User login
Cutis is a peer-reviewed clinical journal for the dermatologist, allergist, and general practitioner published monthly since 1965. Concise clinical articles present the practical side of dermatology, helping physicians to improve patient care. Cutis is referenced in Index Medicus/MEDLINE and is written and edited by industry leaders.
ass lick
assault rifle
balls
ballsac
black jack
bleach
Boko Haram
bondage
causas
cheap
child abuse
cocaine
compulsive behaviors
cost of miracles
cunt
Daech
display network stats
drug paraphernalia
explosion
fart
fda and death
fda AND warn
fda AND warning
fda AND warns
feom
fuck
gambling
gfc
gun
human trafficking
humira AND expensive
illegal
ISIL
ISIS
Islamic caliphate
Islamic state
madvocate
masturbation
mixed martial arts
MMA
molestation
national rifle association
NRA
nsfw
nuccitelli
pedophile
pedophilia
poker
porn
porn
pornography
psychedelic drug
recreational drug
sex slave rings
shit
slot machine
snort
substance abuse
terrorism
terrorist
texarkana
Texas hold 'em
UFC
section[contains(@class, 'nav-hidden')]
section[contains(@class, 'nav-hidden active')
A peer-reviewed, indexed journal for dermatologists with original research, image quizzes, cases and reviews, and columns.
What Do You Want to Be When You Grow Up? Pearls for Postresidency Planning
Dermatology residency training can feel endless at the outset; an arduous intern year followed by 3 years of specialized training. However, I have realized that, within residency, time moves quickly. As I look ahead to postresidency life, I realize that residents are all facing the same question: What do you want to be when you grow up?
You may think you have answered that question already; however, there are many different careers within the field of dermatology and no amount of studying or reading will help you choose the right one. In an attempt to make sense of these choices, I have spoken to many recent dermatology graduates over the last several months to get a sense of how they made their postresidency decisions, and I want to share their pearls.
Pearl: Explore Fellowship Opportunities Early
The first decision is whether or not to pursue a fellowship after residency. There currently are 2 Accreditation Council for Graduate Medical Education–approved fellowships after dermatology residency: dermatopathology and micrographic surgery. Pediatric dermatology is another board-certified fellowship. A list of these training programs and the requirements can be found on the American Board of Dermatology website (www.abderm.org). There also are several nonaccredited fellowships including pediatrics, cosmetics, complex medical dermatology, cutaneous oncology, and rheumatology.
Even if you are not completely committed to pursuing a fellowship, it is beneficial to explore any fellowship options early in residency. Spend extra time in any field you are considering for fellowship and consider research in the field. If there is a fellowship position at your institution, try to rotate there early in residency. Rotations at other institutions can demonstrate your interest and enthusiasm while also helping you to network within your chosen subspecialty. Several of the dermatology interest groups even sponsor rotations at outside institutions, if extra funding is needed. If recent graduates from your program have matched in fellowship, it is always a good idea to reach out to them to get program-specific advice. It takes a lot of time, confidence, and persistence to organize the opportunities that will help you maximize your fellowship potential, but it is well worth the effort.
Fellowships can occur through an official “match,” similar to residency, or can be accepted on a rolling basis. For example, many dermatopathology fellowships can begin accepting applications as early as the summer between the first and second year of residency (www.abderm.org). It is important to get this information early so that you do not miss any application deadlines.
Pearl: Prioritize Where You Want to Practice
If you have decided that fellowship is not for you, then it is time to apply for your first job as a physician. There are several big factors that help narrow the search. It is best to start the search early to allow yourself time and different options. According to the 2016 American Academy of Dermatology database, there currently are approximately 3.4 dermatologists per 100,000 Americans; however, they are unevenly distributed throughout the country. In this study, the researchers found the highest density of dermatologists on the Upper East Side of Manhattan (41.8 per 100,000 dermatologists) compared to Swainsboro, Georgia (0.45 per 100,000 dermatologists).1
With more competition for jobs in areas with a higher concentration of dermatologists, compensation often is lower. There also are many personal factors that contribute to where you want to live and work, and if you prioritize them, it will lead to greater overall satisfaction in postresidency life.
Another large factor to consider is private practice versus academic dermatology. Academic dermatology can provide opportunities for research as well as the opportunity to work with students and residents. As part of a larger hospital system, there often is the opportunity for benefits, such as 401(k) matching, that might be less accessible in small practices.
Pearl: Get Recruiter Recommendations From Your Peers
There are many recruiting services that can help put you in touch with practices that are hiring. These services can be helpful but also can be overwhelming at times, with many emails and telephone calls. In my experience, recent graduates had mixed feelings about recruiting services. Those who had been the happiest with their recruiting experience had often gotten the name of a specific recruiter from someone else in their program who had a positive experience. Mentors at your training institution or beyond also can be a good source of information for job opportunities. It can be helpful to get involved early in the various dermatologic societies and network at academic conferences throughout your training.
Pearl: Talk to Partners and Nonpartners About the Practice’s Philosophy
When picking a private practice for your first job, make sure you get a sense of the philosophy of the practice, including the partners’ goals for the office, the patient population, and the dynamic of the office staff. If there is a cosmetic component, it is important to know what devices are available and which products are sold. It is important to talk to nonpartners at a practice and get a sense of their satisfaction. If you sign the employment contract, you will be in their shoes soon!
Pearl: Have an Attorney Review Your Contract
There are many important topics in your employment contract. After years of medical school loans and resident salary, it is easy to focus only on compensation. However, pay attention to the other aspects of reimbursement including bonuses, benefits, noncompete clauses, and call schedules. Also consider the termination policies. The general advice I have received is to have a lawyer look at your contract. Although it may be tempting to skip the lawyer’s fee and review it yourself, you may actually end up negotiating a contract that benefits you more in the long-run or avoid signing a contract that will limit you.
- Glazer AM, Farberg AS, Winkelmann RR, et al. Analysis of trends in geographic distribution and density of US dermatologists. JAMA Dermatol. 2017;153:322-325.
Dermatology residency training can feel endless at the outset; an arduous intern year followed by 3 years of specialized training. However, I have realized that, within residency, time moves quickly. As I look ahead to postresidency life, I realize that residents are all facing the same question: What do you want to be when you grow up?
You may think you have answered that question already; however, there are many different careers within the field of dermatology and no amount of studying or reading will help you choose the right one. In an attempt to make sense of these choices, I have spoken to many recent dermatology graduates over the last several months to get a sense of how they made their postresidency decisions, and I want to share their pearls.
Pearl: Explore Fellowship Opportunities Early
The first decision is whether or not to pursue a fellowship after residency. There currently are 2 Accreditation Council for Graduate Medical Education–approved fellowships after dermatology residency: dermatopathology and micrographic surgery. Pediatric dermatology is another board-certified fellowship. A list of these training programs and the requirements can be found on the American Board of Dermatology website (www.abderm.org). There also are several nonaccredited fellowships including pediatrics, cosmetics, complex medical dermatology, cutaneous oncology, and rheumatology.
Even if you are not completely committed to pursuing a fellowship, it is beneficial to explore any fellowship options early in residency. Spend extra time in any field you are considering for fellowship and consider research in the field. If there is a fellowship position at your institution, try to rotate there early in residency. Rotations at other institutions can demonstrate your interest and enthusiasm while also helping you to network within your chosen subspecialty. Several of the dermatology interest groups even sponsor rotations at outside institutions, if extra funding is needed. If recent graduates from your program have matched in fellowship, it is always a good idea to reach out to them to get program-specific advice. It takes a lot of time, confidence, and persistence to organize the opportunities that will help you maximize your fellowship potential, but it is well worth the effort.
Fellowships can occur through an official “match,” similar to residency, or can be accepted on a rolling basis. For example, many dermatopathology fellowships can begin accepting applications as early as the summer between the first and second year of residency (www.abderm.org). It is important to get this information early so that you do not miss any application deadlines.
Pearl: Prioritize Where You Want to Practice
If you have decided that fellowship is not for you, then it is time to apply for your first job as a physician. There are several big factors that help narrow the search. It is best to start the search early to allow yourself time and different options. According to the 2016 American Academy of Dermatology database, there currently are approximately 3.4 dermatologists per 100,000 Americans; however, they are unevenly distributed throughout the country. In this study, the researchers found the highest density of dermatologists on the Upper East Side of Manhattan (41.8 per 100,000 dermatologists) compared to Swainsboro, Georgia (0.45 per 100,000 dermatologists).1
With more competition for jobs in areas with a higher concentration of dermatologists, compensation often is lower. There also are many personal factors that contribute to where you want to live and work, and if you prioritize them, it will lead to greater overall satisfaction in postresidency life.
Another large factor to consider is private practice versus academic dermatology. Academic dermatology can provide opportunities for research as well as the opportunity to work with students and residents. As part of a larger hospital system, there often is the opportunity for benefits, such as 401(k) matching, that might be less accessible in small practices.
Pearl: Get Recruiter Recommendations From Your Peers
There are many recruiting services that can help put you in touch with practices that are hiring. These services can be helpful but also can be overwhelming at times, with many emails and telephone calls. In my experience, recent graduates had mixed feelings about recruiting services. Those who had been the happiest with their recruiting experience had often gotten the name of a specific recruiter from someone else in their program who had a positive experience. Mentors at your training institution or beyond also can be a good source of information for job opportunities. It can be helpful to get involved early in the various dermatologic societies and network at academic conferences throughout your training.
Pearl: Talk to Partners and Nonpartners About the Practice’s Philosophy
When picking a private practice for your first job, make sure you get a sense of the philosophy of the practice, including the partners’ goals for the office, the patient population, and the dynamic of the office staff. If there is a cosmetic component, it is important to know what devices are available and which products are sold. It is important to talk to nonpartners at a practice and get a sense of their satisfaction. If you sign the employment contract, you will be in their shoes soon!
Pearl: Have an Attorney Review Your Contract
There are many important topics in your employment contract. After years of medical school loans and resident salary, it is easy to focus only on compensation. However, pay attention to the other aspects of reimbursement including bonuses, benefits, noncompete clauses, and call schedules. Also consider the termination policies. The general advice I have received is to have a lawyer look at your contract. Although it may be tempting to skip the lawyer’s fee and review it yourself, you may actually end up negotiating a contract that benefits you more in the long-run or avoid signing a contract that will limit you.
Dermatology residency training can feel endless at the outset; an arduous intern year followed by 3 years of specialized training. However, I have realized that, within residency, time moves quickly. As I look ahead to postresidency life, I realize that residents are all facing the same question: What do you want to be when you grow up?
You may think you have answered that question already; however, there are many different careers within the field of dermatology and no amount of studying or reading will help you choose the right one. In an attempt to make sense of these choices, I have spoken to many recent dermatology graduates over the last several months to get a sense of how they made their postresidency decisions, and I want to share their pearls.
Pearl: Explore Fellowship Opportunities Early
The first decision is whether or not to pursue a fellowship after residency. There currently are 2 Accreditation Council for Graduate Medical Education–approved fellowships after dermatology residency: dermatopathology and micrographic surgery. Pediatric dermatology is another board-certified fellowship. A list of these training programs and the requirements can be found on the American Board of Dermatology website (www.abderm.org). There also are several nonaccredited fellowships including pediatrics, cosmetics, complex medical dermatology, cutaneous oncology, and rheumatology.
Even if you are not completely committed to pursuing a fellowship, it is beneficial to explore any fellowship options early in residency. Spend extra time in any field you are considering for fellowship and consider research in the field. If there is a fellowship position at your institution, try to rotate there early in residency. Rotations at other institutions can demonstrate your interest and enthusiasm while also helping you to network within your chosen subspecialty. Several of the dermatology interest groups even sponsor rotations at outside institutions, if extra funding is needed. If recent graduates from your program have matched in fellowship, it is always a good idea to reach out to them to get program-specific advice. It takes a lot of time, confidence, and persistence to organize the opportunities that will help you maximize your fellowship potential, but it is well worth the effort.
Fellowships can occur through an official “match,” similar to residency, or can be accepted on a rolling basis. For example, many dermatopathology fellowships can begin accepting applications as early as the summer between the first and second year of residency (www.abderm.org). It is important to get this information early so that you do not miss any application deadlines.
Pearl: Prioritize Where You Want to Practice
If you have decided that fellowship is not for you, then it is time to apply for your first job as a physician. There are several big factors that help narrow the search. It is best to start the search early to allow yourself time and different options. According to the 2016 American Academy of Dermatology database, there currently are approximately 3.4 dermatologists per 100,000 Americans; however, they are unevenly distributed throughout the country. In this study, the researchers found the highest density of dermatologists on the Upper East Side of Manhattan (41.8 per 100,000 dermatologists) compared to Swainsboro, Georgia (0.45 per 100,000 dermatologists).1
With more competition for jobs in areas with a higher concentration of dermatologists, compensation often is lower. There also are many personal factors that contribute to where you want to live and work, and if you prioritize them, it will lead to greater overall satisfaction in postresidency life.
Another large factor to consider is private practice versus academic dermatology. Academic dermatology can provide opportunities for research as well as the opportunity to work with students and residents. As part of a larger hospital system, there often is the opportunity for benefits, such as 401(k) matching, that might be less accessible in small practices.
Pearl: Get Recruiter Recommendations From Your Peers
There are many recruiting services that can help put you in touch with practices that are hiring. These services can be helpful but also can be overwhelming at times, with many emails and telephone calls. In my experience, recent graduates had mixed feelings about recruiting services. Those who had been the happiest with their recruiting experience had often gotten the name of a specific recruiter from someone else in their program who had a positive experience. Mentors at your training institution or beyond also can be a good source of information for job opportunities. It can be helpful to get involved early in the various dermatologic societies and network at academic conferences throughout your training.
Pearl: Talk to Partners and Nonpartners About the Practice’s Philosophy
When picking a private practice for your first job, make sure you get a sense of the philosophy of the practice, including the partners’ goals for the office, the patient population, and the dynamic of the office staff. If there is a cosmetic component, it is important to know what devices are available and which products are sold. It is important to talk to nonpartners at a practice and get a sense of their satisfaction. If you sign the employment contract, you will be in their shoes soon!
Pearl: Have an Attorney Review Your Contract
There are many important topics in your employment contract. After years of medical school loans and resident salary, it is easy to focus only on compensation. However, pay attention to the other aspects of reimbursement including bonuses, benefits, noncompete clauses, and call schedules. Also consider the termination policies. The general advice I have received is to have a lawyer look at your contract. Although it may be tempting to skip the lawyer’s fee and review it yourself, you may actually end up negotiating a contract that benefits you more in the long-run or avoid signing a contract that will limit you.
- Glazer AM, Farberg AS, Winkelmann RR, et al. Analysis of trends in geographic distribution and density of US dermatologists. JAMA Dermatol. 2017;153:322-325.
- Glazer AM, Farberg AS, Winkelmann RR, et al. Analysis of trends in geographic distribution and density of US dermatologists. JAMA Dermatol. 2017;153:322-325.

Hyaluronic Acid for Lip Rejuvenation

Yellow-Orange Hairless Plaque on the Scalp
The Diagnosis: Nevus Sebaceous
The patient presented with a typical solitary scalp lesion characteristic of nevus sebaceous (NS). The lesion was present at birth as a flat and smooth hairless plaque; however, over time it became more thickened and noticeable, which prompted the parents to seek medical advice.
Nevus sebaceous, also known as NS of Jadassohn, is a benign congenital hamartoma of the sebaceous gland that usually is present at birth and frequently involves the scalp and/or the face. The classic NS lesion is solitary and appears as a well-circumscribed, waxy, yellow-orange or tan, hairless plaque. Despite the presence of these lesions at birth, they may not be noted until early childhood or rarely until adulthood. Generally, the lesion tends to thicken and become more verrucous and velvety over time, particularly around the time of reaching puberty.1 Clinically, NS lesions vary in size from 1 cm to several centimeters. Lesions initially tend to grow proportionately with the child until puberty when they become notably thicker, greasier, and verrucous or nodular under hormonal influences. The yellow discoloration of the lesion is due to sebaceous gland secretion, and the characteristic color usually becomes less evident with age.
Nevus sebaceous occurs in approximately 0.3% of newborns and tends to be sporadic in nature; however, rare familial forms have been reported.2,3 Nevus sebaceous can present as multiple nevi that tend to be extensive and distributed along the Blaschko lines, and they usually are associated with neurologic, ocular, or skeletal defects. Involvement of the central nervous system frequently is associated with large sebaceous nevi located on the face or scalp. This association has been termed NS syndrome.4 Neurologic abnormalities associated with NS syndrome include seizures, mental retardation, and hemimegalencephaly.5 Ocular findings most communally associated with the syndrome are choristomas and colobomas.6-8
There are several benign and malignant epithelial neoplasms that may develop within sebaceous nevi. Benign tumors include trichoblastoma, syringocystadenoma papilliferum, trichilemmoma, sebaceoma, nodular hidradenoma, and hidrocystoma.1,8,9 Malignant neoplasms include basal cell carcinoma (BCC), apocrine carcinoma, sebaceous carcinoma, and squamous cell carcinoma. The lifetime risk of malignancy in NS is unknown. In an extensive literature review by Moody et al10 of 4923 cases of NS for the development of secondary benign and malignant neoplasms, 16% developed benign tumors while 8% developed malignant tumors such as BCC. However, subsequent studies suggested that the incidence of BCC may have been overestimated due to misinterpretation of trichoblastoma and may be less than 1%.11-13
Usually the diagnosis of NS is made clinically and rarely a biopsy for histopathologic confirmation may be needed when the diagnosis is uncertain. Typically, these histopathologic findings include immature hair follicles, hyperplastic immature sebaceous glands, dilated apocrine glands, and epidermal hyperplasia.9 For patients with suspected NS syndrome, additional neurologic and ophthalmologic evaluations should be performed including neuroimaging studies, skeletal radiography, and analysis of liver and renal function.14
The current standard of care in treating NS is full-thickness excision. However, the decision should be individualized based on patient age, extension and location of the lesion, concerns about the cosmetic appearance, and the risk for malignancy.
The 2 main reasons to excise NS include concern about malignancy and undesirable cosmetic appearance. Once a malignant lesion develops within NS, it generally is agreed that the tumor and the entire nevus should be removed; however, recommendations vary for excising NS prophylactically to decrease the risk for malignant growths. Because the risk for malignant transformation seems to be lower than previously thought, observation can be a reasonable choice for lesions that are not associated with cosmetic concern.12,13
Photodynamic therapy, CO2 laser resurfacing, and dermabrasion have been reported as alternative therapeutic approaches. However, there is a growing concern on how effective these treatment modalities are in completely removing the lesion and whether the risk for recurrence and potential for neoplasm development remains.1,9
This patient was healthy with normal development and growth and no signs of neurologic or ocular involvement. The parents were counseled about the risk for malignancy and the long-term cosmetic appearance of the lesion. They opted for surgical excision of the lesion at 18 months of age.
- Eisen DB, Michael DJ. Sebaceous lesions and their associated syndromes: part I. J Am Acad Dermatol. 2009;61:549-560; quiz 561-562.
- Happle R, König A. Familial naevus sebaceus may be explained by paradominant transmission. Br J Dermatol. 1999;141:377.
- Hughes SM, Wilkerson AE, Winfield HL, et al. Familial nevus sebaceus in dizygotic male twins. J Am Acad Dermatol. 2006;54(2 suppl):S47-S48.
- Sugarman JL. Epidermal nevus syndromes. Semin Cutan Med Surg. 2007;26:221-230.
- Davies D, Rogers M. Review of neurological manifestations in 196 patients with sebaceous naevi. Australas J Dermatol. 2002;43:20-23.
- Trivedi N, Nehete G. Complex limbal choristoma in linear nevus sebaceous syndrome managed with scleral grafting. Indian J Ophthalmol. 2016;64:692-694.
- Nema N, Singh K, Verma A. Complex limbal choristoma in nevus sebaceous syndrome [published online February 14, 2012]. Pediatr Dermatol. 2012;29:227-229.
- Park JM, Kim DS, Kim J, et al. Epibulbar complex choristoma and hemimegalencephaly in linear sebaceous naevus syndrome [published online July 2, 2009]. Clin Exp Dermatol. 2009;34:E686-E689.
- Simi CM, Rajalakshmi T, Correa M. Clinicopathologic analysis of 21 cases of nevus sebaceus: a retrospective study. Indian J Dermatol Venereol Leprol. 2008;74:625-627.
- Moody MN, Landau JM, Goldberg LH. Nevus sebaceous revisited. Pediatr Dermatol. 2012;29:15-23.
- Cribier B, Scrivener Y, Grosshans E. Tumors arising in nevus sebaceus: a study of 596 cases. J Am Acad Dermatol. 2000;42(2 pt 1):263-268.
- Santibanez-Gallerani A, Marshall D, Duarte AM, et al. Should nevus sebaceus of Jadassohn in children be excised? a study of 757 cases, and literature review. J Craniofac Surg. 2003;14:658-660.
- Rosen H, Schmidt B, Lam HP, et al. Management of nevus sebaceous and the risk of basal cell carcinoma: an 18-year review. Pediatr Dermatol. 2009;26:676-681.
- Brandling-Bennett HA, Morel KD. Epidermal nevi. Pediatr Clin North Am. 2010;57:1177-1198.
