User login
Cutis is a peer-reviewed clinical journal for the dermatologist, allergist, and general practitioner published monthly since 1965. Concise clinical articles present the practical side of dermatology, helping physicians to improve patient care. Cutis is referenced in Index Medicus/MEDLINE and is written and edited by industry leaders.
ass lick
assault rifle
balls
ballsac
black jack
bleach
Boko Haram
bondage
causas
cheap
child abuse
cocaine
compulsive behaviors
cost of miracles
cunt
Daech
display network stats
drug paraphernalia
explosion
fart
fda and death
fda AND warn
fda AND warning
fda AND warns
feom
fuck
gambling
gfc
gun
human trafficking
humira AND expensive
illegal
ISIL
ISIS
Islamic caliphate
Islamic state
madvocate
masturbation
mixed martial arts
MMA
molestation
national rifle association
NRA
nsfw
nuccitelli
pedophile
pedophilia
poker
porn
porn
pornography
psychedelic drug
recreational drug
sex slave rings
shit
slot machine
snort
substance abuse
terrorism
terrorist
texarkana
Texas hold 'em
UFC
section[contains(@class, 'nav-hidden')]
section[contains(@class, 'nav-hidden active')
A peer-reviewed, indexed journal for dermatologists with original research, image quizzes, cases and reviews, and columns.
Enlarging Mass on the Scalp
Enlarging Mass on the Scalp
THE DIAGNOSIS: Malignant Proliferating Trichilemmal Tumor
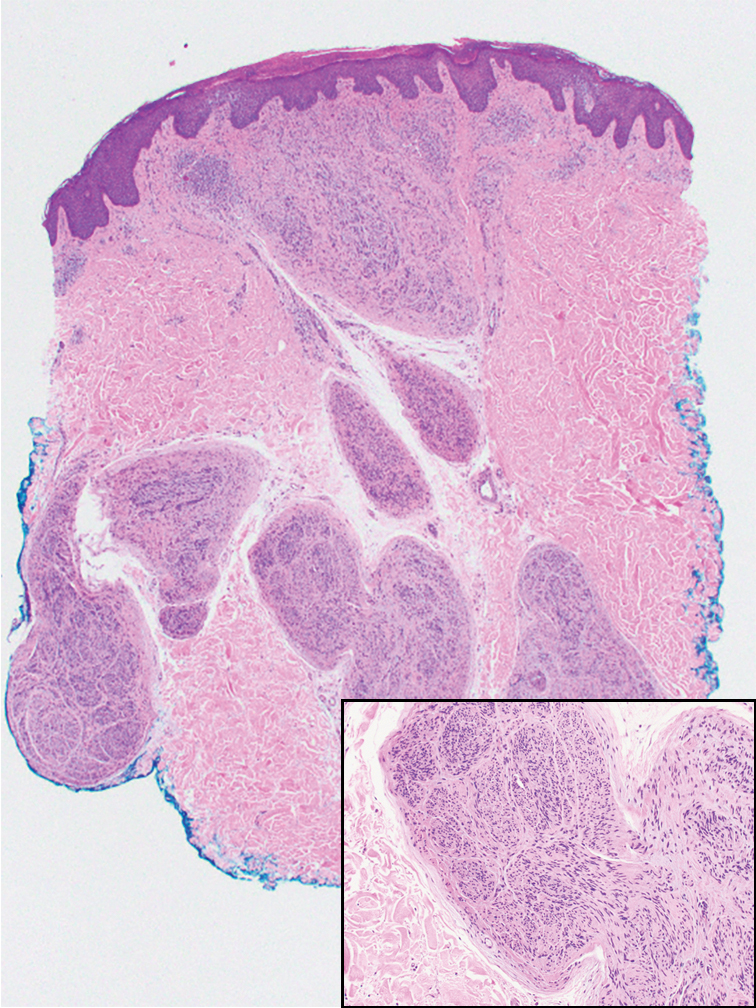
Histologic examination revealed atypical keratinocytes, nuclear pleomorphism, and lobulating epithelial masses with trichilemmal keratinization (Figure). The presence of CD34 positivity, a marker of outer follicular root sheath–derived cells, supported the diagnosis of a malignant proliferating trichilemmal tumor (MPTT). Imaging also revealed signs of bone invasion, further supporting a malignant process. Based on these findings, the patient underwent complete excision of the mass with scalp reconstruction, lymph node dissection, and systemic evaluation for metastases. Final pathology confirmed negative surgical margins and no lymph node involvement. Adjuvant radiation was not required, given the absence of skull invasion or confirmed distant metastasis.
The differential diagnosis for rapidly enlarging scalp tumors can be broad and includes both benign and malignant processes. In this patient, the differential diagnoses included trichilemmal carcinoma, cutaneous squamous cell carcinoma (SCC), sebaceous carcinoma (SC), proliferating trichilemmal tumor (PTT), and MPTT. Due to the notable clinical and histologic overlap among these lesions, definitive diagnosis required histopathologic evaluation in our patient.
Proliferating trichilemmal tumors were first described in 1966 by Wilson-Jones,1 who used the term proliferating epidermoid cysts, noting their distinct histologic features and resemblance to SCC.2 These tumors generally are benign and arise from the isthmus of the outer root sheath of the hair follicle; however, malignant transformation can occur, resulting in a rare entity known as MPTT. This malignant variant was first described in 1983 by Saida et al,3 who emphasized its distinct clinical behavior, including infiltrative growth, high mitotic activity, and potential for local recurrence and metastasis.
A recent literature review identified 60 reported cases of MPTT, with an average patient age of 57 years and a female predominance.4 Clinically, MPTTs often manifest as large (>5 cm) lobulated masses located on sun-exposed, hair-bearing areas of the skin, especially the scalp. These lesions may be flesh-colored to pink and often exhibit ulceration, necrosis, or calcification.5 Typically, MPTTs follow a biphasic course, beginning with a slow-growing phase followed by a period of rapid growth. Due to their aggressive behavior and resemblance to other cutaneous malignancies, accurate differentiation of MPTT from benign PTTs, cutaneous SCCs, SCs, and trichilemmal carcinomas is critical.
Malignant proliferating trichilemmal tumors demonstrate a substantially higher metastatic potential than either benign PTTs or cutaneous SCCs. While cutaneous SCCs carry a metastasis rate of approximately 1.9% to 2.6%, MPTTs carry a considerably higher rate of approximately 25.0%.6 Regional lymphatic spread is the most common route of dissemination, making comprehensive lymph node assessment—both radiographic and clinical—an important component of tumor staging. When lymph node involvement is suspected, surgical dissection may be indicated, along with consideration of adjuvant therapies.
Histopathologically, MPTT is characterized by nuclear atypia, mitotic figures, and lobulated masses of proliferating epithelium showing trichilemmal differentiation and infiltrative growth.4 The presence of CD34 positivity, reflecting outer follicular root sheath differentiation, helps distinguish MPTT from cutaneous SCC and SC, which typically lack this marker.6,7 Immunohistochemistry is therefore a valuable adjunct in differentiating these lesions.
The mainstay of treatment for MPTT is wide local excision with clear margins. Margins of at least 1 cm generally are recommended. Although Mohs micrographic surgery may be used in anatomically sensitive areas, it typically is not preferred due to the potential for skip lesions in MPTT, which may lead to incomplete excision and recurrence.8 In cases with evidence of regional or distant metastasis or when clear margins cannot be achieved confidently, adjunctive treatments such as radiation therapy and systemic chemotherapy may be indicated. Preoperative imaging is used to evaluate for local invasion (skull or bone involvement) and regional lymph node status, which may inform adjuvant therapy postoperatively.
The prognosis for MPTT is variable and depends largely on early recognition, accurate histopathologic diagnosis, complete surgical excision with clear margins, and the presence or absence of metastasis. When the tumor is fully excised with negative margins and no lymph node involvement, the risk for recurrence is substantially reduced; however, MPTT is known for its potential aggressive behavior. Delays in diagnosis or incomplete resection can lead to local recurrence, regional spread, or even distant metastasis. In the literature review discussed previously, the mortality rate of patients with MPTT was 11.7%,4 which is notably higher than that of more common cutaneous malignancies such as cutaneous SCC, which is reported at 1.2%.9
The clinical course of MPTT remains difficult to predict due to its rarity and the limited availability of large-scale studies. Most published data are derived from isolated case reports or small case series, making standardized treatment guidelines challenging. Given this uncertainty, long-term follow-up is strongly recommended to monitor for recurrence or metastatic progression.2
This case highlights the critical role of clinicopathologic correlation in the evaluation of atypical or rapidly growing scalp lesions. The expertise of dermatologists in recognizing atypical presentations, combined with precise histopathologic analysis, including immunohistochemical staining, is vital to ensuring accurate diagnosis and optimal treatment. Early intervention can improve patient outcomes by reducing the risk for local recurrence and metastatic progression as well as the need for more intensive therapies.
- Jones EW. Proliferating epidermoid cysts. Arch Dermatol. 1966;94:11-19.
- Kemaloglu CA, Öztürk M, Aydın B, et al. Malignant proliferating trichilemmal tumor of the scalp: report of 4 cases and a short review of the literature. Case Reports Plast Surg Hand Surg. 2022;9:158-164. doi:10.1080/23320885.2022.2077208
- Saida T, Oohard K, Hori Y, et al. Development of a malignant proliferating trichilemmal cyst in a patient with multiple trichilemmal cysts. Dermatology. 1983;166:203-208. doi:10.1159/000249868
- Abdelhammed MH, Siatecka H, Diwan AH, et al. A rare case of a malignant proliferating trichilemmal tumor: a molecular study harboring potential therapeutic significance and a review of literature. Dermatopathology (Basel). 2024;11:354-363. doi:10.3390 /dermatopathology11040038
- Fronek L, Brahs A, Farsi M, et al. A rare case of trichilemmal carcinoma: histology and management. J Clin Aesthet Dermatol. 2021;14:25-30.
- Osto M, Parry N, Rehman R, et al. Malignant proliferating trichilemmal tumor of the scalp: a systematic review. Am J Dermatopathol. 2021;43:851-866. doi:10.1097/DAD.0000000000001991
- Plaza JA, Mackinnon A, Carrillo L, et al. Role of immunohistochemistry in the diagnosis of sebaceous carcinoma: a clinicopathologic and immunohistochemical study. Am J Dermatopathol. 2015;37:809-821. doi:10.1097/DAD.0000000000000255
- Singh P, Usman A, Motta L, et al. Malignant proliferating trichilemmal tumour. BMJ Case Rep. Published online August 17, 2018. doi:10.1136/bcr-2018-224460
- Ran NA, Granger EE, Brodland DG, et al. Risk factor number and recurrence, metastasis, and disease-related death in cutaneous squamous cell carcinoma. JAMA Dermatol. 2025;161:597-604. doi:10.1001/jamadermatol.2025.0128
THE DIAGNOSIS: Malignant Proliferating Trichilemmal Tumor
Histologic examination revealed atypical keratinocytes, nuclear pleomorphism, and lobulating epithelial masses with trichilemmal keratinization (Figure). The presence of CD34 positivity, a marker of outer follicular root sheath–derived cells, supported the diagnosis of a malignant proliferating trichilemmal tumor (MPTT). Imaging also revealed signs of bone invasion, further supporting a malignant process. Based on these findings, the patient underwent complete excision of the mass with scalp reconstruction, lymph node dissection, and systemic evaluation for metastases. Final pathology confirmed negative surgical margins and no lymph node involvement. Adjuvant radiation was not required, given the absence of skull invasion or confirmed distant metastasis.
The differential diagnosis for rapidly enlarging scalp tumors can be broad and includes both benign and malignant processes. In this patient, the differential diagnoses included trichilemmal carcinoma, cutaneous squamous cell carcinoma (SCC), sebaceous carcinoma (SC), proliferating trichilemmal tumor (PTT), and MPTT. Due to the notable clinical and histologic overlap among these lesions, definitive diagnosis required histopathologic evaluation in our patient.
Proliferating trichilemmal tumors were first described in 1966 by Wilson-Jones,1 who used the term proliferating epidermoid cysts, noting their distinct histologic features and resemblance to SCC.2 These tumors generally are benign and arise from the isthmus of the outer root sheath of the hair follicle; however, malignant transformation can occur, resulting in a rare entity known as MPTT. This malignant variant was first described in 1983 by Saida et al,3 who emphasized its distinct clinical behavior, including infiltrative growth, high mitotic activity, and potential for local recurrence and metastasis.
A recent literature review identified 60 reported cases of MPTT, with an average patient age of 57 years and a female predominance.4 Clinically, MPTTs often manifest as large (>5 cm) lobulated masses located on sun-exposed, hair-bearing areas of the skin, especially the scalp. These lesions may be flesh-colored to pink and often exhibit ulceration, necrosis, or calcification.5 Typically, MPTTs follow a biphasic course, beginning with a slow-growing phase followed by a period of rapid growth. Due to their aggressive behavior and resemblance to other cutaneous malignancies, accurate differentiation of MPTT from benign PTTs, cutaneous SCCs, SCs, and trichilemmal carcinomas is critical.
Malignant proliferating trichilemmal tumors demonstrate a substantially higher metastatic potential than either benign PTTs or cutaneous SCCs. While cutaneous SCCs carry a metastasis rate of approximately 1.9% to 2.6%, MPTTs carry a considerably higher rate of approximately 25.0%.6 Regional lymphatic spread is the most common route of dissemination, making comprehensive lymph node assessment—both radiographic and clinical—an important component of tumor staging. When lymph node involvement is suspected, surgical dissection may be indicated, along with consideration of adjuvant therapies.
Histopathologically, MPTT is characterized by nuclear atypia, mitotic figures, and lobulated masses of proliferating epithelium showing trichilemmal differentiation and infiltrative growth.4 The presence of CD34 positivity, reflecting outer follicular root sheath differentiation, helps distinguish MPTT from cutaneous SCC and SC, which typically lack this marker.6,7 Immunohistochemistry is therefore a valuable adjunct in differentiating these lesions.
The mainstay of treatment for MPTT is wide local excision with clear margins. Margins of at least 1 cm generally are recommended. Although Mohs micrographic surgery may be used in anatomically sensitive areas, it typically is not preferred due to the potential for skip lesions in MPTT, which may lead to incomplete excision and recurrence.8 In cases with evidence of regional or distant metastasis or when clear margins cannot be achieved confidently, adjunctive treatments such as radiation therapy and systemic chemotherapy may be indicated. Preoperative imaging is used to evaluate for local invasion (skull or bone involvement) and regional lymph node status, which may inform adjuvant therapy postoperatively.
The prognosis for MPTT is variable and depends largely on early recognition, accurate histopathologic diagnosis, complete surgical excision with clear margins, and the presence or absence of metastasis. When the tumor is fully excised with negative margins and no lymph node involvement, the risk for recurrence is substantially reduced; however, MPTT is known for its potential aggressive behavior. Delays in diagnosis or incomplete resection can lead to local recurrence, regional spread, or even distant metastasis. In the literature review discussed previously, the mortality rate of patients with MPTT was 11.7%,4 which is notably higher than that of more common cutaneous malignancies such as cutaneous SCC, which is reported at 1.2%.9
The clinical course of MPTT remains difficult to predict due to its rarity and the limited availability of large-scale studies. Most published data are derived from isolated case reports or small case series, making standardized treatment guidelines challenging. Given this uncertainty, long-term follow-up is strongly recommended to monitor for recurrence or metastatic progression.2
This case highlights the critical role of clinicopathologic correlation in the evaluation of atypical or rapidly growing scalp lesions. The expertise of dermatologists in recognizing atypical presentations, combined with precise histopathologic analysis, including immunohistochemical staining, is vital to ensuring accurate diagnosis and optimal treatment. Early intervention can improve patient outcomes by reducing the risk for local recurrence and metastatic progression as well as the need for more intensive therapies.
THE DIAGNOSIS: Malignant Proliferating Trichilemmal Tumor
Histologic examination revealed atypical keratinocytes, nuclear pleomorphism, and lobulating epithelial masses with trichilemmal keratinization (Figure). The presence of CD34 positivity, a marker of outer follicular root sheath–derived cells, supported the diagnosis of a malignant proliferating trichilemmal tumor (MPTT). Imaging also revealed signs of bone invasion, further supporting a malignant process. Based on these findings, the patient underwent complete excision of the mass with scalp reconstruction, lymph node dissection, and systemic evaluation for metastases. Final pathology confirmed negative surgical margins and no lymph node involvement. Adjuvant radiation was not required, given the absence of skull invasion or confirmed distant metastasis.
The differential diagnosis for rapidly enlarging scalp tumors can be broad and includes both benign and malignant processes. In this patient, the differential diagnoses included trichilemmal carcinoma, cutaneous squamous cell carcinoma (SCC), sebaceous carcinoma (SC), proliferating trichilemmal tumor (PTT), and MPTT. Due to the notable clinical and histologic overlap among these lesions, definitive diagnosis required histopathologic evaluation in our patient.
Proliferating trichilemmal tumors were first described in 1966 by Wilson-Jones,1 who used the term proliferating epidermoid cysts, noting their distinct histologic features and resemblance to SCC.2 These tumors generally are benign and arise from the isthmus of the outer root sheath of the hair follicle; however, malignant transformation can occur, resulting in a rare entity known as MPTT. This malignant variant was first described in 1983 by Saida et al,3 who emphasized its distinct clinical behavior, including infiltrative growth, high mitotic activity, and potential for local recurrence and metastasis.
A recent literature review identified 60 reported cases of MPTT, with an average patient age of 57 years and a female predominance.4 Clinically, MPTTs often manifest as large (>5 cm) lobulated masses located on sun-exposed, hair-bearing areas of the skin, especially the scalp. These lesions may be flesh-colored to pink and often exhibit ulceration, necrosis, or calcification.5 Typically, MPTTs follow a biphasic course, beginning with a slow-growing phase followed by a period of rapid growth. Due to their aggressive behavior and resemblance to other cutaneous malignancies, accurate differentiation of MPTT from benign PTTs, cutaneous SCCs, SCs, and trichilemmal carcinomas is critical.
Malignant proliferating trichilemmal tumors demonstrate a substantially higher metastatic potential than either benign PTTs or cutaneous SCCs. While cutaneous SCCs carry a metastasis rate of approximately 1.9% to 2.6%, MPTTs carry a considerably higher rate of approximately 25.0%.6 Regional lymphatic spread is the most common route of dissemination, making comprehensive lymph node assessment—both radiographic and clinical—an important component of tumor staging. When lymph node involvement is suspected, surgical dissection may be indicated, along with consideration of adjuvant therapies.
Histopathologically, MPTT is characterized by nuclear atypia, mitotic figures, and lobulated masses of proliferating epithelium showing trichilemmal differentiation and infiltrative growth.4 The presence of CD34 positivity, reflecting outer follicular root sheath differentiation, helps distinguish MPTT from cutaneous SCC and SC, which typically lack this marker.6,7 Immunohistochemistry is therefore a valuable adjunct in differentiating these lesions.
The mainstay of treatment for MPTT is wide local excision with clear margins. Margins of at least 1 cm generally are recommended. Although Mohs micrographic surgery may be used in anatomically sensitive areas, it typically is not preferred due to the potential for skip lesions in MPTT, which may lead to incomplete excision and recurrence.8 In cases with evidence of regional or distant metastasis or when clear margins cannot be achieved confidently, adjunctive treatments such as radiation therapy and systemic chemotherapy may be indicated. Preoperative imaging is used to evaluate for local invasion (skull or bone involvement) and regional lymph node status, which may inform adjuvant therapy postoperatively.
The prognosis for MPTT is variable and depends largely on early recognition, accurate histopathologic diagnosis, complete surgical excision with clear margins, and the presence or absence of metastasis. When the tumor is fully excised with negative margins and no lymph node involvement, the risk for recurrence is substantially reduced; however, MPTT is known for its potential aggressive behavior. Delays in diagnosis or incomplete resection can lead to local recurrence, regional spread, or even distant metastasis. In the literature review discussed previously, the mortality rate of patients with MPTT was 11.7%,4 which is notably higher than that of more common cutaneous malignancies such as cutaneous SCC, which is reported at 1.2%.9
The clinical course of MPTT remains difficult to predict due to its rarity and the limited availability of large-scale studies. Most published data are derived from isolated case reports or small case series, making standardized treatment guidelines challenging. Given this uncertainty, long-term follow-up is strongly recommended to monitor for recurrence or metastatic progression.2
This case highlights the critical role of clinicopathologic correlation in the evaluation of atypical or rapidly growing scalp lesions. The expertise of dermatologists in recognizing atypical presentations, combined with precise histopathologic analysis, including immunohistochemical staining, is vital to ensuring accurate diagnosis and optimal treatment. Early intervention can improve patient outcomes by reducing the risk for local recurrence and metastatic progression as well as the need for more intensive therapies.
- Jones EW. Proliferating epidermoid cysts. Arch Dermatol. 1966;94:11-19.
- Kemaloglu CA, Öztürk M, Aydın B, et al. Malignant proliferating trichilemmal tumor of the scalp: report of 4 cases and a short review of the literature. Case Reports Plast Surg Hand Surg. 2022;9:158-164. doi:10.1080/23320885.2022.2077208
- Saida T, Oohard K, Hori Y, et al. Development of a malignant proliferating trichilemmal cyst in a patient with multiple trichilemmal cysts. Dermatology. 1983;166:203-208. doi:10.1159/000249868
- Abdelhammed MH, Siatecka H, Diwan AH, et al. A rare case of a malignant proliferating trichilemmal tumor: a molecular study harboring potential therapeutic significance and a review of literature. Dermatopathology (Basel). 2024;11:354-363. doi:10.3390 /dermatopathology11040038
- Fronek L, Brahs A, Farsi M, et al. A rare case of trichilemmal carcinoma: histology and management. J Clin Aesthet Dermatol. 2021;14:25-30.
- Osto M, Parry N, Rehman R, et al. Malignant proliferating trichilemmal tumor of the scalp: a systematic review. Am J Dermatopathol. 2021;43:851-866. doi:10.1097/DAD.0000000000001991
- Plaza JA, Mackinnon A, Carrillo L, et al. Role of immunohistochemistry in the diagnosis of sebaceous carcinoma: a clinicopathologic and immunohistochemical study. Am J Dermatopathol. 2015;37:809-821. doi:10.1097/DAD.0000000000000255
- Singh P, Usman A, Motta L, et al. Malignant proliferating trichilemmal tumour. BMJ Case Rep. Published online August 17, 2018. doi:10.1136/bcr-2018-224460
- Ran NA, Granger EE, Brodland DG, et al. Risk factor number and recurrence, metastasis, and disease-related death in cutaneous squamous cell carcinoma. JAMA Dermatol. 2025;161:597-604. doi:10.1001/jamadermatol.2025.0128
- Jones EW. Proliferating epidermoid cysts. Arch Dermatol. 1966;94:11-19.
- Kemaloglu CA, Öztürk M, Aydın B, et al. Malignant proliferating trichilemmal tumor of the scalp: report of 4 cases and a short review of the literature. Case Reports Plast Surg Hand Surg. 2022;9:158-164. doi:10.1080/23320885.2022.2077208
- Saida T, Oohard K, Hori Y, et al. Development of a malignant proliferating trichilemmal cyst in a patient with multiple trichilemmal cysts. Dermatology. 1983;166:203-208. doi:10.1159/000249868
- Abdelhammed MH, Siatecka H, Diwan AH, et al. A rare case of a malignant proliferating trichilemmal tumor: a molecular study harboring potential therapeutic significance and a review of literature. Dermatopathology (Basel). 2024;11:354-363. doi:10.3390 /dermatopathology11040038
- Fronek L, Brahs A, Farsi M, et al. A rare case of trichilemmal carcinoma: histology and management. J Clin Aesthet Dermatol. 2021;14:25-30.
- Osto M, Parry N, Rehman R, et al. Malignant proliferating trichilemmal tumor of the scalp: a systematic review. Am J Dermatopathol. 2021;43:851-866. doi:10.1097/DAD.0000000000001991
- Plaza JA, Mackinnon A, Carrillo L, et al. Role of immunohistochemistry in the diagnosis of sebaceous carcinoma: a clinicopathologic and immunohistochemical study. Am J Dermatopathol. 2015;37:809-821. doi:10.1097/DAD.0000000000000255
- Singh P, Usman A, Motta L, et al. Malignant proliferating trichilemmal tumour. BMJ Case Rep. Published online August 17, 2018. doi:10.1136/bcr-2018-224460
- Ran NA, Granger EE, Brodland DG, et al. Risk factor number and recurrence, metastasis, and disease-related death in cutaneous squamous cell carcinoma. JAMA Dermatol. 2025;161:597-604. doi:10.1001/jamadermatol.2025.0128
Enlarging Mass on the Scalp
Enlarging Mass on the Scalp
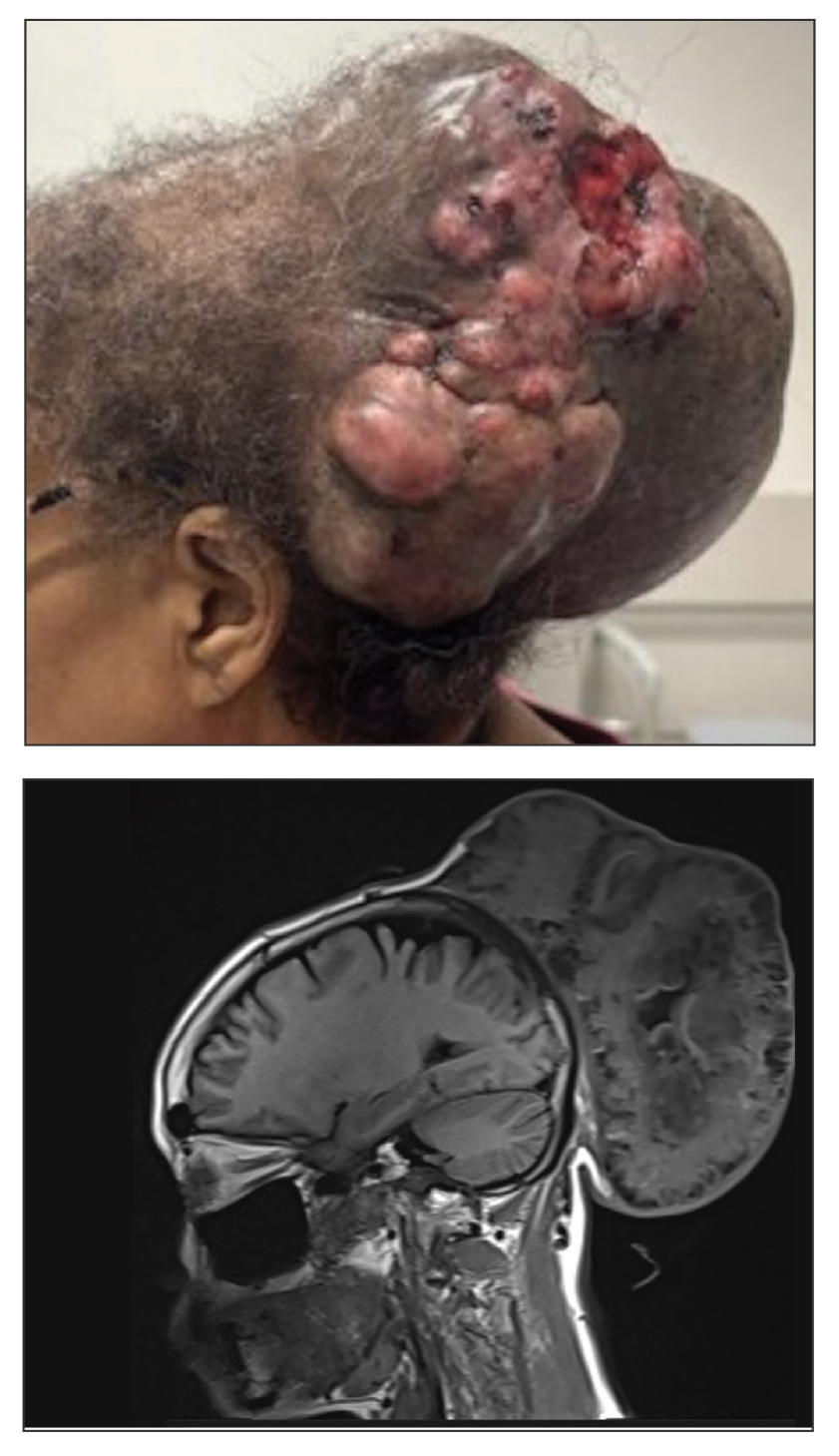
A 61-year-old woman presented to the emergency department with worsening pain and bleeding from a scalp tumor of 16 years’ duration. Initially noted as a small nodule on the left parietal scalp on computed tomography of the head, the mass had grown rapidly in recent years and currently measured 22×10×15 cm. At prior consultations with plastic and general surgery, the patient had declined surgical intervention. At the current presentation, biopsies were performed by plastic surgery, and a dermatopathology consultation was ordered. Histopathology revealed atypical keratinocytes, nuclear pleomorphism, lobulating epithelial masses with trichilemmal keratinization, and CD34 positivity. Subsequent computed tomography and positron emission tomography of the head showed occipital skull erosion and bilateral cervical lymphadenopathy, suggesting metastasis.

Adalimumab in Lichen Planus: A Narrative Review of Treatment and Paradoxical Reactions
Adalimumab in Lichen Planus: A Narrative Review of Treatment and Paradoxical Reactions
Lichen planus (LP) is a chronic inflammatory condition affecting the skin (cutaneous LP), mucous membranes (oral, ocular, or vulvar LP), hair (lichen planopilaris [LPP]), and nails that predominantly occurs in middle-aged adults. Although the true etiology remains unknown, the pathogenesis of LP is thought to involve multiple factors. Several human leukocyte antigen (HLA) alleles have been associated with LP and its variants, including HLA-B27, HLA-B51, HLA-DR1 (cutaneous and oral LP), HLA-DRB1*11, and HLA-DQB1*03 (LPP). Additionally, HLA-Bw57 has been reported to be associated with oral LP in a cohort of British patients.1 In addition to HLA alleles, genetic polymorphisms in cytokines including IL-4, IL-6, IL-18, interferon (IFN) γ, and tumor necrosis factor (TNF) α and its receptor have been found to be associated with LP.2 Beyond genetics, chronic viral infection has been implicated in the development of LP. Systemic infection with the hepatitis C virus has been linked to the development of oral LP by promoting the recruitment of hepatitis C virus–specific CD8+ T cells from peripheral blood to the oral lesions, where they exhibit a terminally differentiated effector status.3 Another report found an association between human herpesvirus 7 (HHV-7) and cutaneous LP; in this study, HHV-7 RNA was detected in plasmacytoid dendritic cells but not T cells and diminished after treatment, providing evidence for dendritic cells being involved in the HHV-7–mediated pathogenesis of cutaneous LP.4 These findings were further corroborated by another study of oral LP patients that found enhanced infiltration of plasmacytoid and myeloid dendritic cells and upregulation in toll-like receptor and IFN-γ signaling.4
In addition to immune cell dysregulation, LP and its variants have been linked to neurogenic inflammation. In oral LP lesions, neurokinin 1 receptor and substance P were highly expressed and demonstrated a positive correlation with the expression of apoptotic marker caspase-3 and proliferation marker Ki-67.5 These results suggest that neuropeptides may be involved in cell proliferation and turnover in oral LP. Similarly, in patients with LPP, substance P was more abundant in affected areas, whereas another neuropeptide, calcitonin gene-related peptide, was more highly expressed in unaffected areas,6 further supporting the pathogenic role of neurogenic inflammation in LP.
A mucosal variant that often goes undiagnosed is vulvar LP. Although no distinct pathologic mechanism for vulvar LP has been established, prior reports found an association with autoantibodies.7,8 In patients with erosive vulvar LP, epidermal-binding basement membrane zone antibodies were detected in epidermal skin biopsies and in circulation with reactivity to bullous pemphigoid antigens 180 (9/11 [81.8%] patients) and 230 (2/11 [18.2%] patients).7 A similar study in patients with vulvar lichen sclerosus found similar proportions of circulating antibodies reactive to bullous pemphigoid antigens 180 (6/7 [85.7%] patients) and 230 (1/7 [14.3%] patients).8 Erosive vulvar LP has been shown to be associated with autoimmune disease (eg, alopecia areata, celiac disease and pernicious anemia),9 which suggests that the previously reported autoreactive antibodies7,8 are secondary to autoimmunity rather than primary drivers of vulvar LP pathogenesis.
Certain medications also have been reported to cause cutaneous lichenoid drug eruptions. Although they can clinically and histologically mimic classic LP, lichenoid drug eruptions are a distinct entity. Common inciting medications include thiazide diuretics, angiotensin-converting enzyme inhibitors, anti-inflammatory drugs, antimalarials, checkpoint inhibitors, antimicrobials, antihypertensives, antidiabetics, and psychiatric drugs. The exact pathologic mechanism of lichenoid drug eruptions currently is unclear but is thought to involve the binding of drug molecules to the cell-surface proteins of the epidermis, creating an antigenic hapten stimulus for CD8+T cells and triggering apoptosis of keratinocytes.1
The clinical severity of LP can range from mild localized disease to widespread and debilitating involvement. Multiple treatment modalities have been developed for management of LP, including topical and intralesional corticosteroids, phototherapy, Janus kinase inhibitors, phosphodiesterase-4 inhibitors, and anti–TNF-α inhibitors. Herein, we provide a narrative review and summary of the use of the TNF-α inhibitor adalimumab as a potential effective treatment for patients with LP.
Methods
We conducted a PubMed search of articles indexed for MEDLINE from 2005 to 2025 using the terms adalimumab AND lichen planus or adalimumab AND lichen. Articles that reported cases of oral LP, cutaneous LP, LPP, or lichenoid eruptions and adalimumab therapy were included in our review. Articles that used non-adalimumab TNF-α inhibitors were excluded. Using the search terms, 2 independent reviewers (M.G. and N.E.) conducted the literature review then screened the articles based on the inclusion and exclusion criteria. Our literature search yielded 40 articles, of which 20 met the criteria for inclusion in our narrative review.
Results
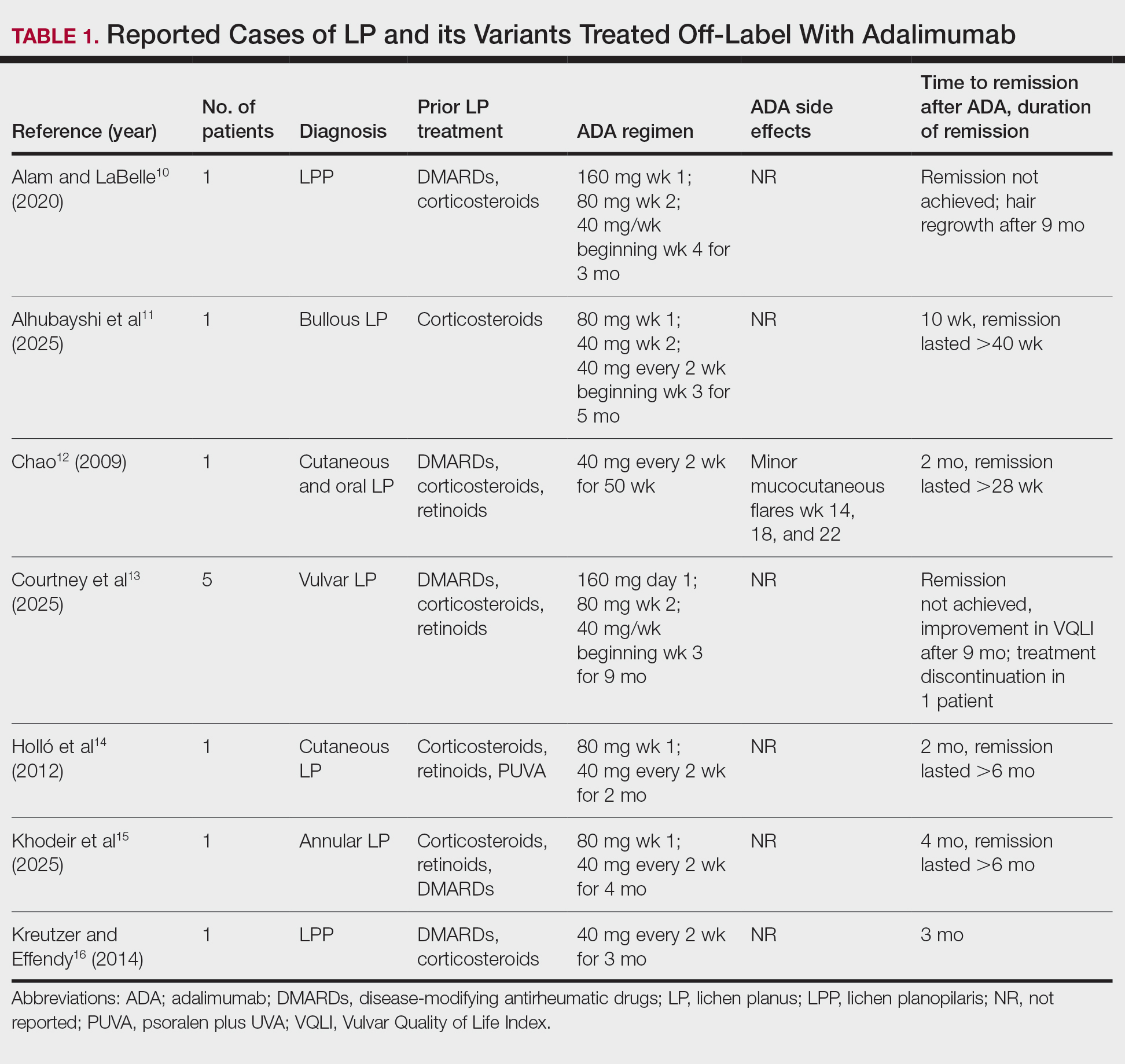
Our literature search yielded 11 patients with LP who were treated with adalimumab across studies (Table 1).10-16 Prior LP treatments included topical corticosteroids (11/11 [100%]), disease-modifying antirheumatic drugs (6/11 [54.5%]), retinoids (4/11 [36.4%]), and psoralen plus UVA (1/11 [36.4%]). Adalimumab was administered subcutaneously following 4 treatment regimens: (1)
Paradoxically, our review of the literature yielded 12 patients in whom adalimumab was associated with lichenoid-type eruptions across 9 studies (Table 2).17-29 The conditions for which these patients were undergoing treatment with adalimumab included ulcerative colitis,17 psoriasis,18,19 Crohn disease,20,26 rheumatoid arthritis,21-23,26 oligoarthritis,24 and ankylosing spondylitis.25 Lichenoid drug eruptions occurred on the legs (5/12 [41.7%]), arms (3/12 [25%]), oral mucosa (2/12 [16.7%]), and forehead or scalp (2/12 [16.7%]). Onset of time to these lichenoid eruptions ranged from 2 weeks to 17 months, with a median of 4 months. Adalimumab was discontinued in 9 (75.0%) patients and was continued in 3 (25.0%). One patient who had an onset of their lichenoid eruption after 17 months of treatment with adalimumab continued to receive adalimumab therapy with the addition of topical corticosteroids, which led to resolution of their oral lesions and partial remission of their cutaneous lesions. In 1 (8.3%) patient with localized buccal lichenoid eruptions, discontinuation of adalimumab on its own was sufficient to completely clear the lesions. Seven patients (7/12 [58.3%]) received topical corticosteroids with minimal (2/12 [16.7%]) or moderate (4/12 [33.3%]) improvement, and 1 (8.3%) patient did not have reported outcomes data. Eosinophils were detected within the adalimumab-associated lichenoid eruptions in 3 (25.0%) patients.17,20,22
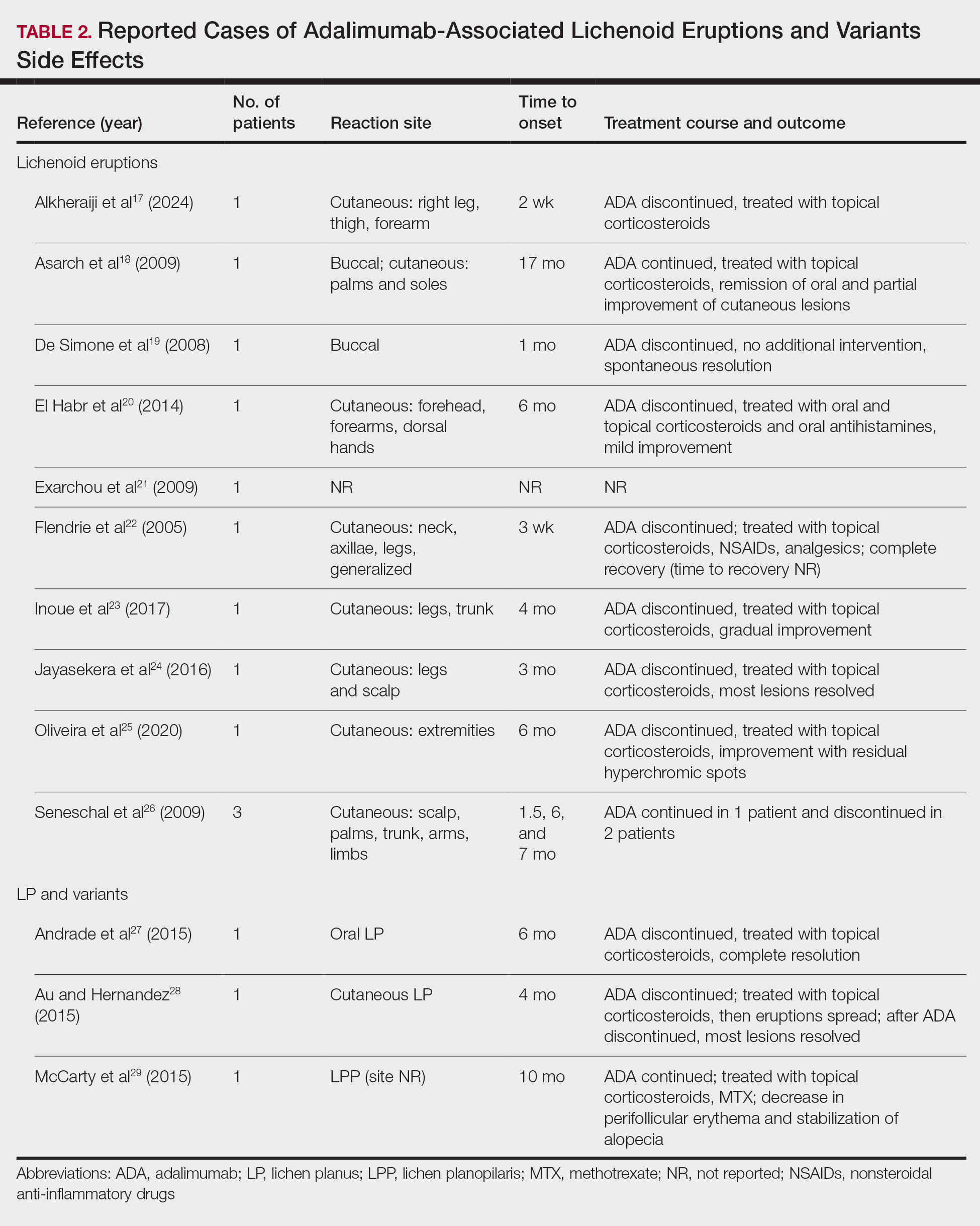
In addition to its association with lichenoid drug eruptions, adalimumab also was reported to induce LPP in a patient who was being treated for Behçet disease,29 oral LP in a patient being treated for Crohn disease,27 and cutaneous LP in a patient being treated for Crohn disease (Table 2).28 Time to onset ranged from 4 to 10 months, with a median of 6 months. Adalimumab was discontinued in 2 of 3 (66.7%) patients and was continued in the other patient (33.3%). After cessation of adalimumab therapy, administration of topical steroids led to complete resolution in the case of associated oral LP. In contrast, in adalimumab-induced cutaneous LP, initial topical corticosteroid treatment led to progression of lesions, which mostly resolved after adalimumab cessation. In 1 patient with LPP in whom adalimumab therapy could not be discontinued, topical corticosteroid and methotrexate therapy reduced the perifollicular erythema and stabilized the alopecia without full remission.
