User login
Cutis is a peer-reviewed clinical journal for the dermatologist, allergist, and general practitioner published monthly since 1965. Concise clinical articles present the practical side of dermatology, helping physicians to improve patient care. Cutis is referenced in Index Medicus/MEDLINE and is written and edited by industry leaders.
ass lick
assault rifle
balls
ballsac
black jack
bleach
Boko Haram
bondage
causas
cheap
child abuse
cocaine
compulsive behaviors
cost of miracles
cunt
Daech
display network stats
drug paraphernalia
explosion
fart
fda and death
fda AND warn
fda AND warning
fda AND warns
feom
fuck
gambling
gfc
gun
human trafficking
humira AND expensive
illegal
ISIL
ISIS
Islamic caliphate
Islamic state
madvocate
masturbation
mixed martial arts
MMA
molestation
national rifle association
NRA
nsfw
nuccitelli
pedophile
pedophilia
poker
porn
porn
pornography
psychedelic drug
recreational drug
sex slave rings
shit
slot machine
snort
substance abuse
terrorism
terrorist
texarkana
Texas hold 'em
UFC
section[contains(@class, 'nav-hidden')]
section[contains(@class, 'nav-hidden active')
A peer-reviewed, indexed journal for dermatologists with original research, image quizzes, cases and reviews, and columns.
Telangiectatic Patch on the Neck
The Diagnosis: Unilateral Nevoid Telangiectasia
Unilateral nevoid telangiectasia (UNT) is an uncommon, or perhaps underreported, cutaneous condition involving telangiectatic patches in a unilateral dermatomal or blaschkoid pattern.1 The condition has been described as either congenital or acquired. Congenital UNT is thought to be a result of somatic mosaicism, whereby a mutation during embryogenesis leads to a distinct population of cells expressing the vascular malformation.1 Congenital UNT has been associated with Becker nevus, which also is thought to be a result of somatic mosaicism, further providing evidence for this theory, though it is unclear whether this finding is incidental.2 The acquired form often is associated with fluctuation of hormones, such as in pregnancy or with oral contraceptive initiation, as well as with hepatic disease as seen in our patient. However, there are many cases of acquired UNT with no implicated underlying disease, alcohol abuse, or hormonal changes, which calls into question if UNT is definitively an estrogen-related condition.3 One study demonstrated an increased level of estrogen and progesterone receptors in affected skin, which may have led to expression of the cutaneous changes at that site.4 More research is needed to elucidate this point, as other studies have not reproduced similar findings.
Congenital UNT occurs more commonly in males, whereas the acquired variant is seen more frequently in females. The third and fourth cervical dermatomes most often are involved.5 Most lesions persist without spontaneous resolution. Treatment options are limited and include pulsed dye laser treatment and makeup application to cover the telangiectatic patches. The main side effect seen with pulsed dye laser treatment is reversible pigmentary changes, with 1 report of textural skin change.6
A biopsy was deemed unnecessary for the clinical diagnosis in our patient because there was a clear explanation for the physical examination findings due to long-standing underlying liver disease. When biopsied, UNT characteristically demonstrates dilated dermal capillaries.5 Our patient elected not to pursue laser therapy but expressed interest in using makeup to camouflage the lesion.
The differential diagnosis includes acquired nevus flammeus, which typically is present on the face and often appears following mechanical or thermal trauma. Angioma serpiginosum most often occurs on the buttocks and legs as small red papules or puncta coalescing into a serpiginous linear arrangement. It often appears in childhood. Angiosarcoma is an aggressive malignancy that often occurs on the head and neck in elderly patients. It is associated with areas of long-standing lymphedema and often appears as a bruiselike lesion. Rosacea typically is not fixed in its clinical appearance and presents as transitory flushing of the head and neck with or without a history of acneform eruptions on the face. It typically is not unilateral.
- Wilkin JK. Unilateral dermatomal superficial telangiectasia. Arch Dermatol. 1984;120:579-580.
- Karakaş M, Durdu M, Sönmezoğlu S, et al. Unilateral nevoid telangiectasia. J Dermatol. 2004;31:109-112.
- Taskapan O, Harmanyeri Y, Sener O, et al. Acquired unilateral nevoid telangiectasia syndrome. Acta Derm Venereol. 1997;77:62-63.
- Uhlin SR, McCarty KS Jr. Unilateral nevoid telangiectatic syndrome: the role of estrogen and progesterone receptors. Arch Dermatol. 1983;119:226-228.
- Derrow AE, Adams BB, Timani S, et al. Acquired unilateral nevoid telangiectasia in a 51-year-old female. Int J Dermatol. 2008;47:1331-1333.
- Sharma VK, Khandpur S. Unilateral nevoid telangiectasia--response to pulsed dye laser. Int J Dermatol. 2006;45:960-964.
The Diagnosis: Unilateral Nevoid Telangiectasia
Unilateral nevoid telangiectasia (UNT) is an uncommon, or perhaps underreported, cutaneous condition involving telangiectatic patches in a unilateral dermatomal or blaschkoid pattern.1 The condition has been described as either congenital or acquired. Congenital UNT is thought to be a result of somatic mosaicism, whereby a mutation during embryogenesis leads to a distinct population of cells expressing the vascular malformation.1 Congenital UNT has been associated with Becker nevus, which also is thought to be a result of somatic mosaicism, further providing evidence for this theory, though it is unclear whether this finding is incidental.2 The acquired form often is associated with fluctuation of hormones, such as in pregnancy or with oral contraceptive initiation, as well as with hepatic disease as seen in our patient. However, there are many cases of acquired UNT with no implicated underlying disease, alcohol abuse, or hormonal changes, which calls into question if UNT is definitively an estrogen-related condition.3 One study demonstrated an increased level of estrogen and progesterone receptors in affected skin, which may have led to expression of the cutaneous changes at that site.4 More research is needed to elucidate this point, as other studies have not reproduced similar findings.
Congenital UNT occurs more commonly in males, whereas the acquired variant is seen more frequently in females. The third and fourth cervical dermatomes most often are involved.5 Most lesions persist without spontaneous resolution. Treatment options are limited and include pulsed dye laser treatment and makeup application to cover the telangiectatic patches. The main side effect seen with pulsed dye laser treatment is reversible pigmentary changes, with 1 report of textural skin change.6
A biopsy was deemed unnecessary for the clinical diagnosis in our patient because there was a clear explanation for the physical examination findings due to long-standing underlying liver disease. When biopsied, UNT characteristically demonstrates dilated dermal capillaries.5 Our patient elected not to pursue laser therapy but expressed interest in using makeup to camouflage the lesion.
The differential diagnosis includes acquired nevus flammeus, which typically is present on the face and often appears following mechanical or thermal trauma. Angioma serpiginosum most often occurs on the buttocks and legs as small red papules or puncta coalescing into a serpiginous linear arrangement. It often appears in childhood. Angiosarcoma is an aggressive malignancy that often occurs on the head and neck in elderly patients. It is associated with areas of long-standing lymphedema and often appears as a bruiselike lesion. Rosacea typically is not fixed in its clinical appearance and presents as transitory flushing of the head and neck with or without a history of acneform eruptions on the face. It typically is not unilateral.
The Diagnosis: Unilateral Nevoid Telangiectasia
Unilateral nevoid telangiectasia (UNT) is an uncommon, or perhaps underreported, cutaneous condition involving telangiectatic patches in a unilateral dermatomal or blaschkoid pattern.1 The condition has been described as either congenital or acquired. Congenital UNT is thought to be a result of somatic mosaicism, whereby a mutation during embryogenesis leads to a distinct population of cells expressing the vascular malformation.1 Congenital UNT has been associated with Becker nevus, which also is thought to be a result of somatic mosaicism, further providing evidence for this theory, though it is unclear whether this finding is incidental.2 The acquired form often is associated with fluctuation of hormones, such as in pregnancy or with oral contraceptive initiation, as well as with hepatic disease as seen in our patient. However, there are many cases of acquired UNT with no implicated underlying disease, alcohol abuse, or hormonal changes, which calls into question if UNT is definitively an estrogen-related condition.3 One study demonstrated an increased level of estrogen and progesterone receptors in affected skin, which may have led to expression of the cutaneous changes at that site.4 More research is needed to elucidate this point, as other studies have not reproduced similar findings.
Congenital UNT occurs more commonly in males, whereas the acquired variant is seen more frequently in females. The third and fourth cervical dermatomes most often are involved.5 Most lesions persist without spontaneous resolution. Treatment options are limited and include pulsed dye laser treatment and makeup application to cover the telangiectatic patches. The main side effect seen with pulsed dye laser treatment is reversible pigmentary changes, with 1 report of textural skin change.6
A biopsy was deemed unnecessary for the clinical diagnosis in our patient because there was a clear explanation for the physical examination findings due to long-standing underlying liver disease. When biopsied, UNT characteristically demonstrates dilated dermal capillaries.5 Our patient elected not to pursue laser therapy but expressed interest in using makeup to camouflage the lesion.
