User login
Cutis is a peer-reviewed clinical journal for the dermatologist, allergist, and general practitioner published monthly since 1965. Concise clinical articles present the practical side of dermatology, helping physicians to improve patient care. Cutis is referenced in Index Medicus/MEDLINE and is written and edited by industry leaders.
ass lick
assault rifle
balls
ballsac
black jack
bleach
Boko Haram
bondage
causas
cheap
child abuse
cocaine
compulsive behaviors
cost of miracles
cunt
Daech
display network stats
drug paraphernalia
explosion
fart
fda and death
fda AND warn
fda AND warning
fda AND warns
feom
fuck
gambling
gfc
gun
human trafficking
humira AND expensive
illegal
ISIL
ISIS
Islamic caliphate
Islamic state
madvocate
masturbation
mixed martial arts
MMA
molestation
national rifle association
NRA
nsfw
nuccitelli
pedophile
pedophilia
poker
porn
porn
pornography
psychedelic drug
recreational drug
sex slave rings
shit
slot machine
snort
substance abuse
terrorism
terrorist
texarkana
Texas hold 'em
UFC
section[contains(@class, 'nav-hidden')]
section[contains(@class, 'nav-hidden active')
A peer-reviewed, indexed journal for dermatologists with original research, image quizzes, cases and reviews, and columns.
Panniculitis, Pancreatitis, and Polyarthritis: A Rare Clinical Syndrome
Pancreatic panniculitis is a rare disease contributing to widespread fat necrosis in patients with underlying pancreatic disorders. This entity was first described in 1883,1 but it was not until 1947 that it was reported in the English-language literature.2 Patients with pancreatitis infrequently develop extrapancreatic manifestations. It has been estimated that only 2% to 3% of patients worldwide with an underlying pancreatic disease develop cutaneous lesions.3 Patients who develop pancreatic panniculitis typically present with tender, edematous, erythematous to brown, subcutaneous nodules on the lower legs with the tendency for spontaneous ulceration. Lesions tend to exude a viscous, yellow-brown, oily substance that represents liquefactive necrosis of enzymatic fat in subcutaneous tissue. Cutaneous lesions may precede, occur simultaneously, or follow the development of an underlying pancreatic disorder. Rarely, patients may develop inflammatory arthritis secondary to intraosseous fat necrosis, completing the triad of findings diagnostic for panniculitis, pancreatitis, and polyarthritis (PPP) syndrome. Although the underlying pancreatic pathology may vary, roughly 80% of cases worldwide have acute/chronic pancreatitis or pancreatic carcinoma, most commonly acinar cell carcinoma.4-6 Less common pancreatic disorders include pancreatic pseudocyst, pancreatic divisum, and vascular pancreatic fistulas.7 Narváez et al8 found that of the 25 cases of PPP syndrome reported in the literature, 68% (17/25) were men, 32% (8/25) were women, 56% (14/25) were younger than 50 years, and 64% (16/25) had a history of prior or current alcohol abuse.
Case Report
A 68-year-old man with a history of hypertension, gastroesophageal reflux disease, chronic pancreatitis of unknown etiology, and arthritis presented to our clinic for evaluation of painful skin nodules on the lower legs of 8 months’ duration, in addition to joint pain and swelling of the metacarpophalangeal (MCP), metatarsophalangeal, and ankle joints. He had a history of numerous hospital admissions over the last 2 years for pancreatitis and was being managed by the rheumatology department for arthritic symptoms.
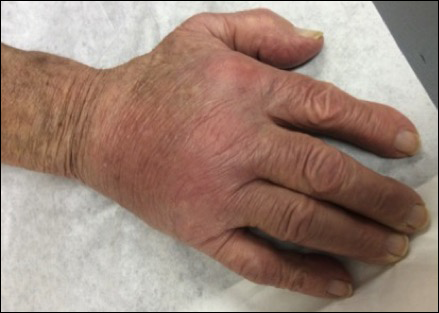
Physical examination revealed multiple 1- to 4-cm, ill-defined, erythematous to brown, subcutaneous nodules on the bilateral lower legs (Figure 1) and right inferomedial thigh that were tender to palpation. Marked erythema and edema of the MCP and metatarsophalangeal joints (Figure 2) and bilateral ankles were observed. Diffuse 2+ pitting edema was present in the bilateral lower extremities, along with areas of hyperpigmentation overlying resolving lesions.

Laboratory data revealed an elevated lipase level (>16,000 U/L [reference range, 31–186 U/L]), amylase level (>4700 U/L [reference range, 27–131 U/L]), erythrocyte sedimentation rate (94 mm/h [reference range, 0–20 mm/h]), and C-reactive protein level (93.5 mg/L [0.08–3.1 mg/L]). The patient had more than 6 episodes of recurrent idiopathic pancreatitis over the last 2 years, though symptoms of abdominal pain were minimal to nonexistent. Liver function tests and alcohol, calcium, and triglyceride levels all were within reference range. Rheumatoid factor and antinuclear antibodies were negative.
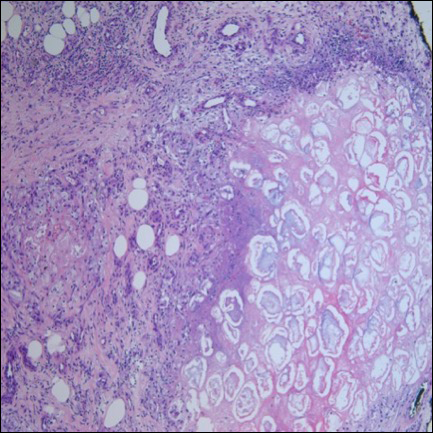
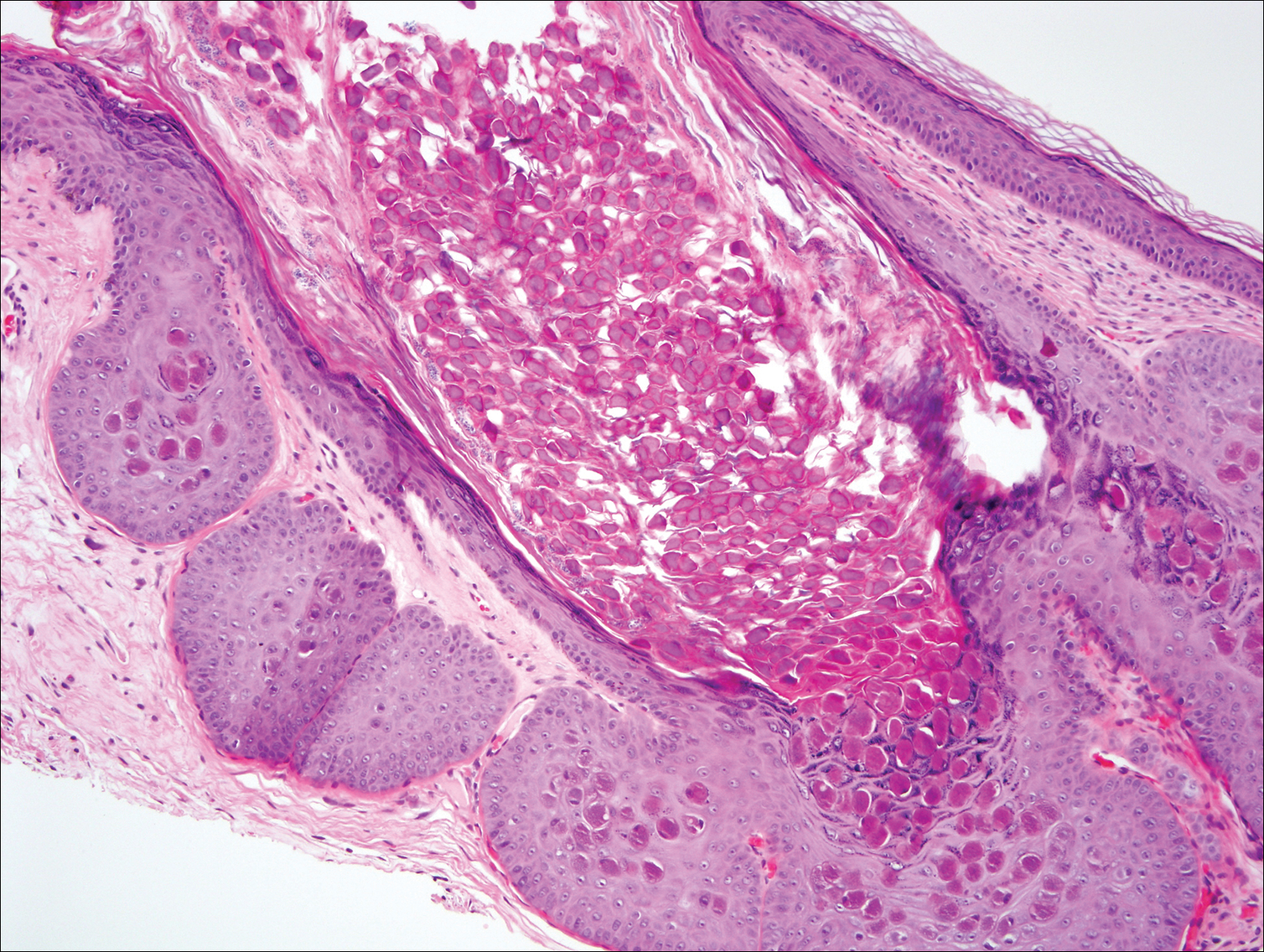
Ultrasonography showed no evidence of cholelithiasis. Computed tomography of the abdomen and pelvis demonstrated a 1.8×1.4-cm hypodense lesion within the pancreatic head with calcifications and mild proximal pancreatic ductal dilatation (Figure 3). However, multiple magnetic resonance cholangiopancreatography examinations and endoscopic ultrasounds with fine-needle aspiration specimens were performed, all negative for malignancy. Computed tomography of the left ankle demonstrated evidence of bony cortical destruction in the lateral aspect of the posterior calcaneus. Bone biopsy specimens demonstrated mild chronic inflammation with no evidence of osteomyelitis. A serum uric acid level was found to be 4.4 mg/dL (reference range, 4.0–8.0 mg/dL) and a joint aspirate demonstrated turbid fluid with lipoid material and no evidence of crystals or organisms on culture. Furthermore, a 4-mm punch biopsy of a nodule on the right leg revealed extensive lobular and septal liquefactive adipocyte necrosis with scattered neutrophils and lymphocytes (Figure 4). Aggregates of fine granular basophilic material were observed with prominent adipocyte degeneration and calcification.

Symptomatic treatment with nonsteroidal anti-inflammatory drugs (NSAIDs) along with intralesional, topical, and oral corticosteroids had proven ineffective in the management of this patient. He was subsequently referred to the surgery department for a pancreaticoduodenectomy (Whipple procedure) with notable improvement in pancreatic enzyme levels, lower leg subcutaneous nodules, and arthritis weeks after surgery.
Comment
A triad of pancreatic panniculitis, pancreatitis, and polyarthritis characterizes a rare entity known as PPP syndrome. Pancreatic panniculitis is a rare form of subcutaneous lobular fat necrosis associated with various underlying pancreatic disorders. Approximately 0.3% to 3.0% of patients with an underlying pancreatic disorder are affected with pancreatic panniculitis.9 Pancreatic panniculitis has been found in roughly 2% to 3% of patients with acute or chronic pancreatitis and pancreatic carcinoma, most commonly the acinar cell type.10 Narváez et al8 reported that nearly two-thirds of patients diagnosed with PPP syndrome have minimal to absent abdominal symptoms that often lead to misdiagnosis and affect the overall prognosis of patients with pancreatic disease. Any delay in the diagnosis of PPP syndrome leads to a worse prognosis, with a mortality rate reported to be approximately 24%.8 Potts et al5 provided a review of 27 patients with pancreatic panniculitis in which all 8 patients with pancreatic carcinoma and 42% (8/19) of patients with pancreatitis died.
Pancreatic panniculitis in the setting of PPP syndrome commonly presents with erythematous to brown, exquisitely tender, edematous, subcutaneous nodules on the lower legs. Lesions can range in size from several millimeters to 5 cm. The subcutaneous nodules may spontaneously ulcerate and exude oily viscous material from the liquefactive necrosis of adipocytes. In approximately 40% of patients, skin lesions are the presenting feature.11 Lesions typically resolve only after the pancreatic inflammation regresses, leaving behind atrophic hyperpigmented scars.3 Other presenting symptoms may include joint pain, pitting edema, and subcutaneous nodules, which can precede the diagnosis by up to 9 months.
The exact pathogenesis of PPP syndrome remains unclear. The most widely recognized hypothesis suggests that pancreatic enzymes (eg, trypsin, amylase, lipase, phospholipase A) released from the damaged pancreas are transported through the bloodstream to distant visceral and soft tissue sites, leading to lipolysis and inflammation to the surrounding subcutis and bone marrow.3 Ferrari et al12 reported this effect as a product of the accumulation of high levels of free fatty acids within the joint space by the action of lipolytic pancreatic enzymes on adipose cell membranes, resulting in acute arthritis.
Histopathologic findings of pancreatic panniculitis vary based on the acuity of the disease. Acute lesions typically demonstrate lobular and septal panniculitis. Szymanski and Bluefarb13 described the pathognomonic histologic findings of focal liquefactive necrosis and anucleate necrotic adipocytes surrounded by a shadowy and thickened cell membrane signifying the characteristic ghost cells. Fine basophilic material also may be seen intermixed with the necrotic adipocytes, representing saponified calcium. A brisk inflammatory infiltrate involving lymphocytes, macrophages, and neutrophils tends to surround the areas of necrotic adipocytes. Chronic lesions often demonstrate a paucity of fat necrosis and ghost cells and more granulomatous infiltrate. Langerhans giant cells, macrophages, and lymphocytes predominate in the subcutaneous fat.
Laboratory findings associated with pancreatic panniculitis may include elevated serum amylase, lipase, and/or trypsin levels. Not all the enzymes have to be elevated simultaneously. On occasion, one enzyme may be within reference range while the others are elevated. Rarely, patients may have an elevated lipase level with no signs of underlying pancreatic disease, which demonstrates that panniculitis does not correlate with the enzyme levels. In all cases of suspected pancreatic panniculitis, a complete laboratory workup is recommended including lipase, amylase, and trypsin serum levels. Eosinophilia may be a prominent finding in patients with pancreatic panniculitis and tends to occur in association with an underlying pancreatic carcinoma. Patients with pancreatic panniculitis associated with pancreatic carcinoma tend to have more severe, diffuse, and persistent subcutaneous nodules that often are refractory to treatment with frequent recurrence. A rare constellation of findings known as Schmid triad is comprised of panniculitis, polyarthritis, and eosinophilia and typically portends a poor prognosis secondary to an underlying pancreatic tumor.14 Cutaneous nodules may predate the diagnosis of pancreatic carcinoma by several months, thus signifying the need for a high index of suspicion in patients with lower leg subcutaneous nodules.
Joint disease most commonly involves the ankles, knees, wrists, and MCP joints.5,6,11 It has been suggested that arthritic symptoms are from periarticular fat necrosis or a direct extension from the necrotic subcutaneous tissue to the adjacent joint space.15 Dahl et al3 reported the composition of joint effusion fluid in 3 patients with PPP syndrome. The aspirate in all 3 patients contained viscous yellow material similar to the necrotic adipose tissue seen draining from subcutaneous nodules. Joint aspirate analysis demonstrated increased concentration of free fatty acids in the joint fluid consistent with severe lipolysis.3
The PPP syndrome acronym may be misleading to physicians, as arthritis is not always polyarticular. Dahl et al3 reported that monoarticular or oligoarticular arthritic symptoms were present in 56% of patients studied. In rare cases, the arthritic symptoms antedated the diagnosis of clinically asymptomatic pancreatic disease. Arthritis can be either symmetric or asymmetric and infrequently follows a chronic course, leading to radiographic lytic lesions and symptoms that often are unresponsive to conventional therapy.16
Treatment of PPP syndrome is largely supportive, with a focus on correcting the underlying pancreatic disease. It is imperative to identify any complicating factors contributing to high levels of circulating pancreatic enzymes. Pseudocysts must be addressed if discovered in these patients, as they often perpetuate the substantial release of pancreatic enzymes into the serum, leading to characteristic subcutaneous fat necrosis and arthritis. Sepsis also is a concern, likely secondary to bacterial colonization of the ulcerated subcutaneous nodules and compromised skin barrier. Nonsteroidal anti-inflammatory drugs and corticosteroids have been used for symptomatic relief but usually are ineffective and have not been shown to reduce the duration of the disease.12,16 Octreotide has been utilized and may potentially reduce pancreatic enzyme secretion leading to improvement in cutaneous and musculoskeletal lesions.17 Plasmapheresis has been used as an adjuvant treatment in patients with persistent hyperamylasemia and hyperlipasemia, but reports are anecdotal. Often reserved for severe disease, cholecystectomy, pancreatic duct removal, and pancreaticoduodenectomy have demonstrated success in the management of chronic pancreatitis and panniculitis. Dahl et al3 reported 2 cases in which cholecystectomy was performed with complete resolution of the skin and pancreatic disease. Our patient was initially treated symptomatically with NSAIDs and corticosteroids but there was no clinical response. The patient eventually underwent a pancreaticoduodenectomy 9 months after the onset of symptoms with complete resolution of joint pain and swelling, greater than 50% resolution of his lower leg subcutaneous nodules, and remarkable reduction in amylase and lipase levels on 1-month follow-up.
Conclusion
Panniculitis, pancreatitis, and polyarthritis syndrome is a rare diagnosis characterized by a triad of pancreatic panniculitis, pancreatitis, and polyarthritis. Adjuvant therapies for PPP syndrome, such as NSAIDs, corticosteroids, plasmapheresis, and octreotide, have been used with equivocal results, but definitive treatment requires correction of the primary pancreatic disorder. More importantly, many pancreatic diseases can cause pancreatic panniculitis, but extensive, refractory, or ulcerated cases could be an early indicator of an occult pancreatic malignancy and should prompt early evaluation with a multidisciplinary approach. This approach should incorporate management from dermatology, internal medicine, rheumatology, gastroenterology, surgery, and primary care.
- Chiari H. Uber die Sogenannte Fettnekrose. Prag Med Wochenschr. 1883;8:285-286, 299-301.
- Blauvelt H. Case of acute pancreatitis with subcutaneous fat necrosis. Br J Surg. 1946;34:207-208.
- Dahl PR, Su D, Cullimore KC, et al. Pancreatic panniculitis. J Am Acad Dermatol. 1995;33:413-417.
- Mullen GT, Caperton EM Jr, Crespin SR, et al. Arthritis and skin lesions resembling erythema nodosum in pancreatic disease. Ann Intern Med. 1968;68:75-87.
- Potts DE, Mass MF, Iseman MD. Syndrome and pancreatic disease, subcutaneous fat necrosis and polyserositis: case report and review of literature. Am J Med. 1975;58:417-423.
- Sorensen EV. Subcutaneous fat necrosis in pancreatic disease: a review and two new case reports. J Clin Gastroenterol. 1988;10:71-75.
- García-Romero D, Vanaclocha F. Pancreatic panniculitis. Dermatol Clin. 2008;26:465-470.
- Narváez J, Bianchi M, Santo P, et al. Pancreatitis, panniculitis, and polyarthritis. Semin Arthritis Rheum. 2010;39:417-423.
- Rongioetti F, Caputo V. Pancreatic panniculitis. G Ital Dermatol Venereol. 2013;148:419-425.
- Poelman SM, Nguyen K. Pancreatic panniculitis associated with acinar cell pancreatic carcinoma. J Cutan Med Surg. 2008;12:38-42.
- Hughes SH, Apisarnthanarax P, Mullins F. Subcutaneous fat necrosis associated with pancreatic disease. Arch Dermatol. 1975:111:506-510.
- Ferrari R, Wendelboe M, Ford PM, et al. Pancreatitis arthritis with periarticular fat necrosis. J Rheumatol. 1993;20:1436-1437.
- Szymanski FJ, Bluefarb SM. Nodular fat necrosis and pancreatic diseases. Arch Dermatol. 1961;83:224-229.
- Beltraminelly HS, Buechner SA, Hausermann P. Pancreatic panniculitis in a patient with an acinar cell cystadenocarcinoma of the pancreas. Dermatology. 2004;208:265-267.
- Burns WA, Matthews MJ, Hamosh M, et al. Lipase-secreting acinar cell carcinoma of the pancreas with polyarthropathy: a light and electron microscopic, histochemical, and biochemical study. Cancer. 1974;33:1002-1009.
- Baron M, Paltiel H, Lander P. Aseptic necrosis of the talus and calcaneal insufficiency fractures in a patient with pancreatitis, subcutaneous fat necrosis, and arthritis. Arthritis Rheum. 1984;27:1309-1313.
- Zundler S, Erber R, Agaimy A, et al. Pancreatic panniculitis in a patient with pancreatic-type acinar cell carcinoma of the liver—case report and review of literature. BMC Cancer. 2016;16:130.
Pancreatic panniculitis is a rare disease contributing to widespread fat necrosis in patients with underlying pancreatic disorders. This entity was first described in 1883,1 but it was not until 1947 that it was reported in the English-language literature.2 Patients with pancreatitis infrequently develop extrapancreatic manifestations. It has been estimated that only 2% to 3% of patients worldwide with an underlying pancreatic disease develop cutaneous lesions.3 Patients who develop pancreatic panniculitis typically present with tender, edematous, erythematous to brown, subcutaneous nodules on the lower legs with the tendency for spontaneous ulceration. Lesions tend to exude a viscous, yellow-brown, oily substance that represents liquefactive necrosis of enzymatic fat in subcutaneous tissue. Cutaneous lesions may precede, occur simultaneously, or follow the development of an underlying pancreatic disorder. Rarely, patients may develop inflammatory arthritis secondary to intraosseous fat necrosis, completing the triad of findings diagnostic for panniculitis, pancreatitis, and polyarthritis (PPP) syndrome. Although the underlying pancreatic pathology may vary, roughly 80% of cases worldwide have acute/chronic pancreatitis or pancreatic carcinoma, most commonly acinar cell carcinoma.4-6 Less common pancreatic disorders include pancreatic pseudocyst, pancreatic divisum, and vascular pancreatic fistulas.7 Narváez et al8 found that of the 25 cases of PPP syndrome reported in the literature, 68% (17/25) were men, 32% (8/25) were women, 56% (14/25) were younger than 50 years, and 64% (16/25) had a history of prior or current alcohol abuse.
Case Report
A 68-year-old man with a history of hypertension, gastroesophageal reflux disease, chronic pancreatitis of unknown etiology, and arthritis presented to our clinic for evaluation of painful skin nodules on the lower legs of 8 months’ duration, in addition to joint pain and swelling of the metacarpophalangeal (MCP), metatarsophalangeal, and ankle joints. He had a history of numerous hospital admissions over the last 2 years for pancreatitis and was being managed by the rheumatology department for arthritic symptoms.
Physical examination revealed multiple 1- to 4-cm, ill-defined, erythematous to brown, subcutaneous nodules on the bilateral lower legs (Figure 1) and right inferomedial thigh that were tender to palpation. Marked erythema and edema of the MCP and metatarsophalangeal joints (Figure 2) and bilateral ankles were observed. Diffuse 2+ pitting edema was present in the bilateral lower extremities, along with areas of hyperpigmentation overlying resolving lesions.
Laboratory data revealed an elevated lipase level (>16,000 U/L [reference range, 31–186 U/L]), amylase level (>4700 U/L [reference range, 27–131 U/L]), erythrocyte sedimentation rate (94 mm/h [reference range, 0–20 mm/h]), and C-reactive protein level (93.5 mg/L [0.08–3.1 mg/L]). The patient had more than 6 episodes of recurrent idiopathic pancreatitis over the last 2 years, though symptoms of abdominal pain were minimal to nonexistent. Liver function tests and alcohol, calcium, and triglyceride levels all were within reference range. Rheumatoid factor and antinuclear antibodies were negative.
Ultrasonography showed no evidence of cholelithiasis. Computed tomography of the abdomen and pelvis demonstrated a 1.8×1.4-cm hypodense lesion within the pancreatic head with calcifications and mild proximal pancreatic ductal dilatation (Figure 3). However, multiple magnetic resonance cholangiopancreatography examinations and endoscopic ultrasounds with fine-needle aspiration specimens were performed, all negative for malignancy. Computed tomography of the left ankle demonstrated evidence of bony cortical destruction in the lateral aspect of the posterior calcaneus. Bone biopsy specimens demonstrated mild chronic inflammation with no evidence of osteomyelitis. A serum uric acid level was found to be 4.4 mg/dL (reference range, 4.0–8.0 mg/dL) and a joint aspirate demonstrated turbid fluid with lipoid material and no evidence of crystals or organisms on culture. Furthermore, a 4-mm punch biopsy of a nodule on the right leg revealed extensive lobular and septal liquefactive adipocyte necrosis with scattered neutrophils and lymphocytes (Figure 4). Aggregates of fine granular basophilic material were observed with prominent adipocyte degeneration and calcification.
Symptomatic treatment with nonsteroidal anti-inflammatory drugs (NSAIDs) along with intralesional, topical, and oral corticosteroids had proven ineffective in the management of this patient. He was subsequently referred to the surgery department for a pancreaticoduodenectomy (Whipple procedure) with notable improvement in pancreatic enzyme levels, lower leg subcutaneous nodules, and arthritis weeks after surgery.
Comment
A triad of pancreatic panniculitis, pancreatitis, and polyarthritis characterizes a rare entity known as PPP syndrome. Pancreatic panniculitis is a rare form of subcutaneous lobular fat necrosis associated with various underlying pancreatic disorders. Approximately 0.3% to 3.0% of patients with an underlying pancreatic disorder are affected with pancreatic panniculitis.9 Pancreatic panniculitis has been found in roughly 2% to 3% of patients with acute or chronic pancreatitis and pancreatic carcinoma, most commonly the acinar cell type.10 Narváez et al8 reported that nearly two-thirds of patients diagnosed with PPP syndrome have minimal to absent abdominal symptoms that often lead to misdiagnosis and affect the overall prognosis of patients with pancreatic disease. Any delay in the diagnosis of PPP syndrome leads to a worse prognosis, with a mortality rate reported to be approximately 24%.8 Potts et al5 provided a review of 27 patients with pancreatic panniculitis in which all 8 patients with pancreatic carcinoma and 42% (8/19) of patients with pancreatitis died.
Pancreatic panniculitis in the setting of PPP syndrome commonly presents with erythematous to brown, exquisitely tender, edematous, subcutaneous nodules on the lower legs. Lesions can range in size from several millimeters to 5 cm. The subcutaneous nodules may spontaneously ulcerate and exude oily viscous material from the liquefactive necrosis of adipocytes. In approximately 40% of patients, skin lesions are the presenting feature.11 Lesions typically resolve only after the pancreatic inflammation regresses, leaving behind atrophic hyperpigmented scars.3 Other presenting symptoms may include joint pain, pitting edema, and subcutaneous nodules, which can precede the diagnosis by up to 9 months.
The exact pathogenesis of PPP syndrome remains unclear. The most widely recognized hypothesis suggests that pancreatic enzymes (eg, trypsin, amylase, lipase, phospholipase A) released from the damaged pancreas are transported through the bloodstream to distant visceral and soft tissue sites, leading to lipolysis and inflammation to the surrounding subcutis and bone marrow.3 Ferrari et al12 reported this effect as a product of the accumulation of high levels of free fatty acids within the joint space by the action of lipolytic pancreatic enzymes on adipose cell membranes, resulting in acute arthritis.
Histopathologic findings of pancreatic panniculitis vary based on the acuity of the disease. Acute lesions typically demonstrate lobular and septal panniculitis. Szymanski and Bluefarb13 described the pathognomonic histologic findings of focal liquefactive necrosis and anucleate necrotic adipocytes surrounded by a shadowy and thickened cell membrane signifying the characteristic ghost cells. Fine basophilic material also may be seen intermixed with the necrotic adipocytes, representing saponified calcium. A brisk inflammatory infiltrate involving lymphocytes, macrophages, and neutrophils tends to surround the areas of necrotic adipocytes. Chronic lesions often demonstrate a paucity of fat necrosis and ghost cells and more granulomatous infiltrate. Langerhans giant cells, macrophages, and lymphocytes predominate in the subcutaneous fat.
Laboratory findings associated with pancreatic panniculitis may include elevated serum amylase, lipase, and/or trypsin levels. Not all the enzymes have to be elevated simultaneously. On occasion, one enzyme may be within reference range while the others are elevated. Rarely, patients may have an elevated lipase level with no signs of underlying pancreatic disease, which demonstrates that panniculitis does not correlate with the enzyme levels. In all cases of suspected pancreatic panniculitis, a complete laboratory workup is recommended including lipase, amylase, and trypsin serum levels. Eosinophilia may be a prominent finding in patients with pancreatic panniculitis and tends to occur in association with an underlying pancreatic carcinoma. Patients with pancreatic panniculitis associated with pancreatic carcinoma tend to have more severe, diffuse, and persistent subcutaneous nodules that often are refractory to treatment with frequent recurrence. A rare constellation of findings known as Schmid triad is comprised of panniculitis, polyarthritis, and eosinophilia and typically portends a poor prognosis secondary to an underlying pancreatic tumor.14 Cutaneous nodules may predate the diagnosis of pancreatic carcinoma by several months, thus signifying the need for a high index of suspicion in patients with lower leg subcutaneous nodules.
Joint disease most commonly involves the ankles, knees, wrists, and MCP joints.5,6,11 It has been suggested that arthritic symptoms are from periarticular fat necrosis or a direct extension from the necrotic subcutaneous tissue to the adjacent joint space.15 Dahl et al3 reported the composition of joint effusion fluid in 3 patients with PPP syndrome. The aspirate in all 3 patients contained viscous yellow material similar to the necrotic adipose tissue seen draining from subcutaneous nodules. Joint aspirate analysis demonstrated increased concentration of free fatty acids in the joint fluid consistent with severe lipolysis.3
The PPP syndrome acronym may be misleading to physicians, as arthritis is not always polyarticular. Dahl et al3 reported that monoarticular or oligoarticular arthritic symptoms were present in 56% of patients studied. In rare cases, the arthritic symptoms antedated the diagnosis of clinically asymptomatic pancreatic disease. Arthritis can be either symmetric or asymmetric and infrequently follows a chronic course, leading to radiographic lytic lesions and symptoms that often are unresponsive to conventional therapy.16
Treatment of PPP syndrome is largely supportive, with a focus on correcting the underlying pancreatic disease. It is imperative to identify any complicating factors contributing to high levels of circulating pancreatic enzymes. Pseudocysts must be addressed if discovered in these patients, as they often perpetuate the substantial release of pancreatic enzymes into the serum, leading to characteristic subcutaneous fat necrosis and arthritis. Sepsis also is a concern, likely secondary to bacterial colonization of the ulcerated subcutaneous nodules and compromised skin barrier. Nonsteroidal anti-inflammatory drugs and corticosteroids have been used for symptomatic relief but usually are ineffective and have not been shown to reduce the duration of the disease.12,16 Octreotide has been utilized and may potentially reduce pancreatic enzyme secretion leading to improvement in cutaneous and musculoskeletal lesions.17 Plasmapheresis has been used as an adjuvant treatment in patients with persistent hyperamylasemia and hyperlipasemia, but reports are anecdotal. Often reserved for severe disease, cholecystectomy, pancreatic duct removal, and pancreaticoduodenectomy have demonstrated success in the management of chronic pancreatitis and panniculitis. Dahl et al3 reported 2 cases in which cholecystectomy was performed with complete resolution of the skin and pancreatic disease. Our patient was initially treated symptomatically with NSAIDs and corticosteroids but there was no clinical response. The patient eventually underwent a pancreaticoduodenectomy 9 months after the onset of symptoms with complete resolution of joint pain and swelling, greater than 50% resolution of his lower leg subcutaneous nodules, and remarkable reduction in amylase and lipase levels on 1-month follow-up.
Conclusion
Panniculitis, pancreatitis, and polyarthritis syndrome is a rare diagnosis characterized by a triad of pancreatic panniculitis, pancreatitis, and polyarthritis. Adjuvant therapies for PPP syndrome, such as NSAIDs, corticosteroids, plasmapheresis, and octreotide, have been used with equivocal results, but definitive treatment requires correction of the primary pancreatic disorder. More importantly, many pancreatic diseases can cause pancreatic panniculitis, but extensive, refractory, or ulcerated cases could be an early indicator of an occult pancreatic malignancy and should prompt early evaluation with a multidisciplinary approach. This approach should incorporate management from dermatology, internal medicine, rheumatology, gastroenterology, surgery, and primary care.
Pancreatic panniculitis is a rare disease contributing to widespread fat necrosis in patients with underlying pancreatic disorders. This entity was first described in 1883,1 but it was not until 1947 that it was reported in the English-language literature.2 Patients with pancreatitis infrequently develop extrapancreatic manifestations. It has been estimated that only 2% to 3% of patients worldwide with an underlying pancreatic disease develop cutaneous lesions.3 Patients who develop pancreatic panniculitis typically present with tender, edematous, erythematous to brown, subcutaneous nodules on the lower legs with the tendency for spontaneous ulceration. Lesions tend to exude a viscous, yellow-brown, oily substance that represents liquefactive necrosis of enzymatic fat in subcutaneous tissue. Cutaneous lesions may precede, occur simultaneously, or follow the development of an underlying pancreatic disorder. Rarely, patients may develop inflammatory arthritis secondary to intraosseous fat necrosis, completing the triad of findings diagnostic for panniculitis, pancreatitis, and polyarthritis (PPP) syndrome. Although the underlying pancreatic pathology may vary, roughly 80% of cases worldwide have acute/chronic pancreatitis or pancreatic carcinoma, most commonly acinar cell carcinoma.4-6 Less common pancreatic disorders include pancreatic pseudocyst, pancreatic divisum, and vascular pancreatic fistulas.7 Narváez et al8 found that of the 25 cases of PPP syndrome reported in the literature, 68% (17/25) were men, 32% (8/25) were women, 56% (14/25) were younger than 50 years, and 64% (16/25) had a history of prior or current alcohol abuse.
Case Report
A 68-year-old man with a history of hypertension, gastroesophageal reflux disease, chronic pancreatitis of unknown etiology, and arthritis presented to our clinic for evaluation of painful skin nodules on the lower legs of 8 months’ duration, in addition to joint pain and swelling of the metacarpophalangeal (MCP), metatarsophalangeal, and ankle joints. He had a history of numerous hospital admissions over the last 2 years for pancreatitis and was being managed by the rheumatology department for arthritic symptoms.
Physical examination revealed multiple 1- to 4-cm, ill-defined, erythematous to brown, subcutaneous nodules on the bilateral lower legs (Figure 1) and right inferomedial thigh that were tender to palpation. Marked erythema and edema of the MCP and metatarsophalangeal joints (Figure 2) and bilateral ankles were observed. Diffuse 2+ pitting edema was present in the bilateral lower extremities, along with areas of hyperpigmentation overlying resolving lesions.
Laboratory data revealed an elevated lipase level (>16,000 U/L [reference range, 31–186 U/L]), amylase level (>4700 U/L [reference range, 27–131 U/L]), erythrocyte sedimentation rate (94 mm/h [reference range, 0–20 mm/h]), and C-reactive protein level (93.5 mg/L [0.08–3.1 mg/L]). The patient had more than 6 episodes of recurrent idiopathic pancreatitis over the last 2 years, though symptoms of abdominal pain were minimal to nonexistent. Liver function tests and alcohol, calcium, and triglyceride levels all were within reference range. Rheumatoid factor and antinuclear antibodies were negative.
Ultrasonography showed no evidence of cholelithiasis. Computed tomography of the abdomen and pelvis demonstrated a 1.8×1.4-cm hypodense lesion within the pancreatic head with calcifications and mild proximal pancreatic ductal dilatation (Figure 3). However, multiple magnetic resonance cholangiopancreatography examinations and endoscopic ultrasounds with fine-needle aspiration specimens were performed, all negative for malignancy. Computed tomography of the left ankle demonstrated evidence of bony cortical destruction in the lateral aspect of the posterior calcaneus. Bone biopsy specimens demonstrated mild chronic inflammation with no evidence of osteomyelitis. A serum uric acid level was found to be 4.4 mg/dL (reference range, 4.0–8.0 mg/dL) and a joint aspirate demonstrated turbid fluid with lipoid material and no evidence of crystals or organisms on culture. Furthermore, a 4-mm punch biopsy of a nodule on the right leg revealed extensive lobular and septal liquefactive adipocyte necrosis with scattered neutrophils and lymphocytes (Figure 4). Aggregates of fine granular basophilic material were observed with prominent adipocyte degeneration and calcification.
Symptomatic treatment with nonsteroidal anti-inflammatory drugs (NSAIDs) along with intralesional, topical, and oral corticosteroids had proven ineffective in the management of this patient. He was subsequently referred to the surgery department for a pancreaticoduodenectomy (Whipple procedure) with notable improvement in pancreatic enzyme levels, lower leg subcutaneous nodules, and arthritis weeks after surgery.
Comment
A triad of pancreatic panniculitis, pancreatitis, and polyarthritis characterizes a rare entity known as PPP syndrome. Pancreatic panniculitis is a rare form of subcutaneous lobular fat necrosis associated with various underlying pancreatic disorders. Approximately 0.3% to 3.0% of patients with an underlying pancreatic disorder are affected with pancreatic panniculitis.9 Pancreatic panniculitis has been found in roughly 2% to 3% of patients with acute or chronic pancreatitis and pancreatic carcinoma, most commonly the acinar cell type.10 Narváez et al8 reported that nearly two-thirds of patients diagnosed with PPP syndrome have minimal to absent abdominal symptoms that often lead to misdiagnosis and affect the overall prognosis of patients with pancreatic disease. Any delay in the diagnosis of PPP syndrome leads to a worse prognosis, with a mortality rate reported to be approximately 24%.8 Potts et al5 provided a review of 27 patients with pancreatic panniculitis in which all 8 patients with pancreatic carcinoma and 42% (8/19) of patients with pancreatitis died.
Pancreatic panniculitis in the setting of PPP syndrome commonly presents with erythematous to brown, exquisitely tender, edematous, subcutaneous nodules on the lower legs. Lesions can range in size from several millimeters to 5 cm. The subcutaneous nodules may spontaneously ulcerate and exude oily viscous material from the liquefactive necrosis of adipocytes. In approximately 40% of patients, skin lesions are the presenting feature.11 Lesions typically resolve only after the pancreatic inflammation regresses, leaving behind atrophic hyperpigmented scars.3 Other presenting symptoms may include joint pain, pitting edema, and subcutaneous nodules, which can precede the diagnosis by up to 9 months.
The exact pathogenesis of PPP syndrome remains unclear. The most widely recognized hypothesis suggests that pancreatic enzymes (eg, trypsin, amylase, lipase, phospholipase A) released from the damaged pancreas are transported through the bloodstream to distant visceral and soft tissue sites, leading to lipolysis and inflammation to the surrounding subcutis and bone marrow.3 Ferrari et al12 reported this effect as a product of the accumulation of high levels of free fatty acids within the joint space by the action of lipolytic pancreatic enzymes on adipose cell membranes, resulting in acute arthritis.
Histopathologic findings of pancreatic panniculitis vary based on the acuity of the disease. Acute lesions typically demonstrate lobular and septal panniculitis. Szymanski and Bluefarb13 described the pathognomonic histologic findings of focal liquefactive necrosis and anucleate necrotic adipocytes surrounded by a shadowy and thickened cell membrane signifying the characteristic ghost cells. Fine basophilic material also may be seen intermixed with the necrotic adipocytes, representing saponified calcium. A brisk inflammatory infiltrate involving lymphocytes, macrophages, and neutrophils tends to surround the areas of necrotic adipocytes. Chronic lesions often demonstrate a paucity of fat necrosis and ghost cells and more granulomatous infiltrate. Langerhans giant cells, macrophages, and lymphocytes predominate in the subcutaneous fat.
Laboratory findings associated with pancreatic panniculitis may include elevated serum amylase, lipase, and/or trypsin levels. Not all the enzymes have to be elevated simultaneously. On occasion, one enzyme may be within reference range while the others are elevated. Rarely, patients may have an elevated lipase level with no signs of underlying pancreatic disease, which demonstrates that panniculitis does not correlate with the enzyme levels. In all cases of suspected pancreatic panniculitis, a complete laboratory workup is recommended including lipase, amylase, and trypsin serum levels. Eosinophilia may be a prominent finding in patients with pancreatic panniculitis and tends to occur in association with an underlying pancreatic carcinoma. Patients with pancreatic panniculitis associated with pancreatic carcinoma tend to have more severe, diffuse, and persistent subcutaneous nodules that often are refractory to treatment with frequent recurrence. A rare constellation of findings known as Schmid triad is comprised of panniculitis, polyarthritis, and eosinophilia and typically portends a poor prognosis secondary to an underlying pancreatic tumor.14 Cutaneous nodules may predate the diagnosis of pancreatic carcinoma by several months, thus signifying the need for a high index of suspicion in patients with lower leg subcutaneous nodules.