Comment
Conventional treatment modalities for LP often include topical corticosteroids as first-line therapy, with systemic corticosteroids, phototherapy, retinoids, or immunosuppressants (eg, cyclosporine or methotrexate) reserved for more severe or widespread disease. Historically, these approaches primarily have aimed to control symptoms rather than achieve long-term resolution; however, novel therapies including biologics and targeted immunomodulators show potential to induce sustained remission and improve quality of life for patients with refractory or mucosal LP.
In all reports where adalimumab was used to treat LP, patients initially received topical corticosteroids. While corticosteroids and other immunosuppressive agents are standard therapies, they often provide only temporary relief and may have an unfavorable side effect profile. Our review highlights the emerging role of adalimumab, a TNF-α inhibitor, in off-label management of LP subtypes, including cutaneous, mucosal, and vulvar LP and LPP. In several small case series and reports, patients treated with adalimumab experienced clinical improvement, including symptom resolution and quality-of-life enhancement, as well as complete remission, indicating a durable response.
The potential benefit of adalimumab in treating LP must be balanced with its paradoxical risk for inducing lichenoid eruptions as well as LP and its variants, as identified in our narrative review that included reports of patients receiving this biologic for other indications.17-29 Since adalimumab is a fully humanized antibody, the development of neutralizing antibodies may not account for drug-induced LP and lichenoid eruptions. Given that it blocks TNF-α, adalimumab may induce these lesions through a cytokine imbalance. This is supported by data demonstrating enhanced type I IFN-related proteins in plaques of patients with psoriasiform lesions treated with TNF-α inhibitors.26 These drug-induced eruptions often resolved or improved with topical corticosteroids after discontinuation, but their occurrence underscores the complexity of therapeutically targeting TNF-α in the management of LP. Our literature review suggests that adalimumab may offer therapeutic benefit in select cases of LP refractory to conventional therapy, especially when systemic control is required. Nonetheless, the risk for LP and lichenoid reactions necessitates cautious use and further investigation.
Conclusion
While the current evidence is limited to case reports and series, adalimumab shows promise as an effective and tolerable off-label treatment for LP, particularly in patients who are unresponsive to conventional immunosuppressive therapies. Remission or clinically significant improvement was achieved in several cases; however, the potential for adalimumab to induce LP and lichenoid eruptions underscores the need for careful patient selection and monitoring. Further prospective studies and larger cohorts are warranted to better define the safety and efficacy of adalimumab in treating LP lesions.
- Boch K, Langan EA, Kridin K, et al. Lichen planus. Front Med (Lausanne). 2021;8:737813.
- Gorouhi F, Davari P, Fazel N. Cutaneous and mucosal lichen planus: a comprehensive review of clinical subtypes, risk factors, diagnosis, and prognosis. ScientificWorldJournal. 2014;2014:742826.
- Pilli M, Penna A, Zerbini A, et al. Oral lichen planus pathogenesis: a role for the HCV-specific cellular immune response. Hepatology. 2002;36:1446-1452.
- Wang Y, Shang S, Sun Q, et al. Increased infiltration of CD11 c+/CD123+ dendritic cell subsets and upregulation of TLR/IFN-α signaling participate in pathogenesis of oral lichen planus. Oral Surg Oral Med Oral Pathol Oral Radiol. 2018;125:459-467.E2.
- González Moles M, Esteban F, Ruiz-Ávila I, et al. A role for the substance P/NK-1 receptor complex in cell proliferation and apoptosis in oral lichen planus. Oral Dis. 2009;15:162-169.
- Doche I, Wilcox GL, Ericson M, et al. Evidence for neurogenic inflammation in lichen planopilaris and frontal fibrosing alopecia pathogenic mechanism. Exp Dermatol. 2020;29:282-285.
- Cooper SM, Dean D, Allen J, et al. Erosive lichen planus of the vulva: weak circulating basement membrane zone antibodies are present. Clin Exp Dermatol. 2005;30:551-556.
- Howard A, Dean D, Cooper S, et al. Circulating basement membrane zone antibodies are found in lichen sclerosus of the vulva. Australas J Dermatol. 2004;45:12-15.
- Cooper SM, Ali I, Baldo M, et al. The association of lichen sclerosus and erosive lichen planus of the vulva with autoimmune disease: a case-control study. Arch Dermatol. 2008;144:1432-1435.
- Alam MS, LaBelle B. Treatment of lichen planopilaris with adalimumab in a patient with hidradenitis suppurativa and rheumatoid arthritis. JAAD Case Rep. 2020;6:219-221.
- Alhubayshi BS, Alnoshan AA, Alhumidi AA, et al. Bullous lichen planus treated with adalimumab: a case report. Case Rep Dermatol. 2025;17:42-47.
- Chao TJ. Adalimumab in the management of cutaneous and oral lichen planus. Cutis. 2009;84:325-328.
- Courtney A, Adamson SR, Veysey E. Adalimumab use in severe recalcitrant vulval lichen sclerosus and vulval lichen planus. J Low Genit Tract Dis. 2025;29:190-194.
- Holló P, Szakonyi J, Kiss D, et al. Successful treatment of lichen planus with adalimumab. Acta Derm Venereol. 2012;92:385-386.
- Khodeir J, Ohanian P, Ohanian M. Successful treatment of annular atrophic lichen planus with adalimumab. Clin Case Rep. 2025;13:E70036.
- Kreutzer K, Effendy I. Therapy-resistant folliculitis decalvans and lichen planopilaris successfully treated with adalimumab. J Dtsch Dermatol Ges. 2014;12:74-76.
- Alkheraiji A, Alotaibi H, Irfan Thalib H. Lichenoid drug eruption secondary to adalimumab: a case report. Cureus. 2024;16:E64013.
- Asarch A, Gottlieb AB, Lee J, et al. Lichen planus-like eruptions: an emerging side effect of tumor necrosis factor-alpha antagonists. J Am Acad Dermatol. 2009;61:104-111.
- De Simone C, Caldarola G, D’Agostino M, et al. Lichenoid reaction induced by adalimumab. J Eur Acad Dermatol Venereol. 2008;22:626-627.
- El Habr C, Meguerian Z, Sammour R. Adalimumab-induced lichenoid drug eruption. J Med Liban. 2014;62:238-240.
- Exarchou SA, Voulgari PV, Markatseli TE, et al. Immune-mediated skin lesions in patients treated with anti-tumour necrosis factor alpha inhibitors. Scand J Rheumatol. 2009;38:328-331.
- Flendrie M, Vissers WH, Creemers MC, et al. Dermatological conditions during TNF-α-blocking therapy in patients with rheumatoid arthritis: a prospective study. Arthritis Res Ther. 2005;7:R666-R676.
- Inoue A, Sawada Y, Yamaguchi T, et al. Lichenoid drug eruption caused by adalimumab: a case report and literature review. Eur J Dermatol. 2017;27:69-70.
- Jayasekera PSA, Walsh ML, Hurrell D, et al. Case report of lichen planopilaris occurring in a pediatric patient receiving a tumor necrosis factor α inhibitor and a review of the literature. Pediatr Dermatol. 2016;33:E143-E146.
- Oliveira SCD, Vasconcelos AHC, Magalhães EPB, et al. Clinical, histopathological and outcome analysis of five patients with lichenoid eruption following anti-tumor necrosis factor-alpha therapy for ankylosing spondylitis: report of one case and review of the literature. Cureus. 2020;12:E10598.
- Seneschal J, Milpied B, Vergier B, et al. Cytokine imbalance with increased production of interferon-alpha in psoriasiform eruptions associated with antitumour necrosis factor-alpha treatments. Br J Dermatol. 2009;161:1081-1088.
- Andrade P, Lopes S, Albuquerque A, et al. Oral lichen planus in IBD patients: a paradoxical adverse effect of anti-TNF-α therapy. Dig Dis Sci. 2015;60:2746-2749.
- Au S, Hernandez C. Paradoxical induction of psoriasis and lichen planus by tumor necrosis factor-α inhibitors. Skinmed. 2015;13:403-405.
- McCarty M, Basile A, Bair B, et al. Lichenoid reactions in association with tumor necrosis factor alpha inhibitors. J Clin Aesthet Dermatol. 2015;8:45-49.
Lichen planus (LP) is a chronic inflammatory condition affecting the skin (cutaneous LP), mucous membranes (oral, ocular, or vulvar LP), hair (lichen planopilaris [LPP]), and nails that predominantly occurs in middle-aged adults. Although the true etiology remains unknown, the pathogenesis of LP is thought to involve multiple factors. Several human leukocyte antigen (HLA) alleles have been associated with LP and its variants, including HLA-B27, HLA-B51, HLA-DR1 (cutaneous and oral LP), HLA-DRB1*11, and HLA-DQB1*03 (LPP). Additionally, HLA-Bw57 has been reported to be associated with oral LP in a cohort of British patients.1 In addition to HLA alleles, genetic polymorphisms in cytokines including IL-4, IL-6, IL-18, interferon (IFN) γ, and tumor necrosis factor (TNF) α and its receptor have been found to be associated with LP.2 Beyond genetics, chronic viral infection has been implicated in the development of LP. Systemic infection with the hepatitis C virus has been linked to the development of oral LP by promoting the recruitment of hepatitis C virus–specific CD8+ T cells from peripheral blood to the oral lesions, where they exhibit a terminally differentiated effector status.3 Another report found an association between human herpesvirus 7 (HHV-7) and cutaneous LP; in this study, HHV-7 RNA was detected in plasmacytoid dendritic cells but not T cells and diminished after treatment, providing evidence for dendritic cells being involved in the HHV-7–mediated pathogenesis of cutaneous LP.4 These findings were further corroborated by another study of oral LP patients that found enhanced infiltration of plasmacytoid and myeloid dendritic cells and upregulation in toll-like receptor and IFN-γ signaling.4
In addition to immune cell dysregulation, LP and its variants have been linked to neurogenic inflammation. In oral LP lesions, neurokinin 1 receptor and substance P were highly expressed and demonstrated a positive correlation with the expression of apoptotic marker caspase-3 and proliferation marker Ki-67.5 These results suggest that neuropeptides may be involved in cell proliferation and turnover in oral LP. Similarly, in patients with LPP, substance P was more abundant in affected areas, whereas another neuropeptide, calcitonin gene-related peptide, was more highly expressed in unaffected areas,6 further supporting the pathogenic role of neurogenic inflammation in LP.
A mucosal variant that often goes undiagnosed is vulvar LP. Although no distinct pathologic mechanism for vulvar LP has been established, prior reports found an association with autoantibodies.7,8 In patients with erosive vulvar LP, epidermal-binding basement membrane zone antibodies were detected in epidermal skin biopsies and in circulation with reactivity to bullous pemphigoid antigens 180 (9/11 [81.8%] patients) and 230 (2/11 [18.2%] patients).7 A similar study in patients with vulvar lichen sclerosus found similar proportions of circulating antibodies reactive to bullous pemphigoid antigens 180 (6/7 [85.7%] patients) and 230 (1/7 [14.3%] patients).8 Erosive vulvar LP has been shown to be associated with autoimmune disease (eg, alopecia areata, celiac disease and pernicious anemia),9 which suggests that the previously reported autoreactive antibodies7,8 are secondary to autoimmunity rather than primary drivers of vulvar LP pathogenesis.
Certain medications also have been reported to cause cutaneous lichenoid drug eruptions. Although they can clinically and histologically mimic classic LP, lichenoid drug eruptions are a distinct entity. Common inciting medications include thiazide diuretics, angiotensin-converting enzyme inhibitors, anti-inflammatory drugs, antimalarials, checkpoint inhibitors, antimicrobials, antihypertensives, antidiabetics, and psychiatric drugs. The exact pathologic mechanism of lichenoid drug eruptions currently is unclear but is thought to involve the binding of drug molecules to the cell-surface proteins of the epidermis, creating an antigenic hapten stimulus for CD8+T cells and triggering apoptosis of keratinocytes.1
The clinical severity of LP can range from mild localized disease to widespread and debilitating involvement. Multiple treatment modalities have been developed for management of LP, including topical and intralesional corticosteroids, phototherapy, Janus kinase inhibitors, phosphodiesterase-4 inhibitors, and anti–TNF-α inhibitors. Herein, we provide a narrative review and summary of the use of the TNF-α inhibitor adalimumab as a potential effective treatment for patients with LP.
Methods
We conducted a PubMed search of articles indexed for MEDLINE from 2005 to 2025 using the terms adalimumab AND lichen planus or adalimumab AND lichen. Articles that reported cases of oral LP, cutaneous LP, LPP, or lichenoid eruptions and adalimumab therapy were included in our review. Articles that used non-adalimumab TNF-α inhibitors were excluded. Using the search terms, 2 independent reviewers (M.G. and N.E.) conducted the literature review then screened the articles based on the inclusion and exclusion criteria. Our literature search yielded 40 articles, of which 20 met the criteria for inclusion in our narrative review.
Results
Our literature search yielded 11 patients with LP who were treated with adalimumab across studies (Table 1).10-16 Prior LP treatments included topical corticosteroids (11/11 [100%]), disease-modifying antirheumatic drugs (6/11 [54.5%]), retinoids (4/11 [36.4%]), and psoralen plus UVA (1/11 [36.4%]). Adalimumab was administered subcutaneously following 4 treatment regimens: (1)
Paradoxically, our review of the literature yielded 12 patients in whom adalimumab was associated with lichenoid-type eruptions across 9 studies (Table 2).17-29 The conditions for which these patients were undergoing treatment with adalimumab included ulcerative colitis,17 psoriasis,18,19 Crohn disease,20,26 rheumatoid arthritis,21-23,26 oligoarthritis,24 and ankylosing spondylitis.25 Lichenoid drug eruptions occurred on the legs (5/12 [41.7%]), arms (3/12 [25%]), oral mucosa (2/12 [16.7%]), and forehead or scalp (2/12 [16.7%]). Onset of time to these lichenoid eruptions ranged from 2 weeks to 17 months, with a median of 4 months. Adalimumab was discontinued in 9 (75.0%) patients and was continued in 3 (25.0%). One patient who had an onset of their lichenoid eruption after 17 months of treatment with adalimumab continued to receive adalimumab therapy with the addition of topical corticosteroids, which led to resolution of their oral lesions and partial remission of their cutaneous lesions. In 1 (8.3%) patient with localized buccal lichenoid eruptions, discontinuation of adalimumab on its own was sufficient to completely clear the lesions. Seven patients (7/12 [58.3%]) received topical corticosteroids with minimal (2/12 [16.7%]) or moderate (4/12 [33.3%]) improvement, and 1 (8.3%) patient did not have reported outcomes data. Eosinophils were detected within the adalimumab-associated lichenoid eruptions in 3 (25.0%) patients.17,20,22
In addition to its association with lichenoid drug eruptions, adalimumab also was reported to induce LPP in a patient who was being treated for Behçet disease,29 oral LP in a patient being treated for Crohn disease,27 and cutaneous LP in a patient being treated for Crohn disease (Table 2).28 Time to onset ranged from 4 to 10 months, with a median of 6 months. Adalimumab was discontinued in 2 of 3 (66.7%) patients and was continued in the other patient (33.3%). After cessation of adalimumab therapy, administration of topical steroids led to complete resolution in the case of associated oral LP. In contrast, in adalimumab-induced cutaneous LP, initial topical corticosteroid treatment led to progression of lesions, which mostly resolved after adalimumab cessation. In 1 patient with LPP in whom adalimumab therapy could not be discontinued, topical corticosteroid and methotrexate therapy reduced the perifollicular erythema and stabilized the alopecia without full remission.
Comment
Conventional treatment modalities for LP often include topical corticosteroids as first-line therapy, with systemic corticosteroids, phototherapy, retinoids, or immunosuppressants (eg, cyclosporine or methotrexate) reserved for more severe or widespread disease. Historically, these approaches primarily have aimed to control symptoms rather than achieve long-term resolution; however, novel therapies including biologics and targeted immunomodulators show potential to induce sustained remission and improve quality of life for patients with refractory or mucosal LP.
In all reports where adalimumab was used to treat LP, patients initially received topical corticosteroids. While corticosteroids and other immunosuppressive agents are standard therapies, they often provide only temporary relief and may have an unfavorable side effect profile. Our review highlights the emerging role of adalimumab, a TNF-α inhibitor, in off-label management of LP subtypes, including cutaneous, mucosal, and vulvar LP and LPP. In several small case series and reports, patients treated with adalimumab experienced clinical improvement, including symptom resolution and quality-of-life enhancement, as well as complete remission, indicating a durable response.
The potential benefit of adalimumab in treating LP must be balanced with its paradoxical risk for inducing lichenoid eruptions as well as LP and its variants, as identified in our narrative review that included reports of patients receiving this biologic for other indications.17-29 Since adalimumab is a fully humanized antibody, the development of neutralizing antibodies may not account for drug-induced LP and lichenoid eruptions. Given that it blocks TNF-α, adalimumab may induce these lesions through a cytokine imbalance. This is supported by data demonstrating enhanced type I IFN-related proteins in plaques of patients with psoriasiform lesions treated with TNF-α inhibitors.26 These drug-induced eruptions often resolved or improved with topical corticosteroids after discontinuation, but their occurrence underscores the complexity of therapeutically targeting TNF-α in the management of LP. Our literature review suggests that adalimumab may offer therapeutic benefit in select cases of LP refractory to conventional therapy, especially when systemic control is required. Nonetheless, the risk for LP and lichenoid reactions necessitates cautious use and further investigation.
Conclusion
While the current evidence is limited to case reports and series, adalimumab shows promise as an effective and tolerable off-label treatment for LP, particularly in patients who are unresponsive to conventional immunosuppressive therapies. Remission or clinically significant improvement was achieved in several cases; however, the potential for adalimumab to induce LP and lichenoid eruptions underscores the need for careful patient selection and monitoring. Further prospective studies and larger cohorts are warranted to better define the safety and efficacy of adalimumab in treating LP lesions.
Lichen planus (LP) is a chronic inflammatory condition affecting the skin (cutaneous LP), mucous membranes (oral, ocular, or vulvar LP), hair (lichen planopilaris [LPP]), and nails that predominantly occurs in middle-aged adults. Although the true etiology remains unknown, the pathogenesis of LP is thought to involve multiple factors. Several human leukocyte antigen (HLA) alleles have been associated with LP and its variants, including HLA-B27, HLA-B51, HLA-DR1 (cutaneous and oral LP), HLA-DRB1*11, and HLA-DQB1*03 (LPP). Additionally, HLA-Bw57 has been reported to be associated with oral LP in a cohort of British patients.1 In addition to HLA alleles, genetic polymorphisms in cytokines including IL-4, IL-6, IL-18, interferon (IFN) γ, and tumor necrosis factor (TNF) α and its receptor have been found to be associated with LP.2 Beyond genetics, chronic viral infection has been implicated in the development of LP. Systemic infection with the hepatitis C virus has been linked to the development of oral LP by promoting the recruitment of hepatitis C virus–specific CD8+ T cells from peripheral blood to the oral lesions, where they exhibit a terminally differentiated effector status.3 Another report found an association between human herpesvirus 7 (HHV-7) and cutaneous LP; in this study, HHV-7 RNA was detected in plasmacytoid dendritic cells but not T cells and diminished after treatment, providing evidence for dendritic cells being involved in the HHV-7–mediated pathogenesis of cutaneous LP.4 These findings were further corroborated by another study of oral LP patients that found enhanced infiltration of plasmacytoid and myeloid dendritic cells and upregulation in toll-like receptor and IFN-γ signaling.4
In addition to immune cell dysregulation, LP and its variants have been linked to neurogenic inflammation. In oral LP lesions, neurokinin 1 receptor and substance P were highly expressed and demonstrated a positive correlation with the expression of apoptotic marker caspase-3 and proliferation marker Ki-67.5 These results suggest that neuropeptides may be involved in cell proliferation and turnover in oral LP. Similarly, in patients with LPP, substance P was more abundant in affected areas, whereas another neuropeptide, calcitonin gene-related peptide, was more highly expressed in unaffected areas,6 further supporting the pathogenic role of neurogenic inflammation in LP.
A mucosal variant that often goes undiagnosed is vulvar LP. Although no distinct pathologic mechanism for vulvar LP has been established, prior reports found an association with autoantibodies.7,8 In patients with erosive vulvar LP, epidermal-binding basement membrane zone antibodies were detected in epidermal skin biopsies and in circulation with reactivity to bullous pemphigoid antigens 180 (9/11 [81.8%] patients) and 230 (2/11 [18.2%] patients).7 A similar study in patients with vulvar lichen sclerosus found similar proportions of circulating antibodies reactive to bullous pemphigoid antigens 180 (6/7 [85.7%] patients) and 230 (1/7 [14.3%] patients).8 Erosive vulvar LP has been shown to be associated with autoimmune disease (eg, alopecia areata, celiac disease and pernicious anemia),9 which suggests that the previously reported autoreactive antibodies7,8 are secondary to autoimmunity rather than primary drivers of vulvar LP pathogenesis.
Certain medications also have been reported to cause cutaneous lichenoid drug eruptions. Although they can clinically and histologically mimic classic LP, lichenoid drug eruptions are a distinct entity. Common inciting medications include thiazide diuretics, angiotensin-converting enzyme inhibitors, anti-inflammatory drugs, antimalarials, checkpoint inhibitors, antimicrobials, antihypertensives, antidiabetics, and psychiatric drugs. The exact pathologic mechanism of lichenoid drug eruptions currently is unclear but is thought to involve the binding of drug molecules to the cell-surface proteins of the epidermis, creating an antigenic hapten stimulus for CD8+T cells and triggering apoptosis of keratinocytes.1
The clinical severity of LP can range from mild localized disease to widespread and debilitating involvement. Multiple treatment modalities have been developed for management of LP, including topical and intralesional corticosteroids, phototherapy, Janus kinase inhibitors, phosphodiesterase-4 inhibitors, and anti–TNF-α inhibitors. Herein, we provide a narrative review and summary of the use of the TNF-α inhibitor adalimumab as a potential effective treatment for patients with LP.
Methods
We conducted a PubMed search of articles indexed for MEDLINE from 2005 to 2025 using the terms adalimumab AND lichen planus or adalimumab AND lichen. Articles that reported cases of oral LP, cutaneous LP, LPP, or lichenoid eruptions and adalimumab therapy were included in our review. Articles that used non-adalimumab TNF-α inhibitors were excluded. Using the search terms, 2 independent reviewers (M.G. and N.E.) conducted the literature review then screened the articles based on the inclusion and exclusion criteria. Our literature search yielded 40 articles, of which 20 met the criteria for inclusion in our narrative review.
Results
Our literature search yielded 11 patients with LP who were treated with adalimumab across studies (Table 1).10-16 Prior LP treatments included topical corticosteroids (11/11 [100%]), disease-modifying antirheumatic drugs (6/11 [54.5%]), retinoids (4/11 [36.4%]), and psoralen plus UVA (1/11 [36.4%]). Adalimumab was administered subcutaneously following 4 treatment regimens: (1)
Paradoxically, our review of the literature yielded 12 patients in whom adalimumab was associated with lichenoid-type eruptions across 9 studies (Table 2).17-29 The conditions for which these patients were undergoing treatment with adalimumab included ulcerative colitis,17 psoriasis,18,19 Crohn disease,20,26 rheumatoid arthritis,21-23,26 oligoarthritis,24 and ankylosing spondylitis.25 Lichenoid drug eruptions occurred on the legs (5/12 [41.7%]), arms (3/12 [25%]), oral mucosa (2/12 [16.7%]), and forehead or scalp (2/12 [16.7%]). Onset of time to these lichenoid eruptions ranged from 2 weeks to 17 months, with a median of 4 months. Adalimumab was discontinued in 9 (75.0%) patients and was continued in 3 (25.0%). One patient who had an onset of their lichenoid eruption after 17 months of treatment with adalimumab continued to receive adalimumab therapy with the addition of topical corticosteroids, which led to resolution of their oral lesions and partial remission of their cutaneous lesions. In 1 (8.3%) patient with localized buccal lichenoid eruptions, discontinuation of adalimumab on its own was sufficient to completely clear the lesions. Seven patients (7/12 [58.3%]) received topical corticosteroids with minimal (2/12 [16.7%]) or moderate (4/12 [33.3%]) improvement, and 1 (8.3%) patient did not have reported outcomes data. Eosinophils were detected within the adalimumab-associated lichenoid eruptions in 3 (25.0%) patients.17,20,22
In addition to its association with lichenoid drug eruptions, adalimumab also was reported to induce LPP in a patient who was being treated for Behçet disease,29 oral LP in a patient being treated for Crohn disease,27 and cutaneous LP in a patient being treated for Crohn disease (Table 2).28 Time to onset ranged from 4 to 10 months, with a median of 6 months. Adalimumab was discontinued in 2 of 3 (66.7%) patients and was continued in the other patient (33.3%). After cessation of adalimumab therapy, administration of topical steroids led to complete resolution in the case of associated oral LP. In contrast, in adalimumab-induced cutaneous LP, initial topical corticosteroid treatment led to progression of lesions, which mostly resolved after adalimumab cessation. In 1 patient with LPP in whom adalimumab therapy could not be discontinued, topical corticosteroid and methotrexate therapy reduced the perifollicular erythema and stabilized the alopecia without full remission.
Comment
Conventional treatment modalities for LP often include topical corticosteroids as first-line therapy, with systemic corticosteroids, phototherapy, retinoids, or immunosuppressants (eg, cyclosporine or methotrexate) reserved for more severe or widespread disease. Historically, these approaches primarily have aimed to control symptoms rather than achieve long-term resolution; however, novel therapies including biologics and targeted immunomodulators show potential to induce sustained remission and improve quality of life for patients with refractory or mucosal LP.
In all reports where adalimumab was used to treat LP, patients initially received topical corticosteroids. While corticosteroids and other immunosuppressive agents are standard therapies, they often provide only temporary relief and may have an unfavorable side effect profile. Our review highlights the emerging role of adalimumab, a TNF-α inhibitor, in off-label management of LP subtypes, including cutaneous, mucosal, and vulvar LP and LPP. In several small case series and reports, patients treated with adalimumab experienced clinical improvement, including symptom resolution and quality-of-life enhancement, as well as complete remission, indicating a durable response.
The potential benefit of adalimumab in treating LP must be balanced with its paradoxical risk for inducing lichenoid eruptions as well as LP and its variants, as identified in our narrative review that included reports of patients receiving this biologic for other indications.17-29 Since adalimumab is a fully humanized antibody, the development of neutralizing antibodies may not account for drug-induced LP and lichenoid eruptions. Given that it blocks TNF-α, adalimumab may induce these lesions through a cytokine imbalance. This is supported by data demonstrating enhanced type I IFN-related proteins in plaques of patients with psoriasiform lesions treated with TNF-α inhibitors.26 These drug-induced eruptions often resolved or improved with topical corticosteroids after discontinuation, but their occurrence underscores the complexity of therapeutically targeting TNF-α in the management of LP. Our literature review suggests that adalimumab may offer therapeutic benefit in select cases of LP refractory to conventional therapy, especially when systemic control is required. Nonetheless, the risk for LP and lichenoid reactions necessitates cautious use and further investigation.
Conclusion
While the current evidence is limited to case reports and series, adalimumab shows promise as an effective and tolerable off-label treatment for LP, particularly in patients who are unresponsive to conventional immunosuppressive therapies. Remission or clinically significant improvement was achieved in several cases; however, the potential for adalimumab to induce LP and lichenoid eruptions underscores the need for careful patient selection and monitoring. Further prospective studies and larger cohorts are warranted to better define the safety and efficacy of adalimumab in treating LP lesions.
- Boch K, Langan EA, Kridin K, et al. Lichen planus. Front Med (Lausanne). 2021;8:737813.
- Gorouhi F, Davari P, Fazel N. Cutaneous and mucosal lichen planus: a comprehensive review of clinical subtypes, risk factors, diagnosis, and prognosis. ScientificWorldJournal. 2014;2014:742826.
- Pilli M, Penna A, Zerbini A, et al. Oral lichen planus pathogenesis: a role for the HCV-specific cellular immune response. Hepatology. 2002;36:1446-1452.
- Wang Y, Shang S, Sun Q, et al. Increased infiltration of CD11 c+/CD123+ dendritic cell subsets and upregulation of TLR/IFN-α signaling participate in pathogenesis of oral lichen planus. Oral Surg Oral Med Oral Pathol Oral Radiol. 2018;125:459-467.E2.
- González Moles M, Esteban F, Ruiz-Ávila I, et al. A role for the substance P/NK-1 receptor complex in cell proliferation and apoptosis in oral lichen planus. Oral Dis. 2009;15:162-169.
- Doche I, Wilcox GL, Ericson M, et al. Evidence for neurogenic inflammation in lichen planopilaris and frontal fibrosing alopecia pathogenic mechanism. Exp Dermatol. 2020;29:282-285.
- Cooper SM, Dean D, Allen J, et al. Erosive lichen planus of the vulva: weak circulating basement membrane zone antibodies are present. Clin Exp Dermatol. 2005;30:551-556.
- Howard A, Dean D, Cooper S, et al. Circulating basement membrane zone antibodies are found in lichen sclerosus of the vulva. Australas J Dermatol. 2004;45:12-15.
- Cooper SM, Ali I, Baldo M, et al. The association of lichen sclerosus and erosive lichen planus of the vulva with autoimmune disease: a case-control study. Arch Dermatol. 2008;144:1432-1435.
- Alam MS, LaBelle B. Treatment of lichen planopilaris with adalimumab in a patient with hidradenitis suppurativa and rheumatoid arthritis. JAAD Case Rep. 2020;6:219-221.
- Alhubayshi BS, Alnoshan AA, Alhumidi AA, et al. Bullous lichen planus treated with adalimumab: a case report. Case Rep Dermatol. 2025;17:42-47.
- Chao TJ. Adalimumab in the management of cutaneous and oral lichen planus. Cutis. 2009;84:325-328.
- Courtney A, Adamson SR, Veysey E. Adalimumab use in severe recalcitrant vulval lichen sclerosus and vulval lichen planus. J Low Genit Tract Dis. 2025;29:190-194.
- Holló P, Szakonyi J, Kiss D, et al. Successful treatment of lichen planus with adalimumab. Acta Derm Venereol. 2012;92:385-386.
- Khodeir J, Ohanian P, Ohanian M. Successful treatment of annular atrophic lichen planus with adalimumab. Clin Case Rep. 2025;13:E70036.
- Kreutzer K, Effendy I. Therapy-resistant folliculitis decalvans and lichen planopilaris successfully treated with adalimumab. J Dtsch Dermatol Ges. 2014;12:74-76.
- Alkheraiji A, Alotaibi H, Irfan Thalib H. Lichenoid drug eruption secondary to adalimumab: a case report. Cureus. 2024;16:E64013.
- Asarch A, Gottlieb AB, Lee J, et al. Lichen planus-like eruptions: an emerging side effect of tumor necrosis factor-alpha antagonists. J Am Acad Dermatol. 2009;61:104-111.
- De Simone C, Caldarola G, D’Agostino M, et al. Lichenoid reaction induced by adalimumab. J Eur Acad Dermatol Venereol. 2008;22:626-627.
- El Habr C, Meguerian Z, Sammour R. Adalimumab-induced lichenoid drug eruption. J Med Liban. 2014;62:238-240.
- Exarchou SA, Voulgari PV, Markatseli TE, et al. Immune-mediated skin lesions in patients treated with anti-tumour necrosis factor alpha inhibitors. Scand J Rheumatol. 2009;38:328-331.
- Flendrie M, Vissers WH, Creemers MC, et al. Dermatological conditions during TNF-α-blocking therapy in patients with rheumatoid arthritis: a prospective study. Arthritis Res Ther. 2005;7:R666-R676.
- Inoue A, Sawada Y, Yamaguchi T, et al. Lichenoid drug eruption caused by adalimumab: a case report and literature review. Eur J Dermatol. 2017;27:69-70.
- Jayasekera PSA, Walsh ML, Hurrell D, et al. Case report of lichen planopilaris occurring in a pediatric patient receiving a tumor necrosis factor α inhibitor and a review of the literature. Pediatr Dermatol. 2016;33:E143-E146.
- Oliveira SCD, Vasconcelos AHC, Magalhães EPB, et al. Clinical, histopathological and outcome analysis of five patients with lichenoid eruption following anti-tumor necrosis factor-alpha therapy for ankylosing spondylitis: report of one case and review of the literature. Cureus. 2020;12:E10598.
- Seneschal J, Milpied B, Vergier B, et al. Cytokine imbalance with increased production of interferon-alpha in psoriasiform eruptions associated with antitumour necrosis factor-alpha treatments. Br J Dermatol. 2009;161:1081-1088.
- Andrade P, Lopes S, Albuquerque A, et al. Oral lichen planus in IBD patients: a paradoxical adverse effect of anti-TNF-α therapy. Dig Dis Sci. 2015;60:2746-2749.
- Au S, Hernandez C. Paradoxical induction of psoriasis and lichen planus by tumor necrosis factor-α inhibitors. Skinmed. 2015;13:403-405.
- McCarty M, Basile A, Bair B, et al. Lichenoid reactions in association with tumor necrosis factor alpha inhibitors. J Clin Aesthet Dermatol. 2015;8:45-49.
- Boch K, Langan EA, Kridin K, et al. Lichen planus. Front Med (Lausanne). 2021;8:737813.
- Gorouhi F, Davari P, Fazel N. Cutaneous and mucosal lichen planus: a comprehensive review of clinical subtypes, risk factors, diagnosis, and prognosis. ScientificWorldJournal. 2014;2014:742826.
- Pilli M, Penna A, Zerbini A, et al. Oral lichen planus pathogenesis: a role for the HCV-specific cellular immune response. Hepatology. 2002;36:1446-1452.
- Wang Y, Shang S, Sun Q, et al. Increased infiltration of CD11 c+/CD123+ dendritic cell subsets and upregulation of TLR/IFN-α signaling participate in pathogenesis of oral lichen planus. Oral Surg Oral Med Oral Pathol Oral Radiol. 2018;125:459-467.E2.
- González Moles M, Esteban F, Ruiz-Ávila I, et al. A role for the substance P/NK-1 receptor complex in cell proliferation and apoptosis in oral lichen planus. Oral Dis. 2009;15:162-169.
- Doche I, Wilcox GL, Ericson M, et al. Evidence for neurogenic inflammation in lichen planopilaris and frontal fibrosing alopecia pathogenic mechanism. Exp Dermatol. 2020;29:282-285.
- Cooper SM, Dean D, Allen J, et al. Erosive lichen planus of the vulva: weak circulating basement membrane zone antibodies are present. Clin Exp Dermatol. 2005;30:551-556.
- Howard A, Dean D, Cooper S, et al. Circulating basement membrane zone antibodies are found in lichen sclerosus of the vulva. Australas J Dermatol. 2004;45:12-15.
- Cooper SM, Ali I, Baldo M, et al. The association of lichen sclerosus and erosive lichen planus of the vulva with autoimmune disease: a case-control study. Arch Dermatol. 2008;144:1432-1435.
- Alam MS, LaBelle B. Treatment of lichen planopilaris with adalimumab in a patient with hidradenitis suppurativa and rheumatoid arthritis. JAAD Case Rep. 2020;6:219-221.
- Alhubayshi BS, Alnoshan AA, Alhumidi AA, et al. Bullous lichen planus treated with adalimumab: a case report. Case Rep Dermatol. 2025;17:42-47.
- Chao TJ. Adalimumab in the management of cutaneous and oral lichen planus. Cutis. 2009;84:325-328.