The differential diagnosis includes acquired nevus flammeus, which typically is present on the face and often appears following mechanical or thermal trauma. Angioma serpiginosum most often occurs on the buttocks and legs as small red papules or puncta coalescing into a serpiginous linear arrangement. It often appears in childhood. Angiosarcoma is an aggressive malignancy that often occurs on the head and neck in elderly patients. It is associated with areas of long-standing lymphedema and often appears as a bruiselike lesion. Rosacea typically is not fixed in its clinical appearance and presents as transitory flushing of the head and neck with or without a history of acneform eruptions on the face. It typically is not unilateral.
- Wilkin JK. Unilateral dermatomal superficial telangiectasia. Arch Dermatol. 1984;120:579-580.
- Karakaş M, Durdu M, Sönmezoğlu S, et al. Unilateral nevoid telangiectasia. J Dermatol. 2004;31:109-112.
- Taskapan O, Harmanyeri Y, Sener O, et al. Acquired unilateral nevoid telangiectasia syndrome. Acta Derm Venereol. 1997;77:62-63.
- Uhlin SR, McCarty KS Jr. Unilateral nevoid telangiectatic syndrome: the role of estrogen and progesterone receptors. Arch Dermatol. 1983;119:226-228.
- Derrow AE, Adams BB, Timani S, et al. Acquired unilateral nevoid telangiectasia in a 51-year-old female. Int J Dermatol. 2008;47:1331-1333.
- Sharma VK, Khandpur S. Unilateral nevoid telangiectasia--response to pulsed dye laser. Int J Dermatol. 2006;45:960-964.
- Wilkin JK. Unilateral dermatomal superficial telangiectasia. Arch Dermatol. 1984;120:579-580.
- Karakaş M, Durdu M, Sönmezoğlu S, et al. Unilateral nevoid telangiectasia. J Dermatol. 2004;31:109-112.
- Taskapan O, Harmanyeri Y, Sener O, et al. Acquired unilateral nevoid telangiectasia syndrome. Acta Derm Venereol. 1997;77:62-63.
- Uhlin SR, McCarty KS Jr. Unilateral nevoid telangiectatic syndrome: the role of estrogen and progesterone receptors. Arch Dermatol. 1983;119:226-228.
- Derrow AE, Adams BB, Timani S, et al. Acquired unilateral nevoid telangiectasia in a 51-year-old female. Int J Dermatol. 2008;47:1331-1333.
- Sharma VK, Khandpur S. Unilateral nevoid telangiectasia--response to pulsed dye laser. Int J Dermatol. 2006;45:960-964.
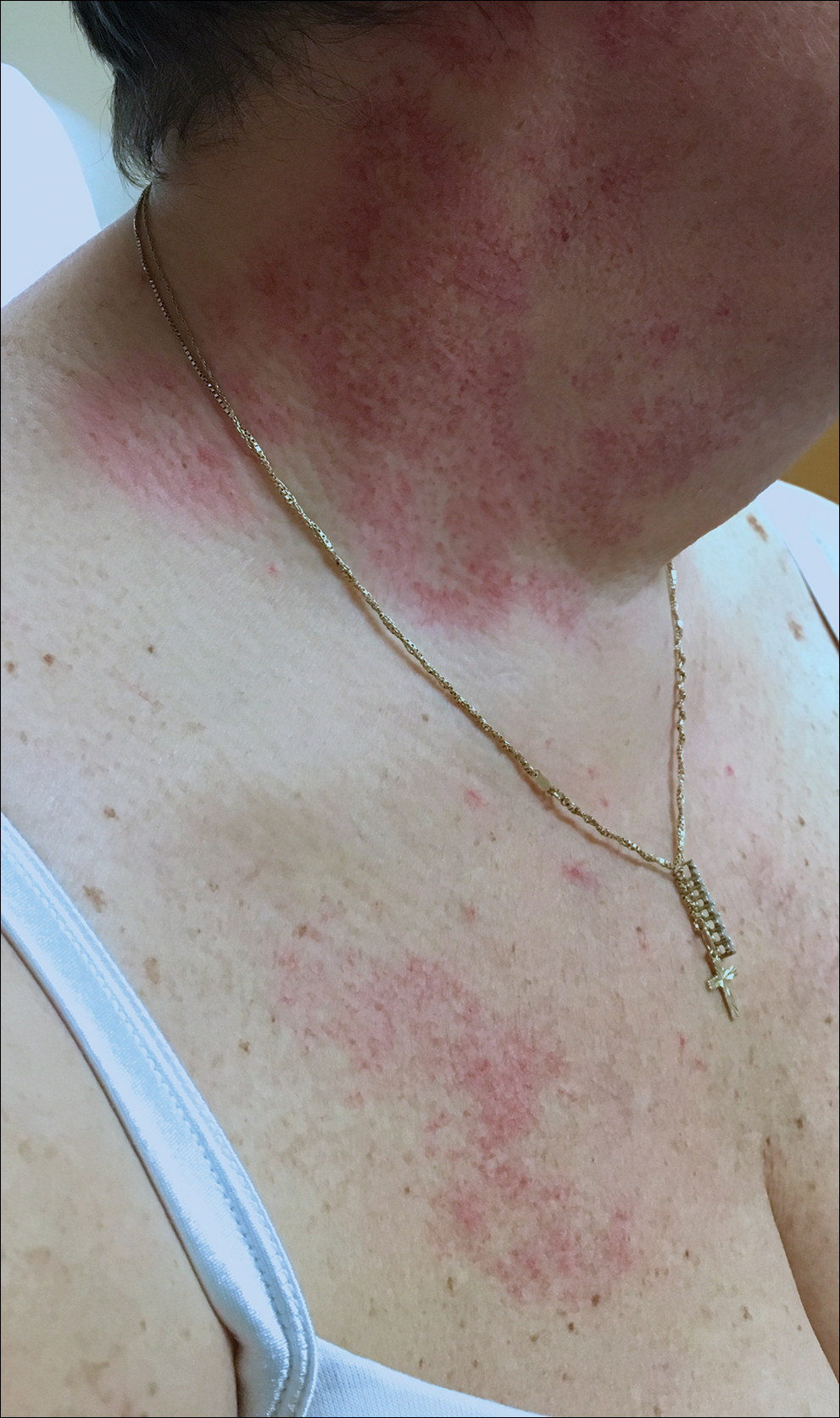
A 55-year-old woman presented to our clinic for a total-body skin examination and was noted to have a completely blanchable telangiectatic patch on the right side of the neck extending down onto the chest and breast. The patient reported that it had been present for 15 years and had slowly expanded in size. The lesion was asymptomatic. Pertinent medical history included cryptogenic cirrhosis of the liver, and she was undergoing a workup for a liver transplant.
Latex Allergy From Biologic Injectable Devices


Friable Erythema and Erosions on the Mouth
The Diagnosis: Radiation Mucositis
The patient was undergoing active radiation therapy for squamous cell carcinoma of the tongue, and according to the oncology team, the findings were in the precise location of radiation exposure. Radiation mucositis is a major and limiting side effect of radiation therapy for head and neck mucosal cancers, and symptom management is critical to ensure completion of the full radiation dose. Although infectious etiologies must be considered, the patient was already on prophylactic antiviral and antibacterial therapies. Moreover, the focal involvement with sparing of more mucosal tissue is atypical for most infections. Fixed drug reactions can present with localized mucosal and nonmucosal inflammation leading to erosion or ulceration. In this case, the only potential culprit was levofloxacin; however, it was initiated 2 days prior, and the patient never had reactions to this medication in the past.
Acute radiation mucositis is a transient but major limiting side effect of radiation therapy. The associated odynophagia, secondary infection, and reduced oral intake often can lead to diminished disease control secondary to treatment interruption and subsequent development of resistant tumor burden. Concurrent chemotherapy and alternated fractionation radiation therapy increase the incidence of mucositis. Trotti et al1 (n=6181) reported that severe mucositis (grades 3 to 4) was found in 56% of patients receiving altered fractionation radiation therapy compared to 34% of patients who received conventional radiation therapy. Other risk factors related to the development of acute radiation mucositis include associated chemotherapy, age (>65 years), poor oral hygiene, diabetes mellitus, and prior periodontal disease.2
Radiation causes direct cellular damage to keratinocytes, leading to ulceration and erythema, as well as keratinocyte stem cells, which interferes with the healing process. Typical symptoms of mucosal radiation injury may include erythema (asymptomatic or causing intolerance of warm foods) that develops at the end of the second week of radiation therapy, focal areas of desquamation that develops in week 3, and confluent mucositis that can further progress to ulceration and necrosis in weeks 4 to 5.2 The development of dysgeusia, which is estimated to occur in 67% of patients receiving radiotherapy and 76% of patients receiving combination therapy, also can contribute to nutritional difficulties and weight loss.3
Avoiding overtreatment by constraining radiation volume and limiting concurrent chemotherapy are important preventative measures. The mainstay for managing mucositis includes symptomatic relief with oral hygiene, topical agents, topical plus systemic analgesia, dietary changes, and treatment of associated infections. Benzydamine, a nonsteroidal anti-inflammatory drug, is not available in the United States but has been shown to effectively improve symptoms.4 Various formulations of topical anesthetics consisting of diphenhydramine with or without corticosteroids, antibiotics, and antifungals help alleviate symptoms of mucositis; however, no single formulation has been studied. Low-level laser therapy also has shown efficacy in managing symptoms of mucositis.5,6 For persistent odynophagia, systemic opioid therapy should be attempted to achieve uninterrupted radiation therapy. Severe mucositis requires balancing risks and benefits of interrupting treatment, as additional damage may cause permanent mucosal injury.
