 3 to 10 minutes. After their feed or during the day, bedbugs will return to their nest in furniture, mattresses, beds, walls, and floors. Bedbug bites appear as small clusters or lines of pruritic erythematous papules with a central hemorrhagic puncta. Other cutaneous symptoms include isolated pruritus, papules, nodules, and bullous eruptions.7 Additional signs of bedbug infestation include black fecal stains in areas of inhabitation as well as actual bedbugs feeding during the day due to overcrowding.
Treatment of pruritic localized cutaneous reactions is supportive and includes antipruritic agents, topical steroids, topical anesthetics, antihistamines, or topical or systemic antibiotics for secondary infections.5-11 Systemic reactions, including anaphylaxis, are treated with epinephrine, antihistamines, and/or corticosteroids, while severe anemia is treated supportively with blood transfusions and iron supplementation.5-11 To prevent reoccurrence, environmental control in the form of nonchemical and chemical treatments is crucial in controlling bedbug infestations.5-11
This case highlights the relevance of a rare but notable morbidity associated with bedbug infestation and the adverse effects of bedbugs on public health. This patient's living situation in a single-room occupancy hotel, poor hygiene, and possible cognitive impairment from his multiple medical conditions may have increased his risk for extreme bedbug infestation. With a good history, physical examination, proper inspection of the patient's belongings, and provider awareness of this epidemic, the severity of this patient's anemia may have been circumvented on the prior hospital admission and follow-up office visit. Once such an infestation is confirmed, a multidisciplinary approach including social work assistance, health services, and pest control is needed to appropriately treat the patient and the environment. Methods in preventing and managing this growing public health problem include improving hygiene, avoiding secondhand goods, and increasing awareness in the identification and proper elimination of bedbugs.5-7
- Venkatachalam PS, Belavady B. Loss of haemoglobin iron due to excessive biting by bed bugs. a possible aetiological factor in the iron deficiency anaemia of infants and children. Trans R Soc Trop Med Hyg. 1962;56:218-221.
- Pritchard MJ, Hwang SW. Severe anemia from bedbugs. CMAJ. 2009;181:287-288.
- Paulke-Korinek M, Széll M, Laferl H, et al. Bed bugs can cause severe anaemia in adults. Parasitol Res. 2012;110:2577-2579.
- Sabou M, Imperiale DG, Andrés E, et al. Bed bugs reproductive life cycle in the clothes of a patient suffering from Alzheimer's disease results in iron deficiency anemia. Parasite. 2013;20:16.
- Studdiford JS, Conniff KM, Trayes KP, et al. Bedbug infestation. Am Fam Physician. 2012;86:653-658.
- Goddard J, deShazo R. Bed bugs (Cimex lectularis) and clinical consequences of their bites. JAMA. 2009;301:1358-1366.
- Bernardeschi C, Le Cleach L, Delaunay P, et al. Bed bug infestation. BMJ. 2013;346:f138.
- Silvia Munoz-Price L, Safdar N, Beier JC, et al. Bed bugs inhealthcare settings. Infect Control Hosp Epidemiol. 2012;33:1137-1142.
- Huntington MK. When bed bugs bite. J Fam Pract. 2012;61:384-388.
- Delaunay P, Blanc V, Del Giudice P, et al. Bedbugs and infectious diseases. Clin Infect Dis. 2011;52:200-212.
- Doggett SL, Dwyer DE, Penas PF, et al. Bed bugs: clinical relevance and control options. Clin Microbiol Rev. 2012;25:164-192.
To the Editor:
A 61-year-old man presented to the emergency department with a rash on the right leg, generalized pruritus, and chest pain. The patient described intermittent exertional pressure-like chest pain over the last few days but had no known prior cardiac history. He also noted worsening edema of the right leg with erythema. Three months prior he had been hospitalized for a similar presentation and was diagnosed with cellulitis of the right leg. The patient was treated with a course of trimethoprim-sulfamethoxazole and permethrin cream for presumed scabies and followed up with dermatology for the persistent generalized pruritic rash and cellulitis. At that time, he was diagnosed with stasis dermatitis with dermatitis neglecta and excoriations. He was educated on general hygiene and treated with triamcinolone, hydrophilic ointment, and pramoxine lotion for pruritus. He also was empirically treated again for scabies.
At the current presentation, preliminary investigation showed profound anemia with a hemoglobin level of 6.2 g/dL (baseline hemoglobin level 3 months prior, 13.1 g/dL). He was subsequently admitted to the general medicine ward for further investigation of severe symptomatic anemia. A medical history revealed moderate chronic obstructive pulmonary disease, hypertension, gastroesophageal reflux disease, xerosis, and fracture of the right ankle following open reduction internal fixation 6 years prior to admission. There was no history of blood loss, antiplatelet agents, or anticoagulants. He was on disability and lived in a single-room occupancy hotel. He did not report any high-risk sexual behaviors or abuse of alcohol or drugs. He actively smoked 1.5 packs of cigarettes per day for the last 30 years. He denied any allergies.
Physical examination revealed the patient was afebrile, nontoxic, disheveled, and in no acute distress. He had anicteric sclera and pale conjunctiva. The right leg appeared more erythematous and edematous compared to the left leg but without warmth or tenderness to palpation. He had innumerable 4- to 5-mm, erythematous, excoriated papules on the skin (Figure). His bed sheets were noted to have multiple rusty-black specks thought to be related to the crusted lesions. Physical examination was otherwise unremarkable.
Laboratory workup revealed severe iron-deficiency anemia without any evidence of hemolysis, marrow suppression, infection, or immune compromise (Table). He had a vitamin B12 deficiency (197 pg/mL [reference range, 239-931 pg/mL]), but we felt it was very unlikely to be responsible for his profound, sudden-onset microcytic anemia. Further evaluation for occult bleeding revealed an unremarkable upper endoscopy with push enteroscopy and colonoscopy. An alternate etiology of the anemia could not be identified.
Subsequently, he reported multiple pruritic bug bites sustained at the hotel room where he resided and continued to note pruritus while hospitalized. Pest control inspected the hospital room and identified bedbugs, Cimex lectularius, among his belongings. Upon further review, his clothes and walker were found to be completely infested with these organisms in different stages of development. Treatment included blood transfusions, iron supplementation, and environmental control of the infested living space both in the hospital and at his residence, with subsequent resolution of symptoms and anemia. Two weeks following discharge, the patient no longer reported pruritus, and his hemoglobin level had returned to baseline.
Over the last decade there has been an exponential resurgence in C lectularius infestations in developed countries attributed to increasing global travel, growing pesticide resistance, lack of public awareness, and inadequate pest control programs. This re-emergence has resulted in a public health problem. Although bedbugs are not known to transmit infectious diseases, severe infestation can result in notable dermatitis, iron-deficiency anemia from chronic blood loss, superinfection, allergic reactions including anaphylaxis in rare cases, and psychologic distress.
Iron-deficiency anemia caused by excessive bedbug biting in infants and children has been documented as early as the 1960s.1 Our knowledge of severe anemia due to bedbug infestation is limited to only 4 cases in the literature, according to a PubMed search of articles indexed for MEDLINE using the terms bedbugs anemia and cimex anemia.1-4 All cases reported bedbug infestations involving personal clothing, belongings, and/or living spaces. Patient concerns at presentation ranged from lethargy and fatigue with pruritic rash to chest pain and syncope with findings of severe microcytic or normocytic anemia (hemoglobin level, 5-8 g/dL). All cases were treated supportively with blood transfusion and iron supplementation, with hemoglobin recovery after several weeks. Environmental extermination also was required to prevent recurrence.1-4 Given that each bedbug blood meal is on average 7 mm3, one would have to incur a minimum of 143,000 bites to experience a blood loss of 1 L.3
The differential diagnosis for a patient with generalized pruritus should be broad and includes dermatologic conditions (eg, xerosis, atopic dermatitis, contact dermatitis, urticaria, dermatophytosis, lichen simplex chronicus, psoriasis, scabies, pediculosis corporis and pubis, other arthropod bites, bullous pemphigoid), systemic disorders (eg, renal disease, diabetes mellitus, thyroid disease, cholestasis, human immunodeficiency virus), malignancy, connective tissue disease, medication side effects, and psychogenic and neuropathic itch.
The diagnosis of C lectularius infestation is confirmed by finding the wingless, reddish brown, flat and ovular arthropod, with adult lengths of 4 to 7 mm, approximately the size of an apple seed.5-11 Bedbugs typically are active at night and feed for 3 to 10 minutes. After their feed or during the day, bedbugs will return to their nest in furniture, mattresses, beds, walls, and floors. Bedbug bites appear as small clusters or lines of pruritic erythematous papules with a central hemorrhagic puncta. Other cutaneous symptoms include isolated pruritus, papules, nodules, and bullous eruptions.7 Additional signs of bedbug infestation include black fecal stains in areas of inhabitation as well as actual bedbugs feeding during the day due to overcrowding.
Treatment of pruritic localized cutaneous reactions is supportive and includes antipruritic agents, topical steroids, topical anesthetics, antihistamines, or topical or systemic antibiotics for secondary infections.5-11 Systemic reactions, including anaphylaxis, are treated with epinephrine, antihistamines, and/or corticosteroids, while severe anemia is treated supportively with blood transfusions and iron supplementation.5-11 To prevent reoccurrence, environmental control in the form of nonchemical and chemical treatments is crucial in controlling bedbug infestations.5-11
This case highlights the relevance of a rare but notable morbidity associated with bedbug infestation and the adverse effects of bedbugs on public health. This patient's living situation in a single-room occupancy hotel, poor hygiene, and possible cognitive impairment from his multiple medical conditions may have increased his risk for extreme bedbug infestation. With a good history, physical examination, proper inspection of the patient's belongings, and provider awareness of this epidemic, the severity of this patient's anemia may have been circumvented on the prior hospital admission and follow-up office visit. Once such an infestation is confirmed, a multidisciplinary approach including social work assistance, health services, and pest control is needed to appropriately treat the patient and the environment. Methods in preventing and managing this growing public health problem include improving hygiene, avoiding secondhand goods, and increasing awareness in the identification and proper elimination of bedbugs.5-7
To the Editor:
A 61-year-old man presented to the emergency department with a rash on the right leg, generalized pruritus, and chest pain. The patient described intermittent exertional pressure-like chest pain over the last few days but had no known prior cardiac history. He also noted worsening edema of the right leg with erythema. Three months prior he had been hospitalized for a similar presentation and was diagnosed with cellulitis of the right leg. The patient was treated with a course of trimethoprim-sulfamethoxazole and permethrin cream for presumed scabies and followed up with dermatology for the persistent generalized pruritic rash and cellulitis. At that time, he was diagnosed with stasis dermatitis with dermatitis neglecta and excoriations. He was educated on general hygiene and treated with triamcinolone, hydrophilic ointment, and pramoxine lotion for pruritus. He also was empirically treated again for scabies.
At the current presentation, preliminary investigation showed profound anemia with a hemoglobin level of 6.2 g/dL (baseline hemoglobin level 3 months prior, 13.1 g/dL). He was subsequently admitted to the general medicine ward for further investigation of severe symptomatic anemia. A medical history revealed moderate chronic obstructive pulmonary disease, hypertension, gastroesophageal reflux disease, xerosis, and fracture of the right ankle following open reduction internal fixation 6 years prior to admission. There was no history of blood loss, antiplatelet agents, or anticoagulants. He was on disability and lived in a single-room occupancy hotel. He did not report any high-risk sexual behaviors or abuse of alcohol or drugs. He actively smoked 1.5 packs of cigarettes per day for the last 30 years. He denied any allergies.
Physical examination revealed the patient was afebrile, nontoxic, disheveled, and in no acute distress. He had anicteric sclera and pale conjunctiva. The right leg appeared more erythematous and edematous compared to the left leg but without warmth or tenderness to palpation. He had innumerable 4- to 5-mm, erythematous, excoriated papules on the skin (Figure). His bed sheets were noted to have multiple rusty-black specks thought to be related to the crusted lesions. Physical examination was otherwise unremarkable.
Laboratory workup revealed severe iron-deficiency anemia without any evidence of hemolysis, marrow suppression, infection, or immune compromise (Table). He had a vitamin B12 deficiency (197 pg/mL [reference range, 239-931 pg/mL]), but we felt it was very unlikely to be responsible for his profound, sudden-onset microcytic anemia. Further evaluation for occult bleeding revealed an unremarkable upper endoscopy with push enteroscopy and colonoscopy. An alternate etiology of the anemia could not be identified.
Subsequently, he reported multiple pruritic bug bites sustained at the hotel room where he resided and continued to note pruritus while hospitalized. Pest control inspected the hospital room and identified bedbugs, Cimex lectularius, among his belongings. Upon further review, his clothes and walker were found to be completely infested with these organisms in different stages of development. Treatment included blood transfusions, iron supplementation, and environmental control of the infested living space both in the hospital and at his residence, with subsequent resolution of symptoms and anemia. Two weeks following discharge, the patient no longer reported pruritus, and his hemoglobin level had returned to baseline.
Over the last decade there has been an exponential resurgence in C lectularius infestations in developed countries attributed to increasing global travel, growing pesticide resistance, lack of public awareness, and inadequate pest control programs. This re-emergence has resulted in a public health problem. Although bedbugs are not known to transmit infectious diseases, severe infestation can result in notable dermatitis, iron-deficiency anemia from chronic blood loss, superinfection, allergic reactions including anaphylaxis in rare cases, and psychologic distress.
Iron-deficiency anemia caused by excessive bedbug biting in infants and children has been documented as early as the 1960s.1 Our knowledge of severe anemia due to bedbug infestation is limited to only 4 cases in the literature, according to a PubMed search of articles indexed for MEDLINE using the terms bedbugs anemia and cimex anemia.1-4 All cases reported bedbug infestations involving personal clothing, belongings, and/or living spaces. Patient concerns at presentation ranged from lethargy and fatigue with pruritic rash to chest pain and syncope with findings of severe microcytic or normocytic anemia (hemoglobin level, 5-8 g/dL). All cases were treated supportively with blood transfusion and iron supplementation, with hemoglobin recovery after several weeks. Environmental extermination also was required to prevent recurrence.1-4 Given that each bedbug blood meal is on average 7 mm3, one would have to incur a minimum of 143,000 bites to experience a blood loss of 1 L.3
The differential diagnosis for a patient with generalized pruritus should be broad and includes dermatologic conditions (eg, xerosis, atopic dermatitis, contact dermatitis, urticaria, dermatophytosis, lichen simplex chronicus, psoriasis, scabies, pediculosis corporis and pubis, other arthropod bites, bullous pemphigoid), systemic disorders (eg, renal disease, diabetes mellitus, thyroid disease, cholestasis, human immunodeficiency virus), malignancy, connective tissue disease, medication side effects, and psychogenic and neuropathic itch.
The diagnosis of C lectularius infestation is confirmed by finding the wingless, reddish brown, flat and ovular arthropod, with adult lengths of 4 to 7 mm, approximately the size of an apple seed.5-11 Bedbugs typically are active at night and feed for 3 to 10 minutes. After their feed or during the day, bedbugs will return to their nest in furniture, mattresses, beds, walls, and floors. Bedbug bites appear as small clusters or lines of pruritic erythematous papules with a central hemorrhagic puncta. Other cutaneous symptoms include isolated pruritus, papules, nodules, and bullous eruptions.7 Additional signs of bedbug infestation include black fecal stains in areas of inhabitation as well as actual bedbugs feeding during the day due to overcrowding.
Treatment of pruritic localized cutaneous reactions is supportive and includes antipruritic agents, topical steroids, topical anesthetics, antihistamines, or topical or systemic antibiotics for secondary infections.5-11 Systemic reactions, including anaphylaxis, are treated with epinephrine, antihistamines, and/or corticosteroids, while severe anemia is treated supportively with blood transfusions and iron supplementation.5-11 To prevent reoccurrence, environmental control in the form of nonchemical and chemical treatments is crucial in controlling bedbug infestations.5-11
This case highlights the relevance of a rare but notable morbidity associated with bedbug infestation and the adverse effects of bedbugs on public health. This patient's living situation in a single-room occupancy hotel, poor hygiene, and possible cognitive impairment from his multiple medical conditions may have increased his risk for extreme bedbug infestation. With a good history, physical examination, proper inspection of the patient's belongings, and provider awareness of this epidemic, the severity of this patient's anemia may have been circumvented on the prior hospital admission and follow-up office visit. Once such an infestation is confirmed, a multidisciplinary approach including social work assistance, health services, and pest control is needed to appropriately treat the patient and the environment. Methods in preventing and managing this growing public health problem include improving hygiene, avoiding secondhand goods, and increasing awareness in the identification and proper elimination of bedbugs.5-7
- Venkatachalam PS, Belavady B. Loss of haemoglobin iron due to excessive biting by bed bugs. a possible aetiological factor in the iron deficiency anaemia of infants and children. Trans R Soc Trop Med Hyg. 1962;56:218-221.
- Pritchard MJ, Hwang SW. Severe anemia from bedbugs. CMAJ. 2009;181:287-288.
- Paulke-Korinek M, Széll M, Laferl H, et al. Bed bugs can cause severe anaemia in adults. Parasitol Res. 2012;110:2577-2579.
- Sabou M, Imperiale DG, Andrés E, et al. Bed bugs reproductive life cycle in the clothes of a patient suffering from Alzheimer's disease results in iron deficiency anemia. Parasite. 2013;20:16.
- Studdiford JS, Conniff KM, Trayes KP, et al. Bedbug infestation. Am Fam Physician. 2012;86:653-658.
- Goddard J, deShazo R. Bed bugs (Cimex lectularis) and clinical consequences of their bites. JAMA. 2009;301:1358-1366.
- Bernardeschi C, Le Cleach L, Delaunay P, et al. Bed bug infestation. BMJ. 2013;346:f138.
- Silvia Munoz-Price L, Safdar N, Beier JC, et al. Bed bugs inhealthcare settings. Infect Control Hosp Epidemiol. 2012;33:1137-1142.
- Huntington MK. When bed bugs bite. J Fam Pract. 2012;61:384-388.
- Delaunay P, Blanc V, Del Giudice P, et al. Bedbugs and infectious diseases. Clin Infect Dis. 2011;52:200-212.
- Doggett SL, Dwyer DE, Penas PF, et al. Bed bugs: clinical relevance and control options. Clin Microbiol Rev. 2012;25:164-192.
- Venkatachalam PS, Belavady B. Loss of haemoglobin iron due to excessive biting by bed bugs. a possible aetiological factor in the iron deficiency anaemia of infants and children. Trans R Soc Trop Med Hyg. 1962;56:218-221.
- Pritchard MJ, Hwang SW. Severe anemia from bedbugs. CMAJ. 2009;181:287-288.
- Paulke-Korinek M, Széll M, Laferl H, et al. Bed bugs can cause severe anaemia in adults. Parasitol Res. 2012;110:2577-2579.
- Sabou M, Imperiale DG, Andrés E, et al. Bed bugs reproductive life cycle in the clothes of a patient suffering from Alzheimer's disease results in iron deficiency anemia. Parasite. 2013;20:16.
- Studdiford JS, Conniff KM, Trayes KP, et al. Bedbug infestation. Am Fam Physician. 2012;86:653-658.
- Goddard J, deShazo R. Bed bugs (Cimex lectularis) and clinical consequences of their bites. JAMA. 2009;301:1358-1366.
- Bernardeschi C, Le Cleach L, Delaunay P, et al. Bed bug infestation. BMJ. 2013;346:f138.
- Silvia Munoz-Price L, Safdar N, Beier JC, et al. Bed bugs inhealthcare settings. Infect Control Hosp Epidemiol. 2012;33:1137-1142.
- Huntington MK. When bed bugs bite. J Fam Pract. 2012;61:384-388.
- Delaunay P, Blanc V, Del Giudice P, et al. Bedbugs and infectious diseases. Clin Infect Dis. 2011;52:200-212.
- Doggett SL, Dwyer DE, Penas PF, et al. Bed bugs: clinical relevance and control options. Clin Microbiol Rev. 2012;25:164-192.
Practice Points
- There has been a resurgence in bedbug (Cimex lectularius) infestations in developed countries.
- Although rare, anemia due to bedbug infestation should be considered in patients presenting with anemia and a widespread pruritic papular eruption.
- A thorough history and physical examination are essential to prevent a delay in diagnosis and avoid a costly and unnecessary workup.
- Successful treatment requires a multidisciplinary approach, which includes medical management, social services, and pest control.
Erythematous Plaques on a Tattoo
The Diagnosis: Epidermodysplasia Verruciformis
Histopathologic examination demonstrated acanthosis and coarse hypergranulosis with enlarged keratinocytes exhibiting blue cytoplasmic discoloration (Figure), which was suggestive of acquired epidermodysplasia verruciformis (EV).
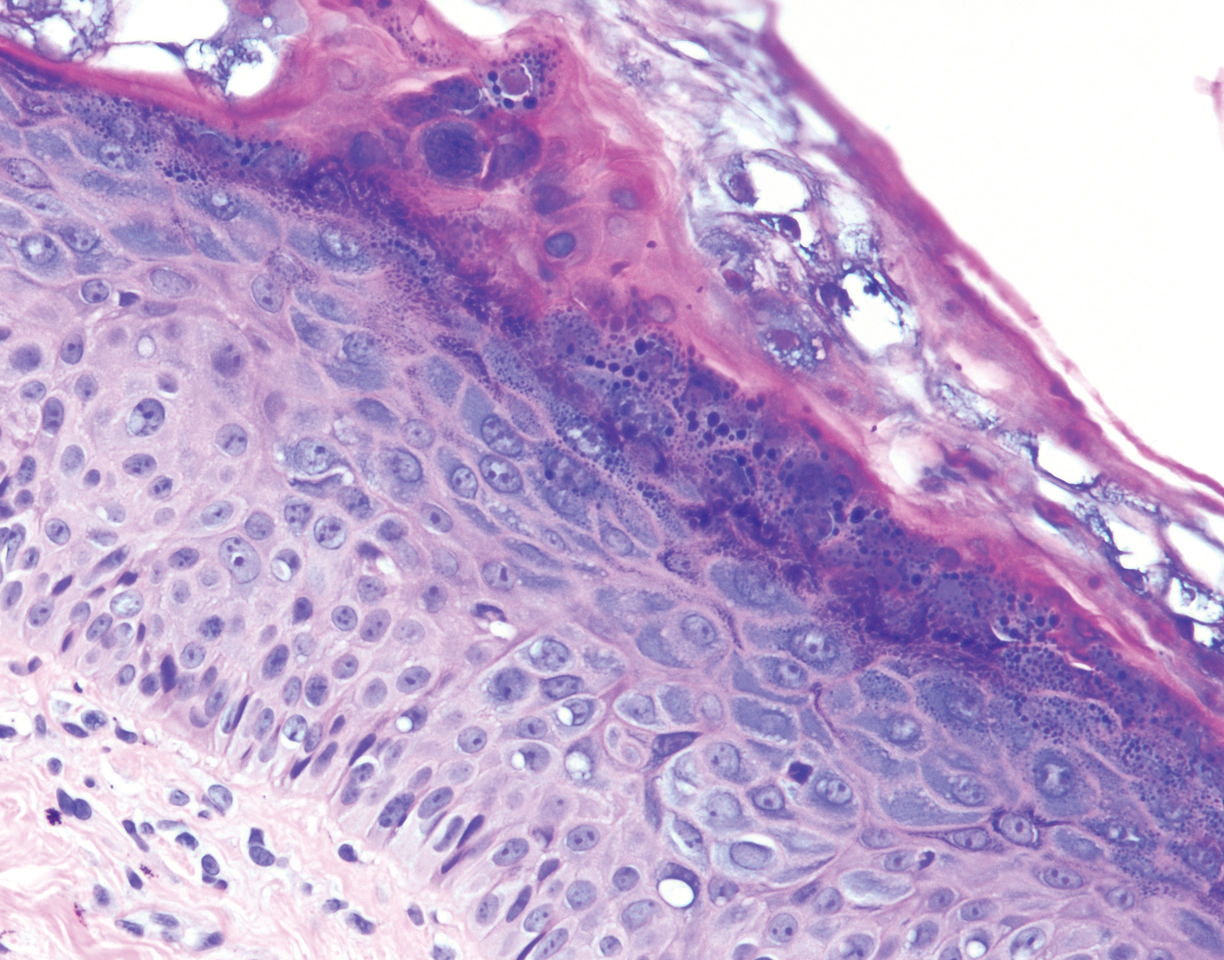
Acquired EV is a rare dermatologic condition associated with specific human papillomavirus (HPV) types that presents with recalcitrant lesions most commonly in the setting of immunosuppression.1 The most common HPV types associated with EV are HPV-5 and -8, but associations with HPV-3, -9, -10, -12, -14, -15, -17, -19 to -25, -36 to -38, -47, and -50 also have been reported.1,2 Acquired EV has been identified in individuals with human immunodeficiency virus, as well as in immunosuppressed patients with organ transplantation, Hodgkin lymphoma, systemic lupus erythematosus, and IgM deficiency, and in patients taking immunosuppressive medications such as tumor necrosis factor α inhibitors.1,3 The diagnosis is clinicopathological with potential polymerase chain reaction studies to identify underlying HPV types.
Acquired EV presents as hypopigmented to red, tinea versicolor-like macules or as verrucous, flat-topped papules on the trunk, arms, and/or legs.4 Histopathology reveals viral epidermal cytopathic changes, blue cytoplasm, and coarse hypogranulosis.4
There is no standardized treatment regimen for acquired EV, and no single approach has proven to yield an efficacious clinical outcome. Topical treatment options include steroids, retinoids, immunomodulators, cryotherapy, and electrosurgery, whereas retinoids or interferon alfa have been used as oral systemic therapy. Photodynamic therapy also has been shown to improve symptoms.3 Combination therapy such as interferon alfa with zidovudine or imiquimod with oral isotretinoin has shown better results than any single treatment.4 Due to the underlying HPV infection and its role in promotion of skin cancer development, lesions can characteristically undergo malignant transformations into Bowen disease but most commonly invasive squamous cell carcinoma (SCC), with initial lesions preferentially affecting sun-exposed areas due to the synergistic effect of UV light with EV-HPV lesions. The EV-HPV strains 5, 8, and 41 carry the highest oncogenic potential.5 Little is known of the true incidence of oncogenicity for acquired EV. Regardless, consistent sun protection and lifelong clinical examinations are critical for prognosis.5
The differential diagnosis of EV presenting in a tattoo includes allergic contact dermatitis, cutaneous sarcoidosis, pityriasis versicolor, and SCC. The pathology is critical to differentiate between these entities. The most frequently reported skin reactions to tattoo ink include inflammatory diseases (eg, allergic contact dermatitis, granulomatous reaction) or infectious diseases (eg, bacterial, viral, fungal).6 Allergic contact dermatitis, typically red pigment, is a common tattoo reaction. The most common histologic feature, however, is spongiosis, which results from intercellular edema. It often is limited to the lower epidermis but may affect the upper layers if the reaction is severe.7 Cutaneous sarcoidosis is a great masquerader that can present in various ways; however, its salient features on pathology are noncaseating granuloma involving the basal cell layer and epithelioid granuloma consisting of Langerhans giant cells.8 Although pityriasis versicolor can present in young immunocompromised adults, histologically salient features are the presence of both spores and hyphae in the stratum corneum.9 Although immunosuppression is a known risk factor for SCC, it is characterized histologically by hyperkeratosis, parakeratosis, and acanthosis with thickened and elongated rete ridges. Scattered atypical cells and frequent mitoses are present.10
- Schultz B, Nguyen CV, Jacobson-Dunlop E. Acquired epidermodysplasia verruciformis in setting of tumor necrosis factor-α inhibitor therapy. J Am Acad Dermatol Case Rep. 2018;4:805-807.
- DeVilliers EM, Fauquet C, Brocker TR, et al. Classification of papillomaviruses. Virology. 2004;324:17-27.
- Zampetti A, Giurdanella F, Manco S, et al. Acquired epidermodysplasia verruciformis: a comprehensive review and a proposal for treatment. Dermatol Surg. 2013;39:974-980.
- Henley JK, Hossler EW. Acquired epidermodysplasia verruciformis occurring in a renal transplant recipient. Cutis. 2017;99:E9-E12.
- Berk DR, Bruckner AL, Lu D. Epidermodysplasia verruciform-like lesions in an HIV patient. Dermatol Online J. 2009;15:1.
- Napolitano M, Megna M, Cappello M, et al. Skin diseases and tattoos: a five-year experience. G Ital Dermatol Venereol. 2018;153:644-648.
- Nixon RL, Mowad CM, Marks JG Jr. Allergic contact dermatitis. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 4th ed. Philadelphia, PA: Elsevier Saunders; 2018:242-259.
- Ferringer T. Granulomatous and histiocytic diseases. In: Elston DM, Ferringer T, Ko C, et al, eds. Dermatopathology. 3rd ed. China: Elsevier; 2019:175-176.
- Elewski BE, Hughey LC, Hunt KM, et al. Fungal diseases. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 4th ed. Philadelphia, PA: Elsevier Saunders; 2018:1329-1346.
- Soyer HP, Rigel DS, McMeniman E. Actinic keratosis, basal cell carcinoma, and squamous cell carcinoma. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 4th ed. Philadelphia, PA: Elsevier Saunders; 2018:1887-1884.
The Diagnosis: Epidermodysplasia Verruciformis
Histopathologic examination demonstrated acanthosis and coarse hypergranulosis with enlarged keratinocytes exhibiting blue cytoplasmic discoloration (Figure), which was suggestive of acquired epidermodysplasia verruciformis (EV).
Acquired EV is a rare dermatologic condition associated with specific human papillomavirus (HPV) types that presents with recalcitrant lesions most commonly in the setting of immunosuppression.1 The most common HPV types associated with EV are HPV-5 and -8, but associations with HPV-3, -9, -10, -12, -14, -15, -17, -19 to -25, -36 to -38, -47, and -50 also have been reported.1,2 Acquired EV has been identified in individuals with human immunodeficiency virus, as well as in immunosuppressed patients with organ transplantation, Hodgkin lymphoma, systemic lupus erythematosus, and IgM deficiency, and in patients taking immunosuppressive medications such as tumor necrosis factor α inhibitors.1,3 The diagnosis is clinicopathological with potential polymerase chain reaction studies to identify underlying HPV types.
Acquired EV presents as hypopigmented to red, tinea versicolor-like macules or as verrucous, flat-topped papules on the trunk, arms, and/or legs.4 Histopathology reveals viral epidermal cytopathic changes, blue cytoplasm, and coarse hypogranulosis.4
There is no standardized treatment regimen for acquired EV, and no single approach has proven to yield an efficacious clinical outcome. Topical treatment options include steroids, retinoids, immunomodulators, cryotherapy, and electrosurgery, whereas retinoids or interferon alfa have been used as oral systemic therapy. Photodynamic therapy also has been shown to improve symptoms.3 Combination therapy such as interferon alfa with zidovudine or imiquimod with oral isotretinoin has shown better results than any single treatment.4 Due to the underlying HPV infection and its role in promotion of skin cancer development, lesions can characteristically undergo malignant transformations into Bowen disease but most commonly invasive squamous cell carcinoma (SCC), with initial lesions preferentially affecting sun-exposed areas due to the synergistic effect of UV light with EV-HPV lesions. The EV-HPV strains 5, 8, and 41 carry the highest oncogenic potential.5 Little is known of the true incidence of oncogenicity for acquired EV. Regardless, consistent sun protection and lifelong clinical examinations are critical for prognosis.5
The differential diagnosis of EV presenting in a tattoo includes allergic contact dermatitis, cutaneous sarcoidosis, pityriasis versicolor, and SCC. The pathology is critical to differentiate between these entities. The most frequently reported skin reactions to tattoo ink include inflammatory diseases (eg, allergic contact dermatitis, granulomatous reaction) or infectious diseases (eg, bacterial, viral, fungal).6 Allergic contact dermatitis, typically red pigment, is a common tattoo reaction. The most common histologic feature, however, is spongiosis, which results from intercellular edema. It often is limited to the lower epidermis but may affect the upper layers if the reaction is severe.7 Cutaneous sarcoidosis is a great masquerader that can present in various ways; however, its salient features on pathology are noncaseating granuloma involving the basal cell layer and epithelioid granuloma consisting of Langerhans giant cells.8 Although pityriasis versicolor can present in young immunocompromised adults, histologically salient features are the presence of both spores and hyphae in the stratum corneum.9 Although immunosuppression is a known risk factor for SCC, it is characterized histologically by hyperkeratosis, parakeratosis, and acanthosis with thickened and elongated rete ridges. Scattered atypical cells and frequent mitoses are present.10
The Diagnosis: Epidermodysplasia Verruciformis
Histopathologic examination demonstrated acanthosis and coarse hypergranulosis with enlarged keratinocytes exhibiting blue cytoplasmic discoloration (Figure), which was suggestive of acquired epidermodysplasia verruciformis (EV).
Acquired EV is a rare dermatologic condition associated with specific human papillomavirus (HPV) types that presents with recalcitrant lesions most commonly in the setting of immunosuppression.1 The most common HPV types associated with EV are HPV-5 and -8, but associations with HPV-3, -9, -10, -12, -14, -15, -17, -19 to -25, -36 to -38, -47, and -50 also have been reported.1,2 Acquired EV has been identified in individuals with human immunodeficiency virus, as well as in immunosuppressed patients with organ transplantation, Hodgkin lymphoma, systemic lupus erythematosus, and IgM deficiency, and in patients taking immunosuppressive medications such as tumor necrosis factor α inhibitors.1,3 The diagnosis is clinicopathological with potential polymerase chain reaction studies to identify underlying HPV types.
Acquired EV presents as hypopigmented to red, tinea versicolor-like macules or as verrucous, flat-topped papules on the trunk, arms, and/or legs.4 Histopathology reveals viral epidermal cytopathic changes, blue cytoplasm, and coarse hypogranulosis.4
There is no standardized treatment regimen for acquired EV, and no single approach has proven to yield an efficacious clinical outcome. Topical treatment options include steroids, retinoids, immunomodulators, cryotherapy, and electrosurgery, whereas retinoids or interferon alfa have been used as oral systemic therapy. Photodynamic therapy also has been shown to improve symptoms.3 Combination therapy such as interferon alfa with zidovudine or imiquimod with oral isotretinoin has shown better results than any single treatment.4 Due to the underlying HPV infection and its role in promotion of skin cancer development, lesions can characteristically undergo malignant transformations into Bowen disease but most commonly invasive squamous cell carcinoma (SCC), with initial lesions preferentially affecting sun-exposed areas due to the synergistic effect of UV light with EV-HPV lesions. The EV-HPV strains 5, 8, and 41 carry the highest oncogenic potential.5 Little is known of the true incidence of oncogenicity for acquired EV. Regardless, consistent sun protection and lifelong clinical examinations are critical for prognosis.5
The differential diagnosis of EV presenting in a tattoo includes allergic contact dermatitis, cutaneous sarcoidosis, pityriasis versicolor, and SCC. The pathology is critical to differentiate between these entities. The most frequently reported skin reactions to tattoo ink include inflammatory diseases (eg, allergic contact dermatitis, granulomatous reaction) or infectious diseases (eg, bacterial, viral, fungal).6 Allergic contact dermatitis, typically red pigment, is a common tattoo reaction. The most common histologic feature, however, is spongiosis, which results from intercellular edema. It often is limited to the lower epidermis but may affect the upper layers if the reaction is severe.7 Cutaneous sarcoidosis is a great masquerader that can present in various ways; however, its salient features on pathology are noncaseating granuloma involving the basal cell layer and epithelioid granuloma consisting of Langerhans giant cells.8 Although pityriasis versicolor can present in young immunocompromised adults, histologically salient features are the presence of both spores and hyphae in the stratum corneum.9 Although immunosuppression is a known risk factor for SCC, it is characterized histologically by hyperkeratosis, parakeratosis, and acanthosis with thickened and elongated rete ridges. Scattered atypical cells and frequent mitoses are present.10
- Schultz B, Nguyen CV, Jacobson-Dunlop E. Acquired epidermodysplasia verruciformis in setting of tumor necrosis factor-α inhibitor therapy. J Am Acad Dermatol Case Rep. 2018;4:805-807.
- DeVilliers EM, Fauquet C, Brocker TR, et al. Classification of papillomaviruses. Virology. 2004;324:17-27.
- Zampetti A, Giurdanella F, Manco S, et al. Acquired epidermodysplasia verruciformis: a comprehensive review and a proposal for treatment. Dermatol Surg. 2013;39:974-980.
