User login
Cutis is a peer-reviewed clinical journal for the dermatologist, allergist, and general practitioner published monthly since 1965. Concise clinical articles present the practical side of dermatology, helping physicians to improve patient care. Cutis is referenced in Index Medicus/MEDLINE and is written and edited by industry leaders.
ass lick
assault rifle
balls
ballsac
black jack
bleach
Boko Haram
bondage
causas
cheap
child abuse
cocaine
compulsive behaviors
cost of miracles
cunt
Daech
display network stats
drug paraphernalia
explosion
fart
fda and death
fda AND warn
fda AND warning
fda AND warns
feom
fuck
gambling
gfc
gun
human trafficking
humira AND expensive
illegal
ISIL
ISIS
Islamic caliphate
Islamic state
madvocate
masturbation
mixed martial arts
MMA
molestation
national rifle association
NRA
nsfw
nuccitelli
pedophile
pedophilia
poker
porn
porn
pornography
psychedelic drug
recreational drug
sex slave rings
shit
slot machine
snort
substance abuse
terrorism
terrorist
texarkana
Texas hold 'em
UFC
section[contains(@class, 'nav-hidden')]
section[contains(@class, 'nav-hidden active')
A peer-reviewed, indexed journal for dermatologists with original research, image quizzes, cases and reviews, and columns.
Acantholytic Anaplastic Extramammary Paget Disease
To the Editor:
Extramammary Paget disease (EMPD) is a rare intraepidermal neoplasm with glandular differentiation that is classically known as a mimicker of Bowen disease (squamous cell carcinoma in situ of the skin) due to their histologic similarities.1,2 However, acantholytic anaplastic EMPD (AAEMPD) is a rare variant that can pose a particularly difficult diagnostic challenge because of its histologic similarity to benign acantholytic disorders and other malignant neoplasms. Major histologic features suggestive of AAEMPD include full-thickness atypia of the epidermis, loss of nuclear polarity, marked cytologic anaplasia, intraepidermal acantholysis, and Paget cells.3 The differential diagnosis of EMPD typically includes Bowen disease and pagetoid Bowen disease, but the acantholytic anaplastic variant more often is confused with intraepidermal acantholytic lesions such as acantholytic dyskeratosis of the genitocrural area, familial benign pemphigus (Hailey-Hailey disease), pemphigus vulgaris, and acantholytic Bowen disease. Immunohistochemistry (IHC) studies to assist in the definitive diagnosis of AAEMPD are strongly advised because of these difficulties in diagnosis.4 Cases of EMPD with an acantholytic appearance have rarely been reported in the literature.5-7
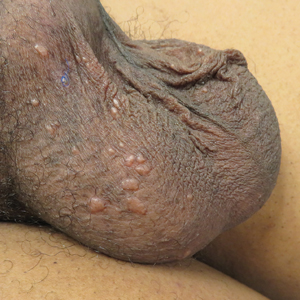
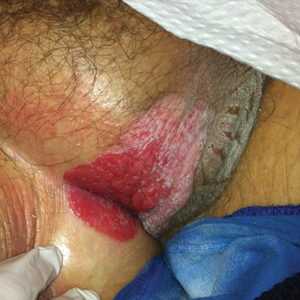
A 78-year-old man with a history of arthritis, heart disease, hypertension, and gastrointestinal disease presented for evaluation of a tender lesion of the right genitocrural crease of 5 years’ duration. He had no history of cutaneous or internal malignancy. Previously the lesion had been treated by dermatology with a variety of topical products including antifungal and antibiotic creams with no improvement. Physical examination revealed a well-defined, 7×5-cm, tender, erythematous, macerated plaque on the right upper inner thigh adjacent to the scrotum with an odor possibly due to secondary infection (Figure 1).
plaque on the right upper inner thigh adjacent to the scrotum.
A biopsy of the lesion was performed, and the specimen was submitted for pathologic examination. Bacterial cultures taken at the time of biopsy revealed polybacterial colonization with Acinetobacter, Morganella, and mixed skin flora. The patient was treated with a 10-day course of oral sulfamethoxazole 800 mg and trimethoprim 160 mg twice daily once culture results returned. The biopsy results were communicated to the patient; however, he subsequently relocated, assumed care at another facility, and has since been lost to follow-up.
The biopsy specimen was examined grossly, serially sectioned, and submitted for routine processing with hematoxylin and eosin, periodic acid–Schiff, and Hale colloidal iron staining. Routine IHC was performed with antibodies to cytokeratin (CK) 7, CK20, carcinoembryonic antigen (CEA), pancytokeratin (CKAE1/AE3), and low- molecular-weight cytokeratin (LMWCK).
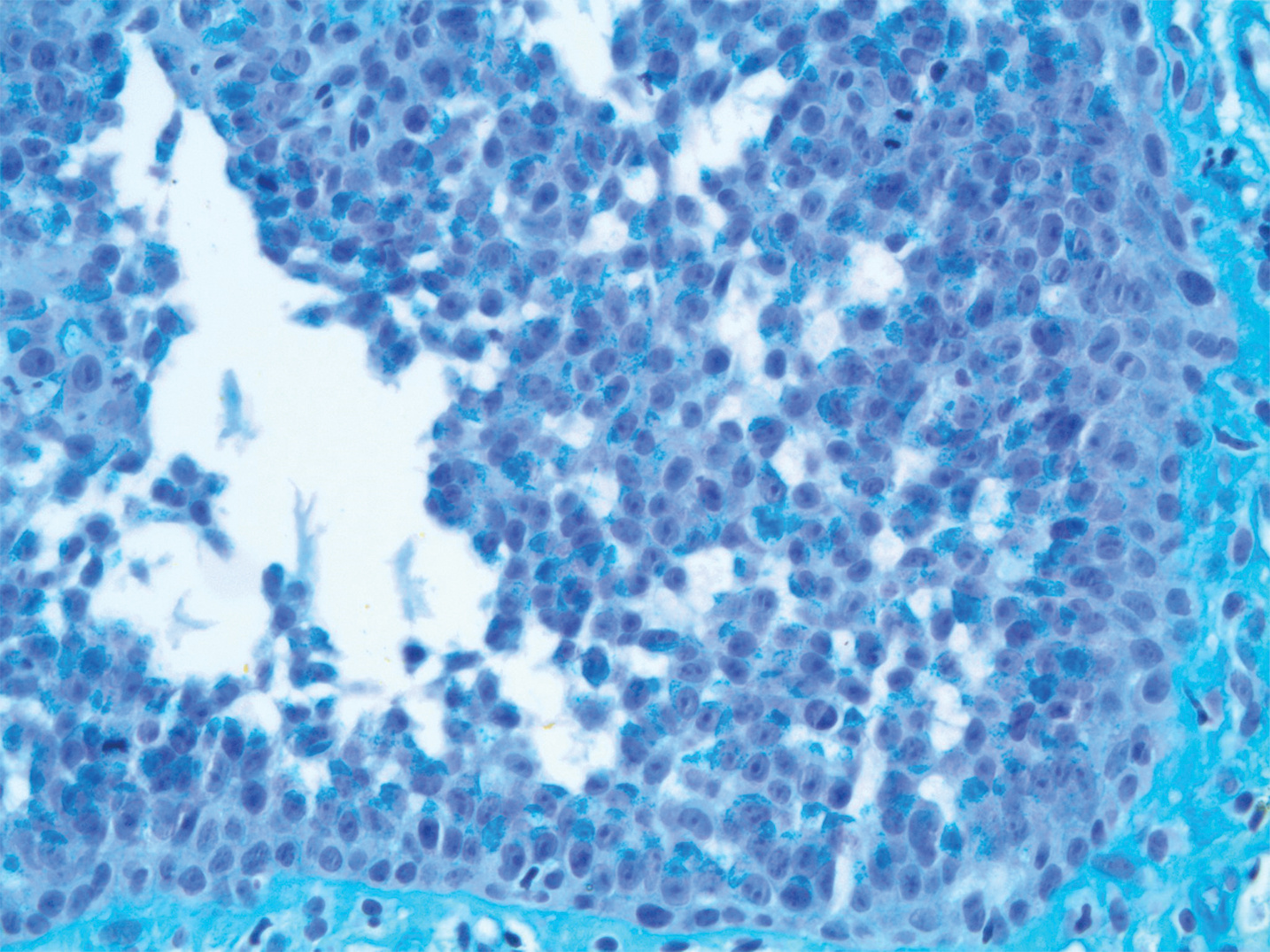
Pathologic examination of the biopsy showed prominent acanthosis of the epidermis composed of a proliferation of epithelial cells with associated full-thickness suprabasal acantholysis (Figure 2A). On inspection at higher magnification, the neoplastic cells demonstrated anaplasia as cytologic atypia with prominent and frequently multiple nucleoli, scant cytoplasm, and a high nuclear to cytoplasmic ratio (Figure 2B). There was a marked increase in mitotic activity with as many as 5 mitotic figures per high-power field. A fairly dense mixed inflammatory infiltrate comprised of lymphocytes, plasma cells, neutrophils, and eosinophils was present in the dermis. No fungal elements were observed on periodic acid–Schiff staining. The vast majority of tumor cells demonstrated moderate to abundant cytoplasmic mucin on Hale colloidal iron staining (Figure 3).
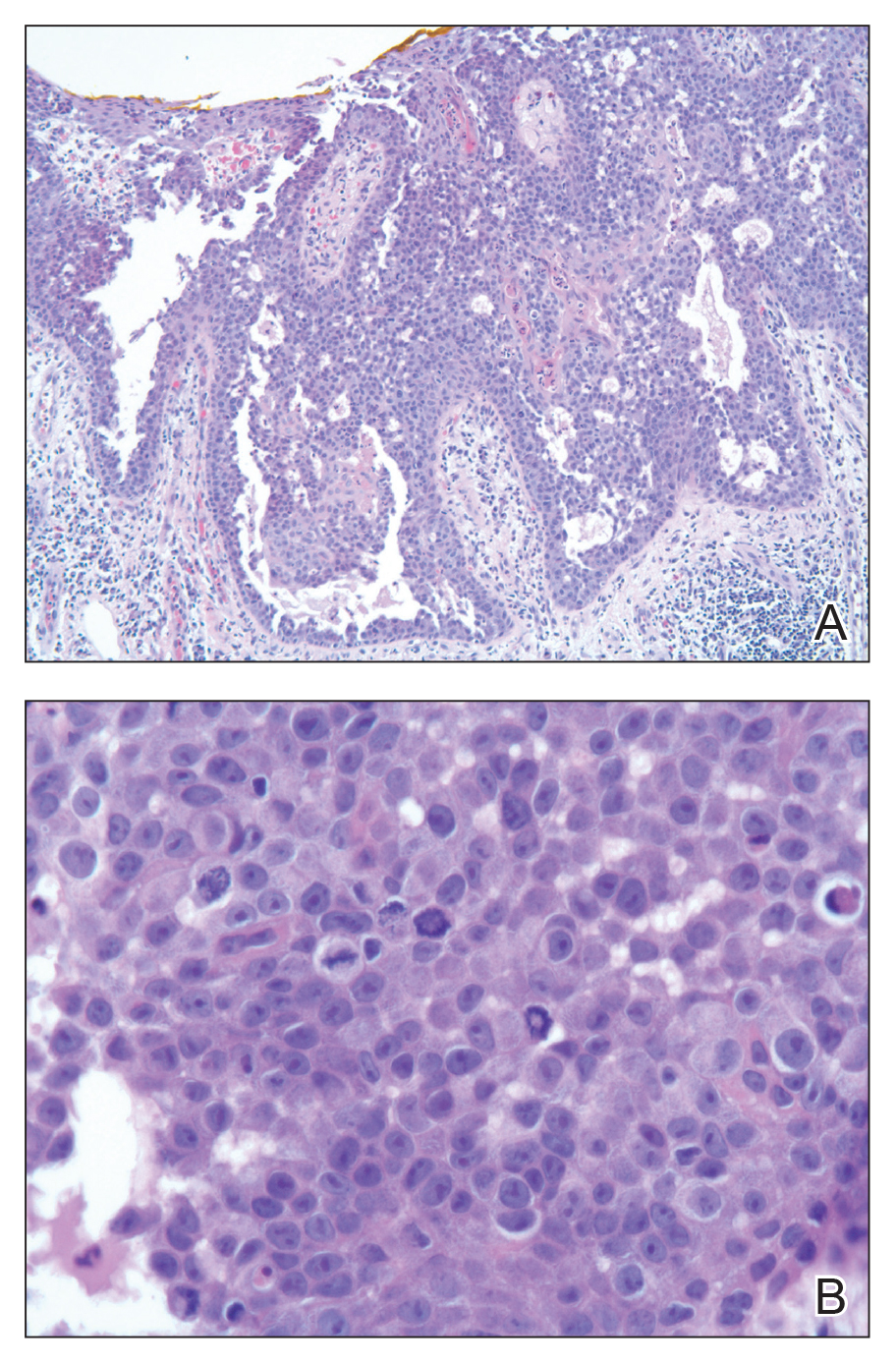
(H&E, original magnification ×400).
Immunohistochemistry staining of tumor cells was positive for CK7, CEA, pancytokeratin (CKAE1/AE3), and LMWCK. The tumor cells were negative for CK20. On the basis of the histopathologic and IHC findings, the patient was diagnosed with AAEMPD.
Extramammary Paget disease is a rare intraepidermal neoplasm with glandular differentiation. The most commonly involved sites are the anogenital areas including the vulvar, perianal, perineal, scrotal, and penile regions, as well as other areas rich in apocrine glands such as the axillae.8 Extramammary Paget disease most commonly originates as a primary intraepidermal neoplasm (type 1 EMPD), but an underlying malignant neoplasm that spreads intraepithelially is seen in a minority of cases (types 2 and 3 EMPD). In the vulva, type 1a refers to cutaneous noninvasive Paget disease, type 1b refers to dermal invasion of Paget disease, type 1c refers to vulvar adenocarcinoma–associated Paget disease, type 2 refers to rectal/anal adenocarcinoma–associated Paget disease, and type 3 refers to urogenital neoplasia–associated Paget disease.9
The acantholytic anaplastic variant of EMPD can be challenging to diagnose because of its similarities to many other lesions, including acantholytic dyskeratosis of the genitocrural area, familial benign pemphigus (Hailey-Hailey disease), pemphigus vulgaris, Bowen disease, pagetoid Bowen disease, and acantholytic Bowen disease. Major histologic features of AAEMPD include full-thickness atypia of the epidermis, loss of nuclear polarity, marked cytologic anaplasia, intraepidermal acantholysis, and Paget cells.3 The acantholytic anaplastic variant of EMPD can be differentiated from other diagnoses using IHC studies, with findings indicative of AAEMPD outlined below.
The proliferative neoplastic cell in EMPD is the Paget cell, which can be identified as a large round cell located in the epidermis with pale-staining cytoplasm, a large nucleus, and sometimes a prominent nucleolus. Paget cells can be distributed singly or in clusters, nests, or glandular structures within the epidermis and adjacent to adnexal structures.10 Extramammary Paget disease can have many patterns, including glandular, acantholysis-like, upper nest, tall nest, budding, and sheetlike.11
Immunohistochemically, Paget cells in EMPD typically express pancytokeratins (CKAE1/AE3), low-molecular-weight/simple epithelial type keratins (CK7, CAM 5.2), sweat gland antigens (epithelial membrane antigen, CEA, gross cystic disease fluid protein 15 [GCDFP15]), mucin 5AC (MUC5AC), and often androgen receptor.12-18 Paget cells contain cytoplasmic mucin and demonstrate prominent cytoplasmic staining with Hale colloidal iron.17 Paget cells typically do not express high-molecular-weight cytokeratin (eg, CK5/6), melanocytic antigens, estrogen receptor, or progesterone receptor.15,18
Immunohistochemical staining has been shown to differ between primary cutaneous (type 1) and secondary (types 2 and 3) EMPD. Primary cutaneous EMPD typically expresses sweat gland markers (CK7+, CK20−, GCDFP15+). Secondary EMPD typically expresses an endodermal phenotype (CK7+, CK20+, GCDFP15−).12
Acantholytic dyskeratosis of the genitocrural area is a rare lesion included in the spectrum of focal acantholytic dyskeratoses described by Ackerman.19 It also has been referred to as papular acantholytic dyskeratosis of the vulva, though histologically similar lesions also have been reported in men.20-22 Histologically, acantholytic dyskeratosis of the genitocrural area has prominent acantholysis and dyskeratosis with corps ronds and grains.19 Familial benign pemphigus (Hailey-Hailey disease) is caused by mutations of the ATP2C1 gene, which encodes for a secretory pathway Ca2+/Mn2+-ATPase pump type 1 (SPCA1) in the Golgi apparatus in keratinocytes.23 Familial benign pemphigus has a histologic appearance similar to acantholytic dyskeratosis of the genitocrural area, but a positive family history of familial benign pemphigus can be used to differentiate the 2 entities from each other due to the autosomal-dominant inheritance pattern of familial benign pemphigus. Both of these disorders can appear similar to AAEMPD because of their extensive intraepidermal acantholysis, but they differ in the lack of Paget cells, intraepidermal atypia, and increased mitotic activity.
Acantholytic Bowen disease is a histologic variant that can be difficult to distinguish from AAEMPD on hematoxylin and eosin–stained sections because of their similar histologic features but can be differentiated by IHC stains.5 Acantholytic Bowen disease expresses high-molecular-weight cytokeratin (eg, CK5/6) but is negative for CK7, CAM 5.2, and CEA. Extramammary Paget disease generally has the opposite pattern: positive staining for CK7, CAM 5.2, and CEA, but negative for high-molecular-weight cytokeratin.13,14,24
Primary cutaneous adenosquamous carcinoma is a rare malignancy of squamous and glandular differentiation known for being locally aggressive and metastatic.25 Histologically, cutaneous adenosquamous carcinoma shows infiltrating nests of neoplastic cells with both squamous and glandular features. It differs notably from AAEMPD in that cutaneous adenosquamous carcinomas tend to arise in the head and arm regions, and their histologic morphology is different. The IHC profiles are similar, with positive staining for CEA, CK7, and mucin; however, they differ in that AAEMPD is negative for high-molecular-weight keratin while cutaneous adenosquamous carcinoma is positive.25
Verrucous carcinoma is an uncommon variant of squamous cell carcinoma with well-differentiated keratinocytes and a blunt pushing border.24 Similar to AAEMPD, this neoplasm can arise in the genital and perineal areas; however, the 2 entities differ considerably in morphology on histologic examination.
Pemphigus vulgaris is an autoimmune intraepidermal blistering disorder of the skin and mucous membranes of which pemphigus vegetans is a subtype.26,27 Pemphigus vulgaris is another diagnosis that can possibly be mimicked by AAEMPD.28 Histologic features of pemphigus vulgaris include intraepidermal acantholysis of keratinocytes immediately above the basal layer of the epidermis. Pemphigus vegetans is similar with the addition of papillomatosis, hyperkeratosis, and an eosinophilic infiltrate.26,27 Immunofluorescence typically demonstrates intercellular C3 and IgG deposits.26 These diseases mimic AAEMPD histologically but differ in their relative lack of atypia and Paget cells.
In summary, we report a case of AAEMPD in a 78-year-old man in whom routine histologic specimens showed marked intraepidermal acantholysis and atypical tumor cells with increased mitoses. The latter finding prompted IHC studies that revealed positive CK7, CEA, pancytokeratin, and LMWCK staining with negative CK20 staining. Hale colloidal iron staining showed moderate to abundant cytoplasmic mucin. The patient was diagnosed with AAEMPD. It is imperative to maintain clinical suspicion for AAEMPD and to examine acantholytic disorders with scrutiny. When there is evidence of atypia or mitoses, use of IHC stains can assist in fully characterizing the lesion.
- Bowen JT. Precancerous dermatosis: a study of two cases of chronic atypical epithelial proliferation. J Cutan Dis. 1912;30:241-255.
- Jones RE Jr, Austin C, Ackerman AB. Extramammary Paget’s disease: a critical reexamination. Am J Dermatopathol. 1979;1:101-132.
- Rayne SC, Santa Cruz DJ. Anaplastic Paget’s disease. Am J Surg Pathol. 1992;16:1085-1091.
- Wang EC, Kwah YC, Tan WP, et al. Extramammary Paget disease: immunohistochemistry is critical to distinguish potential mimickers. Dermatol Online J. 2012;18:4.
- Du X, Yin X, Zhou N, et al. Extramammary Paget’s disease mimicking acantholytic squamous cell carcinoma in situ: a case report. J Cutan Pathol. 2010;37:683.
- Mobini N. Acantholytic anaplastic Paget’s disease. J Cutan Pathol. 2009;36:374-380.
- Oh YJ, Lew BL, Sim WY. Acantholytic anaplastic extramammary Paget’s disease: a case report and review of the literature. Ann Dermatol. 2011;23:226-230.
- Zollo JD, Zeitouni NC. The Roswell Park Cancer Institute experience with extramammary Paget’s disease. Br J Dermatol. 2000;142:59-65.
- Wilkinson EJ, Brown HM. Vulvar Paget disease of urothelial origin: a report of three cases and a proposed classification of vulvar Paget disease. Hum Pathol. 2002;33:549-554.
- Lam C, Funaro D. Extramammary Paget’s disease: summary of current knowledge. Dermatol Clin. 2010;28:807-826.
- Shiomi T, Yoshida Y, Shomori K, et al. Extramammary Paget’s disease: evaluation of the histopathological patterns of Paget cell proliferation in the epidermis. J Dermatol. 2011;38:1054-1057.
- Goldblum JR, Hart WR. Vulvar Paget’s disease: a clinicopathologic and immunohistochemical study of 19 cases. Am J Surg Pathol. 1997;21:1178-1187.
- Alhumaidi A. Practical immunohistochemistry of epithelial skin tumor. Indian J Dermatol Venerol Leprol. 2012;78:698-708.
- Battles O, Page D, Johnson J. Cytokeratins, CEA and mucin histochemistry in the diagnosis and characterization of extramammary Paget’s disease. Am J Clin Pathol. 1997;108:6-12.
- Kanitakis J. Mammary and extramammary Paget’s disease. J Eur Acad Dermatol Venereol. 2007;21:581-590.
- Krishna M. Diagnosis of metastatic neoplasms: an immunohistochemical approach. Arch Pathol Lab Med. 2010;134:207-215.
- Helm KF, Goellner JR, Peters MS. Immunohistochemical stain in extramammary Paget’s disease. Am J Dermatopathol. 1992;14:402-407.
- Liegl B, Horn L, Moinfar F. Androgen receptors are frequently expressed in mammary and extramammary Paget’s disease. Mod Pathol. 2005;18:1283-288.
- Ackerman AB. Focal acantholytic dyskeratosis. Arch Derm. 1972;106:702-706.
- Dittmer CJ, Hornemann A, Rose C, et al. Successful laser therapy of a papular acantholytic dyskeratosis of the vulva: case report and review of literature. Arch Gynecol Obstet. 2010;291:723-725.
- Roh MR, Choi YJ, Lee KG. Papular acantholytic dyskeratosis of the vulva. J Dermatol. 2009;36:427-429.
- Wong KT, Mihm MC Jr. Acantholytic dermatosis localized to genitalia and crural areas of male patients: a report of three cases. J Cutan Pathol. 1994;21:27-32.
- Hu Z, Bonifas JM, Beech J, et al. Mutations in ATP2C1, encoding a calcium pump, cause Hailey-Hailey disease. Nat Genet. 2000; 24:61-65.
- Elston DM. Malignant tumors of the epidermis. In: Elston DM, Ferringer T, eds. Requisites in Dermatology: Dermatopathology. Philadelphia, PA: Elsevier Limited; 2012:53-68.
- Fu JM, McCalmont T, Yu SS. Adenosquamous carcinoma of the skin: a case series. Arch Dermatol. 2009;145:1152-1158.
- Becker BA, Gaspari AA. Pemphigus vulgaris and vegetans. Dermatol Clin. 1993;11:429-452.
- Rados J. Autoimmune blistering diseases: histologic meaning. Clin Dermatol. 2011;29:377-388.
- Kohler S, Smoller BR. A case of extramammary Paget’s disease mimicking pemphigus vulgaris on histologic examination. Dermatology. 1997;195:54-56.
To the Editor:
Extramammary Paget disease (EMPD) is a rare intraepidermal neoplasm with glandular differentiation that is classically known as a mimicker of Bowen disease (squamous cell carcinoma in situ of the skin) due to their histologic similarities.1,2 However, acantholytic anaplastic EMPD (AAEMPD) is a rare variant that can pose a particularly difficult diagnostic challenge because of its histologic similarity to benign acantholytic disorders and other malignant neoplasms. Major histologic features suggestive of AAEMPD include full-thickness atypia of the epidermis, loss of nuclear polarity, marked cytologic anaplasia, intraepidermal acantholysis, and Paget cells.3 The differential diagnosis of EMPD typically includes Bowen disease and pagetoid Bowen disease, but the acantholytic anaplastic variant more often is confused with intraepidermal acantholytic lesions such as acantholytic dyskeratosis of the genitocrural area, familial benign pemphigus (Hailey-Hailey disease), pemphigus vulgaris, and acantholytic Bowen disease. Immunohistochemistry (IHC) studies to assist in the definitive diagnosis of AAEMPD are strongly advised because of these difficulties in diagnosis.4 Cases of EMPD with an acantholytic appearance have rarely been reported in the literature.5-7
A 78-year-old man with a history of arthritis, heart disease, hypertension, and gastrointestinal disease presented for evaluation of a tender lesion of the right genitocrural crease of 5 years’ duration. He had no history of cutaneous or internal malignancy. Previously the lesion had been treated by dermatology with a variety of topical products including antifungal and antibiotic creams with no improvement. Physical examination revealed a well-defined, 7×5-cm, tender, erythematous, macerated plaque on the right upper inner thigh adjacent to the scrotum with an odor possibly due to secondary infection (Figure 1).
plaque on the right upper inner thigh adjacent to the scrotum.
A biopsy of the lesion was performed, and the specimen was submitted for pathologic examination. Bacterial cultures taken at the time of biopsy revealed polybacterial colonization with Acinetobacter, Morganella, and mixed skin flora. The patient was treated with a 10-day course of oral sulfamethoxazole 800 mg and trimethoprim 160 mg twice daily once culture results returned. The biopsy results were communicated to the patient; however, he subsequently relocated, assumed care at another facility, and has since been lost to follow-up.
The biopsy specimen was examined grossly, serially sectioned, and submitted for routine processing with hematoxylin and eosin, periodic acid–Schiff, and Hale colloidal iron staining. Routine IHC was performed with antibodies to cytokeratin (CK) 7, CK20, carcinoembryonic antigen (CEA), pancytokeratin (CKAE1/AE3), and low- molecular-weight cytokeratin (LMWCK).
Pathologic examination of the biopsy showed prominent acanthosis of the epidermis composed of a proliferation of epithelial cells with associated full-thickness suprabasal acantholysis (Figure 2A). On inspection at higher magnification, the neoplastic cells demonstrated anaplasia as cytologic atypia with prominent and frequently multiple nucleoli, scant cytoplasm, and a high nuclear to cytoplasmic ratio (Figure 2B). There was a marked increase in mitotic activity with as many as 5 mitotic figures per high-power field. A fairly dense mixed inflammatory infiltrate comprised of lymphocytes, plasma cells, neutrophils, and eosinophils was present in the dermis. No fungal elements were observed on periodic acid–Schiff staining. The vast majority of tumor cells demonstrated moderate to abundant cytoplasmic mucin on Hale colloidal iron staining (Figure 3).
(H&E, original magnification ×400).
Immunohistochemistry staining of tumor cells was positive for CK7, CEA, pancytokeratin (CKAE1/AE3), and LMWCK. The tumor cells were negative for CK20. On the basis of the histopathologic and IHC findings, the patient was diagnosed with AAEMPD.
Extramammary Paget disease is a rare intraepidermal neoplasm with glandular differentiation. The most commonly involved sites are the anogenital areas including the vulvar, perianal, perineal, scrotal, and penile regions, as well as other areas rich in apocrine glands such as the axillae.8 Extramammary Paget disease most commonly originates as a primary intraepidermal neoplasm (type 1 EMPD), but an underlying malignant neoplasm that spreads intraepithelially is seen in a minority of cases (types 2 and 3 EMPD). In the vulva, type 1a refers to cutaneous noninvasive Paget disease, type 1b refers to dermal invasion of Paget disease, type 1c refers to vulvar adenocarcinoma–associated Paget disease, type 2 refers to rectal/anal adenocarcinoma–associated Paget disease, and type 3 refers to urogenital neoplasia–associated Paget disease.9
The acantholytic anaplastic variant of EMPD can be challenging to diagnose because of its similarities to many other lesions, including acantholytic dyskeratosis of the genitocrural area, familial benign pemphigus (Hailey-Hailey disease), pemphigus vulgaris, Bowen disease, pagetoid Bowen disease, and acantholytic Bowen disease. Major histologic features of AAEMPD include full-thickness atypia of the epidermis, loss of nuclear polarity, marked cytologic anaplasia, intraepidermal acantholysis, and Paget cells.3 The acantholytic anaplastic variant of EMPD can be differentiated from other diagnoses using IHC studies, with findings indicative of AAEMPD outlined below.
The proliferative neoplastic cell in EMPD is the Paget cell, which can be identified as a large round cell located in the epidermis with pale-staining cytoplasm, a large nucleus, and sometimes a prominent nucleolus. Paget cells can be distributed singly or in clusters, nests, or glandular structures within the epidermis and adjacent to adnexal structures.10 Extramammary Paget disease can have many patterns, including glandular, acantholysis-like, upper nest, tall nest, budding, and sheetlike.11
Immunohistochemically, Paget cells in EMPD typically express pancytokeratins (CKAE1/AE3), low-molecular-weight/simple epithelial type keratins (CK7, CAM 5.2), sweat gland antigens (epithelial membrane antigen, CEA, gross cystic disease fluid protein 15 [GCDFP15]), mucin 5AC (MUC5AC), and often androgen receptor.12-18 Paget cells contain cytoplasmic mucin and demonstrate prominent cytoplasmic staining with Hale colloidal iron.17 Paget cells typically do not express high-molecular-weight cytokeratin (eg, CK5/6), melanocytic antigens, estrogen receptor, or progesterone receptor.15,18
Immunohistochemical staining has been shown to differ between primary cutaneous (type 1) and secondary (types 2 and 3) EMPD. Primary cutaneous EMPD typically expresses sweat gland markers (CK7+, CK20−, GCDFP15+). Secondary EMPD typically expresses an endodermal phenotype (CK7+, CK20+, GCDFP15−).12
Acantholytic dyskeratosis of the genitocrural area is a rare lesion included in the spectrum of focal acantholytic dyskeratoses described by Ackerman.19 It also has been referred to as papular acantholytic dyskeratosis of the vulva, though histologically similar lesions also have been reported in men.20-22 Histologically, acantholytic dyskeratosis of the genitocrural area has prominent acantholysis and dyskeratosis with corps ronds and grains.19 Familial benign pemphigus (Hailey-Hailey disease) is caused by mutations of the ATP2C1 gene, which encodes for a secretory pathway Ca2+/Mn2+-ATPase pump type 1 (SPCA1) in the Golgi apparatus in keratinocytes.23 Familial benign pemphigus has a histologic appearance similar to acantholytic dyskeratosis of the genitocrural area, but a positive family history of familial benign pemphigus can be used to differentiate the 2 entities from each other due to the autosomal-dominant inheritance pattern of familial benign pemphigus. Both of these disorders can appear similar to AAEMPD because of their extensive intraepidermal acantholysis, but they differ in the lack of Paget cells, intraepidermal atypia, and increased mitotic activity.
Acantholytic Bowen disease is a histologic variant that can be difficult to distinguish from AAEMPD on hematoxylin and eosin–stained sections because of their similar histologic features but can be differentiated by IHC stains.5 Acantholytic Bowen disease expresses high-molecular-weight cytokeratin (eg, CK5/6) but is negative for CK7, CAM 5.2, and CEA. Extramammary Paget disease generally has the opposite pattern: positive staining for CK7, CAM 5.2, and CEA, but negative for high-molecular-weight cytokeratin.13,14,24
Primary cutaneous adenosquamous carcinoma is a rare malignancy of squamous and glandular differentiation known for being locally aggressive and metastatic.25 Histologically, cutaneous adenosquamous carcinoma shows infiltrating nests of neoplastic cells with both squamous and glandular features. It differs notably from AAEMPD in that cutaneous adenosquamous carcinomas tend to arise in the head and arm regions, and their histologic morphology is different. The IHC profiles are similar, with positive staining for CEA, CK7, and mucin; however, they differ in that AAEMPD is negative for high-molecular-weight keratin while cutaneous adenosquamous carcinoma is positive.25
Verrucous carcinoma is an uncommon variant of squamous cell carcinoma with well-differentiated keratinocytes and a blunt pushing border.24 Similar to AAEMPD, this neoplasm can arise in the genital and perineal areas; however, the 2 entities differ considerably in morphology on histologic examination.
Pemphigus vulgaris is an autoimmune intraepidermal blistering disorder of the skin and mucous membranes of which pemphigus vegetans is a subtype.26,27 Pemphigus vulgaris is another diagnosis that can possibly be mimicked by AAEMPD.28 Histologic features of pemphigus vulgaris include intraepidermal acantholysis of keratinocytes immediately above the basal layer of the epidermis. Pemphigus vegetans is similar with the addition of papillomatosis, hyperkeratosis, and an eosinophilic infiltrate.26,27 Immunofluorescence typically demonstrates intercellular C3 and IgG deposits.26 These diseases mimic AAEMPD histologically but differ in their relative lack of atypia and Paget cells.
In summary, we report a case of AAEMPD in a 78-year-old man in whom routine histologic specimens showed marked intraepidermal acantholysis and atypical tumor cells with increased mitoses. The latter finding prompted IHC studies that revealed positive CK7, CEA, pancytokeratin, and LMWCK staining with negative CK20 staining. Hale colloidal iron staining showed moderate to abundant cytoplasmic mucin. The patient was diagnosed with AAEMPD. It is imperative to maintain clinical suspicion for AAEMPD and to examine acantholytic disorders with scrutiny. When there is evidence of atypia or mitoses, use of IHC stains can assist in fully characterizing the lesion.
To the Editor:
Extramammary Paget disease (EMPD) is a rare intraepidermal neoplasm with glandular differentiation that is classically known as a mimicker of Bowen disease (squamous cell carcinoma in situ of the skin) due to their histologic similarities.1,2 However, acantholytic anaplastic EMPD (AAEMPD) is a rare variant that can pose a particularly difficult diagnostic challenge because of its histologic similarity to benign acantholytic disorders and other malignant neoplasms. Major histologic features suggestive of AAEMPD include full-thickness atypia of the epidermis, loss of nuclear polarity, marked cytologic anaplasia, intraepidermal acantholysis, and Paget cells.3 The differential diagnosis of EMPD typically includes Bowen disease and pagetoid Bowen disease, but the acantholytic anaplastic variant more often is confused with intraepidermal acantholytic lesions such as acantholytic dyskeratosis of the genitocrural area, familial benign pemphigus (Hailey-Hailey disease), pemphigus vulgaris, and acantholytic Bowen disease. Immunohistochemistry (IHC) studies to assist in the definitive diagnosis of AAEMPD are strongly advised because of these difficulties in diagnosis.4 Cases of EMPD with an acantholytic appearance have rarely been reported in the literature.5-7
A 78-year-old man with a history of arthritis, heart disease, hypertension, and gastrointestinal disease presented for evaluation of a tender lesion of the right genitocrural crease of 5 years’ duration. He had no history of cutaneous or internal malignancy. Previously the lesion had been treated by dermatology with a variety of topical products including antifungal and antibiotic creams with no improvement. Physical examination revealed a well-defined, 7×5-cm, tender, erythematous, macerated plaque on the right upper inner thigh adjacent to the scrotum with an odor possibly due to secondary infection (Figure 1).
plaque on the right upper inner thigh adjacent to the scrotum.
A biopsy of the lesion was performed, and the specimen was submitted for pathologic examination. Bacterial cultures taken at the time of biopsy revealed polybacterial colonization with Acinetobacter, Morganella, and mixed skin flora. The patient was treated with a 10-day course of oral sulfamethoxazole 800 mg and trimethoprim 160 mg twice daily once culture results returned. The biopsy results were communicated to the patient; however, he subsequently relocated, assumed care at another facility, and has since been lost to follow-up.
The biopsy specimen was examined grossly, serially sectioned, and submitted for routine processing with hematoxylin and eosin, periodic acid–Schiff, and Hale colloidal iron staining. Routine IHC was performed with antibodies to cytokeratin (CK) 7, CK20, carcinoembryonic antigen (CEA), pancytokeratin (CKAE1/AE3), and low- molecular-weight cytokeratin (LMWCK).
Pathologic examination of the biopsy showed prominent acanthosis of the epidermis composed of a proliferation of epithelial cells with associated full-thickness suprabasal acantholysis (Figure 2A). On inspection at higher magnification, the neoplastic cells demonstrated anaplasia as cytologic atypia with prominent and frequently multiple nucleoli, scant cytoplasm, and a high nuclear to cytoplasmic ratio (Figure 2B). There was a marked increase in mitotic activity with as many as 5 mitotic figures per high-power field. A fairly dense mixed inflammatory infiltrate comprised of lymphocytes, plasma cells, neutrophils, and eosinophils was present in the dermis. No fungal elements were observed on periodic acid–Schiff staining. The vast majority of tumor cells demonstrated moderate to abundant cytoplasmic mucin on Hale colloidal iron staining (Figure 3).
(H&E, original magnification ×400).
Immunohistochemistry staining of tumor cells was positive for CK7, CEA, pancytokeratin (CKAE1/AE3), and LMWCK. The tumor cells were negative for CK20. On the basis of the histopathologic and IHC findings, the patient was diagnosed with AAEMPD.
Extramammary Paget disease is a rare intraepidermal neoplasm with glandular differentiation. The most commonly involved sites are the anogenital areas including the vulvar, perianal, perineal, scrotal, and penile regions, as well as other areas rich in apocrine glands such as the axillae.8 Extramammary Paget disease most commonly originates as a primary intraepidermal neoplasm (type 1 EMPD), but an underlying malignant neoplasm that spreads intraepithelially is seen in a minority of cases (types 2 and 3 EMPD). In the vulva, type 1a refers to cutaneous noninvasive Paget disease, type 1b refers to dermal invasion of Paget disease, type 1c refers to vulvar adenocarcinoma–associated Paget disease, type 2 refers to rectal/anal adenocarcinoma–associated Paget disease, and type 3 refers to urogenital neoplasia–associated Paget disease.9
The acantholytic anaplastic variant of EMPD can be challenging to diagnose because of its similarities to many other lesions, including acantholytic dyskeratosis of the genitocrural area, familial benign pemphigus (Hailey-Hailey disease), pemphigus vulgaris, Bowen disease, pagetoid Bowen disease, and acantholytic Bowen disease. Major histologic features of AAEMPD include full-thickness atypia of the epidermis, loss of nuclear polarity, marked cytologic anaplasia, intraepidermal acantholysis, and Paget cells.3 The acantholytic anaplastic variant of EMPD can be differentiated from other diagnoses using IHC studies, with findings indicative of AAEMPD outlined below.