- Courtney A, Adamson SR, Veysey E. Adalimumab use in severe recalcitrant vulval lichen sclerosus and vulval lichen planus. J Low Genit Tract Dis. 2025;29:190-194.
- Holló P, Szakonyi J, Kiss D, et al. Successful treatment of lichen planus with adalimumab. Acta Derm Venereol. 2012;92:385-386.
- Khodeir J, Ohanian P, Ohanian M. Successful treatment of annular atrophic lichen planus with adalimumab. Clin Case Rep. 2025;13:E70036.
- Kreutzer K, Effendy I. Therapy-resistant folliculitis decalvans and lichen planopilaris successfully treated with adalimumab. J Dtsch Dermatol Ges. 2014;12:74-76.
- Alkheraiji A, Alotaibi H, Irfan Thalib H. Lichenoid drug eruption secondary to adalimumab: a case report. Cureus. 2024;16:E64013.
- Asarch A, Gottlieb AB, Lee J, et al. Lichen planus-like eruptions: an emerging side effect of tumor necrosis factor-alpha antagonists. J Am Acad Dermatol. 2009;61:104-111.
- De Simone C, Caldarola G, D’Agostino M, et al. Lichenoid reaction induced by adalimumab. J Eur Acad Dermatol Venereol. 2008;22:626-627.
- El Habr C, Meguerian Z, Sammour R. Adalimumab-induced lichenoid drug eruption. J Med Liban. 2014;62:238-240.
- Exarchou SA, Voulgari PV, Markatseli TE, et al. Immune-mediated skin lesions in patients treated with anti-tumour necrosis factor alpha inhibitors. Scand J Rheumatol. 2009;38:328-331.
- Flendrie M, Vissers WH, Creemers MC, et al. Dermatological conditions during TNF-α-blocking therapy in patients with rheumatoid arthritis: a prospective study. Arthritis Res Ther. 2005;7:R666-R676.
- Inoue A, Sawada Y, Yamaguchi T, et al. Lichenoid drug eruption caused by adalimumab: a case report and literature review. Eur J Dermatol. 2017;27:69-70.
- Jayasekera PSA, Walsh ML, Hurrell D, et al. Case report of lichen planopilaris occurring in a pediatric patient receiving a tumor necrosis factor α inhibitor and a review of the literature. Pediatr Dermatol. 2016;33:E143-E146.
- Oliveira SCD, Vasconcelos AHC, Magalhães EPB, et al. Clinical, histopathological and outcome analysis of five patients with lichenoid eruption following anti-tumor necrosis factor-alpha therapy for ankylosing spondylitis: report of one case and review of the literature. Cureus. 2020;12:E10598.
- Seneschal J, Milpied B, Vergier B, et al. Cytokine imbalance with increased production of interferon-alpha in psoriasiform eruptions associated with antitumour necrosis factor-alpha treatments. Br J Dermatol. 2009;161:1081-1088.
- Andrade P, Lopes S, Albuquerque A, et al. Oral lichen planus in IBD patients: a paradoxical adverse effect of anti-TNF-α therapy. Dig Dis Sci. 2015;60:2746-2749.
- Au S, Hernandez C. Paradoxical induction of psoriasis and lichen planus by tumor necrosis factor-α inhibitors. Skinmed. 2015;13:403-405.
- McCarty M, Basile A, Bair B, et al. Lichenoid reactions in association with tumor necrosis factor alpha inhibitors. J Clin Aesthet Dermatol. 2015;8:45-49.
Adalimumab in Lichen Planus: A Narrative Review of Treatment and Paradoxical Reactions
Adalimumab in Lichen Planus: A Narrative Review of Treatment and Paradoxical Reactions
Practice Points
- Adalimumab can be beneficial when used off label for treatment of lichen planus in patients who do not respond to conventional therapies, including corticosteroids and immunosuppressants.
- Clinicians should be aware that adalimumab could potentially lead to paradoxical lichenoid eruptions and should monitor patients closely during treatment.
Metastatic Primary Extramammary Paget Disease: A Case Series
Metastatic Primary Extramammary Paget Disease: A Case Series
Extramammary Paget disease (EMPD) is a rare cutaneous malignancy typically seen in apocrine-rich areas, including the axillae and anogenital region. It presents as a slow-growing, erythematous patch or plaque that commonly is misdiagnosed as an infectious or inflammatory condition.1,2 Primary EMPD occurs as a intraepithelial neoplasm, whereas secondary EMPD occurs due to epidermotropic metastases or direct extension of an underlying adenocarcinoma into the skin.1 Most commonly, primary EMPD occurs in situ; however, when present, dermal invasion and metastases from the skin are associated with poorer outcomes.3 Given the rarity of metastatic disease, existing literature is limited to case reports and case series.
We present 2 patients with metastatic primary EMPD who had evidence of invasion on initial biopsy and died secondary to metastatic EMPD. We conducted a comprehensive review of the literature for invasive and metastatic EMPD to highlight key clinicopathologic features, treatment considerations, and the potential for rapid disease progression in cases of invasive EMPD.
Case Series
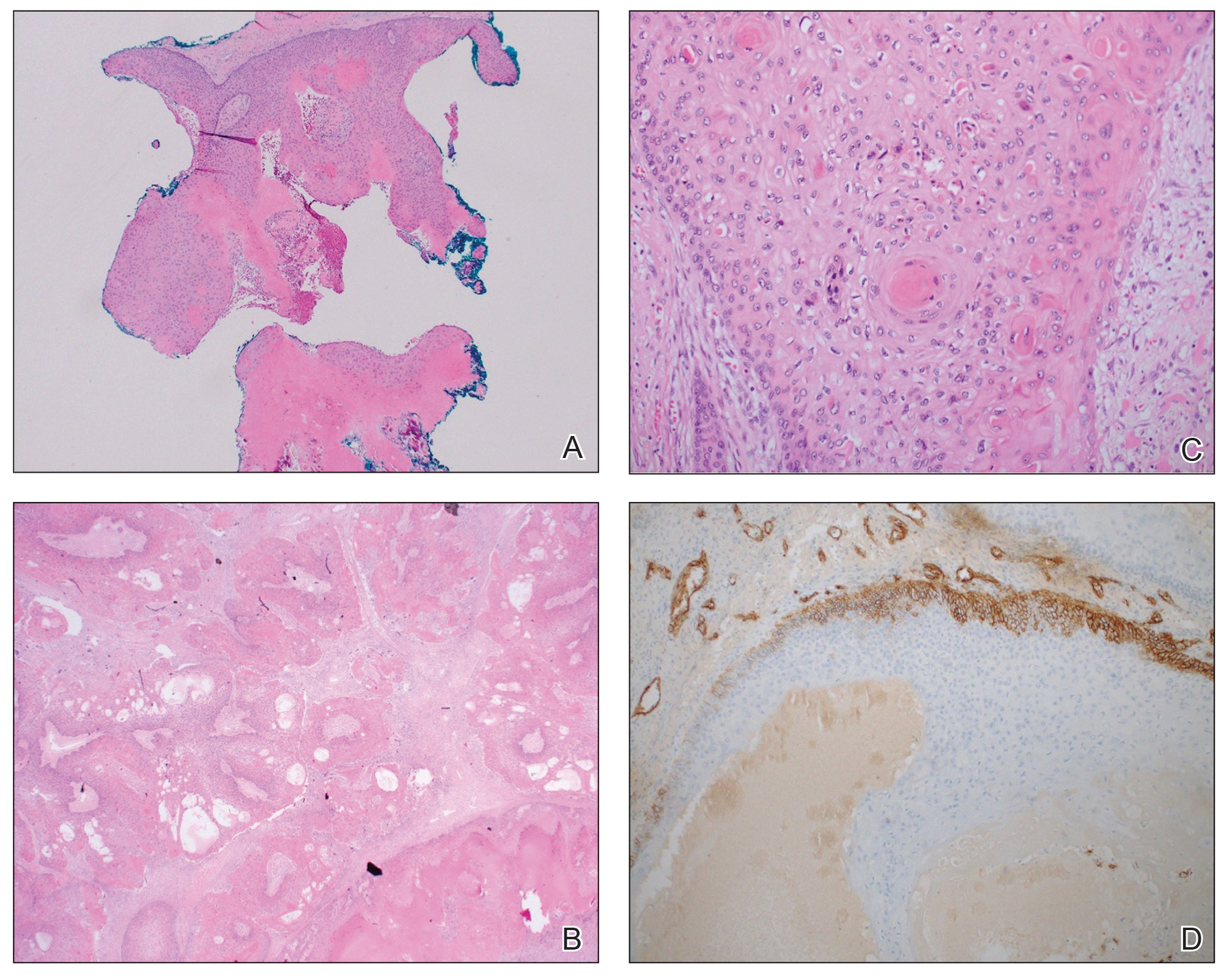
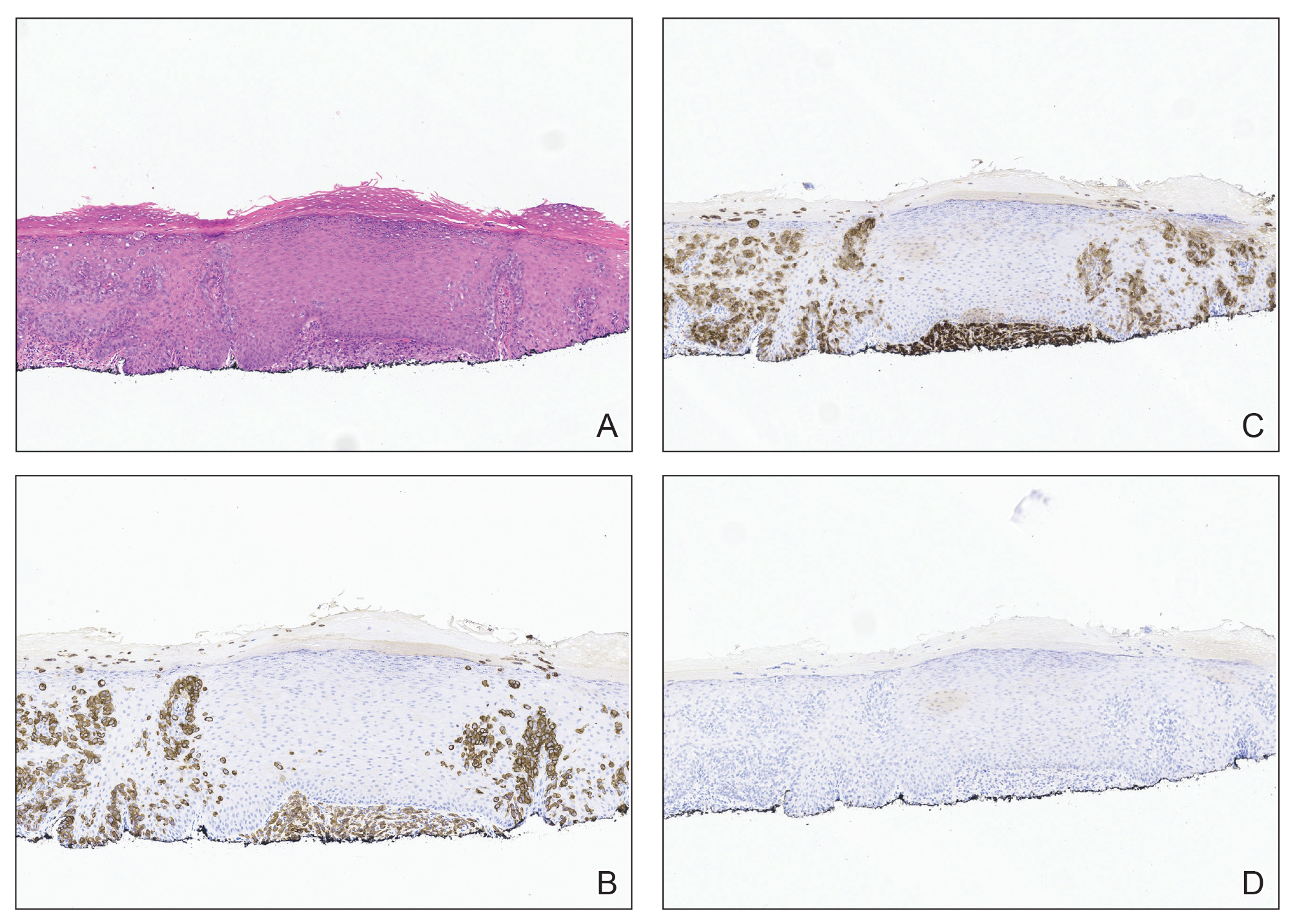
Patient 1—A 68-year-old White man with a history of breast cancer (in remission) presented to our clinic for further management of biopsy-proven scrotal EMPD. Prior to biopsy, he described a 6-month history of worsening scrotal rash treated with topical antifungals, oral antibiotics, and topical steroids due to presumed diagnosis of intertrigo, cellulitis, and dermatitis, respectively. Clinical examination showed indurated, erythematous, ulcerated plaques involving the bilateral groin, genitalia, and perineum (Figure 1). Skin biopsy confirmed a diagnosis of EMPD with both dermal and lymphovascular invasion. An immunohistochemical profile was positive for CK7 and carcinoembryonic antigen (CEA) and negative for CK20 (Figure 2).
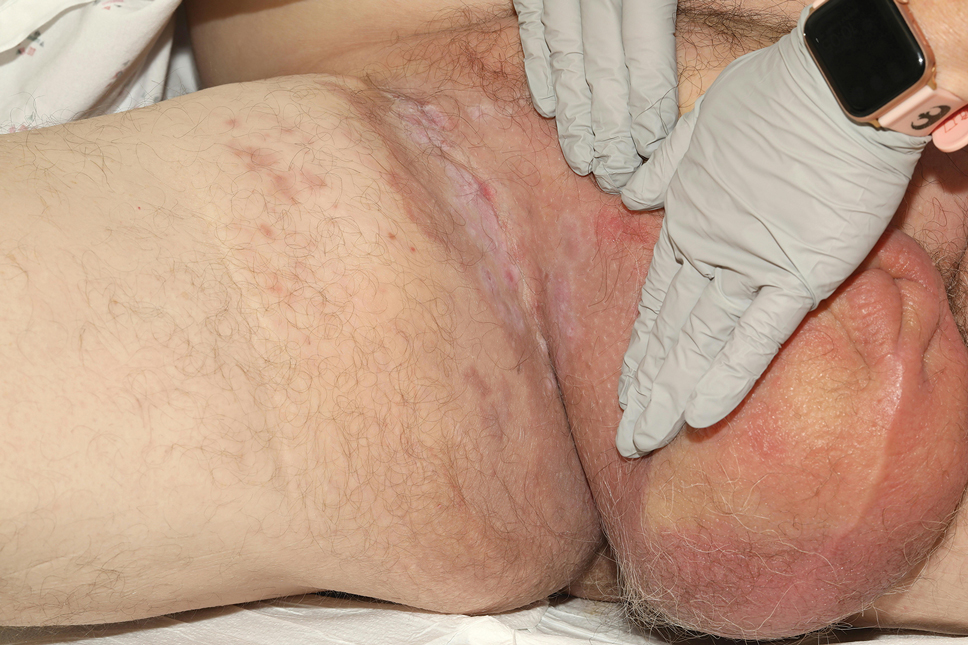
At presentation, the patient had palpable lymphadenopathy and scrotal edema concerning for inguinal and iliac lymph node metastases. Workup for an underlying adenocarcinoma included computed tomography (CT) of the chest, abdomen, and pelvis; urologic consultation with cystoscopy; and a screening colonoscopy. The CT scan revealed multiple enlarged inguinal and external iliac lymph nodes. Fine-needle aspiration revealed CK7- and CEA-positive neoplastic cells consistent with metastatic EMPD. The patient was treated with 6 cycles of carboplatin-paclitaxel, palliative radiation therapy, and pembrolizumab with minimal response to treatment and development of osteolytic vertebral lesions concerning for disease progression. He died 1 year after the initial diagnosis secondary to the disease.
Patient 2—A 79-year-old White man presented for further management of an outside diagnosis of superficially invasive primary EMPD of the bilateral inguinal folds and scrotum that had been present for 5 months prior to biopsy and diagnosis. Clinical examination at initial presentation revealed erythematous patches of the bilateral inguinal folds and scrotum, as well as an erythematous scaling plaque in the right axilla. There was no palpable clinical lymphadenopathy. Biopsy of the axilla and groin were both consistent with invasive EMPD with positive staining for CK7 and negative staining for CK20 and CDX2. Workup for underlying adenocarcinoma with whole-body positron emission tomography/CT, mammography, esophagogastroduodenoscopy, serum CEA, colonoscopy, and cystoscopy were all negative for a metastatic adenocarcinoma. There was no imaging or clinical evidence of lymphadenopathy. Complete circumferential peripheral and deep-margin assessment was performed in a staged manner on both sites, and negative margins were obtained.
Surveillance imaging 6 months after surgery revealed suspicious hepatic lesions. Fine-needle aspiration of the hepatic lesions demonstrated positive staining for CK7 and negative staining for CK20, CDX2, prostate-specific antigen, and thyroid transcription factor 1, consistent with metastatic EMPD. Oncology recommended carboplatin and docetaxel or docetaxel monotherapy chemotherapy. The patient was further managed by an outside oncologist due to ease of travel but died secondary to the disease 15 months following the initial diagnosis.
Comment
Extramammary Paget disease is an uncommon cutaneous malignancy that manifests as pruritic erythematous plaques within apocrine-rich areas such as the genitalia, axillae, or anal region. It most commonly occurs in patients older than 65 years, with White women and Asian men being affected at disproportionately higher rates.1,4 Delay in diagnosis is common, as EMPD can mimic other benign inflammatory or infectious conditions, including contact dermatitis, seborrheic dermatitis, tinea, candidiasis, and eczema.1
Metastatic and multifocal cases of primary EMPD are especially rare. According to a search of PubMed articles indexed for MEDLINE published through December 2023 using the terms extramammary Paget disease, EMPD, neoplasm metastasis, invasive extramammary, and neoplasm invasiveness, we identified 5040 cases of invasive EMPD and 477 cases of metastatic EMPD.5-37 Of the reports that disclosed patient demographic information, 3627 patients were female 1410 were male, and the mean age was 67 years. Sites of metastases included regional lymph nodes, liver, lungs, cervix, bladder, bone, brain, skin, kidney, and adrenal glands
Workup for EMPD—The initial steps for workup of EMPD include a thorough physical examination and lymph node assessment. A skin biopsy also should be performed for patients presenting with refractory, pruritic, and eczematous rashes in apocrine-rich areas to evaluate for EMPD.1 Characterization of large and complex tumors is better achieved through multiple biopsies with particular focus on nodular or thickened areas, as these may indicate invasive disease.2 Primary EMPD is characterized by pagetoid cells with abundant pale cytoplasm proliferating in a single-cell or nested pattern within the epidermis or dermis in invasive disease and often is accompanied by dermal lymphocytic inflammation.1 Immunohistochemistry demonstrates positive staining for CEA, CK7, and CK8, with negative staining for indicators of secondary EMPD including CK20 and CDX2.1,2
As part of the workup, it is critical to distinguish between primary disease and secondary EMPD.1 Beyond skin and clinical lymph node examination, additional workup should be based on age-appropriate and location-directed malignant neoplasm screenings, including colonoscopy, cystoscopy, prostate examination, mammography, and Papanicolaou test. Advanced imaging such as CT, positron emission tomography, or magnetic resonance imaging can be used to assess for metastatic disease if internal malignant neoplasms are present on initial screening or clinical lymphadenopathy is identified.2 Additionally, it can be helpful in the evaluation for nodal disease in cases of invasive EMPD.
The likelihood of associated underlying carcinomas varies depending on the site of involvement.38,39 For example, vulvar involvement constitutes approximately 65% of EMPD cases, with 11% to 20% of cases being associated with underlying gastrointestinal or genitourinary carcinomas. Involvement of the male genitalia, as in our 2 patients, is rare, accounting for approximately 14% of cases, 11% of which are associated with prostate, testicular, and bladder carcinoma. Perianal involvement comprises 20% of EMPD cases and has the greatest risk for underlying malignancy with an incidence of 33% to 86%, the majority of which are rectal or tubo-ovarian cancers.38,39 Consideration of the frequency and types of underlying carcinoma of respective sites of involvement can be helpful when ruling out secondary EMPD.
In both of our patients, palpable lymphadenopathy at the time of original diagnosis and histologic invasive disease on initial biopsy warranted thorough imaging and laboratory workup; there was no evidence of primary malignancy. Given the absence of an underlying carcinoma, both patients were classified as having metastatic primary EMPD.
Assessment of lymphadenopathy is an essential aspect of disease workup, as it is associated with a statistically higher rate of lymph node metastases. A study by Fujisawa et al20 demonstrated that 80% of patients with lymphadenopathy had regional metastases compared to only 15% of patients without clinical lymphadenopathy. The presence of invasive disease also has been shown to correspond with lymph node metastases.40 Ogata et al40 showed that 0% of cases with in situ EMPD had a positive sentinel lymph node biopsy (SLNB) compared to 4% and 43% in cases that showed evidence of microinvasion and dermal invasion, respectively. Lymph node metastases are associated with poor prognosis, with increasingly worse prognosis when there are multiple lymph nodes affected.41 In our case series, patient 1 had lymphadenopathy and both patients had invasive EMPD; they both later developed metastases and died.
Lymphadenopathy should be further investigated with imaging and biopsy or fine-needle aspiration.42 Recent expert consensus guidelines recommended this method of investigation over routine use of SLNB, as there is no evidence that a positive SLNB affects treatment that changes disease-specific survival.2
Treatment of EMPD—Surgical excision of the primary lesion is the first-line treatment of EMPD,1,2 which can be performed by wide local excision; however, studies have demonstrated higher recurrence-free survival with margin-controlled surgery (complete circumferential peripheral and deep margin assessment) or Mohs micrographic surgery (MMS), especially with CK7 immunostaining.2,37,43 The literature on MMS of invasive EMPD is sparse, accounting for 57 patients.25,37,44 Other reports describe management with surgical excision, wide local excision, regional resection, or vulvectomy, in addition to lymph node dissection, radiation therapy (RT), and/or chemotherapy.1-36,39,43-46 Despite the improved outcomes with MMS, the predominance of other surgical approaches in our search suggests that MMS may be currently underutilized for the treatment of invasive or locally advanced EMPD.
Among patients with unresectable disease or distant metastases, management includes RT with curative intent, chemotherapy, or a combination of both.1,2 In our review, 267 cases were treated using RT and 77 with chemotherapy. Radiation therapy is an effective therapeutic option with a reported response rate of 62% to 100% and can be employed as either primary or adjuvant treatment.3 For patients with lymph node metastasis the combination of RT and lymph node dissection has been shown to have improved outcomes compared to lymph node dissection alone, with 1 study showing a 5-year survival of 75% for patients who received adjuvant RT compared to 0% for lymph node dissection alone.45
There are currently no consensus guidelines on the best chemotherapeutic regimen for metastatic EMPD. Several regimens have been reported, including docetaxel monotherapy; low-dose 5-fluorouracil and cisplatin; combination chemotherapy FECOM (5-fluorouracil, epirubicin, carboplatin, vincristine, mitomycin); or combination therapy with cisplatin, epirubicin, and paclitaxel.1
Prognosis of Metastatic EMPD—Because invasive and metastatic EMPD is rare, its natural history is hard to predict. Poor prognosis is associated with nodule formation, tumor thickness, perianal or vaginal involvement, lymphovascular invasion, nodal metastasis, and distant metastasis. The 5-year survival for metastatic EMPD has been reported to be less than 10%.46 Our cases underscore the poor prognostic risk associated with metastatic EMPD.
For all cases of EMPD, close follow-up is warranted. Guidelines recommend physical examination with lymph node assessment every 3 to 6 months for 3 years and every 6 to 12 months for the subsequent 5 years.2 Specific recommendations for follow-up in invasive disease have not yet been described, though the 20% probability of developing an internal malignancy within 5 years after diagnosis and poor prognostic outcomes associated with invasive and metastatic disease support the need for close monitoring.2
Conclusion
Although in situ EMPD often is a slow-growing tumor with good prognosis, invasive disease has high potential to behave aggressively with high morbidity and mortality. Increased awareness and prompt identification of invasive EMPD, expedited comprehensive workup, and early multidisciplinary management might impact patient outcomes.
Acknowledgment—The authors would like to thank Ellen Aaronson, MLIS, AHIP (Mayo Clinic Libraries [Jacksonville, FL]), for creating and conducting the narrative literature search in the MEDLINE database.
- Hashimoto H, Ito T. Current management and treatment of extramammary Paget’s disease. Curr Treat Options Oncol. 2022;23:818-830. doi:10.1007/s11864-021-00923-3
- Kibbi N, Owen JL, Worley B, et al. Evidence-based clinical practice guidelines for extramammary Paget disease. JAMA Oncol. 2022;8:618-628. doi:10.1001/jamaoncol.2021.7148
- Morris CR, Hurst EA. Extramammary Paget’s disease: a review of the literature part II: treatment and prognosis. Dermatol Surg. 2020;46:305-311. doi:10.1097/DSS.0000000000002240
- Merritt BG, Degesys CA, Brodland DG. Extramammary Paget disease. Dermatol Clin. 2019;37:261-267. doi:10.1016/j.det.2019.02.002
- Aroche Gutierrez LL, Holloway SB, Donthi D, et al. Docetaxel treatment for widely metastatic invasive vulvar extramammary Paget’s disease with multifocal bone metastasis. Gynecol Oncol Rep. 2022;45:101114. doi:10.1016/j.gore.2022.101114
- Ueda M, Omori M, Sakai A. Invasive extramammary Paget’s disease with lymph node metastases and high-grade B-cell lymphoma. An Bras Dermatol. 2023;98:414-418. doi:10.1016/j.abd.2022.04.012
- Rathore R, Yadav D, Agarwal S, et al. Primary extra mammary Paget’s disease of vulva, with apocrine adenocarcinoma, signet ring cell differentiation and distant metastasis. J Family Reprod Health. 2020;14:276-280. doi:10.18502/jfrh.v14i4.5213
- Kawahara Y, Umeda Y, Yamaguchi B, et al. Long-term resolution of invasive extramammary Paget’s disease with multiple regional lymph node metastases solely with regional lymph node dissection. J Dermatol. 2021;48:E452-E453. doi:10.1111/1346-8138.16007
- Hanyu T, Fujitani S, Ito A, et al. Brain metastasis from extramammary Paget’s disease. Nagoya J Med Sci. 2020;82:791-798. doi:10.18999/nagjms.82.4.791
- Waki Y, Nobeyama Y, Ogawa T, et al. Case of extramammary Paget’s disease causing pulmonary tumor embolism. J Dermatol. 2020;47:E133-E134. doi:10.1111/1346-8138.15267
- Li ZG, Qin XJ. Extensive invasive extramammary Paget disease evaluated by F-18 FDG PET/CT: a case report. Medicine (Baltimore). 2015;94:E371. doi:10.1097/MD.0000000000000371
- Kato N, Matsue K, Sotodate A, et al. Extramammary Paget’s disease with distant skin metastasis. J Dermatol. 1996;23:408-414. doi:10.1111/j.1346-8138.1996.tb04043.x
- Hosomi M, Miyake O, Matsumiya K, et al. Extramammary Paget’s disease with a large mass in male genitalia: a case report. Article in Japanese. Hinyokika Kiyo. 1989;35:1981-1984.
- Hardy LE, Baxter L, Wan K, et al. Invasive cervical adenocarcinoma arising from extension of recurrent vulval Paget’s disease. BMJ Case Rep. 2020;13e232424. doi:10.1136/bcr-2019-232424
- Onaiwu CO, Ramirez PT, Kamat A, et al. Invasive extramammary Paget’s disease of the bladder diagnosed 18 years after noninvasive extramammary Paget’s disease of the vulva. Gynecol Oncol Case Rep. 2014;8:27-29. doi:10.1016/j.gynor.2014.03.004
- Yao H, Xie M, Fu S, et al. Survival analysis of patients with invasive extramammary Paget disease: implications of anatomic sites. BMC Cancer. 2018;18:403. doi:10.1186/s12885-018-4257-1
- Kato H, Watanabe S, Kariya K, et al. Efficacy of low-dose 5-fluorouracil/cisplatin therapy for invasive extramammary Paget’s disease. J Dermatol. 2018;45:560-563. doi:10.1111/1346-8138.14247
- Yoshino K, Fujisawa Y, Kiyohara Y, et al. Usefulness of docetaxel as first-line chemotherapy for metastatic extramammary Paget’s disease. J Dermatol. 2016;43:633-637. doi:10.1111/1346-8138.13200
- Shu B, Shen XX, Chen P, et al. Primary invasive extramammary Paget disease on penoscrotum: a clinicopathological analysis of 41 cases. Hum Pathol. 2016;47:70-77. doi:10.1016/j.humpath.2015.09.005References
- Fujisawa Y, Yoshino K, Kiyohara Y, et al. The role of sentinel lymph node biopsy in the management of invasive extramammary Paget’s disease: multi-center, retrospective study of 151 patients. J Dermatol Sci. 2015;79:38-42. doi:10.1016/j.jdermsci.2015.03.014
- Dai B, Kong YY, Chang K, et al. Primary invasive carcinoma associated with penoscrotal extramammary Paget’s disease: a clinicopathological analysis of 56 cases. BJU Int. 2015;115:153-160. doi:10.1111/bju.12776
- Shiomi T, Noguchi T, Nakayama H, et al. Clinicopathological study of invasive extramammary Paget’s disease: subgroup comparison according to invasion depth. J Eur Acad Dermatol Venereol. 2013;27:589-592. doi:10.1111/j.1468-3083.2012.04489.x
- Hatta N, Morita R, Yamada M, et al. Sentinel lymph node biopsy in patients with extramammary Paget’s disease. Dermatol Surg. 2004;30:1329-1334. doi:10.1111/j.1524-4725.2004.30377.x
- Karam A, Dorigo O. Treatment outcomes in a large cohort of patients with invasive extramammary Paget’s disease. Gynecol Oncol. 2012;125:346-351. doi:10.1016/j.ygyno.2012.01.032
- Guo L, Liu X, Li H, et al. Clinicopathological features of extramammary Paget’s disease: a report of 75 cases. Article in Chinese. Zhonghua Yi Xue Za Zhi. 2015;95:1751-1754.
- Kilts TP, Long B, Glasgow AE, et al. Invasive vulvar extramammary Paget’s disease in the United States. Gynecol Oncol. 2020;157:649-655. doi:10.1016/j.ygyno.2020.03.018
- Kusatake K, Harada Y, Mizumoto K, et al. Usefulness of sentinel lymph node biopsy for the detection of metastasis in the early stage of extramammary Paget’s disease. Eur J Dermatol. 2015;25:156-161. doi:10.1684/ejd.2015.2534
- Jeong BK, Kim KR. Invasive extramammary Paget disease of the vulva with signet ring cell morphology in a patient with signet ring cell carcinoma of the stomach: report of a case. Int J Gynecol Pathol. 2018;37:147-151. doi:10.1097/PGP.0000000000000405
- Pagnanelli M, De Nardi P, Martella S, et al. Local excision of a mucinous adenocarcinoma of the anal margin (extramammary Paget’s disease) and reconstruction with a bilateral V-Y flap. Case Rep Surg. 2019;2019:9073982. doi:10.1155/2019/9073982
- Sopracordevole F, Di Giuseppe J, De Piero G, et al. Surgical treatment of Paget disease of the vulva: prognostic significance of stromal invasion and surgical margin status. J Low Genit Tract Dis. 2016;20:184-188. doi:10.1097/LGT.0000000000000191
- Evans AT, Neven P. Invasive adenocarcinoma arising in extramammary Paget’s disease of the vulva. Histopathology. 1991;18:355-360. doi:10.1111/j.1365-2559.1991.tb00857.x
- Kitano A, Izumi M, Tamura K, et al. Brain metastasis from cutaneous squamous cell carcinoma coexistent with extramammary Paget’s disease: a case report. Pathol Int. 2019;69:619-625. doi:10.1111/pin.12846
- Miracco C, Francini E, Torre P, et al. Extramammary invasive Paget’s disease and apocrine angiomatous hamartoma: an unusual association. Eur J Dermatol. 2018;28:853-855. doi:10.1684/ejd.2018.3438
- Kambayashi Y, Fujimura T, Ohuchi K, et al. Advanced invasive extramammary Paget’s disease concomitant with cecal cancer possessing rare variant of TP53 single nucleotide polymorphism. Case Rep Oncol. 2019;12:855-860. doi:10.1159/000504339
- Fujimura T, Furudate S, Kambayashi Y, et al. Potential use of bisphosphonates in invasive extramammary Paget’s disease: an immunohistochemical investigation. Clin Dev Immunol. 2013;2013:164982. doi:10.1155/2013/164982
- Kawamura H, Ogata K, Miura H, et al. Patellar metastases. A report of two cases. Int Orthop. 1993;17:57-59. doi:10.1007/BF00195227
- Damavandy AA, Terushkin V, Zitelli JA, et al. Intraoperative immunostaining for cytokeratin-7 during Mohs micrographic surgery demonstrates low local recurrence rates in extramammary Paget’s disease. Dermatol Surg. 2018;44:354-364. doi:10.1097/DSS.0000000000001355
- Morris CR, Hurst EA. Extramammary Paget disease: a review of the literature-part I: history, epidemiology, pathogenesis, presentation, histopathology, and diagnostic work-up. Dermatol Surg. 2020;46:151-158. doi:10.1097/DSS.0000000000002064
- Simonds RM, Segal RJ, Sharma A. Extramammary Paget’s disease: a review of the literature. Int J Dermatol. 2019;58:871-879. doi:10.1111/ijd.14328
- Ogata D, Kiyohara Y, Yoshikawa S, et al. Usefulness of sentinel lymph node biopsy for prognostic prediction in extramammary Paget’s disease. Eur J Dermatol. 2016;26:254-259. doi:10.1684/ejd.2016.2744
- Ohara K, Fujisawa Y, Yoshino K, et al. A proposal for a TNM staging system for extramammary Paget disease: retrospective analysis of 301 patients with invasive primary tumors. J Dermatol Sci. 2016;83:234-239. doi:10.1016/j.jdermsci.2016.06.004
- Fujisawa Y, Yoshino K, Kiyohara Y, et al. The role of sentinel lymph node biopsy in the management of invasive extramammary Paget’s disease: multi-center, retrospective study of 151 patients. J Dermatol Sci. 2015;79:38-42. doi:10.1016/j.jdermsci.2015.03.014
- Kim SJ, Thompson AK, Zubair AS, et al. Surgical treatment and outcomes of patients with extramammary Paget disease: a cohort study. Dermatol Surg. 2017;43:708-714. doi:10.1097/DSS.0000000000001051
- Wollina U. Extensive invasive extramammary Paget’s disease: surgical treatment. J Cutan Aesthet Surg. 2013;6:41-44. doi:10.4103/0974-2077.110098
- Tsutsui K, Takahashi A, Muto Y, et al. Outcomes of lymph node dissection in the treatment of extramammary Paget’s disease: a single-institution study. J Dermatol. 2020;47:512-517. doi:10.1111/1346-8138.15285
- Guercio BJ, Iyer G, Kidwai WZ, et al. Treatment of metastatic extramammary Paget disease with combination ipilimumab and nivolumab: a case report. Case Rep Oncol. 2021;14:430-438. doi:10.1159/000514345
Extramammary Paget disease (EMPD) is a rare cutaneous malignancy typically seen in apocrine-rich areas, including the axillae and anogenital region. It presents as a slow-growing, erythematous patch or plaque that commonly is misdiagnosed as an infectious or inflammatory condition.1,2 Primary EMPD occurs as a intraepithelial neoplasm, whereas secondary EMPD occurs due to epidermotropic metastases or direct extension of an underlying adenocarcinoma into the skin.1 Most commonly, primary EMPD occurs in situ; however, when present, dermal invasion and metastases from the skin are associated with poorer outcomes.3 Given the rarity of metastatic disease, existing literature is limited to case reports and case series.
We present 2 patients with metastatic primary EMPD who had evidence of invasion on initial biopsy and died secondary to metastatic EMPD. We conducted a comprehensive review of the literature for invasive and metastatic EMPD to highlight key clinicopathologic features, treatment considerations, and the potential for rapid disease progression in cases of invasive EMPD.
Case Series
Patient 1—A 68-year-old White man with a history of breast cancer (in remission) presented to our clinic for further management of biopsy-proven scrotal EMPD. Prior to biopsy, he described a 6-month history of worsening scrotal rash treated with topical antifungals, oral antibiotics, and topical steroids due to presumed diagnosis of intertrigo, cellulitis, and dermatitis, respectively. Clinical examination showed indurated, erythematous, ulcerated plaques involving the bilateral groin, genitalia, and perineum (Figure 1). Skin biopsy confirmed a diagnosis of EMPD with both dermal and lymphovascular invasion. An immunohistochemical profile was positive for CK7 and carcinoembryonic antigen (CEA) and negative for CK20 (Figure 2).
At presentation, the patient had palpable lymphadenopathy and scrotal edema concerning for inguinal and iliac lymph node metastases. Workup for an underlying adenocarcinoma included computed tomography (CT) of the chest, abdomen, and pelvis; urologic consultation with cystoscopy; and a screening colonoscopy. The CT scan revealed multiple enlarged inguinal and external iliac lymph nodes. Fine-needle aspiration revealed CK7- and CEA-positive neoplastic cells consistent with metastatic EMPD. The patient was treated with 6 cycles of carboplatin-paclitaxel, palliative radiation therapy, and pembrolizumab with minimal response to treatment and development of osteolytic vertebral lesions concerning for disease progression. He died 1 year after the initial diagnosis secondary to the disease.
Patient 2—A 79-year-old White man presented for further management of an outside diagnosis of superficially invasive primary EMPD of the bilateral inguinal folds and scrotum that had been present for 5 months prior to biopsy and diagnosis. Clinical examination at initial presentation revealed erythematous patches of the bilateral inguinal folds and scrotum, as well as an erythematous scaling plaque in the right axilla. There was no palpable clinical lymphadenopathy. Biopsy of the axilla and groin were both consistent with invasive EMPD with positive staining for CK7 and negative staining for CK20 and CDX2. Workup for underlying adenocarcinoma with whole-body positron emission tomography/CT, mammography, esophagogastroduodenoscopy, serum CEA, colonoscopy, and cystoscopy were all negative for a metastatic adenocarcinoma. There was no imaging or clinical evidence of lymphadenopathy. Complete circumferential peripheral and deep-margin assessment was performed in a staged manner on both sites, and negative margins were obtained.
Surveillance imaging 6 months after surgery revealed suspicious hepatic lesions. Fine-needle aspiration of the hepatic lesions demonstrated positive staining for CK7 and negative staining for CK20, CDX2, prostate-specific antigen, and thyroid transcription factor 1, consistent with metastatic EMPD. Oncology recommended carboplatin and docetaxel or docetaxel monotherapy chemotherapy. The patient was further managed by an outside oncologist due to ease of travel but died secondary to the disease 15 months following the initial diagnosis.