Our patient had adequate symptom control with benzocaine lozenges and a combination mouthwash containing diphenhydramine, nystatin, lidocaine, hydrocortisone, and tetracycline. He required only occasional doses of systemic oxycodone. After a 1-week hospital admission for treatment of the pneumonia, he resumed radiation therapy and completed a full 8-week radiation course.
- Trotti A, Bellm LA, Epstein JB, et al. Mucositis incidence, severity and associated outcomes in patients with head and neck cancer receiving radiotherapy with or without chemotherapy: a systematic literature review. Radiother Oncol. 2003;66:253-262.
- Mallick S, Benson R, Rath GK. Radiation induced oral mucositis: a review of current literature on prevention and management. Eur Arch Otorhinolaryngol. 2016;273:2285-2293.
- Hovan AJ, Williams PM, Stevenson-Moore P, et al; Dysgeusia Section, Oral Care Study Group, Multinational Association of Supportive Care in Cancer (MASCC)/International Society of Oral Oncology (ISOO). A systematic review of dysgeusia induced by cancer therapies. Support Care Cancer. 2010;18:1081-1087.
- Epstein JB, Silverman S, Paggiarino DA, et al. Benzydamine HCl for prophylaxis of radiation‐induced oral mucositis. Cancer. 2001;92:875-885.
- Henke M, Alfonsi M, Foa P, et al. Palifermin decreases severe oral mucositis of patients undergoing postoperative radiochemotherapy for head and neck cancer: a randomized, placebo-controlled trial. J Clin Oncol. 2011;29:2815-2820.
- Bensadoun RJ, Nair RG. Low-level laser therapy in the prevention and treatment of cancer therapy-induced mucositis: 2012 state of the art based on literature review and meta-analysis. Curr Opin Oncol. 2012;24:363-370.
The Diagnosis: Radiation Mucositis
The patient was undergoing active radiation therapy for squamous cell carcinoma of the tongue, and according to the oncology team, the findings were in the precise location of radiation exposure. Radiation mucositis is a major and limiting side effect of radiation therapy for head and neck mucosal cancers, and symptom management is critical to ensure completion of the full radiation dose. Although infectious etiologies must be considered, the patient was already on prophylactic antiviral and antibacterial therapies. Moreover, the focal involvement with sparing of more mucosal tissue is atypical for most infections. Fixed drug reactions can present with localized mucosal and nonmucosal inflammation leading to erosion or ulceration. In this case, the only potential culprit was levofloxacin; however, it was initiated 2 days prior, and the patient never had reactions to this medication in the past.
Acute radiation mucositis is a transient but major limiting side effect of radiation therapy. The associated odynophagia, secondary infection, and reduced oral intake often can lead to diminished disease control secondary to treatment interruption and subsequent development of resistant tumor burden. Concurrent chemotherapy and alternated fractionation radiation therapy increase the incidence of mucositis. Trotti et al1 (n=6181) reported that severe mucositis (grades 3 to 4) was found in 56% of patients receiving altered fractionation radiation therapy compared to 34% of patients who received conventional radiation therapy. Other risk factors related to the development of acute radiation mucositis include associated chemotherapy, age (>65 years), poor oral hygiene, diabetes mellitus, and prior periodontal disease.2
Radiation causes direct cellular damage to keratinocytes, leading to ulceration and erythema, as well as keratinocyte stem cells, which interferes with the healing process. Typical symptoms of mucosal radiation injury may include erythema (asymptomatic or causing intolerance of warm foods) that develops at the end of the second week of radiation therapy, focal areas of desquamation that develops in week 3, and confluent mucositis that can further progress to ulceration and necrosis in weeks 4 to 5.2 The development of dysgeusia, which is estimated to occur in 67% of patients receiving radiotherapy and 76% of patients receiving combination therapy, also can contribute to nutritional difficulties and weight loss.3
Avoiding overtreatment by constraining radiation volume and limiting concurrent chemotherapy are important preventative measures. The mainstay for managing mucositis includes symptomatic relief with oral hygiene, topical agents, topical plus systemic analgesia, dietary changes, and treatment of associated infections. Benzydamine, a nonsteroidal anti-inflammatory drug, is not available in the United States but has been shown to effectively improve symptoms.4 Various formulations of topical anesthetics consisting of diphenhydramine with or without corticosteroids, antibiotics, and antifungals help alleviate symptoms of mucositis; however, no single formulation has been studied. Low-level laser therapy also has shown efficacy in managing symptoms of mucositis.5,6 For persistent odynophagia, systemic opioid therapy should be attempted to achieve uninterrupted radiation therapy. Severe mucositis requires balancing risks and benefits of interrupting treatment, as additional damage may cause permanent mucosal injury.
Our patient had adequate symptom control with benzocaine lozenges and a combination mouthwash containing diphenhydramine, nystatin, lidocaine, hydrocortisone, and tetracycline. He required only occasional doses of systemic oxycodone. After a 1-week hospital admission for treatment of the pneumonia, he resumed radiation therapy and completed a full 8-week radiation course.
The Diagnosis: Radiation Mucositis
The patient was undergoing active radiation therapy for squamous cell carcinoma of the tongue, and according to the oncology team, the findings were in the precise location of radiation exposure. Radiation mucositis is a major and limiting side effect of radiation therapy for head and neck mucosal cancers, and symptom management is critical to ensure completion of the full radiation dose. Although infectious etiologies must be considered, the patient was already on prophylactic antiviral and antibacterial therapies. Moreover, the focal involvement with sparing of more mucosal tissue is atypical for most infections. Fixed drug reactions can present with localized mucosal and nonmucosal inflammation leading to erosion or ulceration. In this case, the only potential culprit was levofloxacin; however, it was initiated 2 days prior, and the patient never had reactions to this medication in the past.
Acute radiation mucositis is a transient but major limiting side effect of radiation therapy. The associated odynophagia, secondary infection, and reduced oral intake often can lead to diminished disease control secondary to treatment interruption and subsequent development of resistant tumor burden. Concurrent chemotherapy and alternated fractionation radiation therapy increase the incidence of mucositis. Trotti et al1 (n=6181) reported that severe mucositis (grades 3 to 4) was found in 56% of patients receiving altered fractionation radiation therapy compared to 34% of patients who received conventional radiation therapy. Other risk factors related to the development of acute radiation mucositis include associated chemotherapy, age (>65 years), poor oral hygiene, diabetes mellitus, and prior periodontal disease.2
Radiation causes direct cellular damage to keratinocytes, leading to ulceration and erythema, as well as keratinocyte stem cells, which interferes with the healing process. Typical symptoms of mucosal radiation injury may include erythema (asymptomatic or causing intolerance of warm foods) that develops at the end of the second week of radiation therapy, focal areas of desquamation that develops in week 3, and confluent mucositis that can further progress to ulceration and necrosis in weeks 4 to 5.2 The development of dysgeusia, which is estimated to occur in 67% of patients receiving radiotherapy and 76% of patients receiving combination therapy, also can contribute to nutritional difficulties and weight loss.3
Avoiding overtreatment by constraining radiation volume and limiting concurrent chemotherapy are important preventative measures. The mainstay for managing mucositis includes symptomatic relief with oral hygiene, topical agents, topical plus systemic analgesia, dietary changes, and treatment of associated infections. Benzydamine, a nonsteroidal anti-inflammatory drug, is not available in the United States but has been shown to effectively improve symptoms.4 Various formulations of topical anesthetics consisting of diphenhydramine with or without corticosteroids, antibiotics, and antifungals help alleviate symptoms of mucositis; however, no single formulation has been studied. Low-level laser therapy also has shown efficacy in managing symptoms of mucositis.5,6 For persistent odynophagia, systemic opioid therapy should be attempted to achieve uninterrupted radiation therapy. Severe mucositis requires balancing risks and benefits of interrupting treatment, as additional damage may cause permanent mucosal injury.
Our patient had adequate symptom control with benzocaine lozenges and a combination mouthwash containing diphenhydramine, nystatin, lidocaine, hydrocortisone, and tetracycline. He required only occasional doses of systemic oxycodone. After a 1-week hospital admission for treatment of the pneumonia, he resumed radiation therapy and completed a full 8-week radiation course.
- Trotti A, Bellm LA, Epstein JB, et al. Mucositis incidence, severity and associated outcomes in patients with head and neck cancer receiving radiotherapy with or without chemotherapy: a systematic literature review. Radiother Oncol. 2003;66:253-262.
- Mallick S, Benson R, Rath GK. Radiation induced oral mucositis: a review of current literature on prevention and management. Eur Arch Otorhinolaryngol. 2016;273:2285-2293.
- Hovan AJ, Williams PM, Stevenson-Moore P, et al; Dysgeusia Section, Oral Care Study Group, Multinational Association of Supportive Care in Cancer (MASCC)/International Society of Oral Oncology (ISOO). A systematic review of dysgeusia induced by cancer therapies. Support Care Cancer. 2010;18:1081-1087.