The proliferative neoplastic cell in EMPD is the Paget cell, which can be identified as a large round cell located in the epidermis with pale-staining cytoplasm, a large nucleus, and sometimes a prominent nucleolus. Paget cells can be distributed singly or in clusters, nests, or glandular structures within the epidermis and adjacent to adnexal structures.10 Extramammary Paget disease can have many patterns, including glandular, acantholysis-like, upper nest, tall nest, budding, and sheetlike.11
Immunohistochemically, Paget cells in EMPD typically express pancytokeratins (CKAE1/AE3), low-molecular-weight/simple epithelial type keratins (CK7, CAM 5.2), sweat gland antigens (epithelial membrane antigen, CEA, gross cystic disease fluid protein 15 [GCDFP15]), mucin 5AC (MUC5AC), and often androgen receptor.12-18 Paget cells contain cytoplasmic mucin and demonstrate prominent cytoplasmic staining with Hale colloidal iron.17 Paget cells typically do not express high-molecular-weight cytokeratin (eg, CK5/6), melanocytic antigens, estrogen receptor, or progesterone receptor.15,18
Immunohistochemical staining has been shown to differ between primary cutaneous (type 1) and secondary (types 2 and 3) EMPD. Primary cutaneous EMPD typically expresses sweat gland markers (CK7+, CK20−, GCDFP15+). Secondary EMPD typically expresses an endodermal phenotype (CK7+, CK20+, GCDFP15−).12
Acantholytic dyskeratosis of the genitocrural area is a rare lesion included in the spectrum of focal acantholytic dyskeratoses described by Ackerman.19 It also has been referred to as papular acantholytic dyskeratosis of the vulva, though histologically similar lesions also have been reported in men.20-22 Histologically, acantholytic dyskeratosis of the genitocrural area has prominent acantholysis and dyskeratosis with corps ronds and grains.19 Familial benign pemphigus (Hailey-Hailey disease) is caused by mutations of the ATP2C1 gene, which encodes for a secretory pathway Ca2+/Mn2+-ATPase pump type 1 (SPCA1) in the Golgi apparatus in keratinocytes.23 Familial benign pemphigus has a histologic appearance similar to acantholytic dyskeratosis of the genitocrural area, but a positive family history of familial benign pemphigus can be used to differentiate the 2 entities from each other due to the autosomal-dominant inheritance pattern of familial benign pemphigus. Both of these disorders can appear similar to AAEMPD because of their extensive intraepidermal acantholysis, but they differ in the lack of Paget cells, intraepidermal atypia, and increased mitotic activity.
Acantholytic Bowen disease is a histologic variant that can be difficult to distinguish from AAEMPD on hematoxylin and eosin–stained sections because of their similar histologic features but can be differentiated by IHC stains.5 Acantholytic Bowen disease expresses high-molecular-weight cytokeratin (eg, CK5/6) but is negative for CK7, CAM 5.2, and CEA. Extramammary Paget disease generally has the opposite pattern: positive staining for CK7, CAM 5.2, and CEA, but negative for high-molecular-weight cytokeratin.13,14,24
Primary cutaneous adenosquamous carcinoma is a rare malignancy of squamous and glandular differentiation known for being locally aggressive and metastatic.25 Histologically, cutaneous adenosquamous carcinoma shows infiltrating nests of neoplastic cells with both squamous and glandular features. It differs notably from AAEMPD in that cutaneous adenosquamous carcinomas tend to arise in the head and arm regions, and their histologic morphology is different. The IHC profiles are similar, with positive staining for CEA, CK7, and mucin; however, they differ in that AAEMPD is negative for high-molecular-weight keratin while cutaneous adenosquamous carcinoma is positive.25
Verrucous carcinoma is an uncommon variant of squamous cell carcinoma with well-differentiated keratinocytes and a blunt pushing border.24 Similar to AAEMPD, this neoplasm can arise in the genital and perineal areas; however, the 2 entities differ considerably in morphology on histologic examination.
Pemphigus vulgaris is an autoimmune intraepidermal blistering disorder of the skin and mucous membranes of which pemphigus vegetans is a subtype.26,27 Pemphigus vulgaris is another diagnosis that can possibly be mimicked by AAEMPD.28 Histologic features of pemphigus vulgaris include intraepidermal acantholysis of keratinocytes immediately above the basal layer of the epidermis. Pemphigus vegetans is similar with the addition of papillomatosis, hyperkeratosis, and an eosinophilic infiltrate.26,27 Immunofluorescence typically demonstrates intercellular C3 and IgG deposits.26 These diseases mimic AAEMPD histologically but differ in their relative lack of atypia and Paget cells.
In summary, we report a case of AAEMPD in a 78-year-old man in whom routine histologic specimens showed marked intraepidermal acantholysis and atypical tumor cells with increased mitoses. The latter finding prompted IHC studies that revealed positive CK7, CEA, pancytokeratin, and LMWCK staining with negative CK20 staining. Hale colloidal iron staining showed moderate to abundant cytoplasmic mucin. The patient was diagnosed with AAEMPD. It is imperative to maintain clinical suspicion for AAEMPD and to examine acantholytic disorders with scrutiny. When there is evidence of atypia or mitoses, use of IHC stains can assist in fully characterizing the lesion.
- Bowen JT. Precancerous dermatosis: a study of two cases of chronic atypical epithelial proliferation. J Cutan Dis. 1912;30:241-255.
- Jones RE Jr, Austin C, Ackerman AB. Extramammary Paget’s disease: a critical reexamination. Am J Dermatopathol. 1979;1:101-132.
- Rayne SC, Santa Cruz DJ. Anaplastic Paget’s disease. Am J Surg Pathol. 1992;16:1085-1091.
- Wang EC, Kwah YC, Tan WP, et al. Extramammary Paget disease: immunohistochemistry is critical to distinguish potential mimickers. Dermatol Online J. 2012;18:4.
- Du X, Yin X, Zhou N, et al. Extramammary Paget’s disease mimicking acantholytic squamous cell carcinoma in situ: a case report. J Cutan Pathol. 2010;37:683.
- Mobini N. Acantholytic anaplastic Paget’s disease. J Cutan Pathol. 2009;36:374-380.
- Oh YJ, Lew BL, Sim WY. Acantholytic anaplastic extramammary Paget’s disease: a case report and review of the literature. Ann Dermatol. 2011;23:226-230.
- Zollo JD, Zeitouni NC. The Roswell Park Cancer Institute experience with extramammary Paget’s disease. Br J Dermatol. 2000;142:59-65.
- Wilkinson EJ, Brown HM. Vulvar Paget disease of urothelial origin: a report of three cases and a proposed classification of vulvar Paget disease. Hum Pathol. 2002;33:549-554.
- Lam C, Funaro D. Extramammary Paget’s disease: summary of current knowledge. Dermatol Clin. 2010;28:807-826.
- Shiomi T, Yoshida Y, Shomori K, et al. Extramammary Paget’s disease: evaluation of the histopathological patterns of Paget cell proliferation in the epidermis. J Dermatol. 2011;38:1054-1057.
- Goldblum JR, Hart WR. Vulvar Paget’s disease: a clinicopathologic and immunohistochemical study of 19 cases. Am J Surg Pathol. 1997;21:1178-1187.
- Alhumaidi A. Practical immunohistochemistry of epithelial skin tumor. Indian J Dermatol Venerol Leprol. 2012;78:698-708.
- Battles O, Page D, Johnson J. Cytokeratins, CEA and mucin histochemistry in the diagnosis and characterization of extramammary Paget’s disease. Am J Clin Pathol. 1997;108:6-12.
- Kanitakis J. Mammary and extramammary Paget’s disease. J Eur Acad Dermatol Venereol. 2007;21:581-590.
- Krishna M. Diagnosis of metastatic neoplasms: an immunohistochemical approach. Arch Pathol Lab Med. 2010;134:207-215.
- Helm KF, Goellner JR, Peters MS. Immunohistochemical stain in extramammary Paget’s disease. Am J Dermatopathol. 1992;14:402-407.
- Liegl B, Horn L, Moinfar F. Androgen receptors are frequently expressed in mammary and extramammary Paget’s disease. Mod Pathol. 2005;18:1283-288.
- Ackerman AB. Focal acantholytic dyskeratosis. Arch Derm. 1972;106:702-706.
- Dittmer CJ, Hornemann A, Rose C, et al. Successful laser therapy of a papular acantholytic dyskeratosis of the vulva: case report and review of literature. Arch Gynecol Obstet. 2010;291:723-725.
- Roh MR, Choi YJ, Lee KG. Papular acantholytic dyskeratosis of the vulva. J Dermatol. 2009;36:427-429.
- Wong KT, Mihm MC Jr. Acantholytic dermatosis localized to genitalia and crural areas of male patients: a report of three cases. J Cutan Pathol. 1994;21:27-32.
- Hu Z, Bonifas JM, Beech J, et al. Mutations in ATP2C1, encoding a calcium pump, cause Hailey-Hailey disease. Nat Genet. 2000; 24:61-65.
- Elston DM. Malignant tumors of the epidermis. In: Elston DM, Ferringer T, eds. Requisites in Dermatology: Dermatopathology. Philadelphia, PA: Elsevier Limited; 2012:53-68.
- Fu JM, McCalmont T, Yu SS. Adenosquamous carcinoma of the skin: a case series. Arch Dermatol. 2009;145:1152-1158.
- Becker BA, Gaspari AA. Pemphigus vulgaris and vegetans. Dermatol Clin. 1993;11:429-452.
- Rados J. Autoimmune blistering diseases: histologic meaning. Clin Dermatol. 2011;29:377-388.
- Kohler S, Smoller BR. A case of extramammary Paget’s disease mimicking pemphigus vulgaris on histologic examination. Dermatology. 1997;195:54-56.
- Bowen JT. Precancerous dermatosis: a study of two cases of chronic atypical epithelial proliferation. J Cutan Dis. 1912;30:241-255.
- Jones RE Jr, Austin C, Ackerman AB. Extramammary Paget’s disease: a critical reexamination. Am J Dermatopathol. 1979;1:101-132.
- Rayne SC, Santa Cruz DJ. Anaplastic Paget’s disease. Am J Surg Pathol. 1992;16:1085-1091.
- Wang EC, Kwah YC, Tan WP, et al. Extramammary Paget disease: immunohistochemistry is critical to distinguish potential mimickers. Dermatol Online J. 2012;18:4.
- Du X, Yin X, Zhou N, et al. Extramammary Paget’s disease mimicking acantholytic squamous cell carcinoma in situ: a case report. J Cutan Pathol. 2010;37:683.
- Mobini N. Acantholytic anaplastic Paget’s disease. J Cutan Pathol. 2009;36:374-380.
- Oh YJ, Lew BL, Sim WY. Acantholytic anaplastic extramammary Paget’s disease: a case report and review of the literature. Ann Dermatol. 2011;23:226-230.
- Zollo JD, Zeitouni NC. The Roswell Park Cancer Institute experience with extramammary Paget’s disease. Br J Dermatol. 2000;142:59-65.
- Wilkinson EJ, Brown HM. Vulvar Paget disease of urothelial origin: a report of three cases and a proposed classification of vulvar Paget disease. Hum Pathol. 2002;33:549-554.
- Lam C, Funaro D. Extramammary Paget’s disease: summary of current knowledge. Dermatol Clin. 2010;28:807-826.
- Shiomi T, Yoshida Y, Shomori K, et al. Extramammary Paget’s disease: evaluation of the histopathological patterns of Paget cell proliferation in the epidermis. J Dermatol. 2011;38:1054-1057.
- Goldblum JR, Hart WR. Vulvar Paget’s disease: a clinicopathologic and immunohistochemical study of 19 cases. Am J Surg Pathol. 1997;21:1178-1187.
- Alhumaidi A. Practical immunohistochemistry of epithelial skin tumor. Indian J Dermatol Venerol Leprol. 2012;78:698-708.
- Battles O, Page D, Johnson J. Cytokeratins, CEA and mucin histochemistry in the diagnosis and characterization of extramammary Paget’s disease. Am J Clin Pathol. 1997;108:6-12.
- Kanitakis J. Mammary and extramammary Paget’s disease. J Eur Acad Dermatol Venereol. 2007;21:581-590.
- Krishna M. Diagnosis of metastatic neoplasms: an immunohistochemical approach. Arch Pathol Lab Med. 2010;134:207-215.
- Helm KF, Goellner JR, Peters MS. Immunohistochemical stain in extramammary Paget’s disease. Am J Dermatopathol. 1992;14:402-407.
- Liegl B, Horn L, Moinfar F. Androgen receptors are frequently expressed in mammary and extramammary Paget’s disease. Mod Pathol. 2005;18:1283-288.
- Ackerman AB. Focal acantholytic dyskeratosis. Arch Derm. 1972;106:702-706.
- Dittmer CJ, Hornemann A, Rose C, et al. Successful laser therapy of a papular acantholytic dyskeratosis of the vulva: case report and review of literature. Arch Gynecol Obstet. 2010;291:723-725.
- Roh MR, Choi YJ, Lee KG. Papular acantholytic dyskeratosis of the vulva. J Dermatol. 2009;36:427-429.
- Wong KT, Mihm MC Jr. Acantholytic dermatosis localized to genitalia and crural areas of male patients: a report of three cases. J Cutan Pathol. 1994;21:27-32.
- Hu Z, Bonifas JM, Beech J, et al. Mutations in ATP2C1, encoding a calcium pump, cause Hailey-Hailey disease. Nat Genet. 2000; 24:61-65.
- Elston DM. Malignant tumors of the epidermis. In: Elston DM, Ferringer T, eds. Requisites in Dermatology: Dermatopathology. Philadelphia, PA: Elsevier Limited; 2012:53-68.
- Fu JM, McCalmont T, Yu SS. Adenosquamous carcinoma of the skin: a case series. Arch Dermatol. 2009;145:1152-1158.
- Becker BA, Gaspari AA. Pemphigus vulgaris and vegetans. Dermatol Clin. 1993;11:429-452.
- Rados J. Autoimmune blistering diseases: histologic meaning. Clin Dermatol. 2011;29:377-388.
- Kohler S, Smoller BR. A case of extramammary Paget’s disease mimicking pemphigus vulgaris on histologic examination. Dermatology. 1997;195:54-56.
Practice Points
- The acantholytic anaplastic variant of extramammary Paget disease (EMPD) can be mimicked by many other entities including Bowen disease, acantholytic dyskeratosis of the genitocrural area, and pemphigus vulgaris.
- A good immunohistochemical panel to evaluate for EMPD includes cytokeratin (CK) 7, pancytokeratin (CKAE1/AE3), CK20, and carcinoembryonic antigen.
Barber’s Sinus Between the Toes of a Female Hairdresser
To the Editor:
Barber’s sinus, or interdigital pilonidal sinus, is an occupational dermatosis with a pathognomonic clinical picture. Nearly all reports of barber’s sinus in the literature have involved the hands of male barbers and hairdressers. We present an uncommon case of barber’s sinus between the toes of a female hairdresser. If left untreated, potential complications of barber’s sinus include abscess formation, cellulitis, lymphangitis, and osteomyelitis. Clinicians should advise patients with an occupational risk of barber’s sinus to wear protective footwear and maintain hygiene in the interdigital spaces.
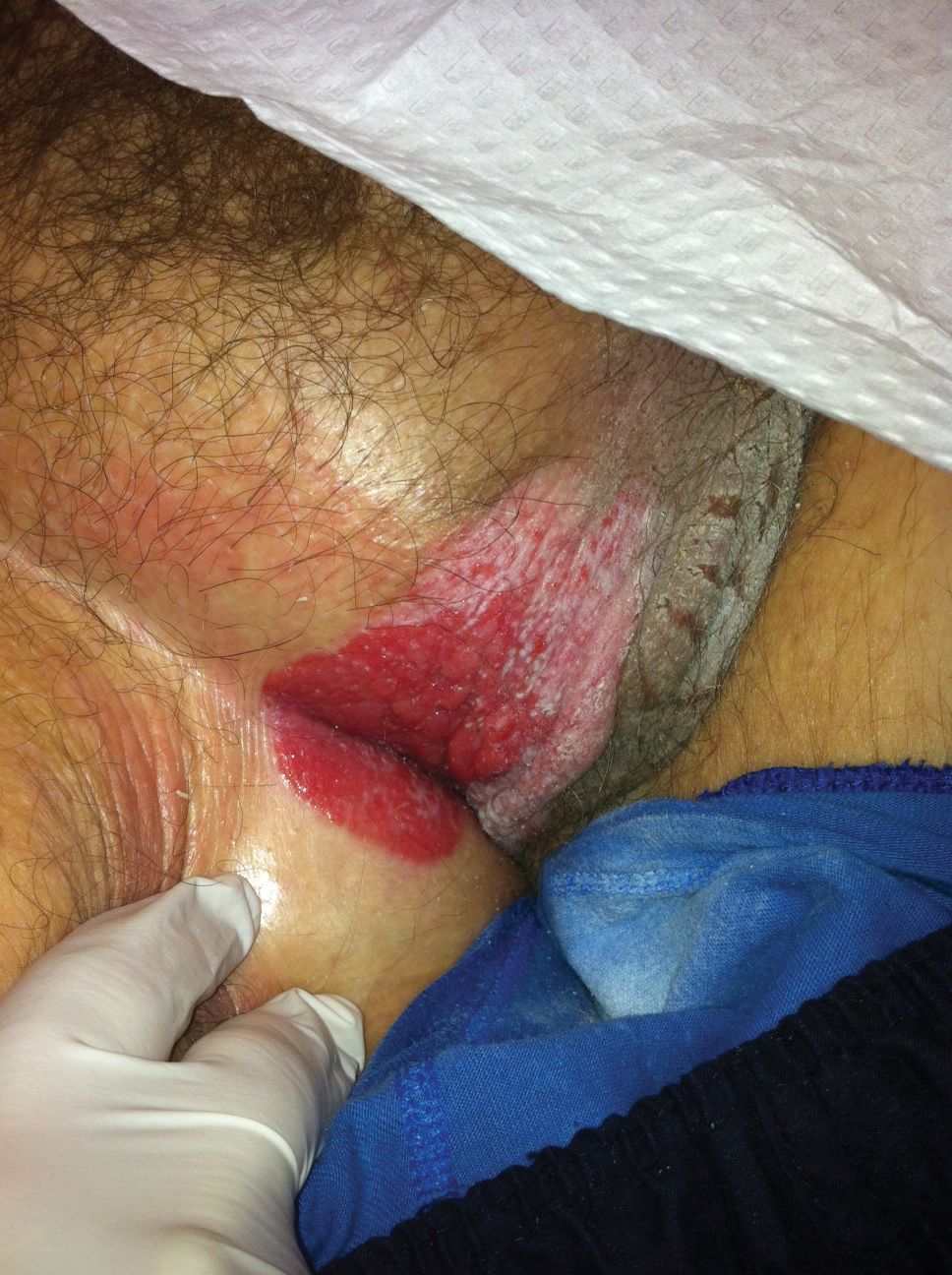
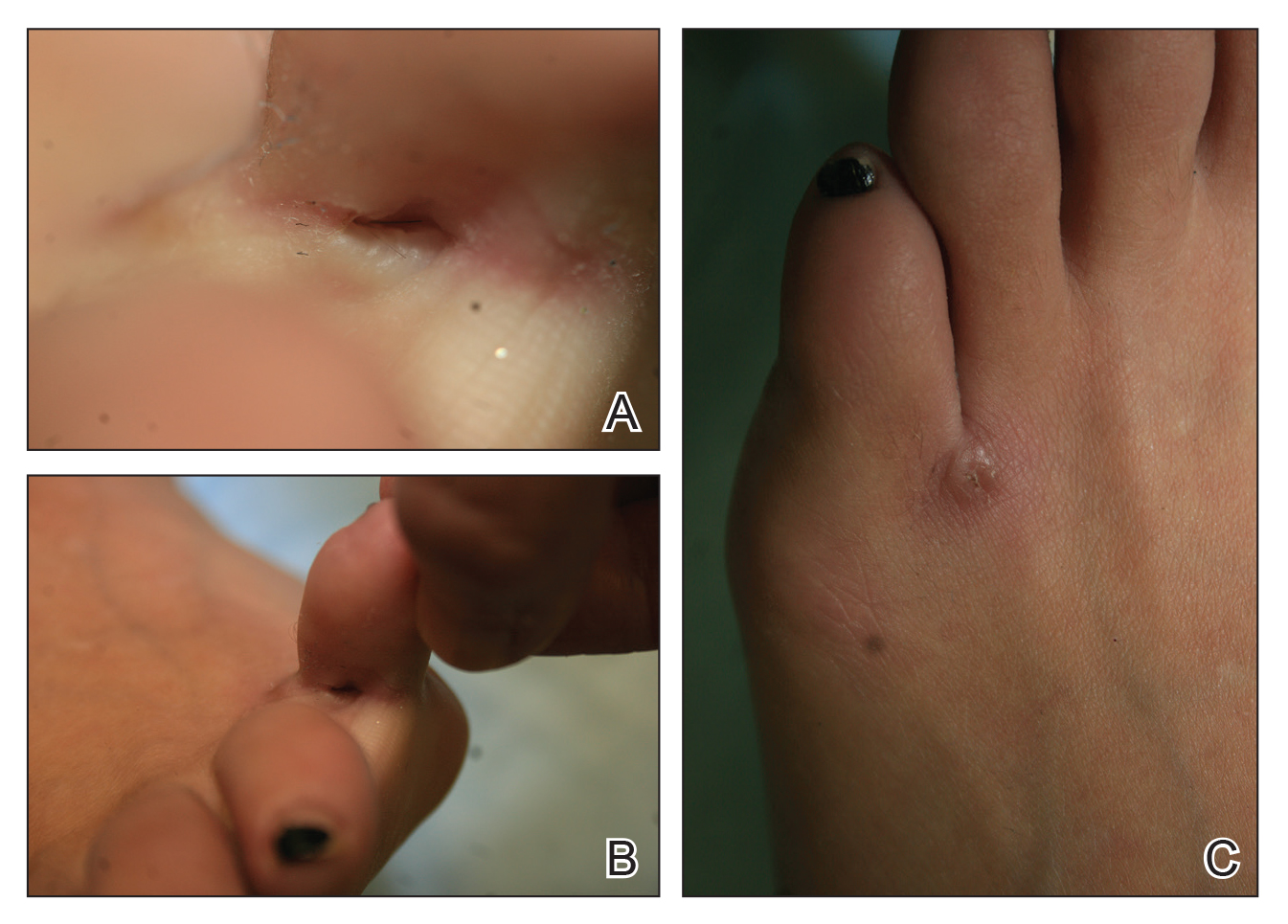
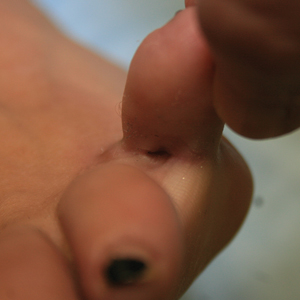
A 23-year-old female hairdresser was referred to our outpatient dermatology clinic by general surgery for evaluation of an asymptomatic interdigital toe lesion of several months’ duration. She was otherwise healthy. Physical examination revealed a 3-mm sinus in the interdigital web space between the fourth and fifth digits of the left foot, creating a partial fistula terminating in an umbilicated pink papule on the dorsal aspect of the interdigital space (Figure). While at work, the patient reported that she usually wore open-toed flip-flops. A diagnosis of barber’s sinus was made clinically. She returned for follow-up to the referring surgeon within 2 months and was offered surgical debridement, but the patient declined treatment, instead opting to wait and monitor for any potential complications. The lesion showed no change in clinical appearance and remained asymptomatic.
Barber’s sinus is caused by sharp fragments of clipped hair that penetrate the fragile interdigital skin and cause a foreign-body reaction. Males are almost exclusively contributory to the reported cases of barber’s sinus in the literature.1,2
The clinical picture of barber’s sinus is pathognomonic, as demonstrated in our case. Other potential diagnoses to consider include atypical mycobacterial infection, deep fungal infection, other foreign-body granuloma, and erosio interdigitalis blastomycetica. Although thorough removal of embedded hair fragments may be curative, most cases require surgical excision, often by curette, and subsequent skin closure. Pathology shows a foreign-body granulomatous reaction to hair fragments. If left untreated, potential complications of barber’s sinus include abscess formation, cellulitis, lymphangitis, and osteomyelitis. This lesion is preventable by maintaining hygiene of the interdigital spaces, use of barrier creams, and wearing protective footwear.3,4
- Efthimiadis C, Kosmidis C, Anthimidis G, et al. Barber’s hair sinus in a female hairdresser: uncommon manifestation of an occupational disease: a case report. Cases J. 2008;1:214.
- O’Neill AC, Purcell EM, Regan PJ. Interdigital pilonidal sinus of the foot [published online May 31, 2009]. Foot (Edinb). 2009;19:227-228.
- Schröder CM, Merk HF, Frank J. Barber’s hair sinus in a female hairdresser: uncommon manifestation of an occupational dermatosis. J Eur Acad Dermatol Venereol. 2006;20:209-211.
- Joseph HL, Gifford H. Barber’s interdigital pilonidal sinus: the incidence, pathology, and pathogenesis. AMA Arch Derm Syphilol. 1954;70:616-624.
To the Editor:
Barber’s sinus, or interdigital pilonidal sinus, is an occupational dermatosis with a pathognomonic clinical picture. Nearly all reports of barber’s sinus in the literature have involved the hands of male barbers and hairdressers. We present an uncommon case of barber’s sinus between the toes of a female hairdresser. If left untreated, potential complications of barber’s sinus include abscess formation, cellulitis, lymphangitis, and osteomyelitis. Clinicians should advise patients with an occupational risk of barber’s sinus to wear protective footwear and maintain hygiene in the interdigital spaces.
A 23-year-old female hairdresser was referred to our outpatient dermatology clinic by general surgery for evaluation of an asymptomatic interdigital toe lesion of several months’ duration. She was otherwise healthy. Physical examination revealed a 3-mm sinus in the interdigital web space between the fourth and fifth digits of the left foot, creating a partial fistula terminating in an umbilicated pink papule on the dorsal aspect of the interdigital space (Figure). While at work, the patient reported that she usually wore open-toed flip-flops. A diagnosis of barber’s sinus was made clinically. She returned for follow-up to the referring surgeon within 2 months and was offered surgical debridement, but the patient declined treatment, instead opting to wait and monitor for any potential complications. The lesion showed no change in clinical appearance and remained asymptomatic.
Barber’s sinus is caused by sharp fragments of clipped hair that penetrate the fragile interdigital skin and cause a foreign-body reaction. Males are almost exclusively contributory to the reported cases of barber’s sinus in the literature.1,2
The clinical picture of barber’s sinus is pathognomonic, as demonstrated in our case. Other potential diagnoses to consider include atypical mycobacterial infection, deep fungal infection, other foreign-body granuloma, and erosio interdigitalis blastomycetica. Although thorough removal of embedded hair fragments may be curative, most cases require surgical excision, often by curette, and subsequent skin closure. Pathology shows a foreign-body granulomatous reaction to hair fragments. If left untreated, potential complications of barber’s sinus include abscess formation, cellulitis, lymphangitis, and osteomyelitis. This lesion is preventable by maintaining hygiene of the interdigital spaces, use of barrier creams, and wearing protective footwear.3,4
To the Editor:
Barber’s sinus, or interdigital pilonidal sinus, is an occupational dermatosis with a pathognomonic clinical picture. Nearly all reports of barber’s sinus in the literature have involved the hands of male barbers and hairdressers. We present an uncommon case of barber’s sinus between the toes of a female hairdresser. If left untreated, potential complications of barber’s sinus include abscess formation, cellulitis, lymphangitis, and osteomyelitis. Clinicians should advise patients with an occupational risk of barber’s sinus to wear protective footwear and maintain hygiene in the interdigital spaces.
A 23-year-old female hairdresser was referred to our outpatient dermatology clinic by general surgery for evaluation of an asymptomatic interdigital toe lesion of several months’ duration. She was otherwise healthy. Physical examination revealed a 3-mm sinus in the interdigital web space between the fourth and fifth digits of the left foot, creating a partial fistula terminating in an umbilicated pink papule on the dorsal aspect of the interdigital space (Figure). While at work, the patient reported that she usually wore open-toed flip-flops. A diagnosis of barber’s sinus was made clinically. She returned for follow-up to the referring surgeon within 2 months and was offered surgical debridement, but the patient declined treatment, instead opting to wait and monitor for any potential complications. The lesion showed no change in clinical appearance and remained asymptomatic.
Barber’s sinus is caused by sharp fragments of clipped hair that penetrate the fragile interdigital skin and cause a foreign-body reaction. Males are almost exclusively contributory to the reported cases of barber’s sinus in the literature.1,2
The clinical picture of barber’s sinus is pathognomonic, as demonstrated in our case. Other potential diagnoses to consider include atypical mycobacterial infection, deep fungal infection, other foreign-body granuloma, and erosio interdigitalis blastomycetica. Although thorough removal of embedded hair fragments may be curative, most cases require surgical excision, often by curette, and subsequent skin closure. Pathology shows a foreign-body granulomatous reaction to hair fragments. If left untreated, potential complications of barber’s sinus include abscess formation, cellulitis, lymphangitis, and osteomyelitis. This lesion is preventable by maintaining hygiene of the interdigital spaces, use of barrier creams, and wearing protective footwear.3,4
- Efthimiadis C, Kosmidis C, Anthimidis G, et al. Barber’s hair sinus in a female hairdresser: uncommon manifestation of an occupational disease: a case report. Cases J. 2008;1:214.
- O’Neill AC, Purcell EM, Regan PJ. Interdigital pilonidal sinus of the foot [published online May 31, 2009]. Foot (Edinb). 2009;19:227-228.
- Schröder CM, Merk HF, Frank J. Barber’s hair sinus in a female hairdresser: uncommon manifestation of an occupational dermatosis. J Eur Acad Dermatol Venereol. 2006;20:209-211.
- Joseph HL, Gifford H. Barber’s interdigital pilonidal sinus: the incidence, pathology, and pathogenesis. AMA Arch Derm Syphilol. 1954;70:616-624.
- Efthimiadis C, Kosmidis C, Anthimidis G, et al. Barber’s hair sinus in a female hairdresser: uncommon manifestation of an occupational disease: a case report. Cases J. 2008;1:214.
- O’Neill AC, Purcell EM, Regan PJ. Interdigital pilonidal sinus of the foot [published online May 31, 2009]. Foot (Edinb). 2009;19:227-228.
- Schröder CM, Merk HF, Frank J. Barber’s hair sinus in a female hairdresser: uncommon manifestation of an occupational dermatosis. J Eur Acad Dermatol Venereol. 2006;20:209-211.
- Joseph HL, Gifford H. Barber’s interdigital pilonidal sinus: the incidence, pathology, and pathogenesis. AMA Arch Derm Syphilol. 1954;70:616-624.
Practice Points
- This case illustrates a disease in which a medical history and simple clinical examination can lead to the diagnosis.
- Patients may value a diagnosis without treatment. A patient with barber’s sinus may be satisfied with watchful waiting.
Streaked Discoloration on the Upper Body
The Diagnosis: Bleomycin-Induced Flagellate Hyperpigmentation
Histopathology of the affected skin demonstrated a slight increase in collagen bundle thickness, a chronic dermal perivascular inflammation, and associated pigment incontinence with dermal melanophages compared to unaffected skin (Figure). CD34 was faintly decreased, and dermal mucin increased in affected skin. This postinflammatory pigmentary alteration with subtle dermal sclerosis had persisted unchanged for more than 5 years after cessation of bleomycin therapy. Topical hydroquinone, physical blocker photoprotection, and laser modalities such as the Q-switched alexandrite (755-nm)/Nd:YAG (1064-nm) and ablative CO2 resurfacing lasers were attempted with minimal overall impact on cosmesis.

Bleomycin is a chemotherapeutic antibiotic that has been commonly used to treat Hodgkin lymphoma, germ cell tumors, and recurrent malignant pleural effusions.1 The drug is inactivated in most tissues by the enzyme bleomycin hydrolase. This enzyme is not present in skin and lung tissue; as a result, these organs are the most common sites of bleomycin toxicity.1 There are a variety of cutaneous effects associated with bleomycin including alopecia, hyperpigmentation, acral erythema, Raynaud phenomenon, and nail dystrophy.2 Flagellate hyperpigmentation is a less common cutaneous toxicity. It is an unusual eruption that appears as whiplike linear streaks on the upper chest and back, limbs, and flanks.3 This cutaneous manifestation was once thought to be specific to bleomycin use; however, it also has been described in dermatomyositis, adult-onset Still disease, and after the ingestion of uncooked or undercooked shiitake mushrooms.4 Flagellate hyperpigmentation also was once thought to be dose dependent; however, it has been described in even very small doses.5 The eruption has been described as independent of the route of drug administration, appearing with intravenous, subcutaneous, and intramuscular bleomycin.2 The association of bleomycin and flagellate hyperpigmentation has been reported since 1970; however, it is less commonly seen in clinical practice with the declining use of bleomycin.1
The exact mechanism for the hyperpigmentation is unknown. It has been proposed that the linear lesions are related to areas of pruritus and subsequent excoriations.1 Dermatographism may be present to a limited extent, but it is unlikely to be a chief cause of flagellate hyperpigmentation, as linear streaks have been reported in the absence of trauma. It also has been proposed that bleomycin has a direct toxic effect on the melanocytes, which stimulates increased melanin secretion.2 The hyperpigmentation also may be due to pigmentary incontinence secondary to inflammation.5 Histopathologic findings usually are varied and nonspecific.2 There may be a deep perivascular lymphocytic infiltrate, which is nonspecific but can be associated with drug-induced pathology.4 Bleomycin also is used to induce localized scleroderma in mouse-model research6 and has been reported to cause localized scleroderma at an infusion site or after an intralesional injection,7,8 which is not typically reported in flagellate erythema, but bleomycin's sclerosing effects may have played a role in the visible and sclerosing atrophy noted in our patient. Yamamoto et al9 reported a similar case of dermal sclerosis induced by bleomycin.
Flagellate hyperpigmentation typically lasts for up to 6 months.3 Patients with cutaneous manifestations from bleomycin therapy usually respond to steroid therapy and discontinuation of the drug. Bleomycin re-exposure should be avoided, as it may cause extension or widespread recurrence of flagellate hyperpigmentation.3 Postinflammatory pigment alteration may persist in patients with darker skin types and in patients with dramatic inciting inflammation.
Atrophoderma of Pasini and Pierini is a form of dermal atrophy that presents with 1 or more sharply demarcated depressed patches. There is some debate whether it is a distinct entity or a primary atrophic morphea.10 Linear atrophoderma of Moulin has a similar morphology with hyperpigmented depressions and "cliff-drop" borders, but these lesions follow the lines of Blaschko.11 Linear morphea initially can present as a linear erythematous streak but more commonly appears as a plaque-type morphea lesion that forms a scarlike band.12 Erythema dyschromicum perstans is an ashy dermatosis characterized by gray or blue-brown macules seen in Fitzpatrick skin types III through V and typically is chronic and progressive.13
- Lee HY, Lim KH, Ryu Y, et al. Bleomycininduced flagellate erythema: a case report and review of the literature. Oncol Lett. 2014;8:933-935.
- Simpson RC, Da Forno P, Nagarajan C, et al. A pruritic rash in a patient with Hodgkin lymphoma. Clin Exp Dermatol. 2011;36:680-682.
- Fyfe AJ, McKay P. Toxicities associated with bleomycin. J R Coll Physicians Edinb. 2010;40:213-215.
- Lu CC, Lu YY, Wang QR, et al. Bleomycin-induced flagellate erythema. Balkan Med J. 2014;31:189-190.
- Abess A, Keel DM, Graham BS. Flagellate hyperpigmentation following intralesional bleomycin treatment of verruca plantaris. Arch Dermatol. 2003;139:337-339.
- Yamamoto T. The bleomycin-induced scleroderma model: what have we learned for scleroderma pathogenesis? Arch Dermatol Res. 2006;297:333-344.
- Kim KH, Yoon TJ, Oh CW, et al. A case of bleomycin-induced scleroderma. J Korean Med Sci. 1996;11:454-456.
- Kerr LD, Spiera H. Scleroderma in association with the use of bleomycin: a report of 3 cases. J Rheumatol. 1992;19:294-296.
- Yamamoto T, Yokozeki H, Nishioka K. Dermal sclerosis in the lesional skin of 'flagellate' erythema (scratch dermatitis) induced by bleomycin. Dermatology. 1998;197:399-400.
- Kencka D, Blaszczyk M, Jablońska S. Atrophoderma Pasini-Pierini is a primary atrophic abortive morphea. Dermatology. 1995;190:203-206.
- Moulin G, Hill MP, Guillaud V, et al. Acquired atrophic pigmented band-like lesions following Blaschko's lines. Ann Dermatol Venereol. 1992;119:729-736.
- Fett N, Werth VP. Update on morphea: part I. epidemiology, clinical presentation, and pathogenesis. J Am Acad Dermatol. 2011;64:217-228.
- Zaynoun S, Rubeiz N, Kibbi AG. Ashy dermatosis--a critical review of literature and a proposed simplified clinical classification. Int J Dermatol. 2008;47:542-544.
The Diagnosis: Bleomycin-Induced Flagellate Hyperpigmentation
Histopathology of the affected skin demonstrated a slight increase in collagen bundle thickness, a chronic dermal perivascular inflammation, and associated pigment incontinence with dermal melanophages compared to unaffected skin (Figure). CD34 was faintly decreased, and dermal mucin increased in affected skin. This postinflammatory pigmentary alteration with subtle dermal sclerosis had persisted unchanged for more than 5 years after cessation of bleomycin therapy. Topical hydroquinone, physical blocker photoprotection, and laser modalities such as the Q-switched alexandrite (755-nm)/Nd:YAG (1064-nm) and ablative CO2 resurfacing lasers were attempted with minimal overall impact on cosmesis.