Comment
Extramammary Paget disease is an uncommon cutaneous malignancy that manifests as pruritic erythematous plaques within apocrine-rich areas such as the genitalia, axillae, or anal region. It most commonly occurs in patients older than 65 years, with White women and Asian men being affected at disproportionately higher rates.1,4 Delay in diagnosis is common, as EMPD can mimic other benign inflammatory or infectious conditions, including contact dermatitis, seborrheic dermatitis, tinea, candidiasis, and eczema.1
Metastatic and multifocal cases of primary EMPD are especially rare. According to a search of PubMed articles indexed for MEDLINE published through December 2023 using the terms extramammary Paget disease, EMPD, neoplasm metastasis, invasive extramammary, and neoplasm invasiveness, we identified 5040 cases of invasive EMPD and 477 cases of metastatic EMPD.5-37 Of the reports that disclosed patient demographic information, 3627 patients were female 1410 were male, and the mean age was 67 years. Sites of metastases included regional lymph nodes, liver, lungs, cervix, bladder, bone, brain, skin, kidney, and adrenal glands
Workup for EMPD—The initial steps for workup of EMPD include a thorough physical examination and lymph node assessment. A skin biopsy also should be performed for patients presenting with refractory, pruritic, and eczematous rashes in apocrine-rich areas to evaluate for EMPD.1 Characterization of large and complex tumors is better achieved through multiple biopsies with particular focus on nodular or thickened areas, as these may indicate invasive disease.2 Primary EMPD is characterized by pagetoid cells with abundant pale cytoplasm proliferating in a single-cell or nested pattern within the epidermis or dermis in invasive disease and often is accompanied by dermal lymphocytic inflammation.1 Immunohistochemistry demonstrates positive staining for CEA, CK7, and CK8, with negative staining for indicators of secondary EMPD including CK20 and CDX2.1,2
As part of the workup, it is critical to distinguish between primary disease and secondary EMPD.1 Beyond skin and clinical lymph node examination, additional workup should be based on age-appropriate and location-directed malignant neoplasm screenings, including colonoscopy, cystoscopy, prostate examination, mammography, and Papanicolaou test. Advanced imaging such as CT, positron emission tomography, or magnetic resonance imaging can be used to assess for metastatic disease if internal malignant neoplasms are present on initial screening or clinical lymphadenopathy is identified.2 Additionally, it can be helpful in the evaluation for nodal disease in cases of invasive EMPD.
The likelihood of associated underlying carcinomas varies depending on the site of involvement.38,39 For example, vulvar involvement constitutes approximately 65% of EMPD cases, with 11% to 20% of cases being associated with underlying gastrointestinal or genitourinary carcinomas. Involvement of the male genitalia, as in our 2 patients, is rare, accounting for approximately 14% of cases, 11% of which are associated with prostate, testicular, and bladder carcinoma. Perianal involvement comprises 20% of EMPD cases and has the greatest risk for underlying malignancy with an incidence of 33% to 86%, the majority of which are rectal or tubo-ovarian cancers.38,39 Consideration of the frequency and types of underlying carcinoma of respective sites of involvement can be helpful when ruling out secondary EMPD.
In both of our patients, palpable lymphadenopathy at the time of original diagnosis and histologic invasive disease on initial biopsy warranted thorough imaging and laboratory workup; there was no evidence of primary malignancy. Given the absence of an underlying carcinoma, both patients were classified as having metastatic primary EMPD.
Assessment of lymphadenopathy is an essential aspect of disease workup, as it is associated with a statistically higher rate of lymph node metastases. A study by Fujisawa et al20 demonstrated that 80% of patients with lymphadenopathy had regional metastases compared to only 15% of patients without clinical lymphadenopathy. The presence of invasive disease also has been shown to correspond with lymph node metastases.40 Ogata et al40 showed that 0% of cases with in situ EMPD had a positive sentinel lymph node biopsy (SLNB) compared to 4% and 43% in cases that showed evidence of microinvasion and dermal invasion, respectively. Lymph node metastases are associated with poor prognosis, with increasingly worse prognosis when there are multiple lymph nodes affected.41 In our case series, patient 1 had lymphadenopathy and both patients had invasive EMPD; they both later developed metastases and died.
Lymphadenopathy should be further investigated with imaging and biopsy or fine-needle aspiration.42 Recent expert consensus guidelines recommended this method of investigation over routine use of SLNB, as there is no evidence that a positive SLNB affects treatment that changes disease-specific survival.2
Treatment of EMPD—Surgical excision of the primary lesion is the first-line treatment of EMPD,1,2 which can be performed by wide local excision; however, studies have demonstrated higher recurrence-free survival with margin-controlled surgery (complete circumferential peripheral and deep margin assessment) or Mohs micrographic surgery (MMS), especially with CK7 immunostaining.2,37,43 The literature on MMS of invasive EMPD is sparse, accounting for 57 patients.25,37,44 Other reports describe management with surgical excision, wide local excision, regional resection, or vulvectomy, in addition to lymph node dissection, radiation therapy (RT), and/or chemotherapy.1-36,39,43-46 Despite the improved outcomes with MMS, the predominance of other surgical approaches in our search suggests that MMS may be currently underutilized for the treatment of invasive or locally advanced EMPD.
Among patients with unresectable disease or distant metastases, management includes RT with curative intent, chemotherapy, or a combination of both.1,2 In our review, 267 cases were treated using RT and 77 with chemotherapy. Radiation therapy is an effective therapeutic option with a reported response rate of 62% to 100% and can be employed as either primary or adjuvant treatment.3 For patients with lymph node metastasis the combination of RT and lymph node dissection has been shown to have improved outcomes compared to lymph node dissection alone, with 1 study showing a 5-year survival of 75% for patients who received adjuvant RT compared to 0% for lymph node dissection alone.45
There are currently no consensus guidelines on the best chemotherapeutic regimen for metastatic EMPD. Several regimens have been reported, including docetaxel monotherapy; low-dose 5-fluorouracil and cisplatin; combination chemotherapy FECOM (5-fluorouracil, epirubicin, carboplatin, vincristine, mitomycin); or combination therapy with cisplatin, epirubicin, and paclitaxel.1
Prognosis of Metastatic EMPD—Because invasive and metastatic EMPD is rare, its natural history is hard to predict. Poor prognosis is associated with nodule formation, tumor thickness, perianal or vaginal involvement, lymphovascular invasion, nodal metastasis, and distant metastasis. The 5-year survival for metastatic EMPD has been reported to be less than 10%.46 Our cases underscore the poor prognostic risk associated with metastatic EMPD.
For all cases of EMPD, close follow-up is warranted. Guidelines recommend physical examination with lymph node assessment every 3 to 6 months for 3 years and every 6 to 12 months for the subsequent 5 years.2 Specific recommendations for follow-up in invasive disease have not yet been described, though the 20% probability of developing an internal malignancy within 5 years after diagnosis and poor prognostic outcomes associated with invasive and metastatic disease support the need for close monitoring.2
Conclusion
Although in situ EMPD often is a slow-growing tumor with good prognosis, invasive disease has high potential to behave aggressively with high morbidity and mortality. Increased awareness and prompt identification of invasive EMPD, expedited comprehensive workup, and early multidisciplinary management might impact patient outcomes.
Acknowledgment—The authors would like to thank Ellen Aaronson, MLIS, AHIP (Mayo Clinic Libraries [Jacksonville, FL]), for creating and conducting the narrative literature search in the MEDLINE database.
Extramammary Paget disease (EMPD) is a rare cutaneous malignancy typically seen in apocrine-rich areas, including the axillae and anogenital region. It presents as a slow-growing, erythematous patch or plaque that commonly is misdiagnosed as an infectious or inflammatory condition.1,2 Primary EMPD occurs as a intraepithelial neoplasm, whereas secondary EMPD occurs due to epidermotropic metastases or direct extension of an underlying adenocarcinoma into the skin.1 Most commonly, primary EMPD occurs in situ; however, when present, dermal invasion and metastases from the skin are associated with poorer outcomes.3 Given the rarity of metastatic disease, existing literature is limited to case reports and case series.
We present 2 patients with metastatic primary EMPD who had evidence of invasion on initial biopsy and died secondary to metastatic EMPD. We conducted a comprehensive review of the literature for invasive and metastatic EMPD to highlight key clinicopathologic features, treatment considerations, and the potential for rapid disease progression in cases of invasive EMPD.
Case Series
Patient 1—A 68-year-old White man with a history of breast cancer (in remission) presented to our clinic for further management of biopsy-proven scrotal EMPD. Prior to biopsy, he described a 6-month history of worsening scrotal rash treated with topical antifungals, oral antibiotics, and topical steroids due to presumed diagnosis of intertrigo, cellulitis, and dermatitis, respectively. Clinical examination showed indurated, erythematous, ulcerated plaques involving the bilateral groin, genitalia, and perineum (Figure 1). Skin biopsy confirmed a diagnosis of EMPD with both dermal and lymphovascular invasion. An immunohistochemical profile was positive for CK7 and carcinoembryonic antigen (CEA) and negative for CK20 (Figure 2).
At presentation, the patient had palpable lymphadenopathy and scrotal edema concerning for inguinal and iliac lymph node metastases. Workup for an underlying adenocarcinoma included computed tomography (CT) of the chest, abdomen, and pelvis; urologic consultation with cystoscopy; and a screening colonoscopy. The CT scan revealed multiple enlarged inguinal and external iliac lymph nodes. Fine-needle aspiration revealed CK7- and CEA-positive neoplastic cells consistent with metastatic EMPD. The patient was treated with 6 cycles of carboplatin-paclitaxel, palliative radiation therapy, and pembrolizumab with minimal response to treatment and development of osteolytic vertebral lesions concerning for disease progression. He died 1 year after the initial diagnosis secondary to the disease.
Patient 2—A 79-year-old White man presented for further management of an outside diagnosis of superficially invasive primary EMPD of the bilateral inguinal folds and scrotum that had been present for 5 months prior to biopsy and diagnosis. Clinical examination at initial presentation revealed erythematous patches of the bilateral inguinal folds and scrotum, as well as an erythematous scaling plaque in the right axilla. There was no palpable clinical lymphadenopathy. Biopsy of the axilla and groin were both consistent with invasive EMPD with positive staining for CK7 and negative staining for CK20 and CDX2. Workup for underlying adenocarcinoma with whole-body positron emission tomography/CT, mammography, esophagogastroduodenoscopy, serum CEA, colonoscopy, and cystoscopy were all negative for a metastatic adenocarcinoma. There was no imaging or clinical evidence of lymphadenopathy. Complete circumferential peripheral and deep-margin assessment was performed in a staged manner on both sites, and negative margins were obtained.
Surveillance imaging 6 months after surgery revealed suspicious hepatic lesions. Fine-needle aspiration of the hepatic lesions demonstrated positive staining for CK7 and negative staining for CK20, CDX2, prostate-specific antigen, and thyroid transcription factor 1, consistent with metastatic EMPD. Oncology recommended carboplatin and docetaxel or docetaxel monotherapy chemotherapy. The patient was further managed by an outside oncologist due to ease of travel but died secondary to the disease 15 months following the initial diagnosis.
Comment
Extramammary Paget disease is an uncommon cutaneous malignancy that manifests as pruritic erythematous plaques within apocrine-rich areas such as the genitalia, axillae, or anal region. It most commonly occurs in patients older than 65 years, with White women and Asian men being affected at disproportionately higher rates.1,4 Delay in diagnosis is common, as EMPD can mimic other benign inflammatory or infectious conditions, including contact dermatitis, seborrheic dermatitis, tinea, candidiasis, and eczema.1
Metastatic and multifocal cases of primary EMPD are especially rare. According to a search of PubMed articles indexed for MEDLINE published through December 2023 using the terms extramammary Paget disease, EMPD, neoplasm metastasis, invasive extramammary, and neoplasm invasiveness, we identified 5040 cases of invasive EMPD and 477 cases of metastatic EMPD.5-37 Of the reports that disclosed patient demographic information, 3627 patients were female 1410 were male, and the mean age was 67 years. Sites of metastases included regional lymph nodes, liver, lungs, cervix, bladder, bone, brain, skin, kidney, and adrenal glands
Workup for EMPD—The initial steps for workup of EMPD include a thorough physical examination and lymph node assessment. A skin biopsy also should be performed for patients presenting with refractory, pruritic, and eczematous rashes in apocrine-rich areas to evaluate for EMPD.1 Characterization of large and complex tumors is better achieved through multiple biopsies with particular focus on nodular or thickened areas, as these may indicate invasive disease.2 Primary EMPD is characterized by pagetoid cells with abundant pale cytoplasm proliferating in a single-cell or nested pattern within the epidermis or dermis in invasive disease and often is accompanied by dermal lymphocytic inflammation.1 Immunohistochemistry demonstrates positive staining for CEA, CK7, and CK8, with negative staining for indicators of secondary EMPD including CK20 and CDX2.1,2
As part of the workup, it is critical to distinguish between primary disease and secondary EMPD.1 Beyond skin and clinical lymph node examination, additional workup should be based on age-appropriate and location-directed malignant neoplasm screenings, including colonoscopy, cystoscopy, prostate examination, mammography, and Papanicolaou test. Advanced imaging such as CT, positron emission tomography, or magnetic resonance imaging can be used to assess for metastatic disease if internal malignant neoplasms are present on initial screening or clinical lymphadenopathy is identified.2 Additionally, it can be helpful in the evaluation for nodal disease in cases of invasive EMPD.
The likelihood of associated underlying carcinomas varies depending on the site of involvement.38,39 For example, vulvar involvement constitutes approximately 65% of EMPD cases, with 11% to 20% of cases being associated with underlying gastrointestinal or genitourinary carcinomas. Involvement of the male genitalia, as in our 2 patients, is rare, accounting for approximately 14% of cases, 11% of which are associated with prostate, testicular, and bladder carcinoma. Perianal involvement comprises 20% of EMPD cases and has the greatest risk for underlying malignancy with an incidence of 33% to 86%, the majority of which are rectal or tubo-ovarian cancers.38,39 Consideration of the frequency and types of underlying carcinoma of respective sites of involvement can be helpful when ruling out secondary EMPD.
In both of our patients, palpable lymphadenopathy at the time of original diagnosis and histologic invasive disease on initial biopsy warranted thorough imaging and laboratory workup; there was no evidence of primary malignancy. Given the absence of an underlying carcinoma, both patients were classified as having metastatic primary EMPD.
Assessment of lymphadenopathy is an essential aspect of disease workup, as it is associated with a statistically higher rate of lymph node metastases. A study by Fujisawa et al20 demonstrated that 80% of patients with lymphadenopathy had regional metastases compared to only 15% of patients without clinical lymphadenopathy. The presence of invasive disease also has been shown to correspond with lymph node metastases.40 Ogata et al40 showed that 0% of cases with in situ EMPD had a positive sentinel lymph node biopsy (SLNB) compared to 4% and 43% in cases that showed evidence of microinvasion and dermal invasion, respectively. Lymph node metastases are associated with poor prognosis, with increasingly worse prognosis when there are multiple lymph nodes affected.41 In our case series, patient 1 had lymphadenopathy and both patients had invasive EMPD; they both later developed metastases and died.
Lymphadenopathy should be further investigated with imaging and biopsy or fine-needle aspiration.42 Recent expert consensus guidelines recommended this method of investigation over routine use of SLNB, as there is no evidence that a positive SLNB affects treatment that changes disease-specific survival.2
Treatment of EMPD—Surgical excision of the primary lesion is the first-line treatment of EMPD,1,2 which can be performed by wide local excision; however, studies have demonstrated higher recurrence-free survival with margin-controlled surgery (complete circumferential peripheral and deep margin assessment) or Mohs micrographic surgery (MMS), especially with CK7 immunostaining.2,37,43 The literature on MMS of invasive EMPD is sparse, accounting for 57 patients.25,37,44 Other reports describe management with surgical excision, wide local excision, regional resection, or vulvectomy, in addition to lymph node dissection, radiation therapy (RT), and/or chemotherapy.1-36,39,43-46 Despite the improved outcomes with MMS, the predominance of other surgical approaches in our search suggests that MMS may be currently underutilized for the treatment of invasive or locally advanced EMPD.
Among patients with unresectable disease or distant metastases, management includes RT with curative intent, chemotherapy, or a combination of both.1,2 In our review, 267 cases were treated using RT and 77 with chemotherapy. Radiation therapy is an effective therapeutic option with a reported response rate of 62% to 100% and can be employed as either primary or adjuvant treatment.3 For patients with lymph node metastasis the combination of RT and lymph node dissection has been shown to have improved outcomes compared to lymph node dissection alone, with 1 study showing a 5-year survival of 75% for patients who received adjuvant RT compared to 0% for lymph node dissection alone.45
There are currently no consensus guidelines on the best chemotherapeutic regimen for metastatic EMPD. Several regimens have been reported, including docetaxel monotherapy; low-dose 5-fluorouracil and cisplatin; combination chemotherapy FECOM (5-fluorouracil, epirubicin, carboplatin, vincristine, mitomycin); or combination therapy with cisplatin, epirubicin, and paclitaxel.1
Prognosis of Metastatic EMPD—Because invasive and metastatic EMPD is rare, its natural history is hard to predict. Poor prognosis is associated with nodule formation, tumor thickness, perianal or vaginal involvement, lymphovascular invasion, nodal metastasis, and distant metastasis. The 5-year survival for metastatic EMPD has been reported to be less than 10%.46 Our cases underscore the poor prognostic risk associated with metastatic EMPD.
For all cases of EMPD, close follow-up is warranted. Guidelines recommend physical examination with lymph node assessment every 3 to 6 months for 3 years and every 6 to 12 months for the subsequent 5 years.2 Specific recommendations for follow-up in invasive disease have not yet been described, though the 20% probability of developing an internal malignancy within 5 years after diagnosis and poor prognostic outcomes associated with invasive and metastatic disease support the need for close monitoring.2
Conclusion
Although in situ EMPD often is a slow-growing tumor with good prognosis, invasive disease has high potential to behave aggressively with high morbidity and mortality. Increased awareness and prompt identification of invasive EMPD, expedited comprehensive workup, and early multidisciplinary management might impact patient outcomes.
Acknowledgment—The authors would like to thank Ellen Aaronson, MLIS, AHIP (Mayo Clinic Libraries [Jacksonville, FL]), for creating and conducting the narrative literature search in the MEDLINE database.
- Hashimoto H, Ito T. Current management and treatment of extramammary Paget’s disease. Curr Treat Options Oncol. 2022;23:818-830. doi:10.1007/s11864-021-00923-3
- Kibbi N, Owen JL, Worley B, et al. Evidence-based clinical practice guidelines for extramammary Paget disease. JAMA Oncol. 2022;8:618-628. doi:10.1001/jamaoncol.2021.7148
- Morris CR, Hurst EA. Extramammary Paget’s disease: a review of the literature part II: treatment and prognosis. Dermatol Surg. 2020;46:305-311. doi:10.1097/DSS.0000000000002240
- Merritt BG, Degesys CA, Brodland DG. Extramammary Paget disease. Dermatol Clin. 2019;37:261-267. doi:10.1016/j.det.2019.02.002
- Aroche Gutierrez LL, Holloway SB, Donthi D, et al. Docetaxel treatment for widely metastatic invasive vulvar extramammary Paget’s disease with multifocal bone metastasis. Gynecol Oncol Rep. 2022;45:101114. doi:10.1016/j.gore.2022.101114
- Ueda M, Omori M, Sakai A. Invasive extramammary Paget’s disease with lymph node metastases and high-grade B-cell lymphoma. An Bras Dermatol. 2023;98:414-418. doi:10.1016/j.abd.2022.04.012
- Rathore R, Yadav D, Agarwal S, et al. Primary extra mammary Paget’s disease of vulva, with apocrine adenocarcinoma, signet ring cell differentiation and distant metastasis. J Family Reprod Health. 2020;14:276-280. doi:10.18502/jfrh.v14i4.5213
- Kawahara Y, Umeda Y, Yamaguchi B, et al. Long-term resolution of invasive extramammary Paget’s disease with multiple regional lymph node metastases solely with regional lymph node dissection. J Dermatol. 2021;48:E452-E453. doi:10.1111/1346-8138.16007
- Hanyu T, Fujitani S, Ito A, et al. Brain metastasis from extramammary Paget’s disease. Nagoya J Med Sci. 2020;82:791-798. doi:10.18999/nagjms.82.4.791
- Waki Y, Nobeyama Y, Ogawa T, et al. Case of extramammary Paget’s disease causing pulmonary tumor embolism. J Dermatol. 2020;47:E133-E134. doi:10.1111/1346-8138.15267
- Li ZG, Qin XJ. Extensive invasive extramammary Paget disease evaluated by F-18 FDG PET/CT: a case report. Medicine (Baltimore). 2015;94:E371. doi:10.1097/MD.0000000000000371
- Kato N, Matsue K, Sotodate A, et al. Extramammary Paget’s disease with distant skin metastasis. J Dermatol. 1996;23:408-414. doi:10.1111/j.1346-8138.1996.tb04043.x
- Hosomi M, Miyake O, Matsumiya K, et al. Extramammary Paget’s disease with a large mass in male genitalia: a case report. Article in Japanese. Hinyokika Kiyo. 1989;35:1981-1984.
- Hardy LE, Baxter L, Wan K, et al. Invasive cervical adenocarcinoma arising from extension of recurrent vulval Paget’s disease. BMJ Case Rep. 2020;13e232424. doi:10.1136/bcr-2019-232424
- Onaiwu CO, Ramirez PT, Kamat A, et al. Invasive extramammary Paget’s disease of the bladder diagnosed 18 years after noninvasive extramammary Paget’s disease of the vulva. Gynecol Oncol Case Rep. 2014;8:27-29. doi:10.1016/j.gynor.2014.03.004
- Yao H, Xie M, Fu S, et al. Survival analysis of patients with invasive extramammary Paget disease: implications of anatomic sites. BMC Cancer. 2018;18:403. doi:10.1186/s12885-018-4257-1
- Kato H, Watanabe S, Kariya K, et al. Efficacy of low-dose 5-fluorouracil/cisplatin therapy for invasive extramammary Paget’s disease. J Dermatol. 2018;45:560-563. doi:10.1111/1346-8138.14247
- Yoshino K, Fujisawa Y, Kiyohara Y, et al. Usefulness of docetaxel as first-line chemotherapy for metastatic extramammary Paget’s disease. J Dermatol. 2016;43:633-637. doi:10.1111/1346-8138.13200
- Shu B, Shen XX, Chen P, et al. Primary invasive extramammary Paget disease on penoscrotum: a clinicopathological analysis of 41 cases. Hum Pathol. 2016;47:70-77. doi:10.1016/j.humpath.2015.09.005References
- Fujisawa Y, Yoshino K, Kiyohara Y, et al. The role of sentinel lymph node biopsy in the management of invasive extramammary Paget’s disease: multi-center, retrospective study of 151 patients. J Dermatol Sci. 2015;79:38-42. doi:10.1016/j.jdermsci.2015.03.014
- Dai B, Kong YY, Chang K, et al. Primary invasive carcinoma associated with penoscrotal extramammary Paget’s disease: a clinicopathological analysis of 56 cases. BJU Int. 2015;115:153-160. doi:10.1111/bju.12776
- Shiomi T, Noguchi T, Nakayama H, et al. Clinicopathological study of invasive extramammary Paget’s disease: subgroup comparison according to invasion depth. J Eur Acad Dermatol Venereol. 2013;27:589-592. doi:10.1111/j.1468-3083.2012.04489.x
- Hatta N, Morita R, Yamada M, et al. Sentinel lymph node biopsy in patients with extramammary Paget’s disease. Dermatol Surg. 2004;30:1329-1334. doi:10.1111/j.1524-4725.2004.30377.x
- Karam A, Dorigo O. Treatment outcomes in a large cohort of patients with invasive extramammary Paget’s disease. Gynecol Oncol. 2012;125:346-351. doi:10.1016/j.ygyno.2012.01.032
- Guo L, Liu X, Li H, et al. Clinicopathological features of extramammary Paget’s disease: a report of 75 cases. Article in Chinese. Zhonghua Yi Xue Za Zhi. 2015;95:1751-1754.
- Kilts TP, Long B, Glasgow AE, et al. Invasive vulvar extramammary Paget’s disease in the United States. Gynecol Oncol. 2020;157:649-655. doi:10.1016/j.ygyno.2020.03.018
- Kusatake K, Harada Y, Mizumoto K, et al. Usefulness of sentinel lymph node biopsy for the detection of metastasis in the early stage of extramammary Paget’s disease. Eur J Dermatol. 2015;25:156-161. doi:10.1684/ejd.2015.2534
- Jeong BK, Kim KR. Invasive extramammary Paget disease of the vulva with signet ring cell morphology in a patient with signet ring cell carcinoma of the stomach: report of a case. Int J Gynecol Pathol. 2018;37:147-151. doi:10.1097/PGP.0000000000000405
- Pagnanelli M, De Nardi P, Martella S, et al. Local excision of a mucinous adenocarcinoma of the anal margin (extramammary Paget’s disease) and reconstruction with a bilateral V-Y flap. Case Rep Surg. 2019;2019:9073982. doi:10.1155/2019/9073982
- Sopracordevole F, Di Giuseppe J, De Piero G, et al. Surgical treatment of Paget disease of the vulva: prognostic significance of stromal invasion and surgical margin status. J Low Genit Tract Dis. 2016;20:184-188. doi:10.1097/LGT.0000000000000191
- Evans AT, Neven P. Invasive adenocarcinoma arising in extramammary Paget’s disease of the vulva. Histopathology. 1991;18:355-360. doi:10.1111/j.1365-2559.1991.tb00857.x
- Kitano A, Izumi M, Tamura K, et al. Brain metastasis from cutaneous squamous cell carcinoma coexistent with extramammary Paget’s disease: a case report. Pathol Int. 2019;69:619-625. doi:10.1111/pin.12846
- Miracco C, Francini E, Torre P, et al. Extramammary invasive Paget’s disease and apocrine angiomatous hamartoma: an unusual association. Eur J Dermatol. 2018;28:853-855. doi:10.1684/ejd.2018.3438
- Kambayashi Y, Fujimura T, Ohuchi K, et al. Advanced invasive extramammary Paget’s disease concomitant with cecal cancer possessing rare variant of TP53 single nucleotide polymorphism. Case Rep Oncol. 2019;12:855-860. doi:10.1159/000504339
- Fujimura T, Furudate S, Kambayashi Y, et al. Potential use of bisphosphonates in invasive extramammary Paget’s disease: an immunohistochemical investigation. Clin Dev Immunol. 2013;2013:164982. doi:10.1155/2013/164982
- Kawamura H, Ogata K, Miura H, et al. Patellar metastases. A report of two cases. Int Orthop. 1993;17:57-59. doi:10.1007/BF00195227
- Damavandy AA, Terushkin V, Zitelli JA, et al. Intraoperative immunostaining for cytokeratin-7 during Mohs micrographic surgery demonstrates low local recurrence rates in extramammary Paget’s disease. Dermatol Surg. 2018;44:354-364. doi:10.1097/DSS.0000000000001355
- Morris CR, Hurst EA. Extramammary Paget disease: a review of the literature-part I: history, epidemiology, pathogenesis, presentation, histopathology, and diagnostic work-up. Dermatol Surg. 2020;46:151-158. doi:10.1097/DSS.0000000000002064
- Simonds RM, Segal RJ, Sharma A. Extramammary Paget’s disease: a review of the literature. Int J Dermatol. 2019;58:871-879. doi:10.1111/ijd.14328
- Ogata D, Kiyohara Y, Yoshikawa S, et al. Usefulness of sentinel lymph node biopsy for prognostic prediction in extramammary Paget’s disease. Eur J Dermatol. 2016;26:254-259. doi:10.1684/ejd.2016.2744
- Ohara K, Fujisawa Y, Yoshino K, et al. A proposal for a TNM staging system for extramammary Paget disease: retrospective analysis of 301 patients with invasive primary tumors. J Dermatol Sci. 2016;83:234-239. doi:10.1016/j.jdermsci.2016.06.004
- Fujisawa Y, Yoshino K, Kiyohara Y, et al. The role of sentinel lymph node biopsy in the management of invasive extramammary Paget’s disease: multi-center, retrospective study of 151 patients. J Dermatol Sci. 2015;79:38-42. doi:10.1016/j.jdermsci.2015.03.014
- Kim SJ, Thompson AK, Zubair AS, et al. Surgical treatment and outcomes of patients with extramammary Paget disease: a cohort study. Dermatol Surg. 2017;43:708-714. doi:10.1097/DSS.0000000000001051
- Wollina U. Extensive invasive extramammary Paget’s disease: surgical treatment. J Cutan Aesthet Surg. 2013;6:41-44. doi:10.4103/0974-2077.110098
- Tsutsui K, Takahashi A, Muto Y, et al. Outcomes of lymph node dissection in the treatment of extramammary Paget’s disease: a single-institution study. J Dermatol. 2020;47:512-517. doi:10.1111/1346-8138.15285
- Guercio BJ, Iyer G, Kidwai WZ, et al. Treatment of metastatic extramammary Paget disease with combination ipilimumab and nivolumab: a case report. Case Rep Oncol. 2021;14:430-438. doi:10.1159/000514345
- Hashimoto H, Ito T. Current management and treatment of extramammary Paget’s disease. Curr Treat Options Oncol. 2022;23:818-830. doi:10.1007/s11864-021-00923-3
- Kibbi N, Owen JL, Worley B, et al. Evidence-based clinical practice guidelines for extramammary Paget disease. JAMA Oncol. 2022;8:618-628. doi:10.1001/jamaoncol.2021.7148
- Morris CR, Hurst EA. Extramammary Paget’s disease: a review of the literature part II: treatment and prognosis. Dermatol Surg. 2020;46:305-311. doi:10.1097/DSS.0000000000002240
- Merritt BG, Degesys CA, Brodland DG. Extramammary Paget disease. Dermatol Clin. 2019;37:261-267. doi:10.1016/j.det.2019.02.002
- Aroche Gutierrez LL, Holloway SB, Donthi D, et al. Docetaxel treatment for widely metastatic invasive vulvar extramammary Paget’s disease with multifocal bone metastasis. Gynecol Oncol Rep. 2022;45:101114. doi:10.1016/j.gore.2022.101114
- Ueda M, Omori M, Sakai A. Invasive extramammary Paget’s disease with lymph node metastases and high-grade B-cell lymphoma. An Bras Dermatol. 2023;98:414-418. doi:10.1016/j.abd.2022.04.012
- Rathore R, Yadav D, Agarwal S, et al. Primary extra mammary Paget’s disease of vulva, with apocrine adenocarcinoma, signet ring cell differentiation and distant metastasis. J Family Reprod Health. 2020;14:276-280. doi:10.18502/jfrh.v14i4.5213
- Kawahara Y, Umeda Y, Yamaguchi B, et al. Long-term resolution of invasive extramammary Paget’s disease with multiple regional lymph node metastases solely with regional lymph node dissection. J Dermatol. 2021;48:E452-E453. doi:10.1111/1346-8138.16007
- Hanyu T, Fujitani S, Ito A, et al. Brain metastasis from extramammary Paget’s disease. Nagoya J Med Sci. 2020;82:791-798. doi:10.18999/nagjms.82.4.791
- Waki Y, Nobeyama Y, Ogawa T, et al. Case of extramammary Paget’s disease causing pulmonary tumor embolism. J Dermatol. 2020;47:E133-E134. doi:10.1111/1346-8138.15267
- Li ZG, Qin XJ. Extensive invasive extramammary Paget disease evaluated by F-18 FDG PET/CT: a case report. Medicine (Baltimore). 2015;94:E371. doi:10.1097/MD.0000000000000371
- Kato N, Matsue K, Sotodate A, et al. Extramammary Paget’s disease with distant skin metastasis. J Dermatol. 1996;23:408-414. doi:10.1111/j.1346-8138.1996.tb04043.x
- Hosomi M, Miyake O, Matsumiya K, et al. Extramammary Paget’s disease with a large mass in male genitalia: a case report. Article in Japanese. Hinyokika Kiyo. 1989;35:1981-1984.
- Hardy LE, Baxter L, Wan K, et al. Invasive cervical adenocarcinoma arising from extension of recurrent vulval Paget’s disease. BMJ Case Rep. 2020;13e232424. doi:10.1136/bcr-2019-232424
- Onaiwu CO, Ramirez PT, Kamat A, et al. Invasive extramammary Paget’s disease of the bladder diagnosed 18 years after noninvasive extramammary Paget’s disease of the vulva. Gynecol Oncol Case Rep. 2014;8:27-29. doi:10.1016/j.gynor.2014.03.004
- Yao H, Xie M, Fu S, et al. Survival analysis of patients with invasive extramammary Paget disease: implications of anatomic sites. BMC Cancer. 2018;18:403. doi:10.1186/s12885-018-4257-1
- Kato H, Watanabe S, Kariya K, et al. Efficacy of low-dose 5-fluorouracil/cisplatin therapy for invasive extramammary Paget’s disease. J Dermatol. 2018;45:560-563. doi:10.1111/1346-8138.14247
- Yoshino K, Fujisawa Y, Kiyohara Y, et al. Usefulness of docetaxel as first-line chemotherapy for metastatic extramammary Paget’s disease. J Dermatol. 2016;43:633-637. doi:10.1111/1346-8138.13200
- Shu B, Shen XX, Chen P, et al. Primary invasive extramammary Paget disease on penoscrotum: a clinicopathological analysis of 41 cases. Hum Pathol. 2016;47:70-77. doi:10.1016/j.humpath.2015.09.005References
- Fujisawa Y, Yoshino K, Kiyohara Y, et al. The role of sentinel lymph node biopsy in the management of invasive extramammary Paget’s disease: multi-center, retrospective study of 151 patients. J Dermatol Sci. 2015;79:38-42. doi:10.1016/j.jdermsci.2015.03.014
- Dai B, Kong YY, Chang K, et al. Primary invasive carcinoma associated with penoscrotal extramammary Paget’s disease: a clinicopathological analysis of 56 cases. BJU Int. 2015;115:153-160. doi:10.1111/bju.12776
- Shiomi T, Noguchi T, Nakayama H, et al. Clinicopathological study of invasive extramammary Paget’s disease: subgroup comparison according to invasion depth. J Eur Acad Dermatol Venereol. 2013;27:589-592. doi:10.1111/j.1468-3083.2012.04489.x
- Hatta N, Morita R, Yamada M, et al. Sentinel lymph node biopsy in patients with extramammary Paget’s disease. Dermatol Surg. 2004;30:1329-1334. doi:10.1111/j.1524-4725.2004.30377.x
- Karam A, Dorigo O. Treatment outcomes in a large cohort of patients with invasive extramammary Paget’s disease. Gynecol Oncol. 2012;125:346-351. doi:10.1016/j.ygyno.2012.01.032
- Guo L, Liu X, Li H, et al. Clinicopathological features of extramammary Paget’s disease: a report of 75 cases. Article in Chinese. Zhonghua Yi Xue Za Zhi. 2015;95:1751-1754.
- Kilts TP, Long B, Glasgow AE, et al. Invasive vulvar extramammary Paget’s disease in the United States. Gynecol Oncol. 2020;157:649-655. doi:10.1016/j.ygyno.2020.03.018
- Kusatake K, Harada Y, Mizumoto K, et al. Usefulness of sentinel lymph node biopsy for the detection of metastasis in the early stage of extramammary Paget’s disease. Eur J Dermatol. 2015;25:156-161. doi:10.1684/ejd.2015.2534
- Jeong BK, Kim KR. Invasive extramammary Paget disease of the vulva with signet ring cell morphology in a patient with signet ring cell carcinoma of the stomach: report of a case. Int J Gynecol Pathol. 2018;37:147-151. doi:10.1097/PGP.0000000000000405
- Pagnanelli M, De Nardi P, Martella S, et al. Local excision of a mucinous adenocarcinoma of the anal margin (extramammary Paget’s disease) and reconstruction with a bilateral V-Y flap. Case Rep Surg. 2019;2019:9073982. doi:10.1155/2019/9073982
- Sopracordevole F, Di Giuseppe J, De Piero G, et al. Surgical treatment of Paget disease of the vulva: prognostic significance of stromal invasion and surgical margin status. J Low Genit Tract Dis. 2016;20:184-188. doi:10.1097/LGT.0000000000000191
- Evans AT, Neven P. Invasive adenocarcinoma arising in extramammary Paget’s disease of the vulva. Histopathology. 1991;18:355-360. doi:10.1111/j.1365-2559.1991.tb00857.x
- Kitano A, Izumi M, Tamura K, et al. Brain metastasis from cutaneous squamous cell carcinoma coexistent with extramammary Paget’s disease: a case report. Pathol Int. 2019;69:619-625. doi:10.1111/pin.12846
- Miracco C, Francini E, Torre P, et al. Extramammary invasive Paget’s disease and apocrine angiomatous hamartoma: an unusual association. Eur J Dermatol. 2018;28:853-855. doi:10.1684/ejd.2018.3438
- Kambayashi Y, Fujimura T, Ohuchi K, et al. Advanced invasive extramammary Paget’s disease concomitant with cecal cancer possessing rare variant of TP53 single nucleotide polymorphism. Case Rep Oncol. 2019;12:855-860. doi:10.1159/000504339
- Fujimura T, Furudate S, Kambayashi Y, et al. Potential use of bisphosphonates in invasive extramammary Paget’s disease: an immunohistochemical investigation. Clin Dev Immunol. 2013;2013:164982. doi:10.1155/2013/164982
- Kawamura H, Ogata K, Miura H, et al. Patellar metastases. A report of two cases. Int Orthop. 1993;17:57-59. doi:10.1007/BF00195227
- Damavandy AA, Terushkin V, Zitelli JA, et al. Intraoperative immunostaining for cytokeratin-7 during Mohs micrographic surgery demonstrates low local recurrence rates in extramammary Paget’s disease. Dermatol Surg. 2018;44:354-364. doi:10.1097/DSS.0000000000001355
- Morris CR, Hurst EA. Extramammary Paget disease: a review of the literature-part I: history, epidemiology, pathogenesis, presentation, histopathology, and diagnostic work-up. Dermatol Surg. 2020;46:151-158. doi:10.1097/DSS.0000000000002064
- Simonds RM, Segal RJ, Sharma A. Extramammary Paget’s disease: a review of the literature. Int J Dermatol. 2019;58:871-879. doi:10.1111/ijd.14328
- Ogata D, Kiyohara Y, Yoshikawa S, et al. Usefulness of sentinel lymph node biopsy for prognostic prediction in extramammary Paget’s disease. Eur J Dermatol. 2016;26:254-259. doi:10.1684/ejd.2016.2744
- Ohara K, Fujisawa Y, Yoshino K, et al. A proposal for a TNM staging system for extramammary Paget disease: retrospective analysis of 301 patients with invasive primary tumors. J Dermatol Sci. 2016;83:234-239. doi:10.1016/j.jdermsci.2016.06.004
- Fujisawa Y, Yoshino K, Kiyohara Y, et al. The role of sentinel lymph node biopsy in the management of invasive extramammary Paget’s disease: multi-center, retrospective study of 151 patients. J Dermatol Sci. 2015;79:38-42. doi:10.1016/j.jdermsci.2015.03.014
- Kim SJ, Thompson AK, Zubair AS, et al. Surgical treatment and outcomes of patients with extramammary Paget disease: a cohort study. Dermatol Surg. 2017;43:708-714. doi:10.1097/DSS.0000000000001051
- Wollina U. Extensive invasive extramammary Paget’s disease: surgical treatment. J Cutan Aesthet Surg. 2013;6:41-44. doi:10.4103/0974-2077.110098
- Tsutsui K, Takahashi A, Muto Y, et al. Outcomes of lymph node dissection in the treatment of extramammary Paget’s disease: a single-institution study. J Dermatol. 2020;47:512-517. doi:10.1111/1346-8138.15285
- Guercio BJ, Iyer G, Kidwai WZ, et al. Treatment of metastatic extramammary Paget disease with combination ipilimumab and nivolumab: a case report. Case Rep Oncol. 2021;14:430-438. doi:10.1159/000514345
Metastatic Primary Extramammary Paget Disease: A Case Series
Metastatic Primary Extramammary Paget Disease: A Case Series
Practice Points
- Invasive primary extramammary Paget disease has a higher risk for lymph node metastasis.
- Consider extramammary Paget disease in patients presenting with erythematous pruritic plaques in apocrine-rich areas that fail to respond to topical steroids or antifungals.
- Prompt diagnosis can expedite comprehensive malignancy work-up and multidisciplinary management, potentially impacting patient outcomes.