The Diagnosis: Nevus Sebaceous
The patient presented with a typical solitary scalp lesion characteristic of nevus sebaceous (NS). The lesion was present at birth as a flat and smooth hairless plaque; however, over time it became more thickened and noticeable, which prompted the parents to seek medical advice.
Nevus sebaceous, also known as NS of Jadassohn, is a benign congenital hamartoma of the sebaceous gland that usually is present at birth and frequently involves the scalp and/or the face. The classic NS lesion is solitary and appears as a well-circumscribed, waxy, yellow-orange or tan, hairless plaque. Despite the presence of these lesions at birth, they may not be noted until early childhood or rarely until adulthood. Generally, the lesion tends to thicken and become more verrucous and velvety over time, particularly around the time of reaching puberty.1 Clinically, NS lesions vary in size from 1 cm to several centimeters. Lesions initially tend to grow proportionately with the child until puberty when they become notably thicker, greasier, and verrucous or nodular under hormonal influences. The yellow discoloration of the lesion is due to sebaceous gland secretion, and the characteristic color usually becomes less evident with age.
Nevus sebaceous occurs in approximately 0.3% of newborns and tends to be sporadic in nature; however, rare familial forms have been reported.2,3 Nevus sebaceous can present as multiple nevi that tend to be extensive and distributed along the Blaschko lines, and they usually are associated with neurologic, ocular, or skeletal defects. Involvement of the central nervous system frequently is associated with large sebaceous nevi located on the face or scalp. This association has been termed NS syndrome.4 Neurologic abnormalities associated with NS syndrome include seizures, mental retardation, and hemimegalencephaly.5 Ocular findings most communally associated with the syndrome are choristomas and colobomas.6-8
There are several benign and malignant epithelial neoplasms that may develop within sebaceous nevi. Benign tumors include trichoblastoma, syringocystadenoma papilliferum, trichilemmoma, sebaceoma, nodular hidradenoma, and hidrocystoma.1,8,9 Malignant neoplasms include basal cell carcinoma (BCC), apocrine carcinoma, sebaceous carcinoma, and squamous cell carcinoma. The lifetime risk of malignancy in NS is unknown. In an extensive literature review by Moody et al10 of 4923 cases of NS for the development of secondary benign and malignant neoplasms, 16% developed benign tumors while 8% developed malignant tumors such as BCC. However, subsequent studies suggested that the incidence of BCC may have been overestimated due to misinterpretation of trichoblastoma and may be less than 1%.11-13
Usually the diagnosis of NS is made clinically and rarely a biopsy for histopathologic confirmation may be needed when the diagnosis is uncertain. Typically, these histopathologic findings include immature hair follicles, hyperplastic immature sebaceous glands, dilated apocrine glands, and epidermal hyperplasia.9 For patients with suspected NS syndrome, additional neurologic and ophthalmologic evaluations should be performed including neuroimaging studies, skeletal radiography, and analysis of liver and renal function.14
The current standard of care in treating NS is full-thickness excision. However, the decision should be individualized based on patient age, extension and location of the lesion, concerns about the cosmetic appearance, and the risk for malignancy.
The 2 main reasons to excise NS include concern about malignancy and undesirable cosmetic appearance. Once a malignant lesion develops within NS, it generally is agreed that the tumor and the entire nevus should be removed; however, recommendations vary for excising NS prophylactically to decrease the risk for malignant growths. Because the risk for malignant transformation seems to be lower than previously thought, observation can be a reasonable choice for lesions that are not associated with cosmetic concern.12,13
Photodynamic therapy, CO2 laser resurfacing, and dermabrasion have been reported as alternative therapeutic approaches. However, there is a growing concern on how effective these treatment modalities are in completely removing the lesion and whether the risk for recurrence and potential for neoplasm development remains.1,9
This patient was healthy with normal development and growth and no signs of neurologic or ocular involvement. The parents were counseled about the risk for malignancy and the long-term cosmetic appearance of the lesion. They opted for surgical excision of the lesion at 18 months of age.
The Diagnosis: Nevus Sebaceous
The patient presented with a typical solitary scalp lesion characteristic of nevus sebaceous (NS). The lesion was present at birth as a flat and smooth hairless plaque; however, over time it became more thickened and noticeable, which prompted the parents to seek medical advice.
Nevus sebaceous, also known as NS of Jadassohn, is a benign congenital hamartoma of the sebaceous gland that usually is present at birth and frequently involves the scalp and/or the face. The classic NS lesion is solitary and appears as a well-circumscribed, waxy, yellow-orange or tan, hairless plaque. Despite the presence of these lesions at birth, they may not be noted until early childhood or rarely until adulthood. Generally, the lesion tends to thicken and become more verrucous and velvety over time, particularly around the time of reaching puberty.1 Clinically, NS lesions vary in size from 1 cm to several centimeters. Lesions initially tend to grow proportionately with the child until puberty when they become notably thicker, greasier, and verrucous or nodular under hormonal influences. The yellow discoloration of the lesion is due to sebaceous gland secretion, and the characteristic color usually becomes less evident with age.
Nevus sebaceous occurs in approximately 0.3% of newborns and tends to be sporadic in nature; however, rare familial forms have been reported.2,3 Nevus sebaceous can present as multiple nevi that tend to be extensive and distributed along the Blaschko lines, and they usually are associated with neurologic, ocular, or skeletal defects. Involvement of the central nervous system frequently is associated with large sebaceous nevi located on the face or scalp. This association has been termed NS syndrome.4 Neurologic abnormalities associated with NS syndrome include seizures, mental retardation, and hemimegalencephaly.5 Ocular findings most communally associated with the syndrome are choristomas and colobomas.6-8
There are several benign and malignant epithelial neoplasms that may develop within sebaceous nevi. Benign tumors include trichoblastoma, syringocystadenoma papilliferum, trichilemmoma, sebaceoma, nodular hidradenoma, and hidrocystoma.1,8,9 Malignant neoplasms include basal cell carcinoma (BCC), apocrine carcinoma, sebaceous carcinoma, and squamous cell carcinoma. The lifetime risk of malignancy in NS is unknown. In an extensive literature review by Moody et al10 of 4923 cases of NS for the development of secondary benign and malignant neoplasms, 16% developed benign tumors while 8% developed malignant tumors such as BCC. However, subsequent studies suggested that the incidence of BCC may have been overestimated due to misinterpretation of trichoblastoma and may be less than 1%.11-13
Usually the diagnosis of NS is made clinically and rarely a biopsy for histopathologic confirmation may be needed when the diagnosis is uncertain. Typically, these histopathologic findings include immature hair follicles, hyperplastic immature sebaceous glands, dilated apocrine glands, and epidermal hyperplasia.9 For patients with suspected NS syndrome, additional neurologic and ophthalmologic evaluations should be performed including neuroimaging studies, skeletal radiography, and analysis of liver and renal function.14
The current standard of care in treating NS is full-thickness excision. However, the decision should be individualized based on patient age, extension and location of the lesion, concerns about the cosmetic appearance, and the risk for malignancy.
The 2 main reasons to excise NS include concern about malignancy and undesirable cosmetic appearance. Once a malignant lesion develops within NS, it generally is agreed that the tumor and the entire nevus should be removed; however, recommendations vary for excising NS prophylactically to decrease the risk for malignant growths. Because the risk for malignant transformation seems to be lower than previously thought, observation can be a reasonable choice for lesions that are not associated with cosmetic concern.12,13
Photodynamic therapy, CO2 laser resurfacing, and dermabrasion have been reported as alternative therapeutic approaches. However, there is a growing concern on how effective these treatment modalities are in completely removing the lesion and whether the risk for recurrence and potential for neoplasm development remains.1,9
This patient was healthy with normal development and growth and no signs of neurologic or ocular involvement. The parents were counseled about the risk for malignancy and the long-term cosmetic appearance of the lesion. They opted for surgical excision of the lesion at 18 months of age.
- Eisen DB, Michael DJ. Sebaceous lesions and their associated syndromes: part I. J Am Acad Dermatol. 2009;61:549-560; quiz 561-562.
- Happle R, König A. Familial naevus sebaceus may be explained by paradominant transmission. Br J Dermatol. 1999;141:377.
- Hughes SM, Wilkerson AE, Winfield HL, et al. Familial nevus sebaceus in dizygotic male twins. J Am Acad Dermatol. 2006;54(2 suppl):S47-S48.
- Sugarman JL. Epidermal nevus syndromes. Semin Cutan Med Surg. 2007;26:221-230.
- Davies D, Rogers M. Review of neurological manifestations in 196 patients with sebaceous naevi. Australas J Dermatol. 2002;43:20-23.
- Trivedi N, Nehete G. Complex limbal choristoma in linear nevus sebaceous syndrome managed with scleral grafting. Indian J Ophthalmol. 2016;64:692-694.
- Nema N, Singh K, Verma A. Complex limbal choristoma in nevus sebaceous syndrome [published online February 14, 2012]. Pediatr Dermatol. 2012;29:227-229.
- Park JM, Kim DS, Kim J, et al. Epibulbar complex choristoma and hemimegalencephaly in linear sebaceous naevus syndrome [published online July 2, 2009]. Clin Exp Dermatol. 2009;34:E686-E689.
- Simi CM, Rajalakshmi T, Correa M. Clinicopathologic analysis of 21 cases of nevus sebaceus: a retrospective study. Indian J Dermatol Venereol Leprol. 2008;74:625-627.
- Moody MN, Landau JM, Goldberg LH. Nevus sebaceous revisited. Pediatr Dermatol. 2012;29:15-23.
- Cribier B, Scrivener Y, Grosshans E. Tumors arising in nevus sebaceus: a study of 596 cases. J Am Acad Dermatol. 2000;42(2 pt 1):263-268.
- Santibanez-Gallerani A, Marshall D, Duarte AM, et al. Should nevus sebaceus of Jadassohn in children be excised? a study of 757 cases, and literature review. J Craniofac Surg. 2003;14:658-660.
- Rosen H, Schmidt B, Lam HP, et al. Management of nevus sebaceous and the risk of basal cell carcinoma: an 18-year review. Pediatr Dermatol. 2009;26:676-681.
- Brandling-Bennett HA, Morel KD. Epidermal nevi. Pediatr Clin North Am. 2010;57:1177-1198.
- Eisen DB, Michael DJ. Sebaceous lesions and their associated syndromes: part I. J Am Acad Dermatol. 2009;61:549-560; quiz 561-562.
- Happle R, König A. Familial naevus sebaceus may be explained by paradominant transmission. Br J Dermatol. 1999;141:377.
- Hughes SM, Wilkerson AE, Winfield HL, et al. Familial nevus sebaceus in dizygotic male twins. J Am Acad Dermatol. 2006;54(2 suppl):S47-S48.
- Sugarman JL. Epidermal nevus syndromes. Semin Cutan Med Surg. 2007;26:221-230.
- Davies D, Rogers M. Review of neurological manifestations in 196 patients with sebaceous naevi. Australas J Dermatol. 2002;43:20-23.
- Trivedi N, Nehete G. Complex limbal choristoma in linear nevus sebaceous syndrome managed with scleral grafting. Indian J Ophthalmol. 2016;64:692-694.
- Nema N, Singh K, Verma A. Complex limbal choristoma in nevus sebaceous syndrome [published online February 14, 2012]. Pediatr Dermatol. 2012;29:227-229.
- Park JM, Kim DS, Kim J, et al. Epibulbar complex choristoma and hemimegalencephaly in linear sebaceous naevus syndrome [published online July 2, 2009]. Clin Exp Dermatol. 2009;34:E686-E689.
- Simi CM, Rajalakshmi T, Correa M. Clinicopathologic analysis of 21 cases of nevus sebaceus: a retrospective study. Indian J Dermatol Venereol Leprol. 2008;74:625-627.
- Moody MN, Landau JM, Goldberg LH. Nevus sebaceous revisited. Pediatr Dermatol. 2012;29:15-23.
- Cribier B, Scrivener Y, Grosshans E. Tumors arising in nevus sebaceus: a study of 596 cases. J Am Acad Dermatol. 2000;42(2 pt 1):263-268.
- Santibanez-Gallerani A, Marshall D, Duarte AM, et al. Should nevus sebaceus of Jadassohn in children be excised? a study of 757 cases, and literature review. J Craniofac Surg. 2003;14:658-660.
- Rosen H, Schmidt B, Lam HP, et al. Management of nevus sebaceous and the risk of basal cell carcinoma: an 18-year review. Pediatr Dermatol. 2009;26:676-681.
- Brandling-Bennett HA, Morel KD. Epidermal nevi. Pediatr Clin North Am. 2010;57:1177-1198.
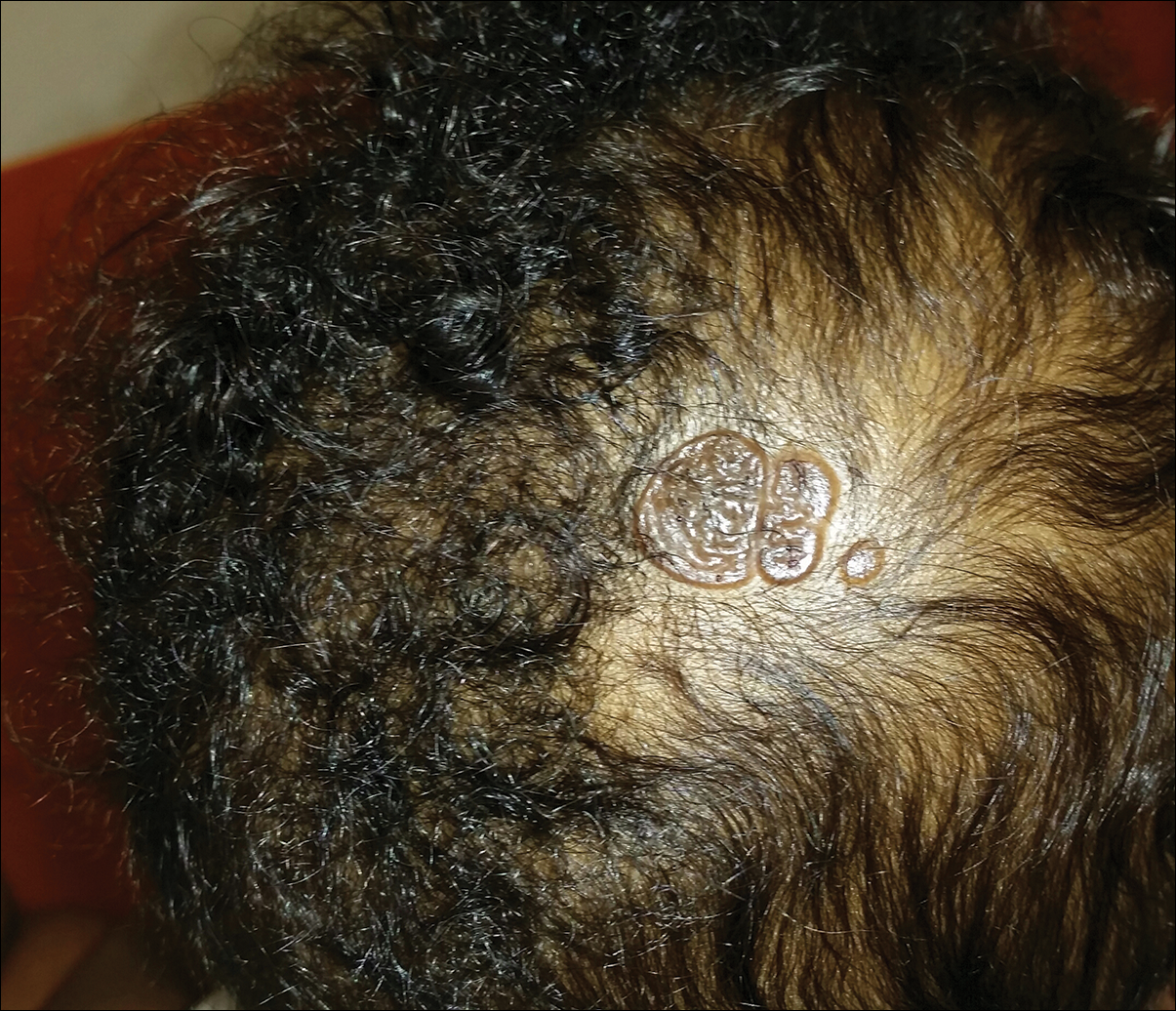
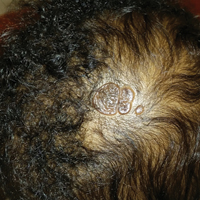
An otherwise healthy 13-month-old boy presented with a well-circumscribed, 3×4-cm, yellow-orange plaque with a verrucous velvety surface on the right side of the posterior scalp. The patient was born at 33 weeks' gestation and had an uneventful perinatal course with a normal head ultrasound at 4 days of age. The lesion had been present since birth and initially was comprised of waxy, yellow-orange, hairless plaques that became more thickened and noticeable over time. The mother recalled that the surface of the plaque initially was flat and smooth but gradually became bumpier and greasier in consistency in the months prior to presentation. The patient was otherwise asymptomatic.
Differentiating Trigeminal Motor Neuropathy and Progressive Hemifacial Atrophy
To the Editor:
Trigeminal motor neuropathy is a rare condition presenting with muscle weakness and atrophy in the distribution of the trigeminal nerve without sensory changes. We present a challenging case with clinical features that mimic progressive hemifacial atrophy (PHA), a disease characterized by slowly progressive, unilateral facial atrophy that can be accompanied by inflammation and sclerosis as early features.
A 55-year-old man presented with right-sided ptosis and progressive right-sided facial atrophy of 4 years’ duration. A clinical diagnosis of PHA was made by the rheumatology department, and the patient was referred to the dermatology department for further evaluation. Examination at presentation revealed right-sided subcutaneous atrophy of the cheek, temple, and forehead extending to the scalp with absence of sclerosis, pigmentary alteration, or typical linear morphea lesions (Figures 1 and 2). The patient had no sensory changes in the affected area.

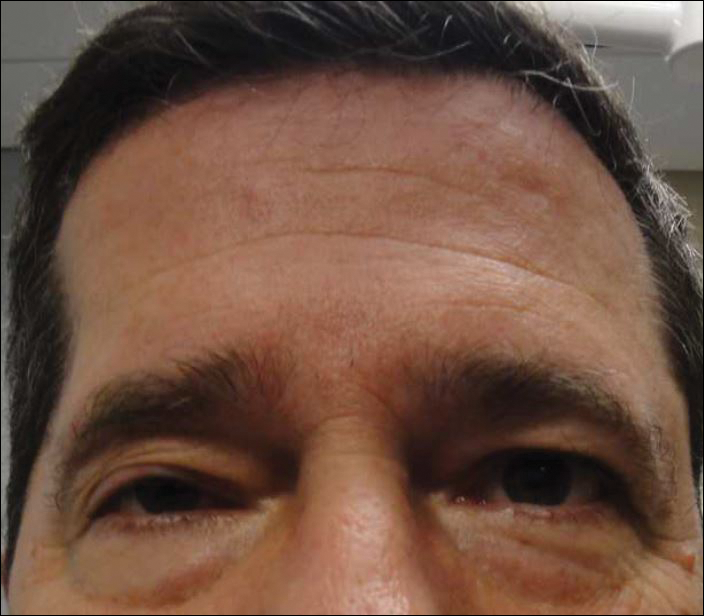
Workup by the dermatology department included magnetic resonance imaging (MRI) of the face and scalp, which demonstrated denervation muscle atrophy exclusively in the distribution of the third branch of the right trigeminal nerve, including severe atrophy of the right temporalis and masseter muscles and moderate atrophy of the pterygoid muscles. No signs of inflammation, fibrosis, or atrophy of the skin or subcutaneous fat were found, ruling out a diagnosis of PHA.
The patient was referred to the neurology department where he was found to have a normal neurologic examination with the exception of right-sided ptosis and temporalis and masseter muscle atrophy. Notably, the patient had normal sensation in the distribution of the trigeminal nerve and normal strength of the masseter and temporalis muscles.
An extensive workup by the neurology department was completed, including magnetic resonance angiography, eyeblink testing, and testing for causes of neuropathies (eg, infectious, autoimmune, vitamin deficiencies, toxin related). Of note, magnetic resonance angiography showed no abnormalities within the cavernous sinus or trigeminal cave but showed potential vascular compression of the trigeminal nerve, which was believed to be an incidental finding. The remainder of the workup was unremarkable. Based on muscle denervation atrophy in the distribution of the third branch of the trigeminal nerve in the absence of sensory symptoms or deficits, the patient’s presentation was consistent with trigeminal motor neuropathy.
In reported cases, the pathogenesis of trigeminal motor neuropathy is attributed to tumors, trauma, stroke, viral infection, and autoimmune reaction.1-6 In other reported cases the cause is unknown,6-8 as was the case in our patient. Magnetic resonance angiography revealed potential vascular compression of the trigeminal nerve, which has been previously reported to cause trigeminal neuropathy.9 However, patients with trigeminal neuropathy presented with sensory changes in the distribution of the trigeminal nerve as opposed to motor symptoms and muscle atrophy.
