User login
Bringing you the latest news, research and reviews, exclusive interviews, podcasts, quizzes, and more.
div[contains(@class, 'header__large-screen')]
div[contains(@class, 'read-next-article')]
div[contains(@class, 'nav-primary')]
nav[contains(@class, 'nav-primary')]
section[contains(@class, 'footer-nav-section-wrapper')]
footer[@id='footer']
div[contains(@class, 'main-prefix')]
section[contains(@class, 'nav-hidden')]
div[contains(@class, 'ce-card-content')]
nav[contains(@class, 'nav-ce-stack')]
FAVOR III China: QFR-guided PCI shows advantage over angiography
Percutaneous coronary intervention (PCI) guided by quantitative flow ratio (QFR) lesion assessment provided better clinical outcomes than visual assessment of the angiogram in the sham-controlled FAVOR III China study.
PCI success rates were about 95% with both strategies; however, QFR guidance was associated with fewer major adverse cardiac events (MACE) at 1 year, use of fewer stents, less contrast medium exposure, and fewer procedural complications.
“The simplicity and safety of QFR compared with wire-based physiologic measurements should facilitate the adoption of physiologic lesion assessment into routine clinical practice,” co–primary investigator Bo Xu, MBBS, Fuwai Hospital, Beijing, said.
The results were presented at Transcatheter Cardiovascular Therapeutics (TCT) 2021, held online and in Orlando, and published simultaneously in The Lancet.
Although pressure wire–based physiological assessment with fractional flow reserve (FFR) and instantaneous wave-free ratio (IFR) more accurately identify flow-limiting lesions than standard angiography and have been shown to improve outcomes after PCI, the authors note that it’s underused in practice because of prolonged procedural time, potential pressure wire complications, and side effects from hyperemic agents.
QFR, however, is derived from 3-dimensional coronary artery reconstruction and computational fluid dynamics from the angiogram, so FFR can be estimated without the need for a pressure wire or hyperemic drugs.
FAVOR III China was designed statistically for superiority and enrolled 3,847 patients with stable or unstable angina or a myocardial infarction (MI) at least 72 hours before screening if they had at least one coronary lesion with a diameter stenosis of 50% to 90% and a reference vessel diameter of at least 2.5 mm. The intention-to-treat population included 3,825 patients (mean age, 62.7 years; 29.4% female).
In the QFR group, QFR was measured in all coronary arteries with a lesion but PCI performed only in lesions with a QFR of at least 0.80 or diameter stenosis greater than 90%. Two angiographic imaging runs were taken and the data transmitted to the AngioPlus system (Pulse Medical Imaging Technology) by a local network of sites for QFR calculation.
PCI in the angiography-guided group was performed on the basis of visual angiographic assessment only. A 10-minute delay was used in both groups to preserve masking.
The primary endpoint of 1-year MACE, a composite of all-cause death, MI, or ischemia-driven revascularization, occurred in 5.8% of the QFR-guided group and 8.8% of the angiography-guided group (hazard ratio, 0.65; 95% CI, 0.51-0.83; P = .0004).
The curves separated within 48 hours, driven largely by fewer MIs (3.4% vs. 5.7%; P = .0008) and ischemia-driven revascularizations (2.0% vs. 3.1%; P = .0078) in the QFR-guided group, Mr. Xu said.
The major secondary endpoint of MACE excluding periprocedural MI occurred in 3.1% of QFR-guided patients and 4.8% of angiography-guided patients (HR, 0.64; 95% CI, 0.46-0.89; P = .0073).
The prerandomization revascularization plan was changed in 23.3% of patients with QFR and only 6.2% in the angiography group (P < .0001), mainly due to deferral of treatment of at least one vessel originally planned for PCI (19.6% vs. 5.2%; P < .0001).
“I think in the next guideline they will change the recommendation, not just to include FFR and IFR, but also to include QFR,” Giuseppe Tarantini, MD, PhD, University of Padua, Italy, said during a press briefing on the study.
“This is a milestone in our community, not only because it is easier to use compared to the other lesion-specific indexes like FFR, IFR, but also for the need to expand the use of physiology in the setting of interventional cardiology,” he added.
In an accompanying commentary, Robert A. Byrne, MBBCh, PhD, and Laurna McGovern, MBBCh, both from the Cardiovascular Research Institute Dublin, say the results are “relevant for cardiovascular disease researchers and clinicians and an important step forward for the field of angiography-derived flow measurements for guidance of PCI.”
They point out, however, that the control group did not receive pressure wire–guided PCI, which is the standard of care in contemporary practice and out of step with clinical practice guidelines, thus limiting external validity.
They also note that experiences to date suggest that up to 20% of patients may be unsuitable for the algorithm analysis because of coronary anatomy, presence of overlapping vessels, and insufficient image quality.
Commenting for this news organization, David E. Kandzari, MD, chief of the Piedmont Heart Institute, Atlanta, said “the technology isn’t readily available in catheterization labs today. Could it be assimilated into the cath labs at one point in the near term? I think absolutely, and that would be a welcome addition to expedite the procedure itself.”
Nevertheless, he said the results “need to be externally validated too, with what is the gold standard today of FFR in a larger experience.”
Session moderator Gregg W. Stone, MD, Icahn School of Medicine at Mount Sinai, New York, said FAVOR III China has “advanced our knowledge” but pointed out that the ongoing randomized FAVOR III Europe Japan study is directly comparing QFR with invasive pressure-wire assessed FFR. The estimated primary completion date for that study is Dec. 31.
The study was supported by grants from the Beijing Municipal Science and Technology Commission, Chinese Academy of Medical Sciences, and the National Clinical Research Center for Cardiovascular Diseases, Fuwai Hospital. Dr. Byrne reported institutional research or educational funding from Abbott Vascular, Biosensors, Biotronik, and Boston Scientific. Ms. McGovern has disclosed no relevant financial relationships. Dr. Kandzari reported minor consulting honoraria from the interventional device industry and institutional research grant support.
A version of this article first appeared on Medscape.com.
Percutaneous coronary intervention (PCI) guided by quantitative flow ratio (QFR) lesion assessment provided better clinical outcomes than visual assessment of the angiogram in the sham-controlled FAVOR III China study.
PCI success rates were about 95% with both strategies; however, QFR guidance was associated with fewer major adverse cardiac events (MACE) at 1 year, use of fewer stents, less contrast medium exposure, and fewer procedural complications.
“The simplicity and safety of QFR compared with wire-based physiologic measurements should facilitate the adoption of physiologic lesion assessment into routine clinical practice,” co–primary investigator Bo Xu, MBBS, Fuwai Hospital, Beijing, said.
The results were presented at Transcatheter Cardiovascular Therapeutics (TCT) 2021, held online and in Orlando, and published simultaneously in The Lancet.
Although pressure wire–based physiological assessment with fractional flow reserve (FFR) and instantaneous wave-free ratio (IFR) more accurately identify flow-limiting lesions than standard angiography and have been shown to improve outcomes after PCI, the authors note that it’s underused in practice because of prolonged procedural time, potential pressure wire complications, and side effects from hyperemic agents.
QFR, however, is derived from 3-dimensional coronary artery reconstruction and computational fluid dynamics from the angiogram, so FFR can be estimated without the need for a pressure wire or hyperemic drugs.
FAVOR III China was designed statistically for superiority and enrolled 3,847 patients with stable or unstable angina or a myocardial infarction (MI) at least 72 hours before screening if they had at least one coronary lesion with a diameter stenosis of 50% to 90% and a reference vessel diameter of at least 2.5 mm. The intention-to-treat population included 3,825 patients (mean age, 62.7 years; 29.4% female).
In the QFR group, QFR was measured in all coronary arteries with a lesion but PCI performed only in lesions with a QFR of at least 0.80 or diameter stenosis greater than 90%. Two angiographic imaging runs were taken and the data transmitted to the AngioPlus system (Pulse Medical Imaging Technology) by a local network of sites for QFR calculation.
PCI in the angiography-guided group was performed on the basis of visual angiographic assessment only. A 10-minute delay was used in both groups to preserve masking.
The primary endpoint of 1-year MACE, a composite of all-cause death, MI, or ischemia-driven revascularization, occurred in 5.8% of the QFR-guided group and 8.8% of the angiography-guided group (hazard ratio, 0.65; 95% CI, 0.51-0.83; P = .0004).
The curves separated within 48 hours, driven largely by fewer MIs (3.4% vs. 5.7%; P = .0008) and ischemia-driven revascularizations (2.0% vs. 3.1%; P = .0078) in the QFR-guided group, Mr. Xu said.
The major secondary endpoint of MACE excluding periprocedural MI occurred in 3.1% of QFR-guided patients and 4.8% of angiography-guided patients (HR, 0.64; 95% CI, 0.46-0.89; P = .0073).
The prerandomization revascularization plan was changed in 23.3% of patients with QFR and only 6.2% in the angiography group (P < .0001), mainly due to deferral of treatment of at least one vessel originally planned for PCI (19.6% vs. 5.2%; P < .0001).
“I think in the next guideline they will change the recommendation, not just to include FFR and IFR, but also to include QFR,” Giuseppe Tarantini, MD, PhD, University of Padua, Italy, said during a press briefing on the study.
“This is a milestone in our community, not only because it is easier to use compared to the other lesion-specific indexes like FFR, IFR, but also for the need to expand the use of physiology in the setting of interventional cardiology,” he added.
In an accompanying commentary, Robert A. Byrne, MBBCh, PhD, and Laurna McGovern, MBBCh, both from the Cardiovascular Research Institute Dublin, say the results are “relevant for cardiovascular disease researchers and clinicians and an important step forward for the field of angiography-derived flow measurements for guidance of PCI.”
They point out, however, that the control group did not receive pressure wire–guided PCI, which is the standard of care in contemporary practice and out of step with clinical practice guidelines, thus limiting external validity.
They also note that experiences to date suggest that up to 20% of patients may be unsuitable for the algorithm analysis because of coronary anatomy, presence of overlapping vessels, and insufficient image quality.
Commenting for this news organization, David E. Kandzari, MD, chief of the Piedmont Heart Institute, Atlanta, said “the technology isn’t readily available in catheterization labs today. Could it be assimilated into the cath labs at one point in the near term? I think absolutely, and that would be a welcome addition to expedite the procedure itself.”
Nevertheless, he said the results “need to be externally validated too, with what is the gold standard today of FFR in a larger experience.”
Session moderator Gregg W. Stone, MD, Icahn School of Medicine at Mount Sinai, New York, said FAVOR III China has “advanced our knowledge” but pointed out that the ongoing randomized FAVOR III Europe Japan study is directly comparing QFR with invasive pressure-wire assessed FFR. The estimated primary completion date for that study is Dec. 31.
The study was supported by grants from the Beijing Municipal Science and Technology Commission, Chinese Academy of Medical Sciences, and the National Clinical Research Center for Cardiovascular Diseases, Fuwai Hospital. Dr. Byrne reported institutional research or educational funding from Abbott Vascular, Biosensors, Biotronik, and Boston Scientific. Ms. McGovern has disclosed no relevant financial relationships. Dr. Kandzari reported minor consulting honoraria from the interventional device industry and institutional research grant support.
A version of this article first appeared on Medscape.com.
Percutaneous coronary intervention (PCI) guided by quantitative flow ratio (QFR) lesion assessment provided better clinical outcomes than visual assessment of the angiogram in the sham-controlled FAVOR III China study.
PCI success rates were about 95% with both strategies; however, QFR guidance was associated with fewer major adverse cardiac events (MACE) at 1 year, use of fewer stents, less contrast medium exposure, and fewer procedural complications.
“The simplicity and safety of QFR compared with wire-based physiologic measurements should facilitate the adoption of physiologic lesion assessment into routine clinical practice,” co–primary investigator Bo Xu, MBBS, Fuwai Hospital, Beijing, said.
The results were presented at Transcatheter Cardiovascular Therapeutics (TCT) 2021, held online and in Orlando, and published simultaneously in The Lancet.
Although pressure wire–based physiological assessment with fractional flow reserve (FFR) and instantaneous wave-free ratio (IFR) more accurately identify flow-limiting lesions than standard angiography and have been shown to improve outcomes after PCI, the authors note that it’s underused in practice because of prolonged procedural time, potential pressure wire complications, and side effects from hyperemic agents.
QFR, however, is derived from 3-dimensional coronary artery reconstruction and computational fluid dynamics from the angiogram, so FFR can be estimated without the need for a pressure wire or hyperemic drugs.
FAVOR III China was designed statistically for superiority and enrolled 3,847 patients with stable or unstable angina or a myocardial infarction (MI) at least 72 hours before screening if they had at least one coronary lesion with a diameter stenosis of 50% to 90% and a reference vessel diameter of at least 2.5 mm. The intention-to-treat population included 3,825 patients (mean age, 62.7 years; 29.4% female).
In the QFR group, QFR was measured in all coronary arteries with a lesion but PCI performed only in lesions with a QFR of at least 0.80 or diameter stenosis greater than 90%. Two angiographic imaging runs were taken and the data transmitted to the AngioPlus system (Pulse Medical Imaging Technology) by a local network of sites for QFR calculation.
PCI in the angiography-guided group was performed on the basis of visual angiographic assessment only. A 10-minute delay was used in both groups to preserve masking.
The primary endpoint of 1-year MACE, a composite of all-cause death, MI, or ischemia-driven revascularization, occurred in 5.8% of the QFR-guided group and 8.8% of the angiography-guided group (hazard ratio, 0.65; 95% CI, 0.51-0.83; P = .0004).
The curves separated within 48 hours, driven largely by fewer MIs (3.4% vs. 5.7%; P = .0008) and ischemia-driven revascularizations (2.0% vs. 3.1%; P = .0078) in the QFR-guided group, Mr. Xu said.
The major secondary endpoint of MACE excluding periprocedural MI occurred in 3.1% of QFR-guided patients and 4.8% of angiography-guided patients (HR, 0.64; 95% CI, 0.46-0.89; P = .0073).
The prerandomization revascularization plan was changed in 23.3% of patients with QFR and only 6.2% in the angiography group (P < .0001), mainly due to deferral of treatment of at least one vessel originally planned for PCI (19.6% vs. 5.2%; P < .0001).
“I think in the next guideline they will change the recommendation, not just to include FFR and IFR, but also to include QFR,” Giuseppe Tarantini, MD, PhD, University of Padua, Italy, said during a press briefing on the study.
“This is a milestone in our community, not only because it is easier to use compared to the other lesion-specific indexes like FFR, IFR, but also for the need to expand the use of physiology in the setting of interventional cardiology,” he added.
In an accompanying commentary, Robert A. Byrne, MBBCh, PhD, and Laurna McGovern, MBBCh, both from the Cardiovascular Research Institute Dublin, say the results are “relevant for cardiovascular disease researchers and clinicians and an important step forward for the field of angiography-derived flow measurements for guidance of PCI.”
They point out, however, that the control group did not receive pressure wire–guided PCI, which is the standard of care in contemporary practice and out of step with clinical practice guidelines, thus limiting external validity.
They also note that experiences to date suggest that up to 20% of patients may be unsuitable for the algorithm analysis because of coronary anatomy, presence of overlapping vessels, and insufficient image quality.
Commenting for this news organization, David E. Kandzari, MD, chief of the Piedmont Heart Institute, Atlanta, said “the technology isn’t readily available in catheterization labs today. Could it be assimilated into the cath labs at one point in the near term? I think absolutely, and that would be a welcome addition to expedite the procedure itself.”
Nevertheless, he said the results “need to be externally validated too, with what is the gold standard today of FFR in a larger experience.”
Session moderator Gregg W. Stone, MD, Icahn School of Medicine at Mount Sinai, New York, said FAVOR III China has “advanced our knowledge” but pointed out that the ongoing randomized FAVOR III Europe Japan study is directly comparing QFR with invasive pressure-wire assessed FFR. The estimated primary completion date for that study is Dec. 31.
The study was supported by grants from the Beijing Municipal Science and Technology Commission, Chinese Academy of Medical Sciences, and the National Clinical Research Center for Cardiovascular Diseases, Fuwai Hospital. Dr. Byrne reported institutional research or educational funding from Abbott Vascular, Biosensors, Biotronik, and Boston Scientific. Ms. McGovern has disclosed no relevant financial relationships. Dr. Kandzari reported minor consulting honoraria from the interventional device industry and institutional research grant support.
A version of this article first appeared on Medscape.com.
Does zinc really help treat colds?
A new study published in BMJ Open adds to the evidence that zinc is effective against viral respiratory infections, such as colds.
Jennifer Hunter, PhD, BMed, of Western Sydney University’s NICM Health Research Institute, New South Wales, Australia, and colleagues conducted a meta-analysis of 28 randomized controlled trials (RCTs). They searched 17 English and Chinese databases to identify the trials and then used the Cochrane rapid review technique for the analysis.
The trials included 5,446 adults who had received zinc in a variety of formulations and routes — oral, sublingual, and nasal spray. The researchers separately analyzed whether zinc prevented or treated respiratory tract infections (RTIs)
Oral or intranasal zinc prevented five RTIs per 100 person-months (95% CI, 1 – 8; numbers needed to treat, 20). There was a 32% lower relative risk (RR) of developing mild to moderate symptoms consistent with a viral RTI.
Use of zinc was also associated with an 87% lower risk of developing moderately severe symptoms (incidence rate ratio, 0.13; 95% CI, 0.04 – 0.38) and a 28% lower risk of developing milder symptoms. The largest reductions in RR were for moderately severe symptoms consistent with an influenza-like illness.
Symptoms resolved 2 days earlier with sublingual or intranasal zinc compared with placebo (95% CI, 0.61 – 3.50; very low-certainty quality of evidence). There were clinically significant reductions in day 3 symptom severity scores (mean difference, -1.20 points; 95% CI, -0.66 to -1.74; low-certainty quality of evidence) but not in overall symptom severity. Participants who used sublingual or topical nasal zinc early in the course of illness were 1.8 times more likely to recover before those who used a placebo.
However, the investigators found no benefit of zinc when patients were inoculated with rhinovirus; there was no reduction in the risk of developing a cold. Asked about this disparity, Dr. Hunter said, “It might well be that when inoculating people to make sure they get infected, you give them a really high dose of the virus. [This] doesn’t really mimic what happens in the real world.”
On the downside of supplemental zinc, there were more side effects among those who used zinc, including nausea or gastrointestinal discomfort, mouth irritation, or soreness from sublingual lozenges (RR, 1.41; 95% CI, 1.17 – 1.69; number needed to harm, 7; moderate-certainty quality of evidence). The risk for a serious adverse event, such as loss of smell or copper deficiency, was low. Although not found in these studies, postmarketing studies have found that there is a risk for severe and in some cases permanent loss of smell associated with the use of nasal gels or sprays containing zinc. Three such products were recalled from the market.
The trial could not provide answers about the comparative efficacy of different types of zinc formulations, nor could the investigators recommend specific doses. The trial was not designed to assess zinc for the prevention or treatment of COVID-19.
Asked for independent comment, pediatrician Aamer Imdad, MBBS, assistant professor at the State University of New York Upstate Medical University, Syracuse, told this news organization, “It’s a very comprehensive review for zinc-related studies in adults” but was challenging because of the “significant clinical heterogeneity in the population.”
Dr. Imdad explained that zinc has “absolutely” been shown to be effective for children with diarrhea. The World Health Organization has recommended it since 2004. “The way it works in diarrhea is that it helps with the regeneration of the epithelium.... It also improves the immunity itself, especially the cell-mediated immunity.” He raised the question of whether it might work similarly in the respiratory tract. Dr. Imdad has a long-standing interest in the use of zinc for pediatric infections. Regarding this study, he concluded, “I think we still need to know the nuts and bolts of this intervention before we can recommend it more specifically.”
Dr. Hunter said, “We don’t have any high-quality studies that have evaluated zinc orally as treatment once you’re actually infected and have symptoms of the cold or influenza, or COVID.”
Asked about zinc’s possible role, Dr. Hunter said, “So I do think it gives us a viable alternative. More people are going, ‘What can I do?’ And you know as well as I do people come to you, and [they say], ‘Well, just give me something. Even if it’s a day or a little bit of symptom relief, anything to make me feel better that isn’t going to hurt me and doesn’t have any major risks.’ So I think in the short term, clinicians and consumers can consider trying it.”
Dr. Hunter was not keen on giving zinc to family members after they develop an RTI: “Consider it. But I don’t think we have enough evidence to say definitely yes.” But she does see a potential role for “people who are at risk of suboptimal zinc absorption, like people who are taking a variety of pharmaceuticals [notably proton pump inhibitors] that block or reduce the absorption of zinc, people with a whole lot of the chronic diseases that we know are associated with an increased risk of worse outcomes from respiratory viral infections, and older adults. Yes, I think [for] those high-risk groups, you could consider using zinc, either in a moderate dose longer term or in a higher dose for very short bursts of, like, 1 to 2 weeks.”
Dr. Hunter concluded, “Up until now, we all commonly thought that zinc’s role was only for people who were zinc deficient, and now we’ve got some signals pointing towards its potential role as an anti-infective and anti-inflammatory agent in people who don’t have zinc deficiency.”
But both Dr. Hunter and Dr. Imdad emphasized that zinc is not a game changer. There is a hint that it produces a small benefit in prevention and may slightly shorten the duration of RTIs. More research is needed.
Dr. Hunter has received payment for providing expert advice about traditional, complementary, and integrative medicine, including nutraceuticals, to industry, government bodies, and nongovernmental organizations and has spoken at workshops, seminars, and conferences for which registration, travel, and/or accommodation has been paid for by the organizers. Dr. Imdad has disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
A new study published in BMJ Open adds to the evidence that zinc is effective against viral respiratory infections, such as colds.