Severe Cutaneous Adverse Reactions in the Setting of Antineoplastic Therapy: A Single-Institution Retrospective Study
Severe Cutaneous Adverse Reactions in the Setting of Antineoplastic Therapy: A Single-Institution Retrospective Study
To the Editor:
Severe cutaneous adverse reactions (SCARs) are rare, life-threatening reactions that include acute generalized exanthematous pustulosis (AGEP), drug reaction with eosinophilia and systemic symptoms (DRESS), and Stevens-Johnson syndrome (SJS)/toxic epidermal necrolysis (TEN).1 In addition to being associated with commonly implicated medications, SCARs also may occur in the setting of antineoplastic therapy.2,3 Although antineoplastic-associated SCARs have been described, diagnosis can be difficult due to varying latency periods and atypical clinical features, such as those observed with BRAF inhibitor–related DRESS during immunotherapy.4 Severe cutaneous adverse reactions can increase morbidity and mortality in the oncologic patient population due to both the clinical sequelae from the cutaneous reaction and the potential to interrupt cancer treatment.
The aim of this study was to evaluate the clinical characteristics, outcomes, and impact on cancer treatment among patients diagnosed with a SCAR while receiving active therapy for malignancy. We conducted a retrospective chart review of electronic medical records at Yale New Haven Hospital (New Haven, Connecticut) from 2013 to 2023, identifying patients receiving antineoplastic therapy who were diagnosed with a SCAR. Cases were identified through a search of the electronic medical record performed by the joint data analytics team using the keywords DRESS, SJS, TEN, AGEP, and generalized bullous fixed drug eruption, along with spelling variations (both abbreviations and full terms), in addition to manual review by one of the authors (K.V.) of the inpatient dermatology consultation log and dermatopathology database. Only patients for whom an antineoplastic agent was identified as a high-probability culprit by the dermatology and/or oncology teams were included.
In total, 20 patients (11 female, 9 male) were identified as having an antineoplastic-associated SCAR. All patients had metastatic or advanced disease. We identified 2 (10%) cases of AGEP, 16 (80%) cases of DRESS, and 3 (15%) cases of SJS/TEN. One patient on immunotherapy had 2 distinct SCARs (AGEP, DRESS) at different time points. Table 1 describes patient and SCAR characteristics as well as impact on cancer treatment. The median (interquartile range [IQR]) latency period for AGEP was 7.5 (4-11) days. The median (IQR) latency period for 13 of the 16 (81%) DRESS cases was 14 (10-32) days. For 3 DRESS cases with a potential second-hit phenomenon in the setting of current or antecedent immunotherapy,5 the median (IQR) latency period was 122 (96-426) days for the immunotherapy drug and 28 (21-52) days for the drug culprit. The median (IQR) latency period for SJS/TEN was 23 (20-27) days.

Patients received treatment with combination systemic corticosteroids and topical corticosteroids in 13 (65%) cases, systemic corticosteroid monotherapy in 6 (30%) cases, or combination systemic corticosteroids and etanercept in 1 (5%) case. All patients experienced resolution of the SCAR and survived to hospital discharge. Most (17/20 [85%]) patients experienced interruption or discontinuation of cancer treatment. Table 2 describes the implicated antineoplastic therapies, which included chemotherapy (3 DRESS, 1 SJS/TEN), hormonal therapy (1 DRESS), immunotherapy (1 AGEP, 4 DRESS), and targeted therapy (1 AGEP, 8 DRESS, 2 SJS/TEN).

Limitations of this study include the retrospective study design, the small sample size, and the challenge of drug culprit identification in oncologic patients on multiple high-probability medications.
Though rare, SCARs can be encountered in patients on antineoplastic therapy with a wide range of drug culprits. In our cohort, SCARs occurred with various antineoplastic agents, including chemotherapy, hormonal therapy, immunotherapy, and targeted therapy. The most common antineoplastic-associated SCAR was DRESS, which had the widest latency period in the setting of a potential second-hit phenomenon with another drug culprit. Although we did not observe any cases of SJS/TEN in the immunotherapy category, it is important to consider progressive immunotherapy-related mucocutaneous eruption in the differential diagnosis. Fortunately, all patients survived to hospital discharge and experienced SCAR resolution with systemic treatment; however, most patients experienced interruption of cancer therapy, which has the potential to affect oncologic outcomes. This interruption is not uncommon, as rechallenge of an antineoplastic agent in patients with a therapy-related SCAR generally is not recommended. The awareness and prompt management of SCARs in a patient on treatment for malignancy are critical in order to minimize negative outcomes in this vulnerable patient population.
- Duong TA, Valeyrie-Allanore L, Wolkenstein P, et al. Severe cutaneous adverse reactions to drugs. Lancet. 2017;390: 1996-2011.
- Chen CB, Wu MY, Ng CY, et al. Severe cutaneous adverse reactions induced by targeted anticancer therapies and immunotherapies. Cancer Manag Res. 2018;10:1259-1273.
- Ng CY, Chen CB, Wu MY, et al. Anticancer drugs induced severe adverse cutaneous drug reactions: an updated review on the risks associated with anticancer targeted therapy or immunotherapies. J Immunol Res. 2018;2018:5376476.
- Maloney NJ, Rana J, Yang JJ, et al. Clinical features of druginduced hypersensitivity syndrome to BRAF inhibitors with and without previous immune checkpoint inhibition: a review. Support Care Cancer. 2022;30:2839-2851.
- Hammond S, Olsson-Brown A, Grice S, et al. Does immune checkpoint inhibitor therapy increase the frequency of adverse reactions to concomitant medications? Clin Exp Allergy. 2022;52:600-603.
To the Editor:
Severe cutaneous adverse reactions (SCARs) are rare, life-threatening reactions that include acute generalized exanthematous pustulosis (AGEP), drug reaction with eosinophilia and systemic symptoms (DRESS), and Stevens-Johnson syndrome (SJS)/toxic epidermal necrolysis (TEN).1 In addition to being associated with commonly implicated medications, SCARs also may occur in the setting of antineoplastic therapy.2,3 Although antineoplastic-associated SCARs have been described, diagnosis can be difficult due to varying latency periods and atypical clinical features, such as those observed with BRAF inhibitor–related DRESS during immunotherapy.4 Severe cutaneous adverse reactions can increase morbidity and mortality in the oncologic patient population due to both the clinical sequelae from the cutaneous reaction and the potential to interrupt cancer treatment.
The aim of this study was to evaluate the clinical characteristics, outcomes, and impact on cancer treatment among patients diagnosed with a SCAR while receiving active therapy for malignancy. We conducted a retrospective chart review of electronic medical records at Yale New Haven Hospital (New Haven, Connecticut) from 2013 to 2023, identifying patients receiving antineoplastic therapy who were diagnosed with a SCAR. Cases were identified through a search of the electronic medical record performed by the joint data analytics team using the keywords DRESS, SJS, TEN, AGEP, and generalized bullous fixed drug eruption, along with spelling variations (both abbreviations and full terms), in addition to manual review by one of the authors (K.V.) of the inpatient dermatology consultation log and dermatopathology database. Only patients for whom an antineoplastic agent was identified as a high-probability culprit by the dermatology and/or oncology teams were included.
In total, 20 patients (11 female, 9 male) were identified as having an antineoplastic-associated SCAR. All patients had metastatic or advanced disease. We identified 2 (10%) cases of AGEP, 16 (80%) cases of DRESS, and 3 (15%) cases of SJS/TEN. One patient on immunotherapy had 2 distinct SCARs (AGEP, DRESS) at different time points. Table 1 describes patient and SCAR characteristics as well as impact on cancer treatment. The median (interquartile range [IQR]) latency period for AGEP was 7.5 (4-11) days. The median (IQR) latency period for 13 of the 16 (81%) DRESS cases was 14 (10-32) days. For 3 DRESS cases with a potential second-hit phenomenon in the setting of current or antecedent immunotherapy,5 the median (IQR) latency period was 122 (96-426) days for the immunotherapy drug and 28 (21-52) days for the drug culprit. The median (IQR) latency period for SJS/TEN was 23 (20-27) days.
Patients received treatment with combination systemic corticosteroids and topical corticosteroids in 13 (65%) cases, systemic corticosteroid monotherapy in 6 (30%) cases, or combination systemic corticosteroids and etanercept in 1 (5%) case. All patients experienced resolution of the SCAR and survived to hospital discharge. Most (17/20 [85%]) patients experienced interruption or discontinuation of cancer treatment. Table 2 describes the implicated antineoplastic therapies, which included chemotherapy (3 DRESS, 1 SJS/TEN), hormonal therapy (1 DRESS), immunotherapy (1 AGEP, 4 DRESS), and targeted therapy (1 AGEP, 8 DRESS, 2 SJS/TEN).
Limitations of this study include the retrospective study design, the small sample size, and the challenge of drug culprit identification in oncologic patients on multiple high-probability medications.
Though rare, SCARs can be encountered in patients on antineoplastic therapy with a wide range of drug culprits. In our cohort, SCARs occurred with various antineoplastic agents, including chemotherapy, hormonal therapy, immunotherapy, and targeted therapy. The most common antineoplastic-associated SCAR was DRESS, which had the widest latency period in the setting of a potential second-hit phenomenon with another drug culprit. Although we did not observe any cases of SJS/TEN in the immunotherapy category, it is important to consider progressive immunotherapy-related mucocutaneous eruption in the differential diagnosis. Fortunately, all patients survived to hospital discharge and experienced SCAR resolution with systemic treatment; however, most patients experienced interruption of cancer therapy, which has the potential to affect oncologic outcomes. This interruption is not uncommon, as rechallenge of an antineoplastic agent in patients with a therapy-related SCAR generally is not recommended. The awareness and prompt management of SCARs in a patient on treatment for malignancy are critical in order to minimize negative outcomes in this vulnerable patient population.
To the Editor:
Severe cutaneous adverse reactions (SCARs) are rare, life-threatening reactions that include acute generalized exanthematous pustulosis (AGEP), drug reaction with eosinophilia and systemic symptoms (DRESS), and Stevens-Johnson syndrome (SJS)/toxic epidermal necrolysis (TEN).1 In addition to being associated with commonly implicated medications, SCARs also may occur in the setting of antineoplastic therapy.2,3 Although antineoplastic-associated SCARs have been described, diagnosis can be difficult due to varying latency periods and atypical clinical features, such as those observed with BRAF inhibitor–related DRESS during immunotherapy.4 Severe cutaneous adverse reactions can increase morbidity and mortality in the oncologic patient population due to both the clinical sequelae from the cutaneous reaction and the potential to interrupt cancer treatment.
The aim of this study was to evaluate the clinical characteristics, outcomes, and impact on cancer treatment among patients diagnosed with a SCAR while receiving active therapy for malignancy. We conducted a retrospective chart review of electronic medical records at Yale New Haven Hospital (New Haven, Connecticut) from 2013 to 2023, identifying patients receiving antineoplastic therapy who were diagnosed with a SCAR. Cases were identified through a search of the electronic medical record performed by the joint data analytics team using the keywords DRESS, SJS, TEN, AGEP, and generalized bullous fixed drug eruption, along with spelling variations (both abbreviations and full terms), in addition to manual review by one of the authors (K.V.) of the inpatient dermatology consultation log and dermatopathology database. Only patients for whom an antineoplastic agent was identified as a high-probability culprit by the dermatology and/or oncology teams were included.
In total, 20 patients (11 female, 9 male) were identified as having an antineoplastic-associated SCAR. All patients had metastatic or advanced disease. We identified 2 (10%) cases of AGEP, 16 (80%) cases of DRESS, and 3 (15%) cases of SJS/TEN. One patient on immunotherapy had 2 distinct SCARs (AGEP, DRESS) at different time points. Table 1 describes patient and SCAR characteristics as well as impact on cancer treatment. The median (interquartile range [IQR]) latency period for AGEP was 7.5 (4-11) days. The median (IQR) latency period for 13 of the 16 (81%) DRESS cases was 14 (10-32) days. For 3 DRESS cases with a potential second-hit phenomenon in the setting of current or antecedent immunotherapy,5 the median (IQR) latency period was 122 (96-426) days for the immunotherapy drug and 28 (21-52) days for the drug culprit. The median (IQR) latency period for SJS/TEN was 23 (20-27) days.
Patients received treatment with combination systemic corticosteroids and topical corticosteroids in 13 (65%) cases, systemic corticosteroid monotherapy in 6 (30%) cases, or combination systemic corticosteroids and etanercept in 1 (5%) case. All patients experienced resolution of the SCAR and survived to hospital discharge. Most (17/20 [85%]) patients experienced interruption or discontinuation of cancer treatment. Table 2 describes the implicated antineoplastic therapies, which included chemotherapy (3 DRESS, 1 SJS/TEN), hormonal therapy (1 DRESS), immunotherapy (1 AGEP, 4 DRESS), and targeted therapy (1 AGEP, 8 DRESS, 2 SJS/TEN).
Limitations of this study include the retrospective study design, the small sample size, and the challenge of drug culprit identification in oncologic patients on multiple high-probability medications.
Though rare, SCARs can be encountered in patients on antineoplastic therapy with a wide range of drug culprits. In our cohort, SCARs occurred with various antineoplastic agents, including chemotherapy, hormonal therapy, immunotherapy, and targeted therapy. The most common antineoplastic-associated SCAR was DRESS, which had the widest latency period in the setting of a potential second-hit phenomenon with another drug culprit. Although we did not observe any cases of SJS/TEN in the immunotherapy category, it is important to consider progressive immunotherapy-related mucocutaneous eruption in the differential diagnosis. Fortunately, all patients survived to hospital discharge and experienced SCAR resolution with systemic treatment; however, most patients experienced interruption of cancer therapy, which has the potential to affect oncologic outcomes. This interruption is not uncommon, as rechallenge of an antineoplastic agent in patients with a therapy-related SCAR generally is not recommended. The awareness and prompt management of SCARs in a patient on treatment for malignancy are critical in order to minimize negative outcomes in this vulnerable patient population.
- Duong TA, Valeyrie-Allanore L, Wolkenstein P, et al. Severe cutaneous adverse reactions to drugs. Lancet. 2017;390: 1996-2011.
- Chen CB, Wu MY, Ng CY, et al. Severe cutaneous adverse reactions induced by targeted anticancer therapies and immunotherapies. Cancer Manag Res. 2018;10:1259-1273.
- Ng CY, Chen CB, Wu MY, et al. Anticancer drugs induced severe adverse cutaneous drug reactions: an updated review on the risks associated with anticancer targeted therapy or immunotherapies. J Immunol Res. 2018;2018:5376476.
- Maloney NJ, Rana J, Yang JJ, et al. Clinical features of druginduced hypersensitivity syndrome to BRAF inhibitors with and without previous immune checkpoint inhibition: a review. Support Care Cancer. 2022;30:2839-2851.
- Hammond S, Olsson-Brown A, Grice S, et al. Does immune checkpoint inhibitor therapy increase the frequency of adverse reactions to concomitant medications? Clin Exp Allergy. 2022;52:600-603.
- Duong TA, Valeyrie-Allanore L, Wolkenstein P, et al. Severe cutaneous adverse reactions to drugs. Lancet. 2017;390: 1996-2011.
- Chen CB, Wu MY, Ng CY, et al. Severe cutaneous adverse reactions induced by targeted anticancer therapies and immunotherapies. Cancer Manag Res. 2018;10:1259-1273.
- Ng CY, Chen CB, Wu MY, et al. Anticancer drugs induced severe adverse cutaneous drug reactions: an updated review on the risks associated with anticancer targeted therapy or immunotherapies. J Immunol Res. 2018;2018:5376476.
- Maloney NJ, Rana J, Yang JJ, et al. Clinical features of druginduced hypersensitivity syndrome to BRAF inhibitors with and without previous immune checkpoint inhibition: a review. Support Care Cancer. 2022;30:2839-2851.
- Hammond S, Olsson-Brown A, Grice S, et al. Does immune checkpoint inhibitor therapy increase the frequency of adverse reactions to concomitant medications? Clin Exp Allergy. 2022;52:600-603.
Severe Cutaneous Adverse Reactions in the Setting of Antineoplastic Therapy: A Single-Institution Retrospective Study
Severe Cutaneous Adverse Reactions in the Setting of Antineoplastic Therapy: A Single-Institution Retrospective Study
Practice Points
- Clinicians should be aware of the occurrence of severe cutaneous adverse reactions (SCARs) in patients on antineoplastic therapy to prevent delays in treatment and improve patient outcomes.
- Rapid initiation of treatment can be effective in resolving SCARs and ensuring full recovery.
- Close coordination between dermatology and oncology teams is crucial to manage SCARs while minimizing cancer treatment interruptions.
Adverse Events Associated With At-Home Microcurrent Facial Devices
Adverse Events Associated With At-Home Microcurrent Facial Devices
To the Editor:
At-home microcurrent facial devices have gained rapid popularity for cosmetic rejuvenation, promising improvements in skin tone, contour, and collagen production.¹ In particular, the post–COVID-19 era has seen a surge in at-home beauty practices driven by social media influence, with the global microcurrent facial market estimated at $372.9 million in 2022 and projected to grow at a compound annual growth rate of 7.3% through 2030.1 Microcurrent devices deliver low-level electrical currents to the skin and underlying muscles. Given the limited exploration of the long-term safety, we aimed to collate existing data and identify trends in reports of adverse events (AEs) associated with these microcurrent devices.
On April 15, 2025, the US Food and Drug Administration’s Manufacturer and User Facility Device Experience (MAUDE) database was queried for medical device reports from January 1, 2013, through March 31, 2025, using product names and keywords including NuFACE, TheraFace, FOREO, and microcurrent device. Search terms were limited to brands for which complaint data existed in the MAUDE database at the time of query. To ensure accuracy, reports were manually reviewed to eliminate duplicates and irrelevant entries.
A total of 28 unique AE reports associated with at-home microcurrent devices were identified (eTable). The majority involved NuFACE devices (ie, NuFACE Trinity, NuFACE Mini, and NuFACE Trinity+)(NuFACE)(n=25), followed by the TheraFace PRO (Therabody, Inc)(n=2) and the FOREO BEAR (FOREO)(n=1). The most frequently documented AEs associated with the NuFACE devices included arrhythmia (7/25 [28%]), pain (6/25 [24%]), dizziness (4/25 [16%]), headache (4/25 [16%]), and inflammation (4/25 [16%]). There was 1 (4%) case of retinal detachment. The TheraFace PRO was associated with device overheating (2/2 [100%]), and the FOREO BEAR was associated with facial deformity/disfigurement (1/1 [100%]).

While microcurrent therapy is widely marketed to consumers through social media influencers and at-home beauty platforms,1 randomized controlled trials (RCTs) evaluating AEs related to use of this technology are lacking, possibly due to nonstringent regulation of nonprescription cosmetic devices.² Contrary to our findings, RCTs of microcurrent devices have reported minimal or no AEs; for instance, an RCT evaluating 56 participants treated 5 times weekly for 12 weeks with a microcurrent device that was not included in our analysis reported only mild erythema in all experimental group participants.2 In another RCT of 30 participants, 15 of whom were treated with a microcurrent device and 15 with placebo for 30 minutes once daily over a period of 10 days, no AEs were reported.3 A cohort analysis of 34 patients also provided preliminary evidence supporting the use of microcurrent therapy for chronic back and neck pain, beyond its cosmetic applications.4 Despite the lack of reported AEs in the literature, there is a notable absence of large-scale, rigorous studies on this topic.
Our analysis was subject to the limitations of the MAUDE database, in which reports of severe AEs are more likely to be reported than transient ones. Additionally, the small sample size and lack of a known denominator make it difficult to compare frequencies of AEs among different microcurrent tools. The products chosen for this study were the select few that reported complaint data, but there is a large existing market of devices that may be associated with AEs that have yet to be reported, potentially because of their novelty.
Our findings suggest that, despite their over-the-counter availability, microcurrent facial devices may carry major risks—particularly in at-home settings. While short-term studies have highlighted potential benefits, the small sample sizes and limited follow-up make it difficult to comprehensively characterize long-term safety risks. Among available studies on microcurrent beauty treatments, the longest follow-up was only 12 weeks.2 Our findings support the need for further large-scale and longitudinal studies to evaluate both the efficacy and safety of at-home microcurrent therapy, especially with increasing consumer interest. The diversity of the products available adds to the challenge of broad safety guidelines, in addition to the lack of long-term clinical studies.
- Microcurrent Facial Market Size, Growth & Trends Report 2030. Grand View Research. Published 2023. Accessed March 3, 2026. https://www.grandviewresearch.com/industry-analysis/microcurrent-facial-market-report
- Bu P, Duan R, Luo J, et al. Development of home beauty devices for facial rejuvenation: establishment of efficacy evaluation system. Clin Cosmet Investig Dermatol. 2024;17:553-563.
- Jain S, Arora M. Effect of microcurrent facial muscle toning on fine wrinkles & firmness of face. IAMR J Physiother. 2012;1:13-19.
- Armstrong K, Gokal R, Chevalier A, et al. Microcurrent point stimulation applied to lower back acupuncture points for the treatment of nonspecific neck pain. J Altern Complement Med. 2017;23:295-299.
To the Editor:
At-home microcurrent facial devices have gained rapid popularity for cosmetic rejuvenation, promising improvements in skin tone, contour, and collagen production.¹ In particular, the post–COVID-19 era has seen a surge in at-home beauty practices driven by social media influence, with the global microcurrent facial market estimated at $372.9 million in 2022 and projected to grow at a compound annual growth rate of 7.3% through 2030.1 Microcurrent devices deliver low-level electrical currents to the skin and underlying muscles. Given the limited exploration of the long-term safety, we aimed to collate existing data and identify trends in reports of adverse events (AEs) associated with these microcurrent devices.
On April 15, 2025, the US Food and Drug Administration’s Manufacturer and User Facility Device Experience (MAUDE) database was queried for medical device reports from January 1, 2013, through March 31, 2025, using product names and keywords including NuFACE, TheraFace, FOREO, and microcurrent device. Search terms were limited to brands for which complaint data existed in the MAUDE database at the time of query. To ensure accuracy, reports were manually reviewed to eliminate duplicates and irrelevant entries.
A total of 28 unique AE reports associated with at-home microcurrent devices were identified (eTable). The majority involved NuFACE devices (ie, NuFACE Trinity, NuFACE Mini, and NuFACE Trinity+)(NuFACE)(n=25), followed by the TheraFace PRO (Therabody, Inc)(n=2) and the FOREO BEAR (FOREO)(n=1). The most frequently documented AEs associated with the NuFACE devices included arrhythmia (7/25 [28%]), pain (6/25 [24%]), dizziness (4/25 [16%]), headache (4/25 [16%]), and inflammation (4/25 [16%]). There was 1 (4%) case of retinal detachment. The TheraFace PRO was associated with device overheating (2/2 [100%]), and the FOREO BEAR was associated with facial deformity/disfigurement (1/1 [100%]).
While microcurrent therapy is widely marketed to consumers through social media influencers and at-home beauty platforms,1 randomized controlled trials (RCTs) evaluating AEs related to use of this technology are lacking, possibly due to nonstringent regulation of nonprescription cosmetic devices.² Contrary to our findings, RCTs of microcurrent devices have reported minimal or no AEs; for instance, an RCT evaluating 56 participants treated 5 times weekly for 12 weeks with a microcurrent device that was not included in our analysis reported only mild erythema in all experimental group participants.2 In another RCT of 30 participants, 15 of whom were treated with a microcurrent device and 15 with placebo for 30 minutes once daily over a period of 10 days, no AEs were reported.3 A cohort analysis of 34 patients also provided preliminary evidence supporting the use of microcurrent therapy for chronic back and neck pain, beyond its cosmetic applications.4 Despite the lack of reported AEs in the literature, there is a notable absence of large-scale, rigorous studies on this topic.
Our analysis was subject to the limitations of the MAUDE database, in which reports of severe AEs are more likely to be reported than transient ones. Additionally, the small sample size and lack of a known denominator make it difficult to compare frequencies of AEs among different microcurrent tools. The products chosen for this study were the select few that reported complaint data, but there is a large existing market of devices that may be associated with AEs that have yet to be reported, potentially because of their novelty.
Our findings suggest that, despite their over-the-counter availability, microcurrent facial devices may carry major risks—particularly in at-home settings. While short-term studies have highlighted potential benefits, the small sample sizes and limited follow-up make it difficult to comprehensively characterize long-term safety risks. Among available studies on microcurrent beauty treatments, the longest follow-up was only 12 weeks.2 Our findings support the need for further large-scale and longitudinal studies to evaluate both the efficacy and safety of at-home microcurrent therapy, especially with increasing consumer interest. The diversity of the products available adds to the challenge of broad safety guidelines, in addition to the lack of long-term clinical studies.
To the Editor:
At-home microcurrent facial devices have gained rapid popularity for cosmetic rejuvenation, promising improvements in skin tone, contour, and collagen production.¹ In particular, the post–COVID-19 era has seen a surge in at-home beauty practices driven by social media influence, with the global microcurrent facial market estimated at $372.9 million in 2022 and projected to grow at a compound annual growth rate of 7.3% through 2030.1 Microcurrent devices deliver low-level electrical currents to the skin and underlying muscles. Given the limited exploration of the long-term safety, we aimed to collate existing data and identify trends in reports of adverse events (AEs) associated with these microcurrent devices.
On April 15, 2025, the US Food and Drug Administration’s Manufacturer and User Facility Device Experience (MAUDE) database was queried for medical device reports from January 1, 2013, through March 31, 2025, using product names and keywords including NuFACE, TheraFace, FOREO, and microcurrent device. Search terms were limited to brands for which complaint data existed in the MAUDE database at the time of query. To ensure accuracy, reports were manually reviewed to eliminate duplicates and irrelevant entries.
A total of 28 unique AE reports associated with at-home microcurrent devices were identified (eTable). The majority involved NuFACE devices (ie, NuFACE Trinity, NuFACE Mini, and NuFACE Trinity+)(NuFACE)(n=25), followed by the TheraFace PRO (Therabody, Inc)(n=2) and the FOREO BEAR (FOREO)(n=1). The most frequently documented AEs associated with the NuFACE devices included arrhythmia (7/25 [28%]), pain (6/25 [24%]), dizziness (4/25 [16%]), headache (4/25 [16%]), and inflammation (4/25 [16%]). There was 1 (4%) case of retinal detachment. The TheraFace PRO was associated with device overheating (2/2 [100%]), and the FOREO BEAR was associated with facial deformity/disfigurement (1/1 [100%]).
While microcurrent therapy is widely marketed to consumers through social media influencers and at-home beauty platforms,1 randomized controlled trials (RCTs) evaluating AEs related to use of this technology are lacking, possibly due to nonstringent regulation of nonprescription cosmetic devices.² Contrary to our findings, RCTs of microcurrent devices have reported minimal or no AEs; for instance, an RCT evaluating 56 participants treated 5 times weekly for 12 weeks with a microcurrent device that was not included in our analysis reported only mild erythema in all experimental group participants.2 In another RCT of 30 participants, 15 of whom were treated with a microcurrent device and 15 with placebo for 30 minutes once daily over a period of 10 days, no AEs were reported.3 A cohort analysis of 34 patients also provided preliminary evidence supporting the use of microcurrent therapy for chronic back and neck pain, beyond its cosmetic applications.4 Despite the lack of reported AEs in the literature, there is a notable absence of large-scale, rigorous studies on this topic.
Our analysis was subject to the limitations of the MAUDE database, in which reports of severe AEs are more likely to be reported than transient ones. Additionally, the small sample size and lack of a known denominator make it difficult to compare frequencies of AEs among different microcurrent tools. The products chosen for this study were the select few that reported complaint data, but there is a large existing market of devices that may be associated with AEs that have yet to be reported, potentially because of their novelty.
Our findings suggest that, despite their over-the-counter availability, microcurrent facial devices may carry major risks—particularly in at-home settings. While short-term studies have highlighted potential benefits, the small sample sizes and limited follow-up make it difficult to comprehensively characterize long-term safety risks. Among available studies on microcurrent beauty treatments, the longest follow-up was only 12 weeks.2 Our findings support the need for further large-scale and longitudinal studies to evaluate both the efficacy and safety of at-home microcurrent therapy, especially with increasing consumer interest. The diversity of the products available adds to the challenge of broad safety guidelines, in addition to the lack of long-term clinical studies.
- Microcurrent Facial Market Size, Growth & Trends Report 2030. Grand View Research. Published 2023. Accessed March 3, 2026. https://www.grandviewresearch.com/industry-analysis/microcurrent-facial-market-report
- Bu P, Duan R, Luo J, et al. Development of home beauty devices for facial rejuvenation: establishment of efficacy evaluation system. Clin Cosmet Investig Dermatol. 2024;17:553-563.
- Jain S, Arora M. Effect of microcurrent facial muscle toning on fine wrinkles & firmness of face. IAMR J Physiother. 2012;1:13-19.
- Armstrong K, Gokal R, Chevalier A, et al. Microcurrent point stimulation applied to lower back acupuncture points for the treatment of nonspecific neck pain. J Altern Complement Med. 2017;23:295-299.
- Microcurrent Facial Market Size, Growth & Trends Report 2030. Grand View Research. Published 2023. Accessed March 3, 2026. https://www.grandviewresearch.com/industry-analysis/microcurrent-facial-market-report
- Bu P, Duan R, Luo J, et al. Development of home beauty devices for facial rejuvenation: establishment of efficacy evaluation system. Clin Cosmet Investig Dermatol. 2024;17:553-563.
- Jain S, Arora M. Effect of microcurrent facial muscle toning on fine wrinkles & firmness of face. IAMR J Physiother. 2012;1:13-19.
- Armstrong K, Gokal R, Chevalier A, et al. Microcurrent point stimulation applied to lower back acupuncture points for the treatment of nonspecific neck pain. J Altern Complement Med. 2017;23:295-299.
Adverse Events Associated With At-Home Microcurrent Facial Devices
Adverse Events Associated With At-Home Microcurrent Facial Devices
PRACTICE POINTS
- At-home microcurrent facial devices have been associated with serious adverse events, including arrhythmia, pain, dizziness, and retinal detachment, based on US Food and Drug Administration Manufacturer and User Facility Device Experience database reports, underscoring the importance of counseling patients about potential risks prior to use.
- Existing randomized controlled trials of microcurrent devices are limited by small sample sizes and short follow-up periods (maximum 12 weeks), making it difficult to characterize the long-term safety profile of these increasingly popular devices.
- Dermatologists should be aware that the largely unregulated at-home microcurrent device market lacks robust, large-scale safety data. Patients, particularly those with cardiac conditions or implanted electrical devices, should be advised to consult a physician before use.

Optimizing Patch Testing in Clinical Practice: Insights From Amber Reck Atwater, MD
Optimizing Patch Testing in Clinical Practice: Insights From Amber Reck Atwater, MD
What evidence exists on the impact of systemic immunosuppressants, biologics, and small-molecule inhibitors on patch test sensitivity and specificity?
DR. ATWATER: Guidance on this topic recently was published by the North American Contact Dermatitis Group (NACDG) in the Journal of the American Academy of Dermatology in June 2025. The article outlined expert recommendations on whether systemic immunosuppressants, biologics, and small-molecule inhibitors should be held before patch testing, how long they should be withheld, and the maximum recommended doses that can be used during testing.
How can dermatologists perform patch testing or use alternative diagnostic strategies when systemic therapy cannot safely be withheld?
DR. ATWATER: When systemic therapy cannot safely be withheld and patch testing is needed for diagnostic purposes, I typically proceed with the understanding that there is risk of false-negative reactions. If the patient has dermatitis on systemic therapy, it suggests that an allergic response on patch testing is also possible. I generally follow the NACDG guidelines mentioned above, and I hold systemic medications during the week of testing, when possible. If the patient has diffuse dermatitis on systemic therapy and their skin is not clear enough to proceed, I prescribe a prednisone taper and patch test on 10 mg per day for the entire week of testing. In patients taking systemic medications, I typically consider doubtful (+/−) patch test reactions to be the equivalent of a positive (1+) reaction.
One alternative diagnostic strategy is to create a safe list that avoids common allergens and have the patient use only products on this list. If their skin clears with avoidance, it suggests that they may have a contact allergy, and you can proceed with patch testing.
In patients with a convincing history of contact dermatitis but a negative patch test, what are the most common causes of false negatives, and how do you distinguish those from true negatives?
DR. ATWATER: In this setting, the most common cause of a false-negative patch test is not testing the correct allergens. This may occur when too few allergens are tested or when relevant allergens are not tested. Other potential causes of false negatives are incorrect timing of allergen exposure and readings, inadequate allergen adherence, expired allergens, and testing with the incorrect vehicle or concentration of allergen. Some immunosuppressant medications also can cause a false-negative patch test. The only way to distinguish false negatives from true negatives is to be aware of these potential pitfalls and continuously work to avoid errors whenever possible.
What technical and practical factors most influence false negatives/positives, and what steps do you recommend to standardize and improve test yield?
DR. ATWATER: Not testing the correct allergens is a potential pitfall in patch testing. For example, when comparing the 35 allergens in the T.R.U.E. Test (thin layer rapid-use epicutaneous test) to the 80 allergens tested by the NACDG in 2021 to 2022, up to 48% of NACDG allergens are missed when testing with only the T.R.U.E. Test. This argues for comprehensive patch testing and testing of at least 80 to 90 allergens whenever possible. Another example is failure to test allergens relevant to occupational exposures, such as in the case of a hairdresser or nail technician. When patches are not applied for the correct period (48 hours) and the final reading isn’t completed in the recommended timeframe (96 to 168 hours), there is increased risk for false negatives and positives. Both false negatives and positives can occur if you complete your final reading too early, whereas false negatives may be more likely if you complete your final reading late. Poor allergen adherence, which can be caused by hair, sweat, poor tape application, water, and exercise, also can result in false negatives. Allergen concentration that is too low to elicit a reaction, as well as too little allergen placed into the chamber, also could increase the risk of false negatives. Too much allergen in the chamber or too high a concentration of the allergen can result in false positives.
Topical medications applied to the patch test site prior to or during patch testing, as well as phototherapy, sun exposure, intramuscular triamcinolone, immunosuppressants, biologics, and small-molecule inhibitors can suppress the immune response to allergen exposure.
To identify true positive patch test reactions, use side lighting and palpate the skin. Be aware of the appearance of irritant reactions, patch test reaction variants such as follicular reactions, and the poral reaction, which can be seen with cobalt. Strong knowledge of how to read patch test reactions will decrease your risk of false-positive and -negative reactions. Training and protocols are vital for standardization and accurate patch testing. We train our staff on the technicalities of patch testing and utilize patch test orders and checklists in our office. We take photos to confirm application sites and visually track reactions between visits. We also provide verbal and written patch test care instructions for our patients and reinforce instructions at each clinic visit.
What are your top practical tips for dermatologists to maximize diagnostic accuracy and patient safety?
DR. ATWATER: My first tip is to develop patch test protocols that are followed by staff and physicians—every time—for every patient. My second tip is to make sure you understand and are comfortable with the patch test process. There are several great patch test resources that can help, including Introduction to Patch Testing, a recently developed CME module in the AAD Learning Center (https://learning.aad.org/Listing/Introduction-to-Patch- Testing-20366). There also are patch test training courses and other resources offered by the American Contact Dermatitis Society (https://www.contactderm.org/).
What single guideline change would most improve patch testing?
DR. ATWATER: The single guideline change that would most improve the practice of patch testing is removal of payer limitations on the number of patches that can be applied per day. For many payers in the United States, this limit is 80 to 90 allergens, and it is sometimes lower. Limits on the number of allergens that can be applied per day may result in false-negative patch testing (when limits are applied), patient inconvenience (if testing is completed over 2 different application days), and insufficient reimbursement (if more allergens are tested than the limit allows).
What evidence exists on the impact of systemic immunosuppressants, biologics, and small-molecule inhibitors on patch test sensitivity and specificity?
DR. ATWATER: Guidance on this topic recently was published by the North American Contact Dermatitis Group (NACDG) in the Journal of the American Academy of Dermatology in June 2025. The article outlined expert recommendations on whether systemic immunosuppressants, biologics, and small-molecule inhibitors should be held before patch testing, how long they should be withheld, and the maximum recommended doses that can be used during testing.
How can dermatologists perform patch testing or use alternative diagnostic strategies when systemic therapy cannot safely be withheld?
DR. ATWATER: When systemic therapy cannot safely be withheld and patch testing is needed for diagnostic purposes, I typically proceed with the understanding that there is risk of false-negative reactions. If the patient has dermatitis on systemic therapy, it suggests that an allergic response on patch testing is also possible. I generally follow the NACDG guidelines mentioned above, and I hold systemic medications during the week of testing, when possible. If the patient has diffuse dermatitis on systemic therapy and their skin is not clear enough to proceed, I prescribe a prednisone taper and patch test on 10 mg per day for the entire week of testing. In patients taking systemic medications, I typically consider doubtful (+/−) patch test reactions to be the equivalent of a positive (1+) reaction.
One alternative diagnostic strategy is to create a safe list that avoids common allergens and have the patient use only products on this list. If their skin clears with avoidance, it suggests that they may have a contact allergy, and you can proceed with patch testing.
In patients with a convincing history of contact dermatitis but a negative patch test, what are the most common causes of false negatives, and how do you distinguish those from true negatives?
DR. ATWATER: In this setting, the most common cause of a false-negative patch test is not testing the correct allergens. This may occur when too few allergens are tested or when relevant allergens are not tested. Other potential causes of false negatives are incorrect timing of allergen exposure and readings, inadequate allergen adherence, expired allergens, and testing with the incorrect vehicle or concentration of allergen. Some immunosuppressant medications also can cause a false-negative patch test. The only way to distinguish false negatives from true negatives is to be aware of these potential pitfalls and continuously work to avoid errors whenever possible.
What technical and practical factors most influence false negatives/positives, and what steps do you recommend to standardize and improve test yield?
DR. ATWATER: Not testing the correct allergens is a potential pitfall in patch testing. For example, when comparing the 35 allergens in the T.R.U.E. Test (thin layer rapid-use epicutaneous test) to the 80 allergens tested by the NACDG in 2021 to 2022, up to 48% of NACDG allergens are missed when testing with only the T.R.U.E. Test. This argues for comprehensive patch testing and testing of at least 80 to 90 allergens whenever possible. Another example is failure to test allergens relevant to occupational exposures, such as in the case of a hairdresser or nail technician. When patches are not applied for the correct period (48 hours) and the final reading isn’t completed in the recommended timeframe (96 to 168 hours), there is increased risk for false negatives and positives. Both false negatives and positives can occur if you complete your final reading too early, whereas false negatives may be more likely if you complete your final reading late. Poor allergen adherence, which can be caused by hair, sweat, poor tape application, water, and exercise, also can result in false negatives. Allergen concentration that is too low to elicit a reaction, as well as too little allergen placed into the chamber, also could increase the risk of false negatives. Too much allergen in the chamber or too high a concentration of the allergen can result in false positives.
Topical medications applied to the patch test site prior to or during patch testing, as well as phototherapy, sun exposure, intramuscular triamcinolone, immunosuppressants, biologics, and small-molecule inhibitors can suppress the immune response to allergen exposure.
To identify true positive patch test reactions, use side lighting and palpate the skin. Be aware of the appearance of irritant reactions, patch test reaction variants such as follicular reactions, and the poral reaction, which can be seen with cobalt. Strong knowledge of how to read patch test reactions will decrease your risk of false-positive and -negative reactions. Training and protocols are vital for standardization and accurate patch testing. We train our staff on the technicalities of patch testing and utilize patch test orders and checklists in our office. We take photos to confirm application sites and visually track reactions between visits. We also provide verbal and written patch test care instructions for our patients and reinforce instructions at each clinic visit.
What are your top practical tips for dermatologists to maximize diagnostic accuracy and patient safety?