Bleomycin is a chemotherapeutic antibiotic that has been commonly used to treat Hodgkin lymphoma, germ cell tumors, and recurrent malignant pleural effusions.1 The drug is inactivated in most tissues by the enzyme bleomycin hydrolase. This enzyme is not present in skin and lung tissue; as a result, these organs are the most common sites of bleomycin toxicity.1 There are a variety of cutaneous effects associated with bleomycin including alopecia, hyperpigmentation, acral erythema, Raynaud phenomenon, and nail dystrophy.2 Flagellate hyperpigmentation is a less common cutaneous toxicity. It is an unusual eruption that appears as whiplike linear streaks on the upper chest and back, limbs, and flanks.3 This cutaneous manifestation was once thought to be specific to bleomycin use; however, it also has been described in dermatomyositis, adult-onset Still disease, and after the ingestion of uncooked or undercooked shiitake mushrooms.4 Flagellate hyperpigmentation also was once thought to be dose dependent; however, it has been described in even very small doses.5 The eruption has been described as independent of the route of drug administration, appearing with intravenous, subcutaneous, and intramuscular bleomycin.2 The association of bleomycin and flagellate hyperpigmentation has been reported since 1970; however, it is less commonly seen in clinical practice with the declining use of bleomycin.1
The exact mechanism for the hyperpigmentation is unknown. It has been proposed that the linear lesions are related to areas of pruritus and subsequent excoriations.1 Dermatographism may be present to a limited extent, but it is unlikely to be a chief cause of flagellate hyperpigmentation, as linear streaks have been reported in the absence of trauma. It also has been proposed that bleomycin has a direct toxic effect on the melanocytes, which stimulates increased melanin secretion.2 The hyperpigmentation also may be due to pigmentary incontinence secondary to inflammation.5 Histopathologic findings usually are varied and nonspecific.2 There may be a deep perivascular lymphocytic infiltrate, which is nonspecific but can be associated with drug-induced pathology.4 Bleomycin also is used to induce localized scleroderma in mouse-model research6 and has been reported to cause localized scleroderma at an infusion site or after an intralesional injection,7,8 which is not typically reported in flagellate erythema, but bleomycin's sclerosing effects may have played a role in the visible and sclerosing atrophy noted in our patient. Yamamoto et al9 reported a similar case of dermal sclerosis induced by bleomycin.
Flagellate hyperpigmentation typically lasts for up to 6 months.3 Patients with cutaneous manifestations from bleomycin therapy usually respond to steroid therapy and discontinuation of the drug. Bleomycin re-exposure should be avoided, as it may cause extension or widespread recurrence of flagellate hyperpigmentation.3 Postinflammatory pigment alteration may persist in patients with darker skin types and in patients with dramatic inciting inflammation.
Atrophoderma of Pasini and Pierini is a form of dermal atrophy that presents with 1 or more sharply demarcated depressed patches. There is some debate whether it is a distinct entity or a primary atrophic morphea.10 Linear atrophoderma of Moulin has a similar morphology with hyperpigmented depressions and "cliff-drop" borders, but these lesions follow the lines of Blaschko.11 Linear morphea initially can present as a linear erythematous streak but more commonly appears as a plaque-type morphea lesion that forms a scarlike band.12 Erythema dyschromicum perstans is an ashy dermatosis characterized by gray or blue-brown macules seen in Fitzpatrick skin types III through V and typically is chronic and progressive.13
The Diagnosis: Bleomycin-Induced Flagellate Hyperpigmentation
Histopathology of the affected skin demonstrated a slight increase in collagen bundle thickness, a chronic dermal perivascular inflammation, and associated pigment incontinence with dermal melanophages compared to unaffected skin (Figure). CD34 was faintly decreased, and dermal mucin increased in affected skin. This postinflammatory pigmentary alteration with subtle dermal sclerosis had persisted unchanged for more than 5 years after cessation of bleomycin therapy. Topical hydroquinone, physical blocker photoprotection, and laser modalities such as the Q-switched alexandrite (755-nm)/Nd:YAG (1064-nm) and ablative CO2 resurfacing lasers were attempted with minimal overall impact on cosmesis.
Bleomycin is a chemotherapeutic antibiotic that has been commonly used to treat Hodgkin lymphoma, germ cell tumors, and recurrent malignant pleural effusions.1 The drug is inactivated in most tissues by the enzyme bleomycin hydrolase. This enzyme is not present in skin and lung tissue; as a result, these organs are the most common sites of bleomycin toxicity.1 There are a variety of cutaneous effects associated with bleomycin including alopecia, hyperpigmentation, acral erythema, Raynaud phenomenon, and nail dystrophy.2 Flagellate hyperpigmentation is a less common cutaneous toxicity. It is an unusual eruption that appears as whiplike linear streaks on the upper chest and back, limbs, and flanks.3 This cutaneous manifestation was once thought to be specific to bleomycin use; however, it also has been described in dermatomyositis, adult-onset Still disease, and after the ingestion of uncooked or undercooked shiitake mushrooms.4 Flagellate hyperpigmentation also was once thought to be dose dependent; however, it has been described in even very small doses.5 The eruption has been described as independent of the route of drug administration, appearing with intravenous, subcutaneous, and intramuscular bleomycin.2 The association of bleomycin and flagellate hyperpigmentation has been reported since 1970; however, it is less commonly seen in clinical practice with the declining use of bleomycin.1
The exact mechanism for the hyperpigmentation is unknown. It has been proposed that the linear lesions are related to areas of pruritus and subsequent excoriations.1 Dermatographism may be present to a limited extent, but it is unlikely to be a chief cause of flagellate hyperpigmentation, as linear streaks have been reported in the absence of trauma. It also has been proposed that bleomycin has a direct toxic effect on the melanocytes, which stimulates increased melanin secretion.2 The hyperpigmentation also may be due to pigmentary incontinence secondary to inflammation.5 Histopathologic findings usually are varied and nonspecific.2 There may be a deep perivascular lymphocytic infiltrate, which is nonspecific but can be associated with drug-induced pathology.4 Bleomycin also is used to induce localized scleroderma in mouse-model research6 and has been reported to cause localized scleroderma at an infusion site or after an intralesional injection,7,8 which is not typically reported in flagellate erythema, but bleomycin's sclerosing effects may have played a role in the visible and sclerosing atrophy noted in our patient. Yamamoto et al9 reported a similar case of dermal sclerosis induced by bleomycin.
Flagellate hyperpigmentation typically lasts for up to 6 months.3 Patients with cutaneous manifestations from bleomycin therapy usually respond to steroid therapy and discontinuation of the drug. Bleomycin re-exposure should be avoided, as it may cause extension or widespread recurrence of flagellate hyperpigmentation.3 Postinflammatory pigment alteration may persist in patients with darker skin types and in patients with dramatic inciting inflammation.
Atrophoderma of Pasini and Pierini is a form of dermal atrophy that presents with 1 or more sharply demarcated depressed patches. There is some debate whether it is a distinct entity or a primary atrophic morphea.10 Linear atrophoderma of Moulin has a similar morphology with hyperpigmented depressions and "cliff-drop" borders, but these lesions follow the lines of Blaschko.11 Linear morphea initially can present as a linear erythematous streak but more commonly appears as a plaque-type morphea lesion that forms a scarlike band.12 Erythema dyschromicum perstans is an ashy dermatosis characterized by gray or blue-brown macules seen in Fitzpatrick skin types III through V and typically is chronic and progressive.13
- Lee HY, Lim KH, Ryu Y, et al. Bleomycininduced flagellate erythema: a case report and review of the literature. Oncol Lett. 2014;8:933-935.
- Simpson RC, Da Forno P, Nagarajan C, et al. A pruritic rash in a patient with Hodgkin lymphoma. Clin Exp Dermatol. 2011;36:680-682.
- Fyfe AJ, McKay P. Toxicities associated with bleomycin. J R Coll Physicians Edinb. 2010;40:213-215.
- Lu CC, Lu YY, Wang QR, et al. Bleomycin-induced flagellate erythema. Balkan Med J. 2014;31:189-190.
- Abess A, Keel DM, Graham BS. Flagellate hyperpigmentation following intralesional bleomycin treatment of verruca plantaris. Arch Dermatol. 2003;139:337-339.
- Yamamoto T. The bleomycin-induced scleroderma model: what have we learned for scleroderma pathogenesis? Arch Dermatol Res. 2006;297:333-344.
- Kim KH, Yoon TJ, Oh CW, et al. A case of bleomycin-induced scleroderma. J Korean Med Sci. 1996;11:454-456.
- Kerr LD, Spiera H. Scleroderma in association with the use of bleomycin: a report of 3 cases. J Rheumatol. 1992;19:294-296.
- Yamamoto T, Yokozeki H, Nishioka K. Dermal sclerosis in the lesional skin of 'flagellate' erythema (scratch dermatitis) induced by bleomycin. Dermatology. 1998;197:399-400.
- Kencka D, Blaszczyk M, Jablońska S. Atrophoderma Pasini-Pierini is a primary atrophic abortive morphea. Dermatology. 1995;190:203-206.
- Moulin G, Hill MP, Guillaud V, et al. Acquired atrophic pigmented band-like lesions following Blaschko's lines. Ann Dermatol Venereol. 1992;119:729-736.
- Fett N, Werth VP. Update on morphea: part I. epidemiology, clinical presentation, and pathogenesis. J Am Acad Dermatol. 2011;64:217-228.
- Zaynoun S, Rubeiz N, Kibbi AG. Ashy dermatosis--a critical review of literature and a proposed simplified clinical classification. Int J Dermatol. 2008;47:542-544.
- Lee HY, Lim KH, Ryu Y, et al. Bleomycininduced flagellate erythema: a case report and review of the literature. Oncol Lett. 2014;8:933-935.
- Simpson RC, Da Forno P, Nagarajan C, et al. A pruritic rash in a patient with Hodgkin lymphoma. Clin Exp Dermatol. 2011;36:680-682.
- Fyfe AJ, McKay P. Toxicities associated with bleomycin. J R Coll Physicians Edinb. 2010;40:213-215.
- Lu CC, Lu YY, Wang QR, et al. Bleomycin-induced flagellate erythema. Balkan Med J. 2014;31:189-190.
- Abess A, Keel DM, Graham BS. Flagellate hyperpigmentation following intralesional bleomycin treatment of verruca plantaris. Arch Dermatol. 2003;139:337-339.
- Yamamoto T. The bleomycin-induced scleroderma model: what have we learned for scleroderma pathogenesis? Arch Dermatol Res. 2006;297:333-344.
- Kim KH, Yoon TJ, Oh CW, et al. A case of bleomycin-induced scleroderma. J Korean Med Sci. 1996;11:454-456.
- Kerr LD, Spiera H. Scleroderma in association with the use of bleomycin: a report of 3 cases. J Rheumatol. 1992;19:294-296.
- Yamamoto T, Yokozeki H, Nishioka K. Dermal sclerosis in the lesional skin of 'flagellate' erythema (scratch dermatitis) induced by bleomycin. Dermatology. 1998;197:399-400.
- Kencka D, Blaszczyk M, Jablońska S. Atrophoderma Pasini-Pierini is a primary atrophic abortive morphea. Dermatology. 1995;190:203-206.
- Moulin G, Hill MP, Guillaud V, et al. Acquired atrophic pigmented band-like lesions following Blaschko's lines. Ann Dermatol Venereol. 1992;119:729-736.
- Fett N, Werth VP. Update on morphea: part I. epidemiology, clinical presentation, and pathogenesis. J Am Acad Dermatol. 2011;64:217-228.
- Zaynoun S, Rubeiz N, Kibbi AG. Ashy dermatosis--a critical review of literature and a proposed simplified clinical classification. Int J Dermatol. 2008;47:542-544.

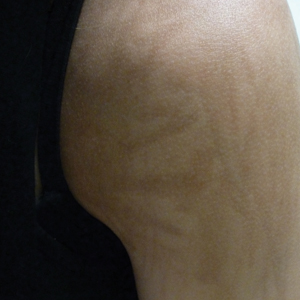
An 18-year-old woman presented to our dermatology clinic with persistent diffuse discoloration on the upper body of more than 5 years’ duration. Her medical history was notable for primary mediastinal classical Hodgkin lymphoma treated with ABVE-PC (doxorubicin, bleomycin, vincristine, etoposide, prednisone, cyclophosphamide) chemotherapy and 22 Gy radiation therapy to the chest 5 years prior. She reported the initial onset of diffuse pruritus with associated scratching and persistent skin discoloration while receiving a course of chemotherapy. Physical examination revealed numerous thin, flagellate, faintly hyperpigmented streaks with subtle atrophy in a parallel configuration on the bilateral shoulders (top), upper back (bottom), and abdomen. Punch biopsies (5 mm) of both affected and unaffected skin on the left side of the lateral upper back were performed.
Management of Refractory Pain From Hereditary Cutaneous Leiomyomas With Nifedipine and Gabapentin
To the Editor:
Leiomyomas are benign smooth muscle tumors. There are 3 types of cutaneous leiomyomas: (1) piloleiomyomas, arising from the arrector pili muscles; (2) angioleiomyomas, arising from the muscles surrounding dermal blood vessels; and (3) leiomyomas of the external genitalia, arising from the dartoic, vulvar, or mammary smooth muscles.1 There is no gender predilection for cutaneous leiomyomas, and lesions present on average at approximately 40 to 45 years of age.2
Piloleiomyomas are the most common type of cutaneous leiomyomas and typically present as red-brown papules and nodules on the trunk, arms, and legs.3 Piloleiomyomas often are associated with spontaneous or induced pain (eg, with cold exposure). The pain associated with piloleiomyomas can be severely debilitating to patients and may have a considerable impact on their quality of life.
A 40-year-old woman presented to our clinic with numerous widespread, painful, red-brown papules and nodules on the head, neck, chest, abdomen, back, arms, and legs of 6 years’ duration that were increasing in number (Figure 1). She had a history of uterine leiomyomas and type 2 renal papillary carcinoma following a left nephrectomy at 38 years of age. The patient’s mother had a history of similar skin lesions as well as uterine cancer. Multiple excisional biopsies were performed, all of which showed piloleiomyomas on histopathology (Figure 2). The pain associated with the patient’s extensive cutaneous leiomyomas considerably impaired her quality of life. Although she experienced pain in all affected areas of the body, the pain was the worst in the upper arms. She reported having requested a nerve ablation procedure from an outside pain management clinic, which was denied for unknown reasons.


Two years prior to the current presentation, the patient had been treated by a pain management specialist with gabapentin 300 mg twice daily as needed for pain associated with leiomyomas. The patient followed this regimen approximately 3 times weekly for the preceding 1 to 2 years with reduction in her pain symptoms; however, the painful episodes became more frequent and severe over time. The patient reported being unable to further increase the gabapentin dosing frequency because it made her too drowsy and impacted her ability to work a job that required heavy lifting. Thus, the patient requested additional therapy and was subsequently treated at our clinic with numerous excisional biopsies of the most painful lesions during the 2 years prior to her current presentation.
When the patient re-presented to our clinic, she requested additional lesion excisions given that she had experienced some pain relief from this treatment modality in the past; however, these prior excisions only resulted in local pain relief limited to the site of the excision. Because of the extent of the lesions and the patient’s inability to tolerate pain from the lidocaine injections, we did not feel multiple excisions were a practical treatment option. The patient subsequently was offered a trial of cryotherapy for symptom relief based on a reported case in which this modality was successfully used.4 After discussing the risks and benefits associated with this treatment, cryotherapy was attempted on a few of the leiomyomas on the patient’s right shoulder; however, she experienced severe pain during cryotherapy treatment, and the procedure had to be aborted.
We then increased the patient’s gabapentin regimen to 300 mg in the morning and 600 mg in the evening, as tolerated. The patient reported that she was better able to tolerate the sedating side effects of the increased dose of gabapentin because she had stopped working due to her severe pain episodes. We also added oral nifedipine 10 mg 3 times daily, as needed. Within 30 minutes of starting this treatment regimen, the pain associated with the lesions remarkably improved (10/10 severity before starting treatment vs 3/10 after starting treatment). Her pain levels remained stable (3/10 severity) during 3 weeks of treatment with this combination regimen, but unfortunately she developed headaches and malaise, which she associated with the nifedipine at the 3 times daily dose.
The patient was able to better tolerate the nifedipine after reducing the dose to once daily on an as-needed basis. On average, the patient took nifedipine once every 3 days; however, she reported that she had to periodically increase the frequency of the nifedipine to once daily for up to 2 weeks at a time for periods of more frequent pain flares. The patient reported a consistent pattern of the breakthrough symptoms rapidly improving with each dose of nifedipine, though she did feel that taking consistent gabapentin enhanced baseline symptom control. The patient also noticed on a few occasions when she did not have access to her nifedipine that her pain would flare to 10/10 severity and would decrease to 4/10 severity 30 minutes after restarting nifedipine at 10 mg once daily. She experienced breakthrough pain due to exacerbating factors including her menstrual cycle; exposure to the sun and cold temperatures or water; excessive physical activity; and mild trauma. Due to exacerbations from sun exposure, the patient often wore long-sleeved shirts, which helped reduce the severity of the pain episodes while she was outdoors.
The exact mechanism for the pain associated with cutaneous leiomyomas is unknown but is thought to be due to infringement of the lesion on the surrounding cutaneous nerves. In addition, norepinephrine activates alpha receptors on the smooth muscle to contract through an influx of ions such as calcium. When smooth muscle contracts, the compression of nerves likely is worsened.
There are a limited number of case reports in the literature that have demonstrated successful treatment of the pain associated with cutaneous leiomyomas. Previously reported treatment modalities have included phenoxybenzamine, an alpha-blocking agent that may reduce pain through its antiadrenergic effects2; nitroglycerin, a venous and arterial dilator that may reduce pain by decreasing muscle oxygen requirements2; gabapentin, an antiepileptic and analgesic medication with structural similarity to the gamma-aminobutyric acid neurotransmitter for which the exact mechanism of action is unknown3; botulinum toxin, a neuromuscular blocker that prevents the release of presynaptic acetylcholine and may decrease neuropathic pain by reducing hyperactive nerves5,6; hyoscine butylbromide and topical hyoscine hydrobromide, both antispasmodics that may reduce pain through their anticholinergic effects, which relax smooth muscle7,8; and the CO2 laser, a treatment that has been utilized for its resurfacing, excisional, and ablative properties.9,10
Calcium channel blockers such as amlodipine, verapamil, and nifedipine also have been used to treat the pain associated with piloleiomyomas.11 Calcium ion channel antagonists inhibit the influx of calcium ions across the cell membrane; therefore, nifedipine and other calcium channel blockers may prevent the smooth muscle contraction that is hypothesized to cause pain in patients with cutaneous leiomyomas.12
Mean plasma concentration of nifedipine has been shown to reach maximum values of 160 +/− 49 µg/L after 30 to 60 minutes following oral administration of 10 mg of nifedipine.13 After 8 hours, the mean concentration drops to 3.4 +/− 1.2 µg/L. The clinical response in our patient appeared consistent with the reported pharmacokinetics of the drug, as she was able to consistently obtain considerable reduction in her pain symptoms within 30 minutes of starting nifedipine, coinciding with the period of time it takes for the nifedipine to reach maximum plasma concentrations.13
Interestingly, our patient had worsening pain episodes associated with sun exposure, which typically is not reported as one of the usual triggers for cutaneous leiomyomas. We are not aware of any described mechanisms that would explain this phenomenon.
Importantly, any patient presenting with multiple cutaneous and uterine (if female) leiomyomas should be screened for hereditary leiomyomatosis and renal cell carcinoma syndrome (HLRCC), an autosomal-dominant disorder linked to a mutation in the fumarate hydratase tumor suppressor gene. Clinically, HLRCC patients typically present with multiple cutaneous leiomyomas, uterine leiomyomas, and renal cell cancer (most often type 2 papillary renal cell carcinoma).14 Hereditary leiomyomatosis and renal cell carcinoma syndrome (also known as multiple cutaneous and uterine leiomyomatosis syndrome) previously was thought to be a separate disease entity from Reed syndrome; however, after the same mutation in the fumarate hydratase tumor suppressor gene was found to be responsible for both Reed syndrome and HLRCC, they are now thought to be the same disease process.15
Diagnosis of HLRCC is likely when the patient meets the major criterion of multiple cutaneous piloleiomyomas confirmed histopathologically. Clinical diagnosis of HLRCC is suspected if 2 or more of the following minor criteria are present: type 2 papillary renal cell carcinoma before 40 years of age; onset of severely symptomatic (requiring surgery) uterine fibroids before 40 years of age in females; and first-degree family member who meets 1 or more of these criteria.15 At the time of presentation, the patient met clinical criteria for HLRCC, including multiple cutaneous leiomyomas (major criterion) and type 2 papillary renal cell carcinoma before 40 years of age (minor criterion). The patient also had a history of uterine leiomyomas, but these lesions did not fulfill the criterion of being severely symptomatic requiring surgery. Furthermore, the patient’s mother had similar cutaneous leiomyomas and a history of uterine cancer, which fulfilled additional minor criterion, consistent with an autosomal-dominant inheritance pattern (with variable penetrance) seen in HLRCC. An important issue for counseling and monitoring patients is that premenopausal women with HLRCC are at an increased risk of developing uterine leiomyosarcoma.15 Our patient followed up with an oncologist for tumor surveillance and subsequently underwent genetic testing, which revealed a mutation in the fumarate hydratase gene.
Treatment of painful cutaneous leiomyomas, particularly in patients with HLRCC, remains a therapeutic challenge. Although surgical and/or destructive treatments can provide pain relief for patients who have a limited number of lesions, these options are impracticable when a patient has numerous widespread leiomyomas; therefore, systemic therapies may be more beneficial. Clinicians should be aware of nifedipine, which may be used in combination with gabapentin as a viable treatment option in the management of acute and breakthrough pain associated with cutaneous leiomyomas.
Acknowledgment
The a
- Holst VA, Junkins-Hopkins JM, Elenitsas R. Cutaneous smooth muscle neoplasms: clinical features, histologic findings, and treatment options. J Am Acad Dermatol. 2002;46:477-494.
- Raj S, Calonje E, Kraus M, et al. Cutaneous pilar leiomyoma: clinicopathologic analysis of 53 lesions in 45 patients. Am J Dermatopathol. 1997;19:2-9.
- Alam M, Rabinowitz AD, Engler DE. Gabapentin treatment of multiple piloleiomyoma-related pain. J Am Acad Dermatol. 2002;46:S27-S29.
- Basendwh MA, Fatani M, Baltow B. Reed’s syndrome: a case of multiple cutaneous leiomyomas treated with liquid nitrogen cryotherapy. Case Rep Dermatol. 2016;8:65-70.
- Sifaki MK, Krueger-Krasagakis S, Koutsopoulos A, et al. Botulinum toxin type A–treatment of a patient with multiple cutaneous piloleiomyomas. Dermatology. 2008;218:44-47.
- Onder M, Adıs¸en E. A new indication of botulinum toxin: leiomyoma-related pain. J Am Acad Dermatol. 2009;60:325-328.
- Kaliyadan F, Manoj J, Dharmaratnam AD. Multiple cutaneous leiomyomas: pain relief with pulsed hysocine butyl bromide. Indian J Dermatol. 2009;54:72.
- Archer CB, Whittaker S, Greaves MW. Pharmacological modulation of cold‐induced pain in cutaneous leiomyomata. Br J Dermatol. 1988;118:255-260.
- Christenson LJ, Smith K, Arpey CJ. Treatment of multiple cutaneous leiomyomas with CO2 laser ablation. Dermatol Surg. 2000;26:319-322.
- Michajłowski I, Błaz˙ewicz I, Karpinsky G, et al. Successful treatment of multiple cutaneous leiomyomas with carbon dioxide laser ablation. Postepy Dermatol Alergol. 2015;32:480-482.
- Archer CB, Greaves MW. Assessment of treatment for painful cutaneous leiomyomas. J Am Acad Dermatol. 1987;17:141-142.
- Thompson JA. Therapy for painful cutaneous leiomyomas. J Am Acad Dermatol. 1985;13:865-867.
- Raemsch KD, Sommer J. Pharmacokinetics and metabolism of nifedipine. Hypertension. 1983;5(4 pt 2):II18-II24.
- Toro JR, Nickerson ML, Wei MH, et al. Mutations in the fumarate hydratase gene cause hereditary leiomyomatosis and renal cell cancer in families in North America. Am J Hum Genet. 2003;73:95-106.
- Smit DL, Mensenkamp AR, Badeloe S, et al. Hereditary leiomyomatosis and renal cell cancer in families referred for fumarate hydratase germline mutation analysis. Clin Genet. 2011;79:49-59.
To the Editor:
Leiomyomas are benign smooth muscle tumors. There are 3 types of cutaneous leiomyomas: (1) piloleiomyomas, arising from the arrector pili muscles; (2) angioleiomyomas, arising from the muscles surrounding dermal blood vessels; and (3) leiomyomas of the external genitalia, arising from the dartoic, vulvar, or mammary smooth muscles.1 There is no gender predilection for cutaneous leiomyomas, and lesions present on average at approximately 40 to 45 years of age.2
Piloleiomyomas are the most common type of cutaneous leiomyomas and typically present as red-brown papules and nodules on the trunk, arms, and legs.3 Piloleiomyomas often are associated with spontaneous or induced pain (eg, with cold exposure). The pain associated with piloleiomyomas can be severely debilitating to patients and may have a considerable impact on their quality of life.
A 40-year-old woman presented to our clinic with numerous widespread, painful, red-brown papules and nodules on the head, neck, chest, abdomen, back, arms, and legs of 6 years’ duration that were increasing in number (Figure 1). She had a history of uterine leiomyomas and type 2 renal papillary carcinoma following a left nephrectomy at 38 years of age. The patient’s mother had a history of similar skin lesions as well as uterine cancer. Multiple excisional biopsies were performed, all of which showed piloleiomyomas on histopathology (Figure 2). The pain associated with the patient’s extensive cutaneous leiomyomas considerably impaired her quality of life. Although she experienced pain in all affected areas of the body, the pain was the worst in the upper arms. She reported having requested a nerve ablation procedure from an outside pain management clinic, which was denied for unknown reasons.
Two years prior to the current presentation, the patient had been treated by a pain management specialist with gabapentin 300 mg twice daily as needed for pain associated with leiomyomas. The patient followed this regimen approximately 3 times weekly for the preceding 1 to 2 years with reduction in her pain symptoms; however, the painful episodes became more frequent and severe over time. The patient reported being unable to further increase the gabapentin dosing frequency because it made her too drowsy and impacted her ability to work a job that required heavy lifting. Thus, the patient requested additional therapy and was subsequently treated at our clinic with numerous excisional biopsies of the most painful lesions during the 2 years prior to her current presentation.
When the patient re-presented to our clinic, she requested additional lesion excisions given that she had experienced some pain relief from this treatment modality in the past; however, these prior excisions only resulted in local pain relief limited to the site of the excision. Because of the extent of the lesions and the patient’s inability to tolerate pain from the lidocaine injections, we did not feel multiple excisions were a practical treatment option. The patient subsequently was offered a trial of cryotherapy for symptom relief based on a reported case in which this modality was successfully used.4 After discussing the risks and benefits associated with this treatment, cryotherapy was attempted on a few of the leiomyomas on the patient’s right shoulder; however, she experienced severe pain during cryotherapy treatment, and the procedure had to be aborted.
We then increased the patient’s gabapentin regimen to 300 mg in the morning and 600 mg in the evening, as tolerated. The patient reported that she was better able to tolerate the sedating side effects of the increased dose of gabapentin because she had stopped working due to her severe pain episodes. We also added oral nifedipine 10 mg 3 times daily, as needed. Within 30 minutes of starting this treatment regimen, the pain associated with the lesions remarkably improved (10/10 severity before starting treatment vs 3/10 after starting treatment). Her pain levels remained stable (3/10 severity) during 3 weeks of treatment with this combination regimen, but unfortunately she developed headaches and malaise, which she associated with the nifedipine at the 3 times daily dose.
The patient was able to better tolerate the nifedipine after reducing the dose to once daily on an as-needed basis. On average, the patient took nifedipine once every 3 days; however, she reported that she had to periodically increase the frequency of the nifedipine to once daily for up to 2 weeks at a time for periods of more frequent pain flares. The patient reported a consistent pattern of the breakthrough symptoms rapidly improving with each dose of nifedipine, though she did feel that taking consistent gabapentin enhanced baseline symptom control. The patient also noticed on a few occasions when she did not have access to her nifedipine that her pain would flare to 10/10 severity and would decrease to 4/10 severity 30 minutes after restarting nifedipine at 10 mg once daily. She experienced breakthrough pain due to exacerbating factors including her menstrual cycle; exposure to the sun and cold temperatures or water; excessive physical activity; and mild trauma. Due to exacerbations from sun exposure, the patient often wore long-sleeved shirts, which helped reduce the severity of the pain episodes while she was outdoors.
The exact mechanism for the pain associated with cutaneous leiomyomas is unknown but is thought to be due to infringement of the lesion on the surrounding cutaneous nerves. In addition, norepinephrine activates alpha receptors on the smooth muscle to contract through an influx of ions such as calcium. When smooth muscle contracts, the compression of nerves likely is worsened.
There are a limited number of case reports in the literature that have demonstrated successful treatment of the pain associated with cutaneous leiomyomas. Previously reported treatment modalities have included phenoxybenzamine, an alpha-blocking agent that may reduce pain through its antiadrenergic effects2; nitroglycerin, a venous and arterial dilator that may reduce pain by decreasing muscle oxygen requirements2; gabapentin, an antiepileptic and analgesic medication with structural similarity to the gamma-aminobutyric acid neurotransmitter for which the exact mechanism of action is unknown3; botulinum toxin, a neuromuscular blocker that prevents the release of presynaptic acetylcholine and may decrease neuropathic pain by reducing hyperactive nerves5,6; hyoscine butylbromide and topical hyoscine hydrobromide, both antispasmodics that may reduce pain through their anticholinergic effects, which relax smooth muscle7,8; and the CO2 laser, a treatment that has been utilized for its resurfacing, excisional, and ablative properties.9,10
Calcium channel blockers such as amlodipine, verapamil, and nifedipine also have been used to treat the pain associated with piloleiomyomas.11 Calcium ion channel antagonists inhibit the influx of calcium ions across the cell membrane; therefore, nifedipine and other calcium channel blockers may prevent the smooth muscle contraction that is hypothesized to cause pain in patients with cutaneous leiomyomas.12
Mean plasma concentration of nifedipine has been shown to reach maximum values of 160 +/− 49 µg/L after 30 to 60 minutes following oral administration of 10 mg of nifedipine.13 After 8 hours, the mean concentration drops to 3.4 +/− 1.2 µg/L. The clinical response in our patient appeared consistent with the reported pharmacokinetics of the drug, as she was able to consistently obtain considerable reduction in her pain symptoms within 30 minutes of starting nifedipine, coinciding with the period of time it takes for the nifedipine to reach maximum plasma concentrations.13
Interestingly, our patient had worsening pain episodes associated with sun exposure, which typically is not reported as one of the usual triggers for cutaneous leiomyomas. We are not aware of any described mechanisms that would explain this phenomenon.
Importantly, any patient presenting with multiple cutaneous and uterine (if female) leiomyomas should be screened for hereditary leiomyomatosis and renal cell carcinoma syndrome (HLRCC), an autosomal-dominant disorder linked to a mutation in the fumarate hydratase tumor suppressor gene. Clinically, HLRCC patients typically present with multiple cutaneous leiomyomas, uterine leiomyomas, and renal cell cancer (most often type 2 papillary renal cell carcinoma).14 Hereditary leiomyomatosis and renal cell carcinoma syndrome (also known as multiple cutaneous and uterine leiomyomatosis syndrome) previously was thought to be a separate disease entity from Reed syndrome; however, after the same mutation in the fumarate hydratase tumor suppressor gene was found to be responsible for both Reed syndrome and HLRCC, they are now thought to be the same disease process.15
Diagnosis of HLRCC is likely when the patient meets the major criterion of multiple cutaneous piloleiomyomas confirmed histopathologically. Clinical diagnosis of HLRCC is suspected if 2 or more of the following minor criteria are present: type 2 papillary renal cell carcinoma before 40 years of age; onset of severely symptomatic (requiring surgery) uterine fibroids before 40 years of age in females; and first-degree family member who meets 1 or more of these criteria.15 At the time of presentation, the patient met clinical criteria for HLRCC, including multiple cutaneous leiomyomas (major criterion) and type 2 papillary renal cell carcinoma before 40 years of age (minor criterion). The patient also had a history of uterine leiomyomas, but these lesions did not fulfill the criterion of being severely symptomatic requiring surgery. Furthermore, the patient’s mother had similar cutaneous leiomyomas and a history of uterine cancer, which fulfilled additional minor criterion, consistent with an autosomal-dominant inheritance pattern (with variable penetrance) seen in HLRCC. An important issue for counseling and monitoring patients is that premenopausal women with HLRCC are at an increased risk of developing uterine leiomyosarcoma.15 Our patient followed up with an oncologist for tumor surveillance and subsequently underwent genetic testing, which revealed a mutation in the fumarate hydratase gene.
Treatment of painful cutaneous leiomyomas, particularly in patients with HLRCC, remains a therapeutic challenge. Although surgical and/or destructive treatments can provide pain relief for patients who have a limited number of lesions, these options are impracticable when a patient has numerous widespread leiomyomas; therefore, systemic therapies may be more beneficial. Clinicians should be aware of nifedipine, which may be used in combination with gabapentin as a viable treatment option in the management of acute and breakthrough pain associated with cutaneous leiomyomas.
Acknowledgment
The a
To the Editor:
Leiomyomas are benign smooth muscle tumors. There are 3 types of cutaneous leiomyomas: (1) piloleiomyomas, arising from the arrector pili muscles; (2) angioleiomyomas, arising from the muscles surrounding dermal blood vessels; and (3) leiomyomas of the external genitalia, arising from the dartoic, vulvar, or mammary smooth muscles.1 There is no gender predilection for cutaneous leiomyomas, and lesions present on average at approximately 40 to 45 years of age.2
Piloleiomyomas are the most common type of cutaneous leiomyomas and typically present as red-brown papules and nodules on the trunk, arms, and legs.3 Piloleiomyomas often are associated with spontaneous or induced pain (eg, with cold exposure). The pain associated with piloleiomyomas can be severely debilitating to patients and may have a considerable impact on their quality of life.
A 40-year-old woman presented to our clinic with numerous widespread, painful, red-brown papules and nodules on the head, neck, chest, abdomen, back, arms, and legs of 6 years’ duration that were increasing in number (Figure 1). She had a history of uterine leiomyomas and type 2 renal papillary carcinoma following a left nephrectomy at 38 years of age. The patient’s mother had a history of similar skin lesions as well as uterine cancer. Multiple excisional biopsies were performed, all of which showed piloleiomyomas on histopathology (Figure 2). The pain associated with the patient’s extensive cutaneous leiomyomas considerably impaired her quality of life. Although she experienced pain in all affected areas of the body, the pain was the worst in the upper arms. She reported having requested a nerve ablation procedure from an outside pain management clinic, which was denied for unknown reasons.
Two years prior to the current presentation, the patient had been treated by a pain management specialist with gabapentin 300 mg twice daily as needed for pain associated with leiomyomas. The patient followed this regimen approximately 3 times weekly for the preceding 1 to 2 years with reduction in her pain symptoms; however, the painful episodes became more frequent and severe over time. The patient reported being unable to further increase the gabapentin dosing frequency because it made her too drowsy and impacted her ability to work a job that required heavy lifting. Thus, the patient requested additional therapy and was subsequently treated at our clinic with numerous excisional biopsies of the most painful lesions during the 2 years prior to her current presentation.
When the patient re-presented to our clinic, she requested additional lesion excisions given that she had experienced some pain relief from this treatment modality in the past; however, these prior excisions only resulted in local pain relief limited to the site of the excision. Because of the extent of the lesions and the patient’s inability to tolerate pain from the lidocaine injections, we did not feel multiple excisions were a practical treatment option. The patient subsequently was offered a trial of cryotherapy for symptom relief based on a reported case in which this modality was successfully used.4 After discussing the risks and benefits associated with this treatment, cryotherapy was attempted on a few of the leiomyomas on the patient’s right shoulder; however, she experienced severe pain during cryotherapy treatment, and the procedure had to be aborted.
We then increased the patient’s gabapentin regimen to 300 mg in the morning and 600 mg in the evening, as tolerated. The patient reported that she was better able to tolerate the sedating side effects of the increased dose of gabapentin because she had stopped working due to her severe pain episodes. We also added oral nifedipine 10 mg 3 times daily, as needed. Within 30 minutes of starting this treatment regimen, the pain associated with the lesions remarkably improved (10/10 severity before starting treatment vs 3/10 after starting treatment). Her pain levels remained stable (3/10 severity) during 3 weeks of treatment with this combination regimen, but unfortunately she developed headaches and malaise, which she associated with the nifedipine at the 3 times daily dose.
The patient was able to better tolerate the nifedipine after reducing the dose to once daily on an as-needed basis. On average, the patient took nifedipine once every 3 days; however, she reported that she had to periodically increase the frequency of the nifedipine to once daily for up to 2 weeks at a time for periods of more frequent pain flares. The patient reported a consistent pattern of the breakthrough symptoms rapidly improving with each dose of nifedipine, though she did feel that taking consistent gabapentin enhanced baseline symptom control. The patient also noticed on a few occasions when she did not have access to her nifedipine that her pain would flare to 10/10 severity and would decrease to 4/10 severity 30 minutes after restarting nifedipine at 10 mg once daily. She experienced breakthrough pain due to exacerbating factors including her menstrual cycle; exposure to the sun and cold temperatures or water; excessive physical activity; and mild trauma. Due to exacerbations from sun exposure, the patient often wore long-sleeved shirts, which helped reduce the severity of the pain episodes while she was outdoors.
The exact mechanism for the pain associated with cutaneous leiomyomas is unknown but is thought to be due to infringement of the lesion on the surrounding cutaneous nerves. In addition, norepinephrine activates alpha receptors on the smooth muscle to contract through an influx of ions such as calcium. When smooth muscle contracts, the compression of nerves likely is worsened.
There are a limited number of case reports in the literature that have demonstrated successful treatment of the pain associated with cutaneous leiomyomas. Previously reported treatment modalities have included phenoxybenzamine, an alpha-blocking agent that may reduce pain through its antiadrenergic effects2; nitroglycerin, a venous and arterial dilator that may reduce pain by decreasing muscle oxygen requirements2; gabapentin, an antiepileptic and analgesic medication with structural similarity to the gamma-aminobutyric acid neurotransmitter for which the exact mechanism of action is unknown3; botulinum toxin, a neuromuscular blocker that prevents the release of presynaptic acetylcholine and may decrease neuropathic pain by reducing hyperactive nerves5,6; hyoscine butylbromide and topical hyoscine hydrobromide, both antispasmodics that may reduce pain through their anticholinergic effects, which relax smooth muscle7,8; and the CO2 laser, a treatment that has been utilized for its resurfacing, excisional, and ablative properties.9,10
Calcium channel blockers such as amlodipine, verapamil, and nifedipine also have been used to treat the pain associated with piloleiomyomas.11 Calcium ion channel antagonists inhibit the influx of calcium ions across the cell membrane; therefore, nifedipine and other calcium channel blockers may prevent the smooth muscle contraction that is hypothesized to cause pain in patients with cutaneous leiomyomas.12
Mean plasma concentration of nifedipine has been shown to reach maximum values of 160 +/− 49 µg/L after 30 to 60 minutes following oral administration of 10 mg of nifedipine.13 After 8 hours, the mean concentration drops to 3.4 +/− 1.2 µg/L. The clinical response in our patient appeared consistent with the reported pharmacokinetics of the drug, as she was able to consistently obtain considerable reduction in her pain symptoms within 30 minutes of starting nifedipine, coinciding with the period of time it takes for the nifedipine to reach maximum plasma concentrations.13
Interestingly, our patient had worsening pain episodes associated with sun exposure, which typically is not reported as one of the usual triggers for cutaneous leiomyomas. We are not aware of any described mechanisms that would explain this phenomenon.