- Epstein JB, Silverman S, Paggiarino DA, et al. Benzydamine HCl for prophylaxis of radiation‐induced oral mucositis. Cancer. 2001;92:875-885.
- Henke M, Alfonsi M, Foa P, et al. Palifermin decreases severe oral mucositis of patients undergoing postoperative radiochemotherapy for head and neck cancer: a randomized, placebo-controlled trial. J Clin Oncol. 2011;29:2815-2820.
- Bensadoun RJ, Nair RG. Low-level laser therapy in the prevention and treatment of cancer therapy-induced mucositis: 2012 state of the art based on literature review and meta-analysis. Curr Opin Oncol. 2012;24:363-370.
- Trotti A, Bellm LA, Epstein JB, et al. Mucositis incidence, severity and associated outcomes in patients with head and neck cancer receiving radiotherapy with or without chemotherapy: a systematic literature review. Radiother Oncol. 2003;66:253-262.
- Mallick S, Benson R, Rath GK. Radiation induced oral mucositis: a review of current literature on prevention and management. Eur Arch Otorhinolaryngol. 2016;273:2285-2293.
- Hovan AJ, Williams PM, Stevenson-Moore P, et al; Dysgeusia Section, Oral Care Study Group, Multinational Association of Supportive Care in Cancer (MASCC)/International Society of Oral Oncology (ISOO). A systematic review of dysgeusia induced by cancer therapies. Support Care Cancer. 2010;18:1081-1087.
- Epstein JB, Silverman S, Paggiarino DA, et al. Benzydamine HCl for prophylaxis of radiation‐induced oral mucositis. Cancer. 2001;92:875-885.
- Henke M, Alfonsi M, Foa P, et al. Palifermin decreases severe oral mucositis of patients undergoing postoperative radiochemotherapy for head and neck cancer: a randomized, placebo-controlled trial. J Clin Oncol. 2011;29:2815-2820.
- Bensadoun RJ, Nair RG. Low-level laser therapy in the prevention and treatment of cancer therapy-induced mucositis: 2012 state of the art based on literature review and meta-analysis. Curr Opin Oncol. 2012;24:363-370.
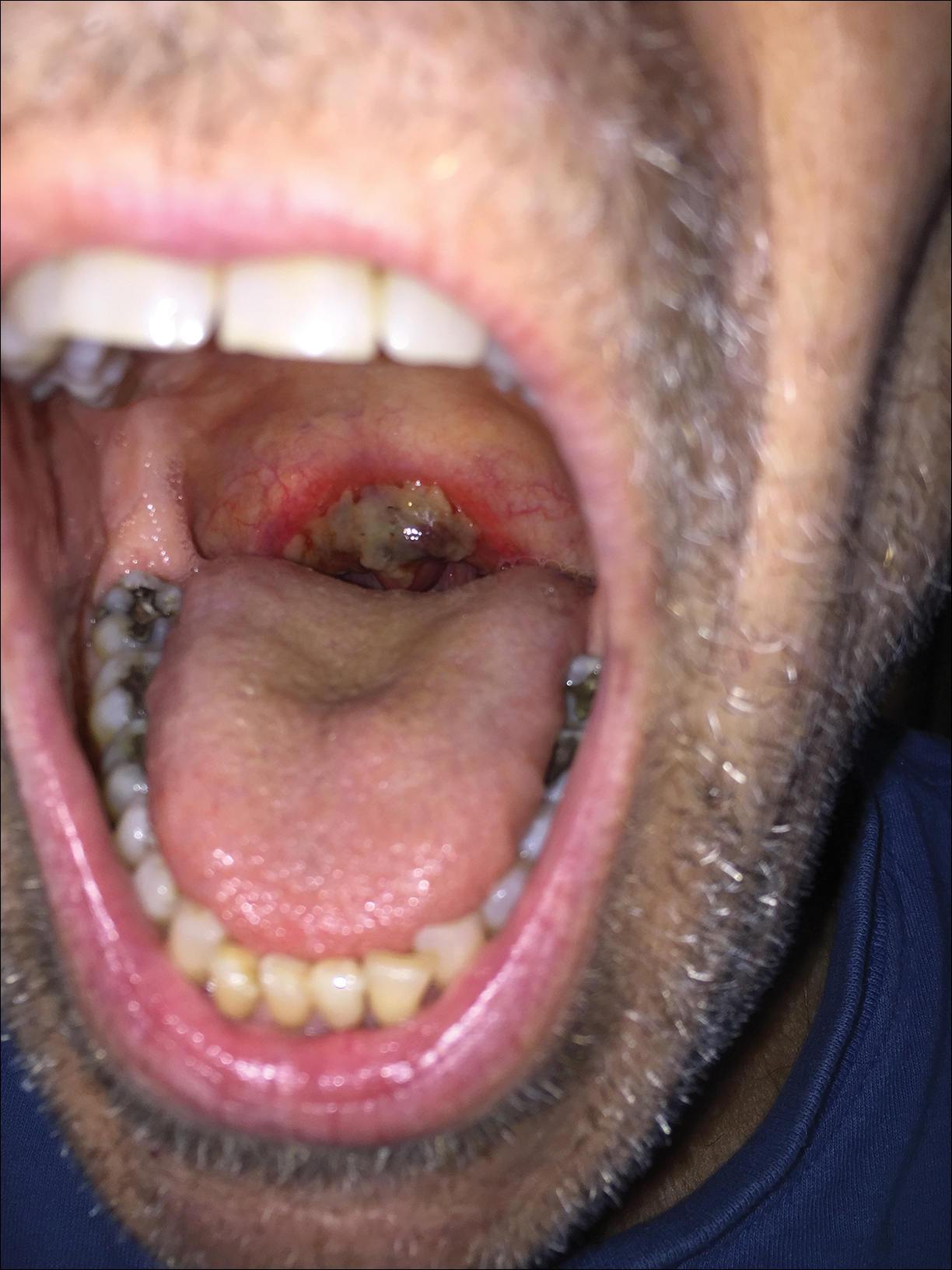
A 68-year-old man with squamous cell carcinoma of the tongue presented with a sore throat and odynophagia of 4 days' duration. At the time he was undergoing radiation therapy for the squamous cell carcinoma, and multiple myeloma was being actively treated with carfilzomib and pomalidomide. At the time of symptom onset he also was undergoing treatment with levofloxacin for community-acquired pneumonia. On day 2 of antibiotic therapy he noted pain with swallowing and an intolerance to warm foods. He was unaware of any new rash or lesions of the lips or mouth. He denied dysgeusia, changes in speech, bleeding, trauma, or recent smoking. He was taking prophylactic acyclovir and trimethoprim-sulfamethoxazole due to chemotherapy. Physical examination revealed a posterior oropharynx and uvula with well-defined friable erythema and erosions covered by white patches. There was no mucosal ulceration and no notable skin findings. The remainder of the physical examination was unremarkable.
Atrophodermalike Guttate Morphea
To the Editor:
Morphea, atrophoderma, guttate lichen sclerosus et atrophicus (LS&A), anetoderma, and their subtypes are inflammatory processes ultimately leading to dermal remodeling. We report a case of a scaly, hypopigmented, macular rash that clinically appeared as an entity along the morphea-atrophoderma spectrum and demonstrated unique histopathologic changes in both collagen and elastin confined to the upper reticular and papillary dermis. This case is a potentially rare variant representing a combination of clinical and microscopic findings.
A 29-year-old woman presented for an increasing number of white spots distributed on the trunk, arms, and legs. She denied local and systemic symptoms. The patient reported that she was stung by 100 wasps 23 years prior. Following the assault, her grandmother placed chewed tobacco leaves atop the painful erythematous wheals and flares. Upon resolution, hypopigmented macules and patches remained in their place. The patient denied associated symptoms or new lesions; she did not seek care at that time.
In her early 20s, the patient noted new, similarly distributed hypopigmented macules and patches without associated arthropod assault. She was treated by an outside dermatologist without result for presumed tinea versicolor. A follow-up superficial shave biopsy cited subtle psoriasiform dermatitis. Topical steroids did not improve the lesions. Her medical history also was remarkable for a reportedly unprovoked complete rotator cuff tear.
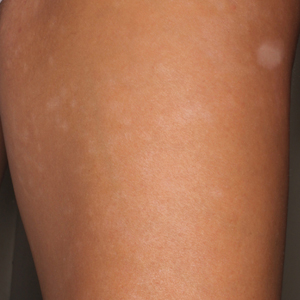
Physical examination revealed 0.5- to 2.0-cm, ill-defined, perifollicular and nonfollicular, slightly scaly macules and patches on the trunk, arms, and legs. There was no follicular plugging (Figure 1A). The hands, feet, face, and mucosal surfaces were spared. She had no family history of similar lesions. Although atrophic in appearance, a single lesion on the left thigh was palpably depressed (Figure 1B). Serology demonstrated a normal complete blood cell count and comprehensive metabolic panel, and negative Lyme titers. Light therapy and topical steroids failed to improve the lesions; calcipotriene cream 0.005% made the lesions erythematous and pruritic.
 as well as hypopigmented macules and a minimally depressed patch on the left thigh (B).")
