User login
MDedge Daily News: Moving toward a universal flu vaccine
Federal scientists advance a plan for a universal flu vaccine, making health care affordable has broad support, some encouraging news about proton pump inhibitors and myocardial infarction, and how obesity affects cardiovascular risk.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
Listen to the MDedge Daily News podcast for all the details on today’s top news.
Federal scientists advance a plan for a universal flu vaccine, making health care affordable has broad support, some encouraging news about proton pump inhibitors and myocardial infarction, and how obesity affects cardiovascular risk.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
Listen to the MDedge Daily News podcast for all the details on today’s top news.
Federal scientists advance a plan for a universal flu vaccine, making health care affordable has broad support, some encouraging news about proton pump inhibitors and myocardial infarction, and how obesity affects cardiovascular risk.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
Listen to the MDedge Daily News podcast for all the details on today’s top news.

HSCT approach provides ‘excellent’ survival in FA

SALT LAKE CITY—A “risk-adjusted” approach leads to “excellent” survival in patients with Fanconi anemia (FA) undergoing alternative donor hematopoietic stem cell transplant (HSCT), according to a speaker at the 2018 BMT Tandem Meetings.
All FA patients who received personalized doses of busulfan in place of total body irradiation (TBI) were alive and disease-free after undergoing HSCT for bone marrow failure or myelodysplastic syndrome (MDS).
None of the patients developed graft-vs-host disease (GVHD), and the most common toxicity was viral infection.
Parinda A. Mehta, MD, of Cincinnati Children’s Hospital Medical Center in Ohio, presented these results at this year’s BMT Tandem Meetings as abstract 109.*
“We all know that inherent chemotherapy and radiation sensitivity makes transplant for patients with Fanconi anemia quite challenging,” Dr Mehta began. “In our recently published, prospective, multi-institutional study, we showed excellent outcomes of alternative donor transplant in patients with Fanconi anemia without using radiation.”
“In that study,** TBI was replaced by pharmacokinetically adjusted busulfan. It proved that, yes, we can do alternative donor transplant successfully without radiation by showing an overall survival of 80% for a total of 45 patients. We were quite ecstatic to see these numbers.”
The study also showed that younger patients fared better with this regimen, and younger patients did best with the lowest dose of busulfan tested (0.6 mg/kg vs 0.8 to 1.0 mg/kg). In addition, patients who underwent HSCT for bone marrow failure had better outcomes than patients who had MDS.
This led Dr Mehta and her colleagues to hypothesize that adjusting busulfan dosing based on a patient’s age and disease status at HSCT could minimize toxicity and improve outcomes.
Patients
The researchers tested their theory in 22 FA patients. They had a median age of 7 (range, 4-27), and most (n=13) were female.
Twelve patients had pancytopenia, 6 had severe single-lineage cytopenia, 3 had low-grade MDS, and 1 patient had acute myeloid leukemia (AML).
Eighteen patients had a history of transfusions, and 3 had a history of androgen use.
Treatment
The preparative regimen consisted of 4 doses of busulfan (every 12 hours on day -7 to -6), followed by cyclophosphamide at 10 mg/kg/day (on day -5 to -2), fludarabine at 35 mg/m2/day (on day -5 to -2), and rabbit antithymocyte globulin at 2.5 mg/kg/day (on day -5 to -2).
Busulfan doses were adjusted according to age and disease status.
Children (age 18 and younger) with bone marrow failure received busulfan at 0.6 to 0.8 mg/kg. Children with MDS/AML received busulfan at 0.8 to 1.0 mg/kg. Adults (19 and older) received the lowest dose of busulfan—0.4 mg/kg—regardless of disease status.
“At the first sight, this will look counterintuitive . . . ,” Dr Mehta said. “However, based on our previous experience, in general and also from results of our previous study, this was specifically designed to avoid upfront TRM [transplant-related mortality] for these adult patients.”
All 22 patients received CD34-selected, T-cell-depleted peripheral blood stem cells from unrelated donors. Eleven patients received a fully matched graft (10/10), 8 patients had a 9/10 match, and 3 had an 8/10 match.
The median number of CD34+ cells/kg was 23.9 x 106 (range, 4.9-76.6), and the median number of CD3 cells/kg was 1 x 104 (range, 0.003-3.1).
T-cell depletion was the only GVHD prophylaxis used.
Patients with MDS/AML could receive azacitidine at day 42 after HSCT, an option intended to prevent relapse in these patients.
Toxicity
There were no cases of acute or chronic GVHD.
Toxicities included infections (n=24), oral mucositis (n=14), hyperbilirubinemia (n=2), pulmonary hemorrhage (n=1), and sinusoidal obstruction syndrome (n=1).
There were 20 viral infections, 4 bacterial infections, and no fungal infections. Viral infections included BK virus (n=7), cytomegalovirus (n=6), Epstein-Barr virus (n=6), and adenovirus (n=1).
Dr Mehta noted that viral infections are “not unexpected in a T-cell-depleted graft setting.”
“Because we know this complication [can occur], and we worry about our patients, one of the things that, in recent years, we have done is, we manufacture viral-specific CTLs [cytotoxic T lymphocytes] for all of these patients ahead of time whenever possible,” she said.
“To give you an example, 19 out of these 20 patients’ viral infections—or rather, viremias—are completely under control with the use of either antivirals or donor-specific CTLs, including a third-party CTL in one of the patients.”
Response and survival
All 22 patients engrafted. The median time to neutrophil engraftment was 9 days (range, 8-10), and the median time to platelet engraftment was 16 days (range, 11-40).
Twenty-one of the 22 patients (95%) were alive and disease-free at last follow-up. The median follow-up was 21 months (range, 6-44).
The single AML patient achieved remission but died of post-transplant lymphoproliferative disorder (PTLD) on day 202 after HSCT. Dr Mehta said this was due to partial loss of follow-up and noncompliance with medical recommendations during PTLD treatment.
The AML patient also had “significant upfront toxicity” but “recovered very nicely,” according to Dr Mehta. He had severe mucositis, herpetic stomatitis, and sinusoidal obstruction syndrome that responded to defibrotide.
“Overall, we are quite excited to see 95% overall survival for this cohort and conclude that the current risk-adjusted approach leads to excellent overall survival and disease-free survival in patients undergoing alternative donor transplant either for marrow failure or MDS/AML,” Dr Mehta said.
“Enrollment is ongoing, and we hope to see continued success in patients with MDS/AML as well as in adult patients.”
*Data in the abstract differ from the presentation.
**Mehta PA et al. Radiation-free, alternative donor HCT for Fanconi anemia patients: results from a prospective multi-institutional study. Blood 2017; doi: https://doi.org/10.1182/blood-2016-09-743112.
SALT LAKE CITY—A “risk-adjusted” approach leads to “excellent” survival in patients with Fanconi anemia (FA) undergoing alternative donor hematopoietic stem cell transplant (HSCT), according to a speaker at the 2018 BMT Tandem Meetings.
All FA patients who received personalized doses of busulfan in place of total body irradiation (TBI) were alive and disease-free after undergoing HSCT for bone marrow failure or myelodysplastic syndrome (MDS).
None of the patients developed graft-vs-host disease (GVHD), and the most common toxicity was viral infection.
Parinda A. Mehta, MD, of Cincinnati Children’s Hospital Medical Center in Ohio, presented these results at this year’s BMT Tandem Meetings as abstract 109.*
“We all know that inherent chemotherapy and radiation sensitivity makes transplant for patients with Fanconi anemia quite challenging,” Dr Mehta began. “In our recently published, prospective, multi-institutional study, we showed excellent outcomes of alternative donor transplant in patients with Fanconi anemia without using radiation.”
“In that study,** TBI was replaced by pharmacokinetically adjusted busulfan. It proved that, yes, we can do alternative donor transplant successfully without radiation by showing an overall survival of 80% for a total of 45 patients. We were quite ecstatic to see these numbers.”
The study also showed that younger patients fared better with this regimen, and younger patients did best with the lowest dose of busulfan tested (0.6 mg/kg vs 0.8 to 1.0 mg/kg). In addition, patients who underwent HSCT for bone marrow failure had better outcomes than patients who had MDS.
This led Dr Mehta and her colleagues to hypothesize that adjusting busulfan dosing based on a patient’s age and disease status at HSCT could minimize toxicity and improve outcomes.
Patients
The researchers tested their theory in 22 FA patients. They had a median age of 7 (range, 4-27), and most (n=13) were female.
Twelve patients had pancytopenia, 6 had severe single-lineage cytopenia, 3 had low-grade MDS, and 1 patient had acute myeloid leukemia (AML).
Eighteen patients had a history of transfusions, and 3 had a history of androgen use.
Treatment
The preparative regimen consisted of 4 doses of busulfan (every 12 hours on day -7 to -6), followed by cyclophosphamide at 10 mg/kg/day (on day -5 to -2), fludarabine at 35 mg/m2/day (on day -5 to -2), and rabbit antithymocyte globulin at 2.5 mg/kg/day (on day -5 to -2).
Busulfan doses were adjusted according to age and disease status.
Children (age 18 and younger) with bone marrow failure received busulfan at 0.6 to 0.8 mg/kg. Children with MDS/AML received busulfan at 0.8 to 1.0 mg/kg. Adults (19 and older) received the lowest dose of busulfan—0.4 mg/kg—regardless of disease status.
“At the first sight, this will look counterintuitive . . . ,” Dr Mehta said. “However, based on our previous experience, in general and also from results of our previous study, this was specifically designed to avoid upfront TRM [transplant-related mortality] for these adult patients.”
All 22 patients received CD34-selected, T-cell-depleted peripheral blood stem cells from unrelated donors. Eleven patients received a fully matched graft (10/10), 8 patients had a 9/10 match, and 3 had an 8/10 match.
The median number of CD34+ cells/kg was 23.9 x 106 (range, 4.9-76.6), and the median number of CD3 cells/kg was 1 x 104 (range, 0.003-3.1).
T-cell depletion was the only GVHD prophylaxis used.
Patients with MDS/AML could receive azacitidine at day 42 after HSCT, an option intended to prevent relapse in these patients.
Toxicity
There were no cases of acute or chronic GVHD.
Toxicities included infections (n=24), oral mucositis (n=14), hyperbilirubinemia (n=2), pulmonary hemorrhage (n=1), and sinusoidal obstruction syndrome (n=1).
There were 20 viral infections, 4 bacterial infections, and no fungal infections. Viral infections included BK virus (n=7), cytomegalovirus (n=6), Epstein-Barr virus (n=6), and adenovirus (n=1).
Dr Mehta noted that viral infections are “not unexpected in a T-cell-depleted graft setting.”
“Because we know this complication [can occur], and we worry about our patients, one of the things that, in recent years, we have done is, we manufacture viral-specific CTLs [cytotoxic T lymphocytes] for all of these patients ahead of time whenever possible,” she said.
“To give you an example, 19 out of these 20 patients’ viral infections—or rather, viremias—are completely under control with the use of either antivirals or donor-specific CTLs, including a third-party CTL in one of the patients.”
Response and survival
All 22 patients engrafted. The median time to neutrophil engraftment was 9 days (range, 8-10), and the median time to platelet engraftment was 16 days (range, 11-40).
Twenty-one of the 22 patients (95%) were alive and disease-free at last follow-up. The median follow-up was 21 months (range, 6-44).
The single AML patient achieved remission but died of post-transplant lymphoproliferative disorder (PTLD) on day 202 after HSCT. Dr Mehta said this was due to partial loss of follow-up and noncompliance with medical recommendations during PTLD treatment.
The AML patient also had “significant upfront toxicity” but “recovered very nicely,” according to Dr Mehta. He had severe mucositis, herpetic stomatitis, and sinusoidal obstruction syndrome that responded to defibrotide.
“Overall, we are quite excited to see 95% overall survival for this cohort and conclude that the current risk-adjusted approach leads to excellent overall survival and disease-free survival in patients undergoing alternative donor transplant either for marrow failure or MDS/AML,” Dr Mehta said.
“Enrollment is ongoing, and we hope to see continued success in patients with MDS/AML as well as in adult patients.”
*Data in the abstract differ from the presentation.
**Mehta PA et al. Radiation-free, alternative donor HCT for Fanconi anemia patients: results from a prospective multi-institutional study. Blood 2017; doi: https://doi.org/10.1182/blood-2016-09-743112.
SALT LAKE CITY—A “risk-adjusted” approach leads to “excellent” survival in patients with Fanconi anemia (FA) undergoing alternative donor hematopoietic stem cell transplant (HSCT), according to a speaker at the 2018 BMT Tandem Meetings.
All FA patients who received personalized doses of busulfan in place of total body irradiation (TBI) were alive and disease-free after undergoing HSCT for bone marrow failure or myelodysplastic syndrome (MDS).
None of the patients developed graft-vs-host disease (GVHD), and the most common toxicity was viral infection.
Parinda A. Mehta, MD, of Cincinnati Children’s Hospital Medical Center in Ohio, presented these results at this year’s BMT Tandem Meetings as abstract 109.*
“We all know that inherent chemotherapy and radiation sensitivity makes transplant for patients with Fanconi anemia quite challenging,” Dr Mehta began. “In our recently published, prospective, multi-institutional study, we showed excellent outcomes of alternative donor transplant in patients with Fanconi anemia without using radiation.”
“In that study,** TBI was replaced by pharmacokinetically adjusted busulfan. It proved that, yes, we can do alternative donor transplant successfully without radiation by showing an overall survival of 80% for a total of 45 patients. We were quite ecstatic to see these numbers.”
The study also showed that younger patients fared better with this regimen, and younger patients did best with the lowest dose of busulfan tested (0.6 mg/kg vs 0.8 to 1.0 mg/kg). In addition, patients who underwent HSCT for bone marrow failure had better outcomes than patients who had MDS.
This led Dr Mehta and her colleagues to hypothesize that adjusting busulfan dosing based on a patient’s age and disease status at HSCT could minimize toxicity and improve outcomes.
Patients
The researchers tested their theory in 22 FA patients. They had a median age of 7 (range, 4-27), and most (n=13) were female.
Twelve patients had pancytopenia, 6 had severe single-lineage cytopenia, 3 had low-grade MDS, and 1 patient had acute myeloid leukemia (AML).
Eighteen patients had a history of transfusions, and 3 had a history of androgen use.
Treatment
The preparative regimen consisted of 4 doses of busulfan (every 12 hours on day -7 to -6), followed by cyclophosphamide at 10 mg/kg/day (on day -5 to -2), fludarabine at 35 mg/m2/day (on day -5 to -2), and rabbit antithymocyte globulin at 2.5 mg/kg/day (on day -5 to -2).
Busulfan doses were adjusted according to age and disease status.
Children (age 18 and younger) with bone marrow failure received busulfan at 0.6 to 0.8 mg/kg. Children with MDS/AML received busulfan at 0.8 to 1.0 mg/kg. Adults (19 and older) received the lowest dose of busulfan—0.4 mg/kg—regardless of disease status.
“At the first sight, this will look counterintuitive . . . ,” Dr Mehta said. “However, based on our previous experience, in general and also from results of our previous study, this was specifically designed to avoid upfront TRM [transplant-related mortality] for these adult patients.”
All 22 patients received CD34-selected, T-cell-depleted peripheral blood stem cells from unrelated donors. Eleven patients received a fully matched graft (10/10), 8 patients had a 9/10 match, and 3 had an 8/10 match.
The median number of CD34+ cells/kg was 23.9 x 106 (range, 4.9-76.6), and the median number of CD3 cells/kg was 1 x 104 (range, 0.003-3.1).
T-cell depletion was the only GVHD prophylaxis used.
Patients with MDS/AML could receive azacitidine at day 42 after HSCT, an option intended to prevent relapse in these patients.
Toxicity
There were no cases of acute or chronic GVHD.
Toxicities included infections (n=24), oral mucositis (n=14), hyperbilirubinemia (n=2), pulmonary hemorrhage (n=1), and sinusoidal obstruction syndrome (n=1).
There were 20 viral infections, 4 bacterial infections, and no fungal infections. Viral infections included BK virus (n=7), cytomegalovirus (n=6), Epstein-Barr virus (n=6), and adenovirus (n=1).
Dr Mehta noted that viral infections are “not unexpected in a T-cell-depleted graft setting.”
“Because we know this complication [can occur], and we worry about our patients, one of the things that, in recent years, we have done is, we manufacture viral-specific CTLs [cytotoxic T lymphocytes] for all of these patients ahead of time whenever possible,” she said.
“To give you an example, 19 out of these 20 patients’ viral infections—or rather, viremias—are completely under control with the use of either antivirals or donor-specific CTLs, including a third-party CTL in one of the patients.”
Response and survival
All 22 patients engrafted. The median time to neutrophil engraftment was 9 days (range, 8-10), and the median time to platelet engraftment was 16 days (range, 11-40).
Twenty-one of the 22 patients (95%) were alive and disease-free at last follow-up. The median follow-up was 21 months (range, 6-44).
The single AML patient achieved remission but died of post-transplant lymphoproliferative disorder (PTLD) on day 202 after HSCT. Dr Mehta said this was due to partial loss of follow-up and noncompliance with medical recommendations during PTLD treatment.
The AML patient also had “significant upfront toxicity” but “recovered very nicely,” according to Dr Mehta. He had severe mucositis, herpetic stomatitis, and sinusoidal obstruction syndrome that responded to defibrotide.
“Overall, we are quite excited to see 95% overall survival for this cohort and conclude that the current risk-adjusted approach leads to excellent overall survival and disease-free survival in patients undergoing alternative donor transplant either for marrow failure or MDS/AML,” Dr Mehta said.
“Enrollment is ongoing, and we hope to see continued success in patients with MDS/AML as well as in adult patients.”
*Data in the abstract differ from the presentation.
**Mehta PA et al. Radiation-free, alternative donor HCT for Fanconi anemia patients: results from a prospective multi-institutional study. Blood 2017; doi: https://doi.org/10.1182/blood-2016-09-743112.
EC approves emicizumab for hemophilia A with inhibitors

The European Commission (EC) has granted marketing authorization for emicizumab (Hemlibra®), a bispecific factor IXa- and factor X-directed antibody.
This means emicizumab is approved for use in the European Union for routine prophylaxis of bleeding episodes in patients of all ages who have hemophilia A and factor VIII inhibitors.
The recommended dose of emicizumab is 3 mg/kg once a week for the first 4 weeks, followed by 1.5 mg/kg once a week.
Emicizumab is designed to bring together factor IXa and factor X, proteins required to activate the natural coagulation cascade and restore the blood clotting process for patients with hemophilia A.
Emicizumab was created by Chugai Pharmaceutical Co., Ltd. and is being co-developed by Chugai, Roche, and Genentech.
The EC’s decision to authorize marketing of emicizumab is based on results from a pair of phase 3 studies—HAVEN 1 and HAVEN 2.
Results from HAVEN 1 were published in NEJM and presented at the 26th ISTH Congress in July 2017. Updated results from HAVEN 2 were presented at the 2017 ASH Annual Meeting in December.
HAVEN 1
This study enrolled 109 patients (age 12 and older) with hemophilia A and factor VIII inhibitors who were previously treated with bypassing agents (BPAs) on-demand or as prophylaxis.
The patients were randomized to receive emicizumab prophylaxis or no prophylaxis. On-demand treatment of breakthrough bleeds with BPAs was allowed.