- Henley JK, Hossler EW. Acquired epidermodysplasia verruciformis occurring in a renal transplant recipient. Cutis. 2017;99:E9-E12.
- Berk DR, Bruckner AL, Lu D. Epidermodysplasia verruciform-like lesions in an HIV patient. Dermatol Online J. 2009;15:1.
- Napolitano M, Megna M, Cappello M, et al. Skin diseases and tattoos: a five-year experience. G Ital Dermatol Venereol. 2018;153:644-648.
- Nixon RL, Mowad CM, Marks JG Jr. Allergic contact dermatitis. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 4th ed. Philadelphia, PA: Elsevier Saunders; 2018:242-259.
- Ferringer T. Granulomatous and histiocytic diseases. In: Elston DM, Ferringer T, Ko C, et al, eds. Dermatopathology. 3rd ed. China: Elsevier; 2019:175-176.
- Elewski BE, Hughey LC, Hunt KM, et al. Fungal diseases. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 4th ed. Philadelphia, PA: Elsevier Saunders; 2018:1329-1346.
- Soyer HP, Rigel DS, McMeniman E. Actinic keratosis, basal cell carcinoma, and squamous cell carcinoma. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 4th ed. Philadelphia, PA: Elsevier Saunders; 2018:1887-1884.
- Schultz B, Nguyen CV, Jacobson-Dunlop E. Acquired epidermodysplasia verruciformis in setting of tumor necrosis factor-α inhibitor therapy. J Am Acad Dermatol Case Rep. 2018;4:805-807.
- DeVilliers EM, Fauquet C, Brocker TR, et al. Classification of papillomaviruses. Virology. 2004;324:17-27.
- Zampetti A, Giurdanella F, Manco S, et al. Acquired epidermodysplasia verruciformis: a comprehensive review and a proposal for treatment. Dermatol Surg. 2013;39:974-980.
- Henley JK, Hossler EW. Acquired epidermodysplasia verruciformis occurring in a renal transplant recipient. Cutis. 2017;99:E9-E12.
- Berk DR, Bruckner AL, Lu D. Epidermodysplasia verruciform-like lesions in an HIV patient. Dermatol Online J. 2009;15:1.
- Napolitano M, Megna M, Cappello M, et al. Skin diseases and tattoos: a five-year experience. G Ital Dermatol Venereol. 2018;153:644-648.
- Nixon RL, Mowad CM, Marks JG Jr. Allergic contact dermatitis. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 4th ed. Philadelphia, PA: Elsevier Saunders; 2018:242-259.
- Ferringer T. Granulomatous and histiocytic diseases. In: Elston DM, Ferringer T, Ko C, et al, eds. Dermatopathology. 3rd ed. China: Elsevier; 2019:175-176.
- Elewski BE, Hughey LC, Hunt KM, et al. Fungal diseases. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 4th ed. Philadelphia, PA: Elsevier Saunders; 2018:1329-1346.
- Soyer HP, Rigel DS, McMeniman E. Actinic keratosis, basal cell carcinoma, and squamous cell carcinoma. In: Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 4th ed. Philadelphia, PA: Elsevier Saunders; 2018:1887-1884.
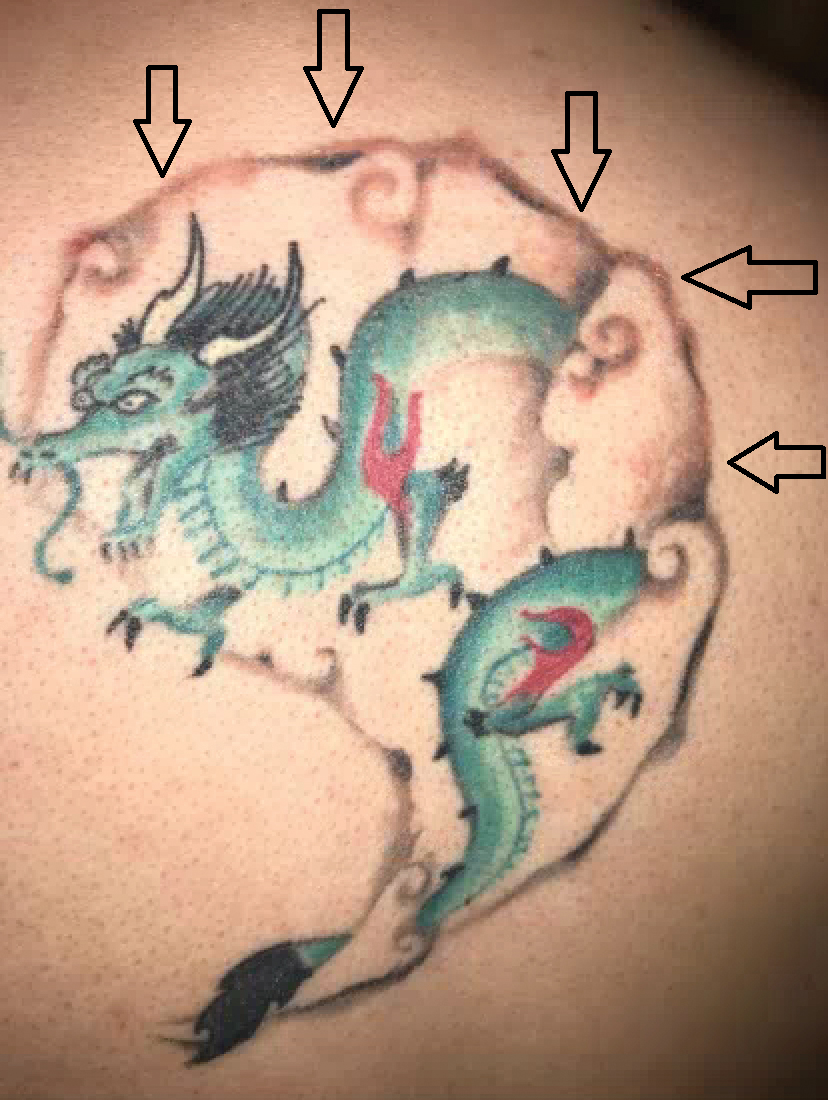
A 29-year-old man presented with increased redness, dryness, and pruritus at the periphery of a tattoo (arrows) on the upper back of 4 months' duration. He was diagnosed with human immunodeficiency virus 8 months prior to presentation and had a history of cystic fibrosis, eczema, and genital molluscum contagiosum. Laboratory analysis 1 month prior revealed a CD4 count of 42 cells/mm3 (reference range, 500-1200 cells/mm3), and the viral load was 2388 copies/mL (reference range, 20-10,000,000 copies/mL). Physical examination revealed multiple erythematous, eczematous, linear plaques along the dark gray lines of the tattoo. A 1.1.2 ×0.7.2 ×0.1-cm shave biopsy specimen was obtained. After the biopsy, tretinoin cream 0.1% and betamethasone dipropionate ointment 0.05% were prescribed to be alternately applied on the tattoo lesions until resolution.
Raynaud Phenomenon of the Nipple Successfully Treated With Nifedipine and Gabapentin
To the Editor:
Raynaud phenomenon is characterized by vasospasm of arterioles causing intermittent ischemia of the digits. The characteristic triphasic color change presents first as a dramatic change in skin color from normal to white, as the vasoconstriction causes pallor secondary to ischemia. This change is followed by a blue appearance, as cyanosis results from the deoxygenated venous blood. Finally, reflex vasodilation and reperfusion manifest as a red color from erythema. Several cases have been reported describing Raynaud phenomenon affecting the nipples of breastfeeding women.1-5 This vasospasm results in episodic nipple pain manifesting from breastfeeding and exposure to cold. If it is not appropriately treated, the pain’s severity causes affected women to stop breastfeeding. We report a case of vasospasm of the nipple in which the patient experienced nipple pain and a separate lancinating pain that radiated through the breasts.
A 36-year-old woman presented with excruciating nipple and breast pain 3 weeks after delivering her first child. She had no history of smoking or Raynaud phenomenon. The nipple pain was triggered upon breastfeeding and exposure to cold. During these episodes, the nipples would initially blanch white, then turn purple and finally a deep red. The patient also experienced an episodic excruciating lancinating pain of the breast that would randomly and spontaneously radiate through either breast several times per day for 15 to 30 seconds. A workup including an antinuclear antibody test, complete blood cell count with differential, and comprehensive metabolic panel all were within reference range.
The patient was diagnosed with nipple vasospasm. Partial relief of nipple pain occurred after treatment with 30 mg daily of nifedipine; 60 mg daily resulted in complete control, allowing the patient to breastfeed without discomfort, but the lancinating pain continued unabated. The patient could not discontinue breastfeeding because her child was intolerant to formula. She became despondent, as she could find no relief from the pain that she found to be intolerable. Because the patient’s description was reminiscent of the lancinating pain seen in postherpetic neuralgia, a trial of pregabalin was prescribed. A dosage of 75 mg twice daily resulted in near-complete resolution of the pain. After 3 months, the patient successfully weaned her child from breast milk to formula, and the nipple and breast pain promptly resolved. The baby experienced no adverse effects from the patient’s use of pregabalin.
This condition was first described by Gunther1 in 1970 as initial blanching of the nipple followed by a mulberry color. It was termed psychosomatic sore nipples.1 Lawlor-Smith and Lawlor-Smith2 described the condition in 1997 and termed it vasospasm of the nipple. They reported 5 patients who experienced debilitating nipple pain as well as the triphasic color change of Raynaud phenomenon or a biphasic color change (white and blue). Two patients had a history of Raynaud phenomenon affecting the digits before their first pregnancy.2 Anderson et al3 presented 12 breastfeeding women with Raynaud phenomenon of the nipple; only 1 patient had a history of Raynaud phenomenon. In this series, all 6 women who chose to try nifedipine responded well to the drug.3
Raynaud phenomenon of the nipple also has been reported to be associated with the use of labetalol.4 In this case, the patient had a history of Raynaud phenomenon affecting the toes and nipples on cold days. In 2 subsequent pregnancies she was treated with labetalol for pregnancy-induced hypertension, which resulted in severe nipple pain with each pregnancy unrelated to cold weather. Unlike other cases, this patient experienced antenatal symptoms in addition to the typical postnatal symptoms. The nipple pain resolved with discontinuation of the labetalol.4
Barrett et al5 conducted a retrospective review of medical records of 88 breastfeeding mothers who presented with nipple pain and dermatitis. They defined the criteria for Raynaud phenomenon of the nipple as chronic deep breast pain (in general lasting >4 weeks) that responded to therapy for the condition and had at least 2 of the following characteristics: (1) observed or self-reported color changes of the nipple, especially with cold exposure (white, blue, or red); (2) cold sensitivity or color changes of the hands or feet with cold exposure; or (3) failed therapy with oral antifungals. Using these criteria, they diagnosed 22 women (25%) with Raynaud phenomenon of the nipple; 20 (91%) reported a history of cold sensitivity or color change of acral surfaces. Of 12 patients who received and tolerated nifedipine use, 10 (83%) reported decreased pain or complete resolution. This series described breast or nipple pain, whereas other reported cases only described nipple pain. The authors described a sharp, shooting, or stabbing pain—qualifications not previously noted.5 Our patient experienced both nipple pain and a lancinating breast pain consistent with the cases reported by Barrett et al.5
The nipple pain and treatment response in our patient was typical of previously reported cases of vasospasm of the nipple in breastfeeding women; however, Barrett et al5 did not describe individual patients who exhibited the dual nature of the pain described in our patient. The nipple pain experienced during breastfeeding in our patient was successfully treated with nifedipine. We report the successful treatment of the separate lancinating pain with pregabalin.
- Gunther M. Infant Feeding. London, United Kingdom: Methuen; 1970.
- Lawlor-Smith L, Lawlor-Smith C. Vasospasm of the nipple—a manifestation of Raynaud’s phenomenon: case reports. BMJ. 1997;314:644-645.
- Anderson JE, Held N, Wright K. Raynaud phenomenon of the nipple: a treatable cause of painful breastfeeding. Pediatrics. 2004;113:360-364.
- McGuinness N, Cording V. Raynaud’s phenomenon of the nipple associated with labetalol use. J Hum Lact. 2013;29:17-19.
- Barrett ME, Heller MM, Stone HF, et al. Raynaud phenomenon of the nipple in breastfeeding mothers: an underdiagnosed cause of nipple pain. JAMA Dermatol. 2013;149:300-306.
To the Editor:
Raynaud phenomenon is characterized by vasospasm of arterioles causing intermittent ischemia of the digits. The characteristic triphasic color change presents first as a dramatic change in skin color from normal to white, as the vasoconstriction causes pallor secondary to ischemia. This change is followed by a blue appearance, as cyanosis results from the deoxygenated venous blood. Finally, reflex vasodilation and reperfusion manifest as a red color from erythema. Several cases have been reported describing Raynaud phenomenon affecting the nipples of breastfeeding women.1-5 This vasospasm results in episodic nipple pain manifesting from breastfeeding and exposure to cold. If it is not appropriately treated, the pain’s severity causes affected women to stop breastfeeding. We report a case of vasospasm of the nipple in which the patient experienced nipple pain and a separate lancinating pain that radiated through the breasts.
A 36-year-old woman presented with excruciating nipple and breast pain 3 weeks after delivering her first child. She had no history of smoking or Raynaud phenomenon. The nipple pain was triggered upon breastfeeding and exposure to cold. During these episodes, the nipples would initially blanch white, then turn purple and finally a deep red. The patient also experienced an episodic excruciating lancinating pain of the breast that would randomly and spontaneously radiate through either breast several times per day for 15 to 30 seconds. A workup including an antinuclear antibody test, complete blood cell count with differential, and comprehensive metabolic panel all were within reference range.
The patient was diagnosed with nipple vasospasm. Partial relief of nipple pain occurred after treatment with 30 mg daily of nifedipine; 60 mg daily resulted in complete control, allowing the patient to breastfeed without discomfort, but the lancinating pain continued unabated. The patient could not discontinue breastfeeding because her child was intolerant to formula. She became despondent, as she could find no relief from the pain that she found to be intolerable. Because the patient’s description was reminiscent of the lancinating pain seen in postherpetic neuralgia, a trial of pregabalin was prescribed. A dosage of 75 mg twice daily resulted in near-complete resolution of the pain. After 3 months, the patient successfully weaned her child from breast milk to formula, and the nipple and breast pain promptly resolved. The baby experienced no adverse effects from the patient’s use of pregabalin.
This condition was first described by Gunther1 in 1970 as initial blanching of the nipple followed by a mulberry color. It was termed psychosomatic sore nipples.1 Lawlor-Smith and Lawlor-Smith2 described the condition in 1997 and termed it vasospasm of the nipple. They reported 5 patients who experienced debilitating nipple pain as well as the triphasic color change of Raynaud phenomenon or a biphasic color change (white and blue). Two patients had a history of Raynaud phenomenon affecting the digits before their first pregnancy.2 Anderson et al3 presented 12 breastfeeding women with Raynaud phenomenon of the nipple; only 1 patient had a history of Raynaud phenomenon. In this series, all 6 women who chose to try nifedipine responded well to the drug.3
Raynaud phenomenon of the nipple also has been reported to be associated with the use of labetalol.4 In this case, the patient had a history of Raynaud phenomenon affecting the toes and nipples on cold days. In 2 subsequent pregnancies she was treated with labetalol for pregnancy-induced hypertension, which resulted in severe nipple pain with each pregnancy unrelated to cold weather. Unlike other cases, this patient experienced antenatal symptoms in addition to the typical postnatal symptoms. The nipple pain resolved with discontinuation of the labetalol.4
Barrett et al5 conducted a retrospective review of medical records of 88 breastfeeding mothers who presented with nipple pain and dermatitis. They defined the criteria for Raynaud phenomenon of the nipple as chronic deep breast pain (in general lasting >4 weeks) that responded to therapy for the condition and had at least 2 of the following characteristics: (1) observed or self-reported color changes of the nipple, especially with cold exposure (white, blue, or red); (2) cold sensitivity or color changes of the hands or feet with cold exposure; or (3) failed therapy with oral antifungals. Using these criteria, they diagnosed 22 women (25%) with Raynaud phenomenon of the nipple; 20 (91%) reported a history of cold sensitivity or color change of acral surfaces. Of 12 patients who received and tolerated nifedipine use, 10 (83%) reported decreased pain or complete resolution. This series described breast or nipple pain, whereas other reported cases only described nipple pain. The authors described a sharp, shooting, or stabbing pain—qualifications not previously noted.5 Our patient experienced both nipple pain and a lancinating breast pain consistent with the cases reported by Barrett et al.5
The nipple pain and treatment response in our patient was typical of previously reported cases of vasospasm of the nipple in breastfeeding women; however, Barrett et al5 did not describe individual patients who exhibited the dual nature of the pain described in our patient. The nipple pain experienced during breastfeeding in our patient was successfully treated with nifedipine. We report the successful treatment of the separate lancinating pain with pregabalin.
To the Editor:
Raynaud phenomenon is characterized by vasospasm of arterioles causing intermittent ischemia of the digits. The characteristic triphasic color change presents first as a dramatic change in skin color from normal to white, as the vasoconstriction causes pallor secondary to ischemia. This change is followed by a blue appearance, as cyanosis results from the deoxygenated venous blood. Finally, reflex vasodilation and reperfusion manifest as a red color from erythema. Several cases have been reported describing Raynaud phenomenon affecting the nipples of breastfeeding women.1-5 This vasospasm results in episodic nipple pain manifesting from breastfeeding and exposure to cold. If it is not appropriately treated, the pain’s severity causes affected women to stop breastfeeding. We report a case of vasospasm of the nipple in which the patient experienced nipple pain and a separate lancinating pain that radiated through the breasts.
A 36-year-old woman presented with excruciating nipple and breast pain 3 weeks after delivering her first child. She had no history of smoking or Raynaud phenomenon. The nipple pain was triggered upon breastfeeding and exposure to cold. During these episodes, the nipples would initially blanch white, then turn purple and finally a deep red. The patient also experienced an episodic excruciating lancinating pain of the breast that would randomly and spontaneously radiate through either breast several times per day for 15 to 30 seconds. A workup including an antinuclear antibody test, complete blood cell count with differential, and comprehensive metabolic panel all were within reference range.
The patient was diagnosed with nipple vasospasm. Partial relief of nipple pain occurred after treatment with 30 mg daily of nifedipine; 60 mg daily resulted in complete control, allowing the patient to breastfeed without discomfort, but the lancinating pain continued unabated. The patient could not discontinue breastfeeding because her child was intolerant to formula. She became despondent, as she could find no relief from the pain that she found to be intolerable. Because the patient’s description was reminiscent of the lancinating pain seen in postherpetic neuralgia, a trial of pregabalin was prescribed. A dosage of 75 mg twice daily resulted in near-complete resolution of the pain. After 3 months, the patient successfully weaned her child from breast milk to formula, and the nipple and breast pain promptly resolved. The baby experienced no adverse effects from the patient’s use of pregabalin.
This condition was first described by Gunther1 in 1970 as initial blanching of the nipple followed by a mulberry color. It was termed psychosomatic sore nipples.1 Lawlor-Smith and Lawlor-Smith2 described the condition in 1997 and termed it vasospasm of the nipple. They reported 5 patients who experienced debilitating nipple pain as well as the triphasic color change of Raynaud phenomenon or a biphasic color change (white and blue). Two patients had a history of Raynaud phenomenon affecting the digits before their first pregnancy.2 Anderson et al3 presented 12 breastfeeding women with Raynaud phenomenon of the nipple; only 1 patient had a history of Raynaud phenomenon. In this series, all 6 women who chose to try nifedipine responded well to the drug.3
Raynaud phenomenon of the nipple also has been reported to be associated with the use of labetalol.4 In this case, the patient had a history of Raynaud phenomenon affecting the toes and nipples on cold days. In 2 subsequent pregnancies she was treated with labetalol for pregnancy-induced hypertension, which resulted in severe nipple pain with each pregnancy unrelated to cold weather. Unlike other cases, this patient experienced antenatal symptoms in addition to the typical postnatal symptoms. The nipple pain resolved with discontinuation of the labetalol.4
Barrett et al5 conducted a retrospective review of medical records of 88 breastfeeding mothers who presented with nipple pain and dermatitis. They defined the criteria for Raynaud phenomenon of the nipple as chronic deep breast pain (in general lasting >4 weeks) that responded to therapy for the condition and had at least 2 of the following characteristics: (1) observed or self-reported color changes of the nipple, especially with cold exposure (white, blue, or red); (2) cold sensitivity or color changes of the hands or feet with cold exposure; or (3) failed therapy with oral antifungals. Using these criteria, they diagnosed 22 women (25%) with Raynaud phenomenon of the nipple; 20 (91%) reported a history of cold sensitivity or color change of acral surfaces. Of 12 patients who received and tolerated nifedipine use, 10 (83%) reported decreased pain or complete resolution. This series described breast or nipple pain, whereas other reported cases only described nipple pain. The authors described a sharp, shooting, or stabbing pain—qualifications not previously noted.5 Our patient experienced both nipple pain and a lancinating breast pain consistent with the cases reported by Barrett et al.5
The nipple pain and treatment response in our patient was typical of previously reported cases of vasospasm of the nipple in breastfeeding women; however, Barrett et al5 did not describe individual patients who exhibited the dual nature of the pain described in our patient. The nipple pain experienced during breastfeeding in our patient was successfully treated with nifedipine. We report the successful treatment of the separate lancinating pain with pregabalin.
- Gunther M. Infant Feeding. London, United Kingdom: Methuen; 1970.
- Lawlor-Smith L, Lawlor-Smith C. Vasospasm of the nipple—a manifestation of Raynaud’s phenomenon: case reports. BMJ. 1997;314:644-645.
- Anderson JE, Held N, Wright K. Raynaud phenomenon of the nipple: a treatable cause of painful breastfeeding. Pediatrics. 2004;113:360-364.
- McGuinness N, Cording V. Raynaud’s phenomenon of the nipple associated with labetalol use. J Hum Lact. 2013;29:17-19.
- Barrett ME, Heller MM, Stone HF, et al. Raynaud phenomenon of the nipple in breastfeeding mothers: an underdiagnosed cause of nipple pain. JAMA Dermatol. 2013;149:300-306.
- Gunther M. Infant Feeding. London, United Kingdom: Methuen; 1970.
- Lawlor-Smith L, Lawlor-Smith C. Vasospasm of the nipple—a manifestation of Raynaud’s phenomenon: case reports. BMJ. 1997;314:644-645.
- Anderson JE, Held N, Wright K. Raynaud phenomenon of the nipple: a treatable cause of painful breastfeeding. Pediatrics. 2004;113:360-364.
- McGuinness N, Cording V. Raynaud’s phenomenon of the nipple associated with labetalol use. J Hum Lact. 2013;29:17-19.
- Barrett ME, Heller MM, Stone HF, et al. Raynaud phenomenon of the nipple in breastfeeding mothers: an underdiagnosed cause of nipple pain. JAMA Dermatol. 2013;149:300-306.
Practice Points
- Raynaud phenomenon of the nipple may be accompanied by lancinating pain of the breast in addition to nipple pain reminiscent of postherpetic neuralgia.
- Associated breast pain is particularly distressing for breastfeeding women, particularly primiparous mothers with children intolerant to formula.
- In women with Raynaud phenomenon accompanied by lancinating breast pain, consider a trial of pregabalin.
Radiation Recall Dermatitis Triggered by Prednisone
To the Editor:
A 69-year-old woman presented to the allergy clinic for evaluation of a rash on the left breast. The patient had a history of breast cancer that was treated with a lumpectomy followed by external beam radiation therapy (total dose, 6000 cGy) to the lateral aspect of the left breast approximately 4 years prior. She developed acute breast dermatitis from the radiation, which was self-treated with over-the-counter hydrocortisone cream. The patient subsequently developed a blistering skin eruption over the area where she applied the cream. She did not recall the subtype of hydrocortisone she used (butyrate and acetate are available over-the-counter). She discontinued the hydrocortisone and was started on triamcinolone cream 0.1%, which was well tolerated, and the rash resolved.
The patient had a history of a similar reaction to hydrocortisone butyrate after blepharoplasty approximately 10 years prior to the current presentation, characterized by facial erythema, pruritus, and blistering. A patch test confirmed reactivity to hydrocortisone-17-butyrate and tixocortol pivalate. However, a skin-prick test for hydrocortisone acetate cream 1% was negative.
Subsequently, the patient developed acute-onset dyspepsia, gnawing epigastric pain, regurgitation, and bloating. A diagnosis of eosinophilic gastritis was established via biopsy, which found increased eosinophils in the lamina propria (>50 eosinophils per high-power field). Helicobacter pylori was not identified. She was started on the proton-pump inhibitor dexlansoprazole but symptoms did not improve. Her other medications included benazepril, alprazolam as needed, vitamin D, and magnesium. The patient subsequently was started on a trial of oral prednisone 40 mg/d. Three days after initiation, she developed an erythematous macular rash over the left breast.
The next day she presented to the allergy clinic. Physical examination of the left breast revealed a 20×10-cm, nipple-sparing patch of well-demarcated erythema without fluctuance or overlying lesions. The area of erythema overlapped with the prior radiation field based on radiation marker tattoos and the lumpectomy scar (Figure). There was no evidence to suggest inflammation of deeper tissue or the pectoral muscles. Vital signs were normal, and the remainder of the examination was unremarkable, including breast, lymph node, and complete skin examinations.
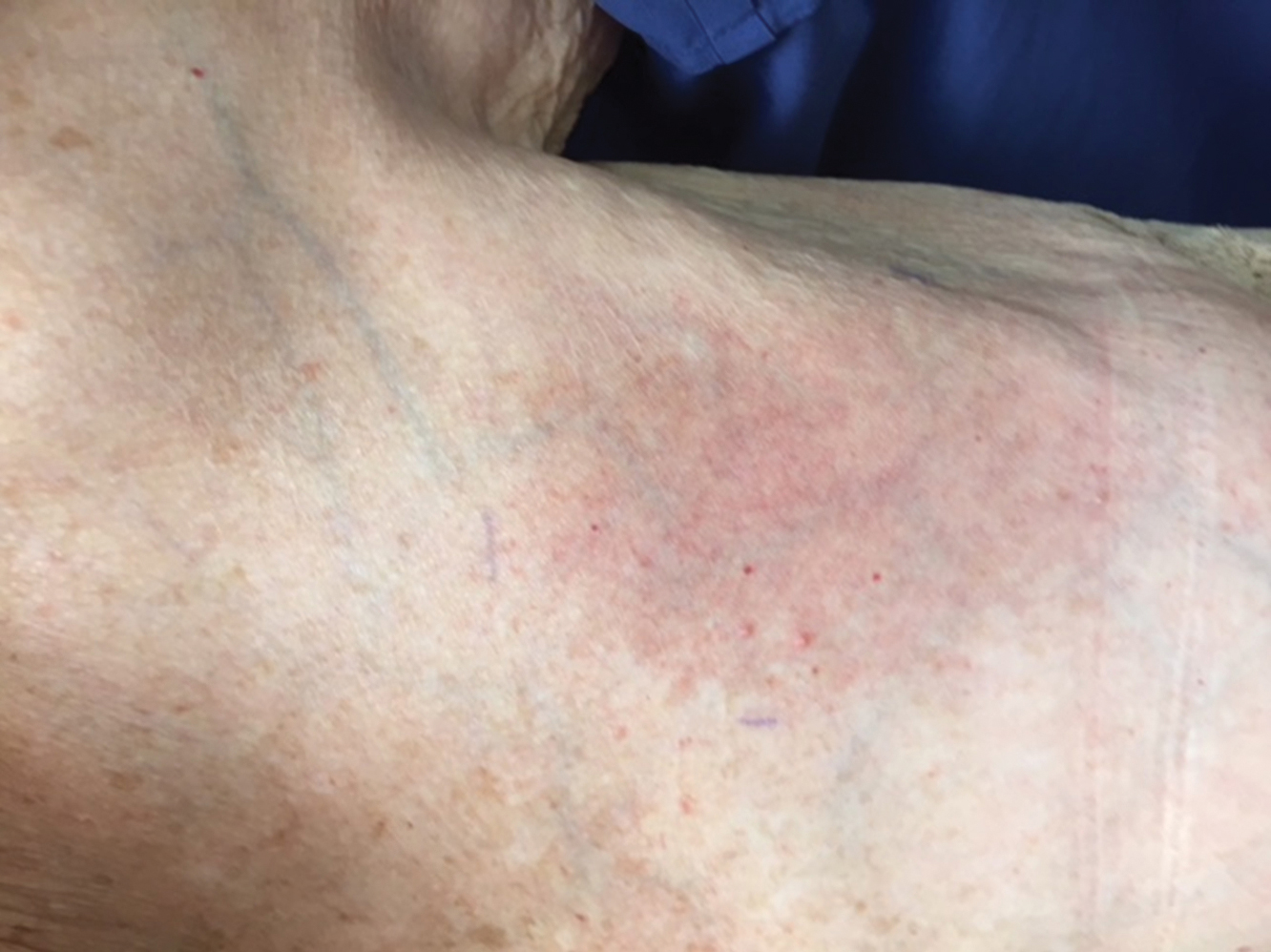
At evaluation, the differential diagnosis included contact dermatitis, fixed drug eruption, infection, tumor recurrence with overlying skin changes, and radiation recall dermatitis. Given that the dermatitis had developed at the site of previously irradiated skin in the absence of fever or an associated mass, the presentation was thought to be most consistent with radiation recall dermatitis.
Oral prednisone was discontinued, and the dermatitis spontaneously improved in a few weeks. Given the patient’s test results and prior tolerance to triamcinolone, eosinophilic gastroenteritis was treated with triamcinolone acetonide 40 mg via intramuscular injection, which was well tolerated.
Radiation recall dermatitis is an acute inflammatory reaction over an area of skin that was previously irradiated. It is most often triggered by chemotherapy agents and occurs in as many as 9% of patients who receive chemotherapy after radiation.1 Commonly implicated chemotherapy agents include anthracyclines, taxanes, antimetabolites, and alkylating agents. Newer targeted cancer treatments also have been reported to trigger radiation recall dermatitis, including epidermal growth factor receptor inhibitors, vascular endothelial growth factor receptor inhibitors, mammalian target of rapamycin inhibitors, and anti–programmed cell death protein 1 monoclonal antibodies.2-5 Radiation recall dermatitis also has been reported to be triggered by intravenous contrast dye.6
The clinical presentation of radiation recall dermatitis ranges from mild rash to skin necrosis and desquamation. Patients often report pruritus or pain in the affected area. The US National Cancer Institute’s Common Terminology Criteria for Adverse Events (CTCAE) includes a 5-point scale for grading the severity of radiation recall dermatitis: grade 1, faint erythema or dry desquamation; grade 2, moderate to brisk erythema or patchy moist desquamation, mostly confined to skin folds and creases; grade 3, moist desquamation in areas other than skin folds and creases, with bleeding induced by minor trauma or abrasion; grade 4, skin necrosis or ulceration of full-thickness dermis, with spontaneous bleeding; grade 5, death.7 Based on these criteria, our patient had grade 2 radiation recall dermatitis.
In addition to cutaneous inflammation, additional sites can be inflamed, including the gastrointestinal tract, lungs, and oral mucosa. Cases of myocarditis, sialadenitis, and cystitis also have been reported.⁷
Radiation recall dermatitis can occur even if dermatitis did not occur upon initial treatment. The inflammatory reaction can occur weeks or years after initial irradiation. A study evaluating targeted chemotherapy agents found the median time from initiation of chemotherapy to radiation recall dermatitis was 16.9 weeks (range, 1–86.9 weeks). Inflammation usually lasts approximately 1 to 2 weeks but has been reported to persist as long as 14 weeks.8 Withdrawal of the offending agent in addition to administration of corticosteroids or nonsteroidal anti-inflammatory agents typically results in clinical improvement. Histology on skin biopsy is nonspecific and can reveal mixed infiltrates.7
The pathophysiology of radiation recall dermatitis remains unknown; the condition might be an idiosyncratic drug reaction. It has been hypothesized that prior radiation lowers the threshold for an inflammatory reaction, an example of Ruocco immunocompromised cutaneous districts, in which a prior injury at a cutaneous site increases the likelihood of opportunistic infection, tumor, and immune reactions.9 Because radiation can induce expression of inflammatory cytokines, such as IL-1, IL-6, platelet-derived growth factor β, and tumor necrosis factor α, cells in irradiated areas can continue to secrete low levels of these cytokines after radiation therapy, thus priming an inflammatory reaction in the future.10 An alternative theory is that radiation induces mutations within surviving stem cells, rendering them unable to tolerate or unusually sensitive to subsequent chemotherapy and cytotoxic drugs. However, this premise would not explain how noncytotoxic drugs also can trigger radiation recall dermatitis, as described in our case.11
Prednisone-triggered radiation recall dermatitis is curious, as corticosteroids are used to treat the condition. Corticosteroids are classified by their chemical structure, and patch testing can be used to distinguish allergies across the various classes. Hydrocortisone acetate,
In contrast, triamcinolone is a class B steroid, which has a C16,17-cis-diol or -ketal. Other than budesonide, which can cross-react with D2 steroids, class B steroids do not cross-react with hydrocortisone or prednisone. Triamcinolone does not usually cross-react with D2 corticosteroids, which likely explains why our patient was later able to tolerate triamcinolone to treat eosinophilic gastrointestinal tract disease.
In summary, we present a case of radiation recall dermatitis triggered by prednisone. Radiation can prime an area for a future inflammatory response by upregulating proinflammatory cytokines or triggering stem cell mutation. In our case, clinical reactivity to hydrocortisone-17-butyrate and sensitization to tixocortol pivalate via patch testing could have increased the likelihood of a reaction with prednisone use due to cross-reactivity. This case instructs dermatologists, allergists, and oncologists to be aware of prednisone as a potential trigger of radiation recall dermatitis.
- Kodym E, Kalinska R, Ehringfeld C, et al. Frequency of radiation recall dermatitis in adult cancer patients. Onkologie. 2005;28:18-21.
- Seidel C, Janssen S, Karstens JH, et al. Recall pneumonitis during systemic treatment with sunitinib. Ann Oncol. 2010;21:2119-2120.
- Togashi Y, Masago K, Mishima M, et al. A case of radiation recall pneumonitis induced by erlotinib, which can be related to high plasma concentration. J Thorac Oncol. 2010;5:924-925.
- Bourgier C, Massard C, Moldovan C, et al. Total recall of radiotherapy with mTOR inhibitors: a novel and potentially frequent side-effect? Ann Oncol. 2011;22:485-486.
- Korman AM, Tyler KH, Kaffenberger BH. Radiation recall dermatitis associated with nivolumab for metastatic malignant melanoma. Int J Dermatol. 2017;56:e75-e77.
- Lau SKM, Rahimi A. Radiation recall precipitated by iodinated nonionic contrast. Pract Radiat Oncol. 2015;5:263-266.
- US Department of Health and Human Services. Common Terminology Criteria for Adverse Events (CTCAE) Version 5.0. https://ctep.cancer.gov/protocoldevelopment/electronic
_applications/docs/CTCAE_v5_Quick_Reference_8.5x11.pdf. Published November 27, 2017. Accessed June 10, 2020.] - Levy A, Hollebecque A, Bourgier C, et al. Targeted therapy-induced radiation recall. Eur J Cancer. 2013;49:1662-1668.
- Piccolo V, Baroni A, Russo T, et al. Ruocco’s immunocompromised cutaneous district. Int J Dermatol. 2016;55:135-141.
- Johnson CJ, Piedboeuf P, Rubin P, et al. Early and persistent alterations in the expression of interleukin-1 alpha, interleukin-1 beta and tumour necrosis factor alpha mRNA levels in fibrosis-resistant and sensitive mice after thoracic irradiation. Radiat Res. 1996;145:762-767.
- Azira D, Magné N, Zouhair A, et al. Radiation recall: a well recognized but neglected phenomenon. Cancer Treat Rev. 2005;31:555-570.
- Jacob SE, Steele T. Corticosteroid classes: a quick reference guide including patch test substances and cross-reactivity. J Am Acad Dermatol. 2006;54:723-727.
To the Editor:
A 69-year-old woman presented to the allergy clinic for evaluation of a rash on the left breast. The patient had a history of breast cancer that was treated with a lumpectomy followed by external beam radiation therapy (total dose, 6000 cGy) to the lateral aspect of the left breast approximately 4 years prior. She developed acute breast dermatitis from the radiation, which was self-treated with over-the-counter hydrocortisone cream. The patient subsequently developed a blistering skin eruption over the area where she applied the cream. She did not recall the subtype of hydrocortisone she used (butyrate and acetate are available over-the-counter). She discontinued the hydrocortisone and was started on triamcinolone cream 0.1%, which was well tolerated, and the rash resolved.