Jennifer Hunter, PhD, BMed, of Western Sydney University’s NICM Health Research Institute, New South Wales, Australia, and colleagues conducted a meta-analysis of 28 randomized controlled trials (RCTs). They searched 17 English and Chinese databases to identify the trials and then used the Cochrane rapid review technique for the analysis.
The trials included 5,446 adults who had received zinc in a variety of formulations and routes — oral, sublingual, and nasal spray. The researchers separately analyzed whether zinc prevented or treated respiratory tract infections (RTIs)
Oral or intranasal zinc prevented five RTIs per 100 person-months (95% CI, 1 – 8; numbers needed to treat, 20). There was a 32% lower relative risk (RR) of developing mild to moderate symptoms consistent with a viral RTI.
Use of zinc was also associated with an 87% lower risk of developing moderately severe symptoms (incidence rate ratio, 0.13; 95% CI, 0.04 – 0.38) and a 28% lower risk of developing milder symptoms. The largest reductions in RR were for moderately severe symptoms consistent with an influenza-like illness.
Symptoms resolved 2 days earlier with sublingual or intranasal zinc compared with placebo (95% CI, 0.61 – 3.50; very low-certainty quality of evidence). There were clinically significant reductions in day 3 symptom severity scores (mean difference, -1.20 points; 95% CI, -0.66 to -1.74; low-certainty quality of evidence) but not in overall symptom severity. Participants who used sublingual or topical nasal zinc early in the course of illness were 1.8 times more likely to recover before those who used a placebo.
However, the investigators found no benefit of zinc when patients were inoculated with rhinovirus; there was no reduction in the risk of developing a cold. Asked about this disparity, Dr. Hunter said, “It might well be that when inoculating people to make sure they get infected, you give them a really high dose of the virus. [This] doesn’t really mimic what happens in the real world.”
On the downside of supplemental zinc, there were more side effects among those who used zinc, including nausea or gastrointestinal discomfort, mouth irritation, or soreness from sublingual lozenges (RR, 1.41; 95% CI, 1.17 – 1.69; number needed to harm, 7; moderate-certainty quality of evidence). The risk for a serious adverse event, such as loss of smell or copper deficiency, was low. Although not found in these studies, postmarketing studies have found that there is a risk for severe and in some cases permanent loss of smell associated with the use of nasal gels or sprays containing zinc. Three such products were recalled from the market.
The trial could not provide answers about the comparative efficacy of different types of zinc formulations, nor could the investigators recommend specific doses. The trial was not designed to assess zinc for the prevention or treatment of COVID-19.
Asked for independent comment, pediatrician Aamer Imdad, MBBS, assistant professor at the State University of New York Upstate Medical University, Syracuse, told this news organization, “It’s a very comprehensive review for zinc-related studies in adults” but was challenging because of the “significant clinical heterogeneity in the population.”
Dr. Imdad explained that zinc has “absolutely” been shown to be effective for children with diarrhea. The World Health Organization has recommended it since 2004. “The way it works in diarrhea is that it helps with the regeneration of the epithelium.... It also improves the immunity itself, especially the cell-mediated immunity.” He raised the question of whether it might work similarly in the respiratory tract. Dr. Imdad has a long-standing interest in the use of zinc for pediatric infections. Regarding this study, he concluded, “I think we still need to know the nuts and bolts of this intervention before we can recommend it more specifically.”
Dr. Hunter said, “We don’t have any high-quality studies that have evaluated zinc orally as treatment once you’re actually infected and have symptoms of the cold or influenza, or COVID.”
Asked about zinc’s possible role, Dr. Hunter said, “So I do think it gives us a viable alternative. More people are going, ‘What can I do?’ And you know as well as I do people come to you, and [they say], ‘Well, just give me something. Even if it’s a day or a little bit of symptom relief, anything to make me feel better that isn’t going to hurt me and doesn’t have any major risks.’ So I think in the short term, clinicians and consumers can consider trying it.”
Dr. Hunter was not keen on giving zinc to family members after they develop an RTI: “Consider it. But I don’t think we have enough evidence to say definitely yes.” But she does see a potential role for “people who are at risk of suboptimal zinc absorption, like people who are taking a variety of pharmaceuticals [notably proton pump inhibitors] that block or reduce the absorption of zinc, people with a whole lot of the chronic diseases that we know are associated with an increased risk of worse outcomes from respiratory viral infections, and older adults. Yes, I think [for] those high-risk groups, you could consider using zinc, either in a moderate dose longer term or in a higher dose for very short bursts of, like, 1 to 2 weeks.”
Dr. Hunter concluded, “Up until now, we all commonly thought that zinc’s role was only for people who were zinc deficient, and now we’ve got some signals pointing towards its potential role as an anti-infective and anti-inflammatory agent in people who don’t have zinc deficiency.”
But both Dr. Hunter and Dr. Imdad emphasized that zinc is not a game changer. There is a hint that it produces a small benefit in prevention and may slightly shorten the duration of RTIs. More research is needed.
Dr. Hunter has received payment for providing expert advice about traditional, complementary, and integrative medicine, including nutraceuticals, to industry, government bodies, and nongovernmental organizations and has spoken at workshops, seminars, and conferences for which registration, travel, and/or accommodation has been paid for by the organizers. Dr. Imdad has disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
A new study published in BMJ Open adds to the evidence that zinc is effective against viral respiratory infections, such as colds.
Jennifer Hunter, PhD, BMed, of Western Sydney University’s NICM Health Research Institute, New South Wales, Australia, and colleagues conducted a meta-analysis of 28 randomized controlled trials (RCTs). They searched 17 English and Chinese databases to identify the trials and then used the Cochrane rapid review technique for the analysis.
The trials included 5,446 adults who had received zinc in a variety of formulations and routes — oral, sublingual, and nasal spray. The researchers separately analyzed whether zinc prevented or treated respiratory tract infections (RTIs)
Oral or intranasal zinc prevented five RTIs per 100 person-months (95% CI, 1 – 8; numbers needed to treat, 20). There was a 32% lower relative risk (RR) of developing mild to moderate symptoms consistent with a viral RTI.
Use of zinc was also associated with an 87% lower risk of developing moderately severe symptoms (incidence rate ratio, 0.13; 95% CI, 0.04 – 0.38) and a 28% lower risk of developing milder symptoms. The largest reductions in RR were for moderately severe symptoms consistent with an influenza-like illness.
Symptoms resolved 2 days earlier with sublingual or intranasal zinc compared with placebo (95% CI, 0.61 – 3.50; very low-certainty quality of evidence). There were clinically significant reductions in day 3 symptom severity scores (mean difference, -1.20 points; 95% CI, -0.66 to -1.74; low-certainty quality of evidence) but not in overall symptom severity. Participants who used sublingual or topical nasal zinc early in the course of illness were 1.8 times more likely to recover before those who used a placebo.
However, the investigators found no benefit of zinc when patients were inoculated with rhinovirus; there was no reduction in the risk of developing a cold. Asked about this disparity, Dr. Hunter said, “It might well be that when inoculating people to make sure they get infected, you give them a really high dose of the virus. [This] doesn’t really mimic what happens in the real world.”
On the downside of supplemental zinc, there were more side effects among those who used zinc, including nausea or gastrointestinal discomfort, mouth irritation, or soreness from sublingual lozenges (RR, 1.41; 95% CI, 1.17 – 1.69; number needed to harm, 7; moderate-certainty quality of evidence). The risk for a serious adverse event, such as loss of smell or copper deficiency, was low. Although not found in these studies, postmarketing studies have found that there is a risk for severe and in some cases permanent loss of smell associated with the use of nasal gels or sprays containing zinc. Three such products were recalled from the market.
The trial could not provide answers about the comparative efficacy of different types of zinc formulations, nor could the investigators recommend specific doses. The trial was not designed to assess zinc for the prevention or treatment of COVID-19.
Asked for independent comment, pediatrician Aamer Imdad, MBBS, assistant professor at the State University of New York Upstate Medical University, Syracuse, told this news organization, “It’s a very comprehensive review for zinc-related studies in adults” but was challenging because of the “significant clinical heterogeneity in the population.”
Dr. Imdad explained that zinc has “absolutely” been shown to be effective for children with diarrhea. The World Health Organization has recommended it since 2004. “The way it works in diarrhea is that it helps with the regeneration of the epithelium.... It also improves the immunity itself, especially the cell-mediated immunity.” He raised the question of whether it might work similarly in the respiratory tract. Dr. Imdad has a long-standing interest in the use of zinc for pediatric infections. Regarding this study, he concluded, “I think we still need to know the nuts and bolts of this intervention before we can recommend it more specifically.”
Dr. Hunter said, “We don’t have any high-quality studies that have evaluated zinc orally as treatment once you’re actually infected and have symptoms of the cold or influenza, or COVID.”
Asked about zinc’s possible role, Dr. Hunter said, “So I do think it gives us a viable alternative. More people are going, ‘What can I do?’ And you know as well as I do people come to you, and [they say], ‘Well, just give me something. Even if it’s a day or a little bit of symptom relief, anything to make me feel better that isn’t going to hurt me and doesn’t have any major risks.’ So I think in the short term, clinicians and consumers can consider trying it.”
Dr. Hunter was not keen on giving zinc to family members after they develop an RTI: “Consider it. But I don’t think we have enough evidence to say definitely yes.” But she does see a potential role for “people who are at risk of suboptimal zinc absorption, like people who are taking a variety of pharmaceuticals [notably proton pump inhibitors] that block or reduce the absorption of zinc, people with a whole lot of the chronic diseases that we know are associated with an increased risk of worse outcomes from respiratory viral infections, and older adults. Yes, I think [for] those high-risk groups, you could consider using zinc, either in a moderate dose longer term or in a higher dose for very short bursts of, like, 1 to 2 weeks.”
Dr. Hunter concluded, “Up until now, we all commonly thought that zinc’s role was only for people who were zinc deficient, and now we’ve got some signals pointing towards its potential role as an anti-infective and anti-inflammatory agent in people who don’t have zinc deficiency.”
But both Dr. Hunter and Dr. Imdad emphasized that zinc is not a game changer. There is a hint that it produces a small benefit in prevention and may slightly shorten the duration of RTIs. More research is needed.
Dr. Hunter has received payment for providing expert advice about traditional, complementary, and integrative medicine, including nutraceuticals, to industry, government bodies, and nongovernmental organizations and has spoken at workshops, seminars, and conferences for which registration, travel, and/or accommodation has been paid for by the organizers. Dr. Imdad has disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM BMJ OPEN
COVID vaccines’ protection dropped sharply over 6 months: Study
, a study of almost 800,000 veterans found.
The study, published in the journal Science ., says the three vaccines offered about the same protection against the virus in March, when the Delta variant was first detected in the United States, but that changed 6 months later.
The Moderna two-dose vaccine went from being 89% effective in March to 58% effective in September, according to a story about the study in theLos Angeles Times.
Meanwhile, the Pfizer/BioNTech vaccine went from being 87% effective to 45% effective over the same time period.
The Johnson & Johnson vaccine showed the biggest drop -- from 86% effectiveness to 13% over those 6 months.
“In summary, although vaccination remains protective against SARS-CoV-2 infection, protection waned as the Delta variant emerged in the U.S., and this decline did not differ by age,” the study said.
The three vaccines also lost effectiveness in the ability to protect against death in veterans 65 and over after only 3 months, the Los Angeles Times reported.
Compared to unvaccinated veterans in that age group, veterans who got the Moderna vaccine and had a breakthrough case were 76% less likely to die of COVID-19 by July.
The protection was 70% for Pfizer/BioNTech vaccine recipients and 52% for J&J vaccine recipients for the same age group, compared to unvaccinated veterans, according to the newspaper.
For veterans under 65, the protectiveness against a fatal case of COVID was 84% for Pfizer/BioNTech recipients, 82% for Moderna recipients, and 73% for J&J recipients, compared to unvaccinated veterans in that age group.
The study confirms the need for booster vaccines and protective measures such as vaccine passports, vaccine mandates, masking, hand-washing, and social distancing, the researchers said.
Of the veterans studied, about 500,000 were vaccinated and 300,000 were not. Researchers noted that the study population had 6 times as many men as women. About 48% of the study group was 65 or older, 29% was 50-64, while 24% was under 50.
Researchers from the Public Health Institute in Oakland, the Veterans Affairs Medical Center in San Francisco, and the University of Texas Health Science Center conducted the study.
A version of this article first appeared on WebMD.com.
, a study of almost 800,000 veterans found.
The study, published in the journal Science ., says the three vaccines offered about the same protection against the virus in March, when the Delta variant was first detected in the United States, but that changed 6 months later.
The Moderna two-dose vaccine went from being 89% effective in March to 58% effective in September, according to a story about the study in theLos Angeles Times.
Meanwhile, the Pfizer/BioNTech vaccine went from being 87% effective to 45% effective over the same time period.
The Johnson & Johnson vaccine showed the biggest drop -- from 86% effectiveness to 13% over those 6 months.
“In summary, although vaccination remains protective against SARS-CoV-2 infection, protection waned as the Delta variant emerged in the U.S., and this decline did not differ by age,” the study said.
The three vaccines also lost effectiveness in the ability to protect against death in veterans 65 and over after only 3 months, the Los Angeles Times reported.
Compared to unvaccinated veterans in that age group, veterans who got the Moderna vaccine and had a breakthrough case were 76% less likely to die of COVID-19 by July.
The protection was 70% for Pfizer/BioNTech vaccine recipients and 52% for J&J vaccine recipients for the same age group, compared to unvaccinated veterans, according to the newspaper.
For veterans under 65, the protectiveness against a fatal case of COVID was 84% for Pfizer/BioNTech recipients, 82% for Moderna recipients, and 73% for J&J recipients, compared to unvaccinated veterans in that age group.
The study confirms the need for booster vaccines and protective measures such as vaccine passports, vaccine mandates, masking, hand-washing, and social distancing, the researchers said.
Of the veterans studied, about 500,000 were vaccinated and 300,000 were not. Researchers noted that the study population had 6 times as many men as women. About 48% of the study group was 65 or older, 29% was 50-64, while 24% was under 50.
Researchers from the Public Health Institute in Oakland, the Veterans Affairs Medical Center in San Francisco, and the University of Texas Health Science Center conducted the study.
A version of this article first appeared on WebMD.com.
, a study of almost 800,000 veterans found.
The study, published in the journal Science ., says the three vaccines offered about the same protection against the virus in March, when the Delta variant was first detected in the United States, but that changed 6 months later.
The Moderna two-dose vaccine went from being 89% effective in March to 58% effective in September, according to a story about the study in theLos Angeles Times.
Meanwhile, the Pfizer/BioNTech vaccine went from being 87% effective to 45% effective over the same time period.
The Johnson & Johnson vaccine showed the biggest drop -- from 86% effectiveness to 13% over those 6 months.
“In summary, although vaccination remains protective against SARS-CoV-2 infection, protection waned as the Delta variant emerged in the U.S., and this decline did not differ by age,” the study said.
The three vaccines also lost effectiveness in the ability to protect against death in veterans 65 and over after only 3 months, the Los Angeles Times reported.
Compared to unvaccinated veterans in that age group, veterans who got the Moderna vaccine and had a breakthrough case were 76% less likely to die of COVID-19 by July.
The protection was 70% for Pfizer/BioNTech vaccine recipients and 52% for J&J vaccine recipients for the same age group, compared to unvaccinated veterans, according to the newspaper.
For veterans under 65, the protectiveness against a fatal case of COVID was 84% for Pfizer/BioNTech recipients, 82% for Moderna recipients, and 73% for J&J recipients, compared to unvaccinated veterans in that age group.
The study confirms the need for booster vaccines and protective measures such as vaccine passports, vaccine mandates, masking, hand-washing, and social distancing, the researchers said.
Of the veterans studied, about 500,000 were vaccinated and 300,000 were not. Researchers noted that the study population had 6 times as many men as women. About 48% of the study group was 65 or older, 29% was 50-64, while 24% was under 50.
Researchers from the Public Health Institute in Oakland, the Veterans Affairs Medical Center in San Francisco, and the University of Texas Health Science Center conducted the study.
A version of this article first appeared on WebMD.com.
FROM SCIENCE
Pfizer says its COVID-19 pill is highly effective
, according to the drug’s maker, Pfizer.
The drug -- called Paxlovid -- was 89% effective, compared to a placebo, at preventing hospitalization or death in patients with COVID-19 who were at high risk of severe complications. The company says it plans to ask the FDA to authorize the drug for emergency use.
The medication appears to work so well that Pfizer has stopped enrollment in the trial of the drug, which works by blocking an enzyme called a protease that the new coronavirus needs to make more copies of itself.
Stopping a clinical trial is a rare action that’s typically taken when a therapy appears to be very effective or clearly dangerous. In both those cases, it’s considered unethical to continue a clinical trial where people are randomly assigned either an active drug or a placebo, when safer or more effective options are available to them.
In this case, the company said in a news release that the move was recommended by an independent panel of advisers who are overseeing the trial, called a data safety monitoring committee, and done in consultation with the FDA.
“Today’s news is a real game-changer in the global efforts to halt the devastation of this pandemic,” said Albert Bourla, PhD, Pfizer chairman and chief executive officer. “These data suggest that our oral antiviral candidate, if approved or authorized by regulatory authorities, has the potential to save patients’ lives, reduce the severity of COVID-19 infections, and eliminate up to nine out of ten hospitalizations.”
In a randomized clinical trial that included more than 1,900 patients who tested positive for COVID-19 and were at risk for having severe complications for their infections, those who received Paxlovid within 3 days of the start of their symptoms were 89% less likely to be hospitalized than those who got a placebo pill -- three patients out of 389 who got the drug were hospitalized, compared with 27 out of 385 who got the placebo. Among patients who got the drug within 5 days of the start of their symptoms, six out of 607 were hospitalized within 28 days, compared to 41 out of 612 who got the placebo.
There were no deaths over the course of a month in patients who took Paxlovid, but 10 deaths in the group that got the placebo.
The news comes on the heels of an announcement in October by the drug company Merck that its experimental antiviral pill, molnupiravir, reduced the risk of hospitalization or death by 50% in patients with mild to moderate COVID, compared to a placebo.
The United Kingdom became the first country to authorize the use of molnupiravir, which is brand-named Lagevrio.
Stephen Griffin, PhD, an associate professor of medicine at the University of Leeds, hailed the success of both new antiviral pills.
“They both demonstrate that, with appropriate investment, the development of bespoke direct-acting antiviral drugs targeting SARS-CoV2 was eminently feasible and has ultimately proven far more successful than repurposing other drugs with questionable antiviral effects,” said Dr. Griffin, who was not involved in the development of either drug.
“The success of these antivirals potentially marks a new era in our ability to prevent the severe consequences of SARS-CoV2 infection, and is also a vital element for the care of clinically vulnerable people who may be unable to either receive or respond to vaccines,” he said.
A version of this article first appeared on WebMD.com.
, according to the drug’s maker, Pfizer.
The drug -- called Paxlovid -- was 89% effective, compared to a placebo, at preventing hospitalization or death in patients with COVID-19 who were at high risk of severe complications. The company says it plans to ask the FDA to authorize the drug for emergency use.
The medication appears to work so well that Pfizer has stopped enrollment in the trial of the drug, which works by blocking an enzyme called a protease that the new coronavirus needs to make more copies of itself.
Stopping a clinical trial is a rare action that’s typically taken when a therapy appears to be very effective or clearly dangerous. In both those cases, it’s considered unethical to continue a clinical trial where people are randomly assigned either an active drug or a placebo, when safer or more effective options are available to them.
In this case, the company said in a news release that the move was recommended by an independent panel of advisers who are overseeing the trial, called a data safety monitoring committee, and done in consultation with the FDA.
“Today’s news is a real game-changer in the global efforts to halt the devastation of this pandemic,” said Albert Bourla, PhD, Pfizer chairman and chief executive officer. “These data suggest that our oral antiviral candidate, if approved or authorized by regulatory authorities, has the potential to save patients’ lives, reduce the severity of COVID-19 infections, and eliminate up to nine out of ten hospitalizations.”
In a randomized clinical trial that included more than 1,900 patients who tested positive for COVID-19 and were at risk for having severe complications for their infections, those who received Paxlovid within 3 days of the start of their symptoms were 89% less likely to be hospitalized than those who got a placebo pill -- three patients out of 389 who got the drug were hospitalized, compared with 27 out of 385 who got the placebo. Among patients who got the drug within 5 days of the start of their symptoms, six out of 607 were hospitalized within 28 days, compared to 41 out of 612 who got the placebo.
There were no deaths over the course of a month in patients who took Paxlovid, but 10 deaths in the group that got the placebo.
The news comes on the heels of an announcement in October by the drug company Merck that its experimental antiviral pill, molnupiravir, reduced the risk of hospitalization or death by 50% in patients with mild to moderate COVID, compared to a placebo.
The United Kingdom became the first country to authorize the use of molnupiravir, which is brand-named Lagevrio.
Stephen Griffin, PhD, an associate professor of medicine at the University of Leeds, hailed the success of both new antiviral pills.
“They both demonstrate that, with appropriate investment, the development of bespoke direct-acting antiviral drugs targeting SARS-CoV2 was eminently feasible and has ultimately proven far more successful than repurposing other drugs with questionable antiviral effects,” said Dr. Griffin, who was not involved in the development of either drug.
“The success of these antivirals potentially marks a new era in our ability to prevent the severe consequences of SARS-CoV2 infection, and is also a vital element for the care of clinically vulnerable people who may be unable to either receive or respond to vaccines,” he said.
A version of this article first appeared on WebMD.com.
, according to the drug’s maker, Pfizer.
The drug -- called Paxlovid -- was 89% effective, compared to a placebo, at preventing hospitalization or death in patients with COVID-19 who were at high risk of severe complications. The company says it plans to ask the FDA to authorize the drug for emergency use.