There was a significant reduction in treated bleeds of 87% with emicizumab prophylaxis compared to no prophylaxis (95% CI: 72.3; 94.3, P<0.0001). And there was an 80% reduction in all bleeds with emicizumab (95% CI: 62.5; 89.8, P<0.0001).
Adverse events (AEs) occurring in at least 5% of patients treated with emicizumab were local injection site reactions, headache, fatigue, upper respiratory tract infection, and arthralgia.
Two patients experienced thromboembolic events (TEs), and 3 had thrombotic microangiopathy (TMA) while receiving emicizumab prophylaxis and more than 100 u/kg/day of activated prothrombin complex concentrate, on average, for 24 hours or more before the event. Two of these patients had also received recombinant factor VIIa.
Neither TE required anticoagulation therapy, and 1 patient restarted emicizumab. The cases of TMA observed were transient, and 1 patient restarted emicizumab.
HAVEN 2
In this single-arm trial, researchers evaluated emicizumab prophylaxis in 60 patients, ages 1 to 17, who had hemophilia A with factor VIII inhibitors.
The efficacy analysis included 57 patients who were younger than 12. The 3 older patients were only included in the safety analysis.
Of the 57 patients, 64.9% had 0 bleeds, 94.7% had 0 treated bleeds, and 98.2% had 0 treated spontaneous bleeds and 0 treated joint bleeds. None of the patients had treated target joint bleeds.
Forty patients had a total of 201 AEs. The most common of these were viral upper respiratory tract infections (16.7%) and injection site reactions (16.7%).
There were no TEs or TMA events, and none of the patients tested positive for anti-drug antibodies. None of the 7 serious AEs in this trial were considered treatment-related.
The European Commission (EC) has granted marketing authorization for emicizumab (Hemlibra®), a bispecific factor IXa- and factor X-directed antibody.
This means emicizumab is approved for use in the European Union for routine prophylaxis of bleeding episodes in patients of all ages who have hemophilia A and factor VIII inhibitors.
The recommended dose of emicizumab is 3 mg/kg once a week for the first 4 weeks, followed by 1.5 mg/kg once a week.
Emicizumab is designed to bring together factor IXa and factor X, proteins required to activate the natural coagulation cascade and restore the blood clotting process for patients with hemophilia A.
Emicizumab was created by Chugai Pharmaceutical Co., Ltd. and is being co-developed by Chugai, Roche, and Genentech.
The EC’s decision to authorize marketing of emicizumab is based on results from a pair of phase 3 studies—HAVEN 1 and HAVEN 2.
Results from HAVEN 1 were published in NEJM and presented at the 26th ISTH Congress in July 2017. Updated results from HAVEN 2 were presented at the 2017 ASH Annual Meeting in December.
HAVEN 1
This study enrolled 109 patients (age 12 and older) with hemophilia A and factor VIII inhibitors who were previously treated with bypassing agents (BPAs) on-demand or as prophylaxis.
The patients were randomized to receive emicizumab prophylaxis or no prophylaxis. On-demand treatment of breakthrough bleeds with BPAs was allowed.
There was a significant reduction in treated bleeds of 87% with emicizumab prophylaxis compared to no prophylaxis (95% CI: 72.3; 94.3, P<0.0001). And there was an 80% reduction in all bleeds with emicizumab (95% CI: 62.5; 89.8, P<0.0001).
Adverse events (AEs) occurring in at least 5% of patients treated with emicizumab were local injection site reactions, headache, fatigue, upper respiratory tract infection, and arthralgia.
Two patients experienced thromboembolic events (TEs), and 3 had thrombotic microangiopathy (TMA) while receiving emicizumab prophylaxis and more than 100 u/kg/day of activated prothrombin complex concentrate, on average, for 24 hours or more before the event. Two of these patients had also received recombinant factor VIIa.
Neither TE required anticoagulation therapy, and 1 patient restarted emicizumab. The cases of TMA observed were transient, and 1 patient restarted emicizumab.
HAVEN 2
In this single-arm trial, researchers evaluated emicizumab prophylaxis in 60 patients, ages 1 to 17, who had hemophilia A with factor VIII inhibitors.
The efficacy analysis included 57 patients who were younger than 12. The 3 older patients were only included in the safety analysis.
Of the 57 patients, 64.9% had 0 bleeds, 94.7% had 0 treated bleeds, and 98.2% had 0 treated spontaneous bleeds and 0 treated joint bleeds. None of the patients had treated target joint bleeds.
Forty patients had a total of 201 AEs. The most common of these were viral upper respiratory tract infections (16.7%) and injection site reactions (16.7%).
There were no TEs or TMA events, and none of the patients tested positive for anti-drug antibodies. None of the 7 serious AEs in this trial were considered treatment-related.
The European Commission (EC) has granted marketing authorization for emicizumab (Hemlibra®), a bispecific factor IXa- and factor X-directed antibody.
This means emicizumab is approved for use in the European Union for routine prophylaxis of bleeding episodes in patients of all ages who have hemophilia A and factor VIII inhibitors.
The recommended dose of emicizumab is 3 mg/kg once a week for the first 4 weeks, followed by 1.5 mg/kg once a week.
Emicizumab is designed to bring together factor IXa and factor X, proteins required to activate the natural coagulation cascade and restore the blood clotting process for patients with hemophilia A.
Emicizumab was created by Chugai Pharmaceutical Co., Ltd. and is being co-developed by Chugai, Roche, and Genentech.
The EC’s decision to authorize marketing of emicizumab is based on results from a pair of phase 3 studies—HAVEN 1 and HAVEN 2.
Results from HAVEN 1 were published in NEJM and presented at the 26th ISTH Congress in July 2017. Updated results from HAVEN 2 were presented at the 2017 ASH Annual Meeting in December.
HAVEN 1
This study enrolled 109 patients (age 12 and older) with hemophilia A and factor VIII inhibitors who were previously treated with bypassing agents (BPAs) on-demand or as prophylaxis.
The patients were randomized to receive emicizumab prophylaxis or no prophylaxis. On-demand treatment of breakthrough bleeds with BPAs was allowed.
There was a significant reduction in treated bleeds of 87% with emicizumab prophylaxis compared to no prophylaxis (95% CI: 72.3; 94.3, P<0.0001). And there was an 80% reduction in all bleeds with emicizumab (95% CI: 62.5; 89.8, P<0.0001).
Adverse events (AEs) occurring in at least 5% of patients treated with emicizumab were local injection site reactions, headache, fatigue, upper respiratory tract infection, and arthralgia.
Two patients experienced thromboembolic events (TEs), and 3 had thrombotic microangiopathy (TMA) while receiving emicizumab prophylaxis and more than 100 u/kg/day of activated prothrombin complex concentrate, on average, for 24 hours or more before the event. Two of these patients had also received recombinant factor VIIa.
Neither TE required anticoagulation therapy, and 1 patient restarted emicizumab. The cases of TMA observed were transient, and 1 patient restarted emicizumab.
HAVEN 2
In this single-arm trial, researchers evaluated emicizumab prophylaxis in 60 patients, ages 1 to 17, who had hemophilia A with factor VIII inhibitors.
The efficacy analysis included 57 patients who were younger than 12. The 3 older patients were only included in the safety analysis.
Of the 57 patients, 64.9% had 0 bleeds, 94.7% had 0 treated bleeds, and 98.2% had 0 treated spontaneous bleeds and 0 treated joint bleeds. None of the patients had treated target joint bleeds.
Forty patients had a total of 201 AEs. The most common of these were viral upper respiratory tract infections (16.7%) and injection site reactions (16.7%).
There were no TEs or TMA events, and none of the patients tested positive for anti-drug antibodies. None of the 7 serious AEs in this trial were considered treatment-related.
Use of RBC, plasma transfusions may be declining in US
A study of US hospital discharges revealed a decrease in the use of red blood cell (RBC) and plasma transfusions—but not platelet transfusions—in recent years.
Researchers examined a sample of US hospital inpatient discharges from 1993 to 2014.
Overall, platelet, plasma, and RBC transfusions increased from 1993 to 2010.
However, from 2011 to 2014, there was a significant decrease in both plasma and RBC transfusions. Platelet transfusions remained stable over that period.
Aaron A. R. Tobian, MD, PhD, of Johns Hopkins University in Baltimore, Maryland, and his colleagues reported these findings in a letter to JAMA.
The researchers analyzed data from the National Inpatient Sample, which uses a stratified probability sample of 20% of all inpatient discharges in the US.
The team looked at transfusion trends from 1993 to 2014 but focused on trends from 2011 to 2014 because there was an inflection point in RBC transfusion in 2011. They used multivariable Poisson regression to estimate adjusted risk ratios (aRRs) comparing the risk of transfusion in 2011 and 2014.
The researchers found that RBC transfusions decreased from 6.8% in 2011 to 5.7% in 2014 (aRR=0.83; 95% confidence interval [CI], 0.78-0.88; P<0.001).
Plasma transfusions decreased from 1.0% to 0.87% (aRR=0.87; 95% CI, 0.80-0.95; P=0.003). And platelet transfusions remained stable (aRR=0.99; 95% CI, 0.89-1.10; P=0.93).
The researchers also conducted subgroup analyses to explore trends in RBC transfusion. They found that, from 2011 to 2014, there were significant reductions in RBC transfusions regardless of patients’ sex, race/ethnicity, risk severity, payer type, and admission type.
However, there was no significant reduction in RBC transfusions among patients younger than 18 years of age or in private investor–owned hospitals.
The researchers said the diagnostic coding used for this study was carried out primarily for billing purposes, and it was not possible to verify its accuracy. They also noted that the study only covered inpatient transfusions, so the results may not be generalizable to outpatient transfusions.
A study of US hospital discharges revealed a decrease in the use of red blood cell (RBC) and plasma transfusions—but not platelet transfusions—in recent years.
Researchers examined a sample of US hospital inpatient discharges from 1993 to 2014.
Overall, platelet, plasma, and RBC transfusions increased from 1993 to 2010.
However, from 2011 to 2014, there was a significant decrease in both plasma and RBC transfusions. Platelet transfusions remained stable over that period.
Aaron A. R. Tobian, MD, PhD, of Johns Hopkins University in Baltimore, Maryland, and his colleagues reported these findings in a letter to JAMA.
The researchers analyzed data from the National Inpatient Sample, which uses a stratified probability sample of 20% of all inpatient discharges in the US.
The team looked at transfusion trends from 1993 to 2014 but focused on trends from 2011 to 2014 because there was an inflection point in RBC transfusion in 2011. They used multivariable Poisson regression to estimate adjusted risk ratios (aRRs) comparing the risk of transfusion in 2011 and 2014.
The researchers found that RBC transfusions decreased from 6.8% in 2011 to 5.7% in 2014 (aRR=0.83; 95% confidence interval [CI], 0.78-0.88; P<0.001).
Plasma transfusions decreased from 1.0% to 0.87% (aRR=0.87; 95% CI, 0.80-0.95; P=0.003). And platelet transfusions remained stable (aRR=0.99; 95% CI, 0.89-1.10; P=0.93).
The researchers also conducted subgroup analyses to explore trends in RBC transfusion. They found that, from 2011 to 2014, there were significant reductions in RBC transfusions regardless of patients’ sex, race/ethnicity, risk severity, payer type, and admission type.
However, there was no significant reduction in RBC transfusions among patients younger than 18 years of age or in private investor–owned hospitals.
The researchers said the diagnostic coding used for this study was carried out primarily for billing purposes, and it was not possible to verify its accuracy. They also noted that the study only covered inpatient transfusions, so the results may not be generalizable to outpatient transfusions.
A study of US hospital discharges revealed a decrease in the use of red blood cell (RBC) and plasma transfusions—but not platelet transfusions—in recent years.
Researchers examined a sample of US hospital inpatient discharges from 1993 to 2014.
Overall, platelet, plasma, and RBC transfusions increased from 1993 to 2010.
However, from 2011 to 2014, there was a significant decrease in both plasma and RBC transfusions. Platelet transfusions remained stable over that period.
Aaron A. R. Tobian, MD, PhD, of Johns Hopkins University in Baltimore, Maryland, and his colleagues reported these findings in a letter to JAMA.
The researchers analyzed data from the National Inpatient Sample, which uses a stratified probability sample of 20% of all inpatient discharges in the US.
The team looked at transfusion trends from 1993 to 2014 but focused on trends from 2011 to 2014 because there was an inflection point in RBC transfusion in 2011. They used multivariable Poisson regression to estimate adjusted risk ratios (aRRs) comparing the risk of transfusion in 2011 and 2014.
The researchers found that RBC transfusions decreased from 6.8% in 2011 to 5.7% in 2014 (aRR=0.83; 95% confidence interval [CI], 0.78-0.88; P<0.001).
Plasma transfusions decreased from 1.0% to 0.87% (aRR=0.87; 95% CI, 0.80-0.95; P=0.003). And platelet transfusions remained stable (aRR=0.99; 95% CI, 0.89-1.10; P=0.93).
The researchers also conducted subgroup analyses to explore trends in RBC transfusion. They found that, from 2011 to 2014, there were significant reductions in RBC transfusions regardless of patients’ sex, race/ethnicity, risk severity, payer type, and admission type.
However, there was no significant reduction in RBC transfusions among patients younger than 18 years of age or in private investor–owned hospitals.
The researchers said the diagnostic coding used for this study was carried out primarily for billing purposes, and it was not possible to verify its accuracy. They also noted that the study only covered inpatient transfusions, so the results may not be generalizable to outpatient transfusions.
Abstract: Please don't give hormone therapy a D recommendation or state that "harms far outweigh benefits for prevention of chronic disease"
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
Pinkerton, J. V., et al, Menopause 24(10):1099, October 2017
The authors, writing for the North American Menopause Society (NAMS), protest the recent guideline recommendations regarding postmenopausal hormone therapy (HT) from the US Preventive Services Task Force (USPSTF). The USPSTF draft guidance gives HT (both combined regimens and estrogen alone) a D recommendation for preventing chronic disease because of harms outweighing benefits. The NAMS panel contends that the USPSTF opinion does not distinguish between approved and unapproved indications for HT. They note that the conclusions are based on the Women’s Health Initiative (WHI) trial, from which certain patient populations who may benefit (bothersome hot flashes, high osteoporosis risk, genitourinary symptoms) were omitted. The WHI tested a single dose of a single formulation in women having an average age of 63 who were 13 years past menopause. According to NAMS, the USPSTF should acknowledge that HT relieves vasomotor symptoms, helps prevent bone loss and fracture, and may improve quality of life in women younger than 60 years who take HT within ten years of menopause. Women younger than 60 years who have had a hysterectomy benefit from early estrogen monotherapy, having a lower incidence of breast cancer, cardiovascular disease and mortality. Further, the panel feels that the USPSTF should state that the findings of harm were based on higher-dose formulations, and that the results may not apply to lower doses, different and newer formulations (e.g., natural progesterone, bazedoxifene), and different dosing routes (e.g., transdermal administration). NAMS agrees that HT does not prevent heart disease, but indicates that its use (and insurance coverage) should not be precluded for women with early menopause, bothersome hot flashes, genitourinary symptoms and/or a high risk of fracture. 12 references
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
Pinkerton, J. V., et al, Menopause 24(10):1099, October 2017
The authors, writing for the North American Menopause Society (NAMS), protest the recent guideline recommendations regarding postmenopausal hormone therapy (HT) from the US Preventive Services Task Force (USPSTF). The USPSTF draft guidance gives HT (both combined regimens and estrogen alone) a D recommendation for preventing chronic disease because of harms outweighing benefits. The NAMS panel contends that the USPSTF opinion does not distinguish between approved and unapproved indications for HT. They note that the conclusions are based on the Women’s Health Initiative (WHI) trial, from which certain patient populations who may benefit (bothersome hot flashes, high osteoporosis risk, genitourinary symptoms) were omitted. The WHI tested a single dose of a single formulation in women having an average age of 63 who were 13 years past menopause. According to NAMS, the USPSTF should acknowledge that HT relieves vasomotor symptoms, helps prevent bone loss and fracture, and may improve quality of life in women younger than 60 years who take HT within ten years of menopause. Women younger than 60 years who have had a hysterectomy benefit from early estrogen monotherapy, having a lower incidence of breast cancer, cardiovascular disease and mortality. Further, the panel feels that the USPSTF should state that the findings of harm were based on higher-dose formulations, and that the results may not apply to lower doses, different and newer formulations (e.g., natural progesterone, bazedoxifene), and different dosing routes (e.g., transdermal administration). NAMS agrees that HT does not prevent heart disease, but indicates that its use (and insurance coverage) should not be precluded for women with early menopause, bothersome hot flashes, genitourinary symptoms and/or a high risk of fracture. 12 references
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
Pinkerton, J. V., et al, Menopause 24(10):1099, October 2017
The authors, writing for the North American Menopause Society (NAMS), protest the recent guideline recommendations regarding postmenopausal hormone therapy (HT) from the US Preventive Services Task Force (USPSTF). The USPSTF draft guidance gives HT (both combined regimens and estrogen alone) a D recommendation for preventing chronic disease because of harms outweighing benefits. The NAMS panel contends that the USPSTF opinion does not distinguish between approved and unapproved indications for HT. They note that the conclusions are based on the Women’s Health Initiative (WHI) trial, from which certain patient populations who may benefit (bothersome hot flashes, high osteoporosis risk, genitourinary symptoms) were omitted. The WHI tested a single dose of a single formulation in women having an average age of 63 who were 13 years past menopause. According to NAMS, the USPSTF should acknowledge that HT relieves vasomotor symptoms, helps prevent bone loss and fracture, and may improve quality of life in women younger than 60 years who take HT within ten years of menopause. Women younger than 60 years who have had a hysterectomy benefit from early estrogen monotherapy, having a lower incidence of breast cancer, cardiovascular disease and mortality. Further, the panel feels that the USPSTF should state that the findings of harm were based on higher-dose formulations, and that the results may not apply to lower doses, different and newer formulations (e.g., natural progesterone, bazedoxifene), and different dosing routes (e.g., transdermal administration). NAMS agrees that HT does not prevent heart disease, but indicates that its use (and insurance coverage) should not be precluded for women with early menopause, bothersome hot flashes, genitourinary symptoms and/or a high risk of fracture. 12 references
Learn more about the Primary Care Medical Abstracts and podcasts, for which you can earn up to 9 CME credits per month.
Copyright © The Center for Medical Education

Woman, 57, With Painful, Swollen Ankle
IN THIS ARTICLE
- Diagnosis
- Treatment
- Care outcome
A 57-year-old horticulturist is working on a ladder leaned up against a tree trunk when the ladder slips, causing her to fall six feet onto concrete. Her right foot and ankle sustain the force of the fall; she is in excruciating pain and unable to bear weight on the foot. She is immediately transported to a local emergency department for evaluation.
Physical exam reveals a tearful middle-aged female in moderate distress and acute pain. There is moderate swelling of the right medial and lateral malleolus, as well as the midfoot, with blue and purple discoloration on the medial and lateral malleolus. Radiographs of the right ankle identify nondisplaced fractures of the distal fibula and tibia. Foot x-rays are unremarkable. A splint is ordered. The patient is given crutches (non-weight-bearing status), pain medication, and a referral to orthopedics.