We present a case of trigeminal motor neuropathy presenting as PHA. Progressive hemifacial atrophy is a rare, slowly progressive disease characterized by unilateral atrophy of the skin, subcutis, muscle, and bony structures of the face. Onset usually is during childhood, though later onset has been reported.10 The pathogenesis of PHA is not well understood, though trauma, infection, immune-mediated causes, sympathetic dysfunction, and metabolic dysfunction have been proposed.11 Diagnosis of PHA typically is based on clinical presentation, but histology and imaging are useful. In contrast to trigeminal motor neuropathy, MRI findings in PHA demonstrate involvement of the skin.12
Differentiation between PHA and trigeminal motor neuropathy is important because treatment differs. Treatment of trigeminal motor neuropathy depends on the etiology and may include removal of underlying neoplasms, while treatment of PHA depends on disease activity. The initial goal when treating PHA is to improve symptoms and slow disease progression; immunosuppressants may be considered. Facial reconstruction is an option when PHA is stable.
In this case, the features differentiating trigeminal motor neuropathy from PHA include age of onset and MRI as well as clinical findings of muscle atrophy limited to the distribution of the third branch of the trigeminal nerve. Although PHA is a rare disorder, this case demonstrates the importance of including trigeminal motor neuropathy in the differential diagnosis.
- Beydoun SR. Unilateral trigeminal motor neuropathy as a presenting feature of neurofibromatosis type 2 (NF2). Muscle Nerve. 1993;16:1136-1137.
- Kang YK, Lee EH, Hwang M. Pure trigeminal motor neuropathy: a case report. Arch Phys Med Rehabil. 2000;81:995-998.
- Kim DH, Kim JK, Kang JY. Pure motor trigeminal neuropathy in a woman with tegmental pontine infarction. J Clin Neurosci. 2013;20:1792-1794.
- Ko KF, Chan KL. A case of isolated pure trigeminal motor neuropathy. Clin Neurol Neurosurg. 1995;97:199-200.
- Park KS, Chung JM, Jeon BS, et al. Unilateral trigeminal mandibular motor neuropathy caused by tumor in the foramen ovale. J Clin Neurol. 2006;2:194-197.
- Chia LG. Pure trigeminal motor neuropathy. Br Med J (Clin Res Ed). 1988;296:609-610.
- Braun JS, Hahn K, Bauknecht HC, et al. Progressive facial asymmetry due to trigeminal motor neuropathy. Eur Neurol. 2006;55:96-98.
- Chiba M, Echigo S. Unilateral atrophy of the masticatory muscles and mandibular ramus due to pure trigeminal motor neuropathy: a case report. Oral Surg Oral Med Oral Pathol Oral Radiol. 2012;113:E30-E34.
- Jannetta PJ, Robbins LJ. Trigeminal neuropathy—new observations. Neurosurgery. 1980;7:347-351.
- Stone J. Parry-Romberg syndrome: a global survey of 205 patients using the Internet. Neurology. 2003;61:674-676.
- El-Kehdy J, Abbas O, Rubeiz N. A review of Parry-Romberg syndrome. J Am Acad Dermatol. 2012;67:769-784.
- Taylor HM, Robinson R, Cox T. Progressive facial hemiatrophy: MRI appearances. Dev Med Child Neurol. 1997;39:484-486.
To the Editor:
Trigeminal motor neuropathy is a rare condition presenting with muscle weakness and atrophy in the distribution of the trigeminal nerve without sensory changes. We present a challenging case with clinical features that mimic progressive hemifacial atrophy (PHA), a disease characterized by slowly progressive, unilateral facial atrophy that can be accompanied by inflammation and sclerosis as early features.
A 55-year-old man presented with right-sided ptosis and progressive right-sided facial atrophy of 4 years’ duration. A clinical diagnosis of PHA was made by the rheumatology department, and the patient was referred to the dermatology department for further evaluation. Examination at presentation revealed right-sided subcutaneous atrophy of the cheek, temple, and forehead extending to the scalp with absence of sclerosis, pigmentary alteration, or typical linear morphea lesions (Figures 1 and 2). The patient had no sensory changes in the affected area.
Workup by the dermatology department included magnetic resonance imaging (MRI) of the face and scalp, which demonstrated denervation muscle atrophy exclusively in the distribution of the third branch of the right trigeminal nerve, including severe atrophy of the right temporalis and masseter muscles and moderate atrophy of the pterygoid muscles. No signs of inflammation, fibrosis, or atrophy of the skin or subcutaneous fat were found, ruling out a diagnosis of PHA.
The patient was referred to the neurology department where he was found to have a normal neurologic examination with the exception of right-sided ptosis and temporalis and masseter muscle atrophy. Notably, the patient had normal sensation in the distribution of the trigeminal nerve and normal strength of the masseter and temporalis muscles.
An extensive workup by the neurology department was completed, including magnetic resonance angiography, eyeblink testing, and testing for causes of neuropathies (eg, infectious, autoimmune, vitamin deficiencies, toxin related). Of note, magnetic resonance angiography showed no abnormalities within the cavernous sinus or trigeminal cave but showed potential vascular compression of the trigeminal nerve, which was believed to be an incidental finding. The remainder of the workup was unremarkable. Based on muscle denervation atrophy in the distribution of the third branch of the trigeminal nerve in the absence of sensory symptoms or deficits, the patient’s presentation was consistent with trigeminal motor neuropathy.
In reported cases, the pathogenesis of trigeminal motor neuropathy is attributed to tumors, trauma, stroke, viral infection, and autoimmune reaction.1-6 In other reported cases the cause is unknown,6-8 as was the case in our patient. Magnetic resonance angiography revealed potential vascular compression of the trigeminal nerve, which has been previously reported to cause trigeminal neuropathy.9 However, patients with trigeminal neuropathy presented with sensory changes in the distribution of the trigeminal nerve as opposed to motor symptoms and muscle atrophy.
We present a case of trigeminal motor neuropathy presenting as PHA. Progressive hemifacial atrophy is a rare, slowly progressive disease characterized by unilateral atrophy of the skin, subcutis, muscle, and bony structures of the face. Onset usually is during childhood, though later onset has been reported.10 The pathogenesis of PHA is not well understood, though trauma, infection, immune-mediated causes, sympathetic dysfunction, and metabolic dysfunction have been proposed.11 Diagnosis of PHA typically is based on clinical presentation, but histology and imaging are useful. In contrast to trigeminal motor neuropathy, MRI findings in PHA demonstrate involvement of the skin.12
Differentiation between PHA and trigeminal motor neuropathy is important because treatment differs. Treatment of trigeminal motor neuropathy depends on the etiology and may include removal of underlying neoplasms, while treatment of PHA depends on disease activity. The initial goal when treating PHA is to improve symptoms and slow disease progression; immunosuppressants may be considered. Facial reconstruction is an option when PHA is stable.
In this case, the features differentiating trigeminal motor neuropathy from PHA include age of onset and MRI as well as clinical findings of muscle atrophy limited to the distribution of the third branch of the trigeminal nerve. Although PHA is a rare disorder, this case demonstrates the importance of including trigeminal motor neuropathy in the differential diagnosis.
To the Editor:
Trigeminal motor neuropathy is a rare condition presenting with muscle weakness and atrophy in the distribution of the trigeminal nerve without sensory changes. We present a challenging case with clinical features that mimic progressive hemifacial atrophy (PHA), a disease characterized by slowly progressive, unilateral facial atrophy that can be accompanied by inflammation and sclerosis as early features.
A 55-year-old man presented with right-sided ptosis and progressive right-sided facial atrophy of 4 years’ duration. A clinical diagnosis of PHA was made by the rheumatology department, and the patient was referred to the dermatology department for further evaluation. Examination at presentation revealed right-sided subcutaneous atrophy of the cheek, temple, and forehead extending to the scalp with absence of sclerosis, pigmentary alteration, or typical linear morphea lesions (Figures 1 and 2). The patient had no sensory changes in the affected area.
Workup by the dermatology department included magnetic resonance imaging (MRI) of the face and scalp, which demonstrated denervation muscle atrophy exclusively in the distribution of the third branch of the right trigeminal nerve, including severe atrophy of the right temporalis and masseter muscles and moderate atrophy of the pterygoid muscles. No signs of inflammation, fibrosis, or atrophy of the skin or subcutaneous fat were found, ruling out a diagnosis of PHA.
The patient was referred to the neurology department where he was found to have a normal neurologic examination with the exception of right-sided ptosis and temporalis and masseter muscle atrophy. Notably, the patient had normal sensation in the distribution of the trigeminal nerve and normal strength of the masseter and temporalis muscles.
An extensive workup by the neurology department was completed, including magnetic resonance angiography, eyeblink testing, and testing for causes of neuropathies (eg, infectious, autoimmune, vitamin deficiencies, toxin related). Of note, magnetic resonance angiography showed no abnormalities within the cavernous sinus or trigeminal cave but showed potential vascular compression of the trigeminal nerve, which was believed to be an incidental finding. The remainder of the workup was unremarkable. Based on muscle denervation atrophy in the distribution of the third branch of the trigeminal nerve in the absence of sensory symptoms or deficits, the patient’s presentation was consistent with trigeminal motor neuropathy.
In reported cases, the pathogenesis of trigeminal motor neuropathy is attributed to tumors, trauma, stroke, viral infection, and autoimmune reaction.1-6 In other reported cases the cause is unknown,6-8 as was the case in our patient. Magnetic resonance angiography revealed potential vascular compression of the trigeminal nerve, which has been previously reported to cause trigeminal neuropathy.9 However, patients with trigeminal neuropathy presented with sensory changes in the distribution of the trigeminal nerve as opposed to motor symptoms and muscle atrophy.
We present a case of trigeminal motor neuropathy presenting as PHA. Progressive hemifacial atrophy is a rare, slowly progressive disease characterized by unilateral atrophy of the skin, subcutis, muscle, and bony structures of the face. Onset usually is during childhood, though later onset has been reported.10 The pathogenesis of PHA is not well understood, though trauma, infection, immune-mediated causes, sympathetic dysfunction, and metabolic dysfunction have been proposed.11 Diagnosis of PHA typically is based on clinical presentation, but histology and imaging are useful. In contrast to trigeminal motor neuropathy, MRI findings in PHA demonstrate involvement of the skin.12
Differentiation between PHA and trigeminal motor neuropathy is important because treatment differs. Treatment of trigeminal motor neuropathy depends on the etiology and may include removal of underlying neoplasms, while treatment of PHA depends on disease activity. The initial goal when treating PHA is to improve symptoms and slow disease progression; immunosuppressants may be considered. Facial reconstruction is an option when PHA is stable.
In this case, the features differentiating trigeminal motor neuropathy from PHA include age of onset and MRI as well as clinical findings of muscle atrophy limited to the distribution of the third branch of the trigeminal nerve. Although PHA is a rare disorder, this case demonstrates the importance of including trigeminal motor neuropathy in the differential diagnosis.
- Beydoun SR. Unilateral trigeminal motor neuropathy as a presenting feature of neurofibromatosis type 2 (NF2). Muscle Nerve. 1993;16:1136-1137.
- Kang YK, Lee EH, Hwang M. Pure trigeminal motor neuropathy: a case report. Arch Phys Med Rehabil. 2000;81:995-998.
- Kim DH, Kim JK, Kang JY. Pure motor trigeminal neuropathy in a woman with tegmental pontine infarction. J Clin Neurosci. 2013;20:1792-1794.
- Ko KF, Chan KL. A case of isolated pure trigeminal motor neuropathy. Clin Neurol Neurosurg. 1995;97:199-200.
- Park KS, Chung JM, Jeon BS, et al. Unilateral trigeminal mandibular motor neuropathy caused by tumor in the foramen ovale. J Clin Neurol. 2006;2:194-197.
- Chia LG. Pure trigeminal motor neuropathy. Br Med J (Clin Res Ed). 1988;296:609-610.
- Braun JS, Hahn K, Bauknecht HC, et al. Progressive facial asymmetry due to trigeminal motor neuropathy. Eur Neurol. 2006;55:96-98.
- Chiba M, Echigo S. Unilateral atrophy of the masticatory muscles and mandibular ramus due to pure trigeminal motor neuropathy: a case report. Oral Surg Oral Med Oral Pathol Oral Radiol. 2012;113:E30-E34.
- Jannetta PJ, Robbins LJ. Trigeminal neuropathy—new observations. Neurosurgery. 1980;7:347-351.
- Stone J. Parry-Romberg syndrome: a global survey of 205 patients using the Internet. Neurology. 2003;61:674-676.
- El-Kehdy J, Abbas O, Rubeiz N. A review of Parry-Romberg syndrome. J Am Acad Dermatol. 2012;67:769-784.
- Taylor HM, Robinson R, Cox T. Progressive facial hemiatrophy: MRI appearances. Dev Med Child Neurol. 1997;39:484-486.
- Beydoun SR. Unilateral trigeminal motor neuropathy as a presenting feature of neurofibromatosis type 2 (NF2). Muscle Nerve. 1993;16:1136-1137.
- Kang YK, Lee EH, Hwang M. Pure trigeminal motor neuropathy: a case report. Arch Phys Med Rehabil. 2000;81:995-998.
- Kim DH, Kim JK, Kang JY. Pure motor trigeminal neuropathy in a woman with tegmental pontine infarction. J Clin Neurosci. 2013;20:1792-1794.
- Ko KF, Chan KL. A case of isolated pure trigeminal motor neuropathy. Clin Neurol Neurosurg. 1995;97:199-200.
- Park KS, Chung JM, Jeon BS, et al. Unilateral trigeminal mandibular motor neuropathy caused by tumor in the foramen ovale. J Clin Neurol. 2006;2:194-197.
- Chia LG. Pure trigeminal motor neuropathy. Br Med J (Clin Res Ed). 1988;296:609-610.
- Braun JS, Hahn K, Bauknecht HC, et al. Progressive facial asymmetry due to trigeminal motor neuropathy. Eur Neurol. 2006;55:96-98.
- Chiba M, Echigo S. Unilateral atrophy of the masticatory muscles and mandibular ramus due to pure trigeminal motor neuropathy: a case report. Oral Surg Oral Med Oral Pathol Oral Radiol. 2012;113:E30-E34.
- Jannetta PJ, Robbins LJ. Trigeminal neuropathy—new observations. Neurosurgery. 1980;7:347-351.
- Stone J. Parry-Romberg syndrome: a global survey of 205 patients using the Internet. Neurology. 2003;61:674-676.
- El-Kehdy J, Abbas O, Rubeiz N. A review of Parry-Romberg syndrome. J Am Acad Dermatol. 2012;67:769-784.
- Taylor HM, Robinson R, Cox T. Progressive facial hemiatrophy: MRI appearances. Dev Med Child Neurol. 1997;39:484-486.
Practice Points
- The differential diagnosis of progressive hemifacial atrophy includes disorders of the trigeminal nerve.
- Trigeminal motor neuropathy presents with muscle weakness and atrophy without involvement of the skin, subcutis, or bone.
Pearls in Dermatology: 2017
The Pearls in Dermatology collection consists of our popular pearls from the year in one convenient file. Topics include:
- Nail psoriasis and psoriasis on the hands and feet
- Genital wart treatment
- Isotretinoin for acne
- Cosmeceuticals for rosacea
- Surgical technique with the flexible scalpel blade
Editor’s Commentary provided by Vincent A. DeLeo, MD, Editor-in-Chief, Cutis.
Save this collection, print it, and/or share it with your colleagues. We hope this comprehensive collection will positively impact how you manage patients.

The Pearls in Dermatology collection consists of our popular pearls from the year in one convenient file. Topics include:
- Nail psoriasis and psoriasis on the hands and feet
- Genital wart treatment
- Isotretinoin for acne
- Cosmeceuticals for rosacea
- Surgical technique with the flexible scalpel blade
Editor’s Commentary provided by Vincent A. DeLeo, MD, Editor-in-Chief, Cutis.
Save this collection, print it, and/or share it with your colleagues. We hope this comprehensive collection will positively impact how you manage patients.
The Pearls in Dermatology collection consists of our popular pearls from the year in one convenient file. Topics include:
- Nail psoriasis and psoriasis on the hands and feet
- Genital wart treatment
- Isotretinoin for acne
- Cosmeceuticals for rosacea
- Surgical technique with the flexible scalpel blade
Editor’s Commentary provided by Vincent A. DeLeo, MD, Editor-in-Chief, Cutis.
Save this collection, print it, and/or share it with your colleagues. We hope this comprehensive collection will positively impact how you manage patients.

Local Depigmentation of a Tattoo
The Diagnosis: Dermatofibroma
On dermoscopy, a central stellate, white, scarlike patch was seen (Figure). On both legs the patient had several additional brown 5- to 7-mm papules with similar dermoscopic features.
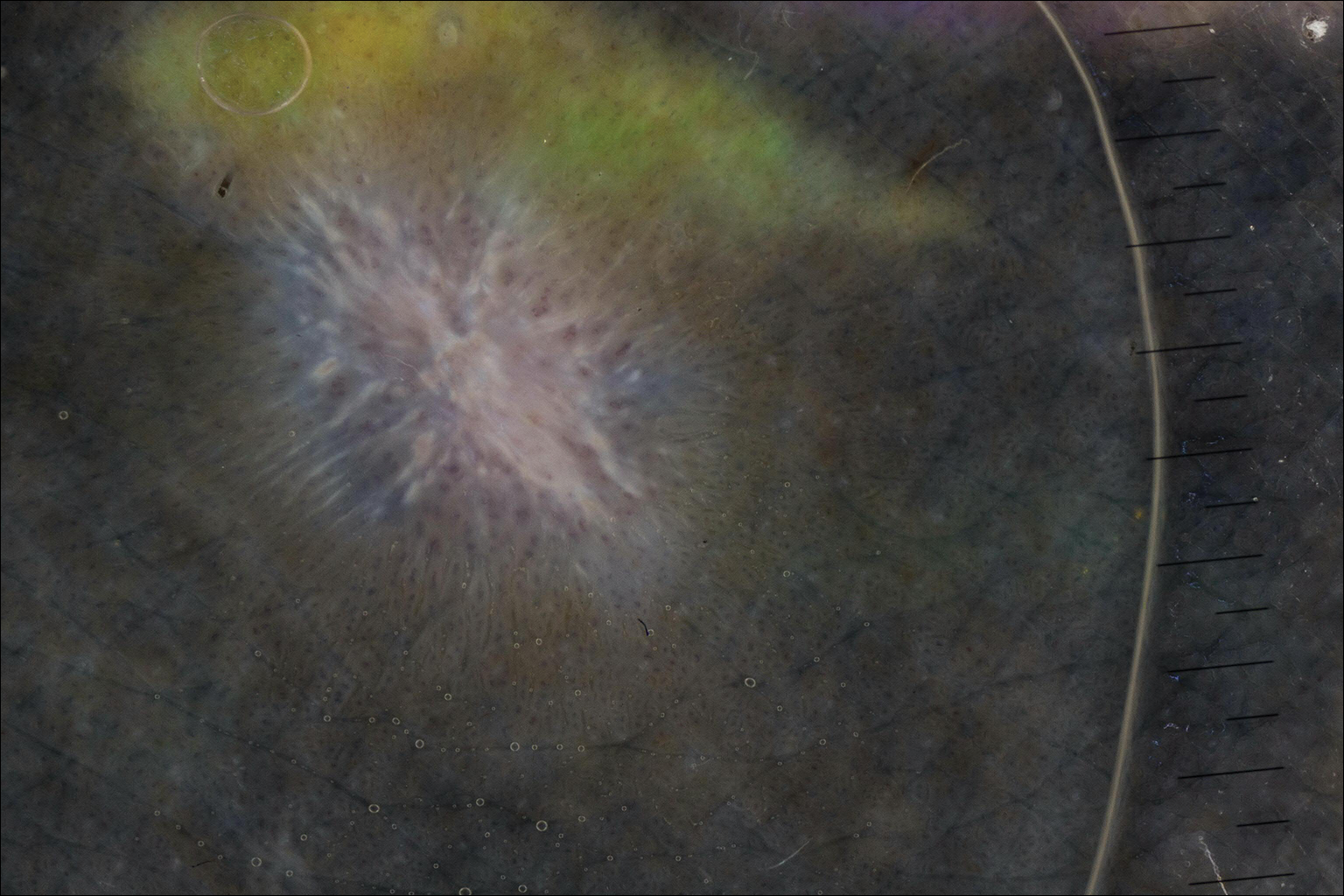
Dermatofibromas are common benign fibrosing tumors that appear as firm papules or plaques with variable color, commonly on the legs. Typically, lateral compression of a dermatofibroma causes downward displacement, called a positive dimple sign. On histology, fibroblasts and myofibroblasts can be seen as short intersecting fascicles with variable inflammatory cells and induction of adjacent structure hyperplasia. The etiology of dermatofibromas is unclear, though some are thought to be secondary to trauma or arthropod bites.1 Because these tumors are benign, the correct diagnosis can avoid unnecessary biopsies or other procedures.
The dermoscopic features of dermatofibromas have been well established.2 As perhaps the most easily identified structure, scarlike patches were seen in as many as 92% (22/24) of dermatofibromas in one study by Ferarri et al,3 while pigment networks also are commonly seen.2 In our case, given the surrounding dense tattoo deposition, it was difficult to ascertain any pigment network. However, the scarlike central patch was clearly apparent by dermoscopy.
Because dermatofibromas are hypothesized to be secondary to trauma, presumably applying tattoos also may cause dermatofibromas. Limited cases have described dermatofibromas arising in tattoos applied several months to years prior.4-6 No prior cases utilized dermoscopy. In our case, clinical examination and dermoscopy clearly demonstrated features consistent with a dermatofibroma, and the patient had more characteristic dermatofibromas scattered elsewhere on both legs. The patient was reassured that the lesions were benign and that the depigmentation was likely secondary to the process of dermatofibroma growth. She declined any treatment.