Joint disease most commonly involves the ankles, knees, wrists, and MCP joints.5,6,11 It has been suggested that arthritic symptoms are from periarticular fat necrosis or a direct extension from the necrotic subcutaneous tissue to the adjacent joint space.15 Dahl et al3 reported the composition of joint effusion fluid in 3 patients with PPP syndrome. The aspirate in all 3 patients contained viscous yellow material similar to the necrotic adipose tissue seen draining from subcutaneous nodules. Joint aspirate analysis demonstrated increased concentration of free fatty acids in the joint fluid consistent with severe lipolysis.3
The PPP syndrome acronym may be misleading to physicians, as arthritis is not always polyarticular. Dahl et al3 reported that monoarticular or oligoarticular arthritic symptoms were present in 56% of patients studied. In rare cases, the arthritic symptoms antedated the diagnosis of clinically asymptomatic pancreatic disease. Arthritis can be either symmetric or asymmetric and infrequently follows a chronic course, leading to radiographic lytic lesions and symptoms that often are unresponsive to conventional therapy.16
Treatment of PPP syndrome is largely supportive, with a focus on correcting the underlying pancreatic disease. It is imperative to identify any complicating factors contributing to high levels of circulating pancreatic enzymes. Pseudocysts must be addressed if discovered in these patients, as they often perpetuate the substantial release of pancreatic enzymes into the serum, leading to characteristic subcutaneous fat necrosis and arthritis. Sepsis also is a concern, likely secondary to bacterial colonization of the ulcerated subcutaneous nodules and compromised skin barrier. Nonsteroidal anti-inflammatory drugs and corticosteroids have been used for symptomatic relief but usually are ineffective and have not been shown to reduce the duration of the disease.12,16 Octreotide has been utilized and may potentially reduce pancreatic enzyme secretion leading to improvement in cutaneous and musculoskeletal lesions.17 Plasmapheresis has been used as an adjuvant treatment in patients with persistent hyperamylasemia and hyperlipasemia, but reports are anecdotal. Often reserved for severe disease, cholecystectomy, pancreatic duct removal, and pancreaticoduodenectomy have demonstrated success in the management of chronic pancreatitis and panniculitis. Dahl et al3 reported 2 cases in which cholecystectomy was performed with complete resolution of the skin and pancreatic disease. Our patient was initially treated symptomatically with NSAIDs and corticosteroids but there was no clinical response. The patient eventually underwent a pancreaticoduodenectomy 9 months after the onset of symptoms with complete resolution of joint pain and swelling, greater than 50% resolution of his lower leg subcutaneous nodules, and remarkable reduction in amylase and lipase levels on 1-month follow-up.
Conclusion
Panniculitis, pancreatitis, and polyarthritis syndrome is a rare diagnosis characterized by a triad of pancreatic panniculitis, pancreatitis, and polyarthritis. Adjuvant therapies for PPP syndrome, such as NSAIDs, corticosteroids, plasmapheresis, and octreotide, have been used with equivocal results, but definitive treatment requires correction of the primary pancreatic disorder. More importantly, many pancreatic diseases can cause pancreatic panniculitis, but extensive, refractory, or ulcerated cases could be an early indicator of an occult pancreatic malignancy and should prompt early evaluation with a multidisciplinary approach. This approach should incorporate management from dermatology, internal medicine, rheumatology, gastroenterology, surgery, and primary care.
- Chiari H. Uber die Sogenannte Fettnekrose. Prag Med Wochenschr. 1883;8:285-286, 299-301.
- Blauvelt H. Case of acute pancreatitis with subcutaneous fat necrosis. Br J Surg. 1946;34:207-208.
- Dahl PR, Su D, Cullimore KC, et al. Pancreatic panniculitis. J Am Acad Dermatol. 1995;33:413-417.
- Mullen GT, Caperton EM Jr, Crespin SR, et al. Arthritis and skin lesions resembling erythema nodosum in pancreatic disease. Ann Intern Med. 1968;68:75-87.
- Potts DE, Mass MF, Iseman MD. Syndrome and pancreatic disease, subcutaneous fat necrosis and polyserositis: case report and review of literature. Am J Med. 1975;58:417-423.
- Sorensen EV. Subcutaneous fat necrosis in pancreatic disease: a review and two new case reports. J Clin Gastroenterol. 1988;10:71-75.
- García-Romero D, Vanaclocha F. Pancreatic panniculitis. Dermatol Clin. 2008;26:465-470.
- Narváez J, Bianchi M, Santo P, et al. Pancreatitis, panniculitis, and polyarthritis. Semin Arthritis Rheum. 2010;39:417-423.
- Rongioetti F, Caputo V. Pancreatic panniculitis. G Ital Dermatol Venereol. 2013;148:419-425.
- Poelman SM, Nguyen K. Pancreatic panniculitis associated with acinar cell pancreatic carcinoma. J Cutan Med Surg. 2008;12:38-42.
- Hughes SH, Apisarnthanarax P, Mullins F. Subcutaneous fat necrosis associated with pancreatic disease. Arch Dermatol. 1975:111:506-510.
- Ferrari R, Wendelboe M, Ford PM, et al. Pancreatitis arthritis with periarticular fat necrosis. J Rheumatol. 1993;20:1436-1437.
- Szymanski FJ, Bluefarb SM. Nodular fat necrosis and pancreatic diseases. Arch Dermatol. 1961;83:224-229.
- Beltraminelly HS, Buechner SA, Hausermann P. Pancreatic panniculitis in a patient with an acinar cell cystadenocarcinoma of the pancreas. Dermatology. 2004;208:265-267.
- Burns WA, Matthews MJ, Hamosh M, et al. Lipase-secreting acinar cell carcinoma of the pancreas with polyarthropathy: a light and electron microscopic, histochemical, and biochemical study. Cancer. 1974;33:1002-1009.
- Baron M, Paltiel H, Lander P. Aseptic necrosis of the talus and calcaneal insufficiency fractures in a patient with pancreatitis, subcutaneous fat necrosis, and arthritis. Arthritis Rheum. 1984;27:1309-1313.
- Zundler S, Erber R, Agaimy A, et al. Pancreatic panniculitis in a patient with pancreatic-type acinar cell carcinoma of the liver—case report and review of literature. BMC Cancer. 2016;16:130.
- Chiari H. Uber die Sogenannte Fettnekrose. Prag Med Wochenschr. 1883;8:285-286, 299-301.
- Blauvelt H. Case of acute pancreatitis with subcutaneous fat necrosis. Br J Surg. 1946;34:207-208.
- Dahl PR, Su D, Cullimore KC, et al. Pancreatic panniculitis. J Am Acad Dermatol. 1995;33:413-417.
- Mullen GT, Caperton EM Jr, Crespin SR, et al. Arthritis and skin lesions resembling erythema nodosum in pancreatic disease. Ann Intern Med. 1968;68:75-87.
- Potts DE, Mass MF, Iseman MD. Syndrome and pancreatic disease, subcutaneous fat necrosis and polyserositis: case report and review of literature. Am J Med. 1975;58:417-423.
- Sorensen EV. Subcutaneous fat necrosis in pancreatic disease: a review and two new case reports. J Clin Gastroenterol. 1988;10:71-75.
- García-Romero D, Vanaclocha F. Pancreatic panniculitis. Dermatol Clin. 2008;26:465-470.
- Narváez J, Bianchi M, Santo P, et al. Pancreatitis, panniculitis, and polyarthritis. Semin Arthritis Rheum. 2010;39:417-423.
- Rongioetti F, Caputo V. Pancreatic panniculitis. G Ital Dermatol Venereol. 2013;148:419-425.
- Poelman SM, Nguyen K. Pancreatic panniculitis associated with acinar cell pancreatic carcinoma. J Cutan Med Surg. 2008;12:38-42.
- Hughes SH, Apisarnthanarax P, Mullins F. Subcutaneous fat necrosis associated with pancreatic disease. Arch Dermatol. 1975:111:506-510.
- Ferrari R, Wendelboe M, Ford PM, et al. Pancreatitis arthritis with periarticular fat necrosis. J Rheumatol. 1993;20:1436-1437.
- Szymanski FJ, Bluefarb SM. Nodular fat necrosis and pancreatic diseases. Arch Dermatol. 1961;83:224-229.
- Beltraminelly HS, Buechner SA, Hausermann P. Pancreatic panniculitis in a patient with an acinar cell cystadenocarcinoma of the pancreas. Dermatology. 2004;208:265-267.
- Burns WA, Matthews MJ, Hamosh M, et al. Lipase-secreting acinar cell carcinoma of the pancreas with polyarthropathy: a light and electron microscopic, histochemical, and biochemical study. Cancer. 1974;33:1002-1009.
- Baron M, Paltiel H, Lander P. Aseptic necrosis of the talus and calcaneal insufficiency fractures in a patient with pancreatitis, subcutaneous fat necrosis, and arthritis. Arthritis Rheum. 1984;27:1309-1313.
- Zundler S, Erber R, Agaimy A, et al. Pancreatic panniculitis in a patient with pancreatic-type acinar cell carcinoma of the liver—case report and review of literature. BMC Cancer. 2016;16:130.
Practice Points
- Recognition of skin lesions in a patient with a history of pancreatitis may represent a rare entity known as pancreatic panniculitis.
- Panniculitis, pancreatitis, and polyarthritis (PPP) syndrome is a rare diagnosis characterized by a triad of pancreatic panniculitis, pancreatitis, and polyarthritis.
- A rare constellation of findings known as Schmid triad is comprised of panniculitis, polyarthritis, and eosinophilia and typically portends a poor prognosis secondary to an underlying pancreatic tumor.
- These findings should prompt early evaluation with a multidisciplinary approach.
Molluscum Contagiosum in Immunocompromised Patients: AIDS Presenting as Molluscum Contagiosum in a Patient With Psoriasis on Biologic Therapy
Molluscum contagiosum (MC) is a double-stranded DNA virus of the Poxviridae family, which commonly infects human keratinocytes resulting in small, umbilicated, flesh-colored papules. The greatest incidence of MC is seen in the pediatric population and sexually active young adults, and it is considered a self-limited disease in immunocompetent individuals.1 With the emergence of the human immunodeficiency virus (HIV) and subsequent AIDS epidemic in the 1980s, a new population of immunocompromised individuals has been observed to be increasingly susceptible to MC with an atypical clinical presentation and a recalcitrant disease course.2 Although the increased prevalence of MC in the HIV population has been well-documented, it has been observed in other disease states or iatrogenically induced immunosuppression due to a deficiency in function or absolute number of T lymphocytes.
We present a case of a patient with long-standing psoriasis on biologic therapy who presented with MC with a subsequent workup that revealed AIDS. This case reiterates the importance of MC as a potential indicator of underlying immunosuppression. We review the literature to evaluate the occurrence of MC in immunosuppressed patients.
Case Report
A 33-year-old man initially presented for evaluation of severe plaque-type psoriasis associated with pain, erythema, and swelling of the joints of the hands of 10 years’ duration. He was started on methotrexate 5 mg weekly and topical corticosteroids but was unable to tolerate methotrexate due to headaches. He also had difficulty affording topical medications and adjunctive phototherapy. The patient was sporadically seen in follow-up with persistence of psoriatic plaques involving up to 60% body surface area (BSA) with the only treatment consisting of occasional topical steroids. Five years later, the patient was restarted on methotrexate 5 to 7.5 mg weekly, which resulted in moderate improvement. However, because of persistent elevation of liver enzymes, this treatment was stopped. Several months later he was evaluated for treatment with a biologic agent, and after a negative tuberculin skin test, he began treatment with etanercept 50 mg subcutaneous injection twice weekly, which provided notable improvement and allowed for reduction of dose frequency to once weekly.
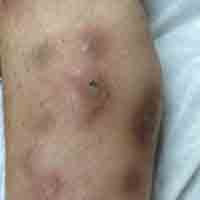
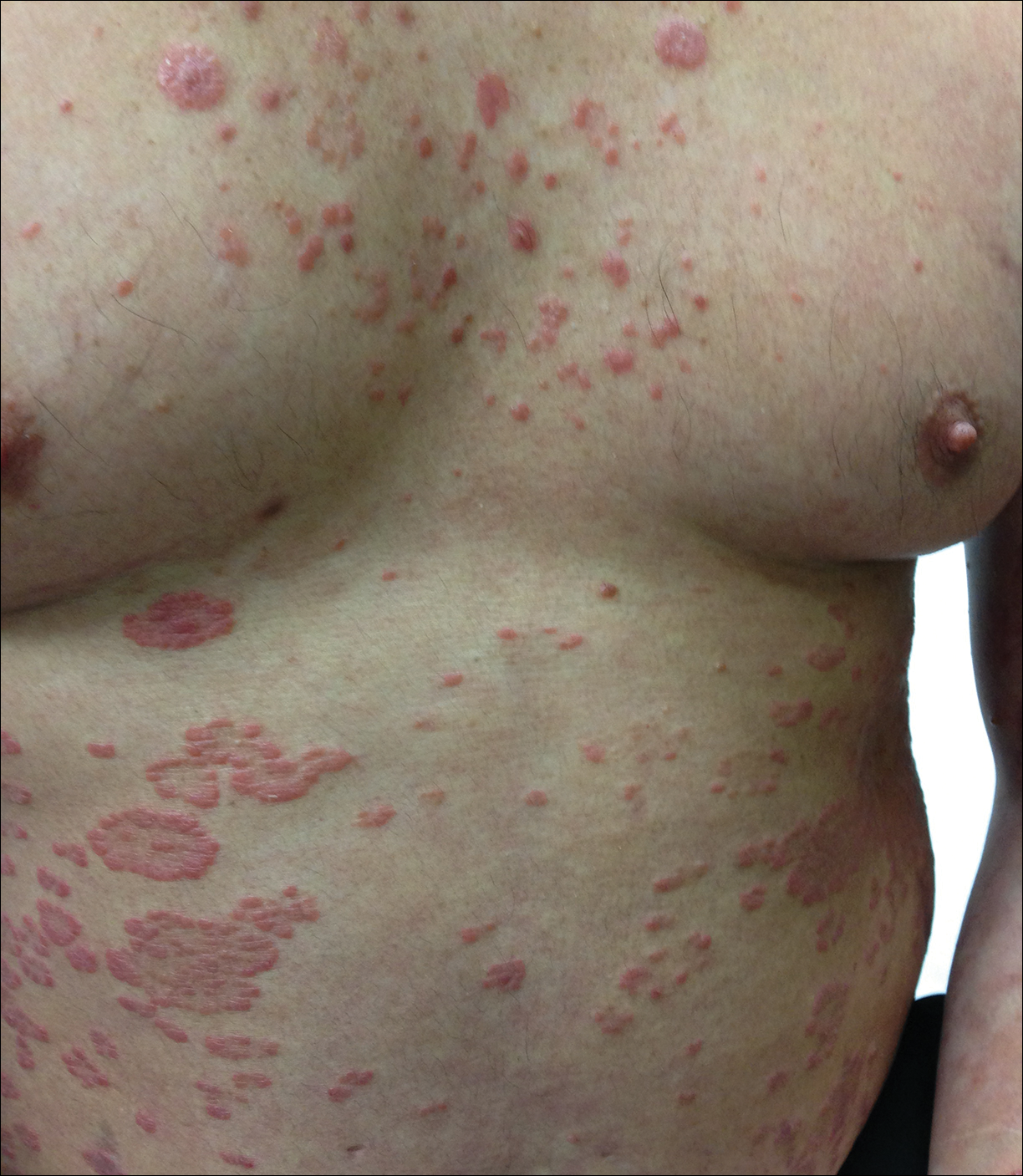
At follow-up 1 year later, the patient had continued improvement of psoriasis with approximately 30% BSA on a treatment regimen of etanercept 50 mg weekly injection and topical corticosteroids. However, on physical examination, there were multiple small semitranslucent papules with telangiectases on the chest and upper back (Figure 1). Biopsy of a representative papule on the chest revealed MC (Figure 2). The patient was subsequently advised to stop etanercept and to return immediately to the clinic for HIV testing. He returned for follow-up 3 months later with pronounced worsening of disease and a new onset of blurred vision of the right eye. Cutaneous examination revealed numerous large erythematous plaques with superficial scale and cerebriform surface on the chest, back, abdomen, and upper and lower extremities involving 80% BSA (Figure 3). Biopsy of a plaque demonstrated psoriasiform dermatitis with neutrophils and parakeratosis consistent with psoriasis. Extensive blood work was notable for reactive HIV antibody and lymphopenia, CD4 lymphocyte count of 60 cells/mm3, and an HIV viral load of 247,000 copies/mL, meeting diagnostic criteria for AIDS. Additionally, ophthalmologic evaluation revealed toxoplasma retinitis. Upon initiation of highly active antiretroviral therapy (HAART) and continued use of topical corticosteroids, the patient experienced notable improvement of disease severity with approximately 20% BSA.
Comment
Molluscum contagiosum is a common skin infection. Among patients with HIV and other types of impaired cellular immunity, the prevalence of MC is estimated to be as high as 20%.3 The MC poxvirus survives and proliferates within the epidermis by interfering with tumor necrosis factor–induced apoptosis of virally infected cells; therefore, intact cell-mediated immunity is an important component of prevention and clearance of poxvirus infections. In immunocompromised patients, the presentation of MC varies widely, and the disease is often difficult to eradicate. This review will highlight the prevalence, presentation, and treatment of MC in the context of immunosuppressed states.
HIV/AIDS
Molluscum contagiosum in HIV-positive patients was first recognized in 1983,2 and its prevalence is estimated to range from 5% to 18% in AIDS patients.3 Molluscum contagiosum is a clinical sign of HIV progression, and its incidence appears to increase with reduced immune function (ie, a CD4 cell count <200/mm3).3 In a study of 456 patients with HIV-associated skin disorders, the majority of patients with MC had notable immunosuppression with a median survival time of 12 months. Thus, MC was not an independent prognostic marker but a clinical indicator of markedly reduced immune status.4
Molluscum contagiosum is transmitted in both sexual and nonsexual patterns in HIV-positive individuals, with the distribution of the latter involving primarily the face and neck. Although it may present with typical umbilicated papules, MC has a wide range of atypical clinical presentations in patients with AIDS that can make it difficult to diagnose. Complicated cases of eyelid MC have been reported in advanced HIV in both adults and children, resulting in obstruction of vision due to large lesions (up to 2 cm) or hundreds of confluent lesions.5 Giant MC, which appears as large exophytic nodules, is another presentation that has been frequently described in patients with advanced HIV. In these patients, the lesions often are too voluminous for conservative therapy and require excision.6 Atypical MC lesions also can resemble other dermatologic conditions, including condyloma acuminatum,7 nevus sebaceous of Jadassohn, ecthyma,8 and cutaneous horns,9,10 as well as other bacterial and fungal infections in HIV-positive patients, such as cutaneous Cryptococcus neoformans,11,12 disseminated histoplasmosis,13 and infections caused by Penicillium marneffei14 and Bartonella henselae.15 In most cases of MC in HIV-positive patients, diagnosis is dependent on the examination of biopsy specimens, which maintain the same histopathologic features regardless of immune status.
The management of MC in patients with HIV/AIDS is difficult. Molluscum contagiosum has shown no evidence of spontaneous resolution in patients with HIV, and treatment with one modality is often insufficient. Treatment is most successful when a combination approach is utilized with destructive procedures (eg, curettage, cryosurgery) and adjunctive agents (eg, retinoids, cantharidin, trichloroacetic acid). Imiquimod and cidofovir have been used off label for MC in AIDS patients.16 Imiquimod, which is used to treat genital warts, another cutaneous viral infection seen in patients with HIV, has demonstrated efficacy in treating MC.16 In a randomized controlled trial comparing imiquimod cream 5% to cryotherapy for MC in healthy children, imiquimod was slow acting but better suited than cryotherapy for patients with eruptions of many small lesions.17 For HIV patients, numerous reports have described successful treatment of disseminated or recalcitrant MC with topical imiquimod.18-20 Cidofovir, an antiviral used to treat cytomegalovirus retinitis in patients with AIDS, is a promising antiviral agent against the poxvirus family. In a study of viral DNA polymerase genes of MC virus, cidofovir inhibited MC virus DNA polymerase activity.21 It has been used in both topical (1% to 3%) and intravenous form to successfully treat recalcitrant and exuberant giant MC.6,22 However, the use of cidofovir is limited by its high costs, especially when compounded into a topical formulation.23
From a systemic standpoint, numerous reports have shown that treating the underlying HIV by optimizing HAART is the most important first step in clearing MC.24-27 However, a special concern regarding the initiation of HAART in patients with MC as well as a markedly impaired immune function is the development of an inflammatory reaction called immune reconstitution inflammatory syndrome (IRIS). This reaction is thought to be a result of immune recovery in severely immunosuppressed patients. During the initial phase of reconstitution when CD4 lymphocyte counts rise and viral load decreases, IRIS occurs due to an inflammatory reaction to microbial and autoimmune antigens, leading to temporary clinical deterioration.28 The incidence has been reported in up to 25% of patients starting HAART, and 52% to 78% of IRIS cases involve dermatologic manifestations such as varicella-zoster virus, cytomegalovirus infections, genital warts, and MC.29,30 In a cohort study of 199 patients, 2% of patients developed MC within 6 months of initiating HAART.31 In a case of exuberant MC lesions after beginning HAART, the lesions spontaneously resolved with the progression of immune reconstitution.28
Malignancies
Patients with hematologic malignancies such as lymphoma and leukemia comprise another subset of patients at risk for atypical presentations of MC. Molluscum contagiosum has been described in patients with hematologic malignancies such as adult T-cell leukemia/lymphoma, multiple myeloma, chronic myeloid leukemia, acute lymphoblastic leukemia, lymphomatoid papulosis, and non-Hodgkin lymphoma. In a review of MC in children with cancer, 0.5% were diagnosed with MC.32,33 Reports also have documented eruptive MC in the presence of solid organ cancers, including lung cancer.34
In patients with malignancies, the differential diagnosis should include other common dermatologic conditions such as varicella, herpes simplex, papillomas, pyoderma, and cutaneous cryptococcosis, as well as MC. Similar to HIV-positive patients, the lesions of MC described in patients with malignancies do not tend to spontaneously resolve. In a report of a pediatric patient with acute lymphoblastic leukemia, MC presented as an ulcerated lesion without any classic features, requiring biopsy for definitive diagnosis. Only partial resolution was achieved with cryotherapy and crusting of the lesion in an attempt to slow the progression.35 In a series of 5 children with hematologic malignancies and MC, little improvement was noted after treatment with surgical scraping, liquid nitrogen, and salicylic acid ointment 5%. Similar to patients with HIV, improvement of immune status and function help clear the disease, and patients who reach remission and discontinue chemotherapeutic agents have a higher rate of spontaneous resolution of previously recalcitrant MC lesions.36
Transplant Patients
Molluscum contagiosum in transplant patients has features similar to patients with HIV/AIDS. In organ transplant recipients, there is an increased risk for cutaneous disease from iatrogenic immunosuppression or immunosuppression through infectious or neoplastic processes.37 As in other immunocompromised populations, MC often has an atypical presentation in transplant patients with more extensive involvement and recalcitrant, rapidly recurring lesions.
In a review of 145 pediatric organ transplant recipients, MC was the fourth most common skin infection after verruca vulgaris, tinea versicolor, and herpes simplex/zoster. Affecting 7% of patients, the majority of patients demonstrated clinically typical lesions; however, the disease was difficult to eradicate if multiple lesions were present.37 In other reports in adults, fulminant and giant MC have been described after renal and other solid organ transplants.38,39 Molluscum contagiosum also has been reported to mimic other skin diseases in transplant patients including tinea barbae40 and nodular basal cell carcinomas.41
The standard treatments are identical to those used in patients with HIV, including ablative methods via liquid nitrogen, electrocautery, cantharidin, trichloroacetic acid, and topical retinoids. Similar to MC in other immunocompromised states, treatment can be difficult and usually requires multiple modalities. For children, imiquimod cream 5% has been recommended due to high clearance rates (up to 92%) and the painless nature of the treatment.42,43
Other Iatrogenic Immunosuppressive States
Immunosuppression through the use of steroids, chemotherapeutic agents, and biologic drugs often is the result of treatment of various diseases. In patients with psoriasis treated with systemic immunosuppressive agents, there are numerous reports that describe the appearance of eruptive MC in association with methotrexate, cyclosporine, and biologics. Methotrexate acts as an immunosuppressive agent by binding to dihydrofolate reductase, which inhibits DNA synthesis in immunologically competent cells.44 It also may block host defense mechanisms against MC by suppressing the expression of serum inflammatory cytokines such as tumor necrosis factor α (TNF-α) and IFN-γ and suppressing the activity of TNF-α inducing apoptosis of virus-infected cells. Cyclosporine used in conjunction with methotrexate may exacerbate the insult to the immune system by inhibiting the production of IFN-γ.45 Biologics are an emerging class of drugs that have demonstrated efficacy in moderate to severe psoriasis by inhibiting TNF-α or other inflammatory molecules. Several published reports have described eruptive or atypical MC in patients on biologic medications. In one case, within 2 weeks after initiation of infliximab, a monoclonal antibody against TNF-α, a patient developed an eruption of MC involving the entire body.46 In another report, an anti–TNF-α agent for rheumatoid arthritis was associated with atypical MC with eyelid lesions.47
There are other skin disorders treated with immunosuppressive agents that also have been associated with MC. In a patient with pemphigus vulgaris treated with prednisolone, pimecrolimus, and azathioprine, MC lesions were observed on the face and within healed pemphigus vulgaris sites.48 Pimecrolimus and tacrolimus, corticosteroid-sparing agents, suppress cell-mediated immunity and inhibit inflammatory cytokines such as IL-2. The infection resolved with a gradual tapering of immunosuppressive therapy and 10 sessions of cryotherapy.48 In a case of topical pimecrolimus for pityriasis alba, the patient developed biopsy-proven MC within 2 weeks of initiating treatment in the areas that were treated with tacrolimus.49
In nontransplant patients with iatrogenic immunosuppression, MC treatment has not been documented to be as challenging as in patients with inherent immunosuppression. Most patients respond to either withdrawal of the drug alone or to simple ablative treatments such as cryotherapy.45,46,48 This important difference is most likely due to the presence of an otherwise intact immune system.
Conclusion
This case describes the appearance of MC in a patient with psoriasis treated with a TNF-α inhibitor who was ultimately diagnosed with AIDS. Although atypical MC infections have been documented in patients with psoriasis undergoing treatment with biologics, it is thought to be more common for MC to occur in more remarkably immunocompromised states such as AIDS. Thus, the persistence and progression of MC in our patient despite discontinuation of etanercept suggested a separate underlying process. Subsequent workup led to the diagnosis of AIDS along with the opportunistic ocular infection of toxoplasmosis retinitis. This clinical sequence consisting of psoriasis treated with a biologic agent, development of MC, and subsequent diagnosis of AIDS is unique and clinically significant to dermatologists. The presentation of psoriasis in patients with HIV can be diverse with different levels of severity and atypical clinical features. In many cases, HIV is known to exacerbate the classic clinical presentation of psoriasis. However, there are other particular presentations of psoriasis in HIV patients that have been observed, which include a predilection for scalp lesions, palmoplantar keratoderma, flexural involvement, and higher levels of immunodeficiency.50 Although tuberculin skin tests are required prior to initiating biologic therapy due to the potential for disease reactivation, there are no requirements for HIV antibody testing. In cases of severe recalcitrant psoriasis, an HIV test should be ordered during the workup to establish an early diagnosis so that an HIV-positive patient can avoid poor outcomes from either the disease processes, the use of certain therapeutic agents, or both. Furthermore, the benefit of avoiding possible harm to the patient and potential legal action outweighs the cost of performing surveillance HIV testing in this subset of patients. Thus, due to the potential additive immunosuppressive effect of HIV with biologic therapy, providers should always assess for risk factors and consider testing for HIV in all patients before initiating treatment with immunosuppressive agents such as biologics.
- Dohil MA, Lin P, Lee J, et al. The epidemiology of molluscum contagiosum in children. J Am Acad Dermatol. 2006;54:47-54.
- Reichert CM, O’Leary TJ, Levens DL, et al. Autopsy pathology in the acquired immune deficiency syndrome. Am J Pathol. 1983;112:357-382.
- Czelusta A, Yen-Moore A, Van der Straten M, et al. An overview of sexually transmitted diseases. Part III. Sexually transmitted diseases in HIV-infected patients. J Am Acad Dermatol. 2000;43:409-432.
- Husak R, Garbe C, Orfanos CE. Mollusca contagiosa in HIV infection. Clinical manifestation, relation to immune status and prognostic value in 39 patients [in German]. Hautarzt. 1997;48:103-109.
- Averbuch D, Jaouni T, Pe’er J, et al. Confluent molluscum contagiosum covering the eyelids of an HIV-positive child. Clin Exp Ophthalmol. 2009;37:525-527.
- Erickson C, Driscoll M, Gaspari A. Efficacy of intravenous cidofovir in the treatment of giant molluscum contagiosum in a patient with human immunodeficiency virus. Arch Dermatol. 2011;147:652-654.
- Mastrolorenzo A, Urbano FG, Salimbeni L, et al. Atypical molluscum contagiosum infection in an HIV-infected patient. Int J Dermatol. 1998;37:378-380.
- Itin PH, Gilli L. Molluscum contagiosum mimicking sebaceous nevus of Jadassohn, ecthyma and giant condylomata acuminata in HIV-infected patients. Dermatology. 1994;189:396-398.
- Sim JH, Lee ES. Molluscum contagiosum presenting as a cutaneous horn. Ann Dermatol. 2011;23:262-263.
- Manchanda Y, Sethuraman G, Paderwani PP, et al. Molluscum contagiosum presenting as penile horn in an HIV positive patient. Sex Transm Infect. 2005;81:183-184.
- Miller SJ. Cutaneous cryptococcus resembling molluscum contagiosum in a patient with acquired immunodeficiency syndrome. Cutis. 1988;41:411-412.
- Sornum A. A mistaken diagnosis of molluscum contagiosum in a HIV-positive patient in rural South Africa. BMJ Case Rep. 2012;14.
- Corti M, Villafañe MF, Palmieri O, et al. Rupioid histoplasmosis: first case reported in an AIDS patient in Argentina. Rev Inst Med Trop Sao Paulo. 2010;52:279-280.
- Saikia L, Nath R, Hazarika D, et al. Atypical cutaneous lesions of Penicillium marneffei infection as a manifestation of the immune reconstitution inflammatory syndrome after highly active antiretroviral therapy. Indian J Dermatol Venereol Leprol. 2010;76:45-48.
- de Souza JA. Molluscum or a mimic? Am J Med. 2006;119:927-929.
- Conant MA. Immunomodulatory therapy in the management of viral infections in patients with HIV infection. J Am Acad Dermatol. 2000;43:S27-S30.
- Gamble RG, Echols KF, Dellavalle RP. Imiquimod vs cryotherapy for molluscum contagiosum: a randomized controlled trial. Arch Dermatol. 2012;148:109-112.
- Brown CW Jr, O’Donoghue M, Moore J, et al. Recalcitrant molluscum contagiosum in an HIV-afflicted male treated successfully with topical imiquimod. Cutis. 2000;65:363-366.
- Strauss RM, Doyle EL, Mohsen AH, et al. Successful treatment of molluscum contagiosum with topical imiquimod in a severely immunocompromised HIV-positive patient. Int J STD AIDS. 2001;12:264-266.
- Theiler M, Kempf W, Kerl K, et al. Disseminated molluscum contagiosum in a HIV-positive child. improvement after therapy with 5% imiquimod. J Dermatol Case Rep. 2011;5:19-23.
- Watanabe T, Tamaki K. Cidofovir diphosphate inhibits molluscum contagiosum virus DNA polymerase activity. J Invest Dermatol. 2008;128:1327-1329.
- Calista D. Topical cidofovir for severe cutaneous human papillomavirus and molluscum contagiosum infections in patients with HIV/AIDS. a pilot study. J Eur Acad Dermatol Venereol. 2000;14:484-488.
- Toro JR, Sanchez S, Turiansky G, et al. Topical cidofovir for the treatment of dermatologic conditions: verruca, condyloma, intraepithelial neoplasia, herpes simplex and its potential use in smallpox. Dermatol Clin. 2003;21:301-309.
- Calista D, Boschini A, Landi G. Resolution of disseminated molluscum contagiosum with highly active anti-retroviral therapy (HAART) in patients with AIDS. Eur J Dermatol. 1999;9:211-213.
- Cattelan AM, Sasset L, Corti L, et al. A complete remission of recalcitrant molluscum contagiosum in an AIDS patient following highly active antiretroviral therapy (HAART). J Infect. 1999;38:58-60.
- Sen S, Bhaumik P. Resolution of giant molluscum contagiosum with antiretroviral therapy. Indian J Dermatol Venereol Leprol. 2008;74:267-268.
- Sen S, Goswami BK, Karjyi N, et al. Disfiguring molluscum contagiosum in a HIV-positive patient responding to antiretroviral therapy. Indian J Dermatol. 2009;54:180-182.
- Pereira B, Fernandes C, Nachiambo E, et al. Exuberant molluscum contagiosum as a manifestation of the immune reconstitution inflammatory syndrome. Dermatol Online J. 2007;13:6.
- Osei-Sekyere B, Karstaedt AS. Immune reconstitution inflammatory syndrome involving the skin. Clin Exp Dermatol. 2010;35:477-481.
- Sung KU, Lee HE, Choi WR, et al. Molluscum contagiosum as a skin manifestation of immune reconstitution inflammatory syndrome in an AIDS patient who is receiving HAART. Korean J Fam Med. 2012;33:182-185.
- Ratnam I, Chiu C, Kandala NB, et al. Incidence and risk factors for immune reconstitution inflammatory syndrome in an ethnically diverse HIV type 1-infected cohort. Clin Infect Dis. 2006;42:418-427.
- Chen KW, Yang CF, Huang CT, et al. Molluscum contagiosum in a patient with adult T-cell leukaemia/lymphoma. Br J Haematol. 2011;155:286.
- Fernandez KH, Bream M, Ali MA, et al. Investigation of molluscum contagiosum virus, orf and other parapoxviruses in lymphomatoid papulosis. J Am Acad Dermatol. 2013;68:1046-1047.
- Nakamura-Wakatsuki T, Kato Y, Miura T, et al. Eruptive molluscum contagiosums in a patient with rheumatoid arthritis and lung cancer. Rheumatol Int. 2011;31:1117-1118.
- Ozyürek E, Sentürk N, Kefeli M, et al. Ulcerating molluscum contagiosum in a boy with relapsed acute lymphoblastic leukemia. J Pediatr Hematol Oncol. 2011;33:E114-E116.
- Hughes WT, Parham DM. Molluscum contagiosum in children with cancer or acquired immunodeficiency syndrome. Pediatr Infect Dis J. 1991;10:152-156.
- Euvrard S, Kanitakis J, Cochat P, et al. Skin diseases in children with organ transplants. J Am Acad Dermatol. 2001;44:932-939.
- Gardner LS, Ormond PJ. Treatment of multiple giant molluscum contagiosum in a renal transplant patient with imiquimod 5% cream. Clin Exp Dermatol. 2006;31:452-453.
- Mansur AT, Göktay F, Gündüz S, et al. Multiple giant molluscum contagiosum in a renal transplant recipient. Transpl Infect Dis. 2004;6:120-123.
- Feldmeyer L, Kamarashev J, Boehler A, et al. Molluscum contagiosum folliculitis mimicking tinea barbae in a lung transplant recipient. J Am Acad Dermatol. 2010;63:169-171.
- Tas¸kapan O, Yenicesu M, Aksu A. A giant solitary molluscum contagiosum, resembling nodular basal cell carcinoma, in a renal transplant recipient. Acta Derm Venereol. 1996;76:247-248.
- Tan HH, Goh CL. Viral infections affecting the skin in organ transplant recipients: epidemiology and current management strategies. Am J Clin Dermatol. 2006;7:13-29.
- Al-Mutairi N, Al-Doukhi A, Al-Farag S, et al. Comparative study on the efficacy, safety, and acceptability of imiquimod 5% cream versus cryotherapy for molluscum contagiosum in children. Pediatr Dermatol. 2010;27:388-394.
- Lim KS, Foo CC. Disseminated molluscum contagiosum in a patient with chronic plaque psoriasis taking methotrexate. Clin Exp Dermatol. 2007;32:591-593.
- Fotiadou C, Lazaridou E, Lekkas D, et al. Disseminated, eruptive molluscum contagiosum lesions in a psoriasis patient under treatment with methotrexate and cyclosporine. Eur J Dermatol. 2012;22:147-148.
- Antoniou C, Kosmadaki MG, Stratigos AJ, et al. Genital HPV lesions and molluscum contagiosum occurring in patients receiving anti-TNF-alpha therapy. Dermatology. 2008;216:364-365.
- Cursiefen C, Grunke M, Dechant C, et al. Multiple bilateral eyelid molluscum contagiosum lesions associated with TNFalpha-antibody and methotrexate therapy. Am J Ophthalmol. 2002;134:270-271.
- Heng YK, Lee JS, Neoh CY. Verrucous plaques in a pemphigus vulgaris patient on immunosuppressive therapy. Int J Dermatol. 2012;51:1044-1046.
- Goksugur N, Ozbostanci B, Goksugur SB. Molluscum contagiosum infection associated with pimecrolimus use in pityriasis alba. Pediatr Dermatol. 2007;24:E63-E65.
- Fernandes S, Pinto GM, Cardoso J. Particular clinical presentations of psoriasis in HIV patients. Int J STD AIDS. 2011;22:653-654.
Molluscum contagiosum (MC) is a double-stranded DNA virus of the Poxviridae family, which commonly infects human keratinocytes resulting in small, umbilicated, flesh-colored papules. The greatest incidence of MC is seen in the pediatric population and sexually active young adults, and it is considered a self-limited disease in immunocompetent individuals.1 With the emergence of the human immunodeficiency virus (HIV) and subsequent AIDS epidemic in the 1980s, a new population of immunocompromised individuals has been observed to be increasingly susceptible to MC with an atypical clinical presentation and a recalcitrant disease course.2 Although the increased prevalence of MC in the HIV population has been well-documented, it has been observed in other disease states or iatrogenically induced immunosuppression due to a deficiency in function or absolute number of T lymphocytes.
We present a case of a patient with long-standing psoriasis on biologic therapy who presented with MC with a subsequent workup that revealed AIDS. This case reiterates the importance of MC as a potential indicator of underlying immunosuppression. We review the literature to evaluate the occurrence of MC in immunosuppressed patients.
Case Report
A 33-year-old man initially presented for evaluation of severe plaque-type psoriasis associated with pain, erythema, and swelling of the joints of the hands of 10 years’ duration. He was started on methotrexate 5 mg weekly and topical corticosteroids but was unable to tolerate methotrexate due to headaches. He also had difficulty affording topical medications and adjunctive phototherapy. The patient was sporadically seen in follow-up with persistence of psoriatic plaques involving up to 60% body surface area (BSA) with the only treatment consisting of occasional topical steroids. Five years later, the patient was restarted on methotrexate 5 to 7.5 mg weekly, which resulted in moderate improvement. However, because of persistent elevation of liver enzymes, this treatment was stopped. Several months later he was evaluated for treatment with a biologic agent, and after a negative tuberculin skin test, he began treatment with etanercept 50 mg subcutaneous injection twice weekly, which provided notable improvement and allowed for reduction of dose frequency to once weekly.
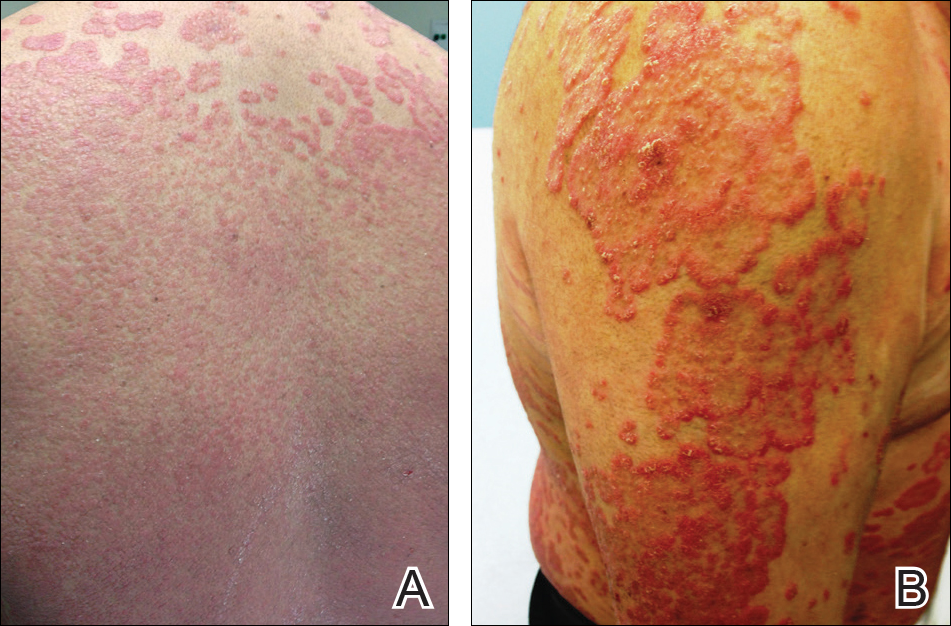
At follow-up 1 year later, the patient had continued improvement of psoriasis with approximately 30% BSA on a treatment regimen of etanercept 50 mg weekly injection and topical corticosteroids. However, on physical examination, there were multiple small semitranslucent papules with telangiectases on the chest and upper back (Figure 1). Biopsy of a representative papule on the chest revealed MC (Figure 2). The patient was subsequently advised to stop etanercept and to return immediately to the clinic for HIV testing. He returned for follow-up 3 months later with pronounced worsening of disease and a new onset of blurred vision of the right eye. Cutaneous examination revealed numerous large erythematous plaques with superficial scale and cerebriform surface on the chest, back, abdomen, and upper and lower extremities involving 80% BSA (Figure 3). Biopsy of a plaque demonstrated psoriasiform dermatitis with neutrophils and parakeratosis consistent with psoriasis. Extensive blood work was notable for reactive HIV antibody and lymphopenia, CD4 lymphocyte count of 60 cells/mm3, and an HIV viral load of 247,000 copies/mL, meeting diagnostic criteria for AIDS. Additionally, ophthalmologic evaluation revealed toxoplasma retinitis. Upon initiation of highly active antiretroviral therapy (HAART) and continued use of topical corticosteroids, the patient experienced notable improvement of disease severity with approximately 20% BSA.