A biopsy from a flank lesion demonstrated a normal epithelium without thinning, a normal basal melanocyte population, and minimally effaced rete ridges. Thin collagen bundles were noted in the upper reticular and papillary dermis with associated fibroplasia (Figure 2). Verhoeff-van Gieson stain revealed decreased and fragmented elastin filaments in the same dermal distribution as the changed collagen (Figure 3). There was no evidence of primary inflammatory disease. The dermis was thinned. Periodic acid–Schiff stain confirmed the absence of hyphae and spores.
(H&E, original magnification ×40)...")
(Verhoeff-van Gieson, original magnification ×40). Normal elastin network in the lower reticular dermis; note the normal size of elastin fibers (B)(Verhoeff-van Gieson, original magnification ×200).")
The relevant findings in our patient including the following: (1) onset of hypopigmented macules and patches following resolution of a toxic insult; (2) initially stable number of lesions that progressed in number but not size; (3) thinned collagen associated with fibroplasia in the upper reticular and papillary dermis; (4) decreased number and fragmentation of elastin filaments confined to the same region; (5) no congenital lesions or similar lesions in family members; and (6) a complete rotator cuff tear with no findings of a systemic connective-tissue disorder such as Ehlers-Danlos syndrome.
We performed a literature search of PubMed articles indexed for MEDLINE using combinations of the terms atrophic, hypopigmented, white, spot disease, confetti-like, guttate, macules, atrophoderma, morphea, anetoderma, elastin, and collagen to identify potentially similar reports of guttate hypopigmented macules demonstrating changes of the collagen and elastin in the papillary and upper reticular dermis. Some variants, namely atrophoderma of Pasini and Pierini (APP), guttate morphea, and superficial morphea, demonstrate similar clinical and histopathologic findings.
Findings similar to our case were documented in case reports of 2 women (aged 34 and 42 years)1 presenting with asymptomatic, atrophic, well-demarcated, shiny, hypopigmented macules over the trunk and upper extremities, which demonstrated a thinned epidermis with coarse hyalinized collagen bundles in the mid and lower dermis. There was upper and diffuse dermal elastolysis (patient 1 and patient 2, respectively).1 Our patient’s lesions were hypopigmented and atrophic in appearance but were slightly scaly and also involved the extremities. Distinct from these patient reports, histopathology from our case demonstrated thin packed collagen bundles and decreased fragmented elastin filaments confined to the upper reticular and papillary dermis.
Plaque morphea is the most common type of localized scleroderma.2 The subtype APP demonstrates round to ovoid, gray-brown depressions with cliff-drop borders. They may appear flesh colored or hypopigmented.3,4 These sclerodermoid lesions lack the violaceous border classic to morphea. Sclerosis and induration also are typically absent.5 Clinically, our patient’s macules resembled this entity. Histopathologically, APP shows normal epithelium with an increased basal layer pigmentation; preserved adnexal structures; and mid to lower dermal collagen edema, clumping, and homogenization.3,4 Elastic fibers classically are unchanged, with exceptions.6-11 Changes in the collagen and elastin of our patient were unlike those reported in APP, which occur in the mid to lower dermis.
Guttate morphea demonstrates small, pale, minimally indurated, coin-shaped lesions on the trunk. Histopathology reveals less sclerosis and more edema, resembling LS&A.12 The earliest descriptions of this entity describe 3 stages: ivory/chalk white, scaly, and atrophic. Follicular plugging (absent in this patient) and fine scale can exist at any stage.13,14 Flattened rete ridges mark an otherwise preserved epidermis; hyalinized collagen typically is superficial and demonstrates less sclerosis yet increased edema.12-14 Fewer elastic fibers typically are present compared to normal skin. Changes seen in this entity are more superficial, as with our patient, than classic scleroderma. However, classic edema was not found in our patient’s biopsy specimen.
Superficial morphea, occurring predominantly in females, presents with hyperpigmented or hypopigmented patches having minimal to no induration. The lesions typically are asymptomatic. Histopathologically, collagen deposition and inflammation are confined to the superficial dermis without homogenization associated with LS&A, findings that were consistent with this patient’s biopsy.15,16 However, similar to other morpheaform variants, elastic fibers are unchanged.15 Verhoeff-van Gieson stain of the biopsy (Figure 3) showed the decreased and fragmented elastin network in the upper reticular and papillary dermis, making this entity less compatible.
Guttate LS&A may present with interfollicular, bluish white macules or papules coalescing into patches or plaques. Lesions evolve to reveal atrophic thin skin with follicular plugging. Histology demonstrates a thinned epidermis with orthohypokeratosis marked by flattened rete ridges. The dermis reveals short hyalinized collagen fibrils with a loss of elastic fibers in the papillary and upper reticular dermis, giving a homogenized appearance. Early disease is marked by an inflammatory infiltrate.17 Most of these findings are consistent with our patient’s pathology, which was confined to the upper dermis. Lacking, however, were characteristic findings of LS&A, including upper dermal homogenization, near-total effacement of rete ridges, orthokeratosis, and vacuolar degeneration at the dermoepidermal junction. As such, this entity is less compatible.
Atrophoderma elastolyticum discretum has clinical features of atrophoderma with elastolytic histopathologic findings.1 Anetoderma presents with outpouchings of atrophic skin with a surrounding ring of normal tissue. Histopathologically, this entity shows normal collagen with elastolysis; there also is a decrease in desmosine, an elastin cross-linker.1,3 Neither the clinical nor histopathologic findings in this patient matched these 2 entities.
The reported chronologic association of these lesions with an arthropod assault raised suspicion to their association with toxic insult or postinflammatory changes. One study reported mechanical trauma, including insect bites, as a possible inciting factor of morphea.11 These data, gathered from patient surveys, reported trauma associated to lesion development.1,17 A review of the literature regarding atrophoderma, morphea, and LS&A failed to identify pathogenic changes seen in this patient following initial trauma. Moreover, although it is difficult to prove causality in the formation of the original hypopigmented spots, the development of identical spots in a similar distribution without further trauma suggests against these etiologies to fully explain her lesions. Nonetheless, circumstance makes it difficult to prove whether the original arthropod insult spurred a smoldering reactive process that caused the newer lesions.
Hereditary connective-tissue disorders also were considered in the differential diagnosis. Because of the patient’s history of an unprovoked complete rotator cuff tear, Ehlers-Danlos syndrome was considered; however, the remainder of her examination was normal, making a syndromic systemic disorder a less likely etiology.Because of the distinct clinical and histopathologic findings, this case may represent a rare and previously unreported variant of morphea. Clinically, these hypopigmented macules and patches exist somewhere along the morphea-atrophoderma spectrum. Histopathologic findings do not conform to prior reports. The name atrophodermalike guttate morphea may be an appropriate appellation. It is possible this presentation represents a variant of what dermatologists have referred to as white spot disease.18 We hope that this case may bring others to discussion, allowing for the identification of a more precise entity and etiology so that patients may receive more directed therapy.
- Aksoy B, Ustün H, Gulbahce R, et al. Confetti-like macular atrophy: a new entity? J Dermatol. 2009;36:592-597.
- Uitto J, Santa Cruz DJ, Bauer EA, et al. Morphea and lichen sclerosus et atrophicus. clinical and histopathologic studies in patients with combined features. J Am Acad Dermatol. 1980;3:271-279.
- Buechner SA, Rufli T. Atrophoderma of Pasini and Pierini. clinical and histopathologic findings and antibodies to Borrelia burgdorferi in thirty-four patients. J Am Acad Dermatol. 1994;30:441-446.
- Saleh Z, Abbas O, Dahdah MJ, et al. Atrophoderma of Pasini and Pierini: a clinical and histopathological study. J Cutan Pathol. 2008;35:1108-1114.
- Canizares O, Sachs PM, Jaimovich L, et al. Idiopathic atrophoderma of Pasini and Pierini. Arch Dermatol. 1958;77:42-58; discussion 58-60.
- Pullara TJ, Lober CW, Fenske NA. Idiopathic atrophoderma of Pasini and Pierini. Int J Dermatol. 1984;23:643-645.
- Jablonska S, Szczepanski A. Atrophoderma Pasini-Pierini: is it an entity? Dermatologica. 1962;125:226-242.
- Ang G, Hyde PM, Lee JB. Unilateral congenital linear atrophoderma of the leg. Pediatr Dermatol. 2005;22:350-354.
- Miteva L, Kadurina M. Unilateral idiopathic atrophoderma of Pasini and Pierini. Int J Dermatol. 2006;45:1391-1393.
- Kee CE, Brothers WS, New W. Idiopathic atrophoderma of Pasini and Pierini with coexistent morphea. a case report. Arch Dermatol. 1960;82:100-103.
- Zulian F, Athreya BH, Laxer R, et al. Juvenile localized scleroderma: clinical and epidemiological features in 750 children. an international study. Rheumatology. 2006;45:614-620.
- Winkelmann RK. Localized cutaneous scleroderma. Semin Dermatol. 1985;4:90-103.
- Dore SE. Two cases of morphoea guttata. Proc R Soc Med. 1918;11:26-28.
- Dore SE. Guttate morphoea. Proc R Soc Med. 1919;12:3-5.
- McNiff JM, Glusac EJ, Lazova RZ, et al. Morphea limited to the superficial reticular dermis: an underrecognized histologic phenomenon. Am J Dermatopathol. 1999;21:315-319.