The medication appears to work so well that Pfizer has stopped enrollment in the trial of the drug, which works by blocking an enzyme called a protease that the new coronavirus needs to make more copies of itself.
Stopping a clinical trial is a rare action that’s typically taken when a therapy appears to be very effective or clearly dangerous. In both those cases, it’s considered unethical to continue a clinical trial where people are randomly assigned either an active drug or a placebo, when safer or more effective options are available to them.
In this case, the company said in a news release that the move was recommended by an independent panel of advisers who are overseeing the trial, called a data safety monitoring committee, and done in consultation with the FDA.
“Today’s news is a real game-changer in the global efforts to halt the devastation of this pandemic,” said Albert Bourla, PhD, Pfizer chairman and chief executive officer. “These data suggest that our oral antiviral candidate, if approved or authorized by regulatory authorities, has the potential to save patients’ lives, reduce the severity of COVID-19 infections, and eliminate up to nine out of ten hospitalizations.”
In a randomized clinical trial that included more than 1,900 patients who tested positive for COVID-19 and were at risk for having severe complications for their infections, those who received Paxlovid within 3 days of the start of their symptoms were 89% less likely to be hospitalized than those who got a placebo pill -- three patients out of 389 who got the drug were hospitalized, compared with 27 out of 385 who got the placebo. Among patients who got the drug within 5 days of the start of their symptoms, six out of 607 were hospitalized within 28 days, compared to 41 out of 612 who got the placebo.
There were no deaths over the course of a month in patients who took Paxlovid, but 10 deaths in the group that got the placebo.
The news comes on the heels of an announcement in October by the drug company Merck that its experimental antiviral pill, molnupiravir, reduced the risk of hospitalization or death by 50% in patients with mild to moderate COVID, compared to a placebo.
The United Kingdom became the first country to authorize the use of molnupiravir, which is brand-named Lagevrio.
Stephen Griffin, PhD, an associate professor of medicine at the University of Leeds, hailed the success of both new antiviral pills.
“They both demonstrate that, with appropriate investment, the development of bespoke direct-acting antiviral drugs targeting SARS-CoV2 was eminently feasible and has ultimately proven far more successful than repurposing other drugs with questionable antiviral effects,” said Dr. Griffin, who was not involved in the development of either drug.
“The success of these antivirals potentially marks a new era in our ability to prevent the severe consequences of SARS-CoV2 infection, and is also a vital element for the care of clinically vulnerable people who may be unable to either receive or respond to vaccines,” he said.
A version of this article first appeared on WebMD.com.
How applicable is ISCHEMIA trial to U.S. clinical practice?
The applicability of the results of the ISCHEMIA trial to real-world clinical practice in the United States has been called into question by a new study showing that less than a third of U.S. patients with stable ischemic heart disease (IHD) who currently undergo intervention would meet the trial’s inclusion criteria.
The ISCHEMIA trial, first reported in 2019, showed that, for stable patients with moderate to severe ischemia, an invasive approach using percutaneous coronary intervention (PCI) did not significantly reduce major cardiovascular events after a median 3.2 years of follow-up, compared with a conservative medical strategy.
For the current study, a group of interventionalists analyzed contemporary U.S. data on patients undergoing PCI and found that a large proportion of patients receiving PCI for stable ischemic heart disease in the United States would not have met criteria of the ISCHEMIA trial population.
The study was published online Nov. 1 in JACC: Cardiovascular Interventions).
“While ISCHEMIA was a very well-conducted trial, our results show that it only applies to about one-third of stable IHD patients undergoing intervention in U.S. clinical practice in the real world. In this group, while ISCHEMIA did not show a reduction in event rate in the intervention group, there was a reduction in symptoms,” lead author Saurav Chatterjee, MD, Long Island Jewish Medical Center, New York, told this news organization.
“But ISCHEMIA did not really answer the question for 67% of stable IHD in current U.S. practice. We may be abIe to defer PCI in these patients, but we don’t know that from the ISCHEMIA trial, as these patients were not included in the trial,” Chatterjee said.
“There is some concern that people will accept the ISCHEMIA results as being universal, but we cannot apply these results to all stable IHD patients who currently undergo intervention,” he added. “We believe that patients who do not fall into the ISCHEMIA population need a nuanced individual approach, taking into account symptom burden and patient preferences.”
In the new report, Dr. Chatterjee and associates note that the applicability of the ISCHEMIA findings to contemporary practice has been questioned by some, because of the exclusion of a significant proportion of patients that are routinely considered for revascularization, both within and outside of the United States.
They point out that the ISCHEMIA trial recruited 16.5% of its participants from the United States, and the proportion of patients in contemporary U.S. practice that would have qualified for the trial is not clear.
They therefore examined the proportion of stable IHD patients meeting inclusion criteria for the ISCHEMIA trial in a U.S. nationwide PCI registry.
The researchers used data from the National Cardiovascular Data Registry (NCDR) CathPCI Registry, which includes patients undergoing PCI at 1,662 institutions and accounts for more than 90% of PCI-capable hospitals in the United States.
All PCI procedures performed at institutions participating in the NCDR CathPCI Registry from October 2017 to June 2019 were identified. Patients presenting with acute coronary syndrome (ACS), cardiogenic shock, or cardiac arrest were excluded, as there is significant evidence in favor of revascularization in these groups, and they were not included in the ISCHEMIA trial.
Subsequently, all remaining stable IHD patients were classified into one of four groups.
- ISCHEMIA-like: These patients had intermediate- or high-risk findings on a stress test but no high-risk features that would have excluded enrollment into the ISCHEMIA trial
- High risk: This group comprised patients with stable IHD and left ventricular ejection fraction less than 35%, significant unprotected left main stenosis (>50%), preexisting dialysis, recent heart-failure exacerbation, or heart transplant. These patients would have met exclusion criteria for the ISCHEMIA trial
- Low risk: This group included patients with stable and negative or low-risk findings on stress test and would have met exclusion criteria for the ISCHEMIA trial
- Not classifiable: This group comprised patients with stable IHD not fitting any of the other cohorts, including no stress test or extent of ischemia not reported on stress testing. These patients would have not had enough information to clearly meet inclusion or exclusion criteria for the ISCHEMIA trial
Results showed that during the study period 927,011 patients underwent PCI as recorded in the NCDR CathPCI Registry. Of these, 58% had ACS, cardiogenic shock, or cardiac arrest and were excluded; the remaining 388,212 patients who underwent PCI for stable IHD comprised the study population.
Of these, 125,302 (32.28%) had a moderate- or high-risk stress test without high-risk anatomic or clinical features and met ISCHEMIA trial inclusion criteria.
Among stable IHD patients not meeting ISCHEMIA trial inclusion criteria, 71,852 (18.51%) had high-risk criteria that would have excluded them from the ISCHEMIA trial, a total of 67,159 (17.29%) patients had low-risk criteria that would have excluded them from the ISCHEMIA trial, and 123,899 (31.92%) were unclassifiable, either owing to lack of stress testing or the extent of ischemia not being reported on stress testing.
The authors suggest that the unclassifiable patients appear to represent a “higher-risk” population than those closely resembling the ISCHEMIA trial population, with more prior myocardial infarction and heart failure.
ISCHEMIA investigators respond
In an accompanying editorial, ISCHEMIA investigators David J. Maron, MD, Stanford (Calif.) University, and Sripal Bangalore, MD, and Judith S. Hochman, MD, New York University, argue that many of the patients highlighted by Dr. Chatterjee and associates were excluded from the ISCHEMIA trial for good reason.
They explain that ISCHEMIA was designed under the premise that prior stable IHD strategy trials such as COURAGE and BARI 2D included lower-risk patients, and the remaining gap was to evaluate the utility of invasive management in those at higher risk with moderate or severe stress-induced ischemia.
They point out that, among the NCDR patients with stable IHD in the current study by Dr. Chatterjee and associates who did not meet ISCHEMIA entry criteria, 18.5% had high-risk features, including 35.2% with left main coronary artery disease, 43.7% with left ventricular systolic dysfunction, and 16.8% with end-stage renal disease.
Although ISCHEMIA results do not apply to patients who were excluded from the trial, there is little controversy regarding the benefit of revascularization in patients with stable IHD with left main coronary artery disease or left ventricular ejection fraction <35%, which is why they were excluded from ISCHEMIA, the editorialists note.
They also report that patients with end-stage renal disease, who were also designated as not meeting ISCHEMIA inclusion criteria, were included in the companion ISCHEMIA CKD trial.
They further point out that, at the other end of the risk spectrum, 17.3% of stable IHD patients in the current analysis had negative or low-risk functional testing, and these patients were excluded from ISCHEMIA because they were shown in COURAGE and BARI 2D to not benefit from revascularization, and they do not meet guideline recommendations for elective PCI in the absence of symptoms.
Of the 31.9% of stable IHD patients who had missing data on ischemic burden, the ISCHEMIA investigators say that some of these would have qualified for the trial, although it is not possible to say how many. They suggest a conservative estimate of 50%.
Taking these arguments into account, the editorialists recalculated the proportion of NCDR PCI patients with stable IHD who would have been included in ISCHEMIA as between 62.1% and 68.6% of patients.
They say the current NCDR analysis by Dr. Chatterjee and associates should be interpreted as indicating, at worst, that the ISCHEMIA trial results apply to only 32% of patients undergoing elective PCI in the United States, and at best “that the results apply to a far higher proportion, excluding only those at high risk (18.5%) or with unacceptable symptoms despite maximal medical therapy (percentage unknown), for whom PCI is clearly indicated.”
The editorialists conclude: “The purpose of the analysis by Chatterjee et al. is to inform the cardiovascular community of the proportion of patients with stable IHD in clinical practice who would have been excluded from ISCHEMIA without regard for the logic of each exclusion criterion. The purpose of this editorial is to provide context for the analysis, admittedly from the perspective of ISCHEMIA investigators, with the hope that this helps readers clearly see the relevance of the trial to patients under their care.”
They add: “For practical and ethical reasons, ISCHEMIA excluded stable patients with high-risk features, angina inadequately controlled by medication, and low-risk features who do not meet evidence-based guidelines for revascularization. That leaves a large percentage of patients for whom the ISCHEMIA trial is highly relevant; exactly what percentage on the basis of NCDR data is hard to say.”
The ISCHEMIA trial was supported by the National Heart, Lung, and Blood Institute.
A version of this article first appeared on Medscape.com.
The applicability of the results of the ISCHEMIA trial to real-world clinical practice in the United States has been called into question by a new study showing that less than a third of U.S. patients with stable ischemic heart disease (IHD) who currently undergo intervention would meet the trial’s inclusion criteria.
The ISCHEMIA trial, first reported in 2019, showed that, for stable patients with moderate to severe ischemia, an invasive approach using percutaneous coronary intervention (PCI) did not significantly reduce major cardiovascular events after a median 3.2 years of follow-up, compared with a conservative medical strategy.
For the current study, a group of interventionalists analyzed contemporary U.S. data on patients undergoing PCI and found that a large proportion of patients receiving PCI for stable ischemic heart disease in the United States would not have met criteria of the ISCHEMIA trial population.
The study was published online Nov. 1 in JACC: Cardiovascular Interventions).
“While ISCHEMIA was a very well-conducted trial, our results show that it only applies to about one-third of stable IHD patients undergoing intervention in U.S. clinical practice in the real world. In this group, while ISCHEMIA did not show a reduction in event rate in the intervention group, there was a reduction in symptoms,” lead author Saurav Chatterjee, MD, Long Island Jewish Medical Center, New York, told this news organization.
“But ISCHEMIA did not really answer the question for 67% of stable IHD in current U.S. practice. We may be abIe to defer PCI in these patients, but we don’t know that from the ISCHEMIA trial, as these patients were not included in the trial,” Chatterjee said.
“There is some concern that people will accept the ISCHEMIA results as being universal, but we cannot apply these results to all stable IHD patients who currently undergo intervention,” he added. “We believe that patients who do not fall into the ISCHEMIA population need a nuanced individual approach, taking into account symptom burden and patient preferences.”
In the new report, Dr. Chatterjee and associates note that the applicability of the ISCHEMIA findings to contemporary practice has been questioned by some, because of the exclusion of a significant proportion of patients that are routinely considered for revascularization, both within and outside of the United States.
They point out that the ISCHEMIA trial recruited 16.5% of its participants from the United States, and the proportion of patients in contemporary U.S. practice that would have qualified for the trial is not clear.
They therefore examined the proportion of stable IHD patients meeting inclusion criteria for the ISCHEMIA trial in a U.S. nationwide PCI registry.
The researchers used data from the National Cardiovascular Data Registry (NCDR) CathPCI Registry, which includes patients undergoing PCI at 1,662 institutions and accounts for more than 90% of PCI-capable hospitals in the United States.
All PCI procedures performed at institutions participating in the NCDR CathPCI Registry from October 2017 to June 2019 were identified. Patients presenting with acute coronary syndrome (ACS), cardiogenic shock, or cardiac arrest were excluded, as there is significant evidence in favor of revascularization in these groups, and they were not included in the ISCHEMIA trial.
Subsequently, all remaining stable IHD patients were classified into one of four groups.
- ISCHEMIA-like: These patients had intermediate- or high-risk findings on a stress test but no high-risk features that would have excluded enrollment into the ISCHEMIA trial
- High risk: This group comprised patients with stable IHD and left ventricular ejection fraction less than 35%, significant unprotected left main stenosis (>50%), preexisting dialysis, recent heart-failure exacerbation, or heart transplant. These patients would have met exclusion criteria for the ISCHEMIA trial
- Low risk: This group included patients with stable and negative or low-risk findings on stress test and would have met exclusion criteria for the ISCHEMIA trial
- Not classifiable: This group comprised patients with stable IHD not fitting any of the other cohorts, including no stress test or extent of ischemia not reported on stress testing. These patients would have not had enough information to clearly meet inclusion or exclusion criteria for the ISCHEMIA trial
Results showed that during the study period 927,011 patients underwent PCI as recorded in the NCDR CathPCI Registry. Of these, 58% had ACS, cardiogenic shock, or cardiac arrest and were excluded; the remaining 388,212 patients who underwent PCI for stable IHD comprised the study population.
Of these, 125,302 (32.28%) had a moderate- or high-risk stress test without high-risk anatomic or clinical features and met ISCHEMIA trial inclusion criteria.
Among stable IHD patients not meeting ISCHEMIA trial inclusion criteria, 71,852 (18.51%) had high-risk criteria that would have excluded them from the ISCHEMIA trial, a total of 67,159 (17.29%) patients had low-risk criteria that would have excluded them from the ISCHEMIA trial, and 123,899 (31.92%) were unclassifiable, either owing to lack of stress testing or the extent of ischemia not being reported on stress testing.
The authors suggest that the unclassifiable patients appear to represent a “higher-risk” population than those closely resembling the ISCHEMIA trial population, with more prior myocardial infarction and heart failure.
ISCHEMIA investigators respond
In an accompanying editorial, ISCHEMIA investigators David J. Maron, MD, Stanford (Calif.) University, and Sripal Bangalore, MD, and Judith S. Hochman, MD, New York University, argue that many of the patients highlighted by Dr. Chatterjee and associates were excluded from the ISCHEMIA trial for good reason.
They explain that ISCHEMIA was designed under the premise that prior stable IHD strategy trials such as COURAGE and BARI 2D included lower-risk patients, and the remaining gap was to evaluate the utility of invasive management in those at higher risk with moderate or severe stress-induced ischemia.
They point out that, among the NCDR patients with stable IHD in the current study by Dr. Chatterjee and associates who did not meet ISCHEMIA entry criteria, 18.5% had high-risk features, including 35.2% with left main coronary artery disease, 43.7% with left ventricular systolic dysfunction, and 16.8% with end-stage renal disease.
Although ISCHEMIA results do not apply to patients who were excluded from the trial, there is little controversy regarding the benefit of revascularization in patients with stable IHD with left main coronary artery disease or left ventricular ejection fraction <35%, which is why they were excluded from ISCHEMIA, the editorialists note.
They also report that patients with end-stage renal disease, who were also designated as not meeting ISCHEMIA inclusion criteria, were included in the companion ISCHEMIA CKD trial.
They further point out that, at the other end of the risk spectrum, 17.3% of stable IHD patients in the current analysis had negative or low-risk functional testing, and these patients were excluded from ISCHEMIA because they were shown in COURAGE and BARI 2D to not benefit from revascularization, and they do not meet guideline recommendations for elective PCI in the absence of symptoms.
Of the 31.9% of stable IHD patients who had missing data on ischemic burden, the ISCHEMIA investigators say that some of these would have qualified for the trial, although it is not possible to say how many. They suggest a conservative estimate of 50%.
Taking these arguments into account, the editorialists recalculated the proportion of NCDR PCI patients with stable IHD who would have been included in ISCHEMIA as between 62.1% and 68.6% of patients.
They say the current NCDR analysis by Dr. Chatterjee and associates should be interpreted as indicating, at worst, that the ISCHEMIA trial results apply to only 32% of patients undergoing elective PCI in the United States, and at best “that the results apply to a far higher proportion, excluding only those at high risk (18.5%) or with unacceptable symptoms despite maximal medical therapy (percentage unknown), for whom PCI is clearly indicated.”
The editorialists conclude: “The purpose of the analysis by Chatterjee et al. is to inform the cardiovascular community of the proportion of patients with stable IHD in clinical practice who would have been excluded from ISCHEMIA without regard for the logic of each exclusion criterion. The purpose of this editorial is to provide context for the analysis, admittedly from the perspective of ISCHEMIA investigators, with the hope that this helps readers clearly see the relevance of the trial to patients under their care.”
They add: “For practical and ethical reasons, ISCHEMIA excluded stable patients with high-risk features, angina inadequately controlled by medication, and low-risk features who do not meet evidence-based guidelines for revascularization. That leaves a large percentage of patients for whom the ISCHEMIA trial is highly relevant; exactly what percentage on the basis of NCDR data is hard to say.”
The ISCHEMIA trial was supported by the National Heart, Lung, and Blood Institute.
A version of this article first appeared on Medscape.com.
The applicability of the results of the ISCHEMIA trial to real-world clinical practice in the United States has been called into question by a new study showing that less than a third of U.S. patients with stable ischemic heart disease (IHD) who currently undergo intervention would meet the trial’s inclusion criteria.
The ISCHEMIA trial, first reported in 2019, showed that, for stable patients with moderate to severe ischemia, an invasive approach using percutaneous coronary intervention (PCI) did not significantly reduce major cardiovascular events after a median 3.2 years of follow-up, compared with a conservative medical strategy.
For the current study, a group of interventionalists analyzed contemporary U.S. data on patients undergoing PCI and found that a large proportion of patients receiving PCI for stable ischemic heart disease in the United States would not have met criteria of the ISCHEMIA trial population.
The study was published online Nov. 1 in JACC: Cardiovascular Interventions).
“While ISCHEMIA was a very well-conducted trial, our results show that it only applies to about one-third of stable IHD patients undergoing intervention in U.S. clinical practice in the real world. In this group, while ISCHEMIA did not show a reduction in event rate in the intervention group, there was a reduction in symptoms,” lead author Saurav Chatterjee, MD, Long Island Jewish Medical Center, New York, told this news organization.
“But ISCHEMIA did not really answer the question for 67% of stable IHD in current U.S. practice. We may be abIe to defer PCI in these patients, but we don’t know that from the ISCHEMIA trial, as these patients were not included in the trial,” Chatterjee said.
“There is some concern that people will accept the ISCHEMIA results as being universal, but we cannot apply these results to all stable IHD patients who currently undergo intervention,” he added. “We believe that patients who do not fall into the ISCHEMIA population need a nuanced individual approach, taking into account symptom burden and patient preferences.”
In the new report, Dr. Chatterjee and associates note that the applicability of the ISCHEMIA findings to contemporary practice has been questioned by some, because of the exclusion of a significant proportion of patients that are routinely considered for revascularization, both within and outside of the United States.
They point out that the ISCHEMIA trial recruited 16.5% of its participants from the United States, and the proportion of patients in contemporary U.S. practice that would have qualified for the trial is not clear.
They therefore examined the proportion of stable IHD patients meeting inclusion criteria for the ISCHEMIA trial in a U.S. nationwide PCI registry.
The researchers used data from the National Cardiovascular Data Registry (NCDR) CathPCI Registry, which includes patients undergoing PCI at 1,662 institutions and accounts for more than 90% of PCI-capable hospitals in the United States.
All PCI procedures performed at institutions participating in the NCDR CathPCI Registry from October 2017 to June 2019 were identified. Patients presenting with acute coronary syndrome (ACS), cardiogenic shock, or cardiac arrest were excluded, as there is significant evidence in favor of revascularization in these groups, and they were not included in the ISCHEMIA trial.
Subsequently, all remaining stable IHD patients were classified into one of four groups.
- ISCHEMIA-like: These patients had intermediate- or high-risk findings on a stress test but no high-risk features that would have excluded enrollment into the ISCHEMIA trial
- High risk: This group comprised patients with stable IHD and left ventricular ejection fraction less than 35%, significant unprotected left main stenosis (>50%), preexisting dialysis, recent heart-failure exacerbation, or heart transplant. These patients would have met exclusion criteria for the ISCHEMIA trial
- Low risk: This group included patients with stable and negative or low-risk findings on stress test and would have met exclusion criteria for the ISCHEMIA trial
- Not classifiable: This group comprised patients with stable IHD not fitting any of the other cohorts, including no stress test or extent of ischemia not reported on stress testing. These patients would have not had enough information to clearly meet inclusion or exclusion criteria for the ISCHEMIA trial
Results showed that during the study period 927,011 patients underwent PCI as recorded in the NCDR CathPCI Registry. Of these, 58% had ACS, cardiogenic shock, or cardiac arrest and were excluded; the remaining 388,212 patients who underwent PCI for stable IHD comprised the study population.