The patient had a history of a similar reaction to hydrocortisone butyrate after blepharoplasty approximately 10 years prior to the current presentation, characterized by facial erythema, pruritus, and blistering. A patch test confirmed reactivity to hydrocortisone-17-butyrate and tixocortol pivalate. However, a skin-prick test for hydrocortisone acetate cream 1% was negative.
Subsequently, the patient developed acute-onset dyspepsia, gnawing epigastric pain, regurgitation, and bloating. A diagnosis of eosinophilic gastritis was established via biopsy, which found increased eosinophils in the lamina propria (>50 eosinophils per high-power field). Helicobacter pylori was not identified. She was started on the proton-pump inhibitor dexlansoprazole but symptoms did not improve. Her other medications included benazepril, alprazolam as needed, vitamin D, and magnesium. The patient subsequently was started on a trial of oral prednisone 40 mg/d. Three days after initiation, she developed an erythematous macular rash over the left breast.
The next day she presented to the allergy clinic. Physical examination of the left breast revealed a 20×10-cm, nipple-sparing patch of well-demarcated erythema without fluctuance or overlying lesions. The area of erythema overlapped with the prior radiation field based on radiation marker tattoos and the lumpectomy scar (Figure). There was no evidence to suggest inflammation of deeper tissue or the pectoral muscles. Vital signs were normal, and the remainder of the examination was unremarkable, including breast, lymph node, and complete skin examinations.
At evaluation, the differential diagnosis included contact dermatitis, fixed drug eruption, infection, tumor recurrence with overlying skin changes, and radiation recall dermatitis. Given that the dermatitis had developed at the site of previously irradiated skin in the absence of fever or an associated mass, the presentation was thought to be most consistent with radiation recall dermatitis.
Oral prednisone was discontinued, and the dermatitis spontaneously improved in a few weeks. Given the patient’s test results and prior tolerance to triamcinolone, eosinophilic gastroenteritis was treated with triamcinolone acetonide 40 mg via intramuscular injection, which was well tolerated.
Radiation recall dermatitis is an acute inflammatory reaction over an area of skin that was previously irradiated. It is most often triggered by chemotherapy agents and occurs in as many as 9% of patients who receive chemotherapy after radiation.1 Commonly implicated chemotherapy agents include anthracyclines, taxanes, antimetabolites, and alkylating agents. Newer targeted cancer treatments also have been reported to trigger radiation recall dermatitis, including epidermal growth factor receptor inhibitors, vascular endothelial growth factor receptor inhibitors, mammalian target of rapamycin inhibitors, and anti–programmed cell death protein 1 monoclonal antibodies.2-5 Radiation recall dermatitis also has been reported to be triggered by intravenous contrast dye.6
The clinical presentation of radiation recall dermatitis ranges from mild rash to skin necrosis and desquamation. Patients often report pruritus or pain in the affected area. The US National Cancer Institute’s Common Terminology Criteria for Adverse Events (CTCAE) includes a 5-point scale for grading the severity of radiation recall dermatitis: grade 1, faint erythema or dry desquamation; grade 2, moderate to brisk erythema or patchy moist desquamation, mostly confined to skin folds and creases; grade 3, moist desquamation in areas other than skin folds and creases, with bleeding induced by minor trauma or abrasion; grade 4, skin necrosis or ulceration of full-thickness dermis, with spontaneous bleeding; grade 5, death.7 Based on these criteria, our patient had grade 2 radiation recall dermatitis.
In addition to cutaneous inflammation, additional sites can be inflamed, including the gastrointestinal tract, lungs, and oral mucosa. Cases of myocarditis, sialadenitis, and cystitis also have been reported.⁷
Radiation recall dermatitis can occur even if dermatitis did not occur upon initial treatment. The inflammatory reaction can occur weeks or years after initial irradiation. A study evaluating targeted chemotherapy agents found the median time from initiation of chemotherapy to radiation recall dermatitis was 16.9 weeks (range, 1–86.9 weeks). Inflammation usually lasts approximately 1 to 2 weeks but has been reported to persist as long as 14 weeks.8 Withdrawal of the offending agent in addition to administration of corticosteroids or nonsteroidal anti-inflammatory agents typically results in clinical improvement. Histology on skin biopsy is nonspecific and can reveal mixed infiltrates.7
The pathophysiology of radiation recall dermatitis remains unknown; the condition might be an idiosyncratic drug reaction. It has been hypothesized that prior radiation lowers the threshold for an inflammatory reaction, an example of Ruocco immunocompromised cutaneous districts, in which a prior injury at a cutaneous site increases the likelihood of opportunistic infection, tumor, and immune reactions.9 Because radiation can induce expression of inflammatory cytokines, such as IL-1, IL-6, platelet-derived growth factor β, and tumor necrosis factor α, cells in irradiated areas can continue to secrete low levels of these cytokines after radiation therapy, thus priming an inflammatory reaction in the future.10 An alternative theory is that radiation induces mutations within surviving stem cells, rendering them unable to tolerate or unusually sensitive to subsequent chemotherapy and cytotoxic drugs. However, this premise would not explain how noncytotoxic drugs also can trigger radiation recall dermatitis, as described in our case.11
Prednisone-triggered radiation recall dermatitis is curious, as corticosteroids are used to treat the condition. Corticosteroids are classified by their chemical structure, and patch testing can be used to distinguish allergies across the various classes. Hydrocortisone acetate,
In contrast, triamcinolone is a class B steroid, which has a C16,17-cis-diol or -ketal. Other than budesonide, which can cross-react with D2 steroids, class B steroids do not cross-react with hydrocortisone or prednisone. Triamcinolone does not usually cross-react with D2 corticosteroids, which likely explains why our patient was later able to tolerate triamcinolone to treat eosinophilic gastrointestinal tract disease.
In summary, we present a case of radiation recall dermatitis triggered by prednisone. Radiation can prime an area for a future inflammatory response by upregulating proinflammatory cytokines or triggering stem cell mutation. In our case, clinical reactivity to hydrocortisone-17-butyrate and sensitization to tixocortol pivalate via patch testing could have increased the likelihood of a reaction with prednisone use due to cross-reactivity. This case instructs dermatologists, allergists, and oncologists to be aware of prednisone as a potential trigger of radiation recall dermatitis.
To the Editor:
A 69-year-old woman presented to the allergy clinic for evaluation of a rash on the left breast. The patient had a history of breast cancer that was treated with a lumpectomy followed by external beam radiation therapy (total dose, 6000 cGy) to the lateral aspect of the left breast approximately 4 years prior. She developed acute breast dermatitis from the radiation, which was self-treated with over-the-counter hydrocortisone cream. The patient subsequently developed a blistering skin eruption over the area where she applied the cream. She did not recall the subtype of hydrocortisone she used (butyrate and acetate are available over-the-counter). She discontinued the hydrocortisone and was started on triamcinolone cream 0.1%, which was well tolerated, and the rash resolved.
The patient had a history of a similar reaction to hydrocortisone butyrate after blepharoplasty approximately 10 years prior to the current presentation, characterized by facial erythema, pruritus, and blistering. A patch test confirmed reactivity to hydrocortisone-17-butyrate and tixocortol pivalate. However, a skin-prick test for hydrocortisone acetate cream 1% was negative.
Subsequently, the patient developed acute-onset dyspepsia, gnawing epigastric pain, regurgitation, and bloating. A diagnosis of eosinophilic gastritis was established via biopsy, which found increased eosinophils in the lamina propria (>50 eosinophils per high-power field). Helicobacter pylori was not identified. She was started on the proton-pump inhibitor dexlansoprazole but symptoms did not improve. Her other medications included benazepril, alprazolam as needed, vitamin D, and magnesium. The patient subsequently was started on a trial of oral prednisone 40 mg/d. Three days after initiation, she developed an erythematous macular rash over the left breast.
The next day she presented to the allergy clinic. Physical examination of the left breast revealed a 20×10-cm, nipple-sparing patch of well-demarcated erythema without fluctuance or overlying lesions. The area of erythema overlapped with the prior radiation field based on radiation marker tattoos and the lumpectomy scar (Figure). There was no evidence to suggest inflammation of deeper tissue or the pectoral muscles. Vital signs were normal, and the remainder of the examination was unremarkable, including breast, lymph node, and complete skin examinations.
At evaluation, the differential diagnosis included contact dermatitis, fixed drug eruption, infection, tumor recurrence with overlying skin changes, and radiation recall dermatitis. Given that the dermatitis had developed at the site of previously irradiated skin in the absence of fever or an associated mass, the presentation was thought to be most consistent with radiation recall dermatitis.
Oral prednisone was discontinued, and the dermatitis spontaneously improved in a few weeks. Given the patient’s test results and prior tolerance to triamcinolone, eosinophilic gastroenteritis was treated with triamcinolone acetonide 40 mg via intramuscular injection, which was well tolerated.
Radiation recall dermatitis is an acute inflammatory reaction over an area of skin that was previously irradiated. It is most often triggered by chemotherapy agents and occurs in as many as 9% of patients who receive chemotherapy after radiation.1 Commonly implicated chemotherapy agents include anthracyclines, taxanes, antimetabolites, and alkylating agents. Newer targeted cancer treatments also have been reported to trigger radiation recall dermatitis, including epidermal growth factor receptor inhibitors, vascular endothelial growth factor receptor inhibitors, mammalian target of rapamycin inhibitors, and anti–programmed cell death protein 1 monoclonal antibodies.2-5 Radiation recall dermatitis also has been reported to be triggered by intravenous contrast dye.6
The clinical presentation of radiation recall dermatitis ranges from mild rash to skin necrosis and desquamation. Patients often report pruritus or pain in the affected area. The US National Cancer Institute’s Common Terminology Criteria for Adverse Events (CTCAE) includes a 5-point scale for grading the severity of radiation recall dermatitis: grade 1, faint erythema or dry desquamation; grade 2, moderate to brisk erythema or patchy moist desquamation, mostly confined to skin folds and creases; grade 3, moist desquamation in areas other than skin folds and creases, with bleeding induced by minor trauma or abrasion; grade 4, skin necrosis or ulceration of full-thickness dermis, with spontaneous bleeding; grade 5, death.7 Based on these criteria, our patient had grade 2 radiation recall dermatitis.
In addition to cutaneous inflammation, additional sites can be inflamed, including the gastrointestinal tract, lungs, and oral mucosa. Cases of myocarditis, sialadenitis, and cystitis also have been reported.⁷
Radiation recall dermatitis can occur even if dermatitis did not occur upon initial treatment. The inflammatory reaction can occur weeks or years after initial irradiation. A study evaluating targeted chemotherapy agents found the median time from initiation of chemotherapy to radiation recall dermatitis was 16.9 weeks (range, 1–86.9 weeks). Inflammation usually lasts approximately 1 to 2 weeks but has been reported to persist as long as 14 weeks.8 Withdrawal of the offending agent in addition to administration of corticosteroids or nonsteroidal anti-inflammatory agents typically results in clinical improvement. Histology on skin biopsy is nonspecific and can reveal mixed infiltrates.7
The pathophysiology of radiation recall dermatitis remains unknown; the condition might be an idiosyncratic drug reaction. It has been hypothesized that prior radiation lowers the threshold for an inflammatory reaction, an example of Ruocco immunocompromised cutaneous districts, in which a prior injury at a cutaneous site increases the likelihood of opportunistic infection, tumor, and immune reactions.9 Because radiation can induce expression of inflammatory cytokines, such as IL-1, IL-6, platelet-derived growth factor β, and tumor necrosis factor α, cells in irradiated areas can continue to secrete low levels of these cytokines after radiation therapy, thus priming an inflammatory reaction in the future.10 An alternative theory is that radiation induces mutations within surviving stem cells, rendering them unable to tolerate or unusually sensitive to subsequent chemotherapy and cytotoxic drugs. However, this premise would not explain how noncytotoxic drugs also can trigger radiation recall dermatitis, as described in our case.11
Prednisone-triggered radiation recall dermatitis is curious, as corticosteroids are used to treat the condition. Corticosteroids are classified by their chemical structure, and patch testing can be used to distinguish allergies across the various classes. Hydrocortisone acetate,
In contrast, triamcinolone is a class B steroid, which has a C16,17-cis-diol or -ketal. Other than budesonide, which can cross-react with D2 steroids, class B steroids do not cross-react with hydrocortisone or prednisone. Triamcinolone does not usually cross-react with D2 corticosteroids, which likely explains why our patient was later able to tolerate triamcinolone to treat eosinophilic gastrointestinal tract disease.
In summary, we present a case of radiation recall dermatitis triggered by prednisone. Radiation can prime an area for a future inflammatory response by upregulating proinflammatory cytokines or triggering stem cell mutation. In our case, clinical reactivity to hydrocortisone-17-butyrate and sensitization to tixocortol pivalate via patch testing could have increased the likelihood of a reaction with prednisone use due to cross-reactivity. This case instructs dermatologists, allergists, and oncologists to be aware of prednisone as a potential trigger of radiation recall dermatitis.
- Kodym E, Kalinska R, Ehringfeld C, et al. Frequency of radiation recall dermatitis in adult cancer patients. Onkologie. 2005;28:18-21.
- Seidel C, Janssen S, Karstens JH, et al. Recall pneumonitis during systemic treatment with sunitinib. Ann Oncol. 2010;21:2119-2120.
- Togashi Y, Masago K, Mishima M, et al. A case of radiation recall pneumonitis induced by erlotinib, which can be related to high plasma concentration. J Thorac Oncol. 2010;5:924-925.
- Bourgier C, Massard C, Moldovan C, et al. Total recall of radiotherapy with mTOR inhibitors: a novel and potentially frequent side-effect? Ann Oncol. 2011;22:485-486.
- Korman AM, Tyler KH, Kaffenberger BH. Radiation recall dermatitis associated with nivolumab for metastatic malignant melanoma. Int J Dermatol. 2017;56:e75-e77.
- Lau SKM, Rahimi A. Radiation recall precipitated by iodinated nonionic contrast. Pract Radiat Oncol. 2015;5:263-266.
- US Department of Health and Human Services. Common Terminology Criteria for Adverse Events (CTCAE) Version 5.0. https://ctep.cancer.gov/protocoldevelopment/electronic
_applications/docs/CTCAE_v5_Quick_Reference_8.5x11.pdf. Published November 27, 2017. Accessed June 10, 2020.] - Levy A, Hollebecque A, Bourgier C, et al. Targeted therapy-induced radiation recall. Eur J Cancer. 2013;49:1662-1668.
- Piccolo V, Baroni A, Russo T, et al. Ruocco’s immunocompromised cutaneous district. Int J Dermatol. 2016;55:135-141.
- Johnson CJ, Piedboeuf P, Rubin P, et al. Early and persistent alterations in the expression of interleukin-1 alpha, interleukin-1 beta and tumour necrosis factor alpha mRNA levels in fibrosis-resistant and sensitive mice after thoracic irradiation. Radiat Res. 1996;145:762-767.
- Azira D, Magné N, Zouhair A, et al. Radiation recall: a well recognized but neglected phenomenon. Cancer Treat Rev. 2005;31:555-570.
- Jacob SE, Steele T. Corticosteroid classes: a quick reference guide including patch test substances and cross-reactivity. J Am Acad Dermatol. 2006;54:723-727.
- Kodym E, Kalinska R, Ehringfeld C, et al. Frequency of radiation recall dermatitis in adult cancer patients. Onkologie. 2005;28:18-21.
- Seidel C, Janssen S, Karstens JH, et al. Recall pneumonitis during systemic treatment with sunitinib. Ann Oncol. 2010;21:2119-2120.
- Togashi Y, Masago K, Mishima M, et al. A case of radiation recall pneumonitis induced by erlotinib, which can be related to high plasma concentration. J Thorac Oncol. 2010;5:924-925.
- Bourgier C, Massard C, Moldovan C, et al. Total recall of radiotherapy with mTOR inhibitors: a novel and potentially frequent side-effect? Ann Oncol. 2011;22:485-486.
- Korman AM, Tyler KH, Kaffenberger BH. Radiation recall dermatitis associated with nivolumab for metastatic malignant melanoma. Int J Dermatol. 2017;56:e75-e77.
- Lau SKM, Rahimi A. Radiation recall precipitated by iodinated nonionic contrast. Pract Radiat Oncol. 2015;5:263-266.
- US Department of Health and Human Services. Common Terminology Criteria for Adverse Events (CTCAE) Version 5.0. https://ctep.cancer.gov/protocoldevelopment/electronic
_applications/docs/CTCAE_v5_Quick_Reference_8.5x11.pdf. Published November 27, 2017. Accessed June 10, 2020.] - Levy A, Hollebecque A, Bourgier C, et al. Targeted therapy-induced radiation recall. Eur J Cancer. 2013;49:1662-1668.
- Piccolo V, Baroni A, Russo T, et al. Ruocco’s immunocompromised cutaneous district. Int J Dermatol. 2016;55:135-141.
- Johnson CJ, Piedboeuf P, Rubin P, et al. Early and persistent alterations in the expression of interleukin-1 alpha, interleukin-1 beta and tumour necrosis factor alpha mRNA levels in fibrosis-resistant and sensitive mice after thoracic irradiation. Radiat Res. 1996;145:762-767.
- Azira D, Magné N, Zouhair A, et al. Radiation recall: a well recognized but neglected phenomenon. Cancer Treat Rev. 2005;31:555-570.
- Jacob SE, Steele T. Corticosteroid classes: a quick reference guide including patch test substances and cross-reactivity. J Am Acad Dermatol. 2006;54:723-727.
Practice Points
- Consider the diagnosis of radiation recall dermatitis for a skin eruption that occurs in the same location as prior radiation exposure.
- Prednisone may be a trigger for radiation recall dermatitis in patients with sensitization to cross-reactive topical steroids such as tixocortol pivalate.
- Radiation therapy may prime the skin for a future inflammatory response by upregulating proinflammatory cytokines that persist after the conclusion of treatment.
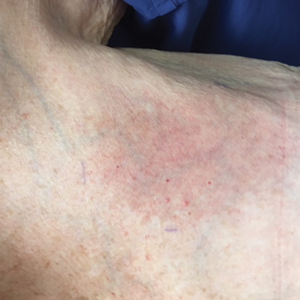
Mycosis Fungoides Manifesting as a Morbilliform Eruption Mimicking a Viral Exanthem
To the Editor:
Mycosis fungoides (MF) is the most common type of primary cutaneous lymphoma, occurring in approximately 4 of 1 million individuals per year in the United States.1 It classically occurs in patch, plaque, and tumor stages with lesions preferentially occurring on regions of the body spared from sun exposure2; however, MF is known to have variable presentations and has been reported to imitate at least 25 other dermatoses.3 This case describes MF as a morbilliform eruption mimicking a viral exanthem.
A 30-year-old man with a 12-year history of nodular sclerosing Hodgkin lymphoma (HL) presented with a widespread rash of 2 weeks’ duration. At the time of diagnosis of HL, the patient had several slightly enlarged, hyperdense, bilateral inguinal lymph nodes seen on positron emission tomography–computed tomography. He achieved complete remission 11 years prior after 6 cycles of ABVD (doxorubicin-bleomycin-vinblastine-dacarbazine) chemotherapy. He initially presented to us prior to starting chemotherapy for evaluation of what he described as eczema on the bilateral arms and legs that had been present for 10 years. Findings from a skin biopsy of an erythematous scaling patch on the left lateral thigh were consistent with MF. One year later, new lesions on the left lateral thigh were clinically and histologically consistent with lymphomatoid papulosis (LyP).
At the current presentation, the patient denied any changes in medications, which consisted of topical clobetasol, triamcinolone, and mupirocin; however, he reported that his young child had recently been diagnosed with bronchitis and impetigo. Physical examination revealed pink-orange macules and papules on the anterior and posterior trunk, medial upper arms, and bilateral legs involving 18% of the body surface area. A complete blood cell count showed no leukocytosis or left shift. A respiratory viral panel was positive for human metapneumovirus. Two weeks later, the patient noted improvement of the rash with use of topical triamcinolone.
Four months later, the rash still had not completely resolved and now involved 50% of the body surface area. A punch biopsy of the left lower abdomen demonstrated an atypical lymphoid infiltrate with focal epidermotropism and predominance of CD4 over CD8 cells (approximately 4:1 ratio), and CD30 labeled rare cells. Polymerase chain reaction analysis of the biopsy revealed monoclonal T-cell receptor gamma chain gene rearrangement. Taken together, the findings were consistent with MF. The patient started narrowband UVB phototherapy and completed a total of 25 treatments, reaching a maximum 4-minute dose, with minimal improvement.
Three months later, the patient had 90% body surface area involvement and started treatment with intramuscular interferon alfa-2b at 1 million units 3 times weekly. He noticed improvement within the first week of treatment and reported that his skin was clear until 5 months later when he woke up one morning with a morbilliform eruption on the anterior trunk, thighs, and upper arms (Figure 1). Biopsy from the right thigh showed an infiltrate of CD3+ lymphocytes with a predominance of CD4 over CD8 cells (approximately 6:1 ratio), both in the dermis and epidermis (Figure 2). CD30 highlighted approximately 10% of cells (Figure 3). Findings again were consistent with MF. Flow cytometry was negative for peripheral blood involvement.
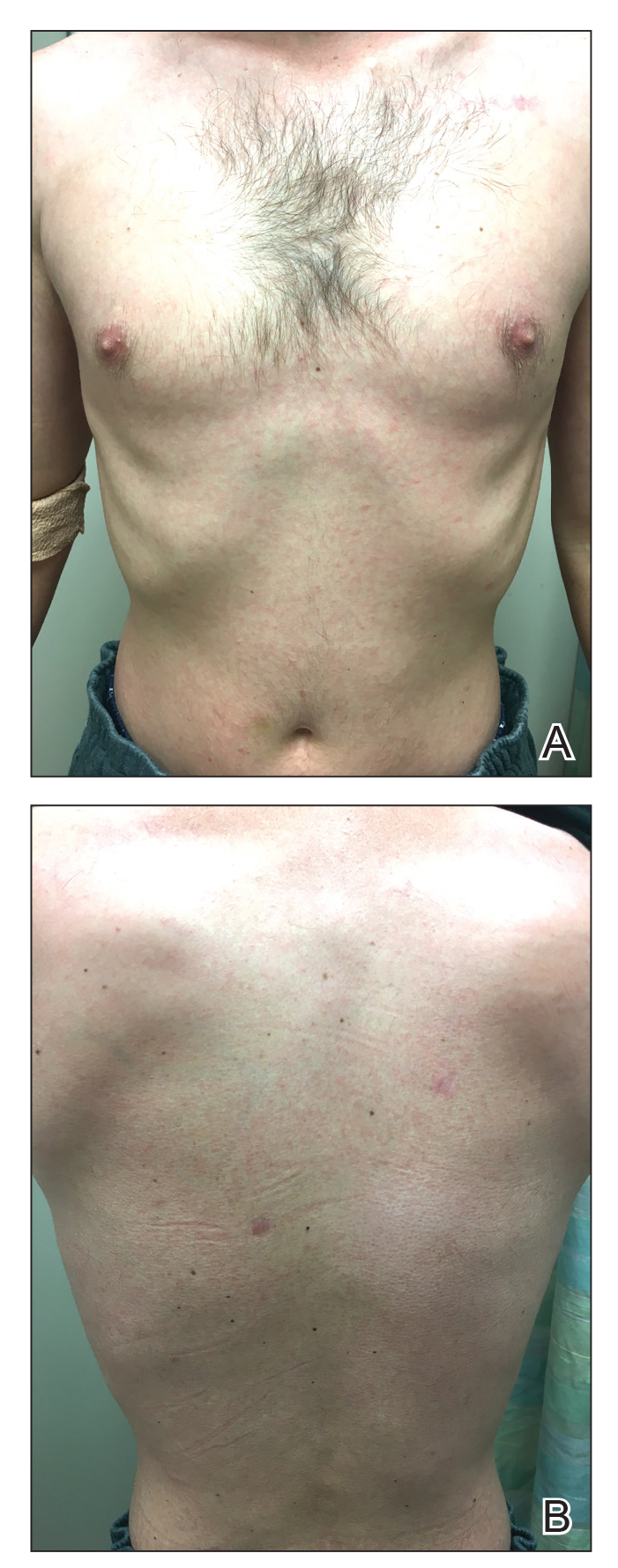
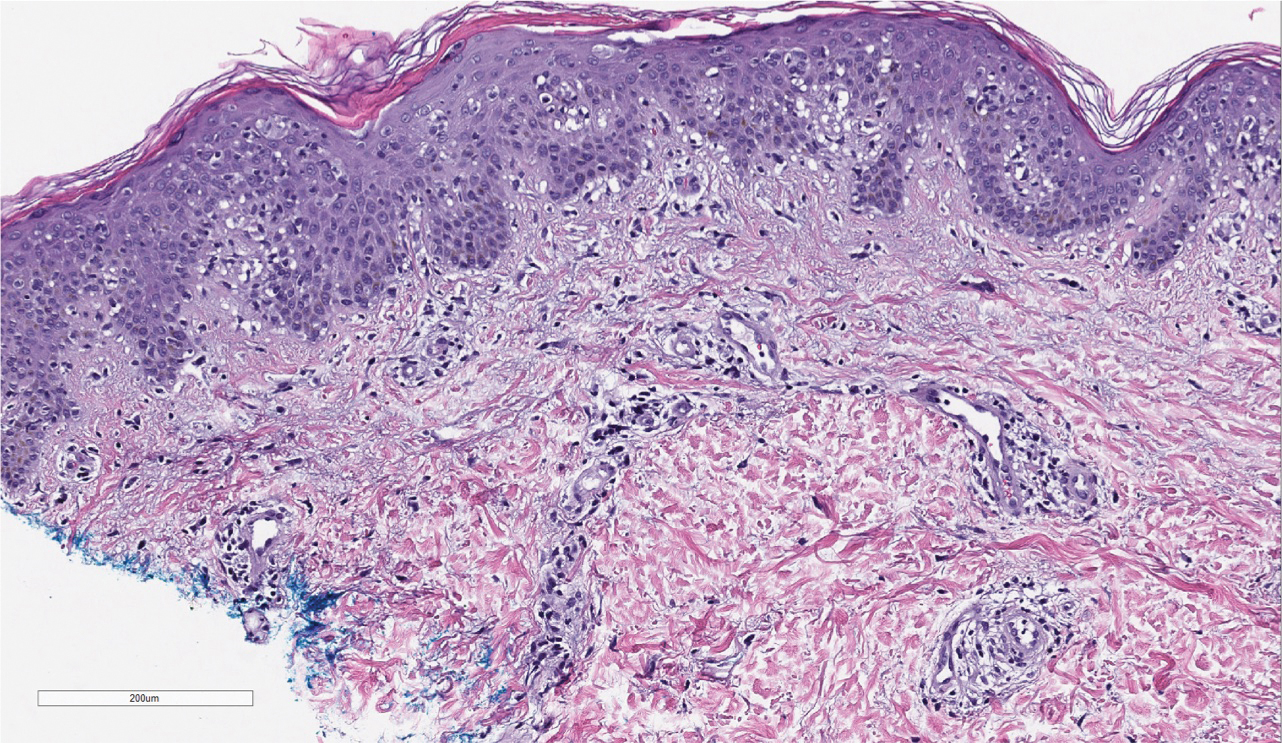
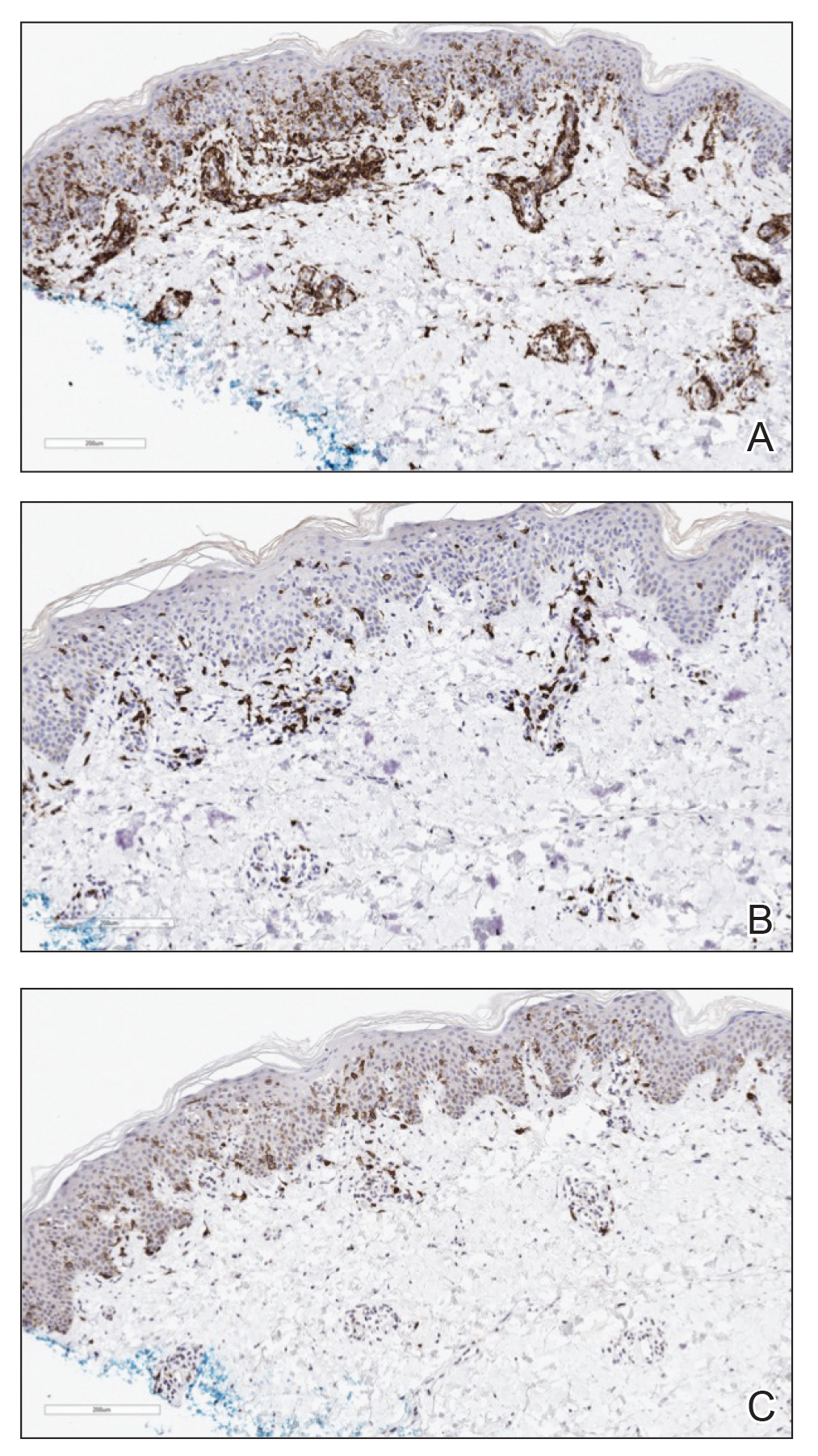
Three months later, the patient reported enlargement of several left inguinal nodes. Fine needle aspiration of 1 node demonstrated an atypical lymphoid proliferation consistent with MF. Positron emission tomography–computed tomography showed several mildly enlarged inguinal lymph nodes, which were unchanged from the initial diagnosis of HL. There were no hypermetabolic lesions. One month later, the patient started extracorporeal electrophoresis in addition to interferon alfa-2b with notable improvement of the rash. The rash later recurred after completion of these treatments and continues to have a waxing and waning course. It is currently managed with triamcinolone cream only.
At the time of the initial diagnosis of MF, the patient’s lesions appeared as eczematous patches on the face, abdomen, buttocks, and legs. Based on the history of a sick child at home, viral panel positive for human metapneumovirus, and clinical appearance, a viral exanthem was considered to be a likely explanation for the patient’s new-onset morbilliform eruption rash occurring 12 years later. A drug reaction also was considered in the differential based on the appearance of the rash; however, it was deemed less likely because the patient reported no changes in his medications at the time of rash onset. Persistence of the eruption for many months was less consistent with a reactive condition. A biopsy demonstrated the rash to be histologically consistent with MF. This patient was a rare case of MF manifesting as a morbilliform eruption mimicking a viral exanthem.
Various inflammatory conditions, including drug eruptions and lichen sclerosus et atrophicus, may mimic MF, not only based on their histophenotypic findings but also occasionally clonal proliferation by molecular study.4,5 In our patient, one consideration was the possibility of a viral infection mimicking MF; however, biopsies showed both definite histophenotypic features of MF and clonality. More importantly, subsequent biopsy also revealed similar findings by morphology, immunohistochemical study, and T-cell gene rearrangement study, confirming the diagnosis of MF.
Another interesting feature of our case was the occurrence of HL, LyP, and MF in the same patient. Lymphomatoid papulosis is a chronic condition characterized by self-healing lesions and histologic features suggestive of malignancy that lies within a spectrum of primary cutaneous CD30+ lymphoproliferative disorders. There is a known association between LyP and an increased incidence of lymphomas, including MF and HL.1 In a 2016 study, lymphomas occurred in 52% of patients with LyP (N=180), with MF being the most frequently associated lymphoma.6 Notably, biopsies consistent with both HL and MF, respectively, in our patient were positive for the CD30 marker. Patients with HL also are at increased risk for developing other malignancies, with the risk of leukemias and non-HLs greater than that of solid tumors.5 There have been multiple reported cases of HL and MF occurring in the same patient and at least one prior reported case of LyP, HL, and MF occurring in the same patient.6,7
This case highlights the myriad presentations of MF and describes an unusual case of MF manifesting as a morbilliform eruption mimicking a viral exanthem.
- de la Garza Bravo MM, Patel KP, Loghavi S, et al. Shared clonality in distinctive lesions of lymphomatoid papulosis and mycosis fungoides occurring in the same patients suggests a common origin [published online December 31, 2014]. Hum Pathol. 2015;46:558-569.
- Howard MS, Smoller BR. Mycosis fungoides: classic disease and variant presentations. Semin Cutan Med Surg. 2000;19:91-99.
- Zackheim HS, Mccalmont TH. Mycosis fungoides: the great imitator. J Am Acad Dermatol. 2002;47:914-918.
- Suchak R, Verdolini R, Robson A, et al. Extragenital lichen sclerosus et atrophicus mimicking cutaneous T-cell lymphoma: report of a case. J Cutan Pathol. 2010;37:982-986.
- Sarantopoulos GP, Palla B, Said J, et al. Mimics of cutaneous lymphoma: report of the 2011 Society for Hematopathology/European Association for Haematopathology workshop. Am J Clin Pathol. 2013;139:536-551.
- Wieser I, Oh CW, Talpur R, et al. Lymphomatoid papulosis: treatment response and associated lymphomas in a study of 180 patients. J Am Acad Dermatol. 2016;74:59-67.
- Sont JK, van Stiphout WA, Noordijk EM, et al. Increased risk of second cancers in managing Hodgkins disease: the 20-year Leiden experience. Ann Hematol. 1992;65:213-218.
To the Editor:
Mycosis fungoides (MF) is the most common type of primary cutaneous lymphoma, occurring in approximately 4 of 1 million individuals per year in the United States.1 It classically occurs in patch, plaque, and tumor stages with lesions preferentially occurring on regions of the body spared from sun exposure2; however, MF is known to have variable presentations and has been reported to imitate at least 25 other dermatoses.3 This case describes MF as a morbilliform eruption mimicking a viral exanthem.
A 30-year-old man with a 12-year history of nodular sclerosing Hodgkin lymphoma (HL) presented with a widespread rash of 2 weeks’ duration. At the time of diagnosis of HL, the patient had several slightly enlarged, hyperdense, bilateral inguinal lymph nodes seen on positron emission tomography–computed tomography. He achieved complete remission 11 years prior after 6 cycles of ABVD (doxorubicin-bleomycin-vinblastine-dacarbazine) chemotherapy. He initially presented to us prior to starting chemotherapy for evaluation of what he described as eczema on the bilateral arms and legs that had been present for 10 years. Findings from a skin biopsy of an erythematous scaling patch on the left lateral thigh were consistent with MF. One year later, new lesions on the left lateral thigh were clinically and histologically consistent with lymphomatoid papulosis (LyP).
At the current presentation, the patient denied any changes in medications, which consisted of topical clobetasol, triamcinolone, and mupirocin; however, he reported that his young child had recently been diagnosed with bronchitis and impetigo. Physical examination revealed pink-orange macules and papules on the anterior and posterior trunk, medial upper arms, and bilateral legs involving 18% of the body surface area. A complete blood cell count showed no leukocytosis or left shift. A respiratory viral panel was positive for human metapneumovirus. Two weeks later, the patient noted improvement of the rash with use of topical triamcinolone.