- Jacobson L, Palazij R, Jaworsky C. Superficial morphea. J Am Acad Dermatol. 2003;49:323-325.
- Bolognia J, Jorizzo JL, Rapini RP, eds. Dermatology. 2nd ed. London, England: Mosby Elsevier; 2007.
- Bunch JL. White-spot disease (morphoea guttata). Proc R Soc Med. 1919;12:24-27.
To the Editor:
Morphea, atrophoderma, guttate lichen sclerosus et atrophicus (LS&A), anetoderma, and their subtypes are inflammatory processes ultimately leading to dermal remodeling. We report a case of a scaly, hypopigmented, macular rash that clinically appeared as an entity along the morphea-atrophoderma spectrum and demonstrated unique histopathologic changes in both collagen and elastin confined to the upper reticular and papillary dermis. This case is a potentially rare variant representing a combination of clinical and microscopic findings.
A 29-year-old woman presented for an increasing number of white spots distributed on the trunk, arms, and legs. She denied local and systemic symptoms. The patient reported that she was stung by 100 wasps 23 years prior. Following the assault, her grandmother placed chewed tobacco leaves atop the painful erythematous wheals and flares. Upon resolution, hypopigmented macules and patches remained in their place. The patient denied associated symptoms or new lesions; she did not seek care at that time.
In her early 20s, the patient noted new, similarly distributed hypopigmented macules and patches without associated arthropod assault. She was treated by an outside dermatologist without result for presumed tinea versicolor. A follow-up superficial shave biopsy cited subtle psoriasiform dermatitis. Topical steroids did not improve the lesions. Her medical history also was remarkable for a reportedly unprovoked complete rotator cuff tear.
Physical examination revealed 0.5- to 2.0-cm, ill-defined, perifollicular and nonfollicular, slightly scaly macules and patches on the trunk, arms, and legs. There was no follicular plugging (Figure 1A). The hands, feet, face, and mucosal surfaces were spared. She had no family history of similar lesions. Although atrophic in appearance, a single lesion on the left thigh was palpably depressed (Figure 1B). Serology demonstrated a normal complete blood cell count and comprehensive metabolic panel, and negative Lyme titers. Light therapy and topical steroids failed to improve the lesions; calcipotriene cream 0.005% made the lesions erythematous and pruritic.
A biopsy from a flank lesion demonstrated a normal epithelium without thinning, a normal basal melanocyte population, and minimally effaced rete ridges. Thin collagen bundles were noted in the upper reticular and papillary dermis with associated fibroplasia (Figure 2). Verhoeff-van Gieson stain revealed decreased and fragmented elastin filaments in the same dermal distribution as the changed collagen (Figure 3). There was no evidence of primary inflammatory disease. The dermis was thinned. Periodic acid–Schiff stain confirmed the absence of hyphae and spores.
The relevant findings in our patient including the following: (1) onset of hypopigmented macules and patches following resolution of a toxic insult; (2) initially stable number of lesions that progressed in number but not size; (3) thinned collagen associated with fibroplasia in the upper reticular and papillary dermis; (4) decreased number and fragmentation of elastin filaments confined to the same region; (5) no congenital lesions or similar lesions in family members; and (6) a complete rotator cuff tear with no findings of a systemic connective-tissue disorder such as Ehlers-Danlos syndrome.
We performed a literature search of PubMed articles indexed for MEDLINE using combinations of the terms atrophic, hypopigmented, white, spot disease, confetti-like, guttate, macules, atrophoderma, morphea, anetoderma, elastin, and collagen to identify potentially similar reports of guttate hypopigmented macules demonstrating changes of the collagen and elastin in the papillary and upper reticular dermis. Some variants, namely atrophoderma of Pasini and Pierini (APP), guttate morphea, and superficial morphea, demonstrate similar clinical and histopathologic findings.
Findings similar to our case were documented in case reports of 2 women (aged 34 and 42 years)1 presenting with asymptomatic, atrophic, well-demarcated, shiny, hypopigmented macules over the trunk and upper extremities, which demonstrated a thinned epidermis with coarse hyalinized collagen bundles in the mid and lower dermis. There was upper and diffuse dermal elastolysis (patient 1 and patient 2, respectively).1 Our patient’s lesions were hypopigmented and atrophic in appearance but were slightly scaly and also involved the extremities. Distinct from these patient reports, histopathology from our case demonstrated thin packed collagen bundles and decreased fragmented elastin filaments confined to the upper reticular and papillary dermis.
Plaque morphea is the most common type of localized scleroderma.2 The subtype APP demonstrates round to ovoid, gray-brown depressions with cliff-drop borders. They may appear flesh colored or hypopigmented.3,4 These sclerodermoid lesions lack the violaceous border classic to morphea. Sclerosis and induration also are typically absent.5 Clinically, our patient’s macules resembled this entity. Histopathologically, APP shows normal epithelium with an increased basal layer pigmentation; preserved adnexal structures; and mid to lower dermal collagen edema, clumping, and homogenization.3,4 Elastic fibers classically are unchanged, with exceptions.6-11 Changes in the collagen and elastin of our patient were unlike those reported in APP, which occur in the mid to lower dermis.
Guttate morphea demonstrates small, pale, minimally indurated, coin-shaped lesions on the trunk. Histopathology reveals less sclerosis and more edema, resembling LS&A.12 The earliest descriptions of this entity describe 3 stages: ivory/chalk white, scaly, and atrophic. Follicular plugging (absent in this patient) and fine scale can exist at any stage.13,14 Flattened rete ridges mark an otherwise preserved epidermis; hyalinized collagen typically is superficial and demonstrates less sclerosis yet increased edema.12-14 Fewer elastic fibers typically are present compared to normal skin. Changes seen in this entity are more superficial, as with our patient, than classic scleroderma. However, classic edema was not found in our patient’s biopsy specimen.
Superficial morphea, occurring predominantly in females, presents with hyperpigmented or hypopigmented patches having minimal to no induration. The lesions typically are asymptomatic. Histopathologically, collagen deposition and inflammation are confined to the superficial dermis without homogenization associated with LS&A, findings that were consistent with this patient’s biopsy.15,16 However, similar to other morpheaform variants, elastic fibers are unchanged.15 Verhoeff-van Gieson stain of the biopsy (Figure 3) showed the decreased and fragmented elastin network in the upper reticular and papillary dermis, making this entity less compatible.
Guttate LS&A may present with interfollicular, bluish white macules or papules coalescing into patches or plaques. Lesions evolve to reveal atrophic thin skin with follicular plugging. Histology demonstrates a thinned epidermis with orthohypokeratosis marked by flattened rete ridges. The dermis reveals short hyalinized collagen fibrils with a loss of elastic fibers in the papillary and upper reticular dermis, giving a homogenized appearance. Early disease is marked by an inflammatory infiltrate.17 Most of these findings are consistent with our patient’s pathology, which was confined to the upper dermis. Lacking, however, were characteristic findings of LS&A, including upper dermal homogenization, near-total effacement of rete ridges, orthokeratosis, and vacuolar degeneration at the dermoepidermal junction. As such, this entity is less compatible.
Atrophoderma elastolyticum discretum has clinical features of atrophoderma with elastolytic histopathologic findings.1 Anetoderma presents with outpouchings of atrophic skin with a surrounding ring of normal tissue. Histopathologically, this entity shows normal collagen with elastolysis; there also is a decrease in desmosine, an elastin cross-linker.1,3 Neither the clinical nor histopathologic findings in this patient matched these 2 entities.
The reported chronologic association of these lesions with an arthropod assault raised suspicion to their association with toxic insult or postinflammatory changes. One study reported mechanical trauma, including insect bites, as a possible inciting factor of morphea.11 These data, gathered from patient surveys, reported trauma associated to lesion development.1,17 A review of the literature regarding atrophoderma, morphea, and LS&A failed to identify pathogenic changes seen in this patient following initial trauma. Moreover, although it is difficult to prove causality in the formation of the original hypopigmented spots, the development of identical spots in a similar distribution without further trauma suggests against these etiologies to fully explain her lesions. Nonetheless, circumstance makes it difficult to prove whether the original arthropod insult spurred a smoldering reactive process that caused the newer lesions.
Hereditary connective-tissue disorders also were considered in the differential diagnosis. Because of the patient’s history of an unprovoked complete rotator cuff tear, Ehlers-Danlos syndrome was considered; however, the remainder of her examination was normal, making a syndromic systemic disorder a less likely etiology.Because of the distinct clinical and histopathologic findings, this case may represent a rare and previously unreported variant of morphea. Clinically, these hypopigmented macules and patches exist somewhere along the morphea-atrophoderma spectrum. Histopathologic findings do not conform to prior reports. The name atrophodermalike guttate morphea may be an appropriate appellation. It is possible this presentation represents a variant of what dermatologists have referred to as white spot disease.18 We hope that this case may bring others to discussion, allowing for the identification of a more precise entity and etiology so that patients may receive more directed therapy.
To the Editor:
Morphea, atrophoderma, guttate lichen sclerosus et atrophicus (LS&A), anetoderma, and their subtypes are inflammatory processes ultimately leading to dermal remodeling. We report a case of a scaly, hypopigmented, macular rash that clinically appeared as an entity along the morphea-atrophoderma spectrum and demonstrated unique histopathologic changes in both collagen and elastin confined to the upper reticular and papillary dermis. This case is a potentially rare variant representing a combination of clinical and microscopic findings.