- Bolognia J, Jorizzo JL, Schaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Saunders; 2012.
- Zaballos P, Puig S, Llambrich A, et al. Dermoscopy of dermatofibromas: a prospective morphological study of 412 cases. Arch Dermatol. 2008;144:75-83.
- Ferrari A, Soyer HP, Peris K, et al. Central white scarlike patch: a dermatoscopic clue for the diagnosis of dermatofibroma. J Am Acad Dermatol. 2000;43:1123-1125.
- Kluger N, Cotten H, Magana C, et al. Dermatofibroma occurring within a tattoo: report of two cases. J Cutan Pathol. 2008;35:696-698.
- Lobato-Berezo A, Churruca-Grijelmo M, Martínez-Pérez M, et al. Dermatofibroma arising within a black tattoo [published online September 23, 2014]. Case Rep Dermatol Med. 2014;2014:745304.
- Bittencourt Mde J, Miranda MF, Parijós AM, et al. Dermatofibroma in a black tattoo: report of a case. An Bras Dermatol. 2013;88:614-616.
The Diagnosis: Dermatofibroma
On dermoscopy, a central stellate, white, scarlike patch was seen (Figure). On both legs the patient had several additional brown 5- to 7-mm papules with similar dermoscopic features.
Dermatofibromas are common benign fibrosing tumors that appear as firm papules or plaques with variable color, commonly on the legs. Typically, lateral compression of a dermatofibroma causes downward displacement, called a positive dimple sign. On histology, fibroblasts and myofibroblasts can be seen as short intersecting fascicles with variable inflammatory cells and induction of adjacent structure hyperplasia. The etiology of dermatofibromas is unclear, though some are thought to be secondary to trauma or arthropod bites.1 Because these tumors are benign, the correct diagnosis can avoid unnecessary biopsies or other procedures.
The dermoscopic features of dermatofibromas have been well established.2 As perhaps the most easily identified structure, scarlike patches were seen in as many as 92% (22/24) of dermatofibromas in one study by Ferarri et al,3 while pigment networks also are commonly seen.2 In our case, given the surrounding dense tattoo deposition, it was difficult to ascertain any pigment network. However, the scarlike central patch was clearly apparent by dermoscopy.
Because dermatofibromas are hypothesized to be secondary to trauma, presumably applying tattoos also may cause dermatofibromas. Limited cases have described dermatofibromas arising in tattoos applied several months to years prior.4-6 No prior cases utilized dermoscopy. In our case, clinical examination and dermoscopy clearly demonstrated features consistent with a dermatofibroma, and the patient had more characteristic dermatofibromas scattered elsewhere on both legs. The patient was reassured that the lesions were benign and that the depigmentation was likely secondary to the process of dermatofibroma growth. She declined any treatment.
The Diagnosis: Dermatofibroma
On dermoscopy, a central stellate, white, scarlike patch was seen (Figure). On both legs the patient had several additional brown 5- to 7-mm papules with similar dermoscopic features.
Dermatofibromas are common benign fibrosing tumors that appear as firm papules or plaques with variable color, commonly on the legs. Typically, lateral compression of a dermatofibroma causes downward displacement, called a positive dimple sign. On histology, fibroblasts and myofibroblasts can be seen as short intersecting fascicles with variable inflammatory cells and induction of adjacent structure hyperplasia. The etiology of dermatofibromas is unclear, though some are thought to be secondary to trauma or arthropod bites.1 Because these tumors are benign, the correct diagnosis can avoid unnecessary biopsies or other procedures.
The dermoscopic features of dermatofibromas have been well established.2 As perhaps the most easily identified structure, scarlike patches were seen in as many as 92% (22/24) of dermatofibromas in one study by Ferarri et al,3 while pigment networks also are commonly seen.2 In our case, given the surrounding dense tattoo deposition, it was difficult to ascertain any pigment network. However, the scarlike central patch was clearly apparent by dermoscopy.
Because dermatofibromas are hypothesized to be secondary to trauma, presumably applying tattoos also may cause dermatofibromas. Limited cases have described dermatofibromas arising in tattoos applied several months to years prior.4-6 No prior cases utilized dermoscopy. In our case, clinical examination and dermoscopy clearly demonstrated features consistent with a dermatofibroma, and the patient had more characteristic dermatofibromas scattered elsewhere on both legs. The patient was reassured that the lesions were benign and that the depigmentation was likely secondary to the process of dermatofibroma growth. She declined any treatment.
- Bolognia J, Jorizzo JL, Schaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Saunders; 2012.
- Zaballos P, Puig S, Llambrich A, et al. Dermoscopy of dermatofibromas: a prospective morphological study of 412 cases. Arch Dermatol. 2008;144:75-83.
- Ferrari A, Soyer HP, Peris K, et al. Central white scarlike patch: a dermatoscopic clue for the diagnosis of dermatofibroma. J Am Acad Dermatol. 2000;43:1123-1125.
- Kluger N, Cotten H, Magana C, et al. Dermatofibroma occurring within a tattoo: report of two cases. J Cutan Pathol. 2008;35:696-698.
- Lobato-Berezo A, Churruca-Grijelmo M, Martínez-Pérez M, et al. Dermatofibroma arising within a black tattoo [published online September 23, 2014]. Case Rep Dermatol Med. 2014;2014:745304.
- Bittencourt Mde J, Miranda MF, Parijós AM, et al. Dermatofibroma in a black tattoo: report of a case. An Bras Dermatol. 2013;88:614-616.
- Bolognia J, Jorizzo JL, Schaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Saunders; 2012.
- Zaballos P, Puig S, Llambrich A, et al. Dermoscopy of dermatofibromas: a prospective morphological study of 412 cases. Arch Dermatol. 2008;144:75-83.
- Ferrari A, Soyer HP, Peris K, et al. Central white scarlike patch: a dermatoscopic clue for the diagnosis of dermatofibroma. J Am Acad Dermatol. 2000;43:1123-1125.
- Kluger N, Cotten H, Magana C, et al. Dermatofibroma occurring within a tattoo: report of two cases. J Cutan Pathol. 2008;35:696-698.
- Lobato-Berezo A, Churruca-Grijelmo M, Martínez-Pérez M, et al. Dermatofibroma arising within a black tattoo [published online September 23, 2014]. Case Rep Dermatol Med. 2014;2014:745304.
- Bittencourt Mde J, Miranda MF, Parijós AM, et al. Dermatofibroma in a black tattoo: report of a case. An Bras Dermatol. 2013;88:614-616.
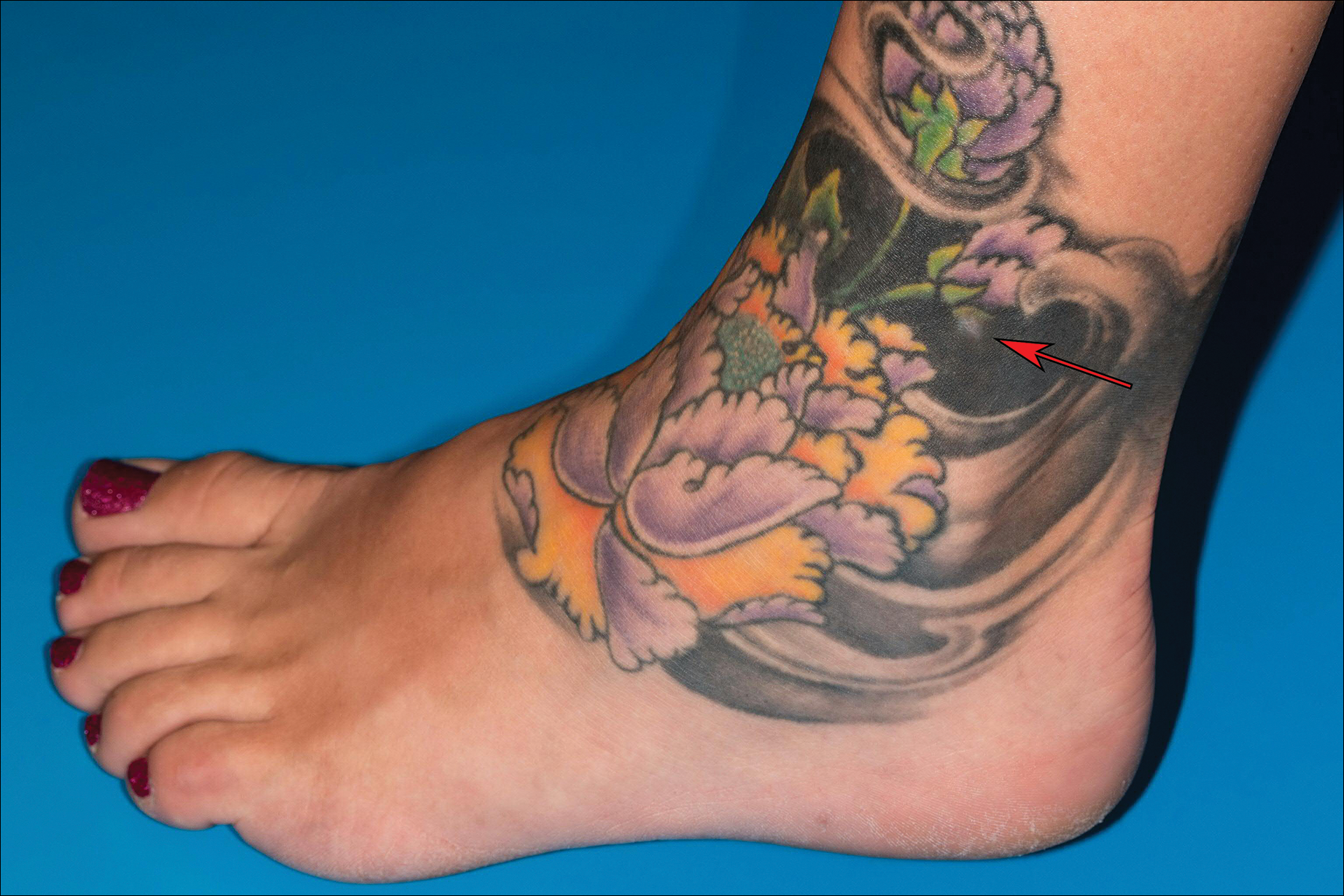
A 41-year-old woman presented with loss of pigment in a tattoo on the left ankle. The tattoo was initially placed several years prior to presentation. For an uncertain amount of time, she had noticed a small palpable whitish area with loss of tattoo pigment. There was no corresponding pain, pruritis, or other symptoms. Her dermatologic history was notable only for keratosis pilaris. Physical examination showed an approximately 7-mm whitish firm papule on the lateral aspect of the left ankle, clearly visible in an otherwise green-black area of the tattoo (arrow). The lesion displaced downward with lateral compression.
Debunking Atopic Dermatitis Myths: Should Patients Avoid Products With Parabens?
Myth: Parabens are dangerous
Some atopic dermatitis (AD) patients may be misinformed by reports that parabens have estrogenic and antiandrogenic effects and may be involved in carcinogenesis via endocrine modulation. Although in Europe some parabens have been banned or restricted, in the United States there are no regulations against the use of parabens in cosmetics. Dermatologists must acknowledge that their AD patients may have concerns about cosmetic products and they must be prepared to dispel any myths.
Parabens such as methylparaben, propylparaben, butylparaben, and ethylparaben are common in cosmetics such as moisturizers. Parabens have protective properties to prevent the growth of harmful bacteria and mold. According to the US Food and Drug Administration, “scientists continue to review published studies on the safety of parabens. At this time, we do not have information showing that parabens as they are used in cosmetics have an effect on human health. . . . If we determine that a health hazard exists, we will advise the industry and the public.”
Here are some important facts to note for patients, based on a research article published in Cosmetics & Toiletries in June 2017:
- Parabens are not toxic at the concentrations used in personal care products
- Parabens are not genotoxic or carcinogenic
- Parabens are readily excreted in urine and do not accumulate in tissues
In patients with chronic dermatitis, the Cosmetic Ingredient Review Expert Panel reported that parabens generally induce sensitization in less than 4% of patients. The panel concluded that they can support the safety of cosmetic products in which parabens are used as preservatives.
In fact, one study published in the Journal of the American Academy of Dermatology found that AD patients were not predisposed to allergies to parabens, formaldehyde, or diazolidinyl urea, but they were more likely to have allergic reactions to formaldehyde releasers. As a result, AD patients should choose moisturizers containing parabens and should have no fears about using them.
Expert Commentary
In general I recommend paraben-containing cleansers and emollients on a daily basis in practice. However, patient concerns exist due to negative online content easily accessed and fear can prevent usage of agents. Therefore, I am also open to offering options lacking in parabens, such as coconut oil.
—Nanette B. Silverberg, MD (New York, New York)
Doyle K. Some skin creams bad news for eczema. Reuters. December 12, 2013. https://www.reuters.com/article/us-skin-creams-eczema/some-skin-creams-bad-news-for-eczema-idUSBRE9BB14720131212. Accessed January 12, 2018.
Final amended report on the safety assessment of Methylparaben, Ethylparaben, Propylparaben, Isopropylparaben, Butylparaben, Isobutylparaben, and Benzylparaben as used in cosmetic products. Int J Toxicol. 2008;27(suppl 4):1-82.
Krowka JF, Loretz L, Geis PA, et al. Preserving the facts on parabens: an overview of these important tools of the trade. Cosmetics & Toiletries. June 1, 2017. http://www.cosmeticsandtoiletries.com/research/chemistry/Preserving-the-Facts-on-Parabens-An-Overview-of-These-Important-Tools-of-the-Trade-425784294.html. Accessed January 12, 2018.
Parabens in cosmetics. US Food and Drug Administration website. https://www.fda.gov/Cosmetics/ProductsIngredients/Ingredients/ucm128042.html. Accessed January 12, 2018.
Sasseville D, Alfalah M, Lacroix JP. “Parabenoia” debunked, or “who’s afraid of parabens?” Dermatitis. 2015;26:254-259.
Shaughnessy CN, Malajian D, Belsito DV. Cutaneous delayed-type hypersensitivity in patients with atopic dermatitis: reactivity to topical preservatives. J Am Acad Dermatol. 2014;70:102-107.
Myth: Parabens are dangerous
Some atopic dermatitis (AD) patients may be misinformed by reports that parabens have estrogenic and antiandrogenic effects and may be involved in carcinogenesis via endocrine modulation. Although in Europe some parabens have been banned or restricted, in the United States there are no regulations against the use of parabens in cosmetics. Dermatologists must acknowledge that their AD patients may have concerns about cosmetic products and they must be prepared to dispel any myths.
Parabens such as methylparaben, propylparaben, butylparaben, and ethylparaben are common in cosmetics such as moisturizers. Parabens have protective properties to prevent the growth of harmful bacteria and mold. According to the US Food and Drug Administration, “scientists continue to review published studies on the safety of parabens. At this time, we do not have information showing that parabens as they are used in cosmetics have an effect on human health. . . . If we determine that a health hazard exists, we will advise the industry and the public.”
Here are some important facts to note for patients, based on a research article published in Cosmetics & Toiletries in June 2017:
- Parabens are not toxic at the concentrations used in personal care products
- Parabens are not genotoxic or carcinogenic
- Parabens are readily excreted in urine and do not accumulate in tissues
In patients with chronic dermatitis, the Cosmetic Ingredient Review Expert Panel reported that parabens generally induce sensitization in less than 4% of patients. The panel concluded that they can support the safety of cosmetic products in which parabens are used as preservatives.
In fact, one study published in the Journal of the American Academy of Dermatology found that AD patients were not predisposed to allergies to parabens, formaldehyde, or diazolidinyl urea, but they were more likely to have allergic reactions to formaldehyde releasers. As a result, AD patients should choose moisturizers containing parabens and should have no fears about using them.
Expert Commentary
In general I recommend paraben-containing cleansers and emollients on a daily basis in practice. However, patient concerns exist due to negative online content easily accessed and fear can prevent usage of agents. Therefore, I am also open to offering options lacking in parabens, such as coconut oil.
—Nanette B. Silverberg, MD (New York, New York)
Myth: Parabens are dangerous
Some atopic dermatitis (AD) patients may be misinformed by reports that parabens have estrogenic and antiandrogenic effects and may be involved in carcinogenesis via endocrine modulation. Although in Europe some parabens have been banned or restricted, in the United States there are no regulations against the use of parabens in cosmetics. Dermatologists must acknowledge that their AD patients may have concerns about cosmetic products and they must be prepared to dispel any myths.
Parabens such as methylparaben, propylparaben, butylparaben, and ethylparaben are common in cosmetics such as moisturizers. Parabens have protective properties to prevent the growth of harmful bacteria and mold. According to the US Food and Drug Administration, “scientists continue to review published studies on the safety of parabens. At this time, we do not have information showing that parabens as they are used in cosmetics have an effect on human health. . . . If we determine that a health hazard exists, we will advise the industry and the public.”
Here are some important facts to note for patients, based on a research article published in Cosmetics & Toiletries in June 2017:
- Parabens are not toxic at the concentrations used in personal care products
- Parabens are not genotoxic or carcinogenic
- Parabens are readily excreted in urine and do not accumulate in tissues
In patients with chronic dermatitis, the Cosmetic Ingredient Review Expert Panel reported that parabens generally induce sensitization in less than 4% of patients. The panel concluded that they can support the safety of cosmetic products in which parabens are used as preservatives.
In fact, one study published in the Journal of the American Academy of Dermatology found that AD patients were not predisposed to allergies to parabens, formaldehyde, or diazolidinyl urea, but they were more likely to have allergic reactions to formaldehyde releasers. As a result, AD patients should choose moisturizers containing parabens and should have no fears about using them.
Expert Commentary
In general I recommend paraben-containing cleansers and emollients on a daily basis in practice. However, patient concerns exist due to negative online content easily accessed and fear can prevent usage of agents. Therefore, I am also open to offering options lacking in parabens, such as coconut oil.
—Nanette B. Silverberg, MD (New York, New York)
Doyle K. Some skin creams bad news for eczema. Reuters. December 12, 2013. https://www.reuters.com/article/us-skin-creams-eczema/some-skin-creams-bad-news-for-eczema-idUSBRE9BB14720131212. Accessed January 12, 2018.
Final amended report on the safety assessment of Methylparaben, Ethylparaben, Propylparaben, Isopropylparaben, Butylparaben, Isobutylparaben, and Benzylparaben as used in cosmetic products. Int J Toxicol. 2008;27(suppl 4):1-82.
Krowka JF, Loretz L, Geis PA, et al. Preserving the facts on parabens: an overview of these important tools of the trade. Cosmetics & Toiletries. June 1, 2017. http://www.cosmeticsandtoiletries.com/research/chemistry/Preserving-the-Facts-on-Parabens-An-Overview-of-These-Important-Tools-of-the-Trade-425784294.html. Accessed January 12, 2018.
Parabens in cosmetics. US Food and Drug Administration website. https://www.fda.gov/Cosmetics/ProductsIngredients/Ingredients/ucm128042.html. Accessed January 12, 2018.
Sasseville D, Alfalah M, Lacroix JP. “Parabenoia” debunked, or “who’s afraid of parabens?” Dermatitis. 2015;26:254-259.
Shaughnessy CN, Malajian D, Belsito DV. Cutaneous delayed-type hypersensitivity in patients with atopic dermatitis: reactivity to topical preservatives. J Am Acad Dermatol. 2014;70:102-107.
Doyle K. Some skin creams bad news for eczema. Reuters. December 12, 2013. https://www.reuters.com/article/us-skin-creams-eczema/some-skin-creams-bad-news-for-eczema-idUSBRE9BB14720131212. Accessed January 12, 2018.
Final amended report on the safety assessment of Methylparaben, Ethylparaben, Propylparaben, Isopropylparaben, Butylparaben, Isobutylparaben, and Benzylparaben as used in cosmetic products. Int J Toxicol. 2008;27(suppl 4):1-82.
Krowka JF, Loretz L, Geis PA, et al. Preserving the facts on parabens: an overview of these important tools of the trade. Cosmetics & Toiletries. June 1, 2017. http://www.cosmeticsandtoiletries.com/research/chemistry/Preserving-the-Facts-on-Parabens-An-Overview-of-These-Important-Tools-of-the-Trade-425784294.html. Accessed January 12, 2018.
Parabens in cosmetics. US Food and Drug Administration website. https://www.fda.gov/Cosmetics/ProductsIngredients/Ingredients/ucm128042.html. Accessed January 12, 2018.
Sasseville D, Alfalah M, Lacroix JP. “Parabenoia” debunked, or “who’s afraid of parabens?” Dermatitis. 2015;26:254-259.
Shaughnessy CN, Malajian D, Belsito DV. Cutaneous delayed-type hypersensitivity in patients with atopic dermatitis: reactivity to topical preservatives. J Am Acad Dermatol. 2014;70:102-107.

Disfiguring Ulcerative Neutrophilic Dermatosis Secondary to Doxycycline and Isotretinoin in an Adolescent Boy With Acne Conglobata
Acne fulminans is an uncommon and debilitating disease that presents as an acute eruption of nodular and ulcerative acne lesions with associated systemic symptoms.1,2 Although its underlying pathophysiology is not well understood, it occurs commonly during treatment of severe acne (eg, acne conglobata) with isotretinoin in young adolescent males.3 Zaba et al4 indicated that an underlying genetic disorder, increase in serum androgen levels, or presence of autoimmune disorders may contribute to the development of acne fulminans.