Importantly, any patient presenting with multiple cutaneous and uterine (if female) leiomyomas should be screened for hereditary leiomyomatosis and renal cell carcinoma syndrome (HLRCC), an autosomal-dominant disorder linked to a mutation in the fumarate hydratase tumor suppressor gene. Clinically, HLRCC patients typically present with multiple cutaneous leiomyomas, uterine leiomyomas, and renal cell cancer (most often type 2 papillary renal cell carcinoma).14 Hereditary leiomyomatosis and renal cell carcinoma syndrome (also known as multiple cutaneous and uterine leiomyomatosis syndrome) previously was thought to be a separate disease entity from Reed syndrome; however, after the same mutation in the fumarate hydratase tumor suppressor gene was found to be responsible for both Reed syndrome and HLRCC, they are now thought to be the same disease process.15
Diagnosis of HLRCC is likely when the patient meets the major criterion of multiple cutaneous piloleiomyomas confirmed histopathologically. Clinical diagnosis of HLRCC is suspected if 2 or more of the following minor criteria are present: type 2 papillary renal cell carcinoma before 40 years of age; onset of severely symptomatic (requiring surgery) uterine fibroids before 40 years of age in females; and first-degree family member who meets 1 or more of these criteria.15 At the time of presentation, the patient met clinical criteria for HLRCC, including multiple cutaneous leiomyomas (major criterion) and type 2 papillary renal cell carcinoma before 40 years of age (minor criterion). The patient also had a history of uterine leiomyomas, but these lesions did not fulfill the criterion of being severely symptomatic requiring surgery. Furthermore, the patient’s mother had similar cutaneous leiomyomas and a history of uterine cancer, which fulfilled additional minor criterion, consistent with an autosomal-dominant inheritance pattern (with variable penetrance) seen in HLRCC. An important issue for counseling and monitoring patients is that premenopausal women with HLRCC are at an increased risk of developing uterine leiomyosarcoma.15 Our patient followed up with an oncologist for tumor surveillance and subsequently underwent genetic testing, which revealed a mutation in the fumarate hydratase gene.
Treatment of painful cutaneous leiomyomas, particularly in patients with HLRCC, remains a therapeutic challenge. Although surgical and/or destructive treatments can provide pain relief for patients who have a limited number of lesions, these options are impracticable when a patient has numerous widespread leiomyomas; therefore, systemic therapies may be more beneficial. Clinicians should be aware of nifedipine, which may be used in combination with gabapentin as a viable treatment option in the management of acute and breakthrough pain associated with cutaneous leiomyomas.
Acknowledgment
The a
- Holst VA, Junkins-Hopkins JM, Elenitsas R. Cutaneous smooth muscle neoplasms: clinical features, histologic findings, and treatment options. J Am Acad Dermatol. 2002;46:477-494.
- Raj S, Calonje E, Kraus M, et al. Cutaneous pilar leiomyoma: clinicopathologic analysis of 53 lesions in 45 patients. Am J Dermatopathol. 1997;19:2-9.
- Alam M, Rabinowitz AD, Engler DE. Gabapentin treatment of multiple piloleiomyoma-related pain. J Am Acad Dermatol. 2002;46:S27-S29.
- Basendwh MA, Fatani M, Baltow B. Reed’s syndrome: a case of multiple cutaneous leiomyomas treated with liquid nitrogen cryotherapy. Case Rep Dermatol. 2016;8:65-70.
- Sifaki MK, Krueger-Krasagakis S, Koutsopoulos A, et al. Botulinum toxin type A–treatment of a patient with multiple cutaneous piloleiomyomas. Dermatology. 2008;218:44-47.
- Onder M, Adıs¸en E. A new indication of botulinum toxin: leiomyoma-related pain. J Am Acad Dermatol. 2009;60:325-328.
- Kaliyadan F, Manoj J, Dharmaratnam AD. Multiple cutaneous leiomyomas: pain relief with pulsed hysocine butyl bromide. Indian J Dermatol. 2009;54:72.
- Archer CB, Whittaker S, Greaves MW. Pharmacological modulation of cold‐induced pain in cutaneous leiomyomata. Br J Dermatol. 1988;118:255-260.
- Christenson LJ, Smith K, Arpey CJ. Treatment of multiple cutaneous leiomyomas with CO2 laser ablation. Dermatol Surg. 2000;26:319-322.
- Michajłowski I, Błaz˙ewicz I, Karpinsky G, et al. Successful treatment of multiple cutaneous leiomyomas with carbon dioxide laser ablation. Postepy Dermatol Alergol. 2015;32:480-482.
- Archer CB, Greaves MW. Assessment of treatment for painful cutaneous leiomyomas. J Am Acad Dermatol. 1987;17:141-142.
- Thompson JA. Therapy for painful cutaneous leiomyomas. J Am Acad Dermatol. 1985;13:865-867.
- Raemsch KD, Sommer J. Pharmacokinetics and metabolism of nifedipine. Hypertension. 1983;5(4 pt 2):II18-II24.
- Toro JR, Nickerson ML, Wei MH, et al. Mutations in the fumarate hydratase gene cause hereditary leiomyomatosis and renal cell cancer in families in North America. Am J Hum Genet. 2003;73:95-106.
- Smit DL, Mensenkamp AR, Badeloe S, et al. Hereditary leiomyomatosis and renal cell cancer in families referred for fumarate hydratase germline mutation analysis. Clin Genet. 2011;79:49-59.
- Holst VA, Junkins-Hopkins JM, Elenitsas R. Cutaneous smooth muscle neoplasms: clinical features, histologic findings, and treatment options. J Am Acad Dermatol. 2002;46:477-494.
- Raj S, Calonje E, Kraus M, et al. Cutaneous pilar leiomyoma: clinicopathologic analysis of 53 lesions in 45 patients. Am J Dermatopathol. 1997;19:2-9.
- Alam M, Rabinowitz AD, Engler DE. Gabapentin treatment of multiple piloleiomyoma-related pain. J Am Acad Dermatol. 2002;46:S27-S29.
- Basendwh MA, Fatani M, Baltow B. Reed’s syndrome: a case of multiple cutaneous leiomyomas treated with liquid nitrogen cryotherapy. Case Rep Dermatol. 2016;8:65-70.
- Sifaki MK, Krueger-Krasagakis S, Koutsopoulos A, et al. Botulinum toxin type A–treatment of a patient with multiple cutaneous piloleiomyomas. Dermatology. 2008;218:44-47.
- Onder M, Adıs¸en E. A new indication of botulinum toxin: leiomyoma-related pain. J Am Acad Dermatol. 2009;60:325-328.
- Kaliyadan F, Manoj J, Dharmaratnam AD. Multiple cutaneous leiomyomas: pain relief with pulsed hysocine butyl bromide. Indian J Dermatol. 2009;54:72.
- Archer CB, Whittaker S, Greaves MW. Pharmacological modulation of cold‐induced pain in cutaneous leiomyomata. Br J Dermatol. 1988;118:255-260.
- Christenson LJ, Smith K, Arpey CJ. Treatment of multiple cutaneous leiomyomas with CO2 laser ablation. Dermatol Surg. 2000;26:319-322.
- Michajłowski I, Błaz˙ewicz I, Karpinsky G, et al. Successful treatment of multiple cutaneous leiomyomas with carbon dioxide laser ablation. Postepy Dermatol Alergol. 2015;32:480-482.
- Archer CB, Greaves MW. Assessment of treatment for painful cutaneous leiomyomas. J Am Acad Dermatol. 1987;17:141-142.
- Thompson JA. Therapy for painful cutaneous leiomyomas. J Am Acad Dermatol. 1985;13:865-867.
- Raemsch KD, Sommer J. Pharmacokinetics and metabolism of nifedipine. Hypertension. 1983;5(4 pt 2):II18-II24.
- Toro JR, Nickerson ML, Wei MH, et al. Mutations in the fumarate hydratase gene cause hereditary leiomyomatosis and renal cell cancer in families in North America. Am J Hum Genet. 2003;73:95-106.
- Smit DL, Mensenkamp AR, Badeloe S, et al. Hereditary leiomyomatosis and renal cell cancer in families referred for fumarate hydratase germline mutation analysis. Clin Genet. 2011;79:49-59.
Practice Points
- Cutaneous leiomyomas (piloleiomyomas) are benign smooth muscle tumors derived from the arrector pili muscle.
- Patients presenting with multiple cutaneous leiomyomas should be evaluated for hereditary leiomyomatosis and renal cell carcinoma syndrome, an autosomal-dominant disorder, which also predisposes to the development of symptomatic uterine fibroids and uterine leiomyosarcoma.
- Cutaneous leiomyomas may be a source of considerable pain, which may respond to treatment with nifedipine in combination with gabapentin.

Symmetrical Pruriginous Nasal Rash
The Diagnosis: Irritant Contact Dermatitis
A slang term for volatile alkyl nitrites, poppers are inhaled for recreational purposes. They produce rapid-onset euphoria and sexual arousal, as well as relax anal and vaginal sphincters, facilitating sexual intercourse. Alkyl nitrites initially were developed to treat coronary disease and angina but were replaced by more potent drugs.1 Because of their psychoactive effects and smooth muscle relaxation properties, they are widely used by homosexual and bisexual men.1-3 The term poppers was originated by the sound generated when the glass vials are crushed; currently, they also may be found in other formats.1
Nausea, hypotension, and headache are mild common adverse effects of volatile alkyl nitrites1; cardiac arrhythmia, oxidative hemolysis,4 and poppers maculopathy5,6 with permanent eye damage also have been reported.7 On the skin, volatile alkyl nitrites induce irritant contact dermatitis that heals without scarring, characteristically involving the face and upper thoracic region, as they are volatile vapors.2 However, the reaction can occur elsewhere. There have been reports of contact dermatitis on other locations, such as the thigh or the ankle, due to vials broken while stored in pockets or on the cuff of the socks.1 There also is a report of irritant contact dermatitis manifesting as a penile ulcer.3 Albeit rare, allergic contact dermatitis to volatile alkyl nitrites and other nitrites also can occur.8
The abuse of alkyl nitrites may increase the risk for sexually transmitted infections (STIs), as they may decrease safer sexual practices and increase the propensity to engage in risky sexual behavior. It has been suggested to screen for STIs in patients with history of volatile alkyl nitrite use. In the past, volatile alkyl nitrites were believed to be a potential vector of human immunodeficiency virus.9 Other popular drugs used in social context or "club drugs," such as 3,4-methylenedioxymethamphetamine, gamma hydroxybutyrate, methamphetamine, and ketamine, do not produce irritant dermatitis as an adverse cutaneous reaction.10 The differential diagnosis in our patient included herpes simplex virus and contagious impetigo1 as well as bullous lupus erythematosus and periorificial dermatitis; however, the clinical picture, acute onset of the reaction, and the patient's medical history were critical in making the correct diagnosis.
The patient was treated with topical hydrocortisone and fusidic acid cream twice daily for 7 days with complete response. Sexually transmitted infection screening was unremarkable. We suggest performing an STI workup on patients with history of volatile alkyl nitrite use.
- Schauber J, Herzinger T. 'Poppers' dermatitis. Clin Exp Dermatol. 2012;37:587-588.
- Foroozan M, Studer M, Splingard B, et al. Facial dermatitis due to inhalation of poppers [in French]. Ann Dermatol Venereol. 2009;136:298-299.
- Latini A, Lora V, Zaccarelli M, et al. Unusual presentation of poppers dermatitis. JAMA Dermatol. 2017;153:233-234.
- Shortt J, Polizzotto MN, Opat SS, et al. Oxidative haemolysis due to 'poppers'. Br J Haematol. 2008;142:328.
- Davies AJ, Kelly SP, Naylor SG, et al. Adverse ophthalmic reaction in poppers users: case series of 'poppers maculopathy'. Eye (Lond). 2012;26:1479-1486.
- Davies AJ, Kelly SP, Bhatt PR. 'Poppers maculopathy'--an emerging ophthalmic reaction to recreational substance abuse. Eye (Lond). 2012;26:888.
- Vignal-Clermont C, Audo I, Sahel JA, et al. Poppers-associated retinal toxicity. N Engl J Med. 2010;363:1583-1585.
- Bos JD, Jansen FC, Timmer JG. Allergic contact dermatitis to amyl nitrite ('poppers'). Contact Dermatitis. 1985;12:109.
- Stratford M, Wilson PD. Agitation effects on microbial cell-cell interactions. Lett Appl Microbiol. 1990;11:1-6.
- Romanelli F, Smith KM, Thornton AC, et al. Poppers: epidemiology and clinical management of inhaled nitrite abuse. Pharmacotherapy. 2004;24:69-78.
The Diagnosis: Irritant Contact Dermatitis
A slang term for volatile alkyl nitrites, poppers are inhaled for recreational purposes. They produce rapid-onset euphoria and sexual arousal, as well as relax anal and vaginal sphincters, facilitating sexual intercourse. Alkyl nitrites initially were developed to treat coronary disease and angina but were replaced by more potent drugs.1 Because of their psychoactive effects and smooth muscle relaxation properties, they are widely used by homosexual and bisexual men.1-3 The term poppers was originated by the sound generated when the glass vials are crushed; currently, they also may be found in other formats.1
Nausea, hypotension, and headache are mild common adverse effects of volatile alkyl nitrites1; cardiac arrhythmia, oxidative hemolysis,4 and poppers maculopathy5,6 with permanent eye damage also have been reported.7 On the skin, volatile alkyl nitrites induce irritant contact dermatitis that heals without scarring, characteristically involving the face and upper thoracic region, as they are volatile vapors.2 However, the reaction can occur elsewhere. There have been reports of contact dermatitis on other locations, such as the thigh or the ankle, due to vials broken while stored in pockets or on the cuff of the socks.1 There also is a report of irritant contact dermatitis manifesting as a penile ulcer.3 Albeit rare, allergic contact dermatitis to volatile alkyl nitrites and other nitrites also can occur.8
The abuse of alkyl nitrites may increase the risk for sexually transmitted infections (STIs), as they may decrease safer sexual practices and increase the propensity to engage in risky sexual behavior. It has been suggested to screen for STIs in patients with history of volatile alkyl nitrite use. In the past, volatile alkyl nitrites were believed to be a potential vector of human immunodeficiency virus.9 Other popular drugs used in social context or "club drugs," such as 3,4-methylenedioxymethamphetamine, gamma hydroxybutyrate, methamphetamine, and ketamine, do not produce irritant dermatitis as an adverse cutaneous reaction.10 The differential diagnosis in our patient included herpes simplex virus and contagious impetigo1 as well as bullous lupus erythematosus and periorificial dermatitis; however, the clinical picture, acute onset of the reaction, and the patient's medical history were critical in making the correct diagnosis.
The patient was treated with topical hydrocortisone and fusidic acid cream twice daily for 7 days with complete response. Sexually transmitted infection screening was unremarkable. We suggest performing an STI workup on patients with history of volatile alkyl nitrite use.
The Diagnosis: Irritant Contact Dermatitis
A slang term for volatile alkyl nitrites, poppers are inhaled for recreational purposes. They produce rapid-onset euphoria and sexual arousal, as well as relax anal and vaginal sphincters, facilitating sexual intercourse. Alkyl nitrites initially were developed to treat coronary disease and angina but were replaced by more potent drugs.1 Because of their psychoactive effects and smooth muscle relaxation properties, they are widely used by homosexual and bisexual men.1-3 The term poppers was originated by the sound generated when the glass vials are crushed; currently, they also may be found in other formats.1
Nausea, hypotension, and headache are mild common adverse effects of volatile alkyl nitrites1; cardiac arrhythmia, oxidative hemolysis,4 and poppers maculopathy5,6 with permanent eye damage also have been reported.7 On the skin, volatile alkyl nitrites induce irritant contact dermatitis that heals without scarring, characteristically involving the face and upper thoracic region, as they are volatile vapors.2 However, the reaction can occur elsewhere. There have been reports of contact dermatitis on other locations, such as the thigh or the ankle, due to vials broken while stored in pockets or on the cuff of the socks.1 There also is a report of irritant contact dermatitis manifesting as a penile ulcer.3 Albeit rare, allergic contact dermatitis to volatile alkyl nitrites and other nitrites also can occur.8
The abuse of alkyl nitrites may increase the risk for sexually transmitted infections (STIs), as they may decrease safer sexual practices and increase the propensity to engage in risky sexual behavior. It has been suggested to screen for STIs in patients with history of volatile alkyl nitrite use. In the past, volatile alkyl nitrites were believed to be a potential vector of human immunodeficiency virus.9 Other popular drugs used in social context or "club drugs," such as 3,4-methylenedioxymethamphetamine, gamma hydroxybutyrate, methamphetamine, and ketamine, do not produce irritant dermatitis as an adverse cutaneous reaction.10 The differential diagnosis in our patient included herpes simplex virus and contagious impetigo1 as well as bullous lupus erythematosus and periorificial dermatitis; however, the clinical picture, acute onset of the reaction, and the patient's medical history were critical in making the correct diagnosis.
The patient was treated with topical hydrocortisone and fusidic acid cream twice daily for 7 days with complete response. Sexually transmitted infection screening was unremarkable. We suggest performing an STI workup on patients with history of volatile alkyl nitrite use.
- Schauber J, Herzinger T. 'Poppers' dermatitis. Clin Exp Dermatol. 2012;37:587-588.
- Foroozan M, Studer M, Splingard B, et al. Facial dermatitis due to inhalation of poppers [in French]. Ann Dermatol Venereol. 2009;136:298-299.
- Latini A, Lora V, Zaccarelli M, et al. Unusual presentation of poppers dermatitis. JAMA Dermatol. 2017;153:233-234.
- Shortt J, Polizzotto MN, Opat SS, et al. Oxidative haemolysis due to 'poppers'. Br J Haematol. 2008;142:328.
- Davies AJ, Kelly SP, Naylor SG, et al. Adverse ophthalmic reaction in poppers users: case series of 'poppers maculopathy'. Eye (Lond). 2012;26:1479-1486.
- Davies AJ, Kelly SP, Bhatt PR. 'Poppers maculopathy'--an emerging ophthalmic reaction to recreational substance abuse. Eye (Lond). 2012;26:888.
- Vignal-Clermont C, Audo I, Sahel JA, et al. Poppers-associated retinal toxicity. N Engl J Med. 2010;363:1583-1585.
- Bos JD, Jansen FC, Timmer JG. Allergic contact dermatitis to amyl nitrite ('poppers'). Contact Dermatitis. 1985;12:109.
- Stratford M, Wilson PD. Agitation effects on microbial cell-cell interactions. Lett Appl Microbiol. 1990;11:1-6.
- Romanelli F, Smith KM, Thornton AC, et al. Poppers: epidemiology and clinical management of inhaled nitrite abuse. Pharmacotherapy. 2004;24:69-78.
- Schauber J, Herzinger T. 'Poppers' dermatitis. Clin Exp Dermatol. 2012;37:587-588.
- Foroozan M, Studer M, Splingard B, et al. Facial dermatitis due to inhalation of poppers [in French]. Ann Dermatol Venereol. 2009;136:298-299.
- Latini A, Lora V, Zaccarelli M, et al. Unusual presentation of poppers dermatitis. JAMA Dermatol. 2017;153:233-234.
- Shortt J, Polizzotto MN, Opat SS, et al. Oxidative haemolysis due to 'poppers'. Br J Haematol. 2008;142:328.
- Davies AJ, Kelly SP, Naylor SG, et al. Adverse ophthalmic reaction in poppers users: case series of 'poppers maculopathy'. Eye (Lond). 2012;26:1479-1486.
- Davies AJ, Kelly SP, Bhatt PR. 'Poppers maculopathy'--an emerging ophthalmic reaction to recreational substance abuse. Eye (Lond). 2012;26:888.
- Vignal-Clermont C, Audo I, Sahel JA, et al. Poppers-associated retinal toxicity. N Engl J Med. 2010;363:1583-1585.
- Bos JD, Jansen FC, Timmer JG. Allergic contact dermatitis to amyl nitrite ('poppers'). Contact Dermatitis. 1985;12:109.
- Stratford M, Wilson PD. Agitation effects on microbial cell-cell interactions. Lett Appl Microbiol. 1990;11:1-6.
- Romanelli F, Smith KM, Thornton AC, et al. Poppers: epidemiology and clinical management of inhaled nitrite abuse. Pharmacotherapy. 2004;24:69-78.
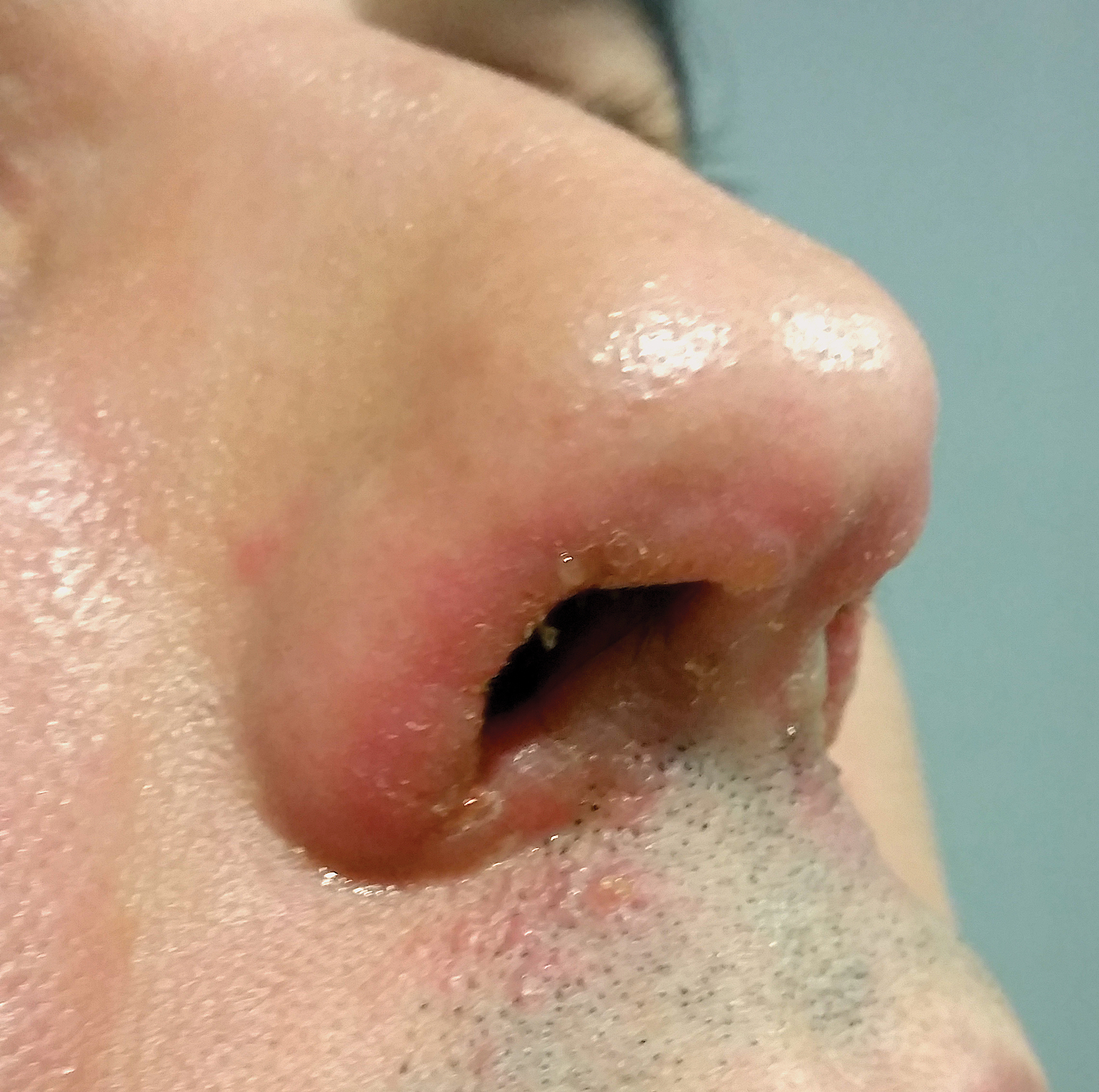
A 44-year-old man was referred to the department of dermatology for a pruriginous nasal rash. Physical examination revealed vesicles with clear content and crusts symmetrically in both nostrils and philtra. The remainder of the examination was otherwise unremarkable. The patient reported inhalation of poppers the prior night during a party. No history of connective tissue diseases was present. The patient was in overall good health with no fever or chills.
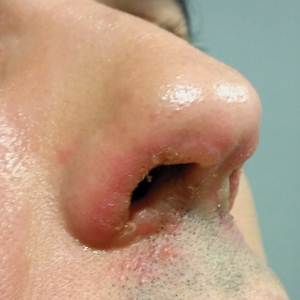
UV Radiation Exposure in Welders: Impact on the Skin and Eyes
Although solar radiation is the most commonly recognized source of UV radiation (UVR), occupational exposures can contribute due to the intensity and chronicity of exposure. Arc welding is a process whereby metal is fused together by heat produced from an electric arc. The electric arc that forms between the electrode and the base metal emits radiation in the full UV spectrum including UVA (400–315 nm), UVB (315–290 nm), and UVC (290–100 nm) wavelengths. Welders, therefore, have an increased risk for broad-spectrum, intense exposure to UVR, which may play a notable role in UV-related skin disease without proper protection. We report 3 welders with skin disease attributed to occupational exposure to UVR.
Case Reports
Patient 1
A 41-year-old man presented for evaluation of treatment-resistant cutaneous lupus. During the 10-year disease course, the patient was treated by both dermatologists and rheumatologists with frequent exacerbations and poor disease control. At the time of presentation, treatment with hydroxychloroquine 200 mg twice daily, azathioprine 50 mg twice daily, intramuscular methylprednisolone acetateinjectable suspension 40 mg, and prednisone 20 mg daily was failing. Physical examination revealed polycyclic erythematous plaques typical of subacute cutaneous lupus erythematosus. A skin biopsy confirmed the diagnosis. Upon further discussion of exacerbating risk factors, the patient noted UVR exposure while working as a welder. Although he had been previously told to avoid sunlight, he did not realize that this recommendation included all forms of UV light. Once this work exposure was eliminated, he was restarted on hydroxychloroquine 200 mg twice daily and topical steroids, and he responded with complete and sustained clearance of disease. When he returned to welding, utilization of sunscreen and sun-protective clothing enabled him to maintain control of his subacute cutaneous lupus erythematosus on oral hydroxychloroquine 200 mg twice daily and topical steroids.
Patient 2
A 55-year-old man presented with numerous actinic keratoses and persistent erythema in a well-demarcated area involving the forehead, temples, and lateral cheeks but sparing the periorbital area. The patient also experienced UVR exposure from welding (up to 4 to 5 times per week during his career spanning more than 20 years). He cited frequent burns in areas where his protective equipment did not cover his skin. He also reported that he often forgoes wearing protective equipment, even though it is available, and only uses safety goggles due to the extreme heat of the working environment as well as the awkwardness of wearing full protective gear while performing certain aspects of the job.
Patient 3
A 63-year-old man presented with a growth on the left side of the upper forehead. A biopsy revealed a squamous cell carcinoma, keratoacanthoma type. He worked as a welder for 40 years until retiring 1 year prior to presentation. He welded daily and always wore a tall face shield. Although the face shield covered most of his face, the scalp and some parts of the upper face were not well protected. In addition to the keratoacanthoma, which presented just outside of the area protected by the face shield, the patient had numerous actinic keratoses on the scalp.
Comment
Welding and UVR Exposure
Arc welders endure large amounts of UVR exposure, which is substantial enough to have notable health effects. The duration of exposure, electrical current used, angle of exposure, amount of ventilation, and the distance from the welding arc play a role in overall UVR exposure.1,2 Maximum permissible exposure (MPE) limits to UVR have been set by the International Commission on Non-Ionizing Radiation Protection and the National Institute for Occupational Safety and Health.3,4 The quantity of radiation produced by the arc allows for an exposure time of only a few seconds to minutes before surpassing MPE to UV light.1,5 Welders are exposed to total-body UVR doses up to 3000 times the MPE, and mean cumulative exposure calculated over an 8-hour workday can reach 9795 mJ/cm2.6
Workers in close proximity to welders also receive large UVR doses and may not be aware of its hazardous effects. Nearby nonwelders can be exposed to 13 times the MPE of UVR.6 At distances up to 10 m from the arc, the irradiance is large enough to reach MPE to UVR in less than 3 hours.1
Skin and Eye Damage From Welding
Exposure to UVR produced by the welding arc may lead to acute skin or eye reactions, chronic skin or eye disorders, or exacerbation of photosensitive diseases. Common acute problems are photokeratoconjunctivitis (welder’s flash) and skin erythema.7,8
Actinic elastosis, actinic keratoses, ocular melanoma, and photosensitive diseases represent a spectrum of disorders that can present from chronic UV exposure in welders. In a study by Emmett et al7 of 152 welders and 58 controls, actinic elastosis was found to be more frequent in welders than controls. Cases of basal cell carcinoma and squamous cell carcinoma also have been reported in welders.9,10 However, in the study by Emmett et al,7 a statistically significant correlation between welding and skin cancer was not documented. There were limitations in the study, such as small sample size and a young average age of welders.7 Future studies may be needed to further clarify the risk for skin cancer in welders.
Although there is no clear association with skin cancer, an increased risk of ocular melanoma in welders is more clearly established. A meta-analysis of 5 studies found that welding was a significant risk factor for ocular melanoma, with an odds ratio of 2.05 (95% confidence interval, 1.20-3.51).11 Other reported eye damage from chronic UVR exposure includes cataracts, chronic conjunctivitis, and retinal damage.12,13
Case reports of the following photosensitive diseases have been reported to be exacerbated or caused by UV light exposure in welders: discoid lupus erythematosus14; photodermatitis15; broadband photosensitivity with decreased minimal erythema dose to UVA, UVB, and UVC16; UVC-exacerbated atopic dermatitis17; polymorphous light eruption–like skin eruption18; and UVA-induced photoallergy to hydrochlorothiazide and ramipril.19
Prevention of Occupational Exposure to UVR
Occupational Safety and Health Administration guidelines protect workers from excessive exposure to UVR with personal protective equipment (PPE). In addition to UVR protection, PPE needs to protect welders from other risks including trauma from welding debris (slag), fires, electrical burns, and fumes. Online resources from the National Ag Safety Database,20 the American Welding Society,21 and Occupational Safety and Health Administration22,23 are available. These resources advise welders to work in ventilated areas with respirators specific for the metal being welded and to wear clothing and gloves that are not only fire retardant but also UV resistant.20-23 Additional PPE should protect the head, face, and eyes.
Unfortunately, even workers well trained in prevention guidelines may not adequately protect themselves. Some welders forego PPE due to heat, thus exposing themselves to UVR damage in areas that are normally covered. Welders also may forego equipment when working on jobs requiring more detailed welds where clothing, masks, and glasses may be overly bulky and inhibit the worker’s precision. Nontraditional welders, such as artisans or handymen, may not have workplace safety education to be aware of UVR emitted from welding and may not have readily available PPE.
The Figure portrays an amateur welder working without full PPE. Although he is wearing a face shield, he is not wearing fire-retardant clothing, lacks full protective garments, and has no ventilation system.

Conclusion
It is important to recognize welding as an occupation with notable exposure to UVR. Personal protective equipment should be the mainstay of prevention. Sunscreen is a useful adjunct but does not cover UVC that is emitted in the welding arc. Screens and welding blankets can be placed around welders to contain UVR and limit nonwelder exposure. Although UVR hazards should be regulated in the workplace as part of regular safety reviews, the clinician can play a role in recognizing this source of UVR in skin disease and in encouraging the use of PPE.
- Okuno T, Ojima J, Saito H. Ultraviolet radiation emitted by CO(2) arc welding. Ann Occup Hyg. 2001;45:597-601.
- Peng CY, Liu HH, Chang CP, et al. Evaluation and monitoring of UVR in shield metal ARC welding processing. Health Phys. 2007;93:101-108.
- The National Institute for Occupational Safety and Health. Criteria for a recommended standard: occupational exposure to ultraviolet radiation. DHHS (NIOSH) publication 73-11009. https://www.cdc.gov/niosh/docs/73-11009/. Updated June 6, 2014. Accessed September 6, 2019.
- International Commission on Non-Ionizing Radiation Protection. Guidelines on limits of exposure to ultraviolet radiation of wavelengths between 180 nm and 400 nm (incoherent optical radiation). Health Phys. 2004;87:171-186.
- Peng CY, Lan CH, Juang YJ, et al. Exposure assessment of aluminum arc welding radiation. Health Phys. 2007;93:298-306.
- Tenkate TD, Collins MJ. Personal ultraviolet radiation exposure of workers in a welding environment. Am Ind Hyg Assoc J. 1997;58:33-38.
- Emmett EA, Buncher CR, Suskind RB, et al. Skin and eye diseases among arc welders and those exposed to welding operations. J Occup Med. 1981;23:85-90.
- Bruze M, Hindsén M, Trulsson L. Dermatitis with an unusual explanation in a welder. Acta Derm Venereol. 1994;74:380-382.
- Donoghue AM, Sinclair MJ. Basal cell carcinoma after frequent episodes of cutaneous erythema and peeling induced by welding. Occup Environ Med. 1999;56:646.
- Currie CL, Monk BE. Welding and non-melanoma skin cancer. Clin Exp Dermatol. 2000;25:28-29.
- Shah CP, Weis E, Lajous M, et al. Intermittent and chronic ultraviolet light exposure and uveal melanoma: a meta-analysis. Ophthalmology. 2005;112:1599-1607.
- Yang X, Shao D, Ding X, et al. Chronic phototoxic maculopathy caused by welding arc in occupational welders. Can J Ophthalmol. 2012;47:45-50.
- Davies KG, Asanga U, Nku CO, et al. Effect of chronic exposure to welding light on Calabar welders. Niger J Physiol Sci. 2007;22:55-58.
- Wozniak KD. Erythematodes chronicus discoides as an occupational disease in an electric welder [in German]. Berufs-Dermatosen. 1971;19:187-196.
- Shehade SA, Roberts PJ, Diffey BL, et al. Photodermatitis due to spot welding. Br J Dermatol. 1987;117:117-119.
- Roelandts R, Huys I. Broad-band and persistent photosensitivity following accidental ultraviolet C overexposure. Photodermatol Photoimmunol Photomed. 1993;9:144-146.
- Elsner P, Hassam S. Occupational UVC-induced exacerbation of atopic dermatitis in a welder. Contact Dermatitis. 1996;35:180-181.
- Majoie IM, van Weelden H, Sybesma IM, et al. Polymorphous light eruption-like skin lesions in welders caused by ultraviolet C light. J Am Acad Dermatol. 2010;62:150-151.
- Wagner SN, Welke F, Goos M. Occupational UVA-induced allergic photodermatitis in a welder due to hydrochlorothiazide and ramipril. Contact Dermatitis. 2000;43:245-246.
- Fluegel L, Rein BK. Arc welding safety. National Ag Safety Database website. http://nasdonline.org/1083/d000873/arc-welding-safety.html. Published May 1989. Accessed September 6, 2019.
- American Welding Society. Personal protective equipment (PPE) for welding and cutting. Fact sheet no. 33-04/14. http://www.aws.org/technical/facts/FACT-33_2014.pdf. Published April 2014. Accessed September 6, 2019.
- Occupational Safety and Health Administration. Eye protection against radiant energy during welding and cutting in shipyard employment. https://www.osha.gov/Publications/OSHAfactsheet-eyeprotection-during-welding.pdf. Published January 2012. Accessed September 6, 2019.
- Occupational Safety and Health Administration. Welding, cutting, and brazing. https://www.osha.gov/SLTC/weldingcuttingbrazing/standards.html. Accessed September 10, 2019.
Although solar radiation is the most commonly recognized source of UV radiation (UVR), occupational exposures can contribute due to the intensity and chronicity of exposure. Arc welding is a process whereby metal is fused together by heat produced from an electric arc. The electric arc that forms between the electrode and the base metal emits radiation in the full UV spectrum including UVA (400–315 nm), UVB (315–290 nm), and UVC (290–100 nm) wavelengths. Welders, therefore, have an increased risk for broad-spectrum, intense exposure to UVR, which may play a notable role in UV-related skin disease without proper protection. We report 3 welders with skin disease attributed to occupational exposure to UVR.
Case Reports
Patient 1
A 41-year-old man presented for evaluation of treatment-resistant cutaneous lupus. During the 10-year disease course, the patient was treated by both dermatologists and rheumatologists with frequent exacerbations and poor disease control. At the time of presentation, treatment with hydroxychloroquine 200 mg twice daily, azathioprine 50 mg twice daily, intramuscular methylprednisolone acetateinjectable suspension 40 mg, and prednisone 20 mg daily was failing. Physical examination revealed polycyclic erythematous plaques typical of subacute cutaneous lupus erythematosus. A skin biopsy confirmed the diagnosis. Upon further discussion of exacerbating risk factors, the patient noted UVR exposure while working as a welder. Although he had been previously told to avoid sunlight, he did not realize that this recommendation included all forms of UV light. Once this work exposure was eliminated, he was restarted on hydroxychloroquine 200 mg twice daily and topical steroids, and he responded with complete and sustained clearance of disease. When he returned to welding, utilization of sunscreen and sun-protective clothing enabled him to maintain control of his subacute cutaneous lupus erythematosus on oral hydroxychloroquine 200 mg twice daily and topical steroids.
Patient 2
A 55-year-old man presented with numerous actinic keratoses and persistent erythema in a well-demarcated area involving the forehead, temples, and lateral cheeks but sparing the periorbital area. The patient also experienced UVR exposure from welding (up to 4 to 5 times per week during his career spanning more than 20 years). He cited frequent burns in areas where his protective equipment did not cover his skin. He also reported that he often forgoes wearing protective equipment, even though it is available, and only uses safety goggles due to the extreme heat of the working environment as well as the awkwardness of wearing full protective gear while performing certain aspects of the job.
Patient 3
A 63-year-old man presented with a growth on the left side of the upper forehead. A biopsy revealed a squamous cell carcinoma, keratoacanthoma type. He worked as a welder for 40 years until retiring 1 year prior to presentation. He welded daily and always wore a tall face shield. Although the face shield covered most of his face, the scalp and some parts of the upper face were not well protected. In addition to the keratoacanthoma, which presented just outside of the area protected by the face shield, the patient had numerous actinic keratoses on the scalp.