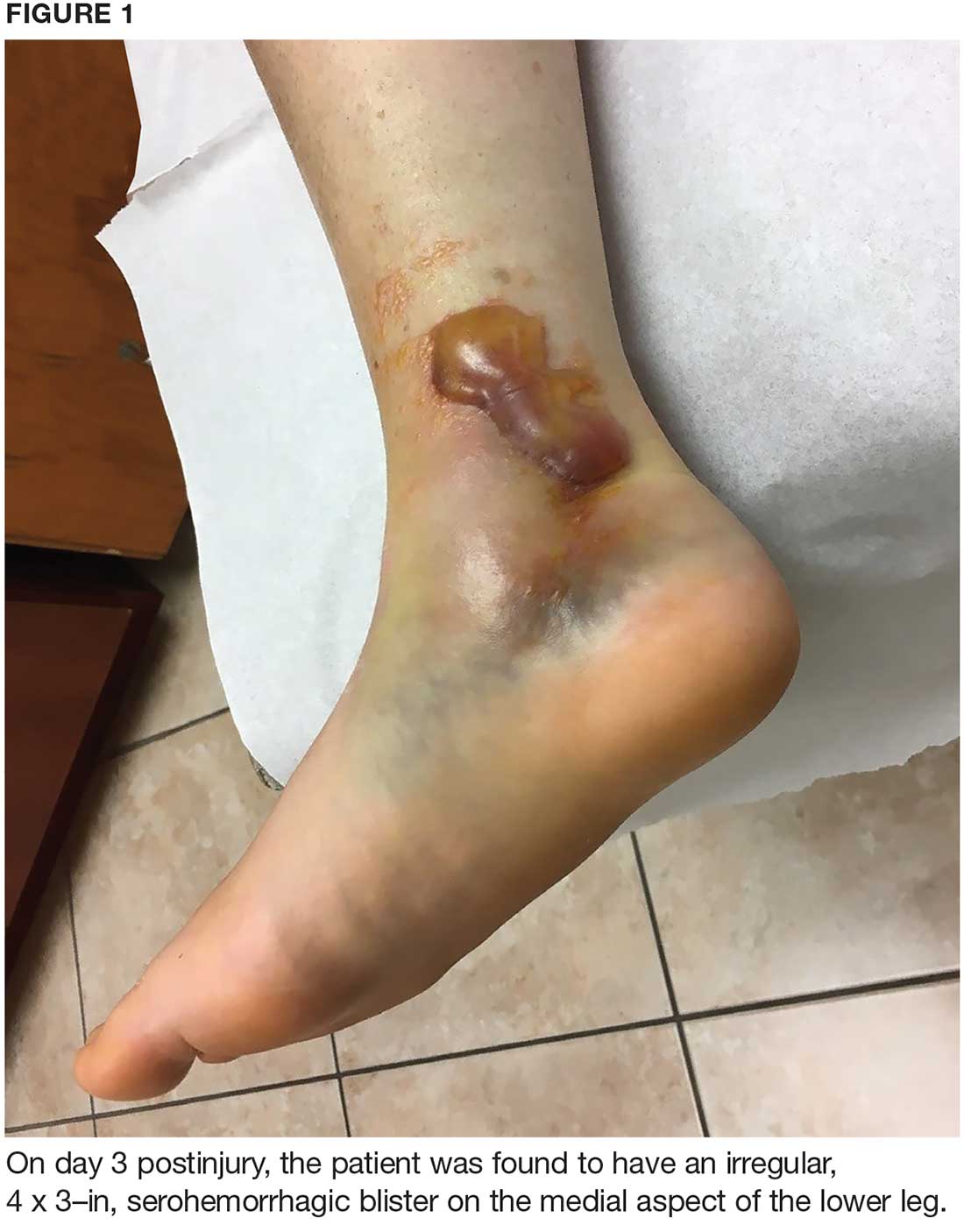
On day 3, the patient presents to orthopedics, where the splint is removed. An irregular, 4 × 3–in (at largest diameter), serohemorrhagic blister is discovered on the medial aspect of the lower leg, above the right malleolus (see Figure 1). Multiple 1- to 3-mm vesicles surround much of the anterior border. Moderate edema is noted from the top of the lesion to the midfoot, concentrated around the lateral and medial malleolus. Extensive blue, purple, and black discoloration is seen below the malleolus. The patient is diagnosed with a fracture blister.
DISCUSSION
Fracture blisters are taut, bullous, subepidermal vesicles that can accompany fractures or severe twisting injuries. They overlie markedly edematous soft tissue and histologically resemble a second-degree burn.1,2
Physiologically, blisters are caused by increased interstitial pressure due to swelling, with subsequent increased filtration pressure and colloid osmotic pressure in the epidermal gap.3 This causes a disruption that allows fluid to move into the weakened area.3 Areas most at risk for fracture blister formation are those with tight, closely adhered skin without muscle or enveloping fascia, where there is less soft tissue between the skin and bone prominences (eg, ankle, elbow, foot, distal tibia).2-4
Approximately 3% of all patients with acute fractures requiring hospitalization develop a fracture blister.4 Any condition that predisposes a patient to poor wound healing (eg, peripheral vascular disease, diabetes, hypertension) increases risk for a fracture blister.2 Recognizing which patients are at greatest risk is vital, as implementing prevention strategies and intervening when fracture blisters do form can help decrease complications—including infection and delayed surgery—and improve fracture resolution. In this patient’s case, the extent of the injury and force of the fall caused the fracture blister to form.
Diagnosis
Diagnosis of a fracture blister is based on clinical presentation. There are two types: hemorrhagic blisters and clear fluid-filled blisters. Hemorrhagic blisters indicate more severe injury and longer healing time (approximately 16 d), while clear fluid-filled blisters demonstrate minimal injury and therefore are quicker to heal.2,4
The differential diagnosis for fracture blisters includes friction blisters and disorders such as epidermolysis bullosa and bullous pemphigoid. Friction blisters form when the epidermis is subjected to repeated friction or shear forces (eg, from a cast or splint).5,6 These forces mechanically separate epidermal cells at the stratum spinosum layer.7 The pressure that moves across the skin forces fluid into the deeper open spaces, filling them but leaving the surface layer intact.1
Epidermolysis bullosa (EB) is a group of rare inherited cutaneous and mucus membrane disorders. EB involves fragility and detachment of subepithelial tissues, which results in blistering and erosions.8,9 The blisters tend to develop in areas subject to minor trauma, such as the extensor aspects of the elbows and the dorsal aspects of the hands and feet.9 They can also be triggered by exposure to heat, friction, scratching, and adhesive tape.10
Bullous pemphigoid, a chronic autoimmune skin disorder, is characterized by pruritic, bullous lesions. When IgG autoantibodies bind to certain hemidesmosomal antigens, complement activation causes a subepidermal blister.11While bullous pemphigoid most commonly affects those older than 60, it can also occur in children. Diagnosis is confirmed by skin biopsy and immunofluorescence testing.11
Treatment and management
Although several recommendations have been published, there is no gold standard and treatment of fracture blisters remains controversial. Early surgical intervention for fractures could decrease the incidence of fracture blisters.1,3
The goal of treatment is to achieve re-epithelialization of the dermis.3,12,13 Once a blister forms, management techniques vary. Some recommend keeping closed blisters covered with a dry dressing to protect them from damage.3 Strauss et al recommend unroofing to avoid traumatic rupture; however, this does increase risk for infection.12 Recommendations differ depending on provider preference and each patient’s individual situation.
Elective unroofing of a blister is typically followed with one of several treatment options. These include covering the open blister with a topical antibiotic cream (eg, silver sulfadiazine 2%); applying a nonadherent, occlusive bismuth-tribromophenate-petroleum gauze dressing; or elevating and immobilizing the affected extremity.12,13
Treatment of spontaneously ruptured fracture blisters entails
- Unroofing the blister completely and applying a topical antimicrobial (eg, silver sulfadiazine, polymyxin B, neomycin, bacitracin).
- Applying a hydrocolloid dressing to keep the environment moist.
- Using a first-aid gel containing melaleuca (tea tree) oil.
- Initiating prophylactic oral antibiotics.
- Using whirlpool treatments.
- Elevating and immobilizing the affected extremity.3,12,14
OUTCOME FOR THE CASE PATIENT
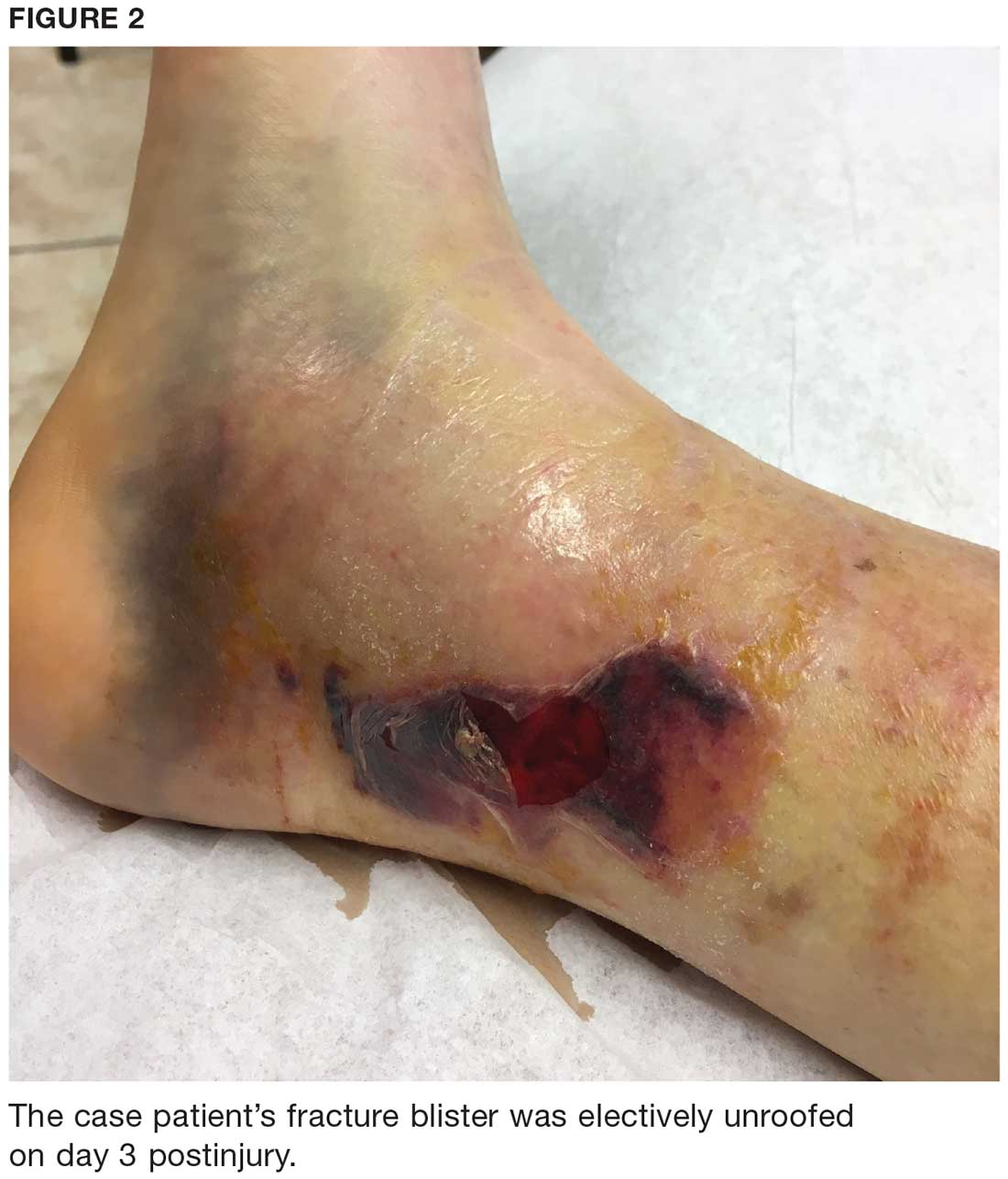
The fracture blister was electively unroofed (see Figure 2) based on provider preference. The patient was instructed to clean the wound daily and apply topical cream (silver sulfadiazine 2% bid) to the wound and cover it with gauze. The patient was made non-weight-bearing to the right lower extremity. Continuous elevation was highly encouraged except for bathing and restroom use, and an NSAID was recommended as needed for pain. She was reassessed the following day and, due to partial refilling, the blister required additional unroofing. The patient was instructed to resume previous wound care orders.
No surgical intervention was required. CT of the right foot and ankle without contrast (performed on day 4 postinjury) confirmed a nondisplaced transverse fracture of the medial malleolus and a sagittal avulsion fracture of the anterior-inferior lateral malleolus. Multiple smaller fracture fragments were noted posterior and medial to the medial malleolus as well as inferiorly along the course of the deltoid ligament. There was a small, nondisplaced avulsion fracture of the medial malleolus at the anterolateral and posterolateral tibial plafond.
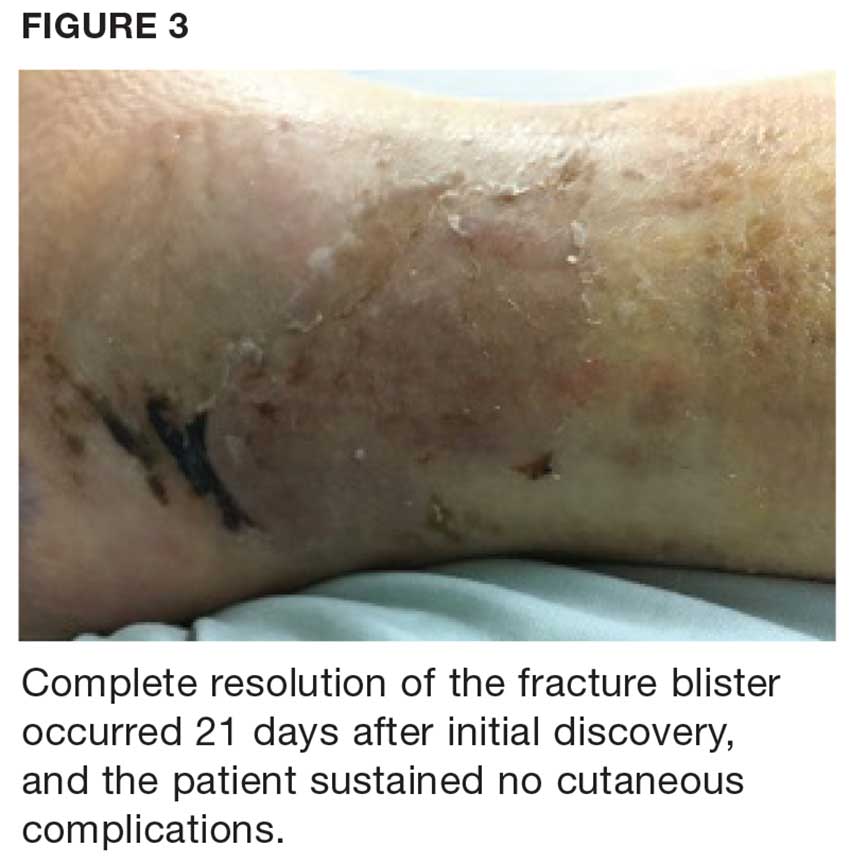
Due to the extent of the swelling, multiple fractures, and blister formation, the patient was essentially bed bound for the first three weeks; complete resolution of the fracture blister occurred 21 days after initial discovery (see Figure 3). The patient did not experience cutaneous complications. Her lower extremity was then casted in a short-leg removable cast for 10 weeks. She underwent physical therapy, and after 12 weeks, the patient was weight-bearing and was discharged from orthopedics. The patient reported refractory pain and swelling for an additional eight weeks following injury, warranting daily ibuprofen.
CONCLUSION
Fracture blisters are rare, and experience and knowledge about them in primary care is lacking. But clinicians need to be able to identify, diagnose, and refer at-risk patients to orthopedics in a timely manner.
Current management and treatment recommendations are inconsistent. Treatment varies depending on the site, severity, type, and status of the blister and the overall health of the patient. Fracture blisters may be left intact, electively unroofed, or treated after spontaneous rupture. More research is needed to clarify management recommendations, specifically regarding the decision to unroof a blister or leave it intact. Early surgical intervention may prevent the development of a fracture blister.
1. Wallace GF, Sullivan J. Fracture blisters. Clin Podiatr Med Surg. 1995;12(4):801-811.
2. Halawi MJ. Fracture blisters after primary total knee arthroplasty. Am J Orthop. 2015; 44(8):E291-E293.
3. McCann S, Gruen G. Fracture blisters: a review of the literature. Orthop Nurs. 1997; 16(2):17-24.
4. Uebbing CM, Walsh M, Miller JB, et al. Fracture blister. West J Emerg Med. 2011; 12(1):131-133.
5. Kirkham S, Lam S, Nester C, Hashmi F. The effect of hydration on the risk of friction blister formation on the heel of the foot. Skin Res Tech. 2014;20:246-253.
6. Boyd A, Benjamin H, Asplund C. Principles of casting and splinting. Am Fam Physician. 2009;79(1):16-24.
7. Knapik J, Reynolds K, Duplantis K, Jones B. Friction blisters. Pathophysiology, prevention and treatment. Sports Med. 1995; 20(3):136-147.
8. Iranzo P, Herrero-González JE, Mascaró-Galy JM, et al. Epidermolysis bullosa acquisita: a retrospective analysis of 12 patients evaluated in four tertiary hospitals in Spain. Br J Dermatol. 2014;171(5):1022-1030.
9. Peraza DM. Epidermolysis bullosa acquisita. Merck Manual Professional Version. August 2016. www.merckmanuals.com/professional/dermatologic-disorders/bullous-diseases/epidermolysis-bullosa-acquisita. Accessed January 26, 2018.
10. Lyons F, Ousley L. Dermatology for the Advanced Practice Nurse. New York, NY: Springer; 2015.
11. Peraza D. Bullous pemphigoid. Merck Manual Professional Version. August 2016. www.merckmanuals.com/professional/dermatologic-disorders/bullous-diseases/bullous-pemphigoid. Accessed January 26, 2018.
12. Strauss EJ, Petrucelli G, Bong M, et al. Blisters associated with lower-extremity fracture: Results of a prospective treatment protocol. J Orthop Trauma. 2006;20(9): 618-622.
13. Tolpinrud WL, Rebolledo BJ, Lorich DG, Grossman ME. A case of extensive fracture bullae: a multidisciplinary approach for acute management. JAAD Case Rep. 2015;1(3):132-135.
14. Cox H, Nealon L. Case report: the use of Burnaid Gel on fracture blisters. Wound Practice and Research. 2008;16(1):32-36.
IN THIS ARTICLE
- Diagnosis
- Treatment
- Care outcome
A 57-year-old horticulturist is working on a ladder leaned up against a tree trunk when the ladder slips, causing her to fall six feet onto concrete. Her right foot and ankle sustain the force of the fall; she is in excruciating pain and unable to bear weight on the foot. She is immediately transported to a local emergency department for evaluation.
Physical exam reveals a tearful middle-aged female in moderate distress and acute pain. There is moderate swelling of the right medial and lateral malleolus, as well as the midfoot, with blue and purple discoloration on the medial and lateral malleolus. Radiographs of the right ankle identify nondisplaced fractures of the distal fibula and tibia. Foot x-rays are unremarkable. A splint is ordered. The patient is given crutches (non-weight-bearing status), pain medication, and a referral to orthopedics.
On day 3, the patient presents to orthopedics, where the splint is removed. An irregular, 4 × 3–in (at largest diameter), serohemorrhagic blister is discovered on the medial aspect of the lower leg, above the right malleolus (see Figure 1). Multiple 1- to 3-mm vesicles surround much of the anterior border. Moderate edema is noted from the top of the lesion to the midfoot, concentrated around the lateral and medial malleolus. Extensive blue, purple, and black discoloration is seen below the malleolus. The patient is diagnosed with a fracture blister.