Isotretinoin and doxycycline also can potentially induce development of neutrophilic dermatoses including Sweet syndrome and pyoderma gangrenosum in patients with severe acne lesions, which can be clinically similar to an acne fulminans eruption. The neutrophilic dermatosis is characterized by the acute appearance of painful ulcerative papulonodules accompanied by systemic symptoms including fever and leukocytosis.
Case Report
A 13-year-old adolescent boy was initially assessed by his family physician 2 months prior and started on oral doxycycline 100 mg twice daily for acne conglobata on the back. Unfortunately, the acne lesions, especially those on the upper back (Figure 1), started getting worse after 1 month of treatment with doxycycline; thus, he subsequently was switched to oral isotretinoin 0.5 mg/kg once daily. Less than 2 weeks later, the acne lesions worsened, and the patient also developed severe generalized arthralgia, myalgia, and fever (>38.3°C). He acutely developed hundreds of ulcerative plaques covering the entire trunk, upper extremities, face, and neck.
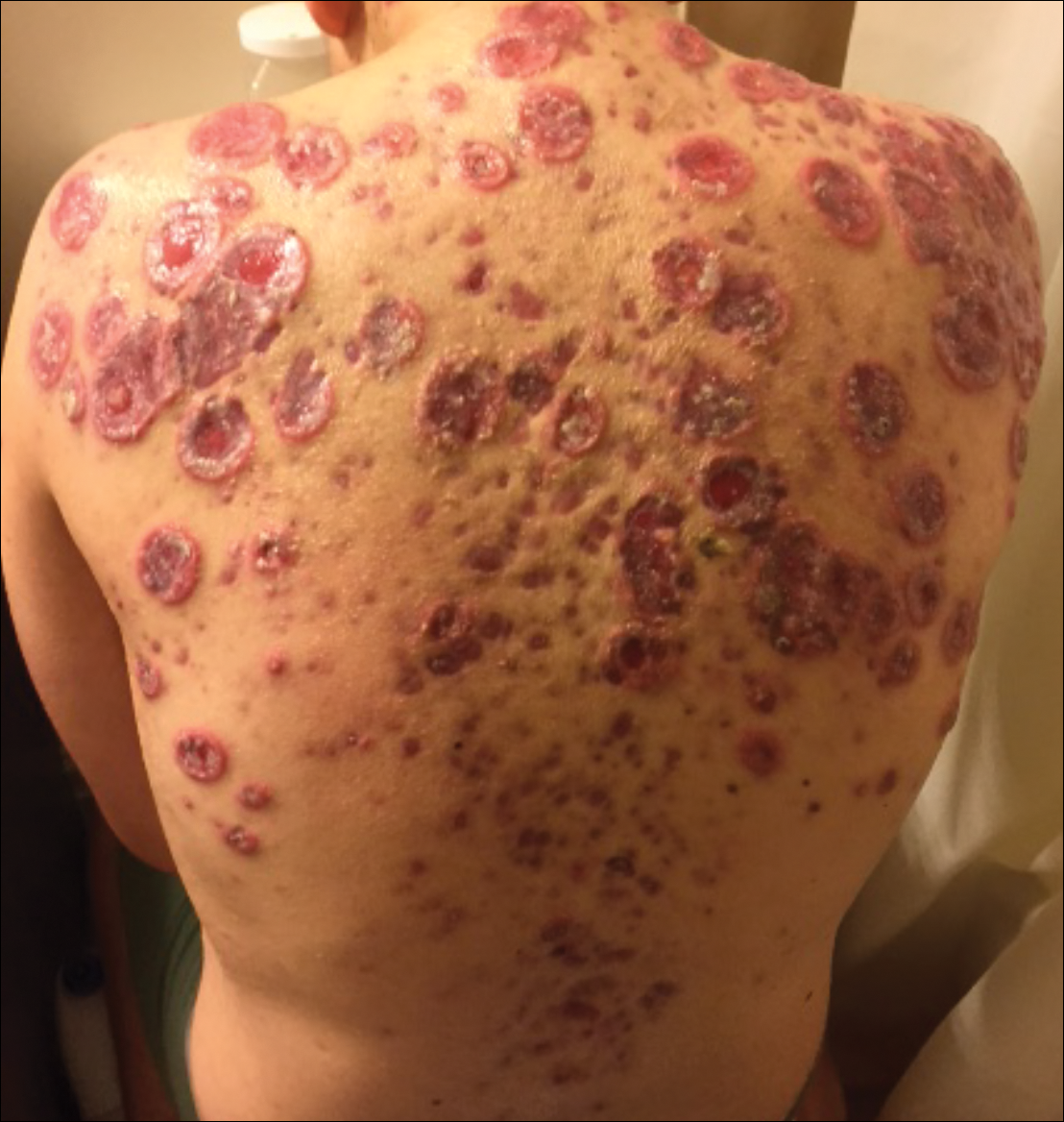
He was admitted to the Stollery Children’s Hospital (Edmonton, Alberta, Canada) and was assessed by the dermatology, rheumatology, and general pediatric teams (Figure 2). He initially was investigated for the potential presence of autoinflammatory disorders, such as PAPA syndrome (pyogenic arthritis, pyoderma gangrenosum, acne) and SAPHO syndrome (synovitis, acne, pustulosis, hyperostosis, osteitis).
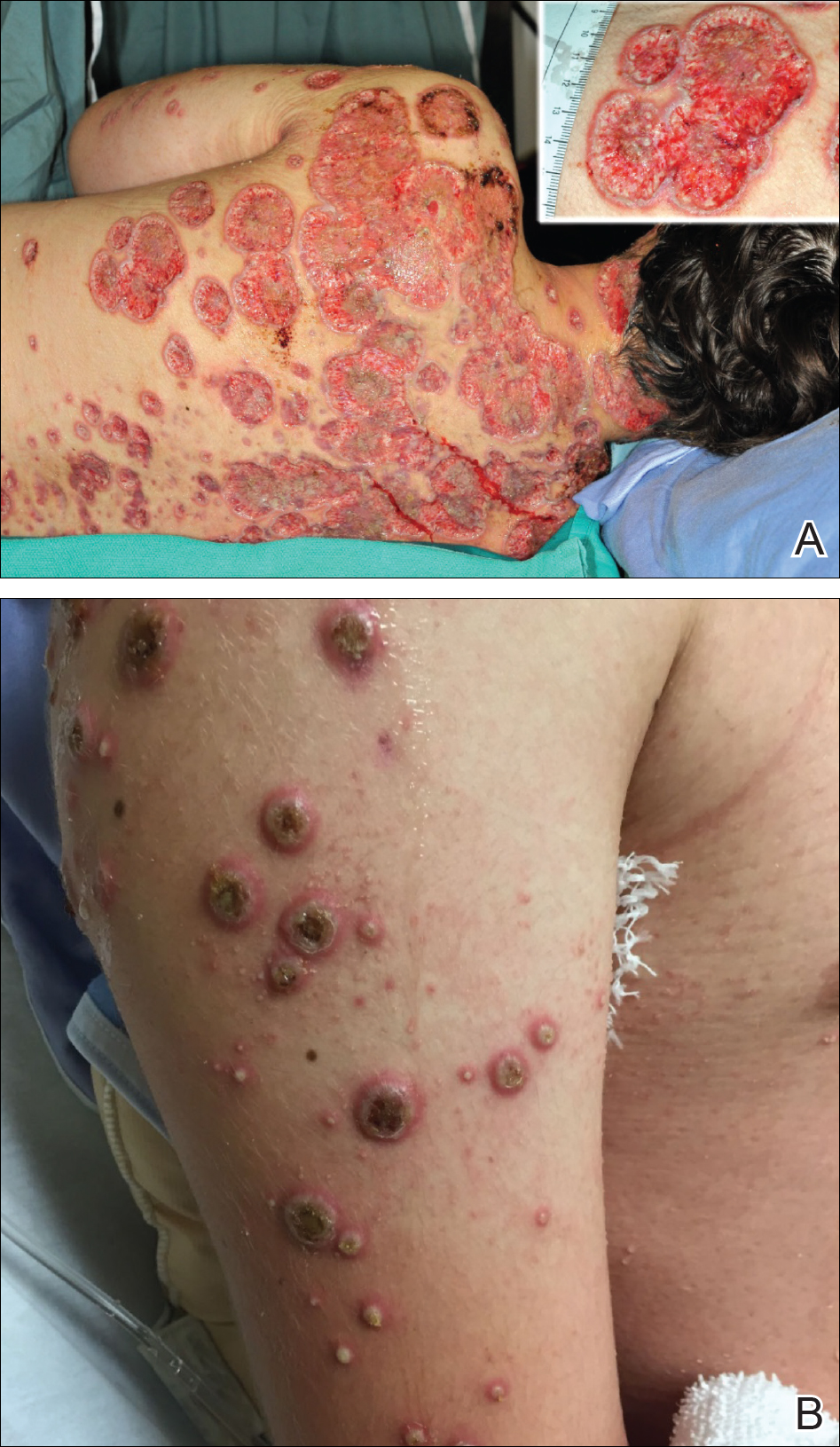
The patient initially was treated with prednisone 30 mg once daily for 3 weeks; dapsone 50 mg once daily and colchicine 0.6 mg twice daily were added while attempting to slowly wean off the prednisone (starting at 30 mg daily and reducing by 5 mg every other week). An attempt to discontinue the prednisone after 2 months was followed by immediate recurrence of the lesions (Figure 3), and the prednisone was restarted for another month. He was subsequently switched to oral cyclosporine 5 mg/kg once daily and achieved considerable improvement in his skin condition (Figure 4).
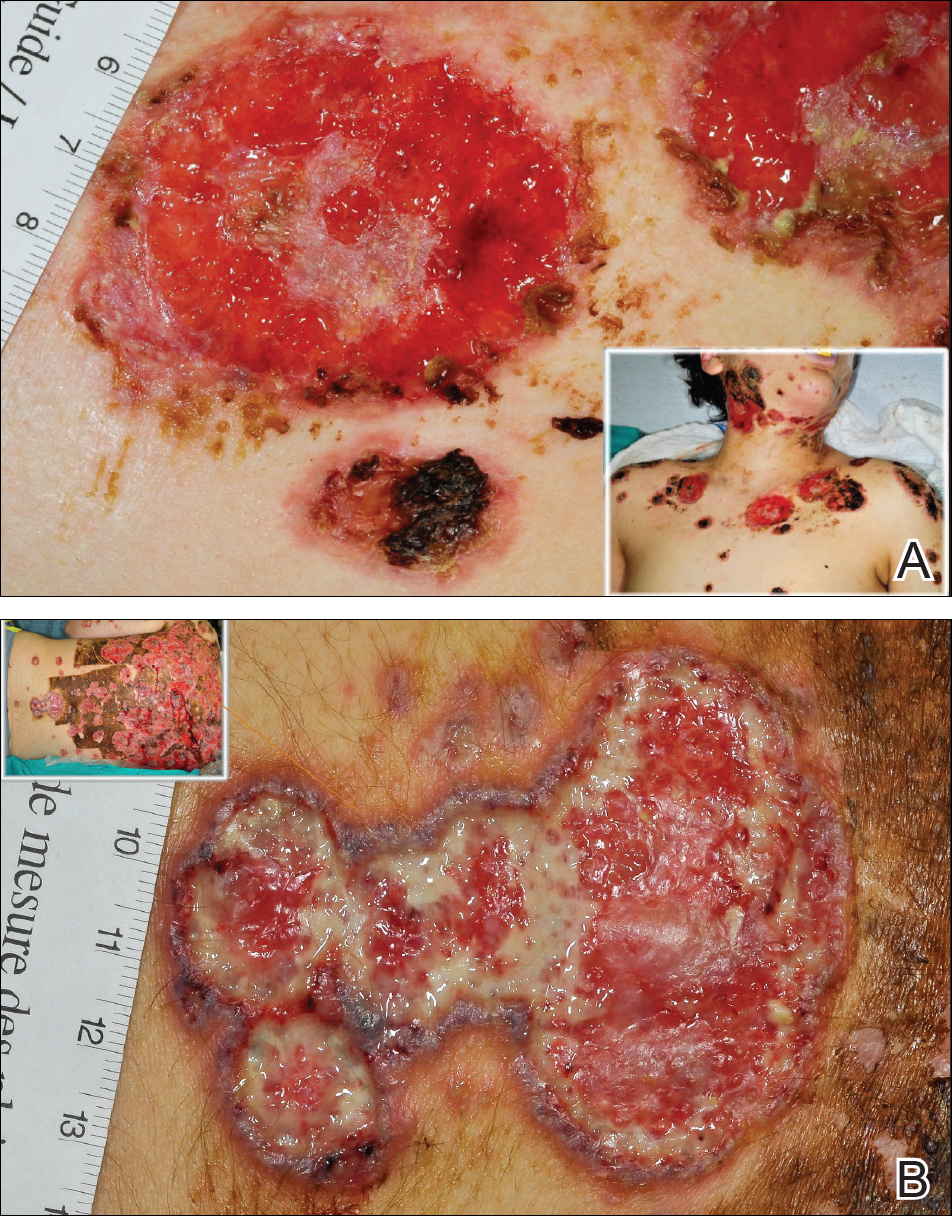
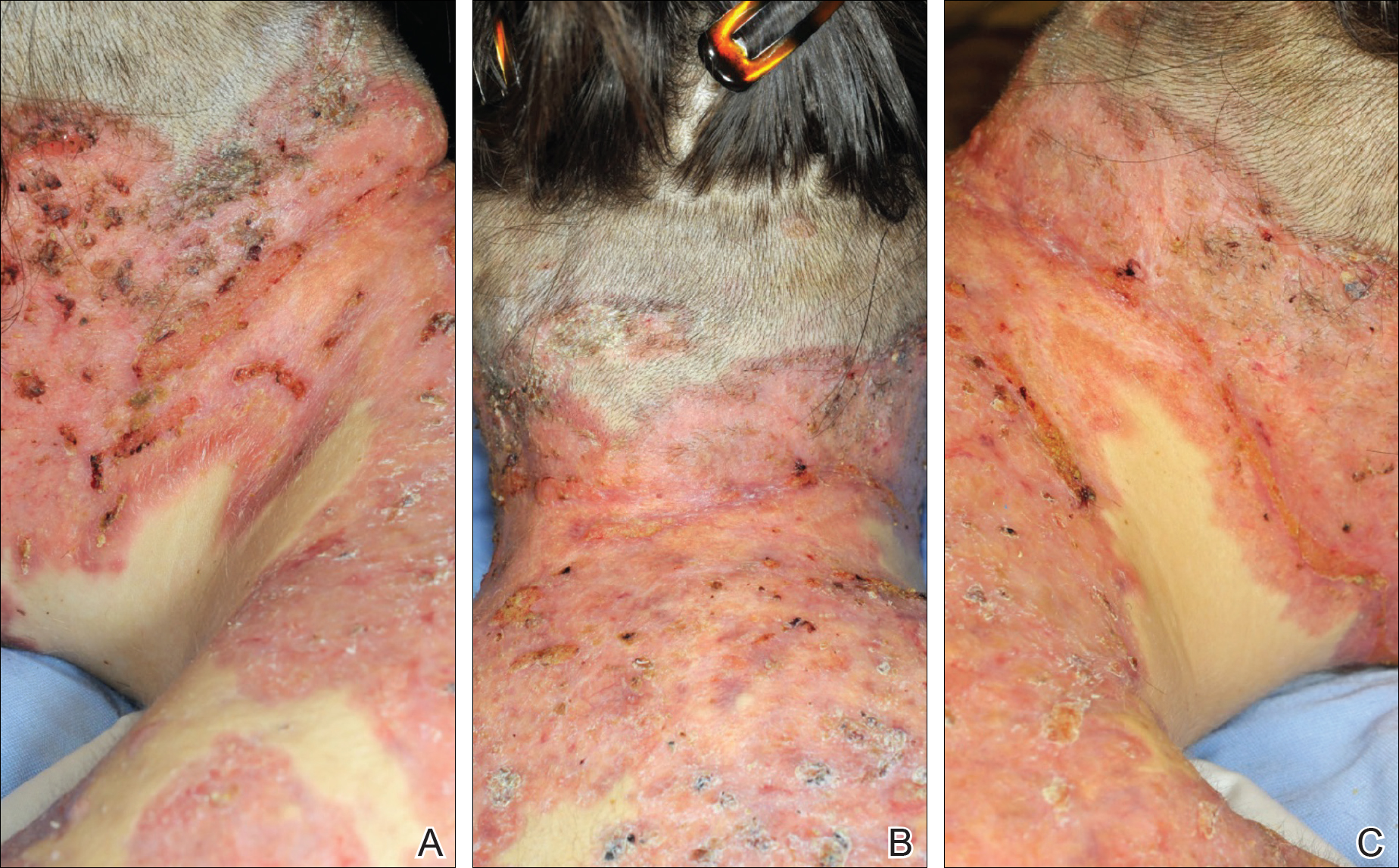
Comment
Thomson and Cunliffe5 reported a small case series of 11 young male patients with a mean age of 17 years who presented with severe worsening of their acne eruptions after taking isotretinoin, and they all responded well to an oral steroid. In another study, Bottomley and Cunliffe6 indicated that young male patients with notable acne on the trunk who are receiving a minimum dose of 0.5 mg/kg once daily of isotretinoin are at considerable risk for severe worsening of their skin condition.
Although severe worsening of acne lesions leading to acne fulminans or neutrophilic dermatosis secondary to isotretinoin or even doxycycline use is a rare entity, precautionary steps should be taken prior to treating acne conglobata patients with these agents. A review of PubMed articles indexed for MEDLINE using the terms acne, acne conglobata, and doxycycline revealed 2 prior cases of worsening acne in patients treated with doxycycline.7,8 Therefore, any patient presenting with acute worsening of an acne eruption while being treated with isotretinoin or doxycycline needs to be assessed for potential diagnosis of drug-induced acne fulminans or neutrophilic dermatosis.
It has been clearly documented in the literature that both doxycycline and isotretinoin can induce or exacerbate neutrophilic dermatoses in patients with severe underlying acne.6-8 The presentation may be mistaken for worsening acne, leading to inappropriate initiation or increase in the dose of isotretinoin therapy and worsening of the disease with potentially devastating disfiguring consequences. These patients tend to respond well to high-dose oral steroids alone or in combination with dapsone. A slow steroid taper over several months is recommended due to a high tendency for recurrence.
- Grando LR, Leite OG, Cestari TF. Pseudo-acne fulminans associated with oral isotretinoin. An Bras Dermatol. 2014;89:657-659.
- Burns RE, Colville JM. Acne conglobata with septicemia. Arch Dermatol. 1959;79:361-363.
- Karvonen SL. Acne fulminans: report of clinical findings and treatment of twenty-four patients. J Am Acad Dermatol. 1993;28:572-579.
- Zaba R, Schwartz R, Jarmuda S, et al. Acne fulminans: explosive systemic form of acne. J Eur Acad Dermatol Venereol. 2011;25:501-507.
- Thomson KF, Cunliffe WJ. Acne fulminans ‘sine fulminans.’ Clin Exp Dermatol. 2000;25:299-301.
- Bottomley WW, Cunliffe WJ. Severe flares of acne following isotretinoin: large closed comedones (macrocomedones) are a risk factor. Acta Derm Venereol. 1993;73:74.
- Weinstein M, Laxer R, Debosz J, et al. Doxycycline-induced cutaneous inflammation with systemic symptoms in a patient with acne vulgaris. J Cutan Med Surg. 2013;17:283-286.
- Yeo PM, Koh WL, Ang CC, et al. Paradoxical worsening of truncal acne with doxycycline. Ann Acad Med Singapore. 2016;45:430-431.
Acne fulminans is an uncommon and debilitating disease that presents as an acute eruption of nodular and ulcerative acne lesions with associated systemic symptoms.1,2 Although its underlying pathophysiology is not well understood, it occurs commonly during treatment of severe acne (eg, acne conglobata) with isotretinoin in young adolescent males.3 Zaba et al4 indicated that an underlying genetic disorder, increase in serum androgen levels, or presence of autoimmune disorders may contribute to the development of acne fulminans.
Isotretinoin and doxycycline also can potentially induce development of neutrophilic dermatoses including Sweet syndrome and pyoderma gangrenosum in patients with severe acne lesions, which can be clinically similar to an acne fulminans eruption. The neutrophilic dermatosis is characterized by the acute appearance of painful ulcerative papulonodules accompanied by systemic symptoms including fever and leukocytosis.
Case Report
A 13-year-old adolescent boy was initially assessed by his family physician 2 months prior and started on oral doxycycline 100 mg twice daily for acne conglobata on the back. Unfortunately, the acne lesions, especially those on the upper back (Figure 1), started getting worse after 1 month of treatment with doxycycline; thus, he subsequently was switched to oral isotretinoin 0.5 mg/kg once daily. Less than 2 weeks later, the acne lesions worsened, and the patient also developed severe generalized arthralgia, myalgia, and fever (>38.3°C). He acutely developed hundreds of ulcerative plaques covering the entire trunk, upper extremities, face, and neck.