Of these, 125,302 (32.28%) had a moderate- or high-risk stress test without high-risk anatomic or clinical features and met ISCHEMIA trial inclusion criteria.
Among stable IHD patients not meeting ISCHEMIA trial inclusion criteria, 71,852 (18.51%) had high-risk criteria that would have excluded them from the ISCHEMIA trial, a total of 67,159 (17.29%) patients had low-risk criteria that would have excluded them from the ISCHEMIA trial, and 123,899 (31.92%) were unclassifiable, either owing to lack of stress testing or the extent of ischemia not being reported on stress testing.
The authors suggest that the unclassifiable patients appear to represent a “higher-risk” population than those closely resembling the ISCHEMIA trial population, with more prior myocardial infarction and heart failure.
ISCHEMIA investigators respond
In an accompanying editorial, ISCHEMIA investigators David J. Maron, MD, Stanford (Calif.) University, and Sripal Bangalore, MD, and Judith S. Hochman, MD, New York University, argue that many of the patients highlighted by Dr. Chatterjee and associates were excluded from the ISCHEMIA trial for good reason.
They explain that ISCHEMIA was designed under the premise that prior stable IHD strategy trials such as COURAGE and BARI 2D included lower-risk patients, and the remaining gap was to evaluate the utility of invasive management in those at higher risk with moderate or severe stress-induced ischemia.
They point out that, among the NCDR patients with stable IHD in the current study by Dr. Chatterjee and associates who did not meet ISCHEMIA entry criteria, 18.5% had high-risk features, including 35.2% with left main coronary artery disease, 43.7% with left ventricular systolic dysfunction, and 16.8% with end-stage renal disease.
Although ISCHEMIA results do not apply to patients who were excluded from the trial, there is little controversy regarding the benefit of revascularization in patients with stable IHD with left main coronary artery disease or left ventricular ejection fraction <35%, which is why they were excluded from ISCHEMIA, the editorialists note.
They also report that patients with end-stage renal disease, who were also designated as not meeting ISCHEMIA inclusion criteria, were included in the companion ISCHEMIA CKD trial.
They further point out that, at the other end of the risk spectrum, 17.3% of stable IHD patients in the current analysis had negative or low-risk functional testing, and these patients were excluded from ISCHEMIA because they were shown in COURAGE and BARI 2D to not benefit from revascularization, and they do not meet guideline recommendations for elective PCI in the absence of symptoms.
Of the 31.9% of stable IHD patients who had missing data on ischemic burden, the ISCHEMIA investigators say that some of these would have qualified for the trial, although it is not possible to say how many. They suggest a conservative estimate of 50%.
Taking these arguments into account, the editorialists recalculated the proportion of NCDR PCI patients with stable IHD who would have been included in ISCHEMIA as between 62.1% and 68.6% of patients.
They say the current NCDR analysis by Dr. Chatterjee and associates should be interpreted as indicating, at worst, that the ISCHEMIA trial results apply to only 32% of patients undergoing elective PCI in the United States, and at best “that the results apply to a far higher proportion, excluding only those at high risk (18.5%) or with unacceptable symptoms despite maximal medical therapy (percentage unknown), for whom PCI is clearly indicated.”
The editorialists conclude: “The purpose of the analysis by Chatterjee et al. is to inform the cardiovascular community of the proportion of patients with stable IHD in clinical practice who would have been excluded from ISCHEMIA without regard for the logic of each exclusion criterion. The purpose of this editorial is to provide context for the analysis, admittedly from the perspective of ISCHEMIA investigators, with the hope that this helps readers clearly see the relevance of the trial to patients under their care.”
They add: “For practical and ethical reasons, ISCHEMIA excluded stable patients with high-risk features, angina inadequately controlled by medication, and low-risk features who do not meet evidence-based guidelines for revascularization. That leaves a large percentage of patients for whom the ISCHEMIA trial is highly relevant; exactly what percentage on the basis of NCDR data is hard to say.”
The ISCHEMIA trial was supported by the National Heart, Lung, and Blood Institute.
A version of this article first appeared on Medscape.com.
FFR-guided PCI falls short vs. surgery in multivessel disease: FAME 3
Coronary stenting guided by fractional flow reserve (FFR) readings, considered to reflect the targeted lesion’s functional impact, was no match for coronary bypass surgery (CABG) in patients with multivessel disease (MVD) in a major international randomized trial.

Indeed, FFR-guided percutaneous coronary intervention (PCI) using one of the latest drug-eluting stents (DES) seemed to perform poorly in the trial, compared with surgery, apparently upping the risk for clinical events by 50% over 1 year.
Designed statistically for noninferiority, the third Fractional Flow Reserve Versus Angiography for Multivessel Evaluation (FAME 3) trial, with 1,500 randomized patients, showed that FFR-guided PCI was “not noninferior” to CABG. Of those randomized to PCI, 10.6% met the 1-year primary endpoint of major adverse cardiac or cerebrovascular events (MACCE), compared with only 6.9% of patients assigned to CABG.
The trial enrolled only patients with three-vessel coronary disease with no left-main coronary artery involvement, who were declared by their institution’s multidisciplinary heart team to be appropriate for either form of revascularization.
One of the roles of FFR for PCI guidance is to identify significant lesions “that are underrecognized by the angiogram,” which is less likely to happen in patients with very complex coronary anatomy, study chair William F. Fearon, MD, Stanford (Calif.) University, said in an interview.
“That’s what we saw in a subgroup analysis based on SYNTAX score,” an index of lesion complexity. “In patients with very high SYNTAX scores, CABG outperformed FFR-guided PCI. But if you look at patients with low SYNTAX scores, actually, FFR-guided PCI outperformed CABG for 1-year MACCE.”
Dr. Fearon is lead author on the study’s Nov. 4, 2021, publication in the New England Journal of Medicine, its release timed to coincide with his presentation of the trial at the Transcatheter Cardiovascular Therapeutics annual meeting, held virtually and live in Orlando and sponsored by the Cardiovascular Research Foundation.
He noted that FAME-3 “wasn’t designed or powered to test for superiority,” so its results do not imply CABG is superior to FFR-PCI in patients with MVD, and remains “inconclusive” on that question.
“I think what this study does is provide both the physician and patients more contemporary data and information on options and expected outcomes in multivessel disease. So if you are a patient who has less complex disease, I think you can feel comfortable that you will get an equivalent result with FFR-guided PCI.” But, at least based on FAME-3, Dr. Fearon said, CABG provides better outcomes in patients with more complex disease.
“I think there are still patients that look at trade-offs. Some patients will accept a higher event rate in order to avoid a long recovery, and vice versa.” So the trial may allow patients and physicians to make more informed decisions, he said.
A main message of FAME-3 “is that we’re getting very good results with three-vessel PCI, but better results with surgery,” Ran Kornowski, MD, Rabin Medical Center, Petah Tikva, Israel, and Tel Aviv University, said as a discussant following Dr. Fearon’s presentation of the trial. The subanalysis by SYNTAX score, he agreed, probably could be used as part of shared decision-making with patients.
Not all that surprising
“It’s a well-designed study, with a lot of patients,” said surgeon Frank W. Sellke, MD, of Rhode Island Hospital, Miriam Hospital, and Brown University, all in Providence.
“I don’t think it’s all that surprising,” he said in an interview. “It’s very consistent with what other studies have shown, that for three-vessel disease, surgery tends to have the edge,” even when pitted against FFR-guided PCI.
Indeed, pressure-wire FFR-PCI has a spotty history, even as an alternative to standard angiography-based PCI. For example, it has performed well in registry and other cohort studies but showed no advantage in the all-comers RIPCORD-2 trial or in the setting of complete revascularization PCI for acute MI in FLOWER-MI. And it emitted an increased-mortality signal in the prematurely halted FUTURE trial.
In FAME-3, “the 1-year follow-up was the best chance for FFR-PCI to be noninferior to CABG. The CABG advantage is only going to get better with time if prior experience and pathobiology is true,” Sanjay Kaul, MD, Cedars-Sinai Medical Center, Los Angeles, said in an interview.
Overall, “the quality and quantity of evidence is insufficient to support FFR-guided PCI” in patients with complex coronary artery disease (CAD), he said. “I would also argue that the evidence for FFR-guided PCI for simple CAD is also not high quality.”
Dr. Kaul also blasted the claim that FFR-PCI was seen to perform better against CABG in patients with low SYNTAX scores. “In general, one cannot use a positive subgroup in a null or negative trial, as is the case with FAME-3, to ‘rescue’ the treatment intervention.” Such a positive subgroup finding, he said, “would at best be deemed hypothesis-generating and not hypothesis validating.”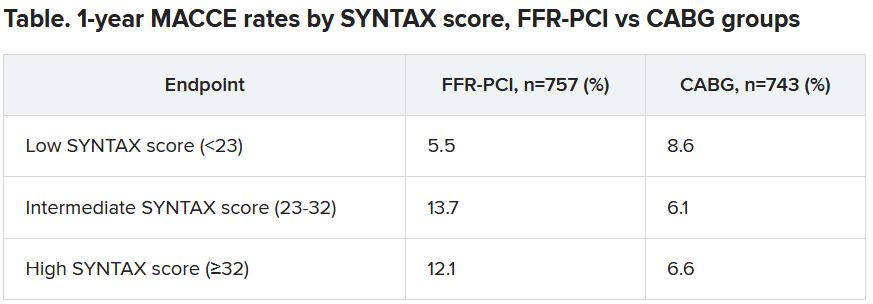
Dr. Fearon agreed that the subgroup analysis by SYNTAX score, though prespecified, was only hypothesis generating. “But I think that other studies have shown the same thing – that in less complex disease, the two strategies appear to perform in a similar fashion.”
The FAME-3 trial’s 1,500 patients were randomly assigned at 48 centers to undergo standard CABG or FFR-guided PCI with Resolute Integrity (Medtronic) zotarolimus-eluting DES. Lesions with a pressure-wire FFR of 0.80 or less were stented and those with higher FFR readings were deferred.
The 1-year hazard ratio for the primary endpoint—a composite of death from any cause, MI, stroke, or repeat revascularization – was 1.5 (95% confidence interval, 1.1-2.2) with a noninferiority P value of .35 for the comparison of FFR-PCI versus CABG.
FFR-guided PCI fared significantly better than CABG for some safety endpoints, including major bleeding (1.6% vs 3.8%, P < .01), arrhythmia including atrial fibrillation (2.4% vs. 14.1%, P < .001), acute kidney injury (0.1% vs 0.9%, P < .04), and 30-day rehospitalization (5.5% vs 10.2%, P < .001).
Did the primary endpoint favor CABG?
At a media briefing prior to Dr. Fearon’s TCT 2021 presentation of the trail, Roxana Mehran, MD, Icahn School of Medicine at Mount Sinai, New York, proposed that the inclusion of repeat revascularization in the trial’s composite primary endpoint tilted the outcome in favor of CABG. “To me, the FAME-3 results are predictable because repeat revascularization is in the equation.”

It’s well recognized that the endpoint is less likely after CABG than PCI. The latter treats focal lesions that are a limited part of a coronary artery in which CAD is still likely progressing. CABG, on the other hand, can bypass longer segments of diseased artery.
Indeed, as Dr. Fearon reported, the rates of death, MI, or stroke excluding repeat revascularization were 7.3% with FFR-PCI and 5.2% for CABG, for an HR of 1.4 (95% CI, 0.9-2.1).
Dr. Mehran also proposed that intravascular-ultrasound (IVUS) guidance, had it been part of the trial, could potentially have boosted the performance of FFR-PCI.
Repeat revascularization, Dr. Kaul agreed, “should not have been included” in the trial’s primary endpoint. It had been added “to amplify events and to minimize sample size. Not including revascularization would render the sample size prohibitive. There is always give and take in designing clinical trials.”
And he agreed that “IVUS-based PCI optimization would have further improved PCI outcomes.” However, “IVUS plus FFR adds to the procedural burden and limited resources available.” Dr. Fearon said when interviewed that the trial’s definition of procedural MI, a component of the primary endpoint, might potentially be seen as controversial. Procedural MIs in both the PCI and CABG groups were required to meet the standards of CABG-related type-5 MI according to the third and fourth Universal Definitions. The had also had to be accompanied by “a significant finding like new Q waves or a new wall-motion abnormality on echocardiography,” he said.
“That’s fairly strict. Because of that, we had a low rate of periprocedural MI and it was similar between the two groups, around 1.5% in both arms.”
FAME-3 was funded by Medtronic and Abbott Vascular. Dr. Kaul disclosed no relevant financial relationships. Dr. Kornowsky receives royalties from or holds intellectual property rights with CathWorks. Dr. Mehran disclosed financial ties to numerous pharmaceutical and device companies, and that she, her spouse, or her institution hold equity in Elixir Medical, Applied Therapeutics, and ControlRad.
A version of this article first appeared on Medscape.com.
Coronary stenting guided by fractional flow reserve (FFR) readings, considered to reflect the targeted lesion’s functional impact, was no match for coronary bypass surgery (CABG) in patients with multivessel disease (MVD) in a major international randomized trial.
Indeed, FFR-guided percutaneous coronary intervention (PCI) using one of the latest drug-eluting stents (DES) seemed to perform poorly in the trial, compared with surgery, apparently upping the risk for clinical events by 50% over 1 year.
Designed statistically for noninferiority, the third Fractional Flow Reserve Versus Angiography for Multivessel Evaluation (FAME 3) trial, with 1,500 randomized patients, showed that FFR-guided PCI was “not noninferior” to CABG. Of those randomized to PCI, 10.6% met the 1-year primary endpoint of major adverse cardiac or cerebrovascular events (MACCE), compared with only 6.9% of patients assigned to CABG.
The trial enrolled only patients with three-vessel coronary disease with no left-main coronary artery involvement, who were declared by their institution’s multidisciplinary heart team to be appropriate for either form of revascularization.
One of the roles of FFR for PCI guidance is to identify significant lesions “that are underrecognized by the angiogram,” which is less likely to happen in patients with very complex coronary anatomy, study chair William F. Fearon, MD, Stanford (Calif.) University, said in an interview.
“That’s what we saw in a subgroup analysis based on SYNTAX score,” an index of lesion complexity. “In patients with very high SYNTAX scores, CABG outperformed FFR-guided PCI. But if you look at patients with low SYNTAX scores, actually, FFR-guided PCI outperformed CABG for 1-year MACCE.”
Dr. Fearon is lead author on the study’s Nov. 4, 2021, publication in the New England Journal of Medicine, its release timed to coincide with his presentation of the trial at the Transcatheter Cardiovascular Therapeutics annual meeting, held virtually and live in Orlando and sponsored by the Cardiovascular Research Foundation.
He noted that FAME-3 “wasn’t designed or powered to test for superiority,” so its results do not imply CABG is superior to FFR-PCI in patients with MVD, and remains “inconclusive” on that question.
“I think what this study does is provide both the physician and patients more contemporary data and information on options and expected outcomes in multivessel disease. So if you are a patient who has less complex disease, I think you can feel comfortable that you will get an equivalent result with FFR-guided PCI.” But, at least based on FAME-3, Dr. Fearon said, CABG provides better outcomes in patients with more complex disease.
“I think there are still patients that look at trade-offs. Some patients will accept a higher event rate in order to avoid a long recovery, and vice versa.” So the trial may allow patients and physicians to make more informed decisions, he said.
A main message of FAME-3 “is that we’re getting very good results with three-vessel PCI, but better results with surgery,” Ran Kornowski, MD, Rabin Medical Center, Petah Tikva, Israel, and Tel Aviv University, said as a discussant following Dr. Fearon’s presentation of the trial. The subanalysis by SYNTAX score, he agreed, probably could be used as part of shared decision-making with patients.
Not all that surprising
“It’s a well-designed study, with a lot of patients,” said surgeon Frank W. Sellke, MD, of Rhode Island Hospital, Miriam Hospital, and Brown University, all in Providence.
“I don’t think it’s all that surprising,” he said in an interview. “It’s very consistent with what other studies have shown, that for three-vessel disease, surgery tends to have the edge,” even when pitted against FFR-guided PCI.
Indeed, pressure-wire FFR-PCI has a spotty history, even as an alternative to standard angiography-based PCI. For example, it has performed well in registry and other cohort studies but showed no advantage in the all-comers RIPCORD-2 trial or in the setting of complete revascularization PCI for acute MI in FLOWER-MI. And it emitted an increased-mortality signal in the prematurely halted FUTURE trial.
In FAME-3, “the 1-year follow-up was the best chance for FFR-PCI to be noninferior to CABG. The CABG advantage is only going to get better with time if prior experience and pathobiology is true,” Sanjay Kaul, MD, Cedars-Sinai Medical Center, Los Angeles, said in an interview.
Overall, “the quality and quantity of evidence is insufficient to support FFR-guided PCI” in patients with complex coronary artery disease (CAD), he said. “I would also argue that the evidence for FFR-guided PCI for simple CAD is also not high quality.”
Dr. Kaul also blasted the claim that FFR-PCI was seen to perform better against CABG in patients with low SYNTAX scores. “In general, one cannot use a positive subgroup in a null or negative trial, as is the case with FAME-3, to ‘rescue’ the treatment intervention.” Such a positive subgroup finding, he said, “would at best be deemed hypothesis-generating and not hypothesis validating.”
Dr. Fearon agreed that the subgroup analysis by SYNTAX score, though prespecified, was only hypothesis generating. “But I think that other studies have shown the same thing – that in less complex disease, the two strategies appear to perform in a similar fashion.”
The FAME-3 trial’s 1,500 patients were randomly assigned at 48 centers to undergo standard CABG or FFR-guided PCI with Resolute Integrity (Medtronic) zotarolimus-eluting DES. Lesions with a pressure-wire FFR of 0.80 or less were stented and those with higher FFR readings were deferred.
The 1-year hazard ratio for the primary endpoint—a composite of death from any cause, MI, stroke, or repeat revascularization – was 1.5 (95% confidence interval, 1.1-2.2) with a noninferiority P value of .35 for the comparison of FFR-PCI versus CABG.
FFR-guided PCI fared significantly better than CABG for some safety endpoints, including major bleeding (1.6% vs 3.8%, P < .01), arrhythmia including atrial fibrillation (2.4% vs. 14.1%, P < .001), acute kidney injury (0.1% vs 0.9%, P < .04), and 30-day rehospitalization (5.5% vs 10.2%, P < .001).
Did the primary endpoint favor CABG?
At a media briefing prior to Dr. Fearon’s TCT 2021 presentation of the trail, Roxana Mehran, MD, Icahn School of Medicine at Mount Sinai, New York, proposed that the inclusion of repeat revascularization in the trial’s composite primary endpoint tilted the outcome in favor of CABG. “To me, the FAME-3 results are predictable because repeat revascularization is in the equation.”
It’s well recognized that the endpoint is less likely after CABG than PCI. The latter treats focal lesions that are a limited part of a coronary artery in which CAD is still likely progressing. CABG, on the other hand, can bypass longer segments of diseased artery.
Indeed, as Dr. Fearon reported, the rates of death, MI, or stroke excluding repeat revascularization were 7.3% with FFR-PCI and 5.2% for CABG, for an HR of 1.4 (95% CI, 0.9-2.1).
Dr. Mehran also proposed that intravascular-ultrasound (IVUS) guidance, had it been part of the trial, could potentially have boosted the performance of FFR-PCI.
Repeat revascularization, Dr. Kaul agreed, “should not have been included” in the trial’s primary endpoint. It had been added “to amplify events and to minimize sample size. Not including revascularization would render the sample size prohibitive. There is always give and take in designing clinical trials.”
And he agreed that “IVUS-based PCI optimization would have further improved PCI outcomes.” However, “IVUS plus FFR adds to the procedural burden and limited resources available.” Dr. Fearon said when interviewed that the trial’s definition of procedural MI, a component of the primary endpoint, might potentially be seen as controversial. Procedural MIs in both the PCI and CABG groups were required to meet the standards of CABG-related type-5 MI according to the third and fourth Universal Definitions. The had also had to be accompanied by “a significant finding like new Q waves or a new wall-motion abnormality on echocardiography,” he said.
“That’s fairly strict. Because of that, we had a low rate of periprocedural MI and it was similar between the two groups, around 1.5% in both arms.”
FAME-3 was funded by Medtronic and Abbott Vascular. Dr. Kaul disclosed no relevant financial relationships. Dr. Kornowsky receives royalties from or holds intellectual property rights with CathWorks. Dr. Mehran disclosed financial ties to numerous pharmaceutical and device companies, and that she, her spouse, or her institution hold equity in Elixir Medical, Applied Therapeutics, and ControlRad.
A version of this article first appeared on Medscape.com.
Coronary stenting guided by fractional flow reserve (FFR) readings, considered to reflect the targeted lesion’s functional impact, was no match for coronary bypass surgery (CABG) in patients with multivessel disease (MVD) in a major international randomized trial.
Indeed, FFR-guided percutaneous coronary intervention (PCI) using one of the latest drug-eluting stents (DES) seemed to perform poorly in the trial, compared with surgery, apparently upping the risk for clinical events by 50% over 1 year.
Designed statistically for noninferiority, the third Fractional Flow Reserve Versus Angiography for Multivessel Evaluation (FAME 3) trial, with 1,500 randomized patients, showed that FFR-guided PCI was “not noninferior” to CABG. Of those randomized to PCI, 10.6% met the 1-year primary endpoint of major adverse cardiac or cerebrovascular events (MACCE), compared with only 6.9% of patients assigned to CABG.