A 29-year-old woman presented for an increasing number of white spots distributed on the trunk, arms, and legs. She denied local and systemic symptoms. The patient reported that she was stung by 100 wasps 23 years prior. Following the assault, her grandmother placed chewed tobacco leaves atop the painful erythematous wheals and flares. Upon resolution, hypopigmented macules and patches remained in their place. The patient denied associated symptoms or new lesions; she did not seek care at that time.
In her early 20s, the patient noted new, similarly distributed hypopigmented macules and patches without associated arthropod assault. She was treated by an outside dermatologist without result for presumed tinea versicolor. A follow-up superficial shave biopsy cited subtle psoriasiform dermatitis. Topical steroids did not improve the lesions. Her medical history also was remarkable for a reportedly unprovoked complete rotator cuff tear.
Physical examination revealed 0.5- to 2.0-cm, ill-defined, perifollicular and nonfollicular, slightly scaly macules and patches on the trunk, arms, and legs. There was no follicular plugging (Figure 1A). The hands, feet, face, and mucosal surfaces were spared. She had no family history of similar lesions. Although atrophic in appearance, a single lesion on the left thigh was palpably depressed (Figure 1B). Serology demonstrated a normal complete blood cell count and comprehensive metabolic panel, and negative Lyme titers. Light therapy and topical steroids failed to improve the lesions; calcipotriene cream 0.005% made the lesions erythematous and pruritic.
A biopsy from a flank lesion demonstrated a normal epithelium without thinning, a normal basal melanocyte population, and minimally effaced rete ridges. Thin collagen bundles were noted in the upper reticular and papillary dermis with associated fibroplasia (Figure 2). Verhoeff-van Gieson stain revealed decreased and fragmented elastin filaments in the same dermal distribution as the changed collagen (Figure 3). There was no evidence of primary inflammatory disease. The dermis was thinned. Periodic acid–Schiff stain confirmed the absence of hyphae and spores.
The relevant findings in our patient including the following: (1) onset of hypopigmented macules and patches following resolution of a toxic insult; (2) initially stable number of lesions that progressed in number but not size; (3) thinned collagen associated with fibroplasia in the upper reticular and papillary dermis; (4) decreased number and fragmentation of elastin filaments confined to the same region; (5) no congenital lesions or similar lesions in family members; and (6) a complete rotator cuff tear with no findings of a systemic connective-tissue disorder such as Ehlers-Danlos syndrome.
We performed a literature search of PubMed articles indexed for MEDLINE using combinations of the terms atrophic, hypopigmented, white, spot disease, confetti-like, guttate, macules, atrophoderma, morphea, anetoderma, elastin, and collagen to identify potentially similar reports of guttate hypopigmented macules demonstrating changes of the collagen and elastin in the papillary and upper reticular dermis. Some variants, namely atrophoderma of Pasini and Pierini (APP), guttate morphea, and superficial morphea, demonstrate similar clinical and histopathologic findings.
Findings similar to our case were documented in case reports of 2 women (aged 34 and 42 years)1 presenting with asymptomatic, atrophic, well-demarcated, shiny, hypopigmented macules over the trunk and upper extremities, which demonstrated a thinned epidermis with coarse hyalinized collagen bundles in the mid and lower dermis. There was upper and diffuse dermal elastolysis (patient 1 and patient 2, respectively).1 Our patient’s lesions were hypopigmented and atrophic in appearance but were slightly scaly and also involved the extremities. Distinct from these patient reports, histopathology from our case demonstrated thin packed collagen bundles and decreased fragmented elastin filaments confined to the upper reticular and papillary dermis.
Plaque morphea is the most common type of localized scleroderma.2 The subtype APP demonstrates round to ovoid, gray-brown depressions with cliff-drop borders. They may appear flesh colored or hypopigmented.3,4 These sclerodermoid lesions lack the violaceous border classic to morphea. Sclerosis and induration also are typically absent.5 Clinically, our patient’s macules resembled this entity. Histopathologically, APP shows normal epithelium with an increased basal layer pigmentation; preserved adnexal structures; and mid to lower dermal collagen edema, clumping, and homogenization.3,4 Elastic fibers classically are unchanged, with exceptions.6-11 Changes in the collagen and elastin of our patient were unlike those reported in APP, which occur in the mid to lower dermis.
Guttate morphea demonstrates small, pale, minimally indurated, coin-shaped lesions on the trunk. Histopathology reveals less sclerosis and more edema, resembling LS&A.12 The earliest descriptions of this entity describe 3 stages: ivory/chalk white, scaly, and atrophic. Follicular plugging (absent in this patient) and fine scale can exist at any stage.13,14 Flattened rete ridges mark an otherwise preserved epidermis; hyalinized collagen typically is superficial and demonstrates less sclerosis yet increased edema.12-14 Fewer elastic fibers typically are present compared to normal skin. Changes seen in this entity are more superficial, as with our patient, than classic scleroderma. However, classic edema was not found in our patient’s biopsy specimen.
Superficial morphea, occurring predominantly in females, presents with hyperpigmented or hypopigmented patches having minimal to no induration. The lesions typically are asymptomatic. Histopathologically, collagen deposition and inflammation are confined to the superficial dermis without homogenization associated with LS&A, findings that were consistent with this patient’s biopsy.15,16 However, similar to other morpheaform variants, elastic fibers are unchanged.15 Verhoeff-van Gieson stain of the biopsy (Figure 3) showed the decreased and fragmented elastin network in the upper reticular and papillary dermis, making this entity less compatible.
Guttate LS&A may present with interfollicular, bluish white macules or papules coalescing into patches or plaques. Lesions evolve to reveal atrophic thin skin with follicular plugging. Histology demonstrates a thinned epidermis with orthohypokeratosis marked by flattened rete ridges. The dermis reveals short hyalinized collagen fibrils with a loss of elastic fibers in the papillary and upper reticular dermis, giving a homogenized appearance. Early disease is marked by an inflammatory infiltrate.17 Most of these findings are consistent with our patient’s pathology, which was confined to the upper dermis. Lacking, however, were characteristic findings of LS&A, including upper dermal homogenization, near-total effacement of rete ridges, orthokeratosis, and vacuolar degeneration at the dermoepidermal junction. As such, this entity is less compatible.
Atrophoderma elastolyticum discretum has clinical features of atrophoderma with elastolytic histopathologic findings.1 Anetoderma presents with outpouchings of atrophic skin with a surrounding ring of normal tissue. Histopathologically, this entity shows normal collagen with elastolysis; there also is a decrease in desmosine, an elastin cross-linker.1,3 Neither the clinical nor histopathologic findings in this patient matched these 2 entities.
The reported chronologic association of these lesions with an arthropod assault raised suspicion to their association with toxic insult or postinflammatory changes. One study reported mechanical trauma, including insect bites, as a possible inciting factor of morphea.11 These data, gathered from patient surveys, reported trauma associated to lesion development.1,17 A review of the literature regarding atrophoderma, morphea, and LS&A failed to identify pathogenic changes seen in this patient following initial trauma. Moreover, although it is difficult to prove causality in the formation of the original hypopigmented spots, the development of identical spots in a similar distribution without further trauma suggests against these etiologies to fully explain her lesions. Nonetheless, circumstance makes it difficult to prove whether the original arthropod insult spurred a smoldering reactive process that caused the newer lesions.
Hereditary connective-tissue disorders also were considered in the differential diagnosis. Because of the patient’s history of an unprovoked complete rotator cuff tear, Ehlers-Danlos syndrome was considered; however, the remainder of her examination was normal, making a syndromic systemic disorder a less likely etiology.Because of the distinct clinical and histopathologic findings, this case may represent a rare and previously unreported variant of morphea. Clinically, these hypopigmented macules and patches exist somewhere along the morphea-atrophoderma spectrum. Histopathologic findings do not conform to prior reports. The name atrophodermalike guttate morphea may be an appropriate appellation. It is possible this presentation represents a variant of what dermatologists have referred to as white spot disease.18 We hope that this case may bring others to discussion, allowing for the identification of a more precise entity and etiology so that patients may receive more directed therapy.
- Aksoy B, Ustün H, Gulbahce R, et al. Confetti-like macular atrophy: a new entity? J Dermatol. 2009;36:592-597.
- Uitto J, Santa Cruz DJ, Bauer EA, et al. Morphea and lichen sclerosus et atrophicus. clinical and histopathologic studies in patients with combined features. J Am Acad Dermatol. 1980;3:271-279.
- Buechner SA, Rufli T. Atrophoderma of Pasini and Pierini. clinical and histopathologic findings and antibodies to Borrelia burgdorferi in thirty-four patients. J Am Acad Dermatol. 1994;30:441-446.
- Saleh Z, Abbas O, Dahdah MJ, et al. Atrophoderma of Pasini and Pierini: a clinical and histopathological study. J Cutan Pathol. 2008;35:1108-1114.
- Canizares O, Sachs PM, Jaimovich L, et al. Idiopathic atrophoderma of Pasini and Pierini. Arch Dermatol. 1958;77:42-58; discussion 58-60.