He was admitted to the Stollery Children’s Hospital (Edmonton, Alberta, Canada) and was assessed by the dermatology, rheumatology, and general pediatric teams (Figure 2). He initially was investigated for the potential presence of autoinflammatory disorders, such as PAPA syndrome (pyogenic arthritis, pyoderma gangrenosum, acne) and SAPHO syndrome (synovitis, acne, pustulosis, hyperostosis, osteitis).
The patient initially was treated with prednisone 30 mg once daily for 3 weeks; dapsone 50 mg once daily and colchicine 0.6 mg twice daily were added while attempting to slowly wean off the prednisone (starting at 30 mg daily and reducing by 5 mg every other week). An attempt to discontinue the prednisone after 2 months was followed by immediate recurrence of the lesions (Figure 3), and the prednisone was restarted for another month. He was subsequently switched to oral cyclosporine 5 mg/kg once daily and achieved considerable improvement in his skin condition (Figure 4).
Comment
Thomson and Cunliffe5 reported a small case series of 11 young male patients with a mean age of 17 years who presented with severe worsening of their acne eruptions after taking isotretinoin, and they all responded well to an oral steroid. In another study, Bottomley and Cunliffe6 indicated that young male patients with notable acne on the trunk who are receiving a minimum dose of 0.5 mg/kg once daily of isotretinoin are at considerable risk for severe worsening of their skin condition.
Although severe worsening of acne lesions leading to acne fulminans or neutrophilic dermatosis secondary to isotretinoin or even doxycycline use is a rare entity, precautionary steps should be taken prior to treating acne conglobata patients with these agents. A review of PubMed articles indexed for MEDLINE using the terms acne, acne conglobata, and doxycycline revealed 2 prior cases of worsening acne in patients treated with doxycycline.7,8 Therefore, any patient presenting with acute worsening of an acne eruption while being treated with isotretinoin or doxycycline needs to be assessed for potential diagnosis of drug-induced acne fulminans or neutrophilic dermatosis.
It has been clearly documented in the literature that both doxycycline and isotretinoin can induce or exacerbate neutrophilic dermatoses in patients with severe underlying acne.6-8 The presentation may be mistaken for worsening acne, leading to inappropriate initiation or increase in the dose of isotretinoin therapy and worsening of the disease with potentially devastating disfiguring consequences. These patients tend to respond well to high-dose oral steroids alone or in combination with dapsone. A slow steroid taper over several months is recommended due to a high tendency for recurrence.
Acne fulminans is an uncommon and debilitating disease that presents as an acute eruption of nodular and ulcerative acne lesions with associated systemic symptoms.1,2 Although its underlying pathophysiology is not well understood, it occurs commonly during treatment of severe acne (eg, acne conglobata) with isotretinoin in young adolescent males.3 Zaba et al4 indicated that an underlying genetic disorder, increase in serum androgen levels, or presence of autoimmune disorders may contribute to the development of acne fulminans.
Isotretinoin and doxycycline also can potentially induce development of neutrophilic dermatoses including Sweet syndrome and pyoderma gangrenosum in patients with severe acne lesions, which can be clinically similar to an acne fulminans eruption. The neutrophilic dermatosis is characterized by the acute appearance of painful ulcerative papulonodules accompanied by systemic symptoms including fever and leukocytosis.
Case Report
A 13-year-old adolescent boy was initially assessed by his family physician 2 months prior and started on oral doxycycline 100 mg twice daily for acne conglobata on the back. Unfortunately, the acne lesions, especially those on the upper back (Figure 1), started getting worse after 1 month of treatment with doxycycline; thus, he subsequently was switched to oral isotretinoin 0.5 mg/kg once daily. Less than 2 weeks later, the acne lesions worsened, and the patient also developed severe generalized arthralgia, myalgia, and fever (>38.3°C). He acutely developed hundreds of ulcerative plaques covering the entire trunk, upper extremities, face, and neck.
He was admitted to the Stollery Children’s Hospital (Edmonton, Alberta, Canada) and was assessed by the dermatology, rheumatology, and general pediatric teams (Figure 2). He initially was investigated for the potential presence of autoinflammatory disorders, such as PAPA syndrome (pyogenic arthritis, pyoderma gangrenosum, acne) and SAPHO syndrome (synovitis, acne, pustulosis, hyperostosis, osteitis).
The patient initially was treated with prednisone 30 mg once daily for 3 weeks; dapsone 50 mg once daily and colchicine 0.6 mg twice daily were added while attempting to slowly wean off the prednisone (starting at 30 mg daily and reducing by 5 mg every other week). An attempt to discontinue the prednisone after 2 months was followed by immediate recurrence of the lesions (Figure 3), and the prednisone was restarted for another month. He was subsequently switched to oral cyclosporine 5 mg/kg once daily and achieved considerable improvement in his skin condition (Figure 4).
Comment
Thomson and Cunliffe5 reported a small case series of 11 young male patients with a mean age of 17 years who presented with severe worsening of their acne eruptions after taking isotretinoin, and they all responded well to an oral steroid. In another study, Bottomley and Cunliffe6 indicated that young male patients with notable acne on the trunk who are receiving a minimum dose of 0.5 mg/kg once daily of isotretinoin are at considerable risk for severe worsening of their skin condition.
Although severe worsening of acne lesions leading to acne fulminans or neutrophilic dermatosis secondary to isotretinoin or even doxycycline use is a rare entity, precautionary steps should be taken prior to treating acne conglobata patients with these agents. A review of PubMed articles indexed for MEDLINE using the terms acne, acne conglobata, and doxycycline revealed 2 prior cases of worsening acne in patients treated with doxycycline.7,8 Therefore, any patient presenting with acute worsening of an acne eruption while being treated with isotretinoin or doxycycline needs to be assessed for potential diagnosis of drug-induced acne fulminans or neutrophilic dermatosis.
It has been clearly documented in the literature that both doxycycline and isotretinoin can induce or exacerbate neutrophilic dermatoses in patients with severe underlying acne.6-8 The presentation may be mistaken for worsening acne, leading to inappropriate initiation or increase in the dose of isotretinoin therapy and worsening of the disease with potentially devastating disfiguring consequences. These patients tend to respond well to high-dose oral steroids alone or in combination with dapsone. A slow steroid taper over several months is recommended due to a high tendency for recurrence.
- Grando LR, Leite OG, Cestari TF. Pseudo-acne fulminans associated with oral isotretinoin. An Bras Dermatol. 2014;89:657-659.
- Burns RE, Colville JM. Acne conglobata with septicemia. Arch Dermatol. 1959;79:361-363.
- Karvonen SL. Acne fulminans: report of clinical findings and treatment of twenty-four patients. J Am Acad Dermatol. 1993;28:572-579.
- Zaba R, Schwartz R, Jarmuda S, et al. Acne fulminans: explosive systemic form of acne. J Eur Acad Dermatol Venereol. 2011;25:501-507.
- Thomson KF, Cunliffe WJ. Acne fulminans ‘sine fulminans.’ Clin Exp Dermatol. 2000;25:299-301.
- Bottomley WW, Cunliffe WJ. Severe flares of acne following isotretinoin: large closed comedones (macrocomedones) are a risk factor. Acta Derm Venereol. 1993;73:74.
- Weinstein M, Laxer R, Debosz J, et al. Doxycycline-induced cutaneous inflammation with systemic symptoms in a patient with acne vulgaris. J Cutan Med Surg. 2013;17:283-286.
- Yeo PM, Koh WL, Ang CC, et al. Paradoxical worsening of truncal acne with doxycycline. Ann Acad Med Singapore. 2016;45:430-431.
- Grando LR, Leite OG, Cestari TF. Pseudo-acne fulminans associated with oral isotretinoin. An Bras Dermatol. 2014;89:657-659.
- Burns RE, Colville JM. Acne conglobata with septicemia. Arch Dermatol. 1959;79:361-363.
- Karvonen SL. Acne fulminans: report of clinical findings and treatment of twenty-four patients. J Am Acad Dermatol. 1993;28:572-579.
- Zaba R, Schwartz R, Jarmuda S, et al. Acne fulminans: explosive systemic form of acne. J Eur Acad Dermatol Venereol. 2011;25:501-507.
- Thomson KF, Cunliffe WJ. Acne fulminans ‘sine fulminans.’ Clin Exp Dermatol. 2000;25:299-301.
- Bottomley WW, Cunliffe WJ. Severe flares of acne following isotretinoin: large closed comedones (macrocomedones) are a risk factor. Acta Derm Venereol. 1993;73:74.
- Weinstein M, Laxer R, Debosz J, et al. Doxycycline-induced cutaneous inflammation with systemic symptoms in a patient with acne vulgaris. J Cutan Med Surg. 2013;17:283-286.
- Yeo PM, Koh WL, Ang CC, et al. Paradoxical worsening of truncal acne with doxycycline. Ann Acad Med Singapore. 2016;45:430-431.
Resident Pearl
- Doxycycline and isotretinoin have been widely used for treatment of inflammatory and nodulocystic acne. Although outstanding results can be achieved, paradoxical worsening of acne while starting these medications has been described. In patients with severe acne (ie, acne conglobata), initiation of doxycycline and especially isotretinoin at regular dosages as the sole treatment can impose devastating risks on the patient. These patients are best treated with a combination of low-dose isotretinoin (at the beginning) with a moderate dose of steroids, which should be gradually tapered while the isotretinoin dose is increased to 0.5 to 1 mg/kg once daily.

Nonmalignant Cutaneous Findings Associated With Vemurafenib
To the Editor:
A 53-year-old woman was referred by her oncologist to our dermatology office with lesions on the face and body that presented 8 days after starting vemurafenib 960 mg twice daily for metastatic melanoma. The patient denied any symptoms from the lesions but was concerned they would spread to cover her entire face and body.
The patient's medical history included a diagnosis of metastatic melanoma 6 years prior to presentation. She stated that the primary cutaneous melanoma site was unknown. The patient had endured numerous surgeries to excise lymph node tumors, with some lesions up to 3 cm. The patient recently started vemurafenib, a treatment for BRAF V600E mutation-positive metastatic melanoma. The patient's personal history was notable for hepatitis A, B, and C, and her family history revealed her mother had metastatic lung cancer.
Physical examination revealed numerous 2- to 3-mm, round-oval, flesh-colored to light-brown papules on the cheeks, chest, abdomen (Figure 1), back, and both arms and legs. Some papules were inflamed and some had a stuck-on appearance. Lesions on the chest between the breasts and inframammary region were slightly inflamed. Two skin biopsies were performed. Biopsy of the lesion on the right lateral back revealed solar lentigo, early macular seborrheic keratosis, and a focus of inflamed mild solar keratosis. The dermis showed a mild superficial perivascular and interstitial inflammatory infiltrate composed mostly of lymphocytes, histiocytes, and eosinophils. There were occasional melanophages present (Figure 2). Biopsy of the lesion between the breasts revealed inflamed verrucous seborrheic keratosis (Figure 3).


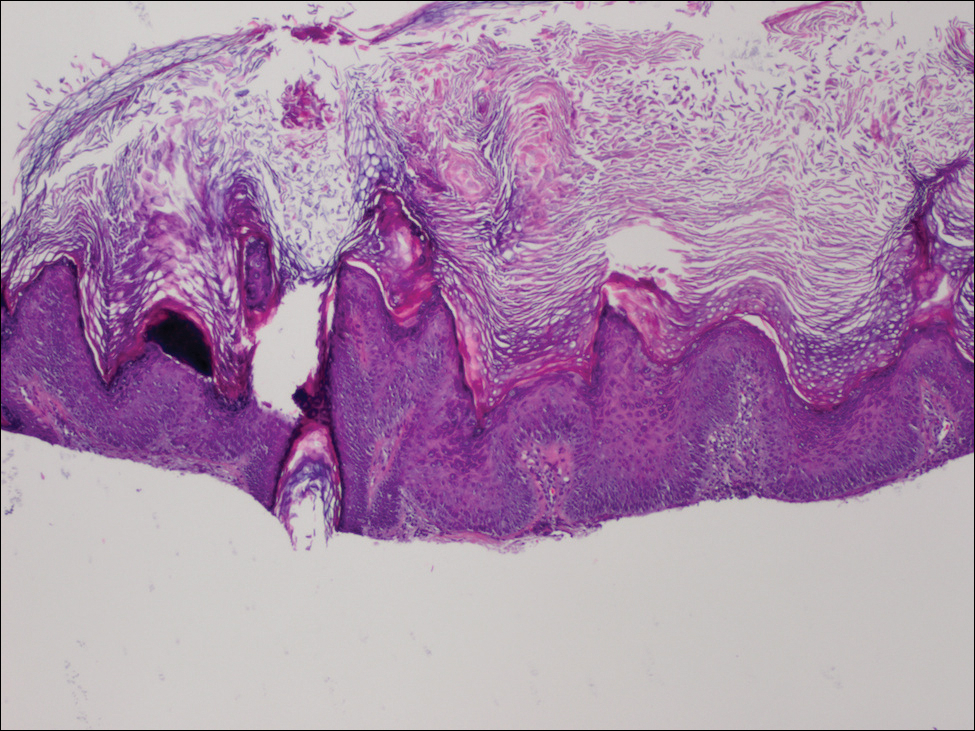
We treated the lesion on the right lateral back with cycles of cryotherapy and explained to the patient that the lesion between the breasts was benign. We also reiterated to the patient the importance of wearing sun-protective clothing and UVA/UVB sunblock with a sun protection factor of 30 or higher.
Our patient was diagnosed with pneumonia and subsequently had to discontinue vemurafenib. During the period of nontreatment, the keratotic lesions cleared with postinflammatory hyperpigmentation and no epidermal changes, which showed a possible inference of a direct relationship between the vemurafenib and the appearance of the nonmalignant cutaneous lesions. Although this report only represents 1 patient, other patients possibly can benefit from a modified dose of vemurafenib, which either would resolve or lessen the quantity of these lesions.
Vemurafenib is the first US Food and Drug Administration-approved treatment for nonresectable metastatic melanoma with the BRAF V600E mutation as detected by a US Food and Drug Administration-approved test.1,2 Mutated BRAF is present in approximately 60% of cutaneous melanomas.3 Vemurafenib targets the oncogenic BRAF V600E making the protein inactive, thus inhibiting cell proliferation and leading to apoptosis and shrinkage of the metastatic tumors.3-5 Vemurafenib has a response rate of more than 50% and is associated with rapid improvement in quality of life.3
Cutaneous side effects include increased incidence of squamous cell carcinoma and keratoacanthomas, appearing approximately 7 to 8 weeks after starting vemurafenib.4 The incidence of these lesions increases in patients 65 years and older and in patients with prior skin cancer and chronic sun exposure. The paradoxical activation of the mitogen-activated protein kinase pathway by mutant BRAF-selective inhibitors provides an explanation of the induction of squamous cell carcinomas.4 Prior to the initiation of vemurafenib, all patients should receive a total-body skin examination and every 2 months thereafter while on treatment. After discontinuation of the medicine, the patient should continue to receive total-body skin evaluations every 6 months indefinitely.
Patients should be aware of the potential for mild to severe photosensitivity reactions. They should be advised to limit their sun exposure time and to wear sun-protective clothing when outdoors. The use of broad-spectrum UVA/UVB sunscreen and lip protectant with a sun protection factor of 30 or higher also should be stressed.6,7 Patients should be aware that UVA rays penetrate glass; therefore, UV-protective clothing should be worn throughout the day and during all seasons.7
In clinical trials of vemurafenib, Stevens-Johnson syndrome and toxic epidermal necrolysis was reported in 2 patients.8,9 Clinical trials also reported patients developing new primary malignant melanoma lesions.10 These findings further emphasize the need for patients to undergo total-body skin examinations during and after treatment.
Other possible dermatologic reactions include a generalized rash, erythema, alopecia, and pruritus.2,3 The development of benign growths associated with patients on vemurafenib include follicular plugging seen in keratosis pilaris, palmar and plantar hyperkeratosis, seborrheic dermatitis-like rashes, verrucous keratosis, and acantholytic dyskeratosis.8,11,12
We report a case of nonmalignant growths occurring 8 days after starting vemurafenib. This case illustrates potential cutaneous adverse reactions that were benign yet still of great concern to our patient. Many of these nonmalignant cutaneous findings are associated with abnormal follicular keratinization thought to be secondary to abnormal signaling of the mitogen-activated protein kinase pathway that occurs with the use of BRAF inhibitors.8 Although in this case malignant lesions were not discovered, the need for total-body skin examinations exists during all stages of treatment. Supportive care and reassurance should be given to patients along with local treatments including topical therapies (steroids, retinoids), cryotherapy, and biopsies or excisions when necessary.13,14
- Holstein S, Hohl R. Therapeutic additions and possible deletions in oncology in 2011. Clin Pharmacol Ther. 2011;91:15-17.
- Zambon A, Niculescu-Dovaz I, Niculescu-Dovaz D, et al. Small molecule inhibitors of BRAF in clinical trials. Bioorg Med Chem Lett. 2012;22:789-792.
- Luke JJ, Hodi FS. Vemurafenib and BRAF inhibition: a new class of treatment for metastatic melanoma [published online November 14, 2011]. Clin Cancer Res. 2012;18:9-14.
- Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010; 363:809-819.
- Tsai J, Lee JT, Wang W, et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc Natl Acad Sci USA. 2008;105:3041-3046.
- Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507-2516.
- Dummer R, Rinderknecht J, Goldinger SM. Ultraviolet A and photosensitivity during vemuranefib therapy. N Engl J Med. 2012;366:480-481.
- Bovd KP, Vincent B, Andrea A, et al. Nonmalignant cutaneous findings associated with vemurafenib use in patients with metastatic melanoma. J Am Acad Dermatol. 2012;67:1375-1379.
- Wang CM, Fleming KF Hsu S. A case of vemurafenib-induced keratosis pilaris-like eruption. Dermatol Online J. 2012;18:7.
- Zimmer L, Hillen U, Livingstone E, et al. Atypical melanocytic proliferations and new primary melanomas in patients with advanced melanoma undergoing selective BRAF inhibition. J Clin Oncol. 2012;30:2375-2383.
- Huang V, Hepper D, Anadkat M, et al. Cutaneous toxic effects associated with vemurafenib and inhibition of the BRAF pathway. Arch Dermatol. 2012;148:628-633.
- Gupta M, Huang V, Linette G, et al. Unusual complication of vemurafenib treatment of metastatic melanoma: exacerbation of acantholytic dyskeratosis complicated by Kaposi varicelliform eruption. Arch Dermatol. 2012;148:966-968;
- Sinha R, Edmonds K, Newton-Bishop JA, et al. Cutaneous adverse events associated with vemurafenib in patients with metastatic melanoma: practical advice on diagnosis, preventions and management of the main treatment related skin toxicities. Br J Dermatol. 2012;167:987-994.
- Boussemart L, Routier E, Mateus C, et al. Prospective study of cutaneous side effects associated with the BRAF inhibitor vemurafenib: a study of 42 patients. Ann Oncol. 2013;24:1691-1697.
To the Editor:
A 53-year-old woman was referred by her oncologist to our dermatology office with lesions on the face and body that presented 8 days after starting vemurafenib 960 mg twice daily for metastatic melanoma. The patient denied any symptoms from the lesions but was concerned they would spread to cover her entire face and body.
The patient's medical history included a diagnosis of metastatic melanoma 6 years prior to presentation. She stated that the primary cutaneous melanoma site was unknown. The patient had endured numerous surgeries to excise lymph node tumors, with some lesions up to 3 cm. The patient recently started vemurafenib, a treatment for BRAF V600E mutation-positive metastatic melanoma. The patient's personal history was notable for hepatitis A, B, and C, and her family history revealed her mother had metastatic lung cancer.
Physical examination revealed numerous 2- to 3-mm, round-oval, flesh-colored to light-brown papules on the cheeks, chest, abdomen (Figure 1), back, and both arms and legs. Some papules were inflamed and some had a stuck-on appearance. Lesions on the chest between the breasts and inframammary region were slightly inflamed. Two skin biopsies were performed. Biopsy of the lesion on the right lateral back revealed solar lentigo, early macular seborrheic keratosis, and a focus of inflamed mild solar keratosis. The dermis showed a mild superficial perivascular and interstitial inflammatory infiltrate composed mostly of lymphocytes, histiocytes, and eosinophils. There were occasional melanophages present (Figure 2). Biopsy of the lesion between the breasts revealed inflamed verrucous seborrheic keratosis (Figure 3).
We treated the lesion on the right lateral back with cycles of cryotherapy and explained to the patient that the lesion between the breasts was benign. We also reiterated to the patient the importance of wearing sun-protective clothing and UVA/UVB sunblock with a sun protection factor of 30 or higher.
Our patient was diagnosed with pneumonia and subsequently had to discontinue vemurafenib. During the period of nontreatment, the keratotic lesions cleared with postinflammatory hyperpigmentation and no epidermal changes, which showed a possible inference of a direct relationship between the vemurafenib and the appearance of the nonmalignant cutaneous lesions. Although this report only represents 1 patient, other patients possibly can benefit from a modified dose of vemurafenib, which either would resolve or lessen the quantity of these lesions.