The trial enrolled only patients with three-vessel coronary disease with no left-main coronary artery involvement, who were declared by their institution’s multidisciplinary heart team to be appropriate for either form of revascularization.
One of the roles of FFR for PCI guidance is to identify significant lesions “that are underrecognized by the angiogram,” which is less likely to happen in patients with very complex coronary anatomy, study chair William F. Fearon, MD, Stanford (Calif.) University, said in an interview.
“That’s what we saw in a subgroup analysis based on SYNTAX score,” an index of lesion complexity. “In patients with very high SYNTAX scores, CABG outperformed FFR-guided PCI. But if you look at patients with low SYNTAX scores, actually, FFR-guided PCI outperformed CABG for 1-year MACCE.”
Dr. Fearon is lead author on the study’s Nov. 4, 2021, publication in the New England Journal of Medicine, its release timed to coincide with his presentation of the trial at the Transcatheter Cardiovascular Therapeutics annual meeting, held virtually and live in Orlando and sponsored by the Cardiovascular Research Foundation.
He noted that FAME-3 “wasn’t designed or powered to test for superiority,” so its results do not imply CABG is superior to FFR-PCI in patients with MVD, and remains “inconclusive” on that question.
“I think what this study does is provide both the physician and patients more contemporary data and information on options and expected outcomes in multivessel disease. So if you are a patient who has less complex disease, I think you can feel comfortable that you will get an equivalent result with FFR-guided PCI.” But, at least based on FAME-3, Dr. Fearon said, CABG provides better outcomes in patients with more complex disease.
“I think there are still patients that look at trade-offs. Some patients will accept a higher event rate in order to avoid a long recovery, and vice versa.” So the trial may allow patients and physicians to make more informed decisions, he said.
A main message of FAME-3 “is that we’re getting very good results with three-vessel PCI, but better results with surgery,” Ran Kornowski, MD, Rabin Medical Center, Petah Tikva, Israel, and Tel Aviv University, said as a discussant following Dr. Fearon’s presentation of the trial. The subanalysis by SYNTAX score, he agreed, probably could be used as part of shared decision-making with patients.
Not all that surprising
“It’s a well-designed study, with a lot of patients,” said surgeon Frank W. Sellke, MD, of Rhode Island Hospital, Miriam Hospital, and Brown University, all in Providence.
“I don’t think it’s all that surprising,” he said in an interview. “It’s very consistent with what other studies have shown, that for three-vessel disease, surgery tends to have the edge,” even when pitted against FFR-guided PCI.
Indeed, pressure-wire FFR-PCI has a spotty history, even as an alternative to standard angiography-based PCI. For example, it has performed well in registry and other cohort studies but showed no advantage in the all-comers RIPCORD-2 trial or in the setting of complete revascularization PCI for acute MI in FLOWER-MI. And it emitted an increased-mortality signal in the prematurely halted FUTURE trial.
In FAME-3, “the 1-year follow-up was the best chance for FFR-PCI to be noninferior to CABG. The CABG advantage is only going to get better with time if prior experience and pathobiology is true,” Sanjay Kaul, MD, Cedars-Sinai Medical Center, Los Angeles, said in an interview.
Overall, “the quality and quantity of evidence is insufficient to support FFR-guided PCI” in patients with complex coronary artery disease (CAD), he said. “I would also argue that the evidence for FFR-guided PCI for simple CAD is also not high quality.”
Dr. Kaul also blasted the claim that FFR-PCI was seen to perform better against CABG in patients with low SYNTAX scores. “In general, one cannot use a positive subgroup in a null or negative trial, as is the case with FAME-3, to ‘rescue’ the treatment intervention.” Such a positive subgroup finding, he said, “would at best be deemed hypothesis-generating and not hypothesis validating.”
Dr. Fearon agreed that the subgroup analysis by SYNTAX score, though prespecified, was only hypothesis generating. “But I think that other studies have shown the same thing – that in less complex disease, the two strategies appear to perform in a similar fashion.”
The FAME-3 trial’s 1,500 patients were randomly assigned at 48 centers to undergo standard CABG or FFR-guided PCI with Resolute Integrity (Medtronic) zotarolimus-eluting DES. Lesions with a pressure-wire FFR of 0.80 or less were stented and those with higher FFR readings were deferred.
The 1-year hazard ratio for the primary endpoint—a composite of death from any cause, MI, stroke, or repeat revascularization – was 1.5 (95% confidence interval, 1.1-2.2) with a noninferiority P value of .35 for the comparison of FFR-PCI versus CABG.
FFR-guided PCI fared significantly better than CABG for some safety endpoints, including major bleeding (1.6% vs 3.8%, P < .01), arrhythmia including atrial fibrillation (2.4% vs. 14.1%, P < .001), acute kidney injury (0.1% vs 0.9%, P < .04), and 30-day rehospitalization (5.5% vs 10.2%, P < .001).
Did the primary endpoint favor CABG?
At a media briefing prior to Dr. Fearon’s TCT 2021 presentation of the trail, Roxana Mehran, MD, Icahn School of Medicine at Mount Sinai, New York, proposed that the inclusion of repeat revascularization in the trial’s composite primary endpoint tilted the outcome in favor of CABG. “To me, the FAME-3 results are predictable because repeat revascularization is in the equation.”
It’s well recognized that the endpoint is less likely after CABG than PCI. The latter treats focal lesions that are a limited part of a coronary artery in which CAD is still likely progressing. CABG, on the other hand, can bypass longer segments of diseased artery.
Indeed, as Dr. Fearon reported, the rates of death, MI, or stroke excluding repeat revascularization were 7.3% with FFR-PCI and 5.2% for CABG, for an HR of 1.4 (95% CI, 0.9-2.1).
Dr. Mehran also proposed that intravascular-ultrasound (IVUS) guidance, had it been part of the trial, could potentially have boosted the performance of FFR-PCI.
Repeat revascularization, Dr. Kaul agreed, “should not have been included” in the trial’s primary endpoint. It had been added “to amplify events and to minimize sample size. Not including revascularization would render the sample size prohibitive. There is always give and take in designing clinical trials.”
And he agreed that “IVUS-based PCI optimization would have further improved PCI outcomes.” However, “IVUS plus FFR adds to the procedural burden and limited resources available.” Dr. Fearon said when interviewed that the trial’s definition of procedural MI, a component of the primary endpoint, might potentially be seen as controversial. Procedural MIs in both the PCI and CABG groups were required to meet the standards of CABG-related type-5 MI according to the third and fourth Universal Definitions. The had also had to be accompanied by “a significant finding like new Q waves or a new wall-motion abnormality on echocardiography,” he said.
“That’s fairly strict. Because of that, we had a low rate of periprocedural MI and it was similar between the two groups, around 1.5% in both arms.”
FAME-3 was funded by Medtronic and Abbott Vascular. Dr. Kaul disclosed no relevant financial relationships. Dr. Kornowsky receives royalties from or holds intellectual property rights with CathWorks. Dr. Mehran disclosed financial ties to numerous pharmaceutical and device companies, and that she, her spouse, or her institution hold equity in Elixir Medical, Applied Therapeutics, and ControlRad.
A version of this article first appeared on Medscape.com.
SUGAR trial finds superior stent for those with diabetes and CAD
Superiority shown on TLF endpoint
Designed to show noninferiority for treatment of coronary artery disease (CAD) in patients with diabetes, a head-to-head comparison of contemporary stents ended up showing that one was superior to the for the primary endpoint of target lesion failure (TLF).

In the superiority analysis, the 35% relative reduction in the risk of TLF at 1 year for the Cre8 EVO (Alvimedica) stent relative to the Resolute Onyx (Medtronic) device reached significance, according to Rafael Romaguera, MD, PhD, an interventional cardiologist at the Bellvitge University Hospital, Barcelona.
At 1 year, the rates of TLF were 7.2% and 10.5% for the Cre8 EVO and Resolute Onyx stents, respectively. On the basis of noninferiority, the 3.73% reduction in TLF at 1 year among those receiving the Cre8 EVO device provided a highly significant confirmation of noninferiority (P < .001) and triggered the preplanned superiority analysis.
When the significant advantage on the TLF endpoint (P = .03) was broken down into its components, the Cre8 EVO stent was linked to numerically lower rates of cardiac death (2.1% vs. 2.7%), target vessel MI (5.3% vs. 7.2%), and target lesion revascularization (2.4% vs. 3.9%), according to the SUGAR (Second-Generation Drug-Eluting Stents in Diabetes) trial results presented at the Transcatheter Cardiovascular Therapeutics annual meeting, held virtually and live in Orlando and sponsored by the Cardiovascular Research Foundation.
In a previous study comparing these devices, called the ReCre8 trial, the rates of TLF in an all-comer CAD population were similar at 1 year. When an updated 3-year analysis was presented earlier in 2021 at the Cardiovascular Research Technologies meeting, they remained similar.
Diabetes-centered trial was unmet need
The rationale for conducting a new trial limited to patients with diabetes was based on the greater risk in this population, according to Dr. Romaguera. He cited data that indicate the risk of major adverse cardiac events are about two times higher 2 years after stent implantation in patients with diabetes relative to those without, even when contemporary drug-eluting stents are used.
Both the Cre8 EVO and Resolute Onyx stent are drug eluting and employ contemporary architecture that provides the basis for marketing claims that they are suitable for complex patients; but they have differences.
“There are three features that I think differentiate the Cre8 EVO stent,” Dr. Romaguera reported at the meeting, sponsored by the Cardiovascular Research Foundation.
One is the absence of polymer, which contrasts with the permanent polymer of the Resolute device. This feature affects the dissolution of the anti-inflammatory drug and might be one explanation for the greater protection from ischemic events, according to Dr. Romaguera.
Another is the thickness of the struts, which range from 70 to 80 mm for the Cre8 EVO device and from 92 to 102 mm for the Resolute Onyx device. In experimental studies, strut thickness has been associated with greater risk of thrombus formation, although it is unclear if this modest difference is clinically significant.
Also important, the Cre8 EVO device employs sirolimus for an anti-inflammatory effect, while the Resolute Onyx elutes zotarolimus. Again, experimental evidence suggests a greater anti-inflammatory effect reduces the need for dual-antiplatelet therapy (DAPT); that might offer a relative advantage in patients with an elevated risk of bleeding.
It is not clear whether all of these features contribute to the better results observed in this trial in diabetes patients, but Dr. Romaguera indicated that the lower risk of TLF with Cre8 EVO is not just statistically significant but also clinically meaningful.
In SUGAR, which included 23 centers in Spain, 1,175 patients with confirmed diabetes scheduled for percutaneous intervention (PCI) were randomized to one of the two stents. The study was purposely designed with very few exclusion criteria.
SUGAR trial employed all-comer design
“This was an all-comer design and there was no limitation in regard to clinical presentation, complexity, number of lesions, or other disease features,” said Dr. Romaguera. The major exclusions were a life expectancy of less than 2 years and a contraindication to taking DAPT for at least 1 month,
The patients were almost equally divided between those who had a non–ST-segment elevation MI) and those with chronic coronary artery disease, but patients with a STEMI, representing about 12% of the population, were included. Almost all of the patients (about 95%) had type 2 diabetes; nearly one-third were on insulin at the time of randomization.
According to Dr. Romaguera, “SUGAR is the first powered trial to compare new-generation drug-eluting stents in patients with diabetes,” and he emphasized the all-comer design in supporting its clinical relevance.
Several of those participating in discussion of the trial during the late-breaker session agreed. Although the moderator, Gregg Stone, MD, of the Icahn School of Medicine at Mount Sinai, New York, expressed surprise that the trial “actually demonstrated superiority” given the difficulty of showing a difference between modern stents, he called the findings “remarkable.”
Others seemed to suggest that it would alter their practice.
“This study is sweet like sugar for us, because now we have a stent that is dedicated and fitted for the diabetic population,” said Gennaro Sardella, MD, of Sapienza University of Rome.
For Marc Etienne Jolicoeur, MD, an interventional cardiologist associated with Duke University, Durham, N.C., one of the impressive findings was the early separation of the curves in favor of Cre8 EVO. Calling SUGAR a “fantastic trial,” he indicated that the progressive advantage over time reinforced his impression that the difference is real.
However, David Kandzari, MD, director of interventional cardiology, Piedmont Hart Institute, Atlanta, was more circumspect. He did not express any criticisms of the trial, but he called for “a larger evidence base” before declaring the Cre8 EVO device a standard of care for patients with diabetes undergoing PCI.
The SUGAR results were published in the European Heart Journal at the time of presentation at the meeting.
The trial was funded by the Spanish Society of Cardiology. Dr. Romaguera reported financial relationships with Biotronik and Boston Scientific. Dr. Stone, has financial relationships with more than 10 pharmaceutical companies, including those developing devices used in PCI. Dr. Sardella and Dr. Jolicoeur reported no financial relationships relevant to this topic. Dr. Kandzari reported financial relationships with Ablative Solutions and Medtronic.
Superiority shown on TLF endpoint
Superiority shown on TLF endpoint
Designed to show noninferiority for treatment of coronary artery disease (CAD) in patients with diabetes, a head-to-head comparison of contemporary stents ended up showing that one was superior to the for the primary endpoint of target lesion failure (TLF).
In the superiority analysis, the 35% relative reduction in the risk of TLF at 1 year for the Cre8 EVO (Alvimedica) stent relative to the Resolute Onyx (Medtronic) device reached significance, according to Rafael Romaguera, MD, PhD, an interventional cardiologist at the Bellvitge University Hospital, Barcelona.
At 1 year, the rates of TLF were 7.2% and 10.5% for the Cre8 EVO and Resolute Onyx stents, respectively. On the basis of noninferiority, the 3.73% reduction in TLF at 1 year among those receiving the Cre8 EVO device provided a highly significant confirmation of noninferiority (P < .001) and triggered the preplanned superiority analysis.
When the significant advantage on the TLF endpoint (P = .03) was broken down into its components, the Cre8 EVO stent was linked to numerically lower rates of cardiac death (2.1% vs. 2.7%), target vessel MI (5.3% vs. 7.2%), and target lesion revascularization (2.4% vs. 3.9%), according to the SUGAR (Second-Generation Drug-Eluting Stents in Diabetes) trial results presented at the Transcatheter Cardiovascular Therapeutics annual meeting, held virtually and live in Orlando and sponsored by the Cardiovascular Research Foundation.
In a previous study comparing these devices, called the ReCre8 trial, the rates of TLF in an all-comer CAD population were similar at 1 year. When an updated 3-year analysis was presented earlier in 2021 at the Cardiovascular Research Technologies meeting, they remained similar.
Diabetes-centered trial was unmet need
The rationale for conducting a new trial limited to patients with diabetes was based on the greater risk in this population, according to Dr. Romaguera. He cited data that indicate the risk of major adverse cardiac events are about two times higher 2 years after stent implantation in patients with diabetes relative to those without, even when contemporary drug-eluting stents are used.
Both the Cre8 EVO and Resolute Onyx stent are drug eluting and employ contemporary architecture that provides the basis for marketing claims that they are suitable for complex patients; but they have differences.
“There are three features that I think differentiate the Cre8 EVO stent,” Dr. Romaguera reported at the meeting, sponsored by the Cardiovascular Research Foundation.
One is the absence of polymer, which contrasts with the permanent polymer of the Resolute device. This feature affects the dissolution of the anti-inflammatory drug and might be one explanation for the greater protection from ischemic events, according to Dr. Romaguera.
Another is the thickness of the struts, which range from 70 to 80 mm for the Cre8 EVO device and from 92 to 102 mm for the Resolute Onyx device. In experimental studies, strut thickness has been associated with greater risk of thrombus formation, although it is unclear if this modest difference is clinically significant.
Also important, the Cre8 EVO device employs sirolimus for an anti-inflammatory effect, while the Resolute Onyx elutes zotarolimus. Again, experimental evidence suggests a greater anti-inflammatory effect reduces the need for dual-antiplatelet therapy (DAPT); that might offer a relative advantage in patients with an elevated risk of bleeding.
It is not clear whether all of these features contribute to the better results observed in this trial in diabetes patients, but Dr. Romaguera indicated that the lower risk of TLF with Cre8 EVO is not just statistically significant but also clinically meaningful.
In SUGAR, which included 23 centers in Spain, 1,175 patients with confirmed diabetes scheduled for percutaneous intervention (PCI) were randomized to one of the two stents. The study was purposely designed with very few exclusion criteria.
SUGAR trial employed all-comer design
“This was an all-comer design and there was no limitation in regard to clinical presentation, complexity, number of lesions, or other disease features,” said Dr. Romaguera. The major exclusions were a life expectancy of less than 2 years and a contraindication to taking DAPT for at least 1 month,
The patients were almost equally divided between those who had a non–ST-segment elevation MI) and those with chronic coronary artery disease, but patients with a STEMI, representing about 12% of the population, were included. Almost all of the patients (about 95%) had type 2 diabetes; nearly one-third were on insulin at the time of randomization.
According to Dr. Romaguera, “SUGAR is the first powered trial to compare new-generation drug-eluting stents in patients with diabetes,” and he emphasized the all-comer design in supporting its clinical relevance.
Several of those participating in discussion of the trial during the late-breaker session agreed. Although the moderator, Gregg Stone, MD, of the Icahn School of Medicine at Mount Sinai, New York, expressed surprise that the trial “actually demonstrated superiority” given the difficulty of showing a difference between modern stents, he called the findings “remarkable.”
Others seemed to suggest that it would alter their practice.
“This study is sweet like sugar for us, because now we have a stent that is dedicated and fitted for the diabetic population,” said Gennaro Sardella, MD, of Sapienza University of Rome.
For Marc Etienne Jolicoeur, MD, an interventional cardiologist associated with Duke University, Durham, N.C., one of the impressive findings was the early separation of the curves in favor of Cre8 EVO. Calling SUGAR a “fantastic trial,” he indicated that the progressive advantage over time reinforced his impression that the difference is real.
However, David Kandzari, MD, director of interventional cardiology, Piedmont Hart Institute, Atlanta, was more circumspect. He did not express any criticisms of the trial, but he called for “a larger evidence base” before declaring the Cre8 EVO device a standard of care for patients with diabetes undergoing PCI.
The SUGAR results were published in the European Heart Journal at the time of presentation at the meeting.
The trial was funded by the Spanish Society of Cardiology. Dr. Romaguera reported financial relationships with Biotronik and Boston Scientific. Dr. Stone, has financial relationships with more than 10 pharmaceutical companies, including those developing devices used in PCI. Dr. Sardella and Dr. Jolicoeur reported no financial relationships relevant to this topic. Dr. Kandzari reported financial relationships with Ablative Solutions and Medtronic.
Designed to show noninferiority for treatment of coronary artery disease (CAD) in patients with diabetes, a head-to-head comparison of contemporary stents ended up showing that one was superior to the for the primary endpoint of target lesion failure (TLF).
In the superiority analysis, the 35% relative reduction in the risk of TLF at 1 year for the Cre8 EVO (Alvimedica) stent relative to the Resolute Onyx (Medtronic) device reached significance, according to Rafael Romaguera, MD, PhD, an interventional cardiologist at the Bellvitge University Hospital, Barcelona.
At 1 year, the rates of TLF were 7.2% and 10.5% for the Cre8 EVO and Resolute Onyx stents, respectively. On the basis of noninferiority, the 3.73% reduction in TLF at 1 year among those receiving the Cre8 EVO device provided a highly significant confirmation of noninferiority (P < .001) and triggered the preplanned superiority analysis.
When the significant advantage on the TLF endpoint (P = .03) was broken down into its components, the Cre8 EVO stent was linked to numerically lower rates of cardiac death (2.1% vs. 2.7%), target vessel MI (5.3% vs. 7.2%), and target lesion revascularization (2.4% vs. 3.9%), according to the SUGAR (Second-Generation Drug-Eluting Stents in Diabetes) trial results presented at the Transcatheter Cardiovascular Therapeutics annual meeting, held virtually and live in Orlando and sponsored by the Cardiovascular Research Foundation.
In a previous study comparing these devices, called the ReCre8 trial, the rates of TLF in an all-comer CAD population were similar at 1 year. When an updated 3-year analysis was presented earlier in 2021 at the Cardiovascular Research Technologies meeting, they remained similar.
Diabetes-centered trial was unmet need
The rationale for conducting a new trial limited to patients with diabetes was based on the greater risk in this population, according to Dr. Romaguera. He cited data that indicate the risk of major adverse cardiac events are about two times higher 2 years after stent implantation in patients with diabetes relative to those without, even when contemporary drug-eluting stents are used.
Both the Cre8 EVO and Resolute Onyx stent are drug eluting and employ contemporary architecture that provides the basis for marketing claims that they are suitable for complex patients; but they have differences.
“There are three features that I think differentiate the Cre8 EVO stent,” Dr. Romaguera reported at the meeting, sponsored by the Cardiovascular Research Foundation.
One is the absence of polymer, which contrasts with the permanent polymer of the Resolute device. This feature affects the dissolution of the anti-inflammatory drug and might be one explanation for the greater protection from ischemic events, according to Dr. Romaguera.
Another is the thickness of the struts, which range from 70 to 80 mm for the Cre8 EVO device and from 92 to 102 mm for the Resolute Onyx device. In experimental studies, strut thickness has been associated with greater risk of thrombus formation, although it is unclear if this modest difference is clinically significant.
Also important, the Cre8 EVO device employs sirolimus for an anti-inflammatory effect, while the Resolute Onyx elutes zotarolimus. Again, experimental evidence suggests a greater anti-inflammatory effect reduces the need for dual-antiplatelet therapy (DAPT); that might offer a relative advantage in patients with an elevated risk of bleeding.
It is not clear whether all of these features contribute to the better results observed in this trial in diabetes patients, but Dr. Romaguera indicated that the lower risk of TLF with Cre8 EVO is not just statistically significant but also clinically meaningful.