Comment
Molluscum contagiosum is a common skin infection. Among patients with HIV and other types of impaired cellular immunity, the prevalence of MC is estimated to be as high as 20%.3 The MC poxvirus survives and proliferates within the epidermis by interfering with tumor necrosis factor–induced apoptosis of virally infected cells; therefore, intact cell-mediated immunity is an important component of prevention and clearance of poxvirus infections. In immunocompromised patients, the presentation of MC varies widely, and the disease is often difficult to eradicate. This review will highlight the prevalence, presentation, and treatment of MC in the context of immunosuppressed states.
HIV/AIDS
Molluscum contagiosum in HIV-positive patients was first recognized in 1983,2 and its prevalence is estimated to range from 5% to 18% in AIDS patients.3 Molluscum contagiosum is a clinical sign of HIV progression, and its incidence appears to increase with reduced immune function (ie, a CD4 cell count <200/mm3).3 In a study of 456 patients with HIV-associated skin disorders, the majority of patients with MC had notable immunosuppression with a median survival time of 12 months. Thus, MC was not an independent prognostic marker but a clinical indicator of markedly reduced immune status.4
Molluscum contagiosum is transmitted in both sexual and nonsexual patterns in HIV-positive individuals, with the distribution of the latter involving primarily the face and neck. Although it may present with typical umbilicated papules, MC has a wide range of atypical clinical presentations in patients with AIDS that can make it difficult to diagnose. Complicated cases of eyelid MC have been reported in advanced HIV in both adults and children, resulting in obstruction of vision due to large lesions (up to 2 cm) or hundreds of confluent lesions.5 Giant MC, which appears as large exophytic nodules, is another presentation that has been frequently described in patients with advanced HIV. In these patients, the lesions often are too voluminous for conservative therapy and require excision.6 Atypical MC lesions also can resemble other dermatologic conditions, including condyloma acuminatum,7 nevus sebaceous of Jadassohn, ecthyma,8 and cutaneous horns,9,10 as well as other bacterial and fungal infections in HIV-positive patients, such as cutaneous Cryptococcus neoformans,11,12 disseminated histoplasmosis,13 and infections caused by Penicillium marneffei14 and Bartonella henselae.15 In most cases of MC in HIV-positive patients, diagnosis is dependent on the examination of biopsy specimens, which maintain the same histopathologic features regardless of immune status.
The management of MC in patients with HIV/AIDS is difficult. Molluscum contagiosum has shown no evidence of spontaneous resolution in patients with HIV, and treatment with one modality is often insufficient. Treatment is most successful when a combination approach is utilized with destructive procedures (eg, curettage, cryosurgery) and adjunctive agents (eg, retinoids, cantharidin, trichloroacetic acid). Imiquimod and cidofovir have been used off label for MC in AIDS patients.16 Imiquimod, which is used to treat genital warts, another cutaneous viral infection seen in patients with HIV, has demonstrated efficacy in treating MC.16 In a randomized controlled trial comparing imiquimod cream 5% to cryotherapy for MC in healthy children, imiquimod was slow acting but better suited than cryotherapy for patients with eruptions of many small lesions.17 For HIV patients, numerous reports have described successful treatment of disseminated or recalcitrant MC with topical imiquimod.18-20 Cidofovir, an antiviral used to treat cytomegalovirus retinitis in patients with AIDS, is a promising antiviral agent against the poxvirus family. In a study of viral DNA polymerase genes of MC virus, cidofovir inhibited MC virus DNA polymerase activity.21 It has been used in both topical (1% to 3%) and intravenous form to successfully treat recalcitrant and exuberant giant MC.6,22 However, the use of cidofovir is limited by its high costs, especially when compounded into a topical formulation.23
From a systemic standpoint, numerous reports have shown that treating the underlying HIV by optimizing HAART is the most important first step in clearing MC.24-27 However, a special concern regarding the initiation of HAART in patients with MC as well as a markedly impaired immune function is the development of an inflammatory reaction called immune reconstitution inflammatory syndrome (IRIS). This reaction is thought to be a result of immune recovery in severely immunosuppressed patients. During the initial phase of reconstitution when CD4 lymphocyte counts rise and viral load decreases, IRIS occurs due to an inflammatory reaction to microbial and autoimmune antigens, leading to temporary clinical deterioration.28 The incidence has been reported in up to 25% of patients starting HAART, and 52% to 78% of IRIS cases involve dermatologic manifestations such as varicella-zoster virus, cytomegalovirus infections, genital warts, and MC.29,30 In a cohort study of 199 patients, 2% of patients developed MC within 6 months of initiating HAART.31 In a case of exuberant MC lesions after beginning HAART, the lesions spontaneously resolved with the progression of immune reconstitution.28
Malignancies
Patients with hematologic malignancies such as lymphoma and leukemia comprise another subset of patients at risk for atypical presentations of MC. Molluscum contagiosum has been described in patients with hematologic malignancies such as adult T-cell leukemia/lymphoma, multiple myeloma, chronic myeloid leukemia, acute lymphoblastic leukemia, lymphomatoid papulosis, and non-Hodgkin lymphoma. In a review of MC in children with cancer, 0.5% were diagnosed with MC.32,33 Reports also have documented eruptive MC in the presence of solid organ cancers, including lung cancer.34
In patients with malignancies, the differential diagnosis should include other common dermatologic conditions such as varicella, herpes simplex, papillomas, pyoderma, and cutaneous cryptococcosis, as well as MC. Similar to HIV-positive patients, the lesions of MC described in patients with malignancies do not tend to spontaneously resolve. In a report of a pediatric patient with acute lymphoblastic leukemia, MC presented as an ulcerated lesion without any classic features, requiring biopsy for definitive diagnosis. Only partial resolution was achieved with cryotherapy and crusting of the lesion in an attempt to slow the progression.35 In a series of 5 children with hematologic malignancies and MC, little improvement was noted after treatment with surgical scraping, liquid nitrogen, and salicylic acid ointment 5%. Similar to patients with HIV, improvement of immune status and function help clear the disease, and patients who reach remission and discontinue chemotherapeutic agents have a higher rate of spontaneous resolution of previously recalcitrant MC lesions.36
Transplant Patients
Molluscum contagiosum in transplant patients has features similar to patients with HIV/AIDS. In organ transplant recipients, there is an increased risk for cutaneous disease from iatrogenic immunosuppression or immunosuppression through infectious or neoplastic processes.37 As in other immunocompromised populations, MC often has an atypical presentation in transplant patients with more extensive involvement and recalcitrant, rapidly recurring lesions.
In a review of 145 pediatric organ transplant recipients, MC was the fourth most common skin infection after verruca vulgaris, tinea versicolor, and herpes simplex/zoster. Affecting 7% of patients, the majority of patients demonstrated clinically typical lesions; however, the disease was difficult to eradicate if multiple lesions were present.37 In other reports in adults, fulminant and giant MC have been described after renal and other solid organ transplants.38,39 Molluscum contagiosum also has been reported to mimic other skin diseases in transplant patients including tinea barbae40 and nodular basal cell carcinomas.41
The standard treatments are identical to those used in patients with HIV, including ablative methods via liquid nitrogen, electrocautery, cantharidin, trichloroacetic acid, and topical retinoids. Similar to MC in other immunocompromised states, treatment can be difficult and usually requires multiple modalities. For children, imiquimod cream 5% has been recommended due to high clearance rates (up to 92%) and the painless nature of the treatment.42,43
Other Iatrogenic Immunosuppressive States
Immunosuppression through the use of steroids, chemotherapeutic agents, and biologic drugs often is the result of treatment of various diseases. In patients with psoriasis treated with systemic immunosuppressive agents, there are numerous reports that describe the appearance of eruptive MC in association with methotrexate, cyclosporine, and biologics. Methotrexate acts as an immunosuppressive agent by binding to dihydrofolate reductase, which inhibits DNA synthesis in immunologically competent cells.44 It also may block host defense mechanisms against MC by suppressing the expression of serum inflammatory cytokines such as tumor necrosis factor α (TNF-α) and IFN-γ and suppressing the activity of TNF-α inducing apoptosis of virus-infected cells. Cyclosporine used in conjunction with methotrexate may exacerbate the insult to the immune system by inhibiting the production of IFN-γ.45 Biologics are an emerging class of drugs that have demonstrated efficacy in moderate to severe psoriasis by inhibiting TNF-α or other inflammatory molecules. Several published reports have described eruptive or atypical MC in patients on biologic medications. In one case, within 2 weeks after initiation of infliximab, a monoclonal antibody against TNF-α, a patient developed an eruption of MC involving the entire body.46 In another report, an anti–TNF-α agent for rheumatoid arthritis was associated with atypical MC with eyelid lesions.47
There are other skin disorders treated with immunosuppressive agents that also have been associated with MC. In a patient with pemphigus vulgaris treated with prednisolone, pimecrolimus, and azathioprine, MC lesions were observed on the face and within healed pemphigus vulgaris sites.48 Pimecrolimus and tacrolimus, corticosteroid-sparing agents, suppress cell-mediated immunity and inhibit inflammatory cytokines such as IL-2. The infection resolved with a gradual tapering of immunosuppressive therapy and 10 sessions of cryotherapy.48 In a case of topical pimecrolimus for pityriasis alba, the patient developed biopsy-proven MC within 2 weeks of initiating treatment in the areas that were treated with tacrolimus.49
In nontransplant patients with iatrogenic immunosuppression, MC treatment has not been documented to be as challenging as in patients with inherent immunosuppression. Most patients respond to either withdrawal of the drug alone or to simple ablative treatments such as cryotherapy.45,46,48 This important difference is most likely due to the presence of an otherwise intact immune system.
Conclusion
This case describes the appearance of MC in a patient with psoriasis treated with a TNF-α inhibitor who was ultimately diagnosed with AIDS. Although atypical MC infections have been documented in patients with psoriasis undergoing treatment with biologics, it is thought to be more common for MC to occur in more remarkably immunocompromised states such as AIDS. Thus, the persistence and progression of MC in our patient despite discontinuation of etanercept suggested a separate underlying process. Subsequent workup led to the diagnosis of AIDS along with the opportunistic ocular infection of toxoplasmosis retinitis. This clinical sequence consisting of psoriasis treated with a biologic agent, development of MC, and subsequent diagnosis of AIDS is unique and clinically significant to dermatologists. The presentation of psoriasis in patients with HIV can be diverse with different levels of severity and atypical clinical features. In many cases, HIV is known to exacerbate the classic clinical presentation of psoriasis. However, there are other particular presentations of psoriasis in HIV patients that have been observed, which include a predilection for scalp lesions, palmoplantar keratoderma, flexural involvement, and higher levels of immunodeficiency.50 Although tuberculin skin tests are required prior to initiating biologic therapy due to the potential for disease reactivation, there are no requirements for HIV antibody testing. In cases of severe recalcitrant psoriasis, an HIV test should be ordered during the workup to establish an early diagnosis so that an HIV-positive patient can avoid poor outcomes from either the disease processes, the use of certain therapeutic agents, or both. Furthermore, the benefit of avoiding possible harm to the patient and potential legal action outweighs the cost of performing surveillance HIV testing in this subset of patients. Thus, due to the potential additive immunosuppressive effect of HIV with biologic therapy, providers should always assess for risk factors and consider testing for HIV in all patients before initiating treatment with immunosuppressive agents such as biologics.
Molluscum contagiosum (MC) is a double-stranded DNA virus of the Poxviridae family, which commonly infects human keratinocytes resulting in small, umbilicated, flesh-colored papules. The greatest incidence of MC is seen in the pediatric population and sexually active young adults, and it is considered a self-limited disease in immunocompetent individuals.1 With the emergence of the human immunodeficiency virus (HIV) and subsequent AIDS epidemic in the 1980s, a new population of immunocompromised individuals has been observed to be increasingly susceptible to MC with an atypical clinical presentation and a recalcitrant disease course.2 Although the increased prevalence of MC in the HIV population has been well-documented, it has been observed in other disease states or iatrogenically induced immunosuppression due to a deficiency in function or absolute number of T lymphocytes.
We present a case of a patient with long-standing psoriasis on biologic therapy who presented with MC with a subsequent workup that revealed AIDS. This case reiterates the importance of MC as a potential indicator of underlying immunosuppression. We review the literature to evaluate the occurrence of MC in immunosuppressed patients.
Case Report
A 33-year-old man initially presented for evaluation of severe plaque-type psoriasis associated with pain, erythema, and swelling of the joints of the hands of 10 years’ duration. He was started on methotrexate 5 mg weekly and topical corticosteroids but was unable to tolerate methotrexate due to headaches. He also had difficulty affording topical medications and adjunctive phototherapy. The patient was sporadically seen in follow-up with persistence of psoriatic plaques involving up to 60% body surface area (BSA) with the only treatment consisting of occasional topical steroids. Five years later, the patient was restarted on methotrexate 5 to 7.5 mg weekly, which resulted in moderate improvement. However, because of persistent elevation of liver enzymes, this treatment was stopped. Several months later he was evaluated for treatment with a biologic agent, and after a negative tuberculin skin test, he began treatment with etanercept 50 mg subcutaneous injection twice weekly, which provided notable improvement and allowed for reduction of dose frequency to once weekly.
At follow-up 1 year later, the patient had continued improvement of psoriasis with approximately 30% BSA on a treatment regimen of etanercept 50 mg weekly injection and topical corticosteroids. However, on physical examination, there were multiple small semitranslucent papules with telangiectases on the chest and upper back (Figure 1). Biopsy of a representative papule on the chest revealed MC (Figure 2). The patient was subsequently advised to stop etanercept and to return immediately to the clinic for HIV testing. He returned for follow-up 3 months later with pronounced worsening of disease and a new onset of blurred vision of the right eye. Cutaneous examination revealed numerous large erythematous plaques with superficial scale and cerebriform surface on the chest, back, abdomen, and upper and lower extremities involving 80% BSA (Figure 3). Biopsy of a plaque demonstrated psoriasiform dermatitis with neutrophils and parakeratosis consistent with psoriasis. Extensive blood work was notable for reactive HIV antibody and lymphopenia, CD4 lymphocyte count of 60 cells/mm3, and an HIV viral load of 247,000 copies/mL, meeting diagnostic criteria for AIDS. Additionally, ophthalmologic evaluation revealed toxoplasma retinitis. Upon initiation of highly active antiretroviral therapy (HAART) and continued use of topical corticosteroids, the patient experienced notable improvement of disease severity with approximately 20% BSA.
Comment
Molluscum contagiosum is a common skin infection. Among patients with HIV and other types of impaired cellular immunity, the prevalence of MC is estimated to be as high as 20%.3 The MC poxvirus survives and proliferates within the epidermis by interfering with tumor necrosis factor–induced apoptosis of virally infected cells; therefore, intact cell-mediated immunity is an important component of prevention and clearance of poxvirus infections. In immunocompromised patients, the presentation of MC varies widely, and the disease is often difficult to eradicate. This review will highlight the prevalence, presentation, and treatment of MC in the context of immunosuppressed states.
HIV/AIDS
Molluscum contagiosum in HIV-positive patients was first recognized in 1983,2 and its prevalence is estimated to range from 5% to 18% in AIDS patients.3 Molluscum contagiosum is a clinical sign of HIV progression, and its incidence appears to increase with reduced immune function (ie, a CD4 cell count <200/mm3).3 In a study of 456 patients with HIV-associated skin disorders, the majority of patients with MC had notable immunosuppression with a median survival time of 12 months. Thus, MC was not an independent prognostic marker but a clinical indicator of markedly reduced immune status.4
Molluscum contagiosum is transmitted in both sexual and nonsexual patterns in HIV-positive individuals, with the distribution of the latter involving primarily the face and neck. Although it may present with typical umbilicated papules, MC has a wide range of atypical clinical presentations in patients with AIDS that can make it difficult to diagnose. Complicated cases of eyelid MC have been reported in advanced HIV in both adults and children, resulting in obstruction of vision due to large lesions (up to 2 cm) or hundreds of confluent lesions.5 Giant MC, which appears as large exophytic nodules, is another presentation that has been frequently described in patients with advanced HIV. In these patients, the lesions often are too voluminous for conservative therapy and require excision.6 Atypical MC lesions also can resemble other dermatologic conditions, including condyloma acuminatum,7 nevus sebaceous of Jadassohn, ecthyma,8 and cutaneous horns,9,10 as well as other bacterial and fungal infections in HIV-positive patients, such as cutaneous Cryptococcus neoformans,11,12 disseminated histoplasmosis,13 and infections caused by Penicillium marneffei14 and Bartonella henselae.15 In most cases of MC in HIV-positive patients, diagnosis is dependent on the examination of biopsy specimens, which maintain the same histopathologic features regardless of immune status.
The management of MC in patients with HIV/AIDS is difficult. Molluscum contagiosum has shown no evidence of spontaneous resolution in patients with HIV, and treatment with one modality is often insufficient. Treatment is most successful when a combination approach is utilized with destructive procedures (eg, curettage, cryosurgery) and adjunctive agents (eg, retinoids, cantharidin, trichloroacetic acid). Imiquimod and cidofovir have been used off label for MC in AIDS patients.16 Imiquimod, which is used to treat genital warts, another cutaneous viral infection seen in patients with HIV, has demonstrated efficacy in treating MC.16 In a randomized controlled trial comparing imiquimod cream 5% to cryotherapy for MC in healthy children, imiquimod was slow acting but better suited than cryotherapy for patients with eruptions of many small lesions.17 For HIV patients, numerous reports have described successful treatment of disseminated or recalcitrant MC with topical imiquimod.18-20 Cidofovir, an antiviral used to treat cytomegalovirus retinitis in patients with AIDS, is a promising antiviral agent against the poxvirus family. In a study of viral DNA polymerase genes of MC virus, cidofovir inhibited MC virus DNA polymerase activity.21 It has been used in both topical (1% to 3%) and intravenous form to successfully treat recalcitrant and exuberant giant MC.6,22 However, the use of cidofovir is limited by its high costs, especially when compounded into a topical formulation.23
From a systemic standpoint, numerous reports have shown that treating the underlying HIV by optimizing HAART is the most important first step in clearing MC.24-27 However, a special concern regarding the initiation of HAART in patients with MC as well as a markedly impaired immune function is the development of an inflammatory reaction called immune reconstitution inflammatory syndrome (IRIS). This reaction is thought to be a result of immune recovery in severely immunosuppressed patients. During the initial phase of reconstitution when CD4 lymphocyte counts rise and viral load decreases, IRIS occurs due to an inflammatory reaction to microbial and autoimmune antigens, leading to temporary clinical deterioration.28 The incidence has been reported in up to 25% of patients starting HAART, and 52% to 78% of IRIS cases involve dermatologic manifestations such as varicella-zoster virus, cytomegalovirus infections, genital warts, and MC.29,30 In a cohort study of 199 patients, 2% of patients developed MC within 6 months of initiating HAART.31 In a case of exuberant MC lesions after beginning HAART, the lesions spontaneously resolved with the progression of immune reconstitution.28
Malignancies
Patients with hematologic malignancies such as lymphoma and leukemia comprise another subset of patients at risk for atypical presentations of MC. Molluscum contagiosum has been described in patients with hematologic malignancies such as adult T-cell leukemia/lymphoma, multiple myeloma, chronic myeloid leukemia, acute lymphoblastic leukemia, lymphomatoid papulosis, and non-Hodgkin lymphoma. In a review of MC in children with cancer, 0.5% were diagnosed with MC.32,33 Reports also have documented eruptive MC in the presence of solid organ cancers, including lung cancer.34
In patients with malignancies, the differential diagnosis should include other common dermatologic conditions such as varicella, herpes simplex, papillomas, pyoderma, and cutaneous cryptococcosis, as well as MC. Similar to HIV-positive patients, the lesions of MC described in patients with malignancies do not tend to spontaneously resolve. In a report of a pediatric patient with acute lymphoblastic leukemia, MC presented as an ulcerated lesion without any classic features, requiring biopsy for definitive diagnosis. Only partial resolution was achieved with cryotherapy and crusting of the lesion in an attempt to slow the progression.35 In a series of 5 children with hematologic malignancies and MC, little improvement was noted after treatment with surgical scraping, liquid nitrogen, and salicylic acid ointment 5%. Similar to patients with HIV, improvement of immune status and function help clear the disease, and patients who reach remission and discontinue chemotherapeutic agents have a higher rate of spontaneous resolution of previously recalcitrant MC lesions.36
Transplant Patients
Molluscum contagiosum in transplant patients has features similar to patients with HIV/AIDS. In organ transplant recipients, there is an increased risk for cutaneous disease from iatrogenic immunosuppression or immunosuppression through infectious or neoplastic processes.37 As in other immunocompromised populations, MC often has an atypical presentation in transplant patients with more extensive involvement and recalcitrant, rapidly recurring lesions.
In a review of 145 pediatric organ transplant recipients, MC was the fourth most common skin infection after verruca vulgaris, tinea versicolor, and herpes simplex/zoster. Affecting 7% of patients, the majority of patients demonstrated clinically typical lesions; however, the disease was difficult to eradicate if multiple lesions were present.37 In other reports in adults, fulminant and giant MC have been described after renal and other solid organ transplants.38,39 Molluscum contagiosum also has been reported to mimic other skin diseases in transplant patients including tinea barbae40 and nodular basal cell carcinomas.41
The standard treatments are identical to those used in patients with HIV, including ablative methods via liquid nitrogen, electrocautery, cantharidin, trichloroacetic acid, and topical retinoids. Similar to MC in other immunocompromised states, treatment can be difficult and usually requires multiple modalities. For children, imiquimod cream 5% has been recommended due to high clearance rates (up to 92%) and the painless nature of the treatment.42,43
Other Iatrogenic Immunosuppressive States
Immunosuppression through the use of steroids, chemotherapeutic agents, and biologic drugs often is the result of treatment of various diseases. In patients with psoriasis treated with systemic immunosuppressive agents, there are numerous reports that describe the appearance of eruptive MC in association with methotrexate, cyclosporine, and biologics. Methotrexate acts as an immunosuppressive agent by binding to dihydrofolate reductase, which inhibits DNA synthesis in immunologically competent cells.44 It also may block host defense mechanisms against MC by suppressing the expression of serum inflammatory cytokines such as tumor necrosis factor α (TNF-α) and IFN-γ and suppressing the activity of TNF-α inducing apoptosis of virus-infected cells. Cyclosporine used in conjunction with methotrexate may exacerbate the insult to the immune system by inhibiting the production of IFN-γ.45 Biologics are an emerging class of drugs that have demonstrated efficacy in moderate to severe psoriasis by inhibiting TNF-α or other inflammatory molecules. Several published reports have described eruptive or atypical MC in patients on biologic medications. In one case, within 2 weeks after initiation of infliximab, a monoclonal antibody against TNF-α, a patient developed an eruption of MC involving the entire body.46 In another report, an anti–TNF-α agent for rheumatoid arthritis was associated with atypical MC with eyelid lesions.47
There are other skin disorders treated with immunosuppressive agents that also have been associated with MC. In a patient with pemphigus vulgaris treated with prednisolone, pimecrolimus, and azathioprine, MC lesions were observed on the face and within healed pemphigus vulgaris sites.48 Pimecrolimus and tacrolimus, corticosteroid-sparing agents, suppress cell-mediated immunity and inhibit inflammatory cytokines such as IL-2. The infection resolved with a gradual tapering of immunosuppressive therapy and 10 sessions of cryotherapy.48 In a case of topical pimecrolimus for pityriasis alba, the patient developed biopsy-proven MC within 2 weeks of initiating treatment in the areas that were treated with tacrolimus.49
In nontransplant patients with iatrogenic immunosuppression, MC treatment has not been documented to be as challenging as in patients with inherent immunosuppression. Most patients respond to either withdrawal of the drug alone or to simple ablative treatments such as cryotherapy.45,46,48 This important difference is most likely due to the presence of an otherwise intact immune system.
Conclusion
This case describes the appearance of MC in a patient with psoriasis treated with a TNF-α inhibitor who was ultimately diagnosed with AIDS. Although atypical MC infections have been documented in patients with psoriasis undergoing treatment with biologics, it is thought to be more common for MC to occur in more remarkably immunocompromised states such as AIDS. Thus, the persistence and progression of MC in our patient despite discontinuation of etanercept suggested a separate underlying process. Subsequent workup led to the diagnosis of AIDS along with the opportunistic ocular infection of toxoplasmosis retinitis. This clinical sequence consisting of psoriasis treated with a biologic agent, development of MC, and subsequent diagnosis of AIDS is unique and clinically significant to dermatologists. The presentation of psoriasis in patients with HIV can be diverse with different levels of severity and atypical clinical features. In many cases, HIV is known to exacerbate the classic clinical presentation of psoriasis. However, there are other particular presentations of psoriasis in HIV patients that have been observed, which include a predilection for scalp lesions, palmoplantar keratoderma, flexural involvement, and higher levels of immunodeficiency.50 Although tuberculin skin tests are required prior to initiating biologic therapy due to the potential for disease reactivation, there are no requirements for HIV antibody testing. In cases of severe recalcitrant psoriasis, an HIV test should be ordered during the workup to establish an early diagnosis so that an HIV-positive patient can avoid poor outcomes from either the disease processes, the use of certain therapeutic agents, or both. Furthermore, the benefit of avoiding possible harm to the patient and potential legal action outweighs the cost of performing surveillance HIV testing in this subset of patients. Thus, due to the potential additive immunosuppressive effect of HIV with biologic therapy, providers should always assess for risk factors and consider testing for HIV in all patients before initiating treatment with immunosuppressive agents such as biologics.
- Dohil MA, Lin P, Lee J, et al. The epidemiology of molluscum contagiosum in children. J Am Acad Dermatol. 2006;54:47-54.
- Reichert CM, O’Leary TJ, Levens DL, et al. Autopsy pathology in the acquired immune deficiency syndrome. Am J Pathol. 1983;112:357-382.
- Czelusta A, Yen-Moore A, Van der Straten M, et al. An overview of sexually transmitted diseases. Part III. Sexually transmitted diseases in HIV-infected patients. J Am Acad Dermatol. 2000;43:409-432.
- Husak R, Garbe C, Orfanos CE. Mollusca contagiosa in HIV infection. Clinical manifestation, relation to immune status and prognostic value in 39 patients [in German]. Hautarzt. 1997;48:103-109.
- Averbuch D, Jaouni T, Pe’er J, et al. Confluent molluscum contagiosum covering the eyelids of an HIV-positive child. Clin Exp Ophthalmol. 2009;37:525-527.
- Erickson C, Driscoll M, Gaspari A. Efficacy of intravenous cidofovir in the treatment of giant molluscum contagiosum in a patient with human immunodeficiency virus. Arch Dermatol. 2011;147:652-654.
- Mastrolorenzo A, Urbano FG, Salimbeni L, et al. Atypical molluscum contagiosum infection in an HIV-infected patient. Int J Dermatol. 1998;37:378-380.
- Itin PH, Gilli L. Molluscum contagiosum mimicking sebaceous nevus of Jadassohn, ecthyma and giant condylomata acuminata in HIV-infected patients. Dermatology. 1994;189:396-398.
- Sim JH, Lee ES. Molluscum contagiosum presenting as a cutaneous horn. Ann Dermatol. 2011;23:262-263.
- Manchanda Y, Sethuraman G, Paderwani PP, et al. Molluscum contagiosum presenting as penile horn in an HIV positive patient. Sex Transm Infect. 2005;81:183-184.
- Miller SJ. Cutaneous cryptococcus resembling molluscum contagiosum in a patient with acquired immunodeficiency syndrome. Cutis. 1988;41:411-412.
- Sornum A. A mistaken diagnosis of molluscum contagiosum in a HIV-positive patient in rural South Africa. BMJ Case Rep. 2012;14.
- Corti M, Villafañe MF, Palmieri O, et al. Rupioid histoplasmosis: first case reported in an AIDS patient in Argentina. Rev Inst Med Trop Sao Paulo. 2010;52:279-280.
- Saikia L, Nath R, Hazarika D, et al. Atypical cutaneous lesions of Penicillium marneffei infection as a manifestation of the immune reconstitution inflammatory syndrome after highly active antiretroviral therapy. Indian J Dermatol Venereol Leprol. 2010;76:45-48.
- de Souza JA. Molluscum or a mimic? Am J Med. 2006;119:927-929.
- Conant MA. Immunomodulatory therapy in the management of viral infections in patients with HIV infection. J Am Acad Dermatol. 2000;43:S27-S30.
- Gamble RG, Echols KF, Dellavalle RP. Imiquimod vs cryotherapy for molluscum contagiosum: a randomized controlled trial. Arch Dermatol. 2012;148:109-112.
- Brown CW Jr, O’Donoghue M, Moore J, et al. Recalcitrant molluscum contagiosum in an HIV-afflicted male treated successfully with topical imiquimod. Cutis. 2000;65:363-366.
- Strauss RM, Doyle EL, Mohsen AH, et al. Successful treatment of molluscum contagiosum with topical imiquimod in a severely immunocompromised HIV-positive patient. Int J STD AIDS. 2001;12:264-266.
- Theiler M, Kempf W, Kerl K, et al. Disseminated molluscum contagiosum in a HIV-positive child. improvement after therapy with 5% imiquimod. J Dermatol Case Rep. 2011;5:19-23.
- Watanabe T, Tamaki K. Cidofovir diphosphate inhibits molluscum contagiosum virus DNA polymerase activity. J Invest Dermatol. 2008;128:1327-1329.
- Calista D. Topical cidofovir for severe cutaneous human papillomavirus and molluscum contagiosum infections in patients with HIV/AIDS. a pilot study. J Eur Acad Dermatol Venereol. 2000;14:484-488.
- Toro JR, Sanchez S, Turiansky G, et al. Topical cidofovir for the treatment of dermatologic conditions: verruca, condyloma, intraepithelial neoplasia, herpes simplex and its potential use in smallpox. Dermatol Clin. 2003;21:301-309.
- Calista D, Boschini A, Landi G. Resolution of disseminated molluscum contagiosum with highly active anti-retroviral therapy (HAART) in patients with AIDS. Eur J Dermatol. 1999;9:211-213.
- Cattelan AM, Sasset L, Corti L, et al. A complete remission of recalcitrant molluscum contagiosum in an AIDS patient following highly active antiretroviral therapy (HAART). J Infect. 1999;38:58-60.
- Sen S, Bhaumik P. Resolution of giant molluscum contagiosum with antiretroviral therapy. Indian J Dermatol Venereol Leprol. 2008;74:267-268.
- Sen S, Goswami BK, Karjyi N, et al. Disfiguring molluscum contagiosum in a HIV-positive patient responding to antiretroviral therapy. Indian J Dermatol. 2009;54:180-182.
- Pereira B, Fernandes C, Nachiambo E, et al. Exuberant molluscum contagiosum as a manifestation of the immune reconstitution inflammatory syndrome. Dermatol Online J. 2007;13:6.
- Osei-Sekyere B, Karstaedt AS. Immune reconstitution inflammatory syndrome involving the skin. Clin Exp Dermatol. 2010;35:477-481.
- Sung KU, Lee HE, Choi WR, et al. Molluscum contagiosum as a skin manifestation of immune reconstitution inflammatory syndrome in an AIDS patient who is receiving HAART. Korean J Fam Med. 2012;33:182-185.
- Ratnam I, Chiu C, Kandala NB, et al. Incidence and risk factors for immune reconstitution inflammatory syndrome in an ethnically diverse HIV type 1-infected cohort. Clin Infect Dis. 2006;42:418-427.
- Chen KW, Yang CF, Huang CT, et al. Molluscum contagiosum in a patient with adult T-cell leukaemia/lymphoma. Br J Haematol. 2011;155:286.
- Fernandez KH, Bream M, Ali MA, et al. Investigation of molluscum contagiosum virus, orf and other parapoxviruses in lymphomatoid papulosis. J Am Acad Dermatol. 2013;68:1046-1047.
- Nakamura-Wakatsuki T, Kato Y, Miura T, et al. Eruptive molluscum contagiosums in a patient with rheumatoid arthritis and lung cancer. Rheumatol Int. 2011;31:1117-1118.
- Ozyürek E, Sentürk N, Kefeli M, et al. Ulcerating molluscum contagiosum in a boy with relapsed acute lymphoblastic leukemia. J Pediatr Hematol Oncol. 2011;33:E114-E116.
- Hughes WT, Parham DM. Molluscum contagiosum in children with cancer or acquired immunodeficiency syndrome. Pediatr Infect Dis J. 1991;10:152-156.
- Euvrard S, Kanitakis J, Cochat P, et al. Skin diseases in children with organ transplants. J Am Acad Dermatol. 2001;44:932-939.
- Gardner LS, Ormond PJ. Treatment of multiple giant molluscum contagiosum in a renal transplant patient with imiquimod 5% cream. Clin Exp Dermatol. 2006;31:452-453.
- Mansur AT, Göktay F, Gündüz S, et al. Multiple giant molluscum contagiosum in a renal transplant recipient. Transpl Infect Dis. 2004;6:120-123.
- Feldmeyer L, Kamarashev J, Boehler A, et al. Molluscum contagiosum folliculitis mimicking tinea barbae in a lung transplant recipient. J Am Acad Dermatol. 2010;63:169-171.
- Tas¸kapan O, Yenicesu M, Aksu A. A giant solitary molluscum contagiosum, resembling nodular basal cell carcinoma, in a renal transplant recipient. Acta Derm Venereol. 1996;76:247-248.
- Tan HH, Goh CL. Viral infections affecting the skin in organ transplant recipients: epidemiology and current management strategies. Am J Clin Dermatol. 2006;7:13-29.
- Al-Mutairi N, Al-Doukhi A, Al-Farag S, et al. Comparative study on the efficacy, safety, and acceptability of imiquimod 5% cream versus cryotherapy for molluscum contagiosum in children. Pediatr Dermatol. 2010;27:388-394.
- Lim KS, Foo CC. Disseminated molluscum contagiosum in a patient with chronic plaque psoriasis taking methotrexate. Clin Exp Dermatol. 2007;32:591-593.
- Fotiadou C, Lazaridou E, Lekkas D, et al. Disseminated, eruptive molluscum contagiosum lesions in a psoriasis patient under treatment with methotrexate and cyclosporine. Eur J Dermatol. 2012;22:147-148.
- Antoniou C, Kosmadaki MG, Stratigos AJ, et al. Genital HPV lesions and molluscum contagiosum occurring in patients receiving anti-TNF-alpha therapy. Dermatology. 2008;216:364-365.
- Cursiefen C, Grunke M, Dechant C, et al. Multiple bilateral eyelid molluscum contagiosum lesions associated with TNFalpha-antibody and methotrexate therapy. Am J Ophthalmol. 2002;134:270-271.
- Heng YK, Lee JS, Neoh CY. Verrucous plaques in a pemphigus vulgaris patient on immunosuppressive therapy. Int J Dermatol. 2012;51:1044-1046.
- Goksugur N, Ozbostanci B, Goksugur SB. Molluscum contagiosum infection associated with pimecrolimus use in pityriasis alba. Pediatr Dermatol. 2007;24:E63-E65.
- Fernandes S, Pinto GM, Cardoso J. Particular clinical presentations of psoriasis in HIV patients. Int J STD AIDS. 2011;22:653-654.
- Dohil MA, Lin P, Lee J, et al. The epidemiology of molluscum contagiosum in children. J Am Acad Dermatol. 2006;54:47-54.
- Reichert CM, O’Leary TJ, Levens DL, et al. Autopsy pathology in the acquired immune deficiency syndrome. Am J Pathol. 1983;112:357-382.
- Czelusta A, Yen-Moore A, Van der Straten M, et al. An overview of sexually transmitted diseases. Part III. Sexually transmitted diseases in HIV-infected patients. J Am Acad Dermatol. 2000;43:409-432.
- Husak R, Garbe C, Orfanos CE. Mollusca contagiosa in HIV infection. Clinical manifestation, relation to immune status and prognostic value in 39 patients [in German]. Hautarzt. 1997;48:103-109.
- Averbuch D, Jaouni T, Pe’er J, et al. Confluent molluscum contagiosum covering the eyelids of an HIV-positive child. Clin Exp Ophthalmol. 2009;37:525-527.
- Erickson C, Driscoll M, Gaspari A. Efficacy of intravenous cidofovir in the treatment of giant molluscum contagiosum in a patient with human immunodeficiency virus. Arch Dermatol. 2011;147:652-654.
- Mastrolorenzo A, Urbano FG, Salimbeni L, et al. Atypical molluscum contagiosum infection in an HIV-infected patient. Int J Dermatol. 1998;37:378-380.
- Itin PH, Gilli L. Molluscum contagiosum mimicking sebaceous nevus of Jadassohn, ecthyma and giant condylomata acuminata in HIV-infected patients. Dermatology. 1994;189:396-398.
- Sim JH, Lee ES. Molluscum contagiosum presenting as a cutaneous horn. Ann Dermatol. 2011;23:262-263.
- Manchanda Y, Sethuraman G, Paderwani PP, et al. Molluscum contagiosum presenting as penile horn in an HIV positive patient. Sex Transm Infect. 2005;81:183-184.
- Miller SJ. Cutaneous cryptococcus resembling molluscum contagiosum in a patient with acquired immunodeficiency syndrome. Cutis. 1988;41:411-412.
- Sornum A. A mistaken diagnosis of molluscum contagiosum in a HIV-positive patient in rural South Africa. BMJ Case Rep. 2012;14.
- Corti M, Villafañe MF, Palmieri O, et al. Rupioid histoplasmosis: first case reported in an AIDS patient in Argentina. Rev Inst Med Trop Sao Paulo. 2010;52:279-280.
- Saikia L, Nath R, Hazarika D, et al. Atypical cutaneous lesions of Penicillium marneffei infection as a manifestation of the immune reconstitution inflammatory syndrome after highly active antiretroviral therapy. Indian J Dermatol Venereol Leprol. 2010;76:45-48.
- de Souza JA. Molluscum or a mimic? Am J Med. 2006;119:927-929.
- Conant MA. Immunomodulatory therapy in the management of viral infections in patients with HIV infection. J Am Acad Dermatol. 2000;43:S27-S30.
- Gamble RG, Echols KF, Dellavalle RP. Imiquimod vs cryotherapy for molluscum contagiosum: a randomized controlled trial. Arch Dermatol. 2012;148:109-112.
- Brown CW Jr, O’Donoghue M, Moore J, et al. Recalcitrant molluscum contagiosum in an HIV-afflicted male treated successfully with topical imiquimod. Cutis. 2000;65:363-366.
- Strauss RM, Doyle EL, Mohsen AH, et al. Successful treatment of molluscum contagiosum with topical imiquimod in a severely immunocompromised HIV-positive patient. Int J STD AIDS. 2001;12:264-266.
- Theiler M, Kempf W, Kerl K, et al. Disseminated molluscum contagiosum in a HIV-positive child. improvement after therapy with 5% imiquimod. J Dermatol Case Rep. 2011;5:19-23.
- Watanabe T, Tamaki K. Cidofovir diphosphate inhibits molluscum contagiosum virus DNA polymerase activity. J Invest Dermatol. 2008;128:1327-1329.
- Calista D. Topical cidofovir for severe cutaneous human papillomavirus and molluscum contagiosum infections in patients with HIV/AIDS. a pilot study. J Eur Acad Dermatol Venereol. 2000;14:484-488.
- Toro JR, Sanchez S, Turiansky G, et al. Topical cidofovir for the treatment of dermatologic conditions: verruca, condyloma, intraepithelial neoplasia, herpes simplex and its potential use in smallpox. Dermatol Clin. 2003;21:301-309.
- Calista D, Boschini A, Landi G. Resolution of disseminated molluscum contagiosum with highly active anti-retroviral therapy (HAART) in patients with AIDS. Eur J Dermatol. 1999;9:211-213.
- Cattelan AM, Sasset L, Corti L, et al. A complete remission of recalcitrant molluscum contagiosum in an AIDS patient following highly active antiretroviral therapy (HAART). J Infect. 1999;38:58-60.
- Sen S, Bhaumik P. Resolution of giant molluscum contagiosum with antiretroviral therapy. Indian J Dermatol Venereol Leprol. 2008;74:267-268.
- Sen S, Goswami BK, Karjyi N, et al. Disfiguring molluscum contagiosum in a HIV-positive patient responding to antiretroviral therapy. Indian J Dermatol. 2009;54:180-182.