DR. ATWATER: My first tip is to develop patch test protocols that are followed by staff and physicians—every time—for every patient. My second tip is to make sure you understand and are comfortable with the patch test process. There are several great patch test resources that can help, including Introduction to Patch Testing, a recently developed CME module in the AAD Learning Center (https://learning.aad.org/Listing/Introduction-to-Patch- Testing-20366). There also are patch test training courses and other resources offered by the American Contact Dermatitis Society (https://www.contactderm.org/).
What single guideline change would most improve patch testing?
DR. ATWATER: The single guideline change that would most improve the practice of patch testing is removal of payer limitations on the number of patches that can be applied per day. For many payers in the United States, this limit is 80 to 90 allergens, and it is sometimes lower. Limits on the number of allergens that can be applied per day may result in false-negative patch testing (when limits are applied), patient inconvenience (if testing is completed over 2 different application days), and insufficient reimbursement (if more allergens are tested than the limit allows).
What evidence exists on the impact of systemic immunosuppressants, biologics, and small-molecule inhibitors on patch test sensitivity and specificity?
DR. ATWATER: Guidance on this topic recently was published by the North American Contact Dermatitis Group (NACDG) in the Journal of the American Academy of Dermatology in June 2025. The article outlined expert recommendations on whether systemic immunosuppressants, biologics, and small-molecule inhibitors should be held before patch testing, how long they should be withheld, and the maximum recommended doses that can be used during testing.
How can dermatologists perform patch testing or use alternative diagnostic strategies when systemic therapy cannot safely be withheld?
DR. ATWATER: When systemic therapy cannot safely be withheld and patch testing is needed for diagnostic purposes, I typically proceed with the understanding that there is risk of false-negative reactions. If the patient has dermatitis on systemic therapy, it suggests that an allergic response on patch testing is also possible. I generally follow the NACDG guidelines mentioned above, and I hold systemic medications during the week of testing, when possible. If the patient has diffuse dermatitis on systemic therapy and their skin is not clear enough to proceed, I prescribe a prednisone taper and patch test on 10 mg per day for the entire week of testing. In patients taking systemic medications, I typically consider doubtful (+/−) patch test reactions to be the equivalent of a positive (1+) reaction.
One alternative diagnostic strategy is to create a safe list that avoids common allergens and have the patient use only products on this list. If their skin clears with avoidance, it suggests that they may have a contact allergy, and you can proceed with patch testing.
In patients with a convincing history of contact dermatitis but a negative patch test, what are the most common causes of false negatives, and how do you distinguish those from true negatives?
DR. ATWATER: In this setting, the most common cause of a false-negative patch test is not testing the correct allergens. This may occur when too few allergens are tested or when relevant allergens are not tested. Other potential causes of false negatives are incorrect timing of allergen exposure and readings, inadequate allergen adherence, expired allergens, and testing with the incorrect vehicle or concentration of allergen. Some immunosuppressant medications also can cause a false-negative patch test. The only way to distinguish false negatives from true negatives is to be aware of these potential pitfalls and continuously work to avoid errors whenever possible.
What technical and practical factors most influence false negatives/positives, and what steps do you recommend to standardize and improve test yield?
DR. ATWATER: Not testing the correct allergens is a potential pitfall in patch testing. For example, when comparing the 35 allergens in the T.R.U.E. Test (thin layer rapid-use epicutaneous test) to the 80 allergens tested by the NACDG in 2021 to 2022, up to 48% of NACDG allergens are missed when testing with only the T.R.U.E. Test. This argues for comprehensive patch testing and testing of at least 80 to 90 allergens whenever possible. Another example is failure to test allergens relevant to occupational exposures, such as in the case of a hairdresser or nail technician. When patches are not applied for the correct period (48 hours) and the final reading isn’t completed in the recommended timeframe (96 to 168 hours), there is increased risk for false negatives and positives. Both false negatives and positives can occur if you complete your final reading too early, whereas false negatives may be more likely if you complete your final reading late. Poor allergen adherence, which can be caused by hair, sweat, poor tape application, water, and exercise, also can result in false negatives. Allergen concentration that is too low to elicit a reaction, as well as too little allergen placed into the chamber, also could increase the risk of false negatives. Too much allergen in the chamber or too high a concentration of the allergen can result in false positives.
Topical medications applied to the patch test site prior to or during patch testing, as well as phototherapy, sun exposure, intramuscular triamcinolone, immunosuppressants, biologics, and small-molecule inhibitors can suppress the immune response to allergen exposure.
To identify true positive patch test reactions, use side lighting and palpate the skin. Be aware of the appearance of irritant reactions, patch test reaction variants such as follicular reactions, and the poral reaction, which can be seen with cobalt. Strong knowledge of how to read patch test reactions will decrease your risk of false-positive and -negative reactions. Training and protocols are vital for standardization and accurate patch testing. We train our staff on the technicalities of patch testing and utilize patch test orders and checklists in our office. We take photos to confirm application sites and visually track reactions between visits. We also provide verbal and written patch test care instructions for our patients and reinforce instructions at each clinic visit.
What are your top practical tips for dermatologists to maximize diagnostic accuracy and patient safety?
DR. ATWATER: My first tip is to develop patch test protocols that are followed by staff and physicians—every time—for every patient. My second tip is to make sure you understand and are comfortable with the patch test process. There are several great patch test resources that can help, including Introduction to Patch Testing, a recently developed CME module in the AAD Learning Center (https://learning.aad.org/Listing/Introduction-to-Patch- Testing-20366). There also are patch test training courses and other resources offered by the American Contact Dermatitis Society (https://www.contactderm.org/).
What single guideline change would most improve patch testing?
DR. ATWATER: The single guideline change that would most improve the practice of patch testing is removal of payer limitations on the number of patches that can be applied per day. For many payers in the United States, this limit is 80 to 90 allergens, and it is sometimes lower. Limits on the number of allergens that can be applied per day may result in false-negative patch testing (when limits are applied), patient inconvenience (if testing is completed over 2 different application days), and insufficient reimbursement (if more allergens are tested than the limit allows).
Optimizing Patch Testing in Clinical Practice: Insights From Amber Reck Atwater, MD
Optimizing Patch Testing in Clinical Practice: Insights From Amber Reck Atwater, MD

A Hybrid Suture Technique: Suture Modification With Dental Roll Insertion
A Hybrid Suture Technique: Suture Modification With Dental Roll Insertion
Practice Gap
If not cared for properly, epidermal suture knots can cause discomfort, skin irritation, and an increased risk for infection. There is limited guidance on a simple adaptable method to reduce tram-track marks from epidermal sutures exerting pressure on the epidermis while still facilitating healing in dermatologic procedures such as excision of cysts or lipomas. We present a hybrid suture method that combines elements of traditional simple interrupted and retention sutures with a layer of sterile, absorbent rolled gauze or a dental roll placed beneath the suture knots.
The Technique
Traditional epidermal sutures concentrate pressure at the knot, increasing the risk for tram-track marks and patient discomfort. To address this, we developed a hybrid technique combining simple interrupted sutures with a sterile dental roll beneath the knots to reduce pressure, protect the wound, and promote comfortable wound healing.
After excision of a cyst, we approximated the wound edges with buried vertical mattress sutures for eversion (a set-back buried dermal suture also may be used). The sutures initially were placed loosely but were left untied (eFigure 1A). A sterile dental roll with sterile petrolatum on the underside was positioned over the wound before the knots were secured, similar to a bolster dressing (eFigures 1B and 1C). The dressing then was covered and left in place for 24 to 48 hours. After removal of the dressing, no bandage was needed because the wound was clean and hemostatic and the dental roll had absorbed minimal drainage and protected the incision edges during the initial healing period. The patient applied petrolatum daily to prevent the dental roll from drying out. Sutures and the bolster were removed at 14 days without complications or complaints.
Rolled gauze may be used as an alternative to the dental roll. To maintain a clean surgical field, nonsterile gauze may be soaked in a disinfectant (eg, alcohol) and wrung out to remove excess moisture before placement on the skin. The side of the gauze in contact with the skin also should be lubricated with petroleum jelly to prevent sticking. If the sutures slip during knot tying, one end can be secured with a needle driver or hemostat. Patients should be advised to keep the dental roll dry to prevent maceration and promote optimal wound healing, but minor dampness is permissible if followed by air-drying.
This suturing method is most suitable for low- to moderate-tension closures such as cyst or lipoma excisions. The serosanguinous drainage can be absorbed by the gauze or dental roll while pressure is simultaneously applied to the wound. We do not recommend this technique for high-tension wounds in which large surface areas are removed (eg, skin cancer excisions on the posterior shoulder that require wide margins). Close monitoring of the wound for dehiscence is needed. As the sutures stretch and swelling decreases, the pressure is distributed accordingly without excessive compression to the wound line. Depending on the location, the sutures and dental roll can be removed in 7 to 14 days.
Practice Implications
Placing a dental roll or rolled gauze beneath suture knots can prevent tram-track scarring by eliminating direct knot-to-skin contact (eFigure 2).1,2 This technique distributes tension evenly, reduces the risk for wound edge necrosis, and absorbs serosanguinous drainage while providing hemostasis. The modification is quick, inexpensive, and especially beneficial for patients who may struggle with complex wound care, maintaining a clean environment until sutures are removed.
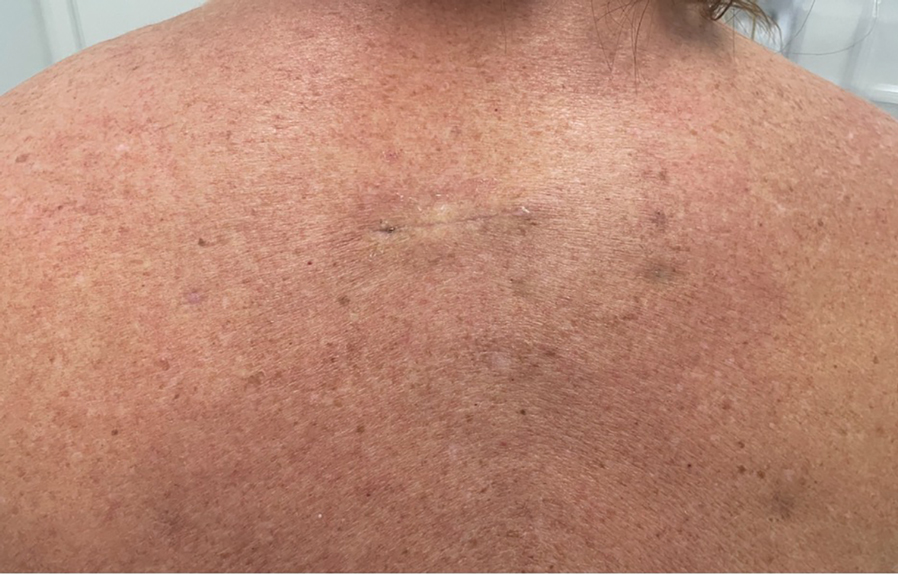
- Li E, Zhang T, Ma Q, et al. Effect of modified Allgöwer-Donati suture technique on wound cosmetics in spinal surgery. Orthop Surg. 2022;14:678-685. doi:10.1111/os.13188
- Park L, Lum ZC, Joo JS. Allgöwer-Donati suture: a technique to preserve skin microcirculation in dermatologic surgery. J Am Acad Dermatol. 2024;90:E129-E130. doi:10.1016/j.jaad.2023.05.052
Practice Gap
If not cared for properly, epidermal suture knots can cause discomfort, skin irritation, and an increased risk for infection. There is limited guidance on a simple adaptable method to reduce tram-track marks from epidermal sutures exerting pressure on the epidermis while still facilitating healing in dermatologic procedures such as excision of cysts or lipomas. We present a hybrid suture method that combines elements of traditional simple interrupted and retention sutures with a layer of sterile, absorbent rolled gauze or a dental roll placed beneath the suture knots.
The Technique
Traditional epidermal sutures concentrate pressure at the knot, increasing the risk for tram-track marks and patient discomfort. To address this, we developed a hybrid technique combining simple interrupted sutures with a sterile dental roll beneath the knots to reduce pressure, protect the wound, and promote comfortable wound healing.
After excision of a cyst, we approximated the wound edges with buried vertical mattress sutures for eversion (a set-back buried dermal suture also may be used). The sutures initially were placed loosely but were left untied (eFigure 1A). A sterile dental roll with sterile petrolatum on the underside was positioned over the wound before the knots were secured, similar to a bolster dressing (eFigures 1B and 1C). The dressing then was covered and left in place for 24 to 48 hours. After removal of the dressing, no bandage was needed because the wound was clean and hemostatic and the dental roll had absorbed minimal drainage and protected the incision edges during the initial healing period. The patient applied petrolatum daily to prevent the dental roll from drying out. Sutures and the bolster were removed at 14 days without complications or complaints.
Rolled gauze may be used as an alternative to the dental roll. To maintain a clean surgical field, nonsterile gauze may be soaked in a disinfectant (eg, alcohol) and wrung out to remove excess moisture before placement on the skin. The side of the gauze in contact with the skin also should be lubricated with petroleum jelly to prevent sticking. If the sutures slip during knot tying, one end can be secured with a needle driver or hemostat. Patients should be advised to keep the dental roll dry to prevent maceration and promote optimal wound healing, but minor dampness is permissible if followed by air-drying.
This suturing method is most suitable for low- to moderate-tension closures such as cyst or lipoma excisions. The serosanguinous drainage can be absorbed by the gauze or dental roll while pressure is simultaneously applied to the wound. We do not recommend this technique for high-tension wounds in which large surface areas are removed (eg, skin cancer excisions on the posterior shoulder that require wide margins). Close monitoring of the wound for dehiscence is needed. As the sutures stretch and swelling decreases, the pressure is distributed accordingly without excessive compression to the wound line. Depending on the location, the sutures and dental roll can be removed in 7 to 14 days.
Practice Implications
Placing a dental roll or rolled gauze beneath suture knots can prevent tram-track scarring by eliminating direct knot-to-skin contact (eFigure 2).1,2 This technique distributes tension evenly, reduces the risk for wound edge necrosis, and absorbs serosanguinous drainage while providing hemostasis. The modification is quick, inexpensive, and especially beneficial for patients who may struggle with complex wound care, maintaining a clean environment until sutures are removed.
Practice Gap
If not cared for properly, epidermal suture knots can cause discomfort, skin irritation, and an increased risk for infection. There is limited guidance on a simple adaptable method to reduce tram-track marks from epidermal sutures exerting pressure on the epidermis while still facilitating healing in dermatologic procedures such as excision of cysts or lipomas. We present a hybrid suture method that combines elements of traditional simple interrupted and retention sutures with a layer of sterile, absorbent rolled gauze or a dental roll placed beneath the suture knots.
The Technique
Traditional epidermal sutures concentrate pressure at the knot, increasing the risk for tram-track marks and patient discomfort. To address this, we developed a hybrid technique combining simple interrupted sutures with a sterile dental roll beneath the knots to reduce pressure, protect the wound, and promote comfortable wound healing.
After excision of a cyst, we approximated the wound edges with buried vertical mattress sutures for eversion (a set-back buried dermal suture also may be used). The sutures initially were placed loosely but were left untied (eFigure 1A). A sterile dental roll with sterile petrolatum on the underside was positioned over the wound before the knots were secured, similar to a bolster dressing (eFigures 1B and 1C). The dressing then was covered and left in place for 24 to 48 hours. After removal of the dressing, no bandage was needed because the wound was clean and hemostatic and the dental roll had absorbed minimal drainage and protected the incision edges during the initial healing period. The patient applied petrolatum daily to prevent the dental roll from drying out. Sutures and the bolster were removed at 14 days without complications or complaints.
Rolled gauze may be used as an alternative to the dental roll. To maintain a clean surgical field, nonsterile gauze may be soaked in a disinfectant (eg, alcohol) and wrung out to remove excess moisture before placement on the skin. The side of the gauze in contact with the skin also should be lubricated with petroleum jelly to prevent sticking. If the sutures slip during knot tying, one end can be secured with a needle driver or hemostat. Patients should be advised to keep the dental roll dry to prevent maceration and promote optimal wound healing, but minor dampness is permissible if followed by air-drying.
This suturing method is most suitable for low- to moderate-tension closures such as cyst or lipoma excisions. The serosanguinous drainage can be absorbed by the gauze or dental roll while pressure is simultaneously applied to the wound. We do not recommend this technique for high-tension wounds in which large surface areas are removed (eg, skin cancer excisions on the posterior shoulder that require wide margins). Close monitoring of the wound for dehiscence is needed. As the sutures stretch and swelling decreases, the pressure is distributed accordingly without excessive compression to the wound line. Depending on the location, the sutures and dental roll can be removed in 7 to 14 days.
Practice Implications
Placing a dental roll or rolled gauze beneath suture knots can prevent tram-track scarring by eliminating direct knot-to-skin contact (eFigure 2).1,2 This technique distributes tension evenly, reduces the risk for wound edge necrosis, and absorbs serosanguinous drainage while providing hemostasis. The modification is quick, inexpensive, and especially beneficial for patients who may struggle with complex wound care, maintaining a clean environment until sutures are removed.
- Li E, Zhang T, Ma Q, et al. Effect of modified Allgöwer-Donati suture technique on wound cosmetics in spinal surgery. Orthop Surg. 2022;14:678-685. doi:10.1111/os.13188
- Park L, Lum ZC, Joo JS. Allgöwer-Donati suture: a technique to preserve skin microcirculation in dermatologic surgery. J Am Acad Dermatol. 2024;90:E129-E130. doi:10.1016/j.jaad.2023.05.052
- Li E, Zhang T, Ma Q, et al. Effect of modified Allgöwer-Donati suture technique on wound cosmetics in spinal surgery. Orthop Surg. 2022;14:678-685. doi:10.1111/os.13188
- Park L, Lum ZC, Joo JS. Allgöwer-Donati suture: a technique to preserve skin microcirculation in dermatologic surgery. J Am Acad Dermatol. 2024;90:E129-E130. doi:10.1016/j.jaad.2023.05.052
A Hybrid Suture Technique: Suture Modification With Dental Roll Insertion
A Hybrid Suture Technique: Suture Modification With Dental Roll Insertion

Cutaneous Manifestations of Neglected Infectious Diseases in US Military Personnel
Cutaneous Manifestations of Neglected Infectious Diseases in US Military Personnel
Infectious diseases historically have posed major challenges to the operations and health of military forces. In recent conflicts, nonbattle injuries including infections have caused more evacuations than combat trauma.1 Deployment to endemic regions, poor sanitation, and trauma increase susceptibility to both common and rare infections, many of which have cutaneous manifestations.
Surveillance programs such as the Armed Forces Health Surveillance Division serve a critical role in monitoring outbreaks among deployed personnel.2 Cutaneous manifestations of systemic disease often serve as early clinical indicators, especially in settings with limited diagnostic resources. This review describes rarely encountered infectious agents for which US military personnel are at increased risk and outlines management strategies for clinicians practicing in austere environments.
EPIDEMIOLOGIC RISK FACTORS IN MILITARY POPULATIONS
United States military personnel face an elevated risk for infectious diseases when deployed in tropical and subtropical regions where endemic pathogens are prevalent. Exposure to soil, water, and insect vectors facilitates transmission. Direct exposure during combat or training combined with high-density housing, combat-related trauma, and constraints on hygiene access during operations increases infection risk.3
REGION-SPECIFIC PATHOGENS
Middle East
Leishmania species—Leishmania, a protozoa transmitted via sand fly bites, has caused multiple documented outbreaks among US troops in Iraq and Afghanistan, with a reported incidence of 14%.4 Leishmaniasis can present as 3 main clinical variants: cutaneous, visceral, and mucocutaneous. Cutaneous leishmaniasis typically manifests as painless ulcers covered with hemorrhagic crusts on exposed regions of the body. While typically self-limited, lesions frequently result in irreversible scarring. Many Leishmania species respond well to antimonials such as sodium stibogluconate. Preventive measures include wearing protective clothing and sleeping inside insecticide-treated bed nets.5
Coxiella burnetii—Coxiella burnetii transmits through inhalation of aerosolized particles originating from the urine, feces, birth products, or milk of infected bovine. In 2003, a small number of cases were identified in US service members exposed to livestock while serving in Iraq.6 Outbreaks also occurred during World War II, but it is unclear whether they were caused by naturally occurring C burnetii or biowarfare.7 Though primarily a systemic illness with severe pneumonia, Q fever may manifest with an associated purpuric or morbilliform rash.8 Doxycycline is recommended both for treatment and empiric coverage.6
Acinetobacter baumannii—This multidrug-resistant organism is known to infect combat wounds and is associated with nosocomial outbreaks in military hospitals. Studies suggest environmental contamination and health care transmission contribute substantially to outbreaks in military hospitals.9 Cutaneous manifestations can include cellulitis with a peau d’orange appearance or necrotizing fasciitis; however, pneumonia and bacteremia have been reported. Prompt culture and antibiotic initiation with debridement are essential for treatment.10 Antibiotic stewardship and strict infection control are critical to prevent outbreaks and limit resistance.9
Africa
Plasmodium species—Malaria remains a life-threatening disease found in tropical and subtropical areas around the world. Despite preventive measures, 30 cases among US service members were reported in 2024.11 Cutaneous findings include purpura fulminans, petechiae, acral necrosis, or reticulated erythema.12 Service members stationed in endemic areas should take prophylactic antimalarials. Symptoms include fevers, headaches, and malaise, with possible rapid deterioration.13
Mycobacterium ulcerans—Mycobacterium ulcerans causes extensive necrotic ulcers—commonly known as Buruli ulcers—which generally begin as a nodule, plaque, papule, or edematous lesion, eventually progressing to extensive ulceration. Despite no documented cases of US personnel contracting Buruli ulcers, those stationed in endemic regions remain at risk. Environmental reservoirs of M ulcerans are unknown, but its DNA has been isolated from water sources.14,15 These ulcers take months to heal, making wound management and antimycobacterial therapy essential. Primary preventive measures include avoidance of swimming in rivers or agricultural work in endemic areas.14
Mpox Virus—During the 2022 mpox outbreak, male service members who engaged in sexual activity with other men were at the highest risk, with 88.8% of infected service members reporting this practice.16 While the virus is endemic to Africa, 89.0% of cases were reported from service members stationed in the United States.17 Typical infection results in fever, headache, lymphadenopathy, and myalgias, followed by a facial rash that spreads over the body, palms, and soles. Safe-sex practices help prevent transmission, and there is a vaccine available for high-risk patients.16
Southeast Asia
Leptospira species—Leptospira is an aerobic spirochete found in tropical regions worldwide. Transmission occurs when water contaminated with urine from infected animals exposes humans to the organism. Infection manifests as a mild febrile illness, though approximately 10% of patients develop Weil syndrome, consisting of conjunctival suffusion, jaundice, and acute kidney injury. Treatment and prophylaxis include doxycycline, but severe disease warrants intravenous antibiotics.17,18 A 2014 outbreak among Marines in Japan highlighted poor prophylactic compliance as a key risk factor.19 Proper education for those at risk is essential to prevent future outbreaks.
Mycobacterium leprae—Leprosy is an acid-fast mycobacterium that remains endemic in the Pacific Islands and Southeast Asia. Case reports of US service members diagnosed with leprosy exist, though only in patients who emigrated from endemic areas.20 This disease has a spectrum of manifestations depending on the immune response, with tuberculoid leprosy showing a cell-mediated (T helper 1) response and lepromatous leprosy having more of a humoral (T helper 2) response.21 It manifests with hypopigmented anesthetic macules and peripheral neuropathy. Diagnosis is made by skin biopsy, which shows perineural lymphohistiocytic inflammation and non-necrotizing granulomas.20 The infection typically is curable with a multidrug regimen.21
Strongyloides stercoralis—This nematode causes infection by transdermal penetration of bare feet. They then migrate to the lungs where the patient coughs and swallows the nematode into the gastrointestinal tract. Strongyloides stercoralis autoinfect by penetrating the intestinal wall, resulting in chronic digestive, respiratory, and cutaneous symptoms. Worldwide prevalence of S stercoralis infection is estimated to be 10% to 40%, with foreign-born US military members at increased risk compared to the general military population.22,23 Larva currens may manifest with a pruritic erythematous plaque at the site of penetration that progresses to an intensely pruritic, creeping dermatitis as the nematode migrates under the skin. Avoidance of barefoot soil exposure and treatment with ivermectin are effective preventive and therapeutic measures.23
South America
Ancylostoma braziliense—Found throughout the subtropical world, this hookworm primarily infects dogs and cats and is found in their stool. Larva currens has a similar manifestation and life cycle to cutaneous larva migrans, but autoinfection does not occur. Transmission occurs similarly to S stercoralis and responds well to oral albendazole or ivermectin; however, the infection is self-limited.24 Military cases have been reported,25 though overall morbidity remains poorly characterized.
Dengue Virus—An arbovirus transmitted by Aedes mosquitoes, dengue remains a major military threat. Service members in the Vietnam War experienced an attack rate as high as 80%.26,27 Infection often manifests with retro-orbital pain and a morbilliform rash that occurs 2 to 5 days after fever, though severe cases may progress to hemorrhagic dengue with skin petechiae or ecchymosis.28 Immediate intervention is essential in symptomatic patients to prevent life-threatening progression. There are no dengue vaccines approved by the US Food and Drug Administration for adults, thus military personnel in endemic areas remain at risk.27
Trypanosoma cruzi—Chagas disease is transmitted when a reduviid infected with T cruzi bites and defecates on the patient’s skin. A skin nodule (chagoma) or painless eyelid edema (Romaña sign) may appear at the site of parasite entry. Chronic disease may result in dilated cardiomyopathy.29 Several cases of Chagas disease have been reported in South American military operations, including an outbreak in 9 Columbian military personnel.30 Cases in the southwestern United States have recently emerged, emphasizing the need for increased awareness.31 Proper insect repellent helps to ward off reduviid bugs. Nifurtimox and benznidazole are the only drugs with proven efficacy against T cruzi.29
Continental United States of America
Coccidioides immitis—The first reported case of coccidiomycosis was described in 1892 in a service member with debilitating masses and ulcers.32 Endemic to arid regions of the western United States, coccidioidomycosis affects military trainees at rates up to 32% annually in high-risk settings.33 Primary infection occurs in the lungs and may spread hematologically. The fungi prefer dry desert soils, which may aerosolize during military maneuvers. Coccidioidomycosis occasionally causes erythema nodosum, and diffuse infection shows verrucous plaques, ulcers, or abscesses. Dust avoidance and mask wearing are advised for those in endemic regions. Ketoconazole and amphotericin B are the only treatments approved by the US Food and Drug Administration.32 When starting immunosuppressive drugs, clinicians should inquire if patients have previously been stationed in Coccidioides-endemic areas, such as Fort Irwin, California, to avoid reactivation of the fungi.33
Francisella tularensis—Acquired via ticks or contact with wild animals, tularemia causes an ulceroglandular disease with regional lymphadenopathy. Inoculation requires as few as 10 to 25 organisms; thus it is considered a Category A agent for bioterror.34 Natural outbreaks have occurred during war times, most recently during the civil wars in Bosnia and Kosovo.35 Patients may present with a painful ulcer that enlarges to form a plaque with raised borders. Personnel in wooded areas should use tick precautions and handle wild animals cautiously. Treatment includes gentamicin for severe disease, with tetracyclines effective in mild cases.34
PREVENTION AND MANAGEMENT STRATEGIES IN AUSTERE SETTINGS
For health care professionals practicing in military settings, austere environments can provide a challenge for diagnosis of neglected diseases. Despite a lack of advanced diagnostic tools, practical options can be applied to the diagnostic process; for example, teledermatology is utilized for treatment of service members deployed to remote environments.36
Management of uncommon infectious diseases in military personnel often requires treatments outside those practiced in domestic clinics. Field management may indicate prompt empiric therapy while balancing the risks of overtreatment against those of missed diagnoses37; however, medical evacuation to a higher level of care may be indicated in patients with severe presentations to expedite diagnosis and treatment.38
Prevention remains the primary goal to avoid local outbreaks. Long-sleeved uniforms, DEET (N, N-diethyl-meta-toluamide)–based repellents, permethrin-impregnated clothing, and bed nets are effective for vector protection. Prophylactic medications and vaccinations often are provided when personnel are deployed to endemic locations.39
Onsite entomology teams also can provide surveillance of the local insect populations. They may contribute to vector control through insecticide application and environmental modification. The Armed Forces Health Surveillance Division and the Global Emerging Infections Surveillance Program monitor infectious threats in real time to locate any potential outbreaks, guiding operational responses.40
FINAL THOUGHTS
Dermatologic signs often provide early evidence of infection in military personnel. With increasing antimicrobial resistance and the emergence of new pathogens, it is imperative for clinicians treating members of the military to recognize cutaneous signs, employ efficient diagnostic strategies, and encourage proactive prevention. A collaborative approach spanning dermatology, infectious disease, and public health is essential to protect the modern service member.
- Murray CK. Infectious disease complications of combat-related injuries. Crit Care Med. 2008;36(7 suppl):S358-S364. doi:10.1097/CCM.0b013e31817e2ffc
- Armed Forces Health Surveillance Division. AFHSD Annual Report. Defense Health Agency; 2023. Accessed March 5, 2026. https://www.health.mil/Reference-Center/Reports/2024/09/19/AFHSD-Annual-Report-2023
- Murray CK, Yun HC, Markelz AE, et al. Operation United Assistance: infectious disease threats to deployed military personnel. Military Medicine. 2015;180:626-651. doi:10.7205/MILMED-D-14-00691
- Niba Rawlings N, Bailey M, Courtenay O. Leishmaniasis in deployed military populations: a systematic review and meta-analysis. PLoS Negl Trop Dis. 2025;19:E0012680. doi:10.1371/journal.pntd.0012680
- de Vries HJC, Schallig HD. Cutaneous leishmaniasis: a 2022 updated narrative review into diagnosis and management developments. Am J Clin Dermatol. 2022;23:823-840. doi:10.1007/s40257-022-00726-8
- Anderson AD, Smoak B, Shuping E, et al. Q fever and the US military. Emerg Infect Dis. 2005;11:1320-1322. doi:10.3201/eid1108.050314
- Madariaga MG, Rezai K, Trenholme GM, et al. Q fever: a biological weapon in your backyard. Lancet Infect Dis. 2003;3:709-721. doi:10.1016/S1473-3099(03)00804-1
- Uribe Pulido N, Escorcia García C, Cabrera Orrego R, et al. Acute Q fever with dermatologic manifestations, molecular diagnosis, and no seroconversion. Open Forum Infect Dis. 2021;8:ofab458. doi:10.1093/ofid/ofab458
- Scott P, Deye G, Srinivasan A, et al. An outbreak of multidrug-resistant acinetobacter baumannii-calcoaceticus complex infection in the US Military Health Care System associated with military operations in Iraq. Clin Infect Dis. 2007;44:1577-1584. doi:10.1086/518170
- Guerrero DM, Perez F, Conger NG, et al. Acinetobacter baumannii-associated skin and soft tissue infections: recognizing a broadening spectrum of disease. Surg Infect (Larchmt). 2010;11:49-57. doi:10.1089/sur.2009.022
- Armed Forces Health Surveillance Division. Malaria among members of the US Armed Forces, 2024. MSMR. 2025;32:22-28.
- Farkouh CS, Abdi P, Amatul-Hadi F, et al. Cutaneous manifestations of malaria and their prognostic windows: a narrative review. Cureus. 2023;15:E41706. doi:10.7759/cureus.41706
- Shahbodaghi SD, Rathjen NA. Malaria: prevention, diagnosis, and treatment. Am Fam Physician. 2022;106:270-278.
- Yotsu RR, Suzuki K, Simmonds RE, et al. Buruli ulcer: a review of the current knowledge. Curr Trop Med Rep. 2018;5:247-256. doi:10.1007/s40475-018-0166-2
- Portaels F, Meyers WM, Ablordey A, et al. First cultivation and characterization of Mycobacterium ulcerans from the environment. PLoS Negl Trop Dis. 2008;2:E178. doi:10.1371/journal.pntd.0000178
- Metcalf-Kelly M, Garrison M, Stidham R. Characteristics of mpox cases diagnosed in Military Health System beneficiaries, May 2022-April 2024. MSMR. 2024;31:7-11.
- Rajapakse S. Leptospirosis: clinical aspects. Clin Med (Lond). 2022;22:14-17. doi:10.7861/clinmed.2021-0784
- Heath CW, Alexander AD, Galton MM. Leptospirosis in the United States: a of 483 cases in man, 1949–1961. N Engl J Med. 1965;273:857-864. doi:10.1056/NEJM196510142731606
- Mason V. Mystery outbreak investigation 2014—Leptospirosis licerasiae. November 17, 2017. Accessed March 5, 2026. https://usupulse.blogspot.com/2017/11/mystery-outbreak-investigation-2014.html
- Berjohn CM, DuPlessis CA, Tieu K, et al. Multibacillary leprosy in an active duty military member. Emerg Infect Dis. 2015;21:1077-1078. doi:10.3201/eid2106.141666
- Scollard DM, Adams LB, Gillis TP, et al. The continuing challenges of leprosy. Clin Microbiol Rev. 2006;19:338-381. doi:10.1128/CMR.19.2.338-381.2006
- Schär F, Trostdorf U, Giardina F, et al. Strongyloides stercoralis: global distribution and risk factors. PLoS Negl Trop Dis. 2013;7:E2288. doi:10.1371/journal.pntd.0002288
- Taheri B, Kuo HC, Hockenbury N, et al. Strongyloides stercoralis in the US Military Health System. Open Forum Infect Dis. 2023;10:ofad127. doi:10.1093/ofid/ofad127
- Bowman DD, Montgomery SP, Zajac AM, et al. Hookworms of dogs and cats as agents of cutaneous larva migrans. Trends Parasitol. 2010;26:162-167. doi:10.1016/j.pt.2010.01.005
- Inglis DM, Bailey MS. Unusual presentations of cutaneous larva migrans in British military personnel. BMJ Mil Health. 2023;169:E78-E81. doi:10.1136/bmjmilitary-2020-001677
- Halstead SB, Udomsakdi S, Singharaj P, et al. Dengue and chikungunya virus infection in man in Thailand, 1962–1964. 3. clinical, epidemiologic, and virologic observations on disease in non-indigenous white persons. Am J Trop Med Hyg. 1969;18:984-996. doi:10.4269/ajtmh.1969.18.984
- Gibbons RV, Streitz M, Babina T, et al. Dengue and US military operations from the Spanish-American War through today. Emerg Infect Dis. 2012;18:623-630. doi:10.3201/eid1804.110134
- Guzman MG, Harris E. Dengue. Lancet. 2015;385:453-465. doi:10.1016/S0140-6736(14)60572-9
- Bern C. Chagas’ disease. N Engl J Med. 2015;373:456-466. doi:10.1056/NEJMra1410150
- Vergara HD, Gómez CH, Faccini-Martínez ÁA, et al. Acute Chagas disease outbreak among military personnel, Colombia, 2021. Emerg Infect Dis. 2023;29:1882-1885. doi:10.3201/eid2909.230886
- Harris N, Woc-Colburn L, Gunter SM, et al. Autochthonous Chagas disease in the southern United States: a case report of suspected residential and military exposures. Zoonoses Public Health. 2017;64:491-493. doi:10.1111/zph.12360
- Crum NF. Coccidioidomycosis: a contemporary review. Infect Dis Ther. 2022;11:713-742. doi:10.1007/s40121-022-00606-y
- Crum NF, Potter M, Pappagianis D. Seroincidence of coccidioidomycosis during military desert training exercises. J Clin Microbiol. 2004;42:4552-4555. doi:10.1128/JCM.42.10.4552-4555.2004
- Antonello RM, Giacomelli A, Riccardi N. Tularemia for clinicians: an up-to-date review on epidemiology, diagnosis, prevention and treatment. Eur J Intern Med. 2025;135:25-32. doi:10.1016/j.ejim.2025.03.013
- Nelson CA, Sjöstedt A. Tularemia: a storied history, an ongoing threat. Clin Infect Dis. 2024;78(supplement_1):S1-S3. doi:10.1093/cid/ciad681
- Pendlebury GA, Roman J, Shrivastava V, et al. A call to action: evidence for the military integration of teledermoscopy in a pandemic era. Dermatopathology (Basel). 2022;9:327-342. doi:10.3390/dermatopathology9040039
- Bhagchandani R, Singhi S, Peter JV, et al. Tropical fevers: management guidelines. Indian J Crit Care Med. 2014;18:62-69. doi:10.4103/0972-5229.126074
- Joint Chiefs of Staff. Joint Publication 4-02: Joint Health Services. December 11, 2017. Accessed March 5, 2026. https://cdmrp.health.mil/pubs/pdf/Joint%20Health%20Services%20Publication%20JP%204-02.pdf
- Armed Services Pest Management Board. Technical Guide No. 36: Personal Protective Measures Against Insects and Other Arthropods of Military Significance. Updated November 2015. Accessed March 5, 2026. https://www.acq.osd.mil/eie/afpmb/docs/techguides/tg36.pdf
- Global Emerging Infections Surveillance. Armed Forces Health Surveillance Division Annual Report 2024. Defense Health Agency; 2024:15-28. Accessed March 17, 2026. https://www.health.mil/Reference-Center/Reports/2026/01/05/AFHSD-Annual-Report-2024
Infectious diseases historically have posed major challenges to the operations and health of military forces. In recent conflicts, nonbattle injuries including infections have caused more evacuations than combat trauma.1 Deployment to endemic regions, poor sanitation, and trauma increase susceptibility to both common and rare infections, many of which have cutaneous manifestations.
Surveillance programs such as the Armed Forces Health Surveillance Division serve a critical role in monitoring outbreaks among deployed personnel.2 Cutaneous manifestations of systemic disease often serve as early clinical indicators, especially in settings with limited diagnostic resources. This review describes rarely encountered infectious agents for which US military personnel are at increased risk and outlines management strategies for clinicians practicing in austere environments.
EPIDEMIOLOGIC RISK FACTORS IN MILITARY POPULATIONS
United States military personnel face an elevated risk for infectious diseases when deployed in tropical and subtropical regions where endemic pathogens are prevalent. Exposure to soil, water, and insect vectors facilitates transmission. Direct exposure during combat or training combined with high-density housing, combat-related trauma, and constraints on hygiene access during operations increases infection risk.3
REGION-SPECIFIC PATHOGENS
Middle East
Leishmania species—Leishmania, a protozoa transmitted via sand fly bites, has caused multiple documented outbreaks among US troops in Iraq and Afghanistan, with a reported incidence of 14%.4 Leishmaniasis can present as 3 main clinical variants: cutaneous, visceral, and mucocutaneous. Cutaneous leishmaniasis typically manifests as painless ulcers covered with hemorrhagic crusts on exposed regions of the body. While typically self-limited, lesions frequently result in irreversible scarring. Many Leishmania species respond well to antimonials such as sodium stibogluconate. Preventive measures include wearing protective clothing and sleeping inside insecticide-treated bed nets.5
Coxiella burnetii—Coxiella burnetii transmits through inhalation of aerosolized particles originating from the urine, feces, birth products, or milk of infected bovine. In 2003, a small number of cases were identified in US service members exposed to livestock while serving in Iraq.6 Outbreaks also occurred during World War II, but it is unclear whether they were caused by naturally occurring C burnetii or biowarfare.7 Though primarily a systemic illness with severe pneumonia, Q fever may manifest with an associated purpuric or morbilliform rash.8 Doxycycline is recommended both for treatment and empiric coverage.6
Acinetobacter baumannii—This multidrug-resistant organism is known to infect combat wounds and is associated with nosocomial outbreaks in military hospitals. Studies suggest environmental contamination and health care transmission contribute substantially to outbreaks in military hospitals.9 Cutaneous manifestations can include cellulitis with a peau d’orange appearance or necrotizing fasciitis; however, pneumonia and bacteremia have been reported. Prompt culture and antibiotic initiation with debridement are essential for treatment.10 Antibiotic stewardship and strict infection control are critical to prevent outbreaks and limit resistance.9
Africa
Plasmodium species—Malaria remains a life-threatening disease found in tropical and subtropical areas around the world. Despite preventive measures, 30 cases among US service members were reported in 2024.11 Cutaneous findings include purpura fulminans, petechiae, acral necrosis, or reticulated erythema.12 Service members stationed in endemic areas should take prophylactic antimalarials. Symptoms include fevers, headaches, and malaise, with possible rapid deterioration.13
Mycobacterium ulcerans—Mycobacterium ulcerans causes extensive necrotic ulcers—commonly known as Buruli ulcers—which generally begin as a nodule, plaque, papule, or edematous lesion, eventually progressing to extensive ulceration. Despite no documented cases of US personnel contracting Buruli ulcers, those stationed in endemic regions remain at risk. Environmental reservoirs of M ulcerans are unknown, but its DNA has been isolated from water sources.14,15 These ulcers take months to heal, making wound management and antimycobacterial therapy essential. Primary preventive measures include avoidance of swimming in rivers or agricultural work in endemic areas.14
Mpox Virus—During the 2022 mpox outbreak, male service members who engaged in sexual activity with other men were at the highest risk, with 88.8% of infected service members reporting this practice.16 While the virus is endemic to Africa, 89.0% of cases were reported from service members stationed in the United States.17 Typical infection results in fever, headache, lymphadenopathy, and myalgias, followed by a facial rash that spreads over the body, palms, and soles. Safe-sex practices help prevent transmission, and there is a vaccine available for high-risk patients.16
Southeast Asia
Leptospira species—Leptospira is an aerobic spirochete found in tropical regions worldwide. Transmission occurs when water contaminated with urine from infected animals exposes humans to the organism. Infection manifests as a mild febrile illness, though approximately 10% of patients develop Weil syndrome, consisting of conjunctival suffusion, jaundice, and acute kidney injury. Treatment and prophylaxis include doxycycline, but severe disease warrants intravenous antibiotics.17,18 A 2014 outbreak among Marines in Japan highlighted poor prophylactic compliance as a key risk factor.19 Proper education for those at risk is essential to prevent future outbreaks.