In SUGAR, which included 23 centers in Spain, 1,175 patients with confirmed diabetes scheduled for percutaneous intervention (PCI) were randomized to one of the two stents. The study was purposely designed with very few exclusion criteria.
SUGAR trial employed all-comer design
“This was an all-comer design and there was no limitation in regard to clinical presentation, complexity, number of lesions, or other disease features,” said Dr. Romaguera. The major exclusions were a life expectancy of less than 2 years and a contraindication to taking DAPT for at least 1 month,
The patients were almost equally divided between those who had a non–ST-segment elevation MI) and those with chronic coronary artery disease, but patients with a STEMI, representing about 12% of the population, were included. Almost all of the patients (about 95%) had type 2 diabetes; nearly one-third were on insulin at the time of randomization.
According to Dr. Romaguera, “SUGAR is the first powered trial to compare new-generation drug-eluting stents in patients with diabetes,” and he emphasized the all-comer design in supporting its clinical relevance.
Several of those participating in discussion of the trial during the late-breaker session agreed. Although the moderator, Gregg Stone, MD, of the Icahn School of Medicine at Mount Sinai, New York, expressed surprise that the trial “actually demonstrated superiority” given the difficulty of showing a difference between modern stents, he called the findings “remarkable.”
Others seemed to suggest that it would alter their practice.
“This study is sweet like sugar for us, because now we have a stent that is dedicated and fitted for the diabetic population,” said Gennaro Sardella, MD, of Sapienza University of Rome.
For Marc Etienne Jolicoeur, MD, an interventional cardiologist associated with Duke University, Durham, N.C., one of the impressive findings was the early separation of the curves in favor of Cre8 EVO. Calling SUGAR a “fantastic trial,” he indicated that the progressive advantage over time reinforced his impression that the difference is real.
However, David Kandzari, MD, director of interventional cardiology, Piedmont Hart Institute, Atlanta, was more circumspect. He did not express any criticisms of the trial, but he called for “a larger evidence base” before declaring the Cre8 EVO device a standard of care for patients with diabetes undergoing PCI.
The SUGAR results were published in the European Heart Journal at the time of presentation at the meeting.
The trial was funded by the Spanish Society of Cardiology. Dr. Romaguera reported financial relationships with Biotronik and Boston Scientific. Dr. Stone, has financial relationships with more than 10 pharmaceutical companies, including those developing devices used in PCI. Dr. Sardella and Dr. Jolicoeur reported no financial relationships relevant to this topic. Dr. Kandzari reported financial relationships with Ablative Solutions and Medtronic.
FROM TCT 2021
Ivermectin–COVID-19 study retracted; authors blame file mix-up
The paper, “Effects of a Single Dose of Ivermectin on Viral and Clinical Outcomes in Asymptomatic SARS-CoV-2 Infected Subjects: A Pilot Clinical Trial in Lebanon,” appeared in the journal Viruses in May. According to the abstract: “A randomized controlled trial was conducted in 100 asymptomatic Lebanese subjects that have tested positive for SARS-CoV2. Fifty patients received standard preventive treatment, mainly supplements, and the experimental group received a single dose (according to body weight) of ivermectin, in addition to the same supplements the control group received.”
Results results results … and: “Ivermectin appears to be efficacious in providing clinical benefits in a randomized treatment of asymptomatic SARS-CoV-2-positive subjects, effectively resulting in fewer symptoms, lower viral load and reduced hospital admissions. However, larger-scale trials are warranted for this conclusion to be further cemented.”
However, in early October, the BBC reported — in a larger piece about the concerns about ivermectin-Covid-19 research — that the study “was found to have blocks of details of 11 patients that had been copied and pasted repeatedly – suggesting many of the trial’s apparent patients didn’t really exist.”
The study’s authors told the BBC that the ‘original set of data was rigged, sabotaged or mistakenly entered in the final file’ and that they have submitted a retraction to the scientific journal which published it.
That’s not quite what the retraction notice states: “The journal retracts the article, Effects of a Single Dose of Ivermectin on Viral and Clinical Outcomes in Asymptomatic SARS-CoV-2 Infected Subjects: A Pilot Clinical Trial in Lebanon [ 1 ], cited above. Following publication, the authors contacted the editorial office regarding an error between files used for the statistical analysis. Adhering to our complaints procedure, an investigation was conducted that confirmed the error reported by the authors.
This retraction was approved by the Editor in Chief of the journal. The authors agreed to this retraction.”
Ali Samaha, of Lebanese University in Beirut, and the lead author of the study, told us: “It was brought to our attention that we have used wrong file for our paper. We informed immediately the journal and we have run investigations. After revising the raw data we realised that a file that was used to train a research assistant was sent by mistake for analysis. Re-analysing the original data , the conclusions of the paper remained valid. For our transparency we asked for retraction.”
About that BBC report? Samaha said: “The BBC article was generated before the report of independent reviewers who confirmed an innocent mistake by using wrong file.”
Samaha added that he and his colleagues are now considering whether to resubmit the paper.
The article has been cited four times, according to Clarivate Analytics’ Web of Science — including in this meta-analysis published in June in the American Journal of Therapeutics , which concluded that: “Moderate-certainty evidence finds that large reductions in COVID-19 deaths are possible using ivermectin. Using ivermectin early in the clinical course may reduce numbers progressing to severe disease. The apparent safety and low cost suggest that ivermectin is likely to have a significant impact on the SARS-CoV-2 pandemic globally.”
That article was a social media darling, receiving more than 45,000 tweets and pickups in 90 news outlets, according to Altmetrics, which ranks it No. 7 among all papers published at that time.
A version of this article first appeared on Retraction Watch.
The paper, “Effects of a Single Dose of Ivermectin on Viral and Clinical Outcomes in Asymptomatic SARS-CoV-2 Infected Subjects: A Pilot Clinical Trial in Lebanon,” appeared in the journal Viruses in May. According to the abstract: “A randomized controlled trial was conducted in 100 asymptomatic Lebanese subjects that have tested positive for SARS-CoV2. Fifty patients received standard preventive treatment, mainly supplements, and the experimental group received a single dose (according to body weight) of ivermectin, in addition to the same supplements the control group received.”
Results results results … and: “Ivermectin appears to be efficacious in providing clinical benefits in a randomized treatment of asymptomatic SARS-CoV-2-positive subjects, effectively resulting in fewer symptoms, lower viral load and reduced hospital admissions. However, larger-scale trials are warranted for this conclusion to be further cemented.”
However, in early October, the BBC reported — in a larger piece about the concerns about ivermectin-Covid-19 research — that the study “was found to have blocks of details of 11 patients that had been copied and pasted repeatedly – suggesting many of the trial’s apparent patients didn’t really exist.”
The study’s authors told the BBC that the ‘original set of data was rigged, sabotaged or mistakenly entered in the final file’ and that they have submitted a retraction to the scientific journal which published it.
That’s not quite what the retraction notice states: “The journal retracts the article, Effects of a Single Dose of Ivermectin on Viral and Clinical Outcomes in Asymptomatic SARS-CoV-2 Infected Subjects: A Pilot Clinical Trial in Lebanon [ 1 ], cited above. Following publication, the authors contacted the editorial office regarding an error between files used for the statistical analysis. Adhering to our complaints procedure, an investigation was conducted that confirmed the error reported by the authors.
This retraction was approved by the Editor in Chief of the journal. The authors agreed to this retraction.”
Ali Samaha, of Lebanese University in Beirut, and the lead author of the study, told us: “It was brought to our attention that we have used wrong file for our paper. We informed immediately the journal and we have run investigations. After revising the raw data we realised that a file that was used to train a research assistant was sent by mistake for analysis. Re-analysing the original data , the conclusions of the paper remained valid. For our transparency we asked for retraction.”
About that BBC report? Samaha said: “The BBC article was generated before the report of independent reviewers who confirmed an innocent mistake by using wrong file.”
Samaha added that he and his colleagues are now considering whether to resubmit the paper.
The article has been cited four times, according to Clarivate Analytics’ Web of Science — including in this meta-analysis published in June in the American Journal of Therapeutics , which concluded that: “Moderate-certainty evidence finds that large reductions in COVID-19 deaths are possible using ivermectin. Using ivermectin early in the clinical course may reduce numbers progressing to severe disease. The apparent safety and low cost suggest that ivermectin is likely to have a significant impact on the SARS-CoV-2 pandemic globally.”
That article was a social media darling, receiving more than 45,000 tweets and pickups in 90 news outlets, according to Altmetrics, which ranks it No. 7 among all papers published at that time.
A version of this article first appeared on Retraction Watch.
The paper, “Effects of a Single Dose of Ivermectin on Viral and Clinical Outcomes in Asymptomatic SARS-CoV-2 Infected Subjects: A Pilot Clinical Trial in Lebanon,” appeared in the journal Viruses in May. According to the abstract: “A randomized controlled trial was conducted in 100 asymptomatic Lebanese subjects that have tested positive for SARS-CoV2. Fifty patients received standard preventive treatment, mainly supplements, and the experimental group received a single dose (according to body weight) of ivermectin, in addition to the same supplements the control group received.”
Results results results … and: “Ivermectin appears to be efficacious in providing clinical benefits in a randomized treatment of asymptomatic SARS-CoV-2-positive subjects, effectively resulting in fewer symptoms, lower viral load and reduced hospital admissions. However, larger-scale trials are warranted for this conclusion to be further cemented.”
However, in early October, the BBC reported — in a larger piece about the concerns about ivermectin-Covid-19 research — that the study “was found to have blocks of details of 11 patients that had been copied and pasted repeatedly – suggesting many of the trial’s apparent patients didn’t really exist.”
The study’s authors told the BBC that the ‘original set of data was rigged, sabotaged or mistakenly entered in the final file’ and that they have submitted a retraction to the scientific journal which published it.
That’s not quite what the retraction notice states: “The journal retracts the article, Effects of a Single Dose of Ivermectin on Viral and Clinical Outcomes in Asymptomatic SARS-CoV-2 Infected Subjects: A Pilot Clinical Trial in Lebanon [ 1 ], cited above. Following publication, the authors contacted the editorial office regarding an error between files used for the statistical analysis. Adhering to our complaints procedure, an investigation was conducted that confirmed the error reported by the authors.
This retraction was approved by the Editor in Chief of the journal. The authors agreed to this retraction.”
Ali Samaha, of Lebanese University in Beirut, and the lead author of the study, told us: “It was brought to our attention that we have used wrong file for our paper. We informed immediately the journal and we have run investigations. After revising the raw data we realised that a file that was used to train a research assistant was sent by mistake for analysis. Re-analysing the original data , the conclusions of the paper remained valid. For our transparency we asked for retraction.”
About that BBC report? Samaha said: “The BBC article was generated before the report of independent reviewers who confirmed an innocent mistake by using wrong file.”
Samaha added that he and his colleagues are now considering whether to resubmit the paper.
The article has been cited four times, according to Clarivate Analytics’ Web of Science — including in this meta-analysis published in June in the American Journal of Therapeutics , which concluded that: “Moderate-certainty evidence finds that large reductions in COVID-19 deaths are possible using ivermectin. Using ivermectin early in the clinical course may reduce numbers progressing to severe disease. The apparent safety and low cost suggest that ivermectin is likely to have a significant impact on the SARS-CoV-2 pandemic globally.”
That article was a social media darling, receiving more than 45,000 tweets and pickups in 90 news outlets, according to Altmetrics, which ranks it No. 7 among all papers published at that time.
A version of this article first appeared on Retraction Watch.
An MD’s nightmare began with reporting her manic episode to the medical board
Susan Haney, MD, a board-certified emergency physician in Coos Bay, Ore., was 2 years into her career when she had her first manic episode, likely a side effect of the steroid prednisone, which she had been prescribed for an asthma flare-up. Her boss at Bay Area Hospital told her that if she wanted to return to work, she would need to have written clearance from the medical board.
In retrospect, Dr. Haney says, “I don’t think they had any idea of what they would set in motion.”
Dr. Haney says the Oregon Medical Board posted her name and the nondisciplinary action on their website and in their newsletter. Her local newspaper read it and ran a story about her. “They effectively announced my mental illness to the general public despite my objections,” she says.
During the next decade, she had two more manic episodes, and more board investigations and actions followed. Despite being cleared for work each time, Dr. Haney says the board actions decimated her career in emergency medicine and her income, which is about half of what she would have earned by now. She is frustrated, sad, and angry about what happened but considers herself lucky to be practicing medicine in urgent care.
Being investigated is scary
After her first manic episode in 2006, Dr. Haney contacted the board’s medical director, a retired general surgeon, who told her the only way the board would authorize her return to work was if she agreed to open a board investigation.
She gave them the green light because she thought she had nothing to fear – she was cooperating fully and wasn’t impaired. Now Dr. Haney says she was naive. “The board is not your friend,” she says.
Dr. Haney was also anxious to return to work. She worked in a seven-person emergency department, and two colleagues were on maternity leave or medical leave.
“My colleagues kept calling asking me when I was going to return to work, and I kept saying, ‘I don’t know because the board won’t tell me,’ “ she says.
She was also feeling a lot of financial pressure. She was 2 years out of residency, owed $100,000 in student loans, and had just bought a house.
“I was really scared – I didn’t know how long this would last or if they would let me return to work. Early on, I even got a fitness for duty evaluation from the state’s consulting psychiatrist, who cleared me for work, and the board still wouldn’t let me return. They told me I had to go through their bureaucracy and a board meeting, which didn’t make sense to me.”
Dr. Haney consented to give the board’s investigative staff access to her medical records because she feared that if she challenged them, they would suspend or revoke her license immediately.
After investigating her for 4 months, the board cleared Dr. Haney to return to work at Bay Area Hospital. She agreed to the board’s “corrective action” terms: She would continue to receive psychiatric care, maintain a physician-patient relationship with a primary care physician, and enroll in the Health Physicians Program (HPP) for substance abuse monitoring.
Dr. Haney suspects that the board investigation damaged her reputation at work. “Before this, my work evaluations were consistently excellent. Afterwards, they were all adequate. I don’t think that was a coincidence.”
Worst time of her life
Five years later, after taking prednisone for another asthma flare-up, Dr. Haney had a more severe manic episode and was hospitalized.
The consulting psychiatrist who evaluated her reported her case to the medical board, stating she had bipolar disorder, was mentally incompetent, and shouldn’t be practicing medicine. The board opened a second investigation of her in 2012, which lasted 4 months.
Dr. Haney had quit her job at Bay Area Hospital in 2011 because she was pregnant and was planning to take a year off to care for the baby at home.
“That was the worst time of my life. I lost the baby at 4 months, I wasn’t working, and now I was under investigation by the board again,” she says.
The board issued an “interim stipulated order” that required that she be monitored regularly for mental illness and substance abuse by the Health Professionals Services Program (HPSP) for 2 years. “The board accused me of abusing prednisone, which I wasn’t. I was using it as prescribed and medically indicated,” she said.
The board order was reported to the National Practitioner Databank and is now permanently in her record. Although the board cleared her to work, she could not find a permanent job in a hospital emergency department.
“The repeated ‘nondisciplinary’ public board orders have had the same net impact on my career as if I had been disciplined for killing or harming my patients. For all intents and purposes, people treat it as a disciplinary action for the rest of your career,” she said.
To keep afloat financially, she found locum tenens work in local emergency departments until 2019.
Mental health toll
Dr. Haney feels that the stress of repeated board investigations has affected her mental health. “Both times this happened, it made my mental health worse, made the mania worse, and subsequent depression worse.”
Particularly distressing to her was the fact that the administrative staff who investigated her were attorneys and persons in law enforcement, rather than medical professionals with mental health training.
“I was required to disclose intimate personal details of my psychological and psychiatric history to anybody at the board who requested them. These investigators were asking me about my childhood history. That was traumatic and none of their business!”
Dr. Haney had quietly managed episodes of major depression since she was in her early 20s with the help of a psychiatrist. Her third episode of mania, which occurred in 2014, triggered a more severe depression, which she says deepened when she learned that the HPSP had notified the board about her manic symptoms and that she would not be released from the 2-year monitoring contract. When the board notified her 2 weeks later that they were opening another investigation, Dr. Haney says she had an emotional crisis, attempted suicide, and was briefly hospitalized. Several weeks later, she decided to take a mood stabilizer, which she continues to take.
The board’s 2015 corrective action agreement required Dr. Haney to practice medicine only in settings that the board’s medical director preapproved and to obtain a preapproved monitoring health care provider who would send quarterly reports to the medical director. Dr. Haney says the “nondisciplinary” action agreement was also reported to the National Practitioner Data Bank.
She also agreed to ongoing monitoring by the HPSP for mental illness and substance abuse, which involved random drug testing. When she didn’t call in one day in 2019 and missed a scheduled test, the board opened another investigation on her that lasted 7 months until July 2020. Dr. Haney said this was despite three subsequent negative tests.
Dr. Haney believes that the “open investigation” doomed a job offer from a hospital emergency department in the Virgin Islands. “I had passed all the required credentialing and explained previous board orders. They pulled the rug from under me 1 week before I was supposed to move there,” says Dr. Haney.
Her license was inactivated again because she hadn’t practiced medicine for a year, which she says was a new board policy. Although Dr. Haney says the medical director reactivated her license after talking with her, “By the time I was able to apply emergency medicine jobs, no one was interested in me anymore.”
Financial toll
Dr. Haney started her medical career when she was 42 as a second career. She says the board investigations and actions have resulted in a significant loss of work and income. “I have only worked 14 of the past 17 years as a doctor. I live cheaply because I never know how much longer my career will last,” says Dr. Haney.
The ordeal has devastated her finances. She has shelled out at least $200,000 in legal fees – she hired an attorney in 2007 and filed a lawsuit against the board in Oregon district court alleging that members had violated several of her rights. The district judge sided with the state medical board, and it was upheld on appeal in 2012, referring to state laws that gave the board absolute immunity from civil lawsuits. “I had no legal recourse to contest their decisions, no matter how injurious or unjust,” says Dr. Haney.
She has also shelled out at least $100,000 to be evaluated and monitored by the health physician program (now HPSP) for several years. Physicians who agree to be monitored by these health programs have to pay their fees. The board finally agreed last July to end her HPSP participation.
Dr. Haney also filed a complaint in 2007 with the federal Department of Health & Human Services Office for Civil Rights, alleging that the board violated her civil rights under the Americans with Disabilities Act. She says that her lawsuit and the OCR investigation of the board enabled her to withdraw from the HPP in good standing in 2008..
What would she have done differently?
She regrets not hiring an attorney earlier because “most likely the board action would not have been made public. It snowballed after that -- any mistake I made in my career was viewed in the lens of potential impairment.”
She also regrets telling her employer about the nature of her illness and reporting it to the board. A psychiatrist she saw later shared advice he gives to other patients who want to remain anonymous: get help but go out of town, use a false name, and pay cash.
“I wish I had that advice when all this started. That was the best way to protect my career,” says Dr. Haney.
Protecting the public?
The Oregon Medical Board declined to comment on Dr. Haney’s experience because investigations are confidential, but the executive director, Nicole Krishnaswami, JD, answered questions in an email about how the current board operates.
She says the board has 11 medical professionals and employs a medical director and expert consultants in specialty-specific fields. MDs with mental health training are involved in investigating/reviewing cases involving doctors with mental illnesses.
“State medical boards have a responsibility to protect and inform the public. State laws further require state agencies to provide access and transparency regarding the board’s official actions. If the board receives a complaint that a licensee is impaired and thus unable to safely practice, the board has a responsibility to investigate and ensure the licensee is practicing medicine safely,” Ms. Krishnaswami said.
The HPSP is the monitoring program established by state law to provide oversight in order to ensure that licensees are not practicing while impaired. HPSP is separate from the board and the board adopted a statement outlining its perspective on the program in support of doctors with substance abuse and mental health disorder.
The board also founded the Oregon Wellness Program, which provides free, confidential counseling to all Oregon-licensed physicians and physician assistants.
Stigma continues
Dr. Haney feels there is huge stigma associated with mental illness in the medical profession. “If I had cancer twice, I wouldn’t have been put in this position and would be at the peak of my career,” she says.
Nearly half of the 862 emergency medicine physicians surveyed last October said they were reluctant to seek mental health treatment. The reasons included fear of professional repercussions and stigma in the workplace. Several physicians said they were concerned about potentially having to report the treatment on medical license applications in the future, according to a survey by the American College of Emergency Physicians.
In addition, 26% of the more than 12,000 physicians who responded to a Medscape survey last year said they didn’t want to risk disclosure (20%) or that they distrusted mental health professionals (6%).
Another physician fights back
Steven Miles, MD, an award-winning professor emeritus of medicine and bioethics at the Center for Bioethics at the University of Minnesota, in Minneapolis, understands their reluctance. In 1996, he disclosed on his license renewal application that he had recently been diagnosed with a mainly depressive type of bipolar disorder and was in treatment. He had already told his employer, who was supportive.
That set off a 14-month investigation of him by the Minnesota Board of Medical Practice. Dr. Miles and his psychiatrist refused to release his confidential records to a panel of physicians, most of whom had no expertise in mental health care. He also filed a federal claim that the board’s requests violated the ADA, and he won the case.
“Had the board given me evidence of impaired ability to practice with ordinary skill and safety, I would have cooperated. Instead, they proposed a course of action, which would have degraded the privacy of my relationship with my psychiatrist and arguably increased the barrier to getting proper care and the risk of impairment,” he said.
The board kept renewing his license, and Dr. Miles continued to work full time. “I was empowered and protected by my stature in the field at the time my mental illness was diagnosed. Early-career physicians do not yet have that protection and should be very careful of disclosing, given the still widespread stigma of mental illnesses,” he said.