Four months later, the rash still had not completely resolved and now involved 50% of the body surface area. A punch biopsy of the left lower abdomen demonstrated an atypical lymphoid infiltrate with focal epidermotropism and predominance of CD4 over CD8 cells (approximately 4:1 ratio), and CD30 labeled rare cells. Polymerase chain reaction analysis of the biopsy revealed monoclonal T-cell receptor gamma chain gene rearrangement. Taken together, the findings were consistent with MF. The patient started narrowband UVB phototherapy and completed a total of 25 treatments, reaching a maximum 4-minute dose, with minimal improvement.
Three months later, the patient had 90% body surface area involvement and started treatment with intramuscular interferon alfa-2b at 1 million units 3 times weekly. He noticed improvement within the first week of treatment and reported that his skin was clear until 5 months later when he woke up one morning with a morbilliform eruption on the anterior trunk, thighs, and upper arms (Figure 1). Biopsy from the right thigh showed an infiltrate of CD3+ lymphocytes with a predominance of CD4 over CD8 cells (approximately 6:1 ratio), both in the dermis and epidermis (Figure 2). CD30 highlighted approximately 10% of cells (Figure 3). Findings again were consistent with MF. Flow cytometry was negative for peripheral blood involvement.
Three months later, the patient reported enlargement of several left inguinal nodes. Fine needle aspiration of 1 node demonstrated an atypical lymphoid proliferation consistent with MF. Positron emission tomography–computed tomography showed several mildly enlarged inguinal lymph nodes, which were unchanged from the initial diagnosis of HL. There were no hypermetabolic lesions. One month later, the patient started extracorporeal electrophoresis in addition to interferon alfa-2b with notable improvement of the rash. The rash later recurred after completion of these treatments and continues to have a waxing and waning course. It is currently managed with triamcinolone cream only.
At the time of the initial diagnosis of MF, the patient’s lesions appeared as eczematous patches on the face, abdomen, buttocks, and legs. Based on the history of a sick child at home, viral panel positive for human metapneumovirus, and clinical appearance, a viral exanthem was considered to be a likely explanation for the patient’s new-onset morbilliform eruption rash occurring 12 years later. A drug reaction also was considered in the differential based on the appearance of the rash; however, it was deemed less likely because the patient reported no changes in his medications at the time of rash onset. Persistence of the eruption for many months was less consistent with a reactive condition. A biopsy demonstrated the rash to be histologically consistent with MF. This patient was a rare case of MF manifesting as a morbilliform eruption mimicking a viral exanthem.
Various inflammatory conditions, including drug eruptions and lichen sclerosus et atrophicus, may mimic MF, not only based on their histophenotypic findings but also occasionally clonal proliferation by molecular study.4,5 In our patient, one consideration was the possibility of a viral infection mimicking MF; however, biopsies showed both definite histophenotypic features of MF and clonality. More importantly, subsequent biopsy also revealed similar findings by morphology, immunohistochemical study, and T-cell gene rearrangement study, confirming the diagnosis of MF.
Another interesting feature of our case was the occurrence of HL, LyP, and MF in the same patient. Lymphomatoid papulosis is a chronic condition characterized by self-healing lesions and histologic features suggestive of malignancy that lies within a spectrum of primary cutaneous CD30+ lymphoproliferative disorders. There is a known association between LyP and an increased incidence of lymphomas, including MF and HL.1 In a 2016 study, lymphomas occurred in 52% of patients with LyP (N=180), with MF being the most frequently associated lymphoma.6 Notably, biopsies consistent with both HL and MF, respectively, in our patient were positive for the CD30 marker. Patients with HL also are at increased risk for developing other malignancies, with the risk of leukemias and non-HLs greater than that of solid tumors.5 There have been multiple reported cases of HL and MF occurring in the same patient and at least one prior reported case of LyP, HL, and MF occurring in the same patient.6,7
This case highlights the myriad presentations of MF and describes an unusual case of MF manifesting as a morbilliform eruption mimicking a viral exanthem.
To the Editor:
Mycosis fungoides (MF) is the most common type of primary cutaneous lymphoma, occurring in approximately 4 of 1 million individuals per year in the United States.1 It classically occurs in patch, plaque, and tumor stages with lesions preferentially occurring on regions of the body spared from sun exposure2; however, MF is known to have variable presentations and has been reported to imitate at least 25 other dermatoses.3 This case describes MF as a morbilliform eruption mimicking a viral exanthem.
A 30-year-old man with a 12-year history of nodular sclerosing Hodgkin lymphoma (HL) presented with a widespread rash of 2 weeks’ duration. At the time of diagnosis of HL, the patient had several slightly enlarged, hyperdense, bilateral inguinal lymph nodes seen on positron emission tomography–computed tomography. He achieved complete remission 11 years prior after 6 cycles of ABVD (doxorubicin-bleomycin-vinblastine-dacarbazine) chemotherapy. He initially presented to us prior to starting chemotherapy for evaluation of what he described as eczema on the bilateral arms and legs that had been present for 10 years. Findings from a skin biopsy of an erythematous scaling patch on the left lateral thigh were consistent with MF. One year later, new lesions on the left lateral thigh were clinically and histologically consistent with lymphomatoid papulosis (LyP).
At the current presentation, the patient denied any changes in medications, which consisted of topical clobetasol, triamcinolone, and mupirocin; however, he reported that his young child had recently been diagnosed with bronchitis and impetigo. Physical examination revealed pink-orange macules and papules on the anterior and posterior trunk, medial upper arms, and bilateral legs involving 18% of the body surface area. A complete blood cell count showed no leukocytosis or left shift. A respiratory viral panel was positive for human metapneumovirus. Two weeks later, the patient noted improvement of the rash with use of topical triamcinolone.
Four months later, the rash still had not completely resolved and now involved 50% of the body surface area. A punch biopsy of the left lower abdomen demonstrated an atypical lymphoid infiltrate with focal epidermotropism and predominance of CD4 over CD8 cells (approximately 4:1 ratio), and CD30 labeled rare cells. Polymerase chain reaction analysis of the biopsy revealed monoclonal T-cell receptor gamma chain gene rearrangement. Taken together, the findings were consistent with MF. The patient started narrowband UVB phototherapy and completed a total of 25 treatments, reaching a maximum 4-minute dose, with minimal improvement.
Three months later, the patient had 90% body surface area involvement and started treatment with intramuscular interferon alfa-2b at 1 million units 3 times weekly. He noticed improvement within the first week of treatment and reported that his skin was clear until 5 months later when he woke up one morning with a morbilliform eruption on the anterior trunk, thighs, and upper arms (Figure 1). Biopsy from the right thigh showed an infiltrate of CD3+ lymphocytes with a predominance of CD4 over CD8 cells (approximately 6:1 ratio), both in the dermis and epidermis (Figure 2). CD30 highlighted approximately 10% of cells (Figure 3). Findings again were consistent with MF. Flow cytometry was negative for peripheral blood involvement.
Three months later, the patient reported enlargement of several left inguinal nodes. Fine needle aspiration of 1 node demonstrated an atypical lymphoid proliferation consistent with MF. Positron emission tomography–computed tomography showed several mildly enlarged inguinal lymph nodes, which were unchanged from the initial diagnosis of HL. There were no hypermetabolic lesions. One month later, the patient started extracorporeal electrophoresis in addition to interferon alfa-2b with notable improvement of the rash. The rash later recurred after completion of these treatments and continues to have a waxing and waning course. It is currently managed with triamcinolone cream only.
At the time of the initial diagnosis of MF, the patient’s lesions appeared as eczematous patches on the face, abdomen, buttocks, and legs. Based on the history of a sick child at home, viral panel positive for human metapneumovirus, and clinical appearance, a viral exanthem was considered to be a likely explanation for the patient’s new-onset morbilliform eruption rash occurring 12 years later. A drug reaction also was considered in the differential based on the appearance of the rash; however, it was deemed less likely because the patient reported no changes in his medications at the time of rash onset. Persistence of the eruption for many months was less consistent with a reactive condition. A biopsy demonstrated the rash to be histologically consistent with MF. This patient was a rare case of MF manifesting as a morbilliform eruption mimicking a viral exanthem.
Various inflammatory conditions, including drug eruptions and lichen sclerosus et atrophicus, may mimic MF, not only based on their histophenotypic findings but also occasionally clonal proliferation by molecular study.4,5 In our patient, one consideration was the possibility of a viral infection mimicking MF; however, biopsies showed both definite histophenotypic features of MF and clonality. More importantly, subsequent biopsy also revealed similar findings by morphology, immunohistochemical study, and T-cell gene rearrangement study, confirming the diagnosis of MF.
Another interesting feature of our case was the occurrence of HL, LyP, and MF in the same patient. Lymphomatoid papulosis is a chronic condition characterized by self-healing lesions and histologic features suggestive of malignancy that lies within a spectrum of primary cutaneous CD30+ lymphoproliferative disorders. There is a known association between LyP and an increased incidence of lymphomas, including MF and HL.1 In a 2016 study, lymphomas occurred in 52% of patients with LyP (N=180), with MF being the most frequently associated lymphoma.6 Notably, biopsies consistent with both HL and MF, respectively, in our patient were positive for the CD30 marker. Patients with HL also are at increased risk for developing other malignancies, with the risk of leukemias and non-HLs greater than that of solid tumors.5 There have been multiple reported cases of HL and MF occurring in the same patient and at least one prior reported case of LyP, HL, and MF occurring in the same patient.6,7
This case highlights the myriad presentations of MF and describes an unusual case of MF manifesting as a morbilliform eruption mimicking a viral exanthem.
- de la Garza Bravo MM, Patel KP, Loghavi S, et al. Shared clonality in distinctive lesions of lymphomatoid papulosis and mycosis fungoides occurring in the same patients suggests a common origin [published online December 31, 2014]. Hum Pathol. 2015;46:558-569.
- Howard MS, Smoller BR. Mycosis fungoides: classic disease and variant presentations. Semin Cutan Med Surg. 2000;19:91-99.
- Zackheim HS, Mccalmont TH. Mycosis fungoides: the great imitator. J Am Acad Dermatol. 2002;47:914-918.
- Suchak R, Verdolini R, Robson A, et al. Extragenital lichen sclerosus et atrophicus mimicking cutaneous T-cell lymphoma: report of a case. J Cutan Pathol. 2010;37:982-986.
- Sarantopoulos GP, Palla B, Said J, et al. Mimics of cutaneous lymphoma: report of the 2011 Society for Hematopathology/European Association for Haematopathology workshop. Am J Clin Pathol. 2013;139:536-551.
- Wieser I, Oh CW, Talpur R, et al. Lymphomatoid papulosis: treatment response and associated lymphomas in a study of 180 patients. J Am Acad Dermatol. 2016;74:59-67.
- Sont JK, van Stiphout WA, Noordijk EM, et al. Increased risk of second cancers in managing Hodgkins disease: the 20-year Leiden experience. Ann Hematol. 1992;65:213-218.
- de la Garza Bravo MM, Patel KP, Loghavi S, et al. Shared clonality in distinctive lesions of lymphomatoid papulosis and mycosis fungoides occurring in the same patients suggests a common origin [published online December 31, 2014]. Hum Pathol. 2015;46:558-569.
- Howard MS, Smoller BR. Mycosis fungoides: classic disease and variant presentations. Semin Cutan Med Surg. 2000;19:91-99.
- Zackheim HS, Mccalmont TH. Mycosis fungoides: the great imitator. J Am Acad Dermatol. 2002;47:914-918.
- Suchak R, Verdolini R, Robson A, et al. Extragenital lichen sclerosus et atrophicus mimicking cutaneous T-cell lymphoma: report of a case. J Cutan Pathol. 2010;37:982-986.
- Sarantopoulos GP, Palla B, Said J, et al. Mimics of cutaneous lymphoma: report of the 2011 Society for Hematopathology/European Association for Haematopathology workshop. Am J Clin Pathol. 2013;139:536-551.
- Wieser I, Oh CW, Talpur R, et al. Lymphomatoid papulosis: treatment response and associated lymphomas in a study of 180 patients. J Am Acad Dermatol. 2016;74:59-67.
- Sont JK, van Stiphout WA, Noordijk EM, et al. Increased risk of second cancers in managing Hodgkins disease: the 20-year Leiden experience. Ann Hematol. 1992;65:213-218.
Practice Points
- Mycosis fungoides classically occurs in patch, plaque, and tumor stages, with lesions preferentially occurring on regions of the body spared from sun exposure; however, the condition may present atypically, mimicking a variety of other conditions.
- Lymphomatoid papulosis exists within a spectrum of primary cutaneous CD30+ lymphoproliferative disorders and is associated with increased incidence of lymphomas.
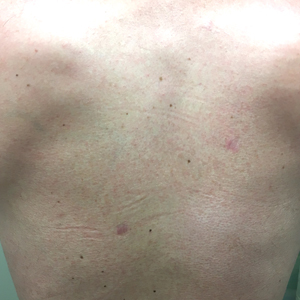
Severe Gingival Swelling and Erythema
The Diagnosis: Plasma Cell Gingivitis
Microscopic analysis demonstrated an acanthotic stratified squamous epithelium with an edematous fibrous stroma containing dense perivascular infiltrates of plasma cells and lymphocytes (Figure 1). Immunohistochemical analysis with kappa, lambda, and CD79a immunostains indicated a polyclonal proliferation of plasma cells that excluded monoclonal plasma cell neoplasia (Figure 2). Direct immunofluorescence (DIF) was negative. Serum enzyme-linked immunosorbent assay for bullous pemphigoid 180 and 230 antibodies as well as desmoglein 1 and 3 antibodies was normal. The cumulative findings were consistent with plasma cell gingivitis (PCG). It was recommended that the patient avoid possible foods (eg, citrus) and oral hygiene products (eg, mint-flavored toothpaste) that could trigger PCG. With patient compliance to an elimination diet for 3 months, the condition resolved (Figure 3).
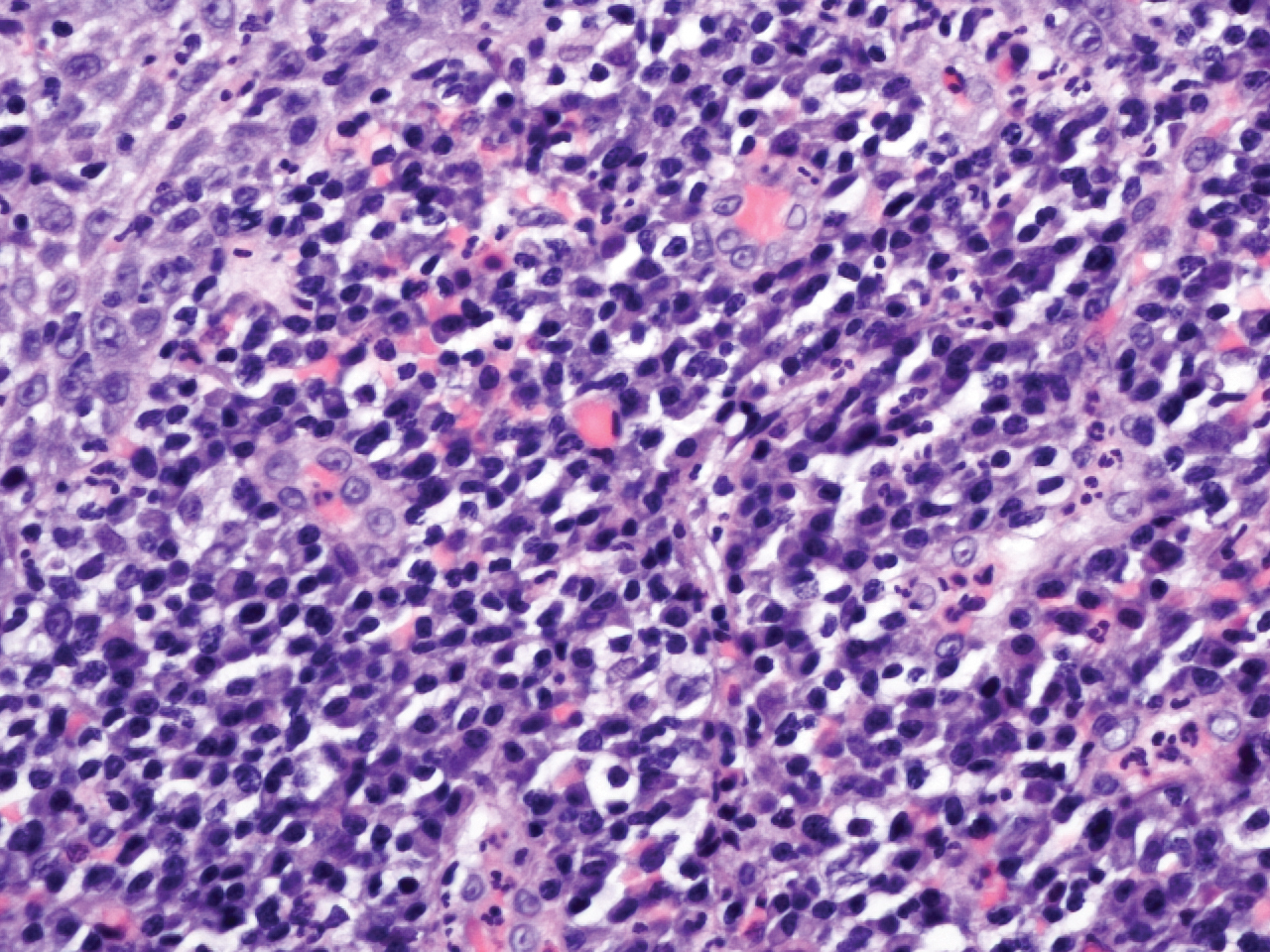
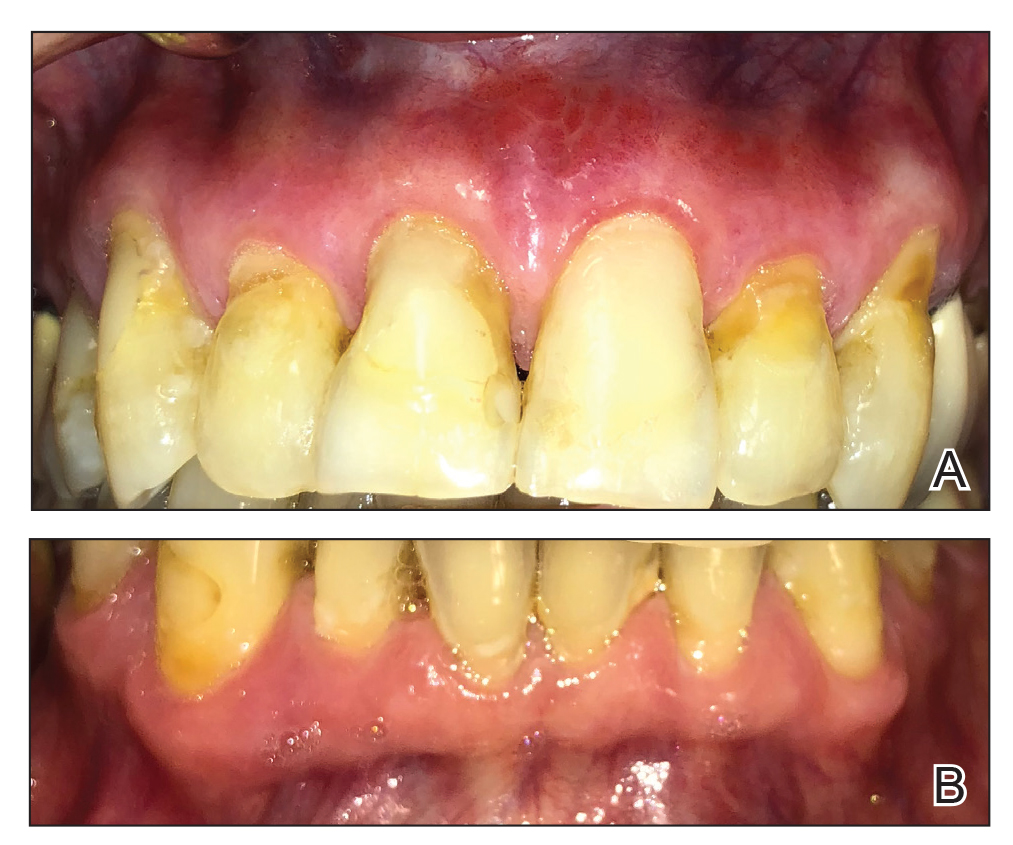
Plasma cell gingivitis is a rare condition characterized by generalized edema and erythema of the attached gingiva. It was described in the 1960s and classified into 3 types based on etiology: (1) hypersensitivity (most common), (2) neoplastic, and (3) PCG of unknown origin.1,2 Spices, herbs, and flavoring agents are implicated as potential triggers of hypersensitivity PCG, while neoplastic PCG is associated with monoclonal plasma cell neoplasms, such as multiple myeloma and extramedullary plasmacytoma.2,3 Histologically, a diffuse subepithelial infiltrate of a polyclonal mixture of plasma cells typically is observed in hypersensitivity PCG.3 The plasma cell infiltration in hypersensitivity PCG is a benign reactive process without known risk for development of plasma cell malignancy, but the presence of a notable number of plasma cells may require special tissue staining to rule out the possibility of associated neoplasia.2,3 There are no standardized protocols for management of PCG.4 Elimination of potential allergens, including flavored oral hygiene products, may result in resolution of hypersensitivity PCG lesions, as exemplified in our patient.1 Neoplastic PCG responds to treatment of the underlying malignancy.5 Topical, intralesional, and/or systemic steroids may be considered in symptomatic cases of PCG.4
Clinical presentation of PCG can mimic immune-mediated mucocutaneous diseases such as mucous membrane pemphigoid (MMP), pemphigus vulgaris (PV), and oral lichen planus; microscopic analysis is needed to establish the diagnosis.6 Mucous membrane pemphigoid is a chronic autoimmune blistering disease involving the mucous membranes with possible cutaneous involvement. It is characterized by a complement-mediated autoantibody process against one or several antigens in the epithelial basement membrane. The oral mucosa is involved in 85% of MMP patients, and 65% of patients experience complications involving the ocular conjunctiva. Intraorally, MMP typically manifests as painful erosions, ulcerations, desquamative gingivitis, and/or occasionally intact blisters. Ocular complications include conjunctivitis and corneal erosions that often scar, resulting in blindness in approximately 15% of patients with ocular involvement. Microscopic features of MMP classically exhibit subepithelial separation with a mixed inflammatory cell infiltrate on routine analysis and linear deposition of IgG, IgA, or C3 within the basement membrane zone on DIF. Treatment of MMP involves topical or systemic immunosuppressants to control symptoms, minimize complications, and alter disease progression.6
Pemphigus vulgaris is an autoimmune vesiculobullous disease that affects the oral mucosa with or without cutaneous involvement.7 Desmogleins 1 and 3, transmembrane glycoproteins of desmosomes that convene cell-to-cell adhesion, are identified as antigens in PV. Antibodies against these desmoglein proteins result in intraepithelial separation, which leads to blister formation.7 Oral manifestations of PV include mucosal erosions and ulcerations as well as desquamative gingivitis. Bullae rarely are seen in the oral cavity, as they tend to rupture, leaving nonhealing ulcerations.8 Histologically, PV is characterized by acantholysis of the suprabasal cell layers with an intact basement membrane zone on routine examination. The distinctive microscopic feature of PV is the detection of cell surface-bound IgG within the epidermis on DIF.7 Treatment of PV may include topical and/or systemic corticosteroids and other immunosuppressants. Rituximab, a monoclonal antibody, has been successful in the management of PV.8
Oral lichen planus is a T-cell mediated autoimmune condition that leads to subepithelial lymphocytic infiltration and excessive keratinocyte apoptosis.9 Women typically are affected more often than men, and 75% of patients also have cutaneous manifestations of the condition. Desquamation and/or erythema of the gingiva may be the initial manifestation of oral lichen planus.9 Other commonly involved sites include the buccal mucosa, tongue, and palate. Biopsy of affected tissues typically demonstrates degeneration of the basal cell layer with subjacent bandlike lymphocytic infiltration on routine staining. Linear fibrinogen at the basement membrane zone usually is observed on DIF. Topical corticosteroids are considered first-line therapy, but systemic therapy including corticosteroids, steroid-sparing agents, or immunomodulators may be used in severe cases.9
There are 3 variants of plasma cell neoplasms including multiple myeloma, medullary plasmacytoma (also known as solitary bone plasmacytoma), and extramedullary plasmacytoma (EMP).10 Extramedullary plasmacytoma, sometimes referred to as extraosseous plasmacytoma, is described as a solitary or multiple plasma cell neoplasm contained in the soft tissue. Its occurrence is rare, accounting for only 3% of plasma cell neoplasms. Approximately 90% of EMPs affect the head and neck region, and males are affected 4 times more often than females. The oral cavity is one of the sites of clinical presentation; the gingival tissue infrequently is affected. When EMP affects the gingiva, it can mimic any form of gingivitis as well as other benign inflammatory conditions, such as pyogenic granuloma. Biopsy is the gold standard diagnostic method for differentiating EMP from other conditions, and specific immunohistochemical stains are essential for the diagnosis. Extramedullary plasmacytoma has the best prognosis among plasma cell neoplasms, despite the risk for progression to multiple myeloma. Extramedullary plasmacytoma lesions are very sensitive to radiotherapy, and the 10-year survival rate is approximately 70%.10
- Sollecito TP, Greenberg MS. Plasma cell gingivitis: report of two cases. Oral Surg Oral Med Oral Pathol. 1992;73:690-693.
- Gargiulo AV, Ladone JA, Ladone PA, et al. Case report: plasma cell gingivitis A. CDS Rev. 1995;88:22-23.
- Abhishek K, Rashmi J. Plasma cell gingivitis associated with inflammatory cheilitis: a report on a rare case. Ethiop J Health Sci. 2013;23:183-187.
- Arduino PG, D'Aiuto F, Cavallito C, et al. Professional oral hygiene as a therapeutic option for pediatric patients with plasma cell gingivitis: preliminary results of a prospective case series. J Periodontol. 2011;82:1670-1675.
- Nayak A, Nayak MT. Multiple myeloma with an unusual oral presentation. J Exp Ther Oncol. 2016;11:199-206.
- Xu HH, Werth VP, Parisi E, et al. Mucous membrane pemphigoid. Dent Clin North Am. 2013;57:611-630.
- Hammers CM, Stanley JR. Mechanisms of disease: pemphigus and bullous pemphigoid. Ann Rev Pathol. 2016;11:75-97.
- Cizenski JD, Michel P, Watson IT, et al. Spectrum of orocutaneous disease associations: immune-mediated conditions. J Am Acad Dermatol. 2017;77:795-806.
- Stoopler ET, Sollecito TP. Recurrent gingival and oral mucosal lesions. JAMA. 2014;312:1794-1795.
- Nair SK, Faizuddin M, Jayanthi D, et al. Extramedullary plasmacytoma of gingiva and soft tissue in neck. J Clin Diagn Res. 2014;8:ZD16-ZD18.
The Diagnosis: Plasma Cell Gingivitis
Microscopic analysis demonstrated an acanthotic stratified squamous epithelium with an edematous fibrous stroma containing dense perivascular infiltrates of plasma cells and lymphocytes (Figure 1). Immunohistochemical analysis with kappa, lambda, and CD79a immunostains indicated a polyclonal proliferation of plasma cells that excluded monoclonal plasma cell neoplasia (Figure 2). Direct immunofluorescence (DIF) was negative. Serum enzyme-linked immunosorbent assay for bullous pemphigoid 180 and 230 antibodies as well as desmoglein 1 and 3 antibodies was normal. The cumulative findings were consistent with plasma cell gingivitis (PCG). It was recommended that the patient avoid possible foods (eg, citrus) and oral hygiene products (eg, mint-flavored toothpaste) that could trigger PCG. With patient compliance to an elimination diet for 3 months, the condition resolved (Figure 3).
Plasma cell gingivitis is a rare condition characterized by generalized edema and erythema of the attached gingiva. It was described in the 1960s and classified into 3 types based on etiology: (1) hypersensitivity (most common), (2) neoplastic, and (3) PCG of unknown origin.1,2 Spices, herbs, and flavoring agents are implicated as potential triggers of hypersensitivity PCG, while neoplastic PCG is associated with monoclonal plasma cell neoplasms, such as multiple myeloma and extramedullary plasmacytoma.2,3 Histologically, a diffuse subepithelial infiltrate of a polyclonal mixture of plasma cells typically is observed in hypersensitivity PCG.3 The plasma cell infiltration in hypersensitivity PCG is a benign reactive process without known risk for development of plasma cell malignancy, but the presence of a notable number of plasma cells may require special tissue staining to rule out the possibility of associated neoplasia.2,3 There are no standardized protocols for management of PCG.4 Elimination of potential allergens, including flavored oral hygiene products, may result in resolution of hypersensitivity PCG lesions, as exemplified in our patient.1 Neoplastic PCG responds to treatment of the underlying malignancy.5 Topical, intralesional, and/or systemic steroids may be considered in symptomatic cases of PCG.4
Clinical presentation of PCG can mimic immune-mediated mucocutaneous diseases such as mucous membrane pemphigoid (MMP), pemphigus vulgaris (PV), and oral lichen planus; microscopic analysis is needed to establish the diagnosis.6 Mucous membrane pemphigoid is a chronic autoimmune blistering disease involving the mucous membranes with possible cutaneous involvement. It is characterized by a complement-mediated autoantibody process against one or several antigens in the epithelial basement membrane. The oral mucosa is involved in 85% of MMP patients, and 65% of patients experience complications involving the ocular conjunctiva. Intraorally, MMP typically manifests as painful erosions, ulcerations, desquamative gingivitis, and/or occasionally intact blisters. Ocular complications include conjunctivitis and corneal erosions that often scar, resulting in blindness in approximately 15% of patients with ocular involvement. Microscopic features of MMP classically exhibit subepithelial separation with a mixed inflammatory cell infiltrate on routine analysis and linear deposition of IgG, IgA, or C3 within the basement membrane zone on DIF. Treatment of MMP involves topical or systemic immunosuppressants to control symptoms, minimize complications, and alter disease progression.6
Pemphigus vulgaris is an autoimmune vesiculobullous disease that affects the oral mucosa with or without cutaneous involvement.7 Desmogleins 1 and 3, transmembrane glycoproteins of desmosomes that convene cell-to-cell adhesion, are identified as antigens in PV. Antibodies against these desmoglein proteins result in intraepithelial separation, which leads to blister formation.7 Oral manifestations of PV include mucosal erosions and ulcerations as well as desquamative gingivitis. Bullae rarely are seen in the oral cavity, as they tend to rupture, leaving nonhealing ulcerations.8 Histologically, PV is characterized by acantholysis of the suprabasal cell layers with an intact basement membrane zone on routine examination. The distinctive microscopic feature of PV is the detection of cell surface-bound IgG within the epidermis on DIF.7 Treatment of PV may include topical and/or systemic corticosteroids and other immunosuppressants. Rituximab, a monoclonal antibody, has been successful in the management of PV.8
Oral lichen planus is a T-cell mediated autoimmune condition that leads to subepithelial lymphocytic infiltration and excessive keratinocyte apoptosis.9 Women typically are affected more often than men, and 75% of patients also have cutaneous manifestations of the condition. Desquamation and/or erythema of the gingiva may be the initial manifestation of oral lichen planus.9 Other commonly involved sites include the buccal mucosa, tongue, and palate. Biopsy of affected tissues typically demonstrates degeneration of the basal cell layer with subjacent bandlike lymphocytic infiltration on routine staining. Linear fibrinogen at the basement membrane zone usually is observed on DIF. Topical corticosteroids are considered first-line therapy, but systemic therapy including corticosteroids, steroid-sparing agents, or immunomodulators may be used in severe cases.9
There are 3 variants of plasma cell neoplasms including multiple myeloma, medullary plasmacytoma (also known as solitary bone plasmacytoma), and extramedullary plasmacytoma (EMP).10 Extramedullary plasmacytoma, sometimes referred to as extraosseous plasmacytoma, is described as a solitary or multiple plasma cell neoplasm contained in the soft tissue. Its occurrence is rare, accounting for only 3% of plasma cell neoplasms. Approximately 90% of EMPs affect the head and neck region, and males are affected 4 times more often than females. The oral cavity is one of the sites of clinical presentation; the gingival tissue infrequently is affected. When EMP affects the gingiva, it can mimic any form of gingivitis as well as other benign inflammatory conditions, such as pyogenic granuloma. Biopsy is the gold standard diagnostic method for differentiating EMP from other conditions, and specific immunohistochemical stains are essential for the diagnosis. Extramedullary plasmacytoma has the best prognosis among plasma cell neoplasms, despite the risk for progression to multiple myeloma. Extramedullary plasmacytoma lesions are very sensitive to radiotherapy, and the 10-year survival rate is approximately 70%.10
The Diagnosis: Plasma Cell Gingivitis
Microscopic analysis demonstrated an acanthotic stratified squamous epithelium with an edematous fibrous stroma containing dense perivascular infiltrates of plasma cells and lymphocytes (Figure 1). Immunohistochemical analysis with kappa, lambda, and CD79a immunostains indicated a polyclonal proliferation of plasma cells that excluded monoclonal plasma cell neoplasia (Figure 2). Direct immunofluorescence (DIF) was negative. Serum enzyme-linked immunosorbent assay for bullous pemphigoid 180 and 230 antibodies as well as desmoglein 1 and 3 antibodies was normal. The cumulative findings were consistent with plasma cell gingivitis (PCG). It was recommended that the patient avoid possible foods (eg, citrus) and oral hygiene products (eg, mint-flavored toothpaste) that could trigger PCG. With patient compliance to an elimination diet for 3 months, the condition resolved (Figure 3).
Plasma cell gingivitis is a rare condition characterized by generalized edema and erythema of the attached gingiva. It was described in the 1960s and classified into 3 types based on etiology: (1) hypersensitivity (most common), (2) neoplastic, and (3) PCG of unknown origin.1,2 Spices, herbs, and flavoring agents are implicated as potential triggers of hypersensitivity PCG, while neoplastic PCG is associated with monoclonal plasma cell neoplasms, such as multiple myeloma and extramedullary plasmacytoma.2,3 Histologically, a diffuse subepithelial infiltrate of a polyclonal mixture of plasma cells typically is observed in hypersensitivity PCG.3 The plasma cell infiltration in hypersensitivity PCG is a benign reactive process without known risk for development of plasma cell malignancy, but the presence of a notable number of plasma cells may require special tissue staining to rule out the possibility of associated neoplasia.2,3 There are no standardized protocols for management of PCG.4 Elimination of potential allergens, including flavored oral hygiene products, may result in resolution of hypersensitivity PCG lesions, as exemplified in our patient.1 Neoplastic PCG responds to treatment of the underlying malignancy.5 Topical, intralesional, and/or systemic steroids may be considered in symptomatic cases of PCG.4
Clinical presentation of PCG can mimic immune-mediated mucocutaneous diseases such as mucous membrane pemphigoid (MMP), pemphigus vulgaris (PV), and oral lichen planus; microscopic analysis is needed to establish the diagnosis.6 Mucous membrane pemphigoid is a chronic autoimmune blistering disease involving the mucous membranes with possible cutaneous involvement. It is characterized by a complement-mediated autoantibody process against one or several antigens in the epithelial basement membrane. The oral mucosa is involved in 85% of MMP patients, and 65% of patients experience complications involving the ocular conjunctiva. Intraorally, MMP typically manifests as painful erosions, ulcerations, desquamative gingivitis, and/or occasionally intact blisters. Ocular complications include conjunctivitis and corneal erosions that often scar, resulting in blindness in approximately 15% of patients with ocular involvement. Microscopic features of MMP classically exhibit subepithelial separation with a mixed inflammatory cell infiltrate on routine analysis and linear deposition of IgG, IgA, or C3 within the basement membrane zone on DIF. Treatment of MMP involves topical or systemic immunosuppressants to control symptoms, minimize complications, and alter disease progression.6
Pemphigus vulgaris is an autoimmune vesiculobullous disease that affects the oral mucosa with or without cutaneous involvement.7 Desmogleins 1 and 3, transmembrane glycoproteins of desmosomes that convene cell-to-cell adhesion, are identified as antigens in PV. Antibodies against these desmoglein proteins result in intraepithelial separation, which leads to blister formation.7 Oral manifestations of PV include mucosal erosions and ulcerations as well as desquamative gingivitis. Bullae rarely are seen in the oral cavity, as they tend to rupture, leaving nonhealing ulcerations.8 Histologically, PV is characterized by acantholysis of the suprabasal cell layers with an intact basement membrane zone on routine examination. The distinctive microscopic feature of PV is the detection of cell surface-bound IgG within the epidermis on DIF.7 Treatment of PV may include topical and/or systemic corticosteroids and other immunosuppressants. Rituximab, a monoclonal antibody, has been successful in the management of PV.8
Oral lichen planus is a T-cell mediated autoimmune condition that leads to subepithelial lymphocytic infiltration and excessive keratinocyte apoptosis.9 Women typically are affected more often than men, and 75% of patients also have cutaneous manifestations of the condition. Desquamation and/or erythema of the gingiva may be the initial manifestation of oral lichen planus.9 Other commonly involved sites include the buccal mucosa, tongue, and palate. Biopsy of affected tissues typically demonstrates degeneration of the basal cell layer with subjacent bandlike lymphocytic infiltration on routine staining. Linear fibrinogen at the basement membrane zone usually is observed on DIF. Topical corticosteroids are considered first-line therapy, but systemic therapy including corticosteroids, steroid-sparing agents, or immunomodulators may be used in severe cases.9
There are 3 variants of plasma cell neoplasms including multiple myeloma, medullary plasmacytoma (also known as solitary bone plasmacytoma), and extramedullary plasmacytoma (EMP).10 Extramedullary plasmacytoma, sometimes referred to as extraosseous plasmacytoma, is described as a solitary or multiple plasma cell neoplasm contained in the soft tissue. Its occurrence is rare, accounting for only 3% of plasma cell neoplasms. Approximately 90% of EMPs affect the head and neck region, and males are affected 4 times more often than females. The oral cavity is one of the sites of clinical presentation; the gingival tissue infrequently is affected. When EMP affects the gingiva, it can mimic any form of gingivitis as well as other benign inflammatory conditions, such as pyogenic granuloma. Biopsy is the gold standard diagnostic method for differentiating EMP from other conditions, and specific immunohistochemical stains are essential for the diagnosis. Extramedullary plasmacytoma has the best prognosis among plasma cell neoplasms, despite the risk for progression to multiple myeloma. Extramedullary plasmacytoma lesions are very sensitive to radiotherapy, and the 10-year survival rate is approximately 70%.10
- Sollecito TP, Greenberg MS. Plasma cell gingivitis: report of two cases. Oral Surg Oral Med Oral Pathol. 1992;73:690-693.