- Pullara TJ, Lober CW, Fenske NA. Idiopathic atrophoderma of Pasini and Pierini. Int J Dermatol. 1984;23:643-645.
- Jablonska S, Szczepanski A. Atrophoderma Pasini-Pierini: is it an entity? Dermatologica. 1962;125:226-242.
- Ang G, Hyde PM, Lee JB. Unilateral congenital linear atrophoderma of the leg. Pediatr Dermatol. 2005;22:350-354.
- Miteva L, Kadurina M. Unilateral idiopathic atrophoderma of Pasini and Pierini. Int J Dermatol. 2006;45:1391-1393.
- Kee CE, Brothers WS, New W. Idiopathic atrophoderma of Pasini and Pierini with coexistent morphea. a case report. Arch Dermatol. 1960;82:100-103.
- Zulian F, Athreya BH, Laxer R, et al. Juvenile localized scleroderma: clinical and epidemiological features in 750 children. an international study. Rheumatology. 2006;45:614-620.
- Winkelmann RK. Localized cutaneous scleroderma. Semin Dermatol. 1985;4:90-103.
- Dore SE. Two cases of morphoea guttata. Proc R Soc Med. 1918;11:26-28.
- Dore SE. Guttate morphoea. Proc R Soc Med. 1919;12:3-5.
- McNiff JM, Glusac EJ, Lazova RZ, et al. Morphea limited to the superficial reticular dermis: an underrecognized histologic phenomenon. Am J Dermatopathol. 1999;21:315-319.
- Jacobson L, Palazij R, Jaworsky C. Superficial morphea. J Am Acad Dermatol. 2003;49:323-325.
- Bolognia J, Jorizzo JL, Rapini RP, eds. Dermatology. 2nd ed. London, England: Mosby Elsevier; 2007.
- Bunch JL. White-spot disease (morphoea guttata). Proc R Soc Med. 1919;12:24-27.
- Aksoy B, Ustün H, Gulbahce R, et al. Confetti-like macular atrophy: a new entity? J Dermatol. 2009;36:592-597.
- Uitto J, Santa Cruz DJ, Bauer EA, et al. Morphea and lichen sclerosus et atrophicus. clinical and histopathologic studies in patients with combined features. J Am Acad Dermatol. 1980;3:271-279.
- Buechner SA, Rufli T. Atrophoderma of Pasini and Pierini. clinical and histopathologic findings and antibodies to Borrelia burgdorferi in thirty-four patients. J Am Acad Dermatol. 1994;30:441-446.
- Saleh Z, Abbas O, Dahdah MJ, et al. Atrophoderma of Pasini and Pierini: a clinical and histopathological study. J Cutan Pathol. 2008;35:1108-1114.
- Canizares O, Sachs PM, Jaimovich L, et al. Idiopathic atrophoderma of Pasini and Pierini. Arch Dermatol. 1958;77:42-58; discussion 58-60.
- Pullara TJ, Lober CW, Fenske NA. Idiopathic atrophoderma of Pasini and Pierini. Int J Dermatol. 1984;23:643-645.
- Jablonska S, Szczepanski A. Atrophoderma Pasini-Pierini: is it an entity? Dermatologica. 1962;125:226-242.
- Ang G, Hyde PM, Lee JB. Unilateral congenital linear atrophoderma of the leg. Pediatr Dermatol. 2005;22:350-354.
- Miteva L, Kadurina M. Unilateral idiopathic atrophoderma of Pasini and Pierini. Int J Dermatol. 2006;45:1391-1393.
- Kee CE, Brothers WS, New W. Idiopathic atrophoderma of Pasini and Pierini with coexistent morphea. a case report. Arch Dermatol. 1960;82:100-103.
- Zulian F, Athreya BH, Laxer R, et al. Juvenile localized scleroderma: clinical and epidemiological features in 750 children. an international study. Rheumatology. 2006;45:614-620.
- Winkelmann RK. Localized cutaneous scleroderma. Semin Dermatol. 1985;4:90-103.
- Dore SE. Two cases of morphoea guttata. Proc R Soc Med. 1918;11:26-28.
- Dore SE. Guttate morphoea. Proc R Soc Med. 1919;12:3-5.
- McNiff JM, Glusac EJ, Lazova RZ, et al. Morphea limited to the superficial reticular dermis: an underrecognized histologic phenomenon. Am J Dermatopathol. 1999;21:315-319.
- Jacobson L, Palazij R, Jaworsky C. Superficial morphea. J Am Acad Dermatol. 2003;49:323-325.
- Bolognia J, Jorizzo JL, Rapini RP, eds. Dermatology. 2nd ed. London, England: Mosby Elsevier; 2007.
- Bunch JL. White-spot disease (morphoea guttata). Proc R Soc Med. 1919;12:24-27.
Practice Points
- Atrophodermalike guttate morphea is a potentially underreported or undescribed entity consisting of a combination of clinicopathologic features.
- Widespread hypopigmented macules on the trunk and extremities marked by thinned collagen, fibroplasia, and altered fragmented elastin in the papillary dermis and upper reticular dermis are the key features.
- Atrophoderma, morphea, and lichen sclerosus et atrophicus should be ruled out during clinical workup.
Aplastic Anemia and MDS International Foundation (AAMDSIF)
AAMDSIF will be hosting seven “Living with Aplastic Anemia, MDS, PNH” patient and family conferences in cities around the country this year. Each free event offers opportunities to learn from leading medical experts and to connect with other patients and caregivers. More.
AAMDSIF will be hosting seven “Living with Aplastic Anemia, MDS, PNH” patient and family conferences in cities around the country this year. Each free event offers opportunities to learn from leading medical experts and to connect with other patients and caregivers. More.
AAMDSIF will be hosting seven “Living with Aplastic Anemia, MDS, PNH” patient and family conferences in cities around the country this year. Each free event offers opportunities to learn from leading medical experts and to connect with other patients and caregivers. More.
Cure SMA
Cure SMA is conducting a survey of pediatricians to assess baseline awareness regarding spinal muscular atrophy. Support Cure SMA and take the survey! More.
Cure SMA is conducting a survey of pediatricians to assess baseline awareness regarding spinal muscular atrophy. Support Cure SMA and take the survey! More.
Cure SMA is conducting a survey of pediatricians to assess baseline awareness regarding spinal muscular atrophy. Support Cure SMA and take the survey! More.
CurePSP
The First International Symposium on PSP and CBD will take place in London Oct. 25-26, 2018, in conjunction with CurePSP and the PSP Association. The latest research updates will be shared through scientific and clinical presentations and poster sessions. View a draft agenda and register now. CurePSP sponsors and organizes family conferences around the US, providing opportunities to learn more about PSP, CBD, and MSA. Join CurePSP and the Mayo Clinic in Rochester, MD, on June 29-30, 2018. More.
The First International Symposium on PSP and CBD will take place in London Oct. 25-26, 2018, in conjunction with CurePSP and the PSP Association. The latest research updates will be shared through scientific and clinical presentations and poster sessions. View a draft agenda and register now. CurePSP sponsors and organizes family conferences around the US, providing opportunities to learn more about PSP, CBD, and MSA. Join CurePSP and the Mayo Clinic in Rochester, MD, on June 29-30, 2018. More.
The First International Symposium on PSP and CBD will take place in London Oct. 25-26, 2018, in conjunction with CurePSP and the PSP Association. The latest research updates will be shared through scientific and clinical presentations and poster sessions. View a draft agenda and register now. CurePSP sponsors and organizes family conferences around the US, providing opportunities to learn more about PSP, CBD, and MSA. Join CurePSP and the Mayo Clinic in Rochester, MD, on June 29-30, 2018. More.
Ehlers-Danlos Society
The Ehlers-Danlos Society will hold an International Symposium Sept. 26-29, 2018, in Ghent, Belgium. Clinicians and researchers are encouraged to attend to discuss the molecular, pathogenic, clinical, and management aspects of all types of Ehlers-Danlos syndromes. More.
The Ehlers-Danlos Society will hold an International Symposium Sept. 26-29, 2018, in Ghent, Belgium. Clinicians and researchers are encouraged to attend to discuss the molecular, pathogenic, clinical, and management aspects of all types of Ehlers-Danlos syndromes. More.
The Ehlers-Danlos Society will hold an International Symposium Sept. 26-29, 2018, in Ghent, Belgium. Clinicians and researchers are encouraged to attend to discuss the molecular, pathogenic, clinical, and management aspects of all types of Ehlers-Danlos syndromes. More.
A Peek at Our August 2018 Issue
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel

{kind=link}
Osteogenesis Imperfecta Foundation (OIF)
The OIF will host its biennial national conference in Baltimore July 13-15, 2018. The program will feature sessions on medical care and practical living, forums with leading experts in osteogenesis imperfect care and research, and other opportunities. More.
The OIF will host its biennial national conference in Baltimore July 13-15, 2018. The program will feature sessions on medical care and practical living, forums with leading experts in osteogenesis imperfect care and research, and other opportunities. More.
The OIF will host its biennial national conference in Baltimore July 13-15, 2018. The program will feature sessions on medical care and practical living, forums with leading experts in osteogenesis imperfect care and research, and other opportunities. More.