Comment
Welding and UVR Exposure
Arc welders endure large amounts of UVR exposure, which is substantial enough to have notable health effects. The duration of exposure, electrical current used, angle of exposure, amount of ventilation, and the distance from the welding arc play a role in overall UVR exposure.1,2 Maximum permissible exposure (MPE) limits to UVR have been set by the International Commission on Non-Ionizing Radiation Protection and the National Institute for Occupational Safety and Health.3,4 The quantity of radiation produced by the arc allows for an exposure time of only a few seconds to minutes before surpassing MPE to UV light.1,5 Welders are exposed to total-body UVR doses up to 3000 times the MPE, and mean cumulative exposure calculated over an 8-hour workday can reach 9795 mJ/cm2.6
Workers in close proximity to welders also receive large UVR doses and may not be aware of its hazardous effects. Nearby nonwelders can be exposed to 13 times the MPE of UVR.6 At distances up to 10 m from the arc, the irradiance is large enough to reach MPE to UVR in less than 3 hours.1
Skin and Eye Damage From Welding
Exposure to UVR produced by the welding arc may lead to acute skin or eye reactions, chronic skin or eye disorders, or exacerbation of photosensitive diseases. Common acute problems are photokeratoconjunctivitis (welder’s flash) and skin erythema.7,8
Actinic elastosis, actinic keratoses, ocular melanoma, and photosensitive diseases represent a spectrum of disorders that can present from chronic UV exposure in welders. In a study by Emmett et al7 of 152 welders and 58 controls, actinic elastosis was found to be more frequent in welders than controls. Cases of basal cell carcinoma and squamous cell carcinoma also have been reported in welders.9,10 However, in the study by Emmett et al,7 a statistically significant correlation between welding and skin cancer was not documented. There were limitations in the study, such as small sample size and a young average age of welders.7 Future studies may be needed to further clarify the risk for skin cancer in welders.
Although there is no clear association with skin cancer, an increased risk of ocular melanoma in welders is more clearly established. A meta-analysis of 5 studies found that welding was a significant risk factor for ocular melanoma, with an odds ratio of 2.05 (95% confidence interval, 1.20-3.51).11 Other reported eye damage from chronic UVR exposure includes cataracts, chronic conjunctivitis, and retinal damage.12,13
Case reports of the following photosensitive diseases have been reported to be exacerbated or caused by UV light exposure in welders: discoid lupus erythematosus14; photodermatitis15; broadband photosensitivity with decreased minimal erythema dose to UVA, UVB, and UVC16; UVC-exacerbated atopic dermatitis17; polymorphous light eruption–like skin eruption18; and UVA-induced photoallergy to hydrochlorothiazide and ramipril.19
Prevention of Occupational Exposure to UVR
Occupational Safety and Health Administration guidelines protect workers from excessive exposure to UVR with personal protective equipment (PPE). In addition to UVR protection, PPE needs to protect welders from other risks including trauma from welding debris (slag), fires, electrical burns, and fumes. Online resources from the National Ag Safety Database,20 the American Welding Society,21 and Occupational Safety and Health Administration22,23 are available. These resources advise welders to work in ventilated areas with respirators specific for the metal being welded and to wear clothing and gloves that are not only fire retardant but also UV resistant.20-23 Additional PPE should protect the head, face, and eyes.
Unfortunately, even workers well trained in prevention guidelines may not adequately protect themselves. Some welders forego PPE due to heat, thus exposing themselves to UVR damage in areas that are normally covered. Welders also may forego equipment when working on jobs requiring more detailed welds where clothing, masks, and glasses may be overly bulky and inhibit the worker’s precision. Nontraditional welders, such as artisans or handymen, may not have workplace safety education to be aware of UVR emitted from welding and may not have readily available PPE.
The Figure portrays an amateur welder working without full PPE. Although he is wearing a face shield, he is not wearing fire-retardant clothing, lacks full protective garments, and has no ventilation system.
Conclusion
It is important to recognize welding as an occupation with notable exposure to UVR. Personal protective equipment should be the mainstay of prevention. Sunscreen is a useful adjunct but does not cover UVC that is emitted in the welding arc. Screens and welding blankets can be placed around welders to contain UVR and limit nonwelder exposure. Although UVR hazards should be regulated in the workplace as part of regular safety reviews, the clinician can play a role in recognizing this source of UVR in skin disease and in encouraging the use of PPE.
Although solar radiation is the most commonly recognized source of UV radiation (UVR), occupational exposures can contribute due to the intensity and chronicity of exposure. Arc welding is a process whereby metal is fused together by heat produced from an electric arc. The electric arc that forms between the electrode and the base metal emits radiation in the full UV spectrum including UVA (400–315 nm), UVB (315–290 nm), and UVC (290–100 nm) wavelengths. Welders, therefore, have an increased risk for broad-spectrum, intense exposure to UVR, which may play a notable role in UV-related skin disease without proper protection. We report 3 welders with skin disease attributed to occupational exposure to UVR.
Case Reports
Patient 1
A 41-year-old man presented for evaluation of treatment-resistant cutaneous lupus. During the 10-year disease course, the patient was treated by both dermatologists and rheumatologists with frequent exacerbations and poor disease control. At the time of presentation, treatment with hydroxychloroquine 200 mg twice daily, azathioprine 50 mg twice daily, intramuscular methylprednisolone acetateinjectable suspension 40 mg, and prednisone 20 mg daily was failing. Physical examination revealed polycyclic erythematous plaques typical of subacute cutaneous lupus erythematosus. A skin biopsy confirmed the diagnosis. Upon further discussion of exacerbating risk factors, the patient noted UVR exposure while working as a welder. Although he had been previously told to avoid sunlight, he did not realize that this recommendation included all forms of UV light. Once this work exposure was eliminated, he was restarted on hydroxychloroquine 200 mg twice daily and topical steroids, and he responded with complete and sustained clearance of disease. When he returned to welding, utilization of sunscreen and sun-protective clothing enabled him to maintain control of his subacute cutaneous lupus erythematosus on oral hydroxychloroquine 200 mg twice daily and topical steroids.
Patient 2
A 55-year-old man presented with numerous actinic keratoses and persistent erythema in a well-demarcated area involving the forehead, temples, and lateral cheeks but sparing the periorbital area. The patient also experienced UVR exposure from welding (up to 4 to 5 times per week during his career spanning more than 20 years). He cited frequent burns in areas where his protective equipment did not cover his skin. He also reported that he often forgoes wearing protective equipment, even though it is available, and only uses safety goggles due to the extreme heat of the working environment as well as the awkwardness of wearing full protective gear while performing certain aspects of the job.
Patient 3
A 63-year-old man presented with a growth on the left side of the upper forehead. A biopsy revealed a squamous cell carcinoma, keratoacanthoma type. He worked as a welder for 40 years until retiring 1 year prior to presentation. He welded daily and always wore a tall face shield. Although the face shield covered most of his face, the scalp and some parts of the upper face were not well protected. In addition to the keratoacanthoma, which presented just outside of the area protected by the face shield, the patient had numerous actinic keratoses on the scalp.
Comment
Welding and UVR Exposure
Arc welders endure large amounts of UVR exposure, which is substantial enough to have notable health effects. The duration of exposure, electrical current used, angle of exposure, amount of ventilation, and the distance from the welding arc play a role in overall UVR exposure.1,2 Maximum permissible exposure (MPE) limits to UVR have been set by the International Commission on Non-Ionizing Radiation Protection and the National Institute for Occupational Safety and Health.3,4 The quantity of radiation produced by the arc allows for an exposure time of only a few seconds to minutes before surpassing MPE to UV light.1,5 Welders are exposed to total-body UVR doses up to 3000 times the MPE, and mean cumulative exposure calculated over an 8-hour workday can reach 9795 mJ/cm2.6
Workers in close proximity to welders also receive large UVR doses and may not be aware of its hazardous effects. Nearby nonwelders can be exposed to 13 times the MPE of UVR.6 At distances up to 10 m from the arc, the irradiance is large enough to reach MPE to UVR in less than 3 hours.1
Skin and Eye Damage From Welding
Exposure to UVR produced by the welding arc may lead to acute skin or eye reactions, chronic skin or eye disorders, or exacerbation of photosensitive diseases. Common acute problems are photokeratoconjunctivitis (welder’s flash) and skin erythema.7,8
Actinic elastosis, actinic keratoses, ocular melanoma, and photosensitive diseases represent a spectrum of disorders that can present from chronic UV exposure in welders. In a study by Emmett et al7 of 152 welders and 58 controls, actinic elastosis was found to be more frequent in welders than controls. Cases of basal cell carcinoma and squamous cell carcinoma also have been reported in welders.9,10 However, in the study by Emmett et al,7 a statistically significant correlation between welding and skin cancer was not documented. There were limitations in the study, such as small sample size and a young average age of welders.7 Future studies may be needed to further clarify the risk for skin cancer in welders.
Although there is no clear association with skin cancer, an increased risk of ocular melanoma in welders is more clearly established. A meta-analysis of 5 studies found that welding was a significant risk factor for ocular melanoma, with an odds ratio of 2.05 (95% confidence interval, 1.20-3.51).11 Other reported eye damage from chronic UVR exposure includes cataracts, chronic conjunctivitis, and retinal damage.12,13
Case reports of the following photosensitive diseases have been reported to be exacerbated or caused by UV light exposure in welders: discoid lupus erythematosus14; photodermatitis15; broadband photosensitivity with decreased minimal erythema dose to UVA, UVB, and UVC16; UVC-exacerbated atopic dermatitis17; polymorphous light eruption–like skin eruption18; and UVA-induced photoallergy to hydrochlorothiazide and ramipril.19
Prevention of Occupational Exposure to UVR
Occupational Safety and Health Administration guidelines protect workers from excessive exposure to UVR with personal protective equipment (PPE). In addition to UVR protection, PPE needs to protect welders from other risks including trauma from welding debris (slag), fires, electrical burns, and fumes. Online resources from the National Ag Safety Database,20 the American Welding Society,21 and Occupational Safety and Health Administration22,23 are available. These resources advise welders to work in ventilated areas with respirators specific for the metal being welded and to wear clothing and gloves that are not only fire retardant but also UV resistant.20-23 Additional PPE should protect the head, face, and eyes.
Unfortunately, even workers well trained in prevention guidelines may not adequately protect themselves. Some welders forego PPE due to heat, thus exposing themselves to UVR damage in areas that are normally covered. Welders also may forego equipment when working on jobs requiring more detailed welds where clothing, masks, and glasses may be overly bulky and inhibit the worker’s precision. Nontraditional welders, such as artisans or handymen, may not have workplace safety education to be aware of UVR emitted from welding and may not have readily available PPE.
The Figure portrays an amateur welder working without full PPE. Although he is wearing a face shield, he is not wearing fire-retardant clothing, lacks full protective garments, and has no ventilation system.
Conclusion
It is important to recognize welding as an occupation with notable exposure to UVR. Personal protective equipment should be the mainstay of prevention. Sunscreen is a useful adjunct but does not cover UVC that is emitted in the welding arc. Screens and welding blankets can be placed around welders to contain UVR and limit nonwelder exposure. Although UVR hazards should be regulated in the workplace as part of regular safety reviews, the clinician can play a role in recognizing this source of UVR in skin disease and in encouraging the use of PPE.
- Okuno T, Ojima J, Saito H. Ultraviolet radiation emitted by CO(2) arc welding. Ann Occup Hyg. 2001;45:597-601.
- Peng CY, Liu HH, Chang CP, et al. Evaluation and monitoring of UVR in shield metal ARC welding processing. Health Phys. 2007;93:101-108.
- The National Institute for Occupational Safety and Health. Criteria for a recommended standard: occupational exposure to ultraviolet radiation. DHHS (NIOSH) publication 73-11009. https://www.cdc.gov/niosh/docs/73-11009/. Updated June 6, 2014. Accessed September 6, 2019.
- International Commission on Non-Ionizing Radiation Protection. Guidelines on limits of exposure to ultraviolet radiation of wavelengths between 180 nm and 400 nm (incoherent optical radiation). Health Phys. 2004;87:171-186.
- Peng CY, Lan CH, Juang YJ, et al. Exposure assessment of aluminum arc welding radiation. Health Phys. 2007;93:298-306.
- Tenkate TD, Collins MJ. Personal ultraviolet radiation exposure of workers in a welding environment. Am Ind Hyg Assoc J. 1997;58:33-38.
- Emmett EA, Buncher CR, Suskind RB, et al. Skin and eye diseases among arc welders and those exposed to welding operations. J Occup Med. 1981;23:85-90.
- Bruze M, Hindsén M, Trulsson L. Dermatitis with an unusual explanation in a welder. Acta Derm Venereol. 1994;74:380-382.
- Donoghue AM, Sinclair MJ. Basal cell carcinoma after frequent episodes of cutaneous erythema and peeling induced by welding. Occup Environ Med. 1999;56:646.
- Currie CL, Monk BE. Welding and non-melanoma skin cancer. Clin Exp Dermatol. 2000;25:28-29.
- Shah CP, Weis E, Lajous M, et al. Intermittent and chronic ultraviolet light exposure and uveal melanoma: a meta-analysis. Ophthalmology. 2005;112:1599-1607.
- Yang X, Shao D, Ding X, et al. Chronic phototoxic maculopathy caused by welding arc in occupational welders. Can J Ophthalmol. 2012;47:45-50.
- Davies KG, Asanga U, Nku CO, et al. Effect of chronic exposure to welding light on Calabar welders. Niger J Physiol Sci. 2007;22:55-58.
- Wozniak KD. Erythematodes chronicus discoides as an occupational disease in an electric welder [in German]. Berufs-Dermatosen. 1971;19:187-196.
- Shehade SA, Roberts PJ, Diffey BL, et al. Photodermatitis due to spot welding. Br J Dermatol. 1987;117:117-119.
- Roelandts R, Huys I. Broad-band and persistent photosensitivity following accidental ultraviolet C overexposure. Photodermatol Photoimmunol Photomed. 1993;9:144-146.
- Elsner P, Hassam S. Occupational UVC-induced exacerbation of atopic dermatitis in a welder. Contact Dermatitis. 1996;35:180-181.
- Majoie IM, van Weelden H, Sybesma IM, et al. Polymorphous light eruption-like skin lesions in welders caused by ultraviolet C light. J Am Acad Dermatol. 2010;62:150-151.
- Wagner SN, Welke F, Goos M. Occupational UVA-induced allergic photodermatitis in a welder due to hydrochlorothiazide and ramipril. Contact Dermatitis. 2000;43:245-246.
- Fluegel L, Rein BK. Arc welding safety. National Ag Safety Database website. http://nasdonline.org/1083/d000873/arc-welding-safety.html. Published May 1989. Accessed September 6, 2019.
- American Welding Society. Personal protective equipment (PPE) for welding and cutting. Fact sheet no. 33-04/14. http://www.aws.org/technical/facts/FACT-33_2014.pdf. Published April 2014. Accessed September 6, 2019.
- Occupational Safety and Health Administration. Eye protection against radiant energy during welding and cutting in shipyard employment. https://www.osha.gov/Publications/OSHAfactsheet-eyeprotection-during-welding.pdf. Published January 2012. Accessed September 6, 2019.
- Occupational Safety and Health Administration. Welding, cutting, and brazing. https://www.osha.gov/SLTC/weldingcuttingbrazing/standards.html. Accessed September 10, 2019.
- Okuno T, Ojima J, Saito H. Ultraviolet radiation emitted by CO(2) arc welding. Ann Occup Hyg. 2001;45:597-601.
- Peng CY, Liu HH, Chang CP, et al. Evaluation and monitoring of UVR in shield metal ARC welding processing. Health Phys. 2007;93:101-108.
- The National Institute for Occupational Safety and Health. Criteria for a recommended standard: occupational exposure to ultraviolet radiation. DHHS (NIOSH) publication 73-11009. https://www.cdc.gov/niosh/docs/73-11009/. Updated June 6, 2014. Accessed September 6, 2019.
- International Commission on Non-Ionizing Radiation Protection. Guidelines on limits of exposure to ultraviolet radiation of wavelengths between 180 nm and 400 nm (incoherent optical radiation). Health Phys. 2004;87:171-186.
- Peng CY, Lan CH, Juang YJ, et al. Exposure assessment of aluminum arc welding radiation. Health Phys. 2007;93:298-306.
- Tenkate TD, Collins MJ. Personal ultraviolet radiation exposure of workers in a welding environment. Am Ind Hyg Assoc J. 1997;58:33-38.
- Emmett EA, Buncher CR, Suskind RB, et al. Skin and eye diseases among arc welders and those exposed to welding operations. J Occup Med. 1981;23:85-90.
- Bruze M, Hindsén M, Trulsson L. Dermatitis with an unusual explanation in a welder. Acta Derm Venereol. 1994;74:380-382.
- Donoghue AM, Sinclair MJ. Basal cell carcinoma after frequent episodes of cutaneous erythema and peeling induced by welding. Occup Environ Med. 1999;56:646.
- Currie CL, Monk BE. Welding and non-melanoma skin cancer. Clin Exp Dermatol. 2000;25:28-29.
- Shah CP, Weis E, Lajous M, et al. Intermittent and chronic ultraviolet light exposure and uveal melanoma: a meta-analysis. Ophthalmology. 2005;112:1599-1607.
- Yang X, Shao D, Ding X, et al. Chronic phototoxic maculopathy caused by welding arc in occupational welders. Can J Ophthalmol. 2012;47:45-50.
- Davies KG, Asanga U, Nku CO, et al. Effect of chronic exposure to welding light on Calabar welders. Niger J Physiol Sci. 2007;22:55-58.
- Wozniak KD. Erythematodes chronicus discoides as an occupational disease in an electric welder [in German]. Berufs-Dermatosen. 1971;19:187-196.
- Shehade SA, Roberts PJ, Diffey BL, et al. Photodermatitis due to spot welding. Br J Dermatol. 1987;117:117-119.
- Roelandts R, Huys I. Broad-band and persistent photosensitivity following accidental ultraviolet C overexposure. Photodermatol Photoimmunol Photomed. 1993;9:144-146.
- Elsner P, Hassam S. Occupational UVC-induced exacerbation of atopic dermatitis in a welder. Contact Dermatitis. 1996;35:180-181.
- Majoie IM, van Weelden H, Sybesma IM, et al. Polymorphous light eruption-like skin lesions in welders caused by ultraviolet C light. J Am Acad Dermatol. 2010;62:150-151.
- Wagner SN, Welke F, Goos M. Occupational UVA-induced allergic photodermatitis in a welder due to hydrochlorothiazide and ramipril. Contact Dermatitis. 2000;43:245-246.
- Fluegel L, Rein BK. Arc welding safety. National Ag Safety Database website. http://nasdonline.org/1083/d000873/arc-welding-safety.html. Published May 1989. Accessed September 6, 2019.
- American Welding Society. Personal protective equipment (PPE) for welding and cutting. Fact sheet no. 33-04/14. http://www.aws.org/technical/facts/FACT-33_2014.pdf. Published April 2014. Accessed September 6, 2019.
- Occupational Safety and Health Administration. Eye protection against radiant energy during welding and cutting in shipyard employment. https://www.osha.gov/Publications/OSHAfactsheet-eyeprotection-during-welding.pdf. Published January 2012. Accessed September 6, 2019.
- Occupational Safety and Health Administration. Welding, cutting, and brazing. https://www.osha.gov/SLTC/weldingcuttingbrazing/standards.html. Accessed September 10, 2019.
Practice Points
- Arc welding can be a major source of UV radiation exposure.
- Welders should be advised to work with proper ventilation and with welding masks, clothing, and gloves that not only are fire retardant but also are UV resistant.

Role of the Nervous System in Psoriasis
1. Amanat M, Salehi M, Rezaei N. Neurological and psychiatric disorders in psoriasis. Rev Neurosci. 2018;29:805-813.
2. Eberle FC, Brück J, Holstein J, et al. Recent advances in understanding psoriasis [published April 28, 2016]. F1000Res. doi:10.12688/f1000research.7927.1.
3. Lee EB, Reynolds KA, Pithadia DJ, et al. Clearance of psoriasis after ischemic stroke. Cutis. 2019;103:74-76.
4. Zhu TH, Nakamura M, Farahnik B, et al. The role of the nervous system in the pathophysiology of psoriasis: a review of cases of psoriasis remission or improvement following denervation injury. Am J Clin Dermatol. 2016;17:257-263.
5. Raychaudhuri SP, Farber EM. Neuroimmunologic aspects of psoriasis. Cutis. 2000;66:357-362.
6. Kwon CW, Fried RG, Nousari Y, et al. Psoriasis: psychosomatic, somatopsychic, or both? Clin Dermatol. 2018;36:698-703.
7. Lotti T, D’Erme AM, Hercogová J. The role of neuropeptides in the control of regional immunity. Clin Dermatol. 2014;32:633-645.
8. Hall JM, Cruser D, Podawiltz A, et al. Psychological stress and the cutaneous immune response: roles of the HPA axis and the sympathetic nervous system in atopic dermatitis and psoriasis [published online August 30, 2012]. Dermatol Res Pract. 2012;2012:403908.
9. Raychaudhuri SK, Raychaudhuri SP. NGF and its receptor system: a new dimension in the pathogenesis of psoriasis and psoriatic arthritis. Ann N Y Acad Sci. 2009;1173:470-477.
10. Glaser R, Kiecolt-Glaser JK. Stress-induced immune dysfunction: implications for health. Nat Rev Immunol. 2005;5:243-251.
11. Levi-Montalcini R, Skaper SD, Dal Toso R, et al. Nerve growth factor: from neurotrophin to neurokine. Trends Neurosci. 1996;19:514-520.
12. Harvima IT, Viinamäki H, Naukkarinen A, et al. Association of cutaneous mast cells and sensory nerves with psychic stress in psoriasis. Psychother Psychosom. 1993;60:168-176.
13. He Y, Ding G, Wang X, et al. Calcitonin gene‐related peptide in Langerhans cells in psoriatic plaque lesions. Chin Med J (Engl). 2000;113:747-751.
14. Chu DQ, Choy M, Foster P, et al. A comparative study of the ability of calcitonin gene‐related peptide and adrenomedullin13–52 to modulate microvascular but not thermal hyperalgesia responses. Br J Pharmacol. 2000;130:1589-1596.
15. Al’Abadie MS, Senior HJ, Bleehen SS, et al. Neuropeptides and general neuronal marker in psoriasis—an immunohistochemical study. Clin Exp Dermatol. 1995;20:384-389.
16. Farber EM, Nickoloff BJ, Recht B, et al. Stress, symmetry, and psoriasis: possible role of neuropeptides. J Am Acad Dermatol. 1986;14(2, pt 1):305-311.
17. Pincelli C, Fantini F, Romualdi P, et al. Substance P is diminished and vasoactive intestinal peptide is augmented in psoriatic lesions and these peptides exert disparate effects on the proliferation of cultured human keratinocytes. J Invest Dermatol. 1992;98:421-427.
18. Raychaudhuri SP, Jiang WY, Farber EM. Psoriatic keratinocytes express high levels of nerve growth factor. Acta Derm Venereol. 1998;78:84-86.
19. Pincelli C. Nerve growth factor and keratinocytes: a role in psoriasis. Eur J Dermatol. 2000;10:85-90.
20. Sagi L, Trau H. The Koebner phenomenon. Clin Dermatol. 2011;29:231-236.
21. Nakamura M, Toyoda M, Morohashi M. Pruritogenic mediators in psoriasis vulgaris: comparative evaluation of itch-associated cutaneous factors. Br J Dermatol. 2003;149:718-730.
22. Stratigos AJ, Katoulis AK, Stavrianeas NG. Spontaneous clearing of psoriasis after stroke. J Am Acad Dermatol. 1998;38(5, pt 1):768-770.
23. Wang TS, Tsai TF. Psoriasis sparing the lower limb with postpoliomyelitis residual paralysis. Br J Dermatol. 2014;171:429-431.
24. Weiner SR, Bassett LW, Reichman RP. Protective effect of poliomyelitis on psoriatic arthritis. Arthritis Rheum. 1985;28:703-706.
25. Ostrowski SM, Belkai A, Loyd CM, et al. Cutaneous denervation of psoriasiform mouse skin improves acanthosis and inflammation in a sensory neuropeptide-dependent manner. J Invest Dermatol. 2011;131:1530-1538.
26. Farber EM, Lanigan SW, Boer J. The role of cutaneous sensory nerves in the maintenance of psoriasis. Int J Dermatol. 1990;29:418-420.
27. Dewing SB. Remission of psoriasis associated with cutaneous nerve section. Arch Dermatol. 1971;104:220-221.
28. Perlman HH. Remission of psoriasis vulgaris from the use of nerve-blocking agents. Arch Dermatol. 1972;105:128-129.
1. Amanat M, Salehi M, Rezaei N. Neurological and psychiatric disorders in psoriasis. Rev Neurosci. 2018;29:805-813.
2. Eberle FC, Brück J, Holstein J, et al. Recent advances in understanding psoriasis [published April 28, 2016]. F1000Res. doi:10.12688/f1000research.7927.1.
3. Lee EB, Reynolds KA, Pithadia DJ, et al. Clearance of psoriasis after ischemic stroke. Cutis. 2019;103:74-76.
4. Zhu TH, Nakamura M, Farahnik B, et al. The role of the nervous system in the pathophysiology of psoriasis: a review of cases of psoriasis remission or improvement following denervation injury. Am J Clin Dermatol. 2016;17:257-263.
5. Raychaudhuri SP, Farber EM. Neuroimmunologic aspects of psoriasis. Cutis. 2000;66:357-362.
6. Kwon CW, Fried RG, Nousari Y, et al. Psoriasis: psychosomatic, somatopsychic, or both? Clin Dermatol. 2018;36:698-703.
7. Lotti T, D’Erme AM, Hercogová J. The role of neuropeptides in the control of regional immunity. Clin Dermatol. 2014;32:633-645.
8. Hall JM, Cruser D, Podawiltz A, et al. Psychological stress and the cutaneous immune response: roles of the HPA axis and the sympathetic nervous system in atopic dermatitis and psoriasis [published online August 30, 2012]. Dermatol Res Pract. 2012;2012:403908.
9. Raychaudhuri SK, Raychaudhuri SP. NGF and its receptor system: a new dimension in the pathogenesis of psoriasis and psoriatic arthritis. Ann N Y Acad Sci. 2009;1173:470-477.
10. Glaser R, Kiecolt-Glaser JK. Stress-induced immune dysfunction: implications for health. Nat Rev Immunol. 2005;5:243-251.
11. Levi-Montalcini R, Skaper SD, Dal Toso R, et al. Nerve growth factor: from neurotrophin to neurokine. Trends Neurosci. 1996;19:514-520.
12. Harvima IT, Viinamäki H, Naukkarinen A, et al. Association of cutaneous mast cells and sensory nerves with psychic stress in psoriasis. Psychother Psychosom. 1993;60:168-176.
13. He Y, Ding G, Wang X, et al. Calcitonin gene‐related peptide in Langerhans cells in psoriatic plaque lesions. Chin Med J (Engl). 2000;113:747-751.
14. Chu DQ, Choy M, Foster P, et al. A comparative study of the ability of calcitonin gene‐related peptide and adrenomedullin13–52 to modulate microvascular but not thermal hyperalgesia responses. Br J Pharmacol. 2000;130:1589-1596.
15. Al’Abadie MS, Senior HJ, Bleehen SS, et al. Neuropeptides and general neuronal marker in psoriasis—an immunohistochemical study. Clin Exp Dermatol. 1995;20:384-389.
16. Farber EM, Nickoloff BJ, Recht B, et al. Stress, symmetry, and psoriasis: possible role of neuropeptides. J Am Acad Dermatol. 1986;14(2, pt 1):305-311.
17. Pincelli C, Fantini F, Romualdi P, et al. Substance P is diminished and vasoactive intestinal peptide is augmented in psoriatic lesions and these peptides exert disparate effects on the proliferation of cultured human keratinocytes. J Invest Dermatol. 1992;98:421-427.
18. Raychaudhuri SP, Jiang WY, Farber EM. Psoriatic keratinocytes express high levels of nerve growth factor. Acta Derm Venereol. 1998;78:84-86.
19. Pincelli C. Nerve growth factor and keratinocytes: a role in psoriasis. Eur J Dermatol. 2000;10:85-90.
20. Sagi L, Trau H. The Koebner phenomenon. Clin Dermatol. 2011;29:231-236.
21. Nakamura M, Toyoda M, Morohashi M. Pruritogenic mediators in psoriasis vulgaris: comparative evaluation of itch-associated cutaneous factors. Br J Dermatol. 2003;149:718-730.
22. Stratigos AJ, Katoulis AK, Stavrianeas NG. Spontaneous clearing of psoriasis after stroke. J Am Acad Dermatol. 1998;38(5, pt 1):768-770.
23. Wang TS, Tsai TF. Psoriasis sparing the lower limb with postpoliomyelitis residual paralysis. Br J Dermatol. 2014;171:429-431.
24. Weiner SR, Bassett LW, Reichman RP. Protective effect of poliomyelitis on psoriatic arthritis. Arthritis Rheum. 1985;28:703-706.
25. Ostrowski SM, Belkai A, Loyd CM, et al. Cutaneous denervation of psoriasiform mouse skin improves acanthosis and inflammation in a sensory neuropeptide-dependent manner. J Invest Dermatol. 2011;131:1530-1538.
26. Farber EM, Lanigan SW, Boer J. The role of cutaneous sensory nerves in the maintenance of psoriasis. Int J Dermatol. 1990;29:418-420.
27. Dewing SB. Remission of psoriasis associated with cutaneous nerve section. Arch Dermatol. 1971;104:220-221.
28. Perlman HH. Remission of psoriasis vulgaris from the use of nerve-blocking agents. Arch Dermatol. 1972;105:128-129.
1. Amanat M, Salehi M, Rezaei N. Neurological and psychiatric disorders in psoriasis. Rev Neurosci. 2018;29:805-813.
2. Eberle FC, Brück J, Holstein J, et al. Recent advances in understanding psoriasis [published April 28, 2016]. F1000Res. doi:10.12688/f1000research.7927.1.
3. Lee EB, Reynolds KA, Pithadia DJ, et al. Clearance of psoriasis after ischemic stroke. Cutis. 2019;103:74-76.
4. Zhu TH, Nakamura M, Farahnik B, et al. The role of the nervous system in the pathophysiology of psoriasis: a review of cases of psoriasis remission or improvement following denervation injury. Am J Clin Dermatol. 2016;17:257-263.
5. Raychaudhuri SP, Farber EM. Neuroimmunologic aspects of psoriasis. Cutis. 2000;66:357-362.
6. Kwon CW, Fried RG, Nousari Y, et al. Psoriasis: psychosomatic, somatopsychic, or both? Clin Dermatol. 2018;36:698-703.
7. Lotti T, D’Erme AM, Hercogová J. The role of neuropeptides in the control of regional immunity. Clin Dermatol. 2014;32:633-645.
8. Hall JM, Cruser D, Podawiltz A, et al. Psychological stress and the cutaneous immune response: roles of the HPA axis and the sympathetic nervous system in atopic dermatitis and psoriasis [published online August 30, 2012]. Dermatol Res Pract. 2012;2012:403908.
9. Raychaudhuri SK, Raychaudhuri SP. NGF and its receptor system: a new dimension in the pathogenesis of psoriasis and psoriatic arthritis. Ann N Y Acad Sci. 2009;1173:470-477.
10. Glaser R, Kiecolt-Glaser JK. Stress-induced immune dysfunction: implications for health. Nat Rev Immunol. 2005;5:243-251.
11. Levi-Montalcini R, Skaper SD, Dal Toso R, et al. Nerve growth factor: from neurotrophin to neurokine. Trends Neurosci. 1996;19:514-520.
12. Harvima IT, Viinamäki H, Naukkarinen A, et al. Association of cutaneous mast cells and sensory nerves with psychic stress in psoriasis. Psychother Psychosom. 1993;60:168-176.
13. He Y, Ding G, Wang X, et al. Calcitonin gene‐related peptide in Langerhans cells in psoriatic plaque lesions. Chin Med J (Engl). 2000;113:747-751.
14. Chu DQ, Choy M, Foster P, et al. A comparative study of the ability of calcitonin gene‐related peptide and adrenomedullin13–52 to modulate microvascular but not thermal hyperalgesia responses. Br J Pharmacol. 2000;130:1589-1596.
15. Al’Abadie MS, Senior HJ, Bleehen SS, et al. Neuropeptides and general neuronal marker in psoriasis—an immunohistochemical study. Clin Exp Dermatol. 1995;20:384-389.
16. Farber EM, Nickoloff BJ, Recht B, et al. Stress, symmetry, and psoriasis: possible role of neuropeptides. J Am Acad Dermatol. 1986;14(2, pt 1):305-311.
17. Pincelli C, Fantini F, Romualdi P, et al. Substance P is diminished and vasoactive intestinal peptide is augmented in psoriatic lesions and these peptides exert disparate effects on the proliferation of cultured human keratinocytes. J Invest Dermatol. 1992;98:421-427.
18. Raychaudhuri SP, Jiang WY, Farber EM. Psoriatic keratinocytes express high levels of nerve growth factor. Acta Derm Venereol. 1998;78:84-86.
19. Pincelli C. Nerve growth factor and keratinocytes: a role in psoriasis. Eur J Dermatol. 2000;10:85-90.
20. Sagi L, Trau H. The Koebner phenomenon. Clin Dermatol. 2011;29:231-236.
21. Nakamura M, Toyoda M, Morohashi M. Pruritogenic mediators in psoriasis vulgaris: comparative evaluation of itch-associated cutaneous factors. Br J Dermatol. 2003;149:718-730.
22. Stratigos AJ, Katoulis AK, Stavrianeas NG. Spontaneous clearing of psoriasis after stroke. J Am Acad Dermatol. 1998;38(5, pt 1):768-770.
23. Wang TS, Tsai TF. Psoriasis sparing the lower limb with postpoliomyelitis residual paralysis. Br J Dermatol. 2014;171:429-431.
24. Weiner SR, Bassett LW, Reichman RP. Protective effect of poliomyelitis on psoriatic arthritis. Arthritis Rheum. 1985;28:703-706.
25. Ostrowski SM, Belkai A, Loyd CM, et al. Cutaneous denervation of psoriasiform mouse skin improves acanthosis and inflammation in a sensory neuropeptide-dependent manner. J Invest Dermatol. 2011;131:1530-1538.
26. Farber EM, Lanigan SW, Boer J. The role of cutaneous sensory nerves in the maintenance of psoriasis. Int J Dermatol. 1990;29:418-420.
27. Dewing SB. Remission of psoriasis associated with cutaneous nerve section. Arch Dermatol. 1971;104:220-221.
28. Perlman HH. Remission of psoriasis vulgaris from the use of nerve-blocking agents. Arch Dermatol. 1972;105:128-129.
Mycobacterium haemophilum: A Challenging Treatment Dilemma in an Immunocompromised Patient
To the Editor:
The increase in nontuberculous mycobacteria (NTM) infections over the last 3 decades likely is multifaceted, including increased clinical awareness, improved laboratory diagnostics, growing numbers of immunocompromised patients, and an aging population.1,2 Historically, the majority of mycobacteria-related diseases are due to Mycobacterium tuberculosis, Mycobacterium bovis, and Mycobacterium leprae.3
Mycobacterium haemophilum is a slow-growing acid-fast bacillus (AFB) that differs from other Mycobacterium species in that it requires iron-supplemented media and incubation temperatures of 30°C to 32°C for culture. As these requirements for growth are not standard for AFB cultures, M haemophilum infection may be underrecognized and underreported.3Mycobacterium haemophilum infections largely are cutaneous and generally are seen in AIDS patients and bone marrow transplant recipients who are iatrogenically immunosuppressed.4,5 No species-specific treatment guidelines exist2; however, triple-drug therapy combining a macrolide, rifamycin, and a quinolone for a minimum of 12 months often is recommended.
A 64-year-old man with a history of coronary artery disease, hypertension, hyperlipidemia, and acute myelogenous leukemia (AML) underwent allogenic stem cell transplantation. His posttransplant course was complicated by multiple deep vein thromboses, hypogammaglobulinemia, and graft-vs-host disease (GVHD) of the skin and gastrointestinal tract that manifested as chronic diarrhea, which was managed with chronic prednisone. Thirteen months after the transplant, the patient presented to his outpatient oncologist (M.K.) for evaluation of painless, nonpruritic, erythematous papules and nodules that had emerged on the right side of the chest, right arm, and left leg of approximately 2 weeks’ duration.
On review of systems by oncology, the patient denied any fevers, chills, or night sweats but noted chronic loose nonbloody stools without abdominal pain, likely related to the GVHD. The patient’s medications included prednisone 20 mg once daily, fluconazole, amitriptyline, atovaquone, budesonide, dabigatran, metoprolol, pantoprazole, rosuvastatin, senna glycoside, spironolactone, tramadol, and valacyclovir.
Physical examination revealed multiple singular erythematous nodules on the right side of the chest (Figure 1A), right arm (Figure 1B), and left leg. There was no regional lymphadenopathy. The patient was afebrile and hemodynamically stable. A biopsy of the arm performed to rule out leukemia cutis revealed a granulomatous dermatitis with numerous AFB (Figures 2A and 2B), which were confirmed on Ziehl-Neelsen staining (Figures 2C and 2D). The presence of AFB raised concern for a disseminated mycobacterial infection. The patient was admitted to our institution approximately 1 week after the outpatient biopsy was performed. He was evaluated by infectious diseases (B.H.) and was recommended for repeat biopsy with AFB culture and for initiation of intravenous antibiotics.