Mycobacterium leprae—Leprosy is an acid-fast mycobacterium that remains endemic in the Pacific Islands and Southeast Asia. Case reports of US service members diagnosed with leprosy exist, though only in patients who emigrated from endemic areas.20 This disease has a spectrum of manifestations depending on the immune response, with tuberculoid leprosy showing a cell-mediated (T helper 1) response and lepromatous leprosy having more of a humoral (T helper 2) response.21 It manifests with hypopigmented anesthetic macules and peripheral neuropathy. Diagnosis is made by skin biopsy, which shows perineural lymphohistiocytic inflammation and non-necrotizing granulomas.20 The infection typically is curable with a multidrug regimen.21
Strongyloides stercoralis—This nematode causes infection by transdermal penetration of bare feet. They then migrate to the lungs where the patient coughs and swallows the nematode into the gastrointestinal tract. Strongyloides stercoralis autoinfect by penetrating the intestinal wall, resulting in chronic digestive, respiratory, and cutaneous symptoms. Worldwide prevalence of S stercoralis infection is estimated to be 10% to 40%, with foreign-born US military members at increased risk compared to the general military population.22,23 Larva currens may manifest with a pruritic erythematous plaque at the site of penetration that progresses to an intensely pruritic, creeping dermatitis as the nematode migrates under the skin. Avoidance of barefoot soil exposure and treatment with ivermectin are effective preventive and therapeutic measures.23
South America
Ancylostoma braziliense—Found throughout the subtropical world, this hookworm primarily infects dogs and cats and is found in their stool. Larva currens has a similar manifestation and life cycle to cutaneous larva migrans, but autoinfection does not occur. Transmission occurs similarly to S stercoralis and responds well to oral albendazole or ivermectin; however, the infection is self-limited.24 Military cases have been reported,25 though overall morbidity remains poorly characterized.
Dengue Virus—An arbovirus transmitted by Aedes mosquitoes, dengue remains a major military threat. Service members in the Vietnam War experienced an attack rate as high as 80%.26,27 Infection often manifests with retro-orbital pain and a morbilliform rash that occurs 2 to 5 days after fever, though severe cases may progress to hemorrhagic dengue with skin petechiae or ecchymosis.28 Immediate intervention is essential in symptomatic patients to prevent life-threatening progression. There are no dengue vaccines approved by the US Food and Drug Administration for adults, thus military personnel in endemic areas remain at risk.27
Trypanosoma cruzi—Chagas disease is transmitted when a reduviid infected with T cruzi bites and defecates on the patient’s skin. A skin nodule (chagoma) or painless eyelid edema (Romaña sign) may appear at the site of parasite entry. Chronic disease may result in dilated cardiomyopathy.29 Several cases of Chagas disease have been reported in South American military operations, including an outbreak in 9 Columbian military personnel.30 Cases in the southwestern United States have recently emerged, emphasizing the need for increased awareness.31 Proper insect repellent helps to ward off reduviid bugs. Nifurtimox and benznidazole are the only drugs with proven efficacy against T cruzi.29
Continental United States of America
Coccidioides immitis—The first reported case of coccidiomycosis was described in 1892 in a service member with debilitating masses and ulcers.32 Endemic to arid regions of the western United States, coccidioidomycosis affects military trainees at rates up to 32% annually in high-risk settings.33 Primary infection occurs in the lungs and may spread hematologically. The fungi prefer dry desert soils, which may aerosolize during military maneuvers. Coccidioidomycosis occasionally causes erythema nodosum, and diffuse infection shows verrucous plaques, ulcers, or abscesses. Dust avoidance and mask wearing are advised for those in endemic regions. Ketoconazole and amphotericin B are the only treatments approved by the US Food and Drug Administration.32 When starting immunosuppressive drugs, clinicians should inquire if patients have previously been stationed in Coccidioides-endemic areas, such as Fort Irwin, California, to avoid reactivation of the fungi.33
Francisella tularensis—Acquired via ticks or contact with wild animals, tularemia causes an ulceroglandular disease with regional lymphadenopathy. Inoculation requires as few as 10 to 25 organisms; thus it is considered a Category A agent for bioterror.34 Natural outbreaks have occurred during war times, most recently during the civil wars in Bosnia and Kosovo.35 Patients may present with a painful ulcer that enlarges to form a plaque with raised borders. Personnel in wooded areas should use tick precautions and handle wild animals cautiously. Treatment includes gentamicin for severe disease, with tetracyclines effective in mild cases.34
PREVENTION AND MANAGEMENT STRATEGIES IN AUSTERE SETTINGS
For health care professionals practicing in military settings, austere environments can provide a challenge for diagnosis of neglected diseases. Despite a lack of advanced diagnostic tools, practical options can be applied to the diagnostic process; for example, teledermatology is utilized for treatment of service members deployed to remote environments.36
Management of uncommon infectious diseases in military personnel often requires treatments outside those practiced in domestic clinics. Field management may indicate prompt empiric therapy while balancing the risks of overtreatment against those of missed diagnoses37; however, medical evacuation to a higher level of care may be indicated in patients with severe presentations to expedite diagnosis and treatment.38
Prevention remains the primary goal to avoid local outbreaks. Long-sleeved uniforms, DEET (N, N-diethyl-meta-toluamide)–based repellents, permethrin-impregnated clothing, and bed nets are effective for vector protection. Prophylactic medications and vaccinations often are provided when personnel are deployed to endemic locations.39
Onsite entomology teams also can provide surveillance of the local insect populations. They may contribute to vector control through insecticide application and environmental modification. The Armed Forces Health Surveillance Division and the Global Emerging Infections Surveillance Program monitor infectious threats in real time to locate any potential outbreaks, guiding operational responses.40
FINAL THOUGHTS
Dermatologic signs often provide early evidence of infection in military personnel. With increasing antimicrobial resistance and the emergence of new pathogens, it is imperative for clinicians treating members of the military to recognize cutaneous signs, employ efficient diagnostic strategies, and encourage proactive prevention. A collaborative approach spanning dermatology, infectious disease, and public health is essential to protect the modern service member.
Infectious diseases historically have posed major challenges to the operations and health of military forces. In recent conflicts, nonbattle injuries including infections have caused more evacuations than combat trauma.1 Deployment to endemic regions, poor sanitation, and trauma increase susceptibility to both common and rare infections, many of which have cutaneous manifestations.
Surveillance programs such as the Armed Forces Health Surveillance Division serve a critical role in monitoring outbreaks among deployed personnel.2 Cutaneous manifestations of systemic disease often serve as early clinical indicators, especially in settings with limited diagnostic resources. This review describes rarely encountered infectious agents for which US military personnel are at increased risk and outlines management strategies for clinicians practicing in austere environments.
EPIDEMIOLOGIC RISK FACTORS IN MILITARY POPULATIONS
United States military personnel face an elevated risk for infectious diseases when deployed in tropical and subtropical regions where endemic pathogens are prevalent. Exposure to soil, water, and insect vectors facilitates transmission. Direct exposure during combat or training combined with high-density housing, combat-related trauma, and constraints on hygiene access during operations increases infection risk.3
REGION-SPECIFIC PATHOGENS
Middle East
Leishmania species—Leishmania, a protozoa transmitted via sand fly bites, has caused multiple documented outbreaks among US troops in Iraq and Afghanistan, with a reported incidence of 14%.4 Leishmaniasis can present as 3 main clinical variants: cutaneous, visceral, and mucocutaneous. Cutaneous leishmaniasis typically manifests as painless ulcers covered with hemorrhagic crusts on exposed regions of the body. While typically self-limited, lesions frequently result in irreversible scarring. Many Leishmania species respond well to antimonials such as sodium stibogluconate. Preventive measures include wearing protective clothing and sleeping inside insecticide-treated bed nets.5
Coxiella burnetii—Coxiella burnetii transmits through inhalation of aerosolized particles originating from the urine, feces, birth products, or milk of infected bovine. In 2003, a small number of cases were identified in US service members exposed to livestock while serving in Iraq.6 Outbreaks also occurred during World War II, but it is unclear whether they were caused by naturally occurring C burnetii or biowarfare.7 Though primarily a systemic illness with severe pneumonia, Q fever may manifest with an associated purpuric or morbilliform rash.8 Doxycycline is recommended both for treatment and empiric coverage.6
Acinetobacter baumannii—This multidrug-resistant organism is known to infect combat wounds and is associated with nosocomial outbreaks in military hospitals. Studies suggest environmental contamination and health care transmission contribute substantially to outbreaks in military hospitals.9 Cutaneous manifestations can include cellulitis with a peau d’orange appearance or necrotizing fasciitis; however, pneumonia and bacteremia have been reported. Prompt culture and antibiotic initiation with debridement are essential for treatment.10 Antibiotic stewardship and strict infection control are critical to prevent outbreaks and limit resistance.9
Africa
Plasmodium species—Malaria remains a life-threatening disease found in tropical and subtropical areas around the world. Despite preventive measures, 30 cases among US service members were reported in 2024.11 Cutaneous findings include purpura fulminans, petechiae, acral necrosis, or reticulated erythema.12 Service members stationed in endemic areas should take prophylactic antimalarials. Symptoms include fevers, headaches, and malaise, with possible rapid deterioration.13
Mycobacterium ulcerans—Mycobacterium ulcerans causes extensive necrotic ulcers—commonly known as Buruli ulcers—which generally begin as a nodule, plaque, papule, or edematous lesion, eventually progressing to extensive ulceration. Despite no documented cases of US personnel contracting Buruli ulcers, those stationed in endemic regions remain at risk. Environmental reservoirs of M ulcerans are unknown, but its DNA has been isolated from water sources.14,15 These ulcers take months to heal, making wound management and antimycobacterial therapy essential. Primary preventive measures include avoidance of swimming in rivers or agricultural work in endemic areas.14
Mpox Virus—During the 2022 mpox outbreak, male service members who engaged in sexual activity with other men were at the highest risk, with 88.8% of infected service members reporting this practice.16 While the virus is endemic to Africa, 89.0% of cases were reported from service members stationed in the United States.17 Typical infection results in fever, headache, lymphadenopathy, and myalgias, followed by a facial rash that spreads over the body, palms, and soles. Safe-sex practices help prevent transmission, and there is a vaccine available for high-risk patients.16
Southeast Asia
Leptospira species—Leptospira is an aerobic spirochete found in tropical regions worldwide. Transmission occurs when water contaminated with urine from infected animals exposes humans to the organism. Infection manifests as a mild febrile illness, though approximately 10% of patients develop Weil syndrome, consisting of conjunctival suffusion, jaundice, and acute kidney injury. Treatment and prophylaxis include doxycycline, but severe disease warrants intravenous antibiotics.17,18 A 2014 outbreak among Marines in Japan highlighted poor prophylactic compliance as a key risk factor.19 Proper education for those at risk is essential to prevent future outbreaks.
Mycobacterium leprae—Leprosy is an acid-fast mycobacterium that remains endemic in the Pacific Islands and Southeast Asia. Case reports of US service members diagnosed with leprosy exist, though only in patients who emigrated from endemic areas.20 This disease has a spectrum of manifestations depending on the immune response, with tuberculoid leprosy showing a cell-mediated (T helper 1) response and lepromatous leprosy having more of a humoral (T helper 2) response.21 It manifests with hypopigmented anesthetic macules and peripheral neuropathy. Diagnosis is made by skin biopsy, which shows perineural lymphohistiocytic inflammation and non-necrotizing granulomas.20 The infection typically is curable with a multidrug regimen.21
Strongyloides stercoralis—This nematode causes infection by transdermal penetration of bare feet. They then migrate to the lungs where the patient coughs and swallows the nematode into the gastrointestinal tract. Strongyloides stercoralis autoinfect by penetrating the intestinal wall, resulting in chronic digestive, respiratory, and cutaneous symptoms. Worldwide prevalence of S stercoralis infection is estimated to be 10% to 40%, with foreign-born US military members at increased risk compared to the general military population.22,23 Larva currens may manifest with a pruritic erythematous plaque at the site of penetration that progresses to an intensely pruritic, creeping dermatitis as the nematode migrates under the skin. Avoidance of barefoot soil exposure and treatment with ivermectin are effective preventive and therapeutic measures.23
South America
Ancylostoma braziliense—Found throughout the subtropical world, this hookworm primarily infects dogs and cats and is found in their stool. Larva currens has a similar manifestation and life cycle to cutaneous larva migrans, but autoinfection does not occur. Transmission occurs similarly to S stercoralis and responds well to oral albendazole or ivermectin; however, the infection is self-limited.24 Military cases have been reported,25 though overall morbidity remains poorly characterized.
Dengue Virus—An arbovirus transmitted by Aedes mosquitoes, dengue remains a major military threat. Service members in the Vietnam War experienced an attack rate as high as 80%.26,27 Infection often manifests with retro-orbital pain and a morbilliform rash that occurs 2 to 5 days after fever, though severe cases may progress to hemorrhagic dengue with skin petechiae or ecchymosis.28 Immediate intervention is essential in symptomatic patients to prevent life-threatening progression. There are no dengue vaccines approved by the US Food and Drug Administration for adults, thus military personnel in endemic areas remain at risk.27
Trypanosoma cruzi—Chagas disease is transmitted when a reduviid infected with T cruzi bites and defecates on the patient’s skin. A skin nodule (chagoma) or painless eyelid edema (Romaña sign) may appear at the site of parasite entry. Chronic disease may result in dilated cardiomyopathy.29 Several cases of Chagas disease have been reported in South American military operations, including an outbreak in 9 Columbian military personnel.30 Cases in the southwestern United States have recently emerged, emphasizing the need for increased awareness.31 Proper insect repellent helps to ward off reduviid bugs. Nifurtimox and benznidazole are the only drugs with proven efficacy against T cruzi.29
Continental United States of America
Coccidioides immitis—The first reported case of coccidiomycosis was described in 1892 in a service member with debilitating masses and ulcers.32 Endemic to arid regions of the western United States, coccidioidomycosis affects military trainees at rates up to 32% annually in high-risk settings.33 Primary infection occurs in the lungs and may spread hematologically. The fungi prefer dry desert soils, which may aerosolize during military maneuvers. Coccidioidomycosis occasionally causes erythema nodosum, and diffuse infection shows verrucous plaques, ulcers, or abscesses. Dust avoidance and mask wearing are advised for those in endemic regions. Ketoconazole and amphotericin B are the only treatments approved by the US Food and Drug Administration.32 When starting immunosuppressive drugs, clinicians should inquire if patients have previously been stationed in Coccidioides-endemic areas, such as Fort Irwin, California, to avoid reactivation of the fungi.33
Francisella tularensis—Acquired via ticks or contact with wild animals, tularemia causes an ulceroglandular disease with regional lymphadenopathy. Inoculation requires as few as 10 to 25 organisms; thus it is considered a Category A agent for bioterror.34 Natural outbreaks have occurred during war times, most recently during the civil wars in Bosnia and Kosovo.35 Patients may present with a painful ulcer that enlarges to form a plaque with raised borders. Personnel in wooded areas should use tick precautions and handle wild animals cautiously. Treatment includes gentamicin for severe disease, with tetracyclines effective in mild cases.34
PREVENTION AND MANAGEMENT STRATEGIES IN AUSTERE SETTINGS
For health care professionals practicing in military settings, austere environments can provide a challenge for diagnosis of neglected diseases. Despite a lack of advanced diagnostic tools, practical options can be applied to the diagnostic process; for example, teledermatology is utilized for treatment of service members deployed to remote environments.36
Management of uncommon infectious diseases in military personnel often requires treatments outside those practiced in domestic clinics. Field management may indicate prompt empiric therapy while balancing the risks of overtreatment against those of missed diagnoses37; however, medical evacuation to a higher level of care may be indicated in patients with severe presentations to expedite diagnosis and treatment.38
Prevention remains the primary goal to avoid local outbreaks. Long-sleeved uniforms, DEET (N, N-diethyl-meta-toluamide)–based repellents, permethrin-impregnated clothing, and bed nets are effective for vector protection. Prophylactic medications and vaccinations often are provided when personnel are deployed to endemic locations.39
Onsite entomology teams also can provide surveillance of the local insect populations. They may contribute to vector control through insecticide application and environmental modification. The Armed Forces Health Surveillance Division and the Global Emerging Infections Surveillance Program monitor infectious threats in real time to locate any potential outbreaks, guiding operational responses.40
FINAL THOUGHTS
Dermatologic signs often provide early evidence of infection in military personnel. With increasing antimicrobial resistance and the emergence of new pathogens, it is imperative for clinicians treating members of the military to recognize cutaneous signs, employ efficient diagnostic strategies, and encourage proactive prevention. A collaborative approach spanning dermatology, infectious disease, and public health is essential to protect the modern service member.
- Murray CK. Infectious disease complications of combat-related injuries. Crit Care Med. 2008;36(7 suppl):S358-S364. doi:10.1097/CCM.0b013e31817e2ffc
- Armed Forces Health Surveillance Division. AFHSD Annual Report. Defense Health Agency; 2023. Accessed March 5, 2026. https://www.health.mil/Reference-Center/Reports/2024/09/19/AFHSD-Annual-Report-2023
- Murray CK, Yun HC, Markelz AE, et al. Operation United Assistance: infectious disease threats to deployed military personnel. Military Medicine. 2015;180:626-651. doi:10.7205/MILMED-D-14-00691
- Niba Rawlings N, Bailey M, Courtenay O. Leishmaniasis in deployed military populations: a systematic review and meta-analysis. PLoS Negl Trop Dis. 2025;19:E0012680. doi:10.1371/journal.pntd.0012680
- de Vries HJC, Schallig HD. Cutaneous leishmaniasis: a 2022 updated narrative review into diagnosis and management developments. Am J Clin Dermatol. 2022;23:823-840. doi:10.1007/s40257-022-00726-8
- Anderson AD, Smoak B, Shuping E, et al. Q fever and the US military. Emerg Infect Dis. 2005;11:1320-1322. doi:10.3201/eid1108.050314
- Madariaga MG, Rezai K, Trenholme GM, et al. Q fever: a biological weapon in your backyard. Lancet Infect Dis. 2003;3:709-721. doi:10.1016/S1473-3099(03)00804-1
- Uribe Pulido N, Escorcia García C, Cabrera Orrego R, et al. Acute Q fever with dermatologic manifestations, molecular diagnosis, and no seroconversion. Open Forum Infect Dis. 2021;8:ofab458. doi:10.1093/ofid/ofab458
- Scott P, Deye G, Srinivasan A, et al. An outbreak of multidrug-resistant acinetobacter baumannii-calcoaceticus complex infection in the US Military Health Care System associated with military operations in Iraq. Clin Infect Dis. 2007;44:1577-1584. doi:10.1086/518170
- Guerrero DM, Perez F, Conger NG, et al. Acinetobacter baumannii-associated skin and soft tissue infections: recognizing a broadening spectrum of disease. Surg Infect (Larchmt). 2010;11:49-57. doi:10.1089/sur.2009.022
- Armed Forces Health Surveillance Division. Malaria among members of the US Armed Forces, 2024. MSMR. 2025;32:22-28.
- Farkouh CS, Abdi P, Amatul-Hadi F, et al. Cutaneous manifestations of malaria and their prognostic windows: a narrative review. Cureus. 2023;15:E41706. doi:10.7759/cureus.41706
- Shahbodaghi SD, Rathjen NA. Malaria: prevention, diagnosis, and treatment. Am Fam Physician. 2022;106:270-278.
- Yotsu RR, Suzuki K, Simmonds RE, et al. Buruli ulcer: a review of the current knowledge. Curr Trop Med Rep. 2018;5:247-256. doi:10.1007/s40475-018-0166-2
- Portaels F, Meyers WM, Ablordey A, et al. First cultivation and characterization of Mycobacterium ulcerans from the environment. PLoS Negl Trop Dis. 2008;2:E178. doi:10.1371/journal.pntd.0000178
- Metcalf-Kelly M, Garrison M, Stidham R. Characteristics of mpox cases diagnosed in Military Health System beneficiaries, May 2022-April 2024. MSMR. 2024;31:7-11.
- Rajapakse S. Leptospirosis: clinical aspects. Clin Med (Lond). 2022;22:14-17. doi:10.7861/clinmed.2021-0784
- Heath CW, Alexander AD, Galton MM. Leptospirosis in the United States: a of 483 cases in man, 1949–1961. N Engl J Med. 1965;273:857-864. doi:10.1056/NEJM196510142731606
- Mason V. Mystery outbreak investigation 2014—Leptospirosis licerasiae. November 17, 2017. Accessed March 5, 2026. https://usupulse.blogspot.com/2017/11/mystery-outbreak-investigation-2014.html
- Berjohn CM, DuPlessis CA, Tieu K, et al. Multibacillary leprosy in an active duty military member. Emerg Infect Dis. 2015;21:1077-1078. doi:10.3201/eid2106.141666
- Scollard DM, Adams LB, Gillis TP, et al. The continuing challenges of leprosy. Clin Microbiol Rev. 2006;19:338-381. doi:10.1128/CMR.19.2.338-381.2006
- Schär F, Trostdorf U, Giardina F, et al. Strongyloides stercoralis: global distribution and risk factors. PLoS Negl Trop Dis. 2013;7:E2288. doi:10.1371/journal.pntd.0002288
- Taheri B, Kuo HC, Hockenbury N, et al. Strongyloides stercoralis in the US Military Health System. Open Forum Infect Dis. 2023;10:ofad127. doi:10.1093/ofid/ofad127
- Bowman DD, Montgomery SP, Zajac AM, et al. Hookworms of dogs and cats as agents of cutaneous larva migrans. Trends Parasitol. 2010;26:162-167. doi:10.1016/j.pt.2010.01.005
- Inglis DM, Bailey MS. Unusual presentations of cutaneous larva migrans in British military personnel. BMJ Mil Health. 2023;169:E78-E81. doi:10.1136/bmjmilitary-2020-001677
- Halstead SB, Udomsakdi S, Singharaj P, et al. Dengue and chikungunya virus infection in man in Thailand, 1962–1964. 3. clinical, epidemiologic, and virologic observations on disease in non-indigenous white persons. Am J Trop Med Hyg. 1969;18:984-996. doi:10.4269/ajtmh.1969.18.984
- Gibbons RV, Streitz M, Babina T, et al. Dengue and US military operations from the Spanish-American War through today. Emerg Infect Dis. 2012;18:623-630. doi:10.3201/eid1804.110134
- Guzman MG, Harris E. Dengue. Lancet. 2015;385:453-465. doi:10.1016/S0140-6736(14)60572-9
- Bern C. Chagas’ disease. N Engl J Med. 2015;373:456-466. doi:10.1056/NEJMra1410150
- Vergara HD, Gómez CH, Faccini-Martínez ÁA, et al. Acute Chagas disease outbreak among military personnel, Colombia, 2021. Emerg Infect Dis. 2023;29:1882-1885. doi:10.3201/eid2909.230886
- Harris N, Woc-Colburn L, Gunter SM, et al. Autochthonous Chagas disease in the southern United States: a case report of suspected residential and military exposures. Zoonoses Public Health. 2017;64:491-493. doi:10.1111/zph.12360
- Crum NF. Coccidioidomycosis: a contemporary review. Infect Dis Ther. 2022;11:713-742. doi:10.1007/s40121-022-00606-y
- Crum NF, Potter M, Pappagianis D. Seroincidence of coccidioidomycosis during military desert training exercises. J Clin Microbiol. 2004;42:4552-4555. doi:10.1128/JCM.42.10.4552-4555.2004
- Antonello RM, Giacomelli A, Riccardi N. Tularemia for clinicians: an up-to-date review on epidemiology, diagnosis, prevention and treatment. Eur J Intern Med. 2025;135:25-32. doi:10.1016/j.ejim.2025.03.013
- Nelson CA, Sjöstedt A. Tularemia: a storied history, an ongoing threat. Clin Infect Dis. 2024;78(supplement_1):S1-S3. doi:10.1093/cid/ciad681
- Pendlebury GA, Roman J, Shrivastava V, et al. A call to action: evidence for the military integration of teledermoscopy in a pandemic era. Dermatopathology (Basel). 2022;9:327-342. doi:10.3390/dermatopathology9040039
- Bhagchandani R, Singhi S, Peter JV, et al. Tropical fevers: management guidelines. Indian J Crit Care Med. 2014;18:62-69. doi:10.4103/0972-5229.126074
- Joint Chiefs of Staff. Joint Publication 4-02: Joint Health Services. December 11, 2017. Accessed March 5, 2026. https://cdmrp.health.mil/pubs/pdf/Joint%20Health%20Services%20Publication%20JP%204-02.pdf
- Armed Services Pest Management Board. Technical Guide No. 36: Personal Protective Measures Against Insects and Other Arthropods of Military Significance. Updated November 2015. Accessed March 5, 2026. https://www.acq.osd.mil/eie/afpmb/docs/techguides/tg36.pdf
- Global Emerging Infections Surveillance. Armed Forces Health Surveillance Division Annual Report 2024. Defense Health Agency; 2024:15-28. Accessed March 17, 2026. https://www.health.mil/Reference-Center/Reports/2026/01/05/AFHSD-Annual-Report-2024
- Murray CK. Infectious disease complications of combat-related injuries. Crit Care Med. 2008;36(7 suppl):S358-S364. doi:10.1097/CCM.0b013e31817e2ffc
- Armed Forces Health Surveillance Division. AFHSD Annual Report. Defense Health Agency; 2023. Accessed March 5, 2026. https://www.health.mil/Reference-Center/Reports/2024/09/19/AFHSD-Annual-Report-2023
- Murray CK, Yun HC, Markelz AE, et al. Operation United Assistance: infectious disease threats to deployed military personnel. Military Medicine. 2015;180:626-651. doi:10.7205/MILMED-D-14-00691
- Niba Rawlings N, Bailey M, Courtenay O. Leishmaniasis in deployed military populations: a systematic review and meta-analysis. PLoS Negl Trop Dis. 2025;19:E0012680. doi:10.1371/journal.pntd.0012680
- de Vries HJC, Schallig HD. Cutaneous leishmaniasis: a 2022 updated narrative review into diagnosis and management developments. Am J Clin Dermatol. 2022;23:823-840. doi:10.1007/s40257-022-00726-8
- Anderson AD, Smoak B, Shuping E, et al. Q fever and the US military. Emerg Infect Dis. 2005;11:1320-1322. doi:10.3201/eid1108.050314
- Madariaga MG, Rezai K, Trenholme GM, et al. Q fever: a biological weapon in your backyard. Lancet Infect Dis. 2003;3:709-721. doi:10.1016/S1473-3099(03)00804-1
- Uribe Pulido N, Escorcia García C, Cabrera Orrego R, et al. Acute Q fever with dermatologic manifestations, molecular diagnosis, and no seroconversion. Open Forum Infect Dis. 2021;8:ofab458. doi:10.1093/ofid/ofab458
- Scott P, Deye G, Srinivasan A, et al. An outbreak of multidrug-resistant acinetobacter baumannii-calcoaceticus complex infection in the US Military Health Care System associated with military operations in Iraq. Clin Infect Dis. 2007;44:1577-1584. doi:10.1086/518170
- Guerrero DM, Perez F, Conger NG, et al. Acinetobacter baumannii-associated skin and soft tissue infections: recognizing a broadening spectrum of disease. Surg Infect (Larchmt). 2010;11:49-57. doi:10.1089/sur.2009.022
- Armed Forces Health Surveillance Division. Malaria among members of the US Armed Forces, 2024. MSMR. 2025;32:22-28.
- Farkouh CS, Abdi P, Amatul-Hadi F, et al. Cutaneous manifestations of malaria and their prognostic windows: a narrative review. Cureus. 2023;15:E41706. doi:10.7759/cureus.41706
- Shahbodaghi SD, Rathjen NA. Malaria: prevention, diagnosis, and treatment. Am Fam Physician. 2022;106:270-278.
- Yotsu RR, Suzuki K, Simmonds RE, et al. Buruli ulcer: a review of the current knowledge. Curr Trop Med Rep. 2018;5:247-256. doi:10.1007/s40475-018-0166-2
- Portaels F, Meyers WM, Ablordey A, et al. First cultivation and characterization of Mycobacterium ulcerans from the environment. PLoS Negl Trop Dis. 2008;2:E178. doi:10.1371/journal.pntd.0000178
- Metcalf-Kelly M, Garrison M, Stidham R. Characteristics of mpox cases diagnosed in Military Health System beneficiaries, May 2022-April 2024. MSMR. 2024;31:7-11.
- Rajapakse S. Leptospirosis: clinical aspects. Clin Med (Lond). 2022;22:14-17. doi:10.7861/clinmed.2021-0784
- Heath CW, Alexander AD, Galton MM. Leptospirosis in the United States: a of 483 cases in man, 1949–1961. N Engl J Med. 1965;273:857-864. doi:10.1056/NEJM196510142731606
- Mason V. Mystery outbreak investigation 2014—Leptospirosis licerasiae. November 17, 2017. Accessed March 5, 2026. https://usupulse.blogspot.com/2017/11/mystery-outbreak-investigation-2014.html
- Berjohn CM, DuPlessis CA, Tieu K, et al. Multibacillary leprosy in an active duty military member. Emerg Infect Dis. 2015;21:1077-1078. doi:10.3201/eid2106.141666
- Scollard DM, Adams LB, Gillis TP, et al. The continuing challenges of leprosy. Clin Microbiol Rev. 2006;19:338-381. doi:10.1128/CMR.19.2.338-381.2006
- Schär F, Trostdorf U, Giardina F, et al. Strongyloides stercoralis: global distribution and risk factors. PLoS Negl Trop Dis. 2013;7:E2288. doi:10.1371/journal.pntd.0002288
- Taheri B, Kuo HC, Hockenbury N, et al. Strongyloides stercoralis in the US Military Health System. Open Forum Infect Dis. 2023;10:ofad127. doi:10.1093/ofid/ofad127
- Bowman DD, Montgomery SP, Zajac AM, et al. Hookworms of dogs and cats as agents of cutaneous larva migrans. Trends Parasitol. 2010;26:162-167. doi:10.1016/j.pt.2010.01.005
- Inglis DM, Bailey MS. Unusual presentations of cutaneous larva migrans in British military personnel. BMJ Mil Health. 2023;169:E78-E81. doi:10.1136/bmjmilitary-2020-001677
- Halstead SB, Udomsakdi S, Singharaj P, et al. Dengue and chikungunya virus infection in man in Thailand, 1962–1964. 3. clinical, epidemiologic, and virologic observations on disease in non-indigenous white persons. Am J Trop Med Hyg. 1969;18:984-996. doi:10.4269/ajtmh.1969.18.984
- Gibbons RV, Streitz M, Babina T, et al. Dengue and US military operations from the Spanish-American War through today. Emerg Infect Dis. 2012;18:623-630. doi:10.3201/eid1804.110134
- Guzman MG, Harris E. Dengue. Lancet. 2015;385:453-465. doi:10.1016/S0140-6736(14)60572-9
- Bern C. Chagas’ disease. N Engl J Med. 2015;373:456-466. doi:10.1056/NEJMra1410150
- Vergara HD, Gómez CH, Faccini-Martínez ÁA, et al. Acute Chagas disease outbreak among military personnel, Colombia, 2021. Emerg Infect Dis. 2023;29:1882-1885. doi:10.3201/eid2909.230886
- Harris N, Woc-Colburn L, Gunter SM, et al. Autochthonous Chagas disease in the southern United States: a case report of suspected residential and military exposures. Zoonoses Public Health. 2017;64:491-493. doi:10.1111/zph.12360
- Crum NF. Coccidioidomycosis: a contemporary review. Infect Dis Ther. 2022;11:713-742. doi:10.1007/s40121-022-00606-y
- Crum NF, Potter M, Pappagianis D. Seroincidence of coccidioidomycosis during military desert training exercises. J Clin Microbiol. 2004;42:4552-4555. doi:10.1128/JCM.42.10.4552-4555.2004
- Antonello RM, Giacomelli A, Riccardi N. Tularemia for clinicians: an up-to-date review on epidemiology, diagnosis, prevention and treatment. Eur J Intern Med. 2025;135:25-32. doi:10.1016/j.ejim.2025.03.013
- Nelson CA, Sjöstedt A. Tularemia: a storied history, an ongoing threat. Clin Infect Dis. 2024;78(supplement_1):S1-S3. doi:10.1093/cid/ciad681
- Pendlebury GA, Roman J, Shrivastava V, et al. A call to action: evidence for the military integration of teledermoscopy in a pandemic era. Dermatopathology (Basel). 2022;9:327-342. doi:10.3390/dermatopathology9040039
- Bhagchandani R, Singhi S, Peter JV, et al. Tropical fevers: management guidelines. Indian J Crit Care Med. 2014;18:62-69. doi:10.4103/0972-5229.126074
- Joint Chiefs of Staff. Joint Publication 4-02: Joint Health Services. December 11, 2017. Accessed March 5, 2026. https://cdmrp.health.mil/pubs/pdf/Joint%20Health%20Services%20Publication%20JP%204-02.pdf
- Armed Services Pest Management Board. Technical Guide No. 36: Personal Protective Measures Against Insects and Other Arthropods of Military Significance. Updated November 2015. Accessed March 5, 2026. https://www.acq.osd.mil/eie/afpmb/docs/techguides/tg36.pdf
- Global Emerging Infections Surveillance. Armed Forces Health Surveillance Division Annual Report 2024. Defense Health Agency; 2024:15-28. Accessed March 17, 2026. https://www.health.mil/Reference-Center/Reports/2026/01/05/AFHSD-Annual-Report-2024
Cutaneous Manifestations of Neglected Infectious Diseases in US Military Personnel
Cutaneous Manifestations of Neglected Infectious Diseases in US Military Personnel
Practice Points
- Military personnel stationed overseas are at risk for encountering infectious organisms that are not regularly observed domestically—many of which have cutaneous manifestations.
- Health care professionals treating military personnel should consider uncommonly encountered infections in the differential diagnosis for certain dermatologic presentations.
- Clinicians should inquire if patients have been stationed in Coccidioides immitis–endemic areas prior to the initiation of immunosuppression.

Solitary Papule on the Upper Back
Solitary Papule on the Upper Back
THE DIAGNOSIS: Plexiform Palisaded Encapsulated Neuroma
Microscopically, there was a superficial to deep dermal proliferation of tapered spindle cells in fascicles that were well circumscribed in nodules throughout the dermis with pale background stroma, mild mucin, and a thin capsule. The tapered spindle cells stained positive for SOX-10 and negative for Melan-A (Figure 1A). Staining for epithelial membrane antigen highlighted delicate cells around the periphery of the nodules, consistent with perineurium (Figure 1B). A diagnosis of plexiform palisaded encapsulated neuroma was made. No additional treatment was pursued due to the benign nature of the condition.
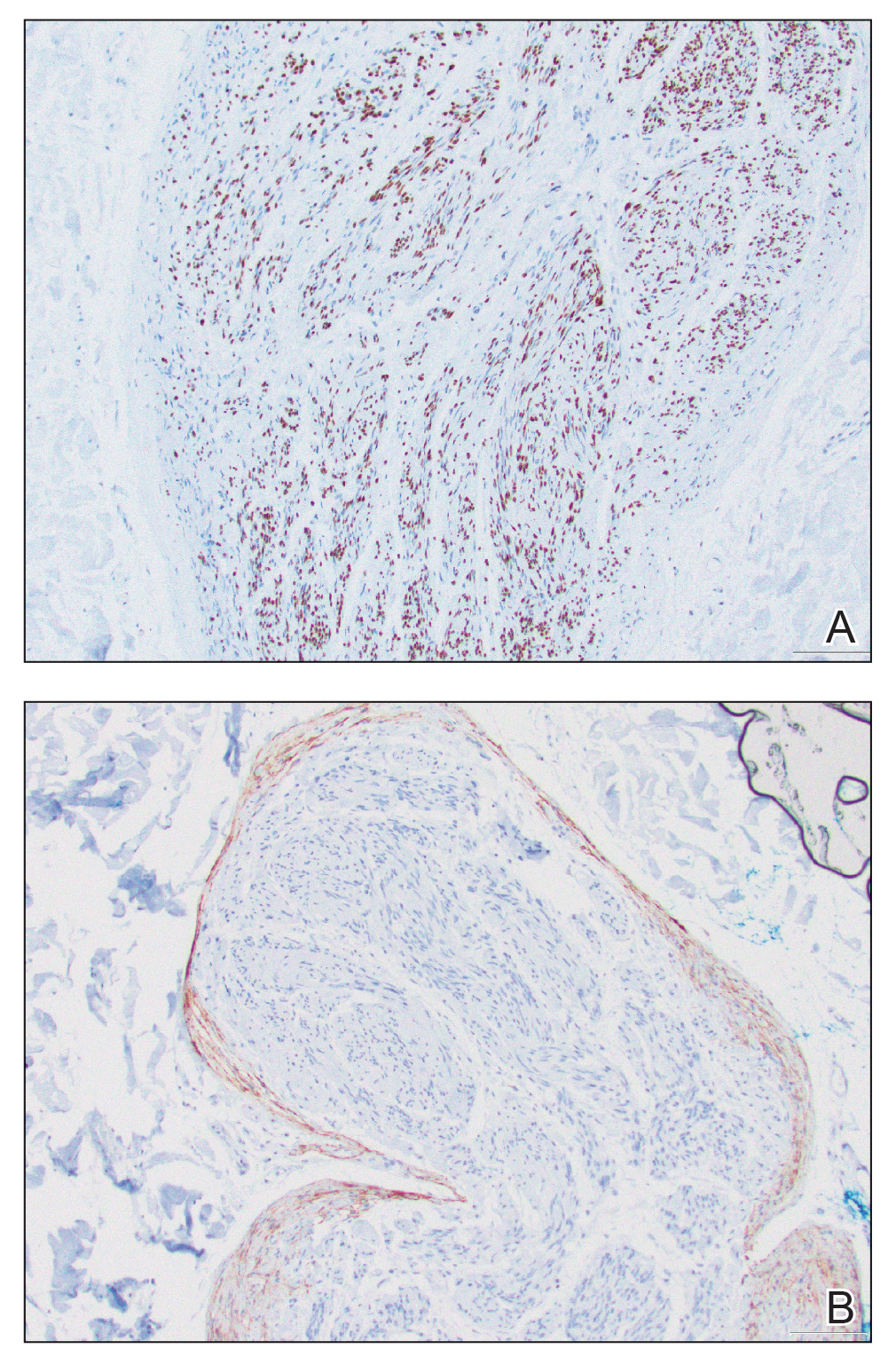
Palisaded encapsulated neuroma (PEN), also referred to as solitary circumscribed neuroma,1 is a benign, generally solitary neurogenic tumor that manifests predominantly on the skin, particularly in areas of frequent outside trauma such as the face. Lesions also may occur on mucosal and acral sites.2 First described by Reed et al3 in 1972, PEN characteristically manifests as a well-circumscribed, dermal nodule with a distinctive palisading pattern of Schwann cells and axons within a delicate perineurial capsule, the latter of which may be incomplete.3 Palisaded encapsulated neuroma frequently exhibits clefting between the tumor and the surrounding dermis. While PEN generally is sporadic, rare cases have been reported in association with Cowden syndrome and neurofibromatosis type 2.4,5
While the nodular growth pattern is most common, PEN also may present in epithelioid, plexiform, multinodular, or fungating subtypes.6 The plexiform subtype of PEN is rare. It has a complex growth pattern and a tendency to involve multiple adjacent nerve bundles in a plexiform arrangement.6,7 In two independent reviews characterizing the predominant growth patterns of PEN, nonnodular growth patterns were observed in a minority of the 85 cases: 18.8% (16/85) were plexiform, 7.1% (6/85) were multinodular or multilobular, and 7.1% (6/85) were fungating.6,7
The clinical presentation of plexiform PEN often includes a painless, slow-growing mass, and it predominantly occurs in middle-aged adults.2 Immunohistochemical staining reveals diffuse positivity for SOX-10 and S-100, which highlights the neural origin of the tumor.6 This variant, like conventional PEN, lacks notable atypia or mitotic activity.