- Pereira B, Fernandes C, Nachiambo E, et al. Exuberant molluscum contagiosum as a manifestation of the immune reconstitution inflammatory syndrome. Dermatol Online J. 2007;13:6.
- Osei-Sekyere B, Karstaedt AS. Immune reconstitution inflammatory syndrome involving the skin. Clin Exp Dermatol. 2010;35:477-481.
- Sung KU, Lee HE, Choi WR, et al. Molluscum contagiosum as a skin manifestation of immune reconstitution inflammatory syndrome in an AIDS patient who is receiving HAART. Korean J Fam Med. 2012;33:182-185.
- Ratnam I, Chiu C, Kandala NB, et al. Incidence and risk factors for immune reconstitution inflammatory syndrome in an ethnically diverse HIV type 1-infected cohort. Clin Infect Dis. 2006;42:418-427.
- Chen KW, Yang CF, Huang CT, et al. Molluscum contagiosum in a patient with adult T-cell leukaemia/lymphoma. Br J Haematol. 2011;155:286.
- Fernandez KH, Bream M, Ali MA, et al. Investigation of molluscum contagiosum virus, orf and other parapoxviruses in lymphomatoid papulosis. J Am Acad Dermatol. 2013;68:1046-1047.
- Nakamura-Wakatsuki T, Kato Y, Miura T, et al. Eruptive molluscum contagiosums in a patient with rheumatoid arthritis and lung cancer. Rheumatol Int. 2011;31:1117-1118.
- Ozyürek E, Sentürk N, Kefeli M, et al. Ulcerating molluscum contagiosum in a boy with relapsed acute lymphoblastic leukemia. J Pediatr Hematol Oncol. 2011;33:E114-E116.
- Hughes WT, Parham DM. Molluscum contagiosum in children with cancer or acquired immunodeficiency syndrome. Pediatr Infect Dis J. 1991;10:152-156.
- Euvrard S, Kanitakis J, Cochat P, et al. Skin diseases in children with organ transplants. J Am Acad Dermatol. 2001;44:932-939.
- Gardner LS, Ormond PJ. Treatment of multiple giant molluscum contagiosum in a renal transplant patient with imiquimod 5% cream. Clin Exp Dermatol. 2006;31:452-453.
- Mansur AT, Göktay F, Gündüz S, et al. Multiple giant molluscum contagiosum in a renal transplant recipient. Transpl Infect Dis. 2004;6:120-123.
- Feldmeyer L, Kamarashev J, Boehler A, et al. Molluscum contagiosum folliculitis mimicking tinea barbae in a lung transplant recipient. J Am Acad Dermatol. 2010;63:169-171.
- Tas¸kapan O, Yenicesu M, Aksu A. A giant solitary molluscum contagiosum, resembling nodular basal cell carcinoma, in a renal transplant recipient. Acta Derm Venereol. 1996;76:247-248.
- Tan HH, Goh CL. Viral infections affecting the skin in organ transplant recipients: epidemiology and current management strategies. Am J Clin Dermatol. 2006;7:13-29.
- Al-Mutairi N, Al-Doukhi A, Al-Farag S, et al. Comparative study on the efficacy, safety, and acceptability of imiquimod 5% cream versus cryotherapy for molluscum contagiosum in children. Pediatr Dermatol. 2010;27:388-394.
- Lim KS, Foo CC. Disseminated molluscum contagiosum in a patient with chronic plaque psoriasis taking methotrexate. Clin Exp Dermatol. 2007;32:591-593.
- Fotiadou C, Lazaridou E, Lekkas D, et al. Disseminated, eruptive molluscum contagiosum lesions in a psoriasis patient under treatment with methotrexate and cyclosporine. Eur J Dermatol. 2012;22:147-148.
- Antoniou C, Kosmadaki MG, Stratigos AJ, et al. Genital HPV lesions and molluscum contagiosum occurring in patients receiving anti-TNF-alpha therapy. Dermatology. 2008;216:364-365.
- Cursiefen C, Grunke M, Dechant C, et al. Multiple bilateral eyelid molluscum contagiosum lesions associated with TNFalpha-antibody and methotrexate therapy. Am J Ophthalmol. 2002;134:270-271.
- Heng YK, Lee JS, Neoh CY. Verrucous plaques in a pemphigus vulgaris patient on immunosuppressive therapy. Int J Dermatol. 2012;51:1044-1046.
- Goksugur N, Ozbostanci B, Goksugur SB. Molluscum contagiosum infection associated with pimecrolimus use in pityriasis alba. Pediatr Dermatol. 2007;24:E63-E65.
- Fernandes S, Pinto GM, Cardoso J. Particular clinical presentations of psoriasis in HIV patients. Int J STD AIDS. 2011;22:653-654.
Practice Points
- Molluscum contagiosum (MC) is highly prevalent and can have a wide range of atypical clinical presentations in patients with impaired cellular immunity (eg, human immunodeficiency virus [HIV]).
- Treatment of MC should include destructive procedures, if possible, as well as adjunctive agents such as topical retinoids, cantharidin, trichloroacetic acid, imiquimod, or cidofovir.
- Clinicians should consider screening patients with severe recalcitrant psoriasis for HIV to avoid poor outcomes from therapeutic agents.
Nail-Patella Syndrome: Clinical Clues for Making the Diagnosis
Nail-patella syndrome (NPS), also known as hereditary osteo-onychodysplasia syndrome, is a rare autosomal-dominant disorder with an estimated incidence of 1 per 50,000 individuals in the United States. Nail-patella syndrome presents due to a heterozygous loss-of-function mutation in the LIM homeobox transcription factor 1 beta gene, LMX1B, on chromosome 9q34.1 LMX1B gene mutations are fully penetrant, but there is variable expressivity, even within families.2
Case Report
A 69-year-old man presented to the dermatology clinic for a routine skin cancer screening. The patient’s history was remarkable for dystrophic fingernails and toenails since birth. In his 20s he developed progressively worsening instability of the left knee and chronic back pain due to scoliosis, lumbar lordosis, and spinal disc herniation. Since then, he underwent knee surgery and 7 back surgeries for rheumatologic disease. His medical history also was remarkable for osteoporosis, hypertension, and glaucoma. Family history was notable for similar findings in the patient’s sister; mother; and maternal aunt, uncle, and grandmother, all with varying disease severity.
Physical examination was remarkable for bilateral fingernail hypoplasia that was most prominent on the thumb, with improvement in each nail on progression toward the fifth digit (Figure 1A). Triangular fingernail lunulae, longitudinal ridging, and nail splitting were present (Figure 1A and 1B). Hypoplastic crumbly toenails also were appreciated (Figure 1C). Skin creases over the distal interphalangeal joints of the fingers and toes were conspicuously absent. Limited range of motion was noted in multiple joints, with profound limitation of bilateral elbow extension. Review of prior imaging reports revealed bilateral iliac horns as well as left patellar absence and right patellar hypoplasia (Figure 2). Urinalysis was remarkable for proteinuria and microscopic hematuria. Given the constellation of examination findings and positive family history, a diagnosis of NPS was made.
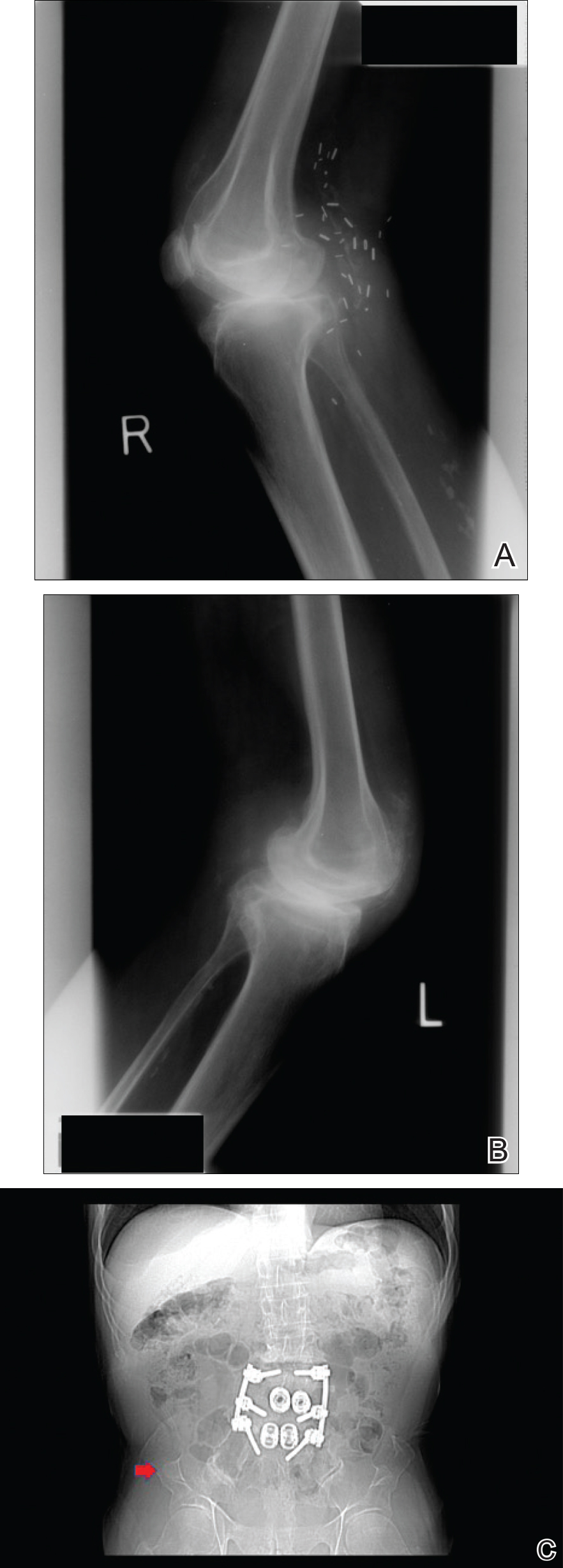
Comment
Nail-patella syndrome is characterized by variable dermatologic, neurologic, nephrogenic, ophthalmologic, and orthopedic clinical manifestations.3 Almost all patients with NPS have bilateral and symmetric nail changes, including absent or hypoplastic nails with ridging, splitting, or discoloration and triangular-shaped lunulae.1,4 Nail findings are the most consistent findings of NPS, as they are present in more than 98% of patients.5 The thumb often is the most severely affected nail, with improvement appreciated on progression toward the fifth digit, as seen in our patient (Figure 1A).5 Each individual nail usually is more severely affected on its ulnar side. When toenails are involved, the abnormalities tend to be less severe, and the little toenail is most commonly affected. Distal digital changes also are observed in almost all patients. Loss of dorsal creases in the skin overlying the distal interphalangeal joints can be considered as a diagnostic clue.3,4
There are a variety of orthopedic manifestations of NPS. Hypoplastic or absent patellae leading to recurrent subluxations or dislocations is a common finding.4 Bilateral symmetric bone formations (horns) arising from the iliac crest are pathognomonic but only found on radiography 70% of the time.6 Occasionally these protuberances can be palpated on physical examination,5 though this finding was not appreciated in our patient. Dysplasia of the elbows may result in limited elbow extension and limited pronation and supination. Early degenerative arthritis, lumbar lordosis, and scoliosis also are not uncommon. In addition, skeletal integrity is compromised, leading to early osteoporosis and increased risk for fractures.5
Nephropathy develops in approximately 30% to 40% of patients and is a major determinant of mortality in these patients.2 Mutations in the LMX1B gene lead to abnormal development of podocytes and reduction in collagen in the glomerular basement membrane. The first sign of renal involvement usually is proteinuria, with or without microscopic hematuria. As in our patient, many patients develop hypertension. Patients may progress to develop nephrotic syndrome and end-stage renal failure (5%–10%).7 Death from NPS-related nephropathy has occurred, even in childhood.4,5
Primary open-angle glaucoma has been recognized as a feature of NPS.8 It is the most frequent ocular abnormality observed, followed by ocular hypertension and Lester sign of the iris.3,5 These conditions also are more common in younger patients with NPS than in the general population.5 Important neurologic findings include epilepsy, peripheral neuropathy, attention deficit disorder, major depressive disorder, and vasomotor problems.9
Our case highlights the importance of recognizing this rare condition to provide a multidisciplinary approach to care that addresses all aspects of LMX1B-associated disease in affected individuals. Nail findings may be the first clue to the need for additional screenings in these patients. Nail-patella syndrome patients should undergo thorough ophthalmologic examinations every 2 years, including measurement of intraocular pressure, examination of the optic disc, and assessment of visual fields. Given the variability in severity of joint problems and the unpredictable anatomy of the joints, magnetic resonance imaging of the joints is recommended prior to orthopedic intervention. Most importantly, physicians should recognize this genodermatosis to implement periodic screenings for renal disease, as up to 40% of NPS patients develop kidney failure. Annual blood pressure measurements, urinalysis, and measurement of the protein to creatinine ratio in the urine are recommended. For patients with end-stage renal failure, renal transplantation results in cure of nephropathy and may even result in nail regrowth.10 Further, this case is notable in that it describes a patient with NPS who is older than most other individuals presenting with the condition, thereby revealing novel information about NPS in its more advanced stages.
- Harita Y, Kitanaka S, Isojima T, et al. Spectrum of LMX1B mutations: from nail-patella syndrome to isolated nephropathy [published online July 23, 2016]. Pediatr Nephrol. doi:10.1007/s00467-016-3462-x.
- Ghoumid J, Petit F, Holder-Espinasse M, et al. Nail-patella syndrome: clinical and molecular data in 55 families raising the hypothesis of a genetic heterogeneity [published online April 22, 2015]. Eur J Hum Genet. 2016;24:44-50.
- Tong SY, Luk HM, Tong TM, et al. The nail points to the diagnosis. Fong disease or hereditary osteo-onychodysplasia. Hong Kong Med J. 2015;21:573.e3-573.e5.
- Figueroa-Silva O, Vicente A, Agudo A, et al. Nail-patella syndrome: report of 11 pediatric cases. J Eur Acad Dermatol Venereol. 2016;30:1614-1617.
- Sweeney E, Fryer A, Mountford R, et al. Nail patella syndrome: a review of the phenotype aided by developmental biology. J Med Genet. 2003;40:153-162.
- Tigchelaar S, Lenting A, Bongers EM, et al. Nail patella syndrome: knee symptoms and surgical outcomes. a questionnaire-based survey [published online November 17, 2015]. Orthop Traumatol Surg Res. 2015;101:959-962.
- Lemley KV. Kidney disease in nail-patella syndrome [published online June 6, 2008]. Pediatr Nephrol. 2009;24:2345-2354.
- Sweeney E, Hoover-Fong JE, McIntosh I. Nail-patella syndrome. In: Adam MP, Ardinger HH, Pagon RA, et al, eds. GeneReviews. Seattle, WA: University of Washington, Seattle; 2003. https://www.ncbi.nlm.nih.gov/books/NBK1132/. Updated November 13, 2014. Accessed January 30, 2018.
- Lopez-Arvizu C, Sparrow EP, Strube MJ, et al. Increased symptoms of attention deficit hyperactivity disorder and major depressive disorder symptoms in nail-patella syndrome: potential association with LMX1B loss-of-function [published online November 2, 2010]. Am J Med Genet B Neuropsychiatr Genet. 2011;156B:59-66.
- Chan PC, Chan KW, Cheng IK, et al. Living-related renal transplantation in a patient with nail-patella syndrome. Nephron. 1988;50:164-166.
Nail-patella syndrome (NPS), also known as hereditary osteo-onychodysplasia syndrome, is a rare autosomal-dominant disorder with an estimated incidence of 1 per 50,000 individuals in the United States. Nail-patella syndrome presents due to a heterozygous loss-of-function mutation in the LIM homeobox transcription factor 1 beta gene, LMX1B, on chromosome 9q34.1 LMX1B gene mutations are fully penetrant, but there is variable expressivity, even within families.2
Case Report
A 69-year-old man presented to the dermatology clinic for a routine skin cancer screening. The patient’s history was remarkable for dystrophic fingernails and toenails since birth. In his 20s he developed progressively worsening instability of the left knee and chronic back pain due to scoliosis, lumbar lordosis, and spinal disc herniation. Since then, he underwent knee surgery and 7 back surgeries for rheumatologic disease. His medical history also was remarkable for osteoporosis, hypertension, and glaucoma. Family history was notable for similar findings in the patient’s sister; mother; and maternal aunt, uncle, and grandmother, all with varying disease severity.
Physical examination was remarkable for bilateral fingernail hypoplasia that was most prominent on the thumb, with improvement in each nail on progression toward the fifth digit (Figure 1A). Triangular fingernail lunulae, longitudinal ridging, and nail splitting were present (Figure 1A and 1B). Hypoplastic crumbly toenails also were appreciated (Figure 1C). Skin creases over the distal interphalangeal joints of the fingers and toes were conspicuously absent. Limited range of motion was noted in multiple joints, with profound limitation of bilateral elbow extension. Review of prior imaging reports revealed bilateral iliac horns as well as left patellar absence and right patellar hypoplasia (Figure 2). Urinalysis was remarkable for proteinuria and microscopic hematuria. Given the constellation of examination findings and positive family history, a diagnosis of NPS was made.
Comment
Nail-patella syndrome is characterized by variable dermatologic, neurologic, nephrogenic, ophthalmologic, and orthopedic clinical manifestations.3 Almost all patients with NPS have bilateral and symmetric nail changes, including absent or hypoplastic nails with ridging, splitting, or discoloration and triangular-shaped lunulae.1,4 Nail findings are the most consistent findings of NPS, as they are present in more than 98% of patients.5 The thumb often is the most severely affected nail, with improvement appreciated on progression toward the fifth digit, as seen in our patient (Figure 1A).5 Each individual nail usually is more severely affected on its ulnar side. When toenails are involved, the abnormalities tend to be less severe, and the little toenail is most commonly affected. Distal digital changes also are observed in almost all patients. Loss of dorsal creases in the skin overlying the distal interphalangeal joints can be considered as a diagnostic clue.3,4
There are a variety of orthopedic manifestations of NPS. Hypoplastic or absent patellae leading to recurrent subluxations or dislocations is a common finding.4 Bilateral symmetric bone formations (horns) arising from the iliac crest are pathognomonic but only found on radiography 70% of the time.6 Occasionally these protuberances can be palpated on physical examination,5 though this finding was not appreciated in our patient. Dysplasia of the elbows may result in limited elbow extension and limited pronation and supination. Early degenerative arthritis, lumbar lordosis, and scoliosis also are not uncommon. In addition, skeletal integrity is compromised, leading to early osteoporosis and increased risk for fractures.5
Nephropathy develops in approximately 30% to 40% of patients and is a major determinant of mortality in these patients.2 Mutations in the LMX1B gene lead to abnormal development of podocytes and reduction in collagen in the glomerular basement membrane. The first sign of renal involvement usually is proteinuria, with or without microscopic hematuria. As in our patient, many patients develop hypertension. Patients may progress to develop nephrotic syndrome and end-stage renal failure (5%–10%).7 Death from NPS-related nephropathy has occurred, even in childhood.4,5
Primary open-angle glaucoma has been recognized as a feature of NPS.8 It is the most frequent ocular abnormality observed, followed by ocular hypertension and Lester sign of the iris.3,5 These conditions also are more common in younger patients with NPS than in the general population.5 Important neurologic findings include epilepsy, peripheral neuropathy, attention deficit disorder, major depressive disorder, and vasomotor problems.9
Our case highlights the importance of recognizing this rare condition to provide a multidisciplinary approach to care that addresses all aspects of LMX1B-associated disease in affected individuals. Nail findings may be the first clue to the need for additional screenings in these patients. Nail-patella syndrome patients should undergo thorough ophthalmologic examinations every 2 years, including measurement of intraocular pressure, examination of the optic disc, and assessment of visual fields. Given the variability in severity of joint problems and the unpredictable anatomy of the joints, magnetic resonance imaging of the joints is recommended prior to orthopedic intervention. Most importantly, physicians should recognize this genodermatosis to implement periodic screenings for renal disease, as up to 40% of NPS patients develop kidney failure. Annual blood pressure measurements, urinalysis, and measurement of the protein to creatinine ratio in the urine are recommended. For patients with end-stage renal failure, renal transplantation results in cure of nephropathy and may even result in nail regrowth.10 Further, this case is notable in that it describes a patient with NPS who is older than most other individuals presenting with the condition, thereby revealing novel information about NPS in its more advanced stages.
Nail-patella syndrome (NPS), also known as hereditary osteo-onychodysplasia syndrome, is a rare autosomal-dominant disorder with an estimated incidence of 1 per 50,000 individuals in the United States. Nail-patella syndrome presents due to a heterozygous loss-of-function mutation in the LIM homeobox transcription factor 1 beta gene, LMX1B, on chromosome 9q34.1 LMX1B gene mutations are fully penetrant, but there is variable expressivity, even within families.2
Case Report
A 69-year-old man presented to the dermatology clinic for a routine skin cancer screening. The patient’s history was remarkable for dystrophic fingernails and toenails since birth. In his 20s he developed progressively worsening instability of the left knee and chronic back pain due to scoliosis, lumbar lordosis, and spinal disc herniation. Since then, he underwent knee surgery and 7 back surgeries for rheumatologic disease. His medical history also was remarkable for osteoporosis, hypertension, and glaucoma. Family history was notable for similar findings in the patient’s sister; mother; and maternal aunt, uncle, and grandmother, all with varying disease severity.
Physical examination was remarkable for bilateral fingernail hypoplasia that was most prominent on the thumb, with improvement in each nail on progression toward the fifth digit (Figure 1A). Triangular fingernail lunulae, longitudinal ridging, and nail splitting were present (Figure 1A and 1B). Hypoplastic crumbly toenails also were appreciated (Figure 1C). Skin creases over the distal interphalangeal joints of the fingers and toes were conspicuously absent. Limited range of motion was noted in multiple joints, with profound limitation of bilateral elbow extension. Review of prior imaging reports revealed bilateral iliac horns as well as left patellar absence and right patellar hypoplasia (Figure 2). Urinalysis was remarkable for proteinuria and microscopic hematuria. Given the constellation of examination findings and positive family history, a diagnosis of NPS was made.
Comment
Nail-patella syndrome is characterized by variable dermatologic, neurologic, nephrogenic, ophthalmologic, and orthopedic clinical manifestations.3 Almost all patients with NPS have bilateral and symmetric nail changes, including absent or hypoplastic nails with ridging, splitting, or discoloration and triangular-shaped lunulae.1,4 Nail findings are the most consistent findings of NPS, as they are present in more than 98% of patients.5 The thumb often is the most severely affected nail, with improvement appreciated on progression toward the fifth digit, as seen in our patient (Figure 1A).5 Each individual nail usually is more severely affected on its ulnar side. When toenails are involved, the abnormalities tend to be less severe, and the little toenail is most commonly affected. Distal digital changes also are observed in almost all patients. Loss of dorsal creases in the skin overlying the distal interphalangeal joints can be considered as a diagnostic clue.3,4
There are a variety of orthopedic manifestations of NPS. Hypoplastic or absent patellae leading to recurrent subluxations or dislocations is a common finding.4 Bilateral symmetric bone formations (horns) arising from the iliac crest are pathognomonic but only found on radiography 70% of the time.6 Occasionally these protuberances can be palpated on physical examination,5 though this finding was not appreciated in our patient. Dysplasia of the elbows may result in limited elbow extension and limited pronation and supination. Early degenerative arthritis, lumbar lordosis, and scoliosis also are not uncommon. In addition, skeletal integrity is compromised, leading to early osteoporosis and increased risk for fractures.5
Nephropathy develops in approximately 30% to 40% of patients and is a major determinant of mortality in these patients.2 Mutations in the LMX1B gene lead to abnormal development of podocytes and reduction in collagen in the glomerular basement membrane. The first sign of renal involvement usually is proteinuria, with or without microscopic hematuria. As in our patient, many patients develop hypertension. Patients may progress to develop nephrotic syndrome and end-stage renal failure (5%–10%).7 Death from NPS-related nephropathy has occurred, even in childhood.4,5
Primary open-angle glaucoma has been recognized as a feature of NPS.8 It is the most frequent ocular abnormality observed, followed by ocular hypertension and Lester sign of the iris.3,5 These conditions also are more common in younger patients with NPS than in the general population.5 Important neurologic findings include epilepsy, peripheral neuropathy, attention deficit disorder, major depressive disorder, and vasomotor problems.9
Our case highlights the importance of recognizing this rare condition to provide a multidisciplinary approach to care that addresses all aspects of LMX1B-associated disease in affected individuals. Nail findings may be the first clue to the need for additional screenings in these patients. Nail-patella syndrome patients should undergo thorough ophthalmologic examinations every 2 years, including measurement of intraocular pressure, examination of the optic disc, and assessment of visual fields. Given the variability in severity of joint problems and the unpredictable anatomy of the joints, magnetic resonance imaging of the joints is recommended prior to orthopedic intervention. Most importantly, physicians should recognize this genodermatosis to implement periodic screenings for renal disease, as up to 40% of NPS patients develop kidney failure. Annual blood pressure measurements, urinalysis, and measurement of the protein to creatinine ratio in the urine are recommended. For patients with end-stage renal failure, renal transplantation results in cure of nephropathy and may even result in nail regrowth.10 Further, this case is notable in that it describes a patient with NPS who is older than most other individuals presenting with the condition, thereby revealing novel information about NPS in its more advanced stages.
- Harita Y, Kitanaka S, Isojima T, et al. Spectrum of LMX1B mutations: from nail-patella syndrome to isolated nephropathy [published online July 23, 2016]. Pediatr Nephrol. doi:10.1007/s00467-016-3462-x.
- Ghoumid J, Petit F, Holder-Espinasse M, et al. Nail-patella syndrome: clinical and molecular data in 55 families raising the hypothesis of a genetic heterogeneity [published online April 22, 2015]. Eur J Hum Genet. 2016;24:44-50.
- Tong SY, Luk HM, Tong TM, et al. The nail points to the diagnosis. Fong disease or hereditary osteo-onychodysplasia. Hong Kong Med J. 2015;21:573.e3-573.e5.
- Figueroa-Silva O, Vicente A, Agudo A, et al. Nail-patella syndrome: report of 11 pediatric cases. J Eur Acad Dermatol Venereol. 2016;30:1614-1617.
- Sweeney E, Fryer A, Mountford R, et al. Nail patella syndrome: a review of the phenotype aided by developmental biology. J Med Genet. 2003;40:153-162.
- Tigchelaar S, Lenting A, Bongers EM, et al. Nail patella syndrome: knee symptoms and surgical outcomes. a questionnaire-based survey [published online November 17, 2015]. Orthop Traumatol Surg Res. 2015;101:959-962.
- Lemley KV. Kidney disease in nail-patella syndrome [published online June 6, 2008]. Pediatr Nephrol. 2009;24:2345-2354.
- Sweeney E, Hoover-Fong JE, McIntosh I. Nail-patella syndrome. In: Adam MP, Ardinger HH, Pagon RA, et al, eds. GeneReviews. Seattle, WA: University of Washington, Seattle; 2003. https://www.ncbi.nlm.nih.gov/books/NBK1132/. Updated November 13, 2014. Accessed January 30, 2018.
- Lopez-Arvizu C, Sparrow EP, Strube MJ, et al. Increased symptoms of attention deficit hyperactivity disorder and major depressive disorder symptoms in nail-patella syndrome: potential association with LMX1B loss-of-function [published online November 2, 2010]. Am J Med Genet B Neuropsychiatr Genet. 2011;156B:59-66.
- Chan PC, Chan KW, Cheng IK, et al. Living-related renal transplantation in a patient with nail-patella syndrome. Nephron. 1988;50:164-166.
- Harita Y, Kitanaka S, Isojima T, et al. Spectrum of LMX1B mutations: from nail-patella syndrome to isolated nephropathy [published online July 23, 2016]. Pediatr Nephrol. doi:10.1007/s00467-016-3462-x.
- Ghoumid J, Petit F, Holder-Espinasse M, et al. Nail-patella syndrome: clinical and molecular data in 55 families raising the hypothesis of a genetic heterogeneity [published online April 22, 2015]. Eur J Hum Genet. 2016;24:44-50.
- Tong SY, Luk HM, Tong TM, et al. The nail points to the diagnosis. Fong disease or hereditary osteo-onychodysplasia. Hong Kong Med J. 2015;21:573.e3-573.e5.
- Figueroa-Silva O, Vicente A, Agudo A, et al. Nail-patella syndrome: report of 11 pediatric cases. J Eur Acad Dermatol Venereol. 2016;30:1614-1617.
- Sweeney E, Fryer A, Mountford R, et al. Nail patella syndrome: a review of the phenotype aided by developmental biology. J Med Genet. 2003;40:153-162.
- Tigchelaar S, Lenting A, Bongers EM, et al. Nail patella syndrome: knee symptoms and surgical outcomes. a questionnaire-based survey [published online November 17, 2015]. Orthop Traumatol Surg Res. 2015;101:959-962.
- Lemley KV. Kidney disease in nail-patella syndrome [published online June 6, 2008]. Pediatr Nephrol. 2009;24:2345-2354.
- Sweeney E, Hoover-Fong JE, McIntosh I. Nail-patella syndrome. In: Adam MP, Ardinger HH, Pagon RA, et al, eds. GeneReviews. Seattle, WA: University of Washington, Seattle; 2003. https://www.ncbi.nlm.nih.gov/books/NBK1132/. Updated November 13, 2014. Accessed January 30, 2018.
- Lopez-Arvizu C, Sparrow EP, Strube MJ, et al. Increased symptoms of attention deficit hyperactivity disorder and major depressive disorder symptoms in nail-patella syndrome: potential association with LMX1B loss-of-function [published online November 2, 2010]. Am J Med Genet B Neuropsychiatr Genet. 2011;156B:59-66.
- Chan PC, Chan KW, Cheng IK, et al. Living-related renal transplantation in a patient with nail-patella syndrome. Nephron. 1988;50:164-166.
Practice Points
- Nail-patella syndrome (NPS) is a multisystem disease.
- Nail findings (eg, triangular lunulae) may be the first clue to NPS and should prompt investigation of associated renal, ocular, neurologic, skeletal, and orthopedic abnormalities.
- Early intervention and a multidisciplinary approach to care can improve morbidity and mortality in patients with NPS.
Melkersson-Rosenthal Syndrome Successfully Treated With Adalimumab
Melkersson-Rosenthal syndrome (MRS) is a rare condition comprised of unilateral peripheral facial nerve palsy, episodic or progressive facial edema, and lingua plicata (also known as fissured tongue). Melkersson-Rosenthal syndrome is a subtype of orofacial granulomatosis and often is mistaken for angioedema or pseudoangioedema due to the swelling of the lips and eyelids. We present a case of MRS that cleared in response to adalimumab therapy.
Case Report
A 69-year-old woman presented to our dermatology clinic with facial edema and a fissured tongue of 4 years’ duration. These symptoms had failed to improve with doxycycline, tacrolimus ointment 0.1%, and cortisone injections of the upper lip, as well as a balsam-free diet, fragrance-free skin products, and flavor-free toothpaste prescribed by multiple physicians over 4 years. Two weeks prior to the current presentation the patient developed left facial nerve palsy that was diagnosed by an outside physician as Bell palsy, and the patient completed a 7-day course of prednisone 1 day prior to presentation. The patient’s medical history was remarkable for type 2 diabetes mellitus controlled with metformin, hyperlipidemia controlled with ezetimibe-simvastatin, and psoriasis. She reported no family history of autoimmune or dermatologic disorders and denied any fever, unintentional weight loss, nausea, vomiting, or diarrhea.
On physical examination, the patient had considerable perioral edema and erythema without warmth or tenderness (Figure 1A). The tongue was fissured with notable scalloping at the lateral margins (Figure 1B). There were no aphthous ulcers or lymphadenopathy, and the remainder of the neurologic examination was normal. The patient had erythematous plaques with scaling on the bilateral elbows. Cardiopulmonary, musculoskeletal, and abdominal examinations were otherwise normal.
Laboratory data revealed an elevated white blood cell count of 17,500/µL (reference range, 4500–11,000/µL), an elevated absolute neutrophil count of 14,018/µL (reference range, 0–700/µL), and an absolute eosinophil count of 0/µL (reference range, 0–450/µL), with the rest of the complete blood cell count within reference range. A basic metabolic panel showed an elevated glucose level of 326 mg/dL (reference range, 70–110 mg/dL), consistent with diabetes and most likely exacerbated by the recent steroid course. A lipid panel was consistent with diagnosed hyperlipidemia (total cholesterol, 236 mg/dL [reference range, <200 mg/dL]; low-density lipoprotein, 134 mg/dL [reference range, 10–30 mg/dL]; triglycerides, 188 mg/dL [reference range, <160 mg/dL]). Hepatitis B and C tests were negative. A punch biopsy of the buccal and labial mucosa was taken, revealing a parakeratinized stratified squamous epithelium with an unusual pattern of surface keratinization with foci of intracellular and extracellular edema in the spinous layer. The underlying fibrous connective tissue was edematous with infiltrates of lymphocytes, mast cells, macrophages, and a few plasma cells. The pathology report listed the diagnosis as nonspecific “chronic mucositis,” with a list of differential diagnoses that included angioedema, hypersensitivity reaction, or other possible autoimmune disorders.
On consideration of these differential diagnoses, it was felt most likely to be MRS, which remains a primarily clinical diagnosis characterized by the triad of symptoms seen in this patient. Treatment of this condition emphasizes inflammation, and steroid therapy often is utilized, as it was in our patient. After the diagnosis of MRS was made, the patient received adalimumab 80 mg subcutaneously on day 1 and 40 mg on day 8 as a loading dose; she subsequently began a course of subcutaneous injections of adalimumab 40 mg once every other week for treatment of psoriasis with the goal of simultaneously treating the MRS. The symptoms did not completely resolve at this dose, so it was increased to 40 mg once weekly. The patient reported that the facial edema, lingua plicata, and facial nerve palsy resolved concomitantly over approximately 3 months with greater improvement at 5 months (Figure 2). The patient has had no relapses as of the last follow-up at 11 months.
Comment
Melkersson-Rosenthal syndrome usually presents sporadically, though there are reports of familial association,1-3 and only 8% to 25% of patients worldwide present with the complete triad of symptoms.4 The pathogenesis of the syndrome is controversial. Granulomatous changes have been found in patients experiencing chronic edema. However, according to Zimmer et al5 in a study of 42 MRS patients, only 46% (19/42) had granulomatous changes; 36% (15/42) had nonspecific inflammation, 11% (5/42) had incidental findings, and 7% (3/42) showed no histopathologic abnormalities. Granulomatous cheilitis is a subtype of orofacial granulomatosis, an idiopathic process that causes swelling of the face and lips as well as intraoral swelling and ulceration. Orofacial granulomatosis is referred to as granulomatous cheilitis when the lip is involved. Melkersson-Rosenthal syndrome is another subtype of orofacial granulomatosis that includes facial palsy and fissured tongue.6,7
In a clinical study of 7 patients with MRS, Liu and Yu1 found 3 (42%) patients to have dysarthria, dysphagia, and tongue muscle atrophy; 1 patient to have migrainelike headaches; 1 patient to have decreased vision and an ocular movement disorder; 1 patient to have ipsilateral hearing loss; and 1 patient to lack any other symptoms. Halevy et al8 suggested a possible association of MRS with psoriasis. In their review of 12 patients, 1 (8%) had psoriatic arthritis, 2 (17%) had skin biopsy–proven psoriasis, and 3 (25%) had a family history of psoriasis.8 Because the disease is quite rare, it is difficult to determine other symptoms that may be associated with the disease.
Tumor necrosis factor α (TNF-α) is needed for granuloma formation, and TNF-α antagonists have been used to treat a number of granulomatous conditions including Crohn disease and sarcoidosis.9-11 Two case reports indicate that infliximab, a mouse/human chimeric monoclonal antibody to TNF-α, has been used successfully to clear MRS.12,13 One report cited the use of adalimumab for maintenance therapy of MRS,12 and more recently, adalimumab has been reported for refractory MRS.14 However, there currently are no known reports regarding the efficacy of adalimumab as a first-line treatment of MRS.
Adalimumab is a fully human monoclonal antibody to TNF-α, which is administered via subcutaneous injections. Infliximab must be administered at an infusion center, making treatment logistically more difficult for patients, and can be associated with the development of infusion reactions, though the exact data on infusion reactions are difficult to estimate due to variations in reporting.15,16
In 2014, Stein et al
Conclusion
We present a case of a 69-year-old woman who presented with facial nerve palsy, facial edema, and a fissured tongue, which is the classic triad of MRS, and all 3 symptoms improved with adalimumab.
- Liu R, Yu S. Melk
ersson-Rosenthal syndrome: a review of seven patients [published online May 7, 2013]. J Clin Neurosci. 2013;20:993-995. - Sun B, Zhou C, Han Z. Facial palsy in Melkersson-Rosenthal syndrome and Bell’s palsy: familial history and recurrence tendency [published online August 13, 2014]. Ann Otol Rhinol Laryngol. 2015;124:107-109.
- Meisel-Stosiek M, Hornstein OP, Stosiek N. Family study on Melkersson-Rosenthal syndrome. some hereditary aspects of the disease and review of literature. Acta Derm Venereol. 1990;70:221-226.
- Sciubba JJ, Said-Al-Naief N. Orofacial granulomatosis: presentation, pathology and management of 13 cases. J Oral Pathol Med. 2003;32:576-585.
- Zimmer WM, Rogers
RS 3rd, Reeve CM, et al. Orofacial manifestations of Melkersson-Rosenthal syndrome. a study of 42 patients and review of 220 cases from the literature. Oral Surg Oral Med Oral Pathol. 1992;74:610-619. - Critchlow WA, Cha
ng D. Cheilitis granulomatosa: a review [published online September 22, 2013]. Head Neck Pathol. 2014;8:209-213. - Allen CM, Camisa
C. Oral disease. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Philadelphia, PA: Elsevier; 2012:1157-1160. - Halevy S, Shalom
G, Trattner A, et al. Melkersson-Rosenthal syndrome: a possible association with psoriasis. J Am Acad Dermatol. 2012;67:795-796. - Algood HM, Lin P
L, Flynn JL. Tumor necrosis factor and chemokine interactions in the formation and maintenance of granulomas in tuberculosis. Clin Infect Dis. 2005;41(suppl 3):S189-S193. - Yee AM, Pochapin
MB. Treatment of complicated sarcoidosis with infliximab anti-tumor necrosis factor-alpha therapy. Ann Intern Med. 2001;135:27-31. - Targan SR, Hanau
er SB, van Deventer SJ, et al. A short-term study of chimeric monoclonal antibody cA2 to tumor necrosis factor alpha for Crohn’s disease. Crohn’s Disease cA2 Study Group. N Engl J Med. 1997;337:1029-1035. - Kakimoto C, Spar
ks C, White AA. Melkersson-Rosenthal syndrome: a form of pseudoangioedema. Ann Allergy Asthma Immunol. 2007;99:185-189. - Wickramasinghe N
, Gunasekara CN, Fernando WS, et al. Vulvitis granulomatosa, Melkersson-Rosenthal syndrome, and Crohn’s disease: dramatic response to infliximab therapy. Int J Dermatol. 2012;51:966-968. - Stein J, Paulke
A, Schacher B, et al. An extraordinary form of the Melkersson-Rosenthal syndrome successfully treated with the tumour necrosis factor-α blocker adalimumab [published online May 14, 2014]. BMJ Case Rep. doi:10.1136/bcr-2014-204674. - Cheifetz A, Smedley M, Martin S, et al. The incidence and management of infusion reactions to infliximab: a large center experience. Am J Gastroenterol. 2003;98:1315-1324.
- Choquette D, Faraawi R, Chow A, et al. Incidence and management of infusion reactions to infliximab in a prospective real-world community registry. J Rheumatol. 2015;42:1105-1111.
- Ruiz Villaverde R, Sánchez Cano D. Successful treatment of granulomatous cheilitis with adalimumab. Int J Dermatol. 2012;51:118-120.
Melkersson-Rosenthal syndrome (MRS) is a rare condition comprised of unilateral peripheral facial nerve palsy, episodic or progressive facial edema, and lingua plicata (also known as fissured tongue). Melkersson-Rosenthal syndrome is a subtype of orofacial granulomatosis and often is mistaken for angioedema or pseudoangioedema due to the swelling of the lips and eyelids. We present a case of MRS that cleared in response to adalimumab therapy.
Case Report
A 69-year-old woman presented to our dermatology clinic with facial edema and a fissured tongue of 4 years’ duration. These symptoms had failed to improve with doxycycline, tacrolimus ointment 0.1%, and cortisone injections of the upper lip, as well as a balsam-free diet, fragrance-free skin products, and flavor-free toothpaste prescribed by multiple physicians over 4 years. Two weeks prior to the current presentation the patient developed left facial nerve palsy that was diagnosed by an outside physician as Bell palsy, and the patient completed a 7-day course of prednisone 1 day prior to presentation. The patient’s medical history was remarkable for type 2 diabetes mellitus controlled with metformin, hyperlipidemia controlled with ezetimibe-simvastatin, and psoriasis. She reported no family history of autoimmune or dermatologic disorders and denied any fever, unintentional weight loss, nausea, vomiting, or diarrhea.