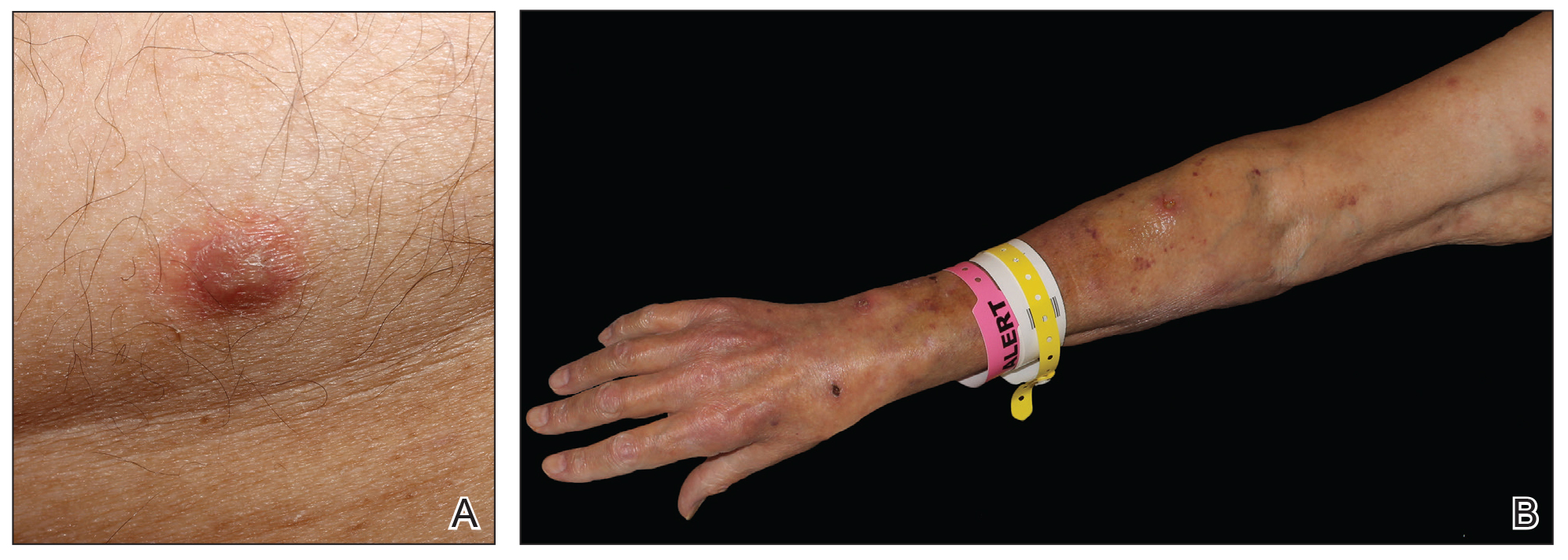

The patient was evaluated by the dermatology consultation service on hospital day 1. At the time of consultation, the lesions were still painless but had enlarged. Two new satellite lesions were noted on his other extremities. Due to the widespread distribution of the lesions, there was concern for disseminated disease. The relatively rapid onset of new lesions increased concern for infection with rapid-growing mycobacteria, including Mycobacterium abscessus, Mycobacterium fortuitum, and Mycobacterium chelonae. A detailed history revealed that the patient’s wife had a fish tank, which supported the inclusion of Mycobacterium marinum in the differential; however, further questioning revealed that the patient never came in contact with the aquarium water. The initial outpatient biopsy had not been sent for culture. Following inpatient biopsy, the patient was initiated on empiric antimycobacterials, including imipenem, amikacin, clarithromycin, and levofloxacin. Computed tomography of the head was negative for cerebral involvement.
Acid-fast bacilli blood cultures were drawn per the recommendation from infectious diseases in an attempt to confirm disseminated disease; however, blood cultures remained negative. Tissue biopsy from the right arm was sent for AFB staining and culture. Many AFB were identified on microscopy, and growth was observed in the mycobacterial growth indicator tube after 6 days of incubation. The DNA probe was negative for M tuberculosis complex or Mycobacterium avium complex.
The patient was discharged on hospital day 6 on empiric therapy for rapid-growing mycobacteria while cultures were pending. The empiric regimen included intravenous imipenem 1 g every 6 hours, intravenous amikacin 1 g once daily, clarithromycin 500 mg every 12 hours, and levofloxacin 750 mg once daily. All solid media cultures were negative at the time of discharge.
The biopsy specimen proved difficult to culture on solid media using traditional methods. Three weeks after the inpatient biopsy, the microbiology laboratory reported that growth was observed on solid media that was incubated at 30°C and supplemented with iron. These findings were not characteristic of a rapidly growing mycobacteria (eg, M fortuitum, M chelonae, M abscessus) or M marinum but raised concern for infectionwith M haemophilum. Antimycobacterial treatment was adjusted to amikacin, clarithromycin, levofloxacin, and rifabutin.
Six weeks after the inpatient skin biopsy, final speciation confirmed infection with M haemophilum. The isolate proved susceptible to amikacin (minimal inhibitory concentration [MIC], 16), clarithromycin (MIC, 0.12), linezolid (MIC, <1), moxifloxacin (MIC, 0.5), rifabutin (MIC, <0.25), and trimethoprim-sulfamethoxazole (MIC, 0.5/9.5). The isolate was resistant to ciprofloxacin (MIC, 4), ethambutol (MIC, >16), and rifampin (MIC, 2). Based on these findings, an infectious disease specialist modified the treatment regimen to azithromycin 600 mg once daily, moxifloxacin 400 mg once daily, and rifabutin 300 mg once daily. Azithromycin was substituted for clarithromycin in an attempt to minimize the gastrointestinal side effects of the antibiotics. The infectious disease specialist was concerned that the clarithromycin could exacerbate the patient’s chronic GVHD-associated diarrhea, which posed a challenge to the oncologist, who was attempting to manage the patient’s GVHD and minimize the use of additional prednisone. At the time of this change, the patient was doing well clinically and denied any active skin lesions.
Four months later, he developed new left-sided neck swelling. Computed tomography revealed nonspecific enhancement involving the skin and superficial subcutaneous tissues in the left anterior neck. He was referred to otolaryngology given concern for recurrent infection vs leukemia cutis. He underwent excisional biopsy. Pathology was negative for malignancy but demonstrated subcutaneous necrotizing granulomatous inflammation with a positive AFB stain. Tissue AFB cultures revealed moderate AFB on direct stain, but there was no AFB growth at 12 weeks. Clarithromycin was restarted in place of azithromycin to increase the potency of the antimycobacterial regimen. Cultures from this neck biopsy were negative after 12 weeks of incubation.
In addition to this change in antibiotic coverage, the patient’s medical oncologist tapered the patient’s immunosuppression considerably. The patient subsequently completed 12 months of therapy with clarithromycin, moxifloxacin, and rifabutin starting from the time of the neck biopsy. He remained free of recurrence of mycobacterial infection for nearly 2 years until he died from an unrelated illness.
Nontuberculous mycobacteria are an ubiquitous environmental group.2 Sources include soil and natural water (M avium), fish tanks and swimming pools (M marinum), and tap water and occasionally domestic animals (Mycobacterium kansasii). Additionally, rapidly growing NTM such as M abscessus, M chelonae, and M fortuitum have been isolated from soil and natural water supplies.3
Mycobacterium haemophilum is a fastidious organism with a predilection for skin of the chest and extremities. Iatrogenically or inherently immunocompromised patients are most commonly affected6-11; however, there also have been reports in healthy patients.12,13 Infections typically present as painless erythematous papules or nodules that eventually suppurate, ulcerate, and become painful. Presentations involving Fitz-Hugh–Curtis syndrome,13 new B-cell lymphoma,10 and lymphadenitis12 also have been described. Beyond cutaneous involvement, M haemophilum has been cultured from bone, the synovium, the lungs, and the central nervous system.4,9 The majority of morbidities occur in patients with lung involvement.4 Therefore, even patients presenting with isolated cutaneous disease require close follow-up.
Mycobacterium haemophilum is a slowly proliferating organism that is unable to grow in standard egg-potato (Lowenstein-Jensen) medium or agar base (Middlebrook 7H10 or 7H11 agar) without iron supplementation (ferric ammonium citrate, hemin, or hemoglobin). It also requires temperatures of 30°C to 32°C for growth. Its iron requisite is unique, but species such as M marinum and Mycobacterium ulcerans also share reduced temperature requirements. Without a high index of suspicion, growth often is absent because standard Mycobacterium culture techniques will not foster organism growth. Our case demonstrated that special culture instructions must be relayed to the laboratory, even in the face of positive AFB smears. Failure to request hemin and modified incubation temperatures may have contributed to the negative AFB blood culture in our patient.
Due to the relatively rare incidence of M haemophilum infection, there are no known randomized controlled trials guiding antibiotic regimens. Infectious disease specialists often treat empirically with triple-drug therapy derived from locally reported species susceptibilities. The largest case series to date did not identify resistance to amikacin, ciprofloxacin, or clarithromycin.4 Our case identified a novel finding of ciprofloxacin and rifampin resistance, which may highlight the emergence of a newly resistant strain of M haemophilum. Of note, one case of rifampin resistance has been reported, but the culture was drawn from a postmortem specimen in the setting of previously rifampin-sensitive isolates.4 Empiric therapies should be guided by hospital susceptibility reports and expert consultation.
Coinfection with 2 or more NTM—including M tuberculosis, M leprae, and M fortuitum—has been reported.8,14 Temporally distinct coinfections with M leprae and M haemophilum also have been described.15 Thus, practitioners should have a low threshold for repeat cultures in the context of new cutaneous nodules or granulomas, not only to detect concomitant infections but also to identify resistance patterns that might explain recurrent or recalcitrant disease. Immune reconstitution inflammatory syndrome also must be considered with new or worsening lesions, especially in the first months of therapy, as this is a common occurrence when immunosuppressive regimens are tapered to help manage infections.
In conclusion, M haemophilum is an underrecognized infection that presents as cutaneous nodules or lymphadenitis in immunocompromised or healthy individuals. Diagnosis requires a high index of suspicion because its unique growth requirements necessitate special laboratory techniques. Our case represents a classic presentation of this NTM infection in a patient with AML following allogenic stem cell transplantation. Repeat cultures, workup of potentially disseminated infections, and close follow-up are requisite to minimizing morbidity and mortality. A multidisciplinary approach involving infectious disease, medical oncology, radiology, and dermatology best manages this type of infection.
- Sheu LC, Tran TM, Jarlsberg LG, et al. Non-tuberculous mycobacterial infections at San Francisco General Hospital. Clin Respir J. 2015;9:436-442.
- Knoll BM. Update on nontuberculous mycobacterial infections in solid organ and hematopoietic stem cell transplant recipients. Curr Infect Dis Rep. 2014;16:421.
- Diagnosis and treatment of disease caused by nontuberculous mycobacteria. this official statement of the American Thoracic Society was approved by the Board of Directors, March 1997. Medical Section of the American Lung Association. Am J Respir Crit Care Med. 1997;156(2 pt 2):S1-S25.
- Shah MK, Sebti A, Kiehn TE, et al. Mycobacterium haemophilum in immunocompromised patients. Clin Infect Dis. 2001;33:330-337.
- Griffiths DE, Aksamit T, Brown-Elliott BA. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. 2007;175:367-416.
- Copeland NK, Arora NS, Ferguson TM. Mycobacterium haemophilum masquerading as leprosy in a renal transplant patient [published online November 28, 2013]. Case Rep Dermatol Med. 2013;2013:793127.
- Aslam A, Green RL, Motta L, et al. Cutaneous Mycobacterium haemophilum infection in a patient receiving infliximab for psoriasis. Br J Dermatol. 2013;168:446-447.
- Agrawal S, Sharma A. Dual mycobacterial infection in the setting of leflunomide treatment for rheumatoid arthritis. Ann Rheum Dis. 2007;66:277.
- Buppajarntham A, Apisarnthanarak A, Rutjanawech S, et al. Central nervous system infection due to Mycobacterium haemophilum in a patient with acquired immunodeficiency syndrome. Int J STD AIDS. 2015;26:288-290.
- Doherty T, Lynn M, Cavazza A, et al. Mycobacterium haemophilum as the initial presentation of a B-cell lymphoma in a liver transplant patient [published online January 12, 2014]. Case Rep Rheumatol. 2014;2014:742978.
- Ducharlet K, Murphy C, Tan SJ, et al. Recurrent Mycobacterium haemophilum in a renal transplant recipient. Nephrology (Carlton). 2014;(19 suppl 1):14-17.
- Dawson DJ, Blacklock ZM, Kane DW. Mycobacterium haemophilum causing lymphadenitis in an otherwise healthy child. Med J Aust. 1981;2:289-290.
- Jang HY, Burbelo PD, Chae YS, et al. Nontuberculous mycobacterial infection in a clinical presentation of Fitz-Hugh-Curtis syndrome: a case report with multigene diagnostic approach. BMC Womens Health. 2014;14:95.
- Scollard DM, Stryjewska BM, Prestigiacomo JF, et al. Hansen’s disease (leprosy) complicated by secondary mycobacterial infection. J Am Acad Dermatol. 2011;64:593-596.
- SoRelle JA, Beal SG, Scollard DM, et al. Mycobacterium leprae and Mycobacterium haemophilum co-infection in an iatrogenically immunosuppressed patient. Diagn Microbiol Infect Dis. 2014;78:494-496.
To the Editor:
The increase in nontuberculous mycobacteria (NTM) infections over the last 3 decades likely is multifaceted, including increased clinical awareness, improved laboratory diagnostics, growing numbers of immunocompromised patients, and an aging population.1,2 Historically, the majority of mycobacteria-related diseases are due to Mycobacterium tuberculosis, Mycobacterium bovis, and Mycobacterium leprae.3
Mycobacterium haemophilum is a slow-growing acid-fast bacillus (AFB) that differs from other Mycobacterium species in that it requires iron-supplemented media and incubation temperatures of 30°C to 32°C for culture. As these requirements for growth are not standard for AFB cultures, M haemophilum infection may be underrecognized and underreported.3Mycobacterium haemophilum infections largely are cutaneous and generally are seen in AIDS patients and bone marrow transplant recipients who are iatrogenically immunosuppressed.4,5 No species-specific treatment guidelines exist2; however, triple-drug therapy combining a macrolide, rifamycin, and a quinolone for a minimum of 12 months often is recommended.
A 64-year-old man with a history of coronary artery disease, hypertension, hyperlipidemia, and acute myelogenous leukemia (AML) underwent allogenic stem cell transplantation. His posttransplant course was complicated by multiple deep vein thromboses, hypogammaglobulinemia, and graft-vs-host disease (GVHD) of the skin and gastrointestinal tract that manifested as chronic diarrhea, which was managed with chronic prednisone. Thirteen months after the transplant, the patient presented to his outpatient oncologist (M.K.) for evaluation of painless, nonpruritic, erythematous papules and nodules that had emerged on the right side of the chest, right arm, and left leg of approximately 2 weeks’ duration.
On review of systems by oncology, the patient denied any fevers, chills, or night sweats but noted chronic loose nonbloody stools without abdominal pain, likely related to the GVHD. The patient’s medications included prednisone 20 mg once daily, fluconazole, amitriptyline, atovaquone, budesonide, dabigatran, metoprolol, pantoprazole, rosuvastatin, senna glycoside, spironolactone, tramadol, and valacyclovir.
Physical examination revealed multiple singular erythematous nodules on the right side of the chest (Figure 1A), right arm (Figure 1B), and left leg. There was no regional lymphadenopathy. The patient was afebrile and hemodynamically stable. A biopsy of the arm performed to rule out leukemia cutis revealed a granulomatous dermatitis with numerous AFB (Figures 2A and 2B), which were confirmed on Ziehl-Neelsen staining (Figures 2C and 2D). The presence of AFB raised concern for a disseminated mycobacterial infection. The patient was admitted to our institution approximately 1 week after the outpatient biopsy was performed. He was evaluated by infectious diseases (B.H.) and was recommended for repeat biopsy with AFB culture and for initiation of intravenous antibiotics.
The patient was evaluated by the dermatology consultation service on hospital day 1. At the time of consultation, the lesions were still painless but had enlarged. Two new satellite lesions were noted on his other extremities. Due to the widespread distribution of the lesions, there was concern for disseminated disease. The relatively rapid onset of new lesions increased concern for infection with rapid-growing mycobacteria, including Mycobacterium abscessus, Mycobacterium fortuitum, and Mycobacterium chelonae. A detailed history revealed that the patient’s wife had a fish tank, which supported the inclusion of Mycobacterium marinum in the differential; however, further questioning revealed that the patient never came in contact with the aquarium water. The initial outpatient biopsy had not been sent for culture. Following inpatient biopsy, the patient was initiated on empiric antimycobacterials, including imipenem, amikacin, clarithromycin, and levofloxacin. Computed tomography of the head was negative for cerebral involvement.
Acid-fast bacilli blood cultures were drawn per the recommendation from infectious diseases in an attempt to confirm disseminated disease; however, blood cultures remained negative. Tissue biopsy from the right arm was sent for AFB staining and culture. Many AFB were identified on microscopy, and growth was observed in the mycobacterial growth indicator tube after 6 days of incubation. The DNA probe was negative for M tuberculosis complex or Mycobacterium avium complex.
The patient was discharged on hospital day 6 on empiric therapy for rapid-growing mycobacteria while cultures were pending. The empiric regimen included intravenous imipenem 1 g every 6 hours, intravenous amikacin 1 g once daily, clarithromycin 500 mg every 12 hours, and levofloxacin 750 mg once daily. All solid media cultures were negative at the time of discharge.
The biopsy specimen proved difficult to culture on solid media using traditional methods. Three weeks after the inpatient biopsy, the microbiology laboratory reported that growth was observed on solid media that was incubated at 30°C and supplemented with iron. These findings were not characteristic of a rapidly growing mycobacteria (eg, M fortuitum, M chelonae, M abscessus) or M marinum but raised concern for infectionwith M haemophilum. Antimycobacterial treatment was adjusted to amikacin, clarithromycin, levofloxacin, and rifabutin.
Six weeks after the inpatient skin biopsy, final speciation confirmed infection with M haemophilum. The isolate proved susceptible to amikacin (minimal inhibitory concentration [MIC], 16), clarithromycin (MIC, 0.12), linezolid (MIC, <1), moxifloxacin (MIC, 0.5), rifabutin (MIC, <0.25), and trimethoprim-sulfamethoxazole (MIC, 0.5/9.5). The isolate was resistant to ciprofloxacin (MIC, 4), ethambutol (MIC, >16), and rifampin (MIC, 2). Based on these findings, an infectious disease specialist modified the treatment regimen to azithromycin 600 mg once daily, moxifloxacin 400 mg once daily, and rifabutin 300 mg once daily. Azithromycin was substituted for clarithromycin in an attempt to minimize the gastrointestinal side effects of the antibiotics. The infectious disease specialist was concerned that the clarithromycin could exacerbate the patient’s chronic GVHD-associated diarrhea, which posed a challenge to the oncologist, who was attempting to manage the patient’s GVHD and minimize the use of additional prednisone. At the time of this change, the patient was doing well clinically and denied any active skin lesions.
Four months later, he developed new left-sided neck swelling. Computed tomography revealed nonspecific enhancement involving the skin and superficial subcutaneous tissues in the left anterior neck. He was referred to otolaryngology given concern for recurrent infection vs leukemia cutis. He underwent excisional biopsy. Pathology was negative for malignancy but demonstrated subcutaneous necrotizing granulomatous inflammation with a positive AFB stain. Tissue AFB cultures revealed moderate AFB on direct stain, but there was no AFB growth at 12 weeks. Clarithromycin was restarted in place of azithromycin to increase the potency of the antimycobacterial regimen. Cultures from this neck biopsy were negative after 12 weeks of incubation.
In addition to this change in antibiotic coverage, the patient’s medical oncologist tapered the patient’s immunosuppression considerably. The patient subsequently completed 12 months of therapy with clarithromycin, moxifloxacin, and rifabutin starting from the time of the neck biopsy. He remained free of recurrence of mycobacterial infection for nearly 2 years until he died from an unrelated illness.
Nontuberculous mycobacteria are an ubiquitous environmental group.2 Sources include soil and natural water (M avium), fish tanks and swimming pools (M marinum), and tap water and occasionally domestic animals (Mycobacterium kansasii). Additionally, rapidly growing NTM such as M abscessus, M chelonae, and M fortuitum have been isolated from soil and natural water supplies.3
Mycobacterium haemophilum is a fastidious organism with a predilection for skin of the chest and extremities. Iatrogenically or inherently immunocompromised patients are most commonly affected6-11; however, there also have been reports in healthy patients.12,13 Infections typically present as painless erythematous papules or nodules that eventually suppurate, ulcerate, and become painful. Presentations involving Fitz-Hugh–Curtis syndrome,13 new B-cell lymphoma,10 and lymphadenitis12 also have been described. Beyond cutaneous involvement, M haemophilum has been cultured from bone, the synovium, the lungs, and the central nervous system.4,9 The majority of morbidities occur in patients with lung involvement.4 Therefore, even patients presenting with isolated cutaneous disease require close follow-up.
Mycobacterium haemophilum is a slowly proliferating organism that is unable to grow in standard egg-potato (Lowenstein-Jensen) medium or agar base (Middlebrook 7H10 or 7H11 agar) without iron supplementation (ferric ammonium citrate, hemin, or hemoglobin). It also requires temperatures of 30°C to 32°C for growth. Its iron requisite is unique, but species such as M marinum and Mycobacterium ulcerans also share reduced temperature requirements. Without a high index of suspicion, growth often is absent because standard Mycobacterium culture techniques will not foster organism growth. Our case demonstrated that special culture instructions must be relayed to the laboratory, even in the face of positive AFB smears. Failure to request hemin and modified incubation temperatures may have contributed to the negative AFB blood culture in our patient.
Due to the relatively rare incidence of M haemophilum infection, there are no known randomized controlled trials guiding antibiotic regimens. Infectious disease specialists often treat empirically with triple-drug therapy derived from locally reported species susceptibilities. The largest case series to date did not identify resistance to amikacin, ciprofloxacin, or clarithromycin.4 Our case identified a novel finding of ciprofloxacin and rifampin resistance, which may highlight the emergence of a newly resistant strain of M haemophilum. Of note, one case of rifampin resistance has been reported, but the culture was drawn from a postmortem specimen in the setting of previously rifampin-sensitive isolates.4 Empiric therapies should be guided by hospital susceptibility reports and expert consultation.
Coinfection with 2 or more NTM—including M tuberculosis, M leprae, and M fortuitum—has been reported.8,14 Temporally distinct coinfections with M leprae and M haemophilum also have been described.15 Thus, practitioners should have a low threshold for repeat cultures in the context of new cutaneous nodules or granulomas, not only to detect concomitant infections but also to identify resistance patterns that might explain recurrent or recalcitrant disease. Immune reconstitution inflammatory syndrome also must be considered with new or worsening lesions, especially in the first months of therapy, as this is a common occurrence when immunosuppressive regimens are tapered to help manage infections.
In conclusion, M haemophilum is an underrecognized infection that presents as cutaneous nodules or lymphadenitis in immunocompromised or healthy individuals. Diagnosis requires a high index of suspicion because its unique growth requirements necessitate special laboratory techniques. Our case represents a classic presentation of this NTM infection in a patient with AML following allogenic stem cell transplantation. Repeat cultures, workup of potentially disseminated infections, and close follow-up are requisite to minimizing morbidity and mortality. A multidisciplinary approach involving infectious disease, medical oncology, radiology, and dermatology best manages this type of infection.
To the Editor:
The increase in nontuberculous mycobacteria (NTM) infections over the last 3 decades likely is multifaceted, including increased clinical awareness, improved laboratory diagnostics, growing numbers of immunocompromised patients, and an aging population.1,2 Historically, the majority of mycobacteria-related diseases are due to Mycobacterium tuberculosis, Mycobacterium bovis, and Mycobacterium leprae.3
Mycobacterium haemophilum is a slow-growing acid-fast bacillus (AFB) that differs from other Mycobacterium species in that it requires iron-supplemented media and incubation temperatures of 30°C to 32°C for culture. As these requirements for growth are not standard for AFB cultures, M haemophilum infection may be underrecognized and underreported.3Mycobacterium haemophilum infections largely are cutaneous and generally are seen in AIDS patients and bone marrow transplant recipients who are iatrogenically immunosuppressed.4,5 No species-specific treatment guidelines exist2; however, triple-drug therapy combining a macrolide, rifamycin, and a quinolone for a minimum of 12 months often is recommended.
A 64-year-old man with a history of coronary artery disease, hypertension, hyperlipidemia, and acute myelogenous leukemia (AML) underwent allogenic stem cell transplantation. His posttransplant course was complicated by multiple deep vein thromboses, hypogammaglobulinemia, and graft-vs-host disease (GVHD) of the skin and gastrointestinal tract that manifested as chronic diarrhea, which was managed with chronic prednisone. Thirteen months after the transplant, the patient presented to his outpatient oncologist (M.K.) for evaluation of painless, nonpruritic, erythematous papules and nodules that had emerged on the right side of the chest, right arm, and left leg of approximately 2 weeks’ duration.
On review of systems by oncology, the patient denied any fevers, chills, or night sweats but noted chronic loose nonbloody stools without abdominal pain, likely related to the GVHD. The patient’s medications included prednisone 20 mg once daily, fluconazole, amitriptyline, atovaquone, budesonide, dabigatran, metoprolol, pantoprazole, rosuvastatin, senna glycoside, spironolactone, tramadol, and valacyclovir.
Physical examination revealed multiple singular erythematous nodules on the right side of the chest (Figure 1A), right arm (Figure 1B), and left leg. There was no regional lymphadenopathy. The patient was afebrile and hemodynamically stable. A biopsy of the arm performed to rule out leukemia cutis revealed a granulomatous dermatitis with numerous AFB (Figures 2A and 2B), which were confirmed on Ziehl-Neelsen staining (Figures 2C and 2D). The presence of AFB raised concern for a disseminated mycobacterial infection. The patient was admitted to our institution approximately 1 week after the outpatient biopsy was performed. He was evaluated by infectious diseases (B.H.) and was recommended for repeat biopsy with AFB culture and for initiation of intravenous antibiotics.
The patient was evaluated by the dermatology consultation service on hospital day 1. At the time of consultation, the lesions were still painless but had enlarged. Two new satellite lesions were noted on his other extremities. Due to the widespread distribution of the lesions, there was concern for disseminated disease. The relatively rapid onset of new lesions increased concern for infection with rapid-growing mycobacteria, including Mycobacterium abscessus, Mycobacterium fortuitum, and Mycobacterium chelonae. A detailed history revealed that the patient’s wife had a fish tank, which supported the inclusion of Mycobacterium marinum in the differential; however, further questioning revealed that the patient never came in contact with the aquarium water. The initial outpatient biopsy had not been sent for culture. Following inpatient biopsy, the patient was initiated on empiric antimycobacterials, including imipenem, amikacin, clarithromycin, and levofloxacin. Computed tomography of the head was negative for cerebral involvement.
Acid-fast bacilli blood cultures were drawn per the recommendation from infectious diseases in an attempt to confirm disseminated disease; however, blood cultures remained negative. Tissue biopsy from the right arm was sent for AFB staining and culture. Many AFB were identified on microscopy, and growth was observed in the mycobacterial growth indicator tube after 6 days of incubation. The DNA probe was negative for M tuberculosis complex or Mycobacterium avium complex.
The patient was discharged on hospital day 6 on empiric therapy for rapid-growing mycobacteria while cultures were pending. The empiric regimen included intravenous imipenem 1 g every 6 hours, intravenous amikacin 1 g once daily, clarithromycin 500 mg every 12 hours, and levofloxacin 750 mg once daily. All solid media cultures were negative at the time of discharge.
The biopsy specimen proved difficult to culture on solid media using traditional methods. Three weeks after the inpatient biopsy, the microbiology laboratory reported that growth was observed on solid media that was incubated at 30°C and supplemented with iron. These findings were not characteristic of a rapidly growing mycobacteria (eg, M fortuitum, M chelonae, M abscessus) or M marinum but raised concern for infectionwith M haemophilum. Antimycobacterial treatment was adjusted to amikacin, clarithromycin, levofloxacin, and rifabutin.
Six weeks after the inpatient skin biopsy, final speciation confirmed infection with M haemophilum. The isolate proved susceptible to amikacin (minimal inhibitory concentration [MIC], 16), clarithromycin (MIC, 0.12), linezolid (MIC, <1), moxifloxacin (MIC, 0.5), rifabutin (MIC, <0.25), and trimethoprim-sulfamethoxazole (MIC, 0.5/9.5). The isolate was resistant to ciprofloxacin (MIC, 4), ethambutol (MIC, >16), and rifampin (MIC, 2). Based on these findings, an infectious disease specialist modified the treatment regimen to azithromycin 600 mg once daily, moxifloxacin 400 mg once daily, and rifabutin 300 mg once daily. Azithromycin was substituted for clarithromycin in an attempt to minimize the gastrointestinal side effects of the antibiotics. The infectious disease specialist was concerned that the clarithromycin could exacerbate the patient’s chronic GVHD-associated diarrhea, which posed a challenge to the oncologist, who was attempting to manage the patient’s GVHD and minimize the use of additional prednisone. At the time of this change, the patient was doing well clinically and denied any active skin lesions.
Four months later, he developed new left-sided neck swelling. Computed tomography revealed nonspecific enhancement involving the skin and superficial subcutaneous tissues in the left anterior neck. He was referred to otolaryngology given concern for recurrent infection vs leukemia cutis. He underwent excisional biopsy. Pathology was negative for malignancy but demonstrated subcutaneous necrotizing granulomatous inflammation with a positive AFB stain. Tissue AFB cultures revealed moderate AFB on direct stain, but there was no AFB growth at 12 weeks. Clarithromycin was restarted in place of azithromycin to increase the potency of the antimycobacterial regimen. Cultures from this neck biopsy were negative after 12 weeks of incubation.
In addition to this change in antibiotic coverage, the patient’s medical oncologist tapered the patient’s immunosuppression considerably. The patient subsequently completed 12 months of therapy with clarithromycin, moxifloxacin, and rifabutin starting from the time of the neck biopsy. He remained free of recurrence of mycobacterial infection for nearly 2 years until he died from an unrelated illness.
Nontuberculous mycobacteria are an ubiquitous environmental group.2 Sources include soil and natural water (M avium), fish tanks and swimming pools (M marinum), and tap water and occasionally domestic animals (Mycobacterium kansasii). Additionally, rapidly growing NTM such as M abscessus, M chelonae, and M fortuitum have been isolated from soil and natural water supplies.3
Mycobacterium haemophilum is a fastidious organism with a predilection for skin of the chest and extremities. Iatrogenically or inherently immunocompromised patients are most commonly affected6-11; however, there also have been reports in healthy patients.12,13 Infections typically present as painless erythematous papules or nodules that eventually suppurate, ulcerate, and become painful. Presentations involving Fitz-Hugh–Curtis syndrome,13 new B-cell lymphoma,10 and lymphadenitis12 also have been described. Beyond cutaneous involvement, M haemophilum has been cultured from bone, the synovium, the lungs, and the central nervous system.4,9 The majority of morbidities occur in patients with lung involvement.4 Therefore, even patients presenting with isolated cutaneous disease require close follow-up.
Mycobacterium haemophilum is a slowly proliferating organism that is unable to grow in standard egg-potato (Lowenstein-Jensen) medium or agar base (Middlebrook 7H10 or 7H11 agar) without iron supplementation (ferric ammonium citrate, hemin, or hemoglobin). It also requires temperatures of 30°C to 32°C for growth. Its iron requisite is unique, but species such as M marinum and Mycobacterium ulcerans also share reduced temperature requirements. Without a high index of suspicion, growth often is absent because standard Mycobacterium culture techniques will not foster organism growth. Our case demonstrated that special culture instructions must be relayed to the laboratory, even in the face of positive AFB smears. Failure to request hemin and modified incubation temperatures may have contributed to the negative AFB blood culture in our patient.
Due to the relatively rare incidence of M haemophilum infection, there are no known randomized controlled trials guiding antibiotic regimens. Infectious disease specialists often treat empirically with triple-drug therapy derived from locally reported species susceptibilities. The largest case series to date did not identify resistance to amikacin, ciprofloxacin, or clarithromycin.4 Our case identified a novel finding of ciprofloxacin and rifampin resistance, which may highlight the emergence of a newly resistant strain of M haemophilum. Of note, one case of rifampin resistance has been reported, but the culture was drawn from a postmortem specimen in the setting of previously rifampin-sensitive isolates.4 Empiric therapies should be guided by hospital susceptibility reports and expert consultation.
Coinfection with 2 or more NTM—including M tuberculosis, M leprae, and M fortuitum—has been reported.8,14 Temporally distinct coinfections with M leprae and M haemophilum also have been described.15 Thus, practitioners should have a low threshold for repeat cultures in the context of new cutaneous nodules or granulomas, not only to detect concomitant infections but also to identify resistance patterns that might explain recurrent or recalcitrant disease. Immune reconstitution inflammatory syndrome also must be considered with new or worsening lesions, especially in the first months of therapy, as this is a common occurrence when immunosuppressive regimens are tapered to help manage infections.
In conclusion, M haemophilum is an underrecognized infection that presents as cutaneous nodules or lymphadenitis in immunocompromised or healthy individuals. Diagnosis requires a high index of suspicion because its unique growth requirements necessitate special laboratory techniques. Our case represents a classic presentation of this NTM infection in a patient with AML following allogenic stem cell transplantation. Repeat cultures, workup of potentially disseminated infections, and close follow-up are requisite to minimizing morbidity and mortality. A multidisciplinary approach involving infectious disease, medical oncology, radiology, and dermatology best manages this type of infection.
- Sheu LC, Tran TM, Jarlsberg LG, et al. Non-tuberculous mycobacterial infections at San Francisco General Hospital. Clin Respir J. 2015;9:436-442.
- Knoll BM. Update on nontuberculous mycobacterial infections in solid organ and hematopoietic stem cell transplant recipients. Curr Infect Dis Rep. 2014;16:421.
- Diagnosis and treatment of disease caused by nontuberculous mycobacteria. this official statement of the American Thoracic Society was approved by the Board of Directors, March 1997. Medical Section of the American Lung Association. Am J Respir Crit Care Med. 1997;156(2 pt 2):S1-S25.
- Shah MK, Sebti A, Kiehn TE, et al. Mycobacterium haemophilum in immunocompromised patients. Clin Infect Dis. 2001;33:330-337.
- Griffiths DE, Aksamit T, Brown-Elliott BA. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. 2007;175:367-416.
- Copeland NK, Arora NS, Ferguson TM. Mycobacterium haemophilum masquerading as leprosy in a renal transplant patient [published online November 28, 2013]. Case Rep Dermatol Med. 2013;2013:793127.
- Aslam A, Green RL, Motta L, et al. Cutaneous Mycobacterium haemophilum infection in a patient receiving infliximab for psoriasis. Br J Dermatol. 2013;168:446-447.
- Agrawal S, Sharma A. Dual mycobacterial infection in the setting of leflunomide treatment for rheumatoid arthritis. Ann Rheum Dis. 2007;66:277.
- Buppajarntham A, Apisarnthanarak A, Rutjanawech S, et al. Central nervous system infection due to Mycobacterium haemophilum in a patient with acquired immunodeficiency syndrome. Int J STD AIDS. 2015;26:288-290.
- Doherty T, Lynn M, Cavazza A, et al. Mycobacterium haemophilum as the initial presentation of a B-cell lymphoma in a liver transplant patient [published online January 12, 2014]. Case Rep Rheumatol. 2014;2014:742978.
- Ducharlet K, Murphy C, Tan SJ, et al. Recurrent Mycobacterium haemophilum in a renal transplant recipient. Nephrology (Carlton). 2014;(19 suppl 1):14-17.
- Dawson DJ, Blacklock ZM, Kane DW. Mycobacterium haemophilum causing lymphadenitis in an otherwise healthy child. Med J Aust. 1981;2:289-290.
- Jang HY, Burbelo PD, Chae YS, et al. Nontuberculous mycobacterial infection in a clinical presentation of Fitz-Hugh-Curtis syndrome: a case report with multigene diagnostic approach. BMC Womens Health. 2014;14:95.
- Scollard DM, Stryjewska BM, Prestigiacomo JF, et al. Hansen’s disease (leprosy) complicated by secondary mycobacterial infection. J Am Acad Dermatol. 2011;64:593-596.
- SoRelle JA, Beal SG, Scollard DM, et al. Mycobacterium leprae and Mycobacterium haemophilum co-infection in an iatrogenically immunosuppressed patient. Diagn Microbiol Infect Dis. 2014;78:494-496.
- Sheu LC, Tran TM, Jarlsberg LG, et al. Non-tuberculous mycobacterial infections at San Francisco General Hospital. Clin Respir J. 2015;9:436-442.
- Knoll BM. Update on nontuberculous mycobacterial infections in solid organ and hematopoietic stem cell transplant recipients. Curr Infect Dis Rep. 2014;16:421.
- Diagnosis and treatment of disease caused by nontuberculous mycobacteria. this official statement of the American Thoracic Society was approved by the Board of Directors, March 1997. Medical Section of the American Lung Association. Am J Respir Crit Care Med. 1997;156(2 pt 2):S1-S25.
- Shah MK, Sebti A, Kiehn TE, et al. Mycobacterium haemophilum in immunocompromised patients. Clin Infect Dis. 2001;33:330-337.
- Griffiths DE, Aksamit T, Brown-Elliott BA. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med. 2007;175:367-416.
- Copeland NK, Arora NS, Ferguson TM. Mycobacterium haemophilum masquerading as leprosy in a renal transplant patient [published online November 28, 2013]. Case Rep Dermatol Med. 2013;2013:793127.
- Aslam A, Green RL, Motta L, et al. Cutaneous Mycobacterium haemophilum infection in a patient receiving infliximab for psoriasis. Br J Dermatol. 2013;168:446-447.
- Agrawal S, Sharma A. Dual mycobacterial infection in the setting of leflunomide treatment for rheumatoid arthritis. Ann Rheum Dis. 2007;66:277.
- Buppajarntham A, Apisarnthanarak A, Rutjanawech S, et al. Central nervous system infection due to Mycobacterium haemophilum in a patient with acquired immunodeficiency syndrome. Int J STD AIDS. 2015;26:288-290.
- Doherty T, Lynn M, Cavazza A, et al. Mycobacterium haemophilum as the initial presentation of a B-cell lymphoma in a liver transplant patient [published online January 12, 2014]. Case Rep Rheumatol. 2014;2014:742978.
- Ducharlet K, Murphy C, Tan SJ, et al. Recurrent Mycobacterium haemophilum in a renal transplant recipient. Nephrology (Carlton). 2014;(19 suppl 1):14-17.
- Dawson DJ, Blacklock ZM, Kane DW. Mycobacterium haemophilum causing lymphadenitis in an otherwise healthy child. Med J Aust. 1981;2:289-290.
- Jang HY, Burbelo PD, Chae YS, et al. Nontuberculous mycobacterial infection in a clinical presentation of Fitz-Hugh-Curtis syndrome: a case report with multigene diagnostic approach. BMC Womens Health. 2014;14:95.
- Scollard DM, Stryjewska BM, Prestigiacomo JF, et al. Hansen’s disease (leprosy) complicated by secondary mycobacterial infection. J Am Acad Dermatol. 2011;64:593-596.
- SoRelle JA, Beal SG, Scollard DM, et al. Mycobacterium leprae and Mycobacterium haemophilum co-infection in an iatrogenically immunosuppressed patient. Diagn Microbiol Infect Dis. 2014;78:494-496.
Practice Points
- Mycobacterium haemophilum is a slow-growing acid-fast bacillus that requires iron-supplemented media and incubation temperatures of 30°C to 32°C for culture. Because these requirements for growth are not standard for acid-fast bacteria cultures, M haemophilum infection may be underrecognized and underreported.
- There are no species-specific treatment guidelines, but extended course of treatment with multiple active antibacterials typically is recommended.