Vemurafenib is the first US Food and Drug Administration-approved treatment for nonresectable metastatic melanoma with the BRAF V600E mutation as detected by a US Food and Drug Administration-approved test.1,2 Mutated BRAF is present in approximately 60% of cutaneous melanomas.3 Vemurafenib targets the oncogenic BRAF V600E making the protein inactive, thus inhibiting cell proliferation and leading to apoptosis and shrinkage of the metastatic tumors.3-5 Vemurafenib has a response rate of more than 50% and is associated with rapid improvement in quality of life.3
Cutaneous side effects include increased incidence of squamous cell carcinoma and keratoacanthomas, appearing approximately 7 to 8 weeks after starting vemurafenib.4 The incidence of these lesions increases in patients 65 years and older and in patients with prior skin cancer and chronic sun exposure. The paradoxical activation of the mitogen-activated protein kinase pathway by mutant BRAF-selective inhibitors provides an explanation of the induction of squamous cell carcinomas.4 Prior to the initiation of vemurafenib, all patients should receive a total-body skin examination and every 2 months thereafter while on treatment. After discontinuation of the medicine, the patient should continue to receive total-body skin evaluations every 6 months indefinitely.
Patients should be aware of the potential for mild to severe photosensitivity reactions. They should be advised to limit their sun exposure time and to wear sun-protective clothing when outdoors. The use of broad-spectrum UVA/UVB sunscreen and lip protectant with a sun protection factor of 30 or higher also should be stressed.6,7 Patients should be aware that UVA rays penetrate glass; therefore, UV-protective clothing should be worn throughout the day and during all seasons.7
In clinical trials of vemurafenib, Stevens-Johnson syndrome and toxic epidermal necrolysis was reported in 2 patients.8,9 Clinical trials also reported patients developing new primary malignant melanoma lesions.10 These findings further emphasize the need for patients to undergo total-body skin examinations during and after treatment.
Other possible dermatologic reactions include a generalized rash, erythema, alopecia, and pruritus.2,3 The development of benign growths associated with patients on vemurafenib include follicular plugging seen in keratosis pilaris, palmar and plantar hyperkeratosis, seborrheic dermatitis-like rashes, verrucous keratosis, and acantholytic dyskeratosis.8,11,12
We report a case of nonmalignant growths occurring 8 days after starting vemurafenib. This case illustrates potential cutaneous adverse reactions that were benign yet still of great concern to our patient. Many of these nonmalignant cutaneous findings are associated with abnormal follicular keratinization thought to be secondary to abnormal signaling of the mitogen-activated protein kinase pathway that occurs with the use of BRAF inhibitors.8 Although in this case malignant lesions were not discovered, the need for total-body skin examinations exists during all stages of treatment. Supportive care and reassurance should be given to patients along with local treatments including topical therapies (steroids, retinoids), cryotherapy, and biopsies or excisions when necessary.13,14
To the Editor:
A 53-year-old woman was referred by her oncologist to our dermatology office with lesions on the face and body that presented 8 days after starting vemurafenib 960 mg twice daily for metastatic melanoma. The patient denied any symptoms from the lesions but was concerned they would spread to cover her entire face and body.
The patient's medical history included a diagnosis of metastatic melanoma 6 years prior to presentation. She stated that the primary cutaneous melanoma site was unknown. The patient had endured numerous surgeries to excise lymph node tumors, with some lesions up to 3 cm. The patient recently started vemurafenib, a treatment for BRAF V600E mutation-positive metastatic melanoma. The patient's personal history was notable for hepatitis A, B, and C, and her family history revealed her mother had metastatic lung cancer.
Physical examination revealed numerous 2- to 3-mm, round-oval, flesh-colored to light-brown papules on the cheeks, chest, abdomen (Figure 1), back, and both arms and legs. Some papules were inflamed and some had a stuck-on appearance. Lesions on the chest between the breasts and inframammary region were slightly inflamed. Two skin biopsies were performed. Biopsy of the lesion on the right lateral back revealed solar lentigo, early macular seborrheic keratosis, and a focus of inflamed mild solar keratosis. The dermis showed a mild superficial perivascular and interstitial inflammatory infiltrate composed mostly of lymphocytes, histiocytes, and eosinophils. There were occasional melanophages present (Figure 2). Biopsy of the lesion between the breasts revealed inflamed verrucous seborrheic keratosis (Figure 3).
We treated the lesion on the right lateral back with cycles of cryotherapy and explained to the patient that the lesion between the breasts was benign. We also reiterated to the patient the importance of wearing sun-protective clothing and UVA/UVB sunblock with a sun protection factor of 30 or higher.
Our patient was diagnosed with pneumonia and subsequently had to discontinue vemurafenib. During the period of nontreatment, the keratotic lesions cleared with postinflammatory hyperpigmentation and no epidermal changes, which showed a possible inference of a direct relationship between the vemurafenib and the appearance of the nonmalignant cutaneous lesions. Although this report only represents 1 patient, other patients possibly can benefit from a modified dose of vemurafenib, which either would resolve or lessen the quantity of these lesions.
Vemurafenib is the first US Food and Drug Administration-approved treatment for nonresectable metastatic melanoma with the BRAF V600E mutation as detected by a US Food and Drug Administration-approved test.1,2 Mutated BRAF is present in approximately 60% of cutaneous melanomas.3 Vemurafenib targets the oncogenic BRAF V600E making the protein inactive, thus inhibiting cell proliferation and leading to apoptosis and shrinkage of the metastatic tumors.3-5 Vemurafenib has a response rate of more than 50% and is associated with rapid improvement in quality of life.3
Cutaneous side effects include increased incidence of squamous cell carcinoma and keratoacanthomas, appearing approximately 7 to 8 weeks after starting vemurafenib.4 The incidence of these lesions increases in patients 65 years and older and in patients with prior skin cancer and chronic sun exposure. The paradoxical activation of the mitogen-activated protein kinase pathway by mutant BRAF-selective inhibitors provides an explanation of the induction of squamous cell carcinomas.4 Prior to the initiation of vemurafenib, all patients should receive a total-body skin examination and every 2 months thereafter while on treatment. After discontinuation of the medicine, the patient should continue to receive total-body skin evaluations every 6 months indefinitely.
Patients should be aware of the potential for mild to severe photosensitivity reactions. They should be advised to limit their sun exposure time and to wear sun-protective clothing when outdoors. The use of broad-spectrum UVA/UVB sunscreen and lip protectant with a sun protection factor of 30 or higher also should be stressed.6,7 Patients should be aware that UVA rays penetrate glass; therefore, UV-protective clothing should be worn throughout the day and during all seasons.7
In clinical trials of vemurafenib, Stevens-Johnson syndrome and toxic epidermal necrolysis was reported in 2 patients.8,9 Clinical trials also reported patients developing new primary malignant melanoma lesions.10 These findings further emphasize the need for patients to undergo total-body skin examinations during and after treatment.
Other possible dermatologic reactions include a generalized rash, erythema, alopecia, and pruritus.2,3 The development of benign growths associated with patients on vemurafenib include follicular plugging seen in keratosis pilaris, palmar and plantar hyperkeratosis, seborrheic dermatitis-like rashes, verrucous keratosis, and acantholytic dyskeratosis.8,11,12
We report a case of nonmalignant growths occurring 8 days after starting vemurafenib. This case illustrates potential cutaneous adverse reactions that were benign yet still of great concern to our patient. Many of these nonmalignant cutaneous findings are associated with abnormal follicular keratinization thought to be secondary to abnormal signaling of the mitogen-activated protein kinase pathway that occurs with the use of BRAF inhibitors.8 Although in this case malignant lesions were not discovered, the need for total-body skin examinations exists during all stages of treatment. Supportive care and reassurance should be given to patients along with local treatments including topical therapies (steroids, retinoids), cryotherapy, and biopsies or excisions when necessary.13,14
- Holstein S, Hohl R. Therapeutic additions and possible deletions in oncology in 2011. Clin Pharmacol Ther. 2011;91:15-17.
- Zambon A, Niculescu-Dovaz I, Niculescu-Dovaz D, et al. Small molecule inhibitors of BRAF in clinical trials. Bioorg Med Chem Lett. 2012;22:789-792.
- Luke JJ, Hodi FS. Vemurafenib and BRAF inhibition: a new class of treatment for metastatic melanoma [published online November 14, 2011]. Clin Cancer Res. 2012;18:9-14.
- Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010; 363:809-819.
- Tsai J, Lee JT, Wang W, et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc Natl Acad Sci USA. 2008;105:3041-3046.
- Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507-2516.
- Dummer R, Rinderknecht J, Goldinger SM. Ultraviolet A and photosensitivity during vemuranefib therapy. N Engl J Med. 2012;366:480-481.
- Bovd KP, Vincent B, Andrea A, et al. Nonmalignant cutaneous findings associated with vemurafenib use in patients with metastatic melanoma. J Am Acad Dermatol. 2012;67:1375-1379.
- Wang CM, Fleming KF Hsu S. A case of vemurafenib-induced keratosis pilaris-like eruption. Dermatol Online J. 2012;18:7.
- Zimmer L, Hillen U, Livingstone E, et al. Atypical melanocytic proliferations and new primary melanomas in patients with advanced melanoma undergoing selective BRAF inhibition. J Clin Oncol. 2012;30:2375-2383.
- Huang V, Hepper D, Anadkat M, et al. Cutaneous toxic effects associated with vemurafenib and inhibition of the BRAF pathway. Arch Dermatol. 2012;148:628-633.
- Gupta M, Huang V, Linette G, et al. Unusual complication of vemurafenib treatment of metastatic melanoma: exacerbation of acantholytic dyskeratosis complicated by Kaposi varicelliform eruption. Arch Dermatol. 2012;148:966-968;
- Sinha R, Edmonds K, Newton-Bishop JA, et al. Cutaneous adverse events associated with vemurafenib in patients with metastatic melanoma: practical advice on diagnosis, preventions and management of the main treatment related skin toxicities. Br J Dermatol. 2012;167:987-994.
- Boussemart L, Routier E, Mateus C, et al. Prospective study of cutaneous side effects associated with the BRAF inhibitor vemurafenib: a study of 42 patients. Ann Oncol. 2013;24:1691-1697.
- Holstein S, Hohl R. Therapeutic additions and possible deletions in oncology in 2011. Clin Pharmacol Ther. 2011;91:15-17.
- Zambon A, Niculescu-Dovaz I, Niculescu-Dovaz D, et al. Small molecule inhibitors of BRAF in clinical trials. Bioorg Med Chem Lett. 2012;22:789-792.
- Luke JJ, Hodi FS. Vemurafenib and BRAF inhibition: a new class of treatment for metastatic melanoma [published online November 14, 2011]. Clin Cancer Res. 2012;18:9-14.
- Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010; 363:809-819.
- Tsai J, Lee JT, Wang W, et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc Natl Acad Sci USA. 2008;105:3041-3046.
- Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507-2516.
- Dummer R, Rinderknecht J, Goldinger SM. Ultraviolet A and photosensitivity during vemuranefib therapy. N Engl J Med. 2012;366:480-481.
- Bovd KP, Vincent B, Andrea A, et al. Nonmalignant cutaneous findings associated with vemurafenib use in patients with metastatic melanoma. J Am Acad Dermatol. 2012;67:1375-1379.
- Wang CM, Fleming KF Hsu S. A case of vemurafenib-induced keratosis pilaris-like eruption. Dermatol Online J. 2012;18:7.
- Zimmer L, Hillen U, Livingstone E, et al. Atypical melanocytic proliferations and new primary melanomas in patients with advanced melanoma undergoing selective BRAF inhibition. J Clin Oncol. 2012;30:2375-2383.
- Huang V, Hepper D, Anadkat M, et al. Cutaneous toxic effects associated with vemurafenib and inhibition of the BRAF pathway. Arch Dermatol. 2012;148:628-633.
- Gupta M, Huang V, Linette G, et al. Unusual complication of vemurafenib treatment of metastatic melanoma: exacerbation of acantholytic dyskeratosis complicated by Kaposi varicelliform eruption. Arch Dermatol. 2012;148:966-968;
- Sinha R, Edmonds K, Newton-Bishop JA, et al. Cutaneous adverse events associated with vemurafenib in patients with metastatic melanoma: practical advice on diagnosis, preventions and management of the main treatment related skin toxicities. Br J Dermatol. 2012;167:987-994.
- Boussemart L, Routier E, Mateus C, et al. Prospective study of cutaneous side effects associated with the BRAF inhibitor vemurafenib: a study of 42 patients. Ann Oncol. 2013;24:1691-1697.
Practice Points
- Prior to starting a BRAF inhibitor, clinicians should perform a baseline total-body skin examination and follow-up every 2 months.
- Take photographs of the patient's entire body on initial total-body skin examination.
- Encourage sun protection for exposed areas on the body in all seasons.
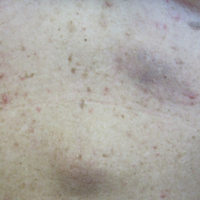
Graft-versus-host Disease Presenting Along Blaschko Lines: Cutaneous Mosaicism
To the Editor:
Graft-versus-host disease (GVHD) is a common and serious complication seen most often with bone marrow transplantation and peripheral blood stem cell transplantation. With these therapies, functional lymphoid cells are transferred from an immunocompetent donor into a nongenetically identical recipient, or "host." Because of the allogeneic nature of these transplants, the transplanted lymphoid cells have a high potential to recognize and treat the host's cells as foreign, and the resultant clinical and pathologic picture is that of GVHD. The primary organ systems affected in this immune response are the skin, gastrointestinal tract, and hepatobiliary system.1,2 Cutaneous manifestations are by far the most common.3
Although notable gains have been made in elucidating the causes, risk factors, and mechanisms that result in the clinical picture of GVHD, gaps in our knowledge and understanding still exist. Our patient represents a unique case of unilateral GVHD occurring along Blaschko lines, which has important implications for both recognizing and understanding the pathogenesis of GVHD.
A 35-year-old woman was diagnosed with stage IV follicular lymphoma and received various chemotherapy regimens over the next 4 years. Unfortunately, her disease progressed despite treatment. At 39 years of age, she underwent a nonmyeloablative allogeneic peripheral blood stem cell transplantation from a single HLA-mismatched sibling. She was placed on prednisone and cyclosporine for immunosuppression. High-dose acyclovir prophylaxis also was initiated given her history of zoster affecting the right C3 dermatome. Successful engraftment was achieved, with molecular studies showing 100% of cells following transplantation were of donor origin. Restaging at 1 and 2 years following transplantation found her to be in complete remission.
At 2 years following transplantation, she began a slow taper of immunosuppressive medications. She was successfully weaned off prednisone and continued to gradually reduce the cyclosporine dose. Toward the end of the cyclosporine taper 3 months later, she developed a pruritic eruption on the left proximal arm.
She was seen in a bone marrow transplant clinic 4 weeks after the rash developed. On examination, she had multiple, violaceous, lichenoid papules coalescing into linear bandlike plaques. One plaque extended along the left upper arm and 2 others encircled the left hemithorax, respecting the midline. She was treated empirically for zoster with valacyclovir 1 g 3 times daily based on the presumed dermatomal distribution of the eruption. Despite treatment, the rash progressed, and she developed fever. Eight days later, she was admitted with concern for disseminated zoster (Figure). Viral tissue cultures and polymerase chain reaction analysis of the lesions were negative for varicella-zoster virus and herpes simplex viruses 1 and 2. Biopsies of skin lesions on the arm and trunk were both consistent with GVHD.
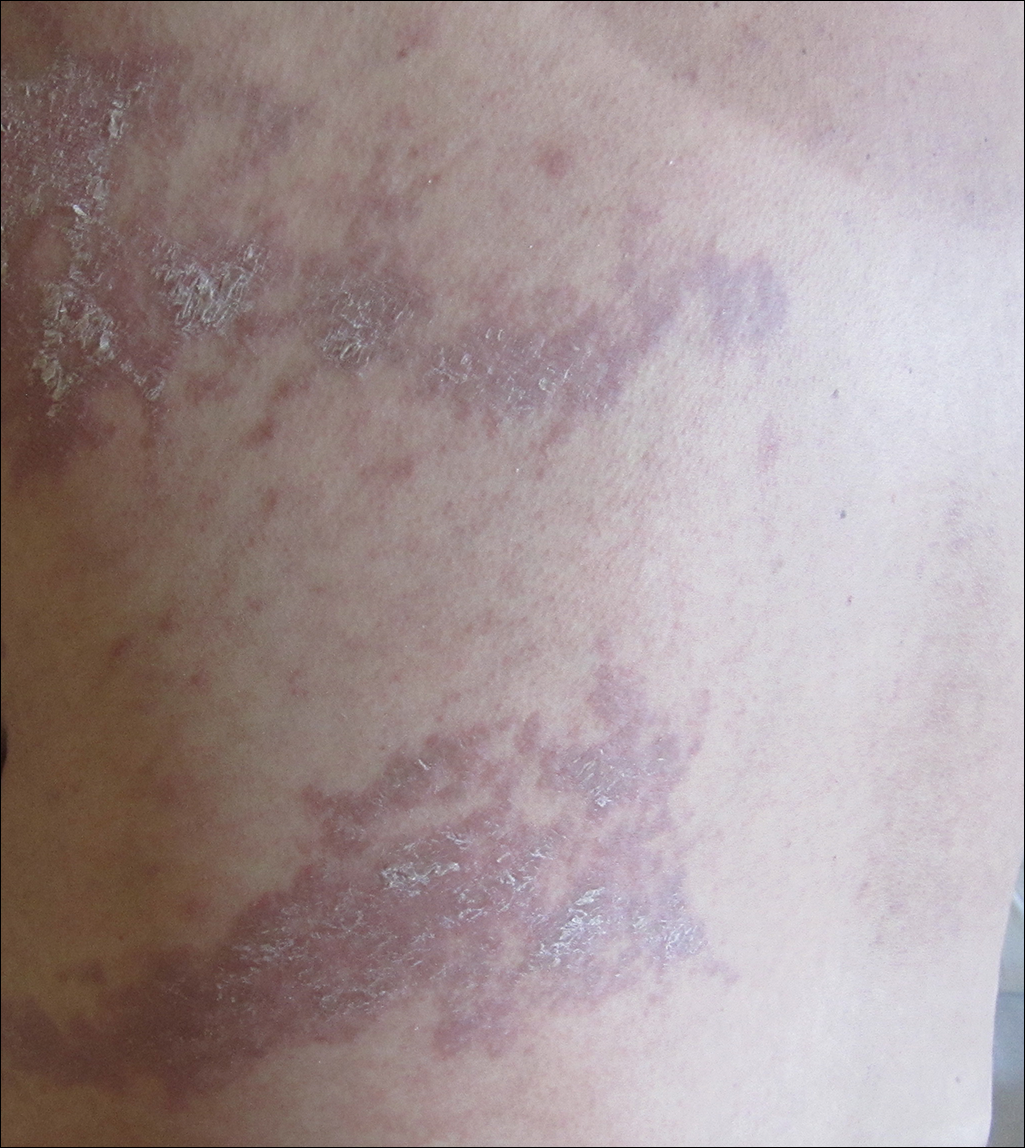
Given the clinical history, characteristic lesion morphology, and distinct linear distribution along with histopathological confirmation, a diagnosis of GVHD along Blaschko lines was made. Recognizing the cause to be immunogenic rather than infectious, immunosuppressive medications were started. In addition to increasing the prednisone and cyclosporine back to therapeutic levels, she received weekly methylprednisolone. With treatment, she showed gradual but marked improvement.
Six cases of linear GVHD have occurred as an isotopic response along dermatomes previously affected by varicella-zoster virus.4-8 These cases give credence to the idea that a cutaneous viral infection may alter the skin through unknown mechanisms, predisposing it to become affected by GVHD. Notably, this phenomenon occurred despite absence of a persistent viral genome when assessed using polymerase chain reaction analysis.4
An additional 3 cases of GVHD occurring in a dermatomal distribution without any prior infections in those areas have been reported.9,10 Of note, 2 of 3 patients did have episodes of zoster occur at other sites following transplantation and did not develop GVHD symptoms in any of those locations.9 Interestingly, controversy exists as to whether the distribution of these lesions was dermatomal or followed Blaschko lines.11
Two cases of linear GVHD have been reported in which lesions were identified as occurring along Blaschko lines.12,13 The lines of Blaschko, first described in 1901, correspond to cellular migration patterns during embryological development.14 Postzygotic mutations causing epidermal cell mosaicism may result in skin disorders occurring in segmental areas defined by the Blaschko lines.15-17 Accordingly, the Blaschko-linear pattern in GVHD suggests cellular mosaicism as the etiology in this case. Although the host's immune system develops immunotolerance to both cellular lineages during maturation, transplanted lymphoid cells from a nongenetically identical sibling may identify just one of the cell lines as nonself, producing a selective pattern of GVHD18 confined to the distribution of the genetically disparate cell line, which occurs along the lines of Blaschko in the skin. Candidate genes for mutations that would produce a mosaic following transplant GVHD include any of the 25 to 30 known minor histocompatibility antigens (or any of the several hundred yet to be found).19 Although well established for monogenic dominant disorders, in 2007 it was recognized that a postzygotic mutation can cause many complex polygenetic disorders, including GVHD, to manifest in a limited segmental pattern. This understanding, along with retrospective case review, has brought into question previously reported "dermatomal" or "zosteriform" presentations of GVHD, asserting that the linear patterns were misidentified and thus inappropriately attributed to a postviral response.20 Recognition of the Blaschko-linear distribution holds significance in both identifying lesion etiology and understanding disease pathogenesis and treatment.