On physical examination, the patient had considerable perioral edema and erythema without warmth or tenderness (Figure 1A). The tongue was fissured with notable scalloping at the lateral margins (Figure 1B). There were no aphthous ulcers or lymphadenopathy, and the remainder of the neurologic examination was normal. The patient had erythematous plaques with scaling on the bilateral elbows. Cardiopulmonary, musculoskeletal, and abdominal examinations were otherwise normal.
Laboratory data revealed an elevated white blood cell count of 17,500/µL (reference range, 4500–11,000/µL), an elevated absolute neutrophil count of 14,018/µL (reference range, 0–700/µL), and an absolute eosinophil count of 0/µL (reference range, 0–450/µL), with the rest of the complete blood cell count within reference range. A basic metabolic panel showed an elevated glucose level of 326 mg/dL (reference range, 70–110 mg/dL), consistent with diabetes and most likely exacerbated by the recent steroid course. A lipid panel was consistent with diagnosed hyperlipidemia (total cholesterol, 236 mg/dL [reference range, <200 mg/dL]; low-density lipoprotein, 134 mg/dL [reference range, 10–30 mg/dL]; triglycerides, 188 mg/dL [reference range, <160 mg/dL]). Hepatitis B and C tests were negative. A punch biopsy of the buccal and labial mucosa was taken, revealing a parakeratinized stratified squamous epithelium with an unusual pattern of surface keratinization with foci of intracellular and extracellular edema in the spinous layer. The underlying fibrous connective tissue was edematous with infiltrates of lymphocytes, mast cells, macrophages, and a few plasma cells. The pathology report listed the diagnosis as nonspecific “chronic mucositis,” with a list of differential diagnoses that included angioedema, hypersensitivity reaction, or other possible autoimmune disorders.
On consideration of these differential diagnoses, it was felt most likely to be MRS, which remains a primarily clinical diagnosis characterized by the triad of symptoms seen in this patient. Treatment of this condition emphasizes inflammation, and steroid therapy often is utilized, as it was in our patient. After the diagnosis of MRS was made, the patient received adalimumab 80 mg subcutaneously on day 1 and 40 mg on day 8 as a loading dose; she subsequently began a course of subcutaneous injections of adalimumab 40 mg once every other week for treatment of psoriasis with the goal of simultaneously treating the MRS. The symptoms did not completely resolve at this dose, so it was increased to 40 mg once weekly. The patient reported that the facial edema, lingua plicata, and facial nerve palsy resolved concomitantly over approximately 3 months with greater improvement at 5 months (Figure 2). The patient has had no relapses as of the last follow-up at 11 months.
Comment
Melkersson-Rosenthal syndrome usually presents sporadically, though there are reports of familial association,1-3 and only 8% to 25% of patients worldwide present with the complete triad of symptoms.4 The pathogenesis of the syndrome is controversial. Granulomatous changes have been found in patients experiencing chronic edema. However, according to Zimmer et al5 in a study of 42 MRS patients, only 46% (19/42) had granulomatous changes; 36% (15/42) had nonspecific inflammation, 11% (5/42) had incidental findings, and 7% (3/42) showed no histopathologic abnormalities. Granulomatous cheilitis is a subtype of orofacial granulomatosis, an idiopathic process that causes swelling of the face and lips as well as intraoral swelling and ulceration. Orofacial granulomatosis is referred to as granulomatous cheilitis when the lip is involved. Melkersson-Rosenthal syndrome is another subtype of orofacial granulomatosis that includes facial palsy and fissured tongue.6,7
In a clinical study of 7 patients with MRS, Liu and Yu1 found 3 (42%) patients to have dysarthria, dysphagia, and tongue muscle atrophy; 1 patient to have migrainelike headaches; 1 patient to have decreased vision and an ocular movement disorder; 1 patient to have ipsilateral hearing loss; and 1 patient to lack any other symptoms. Halevy et al8 suggested a possible association of MRS with psoriasis. In their review of 12 patients, 1 (8%) had psoriatic arthritis, 2 (17%) had skin biopsy–proven psoriasis, and 3 (25%) had a family history of psoriasis.8 Because the disease is quite rare, it is difficult to determine other symptoms that may be associated with the disease.
Tumor necrosis factor α (TNF-α) is needed for granuloma formation, and TNF-α antagonists have been used to treat a number of granulomatous conditions including Crohn disease and sarcoidosis.9-11 Two case reports indicate that infliximab, a mouse/human chimeric monoclonal antibody to TNF-α, has been used successfully to clear MRS.12,13 One report cited the use of adalimumab for maintenance therapy of MRS,12 and more recently, adalimumab has been reported for refractory MRS.14 However, there currently are no known reports regarding the efficacy of adalimumab as a first-line treatment of MRS.
Adalimumab is a fully human monoclonal antibody to TNF-α, which is administered via subcutaneous injections. Infliximab must be administered at an infusion center, making treatment logistically more difficult for patients, and can be associated with the development of infusion reactions, though the exact data on infusion reactions are difficult to estimate due to variations in reporting.15,16
In 2014, Stein et al
Conclusion
We present a case of a 69-year-old woman who presented with facial nerve palsy, facial edema, and a fissured tongue, which is the classic triad of MRS, and all 3 symptoms improved with adalimumab.
Melkersson-Rosenthal syndrome (MRS) is a rare condition comprised of unilateral peripheral facial nerve palsy, episodic or progressive facial edema, and lingua plicata (also known as fissured tongue). Melkersson-Rosenthal syndrome is a subtype of orofacial granulomatosis and often is mistaken for angioedema or pseudoangioedema due to the swelling of the lips and eyelids. We present a case of MRS that cleared in response to adalimumab therapy.
Case Report
A 69-year-old woman presented to our dermatology clinic with facial edema and a fissured tongue of 4 years’ duration. These symptoms had failed to improve with doxycycline, tacrolimus ointment 0.1%, and cortisone injections of the upper lip, as well as a balsam-free diet, fragrance-free skin products, and flavor-free toothpaste prescribed by multiple physicians over 4 years. Two weeks prior to the current presentation the patient developed left facial nerve palsy that was diagnosed by an outside physician as Bell palsy, and the patient completed a 7-day course of prednisone 1 day prior to presentation. The patient’s medical history was remarkable for type 2 diabetes mellitus controlled with metformin, hyperlipidemia controlled with ezetimibe-simvastatin, and psoriasis. She reported no family history of autoimmune or dermatologic disorders and denied any fever, unintentional weight loss, nausea, vomiting, or diarrhea.
On physical examination, the patient had considerable perioral edema and erythema without warmth or tenderness (Figure 1A). The tongue was fissured with notable scalloping at the lateral margins (Figure 1B). There were no aphthous ulcers or lymphadenopathy, and the remainder of the neurologic examination was normal. The patient had erythematous plaques with scaling on the bilateral elbows. Cardiopulmonary, musculoskeletal, and abdominal examinations were otherwise normal.
Laboratory data revealed an elevated white blood cell count of 17,500/µL (reference range, 4500–11,000/µL), an elevated absolute neutrophil count of 14,018/µL (reference range, 0–700/µL), and an absolute eosinophil count of 0/µL (reference range, 0–450/µL), with the rest of the complete blood cell count within reference range. A basic metabolic panel showed an elevated glucose level of 326 mg/dL (reference range, 70–110 mg/dL), consistent with diabetes and most likely exacerbated by the recent steroid course. A lipid panel was consistent with diagnosed hyperlipidemia (total cholesterol, 236 mg/dL [reference range, <200 mg/dL]; low-density lipoprotein, 134 mg/dL [reference range, 10–30 mg/dL]; triglycerides, 188 mg/dL [reference range, <160 mg/dL]). Hepatitis B and C tests were negative. A punch biopsy of the buccal and labial mucosa was taken, revealing a parakeratinized stratified squamous epithelium with an unusual pattern of surface keratinization with foci of intracellular and extracellular edema in the spinous layer. The underlying fibrous connective tissue was edematous with infiltrates of lymphocytes, mast cells, macrophages, and a few plasma cells. The pathology report listed the diagnosis as nonspecific “chronic mucositis,” with a list of differential diagnoses that included angioedema, hypersensitivity reaction, or other possible autoimmune disorders.
On consideration of these differential diagnoses, it was felt most likely to be MRS, which remains a primarily clinical diagnosis characterized by the triad of symptoms seen in this patient. Treatment of this condition emphasizes inflammation, and steroid therapy often is utilized, as it was in our patient. After the diagnosis of MRS was made, the patient received adalimumab 80 mg subcutaneously on day 1 and 40 mg on day 8 as a loading dose; she subsequently began a course of subcutaneous injections of adalimumab 40 mg once every other week for treatment of psoriasis with the goal of simultaneously treating the MRS. The symptoms did not completely resolve at this dose, so it was increased to 40 mg once weekly. The patient reported that the facial edema, lingua plicata, and facial nerve palsy resolved concomitantly over approximately 3 months with greater improvement at 5 months (Figure 2). The patient has had no relapses as of the last follow-up at 11 months.
Comment
Melkersson-Rosenthal syndrome usually presents sporadically, though there are reports of familial association,1-3 and only 8% to 25% of patients worldwide present with the complete triad of symptoms.4 The pathogenesis of the syndrome is controversial. Granulomatous changes have been found in patients experiencing chronic edema. However, according to Zimmer et al5 in a study of 42 MRS patients, only 46% (19/42) had granulomatous changes; 36% (15/42) had nonspecific inflammation, 11% (5/42) had incidental findings, and 7% (3/42) showed no histopathologic abnormalities. Granulomatous cheilitis is a subtype of orofacial granulomatosis, an idiopathic process that causes swelling of the face and lips as well as intraoral swelling and ulceration. Orofacial granulomatosis is referred to as granulomatous cheilitis when the lip is involved. Melkersson-Rosenthal syndrome is another subtype of orofacial granulomatosis that includes facial palsy and fissured tongue.6,7
In a clinical study of 7 patients with MRS, Liu and Yu1 found 3 (42%) patients to have dysarthria, dysphagia, and tongue muscle atrophy; 1 patient to have migrainelike headaches; 1 patient to have decreased vision and an ocular movement disorder; 1 patient to have ipsilateral hearing loss; and 1 patient to lack any other symptoms. Halevy et al8 suggested a possible association of MRS with psoriasis. In their review of 12 patients, 1 (8%) had psoriatic arthritis, 2 (17%) had skin biopsy–proven psoriasis, and 3 (25%) had a family history of psoriasis.8 Because the disease is quite rare, it is difficult to determine other symptoms that may be associated with the disease.
Tumor necrosis factor α (TNF-α) is needed for granuloma formation, and TNF-α antagonists have been used to treat a number of granulomatous conditions including Crohn disease and sarcoidosis.9-11 Two case reports indicate that infliximab, a mouse/human chimeric monoclonal antibody to TNF-α, has been used successfully to clear MRS.12,13 One report cited the use of adalimumab for maintenance therapy of MRS,12 and more recently, adalimumab has been reported for refractory MRS.14 However, there currently are no known reports regarding the efficacy of adalimumab as a first-line treatment of MRS.
Adalimumab is a fully human monoclonal antibody to TNF-α, which is administered via subcutaneous injections. Infliximab must be administered at an infusion center, making treatment logistically more difficult for patients, and can be associated with the development of infusion reactions, though the exact data on infusion reactions are difficult to estimate due to variations in reporting.15,16
In 2014, Stein et al
Conclusion
We present a case of a 69-year-old woman who presented with facial nerve palsy, facial edema, and a fissured tongue, which is the classic triad of MRS, and all 3 symptoms improved with adalimumab.
- Liu R, Yu S. Melk
ersson-Rosenthal syndrome: a review of seven patients [published online May 7, 2013]. J Clin Neurosci. 2013;20:993-995. - Sun B, Zhou C, Han Z. Facial palsy in Melkersson-Rosenthal syndrome and Bell’s palsy: familial history and recurrence tendency [published online August 13, 2014]. Ann Otol Rhinol Laryngol. 2015;124:107-109.
- Meisel-Stosiek M, Hornstein OP, Stosiek N. Family study on Melkersson-Rosenthal syndrome. some hereditary aspects of the disease and review of literature. Acta Derm Venereol. 1990;70:221-226.
- Sciubba JJ, Said-Al-Naief N. Orofacial granulomatosis: presentation, pathology and management of 13 cases. J Oral Pathol Med. 2003;32:576-585.
- Zimmer WM, Rogers
RS 3rd, Reeve CM, et al. Orofacial manifestations of Melkersson-Rosenthal syndrome. a study of 42 patients and review of 220 cases from the literature. Oral Surg Oral Med Oral Pathol. 1992;74:610-619. - Critchlow WA, Cha
ng D. Cheilitis granulomatosa: a review [published online September 22, 2013]. Head Neck Pathol. 2014;8:209-213. - Allen CM, Camisa
C. Oral disease. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Philadelphia, PA: Elsevier; 2012:1157-1160. - Halevy S, Shalom
G, Trattner A, et al. Melkersson-Rosenthal syndrome: a possible association with psoriasis. J Am Acad Dermatol. 2012;67:795-796. - Algood HM, Lin P
L, Flynn JL. Tumor necrosis factor and chemokine interactions in the formation and maintenance of granulomas in tuberculosis. Clin Infect Dis. 2005;41(suppl 3):S189-S193. - Yee AM, Pochapin
MB. Treatment of complicated sarcoidosis with infliximab anti-tumor necrosis factor-alpha therapy. Ann Intern Med. 2001;135:27-31. - Targan SR, Hanau
er SB, van Deventer SJ, et al. A short-term study of chimeric monoclonal antibody cA2 to tumor necrosis factor alpha for Crohn’s disease. Crohn’s Disease cA2 Study Group. N Engl J Med. 1997;337:1029-1035. - Kakimoto C, Spar
ks C, White AA. Melkersson-Rosenthal syndrome: a form of pseudoangioedema. Ann Allergy Asthma Immunol. 2007;99:185-189. - Wickramasinghe N
, Gunasekara CN, Fernando WS, et al. Vulvitis granulomatosa, Melkersson-Rosenthal syndrome, and Crohn’s disease: dramatic response to infliximab therapy. Int J Dermatol. 2012;51:966-968. - Stein J, Paulke
A, Schacher B, et al. An extraordinary form of the Melkersson-Rosenthal syndrome successfully treated with the tumour necrosis factor-α blocker adalimumab [published online May 14, 2014]. BMJ Case Rep. doi:10.1136/bcr-2014-204674. - Cheifetz A, Smedley M, Martin S, et al. The incidence and management of infusion reactions to infliximab: a large center experience. Am J Gastroenterol. 2003;98:1315-1324.
- Choquette D, Faraawi R, Chow A, et al. Incidence and management of infusion reactions to infliximab in a prospective real-world community registry. J Rheumatol. 2015;42:1105-1111.
- Ruiz Villaverde R, Sánchez Cano D. Successful treatment of granulomatous cheilitis with adalimumab. Int J Dermatol. 2012;51:118-120.
- Liu R, Yu S. Melk
ersson-Rosenthal syndrome: a review of seven patients [published online May 7, 2013]. J Clin Neurosci. 2013;20:993-995. - Sun B, Zhou C, Han Z. Facial palsy in Melkersson-Rosenthal syndrome and Bell’s palsy: familial history and recurrence tendency [published online August 13, 2014]. Ann Otol Rhinol Laryngol. 2015;124:107-109.
- Meisel-Stosiek M, Hornstein OP, Stosiek N. Family study on Melkersson-Rosenthal syndrome. some hereditary aspects of the disease and review of literature. Acta Derm Venereol. 1990;70:221-226.
- Sciubba JJ, Said-Al-Naief N. Orofacial granulomatosis: presentation, pathology and management of 13 cases. J Oral Pathol Med. 2003;32:576-585.
- Zimmer WM, Rogers
RS 3rd, Reeve CM, et al. Orofacial manifestations of Melkersson-Rosenthal syndrome. a study of 42 patients and review of 220 cases from the literature. Oral Surg Oral Med Oral Pathol. 1992;74:610-619. - Critchlow WA, Cha
ng D. Cheilitis granulomatosa: a review [published online September 22, 2013]. Head Neck Pathol. 2014;8:209-213. - Allen CM, Camisa
C. Oral disease. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. Philadelphia, PA: Elsevier; 2012:1157-1160. - Halevy S, Shalom
G, Trattner A, et al. Melkersson-Rosenthal syndrome: a possible association with psoriasis. J Am Acad Dermatol. 2012;67:795-796. - Algood HM, Lin P
L, Flynn JL. Tumor necrosis factor and chemokine interactions in the formation and maintenance of granulomas in tuberculosis. Clin Infect Dis. 2005;41(suppl 3):S189-S193. - Yee AM, Pochapin
MB. Treatment of complicated sarcoidosis with infliximab anti-tumor necrosis factor-alpha therapy. Ann Intern Med. 2001;135:27-31. - Targan SR, Hanau
er SB, van Deventer SJ, et al. A short-term study of chimeric monoclonal antibody cA2 to tumor necrosis factor alpha for Crohn’s disease. Crohn’s Disease cA2 Study Group. N Engl J Med. 1997;337:1029-1035. - Kakimoto C, Spar
ks C, White AA. Melkersson-Rosenthal syndrome: a form of pseudoangioedema. Ann Allergy Asthma Immunol. 2007;99:185-189. - Wickramasinghe N
, Gunasekara CN, Fernando WS, et al. Vulvitis granulomatosa, Melkersson-Rosenthal syndrome, and Crohn’s disease: dramatic response to infliximab therapy. Int J Dermatol. 2012;51:966-968. - Stein J, Paulke
A, Schacher B, et al. An extraordinary form of the Melkersson-Rosenthal syndrome successfully treated with the tumour necrosis factor-α blocker adalimumab [published online May 14, 2014]. BMJ Case Rep. doi:10.1136/bcr-2014-204674. - Cheifetz A, Smedley M, Martin S, et al. The incidence and management of infusion reactions to infliximab: a large center experience. Am J Gastroenterol. 2003;98:1315-1324.
- Choquette D, Faraawi R, Chow A, et al. Incidence and management of infusion reactions to infliximab in a prospective real-world community registry. J Rheumatol. 2015;42:1105-1111.
- Ruiz Villaverde R, Sánchez Cano D. Successful treatment of granulomatous cheilitis with adalimumab. Int J Dermatol. 2012;51:118-120.
Practice Points
- The classical triad of Melkersson-Rosenthal syndrome (MRS), which includes facial nerve palsy, facial edema, and lingua plicata, can present gradually over time and should therefore be kept in the differential of cheilitis.
- Tumor necrosis factor α therapy may play a crucial role in rare granulomatous diseases, including MRS.
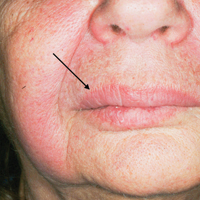
Debunking Psoriasis Myths: How Long Do Patients Have to Wait to See Results With Biologics?
Myth: Biologics Work Slowly
Biologics have demonstrated efficacy in psoriasis and often are used in psoriasis patients who have not achieved desired results with other treatments, patients who have had intolerable side effects from other treatments, and patients with concurrent diseases that preclude the use of systemic therapies. Because of the quality-of-life impact of psoriasis, patients look for quick clearance of their symptoms, but can biologics deliver fast results or do they work slowly?
Biologics such as etanercept and adalimumab block tumor necrosis factor α signaling, while ustekinumab targets IL-12 and IL-23 and others target IL-17. Some patients may begin to see improvement in skin lesions within 1 month of initiating biologic therapies because they target specific proinflammatory pathways that are critical to the pathogenesis of psoriasis, but response time varies among patients and specific therapy used.
The psoriasis area and severity index (PASI) measures psoriasis treatment success. Based on the American Academy of Dermatology’s guidelines of care for the management of psoriasis and psoriatic arthritis published in 2008, short-term response was achieved in 10 to 14 weeks for the following biologics:
- Adalimumab: 80% of patients achieved PASI 75 at week 12
- Etanercept: 49% of patients given 50 mg twice weekly achieved PASI 75 at 12 weeks; 34% of patients given 25 mg twice weekly achieved PASI 75 at 12 weeks
- Infliximab: 80% of patients achieved PASI 75 at week 10
Of the newer biologics, Premier Research recently noted that PASI 75 was achieved after 12 weeks with the following biologics:
- Brodalumab: 83% after 12 weeks
- Ixekizumab: 90% after 12 weeks
- Secukinumab: 80% after 12 weeks
- Ustekinumab: 70% after 12 weeks
There are a variety of factors to consider when determining which biologic to use for a psoriasis patient. These data may help in the decision process. However, dermatologists must educate psoriasis patients with a high body mass index that their disease may take longer to respond and may need combination therapy for optimal clearance.
Expert Commentary
All of the biologics, especially the IL-17 inhibitors, work very quickly to clear psoriasis. The only way they work “slowly” is that it may take time (usually a few days) for the payers to approve biologic prescriptions.
—Jashin J. Wu, MD (Los Angeles, California)
Biologics. DermNet New Zealand website. https://www.dermnetnz.org/topics/biologics/. Accessed February 6, 2018.
Biologics in psoriasis treatment. Premier Research website. https://premier-research.com/perspectives-biologics-psoriasis-treatment/. Published May 9, 2017. Accessed February 6, 2018.
Menter A, Gottlieb A, Feldman SR, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: section 1. Overview of psoriasis and guidelines of care for the treatment of psoriasis with biologics. J Am Acad Dermatol. 2008;58:826-850.
Vilarrasa E, Notario J, Bordas X, et al. ORBIT (Outcome and Retention Rate of Biologic Treatments for Psoriasis): a retrospective observational study on biologic drug survival in daily practice. J Am Acad Dermatol. 2016;74:1066-1072.
Myth: Biologics Work Slowly
Biologics have demonstrated efficacy in psoriasis and often are used in psoriasis patients who have not achieved desired results with other treatments, patients who have had intolerable side effects from other treatments, and patients with concurrent diseases that preclude the use of systemic therapies. Because of the quality-of-life impact of psoriasis, patients look for quick clearance of their symptoms, but can biologics deliver fast results or do they work slowly?
Biologics such as etanercept and adalimumab block tumor necrosis factor α signaling, while ustekinumab targets IL-12 and IL-23 and others target IL-17. Some patients may begin to see improvement in skin lesions within 1 month of initiating biologic therapies because they target specific proinflammatory pathways that are critical to the pathogenesis of psoriasis, but response time varies among patients and specific therapy used.
The psoriasis area and severity index (PASI) measures psoriasis treatment success. Based on the American Academy of Dermatology’s guidelines of care for the management of psoriasis and psoriatic arthritis published in 2008, short-term response was achieved in 10 to 14 weeks for the following biologics:
- Adalimumab: 80% of patients achieved PASI 75 at week 12
- Etanercept: 49% of patients given 50 mg twice weekly achieved PASI 75 at 12 weeks; 34% of patients given 25 mg twice weekly achieved PASI 75 at 12 weeks
- Infliximab: 80% of patients achieved PASI 75 at week 10
Of the newer biologics, Premier Research recently noted that PASI 75 was achieved after 12 weeks with the following biologics:
- Brodalumab: 83% after 12 weeks
- Ixekizumab: 90% after 12 weeks
- Secukinumab: 80% after 12 weeks
- Ustekinumab: 70% after 12 weeks
There are a variety of factors to consider when determining which biologic to use for a psoriasis patient. These data may help in the decision process. However, dermatologists must educate psoriasis patients with a high body mass index that their disease may take longer to respond and may need combination therapy for optimal clearance.
Expert Commentary
All of the biologics, especially the IL-17 inhibitors, work very quickly to clear psoriasis. The only way they work “slowly” is that it may take time (usually a few days) for the payers to approve biologic prescriptions.
—Jashin J. Wu, MD (Los Angeles, California)
Myth: Biologics Work Slowly
Biologics have demonstrated efficacy in psoriasis and often are used in psoriasis patients who have not achieved desired results with other treatments, patients who have had intolerable side effects from other treatments, and patients with concurrent diseases that preclude the use of systemic therapies. Because of the quality-of-life impact of psoriasis, patients look for quick clearance of their symptoms, but can biologics deliver fast results or do they work slowly?
Biologics such as etanercept and adalimumab block tumor necrosis factor α signaling, while ustekinumab targets IL-12 and IL-23 and others target IL-17. Some patients may begin to see improvement in skin lesions within 1 month of initiating biologic therapies because they target specific proinflammatory pathways that are critical to the pathogenesis of psoriasis, but response time varies among patients and specific therapy used.
The psoriasis area and severity index (PASI) measures psoriasis treatment success. Based on the American Academy of Dermatology’s guidelines of care for the management of psoriasis and psoriatic arthritis published in 2008, short-term response was achieved in 10 to 14 weeks for the following biologics:
- Adalimumab: 80% of patients achieved PASI 75 at week 12
- Etanercept: 49% of patients given 50 mg twice weekly achieved PASI 75 at 12 weeks; 34% of patients given 25 mg twice weekly achieved PASI 75 at 12 weeks
- Infliximab: 80% of patients achieved PASI 75 at week 10
Of the newer biologics, Premier Research recently noted that PASI 75 was achieved after 12 weeks with the following biologics:
- Brodalumab: 83% after 12 weeks
- Ixekizumab: 90% after 12 weeks
- Secukinumab: 80% after 12 weeks
- Ustekinumab: 70% after 12 weeks
There are a variety of factors to consider when determining which biologic to use for a psoriasis patient. These data may help in the decision process. However, dermatologists must educate psoriasis patients with a high body mass index that their disease may take longer to respond and may need combination therapy for optimal clearance.
Expert Commentary
All of the biologics, especially the IL-17 inhibitors, work very quickly to clear psoriasis. The only way they work “slowly” is that it may take time (usually a few days) for the payers to approve biologic prescriptions.
—Jashin J. Wu, MD (Los Angeles, California)
Biologics. DermNet New Zealand website. https://www.dermnetnz.org/topics/biologics/. Accessed February 6, 2018.
Biologics in psoriasis treatment. Premier Research website. https://premier-research.com/perspectives-biologics-psoriasis-treatment/. Published May 9, 2017. Accessed February 6, 2018.
Menter A, Gottlieb A, Feldman SR, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: section 1. Overview of psoriasis and guidelines of care for the treatment of psoriasis with biologics. J Am Acad Dermatol. 2008;58:826-850.
Vilarrasa E, Notario J, Bordas X, et al. ORBIT (Outcome and Retention Rate of Biologic Treatments for Psoriasis): a retrospective observational study on biologic drug survival in daily practice. J Am Acad Dermatol. 2016;74:1066-1072.
Biologics. DermNet New Zealand website. https://www.dermnetnz.org/topics/biologics/. Accessed February 6, 2018.
Biologics in psoriasis treatment. Premier Research website. https://premier-research.com/perspectives-biologics-psoriasis-treatment/. Published May 9, 2017. Accessed February 6, 2018.
Menter A, Gottlieb A, Feldman SR, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: section 1. Overview of psoriasis and guidelines of care for the treatment of psoriasis with biologics. J Am Acad Dermatol. 2008;58:826-850.
Vilarrasa E, Notario J, Bordas X, et al. ORBIT (Outcome and Retention Rate of Biologic Treatments for Psoriasis): a retrospective observational study on biologic drug survival in daily practice. J Am Acad Dermatol. 2016;74:1066-1072.

Enlarging Red Papulonodule on the Chest
The Diagnosis: Metastatic Renal Cell Carcinoma
Histopathologic examination of the punch biopsy demonstrated epithelioid cells with abundant clear cytoplasm and numerous chicken wire-like vascular channels consistent with a diagnosis of cutaneous metastasis of renal cell carcinoma (RCC)(Figure). Collateral history revealed that 8 years prior, the patient had been diagnosed with clear cell RCC, stage III (T3aN0M0). At that time, he was treated with radical nephrectomy, which was considered curative. He remained disease free until several months prior to the development of the cutaneous lesion when he was found to have pulmonary and cerebral metastases with biopsies showing metastatic RCC. He was treated with lobectomy and Gamma Knife radiation for the lung and cerebral metastases, respectively. His oncologist planned to initiate therapy with the multikinase inhibitor sunitinib, which inhibits vascular endothelial growth factor (VEGF) signaling. Unfortunately, the patient died prior to treatment due to overwhelming tumor burden.
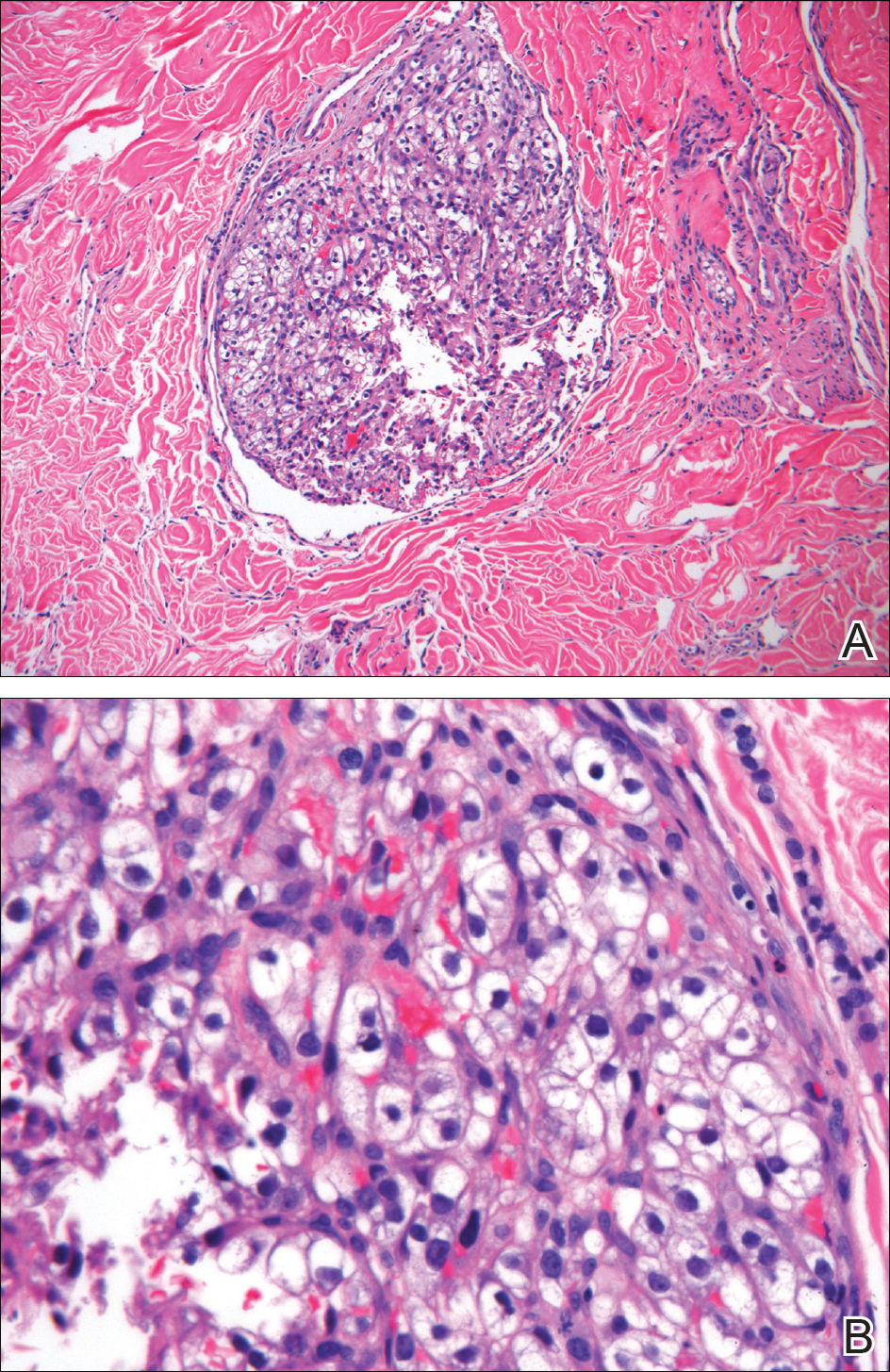
Clear cell RCC, the most common renal malignancy, presents with metastatic disease at the time of diagnosis in 21% of patients.1 An additional 20% of patients with localized disease develop metastases within several years of receiving a nephrectomy without adjuvant therapy, which is standard treatment for stage I to stage III disease.1,2 Metastatic RCC most frequently targets the lungs, bone, liver, and brain, though virtually any organ can be involved. Cutaneous involvement is estimated to occur in 3.3% of RCC cases,3 accounting for only 1.4% of cutaneous metastases overall.4 The risk for developing cutaneous metastases is greatest within 3 years following nephrectomy.3 However, our patient demonstrates that metastasis of RCC to skin can be long delayed (>5 years) despite an initial diagnosis of localized disease.
Cutaneous RCC classically presents as a painless firm papulonodule with a deep red or purple color due to its high vascularity.4 Several retrospective studies have identified the scalp as the most frequent site of cutaneous involvement, followed by the chest, abdomen, and nephrectomy scar.3,4 The differential diagnosis includes other vascular lesions such as pyogenic granuloma, hemangioma, angiosarcoma, bacillary angiomatosis, and Kaposi sarcoma. Diagnosis usually is easily confirmed histologically. Proliferative nests of epithelioid cells with clear cell morphology are surrounded by delicately branching vessels referred to as chicken wire-like vasculature. Immunohistochemical studies demonstrate positivity for pan-cytokeratin, vimentin, and CD-10, and negativity for p63 and cytokeratins 5 and 6, helping to confirm the diagnosis in more challenging cases, especially when there is no known history of primary RCC.5
If cutaneous metastasis of RCC is diagnosed, a chest and abdominal computed tomography scan as well as serum alkaline phosphatase test are warranted, as up to 90% of patients with RCC in the skin have additional lesions in at least 1 other site such as the lungs, bones, or liver.3 Management of metastatic RCC includes surgical excision if a single metastasis is found and either immunotherapy with high-dose IL-2 or an anti-programmed cell death inhibitor. Patients with progressive disease also may receive targeted anti-VEGF inhibitors (eg, axitinib, pazopanib, sunitinib), which have been shown to increase progression-free survival in metastatic RCC.6-8 Interestingly, some evidence suggests severely delayed recurrence of RCC (>5 years following nephrectomy) may predict better response to systemic therapy.9
This case of severely delayed metastasis of RCC 8 years after nephrectomy raises the question of whether routine surveillance for RCC recurrence should continue beyond 5 years. It also underscores the need for further studies to determine the utility of postsurgical adjuvant therapy for localized disease (stages I-III). A randomized clinical trial showed no significant difference in disease-free survival when the multikinase inhibitors sunitinib and sorafenib were used as adjuvant therapy.10 The randomized, placebo-controlled PROTECT trial showed no significant difference in disease-free survival between the VEGF inhibitor pazopanib and placebo when used as adjuvant therapy.11 However, trials are ongoing to investigate a potential survival advantage of adjuvant therapy with the VEGF receptor inhibitor axitinib and the mammalian target of rapamycin inhibitor everolimus.
- Dabestani S, Thorstenson A, Lindblad P, et al. Renal cell carcinoma recurrences and metastases in primary non-metastatic patients: a population-based study. World J Urol. 2016;34:1081-1086.
- Ljungberg B, Campbell SC, Choi HY, et al. The epidemiology of renal cell carcinoma. Eur Urol. 2011;60:615-621.
- Dorairajan LN, Hemal AK, Aron M, et al. Cutaneous metastases in renal cell carcinoma. Urol Int. 1999;63:164-167.
- Lookingbill DP, Spangler N, Helm KF. Cutaneous metastases in patients with metastatic carcinoma: a retrospective study of 4020 patients. J Am Acad Dermatol. 1993;29(2, pt 1):228-236.
- Sariya D, Ruth K, Adams-McDonnell R, et al. Clinicopathologic correlation of cutaneous metastases: experience from a cancer center. Arch Dermatol. 2007;143:613-620.
- Sternberg CN, Davis ID, Mardiak J, et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: results of a randomized phase III trial. J Clin Oncol. 2010;28:1061-1068.
- Motzer RJ, Hutson TE, Tomczak P, et al. Overall survival and updated results for sunitinib compared with interferon alfa in patients with metastatic renal cell carcinoma. J Clin Oncol. 2009;27:3584-3590.
- Rini BI, Grunwald V, Fishman MN, et al. Axitinib for first-line metastatic renal cell carcinoma (mRCC): overall efficacy and pharmacokinetic (PK) analyses from a randomized phase II study. J Clin Oncol. 2012;30(suppl). doi:10.1200/jco.2012.30.15_suppl.4503.
- Ficarra V, Novara G. Characterizing late recurrence of renal cell carcinoma. Nat Rev Urol. 2013;10:687-689.
- Haas NB, Manola J, Uzzo RG, et al. Adjuvant sunitinib or sorafenib for high-risk, non-metastatic renal-cell carcinoma (ECOG-ACRIN E2805): a double-blind, placebo-controlled, randomised, phase 3 trial [published online March 9, 2016]. Lancet. 2016;387:2008-2016.
- Motzer RJ, Haas NB, Donskov F, et al; PROTECT investigators. Randomized phase III trial of adjuvant pazopanib versus placebo after nephrectomy in patients with localized or locally advanced renal cell carcinoma [published online September 13, 2017]. J Clin Oncol. 2017;35:3916-3923.
The Diagnosis: Metastatic Renal Cell Carcinoma
Histopathologic examination of the punch biopsy demonstrated epithelioid cells with abundant clear cytoplasm and numerous chicken wire-like vascular channels consistent with a diagnosis of cutaneous metastasis of renal cell carcinoma (RCC)(Figure). Collateral history revealed that 8 years prior, the patient had been diagnosed with clear cell RCC, stage III (T3aN0M0). At that time, he was treated with radical nephrectomy, which was considered curative. He remained disease free until several months prior to the development of the cutaneous lesion when he was found to have pulmonary and cerebral metastases with biopsies showing metastatic RCC. He was treated with lobectomy and Gamma Knife radiation for the lung and cerebral metastases, respectively. His oncologist planned to initiate therapy with the multikinase inhibitor sunitinib, which inhibits vascular endothelial growth factor (VEGF) signaling. Unfortunately, the patient died prior to treatment due to overwhelming tumor burden.
Clear cell RCC, the most common renal malignancy, presents with metastatic disease at the time of diagnosis in 21% of patients.1 An additional 20% of patients with localized disease develop metastases within several years of receiving a nephrectomy without adjuvant therapy, which is standard treatment for stage I to stage III disease.1,2 Metastatic RCC most frequently targets the lungs, bone, liver, and brain, though virtually any organ can be involved. Cutaneous involvement is estimated to occur in 3.3% of RCC cases,3 accounting for only 1.4% of cutaneous metastases overall.4 The risk for developing cutaneous metastases is greatest within 3 years following nephrectomy.3 However, our patient demonstrates that metastasis of RCC to skin can be long delayed (>5 years) despite an initial diagnosis of localized disease.
Cutaneous RCC classically presents as a painless firm papulonodule with a deep red or purple color due to its high vascularity.4 Several retrospective studies have identified the scalp as the most frequent site of cutaneous involvement, followed by the chest, abdomen, and nephrectomy scar.3,4 The differential diagnosis includes other vascular lesions such as pyogenic granuloma, hemangioma, angiosarcoma, bacillary angiomatosis, and Kaposi sarcoma. Diagnosis usually is easily confirmed histologically. Proliferative nests of epithelioid cells with clear cell morphology are surrounded by delicately branching vessels referred to as chicken wire-like vasculature. Immunohistochemical studies demonstrate positivity for pan-cytokeratin, vimentin, and CD-10, and negativity for p63 and cytokeratins 5 and 6, helping to confirm the diagnosis in more challenging cases, especially when there is no known history of primary RCC.5
If cutaneous metastasis of RCC is diagnosed, a chest and abdominal computed tomography scan as well as serum alkaline phosphatase test are warranted, as up to 90% of patients with RCC in the skin have additional lesions in at least 1 other site such as the lungs, bones, or liver.3 Management of metastatic RCC includes surgical excision if a single metastasis is found and either immunotherapy with high-dose IL-2 or an anti-programmed cell death inhibitor. Patients with progressive disease also may receive targeted anti-VEGF inhibitors (eg, axitinib, pazopanib, sunitinib), which have been shown to increase progression-free survival in metastatic RCC.6-8 Interestingly, some evidence suggests severely delayed recurrence of RCC (>5 years following nephrectomy) may predict better response to systemic therapy.9
This case of severely delayed metastasis of RCC 8 years after nephrectomy raises the question of whether routine surveillance for RCC recurrence should continue beyond 5 years. It also underscores the need for further studies to determine the utility of postsurgical adjuvant therapy for localized disease (stages I-III). A randomized clinical trial showed no significant difference in disease-free survival when the multikinase inhibitors sunitinib and sorafenib were used as adjuvant therapy.10 The randomized, placebo-controlled PROTECT trial showed no significant difference in disease-free survival between the VEGF inhibitor pazopanib and placebo when used as adjuvant therapy.11 However, trials are ongoing to investigate a potential survival advantage of adjuvant therapy with the VEGF receptor inhibitor axitinib and the mammalian target of rapamycin inhibitor everolimus.