- Gargiulo AV, Ladone JA, Ladone PA, et al. Case report: plasma cell gingivitis A. CDS Rev. 1995;88:22-23.
- Abhishek K, Rashmi J. Plasma cell gingivitis associated with inflammatory cheilitis: a report on a rare case. Ethiop J Health Sci. 2013;23:183-187.
- Arduino PG, D'Aiuto F, Cavallito C, et al. Professional oral hygiene as a therapeutic option for pediatric patients with plasma cell gingivitis: preliminary results of a prospective case series. J Periodontol. 2011;82:1670-1675.
- Nayak A, Nayak MT. Multiple myeloma with an unusual oral presentation. J Exp Ther Oncol. 2016;11:199-206.
- Xu HH, Werth VP, Parisi E, et al. Mucous membrane pemphigoid. Dent Clin North Am. 2013;57:611-630.
- Hammers CM, Stanley JR. Mechanisms of disease: pemphigus and bullous pemphigoid. Ann Rev Pathol. 2016;11:75-97.
- Cizenski JD, Michel P, Watson IT, et al. Spectrum of orocutaneous disease associations: immune-mediated conditions. J Am Acad Dermatol. 2017;77:795-806.
- Stoopler ET, Sollecito TP. Recurrent gingival and oral mucosal lesions. JAMA. 2014;312:1794-1795.
- Nair SK, Faizuddin M, Jayanthi D, et al. Extramedullary plasmacytoma of gingiva and soft tissue in neck. J Clin Diagn Res. 2014;8:ZD16-ZD18.
- Sollecito TP, Greenberg MS. Plasma cell gingivitis: report of two cases. Oral Surg Oral Med Oral Pathol. 1992;73:690-693.
- Gargiulo AV, Ladone JA, Ladone PA, et al. Case report: plasma cell gingivitis A. CDS Rev. 1995;88:22-23.
- Abhishek K, Rashmi J. Plasma cell gingivitis associated with inflammatory cheilitis: a report on a rare case. Ethiop J Health Sci. 2013;23:183-187.
- Arduino PG, D'Aiuto F, Cavallito C, et al. Professional oral hygiene as a therapeutic option for pediatric patients with plasma cell gingivitis: preliminary results of a prospective case series. J Periodontol. 2011;82:1670-1675.
- Nayak A, Nayak MT. Multiple myeloma with an unusual oral presentation. J Exp Ther Oncol. 2016;11:199-206.
- Xu HH, Werth VP, Parisi E, et al. Mucous membrane pemphigoid. Dent Clin North Am. 2013;57:611-630.
- Hammers CM, Stanley JR. Mechanisms of disease: pemphigus and bullous pemphigoid. Ann Rev Pathol. 2016;11:75-97.
- Cizenski JD, Michel P, Watson IT, et al. Spectrum of orocutaneous disease associations: immune-mediated conditions. J Am Acad Dermatol. 2017;77:795-806.
- Stoopler ET, Sollecito TP. Recurrent gingival and oral mucosal lesions. JAMA. 2014;312:1794-1795.
- Nair SK, Faizuddin M, Jayanthi D, et al. Extramedullary plasmacytoma of gingiva and soft tissue in neck. J Clin Diagn Res. 2014;8:ZD16-ZD18.
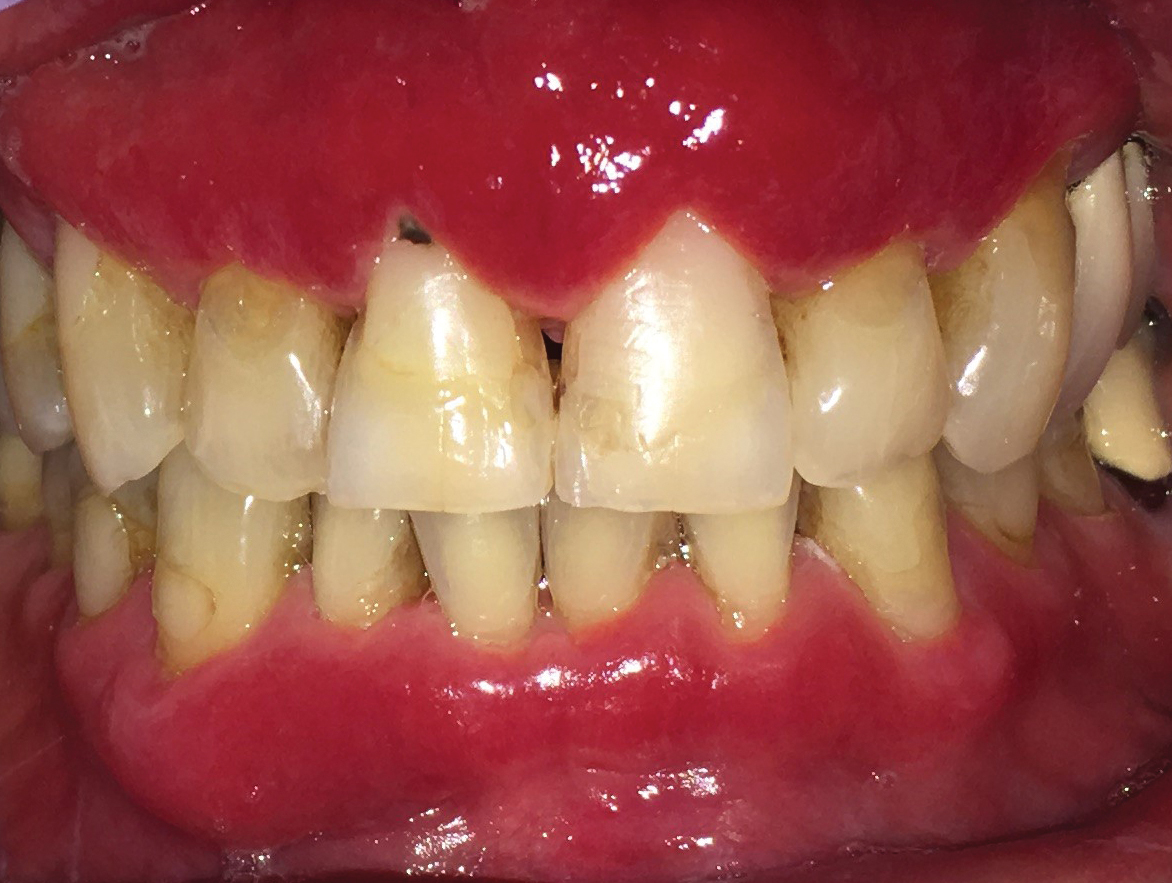
A 62-year-old man presented to an oral medicine specialist with gingival inflammation of at least 1 year's duration. He reported mild discomfort when consuming spicy foods and denied associated extraoral lesions. His medical history revealed hypertension, hypothyroidism, and psoriasis. Medications included lisinopril 10 mg and levothyroxine 100 µg daily. No known drug allergies were reported. His family and social history were noncontributory, and a detailed review of systems was unremarkable. Extraoral examination revealed no lymphadenopathy, salivary gland enlargement, or thyromegaly. Intraoral examination revealed diffuse enlargement of the maxillary and mandibular gingiva accompanied by severe erythema and bleeding on provocation. A 3-mm punch biopsy of the gingiva was performed for routine analysis and direct immunofluorescence.
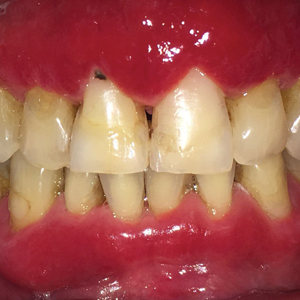
Disseminated Erythema Induratum in a Patient With a History of Tuberculosis
To the Editor:
Erythema induratum, also known as nodular vasculitis, is a panniculitis that usually affects the lower extremities in middle-aged women. Classically, it has been described as a delayed-type hypersensitivity reaction to Mycobacterium tuberculosis, also known as a tuberculid.1,2 Other infections, however, also have been implicated as causes of erythema induratum, including bacillus Calmette-Guérin (BCG), the attenuated form of Mycobacterium bovis, which commonly is used for tuberculosis vaccination. Medications also may cause erythema induratum. The characteristic distribution of the nodules on the posterior calves helps to distinguish erythema induratum from other panniculitides. A PubMed search of articles indexed for MEDLINE using the term disseminated erythema induratum revealed few case reports documenting nodules on the arms, thighs, or chest, and only 1 case report of disseminated erythema induratum.3-8 We describe a rare combination of disseminated erythema induratum in a patient with remote exposure to tuberculosis and recent BCG exposure.
An 88-year-old woman presented for evaluation of violaceous, minimally tender, nonulcerated, subcutaneous nodules on the legs, arms, and trunk of several weeks’ duration (Figure 1). She had a remote history of tuberculosis as a child, prior to the advent of modern antituberculosis regimens. Her medical history also included hypertension, breast cancer treated with lymph node dissection, gastroesophageal reflux disease, and bladder cancer treated with intravesical BCG 10 years prior to the onset of the nodules. She reported minimal coughing and a 25-lb weight loss over the last year, but she denied night sweats, fever, or chills.
Workup included a biopsy, which showed a dense inflammatory infiltrate within the septae and lobules of the subcutaneous tissue (Figure 2A). Foci of necrosis were seen within the fat lobules (Figure 2B). The histologic diagnosis was erythema induratum. Tissue cultures for bacteria, fungi, and atypical mycobacteria were negative. Mycobacterium tuberculosis polymerase chain reaction (PCR) analysis also was negative. An IFN-γ release assay test was positive for infection with M tuberculosis, suggesting that the erythema induratum was due to tuberculosis rather than BCG exposure. A chest radiograph demonstrated a 22-mm nodule in the left lung (unchanged from a prior film) and a new 10-mm nodule in the left upper lobe.
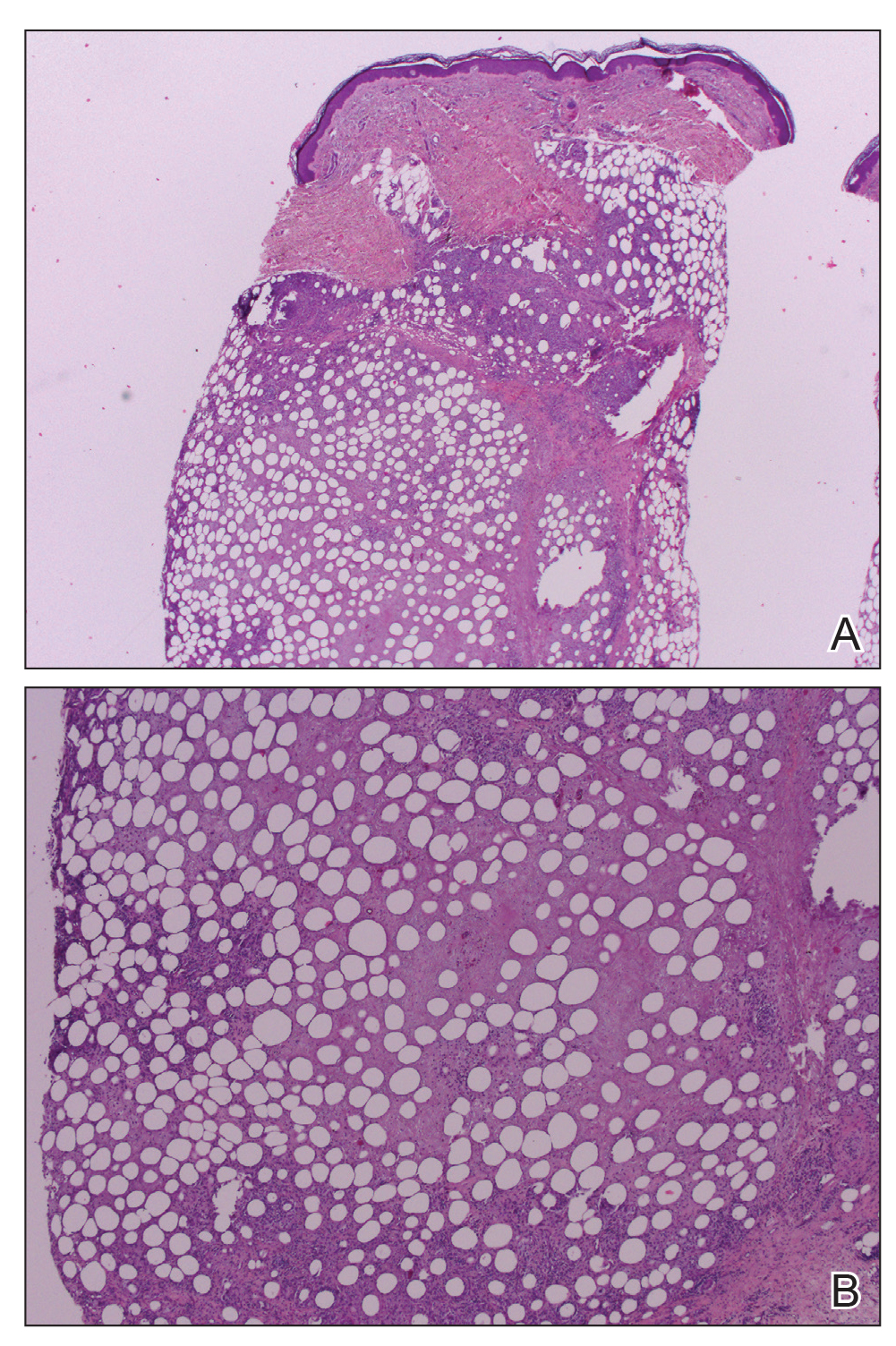
The patient was referred to an infectious disease specialist who concurred that the erythema induratum and the new lung nodule likely represented a reactivation of tuberculosis. Sputum samples were found to be smear and culture negative for mycobacteria, but due to high clinical suspicion, she was started on a 4-drug tuberculosis regimen of isoniazid, rifampin, pyrazinamide, and ethambutol. Some lesions had started to improve prior to the institution of therapy; after initiation of treatment, all lesions resolved within 4 weeks of starting treatment without recurrence.
Erythema induratum was first described by Bazin9 in 1861. The disorder usually occurs in middle-aged women and is characterized by violaceous ulcerative plaques that classically present on the lower extremities, especially the calves. When the eruption occurs due to a nontuberculous etiology, the term nodular vasculitis is used.1,5 The distinction largely is historical, as most dermatologists today recognize erythema induratum and nodular vasculitis to be the same entity. Examples of nontuberculous causes include infections such as Nocardia, Pseudomonas, Fusarium, or other Mycobacterium species.10 Medications such as propylthiouracil also have been implicated.11 The classification of erythema induratum as a tuberculid suggests that the nodules are a reaction pattern rather than a primary infection, though the term tuberculid may be imprecise. The differential diagnosis of violaceous nodules on the lower extremities and trunk is broad and includes erythema nodosum, cutaneous polyarteritis nodosa, pancreatic panniculitis, subcutaneous T-cell lymphoma, and lupus profundus.1,11,12
Histologically, lesions classically demonstrate a mostly lobular panniculitis with varying degrees of septal fibrosis and focal necrosis. Neutrophils may predominate early, while adipocyte necrosis, epithelioid histiocytes, multinucleated giant cells, and lymphocytes may be found in older lesions. The presence of vasculitis as a requisite diagnostic criterion remains controversial.1,12
The incidence of erythema induratum has decreased since multidrug tuberculosis treatment has become more widespread.3 Our case displayed the disseminated variant of erythema induratum, an even rarer clinical entity.8 Interestingly, our patient had a history of tuberculosis and exposure to BCG prior to the development of lesions. Case reports have documented erythema induratum after BCG exposure but less frequently than in cases associated with tuberculosis.3,13
The use of BCG vaccines has necessitated the need for a more precise method of determining tuberculosis activity. The tuberculin skin test reacts positively with a history of BCG exposure, rendering it an inadequate test in a patient who is suspected of having an active or latent M tuberculosis infection.13,14 IFN-γ release assays are more specific in detecting latent or active tuberculosis than the tuberculin skin test. Such assays use early secretory antigenic target 6 and cultured filtrate protein 10 as antigens to determine sensitization to M tuberculosis.13,15 These antigens are not produced by BCG or Mycobacterium avium; however, other mycobacteria such as Mycobacterium marinum, Mycobacterium kansasii, and some strains of M bovis produce the aforementioned antigens, and exposure to these microbes may be confounding.13 Importantly, positive IFN-γ release assay results also have been documented after BCG exposure but occur at a much lower frequency than for tuberculosis.15 Thus, the combination of the positive IFN-γ release assay and new chest radiograph nodule in our patient provided strong evidence of reactivated tuberculosis as the precipitating cause of her skin disease.
Despite her negative PCR study, our patient’s presentation remains consistent with the diagnosis of disseminated erythema induratum.13,15 The value of PCR studies in establishing the diagnosis remains to be determined. Case reports have described positive PCR results detecting M tuberculosis in panniculitic nodules, suggesting that trace amounts of the organism are present in lesional tissue despite the negative culture result and immunostains.1 Tuberculid reactions, including lichen scrofulosorum, papulonecrotic tuberculid, and erythema induratum, historically are defined by the lack of positive cultures and immunostains, making positive PCR results difficult to reconcile pathophysiologically.1,13 Therefore, use of the term tuberculid altogether as a descriptor for pathogenesis of this disease may need to be avoided.16 Postulated explanations for the relationship of tuberculid diseases and negative cultures and immunostains include the presence of a small number of bacilli that escape routine laboratory detection, early destruction of organisms, or a reaction to circulating M tuberculosis fragments.2 Regardless, until the pathophysiology of erythema induratum has been fully elucidated, the value of PCR remains unclear.
Disseminated erythema induratum, an exceptionally rare variant of panniculitis, may be seen in patients with a remote history of M tuberculosis exposure and/or recent therapeutic BCG exposure. It is imperative to rule out active tuberculosis, especially in elderly patients whose disease predated the advent of modern antituberculosis therapy. Using an IFN-γ release assay in addition to chest radiographs and other clinical stigmata allows differentiation of the etiology of erythema induratum in those patients with tuberculosis who also were treated with BCG.
- Mascaro JM, Basalga E. Erythema induratum of Bazin. Dermatol Clin. 2008;28:439-445.
- Lighter J, Tse DB, Li Y, et al. Erythema induratum of Bazin in a child: evidence for a cell-mediated hyper-response to Mycobacterium tuberculosis. Pediatr Infect Dis J. 2009;28:326-328.
- Inoue T, Fukumoto T, Ansai S, et al. Erythema induratum of Bazin in an infant after bacilli Calmette-Guerin vaccination. J Dermatol. 2006;33:268-272.
- Degonda Halter M, Nebiker P, Hug B, et al. Atypical erythema induratum Bazin with tuberculous osteomyelitis. Internist. 2006;47:853-856.
- Gilchrist H, Patterson JW. Erythema nodosum and erythema induratum (nodular vasculitis): diagnosis and management. Dermatol Ther. 2010;23:320-327.
- Sharma S, Sehgal VN, Bhattacharya SN, et al. Clinicopathologic spectrum of cutaneous tuberculosis: a retrospective analysis of 165 Indians. Am J Dermatopathol. 2015;37:444-450.
- Sethuraman G, Ramesh V. Cutaneous tuberculosis in children. Pediatr Dermatol. 2013;30:7-16.
- Teramura K, Fujimoto N, Nakanishi G, et al. Disseminated erythema induratum of Bazin. Eur J Dermatol. 2014;24:697-698.
- Bazin E. Extrait des Lecons Théoretiques et Cliniques sur le Scrofule. 2nd ed. Paris, France: Delhaye; 1861.
- Campbell SM, Winkelmann RR, Sammons DL. Erythema induratum caused by Mycobacterium chelonei in an immunocompetent patient. J Clin Aesthet Dermatol. 2013;6:38-40.
- Patterson JW. Panniculitis. In: Bolognia JL, Jorizzo J, Rapini RP, et al, eds. Dermatology. Barcelona, Spain: Mosby Elsevier; 2012:1641-1662.
- Segura S, Pujol R, Trinidade F, et al. Vasculitis in erythema induratum of Bazin: a histopathologic study of 101 biopsy specimens from 86 patients. J Am Acad Dermatol. 2008;59:839-851.
- Vera-Kellet C, Peters L, Elwood K, et al. Usefulness of interferon-γ release assays in the diagnosis of erythema induratum. Arch Dermatol. 2011;147:949-952.
- Prajapati V, Steed M, Grewal P, et al. Erythema induratum: case series illustrating the utility of the interferon-γ release assay in determining the association with tuberculosis. J Cutan Med Surg. 2013;17:S6-S11.
- Sim JH, Whang KU. Application of the QuantiFERON-Gold TB test in erythema induratum. J Dermatolog Treat. 2014;25:260-263.
- Wiebels D, Turnbull K, Steinkraus V, et al. Erythema induratum Bazin.”tuberculid” or tuberculosis? [in German]. Hautarzt. 2007;58:237-240.
To the Editor:
Erythema induratum, also known as nodular vasculitis, is a panniculitis that usually affects the lower extremities in middle-aged women. Classically, it has been described as a delayed-type hypersensitivity reaction to Mycobacterium tuberculosis, also known as a tuberculid.1,2 Other infections, however, also have been implicated as causes of erythema induratum, including bacillus Calmette-Guérin (BCG), the attenuated form of Mycobacterium bovis, which commonly is used for tuberculosis vaccination. Medications also may cause erythema induratum. The characteristic distribution of the nodules on the posterior calves helps to distinguish erythema induratum from other panniculitides. A PubMed search of articles indexed for MEDLINE using the term disseminated erythema induratum revealed few case reports documenting nodules on the arms, thighs, or chest, and only 1 case report of disseminated erythema induratum.3-8 We describe a rare combination of disseminated erythema induratum in a patient with remote exposure to tuberculosis and recent BCG exposure.
An 88-year-old woman presented for evaluation of violaceous, minimally tender, nonulcerated, subcutaneous nodules on the legs, arms, and trunk of several weeks’ duration (Figure 1). She had a remote history of tuberculosis as a child, prior to the advent of modern antituberculosis regimens. Her medical history also included hypertension, breast cancer treated with lymph node dissection, gastroesophageal reflux disease, and bladder cancer treated with intravesical BCG 10 years prior to the onset of the nodules. She reported minimal coughing and a 25-lb weight loss over the last year, but she denied night sweats, fever, or chills.
Workup included a biopsy, which showed a dense inflammatory infiltrate within the septae and lobules of the subcutaneous tissue (Figure 2A). Foci of necrosis were seen within the fat lobules (Figure 2B). The histologic diagnosis was erythema induratum. Tissue cultures for bacteria, fungi, and atypical mycobacteria were negative. Mycobacterium tuberculosis polymerase chain reaction (PCR) analysis also was negative. An IFN-γ release assay test was positive for infection with M tuberculosis, suggesting that the erythema induratum was due to tuberculosis rather than BCG exposure. A chest radiograph demonstrated a 22-mm nodule in the left lung (unchanged from a prior film) and a new 10-mm nodule in the left upper lobe.
The patient was referred to an infectious disease specialist who concurred that the erythema induratum and the new lung nodule likely represented a reactivation of tuberculosis. Sputum samples were found to be smear and culture negative for mycobacteria, but due to high clinical suspicion, she was started on a 4-drug tuberculosis regimen of isoniazid, rifampin, pyrazinamide, and ethambutol. Some lesions had started to improve prior to the institution of therapy; after initiation of treatment, all lesions resolved within 4 weeks of starting treatment without recurrence.
Erythema induratum was first described by Bazin9 in 1861. The disorder usually occurs in middle-aged women and is characterized by violaceous ulcerative plaques that classically present on the lower extremities, especially the calves. When the eruption occurs due to a nontuberculous etiology, the term nodular vasculitis is used.1,5 The distinction largely is historical, as most dermatologists today recognize erythema induratum and nodular vasculitis to be the same entity. Examples of nontuberculous causes include infections such as Nocardia, Pseudomonas, Fusarium, or other Mycobacterium species.10 Medications such as propylthiouracil also have been implicated.11 The classification of erythema induratum as a tuberculid suggests that the nodules are a reaction pattern rather than a primary infection, though the term tuberculid may be imprecise. The differential diagnosis of violaceous nodules on the lower extremities and trunk is broad and includes erythema nodosum, cutaneous polyarteritis nodosa, pancreatic panniculitis, subcutaneous T-cell lymphoma, and lupus profundus.1,11,12
Histologically, lesions classically demonstrate a mostly lobular panniculitis with varying degrees of septal fibrosis and focal necrosis. Neutrophils may predominate early, while adipocyte necrosis, epithelioid histiocytes, multinucleated giant cells, and lymphocytes may be found in older lesions. The presence of vasculitis as a requisite diagnostic criterion remains controversial.1,12
The incidence of erythema induratum has decreased since multidrug tuberculosis treatment has become more widespread.3 Our case displayed the disseminated variant of erythema induratum, an even rarer clinical entity.8 Interestingly, our patient had a history of tuberculosis and exposure to BCG prior to the development of lesions. Case reports have documented erythema induratum after BCG exposure but less frequently than in cases associated with tuberculosis.3,13
The use of BCG vaccines has necessitated the need for a more precise method of determining tuberculosis activity. The tuberculin skin test reacts positively with a history of BCG exposure, rendering it an inadequate test in a patient who is suspected of having an active or latent M tuberculosis infection.13,14 IFN-γ release assays are more specific in detecting latent or active tuberculosis than the tuberculin skin test. Such assays use early secretory antigenic target 6 and cultured filtrate protein 10 as antigens to determine sensitization to M tuberculosis.13,15 These antigens are not produced by BCG or Mycobacterium avium; however, other mycobacteria such as Mycobacterium marinum, Mycobacterium kansasii, and some strains of M bovis produce the aforementioned antigens, and exposure to these microbes may be confounding.13 Importantly, positive IFN-γ release assay results also have been documented after BCG exposure but occur at a much lower frequency than for tuberculosis.15 Thus, the combination of the positive IFN-γ release assay and new chest radiograph nodule in our patient provided strong evidence of reactivated tuberculosis as the precipitating cause of her skin disease.
Despite her negative PCR study, our patient’s presentation remains consistent with the diagnosis of disseminated erythema induratum.13,15 The value of PCR studies in establishing the diagnosis remains to be determined. Case reports have described positive PCR results detecting M tuberculosis in panniculitic nodules, suggesting that trace amounts of the organism are present in lesional tissue despite the negative culture result and immunostains.1 Tuberculid reactions, including lichen scrofulosorum, papulonecrotic tuberculid, and erythema induratum, historically are defined by the lack of positive cultures and immunostains, making positive PCR results difficult to reconcile pathophysiologically.1,13 Therefore, use of the term tuberculid altogether as a descriptor for pathogenesis of this disease may need to be avoided.16 Postulated explanations for the relationship of tuberculid diseases and negative cultures and immunostains include the presence of a small number of bacilli that escape routine laboratory detection, early destruction of organisms, or a reaction to circulating M tuberculosis fragments.2 Regardless, until the pathophysiology of erythema induratum has been fully elucidated, the value of PCR remains unclear.
Disseminated erythema induratum, an exceptionally rare variant of panniculitis, may be seen in patients with a remote history of M tuberculosis exposure and/or recent therapeutic BCG exposure. It is imperative to rule out active tuberculosis, especially in elderly patients whose disease predated the advent of modern antituberculosis therapy. Using an IFN-γ release assay in addition to chest radiographs and other clinical stigmata allows differentiation of the etiology of erythema induratum in those patients with tuberculosis who also were treated with BCG.
To the Editor:
Erythema induratum, also known as nodular vasculitis, is a panniculitis that usually affects the lower extremities in middle-aged women. Classically, it has been described as a delayed-type hypersensitivity reaction to Mycobacterium tuberculosis, also known as a tuberculid.1,2 Other infections, however, also have been implicated as causes of erythema induratum, including bacillus Calmette-Guérin (BCG), the attenuated form of Mycobacterium bovis, which commonly is used for tuberculosis vaccination. Medications also may cause erythema induratum. The characteristic distribution of the nodules on the posterior calves helps to distinguish erythema induratum from other panniculitides. A PubMed search of articles indexed for MEDLINE using the term disseminated erythema induratum revealed few case reports documenting nodules on the arms, thighs, or chest, and only 1 case report of disseminated erythema induratum.3-8 We describe a rare combination of disseminated erythema induratum in a patient with remote exposure to tuberculosis and recent BCG exposure.
An 88-year-old woman presented for evaluation of violaceous, minimally tender, nonulcerated, subcutaneous nodules on the legs, arms, and trunk of several weeks’ duration (Figure 1). She had a remote history of tuberculosis as a child, prior to the advent of modern antituberculosis regimens. Her medical history also included hypertension, breast cancer treated with lymph node dissection, gastroesophageal reflux disease, and bladder cancer treated with intravesical BCG 10 years prior to the onset of the nodules. She reported minimal coughing and a 25-lb weight loss over the last year, but she denied night sweats, fever, or chills.
Workup included a biopsy, which showed a dense inflammatory infiltrate within the septae and lobules of the subcutaneous tissue (Figure 2A). Foci of necrosis were seen within the fat lobules (Figure 2B). The histologic diagnosis was erythema induratum. Tissue cultures for bacteria, fungi, and atypical mycobacteria were negative. Mycobacterium tuberculosis polymerase chain reaction (PCR) analysis also was negative. An IFN-γ release assay test was positive for infection with M tuberculosis, suggesting that the erythema induratum was due to tuberculosis rather than BCG exposure. A chest radiograph demonstrated a 22-mm nodule in the left lung (unchanged from a prior film) and a new 10-mm nodule in the left upper lobe.
The patient was referred to an infectious disease specialist who concurred that the erythema induratum and the new lung nodule likely represented a reactivation of tuberculosis. Sputum samples were found to be smear and culture negative for mycobacteria, but due to high clinical suspicion, she was started on a 4-drug tuberculosis regimen of isoniazid, rifampin, pyrazinamide, and ethambutol. Some lesions had started to improve prior to the institution of therapy; after initiation of treatment, all lesions resolved within 4 weeks of starting treatment without recurrence.
Erythema induratum was first described by Bazin9 in 1861. The disorder usually occurs in middle-aged women and is characterized by violaceous ulcerative plaques that classically present on the lower extremities, especially the calves. When the eruption occurs due to a nontuberculous etiology, the term nodular vasculitis is used.1,5 The distinction largely is historical, as most dermatologists today recognize erythema induratum and nodular vasculitis to be the same entity. Examples of nontuberculous causes include infections such as Nocardia, Pseudomonas, Fusarium, or other Mycobacterium species.10 Medications such as propylthiouracil also have been implicated.11 The classification of erythema induratum as a tuberculid suggests that the nodules are a reaction pattern rather than a primary infection, though the term tuberculid may be imprecise. The differential diagnosis of violaceous nodules on the lower extremities and trunk is broad and includes erythema nodosum, cutaneous polyarteritis nodosa, pancreatic panniculitis, subcutaneous T-cell lymphoma, and lupus profundus.1,11,12
Histologically, lesions classically demonstrate a mostly lobular panniculitis with varying degrees of septal fibrosis and focal necrosis. Neutrophils may predominate early, while adipocyte necrosis, epithelioid histiocytes, multinucleated giant cells, and lymphocytes may be found in older lesions. The presence of vasculitis as a requisite diagnostic criterion remains controversial.1,12
The incidence of erythema induratum has decreased since multidrug tuberculosis treatment has become more widespread.3 Our case displayed the disseminated variant of erythema induratum, an even rarer clinical entity.8 Interestingly, our patient had a history of tuberculosis and exposure to BCG prior to the development of lesions. Case reports have documented erythema induratum after BCG exposure but less frequently than in cases associated with tuberculosis.3,13
The use of BCG vaccines has necessitated the need for a more precise method of determining tuberculosis activity. The tuberculin skin test reacts positively with a history of BCG exposure, rendering it an inadequate test in a patient who is suspected of having an active or latent M tuberculosis infection.13,14 IFN-γ release assays are more specific in detecting latent or active tuberculosis than the tuberculin skin test. Such assays use early secretory antigenic target 6 and cultured filtrate protein 10 as antigens to determine sensitization to M tuberculosis.13,15 These antigens are not produced by BCG or Mycobacterium avium; however, other mycobacteria such as Mycobacterium marinum, Mycobacterium kansasii, and some strains of M bovis produce the aforementioned antigens, and exposure to these microbes may be confounding.13 Importantly, positive IFN-γ release assay results also have been documented after BCG exposure but occur at a much lower frequency than for tuberculosis.15 Thus, the combination of the positive IFN-γ release assay and new chest radiograph nodule in our patient provided strong evidence of reactivated tuberculosis as the precipitating cause of her skin disease.
Despite her negative PCR study, our patient’s presentation remains consistent with the diagnosis of disseminated erythema induratum.13,15 The value of PCR studies in establishing the diagnosis remains to be determined. Case reports have described positive PCR results detecting M tuberculosis in panniculitic nodules, suggesting that trace amounts of the organism are present in lesional tissue despite the negative culture result and immunostains.1 Tuberculid reactions, including lichen scrofulosorum, papulonecrotic tuberculid, and erythema induratum, historically are defined by the lack of positive cultures and immunostains, making positive PCR results difficult to reconcile pathophysiologically.1,13 Therefore, use of the term tuberculid altogether as a descriptor for pathogenesis of this disease may need to be avoided.16 Postulated explanations for the relationship of tuberculid diseases and negative cultures and immunostains include the presence of a small number of bacilli that escape routine laboratory detection, early destruction of organisms, or a reaction to circulating M tuberculosis fragments.2 Regardless, until the pathophysiology of erythema induratum has been fully elucidated, the value of PCR remains unclear.
Disseminated erythema induratum, an exceptionally rare variant of panniculitis, may be seen in patients with a remote history of M tuberculosis exposure and/or recent therapeutic BCG exposure. It is imperative to rule out active tuberculosis, especially in elderly patients whose disease predated the advent of modern antituberculosis therapy. Using an IFN-γ release assay in addition to chest radiographs and other clinical stigmata allows differentiation of the etiology of erythema induratum in those patients with tuberculosis who also were treated with BCG.
- Mascaro JM, Basalga E. Erythema induratum of Bazin. Dermatol Clin. 2008;28:439-445.