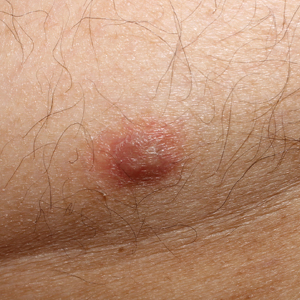
Psoriasiform Drug Eruption Secondary to Sorafenib: Case Series and Review of the Literature
The expanded use of targeted anticancer agents such as sorafenib has revealed a growing spectrum of adverse cutaneous eruptions. We describe 3 patients with sorafenib-induced psoriasiform dermatitis and review the literature of only 10 other similar reported cases based on a search of PubMed, Web of Science, and American Society of Clinical Oncology abstracts using the terms psoriasis or psoriasiform dermatitis and sorafenib.1-10 We seek to increase awareness of this particular drug eruption in response to sorafenib and to describe potential effective treatment options, especially when sorafenib cannot be discontinued.
Case Reports
Patient 1
A 68-year-old man with chronic hepatitis B infection and hepatocellular carcinoma (HCC) was started on sorafenib 400 mg daily. After 2 months of treatment, he developed painful hyperkeratotic lesions on the bilateral palms and soles with formation of calluses and superficial blisters on an erythematous base that was consistent with hand-foot skin reaction (HFSR). He also had numerous erythematous thin papules and plaques with adherent white scale and yellow crust on the bilateral thighs, lower legs, forearms, dorsal hands, abdomen, back, and buttocks (Figure 1). He had no personal or family history of psoriasis, and blood tests were unremarkable. Histologic analysis of punch biopsies from the buttocks and right leg revealed focal parakeratosis with neutrophils and serous crust, acanthosis, mild spongiosis, and lymphocytes at the dermoepidermal junction and surrounding dermal vessels, consistent with psoriasiform dermatitis (Figure 2). Sorafenib was discontinued and the eruption began to resolve within a week. A lower dose of sorafenib (200 mg daily) was attempted and the psoriasiform eruption recurred.
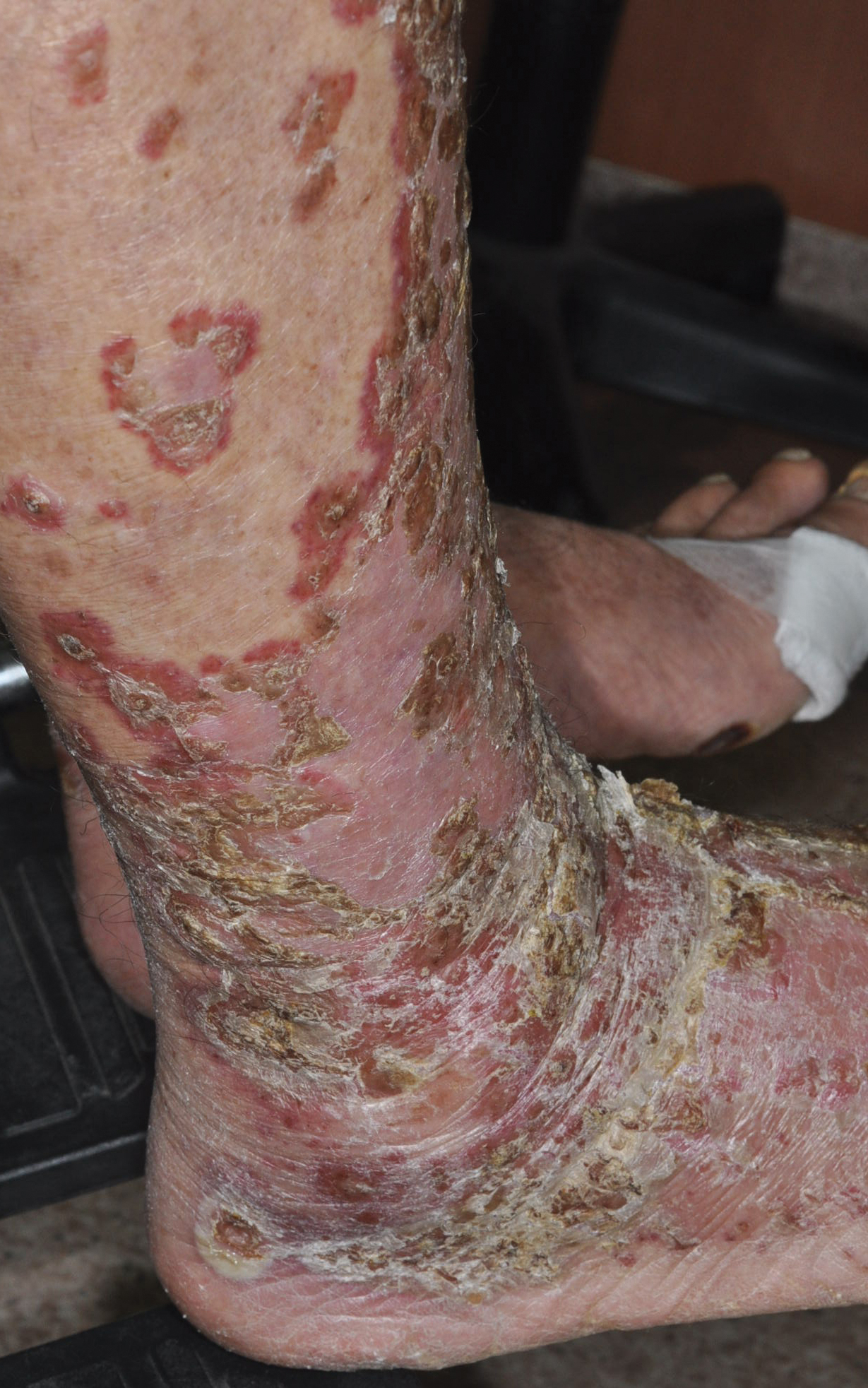
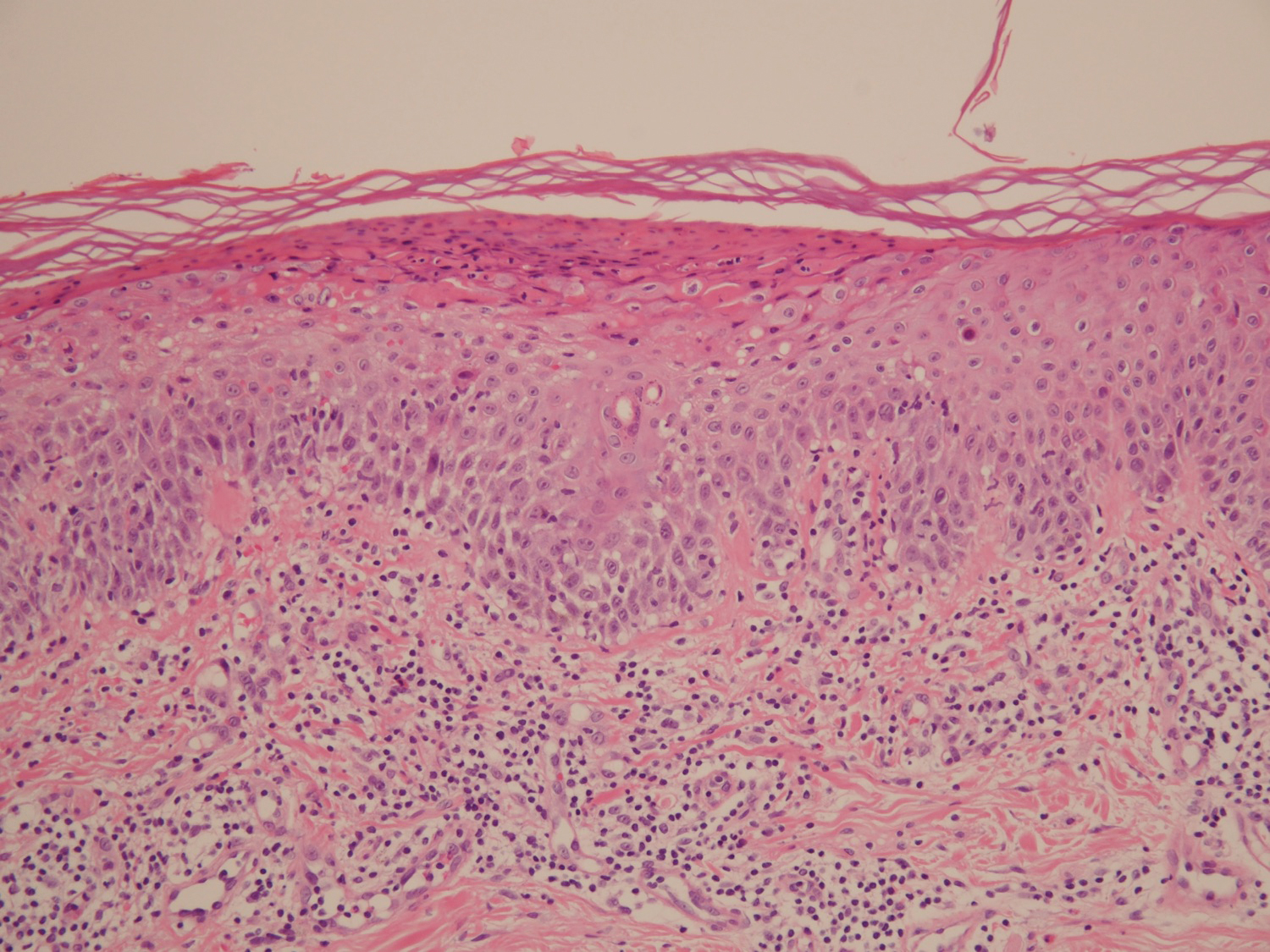
Patient 2
An 82-year-old man with chronic hepatitis B infection and HCC with lung metastasis was treated with sorafenib 400 mg daily. One week after treatment, he developed painful, thick, erythematous lesions on acral surfaces, consistent with HFSR. The sorafenib dose was decreased to 200 mg daily and HFSR resolved. Four months later, he developed well-demarcated, erythematous, scaly plaques with peripheral pustules on the right thigh (Figure 3) and right shin. He had no personal or family history of psoriasis, and blood tests were unremarkable. Samples from the pustules were taken for bacterial culture and fungal stain, but both were negative. Histologic analysis of a punch biopsy from the right thigh revealed necrotic parakeratosis, spongiform pustules, mild acanthosis, and a perivascular lymphocytic infiltrate with many neutrophils in the dermis. These findings suggested a diagnosis of pustular psoriasis, pustular drug eruption, or acute generalized exanthematous pustulosis. Treatment was initiated with mometasone cream. The patient subsequently developed hemoptysis and ascites from sorafenib. Sorafenib was discontinued and his skin eruption gradually resolved.
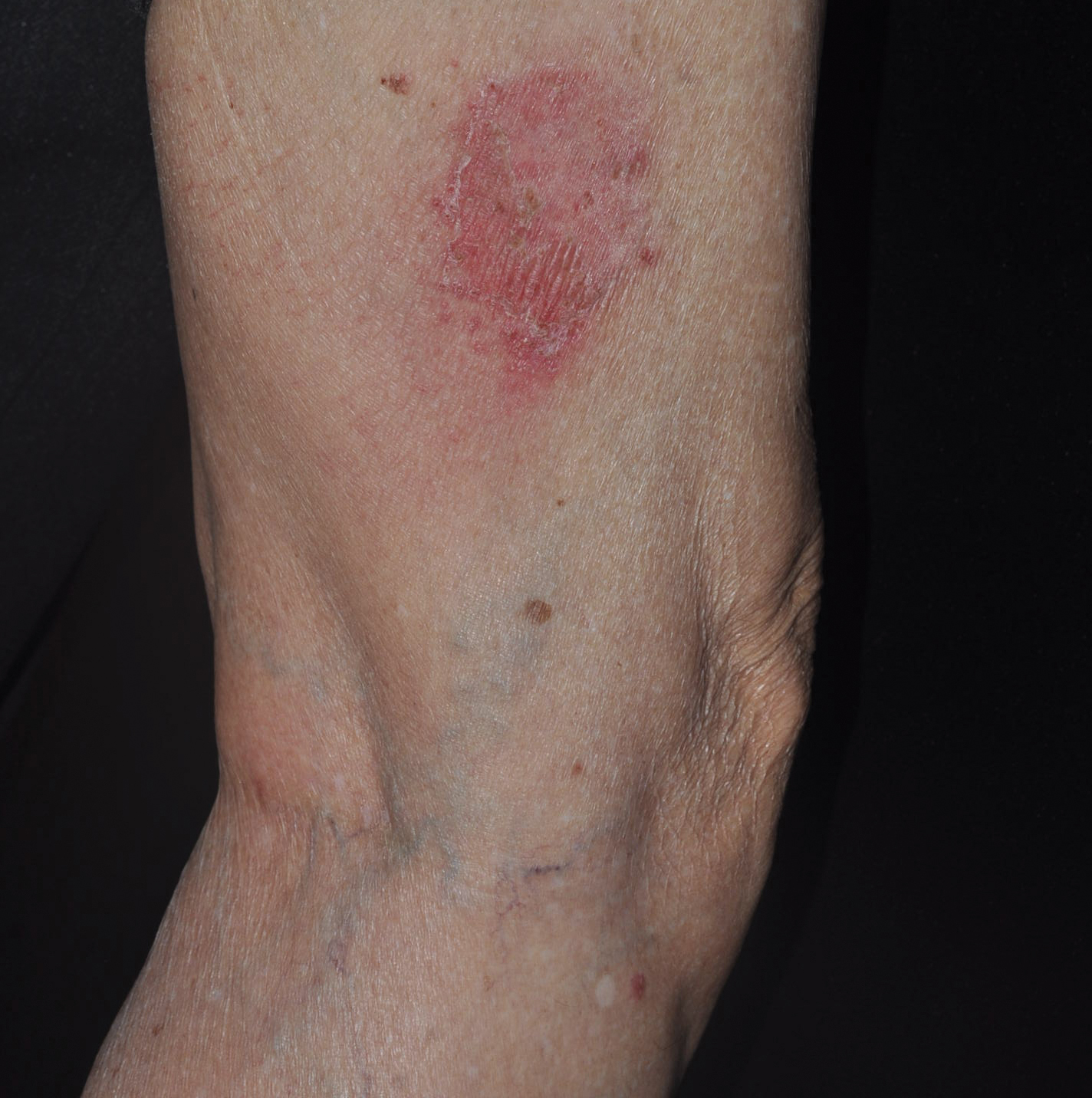
Patient 3
A 45-year-old woman with history of acute myeloid leukemia (AML) was started on sorafenib 200 mg twice daily as part of a clinical pilot study to maintain remission following an allogeneic bone marrow transplant. Four months after beginning sorafenib, the patient developed multiple well-defined, erythematous, thin papules and plaques with overlying flaky white scale on the bilateral upper extremities and trunk and scattered on the bilateral upper thighs (Figure 4) along with abdominal pain. Her other medical history, physical findings, and laboratory results were unremarkable, and there was no personal or family history of psoriasis. Her oncologist suspected that the eruption and symptoms were due to sorafenib and reduced the dose to 200 mg daily. Histologic analysis of a punch biopsy specimen revealed subcorneal neutrophilic collections with mild spongiosis and mild perivascular inflammatory infiltrate composed of lymphocytes and neutrophils (Figure 5). Direct immunofluorescence was negative for antibody or complement deposition. A bone marrow biopsy was negative for AML recurrence. The patient was continued on sorafenib to prevent AML recurrence, and she was started on triamcinolone cream 0.1% twice daily. Two weeks later, the eruption worsened and the patient was started on oral hydroxyzine for pruritus and narrowband UVB (NB-UVB) phototherapy 3 times a week. After 9 applications of NB-UVB phototherapy, there was complete resolution of the eruption.
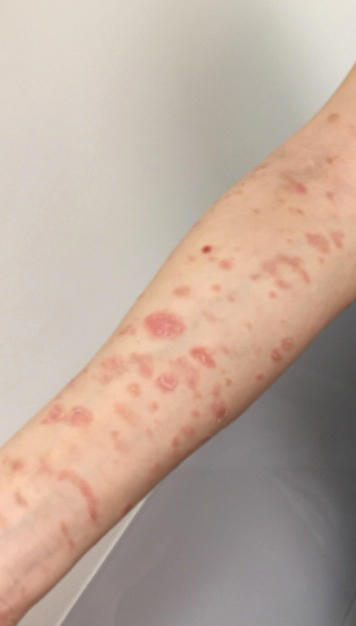
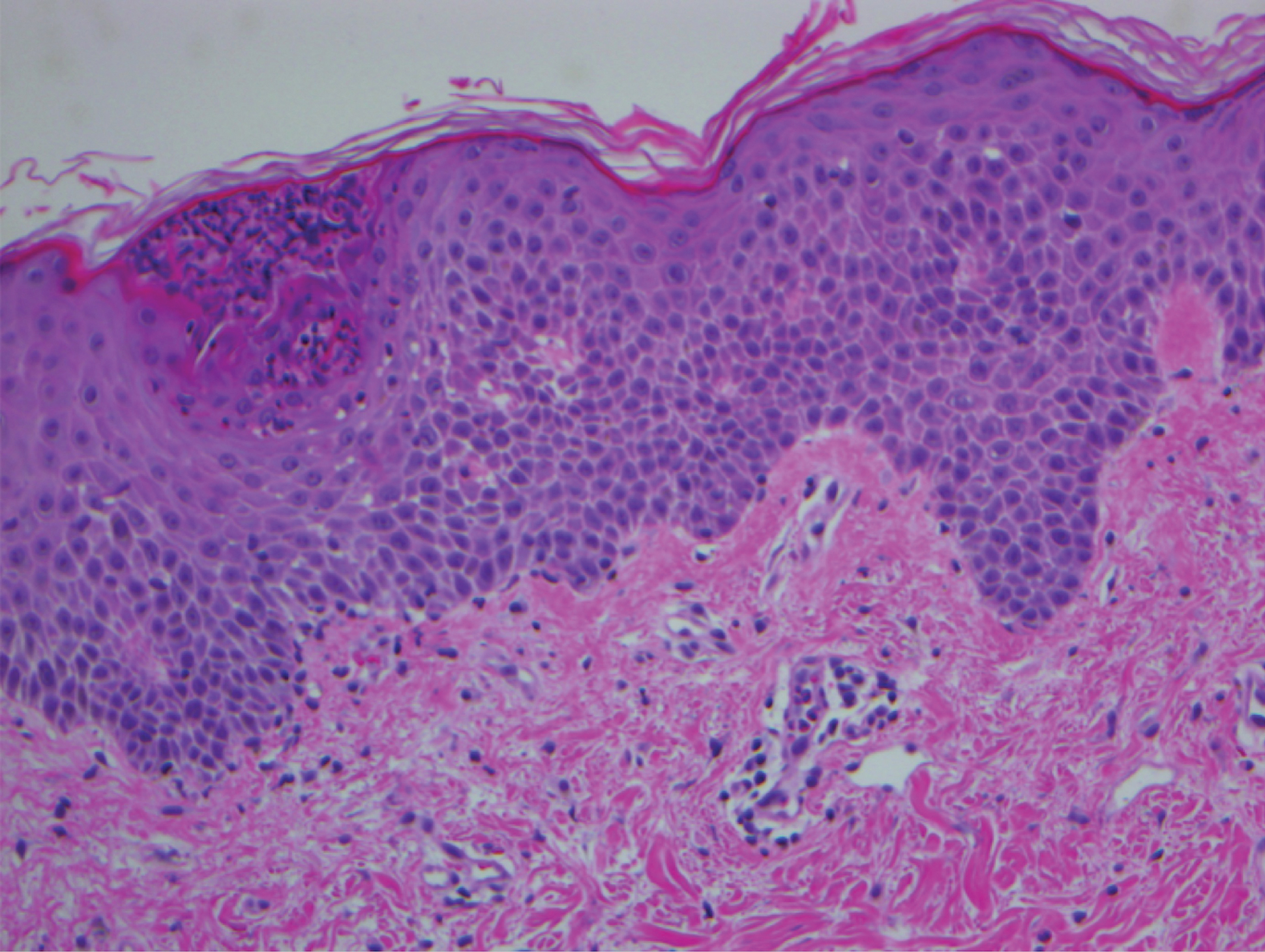
Comment
Sorafenib is an oral tyrosine kinase inhibitor that blocks tumor cell proliferation and angiogenesis due to its activity against vascular endothelial growth factor (VEGF) receptor, platelet-derived growth factor receptor, stem cell growth factor receptor, and rapidly accelerated fibrosarcoma kinases.11 It is primarily used for the treatment of solid tumors, such as advanced renal cell carcinoma, unresectable HCC, and thyroid carcinoma, and more recently has been expanded for treatment of AML due to potential inhibition of FMS-like tyrosine kinase 3 receptor. Although dermatologic toxicity is a common adverse event during treatment with sorafenib,11 reports of psoriasiform drug eruptions are rare.
Review of Cases
Based on our literature search, there are 10 previously reported cases of psoriasiform drug eruption secondary to sorafenib. Of the 13 total cases (including the 3 patients in this report), 7 patients had a history of psoriasis; most were middle-aged men; and the treatment with sorafenib was for solid tumors, primarily HCC with the exception of patient 3 from the current report who was treated for AML (Table). In all cases, the dose of sorafenib ranged from 200 to 800 mg daily. In 5 cases, HFSR preceded (as with patient 2 in the current report) or presented concurrently (as with patient 1 in the current report) with the onset of psoriasiform rash.1,3,5
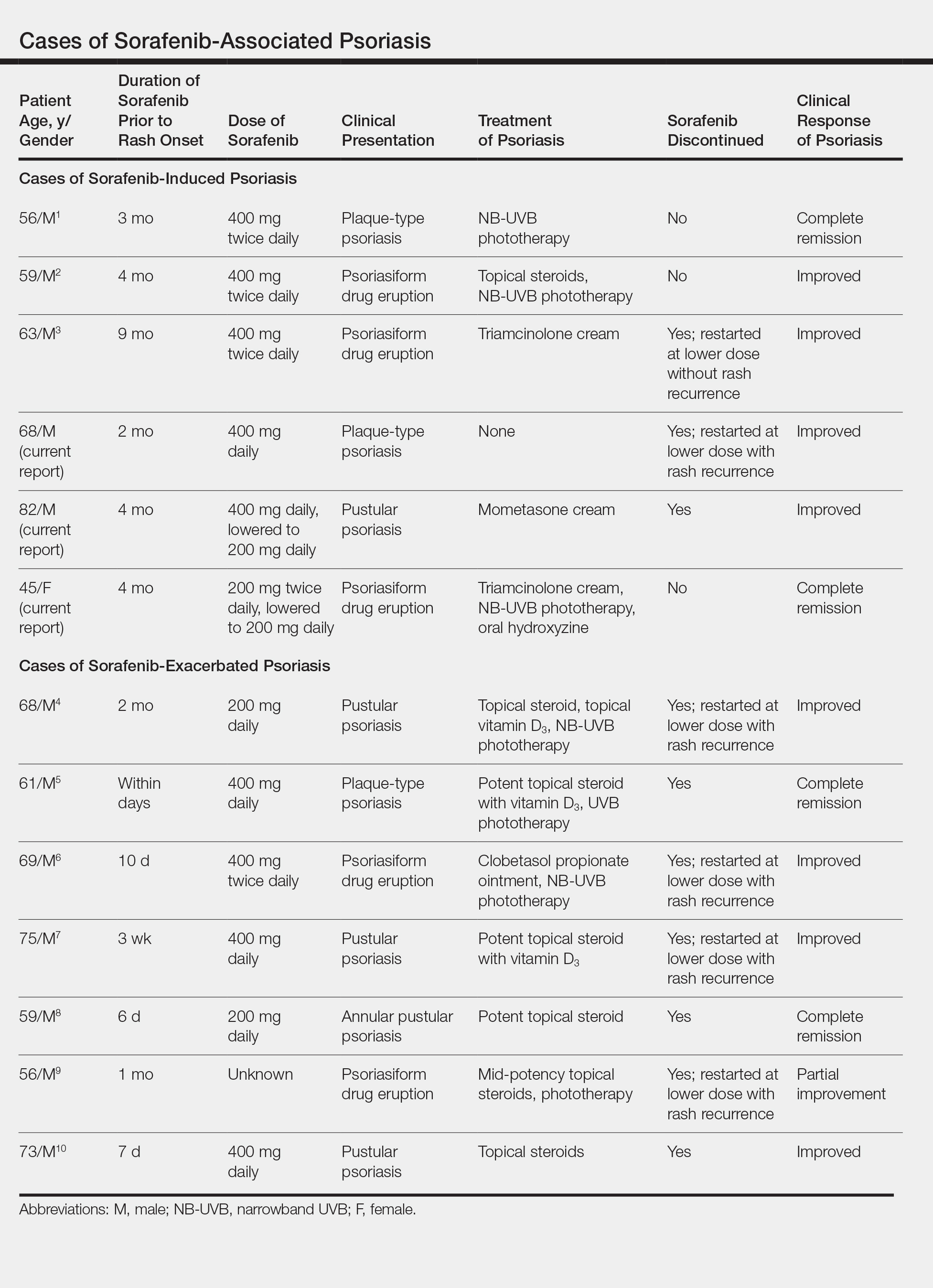
Of the 13 total cases, patients with a history of psoriasis generally developed the eruption in a shorter period of time after starting sorafenib (eg, days to 2 months) compared to those without a history of psoriasis (eg, 2 to 9 months)(Table), suggesting that patients with preexisting psoriasis more rapidly developed the drug eruption than patients without a history. In these patients with a history of psoriasis, all had long-standing mild to moderate stable plaque psoriasis, with the exception of 1 case in which the type of psoriasis was not described (Table).7 The presentation of the drug eruption following sorafenib varied from psoriasiform drug eruption (5 patients, including patient 3),2,3,6,9 pustular psoriasis (5 patients, including patient 2),4,7,8,10 and plaque psoriasis (3 patients, including patient 1).1,5 Interestingly, 5 of 6 patients with a history of plaque psoriasis presented with pustular psoriasis or psoriasiform drug eruption after treatment with sorafenib.4-6,8-10 These results suggest a causal relationship between sorafenib and exacerbation of preexisting psoriasis.
In the 13 total cases, treatments included mid- to high-potency topical steroids (10 cases), UVB or NB-UVB phototherapy (7 cases), and discontinuation of sorafenib (10 cases)(Table). All of these treatments led to improvement of the eruption with the exception of 1 case in which hand involvement was recalcitrant to therapy.9 Of the 10 cases in which sorafenib was discontinued, rechallenge at a lower dose was performed in 6 cases (including patient 1)3,4,6,7,9 with recurrence of psoriasiform rash seen in 5 cases (including patient 1)(Table).4,6,7,9 These data strongly implicate sorafenib as the direct cause of these psoriasiform eruptions. In the 3 cases in which sorafenib was not discontinued (including patient 3), there was notable improvement of the eruption with NB-UVB phototherapy.1,2
Vascular endothelial growth factor is overexpressed on psoriatic keratinocytes, contributes to epidermal hyperplasia, and induces angiogenesis in the dermis.12 The development of psoriasiform eruptions in patients treated with sorafenib seems paradoxical, as this drug has been considered as potential therapy for psoriasis due to its ability to block VEGF receptor signaling. Indeed, an improvement of psoriasis has been reported in 1 case of a patient treated with sorafenib13 and in multiple patients with psoriasis treated with other VEGF antagonists (eg, bevacizumab).14 The underlying mechanisms by which sorafenib induced or exacerbated psoriasis are not entirely clear. Palmoplantar hyperkeratosis, keratosis pilaris–like eruption, multiple cysts, eruptive keratoacanthomas, and squamous cell carcinoma have been described in patients treated with sorafenib, supporting the hypothesis that treatment with sorafenib alters keratinocyte proliferation and differentiation.15 In addition, B-Raf inhibitors such as imatinib are known to induce or exacerbate psoriasiform dermatitis.16 The activity of sorafenib resulting in psoriasis may be specific to RAF kinase inhibition, as there are no reports in the literature that describe psoriasiform dermatitis with agents that preferentially block other sorafenib targets such as VEGF receptor, stem cell growth factor receptor, or platelet-derived growth factor receptor. Future studies are needed to fully elucidate the underlying mechanisms by which sorafenib induces or exacerbates psoriasiform dermatitis and whether the severity of the drug eruption correlates with the antitumor efficacy of sorafenib.
Conclusion
Although psoriasiform drug eruptions secondary to sorafenib are not life-threatening, they impact quality of life with associated pain, pruritus, infection, and limitation of daily activities. Dose reduction or discontinuation of sorafenib resulted in resolution of the psoriasiform dermatitis; however, as demonstrated in 3 cases (including patient 3),1,2 psoriasiform dermatitis can be managed while maintaining the patient on sorafenib so that treatment of the malignancy is not compromised.
- Hung CT, Chiang CP, Wu BY. Sorafenib-induced psoriasis and hand-foot skin reaction responded dramatically to systemic narrowband ultraviolet B phototherapy. J Dermatol. 2012;39:1076-1077.
- González-López M, Yáñez S, Val-Bernal JF, et al. Psoriasiform skin eruption associated with sorafenib therapy. Indian J Dermatol Venereol Leprol. 2011;77:614-615.
- Diamantis ML, Chon SY. Sorafenib-induced psoriasiform eruption in a patient with metastatic thyroid carcinoma. J Drugs Dermatol. 2010;9:169-171.
- Hsu MC, Chen CC. Psoriasis flare-ups following sorafenib therapy: a rare case. Dermatologica Sin. 2016;34:148-150.
- Yiu ZZ, Ali FR, Griffiths CE. Paradoxical exacerbation of chronic plaque psoriasis by sorafenib. Clin Exp Dermatol. 2016;41:407-409.
- I˙lknur T, Akarsu S, Çarsanbali S, et al. Sorafenib-associated psoriasiform eruption in a patient with hepatocellular carcinoma. J Drugs Dermatol. 2014;13:899-900.
- Maki N, Komine M, Takatsuka Y, et al. Pustular eruption induced by sorafenib in a case of psoriasis vulgaris. J Dermatol. 2013;40:299-300.
- Du-Thanh A, Girard C, Pageaux GP, et al. Sorafenib-induced annular pustular psoriasis (Milian-Katchoura type). Eur J Dermatol. 2013;23:900-901.
- Laquer V, Saedi N, Dann F, et al. Sorafenib-associated psoriasiform skin changes. Cutis. 2010;85:301-302.
- Ohashi T, Yamamoto T. Exacerbation of psoriasis with pustulation by sorafenib in a patient with metastatic hepatocellular carcinoma. Indian J Dermatol. 2019;64:75-77.
- Chu D, Lacouture ME, Fillos T, et al. Risk of hand-foot skin reaction with sorafenib: a systematic review and meta-analysis. Acta Oncol (Madr). 2008;47:176-186.
- Canavese M, Altruda F, Ruzicka T, et al. Vascular endothelial growth factor (VEGF) in the pathogenesis of psoriasis--a possible target for novel therapies? J Dermatol Sci. 2010;58:171-176.
- Fournier C, Tisman G. Sorafenib-associated remission of psoriasis in hypernephroma: case report. Dermatol Online J. 2010;16:17.
- Akman A, Yilmaz E, Mutlu H, et al. Complete remission of psoriasis following bevacizumab therapy for colon cancer. Clin Exp Dermatol. 2009;34:E202-E204.
- Kong HH, Turner ML. Array of cutaneous adverse effects associated with sorafenib. J Am Acad Dermatol. 2009;61:360-361.
- Atalay F, Kızılkılıç E, Ada RS. Imatinib-induced psoriasis. Turk J Haematol. 2013;30:216-218.
The expanded use of targeted anticancer agents such as sorafenib has revealed a growing spectrum of adverse cutaneous eruptions. We describe 3 patients with sorafenib-induced psoriasiform dermatitis and review the literature of only 10 other similar reported cases based on a search of PubMed, Web of Science, and American Society of Clinical Oncology abstracts using the terms psoriasis or psoriasiform dermatitis and sorafenib.1-10 We seek to increase awareness of this particular drug eruption in response to sorafenib and to describe potential effective treatment options, especially when sorafenib cannot be discontinued.
Case Reports
Patient 1
A 68-year-old man with chronic hepatitis B infection and hepatocellular carcinoma (HCC) was started on sorafenib 400 mg daily. After 2 months of treatment, he developed painful hyperkeratotic lesions on the bilateral palms and soles with formation of calluses and superficial blisters on an erythematous base that was consistent with hand-foot skin reaction (HFSR). He also had numerous erythematous thin papules and plaques with adherent white scale and yellow crust on the bilateral thighs, lower legs, forearms, dorsal hands, abdomen, back, and buttocks (Figure 1). He had no personal or family history of psoriasis, and blood tests were unremarkable. Histologic analysis of punch biopsies from the buttocks and right leg revealed focal parakeratosis with neutrophils and serous crust, acanthosis, mild spongiosis, and lymphocytes at the dermoepidermal junction and surrounding dermal vessels, consistent with psoriasiform dermatitis (Figure 2). Sorafenib was discontinued and the eruption began to resolve within a week. A lower dose of sorafenib (200 mg daily) was attempted and the psoriasiform eruption recurred.
Patient 2
An 82-year-old man with chronic hepatitis B infection and HCC with lung metastasis was treated with sorafenib 400 mg daily. One week after treatment, he developed painful, thick, erythematous lesions on acral surfaces, consistent with HFSR. The sorafenib dose was decreased to 200 mg daily and HFSR resolved. Four months later, he developed well-demarcated, erythematous, scaly plaques with peripheral pustules on the right thigh (Figure 3) and right shin. He had no personal or family history of psoriasis, and blood tests were unremarkable. Samples from the pustules were taken for bacterial culture and fungal stain, but both were negative. Histologic analysis of a punch biopsy from the right thigh revealed necrotic parakeratosis, spongiform pustules, mild acanthosis, and a perivascular lymphocytic infiltrate with many neutrophils in the dermis. These findings suggested a diagnosis of pustular psoriasis, pustular drug eruption, or acute generalized exanthematous pustulosis. Treatment was initiated with mometasone cream. The patient subsequently developed hemoptysis and ascites from sorafenib. Sorafenib was discontinued and his skin eruption gradually resolved.
Patient 3
A 45-year-old woman with history of acute myeloid leukemia (AML) was started on sorafenib 200 mg twice daily as part of a clinical pilot study to maintain remission following an allogeneic bone marrow transplant. Four months after beginning sorafenib, the patient developed multiple well-defined, erythematous, thin papules and plaques with overlying flaky white scale on the bilateral upper extremities and trunk and scattered on the bilateral upper thighs (Figure 4) along with abdominal pain. Her other medical history, physical findings, and laboratory results were unremarkable, and there was no personal or family history of psoriasis. Her oncologist suspected that the eruption and symptoms were due to sorafenib and reduced the dose to 200 mg daily. Histologic analysis of a punch biopsy specimen revealed subcorneal neutrophilic collections with mild spongiosis and mild perivascular inflammatory infiltrate composed of lymphocytes and neutrophils (Figure 5). Direct immunofluorescence was negative for antibody or complement deposition. A bone marrow biopsy was negative for AML recurrence. The patient was continued on sorafenib to prevent AML recurrence, and she was started on triamcinolone cream 0.1% twice daily. Two weeks later, the eruption worsened and the patient was started on oral hydroxyzine for pruritus and narrowband UVB (NB-UVB) phototherapy 3 times a week. After 9 applications of NB-UVB phototherapy, there was complete resolution of the eruption.
Comment
Sorafenib is an oral tyrosine kinase inhibitor that blocks tumor cell proliferation and angiogenesis due to its activity against vascular endothelial growth factor (VEGF) receptor, platelet-derived growth factor receptor, stem cell growth factor receptor, and rapidly accelerated fibrosarcoma kinases.11 It is primarily used for the treatment of solid tumors, such as advanced renal cell carcinoma, unresectable HCC, and thyroid carcinoma, and more recently has been expanded for treatment of AML due to potential inhibition of FMS-like tyrosine kinase 3 receptor. Although dermatologic toxicity is a common adverse event during treatment with sorafenib,11 reports of psoriasiform drug eruptions are rare.
Review of Cases
Based on our literature search, there are 10 previously reported cases of psoriasiform drug eruption secondary to sorafenib. Of the 13 total cases (including the 3 patients in this report), 7 patients had a history of psoriasis; most were middle-aged men; and the treatment with sorafenib was for solid tumors, primarily HCC with the exception of patient 3 from the current report who was treated for AML (Table). In all cases, the dose of sorafenib ranged from 200 to 800 mg daily. In 5 cases, HFSR preceded (as with patient 2 in the current report) or presented concurrently (as with patient 1 in the current report) with the onset of psoriasiform rash.1,3,5
Of the 13 total cases, patients with a history of psoriasis generally developed the eruption in a shorter period of time after starting sorafenib (eg, days to 2 months) compared to those without a history of psoriasis (eg, 2 to 9 months)(Table), suggesting that patients with preexisting psoriasis more rapidly developed the drug eruption than patients without a history. In these patients with a history of psoriasis, all had long-standing mild to moderate stable plaque psoriasis, with the exception of 1 case in which the type of psoriasis was not described (Table).7 The presentation of the drug eruption following sorafenib varied from psoriasiform drug eruption (5 patients, including patient 3),2,3,6,9 pustular psoriasis (5 patients, including patient 2),4,7,8,10 and plaque psoriasis (3 patients, including patient 1).1,5 Interestingly, 5 of 6 patients with a history of plaque psoriasis presented with pustular psoriasis or psoriasiform drug eruption after treatment with sorafenib.4-6,8-10 These results suggest a causal relationship between sorafenib and exacerbation of preexisting psoriasis.
In the 13 total cases, treatments included mid- to high-potency topical steroids (10 cases), UVB or NB-UVB phototherapy (7 cases), and discontinuation of sorafenib (10 cases)(Table). All of these treatments led to improvement of the eruption with the exception of 1 case in which hand involvement was recalcitrant to therapy.9 Of the 10 cases in which sorafenib was discontinued, rechallenge at a lower dose was performed in 6 cases (including patient 1)3,4,6,7,9 with recurrence of psoriasiform rash seen in 5 cases (including patient 1)(Table).4,6,7,9 These data strongly implicate sorafenib as the direct cause of these psoriasiform eruptions. In the 3 cases in which sorafenib was not discontinued (including patient 3), there was notable improvement of the eruption with NB-UVB phototherapy.1,2
Vascular endothelial growth factor is overexpressed on psoriatic keratinocytes, contributes to epidermal hyperplasia, and induces angiogenesis in the dermis.12 The development of psoriasiform eruptions in patients treated with sorafenib seems paradoxical, as this drug has been considered as potential therapy for psoriasis due to its ability to block VEGF receptor signaling. Indeed, an improvement of psoriasis has been reported in 1 case of a patient treated with sorafenib13 and in multiple patients with psoriasis treated with other VEGF antagonists (eg, bevacizumab).14 The underlying mechanisms by which sorafenib induced or exacerbated psoriasis are not entirely clear. Palmoplantar hyperkeratosis, keratosis pilaris–like eruption, multiple cysts, eruptive keratoacanthomas, and squamous cell carcinoma have been described in patients treated with sorafenib, supporting the hypothesis that treatment with sorafenib alters keratinocyte proliferation and differentiation.15 In addition, B-Raf inhibitors such as imatinib are known to induce or exacerbate psoriasiform dermatitis.16 The activity of sorafenib resulting in psoriasis may be specific to RAF kinase inhibition, as there are no reports in the literature that describe psoriasiform dermatitis with agents that preferentially block other sorafenib targets such as VEGF receptor, stem cell growth factor receptor, or platelet-derived growth factor receptor. Future studies are needed to fully elucidate the underlying mechanisms by which sorafenib induces or exacerbates psoriasiform dermatitis and whether the severity of the drug eruption correlates with the antitumor efficacy of sorafenib.