The Diagnosis: Metastatic Renal Cell Carcinoma
Histopathologic examination of the punch biopsy demonstrated epithelioid cells with abundant clear cytoplasm and numerous chicken wire-like vascular channels consistent with a diagnosis of cutaneous metastasis of renal cell carcinoma (RCC)(Figure). Collateral history revealed that 8 years prior, the patient had been diagnosed with clear cell RCC, stage III (T3aN0M0). At that time, he was treated with radical nephrectomy, which was considered curative. He remained disease free until several months prior to the development of the cutaneous lesion when he was found to have pulmonary and cerebral metastases with biopsies showing metastatic RCC. He was treated with lobectomy and Gamma Knife radiation for the lung and cerebral metastases, respectively. His oncologist planned to initiate therapy with the multikinase inhibitor sunitinib, which inhibits vascular endothelial growth factor (VEGF) signaling. Unfortunately, the patient died prior to treatment due to overwhelming tumor burden.
Clear cell RCC, the most common renal malignancy, presents with metastatic disease at the time of diagnosis in 21% of patients.1 An additional 20% of patients with localized disease develop metastases within several years of receiving a nephrectomy without adjuvant therapy, which is standard treatment for stage I to stage III disease.1,2 Metastatic RCC most frequently targets the lungs, bone, liver, and brain, though virtually any organ can be involved. Cutaneous involvement is estimated to occur in 3.3% of RCC cases,3 accounting for only 1.4% of cutaneous metastases overall.4 The risk for developing cutaneous metastases is greatest within 3 years following nephrectomy.3 However, our patient demonstrates that metastasis of RCC to skin can be long delayed (>5 years) despite an initial diagnosis of localized disease.
Cutaneous RCC classically presents as a painless firm papulonodule with a deep red or purple color due to its high vascularity.4 Several retrospective studies have identified the scalp as the most frequent site of cutaneous involvement, followed by the chest, abdomen, and nephrectomy scar.3,4 The differential diagnosis includes other vascular lesions such as pyogenic granuloma, hemangioma, angiosarcoma, bacillary angiomatosis, and Kaposi sarcoma. Diagnosis usually is easily confirmed histologically. Proliferative nests of epithelioid cells with clear cell morphology are surrounded by delicately branching vessels referred to as chicken wire-like vasculature. Immunohistochemical studies demonstrate positivity for pan-cytokeratin, vimentin, and CD-10, and negativity for p63 and cytokeratins 5 and 6, helping to confirm the diagnosis in more challenging cases, especially when there is no known history of primary RCC.5
If cutaneous metastasis of RCC is diagnosed, a chest and abdominal computed tomography scan as well as serum alkaline phosphatase test are warranted, as up to 90% of patients with RCC in the skin have additional lesions in at least 1 other site such as the lungs, bones, or liver.3 Management of metastatic RCC includes surgical excision if a single metastasis is found and either immunotherapy with high-dose IL-2 or an anti-programmed cell death inhibitor. Patients with progressive disease also may receive targeted anti-VEGF inhibitors (eg, axitinib, pazopanib, sunitinib), which have been shown to increase progression-free survival in metastatic RCC.6-8 Interestingly, some evidence suggests severely delayed recurrence of RCC (>5 years following nephrectomy) may predict better response to systemic therapy.9
This case of severely delayed metastasis of RCC 8 years after nephrectomy raises the question of whether routine surveillance for RCC recurrence should continue beyond 5 years. It also underscores the need for further studies to determine the utility of postsurgical adjuvant therapy for localized disease (stages I-III). A randomized clinical trial showed no significant difference in disease-free survival when the multikinase inhibitors sunitinib and sorafenib were used as adjuvant therapy.10 The randomized, placebo-controlled PROTECT trial showed no significant difference in disease-free survival between the VEGF inhibitor pazopanib and placebo when used as adjuvant therapy.11 However, trials are ongoing to investigate a potential survival advantage of adjuvant therapy with the VEGF receptor inhibitor axitinib and the mammalian target of rapamycin inhibitor everolimus.
- Dabestani S, Thorstenson A, Lindblad P, et al. Renal cell carcinoma recurrences and metastases in primary non-metastatic patients: a population-based study. World J Urol. 2016;34:1081-1086.
- Ljungberg B, Campbell SC, Choi HY, et al. The epidemiology of renal cell carcinoma. Eur Urol. 2011;60:615-621.
- Dorairajan LN, Hemal AK, Aron M, et al. Cutaneous metastases in renal cell carcinoma. Urol Int. 1999;63:164-167.
- Lookingbill DP, Spangler N, Helm KF. Cutaneous metastases in patients with metastatic carcinoma: a retrospective study of 4020 patients. J Am Acad Dermatol. 1993;29(2, pt 1):228-236.
- Sariya D, Ruth K, Adams-McDonnell R, et al. Clinicopathologic correlation of cutaneous metastases: experience from a cancer center. Arch Dermatol. 2007;143:613-620.
- Sternberg CN, Davis ID, Mardiak J, et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: results of a randomized phase III trial. J Clin Oncol. 2010;28:1061-1068.
- Motzer RJ, Hutson TE, Tomczak P, et al. Overall survival and updated results for sunitinib compared with interferon alfa in patients with metastatic renal cell carcinoma. J Clin Oncol. 2009;27:3584-3590.
- Rini BI, Grunwald V, Fishman MN, et al. Axitinib for first-line metastatic renal cell carcinoma (mRCC): overall efficacy and pharmacokinetic (PK) analyses from a randomized phase II study. J Clin Oncol. 2012;30(suppl). doi:10.1200/jco.2012.30.15_suppl.4503.
- Ficarra V, Novara G. Characterizing late recurrence of renal cell carcinoma. Nat Rev Urol. 2013;10:687-689.
- Haas NB, Manola J, Uzzo RG, et al. Adjuvant sunitinib or sorafenib for high-risk, non-metastatic renal-cell carcinoma (ECOG-ACRIN E2805): a double-blind, placebo-controlled, randomised, phase 3 trial [published online March 9, 2016]. Lancet. 2016;387:2008-2016.
- Motzer RJ, Haas NB, Donskov F, et al; PROTECT investigators. Randomized phase III trial of adjuvant pazopanib versus placebo after nephrectomy in patients with localized or locally advanced renal cell carcinoma [published online September 13, 2017]. J Clin Oncol. 2017;35:3916-3923.
- Dabestani S, Thorstenson A, Lindblad P, et al. Renal cell carcinoma recurrences and metastases in primary non-metastatic patients: a population-based study. World J Urol. 2016;34:1081-1086.
- Ljungberg B, Campbell SC, Choi HY, et al. The epidemiology of renal cell carcinoma. Eur Urol. 2011;60:615-621.
- Dorairajan LN, Hemal AK, Aron M, et al. Cutaneous metastases in renal cell carcinoma. Urol Int. 1999;63:164-167.
- Lookingbill DP, Spangler N, Helm KF. Cutaneous metastases in patients with metastatic carcinoma: a retrospective study of 4020 patients. J Am Acad Dermatol. 1993;29(2, pt 1):228-236.
- Sariya D, Ruth K, Adams-McDonnell R, et al. Clinicopathologic correlation of cutaneous metastases: experience from a cancer center. Arch Dermatol. 2007;143:613-620.
- Sternberg CN, Davis ID, Mardiak J, et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: results of a randomized phase III trial. J Clin Oncol. 2010;28:1061-1068.
- Motzer RJ, Hutson TE, Tomczak P, et al. Overall survival and updated results for sunitinib compared with interferon alfa in patients with metastatic renal cell carcinoma. J Clin Oncol. 2009;27:3584-3590.
- Rini BI, Grunwald V, Fishman MN, et al. Axitinib for first-line metastatic renal cell carcinoma (mRCC): overall efficacy and pharmacokinetic (PK) analyses from a randomized phase II study. J Clin Oncol. 2012;30(suppl). doi:10.1200/jco.2012.30.15_suppl.4503.
- Ficarra V, Novara G. Characterizing late recurrence of renal cell carcinoma. Nat Rev Urol. 2013;10:687-689.
- Haas NB, Manola J, Uzzo RG, et al. Adjuvant sunitinib or sorafenib for high-risk, non-metastatic renal-cell carcinoma (ECOG-ACRIN E2805): a double-blind, placebo-controlled, randomised, phase 3 trial [published online March 9, 2016]. Lancet. 2016;387:2008-2016.
- Motzer RJ, Haas NB, Donskov F, et al; PROTECT investigators. Randomized phase III trial of adjuvant pazopanib versus placebo after nephrectomy in patients with localized or locally advanced renal cell carcinoma [published online September 13, 2017]. J Clin Oncol. 2017;35:3916-3923.
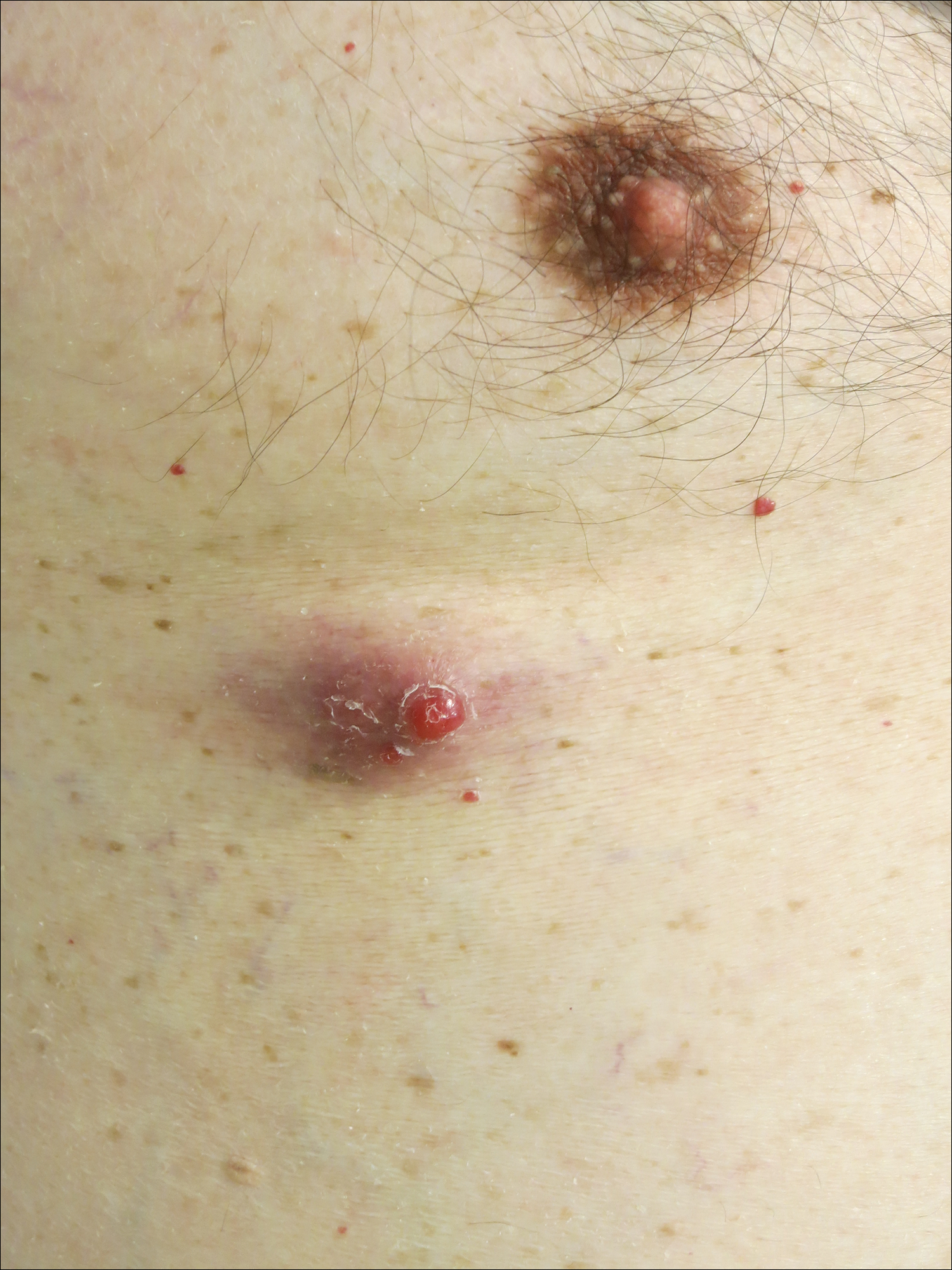
A man in his 60s presented with a subcutaneous nodule on the right side of the chest. Due to impaired mental status, he was unable to describe the precise age of the lesion, but his wife reported it had been present at least several weeks. She recently noted a new, bright red growth on top of the nodule. The lesion was asymptomatic but seemed to be growing in size. Physical examination revealed a 3-cm firm fixed nodule on the right side of the chest with an overlying, exophytic bright red papule. No similar lesions were found elsewhere on physical examination. A punch biopsy of the lesion was performed.
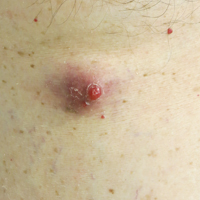
A New Era for Physician-Patient Communication in Dermatology
The physician-patient relationship is an important component of patient care. In the last few years a new paradigm has emerged of instant communication. Because dermatologic diagnosis is visual, many patients feel that making a correct diagnosis is as easy as taking a quick look. The availability of smartphone photography and easy ways to get in touch with dermatologists have created a new reality in physician-patient communication, which sometimes may be abused. We conducted an email survey to assess the attitudes of Chilean dermatologists regarding new methods of communication with their patients.
A survey of 16 questions was distributed to all 343 members of the Chilean Society of Dermatology and Venerology from July 2016 to August 2016. A total of 147 (42.9%) dermatologists completed the survey. When asked if they use personal and direct communication with their patients outside of an office visit, 39% of respondents said always, 41% said sometimes, 17% said only in some circumstances, and 3% said never. Regarding the method of communication, 79% used personal email, 59% used mobile phones, 35% used corporate email, and 34% used text messages. Among respondents who gave their personal email address and phone number to patients, the primary reason stated was to be available for any kind of emergency (67%), for patient follow-up (57%), and for patients to feel close to their dermatologist (28%).
Sixty-nine percent of respondents said patients occasionally have requested to receive a diagnosis via a mobile messaging application, social networks, and email. Of them, 22% said they were very annoyed by these requests. When dermatologists were asked if these instant types of communication improved their relationship with patients, 30% said it does help and 36% said it does not; 30% said they do not know and 4% did not respond. If patients used personal methods of communication to contact their dermatologist that was considered outside of physician-patient boundaries, 63% of physician respondents said they kindly directed patients to formal ways of communication and 15% did not respond to such requests; 22% responded by informal methods of communication. Eighty-one percent of all respondents felt the limits of formal communication between physicians and patients have been surpassed.
To improve the quality of health care, many clinicians use modern methods of communication with their patients. Today, patients can turn to their physicians for medical advice by mobile phone or email. We attempted to characterize the attitudes of Chilean dermatologists regarding new ways of communicating with patients. Our results are similar to other studies. One analysis of primary care physicians in Geneva, Switzerland (N=372), showed that 72% gave their personal email address and 74% gave their mobile phone number to patients. The latter is higher than what was found in our study (59%), which may be explained by the fact that primary care physicians may need to maintain closer contact with their patients.1
In another study performed in primary care physicians in Israel, physicians preferred to provide their mobile phone number rather than their personal email address because they felt that email communication was more likely to lead miscommunication than a phone call.2 There are few reports on this subject in the international literature, and we believe cultural differences may be important when physicians confront these issues.
In general, patient satisfaction is high when patients can contact their physician by phone or email; however, new immediate forms of communication may lead to physician burnout, as patients expect immediate responses and solutions to their requests and healthy physician-patient boundaries may be surpassed. It is important to educate both patients and physicians on how these new tools may be properly used on both sides. New boundaries must be set.
- Dash J, Haller DM, Sommer J, et al. Use of email, cell phone and text message between patients and primary-care physicians: cross-sectional study in a French-speaking part of Switzerland. BMC Health Serv Res. 2016;16:549.
- Peleg R, Avdalimov A, Freud T. Providing cell phone numbers and email addresses to patients: the physician’s perspective. BMC Res Notes. 2011;4:76.
The physician-patient relationship is an important component of patient care. In the last few years a new paradigm has emerged of instant communication. Because dermatologic diagnosis is visual, many patients feel that making a correct diagnosis is as easy as taking a quick look. The availability of smartphone photography and easy ways to get in touch with dermatologists have created a new reality in physician-patient communication, which sometimes may be abused. We conducted an email survey to assess the attitudes of Chilean dermatologists regarding new methods of communication with their patients.
A survey of 16 questions was distributed to all 343 members of the Chilean Society of Dermatology and Venerology from July 2016 to August 2016. A total of 147 (42.9%) dermatologists completed the survey. When asked if they use personal and direct communication with their patients outside of an office visit, 39% of respondents said always, 41% said sometimes, 17% said only in some circumstances, and 3% said never. Regarding the method of communication, 79% used personal email, 59% used mobile phones, 35% used corporate email, and 34% used text messages. Among respondents who gave their personal email address and phone number to patients, the primary reason stated was to be available for any kind of emergency (67%), for patient follow-up (57%), and for patients to feel close to their dermatologist (28%).
Sixty-nine percent of respondents said patients occasionally have requested to receive a diagnosis via a mobile messaging application, social networks, and email. Of them, 22% said they were very annoyed by these requests. When dermatologists were asked if these instant types of communication improved their relationship with patients, 30% said it does help and 36% said it does not; 30% said they do not know and 4% did not respond. If patients used personal methods of communication to contact their dermatologist that was considered outside of physician-patient boundaries, 63% of physician respondents said they kindly directed patients to formal ways of communication and 15% did not respond to such requests; 22% responded by informal methods of communication. Eighty-one percent of all respondents felt the limits of formal communication between physicians and patients have been surpassed.
To improve the quality of health care, many clinicians use modern methods of communication with their patients. Today, patients can turn to their physicians for medical advice by mobile phone or email. We attempted to characterize the attitudes of Chilean dermatologists regarding new ways of communicating with patients. Our results are similar to other studies. One analysis of primary care physicians in Geneva, Switzerland (N=372), showed that 72% gave their personal email address and 74% gave their mobile phone number to patients. The latter is higher than what was found in our study (59%), which may be explained by the fact that primary care physicians may need to maintain closer contact with their patients.1
In another study performed in primary care physicians in Israel, physicians preferred to provide their mobile phone number rather than their personal email address because they felt that email communication was more likely to lead miscommunication than a phone call.2 There are few reports on this subject in the international literature, and we believe cultural differences may be important when physicians confront these issues.
In general, patient satisfaction is high when patients can contact their physician by phone or email; however, new immediate forms of communication may lead to physician burnout, as patients expect immediate responses and solutions to their requests and healthy physician-patient boundaries may be surpassed. It is important to educate both patients and physicians on how these new tools may be properly used on both sides. New boundaries must be set.
The physician-patient relationship is an important component of patient care. In the last few years a new paradigm has emerged of instant communication. Because dermatologic diagnosis is visual, many patients feel that making a correct diagnosis is as easy as taking a quick look. The availability of smartphone photography and easy ways to get in touch with dermatologists have created a new reality in physician-patient communication, which sometimes may be abused. We conducted an email survey to assess the attitudes of Chilean dermatologists regarding new methods of communication with their patients.
A survey of 16 questions was distributed to all 343 members of the Chilean Society of Dermatology and Venerology from July 2016 to August 2016. A total of 147 (42.9%) dermatologists completed the survey. When asked if they use personal and direct communication with their patients outside of an office visit, 39% of respondents said always, 41% said sometimes, 17% said only in some circumstances, and 3% said never. Regarding the method of communication, 79% used personal email, 59% used mobile phones, 35% used corporate email, and 34% used text messages. Among respondents who gave their personal email address and phone number to patients, the primary reason stated was to be available for any kind of emergency (67%), for patient follow-up (57%), and for patients to feel close to their dermatologist (28%).
Sixty-nine percent of respondents said patients occasionally have requested to receive a diagnosis via a mobile messaging application, social networks, and email. Of them, 22% said they were very annoyed by these requests. When dermatologists were asked if these instant types of communication improved their relationship with patients, 30% said it does help and 36% said it does not; 30% said they do not know and 4% did not respond. If patients used personal methods of communication to contact their dermatologist that was considered outside of physician-patient boundaries, 63% of physician respondents said they kindly directed patients to formal ways of communication and 15% did not respond to such requests; 22% responded by informal methods of communication. Eighty-one percent of all respondents felt the limits of formal communication between physicians and patients have been surpassed.
To improve the quality of health care, many clinicians use modern methods of communication with their patients. Today, patients can turn to their physicians for medical advice by mobile phone or email. We attempted to characterize the attitudes of Chilean dermatologists regarding new ways of communicating with patients. Our results are similar to other studies. One analysis of primary care physicians in Geneva, Switzerland (N=372), showed that 72% gave their personal email address and 74% gave their mobile phone number to patients. The latter is higher than what was found in our study (59%), which may be explained by the fact that primary care physicians may need to maintain closer contact with their patients.1
In another study performed in primary care physicians in Israel, physicians preferred to provide their mobile phone number rather than their personal email address because they felt that email communication was more likely to lead miscommunication than a phone call.2 There are few reports on this subject in the international literature, and we believe cultural differences may be important when physicians confront these issues.
In general, patient satisfaction is high when patients can contact their physician by phone or email; however, new immediate forms of communication may lead to physician burnout, as patients expect immediate responses and solutions to their requests and healthy physician-patient boundaries may be surpassed. It is important to educate both patients and physicians on how these new tools may be properly used on both sides. New boundaries must be set.
- Dash J, Haller DM, Sommer J, et al. Use of email, cell phone and text message between patients and primary-care physicians: cross-sectional study in a French-speaking part of Switzerland. BMC Health Serv Res. 2016;16:549.
- Peleg R, Avdalimov A, Freud T. Providing cell phone numbers and email addresses to patients: the physician’s perspective. BMC Res Notes. 2011;4:76.
- Dash J, Haller DM, Sommer J, et al. Use of email, cell phone and text message between patients and primary-care physicians: cross-sectional study in a French-speaking part of Switzerland. BMC Health Serv Res. 2016;16:549.
- Peleg R, Avdalimov A, Freud T. Providing cell phone numbers and email addresses to patients: the physician’s perspective. BMC Res Notes. 2011;4:76.
Blueberry Muffin Rash Secondary to Hereditary Spherocytosis
The term blueberry muffin rash historically was used to describe the cutaneous manifestations observed in congenital rubella. The term traditionally describes the result of a postnatal dermal extramedullary hematopoiesis. Today, TORCH (toxoplasmosis, other agents, rubella, cytomegalovirus, herpes) infections and plasma cell dyscrasias are all potential causes of extramedullary hematopoiesis. Herein, we present a unique case of a neonate born with a blueberry muffin rash secondary to extramedullary hematopoiesis induced by hereditary spherocytosis.
Case Report
The dermatology department was consulted to evaluate a 2-day-old male neonate born with a “rash.” The patient was born to a 34-year-old gravida 3, para 2, woman at 39 weeks’ gestation. The mother’s prenatal laboratory values were within reference range and ultrasounds were normal, and she was compliant with her prenatal care. She underwent a normal spontaneous vaginal delivery 3 hours after rupture of membranes without complication. The amniotic fluid and umbilical cord both were clear. There was no use of forceps or any other external aiding devices during the delivery. At the time of delivery, the consulting physician noted that the patient had “skin lesions from head to toe.”
The patient’s parents reported that the rash did not seem to cause any discomfort for the patient. In the 24 hours after birth, the parents reported that the erythema seemed to slightly fade. Physical examination revealed many scattered erythematous to violaceous, nonblanching papulonodules affecting the scalp (Figure 1), face, arms, hands (Figure 2A), back (Figure 2B), buttocks, legs, and feet. Some of the papulonodules were soft while others were firm and indurated. Several lesions had a yellowish hue with some overlying crust. There was no mucosal, genital, or ocular involvement. No erosions, ulcerations, petechiae, ecchymoses, or hepatosplenomegaly were noted on examination.
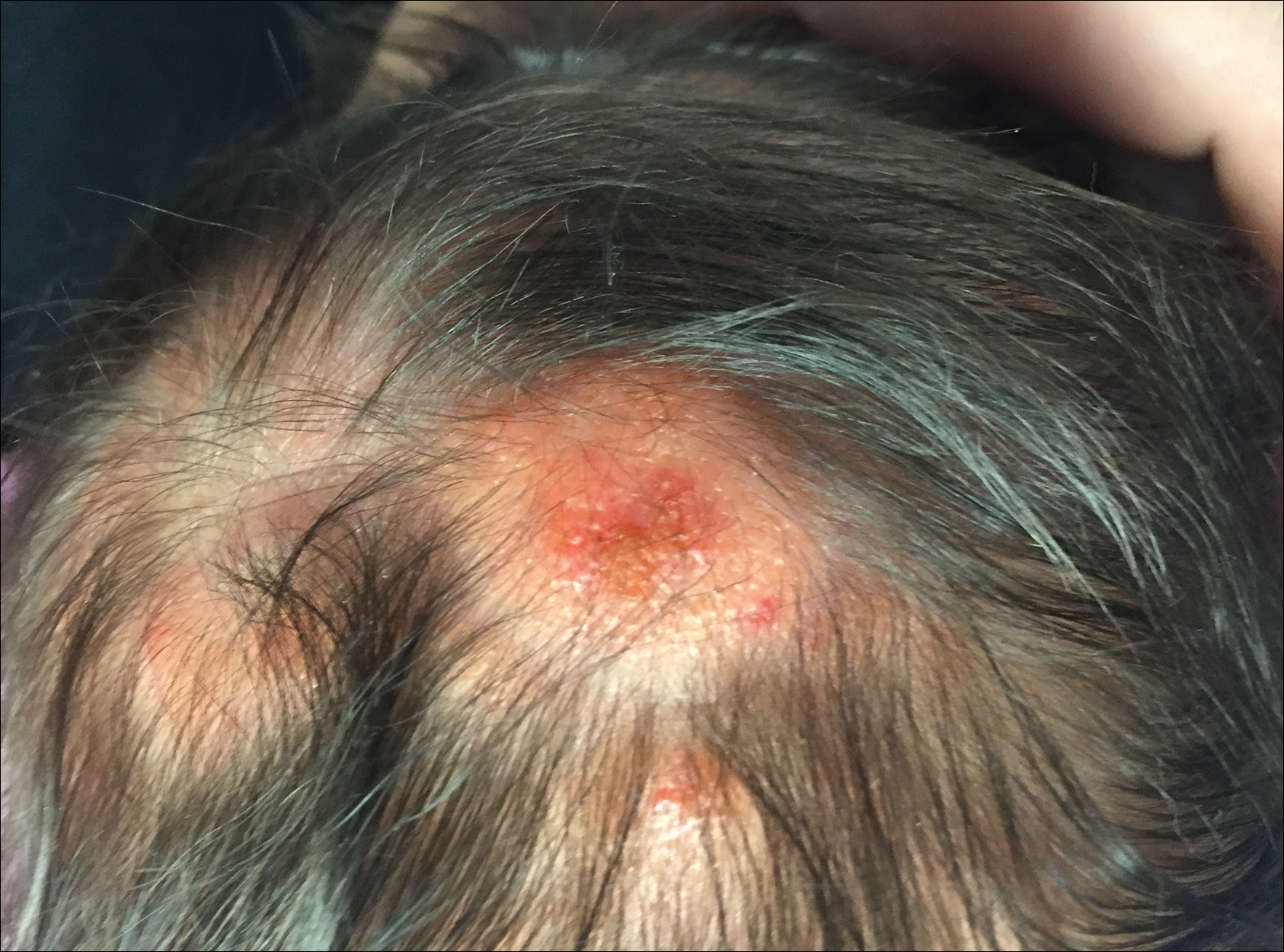

The patient was otherwise healthy with an Apgar score of 8/9 at 1 and 5 minutes. His birth weight, length, and head circumference were within normal limits. There was no evidence of ABO blood group or Rhesus factor incompatibility. His temperature, vital signs, laboratory values (including calcium level and TORCH titers, which included cytomegalovirus, rubella, toxoplasmosis, and herpes simplex virus), and review of systems all were within reference range. A bone survey of the skull, spine, ribs, arms, pelvis, legs, and feet was within normal limits.
The mother’s placenta was sent for pathology and revealed a lymphoplasmacytic chronic deciduitis and acute subchorionitis consistent with a nonspecific inflammatory response, unlikely to be from an infectious etiology.
A 4-mm punch biopsy was taken from the left thigh and revealed a predominately lymphocytic infiltrate with rare eosinophils and erythrocyte precursors (Figure 3). Immunohistochemical staining was performed showing that the majority of the lymphocytes represented T lymphocytes, which stained positive for CD45 and CD3 and negative for S-100, CD1a, CD30, and CD117. There were scattered CD34+ cells, and scattered cells stained positive for myeloperoxidase. No significant CD20 immunoreactivity was noted. There were scattered eosinophils and rare normoblasts but no megakaryocytes. A complete blood cell count (CBC) with differential and reticulocyte count was within reference range.
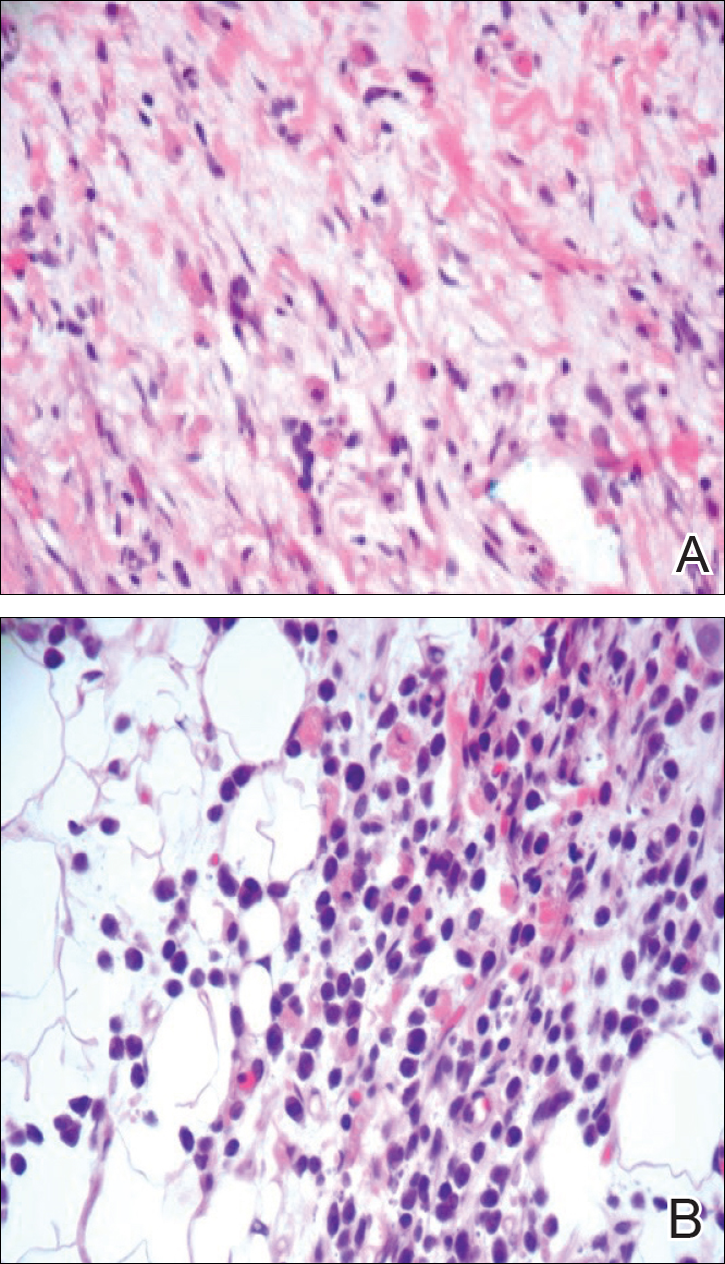
At 1-, 3-, 8-, 12-, and 28-week follow-up visits, the patient continued to grow and feed appropriately. No new lesions developed during this time, and the preexisting lesions continued to fade into slightly hyperpigmented patches without induration (Figure 4). At 6 months of age, a CBC performed at the time of an upper respiratory infection and otitis media revealed normocytic anemia with a hemoglobin level of 9.9 g/dL (reference range, 14.0–17.5 g/dL), a reticulocyte count of 0.8% (reference range, 0.5%–1.5%), and a lactate dehydrogenase level of 424 U/L (reference range, 100–200 U/L). All red blood cell (RBC) indices were within reference range. Flow cytometry, eosin-5-maleimide, and ektacytometry were performed with results consistent with mild hereditary spherocytosis.
Comment
Dermal extramedullary hematopoiesis is a normal component of embryologic development up until the fifth month of gestation.1 The term blueberry muffin rash typically is used to describe the cutaneous manifestations of extramedullary hematopoiesis, which commonly is caused by a TORCH infection or hematologic dyscrasia.2 It has been suggested that the term be expanded to include neoplastic processes (eg, neuroblastomas) and vascular processes (eg, multiple hemangiomas, blue rubber bleb nevus syndrome, glomangiomas, multifocal lymphangionendotheliomatosis), which although not associated with an extramedullary hematopoiesis, can clinically resemble a blueberry muffin rash.
Because of the potential for serious systemic complications, a cause must be sought for all newborns presenting with a blueberry muffin rash. Our patient’s lack of cardiovascular, otic, and ocular involvement combined with a negative TORCH screen and normal CBC strongly suggested against a TORCH infection. In addition, a normal bone survey and CBC, as well as a lack of petechiae, ecchymoses, and hepatosplenomegaly, were evidence against congenital leukemia.3 With the spontaneously resolving lesions and apparent clinical resolution, a bone marrow biopsy was not performed. The skin biopsy revealed negative staining for S-100 and CD1a, making the diagnosis of congenital self-healing reticulohistiocytosis unlikely. No panniculitis was noted and calcium levels were normal, ruling out subcutaneous fat necrosis of the newborn. The predominantly T-cell lymphocytic infiltrate demonstrated on skin biopsy led us to a differential diagnosis of aleukemic leukemia cutis versus idiopathic dermal extramedullary hematopoiesis; however, normocytic anemia was later identified when the patient’s hemoglobin level dropped to 9.9 g/dL. The abnormal eosin-5-maleimide and ektacytometry results unmasked a hereditary spherocytosis.
Hereditary spherocytosis typically is inherited in an autosomal-dominant manner and may be caused by mutations in ankyrin-1, band 3, spectrin, or protein 4.2 on the erythrocyte membrane. It is the third leading cause of hemolytic anemia in newborns and the leading cause of direct Coombs-negative hemolytic anemia requiring blood transfusion in neonates. It is most common in neonates of Northern European ancestry, affecting 1 in every 1000 to 2000 births.4 Presentation may range from asymptomatic to severe anemia with hydrops fetalis. Most neonates have an elevated mean corpuscular hemoglobin and low mean corpuscular volume. Acute illness may cause hemolytic or aplastic crises, possibly explaining our patient’s normocytic anemia discovered on a CBC during an episode of an upper respiratory infection and otitis media.
Treatment options for hereditary spherocytosis include phototherapy for jaundiced neonates, folate supplementation, packed erythrocyte transfusions for symptomatic anemia, and recombinant erythropoietin in neonates.4 Splenectomy is curative for the majority of patients and requires immunization against Streptococcus pneumoniae, Haemophilus influenzae type b, and Neisseria meningitidis several weeks preoperatively. Patients with symptomatic gallstones may be treated with cholecystectomy at the time of splenectomy or by laparoscopic cholecystectomy, endoscopic sphincterotomy, cholecystostomy, or extracorporeal cholecystolithotripsy.5
Although a PubMed search of articles indexed for MEDLINE using the terms dermal hematopoiesis, extramedullary hematopoiesis, hereditary spherocytosis, and blueberry muffin rash yielded only 1 other known case of blueberry muffin rash caused by hereditary spherocytosis,6 other case reports demonstrate extramedullary hematopoiesis in hereditary spherocytosis patients in locations other than the skin. Calhoun et al7 described a case of a 9-year-old boy with hereditary spherocytosis who presented with jaundice. Pathologic examination revealed a 5-cm suprarenal mass demonstrating extramedullary hematopoiesis.7 A case reported by Xiros et al8 described a 64-year-old man with a history of hereditary spherocytosis who presented with hemothorax from paravertebral extramedullary hematopoiesis. De Backer et al9 reported a case of a 60-year-old man diagnosed with hereditary spherocytosis after an abnormal CBC who was subsequently found to have paravertebral masses containing extramedullary hematopoiesis.
There is one known case of a blueberry muffin rash caused by hereditary spherocytosis.6 A female neonate was born at 38 weeks’ gestation with multiple petechiae and faint purpuric papules. Initial complications included intracranial ventricular hemorrhage, hyperbilirubinemia, and anemia requiring blood transfusions on the first day of life. TORCH titers were negative and a skin biopsy demonstrated a diffuse infiltrate of mature RBCs, normoblasts, and pronormoblasts in the reticular dermis. She was healthy until 3 months of age when she had several days of vomiting and diarrhea. Laboratory workup revealed a hematocrit level of 20.5% (reference range, 41%–50%); a reticulocyte count of 22.6% (reference range, 0.5%–1.5%); and a peripheral blood smear demonstrating polychromatophilia, anisocytosis, and spherocytosis. She was then diagnosed with hereditary spherocytosis.6
Hereditary spherocytosis is a known, albeit rare, cause of extramedullary hematopoiesis presenting as blueberry muffin rash. Patients with mild hereditary spherocytosis may have a compensated hemolysis without anemia or spherocytes on peripheral smear, which may explain the lack of severe hemolytic anemia or RBC-predominant pathology in our patient.5 Argyle and Zone6 proposed that severe hemolysis and hypoxia were the cause of extramedullary hematopoiesis in their patient. Because our patient did not experience a notable hemolytic episode until he had an upper respiratory infection and otitis media at 6 months of age, the pathophysiology is less clear; a compensated hemolytic process may underlie the extramedullary hematopoiesis and normal RBC indices.
Regardless of the precise cause of extramedullary hematopoiesis in our patient, this case of a T lymphocyte–dominant cutaneous infiltrate in a patient with mild hereditary spherocytosis is exceptionally rare and leads us to consider that perhaps there are causes of this pathology that are unknown to us.
- Zhang IH, Zane LT, Braun BS, et al. Congenital leukemia cutis with subsequent development of leukemia. J Am Acad Dermatol. 2006;54(2 suppl):S22–S27.
- Karmegaraj B, Vijayakumar S, Ramanathan R, et al. Extramedullary haematopoiesis resembling a blueberry muffin, in a neonate. BMJ Case Rep. pii: bcr2014208473. doi: 10.1136/bcr-2014-208473.
- Handler MZ, Schwartz RA. Neonatal leukaemia cutis. J Eur Acad Dermatol Venereol. 2015;29:1884-1889.
- Christensen RD, Yaish HM, Gallagher PG. A pediatrician’s practical guide to diagnosing and treating hereditary spherocytosis in neonates. Pediatrics. 2015;135:1107-1114.
- Perrotta S, Gallagher PG, Mohandas N. Hereditary spherocytosis. Lancet. 2008;372:1411-1426.
- Argyle JC, Zone JJ. Dermal erythropoiesis in a neonate. Arch Dermatol. 1981;117:492-494.
- Calhoun SK, Murphy RC, Shariati N, et al. Extramedullary hematopoiesis in a child with hereditary spherocytosis: an uncommon cause of an adrenal mass. Pediatr Radiol. 2001;31:879-881.
- Xiros N, Economopoulos T, Papageorgiou E, et al. Massive hemothorax due to intrathoracic extramedullary hematopoiesis in a patient with hereditary spherocytosis. Ann Hematol. 2001;80:38-40.
- De Backer AI, Zachée P, Vanschoubroeck IJ, et al. Extramedullary paraspinal hematopoiesis in hereditary spherocytosis. JBR-BTR. 2002;85:206-208.
The term blueberry muffin rash historically was used to describe the cutaneous manifestations observed in congenital rubella. The term traditionally describes the result of a postnatal dermal extramedullary hematopoiesis. Today, TORCH (toxoplasmosis, other agents, rubella, cytomegalovirus, herpes) infections and plasma cell dyscrasias are all potential causes of extramedullary hematopoiesis. Herein, we present a unique case of a neonate born with a blueberry muffin rash secondary to extramedullary hematopoiesis induced by hereditary spherocytosis.
Case Report
The dermatology department was consulted to evaluate a 2-day-old male neonate born with a “rash.” The patient was born to a 34-year-old gravida 3, para 2, woman at 39 weeks’ gestation. The mother’s prenatal laboratory values were within reference range and ultrasounds were normal, and she was compliant with her prenatal care. She underwent a normal spontaneous vaginal delivery 3 hours after rupture of membranes without complication. The amniotic fluid and umbilical cord both were clear. There was no use of forceps or any other external aiding devices during the delivery. At the time of delivery, the consulting physician noted that the patient had “skin lesions from head to toe.”
The patient’s parents reported that the rash did not seem to cause any discomfort for the patient. In the 24 hours after birth, the parents reported that the erythema seemed to slightly fade. Physical examination revealed many scattered erythematous to violaceous, nonblanching papulonodules affecting the scalp (Figure 1), face, arms, hands (Figure 2A), back (Figure 2B), buttocks, legs, and feet. Some of the papulonodules were soft while others were firm and indurated. Several lesions had a yellowish hue with some overlying crust. There was no mucosal, genital, or ocular involvement. No erosions, ulcerations, petechiae, ecchymoses, or hepatosplenomegaly were noted on examination.