DISCUSSION
Fracture blisters are taut, bullous, subepidermal vesicles that can accompany fractures or severe twisting injuries. They overlie markedly edematous soft tissue and histologically resemble a second-degree burn.1,2
Physiologically, blisters are caused by increased interstitial pressure due to swelling, with subsequent increased filtration pressure and colloid osmotic pressure in the epidermal gap.3 This causes a disruption that allows fluid to move into the weakened area.3 Areas most at risk for fracture blister formation are those with tight, closely adhered skin without muscle or enveloping fascia, where there is less soft tissue between the skin and bone prominences (eg, ankle, elbow, foot, distal tibia).2-4
Approximately 3% of all patients with acute fractures requiring hospitalization develop a fracture blister.4 Any condition that predisposes a patient to poor wound healing (eg, peripheral vascular disease, diabetes, hypertension) increases risk for a fracture blister.2 Recognizing which patients are at greatest risk is vital, as implementing prevention strategies and intervening when fracture blisters do form can help decrease complications—including infection and delayed surgery—and improve fracture resolution. In this patient’s case, the extent of the injury and force of the fall caused the fracture blister to form.
Diagnosis
Diagnosis of a fracture blister is based on clinical presentation. There are two types: hemorrhagic blisters and clear fluid-filled blisters. Hemorrhagic blisters indicate more severe injury and longer healing time (approximately 16 d), while clear fluid-filled blisters demonstrate minimal injury and therefore are quicker to heal.2,4
The differential diagnosis for fracture blisters includes friction blisters and disorders such as epidermolysis bullosa and bullous pemphigoid. Friction blisters form when the epidermis is subjected to repeated friction or shear forces (eg, from a cast or splint).5,6 These forces mechanically separate epidermal cells at the stratum spinosum layer.7 The pressure that moves across the skin forces fluid into the deeper open spaces, filling them but leaving the surface layer intact.1
Epidermolysis bullosa (EB) is a group of rare inherited cutaneous and mucus membrane disorders. EB involves fragility and detachment of subepithelial tissues, which results in blistering and erosions.8,9 The blisters tend to develop in areas subject to minor trauma, such as the extensor aspects of the elbows and the dorsal aspects of the hands and feet.9 They can also be triggered by exposure to heat, friction, scratching, and adhesive tape.10
Bullous pemphigoid, a chronic autoimmune skin disorder, is characterized by pruritic, bullous lesions. When IgG autoantibodies bind to certain hemidesmosomal antigens, complement activation causes a subepidermal blister.11While bullous pemphigoid most commonly affects those older than 60, it can also occur in children. Diagnosis is confirmed by skin biopsy and immunofluorescence testing.11
Treatment and management
Although several recommendations have been published, there is no gold standard and treatment of fracture blisters remains controversial. Early surgical intervention for fractures could decrease the incidence of fracture blisters.1,3
The goal of treatment is to achieve re-epithelialization of the dermis.3,12,13 Once a blister forms, management techniques vary. Some recommend keeping closed blisters covered with a dry dressing to protect them from damage.3 Strauss et al recommend unroofing to avoid traumatic rupture; however, this does increase risk for infection.12 Recommendations differ depending on provider preference and each patient’s individual situation.
Elective unroofing of a blister is typically followed with one of several treatment options. These include covering the open blister with a topical antibiotic cream (eg, silver sulfadiazine 2%); applying a nonadherent, occlusive bismuth-tribromophenate-petroleum gauze dressing; or elevating and immobilizing the affected extremity.12,13
Treatment of spontaneously ruptured fracture blisters entails
- Unroofing the blister completely and applying a topical antimicrobial (eg, silver sulfadiazine, polymyxin B, neomycin, bacitracin).
- Applying a hydrocolloid dressing to keep the environment moist.
- Using a first-aid gel containing melaleuca (tea tree) oil.
- Initiating prophylactic oral antibiotics.
- Using whirlpool treatments.
- Elevating and immobilizing the affected extremity.3,12,14
OUTCOME FOR THE CASE PATIENT
The fracture blister was electively unroofed (see Figure 2) based on provider preference. The patient was instructed to clean the wound daily and apply topical cream (silver sulfadiazine 2% bid) to the wound and cover it with gauze. The patient was made non-weight-bearing to the right lower extremity. Continuous elevation was highly encouraged except for bathing and restroom use, and an NSAID was recommended as needed for pain. She was reassessed the following day and, due to partial refilling, the blister required additional unroofing. The patient was instructed to resume previous wound care orders.
No surgical intervention was required. CT of the right foot and ankle without contrast (performed on day 4 postinjury) confirmed a nondisplaced transverse fracture of the medial malleolus and a sagittal avulsion fracture of the anterior-inferior lateral malleolus. Multiple smaller fracture fragments were noted posterior and medial to the medial malleolus as well as inferiorly along the course of the deltoid ligament. There was a small, nondisplaced avulsion fracture of the medial malleolus at the anterolateral and posterolateral tibial plafond.
Due to the extent of the swelling, multiple fractures, and blister formation, the patient was essentially bed bound for the first three weeks; complete resolution of the fracture blister occurred 21 days after initial discovery (see Figure 3). The patient did not experience cutaneous complications. Her lower extremity was then casted in a short-leg removable cast for 10 weeks. She underwent physical therapy, and after 12 weeks, the patient was weight-bearing and was discharged from orthopedics. The patient reported refractory pain and swelling for an additional eight weeks following injury, warranting daily ibuprofen.
CONCLUSION
Fracture blisters are rare, and experience and knowledge about them in primary care is lacking. But clinicians need to be able to identify, diagnose, and refer at-risk patients to orthopedics in a timely manner.
Current management and treatment recommendations are inconsistent. Treatment varies depending on the site, severity, type, and status of the blister and the overall health of the patient. Fracture blisters may be left intact, electively unroofed, or treated after spontaneous rupture. More research is needed to clarify management recommendations, specifically regarding the decision to unroof a blister or leave it intact. Early surgical intervention may prevent the development of a fracture blister.
IN THIS ARTICLE
- Diagnosis
- Treatment
- Care outcome
A 57-year-old horticulturist is working on a ladder leaned up against a tree trunk when the ladder slips, causing her to fall six feet onto concrete. Her right foot and ankle sustain the force of the fall; she is in excruciating pain and unable to bear weight on the foot. She is immediately transported to a local emergency department for evaluation.
Physical exam reveals a tearful middle-aged female in moderate distress and acute pain. There is moderate swelling of the right medial and lateral malleolus, as well as the midfoot, with blue and purple discoloration on the medial and lateral malleolus. Radiographs of the right ankle identify nondisplaced fractures of the distal fibula and tibia. Foot x-rays are unremarkable. A splint is ordered. The patient is given crutches (non-weight-bearing status), pain medication, and a referral to orthopedics.
On day 3, the patient presents to orthopedics, where the splint is removed. An irregular, 4 × 3–in (at largest diameter), serohemorrhagic blister is discovered on the medial aspect of the lower leg, above the right malleolus (see Figure 1). Multiple 1- to 3-mm vesicles surround much of the anterior border. Moderate edema is noted from the top of the lesion to the midfoot, concentrated around the lateral and medial malleolus. Extensive blue, purple, and black discoloration is seen below the malleolus. The patient is diagnosed with a fracture blister.
DISCUSSION
Fracture blisters are taut, bullous, subepidermal vesicles that can accompany fractures or severe twisting injuries. They overlie markedly edematous soft tissue and histologically resemble a second-degree burn.1,2
Physiologically, blisters are caused by increased interstitial pressure due to swelling, with subsequent increased filtration pressure and colloid osmotic pressure in the epidermal gap.3 This causes a disruption that allows fluid to move into the weakened area.3 Areas most at risk for fracture blister formation are those with tight, closely adhered skin without muscle or enveloping fascia, where there is less soft tissue between the skin and bone prominences (eg, ankle, elbow, foot, distal tibia).2-4
Approximately 3% of all patients with acute fractures requiring hospitalization develop a fracture blister.4 Any condition that predisposes a patient to poor wound healing (eg, peripheral vascular disease, diabetes, hypertension) increases risk for a fracture blister.2 Recognizing which patients are at greatest risk is vital, as implementing prevention strategies and intervening when fracture blisters do form can help decrease complications—including infection and delayed surgery—and improve fracture resolution. In this patient’s case, the extent of the injury and force of the fall caused the fracture blister to form.
Diagnosis
Diagnosis of a fracture blister is based on clinical presentation. There are two types: hemorrhagic blisters and clear fluid-filled blisters. Hemorrhagic blisters indicate more severe injury and longer healing time (approximately 16 d), while clear fluid-filled blisters demonstrate minimal injury and therefore are quicker to heal.2,4
The differential diagnosis for fracture blisters includes friction blisters and disorders such as epidermolysis bullosa and bullous pemphigoid. Friction blisters form when the epidermis is subjected to repeated friction or shear forces (eg, from a cast or splint).5,6 These forces mechanically separate epidermal cells at the stratum spinosum layer.7 The pressure that moves across the skin forces fluid into the deeper open spaces, filling them but leaving the surface layer intact.1
Epidermolysis bullosa (EB) is a group of rare inherited cutaneous and mucus membrane disorders. EB involves fragility and detachment of subepithelial tissues, which results in blistering and erosions.8,9 The blisters tend to develop in areas subject to minor trauma, such as the extensor aspects of the elbows and the dorsal aspects of the hands and feet.9 They can also be triggered by exposure to heat, friction, scratching, and adhesive tape.10
Bullous pemphigoid, a chronic autoimmune skin disorder, is characterized by pruritic, bullous lesions. When IgG autoantibodies bind to certain hemidesmosomal antigens, complement activation causes a subepidermal blister.11While bullous pemphigoid most commonly affects those older than 60, it can also occur in children. Diagnosis is confirmed by skin biopsy and immunofluorescence testing.11
Treatment and management
Although several recommendations have been published, there is no gold standard and treatment of fracture blisters remains controversial. Early surgical intervention for fractures could decrease the incidence of fracture blisters.1,3
The goal of treatment is to achieve re-epithelialization of the dermis.3,12,13 Once a blister forms, management techniques vary. Some recommend keeping closed blisters covered with a dry dressing to protect them from damage.3 Strauss et al recommend unroofing to avoid traumatic rupture; however, this does increase risk for infection.12 Recommendations differ depending on provider preference and each patient’s individual situation.
Elective unroofing of a blister is typically followed with one of several treatment options. These include covering the open blister with a topical antibiotic cream (eg, silver sulfadiazine 2%); applying a nonadherent, occlusive bismuth-tribromophenate-petroleum gauze dressing; or elevating and immobilizing the affected extremity.12,13
Treatment of spontaneously ruptured fracture blisters entails
- Unroofing the blister completely and applying a topical antimicrobial (eg, silver sulfadiazine, polymyxin B, neomycin, bacitracin).
- Applying a hydrocolloid dressing to keep the environment moist.
- Using a first-aid gel containing melaleuca (tea tree) oil.
- Initiating prophylactic oral antibiotics.
- Using whirlpool treatments.
- Elevating and immobilizing the affected extremity.3,12,14
OUTCOME FOR THE CASE PATIENT
The fracture blister was electively unroofed (see Figure 2) based on provider preference. The patient was instructed to clean the wound daily and apply topical cream (silver sulfadiazine 2% bid) to the wound and cover it with gauze. The patient was made non-weight-bearing to the right lower extremity. Continuous elevation was highly encouraged except for bathing and restroom use, and an NSAID was recommended as needed for pain. She was reassessed the following day and, due to partial refilling, the blister required additional unroofing. The patient was instructed to resume previous wound care orders.
No surgical intervention was required. CT of the right foot and ankle without contrast (performed on day 4 postinjury) confirmed a nondisplaced transverse fracture of the medial malleolus and a sagittal avulsion fracture of the anterior-inferior lateral malleolus. Multiple smaller fracture fragments were noted posterior and medial to the medial malleolus as well as inferiorly along the course of the deltoid ligament. There was a small, nondisplaced avulsion fracture of the medial malleolus at the anterolateral and posterolateral tibial plafond.
Due to the extent of the swelling, multiple fractures, and blister formation, the patient was essentially bed bound for the first three weeks; complete resolution of the fracture blister occurred 21 days after initial discovery (see Figure 3). The patient did not experience cutaneous complications. Her lower extremity was then casted in a short-leg removable cast for 10 weeks. She underwent physical therapy, and after 12 weeks, the patient was weight-bearing and was discharged from orthopedics. The patient reported refractory pain and swelling for an additional eight weeks following injury, warranting daily ibuprofen.
CONCLUSION
Fracture blisters are rare, and experience and knowledge about them in primary care is lacking. But clinicians need to be able to identify, diagnose, and refer at-risk patients to orthopedics in a timely manner.
Current management and treatment recommendations are inconsistent. Treatment varies depending on the site, severity, type, and status of the blister and the overall health of the patient. Fracture blisters may be left intact, electively unroofed, or treated after spontaneous rupture. More research is needed to clarify management recommendations, specifically regarding the decision to unroof a blister or leave it intact. Early surgical intervention may prevent the development of a fracture blister.
1. Wallace GF, Sullivan J. Fracture blisters. Clin Podiatr Med Surg. 1995;12(4):801-811.
2. Halawi MJ. Fracture blisters after primary total knee arthroplasty. Am J Orthop. 2015; 44(8):E291-E293.
3. McCann S, Gruen G. Fracture blisters: a review of the literature. Orthop Nurs. 1997; 16(2):17-24.
4. Uebbing CM, Walsh M, Miller JB, et al. Fracture blister. West J Emerg Med. 2011; 12(1):131-133.
5. Kirkham S, Lam S, Nester C, Hashmi F. The effect of hydration on the risk of friction blister formation on the heel of the foot. Skin Res Tech. 2014;20:246-253.
6. Boyd A, Benjamin H, Asplund C. Principles of casting and splinting. Am Fam Physician. 2009;79(1):16-24.
7. Knapik J, Reynolds K, Duplantis K, Jones B. Friction blisters. Pathophysiology, prevention and treatment. Sports Med. 1995; 20(3):136-147.
8. Iranzo P, Herrero-González JE, Mascaró-Galy JM, et al. Epidermolysis bullosa acquisita: a retrospective analysis of 12 patients evaluated in four tertiary hospitals in Spain. Br J Dermatol. 2014;171(5):1022-1030.
9. Peraza DM. Epidermolysis bullosa acquisita. Merck Manual Professional Version. August 2016. www.merckmanuals.com/professional/dermatologic-disorders/bullous-diseases/epidermolysis-bullosa-acquisita. Accessed January 26, 2018.
10. Lyons F, Ousley L. Dermatology for the Advanced Practice Nurse. New York, NY: Springer; 2015.
11. Peraza D. Bullous pemphigoid. Merck Manual Professional Version. August 2016. www.merckmanuals.com/professional/dermatologic-disorders/bullous-diseases/bullous-pemphigoid. Accessed January 26, 2018.
12. Strauss EJ, Petrucelli G, Bong M, et al. Blisters associated with lower-extremity fracture: Results of a prospective treatment protocol. J Orthop Trauma. 2006;20(9): 618-622.
13. Tolpinrud WL, Rebolledo BJ, Lorich DG, Grossman ME. A case of extensive fracture bullae: a multidisciplinary approach for acute management. JAAD Case Rep. 2015;1(3):132-135.
14. Cox H, Nealon L. Case report: the use of Burnaid Gel on fracture blisters. Wound Practice and Research. 2008;16(1):32-36.
1. Wallace GF, Sullivan J. Fracture blisters. Clin Podiatr Med Surg. 1995;12(4):801-811.
2. Halawi MJ. Fracture blisters after primary total knee arthroplasty. Am J Orthop. 2015; 44(8):E291-E293.
3. McCann S, Gruen G. Fracture blisters: a review of the literature. Orthop Nurs. 1997; 16(2):17-24.
4. Uebbing CM, Walsh M, Miller JB, et al. Fracture blister. West J Emerg Med. 2011; 12(1):131-133.
5. Kirkham S, Lam S, Nester C, Hashmi F. The effect of hydration on the risk of friction blister formation on the heel of the foot. Skin Res Tech. 2014;20:246-253.
6. Boyd A, Benjamin H, Asplund C. Principles of casting and splinting. Am Fam Physician. 2009;79(1):16-24.
7. Knapik J, Reynolds K, Duplantis K, Jones B. Friction blisters. Pathophysiology, prevention and treatment. Sports Med. 1995; 20(3):136-147.
8. Iranzo P, Herrero-González JE, Mascaró-Galy JM, et al. Epidermolysis bullosa acquisita: a retrospective analysis of 12 patients evaluated in four tertiary hospitals in Spain. Br J Dermatol. 2014;171(5):1022-1030.
9. Peraza DM. Epidermolysis bullosa acquisita. Merck Manual Professional Version. August 2016. www.merckmanuals.com/professional/dermatologic-disorders/bullous-diseases/epidermolysis-bullosa-acquisita. Accessed January 26, 2018.
10. Lyons F, Ousley L. Dermatology for the Advanced Practice Nurse. New York, NY: Springer; 2015.
11. Peraza D. Bullous pemphigoid. Merck Manual Professional Version. August 2016. www.merckmanuals.com/professional/dermatologic-disorders/bullous-diseases/bullous-pemphigoid. Accessed January 26, 2018.
12. Strauss EJ, Petrucelli G, Bong M, et al. Blisters associated with lower-extremity fracture: Results of a prospective treatment protocol. J Orthop Trauma. 2006;20(9): 618-622.
13. Tolpinrud WL, Rebolledo BJ, Lorich DG, Grossman ME. A case of extensive fracture bullae: a multidisciplinary approach for acute management. JAAD Case Rep. 2015;1(3):132-135.
14. Cox H, Nealon L. Case report: the use of Burnaid Gel on fracture blisters. Wound Practice and Research. 2008;16(1):32-36.
Consider drug holidays for BCC patients on hedgehog inhibitors
KAUAI, HAWAII – according to Kishwer Nehal, MD, director of Mohs micrographic and dermatologic surgery at Memorial Sloan Kettering Cancer Center, New York.
That’s important because, although some patients have a good response to vismodegib, more – about 80% – have side effects that make it necessary to stop treatment, including muscle spasms and weight loss, among other problems. Side effects often come on quickly and can become intolerable after a few months of treatment, so physicians have looked for alternative dosing regimens to hold them off, with some success.
Compared with those on continuous dosing, fewer patients on intermittent dosing discontinued treatment for adverse events (23% versus 31%). Patients on intermittent dosing also experienced fewer grade 3 adverse events (31% versus 44%) and were on treatment for a longer period of time (a median of 71.4 weeks versus 37.6 weeks).
Meanwhile, among those on intermittent dosing, the number of BCCs was reduced in more than half of the patients in both interrupted treatment groups, but more so in the 12-weeks-on/8-weeks-off group (Lancet Oncol. 2017 Mar; 18[3]:404-12).
Other treatment options are being explored for vismodegib, as well as for sonidegib (Odomzo), another hedgehog signaling pathway inhibitor approved for advanced BCC. Ongoing trials are looking at the use of hedgehog inhibitors with radiation, and for shrinking tumors before surgery, Dr. Nehal said
For now, however, surgery remains the mainstay of treatment for BCC; both biologics are indicated for when other treatments fail or are not feasible. For high-risk BCC (meaning high risk for recurrence, based on infiltrative or poorly defined histology, perineural or bony involvement, or location on the face, for instance), “surgery with clear margins remains the goal and is the most effective treatment. For a high-risk [BCC], you pretty much need surgery,” she said.