- Lighter J, Tse DB, Li Y, et al. Erythema induratum of Bazin in a child: evidence for a cell-mediated hyper-response to Mycobacterium tuberculosis. Pediatr Infect Dis J. 2009;28:326-328.
- Inoue T, Fukumoto T, Ansai S, et al. Erythema induratum of Bazin in an infant after bacilli Calmette-Guerin vaccination. J Dermatol. 2006;33:268-272.
- Degonda Halter M, Nebiker P, Hug B, et al. Atypical erythema induratum Bazin with tuberculous osteomyelitis. Internist. 2006;47:853-856.
- Gilchrist H, Patterson JW. Erythema nodosum and erythema induratum (nodular vasculitis): diagnosis and management. Dermatol Ther. 2010;23:320-327.
- Sharma S, Sehgal VN, Bhattacharya SN, et al. Clinicopathologic spectrum of cutaneous tuberculosis: a retrospective analysis of 165 Indians. Am J Dermatopathol. 2015;37:444-450.
- Sethuraman G, Ramesh V. Cutaneous tuberculosis in children. Pediatr Dermatol. 2013;30:7-16.
- Teramura K, Fujimoto N, Nakanishi G, et al. Disseminated erythema induratum of Bazin. Eur J Dermatol. 2014;24:697-698.
- Bazin E. Extrait des Lecons Théoretiques et Cliniques sur le Scrofule. 2nd ed. Paris, France: Delhaye; 1861.
- Campbell SM, Winkelmann RR, Sammons DL. Erythema induratum caused by Mycobacterium chelonei in an immunocompetent patient. J Clin Aesthet Dermatol. 2013;6:38-40.
- Patterson JW. Panniculitis. In: Bolognia JL, Jorizzo J, Rapini RP, et al, eds. Dermatology. Barcelona, Spain: Mosby Elsevier; 2012:1641-1662.
- Segura S, Pujol R, Trinidade F, et al. Vasculitis in erythema induratum of Bazin: a histopathologic study of 101 biopsy specimens from 86 patients. J Am Acad Dermatol. 2008;59:839-851.
- Vera-Kellet C, Peters L, Elwood K, et al. Usefulness of interferon-γ release assays in the diagnosis of erythema induratum. Arch Dermatol. 2011;147:949-952.
- Prajapati V, Steed M, Grewal P, et al. Erythema induratum: case series illustrating the utility of the interferon-γ release assay in determining the association with tuberculosis. J Cutan Med Surg. 2013;17:S6-S11.
- Sim JH, Whang KU. Application of the QuantiFERON-Gold TB test in erythema induratum. J Dermatolog Treat. 2014;25:260-263.
- Wiebels D, Turnbull K, Steinkraus V, et al. Erythema induratum Bazin.”tuberculid” or tuberculosis? [in German]. Hautarzt. 2007;58:237-240.
- Mascaro JM, Basalga E. Erythema induratum of Bazin. Dermatol Clin. 2008;28:439-445.
- Lighter J, Tse DB, Li Y, et al. Erythema induratum of Bazin in a child: evidence for a cell-mediated hyper-response to Mycobacterium tuberculosis. Pediatr Infect Dis J. 2009;28:326-328.
- Inoue T, Fukumoto T, Ansai S, et al. Erythema induratum of Bazin in an infant after bacilli Calmette-Guerin vaccination. J Dermatol. 2006;33:268-272.
- Degonda Halter M, Nebiker P, Hug B, et al. Atypical erythema induratum Bazin with tuberculous osteomyelitis. Internist. 2006;47:853-856.
- Gilchrist H, Patterson JW. Erythema nodosum and erythema induratum (nodular vasculitis): diagnosis and management. Dermatol Ther. 2010;23:320-327.
- Sharma S, Sehgal VN, Bhattacharya SN, et al. Clinicopathologic spectrum of cutaneous tuberculosis: a retrospective analysis of 165 Indians. Am J Dermatopathol. 2015;37:444-450.
- Sethuraman G, Ramesh V. Cutaneous tuberculosis in children. Pediatr Dermatol. 2013;30:7-16.
- Teramura K, Fujimoto N, Nakanishi G, et al. Disseminated erythema induratum of Bazin. Eur J Dermatol. 2014;24:697-698.
- Bazin E. Extrait des Lecons Théoretiques et Cliniques sur le Scrofule. 2nd ed. Paris, France: Delhaye; 1861.
- Campbell SM, Winkelmann RR, Sammons DL. Erythema induratum caused by Mycobacterium chelonei in an immunocompetent patient. J Clin Aesthet Dermatol. 2013;6:38-40.
- Patterson JW. Panniculitis. In: Bolognia JL, Jorizzo J, Rapini RP, et al, eds. Dermatology. Barcelona, Spain: Mosby Elsevier; 2012:1641-1662.
- Segura S, Pujol R, Trinidade F, et al. Vasculitis in erythema induratum of Bazin: a histopathologic study of 101 biopsy specimens from 86 patients. J Am Acad Dermatol. 2008;59:839-851.
- Vera-Kellet C, Peters L, Elwood K, et al. Usefulness of interferon-γ release assays in the diagnosis of erythema induratum. Arch Dermatol. 2011;147:949-952.
- Prajapati V, Steed M, Grewal P, et al. Erythema induratum: case series illustrating the utility of the interferon-γ release assay in determining the association with tuberculosis. J Cutan Med Surg. 2013;17:S6-S11.
- Sim JH, Whang KU. Application of the QuantiFERON-Gold TB test in erythema induratum. J Dermatolog Treat. 2014;25:260-263.
- Wiebels D, Turnbull K, Steinkraus V, et al. Erythema induratum Bazin.”tuberculid” or tuberculosis? [in German]. Hautarzt. 2007;58:237-240.
Practice Points
- Erythema induratum is an uncommon panniculitis attributed to a delayed-type hypersensitivity reaction, classically to Mycobacterium tuberculosis.
- The workup for such patients with exposure to both M tuberculosis and bacillus Calmette-Guérin should include IFN-11γ release assays.
- Clinicians should be aware of the disseminated variant of erythema induratum and the laboratory testing needed to establish a cause and help direct treatment.
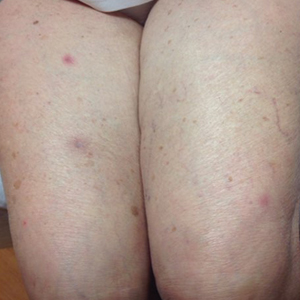
Anti–PD1 Immune Checkpoint Inhibitor–Induced Bullous Pemphigoid in Metastatic Melanoma and Non–Small Cell Lung Cancer
Immune checkpoint inhibitors are used for a variety of advanced malignancies, including melanoma, non–small cell lung cancer, urothelial cancer, and renal cell carcinoma. Anti–programmed cell death 1 (PD1) targeted therapies, such as pembrolizumab and nivolumab, are improving patient survival. This class of immunotherapy is revolutionary but is associated with autoimmune adverse effects. A rare but increasingly reported adverse effect of anti-PD1 therapy is bullous pemphigoid (BP), an autoimmune blistering disease directed against
High clinical suspicion, early diagnosis, and proper management of immunotherapy-related BP are imperative for keeping patients on life-prolonging treatment. We present 3 cases of BP secondary to anti-PD1 immunotherapy in patients with melanoma or non–small cell lung cancer to highlight the diagnosis and treatment of BP as well as emphasize the importance of the dermatologist in the care of patients with immunotherapy-related skin disease.
Case Reports
Patient 1
A 72-year-old woman with metastatic BRAF-mutated melanoma from an unknown primary site presented with intensely pruritic papules on the back, chest, and extremities of 4 months’ duration. She described her symptoms as insidious in onset and refractory to clobetasol ointment, oral diphenhydramine, and over-the-counter anti-itch creams. The patient had been treated with oral dabrafenib 150 mg twice daily and trametinib 2 mg/d but was switched to pembrolizumab when the disease progressed. After 8 months, she had a complete radiologic response to pembrolizumab 2 mg/kg every 3 weeks, which was discontinued in favor of observation 3 months prior to presentation to dermatology.
At the current presentation, physical examination revealed innumerable erythematous, excoriated, 2- to 4-mm, red papules diffusely scattered on the upper back, chest, abdomen, and thighs, with one 8×4-mm vesicle on the right side of the upper back (Figure 1). Discrete areas of depigmented macules, consistent with vitiligo, coalesced into patches on the legs, thighs, arms, and back. The patient was started on a 3-week oral prednisone taper for symptom relief. A hematoxylin and eosin (H&E)–stained punch biopsy of the back revealed a subepidermal split with eosinophils and a dense eosinophilic infiltrate in the dermis (Figure 2). Direct immunofluorescence (DIF) studies from a specimen adjacent to the biopsy collected for H&E staining showed linear deposition of IgA, IgG, and C3 along the dermoepidermal junction (Figure 3). Histologic findings were consistent with BP.
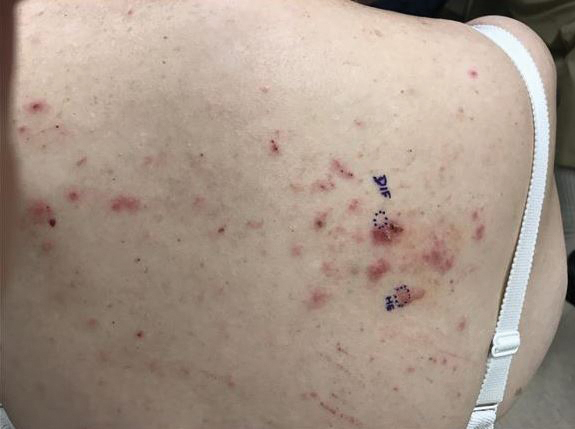
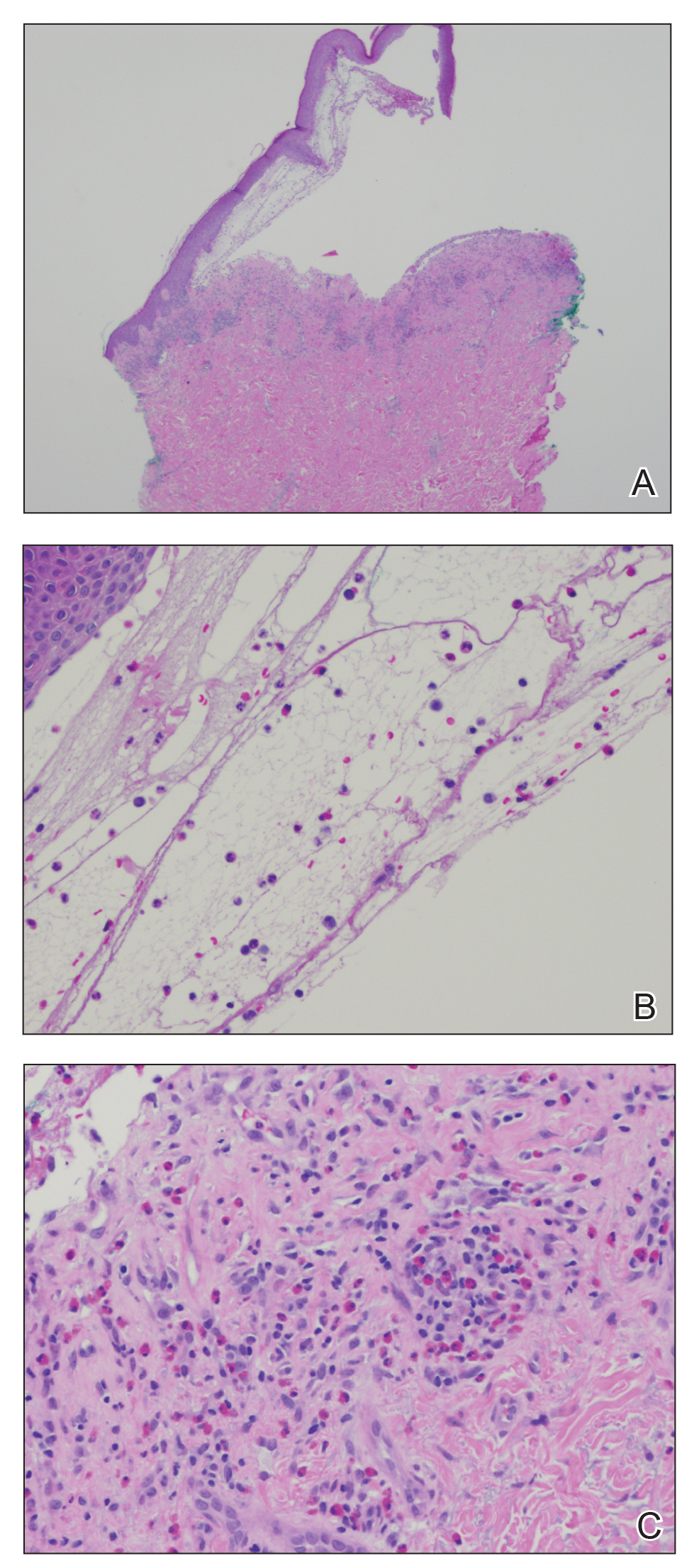
The patient was started on doxycycline 100 mg twice daily and clobetasol ointment 0.05% once daily to supplement the prednisone taper. At 3-week follow-up, she reported pruritus and a few erythematous macules but no new bullae. At 12 weeks, some papules persisted; however, the patient was averse to using systemic agents and decided that symptoms were adequately controlled with clobetasol ointment and oral doxycycline.
Because the patient currently remains in clinical and radiologic remission, anti-PD1 immune checkpoint inhibitors have not been restarted but remain an option for the future if disease recurs
Patient 2
An 82-year-old man with a history of stage IIC desmoplastic melanoma presented to dermatology with an intensely pruritic eruption on the legs, arms, waist, upper torso, and scalp of 3 weeks’ duration. Clobetasol ointment had provided minimal relief.
Six months prior to presenting to dermatology, the patient underwent immunotherapy with 4 cycles of ipilimumab 200 mg intravenous (IV) and nivolumab 240 mg IV every 2 weeks, receiving ipilimumab during the first cycle only because of a lack of availability at the pharmacy. He then received nivolumab 240 mg IV every 2 weeks as maintenance therapy. After the second dose of nivolumab maintenance therapy, however, he developed generalized bullae and pruritus. Dermatology was consulted during an oncology appointment, and his oncologist decided to hold nivolumab.
Physical examination revealed generalized tense and eroded bullae covering more than 50% of the body surface area and affecting the scalp, arms, legs, torso, and buttocks. Two punch biopsies were obtained. Hematoxylin and eosin staining revealed a subepidermal split with predominantly eosinophils and scattered neutrophils. Direct immunofluorescence studies showed linear deposition of IgG, IgA, and C3 along the dermoepidermal junction, consistent with BP.
The patient’s BP was difficult to control, requiring several hospital admissions for wound care, high-dose systemic steroids, and initiation of mycophenolate mofetil. After 4 months of waxing and waning symptoms, the BP was controlled with mycophenolate mofetil 1500 mg/d; clobetasol ointment 0.05%; and diphenhydramine for pruritus. Due to the prolonged recovery and severity of BP, the patient’s oncologist deemed that he was not a candidate for future immunotherapy.
Patient 3
A 68-year-old man with PD1-negative, metastatic, well-differentiated squamous cell carcinoma of the lung presented to dermatology with a pruritic rash of 3 weeks’ duration. He had been receiving nivolumab for 2 years after disease progressed on prior chemotherapies and experienced several grade 1 or grade 2 nivolumab-induced autoimmune reactions including thyroiditis, dermatitis, and nephritis, for which he was taking prednisone 5 mg/d for suppression.
Physical examination revealed psoriasiform pink plaques on the arms, chest, and legs. The differential diagnosis at the time favored psoriasiform dermatitis over lichenoid dermatitis. A punch biopsy revealed psoriasiform dermatitis. The patient was prescribed fluocinonide ointment 0.05% daily. His plaques improved with topical steroids.
The patient returned approximately 1 month later with a report of a new blistering rash on the legs. Physical examination revealed interval improvement of the psoriasiform plaques on the scalp, torso, and extremities, but tense bullae were seen on the thighs, with surrounding superficial erosions at sites of recent bullae. Punch biopsies of the skin for H&E staining and DIF showed BP.
Prednisone was increased to 50 mg/d for a 3-week taper. Doxycycline 100 mg twice daily was started. The patient’s skin disease continued to be difficult to control with therapy; nivolumab was held by his oncologist.
Comment
Immunotherapy with immune checkpoint blockade represents a successful application of immune recognition to treat metastatic cancers, including melanoma, non–small cell lung cancer, urothelial cancer, and renal cell carcinoma.
Anti-PD1 targeted therapies improve survival in solid and hematologic malignancies, with a response rate as high as 40% in melanoma.2 Although these medications can prolong survival, many are associated with loss of self-tolerance and severe autoimmunelike events that can limit therapy.3 An exception is PD1-induced vitiligo, which patient 1 developed and has been associated with a better response to therapy.4
Anti-PD1–induced BP is a newly reported adverse effect. In its early stages, BP can be difficult to differentiate from eczematous or urticarial dermatitis.5-8 Discontinuation of immunotherapy has been reported in more than 70% of patients who develop BP.1 There are reports of successful treatment of BP with a course of a PD1 inhibitor,9 but 2 of our patients had severe BP that led to discontinuation of immunotherapy.
Consider Prescreening
Given that development of BP often leads to cessation of therapy, identifying patients at risk prior to starting an immune checkpoint inhibitor might have clinical utility. Biopsy with DIF is the gold standard for diagnosis, but serologic testing can be a useful adjunct because enzyme-linked immunosorbent assay for BP antigen 1 and BP antigen 2 has a reported sensitivity and specificity of 87% and 98%, respectively.10 Serologic testing prior to starting therapy with an immune checkpoint inhibitor can provide a baseline for patients. A rise in titer, in conjunction with onset of a rash, might aid in earlier diagnosis, particularly because urticarial BP can be difficult to diagnose clinically.
Further study on the utility vs cost-benefit of these screening modalities is warranted. Their predictive utility might be limited, however, and positive serologic test results might have unanticipated consequences, such as hesitation in treating patients, thus leading to a delay in therapy or access to these medications.
Conclusion
The expanding use of immune checkpoint inhibitors is increasing survival in patients with metastatic melanoma and other malignancies. Adverse effects are part of the continuum of immune system stimulation, with overstimulation resulting in dermatitis; thyroiditis; pneumonitis; and less commonly hypophysitis, vitiligo, and colitis.
Rarely, immune checkpoint inhibition induces BP. Development of BP leads to discontinuation of therapy in more than half of reported cases due to lack of adequate treatment for this skin disease and its impact on quality of life. Therefore, quick diagnosis of BP in patients on immunotherapy and successful management techniques can prevent discontinuation of these lifesaving cancer therapies. For that reason, dermatologists play an important role in the management of patients on immune checkpoint inhibitors for cancer.
- Lopez AT, Khanna T, Antonov N, et al. A review of bullous pemphigoid associated with PD-1 and PD-L1 inhibitors. Int J Dermatol. 2018;57:664-669.
- Márquez-Rodas, I, Cerezuela P, Soria A, et al. Immune checkpoint inhibitors: therapeutic advances in melanoma. Ann Transl Med. 2015;3:267.
- Friedman CF, Proverbs-Singh TA, Postow MA. Treatment of the immune-related adverse effects of immune checkpoint inhibitors a review. JAMA Oncol. 2016;2:1346-1353.
- Hua C, Boussemart L, Mateus C, et al. Association of vitiligo with tumor response in patients with metastatic melanoma treated with pembrolizumab. JAMA Dermatol. 2016;152:45-51.
- Hwang SJE, Carlos G, Chou S, et al. Bullous pemphigoid, an autoantibody-mediated disease, is a novel immune-related adverse event in patients treated with anti-programmed cell death 1 antibodies. Melanoma Res. 2016;26:413-416.
- Damsky W, Kole L, Tomayko MM. Development of bullous pemphigoid during nivolumab therapy. JAAD Case Rep. 2016;2:442-444.
- Garje R, Chau JJ, Chung J, et al. Acute flare of bullous pemphigus with pembrolizumab used for treatment of metastatic urothelial cancer. J Immunother. 2018;41:42-44.
- Ito M, Hoashi T, Endo Y, et al. Atypical pemphigus developed in a patient with urothelial carcinoma treated with nivolumab. J Dermatol. 2019;46:e90-e92.
- Chen W-S, Tetzlaff MT, Diwan H, et al. Suprabasal acantholytic dermatologic toxicities associated checkpoint inhibitor therapy: a spectrum of immune reactions from paraneoplastic pemphigus-like to Grover-like lesions. J Cutan Pathol. 2018;45:764-773.
- Muglia C, Bronsnick T, Kirkorian AY, et al. Questioning the specificity and sensitivity of ELISA for bullous pemphigoid diagnosis. Cutis. 2017;99:E27-E30.
Immune checkpoint inhibitors are used for a variety of advanced malignancies, including melanoma, non–small cell lung cancer, urothelial cancer, and renal cell carcinoma. Anti–programmed cell death 1 (PD1) targeted therapies, such as pembrolizumab and nivolumab, are improving patient survival. This class of immunotherapy is revolutionary but is associated with autoimmune adverse effects. A rare but increasingly reported adverse effect of anti-PD1 therapy is bullous pemphigoid (BP), an autoimmune blistering disease directed against
High clinical suspicion, early diagnosis, and proper management of immunotherapy-related BP are imperative for keeping patients on life-prolonging treatment. We present 3 cases of BP secondary to anti-PD1 immunotherapy in patients with melanoma or non–small cell lung cancer to highlight the diagnosis and treatment of BP as well as emphasize the importance of the dermatologist in the care of patients with immunotherapy-related skin disease.
Case Reports
Patient 1
A 72-year-old woman with metastatic BRAF-mutated melanoma from an unknown primary site presented with intensely pruritic papules on the back, chest, and extremities of 4 months’ duration. She described her symptoms as insidious in onset and refractory to clobetasol ointment, oral diphenhydramine, and over-the-counter anti-itch creams. The patient had been treated with oral dabrafenib 150 mg twice daily and trametinib 2 mg/d but was switched to pembrolizumab when the disease progressed. After 8 months, she had a complete radiologic response to pembrolizumab 2 mg/kg every 3 weeks, which was discontinued in favor of observation 3 months prior to presentation to dermatology.
At the current presentation, physical examination revealed innumerable erythematous, excoriated, 2- to 4-mm, red papules diffusely scattered on the upper back, chest, abdomen, and thighs, with one 8×4-mm vesicle on the right side of the upper back (Figure 1). Discrete areas of depigmented macules, consistent with vitiligo, coalesced into patches on the legs, thighs, arms, and back. The patient was started on a 3-week oral prednisone taper for symptom relief. A hematoxylin and eosin (H&E)–stained punch biopsy of the back revealed a subepidermal split with eosinophils and a dense eosinophilic infiltrate in the dermis (Figure 2). Direct immunofluorescence (DIF) studies from a specimen adjacent to the biopsy collected for H&E staining showed linear deposition of IgA, IgG, and C3 along the dermoepidermal junction (Figure 3). Histologic findings were consistent with BP.
The patient was started on doxycycline 100 mg twice daily and clobetasol ointment 0.05% once daily to supplement the prednisone taper. At 3-week follow-up, she reported pruritus and a few erythematous macules but no new bullae. At 12 weeks, some papules persisted; however, the patient was averse to using systemic agents and decided that symptoms were adequately controlled with clobetasol ointment and oral doxycycline.
Because the patient currently remains in clinical and radiologic remission, anti-PD1 immune checkpoint inhibitors have not been restarted but remain an option for the future if disease recurs
Patient 2
An 82-year-old man with a history of stage IIC desmoplastic melanoma presented to dermatology with an intensely pruritic eruption on the legs, arms, waist, upper torso, and scalp of 3 weeks’ duration. Clobetasol ointment had provided minimal relief.
Six months prior to presenting to dermatology, the patient underwent immunotherapy with 4 cycles of ipilimumab 200 mg intravenous (IV) and nivolumab 240 mg IV every 2 weeks, receiving ipilimumab during the first cycle only because of a lack of availability at the pharmacy. He then received nivolumab 240 mg IV every 2 weeks as maintenance therapy. After the second dose of nivolumab maintenance therapy, however, he developed generalized bullae and pruritus. Dermatology was consulted during an oncology appointment, and his oncologist decided to hold nivolumab.
Physical examination revealed generalized tense and eroded bullae covering more than 50% of the body surface area and affecting the scalp, arms, legs, torso, and buttocks. Two punch biopsies were obtained. Hematoxylin and eosin staining revealed a subepidermal split with predominantly eosinophils and scattered neutrophils. Direct immunofluorescence studies showed linear deposition of IgG, IgA, and C3 along the dermoepidermal junction, consistent with BP.
The patient’s BP was difficult to control, requiring several hospital admissions for wound care, high-dose systemic steroids, and initiation of mycophenolate mofetil. After 4 months of waxing and waning symptoms, the BP was controlled with mycophenolate mofetil 1500 mg/d; clobetasol ointment 0.05%; and diphenhydramine for pruritus. Due to the prolonged recovery and severity of BP, the patient’s oncologist deemed that he was not a candidate for future immunotherapy.
Patient 3
A 68-year-old man with PD1-negative, metastatic, well-differentiated squamous cell carcinoma of the lung presented to dermatology with a pruritic rash of 3 weeks’ duration. He had been receiving nivolumab for 2 years after disease progressed on prior chemotherapies and experienced several grade 1 or grade 2 nivolumab-induced autoimmune reactions including thyroiditis, dermatitis, and nephritis, for which he was taking prednisone 5 mg/d for suppression.
Physical examination revealed psoriasiform pink plaques on the arms, chest, and legs. The differential diagnosis at the time favored psoriasiform dermatitis over lichenoid dermatitis. A punch biopsy revealed psoriasiform dermatitis. The patient was prescribed fluocinonide ointment 0.05% daily. His plaques improved with topical steroids.
The patient returned approximately 1 month later with a report of a new blistering rash on the legs. Physical examination revealed interval improvement of the psoriasiform plaques on the scalp, torso, and extremities, but tense bullae were seen on the thighs, with surrounding superficial erosions at sites of recent bullae. Punch biopsies of the skin for H&E staining and DIF showed BP.
Prednisone was increased to 50 mg/d for a 3-week taper. Doxycycline 100 mg twice daily was started. The patient’s skin disease continued to be difficult to control with therapy; nivolumab was held by his oncologist.
Comment
Immunotherapy with immune checkpoint blockade represents a successful application of immune recognition to treat metastatic cancers, including melanoma, non–small cell lung cancer, urothelial cancer, and renal cell carcinoma.
Anti-PD1 targeted therapies improve survival in solid and hematologic malignancies, with a response rate as high as 40% in melanoma.2 Although these medications can prolong survival, many are associated with loss of self-tolerance and severe autoimmunelike events that can limit therapy.3 An exception is PD1-induced vitiligo, which patient 1 developed and has been associated with a better response to therapy.4
Anti-PD1–induced BP is a newly reported adverse effect. In its early stages, BP can be difficult to differentiate from eczematous or urticarial dermatitis.5-8 Discontinuation of immunotherapy has been reported in more than 70% of patients who develop BP.1 There are reports of successful treatment of BP with a course of a PD1 inhibitor,9 but 2 of our patients had severe BP that led to discontinuation of immunotherapy.
Consider Prescreening
Given that development of BP often leads to cessation of therapy, identifying patients at risk prior to starting an immune checkpoint inhibitor might have clinical utility. Biopsy with DIF is the gold standard for diagnosis, but serologic testing can be a useful adjunct because enzyme-linked immunosorbent assay for BP antigen 1 and BP antigen 2 has a reported sensitivity and specificity of 87% and 98%, respectively.10 Serologic testing prior to starting therapy with an immune checkpoint inhibitor can provide a baseline for patients. A rise in titer, in conjunction with onset of a rash, might aid in earlier diagnosis, particularly because urticarial BP can be difficult to diagnose clinically.
Further study on the utility vs cost-benefit of these screening modalities is warranted. Their predictive utility might be limited, however, and positive serologic test results might have unanticipated consequences, such as hesitation in treating patients, thus leading to a delay in therapy or access to these medications.
Conclusion
The expanding use of immune checkpoint inhibitors is increasing survival in patients with metastatic melanoma and other malignancies. Adverse effects are part of the continuum of immune system stimulation, with overstimulation resulting in dermatitis; thyroiditis; pneumonitis; and less commonly hypophysitis, vitiligo, and colitis.
Rarely, immune checkpoint inhibition induces BP. Development of BP leads to discontinuation of therapy in more than half of reported cases due to lack of adequate treatment for this skin disease and its impact on quality of life. Therefore, quick diagnosis of BP in patients on immunotherapy and successful management techniques can prevent discontinuation of these lifesaving cancer therapies. For that reason, dermatologists play an important role in the management of patients on immune checkpoint inhibitors for cancer.
Immune checkpoint inhibitors are used for a variety of advanced malignancies, including melanoma, non–small cell lung cancer, urothelial cancer, and renal cell carcinoma. Anti–programmed cell death 1 (PD1) targeted therapies, such as pembrolizumab and nivolumab, are improving patient survival. This class of immunotherapy is revolutionary but is associated with autoimmune adverse effects. A rare but increasingly reported adverse effect of anti-PD1 therapy is bullous pemphigoid (BP), an autoimmune blistering disease directed against
High clinical suspicion, early diagnosis, and proper management of immunotherapy-related BP are imperative for keeping patients on life-prolonging treatment. We present 3 cases of BP secondary to anti-PD1 immunotherapy in patients with melanoma or non–small cell lung cancer to highlight the diagnosis and treatment of BP as well as emphasize the importance of the dermatologist in the care of patients with immunotherapy-related skin disease.
Case Reports
Patient 1
A 72-year-old woman with metastatic BRAF-mutated melanoma from an unknown primary site presented with intensely pruritic papules on the back, chest, and extremities of 4 months’ duration. She described her symptoms as insidious in onset and refractory to clobetasol ointment, oral diphenhydramine, and over-the-counter anti-itch creams. The patient had been treated with oral dabrafenib 150 mg twice daily and trametinib 2 mg/d but was switched to pembrolizumab when the disease progressed. After 8 months, she had a complete radiologic response to pembrolizumab 2 mg/kg every 3 weeks, which was discontinued in favor of observation 3 months prior to presentation to dermatology.
At the current presentation, physical examination revealed innumerable erythematous, excoriated, 2- to 4-mm, red papules diffusely scattered on the upper back, chest, abdomen, and thighs, with one 8×4-mm vesicle on the right side of the upper back (Figure 1). Discrete areas of depigmented macules, consistent with vitiligo, coalesced into patches on the legs, thighs, arms, and back. The patient was started on a 3-week oral prednisone taper for symptom relief. A hematoxylin and eosin (H&E)–stained punch biopsy of the back revealed a subepidermal split with eosinophils and a dense eosinophilic infiltrate in the dermis (Figure 2). Direct immunofluorescence (DIF) studies from a specimen adjacent to the biopsy collected for H&E staining showed linear deposition of IgA, IgG, and C3 along the dermoepidermal junction (Figure 3). Histologic findings were consistent with BP.
The patient was started on doxycycline 100 mg twice daily and clobetasol ointment 0.05% once daily to supplement the prednisone taper. At 3-week follow-up, she reported pruritus and a few erythematous macules but no new bullae. At 12 weeks, some papules persisted; however, the patient was averse to using systemic agents and decided that symptoms were adequately controlled with clobetasol ointment and oral doxycycline.
Because the patient currently remains in clinical and radiologic remission, anti-PD1 immune checkpoint inhibitors have not been restarted but remain an option for the future if disease recurs
Patient 2
An 82-year-old man with a history of stage IIC desmoplastic melanoma presented to dermatology with an intensely pruritic eruption on the legs, arms, waist, upper torso, and scalp of 3 weeks’ duration. Clobetasol ointment had provided minimal relief.
Six months prior to presenting to dermatology, the patient underwent immunotherapy with 4 cycles of ipilimumab 200 mg intravenous (IV) and nivolumab 240 mg IV every 2 weeks, receiving ipilimumab during the first cycle only because of a lack of availability at the pharmacy. He then received nivolumab 240 mg IV every 2 weeks as maintenance therapy. After the second dose of nivolumab maintenance therapy, however, he developed generalized bullae and pruritus. Dermatology was consulted during an oncology appointment, and his oncologist decided to hold nivolumab.
Physical examination revealed generalized tense and eroded bullae covering more than 50% of the body surface area and affecting the scalp, arms, legs, torso, and buttocks. Two punch biopsies were obtained. Hematoxylin and eosin staining revealed a subepidermal split with predominantly eosinophils and scattered neutrophils. Direct immunofluorescence studies showed linear deposition of IgG, IgA, and C3 along the dermoepidermal junction, consistent with BP.
The patient’s BP was difficult to control, requiring several hospital admissions for wound care, high-dose systemic steroids, and initiation of mycophenolate mofetil. After 4 months of waxing and waning symptoms, the BP was controlled with mycophenolate mofetil 1500 mg/d; clobetasol ointment 0.05%; and diphenhydramine for pruritus. Due to the prolonged recovery and severity of BP, the patient’s oncologist deemed that he was not a candidate for future immunotherapy.
Patient 3
A 68-year-old man with PD1-negative, metastatic, well-differentiated squamous cell carcinoma of the lung presented to dermatology with a pruritic rash of 3 weeks’ duration. He had been receiving nivolumab for 2 years after disease progressed on prior chemotherapies and experienced several grade 1 or grade 2 nivolumab-induced autoimmune reactions including thyroiditis, dermatitis, and nephritis, for which he was taking prednisone 5 mg/d for suppression.
Physical examination revealed psoriasiform pink plaques on the arms, chest, and legs. The differential diagnosis at the time favored psoriasiform dermatitis over lichenoid dermatitis. A punch biopsy revealed psoriasiform dermatitis. The patient was prescribed fluocinonide ointment 0.05% daily. His plaques improved with topical steroids.
The patient returned approximately 1 month later with a report of a new blistering rash on the legs. Physical examination revealed interval improvement of the psoriasiform plaques on the scalp, torso, and extremities, but tense bullae were seen on the thighs, with surrounding superficial erosions at sites of recent bullae. Punch biopsies of the skin for H&E staining and DIF showed BP.
Prednisone was increased to 50 mg/d for a 3-week taper. Doxycycline 100 mg twice daily was started. The patient’s skin disease continued to be difficult to control with therapy; nivolumab was held by his oncologist.
Comment
Immunotherapy with immune checkpoint blockade represents a successful application of immune recognition to treat metastatic cancers, including melanoma, non–small cell lung cancer, urothelial cancer, and renal cell carcinoma.
Anti-PD1 targeted therapies improve survival in solid and hematologic malignancies, with a response rate as high as 40% in melanoma.2 Although these medications can prolong survival, many are associated with loss of self-tolerance and severe autoimmunelike events that can limit therapy.3 An exception is PD1-induced vitiligo, which patient 1 developed and has been associated with a better response to therapy.4
Anti-PD1–induced BP is a newly reported adverse effect. In its early stages, BP can be difficult to differentiate from eczematous or urticarial dermatitis.5-8 Discontinuation of immunotherapy has been reported in more than 70% of patients who develop BP.1 There are reports of successful treatment of BP with a course of a PD1 inhibitor,9 but 2 of our patients had severe BP that led to discontinuation of immunotherapy.
Consider Prescreening
Given that development of BP often leads to cessation of therapy, identifying patients at risk prior to starting an immune checkpoint inhibitor might have clinical utility. Biopsy with DIF is the gold standard for diagnosis, but serologic testing can be a useful adjunct because enzyme-linked immunosorbent assay for BP antigen 1 and BP antigen 2 has a reported sensitivity and specificity of 87% and 98%, respectively.10 Serologic testing prior to starting therapy with an immune checkpoint inhibitor can provide a baseline for patients. A rise in titer, in conjunction with onset of a rash, might aid in earlier diagnosis, particularly because urticarial BP can be difficult to diagnose clinically.
Further study on the utility vs cost-benefit of these screening modalities is warranted. Their predictive utility might be limited, however, and positive serologic test results might have unanticipated consequences, such as hesitation in treating patients, thus leading to a delay in therapy or access to these medications.
Conclusion
The expanding use of immune checkpoint inhibitors is increasing survival in patients with metastatic melanoma and other malignancies. Adverse effects are part of the continuum of immune system stimulation, with overstimulation resulting in dermatitis; thyroiditis; pneumonitis; and less commonly hypophysitis, vitiligo, and colitis.