Palisaded encapsulated neuroma, regardless of subtype, has an excellent prognosis, with no known cases of malignant transformation, and surgical excision with clear margins is curative.8
The differential diagnosis for plexiform PEN includes plexiform variants of neurofibroma and schwannoma, traumatic neuroma, and malignant peripheral nerve sheath tumor.
Neurofibromas are nonencapsulated lesions composed of spindle cells with wavy nuclei dispersed in a myxoid background.8 Neurofibromas can manifest in various locations throughout the body, including the skin, subcutaneous tissues, and internal organs. They are slow-growing tumors but may accelerate during periods of hormonal changes, such as pregnancy and puberty, or in cases of malignant transformation.8 Although plexiform neurofibromas are benign, malignant transformation can occur, particularly in patients with neurofibromatosis type 1 (NF1).8,9 Neurofibromas may assume one of 3 growth patterns: localized, diffuse, or plexiform.8 Plexiform neurofibromas exhibit a multinodular, ropelike growth pattern with a mix of Schwann cells and fibroblasts (Figure 2).8,9 These lesions are pathognomonic for NF1 and can infiltrate the surrounding tissue. They may involve large nerve trunks, leading to a more complex growth pattern compared to solitary neurofibromas.8,9 The plexiform variants of both neurofibromas and PEN demonstrate a multinodular growth pattern; however, plexiform neurofibromas are nonencapsulated and show a more diffuse infiltrative nature, whereas plexiform PEN remains well circumscribed. Additionally, plexiform neurofibromas are associated with NF1, while plexiform PEN lacks this genetic association.
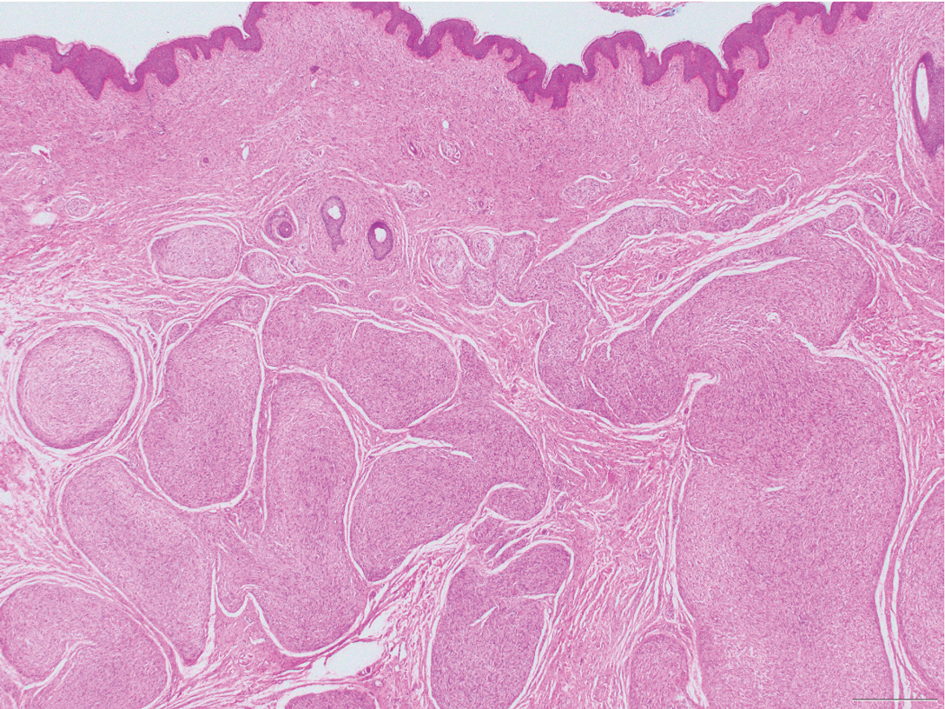
Schwannomas are encapsulated tumors that originate from the outer sheath of peripheral nerves, usually positioned eccentrically to the nerve fibers. Schwannomas are characterized by Antoni A and Antoni B areas, which usually are absent in PEN. Antoni A areas are composed of compact spindle cells arranged in palisades with Verocay bodies, while Antoni B areas are more loosely arranged and have a myxoid background (Figure 3).8,9 Schwannomas stain positive for S-100 and often show degenerative changes such as cystic degeneration or calcification, particularly in larger lesions.8,9 Plexiform schwannoma is a rare variant of schwannoma, and while it carries a substantial risk for local recurrence with rates as high as 50%, it has not been shown to possess malignant or metastatic potential.10 Unlike PEN, schwannomas have a consistent capsule but share S-100 positivity with PEN. Verocay bodies occasionally can be observed in PENs, with studies reporting their presence in 20% to 36% of cases.7,11,12 Additionally, some schwannomas may exhibit few Verocay bodies or poorly developed forms, which can make histopathologic distinction more challenging.7,11,12
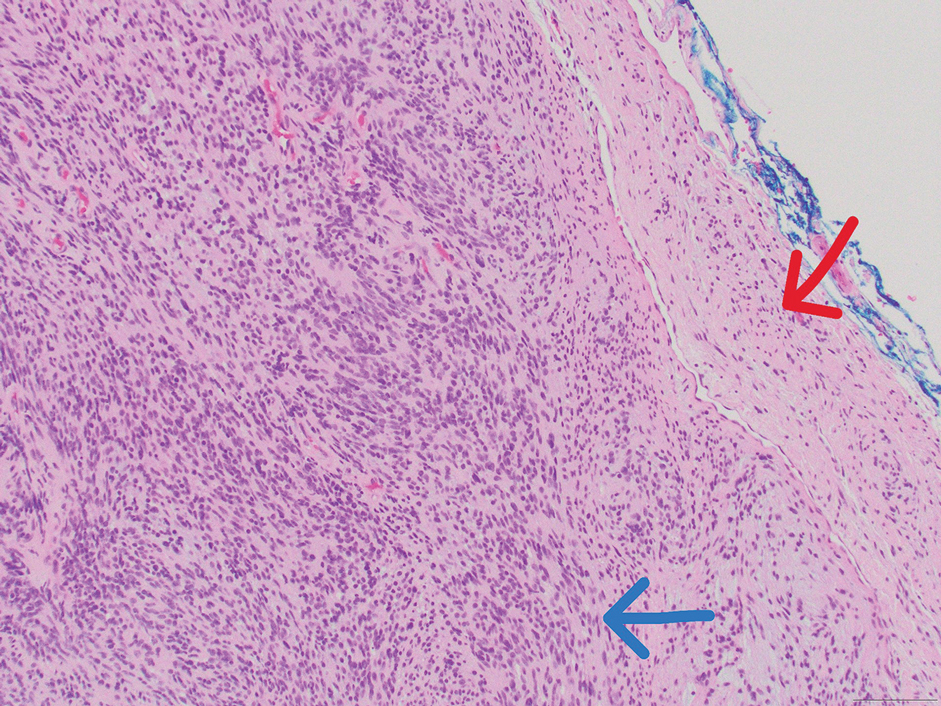
Traumatic neuromas result from nerve regeneration following any type of outside trauma that is deep enough to cause nerve injury. The lesion often is painful and associated with prior trauma or surgery. Under optimal conditions, the severed ends of a nerve reconnect through the orderly growth of axons from the proximal stump to the distal stump, guided by tubes formed by proliferating Schwann cells. If the nerve ends are not properly aligned or if the distal stump is absent, the axons may proliferate in a disorganized manner at the proximal stump, resulting in the formation of a traumatic neuroma.8 Histologically, these lesions exhibit disorganized, proliferating nerve fibers intermixed with fibrous stroma.8,13 The nerve fibers are not encapsulated, and there is an irregular arrangement of axons and Schwann cells (Figure 4).8,13 Unlike PEN, which usually is encapsulated and well organized with fascicular architecture, traumatic neuromas exhibit a disorganized, haphazard arrangement of neural elements and lack a capsule.8 Clinically, traumatic neuromas also are more likely to be painful.
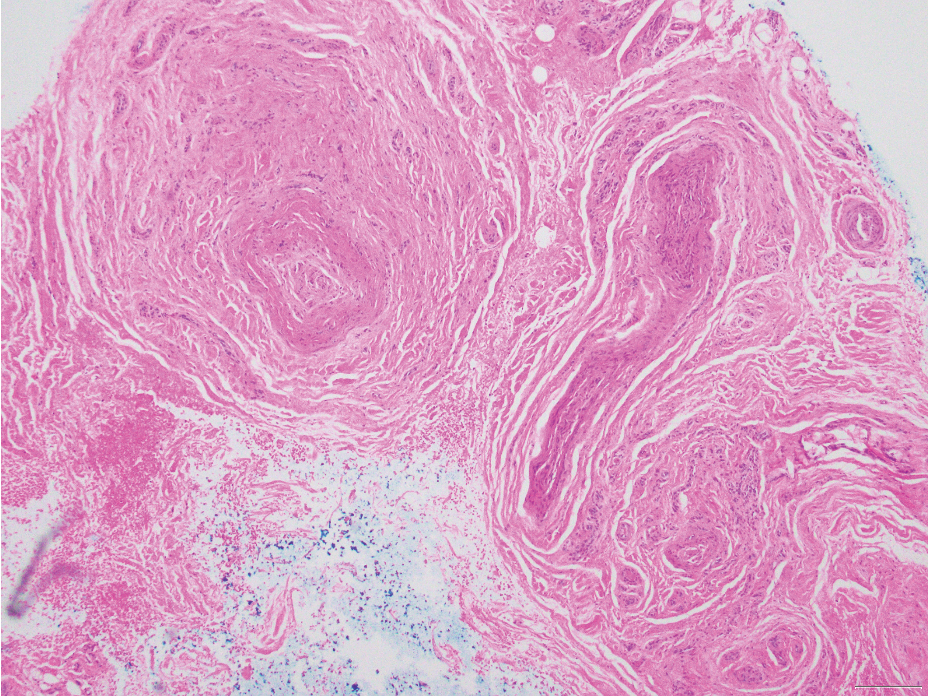
Malignant peripheral nerve sheath tumors are aggressive malignant spindle-cell tumors that may be associated with NF1 or occur sporadically.9,14 The spindle cells are arranged in fascicles, and these tumors can have areas of necrosis, hemorrhage, and high mitotic activity.9,15 The spindle cells may be arranged in a herringbone pattern, and alternating areas of hypocellularity and hypercellularity impart a marbled appearance (Figure 5).16 Malignant peripheral nerve sheath tumors frequently exhibit inactivation of the SWI/SNF-related, matrix- associated, actin-dependent regulator of chromatin subfamily B member 1 gene and loss of integrase interactor 1 protein. Transformation from plexiform neurofibroma to malignant peripheral nerve sheath tumor frequently is accompanied by progressive genomic changes.17 Malignant peripheral nerve sheath tumors differ substantially from PEN in their aggressive histologic features, including nuclear atypia and mitotic figures, which are absent in PEN.
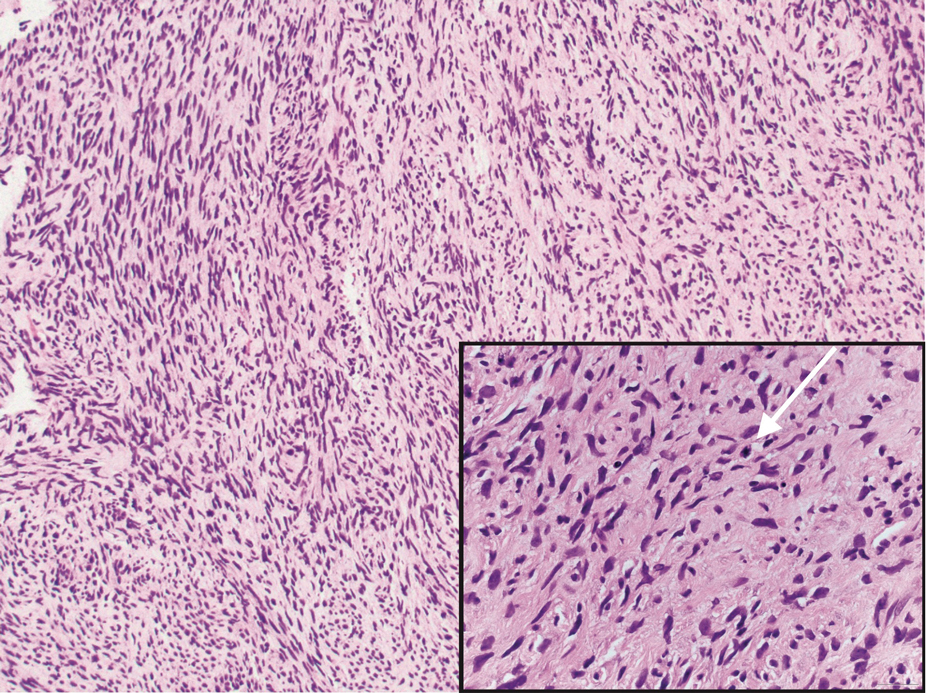
- Fletcher CD. Solitary circumscribed neuroma of the skin (so-called palisaded, encapsulated neuroma). a clinicopathologic and immunohistochemical study. Am J Surg Pathol. 1989;13:574-580. doi:10.1097/00000478-198907000-00005
- Jokinen CH, Ragsdale BD, Argenyi ZB. Expanding the clinicopathologic spectrum of palisaded encapsulated neuroma. J Cutan Pathol. 2010;37:43-48. doi:10.1111/j.1600-0560.2009.01380.x
- Reed RJ, Fine RM, Meltzer HD. Palisaded encapsulated neuromas of the skin. Arch Dermatol. 1972;106:865-870.
- Harris E, Mir A. Acral plexiform palisaded encapsulated neuromas as the initial cutaneous manifestation of Cowden syndrome. Pediatr Dermatol. 2017;34:E219-E220. doi:10.1111/pde.13161
- Arole V, Shaker N, Kim LR, et al. Multiple cutaneous solitary circumscribed neuroma in a patient with neurofibromatosis type 2: an “incidentaloma” or new association?. Int J Surg Pathol. 2023;31:734-737. doi:10.1177/10668969221120782
- Argenyi ZB, Cooper PH, Cruz DS. Plexiform and other unusual variants of palisaded encapsulated neuroma. J Cutan Pathol. 1993;20:34-39. doi:10.1111/j.1600-0560.1993.tb01246.x
- Leblebici C, Savli TC, Yeni B, et al. Palisaded encapsulated (solitary circumscribed) neuroma: a review of 30 cases. Int J Surg Pathol. 2019;27:506-514. doi:10.1177/1066896919833172
- Weiss SW, Goldblum JR. Enzinger and Weiss’s Soft Tissue Tumors. 6th ed. Elsevier Saunders; 2014.
- Rodriguez FJ, Folpe AL, Giannini C, et al. Pathology of peripheral nerve sheath tumors: diagnostic overview and update on selected diagnostic problems. Acta Neuropathol. 2012;123:295-319. doi:10.1007 /s00401-012-0954-z
- Berg JC, Scheithauer BW, Spinner RJ, et al. Plexiform schwannoma: a clinicopathologic overview with emphasis on the head and neck region. Hum Pathol. 2008;39:633-640. doi:10.1016 /j.humpath.2007.10.029
- Koutlas IG, Scheithauer BW. Palisaded encapsulated (“solitary circumscribed”) neuroma of the oral cavity: a review of 55 cases. Head Neck Pathol. 2010;4:15-26. doi:10.1007/s12105-010-0162-x
- Kossard S, Kumar A, Wilkinson B. Neural spectrum: palisaded encapsulated neuroma and verocay body poor dermal schwannoma. J Cutan Pathol. 1999;26:31-36. doi:10.1111/j.1600-0560.1999 .tb01787.x
- Yang H, Dong Y, Wang Z, et al. Traumatic neuromas of peripheral nerves: diagnosis, management and future perspectives. Front Neurol. 2023;13:1039529. doi:10.3389/fneur.2022.1039529
- Knight SWE, Knight TE, Santiago T, et al. Malignant peripheral nerve sheath tumors-a comprehensive review of pathophysiology, diagnosis, and multidisciplinary management. Children (Basel). 2022;9:38. doi:10.3390/children9010038
- Perry A, Gutmann DH. Malignant peripheral nerve sheath tumors: clinical and genetic aspects of pathogenesis. Clin Neuropathol. 2000;19:105-114.
- Lindberg G, Lucas D, Cassarino D, et al, eds. Diagnostic Pathology: Soft Tissue Tumors. 3rd ed. Elsevier; 2023.
- Miettinen MM, Antonescu CR, Fletcher CDM, et al. Histopathologic evaluation of atypical neurofibromatous tumors and their transformation into malignant peripheral nerve sheath tumor in patients with neurofibromatosis 1-a consensus overview. Hum Pathol. 2017;67:1-10. doi:10.1016/j.humpath.2017.05.010
THE DIAGNOSIS: Plexiform Palisaded Encapsulated Neuroma
Microscopically, there was a superficial to deep dermal proliferation of tapered spindle cells in fascicles that were well circumscribed in nodules throughout the dermis with pale background stroma, mild mucin, and a thin capsule. The tapered spindle cells stained positive for SOX-10 and negative for Melan-A (Figure 1A). Staining for epithelial membrane antigen highlighted delicate cells around the periphery of the nodules, consistent with perineurium (Figure 1B). A diagnosis of plexiform palisaded encapsulated neuroma was made. No additional treatment was pursued due to the benign nature of the condition.
Palisaded encapsulated neuroma (PEN), also referred to as solitary circumscribed neuroma,1 is a benign, generally solitary neurogenic tumor that manifests predominantly on the skin, particularly in areas of frequent outside trauma such as the face. Lesions also may occur on mucosal and acral sites.2 First described by Reed et al3 in 1972, PEN characteristically manifests as a well-circumscribed, dermal nodule with a distinctive palisading pattern of Schwann cells and axons within a delicate perineurial capsule, the latter of which may be incomplete.3 Palisaded encapsulated neuroma frequently exhibits clefting between the tumor and the surrounding dermis. While PEN generally is sporadic, rare cases have been reported in association with Cowden syndrome and neurofibromatosis type 2.4,5
While the nodular growth pattern is most common, PEN also may present in epithelioid, plexiform, multinodular, or fungating subtypes.6 The plexiform subtype of PEN is rare. It has a complex growth pattern and a tendency to involve multiple adjacent nerve bundles in a plexiform arrangement.6,7 In two independent reviews characterizing the predominant growth patterns of PEN, nonnodular growth patterns were observed in a minority of the 85 cases: 18.8% (16/85) were plexiform, 7.1% (6/85) were multinodular or multilobular, and 7.1% (6/85) were fungating.6,7
The clinical presentation of plexiform PEN often includes a painless, slow-growing mass, and it predominantly occurs in middle-aged adults.2 Immunohistochemical staining reveals diffuse positivity for SOX-10 and S-100, which highlights the neural origin of the tumor.6 This variant, like conventional PEN, lacks notable atypia or mitotic activity.
Palisaded encapsulated neuroma, regardless of subtype, has an excellent prognosis, with no known cases of malignant transformation, and surgical excision with clear margins is curative.8
The differential diagnosis for plexiform PEN includes plexiform variants of neurofibroma and schwannoma, traumatic neuroma, and malignant peripheral nerve sheath tumor.
Neurofibromas are nonencapsulated lesions composed of spindle cells with wavy nuclei dispersed in a myxoid background.8 Neurofibromas can manifest in various locations throughout the body, including the skin, subcutaneous tissues, and internal organs. They are slow-growing tumors but may accelerate during periods of hormonal changes, such as pregnancy and puberty, or in cases of malignant transformation.8 Although plexiform neurofibromas are benign, malignant transformation can occur, particularly in patients with neurofibromatosis type 1 (NF1).8,9 Neurofibromas may assume one of 3 growth patterns: localized, diffuse, or plexiform.8 Plexiform neurofibromas exhibit a multinodular, ropelike growth pattern with a mix of Schwann cells and fibroblasts (Figure 2).8,9 These lesions are pathognomonic for NF1 and can infiltrate the surrounding tissue. They may involve large nerve trunks, leading to a more complex growth pattern compared to solitary neurofibromas.8,9 The plexiform variants of both neurofibromas and PEN demonstrate a multinodular growth pattern; however, plexiform neurofibromas are nonencapsulated and show a more diffuse infiltrative nature, whereas plexiform PEN remains well circumscribed. Additionally, plexiform neurofibromas are associated with NF1, while plexiform PEN lacks this genetic association.
Schwannomas are encapsulated tumors that originate from the outer sheath of peripheral nerves, usually positioned eccentrically to the nerve fibers. Schwannomas are characterized by Antoni A and Antoni B areas, which usually are absent in PEN. Antoni A areas are composed of compact spindle cells arranged in palisades with Verocay bodies, while Antoni B areas are more loosely arranged and have a myxoid background (Figure 3).8,9 Schwannomas stain positive for S-100 and often show degenerative changes such as cystic degeneration or calcification, particularly in larger lesions.8,9 Plexiform schwannoma is a rare variant of schwannoma, and while it carries a substantial risk for local recurrence with rates as high as 50%, it has not been shown to possess malignant or metastatic potential.10 Unlike PEN, schwannomas have a consistent capsule but share S-100 positivity with PEN. Verocay bodies occasionally can be observed in PENs, with studies reporting their presence in 20% to 36% of cases.7,11,12 Additionally, some schwannomas may exhibit few Verocay bodies or poorly developed forms, which can make histopathologic distinction more challenging.7,11,12
Traumatic neuromas result from nerve regeneration following any type of outside trauma that is deep enough to cause nerve injury. The lesion often is painful and associated with prior trauma or surgery. Under optimal conditions, the severed ends of a nerve reconnect through the orderly growth of axons from the proximal stump to the distal stump, guided by tubes formed by proliferating Schwann cells. If the nerve ends are not properly aligned or if the distal stump is absent, the axons may proliferate in a disorganized manner at the proximal stump, resulting in the formation of a traumatic neuroma.8 Histologically, these lesions exhibit disorganized, proliferating nerve fibers intermixed with fibrous stroma.8,13 The nerve fibers are not encapsulated, and there is an irregular arrangement of axons and Schwann cells (Figure 4).8,13 Unlike PEN, which usually is encapsulated and well organized with fascicular architecture, traumatic neuromas exhibit a disorganized, haphazard arrangement of neural elements and lack a capsule.8 Clinically, traumatic neuromas also are more likely to be painful.
Malignant peripheral nerve sheath tumors are aggressive malignant spindle-cell tumors that may be associated with NF1 or occur sporadically.9,14 The spindle cells are arranged in fascicles, and these tumors can have areas of necrosis, hemorrhage, and high mitotic activity.9,15 The spindle cells may be arranged in a herringbone pattern, and alternating areas of hypocellularity and hypercellularity impart a marbled appearance (Figure 5).16 Malignant peripheral nerve sheath tumors frequently exhibit inactivation of the SWI/SNF-related, matrix- associated, actin-dependent regulator of chromatin subfamily B member 1 gene and loss of integrase interactor 1 protein. Transformation from plexiform neurofibroma to malignant peripheral nerve sheath tumor frequently is accompanied by progressive genomic changes.17 Malignant peripheral nerve sheath tumors differ substantially from PEN in their aggressive histologic features, including nuclear atypia and mitotic figures, which are absent in PEN.
THE DIAGNOSIS: Plexiform Palisaded Encapsulated Neuroma
Microscopically, there was a superficial to deep dermal proliferation of tapered spindle cells in fascicles that were well circumscribed in nodules throughout the dermis with pale background stroma, mild mucin, and a thin capsule. The tapered spindle cells stained positive for SOX-10 and negative for Melan-A (Figure 1A). Staining for epithelial membrane antigen highlighted delicate cells around the periphery of the nodules, consistent with perineurium (Figure 1B). A diagnosis of plexiform palisaded encapsulated neuroma was made. No additional treatment was pursued due to the benign nature of the condition.
Palisaded encapsulated neuroma (PEN), also referred to as solitary circumscribed neuroma,1 is a benign, generally solitary neurogenic tumor that manifests predominantly on the skin, particularly in areas of frequent outside trauma such as the face. Lesions also may occur on mucosal and acral sites.2 First described by Reed et al3 in 1972, PEN characteristically manifests as a well-circumscribed, dermal nodule with a distinctive palisading pattern of Schwann cells and axons within a delicate perineurial capsule, the latter of which may be incomplete.3 Palisaded encapsulated neuroma frequently exhibits clefting between the tumor and the surrounding dermis. While PEN generally is sporadic, rare cases have been reported in association with Cowden syndrome and neurofibromatosis type 2.4,5
While the nodular growth pattern is most common, PEN also may present in epithelioid, plexiform, multinodular, or fungating subtypes.6 The plexiform subtype of PEN is rare. It has a complex growth pattern and a tendency to involve multiple adjacent nerve bundles in a plexiform arrangement.6,7 In two independent reviews characterizing the predominant growth patterns of PEN, nonnodular growth patterns were observed in a minority of the 85 cases: 18.8% (16/85) were plexiform, 7.1% (6/85) were multinodular or multilobular, and 7.1% (6/85) were fungating.6,7
The clinical presentation of plexiform PEN often includes a painless, slow-growing mass, and it predominantly occurs in middle-aged adults.2 Immunohistochemical staining reveals diffuse positivity for SOX-10 and S-100, which highlights the neural origin of the tumor.6 This variant, like conventional PEN, lacks notable atypia or mitotic activity.
Palisaded encapsulated neuroma, regardless of subtype, has an excellent prognosis, with no known cases of malignant transformation, and surgical excision with clear margins is curative.8
The differential diagnosis for plexiform PEN includes plexiform variants of neurofibroma and schwannoma, traumatic neuroma, and malignant peripheral nerve sheath tumor.
Neurofibromas are nonencapsulated lesions composed of spindle cells with wavy nuclei dispersed in a myxoid background.8 Neurofibromas can manifest in various locations throughout the body, including the skin, subcutaneous tissues, and internal organs. They are slow-growing tumors but may accelerate during periods of hormonal changes, such as pregnancy and puberty, or in cases of malignant transformation.8 Although plexiform neurofibromas are benign, malignant transformation can occur, particularly in patients with neurofibromatosis type 1 (NF1).8,9 Neurofibromas may assume one of 3 growth patterns: localized, diffuse, or plexiform.8 Plexiform neurofibromas exhibit a multinodular, ropelike growth pattern with a mix of Schwann cells and fibroblasts (Figure 2).8,9 These lesions are pathognomonic for NF1 and can infiltrate the surrounding tissue. They may involve large nerve trunks, leading to a more complex growth pattern compared to solitary neurofibromas.8,9 The plexiform variants of both neurofibromas and PEN demonstrate a multinodular growth pattern; however, plexiform neurofibromas are nonencapsulated and show a more diffuse infiltrative nature, whereas plexiform PEN remains well circumscribed. Additionally, plexiform neurofibromas are associated with NF1, while plexiform PEN lacks this genetic association.
Schwannomas are encapsulated tumors that originate from the outer sheath of peripheral nerves, usually positioned eccentrically to the nerve fibers. Schwannomas are characterized by Antoni A and Antoni B areas, which usually are absent in PEN. Antoni A areas are composed of compact spindle cells arranged in palisades with Verocay bodies, while Antoni B areas are more loosely arranged and have a myxoid background (Figure 3).8,9 Schwannomas stain positive for S-100 and often show degenerative changes such as cystic degeneration or calcification, particularly in larger lesions.8,9 Plexiform schwannoma is a rare variant of schwannoma, and while it carries a substantial risk for local recurrence with rates as high as 50%, it has not been shown to possess malignant or metastatic potential.10 Unlike PEN, schwannomas have a consistent capsule but share S-100 positivity with PEN. Verocay bodies occasionally can be observed in PENs, with studies reporting their presence in 20% to 36% of cases.7,11,12 Additionally, some schwannomas may exhibit few Verocay bodies or poorly developed forms, which can make histopathologic distinction more challenging.7,11,12
Traumatic neuromas result from nerve regeneration following any type of outside trauma that is deep enough to cause nerve injury. The lesion often is painful and associated with prior trauma or surgery. Under optimal conditions, the severed ends of a nerve reconnect through the orderly growth of axons from the proximal stump to the distal stump, guided by tubes formed by proliferating Schwann cells. If the nerve ends are not properly aligned or if the distal stump is absent, the axons may proliferate in a disorganized manner at the proximal stump, resulting in the formation of a traumatic neuroma.8 Histologically, these lesions exhibit disorganized, proliferating nerve fibers intermixed with fibrous stroma.8,13 The nerve fibers are not encapsulated, and there is an irregular arrangement of axons and Schwann cells (Figure 4).8,13 Unlike PEN, which usually is encapsulated and well organized with fascicular architecture, traumatic neuromas exhibit a disorganized, haphazard arrangement of neural elements and lack a capsule.8 Clinically, traumatic neuromas also are more likely to be painful.
Malignant peripheral nerve sheath tumors are aggressive malignant spindle-cell tumors that may be associated with NF1 or occur sporadically.9,14 The spindle cells are arranged in fascicles, and these tumors can have areas of necrosis, hemorrhage, and high mitotic activity.9,15 The spindle cells may be arranged in a herringbone pattern, and alternating areas of hypocellularity and hypercellularity impart a marbled appearance (Figure 5).16 Malignant peripheral nerve sheath tumors frequently exhibit inactivation of the SWI/SNF-related, matrix- associated, actin-dependent regulator of chromatin subfamily B member 1 gene and loss of integrase interactor 1 protein. Transformation from plexiform neurofibroma to malignant peripheral nerve sheath tumor frequently is accompanied by progressive genomic changes.17 Malignant peripheral nerve sheath tumors differ substantially from PEN in their aggressive histologic features, including nuclear atypia and mitotic figures, which are absent in PEN.
- Fletcher CD. Solitary circumscribed neuroma of the skin (so-called palisaded, encapsulated neuroma). a clinicopathologic and immunohistochemical study. Am J Surg Pathol. 1989;13:574-580. doi:10.1097/00000478-198907000-00005
- Jokinen CH, Ragsdale BD, Argenyi ZB. Expanding the clinicopathologic spectrum of palisaded encapsulated neuroma. J Cutan Pathol. 2010;37:43-48. doi:10.1111/j.1600-0560.2009.01380.x
- Reed RJ, Fine RM, Meltzer HD. Palisaded encapsulated neuromas of the skin. Arch Dermatol. 1972;106:865-870.
- Harris E, Mir A. Acral plexiform palisaded encapsulated neuromas as the initial cutaneous manifestation of Cowden syndrome. Pediatr Dermatol. 2017;34:E219-E220. doi:10.1111/pde.13161
- Arole V, Shaker N, Kim LR, et al. Multiple cutaneous solitary circumscribed neuroma in a patient with neurofibromatosis type 2: an “incidentaloma” or new association?. Int J Surg Pathol. 2023;31:734-737. doi:10.1177/10668969221120782
- Argenyi ZB, Cooper PH, Cruz DS. Plexiform and other unusual variants of palisaded encapsulated neuroma. J Cutan Pathol. 1993;20:34-39. doi:10.1111/j.1600-0560.1993.tb01246.x
- Leblebici C, Savli TC, Yeni B, et al. Palisaded encapsulated (solitary circumscribed) neuroma: a review of 30 cases. Int J Surg Pathol. 2019;27:506-514. doi:10.1177/1066896919833172
- Weiss SW, Goldblum JR. Enzinger and Weiss’s Soft Tissue Tumors. 6th ed. Elsevier Saunders; 2014.
- Rodriguez FJ, Folpe AL, Giannini C, et al. Pathology of peripheral nerve sheath tumors: diagnostic overview and update on selected diagnostic problems. Acta Neuropathol. 2012;123:295-319. doi:10.1007 /s00401-012-0954-z
- Berg JC, Scheithauer BW, Spinner RJ, et al. Plexiform schwannoma: a clinicopathologic overview with emphasis on the head and neck region. Hum Pathol. 2008;39:633-640. doi:10.1016 /j.humpath.2007.10.029
- Koutlas IG, Scheithauer BW. Palisaded encapsulated (“solitary circumscribed”) neuroma of the oral cavity: a review of 55 cases. Head Neck Pathol. 2010;4:15-26. doi:10.1007/s12105-010-0162-x
- Kossard S, Kumar A, Wilkinson B. Neural spectrum: palisaded encapsulated neuroma and verocay body poor dermal schwannoma. J Cutan Pathol. 1999;26:31-36. doi:10.1111/j.1600-0560.1999 .tb01787.x
- Yang H, Dong Y, Wang Z, et al. Traumatic neuromas of peripheral nerves: diagnosis, management and future perspectives. Front Neurol. 2023;13:1039529. doi:10.3389/fneur.2022.1039529
- Knight SWE, Knight TE, Santiago T, et al. Malignant peripheral nerve sheath tumors-a comprehensive review of pathophysiology, diagnosis, and multidisciplinary management. Children (Basel). 2022;9:38. doi:10.3390/children9010038
- Perry A, Gutmann DH. Malignant peripheral nerve sheath tumors: clinical and genetic aspects of pathogenesis. Clin Neuropathol. 2000;19:105-114.
- Lindberg G, Lucas D, Cassarino D, et al, eds. Diagnostic Pathology: Soft Tissue Tumors. 3rd ed. Elsevier; 2023.
- Miettinen MM, Antonescu CR, Fletcher CDM, et al. Histopathologic evaluation of atypical neurofibromatous tumors and their transformation into malignant peripheral nerve sheath tumor in patients with neurofibromatosis 1-a consensus overview. Hum Pathol. 2017;67:1-10. doi:10.1016/j.humpath.2017.05.010
- Fletcher CD. Solitary circumscribed neuroma of the skin (so-called palisaded, encapsulated neuroma). a clinicopathologic and immunohistochemical study. Am J Surg Pathol. 1989;13:574-580. doi:10.1097/00000478-198907000-00005
- Jokinen CH, Ragsdale BD, Argenyi ZB. Expanding the clinicopathologic spectrum of palisaded encapsulated neuroma. J Cutan Pathol. 2010;37:43-48. doi:10.1111/j.1600-0560.2009.01380.x
- Reed RJ, Fine RM, Meltzer HD. Palisaded encapsulated neuromas of the skin. Arch Dermatol. 1972;106:865-870.
- Harris E, Mir A. Acral plexiform palisaded encapsulated neuromas as the initial cutaneous manifestation of Cowden syndrome. Pediatr Dermatol. 2017;34:E219-E220. doi:10.1111/pde.13161
- Arole V, Shaker N, Kim LR, et al. Multiple cutaneous solitary circumscribed neuroma in a patient with neurofibromatosis type 2: an “incidentaloma” or new association?. Int J Surg Pathol. 2023;31:734-737. doi:10.1177/10668969221120782
- Argenyi ZB, Cooper PH, Cruz DS. Plexiform and other unusual variants of palisaded encapsulated neuroma. J Cutan Pathol. 1993;20:34-39. doi:10.1111/j.1600-0560.1993.tb01246.x
- Leblebici C, Savli TC, Yeni B, et al. Palisaded encapsulated (solitary circumscribed) neuroma: a review of 30 cases. Int J Surg Pathol. 2019;27:506-514. doi:10.1177/1066896919833172
- Weiss SW, Goldblum JR. Enzinger and Weiss’s Soft Tissue Tumors. 6th ed. Elsevier Saunders; 2014.
- Rodriguez FJ, Folpe AL, Giannini C, et al. Pathology of peripheral nerve sheath tumors: diagnostic overview and update on selected diagnostic problems. Acta Neuropathol. 2012;123:295-319. doi:10.1007 /s00401-012-0954-z
- Berg JC, Scheithauer BW, Spinner RJ, et al. Plexiform schwannoma: a clinicopathologic overview with emphasis on the head and neck region. Hum Pathol. 2008;39:633-640. doi:10.1016 /j.humpath.2007.10.029
- Koutlas IG, Scheithauer BW. Palisaded encapsulated (“solitary circumscribed”) neuroma of the oral cavity: a review of 55 cases. Head Neck Pathol. 2010;4:15-26. doi:10.1007/s12105-010-0162-x
- Kossard S, Kumar A, Wilkinson B. Neural spectrum: palisaded encapsulated neuroma and verocay body poor dermal schwannoma. J Cutan Pathol. 1999;26:31-36. doi:10.1111/j.1600-0560.1999 .tb01787.x
- Yang H, Dong Y, Wang Z, et al. Traumatic neuromas of peripheral nerves: diagnosis, management and future perspectives. Front Neurol. 2023;13:1039529. doi:10.3389/fneur.2022.1039529
- Knight SWE, Knight TE, Santiago T, et al. Malignant peripheral nerve sheath tumors-a comprehensive review of pathophysiology, diagnosis, and multidisciplinary management. Children (Basel). 2022;9:38. doi:10.3390/children9010038
- Perry A, Gutmann DH. Malignant peripheral nerve sheath tumors: clinical and genetic aspects of pathogenesis. Clin Neuropathol. 2000;19:105-114.
- Lindberg G, Lucas D, Cassarino D, et al, eds. Diagnostic Pathology: Soft Tissue Tumors. 3rd ed. Elsevier; 2023.
- Miettinen MM, Antonescu CR, Fletcher CDM, et al. Histopathologic evaluation of atypical neurofibromatous tumors and their transformation into malignant peripheral nerve sheath tumor in patients with neurofibromatosis 1-a consensus overview. Hum Pathol. 2017;67:1-10. doi:10.1016/j.humpath.2017.05.010
Solitary Papule on the Upper Back
Solitary Papule on the Upper Back
An 88-year-old woman presented to the dermatology clinic with an asymptomatic papule on the left upper back of unknown duration. The patient reported that her medical history was negative for eczematous dermatitis, hypertension, and osteoarthritis. Physical examination revealed a firm, well-circumscribed, flesh-colored, 6-mm papule with no overlying scale or ulceration. No other concerning lesions were noted on full skin examination. A punch biopsy of the papule was performed.