Recurrence is less likely with Mohs surgery than with standard excision. When Mohs isn’t available, “you should wait for the pathology report before reconstruction,” she said.
“Radiation for high-risk [BCC] is really reserved for nonsurgical candidates,” Dr. Nehal commented. There are only two scenarios to consider radiation in high-risk BCC, “and they really have no proven benefit in any sort of prospective trial. One is if you cannot, after exhaustive surgery, clear your very high risk [BCC].” The other is if there is “really large nerve involvement, greater than 0.1 mm, or such extensive perineural involvement that surgery is unlikely to be successful,” she said.
Dr. Nehal had no relevant disclosures. SDEF/Global Academy for Medical Education and this news organization are owned by the same parent company.
KAUAI, HAWAII – according to Kishwer Nehal, MD, director of Mohs micrographic and dermatologic surgery at Memorial Sloan Kettering Cancer Center, New York.
That’s important because, although some patients have a good response to vismodegib, more – about 80% – have side effects that make it necessary to stop treatment, including muscle spasms and weight loss, among other problems. Side effects often come on quickly and can become intolerable after a few months of treatment, so physicians have looked for alternative dosing regimens to hold them off, with some success.
Compared with those on continuous dosing, fewer patients on intermittent dosing discontinued treatment for adverse events (23% versus 31%). Patients on intermittent dosing also experienced fewer grade 3 adverse events (31% versus 44%) and were on treatment for a longer period of time (a median of 71.4 weeks versus 37.6 weeks).
Meanwhile, among those on intermittent dosing, the number of BCCs was reduced in more than half of the patients in both interrupted treatment groups, but more so in the 12-weeks-on/8-weeks-off group (Lancet Oncol. 2017 Mar; 18[3]:404-12).
Other treatment options are being explored for vismodegib, as well as for sonidegib (Odomzo), another hedgehog signaling pathway inhibitor approved for advanced BCC. Ongoing trials are looking at the use of hedgehog inhibitors with radiation, and for shrinking tumors before surgery, Dr. Nehal said
For now, however, surgery remains the mainstay of treatment for BCC; both biologics are indicated for when other treatments fail or are not feasible. For high-risk BCC (meaning high risk for recurrence, based on infiltrative or poorly defined histology, perineural or bony involvement, or location on the face, for instance), “surgery with clear margins remains the goal and is the most effective treatment. For a high-risk [BCC], you pretty much need surgery,” she said.
Recurrence is less likely with Mohs surgery than with standard excision. When Mohs isn’t available, “you should wait for the pathology report before reconstruction,” she said.
“Radiation for high-risk [BCC] is really reserved for nonsurgical candidates,” Dr. Nehal commented. There are only two scenarios to consider radiation in high-risk BCC, “and they really have no proven benefit in any sort of prospective trial. One is if you cannot, after exhaustive surgery, clear your very high risk [BCC].” The other is if there is “really large nerve involvement, greater than 0.1 mm, or such extensive perineural involvement that surgery is unlikely to be successful,” she said.
Dr. Nehal had no relevant disclosures. SDEF/Global Academy for Medical Education and this news organization are owned by the same parent company.
KAUAI, HAWAII – according to Kishwer Nehal, MD, director of Mohs micrographic and dermatologic surgery at Memorial Sloan Kettering Cancer Center, New York.
That’s important because, although some patients have a good response to vismodegib, more – about 80% – have side effects that make it necessary to stop treatment, including muscle spasms and weight loss, among other problems. Side effects often come on quickly and can become intolerable after a few months of treatment, so physicians have looked for alternative dosing regimens to hold them off, with some success.
Compared with those on continuous dosing, fewer patients on intermittent dosing discontinued treatment for adverse events (23% versus 31%). Patients on intermittent dosing also experienced fewer grade 3 adverse events (31% versus 44%) and were on treatment for a longer period of time (a median of 71.4 weeks versus 37.6 weeks).
Meanwhile, among those on intermittent dosing, the number of BCCs was reduced in more than half of the patients in both interrupted treatment groups, but more so in the 12-weeks-on/8-weeks-off group (Lancet Oncol. 2017 Mar; 18[3]:404-12).
Other treatment options are being explored for vismodegib, as well as for sonidegib (Odomzo), another hedgehog signaling pathway inhibitor approved for advanced BCC. Ongoing trials are looking at the use of hedgehog inhibitors with radiation, and for shrinking tumors before surgery, Dr. Nehal said
For now, however, surgery remains the mainstay of treatment for BCC; both biologics are indicated for when other treatments fail or are not feasible. For high-risk BCC (meaning high risk for recurrence, based on infiltrative or poorly defined histology, perineural or bony involvement, or location on the face, for instance), “surgery with clear margins remains the goal and is the most effective treatment. For a high-risk [BCC], you pretty much need surgery,” she said.
Recurrence is less likely with Mohs surgery than with standard excision. When Mohs isn’t available, “you should wait for the pathology report before reconstruction,” she said.
“Radiation for high-risk [BCC] is really reserved for nonsurgical candidates,” Dr. Nehal commented. There are only two scenarios to consider radiation in high-risk BCC, “and they really have no proven benefit in any sort of prospective trial. One is if you cannot, after exhaustive surgery, clear your very high risk [BCC].” The other is if there is “really large nerve involvement, greater than 0.1 mm, or such extensive perineural involvement that surgery is unlikely to be successful,” she said.
Dr. Nehal had no relevant disclosures. SDEF/Global Academy for Medical Education and this news organization are owned by the same parent company.
EXPERT ANALYSIS FROM SDEF HAWAII DERMATOLOGY SEMINAR
Pain, opioids and addiction
In the year 2017, a plethora of articles and commentaries on the “opioid crisis” have appeared in major medical journals, alongside the ongoing hyperbole seen daily in the lay media. But the pressing concern remains: How best to manage patients who are 1.) already taking opioids and 2.) those newly requesting relief of serious and chronic pain.
Opioid for Pain and Its Misuse
In this article by Volkow and Collins, both of whom are titans in neuroscience, we are reminded that despite all the warnings, opioids are being widely prescribed in the U.S. In a weighted national sample of over 50,000 adults, the investigators concluded that more than one-third of the adult population has taken an opioid at some point during 2015. Among these, 12.5% confirmed that they misused the drug, e.g., used them without a prescription or in any way contrary to the prescribed directions. Of these, 16.7% developed an opioid-use disorder, as defined in the DSM-IV.
In response, Volkow and Collins note that an increasing number of clinicians are attempting to control chronic or intractable pain with new anticonvulsants such as Pregabalin (Lyrica) and Gabapentin. Yet, these drugs have only been shown to be effective only for fibromyalgia and certain forms of neurogenic pain. In addition, the authors note that a multidisciplinary workgroup convened by the NIH Office of Disease Prevention (2014) found that there had been no randomized trials to evaluate the efficacy of long-term (>1 year) opioid treatment. Accordingly, the authors recommend short-term strategy to develop abuse-deterrent formulations that can minimize diversion and misuse.
What About Cannabis?
In a 2017 report from the National Academies of Sciences, Engineering, and Medicine, substantial evidence supports the effectiveness of cannabinoids in treating some types of pain. However, again there is scant research on phytocannabinoids as medicine. In addition, there are abundant research and legitimate concerns related to cognitive, motor and motivational impairment and the effects on brain development. However, the therapeutic potential of cannabinoids and mediators of the abundant endocannabinoid system warrants further exploration for alternatives to opioids.
Lastly, non-pharmacologic interventions, including behavioral, self-management interventions, may play an important role in pain management. The initiative described by the authors supports partnerships between the NIH and pharmaceutical and biotechnology companies to hasten medication and device development.
Why Does This Matter?
If these data are true, and one-third of the U.S. adult population suffers from chronic pain, we are duty bound to find therapeutic options with less risk, addictive potential and mortality. As I have argued for nearly 40 years, basic and translational research is desperately needed, as clinicians are in a conundrum between the worthy goals of alleviating pain and suffering and decreasing the risk for addiction and mortality. We can, and must do better.
Volkow ND, Collins FS. The Role of Science in Addressing the Opioid Crisis. N Engl J Med. 2017;377(4):391-394.
In the year 2017, a plethora of articles and commentaries on the “opioid crisis” have appeared in major medical journals, alongside the ongoing hyperbole seen daily in the lay media. But the pressing concern remains: How best to manage patients who are 1.) already taking opioids and 2.) those newly requesting relief of serious and chronic pain.
Opioid for Pain and Its Misuse
In this article by Volkow and Collins, both of whom are titans in neuroscience, we are reminded that despite all the warnings, opioids are being widely prescribed in the U.S. In a weighted national sample of over 50,000 adults, the investigators concluded that more than one-third of the adult population has taken an opioid at some point during 2015. Among these, 12.5% confirmed that they misused the drug, e.g., used them without a prescription or in any way contrary to the prescribed directions. Of these, 16.7% developed an opioid-use disorder, as defined in the DSM-IV.
In response, Volkow and Collins note that an increasing number of clinicians are attempting to control chronic or intractable pain with new anticonvulsants such as Pregabalin (Lyrica) and Gabapentin. Yet, these drugs have only been shown to be effective only for fibromyalgia and certain forms of neurogenic pain. In addition, the authors note that a multidisciplinary workgroup convened by the NIH Office of Disease Prevention (2014) found that there had been no randomized trials to evaluate the efficacy of long-term (>1 year) opioid treatment. Accordingly, the authors recommend short-term strategy to develop abuse-deterrent formulations that can minimize diversion and misuse.
What About Cannabis?
In a 2017 report from the National Academies of Sciences, Engineering, and Medicine, substantial evidence supports the effectiveness of cannabinoids in treating some types of pain. However, again there is scant research on phytocannabinoids as medicine. In addition, there are abundant research and legitimate concerns related to cognitive, motor and motivational impairment and the effects on brain development. However, the therapeutic potential of cannabinoids and mediators of the abundant endocannabinoid system warrants further exploration for alternatives to opioids.
Lastly, non-pharmacologic interventions, including behavioral, self-management interventions, may play an important role in pain management. The initiative described by the authors supports partnerships between the NIH and pharmaceutical and biotechnology companies to hasten medication and device development.
Why Does This Matter?
If these data are true, and one-third of the U.S. adult population suffers from chronic pain, we are duty bound to find therapeutic options with less risk, addictive potential and mortality. As I have argued for nearly 40 years, basic and translational research is desperately needed, as clinicians are in a conundrum between the worthy goals of alleviating pain and suffering and decreasing the risk for addiction and mortality. We can, and must do better.
In the year 2017, a plethora of articles and commentaries on the “opioid crisis” have appeared in major medical journals, alongside the ongoing hyperbole seen daily in the lay media. But the pressing concern remains: How best to manage patients who are 1.) already taking opioids and 2.) those newly requesting relief of serious and chronic pain.
Opioid for Pain and Its Misuse
In this article by Volkow and Collins, both of whom are titans in neuroscience, we are reminded that despite all the warnings, opioids are being widely prescribed in the U.S. In a weighted national sample of over 50,000 adults, the investigators concluded that more than one-third of the adult population has taken an opioid at some point during 2015. Among these, 12.5% confirmed that they misused the drug, e.g., used them without a prescription or in any way contrary to the prescribed directions. Of these, 16.7% developed an opioid-use disorder, as defined in the DSM-IV.
In response, Volkow and Collins note that an increasing number of clinicians are attempting to control chronic or intractable pain with new anticonvulsants such as Pregabalin (Lyrica) and Gabapentin. Yet, these drugs have only been shown to be effective only for fibromyalgia and certain forms of neurogenic pain. In addition, the authors note that a multidisciplinary workgroup convened by the NIH Office of Disease Prevention (2014) found that there had been no randomized trials to evaluate the efficacy of long-term (>1 year) opioid treatment. Accordingly, the authors recommend short-term strategy to develop abuse-deterrent formulations that can minimize diversion and misuse.
What About Cannabis?
In a 2017 report from the National Academies of Sciences, Engineering, and Medicine, substantial evidence supports the effectiveness of cannabinoids in treating some types of pain. However, again there is scant research on phytocannabinoids as medicine. In addition, there are abundant research and legitimate concerns related to cognitive, motor and motivational impairment and the effects on brain development. However, the therapeutic potential of cannabinoids and mediators of the abundant endocannabinoid system warrants further exploration for alternatives to opioids.
Lastly, non-pharmacologic interventions, including behavioral, self-management interventions, may play an important role in pain management. The initiative described by the authors supports partnerships between the NIH and pharmaceutical and biotechnology companies to hasten medication and device development.
Why Does This Matter?
If these data are true, and one-third of the U.S. adult population suffers from chronic pain, we are duty bound to find therapeutic options with less risk, addictive potential and mortality. As I have argued for nearly 40 years, basic and translational research is desperately needed, as clinicians are in a conundrum between the worthy goals of alleviating pain and suffering and decreasing the risk for addiction and mortality. We can, and must do better.
Volkow ND, Collins FS. The Role of Science in Addressing the Opioid Crisis. N Engl J Med. 2017;377(4):391-394.
Volkow ND, Collins FS. The Role of Science in Addressing the Opioid Crisis. N Engl J Med. 2017;377(4):391-394.

Anti-PD-1 therapy with nivolumab in the treatment of metastatic malignant PEComa
Perivascular epithelioid cell neoplasms (PEComas) are an uncommon class of tumors consisting on histology of perivascular epithelioid cells occurring in both localized and metastatic forms at various body sites. The approach to treatment of these tumors generally involves a combination of surgical resection, chemotherapy, and/or radiation therapy.1
Case presentation and summary
A 46-year-old man presented to our institution with a non-tender, slowly enlarging, 8.3 cm mass in his right popliteal fossa. Upon biopsy, the pathologic findings were consistent with an epithelioid malignancy with melanocytic differentiation most consistent with a PEComa. Discussion of the pathologic diagnosis of our patient has been reported by the pathology group at our institution in a separate case report.2
Our patient was initially offered and refused amputation. He was started on therapy with the mechanistic Target of Rapamycin (mTOR) inhibitor everolimus, but was unable to tolerate the side effects after the first week of treatment. He then elected to monitor his symptoms clinically.
Approximately one year after his initial diagnosis, he presented to our facility with sepsis and bleeding from a now fungating tumor on his right knee. At this time, emergent above-knee amputation was performed. Re-staging images now showed the presence of multiple pulmonary nodules in his right lung as well as a lytic rib lesion, a concerning finding for metastatic disease. Video-Assisted Thorascopic Surgery (VATS) and right lower lobe wedge resection were performed and findings confirmed metastatic PEComa.
Given the patient’s intolerance to everolimus, he was started on the growth factor inhibitor, pazopanib. His disease did not progress on pazopanib, and improvement was noted in the dominant pulmonary nodule. Subsequently, however, he developed significant skin irritation and discontinued pazopanib. Repeat imaging approximately 2 months after stopping pazopanib showed significant disease progression.
We elected to start the patient on a non-standard approach to therapy with nivolumab infusions once every 2 weeks and concurrent radiation therapy to the rib lesion. At 2 and 5 months after initiating this treatment approach, CT imaging showed improvement in disease. At 12 months, significant disease response was noted (Figure 1).
The patient is now at 12 months of nivolumab therapy with progression free survival and no new identifiable metastatic lesions. He has been tolerating the medication with minimal side effects and has had an overall improvement in his pain and functional status. He continues to work full time.
Discussion
Our patient’s response presents a unique opportunity to talk about the role of immunotherapy as a treatment modality in patients with PEComa. The efficacy of check-point blockade in soft tissue sarcoma is still unclear predominantly because it is difficult to assess the degree of expression of immunogenic cell surface markers such as programmed cell death protein 1 (PD-1).1,3 Nivolumab has been tried in small cohorts for treatment of soft tissue sarcomas that express PD-1 and results showed some clinical benefit in about half of patients.4 Further, the expression of PD-1 has been assessed in soft tissue sarcomas and has been reported to suggest a negative prognostic role.5
To our knowledge, there has not yet been another reported case of PEComa that has been treated with immunotherapy and achieved a sustained response. Further clinical studies need to be done to assess response to agents such as nivolumab in the treatment of PEComa to bolster our observation that nivolumab is a viable treatment option that may lead to lasting remission. Our patient’s case also brings to light the need for further inquiry into assessing the immune tumor microenvironments, particularly looking at the expression of cell surface proteins such as PD-1, as it ultimately affects treatment options. TSJ
Correspondence
REFERENCES
1. Burgess, Melissa, et al. “Immunotherapy in Sarcoma: Future Horizons.” Current Oncology Reports, vol. 17, no. 11, 2015, doi:10.1007/s11912-015-0476-7.
2. Alnajar, Hussein, et al. “Metastatic Malignant PEComa of the Leg with Identification of ATRX Mutation by next-Generation Sequencing.” Virchows Archiv (2017). https://doi:10.1007/s004280172208-x.
3. Ghosn, Marwan, et al. “Immunotherapies in Sarcoma: Updates and Future Perspectives.” World Journal of Clinical Oncology, vol. 8, no. 2, 2017, p. 145., doi:10.5306/wjco.v8.i2.145.
4. Paoluzzi, L., et al. “Response to Anti-PD1 Therapy with Nivolumab in Metastatic Sarcomas.” Clinical Sarcoma Research, vol. 6, no. 1, 2016, doi:10.1186/s13569-016 0064-0.
5. Kim, Chan, et al. “Prognostic Implications of PD-L1 Expression in Patients with Soft Tissue Sarcoma.” BMC Cancer, BioMed Central 8 July 2016.
Perivascular epithelioid cell neoplasms (PEComas) are an uncommon class of tumors consisting on histology of perivascular epithelioid cells occurring in both localized and metastatic forms at various body sites. The approach to treatment of these tumors generally involves a combination of surgical resection, chemotherapy, and/or radiation therapy.1
Case presentation and summary
A 46-year-old man presented to our institution with a non-tender, slowly enlarging, 8.3 cm mass in his right popliteal fossa. Upon biopsy, the pathologic findings were consistent with an epithelioid malignancy with melanocytic differentiation most consistent with a PEComa. Discussion of the pathologic diagnosis of our patient has been reported by the pathology group at our institution in a separate case report.2
Our patient was initially offered and refused amputation. He was started on therapy with the mechanistic Target of Rapamycin (mTOR) inhibitor everolimus, but was unable to tolerate the side effects after the first week of treatment. He then elected to monitor his symptoms clinically.
Approximately one year after his initial diagnosis, he presented to our facility with sepsis and bleeding from a now fungating tumor on his right knee. At this time, emergent above-knee amputation was performed. Re-staging images now showed the presence of multiple pulmonary nodules in his right lung as well as a lytic rib lesion, a concerning finding for metastatic disease. Video-Assisted Thorascopic Surgery (VATS) and right lower lobe wedge resection were performed and findings confirmed metastatic PEComa.
Given the patient’s intolerance to everolimus, he was started on the growth factor inhibitor, pazopanib. His disease did not progress on pazopanib, and improvement was noted in the dominant pulmonary nodule. Subsequently, however, he developed significant skin irritation and discontinued pazopanib. Repeat imaging approximately 2 months after stopping pazopanib showed significant disease progression.