Rarely, immune checkpoint inhibition induces BP. Development of BP leads to discontinuation of therapy in more than half of reported cases due to lack of adequate treatment for this skin disease and its impact on quality of life. Therefore, quick diagnosis of BP in patients on immunotherapy and successful management techniques can prevent discontinuation of these lifesaving cancer therapies. For that reason, dermatologists play an important role in the management of patients on immune checkpoint inhibitors for cancer.
- Lopez AT, Khanna T, Antonov N, et al. A review of bullous pemphigoid associated with PD-1 and PD-L1 inhibitors. Int J Dermatol. 2018;57:664-669.
- Márquez-Rodas, I, Cerezuela P, Soria A, et al. Immune checkpoint inhibitors: therapeutic advances in melanoma. Ann Transl Med. 2015;3:267.
- Friedman CF, Proverbs-Singh TA, Postow MA. Treatment of the immune-related adverse effects of immune checkpoint inhibitors a review. JAMA Oncol. 2016;2:1346-1353.
- Hua C, Boussemart L, Mateus C, et al. Association of vitiligo with tumor response in patients with metastatic melanoma treated with pembrolizumab. JAMA Dermatol. 2016;152:45-51.
- Hwang SJE, Carlos G, Chou S, et al. Bullous pemphigoid, an autoantibody-mediated disease, is a novel immune-related adverse event in patients treated with anti-programmed cell death 1 antibodies. Melanoma Res. 2016;26:413-416.
- Damsky W, Kole L, Tomayko MM. Development of bullous pemphigoid during nivolumab therapy. JAAD Case Rep. 2016;2:442-444.
- Garje R, Chau JJ, Chung J, et al. Acute flare of bullous pemphigus with pembrolizumab used for treatment of metastatic urothelial cancer. J Immunother. 2018;41:42-44.
- Ito M, Hoashi T, Endo Y, et al. Atypical pemphigus developed in a patient with urothelial carcinoma treated with nivolumab. J Dermatol. 2019;46:e90-e92.
- Chen W-S, Tetzlaff MT, Diwan H, et al. Suprabasal acantholytic dermatologic toxicities associated checkpoint inhibitor therapy: a spectrum of immune reactions from paraneoplastic pemphigus-like to Grover-like lesions. J Cutan Pathol. 2018;45:764-773.
- Muglia C, Bronsnick T, Kirkorian AY, et al. Questioning the specificity and sensitivity of ELISA for bullous pemphigoid diagnosis. Cutis. 2017;99:E27-E30.
- Lopez AT, Khanna T, Antonov N, et al. A review of bullous pemphigoid associated with PD-1 and PD-L1 inhibitors. Int J Dermatol. 2018;57:664-669.
- Márquez-Rodas, I, Cerezuela P, Soria A, et al. Immune checkpoint inhibitors: therapeutic advances in melanoma. Ann Transl Med. 2015;3:267.
- Friedman CF, Proverbs-Singh TA, Postow MA. Treatment of the immune-related adverse effects of immune checkpoint inhibitors a review. JAMA Oncol. 2016;2:1346-1353.
- Hua C, Boussemart L, Mateus C, et al. Association of vitiligo with tumor response in patients with metastatic melanoma treated with pembrolizumab. JAMA Dermatol. 2016;152:45-51.
- Hwang SJE, Carlos G, Chou S, et al. Bullous pemphigoid, an autoantibody-mediated disease, is a novel immune-related adverse event in patients treated with anti-programmed cell death 1 antibodies. Melanoma Res. 2016;26:413-416.
- Damsky W, Kole L, Tomayko MM. Development of bullous pemphigoid during nivolumab therapy. JAAD Case Rep. 2016;2:442-444.
- Garje R, Chau JJ, Chung J, et al. Acute flare of bullous pemphigus with pembrolizumab used for treatment of metastatic urothelial cancer. J Immunother. 2018;41:42-44.
- Ito M, Hoashi T, Endo Y, et al. Atypical pemphigus developed in a patient with urothelial carcinoma treated with nivolumab. J Dermatol. 2019;46:e90-e92.
- Chen W-S, Tetzlaff MT, Diwan H, et al. Suprabasal acantholytic dermatologic toxicities associated checkpoint inhibitor therapy: a spectrum of immune reactions from paraneoplastic pemphigus-like to Grover-like lesions. J Cutan Pathol. 2018;45:764-773.
- Muglia C, Bronsnick T, Kirkorian AY, et al. Questioning the specificity and sensitivity of ELISA for bullous pemphigoid diagnosis. Cutis. 2017;99:E27-E30.
Practice Points
- Anti–programmed cell death 1 (PD1) targeted therapies improve survival in solid and hematologic malignancies but are associated with autoimmune side effects, with bullous pemphigoid (BP) being the newest reported.
- Bullous pemphigoid can develop months into immunotherapy treatment.
- Bullous pemphigoid should be on the differential diagnosis in a patient who is on an anti-PD1 immune checkpoint inhibitor and develops 1 or more of the following: pruritus, dermatitis, and vesicles.
- Early diagnosis of BP is essential for keeping patients on immunotherapy because its severity often results in temporary or permanent discontinuation of treatment.
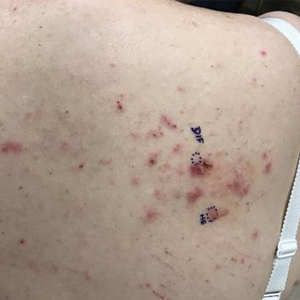
Topical Clobetasol Propionate Treatment and Cutaneous Adverse Effects in Patients With Early-Stage Mycosis Fungoides: An Observational Study
Mycosis fungoides (MF), the most common variant of cutaneous T-cell lymphoma, is a non-Hodgkin lymphoma of T-cell origin that primarily develops in the skin and has a chronic relapsing course. Early-stage MF (stages IA–IIA) is defined as papules, patches, or plaques with limited (if any) lymph node and blood involvement and no visceral involvement.1 Early-stage MF has a favorable prognosis, and first-line treatments are skin-directed therapies including topical corticosteroids (CSs), topical chemotherapy (nitrogen mustard or carmustine), topical retinoids, topical imiquimod, local radiation, or phototherapy.2 Topical CSs are effective in treating early-stage MF and have been widely used for this indication for several decades; however, there are very little data in the literature on topical CS use in MF.3 Superpotent topical CSs have been shown to have a high overall response rate in early-stage MF3; however, cutaneous side effects associated with long-term topical use include cutaneous atrophy, striae formation, skin fragility, and irritation.
The US Food and Drug Administration (FDA) approved bexarotene gel and mechlorethamine gel for topical treatment of cutaneous lesions in patients with stage IA and IB MF in 2000 and 2013, respectively. Although each may be effective in achieving complete or partial response in MF, both agents are associated with cutaneous side effects, mainly irritation and frequent contact hypersensitivity reactions, respectively.4,5 Additionally, their high prices and limited availability are other major drawbacks of treatment.
At our institution, high-potency topical CSs, specifically once or twice daily clobetasol propionate cream 0.05% prescribed as monotherapy for at least several months, remain the mainstay of treatment in patients with limited patches, papules, and plaques covering less than 10% of the skin surface (stage IA). In this study, we aimed to assess the risk of cutaneous side effects in patients with early-stage MF who were treated with long-term, high-potency topical CSs.
Methods
This prospective observational cohort study included patients with early-stage MF who were seen at the Cutaneous Lymphoma Clinic at Memorial Sloan Kettering Cancer Center (MSKCC) in New York, New York, and were started on a superpotent (class I) topical CS (clobetasol propionate cream 0.05%) as monotherapy for MF from July 2016 to July 2017. The diagnosis of MF had to be supported by clinical findings and histopathologic features. All patients were Fitzpatrick skin types I, II, or III. Eligible patients were evaluated for development of CS-induced cutaneous AEs by physical examination and clinical photography of the treated lesions performed at baseline and as part of routine follow-up visits (usually scheduled every 2 to 6 months) at the MSKCC Cutaneous Lymphoma Clinic. Patients’ skin was evaluated clinically for MF activity, atrophy, telangiectasia, purpura, hypopigmentation, and stretch marks (striae). Use of the topical CS was self-reported and also was documented at follow-up visits. Treatment response was defined as follows: complete clinical response (CCR) if the treated lesions resolved completely compared to initial photography; minimal active disease (MAD) if resolution of the vast majority (≥75%) of lesions was seen; and partial response (PR) if some of the lesions resolved (<75%). We analyzed the treatment response rates and adverse effects (AEs). Results were summarized using descriptive statistics.
Results
We identified 13 patients who were started on topical clobetasol propionate as monotherapy for early-stage MF during the study period. Our cohort included 6 males and 7 females aged 36 to 76 years (median age, 61 years). All but 1 participant were diagnosed with stage IA MF (12/13 [92.3%]); of those, 9 (75.0%) had patch-stage disease and 3 (25.0%) presented with plaques. One (7.7%) participant presented with hyperpigmented patches and plaques that involved a little more than 10% of the skin surface (stage IB), and involvement of the hair follicles was noted on histology (folliculotropic MF). All prior treatments were stopped when participants started the superpotent topical CS: 6 (46.2%) participants had been treated with lower-potency topical agents and 1 (7.7%) participant was getting psoralen plus UVA therapy, while the other 6 (46.2%) participants were receiving no therapy for MF prior to starting the study. All participants were prescribed clobetasol propionate cream 0.05% once or twice daily as monotherapy and were instructed to apply it to the MF lesions only, avoiding skin folds and the face. One participant was lost to follow-up, and another stopped using the clobetasol propionate cream after 1.5 months due to local irritation associated with treatment. At their follow-up visits, the other 11 participants were advised to continue with once-daily treatment with clobetasol propionate or were tapered to once every other day, twice weekly, or once weekly depending on their response to treatment and AEs (Table). Participants were advised not to use more than 50 g of clobetasol propionate cream weekly.
All participants responded to the clobetasol propionate cream, and improvement was noted in the treated lesions; however, progression of disease (from stage IA to stage IB) occurred in 1 (8.3%) participant, and phototherapy was added with good response. The participants in our cohort were followed for 4 to 17 months (median, 11.5 months). At the last follow-up visit, all 12 participants showed treatment response: 4 (33.3%) had CCR, 5 (41.7%) had MAD; and 3 (25.0%) had PR. In one participant with a history of partial response to bexarotene gel 1%, daily clobetasol propionate cream 0.05% initially was used alone for 9 months and was later combined with bexarotene gel once weekly, resulting in MAD.
In 7 (58.3%) participants, no AEs to topical clobetasol propionate were recorded. Four (33.3%) participants developed local hypopigmentation at the application site, and 2 (16.7%) developed cutaneous atrophy with local fine wrinkling of the skin (Figure 1); none of the participants developed stretch marks (striae), telangiectases, or skin fragility. One (8.3%) participant developed a petechial rash at the clobetasol propionate application site that resolved once treatment was discontinued and did not recur after restarting clobetasol propionate twice weekly.
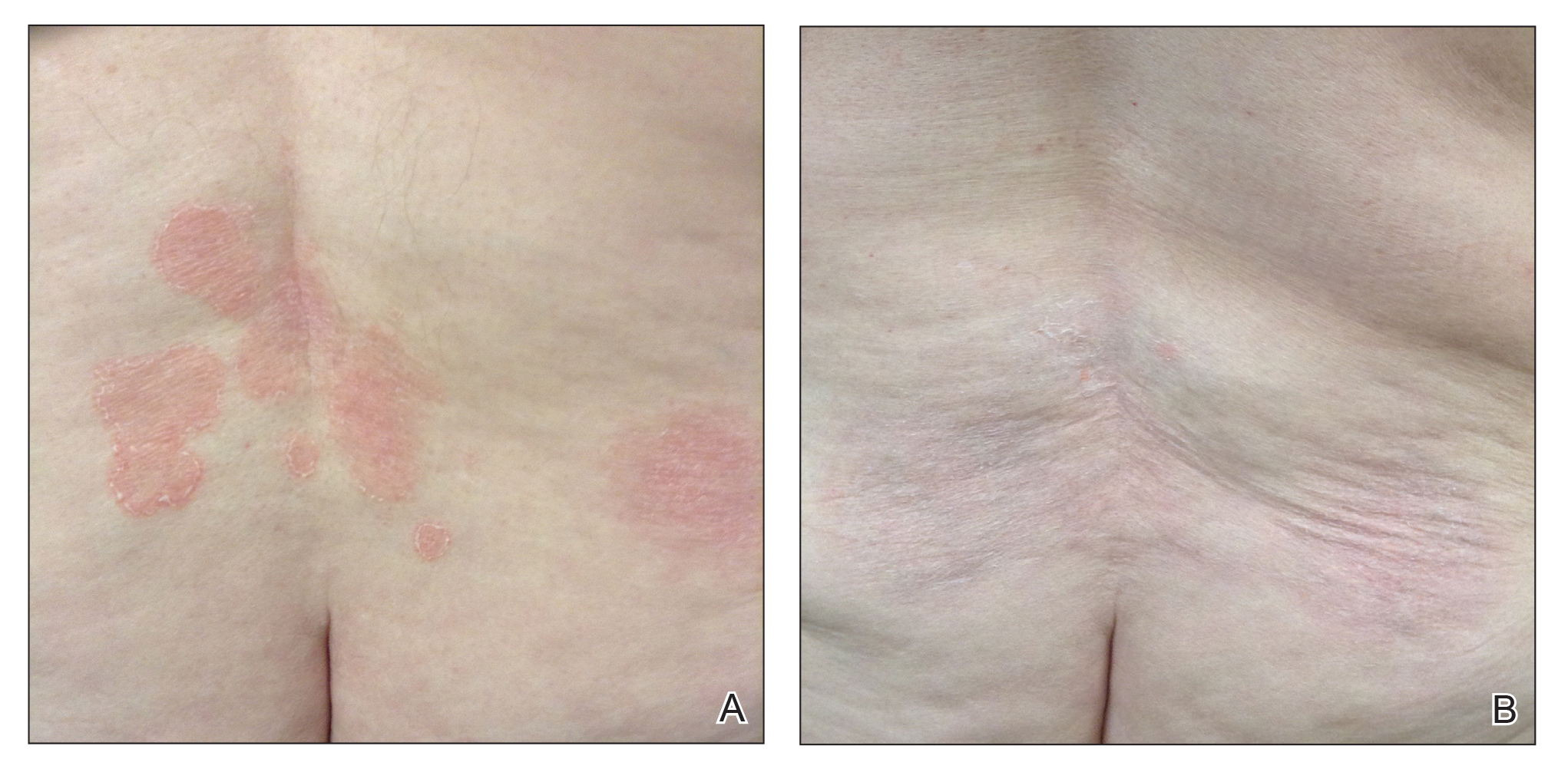
Comment
Topical CSs are the most commonly prescribed agents, either as monotherapy or in combination with other agents, in the treatment of numerous dermatologic conditions, including cutaneous T-cell lymphoma and MF. Cutaneous and systemic AEs have been associated with topical CS use. Local AEs are encountered more frequently and include cutaneous atrophy, striae, telangiectasia, purpura, skin fragility, hypopigmentation, hyperpigmentation, acneform eruptions, and hypertrichosis.6 Factors other than potency of the topical CS agent may affect the development of skin atrophy, including anatomic location, duration of therapy, vehicle, and method and frequency of application.7 The potential for systemic AEs due to percutaneous absorption of high-potency CSs, specifically Cushing syndrome and pathologic adrenal suppression, has been a long-standing concern and led the FDA to recommend limiting the use of superpotent CSs to 50 g weekly for 2 or 4 consecutive weeks.8 However, if using an excess of 50 g weekly is avoided, superpotent topical CSs may be safe to use consecutively for months, perhaps even years, without causing systemic effects.9
The effects of topical CSs in MF include induction of apoptosis; inhibition of lymphocyte binding to the endothelium; and downregulation of transcription factors with decreased cytokines, adhesion molecules, and production of growth factors.2 For patients with limited early-stage MF patches and thin plaques, topical CSs often control the disease for many years and frequently are the only form of therapy required. Intralesional steroids can be effective in treating thicker lesions, such as plaques or tumors.10 In an uncontrolled study, Zackheim et al11 prospectively evaluated the effectiveness and safety of twice-daily use of mainly high-potency topical CSs in 79 patients with MF stages IA to IB and observed an overall response rate of 94%. None of the patients were using systemic agents while being treated with topical CSs. Adverse effects were rare: 2 (2.5%) patients experienced temporary minor irritation from the topical CS, 1 (1.3%) patient developed localized skin atrophy under the breast that resolved several months after she stopped treatment, and 1 (1.3%) patient developed stretch marks on the thighs.11 Zackheim12 later reported treatment of approximately 200 patients with class I topical CSs, and overall response rates were over 90% in stage T1 and over 80% in stage T2 patients. Response to topical CS was reported to be evident within 3 months and often much sooner. Side effects were most likely related to the more prolonged treatment periods. Irritant dermatitis or purpura developed in approximately 10% to 20% of patients, and purpura was seen at the sites of treatment as well as at distant sites. Only a small number of patients developed cutaneous atrophy and striae, which were reversible.12 Successful use of intralesional steroids for treatment-resistant MF was reported in 4 patients who tolerated treatment well without any side effects other than local hypopigmentation in a single patient.13
At MSKCC, the first line of treatment in localized (stage IA) MF in light-skinned individuals most frequently is class I topical CSs, usually clobetasol propionate cream 0.05%. Patients are instructed to apply the cream twice daily on active MF lesions uninterruptedly until completely clear and to avoid using it on the face and in skin folds (axillary, inguinal, and abdominal). Patients are instructed to observe themselves for possible cutaneous AEs related to treatment and to stop or taper treatment if any AEs are noticed. In patients with darker skin, we may recommend other modalities such as narrowband UVB phototherapy for even limited MF disease because of the risk for uneven/hypopigmentation with superpotent CSs.
The current study offers a real-life observation of topical high-potency CSs for treatment of early-stage MF and the associated cutaneous AEs. Local hypopigmentation was identified in 4 participants (33.3%), local skin atrophy was seen in 2 participants (16.7%), and local purpura and irritation were seen in 1 participant each (8.3%). All patients responded to therapy and 75.0% (9/12) achieved CCR or showed only MAD at their last follow-up visit. The limitations of our study were the small number of patients included and the relatively short follow-up period.
In MF patients, patches can present as fine wrinkling of the skin resembling atrophy, which can make it difficult to differentiate active MF from CS-induced atrophy in patients treated with topical CSs (Figure 1) and may have caused us to overestimate the occurrence of this AE. Corticosteroid-induced skin atrophy has been studied mainly in normal skin and to a lesser extent in pathological skin in psoriasis and atopic dermatitis. Some of these studies reported that CS-induced atrophy is reversible, and skin thickness can return to normal after topical application of CS is stopped.7
When hypopigmentation is seen around MF lesions, it is a confirmation that the patient is compliant with the therapy. From our experience, local hypopigmentation due to topical CSs is reversible (Figure 2). In some cases, MF patients have applied topical clobetasol propionate to lesional and surrounding skin, and hypopigmentation can be lessened with more careful limited application. In most cases, after discontinuation or tapering of the therapy, the skin returns to its normal color.
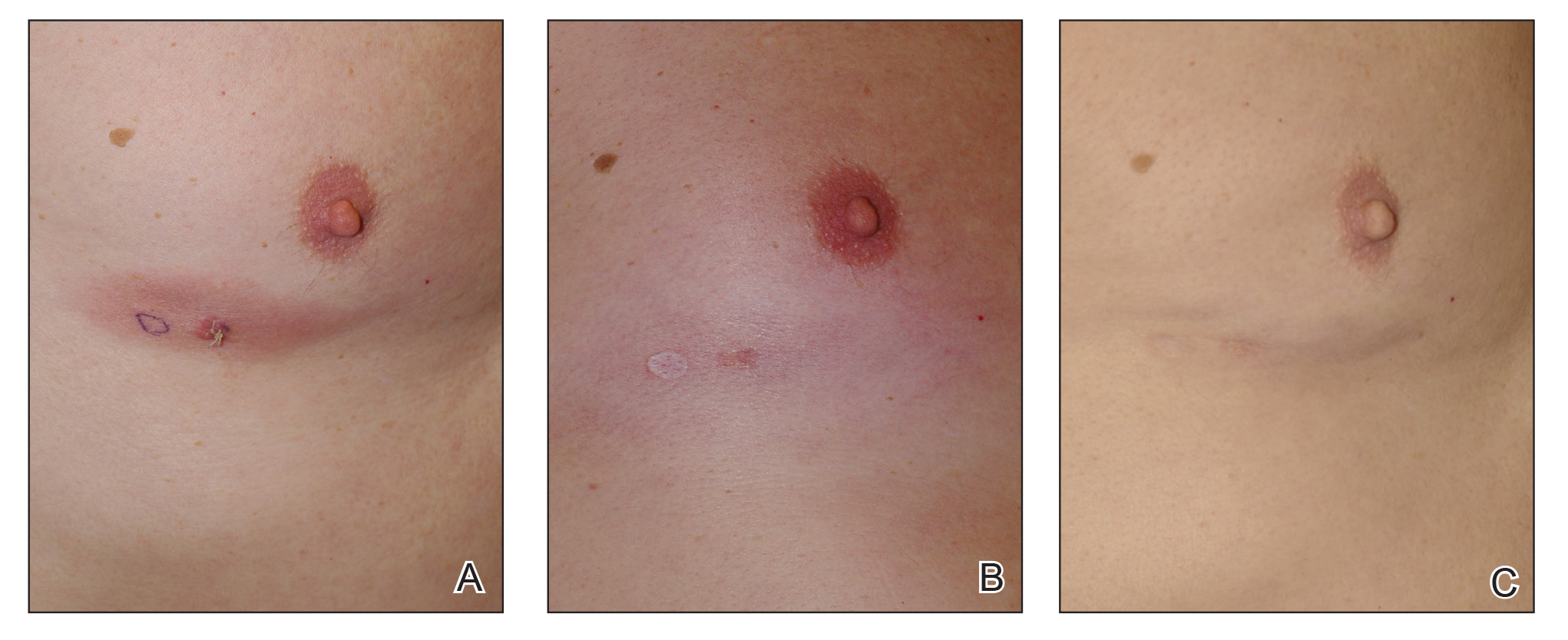
Based on our experience and the results of the current study, we conclude that topical superpotent CSs should remain the first-choice treatment for patients with early-stage MF (stage IA). Although bexarotene gel and mechlorethamine gel are FDA approved for early-stage MF, they are not widely available outside of the United States and are associated with AEs, mainly local skin irritation, rash, and pruritus.4,5 In contrast to bexarotene gel and mechlorethamine gel, topical clobetasol propionate can be used in young children (>12 years) and is classified as pregnancy category C.8
Conclusion
Patients with early-stage MF should be treated with skin-directed therapies, and the choice between different therapeutic options is made based on the physician’s experience with the treatment, patient characteristics, location and morphology of the MF lesions, and the AE profile of the treatment. Based on our experience, superpotent topical CSs are readily available and easily applied, have minor side effects, and remain the mainstay of therapy in patients with stage IA disease. Patients with MF on superpotent topical CS therapy should be monitored periodically and instructed how to identify cutaneous AEs related to treatment.
- Olsen EA, Whittaker S, Kim YH, et al. Clinical end points and response criteria in mycosis fungoides and Sezary syndrome: a consensus statement of the International Society for Cutaneous Lymphomas, the United States Cutaneous Lymphoma Consortium, and the Cutaneous Lymphoma Task Force of the European Organisation for Research and Treatment of Cancer. J Clin Oncol. 2011;29:2598-2607.
- Jawed SI, Myskowski PL, Horwitz S, et al. Primary cutaneous T-cell lymphoma (mycosis fungoides and Sezary syndrome): part II. prognosis, management, and future directions. J Am Acad Dermatol. 2014;70:223.e221-217; quiz 240-222.
- Weberschock T, Strametz R, Lorenz M, et al. Interventions for mycosis fungoides [published online September 12, 2012]. Cochrane Database Syst Rev. doi:10.1002/14651858.CD008946.pub2.
- Heald P, Mehlmauer M, Martin AG, et al. Topical bexarotene therapy for patients with refractory or persistent early-stage cutaneous T-cell lymphoma: results of the phase III clinical trial. J Am Acad Dermatol. 2003;49:801-815.
- Lessin SR, Duvic M, Guitart J, et al. Topical chemotherapy in cutaneous T-cell lymphoma: positive results of a randomized, controlled, multicenter trial testing the efficacy and safety of a novel mechlorethamine, 0.02%, gel in mycosis fungoides. JAMA Dermatol. 2013;149:25-32.
- Tadicherla S, Ross K, Shenefelt PD, et al. Topical corticosteroids in dermatology. J Drugs Dermatol. 2009;8:1093-1105.
- Barnes L, Kaya G, Rollason V. Topical corticosteroid-induced skin atrophy: a comprehensive review. Drug Saf. 2015;38:493-509.
- Temovate E (Clobetasol Propionate) Cream, 0.05% [package insert]. Melville, NY: PharmaDerm, a division of Fougera Pharmaceuticals Inc; 2012.
- Nakamura M, Abrouk M, Zhu H, et al. Update on the systemic risks of superpotent topical steroids. J Drugs Dermatol. 2017;16:643-648.
- Prince HM, Whittaker S, Hoppe RT. How I treat mycosis fungoides and Sezary syndrome. Blood. 2009;114:4337-4353.
- Zackheim HS, Kashani-Sabet M, Amin S. Topical corticosteroids for mycosis fungoides. experience in 79 patients. Arch Dermatol. 1998;134:949-954.
- Zackheim HS. Treatment of patch-stage mycosis fungoides with topical corticosteroids. Dermatol Ther. 2003;16:283-287.
- Liu DY, Shaath T, Rajpara AN, et al. Safe and efficacious use of intralesional steroids for the treatment of focally resistant mycosis fungoides. J Drugs Dermatol. 2015;14:466-471.
Mycosis fungoides (MF), the most common variant of cutaneous T-cell lymphoma, is a non-Hodgkin lymphoma of T-cell origin that primarily develops in the skin and has a chronic relapsing course. Early-stage MF (stages IA–IIA) is defined as papules, patches, or plaques with limited (if any) lymph node and blood involvement and no visceral involvement.1 Early-stage MF has a favorable prognosis, and first-line treatments are skin-directed therapies including topical corticosteroids (CSs), topical chemotherapy (nitrogen mustard or carmustine), topical retinoids, topical imiquimod, local radiation, or phototherapy.2 Topical CSs are effective in treating early-stage MF and have been widely used for this indication for several decades; however, there are very little data in the literature on topical CS use in MF.3 Superpotent topical CSs have been shown to have a high overall response rate in early-stage MF3; however, cutaneous side effects associated with long-term topical use include cutaneous atrophy, striae formation, skin fragility, and irritation.
The US Food and Drug Administration (FDA) approved bexarotene gel and mechlorethamine gel for topical treatment of cutaneous lesions in patients with stage IA and IB MF in 2000 and 2013, respectively. Although each may be effective in achieving complete or partial response in MF, both agents are associated with cutaneous side effects, mainly irritation and frequent contact hypersensitivity reactions, respectively.4,5 Additionally, their high prices and limited availability are other major drawbacks of treatment.
At our institution, high-potency topical CSs, specifically once or twice daily clobetasol propionate cream 0.05% prescribed as monotherapy for at least several months, remain the mainstay of treatment in patients with limited patches, papules, and plaques covering less than 10% of the skin surface (stage IA). In this study, we aimed to assess the risk of cutaneous side effects in patients with early-stage MF who were treated with long-term, high-potency topical CSs.
Methods
This prospective observational cohort study included patients with early-stage MF who were seen at the Cutaneous Lymphoma Clinic at Memorial Sloan Kettering Cancer Center (MSKCC) in New York, New York, and were started on a superpotent (class I) topical CS (clobetasol propionate cream 0.05%) as monotherapy for MF from July 2016 to July 2017. The diagnosis of MF had to be supported by clinical findings and histopathologic features. All patients were Fitzpatrick skin types I, II, or III. Eligible patients were evaluated for development of CS-induced cutaneous AEs by physical examination and clinical photography of the treated lesions performed at baseline and as part of routine follow-up visits (usually scheduled every 2 to 6 months) at the MSKCC Cutaneous Lymphoma Clinic. Patients’ skin was evaluated clinically for MF activity, atrophy, telangiectasia, purpura, hypopigmentation, and stretch marks (striae). Use of the topical CS was self-reported and also was documented at follow-up visits. Treatment response was defined as follows: complete clinical response (CCR) if the treated lesions resolved completely compared to initial photography; minimal active disease (MAD) if resolution of the vast majority (≥75%) of lesions was seen; and partial response (PR) if some of the lesions resolved (<75%). We analyzed the treatment response rates and adverse effects (AEs). Results were summarized using descriptive statistics.
Results
We identified 13 patients who were started on topical clobetasol propionate as monotherapy for early-stage MF during the study period. Our cohort included 6 males and 7 females aged 36 to 76 years (median age, 61 years). All but 1 participant were diagnosed with stage IA MF (12/13 [92.3%]); of those, 9 (75.0%) had patch-stage disease and 3 (25.0%) presented with plaques. One (7.7%) participant presented with hyperpigmented patches and plaques that involved a little more than 10% of the skin surface (stage IB), and involvement of the hair follicles was noted on histology (folliculotropic MF). All prior treatments were stopped when participants started the superpotent topical CS: 6 (46.2%) participants had been treated with lower-potency topical agents and 1 (7.7%) participant was getting psoralen plus UVA therapy, while the other 6 (46.2%) participants were receiving no therapy for MF prior to starting the study. All participants were prescribed clobetasol propionate cream 0.05% once or twice daily as monotherapy and were instructed to apply it to the MF lesions only, avoiding skin folds and the face. One participant was lost to follow-up, and another stopped using the clobetasol propionate cream after 1.5 months due to local irritation associated with treatment. At their follow-up visits, the other 11 participants were advised to continue with once-daily treatment with clobetasol propionate or were tapered to once every other day, twice weekly, or once weekly depending on their response to treatment and AEs (Table). Participants were advised not to use more than 50 g of clobetasol propionate cream weekly.
All participants responded to the clobetasol propionate cream, and improvement was noted in the treated lesions; however, progression of disease (from stage IA to stage IB) occurred in 1 (8.3%) participant, and phototherapy was added with good response. The participants in our cohort were followed for 4 to 17 months (median, 11.5 months). At the last follow-up visit, all 12 participants showed treatment response: 4 (33.3%) had CCR, 5 (41.7%) had MAD; and 3 (25.0%) had PR. In one participant with a history of partial response to bexarotene gel 1%, daily clobetasol propionate cream 0.05% initially was used alone for 9 months and was later combined with bexarotene gel once weekly, resulting in MAD.
In 7 (58.3%) participants, no AEs to topical clobetasol propionate were recorded. Four (33.3%) participants developed local hypopigmentation at the application site, and 2 (16.7%) developed cutaneous atrophy with local fine wrinkling of the skin (Figure 1); none of the participants developed stretch marks (striae), telangiectases, or skin fragility. One (8.3%) participant developed a petechial rash at the clobetasol propionate application site that resolved once treatment was discontinued and did not recur after restarting clobetasol propionate twice weekly.
Comment
Topical CSs are the most commonly prescribed agents, either as monotherapy or in combination with other agents, in the treatment of numerous dermatologic conditions, including cutaneous T-cell lymphoma and MF. Cutaneous and systemic AEs have been associated with topical CS use. Local AEs are encountered more frequently and include cutaneous atrophy, striae, telangiectasia, purpura, skin fragility, hypopigmentation, hyperpigmentation, acneform eruptions, and hypertrichosis.6 Factors other than potency of the topical CS agent may affect the development of skin atrophy, including anatomic location, duration of therapy, vehicle, and method and frequency of application.7 The potential for systemic AEs due to percutaneous absorption of high-potency CSs, specifically Cushing syndrome and pathologic adrenal suppression, has been a long-standing concern and led the FDA to recommend limiting the use of superpotent CSs to 50 g weekly for 2 or 4 consecutive weeks.8 However, if using an excess of 50 g weekly is avoided, superpotent topical CSs may be safe to use consecutively for months, perhaps even years, without causing systemic effects.9
The effects of topical CSs in MF include induction of apoptosis; inhibition of lymphocyte binding to the endothelium; and downregulation of transcription factors with decreased cytokines, adhesion molecules, and production of growth factors.2 For patients with limited early-stage MF patches and thin plaques, topical CSs often control the disease for many years and frequently are the only form of therapy required. Intralesional steroids can be effective in treating thicker lesions, such as plaques or tumors.10 In an uncontrolled study, Zackheim et al11 prospectively evaluated the effectiveness and safety of twice-daily use of mainly high-potency topical CSs in 79 patients with MF stages IA to IB and observed an overall response rate of 94%. None of the patients were using systemic agents while being treated with topical CSs. Adverse effects were rare: 2 (2.5%) patients experienced temporary minor irritation from the topical CS, 1 (1.3%) patient developed localized skin atrophy under the breast that resolved several months after she stopped treatment, and 1 (1.3%) patient developed stretch marks on the thighs.11 Zackheim12 later reported treatment of approximately 200 patients with class I topical CSs, and overall response rates were over 90% in stage T1 and over 80% in stage T2 patients. Response to topical CS was reported to be evident within 3 months and often much sooner. Side effects were most likely related to the more prolonged treatment periods. Irritant dermatitis or purpura developed in approximately 10% to 20% of patients, and purpura was seen at the sites of treatment as well as at distant sites. Only a small number of patients developed cutaneous atrophy and striae, which were reversible.12 Successful use of intralesional steroids for treatment-resistant MF was reported in 4 patients who tolerated treatment well without any side effects other than local hypopigmentation in a single patient.13
At MSKCC, the first line of treatment in localized (stage IA) MF in light-skinned individuals most frequently is class I topical CSs, usually clobetasol propionate cream 0.05%. Patients are instructed to apply the cream twice daily on active MF lesions uninterruptedly until completely clear and to avoid using it on the face and in skin folds (axillary, inguinal, and abdominal). Patients are instructed to observe themselves for possible cutaneous AEs related to treatment and to stop or taper treatment if any AEs are noticed. In patients with darker skin, we may recommend other modalities such as narrowband UVB phototherapy for even limited MF disease because of the risk for uneven/hypopigmentation with superpotent CSs.
The current study offers a real-life observation of topical high-potency CSs for treatment of early-stage MF and the associated cutaneous AEs. Local hypopigmentation was identified in 4 participants (33.3%), local skin atrophy was seen in 2 participants (16.7%), and local purpura and irritation were seen in 1 participant each (8.3%). All patients responded to therapy and 75.0% (9/12) achieved CCR or showed only MAD at their last follow-up visit. The limitations of our study were the small number of patients included and the relatively short follow-up period.
In MF patients, patches can present as fine wrinkling of the skin resembling atrophy, which can make it difficult to differentiate active MF from CS-induced atrophy in patients treated with topical CSs (Figure 1) and may have caused us to overestimate the occurrence of this AE. Corticosteroid-induced skin atrophy has been studied mainly in normal skin and to a lesser extent in pathological skin in psoriasis and atopic dermatitis. Some of these studies reported that CS-induced atrophy is reversible, and skin thickness can return to normal after topical application of CS is stopped.7
When hypopigmentation is seen around MF lesions, it is a confirmation that the patient is compliant with the therapy. From our experience, local hypopigmentation due to topical CSs is reversible (Figure 2). In some cases, MF patients have applied topical clobetasol propionate to lesional and surrounding skin, and hypopigmentation can be lessened with more careful limited application. In most cases, after discontinuation or tapering of the therapy, the skin returns to its normal color.
Based on our experience and the results of the current study, we conclude that topical superpotent CSs should remain the first-choice treatment for patients with early-stage MF (stage IA). Although bexarotene gel and mechlorethamine gel are FDA approved for early-stage MF, they are not widely available outside of the United States and are associated with AEs, mainly local skin irritation, rash, and pruritus.4,5 In contrast to bexarotene gel and mechlorethamine gel, topical clobetasol propionate can be used in young children (>12 years) and is classified as pregnancy category C.8
Conclusion
Patients with early-stage MF should be treated with skin-directed therapies, and the choice between different therapeutic options is made based on the physician’s experience with the treatment, patient characteristics, location and morphology of the MF lesions, and the AE profile of the treatment. Based on our experience, superpotent topical CSs are readily available and easily applied, have minor side effects, and remain the mainstay of therapy in patients with stage IA disease. Patients with MF on superpotent topical CS therapy should be monitored periodically and instructed how to identify cutaneous AEs related to treatment.