His advocacy led to changes
Dr. Miles went public to mobilize support for his ADA claim. He wrote editorials that were published in JAMA and Minnesota Medicine that refer to the American Psychiatric Association’s 1984 position paper, which says that the mandatory disclosure of the physician’s confidential medical record is without merit. Dr. Miles adds that major newspapers ran stories based on his editorials.
The board backed down after Dr. Miles won his ADA case, and it met with him. “I said this is not good stewardship of the medical profession; you are injuring doctors by keeping them from psychiatric care, which is out of line with the medical view of the treatability of depression and that needs to change,” he says.
Dr. Miles says he won a victory because his practice continued. “I also won a victory in the way the board was handling these questions, which was an opening salvo in a process that continues to this day.”
The original form asked whether he had ever been diagnosed with or treated for manic depression, schizophrenia, compulsive gambling, or other psychiatric conditions.
The revised form asks, “Do you have a physical or mental condition that would affect your ability, with or without reasonable accommodation, to provide appropriate care to patients and otherwise perform the essential functions of a practitioner in your area of practice without posing a health or safety risk to your patients? If yes, what accommodations would help you provide appropriate care to patients and perform other essential functions?”
Dr. Miles says that the final wording wasn’t ideal and that it was confusing to physicians. He says this prompted additional changes in wording by the board. Starting in January, applicants will be asked, “Do you currently have any condition that is not being appropriately treated that is likely to impair or adversely affect your ability to practice medicine with reasonable skill and safety in a competent, ethical, and professional manner?” the medical board’s executive director, Ruth M. Martinez, said in an email.
When asked whether the board still investigates physicians who reveal mental illnesses on licensing applications, Ms. Martinez responded, “All disclosures are evaluated to assure that the practitioner is qualified and safe to practice.”
This article was updated 11/4/21.
A version of this article first appeared on Medscape.com.
Susan Haney, MD, a board-certified emergency physician in Coos Bay, Ore., was 2 years into her career when she had her first manic episode, likely a side effect of the steroid prednisone, which she had been prescribed for an asthma flare-up. Her boss at Bay Area Hospital told her that if she wanted to return to work, she would need to have written clearance from the medical board.
In retrospect, Dr. Haney says, “I don’t think they had any idea of what they would set in motion.”
Dr. Haney says the Oregon Medical Board posted her name and the nondisciplinary action on their website and in their newsletter. Her local newspaper read it and ran a story about her. “They effectively announced my mental illness to the general public despite my objections,” she says.
During the next decade, she had two more manic episodes, and more board investigations and actions followed. Despite being cleared for work each time, Dr. Haney says the board actions decimated her career in emergency medicine and her income, which is about half of what she would have earned by now. She is frustrated, sad, and angry about what happened but considers herself lucky to be practicing medicine in urgent care.
Being investigated is scary
After her first manic episode in 2006, Dr. Haney contacted the board’s medical director, a retired general surgeon, who told her the only way the board would authorize her return to work was if she agreed to open a board investigation.
She gave them the green light because she thought she had nothing to fear – she was cooperating fully and wasn’t impaired. Now Dr. Haney says she was naive. “The board is not your friend,” she says.
Dr. Haney was also anxious to return to work. She worked in a seven-person emergency department, and two colleagues were on maternity leave or medical leave.
“My colleagues kept calling asking me when I was going to return to work, and I kept saying, ‘I don’t know because the board won’t tell me,’ “ she says.
She was also feeling a lot of financial pressure. She was 2 years out of residency, owed $100,000 in student loans, and had just bought a house.
“I was really scared – I didn’t know how long this would last or if they would let me return to work. Early on, I even got a fitness for duty evaluation from the state’s consulting psychiatrist, who cleared me for work, and the board still wouldn’t let me return. They told me I had to go through their bureaucracy and a board meeting, which didn’t make sense to me.”
Dr. Haney consented to give the board’s investigative staff access to her medical records because she feared that if she challenged them, they would suspend or revoke her license immediately.
After investigating her for 4 months, the board cleared Dr. Haney to return to work at Bay Area Hospital. She agreed to the board’s “corrective action” terms: She would continue to receive psychiatric care, maintain a physician-patient relationship with a primary care physician, and enroll in the Health Physicians Program (HPP) for substance abuse monitoring.
Dr. Haney suspects that the board investigation damaged her reputation at work. “Before this, my work evaluations were consistently excellent. Afterwards, they were all adequate. I don’t think that was a coincidence.”
Worst time of her life
Five years later, after taking prednisone for another asthma flare-up, Dr. Haney had a more severe manic episode and was hospitalized.
The consulting psychiatrist who evaluated her reported her case to the medical board, stating she had bipolar disorder, was mentally incompetent, and shouldn’t be practicing medicine. The board opened a second investigation of her in 2012, which lasted 4 months.
Dr. Haney had quit her job at Bay Area Hospital in 2011 because she was pregnant and was planning to take a year off to care for the baby at home.
“That was the worst time of my life. I lost the baby at 4 months, I wasn’t working, and now I was under investigation by the board again,” she says.
The board issued an “interim stipulated order” that required that she be monitored regularly for mental illness and substance abuse by the Health Professionals Services Program (HPSP) for 2 years. “The board accused me of abusing prednisone, which I wasn’t. I was using it as prescribed and medically indicated,” she said.
The board order was reported to the National Practitioner Databank and is now permanently in her record. Although the board cleared her to work, she could not find a permanent job in a hospital emergency department.
“The repeated ‘nondisciplinary’ public board orders have had the same net impact on my career as if I had been disciplined for killing or harming my patients. For all intents and purposes, people treat it as a disciplinary action for the rest of your career,” she said.
To keep afloat financially, she found locum tenens work in local emergency departments until 2019.
Mental health toll
Dr. Haney feels that the stress of repeated board investigations has affected her mental health. “Both times this happened, it made my mental health worse, made the mania worse, and subsequent depression worse.”
Particularly distressing to her was the fact that the administrative staff who investigated her were attorneys and persons in law enforcement, rather than medical professionals with mental health training.
“I was required to disclose intimate personal details of my psychological and psychiatric history to anybody at the board who requested them. These investigators were asking me about my childhood history. That was traumatic and none of their business!”
Dr. Haney had quietly managed episodes of major depression since she was in her early 20s with the help of a psychiatrist. Her third episode of mania, which occurred in 2014, triggered a more severe depression, which she says deepened when she learned that the HPSP had notified the board about her manic symptoms and that she would not be released from the 2-year monitoring contract. When the board notified her 2 weeks later that they were opening another investigation, Dr. Haney says she had an emotional crisis, attempted suicide, and was briefly hospitalized. Several weeks later, she decided to take a mood stabilizer, which she continues to take.
The board’s 2015 corrective action agreement required Dr. Haney to practice medicine only in settings that the board’s medical director preapproved and to obtain a preapproved monitoring health care provider who would send quarterly reports to the medical director. Dr. Haney says the “nondisciplinary” action agreement was also reported to the National Practitioner Data Bank.
She also agreed to ongoing monitoring by the HPSP for mental illness and substance abuse, which involved random drug testing. When she didn’t call in one day in 2019 and missed a scheduled test, the board opened another investigation on her that lasted 7 months until July 2020. Dr. Haney said this was despite three subsequent negative tests.
Dr. Haney believes that the “open investigation” doomed a job offer from a hospital emergency department in the Virgin Islands. “I had passed all the required credentialing and explained previous board orders. They pulled the rug from under me 1 week before I was supposed to move there,” says Dr. Haney.
Her license was inactivated again because she hadn’t practiced medicine for a year, which she says was a new board policy. Although Dr. Haney says the medical director reactivated her license after talking with her, “By the time I was able to apply emergency medicine jobs, no one was interested in me anymore.”
Financial toll
Dr. Haney started her medical career when she was 42 as a second career. She says the board investigations and actions have resulted in a significant loss of work and income. “I have only worked 14 of the past 17 years as a doctor. I live cheaply because I never know how much longer my career will last,” says Dr. Haney.
The ordeal has devastated her finances. She has shelled out at least $200,000 in legal fees – she hired an attorney in 2007 and filed a lawsuit against the board in Oregon district court alleging that members had violated several of her rights. The district judge sided with the state medical board, and it was upheld on appeal in 2012, referring to state laws that gave the board absolute immunity from civil lawsuits. “I had no legal recourse to contest their decisions, no matter how injurious or unjust,” says Dr. Haney.
She has also shelled out at least $100,000 to be evaluated and monitored by the health physician program (now HPSP) for several years. Physicians who agree to be monitored by these health programs have to pay their fees. The board finally agreed last July to end her HPSP participation.
Dr. Haney also filed a complaint in 2007 with the federal Department of Health & Human Services Office for Civil Rights, alleging that the board violated her civil rights under the Americans with Disabilities Act. She says that her lawsuit and the OCR investigation of the board enabled her to withdraw from the HPP in good standing in 2008..
What would she have done differently?
She regrets not hiring an attorney earlier because “most likely the board action would not have been made public. It snowballed after that -- any mistake I made in my career was viewed in the lens of potential impairment.”
She also regrets telling her employer about the nature of her illness and reporting it to the board. A psychiatrist she saw later shared advice he gives to other patients who want to remain anonymous: get help but go out of town, use a false name, and pay cash.
“I wish I had that advice when all this started. That was the best way to protect my career,” says Dr. Haney.
Protecting the public?
The Oregon Medical Board declined to comment on Dr. Haney’s experience because investigations are confidential, but the executive director, Nicole Krishnaswami, JD, answered questions in an email about how the current board operates.
She says the board has 11 medical professionals and employs a medical director and expert consultants in specialty-specific fields. MDs with mental health training are involved in investigating/reviewing cases involving doctors with mental illnesses.
“State medical boards have a responsibility to protect and inform the public. State laws further require state agencies to provide access and transparency regarding the board’s official actions. If the board receives a complaint that a licensee is impaired and thus unable to safely practice, the board has a responsibility to investigate and ensure the licensee is practicing medicine safely,” Ms. Krishnaswami said.
The HPSP is the monitoring program established by state law to provide oversight in order to ensure that licensees are not practicing while impaired. HPSP is separate from the board and the board adopted a statement outlining its perspective on the program in support of doctors with substance abuse and mental health disorder.
The board also founded the Oregon Wellness Program, which provides free, confidential counseling to all Oregon-licensed physicians and physician assistants.
Stigma continues
Dr. Haney feels there is huge stigma associated with mental illness in the medical profession. “If I had cancer twice, I wouldn’t have been put in this position and would be at the peak of my career,” she says.
Nearly half of the 862 emergency medicine physicians surveyed last October said they were reluctant to seek mental health treatment. The reasons included fear of professional repercussions and stigma in the workplace. Several physicians said they were concerned about potentially having to report the treatment on medical license applications in the future, according to a survey by the American College of Emergency Physicians.
In addition, 26% of the more than 12,000 physicians who responded to a Medscape survey last year said they didn’t want to risk disclosure (20%) or that they distrusted mental health professionals (6%).
Another physician fights back
Steven Miles, MD, an award-winning professor emeritus of medicine and bioethics at the Center for Bioethics at the University of Minnesota, in Minneapolis, understands their reluctance. In 1996, he disclosed on his license renewal application that he had recently been diagnosed with a mainly depressive type of bipolar disorder and was in treatment. He had already told his employer, who was supportive.
That set off a 14-month investigation of him by the Minnesota Board of Medical Practice. Dr. Miles and his psychiatrist refused to release his confidential records to a panel of physicians, most of whom had no expertise in mental health care. He also filed a federal claim that the board’s requests violated the ADA, and he won the case.
“Had the board given me evidence of impaired ability to practice with ordinary skill and safety, I would have cooperated. Instead, they proposed a course of action, which would have degraded the privacy of my relationship with my psychiatrist and arguably increased the barrier to getting proper care and the risk of impairment,” he said.
The board kept renewing his license, and Dr. Miles continued to work full time. “I was empowered and protected by my stature in the field at the time my mental illness was diagnosed. Early-career physicians do not yet have that protection and should be very careful of disclosing, given the still widespread stigma of mental illnesses,” he said.
His advocacy led to changes
Dr. Miles went public to mobilize support for his ADA claim. He wrote editorials that were published in JAMA and Minnesota Medicine that refer to the American Psychiatric Association’s 1984 position paper, which says that the mandatory disclosure of the physician’s confidential medical record is without merit. Dr. Miles adds that major newspapers ran stories based on his editorials.
The board backed down after Dr. Miles won his ADA case, and it met with him. “I said this is not good stewardship of the medical profession; you are injuring doctors by keeping them from psychiatric care, which is out of line with the medical view of the treatability of depression and that needs to change,” he says.
Dr. Miles says he won a victory because his practice continued. “I also won a victory in the way the board was handling these questions, which was an opening salvo in a process that continues to this day.”
The original form asked whether he had ever been diagnosed with or treated for manic depression, schizophrenia, compulsive gambling, or other psychiatric conditions.
The revised form asks, “Do you have a physical or mental condition that would affect your ability, with or without reasonable accommodation, to provide appropriate care to patients and otherwise perform the essential functions of a practitioner in your area of practice without posing a health or safety risk to your patients? If yes, what accommodations would help you provide appropriate care to patients and perform other essential functions?”
Dr. Miles says that the final wording wasn’t ideal and that it was confusing to physicians. He says this prompted additional changes in wording by the board. Starting in January, applicants will be asked, “Do you currently have any condition that is not being appropriately treated that is likely to impair or adversely affect your ability to practice medicine with reasonable skill and safety in a competent, ethical, and professional manner?” the medical board’s executive director, Ruth M. Martinez, said in an email.
When asked whether the board still investigates physicians who reveal mental illnesses on licensing applications, Ms. Martinez responded, “All disclosures are evaluated to assure that the practitioner is qualified and safe to practice.”
This article was updated 11/4/21.
A version of this article first appeared on Medscape.com.
Susan Haney, MD, a board-certified emergency physician in Coos Bay, Ore., was 2 years into her career when she had her first manic episode, likely a side effect of the steroid prednisone, which she had been prescribed for an asthma flare-up. Her boss at Bay Area Hospital told her that if she wanted to return to work, she would need to have written clearance from the medical board.
In retrospect, Dr. Haney says, “I don’t think they had any idea of what they would set in motion.”
Dr. Haney says the Oregon Medical Board posted her name and the nondisciplinary action on their website and in their newsletter. Her local newspaper read it and ran a story about her. “They effectively announced my mental illness to the general public despite my objections,” she says.
During the next decade, she had two more manic episodes, and more board investigations and actions followed. Despite being cleared for work each time, Dr. Haney says the board actions decimated her career in emergency medicine and her income, which is about half of what she would have earned by now. She is frustrated, sad, and angry about what happened but considers herself lucky to be practicing medicine in urgent care.
Being investigated is scary
After her first manic episode in 2006, Dr. Haney contacted the board’s medical director, a retired general surgeon, who told her the only way the board would authorize her return to work was if she agreed to open a board investigation.
She gave them the green light because she thought she had nothing to fear – she was cooperating fully and wasn’t impaired. Now Dr. Haney says she was naive. “The board is not your friend,” she says.
Dr. Haney was also anxious to return to work. She worked in a seven-person emergency department, and two colleagues were on maternity leave or medical leave.
“My colleagues kept calling asking me when I was going to return to work, and I kept saying, ‘I don’t know because the board won’t tell me,’ “ she says.
She was also feeling a lot of financial pressure. She was 2 years out of residency, owed $100,000 in student loans, and had just bought a house.
“I was really scared – I didn’t know how long this would last or if they would let me return to work. Early on, I even got a fitness for duty evaluation from the state’s consulting psychiatrist, who cleared me for work, and the board still wouldn’t let me return. They told me I had to go through their bureaucracy and a board meeting, which didn’t make sense to me.”
Dr. Haney consented to give the board’s investigative staff access to her medical records because she feared that if she challenged them, they would suspend or revoke her license immediately.
After investigating her for 4 months, the board cleared Dr. Haney to return to work at Bay Area Hospital. She agreed to the board’s “corrective action” terms: She would continue to receive psychiatric care, maintain a physician-patient relationship with a primary care physician, and enroll in the Health Physicians Program (HPP) for substance abuse monitoring.
Dr. Haney suspects that the board investigation damaged her reputation at work. “Before this, my work evaluations were consistently excellent. Afterwards, they were all adequate. I don’t think that was a coincidence.”
Worst time of her life
Five years later, after taking prednisone for another asthma flare-up, Dr. Haney had a more severe manic episode and was hospitalized.
The consulting psychiatrist who evaluated her reported her case to the medical board, stating she had bipolar disorder, was mentally incompetent, and shouldn’t be practicing medicine. The board opened a second investigation of her in 2012, which lasted 4 months.
Dr. Haney had quit her job at Bay Area Hospital in 2011 because she was pregnant and was planning to take a year off to care for the baby at home.
“That was the worst time of my life. I lost the baby at 4 months, I wasn’t working, and now I was under investigation by the board again,” she says.
The board issued an “interim stipulated order” that required that she be monitored regularly for mental illness and substance abuse by the Health Professionals Services Program (HPSP) for 2 years. “The board accused me of abusing prednisone, which I wasn’t. I was using it as prescribed and medically indicated,” she said.
The board order was reported to the National Practitioner Databank and is now permanently in her record. Although the board cleared her to work, she could not find a permanent job in a hospital emergency department.
“The repeated ‘nondisciplinary’ public board orders have had the same net impact on my career as if I had been disciplined for killing or harming my patients. For all intents and purposes, people treat it as a disciplinary action for the rest of your career,” she said.
To keep afloat financially, she found locum tenens work in local emergency departments until 2019.
Mental health toll
Dr. Haney feels that the stress of repeated board investigations has affected her mental health. “Both times this happened, it made my mental health worse, made the mania worse, and subsequent depression worse.”
Particularly distressing to her was the fact that the administrative staff who investigated her were attorneys and persons in law enforcement, rather than medical professionals with mental health training.
“I was required to disclose intimate personal details of my psychological and psychiatric history to anybody at the board who requested them. These investigators were asking me about my childhood history. That was traumatic and none of their business!”
Dr. Haney had quietly managed episodes of major depression since she was in her early 20s with the help of a psychiatrist. Her third episode of mania, which occurred in 2014, triggered a more severe depression, which she says deepened when she learned that the HPSP had notified the board about her manic symptoms and that she would not be released from the 2-year monitoring contract. When the board notified her 2 weeks later that they were opening another investigation, Dr. Haney says she had an emotional crisis, attempted suicide, and was briefly hospitalized. Several weeks later, she decided to take a mood stabilizer, which she continues to take.
The board’s 2015 corrective action agreement required Dr. Haney to practice medicine only in settings that the board’s medical director preapproved and to obtain a preapproved monitoring health care provider who would send quarterly reports to the medical director. Dr. Haney says the “nondisciplinary” action agreement was also reported to the National Practitioner Data Bank.
She also agreed to ongoing monitoring by the HPSP for mental illness and substance abuse, which involved random drug testing. When she didn’t call in one day in 2019 and missed a scheduled test, the board opened another investigation on her that lasted 7 months until July 2020. Dr. Haney said this was despite three subsequent negative tests.
Dr. Haney believes that the “open investigation” doomed a job offer from a hospital emergency department in the Virgin Islands. “I had passed all the required credentialing and explained previous board orders. They pulled the rug from under me 1 week before I was supposed to move there,” says Dr. Haney.
Her license was inactivated again because she hadn’t practiced medicine for a year, which she says was a new board policy. Although Dr. Haney says the medical director reactivated her license after talking with her, “By the time I was able to apply emergency medicine jobs, no one was interested in me anymore.”
Financial toll
Dr. Haney started her medical career when she was 42 as a second career. She says the board investigations and actions have resulted in a significant loss of work and income. “I have only worked 14 of the past 17 years as a doctor. I live cheaply because I never know how much longer my career will last,” says Dr. Haney.
The ordeal has devastated her finances. She has shelled out at least $200,000 in legal fees – she hired an attorney in 2007 and filed a lawsuit against the board in Oregon district court alleging that members had violated several of her rights. The district judge sided with the state medical board, and it was upheld on appeal in 2012, referring to state laws that gave the board absolute immunity from civil lawsuits. “I had no legal recourse to contest their decisions, no matter how injurious or unjust,” says Dr. Haney.
She has also shelled out at least $100,000 to be evaluated and monitored by the health physician program (now HPSP) for several years. Physicians who agree to be monitored by these health programs have to pay their fees. The board finally agreed last July to end her HPSP participation.
Dr. Haney also filed a complaint in 2007 with the federal Department of Health & Human Services Office for Civil Rights, alleging that the board violated her civil rights under the Americans with Disabilities Act. She says that her lawsuit and the OCR investigation of the board enabled her to withdraw from the HPP in good standing in 2008..
What would she have done differently?
She regrets not hiring an attorney earlier because “most likely the board action would not have been made public. It snowballed after that -- any mistake I made in my career was viewed in the lens of potential impairment.”
She also regrets telling her employer about the nature of her illness and reporting it to the board. A psychiatrist she saw later shared advice he gives to other patients who want to remain anonymous: get help but go out of town, use a false name, and pay cash.
“I wish I had that advice when all this started. That was the best way to protect my career,” says Dr. Haney.
Protecting the public?
The Oregon Medical Board declined to comment on Dr. Haney’s experience because investigations are confidential, but the executive director, Nicole Krishnaswami, JD, answered questions in an email about how the current board operates.
She says the board has 11 medical professionals and employs a medical director and expert consultants in specialty-specific fields. MDs with mental health training are involved in investigating/reviewing cases involving doctors with mental illnesses.
“State medical boards have a responsibility to protect and inform the public. State laws further require state agencies to provide access and transparency regarding the board’s official actions. If the board receives a complaint that a licensee is impaired and thus unable to safely practice, the board has a responsibility to investigate and ensure the licensee is practicing medicine safely,” Ms. Krishnaswami said.