We elected to start the patient on a non-standard approach to therapy with nivolumab infusions once every 2 weeks and concurrent radiation therapy to the rib lesion. At 2 and 5 months after initiating this treatment approach, CT imaging showed improvement in disease. At 12 months, significant disease response was noted (Figure 1).
The patient is now at 12 months of nivolumab therapy with progression free survival and no new identifiable metastatic lesions. He has been tolerating the medication with minimal side effects and has had an overall improvement in his pain and functional status. He continues to work full time.
Discussion
Our patient’s response presents a unique opportunity to talk about the role of immunotherapy as a treatment modality in patients with PEComa. The efficacy of check-point blockade in soft tissue sarcoma is still unclear predominantly because it is difficult to assess the degree of expression of immunogenic cell surface markers such as programmed cell death protein 1 (PD-1).1,3 Nivolumab has been tried in small cohorts for treatment of soft tissue sarcomas that express PD-1 and results showed some clinical benefit in about half of patients.4 Further, the expression of PD-1 has been assessed in soft tissue sarcomas and has been reported to suggest a negative prognostic role.5
To our knowledge, there has not yet been another reported case of PEComa that has been treated with immunotherapy and achieved a sustained response. Further clinical studies need to be done to assess response to agents such as nivolumab in the treatment of PEComa to bolster our observation that nivolumab is a viable treatment option that may lead to lasting remission. Our patient’s case also brings to light the need for further inquiry into assessing the immune tumor microenvironments, particularly looking at the expression of cell surface proteins such as PD-1, as it ultimately affects treatment options. TSJ
Correspondence
REFERENCES
1. Burgess, Melissa, et al. “Immunotherapy in Sarcoma: Future Horizons.” Current Oncology Reports, vol. 17, no. 11, 2015, doi:10.1007/s11912-015-0476-7.
2. Alnajar, Hussein, et al. “Metastatic Malignant PEComa of the Leg with Identification of ATRX Mutation by next-Generation Sequencing.” Virchows Archiv (2017). https://doi:10.1007/s004280172208-x.
3. Ghosn, Marwan, et al. “Immunotherapies in Sarcoma: Updates and Future Perspectives.” World Journal of Clinical Oncology, vol. 8, no. 2, 2017, p. 145., doi:10.5306/wjco.v8.i2.145.
4. Paoluzzi, L., et al. “Response to Anti-PD1 Therapy with Nivolumab in Metastatic Sarcomas.” Clinical Sarcoma Research, vol. 6, no. 1, 2016, doi:10.1186/s13569-016 0064-0.
5. Kim, Chan, et al. “Prognostic Implications of PD-L1 Expression in Patients with Soft Tissue Sarcoma.” BMC Cancer, BioMed Central 8 July 2016.
Perivascular epithelioid cell neoplasms (PEComas) are an uncommon class of tumors consisting on histology of perivascular epithelioid cells occurring in both localized and metastatic forms at various body sites. The approach to treatment of these tumors generally involves a combination of surgical resection, chemotherapy, and/or radiation therapy.1
Case presentation and summary
A 46-year-old man presented to our institution with a non-tender, slowly enlarging, 8.3 cm mass in his right popliteal fossa. Upon biopsy, the pathologic findings were consistent with an epithelioid malignancy with melanocytic differentiation most consistent with a PEComa. Discussion of the pathologic diagnosis of our patient has been reported by the pathology group at our institution in a separate case report.2
Our patient was initially offered and refused amputation. He was started on therapy with the mechanistic Target of Rapamycin (mTOR) inhibitor everolimus, but was unable to tolerate the side effects after the first week of treatment. He then elected to monitor his symptoms clinically.
Approximately one year after his initial diagnosis, he presented to our facility with sepsis and bleeding from a now fungating tumor on his right knee. At this time, emergent above-knee amputation was performed. Re-staging images now showed the presence of multiple pulmonary nodules in his right lung as well as a lytic rib lesion, a concerning finding for metastatic disease. Video-Assisted Thorascopic Surgery (VATS) and right lower lobe wedge resection were performed and findings confirmed metastatic PEComa.
Given the patient’s intolerance to everolimus, he was started on the growth factor inhibitor, pazopanib. His disease did not progress on pazopanib, and improvement was noted in the dominant pulmonary nodule. Subsequently, however, he developed significant skin irritation and discontinued pazopanib. Repeat imaging approximately 2 months after stopping pazopanib showed significant disease progression.
We elected to start the patient on a non-standard approach to therapy with nivolumab infusions once every 2 weeks and concurrent radiation therapy to the rib lesion. At 2 and 5 months after initiating this treatment approach, CT imaging showed improvement in disease. At 12 months, significant disease response was noted (Figure 1).
The patient is now at 12 months of nivolumab therapy with progression free survival and no new identifiable metastatic lesions. He has been tolerating the medication with minimal side effects and has had an overall improvement in his pain and functional status. He continues to work full time.
Discussion
Our patient’s response presents a unique opportunity to talk about the role of immunotherapy as a treatment modality in patients with PEComa. The efficacy of check-point blockade in soft tissue sarcoma is still unclear predominantly because it is difficult to assess the degree of expression of immunogenic cell surface markers such as programmed cell death protein 1 (PD-1).1,3 Nivolumab has been tried in small cohorts for treatment of soft tissue sarcomas that express PD-1 and results showed some clinical benefit in about half of patients.4 Further, the expression of PD-1 has been assessed in soft tissue sarcomas and has been reported to suggest a negative prognostic role.5
To our knowledge, there has not yet been another reported case of PEComa that has been treated with immunotherapy and achieved a sustained response. Further clinical studies need to be done to assess response to agents such as nivolumab in the treatment of PEComa to bolster our observation that nivolumab is a viable treatment option that may lead to lasting remission. Our patient’s case also brings to light the need for further inquiry into assessing the immune tumor microenvironments, particularly looking at the expression of cell surface proteins such as PD-1, as it ultimately affects treatment options. TSJ
Correspondence
REFERENCES
1. Burgess, Melissa, et al. “Immunotherapy in Sarcoma: Future Horizons.” Current Oncology Reports, vol. 17, no. 11, 2015, doi:10.1007/s11912-015-0476-7.
2. Alnajar, Hussein, et al. “Metastatic Malignant PEComa of the Leg with Identification of ATRX Mutation by next-Generation Sequencing.” Virchows Archiv (2017). https://doi:10.1007/s004280172208-x.
3. Ghosn, Marwan, et al. “Immunotherapies in Sarcoma: Updates and Future Perspectives.” World Journal of Clinical Oncology, vol. 8, no. 2, 2017, p. 145., doi:10.5306/wjco.v8.i2.145.
4. Paoluzzi, L., et al. “Response to Anti-PD1 Therapy with Nivolumab in Metastatic Sarcomas.” Clinical Sarcoma Research, vol. 6, no. 1, 2016, doi:10.1186/s13569-016 0064-0.
5. Kim, Chan, et al. “Prognostic Implications of PD-L1 Expression in Patients with Soft Tissue Sarcoma.” BMC Cancer, BioMed Central 8 July 2016.
Tumor lysis syndrome in an adolescent with recurrence of abdominal rhabdomyosarcoma: A case report and literature review
Introduction
Tumor lysis syndrome (TLS) is a life-threatening oncologic emergency that results when massive cell breakdown occurs either spontaneously or in response to cytotoxic chemotherapy. TLS is characterized by metabolic derangements, including hyperkalemia and hyperphosphatemia, secondary to the release of intracellular components into the systemic circulatory system. In addition, purine degradation can lead to hyperuricemia, and precipitation of calcium phosphate can result in hypocalcemia. Lactate dehydrogenase (LDH) levels are often elevated, especially in higher risk patients; however, this finding is not a specific marker for TLS.
TLS more commonly occurs in patients with rapidly proliferating hematological malignancies, such as acute leukemias with a high white blood cell count and Burkitt’s lymphoma, and is a relatively rare event in patients with solid malignancies.1-3 It is even more rare in patients with tumor recurrence.
There are few reported cases of TLS in children with solid malignancies. To our knowledge, only one case of TLS has previously been reported in a pediatric patient with abdominal rhabdomyosarcoma. We report the second such case, and what we believe to be the only reported case of TLS occurring in a pediatric patient with recurrence of a solid tumor.
Case Description
A 15-year-old male from Saudi Arabia presented to our hospital with confirmed stage IV abdominal rhabdomyosarcoma and lung metastases diagnosed in 2012. His initial treatment consisted of complete surgical resection, lung irradiation, and chemotherapy with intercalating cycles of ifosfamide/etoposide and vincristine/doxorubicin/cyclophosphamide, as per the COG-ARST0431 high-risk sarcoma protocol (NCT00354744). He completed treatment without any reported TLS in Saudi Arabia in June 2014. He had no residual tumor at the end of therapy, but six months later he was found to have an abdominal recurrence and started treatment with single-agent topotecan chemotherapy. He experienced worsening abdominal distention, pain, and difficulty voiding, prompting his family to seek further treatment options abroad.
The patient was admitted to our hospital in March 2015. Despite being severely malnourished, he was in stable condition. He was noted to have a markedly enlarged, firm, distended abdomen with dilated veins, abdominal and lower back pain, lower extremity pitting edema, and difficulty urinating.
Initial laboratory findings were unremarkable except for elevated levels of BUN (29 mg/dL), creatinine (1.69 mg/dL), and phosphorus (5.6 mg/dL). MRI revealed a large pelvic mass measuring 15.3 x 15.2 x 21.3 centimeters in transverse, anterior-posterior, and craniocaudal dimensions, respectively; with concomitant severe bilateral hydroureternephrosis (FIGURE 1).

FIGURE 1. Sagittal (A) and Axial (B) T2-weighted MR images of the pelvis (prior to initiating therapy) demonstrating a large heterogeneous mass occupying the entire pelvis. There is evidence of edema involving the soft tissues of the perineum (long arrow) and a large associated hydrocele (short arrow).
Three days following admission, the patient’s urine output decreased and his creatinine level rose rapidly. His worsening abdominal distention was attributed to growing tumor bulk and obstructive nephropathy. He required emergency placement of bilateral nephrostomy tubes. Urine output subsequently improved; although, serum creatinine remained persistently elevated.
Given his worsening condition, chemotherapy was begun three days after nephrostomy tube placement with vinorelbine, cyclophosphamide, and temsirolimus, as per COG-ARST0921 (NCT01222715), at renal-adjusted doses. Laboratory studies approximately 24 hours after chemotherapy initiation demonstrated the presence of TLS (TABLE 1). Potassium level was at the upper end of normal at 4.9 mmol/L, calcium level was decreased to 7.1 mg/dL, phosphorus level elevated to 12 mg/dL, uric acid level was markedly elevated to 19.5 mg/dL, and LDH elevated to 662 unit/L. A dose of 0.15 mg/kg of rasburicase was immediately given with a second dose repeated 14 hours later, after which the uric acid level decreased to less than 0.5 mg/dL. Sevelamer, sodium polystyrene, calcium carbonate, and magnesium gluconate were also administered to treat other electrolyte imbalances. The patient remained at clinical baseline throughout, and the TLS laboratory derangements normalized by three days after the TLS diagnosis; LDH level normalized after one week. The patient continued with chemotherapy, per protocol, with no further TLS-related complications. Over subsequent weeks, his tumor continued to shrink dramatically. Pain related to intra-abdominal compression, lower extremity edema, and difficulty voiding resolved.
Discussion
A literature search was performed using Pubmed/Medline and Scopus from 1950 to July 2016 using key words “TLS,” “tumor lysis syndrome,” “pediatric tumor lysis syndrome,” “tumor lysis syndrome in solid malignancies,” “recurrence,” “solid tumor,” “sarcoma,” “rhabdomyosarcoma,” and their combinations. The references of relevant articles were reviewed. Baeksgaard and Sorensen,3 and Vodopivec, et al4 provide an organized review of reported cases of TLS in solid tumors until 2002 and 2011 respectively; their articles are supported by the 2014 literature review by Mirrakhimov, et al.1 Excluding our case, 13 cases of TLS have been described in pediatric patients with solid tumors, with only one occurring in patient with abdominal rhabdomyosarcoma5. Patients’ ages ranged from 2 days to 23 years; the cases are summarized in the following table (TABLE 2). To our knowledge, ours is the first case of TLS reported in association with a pediatric solid tumor recurrence.
It is important to note that the three reported cases of disseminated rhabdomyosarcoma6,7 were initially believed to be hematologic malignancies because of their presentation with lymphadenopathy, metastases to the bone marrow, and spontaneous onset of TLS. Rhabdomyosarcoma with bone marrow involvement without an obvious primary tumor is easily confused with acute leukemia, particularly of the lymphoblastic type.12 However, this disseminated-hematologic presentation of rhabdomyosarcoma differs from the solid abdominal-pelvic tumor, which we describe.
Cairo and Bishop13 categorize patients as either laboratory TLS, depicted by metabolic abnormalities alone, or clinical TLS, occurring when laboratory imbalances lead to significant, life-threatening clinical manifestations. Hyperkalemia may lead to cardiac arrhythmias such as torsades de pointes and cardiac arrest. Obstructive nephropathy can occur from the precipitation of calcium phosphate or uric acid crystals in the renal tubules. Hypocalcemia may cause neuromuscular irritability including tetany, convulsions, and altered mental status.13, 14The 2015 “Guidelines for the management of tumour lysis syndrome in adults and children with haematological malignancies on behalf of the British Committee for Standards in Haematology”4 state there are well-recognized risk factors for the development of TLS including, but not limited to, high tumor burden, tumors with rapid cell turnover, and pre-existing renal impairment. Cairo and Bishop, on behalf of the TLS expert panel consensus of 20102, classify patients as having low-risk disease (LRD), intermediate-risk disease (IRD), or high-risk disease (HRD) based on the risk factors and type of malignancy. All patients with solid tumors are classified into LRD, unless the tumors are bulky or sensitive to chemotherapy, mentioning specifically that neuroblastomas, germ-cell tumors and small cell lung cancers are classified as IRD. Cairo and Bishop take into account the risk factor of renal dysfunction/ involvement, which if present, increases the risk by one level. For example, if the patient has IRD and has renal dysfunction, risk increases to HRD2. However, these guidelines do not mention or address the significance of recurrence in any kind of malignancy with regards to assessing risk for TLS.
The British Committee’s 2015 Guidelines for management of TLS in hematologic malignancies14 provide recommendations for treatment based on the patient’s risk classification (TABLE 3). Children with HRD are recommended to be treated prophylactically with a single dose of 0.2 mg/kg of rasburicase. Patients with IRD are recommended to be offered up to 7 days of allopurinol prophylaxis with increased hydration post initiation of treatment or until risk of TLS has resolved. Patients with LRD are recommended to be managed essentially with close observation. Patients with established TLS should receive rasburicase 0.2 mg/kg/day - duration to depend on clinical response. If the patient is receiving rasburicase, the addition of allopurinol is not recommended, as it has the potential to reduce the effectiveness of rasburicase. Further, rasburicase is to be avoided in patients with glucose-6-phosphate dehydrogenase (G6PD) deficiency14.
Our patient likely developed TLS because of a fast growing tumor that caused significant tumor burden and renal involvement, indicated by an elevated phosphorus level. Despite these risk factors, TLS was not anticipated in the case presented; therefore, a uric acid level was not collected at the time of admission. Review of the literature indicates that the incidence of TLS in a solid tumor recurrence is either unheard of, or is likely under-reported and truly unknown. Further, the TLS expert panel consensus of 20102, which provides guidelines on risk assessment for TLS, does not address the risk of TLS in a malignancy recurrence. The British Committee’s 2015 guidelines14 also do not address hyperuricemia prophylaxis in a solid tumor recurrence.
Our case presents a question regarding the degree of risk for the development of TLS in a solid tumor recurrence. If the guidelines had existed at the time of the case presentation and had been applied, our patient would likely be classified as having IRD because of his renal involvement. This classification would have lead to a different course of management when initiating chemotherapy, likely prevented laboratory TLS, and provided more cost effective treatment, as rasburicase is known to be expensive.
On the other hand, it can also be argued that our patient classifies as LRD, considering the rarity of TLS in a solid tumor recurrence, that the patient had no TLS complication with his initial course of therapy, and also had a normal LDH on admission. LDH is sometimes used to assess risk in hematological malignancies, although it is not used to make the diagnosis of TLS2. However, with such an argument, it is assumed that the risk of TLS in a solid tumor malignancy recurrence, with no previous TLS complication, is less than the risk associated with a new-onset solid tumor malignancy when, truly, the actual risk is not known. Again, the question is raised of the degree of risk for the development of TLS in a case of a malignancy recurrence, and also in a pediatric patient with risk factors.
In our patient’s case, close observation allowed for prompt diagnosis, appropriate treatment of laboratory TLS, and prevented clinical symptoms from developing. However, a screening or baseline uric acid level may have lead to a more conservative approach towards hyperuricemia prophylaxis, similar to treating the patient as IRD. Therefore, we recommend that a screening or baseline uric acid level and LDH level be obtained when initiating chemotherapy, even in patients with LRD.
Our patient was never hyperkalemic, likely because of concomitant administration of furosemide in an attempt to improve his decreased urine output. Hyperuricemia dropped from 19.5 mg/dL to less than 0.5 mg/dL within 24 hours, following two doses of 0.15 mg/kg of rasburicase, confirming the efficacy of this therapy in cases of established TLS, as is recommended by the British Committee’s 2015 guidelines.14
Conclusion
TLS is a relatively rare event in patients with solid malignancies and even more rare in a tumor recurrence. While there is only one previously reported case of TLS occurring in a pediatric patient with abdominal rhabdomyosarcoma, there are not any reported cases to date of TLS occurring in pediatric solid tumor recurrence. This may be because the incidence is truly rare or because cases may be under-reported. Thus, a question is raised regarding the risk for TLS in a solid tumor recurrence, and moreover in a pediatric patient with pre-existing risk factors, such as renal involvement.
TLS remains a life-threatening emergency that can be prevented and reversed if a high index of suspicion is maintained. We recommend all patients with malignancies receiving chemotherapy, especially those with risk factors, have a baseline or screening uric acid and LDH level drawn, as part of the assessment and risk-stratification for TLS which should always be performed. TSJ
Correspondence
References
1. Mirrakhimov AE, Ali AM, Khan M, et al. Tumor lysis syndrome in solid tumors: an up to date review of the literature. Rare Tumors. 2014;6:68-74.
2. Cairo MS, Bertrand C, Reiter A, et al. Recommendation for the evaluation of risk and prophylaxis of tumour lysis syndrome (TLS) in adults and children with malignant diseases: an expert TLS panel consensus. Br J Haematol. 2010;149:578-586.
3. Baeksgaard L, Sorensen JB. Acute tumor lysis syndrome in solid tumors – a case report and review of the literature. Cancer Chemother Pharmacol. 2003;51:187-192.
4. Vodopivec D, Rubio J, Fornoni A, et al. An unusual presentation of tumor lysis syndrome in a patient with advanced gastric adenocarcinoma: case report and literature review. Case Rep Med. 2012;2012:1-12.
5. Khan J, Broadbent VA. Tumor lysis syndrome complicating treatment of widespread metastatic abdominal rhabdomyosarcoma. Pediatr Hematol Oncol. 1993;10:151-155.