Conclusion
Although psoriasiform drug eruptions secondary to sorafenib are not life-threatening, they impact quality of life with associated pain, pruritus, infection, and limitation of daily activities. Dose reduction or discontinuation of sorafenib resulted in resolution of the psoriasiform dermatitis; however, as demonstrated in 3 cases (including patient 3),1,2 psoriasiform dermatitis can be managed while maintaining the patient on sorafenib so that treatment of the malignancy is not compromised.
The expanded use of targeted anticancer agents such as sorafenib has revealed a growing spectrum of adverse cutaneous eruptions. We describe 3 patients with sorafenib-induced psoriasiform dermatitis and review the literature of only 10 other similar reported cases based on a search of PubMed, Web of Science, and American Society of Clinical Oncology abstracts using the terms psoriasis or psoriasiform dermatitis and sorafenib.1-10 We seek to increase awareness of this particular drug eruption in response to sorafenib and to describe potential effective treatment options, especially when sorafenib cannot be discontinued.
Case Reports
Patient 1
A 68-year-old man with chronic hepatitis B infection and hepatocellular carcinoma (HCC) was started on sorafenib 400 mg daily. After 2 months of treatment, he developed painful hyperkeratotic lesions on the bilateral palms and soles with formation of calluses and superficial blisters on an erythematous base that was consistent with hand-foot skin reaction (HFSR). He also had numerous erythematous thin papules and plaques with adherent white scale and yellow crust on the bilateral thighs, lower legs, forearms, dorsal hands, abdomen, back, and buttocks (Figure 1). He had no personal or family history of psoriasis, and blood tests were unremarkable. Histologic analysis of punch biopsies from the buttocks and right leg revealed focal parakeratosis with neutrophils and serous crust, acanthosis, mild spongiosis, and lymphocytes at the dermoepidermal junction and surrounding dermal vessels, consistent with psoriasiform dermatitis (Figure 2). Sorafenib was discontinued and the eruption began to resolve within a week. A lower dose of sorafenib (200 mg daily) was attempted and the psoriasiform eruption recurred.
Patient 2
An 82-year-old man with chronic hepatitis B infection and HCC with lung metastasis was treated with sorafenib 400 mg daily. One week after treatment, he developed painful, thick, erythematous lesions on acral surfaces, consistent with HFSR. The sorafenib dose was decreased to 200 mg daily and HFSR resolved. Four months later, he developed well-demarcated, erythematous, scaly plaques with peripheral pustules on the right thigh (Figure 3) and right shin. He had no personal or family history of psoriasis, and blood tests were unremarkable. Samples from the pustules were taken for bacterial culture and fungal stain, but both were negative. Histologic analysis of a punch biopsy from the right thigh revealed necrotic parakeratosis, spongiform pustules, mild acanthosis, and a perivascular lymphocytic infiltrate with many neutrophils in the dermis. These findings suggested a diagnosis of pustular psoriasis, pustular drug eruption, or acute generalized exanthematous pustulosis. Treatment was initiated with mometasone cream. The patient subsequently developed hemoptysis and ascites from sorafenib. Sorafenib was discontinued and his skin eruption gradually resolved.
Patient 3
A 45-year-old woman with history of acute myeloid leukemia (AML) was started on sorafenib 200 mg twice daily as part of a clinical pilot study to maintain remission following an allogeneic bone marrow transplant. Four months after beginning sorafenib, the patient developed multiple well-defined, erythematous, thin papules and plaques with overlying flaky white scale on the bilateral upper extremities and trunk and scattered on the bilateral upper thighs (Figure 4) along with abdominal pain. Her other medical history, physical findings, and laboratory results were unremarkable, and there was no personal or family history of psoriasis. Her oncologist suspected that the eruption and symptoms were due to sorafenib and reduced the dose to 200 mg daily. Histologic analysis of a punch biopsy specimen revealed subcorneal neutrophilic collections with mild spongiosis and mild perivascular inflammatory infiltrate composed of lymphocytes and neutrophils (Figure 5). Direct immunofluorescence was negative for antibody or complement deposition. A bone marrow biopsy was negative for AML recurrence. The patient was continued on sorafenib to prevent AML recurrence, and she was started on triamcinolone cream 0.1% twice daily. Two weeks later, the eruption worsened and the patient was started on oral hydroxyzine for pruritus and narrowband UVB (NB-UVB) phototherapy 3 times a week. After 9 applications of NB-UVB phototherapy, there was complete resolution of the eruption.
Comment
Sorafenib is an oral tyrosine kinase inhibitor that blocks tumor cell proliferation and angiogenesis due to its activity against vascular endothelial growth factor (VEGF) receptor, platelet-derived growth factor receptor, stem cell growth factor receptor, and rapidly accelerated fibrosarcoma kinases.11 It is primarily used for the treatment of solid tumors, such as advanced renal cell carcinoma, unresectable HCC, and thyroid carcinoma, and more recently has been expanded for treatment of AML due to potential inhibition of FMS-like tyrosine kinase 3 receptor. Although dermatologic toxicity is a common adverse event during treatment with sorafenib,11 reports of psoriasiform drug eruptions are rare.
Review of Cases
Based on our literature search, there are 10 previously reported cases of psoriasiform drug eruption secondary to sorafenib. Of the 13 total cases (including the 3 patients in this report), 7 patients had a history of psoriasis; most were middle-aged men; and the treatment with sorafenib was for solid tumors, primarily HCC with the exception of patient 3 from the current report who was treated for AML (Table). In all cases, the dose of sorafenib ranged from 200 to 800 mg daily. In 5 cases, HFSR preceded (as with patient 2 in the current report) or presented concurrently (as with patient 1 in the current report) with the onset of psoriasiform rash.1,3,5
Of the 13 total cases, patients with a history of psoriasis generally developed the eruption in a shorter period of time after starting sorafenib (eg, days to 2 months) compared to those without a history of psoriasis (eg, 2 to 9 months)(Table), suggesting that patients with preexisting psoriasis more rapidly developed the drug eruption than patients without a history. In these patients with a history of psoriasis, all had long-standing mild to moderate stable plaque psoriasis, with the exception of 1 case in which the type of psoriasis was not described (Table).7 The presentation of the drug eruption following sorafenib varied from psoriasiform drug eruption (5 patients, including patient 3),2,3,6,9 pustular psoriasis (5 patients, including patient 2),4,7,8,10 and plaque psoriasis (3 patients, including patient 1).1,5 Interestingly, 5 of 6 patients with a history of plaque psoriasis presented with pustular psoriasis or psoriasiform drug eruption after treatment with sorafenib.4-6,8-10 These results suggest a causal relationship between sorafenib and exacerbation of preexisting psoriasis.
In the 13 total cases, treatments included mid- to high-potency topical steroids (10 cases), UVB or NB-UVB phototherapy (7 cases), and discontinuation of sorafenib (10 cases)(Table). All of these treatments led to improvement of the eruption with the exception of 1 case in which hand involvement was recalcitrant to therapy.9 Of the 10 cases in which sorafenib was discontinued, rechallenge at a lower dose was performed in 6 cases (including patient 1)3,4,6,7,9 with recurrence of psoriasiform rash seen in 5 cases (including patient 1)(Table).4,6,7,9 These data strongly implicate sorafenib as the direct cause of these psoriasiform eruptions. In the 3 cases in which sorafenib was not discontinued (including patient 3), there was notable improvement of the eruption with NB-UVB phototherapy.1,2
Vascular endothelial growth factor is overexpressed on psoriatic keratinocytes, contributes to epidermal hyperplasia, and induces angiogenesis in the dermis.12 The development of psoriasiform eruptions in patients treated with sorafenib seems paradoxical, as this drug has been considered as potential therapy for psoriasis due to its ability to block VEGF receptor signaling. Indeed, an improvement of psoriasis has been reported in 1 case of a patient treated with sorafenib13 and in multiple patients with psoriasis treated with other VEGF antagonists (eg, bevacizumab).14 The underlying mechanisms by which sorafenib induced or exacerbated psoriasis are not entirely clear. Palmoplantar hyperkeratosis, keratosis pilaris–like eruption, multiple cysts, eruptive keratoacanthomas, and squamous cell carcinoma have been described in patients treated with sorafenib, supporting the hypothesis that treatment with sorafenib alters keratinocyte proliferation and differentiation.15 In addition, B-Raf inhibitors such as imatinib are known to induce or exacerbate psoriasiform dermatitis.16 The activity of sorafenib resulting in psoriasis may be specific to RAF kinase inhibition, as there are no reports in the literature that describe psoriasiform dermatitis with agents that preferentially block other sorafenib targets such as VEGF receptor, stem cell growth factor receptor, or platelet-derived growth factor receptor. Future studies are needed to fully elucidate the underlying mechanisms by which sorafenib induces or exacerbates psoriasiform dermatitis and whether the severity of the drug eruption correlates with the antitumor efficacy of sorafenib.
Conclusion
Although psoriasiform drug eruptions secondary to sorafenib are not life-threatening, they impact quality of life with associated pain, pruritus, infection, and limitation of daily activities. Dose reduction or discontinuation of sorafenib resulted in resolution of the psoriasiform dermatitis; however, as demonstrated in 3 cases (including patient 3),1,2 psoriasiform dermatitis can be managed while maintaining the patient on sorafenib so that treatment of the malignancy is not compromised.
- Hung CT, Chiang CP, Wu BY. Sorafenib-induced psoriasis and hand-foot skin reaction responded dramatically to systemic narrowband ultraviolet B phototherapy. J Dermatol. 2012;39:1076-1077.
- González-López M, Yáñez S, Val-Bernal JF, et al. Psoriasiform skin eruption associated with sorafenib therapy. Indian J Dermatol Venereol Leprol. 2011;77:614-615.
- Diamantis ML, Chon SY. Sorafenib-induced psoriasiform eruption in a patient with metastatic thyroid carcinoma. J Drugs Dermatol. 2010;9:169-171.
- Hsu MC, Chen CC. Psoriasis flare-ups following sorafenib therapy: a rare case. Dermatologica Sin. 2016;34:148-150.
- Yiu ZZ, Ali FR, Griffiths CE. Paradoxical exacerbation of chronic plaque psoriasis by sorafenib. Clin Exp Dermatol. 2016;41:407-409.
- I˙lknur T, Akarsu S, Çarsanbali S, et al. Sorafenib-associated psoriasiform eruption in a patient with hepatocellular carcinoma. J Drugs Dermatol. 2014;13:899-900.
- Maki N, Komine M, Takatsuka Y, et al. Pustular eruption induced by sorafenib in a case of psoriasis vulgaris. J Dermatol. 2013;40:299-300.
- Du-Thanh A, Girard C, Pageaux GP, et al. Sorafenib-induced annular pustular psoriasis (Milian-Katchoura type). Eur J Dermatol. 2013;23:900-901.
- Laquer V, Saedi N, Dann F, et al. Sorafenib-associated psoriasiform skin changes. Cutis. 2010;85:301-302.
- Ohashi T, Yamamoto T. Exacerbation of psoriasis with pustulation by sorafenib in a patient with metastatic hepatocellular carcinoma. Indian J Dermatol. 2019;64:75-77.
- Chu D, Lacouture ME, Fillos T, et al. Risk of hand-foot skin reaction with sorafenib: a systematic review and meta-analysis. Acta Oncol (Madr). 2008;47:176-186.
- Canavese M, Altruda F, Ruzicka T, et al. Vascular endothelial growth factor (VEGF) in the pathogenesis of psoriasis--a possible target for novel therapies? J Dermatol Sci. 2010;58:171-176.
- Fournier C, Tisman G. Sorafenib-associated remission of psoriasis in hypernephroma: case report. Dermatol Online J. 2010;16:17.
- Akman A, Yilmaz E, Mutlu H, et al. Complete remission of psoriasis following bevacizumab therapy for colon cancer. Clin Exp Dermatol. 2009;34:E202-E204.
- Kong HH, Turner ML. Array of cutaneous adverse effects associated with sorafenib. J Am Acad Dermatol. 2009;61:360-361.
- Atalay F, Kızılkılıç E, Ada RS. Imatinib-induced psoriasis. Turk J Haematol. 2013;30:216-218.
- Hung CT, Chiang CP, Wu BY. Sorafenib-induced psoriasis and hand-foot skin reaction responded dramatically to systemic narrowband ultraviolet B phototherapy. J Dermatol. 2012;39:1076-1077.
- González-López M, Yáñez S, Val-Bernal JF, et al. Psoriasiform skin eruption associated with sorafenib therapy. Indian J Dermatol Venereol Leprol. 2011;77:614-615.
- Diamantis ML, Chon SY. Sorafenib-induced psoriasiform eruption in a patient with metastatic thyroid carcinoma. J Drugs Dermatol. 2010;9:169-171.
- Hsu MC, Chen CC. Psoriasis flare-ups following sorafenib therapy: a rare case. Dermatologica Sin. 2016;34:148-150.
- Yiu ZZ, Ali FR, Griffiths CE. Paradoxical exacerbation of chronic plaque psoriasis by sorafenib. Clin Exp Dermatol. 2016;41:407-409.
- I˙lknur T, Akarsu S, Çarsanbali S, et al. Sorafenib-associated psoriasiform eruption in a patient with hepatocellular carcinoma. J Drugs Dermatol. 2014;13:899-900.
- Maki N, Komine M, Takatsuka Y, et al. Pustular eruption induced by sorafenib in a case of psoriasis vulgaris. J Dermatol. 2013;40:299-300.
- Du-Thanh A, Girard C, Pageaux GP, et al. Sorafenib-induced annular pustular psoriasis (Milian-Katchoura type). Eur J Dermatol. 2013;23:900-901.
- Laquer V, Saedi N, Dann F, et al. Sorafenib-associated psoriasiform skin changes. Cutis. 2010;85:301-302.
- Ohashi T, Yamamoto T. Exacerbation of psoriasis with pustulation by sorafenib in a patient with metastatic hepatocellular carcinoma. Indian J Dermatol. 2019;64:75-77.
- Chu D, Lacouture ME, Fillos T, et al. Risk of hand-foot skin reaction with sorafenib: a systematic review and meta-analysis. Acta Oncol (Madr). 2008;47:176-186.
- Canavese M, Altruda F, Ruzicka T, et al. Vascular endothelial growth factor (VEGF) in the pathogenesis of psoriasis--a possible target for novel therapies? J Dermatol Sci. 2010;58:171-176.
- Fournier C, Tisman G. Sorafenib-associated remission of psoriasis in hypernephroma: case report. Dermatol Online J. 2010;16:17.
- Akman A, Yilmaz E, Mutlu H, et al. Complete remission of psoriasis following bevacizumab therapy for colon cancer. Clin Exp Dermatol. 2009;34:E202-E204.
- Kong HH, Turner ML. Array of cutaneous adverse effects associated with sorafenib. J Am Acad Dermatol. 2009;61:360-361.
- Atalay F, Kızılkılıç E, Ada RS. Imatinib-induced psoriasis. Turk J Haematol. 2013;30:216-218.
Practice Points
- The use of targeted anticancer agents continues to expand. With this expansion, the number and type of cutaneous adverse events continues to increase.
- Although sorafenib is known to cause various dermatologic side effects, there are few reports of psoriasiform dermatitis.
- Increased awareness of sorafenib-induced psoriasiform dermatitis and its management is vital to prevent discontinuation of potentially life-saving anticancer therapy.
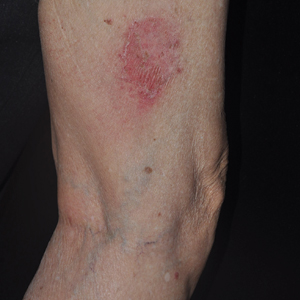
Flesh-Colored Papules on the Scrotum
The Diagnosis: Cutaneous Sarcoidosis
Histologic examination of the shave biopsy showed focal parakeratosis, irregular epidermal hyperplasia, and multiple noncaseating naked granulomas with occasional multinucleated giant cells in the dermis (Figure, A). The granulomas were surrounded by mild lymphocytic infiltration with rare eosinophils (Figure, B). Periodic acid-Schiff and Fite stains were negative for organisms, and polariscopic examination was negative; these findings confirmed the diagnosis of cutaneous sarcoidosis. Topical or intralesional steroids were recommended, but our patient declined treatment given that the lesions were asymptomatic.
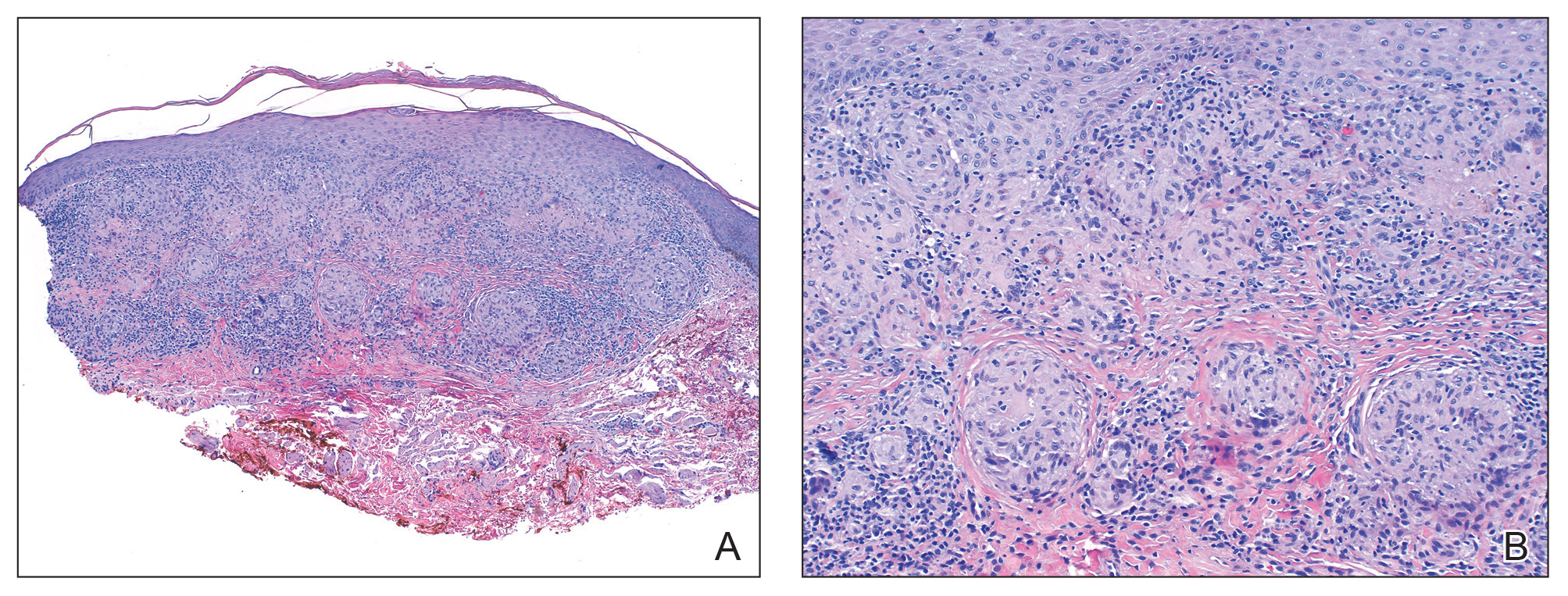
Sarcoidosis is a multisystem granulomatous disease of unknown etiology that affects the skin in approximately 25% of patients.1 Cutaneous lesions manifest in 2 forms: specific and nonspecific. Noncaseating granulomas are considered specific. Nonspecific lesions include erythema nodosum, calcinosis cutis, Sweet syndrome, and nail clubbing. The most common sites of specific sarcoidosis lesions include the face, lips, neck, upper trunk, and extremities. Few cases have reported cutaneous sarcoidosis involving the genitalia; most reports describe vulvar cutaneous sarcoidosis.2-4 Although there have been reports of sarcoidosis involving the epididymis and testes, which presented as scrotal masses, cutaneous scrotal involvement with the skin as the primary site of involvement is rare.5,6 McLaughlin et al5 reported an extensive, pruritic, and eczematous eruption of the scrotum with associated edema and tenderness. Wei et al6 reported cutaneous sarcoidosis in the form of multiple indurated papules involving the penis and scrotum, similar to our case.
Comparing our patient to the case reported by Wei et al,6 both patients had Fitzpatrick skin type V or VI and systemic involvement including pulmonary disease. However, Wei et al6 did not clearly mention if the cutaneous manifestations preceded the diagnosis of systemic sarcoidosis or if they were present at the time of the diagnosis. Our patient developed cutaneous lesions 4 years after being diagnosed with systemic sarcoidosis from hilar lymphadenopathy. In addition to the scrotal lesions, he also had a lesion of lupus pernio presenting as a violaceous to brown plaque on the tip of the nose. Although both patients denied pruritus, the other patient's lesions were painful.6 Wei et al6 mentioned that treatment with topical, intralesional, and systemic steroids failed, and the patient's lesions continued to progress. Generally, topical and intralesional steroids are considered mainstay treatment of cutaneous sarcoidosis despite insufficient data to support their efficacy.7
The differential diagnosis of papules on the scrotum can be broad. Our provisional diagnoses for this particular morphology of small, flesh-colored, shiny, polygonal, flat-topped papules included condyloma acuminatum; lichen planus; idiopathic scrotal calcinosis; steatocystoma multiplex; and sarcoidosis (although uncommon for the site), given the history of pulmonary involvement. We considered a diagnosis of condyloma acuminatum, but the lesions were too shiny and smooth. On histology, condyloma acuminatum shows a hyperkeratotic and parakeratotic stratum corneum, an exophytic growth with marked acanthosis, and superficially located koilocytes. Morphologically, our patient's lesions resembled genital lichen planus. However, Wickham striae were absent, and our patient's lesions were asymptomatic while lesions of lichen planus usually are pruritic. Histologically, lichen planus is characterized by hyperkeratosis, hypergranulosis, sawtooth rete ridges, and lichenoid interface inflammation. Idiopathic scrotal calcinosis also could be included in the differential; however, the lesions would look whiter and firmer than those of our patient, and biopsy will clearly show calcium deposition. Steatocystoma multiplex is another condition that can affect the scrotum, along with the trunk, axillae, extremities, and neck. However, the lesions are expected to discharge oily material if squeezed and have a characteristic corrugated eosinophilic cuticle lining a cyst histologically.
Although it is undetermined if the risk for systemic involvement increases in patients with cutaneous sarcoidosis, evaluation for probable systemic involvement is necessary.1 Because cutaneous sarcoidosis generally can precede any systemic involvement, it would be reasonable to consider skin biopsies in patients who present with atypical wartlike lesions on the scrotum and penis to rule out sarcoidosis.
- Marcoval J, Mañá J, Rubio M. Specific cutaneous lesions in patients with systemic sarcoidosis: relationship to severity and chronicity of disease. Clin Exp Dermatol. 2011;36:739.
- Vera C, Funaro D, Bouffard D. Vulvar sarcoidosis: case report and review of the literature. J Cutan Med Surg. 2013;17:287-290.
- Watkins S, Ismail A, McKay K, et al. Systemic sarcoidosis with unique vulvar involvement. JAMA Dermatol. 2014;150:666-667.
- Pereira IB, Khan A. Sarcoidosis rare cutaneous manifestations: vulval and perianal involvement. J Obstet Gynaecol. 2017;6:1-2.
- McLaughlin SS, Linquist AM, Burnett JW. Cutaneous sarcoidosis of the scrotum: a rare manifestation of systemic disease. Acta Derm Venereol. 2002;82:216-217.
- Wei H, Friedman KA, Rudikoff D. Multiple indurated papules on penis and scrotum. J Cutan Med Surg. 2000;4:202-204.
- Doherty CB, Rosen T. Evidence-based therapy for cutaneous sarcoidosis. Drugs. 2008;68:1361.
The Diagnosis: Cutaneous Sarcoidosis
Histologic examination of the shave biopsy showed focal parakeratosis, irregular epidermal hyperplasia, and multiple noncaseating naked granulomas with occasional multinucleated giant cells in the dermis (Figure, A). The granulomas were surrounded by mild lymphocytic infiltration with rare eosinophils (Figure, B). Periodic acid-Schiff and Fite stains were negative for organisms, and polariscopic examination was negative; these findings confirmed the diagnosis of cutaneous sarcoidosis. Topical or intralesional steroids were recommended, but our patient declined treatment given that the lesions were asymptomatic.
Sarcoidosis is a multisystem granulomatous disease of unknown etiology that affects the skin in approximately 25% of patients.1 Cutaneous lesions manifest in 2 forms: specific and nonspecific. Noncaseating granulomas are considered specific. Nonspecific lesions include erythema nodosum, calcinosis cutis, Sweet syndrome, and nail clubbing. The most common sites of specific sarcoidosis lesions include the face, lips, neck, upper trunk, and extremities. Few cases have reported cutaneous sarcoidosis involving the genitalia; most reports describe vulvar cutaneous sarcoidosis.2-4 Although there have been reports of sarcoidosis involving the epididymis and testes, which presented as scrotal masses, cutaneous scrotal involvement with the skin as the primary site of involvement is rare.5,6 McLaughlin et al5 reported an extensive, pruritic, and eczematous eruption of the scrotum with associated edema and tenderness. Wei et al6 reported cutaneous sarcoidosis in the form of multiple indurated papules involving the penis and scrotum, similar to our case.
Comparing our patient to the case reported by Wei et al,6 both patients had Fitzpatrick skin type V or VI and systemic involvement including pulmonary disease. However, Wei et al6 did not clearly mention if the cutaneous manifestations preceded the diagnosis of systemic sarcoidosis or if they were present at the time of the diagnosis. Our patient developed cutaneous lesions 4 years after being diagnosed with systemic sarcoidosis from hilar lymphadenopathy. In addition to the scrotal lesions, he also had a lesion of lupus pernio presenting as a violaceous to brown plaque on the tip of the nose. Although both patients denied pruritus, the other patient's lesions were painful.6 Wei et al6 mentioned that treatment with topical, intralesional, and systemic steroids failed, and the patient's lesions continued to progress. Generally, topical and intralesional steroids are considered mainstay treatment of cutaneous sarcoidosis despite insufficient data to support their efficacy.7
The differential diagnosis of papules on the scrotum can be broad. Our provisional diagnoses for this particular morphology of small, flesh-colored, shiny, polygonal, flat-topped papules included condyloma acuminatum; lichen planus; idiopathic scrotal calcinosis; steatocystoma multiplex; and sarcoidosis (although uncommon for the site), given the history of pulmonary involvement. We considered a diagnosis of condyloma acuminatum, but the lesions were too shiny and smooth. On histology, condyloma acuminatum shows a hyperkeratotic and parakeratotic stratum corneum, an exophytic growth with marked acanthosis, and superficially located koilocytes. Morphologically, our patient's lesions resembled genital lichen planus. However, Wickham striae were absent, and our patient's lesions were asymptomatic while lesions of lichen planus usually are pruritic. Histologically, lichen planus is characterized by hyperkeratosis, hypergranulosis, sawtooth rete ridges, and lichenoid interface inflammation. Idiopathic scrotal calcinosis also could be included in the differential; however, the lesions would look whiter and firmer than those of our patient, and biopsy will clearly show calcium deposition. Steatocystoma multiplex is another condition that can affect the scrotum, along with the trunk, axillae, extremities, and neck. However, the lesions are expected to discharge oily material if squeezed and have a characteristic corrugated eosinophilic cuticle lining a cyst histologically.
Although it is undetermined if the risk for systemic involvement increases in patients with cutaneous sarcoidosis, evaluation for probable systemic involvement is necessary.1 Because cutaneous sarcoidosis generally can precede any systemic involvement, it would be reasonable to consider skin biopsies in patients who present with atypical wartlike lesions on the scrotum and penis to rule out sarcoidosis.
The Diagnosis: Cutaneous Sarcoidosis
Histologic examination of the shave biopsy showed focal parakeratosis, irregular epidermal hyperplasia, and multiple noncaseating naked granulomas with occasional multinucleated giant cells in the dermis (Figure, A). The granulomas were surrounded by mild lymphocytic infiltration with rare eosinophils (Figure, B). Periodic acid-Schiff and Fite stains were negative for organisms, and polariscopic examination was negative; these findings confirmed the diagnosis of cutaneous sarcoidosis. Topical or intralesional steroids were recommended, but our patient declined treatment given that the lesions were asymptomatic.
Sarcoidosis is a multisystem granulomatous disease of unknown etiology that affects the skin in approximately 25% of patients.1 Cutaneous lesions manifest in 2 forms: specific and nonspecific. Noncaseating granulomas are considered specific. Nonspecific lesions include erythema nodosum, calcinosis cutis, Sweet syndrome, and nail clubbing. The most common sites of specific sarcoidosis lesions include the face, lips, neck, upper trunk, and extremities. Few cases have reported cutaneous sarcoidosis involving the genitalia; most reports describe vulvar cutaneous sarcoidosis.2-4 Although there have been reports of sarcoidosis involving the epididymis and testes, which presented as scrotal masses, cutaneous scrotal involvement with the skin as the primary site of involvement is rare.5,6 McLaughlin et al5 reported an extensive, pruritic, and eczematous eruption of the scrotum with associated edema and tenderness. Wei et al6 reported cutaneous sarcoidosis in the form of multiple indurated papules involving the penis and scrotum, similar to our case.
Comparing our patient to the case reported by Wei et al,6 both patients had Fitzpatrick skin type V or VI and systemic involvement including pulmonary disease. However, Wei et al6 did not clearly mention if the cutaneous manifestations preceded the diagnosis of systemic sarcoidosis or if they were present at the time of the diagnosis. Our patient developed cutaneous lesions 4 years after being diagnosed with systemic sarcoidosis from hilar lymphadenopathy. In addition to the scrotal lesions, he also had a lesion of lupus pernio presenting as a violaceous to brown plaque on the tip of the nose. Although both patients denied pruritus, the other patient's lesions were painful.6 Wei et al6 mentioned that treatment with topical, intralesional, and systemic steroids failed, and the patient's lesions continued to progress. Generally, topical and intralesional steroids are considered mainstay treatment of cutaneous sarcoidosis despite insufficient data to support their efficacy.7
The differential diagnosis of papules on the scrotum can be broad. Our provisional diagnoses for this particular morphology of small, flesh-colored, shiny, polygonal, flat-topped papules included condyloma acuminatum; lichen planus; idiopathic scrotal calcinosis; steatocystoma multiplex; and sarcoidosis (although uncommon for the site), given the history of pulmonary involvement. We considered a diagnosis of condyloma acuminatum, but the lesions were too shiny and smooth. On histology, condyloma acuminatum shows a hyperkeratotic and parakeratotic stratum corneum, an exophytic growth with marked acanthosis, and superficially located koilocytes. Morphologically, our patient's lesions resembled genital lichen planus. However, Wickham striae were absent, and our patient's lesions were asymptomatic while lesions of lichen planus usually are pruritic. Histologically, lichen planus is characterized by hyperkeratosis, hypergranulosis, sawtooth rete ridges, and lichenoid interface inflammation. Idiopathic scrotal calcinosis also could be included in the differential; however, the lesions would look whiter and firmer than those of our patient, and biopsy will clearly show calcium deposition. Steatocystoma multiplex is another condition that can affect the scrotum, along with the trunk, axillae, extremities, and neck. However, the lesions are expected to discharge oily material if squeezed and have a characteristic corrugated eosinophilic cuticle lining a cyst histologically.
Although it is undetermined if the risk for systemic involvement increases in patients with cutaneous sarcoidosis, evaluation for probable systemic involvement is necessary.1 Because cutaneous sarcoidosis generally can precede any systemic involvement, it would be reasonable to consider skin biopsies in patients who present with atypical wartlike lesions on the scrotum and penis to rule out sarcoidosis.
- Marcoval J, Mañá J, Rubio M. Specific cutaneous lesions in patients with systemic sarcoidosis: relationship to severity and chronicity of disease. Clin Exp Dermatol. 2011;36:739.
- Vera C, Funaro D, Bouffard D. Vulvar sarcoidosis: case report and review of the literature. J Cutan Med Surg. 2013;17:287-290.
- Watkins S, Ismail A, McKay K, et al. Systemic sarcoidosis with unique vulvar involvement. JAMA Dermatol. 2014;150:666-667.
- Pereira IB, Khan A. Sarcoidosis rare cutaneous manifestations: vulval and perianal involvement. J Obstet Gynaecol. 2017;6:1-2.
- McLaughlin SS, Linquist AM, Burnett JW. Cutaneous sarcoidosis of the scrotum: a rare manifestation of systemic disease. Acta Derm Venereol. 2002;82:216-217.
- Wei H, Friedman KA, Rudikoff D. Multiple indurated papules on penis and scrotum. J Cutan Med Surg. 2000;4:202-204.
- Doherty CB, Rosen T. Evidence-based therapy for cutaneous sarcoidosis. Drugs. 2008;68:1361.
- Marcoval J, Mañá J, Rubio M. Specific cutaneous lesions in patients with systemic sarcoidosis: relationship to severity and chronicity of disease. Clin Exp Dermatol. 2011;36:739.
- Vera C, Funaro D, Bouffard D. Vulvar sarcoidosis: case report and review of the literature. J Cutan Med Surg. 2013;17:287-290.
- Watkins S, Ismail A, McKay K, et al. Systemic sarcoidosis with unique vulvar involvement. JAMA Dermatol. 2014;150:666-667.
- Pereira IB, Khan A. Sarcoidosis rare cutaneous manifestations: vulval and perianal involvement. J Obstet Gynaecol. 2017;6:1-2.
- McLaughlin SS, Linquist AM, Burnett JW. Cutaneous sarcoidosis of the scrotum: a rare manifestation of systemic disease. Acta Derm Venereol. 2002;82:216-217.
- Wei H, Friedman KA, Rudikoff D. Multiple indurated papules on penis and scrotum. J Cutan Med Surg. 2000;4:202-204.
- Doherty CB, Rosen T. Evidence-based therapy for cutaneous sarcoidosis. Drugs. 2008;68:1361.
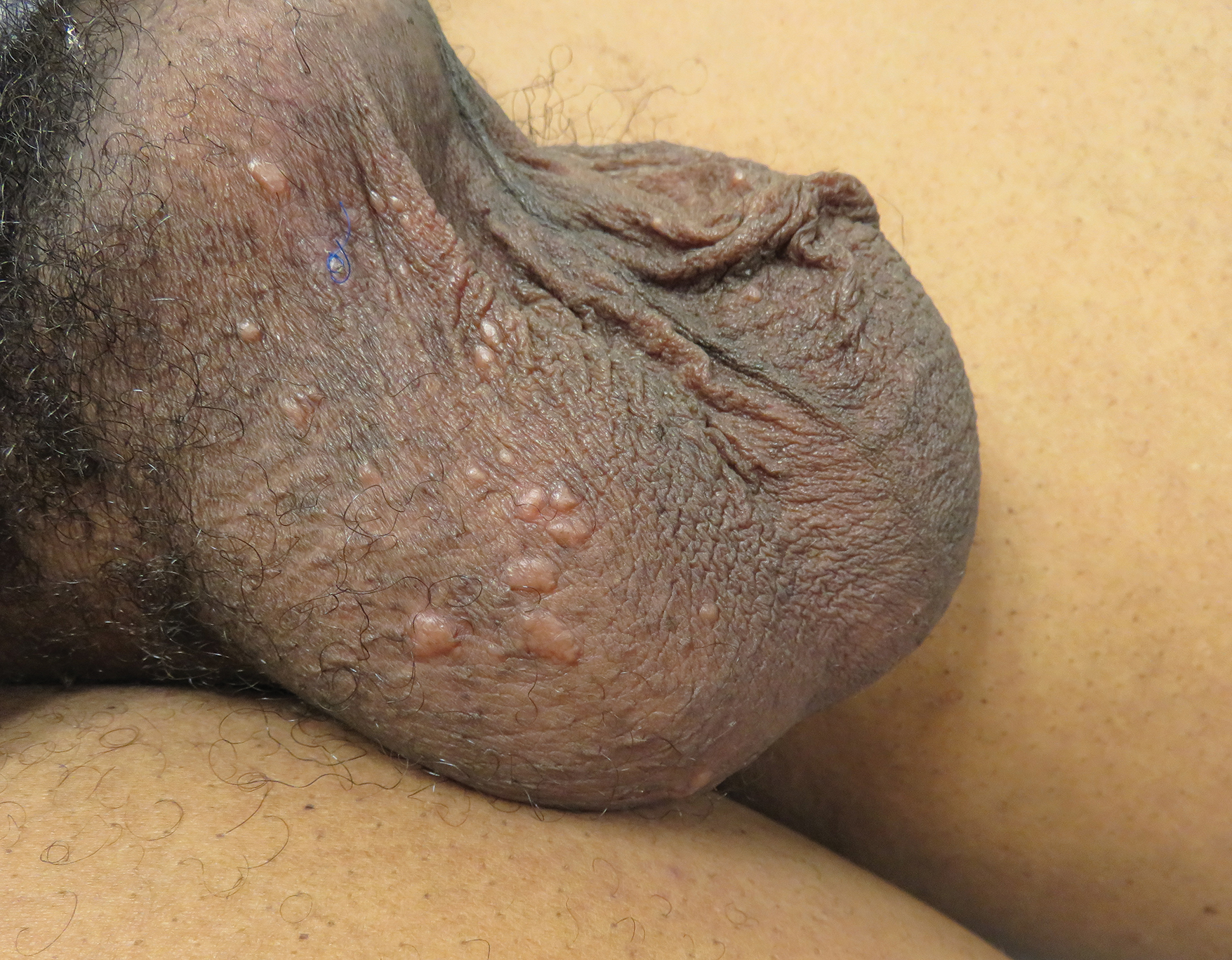
A 44-year-old black man presented with "bumps on the scrotum" of approximately 4 months' duration. They were asymptomatic and untreated. The patient denied extramarital sexual contacts or a history of any sexually transmitted infection. His medical history was notable for sarcoidosis diagnosed 4 years prior to presentation when hilar lymphadenopathy was incidentally found on routine screening. His condition was managed with regular follow-up without treatment. He also had a positive tuberculosis skin test in the past without radiologic evidence of active pulmonary disease. Physical examination revealed multiple 2- to 5-mm, flesh-colored, shiny, polygonal, flat-topped papules spread diffusely over the scrotum. A 1-cm, barely palpable, nonscaly, violaceous to brown plaque also was seen on the tip of the nose. A punch biopsy was taken from a lesion on the scrotum.