Our patient illustrates a case of a Blaschko-linear GVHD. The distinctive pattern of her physical findings strongly favored epidermal cell mosaicism as the etiology of her disease. More than just a phenotypically unique case, it provided further insight into the complex etiology underlying GVHD and iterated the basic concepts of Blaschko lines and genetic alterations in development.
- Thomas ED, Storb R, Clift RA, et al. Bone-marrow transplantation. N Engl J Med. 1975;292:895-902.
- Lee SJ, Vogelsang G, Flowers ME. Chronic graft-versus-host disease. Biol Blood Marrow Transplant. 2003;9:215-233.
- Johnson ML, Farmer ER. Graft versus host reactions in dermatology. J Am Acad Dermatol. 1998;38:369-384.
- Baselga E, Drolet BA, Segura AD, et al. Dermatomal lichenoid chronic graft-vs-host disease following varicella-zoster infection despite absence of viral genome. J Cutan Pathol. 1996;23:576-581.
- Lacour JP, Sirvent N, Monpoux F, et al. Dermatomal chronic cutaneous graft versus host disease at the site of prior herpes zoster. Br J Dermatol. 1999;141:587-589.
- Cordoba S, Fraga J, Bartolome B, et al. Giant cell lichenoid dermatitis within herpes zoster scars in a bone marrow recipient. J Cutan Pathol. 2000;27:255-257.
- Sanli H, Anadolu R, Arat M, et al. Dermatomal lichenoid graft-versus-host disease within herpes zoster scars. Int J Dermatol. 2003;42:562-564.
- Martires KJ, Baird K, Citrin DE, et al. Localization of sclerotic-type chronic graft-versus-host disease to sites of skin injury: potential insight into the mechanism of isomorphic and isotopic responses. Arch Dermatol. 2011;147:1081-1086.
- Freemer CS, Farmer ER, Corio RL, et al. Lichenoid chronic graft-vs-host disease occurring in a dermatomal distribution. Arch Dermatol. 1994;130:70-72.
- Cohen PR, Hymes SR. Linear and dermatomal cutaneous graft-versus-host disease. South Med J. 1994;87:758-761.
- Reisfeld PL. Linear lichenoid graft-vs-host disease. Arch Dermatol. 1994;130:1207-1208.
- Beers B, Kalish RS, Kaye VN, et al. Unilateral linear lichenoid eruption after bone marrow transplantation: an unmasking of tolerance to an abnormal keratinocyte clone? J Am Acad Dermatol. 1993;28(5, pt 2):888-892.
- Wilson BB, Lockman DW. Linear lichenoid graft-vs-host disease. Arch Dermatol. 1994;130:1206-1207.
- Goldberg I, Sprecher E. Patterned disorders in dermatology. Clin Dermatol. 2011;29:498-503.
- Colman SD, Rasmussen SA, Ho VT, et al. Somatic mosaicism in a patient with neurofibromatosis type 1. Am J Hum Genet. 1996;58:484-490.
- Munro CS, Wilkie AO. Epidermal mosaicism producing localised acne: somatic mutation in FGFR2. Lancet. 1998;352:704-705.
- Sakuntabhai A, Dhitavat J, Burge S, et al. Mosaicism for ATP2A2 mutations causes segmental Darier's disease. J Invest Dermatol. 2000;115:1144-1147.
- Dickinson AM, Wang XN, Sviland L, et al. In situ dissection of the graft-versus-host activities of cytotoxic T cells specific for minor histocompatibility antigens. Nat Med. 2002;8:410-414.
- Hansen JA, Chien JW, Warren EH, et al. Defining genetic risk for graft- versus-host disease and mortality following allogeneic hematopoietic stem cell transplantation. Curr Opin Hematol. 2010;17:483-492.
- Happle R. Superimposed segmental manifestation of polygenic skin disorders. J Am Acad Dermatol. 2007;57:690-699.
To the Editor:
Graft-versus-host disease (GVHD) is a common and serious complication seen most often with bone marrow transplantation and peripheral blood stem cell transplantation. With these therapies, functional lymphoid cells are transferred from an immunocompetent donor into a nongenetically identical recipient, or "host." Because of the allogeneic nature of these transplants, the transplanted lymphoid cells have a high potential to recognize and treat the host's cells as foreign, and the resultant clinical and pathologic picture is that of GVHD. The primary organ systems affected in this immune response are the skin, gastrointestinal tract, and hepatobiliary system.1,2 Cutaneous manifestations are by far the most common.3
Although notable gains have been made in elucidating the causes, risk factors, and mechanisms that result in the clinical picture of GVHD, gaps in our knowledge and understanding still exist. Our patient represents a unique case of unilateral GVHD occurring along Blaschko lines, which has important implications for both recognizing and understanding the pathogenesis of GVHD.
A 35-year-old woman was diagnosed with stage IV follicular lymphoma and received various chemotherapy regimens over the next 4 years. Unfortunately, her disease progressed despite treatment. At 39 years of age, she underwent a nonmyeloablative allogeneic peripheral blood stem cell transplantation from a single HLA-mismatched sibling. She was placed on prednisone and cyclosporine for immunosuppression. High-dose acyclovir prophylaxis also was initiated given her history of zoster affecting the right C3 dermatome. Successful engraftment was achieved, with molecular studies showing 100% of cells following transplantation were of donor origin. Restaging at 1 and 2 years following transplantation found her to be in complete remission.
At 2 years following transplantation, she began a slow taper of immunosuppressive medications. She was successfully weaned off prednisone and continued to gradually reduce the cyclosporine dose. Toward the end of the cyclosporine taper 3 months later, she developed a pruritic eruption on the left proximal arm.
She was seen in a bone marrow transplant clinic 4 weeks after the rash developed. On examination, she had multiple, violaceous, lichenoid papules coalescing into linear bandlike plaques. One plaque extended along the left upper arm and 2 others encircled the left hemithorax, respecting the midline. She was treated empirically for zoster with valacyclovir 1 g 3 times daily based on the presumed dermatomal distribution of the eruption. Despite treatment, the rash progressed, and she developed fever. Eight days later, she was admitted with concern for disseminated zoster (Figure). Viral tissue cultures and polymerase chain reaction analysis of the lesions were negative for varicella-zoster virus and herpes simplex viruses 1 and 2. Biopsies of skin lesions on the arm and trunk were both consistent with GVHD.
Given the clinical history, characteristic lesion morphology, and distinct linear distribution along with histopathological confirmation, a diagnosis of GVHD along Blaschko lines was made. Recognizing the cause to be immunogenic rather than infectious, immunosuppressive medications were started. In addition to increasing the prednisone and cyclosporine back to therapeutic levels, she received weekly methylprednisolone. With treatment, she showed gradual but marked improvement.
Six cases of linear GVHD have occurred as an isotopic response along dermatomes previously affected by varicella-zoster virus.4-8 These cases give credence to the idea that a cutaneous viral infection may alter the skin through unknown mechanisms, predisposing it to become affected by GVHD. Notably, this phenomenon occurred despite absence of a persistent viral genome when assessed using polymerase chain reaction analysis.4
An additional 3 cases of GVHD occurring in a dermatomal distribution without any prior infections in those areas have been reported.9,10 Of note, 2 of 3 patients did have episodes of zoster occur at other sites following transplantation and did not develop GVHD symptoms in any of those locations.9 Interestingly, controversy exists as to whether the distribution of these lesions was dermatomal or followed Blaschko lines.11
Two cases of linear GVHD have been reported in which lesions were identified as occurring along Blaschko lines.12,13 The lines of Blaschko, first described in 1901, correspond to cellular migration patterns during embryological development.14 Postzygotic mutations causing epidermal cell mosaicism may result in skin disorders occurring in segmental areas defined by the Blaschko lines.15-17 Accordingly, the Blaschko-linear pattern in GVHD suggests cellular mosaicism as the etiology in this case. Although the host's immune system develops immunotolerance to both cellular lineages during maturation, transplanted lymphoid cells from a nongenetically identical sibling may identify just one of the cell lines as nonself, producing a selective pattern of GVHD18 confined to the distribution of the genetically disparate cell line, which occurs along the lines of Blaschko in the skin. Candidate genes for mutations that would produce a mosaic following transplant GVHD include any of the 25 to 30 known minor histocompatibility antigens (or any of the several hundred yet to be found).19 Although well established for monogenic dominant disorders, in 2007 it was recognized that a postzygotic mutation can cause many complex polygenetic disorders, including GVHD, to manifest in a limited segmental pattern. This understanding, along with retrospective case review, has brought into question previously reported "dermatomal" or "zosteriform" presentations of GVHD, asserting that the linear patterns were misidentified and thus inappropriately attributed to a postviral response.20 Recognition of the Blaschko-linear distribution holds significance in both identifying lesion etiology and understanding disease pathogenesis and treatment.
Our patient illustrates a case of a Blaschko-linear GVHD. The distinctive pattern of her physical findings strongly favored epidermal cell mosaicism as the etiology of her disease. More than just a phenotypically unique case, it provided further insight into the complex etiology underlying GVHD and iterated the basic concepts of Blaschko lines and genetic alterations in development.
To the Editor:
Graft-versus-host disease (GVHD) is a common and serious complication seen most often with bone marrow transplantation and peripheral blood stem cell transplantation. With these therapies, functional lymphoid cells are transferred from an immunocompetent donor into a nongenetically identical recipient, or "host." Because of the allogeneic nature of these transplants, the transplanted lymphoid cells have a high potential to recognize and treat the host's cells as foreign, and the resultant clinical and pathologic picture is that of GVHD. The primary organ systems affected in this immune response are the skin, gastrointestinal tract, and hepatobiliary system.1,2 Cutaneous manifestations are by far the most common.3
Although notable gains have been made in elucidating the causes, risk factors, and mechanisms that result in the clinical picture of GVHD, gaps in our knowledge and understanding still exist. Our patient represents a unique case of unilateral GVHD occurring along Blaschko lines, which has important implications for both recognizing and understanding the pathogenesis of GVHD.
A 35-year-old woman was diagnosed with stage IV follicular lymphoma and received various chemotherapy regimens over the next 4 years. Unfortunately, her disease progressed despite treatment. At 39 years of age, she underwent a nonmyeloablative allogeneic peripheral blood stem cell transplantation from a single HLA-mismatched sibling. She was placed on prednisone and cyclosporine for immunosuppression. High-dose acyclovir prophylaxis also was initiated given her history of zoster affecting the right C3 dermatome. Successful engraftment was achieved, with molecular studies showing 100% of cells following transplantation were of donor origin. Restaging at 1 and 2 years following transplantation found her to be in complete remission.
At 2 years following transplantation, she began a slow taper of immunosuppressive medications. She was successfully weaned off prednisone and continued to gradually reduce the cyclosporine dose. Toward the end of the cyclosporine taper 3 months later, she developed a pruritic eruption on the left proximal arm.
She was seen in a bone marrow transplant clinic 4 weeks after the rash developed. On examination, she had multiple, violaceous, lichenoid papules coalescing into linear bandlike plaques. One plaque extended along the left upper arm and 2 others encircled the left hemithorax, respecting the midline. She was treated empirically for zoster with valacyclovir 1 g 3 times daily based on the presumed dermatomal distribution of the eruption. Despite treatment, the rash progressed, and she developed fever. Eight days later, she was admitted with concern for disseminated zoster (Figure). Viral tissue cultures and polymerase chain reaction analysis of the lesions were negative for varicella-zoster virus and herpes simplex viruses 1 and 2. Biopsies of skin lesions on the arm and trunk were both consistent with GVHD.
Given the clinical history, characteristic lesion morphology, and distinct linear distribution along with histopathological confirmation, a diagnosis of GVHD along Blaschko lines was made. Recognizing the cause to be immunogenic rather than infectious, immunosuppressive medications were started. In addition to increasing the prednisone and cyclosporine back to therapeutic levels, she received weekly methylprednisolone. With treatment, she showed gradual but marked improvement.
Six cases of linear GVHD have occurred as an isotopic response along dermatomes previously affected by varicella-zoster virus.4-8 These cases give credence to the idea that a cutaneous viral infection may alter the skin through unknown mechanisms, predisposing it to become affected by GVHD. Notably, this phenomenon occurred despite absence of a persistent viral genome when assessed using polymerase chain reaction analysis.4
An additional 3 cases of GVHD occurring in a dermatomal distribution without any prior infections in those areas have been reported.9,10 Of note, 2 of 3 patients did have episodes of zoster occur at other sites following transplantation and did not develop GVHD symptoms in any of those locations.9 Interestingly, controversy exists as to whether the distribution of these lesions was dermatomal or followed Blaschko lines.11
Two cases of linear GVHD have been reported in which lesions were identified as occurring along Blaschko lines.12,13 The lines of Blaschko, first described in 1901, correspond to cellular migration patterns during embryological development.14 Postzygotic mutations causing epidermal cell mosaicism may result in skin disorders occurring in segmental areas defined by the Blaschko lines.15-17 Accordingly, the Blaschko-linear pattern in GVHD suggests cellular mosaicism as the etiology in this case. Although the host's immune system develops immunotolerance to both cellular lineages during maturation, transplanted lymphoid cells from a nongenetically identical sibling may identify just one of the cell lines as nonself, producing a selective pattern of GVHD18 confined to the distribution of the genetically disparate cell line, which occurs along the lines of Blaschko in the skin. Candidate genes for mutations that would produce a mosaic following transplant GVHD include any of the 25 to 30 known minor histocompatibility antigens (or any of the several hundred yet to be found).19 Although well established for monogenic dominant disorders, in 2007 it was recognized that a postzygotic mutation can cause many complex polygenetic disorders, including GVHD, to manifest in a limited segmental pattern. This understanding, along with retrospective case review, has brought into question previously reported "dermatomal" or "zosteriform" presentations of GVHD, asserting that the linear patterns were misidentified and thus inappropriately attributed to a postviral response.20 Recognition of the Blaschko-linear distribution holds significance in both identifying lesion etiology and understanding disease pathogenesis and treatment.
Our patient illustrates a case of a Blaschko-linear GVHD. The distinctive pattern of her physical findings strongly favored epidermal cell mosaicism as the etiology of her disease. More than just a phenotypically unique case, it provided further insight into the complex etiology underlying GVHD and iterated the basic concepts of Blaschko lines and genetic alterations in development.
- Thomas ED, Storb R, Clift RA, et al. Bone-marrow transplantation. N Engl J Med. 1975;292:895-902.
- Lee SJ, Vogelsang G, Flowers ME. Chronic graft-versus-host disease. Biol Blood Marrow Transplant. 2003;9:215-233.
- Johnson ML, Farmer ER. Graft versus host reactions in dermatology. J Am Acad Dermatol. 1998;38:369-384.
- Baselga E, Drolet BA, Segura AD, et al. Dermatomal lichenoid chronic graft-vs-host disease following varicella-zoster infection despite absence of viral genome. J Cutan Pathol. 1996;23:576-581.
- Lacour JP, Sirvent N, Monpoux F, et al. Dermatomal chronic cutaneous graft versus host disease at the site of prior herpes zoster. Br J Dermatol. 1999;141:587-589.
- Cordoba S, Fraga J, Bartolome B, et al. Giant cell lichenoid dermatitis within herpes zoster scars in a bone marrow recipient. J Cutan Pathol. 2000;27:255-257.
- Sanli H, Anadolu R, Arat M, et al. Dermatomal lichenoid graft-versus-host disease within herpes zoster scars. Int J Dermatol. 2003;42:562-564.
- Martires KJ, Baird K, Citrin DE, et al. Localization of sclerotic-type chronic graft-versus-host disease to sites of skin injury: potential insight into the mechanism of isomorphic and isotopic responses. Arch Dermatol. 2011;147:1081-1086.
- Freemer CS, Farmer ER, Corio RL, et al. Lichenoid chronic graft-vs-host disease occurring in a dermatomal distribution. Arch Dermatol. 1994;130:70-72.
- Cohen PR, Hymes SR. Linear and dermatomal cutaneous graft-versus-host disease. South Med J. 1994;87:758-761.
- Reisfeld PL. Linear lichenoid graft-vs-host disease. Arch Dermatol. 1994;130:1207-1208.
- Beers B, Kalish RS, Kaye VN, et al. Unilateral linear lichenoid eruption after bone marrow transplantation: an unmasking of tolerance to an abnormal keratinocyte clone? J Am Acad Dermatol. 1993;28(5, pt 2):888-892.
- Wilson BB, Lockman DW. Linear lichenoid graft-vs-host disease. Arch Dermatol. 1994;130:1206-1207.
- Goldberg I, Sprecher E. Patterned disorders in dermatology. Clin Dermatol. 2011;29:498-503.
- Colman SD, Rasmussen SA, Ho VT, et al. Somatic mosaicism in a patient with neurofibromatosis type 1. Am J Hum Genet. 1996;58:484-490.
- Munro CS, Wilkie AO. Epidermal mosaicism producing localised acne: somatic mutation in FGFR2. Lancet. 1998;352:704-705.
- Sakuntabhai A, Dhitavat J, Burge S, et al. Mosaicism for ATP2A2 mutations causes segmental Darier's disease. J Invest Dermatol. 2000;115:1144-1147.
- Dickinson AM, Wang XN, Sviland L, et al. In situ dissection of the graft-versus-host activities of cytotoxic T cells specific for minor histocompatibility antigens. Nat Med. 2002;8:410-414.
- Hansen JA, Chien JW, Warren EH, et al. Defining genetic risk for graft- versus-host disease and mortality following allogeneic hematopoietic stem cell transplantation. Curr Opin Hematol. 2010;17:483-492.
- Happle R. Superimposed segmental manifestation of polygenic skin disorders. J Am Acad Dermatol. 2007;57:690-699.
- Thomas ED, Storb R, Clift RA, et al. Bone-marrow transplantation. N Engl J Med. 1975;292:895-902.
- Lee SJ, Vogelsang G, Flowers ME. Chronic graft-versus-host disease. Biol Blood Marrow Transplant. 2003;9:215-233.
- Johnson ML, Farmer ER. Graft versus host reactions in dermatology. J Am Acad Dermatol. 1998;38:369-384.
- Baselga E, Drolet BA, Segura AD, et al. Dermatomal lichenoid chronic graft-vs-host disease following varicella-zoster infection despite absence of viral genome. J Cutan Pathol. 1996;23:576-581.
- Lacour JP, Sirvent N, Monpoux F, et al. Dermatomal chronic cutaneous graft versus host disease at the site of prior herpes zoster. Br J Dermatol. 1999;141:587-589.
- Cordoba S, Fraga J, Bartolome B, et al. Giant cell lichenoid dermatitis within herpes zoster scars in a bone marrow recipient. J Cutan Pathol. 2000;27:255-257.
- Sanli H, Anadolu R, Arat M, et al. Dermatomal lichenoid graft-versus-host disease within herpes zoster scars. Int J Dermatol. 2003;42:562-564.
- Martires KJ, Baird K, Citrin DE, et al. Localization of sclerotic-type chronic graft-versus-host disease to sites of skin injury: potential insight into the mechanism of isomorphic and isotopic responses. Arch Dermatol. 2011;147:1081-1086.
- Freemer CS, Farmer ER, Corio RL, et al. Lichenoid chronic graft-vs-host disease occurring in a dermatomal distribution. Arch Dermatol. 1994;130:70-72.
- Cohen PR, Hymes SR. Linear and dermatomal cutaneous graft-versus-host disease. South Med J. 1994;87:758-761.
- Reisfeld PL. Linear lichenoid graft-vs-host disease. Arch Dermatol. 1994;130:1207-1208.
- Beers B, Kalish RS, Kaye VN, et al. Unilateral linear lichenoid eruption after bone marrow transplantation: an unmasking of tolerance to an abnormal keratinocyte clone? J Am Acad Dermatol. 1993;28(5, pt 2):888-892.
- Wilson BB, Lockman DW. Linear lichenoid graft-vs-host disease. Arch Dermatol. 1994;130:1206-1207.
- Goldberg I, Sprecher E. Patterned disorders in dermatology. Clin Dermatol. 2011;29:498-503.
- Colman SD, Rasmussen SA, Ho VT, et al. Somatic mosaicism in a patient with neurofibromatosis type 1. Am J Hum Genet. 1996;58:484-490.
- Munro CS, Wilkie AO. Epidermal mosaicism producing localised acne: somatic mutation in FGFR2. Lancet. 1998;352:704-705.
- Sakuntabhai A, Dhitavat J, Burge S, et al. Mosaicism for ATP2A2 mutations causes segmental Darier's disease. J Invest Dermatol. 2000;115:1144-1147.
- Dickinson AM, Wang XN, Sviland L, et al. In situ dissection of the graft-versus-host activities of cytotoxic T cells specific for minor histocompatibility antigens. Nat Med. 2002;8:410-414.
- Hansen JA, Chien JW, Warren EH, et al. Defining genetic risk for graft- versus-host disease and mortality following allogeneic hematopoietic stem cell transplantation. Curr Opin Hematol. 2010;17:483-492.
- Happle R. Superimposed segmental manifestation of polygenic skin disorders. J Am Acad Dermatol. 2007;57:690-699.
Practice Points
- Recognizing the characteristic manners in which different linear dermatoses present can aid in correctly identifying disorders that most commonly present in either a dermatomal or Blaschko-linear-type distribution.
- A blaschkoid-type distribution is the result of cutaneous mosaicism that occurs during embryological development and therefore subsequently produces a unique phenotypical presentation for various genetically influenced skin disorders, including graft-versus-host disease.