6. Bien E, Maciejka-Kapuscinka L, Niedzwiecki M, et al. Childhood rhabdomyosarcoma metastatic to bone marrow presenting with disseminated intravascular coagulation and acute tumour lysis syndrome: review of the literature apropos of two cases. Clin Exp Metastasis. 2010;27:399-407.
7. Patiroglu T, Isik B, Unal E, et al. Cranial metastatic alveolar rhabdomyosarcoma mimicking hematological malignancy in an adolescent boy. Childs Nerv Syst. 2014;30:1737-1741.
8. Hain RD, Rayner L, Weitzman S, et al. Acute tumour lysis syndrome complicating treatment of stage IVS neuroblastoma in infants under six months old. Med Pediatr Oncol. 1994;23:136-139.
9. Kushner BH, LaQuaglia MP, Modak S, et al. Tumor lysis syndrome, neuroblastoma, and correlation between serum lactate dehydrogenase levels and MYCN-amplification. Med Pediatr Oncol. 2003;41:80-82.
10. Bercovitz RS, Greffe BS, Hunger SP. Acute tumor lysis syndrome in a 7-month-old with hepatoblastoma. Curr Opin Pediatr. 2010;22:113-116.
11. Lobe TE, Karkera MS, Custer MD, et al. Fatal refractory hyperkalemia due to tumor lysis during primary resection for hepatoblastoma. J Pediatr Surg. 1990;25:249-250.
12. Sandberg A, Stone J, Czarnecki L, et al. Hematologic Masquerade of Rhabdomyosarcoma. Am J Hematol. 2001;68:51-57
13. Cairo M, Bishop M. Tumour lysis syndrome: new therapeutic strategies and classification. Br J Haematol. 2004;127:3-11.
14. Jones G, Will A, Jackson GH, et al. Guidelines for the management of tumour lysis syndrome in adults and children with haematological malignancies on behalf of the British Committee for Standards in Haematology. Br J Haematol. 2015;169:661-671.
References
1. Mirrakhimov AE, Ali AM, Khan M, et al. Tumor lysis syndrome in solid tumors: an up to date review of the literature. Rare Tumors. 2014;6:68-74.
2. Cairo MS, Bertrand C, Reiter A, et al. Recommendation for the evaluation of risk and prophylaxis of tumour lysis syndrome (TLS) in adults and children with malignant diseases: an expert TLS panel consensus. Br J Haematol. 2010;149:578-586.
3. Baeksgaard L, Sorensen JB. Acute tumor lysis syndrome in solid tumors – a case report and review of the literature. Cancer Chemother Pharmacol. 2003;51:187-192.
4. Vodopivec D, Rubio J, Fornoni A, et al. An unusual presentation of tumor lysis syndrome in a patient with advanced gastric adenocarcinoma: case report and literature review. Case Rep Med. 2012;2012:1-12.
5. Khan J, Broadbent VA. Tumor lysis syndrome complicating treatment of widespread metastatic abdominal rhabdomyosarcoma. Pediatr Hematol Oncol. 1993;10:151-155.
6. Bien E, Maciejka-Kapuscinka L, Niedzwiecki M, et al. Childhood rhabdomyosarcoma metastatic to bone marrow presenting with disseminated intravascular coagulation and acute tumour lysis syndrome: review of the literature apropos of two cases. Clin Exp Metastasis. 2010;27:399-407.
7. Patiroglu T, Isik B, Unal E, et al. Cranial metastatic alveolar rhabdomyosarcoma mimicking hematological malignancy in an adolescent boy. Childs Nerv Syst. 2014;30:1737-1741.
8. Hain RD, Rayner L, Weitzman S, et al. Acute tumour lysis syndrome complicating treatment of stage IVS neuroblastoma in infants under six months old. Med Pediatr Oncol. 1994;23:136-139.
9. Kushner BH, LaQuaglia MP, Modak S, et al. Tumor lysis syndrome, neuroblastoma, and correlation between serum lactate dehydrogenase levels and MYCN-amplification. Med Pediatr Oncol. 2003;41:80-82.
10. Bercovitz RS, Greffe BS, Hunger SP. Acute tumor lysis syndrome in a 7-month-old with hepatoblastoma. Curr Opin Pediatr. 2010;22:113-116.
11. Lobe TE, Karkera MS, Custer MD, et al. Fatal refractory hyperkalemia due to tumor lysis during primary resection for hepatoblastoma. J Pediatr Surg. 1990;25:249-250.
12. Sandberg A, Stone J, Czarnecki L, et al. Hematologic Masquerade of Rhabdomyosarcoma. Am J Hematol. 2001;68:51-57
13. Cairo M, Bishop M. Tumour lysis syndrome: new therapeutic strategies and classification. Br J Haematol. 2004;127:3-11.
14. Jones G, Will A, Jackson GH, et al. Guidelines for the management of tumour lysis syndrome in adults and children with haematological malignancies on behalf of the British Committee for Standards in Haematology. Br J Haematol. 2015;169:661-671.
Introduction
Tumor lysis syndrome (TLS) is a life-threatening oncologic emergency that results when massive cell breakdown occurs either spontaneously or in response to cytotoxic chemotherapy. TLS is characterized by metabolic derangements, including hyperkalemia and hyperphosphatemia, secondary to the release of intracellular components into the systemic circulatory system. In addition, purine degradation can lead to hyperuricemia, and precipitation of calcium phosphate can result in hypocalcemia. Lactate dehydrogenase (LDH) levels are often elevated, especially in higher risk patients; however, this finding is not a specific marker for TLS.
TLS more commonly occurs in patients with rapidly proliferating hematological malignancies, such as acute leukemias with a high white blood cell count and Burkitt’s lymphoma, and is a relatively rare event in patients with solid malignancies.1-3 It is even more rare in patients with tumor recurrence.
There are few reported cases of TLS in children with solid malignancies. To our knowledge, only one case of TLS has previously been reported in a pediatric patient with abdominal rhabdomyosarcoma. We report the second such case, and what we believe to be the only reported case of TLS occurring in a pediatric patient with recurrence of a solid tumor.
Case Description
A 15-year-old male from Saudi Arabia presented to our hospital with confirmed stage IV abdominal rhabdomyosarcoma and lung metastases diagnosed in 2012. His initial treatment consisted of complete surgical resection, lung irradiation, and chemotherapy with intercalating cycles of ifosfamide/etoposide and vincristine/doxorubicin/cyclophosphamide, as per the COG-ARST0431 high-risk sarcoma protocol (NCT00354744). He completed treatment without any reported TLS in Saudi Arabia in June 2014. He had no residual tumor at the end of therapy, but six months later he was found to have an abdominal recurrence and started treatment with single-agent topotecan chemotherapy. He experienced worsening abdominal distention, pain, and difficulty voiding, prompting his family to seek further treatment options abroad.
The patient was admitted to our hospital in March 2015. Despite being severely malnourished, he was in stable condition. He was noted to have a markedly enlarged, firm, distended abdomen with dilated veins, abdominal and lower back pain, lower extremity pitting edema, and difficulty urinating.
Initial laboratory findings were unremarkable except for elevated levels of BUN (29 mg/dL), creatinine (1.69 mg/dL), and phosphorus (5.6 mg/dL). MRI revealed a large pelvic mass measuring 15.3 x 15.2 x 21.3 centimeters in transverse, anterior-posterior, and craniocaudal dimensions, respectively; with concomitant severe bilateral hydroureternephrosis (FIGURE 1).
FIGURE 1. Sagittal (A) and Axial (B) T2-weighted MR images of the pelvis (prior to initiating therapy) demonstrating a large heterogeneous mass occupying the entire pelvis. There is evidence of edema involving the soft tissues of the perineum (long arrow) and a large associated hydrocele (short arrow).
Three days following admission, the patient’s urine output decreased and his creatinine level rose rapidly. His worsening abdominal distention was attributed to growing tumor bulk and obstructive nephropathy. He required emergency placement of bilateral nephrostomy tubes. Urine output subsequently improved; although, serum creatinine remained persistently elevated.
Given his worsening condition, chemotherapy was begun three days after nephrostomy tube placement with vinorelbine, cyclophosphamide, and temsirolimus, as per COG-ARST0921 (NCT01222715), at renal-adjusted doses. Laboratory studies approximately 24 hours after chemotherapy initiation demonstrated the presence of TLS (TABLE 1). Potassium level was at the upper end of normal at 4.9 mmol/L, calcium level was decreased to 7.1 mg/dL, phosphorus level elevated to 12 mg/dL, uric acid level was markedly elevated to 19.5 mg/dL, and LDH elevated to 662 unit/L. A dose of 0.15 mg/kg of rasburicase was immediately given with a second dose repeated 14 hours later, after which the uric acid level decreased to less than 0.5 mg/dL. Sevelamer, sodium polystyrene, calcium carbonate, and magnesium gluconate were also administered to treat other electrolyte imbalances. The patient remained at clinical baseline throughout, and the TLS laboratory derangements normalized by three days after the TLS diagnosis; LDH level normalized after one week. The patient continued with chemotherapy, per protocol, with no further TLS-related complications. Over subsequent weeks, his tumor continued to shrink dramatically. Pain related to intra-abdominal compression, lower extremity edema, and difficulty voiding resolved.
Discussion
A literature search was performed using Pubmed/Medline and Scopus from 1950 to July 2016 using key words “TLS,” “tumor lysis syndrome,” “pediatric tumor lysis syndrome,” “tumor lysis syndrome in solid malignancies,” “recurrence,” “solid tumor,” “sarcoma,” “rhabdomyosarcoma,” and their combinations. The references of relevant articles were reviewed. Baeksgaard and Sorensen,3 and Vodopivec, et al4 provide an organized review of reported cases of TLS in solid tumors until 2002 and 2011 respectively; their articles are supported by the 2014 literature review by Mirrakhimov, et al.1 Excluding our case, 13 cases of TLS have been described in pediatric patients with solid tumors, with only one occurring in patient with abdominal rhabdomyosarcoma5. Patients’ ages ranged from 2 days to 23 years; the cases are summarized in the following table (TABLE 2). To our knowledge, ours is the first case of TLS reported in association with a pediatric solid tumor recurrence.
It is important to note that the three reported cases of disseminated rhabdomyosarcoma6,7 were initially believed to be hematologic malignancies because of their presentation with lymphadenopathy, metastases to the bone marrow, and spontaneous onset of TLS. Rhabdomyosarcoma with bone marrow involvement without an obvious primary tumor is easily confused with acute leukemia, particularly of the lymphoblastic type.12 However, this disseminated-hematologic presentation of rhabdomyosarcoma differs from the solid abdominal-pelvic tumor, which we describe.
Cairo and Bishop13 categorize patients as either laboratory TLS, depicted by metabolic abnormalities alone, or clinical TLS, occurring when laboratory imbalances lead to significant, life-threatening clinical manifestations. Hyperkalemia may lead to cardiac arrhythmias such as torsades de pointes and cardiac arrest. Obstructive nephropathy can occur from the precipitation of calcium phosphate or uric acid crystals in the renal tubules. Hypocalcemia may cause neuromuscular irritability including tetany, convulsions, and altered mental status.13, 14The 2015 “Guidelines for the management of tumour lysis syndrome in adults and children with haematological malignancies on behalf of the British Committee for Standards in Haematology”4 state there are well-recognized risk factors for the development of TLS including, but not limited to, high tumor burden, tumors with rapid cell turnover, and pre-existing renal impairment. Cairo and Bishop, on behalf of the TLS expert panel consensus of 20102, classify patients as having low-risk disease (LRD), intermediate-risk disease (IRD), or high-risk disease (HRD) based on the risk factors and type of malignancy. All patients with solid tumors are classified into LRD, unless the tumors are bulky or sensitive to chemotherapy, mentioning specifically that neuroblastomas, germ-cell tumors and small cell lung cancers are classified as IRD. Cairo and Bishop take into account the risk factor of renal dysfunction/ involvement, which if present, increases the risk by one level. For example, if the patient has IRD and has renal dysfunction, risk increases to HRD2. However, these guidelines do not mention or address the significance of recurrence in any kind of malignancy with regards to assessing risk for TLS.
The British Committee’s 2015 Guidelines for management of TLS in hematologic malignancies14 provide recommendations for treatment based on the patient’s risk classification (TABLE 3). Children with HRD are recommended to be treated prophylactically with a single dose of 0.2 mg/kg of rasburicase. Patients with IRD are recommended to be offered up to 7 days of allopurinol prophylaxis with increased hydration post initiation of treatment or until risk of TLS has resolved. Patients with LRD are recommended to be managed essentially with close observation. Patients with established TLS should receive rasburicase 0.2 mg/kg/day - duration to depend on clinical response. If the patient is receiving rasburicase, the addition of allopurinol is not recommended, as it has the potential to reduce the effectiveness of rasburicase. Further, rasburicase is to be avoided in patients with glucose-6-phosphate dehydrogenase (G6PD) deficiency14.
Our patient likely developed TLS because of a fast growing tumor that caused significant tumor burden and renal involvement, indicated by an elevated phosphorus level. Despite these risk factors, TLS was not anticipated in the case presented; therefore, a uric acid level was not collected at the time of admission. Review of the literature indicates that the incidence of TLS in a solid tumor recurrence is either unheard of, or is likely under-reported and truly unknown. Further, the TLS expert panel consensus of 20102, which provides guidelines on risk assessment for TLS, does not address the risk of TLS in a malignancy recurrence. The British Committee’s 2015 guidelines14 also do not address hyperuricemia prophylaxis in a solid tumor recurrence.
Our case presents a question regarding the degree of risk for the development of TLS in a solid tumor recurrence. If the guidelines had existed at the time of the case presentation and had been applied, our patient would likely be classified as having IRD because of his renal involvement. This classification would have lead to a different course of management when initiating chemotherapy, likely prevented laboratory TLS, and provided more cost effective treatment, as rasburicase is known to be expensive.
On the other hand, it can also be argued that our patient classifies as LRD, considering the rarity of TLS in a solid tumor recurrence, that the patient had no TLS complication with his initial course of therapy, and also had a normal LDH on admission. LDH is sometimes used to assess risk in hematological malignancies, although it is not used to make the diagnosis of TLS2. However, with such an argument, it is assumed that the risk of TLS in a solid tumor malignancy recurrence, with no previous TLS complication, is less than the risk associated with a new-onset solid tumor malignancy when, truly, the actual risk is not known. Again, the question is raised of the degree of risk for the development of TLS in a case of a malignancy recurrence, and also in a pediatric patient with risk factors.
In our patient’s case, close observation allowed for prompt diagnosis, appropriate treatment of laboratory TLS, and prevented clinical symptoms from developing. However, a screening or baseline uric acid level may have lead to a more conservative approach towards hyperuricemia prophylaxis, similar to treating the patient as IRD. Therefore, we recommend that a screening or baseline uric acid level and LDH level be obtained when initiating chemotherapy, even in patients with LRD.
Our patient was never hyperkalemic, likely because of concomitant administration of furosemide in an attempt to improve his decreased urine output. Hyperuricemia dropped from 19.5 mg/dL to less than 0.5 mg/dL within 24 hours, following two doses of 0.15 mg/kg of rasburicase, confirming the efficacy of this therapy in cases of established TLS, as is recommended by the British Committee’s 2015 guidelines.14
Conclusion
TLS is a relatively rare event in patients with solid malignancies and even more rare in a tumor recurrence. While there is only one previously reported case of TLS occurring in a pediatric patient with abdominal rhabdomyosarcoma, there are not any reported cases to date of TLS occurring in pediatric solid tumor recurrence. This may be because the incidence is truly rare or because cases may be under-reported. Thus, a question is raised regarding the risk for TLS in a solid tumor recurrence, and moreover in a pediatric patient with pre-existing risk factors, such as renal involvement.
TLS remains a life-threatening emergency that can be prevented and reversed if a high index of suspicion is maintained. We recommend all patients with malignancies receiving chemotherapy, especially those with risk factors, have a baseline or screening uric acid and LDH level drawn, as part of the assessment and risk-stratification for TLS which should always be performed. TSJ
Correspondence
References
1. Mirrakhimov AE, Ali AM, Khan M, et al. Tumor lysis syndrome in solid tumors: an up to date review of the literature. Rare Tumors. 2014;6:68-74.
2. Cairo MS, Bertrand C, Reiter A, et al. Recommendation for the evaluation of risk and prophylaxis of tumour lysis syndrome (TLS) in adults and children with malignant diseases: an expert TLS panel consensus. Br J Haematol. 2010;149:578-586.
3. Baeksgaard L, Sorensen JB. Acute tumor lysis syndrome in solid tumors – a case report and review of the literature. Cancer Chemother Pharmacol. 2003;51:187-192.
4. Vodopivec D, Rubio J, Fornoni A, et al. An unusual presentation of tumor lysis syndrome in a patient with advanced gastric adenocarcinoma: case report and literature review. Case Rep Med. 2012;2012:1-12.
5. Khan J, Broadbent VA. Tumor lysis syndrome complicating treatment of widespread metastatic abdominal rhabdomyosarcoma. Pediatr Hematol Oncol. 1993;10:151-155.
6. Bien E, Maciejka-Kapuscinka L, Niedzwiecki M, et al. Childhood rhabdomyosarcoma metastatic to bone marrow presenting with disseminated intravascular coagulation and acute tumour lysis syndrome: review of the literature apropos of two cases. Clin Exp Metastasis. 2010;27:399-407.
7. Patiroglu T, Isik B, Unal E, et al. Cranial metastatic alveolar rhabdomyosarcoma mimicking hematological malignancy in an adolescent boy. Childs Nerv Syst. 2014;30:1737-1741.
8. Hain RD, Rayner L, Weitzman S, et al. Acute tumour lysis syndrome complicating treatment of stage IVS neuroblastoma in infants under six months old. Med Pediatr Oncol. 1994;23:136-139.
9. Kushner BH, LaQuaglia MP, Modak S, et al. Tumor lysis syndrome, neuroblastoma, and correlation between serum lactate dehydrogenase levels and MYCN-amplification. Med Pediatr Oncol. 2003;41:80-82.
10. Bercovitz RS, Greffe BS, Hunger SP. Acute tumor lysis syndrome in a 7-month-old with hepatoblastoma. Curr Opin Pediatr. 2010;22:113-116.
11. Lobe TE, Karkera MS, Custer MD, et al. Fatal refractory hyperkalemia due to tumor lysis during primary resection for hepatoblastoma. J Pediatr Surg. 1990;25:249-250.
12. Sandberg A, Stone J, Czarnecki L, et al. Hematologic Masquerade of Rhabdomyosarcoma. Am J Hematol. 2001;68:51-57
13. Cairo M, Bishop M. Tumour lysis syndrome: new therapeutic strategies and classification. Br J Haematol. 2004;127:3-11.
14. Jones G, Will A, Jackson GH, et al. Guidelines for the management of tumour lysis syndrome in adults and children with haematological malignancies on behalf of the British Committee for Standards in Haematology. Br J Haematol. 2015;169:661-671.
Introduction
Tumor lysis syndrome (TLS) is a life-threatening oncologic emergency that results when massive cell breakdown occurs either spontaneously or in response to cytotoxic chemotherapy. TLS is characterized by metabolic derangements, including hyperkalemia and hyperphosphatemia, secondary to the release of intracellular components into the systemic circulatory system. In addition, purine degradation can lead to hyperuricemia, and precipitation of calcium phosphate can result in hypocalcemia. Lactate dehydrogenase (LDH) levels are often elevated, especially in higher risk patients; however, this finding is not a specific marker for TLS.