The patient was otherwise healthy with an Apgar score of 8/9 at 1 and 5 minutes. His birth weight, length, and head circumference were within normal limits. There was no evidence of ABO blood group or Rhesus factor incompatibility. His temperature, vital signs, laboratory values (including calcium level and TORCH titers, which included cytomegalovirus, rubella, toxoplasmosis, and herpes simplex virus), and review of systems all were within reference range. A bone survey of the skull, spine, ribs, arms, pelvis, legs, and feet was within normal limits.
The mother’s placenta was sent for pathology and revealed a lymphoplasmacytic chronic deciduitis and acute subchorionitis consistent with a nonspecific inflammatory response, unlikely to be from an infectious etiology.
A 4-mm punch biopsy was taken from the left thigh and revealed a predominately lymphocytic infiltrate with rare eosinophils and erythrocyte precursors (Figure 3). Immunohistochemical staining was performed showing that the majority of the lymphocytes represented T lymphocytes, which stained positive for CD45 and CD3 and negative for S-100, CD1a, CD30, and CD117. There were scattered CD34+ cells, and scattered cells stained positive for myeloperoxidase. No significant CD20 immunoreactivity was noted. There were scattered eosinophils and rare normoblasts but no megakaryocytes. A complete blood cell count (CBC) with differential and reticulocyte count was within reference range.
At 1-, 3-, 8-, 12-, and 28-week follow-up visits, the patient continued to grow and feed appropriately. No new lesions developed during this time, and the preexisting lesions continued to fade into slightly hyperpigmented patches without induration (Figure 4). At 6 months of age, a CBC performed at the time of an upper respiratory infection and otitis media revealed normocytic anemia with a hemoglobin level of 9.9 g/dL (reference range, 14.0–17.5 g/dL), a reticulocyte count of 0.8% (reference range, 0.5%–1.5%), and a lactate dehydrogenase level of 424 U/L (reference range, 100–200 U/L). All red blood cell (RBC) indices were within reference range. Flow cytometry, eosin-5-maleimide, and ektacytometry were performed with results consistent with mild hereditary spherocytosis.
Comment
Dermal extramedullary hematopoiesis is a normal component of embryologic development up until the fifth month of gestation.1 The term blueberry muffin rash typically is used to describe the cutaneous manifestations of extramedullary hematopoiesis, which commonly is caused by a TORCH infection or hematologic dyscrasia.2 It has been suggested that the term be expanded to include neoplastic processes (eg, neuroblastomas) and vascular processes (eg, multiple hemangiomas, blue rubber bleb nevus syndrome, glomangiomas, multifocal lymphangionendotheliomatosis), which although not associated with an extramedullary hematopoiesis, can clinically resemble a blueberry muffin rash.
Because of the potential for serious systemic complications, a cause must be sought for all newborns presenting with a blueberry muffin rash. Our patient’s lack of cardiovascular, otic, and ocular involvement combined with a negative TORCH screen and normal CBC strongly suggested against a TORCH infection. In addition, a normal bone survey and CBC, as well as a lack of petechiae, ecchymoses, and hepatosplenomegaly, were evidence against congenital leukemia.3 With the spontaneously resolving lesions and apparent clinical resolution, a bone marrow biopsy was not performed. The skin biopsy revealed negative staining for S-100 and CD1a, making the diagnosis of congenital self-healing reticulohistiocytosis unlikely. No panniculitis was noted and calcium levels were normal, ruling out subcutaneous fat necrosis of the newborn. The predominantly T-cell lymphocytic infiltrate demonstrated on skin biopsy led us to a differential diagnosis of aleukemic leukemia cutis versus idiopathic dermal extramedullary hematopoiesis; however, normocytic anemia was later identified when the patient’s hemoglobin level dropped to 9.9 g/dL. The abnormal eosin-5-maleimide and ektacytometry results unmasked a hereditary spherocytosis.
Hereditary spherocytosis typically is inherited in an autosomal-dominant manner and may be caused by mutations in ankyrin-1, band 3, spectrin, or protein 4.2 on the erythrocyte membrane. It is the third leading cause of hemolytic anemia in newborns and the leading cause of direct Coombs-negative hemolytic anemia requiring blood transfusion in neonates. It is most common in neonates of Northern European ancestry, affecting 1 in every 1000 to 2000 births.4 Presentation may range from asymptomatic to severe anemia with hydrops fetalis. Most neonates have an elevated mean corpuscular hemoglobin and low mean corpuscular volume. Acute illness may cause hemolytic or aplastic crises, possibly explaining our patient’s normocytic anemia discovered on a CBC during an episode of an upper respiratory infection and otitis media.
Treatment options for hereditary spherocytosis include phototherapy for jaundiced neonates, folate supplementation, packed erythrocyte transfusions for symptomatic anemia, and recombinant erythropoietin in neonates.4 Splenectomy is curative for the majority of patients and requires immunization against Streptococcus pneumoniae, Haemophilus influenzae type b, and Neisseria meningitidis several weeks preoperatively. Patients with symptomatic gallstones may be treated with cholecystectomy at the time of splenectomy or by laparoscopic cholecystectomy, endoscopic sphincterotomy, cholecystostomy, or extracorporeal cholecystolithotripsy.5
Although a PubMed search of articles indexed for MEDLINE using the terms dermal hematopoiesis, extramedullary hematopoiesis, hereditary spherocytosis, and blueberry muffin rash yielded only 1 other known case of blueberry muffin rash caused by hereditary spherocytosis,6 other case reports demonstrate extramedullary hematopoiesis in hereditary spherocytosis patients in locations other than the skin. Calhoun et al7 described a case of a 9-year-old boy with hereditary spherocytosis who presented with jaundice. Pathologic examination revealed a 5-cm suprarenal mass demonstrating extramedullary hematopoiesis.7 A case reported by Xiros et al8 described a 64-year-old man with a history of hereditary spherocytosis who presented with hemothorax from paravertebral extramedullary hematopoiesis. De Backer et al9 reported a case of a 60-year-old man diagnosed with hereditary spherocytosis after an abnormal CBC who was subsequently found to have paravertebral masses containing extramedullary hematopoiesis.
There is one known case of a blueberry muffin rash caused by hereditary spherocytosis.6 A female neonate was born at 38 weeks’ gestation with multiple petechiae and faint purpuric papules. Initial complications included intracranial ventricular hemorrhage, hyperbilirubinemia, and anemia requiring blood transfusions on the first day of life. TORCH titers were negative and a skin biopsy demonstrated a diffuse infiltrate of mature RBCs, normoblasts, and pronormoblasts in the reticular dermis. She was healthy until 3 months of age when she had several days of vomiting and diarrhea. Laboratory workup revealed a hematocrit level of 20.5% (reference range, 41%–50%); a reticulocyte count of 22.6% (reference range, 0.5%–1.5%); and a peripheral blood smear demonstrating polychromatophilia, anisocytosis, and spherocytosis. She was then diagnosed with hereditary spherocytosis.6
Hereditary spherocytosis is a known, albeit rare, cause of extramedullary hematopoiesis presenting as blueberry muffin rash. Patients with mild hereditary spherocytosis may have a compensated hemolysis without anemia or spherocytes on peripheral smear, which may explain the lack of severe hemolytic anemia or RBC-predominant pathology in our patient.5 Argyle and Zone6 proposed that severe hemolysis and hypoxia were the cause of extramedullary hematopoiesis in their patient. Because our patient did not experience a notable hemolytic episode until he had an upper respiratory infection and otitis media at 6 months of age, the pathophysiology is less clear; a compensated hemolytic process may underlie the extramedullary hematopoiesis and normal RBC indices.
Regardless of the precise cause of extramedullary hematopoiesis in our patient, this case of a T lymphocyte–dominant cutaneous infiltrate in a patient with mild hereditary spherocytosis is exceptionally rare and leads us to consider that perhaps there are causes of this pathology that are unknown to us.
The term blueberry muffin rash historically was used to describe the cutaneous manifestations observed in congenital rubella. The term traditionally describes the result of a postnatal dermal extramedullary hematopoiesis. Today, TORCH (toxoplasmosis, other agents, rubella, cytomegalovirus, herpes) infections and plasma cell dyscrasias are all potential causes of extramedullary hematopoiesis. Herein, we present a unique case of a neonate born with a blueberry muffin rash secondary to extramedullary hematopoiesis induced by hereditary spherocytosis.
Case Report
The dermatology department was consulted to evaluate a 2-day-old male neonate born with a “rash.” The patient was born to a 34-year-old gravida 3, para 2, woman at 39 weeks’ gestation. The mother’s prenatal laboratory values were within reference range and ultrasounds were normal, and she was compliant with her prenatal care. She underwent a normal spontaneous vaginal delivery 3 hours after rupture of membranes without complication. The amniotic fluid and umbilical cord both were clear. There was no use of forceps or any other external aiding devices during the delivery. At the time of delivery, the consulting physician noted that the patient had “skin lesions from head to toe.”
The patient’s parents reported that the rash did not seem to cause any discomfort for the patient. In the 24 hours after birth, the parents reported that the erythema seemed to slightly fade. Physical examination revealed many scattered erythematous to violaceous, nonblanching papulonodules affecting the scalp (Figure 1), face, arms, hands (Figure 2A), back (Figure 2B), buttocks, legs, and feet. Some of the papulonodules were soft while others were firm and indurated. Several lesions had a yellowish hue with some overlying crust. There was no mucosal, genital, or ocular involvement. No erosions, ulcerations, petechiae, ecchymoses, or hepatosplenomegaly were noted on examination.
The patient was otherwise healthy with an Apgar score of 8/9 at 1 and 5 minutes. His birth weight, length, and head circumference were within normal limits. There was no evidence of ABO blood group or Rhesus factor incompatibility. His temperature, vital signs, laboratory values (including calcium level and TORCH titers, which included cytomegalovirus, rubella, toxoplasmosis, and herpes simplex virus), and review of systems all were within reference range. A bone survey of the skull, spine, ribs, arms, pelvis, legs, and feet was within normal limits.
The mother’s placenta was sent for pathology and revealed a lymphoplasmacytic chronic deciduitis and acute subchorionitis consistent with a nonspecific inflammatory response, unlikely to be from an infectious etiology.
A 4-mm punch biopsy was taken from the left thigh and revealed a predominately lymphocytic infiltrate with rare eosinophils and erythrocyte precursors (Figure 3). Immunohistochemical staining was performed showing that the majority of the lymphocytes represented T lymphocytes, which stained positive for CD45 and CD3 and negative for S-100, CD1a, CD30, and CD117. There were scattered CD34+ cells, and scattered cells stained positive for myeloperoxidase. No significant CD20 immunoreactivity was noted. There were scattered eosinophils and rare normoblasts but no megakaryocytes. A complete blood cell count (CBC) with differential and reticulocyte count was within reference range.
At 1-, 3-, 8-, 12-, and 28-week follow-up visits, the patient continued to grow and feed appropriately. No new lesions developed during this time, and the preexisting lesions continued to fade into slightly hyperpigmented patches without induration (Figure 4). At 6 months of age, a CBC performed at the time of an upper respiratory infection and otitis media revealed normocytic anemia with a hemoglobin level of 9.9 g/dL (reference range, 14.0–17.5 g/dL), a reticulocyte count of 0.8% (reference range, 0.5%–1.5%), and a lactate dehydrogenase level of 424 U/L (reference range, 100–200 U/L). All red blood cell (RBC) indices were within reference range. Flow cytometry, eosin-5-maleimide, and ektacytometry were performed with results consistent with mild hereditary spherocytosis.
Comment
Dermal extramedullary hematopoiesis is a normal component of embryologic development up until the fifth month of gestation.1 The term blueberry muffin rash typically is used to describe the cutaneous manifestations of extramedullary hematopoiesis, which commonly is caused by a TORCH infection or hematologic dyscrasia.2 It has been suggested that the term be expanded to include neoplastic processes (eg, neuroblastomas) and vascular processes (eg, multiple hemangiomas, blue rubber bleb nevus syndrome, glomangiomas, multifocal lymphangionendotheliomatosis), which although not associated with an extramedullary hematopoiesis, can clinically resemble a blueberry muffin rash.
Because of the potential for serious systemic complications, a cause must be sought for all newborns presenting with a blueberry muffin rash. Our patient’s lack of cardiovascular, otic, and ocular involvement combined with a negative TORCH screen and normal CBC strongly suggested against a TORCH infection. In addition, a normal bone survey and CBC, as well as a lack of petechiae, ecchymoses, and hepatosplenomegaly, were evidence against congenital leukemia.3 With the spontaneously resolving lesions and apparent clinical resolution, a bone marrow biopsy was not performed. The skin biopsy revealed negative staining for S-100 and CD1a, making the diagnosis of congenital self-healing reticulohistiocytosis unlikely. No panniculitis was noted and calcium levels were normal, ruling out subcutaneous fat necrosis of the newborn. The predominantly T-cell lymphocytic infiltrate demonstrated on skin biopsy led us to a differential diagnosis of aleukemic leukemia cutis versus idiopathic dermal extramedullary hematopoiesis; however, normocytic anemia was later identified when the patient’s hemoglobin level dropped to 9.9 g/dL. The abnormal eosin-5-maleimide and ektacytometry results unmasked a hereditary spherocytosis.
Hereditary spherocytosis typically is inherited in an autosomal-dominant manner and may be caused by mutations in ankyrin-1, band 3, spectrin, or protein 4.2 on the erythrocyte membrane. It is the third leading cause of hemolytic anemia in newborns and the leading cause of direct Coombs-negative hemolytic anemia requiring blood transfusion in neonates. It is most common in neonates of Northern European ancestry, affecting 1 in every 1000 to 2000 births.4 Presentation may range from asymptomatic to severe anemia with hydrops fetalis. Most neonates have an elevated mean corpuscular hemoglobin and low mean corpuscular volume. Acute illness may cause hemolytic or aplastic crises, possibly explaining our patient’s normocytic anemia discovered on a CBC during an episode of an upper respiratory infection and otitis media.
Treatment options for hereditary spherocytosis include phototherapy for jaundiced neonates, folate supplementation, packed erythrocyte transfusions for symptomatic anemia, and recombinant erythropoietin in neonates.4 Splenectomy is curative for the majority of patients and requires immunization against Streptococcus pneumoniae, Haemophilus influenzae type b, and Neisseria meningitidis several weeks preoperatively. Patients with symptomatic gallstones may be treated with cholecystectomy at the time of splenectomy or by laparoscopic cholecystectomy, endoscopic sphincterotomy, cholecystostomy, or extracorporeal cholecystolithotripsy.5
Although a PubMed search of articles indexed for MEDLINE using the terms dermal hematopoiesis, extramedullary hematopoiesis, hereditary spherocytosis, and blueberry muffin rash yielded only 1 other known case of blueberry muffin rash caused by hereditary spherocytosis,6 other case reports demonstrate extramedullary hematopoiesis in hereditary spherocytosis patients in locations other than the skin. Calhoun et al7 described a case of a 9-year-old boy with hereditary spherocytosis who presented with jaundice. Pathologic examination revealed a 5-cm suprarenal mass demonstrating extramedullary hematopoiesis.7 A case reported by Xiros et al8 described a 64-year-old man with a history of hereditary spherocytosis who presented with hemothorax from paravertebral extramedullary hematopoiesis. De Backer et al9 reported a case of a 60-year-old man diagnosed with hereditary spherocytosis after an abnormal CBC who was subsequently found to have paravertebral masses containing extramedullary hematopoiesis.
There is one known case of a blueberry muffin rash caused by hereditary spherocytosis.6 A female neonate was born at 38 weeks’ gestation with multiple petechiae and faint purpuric papules. Initial complications included intracranial ventricular hemorrhage, hyperbilirubinemia, and anemia requiring blood transfusions on the first day of life. TORCH titers were negative and a skin biopsy demonstrated a diffuse infiltrate of mature RBCs, normoblasts, and pronormoblasts in the reticular dermis. She was healthy until 3 months of age when she had several days of vomiting and diarrhea. Laboratory workup revealed a hematocrit level of 20.5% (reference range, 41%–50%); a reticulocyte count of 22.6% (reference range, 0.5%–1.5%); and a peripheral blood smear demonstrating polychromatophilia, anisocytosis, and spherocytosis. She was then diagnosed with hereditary spherocytosis.6
Hereditary spherocytosis is a known, albeit rare, cause of extramedullary hematopoiesis presenting as blueberry muffin rash. Patients with mild hereditary spherocytosis may have a compensated hemolysis without anemia or spherocytes on peripheral smear, which may explain the lack of severe hemolytic anemia or RBC-predominant pathology in our patient.5 Argyle and Zone6 proposed that severe hemolysis and hypoxia were the cause of extramedullary hematopoiesis in their patient. Because our patient did not experience a notable hemolytic episode until he had an upper respiratory infection and otitis media at 6 months of age, the pathophysiology is less clear; a compensated hemolytic process may underlie the extramedullary hematopoiesis and normal RBC indices.
Regardless of the precise cause of extramedullary hematopoiesis in our patient, this case of a T lymphocyte–dominant cutaneous infiltrate in a patient with mild hereditary spherocytosis is exceptionally rare and leads us to consider that perhaps there are causes of this pathology that are unknown to us.
- Zhang IH, Zane LT, Braun BS, et al. Congenital leukemia cutis with subsequent development of leukemia. J Am Acad Dermatol. 2006;54(2 suppl):S22–S27.
- Karmegaraj B, Vijayakumar S, Ramanathan R, et al. Extramedullary haematopoiesis resembling a blueberry muffin, in a neonate. BMJ Case Rep. pii: bcr2014208473. doi: 10.1136/bcr-2014-208473.
- Handler MZ, Schwartz RA. Neonatal leukaemia cutis. J Eur Acad Dermatol Venereol. 2015;29:1884-1889.
- Christensen RD, Yaish HM, Gallagher PG. A pediatrician’s practical guide to diagnosing and treating hereditary spherocytosis in neonates. Pediatrics. 2015;135:1107-1114.
- Perrotta S, Gallagher PG, Mohandas N. Hereditary spherocytosis. Lancet. 2008;372:1411-1426.
- Argyle JC, Zone JJ. Dermal erythropoiesis in a neonate. Arch Dermatol. 1981;117:492-494.
- Calhoun SK, Murphy RC, Shariati N, et al. Extramedullary hematopoiesis in a child with hereditary spherocytosis: an uncommon cause of an adrenal mass. Pediatr Radiol. 2001;31:879-881.
- Xiros N, Economopoulos T, Papageorgiou E, et al. Massive hemothorax due to intrathoracic extramedullary hematopoiesis in a patient with hereditary spherocytosis. Ann Hematol. 2001;80:38-40.
- De Backer AI, Zachée P, Vanschoubroeck IJ, et al. Extramedullary paraspinal hematopoiesis in hereditary spherocytosis. JBR-BTR. 2002;85:206-208.
- Zhang IH, Zane LT, Braun BS, et al. Congenital leukemia cutis with subsequent development of leukemia. J Am Acad Dermatol. 2006;54(2 suppl):S22–S27.
- Karmegaraj B, Vijayakumar S, Ramanathan R, et al. Extramedullary haematopoiesis resembling a blueberry muffin, in a neonate. BMJ Case Rep. pii: bcr2014208473. doi: 10.1136/bcr-2014-208473.
- Handler MZ, Schwartz RA. Neonatal leukaemia cutis. J Eur Acad Dermatol Venereol. 2015;29:1884-1889.
- Christensen RD, Yaish HM, Gallagher PG. A pediatrician’s practical guide to diagnosing and treating hereditary spherocytosis in neonates. Pediatrics. 2015;135:1107-1114.
- Perrotta S, Gallagher PG, Mohandas N. Hereditary spherocytosis. Lancet. 2008;372:1411-1426.
- Argyle JC, Zone JJ. Dermal erythropoiesis in a neonate. Arch Dermatol. 1981;117:492-494.
- Calhoun SK, Murphy RC, Shariati N, et al. Extramedullary hematopoiesis in a child with hereditary spherocytosis: an uncommon cause of an adrenal mass. Pediatr Radiol. 2001;31:879-881.
- Xiros N, Economopoulos T, Papageorgiou E, et al. Massive hemothorax due to intrathoracic extramedullary hematopoiesis in a patient with hereditary spherocytosis. Ann Hematol. 2001;80:38-40.
- De Backer AI, Zachée P, Vanschoubroeck IJ, et al. Extramedullary paraspinal hematopoiesis in hereditary spherocytosis. JBR-BTR. 2002;85:206-208.
Practice Points
- The term blueberry muffin rash is used to describe the clinical presentation of dermal extramedullary hematopoiesis. The common culprits of this rash include a TORCH (toxoplasmosis, other agents, rubella, cytomegalovirus, herpes) infection or hematologic dyscrasia.
- Because of the potential for serious systemic complications, a cause must be sought for all newborns presenting with a blueberry muffin rash.
- Hereditary spherocytosis typically is inherited in an autosomal-dominant manner and may be caused by mutations in ankyrin-1, band 3, spectrin, or protein 4.2 on the erythrocyte membrane. It is the third leading cause of hemolytic anemia in newborns and the leading cause of direct Coombs-negative hemolytic anemia requiring blood transfusion in neonates.
- Treatment options for hereditary spherocytosis include phototherapy for jaundiced neonates, folate supplementation, packed erythrocyte transfusions for symptomatic anemia, and recombinant erythropoietin in neonates.
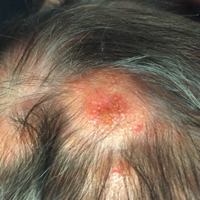
Growing Nodule on the Arm
The Diagnosis: Primary Cutaneous Anaplastic Large Cell Lymphoma
Primary cutaneous CD30+ lymphoproliferative disorders encompass lymphomatoid papulosis and primary cutaneous anaplastic large cell lymphoma (PCALCL) as well as borderline cases. Primary cutaneous anaplastic large cell lymphoma is a rare disease that is more common in white patients with slight male predominance and median age at diagnosis of 61 years.1 Prognosis is excellent, with a 90% survival rate at 10 years. Although lesions spontaneously regress in 6% to 22% of cases, complete resolution is rare.2 Clinically, the classic presentation is a solitary, rapidly growing, flesh-colored, erythematous nodule or plaque on the arms and legs or trunk, often with ulceration. Proper diagnosis requires clinical, histopathologic, and immunophenotypic correlation.
Histopathologic examination of PCALCL typically reveals large, atypical, Reed-Sternberg-like cells most commonly with anaplastic cytomorphology, but pleomorphic or immunoblastic morphology is not uncommon. Cells are in sheets or nodules, diffusely occupying the dermis and often the subcutaneous fat, with more than 75% of large cells expressing CD30.3 In addition to CD30 positivity, immunophenotype is classically CD4+, cutaneous lymphocyte-associated antigen positive, epithelial membrane antigen negative, and anaplastic lymphoma kinase negative; CD2, CD5, and CD3 expression is variable. Interestingly, in our case, there was a minor population of CD8+ cells. CD8 expression is seen in less than 5% of PCALCL cases; this phenotype is associated with an indolent disease with favorable prognosis.3 Of note, anaplastic lymphoma kinase positivity corresponding to a t(2;5) translocation is more suggestive of systemic anaplastic large cell lymphoma with secondary skin involvement and more commonly is seen in children. For reasons possibly related to mediators such as epidermal growth factor or transforming growth factor α from CD30+ cells, epidermal hyperplasia can be seen in PCALCL.4 The subsequent hyperkeratosis, crusting, and ulceration can be difficult to distinguish from lesions such as pyoderma gangrenosum, squamous cell carcinoma, arthropod bite, leukemia cutis, Merkel cell carcinoma (MCC), and metastatic breast cancer.
Skin involvement with leukemia is rare but most commonly is seen in acute myelogenous leukemia, specifically more mature forms such as acute myelomonocytic leukemia and acute monocytic leukemia. Approximately 10% to 20% of acute myelomonocytic leukemia cases have cutaneous involvement.5 Although there is a variety of potential skin lesions, the most common is a red-purple papule or nodule, sometimes with hemorrhage or ulceration, on the head, neck, and trunk. Leukemic infiltrates may arise from sites of prior trauma. Histopathology depends on the type of leukemia; however, general features include a normal epidermis without epidermotropism and perivascular, nodular, or diffuse infiltrate of neoplastic cells in the dermis, often with a Grenz zone (Figure 1). Compared to PCALCL, leukemia cutis shows sparing of the papillary dermis (Grenz zone), and the cells have more cytoplasm and show a different immunophenotype. The cells often are fragile and show crush artifact. Acute myelogenous leukemia often will show cytoplasmic granules; however, immature precursor cells may not have granules. The myeloid cells will stain with myeloperoxidase and chloroacetate. Positivity is seen for CD13, CD33, and CD68. Clinical correlation is important because other diseases with nodular or diffuse infiltrates of small cell infiltrates, such as extramedullary hematopoiesis and lymphoma, appear similar. Acute myelogenous leukemia is associated with neutrophilic dermatoses such as Sweet syndrome and pyoderma gangrenosum. Cutaneous eruption resolves with successful treatment of the leukemia.
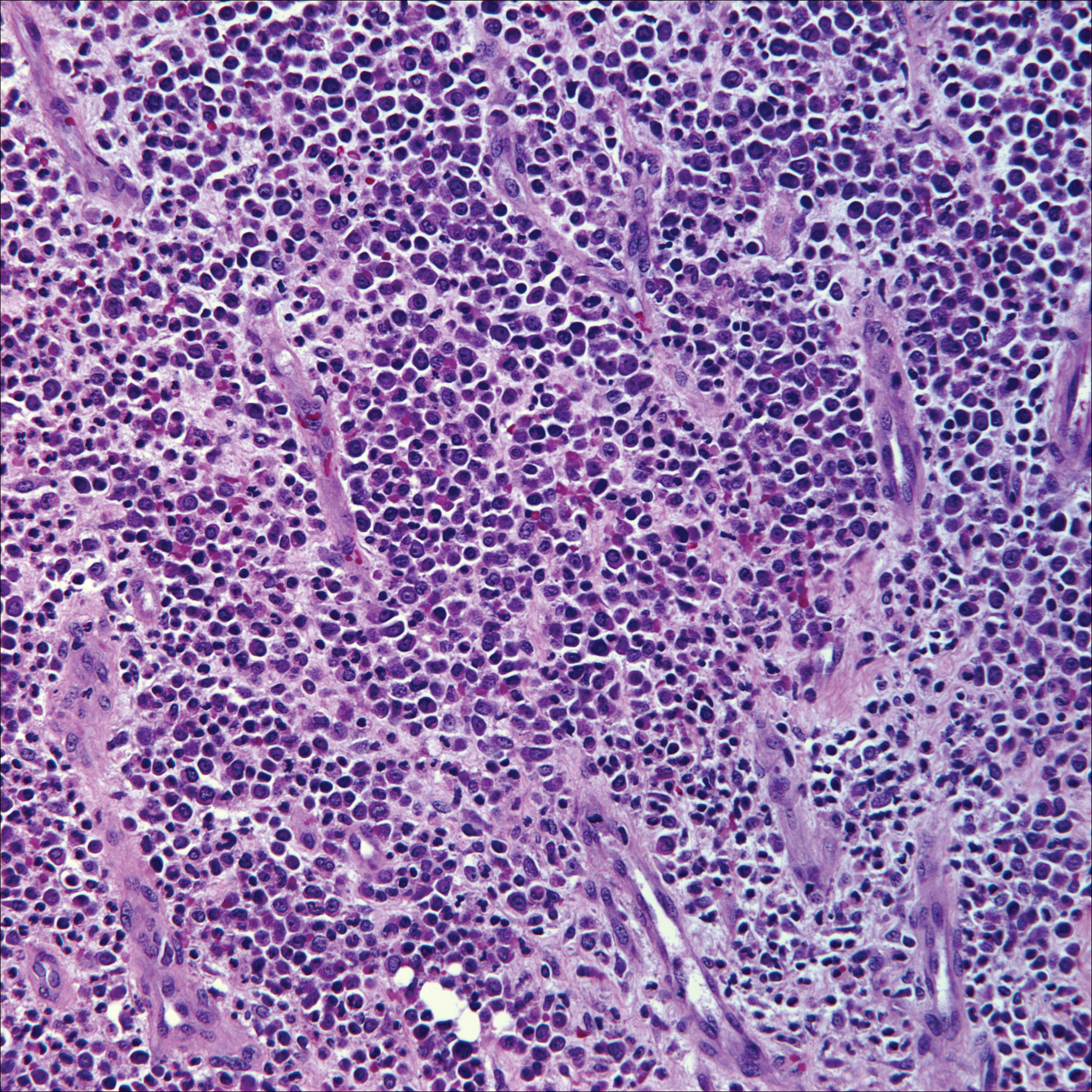
Breast cancer is the most common cancer to metastasize to the skin in women, accounting for 73% of cutaneous metastases, followed by melanoma, which is responsible for 11%.5 The classic presentation is an erythematous patch with spreading borders or a nodule on the trunk. Many cases of metastatic breast cancer with skin involvement may represent direct extension of the cancer into the skin. General histologic clues to cutaneous metastasis include well-circumscribed dermal or subcutaneous nodules of atypical cells with an increase in mitotic activity without connection to the epidermis. Tumor cells may show diffuse, nodular, or single file pattern and may exhibit areas of necrosis. Ductal carcinoma additionally may show ductal or glandular differentiation with surrounding desmoplasia (Figure 2). Immunohistochemistry typically is positive for cytokeratin (CK) 7, estrogen receptor/progesterone receptor, mammaglobin, and gross cystic disease fluid protein-15, and negative for CK20, CK5/6, and thyroid transcription factor-1.
Papulovesicular and nodular lesions appearing as an arthropod bite have been noted in hematologic malignancies, underscoring the importance of histopathology and clinical correlation. Arthropod bites commonly present as red papules, nodules, vesicles, or pustules at the site of the bite. Pseudolymphomatous nodules occasionally develop. Excoriations and further progression to persistent prurigo also may occur. Histopathology shows variable epidermal features including spongiosis, acanthosis, parakeratosis, dermal edema, and superficial and deep perivascular neutrophils (Figure 3). Additionally, lymphocytes sometimes with CD30 positivity may be seen. The presence of eosinophils in interstitial areas, especially in the deep dermis, is a useful clue.
Lack of staining for epithelial and neuroendocrine markers differentiates PCALCL from MCC; specifically CK20, an epithelial marker positive in more than 90% of MCC cases, excludes lymphoma.6 Merkel cell carcinoma presents as a solitary, quickly growing, red and often ulcerated nodule or plaque on the head, neck, or legs of elderly patients. The lesions often are in areas of sun damage. Histopathology classically shows a diffuse dermal infiltrate of monotonous round blue cells with a scant cytoplasmic rim and multiple inconspicuous nucleoli in nests, rosettes, or strands in the dermis. There are frequent mitotic figures. The cells are uniform and 2 to 3 times larger than mature lymphocytes. Single-cell necrosis and crush artifact is common. Epidermotropism or coexisting Bowenoid change also may be observed (Figure 4). The term primary neuroendocrine carcinoma of the skin is preferred over Merkel cell carcinoma because the tumor cells share similar morphology to the specialized touch receptor of the basal layer (Merkel cell), but no direct histogenetic relationship has been established.7,8
Immunohistochemistry is key to diagnosis because MCC stains for both epithelial and neuroendocrine markers. Positivity is seen for neuron-specific enolase, epithelial membrane antigen, neurofilament, synaptophysin, and chromogranin. Because the histology of MCC may resemble small cell carcinoma of the lung, staining for low-molecular-weight keratin such as CK20 and CK7 help to distinguish MCC. Merkel cell carcinoma typically is CK20+ and CK7-, while small cell carcinoma of the lung is the opposite.9 The tumor grows aggressively and metastasis is common, thus surgery is the primary approach, but adjuvant chemotherapy and radiation often are given in addition.
- Yu J, Blitzblau R, Decker R, et al. Analysis of primary CD30+ cutaneous lymphoproliferative disease and survival from the Surveillance, Epidemiology, and End Results database. J Clin Oncol. 2008;26:1483-1488.
- Liu HL, Hoppe RT, Kohler S, et al. CD30+ cutaneous lymphoproliferative disorders: the Stanford experience in lymphomatoid papulosis and primary cutaneous anaplastic large cell lymphoma. J Am Acad Dermatol. 2003;49:1049-1058.
- Nasit JG, Patel SC. Primary cutaneous CD8(+) CD30(+) anaplastic large cell lymphoma: an unusual case with a high Ki-67 index--a short review. Indian J Dermatol. 2015;60:373-377.
- Park J, Lee J, Lim Y, et al. Synchronous occurrence of primary cutaneous anaplastic large cell lymphoma and squamous cell carcinoma. Ann Dermatol. 2016;28:491-494.
- Marks JG Jr, Miller JJ. Lookingbill and Marks' Principles of Dermatology. 5th ed. Philadelphia, PA: Elsevier Saunders; 2013.
- Kudchadkar R, Gonzalez R, Lewis K, et al. A case of Merkel cell carcinoma. Oncology. 2008;22:322-328.
- Ratner D, Nelson BR, Brown MD, et al. Merkel cell carcinoma. J Am Acad Dermatol. 1993;29:143-156.
- Zur Hausen A, Rennspiess D, Winnepenninckx V, et al. Early B-cell differentiation in Merkel cell carcinomas: clues to cellular ancestry [published online April 10, 2013]. Cancer Res. 2013;73:4982-4987.
- Sidiropoulos M, Hanna W, Raphael SJ, et al. Expression of TdT in Merkel cell carcinoma and small cell lung carcinoma. Am J Clin Pathol. 2011;135:831-838.
The Diagnosis: Primary Cutaneous Anaplastic Large Cell Lymphoma
Primary cutaneous CD30+ lymphoproliferative disorders encompass lymphomatoid papulosis and primary cutaneous anaplastic large cell lymphoma (PCALCL) as well as borderline cases. Primary cutaneous anaplastic large cell lymphoma is a rare disease that is more common in white patients with slight male predominance and median age at diagnosis of 61 years.1 Prognosis is excellent, with a 90% survival rate at 10 years. Although lesions spontaneously regress in 6% to 22% of cases, complete resolution is rare.2 Clinically, the classic presentation is a solitary, rapidly growing, flesh-colored, erythematous nodule or plaque on the arms and legs or trunk, often with ulceration. Proper diagnosis requires clinical, histopathologic, and immunophenotypic correlation.
Histopathologic examination of PCALCL typically reveals large, atypical, Reed-Sternberg-like cells most commonly with anaplastic cytomorphology, but pleomorphic or immunoblastic morphology is not uncommon. Cells are in sheets or nodules, diffusely occupying the dermis and often the subcutaneous fat, with more than 75% of large cells expressing CD30.3 In addition to CD30 positivity, immunophenotype is classically CD4+, cutaneous lymphocyte-associated antigen positive, epithelial membrane antigen negative, and anaplastic lymphoma kinase negative; CD2, CD5, and CD3 expression is variable. Interestingly, in our case, there was a minor population of CD8+ cells. CD8 expression is seen in less than 5% of PCALCL cases; this phenotype is associated with an indolent disease with favorable prognosis.3 Of note, anaplastic lymphoma kinase positivity corresponding to a t(2;5) translocation is more suggestive of systemic anaplastic large cell lymphoma with secondary skin involvement and more commonly is seen in children. For reasons possibly related to mediators such as epidermal growth factor or transforming growth factor α from CD30+ cells, epidermal hyperplasia can be seen in PCALCL.4 The subsequent hyperkeratosis, crusting, and ulceration can be difficult to distinguish from lesions such as pyoderma gangrenosum, squamous cell carcinoma, arthropod bite, leukemia cutis, Merkel cell carcinoma (MCC), and metastatic breast cancer.
Skin involvement with leukemia is rare but most commonly is seen in acute myelogenous leukemia, specifically more mature forms such as acute myelomonocytic leukemia and acute monocytic leukemia. Approximately 10% to 20% of acute myelomonocytic leukemia cases have cutaneous involvement.5 Although there is a variety of potential skin lesions, the most common is a red-purple papule or nodule, sometimes with hemorrhage or ulceration, on the head, neck, and trunk. Leukemic infiltrates may arise from sites of prior trauma. Histopathology depends on the type of leukemia; however, general features include a normal epidermis without epidermotropism and perivascular, nodular, or diffuse infiltrate of neoplastic cells in the dermis, often with a Grenz zone (Figure 1). Compared to PCALCL, leukemia cutis shows sparing of the papillary dermis (Grenz zone), and the cells have more cytoplasm and show a different immunophenotype. The cells often are fragile and show crush artifact. Acute myelogenous leukemia often will show cytoplasmic granules; however, immature precursor cells may not have granules. The myeloid cells will stain with myeloperoxidase and chloroacetate. Positivity is seen for CD13, CD33, and CD68. Clinical correlation is important because other diseases with nodular or diffuse infiltrates of small cell infiltrates, such as extramedullary hematopoiesis and lymphoma, appear similar. Acute myelogenous leukemia is associated with neutrophilic dermatoses such as Sweet syndrome and pyoderma gangrenosum. Cutaneous eruption resolves with successful treatment of the leukemia.
Breast cancer is the most common cancer to metastasize to the skin in women, accounting for 73% of cutaneous metastases, followed by melanoma, which is responsible for 11%.5 The classic presentation is an erythematous patch with spreading borders or a nodule on the trunk. Many cases of metastatic breast cancer with skin involvement may represent direct extension of the cancer into the skin. General histologic clues to cutaneous metastasis include well-circumscribed dermal or subcutaneous nodules of atypical cells with an increase in mitotic activity without connection to the epidermis. Tumor cells may show diffuse, nodular, or single file pattern and may exhibit areas of necrosis. Ductal carcinoma additionally may show ductal or glandular differentiation with surrounding desmoplasia (Figure 2). Immunohistochemistry typically is positive for cytokeratin (CK) 7, estrogen receptor/progesterone receptor, mammaglobin, and gross cystic disease fluid protein-15, and negative for CK20, CK5/6, and thyroid transcription factor-1.
Papulovesicular and nodular lesions appearing as an arthropod bite have been noted in hematologic malignancies, underscoring the importance of histopathology and clinical correlation. Arthropod bites commonly present as red papules, nodules, vesicles, or pustules at the site of the bite. Pseudolymphomatous nodules occasionally develop. Excoriations and further progression to persistent prurigo also may occur. Histopathology shows variable epidermal features including spongiosis, acanthosis, parakeratosis, dermal edema, and superficial and deep perivascular neutrophils (Figure 3). Additionally, lymphocytes sometimes with CD30 positivity may be seen. The presence of eosinophils in interstitial areas, especially in the deep dermis, is a useful clue.
Lack of staining for epithelial and neuroendocrine markers differentiates PCALCL from MCC; specifically CK20, an epithelial marker positive in more than 90% of MCC cases, excludes lymphoma.6 Merkel cell carcinoma presents as a solitary, quickly growing, red and often ulcerated nodule or plaque on the head, neck, or legs of elderly patients. The lesions often are in areas of sun damage. Histopathology classically shows a diffuse dermal infiltrate of monotonous round blue cells with a scant cytoplasmic rim and multiple inconspicuous nucleoli in nests, rosettes, or strands in the dermis. There are frequent mitotic figures. The cells are uniform and 2 to 3 times larger than mature lymphocytes. Single-cell necrosis and crush artifact is common. Epidermotropism or coexisting Bowenoid change also may be observed (Figure 4). The term primary neuroendocrine carcinoma of the skin is preferred over Merkel cell carcinoma because the tumor cells share similar morphology to the specialized touch receptor of the basal layer (Merkel cell), but no direct histogenetic relationship has been established.7,8
Immunohistochemistry is key to diagnosis because MCC stains for both epithelial and neuroendocrine markers. Positivity is seen for neuron-specific enolase, epithelial membrane antigen, neurofilament, synaptophysin, and chromogranin. Because the histology of MCC may resemble small cell carcinoma of the lung, staining for low-molecular-weight keratin such as CK20 and CK7 help to distinguish MCC. Merkel cell carcinoma typically is CK20+ and CK7-, while small cell carcinoma of the lung is the opposite.9 The tumor grows aggressively and metastasis is common, thus surgery is the primary approach, but adjuvant chemotherapy and radiation often are given in addition.
The Diagnosis: Primary Cutaneous Anaplastic Large Cell Lymphoma
Primary cutaneous CD30+ lymphoproliferative disorders encompass lymphomatoid papulosis and primary cutaneous anaplastic large cell lymphoma (PCALCL) as well as borderline cases. Primary cutaneous anaplastic large cell lymphoma is a rare disease that is more common in white patients with slight male predominance and median age at diagnosis of 61 years.1 Prognosis is excellent, with a 90% survival rate at 10 years. Although lesions spontaneously regress in 6% to 22% of cases, complete resolution is rare.2 Clinically, the classic presentation is a solitary, rapidly growing, flesh-colored, erythematous nodule or plaque on the arms and legs or trunk, often with ulceration. Proper diagnosis requires clinical, histopathologic, and immunophenotypic correlation.