Mycosis fungoides (MF), the most common variant of cutaneous T-cell lymphoma, is a non-Hodgkin lymphoma of T-cell origin that primarily develops in the skin and has a chronic relapsing course. Early-stage MF (stages IA–IIA) is defined as papules, patches, or plaques with limited (if any) lymph node and blood involvement and no visceral involvement.1 Early-stage MF has a favorable prognosis, and first-line treatments are skin-directed therapies including topical corticosteroids (CSs), topical chemotherapy (nitrogen mustard or carmustine), topical retinoids, topical imiquimod, local radiation, or phototherapy.2 Topical CSs are effective in treating early-stage MF and have been widely used for this indication for several decades; however, there are very little data in the literature on topical CS use in MF.3 Superpotent topical CSs have been shown to have a high overall response rate in early-stage MF3; however, cutaneous side effects associated with long-term topical use include cutaneous atrophy, striae formation, skin fragility, and irritation.
The US Food and Drug Administration (FDA) approved bexarotene gel and mechlorethamine gel for topical treatment of cutaneous lesions in patients with stage IA and IB MF in 2000 and 2013, respectively. Although each may be effective in achieving complete or partial response in MF, both agents are associated with cutaneous side effects, mainly irritation and frequent contact hypersensitivity reactions, respectively.4,5 Additionally, their high prices and limited availability are other major drawbacks of treatment.
At our institution, high-potency topical CSs, specifically once or twice daily clobetasol propionate cream 0.05% prescribed as monotherapy for at least several months, remain the mainstay of treatment in patients with limited patches, papules, and plaques covering less than 10% of the skin surface (stage IA). In this study, we aimed to assess the risk of cutaneous side effects in patients with early-stage MF who were treated with long-term, high-potency topical CSs.
Methods
This prospective observational cohort study included patients with early-stage MF who were seen at the Cutaneous Lymphoma Clinic at Memorial Sloan Kettering Cancer Center (MSKCC) in New York, New York, and were started on a superpotent (class I) topical CS (clobetasol propionate cream 0.05%) as monotherapy for MF from July 2016 to July 2017. The diagnosis of MF had to be supported by clinical findings and histopathologic features. All patients were Fitzpatrick skin types I, II, or III. Eligible patients were evaluated for development of CS-induced cutaneous AEs by physical examination and clinical photography of the treated lesions performed at baseline and as part of routine follow-up visits (usually scheduled every 2 to 6 months) at the MSKCC Cutaneous Lymphoma Clinic. Patients’ skin was evaluated clinically for MF activity, atrophy, telangiectasia, purpura, hypopigmentation, and stretch marks (striae). Use of the topical CS was self-reported and also was documented at follow-up visits. Treatment response was defined as follows: complete clinical response (CCR) if the treated lesions resolved completely compared to initial photography; minimal active disease (MAD) if resolution of the vast majority (≥75%) of lesions was seen; and partial response (PR) if some of the lesions resolved (<75%). We analyzed the treatment response rates and adverse effects (AEs). Results were summarized using descriptive statistics.
Results
We identified 13 patients who were started on topical clobetasol propionate as monotherapy for early-stage MF during the study period. Our cohort included 6 males and 7 females aged 36 to 76 years (median age, 61 years). All but 1 participant were diagnosed with stage IA MF (12/13 [92.3%]); of those, 9 (75.0%) had patch-stage disease and 3 (25.0%) presented with plaques. One (7.7%) participant presented with hyperpigmented patches and plaques that involved a little more than 10% of the skin surface (stage IB), and involvement of the hair follicles was noted on histology (folliculotropic MF). All prior treatments were stopped when participants started the superpotent topical CS: 6 (46.2%) participants had been treated with lower-potency topical agents and 1 (7.7%) participant was getting psoralen plus UVA therapy, while the other 6 (46.2%) participants were receiving no therapy for MF prior to starting the study. All participants were prescribed clobetasol propionate cream 0.05% once or twice daily as monotherapy and were instructed to apply it to the MF lesions only, avoiding skin folds and the face. One participant was lost to follow-up, and another stopped using the clobetasol propionate cream after 1.5 months due to local irritation associated with treatment. At their follow-up visits, the other 11 participants were advised to continue with once-daily treatment with clobetasol propionate or were tapered to once every other day, twice weekly, or once weekly depending on their response to treatment and AEs (Table). Participants were advised not to use more than 50 g of clobetasol propionate cream weekly.
All participants responded to the clobetasol propionate cream, and improvement was noted in the treated lesions; however, progression of disease (from stage IA to stage IB) occurred in 1 (8.3%) participant, and phototherapy was added with good response. The participants in our cohort were followed for 4 to 17 months (median, 11.5 months). At the last follow-up visit, all 12 participants showed treatment response: 4 (33.3%) had CCR, 5 (41.7%) had MAD; and 3 (25.0%) had PR. In one participant with a history of partial response to bexarotene gel 1%, daily clobetasol propionate cream 0.05% initially was used alone for 9 months and was later combined with bexarotene gel once weekly, resulting in MAD.
In 7 (58.3%) participants, no AEs to topical clobetasol propionate were recorded. Four (33.3%) participants developed local hypopigmentation at the application site, and 2 (16.7%) developed cutaneous atrophy with local fine wrinkling of the skin (Figure 1); none of the participants developed stretch marks (striae), telangiectases, or skin fragility. One (8.3%) participant developed a petechial rash at the clobetasol propionate application site that resolved once treatment was discontinued and did not recur after restarting clobetasol propionate twice weekly.
Comment
Topical CSs are the most commonly prescribed agents, either as monotherapy or in combination with other agents, in the treatment of numerous dermatologic conditions, including cutaneous T-cell lymphoma and MF. Cutaneous and systemic AEs have been associated with topical CS use. Local AEs are encountered more frequently and include cutaneous atrophy, striae, telangiectasia, purpura, skin fragility, hypopigmentation, hyperpigmentation, acneform eruptions, and hypertrichosis.6 Factors other than potency of the topical CS agent may affect the development of skin atrophy, including anatomic location, duration of therapy, vehicle, and method and frequency of application.7 The potential for systemic AEs due to percutaneous absorption of high-potency CSs, specifically Cushing syndrome and pathologic adrenal suppression, has been a long-standing concern and led the FDA to recommend limiting the use of superpotent CSs to 50 g weekly for 2 or 4 consecutive weeks.8 However, if using an excess of 50 g weekly is avoided, superpotent topical CSs may be safe to use consecutively for months, perhaps even years, without causing systemic effects.9
The effects of topical CSs in MF include induction of apoptosis; inhibition of lymphocyte binding to the endothelium; and downregulation of transcription factors with decreased cytokines, adhesion molecules, and production of growth factors.2 For patients with limited early-stage MF patches and thin plaques, topical CSs often control the disease for many years and frequently are the only form of therapy required. Intralesional steroids can be effective in treating thicker lesions, such as plaques or tumors.10 In an uncontrolled study, Zackheim et al11 prospectively evaluated the effectiveness and safety of twice-daily use of mainly high-potency topical CSs in 79 patients with MF stages IA to IB and observed an overall response rate of 94%. None of the patients were using systemic agents while being treated with topical CSs. Adverse effects were rare: 2 (2.5%) patients experienced temporary minor irritation from the topical CS, 1 (1.3%) patient developed localized skin atrophy under the breast that resolved several months after she stopped treatment, and 1 (1.3%) patient developed stretch marks on the thighs.11 Zackheim12 later reported treatment of approximately 200 patients with class I topical CSs, and overall response rates were over 90% in stage T1 and over 80% in stage T2 patients. Response to topical CS was reported to be evident within 3 months and often much sooner. Side effects were most likely related to the more prolonged treatment periods. Irritant dermatitis or purpura developed in approximately 10% to 20% of patients, and purpura was seen at the sites of treatment as well as at distant sites. Only a small number of patients developed cutaneous atrophy and striae, which were reversible.12 Successful use of intralesional steroids for treatment-resistant MF was reported in 4 patients who tolerated treatment well without any side effects other than local hypopigmentation in a single patient.13
At MSKCC, the first line of treatment in localized (stage IA) MF in light-skinned individuals most frequently is class I topical CSs, usually clobetasol propionate cream 0.05%. Patients are instructed to apply the cream twice daily on active MF lesions uninterruptedly until completely clear and to avoid using it on the face and in skin folds (axillary, inguinal, and abdominal). Patients are instructed to observe themselves for possible cutaneous AEs related to treatment and to stop or taper treatment if any AEs are noticed. In patients with darker skin, we may recommend other modalities such as narrowband UVB phototherapy for even limited MF disease because of the risk for uneven/hypopigmentation with superpotent CSs.
The current study offers a real-life observation of topical high-potency CSs for treatment of early-stage MF and the associated cutaneous AEs. Local hypopigmentation was identified in 4 participants (33.3%), local skin atrophy was seen in 2 participants (16.7%), and local purpura and irritation were seen in 1 participant each (8.3%). All patients responded to therapy and 75.0% (9/12) achieved CCR or showed only MAD at their last follow-up visit. The limitations of our study were the small number of patients included and the relatively short follow-up period.
In MF patients, patches can present as fine wrinkling of the skin resembling atrophy, which can make it difficult to differentiate active MF from CS-induced atrophy in patients treated with topical CSs (Figure 1) and may have caused us to overestimate the occurrence of this AE. Corticosteroid-induced skin atrophy has been studied mainly in normal skin and to a lesser extent in pathological skin in psoriasis and atopic dermatitis. Some of these studies reported that CS-induced atrophy is reversible, and skin thickness can return to normal after topical application of CS is stopped.7
When hypopigmentation is seen around MF lesions, it is a confirmation that the patient is compliant with the therapy. From our experience, local hypopigmentation due to topical CSs is reversible (Figure 2). In some cases, MF patients have applied topical clobetasol propionate to lesional and surrounding skin, and hypopigmentation can be lessened with more careful limited application. In most cases, after discontinuation or tapering of the therapy, the skin returns to its normal color.
Based on our experience and the results of the current study, we conclude that topical superpotent CSs should remain the first-choice treatment for patients with early-stage MF (stage IA). Although bexarotene gel and mechlorethamine gel are FDA approved for early-stage MF, they are not widely available outside of the United States and are associated with AEs, mainly local skin irritation, rash, and pruritus.4,5 In contrast to bexarotene gel and mechlorethamine gel, topical clobetasol propionate can be used in young children (>12 years) and is classified as pregnancy category C.8
Conclusion
Patients with early-stage MF should be treated with skin-directed therapies, and the choice between different therapeutic options is made based on the physician’s experience with the treatment, patient characteristics, location and morphology of the MF lesions, and the AE profile of the treatment. Based on our experience, superpotent topical CSs are readily available and easily applied, have minor side effects, and remain the mainstay of therapy in patients with stage IA disease. Patients with MF on superpotent topical CS therapy should be monitored periodically and instructed how to identify cutaneous AEs related to treatment.
- Olsen EA, Whittaker S, Kim YH, et al. Clinical end points and response criteria in mycosis fungoides and Sezary syndrome: a consensus statement of the International Society for Cutaneous Lymphomas, the United States Cutaneous Lymphoma Consortium, and the Cutaneous Lymphoma Task Force of the European Organisation for Research and Treatment of Cancer. J Clin Oncol. 2011;29:2598-2607.
- Jawed SI, Myskowski PL, Horwitz S, et al. Primary cutaneous T-cell lymphoma (mycosis fungoides and Sezary syndrome): part II. prognosis, management, and future directions. J Am Acad Dermatol. 2014;70:223.e221-217; quiz 240-222.
- Weberschock T, Strametz R, Lorenz M, et al. Interventions for mycosis fungoides [published online September 12, 2012]. Cochrane Database Syst Rev. doi:10.1002/14651858.CD008946.pub2.
- Heald P, Mehlmauer M, Martin AG, et al. Topical bexarotene therapy for patients with refractory or persistent early-stage cutaneous T-cell lymphoma: results of the phase III clinical trial. J Am Acad Dermatol. 2003;49:801-815.
- Lessin SR, Duvic M, Guitart J, et al. Topical chemotherapy in cutaneous T-cell lymphoma: positive results of a randomized, controlled, multicenter trial testing the efficacy and safety of a novel mechlorethamine, 0.02%, gel in mycosis fungoides. JAMA Dermatol. 2013;149:25-32.
- Tadicherla S, Ross K, Shenefelt PD, et al. Topical corticosteroids in dermatology. J Drugs Dermatol. 2009;8:1093-1105.
- Barnes L, Kaya G, Rollason V. Topical corticosteroid-induced skin atrophy: a comprehensive review. Drug Saf. 2015;38:493-509.
- Temovate E (Clobetasol Propionate) Cream, 0.05% [package insert]. Melville, NY: PharmaDerm, a division of Fougera Pharmaceuticals Inc; 2012.
- Nakamura M, Abrouk M, Zhu H, et al. Update on the systemic risks of superpotent topical steroids. J Drugs Dermatol. 2017;16:643-648.
- Prince HM, Whittaker S, Hoppe RT. How I treat mycosis fungoides and Sezary syndrome. Blood. 2009;114:4337-4353.
- Zackheim HS, Kashani-Sabet M, Amin S. Topical corticosteroids for mycosis fungoides. experience in 79 patients. Arch Dermatol. 1998;134:949-954.
- Zackheim HS. Treatment of patch-stage mycosis fungoides with topical corticosteroids. Dermatol Ther. 2003;16:283-287.
- Liu DY, Shaath T, Rajpara AN, et al. Safe and efficacious use of intralesional steroids for the treatment of focally resistant mycosis fungoides. J Drugs Dermatol. 2015;14:466-471.
- Olsen EA, Whittaker S, Kim YH, et al. Clinical end points and response criteria in mycosis fungoides and Sezary syndrome: a consensus statement of the International Society for Cutaneous Lymphomas, the United States Cutaneous Lymphoma Consortium, and the Cutaneous Lymphoma Task Force of the European Organisation for Research and Treatment of Cancer. J Clin Oncol. 2011;29:2598-2607.
- Jawed SI, Myskowski PL, Horwitz S, et al. Primary cutaneous T-cell lymphoma (mycosis fungoides and Sezary syndrome): part II. prognosis, management, and future directions. J Am Acad Dermatol. 2014;70:223.e221-217; quiz 240-222.
- Weberschock T, Strametz R, Lorenz M, et al. Interventions for mycosis fungoides [published online September 12, 2012]. Cochrane Database Syst Rev. doi:10.1002/14651858.CD008946.pub2.
- Heald P, Mehlmauer M, Martin AG, et al. Topical bexarotene therapy for patients with refractory or persistent early-stage cutaneous T-cell lymphoma: results of the phase III clinical trial. J Am Acad Dermatol. 2003;49:801-815.
- Lessin SR, Duvic M, Guitart J, et al. Topical chemotherapy in cutaneous T-cell lymphoma: positive results of a randomized, controlled, multicenter trial testing the efficacy and safety of a novel mechlorethamine, 0.02%, gel in mycosis fungoides. JAMA Dermatol. 2013;149:25-32.
- Tadicherla S, Ross K, Shenefelt PD, et al. Topical corticosteroids in dermatology. J Drugs Dermatol. 2009;8:1093-1105.
- Barnes L, Kaya G, Rollason V. Topical corticosteroid-induced skin atrophy: a comprehensive review. Drug Saf. 2015;38:493-509.
- Temovate E (Clobetasol Propionate) Cream, 0.05% [package insert]. Melville, NY: PharmaDerm, a division of Fougera Pharmaceuticals Inc; 2012.
- Nakamura M, Abrouk M, Zhu H, et al. Update on the systemic risks of superpotent topical steroids. J Drugs Dermatol. 2017;16:643-648.
- Prince HM, Whittaker S, Hoppe RT. How I treat mycosis fungoides and Sezary syndrome. Blood. 2009;114:4337-4353.
- Zackheim HS, Kashani-Sabet M, Amin S. Topical corticosteroids for mycosis fungoides. experience in 79 patients. Arch Dermatol. 1998;134:949-954.
- Zackheim HS. Treatment of patch-stage mycosis fungoides with topical corticosteroids. Dermatol Ther. 2003;16:283-287.
- Liu DY, Shaath T, Rajpara AN, et al. Safe and efficacious use of intralesional steroids for the treatment of focally resistant mycosis fungoides. J Drugs Dermatol. 2015;14:466-471.
Practice Points
- Topical corticosteroid (CS) treatment is a safe skin-directed therapy that can effectively obtain complete and long-term response in patients with early-stage mycosis fungoides (MF).
- Despite the availability of optional topical treatments in MF, topical superpotent class I CSs are still considered the first-line treatment in patients with limited disease (stage IA).
- Patients using prolonged topical superpotent CSs should be monitored periodically and instructed on how to identify cutaneous adverse effects related to treatment, mainly local hypopigmentation and skin atrophy.
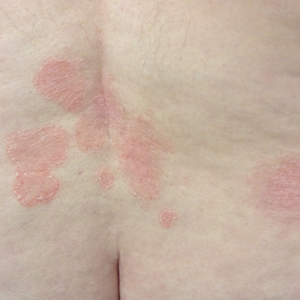
Petechial Rash on the Thighs in an Immunosuppressed Patient
The Diagnosis: Disseminated Strongyloidiasis
Strongyloidiasis is a parasitic infection caused by Strongyloides stercoralis. In the United States it is most prevalent in the Appalachian region. During the filariform larval stage of the parasite's life cycle, larvae from contaminated soil infect the human skin and spread to the intestinal epithelium,1 then the larvae mature into adult female worms that can produce eggs asexually. Rhabditiform larvae hatch from the eggs and are either excreted in the stool or develop into infectious filariform larvae. The latter can cause autoinfection of the intestinal mucosa or nearby skin; in addition, if the larvae enter the bloodstream, they can spread throughout the body and lead to disseminated strongyloidiasis and hyperinfection syndrome.2 This often fatal progression most commonly occurs in immunosuppressed individuals.3 The mortality rate has been reported to be up to 87%.2,4
Fever, abdominal pain, nausea, and diarrhea are clinically common in disseminated strongyloidiasis and hyperinfection syndrome.5 Patients also may exhibit dyspnea, cough, wheezing, and hemoptysis.2 Cutaneous manifestations are rare and typically include pruritus and petechiae.6 Eosinophilia may be present but is not a reliable indicator.1
Our patient displayed several risk factors and an early clinical presentation for disseminated strongyloidiasis and hyperinfection syndrome, which evolved over the course of hospitalization. Clues to the diagnosis included an immunosuppressed state; erythematous pruritic macules at presentation that later developed into reticulated petechial patches; and fever, general abdominal symptoms, and dyspnea. However, the patient's overall physical examination findings were subtle and nonspecific. Additionally, the patient did not display the classic larva currens for strongyloidiasis or the pathognomonic periumbilical thumbprint purpura of disseminated infection,6,7 which may indicate that the latter is a later-stage finding. Although graft-vs-host disease initially was suspected, a third skin biopsy revealed basophilic Strongyloides larvae, extravasated erythrocytes, and mild perivascular inflammation (Figure).
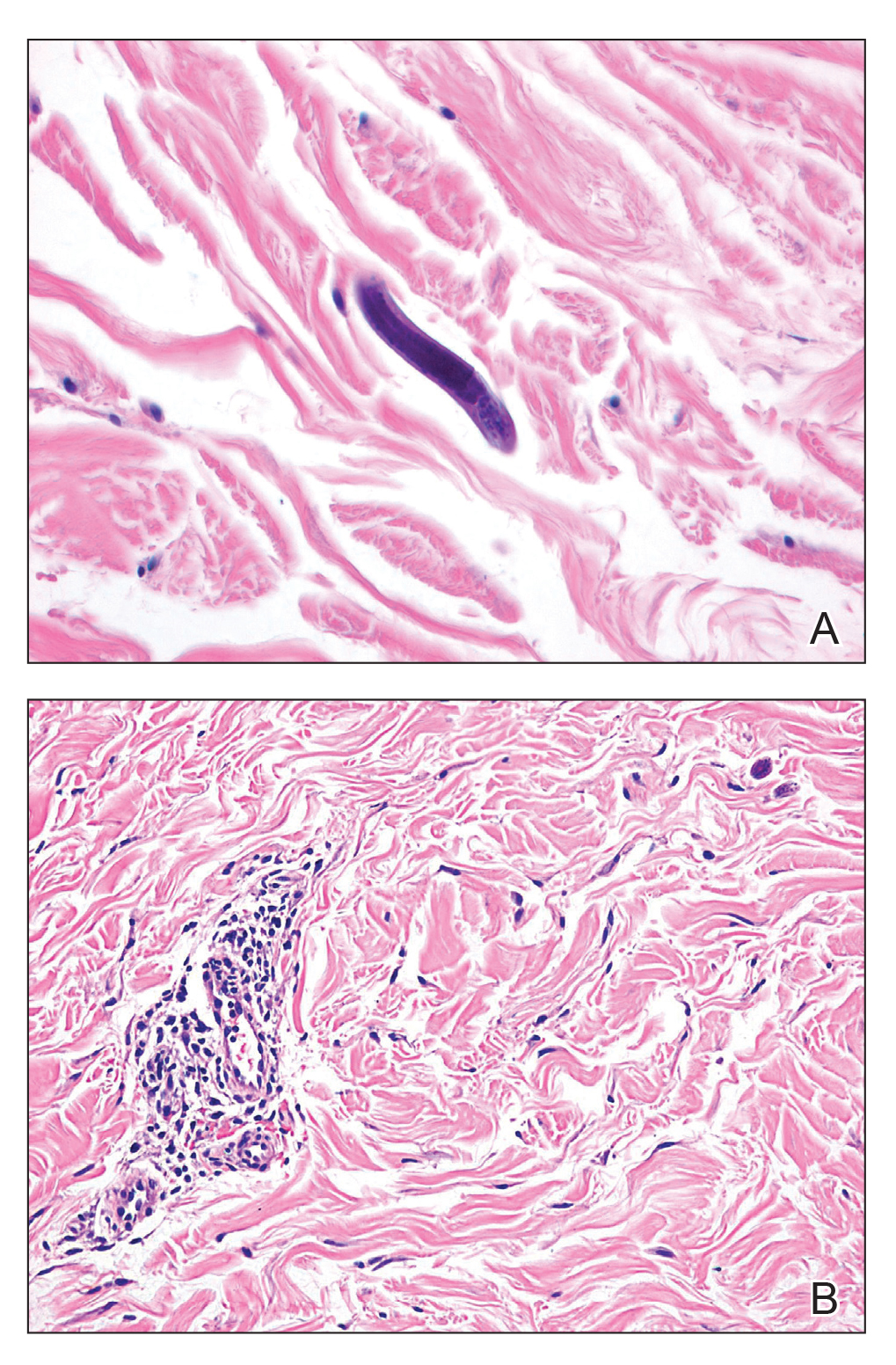
Subsequent gastric aspirates and stool cultures revealed S stercoralis. A bronchoalveolar lavage specimen and serum enzyme-linked immunosorbent assay for Strongyloides antibody were negative. The patient was treated with an extended 16-day course of ivermectin 12 mg daily until gastric aspirates and stool cultures were negative for the parasite. The rash receded by the end of the patient's 32-day hospital stay.
Because of the high mortality rate of untreated disseminated strongyloidiasis and hyperinfection syndrome, early diagnosis and initiation of anthelmintic treatment is vital in improving patient outcomes. As such, the diagnosis of disseminated strongyloidiasis should be considered in any immunosuppressed patient with multisystemic symptoms and/or petechiae. The differential diagnosis includes graft-vs-host disease, drug-induced urticaria, disseminated intravascular coagulation, and other opportunistic parasites.6,8,9
- Concha R, Harrington W Jr, Rogers AI. Intestinal strongyloidiasis: recognition, management, and determinants of outcome. J Clin Gastroenterol. 2005;39:203-211.
- Vadlamudi RS, Chi DS, Krishnaswamy G. Intestinal strongyloidiasis and hyperinfection syndrome. Clin Mol Allergy. 2006;4:8.
- Keiser PB, Nutman TB. Strongyloides stercoralis in the immunocompromised population. Clin Microbiol Rev. 2004;17:208-217.
- Chan FLY, Kennedy B, Nelson R. Fatal Strongyloides hyperinfection syndrome in an immunocompetent adult with review of the literature. Intern Med J. 2018;48:872-875.
- Scowden EB, Schaffner W, Stone WJ. Overwhelming strongyloidiasis: an unappreciated opportunistic infection. Medicine (Baltimore). 1978;57:527-544.
- von Kuster LC, Genta RM. Cutaneous manifestations of strongyloidiasis. Arch Dermatol. 1988;124:1826-1830.
- Weiser JA, Scully BE, Bulman WA, et al. Periumbilical parasitic thumbprint purpura: Strongyloides hyperinfection syndrome acquired from a cadaveric renal transplant. Transpl Infect Dis. 2011;13:58-62.
- Berenson CS, Dobuler KJ, Bia FJ. Fever, petechiae, and pulmonary infiltrates in an immunocompromised Peruvian man. Yale J Biol Med. 1987;60:437-445.
- Ly MN, Bethel SL, Usmani AS, et al. Cutaneous Strongyloides stercoralis infection: an unusual presentation. J Am Acad Dermatol. 2003;49(2 suppl case reports):S157-S160.
The Diagnosis: Disseminated Strongyloidiasis
Strongyloidiasis is a parasitic infection caused by Strongyloides stercoralis. In the United States it is most prevalent in the Appalachian region. During the filariform larval stage of the parasite's life cycle, larvae from contaminated soil infect the human skin and spread to the intestinal epithelium,1 then the larvae mature into adult female worms that can produce eggs asexually. Rhabditiform larvae hatch from the eggs and are either excreted in the stool or develop into infectious filariform larvae. The latter can cause autoinfection of the intestinal mucosa or nearby skin; in addition, if the larvae enter the bloodstream, they can spread throughout the body and lead to disseminated strongyloidiasis and hyperinfection syndrome.2 This often fatal progression most commonly occurs in immunosuppressed individuals.3 The mortality rate has been reported to be up to 87%.2,4
Fever, abdominal pain, nausea, and diarrhea are clinically common in disseminated strongyloidiasis and hyperinfection syndrome.5 Patients also may exhibit dyspnea, cough, wheezing, and hemoptysis.2 Cutaneous manifestations are rare and typically include pruritus and petechiae.6 Eosinophilia may be present but is not a reliable indicator.1
Our patient displayed several risk factors and an early clinical presentation for disseminated strongyloidiasis and hyperinfection syndrome, which evolved over the course of hospitalization. Clues to the diagnosis included an immunosuppressed state; erythematous pruritic macules at presentation that later developed into reticulated petechial patches; and fever, general abdominal symptoms, and dyspnea. However, the patient's overall physical examination findings were subtle and nonspecific. Additionally, the patient did not display the classic larva currens for strongyloidiasis or the pathognomonic periumbilical thumbprint purpura of disseminated infection,6,7 which may indicate that the latter is a later-stage finding. Although graft-vs-host disease initially was suspected, a third skin biopsy revealed basophilic Strongyloides larvae, extravasated erythrocytes, and mild perivascular inflammation (Figure).
Subsequent gastric aspirates and stool cultures revealed S stercoralis. A bronchoalveolar lavage specimen and serum enzyme-linked immunosorbent assay for Strongyloides antibody were negative. The patient was treated with an extended 16-day course of ivermectin 12 mg daily until gastric aspirates and stool cultures were negative for the parasite. The rash receded by the end of the patient's 32-day hospital stay.
Because of the high mortality rate of untreated disseminated strongyloidiasis and hyperinfection syndrome, early diagnosis and initiation of anthelmintic treatment is vital in improving patient outcomes. As such, the diagnosis of disseminated strongyloidiasis should be considered in any immunosuppressed patient with multisystemic symptoms and/or petechiae. The differential diagnosis includes graft-vs-host disease, drug-induced urticaria, disseminated intravascular coagulation, and other opportunistic parasites.6,8,9
The Diagnosis: Disseminated Strongyloidiasis
Strongyloidiasis is a parasitic infection caused by Strongyloides stercoralis. In the United States it is most prevalent in the Appalachian region. During the filariform larval stage of the parasite's life cycle, larvae from contaminated soil infect the human skin and spread to the intestinal epithelium,1 then the larvae mature into adult female worms that can produce eggs asexually. Rhabditiform larvae hatch from the eggs and are either excreted in the stool or develop into infectious filariform larvae. The latter can cause autoinfection of the intestinal mucosa or nearby skin; in addition, if the larvae enter the bloodstream, they can spread throughout the body and lead to disseminated strongyloidiasis and hyperinfection syndrome.2 This often fatal progression most commonly occurs in immunosuppressed individuals.3 The mortality rate has been reported to be up to 87%.2,4
Fever, abdominal pain, nausea, and diarrhea are clinically common in disseminated strongyloidiasis and hyperinfection syndrome.5 Patients also may exhibit dyspnea, cough, wheezing, and hemoptysis.2 Cutaneous manifestations are rare and typically include pruritus and petechiae.6 Eosinophilia may be present but is not a reliable indicator.1
Our patient displayed several risk factors and an early clinical presentation for disseminated strongyloidiasis and hyperinfection syndrome, which evolved over the course of hospitalization. Clues to the diagnosis included an immunosuppressed state; erythematous pruritic macules at presentation that later developed into reticulated petechial patches; and fever, general abdominal symptoms, and dyspnea. However, the patient's overall physical examination findings were subtle and nonspecific. Additionally, the patient did not display the classic larva currens for strongyloidiasis or the pathognomonic periumbilical thumbprint purpura of disseminated infection,6,7 which may indicate that the latter is a later-stage finding. Although graft-vs-host disease initially was suspected, a third skin biopsy revealed basophilic Strongyloides larvae, extravasated erythrocytes, and mild perivascular inflammation (Figure).
Subsequent gastric aspirates and stool cultures revealed S stercoralis. A bronchoalveolar lavage specimen and serum enzyme-linked immunosorbent assay for Strongyloides antibody were negative. The patient was treated with an extended 16-day course of ivermectin 12 mg daily until gastric aspirates and stool cultures were negative for the parasite. The rash receded by the end of the patient's 32-day hospital stay.
Because of the high mortality rate of untreated disseminated strongyloidiasis and hyperinfection syndrome, early diagnosis and initiation of anthelmintic treatment is vital in improving patient outcomes. As such, the diagnosis of disseminated strongyloidiasis should be considered in any immunosuppressed patient with multisystemic symptoms and/or petechiae. The differential diagnosis includes graft-vs-host disease, drug-induced urticaria, disseminated intravascular coagulation, and other opportunistic parasites.6,8,9
- Concha R, Harrington W Jr, Rogers AI. Intestinal strongyloidiasis: recognition, management, and determinants of outcome. J Clin Gastroenterol. 2005;39:203-211.
- Vadlamudi RS, Chi DS, Krishnaswamy G. Intestinal strongyloidiasis and hyperinfection syndrome. Clin Mol Allergy. 2006;4:8.
- Keiser PB, Nutman TB. Strongyloides stercoralis in the immunocompromised population. Clin Microbiol Rev. 2004;17:208-217.
- Chan FLY, Kennedy B, Nelson R. Fatal Strongyloides hyperinfection syndrome in an immunocompetent adult with review of the literature. Intern Med J. 2018;48:872-875.
- Scowden EB, Schaffner W, Stone WJ. Overwhelming strongyloidiasis: an unappreciated opportunistic infection. Medicine (Baltimore). 1978;57:527-544.
- von Kuster LC, Genta RM. Cutaneous manifestations of strongyloidiasis. Arch Dermatol. 1988;124:1826-1830.
- Weiser JA, Scully BE, Bulman WA, et al. Periumbilical parasitic thumbprint purpura: Strongyloides hyperinfection syndrome acquired from a cadaveric renal transplant. Transpl Infect Dis. 2011;13:58-62.
- Berenson CS, Dobuler KJ, Bia FJ. Fever, petechiae, and pulmonary infiltrates in an immunocompromised Peruvian man. Yale J Biol Med. 1987;60:437-445.
- Ly MN, Bethel SL, Usmani AS, et al. Cutaneous Strongyloides stercoralis infection: an unusual presentation. J Am Acad Dermatol. 2003;49(2 suppl case reports):S157-S160.
- Concha R, Harrington W Jr, Rogers AI. Intestinal strongyloidiasis: recognition, management, and determinants of outcome. J Clin Gastroenterol. 2005;39:203-211.
- Vadlamudi RS, Chi DS, Krishnaswamy G. Intestinal strongyloidiasis and hyperinfection syndrome. Clin Mol Allergy. 2006;4:8.
- Keiser PB, Nutman TB. Strongyloides stercoralis in the immunocompromised population. Clin Microbiol Rev. 2004;17:208-217.
- Chan FLY, Kennedy B, Nelson R. Fatal Strongyloides hyperinfection syndrome in an immunocompetent adult with review of the literature. Intern Med J. 2018;48:872-875.
- Scowden EB, Schaffner W, Stone WJ. Overwhelming strongyloidiasis: an unappreciated opportunistic infection. Medicine (Baltimore). 1978;57:527-544.
- von Kuster LC, Genta RM. Cutaneous manifestations of strongyloidiasis. Arch Dermatol. 1988;124:1826-1830.
- Weiser JA, Scully BE, Bulman WA, et al. Periumbilical parasitic thumbprint purpura: Strongyloides hyperinfection syndrome acquired from a cadaveric renal transplant. Transpl Infect Dis. 2011;13:58-62.
- Berenson CS, Dobuler KJ, Bia FJ. Fever, petechiae, and pulmonary infiltrates in an immunocompromised Peruvian man. Yale J Biol Med. 1987;60:437-445.
- Ly MN, Bethel SL, Usmani AS, et al. Cutaneous Strongyloides stercoralis infection: an unusual presentation. J Am Acad Dermatol. 2003;49(2 suppl case reports):S157-S160.
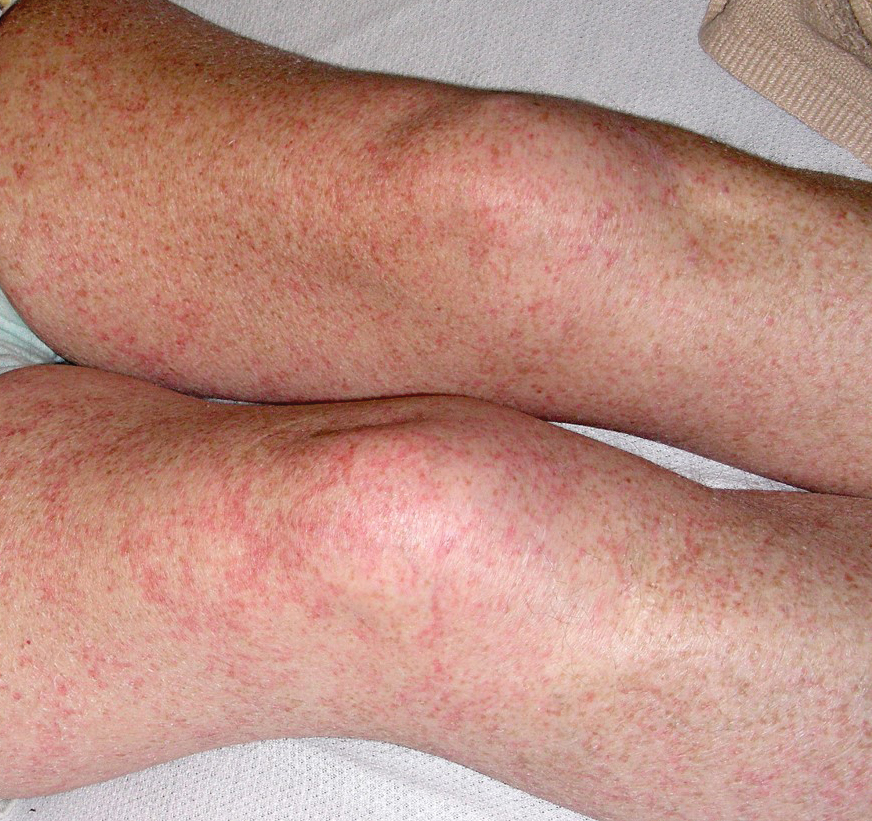
A 48-year-old woman from rural Virginia presented with centrifugally spreading, pruritic, blanchable macules over the lower abdomen and upper thighs noted 4 months after a pancreas transplant. After 3 weeks, the macules coalesced into reticulated nonblanching petechial patches. Fever, dyspnea, increasing xerosis, abdominal pain, and constipation were present. The patient had a medical history of type 1 diabetes mellitus requiring a pancreas transplant. Initial skin biopsy and fluorescence in situ hybridization to test for immune reaction to the XY-donor pancreas were negative. Mild transient eosinophilia was present at admission.