TLS more commonly occurs in patients with rapidly proliferating hematological malignancies, such as acute leukemias with a high white blood cell count and Burkitt’s lymphoma, and is a relatively rare event in patients with solid malignancies.1-3 It is even more rare in patients with tumor recurrence.
There are few reported cases of TLS in children with solid malignancies. To our knowledge, only one case of TLS has previously been reported in a pediatric patient with abdominal rhabdomyosarcoma. We report the second such case, and what we believe to be the only reported case of TLS occurring in a pediatric patient with recurrence of a solid tumor.
Case Description
A 15-year-old male from Saudi Arabia presented to our hospital with confirmed stage IV abdominal rhabdomyosarcoma and lung metastases diagnosed in 2012. His initial treatment consisted of complete surgical resection, lung irradiation, and chemotherapy with intercalating cycles of ifosfamide/etoposide and vincristine/doxorubicin/cyclophosphamide, as per the COG-ARST0431 high-risk sarcoma protocol (NCT00354744). He completed treatment without any reported TLS in Saudi Arabia in June 2014. He had no residual tumor at the end of therapy, but six months later he was found to have an abdominal recurrence and started treatment with single-agent topotecan chemotherapy. He experienced worsening abdominal distention, pain, and difficulty voiding, prompting his family to seek further treatment options abroad.
The patient was admitted to our hospital in March 2015. Despite being severely malnourished, he was in stable condition. He was noted to have a markedly enlarged, firm, distended abdomen with dilated veins, abdominal and lower back pain, lower extremity pitting edema, and difficulty urinating.
Initial laboratory findings were unremarkable except for elevated levels of BUN (29 mg/dL), creatinine (1.69 mg/dL), and phosphorus (5.6 mg/dL). MRI revealed a large pelvic mass measuring 15.3 x 15.2 x 21.3 centimeters in transverse, anterior-posterior, and craniocaudal dimensions, respectively; with concomitant severe bilateral hydroureternephrosis (FIGURE 1).
FIGURE 1. Sagittal (A) and Axial (B) T2-weighted MR images of the pelvis (prior to initiating therapy) demonstrating a large heterogeneous mass occupying the entire pelvis. There is evidence of edema involving the soft tissues of the perineum (long arrow) and a large associated hydrocele (short arrow).
Three days following admission, the patient’s urine output decreased and his creatinine level rose rapidly. His worsening abdominal distention was attributed to growing tumor bulk and obstructive nephropathy. He required emergency placement of bilateral nephrostomy tubes. Urine output subsequently improved; although, serum creatinine remained persistently elevated.
Given his worsening condition, chemotherapy was begun three days after nephrostomy tube placement with vinorelbine, cyclophosphamide, and temsirolimus, as per COG-ARST0921 (NCT01222715), at renal-adjusted doses. Laboratory studies approximately 24 hours after chemotherapy initiation demonstrated the presence of TLS (TABLE 1). Potassium level was at the upper end of normal at 4.9 mmol/L, calcium level was decreased to 7.1 mg/dL, phosphorus level elevated to 12 mg/dL, uric acid level was markedly elevated to 19.5 mg/dL, and LDH elevated to 662 unit/L. A dose of 0.15 mg/kg of rasburicase was immediately given with a second dose repeated 14 hours later, after which the uric acid level decreased to less than 0.5 mg/dL. Sevelamer, sodium polystyrene, calcium carbonate, and magnesium gluconate were also administered to treat other electrolyte imbalances. The patient remained at clinical baseline throughout, and the TLS laboratory derangements normalized by three days after the TLS diagnosis; LDH level normalized after one week. The patient continued with chemotherapy, per protocol, with no further TLS-related complications. Over subsequent weeks, his tumor continued to shrink dramatically. Pain related to intra-abdominal compression, lower extremity edema, and difficulty voiding resolved.
Discussion
A literature search was performed using Pubmed/Medline and Scopus from 1950 to July 2016 using key words “TLS,” “tumor lysis syndrome,” “pediatric tumor lysis syndrome,” “tumor lysis syndrome in solid malignancies,” “recurrence,” “solid tumor,” “sarcoma,” “rhabdomyosarcoma,” and their combinations. The references of relevant articles were reviewed. Baeksgaard and Sorensen,3 and Vodopivec, et al4 provide an organized review of reported cases of TLS in solid tumors until 2002 and 2011 respectively; their articles are supported by the 2014 literature review by Mirrakhimov, et al.1 Excluding our case, 13 cases of TLS have been described in pediatric patients with solid tumors, with only one occurring in patient with abdominal rhabdomyosarcoma5. Patients’ ages ranged from 2 days to 23 years; the cases are summarized in the following table (TABLE 2). To our knowledge, ours is the first case of TLS reported in association with a pediatric solid tumor recurrence.
It is important to note that the three reported cases of disseminated rhabdomyosarcoma6,7 were initially believed to be hematologic malignancies because of their presentation with lymphadenopathy, metastases to the bone marrow, and spontaneous onset of TLS. Rhabdomyosarcoma with bone marrow involvement without an obvious primary tumor is easily confused with acute leukemia, particularly of the lymphoblastic type.12 However, this disseminated-hematologic presentation of rhabdomyosarcoma differs from the solid abdominal-pelvic tumor, which we describe.
Cairo and Bishop13 categorize patients as either laboratory TLS, depicted by metabolic abnormalities alone, or clinical TLS, occurring when laboratory imbalances lead to significant, life-threatening clinical manifestations. Hyperkalemia may lead to cardiac arrhythmias such as torsades de pointes and cardiac arrest. Obstructive nephropathy can occur from the precipitation of calcium phosphate or uric acid crystals in the renal tubules. Hypocalcemia may cause neuromuscular irritability including tetany, convulsions, and altered mental status.13, 14The 2015 “Guidelines for the management of tumour lysis syndrome in adults and children with haematological malignancies on behalf of the British Committee for Standards in Haematology”4 state there are well-recognized risk factors for the development of TLS including, but not limited to, high tumor burden, tumors with rapid cell turnover, and pre-existing renal impairment. Cairo and Bishop, on behalf of the TLS expert panel consensus of 20102, classify patients as having low-risk disease (LRD), intermediate-risk disease (IRD), or high-risk disease (HRD) based on the risk factors and type of malignancy. All patients with solid tumors are classified into LRD, unless the tumors are bulky or sensitive to chemotherapy, mentioning specifically that neuroblastomas, germ-cell tumors and small cell lung cancers are classified as IRD. Cairo and Bishop take into account the risk factor of renal dysfunction/ involvement, which if present, increases the risk by one level. For example, if the patient has IRD and has renal dysfunction, risk increases to HRD2. However, these guidelines do not mention or address the significance of recurrence in any kind of malignancy with regards to assessing risk for TLS.
The British Committee’s 2015 Guidelines for management of TLS in hematologic malignancies14 provide recommendations for treatment based on the patient’s risk classification (TABLE 3). Children with HRD are recommended to be treated prophylactically with a single dose of 0.2 mg/kg of rasburicase. Patients with IRD are recommended to be offered up to 7 days of allopurinol prophylaxis with increased hydration post initiation of treatment or until risk of TLS has resolved. Patients with LRD are recommended to be managed essentially with close observation. Patients with established TLS should receive rasburicase 0.2 mg/kg/day - duration to depend on clinical response. If the patient is receiving rasburicase, the addition of allopurinol is not recommended, as it has the potential to reduce the effectiveness of rasburicase. Further, rasburicase is to be avoided in patients with glucose-6-phosphate dehydrogenase (G6PD) deficiency14.
Our patient likely developed TLS because of a fast growing tumor that caused significant tumor burden and renal involvement, indicated by an elevated phosphorus level. Despite these risk factors, TLS was not anticipated in the case presented; therefore, a uric acid level was not collected at the time of admission. Review of the literature indicates that the incidence of TLS in a solid tumor recurrence is either unheard of, or is likely under-reported and truly unknown. Further, the TLS expert panel consensus of 20102, which provides guidelines on risk assessment for TLS, does not address the risk of TLS in a malignancy recurrence. The British Committee’s 2015 guidelines14 also do not address hyperuricemia prophylaxis in a solid tumor recurrence.
Our case presents a question regarding the degree of risk for the development of TLS in a solid tumor recurrence. If the guidelines had existed at the time of the case presentation and had been applied, our patient would likely be classified as having IRD because of his renal involvement. This classification would have lead to a different course of management when initiating chemotherapy, likely prevented laboratory TLS, and provided more cost effective treatment, as rasburicase is known to be expensive.
On the other hand, it can also be argued that our patient classifies as LRD, considering the rarity of TLS in a solid tumor recurrence, that the patient had no TLS complication with his initial course of therapy, and also had a normal LDH on admission. LDH is sometimes used to assess risk in hematological malignancies, although it is not used to make the diagnosis of TLS2. However, with such an argument, it is assumed that the risk of TLS in a solid tumor malignancy recurrence, with no previous TLS complication, is less than the risk associated with a new-onset solid tumor malignancy when, truly, the actual risk is not known. Again, the question is raised of the degree of risk for the development of TLS in a case of a malignancy recurrence, and also in a pediatric patient with risk factors.
In our patient’s case, close observation allowed for prompt diagnosis, appropriate treatment of laboratory TLS, and prevented clinical symptoms from developing. However, a screening or baseline uric acid level may have lead to a more conservative approach towards hyperuricemia prophylaxis, similar to treating the patient as IRD. Therefore, we recommend that a screening or baseline uric acid level and LDH level be obtained when initiating chemotherapy, even in patients with LRD.
Our patient was never hyperkalemic, likely because of concomitant administration of furosemide in an attempt to improve his decreased urine output. Hyperuricemia dropped from 19.5 mg/dL to less than 0.5 mg/dL within 24 hours, following two doses of 0.15 mg/kg of rasburicase, confirming the efficacy of this therapy in cases of established TLS, as is recommended by the British Committee’s 2015 guidelines.14
Conclusion
TLS is a relatively rare event in patients with solid malignancies and even more rare in a tumor recurrence. While there is only one previously reported case of TLS occurring in a pediatric patient with abdominal rhabdomyosarcoma, there are not any reported cases to date of TLS occurring in pediatric solid tumor recurrence. This may be because the incidence is truly rare or because cases may be under-reported. Thus, a question is raised regarding the risk for TLS in a solid tumor recurrence, and moreover in a pediatric patient with pre-existing risk factors, such as renal involvement.
TLS remains a life-threatening emergency that can be prevented and reversed if a high index of suspicion is maintained. We recommend all patients with malignancies receiving chemotherapy, especially those with risk factors, have a baseline or screening uric acid and LDH level drawn, as part of the assessment and risk-stratification for TLS which should always be performed. TSJ
Correspondence
References
1. Mirrakhimov AE, Ali AM, Khan M, et al. Tumor lysis syndrome in solid tumors: an up to date review of the literature. Rare Tumors. 2014;6:68-74.
2. Cairo MS, Bertrand C, Reiter A, et al. Recommendation for the evaluation of risk and prophylaxis of tumour lysis syndrome (TLS) in adults and children with malignant diseases: an expert TLS panel consensus. Br J Haematol. 2010;149:578-586.
3. Baeksgaard L, Sorensen JB. Acute tumor lysis syndrome in solid tumors – a case report and review of the literature. Cancer Chemother Pharmacol. 2003;51:187-192.
4. Vodopivec D, Rubio J, Fornoni A, et al. An unusual presentation of tumor lysis syndrome in a patient with advanced gastric adenocarcinoma: case report and literature review. Case Rep Med. 2012;2012:1-12.
5. Khan J, Broadbent VA. Tumor lysis syndrome complicating treatment of widespread metastatic abdominal rhabdomyosarcoma. Pediatr Hematol Oncol. 1993;10:151-155.
6. Bien E, Maciejka-Kapuscinka L, Niedzwiecki M, et al. Childhood rhabdomyosarcoma metastatic to bone marrow presenting with disseminated intravascular coagulation and acute tumour lysis syndrome: review of the literature apropos of two cases. Clin Exp Metastasis. 2010;27:399-407.
7. Patiroglu T, Isik B, Unal E, et al. Cranial metastatic alveolar rhabdomyosarcoma mimicking hematological malignancy in an adolescent boy. Childs Nerv Syst. 2014;30:1737-1741.
8. Hain RD, Rayner L, Weitzman S, et al. Acute tumour lysis syndrome complicating treatment of stage IVS neuroblastoma in infants under six months old. Med Pediatr Oncol. 1994;23:136-139.
9. Kushner BH, LaQuaglia MP, Modak S, et al. Tumor lysis syndrome, neuroblastoma, and correlation between serum lactate dehydrogenase levels and MYCN-amplification. Med Pediatr Oncol. 2003;41:80-82.
10. Bercovitz RS, Greffe BS, Hunger SP. Acute tumor lysis syndrome in a 7-month-old with hepatoblastoma. Curr Opin Pediatr. 2010;22:113-116.
11. Lobe TE, Karkera MS, Custer MD, et al. Fatal refractory hyperkalemia due to tumor lysis during primary resection for hepatoblastoma. J Pediatr Surg. 1990;25:249-250.
12. Sandberg A, Stone J, Czarnecki L, et al. Hematologic Masquerade of Rhabdomyosarcoma. Am J Hematol. 2001;68:51-57
13. Cairo M, Bishop M. Tumour lysis syndrome: new therapeutic strategies and classification. Br J Haematol. 2004;127:3-11.
14. Jones G, Will A, Jackson GH, et al. Guidelines for the management of tumour lysis syndrome in adults and children with haematological malignancies on behalf of the British Committee for Standards in Haematology. Br J Haematol. 2015;169:661-671.
References
1. Mirrakhimov AE, Ali AM, Khan M, et al. Tumor lysis syndrome in solid tumors: an up to date review of the literature. Rare Tumors. 2014;6:68-74.
2. Cairo MS, Bertrand C, Reiter A, et al. Recommendation for the evaluation of risk and prophylaxis of tumour lysis syndrome (TLS) in adults and children with malignant diseases: an expert TLS panel consensus. Br J Haematol. 2010;149:578-586.
3. Baeksgaard L, Sorensen JB. Acute tumor lysis syndrome in solid tumors – a case report and review of the literature. Cancer Chemother Pharmacol. 2003;51:187-192.
4. Vodopivec D, Rubio J, Fornoni A, et al. An unusual presentation of tumor lysis syndrome in a patient with advanced gastric adenocarcinoma: case report and literature review. Case Rep Med. 2012;2012:1-12.
5. Khan J, Broadbent VA. Tumor lysis syndrome complicating treatment of widespread metastatic abdominal rhabdomyosarcoma. Pediatr Hematol Oncol. 1993;10:151-155.
6. Bien E, Maciejka-Kapuscinka L, Niedzwiecki M, et al. Childhood rhabdomyosarcoma metastatic to bone marrow presenting with disseminated intravascular coagulation and acute tumour lysis syndrome: review of the literature apropos of two cases. Clin Exp Metastasis. 2010;27:399-407.
7. Patiroglu T, Isik B, Unal E, et al. Cranial metastatic alveolar rhabdomyosarcoma mimicking hematological malignancy in an adolescent boy. Childs Nerv Syst. 2014;30:1737-1741.
8. Hain RD, Rayner L, Weitzman S, et al. Acute tumour lysis syndrome complicating treatment of stage IVS neuroblastoma in infants under six months old. Med Pediatr Oncol. 1994;23:136-139.
9. Kushner BH, LaQuaglia MP, Modak S, et al. Tumor lysis syndrome, neuroblastoma, and correlation between serum lactate dehydrogenase levels and MYCN-amplification. Med Pediatr Oncol. 2003;41:80-82.
10. Bercovitz RS, Greffe BS, Hunger SP. Acute tumor lysis syndrome in a 7-month-old with hepatoblastoma. Curr Opin Pediatr. 2010;22:113-116.
11. Lobe TE, Karkera MS, Custer MD, et al. Fatal refractory hyperkalemia due to tumor lysis during primary resection for hepatoblastoma. J Pediatr Surg. 1990;25:249-250.
12. Sandberg A, Stone J, Czarnecki L, et al. Hematologic Masquerade of Rhabdomyosarcoma. Am J Hematol. 2001;68:51-57
13. Cairo M, Bishop M. Tumour lysis syndrome: new therapeutic strategies and classification. Br J Haematol. 2004;127:3-11.
14. Jones G, Will A, Jackson GH, et al. Guidelines for the management of tumour lysis syndrome in adults and children with haematological malignancies on behalf of the British Committee for Standards in Haematology. Br J Haematol. 2015;169:661-671.
References
1. Mirrakhimov AE, Ali AM, Khan M, et al. Tumor lysis syndrome in solid tumors: an up to date review of the literature. Rare Tumors. 2014;6:68-74.
2. Cairo MS, Bertrand C, Reiter A, et al. Recommendation for the evaluation of risk and prophylaxis of tumour lysis syndrome (TLS) in adults and children with malignant diseases: an expert TLS panel consensus. Br J Haematol. 2010;149:578-586.
3. Baeksgaard L, Sorensen JB. Acute tumor lysis syndrome in solid tumors – a case report and review of the literature. Cancer Chemother Pharmacol. 2003;51:187-192.
4. Vodopivec D, Rubio J, Fornoni A, et al. An unusual presentation of tumor lysis syndrome in a patient with advanced gastric adenocarcinoma: case report and literature review. Case Rep Med. 2012;2012:1-12.
5. Khan J, Broadbent VA. Tumor lysis syndrome complicating treatment of widespread metastatic abdominal rhabdomyosarcoma. Pediatr Hematol Oncol. 1993;10:151-155.
6. Bien E, Maciejka-Kapuscinka L, Niedzwiecki M, et al. Childhood rhabdomyosarcoma metastatic to bone marrow presenting with disseminated intravascular coagulation and acute tumour lysis syndrome: review of the literature apropos of two cases. Clin Exp Metastasis. 2010;27:399-407.
7. Patiroglu T, Isik B, Unal E, et al. Cranial metastatic alveolar rhabdomyosarcoma mimicking hematological malignancy in an adolescent boy. Childs Nerv Syst. 2014;30:1737-1741.
8. Hain RD, Rayner L, Weitzman S, et al. Acute tumour lysis syndrome complicating treatment of stage IVS neuroblastoma in infants under six months old. Med Pediatr Oncol. 1994;23:136-139.
9. Kushner BH, LaQuaglia MP, Modak S, et al. Tumor lysis syndrome, neuroblastoma, and correlation between serum lactate dehydrogenase levels and MYCN-amplification. Med Pediatr Oncol. 2003;41:80-82.
10. Bercovitz RS, Greffe BS, Hunger SP. Acute tumor lysis syndrome in a 7-month-old with hepatoblastoma. Curr Opin Pediatr. 2010;22:113-116.
11. Lobe TE, Karkera MS, Custer MD, et al. Fatal refractory hyperkalemia due to tumor lysis during primary resection for hepatoblastoma. J Pediatr Surg. 1990;25:249-250.
12. Sandberg A, Stone J, Czarnecki L, et al. Hematologic Masquerade of Rhabdomyosarcoma. Am J Hematol. 2001;68:51-57
13. Cairo M, Bishop M. Tumour lysis syndrome: new therapeutic strategies and classification. Br J Haematol. 2004;127:3-11.
14. Jones G, Will A, Jackson GH, et al. Guidelines for the management of tumour lysis syndrome in adults and children with haematological malignancies on behalf of the British Committee for Standards in Haematology. Br J Haematol. 2015;169:661-671.