Histopathologic examination of PCALCL typically reveals large, atypical, Reed-Sternberg-like cells most commonly with anaplastic cytomorphology, but pleomorphic or immunoblastic morphology is not uncommon. Cells are in sheets or nodules, diffusely occupying the dermis and often the subcutaneous fat, with more than 75% of large cells expressing CD30.3 In addition to CD30 positivity, immunophenotype is classically CD4+, cutaneous lymphocyte-associated antigen positive, epithelial membrane antigen negative, and anaplastic lymphoma kinase negative; CD2, CD5, and CD3 expression is variable. Interestingly, in our case, there was a minor population of CD8+ cells. CD8 expression is seen in less than 5% of PCALCL cases; this phenotype is associated with an indolent disease with favorable prognosis.3 Of note, anaplastic lymphoma kinase positivity corresponding to a t(2;5) translocation is more suggestive of systemic anaplastic large cell lymphoma with secondary skin involvement and more commonly is seen in children. For reasons possibly related to mediators such as epidermal growth factor or transforming growth factor α from CD30+ cells, epidermal hyperplasia can be seen in PCALCL.4 The subsequent hyperkeratosis, crusting, and ulceration can be difficult to distinguish from lesions such as pyoderma gangrenosum, squamous cell carcinoma, arthropod bite, leukemia cutis, Merkel cell carcinoma (MCC), and metastatic breast cancer.
Skin involvement with leukemia is rare but most commonly is seen in acute myelogenous leukemia, specifically more mature forms such as acute myelomonocytic leukemia and acute monocytic leukemia. Approximately 10% to 20% of acute myelomonocytic leukemia cases have cutaneous involvement.5 Although there is a variety of potential skin lesions, the most common is a red-purple papule or nodule, sometimes with hemorrhage or ulceration, on the head, neck, and trunk. Leukemic infiltrates may arise from sites of prior trauma. Histopathology depends on the type of leukemia; however, general features include a normal epidermis without epidermotropism and perivascular, nodular, or diffuse infiltrate of neoplastic cells in the dermis, often with a Grenz zone (Figure 1). Compared to PCALCL, leukemia cutis shows sparing of the papillary dermis (Grenz zone), and the cells have more cytoplasm and show a different immunophenotype. The cells often are fragile and show crush artifact. Acute myelogenous leukemia often will show cytoplasmic granules; however, immature precursor cells may not have granules. The myeloid cells will stain with myeloperoxidase and chloroacetate. Positivity is seen for CD13, CD33, and CD68. Clinical correlation is important because other diseases with nodular or diffuse infiltrates of small cell infiltrates, such as extramedullary hematopoiesis and lymphoma, appear similar. Acute myelogenous leukemia is associated with neutrophilic dermatoses such as Sweet syndrome and pyoderma gangrenosum. Cutaneous eruption resolves with successful treatment of the leukemia.
Breast cancer is the most common cancer to metastasize to the skin in women, accounting for 73% of cutaneous metastases, followed by melanoma, which is responsible for 11%.5 The classic presentation is an erythematous patch with spreading borders or a nodule on the trunk. Many cases of metastatic breast cancer with skin involvement may represent direct extension of the cancer into the skin. General histologic clues to cutaneous metastasis include well-circumscribed dermal or subcutaneous nodules of atypical cells with an increase in mitotic activity without connection to the epidermis. Tumor cells may show diffuse, nodular, or single file pattern and may exhibit areas of necrosis. Ductal carcinoma additionally may show ductal or glandular differentiation with surrounding desmoplasia (Figure 2). Immunohistochemistry typically is positive for cytokeratin (CK) 7, estrogen receptor/progesterone receptor, mammaglobin, and gross cystic disease fluid protein-15, and negative for CK20, CK5/6, and thyroid transcription factor-1.
Papulovesicular and nodular lesions appearing as an arthropod bite have been noted in hematologic malignancies, underscoring the importance of histopathology and clinical correlation. Arthropod bites commonly present as red papules, nodules, vesicles, or pustules at the site of the bite. Pseudolymphomatous nodules occasionally develop. Excoriations and further progression to persistent prurigo also may occur. Histopathology shows variable epidermal features including spongiosis, acanthosis, parakeratosis, dermal edema, and superficial and deep perivascular neutrophils (Figure 3). Additionally, lymphocytes sometimes with CD30 positivity may be seen. The presence of eosinophils in interstitial areas, especially in the deep dermis, is a useful clue.
Lack of staining for epithelial and neuroendocrine markers differentiates PCALCL from MCC; specifically CK20, an epithelial marker positive in more than 90% of MCC cases, excludes lymphoma.6 Merkel cell carcinoma presents as a solitary, quickly growing, red and often ulcerated nodule or plaque on the head, neck, or legs of elderly patients. The lesions often are in areas of sun damage. Histopathology classically shows a diffuse dermal infiltrate of monotonous round blue cells with a scant cytoplasmic rim and multiple inconspicuous nucleoli in nests, rosettes, or strands in the dermis. There are frequent mitotic figures. The cells are uniform and 2 to 3 times larger than mature lymphocytes. Single-cell necrosis and crush artifact is common. Epidermotropism or coexisting Bowenoid change also may be observed (Figure 4). The term primary neuroendocrine carcinoma of the skin is preferred over Merkel cell carcinoma because the tumor cells share similar morphology to the specialized touch receptor of the basal layer (Merkel cell), but no direct histogenetic relationship has been established.7,8
Immunohistochemistry is key to diagnosis because MCC stains for both epithelial and neuroendocrine markers. Positivity is seen for neuron-specific enolase, epithelial membrane antigen, neurofilament, synaptophysin, and chromogranin. Because the histology of MCC may resemble small cell carcinoma of the lung, staining for low-molecular-weight keratin such as CK20 and CK7 help to distinguish MCC. Merkel cell carcinoma typically is CK20+ and CK7-, while small cell carcinoma of the lung is the opposite.9 The tumor grows aggressively and metastasis is common, thus surgery is the primary approach, but adjuvant chemotherapy and radiation often are given in addition.
- Yu J, Blitzblau R, Decker R, et al. Analysis of primary CD30+ cutaneous lymphoproliferative disease and survival from the Surveillance, Epidemiology, and End Results database. J Clin Oncol. 2008;26:1483-1488.
- Liu HL, Hoppe RT, Kohler S, et al. CD30+ cutaneous lymphoproliferative disorders: the Stanford experience in lymphomatoid papulosis and primary cutaneous anaplastic large cell lymphoma. J Am Acad Dermatol. 2003;49:1049-1058.
- Nasit JG, Patel SC. Primary cutaneous CD8(+) CD30(+) anaplastic large cell lymphoma: an unusual case with a high Ki-67 index--a short review. Indian J Dermatol. 2015;60:373-377.
- Park J, Lee J, Lim Y, et al. Synchronous occurrence of primary cutaneous anaplastic large cell lymphoma and squamous cell carcinoma. Ann Dermatol. 2016;28:491-494.
- Marks JG Jr, Miller JJ. Lookingbill and Marks' Principles of Dermatology. 5th ed. Philadelphia, PA: Elsevier Saunders; 2013.
- Kudchadkar R, Gonzalez R, Lewis K, et al. A case of Merkel cell carcinoma. Oncology. 2008;22:322-328.
- Ratner D, Nelson BR, Brown MD, et al. Merkel cell carcinoma. J Am Acad Dermatol. 1993;29:143-156.
- Zur Hausen A, Rennspiess D, Winnepenninckx V, et al. Early B-cell differentiation in Merkel cell carcinomas: clues to cellular ancestry [published online April 10, 2013]. Cancer Res. 2013;73:4982-4987.
- Sidiropoulos M, Hanna W, Raphael SJ, et al. Expression of TdT in Merkel cell carcinoma and small cell lung carcinoma. Am J Clin Pathol. 2011;135:831-838.
- Yu J, Blitzblau R, Decker R, et al. Analysis of primary CD30+ cutaneous lymphoproliferative disease and survival from the Surveillance, Epidemiology, and End Results database. J Clin Oncol. 2008;26:1483-1488.
- Liu HL, Hoppe RT, Kohler S, et al. CD30+ cutaneous lymphoproliferative disorders: the Stanford experience in lymphomatoid papulosis and primary cutaneous anaplastic large cell lymphoma. J Am Acad Dermatol. 2003;49:1049-1058.
- Nasit JG, Patel SC. Primary cutaneous CD8(+) CD30(+) anaplastic large cell lymphoma: an unusual case with a high Ki-67 index--a short review. Indian J Dermatol. 2015;60:373-377.
- Park J, Lee J, Lim Y, et al. Synchronous occurrence of primary cutaneous anaplastic large cell lymphoma and squamous cell carcinoma. Ann Dermatol. 2016;28:491-494.
- Marks JG Jr, Miller JJ. Lookingbill and Marks' Principles of Dermatology. 5th ed. Philadelphia, PA: Elsevier Saunders; 2013.
- Kudchadkar R, Gonzalez R, Lewis K, et al. A case of Merkel cell carcinoma. Oncology. 2008;22:322-328.
- Ratner D, Nelson BR, Brown MD, et al. Merkel cell carcinoma. J Am Acad Dermatol. 1993;29:143-156.
- Zur Hausen A, Rennspiess D, Winnepenninckx V, et al. Early B-cell differentiation in Merkel cell carcinomas: clues to cellular ancestry [published online April 10, 2013]. Cancer Res. 2013;73:4982-4987.
- Sidiropoulos M, Hanna W, Raphael SJ, et al. Expression of TdT in Merkel cell carcinoma and small cell lung carcinoma. Am J Clin Pathol. 2011;135:831-838.
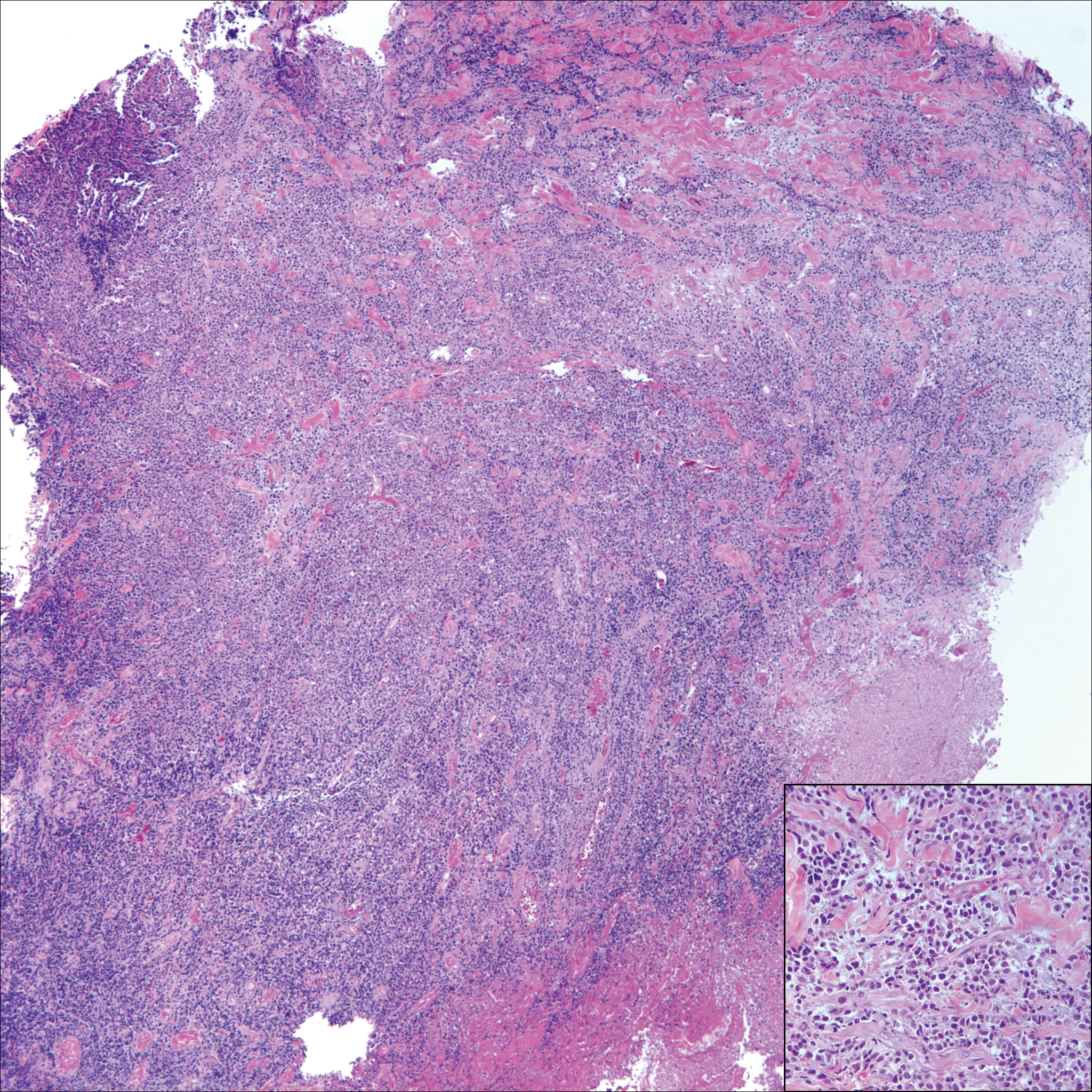
A 65-year-old white woman presented with an asymptomatic bump on the left upper arm of 4 months' duration that arose following a cat scratch. Physical examination was notable for a 35×30-mm, firm, ulcerated, exophytic nodule. Histologic examination demonstrated an ulcerated epidermis and a dense basophilic infiltrate occupying the entire dermis and extending to the subcutaneous tissue. Higher magnification (inset) demonstrated a pleomorphic population of medium- to large-sized discohesive round cells containing variable amounts of slightly eosinophilic cytoplasm, irregular nuclear contours, and prominent nucleoli. Scattered atypical mitotic figures were identified. CD30, CD4, leukocyte common antigen, and Ki-67 immunostains were strongly and diffusely positive. Notable negative stains included anaplastic lymphoma kinase, synaptophysin, epithelial membrane antigen, neuron-specific enolase, CD20, and S-100.
Mobile Medical Apps for Patient Education: A Graded Review of Available Dermatology Apps
According to industry estimates, roughly 64% of US adults were smartphone users in 2015.1 Smartphones enable users to utilize mobile applications (apps) that can perform a variety of functions in many categories, including business, music, photography, entertainment, education, social networking, travel, and lifestyle. The widespread adoption and use of mobile apps has implications for medical practice. Mobile apps have the capability to serve as information sources for patients, educational tools for students, and diagnostic aids for physicians.2 Consequently, a number of medical and health care–oriented apps have already been developed3 and are increasingly utilized by patients and providers.4
Given its visual nature, dermatology is particularly amenable to the integration of mobile medical apps. A study by Brewer et al5 identified more than 229 dermatology-related apps in categories ranging from general dermatology reference, self-surveillance and diagnosis, disease guides, educational aids, sunscreen and UV recommendations, and teledermatology. Patients served as the target audience and principal consumers of more than half of these dermatology apps.5
Mobile medical and health care apps demonstrate great potential for serving as valuable information sources for patients with dermatologic conditions; however, the content, functions, accuracy, and educational value of dermatology mobile apps are not well characterized, making it difficult for patients and health care providers to select and recommend appropriate apps.6 In this study, we created a rubric to objectively grade 44 publicly available mobile dermatology apps with the primary focus of patient education.
Methods
We conducted a search of dermatology-related educational mobile apps that were publicly available via the App Store (Apple Inc) from January 2016 to November 2016. (The pricing, availability, and other features of these apps may have changed since the study period.) The following search terms were used: dermatology, dermoscopy, melanoma, skin cancer, psoriasis, rosacea, acne, eczema, dermal fillers, and Mohs surgery. We excluded apps that were not in English; had a solely commercial focus; were mobile textbooks or scientific journals; were used to provide teledermatology services with no educational purpose; were solely focused on homeopathic, alternative, and/or complementary medicine; or were intended primarily as a reference for students or health care professionals. Our search yielded 44 apps with patient education as a primary objective. The apps were divided into 6 categories based on their focus: general dermatology, cosmetic dermatology, acne, eczema, psoriasis, and skin cancer.
Each app was reviewed using a quantified grading rubric developed by the researchers. In a prior evaluation, Handel7 reviewed 35 health and wellness mobile apps utilizing the categories of ease of use, reliability, quality, scope of information, and aesthetics.4 These criteria were modified and adapted for the purposes of this study, and a 4-point scale was applied to each criterion. The final criteria were (1) educational objectives, (2) content, (3) accuracy, (4) design, and (5) conflict of interest. The quantified grading rubric is described in Table 1.
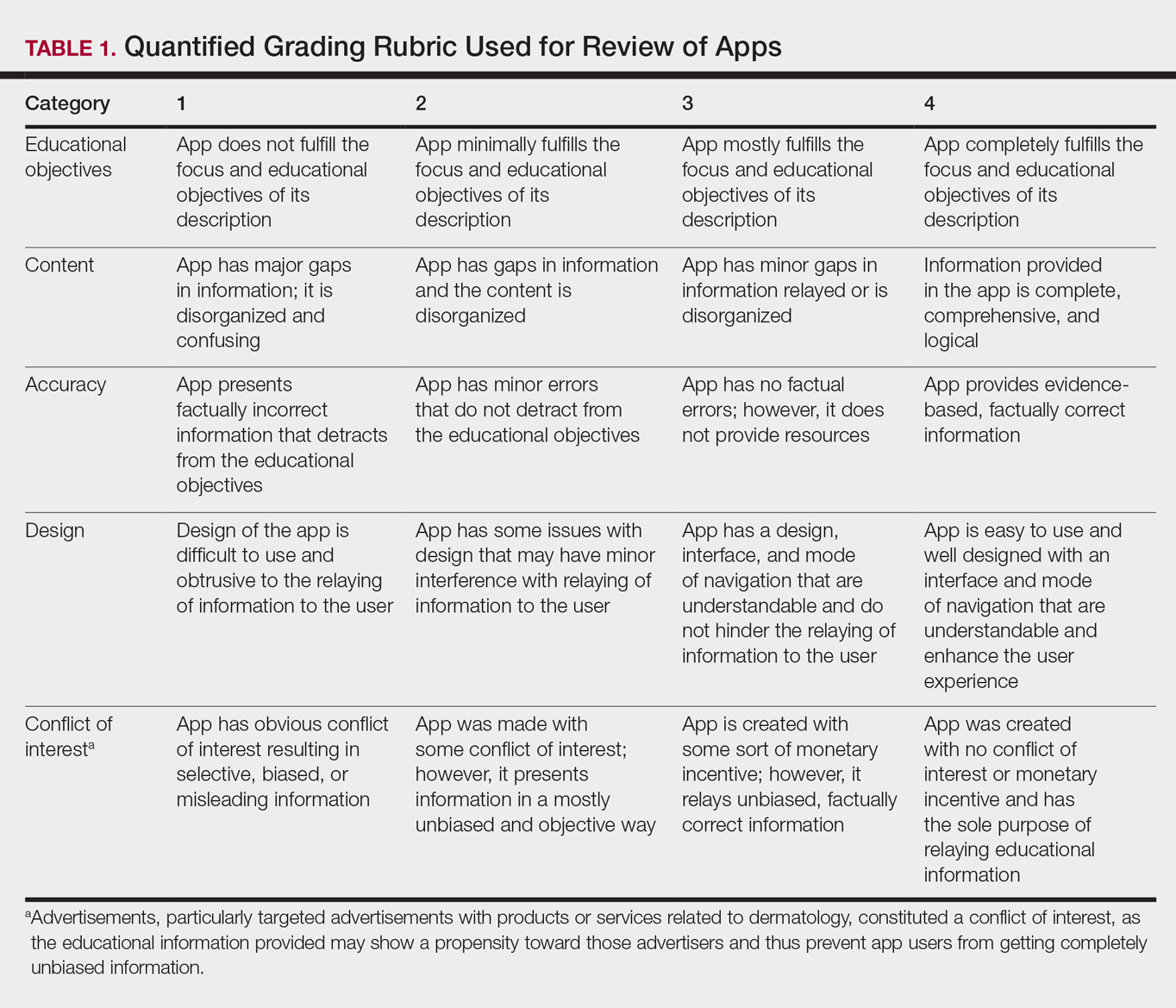
Results
The possible range of scores based on the grading rubric was 5 to 20. The actual range of scores was 8 to 19 (Table 2). The 44 reviewed apps were categorized by topic as acne, cosmetic dermatology, eczema, general dermatology, psoriasis, or skin cancer. A sample of 15 apps selected to represent the distribution of scores and their grading on the rubric are presented in Table 3.
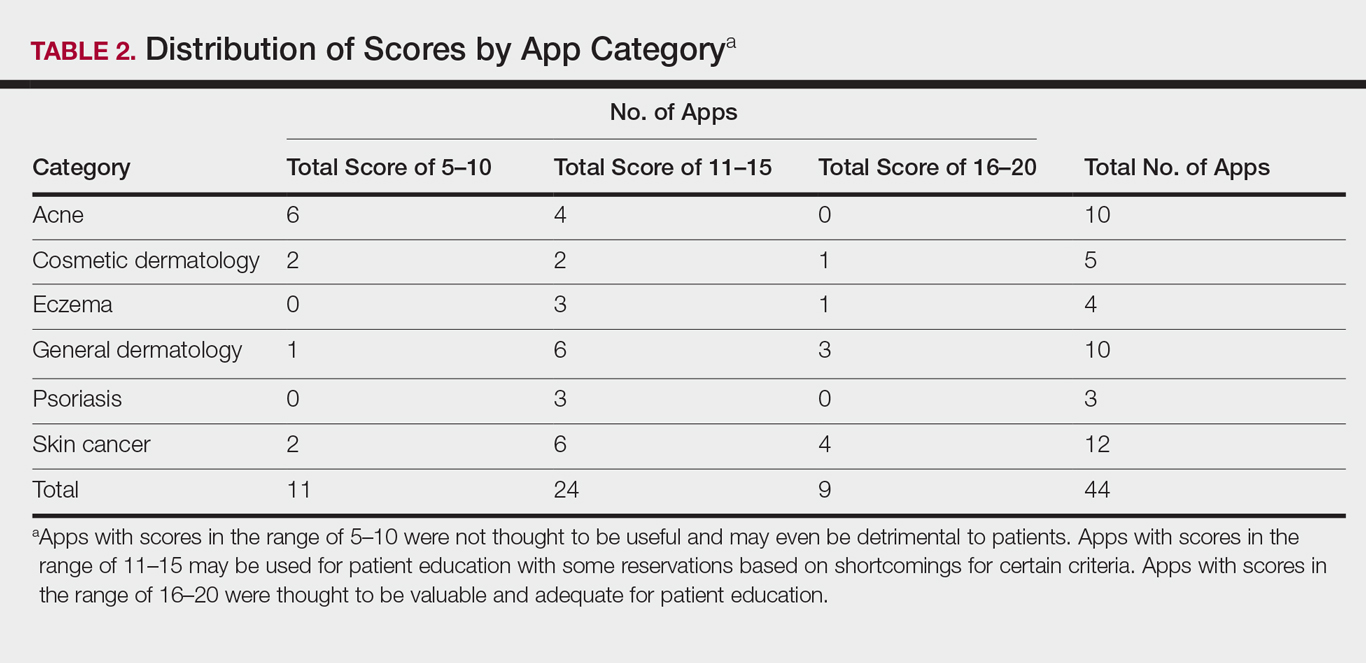
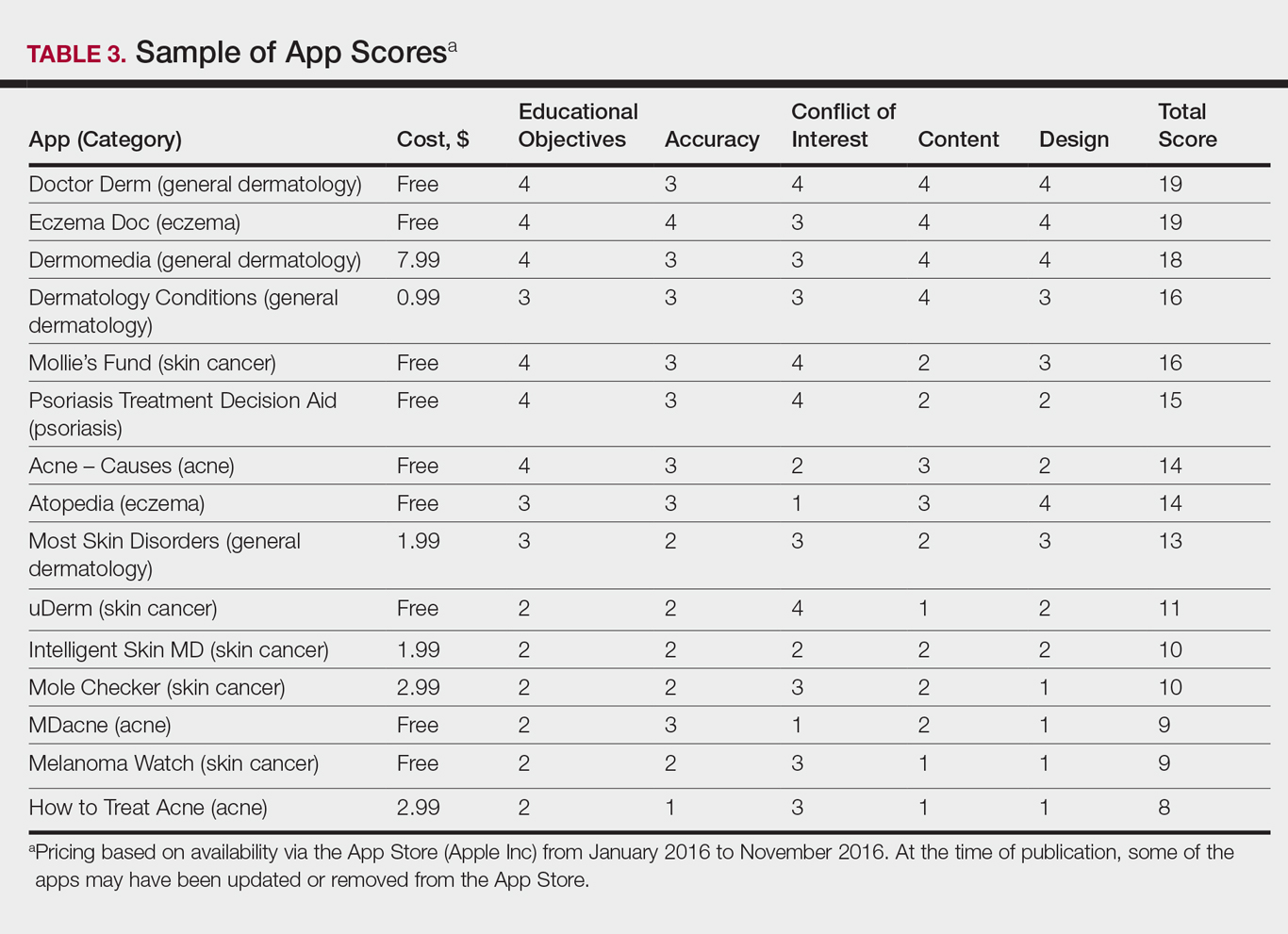
Comment
The number of dermatology-related apps available to mobile users continues to grow at an increasing rate.8 The apps vary in many aspects, including their purpose, scope, intended audience, and goals of the app publisher. In turn, more individuals are turning to mobile apps for medical information,4 especially in dermatology, thus it is necessary to create a systematic way to evaluate the quality and utility of each app to assist users in making informed decisions about which apps will best meet their needs in the midst of a wide array of choices.
For the purpose of this study, an objective rubric was created that can be used to evaluate the quality of medical apps for patient education in dermatology. An app’s adequacy and usefulness for patient education was thought to depend on 3 possible score ranges into which the app could fall based on the grading rubric. An app with a total score in the range of 5 to 10 was not thought to be useful and may even be detrimental to patients. An app with a total score in the range of 11 to 15 may be used for patient education with some reservations based on shortcomings for certain criteria. An app with a score in the range of 16 to 20 was thought to be valuable and adequate for patient education. For example, the How to Treat Acne app received a total score of 8 and therefore would not be recommended to patients based on the grading rubric used in this study. This particular app provided sparse and sometimes inaccurate information, had a confusing user interface, and contained many obstructive advertisements. In contrast, the Eczema Doc app received a total score of 19, which indicates a quality app deemed to be useful for patient information based on the established rubric. This app met all the objectives that it advertised, contained accurate information with verified citation of sources, and was very easy for users to navigate.
Of the 44 graded apps, only 9 (20.5%) received scores in the highest range of 16 to 20, which indicates a need for improvements in mobile dermatology apps intended for patient education. Adopting the grading rubric developed in this study as a standard in the creation of medical apps could have beneficial implications in disseminating accurate, safe, unbiased, and easy-to-understand information to patients.
- Smith A. U.S. smartphone use in 2015. Pew Research Center website. http://www.pewinternet.org/2015/04/01/us-smartphone-use-in-2015. Published April 1, 2015. Accessed August 29, 2017.
- Nilsen W, Kumar S, Shar A, et al. Advancing the science of mHealth. J Health Commun. 2012;17(suppl 1):5-10.
- West DM. How mobile devices are transforming healthcare issues in technology innovation. Issues Technol Innov. 2012;18:1-14.
- Boudreaux ED, Waring ME, Hayes RB, et al. Evaluating and selecting mobile health apps: strategies for healthcare providers and healthcare organizations. Transl Behav Med. 2014;4:363-371.
- Brewer AC, Endly DC, Henley J, et al. Mobile applications in dermatology. JAMA Dermatol. 2013;149:1300-1304.
- Cummings E, Borycki E, Roehrer E. Issues and considerations for healthcare consumers using mobile applications. Stud Health Technol Inform. 2013;183:227-231.
- Handel MJ. mHealth (mobile health)-using apps for health and wellness. Explore. 2011;7:256-261.
- Boulos MN, Brewer AC, Karimkhani C, et al. Mobile medical and health apps: state of the art, concerns, regulatory control and certification. Online J Public Health Inform. 2014;5:229.
According to industry estimates, roughly 64% of US adults were smartphone users in 2015.1 Smartphones enable users to utilize mobile applications (apps) that can perform a variety of functions in many categories, including business, music, photography, entertainment, education, social networking, travel, and lifestyle. The widespread adoption and use of mobile apps has implications for medical practice. Mobile apps have the capability to serve as information sources for patients, educational tools for students, and diagnostic aids for physicians.2 Consequently, a number of medical and health care–oriented apps have already been developed3 and are increasingly utilized by patients and providers.4
Given its visual nature, dermatology is particularly amenable to the integration of mobile medical apps. A study by Brewer et al5 identified more than 229 dermatology-related apps in categories ranging from general dermatology reference, self-surveillance and diagnosis, disease guides, educational aids, sunscreen and UV recommendations, and teledermatology. Patients served as the target audience and principal consumers of more than half of these dermatology apps.5
Mobile medical and health care apps demonstrate great potential for serving as valuable information sources for patients with dermatologic conditions; however, the content, functions, accuracy, and educational value of dermatology mobile apps are not well characterized, making it difficult for patients and health care providers to select and recommend appropriate apps.6 In this study, we created a rubric to objectively grade 44 publicly available mobile dermatology apps with the primary focus of patient education.
Methods
We conducted a search of dermatology-related educational mobile apps that were publicly available via the App Store (Apple Inc) from January 2016 to November 2016. (The pricing, availability, and other features of these apps may have changed since the study period.) The following search terms were used: dermatology, dermoscopy, melanoma, skin cancer, psoriasis, rosacea, acne, eczema, dermal fillers, and Mohs surgery. We excluded apps that were not in English; had a solely commercial focus; were mobile textbooks or scientific journals; were used to provide teledermatology services with no educational purpose; were solely focused on homeopathic, alternative, and/or complementary medicine; or were intended primarily as a reference for students or health care professionals. Our search yielded 44 apps with patient education as a primary objective. The apps were divided into 6 categories based on their focus: general dermatology, cosmetic dermatology, acne, eczema, psoriasis, and skin cancer.
Each app was reviewed using a quantified grading rubric developed by the researchers. In a prior evaluation, Handel7 reviewed 35 health and wellness mobile apps utilizing the categories of ease of use, reliability, quality, scope of information, and aesthetics.4 These criteria were modified and adapted for the purposes of this study, and a 4-point scale was applied to each criterion. The final criteria were (1) educational objectives, (2) content, (3) accuracy, (4) design, and (5) conflict of interest. The quantified grading rubric is described in Table 1.
Results
The possible range of scores based on the grading rubric was 5 to 20. The actual range of scores was 8 to 19 (Table 2). The 44 reviewed apps were categorized by topic as acne, cosmetic dermatology, eczema, general dermatology, psoriasis, or skin cancer. A sample of 15 apps selected to represent the distribution of scores and their grading on the rubric are presented in Table 3.
Comment
The number of dermatology-related apps available to mobile users continues to grow at an increasing rate.8 The apps vary in many aspects, including their purpose, scope, intended audience, and goals of the app publisher. In turn, more individuals are turning to mobile apps for medical information,4 especially in dermatology, thus it is necessary to create a systematic way to evaluate the quality and utility of each app to assist users in making informed decisions about which apps will best meet their needs in the midst of a wide array of choices.
For the purpose of this study, an objective rubric was created that can be used to evaluate the quality of medical apps for patient education in dermatology. An app’s adequacy and usefulness for patient education was thought to depend on 3 possible score ranges into which the app could fall based on the grading rubric. An app with a total score in the range of 5 to 10 was not thought to be useful and may even be detrimental to patients. An app with a total score in the range of 11 to 15 may be used for patient education with some reservations based on shortcomings for certain criteria. An app with a score in the range of 16 to 20 was thought to be valuable and adequate for patient education. For example, the How to Treat Acne app received a total score of 8 and therefore would not be recommended to patients based on the grading rubric used in this study. This particular app provided sparse and sometimes inaccurate information, had a confusing user interface, and contained many obstructive advertisements. In contrast, the Eczema Doc app received a total score of 19, which indicates a quality app deemed to be useful for patient information based on the established rubric. This app met all the objectives that it advertised, contained accurate information with verified citation of sources, and was very easy for users to navigate.
Of the 44 graded apps, only 9 (20.5%) received scores in the highest range of 16 to 20, which indicates a need for improvements in mobile dermatology apps intended for patient education. Adopting the grading rubric developed in this study as a standard in the creation of medical apps could have beneficial implications in disseminating accurate, safe, unbiased, and easy-to-understand information to patients.
According to industry estimates, roughly 64% of US adults were smartphone users in 2015.1 Smartphones enable users to utilize mobile applications (apps) that can perform a variety of functions in many categories, including business, music, photography, entertainment, education, social networking, travel, and lifestyle. The widespread adoption and use of mobile apps has implications for medical practice. Mobile apps have the capability to serve as information sources for patients, educational tools for students, and diagnostic aids for physicians.2 Consequently, a number of medical and health care–oriented apps have already been developed3 and are increasingly utilized by patients and providers.4
Given its visual nature, dermatology is particularly amenable to the integration of mobile medical apps. A study by Brewer et al5 identified more than 229 dermatology-related apps in categories ranging from general dermatology reference, self-surveillance and diagnosis, disease guides, educational aids, sunscreen and UV recommendations, and teledermatology. Patients served as the target audience and principal consumers of more than half of these dermatology apps.5
Mobile medical and health care apps demonstrate great potential for serving as valuable information sources for patients with dermatologic conditions; however, the content, functions, accuracy, and educational value of dermatology mobile apps are not well characterized, making it difficult for patients and health care providers to select and recommend appropriate apps.6 In this study, we created a rubric to objectively grade 44 publicly available mobile dermatology apps with the primary focus of patient education.
Methods
We conducted a search of dermatology-related educational mobile apps that were publicly available via the App Store (Apple Inc) from January 2016 to November 2016. (The pricing, availability, and other features of these apps may have changed since the study period.) The following search terms were used: dermatology, dermoscopy, melanoma, skin cancer, psoriasis, rosacea, acne, eczema, dermal fillers, and Mohs surgery. We excluded apps that were not in English; had a solely commercial focus; were mobile textbooks or scientific journals; were used to provide teledermatology services with no educational purpose; were solely focused on homeopathic, alternative, and/or complementary medicine; or were intended primarily as a reference for students or health care professionals. Our search yielded 44 apps with patient education as a primary objective. The apps were divided into 6 categories based on their focus: general dermatology, cosmetic dermatology, acne, eczema, psoriasis, and skin cancer.
Each app was reviewed using a quantified grading rubric developed by the researchers. In a prior evaluation, Handel7 reviewed 35 health and wellness mobile apps utilizing the categories of ease of use, reliability, quality, scope of information, and aesthetics.4 These criteria were modified and adapted for the purposes of this study, and a 4-point scale was applied to each criterion. The final criteria were (1) educational objectives, (2) content, (3) accuracy, (4) design, and (5) conflict of interest. The quantified grading rubric is described in Table 1.
Results
The possible range of scores based on the grading rubric was 5 to 20. The actual range of scores was 8 to 19 (Table 2). The 44 reviewed apps were categorized by topic as acne, cosmetic dermatology, eczema, general dermatology, psoriasis, or skin cancer. A sample of 15 apps selected to represent the distribution of scores and their grading on the rubric are presented in Table 3.
Comment
The number of dermatology-related apps available to mobile users continues to grow at an increasing rate.8 The apps vary in many aspects, including their purpose, scope, intended audience, and goals of the app publisher. In turn, more individuals are turning to mobile apps for medical information,4 especially in dermatology, thus it is necessary to create a systematic way to evaluate the quality and utility of each app to assist users in making informed decisions about which apps will best meet their needs in the midst of a wide array of choices.
For the purpose of this study, an objective rubric was created that can be used to evaluate the quality of medical apps for patient education in dermatology. An app’s adequacy and usefulness for patient education was thought to depend on 3 possible score ranges into which the app could fall based on the grading rubric. An app with a total score in the range of 5 to 10 was not thought to be useful and may even be detrimental to patients. An app with a total score in the range of 11 to 15 may be used for patient education with some reservations based on shortcomings for certain criteria. An app with a score in the range of 16 to 20 was thought to be valuable and adequate for patient education. For example, the How to Treat Acne app received a total score of 8 and therefore would not be recommended to patients based on the grading rubric used in this study. This particular app provided sparse and sometimes inaccurate information, had a confusing user interface, and contained many obstructive advertisements. In contrast, the Eczema Doc app received a total score of 19, which indicates a quality app deemed to be useful for patient information based on the established rubric. This app met all the objectives that it advertised, contained accurate information with verified citation of sources, and was very easy for users to navigate.
Of the 44 graded apps, only 9 (20.5%) received scores in the highest range of 16 to 20, which indicates a need for improvements in mobile dermatology apps intended for patient education. Adopting the grading rubric developed in this study as a standard in the creation of medical apps could have beneficial implications in disseminating accurate, safe, unbiased, and easy-to-understand information to patients.
- Smith A. U.S. smartphone use in 2015. Pew Research Center website. http://www.pewinternet.org/2015/04/01/us-smartphone-use-in-2015. Published April 1, 2015. Accessed August 29, 2017.
- Nilsen W, Kumar S, Shar A, et al. Advancing the science of mHealth. J Health Commun. 2012;17(suppl 1):5-10.
- West DM. How mobile devices are transforming healthcare issues in technology innovation. Issues Technol Innov. 2012;18:1-14.
- Boudreaux ED, Waring ME, Hayes RB, et al. Evaluating and selecting mobile health apps: strategies for healthcare providers and healthcare organizations. Transl Behav Med. 2014;4:363-371.
- Brewer AC, Endly DC, Henley J, et al. Mobile applications in dermatology. JAMA Dermatol. 2013;149:1300-1304.
- Cummings E, Borycki E, Roehrer E. Issues and considerations for healthcare consumers using mobile applications. Stud Health Technol Inform. 2013;183:227-231.
- Handel MJ. mHealth (mobile health)-using apps for health and wellness. Explore. 2011;7:256-261.
- Boulos MN, Brewer AC, Karimkhani C, et al. Mobile medical and health apps: state of the art, concerns, regulatory control and certification. Online J Public Health Inform. 2014;5:229.
- Smith A. U.S. smartphone use in 2015. Pew Research Center website. http://www.pewinternet.org/2015/04/01/us-smartphone-use-in-2015. Published April 1, 2015. Accessed August 29, 2017.
- Nilsen W, Kumar S, Shar A, et al. Advancing the science of mHealth. J Health Commun. 2012;17(suppl 1):5-10.
- West DM. How mobile devices are transforming healthcare issues in technology innovation. Issues Technol Innov. 2012;18:1-14.
- Boudreaux ED, Waring ME, Hayes RB, et al. Evaluating and selecting mobile health apps: strategies for healthcare providers and healthcare organizations. Transl Behav Med. 2014;4:363-371.
- Brewer AC, Endly DC, Henley J, et al. Mobile applications in dermatology. JAMA Dermatol. 2013;149:1300-1304.
- Cummings E, Borycki E, Roehrer E. Issues and considerations for healthcare consumers using mobile applications. Stud Health Technol Inform. 2013;183:227-231.
- Handel MJ. mHealth (mobile health)-using apps for health and wellness. Explore. 2011;7:256-261.
- Boulos MN, Brewer AC, Karimkhani C, et al. Mobile medical and health apps: state of the art, concerns, regulatory control and certification. Online J Public Health Inform. 2014;5:229.
Practice Points
- Mobile dermatology apps for educational purposes should be objectively reviewed before being used by patients.
- In our study, only 9 (20.5%) of the 44 dermatology apps evaluated were considered adequate for patient information based on our grading criteria.