The HPSP is the monitoring program established by state law to provide oversight in order to ensure that licensees are not practicing while impaired. HPSP is separate from the board and the board adopted a statement outlining its perspective on the program in support of doctors with substance abuse and mental health disorder.
The board also founded the Oregon Wellness Program, which provides free, confidential counseling to all Oregon-licensed physicians and physician assistants.
Stigma continues
Dr. Haney feels there is huge stigma associated with mental illness in the medical profession. “If I had cancer twice, I wouldn’t have been put in this position and would be at the peak of my career,” she says.
Nearly half of the 862 emergency medicine physicians surveyed last October said they were reluctant to seek mental health treatment. The reasons included fear of professional repercussions and stigma in the workplace. Several physicians said they were concerned about potentially having to report the treatment on medical license applications in the future, according to a survey by the American College of Emergency Physicians.
In addition, 26% of the more than 12,000 physicians who responded to a Medscape survey last year said they didn’t want to risk disclosure (20%) or that they distrusted mental health professionals (6%).
Another physician fights back
Steven Miles, MD, an award-winning professor emeritus of medicine and bioethics at the Center for Bioethics at the University of Minnesota, in Minneapolis, understands their reluctance. In 1996, he disclosed on his license renewal application that he had recently been diagnosed with a mainly depressive type of bipolar disorder and was in treatment. He had already told his employer, who was supportive.
That set off a 14-month investigation of him by the Minnesota Board of Medical Practice. Dr. Miles and his psychiatrist refused to release his confidential records to a panel of physicians, most of whom had no expertise in mental health care. He also filed a federal claim that the board’s requests violated the ADA, and he won the case.
“Had the board given me evidence of impaired ability to practice with ordinary skill and safety, I would have cooperated. Instead, they proposed a course of action, which would have degraded the privacy of my relationship with my psychiatrist and arguably increased the barrier to getting proper care and the risk of impairment,” he said.
The board kept renewing his license, and Dr. Miles continued to work full time. “I was empowered and protected by my stature in the field at the time my mental illness was diagnosed. Early-career physicians do not yet have that protection and should be very careful of disclosing, given the still widespread stigma of mental illnesses,” he said.
His advocacy led to changes
Dr. Miles went public to mobilize support for his ADA claim. He wrote editorials that were published in JAMA and Minnesota Medicine that refer to the American Psychiatric Association’s 1984 position paper, which says that the mandatory disclosure of the physician’s confidential medical record is without merit. Dr. Miles adds that major newspapers ran stories based on his editorials.
The board backed down after Dr. Miles won his ADA case, and it met with him. “I said this is not good stewardship of the medical profession; you are injuring doctors by keeping them from psychiatric care, which is out of line with the medical view of the treatability of depression and that needs to change,” he says.
Dr. Miles says he won a victory because his practice continued. “I also won a victory in the way the board was handling these questions, which was an opening salvo in a process that continues to this day.”
The original form asked whether he had ever been diagnosed with or treated for manic depression, schizophrenia, compulsive gambling, or other psychiatric conditions.
The revised form asks, “Do you have a physical or mental condition that would affect your ability, with or without reasonable accommodation, to provide appropriate care to patients and otherwise perform the essential functions of a practitioner in your area of practice without posing a health or safety risk to your patients? If yes, what accommodations would help you provide appropriate care to patients and perform other essential functions?”
Dr. Miles says that the final wording wasn’t ideal and that it was confusing to physicians. He says this prompted additional changes in wording by the board. Starting in January, applicants will be asked, “Do you currently have any condition that is not being appropriately treated that is likely to impair or adversely affect your ability to practice medicine with reasonable skill and safety in a competent, ethical, and professional manner?” the medical board’s executive director, Ruth M. Martinez, said in an email.
When asked whether the board still investigates physicians who reveal mental illnesses on licensing applications, Ms. Martinez responded, “All disclosures are evaluated to assure that the practitioner is qualified and safe to practice.”
This article was updated 11/4/21.
A version of this article first appeared on Medscape.com.
James Bond taken down by an epidemiologist
No, Mr. Bond, I expect you to die
Movie watching usually requires a certain suspension of disbelief, and it’s safe to say James Bond movies require this more than most. Between the impossible gadgets and ludicrous doomsday plans, very few have ever stopped to consider the health risks of the James Bond universe.

Now, however, Bond, James Bond, has met his most formidable opponent: Wouter Graumans, a graduate student in epidemiology from the Netherlands. During a foray to Burkina Faso to study infectious diseases, Mr. Graumans came down with a case of food poisoning, which led him to wonder how 007 is able to trot across this big world of ours without contracting so much as a sinus infection.
Because Mr. Graumans is a man of science and conviction, mere speculation wasn’t enough. He and a group of coauthors wrote an entire paper on the health risks of the James Bond universe.
Doing so required watching over 3,000 minutes of numerous movies and analyzing Bond’s 86 total trips to 46 different countries based on current Centers for Disease Control and Prevention advice for travel to those countries. Time which, the authors state in the abstract, “could easily have been spent on more pressing societal issues or forms of relaxation that are more acceptable in academic circles.”
Naturally, Mr. Bond’s line of work entails exposure to unpleasant things, such as poison, dehydration, heatstroke, and dangerous wildlife (everything from ticks to crocodiles), though oddly enough he never succumbs to any of it. He’s also curiously immune to hangovers, despite rarely drinking anything nonalcoholic. There are also less obvious risks: For one, 007 rarely washes his hands. During one movie, he handles raw chicken to lure away a pack of crocodiles but fails to wash his hands afterward, leaving him at risk for multiple food-borne illnesses.
Of course, we must address the elephant in the bedroom: Mr. Bond’s numerous, er, encounters with women. One would imagine the biggest risk to those women would be from the various STDs that likely course through Bond’s body, but of the 27% who died shortly after … encountering … him, all involved violence, with disease playing no obvious role. Who knows, maybe he’s clean? Stranger things have happened.
The timing of this article may seem a bit suspicious. Was it a PR stunt by the studio? Rest assured, the authors addressed this, noting that they received no funding for the study, and that, “given the futility of its academic value, this is deemed entirely appropriate by all authors.” We love when a punchline writes itself.
How to see Atlanta on $688.35 a day
The world is always changing, so we have to change with it. This week, LOTME becomes a travel guide, and our first stop is the Big A, the Big Peach, Dogwood City, Empire City of the South, Wakanda.
There’s lots to do in Atlanta: Celebrate a World Series win, visit the College Football Hall of Fame or the World of Coca Cola, or take the Stranger Things/Upside Down film locations tour. Serious adventurers, however, get out of the city and go to Emory Decatur Hospital in – you guessed it – Decatur (unofficial motto: “Everything is Greater in Decatur”).
Find the emergency room and ask for Taylor Davis, who will be your personal guide. She’ll show you how to check in at the desk, sit in the waiting room for 7 hours, and then leave without seeing any medical personnel or receiving any sort of attention whatsoever. All the things she did when she went there in July for a head injury.
Ms. Davis told Fox5 Atlanta: “I didn’t get my vitals taken, nobody called my name. I wasn’t seen at all.”
But wait! There’s more! By booking your trip through LOTMEgo* and using the code “Decatur,” you’ll get the Taylor Davis special, which includes a bill/cover charge for $688.35 from the hospital. An Emory Healthcare patient financial services employee told Ms. Davis that “you get charged before you are seen. Not for being seen.”
If all this has you ready to hop in your car (really?), then check out LOTMEgo* on Twittbook and InstaTok. You’ll also find trick-or-treating tips and discounts on haunted hospital tours.
*Does not actually exist
Breaking down the hot flash
Do you ever wonder why we scramble for cold things when we’re feeling nauseous? Whether it’s the cool air that needs to hit your face in the car or a cold, damp towel on the back of your neck, scientists think it could possibly be an evolutionary mechanism at the cellular level.

Motion sickness it’s actually a battle of body temperature, according to an article from LiveScience. Capillaries in the skin dilate, allowing for more blood flow near the skin’s surface and causing core temperature to fall. Once body temperature drops, the hypothalamus, which regulates temperature, tries to do its job by raising body temperature. Thus the hot flash!
The cold compress and cool air help fight the battle by counteracting the hypothalamus, but why the drop in body temperature to begin with?
There are a few theories. Dr. Robert Glatter, an emergency physician at Lenox Hill Hospital in New York, told LiveScience that the lack of oxygen needed in body tissue to survive at lower temperatures could be making it difficult to get oxygen to the body when a person is ill, and is “more likely an adaptive response influenced by poorly understood mechanisms at the cellular level.”
Another theory is that the nausea and body temperature shift is the body’s natural response to help people vomit.
Then there’s the theory of “defensive hypothermia,” which suggests that cold sweats are a possible mechanism to conserve energy so the body can fight off an intruder, which was supported by a 2014 study and a 2016 review.
It’s another one of the body’s many survival tricks.
Teachers were right: Pupils can do the math
Teachers liked to preach that we wouldn’t have calculators with us all the time, but that wound up not being true. Our phones have calculators at the press of a button. But maybe even calculators aren’t always needed because our pupils do more math than you think.

The pupil light reflex – constrict in light and dilate in darkness – is well known, but recent work shows that pupil size is also regulated by cognitive and perceptual factors. By presenting subjects with images of various numbers of dots and measuring pupil size, the investigators were able to show “that numerical information is intrinsically related to perception,” lead author Dr. Elisa Castaldi of Florence University noted in a written statement.
The researchers found that pupils are responsible for important survival techniques. Coauthor David Burr of the University of Sydney and the University of Florence gave an evolutionary perspective: “When we look around, we spontaneously perceive the form, size, movement and colour of a scene. Equally spontaneously, we perceive the number of items before us. This ability, shared with most other animals, is an evolutionary fundamental: It reveals immediately important quantities, such as how many apples there are on the tree, or how many enemies are attacking.”
Useful information, indeed, but our pupils seem to be more interested in the quantity of beers in the refrigerator.
No, Mr. Bond, I expect you to die
Movie watching usually requires a certain suspension of disbelief, and it’s safe to say James Bond movies require this more than most. Between the impossible gadgets and ludicrous doomsday plans, very few have ever stopped to consider the health risks of the James Bond universe.
Now, however, Bond, James Bond, has met his most formidable opponent: Wouter Graumans, a graduate student in epidemiology from the Netherlands. During a foray to Burkina Faso to study infectious diseases, Mr. Graumans came down with a case of food poisoning, which led him to wonder how 007 is able to trot across this big world of ours without contracting so much as a sinus infection.
Because Mr. Graumans is a man of science and conviction, mere speculation wasn’t enough. He and a group of coauthors wrote an entire paper on the health risks of the James Bond universe.
Doing so required watching over 3,000 minutes of numerous movies and analyzing Bond’s 86 total trips to 46 different countries based on current Centers for Disease Control and Prevention advice for travel to those countries. Time which, the authors state in the abstract, “could easily have been spent on more pressing societal issues or forms of relaxation that are more acceptable in academic circles.”
Naturally, Mr. Bond’s line of work entails exposure to unpleasant things, such as poison, dehydration, heatstroke, and dangerous wildlife (everything from ticks to crocodiles), though oddly enough he never succumbs to any of it. He’s also curiously immune to hangovers, despite rarely drinking anything nonalcoholic. There are also less obvious risks: For one, 007 rarely washes his hands. During one movie, he handles raw chicken to lure away a pack of crocodiles but fails to wash his hands afterward, leaving him at risk for multiple food-borne illnesses.
Of course, we must address the elephant in the bedroom: Mr. Bond’s numerous, er, encounters with women. One would imagine the biggest risk to those women would be from the various STDs that likely course through Bond’s body, but of the 27% who died shortly after … encountering … him, all involved violence, with disease playing no obvious role. Who knows, maybe he’s clean? Stranger things have happened.
The timing of this article may seem a bit suspicious. Was it a PR stunt by the studio? Rest assured, the authors addressed this, noting that they received no funding for the study, and that, “given the futility of its academic value, this is deemed entirely appropriate by all authors.” We love when a punchline writes itself.
How to see Atlanta on $688.35 a day
The world is always changing, so we have to change with it. This week, LOTME becomes a travel guide, and our first stop is the Big A, the Big Peach, Dogwood City, Empire City of the South, Wakanda.
There’s lots to do in Atlanta: Celebrate a World Series win, visit the College Football Hall of Fame or the World of Coca Cola, or take the Stranger Things/Upside Down film locations tour. Serious adventurers, however, get out of the city and go to Emory Decatur Hospital in – you guessed it – Decatur (unofficial motto: “Everything is Greater in Decatur”).
Find the emergency room and ask for Taylor Davis, who will be your personal guide. She’ll show you how to check in at the desk, sit in the waiting room for 7 hours, and then leave without seeing any medical personnel or receiving any sort of attention whatsoever. All the things she did when she went there in July for a head injury.
Ms. Davis told Fox5 Atlanta: “I didn’t get my vitals taken, nobody called my name. I wasn’t seen at all.”
But wait! There’s more! By booking your trip through LOTMEgo* and using the code “Decatur,” you’ll get the Taylor Davis special, which includes a bill/cover charge for $688.35 from the hospital. An Emory Healthcare patient financial services employee told Ms. Davis that “you get charged before you are seen. Not for being seen.”
If all this has you ready to hop in your car (really?), then check out LOTMEgo* on Twittbook and InstaTok. You’ll also find trick-or-treating tips and discounts on haunted hospital tours.
*Does not actually exist
Breaking down the hot flash
Do you ever wonder why we scramble for cold things when we’re feeling nauseous? Whether it’s the cool air that needs to hit your face in the car or a cold, damp towel on the back of your neck, scientists think it could possibly be an evolutionary mechanism at the cellular level.
Motion sickness it’s actually a battle of body temperature, according to an article from LiveScience. Capillaries in the skin dilate, allowing for more blood flow near the skin’s surface and causing core temperature to fall. Once body temperature drops, the hypothalamus, which regulates temperature, tries to do its job by raising body temperature. Thus the hot flash!
The cold compress and cool air help fight the battle by counteracting the hypothalamus, but why the drop in body temperature to begin with?
There are a few theories. Dr. Robert Glatter, an emergency physician at Lenox Hill Hospital in New York, told LiveScience that the lack of oxygen needed in body tissue to survive at lower temperatures could be making it difficult to get oxygen to the body when a person is ill, and is “more likely an adaptive response influenced by poorly understood mechanisms at the cellular level.”
Another theory is that the nausea and body temperature shift is the body’s natural response to help people vomit.
Then there’s the theory of “defensive hypothermia,” which suggests that cold sweats are a possible mechanism to conserve energy so the body can fight off an intruder, which was supported by a 2014 study and a 2016 review.
It’s another one of the body’s many survival tricks.
Teachers were right: Pupils can do the math
Teachers liked to preach that we wouldn’t have calculators with us all the time, but that wound up not being true. Our phones have calculators at the press of a button. But maybe even calculators aren’t always needed because our pupils do more math than you think.
The pupil light reflex – constrict in light and dilate in darkness – is well known, but recent work shows that pupil size is also regulated by cognitive and perceptual factors. By presenting subjects with images of various numbers of dots and measuring pupil size, the investigators were able to show “that numerical information is intrinsically related to perception,” lead author Dr. Elisa Castaldi of Florence University noted in a written statement.
The researchers found that pupils are responsible for important survival techniques. Coauthor David Burr of the University of Sydney and the University of Florence gave an evolutionary perspective: “When we look around, we spontaneously perceive the form, size, movement and colour of a scene. Equally spontaneously, we perceive the number of items before us. This ability, shared with most other animals, is an evolutionary fundamental: It reveals immediately important quantities, such as how many apples there are on the tree, or how many enemies are attacking.”
Useful information, indeed, but our pupils seem to be more interested in the quantity of beers in the refrigerator.
No, Mr. Bond, I expect you to die
Movie watching usually requires a certain suspension of disbelief, and it’s safe to say James Bond movies require this more than most. Between the impossible gadgets and ludicrous doomsday plans, very few have ever stopped to consider the health risks of the James Bond universe.
Now, however, Bond, James Bond, has met his most formidable opponent: Wouter Graumans, a graduate student in epidemiology from the Netherlands. During a foray to Burkina Faso to study infectious diseases, Mr. Graumans came down with a case of food poisoning, which led him to wonder how 007 is able to trot across this big world of ours without contracting so much as a sinus infection.
Because Mr. Graumans is a man of science and conviction, mere speculation wasn’t enough. He and a group of coauthors wrote an entire paper on the health risks of the James Bond universe.
Doing so required watching over 3,000 minutes of numerous movies and analyzing Bond’s 86 total trips to 46 different countries based on current Centers for Disease Control and Prevention advice for travel to those countries. Time which, the authors state in the abstract, “could easily have been spent on more pressing societal issues or forms of relaxation that are more acceptable in academic circles.”
Naturally, Mr. Bond’s line of work entails exposure to unpleasant things, such as poison, dehydration, heatstroke, and dangerous wildlife (everything from ticks to crocodiles), though oddly enough he never succumbs to any of it. He’s also curiously immune to hangovers, despite rarely drinking anything nonalcoholic. There are also less obvious risks: For one, 007 rarely washes his hands. During one movie, he handles raw chicken to lure away a pack of crocodiles but fails to wash his hands afterward, leaving him at risk for multiple food-borne illnesses.
Of course, we must address the elephant in the bedroom: Mr. Bond’s numerous, er, encounters with women. One would imagine the biggest risk to those women would be from the various STDs that likely course through Bond’s body, but of the 27% who died shortly after … encountering … him, all involved violence, with disease playing no obvious role. Who knows, maybe he’s clean? Stranger things have happened.
The timing of this article may seem a bit suspicious. Was it a PR stunt by the studio? Rest assured, the authors addressed this, noting that they received no funding for the study, and that, “given the futility of its academic value, this is deemed entirely appropriate by all authors.” We love when a punchline writes itself.
How to see Atlanta on $688.35 a day
The world is always changing, so we have to change with it. This week, LOTME becomes a travel guide, and our first stop is the Big A, the Big Peach, Dogwood City, Empire City of the South, Wakanda.
There’s lots to do in Atlanta: Celebrate a World Series win, visit the College Football Hall of Fame or the World of Coca Cola, or take the Stranger Things/Upside Down film locations tour. Serious adventurers, however, get out of the city and go to Emory Decatur Hospital in – you guessed it – Decatur (unofficial motto: “Everything is Greater in Decatur”).
Find the emergency room and ask for Taylor Davis, who will be your personal guide. She’ll show you how to check in at the desk, sit in the waiting room for 7 hours, and then leave without seeing any medical personnel or receiving any sort of attention whatsoever. All the things she did when she went there in July for a head injury.
Ms. Davis told Fox5 Atlanta: “I didn’t get my vitals taken, nobody called my name. I wasn’t seen at all.”
But wait! There’s more! By booking your trip through LOTMEgo* and using the code “Decatur,” you’ll get the Taylor Davis special, which includes a bill/cover charge for $688.35 from the hospital. An Emory Healthcare patient financial services employee told Ms. Davis that “you get charged before you are seen. Not for being seen.”
If all this has you ready to hop in your car (really?), then check out LOTMEgo* on Twittbook and InstaTok. You’ll also find trick-or-treating tips and discounts on haunted hospital tours.
*Does not actually exist
Breaking down the hot flash
Do you ever wonder why we scramble for cold things when we’re feeling nauseous? Whether it’s the cool air that needs to hit your face in the car or a cold, damp towel on the back of your neck, scientists think it could possibly be an evolutionary mechanism at the cellular level.
Motion sickness it’s actually a battle of body temperature, according to an article from LiveScience. Capillaries in the skin dilate, allowing for more blood flow near the skin’s surface and causing core temperature to fall. Once body temperature drops, the hypothalamus, which regulates temperature, tries to do its job by raising body temperature. Thus the hot flash!
The cold compress and cool air help fight the battle by counteracting the hypothalamus, but why the drop in body temperature to begin with?
There are a few theories. Dr. Robert Glatter, an emergency physician at Lenox Hill Hospital in New York, told LiveScience that the lack of oxygen needed in body tissue to survive at lower temperatures could be making it difficult to get oxygen to the body when a person is ill, and is “more likely an adaptive response influenced by poorly understood mechanisms at the cellular level.”
Another theory is that the nausea and body temperature shift is the body’s natural response to help people vomit.
Then there’s the theory of “defensive hypothermia,” which suggests that cold sweats are a possible mechanism to conserve energy so the body can fight off an intruder, which was supported by a 2014 study and a 2016 review.
It’s another one of the body’s many survival tricks.
Teachers were right: Pupils can do the math
Teachers liked to preach that we wouldn’t have calculators with us all the time, but that wound up not being true. Our phones have calculators at the press of a button. But maybe even calculators aren’t always needed because our pupils do more math than you think.
The pupil light reflex – constrict in light and dilate in darkness – is well known, but recent work shows that pupil size is also regulated by cognitive and perceptual factors. By presenting subjects with images of various numbers of dots and measuring pupil size, the investigators were able to show “that numerical information is intrinsically related to perception,” lead author Dr. Elisa Castaldi of Florence University noted in a written statement.
The researchers found that pupils are responsible for important survival techniques. Coauthor David Burr of the University of Sydney and the University of Florence gave an evolutionary perspective: “When we look around, we spontaneously perceive the form, size, movement and colour of a scene. Equally spontaneously, we perceive the number of items before us. This ability, shared with most other animals, is an evolutionary fundamental: It reveals immediately important quantities, such as how many apples there are on the tree, or how many enemies are attacking.”
Useful information, indeed, but our pupils seem to be more interested in the quantity of beers in the refrigerator.